/

























































































































































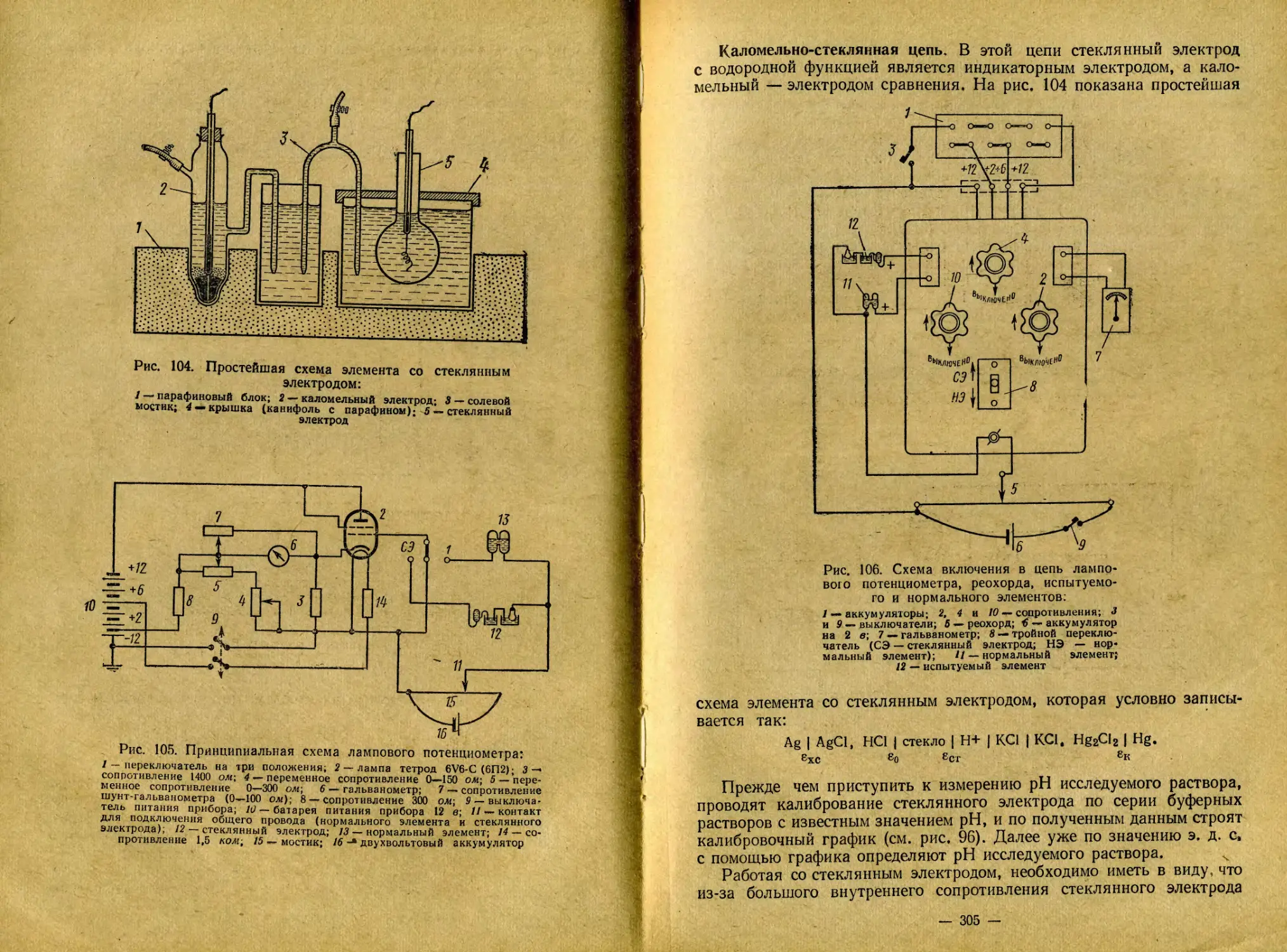









































































































Text
А.И. БОЛДЫРЕВ
ФИЗИЧЕСКАЯ
И КОЛЛОИДНАЯ ХИМИЯ
А. И. БОЛДЫРЕВ
ФИЗИЧЕСКАЯ
И КОЛЛОИДНАЯ ХИМИЯ
Допущено
Министерством сельского хозяйства СССР в качестве учебного пособия для студентов агрономических и агрохимических специальностей сельскохозяйственных вузов
МОСКВА «ВЫСШАЯ ШКОЛА» 1974 г
541
Б79
УДК 541.1+541.18(075)
Рецензенты: проф. Л. А. Николаев (МИИТ) и кафедра химии Херсонского сельскохозяйственного ин-та им. А. Д. Цюрупы (зав. кафедрой проф. Б. 3. Рудой)
Анатолий Иванович Болдырев ФИЗИЧЕСКАЯ И КОЛЛОИДНАЯ ХИМИЯ Редактор Г. С. Гольденберг, Технический редактор Н. Н. Баранова, Художественный редактор Н. Е. Алешина. Художник М. Н. Бакушев, Корректор Р. К. Косинова
Сдано в набор 22/Х — 73 г. Подп. к печати 20/V — 1974 г.
Формат 60Х901/1в. Объем 31,5 печ. л. Бум. тип. № 3
Уч.-изд. л. 35,63 ( усл. печ. л. 31,5) Изд. № ХИМ-474
Тираж 40 000 экз. Цена 1 р. 13 коп.
План выпуска литературы изд-ва «Высшая школа» (вузы и техникумы) на 1974 г. Позиция № 69 Москва, К-51, Неглинная ул., 29/14 Издательство «Высшая школа»
Московская типография № 4 «Союзполиграфпрома» при Государственном койитете Совета Министров СССР по делам издательств, полиграфии и книжной торговли, Москва, И-41, Б. Переяславская» 46 Зак. 560
Болдырев А.И.
Б79 . Физическая и коллоидная химия. Учеб, пособие для с.-х. вузов. М., «Высш, школа», 1974.
504 с. с ил.
Излагается курс физической и коллоидной химии для сельскохозяйственных вузов. Агрегатные состояния вещества, современное уче-. ниё о растворах, явления диффузии и осмоса; тургора и плазмолиза, электропроводность растворов, основы химической термодинамики и термохимии, вопросы химической кинетики и катализа и химических равновесий, электрохимия рассмотрены с точки зрения их приложения в биологии и сельском хозяйстве. Рассмотрены также коллоидно-химические свойства белков, роль свободной воды в коллоидах, коллоидно-химические свойства протоплазмы, свойства коллоидов почвы.
Б 20503-091 69.74
001(01)74
541
© Издательство «Высшая школа», 1974.
ПРЕДИСЛОВИЕ
Предлагаемое пособие «Физическая и коллоидная химия» предназначено для студентов сельскохозяйственных высших учебных заведений и составлено в основном в соответствии с программой, утвержденной Министерством высшего и среднего специального образования СССР. Оно рассчитано на подготовку агрономов всех специальностей, включая агрохимиков и почвоведов.
В связи с широким развитием заочного образования автор при составлении данной книги учитывал специфику самостоятельной работы студентов-заочников, которые практически не имеют возможности про-. слушать систематический курс лекций по данной дисциплине. Поэтому автор стремился излагать материал в наиболее доступной форме, -по возможности не перегружать его математическим аппаратом, иллюстрировать теоретические положения примерами из почвоведения, агрохимии, биохимии, физиологии растений и других смежных наук. Во всех случаях автор свою главную задачу видел в раскрытии физической сущности рассматриваемых теоретических положений, в указании их практического значения и применения в различных областях того сложного биологического процесса, которым является наше технически вооруженное сельское хозяйство.
За последние годы в практике агрохимических и почвенных лабораторий все более широкое распространение получают различные физико-химические методы анализа, знание которых необходимо для агронома-агрохимика и почвоведа. Поэтому автор счел необходимым изложить в специальной главе теоретические основы современных физико-химических методов исследования, а также их применение для анализа растений, почв, удобрений, ядохимикатов и других сельскохозяйственных объектов. Эта глава имеет очень важное значение для студентов факультета почвоведения и агрохимии. На других факультетах она может быть опущена.
Во всех случаях материал, отсутствующий в действующей программе для агрономических специальностей, но необходимый для студентов-агрохимиков и почвоведов, набран мелким шрифтом.
В настоящей книге изложены все основные разделы физической и коллоидной химии, знание которых необходимо студентам для более
— 3 —
углубленного изучения в последующем специальных дисциплин, таких, как агрохимия, почвоведение, биохимия, физиология растений и т. п. Исключение составляет раздел физической химии «Строение атомов и молекул», который не включен в данную книгу, так как он подробно излагается в курсе общей химии.
Автор заранее благодарен всем лицам, которые выскажут ему свои критические замечания по книге. Все эти замечания будут тщательно проанализированы и учтены в дальнейшей работе над книгой.
Автор выражает свою глубокую признательность рецензентам — проф. Л. А. Николаеву и проф. Б. 3. Рудому — за существенные замечания, сделанные при просмотре рукописи.
Л. Болдырев
Часть первая ФИЗИЧЕСКАЯ ХИМИЯ
ВВЕДЕНИЕ
Предмет физической химии и ее значение для промышленности и сельского хозяйства. Физическая химия — наука, объясняющая химические явления на основании физических принципов и законов. В настоящее время она определилась как самостоятельная отрасль науки, обладающая специфическими методами исследования. Физическая химия занимается многосторонним исследованием различных химических реакций и сопутствующих им физических процессов. Как пограничная наука она изучает объект с нескольких сторон, учитывая диалектический характер взаимосвязи и взаимодействия сложных явлений материального мира.
Химические реакции тесно связаны с такими физическими процессами как электрические явления, теплопередача, поглощение или излучение электромагнитных колебаний. Например, химические реакции, протекающие в гальванических элементах и аккумуляторах, являются причиной возникновения электрического тока. Многие химические реакции сопровождаются выделением или поглощением энергии в виде теплоты, возникновение же других реакций обусловлено действием света. Так, поглощение солнечного света зелеными растениями вызывает сложные реакции фотосинтеза, в результате которых из двуокиси углерода и воды .образуются различные органические соединения. Таким образом, физическая химия решает наиболее общие вопросы химии, опираясь на физические законы и методы исследования.
Физическая химия является важной учебной дисциплиной в химических, химико-технологических, горнометаллургических, медицинских, сельскохозяйственных и других высших учебных заведениях.
Физическая химия включает несколько основных разделов, характеризующих направление этой науки и определяющих ее предмет.
Строение вещества. Этот раздел физической химии изучает связь между строением веществ и их физическими и химическими свойствами, а также агрегатные состояния веществ. На основании результатов, полученных физикой при изучении атомов и молекул, а также достижений квантовой механики в этой области за последние годы наблюдаются значительные успехи. Благодаря применению в экспериментальных работах новейших методов молекулярной спектроскопии (включая радиоспектроскопию), а также рентгеноструктурного, электроннографического, электронномикроскопического и других методов исследо
— 5
вания получены новые данные о строении атомов и молекул, о природе сил, действующих между ними.
В учении об агрегатных состояниях рассматриваются вопросы взаимодействия молекул и важнейшие свойства веществ, находящихся в газообразном, жидком и кристаллическом состоянии. Это учение получило свое развитие на базе кинетической теории материи и статистической физики.
Химическая термодинамика. Этот раздел физической химии позволяет на основе законов термодинамики проводить энергетические расчеты химических реакций и химического равновесия, а также определять возможность и направление самопроизвольного течения того или иного химического процесса. Химическая термодинамика изучает фазовые переходы (растворение, испарение, кристаллизацию и др.), адсорбцию и т. п. Важным разделом химической термодинамики является термохимия, которая изучает тепловые эффекты химических реакций. Этот раздел физической химии имеет большое значение в народном хозяйстве, особенно в области промышленного синтеза.
Учение о растворах. В этом разделе изучаются молекулярные структуры растворов, различные их свойства, процессы образования растворов и особенности протекающих в них реакций, а также вопросы растворимости. >
Электрохимия. Рассматривает важнейшие процессы взаимного превращения электрической и химической форм движения материи, а также свойства и строение растворов электролитов, процессы электролиза, работу гальванических элементов, электрохимическую коррозию металлов, электросинтез веществ и др. В настоящее время электрохимические методы исследования и анализа приобретают все большее значение в практике заводских, агрохимических, почвенных и других лабораторий.
Химическая кинетика — это учение о скоростях химических реакций и их зависимости от температуры, давления, концентрации, среды, перемешивания и т. д. Она рассматривает также вопросы катализа гомогенных и гетерогенных химических реакций и способы, позволяющие регулировать и направлять течение различных химических процессов, а также выход продуктов реакции. В этом разделе физической химии рассматривается также механизм действия биологических катализаторов — ферментов.
Фотохимия. Фотохимия изучает процессы воздействия электромагнитных излучений на ход химических превращений. Важными фотохимическими реакциями являются фотосинтез, люминесценция, фотографические и многие другие процессы. Фотохимия тесно связана с учением о строении молекул, а также с химической*кинетикой.
Таковы основные разделы физической химии. Приведенное деление условно, поскольку каждый физико-химический процесс не только многогранен, но и тесно связан с целым рядом других явлений. Марксистско-ленинский диалектический метод учит, что любое явление материального мира необходимо рассматривать в его тесной и неразрывной связи с окружающими явлениями. Этим объясняется и возникновение физической химии и тесная взаимосвязь различных ее разделов.
— ь —
Необходимо учесть, что всякое реальное движение материи сложно и едино, а разделение его на отдельные формы движения условно и относительно. В силу этого границы между основными разделами физической химии также приблизительны и условны.
Указанные разделы не охватывают всех областей физической химии. В последние годы в самостоятельные разделы выделены магнетохимия, радиационная химия, физико-химия высокомолекулярных соединений и др.
Физическая химия не только всесторонне изучает и обобщает материал по различным разделам химии, она объединяет его, анализирует и выводит общие закономерности, лежащие в основе развития вечно движущейся материи. В этом заключается общенаучное значение физической химии. Законы, открываемые ею, широко используются общей химией, биологией, геологией, агрохимией, почвоведением и многими прикладными науками.
Велико значение физической химии и методов ее исследования в развиТйй“Химичёской технологий. Знакомство с физической химией дает инженеру возможность не только глубоко понять сущность химического процесса, лежащего в основе производства, но и сознательно выбирать и регулировать условия, наиболее благоприятные для проведения нужных процессов. Физическая химия позволяет предвидеть направление химической реакции, а также рассчитать теоретически выход ее продуктов.
Физическая химия как наука призвана сыграть в решении проблемы химизации сельского хозяйства одну из первостепенных ролей. Производство новых и высокоэффективных удобрений, разработка и внедрение химических способов борьбы с вредителями и болезнями растений, улучшение водно-физических свойств почвы — эти вопросы могут быть успешно решены лишь на основе знания физической химии. Это убедительно доказали работы советских агрохимиков К. К. Гедрой-ца и Д. Н. Прянишникова. На основании их обширных и разносторонних исследований с применением методов физической химии было создано учение о почвенном поглощающем комплексе, которое получило широкое признание в нашей стране и за рубежом.
В настоящее время все большее распространение получают такие физико-химические методы исследования, как термический, рентгенографический, электронномикроскопический, инфракрасноспектроскопический и многие другие, с помощью которых были получены весьма ценные данные о природе и строении почвенного поглощающего комплекса,. Методы электропроводности, потенциометрии, криоскопии, фотометрии, эмиссионного анализа и другие также широко применяются в решении основных проблем агрономии.
f ^Изучение с помощью физической химии фотохимических реакций позволяет глубже вникать в сущность сложных процессов фотосинтеза.
С Как известно, из всей солнечной энергии, доходящей до поверхности земли, энергия, усваиваемая за счет фотосинтеза всей растительностью земного шара, составляет в среднем только 0,3%. Культурные растения используют солнечную энергию полнее, чем дикая растительность Земли в целом. Используемая ими доля солнечной энер
гии составляет примерно 0,5—1,5%, а для таких культур, как рис, соевые бобы, сахарная свекла, сахарный тростник, кукуруза и некоторых других, 4—5% от общего количества солнечной энергии, попадающей на посевы за вегетационный период. Есть основание считать, что полное раскрытие наукой механизма процесса фотосинтеза и овладение управлением им даст возможность повысить коэффициент использования солнечной энергии растениями в два-три и более раз.
Такие дисциплины, как агрохимия, почвоведение, физиология растений, микробиология, биохимия, земледелие, защита растений и многие другие, широко используют методы и основные теоретические положения физической химии.
Преподавание физической химии в сельскохозяйственных высших учебных заведениях имеет свою специфику. Основной задачей курса является изучение теоретических основ этой науки, знакомство с физико-химическими методами исследования с целью их применения в решении основных вопросов сельского хозяйства.
Роль русских и советски * ученых в развитии физической химии. Основателем физической химии является великий русский ученый М.аВ. Ломоносов (1711 —1765). Ему принадлежит сам термин «физическая химия»; определение задач этой науки было дано им еще в 1752 г., когда он приступил к чтению систематического курса физической химии для студентов Академии наук. Ломоносовым было написано первое учебное пособие «Введение в истинную физическую химию», а также составлена программа экспериментальных работ — «Опыт физической химии» (1754). Под его непосредственным руководством студенты не только проходили лабораторный практикум, но и выполняли дипломные работы по физической химии.
Задачи, которые должна решить физическая химия, Ломоносов сформулировал следующим образом: «Физическая химия есть наука, объясняющая на основании положений и опытов физики то, что происходит в смешанных телах при химических операциях». Это определение физической химии очень близко к современному.
Ломоносов выдвинул и обосновал целый* ряд положений, которые легли в основу физической химии и не потеряли своего значения и сейчас. Им впервые был открыт закон сохранения материи и движения — один из величайших законов природы.
Ломоносов создал стройную кинетическую теорию материи и объяснил теплоту как проявление движения молекул. Он первым указал на невозможность перехода теплоты от холодного тела к горячему, и тем самым вплотную подошел к формулировке второго закона термодинамики. Ломоносов впервые ввел в науку представление о молекулах и установил четкое различие между молекулами н атомами. Он объяснил природу газового состояния, высказал мысль о существовании абсолютного нуля температуры, дал правильное толкование процесса растворения как проявления взаимодействия молекул растворенного вещества с молекулами растворителя, выполнил целый ряд обстоятельных работ по изучению растворов.
Таким образом, Ломоносов является не только основателем физической химии, но и первым в мире физико-химиком.
Русский ученый Г. И. Гесс (1802—1850) профессор Горного института в Петербурге впервые сформулировал основной закон термохимии о постоянстве сумм тепла при химических реакциях. Этот закон, впоследствии названной его именем, следует рассматривать как одно из выражений открытого позднее первого закона термодинамики применительно к химическим реакциям.
Большая заслуга в дальнейшем развитии физической химии принадлежит русскому ученому Н. Н. Бекетову (1826—1911), который с 1865 г. возобновил после Ломоносова чтение курса физической химии; он впервые (1865) дал частную формулировку закона действующих масс. Значительный вклад в разви
— 8 —
тие физической химии внесли работы Бекетова по изучению восстанавливающей способности одних металлов по отношению к другим.
Первый в мире учебник по физической химии также был создан в России Н. Н. Л юб а в и н ы м. Он вышел в 1876—1877 гг. и выдержал два издания.
Ценный вклад в развитие представлений о строении вещества внес основоположник теории химического строения органических соединений А. М. Бутлеров (1828—1886).
Для развития химической науки вообще и для физической химии в частности огромное значение имело открытие Д. И. Менделеевым (1834—1907) периодического закона химических элементов (1869), впоследствии названного его именем. Этот закон позволил на основании знания химических свойств одних элементов предвидеть свойства других. Оценивая это открытие Д. И. Менделеева, Ф. Энгельс писал: «Менделеев, применив бессознательно гегелевский закон о переходе количества в качество, совершил научный подвиг, который смело можно поставить рядом с открытием Леверье, вычислившего орбиту еще Неизвестной планеты — Нептуна»*.
Менделеев является также автором гидратной, теории растворов, на которой основаны современные исследования в области растворов.
Большая роль в развитии сельского хозяйства в России принадлежит трудам Д-. И. Менделеева в области агрохимии. Он впервые поставил задачу широкого использования химии для подъема отечественного сельского хозяйства. Можно с полным правом утверждать: если бы Менделеев не открыл и не разработал периодический закон химических элементов, его имя вошло бы в историю науки и народного хозяйства благодаря замечательным трудам в области сельского хозяйства, особенно в области удобрений.
Известный русский ученый К. А. Тимирязев проходил сельскохозяйственную практику под руководством Д. И. Менделеева, а Д. Н. Прянишников был учеником и последователем К. А. Тимирязева. Благодаря такой преемственности возникло целое направление в отечественной агрохимии, которое сыграло выдающуюся роль в широком творческом проникновении химии в сельское хозяйство, в его всесторонней химизации (выражение Д.’Н. Прянишникова).
Известную роль в развитии физической химии сыграли работы И. А. Каблукова (1857—1942), который, исходя из гидратной теории Д. И. Менделеева, установил явление гидратации ионов электролитов в водных растворах и сущность химического взаимодействия в процессах электролитической диссоциации (1891). Им впервые были выполнены работы по исследованию поведения электролитов в неводных растворах. Каблуков организовал первую кафедру физической химии в сельскохозяйственном вузе и начал читать систематический курс физической химии будущим агрономам.
После Великой Октябрьской социалистической революции физическая химия стала развиваться особенно бурно.
Работы Н. С. Курнакова — создателя физико-химического анализа, Н. Д. Зелинского — основателя научной школы органического катализа, Н. А. Шилова, выполнившего ряд важных работ в области кинетики сопряженных химических реакций и адсорбции растворенных веществ из растворов, а также других ученых заложили прочный фундамент в развитии физической химии. Крупный вклад в развитие физической химии внесли исследования Н. Н. Семенова, разработавшего теорию цепных разветвленных реакций, П. А. Ребиндер, А. Н. Фрумкин, М. М. Дубинин и другие советские ученые в исследованиях, охватывающих область поверхностных явлений и адсорбции. Огромное практическое значение для повышения плодородия почв имели исследования К. К. Гедройна — создателя учения о почвенном поглощающем комплексе, а также Д. Н. Прянишникова.
* Ф. Энгельс. Диалектика природы. Политиздат, 1965, стр. 49.
«Химик без знания физики подобен человеку, который всего искать должен ощупом. И сии две науки так соединены между собой, что одна без другой в совершенстве быть не могут».
/И. В. Ломоносов
Глава I
МОЛЕКУЛЯРНО-КИНЕТИЧЕСКАЯ ТЕОРИЯ ТРЕХ АГРЕГАТНЫХ СОСТОЯНИЙ ВЕЩЕСТВА
§ 1. Агрегатные состояния вещества. Понятие о плазме
В зависимости от внешних условий (температуры и давления) почти каждое вещество может находиться в газообразном, жидком или твердом состоянии. Это есть агрегатные состояния вещества. Агрегатное состояние характеризуется двумя факторами — силами межмолекулярного взаимодействия и кинетической энергией теплового движения молекул.
Вещество в газообразном состоянии обладает способностью беспрепятственно расширяться и стремится занять весь предоставленный ему объем. Молекулы газа находятся на сравнительно больших расстояниях друг от друга, поэтому силы взаимодействия между ними очень, слабые. При повышении давления газы легко изменяют свой объем.
Жидкость принимает форму заключающего ее сосуда, но сохраняет постоянным свой объем. В жидкостях молекулы более сближены, чем в газах. Силы межмолекулярных взаимодействий в жидкостях больше, чем в газах.
Однако молекулы их не закреплены в определенных точках пространства и аналогично газам находятся в хаотическом поступательном движении. При повышении давления жидкости лишь незначительно изменяют свой объем, поскольку этому препятствуют силы электростатического отталкивания, увеличивающиеся при сближении молекул. Иными словами, жидкости являются практически несжимаемыми веществами.
Твердое вещество имеет собственную форму и объем. Силы притяжения между молекулами в твердых телах уравновешиваются силами отталкивания. Этим объясняется геометрически правильное расположение частиц кристалла в определенных точках пространства, образующих пространственную кристаллическую решетку. Частицы твердого тела утрачивают свободу поступательного движения и испытывают только колебательные движения, находясь в узлах кристаллической решетки.
— Ю —
Не все вещества способны находиться во всех трех агрегатных со-
стояниях. Для некоторых веществ возможны только одно или два агрегатных состояния. Так, карбонат кальция СаСО3 практически невоз-
можно получить ни в жидком, ни в газ
разном состояниях, посколь-
ку при нагревании он разлагается на нелетучую окись кальция и газообразную двуокись углерода. Другие вещества при определенных условиях могут находиться одновременно в двух или даже трех агрегатных состояниях. Так, вода при давлении 4,579 мм рт. ст. (6,14 х X 102«/л<2)и температуре 0,0075° С находится в устойчивом равновесии в трех состояниях: твердом (лед), жидком (жидкая вода) и газообразном (водяной пар).
Переход вещества из жидкого состояния в газообразное называется парообразованием, из твердого в газообразное —сублимацией и из твердого в жидкое — плавлением. Обратные процессы перехода соответственно носят название сжижения, десублимации и отвердевания. Все эти процессы, как правило, сопровождаются выделением теплоты (теплоты парообразования, сублимации, плавления и т. д.).
Постепенно изменяя внешние условия (температуру и давление), можно осуществлять переход из одного агрегатного состояния в другое.
С изменением температуры и давления постепенно изменяются расстояния между частицами в веществе, что оказывает влияние на количественную сторону, а на определенном этапе в результате скачка вещество приобретает новое качество, т. е. переходит в новое агрегатное состояние. Агрегатные состояния, — по выражению Энгельса, — узловые точки, где количественное изменение переходит в качественное.
Кроме перечисленных выше трех состояний вещество может находиться в четвертом агрегатном состоянии — плазменном, которое открыто сравнительно недавно. Состояние плазмы возникает в том случае, если на вещество в газообразном состоянии действуют такие сильные ионизирующие факторы, как сверхвысокие температуры (в несколько миллионов градусов), мощные электрические разряды или электромагнитные излучения. При этом происходит разрушение молекул и атомов вещества и превращение его в смесь, состоящую из положительно заряженных ядер и электронов, движущихся с колоссальными скоростями. По этой причине плазму иногда называют электронно-ядерным газом.
Различают два вида плазмы: изотермическую и газоразрядную.
Изотермическая плазма получается при высоких температурах, под влиянием которых имеет место термическая диссоциация атомов вещества, и может существовать неограниченно долго. Такой вид плазмы представляет собой вещество звезд, а также шаровых молний.
Ионосфера Земли — это также особая разновидность плазмы; однако в данном случае ионизациия происходит под влиянием ультрафиолетового излучения Солнца.
Изотермическая плазма играет исключительно важную роль в космических процессах. Три других агрегатных состояния вещества в космическом пространстве являются исключением.
Газоразрядная плазма образуется при электрическом разряде и поэтому устойчива только при наличии электрического поля. Как только
н
прекращается действие внешнего поля, газоразрядная плазма вследствие образования нейтральных атомов из ионов и электронов исчезает в течение 10’5 •—10~4 сек.
Одним из замечательных свойств плазмы является ее высокая электропроводность. Чем выше температура плазмы, тем выше ее электропроводность. В силу этого через плазму можно пропускать токи в сотни тысяч и миллионы ампер.
При прохождении через плазму электрический ток создает вокруг нее сильное магнитное поле, которое сжимает поток электронов и ионов в плазменный шнур. Этим достигается тепловая изоляция плазмы от стенок сосуда. С увеличением силы тока электромагнитное сжатие плазмы проявляется сильнее.
' При пропускании через плазму токов большой величины можно поднять ее температуру до миллиона градусов и выше, а давление — до десятка миллиардов атмосфер. Подобные условия, как известно, благоприятны для проведения термоядерных реакций синтеза.
В Советском Союзе в настоящее время ведутся работы по осуществлению управляемой термоядерной реакции. Пионером в этой области был академик И. В. Курчатов. При условии успешного завершения этих работ человечество получит новый неисчерпаемый источник энергии.
ГАЗООБРАЗНОЕ (ПАРООБРАЗНОЕ) СОСТОЯНИЕ ВЕЩЕСТВА
Сила взаимодействия между молекулами, как известно, зависит от расстояния, на котором они находятся друг от друга. Силы меж-мэлекулярного взаимодействия (так называемые силы когезии) за пределами расстояния большего 10~7 см от центра молекулы настолько ослабевают, что ими можно пренебречь.
Средней кинетической энергии теплового движения молекул газа вполне достаточно, чтобы преодолеть силы когезии (силы межмолекулярного взаимодействия). При столкновении молекул друг с другом, когда одна молекула попадает в силовое поле притяжения другой молекулы, между ними возникает мгновенное когезионное взаимодействие. Однако из-за больших скоростей молекулы не могут сколько-нибудь длительно удерживаться друг возле друга и быстро разлетаются в разные стороны до следующего соударения с другими молекулами и т. д. Число этих соударений в I сек для одной молекулы равно 109. Вот почему газ путем почти беспрепятственной диффузии может равномерно распределяться по всему объему, который ему предоставлен {текучесть газа).
Чем больше давление, тем больше число столкновений молекул газа в 1 сек, а следовательно, и суммарные мгновенные когезионные взаимодействия между ними оказывают большее влияние на свойства данного газа. И наоборот, в состоянии сильного разрежения размеры молекул по сравнению с межмолекулярными расстояниями и силы взаимодействия между молекулами ничтожны.
В силу хаотического движения молекул газа концентрация их в любой части занимаемого газом пространства одинакова, как и плотность его во всей массе. * *
12 —
Однако это справедливо лишь в том случае, если речь идет о средней плотности газа в макрообъеме. В микрообъеме же вследствие хаотичности молекулярного движения могут иметь место значительные отклонения от средней плотности. Это явление называется флуктуацией (лат. f luctuatio — колебание). Флуктуационные отклонения плотности тем больше, чем меньше микрообъем. В отдельных случаях они могут достигать 20% и более по сравнению со средней плотностью газа во всей его массе.
Газы широко распространены в природе и используются в различных отраслях народного хозяйства в качестве топлива, теплоносителей, сырья для химической промышленности, рабочего тела для выполнения механической работы (газовые турбины) и во многих других случаях. Отсюда вытекает необходимость знания законов, которым подчиняются газы.
§ 2. Основные газовые законы
Основные газовые законы выведены для идеального газа.
Идеальным называется газ, находящийся в таком состоянии, при котором можно пренебречь силами межмолекулярного взаимодействия и собственным объемом его молекул.
Свойства идеального газа, таким образом, определяются температурой и давлением, при которых газ находится в данный момент.
Газы, реально существующие в природе (реальные газы), в большей или меньшей степени отступают от газовых законов (см. стр. 23).
Закон Бойля — Мариотта. Объем данной массы газа (V) при постоянной температуре изменяется обратно пропорционально давлению (Р), под которым газ находится:
Vi е
0,1)
или
PiV1C=P2V2.
Аналогичное равенство можно написать и для других значений Р и V:
Р4 V4 = const
Л V!
Л V1 = P2V2c=P3V3c=f
Отбросив индексы, получим
PV=const
(1,2)
при условии, что / = const.
Таким образом, произведение объема газа на его давление при постоянной температуре есть величина постоянная. Величина константы в уравнении (I, 2) зависит от природы газа, его количества и температуры, но не зависит от изменения объема или изменения давления.
На рис. 1 приведено графическое изображение закона Бойля—Мариотта в системе координат Р—V. Кривая в данном случае представляет собой равностороннюю гиперболу, асимптотически приближающую
— 13 —
ся к осям координат. Для любой точки такой гиперболы произведения величин абсциссы на ординату равны между собой. Для точек Л, В, С и D, изображенных на рис. 1, эти произведения выражаются равными площадями соответствующих прямоугольников.
На рис. 2 показано изображение закона Бойля — Мариотта в системе координат PV — Р (V). График представляет собой ряд прямых линий А, В, С и т. д., параллельных оси абсцисс. Из него следует, что для данной температуры произведение PV постоянно и не зависит от изменений давления (или объема ) газа.
Линии, выражающие зависимость изменения объема от давления при постоянной температуре, носят название изотерм. Прямые, характеризующие зависимость произведения PV от Р (или V) при постоянной температуре, также являются изотермами.
Рис. 1. Изотерма идеал ь- Рис. 2.. Изотермы идеаль-
ного газа в координатах кого газа в координатах
Р— V PV— P(V)
' ' s ' 1
Из закона Бойля — Мариотта вытекает следующее: концентрация и плотность данной массы газа изменяются при постоянной температуре прямо пропорционально изменению давления и обратно пропорционально изменению объема.
Таким образом, исходя из уравнения (1,1) можно записать:
С2 Р, 9
^2
d2 “ Р2 ’
(1.3)
(М)
где Сх, С2 и d19 d2 — соответственно концентрации и плотности данной массы газа.
Закон Гей-Люссака. При нагревании данной массы газа на F при постоянном давлении объем его увеличивается на 1/273,15 часть того объема^ каким обладал бы газ при СР С и при том же давлении.
Так, если объем газа при 0° С был V©, при нагревании газа на t градусов стал а прирост объема ДУ, то
Z/«J, !□
— 14 —
или
vz = J/n/l -ь —J— /) . (1,5)
\ 273, 15 J ' '
В этом уравнении величина 1/273,15= а носит название коэффициента термического расширения. Этот коэффициент не зависит от природа идеального газа, его давления, объема и температуры.
Таким образом,
V/=Vo (1 4- а/) при P = const. (1,6)
Если объем газа остается постоянным, то по такому же закону растет и давление
Р/=Ро(1 + се/) при V = const. (1,7)
В этом случае величина а, равная 1/273,15, называется термическим коэффициентом упругости газа.
Математическую зависимость, выражающую закон Гей-Люссака, можно значительно упростить, если в уравнение (1,5) вместо t ввести абсолютную температуру Т.
Учитывая, что
7 =/ + 273,15, (1,8)
преобразуем уравнение (1,5) следующим образом:
vt-v
1 273,15 / 273,15
Отбросив индексы и объединив постоянные величины волну константу, получим V = const Т, при Р = const.
Аналогично можно преобразовать и уравнение (1,7), получив Р = = const Т при V = const.
На основании приведенных уравнений, можно сделать вывод: объем и давление изменяются прямо пропорционально изменению абсолютной температуры газа:
Графически закон Гей-Люссака выражается пучком прямых линий, выходящих из начала координат (изобары и изохоры идеального газа, рис. 3 и 4).
Из закона Гей-Люссака вытекает: плотность и концентрация газа, находящегося под постоянным давлением, обратно пропорциональны абсолютным температурам:
(1, Н)
(1,12)
- 15
Закон Авогадро. В равных объемах различных газов при одинаковой температуре и давлении содержится одинаковое число молекул.
Из закона Авогадро вытекает важное следствие. Число молекул, которое содержится в одной килограмм-молекуле любого газа, есть величина постоянная: NQ — 6,025 *1026 (число Авогадро).
Следовательно, при одинаковых условиях 1 кмоль любого газообразного вещества должен занимать постоянный объем. При нормальных условиях (t = 0° С; Р= 101 325 н!м2) 1 кмоль любого газа занимает объем 22,4 м\ Этой величиной часто пользуются в расчетах.
Рис. 3; Изобары идеального газа (V—T)
Рис. 4. Изохоры идеального газа (Р—Т)
Состояние газа характеризуется тремя величинами: давлением Р. объемом V и температурой Т. Эти три величины связаны уравнением, которое получило название уравнения состояния идеального газа.
Оно выводится путем объединения законов Бойля — Мариотта, Гей-Люссака и Авогадро. Если взять 1 кмоль газа при нормальных условиях (Pq. То и Vo) и нагреть его до определенной температуры Т при toy же давлении, то согласно закону Гей-Люссака объем газа при этой температуре Vt будет равен:
. (ЫЗ)
1 о
Если при постоянной температуре Т изменить давление газа от Pq до любого значения Р. то объем газа также изменится и станет равным V. На основании закона Бойля — Мариотта
PV = P0 vT.
Подставив в это уравнение значение Vt (1,13), получим:
откуда
РУ Рс V.
О, 14)
о
Поскольку Po, Vo и То — величины постоянные, отношение P^VJTq есть также величина постоянная для всех газов, независимо от их химической природы. Эту постоянную величину обозначают буквойR и называют универсальной газовой постоянной. С учетом этого уравнение (1,14) преобразится:
ру
или PV=RT. (I, 15)
Уравнение (1,15) справедливо для 1 кмоль газа. Если в объеме газа будет содержаться п кмоль, то это уравнение будет иметь более общий вид:
PV =nRT. (1,16)
Уравнение (1,16) является основным уравнением газового состояния и называется уравнение^ Клапейрона—Менделеева. Впервые это уравнение было выведено Клапейроном в 1834 г. Д. И. Менделеев в своих работах в 1874 г. указал, что благодаря закону Авогадро уравнение Клапейрона приобретает наибольшую общность, когда оно относится не к обычной весовой единице (грамму или килограмму), а к 1 кмоль газа. *
4исло киломолей газа п можно рассчитать по формуле:
т
где т — масса газа (кг), содержащегося в объеме V при давлении Р и температуре Т\ М — масса киломолекулы газа.
Подставив значение п в уравнение (1,16), получим:
PV = ^-PT, м
откуда
Отношение mlV есть не что иное, как плотность газа d, откуда
МР d~ RT'
Если обе части уравнения (1,16) разделить на объем V, получим:
P=~-RT. (1,18)
Поскольку отношение nlV есть концентрация газа С, то уравнение Клапейрона — Менделеева будет иметь вид:
Р=С/?Т. (1,19)
Численное значение универсальной газовой постоянной зависит от того, в каких единицах измерены нормальное давление Ро и объем Vo одной килограмм-молекулы газа.
— 17
Найдем численное значение константы R, применив уравнение (1,16) для 1 кмоль газа, находящегося при нормальных условиях. Так как п = 1, то
P0V0=Z?T0.
101 325 w/jh2*22,4 ms
-----~ -------= 8,313• 10s джIкмоль град,
273,16
или
R =8,313 дж/моль*град.
Универсальная газовая постоянная R может быть выражена и в других единицах.
Физический смысл универсальной газовой постоянной легко можно установить, если в ее выражение ввести при Р = const два значения температуры 7\ и Т2 и соответствующие им объемы и V2. При условии, что Т2 > Т1У a V2 > Vi, значение универсальной газовой постоянной будет равно:
' (1,20)
“ т2—т
Из этого соотношения следует, что универсальная газовая постоянная есть не что иное, как работа расширения одного киломоля газа при нагревании его на 1Q
при постоянном давлении.
§ 3. Молекулярно-кинетическая теория газов
Основы молекулярно-кинетической теории газов, которая объяснила физический смысл газовых законов, были заложены еще в работах М. В. Ломоносова. В 1744—1748 гг. он разработал теорию атомномолекулярного строения вещества, впервые обосновал кинетическую теорию теплоты и на основании этого объяснил многие не известные до него явления. В XIX в. молекулярно-кинетическая теория газов получила свое дальнейшее развитие в работах Клаузиуса, Максвелла и Больцмана. На новейшем ее этапе эта теория была в современном виде разработана Я. И. Френкелем.
В основе кинетической теории идеальных газов лежат следующие простые допущения.
1. Каждый газ состоит из молекул, которые можно рассматривать как однородные совершенно упругие шарики. Причем, размеры этих шариков-молекул настолько малы по сравнению с межмолекулярными расстояниями, что их можно рассматривать как отдельные материальные точки.
2. Столкновение молекул между собой подчиняется законам удара упругих шаров.
3. Молекулы не взаимодействуют друг с другом, пока не столкнут* ся.
4. Движение молекул в газе хаотично и непрерывно поступательно. Молекула в разные моменты времени обладает самыми разнообраз-
Молекула в разные моменты времени обладает самыми рази , _ ними скоростями как по величине, так и по направлению. Поэтому трудно описать движение отдельной молекулы. Однако в целом к
— 18 —
огромному количеству молекул вследствие беспорядочности их движения можно применять законы теории вероятности.
Известно, что при множестве движущихся молекул (или атомов) число частиц, обладающих скоростью, лежащей в данном интервале значений, остается постоянным. Теоретические подсчеты, произведенные Максвеллом в 1860 г., показали, что молекулы газа по своим скоростям движения при данной температуре распределяются строго определенным образом. В качестве примера приведем данные распределения по скоростям движения для молекул кислорода при 0°С (табл.1).
Таблица 1
Распределение молекул кислорода по скоростям движения при 0°С
Пределы скоростей, м/сек. Процент молекул, обладающих этой скоростью Пределы скоростей, м/сек Процент молекул, обладающих этой скоростью
Меньше 100 1,4 От 400 до 500 20 3
От 100 до 200 8,1 » 500 » 600 15,1
» 200 » 300 16,7 » 600 » 700 9,2
» 300 » 400 21,5 Больше 700 7,7
Как показали исследования, распределение молекул по скоростям
зависит только от температуры; оно не изменяется во времени, хотя каждая отдельная молекула постоянно изменяет свою скорость.
Из данных табл. 1 видно, что примерно половина всех молекул обладает скоростями, близкими к некоторой определенной средней величине. Однако есть молекулы, скорости которых меньше или больше, чем
средняя величина.
На рис. 5 приведены кривые распределения молекул по скоростям их движения. Как видно из рисунка, с повышением температуры максимум кривой смещается в сторону больших скоростей и становится более пологим.
Молекулярно-кинетическая теория объяснила многие свойства . газов, например, стремление их занять возможно больший объем, возникновение давления на стенки сосуда, медленный характер процесса, диффузии, рост давления с повышением температуры и др.
Вследствие многочисленных столкновений друг с другом молеку-
лы газа движутся зигзагообразно, всякий раз проходя в одном направлении очень малый отрезок пути (примерно 10~7 м). Отдельные молекулы вырываются из общего скопления и летят в окружающее простран-
ство, чем и обусловливается стремление любого газа занимать максимальный объем.
Молекулы ударяются о стенки сосуда, создавая тем самым газовое давление. Мерой этого давления является сила ударов движущихся молекул о поверхность в 1 л/2 в 1 сек. С повышением температуры скорость движения молекул увеличивается, вместе с тем увеличивается и
число молекул, ударяющихся о стенки сосуда, т. е. растет давление газа.
— 19 —
В каждой отдельный момент молекула имеет разные скорости как по величине, так и по направлению, но для каждой постоянной средняя скорость движения молекул есть величина постоянная.
Различает среднюю арифметическую и среднюю квадратичную скорости. Средняя арифметическая скорость определяется соотношением:
«а =-------“-------г О» 21)
где па — средняя арифметическая скорость; и19 и2, ...» ип — скорости движения Отдельных молекул газа; п — общее число молекул газа.
Рис. 5. Кривые распределения молекул по скоростям их движения
Средняя квадратичная скорость может быть вычислена путем деления суммы квадратов скоростей отдельных молекул на общее число молекул: «
_ +... 4-и*
U е=---------------I
П
откуда
Применяя к хаотическому движению молекул в газе законы механики, удалось получить основное уравнение, которое связывает объем и давление со средней квадратичной скоростью движения молекул газа:
PV 1/З/Vww2, (1,23)
где У — число молекул газа, находящихся в объеме V, т — масса одной молекулы данного газа.
— 20 —
В случае когда имеется 1 кмоль газа, можно записать основное уравнение:
= l/3/V0ma2, (I, 24)
где Af0 — число Авогадро.
Уравнение (1,24) называется основным, потому что из него можно математически вывести все рассмотренные выше законы идеальных газов, рассчитать кинетическую энергию молекулы, среднюю скорость движения молекул газа и ряд других важных следствий.
Например, нужно вывести закон Бойля—хЧариотта. Для этой цели используем уравнение (1,23): 1 ' >
PV = l/3Nmu2.
Необходимо доказать, что при постоянной температуре PV = const. Для этой цели рассмотрим величины, находящиеся в правой части уравнения (1,23): т — масса молекул — постоянная величина; N— число молекул в данном объеме, постоянно; и — величина, также постоянная при t — const. Таким образом, PV = const при t = const.
Для обоснования других газовых законов найдем связь между абсолютной температурой газа и кинетической энергией его молекулы.
Используем уравнение (1,24) для 1 кмоль газа:
PV = l/3/V0ти\ 1 но при л = 1 PV=PT, тогда
или
РТ=1/ЗЛ'ошп2,
Умножим и разделим правую часть равенства, на 2:
Тогда
2 /Vo
3 ‘ R
~ ^кин — кинетическая энергия
ти*
‘ . 2 ’ ...
> . . . S'
одной молекулы газа, а произведение
то'
—- Д/о = ЕК1И1 (1, 25)
выразит среднюю кинетическую энергию всех молекул, содержащихся в 1 кмоль газа.
Следовательно,
= (1.26)
□Л
или
Еки11=4'А’Г- • (,’27)
Из уравнения (1,27) вытекает, что абсолютная температура пропорциональна кинетической энергии поступательного движения молекул газа. Чем выше температура, тем быстрее движутся молекулы газа, т. е. их кинетическая энергия возрастает. И наоборот, с понижением температуры наблюдается уменьшение кинетической энергии молекул. ’
— 21 —
Из уравнения (1,27) также следует, что если Т = 0, то и Екин = 0. Иными словами, абсолютный нуль приобретает конкретный физический смысл: при этой температуре вовсе прекращается поступательное движение молекул. Однако следует подчеркнуть, что и при абсолютном нуле некоторые виды движения внутри молекул и атомов сохраняются.
Из основного уравнения кинетической теории газов можно вывести и закон Авогадро. Предположим, что имеется два различных газа. Для первого массу молекулы обозначим тъ среднюю квадратичную скорость число молекул Массу молекулы второго газа т2, скорость и?, число молекул N2. Причем, объем V, давление Р и температура Т обоих газов одинаковы. Необходимо доказать, что Nx = Л/2*
Применим для первого и второго газов уравнение (1,23)
pv=-^-Nlml~u2,
о
РУ ==-- N2 m2 uj. О •
Так как Р и V одинаковы, то = N2m2U2- Разделим обе
части этого равенства на 2:
Так как при одинаковой температуре кинетическая энергия молекул одинакова, т. е.
miiii m2ul
2 “ 2 * \
следовательно = N2.
Пользуясь основным уравнением (1,24), можно вывести простое соотношение, чтобы вычислить средние квадратичные скорости движения молекул различных газов для разных температур. При этом необходимо заменить произведение NGm равной ему молекулярной массой М
PV=-~Mu2, но RV—RTi RT=-±-Mu2,
откуда
"'2Ч
Из уравнения (1,28) следует, что средняя квадратичная скорость зависит от температуры и природы газа. Для данного газа при постоянной температуре и является величиной постоянной и выражается в м!сек. Так, для водорода, азота и кислорода средние квадратичные скорости молекул при 0е С соответственно равны 1845, 493 и 461 м1еек.
— 22 —
Эти скорости огромны. Между тем скорости перемещения газов в пространстве сравнительно ничтожны. Это объясняется тем, что молекулы, двигаясь с огромными скоростями, постоянно сталкиваются между собой и перемещаются в пространстве не по прямой, а по сложным зигзагообразным линиям, состоящим из очень коротких отрезков прямой. В результате молекулы незначительно удаляются от своего первоначального положения. Расстояние, проходимое молекулой от столкновения к столкновению, называется свободным пробегом. Эти расстояния в газе разнообразны по своей величине и в практических вычислениях обычно ограничиваются определением только среднего свободного пробега. Длину свободного пробега вычисляют по формуле:
• где L — длина свободного пробега, т — масса молекулы, d — радиус молекулы, S — плотность газа. Из этого уравнения следует, что длина свободного пробега обратно пропорциональна плотности газа, а следовательно, и давлению его. Величина среднего свободного пробега различных газов при нормальных условиях находится в пределах 10~3 см.
§ 4. Реальные газы
Как известно, в поведении различных газов наблюдаются большие отклонения от законов идеальных газов. Эти отклонения тем больше, чем ниже температура и выше давление, при которых находится газ. На рис. 6 заметны отклонения различных газов от закона Бойля— Мариотта при высоких давлениях.
Отклонения реальных газов от идеального состояния обусловлены в основном двумя причинами: силами межмолекулярного взаимодействия (притяжения) и собственным объемом молекул реального газа,
который при высоких давлениях необходимо учитывать. Взаимное притяжение между молекулами приводит к уменьшению расстояния между ними и вызывает уменьшение объема, занимаемого газом, т. е. действует как некоторое добавочное давление, приложенное извне к газу. Это давление получило название внутреннего. Поскольку реальные молекулы газа имеют собственный объем, уменьшение свободного пространства между ними приводит к уменьшению длины свободного пробега молекул.
При высоких давлениях и низких температурах расстояние между
Рис. 6. Отклонения от законов идеальных газов
— 23
iuu £UU эии
К см3/ноль
Рис. 7. И30терМЬ1 двуокиси углерода
молекулами газа настолько уменьшается, а силы межмолекулярного взаимодействия настолько увеличиваются, что газ может перейти в жидкое состояние. Еще в 1823 г. Фарадей, применяя низкие температуры и высокие давления, осуществил сжижение таких газов, как СО2, NH3, Cl2- Однако не все газы ему удалось получить в жидком состоянии. Такие газы, как Н2, N2, О2 и некоторые другие, даже при самых высоких давлениях не переходили в жидкое состояние. Эти газы назвали постоянными газами.
После создания теории сжижения газов благодаря работам Д. И. Менделеева удалось получить и эти газы в жидком состоянии. Согласно
Iсирии, для каждого газа с ществует такая температура, выше которой газ не может быть лг вращен в жидкость ни при каком давлении. Такая Tcmik лучила название критической. При этой температуре для газа требуется наибольшее тическим. I] наоборот, при температурах ниже кр*ии^ ниже температура, тем меньшее давление необходимо для газа. Объем 1 моль г-критическим, объемом, а состояние газа, иаходящег^л иид к ским давлением при критической температуре и занимающего ческий объе^, ,__например, кри-
тическое состояние СО2 характеризуется следующими параметрами: критическая темпрпятх/no qi ----------------- ----- -------
теории, для каждого газа су----” ' ___претемпература по-
। сжижения давление, которое также называется кри-—--------------: ниже критической чем
I сжижения газа при критической температуре называется - — l находящегося под критиче-
__r_. icmiicpaiype и занимающего крити-
ческий объе^ называется критическим состоянием. Например, кри-шир СП* vor'2KT2p:i3yc7Cx параметрами:
критическая температура 3*1,3° С,. критическое давление 72,9 атм, критический объем 0,096 л.
На рис. 7 приведены изотермы двуокиси углерода. Рассмотрим изотерму для 103 с. На ней только участок А В соответствует газовому состоянию, Подчиняющемуся закону Бойля — Мариотта. Участок ВС соответствует состоянию жидкость — пар. Здесь наблюдается резкое уменьшение Объема при постоянном давлении. Участок С соответствует жидкому Состоянию; он не показывает заметного уменьшения объема с повышением давления. При более высоких температурах горизонтальные участки изотерм, соответствующие участку ВС, постепенно уменьшаются и, наконец, при 31,3° С превращаются в точку перегиба К (критическая точка).
— 24 -
Таким образом, СО2 переходит в жидкое состояние при температуре 31,3° С и давлении 72,9 атм. При температурах выше критической (31,3° С) СО2 ни при каких давлениях не перейдет в жидкость.7
Параметры критического состояния для различных газов приведены в табл. 2.
Таблица 2
Параметры критического состояния различных газов
Газ Критическая температура Критическое давление, атм* Критический объем, см*/моль
°C °к
Гелий —267,9 5,2 2,66 58
Водород —239,9 33,2 12,8 65
Азот . —147,1 126,0 33,5 90,0
Окись углерода —140,2 133,0 34,5 93
Аргон — 122,4 150,7 48,0 75,5
Кислород —118,8 154,3 49,7 •74,3
Метан — 82,5 190,7 45,7 95
Двуокись углерода .... 4- 31,3 304,5 72,9 96
Аммиак 132,4 405,5 111,5 72,4
Хлор 144,0 417,1 76,0 124,0
Двуокись серы ...... 157,2 430,3 77,7 123,0
* По СИ атмосфера физическая равна 1,01325* 106 н[м?.
Из таблицы видно, что такие газы, как гелий, кислород, водород, азот, характеризуются очень низкими критическими температурами.
Исходя из теории критического состояния следует, что между паром и газом нет принципиальных различий и что любой газ может быть сжижен при охлаждении его до критической температуры и ниже.
Жидкие газы, как известно, удобнее транспортировать. Они широко применяются в металлургической и химической промышленности, а также в технике и научных лабораториях для получения низких температур и для других целей. Сжижением воздуха с последующей возгонкой получают кислород и азот, которые в дальнейшем используются при получении азотной кислоты и азотных удобрений. При этом сначала синтезируют аммиак из азота и водорода (эти газы находятся в установках для синтеза под давлением в несколько сот атмосфер), а затем уже аммиак окисляют кислородом до получения азотной кислоты и т. д.
Ван-дер-Ваальс (1878) внес соответствующие поправки в уравнение Клапейрона — Менделеева с учетом объема молекул газа и сил взаимодействия между ними. При этом он исходил из следующих соображений. Если взять какой-то сосуд объемом V, в котором находится А’ молекул газа, то любая молекула этого газа на может находиться в тех местах объема сосуда, где находятся остальные N — 1 молекул, т. е. ей доступен не весь объем сосуда, а только часть его, равная V — Ь. Величина несжимаемого пространства b равна, согласно Ван-дер-Ваальсу, учетверенному сплошному объему самих молекул. Уменьшение объема газа происходит, таким образом, за счет сжатия свободного прост
— 25 —
ранства V — b. По этой причине в уравнении состояния идеального газа (1,17) вместо V необходимо взять величину V — 6:
т p(V-b)=—RT, , М
откуда
т RT
лГ * V—b '
(1,30)
Давление реального газа на стенки сосуда прямо пропорционально концентрации молекул С —NlV в слое /, который непосредственно при-
Рис. 8. Сила взаимного притяжения молекул реального газа
мыкает к стенке сосуда (рис. 8). Молекулы, находящиеся в близлежащем слое II, будут притягивать молекулы слоя I и тем самым ослаблять их давление на стенку сосуда на какую-то величину Pt.
Сила взаимного притяжения молекул обоих слоев, по Ван-дер-Ваальсу, зависит от природы притяжения и возрастает прямо пропорционально квадрату объема газа:
/V2 а
Pi=aw=v;’ (,’3,)
где а = aN2 — константа Ван-дер-Ваальса, зависящая от природы газа. Таким образом, для реальных газов в уравнение (1,30) вместо Р надо подставить величину Р + alV\ тогда это уравнение примет вид:
RT а
V—b~~V* ’
Окончательное уравнение Ван-дер-Ваальса для реальных газов бу-, дет
(Р + ^) (V-b)=-^RT. (1,32)
В табл. 3 представлены константы Ван-дер-Ваальса для некоторых газов.
Уравнение Ван-дер-Ваальса значительно точнее отображает состояние реального газа, чем уравнение (1,17), которое было выведено для идеальных газов.
Изучение критических явлений привело к выводу, что газообразное и жидкое состояние вещества различаются лишь по степени агрегации молекул, которая, главным образом, зависит от температуры и давления.
— 26 —
Константы Ван-дер-Ваальса
Таблица 3
1 аз а Ъ . 10} Газ а Ь-10’
н2 0,245 2,67 НС1 3,8 4,1
Не 0,034 2,36 NH3 4,0 3,6
N2 1,38 3,94 С2На 4,4 5,1
О2 1,32 3,12 С2Н4 4,5 5,6
со 1,49 4,00 С12 5,5 4,9
со2 3,60 4,28 О2 6,7 5,6
§ 5. Газовые смеси. Закон Дальтона
В жизни очень часто приходится иметь дело не с чистыми газами, а с их смесями. Особенно широкое распространение газовые смеси получили в технике. Одни из них служат ценным химическим сырьем и используются при синтезе ряда веществ (нефтяные газы, воздух), другие являются хорошим газообразным топливом (природный, доменный, генераторный газы).
Основным законом газовых смесей является закон Дальтона, который имеет следующую формулировку: общее давление газовой смеси ^обш равно сумме парциальных давлений всех входящих в нее газов рх + + Р2 + Рз + ... • т. е.
Робщ — Pi 4" Р‘2, + Рз 4 • • • 4- Рп (Ь 33)
Парциальное давление— это часть общего давления в газовой смеси. Оно равно тому давлению газа, которым он обладал бы, занимая один весь объем смеси.
Как и другие законы идеальных газов, закон Дальтона не выполняется при высоких давлениях и низких температурах.
К каждому отдельному газу, входящему в данную газовую смесь, может быть применено уравнение состояния газа (1,16):
RT RT RT
Р\=Щ- ; р2 = л2-------; p3 = n3- и т. д.,
‘'общ ‘'общ ‘'общ '
где Кобщ — общий объем газовой смеси, пь п2, п3 ... — числа молей отдельных газов в смеси.
Подставив эти выражения в (1,33) и сделав соответствующие преобразования, получим уравнение состояния применительно к смесям идеальных газов:
РГ
Робщ — (^J + • ••) ., 9
или
Робщ общ ХпРТ, где Sn — сумма числа молей газов в смеси.
О, 34)
— 27 —
Помимо парциального давления, для газовых смесей различают парциальный объем каждого из газов v2, v3 и т. д. Парциальный объем — это объем, который занимал бы отдельный газ, входящий в состав смеси идеальных газов, если бы при том же количестве он имел давление и температуру смеси.
Сумма .. парциальных объемов всех идеальных газов, входящих в смесь, равна общему объему смеси:
Тобщ — 4~ ^2 4” 4” • • • 4- Vn • •
Состав газовых смесей обычно выражают в весовых и объемных процентах, а также в мольных долях и мольных процентах.
Выражение состава газовых смесей в объемных процентах широко применяется в газовом анализе.
Мольная доля показывает, какую часть от общего числа молей смеси составляет данный газ. Рассмотрим это на конкретном примере. Предположим, газовая смесь состоит из трех газов, взятых в количестве nt и п2, п3 молей. Мольные доли каждого из газов равны:
”1
ЛГ8=—
П2
«2
«3
где >, п—п^ 4- ^2 4- п3.
Не трудно догадаться, что сумма мольных долей всех газов, входящих в данную смесь, всегда равна единице, т. е.
или
Мольный процент данного газа в смеси равен мольной его доле, увеличенной в 100 раз.
Если разделить почленно приведенные выше уравнения для парциальных давлений газов р19 р2 и р3 на уравнение (1,34), то получим:
Pi _______
Робщ Zuп
Аналогично для р2 и р3.
”2 о .
Р2 — X1 * общ»
Р1 = “х^
— Рг
^1 гобщ •
Рз =
Таким образом, парциальное давление любого компонента газовой смеси равно мольной доле его в смеси, умноженной на общее давление смеси.
§ 6. Воздушный режим почвы
Воздух в почве является ее составной частью, занимая все поры, незанятые водой. Содержание воздуха в почве подвержено постоянным колебаниям, так как оно зависит от влажности и пористости почвы и может варьировать
Таблица 4
Состав атмосферного воздуха и воздуха в почве (об. %)
Газы
Атмосферный воздух
Воздух в почве
Азот N2 .................... . . . .
Кислород О2..........................
Аргон Аг.............................
Двуокись углерода СО2................
Все остальные газы...................
78,08 20,95 0,93 0,03 0,04
78,08—80,24*
20,90—0,0
0,03—20,0
* Азот -f- аргсн.
в пределах 25—80 об. %. Состав воздуха в почве может значительно отличаться от атмосферного, что хорошо подтверждается данными табл. 4.
Из этой таблицы видно, что основными компонентами воздуха почвы являются азот, кислород, аргон и двуокись углерода. Как правило, в почве содержится меньше кислорода и больше СО2. Азот иногда подвергается изменениям — связывается за счет микробиологических процессов, протекающих в почве. Воздух в затопляемых рисовых почвах, а также в болотных и заболоченных почвах, помимо вышерассмотренных компонентов, может в заметных количествах содержать газообразные продукты анаэробного разложения: метан (СН4), водород (Н2), аммиак (NH3) и некоторые другие.
Опыт показывает, что из всех, газов наибольшее значение для жизнедеятельности населяющих почву микроорганизмов имеют кислород и углекислый газ. Потребителями кислорода в почве являются не только микроорганизмы, но и корни растений и все животное население почвы. Причем не исключено участие кислорода в некоторых химических реакциях, протекающих в почвах. Накопление СО2 в почве может происходить за счет биологических процессов (разложение органического вещества), в результате химического разложения карбонатов, а также из грунтовых вод. Соотношение между кислородом и СО2 в конечном итоге зависит не только от скорости потребления и продуцирования этих газов в почве, но и от скорости газообмена между почвенным и атмосферным воздухом. Скорость же газообмена почвы зависит от изменения температуры, барометрического давления, влияния ветра, изменения уровня грунтовых вод и влажности почвы, а также от скорости диффузии газа в порах почвы.
Содержание кислорода и углекислого газа в почвенном воздухе оказывает разностороннее воздействие на свойства по&вы и прямо или косвенно влияет па урожай сельскохозяйственных культур.
При недостатке кислорода ослабляется дыхание растений, что отрицательно сказывается на урожае. Кроме того, при недостатке его в почве развиваются восстановительные анаэробные процессы, приводящие в ряде случаев к накоплению таких токсичных для растений продуктов, как метан и сероводород. Опыт показывает, что при отсутствии в почве свободного кислорода прекращается развитие любых растений. Наиболее благоприятные условия создаются для растений в том случае, если содержание кислорода в почве составляет около 20%. При таком содержании кислорода семена прорастают своевременно и дружно.
СО2 оказывает большое влияние на различные свойства почвы, хотя повышенная концентрация его в почве отрицательно сказывается на урожае растений. Примерно от 40 до 70% всего количества СО2, которое растение усваивает при фотосинтезе, доставляется из почвы. Обогащение приземного слоя почвы СО2 в дневные часы положительно сказывается не только на интенсивности процессов фотосинтеза, но и на урожае растений. Поэтому необходимо проводить специальные мероприятия, направленные на активное образование СО2 в почве.
Углекислый газ, растворяясь в почвенном растворе, оказывает значительное влияние на изменение минеральной части почвы. В таком растворе соединения СаСО3, MgCO3, FeCO3 и др. обладают большей растворимостью, чем в чистой воде.
•— 29 —'
Особенно важно уметь создать оптимальный воздушный режим в районах:-временного избыточного увлажнения почв. В них из-за преобладания восстанови-1: тельных процессов практически весь кислород оказывается связанным, в силу чего растения испытывают в нем острый недостаток. Основная задача, которая неизменно стоит перед агрономами, — это усиление доступа воздуха, а следовательно, и кислорода в почву, проветривание и подсушивание ее. Для этой цели применяется зяблевая вспашка, дренаж и другие агротехнические и мелиоративные приемы. . .
Таким образом, создание оптимального воздушного режима в сочетании с другими благоприятными факторами жизни растений является необходимым условием для получения максимально высоких урожаев сельскохозяйственных культур.
ТВЕРДОЕ АГРЕГАТНОЕ СОСТОЯНИЕ /
§ 7. Признаки твердого состояния
Как известно, при достаточно низкой температуре все вещества переходят в твердое состояние. При этом скорость движения атомов, молекул или ионов, из которых состоит данное вещество, настолько уменьшается, что силы взаимного притяжения, силы сцепления между ними становятся соизмеримыми с силами отталкивания. Тело в результате этого приобретает определеннук форму, которая не изменяется. Кроме того, твердые вещества обладают способностью восстанавливать прежнюю форму после снятия действия сил, направленных на ее изменение, т. е. для твердых веществ характерно явление деформации. По способности к деформации все твердые тела подразделяются на упругие, пластичные и хрупкие.
Частицы твердых тел настолько прочно связаны друг с другом силами взаимного притяжения, что для них исключается поступательное движение и имеет место лишь колебательное движение около определенных точек. Под действием внешних сил эти частицы могут несколько смещаться из своего первоначального положения, но при снятии нагрузки они вновь возвращаются в него обратно. Таким образом, для всякого* твердого вещества характерна не только собственная форма, но и способность к деформации.
Твердые тела обычно подразделяют на кристаллические и аморфные.
Кристаллические вещества имеют четкую внутреннюю структуру, обусловленную правильным расположением частиц в строго определенном периодически повторяющемся порядке. Кроме того, для каждого твердого кристаллического тела существует строго постоянная точка (температура) плавления.
Для кристаллических тел весьма характерно явление анизотропии, сущность которого состоит в том, что кристалл в различных направлениях обладает неодинаковыми свойствами. Такие свойства, как тепло-и электропроводность, механическая прочность, коэффициент теплового расширения, скорость растворения и другие свойства в различных направлениях кристалла различны. Например, слюда сравнительно легко разделяется на пластинки только в одном направлении (параллельно поверхности), в других же направлениях разрушение слюды требует гораздо больших усилий. Если из какого-то кристалла (не куби
— 30 —
ческой формы) выточить’ шар, а затем его нагреть, то шар изменит свою форму и превратится в эллипсоид. Изменение внешней формы тела в данном случае произойдет потому, что коэффициент линейного расширения по различным направлениям кристалла не одинаков.
* Аморфные вещества в отличие от кристаллических не имеют ясно выраженного порядка во взаимном расположении слагающих их частиц (рис. 9). Кроме того, аморфные тела изотропны, т. е. их свойства с( вершенно одинаковы по всем направлениям внутри тела. Эти вещества не имеют постоянной температуры плавления. При нагревании они сначала размягчаются в определенном интервале температур, затем, постепенно уменьшая свою вязкость, переходят в жидкотекучее состо
Рис. 9. Порядок расположения частиц в амор- Рис. 10. Кривые нагревания фном (а) и кристаллическом (б) телах аморфного (/) и кристаллического (2) веществ
яние. При охлаждении эти расплавы вновь могут перейти в твердое состояние без образования кристаллической структуры. На рис. 10 приведены кривые нагревания аморфного (/) и кристаллического (2) вещества.
Аморфные вещества по структуре аналогичны жидкостям и отличаются от них лишь весьма малой подвижностью своих частиц. Поэтому аморфные вещества рассматривают как переохлажденные жидкости. Из-за большого внутреннего трения* переход их в кристаллическое состояние сильно затруднен.
Однако резко противопоставлять аморфные тела кристаллическим не следует, так как многие вещества можно получить как в кристаллическом, так и в аморфном состоянии. Например, кварц SiO2 существует в природе в кристаллическом (горный хрусталь) и аморфном состоянии (опал). Кроме того, современные рентгенографические и электро-пографические исследования показали, что во многих телах, которые раньше считали аморфными (например, аморфные формы кварца или углерода), расположение атомов не является вполне хаотичным. Они содержат мельчайшие зародыши кристаллов размерами 10~6—Ю"7 см.
* В 1013—1015 раз больше, чем у воды, и в миллиарды раз больше, чем у глицерина.
।
— 31 —
И только чрезвычайно высокой вязкостью, которая быстро нарастает при охлаждении вещества, можно объяснить отсутствие дальнейшего развития (роста) этих кристаллов.
С энергетической точки зрения аморфные вещества по сравнению с кристаллическими обладают большим запасом энергии. Об этом свидетельствует хотя бы тот факт, что при кристаллизации твердого вещества происходит заметное выделение тепла. При застывании же расплавленного аморфного вещества никакого выделения тепла не наблюдается. Поскольку аморфное состояние вещества является энергетически менее устойчивым, возникает тенденция к переходу вещества из аморфного состояния в кристаллическое. Этот процесс является чрезвычайно длительным во времени. Так, для перехода стекла в кристаллическое состояние необходимо время в сто и более лет. При этом стекло мутнеет. В процессе кристаллизации внутреннее напряжение в стекле может настолько увеличиться, что оно разрушается без видимых внешних причин. Известны случаи, когда старинные массивные стеклянные предметы вдруг разлетались вдребезги без всякого прикосновения к ним.
§ 8. Внутреннее строение кристаллов и основные типы кристаллических решеток
Весьма тонкие современные методы исследования кристаллического состояния вещества подтвердили, что частицы в кристаллах (атомы, молекулы или ионы) располагаются закономерно, образуя так называемую пространственную решетку кристалла. Внешняя геометрическая форма кристалла теснейшим образом связана с его внутренней структурой. В кристаллической решетке любого тела можно выделить определенную часть, которая носит название элементарной ячейки. Эта ячейка представляет собой наименьший объем кристаллической решетки вещества, который точно отражает его химический состав и все особенности внутренней структуры данного кристалла.
Важнейшей особенностью кристаллических образований является их способность самоограняться. Так, при выделении кристаллического вещества из раствора или из расплавленной массы оно принимает геометрическую форму определенных кристаллов с явно выраженными плоскими гранями. При достаточно сильном ударе более крупные кристаллы распадаются на ряд более мелких кристаллов, которые ограничены плоскостями, пересекающимися между собой под определенным углом. Эта способность кристаллов раскалываться на слои по определенным плоскостям носит название спайности. Как известно, у аморфных тел это свойство отсутствует — поверхность излома их бывает неровной, раковистой.
Во внутреннем строении кристаллов выполняется принцип плотнейшей упаковки частиц, из которых состоит данный кристалл. Под действием сил взаимного притяжения частицы стремятся разместиться как можно ближе друг к другу*. Поэтому наиболее энергетически
* Следует иметь в виду, что при чрезмерном сближении частиц в кристалле проявляются силы отталкивания.
— 32 —
выгодным будет такое взаимное расположение частиц в кристалле, которое отвечает их наиболее плотной упаковке. Промежутки между ними достигают минимума. При этом могут иметь место два случая.
1. Частицы, из которых состоит кристалл, имеют равные или очень близкие по величине радиусы. Этому условию отвечают два типа кри-
сталлических решеток: гексагональная и гранецентрированная кубическая (рис. 11, а и б). В таких решетках степень заполнения объема кристалла частицами составляет 74%. Это максимально плотная упаковка частиц одинакового или близких по величине радиусов. Подобный тип решеток свойствен большинству металлов.
2. Частицы, образующие кристаллы, сильно различаются своими радиусами. Принцип плотнейшей упа-
а
Рис. II. Плотная упаковка одинаковых сфер: а «• гексагональная; б — кубическая
Рис. 12. Структура кристалла хлорида натрия
ковки применим и в этом случае. Частицы более крупных размеров в основном образуют кубическую или гексагональную сетку, а более мелкие частицы занимают свободное пространство между ними. Этот тип решетки характерен для ионных кристаллов, поскольку разные ионы довольно резко отличаются друг от друга по радиусам; например, такова структура кристалла хлорида натрия (рис. 12).
Следует отметить, что наряду с соотношением размеров частиц на структуру кристалла оказывают известное влияние и поляризационные взаимодействия между ними.
С точки зрения структурных элементов и действующих между ними сил различают четыре типа кристаллов: молекулярные, атомные, ионные и металлические.
2
Зак. 560
— 33 —
&-Атон С; Q-Атом О
Рис. 13. Элементарная ячейка кристалла СОг: кубическая решетка; атомы уг-лецфта занимают узлы гранецентрированной яЧ'ейки
Молекулярная решетка. Молекулярные кристаллы имеют в углах пространственной решетки полярные или неполярные молекулы, связанные между собой силами Ван-дер-Ваальса. В качестве примера можно указать на твердую двуокись углерода (сухой лед), нафталин, лед. На рис. 13 показано строение элементарной ячейки твердой двуокиси углерода. Как видим, атомы углерода образуют кубическую решетку с центрированными гранями: атомы кислорода расположены по обе стороны от углерода на отрезках прямых, ориентированных определенным образом относительно ребер элементарной ячейки.
Поскольку силы взаимодействия
> между молекулами сравнительно слабы, то и вещества с данным типом решетки обладают малой твердостью, низкими температурами плавления и кипения. Растворы этих веществ, как правило, имеют сравнительно малую электропроводность.
Атомная решетка. В узлах кристаллических решеток этого типа расположены нейтральные атомы, определенным образом ориентированные в пространстве и связанные ковалентными связями. К числу веществ с атомной решеткой относятся, например, кремний, графит, алмаз, бор и др. Ковалентная связь, как известно, очень прочная,
поэтому все связи в кристалле равноценны и очень прочны. Вещества, образованные атомными решетками, имеют большую твердость, высокую температуру плавления, малую растворимость и малую летучесть.
На рис. 14, а и б приведены схемы строения атомных решеток алмаза и графита. В силу своеобразия структуры графит имеет очень малую прочность связи по плоскостям спайности кристалла, тогда как алмаз обладает огромной твердостью, поскольку все атомы углерода в его кристаллической решетке расположены друг от друга на одинаковом расстоянии.
Ионная решетка. Ионные кристаллы имеют в узлах пространственных решеток положительно и отрицательно заряженные ионы, которые связаны между собой электростатическими силами притяжения одноименных зарядов. Силы взаимодействия в ионных кристаллах весьма значительны, благодаря чему вещества с ионным типом решетки обладают высокой прочностью, высокими температурами плавления и малой летучестью.
Ионные решетки характерны для большинства неорганических соединений (соли, оксиды и другие классы соединений). Многие минералы также имеют ионное строение. Так, кристаллы, имеющие ионную решетку, как правило, хорошо растворимы в воде, а растворы их обладают высокой электропроводностью. В твердом виде ионные кристаллы не проводят электрический ток, так как в них электроны прочно
— 34 —
Рис. 14. Кристаллическая решетка алмаза (а) и графита (б)
АБ _ направление расположения плоскости спайности
2*
— 35 —
удерживаются в атомных орбиталях отдельных ионов. В расплавленном состоянии кристаллические вещества проводят электрический ток, причем электропроводность осуществляется за счет переноса ионов. Электропроводность расплавов является характерным свойством любых ионных структур.
Металлическая решетка. Этот тип кристаллических решеток отличается от всех рассмотренных выше типов структур. Согласно современным представлениям, в узлах пространственной решетки типичных металлов в основном находятся положительно заряженные ионы, упакованные по принципу плотнейшей упаковки шаров, а в промежутках между ними находятся электроны в свободном состоянии. Последние образуют своеобразный «электронный газ», который как бы скрепляет одноименно заряженные ионы металла в плотнейшую крис-
Молекулярные
Атомная
Металлическая
Ионная
Р|ис 15. Плоскостные схемы кристаллических решеток различных типов
таллическую решетку. С другой стороны, и сами электроны удерживаются катионами металла, в силу чего они не могут свободно покинуть кристаллическую решетку. Именно наличием свободных электронов объясняется хорошая электро- и теплопроводность, а также многие химические свойства металлов.
Металлы, как известно, от всех известных природных материалов отличаются высокой прочностью наряду с хорошей пластичностью как в холодном, так ив горячем состоянии. Высокая температура плавления металлов указывает на значительную прочность металлической решетки и также объясняется наличием «электронного газа» в нем.
Под влиянием разности потенциалов электроны в металле начинают передвигаться в определенном направлении, что является причиной возникновения электрического тока. /
На рис. 15 приведены плоскостные схемы всех рассмотренных типов кристаллических решеток. Однако, принимая такую классификацию кристаллов, всегда нужно иметь в виду, что характер разных связей даже в одном и том же кристалле может быть не одинаковыгл и классификационные признаки не всегда четко и хорошо выражены. Наряду с кристаллами, относящимися к одному из четырех рассмотренных видов связи, существуют кристаллы с различными переходными и смешанными формами связи. Это, например, целиком относится к кристаллогидратам, в которых встречаются одновременно ионный тип связи между катионами и анионами соли, ковалентная связь между атомами,
— 36 —
входящими в состав аниона, а также полярные связи внутри молекул воды и ионодипольная связь молекул с ионами.
Значительный интерес представляют кристаллы, образующие так называемые слоистые решетки, которые характерны для графита, слюд и глинистых минералов.
§ 9. Координационное число и энергия кристаллической решетки
Известно, что атом не имеет определенных границ. Вокруг ядра любого атома невозможно описать некую сферу, которая охватила бы все электроны, связанные с ядром. При сближении двух ионов силы отталкивания между ними резко возрастают вблизи определенного значения межатомного расстояния. Исходя из этого, ионы можно рассматривать как соприкасающиеся сферы с характерным для каждого иона (или атома) радиусом.
Относительные размеры элементарных ионов видны из рис. 16, масштаб которого отвечает увеличению примерно в 30 миллионов раз. • Таким образом, современная кристаллохимия исходит из представления, что все ионы и атомы имеют сферическую форму, а пространственная кристаллическая решетка формируется по принципу плотнейшей шаровой упаковки. В каждом кристалле, любая частица, входящая в его состав (молекула, атом или ион), взаимодействует не только с соседней частицей, но одновременно и
окружают. В результате все частицы кристалла оказываются связанными в единую систему, в которой силы взаимного притяжения и отталкивания уравновешены. Этому отвечает состояние наибольшего сближения разноименных ионов и наибольшего удаления одноименных ионов, т. е. ионы занимают в кристалле наиболее устойчивую конфигурацию, соответствующую минимуму потенциальной энергии.
Каждая частица в кристалле (молекула, атом или ион) окружена другими частицами, которые непосредственно с ней взаимодействуют. Число взаимодействующих частиц носит название координационного числа и является характерной величиной для данного типа кристаллической решетки. Координационное число, как правило, имеет значе-
0
Li
Не
Na
Ca
Be
Mg
Rb Sr
Си. Zn
Ag Cd In
о
с о
N
о
Al
Sc
Ga
Si
Ti
Ge
Zr
Sn
1
Ce
cs Ba La
Хе
Рис. 16.
Относительные размеры некоторых ионов
+5
S
Cr
As
Nb
Sb
с другими частицами, которые
Se
Mo
Те
I
ее
— 37 —
пие 3,4,6,8 и обычно тем больше, чем меньшеразличие в размерах ионов (или атомов). При одинаковых размерах ионов координационное число может достигать значения 12, как, например, у металлов. Каждой конфигурации ионов соответствует определенное предельное отношение между радиусом катиона и аниона. При изменении этого отношения изменяется и координационное число и, как результат, тип кристаллической решетки. В табл. 5 показана зависимость между предельным
Таблица 5
Различные типы координации катионов и анионов (по Бетехтину)
Отношение радиуса катиона к радиусу аниона Координационное число Состав элементарной ячейки Форма окружения (а трех различных изображениях)
№+ : О2- 0,15 С4+ :О2~ 0,14 В3+ : О2- 0,17 а NO3 со3 (ВО3 2— 3- Треугольник A
S6+ :О2- 0,261 р:>+ . о2- 0,26 Si'+ : О2- 0,31 д]з+ . о2- 0,43 4 > с/) -Q GO оорР I-* л —- —— V 7 1 1 1 || Тетраэдр. 1 /|\ / I \ \ A-л / » \ )
А13+ : О2- 0,43 Mg2+ . 02- о,57 Fe2+ : О2- 0,62 Cu2+ : О2- 0,62 6 |А1ОС MgO(i FeOe CuOe I9- чЙ/' Октаэдр «хЛо ИSj /'\° У/ 1 1 ['
Na+ : О2- 0,75 Са2+ : О2- 0,80 8 NaOg CaO8 К у 5 1 1— I <4 I
К+ :О2- 1,00 12 ко12 (fl ( » I V— kO Kyfio-акгпаэдр ’ t 1 ) 1 XfT
— 38 —
соотношением радиусов ионов и формой кристалла для бинарных соединений типа АБ.
Однако не всегда кристаллы обладают структурой, отвечающей максимальному координационному числу. Делов том, что соотношение радиусов ионов является не единственным фактором, который обусловливает величину координационного числа и вместе с ним определенную структуру. Во многих случаях существенное влияние на характер связи между частицами в кристалле оказывает поляризуемость ионов.
Прочность и устойчивость кристаллической решетки зависит от сил взаимодействия между образующими ее ионами, атомами или молекулами. Силы взаимодействия между частицами в кристалле характеризуются определенным качеством энергии, которая называется энергией кристаллической решетки.
За величину кристаллической решетки принимают энергию образования 1 моль кристаллов данного вещества из частиц, находившихся до этого в состоянии идеального газа при той же температуре, при которой идет процесс кристаллизации. Иными словами, энергия кристаллической решетки эквивалентна работе, которую нужно затратить для разрушения и удаления ее составных частей на бесконечно большие расстояния. Эту энергию обычно выражают в килоджоулях. Так, энергия ионных кристаллов (для решеток, не содержащих сильно поляризующих или сильно поляризуемых ионов) выражается формулой:
Е-^2563-^ Д , (1,35)
ГК + ГА
где Sn — число ионов, определяющее состав вещества, Wk и Й7Л — -валентности ионов и Гк + га — расстояние между центрами ионов для координационного числа 6.
Частицы, из которые состоит любая кристаллическая решетка, находятся в состоянии непрерывного колебательного движения. Средняя величина энергии колебания всех частиц для каждой температуры постоянна и прямо пропорционально изменяется вместе с ней. Если отдельные частицы приобретают большую кинетическую энергию по сравнению с энергией связи в кристалле, то они могут отрываться от кристаллической решетки и переходить в пар (этот процесс называется сублимацией). С сублимацией связана летучесть твердых тел. Вполне понятно, что чем выше прочность кристаллической решетки, тем меньшей летучестью обладает данно^ вещество, и наоборот. Если средняя колебательная энергия частиц становится достаточно большой, что имеет место при повышении температуры, то происходит полное разрушение решетки — тело плавится.
Растворимость любого кристаллического соединения -также находится в прямой зависимости от прочности его кристаллической решетки. Объясняется это тем, что полярные молекулы растворителя (например, воды) при погружении в него кристалла определенным образом ориентируются около ионов на поверхности кристалла, вызывая тем самым ослабление связи ионов в решетке за счет сил поляризации. Чем меньше энергия кристаллической решетки, тем легче она разрушается
— 39 —
в жидкости. Таким образом, такие важные свойства твердого вещества, как растворимость, летучесть, температура плавления и другие свойства, характеризующие устойчивость соединения, целиком обусловлены энергией кристаллической решетки.
§ 10. Полиморфизм и изоморфизм
Некоторые вещества (как простые, так и сложные) могут существовать в нескольких кристаллических формах, называемых модификациями или полиморфными формами. Это явление впоследствии получило название полиморфизма (греч. poly — много, многое; morphe —-форма; polymorphos—многообразный). Оно было открыто в 1821 г. немецким химиком Митчерлихом.
В молекулярных, ионных, атомных и металлических кристаллах явление полиморфизма встречается довольно часто. Например, СаСО3 образует в природе минералы кальцит и арагонит с одним и тем же химическим составом, но различным внутренним кристаллическим строением. Двуокись титана ТЮ2 также образует в природе два минерала — анатаз и рутил, кристаллы которых отличаются друг от друга.
Перечень подобных примеров можно легко продолжить, поскольку почти все вещества при известных условиях могут быть получены в различных кристаллических модификациях.
Модификации одного и того же вещества обычно обозначают греческими буквами а, |3, у ... в порядке повышения или понижения температуры превращения (например, а- и |3-кварц). Температура, при которой вещество переходит из одной модификации в другую, носит название точки превращения или точки перехода. Так, при 900° С a-Fe самопроизвольно переходит в другую полиморфную форму — |3-Fe и т. д.
Перестройка кристаллической структуры полиморфных вещестц при изменении внешних условий объясняется сильными изменениями взаимной поляризации структурных единиц, из которых состоит кристаллическая решетка.
Явление полиморфизма имеет большое значение в технике. Так, например, а и у-железо очень резко отличаются друг от друга по механическим, магнитным и другим свойствам, причем эта модификация устойчива при температуре свыше 910° С. Если железо, нагретое до температуры выше 910° С, быстро охладить, то его у-структура при этом может сохраниться. В этом и состоит сущность закалки стали. Вновь приобретенная структура, не являющаяся устойчивой для данной температуры, может сохраняться в телах очень и очень длительное время.
Еще один пример. В природе при нормальных условиях существуют две разновидности кристаллического углерода — графит и алмаз. Алмазная модификация углерода является неустойчивой для низких температур, и казалось бы по этой причине алмаз должен превратиться в графит. Однако этот процесс при обычных температурах практически не протекает.
— 40 —
Многие вещества различного химического состава могут образовать кристаллы совершенно одинаковой формы и весьма близкие по внутренней структуре. Это явление также впервые было открыто Э. Митчер-лихом в 1819 г. на солях КН2РО4 и KH2AsO4 и было названо им изомер-физмом.
Ниже приведены изоморфные ряды для некоторых веществ
MgSO4.7Н2О — ZnSO4.7Н2О — N iSO4 • 7Н2О
ПЛИ
CaCOj — MgCO3— MnCO3 и т. д.
Явление изоморфизма широко распространено в природе. Именно этим объясняется огромное разнообразие минералов и их подчас весьма сложный состав (силикаты, алюмосиликаты и многие другие).Изоморфные вещества могут кристаллизоваться совместно, с образованием смешанных кристаллов, например, хромовые и алюминиевые квасцы и др.
Рассматривая явление образования смешанных кристаллов, А. Е. Ферсман дает следующую формулировку правила изоморфизма: элементы могут замещать друг друга в кристаллических постройках., когда они принадлежат к одному и тому же состоянию рететки иногда радиусы их ионов (или атомов) являются близкими друг другу.
На явление изоморфизма большое влияние оказывает также способность ионов к поляризации. При изоморфных замещениях поляризационные свойства взаимозамещаемых ионов должны быть близкими. Если же это условие не выполнено, то даже при равенстве ионных радиусов получить изоморфные смеси не удается.
Огромная заслуга в изучении явления изоморфизма принадлежит советским ученым — ьГристаллохимикам В. И. Вернадскому, А. Е. Ферсману, А. В. Шубникову и другим, а нз зарубежных ученых — Гольдшмидту.
§ 11. Глинистые минералы, их строение, свойства и значение в почвоведении
Почва более чем на девяносто процентов состоит из минеральных компонентов и содержит основной запас питательных веществ для растений. Почва является полидисперсной системой и имеет довольно сложный механический, минералогический и химический состав. В качестве примера в табл. 6 приведен средний химический состав твердой фазы почвы (по А. П. Виноградову).
Как видно из таблицы, почти половина твердой фазы почвы приходится на кислород, одна треть — на кремний, свыше 10% — на алюминий и железо и только 7% — на все остальные элементы. Из всех перечисленных элементов только азот (а также частично углерод, водород, кислород, фосфор и сера) содержится в органической части почвы. Все остальные элементы приходятся на минеральную часть почвы,
— 41
Таблица 6
Средний химический состав твердой фазы почвы
Название элемента Содержание, % Название элемента Содержание, % Название j элемента Содержание, %
Кислород 49,0 Барий 0,05 Галлий (Ю-3)
Кремний 33,0 Стронций 0,03 Олово (10-3)
Алюминий 7,1 Цирконий 0,03 Кобальт 8-10-’
Железо 3,7 Фтор 0,02 Торий . 6-10-4
Углерод 2,0 Хром 0,02 Мышьяк 5-10—*
Кальций 1,3 Хлор 0,01 Иод 5-10-’
Калий 1,3 Ванадий 0,01 Цезий 5-10-*
Натрий 0,6 Рубидий 6-10~3 Молибден 3-10-*
Магний 0,6 Цинк 5-Ю-3 Уран Ы0-4
Водород (0,50) Церий 5-Ю-3 Бериллий (10-’)
Титан 0,46 Никель 4-Ю-3 Германий (10-’]
Азот 0,10 Литий З-10-з Кадмий 5-10-6
Фосфор 0,08 Медь 2-10-3 Селен ыо-в
Сера 0,08 Бор 1-Ю-3 Ртуть (Ю-«)
Марганец 0,08 Свинец 1-Ю-з Радий 8-10-”
которая состоит из большого числа различных минералов в виде частиц, имеющих размеры от миллионных долей миллиметра до 1 мм и более.
Все минералы, содержащиеся в почве, по происхождению подразделяются на первичные и вторичные. Первичные минералы имеют преимущественно магматическое происхождение. Из них наиболее распространены в почвах кварц (окись кремния), полевые шпаты, амфиболы, пироксены и слюды, т. е. минералы, включающие кислородные соединения кремния. Эти минералы составляют основную массу магматических и почвообразующих пород. В почвах первичные минералы обычно присутствуют в виде более или менее крупных частиц, размером от 1 до 0,001 мм, и только очень незначительная часть их имеет более высокую степень дисперсности.
Первичные минералы являются в условиях земной поверхности неустойчивыми соединениями и под действием сил выветривания переходят в более устойчивые соединения — вторичные минералы. Процесс выветривания протекает под влиянием как чисто физических (колебания температуры, ветер, движущая сила во^ы),так и химических и биологических факторов. В результате этого из первичных минералов могут образоваться вторичные минералы простого состава: гидроксиды железа (II) и (III), алюминия, гидроксид кремния и некоторые другие соединения.
Кроме того, в процессе выветривания образуются также вторичные минералы более сложного строения (алюмо- и феррисиликаты). Эти последние более высокодисперсны, чем первичные, и имеют исключительно важное значение в создании основного свойства почвы — ее плодородия.
— 42 —
Все вторичные минералы сложного состава имеют пластинчатое строение и содержат химически связанную воду. Поскольку эти минералы являются важнейшей составной частью различных глин, они получили название глинистых или глинных минералов.
Число глинистых минералов довольно велико, но в почвах наиболее широкое распространение и значение для плодородия имеют в основном три группы минералов: каолинитовая, монтмориллонитовая и гидрослюдистая.
К минералам каолинитовой группы относятся каолинит I Al2Si2O5 (ОН4)1 и. галлуазит [Al2Si2O5 (ОН)4-2Н2О], а также некоторые другие минералы. Каолпнитовые глины содержат примерно 20— 25% илистых частиц (меньше 0,001 мм), из них 5—10% частиц коло-идных размеров (меньше 0,25 микрона). Минералы этой группы довольно часто встречаются во многих типах почв. Они имеют сравнительно небольшую набухаемость и липкость.
Из минералов монтмориллонитовой группы в почвах наиболее распространены монтмориллонит [Al2Si4Ol0 (ОН)2 • пН2О|, бейделлит [Al2Si3О9 (ОН)3 • лН2О1, нонтронит [Fe2Si4O10 (ОН)3 • лН2О] и некоторые другие. Монтмориллонитовые глины обл-адают в отличие от каолинитовых высокой набухаемостыо, липкостью и связностью. Для них весьма характерным признаком является высокая степень дисперсности (до 80% частиц меньше 0,001 мм, из которых 40—45% меньше 0,25 микрона).
Среди глинистых минералов, встречающихся в почвах, большое место принадлежит минералам группы гидрослюд. В эту группу входят гидромусковит (иллит) {КА12 [(Si, А1)4О10] (ОН)2 • /гН2О}, гидробиотит {К (Mg, Fe)3 [(Al, Si)4O10] (OH)2 • /?Н2О} и вермикулит {(Mg, Fe2+, Fe3+)2 [(Al, Si)4O10] (OH)2 • 4H2O}.
Глинистые минералы различаются по структуре.
Кристаллическая решетка различных глинистых минералов построена из одних и тех же элементарных структурных единиц, состоящих из атомов кремния и кислорода, а также из атомов алюминия, кислорода и водорода. Кроме перечисленных выше элементов в состав глинистых минералов могут входить Fe, Mg, К, Мп и др. В подавляющем большинстве глинистые минералы имеют слоистое строение и относятся к слоистым силикатам. Как показали новейшие рентгенографические и электронографические исследования, слои глинистых минералов состоят из сочетания кремнекислородных и кислород-гидроксилалю-миниевых соединений.
Элементарной ячейкой кремнекислородного соединения является тетраэдр (рис. 17), четыре вершины которого заняты анионами О2* (г = 1, 32 А), а в центре этого тетраэдра находится более мелкий катион Si (г = 0,39 А). Тетраэдр [SiOJ4- является основной структурной единицей не только глинистых минералов, но и всех существующих в природе соединений кремния с кислородом. Избыток отрицательных зарядов этой элементарной ячейки может быть нейтрализован присоединением каких-либо катионов или соединением нескольких тетраэдров через вершины, когда кислородный ион оказывается одновременно связанным с двумя ионами кремния.
— 43 —
Рис. 17, Схема строения кремнекислородных тетраэдров (в двух изображениях)
Рис. 18. Лист кремнекислородных тетраэдров (в двух изображениях)
Рис. 19. Схема строения ал^могидроксильных октаэдров (а) и их слои, из которых построена кристаллическая решетка минерала гиббсита (б)
Рис. 20. Схематическое изображение октаэдрических решеток (по Бетехтину): * а —гиббсит А1(ОН)3 (диоктаэдрическое строение); б — брусит Mg(OH)2 (триоктаэдрическое строение). / — Al, Mg; 2 — ОН, расположенные выше плоскости чертежа; 3 — ОН — расположенные ниже плоскости чертежа
— 44 —
Для глинистых минералов наиболее типичным являются такие соединения, в которых кремнекислородные тетраэдры соединены в слои (или листы) циклической структуры (рис. 18). В таком слое на каждые два иона кремния приходится пять ионов кислорода, что соответствует формуле (Si2O5)2“. Кремнекислородныететраэдрические слои могут соединяться со слоем кислород-алюмо-гидроксильных атомов, которые образуют октаэдры (рис. 19, а). В них ион алюминия окружен атомами кислорода и гидроксид-ионами. Алюмогидроксильные октаэдры соединяются так же, как и кремнекислородные тетраэдры — в октаэдрические сетки или слои. Они могут быть построены по аналогии с минералом гиббситом А1 (ОН)3 или бруситом Mg (ОН)2. На рис. 19,6 показана модель структуры кристаллической решетки гиббсита, состоящая из параллельно расположенных октаэдрических слоев.
На рис. 20 показана проекция на плоскость слоя алюмогидроксиль-ных октаэдров. Если в октаэдрическом слое ионами алюминия заняты не все центры октаэдров (рис. 20, а), а лишь 2/3 всех возможных замещений, то этот слой имеет диоктаэдрическое строение. Соединение окта-
[гииратирооан- (мускооит) (гиоратирооанныи} ныи)
0-7 ^-2 • -3 о-4 ©-5 Ъ-6 ф-7
Рис. 21. Схематическое изображение структуры ряда силикатов со слоистой решеткой:
J — кислород; 2 — гидроксилы; 3 — кремний; 4 — кремний, алюминии; 5 — алюминий; 6 — алюминий, магний; 7 — калий
эдров в нем происходит через два гидроксид-иона, причем каждый и
них оказывается связанным с двумя ионами алюминия.
Если же вместо ионов алюминия в октаэдрических слоях буду' находиться ионы Mg24* (или Fe2+), то они благодаря меньшему заряду способны занять все возможные центры в октаэдрах (рис. 20, б). Та
кое строение октаэдрического слоя носит название триоктаэдрического. Глинистые минералы, помимо кремнекислородного тетраэдрического
слоя, могут иметь один из двух октаэдрических
слоев.
Иными
слова
Рис. 22. Перспективная схема каолинитового слоя, показывающая распределение атомов в различных составляющих его решетки '
ми, кристаллическая решетка их образована сочетанием слоев кремнекислородных тетраэдров со слоями алюмогидроксильных октаэдров. Соотношение между этими слоями в глинистых минералах обозначают цифрами 1 : 1,2: 1, 2 : 2 и т. д.
Кремнекислородные и кислорсд-гидроксид-алюминиевые сетки образуют так называемые тетраэдро-октаэдрические слои и пакеты. При соединении тетраэдрического и октаэдрического слоев ионы О2" тетраэдрического слоя, расположенные на вершинах тетраэдров, становятся общими для обоих слоев, т. е. ионы О2" будут служить своеобразными «мостиками» между ионами Si4+ одного слоя и ионами А13+другого слоя. Такая структура наиболее устойчива, так как количество положительных зарядов Si4+ и А13+в этой структуре равно количеству отрицательных зарядов О2" и ОН".
Иногда внутри кремнекислородных тетраэдров часть кремния замещается на алюминий, а в октаэдрических сочетаниях, как уже говорилось, также возможно взаимное изоморфное замещение алюминия, магния, железа и т. д.
На рис. 21 показана схематическая структура некоторых глинистых и сходных минералов. Из этого рисунка видно, что все минералы отличаются один от другого числом сеток (слоев), порядком их чередования, а также характером изоморфных замещений.
Минералы группы каолинита имеют двухслойную кристаллическую решетку, пакеты которой образованы из двух связанных через общие атомы кислорода слоев: слоя кремнекислородных тетраэдров и алюмогидроксильного слоя, имеющего диоктаэдри-
— 46 —
еское строение. Такие двухслойные пакеты чередуются в кристалле с промежутками, придавая ему пластинчатое строение (рис. 22). Каолинит не способен впитывать воду в межпакетные пространства и поэтому не обладает способностью к набуханию. Расстояние между пакетами каолинита является характерным диагностическим признаком его и равно 7,2 А.
-7 I I I I I I I I i । I о
V 2 4 6 8 10 А
Рис. 23. Структура монтмориллонита в проекции вдоль оси а. Гидроксид-ионы обозначены заштрихованными кружками, кислород — белыми
6 0
4 Si
4О,2ОН
4 Al, Мд 40, 20Н
4 Si
6 0.
Минералы монтмориллонитовой группы по своим кристаллохимическим свойствам разделяются на две группы: 1) диоктаэдрические (монтмориллонит, нонтронит, бейделлит и др.) и 2) триоктаэдрические (сапонит, гекторит и др.).
Монтмориллонит относится к трехслойным минералам. Его пакеты состоят из октаэдрического слоя (диоктаэдрического строения), который заключен между двумя тетраэдрическими слоями (рис. 23). Состав этих слоев вследствие изоморфных замещений не постоянен. Кремний тетраэдров также может быть частично замещен на алюминий и железо, а в октаэдрах, кроме ионов алюминия, могут находиться ионы магния. Избыточные заряды слоев, как правило, компенсируются зарядами других одно-, двухвалентных катионов, расположенных между ними.
— 47 —
I I
I
I
i
।
i
I-
i
। н
i
i
।
।
Г;
I
1'
I I К
I I
I i:. । IV I
I I I I
I
i I--i
I
I'.)
I r
I
I
I к I I
3
I-
I
В отличие от каолинита межпакетные расстояния монтмориллонита Я могут изменяться, колеблясь от 9,4 до 21,4 А. Эти расстояния измени- | ются в зависимости от количества воды, находящейся между пакетами. 1 В силу этого монтмориллонит обладает большой способностью к набу- | ханию. I
Другие минералы этой труп? I пы (нонтронит, бейделлит) от-1 личаются от монтмориллонита I своими изоморфными замеще- I ниями. I
Минералы группы! гидрослюд включают гид-1 ромусковит (иллит), гидроби- | отит, вермикулит и другие гид- 1 ратизированные разновидности I слюд. ' 1
Структура иллита подобна • структуре монтмориллонита, с той лишь разницей, что в его ; кристаллической решетке име- | ются многочисленные изоморф- 1 ные замещения. Так, ионА13+в 1 октаэдрических слоях замещен I на ион Fe3+ и ион Mg2+, при- I чем два иона алюминия заме-щаются тремя ионами магния е ; замещением октаэдрических пу- I стот. В иллите нередко два иона | алюминия в октаэдрах замеща- 1 ются на два иона магния, при этом избыточные отрицательные заряды компенсируются ионами j калия, которые размещаются в межпакетных промежутках (рис. | 24). Аналогичное строение имёют и другие гидрослюды. ]
В почвах встречаются также смешанно-слоистые минералы, в ' кристаллических решетках ко- ! торых чередуются октаэдриче-
ские и тетраэдрические слои разных минералов: монтмориллонита с ' иллитом, каолинита с мусковитом и др.
Современные представления о кристаллической структуре глинистых минералов основаны на результатах исследования, полученных с применением новейших методов. Ранее в науке преобладал взгляд на глинистые минералы как на аморфные вещества, которые образуются в результате процессов выветривания.
При написании химических формул минералов (в том числе и глинистых) пользуются следующими обозначениями: кремнекислородные — 48 —
в проек-
их разме-
Рис. 24. Структура мусковита ции вдоль оси а.
Белые кружки по мере увеличения ра обозначают соответственно кремний, алюминий, тетраэдрический кислород, октаэдрический кислород и калий. Заштрихованные кружки —* гидроксилы
тетраэдры заключаются в квадратные скобки; в круглых скобках ставятся катионы октаэдров; катионы между силикатными слоями ставятся перед катионами октаэдров; группы ОН в вершинах октаэдров, к которым не примыкают вершины тетраэдров, ставятся в круглые скобки, после квадратных; вода помещается в конце формулы.
Установлено, что важнейшие физико-химические и водно-физические свойства почвы — емкость поглощения, гидрофильность, связность, липкость, реакция среды и многие другие — находятся в прямой зависимости от минералогического состава. Теперь известно, что доступность для растений тех или иных питательных элементов в значительной мере зависит от вида минералов, содержащихся в почве, и от степени их дисперсности.
Глинистые минералы в основном сосредоточены в илистой (менее 0,001 мм) фракции почв. Составом и строением минералов этой фракции в значительной степени определяется и поглотительная способность почвы по отношению к катионам и анионам. Чем выше емкость поглощения почвы, тем больший запас питательных элементов в ней сосредоточен, следовательно, лучше ее потенциальное плодородие.
Минералы группы монтмориллонита обладают не только наибольшей степенью дисперсности, но и наибольшей поглотительной способностью (100—150 л/г-жв/100г). Эти минералы способны сильно набухать и содержат до 30% связанной воды, которая не может усваиваться растениями. Присутствие минералов монтмориллонитовой группы в почвах всегда положительно сказывается на растениях, обеспечивает большее содержание в них необходимых питательных элементов. Однако почвы, очень богатые монтмориллонитом, имеют невысокую агрономическую ценность. При высыхании таких почв образуются трещины, водопроницаемость их становится неодинаковой, на поверхности образуется прочная корка. Эти отрицательные свойства монтмориллонита особенно сильно проявляются на почвах, бедных гумусом. При достаточном количестве гумуса физико-химические свойства такой почвы значительно улучшаются за счет образования водопрочных органо-минеральных агрегатов. Практика показывает, что добавление в сильно деградированные песчаные почвы глин, содержащих минералы монтмориллонитовой группы, положительно сказывается на плодородии.
Минералы каолинитовой группы по своим свойствам резко отличаются от монтмориллонита. Каолинит обладает очень малой емкостью поглощения (7—10 мг-экв[\№ г); он практически не набухает и содержит весьма незначительное количество воды. Почвы, в которых много этого минерала, вследствие малой емкости поглощения отличаются низким плодородием. Сам каолинит не содержит поглощенных оснований и поэтому не является источником питания для растений. Почвы, содержащие много каолинита, хорошо отзываются на внесение в них калия и других оснований.
Минералы группы гидрослюд чрезвычайно богаты легкодоступным для растений калием (до 6—7%). Емкость поглощения гидрослюд в несколько раз выше, чем у каолинита, но в два-три раза меньше, чем у монтмориллонита. Почвы, содержащие много гидрослюдистых минералов, практически не нуждаются в калийных удобрениях.
— 4Q —
В трудах многих ученых отмечается активное участие глинистых минералов в повышении степени доступности фосфатов почвы, калия и микроэлементов. Наличие в почвах полуторных оксидов, а также токсичного для растений подвижного алюминия обусловлено составом и строением высокодисперсных (в том числе и глинистых) минералов. Таким образом, качественный и количественный состав вторичных минералов имеет одно из первостепенных значений в создании основного свойства почвы — ее плодородия.
ЖИДКОЕ АГРЕГАТНОЕ СОСТОЯНИЕ
§ 12. Характеристика жидкого состояния
Жидкости по своим свойствам занимают промежуточное положение между твердыми телами и газами и сходны как с теми, так и с другими. При низких температурах по некоторым свойствам жидкости сходны с газами: они текучи, не имеют определенной формы, аморфны и изотропны, т. е. однородны по своим свойствам в любом направлении. С другой стороны, жидкости обладают объемной упругостью, как и твердые тела. Они упруго противодействуют не только всестороннему сжатию, но и всестороннему растяжению. Молекулы их стремятся к некоторому упорядоченному расположению в пространстве, т. е. жидкости имеют зачатки кристаллического строения.
Тепловое движение частиц жидкости несколько отличается от такового в твердых телах, так как жидкость обладает более рыхлой структурой. В ней всегда есть свободные места — «дырки», благодаря которым молекулы могут перемещаться, покидая свое место и занимая одну из соседних свободных «дырок». На новом месте каждая молекула в течение некоторого промежутка времени совершает колебательные движения около определенной точки равновесия, затем вновь перемещается в свободную «дырку» ит. д. Таким образом, тепловое движение молекул жидкости совершается в виде сравнительно редких перемещений этих молекул из одних временных положений равновесия в другие и тепловых колебаний в промежутках между перемещениями. Именно по этой причине жидкости отличаются высокой текучестью и принимают форму того сосуда, в котором они находятся.
Средней кинетической энергии молекулы жидкости вполне хватает, чтобы совершать перескоки из одного положения равновесия в другое, но этой энергии явно недостаточно для того, чтобы полностью преодолеть силы взаимодействия окружающих молекул. Из жидкости вырывается лишь небольшое число наиболее быстрых молекул (процесс испарения). Вследствие этого молекулы в жидкости в отличие от газов, располагаются очень близко друг к другу. В большинстве случаев у жидкостей среднее расстояние между отдельными молекулами равно примерно 3 А, радиус же силы межмолекулярного взаимодействия молекулы равен примерно 10 А. Таким образом, тепловые движения молекул жидкости не выходят за пределы действия когезионных сил, поэтому жидкости имеют постоянный объем.
— 50 —
В вопросе внутреннего строения жидкостей до сих пор еще многое остается неясным. На сегодняшний день не представляется возможным предвидеть и рассчитывать различные свойства жидкостей, как это удается во многих случаях для газов и кристаллических веществ. Это объясняется тем, что внутреннее строение жидкостей значительно сложнее
внутреннего строения не только газов, но и кристаллов.
Огромную роль в свойствах жидкостей играет объем молекул, их форма и полярность. Если молекулы жидкости полярны, то происходит ассоциация (объединение) двух или более мо-
лекул в сложный комплекс (рис. 25). В таких _______ -----------\
жидкостях, как вода, жидкий аммиак, боль- - +л~~ ~0С~~
шую роль в ассоциации молекул играет наличие так называемой водородной связи.
Свойства жидкостей в значительной мере зависят от степени ассоциации их молекул. Как показывает опыт, ассоциированные жидкости обладают более высокой температурой кипения, меньшей летучестью. С по-
Рис. 25. Ассоциация
вышением температуры комплексы распада- молекул
ются и тем сильнее, чем слабее силы взаимо-
действия молекул в комплексе.
Существуют и так называемые кристаллические жидкости или жидкие кристаллы, которые, будучи жидкостями, обладают, как и кристаллические вещества, анизотропными свойствами. В качестве примера
можно привести органическое ароматическое соединение, молекула которого имеет сильно вытянутую форму
С2Н5—О—/ ^> —N — N — / С2Н5
'о/
Такая форма затрудняет вращение молекул в жидкости и способствует их более упорядоченному расположению. При плавлении кристаллов таких веществ сначала образуется кристаллическая жидкость, которая при дальнейшем повышении температуры превращается в обычную изотропную жидкость.
§ 13. Поверхностное натяжение и поверхностная энергия
Жидкость всегда стремится принять такую форму, при которой ее поверхность при данном объеме будет наименьшей. Этому условию отвечает шар.
Поверхностный слой жидкости по своим физико-химическим свойствам отличается от ее внутренних слоев. На каждую молекулу внутри жидкости равномерно действуют силы притяжения со стороны окружающих молекул, поэтому силовое поле каждой молекулы внутри жидкости симметрично насыщено (рис. 26). Равнодействующая всех сил притяжения равна нулю. Иначе обстоит дело с молекулами, которые находятся в поверхностном слое жидкости. На них действуют силы притяжения только со стороны молекул, находящихся в нижней полу
— 51
Рис. 26. Схема взаимодействия молекул поверхностного и глубинного слоев жидкости с окружающими молекулами
сфере. Силы, действующие вне жидкости, ничтожны и ими можно пренебречь. В результате этого равнодействующие молекулярных сил уже не равны нулю и направлены вниз. Поверхностные молекулы жидкости находятся под действием сил, стремящихся втянуть их внутрь жидкости. По этой причине поверхность любой жидкости стремится к сокращению.
Наличие у поверхностных молекул жидкости ненасыщенных, неиспользованных сил сцепления является источником избыточной поверхностной энергии, которая также стремится к уменьшению. На поверхности жидкости образуется как бы пленка, которая обладает поверхностным натяжением.
Для того чтобы увеличить поверхность жидкости, необходимо преодолеть силы ее поверхностного натяжения, т. е. затратить некоторое количество работы. Работа, необходимая для увеличения поверхности жидкости на 1 см2, служит мерой поверхностной энергии и называется коэффициентом поверхностного натяжения, или просто поверхностным натяжением. Повер х ностное н атя жен и е
можно рассматривать не только йак работу на единицу поверхности, но и как силу, которая действует на единицу длины (1 см) линии, ограничивающей поверхность жидкости, и направлена в сторону сокращения поверхности. Обозначается поверхностное натяжение греческой буквой а (сигма).
В зависимости от того или иного определения поверхностное натяжение измеряется или в эргах на 1 см2 или в динах на 1 см. Оба измерения численно совпадают, так как 1 эрг/см2 = \дн • cmIcm2 = 1 дн!см. В единицах СИ размерность поверхностного натяжения н • м и поверхностной энергии дж • м ~2. Эти величины также численно равны между собой: дж!м2 = н • м!м2 = н!м.
Таблица 7
Поверхностное натяжение некоторых жидкостей при 20°С
Жидкость о, дин/см Жидкость о, дин/см
Этиловый эфир .... Этиловый спирт .... Метиловый спирт . . . Ацетон Уксусная кислота . . . 17,00 22,30 22,61 23,70 27,63 1 Бензол Сероуглерод ...... Вода Ртуть жидкая 28,88 30,50 72,75 471,60
— 52 —
Для чистых жидкостей поверхностное натяжение зависит от природы жидкости и температуры, а для растворов — от природы растворите-пя> а также от природы и концентрации растворенного вещества (табл. 7).
Как видно из таблицы, жидкие металлы (например ртуть) отличаются большими величинами поверхностного натяжения. Такие органические жидкости, как спирты, эфиры, ацетон, бензол, имеют малые значения о.
Поверхностное натяжение находится в обратной зависимости от температуры. С повышением температуры, как установил Д. И. Менделеев, поверхностное натяжение уменьшается и при критической температуре становится равным нулю. Нулевое значение о при критик ческой температуре объясняется отсутствием мениска жидкости.
Поверхностное натяжение воды при различных температурах
Температура, 0 С .... О 20 40 G0 9 89 100
о, дин/см.............. 75,64 72,75 69,69 66,18 62,75 58,85
Поверхностное натяжение жидкости находится в обратной зависимости от давления пара над ней. Чем выше давление пара, тем меньше величина внутреннего давления жидкости, меньше величина поверхностной энергии и, следовательно, меньше поверхностное натяжение.
Растворенные вещества также изменяют поверхностное натяжение жидкости. Одни из них значительно понижают поверхностное натяжение и потому носят название поверхностно-активных веществ, другие, наоборот, увеличивают поверхностное натяжение и называются поверхностно-неактивными. По отношению к воде поверхностно-активными веществами являются спирты, белки, мыла. Добавка их к воде облегчает смачивание, поэтому при приготовлении растворов некоторых ядохимикатов добавляют поверхностно-активные вещества (например, мьрла) для того, чтобы раствор хорошо смачивал обрабатываемую поверхность. Эффективность ядохимиката при этом значительно повышается.
Поверхностное натяжение жидкости зависит от природы ее молекул. Например, в гомологическом ряду жирных кислот поверхностное натяжение быстро уменьшается с удлинением углеродной цепи —в среднем в 3,2 раза на каждую прибавляемую группу СН2.
Изучение поверхностного натяжения помогает глубже понять различные технологические процессы: смачивание, крашение, эмульгирование, измельчение твердых тел и др.
§ 14. Методы определения поверхностного натяжения »
Существует несколько различных методов определения поверхностного натяжения. Важнейшими из них являются следующие: метод капиллярного поднятия, сталагмометрический метод, метод наибольшего давления в пузырьках, метод отрыва кольца. Остановимся на характеристике этих методов.
Метод капиллярного поднятия. Поверхностным натяжением обусловливает ся поднятие жидкости в капилляре, если она смачивав, .то стенки. Это явление
— 53 —
объясняется тем, что жидкоегь, смачивая стенки, как бы растекаясь по ним, увеличивает поверхностное натяжение. Уменьшение поверхности достигается тем, что жидкость поднимается в капилляре вслед за смачивающим слоем. Высота поднятия жидкости определяется весом столба жидкости, уравновешивающим поверхностное натяжение (рис. 27).
Рис. 27. Капиллярное поднятие жидкости
Связь между высотой поднятия жидкости h в капиллярной трубке, радиусом этой трубки г, плотностью жидкости d и се поверхностным натяжением о выражается уравнением:
Рис. 28. Сталагмометр для определения поверхностного натяжения
• 2л>*о — лг2 hdg.
где§ — ускорение силы тяжести. Из этого уравнения находим поверхностное на тяжение, оно равно:
1 o=—rhdg. (1.37)
На этом принципе основано определение среднего радиуса капиллярных ходов в почве. В стеклянную трубку (длиной до 1,5—2 м и диаметром 1—2 см) насыпают испытуемую почву и один конец трубки обвязывают марлей, чтобы содержимое трубки не высыпалось. Легким постукиванием этим концом о твердый предмет уплотняют почву в трубке. Затем опускают трубку этим концом в сосуд с водой и закрепляют трубку в штативе в вертикальном положении. Под влиянием сил поверхностного натяжения вода будет медленно подниматься по капиллярным ходам почвы, что хорошо заметно по фронту промачивания. После прекращения поднятия воды в трубке измеряют линейкой высоту поднятия и вычисляют средний радиус капилляров в почве по формуле:
2о
Средний радиус капилляров в почве зависит от ее механического состава и структуры. В более рыхлой, свежеобработанной почве частицы менее плотно упакованы и, как результат, радиусы капиллярных ходов большие. Чем меньше
— 54 —
едний радиус капилляров, тем на большую высоту поднимается вода из нижних горизонтов почвы. С этим явлением связана потеря почвенной влаги в весенний пс иод. За зиму почва сильно слеживается, в ней образуется чрезвычайно разветвленная сеть капиллярных ходов с гораздо меньшим по сравнению с рыхлой почвой средним их радиусом. Накопленная за зиму вода поднимается по этим капиллярам, достигает поверхности почвы и бесполезно теряется для культурных растений за счет процесса испарения.
Для сохранения влаги в почве проводят ранневесеннее боронование. Цель этого агроприема заключается в механическом разрушении капиллярных ходов, с тем чтобы уменьшить испаряемость. И чем раньше будет проведено это боронование, тем больше сохранится влаги в почве и выше будет урожай сельскохозяйственных культур.
Метод сталагмометрии (метод подсчета капель). Под действием сил поверхностного натяжения жидкость, вытекающая из отверстия капилляра, принимает форму шарообразной капли. Отрыв капли наступает тогда, когда вес ее преодолевает действие поверхностного натяжения. Метод сталагмометрии (греч. stalag-ша — капля) основан на зависимости между числом капель, получаемых из данного объема жидкости (при свободном ее падении в виде капель), и поверхностным натяжением. ”
Прибор, в котором проводят измерения, называется стал атмометром. Как видно из рис. 28, он представляет собой пипетку, имеющую две метки / и 2, которые служат для начала и конца подсчета капель. Нижняя часть сталагмометра переходит в капилляр, конец которого утолщен и отшлифован для получения одинаковых капель. Для жидкостей с большим поверхностным натяжением отрыв капель более затруднен и образующиеся капли окажутся более крупными, чем для жидкостей с малым поверхностным натяжением. Число капель, вытекающих из объема И, ограниченного метками / и 2, для жидкости с меньшим поверхностным натяжением будет больше, чем для жидкости с большим поверхностным натяжением.
В момент отрыва капли вес ее Р пропорционален поверхностному натяжению о;
(1,39)
Если из объема V вытекает гг капель исследуемой жидкости, имеющей плотность d, то вес капли будет равен:
где g — ускорение силы тяжести. Объединяя уравнения (1,39) и (1,40), получим I Vd
°=T~S-
Определим численные значения о0, п0 и d0 для чистой воды
1 Vd.
(1,42)
Подставив значение 1/К из уравнения (1,42) в уравнение (1,41), получим:
d
а = 00-7--. (1,43)
/ш0
Таким образом, для определения поверхностного натяжения методом сталагмометрии необходимо определить число капель воды (/?0) и число капель исследуемой жидкости (п). Величины плотности воды и испытуемой жидкости, а также поверхностное натяжение чистой воды (о0) берут из таблиц для той температуры, при которой производятся измерения.
Метод наибольшего давления в пузырьках. Этот метод впервые был разработан П. А. Ребиндером. На рис. 29 показан прибор для определения поверхност-
— 55 —
Рис. 29. Установка П. А Ребиндера для определения поверхностного натяжения
него натяжения этим методом. В испытуемую жидкость опускают вертикально трубку 1 с капилляром на конце; через нее продувают пузырьки воздуха при разрежении над исследуемой жидкостью. Это разрежение достигается путем выли-’вания жидкости из аспиратора 2. Измеряемое с помощью манометра 3 давление, которое необходимо создать для проскока пузырьков воздуха, находится в прямой зависимости от поверхностного натяжения испытуемой жидкости и радиуса капилляр-з г:
гР=2о, (1,44)
откуда
o=_LrP< (J ,45)
Соответственно для воды имеем:
1
о =~ гР0. (1,46)
* *-*
Объединяя уравнения (1,45) и (1,46), определим:
о = Оо-^-. (1,47)
* о
Таким образом, измеряя Ро и Р по манометру и зная о0 для воды (72,75 дин!см), можно вычислить поверхностное натяжение испытуемой жидкости на границе жидкость — газ.
В последние годы прибор Ребиндера был значительно усовершенствован. На рис. 30 показан прибор для измерения поверхностного натяжения в модификации G Н. Алешина. В приборе Ребиндера, как известно, проскакивание пузырьков через исследуемую жидкость осуществляется путем создания разрежения воздуха над ней. Прибор же в модификации Алешина работает на сжатии воздуха в стеклянном шарике 1 с помощью воды, которая самотеком поступает через трехходовой кран К из воронки 2. Прибор Алешина более портативен и более удобен в работе.
Метод отрыва кольца. Прибор для определения поверхностного натяжения по методу отрыва кольца изображен на рис. 31. Сущность метода состоит в том,
— 56 —
Рис. 30. Прибор Н. А. Ребиндера в модификации С. Н. Алешина
Рис. 31. Схема прибора для определения поверхностного натяжения методом отрыва кольца:
/ — кольцо, подвешенное на нити; 2—рычаг, соединяющий нить с указателем прибора; 3 — чашка с исследуемой жидкостью; 4 — прибор, показывающий сопротивление в момент отрыва кольца о г исследуемой жидкости
что на поверхность исследуемой жидкости помещают чистое обезжиренное платиновое кольцо /, подвешенное на нити. Сила, которую необходимо приложить, чтобы оторвать это кольцо от поверхности жидкости, пропорциональна поверхностному натяжений) жидкости. Поверхностное натяжение испытуемой жидкости
вычисляют по формуле:
2л (О — /)
(1,48)
где D — внешний диаметр кольца, / — толщина кольца, g — ускорение силы тяжести, Р — сила, необходимая для отрыва кольца от поверхности жидкости.
§ 15. Внутреннее трение (вязкость) жидкостей
Всякое тело при движении испытывает сопротивление среды, в которой оно движется. Если перемешивать стеклянной палочкой воду, сахарный сироп, глицерин, мед и т. п., ощущается сопротивление движению палочки. Сила, противодействующая движению зела, носит название силы трения.
Когла тело испытывает сопротивление движению со стороны своих же частиц, противодействующая сила называется внутренним трением, или вязкостью. Таким образом, вязкость — это внутреннее трение, проявляющееся при относительном движении соседних слоев жидкости и зависящее от сил сцепления (взаимодействия) между молекулами. Во всех жидкостях при перемещении одних слоев относительно других
— 57 —
возникают более или менее значительные силы трения, направленные по касательной к поверхности этих слоев. Сила внутреннего трения F прямо пропорциональна площади S трущихся друг о друга слоев жидкости, скорости их движения dU и обратно пропорциональна расстоянию этих слоев dx один от другого.
(1,49)
(формула Ньютона), где т] — коэффициент пропорциональности.
Если площадь S = 1 см2, — = 1, то F = т] и носит название коэф-dx
фициента вязкости или коэффициента внутреннего трения. Этот коэффициент зависит от природы жидкости и ее температуры. Из уравнения (1,49) определяем:
При выражении силы трения F в динах, dx — в см, dU — в см!сек, aS — в см2, получим:
(П1 = РМ
сек
см2
Поскольку 1 дн = 1 г-см!сек1, единица ющую размерность:
вязкости
т] будет
иметь
следу-
[д//] [сек] Г г • см
(сл!2] сек2
сек
см2
— [г-см"1 • сек"1].
Эта величина вязкости называется пуазом (по имени французского ученого Пуазейля). Нередко применяют меньшую величину—сантипуаз, 1 спз — 0,01 пз. Единица вязкости в системе СИ имеет следующую размерность:
011 =
н
jh2 J |_ м/сек
= [н-сек-м"2} — [кг-сек"1-м"1].
м
Поскольку единица динамической вязкости по СИ в 10 раз больше пуаза, один пуаз будет равен:
1/13 = 0,1 н-сек!м2 — Ъ,\ кг/сек-м.
Таблица 8
Вязкость некоторых жидкостей при 20°С
Жидкость
Г), спз
Жидкость
Г), СПо
Вода ....
Этиловый эфир Бензол . . .
Этиловый спирт
1,005 0,243 0,65
1,19
Серная кислота . . . . Касторовое масло . . . Глицерин ........
Деготь ........
21,6
1000,0
1499,0 ’
3000,0
— 58 —
Вязкость является величиной, характерной для данной жидкости (табл. 8).
Жидкости, подчиняющиеся уравнению (1,49), получили название ньютоновских. Однако есть жидкости, которые не подчиняются этому уравнению — например, растворы высокомолекулярных соединений.
Вязкость жидкостей в значительной степени зависит от температуры: с повышением ее вязкость жидкости понижается (табл. 9).
Величина, обратная вязкости, т. е. 1/т], называется текучестью.
Таблица 9
Влияние температуры на вязкость воды
Температура. °C И, гпь Температура, °C Г], спз
0 1,792 60 0,4688
10 1,3077 70 0,4061
20 1,005 80 0,3565
30 0,8007 90 0,3165
40 0,656 100 0,2838
50 0,5494 —
Как видно из табл. 8, эфир, этиловый спирт являются легкотекучими или легкоподвижными, а глицерин, деготь — труднотекучими, или, иначе, малоподвижными жидкостями.
Значение явления вязкости в природе очень велико. В биологических системах она влияет на протекание ряда важнейших процессов в живом организме. Большую роль вязкость играет в различных технологических процессах в промышленности. В частности, скорость движения различных* жидкостей по трубам в основном зависит от вязкости транспортируемой жидкости.
С понижением вязкости жидкости при нагревании связано повышение электропроводности растворов электролитов (проводников второго рода).
Существует несколько методов определения вязкости, из которых мы рассмотрим наиболее широко распространенные.
Капиллярный метод. Приборы, применяемые для измерения вязкости, называются вискозиметрами. Определение вязкости можно производить по скорости истечения жидкости через трубки малого диаметра (капилляры). Для ламинарного протекания жидкости в капиллярах существует зависимость, которая установлена Пуазейлем:
%>ч\1
(1,51)
где V — объем жидкости, вытекающей из капилляра, см3; г — радиус капилляра, см; Р — сила, под действием которой движется жидкость, дн; t — время протекания, сек; I — длина капилляра, см.
Отсюда
яг4 Pt
11 — 8/И
(1,52)
— 59 —
Как и при определении поверхностного натяжения, пользуются методом сравнения. В качестве стандартной жидкости применяют дистиллированную воду. Если принять, что 1] — коэффициент вязкости испытуемой жидкости, d — ее плотность, t — время истечения, а для воды соответственно обозначить эти величины т]у, d,} и /0, то можно записать:
г) dt
Лэ
(1,53)
откуда
(1,50
Зная время истечения некоторого объема воды (по секундомеру) и такого же объема исследуемой жидкости, а также ее плотность (плотность воды можно принять за единицу), по формуле (1,54) можно определить вязкость исследуемой жидкости. В практике учебных и научных лабораторий наибольшее распространение получил вискозиметр Оствальда, который изображен на рис. 32.
Рис. 32. Стеклянный вискозиметр Оствальда для определения вязкости жидкостей и растворов: /, 2 — метки, ограничивающие объем исследуемой жидкости; 3 — уровень заполнения сталагмометра жидкостью
Рис. 33. Вискозиметр с падающим шариком:
1 — пробирка с исследуемой жидкостью; 2 — термостат; 3 пробирка с шариками; 4 — мешалка; а, б — верхняя и нижняя отметки
Метод падающего шарика. Шарик из известного металла под действием силы тяжести падает в вязкой среде. Падающий шарик встречает значительное сопротивление, величина которого по Стоксу (1851) выражается формулой:
f=fmn]U. (1»55)
В жидкости падающий шарик находится под действием двух противоположных сил. Сверху вниз действует сила тяжести шарика fr, которая равна произведению его объема на его плотность и на ускорение силы тяжести:
— 60 —
л=-4- n,3Dg,
<3
(1,56)
где г — радиус шарика, см, D — плотность шарика, г! см?, g — ускорение силы тяжести.
Снизу вверх на шарик действует сила выталкивания /2, которая по закону Архимеда равна массе вытесненной жидкости в объеме шарика с плотностью d:
4
li=—nr3dg. (1,57)
Учитывая равномерность падения шарика, можем записать:
или
откуда
2r2 g(D~d) 9q -
2 r2g(D—d)
<Г ’ и
(I 58)
(1,59)
(I .СО)
Пользуясь последней формулой, можно определить вязкость жидкостей, находя опытным путем скорость падения шарика определенного веса и радиуса в данной жидкости (рис. 33).
На законе Стокса основан механический анализ почв и грунтов. Если взмутить суспензию и оставить ее затем в спокойном состоянии, взмученные частицы постепенно осядут. Быстрее будут осаждаться более крупные по размерам частицы как более тяжелые. Зная скорость осаждения частиц различного диаметра, можно взять пробы почвенной суспензии с определенной глубины (по истечении различных сроков после взмучивания) и определить механический состав исследуемой почвы.
На указанных зависимостях построена работа сепараторов в молочной промышленности и центрифуг, используемых в биологии и медицине.
Метод вращающего цилиндра. Два концентрических цилиндра погружают вертикально в жидкость так, чтобы между ними находился слой этой жидкости. При вращении внешнего цилиндра с постоянной скоростью жидкость также начинает вращаться и передает вращение внутреннему цилиндру, который подвешен на упругой нити. Измерив угол закручивания внутреннего цилиндра а, а также угловые скорости вращения ш внешнего цилиндра в двух различных жидкостях, для одной из которых коэффициент вязкости I) известен, можно найти вязкость исследуемой жидкости по уравнению:
dx
—
а
со
(1,61)
Конструкции такого типа вискозиметров в настоящее время довольно широко распространены в научных и заводских лабораториях.
§ 16. Испарение и кипение жидкостей
Молекулы жидкости находятся в непрерывном движении. Однако не все они движутся с одинаковыми скоростями. Некоторая часть молекул обладает избыточным по сравнению со средним значением количеством энергии и имеет большую скорость. Эти молекулы, дости
— 61 —
гая поверхности жидкости, способны преодолеть силы притяжения остальных молекул и вылететь из жидкости. Жидкость испаряется. При движении молекул в направлении от жидкости к воздуху скорость и кинетическая энергия их будет уменьшаться за счет преодоления сил притяжения со стороны молекул жидкости внутренних слоев.
Опыт показывает, что силы притяжения между молекулами жидкости при комнатной температуре в несколько раз превышают среднюю кинетическую энергию, которой обладают ее молекулы при данной температуре. По этой причине подавляющее большинство молекул не покидает жидкость, так как в данный момент они не обладают необходи-
5
Рис. 34. Схема, поясняющая испарение жидкости: а — в открытом сосуде; б — в закрытом сосуде
мым количеством энергии. Если бы силы притяжения между молекулами жидкости были не столь значительными, жидкость при комнатной температуре испарялась бы мгновенно, и вещество существовало бы не в жидком, а в газообразном агрегатном состоянии.
Процесс испарения сопровождается охлаждением, так как жидкость при испарении теряет наиболее быстрые («горячие») молекулы, а сами молекулы, преодолевая силы внутреннего притяжения, теряют значительное количество своей кинетической энергии. При нагревании же одновременно с увеличением средней скорости движения молекул жидкости происходит увеличение и числа молекул, покидающих жидкость: испарение увеличивается.
Испарение зависит также от давления и от общей поверхности испарения. Чем ниже давление и больше величина поверхности, тем интенсивнее идет процесс испарения. Если испарение происходит в закрытом сосуде, оно прекращается, когда давление пара достигает некоторой определенной величины, характерной для каждой жидкости и температуры. Над жидкостью в этом случае устанавливается постоян-
— 62 —
Рис.
35. Кривые давления насыщенного пара:
/ — этилового cnupia С2Н5ОН, 2 — воды
пая концентрация пара. Пар, находящийся в равновесии с жидкостью, называется насыщенным, а давление, которое он производит, — давлением насыщенного пара (рис. 34).
Таким образом, при постоянной температуре давление насыщенного пара данного вещества является величиной постоянной и характерной для него. При повышении температуры давление насыщенного пара увеличивается. На рис. 35 изображены кривые давления насыщенного пара для этилового спирта (/) и воды (2). Кривые давления пара оканчиваются в критической точке, когда давление пара равно критическому давлению.
Переход вещества из жидкого состояния в парообразное называется парообразованием. Различают две формы этого процесса: испарение и кипение. В первом случае парообразование происходит только со свободной поверхности жидкости. Во втором— не только с поверхности, но и во всем объеме жидкости путем образования пузырьков пара, и выделения их. Давление насыщенного пара при этом равно внешнему
давлению. Температура, при которой происходит этот процесс, называется температурой кипения.
Температура кипения жидкости изменяется с изменением внешнего давления. Если уменьшить последнее, то давление пара над жидкостью сравняется с внешним давлением при меньших значениях температуры, т. е. жидкость в данном случае будет кипеть при более низкой температуре. Подобное явление можно наблюдать в горах, где давление воздуха значительно ниже нормального атмосферного давления.
Количество теплоты, которое поглощается единицей массы вещества при изотермическом испарении, называют теплотой испарения. Различают удельную теплоту испарения (отнесенную к одному грамму вещества) и мольную теплоту испарения (т. е. теплоту испарения одного моля). Мольная теплота испарения при температурах кипения под атмосферным давлением £кпп различных жидкостей прямо пропорциональна температуре их кипения 7\1Шв градусах абсолютной шкалы:
^-КИП =^КИП ^КИЛ = ^КИП> (К ЙО)
• кии
где /<кип — постоянная величина для неассоциированных жидкостей, приблизительно равная 21—22. Это уравнение позволяет вычислить 7\ип, если LKun известно, и наоборот.
— 63 —
§ 17. Роль воды в живых организмах
В состав любого живого организма, помимо различных солей и органических веществ, обязательно входит вода. Она является той средой, в которой диспергированы важнейшие высокомолекулярные соединения, образующие коллоидные растворы, и протекает большинство реакций обмена. Вода сама принимает непосредственное участие в обмене веществ, входя в качестве необходимого компонента во многие реакции синтеза. В качестве примера можно указать хотя бы на гидролитическое расщепление сложных углеводов, жиров и белков, требующих участия воды.
Количественно вода является основной составной частью любого живого организма (табл. 10).
Таблица 10
* Содержание воды в различных организмах
Объект исследования Содержание годы, % Объект исследования Содержание воды, %
Человек 58—67 Рыбы 67—78
Моллюски 75—80 Медузы 96—99
Травы 75—80 Овощи и фрукты . . . 80
Древесные породы . . . 40—60 Дрожжи 54—83
Плесени ► • 83—85 Бактерии ....... 75—88
Высокое содержание воды свидетельствует о том, что в процессе жизнедеятельности организма она играет важную роль. Вода входит в состав белковых коллоидов и принимает непосредственное участие в построении структур живых клеток и тканей. Кровь, лимфа, спинномозговая жидкость у высокоорганизованных организмов, соки, идущие по сосудам растительных организмов, состоят преимущественно из воды. Испарение воды поверхностью животных или растительных организмов обеспечивает охлаждение их при высокой температуре внешней среды.
Высшие животные очень чувствительны к потере воды. Если в процессе голодания животный организм может перенести почти полную потерю запасов жировых веществ, до 50% всех белковых веществ, то потеря более 10% воды вызывает тяжелые патологические изменения, а потеря 15—20% приводит к гибели.
Не менее важно значение воды и в жизни растений. Содержание воды влияет на направленность ферментативного действия, на интенсивность транспирации, фотосинтеза, дыхания, ростовых процессов и т. д. Количество воды в растении обусловливает скорость тех или иных биологических процессов. Так, интенсивность дыхания зерновых находится в прямой зависимости от содержания влаги в семенах. Опыт показывает, что в начале увеличение влажности повышает интенсивность процесса дыхания на сравнительно незначительную величину. Затем, начиная примерно с 14%, повышение влажности на 1% увеличивает интенсивность дыхания на 150%, последующее увеличение влажности уже повышает интенсивность дыхания на несколько сот процентов. Иными словами, чем выше содержание воды в зерне, тем интенсивнее процесс дыхания.
Интенсивность процесса обмена веществ у высших организмов зависит от . возраста организма: чем моложе организм, тем интенсивнее обмен веществ, и тем больше он содержит воды. Например, эмбрион человека ко второму месяцу развития содержит 97% воды, новорожденный ребенок — 74%, организм взрослого человека содержит 58—67% воды. Та же закономерность проявляется и в отношении отдельных тканей и органов животного организма: особенно богаты водой те органы, которые наиболее интенсивно функционируют. Так, сердце высших животных содержит 79% воды, а скелет — всего лишь 22%. - <
Глава I!
ОСНОВЫ ХИМИЧЕСКОЙ ТЕРМОДИНАМИКИ И ТЕРМОХИМИИ
§ 18. Предмет термодинамики. Основные термодинамические понятия
Термодинамика изучает взаимные превращения различных видов энергии, связанные с переходом энергии между телами в форме теплоты и работы.
Термодинамика базируется на двух основных законах, получивших название первого и второго начал термодинамики. Оба начала выведены из обобщения практического опыта.
Термодинамика включает следующие разделы: общую или физическую термодинамику, изучающую наиболее общие законы превращения энергии; техническую термодинамику, рассматривающую взаимопревращения теплоты и механической работы, происходящие в тепловых машинах; химическую термодинамику, предметом которой являются превращения различных видов энергии при химических реакциях, процессах растворения, испарения, кристаллизации, адсорбции.
Химическая термодинамика изучает не только соотношения между химической и другими видами энергии, но исследует возможности и предел самопроизвольного протекания химического процесса в конкретных условиях. Химическая термодинамика необходима для сознательного управления физико-химическими процессами, лежащими в основе химического производства.
Важность знания термодинамики и используемых ею методов расчета можно видеть на примере разработки способа получения аммиака из водорода и азота по схеме:
N2 -ь ЗН2^ 2NH3 4- 22 ккал.
Химическое равновесие, которое устанавливается между веществами, участвующими в реакции, можно на основании принципа Ле Ша-телье сместить вправо или влево путем изменения давления, температуры и концентрации вещества. Однако попытки получить аммиак на основе реакции его синтеза из водорода и азота долгое время не приводили к положительным результатам: выход аммиака был очень мал. Немецкий ученый Габер, применив количественный термодинамический метод расчета условий равновесия этой реакции, впервые показал зависимость выхода аммиака от давления и температуры, при которых протекает синтез. Термодинамические расчеты привели к выводу, что наиболее благоприятными условиями для максимального выхода аммиака являются давление около 1000 атм и температура в пределах 475— 525° С. Благодаря применению метода термодинамики оказалось возможным спроектировать крупные заводские установки по производству аммиака и азотной кислоты.
3 Зак. 560
— 65 —
Этот исторический пример убедительно свидетельствует о том, что термодинамика позволяет осуществлять теоретический анализ без предварительного проведения эксперимента.
Применение термодинамического метода расчета в технологии различных химических производств оказало в дальнейшем огромное влияние на развитие всей химической промышленности. Термодинамический метод в настоящее время широко применяется в металлургических процессах, при производстве пластмасс, удобрений, химических волокон, при химической переработке топлива. В последние годы получила бурное развитие биологическая термодинамика, где методы термодинамики применяются при изучении процессов, протекающих в растительных и животных организмах.
Однако термодинамический метод исследования физико-химических превращений имеет свои недостатки и ограничения. В частности, предсказывая возможность и полноту прохождения реакции в данных условиях, термодинамика не дает представления о времени, которое необходимо для протекания реакции. Время как параметр, характеризующий интенсивность процесса, не входит в уравнения термодинамики. Термодинамический метод применим только к макросистемам. Им нельзя пользоваться при исследованиях отдельных атомов, молекул, электронов. В силу этого термодинамика не рассматривает микроскопический механизм явлений. Ей чужды модельные представления о структуре вещества и характере движения микроскопических частиц, которые входят в состав материального тела.
Остановимся кратко на характеристике некоторых основных положений термодинамики, с которыми в дальнейшем придется иметь дело.
Тело или совокупность взаимодействующих тел, мысленно обособленные от окружающей среды, называют в термодинамике системой. По величине термодинамические системы могут быть самыми разнообразными: от булавочной головки (или меньше) до солнечной системы (или еще больше). Необходимым условием является только то, чтобы и к минимальным, и к максимальным термодинамическим системам были применимы законы термодинамики. С этой точки зрения отдельные частицы (или небольшое число их), а также вся бесконечная Вселенная в целом не являются термодинамическими системами, потому что к ним не применимы законы термодинамики.
Термодинамические системы подразделяются на простые (химически однородные) и сложные (химически неоднородные), гомогенные (физически однородные) и гетерогенные (физически неоднородные), изолированные (замкнутые) и неизолированные (незамкнутые).
Так, система, состоящая из некоторого количества простого или сложного вещества, носит название простой или химически однородной. Сложной называется такая система, которая состоит из двух или более простых веществ или химических соединений. Например, системы
СаСО3 СаО + О2 или ЗН2 + N2 2NH3 уже являются сложными или химически неоднородными.
— G6 —
Система, внутри которой нет поверхностей раздела, является однородной, или гомогенной (например, раствор каких-либо веществ или газ в замкнутом сосуде). Система неоднородная, или гетерогенная, состоит из нескольких однородных тел, между которыми имеется поверхность раздела (например, кристаллы соли в насыщенном растворе).
Термодинамическая система называется неизолированной, или незамкнутой, если она может получать или отдавать тепло в окружающую среду и производить работу, а внешняя среда — совершать работу над системой. Система является изолированной, или замкнутой, если она не имеет обмена теплом с окружающей средой, а изменение давления внутри системы не влияет на окружающую среду и последняя не может произвести работу над системой.
Однородная часть системы с одинаковыми химическими и термодинамическими свойствами, отделенная от других частей видимой поверхностью раздела, при переходе через которую физические и химические свойства резко меняются, называется фазой. Наименьшее число составных частей системы, с помощью которых можно выразить состав любой ее фазы, называется компонентами системы.
Свойства любой термодинамической системы определяются ее параметрами, или, как их еще называют, независимыми переменными. Все параметры системы подразделяются на две группы. Параметры, которые определяют свойства, зависящие от размеров системы (объем, масса, энтропия), относятся к одной группе. Другую составляют такие параметры, которые не зависят от размеров системы (температура, давление, потенциал, мольный или удельный объем). Свойства системы, определяемые параметрами первой группы, называют экстенсивными, свойствами, а определяемые параметрами второй группы — интенсивными свойствами.
В качестве основных параметров системы выбираются такие, которые могут быть непосредственно измерены и выражают интенсивные свойства системы. Сюда относятся давление, температура и объем. Эти параметры могут быть связаны друг с другом уравнением состояния. Таким образом, термодинамическое состояние системы определяется совокупностью ее термодинамических параметров.
Состояние системы может быть равновесным и неравновесным. Если термодинамические параметры стечением времени не изменяются без каких-либо внешних воздействий на систему, такое состояние ее называется равновесным. Состояние системы будет неравновесным* если ее параметры изменяются при отсутствии воздействия.
Термодинамически равновесное состояние системы является в то же время истинным равновесием. Оно характеризуется тем, что бесконечно малые воздействия на систему вызывают бесконечно малые изменения в ней. Если это условие не выполняется, система будет находиться в ложном (или неустойчивом) равновесии. В качестве примера систем, находящихся в состоянии ложного равновесия, можно назвать пересыщенные растворы, переохлажденные жидкости, переохлажденный пар при обычных условиях.
3*
— 67 —
В каждой такой системе протекает односторонний процесс, посредством которого система стремится перейти из состояния ложного (неустойчивого) равновесия в истинное. Поэтому при малейшем воздействии на неустойчивую систему она за короткий промежуток времени перейдет в состояние истинного равновесия.
По этой причине любая изолированная система с течением времени переходит в термодинамически равновесное состояние и никогда самопроизвольно выйти из этого состояния не может. Параметры только равновесной системы могут иметь строго определенные значения и, следовательно, к таким системам применимы любые уравнения состояния.
Переход термодинамической системы из одного состояния в другое называется в термодинамике процессом. При любом процессе одни параметры системы остаются неизменными, другие изменяются. В зависимости от того, какие параметры при переходе системы из одного состояния в другое остаются постоянными, процессы делятся на изохорические (при постоянном объеме), изобарические (при постоянном давлении), изотермические (при постоянной температуре) и т. д.
§ 19. Энергия и ее виды. Внутренняя энергия системы
Энергия — это мера способности тела совершать работу.
Тело может обладать энергией, которая обусловлена его движением, положением или взаимодействием его частей. В первом случае тело обладает кинетической энергией, во втором — потенциальной.
Кинетическая энергия тела равна произведению его массы на половину квадрата скорости, т. е.
где т — масса тела, U — скорость, Е — кинетическая энергия. Урав- • нение (11,1) справедливо для поступательного движения данного тела. Кинетическая энергия вращательного движения равна:
Е-~-/С02, (11,2)
где со — угловая скорость в радианах в секунду, I — момент инерции, который связан с массой частиц, составляющих данное тело, пц и с их расстоянием rt от оси вращения следующим соотношением:
(П,3)
При этом суммирование производится по всем частицам, из которых состоит тело. Работа, произведенная какой-то силой при приведении тела в движение, измеряется приращением кинетической энергии тела за время ее действия.
Потенциальная энергия измеряется работой, которую произвела бы сила, если бы тело уступило действию этой силы. Разность потенциальных энергий тела при одном его положении и при другом равна
— 68 —
работе (взятой со знаком минус), произведенной теми или иными силами при изменении положения данного тела или его конфигурации. Так, при падении какого-либо тела работа производится силами гравитации, при этом происходит уменьшение потенциальной энергии. Например, тело весом 10 кг падает с высоты 10 см под действием силы тяжести. При этом изменение (в данном случае уменьшение) его потенциальной энергии равно:
—10 000 г- 980,7 см-сек~2' 10 см = —98,07-10в эрг.
Чтобы вновь поднять данное тело на высоту в 10 см, необходимо затратить работу, равную 98,07 • 10G эрг.
Потенциальная энергия любого тела, находясь в скрытой форме, может оставаться неопределенно долгое время без всякого изменения.
Оба вида энергии — кинетическая и потенциальная — могут взаимно переходить друг в друга. Так, если какое-то тело (или частица его) поднимается вверх, то оно теряет кинетическую энергию и приобретает потенциальную. При этом кинетическая энергия переходит в потенциальную по мере того, как производится работа против сил тяготения.
Любая термодинамическая система обладает определенным запасом энергии, которая в термодинамике носит название внутренней энергии. Эта энергия складывается из кинетической и потенциальной энергии составляющих вещество молекул, атомов и субатомных частиц (ядер и электронов). Внутренняя энергия тела (системы) включает также энергию поступательного, вращательного и колебательного движения, а также потенциальную энергию, обусловленную силами притяжения и отталкивания, действующими между атомами, молекулами и субатомными частицами. Обозначается внутренняя энергия буквой U. Она зависит от физического состояния вещества и не зависит от того, каким спосфбом оно приведено в это состояние. Внутренняя энергия вещества является экстенсивным свойством, т. е. зависит от количества рассматриваемого вещества,
Полную внутреннюю энергию системы определить невозможно, наука в настоящее время не располагает такими методами. Однако можно экспериментально определить изменение внутренней энергии системы при переходе ее из одного состояния в другое. Если обозначить через Ul внутреннюю энергию системы в состоянии 1, U2 — ее внутреннюю энергию в состоянии 2, то изменение энергии в процессе перехода этой системы из состояния 1 в состояние 2 будет равно:
(П.4)
Здесь символ А употребляется для обозначения разности, причем всегда из величины, относящейся к конечному состоянию системы, вычитается величина, относящаяся к начальному ее состоянию.
Согласно закону сохранения энергии (см. стр. 70) алгебраическая сумма изменений количеств всех видов энергии в любой изолированной системе равна нулю, т. е.
At/=O.
— 69 —
В изолированной системе сумма всех видов энергии является величиной постоянной, так как она не изменяется в результате взаимодействия частей, составляющих данную систему, т. е.
ХЕ = Е = const. (11,5)
Здесь S — знак суммирования, распространяющийся на все виды энергии, которая заключается в данной изолированной системе.
§ 20. Закон сохранения энергии
Закон сохранения энергии имеет абсолютный характер и является одним из наиболее общих законов природы. Он лежит в основе современного естествознания и представляет собой естественнонаучное выражение неуничтожимости, несотворимости движения и способности взаимного преобразования одного вида движения в другой. Закон сохранения энергии является одним из фундаментальных положений диалектического материализма.
В основных чертах этот всеобщий закон природы впервые четко сформулирован в 1748 г. М. В. Ломоносовым в его письме к Л. Эйлеру. В 1756 г. Ломоносов обосновал законы сохранения материи и энергии экспериментально. Дальнейшее экспериментальное обоснование и конкретизацию этот закон получил в середине XIX в. в работах выдающихся ученых разных стран: Лавуазье, Гесса, Карно, Майера, Джоуля, Гельмгольца.
Результаты, полученные учеными при изучении взаимных превращений тепловой и механической форм энергии, были в дальнейшем распространены на все виды энергии. Исследователи установили, что превращение одного вида энергии в другой имеет строго эквивалентный характер. При исчезновении известного количества одной формы энергии появляется строго определенное количество другой. Эквиваленты различных видов энергии приведены в табл. 11. *
Таблица 11
Соотношение между единицами энергии и работы
Джоули
Эрги
Калории
Ватт-часы
10’7
4,1840
3,6-103
107 4,184-Ю7
3,6-1010
2,39-10“8 0,239
861
2,78-10“11
2,78-10~4
1,16-io-3
Закон сохранения энергии может быть сформулирован следующим образом:
Во всех явлениях природы энергия не может исчезнуть бесследно или возникнуть из ничего. Энергия может только превращаться из одной формы в другую, притом лишь в строго эквивалентных соотношениях.
70 —
§ 21. Первое начало термодинамики
Первое начало (или первый закон) термодинамики и есть закон сохранения энергии. Этот закон выполняется во всех явлениях природы и подтверждается всем опытом человечества. Ни одно из его следствий не находится в противоречии с опытом. Закон сохранения энергии подтверждает положение диалектического материализма о вечности и неуничтожаемости движения, поскольку энергия, по определению Энгельса, есть мера движения при его превращениях из одной формы в другую.
Термодинамика рассматривает преимущественно две формы, в виде которых совершается превращение энергии — теплоту и работу. Поэтому первое начало термодинамики и устанавливает соотношение между тепловой энергией (Q) и работой (Л) при изменении общей энергии системы (Дб/). Изменение общей энергии системы выражается уравнением (II, 4).
Из постоянства запаса внутренней энергии изолированной системы непосредственно вытекает: в любом процессе изменение внутренней энергии какой-нибудь системы равно разности между количеством сообщенной системе теплоты и количеством работы, совершенной системой:
ZX£/=Q—Л. (П,6)
Отсюда получаем:
Q = Л. (П,7)
Это уравнение является математическим выражением первого начала термодинамики, которое в данном случае имеет следующую формулировку: подведенное к системе тепло Q идет на увеличение внутренней энергии системы ина совершение внешней работы Л.
Если изменение, претерпеваемое системой, бесконечно мало, то уравнение (11,7) первого начала термодинамики можно записать в следующем виде:
(11,8)
где 6Q — бесконечно малое количество теплоты (элементарная теплота), поглощаемое системой, dU — бесконечно малое приращение внутренней энергии системы, 6Л — бесконечно малая работа (элементарная работа), совершаемая системой в том же процессе.
При переходе системы из одного состояния в другое внутренняя энергия в одних случаях увеличивается, в других — уменьшается. В соответствии с этим изменение внутренней энергии ДV имеет положительный знак или отрицательный.
При пользовании уравнением первого начала термодинамики (11,8) необходимо, чтобы все величины, входящие в это уравнение, были выражены в одних и тех же единицах энергии; чаще всего их выражают в калориях.
Рассмотрим высказанные выше положения на конкретном примере.
П р и м е р. Найти изменение внутренней энергии при испарении 20 г воды при 20° С, приняв, что водяной пар подчиняется законам идеальных газов и что
— 71 —
объем жидкости незначителен по сравнению с объемом пара. Скрытая теплота парообразования воды при 20° С равна 576 кал!г.
Решение. Сначала определяем объем пара, который образуется при испарении 20 г воды при 20° С и давлении 1 атм:
qRT 20-0,082.293
V — “777“ —-----7------- ~ 26,7 л • атм.
РМ — 1.18
1 л • атм = 24,2 кал. Работа расширяющегося газа, выраженная в калориях, равна:
А = 24,2 • 26,7 = 646 кал.
Количество теплоты, выделенное системой при парообразовании 20 г воды, равно:
Q = 20 • 586 = 11 720 кал.
Убыль внутренней энергии при испарении 20 г воды при 20° С будет равна:
A U = Q — А = 11 720 — 646 = 11 074 кал.
Первое начало термодинамики имеет несколько формулировок, однако все они выражают одну и ту же суть: неуничтожаемость и эквивалентность энергии при взаимных переходах различных видов ее друг в друга:
В изолированной системе сумма всех видов энергии есть величина постоянная.
Вечный двигатель первого рода невозможен, так как невозможно создать такую машину, которая производила бы работу без подведения энергии извне.
Система может переходить из одного состояния в другое различными путями. Но в соответствии с законом сохранения энергии изменение внутренней энергии AU системы не зависит от пути перехода: оно одинаково во всех случаях, если одинаковы начальное и конечное состояние системы. Количество же теплоты и количество работы Л зависят от этого пути. Однако, как бы пи менялись значения Q и А при разных путях перехода системы из одного состояния в другое, их алгебраическая сумма всегда одинакова, если только одинаковы начальное и конечное состояния системы.
Первое начало термодинамики имеет огромное философское значение. Утверждая неуничтожимость энергии, оно тем самым обосновывает и неуничтожимость материи, поскольку энергия без материи существовать не может. Во всех процессах превращения материи неразрывно связаны с превращением энергии.
§ 22. Работа расширение газа при различных термодинамических процессах
Термодинамика изучает процессы изменения состояния газа, протекающие при постоянном объеме (изохорные), при постоянном давлении (изобарные), при постоянной температуре (изотермические), а также процессы, протекающие при отсутствии теплового обмена с окружающей средой (адиабатные).
Рассмотрим, какую работу может производить идеальный газ при расширении. Предположим, что имеется цилиндр с поршнем, двига-
Рис. 36. Работа расширения газа
ютимся без трения; в цилиндре заключен идеальный газ объемом (рис. 36). Площадь поршня S, давление газа р. Сила давления газа на поршень pS. Она уравновешивается гирей и поршнем общего веса Р, т. с.
P=pS.
Если газу сообщить некоторое количество тепла, газ расширится и поднимет поршень на высоту /г. Работа, выполненная газом при расширении, равна произведению давления газа на изменение объема:
ЛА = Ph = pSh.
Поскольку Sh — АV (прирост объема), АЛ = =p&V.
При бесконечно малом приращении объема получим бесконечно малую работу расширения газа:
dA = pdV. (11,9)
Используем уравнение (11,9) для различных термодинамических процессов.
Для изохорного процесса при V = const dV = 0. Следовательно, dA — 0. Таким образом, при изохорных процессах работа расширения газа равна нулю.
Для изобарного процесса р = const. Интегрируя уравнение (11,9) в пределах объемов от до У2, получим:
A = p(V2-V1). (11,10)
При изобарном процессе работа газа против внешнего давления равна произведению давления газа на изменение объема (V2 — VJ.
Из уравнения (11,10) можно установить физический смысл универсальной газовой постоянной R. Для этой цели проделаем следующие операции. Раскроем скобки в уравнении (11,10)
A=pV2-pVY. (II, И)
Применим уравнение состояния (1,16) для 1 моль газа при р = = const для нашего случая:
pV2 — RT2i
При подстановке этих значений в уравнение (11,11) получим:
A = RT2 — RT1 = R (Т2 — Тр. (11,12)
Как видим, работа расширения 1 моль газа при р — const равна значению универсальной газовой постоянной (в дж!молъ • град), умноженной на разность температур.
Если в уравнении (11,12) принять разность Т2 — 7\ = Г, то А = R. Следовательно, универсальная газовая постоянная R представляет собой работу, которую совершает 1 моль идеального газа при нагревании его на 1°.
73 —
Рассмотрим работу расширения газа при изотермическом процессе. При этом Т = const, а^величины р и V являются переменными. Используя уравнение состояния идеального газа pV ~ RT, заменим в уравнении (11,9) переменную величину р другой переменной величиной V:
Р=-^-; dA^RT-^-. (11,13)
Проинтегрировав уравнение (11,13) в пределах объемов от до И2, получим: т2 v2
С dV V?
A=RT \ ~ = RT\\n = RT\n~.
J v -Vi
Ту vt
Таким образом, при изотермическом процессе работа расширения 1 моль идеального газа равна:
Л = 7?Т1п-^-. (11,14)
Поскольку давление газа обратно пропорционально объему, можно определить:
A — RT In — . (11,15)
02
При адиабатном процессе одновременно изменяются давление, температура и объем газа. При этом теплообмен с окружающей средой не успевает произойти (например, при явлении взрыва), т. е. Q = const, a dQ ~ 0.
Для характеристики этих процессов воспользуемся математическим выражением первого начала термодинамики:
dQ~dU + dA; O^dUA-dA,
откуда
dA——dUuj\H А——^U = U1 — *
Следовательно, при адиабатном процессе расширение газа происходит за счет уменьшения внутренней энергии.
Если в уравнение первого начала термодинамики для каждого из рассмотренных термодинамических процессов подставить соответствующее значение работы расширения газа, получим равенства: для изобарного процесса
Qp=-.A(7 + p(V2- Vt); (11,16)
для изохорного процесса
= (11,17)
для изотермического процесса
v2
QT = A = RT\n ~А, • (11,18)
V 1
— 74 —
или
О7. = л = /?Т In—, (11,19)
Р2
поскольку при Т—const At/ = 0.
§ 23. Тепловые эффекты химических реакций
Раздел физической химии, изучающий тепловые изменения при химических реакциях, называется термохимией. Начальные основы термохимии впервые были заложены еще М. В. Ломоносовым. Было установлено, что все химические реакции сопровождаются поглощением или выделением тепловой энергии. Реакции, идущие с выделением тепла, получили название экзотермических, а с поглощением тепла — эндотермических. К реакциям первого типа относятся горение угля, спирта, метана, реакции нейтрализации. Примеры экзотермических реакций: разложение водяного пара, карбоната кальция, гидроксида меди, получение йодистого водорода, окиси азота из элементов.
Количество выделенного или поглощенного тепла при той или иной химической реакции называется тепловым эффектом. Его обычно относят к молю реагирующего вещества и выражают в килокалориях или в джоулях (в системе единиц СИ). В технике иногда тепловые эффекты относят к 1 кг вещества, а для газов — к 1 лА
В химии при обозначении тепловых эффектов применяются знаки, обратные тем, которые используются в термодинамике. В термодинамике подводимая к системе теплота носит название положительной, а теплота, которая выделяется,—отрицательной. В химии же, наоборот, тепловой эффект реакций, которые сопровождаются выделением теплоты, считается положительным, а в случае поглощения теплоты — отрицательным.
В термохимии принято химические процессы записывать в виде так называемых термохимических уравнений, в которых указывается тепловой эффект реакций. Например:
Н2 4- —- О2 = Н2О 4- 68,32 ккал (а)
СН4 4-2О2==СО2 4-2Н2О 4-209,1 ккал (6)
Н2О 4-С=Н2 4-СО—31 ккал (в)
эСОд — СаО 4~ СО2 42,8 ккал (г)
Из уравнений (а) и (б) следует, что в результате сгорания 1 моль водорода или метана выделяется соответственно 68,32 и 209,1 ккал тепла. Уравнения (в) и (г) показывают, что при взаимодействии 1 моль воды с одним грамм-атомом углерода поглощается 31 ккал теплоты, а при разложении 1 моль карбоната кальция поглощается 42,8 ккал теплоты.
То количество теплоты, которое выделяется или поглощается при образовании одного моля сложного вещества из простых веществ в термохимии называют теплотой образования данного вещества. Теплота
— 75
образования всегда дается на одну грамм-молекулу вещества. Так, из уравнения реакции
1 1
— н2+ — С12 = НС1(газ) 4-22 ккал £ &
следует, что при образовании одного моля хлористого водорода теплота образования равна +22 ккал.
Количество теплоты, выделяемое или поглощаемое при разложении одного моля сложного вещества на более простые соединения, называют теплотой разложения. Например, теплота разложения карбоната кальция равна —42,8 ккал. В табл. 12 приведены численные значения теплот образования некоторых химических соединений.
Таблица 12
Теплота образования и растворения некоторых химических соединений (ккал/моль*)
< Вещество Теплота Вещество Теплота
образования растворения образования растворения
AgCI (ij 30,3 . 1 КС1 (т) 104,2 —4,5
AgNO3 (т) 29,4 5,4 KNO3 (г) 118,1 —8,6
А12О3 (т) 393,3 КОН (т) 102,0 13,2
BaSO4 (т) 340,2 — I\oS04 (г) 341,8 -6,4
СаО (т) 151,7 — MgSO4 (т) 313,1 20,3
Са(ОН)2 (т) 236,0 3,0 Na2CO8 (т) 271,0 5,6
Са3(РО4)2 (т) 983,0 — NaCl (т) 98,3 -1,2
СН4 (г) 17,889 NaNO3 (т) 111,7 -5,1
СО (г) 26,42 ** Na.,О (т) 99,5 56,3
СОг (г) 94,05 ' 5,9 NaOH (г) 102,0 10,2
СиО (т) 37,5 NH3 (!) 11,04 8,3
FeO (т) 64,5 — NH4C1 (т) 75,0 -3,9
Fe2O:t (т) 195,2 PbSO4 (т) 219,5
Fe3O4 (т) 266,5' SO, (г) 70,9 8,0
FeS (т) 22,8 — SO3 (г) 94,45
НС1 (г) 22,1 17,94 SO3 (ж) 104,2 41,0
Н,0 (г) 57,798 — SOs (т) 105,2 40,0
г12О (ж) 68,317 —- ZnSO4 (т) 233,5 18,5
* Теплота образования химических соединений из простых веществ дана для стандарт* ных условий (/ = 25° С и Р = 760 мм рт. ст.}.
В зависимости от природы растворителя и растворяемого вещества процесс растворения может сопровождаться выделением или поглощением теплоты. Количество теплоты, выделяющееся или поглощаемое при растворении 1 моль вещества, называют теплотой растворения. Теплота растворения зависит от относительных количеств растворителя и растворенного вещества (табл. 13).
Как видно из таблицы, постоянство теплоты растворения наблюдается, когда на 1 моль растворенного вещества приходится более 300 моль растворителя. Таким образом, теплотой растворения называют количество теплоты, поглощающейся или
— 7 б
Таблица 13
Зависимость теплоты растворения 1 моль NaCI от количества растворителя (воды)
Число молей поды Теплота Разность теплоты
на 1 моль хлори- растворения. растворения, ккал
да натрия ккал на 1 моль воды
9 —0,41 — - .
14 . " —0,56 0,0300
33 —0,84 0,0147
100 -1,01 0,0025
324 —1,03 0,0009
выделяющейся при растворении 1 моль вещества в очень большом количестве растворителя, т. е. растворителя берут столько, чтобы при дальнейшем разбавлении раствора уже не наблюдалось добавочного теплового эффекта.
В термохимических уравнениях воду в качестве растворителя обычно обозначают значком «aq» (от латинского аква — вода). Например
(NaCI) + aq = NaCI • aq —1,2 ккал
(NaOH) 4- aq — NaOH- aq 4- 10 ккал.
При растворении твердого вещества в воде (или каком-то другом растворителе) происходит разрушение кристаллической решетки, что, как известно, связано с затратой энергии. Одновременно с этим растворяемое вещество реагирует с растворителем (гидратация или сольватация), образуя соответственно гидраты или сольваты. Процессы гидратации и сольватации являются процессами экзотермическими. Поэтому теплота растворения складывается из двух слагаемых:
Срасти = ~ Qi 4~ Q2 > (11,20)
где Qj — теплота разрушения кристаллической решетки, Q2 — теплота гидратации или сольватации. Знак теплового эффекта QpaCTB зависит от того, какое из слагаемых больше по абсолютной величине. Если вещество обладает прочной кристаллической решеткой, в процессе растворения разрушение его кристаллической решетки будет проходить с поглощением большего количества тепла, чем его выделяется в процессе гидратации, т. е. > Q2. Теплота растворения в этом случае будет отрицательной. Наоборот, у веществ сильно гидратированных и с менее прочной кристаллической решеткой < Q2. Тепловой эффект растворения в этом случае будет положительным. В табл. 12 приведены тепловые эффекты растворения для некоторых веществ.
Свойство веществ растворяться с выделением или поглощением тепла довольно широко используется в технике. Так, вещества с высоким положительным тепловым эффектом используются в химических грелках, а вещества с отрицательным тепловым эффектом применяют для получения низких температур в холодильной промышленности в виде так называемых криогидратных смесей. В табл. 14 приводится примерный состав наиболее широко распространенных холодильных смесей.
— 77 -
Таблица 14
Примерный состав холодильных смесей
Соли Количество соли на 100 вес. ч. воды, вес. ч. Температура смеси, °C
Ацетат натрия CH3COONa .... 85,0 - 4,7
Роданид калия KCSN 150,0 —23,0
Хлорид аммония NH4Cl 30,0 - 5,1
Нитрат натрия NaNO3 60,0 — 13,5
Хлорид кальция СаС12 143,0 —55,0
Хлорид натрия NaCJ 33,0 —21,0
Опыт показывает, что, как правило, процесс нейтрализации сопровождается положительным тепловым эффектом. Процесс нейтрализации сводится к образованию электрически нейтральной молекулы воды за счет нейтрализации положительно заряженного иона Н1” отрицательно заряженным ионом ОН'. Было замечено, что нейтрализация сильной кислоты сильным основанием в водном растворе дает один и тот же тепловой эффект: около 13,7 ккал на грамм-эквивалент кислоты или основания {закон постоянства теплот нейтрализации). Например
NaOJ4 + HCI=NaCl+H2O4-l3,7 ккал
КОНH2SO4 = ~-K2SO4 4-Н2О 4-13,7 ккал 2а 2а
т. е. для обеих реакций
Н++ОН~ =Н2О 4-13,7 ккал
что более точно можно изобразить в виде:
ОН-4- Н3О+— 2Н2О 4-13,7 ккал
Таким образом, теплотой нейтрализации называется количество теплоты, которое выделяется при взаимодействии грамм-эквивалента кислоты с грамм-эквивалентом щелочи. Закон постоянства теплоты нейтрализации не соблюдается при нейтрализации слабых кислот слабыми основаниями: теплота нейтрализации в этих случаях бывает меньше, чем при взаимодействии сильных кислот и оснований. Это объясняется тем, что в реакциях слабых электролитов на тепловой эффект нейтрализации накладывается теплота диссоциации и другие явления.
При смешивании разбавленных растворов солей теплового эффекта не наблюдается. Так, при взаимодействии в растворе
LiCl 4-KBri±LiBr4-KCl
не наблюдается ни выделения, ни поглощения тепла. Это явление (закон термонейтральности) было открыто Г. И. Гессом в 1841 г. и объясняется тем, что никаких существенных изменений с участвующими в процессе ионами (Li+, К+, С1~ и Вг~) не происходит. Однако для не-
— 78 —
обратимых процессов закон термонейтральности не выполняется. Например, в реакции
AgNO3 4- KCI+AgCl | 4-KNO3
выпадает осадок и имеет место тепловой эффект реакции (теплота осаждения).
Еще одно важное для термохимии понятие — теплота сгорания. Теплота сгорания какого-либо вещества есть количество теплоты,
которое выделяется при полном сгорании 1 моль вещества в токе кислорода. В частности, при сгорании органических веществ углерод окисляется до двуокиси углерода, водород до воды и т. п. Например
QH2 4-“т~ О2==
= 2СО2 + Н2О 4-310,62 ккал.
Теплота сгорания пищевых продуктов в живом организме является источником энергии, за счет которой осуществляется его жизнедеятельность.
Теплоты сгорания определяются сжиганием определенного количества вещества в специальном приборе — калориметрической бомбе. Она была сконструирована в свое время Бертло и позднее усовершенствована Малером (рис. 37).
Рис. 37. Разрез калориметра
Калориметр состоит из замкнутого сосуда, имеющего вид бомбы. Внутренний объем его примерно 650 см?, а толщина стенок не менее 8 мм. Изготовляется такая бомба из специальных сортов мягкой ста-
ли, стойких к действию кислот и кислорода. Навеска вещества в специальном тигле помещается в калориметр, который герметически за-
крывается, после чего в него накачивают кислород под давлением в 25 атм. Сжигание в таких условиях происходит практически мгновенно. Воспламенение исследуемого вещества вызывается нагреванием электрическим током специальной спирали 2, находящейся внутри бомбы. Сама бомба помещается в сосуд 4 определенного объема с водой, в которую погружены мешалка и высокочувствительный термометр 5. Тепло, полученное от сжигания вещества в бомбе, нагревает воду калориметра и по разности температур воды определяют тепловой эффект сгорания.
79 —
§ 24. Основные законы термохимии и термохимические расчеты
Во всех химических явлениях выполняется закон сохранения энергии. Соответственно и все законы термохимии являются следствием ' первого начала термодинамики. I
В 1870 г. Лавуазье и Лаплас установили первый закон термохимии: количество тепла, необходимое для разложения сложного вещества на более простые, равно количеству тепла, выделяющемуся при его образовании из простых веществ.
Так, теплота образования одного моля окиси кальция из кальция и кислорода 151, 7 ккал, т. е.
1 Са О2 = СаО 4-151,7 ккал.
Соответственно для разложения одного моля СаО на кальций и кислород необходимо затратить 151,7 ккал, т. е.
1
СаО = Са + — О2 —151,7 ккал.
2 ' \
Закон Лавуазье — Лапласа является частным случаем закона сохранения энергии. Он выполняется при образовании химических соединений из более сложных веществ. Например, теплота образования Li2CO3 из Li2O и СО2 равна 54,2 ккал. Для разложения же одного моля Li2CO3 на исходные оксиды Li2O и СО2 необходимо затратить также 54,2 ккал.
В 1836 г. Г. И. Гесс установил второй закон термохимии: тепловой эффект химической реакции зависит только от начального и конечного состояния реагирующих веществ и не зависит от пути, по которому реакция протекает.
Этот закон также является частным случаем первого начала термодинамики применительно к химическим реакциям, протекающим в изохорных или изобарных условиях.
Так, двуокись углерода можно получить непосредственно, сжигая углерод в кислороде, или же сначала сжигать его до окиси (СО), а затем уже до двуокиси углерода.
Эти два пути можно изобразить в виде следующей схемы:
СО
Суммарные тепловые эффекты в обоих случаях равны: для первого пути реакции С4-О2 — СО2 4*97,8 ккал для второго пути реакции С 4- — О2=СО 4-29,7 ккал
СО 4--^-О2— СО2 4*98,1 ккал
97,8 ккал
Как видим, тепловой эффект первого процесса равен суммарному тепловому эффекту второго процесса.
Закон Гесса имеет большое практическое применение. Он дает возможность вычислять тепловые эффекты не проводя химических реакций. Этот закон выполняется также- в физиологии и в биохимии. Так, количество теплоты, получаемой от окисления пищевых продуктов в организме в результате целой серии сложных реакций, и количество теплоты, выделяемое при сжигании этих веществ в калориметрической бомбе, оказались тождественными (табл. 15).
Таблица 15
Теплота сгорания 1 г пищевых веществ в животном организме и калориметре
Вещество Теплота сгорания в организме, ккал Теплота сгорания в калориметре, ккал
Углеводы 4,1 4,1
Жиры ’ 9,3 9,3
Белки 4,1 5,7
/
В этой таблице приведены средние данные, так как различные углеводы, белки и жиры имеют свои индивидуальные особенности, не полностью окисляются в организме и т. д. В качестве продукта неполного окисления белков из организма выделяется мочевина. Именно этим объясняется, что при полном сжигании белка в калориметрической бомбе теплоты выделяется больше, чем при окислении его в живом организме.
В термохимических расчетах часто пользуются следствиями, которые непосредственно вытекают из закона Гесса.
Следствие первое. Если совершаются две реакции, приводящие из различных начальных состояний к одинаковым конечным, то разница между тепловыми эффектами представляет тепловой эффект перехода из одного начального состояния в другое. Это следствие используется в термохимических расчетах. Например, тепловые эффекты при сжигании угля высокой степени чистоты, алмаза и графита до двуокиси углерода следующие
Суг 4- Ог=СО2 4-97,8 ккал
Сгр 4- О2 = СО2 4-94,05 ккал
Салм Т °2 = СО2 4- 94,50 ккал.
Пользуясь следствием из закона Гесса, можно рассчитать тепловые эффекты перехода из одного аллотропного состояния в другое. Так, при переходе от угля к графиту выделяется 97,8—94,05 = 3,75 ккал на 1 г-агп\ при переходе от алмаза к графиту 94,50—94,05=0,45 ккал\
81 —
при переходе от графита к алмазу поглощается 94,05—94,50 = = — 0,45 ккал.
Следствие второе. Если совершаются две реакции, приводящие из одинаковых начальных состояний к различным конечным, то разница между их тепловыми эффектами представляет тепловой эффект перехода из одного конечного состояния в другое. Это следствие также используется при расчетах. Например, сжигая углерод и окись углерода до двуокиси углерода, можно опытным путем определить их тепловые эффекты
С 4- С>2 = СО2 4- 97,8 ккал (а)
СО 4- — О2 = СО2 4-68, 1 ккал (б)
Вычитая уравнение (б) из уравнения (а), можно вычислить тепловой эффект реакции сжигания углерода до окиси углерода
С 4- ~~~ О2 =СО 4* 29,7 ккал.
£4
Тепловой эффект этой реакции экспериментальным путем определить очень трудно, потому что невозможно сжечь уголь до окиси углерода без того, чтобы не образовалась частично и двуокись углерода.
Закон Гесса дает возможность определять тепловые эффекты таких реакций, которые или не реализуемы, или не могут быть проведены чисто и до конца. На основании этого закона с термохимическими уравнениями можно производить те же действия, что и с обычными алгебраическими уравнениями. Исходя из этого, только что рассмотренный нами пример можно записать в следующем виде
С4-О2 =СО2 4- 97,8 ккал
СО 4-— О2 = СО2 4- 68,1 ккал £
С—СО + — О2=29,7 ккал ^4
или после перестановки членов уравнения
С+4-О2=СО + 29,7 ккал. 4U4
Пример. Определить тепловой эффект реакции
РЬО 4- SO3 = PbSO4 4~ Q ккал
если известно, что тепловые эффекты образования участвующих в этой реакции веществ равныц РЬО = 52,07 ккал!моль, SO3 «= 94,45 ккал/моль, PbSO4 = = 219,50 ккал!моль.
Решение. Напишем термохимические реакции образования РЬО, SO3 и PbSOa
Pb 4-“Т" О2 = РЬО 4-52,07 ккал (а)
Ъ4 3
$ 4" О2 = SO3 4-94,45 ккал (б)
РЬ 4* 5 4~2О2 — PbSO4 4~ 219,50 ккал. (в)
— 82 —
Далее, вычитая из уравнения (в) уравнения (а) и (б), получим
Pb + S + 2О2 = PbSO4 +219,50 ккал.
— РЬ——-О2 =—РЬО — 52,07 ккал
3
—S О2 = —SO3 — 94,45 ккал £
PbO+SO3 = PbSO4+72,98 ккал '
Таким образом, Q = 72,98 ккал.
§ 25. Второе начало термодинамики. Понятие об энтропии
На основе первого начала термодинамики невозможно установить, в каком направлении и до какого предела будет протекать тот или иной процесс, связанный с превращением энергии. Второе начало термодинамики определяет направление превращения энергии, т. е. указывает, какой процесс и в каком направлении может протекать при данных условиях температуры, давления и концентрации без сообщения энергии извне.
Из повседневных наблюдений над естественными процессами в природе известно, что многие из них проходят самопроизвольно без всяких внешних воздействий только в определенном направлении. Так, вода стекает по склону только вниз, а не наоборот, газ распространяется из области высокого давления в область низкого, теплота передается от более нагретого тела к менее нагретому. В обратном же направлении указанные процессы сами собой идти не могут и потому в этом случае они являются несамопроизвольными.
В самом деле, трудно представить, что вода без приложения энергии извне текла бы вверх по склону, или что теплота будет переходить от тела менее нагретого к более нагретому,1 еще более нагревая его, хотя в принципе это не противоречило бы первому началу термодинамики. Отличительной чертой самопроизвольных процессов является их необратимость.
Опыт показывает, что всякий самопроизвольный процесс можно использовать для получения полезной работы. Например, падающая со склона или уступа вода может вращать мельничное колесо или турбину, расширяющийся газ — двигать поршень машины, химическую реакцию можно использовать как источник энергии. Несамопроизвольные переходы энергии могут происходить только при введении в систему энергии извне. Так, чтобы перекачать воду наверх, перенести теплоту из холодной системы в горячую (как в холодильной машине), сжать газ, необходимо затратить энергию. Поскольку во время протекания процесса всегда имеют место невосполнимые потери энергии в виде теплотыу работа, произведенная системой при самопроизвольном процессе, всегда меньше работы, затраченной на возвращение системы в исходное состояние.
-- 83
Наряду с необратимыми процессами термодинамика рассматривает обратимые процессы, т. е. такие, которые могут идти как в прямом, так и в обратном направлениях при бесконечно малом изменении действующих на систему сил и без изменения работоспособности системы в обоих направлениях. В случае самопроизвольно происходящих изменений примерами таких идеальных обратимых процессов могут служить разрядка батареи через потенциометр, дающий разность потен-
циалов противоположного знака, и расширение газа в идеальном цилиндре с поршнем при медленном изменении противодействующего
давления. Поскольку вполне равновесный процесс практически не-
Однако
осуществим, обратимый процесс есть процесс идеальный.
понятие обратимого процесса широко используется в термодина
мике.
Во-первых, в случае обратимого (равновесного) процесса система совершала бы максимум полезной работы; поэтому, сравнивая величину работы, полученной в реальном процессе, с таковой в обратимом процессе, можно судить об эффективности процесса в прямом и обратном направления^. Во-вторых, реальный процесс (химическую реакцию) можно представить протекающим бесконечно медленно и обратимо, что позволяет наиболее просто и однозначно рассчитать термодинамические свойства системы.
Вопросы обратимости и необратимости различных физических и химических процессов, а также перехода теплоты в работу и работы в теплоту разрешаются вторым началом термодинамики. В этом и состоит его важное практическое значение. Зная направление перехода энер
гии, можно при наличии определенного механизма получить полезную работу.
Второе начало термодинамики, так же как и первое,, было выведено из практического опыта, накопленного человечеством. Оно имеет 'не
сколько равноценных формулировок. Одна из них (постулат Клаузиуса, 1850 г.): теплота не может переходить сама собой от менее на-
гретого тела к более нагретому.
В несколько иной форме это утверждение было высказано М. В. Ломоносовым еще в 1747 г. в знаменитом труде «Размышление о причине
теплоты и холода».
Так как обратный переход теплоты идет сам собой путем теплопроводности, можно утверждать, что процесс теплопроводности необратим.
Другая формулировка: процесс, единственным результатом которого является превращение теплоты в работу, невозможен (т. е. невозможно поглощение системой теплоты из окружающей среды и отдача работы, эквивалентной этой теплоте).
Отсюда следует, что процесс превращения работы в теплоту, например путем трения, необратим.
Третья формулировка второго начала термодинамики: невозможно построить такую периодически действующую машину (вечный двигатель второго рода), все действия которой сводились бы к производству работы за счет соответствующего охлаждения теплового источника.
— 84 —
Рассмотрим работу тепловой машины, т. е. машины, производящей работу за счет теплоты, поглощаемой от какого-то тела — теплоотдат-чика.
Схема работы тепловой машины
/
Из схемы ясно, что не вся теплота Qb получаемая рабочим телом, превращается в работу, а лишь некоторая ее часть А = — Q2.
Другая часть теплоты Q2 передается телу с более низкой температурой — теплоприемнику. Таким образом, сущность работы тепловой машины заключается не только в получении теплоты Qj от теплоотдат-чика и в совершении работы Л, но и в передаче некоторого количества теплоты Q2 теплоприемнику, температура которого ниже, чем температура теплоотдатчика.
Следовательно, теплоту никогда не удается полностью (на сто процентов) превратить в работу. Там, где нет перепада температур, т. е. 7\ = Т2> невозможно превратить теплоту в работу. Если бы это условие не было необходимым, можно было бы использовать для получения полезной работы огромные природные запасы теплоты, заключенные, например, в водах морей и океанов. Однако необходимость иметь для этой цели теплоприемник с более низкой, чем вода в океане, температурой, естественно, сильно ограничивает и технически усложняет подобную возможность.
Чтобы получить математическое выражение второго начала термодинамики, следует более детально рассмотреть действия идеальной тепловой машины. Идеальной тепловой машиной мы называем такую машину, которая работала бы без трения и без потерь тепла. В ней рабочим телом является идеальный газ. Работа машины основана на принципе обратимого термодинамического цикла, называемого циклом Карно.
Цикл — это круговой процесс. Рассматриваемый цикл состоит из четырех последовательно совершающихся "процессов: 1) изотермического расширения; 2) адиабатического расширения; 3) изотермического сжатия; 4) адиабатического сжатия газа.
Все процессы проводятся обратимо, в результате чего газ возвращается в исходное положение.
— 85 —
Пусть количество газа, взятого в качестве рабочего тела, равно одному молю, а начальное состояние его характеризуется температурой Т19 давлением Рг и объемом V± (точка А на рис. 38). г 1
В первом процессе изотермическое расширение газа от объема Vx до объема V2 осуществляется при введении его в соприкосновение с теплоотдатчиком, имеющим температуру 7\.
Допускаем, что масса теплоотдатчика так велика, что его температура в этом процессе заметно не изменяется. Поскольку внутренняя
Рис. 38. Цикл Карно
энергия идеального газа зависит только от температуры, то в данном процессе она остается постоянной, и потому согласно уравнению (11,14) работа расширения ликом ты Qp
производится газом це-за счет поглощения тепло-
V2 Q=A1^RT1\n-^.
у 1
рис. 38 этот процесс пред-
На
ставлен отрезком АВ изотермы 7\. Прекратив в точке В подачу тепла и полностью изолировав газ от теплообмена с окружающей средой, предоставим ему адиабатически расширяться. Работа А 2, совершаемая газом при расширении, происходит целиком за счет уменьшения внутренней энергии, т. е. за счет понижения его температуры.
Пусть последняя достигнет некоторого значения Т2. Объем газа в точке С в данных условиях обозначим через V3. При небольших изменениях температуры теплоемкость газа практически остается величиной постоянной, поэтому изменение внутренней энергии идеального газа в этих условиях равно:
Работа, произведенная газом в этом процессе, в точности равна убыли внутренней энергии Л2 = — ЛС, откуда
Л2 = СГ (Т2 — 7\),
где Су — теплоемкость газа при постоянном объеме.
На рис. 38 этот процесс представлен отрезком ВС адиабаты, конечное состояние — точкой С.
При температуре теплоприемника Т2 в результате соприкосновения газа с теплоприемником осуществляется изотермическое сжатие газа до объема V4, чтобы при последующем адиабатном сжатии газ достиг точно исходной температуры 7\. Вся работа Л3, затрачиваемая на сжатие, переходит в теплоту Q2, которая и отводится в холодильник. Следовательно,
~Q2==A3 = RTZ 1п-£-.
^3
— 86 —
На рис. 38 этот процесс представлен отрезком CD изотермы Т2, конечное состояние — точкой D.
Последний процесс — адиабатное сжатие газа — проведем следующем образом: отъединим газ от теплоприемника и сожмем до объема Vi, приведя его этим в исходное состояние.
В этом процессе Внутренняя энергия газа возрастет на величину, равную затраченной работе сжатия:
Д(7 = cv (7\ — -А4 = &U,
Т- е.
= Cv (Т2 - Л) = - Cv (Л -Т2).
На рис. 38 этот процесс представлен отрезком адиабаты DA. Поскольку рассмотренный процесс в целом является круговым, внутренняя энергия газа в конечном состоянии равна таковой в начальном состоянии. Общее количество теплоты, полученное газом, равно общему количеству произведенной им работы:
Qi — Q2 = А 2 -|- А2 -{- Л3 4~ . (11,21)
С учетом того, что А2 и равны по абсолютному значению, но противоположны по знаку, уравнение (11,21) примет вид:
Qi —Q2 —
После подставления в это уравнение соответствующих значений Ак и А з, получим
= “ + ЯПп -Jr"- <П>22)
V £
»
Используя уравнение адиабаты идеального газа, можно доказать, Г2 ' И, что — — —, V, V.
V2
(П.23)
Эго доказательство основывается на использовании уравнения адиабаты
С 'С '
PV Р' » const идеального газа. Согласно этому уравнению для адиабатных процессов, выражаемых кривыми ВС и DA (рис. 38),
Р2ут = Р, р? (а) и PiVy = p4VV (6J>
где y = CptC0.
Для изотермических процессов, выражаемых кривыми А В и CD (рис. 38), справедливы следующие соотношения:
(в) и P3V3 = P4IZ4 (г).
Подставив в уравнение (а) значения и ^я^з из уравнений (в) и (г), получим;
= P4V4^-' (д).
— 87 —
Разделив уравнение (д) на уравнение (б) и произведя соответствующие сокращения и извлечение корня, получим искомое уравнение:
V2 V3 . Я
Vl * 1
Разделив левую часть равенства (11,23) на Q1? а правую на равную величину RT In V2/Vlf получим: 1
V2
* Vi
V2
или
(И, 24)
41 1 1
Коэффициентом полезного действия тепловой машины г) называется отношение количества полученной работы А к количеству поглощенной теплоты Q:
А
“7Г
В рассмотренном случае А = Qr — Q2, где Q2 — количество теплоты, отданное теплоприемнику, следовательно,
(11,26)
11
На основании этого соотношения второму началу термодинамики можно дать еще и такую формулировку: коэффициент полезного действия тепловой машины не зависит от природы и вида тел, участвующих в процессе, а зависит только лишь от разности температур теплообменника (Тг) и теплоприемника (Т2). 1
Так как 7\ > Т2, то коэффициент полезного действия машин может изменяться в пределах от 0 (при Тг — Т2) до единицы (когда Т2 = = 0° К). Однако получить к. п. д. от тепловой машины 1] = 1 невозможно, так как нельзя создать теплоприемник с температурой, равной абсолютному нулю. Следовательно, даже в идеальном случае существует предел превращения теплоты в работу.
На основе анализа работы идеальной машины Карно и взаимных превращений различных видов энергии друг в друга можно сделать следующий общий вывод: любая форма энергии может полностью перейти в теплоту, но теплота преобразуется в другие формы энергии только частично.
Основываясь на этом, термодинамика доказывает, что внутренняя энергия U не может быть полностью превращена в работу при Т — = const. Поэтому условно запас внутренней энергии системы можно представить в виде двух слагаемых:"
(11,27)
- 88 -
где F — полезная часть внутренней энергии, которая способна произвести работу и которая по предложению Гельмгольца названа свободной энергией, G — непроизводительная часть, так называемая связанная энергия, которая ни при каких условиях не может быть превращена в полезную работу и которая способна переходить только в теплоту и рассеиваться.
Свободная энергия в любой системе заключена в виде потенциальной энергии. По мере совершения системой работы ее энергия убывает. Чем больше система содержит свободной энергии, тем большую работу сна сможет совершить. Так, более разреженный газ содержит меньше свободной энергии и больше связанной, чем сжатый газ при той же температуре. Следовательно, сжатый газ способен совершить больше полезной работы.
Величина непроизводительной, т. е. «обесцененной», части энергии G тем больше, чем меньше разность температур в системе. Мерой такого «обесценения» энергии является термодинамическая функция, зависящая от состояния системы и названная Клаузиусом энтропией (греческое— обращать внутрь). Эта функция обозначается в термодинамике буквой S.
Остановимся несколько подробнее на ее характеристике. Из анализа цикла Карно коэффициент полезного действия
Qi — (?2 — Т%
П Qi Л ’
откуда
Если любой цикл произвольного вида разбить бесконечно большим числом адиабат и изотерм на множеством элементарных циклов Карно, отвечающих бесконечно малым количеством теплоты 6Q, принимаемым в отдаваемым рабочим телом, то для каждого из них можно записать: dQ' 6QJ 6Q-
О 5 " -- 0> ——- ~~~~ И 1 * Д« a
T2 Tz Л T2
ine^Q/T называется приведенной теплотой.
Алгебраическая сумма приведенных теплот цикла равна нулю, т. е.
В пределе (м ос) эта сумма переходит в интеграл №Q1T (интеграл, взятый по замкнутому контуру). В теории интегралов доказывается, что если интеграл по замкнутому контуру равен нулю, то подынтегральное выражение является полным дифференциалом не
— 89 —
которой функции от параметров, определяющих состояние системы. Можно записать:
dS = ~. (11,29)
где S —функция состояния системы S ~ f (Р, V, Т, ...). Именноэта функция (S), введенная Клаузиусом (1850), и была названа энтропией.
Так как dS является полным дифференциалом, то изменение энтропии в каком-либо процессе зависит только от начального и конечного состояний и не зависит от пути перехода'.
(11,30)
где AS — изменение энтропии при переходе системы из состояния (1) в состояние (2).
Выражение (11,29) является математической записью второго начала термодинамики для обратимых процессов. Подставляя в уравнение первого начала термодинамики (11,8) вместо 6Q равную величину TdS из уравнения (11,29), получим аналитическое выражение первого и второго законов термодинамики для обратимых процессов:
dU = TdS—t)A. (11,31)
Наиболее просто изменение энтропии AS определяется для обратимых изотермических процессов. Для них из уравнения (11,29) вытекает, что
AS =—, , (11,32)
т. е. в обратимых изотермическИ/Х процессах изменение энтропии равняется тепловому эффекту процесса, деленному на абсолютную температуру. Изменение энтропии рассчитывается на 1 моль и выражается в калориях на градус. Единица измерения энтропии (так называемая «энтропийная единица», сокращенное, е.) имеет следующее выражение:
1 5. е. — 1 кал/град'моль
(энтропийная единица в СИ выражется в дж/град • моль).
Так, при плавлении льда при 0° С поглощается теплота в количестве 1436,3 кал! моль (мольная теплота плавления). Следовательно, изменение энтропии в этом процессе равно:
1436,3
Лэ ---------^5,26 кал/град• моль.
273,16
До сих пор, когда мы говорили об изменении энтропии, имелись в виду обратимые процессы; установлено, что величина AS для бесконечно мало обратимого процесса выражается уравнением (11,29). Однако, как мы уже знаем, обратимые процессы являются идеальными. Реальные же процессы, протекающие в природе, практически являются необратимыми, так как при любых превращениях часть энергии пере
— 90 —
ходит в теплоту, а последняя, как следует из второго начала термодинамики, может превращаться в другие виды энергии лишь частично.
На основании многочисленных опытов было установлено, что во всяком необратимом процессе коэффициент полезного действия всегда меньше, чем в обратимом процессе, т. е.
в пределе алгебраическая сумма < 0 превратится в f6Q/7 <
< 0.
Из этого следует, что при необратимых процессах будет иметь место неравенство
60
(11,33)
В системах изолированных, т. е. лишенных теплообмена с окружающей средой, процессы протекают без изменения тепловой энергии (6Q = 0). Поэтому в изолированных системах при изотермических и полностью обратимых процессах изменение энтропии (dS) равно нулю.
Если в замкнутых системах при Г — const и I/=const будут идти необратимые процессы, энтропия этих систем будет возрастать.
В изолированных системах самопроизвольно могут протекать только процессы, которые сопровождаются увеличением энтропии. Это означает, что состояние устойчивого термодинамического равновесия в изолированной системе отвечает максимуму энтропии:
AS = гпах
Энтропия изолированной системы ни при каких условиях не может самопроизвольно убывать. Однако из всех предыдущих рассуждений нельзя сделать вывод о том, что процессы, протекающие с уменьшением энтропии, вообще невозможны. Дело в том, что подобные процессы в изолированных системах не могут возникать самопроизвольно, так как для их осуществления необходим определенный теплообмен с окружающей средой.
Для удобства запоминания все сказанное выше об энтропии представим схемой:
Неизолированные системы
Обратимые процессы
Необратимые процессы
Изолированные системы
dS=O;
S — const;
dS > 0;
S растет.
91
Уравнения первого и второго начал термодинамики, пригодные для обратимых и необратимых процессов, можно записать так:
dl/<TdS —6Л;
6Л < TdS—dU.
В этих уравнениях U и S не зависят от пути течения процесса. Работа А зависит ст способа проведения процесса и будет максимальной, когда процесс полностью обратим. Эту максимальную работу обычно обозначают Лтах. В необратимых процессах работа всегда меньше, т. е. А < Лтах, поэтому отношение ЛМтах 1 может служить мерой необратимости.
Больцман и Гиббс показали, что энтропия есть некая средняя величина, которая является функцией неупорядоченности (хаотичности) движения множества молекул, т. е. имеет явно выраженный статистический характер.
С точки зрения молекулярно-кинетических представлений второе начало термодинамики можно сформулировать следующим образом. Все процессы, происходящие в природе, стремятся перейти самопроизвольно от состояния менее вероятного к состоянию более вероятному. Для молекул наиболее вероятным является беспорядочное, хаотичное движение, т. е. тепловое движение. Работа характеризуется более или менее упорядоченным движением частиц, каковое является менее вероятным. Отсюда самопроизвольный переход работы в теплоту можно рассматривать как переход молекулярной системы от упорядоченного движения частиц к более вероятному. В этом случае энтропия выступает как своеобразная мера хаотичности движения частиц в системе, т. е. она имеет статистический характер.
При нагревании хаотичность, беспорядочность движения молекул усиливается и энтропия системы повышается. Например, при нагревании воды в пределах от 0 до 25° С энтропия ее возрастает на 1,58 кал!град • м'оль.
Для сопоставления различных систем пользуются понятием стандартной энтропии, т. е. энтропией, вычисленной для температуры 25° С при нормальном давлении 760 мм рт. ст. Эта температура соответствует абсолютной температуре Т = 298° К и потому стандартная энтропия обозначается S298. Ниже приведены значения стандартной энтропии для некоторых веществ:
СО2 Н2О NaCl Na2CO3 С2Н5ОН
S298, э.е.............. 51,061 16,716 17,30 32,50 38,40
Таким образом, в силу статистического характера второе начало не применимо для системы с одной или малым числом частиц или молекул, т. е. для микрообъемов. Так, например, в газах вследствие хаотичности теплового движения молекул в отдельных микрообъемах плотность может кратковременно оказаться более высокой по сравнению со средней плотностью газа в макрообъеме. Это явление носит название флуктуации. Образование микроучастков с повышенной плотностью газа
— 92 —
протекает самопроизвольно и с уменьшением энтропии, что не согласуется со вторым началом термодинамики. Аналогичные явления могут происходить и в отдельных микрообъемах растворов: концентрация частиц растворенного вещества на какой-то краткий период может оказаться в микрообъеме выше средней концентрации в макрообъеме.
§ 26. Критика теории тепловой смерти Вселенной
Рассматривая Вселенную как изолированную систему, Клаузиус распространил и на нее представление о возрастании энтропии при самопроизвольных процессах. Опыт показывает, что при всех реальных процессах происходит хотя бы частичное преобразование любого вида энергии в теплоту и вместе с тем выравнивание температуры всех взаимодействующих тел. Отсюда* Клаузиус сделал вывод: энтропия Вселенной стремится к максимуму.
Это означает, что в природе идет постоянная деградация энергии. Различные виды энергии могут переходить полностью в тепловую энергию, в то время как тепловая энергия в другие виды энергии может переходить лишь частично. В результате вся энергия обесценивается, так как она постепенно превращается в теплоту, которая представляет собой хаотическое движение молекул. По истечении некоторого промежутка времени во Вселенной, по мнению Клаузиуса и его сторонников, будет существовать только один вид энергии — энергия беспорядочного движения частиц, равномерно распределившихся между телами Вселенной и обладающими одинаковой температурой. При этом исчезнет возможность самопроизвольного возникновения каких-либо процессов, так как энергия уже потеряет свою способность к превращениям. Наступит состояние так называемой «тепловой смерти» Вселенной, т. е. состояние вечного равновесия.
Из этой концепции можно сделать далеко идущие выводы. В самом деле, если неизбежен конец Вселенной, значит она имела и свое начало, а следовательно, существует и какая-то сила, стоящая над природой и способная вызвать ее к жизни, в результате какого-то первичного толчка ... и т. п. '
Эта теория от начала и до конца является сугубо идеалистической, ибо, по меткому замечанию Ф. Энгельса, исходит из молчаливого признания акта творения и творца («мировые часы сначала должны быть заведены»).
Современная наука начисто отвергает ложную концепцию о «тепловой смерти» мира. Колоссальный запас знаний, накопленный человечеством за всю историю его развития, убедительно доказывает и то, что мир бесконечен, и то, что развитие его происходило вечно и вечно будет продолжаться. Основная ошибка гипотезы Клаузиуса заключается в том, что второе начало термодинамики, в отличие от первого начала, не является абсолютным законом природы, а имеет относительный характер. Этот факт был вскрыт в работах Больцмана (1895) и Смо-луховского (1914). Эти ученые показали, что нельзя Вселенную рассматривать как замкнутую изолированную конечную систему, а потому к ней неприменимо второе начало термодинамики. Естественно считать, что при иных условиях существования материи, сильно отличающихся от тех, которые имеют место на Земле, процессы могут протекать и в обратном направлении, т. е. с убыванием энтропии. Об этом свидетельствуют наблюдения астрономов и астрофизиков за рождением новых миров. Кроме того, к явлениям микромира, как известно, второе начало термодинамики также неприменимо.
С философских позиций ложность тезиса о «тепловой смерти» мира была вскрыта еще Ф. Энгельсом (1875—1876) в его классическом труде «Диалектика , природы». В этой работе он отмечает, что закон возрастания энтропии (второе начало термодинамики), распространенный на всю Вселенную, не совместим с законом сохранения и превращения энергии, так как, исходя из теории «тепловой смерти», мы непременно сталкиваемся с качественным уничтожением энергии, т. е. с преобразованием ее в вид, в котором она становится неспособной к обратным превращениям.
Полемизируя со сторонниками «тепловой смерти» Вселенной, Ф. Энгельс рисует замечательную картину вечно движущейся материи: «Вот вечный круговорот, в котором движется материя, — круговорот, который завершает свой путь лишь в такие промежутки времени, для которых наш земной год уже не может
\ - 93 -
служить достаточной единицей измерения; круговорот, в котором время наивысшего развития, время органической жизни и, тем более, время жизни существ, сознающих себя и природу, отмерено столь же скудно, как и то пространство, в пределах которого существует жизнь и самосознание; круговорот, в котором каждая конечная форма существования материи—безразлично, солнце или туманность, отдельное животное или животный вид, химическое соединение или разложение — одинаково преходяща и в котором ничто не вечно, кроме вечно изменяющейся, вечно движущейся материи и законов ее движения и изменения. Но как бы часто и как бы безжалостно ни совершался во времени и в пространстве этот круговорот; сколько бы миллионов солнц и земель ни возникало и ни погибало; как бы долго ни длилось время, пока в какой-нибудь солнечной системе и только на одной планете не создались условия для органической жизни; сколько бы бесчисленных органических существ ни должно было раньше возникнуть и погибнуть, прежде, чем из их среды разовьются животные со способным к мышлению мозгом, находя на короткий срок пригодные для своей жизни условия, чтобы затем быть тоже истребленными без милосердия, — у нас есть уверенность в том, что материя во всех своих превращениях остается вечно одной и той же, что ни один из ее атрибутов никогда не может быть утрачен и что поэтому с той же самой железной необходимостью, с какой она когда-нибудь истребит на Земле свой высший цвет —мыслящий дух, она должна будет его снова породить где-нибудь в другом месте и в другое время»*.
Таким образом, для материалиста ясно, что во вселенной, помимо процессов рассеивания энергии, совершаются процессы и обратного характера.
§ 27. второе начало термодинамики и живые организмы
. Живые организмы являясь телами природы, подчиняются всем ее основным законам. К ним полностью применим закон сохранения и превращения энергии, а также и второе начало термодинамики.
В процессе жизнедеятельности любой организм, растительный и животный, осуществляет постоянный обмен вещества с окружающей средой. Он поглощает в качестве пищи разнообразные вещества, ассимилирует и трансформирует их в составные части своего тела, а затем в процессе диссимиляции разрушает и удаляет в виде отработанных продуктов во внешнюю среду. Многочисленные эксперименты установили, что все эти процессы строго подчиняются закону сохранения материи.
Однако некоторые ученые утверждают, что в отличие от неживых систем организмы являются накопителями энергии — следовательно, в них идут процессы, противоречащие второму началу термодинамики, иными словами, рассматривают живые организмы как системы не энтропические, а эктропические (накопители свободной энергии). Ошибочность подобных вглядов обусловлена тем, что живой организм рассматривается вне связи со средой его обитания. Диалектика же учит, что организм необходимо рассматривать в его тесной и неразрывной связи с окружающей средой.
Действительно, необходимую для процессов жизнедеятельности энергию живой организм черпает из пищевых продуктов, которые являются носителями химической энергии высокого потенциала. При распаде этих веществ в организме эта энергия высвобождается и используется организмом на производство тепла, механическую работу, на реакции различного синтеза. Продукты распада живого организма содержат значительно меньше химической энергии, поэтому с этой точки зрения приложимость второго начала термодинамики к органическому миру не вызывает сомнений.
Но, с другой стороны, живые организмы являются системами открытыми, поэтому, используя энергию обмена, могут сами заряжаться до более высокого потенциала и с этой точки зрения имеет место противоречие второму началу термодинамики. Так, зеленые растения для повышения энергетического потенциала используют солнечную энергию, а животные — энергию, поступающую с пищей. Таким образом, хотя энтропия самого организма может изменяться в любом направлении, т. е. она может уменьшаться за счет непрерывного поглощения сво-
* Ф. Энгельс. Диалектика природы. Политизда!, 1965, стр, 23.
— 94 —
бедной энергии из окружающей среды, энтропия системы организм — среда, взятой в целом, несомненно увеличивается. Это дает основание для общего вывода: для живых организмов, как и для тел неживой природы, полностью выполняются законы термодинамики.
§ 28. Третье начало термодинамики. Принцип минимума свободной энергии
Третье начало термодинамики (или тепловая теорема Нернста, 1907 г.) позволяет вычислить значение свободной энергии, зная тепловой эффект реакции, и таким образом определить направление реакции. Тепловая теорема Нернста имеет следующую формулировку: при абсолютном нуле энтропия любого однородного тела равна нулю. Основные положения тепловой теоремы заключаются в следующем:
1. При абсолютном нуле свободная энергия равна теплоте реакции, т. е.
р о = Qo.
2. Не только при абсолютном нуле, но и при температурах, близких к нему, FT = Qj, т. е.
dF dQ
---= —=0.
dT dT
В 1907 г. Эйнштейн доказал, что теплоемкость твердых тел* при абсолютном нуле (Т — 0) должна быть равна нулю. Благодаря тепловой теореме Нернста оказалось возможным определять абсолютную величину энтропии на основании измерения теплоемкостей при разных температурах. На основании этой теоремы можно вывести известные соотношения, которые позволяют по тепловому эффекту реакции вычислять ее свободную энергию, а зная последнюю, можно предсказать и направление реакции.
Любая термодинамическая система находится в устойчивом состоянии в том случае, если она содержит минимум свободной энергии, максимум энтропии и минимум различий в интенсивности. При изменении этих условий равновесие тотчас же смещается, а в системе самопроизвольно возникают процессы, которые вновь приводят свободную энергию к минимальному для данных условий уровню. Таким образом, состояние системы, соответствующее минимуму свободной энергии, является состоянием устойчивого равновесия при данных условиях:
F — min и АД = 0 (при T=const).
Из всего вышеизложенного можно сделать вывод: в изолированных системах самопроизвольно могут протекать только процессы, направленные в сторону понижения свободной энергии системы.
Принцип минимума свободной энергии имеет исключительно большое значение не только для физической химии, но и для всего естест-
* Напоминаем, что под теплоемкостью подразумевают количество теплоты в калориях, необходимое для нагревания тела на 1° С. Если же это относится к 1 г вещества, то такая теплоемкость называется удельной.
- 95 —
i
вознания. Этот принцип указывает направление процессов в той или иной системе, устанавливает условия термодинамического равновесия, которое может существовать до тех пор, пока один из параметров состояния не нарушит этого равновесия.
Непосредственное определение свободной энергии связано с большими трудностями. В случае неполяризующихся электродов можно выразить свободную энергию химической реакции путем измерения электродвижущей силы. Однако таким способом можно определять величину свободной энергии только в обратимых окислительно-восстановительных системах, где измерение электрического потенциала дает меру химического потенциала окислительно-восстановительных реакций. В других случаях можно использовать соответствующие таблицы, которые содержат величины стандартных свободных энергий теплот образования различных веществ. В табл. 16 приведены эти данные для некоторых веществ.
Таблица 16
Стандартные свободные энергии образования некоторых веществ AZ^s при 25° С и 1 атм
Вещество О AZ298’ ккал/моль Вещество AZ298* ккал /моль Вещество AZ298' ккал/моль
н2о -54,629 Н2 (г) —7,83 А1.2О3 (т) —376,77
Н2О (ж) —56,681 NH3 (г) —3,14 A12SO4 (т) —738,99
на (г) —22,74 СО (г) —32,778 СН,СООН (ж) — 93,80
НВг (г) — 12,50 СО2 (г) —94,242 (СООН)2 (ж) — 166,80
NO (г) + 20,10 SO2 (г) —71,60 С,Н6ОН (ж) - 41,75
NO2 (г) -4-12,28 SO., (г) —88,52 ' "* —
Величину свободной энергии F образования вещества при стандартных условиях (при 7=298° К и постоянном давлении Р—1 атм) называют в термодинамике также потенциалом образования, или изобарным потенциалом и обозначают символом AZ^e (табл. 16).
Изменение изобарного потенциала AZ (или, что то же самое, изменение свободной энергии), отвечающее протеканию какой-нибудь данной химической реакции, равно разности между изобарными потенциалами конечных продуктов реакции и исходных веществ:
AZ = S (щ Z/)koh — ^г)пач* (П»34)
Так, для химической реакции mA + пВ = рС + qD имеем:
AZ-,F=(pZc —(?ZD)—(/rtZA —7iZB), (11,35)
где Zc, ZD, Za и ZB — изобарные потенциалы компонентов. Поскольку значения этих потенциалов не известны, вместо них используют значение изменений изобарного потенциала, происходящих при образовании данного соединения из простых веществ, т. е. значения AZ°29 из табл. 16. В частности, для реакции, приведенной выше, можем написать:
А — pAZo6p: с </AZo6p D) —(mAZO6p. а п^обр. в)* (И >36)
— 96 —
Если AZ реакции имеет отрицательное значение, то реакция идет с выделением энергии. В этом случае она протекает самопроизвольно в данном направлении.
ассмотрим в качестве примера реакцию получения кристаллического сульфата алюминия из А12О3и газообразной трехокиси серы при 25° С (все в стандартных состояниях):
А1гО3 4-3SO3 —Al2 (SO4)3, AZ298~^»
Из данных табл. 16 имеем:
2AI 4-О2 = А12 О3,
2Л! 4-3S 4-6O2 = Al2(SOd)3,
S —Ог = SO3,
AZ298 ~—376,77 ккал/моль
= —738,99 ккал/моль
AZ^ = —88,52 ккал/моль.
Для определения AZ298 реакции воспользуемся уравнением
AZ298 = —738,99 — (-376,77—3 88,52) =—96,66 ккал.
Данная реакция идет самопроизвольно, так как AZ298 этой реакции имеет отрицательное значение.
Таким образом, зная AZ29s для всех веществ, участвующих в реакции, можно рассчитать AZ2gs и самой реакции, а следовательно, и направление этой реакции.
Изобарный потенциал реакции можно определить также через энтропию системы. Этот расчет основан на определении теплоемкости при разных температурах. Количество теплоты, которое поглощается телом, равняется, как известно, произведению его теплоемкости на повышение температуры, т. е.
dQ = CpdT, (11,37)
где Ср—теплоемкость при постоянном давлении. Подставляя выражение dQ в уравнение (11,29), получим:
dQ dT
dS = -^- =CP — = CPd\nT. (11,38)
Интегрируя dS в пределах температур от нуля до Т, имеем: т
Sj = Ср d In Т 4* Sq, о
(П,39)
где So — энтропия при абсолютном нуле.
Согласно третьему началу термодинамики энтропия при абсолютном нуле равна нулю; тогда уравнение (11,39) запишется в таком виде:
1
Ср d In Г. (11,40)
о
4 Зак 5G0
— 97 —
Таким образом, на основе третьего начала термодинамики можно определить абсолютную величину энтропии химического соединения, измеряя теплоемкости вещества при разных температурах*.
Энтропия же химической реакции будет равна разности энтропий реагирующих веществ и продуктов реакции. Другими словами, по этой разности определяется изменение (прирост) энтропии данного химического превращения.
Отсюда, зная теплоту реакции, легко рассчитать изменение свободной энергии:
dF—dU — TdS.
§ 29. Максимальная работа и химическое сродство
Опыт показывает, что способность различных веществ вступать во взаимодействие друг с другом неодинакова. Эту способность веществ вступать в химическое взаимодействие друг с другом с образованием качественно новых веществ называют химическим сродством. Химическое сродство реагирующих веществ определяет их реакционную способность.
Бертло (1867) сформулировал принцип, согласно которому всякая система, способная к химическим превращениям, будет преобразована в такую, образование которой сопровождается наибольшим выделением теплоты (так называемый принцип наибольшей работы). Если бы это было так, то тепловой эффект мог бы являться мерой химического сродства реагирующих веществ.
Однако существование реакций, протекающих с поглощением теплоты, противоречит принципу Бертло. Он имеет ограниченное значение и, в частности, подтвердился только для эндотермических реакций. Таким образом, тепловой эффект реакции не может служить мерой химического сродства.
В 1888 г. Вант-Гофф предложил за меру химического сродства, проявляемого в данной реакции, принимать максимальную работу, иными словами, свободную энергию, выделяющуюся при данном процессе. Максимальная работа** есть наибольшая работа, которую может дать химическая реакция, осуществляемая в условиях полной обратимости. Такие условия практически трудно осуществимы, но к ним можно приблизиться.
В самом деле, с одной стороны, направление любой химической реакции определяется наличием сил химического сродства. С другой, согласно второму началу термодинамики, любой самопроизвольный процесс ( в том числе и химический) идет в направлении, при котором
* Обычно следует учитывать изменение теплоемкости в зависимости от того, идет ли процесс при постоянном давлении или при постоянном объеме. Кроме того, необходимо также учитывать агрегатное состояние вещества. Так, если вещество находится в жидком или газообразном состоянии, то вводится поправка, соответствующая плавлению или испарению.
** Здесь имеется в виду максимально полезная работа химической реакции, т. е. максимальная работа химического процесса за вычетом работы расширения.
— 98 —
система совершает максимальную работу, т. е. в направлении макси* мальной убыли свободной энергии. Сопоставляя оба эти положения, можно сделать следующий вывод: максимальная работа, совершаемая химической системой и равная убыли ее свободной энергии, является мерой химического сродства.
Этот принцип можно записать в виде уравнения:
F1 F2 = L = Лтах, (11,41)
где и F2 — свободная энергия химической системы в начальном и конечном ее состояниях, L — химическое сродство, выраженное в механических или тепловых единицах, Лтах — работа процесса.
Например, для хлорида натрия F = — 384,0577 кдж!моль.
Такая большая убыль свободной энергии химической реакции
1
Na 4- — С12 = NaCl
служит наглядным примером большого химического сродства между Na и С1. Для газообразного йодистого водорода HI при 25° С и давлении 1 атм F=-\-1,3129 кдж!моль. Положительное значение свободной энергии свидетельствует о том, что это соединение малоустойчивое и сравнительно легко распадается на иод и водород.
Принимая во внимание второе начало термодинамики, можно показать, что максимальная работа Лгаах любой химической реакции всегда одна и та же, каким бы путем эта реакция ни протекала, лишь бы процесс был изотермический и обратимый. Иными словами, Лтах для каждого химического процесса зависит только от конечного и начального состояния системы. Если бы при переходе какой-либо системы из одного состояния в другое можно было при одном обратимом процессе совершать большую работу, чем при другом, тоже обратимом, то за счет этого удалось бы реализовать идею вечного двигателя (регре-tuum mobile) второго рода, что невозможно.
Зная максимальную работу процесса, можно определить направление течения той или иной химической реакции. Так, если Лтах больше нуля, т. е. если сродство положительно, то реакция пойдет в прямом направлении, если Лтах = 0, то система будет находиться в состоянии равновесия, если же Лшах меньше нуля, реакция пойдет в обратном направлении.
Глава III
УЧЕНИЕ О РАСТВОРАХ
§ 30. Растворы — физико-химические системы. Концентрация растворов
Растворами называют состоящие из двух или нескольких ве- I ществ гомогенные системы, состав которых мод$ет_ изменяться в до- I вольно широких пределахД Свойства растворов (плотность, темпера- I тура кипения, температура замерзания, вязкость и др.), как правило, j изменяются постоянно, плавно. .
Растворы сходны как с механическими смесями частиц, так и I с индивидуальными химическими соединениями. От первых они отличаются тем, что любой макроскопический объем раствора обладает таким же химическим составом и физическими свойствами, как и вся его масса. От химических соединений растворы отличаются тем, что их состав может изменяться в зависимости от количеств взятых компонентов и они не подчиняются закону кратных отношений. Так, состав , водного раствора хлорида натрия может произвольно меняться в пределах, допустимых его растворимостью. В 100 г воды при 20° С можно растворить любое количество NaCI в пределах от 0 до 36,8 г, что соответствует предельной растворимости соли при данной температуре. Растворы отличаются от химических соединений также и природой связи. Если для химических соединений характерны в основном ионная и ковалентная связи, то для растворов характерны более слабые ван-дер-ваальсовы, а в некоторых случаях и водородные связи. I
В отличие от простого смешивания, при растворении веществ происходит определенное взаимодействие между частицами, образующими раствор. Вещество, которое при растворении не меняет своего агрегатного состояния или же входит в состав раствора в преобладающем количестве, обычно называют растворителем. Необходимо отметить, что понятия «растворитель» и «растворенное вещество» имеют смысл I лишь в том случае, когда концентрация растворенного вещества в растворителе не велика. Если взять раствор, содержащий 50% спирта и 50% воды, то его в одинаковой мере можно рассматривать как раствор 1 спирта в воде и воды в спирте. В подобных случаях удобнее говорить о компонентах раствора.
По агрегатному состоянию растворы делят на три группы: 1) растворы газов в газах (газовые смеси); 2) жидкие растворы; 3) твердые j растворы (изучаются с фазовыми равновесиями).
Примерами твердых растворов являются сплавы различных металлов, а газообразных — воздух.
В данном разделе физической химии будут рассматриваться только жидкие растворы.
Жидкие растворы в свою очередь подразделяются на: растворы газов в жидкостях; растворы жидкостей в жидкостях; растворы твердых тел в жидкостях.
— 100 —
Особое значение имеют водные растворы, так как подавляющее большинство процессов в природе совершается в водной среде. Водные * растворы играют исключительно важную роль во всех процессах, протекающих в почвах, а также в животных и растительных организмах. Все природные воды (морская, речная, воды минеральных источников и т. п.) представляют собой не что иное, как растворы различных солей. Различные биологические жидкости — плазма крови, лимфа, соки растительных организмов и другие также содержат в растворенном состоянии органические и неорганические вещества. Иными словами, растворы — наиболее распространенные системы в природе, и потому учение о растворах является важным разделом физической химии.
Процесс растворения нельзя рассматривать как простое механическое распределение одного вещества в другом. При растворении имеет место физико-химическое взаимодействие растворяемого вещества с молекулами растворителями. Процесс растворения часто сопровождается выделением или поглощением тепла (теплота растворения), а также уменьшением или увеличением объема раствора. Так, растворение серной кислоты или едкого натра в воде сопровождается таким же тепловым эффектом, как и обычные химические реакции. Это сви-детельствует о том, что молекулы (или ионы) растворенного вещества / образуют с молекулами растворителя химические соёдиненияТЭпТсое-динения называют сольватами, а процесс их образования — сольвата-цией\ в случае, когда растворителем является вода, их называют гидратами, а процесс их образования гидратацией.
Когда учение о растворах выделилось в самостоятельный раздел химии, определились две точки зрения на природу растворов, две теории растворов — физическая и химическая.
Основы физической теории растворов были заложены уже во второй половине XIX в. Сванте Аррениусом и Вант-Гоффом. Согласно этой теории процесс растворения рассматривается как чисто физический процесс равномерного распределения частиц растворяемого вещества по всему объему растворителя, который представляет собой некую индифферентную среду. При этом допускают, что никакого взаимодействия между молекулами растворителя и частицами растворенного вещества не существует. Физическая теория растворов подкреплялась тем, что целый ряд свойств растворов — повышение температуры кипения, понижение температуры замерзания, давление пара, осмотическое давление действительно зависят только от концентрации растворенного вещества, но не зависят от его природы. Таким образом, растворы, согласно этой теории, представляются как однородные смеси молекул, в которых состояние растворенного вещества подобно состоянию газа.
Однако по мере накопления фактического материала постепенно физическая теория растворов уступила место так называемой гидратной теории, основоположником которой был Д. И. Менделеев. Значительный вклад в эту теорию позднее внесли И. А. Каблуков, Н. С. Кур-наков и другие советские ученые.
Д. И. Менделеев на основании исследований свойств водных растворов серной кислоты, этилового спирта и других веществ пришел к выводу, что между молекулами компонентов раствора существует
— 101 -
взаимодействие, приводящее к образованию нестойких соединений частиц растворенного вещества с молекулами растворителя Так, в растворах серной кислоты Менделеев установил наличие гидратов следующего состава: H2SO4 е Н2О, H2SO4 • 2Н2О, H2SO4 • 4Н2О и др. Он установил также, что соединение молекул или ионов растворяемого вещества с молекулами растворителя осуществляется главным образом за счет водородной связи или же вследствие электростатического взаимодействия полярных молекул веществ.
Уместно напомнить, что водородная связь — это как бы вторая (побочная) валентность водородного атома, которую он может проявлять по отношению к сильно отрицательным атомам, если основная валентность связывает его с атомом, тоже сильно отрицательным в данной молекуле — фтором, кислородом, азотом. Она образуется вследствие притяжения между ковалентно связанным атомОхМ водорода (протон) и свободными электронами электроотрицательного атома другой молекулы. Этот вид связи свойствен любым агрегатным состояниям вещества.
Необходимо отметить, что Менделеев не отрицал значения физических факторов в процессах растворения; он предсказывал создание общей теории растворов, основанной на учете как физической, так и химической стороны процесса растворения. И действительно, современная теория растворов основана на синтезе «химической» и «физической» точек зрения.
Однако проблема растворов полностью еще не разрешена. Теория растворов еще не позволяет в любом случае предопределять свойства растворов по свойствам их компонентов. Объясняется это чрезвычайно большим многообразием и сложностью взаимодействий между молекулами растворителя и частицами растворенного вещества. Структура растворов, как правило, бывает значительно сложнее строения его отдельно взятых компонентов.
Наиболее изучены разбавленные растворы. В них частицы растворенного вещества настолько отделены друг от друга молекулами растворителя, что взаимодействие между ними выражено очень слабо и природа растворенного вещества практически не оказывает влияния на свойства разбавленного раствора. Аналогично свойствам газа свойства разбавленных растворов не зависят от состава частиц растворенного вещества и их размеров, а только от числа частиц в единице объема, т. е. от концентрации.
Как уже говорилось, состав раствора — концентрация является его важнейшей характеристикой.
Концентрация раствора указывает количество вещества, которое содержится в определенном весовом количестве раствора или растворителя или в определенном объеме раствора. В химии применяются разнообразные способы выражения концентрации растворов; это обусловлено тем, что для разных целей и областей исследования оказываются удобными и рациональными разные способы выражения концентрации. В практической работе важно уметь быстро переходить от одних единиц концентраций к другим.
В технике часто пользуются весовыми процентами. Концентрация, выраженная в весовых процентах (С%), указывает количество весовых
— 102 —
единиц растворенного вещества, содержащихся в 100 весовых единицах раствора, т. е.
с% =— . 100, (III, I)
где т — вес растворенного вещества, г, — вес раствора, г.
Например, если концентрация раствора NaCl равна 12%, то это значит, что 12 весовых единиц NaCl содержится в 100 весовых единицах раствора.
В лабораторной практике в большинстве случаев количество раствора измеряют не в весовых, а в объемных единицах—в миллилитрах. Для перехода от веса раствора к его объему пользуются зависимостью:
У = d
где V — объем раствора, мл, пц — вес раствора, d — плотность раствора. В этом случае процентную концентрацию раствора можно вычислить по формуле:
А/ т
с%- 100 ТГ* (Ш’2)
где С% — процентная концентрация, т — вес растворенного вещества, г, V — объем раствора, мл, d — плотность раствора.
Для выявления соотношения компонентов в растворе в химии пользуются выражением концентрации в мольных долях или в мольных процентах. Мольной долей называется отношение числа молей данного компонента к сумме молей всех компонентов раствора. Так, если смесь состоит из пЛ, п2, ... молей разных компонентов, то выражение для
суммы молей будет иметь вид:
Мольная доля первого компонента Ад = n-Jlmi, второго компонента М2 = n2/S/i и т. д., откуда + N2 + N3 + ... + Nk = 1, т. e. сумма всех мольных долей всех компонентов раствора равна 1. Умножив мольную долю данного компонента на 100, получаем мольный процент.
В аналитической химии наибольшее применение нашли способы выражения концентраций числом молей вещества, содержащихся в литре раствора {молярная концентрация, или молярность) и числом грамм-эквивалентов вещества в литре раствора (нормальная концентрация, или нормальность).
Напомним, что грамм-молекулой, или молем, называется такое количество вещества, вес которого, выраженный в граммах, численно равен молекулярному весу этого вещества. Например, грамм-молекула H2SO4 весит 98 г.
Молярная концентрация ^раствора (молярность) может быть вычислена по формуле:
т
Cm=-mv 1000’ (1П,4)
— 103 — .
где См — молярность раствора, т— вес растворенного вещества, а, М — молекулярный вес растворенного вещества, -V — объем раствора, мл.
Если концентрацию выражают числом молей, содержащихся в 1000 г растворителя, то такие растворы называют моляльными. Например, если говорят: двухмоляльный раствор серной кислоты в воде, то это означает, что два моля серной кислоты растворены в 1000 г воды.
Различие между моляльностью и молярностью разбавленного раствора незначительно. Как известно, молярность раствора изменяется с изменением температуры раствора вследствие расширения или сжатия воды, а моляльность от температуры не зависит. Поэтому, моляльные концентрации применяют в случаях, когда имеют место значительные колебания температуры раствора (например, измерение температур кипения и замерзания растворов в зависимости от их концентрации).
Нормальным называется такой раствор, в литре которого содержится один грамм-эквивалент растворенного вещества*.
Концентрацию вещества, выраженную в единицах нормальности, можно вычислить по формуле:
т
Сп = -^7|000> (Ш,5)
где Сн — нормальность раствора, т — вес растворенного вещества, а, Э — эквивалентный вес растворенного вещества, V — объем раствора, мл.
В аналитической химии особенно удобно выражать концентрации в грамм-эквивалентах, так как все вещества реагируют друг с другом в весовых количествах, пропорциональных их эквивалентам.
В практике применяют и другие способы выражения состава раствора, которые здесь не рассматриваются и выбор которых также определяется соображениями удобства при решении определенных задач. В любом случае, если известны молекулярные веса растворенного вещества и растворителя, а также плотность раствора, то всегда можно перейти от одних единиц измерения концентраций к другим.
§ 31. Сущность процесса растворения
Процесс растворения является сложным физико-химическим процессом, в котором наиболее ярко проявляется взаимодействие между частицами (молекулами или ионами) различной химической природы.
На процессы растворения многих веществ, находящихся в различных агрегатных состояниях, большое влияние оказывает полярность молекул растворителя и растворенного вещества. Она выражается в том, что в силу неравномерного распределения электрических заря
* Напомним, что грамм-эквивалентом называется число граммов вещества, равное его эквиваленту. Эквивалентный вес — это весовое количество вещества, равноценное в реакционном отношении восьми весовым частям кислорода, или 1,008 весовым частям водорода.
— 104 —
дов в одной части молекулы могут преобладать положительные заряды, а в другой отрицательные. Полярность отражается на многих свойствах молекул.
Полярность молекулы можно характеризовать количественно, введя представление о так называемом диполе, под которым следует понимать систему, состоящую из двух электрических зарядов е+ и с, равных по величине и противоположных по знаку,> расположенных на некотором расстоянии I друг от друга. Произведение величины этих зарядов е на расстояние между ними называется дипольным моментом и обозначается через р:
В — el.
(Ш.6)
Величина дипольного момента молекул выражается обычно в электростатических единицах, умноженных на сантиметр. Так как заряд одного электрона равен 4,8 • 10“10 эл.-ст. ед, а расстояния между ядрами соседних атомов в молекуле порядка 10~18 см, то дипольный момент молекул в этих единицах выражается величинами порядка 10“18. Иногда величину 1 • 10~10 эл.-ctn. ед. • см называют единицей Дебая и обозначают буквой D. В системе СИ дипольный момент выражается в к*м (заряд электрона равен 1,6 • 10“ls к, линейные размеры молекул порядка 10"10 м).
Следует отличать полярность молекулы в целом от полярности содержащихся в ней связей. Для двухатомных молекул эти два понятия совпадают. Полярность многоатомных молекул зависит от полярностей отдельных связей и от относительного их расположения в молекуле. У молекул, построенных вполне симметрично, дипольный момент равен нулю, хотя бы отдельные связи в них и были полярными, так как в данном случае происходит полная взаимная компенсация дипольных моментов отдельных связей.
У соединений с несимметричным строением молекулы полной компенсации моментов отдельных связей не происходит. Так, в молекуле воды две полярные связи образуют угол в 104,5°. Расстояние между электрическими зарядами в диполе I = 0,0384 • 10“9 м = = 0,0384 нм. Вследствие этого молекула Н2Ов целом обладает значительным дипольным моментом. Аналогичное явление мы наблюдаем
Таблица 17
Дипольные моменты р (дин 2 • см2) некоторых молекул
Молекула ц • 10п Молекула Т) • 1 О’8
СбНб 0 сн3он 1,68
НС1 1,03 СН3СН.ОН 1,70
NH3 1,46 СН3СООН 1,73
СН.С12 1,55 н2о 1,84
— 105 —
в молекуле аммиака, модель которой представляют в виде тетраэдра, в одном из углов которого помещается азот, а в трех остальных — водородные атомы (рис. 39).
К полярным веществам принадлежит большое число соединений (спирты, кислоты, эфиры, кетоны и др.). В табл. 17 приведены значения дипольных моментов некоторых из них.
Как видно из этой таблицы, дипольный момент равен нулю у неполярных молекул (бензола) и имеет наибольшее значение у воды. Именно в силу этого вода является прекрасным растворителем различных полярных соединений (соли, сахара).
Рис. 39. Тетраэдрическая модель молекулы аммиака
Рис. 40. Различные виды ассоциаций полярных молекул в жидкостях
Полярность любого растворителя может быть охарактеризована величиной его диэлектрической постоянной (или, как ее еще называют, диэлектрической проницаемостью). Она показывает, во сколько раз притяжение или отталкивание между Двумя электрическими зарядами в данной среде меньше, чем в вакууме (в = 1). Чем более полярны молекулы среды (растворителя), тем эта разница больше при прочих равных условиях. В табл. 18 приведены значения диэлектрической постоянной для некоторых веществ.
Таблица 18
Диэлектрическая постоянная (е) некоторых соединений при 20° С
Вещество е * Вещество Е
Вакуум Керосин Бензол Оливковое масло ’ . . Хлороформ 1,0 2,0 2,26 3,0 4,05 Лецитин Ацетон Этанол Глицерин Вода 13,0 21,5 27,8 56,2 80,4
Пользоваться понятием диэлектрической постоянной (е) гораздо удобнее, чем понятием дипольного момента (р), так как последний ха
— 106 —
рактеризует только отдельно взятые молекулы, а е — любые вещества, смеси, растворы разных концентраций.
Диэлектрическая постоянная является очень важной характеристикой любого растворителя. Так, растворимость электролитов находится в прямой зависимости от диэлектрической постоянной жидкости (табл. 19).
Т а б л п ц а 19
Диэлектрические постоянные растворителей и растворимость КС! и NH4C1 при 18—20° С
Растворитель
Диэлектрическая
постоянная
Растворимость, г на 100 г растворителя
Этиловый спирт ............
Метиловый спирт ...........
Глицерин ..................
Вода ......................
27,8
31,2
56,2
80,4
0,0034
0,5
6,4
25,5
0,6
3,4
9,0
27,3
Как правило, жидкости с сильно полярными молекулами обладают высокой диэлектрической проницаемостью. С их диэлектрической проницаемостью тесно связаны такие явления, как ассоциация молекул растворителя, процессы электролитической диссоп нации, явление сольватации и др.
Диэлектрические постоянные некоторых смесей могут быть больше, чем каждой из жидкостей, входящих в состав смеси. Например, при 0° С для воды е — 88,3; для перекиси водорода Н2О2 в = 89,2, а для 36%-ной смеси перекиси водорода с водой 8 = 120. Диэлектрические постоянные жидкостей зависят от температуры: с повышением температуры они уменьшаются. Так, для воды имеем следующие значения е:
/, С° 0 10 15 20 25 30 40 50 60 70 80 90 100
е 88,3 84,3 82,3 80,4 78,5 76,7 73,1 69,8 66,5 63,5 60,5 57,8 55,1
В жидкостях полярные молекулы взаимно ориентируются, образуя ассоциаты различной величины и прочности. Это явление имеет место при растворении веществ, состоящих из полярных молекул, в неполярных растворителях (рис. 40). В качестве примера можно указать такие вещества, как бензойная кислота СсН5СООН, этиловый спирт С2Н5ОН и др., молекулы которых, находясь в растворенном состоянии в бензоле (неполярная жидкость), в большей или меньшей степени ассоциированы по две и более в одну частицу.
В растворе всякая заряженная частица, будь то ион или полярная молекула, окружается сольватной оболочкой, которая состоит из ориентированных соответствующим образом молекул растворителя. Если растворителем является вода, то употребляют термин гидратная оболочка, а само явление носит название гидратации.
— 107 —
Степень гидратации различных ионов и молекул неодинакова и зависит как от величины зарядов этих частиц, так и от их размеров. Чём больше заряд и меньше размеры, т. е. чем выше удельная плотность заряда, тем сильнее выражена гидратация. Таким образом, гидратные оболочки удерживаются электростатическими силами притяжения.
Полярные группы молекул растворенного вещества могут образовать также водородные связи с молекулами воды.
Способностью гидратироваться при растворении обладают не только кристаллические, но также газообразные и жидкие вещества. Гид*
Рис. 41. Схема растворения кристалла NaCl в воде
раты (сольваты) являются соединениями все же менее прочными, чем обычные химические соединения. Они легко могут разрушаться даже при незначительном повышении температуры.
Процесс растворения веществ обусловлен взаимодействием частиц растворенного вещества с молекулами растворителя. Механизм растворения твердых тел в жидкости состоит в основном из трех стадий. В качестве примера рассмотрим растворение кристалла хлорида натрия,'который состоит из электростатически связанных ионов натрия и хлора. Как известно, между ионами Na+ и С1~ имеет место ионная связь, между молекулами воды действуют силы Ван-дер-Ваальса и водородная связь, а между ионами натрия и хлора, с одной стороны, и полярными молекулами воды — с другой стороны, возникает ионно-дипольная связь. Все эти виды связи как бы конкурируют между собой. При погружении кристалла в воду полярные молекулы Н2О ориентируются таким образом, что к иону Na+ они обращены своими отрицательными полюсами, а к иону СК — положительными (рис. 41), т. е. происходит явление гидратации этих ионов.
— 108 —
Ионные кристаллы характеризуются интенсивным внешним электрическим полем. Так, напряжение поля на расстоянии 1 А от поверхности кристаллической решетки NaCl составляет около 1,3 • 10й в/м. Такое поле вполне обеспечивает энергичное притяжение полярных молекул воды к поверхности кристалла.
Образованная в результате гидратации ионно-дипольная связь оказывается прочнее, чем межионная связь Na+ — С1~. В результате теплового движения происходит полный разрыв этой связи у ионов, расположенных у поверхности кристалла.
На второй стадии растворения происходит полная гидратация тех ионов, которые полностью перешли в раствор. Третья стадия растворения — это самопроизвольный процесс диффузии гидратированных понов по всему объему растворителя.
Многие кристаллические вещества, относящиеся к неэлектролитам, также обладают высокой растворимостью в воде, что объясняется присутствием в их молекулах полярных групп, способных гидратироваться.
Из рассмотренного выше механизма растворения видно, что на разрушение кристаллической решетки необходимо затратить какое-то количество энергии. С другой стороны, гидратация ионов (или полярных молекул) растворяемого вещества сопровождается выделением энергии в виде тепла. Таким образом, тепловой эффект растворения является суммой двух слагаемых: а) энергии гидратации (в общем случае— сольватации) и б) энергии кристаллической решетки.
Процесс гидратации всегда сопровождается выделением тепла. Подсчитано, что энергия гидратации составляет примерно 34 ккал!моль в том случае, если к каждому иону присоединяется по одной молекуле воды. Процесс разрушения кристаллической решетки, наоборот, является эндотермическим, т. е. он протекает с поглощением тепла. Суммарный тепловой эффект растворения складывается из алгебраических величин энергии разрушения кристаллической решетки твердого вещества и перевода его в жидкое состояние (т. е. на его растворение), и энергии гидратации. Если затраты энергии на растворение какого-либо вещества больше выделяющейся энергии гидратации, то процесс растворения будет эндотермичным. Если же теплота гидратации больше теплоты, необходимой для разрушения кристаллической решетки, то процесс растворения будет экзотермичным. Так, при растворении едкого натра NaOH температура раствора повышается почти до 100° С, а при растворении роданида аммония NH4SCN понижается до —20° С.
Чем больше диэлектрическая постоянная растворителя, тем выше его способность сольватировать частицы растворенного вещества и тем лучше идет процесс растворения. С другой стороны, чем выше способность частиц растворяемого вещества к сольватации, тем лучше оно растворяется в жидкости. А это в свою очередь зависит от полярности молекул растворяемого вещества.
В отдельных случаях сольватация частиц растворяемого вещества может приводить к образованию довольно прочных комплексов. Так, ионы хрома в воде образуют комплексы состава [Сг (НаО)]3+. Подобное же образование — ион гидроксония Н8О+.
— 109 —
При рааворении некоторых веществ имеет место контрактация — сжатие объеиа системы: объем раствора получается несколько меньше суммы объемов растворяемого вещества и растворителя. Так, при смешивании при 20° С 48 объемов воды с 52 объемами спирта вместо 100 объемов смеси получается только 96,3. Это объясняется взаимодействием спирта и воды с образованием гидратов, а также взаимоуплот-пением их колекул в пространстве.
§ 32. Растворимость газов в жидкостях
Газы при соприкосновении с жидкостью способны растворяться в ней. Растворимость газов зависит от их природы, характера жидкости, а также от температуры и давления. В табл. 20 приведены значения растворимости некоторых газов в воде при 18° С и давлении 1 атм.
Таблица 20
Растворимость различных газов в воде
Г аз Растворимость объемов газа в 1 объеме воды Газ Растворимость объемов газа в 1 объеме воды
Гелий Азот ........ Водород Кислород ...... . Двуокись углерода . 0,01390 0,01698 0,01863 0,03220 0,9280 Хлор Двуокись серы . . . Хлористый водород . Аммиак 2,40 42,36 427,90 748,80
Из табл. 20 видно, что в одном объеме воды растворяется 748,8 объемов аммиака и только 0,017 объема азота.
Высокая растворимость аммиака, хлористого водорода, сернистого газа и хлора объясняется их химическим взаимодействием с водой (например, NH3 или SO2) или диссоциацией на ионы (НС1).
Растворимость одних и тех же газов в различных растворителях разная. В табл. 21 показана зависимость растворимости аммиака от природы растворителя.
Газы, молекулы которых неполярны, растворяются, как правило, лучше в неполярных растворителях. И наоборот, в полярных растворителях лучше растворяются газы, молекулы которых полярны.
Из табл. 21 видно, что растворимость аммиака выше всего в воде как в сильно полярной жидкости, в толуоле же как в неполярном растворителе, растворимость его ничтожна.
Таблица 21
Растворимость NH3 в различных растворителях
Растворитель
Растворимость NH3 в 100 г растворителя, г
Вода .........
Этиловый спирт Диэтиловый эфир
87,6
25,0
2,0 0,048
— НО
На растворимость газов большое влияние оказывают давление и температура. Зависимость растворимости газов от давления выражается законом Генри (1803): растворимость данного газа в жидкости прямо пропорциональна его давлению над жидкостью, т. е.
(III,7)
где С — концентрация газа в жидкости, Р — давление газа над раствором, К — коэффициент пропорциональности, зависящий от природы газа.
Из закона Генри вытекают следующие важные следствия.
1. Поскольку давление газа Р пропорционально концентрации его в газовой фазе Сг, то:
отсюда
7^=^ (1П,8)
Иными словами, отношение концентрации газа, растворенного в жидкости, к концентрации его над раствором при постоянной температуре есть величина постоянная.
2. Объем растворенного газа не зависит от внешнего давления, поскольку при увеличении давления в одинаковое число раз возрастает как концентрация растворенного газа, так и концентрация газа над раствором.
Например, в 100 г воды при 20° С и давлении 760 мм рт. ст. растворяется 0,169 г двуокиси углерода СО2. При увеличении давления вдвое в том же количестве воды растворяется 0,338 г, т. е. масса растворяющегося газа тоже удваивается; при этом плотность газа тоже удваивается, так как 0,338 г этого газа при увеличении давления вдвое займут тот же объем, что и 0,169 а его при 760 мм рт. ст.
Таким образом, растворимость г^за в жидкости прямо пропорциональна его парциальному давлению. Понижение парциального давления приводит к уменьшению растворимости газа. В качестве примера можно взять обычную газированную воду, которая представляет собой приготовленный под давлением СО2 в 760 мм рт. ст. насыщенный водный раствор. При соприкосновении воды с воздухом, в котором парциальное давление СО2 составляет всего 0,2 мм рт. ст., растворенная двуокись углерода будет бурно выделяться.
С изменением растворимости газов от резкой перемены давления связана так называемая «кессонная болезнь». При слишком быстром подъеме с больших глубин водолаза или кессонщика, газы, растворенные в его крови, в результате снижения внешнего давления начинают выделяться (кровь как бы закипает). Пузырьки газа закупоривают мелкие кровеносные сосуды в мозгу и в других жизненно важных органах, что и может явиться причиной тяжелого заболевания или даже гибели человека.
Закон Генри справедлив только для разбавленных растворов и при малых давлениях, т. е. когда газы подчиняются законам идеальных га
111
зов. Газы, вступающие при растворении во взаимодействие с растворителем (НС1, NH3, SO2 и др.), закону Генри не подчиняются.
При растворении смеси газов каждый из них растворяется в количестве, пропорциональном его парциальному давлению над раствором, т. е. закон Генри справедлив для каждой составной части газовой смеси. В силу этого вода богаче кислородом, чем воздух (в воде содержится 34,1 об. % кислорода при 18°С, а в воздухе 21,2 об. %), что имеет чрезвычайно большое значение для организмов, живущих в воде.
Растворимость газов в сильной степени зависит от температуры. Численные значения растворимости некоторых газов в воде при разных температурах и давлении 1 атм приведены в табл. 22.
Таблица 22
Растворимость некоторых газов в воде в зависимости от температуры
Температура, °C Газы
n2 о2 н2 со2 H2S SOg
0 0,0239 0,0489 0,0215 1,710 4,67 79,8
20 0,0164 0,0310 0,0182 0,878 2,58 39,4
40 0,0118 0,0230 0,0164 0,530 1,66 18,8
Как видно из табл. 22, растворимость газов с увеличением температуры уменьшается. Объясняется это следующим .образом. В подавляющем большинстве растворение есть экзотермический процесс сольватации газа молекулами растворителя. Поэтому согласно принципу Ле Шателье, который применим для равновесных систем, растворимость газов будет уменьшаться с нагреванием и увеличиваться при охлаждении.
Количественная зависимость между растворимостью газа и температурой определяется уравнением Клапейрона — Клаузиуса:
X / 1 1 \
Nz ~ R ( Т2 ~ Tt )’ (1П,9)
где и TV2 — растворимости при температурах Т2 и Tlt X — теплота, выделяемая при растворении одного моля газа в его насыщенном растворе, R — универсальная газовая постоянная.
Растворение газов в жидкостях имеет большое значение в практической работе. Так, в ряде случаев при использовании артезианских источников для возделывания затопляемой культуры риса не получают хороших урожаев риса, поскольку в холодной воде содержится значительное количество токсичного для риса сероводорода H2S. Неблагоприятное воздействие на рис оказывает также и пониженная температура самой воды. Если же эту воду, предварительно собранную в неглубокие бассейны, подавать на поля спустя два-три дня, урожаи риса будут значительно выше. Дело в том, что поскольку за этот период вода прогревается, она теряет значительное количество сероводорода.
— 112 —
В лабораторных условиях для освобождения дистиллированной воды от СО2 используют также кипячение и т. д.
Растворимость газов зависит и от наличия посторонних электролитов в растворе. Присутствие электролитов снижает растворимость различных газов в жидкости. Зависимость растворимости газов от присутствия электролитов впервые была установлена И. М. Сеченовым:
/v0
= (1.11,10)
где No и W — растворимость газа соответственно в чистой воде и в растворе соли с концентрацией С, моль!л, К — коэффициент, зависящий от природы газа, электролита и температуры.
На растворимость газов оказывают влияние и другие растворенные вещества. Например, многие неэлектролиты, особенно склонные к образованию сольватов, также уменьшают растворимость газов в воде. На этой же основе живой организм, изменяя состав крови, может в известных пределах регулировать величину растворимости таких газов, как кислород и двуокись углерода.
§ 33. Взаимная растворимость жидкостей
В зависимости от природы жидкости могут смешиваться друг с другом в различных соотношениях: 1) смешиваются друг с другом в любых соотношениях с образованием совершенно однородного раствора (вода и глицерин, вода и этиловый спирт); 2) обладают ограниченной растворимостью друг в друге (вода и анилин, вода и эфир); 3) практически не растворимы друг в друге (вода и бензол, вода и ртуть).
Рассмотрим случай ограниченной растворимости на примере двойной системы анилин — вода. Если в пробирку налить немного анилина, прибавить примерно такое же количество воды и энергично встряхивать ее, пока не получится эмульсия, то после непродолжительного отстаивания жидкость в пробирке образует два слоя: верхний— насыщенный раствор анилина в воде, нижний — насыщенный раствор воды в анилине.
Характерно, что для каждой температуры оба раствора имеют строго определенный равновесный состав, не изменяющийся от прибавления дополнительных количеств воды или анилина. Повышение температуры ведет обычно к увеличению взаимной растворимости и может в конечном счете привести к неограниченному взаимному растворению компонентов друг в друге. Температура, при которой ограниченная растворимость переходит в неограниченную, называется критической температурой растворения.
В табл. 23 приведен химический состав двух слоев, образовавшихся при смешивании анилина с водой, при различных температурах. Из таблицы видно, что при повышении температуры в водном слое будет расти концентрация анилина, а в анилиновом слое—концентрация воды. При температуре 168° С составы обоих растворов становятся равными, граница раздела между слоями исчезает, и уже выше 168° С анилин и вода смешиваются друг с другом в любых соотношениях.
— 113 —
I
Таблица 23
Растворимость анилина в воде
Температура, °C Содержание, с Температура, °C 1 Содержание, г
воды в 100 а анилинового слоя анилина в 100 г водного слоя роды в 10 0г анилинового слоя анилина в 100 г водного слоя
20 5,0 3,3 120 14,9 9,1
40 6,0 3,8 140 20,3 14,9
60 7,8 4,8 160 28,8 24,9
80 8,6 5,7 168 51,4 48,6
100 п,о 7,2
Таким образом, критическая точка растворения для системы вода — анилин численно равна 168° С.
Зависимость состава обоих растворов от температуры гораздо удобнее изображать не в виде табличных данных, а в виде соответствующей диаграммы растворимости. На рис. 42 приведена диаграмма для систе-
Рис. 42. Диаграмма взаимной растворимости воды и анилина
мы вода — анилин. Подобные кривые называются кривыми расслоения. На рис. 42 АК—кривая растворимости воды в анилине; ВК—кривая растворимости анилина в воде. Обе кривые сходятся в точке К, соответствующей критической температуре растворения. Любые точки, лежащие вне кривой А КВ, отвечают гомогенной системе, ненасыщенным растворам компонентов. Точки, лежащие внутри фигуры АКВА, соответствуют системе, состоящей из двух растворов (двух слоев), составы которых определяются точками пересечения соответствующей изотермы с кривой АКВ.
Если взять смесь при температуре 140° С состава х, то она будет однородной. При охлаждении ее до 100° Сона расслаивается, и составы образовавшихся насыщенных растворов будут на диаграмме отвечать
1 очкам а и Ь. Если при 140° С взять смесь состава xlf то она, как видно из диаграммы, также расслаивается на насыщенные растворы, составы которых соответствуют точкам aL и bt.
Взаимная растворимость жидкостей в значительной степени зависит от присутствия третьего компонента, который может оказывать существенное влияние на критическую температуру растворения. Например, тот же самый анилин может неограниченно смешиваться с водой при всех температурах, если в растворе присутствует достаточное количество Lil. Объясняется это тем, что Lil в одинаковой мере хорошо растворим как в анилине, так и в воде. Если же третий компонент хорошо растворим только в одной из жидкостей, взаимная растворимость обеих жидкостей в присутствии этого компонента уменьшается, а следовательно, увеличивается критическая температура растворения. В качестве примера можно указать систему фенол — вода. Критическая температура этой системы может увеличиться на 30° при добавлении к ней 3% хлорида калия.
Иногда введением третьего компонента можно добиться расслоения ранее однородного раствора на два слоя. Например, для выделения этилового спирта из его водного раствора поступают следующим образом. К водному раствору спирта прибавляют К2СО3 и энергично встряхивают. После отстаивания раствор разделяется на два несмешивающих-ся слоя. Один из слоев состоит из почти безводного этилового спирта, а другой — из водного раствора К2СО3.
На принципе использования критической температуры растворения основаны некоторые виды анализов сырья. Например, критические температуры маргарина и коровьего масла существенно отличаются.
§ 34. Растворимость твердых веществ в жидкостях
Растворимость твердых веществ также определяется природой растворителя и растворенного вещества и также зависит от температуры. В отличие от растворимости газов, растворимость твердых тел сравнительно мало изменяется с давлением*.
Различия в растворимости твердых тел можно иллюстрировать примерами. В 100 г воды при 25° С растворяется 257 г AgNO3 и лишь 3 • 1О~20 г Hgl. Вода—хороший растворитель сахара, а спирт его практически не растворяет. Нафталин очень хорошо растворяется в спирте и не растворим в воде.
Следует заметить, что вследствие отсутствия общей теории растворов вопрос о зависимости растворимости от физико-химических свойств растворителя и растворенного вещества еще нельзя считать до конца решенным, и конкретные сведения о растворимости твердых тел целиком основаны на опытных данных.
В настоящее время установлен ряд правил о растворимости веществ, но они не обладают универсальностью, не свободны от различного рода исключений и потому носят в большинстве случаев качест-
* Напомним, что растворимостью данного вещества называется количество его, выраженное в граммах, насыщающее 100 г растворителя.
— 115 —
Рис. 43. Зависимость растворимости некоторых солей в воде от температуры
венный характер. Например, замечено, что полярные растворители, как правило, хорошо растворяют полярные вещества и плохо—неполярные. Неполярные растворители, наоборот, хорошо растворяют неполярные вещества и плохо — полярные. В том случае, если один из компонентов раствора полярен, а второй неполярен, растворимость бывает незначительной.
Растворимость большпнст-
ва твердых тел с повышением температуры увеличивается. Однако бывают и исключения
из этого правила. Так, растворимость СаСгО4 и Са (ОН)2 в воде с повышением температуры уменьшается. Изменение растворимости тел от температуры зависит, как показывает опыт, от теплового эффекта растворения. Согласно принципу Ле Шателье, растворимость вещества увеличивается с температурой, если процесс растворения данного вещества идет с поглощением тепла. И, наоборот, с повыше
нием температуры уменьшается растворимость твердого вещества, если его растворение сопровождается выделением тепла.
Зависимость растворимости от температуры обычно изображают в виде кривых растворимости. На рис. 43 приведены кривые растворимости некоторых солей в воде. Резкий излом на кривой растворимости
сульфата натрия соответствует превращению кристаллогидрата
Na2SO4 • ЮН2О (который устойчив при температуре ниже 32, 383° С)
в безводный Na2SO4 (устойчивый при температуре выше 32,383° С). Растворение кристаллогидрата Na2SO4-10H2O сопровождается поглощением тепла, а растворение безводной соли идет с выделением тепла.
Если соль способна к образованию кристаллогидратов (рис. 44), то химический состав и область существования каждого кристаллогидрата можно сравнительно легко определить по характерным кривым растворимости: каждой точке перехода соответствует излом на кривой растворимости..
Изменением растворимости с температурой часто пользуются для очистки веществ путем перекрис
Рис. 44. Зависимость растворимости ZnCh в воде от температуры
— 116
таллизации. При остывании горячего насыщенного раствора какой-либо соли, загрязненной посторонними примесями, значительная часть этой соли выделится в осадок, а загрязняющие примеси останутся в ^растворе, так как последний даже на холоде не будет насыщенным раствором по отношению к этим примесям. Подобным образом можно очищать любые твердые вещества, растворимость которых сильно зависит от температуры.
Если растворимость вещества мало изменяется с температурой, очистка его путем перекристаллизации становится невозможной. В этом случае насыщенный раствор очищают упариванием, т. е. удаляют из него часть воды. В процессе упаривания некоторая доля очищаемого вещества выкристаллизовывается, а примеси остаются в растворе.
§35. Природные растворы
Вода, как известно, вследствие полярности ее молекул является хорошим растворителем для многих веществ. Она играет исключительно важную роль в геохимических и гидрогеологических процессах земной коры. Природные воды активно участвуют в образовании и разрушении различных минералов. При взаимодействии с твердыми телами вода превращается в раствор, который содержит элементы, входившие ранее в состав этих тел. Растворяя газы атмосферы и перенося их течениями на громадные расстояния, вода выступает в роли регулятора состава воздуха. Достаточно указать, что в воде океанов содержится в восемь раз больше двуокиси углерода, чем в воздухе.
По составу природные растворы Являются исключительно сложными физико-химическими системами. Все пресные воды (с содержанием сухого остатка от 1 г/д и менее), а также минерализованные воды (сухой остаток более 1 г!д) являются природными растворами. Это воды рек, озер, морей, оксанов, почвенные и грунтовые воды, межпластовые, жильные, карстовые, так называемые «ювенильные» воды и т. п. Общее количество воды на земле по приблизительным подсчетам составляет 2 • 1018 т. Причем, около 3/5 этого количества сосредоточено в морях и океанах, остальные 2/5 воды приходится на льды суши, водяной пар атмосферы, а также на воду в составе твердых тел земной коры.
Вода в природе выступает не только как растворитель. Многие природные реакции протекают с ее участием. При растворении многих веществ в воде происходит химическое взаимодействие между ионами растворенного вещества и ионами Н+ и ОН~ воды, сопровождающееся образованием слабых кислот или слабых оснований. Эти реакции получили название гидролитических.
Именно в силу своей высокой активности вода играет исключительно важную роль в химическом выветривании горных пород. Причем активность воды при взаимодействии с горными породами значительно возрастает в присутствии двуокиси углерода. Этому фактору В. Р. Вильямс придавал исключительно важное значение в процессах почвообразования.
Находясь под постоянным воздействием воды, воздуха и резкой смены температур, горные породы дробятся. Воды дождей извлекают из них растворимые составные части и вместе с нерастворимыми частицами, главным образом песка и глины, уносят в реки. Здесь взвешенные частицы сортируются по плотности: сначала отлагается песок, а затем более мелкие глинистые частицы. В течение веков вдоль русла реки образуется мощная залежь, состоящая из песка и глины, а сама река вынуждена прокладывать себе новое русло. На обнажившемся старом русле под влиянием биологических и физико-химических факторов образуется почва и развивается наземная растительность.
Почвенный раствор также относится к числу сложных природных систем. Как известно, растения усваивают питательные вещества, растворенные в форме солей в жидкой части почвы, т. е. в почвенном растворе. Эти соли поступают
— 117 —
в почвенный раствор из минералов, разложившихся остатков растений и животных, а также микроорганизмов. Кроме того, на составе почвенного раствора заметно сказывается внесение органических, минеральных, органо-минеральных и бактериальных удобрений. Иногда в почве содержится избыток ле! кораствори-мых солей — хлоридов и сульфатов натрия и других, которые угнетающе действуют на растения. Поэтому для повышения плодородия почв необходимо удалить из них избыток солей путем промывания или другими мелиоративными приемами.
В твердой части почвы имеется большой запас минеральных веществ, в почвенном же растворе находится лишь небольшая их часть, преимущественно в форме ионов кальция, магния, калия, натрия, железа, алюминия и анионов ряда кислот: фосфорной, серной, азотной, угольной. В почвенных растворах засоленных почв содержится много Cl~, SO^“, Са2+, Mg2+ и Na-ионов (табл. 24).
Таблица 24
Состав почвенного раствора в миллиграмм-эквивалентах на 100 г абсолютно сухой почвы (солончак)
Глубина взятия сбразца, см Ионы
СГ SO2 Na+ Са2+ Mg2+
7— 18 42,4 7,0 36,2 0,6 12,1
18— 37 36,9 6,6 30,0 1,0 10,2
37— 47 22,3 4,6 18,8 0,7 7,3
47—100 33,3 6,3 26,3 1,1 12,1
100—150 19,0 4,7 16,3 1,0 6,4
150—177 21,1 6,2 19,4 1,9 7,9
Чрезвычайно сложный солевой состав имеют воды морей и океанов. Среди растворенных солей в морской воде преобладают хлориды и сульфаты натрия и магния. В табл. 25 приведен средний элементарный состав морской воды в процентах по данным А. П. Виноградова (1944).
Элементарный состав морской воды
Таблица 25
Элемент Содержание, % Элемент Содержание, о/ /0 Элемент Содержание, %
О 85,82 Вг 0,007 N 0,000010
н 10,72 С 0,002 I 0,000005
С1 1,89 Sr 0,001 Р 0,000005
Na 1,06 В 0,0005 Zn 0,000005
Mg 0,14 F 0,0001 Ва 0,000005
s 0,09 Si 0,00005 Fe 0,000005
Са 0,04 Rb 0,00002 Си 0,000002
К 0,038 Li 0,000015 As 0,0000015
Необходимо отметить, что помимо перечисленных в этой таблице элементов, морская вода содержит почти все элементы периодической системы Д. И. Менделеева, но в еще меньших количествах. Так, в одной тонне морской воды находится 0,000004 г золота.
— 118 —
Очень часто природные растворы ведут себя как коллоидно-дисперсные системы с характерными для коллоидных растворов оптическими и физико-хими ческими свойствами. Подобные растворы активно участвуют в образовании коры выветривания почвенного покрова, а также в образовании осадочных пород и руд,
РАЗБАВЛЕННЫЕ РАСТВОРЫ
В конце XIX в. Рауль, Вант-Гофф, Аррениус установили весьма важные закономерности, связывающие концентрацию раствора с осмотическим давлением, давлениш.лш£ьцценноп) пара, температурой кипения и замерзаниям Причем законы Рауля и Вант-Гоффа справедливы для сильно разбавленных растворов неэлектролитов (мольная доля растворенного вещества М 0,005). Растворы, подчиняющиеся этим законам, получили название идеальных растворов. Теория идеальных растворов в настоящее время хорошо разработана. Она отличается простотой, поскольку базируется на том условии, что компоненты идеального раствора не взаимодействуют друг с другом. Идеальные растворы ведут себя как газы и подчиняются законам идеальных газов. Ряд свойств идеальных растворов зависит только от концентрации" растворенного вещества и не зависит от химической природы растворенных молекул. Реальные смеси газов и растворы также имеют ряд общих свойств. Это сходство особенно характерно для сильно разбавленных растворов. Поведение молекул неэлектролита в таком растворе во многих отношениях аналогично поведению идеального газа.
§ 36. Диффузия и осмос в растворах
Как известно, в смесях газов и в растворах частицы равномерно распределяются по всему объему. Например, если на концентрированный раствор сахара осторожно налить слой чистой воды, то молекулы сахара, совершая хаотическое тепловое движение, постепенно равномерно распределяются по всему объему жидкости. Одновременно и молекулы воды проникают в раствор сахара, разбавляя его. Оба эти процесса идут самопроизвольно и до тех пор, пока не произойдет полного выравнивания концентраций сахара во всем объеме раствора. Самопроизвольный процесс переноса вещества, в результате которого устанавливается равновесное распределение концентраций вследствие беспорядочного теплового движения молекул, атомов, ионов в газах, жидкостях или твердых телах, называется диффузией. Диффузия имеет место и при смешивании растворов различных концентраций, а также в твердых телах и газах. Причем, скорость ее в газах наибольшая, а в твердых телах наименьшая.
Как правило, диффузия частиц совершается из области большей их концентрации в область меньшей концентрации, т. е. количество частиц растворенного вещества, проходящих в единицу времени в сторону меньшей концентрации, больше, чем в обратном направлении.
Диффузия может быть выражена количественно. Представим себе, что на некотором расстоянии хг от дна сосуда концентрация растворенного вещества (например, сахара) равна СА, а на расстоянии х3 эта концентрация равна С2. По условию больше С2, а х2 больше
— 119 —
<
хь т. е. раствор является более концентрированным у дна сосуда. В нашем случае градиент концентрации, т. е. изменение концентрации, приходящееся на единицу расстояния, будет равно:
с2—сг __ дс х2 — Xj Дх
Знак минус в этом уравнении вызван тем, что >> С2.
На основании закона Фика количество растворенного вещества mt которое проходит за время t через воображаемую площадь поперечного сечения сосуда S, находящуюся посередине между концентрациями Q и С2, будет равно: V
т = — (III, 12) I
Дх
где D — коэффициент диффузии, численно равный количеству вещества, диффундирующего за единицу времени через I см2 поверхности раздела при градиенте концентрации, равном 1. Для коэффициента диффу- I зии Эйнштейн вывел следующее уравнение:
/?Г 1 1
*7---*
N6 6л ту
• ( • «• .«It
где R — универсальная газовая постоянная, Т — абсолютная тем- Н пература, No — число Авогадро, ц — вязкость растворителя, г — радиус диффундирующих частиц. г
Объединяя уравнения (111,12) и (111,13), получим: ж
ЯТ S АС
•—-----• —(111,14)
откуда
т RT S &С II
-----(Ш,15)
t 6лт]г Ах
где выражение mil — скорость диффузии, т. е. количество растворенного вещества /л, проходящего в единицу времени t через площадь сечения S.
Из уравнения (III, 15) видно, что скорость диффузии возрастает при повышении температуры и градиента концентрации и уменьшается при увеличении вязкости среды и радиуса диффундирующих частиц. Отсюда следует, что вещества с большим молекулярным весом будут иметь сравнительно малые коэффициенты диффузии, что хорошо подтверждается данными табл. 26.
Явление диффузии играет чрезвычайно важную роль в жизнедеятельности организмов, в процессах перемещения питательных веществ и продуктов обмена в тканевых жидкостях. В живых организмах диффузия тесно связана с другими биологическими явлениями. Скорости многих физико-химических процессов в организме зависят в конечном счете от скорости диффузии, т. е. от скорости «доставки сырья» для этих реакций. В свою очередь диффузия в живых организмах регулируется
— 120 —
Таблица 20
Молекулярные веса и коэффициенты диффузии некоторых веществ
Вещество
Молекулярный I ес М
Коэффициент диффузии D
Мочевина.........................
Глицерин ........................
Сахароза ........................
Яичный альбумин..................
Сывороточный глобулин лошади . .
Тиреоглобулин свиньи ............
60
92
342
43 800
67 000
627 000
функциональным состоянием тканей и зависит от их физико-химического строения.
Диффузия может проходить также, если на границе раствора и чистого, растворителя (или двух растворов различной концентрации) поместить полупроницаемую перегородку — мембрану. Полупроницаемые перегородки способны пропускать только молекулы растворителя и не пропускают молекулы растворенного вещества. Свойством полупроницаемости обладают многие природные пленки (стенки клеток живых и растительных организмов, стенки кишечника, протоплазма и др.), а также пленки искусственного происхождения (целлофан, пергамент,— оденки из коллодия, желатины),? Односторонняя самопроизвольная диффузия "молекул растворителя через полупроницаемую мембрану в раствор или из раствора с низкой концентрацией в раствор с высокой концентрацией называется осмосом.
Процесс осмоса очень сложен и природа его в настоящее время еще недостаточно выяснена.
Механизм осмоса может быть различен и зависит в основном от природы мембраны. В одних случаях мембрана взаимодействует с растворителем, образуя с ним непрочные соединения промежуточного характера. В других случаях через мембрану могут свободно проходить только те вещества, которые обладают способностью растворяться в ней. Мембрана может представлять собой просто пористую перегородку с порами определенного размера. Осмос можно наблюдать в специальных приборах, которые называются осмометрами. Простейшая схема осмометра приведена на рис. 45. Основной его деталью является осмометрическая ячейка /, отделенная от сосуда 2 с чистым растворителем полупроницаемой мембраной, пропускающей только молекулы растворителя, но не растворенного вещества. Ячейку с концентрированным раствором погружают в сосуд с растворителем—менее концентрированным раствором. Спустя некоторое время отмечается значительное повышение уровня жидкости в трубке. Объясняется это тем, что с поверхностью мембраны снизу сталкивается больше молекул растворителя, чем сверху, так как сверху часть поверхности занята также молекулами растворенного вещества, не проникающего сквозь мембрану. Поэтому в единицу времени вверх переходит значительно больше молекул растворителя, чем в обратном направлении.
— 121 —
Рис. 45. Простейший осмометр
В этом случае мы сталкиваемся с проявлением одного из общих свойств естественных самопроизвольных процессов: в природе самопроизвольно протекают процессы, направленные в сторону выравнивания в системе температур, давлений, электрических потенциалов, концентраций и т. д. Эти процессы находятся в полном соответствии со вторым началом термодинамики. Концентрация растворителя в сосуде 2 больше, чем в ячейке, и поэтому’ поток растворителя направлен в сторону ячейки. Уровень жидкости в осмометриче-ской трубке будет повышаться, пока гидростатическое давление столба жидкости высотой h не задержит осмос. В этом случае уравновешиваются скорости прохождения растворителя из наружного сосуда в ячейку и из ячейки в наружный раствор, подъем жидкости в осмометре прекращается. Гидростатическое давление, кото-
рое надо приложить к раствору, чтобы задержать осмос, называют осмотическим давлением. Осмотическое
давление растворов обычно измеряют цли рассчитывают по отношению к чистому растворителю. Измеряется осмотическое давление в атмосферах или в миллиметрах ртутного столба (н/л*2).
Как показали исследования, осмотическое давление зависит в
первую очередь от концентрации раствора и может достигать больших значений. Так, 6%-ный раствор сахара имеет осмотическое давление 60 800 н/м2, морская вода 2 840 000 н!м\ рассолы самосадочных озер—более 20 300 000 н!м2.
§ 37. Законы осмотического давления и его биологическое значение
Результаты измерения осмотического давления растворов различной концентрации тростникового сахара и некоторых других веществ, полученные в свое время Пфеффером и де Фризом, позволили Вант-Гоффу (1887) установить законы осмотического давления, применив для обобщения результатов измерений осмотического давления законы термодинамики и молекулярно-кинетическую теорию газов. Вант-Гофф установил, что осмотическое давление сильно разбавленных растворов подчиняется законам идеальных газов. Он показал, что при постоянной температуре осмотическое давление прямо пропорционально концентрации или обратно пропорционально молярному объему растворенного вещества (аналогия с законом Бойля):
Pi С.
Р2 С2 ‘
— 122 —
При данной концентрации растворенного вещества осмотическое давление пропорционально абсолютной температуре (аналогия с законом Гей-Люссака):
При одинаковой температуре и одинаковой концентрации различные вещества имеют одно и то же осмотическое давление (аналогия с законом, Авогадро). Объединив эти законы, он получил уравнение состояния для осмотического давления (уравнение Вант-Гоффа); ‘
т
П= — RT — CRT, (III, 16)
которое по форме совпадает с уравнением Клапейрона — Менделеева. В этом уравнении П — осмотическое давление раствора, т — число молей растворенного вещества, С ~ m/V — молярная концентрация растворенного вещества, Т — абсолютная температура, R — универсальная постоянная, не зависящая от вида растворителя и численно равная газовой постоянной (8,31 н • м/град • моль).
Точные измерения осмотического давления растворов показали, что чем ниже концентрация С, тем точнее соблюдается уравнение Вант-Гоффа. Это уравнение для большинства растворов соблюдается при концентрациях не выше 1 • 10“2 кмоль! м\ При более высоких концентрациях отклонения от закона Вант-Гоффа легче учитывать, если вместо объемно-молярной концентрации С употреблять моляльную tn1:
П^оТ RT -=RT —1009, (111,17)
82 M
где gt — вес растворенного вещества, г, g2 — вес растворителя, г, М — молекулярный вес растворенного вещества.
Объединенный закон Вант-Гоффа имеет следующую формулировку: осмотическое давление разбавленного раствора равно тому газовому давлению, которое производило бы растворенное вещество, если бы оно в виде газа при той же температуре занимало тот же объем, что и раствор.
Таким образом, для осмотического давления в истинных растворах низкомолекулярных веществ имеет значение только число растворенных частиц, но не их вес, размеры или форма.
Осмос имеет большое значение в процессах жизнедеятельности жи-вотных и растений. Он обусловливает поднятие воды по стеблю расте-ния, рост клетки и многие другие явления. Осмотическое давление^ в клетках обусловливает их своеобразную упругость и эластичность, а также способствует сохранению определенной формы стеблями и листьями растений. Каждая живая клетка имеет либо оболочку, либо поверхностный слой протоплазмы, обладающие свойством полупроницаемости. Если клетку поместить в раствор, концентрация которого равна концентрации клеточного сока, то состояние клетки ^не изменится, так как осмотическое давление в клетке и в растворе одинаково.
— 123 —
Рис. 46. Плазмолиз растительных клеток в гипертоническом растзоре
Растворы, обладающие при одинаковых условиях одинаковым ос-Имотическим давлением, получили название изотонических. I
/ / В крепких солевых растворах клетка сморщивается (плазмолиз), v что обусловлено потерей воды в более концентрированный внешний z раствор (рис. 46), так как осмотическое давление внешнего раствора. s ~выше, чем внутри клетки. Такой раствор называется гипертоническим.
В растворе, концентрация которого ниже концентрации клеточного ; сока, клетка всасываехводу, что объясняется более низким, чем в клетке, осмотическим давлением раствора. Такие растворы получили название гипотонических.
Кровь* лимфа, а также любые тканевые жидкости человека и животных представляют собой водные растворы молекул и ионов многих • веществ—органических и минеральных. Эти растворы обладают определенным осмотическим давлением. На- ] пример, осмотическое давление крови равно 800 000 н!м*. Такое же давление имеет 0,9%-ный раствор хлорида нат- Я рия, который является по отношению к крови изотоничным. • i ' I
При медицинском введении в кровь физиологических растворов последние должны быть строго изотоничны с раст- ' вором крови. Физиологические растворы 1 широко применяют в хирургии в качест- ве кровезаменителей. " ’ J
Представление об изотоничных растворах позволяет понять, почему морская рыба не может жить в речной воде, а речная рыба — в мор- | ской. Становится также понятной и причина, по которой растения пу- I стыни не могут произрастать на влажной почве, а полевые растения —г I на сильно засоленных почвах.
Организм человека обладает способностью поддерживать осмотическое давление на постоянном уровне.' При изменении осмотического давления организм стремится восстановить его. Так, если с пищей вводится в организм большое количество растворимых веществ (соль, сахар и др.), то осмотическое давление изменяется и организм сейчас же реагирует на это, стремясь как можно скорее восстановить нормальное- осмотическое давление (изменяется количество и состав слюны, пота, мочи и количество выделяемых паров). Все эти процессы в организме регулируются нервной системой и железами внутренней секреции.
При патологических явлениях в тканях организма могут происходить значительные колебания осмотического давления. Так, в очаге воспаления осмотическое давление тканевого сока у человека может в два-три раза превысить норму.
С осмотическим давлением, как уже отмечалось, тесно связана солеустойчиволъ культурных растений. До сравнительно недавнего времени считали, что солеустойчивость растений определяется только их способностью повышать сосущие силы клеток до превышения осмотического давления почвенного раствора. При этом механизм действия
— 124 —
У
- 'i
солей сводился к частному обезвоживанию клеток вследствие недостаточной водообеспеченности растений, произрастающих на засоленных почвах.
Однако объяснять угнетение растений только с точки зрения осмотической теории не совсем правильно. Более детальные исследования показали, что рост и развитие культурных растений обусловлены не только величиной осмотического давления почвенного раствора, но и специфическим физиологическим действием отдельных ионов, входящих в состав почвенного раствора. Изучение механизма действия солей почвенного раствора на культурные растения позволило в последние годы разработать целый ряд'мероприятий по повышению соле-устойчивости растений. В основу этих мероприятий был положен принцип солевого воздействия на семена перед посевом. Испытания предложенных методов предпосевного повышения солеустойчивости растений показали, что с их помощью можно значительно повысить урожай различных сельскохозяйственных культур на засоленных почвах (табл. 27).
Таблица 27
Влияние предпосевной обработки семян на урожай проса в условиях засоления
Степень засоления
Средняя Средняя
Средняя
Сильная
Сильная
Сильная
Варианты опытов
1. Необработанные семена
2. Семена, обработанные 3%-ным раствором хлорида натрия
3. Семена, обработанные 6%-ным раствором хлорида натрия
1. Необработанные семена
2. Семена, обработанные 3%-ным раствором хлорида натрия
3. Семена, обработанные 6%-ным раствором хлорида натрия
Урожай
Ц/го 1 %
7,0.. 100
10,3 147
9,5 - 135
6,0 100
7,8 130
8,7 145
По данным ряда авторов, свойства солеустойчивости, приобретенные растениями после однократной обработки семян, передаются по наследству.
На использовании осмотического /давления основано консервирование фруктов и овощей. Высокие концентрации сахара или соли в консервате создают высокое осмотическое давление, вследствие чего микроорганизмы погибают в результате плазмолиза.
§ 38. Понижение давления насыщенного пара г растворителя
Давление насыщенного пара жидкости при данной температуре является постоянной величиной. Так, при 20° Сдавление насыщенного пара воды равно 2,319 • 103 н!м\ спирта 5,852 • 103 н!м2, эфира 5,8928 -104 н/м2 и т. д. Испарение является эндотермическим про
цессом, поэтому согласно принципу Ле Шателье повышение температуры сдвигает равновесие в сторону дальнейшего парообразования, т. е. с увеличением температуры давление насыщенного пара также увеличивается.
Опыт показывает, что давление насыщенного пара над жидкостью при постоянной температуре понижается, если в ней растворить некоторое количество другого вещества. Это объясняется действием ван-дер-ваальсовых сил между молекулами растворителя и растворенного вещества. Растворяя небольшое количество какого-либо вещества в растворителе, мы понижаем концентрацию последнего в единице объема и тем самым уменьшаем число молекул растворителя, покидающих поверхность раствора в единицу времени (рис. 47). В результате— давление пара над раствором всегда меньше, чем над чистым растворителем. При этом понижение давления пара тем больше, чем больше концентрация растворенного вещества в растворе.
Первый закон Рауля. Обобщая результаты экспериментальных наблюдений, французский физик Рауль (1887) установил, что давление насыщенного пара растворителя над раствором равно его давлению над чистым растворителем, умноженному на мольную долю растворителя в растворе, т. е.
Р=Ро-----г---, (П1,18)
п+п0
\ ч
где Ро — давление насыщенного пара над чистым растворителем, Р — давление насыщенного пара над раствором, и0 — число грамм-молей растворителя, п — число грамм-молей растворенного вещества. Уравнение (III, 18) получило название первого закона Рауля. На практике чаще всего применяют другую, более удобную форму этого закона. Так, если обе части уравнения (III, 18) разделить на Ро, а затем мольную долю растворителя заменить мольной долей растворенного вещества, то (зная, что ———+ —-— = 1) получим:
/ь 4- nQ п+по
откуда
(П1,19)
Это уравнение тождественно уравнению (III, 18) и читается так: относительное понижение давления насыщенного пара растворителя над раствором равно мольной доле растворенного вещества в растворе. _ Это и есть формулировка первого закона Рауля.
Так как для сильно разбавленных растворов п по сравнению с п0 очень мало, им можно пренебречь, и формула (III, 19) примет вид:
(111,20)
— 126 —
Таблица 28
Понижение давления пара различных растворителей
Растворитель р.-р Р» Растворитель р0-р Ро
Вода ........ Сероуглерод .... Хлороформ 0,0102 0,0105 0,0109 Бензол Эфир диэтиловый . . Спирт метиловый . . 0,0106 0,0096 0,0103
Относительное понижение давления пара для данного раствора не зависит от природы растворенного вещества и растворителя, а также от температуры. Оно зависит лишь от концентрации раствора. В табл. 28 приведены данные относительного понижения давления насыщенного пара над раствором при растворении 1 моль вещества в 100 моль растворителя для некоторых растворителей.
Рис. 47. Схема испарения растворителя: а — из чистого растворителя; б — из раствора
Рис. 48. Схема установки для вычисления соотношения между осмотическим давлением и понижением упругости пара
Как видно из табл. 28, природа растворителя практически не сказывается на относительном понижении давления насыщенного пара, все полученные цифры очень близки между собой.
Растворы, которые строго подчиняются закону Рауля, являются идеальными. Закон Рауля соблюдается тем точнее, чем более разбавлен раствор. По мере повышения концентрации в большинстве растворов возникает отклонение от идеального состояния, особенно в растворах солей, кислот и оснований. Объяснение этому явлению будет дано несколько позднее.
На основании первого закона Рауля можно вычислять молекулярные веса растворенных веществ. Если Ро — упругость пара чистого растворителя, Р — раствора, содержащего w граммов растворенного вещества, т — молекулярный вес растворенного вещества, М — мо-
— 127 —
лекулярный вес растворителя, W — вес растворителя в граммах, то из уравнения (III, 19) получим:
Ро—Р п w/m Mw
Ро я-Мо wlm + w/M Wm + Mxu
ч
откуда
Связь осмотического давления раствора с упругостью его пара. Зная упругость пара над чистым растворителем (Ро) и над раствором (Р), можно вычислить осмотическое давление раствора.
Представим себе осмотическую ячейку /, наполненную раствором и погруженную в сосуд с чистым растворителем 2. Оба сосуда накрыты стеклянным колпаком, из-под которого выкачан воздух (рис.48). Прибор остается некоторое время в покое при постоянной температуре Т. Растворитель поступает через полупроницаемую мембрану в осмотическую ячейку (см. стр. 122), пока давление столба жидкости не уравновесит величину осмотического давления. При установлении равновесия между упругостью пара чистого растворителя Ро и упругостью пара раствора Р будет существовать следующее соотношение:
P = P0-hd, (111,22)
где h — высрта поднятия жидкости в осмометре, d — плотность паров растворителя, т. е. вес 1 см3 пара при давлении Ро. Иными словами, упругость пара на высоте h меньше упругости пара на уровне поверхности чистого растворителя на величину, равную весу столба пара растворителя высотой h.
Поскольку пары подчиняются законам газового состояния, можно сравнительно легко вычислить, чему равняется d\
1000V ’
(Ш,23)
где 7И0 — Молекулярный вес растворителя, V — объем (в литрах), занимаемый одним грамм-молем пара при давлении Ро.
Из уравнения Клапейрона — Менделеева имеем:
1 P0V = PT,
откуда
РТ
-, (111,24)
го
где R — универсальная газовая постоянная, Т — абсолютная температура, Ро — давление пара над чистым растворителем, V — объем, занимаемый молем пара при давлении Ро. Подставим значение V из уравнения (111,24) в уравнение (111,23), тогда:
М0Р0 юооет ’
(III, 25)
где d — плотность пара.
— 128 —
Вводя полученную величину d в уравнение (111,22), получим: —^°₽0 h. (Ш,26)
Осмотическое давление П в осмометре равно весу столба раствора высотой h и сечением в 1 сти2, т. е.
n=hS9 (111,27)
где S — плотность раствора, которую для разбавленных растворов можно принять равной плотности чистого растворителя. Из уравнения (II 1,27) находим h и, подставив его значение в уравнение (111,26), получим:
MqPq 10WRT
(Ш,28)
откуда
' (111,29)
/7 =
rnosRT mq
Эта формула устанавливает зависимость между осмотическим давлением раствора и упругостью его насыщенного пара. Зная упругость пара раствора, легко вычислить осмотическое давление его.
Из первого закона Рауля для сильно разбавленных растворов имеем:
- Ро-Р п
Ро п0 ’
Подставив это выражение в уравнение (111,29), получим:
1000S П = —----RTn.
MQn0
Произведение молекулярного веса растворителя ,М0 на число его молей пс даст нам вес растворителя Wo, т. е.
/1о== WQ.
Подставив это выражение в уравнение (111,30), получим
1000S „
п~ ет“-
(111,30)
(Ш,31)
HI,32)
Нетрудно догадаться, что в уравнении (III,32)
1000S 1
~ v 9
(111,33)
где V — объем растворителя (л).
Подставив значение (111,33) в уравнение (111,32), получим уравнение Вант-Гоффа;
/7=у-/?Т, (111,34)
б Зек. 560 .
Г- 129
или, поскольку С = 1/V, уравнение (111,34) примет вид:
n = nRTG. (111,35)
Таким образом, осмотическое давление раствора находится в прямой зависимости от давления его насыщенного пара. Это еще раз подтверждает, что сильно разбавленные растворы неэлектролитов ведут себя, как газы.
§ 39. Температуры замерзания и кипения разбавленных растворов
Опыт показывает, что температура замерзания и кипения растворов зависит от давления пара над ними. Еще М. В. Ломоносов обнаружил, что растворы замерзают при более низкой и кипят при более высокой температуре, чем чистые растворители. Понижение тем-пературы замерзания раствора связано с понижением давления (упругости) пара растворителя над раствором.
Как известно, жидкость закипает при той температуре, при которой давление ее насыщенного пара становится равным атмосферному давлению] Так, очищенная вода при атмосферном давлении замерзает пртгТемпературе 0Q С и кипит при ЮО^С. Стоит растворить в воде какое-либо вещество/как давление ее пара понизится. Чтобы раствор закипел, необходимо нагреть его до температуры выше 100Q С, ибо только при более высокой температуре давление пара станет равным атмосферному давлению. Чем ^больше. концентрация растворенного вещества, тем при более высокой температуре будет кипеть раствор^
Температура замерзания растворов также отличается от температуры замерзания чистых растворителей. Известно, что^жидкость-замерзает при_той температуре, при. которой давление пара вещества в твердом состоянии становится равным давлению пара этого же веще-ств'сГв жидкогщ состоянии//Например, при 0Q С давление пара льда (613,В н[м2) равно давлению пара воды. Лед и вода могут одновременно сосуществовать друг с другом при температуре, которая носит Название температуры замерзания.
Если взять какой-то водный раствор, то вследствие понижения давления пара при 0° С он будет обладать меньшим, чем 613,3 н!м\ давлением пара. По этой причине лед, опущенный в такой раствор, будет быстро таять. Лишь при некоторой температуре, лежащей ниже 0° С, давление пара над раствором уменьшится настолько, что станет равным давлению пара льда при той же температуре. Таким образом, раствор будет замерзать при более низкой температуре, чем чистый растворитель. Например, раствор, содержащий 10 г поваренной соли в 100 г воды, замерзает, как показывает опыт, не при 0Q С, а при — 13,6° С, морская вода—при —2,5Q С.
Второй закон Рауля. Процессы замерзания и кипения растворов были детально изучены Раулем (1882), который установил закон, впоследствии названный его именем.
Рассмотрим простейший вывод этого закона. На рис. 49 показана диаграмма, выражающая зависимость давления насыщенного пара
— 130 —
от температуры над чистым растворителем и над раствором. Кривая АС показывает повышение давления пара воды с увеличением температуры. Кривая АВ — давление пара льда в зависимости от температуры, а кривая BD показывает повышение давления пара над раствором при возрастании температуры. В точке А происходит пересечение кривых АВ и АС. В этой точке давление пара над раствором и давление пара льда будут одинаковыми, поэтому соответствующая данной точке температура 0° С и будет точкой замерзания чистой воды. Точку А еще называют тройной точкой, так как (три этой температуре одновременно сосуществуют три фазы: жидкая (вода), твердая (лед) и газообразная (пар).
Рис. 49. Кривые равновесных давлений паров льда, воды и водных растворов двух различных концентраций:
/ — растворителя; 2 —раствора I; 3 — раствора II; 4—«льда
Из рис. 49 видно, что давление пара над раствором при 0° С ниже, чем у чистого растворителя (воды), но оно не равно давлению пара льда при той же температуре. Лишь при температуре, ниже нуля (—/), давление пара над раствором уменьшается настолько, что становится равным давлению пара льда при той же температуре. Этому соответствует точка В, которая и является точкой замерзания раствора данной концентрации. При более высоких концентрациях раствора кривые, выражающие зависимость давления пара раствора от температуры, располагаются ниже кривой АС, но параллельно ей.
Обозначим давление пара при 0Q С чистого растворителя через Ро, а раствора при температуре его замерзания (/) — через Р. Далее из точки В параллельно оси абсцисс проведем линию до пересечения с перпендикуляром, опущенным из точки А. Пересечение этих прямых обозначим буквой К (рис. 49).
Из прямоугольного треугольника ВАК определяем:
АК
(111,36)
б*
— 131 —
Из рис. 49 видно, что Л/< — Ро — Р, ВК = h — t= М, где Д/ — понижение температуры замерзания раствора. Подставляя эти значения в уравнение (III, 36), получим:
-—-j—= tga, (HI. 37)
д/
откуда
Д/= Р°~'Р-. (111,38)
tga
Из первого закона Рауля для сильно разбавленных растворов имеем:
Рр—Р п
Р» ~ nQ 9 откуда
Р0-Р=Р0—. (111,39)
По
Подставив выражение (111,39) в уравнение (111,38), получим:
М = Р0 —----—, (111,40)
п0 tg а
где п0 и п — соответственно число молей воды и растворенного вещества. Если через W обозначить вес растворителя (воды) в граммах, а 7И0 — молекулярный вес растворителя, то
IV
= —. (Ш,41)
При подстановке этого выражения в уравнение (111,40) получим:
РрМрП 1
W tg а
(111,42)
Умножим и разделим правую часть этого выражения на 1000, тогда
Ро Мо 1000л 1000 tga ‘ W
(111,43)
Объединив все постоянные величины в уравнении (111,43) в одну по-стоянную д, т. е. К = jooo tg «"9 полУчим следующее выражение урав-нения (111,43):
м=к
1000л
W ‘
(111,44)
D 1000 л г „
Выражение представляет собой не что иное, как моляльность раствора т, т. е.
1000л
(1П.46)
— 132 —
Подставив это выражение в уравнение (III,44), получим окончательное уравнение:
Ы — Кпг. (111^-46)
Это и есть математическое выражение второго закона Рауля: понижение температуры замерзания или повышение температуры кипения растворов прямо пропорционально его моляльной концентрации.
Коэффициент К в уравнении (111,46) носит название криоскопической постоянной (греч. kryos — холод). Она представляет собой величину, характерную для данного растворителя, и показывает понижение температуры замерзания, вызываемое растворением одного моля вещества (неэлектролита) в 1000 г этого растворителя.
В самом деле, из уравнения (111,46) следует, что при т — 1 Д/ = К. Криоскопическая постоянная является постоянной величиной, она не зависит от природы растворенного вещества, а только от природы растворителя. Численные значения криоскопических констант для некоторых растворителей приведены в табл. 29.
Таблица 29
Константы замерзания некоторых растворителей
Растворители
название
формула
Криоскопическая константа К, °C
Вода . . . нао 1,86
Бензол с6нв 5,12
Нафталин СюН8 6,90
Нитробензол C„H5NOa 6,90
t Уксусная кислота .... CHgCOOH 3,90
Анилин CeH5NHa . 5,87
Фенол CeH,OH 7,27
Аналогична криоскопической постоянной константа кипения или эбулиоскопическая постоянная (лат. ebulyo — вскипать). Она характерна для данного растворителя и показывает на сколько градусов повышается температура кипения при растворении одного моля неэлектролита в 1000 г растворителя. Численные значения эбулиоскопических констант кипения приведены в табл. 30.
Таблица 30 Численные значения констант кипения некоторых растворителей
Растворитель
Эбулиоскопическая константа Ккип> °C
Растворитель
Эбулиоскопическая константа Ккип. °C
Вода .................
Бензол ...............
Анилин ...............
0,516 2,57 3,69
Этиловый спирт . . . .
Уксусная кислота . . . Четыреххлористый углерод ..................
1,16
3,10
5,00
133
Напомним, что математическое выражение второго закона Рауля в случае изменения температур кипения растворов будет совершенно аналогично уравнению (II 1,46), только вместо криоскопической постоянной берут эбулиоскопическую константу.
Свойство растворов понижать температуру замерзания воды широко используется в практике для приготовления так называемых антифризов, которые пред-ставляют водные растворы некоторых органических и неорганических веществ. Эти растворы не замерзают при низких температурах и потому широко применяются для охлаждения двигателей автомобилей и тракторов в условиях Крайнего Севера. Например, такой антифриз, как 55%-ный раствор этиленгликоля в воде не замерзает даже при температуре — 40° С.
Понижение температуры замерзания растворов имеет большое значение для живых организмов. Так, клеточный сок организмов представляет собой в основном раствор органических веществ, его температура замерзания лежит ниже нуля, поэтому организмы не погибают при пониженных температурах. Характерно отметить, что зимостойкость растений тесно связана с концентрацией клеточного сока: чем выше концентрация, тем более низкие температуры может переносить культурное растение. Процесс превращения более высокомолекулярных соединений в соединения с меньшим молекулярным весом при наступлении холодов (например, крахмала в углеводы типа глюкозы), протекающий в клетках растений, также вызван стремлением повысить концентрацию клеточного сока. По этой же причине хорошо сохраняются овощи и фрукты при температуре —1° С.
Методы измерения концентрации клеточного сока по температурам замерзания растворов в настоящее время широко используются в селекционной работе при выведении новых зимостойких сортов различных сельскохозяйственных культур.
§ 40. Применение методов криоскопии и эбулиоскопии
Измерение понижения температуры замерзания или кипения раствора позволяет решать целый ряд вопросов, касающихся свойств данного раствора и растворенного вещества. Метод исследования, основанный на измерении понижения температуры замерзания растворов, называется криоскопическим методом, а метод, основанный на измерении температуры повышения кипения растворов, получил название эбулиоскопического метода.
Для измерения температуры замерзания или кипения раствора обычно применяют дифференциальный термометр Бекмана, который изображен на рис. 50. Этот термометр имеет шкалу, разделенную на 5—6 градусов. Каждый градус в свою очередь разделен на десятые и сотые доли градуса, так что с помощью лупы можно брать отсчеты с точностью до 0,002° С. Особенностью термометра Бекмана является то, что он имеет в отличие от обычных ртутных термометров дополнительный резервуар с ртутью на верху капиллярной трубки. Наличие этого резервуара дает возможность менять количество ртути в нижнем резервуаре, что позволяет определять с помощью такого термометра разность температур в широком интервале и использовать его для измерения ' температур замерзания, а также для измерения температур кипения различных растворов.
При определении А/ сначала отмечают, где останавливается столбик ртути при кипении (замерзании) чистого растворителя, а затем после погружения термометра в исследуемую жидкость вновь отмечают температуру кипения (или замерзания) раствора. Разность между этими отсчетами даст А/.
На рис. 51 изображен прибор для определения повышения температуры кипения различных жидкостей. Исследуемый раствор помещают в широкую пробирку 1 с боковой трубкой, которая погружается в сосуд 2. Чтобы не нагревать пробирку 1 непосредственно огнем, сосуд 2 заполняют жидкостью, имеющей по сравнению с исследуемым раствором более высокую температуру кипения. Спиралевидные трубки 3 предназначены для охлаждения образующихся паров жидкости и для обратного их перевода в сосуд.
На рис. 52 изображен прибор, предназначенный для измерения температур понижения замерзания растворов. В приборе исследуемый раствор помещается
— 134 —
в пробирку /, которая вставляется в более широкую пробирку 2, погружаемую в охладительную смесь. Этой смесью набивается обширный сосуд 3, покрытый снаружи термоизоляционным слоем (войлок, пенопласт). С помощью мешалки 4 устраняют возможность переохлаждения растворов.
В лабораторной практике криоскопический метод нашел значительно большее распространение по сравнению с методом эбулиоскопии: измерять точки замерзания растворов при этом значительно проще и безопаснее, чем точки кипения их.
Определение молекулярного веса вещества. Криоскопическим методом часто пользуются для определения молекулярного веса вещества. При выводе уравнения второго закона Рауля было получено (III,-44):
„ 1000л
где Л/ — понижение температуры замерзания раствора, К — криоскопическая постоянная воды (ТС =» 1,86), п — число молей растворенного вещества и W — вес растворителя (воды), г.
1,86), n — число молей растворенного вещества и W —
Если обозначить вес растворенного вещества в граммах через т, его молекулярный вес — через М, то
т
М
(in, 47)
Подставив выражение (111,47) в уравнение (111,44) и решая его относительно Л4, получим окончательную формулу для вычисления молекулярного веса криоскопическим методом:
1000m
ГД/
(111,48)
Например, водный раствор сахара, содержащий 0,17 г сахара в 25 г воды, замерзает при температуре —0,037° С. Определить молекулярный вес сахара. Подставив данные в формулу (111,48), получим:
41 = 1,86
1000 - 0,17
25 • 0,037
Определение осмотического давления растворов. Опыт показывает, что измерение осмотического давления с помощью осмометра Пфеффера связано с целым рядом трудностей. Это измерение слишком длительно и не совсем точно, так как на практике трудно подобрать подходящую мембрану, которая бы обладала идеальной полупроницаемостью. Осмотическое давление обычно измеряется косвенными путями, одним из которых и является метод криоскопии.
В основе этого определения лежат законы Вант-Гоффа и Рауля, т. е.!
fl = RTC и
где С — концентрация раствора, выраженная в моль!л раствора, tn — концентрация раствора, выраженная в моль на 1000 а растворителя. Для разбавленных растворов можно без большой погрешности принять, что т = С. Подставив в уравнение закона Вант-Гоффа вместо С равное значение из закона Рауля, найдем:
n = ^~RT. * (111,49)
Л
После замены буквенных выражений /?, Т и К их численнымц значениями (^ля разбавленных растворов) получим окончательное уравПВйиё для" вычисления QcOdW^ecKofo ДОёни^Т^Жор^
/7 = °-082,273 д; или Л=12,04Д7. (П1.50)
1,86
— 135 —
т! 3
। 11
। ।
Рис. 50. Дифференциальный термометр Бекмана
Определение концентрации раствора. На основании второго закона Рауля можно сравнительно легко вычислить моляльную концентрацию раствора, если известно его понижение температуры замерзания:
М откуда т~-—. (111,51)
К
Таким способом можно определять концентрацию клеточного сока растений и концентрацию почвенного раствора. Необходимо помнить, что найденные этим методом концентрации являются суммарными, т. е. показывают содержание всех веществ, находящихся в растворе, выраженное в моль на 1000 г ВОДЫ.'
С помощью метода криоскопии можно определять осмотический коэффициент Вант-Гоффа и степень электролитической диссоциации слабых электролитов.
Рис. 51. Аппарат для эбулиоскопического определения молекулярных весов
Рис. 52. Аппарат для криоскопического определения молекулярных весов
— 136 —
Глава IV
ЭЛЕКТРОПРОВОДНОСТЬ РАСТВОРОВ
§ 41. Отступление от законов Вант-Гоффа и Рауля в растворах электролитов. Теория электролитической диссоциации
Законы Вант-Гоффа и Рауля справедливы для идеальных растворов, в которых не происходит химическое взаимодействие между компонентами раствора, а также диссоциация или ассоциация молекул растворенного вещества. Однако опыт показывает, что не все растворы подчиняются этим законам. Установлено, что растворы, которые способны проводить электрический ток (электролиты), имеют более высокое, чем по закону Вант-Гоффа, осмотическое давление. Эти растворы кипят при более высокой температуре и замерзают при более низкой, чем это следует из закона Рауля. Такими свойствами обладают растворы солей, кислот и оснований.
Обобщая наблюдения, Вант-Гофф пришел к выводу, что в отношении осмотического давления растворы электролитов ведут себя так, как будто они содержат больше частиц, чем это следует из аналитической концентрации. Исходя из этого, Вант-Гофф внес в уравнение (111,35) для растворов электролитов поправку, получившую название коэффициента Вант-Г
£а или изотонического коэффициента (t). При этом
P = iCRT. (IV, I)
Изотонический коэффициент показывает, во сколько раз наблюдаемое осмотическое давление (Роп) раствора больше теоретически ного (Рвыч) для раствора неэлектролита, т. е.
Эй ГН
вычислен-
(IV,2)
Р выч
Отсюда следует: . к,
, RT С оп_____Соп А^зам. on___________А*кип. оп
*ГСВЫЧ Свыч А/Зам выч А/кип выч
Таким образом, коэффициент i можно найти, если измерить непосредственно осмотическое давление с помощью осмометра Пфеффера или криоскопическим методом, что значительно проще.
Коэффициент Вант-Гоффа для неэлектролитов, растворенных в воде, равен l,ja для электролитов он больше единицы. Значение коэффициента растёт по мере разбавления электролита. Для растворов, в которых имеет место ассоциация молекул растворенного вещества, коэффициент i бывает меньше единицы.
Причину отклонения от законов Вант-Гоффа и Рауля в растворах электролитов впервые разъяснил шведский ученый С. Аррениус (1883—1887) в своей теории электролитической диссоциации. Она основывалась на трех постулатах.
137 —
1. Электролиты обладают способностью при растворении в соответствующих растворителях (например, в воде, к которой первоначально и относилась теория Аррениуса) диссоциировать на противоположно заряженные частицы—ионы. При этом молекулы кислот распадаются на положительные ионы водорода и отрицательные ионы кислотного остатка
H2SO4 = 2H+ 4-SO2”
Молекулы оснований—на положительные ионы металла в отрицательные гидроксид-ионы
кон=к+ + он-
Соли (средние) образуют положительные ионы металла и отрицательные ионы кислотного остатка
КС1 = К+4-С1~
Кислые и основные соли, помимо ионов металлов и кислотного остатка, дают также в растворе ионы водорода или гидроксид-ионы
КН2РО4 = К+ + 2Н+ + РО£ -
AI (ОН) SO4 =А13+H-SO|~ 4-ОН-
Таким образом, электролиты при растворении в воде распадаются на ионы, за счет чего увеличивается число частиц. Это увеличение числа частиц и влияет на осмотическое давление и температуры кипения и замерзания растворов, т. е. свойства электролитов определяются суммой концентраций частиц — ионов и недиссоциированных молекул.
2. Электролиты при растворении диссоциируют на ионы не полностью. Доля молекул, распавшихся в состоянии равновесия на ионы, количественно характеризуется степенью электролитической диссоциации и..обпзначае.т£я, через сс
Степень электролитической диссоциации равна отношению числа молекул и, распавшихся на ионы, к общему числу растворенных молекул N (ионизированных п и неионизированных па)
п п а —— = ------
А п4-ла
Величина степени электролитической диссоциации зависит от природы растворенного вещества и растворителя, а также от концентрации и температуры раствора.
Если вещество не диссоциирует при растворении (и = 0, па = М, а = 0), оно не будет электролитом. Если а близка к единице, то п ж ~ N, и соединение является сильным электролитом. Для многих химических соединений 0 < а 1, а следовательно, п 7V, т. е. они будут слабыми электролитами.
По способности к диссоциации Аррениус разделил все электролиты на три группы: сильные электролиты (а >* 30%), электролиты средней силы (а = 5 — 30%), слабые электролиты (а < 5%). К сильным электролитам были отнесены: соляная, бромистоводородная, иодисто-водородная, азотная, серная, марганцовая кислоты; едкий натр, едкое
(IV, 4)
— 138
кали, гидроокись бария, а также большинство солей. Согласно теории Аррениуса для сильных электролитов характерна значительная диссоциация и, следовательно, хорошая электропроводность.
К слабым электролитам относятся почти все органические кислоты (муравьиная, уксусная, бензойная), цианистоводородная кислота, борная кислота, угольная кислота, сероводородная кислота, гидроокись аммония, вода, а также некоторые соли (HgCl2, CdCl2). Для растворов слабых электролитов характерна очень небольшая величина электропроводности.
К электролитам средней силы относятся фосфорная, мышьяковая, иодная, хромовая, сернистая кислоты и целый ряд других соединений,
3. Силы взаимодействия между ионами отсутствуют и растворы электролитов ведут себя подобно идеальным газовым системам. Это положение автором теории электролитической диссоциации и его последователями прямо не высказывалось, но оно лежит в основе всех ее количественных соотношений.
При помощи трех постулатов теория электролитической дцссоциа-ции смогла объяснить многие свойства раствора, дать их количественную характеристику и истолковать многочисленные факты и закономерности.
В своих работах Аррениус показал, что степень диссоциации электролита а можно связать с коэффициентом Вант-Гоффа i. Приведем простейший вывод этих соотношений. Предположим, что в растворе находилось N молекул электролита, из которых только п продиссо-циировало на ионы. Число непродиссоциированных молекул N — п, а число образовавшихся ионов vn, где v — количество ионов, на которое диссоциирует одна молекула электролита. Тогда число всех частиц в растворе, включая и молекулы, и ионы, будет равно (N—ri) + vn, или после соответствующего преобразования:
(N—ri)+vn — N—п+vn = N+ п (у—1). (IV,5)
Таким образом, мы нашли общее число частиц, находящихся в растворе электролита. Если бы у нас был раствор неэлектролита, то число частиц осталось бы равным N. Осмотическое давление, как уже отмечалось, пропорционально числу частиц. Следовательно, наблюдаемое осмотическое давление Рои пропорционально общему числу частиц N + п (у — 1), теоретическое же Рвыч пропорционально AL Исходя из этого можем записать:
. Рon Con RT_______соп (1У
Р ВЫЧ Свыч RT 6*ВЫЧ
После подстановки в уравнение (IV,6) значений N и N + п (у — 1) получим
z==JL+”^-1)..= A+ Mv-i) (IV7)
1 В этом уравнении выражение n/АГ есть не что иное, как степень диссоциации а. После такой замены выражение (IV,7) примет следующий вид:
1с=1 _|_ ct (v— 1)»
139 —
откуда
(IV,8)
(IV,9)
а =----у.
v—1
Поскольку i = Д/ОП7Д/ВЫЧ, то> подставив это выражение в уравнение (IV, 8), получим формулу для вычисления степени электролитической диссоциации криоскопическим методом:
А^оп, J
I 1 А/выч А/оп— А/выч
а =------=------------= -----------
V—1 V— 1 A/BbI4(v—1)
В качестве примера разберем следующую задачу. Раствор, содержащий 0,85 г хлорида цинка7пС12в 125 г воды, замерзает при температуре— 0,230° С. Определить степень электролитической диссоциации а.
Для решения воспользуемся формулой Рауля (111,48), из которой
А „ lOOOzrz 1000-0,85 Л
Д/выч = К------= 1,86 ----— = 0,093 °C.
выч WM 125-136
Таким образом, если бы раствор ZnCI2 подчинялся закону Рауля, он замерзал бы при температуре—0,093° С, т. е. А/выч = 0,093° С. Подставив численные значения Д/оо и А/Выч в уравнение (IV,9), получим
0,230—0,093
0,093 (2 — 1)
= 0,753 = 75,3%.
Из всего вышеизложенного можно сделать следующие выводы.
I. Растворы электролитов будут изотоничными, если при одинаковой температуре они содержат одинаковое число частиц (молекулы + + ионы) в единице объема.
2. Из двух растворов с одинаковой молярной концентрацией осмотическое давление будет выше в растворе электролита с более высокой степенью диссоциации а.
3. Из двух растворов с одинаковой молярной концентрацией и степенью диссоциации а осмотическое давление будет выше в растворе электролита, диссоциирующего на большее число ионов.
Теория электролитической диссоциации Аррениуса дала возможность объяснить не только причины отклонения растворов электролитов от законов Вант-Гоффа и Рауля, но и объяснить многие особенности химических свойств электролитов (реакции гидролиза, значение концентрации водородных ионов и др.). Однако она имела и ряд недостатков, в частности, не учитывала взаимодействия между ионами в растворе, вызываемого их электрическими зарядами.
И. А. Каблуков (1891), основываясь на гидратной теории растворов Д. И. Менделеева, считал, что нельзя рассматривать раствор как систему, в которой отсутствует взаимодействие частиц растворителя и растворенного вещества. В отличие от Аррениуса, Каблуков утверждал,
— 140 —
что ионы растворенного вещества взаимодействуют с водой, образуя химические соединения—гидраты. В работе «Современные теории растворов (Вант-Гоффа и Аррениуса) в связи с учением о химическом равновесии» И. А. Каблуков писал: «... по нашему мнению, вода, разлагая молекулы растворенного тела, входит с ионами в непрочные соединения, находящиеся в состоянии диссоциации, по мнению же Аррениуса, ионы свободно двигаются, подобно тем отдельным атомам, которые происходят при диссоциации молекул галоидов при высокой температуре». Дальнейшее развитие науки показало правоту взглядов русского ученого.
Впоследствии были установлены и другие расхождения теории электролитической диссоциации с опытом. В частности, было доказано, что так называемая теплота нейтрализации сильных кислот сильными основаниями не зависит от их природы. Она имеет постоянную величину. В качестве примера можно привести следующие реакции:
HNO34-NaOH = NaNO34-H2O4-13,7 ккал
НС1 + КОН = KCI 4- Н2О 4-13,7 ккал
НС1О4 4-Na0H = NaC104 4-Н2О 4-13,7 ккал]
в которых, как видим, выделяется одинаковое количество тепла на каждую грамм-молекулу образующейся воды. Все эти реакции фактически сводятся к реакции образования воды из ионов водорода и гидроксид-ионов:
Н+ 4-ОН~ =Н2О 4-13,7 ккал
Если бы в растворах сильных кислот и щелочей имелись недиссо-циированные молекулы, теплота нейтрализации этих кислот щелочами была бы различной, так как она являлась бы алгебраической суммой теплот двух процессов: диссоциации молекул кислоты и щелочи на ионы и образования молекул воды из ионов Н+ и ОН“. Объяснение этому факту может быть только одно — в растворе сильных кислот и оснований молекулы полностью диссоциированы на ионы. А это уже противоречив теории Аррениуса, так как согласно последней все эти вещества имели, хотя и высокую (более 30%), но не 100%-ную степень электролитической диссоциации, а значит должны были бы иметь в растворе и недиссоциированные молекулы.
Согласно теории Аррениуса степень электролитической диссоциации а, определяющая долю ионизированных молекул в растворе, должна быть при заданных условиях одной и той же (независимо от метода ее измерения). При этом, согласно ее физическому смыслу, она не может быть больше единицы и меньше нуля.
Однако многочисленные экспериментальные данные, полученные разными учеными, противоречили этим положениям теории.
В качестве примера в табл. 31 приведены величины а для растворов соляной кислоты, вычисленные на основании измерений электропроводности (ах) и электродвижущих сил (а2).
Как видно из этой таблицы, расхождения между значениями а, 4 прлученными различными методами, увеличиваются по мере возрастания концентрации НС1, причем в области высоких концентраций а2
* Таблица 31
Степень электролитической диссоциации по данным измерений электропроводности (04) и э. д. с. (а2)
Cj-jci» моль/л «2 СнС1 моль!л «1 а.
0,003 0,986 0,990 3,0 1,402
0,08 0,957 0,880 6,0 — 3,4
0,3 0,903 0,773 16,0 —— 13,2
становится значительно больше единицы. Подобный результат с точки зрения теории Аррениуса представляется невероятным, так как в этом случае на ионы должно распадаться больше молекул, чем их вообще. присутствует в растворе. Следовательно, в данном случае степень электролитической диссоциации а не может иметь того физического смысла, какой ей приписывается теорией Аррениуса.
На основании громадного числа фактов было установлено, что теория электролитической диссоциации приложима только к разбавленным растворам слабых электролитов. Поведение концентрированных растворов слабых электролитов, а также растворов сильных электролитов любых концентраций нельзя описать количественно на основании теории Аррениуса.
Главный недостаток теории электролитической диссоциации, и вместе с тем причина всех ее недостатков, заключается в игнорировании взаимодействия частиц растворенного вещества между собой, а также с молекулами растворителя. Все эти противоречия были в дальнейшем в значительной степени устранены в так называемой теории сильных электролитов.
§ 42. Основные положения теории сильных электролитов
Как уже упоминалось, величина электропроводности сильных электролитов далеко не соответствует полной диссоциации их молекул на ионы. Однако при оптических и спектральных исследованиях растворов сильных электролитов в них не обнаруживается характерных свойств молекул, что отличает эти растворы от растворов слабых электролитов, в которых можно обнаружить недиссоциированные молекулы. Рентгенографическое исследование кристаллов сильных электролитов, например КС1 и NaCI, показало, что эти электролиты даже в твердом агрегатном состоянии не содержат молекул и имеют ионные кристаллические решетки. Однако если принять полную диссоциацию сильных электролитов и этим ограничиться, то совершенно необъяснимы будут другие явления. Например, экспериментально определяемые величины понижения температуры замерзания и повышения температуры кипения оказываются у сильных электролитов меньше, чем следовало бы ожидать при полной диссоциации молекул на ионы. Таким образом, теория электролитической диссоциации уже не могла полностью объяснить всех свойств растворов.
142
В 1923 г. Дебай и Гюккель создали теорию сильных электролитов. В разработку этой теории большой вклад внесли Д. П. Коновалов, И. А. Каблуков, В. А. Кистяков, Л. В. Писаржевский, А. Нойес. Согласно этой теории в растворах сильных электролитов действуют электростатические силы притяжения между разноименными ионами и силы отталкивания — между одноименными. Вокруг каждого иона образуется ионная атмосфера, состоящая из ионов противоположного знака. Каждый из ионов этой атмосферы находится в окружении другой ионной атмосферы. Поэтому раствор сильного электролита можно
рассматривать как систему равномерно распределенных по всему объему сосуда разноименных ионов, каждый из которых находится в центре силового поля, создаваемого окружающими ионами (рис. 53). Тепловое движение постоянно изменяет картину распределения ионов в такой сфере: в ней происходит постоянный ионный обмен. Ввиду того, что радиус ионной атмосферы относительно велик, атмосферы двух соседних ионов пересекаются, в результате чего каждый ион в данный мо
Рис. 53. Модель ионной атмосферы, принятая теорией Дебая и Гюккеля
мент может входить в состав одной или даже нескольких ионных атмосфер других ионов.
Все это обусловливает довольно сложные взаимоотношения между компонентами раствора, которые не могут не сказаться на его свой
ствах.
\ Исходя из того, что сильные электролиты полностью диссоциированы, можно было ожидать, что коэффициент Вант-Гоффа i цля электролита, диссоциирующего, например, на два иона, должен равняться двум не только в разбавленных, но и в достаточно концентрированных растворах. Однако опыты не подтверждают этого. Коэффициент i в растворах сильных электролитов в значительной степени зависит от концентрации электролита, уменьшаясь с увеличением концентрации раствора. Такая зависимость i от концентрации в растворах объясняется взаимодействием ионов между собой.
В электрическом поле постоянного тока ионы в растворах сильных электролитов имеют меньшую подвижность ввиду межионного взаимодействия.
Дело в том, что под влиянием внешнего электрического поля «ионная атмосфера» смещается к одному полюсу, а ион, находящийся в центре этой атмосферы, стремится к другому полюсу. Кроме того, увлекаемые ионами сольватные (гидратные) оболочки также способствуют их торможению. Чем выше концентрация растворов, тем плотнее «ионная атмосфера» и тем медленнее движутся ионы.
143 -
Межионное взаимодействие, а также сольватация ионов уменьшают не только абсолютную скорость их движения, но и осмотическое давление растворов, величину понижения давления пара над ними и т. д. Создается впечатление, что в растворе находится меньше ионов, чем на самом деле. Поэтому величина а является не истинной, а кажущейся степенью электролитической диссоциации сильного электролита.
В слабых электролитах, растворы которых содержат относительно малое количество ионов, взаимодействие последних сравнительно невелико. Кажущаяся степень диссоциации для них практически отвечает истинному значению.
Как известно, уравнения Генри, Вант-Гоффа, Рауля и другие термодинамические уравнения хорошо описывают свойства предельно разбавленных (идеальных) растворов. Свойства растворов сильных электролитов, рассчитанные по этим уравнениям, значительно отличались от фактически наблюдаемых. Многочисленные исследования показали, что введение в эти и другие термодинамические уравнения поправочных коэффициентов, учитывающих отклонение от идеального состояния, нецелесообразно, так как при этом сами уравнения становятся чрезмерно сложными.
Поэтому дальнейшее развитие теории растворов пошло по другому пути. Форма уравнений, описывающих свойства идеальных растворов, была сохранена неизменной, но применимость этих уравнений к реальным системам достигалась тем, что в них вместо обычных величин, характеризующих системы (давления, концентрации), стали использовать величины, заимствованные из опыта. В настоящее время все термодинамические расчеты свойств растворов сильных электролитов строятся на использовании введенной Льюисом величины активности электролита или активности его ионов. Активность определяется как величина, подстановка которой вместо концентрации в термодинамические уравнения, действительные для простейших (идеальных) систем, делает их применимыми к рассматриваемым растворам.
Активности отличаются от концентраций только тем, что в них входят силы взаимодействия, существующие в растворах и не зависящие от природы растворенных частиц, а также от их концентрации. Поэтому активность можно представить как произведение концентрации на некоторый переменный фактор, называемый коэффициентом активности, т. е.
a = fC.
(IV. 10)
где а — активность электролита (или его ионов), С — аналитическая концентрация электролита, / — коэффициент активности, включающий поправку на силы взаимодействия.
Коэффициенты активности можно найти, сравнивая аналитические концентрации с теми величинами, которые следует подставить в уравнение для растворов электролитов, чтобы получить полное соответствие с опытными данными. Обычно их определяют экспериментальным путем по величине осмотического давления, по понижению температуры замерзания, по повышению температуры кипения раствора
— 144
или же путем измерения э. д. с. соответствующей гальванической цепи.
Коэффициент активности, как правило, бывает меньше единицы, лишь при очень большом разбавлении раствора, когда силы взаимодействия между ионами приближаются к нулю, коэффициент активности становится равным единице. В этом случае а ж С, т. е. движение ионов в растворе не стеснено. У сильных электролитов, например, что имеет место только в очень разбавленных растворах при С < < 0,0001 моль/л. В таких растворах расстояние между ионами достаточно большое, и межионные силы не оказывают влияния на скорость их передвижения.
Если коэффициент активности меньше единицы, активность ионов меньше их концентрации, получившейся при диссоциации растворенного вещества:
а
Так, в 0,1 М растворе соляной кислоты активная концентрация Н+ и С1“ получается равной всего Сна • 2/ = 0,1 • 2 • 0,814 — = 0,163 г-ион/л вместо 0,1 • 2 = 0,2 г-ион/л, так как коэффициент активности f для однозарядных ионов Н+ и С!** в 0,1 М растворе НС1 равен 0,814. Активность же ионов Н+ (ан+) в 0,1 М растворе соляной кислоты будет:
= /Сн+ — 0,814-0,1 ==0,0814 г-ион/л.
Необходимо отметить, что при очень больших концентрациях некоторых электролитов [ вновь начинает расти, что объясняется недостатком молекул воды для гидратации всех ионов. Ионы, частично или полностью лишенные гидратной оболочки, особенно легко подвижны. Активность в подобных случаях оказывается выше действительной концентрации частиц, а коэффициент активности f становится больше единицы. В табл. 32 приведены значения коэффициентов активности в зависимости от концентрации электролита в растворе.
Таблица 32
Изменение коэффициентов активности (/) KCI, NaC! и LiCl в зависимости от молярного содержания раствора при 25 °C
Концентрация, моль/кг Изменение f для
КС1 NaCl LIC1
0,001 0,965 0,966 • 0,965
0,01 0,899 0,903 0,901
0,1 0,754 0,778 0,779
0,2 0,712 0,732 0,756
0,5 0,597 0,656 0,757
1,0 0,569 0,670 0,919
2,0 0,571 0,714 1,174
4,0 0,581 0,779 1,554
5,0 0,599 1,019 —
145 —
В водных растворах коэффициент активности данного электролита (или данного иона) зависит в основном от концентраций и валентностей всех присутствующих ионов. Коэффициент активности того или иного вещества может быть определен экспериментально различными методами. Необходимо отметить только, что величину коэффициентов активности отдельных ионов опытным путем определить нельзя, так как всегда результат получается итоговый для растворенного вещества в целом.
Для характеристики зависимости активности иона от концентрации всех находящихся в растворе ионов Льюис ввел понятие об ионной силе раствора электролита. Ионной силой раствора электролита называется величина (р), измеряемая полусуммой произведения концентрации (С) каждого из присутствующих в растворе ионов на квадрат их валентности (г), т. е.
М-=~т“ (21 Ci-}-гг Сг Ч-гз С3 4- ••• 4-^пСп),
или короче
(iv.ll)
Недиссоциированные молекулы, как не имеющие зарядов, в формулу подсчета ионной силы раствора не включаются.
Приведем пример вычисления ионной силы раствора для 0,4 М раствора Na2SO4. Для решения используем формулу (IV, 11):
H=v<12CNa+ +22Cso2-)=4-(l2-0>8 + 22-0-4)=V(°-8+1-6) = ]’2-£ £
С увеличением концентрации раствора сильного электролита количество ионов в растворе возрастает, что приводит к увеличению ионной силы раствора и значительному уменьшению коэффициента активности, а следовательно, и активности всех ионов.
Для разбавленных растворов, ионная сила которых не превышает 0,01, коэффициент активности ионов связан с ионной силой раствора следующим соотношением:
lgf--0,5zaV7, (IV,12)
где z — заряд иона, ар — ионная сила раствора.
Из этой формулы следует, Что чем больше ионная сила раствора, тем меньше коэффициент активности его ионов; если ионные силы двух растворов равны, то коэффициенты активности равновалентных ионов также одинаковы. В табл. 33 приведены средние значения коэффициентов активности ионов в зависимости от величины ионной силы раствора.
Коэффициент активности / широко используется в практике и в теоретических расчетах. Активность ионов (выраженная как и концентрация в г-ион/л) является на самом деле эффективной концентрацией, проявляющей себя при химических реакциях. Обычные же концентрации показывают количество вещества, находящееся в растворе.
— 146 —
Таблица 33
Приближенное значение средних коэффициентов активности в зависимости от ионной силы раствора
Ионная сила Коэффициенты активности ионов
однозарядных двухзарядных трехзарядных четырехзарядных
ЫО-4 0,99 0,95 0,90 0,83
2-10-4 0,98 0,94 0,87 0,77
5.10-4 0,97 0,90 0,80 0,67
ыо-3 0,96 0,86 0,73 0,56
2-10~3 0,95 0,81 0,64 0,45
5-Ю-3 0,92 0,72 0,51 0,30
ыо-2 0,89 0,63 0,39 0,19
2-10-2 0,87 0,57 0,28 0,12
Б-К)-2 0,81 0,44 0,15 0,04
0,1 0,78 0,33 0,08 0,01
0,2 0,70 0,24 0,04 0,003
0,5 0,62 —— — ——
Поэтому при теоретических расчетах химических реакций в растворах в связи, например, с использованием закона действующих масс, необходимо брать не концентрации веществ, находящихся в растворе, а действующие активные массы исходных реагентов и образующихся продуктов реакции, которые в данный момент непосредственно участвуют в химическом процессе.
Теория сильных электролитов, развитая Дебаем и Гюккелем, при большой сложности математического аппарата применима только при концентрациях, не превышающих 0,01—0,05 н. Выводы этой теории хорошо согласуются с экспериментальными данными для очень разбавленных водных растворов. Для более высоких концентраций она оказывается непригодной, чтобы достаточно полно охарактеризовать чрезвычайно сложную картину взаимодействия между частицами, находящимися в растворе.
Плотность ионной атмосферы, ее радиус, скорость возникновения и разрушения сложным образом влияют на термодинамические и электропроводные свойства электролита. Количественно учесть влияние всех этих фактов теория Дебая и Гюккеля была в состоянии только для простейших электролитов и при условии очень сильного разбав-т ления.
В настоящее время установлено, что в более концентрированных растворах между заряженными ионами возникает взаимодействие не только электростатического, но и химического порядка. В частности, было установлено, что в концентрированных растворах электролитов в воде (а в неводных растворителях с низкой диэлектрической постоянной и при умеренных концентрациях электролита) возможно образование ионных пар или ионных двойников. Ионные двойники из положительно и отрицательно заряженных ионов появляются в результате действия чисто кулоновских сил, поэтому они менее прочны, чем недиссоциированные молекулы электролита. Однако связи, удерживающие ионы вместе, достаточно сильны для того,
\ {47 —*
чтобы первоначальные ионы потеряли свою самостоятельность и стали проявлять свойства незаряженных частиц.
Например, образование ионных двойников из исходных индивидуальных ионов при растворении хлорида калия можно схематически представлять следующим образом
К+ -ьС1“=К+ Cl-
Такая ассоциация КС! в воде возможна при концентрации 27 кг-экв!м\
Более поздними исследованиями было установлено, что в концентрированных растворах, кроме незаряженных ионных двойников, можно ожидать также образование ионных тройников, в которых заряды ионов не уничтожаются:
М+А* 4- д- ^мAo-
Il
М+ А~ 4-Mi- =.м, Ai-
Образование ионных тройников можно представить себе также как результат ассоциации двух ионных пар с последующей ионизацией возникших комплексов:
и
М+ А“4-М+ А~--(М + А~)2«МА7 +М +
• М+А“ + М+А*’=,(М+А'“)2 = М2А+4-А“
В настоящее время считается экспериментально доказанным, что наряду с заряженными ионными тройниками в концентрированных растворах электролитов (особенно в неводных растворах) могут присутствовать также незаряженные ассоциированные соединения. Эти соединения могут образоваться по схеме:
М2 А1 -J- МА^г ===(А1. A)g
Образование ионных двойников, тройников и незаряженных комплексов является причиной особого поведения сильных электролитов в неводных средах: уменьшение изотонического коэффициента, снижение осмотического давления, электропроводности и т. д, по сравнению с водными растворами равнозначных концентраций.
Таким образом, современная теория растворов сильных электролитов, развитая на основе представлений Дебая и Гюккеля, еще далека от совершенства. Вывод теоретических уравнений для расчета коэффициентов активности, применимых в широкой области концентраций, требует создания более точных представлений о молекулярном строении электролитов и о природе тех сил, которые наряду с кулоновскими силами и силами теплового движения действуют между всеми частицами раствора.
— 148 —
§ 43. Физиологическое действие ионов.
Ионный антагонизм
Как известно, электролиты при растворении диссоциируют на ионы и тем самым увеличивают общую концентрацию частиц в единице объема раствора. В связи с этим растворы электролитов по сравнению с неэлектролитами обнаруживают повышенный осмотический эффект. Однако роль электролитов в живом организме сводится не только к поддержанию на определенном уровне осмотического давления; ионы в живом организме обладают определенной физиологической активностью, специфическим физиологическим воздействием.
Опыт показывает, что наибольшей физиологической активностью обладают ионы водорода и гидроксид-ионы.
Впервые убедительно показал, что электролиты оказывают на протоплазму клетки специфическое физиологическое действие, биолог Жак Лебу в 1893 г. До него считалось, что стоит поместить какой-либо живущий в воде организм в изотонический раствор, как нормальная жизнедеятельность этого организма будет обеспечена. Однако последующие опыты показали, что одного этого явно недостаточно для нормальной жизнедеятельности организма. При отсутствии некоторых катионов живые ткани и даже организмы в целом теряют способность к возбудимости.
Как показал опыт, организмы, живущие в море, погибают и будучи помещены в изотонический раствор соли, например NaCI. Однако, если в этот раствор добавить небольшое, но точно определенное количество раствора соли с двухвалентным катионом, например, СаС12 (обладающим в чистом виде еще большей ядовитостью), то ядовитое действие как той, так и другой соли будет устранено.
Таким образом, растворы солей с одновалентными и двухвалентными катионами, взятые порознь, действуют на живые организмы как яды.
При смешении этих растворов в определенных соотношениях происходит взаимная нейтрализация их ядовитых свойств. Такая взаимная нейтрализация ядовитых свойств растворов получила название антагонизма ионов.
Наиболее ярко выражен антагонизм между одновалентными и двухвалентными ионами. Сущность этого антагонизма в деталях еще не выяснена, но в основе его лежат изменения коллоидно-химических свойств протоплазмы, особенно ее поверхностных слоев. По-видимому, только определенное соотношение между одно- и двухвалентными ионами обеспечивает нормальную структуру поверхностных слоев протоплазмы и тем самым нормальную проницаемость. Исследования последних лет показали, что в основе антагонизма ионов зачастую лежат и другие причины более глубокого физиологического порядка: противоположное влияние ионов-антагонистов на определенные стороны обмена веществ или на активирование ферментов, конкуренция за участие в органическом комплексе ферментов и т. д.
Так, при изучении особенностей обмена железа в условиях железо-марганцевого хлороза было показано, что нарушение процессов зеленения связано не только с изменением характера использования железа, но и деятельности железосодержащих ферментов. В частности, торможение поступления железа в растение при избытке марганца является результатом конкуренции между двумя указанными элементами за место в простетических группах ферментов.
Антагонистическое действие ионов может быть вызвано и другими причинами. Например, антагонистическое действие молибдена по отношению к железу объясняется тем, что молибден способен образовать комплексы с фосфат-иона-ми, а это в конечном итоге ведет к задержке осаждения железа в виде фосфата. Иными словами, молибден улучшает усвояемость железа.
Отмечено сильное антагонистическое действие кремния по отношению к марганцу. Кремний устраняет токсичность марганца при его избытке, не уменьшая поступления марганца в растение. По-видимому, в этом случае кремний образует какие-то комплексные соединения с марганцем, которые не являются токсичными для растения.
Отмечено также антагонистическое действие кремния по отношению к алюминию. Токсичное действие алюминия на кислых почвах, где оно проявляется особенно остро, подавляется внесением в эти почвы кремнийсодержащих удобрений, что в несколько раз увеличивает выносливость пшеницы к алюминию.
. ~ 149 —
В настоящее время антагонизм ионов, механизм их поступления в растения и другие связанные с этим явлением процессы широко изучаются. Достижения в этой области могут быть использованы в борьбе за повышение урожая сельскохозяйственных культур.
§ 44. Электропроводность растворов. Удельная электропроводность
Под прохождением электрического тока через вещество понимают движение (перенос) электрических зарядов от одного полюса к другому под действием внешнего электрического поля. Способность вещества проводить электрический ток называется электропроводностью.
Различают две основные формы проводимости: электронную и ионную. Электронной проводимостью обладают, например, металлы в твердом и расплавленном состоянии. Электрический ток по этим проводникам передается потоком электронов аналогично потоку газов в трубе в направлении от катода цепи к аноду.
В растворах электролитов перенос электричества осуществляется за счет перемещения ионов. Анионы в электрическом поле движутся к положительно заряженному электроду — аноду, катионы—к отрицательному электроду — катоду. Скорость движения ионов в растворах по сравнению со скоростями движения электронов в металлах мала, поэтому электропроводность, например, меди и серебра примерно в 1 000 000 раз больше электропроводности растворов.
Проводник, по которому течет электрический ток, представляет для него определенное сопротивление. За единицу сопротивления, как известно, принят ом, который представляет собой сопротивление столба ртути длиной 106,3 см и площадью сечения 1 мм2 при 0° С.
Согласно закону Ома сопротивление R прямо пропорционально длине проводника /, обратно пропорционально площади сечения S и зависит от материала:
/?=р4-.
В этом уравнении р (греч. «ро») — удельное сопротивление, т. е. сопротивление проводника, имеющего длину 1 см и сечение в 1 см? (при t = const), которое зависит исключительно от качества материала. Для сравнения в табл. 34 приведены численные значения удельного сопротивления различных проводников.
Таблица 34
Удельное сопротивление различных проводников при. 18° С (ом-см)
• Материал Удельное сопротивление Материал Удельное сопротивление
Серебро 0,00000163 Графит 0,0028
Медь 0,00000174 H2SO4, 5%-ный водный
раствор 4.8
Алюминий 0,00000290 NaOH, 5%-ный водный
Ртуть 0,00009580 раствор 5,08
— 150 —
Значения удельных сопротивлений приведены для одной и той же температуры, поскольку сопротивление проводников зависит от температуры. Эта зависимость для металлов и электролитов противоположна: если сопротивление металлов с повышением температуры увеличивается, то сопротивление растворов электролитов, наоборот, уменьшается (примерно на I—2,5% на каждый градус).
Когда речь идет о растворах электролитов, обычно говорят не о сопротивлении растворов, а об их электропроводности. Мерой электропроводности является количество электричества, выраженное в кулонах, которое за единицу времени проходит через электролит. Таким образом, для растворов электролитов справедливо следующее соотношение:
I = LE, (IV,14)
где / — сила тока, Е — электродвижущая сила (э. д. с.), в\ L —электропроводность электролита. В том случае, когда Е = 1, I = Ц L, как и /, есть сила тока, измеряемая в амперах.
Из курса физики известно, что
7=4-* (,v’15)
t\
где / — сила тока, Е — э. д. с., R — сопротивление. Подставив значение / из уравнения (IV, 15) в уравнение (IV, 14), получим:
'-“-т •
откуда
L=— ом-1. ' (IV, 16)
I к
Таким образом, электропроводность раствора можно характеризовать как величину, обратную его сопротивлению. Подставив в уравнение (IV, 16) значение k из закона Ома (IV, 13), будем иметь:
. £=—-4-. (IV,17)
р 1
где 1/р величина, обратная удельному сопротивлению, называемая удельной электропроводностью. Обозначается она буквой х (греч. «каппа»). С учетом этого обозначения уравнение (IV, 17) примет вид:
L = x-y-. (IV,18)
Если S = 1 см2, а I = 1 см, то L = х
Удельная электропроводность электролита х представляет собой величину, обратную сопротивлению столба раствора длиной в 1 см и площадьк? сечения в 1 см2. Измеряется удельная электропроводность § [ом"1 -см"1]. В табл. 35 приведены значения удельной электропроводности для растворов некоторых электролитов.
— 151 —
Таблица 35
Удельная электропроводность растворов некоторых электролитов при 18° С
Концентрация раствора, % КС1 NaOH H2SO4 NaCI
5 6,9.10-2 0,19 0,21 6,7.10-2
10 0,14 0,13 0,39 0,12
15 0,20 0,54 0,16
Поскольку в растворах электролитов при прохождении электричества ионы перемещаются между электродами и отдают свой заряд только на их поверхности, то в приведенной формуле X обозначает площадь, / — расстояние между электродами.
Например, удельное сопротивление некоторого образца воды при 18° С равно р = 2 • 10°ож • см. Удельная электропроводность этого образца воды будет равна:
1 1
Х= = — = 5- Ю“7 ОЛ!'* 1 • СЛ!"1.
р 2-Ю6
Если мы опустим в эту воду два электрода площадью в 1 см2, то при расстоянии между электродами в 1 см и разности потенциалов в 1 в сила тока будет равна 5 • 10“7с (при 18е С). Электропроводность растворов электролитов зависит от общего числа их ионов в единице объема раствора. Вследствие этого удельная электропроводность электролитов зависит от концентрации раствора. По мере увеличения кон
центрации электролита удельная электропроводность сначала растет,
Концентрация, кмоль-м^
Рис. 54. Зависимость удельной электропроводности х некоторых растворов сильных (H2SO4, КОН, LiCI) и слабых (СН3СООН) электролитов от концентрации
азатем уменьшается, так как вместе с ростом числа ионов уменьшается скорость их перемещения, а также степень диссоциации вещества. Первый фактор действует в растворах сильных электролитов, второй—в растворах слабых электролитов. При достижении определенной концентрации раствора влияние перечисленных факторов становится настолько значительным, что дальнейшее увеличение концентрации приводит к уменьшению электропроводности (рис. 54).
Удельная электропроводность растворов электролитов зависит также от индивидуальных свойств ионов. Дело в том, что количество переносимого ионами электрического тока в растворе электролита зависит не только от числа ионов в единице объема, но и от скорости их движения.
— 152 —
Известно, что различные ионы движутся в электрическом. поле с неодинаковой скоростью. В табл. 36 приведены значения скорости движения некоторых ионов, отнесенные к падению потенциала в 1 в/см (абсолютные скорости движения ионов).
Как видно из табл. 36 скорости движения ионов при прохождении электрического тока в общем очень малы по сравнению со скоростями движения молекул в газах. Так, ион водорода в водной среде движется приблизительно в сто миллионов раз медленнее, чем молекула Н2 в газообразной среде. Объясняется это тем, что ионы в воде гидратированы и при движении испытывают огромное сопротивление со сто-
электропроводимости Н+
Рис. 55. Механизм иона
роны среды (растворителя). Из данных табл. 36 видно, что ионы Н+ и ОН~ обладают по сравнению со всеми другими ионами наибольшими абсолютными скоростями, что нельзя объяснить только малым радиусом ионов Н+ и ОН“. Радиус ОН-иона (1,40 А) соизмерим с радиусами других ионов, ион Н+ в водных растворах существует лишь в виде иона гидроксония Н3О+, радиус которого также сравним с радиусами многих ионов.
Таблица 36
Абсолютные скорости ионов (см* сек) в воде при 18° С и разности потенциалов 1 в!см
Катионы Скорость- 10*“ 4 Анионы Скорость-10 1
н+ 32,7 ОН- 18,7
Li+ 3,5 ci- 6,85
Na+ 4,6 NOT 6,40
К+ 6,75 3 6,95
nh+ 6,70 МпО~ 4 5,60
По современным представлениям, развитым Берналом и Фаулером, избыточные протоны, имеющиеся, например, в водных растворах кислот, не закреплены за определенными молекулами воды, с которыми они образуют ион гидроксония Н3О+, а постоянно перескакивают с одной молекулы на другую. Электрический ток переносится скачкообразным переходом протонов Н+ от ионов Н3О+ к соседним молекулам воды (рис. 55).
—153 —
Вполне понятно, что при таком механизме проводимости скорость иона водорода будет значительно больше, чем других ионов. Совершенно аналогичным переходом протона от молекулы воды к иону ОН~ объясняется кажущееся движение гидроксил-ионов в обратном направлении. Поскольку отрыв протона от молекулы воды происходит с'большим трудом, чем его переход от гидроксоний-иона, подвижность ОН-иона несколько меньше, чем подвижность ионов водорода. Именно этим и объясняется значительно большая электропроводность водных растворов кислот и оснований, чем растворов солей при оди-
• Таблица 37
Удельная электропроводность 0,1 н. раствора КС1 при различных температурах
Температура, °C 10~3, ом y-cm 1
0 7,1295
10 9,3158
20 10,6593
30 14,0996
наковых концентрациях.
Электропроводность растворов зависит также и от заряда ионов: чем он выше, тем большее количество электричества переносит ион с одного электрода на другой. Так, каждый двухзарядный анион отдает аноду два электрона, а однозарядный—только один.
Удельная электропроводность растворов зависит также от температуры. Эта зависимость довольно сложная. При повышении темпе-
ратуры скорость движения ионов возрастает в связи с уменьшением вязкости среды. Кроме того, изменение температуры влияет на степень электролитической диссоциации электролита и тем самым на электропроводность раствора. Повышение температуры на 1° ведет к ускорению движения ионов, а следовательно, к возрастанию электропроводности раствора на 1,5—2,7%.
Табл. 37 иллюстрирует влияние температуры на электропроводность растворов.
Поскольку удельная электропроводность зависит от многих фак
торов, на основе ее изучения не представляется возможным сделать каких-либо выводов общего характера. Поэтому для удобства учета влияния на электропроводность растворов электролитов их концентрации и взаимодействия между ионами Ленцем было введено понятие об эквивалентной электропроводности.
§ 45. Эквивалентная электропроводность растворов
Под эквивалентной электропроводностью понимают электропроводность столба раствора, содержащего 1 г-экв растворенного вещества, заключенного между электродами, находящимися друг от друга на расстоянии в 1 см. Обозначается эквивалентная электропроводность буквой X (греч. «ламбда»), причем индексом внизу показывают обычно объем (в литрах), в котором содержится 1 г-экв электролита. Например, Х10 — эквивалентная электропроводность 0,1 н. раствора электролита, т. е. рйСТЁбрй, содержащего В ГО Л 1 S-Экй pacTSfc peftttoFo вещества.
~ 154 -
Физический смысл эквивалентной электропроводности состоит в следующем. Предположим, что какой-то объем раствора электролита, содержащий 1 г-же растворенного вещества, находится в сосуде с плоскопараллельными стенками А и В (рис. 56), которые пред
ставляют собой платиновые электроды 1 см друг от друга. Если объем данного то его электропроводность удельная, больше, например, 1 см3, то разобьем мысленно этот объем на n-е число кубиков (на рис. 56 этот кубик показан справа), каждый из которых будет иметь удельную электропроводность х. Тогда
и находятся на расстоянии раствора составляет 1 см3, Если же объем раствора
Рис. 57. Зависимость эквива-лентной__ электропроводности X от С для сильных (/—КС1, 2—LiCl) и слабого (3 —
СНзСООН) электролитов
Рис. 56. Схематическое изображение объема электролита между двумя параллельными электродами, поясняющее расчет эквивалентной электропроводимости
суммарная или в данном случае эквивалентная электропроводность всего раствора будет равна:
Х=лх. (IV,19)
Поскольку в каждом кубике содержится 1/п грамм-эквивалентов растворенного вещества, концентрация раствора в этом кубике также будет равна I/м. Отсюда эквивалентная электропроводность будет:
Х=— . (IV,20)
Ci
Учитывая, что Сх = <7/1000, где С — число грамм-эквивалентов’ в 1 л раствора, после подстановки этого выражения в уравнение (IV, 20) получим:
ЮООх
(IV,21)
Поскольку концентрация С — величина обратная разбавлению V, уравнение (IV,21) может быть представлено так:
X=1000xV, (IV,22)
*— 155 *—
где V — разбавление раствора (т. е. объем в литрах, содержащий 1 г-экв электролита).
Таким образом, эквивалентная электропроводность' раствора электролита равна его удельной электропроводности, умноженной на разбавление, выраженное в см3 на 1 г-экв электролита. Отсюда размерность эквивалентной электропроводности выразится в [см2 • ом~х • г-экв-1!. Последний множитель определяется уже самим названием эквивалентной электропроводности. Его иногда опускают, и в качестве единицы измерения эквивалентной электропроводности указывают [см2 • ом-1!, подразумевая на 1 г-экв.
Эквивалентная электропроводность у сильных и слабых электролитов возрастает с увеличением разбавления ( т. е. с уменьшением концентрации раствора) и достигает некоторого предельного значения, которое называется электропроводностью при бесконечном разбавлении. Обозначается оно буквой или Zo. Это явление объясняется тем, что по мере разбавления растворов слабых электролитов растет степень электролитической диссоциации а, для сильных же электролитов увеличивается расстояние между ионами, в результате чего силы взаимного притяжения ослабевают и скорость движения ионов повышается.
В табл. 38 приведены значения эквивалентной электропроводности некоторых электролитов.
Таблица 38
Эквивалентна я электропроводность (см--ом-1-г-экв-1) некоторых электролитов в водных растворах при 25° С
Концентрация раствора, г-экв) Л KCi AgNO; HCI КОН СН3СООН ын4он
1.0 98,3 67,8 301 184 1,32 0,89
0,5 102,4 77,8 327 197 2,01 1,35
0,2 108,0 88,1 342 206 3,24 2,30
0,1 112,0 94,3 351 213 4.60 з,з
0,05 115,8 99,5 360 219 6,48 4,6
0,02 120,0 367 225 10,4 7J
0,01 122,4 107,8 370 228 14,3 9,6
0,005 124,4 110,0 373 230 20,0 13,2
0,002 126,3 112,1 376 233 30,2 20,6
0,001 127,3 113,2 377 234 41 28,0
0,0005 128,1 113,9 1. 1 — 57 38,0
0,0002 128,8 114,6 мм «ж* 80 53
0,0001 129,1 115,0 —— 107 66
Как видно из табл. 38, эквивалентная электропроводность сильных электролитов отличается от электропроводности слабых электролитов не только по величине, но и по характеру ее зависимости от концентрации. Если выразить зависимость X от ]/С~графически, то для слабых электролитов в области больших разбавлений получается кривая, а для сильных — прямая линия (рис. 57). Для разбавленных растворов (не
— 156
выше 0,002 н.) сильных электролитов зависимость X от У С довольно хорошо выражается эмпирическим уравнением:
(IV,23)
Рис. 58. Релаксационное торможение
где а — угловой коэффициент, зависящий от природы растворителя, температуры и валентности электролита. Второй член этого уравнения а У С характеризует уменьшение электропроводности вследствие взаимного торможения ионов, природа которого обусловлена наличием ионных атмосфер, окружающих все находящиеся в растворе ионы. Различают два типа ионного торможения: электрофоретическое и релаксационное.
Электрофоретическое торможение вызвано тем, что при наложении электрического поля катионы и анионы перемещаются в сторону, противоположную движению своих ионных атмосфер. Это сказывается на скорости движения ионов.
С другой стороны, ионная атмосфера по мере движения иона рассеивается и возникает в новом месте не мгновенно. Поскольку при движении иона в электрическом поле ионная атмосфера не успевает полностью еще сформироваться, плотность заряда здесь будет несколько меньше. Позади же иона, наоборот, плотность заряда несколько повышена, так как здесь ионная атмосфера
Вызываемое в результате этих явлений торможение иона носит название релаксационного торможения (рис. 58). Таким образом, эквивалентная электропроводность под влиянием торможений уменьшается с увеличением концентрации электролита.
Эквивалентная электропроводность зависит от температуры. Для
еще полностью не распалась.
большинства электролитов электропроводность увеличивается с повышением температуры, что объясняется увеличением скорости движения ионов в растворе. Это увеличение имеет линейный характер:
=Х18 [1 4-v (Z —18)],
(IV,24)
где и Х18 — эквивалентная электропроводность при температуре t и 18° С, v — температурный коэффициент электропроводности. Увеличение температуры на один градус приводит к возрастанию эквивалентной электропроводности в среднем на 2—2,5%. Вот почему при всех измерениях электропроводности необходимо тщательное термо-статирование.
Для некоторых электролитов электропроводность с увеличением температуры уменьшается, что характерно для неводных растворов и обусловлено уменьшением диэлектрической проницаемости растворителя.
— 157 —
§ 46. Связь эквивалентной электропроводности со степенью диссоциации электролита и скоростями движения ионов
Аррениус вывел формулу для электропроводности растворов, на которой основаны многие теоретические расчеты. Рассмотрим вывод этой формулы на примере бинарного электролита, состоящего из двух однозарядных ионов.
Предположим, что какой-то раствор содержит С г-экв/л растворенного вещества. Степень электролитической диссоциации электролита
рис. 59. Схема сосуда, поясняющая вывод уравнения Аррениуса
равна а, скорости движения катиона и аниона, выраженные в см!сек, соответственно равны wK и на. Весь раствор помещен в сосуд цилиндрической формы с площадью сечения S см2 и находится между электродами, расположенными друг от друга на расстоянии I см (рис. 59).
К электродам приложена постоянная разность потенциалов Е, под действием которой катионы и анионы движутся к противоположно заряженным электродам с определенными скоростями, зависящими от расстояния между электродами I и от величины приложенного к ним напряжения Е. Исходя из этого, можем записать, что скорости движения катионов и анионов в данном случае равны:
Е Е
, wa=(7a f * 4
где UK и Ua — абсолютные скорости катиона и
аниона, т. е. скорости движения ионов при градиенте напряжения в 1 в на 1 см.
Представим себе, что через раствор электролита в течение одной секунды пропущен
электрический ток, и рассмотрим происшедшее перераспределение ионов. Как уже отмечалось, под влиянием разности потенциалов катионы будут двигаться к отрицательно заряженному
электроду — катоду, анионы — к положительному электроду—аноду. Через произвольно выбранную площадь сечения АА± за одну секунду пройдут все катионы» и анионы, отстоящие от этого сечения на
расстояние, численно равное их скорости движения, т. е. на. расстояние и иа сантиметров. Иными словами, через площадь сечения АА± за 1 сек пройдут все катионы, находящиеся в объеме SuK см?, и все анионы, присутствующие в объеме Sua см?.
Нетрудно подсчитать число ионов, находящихся в этих объемах. В самом деле, в 1 см? данного раствора содержится аС/1 ООО ионов, тогда число катионов и анионов, прошедших через выбранную нами площадь сечения AAlt будет равно:
число катионов =SwK
а С
1000 •
158 -
число анионов = Sua
аС
1000
Общее же число катионов и анионов равняется: аС аС SaC
Suk iooo +s“a Tooo = iooo <“«+"a)>
После замены наблюдаемых скоростей движения катионов и анионов через их абсолютные скорости получим:
SaCE общее число ионов = +
1 г-экв ионов переносит в секунду 96 500 к электричества (это так называемое число Фарадея F); всеми ионами, прошедшими через площадь сечения AAlt за одну секунду будет перенесено следующее количество электричества:
SaC Е
w55-T(a«+t/a)‘
Из курса физики знаем, что сила тока / в амперах определяется количеством электричества в кулонах, проходящим через поперечное сечение проводника за 1 сек. Таким образом, сила тока в рассмотренном случае будет равна:
<,v’251
Из закона Ома (IV, 15) и уравнения (IV, 18) имеем:
откуда
xSE
I
(IV,26)
Подставляя выражение I из уравнения
получим:
xSE SaC ~c=f 1000
• (^к + ^а)«
(IV,26) в уравнение (IV,25),
(IV,27)
или, после сокращения: хс=То6оаГ ({7,,+{7а)' (IV,28)
Ввиду того, что X = , после преобразования уравнение (IV,28)
получит окончательный вид: =aE((7K4-t7a)- (IV,29)
Таким образом, эквивалентная электропроводность раствора при данном разбавлении пропорциональна степени электролитической диссо-
— 159 —
циации раствора электролита и сумме абсолютных скоростей катиона? и аниона. Число Фарадея является в данном случае коэффициентов пропорциональности. Я
Уравнение (IV,29) известно в литературе под названием урав-нения Аррениуса. Из этого уравнения вытекает целый ряд очень; важных следствий, которые играют большую роль в теории электро*; проводности растворов. Я
'ЛИ
§ 47. Закон независимости движения ионов J
(закон Кольрауша)
Мы уже упоминали, что эквивалентная электропроводность^ при увеличении разбавления раствора стремится к некоторому пре-| делу, т. е. постоянной величине, характерной для каждого электро-| лита и получившей название эквивалентной электропроводности приз бесконечном разбавлении (^ос). В своих исследованиях Кольрауш! показал, что если сравнить эквивалентные электропроводности раз*| личных электролитов при бесконечном разбавлении, то можно заме-тить интересные закономерности. Например, разность между предель-1 ными электропроводностями растворов сульфата калия и натрия равна: ]
д (К2 SO4) - (Na2 SO4) = 21,1. I
Такая же разность получается между величинами ^других солей,| содержащих те же самые катионы, но различные анионы: , 1
A=X00(KF)-Aoo(NaF)=21,l. 1
С другой стороны, эквивалентные электропроводности при беско-1 нечном разбавлении хлорида и нитрата натрия соответственно равны: ]
(Na Cl) = 109,0 и (NaNO3) = 105,2. 1
Разность между ними Д = 3,8. Точно такая же разность получается, I если брать предельные электропроводности различных солей, 1 имеющих одинаковые катионы, но различные анионы. В табл. 391 приведены некоторые экспериментальные данные Кольрауша. <
* я
Таблица 39
Эквивалентная электропроводность различных электролитов при 18° С (сж2 ом~1‘г-экв~1)
Анионы Катионы Разность Катионы Анионы Разнсстъ
Na + Cl” NO7 о
С1~ 130,1 109,0 21,1 к+ 130,1 126,3 3,8
NOr О 126,3 105,2 21.1 Na+ 109,0 105,2 3,8
Ют О 98,5 77,4 21.1 L j 98,9 95,1 3,8
F- 111,25 90,15 21,1 Т1 + 131,5 127,7 3,8
— 160 —
На основании экспериментальных данных Кольрауш пригнел к выводу, что в разбавленных растворах каждый из ионов обусловливает свою определенную долю эквивалентной электропроводности. Иными словами, эквивалентная электропроводность является аддитивным свойством электролита, т. е. суммой двух независимых величин, а именно суммой электропроводностей катиона и аниона:
(IV,30)
где /к и /а — величины электропроводностей катиона и аниона, которые получили название подвижностей катиона и аниона. В этом и состоит открытый Кольраушем закон независимого перемещения ионов, который формулируется следующим образом: эквивалентная электропроводность при бесконечном разбавлении равна сумме подвижностей катиона и аниона данного электролита.
Из закона Кольрауша вытекает: зная электропроводность трех электролитов, можно вычислить электропроводность четвертого электролита. Предположим, что опытным путем были найдены (КС1), (NaCI) и (KF). Необходимо найти, чему равна (NaF). На.основании закона Кольрауша можем записать:
^оо (К. С1) «=
(NaCI) «=/Na+ •
откуда
^00(KCI)-X00(NaCl)=ZK+-ZNa+. (IV,31)
В то же время для фторида калия можем записать:
' MKF)=zk+-Hf-. / (IV,32)
Вычитая из уравнения (IV,32) уравнение (IV,31), получим:
(KF) (КС1) + (NaCI),=ZNa+ + ZF-. (I V.33>
откуда
^Na+ FZp- «A^fNaF).
Если подставить численные значения эквивалентных электропроводностей при бесконечном разбавлении для соответствующих электролитов, обнаружим, что
(NaF) = 111,25 — 130,1 + 109,0 = 90,25.
Эту же величину можно получить экспериментальным путем, изучая ход кривой эквивалентной электропроводности NaF.
Рассмотрим, каков же физический смысл подвижностей ионов, т. е. /к и /а. Из уравнения Аррениуса (IV,29) ^оо=^(^к+^а).
Если V -> оо, то а -> 1 и X ->
Таким образом, для эквивалентной электропроводности при бесконечном разбавлении можем записать:
X=F(Z4 + Z/a),
6 Зак 560
— 161 -
или
^•=FUK + FUa. (IV,34)
По закону Кольрауша (IV, 20)
^00 = ^ "Ь ^а«
Из сравнения уравнений (IV, 30) и (IV, 34) можем записать:
/к— FU^ и /д — FU%.
(IV,35)
Следовательно, подвижность иона есть не что иное, как произведение его абсолютной скорости на число Фарадея.
Численные значения подвижностей всех ионов в настоящее время экспериментально определены и сведены в специальные таблицы. Примером такой таблицы является табл. 40.
Таблица 40
Подвижность некоторых ионов в водном растворе при 18° С
4 Катионы Анионы
н+ 315 он- 174
’ к+ 64,4 ci- 65,5
Na+ 43,2 NOr 62,0
nh+ 64,0 О F- 46,6
Li+ 33,4 CH3COO- 35,0
Ag+ 1 54,3 IO? 33,9
т Ca2+ 51,0’ — SO2’ 2 ^4 68,3
1 Mg2+ 45,0 4 bo| *— A о w to 1 60,0
4"Fe3+ 43,0 2 C2°4 72,0
1 т Fe2+ . 46,0 -rFe(CN)6~ 110,5
Пользуясь этой таблицей, можно определить электролита путем простого подсчета. Например, для уксусной кислоты СН3СООН равна:
Хоо = /К-}- /а=35 4-315=350 см2•ом'’1 • г-экв”1.
Подвижности ионов зависят также от температуры и от вида растворителя. При повышении температуры подвижность ионов возрастает. Так, для катиона натрия и аниона хлора имеет место следующая температурная зависимость:
Температура, °C ... . О
/Na+'........................... 21,1 ’
IgF............................ 41,6
18 100
42,6 ,152,0
66,3 208,0
— 162 —
Именно этим объясняется повышение электропроводности растворов электролитов с повышением температуры.
Величина эквивалентной электропроводности при бесконечном разбавлении (а следовательно, и подвижности катиона и аниона) для данного электролита зависит от природы растворителя. При переходе от одного растворителя к другому сохраняется постоянным произведение
^1)0=const, (VI, 36)
где т)о — вязкость растворителя. Эта закономерность носит название правила Вальдена — Писаржевского. На основании его были найдены подвижности многих ионов в неводных средах.
Пользуясь таблицами подвижностей, можно легко вычислить абсолютную скорость любого иона из соотношения (IV,35):
я
Г , г,
р и Ua~ р •
Например, Zr+ == 315, (/н+=315/96500 = 32,7 • 10~4 см-сек в*см~1. И, наоборот, зная абсолютную скорость движения иона, легко вычислить его подвижность. ♦ *
§ 48. Определение степени диссоциации слабых электролитов и коэффициента электропроводности сильных электролитов методом электропроводности
Эквивалентная электропроводность электролитов находится в прямой зависимости от разбавления раствора. Аррениус объяснил это явление постепенным увеличением количества ионов в растворе: по мере уменьшения концентрации все большее число молекул растворенного вещества диссоциирует на ионы. Он считал также, что эквивалентная электропроводность раствора при данном разбавлении X пропорциональна степени электролитической диссоциации а электролита в этом растворе.
Ранее для эквивалентной электропроводности X было выведено уравнение Аррениуса (IV,29), откуда и из соотношений (IV,35) получим:
X = a(ZK + /a)- (IV,37)
Подставляя в уравнение (IV,37) значение ZK -4 Za из уравнения закона Кольрауша (IV,30), получим:
откуда
а = 4 =---------. (IV ,38)
Л» /к + /а
Таким образом, степень электролитической диссоциации слабого электролита при данном разбавлении равна отношению эквивалент
I
6*
— 163 —
ной электропроводности при этом разбавлении к эквивалентной электропроводности при бесконечном разбавлении.
Эквивалентная электропроводность X легко может быть определена экспериментальным путем, а можно рассчитать по таблицам, пользуясь уравнением (IV,30).
Для сильных электролитов отношение эквивалентной электропроводности при данном разбавлении к эквивалентной электропроводности при бесконечном разбавлении дает уже не а, а Д — коэффициент электропроводности. Он показывает, во сколько раз действительное значение эквивалентной электропроводности % меньше теоретически соответствующей для данной концентрации электролита, т. е. ’
Д = (IV,39)
**оо
Сильные электролиты диссоциированы полностью, и число ионов в растворе постоянно. Поэтому можно было бы ожидать, что для растворов сильных электролитов X = Однако, как показывает опыт, это наблюдается только при бесконечном разбавлении раствора, когда влияние ионной атмосферы на движущийся катион или анион сильно ослаблено. Только при этих условиях эквивалентная электропроводность достигает своего предельного значения и складывается из электропроводностей /к и /а согласно закону Кольрауша (IV,30).
Таким образом, коэффициент электропроводности сильных электролитов принимает значения меньше единицы не в результате неполной диссоциации, как в случае слабых электролитов, а за счет влияния сил межионного взаимодействия. Иными словами, этот коэффициент обусловливается теми же причинами, что и рассмотренный нами ранее коэффициент активности f.
Величина коэффициента электропроводности зависит от концентрации электролита и его валентного типа. Так, в 0,1 н. растворе 1 — 1-валентного электролита Д = 0,8; для 1—2-валентного электролита /х =' 0,75; для 1—3-валентного электролита той же концентрации /х = 0,4 и т. д. По мере же дальнейшего разбавления эти различия постепенно исчезают.
Следовательно, /\ так же как и а, увеличивается с разбавлением раствора. При достаточном разбавлении /х и а становятся равными единице.
§ 49. Методы определения электропроводности растворов электролитов
Электропроводность растворов электролитов на практике определяют по значению их сопротивления электрическому току, протекающему между двумя погруженными в раствор электродами. Принципиально измерение сопротивления раствора может быть проведено как с помощью постоянного тока, так и переменного. Однако на практике наибольшее распространение получил метод, основанный на использовании переменного тока. Дело в том, что изменение направления тока является лучшим средством для устранения влияния электролиза и поляризации, при этом чем выше частота тока, тем меньше сказываются на электропро
—• 164 —
водности эти явления. Измерение сопротивления объема раствора электролита производят обычно с помощью специального моста сопротивления — моста Коль-рауша. Его принципиальная схема приведена на рис. 60.
Предположим, что ток от источника тока поступает в точку где он разветвляется к точкам b и d, а затем через точку с возвращается к источнику. Подбором сопротивлений Ri,R2u Кз добиваются такого распределения токов в ветвях моста, при котором ток в диагонали моста bd падает до нуля, т. е. напряжение между точками b и d равно нулю. Это возможно в том случае, если падение напряжения на участках ab и ad и соответственно Ьс и de будет одинаково. Такое состояние моста называется балансом. При балан-
соотно-частей
(IV, 40)
се моста возникает следующее шение величин сопротивления схемы:
где /?л — искомое сопротивление раствора электролита, — набор (магазин) стандартных сопротивлений, R2 и R3 — изменяемые части сопротивления (реохорда), которые определяются положением скользящего контакта (движка d). В качестве нуль-инструмента, который фиксирует отсутствие тока между точками bud, обычно используют низкоомный телефон, вибрацион-
Рис. 60. Схема установки для измерения сопротивления растворов электролитов (мост Кольрауша)
ный гальванометр или осциллограф.
Источниками переменного тока,для питания мостов Кольрауша обычно являются ламповые генераторы. Рабочая частота должна быть в пределах от 300
до 5000 гц. х
На рис. 61 показан общий вид простейшей установки для измерения электропроводности растворов, в которой реохордом служит линейка с натянутой на ней проволокой и со скользящим контактом. В качестве нуль-инструмента используются обычные наушники. Длина линейки один метр (градуирована на миллиметры). Реохордная проволока выбирается строго постоянного диаметра и изготовляется из материала, который обладает определенным сопротивлением и не корродирует (например, манганин). Точка баланса моста определяется по минимуму звука в наушниках при передвижении ползунка (скользящего контакта)
по реохорднои струне.
Важное значение при -измерениях сопротивления растворов имеет также кон-
струкция и форма сосуда, в который наливают исследуемый раствор. Как показали опыты, сопротивление раствора электролита зависит не только от концент-
рации электролита, площади электродов в измерительном сосуде и расстояния между ними, но также и от взаимного расположения этих электродов и от объема раствора, потому что в переносе электричества участвует гораздо больший объем раствора, чем тот, который непосредственно находится между электродами (рис. 62). i
Форма, расположение электродов и объем электролита во время всех измерений должны быть строго постоянными. Это достигается тем, что в стеклянный сосуд впаиваются постоянные платиновые электроды, так чтобы нельзя было их деформировать. Для улучшения работы электроды покрывают платиновой чернью, т. е. слоем электролитически осажденной платины.
Для растворов, плохо проводящих электрический ток, электроды располагаются ближе друг к другу и имеют большую поверхность. Для хорошо проводящих растворов применяют электроды с меньшей поверхностью и делают большими расстояния между ними. На рис. 63 показаны различные формы сосудов для измерения сопротивления растворов.
Если расстояние между электродами в сосуде точно равняется 1 см, площадь каждого электрода 1 см2, а в прохождении электрического тока участвует
— 165 —
Рис. 61. Установка для измерения электропроводности растворов:
1 — аккумулятор; 2, 3 — клеммы для подключения аккумулятора; 4 — индукционная катушка; 5 — магазин сопротивлений; 6 — сосуд для испытуемого раствора; 7, 8 — клеммы для включения сосуда 6- 9, 10— клеммы для включения телефона; 11 — телефон; АБ — реохорд с подвижным контактом
только объем раствора, заключенный между электродами, то измеренная в таких условиях электропроводность есть удельная электропроводность раствора. Однако практически изготовить сосуды для измерения удельной электропроводности с такими параметрами очень трудно. Для каждого сосуда предварительно определяют его постоянную. Делается это следующим образом. Из закона Ома известно, что
я=р-4- • о
Подставляя вместо р 1/х, получим:
откуда
(IV,41)
Отношение 1/S в уравнении (IV, 41) и носит название постоянной сосуда и обозначается буквой Z. С учетом этого обозначения уравнение (IV, 41) примет вид:
. (IV,42)
Итак, чтобы найти удельную электропроводность исследуемого раствора, необходимо измерить его сопротивление R с помощью моста Кольрауша, а также знать постоянную сосуда Z, в котором производят измерение.
Постоянную сосуда Z определяют следующим образом: в сосуд для измерения электропроводности наливают раствор, электропроводность которого извест-
I
— 166 —
Рис. 63. Различные формы сосудов для измерения электропроводности растворов
Рис. 62. Направление линий тока в объеме раствора между электродами
на, и измеряют сопротивление /?ст, В качестве такого раствора обычно берут раствор хлорида калия, удельную электропроводность которого для различны» температур см. в табл. 41.
Таблица 41
Удельная электропроводность водных растворов KCI (оя-^см"1) при различных температурах
Температура, ®С 21 —— Удельная электропроводность при концентрации раствора КС1 (г-экв/л)
0 ,1 0,02 0,01
15 0,01048 0,002243 0,001147
16 0,01072 0,002294 0,001173
17 0,01095 0,002345 0,001199
18 0,01119 0,002397 0,001225
19 0,01143 0,002449 0,001251
20 0,01167 0,002501 0,001278
21 0,01191 0,002553 0,001305
22 0,01215 0,002606 0,001332
23 0,01239 0,002659 0,001359
24 0,01264 0,002712 0,001386
25 0,01288 0,002765 0,001417
Измерив сопротивление хлорида калия, по формуле
Z— Крт(IV, 43) вычисляют постоянную сосуда Z.
При измерении сопротивления любых растворов сосуд с раствором обязательно должен находиться в термостате, где поддерживается постоянная температура. На рис. 64 показано устройство водяного термостата емкостью 5—10 л. Основными деталями его являются механическая мешалка 4, терморегулятор 5 и два нагревателя. Один из нагревателей 3 служит для быстрого предварительного поднятия температуры, второй 2 — для автоматического поддержания
167
заданной температуры. Включение и выключение второго нагревателя осуществляется с помощью специального реле, работающего от контактного термометра или от толуолового терморегулятора.
Принцип действия контактного термометра состоит в том, что при повышении температуры выше заданной замыкаются контакты релейного устройства и нагреватель отключается от сети. При размыкании контактов реле автоматически включает нагреватель. Поскольку при размыкании контактов терморегулирующего устройства возникает искра, для ее гашения параллельно этим контактам включают конденсатор на 0,5—1 микрофарад.
Рис. 64. Водяной термостат:
/ — контактный термометр; 2 — нагреватель (лампочка на 50 вт); 3 — дополнительный нагреватель для предварительного поднятия температуры; 4 — мешалка с мотором; 5 — терморегулятор
Определив постоянную сосуда Z по формуле (IV, 42), можно вычислить удельную электропроводность любого исследуемого раствора. Зная же концентрацию и удельную электропроводность раствора, легко можно вычислить и другие величины: эквивалентную электропроводность раствора X, степень его диссоциации а (или — коэффициент электропроводности сильного электролита), а также константу электролитической диссоциации К для слабых электролитов.
В последние годы все большее распространение получают реохордные мосты, выпускаемые промышленностью в одном блоке вместе с нуль-инструментом и источником напряжения (например, мост Р-38). Особое значение приобретает также развитие безэлектродного метода измерения электропроводности, о чем более подробно см. в следующем параграфе.
§ 50. Применение методов измерения электропроводности в лабораторной практике и в агрономии
Измерение электропроводности растворов (так называемая кондуктометрия) позволяет решить целый ряд практических задач. По электропроводности растворов можно определить основность органических кислот, растворимость и произведение растворимости малорастворимых соединений, влажность различных объектов, степень минерализации вод, почв и грунтов. Кроме того, большое зна
— 168 —
чение имеет определение общей или титруемой кислотности методом кондуктометрического титрования. Рассмотрим вкратце все эти методы, основанные на принципе электропроводимости.
Определение основности органических кислот. В свое время метод электропроводности был предложен для определения основности органических кислот. Эмпирическим путем было установлено, что эквивалентные электропроводности солей органических кислот при разбавлениях, равных 1024 и 32 л (Х32 и Х1024), находятся в следующей зависимости:
^1024 ’—’^32 — Ал, (IV,44)
где л — основность кислоты, а А — эмпирический коэффициент, численно равный примерно 10. Это правило много раз было успешно использовано для практических определений основности вновь открываемых кислот.
Определение растворимости малорастворимых соединений. Опыт показывает, что методом электропроводности наиболее удобно определять растворимость галидов ртути, серебра и других соединений. Рассмотрим определение растворимости на примере бромида серебра AgBr. Так как AgBr очень мало растворим, его степень диссоциации а в водном растворе должна быть близка к единице, т. е. недиссоциированная часть AgBr весьма мала по сравнению с диссоциированной. Поэтому недиссоциированной частью можно пренебречь и считать, что а — 1.
Пусть удельная электропроводность воды/ в которой растворяется бромид серебра, равна х= 1,519 • 10"*ъ \ом~1 • см~1 (при 18° С). Удельная электропроводность раствора AgBr в этой воде х= 1,576* 10~6ojw~1 • см~1. Увеличение электропроводности при растворении в воде AgBr обусловлено появлением в растворе (хотя и в малом количестве) ионов Ag+ и Вг”. Таким образом, сам бромид серебра обеспечивает электропроводность, которая равна:
xAg Вг«(1,576—1,519)* 10-6=0,057.
По таблицам подвижностей ионов вычисляем:
^(Ag Br)=/Ag+ +ZBr “ =J27.1
Поскольку для данного случая а « 1, то X « Исходя из этого, можем записать:
1000 х
(IV ,45)
откуда
ЮООх
ос
Подставляя в формулу (IV, 45) численные значения отдельных величин, получим:
0,057.10-6*1000 „ Л
С=-------------------= 4,48* 10“7 г-же/л или С = 0,085 мг!л.
127,1 .
С помощью метода электропроводности легко можно вычислять также и произведение растворимости малорастворимых соединений. Так, для бромида серебра можем записать: [Ag+] « [Вг~] ==> 4,48 * 10“7 г-ион/л. Зная концентрации ионов, нетрудно определить и произведение растворимости (ПР):
nP = [Ag+][Br-] =4,48* Ю-7*4,48-10-7 =2,0.10-^.
Совершенно аналогичным способом определяют растворимость и произведение растворимости других солей.
Кондуктометрический метод определения влажности. На принципе измерения проводимости растворов основано устройство различных влагомеров. В частности, в практике сельского хозяйства за последние годы получили широкое распространение приборы для определения влажности зерна. Принцип работы этих приборов очень прост. Определенный объем пробы зерна помещается в специаль-
— 169 —
яый сосуд между двумя электродами и с помощью моста Кольрауша измеряется сопротивление этой пробы. Чем больше содержится влаги в зерне, тем меньшим Сопротивлением обладает зерно, и наоборот. Шкала прибора градуируется в весовых процентах влажности для каждого вида зерна. Обычно в целях универсальности прибор снабжается несколькими сменными шкалами для измерения влажности зерна различных сельскохозяйственных культур (например, ржи, пшеницы, ячменя, овса, кукурузы). Метод отличается не только быстротой, но и довольно высокой точностью, и потому все приемные и ссыпные пункты зерна в настоящее время оборудованы такими приборами.
Рис. 65. Гипсовый датчик влажности почвы:
1 — гипс; 2 — изоляция; 3 — медная проволока
Рис. 66. Схема коммутации датчиков
В практике сельского хозяйства в настоящее время наибольшее распространение получил кондуктометрический метод определения влажности почвы при помощи датчиков с промежуточной средой из гипса. Это так называемый метод гипсовых блоков; он широко применяется в США, Австралии, в Новой Зеландии, в нашей стране и в других странах.
Сущность метода заключается в следующем. В почву на необходимую глубину помещают на несколько лет датчики, которые представляет собой гипсовые блоки с залитыми в них некорродирующимц электродами (рис. 65). При помощи проводов, проложенных подпахотным горизонтом, все датчики подсоединяются к клеммам коммутационных щитков (рис. 66), которые устанавливаются в таких местах, где они не могут быть повреждены сельскохозяйственными орудиями. Сопротивление гипсового датчика зависит от влажности окружающей среды (почвы): чем выше влажность почвы, тем меньрим сопротивлением он обладает. Измерение сопротивления (следовательно, и влажности почвы) осуществляют переносным прибором, который поочередно подключается к клеммам коммутационного щитка.
— 170 —
Процесс сбора подобной информации с датчиков влажности, установленных на обширных площадях, можно легко автоматизировать, если эту информацию (после соответствующего кодирования) передавать по радиоканалу в соответствующий центр, который прогнозирует и планирует сроки полива сельскохозяйственных культур в различных хозяйствах обслуживаемого района. Для этой цели коммутационные щитки должны быть дополнительно оборудованы недорогой и портативной приемо-передающей аппаратурой, работающей в УКВ-диапа-зоне. Сбор информации осуществляется передвижной станцией управления, поскольку передатчики УКВ-диапазона способны передать информацию на расстояние видимой связи (10—20кл<). В настоящее время в нашей стране уже разрабатываются подобные системы контроля в орошаемом земледелии.
Метод использования описанных датчиков свободен от целого ряда недостатков, присущих измерению влажности методами прямого определения электропроводности почвы.
Определение солесодержания в почвах. Степень засоленности почвы часто является решающим фактором при освоении новых земель и выборе наиболее рациональных методов орошения. На культурных орошаемых массивах контроль за динамикой солей в почве необходим хотя бы для проведения профилактических мероприятий по предупреждению вторичного засоления.
Классический метод характеристики солесодержания по величине плотного остатка имеет два существенных недостатка. Во-первых, при фильтрации водных вытяжек в фильтраты часто переходят органические и минеральные коллоиды, освободиться от которых бывает очень трудно. Во-вторых, фильтрование и выпаривание требует значительных затрат времени и энергии. Более быстрый и надежный способ определения солесодержания в почве основан на измерении электропроводности.
Попытки разработать метод определения солесодержания путем непосредственного измерения электропроводности почвы не увенчались успехом, поскольку электропроводность почвы зависит от многих факторов (в частности, от наличия солей, влажности и других свойств почвы). Поэтому определение солесодержания в настоящее время проводят по величине электропроводности почвенной вытяжки. Приборы, с помощью которых производят определение солей в лабораторных и полевых условиях, получили название солемеров.
Как известно, кондуктометрическим методом измеряют удельную электропроводность х, которая зависит не только от концентрации соли, но и от степени ее диссоциации и скорости движения ионов в электрическом поле, т. е.
х=СосЕ +
где с — концентрация соли, г-же/л, а — степень ее диссоциации (кажущаяся), р — число Фарадея, UK и Ua — скорости движения катионов и анионов при напряжении электрического поля в 1 в/см (так называемая абсолютная скорость иона).
Это-уравнение можно использовать для определения солесодержания, но оно пригодно лишь при известных значениях а, [7К и 1/а. Используя уравнение Аррениуса (IV, 29), получим:
х = С?1, где к — эквивалентная электропроводность раствора.
Водные вытяжки из почв содержат различные соли в довольно сложных сочетаниях, поэтому прямое использование эквивалентных электропроводностей становится невозможным. Наиболее правильно проводить эмпирическую градуировку солемеров в пределах одной почвенной зоны с примерно одинаковым составом солей.
Нашей промышленностью выпускаются различные солемеры — ЛИС-56, ПС-3, солемер «ВСЕГИНГЕО».
На рис. 67 показана принципиальная схема почвенного солемера ПС-3. В этом приборе использована дифференциальная (разностная) схема, отличающаяся высокой чувствительностью.
В двух контурах схемы / и II возникают одинаковые по величине э. д. с. В один контур включен электродный сосуд 1 с исследуемым раствором, в другой—
— 171 —
переменное сопротивление 3. После выпрямления токи из обоих контуров поступают на гальванометр 5, который и показывает разность этих токов. Путем изменения переменного сопротивления 3 можно добиться, чтобы гальванометр показывал нуль. В этом случае выпрямленные токи равны, а сопротивление электродного сосуда равно переменному сопротивлению. В приборе предусмотрено питание от сети переменного тока или от сухих батарей, вмонтированных в прибор.
Обладая высокой производительностью и достаточной степенью точности, кондуктометрические методы определения солесодержания имеют большое значение при возделывании сельскохозяйственных культур на "‘засоленных землях в условиях орошения. Постоянный контроль за мелиоративным состоянием орошаемых земель является важнейшей задачей. Неумелое пользование поливной водой без учета потребности сельскохозяйственных культур в воде, а также без учета физико-химических и водно-физических свойств орошаемой почвы может привести к явлению вторичного засоления почв. Вот почему агроном, вооруженный портативным солемером, может осуществлять своевременный контроль не только за мелиоративным состоянием орошаемых земель, но также и за минерализацией поливных и сбросных вод.
Рис. 67. Принципиальная схема почвенного солемера ПС-3: 1 — электродный сосуд; 2 — трансформатор питания; 3 — переменное отсчетное сопротивление; 4 — выпрямители; 5 — нулевой гальванометр;
6 — потенциометр установки нуля
Кондуктометрическое титрование. Большое практическое значение имеет так называемое кондуктометрическое титрование, сущность которого заключается в том, что концентрацию электролита в растворе определяют измерением электропроводности при титровании. При исследовании мутных и окрашенных растворов, которые невозможно титровать с применением обычных индикаторов, особенно удобно применять кондуктометрическое титрование.
Метод основан на замене ионов титруемого вещества ионами добавляемого реагента в процессе титрования. Поскольку подвижности различных ионов отличаются друг от друга, в точке эквивалентности всегда наблюдается довольно заметное изменение электропроводности. На рис. 68 показаны кривые, полученные в различных случаях кондуктометрического титрования. Кривая / есть результат титрования сильной кислоты сильным основанием или, наоборот, сильного основания сильной кислотой. Минимум на этой кривой обусловлен заменой ионов водорода (или гидроксид-ионов), обладающих -наибольшей подвижностью, менее подвижными катионами (или анионами) образующейся соли по схеме
Н+ +С1-+Na+4-ОН--> Н2 О 4-Na+4-С1~ кислота сснование соль
Подвижный ион водорода кислоты обладает гораздо большей электропроводностью, чем соответствующие катионы соли любого металла. Вполне понятно, что
172 —
в момент, когда вся кислота (или основание) нейтрализована эквивалентным количеством основания (или кислоты), электропроводность раствора будет минимальной. При добавлении избытка основания (или кислоты) в растворе вновь появятся высокоподвижные ионы ОН“ (или Н+), и-электропроводность раствора начнет быстро возрастать.
Кривая 2 на рис. 68 получена в ходе титрования слабой кислоты сильным основанием или слабого основания сильной кислотой. Более крутой подъем прямой после точки эквивалентности обусловлен более высокой диссоциацией образующейся соли по сравнению с диссоциацией титруемой слабой кислоты или слабого основания.
Кривая 3 (рис. 68) соответствует титрованию сильным основанием смеси сильной (а) и слабой (Ь) кислот.
Количество щелочи, которое соответствует наименьшей электропроводности, полностью нейтрализует взятую кислоту. Зная объем взятой для титрования кислоты, а также объем и нормальность раствора щелочи, затраченной на нейтрализацию, легко рассчитать нормальность титруемой кислоты по формуле:
Vo TVo
V^tc=V2^; ^ = 4^, (IV,46)
Рис. 68. Зависимость удельной электропроводности растворов электролита от количества прибавленного реагента при кондуктометрическом тн-. тровании
где Vj — объем раствора кислоты, — нормальность титруемого раствора кислоты, V2 — объем раствора щелочи, пошедший на титрование кислоты и — нормальность щелочи.
Высокочастотная кондуктометрия. В последние годы все большее распространение получает так называемая высокочастотная кондуктометрия. В этом случае используют переменные токи с частотами порядка нескольких миллионов герц. При таких высоких частотах электроды'можно вывести из раствора за пределы измерительной ячейки, в которой проводят измерения. При этом возникает целый ряд преимуществ по сравнению с обычной кондуктометрией. В частности, при использовании высокочастотной .кондуктометрии удается избежать многих осложнений, связанных с обычной кондуктометрией: каталитического действия электродов на реакции в растворах, необходимости применения электродов из дорогого материала, например платины, изменения поверхности электродов в ходе измерений и т. п.
При высокочастотной кондуктометрии раствор помещается в специальную измерительную ячейку из стекла или пластмассы.. Измерительная ячейка располагается между двумя металлическими пластинами, плотно примыкающими к ее стенкам. При высокочастотной кондуктометрии измеряется не электропроводность раствора, а совокупность многих свойств раствора и ячейки, включая и диэлектрическую постоянную. Поэтому интерпретация полученных результатов здесь более сложна, чем при обычной кондуктометрии.
Глава V I
ХИМИЧЕСКАЯ КИНЕТИКА И КАТАЛИЗ. ФОТОХИМИЯ «
§ 51. Химическая кинетика реакций. .Основные положения и понятия i
Химической кинетикой называется учение о скорости химических реакций и ее зависимости от различных факторов—природы и концентрации реагирующих веществ, давления, температуры, катализаторов.
Скорости химических реакций могут значительно различаться. Например, некоторые реакции совершаются в тысячные доли секунды (взрыв), другие же протекают в течение значительно более длительного времени. Так, ржавчина на железе становится заветна уже через несколько часов после того, как оно пришло в контакт с водой или влажным воздухом, а образование антрацита в земной коре протекает в течение сотен и тысяч лет.
Вопрос о скорости химических реакций имеет исключительно большое практическое и теоретическое значение. От скорости реакции в различных технологических процессах зависят производительность и габариты заводской аппаратуры, течение тех или иных биологических процессов, эффективность действия на живой организм различных лекарственных препаратов.
Чтобы получить в единицу времени наибольшее количество вырабатываемого продукта, необходимо максимальное увеличение скоростей реакций, лежащих в основе того или иного химического процесса. С другой стороны, вредные, нежелательные процессы—коррозию металлов, окисление каучука—необходимо как можно больше замедлить. Поэтому изучение законов химической кинетики, открывающее путь к сознательному регулированию скоростей реакций, имеет исключительно важное значение для практики. Управление химическим процессом является главной задачей химической кинетики.
Прежде чем приступить к обсуждению закономерностей, управляющих развитием реакции, остановимся на самом понятии скорости химической реакции.
Под скоростью реакции понимается изменение концентрации реагирующих веществ в единицу времени. При этом концентрацию обычно выражают числом молей вещества на единицу объема реакционной смеси (обычно — на 1 л), время в секундах, минутах и т. п. Чтобы вычислить скорость реакции, необходимо _^нать, насколько измщидась концентрация одного^иэ^еагируьощих-веществ в едйницуП^мени^^На-пример, за какой-то промежуток времени Д/ = Q — ^ концентрация реагирующих веществ уменьшается на —ДС = С\ — С 2 (рис. 69). Тогда средняя скорость реакции за промежуток времени Д/ равна:
- ДС
рср — • (V*1)
— 174 —
' у I
Знак минус в правой части выражения (V,l) означает, что концентрация реагирующих веществ, а следовательно, и скорость химической реакции уменьшается.
Рис. 69. Изменение концентрации вещества в процессе его химического превращения
Средняя скорость не отражает истинной скорости в каждый момент времени, поэтому математически истинную скорость v реакции в данный момент принято выражать отношением бесконечно малого изменения концентрации dC к бесконечно малому отрезку времени dt, в течение которого произошло изменение концентрации:
Если изучается изменение концентрации одного из исходных веществ, то < О, если одного из продуктов реакции, то >0. Скорость реакции всегда считается положительной, а отношение в правой части уравнения может быть или положительным или отрицательным.
Скорость химической реакции по закону действия масс пропорциональна концентрации реагирующего вещества в данный момент.вре-' мени. Так, для реакции типа А -> В скорость реакции может быть выражена следующим кинетическим уравнением:
dCA
V=--—=КСА. . (V,3)
ill
где СА — концентрация вещества А в данный момент, К — коэффициент пропорциональности, который носит название константы скорости химической реакции.
При взаимодействии двух или более веществ, т. е. для реакций типа
mA 4-пВ -> рС,
— 175 —
математическое выражение скорости будет
(V.4)
где СА п Св — концентрации веществ А и В в данный момент времени. Таким образом, скорость химической реакции прямо пропорциональна произведению концентраций реагирующих веществ. Причем стехиометрические коэффициенты входят в уравнение скорости в виде показателя степени при соответствующих концентрациях.
Коэффициент пропорциональности или константа скорости химической реакции не меняется при изменении концентраций А и В. Иногда К называют еще удельной скоростью реакции, т. е. скоростью реакции при концентрациях реагирующих веществ, равных единице. Удельная скорость реакции зависит от природы реагирующих веществ, температуры, присутствия посторонних примесей (в частности катализатора), а также среды, в которой протекает реакция. Численная величина К зависит также от выбора единиц концентрации и времени.
Чем больше константа скорости К, тем быстрее идет реакция.
§ 52. Классификация химических реакций
Все кинетические реакции различают по молекулярности и порядку реакции. Молекулярность реакции определяется числом молекул, участвующих в элементарном акте химического взаимодействия. По этому признаку реакции разделяются на мономолекулярные, бимолекулярные и тримолекулярные.
Мономолскулярными называются такие реакции, в которых в элементарном акте взаимодействия участвует одна молекула, например, реакции разложения или радиоактивного распада:
12-21
Сюда же относятся реакции изомерного превращения сложных молекул в газах и растворах. ' ’
Бимолекулярными называются реакции, в которых в элементарном акте взаимодействия встречаются две молекулы. Например, реакция образования йодистого водорода
Н24-12 = 2Н1
К этому типу реакций относятся также реакции этерификации сложных эфиров и многие другие.
Тримолекулярными называются реакции, в которых в элементарном акте взаимодействия участвуют три молекулы. Например
2NO 4-О2 ==2NO2
Одновременная встреча в элементарном акте взаимодействия даже трех молекул случается довольно редко. Реакции же более высокой молекулярности неизвестны. В тех случаях, когда из химического уравнения следует, что в реакции участвует большее число молекул, про
— 176 —
цесс на самом деле происходит более сложным путем — через две и большее число промежуточных стадий моно- или бимолекулярных реакций.
Каждому типу реакции отвечает свое кинетическое уравнение, ко-t торое выражает зависимость скорости химической реакции от концентрации реагирующих веществ! По форме зависимости v — f (С) различают реакции^первого. второго и третьего порядков. Скорость реакции пёр^Ео^порядка пропорциональна концентрации исходных веществ в первой степени. В реакциях второго и третьего порядков скорость соответственно зависит от концентрации во второй и третьей степени. В общем случае порядком реакции называют сумму показателей степеней, с которыми концентрации реагентов входят в экспериментально найденное кинетическое уравнение. Так, для реакции типа
А Д- П2 В :==^3 “Ь ^4
порядок реакции п == и2-
Казалось бы, порядок реакции легко можно определить по виду стехиометрического уравнения. Однако опыт показывает, что порядок, по которому развивается реакция во времени, часто не совпадает с порядком, определяемым по стехиометрическому уравнению. Иными словами, порядок реакции не всегда совпадает с молекулярностью ее. Лишь в наиболее простых случаях наблюдается это совпадение. Реакция может быть бимолекулярной, но протекать по кинетическому уравнению реакции первого порядка и т. п. Примером могут служить реакции гидролиза уксусноэтилового эфира и тростникового сахара в разбавленном водном растворе
СН3 СООС2 Нь + Н2 О =СН3 СООН + С2 Нй ОН (а)
012 ^22 Оц + Hg О = Св Н|2 Об 4" Об Н12 Об (б)
В этих реакциях концентрация воды изменяется ничтожно мало, и скорость реакции зависит только от изменения концентрации эфира (а) или тростникового сахара (б).,Поэтому кинетика этих бимолекулярных реакций соответствует уравнению кинетики мономолеку-лярных реакций. В общем случае; если в реакции одно из веществ находится в большом избытке, порядок реакции, как правило, снижается.
Причина несовпадения порядка и молекулярности реакции объ-ясняется тем, что стехиометрическое уравнение реакции описывает процесс в целом и не отражает истинного механизма реакции, протекающей через ряд последовательных стадий.
§ 53. Реакции первого порядка
К реакциям первого порядка относятся такие, в которых скорость химической реакции связана с концентрацией (С) реагирующих веществ следующим уравнением: V = КС. или согласно уравнению (V, 3)
— 177 —
Если обозначить через а начальную концентрацию вещества до реакции, а через (а — х) концентрацию вещества в конце реакции, где х — количество грамм-молей вещества, прореагировавшего за время /, и подставить эти значения в уравнение (V,2), получим:
d (а—х) dx ., „
----- — = + -77=к (V.6) at----------------------------------------at
Перепишем уравнение (V,5) в таком виде:
dx
-----=Kdt. (V.,6) а—х
Интегрируя уравнение, получим:
J а —х J
— In (а—х) = Kt 4- const, (V,7)
где const— постоянная интегрирования, которая определяется из условия, что в начале опыта t = 0 и х = 0. Подставив эти значения в уравнение (V,7), найдем:
const = —In а.
Подставляя эту величину в уравнение (V,7), получим: — In (а — х) —К t— In а. Откуда 1 а К=—* In-------------------------------.
t а —х
Перейдя от натуральных логарифмов к десятичным, получим кинетическое уравнение реакции первого порядка в следующем виде:
(V.8)
(V,9)
Это уравнение дает возможность вычислить концентрацию реагирующего вещества в любой момент времени по известной величине константы скорости или найти константу скорости реакции при заданной температуре путем определения концентрации в любой момент времени.
Из уравнения (V,10) следует, что размерность константы скорости реакции первого порядка будет /-1. Скорость реакции первого порядка не зависит от объема (разбавления), в котором протекает реакция. Иными словами, в единицу времени превращению подвергается одна и та же часть вещества.
Наряду с константой скорости реакции первого порядка характеризуют также периодом полураспада т (греч. «тау»), который обозначает время, в течение которого превращается ровно половина взятого количества вещества. Короче говоря, тесть такой промежуток времени, в течение которого первоначальная концентрация вещества а
— 178 —
уменьшается доа/2, т. е. х = а12. Если подставить в уравнение (V, 10) вместо х значение а[2 и вместо t значение т, получим:
Так как Ig 2 — 0,301, то
2,303
Л =------
0,6932
2,303
Ig 2.
(VJ1)
Из уравнения (V, 11) следует, что константа скорости реакции первого порядка находится в обратно пропорциональной зависимости от периода полураспада.
Пример. Константа скорости радиоактивного распада эманации радия равна 463~qq'q ' Р» сек)* Через сколько времени активность эманации будет составлять 20%?
Решение. Поскольку радиоактивный распад идет по уравнению реакции первого порядка, для решения воспользуемся уравнением (V. 10), которое необходимо решить относительно t:
2,303
а
а—х
К
Если в это уравнение подставить лучим:
цифровые значения из условий задачи, по-
Откуда
2,303
463 000
а
2,303 а 463 000 а~ 5
/ = 207 ч.
§ 54. Реакции второго порядка
• Г-
•
К реакциям второго порядка относятся реакции соединения типа А + В С, реакции обмена A + B = C + D, а также реакции разложения и др. Скорость реакции второго порядка определяется уравнением:
4
V=+-^=K(a-z)(&-x), (V.12)
at
где — константа скорости реакции, а — число молей вещества А в начале реакции, b — число молей вещества В в начале реакции, % — число прореагировавших молей.
Здесь возможны два случая. Первый случай—это когда какое-то количество вещества А вступает в реакцию с эквивалентным количеством вещества В, т. е. когда а = Ь. И второй случай более сложный — это когда а Ф Ь. Рассмотрим оба эти случая.
179 —
Первый случай (а—Ь). Поскольку исходные концентрации реагирующих веществ равны между собой, уравнение (V, 12) примет следующий вид:
dx
+ — =К(а-х)\ (V.13)
at
Разделяя переменные и производя интегрирование, получим
= (V.14)
J (а—х)3 J
Откуда
—-— =- Kt 4- const. (V J 5)
а — х
При t = 0 х ~ 0, откуда
const=— а
После подстановки этой величины в уравнение (V, 15) получим;
Откуда
J X * " • t a(Q — x)
(V,16)
(V, 17)
Размерность константы скорости ’ реакции второго порядка Поэтому в отличие от константы скорости первого порядка численное значение К зависит от того, в каких единицах выражены / и С. Если последняя выражена в моль!л 9 а время в сек, то имеет размерность ( моль\~’И сек"1 • ----)
\ л )
Для реакций второго порядка большую роль играет число столкновений, которые происходят в единицу времени между молекулами реагирующих веществ. Число столкновений, в свою очередь, пропорционально числу молекул в единице объема, т. е. концентрации. Таким образом, константа скорости, а следовательно, и скорость реакции второго порядка зависят от разбавления раствора.
Второй случай (а ф 6). Если для реакции взяты неэквивалентные количества реагирующих веществ, скорость реакции выразится так: dx — =К(а-х)(Ь-х). (V ,18)
После разделения переменных получим это уравнение в другом виде:
dx (а—х)(Ь—х)
=Kdt.
(V,19)
180 —
Выражение, стоящее в левой части уравнения (V, 19), можно представить как
i 1 / 1 1
(а—х)(Ь — х) а—b\b — x а — х
После подстановки этого выражения в уравнение (V.19) получим:
\ Kdt.
a—b\b — х а — х )
После интегрирования
—-— |1п (а — х) — In (b —х)[ = /(/ + const.
(V,20)
(V,2I)
Поскольку при t =» 0 х = 0, постоянная интегрирования равна:
const =—-—(In я—lnb). (V ,22)
a—b
Подставляя это выражение в уравнение (V,21), найдем:
1 1
[In (а —ж) —In (Д>— х)| ==/</ 4- (1па — In 6), а—b--------------------------а + Ь
откуда
2,303 Ь(а—х) а—b а (Ь — х)
(V.23)
Это и есть кинетическое уравнение реакции второго порядка. Примером подобной реакции может служить окисление эфиров щелочами
СН3 СООС2 Н6 + ОН - -> СН3 СОО- 4-С2 Н5ОН
Кинетика реакций второго порядка была детально изучена С. Г. Крапивиным еще в 1915 г.
Реакции третьего порядка встречаются очень редко и потому не имеет смысла рассматривать математический вывод их кинетического уравнения.
§ 55. Сложные реакции
Сложными называются реакции, общее кинетическое уравнение которых в отличие от кинетического уравнения простых реакций содержит несколько констант скоростей. К сложным реакциям относятся обратимые, параллельные, последовательные, сопряженные, цепные и другие реакции. Теория всех этих реакций основана на положении, что при протекании в системе одновременно нескольких реакций каждая из них происходит самостоятельно и к каждой из них в отдельности применимы уравнения кинетики простых реакций.
Параллельными реакциями называются такие, которые имеют вид 4 В
к/
\С, т. е. при которых одни и те же исходные вещества, одновре-
— 181 —
менно реагируя, образуют разные продукты. Примером подобного типа реакций является реакция разложения бертолетовой соли КС1О8, которая может идти в двух направлениях:
6КСЮ3
2КС1 + О2
ЗКС1О4 + КС1
Распад некоторых радиоактивных элементов также протекает параллельно по двум реакциям. Более часто параллельные реакции встречаются в органической химии. Например, при нитровании фенола азотной кислотой могут одновременно образоваться орто- и пара-нитрофенолы
1)
2)
ОН
4-Н2О (орто-нитрофенол)
4-НО—NO2->
4-Н2О (лара-нитрофенол)
Последовательными реакциями называются такие реакции, которые протекают через ряд последовательных стадий по схеме:
А->в -> сD...
где каждой буквой обозначается отдельная стадия процесса. Реакции подобного типа широко распространены в природе. В общем случае число ступеней в последовательных реакциях может быть и больше трех, причем каждая из ступеней может быть не мономолекулярной, а более сложной. К реакциям этого типа относятся, в частности, реакции гидролиза сложных эфиров дикарбоновых кислот, или сложных эфиров гликолей, или дигалогенопроизводных и т. д.
Примером последовательных реакций может служить гидролиз рафинозы трисахарида, который происходит через стадии образования дисахарида, а последний образует уже моносахариды:
О Cis Н32 Ole + Н2 О -► С6 Hi2 0g ф- С12 Н22 Ои v рафиноза дисахарид
2) С12 О22 01х 4- Н2 О С6 Н12 06 4- Cg Н12 06 дисахарид моносахариды
Расчет кинетики последовательных реакций в общем виде очень сложен и здесь не рассматривается. Отметим только, что если одна из ступеней обладает значительно меньшей скоростью, чем остальные, то общая скорость реакции определяется скоростью именно этой ступени.
Сопряженными реакциями называются реакции, которые протекают по следующей схеме:
' 1) А4-В-»М
2) A4-C-»N
— 182 —
Реакция 1) может протекать самостоятельно, в то время как реакция 2) проходит при наличии реакции 1). Так, сульфат железа окисляется перекисью водорода независимо от присутствия йодистого водорода. Последний же в чистом виде перекисью водорода не окисляется, но при окислении сульфата железа окисляется одновременно с ним.
- Это явление, получившее название химической индукции, было детально изучено Н. А. Шиловым еще в 1905 г. Вещество А, которое является общим для обеих реакций (в нашем случае перекись водорода), получило название актора, вещество В — акцептора, а вещество С — индуктора.
Обратимыми называются такие реакции, скорость которых равна разности между скоростями прямой и обратной реакций
Примером обратимой реакции может служить реакция образования сложного эфира
сн3 соон + С2 Нь ОН сн3 соос2 н5+Н2О
В этом случае скорость прямой реакции с течением времени убывает, а скорость обратной реакции возрастает до тех пор, пока обе скорости не выравняются и не наступит так называемое состояние динамического равновесия. Константа равновесия данной реакции равна отношению констант скоростей прямой и обратной реакции, т. е.
K = (V.24)
Наряду с рассматриваемыми выше реакциями, механизм которых сравнительно прост, существуют также реакции, в которых взаимодействие осуществляется более сложным путем. Примером подобных реакций являются цепные реакции, которые имеют исключительно большое значение в химической технологии, так как они лежат в основе таких процессов, как полимеризация, крекинг нефти, деление атомного ядра.
К цепным реакциям относится большая группа реакций, протекающих путем образования цепи следующих друг за другом реакций, в которых участвуют активные частицы с ненасыщенными свободными валентностями — так называемые свободные радикалы. Свободные радикалы образуются за счет дополнительного поглощения энергии при разрыве связей в молекуле, при электрическом разряде, при поглощении электромагнитных колебаний, а также за счет других внешних источников энергии.
Свободными радикалами являются валентно-ненасыщенные частицы, которые можно представить как осколки молекул, например, .0 : Н (осколок от Н3О), Н: N : Н (осколок от H3N), Н :S :Н (осколок от H2S) и т. д. К свободным радикалам относятся и свободные атомы. Поскольку свободные радикалы имеют неспаренные электроны (на
— 183 —
схеме валентные электроны показаны точками), они являются чрезвычайно реакционноспособными.
Сущность цепного механизма реакции заключается в том, что активная молекула, реагируя, порождает новую активную молекулу или реакционноспособную частицу (валентно-ненасыщенные свободные атомы или радикалы). Процесс исчезновения и регенерации каждой активной частицы в дальнейшем циклически повторяется много раз и создает цепь превращений, совершающихся частью последовательно, а частью параллельно.
В качестве примера радикально-цепной реакции рассмотрим взаимодействие хлора с водородом
Н24-С1а = 2НС1
При обычной температуре и на рассеянном свету реакция протекает крайне медленно, тогда как нагревание смеси газов или освещение ее прямым солнечным светом сопровождается взрывом. Начало реакции связано с тем, что молекула хлора за счет поглощения кванта энергии (ftv) ультрафиолетовых лучей (или за счет нагревания) диссоциируег на свободные радикалы—атомы хлора
С!2 4- С1 -р С1
Валентно-ненасыщенный атом-радикал -С1 затем реагирует с молекулой водорода, образуя молекулу НС1 и радикал -Н. Последний, взаимодействуя с молекулой С12, дает НО и радикал -С1 и т. д.
Таким образом, превращение исходных веществ в конечный продукт протекает через последовательную цепь элементарных актов, что можно представить следующей схемой:
•СЦ-Н2~»НС14- -Н
С!2-> НО +• О
Н2 * НО -г • Н
С12 -* НС! 4- - О и т д.
Отдельные реакции (элементарные химические акты), лежащие в основе цепной реакции, носят название звеньев цепи.
Стадия первичного образования активных частиц или свободных радикалов носит название зарождения цепи.
Длина цепи определяется числом звеньев элементарных химических процессов, которые связаны между собой и обусловливают возникновение одного активного центра—свободного радикала. В зависимости от природы реагирующих веществ длина цепи может колебаться в широких пределах—от 2—3 звеньев до нескольких тысяч и более.
Так, число звеньев цепи в реакции синтеза хлористого водорода достигает 100 тысяч. Иными словами, на каждый поглощенный квант света образуется до 100 тысяч молекул НС1.
Казалось бы, зародившаяся цепь может развиваться бесконечно, до израсходования всех реагирующих веществ. На самом деле подоб
— 184 —
ные цепи всегда имеют конечную длину. Объясняется это тем, что при любой цепной реакции всегда имеют место процессы, которые разрушают активные центры (свободные радикалы) и тем самым вызывают обрыв цепи. Обрыв цепи может происходить и при потере активной частицей своей активности, например, при столкновении этой частицы со стенкой сосуда.
Скорость цепных реакций зависит от концентрации активных центров и от длины цепей; длина цепи в свою очередь зависит от размеров и формы сосуда, а также от наличия примесей в реакционной смеси. Поэтому скорость цепных реакций очень чувствительна к различного рода примесям, на чем и основан принцип регулирования скорости этих реакций.
Иногда одна активная частица в процессе химической реакции образует две или несколько новых активных частиц. В силу этого закончившийся цикл одной элементарной стадии реакции может дать начало не одному, а двум или более звеньям реакции. Цепь, таким образом, разветвляется, и скорость реакции в этом случае быстро возрастает, зачастую приобретая лавинообразный характер, т. е. характер взрыва. В качестве примера подобной реакции можно рассмотреть реакцию окисления водорода
2Н2 4- О2 = 2Н2О
Данное уравнение отражает лишь начало и конец реакции и совершенно ничего не говорит о механизме ее протекания. Началом цепной реакции в данном случае является взаимодействие между валентнонасыщенными молекулами Н2 и О2 (протекающая при нагревании, электроразряде, коротковолновом излучении)
Н2 4- О2 4- hv =
Он 4-- ОН
Свободные радикалы ОН* легко вступают во взаимодействие с молекулами водорода
но -+Н2 = Н2О4-- Н
Активный атом водорода реагирует с молекулой кислорода, в результате чего возникают две активных частицы с тремя свободными валент-.
ностями: одна у радикала ОН- и две у атома кислорода -О-
Н -+О2=НО О.
Таким образом, в этом процессе происходит все возрастающее увеличение активных центров, т. е. реакция приобретает лавинообразный характер и происходит взрыв. Ниже приведена схема разветвленной
— 185 —
цепи реакции окисления водорода
а
2
НО
2
На+О2—
но.
НО. 4-Н2-
НО♦ Н2
НО-
н2о
Впервые определение цепного механизма реакций через образование активных центров было дано Н. А. Шиловым (1904). Чрезвычайно большой вклад в дело изучения цепных реакций внес Н. Н.Семенов, которому за эти исследования была присуждена Нобелевская премия.
В настоящее время цепные реакции изучены настолько хорошо, что стало осуществимо регулирование скорости этих реакций. Добавляя вещество, легко вступающее во взаимодействие с активными центрами, можно значительно увеличить число обрывов цепей и тем самым затормозить (или же прекратить вовсе) цепную реакцию. Например, добавление всего лишь 0,01 % NC13 (треххлористого азота) к смеси водорода и хлора (Н2 и С12), которые реагируют по типу цепных нарастающих реакций, уменьшает скорость образования хлористого водорода в десятки тысяч раз.
Некоторые добавки увеличивают скорость цепных реакций. Так, добавление сравнительно малых количеств NO в значительной сте-$ пени ускоряет цепные реакции окисления углеводородов. При этом удается значительно понизить температуру процесса окисления, что очень важно, так как сохраняются от сгорания ценные промежуточные продукты — уксусный и муравьиный альдегиды.
На цепных химических реакциях основаны многие технологические процессы — синтез спиртов, кетонов, формалина, уксусной кислоты.
В последнее время исследования показали, что некоторые биологические процессы также протекают по типу цепных реакций, в частности процессы биологического окисления.
• — 186 —
/
§ 56. Влияние температуры на скорость химической реакции
Опыт показывает, что с повышением температуры скорость химической реакции возрастает. В уравнении химической кинетики v = = КСАСв влияние температуры практически сказывается на изменении константы скорости реакции /С. С возрастанием температуры растет величина константы /С, следовательно, увеличивается сама скорость реакции.
Если через Kt обозначить константу скорости данной реакции при температуре /, а через К <+ю° — константу скорости той же реакции при температуре (t + 10°), отношение второй величины к первой даст так называемый температурный коэффициент скорости реакции (?)
^Н-10° Kt
(V,25)
Согласно приближенному (эмпирическому) правилу Вант-Гоффа величина температурного коэффициента у колеблется в пределах 2 4,
т. е. при повышении температуры на 10Q скорость химической реакции возрастает в два — четыре раза. Например, если принять температурный коэффициент равным 2, легко можно подсчитать, что при повышении температуры на 100Q скорость реакции увеличится в 1024 раза, т. е.
б»+100°)==2Ц = 1024. • .(V.26)
К/
Таким образом, увеличение температуры в арифметической прогрессии влечет за собой увеличение скорости химической реакции в геометрической прогрессии.
Согласно правилу Вант-Гоффа температурный коэффициент скорости у для каждой химической, реакции должен являться величиной постоянной. Однако в действительности он сильно уменьшается при повышении температуры, что хорошо видно из рис. 70, где приведены кривые у = f (Г) для реакций образования и разложения йодистого водорода. Повышение температуры на 30° (от 743 до 773Q К) влечет за собой уменьшение температурного коэффициента первой реакции ’ в 1,64 раза, второй — в 1,71 раза. Для этих реакций правило Вант-Гоффа справедливо лишь в сравнительно узком интервале температур.
Более точная зависимость константы скорости химической реакции от температуры была найдена Аррениусом (1889). Уравнение Аррениуса имеет вид:
1пК=В-~ , (V.27)
где /С — константа скорости реакции, А и В — постоянные, характерные для данной реакции, Т — абсолютная температура.
— 187 —
Из уравнения (V,27) видно, что логарифм константы скорости находится в линейной зависимости от обратной температуры, т. е.
\ Т )
(V,28).
Коэффициенты Л и В в уравнении Аррениуса могут быть определены путем решения системы из двух уравнений типа (V,27) для двух тем- . ператур или определены графическим способом. Так, если по экспе
Рис. 70. Зависимость температурного коэффициента скорости реакций от температуры в реакциях образования (/) и разложения
HI (2)
риментальным данным построить график зависимости In К — 1/7\ то угловой коэффициент прямой определяет величину А = tg а, а постоянная В определяется отрезком, отсекаемым прямой на оси ординат (рис. 71). Определив таким путем численные значения постоянных Л и б для данной реакции, можно вычислить константу скорости, а следовательно, и скорость этой реакции при. любой температуре.
Величина А в уравнении (V,27) характеризует так называемую энергию активации Е химического процесса. Поясним смысл энергии активации несколько более подробно.
Скорость любой химической
реакции зависит от числа столкновений реагирующих молекул, так как число столкновений пропорционально концентрациям реагирующих веществ. Однако не все
столкновения молекул сопровожда-
ются взаимодействием. Так, в одном моле HI, находящемся в объеме
1 л при 1 атм и 556° К, происходит примерно 5,5 • 1034 столкновений в 1 сек. А количество иода, который образуется за тот же промежуток времени, показывает, что только одно столкновение из 2 » 1017 возможных сопровождается химической реакцией:
HI 4-Н1=Н2 + 12
Очевидно, скорость реакции зависит не только от числа столкновений, но и от каких-то свойств сталкивающихся молекул. Это явление находит объяснение в теории активации Аррениуса.
Согласно этой теории, реакционноспособны только те молекулы, которые обладают запасом энергии, необходимым для осуществления той или иной реакции, т. е. избыточной энергией по сравнению со средней величиной энергии молекулы. Такие молекулы получили название «активных» молекул. Эта избыточная энергия активной молекулы,
— 188 -
благодаря которой становится возможной химическая реакция, носит название энергии активации. Эту энергию обычно выражают в кал!моль. Энергия активации бывает меньше энергии разрыва связей в молекуле, так как для того чтобы молекула прореагировала, вовсе не требуется полного разры-‘ ва связей, их достаточно лишь ослабить.
Величина энергии активации за
Таблица 42
Энергия активации некоторых реакций
Реакция t-, кал/моль
44,4
43,5
40,0
32,0
26,0
висит от строения молекулы и от
того, в какую реакцию эта молекула вступает. Иными словами, каждая химическая реакция характеризуется свойственной ей величиной энергии активации. В табл. 42 приведены эти величины для некоторых реакций.
Рис. 71. Графический способ нахождения коэффициентов в уравнении Аррениуса по опытным данным
Рис. 72. Изменение энергии в ходе экзотермической реакции*
Энергия активации молекул может быть снижена под воздействием внешних факторов: повышение температуры, лучистой энергии, катализаторов и др. Энергия активации проявляется в активных молекулах по-разному: активные молекулы могут обладать большей скоростью движения, повышенной энергией колебания атомов в молекуле и др.
На рис. 72 показано, что скорость реакции пропорциональна числу активных молекул и зависит от величины энергии активации. На этом рисунке по оси ординат отложена энергия рассматриваемой системы молекул, а по оси абсцисс — ход реакции. Если реакция перехода из состояния I в состояние // идет с выделением тепла, т. е. является экзотермической, то общий запас энергии продуктов реакции будет
— 189 —
меньше, чем исходных веществ. Причем, разность энергетических уровней / и II будет равна тепловому эффекту реакции Q. Энергетический уровень К характеризует то наименьшее количество энергии, которым должны обладать молекулы, чтобы при столкновении друг с другом они прореагировали. Разность между уровнем К и уровнем / , характеризует энергию активации Е± прямой реакции, а разность между уровнями К и II — энергию активации обратной реакции Е2« Таким образом, при переходе из состояния I в состояние II система должна преодолеть энергетический барьер, т. е. должна обладать определенным избытком энергии, чтобы вступить в химическое взаимодействие.
Следовательно, скорость химической реакции зависит от величины энергии активации: чем она больше, тем медленнее будет протекать данная реакция. С другой стороны, чем меньше энергетический барьер реакции, тем большее число молекул будет обладать необходимой избыточной энергией и тем быстрее будет протекать эта реакция. Итак, скорость химической реакции в конечном итоге зависит от соотношения между числом активных и неактивных молекул.
В теории активных соударений Аррениус показал, что количество активных молекул может быть вычислено по закону Максвелла — Больцмана:
Е RT
N акт ~ N общ е » (V. 29)
где NaKT — число активных молекул, Af06m — общее количество молекул, е — основание натуральных логарифмов, Е — энергия активации, Т — абсолютная температура, /? — универсальная газовая постоянная.
Уравнению (V,29) можно придать вид:
Е
RT
K=KQe (V,30)
или после логарифмирования:
1пК=1пК0—(V.31) ' i\ I
где К.—константа скорости реакции при обычных условиях, Ко — константа скорости при условии, что все столкновения приводят к реакции.
Сравнение уравнений (V,27) и (V.31) позволяет выяснить физический смысл констант в уравнении Аррениуса. А = EIRT характеризует энергию активации процессов (Е), а В = In Ко, т. е. равно логарифму числа столкновений за 1 сек в единице объема.
Если известны константы скорости и /С2 при двух температурах 7\ и Т2, можно найти значение Е из уравнения (V,31):
190 —
или, заменяя натуральные логарифмы десятичными, и вводя вместо /? его численное значение 1,987 калЫолъ • град, можно записать:
- Кт Е / 1 1 \
1g---- =------/ — — — |
* КТ1 4,575 Т2)
Таким образом, рост скорости реакции с повышением температуры объясняется тем, что с увеличением температуры увеличивается не только средняя кинетическая энергия молекул, но и одновременно,, как следует из уравнения (V, 27), резко возрастает доля молекул, обладающих энергией выше определенного уровня, т. е. доля активных молекул, способных к реакции. *
§ 57. Влияние температуры на скорость биологических процессов
Биологическая жизнь может существовать в пределах интервала температур от— 100 до +100° С. Для всех биологических процессов брльшое значение имеет не только известный уровень температуры, но и вполне определенный температурный интервал. Если бы, например, в силу каких-либо причин средняя годовая температура на Земле неожиданно повысилась или понизилась всего лишь на 10°, основные формы жизни на ней погибли бы.
Температурные границы активной жизнедеятельности большинства живых организмов еще более узки (примерно от +1 до +45° С). И лишь немногие организмы могут длительно существовать при более высоких или более низких температурах. Так, на Аляске водится насекомое «каменная муха» («веснянка»), которая вполне нормально развивается при 0 °C. Треска проявляет максимум биологической активности при температуре морской воды ниже 0° С. Есть также целая группа живых существ — светящиеся бактерии, споры мха, семена некоторых злаков,—оживающих даже после пребывания при температуре жидкого гелия (—269° С). , . ч
В то время как скорость химических процессов обычно увеличивается с ростом температуры и это увеличение практически ничем не лимитируется, биологические процессы имеют свои температурные границы, за которыми наступает резкое уменьшение их скорости и даже полное прекращение — смерть.
В известной степени тепло повышает жизнедеятельность и ускоряет жизненные процессы, благотворно ‘ влияя на рост и размножение организмов. Но если температура превысит определенный предел, эти процессы быстро подавляются. На биологической кривой можно установить три характерные точки: минимальная температура (около 0 °C); оптимальная и максимальная. В интервале от минимума до максимума температур интенсивность биологического процесса растет, и здесь наблюдается подчинение правилу Вант-Гоффа. При дальнейшем повышении температуры организм погибает. Это положение хорошо иллюстрируется рис. 73, где показана зависимость энергии дыхания зерна пшеницы от температуры.
Температура тела у большинства животных организмов колеблется в пределах 35—40° С. Однако и здесь имеются отступления. Так, температура тела некоторых певчих птиц равна 45° С. Один вид холоднокровных, но «теплостойких» рыб прекрасно себя чувствует в горячих источниках острова Цейлон при температуре около 50° С. Самые «жаростойкие» организмы можно найти в мире бактерий и растений. Как известно, некоторые бактерии выживают при 70° С и выше, убить их удается только при длительном кипячении. В качестве примера можно назвать водоросль хлореллу, которая выдерживает 85° С. Она водится в горячих Источниках Йеллоустонского заповедника в США. Есть бактерии, способные вести нормальную жизнь при температурах выше 100° С.
Основными причинами гибели организмов под влиянием высоких температур является распад белков протоплазмы, а также образование токсичных про-
— 191 —
межуточных и конечных продуктов этого распада. При распаде белков нарушав' ся субмикроскопическая структура протопласта и соответственно координаци происходящих в различных частях протопласта физико-биологических проце< сов, которые регулируются целой системой сопряженно действующих ферменто! Помимо распада белков, при повышенных температурах происходит и инактив; ция ферментов, которая также гибельна для организмов.
В самом деле, если проследить зависимость активности любого фермента с температуры, то можно заметить ту же закономерность, что и на рис. 73, т. е. i же характерные температурные точки: минимум, оптимум и максимум. В качеп ве примера на рис. 74 приведены кривые действия фермента трипсина у разлш
--‘’--1-L -1 I |
Рис. 73. Зависимость энергии дыхания зерна пшеницы от температуры:
/ — влажность зерна 14%; 2 — 16%; 3 — 18%;
4 — 22%
О 20 40 60 t?C
Рис. 74. Активность фермен-та трипсина различных животных в зависимости от температуры:
/ — собака; 2 — окунь; 3 — треска
ных животных. Аналогичную картину дают и другие ферменты. Поскольку в основе жизненных процессов лежит целый ряд последовательных органически свя занных друг с другом биохимических реакций, регулируемых большим числом различных ферментов, то достаточно вывести из строя хотя бы один из этих ферментов (или даже нарушить гармоничность их действия), как наступает дезинтеграция жизненных процессов и, как результат, гибель самого организма. Именно этим и объясняется тот факт, что некоторые организмы имеют температурный минимум, который лежит значительно ниже температуры свертывания белка.
§ 58. Зависимость скорости реакции от катализаторе. Катализ гомогенный и гетерогенный
Скорость химической реакции может регулироваться с помощью катализатора. Вещество, изменяющее скорость химической реакции и остающееся после реакции в неизменном состоянии и количестве, называется катализатором. А само изменение скорости химической реакции в присутствии катализаторов получило название катализа.
Катализ широко распространен в природе. Сам термин впервые был введен еще в XIV в. С глубокой древности, задолго до создания
— 192 —
научных основ химии, человек широко использовал различные каталитические процессы, протекающие с участием биологических катализаторов—ферментов. В качестве примера можно назвать такие процессы, как приготовление алкогольных напитков, сыроварение, выделка кож, хлебопечение и многие другие.
Позднее, в конце XVIII — начале XIX в. в химической литературе появляются сведения о том, что ускорение реакции могут вызывать не только биологические, но и другие катализаторы неорганической природы.
Катализаторами могут быть самые разнообразные вещества в любом из трех агрегатных состояний: кислоты, соли, основания, оксиды, металлы, различные органические и органоминеральные соединения, газообразные вещества. В ряде случаев каталитическое действие оказывают всевозможные примеси (например, пыль), поверхность стенок сосуда, а также продукты реакции.
Если от добавления катализатора к реагирующей смеси скорость химической реакции увеличивается, катализ называют положительным. если же реакция замедляется, то катализ называют отрицательным. Например, добавление двуокиси марганца МпО2 к раствору перекиси водорода Н2О2 резко ускоряет ее разложение, тогда как небольшие количества ацетанилида придают ее концентрированному раствору устойчивость.
Иногда катализатор образуется в ходе химического процесса, и такие реакции называются автокаталитическими, а само явление — автокатализом. При автокатализе скорость химической реакции вначале очень мала, но с Появлением каталитически действующих продуктов реакции скорость быстро нарастает до известного максимума, после чего снова уменьшается. Это замедление реакции вызвано уменьшением концентрации реагирующих веществ. Примером автокаталитической реакции может служить омыление уксусноэтилового эфира в нейтральном растворе
с-н3 соос2 нБ + н2 о сн3 соон + с2 н6 он
В этой реакции катализатором является уксусная кислота, точнее ионы водорода Н+.
В ряде случаев присутствие некоторых веществ замедляет или даже практически полностью подавляет действие катализатора. Такие вещества называются соответственно ингибиторами или каталитическими ядами. Так, небольшая примесь окиси углерода отравляет медный катализатор, каталитическое действие платины сильно отравляется селеном и мышьяком, а для железного катализатора такими ядами являются соединения серы (H2S), кислород и его соединения (СО, Н2О и др.).
Каталитическая активность смеси различных катализаторов иногда значительно превосходит активность отдельных катализаторов. Например, смесь, состоящая из оксидов железа и висмута (Fe2O3 + + Bi2O3), хорошо катализирует окисление аммиака до оксидов азота и широко применяется в производстве азотной кислоты вместо значительно более дорогого катализатора — платины. Каждый из этих окси
7 Зак. 560
193 —
дов, взятый в отдельности, обладает очень малым каталитическим действием.
Увеличение активности катализатора часто наблюдается и при добавлении к катализатору веществ, которые сами по себе являются неактивными. Подобные вещества называются промоторами или активаторами. Так, каталитическая активность никелевого катализатора по отношению к реакции взаимодействия окиси углерода с водородом с образованием метана повышается в сотни раз при добавлении небольших количеств церия, а каталитическая активность V2O5 по отношению. к окислению SO2 также повышается во много раз при добавлении небольших количеств щелочи или сульфатов щелочных металлов. В настоящее время смешанные и промотированные катализаторы широко применяются в технике.
Механизм воздействия катализатора на химическую реакцию находит-свое объяснение в теории промежуточных соединений. Катализатор с одним из реагирующих веществ образует непрочное промежуточное соединение, которое легко реагирует со вторым компонентом реакции. Это положение подтверждается тем, что в ряде случаев удалось выделить соединения катализатора с одним из компонентов реакции. . .
В общем виде для реакции типа А + В -> АВ весь каталитический процесс в присутствии катализатора К можно представить следующими уравнениями:
1) А + К->АК, 2) АК-ЬВ-> АВ4-К. %.
Как видим, катализатор образует неустойчивое промежуточное соединение АК. Скорость данной каталитической реакции в конечном счете зависит от того, как быстро образуется и разлагается это промежуточное соединение. Например, если скорость расщепления промежуточного соединения АК на исходные компоненты намного выше, чем превращение промежуточного соединения в конечный продукт, скорость реакции будет мала. И, наоборот, если скорость превращения промежуточного соединения в конечный продукт значительно больше скорости его разложения, каталитическая реакция будет протекать очень быстро.
Опыт показывает, что подавляющее большинство каталитических процессов протекает со скоростью, имеющей промежуточное значение между этими крайними пределами.
Различные каталитические реакции подразделяются на реакции гомогенного и гетерогенного катализа. В тех случаях, когда катализатор и реагирующие вещества образуют однородную систему (т. е. находятся в одной фазе), мы имеем дело с гомогенным катализом. В качестве примеров можно указать на каталитическое окисление СО до СО2 в присутствии паров воды и окисление SO2 до SO3 в присутствии двуокиси азота NO2. К этому типу каталитических реакций относится и реакция гидролиза растворимых углеводов в водном растворе в присутствии кислоты. Как видим, в первых двух случаях катализатор и катализируемые вещества находятся в газообразном состоянии, в третьем — образуют однородный раствор.
194 — ,
Гомогенный катализ в растворах зачастую обусловливается наличием водородных или гидроксид-ионов. Причем с повышением их концентрации увеличивается и скорость реакции.
Вполне определенную роль в гомогенном катализе могут играть весьма кратковременные сочетания ионов и молекул, образующиеся под действием ионодипольных сил или в результате возникновения так называемых водородных связей. Благодаря этому происходит поляризация молекул реагирующих веществ и, как результат, повышение их реакционной способности.
При гетерогенном катализе катализатор и реагирующие вещества находятся в разных фазах, чаще всего катализатором является твердое тело, а реагирующие вещества находятся в жидком или газообразном состоянии и реакция протекает на поверхности раздела двух фаз, т. е. на поверхности катализатора. Поэтому гетерогенные каталитические процессы часто называют просто контактными, а твердые катализаторы — контактными веществами.
Опыт показывает, что при гетерогенном катализе большое значение имеет структура, химический состав и величина поверхности катализатора. Так, гладкая платиновая пластинка, опущенная в раствор перекиси водорода, практически не вызывает разложения последней. Если же поверхность этой пластинки шероховатая, разложение перекиси водорода протекает заметно. Еще больше увеличится скорость разложения, если в качестве катализатора использовать платину в виде порошка. И, наконец, если к раствору перекиси водорода прибавить еще более раздробленную платину в виде ее коллоидного раствора, то разложение Н2О2 произойдет практически мгновенно в виде взрыва.
Как правило, все гетерогенные каталитические реакции (даже относительно простые из них) протекают в несколько стадий: 1) сближение молекул реагирующих веществ, 2) ориентация молекул на активных центрах катализатора, 3) адсорбция молекул реагирующих веществ, сопровождающаяся деформацией связей в молекулах, 4) химическое превращение адсорбированных (и активированных) молекул, 5) десорбция продуктов реакции, 6) удаление этих продуктов с поверхности катализатора.
В некоторых случаях роль катализатора могут играть стенки сосуда, в котором происходит химическая реакция. Так, заметное взаимодействие водорода с кислородом в стеклянном сосуде происходит при температуре 450° С, а в платиновом сосуде—при более низкой температуре (15—20° С).
В настоящее время каталитические процессы широко используются в промышленности. Сейчас даже трудно назвать крупное производство химической промышленности, где бы не применялись катализаторы. Получение спиртов, альдегидов, аммиака, серной и азотной кислот, переработка каменного угля в жидкое топливо, процессы крекинга йефти при получении моторных топлив, синтез каучука, производство пластмасс, красителей, получение маргарина и других пищевых про-' дуктов—вот далеко не полный перечень процессов, где широко используются катализаторы. В ряде случаев за счет применения катализаторов удается значительно снизить температуру проведения реакции, что поз
7*
— 195 —
воляет уменьшать тепловые затраты и использовать менее жаростойкую аппаратуру, а также устранять нежелательные побочные реакции.
Процессы, в которых катализатор участвует в очень мелкораздробленном состоянии, например в виде частиц коллоидных размеров (менее 10“Б см), получили название микрогетерогенного катализа. К этому типу катализа относятся и процессы, протекающие при участии биологических катализаторов — ферментов.
§ 59. Основные свойства катализаторов и факторы, влияющие на катализ
• Характерной особенностью катализаторов является их избирательность, «специфичность», т. е. способность действовать ускоряюще только на определенную реакцию или группу реакций. Особенно ярко проявляется специфичность у ферментов. Так, фермент, действующий на сахарозу, не гидролизует крахмал.
Иногда применение разных катализаторов приводит к образованию совершенно различных продуктов из одних и тех же исходных веществ. В этом случае каждый катализатор вызывает ускорение реакции только в одном из возможных направлений. Например, реакция взаимодействия окиси углерода с водородом может идти по следующим направлениям в зависимости от применяемого катализатора
СО+н2 —
—>СН2О
—► СН3 ОН
—> высшие спирты
—>сн4
—> твердые парафины
(1)
(2)
(3)
(4)
(5) ит. д.
При использовании в качестве катализатора меди и давлении в пределах 300—400 атм продуктом реакции будет метиловый спирт (2). При применении в качестве катализатора никеля и температуре 250° С реакция будет идти по пути образования метана (4). Если же, помимо меди и давления в 300—400 атм, в систему добавить щелочь, то продуктами реакции явятся высшие спирты (3)..
Теория избирательного действия катализаторов еще не изучена в должной мере. Можно лишь предполагать, что оно зависит в какой-то мере от соответствия между расстояниями атомов в молекуле и активными центрами на поверхности катализатора.
Важным условием катализа является то, что для успешного проведения реакции количество катализатора обычно бывает очень невелико по сравнению с количеством реагирующих веществ. Так, в производстве серной кислоты на 1 вес. ч. катализатора превращается 104 вес. ч. реагирующих веществ, а в производстве азотной кислоты окислением аммиака на 1 вес. ч. катализатора превращается 10е вес. ч. исходных веществ. Для заметного ускорения процесса окисле
— 196 —
ния Na2SO3 в водном растворе достаточно добавить в качестве катализатора CuSO4 в концентрации порядка 10~12.
Важной отличительной чертой катализаторов является их способность снижать энергию активации катализируемой реакции.
В присутствии катализатора образуются промежуточные продукты реакции, в силу чего процесс может протекать при более низкой энергии активации. Причины снижения энергии активации при каталитических процессах во многом еще остаются невыясненными. Опыт показывает, что для разложения перекиси водорода на воду и кислород без катализатора требуется энергия активации 18 000 кал! моль, при использовании в качестве катализатора коллоидной платины энергия активации понижается до 11 700 кал!моль, а при действии фермента каталазы — до 5500 кал!моль.
Большое влияние на скорость каталитической реакции оказывает степень дисперсности катализатора. Увеличение активности катализатора с повышением степени дисперсности его происходит до определенного предела, затем она начинает падать. Объясняется это тем, что при дальнейшем увеличении степени дисперсности катализатора исчезает гетерогенность системы (вместо коллоидного раствора образуется истинный).
Помимо степени дисперсности, на активность катализаторов большое влияние оказывают и другие факторы. Рассмотрим каждый из них в отдельности.
Влияние температуры. С повышением температуры скорость ката-лических реакций увеличивается. Повышение температуры оказывает влияние не только на' активность катализаторов, но иногда и на направление каталитической реакции.
Каждый катализатор проявляет свою максимальную активность в той или иной химической реакции при определенной температуре. Отклонения от этой температуры как в ту, так и в другую сторон/ снижают активность катализатора. Вот почему при экзотермических каталитических процессах, осуществляемых в условиях производства, необходимо все время заботиться об отводе избытка тепла.
Биологические катализаторы — ферменты—также весьма чувствительны к изменению температуры в силу своей белковой природы.
Влияние давления. Как показывает опыт, скорость некоторых каталитических реакций весьма существенно зависит и от давления. Особенно это характерно для реакций, идущих с изменением объема. Увеличение давления положительно сказывается на скорости таких реакций. Некоторые каталитические реакции вообще не могут идти при нормальном давлении, например реакции синтеза высших спиртов и др.
В ряде случаев каталитические реакции, протекающие и без уменьшения объема, увеличивают свою скорость при повышении давления, что, по-видимому, связано с увеличением числа эффективных столкновений реагирующих молекул.
Влияние растворителя. В зависимости от природы растворителя скорость каталитических реакций может довольно заметно изменяться. Полная исчерпывающая теория этого явления в настоящее время еще
не разработана. Можно лишь предполагать, что растворитель каким-то способом активизирует молекулы реагирующих веществ, делает их более реакционноспособными. При этом большое значение имеет полярность молекул растворителя, под влиянием которой молекулы реагирующих веществ переходят в более активную ионизированную форму.
'J
. § 60. Теории гетерогенного катализа
В настоящее время подробно изучены сотни и почти тысячи каталитических реакций, многие из которых лежат в основе важнейших промышленных технологических процессов. Однако, несмотря на это, механизм действия катализаторов во многих случаях еще остается невыясненным.
Для понимания механизма и причин гетерогенного катализа имеют большое значение два общих положения, обоснованных многочисленными опытными данными: 1) катализ связан с адсорбцией реагирующих веществ на поверхности катализатора; 2) в каталитической реакции принимает участие не вся поверхность катализатора, а лишь небольшая ее часть, состоящая из ртдельных участков, называемых активными центрами.
Адсорбция является одной из важных стадий гетерогенного катализа. Причем, при адсорбции на поверхности катализатора происходят заметные изменения в структурно-энергетическом состоянии молекул, которые повышают их реакционную способность. Деформация и ослабление химических связей в молекулах, попавших в сферу действия адсорбционных сил поверхности катализатора, возможны только при хемосорбции. Именно хемосорбция определяет механизм гетерогенного катализа. Хемосорбированные мономолекулярные слои реагирующих веществ могут рассматриваться как специфические поверхностные промежуточные соединения, которые вследствие своей неустойчивости обладают реакционной способностью.
Таким образом, при гетерогенном катализе промежуточные соединения образуются на поверхности катализатора с поверхностно расположенными атомами. Существование активных центров на поверхности катализаторов подтверждается прямыми и косвенными данными. Известно, что для отравления катализатора бывает достаточно весьма малых количеств ядов, что указывает на активность не всей поверхности катализатора, а ее отдельных участков, так называемых активных центров. Неравноценность отдельных участков поверхности катализаторов обнаруживается по фигурам травления, а также методами рентгенографического анализа и электронной микроскопии.
Адсорбция вещества происходит главным образом на этих центрах в силу наличия у них довольно сильного неуравновешенного электри--ческого поля. Все теории адсорбционного гетерогенного катализа сводятся к выяснению роли и строения активных центров, а также энергетического состояния молекул на них.
Наиболее простое объяснение образования активных центров на поверхности твердых катализаторов заключается в наличии неровно
— 198 —
стей на их поверхности. Так, на снимках, полученных с помощью электронного микроскопа, видно, что даже хорошо отполированная блестящая поверхность меди или серебра имеет зубцы и выступы порядка 10-6 — 10~7 см. На рис. 75 показана схема строения поверхности твердого катализатора. Нетрудно догадаться, что атомы твердого вещества, расположенные в углублениях, будут энергетически более уравновешенными по сравнению с атомами, находящимися на выступах шероховатой поверхности катализатора. На этих атомах, имеющих свободное силовое поле, и будет в первую очередь происходить адсорбция реагирующих молекул.
Рис. 75. Активные центры катализатора. Профиль поверхности катализатора (по Тейлору)
В настоящее время имеется целый ряд теорий, объясняющих строение активных центров и явление гетерогенного катализа в целом. Наиболее обоснованными из них являются мультиплетная теория А. А. Баландина; теория активных ансамблей Н. И. Кобозева; электронно-химическая теория С. 3. Рогинского.
Мультиплетная теория. В основе этой теории лежит принцип структурного соответствия между расположением атомов на активных участках поверхности катализатора и строением молекул реагирующего вещества. Она рассматривает, Таким образом, не просто адсорбционное взаимодействие молекул в целом с поверхностью катализатора, а взаимодействие отдельных атомов или атомных групп в молекуле реагирующего вещества (так называемых индексных групп) с определенными геометрически правильными группировками атомов или ионов поверхностного слоя катализатора. Иными словами, активными центрами на поверхности катализатора являются отдельные участки (так называемые мультиплеты) кристаллической решетки катализатора,
— 199 —
имеющие правильную геометрическую конфигурацию, которая зависит от строения всей кристаллической решетки катализатора. Адсорбированная молекула «садится» на такой мультиплет так, что разные ее индексные группы связываются с разными атомами мультиплета. При этом происходит деформация связей адсорбированной молекулы. Величина подобной деформации зависит от расстояния между атомами в мультиплете: с увеличением расстояния химические связи в молекуле становятся более рыхлыми и легко могут разрываться. Ослабление и разрыв связей наблюдаются в том случае, если отдельные атомы, входящие в состав данной молекулы, адсорбируются на разных атомах мультиплета.
Связи между атомами способны замыкаться, если они адсорбируются на одном и том же атоме мультиплета.
В качестве примера рассмотрим реакцию дегидрирования этилового спирта. Согласно мультиплетной теории катализа, эта реакция происходит на двух точках катализатора (дублете). К одному атому дублета (7<2) притягиваются водородные атомы групп СН2 и ОН. К другому атому дублета (/(J притягиваются атом кислорода и углеродный атом группы СН2. В результате такого притяжения связи С — Н и О — Н разрываются и образуются новые связи . Н — Ни С — О в молекулах уксусного альдегида и водорода.
Если обозначить атомы дублета на поверхности катализатора в виде точек и /<2, то схему адсорбции молекулы этилового спирта на дублете можно представить следующим образом: у
Н Ki
СН3С — о—> сн3 сно + н2.
.1____|__
н н
Действие промоторов в теории мультиплетов объясняется тем, что их атомы достраивают и изменяют строение мультиплетов. Действие ядов объясняется прочной адсорбцией их на поверхности мультиплета, что также вызывает нарушение их структурного соответствия.
Теория активных ансамблей. Согласно этой теории носителями каталитической активности являются аморфные (не кристаллические) образования из нескольких атомов на каталитически недеятельной поверхности носителя. Эти атомы, собираясь в небольшие группы — ансамбли (по 2—3 атома), образуют активные центры.
Например, когда катализатором служит железо, нанесенное на уголь, асбест или алюмогель, на поверхности носителя образуются ансамбли, состоящие каждый из нескольких атомов железа. Для синтеза аммиака, например, необходимы ансамбли, состоящие из трех атомов железа.
По теории ансамблей атомы железа могут свободно перемещаться (мигрировать) по поверхности носителя. Однако наличие на поверхности густой сети микротрещин приводит к разделению этой поверхности на отдельные маленькие площадки, внутри которых и может
200 —
I
[1
в серьезной доработке, катализа с точки зрения
Рис. 76. Мозаичная структура поверхности катализатора и образование активных ансамблей (пунктирный контур)
Г\
на окислительно-восстано-
происходить перемещение атомов железа. В этом случае говорят о мозаичной структуре поверхности (рис. 76).
Таким образом, активность и специфичность катализатора обусловливается числом ансамблей и их взаимным расположением. Молекулы ядов, присоединяясь к ансамблям, изменяют те количества свободных атомов в ансамблях, которые оптимальны для проявления каталитического действия.
Электронно-химическая . теория катализа. Эта теория является наиболее современной, но еще нуждается В ней делается попытка объяснить механизм электронных уровней внутри кристаллов катализаторов и на их поверхности. Идея о том, что свободные электроны металла являются причиной каталитической активности, в свое время была предложена А. В. Писаржевским с сотрудниками. Электрические свойства поверхности катализатора оказывают существенное влияние на химические связи адсорбированных ’ или ориентированных молекул, вызывая их деформацию и. тем самым облегчая разрыв и перестройку этих связей.
По характеру взаимодействия реагирующего вещества и катализатора все каталитические реакции с точки зрения электронной теории подразделяются
вительные и кислотно-щелочные. В реакциях первого типа происходит переход электронов на катализатор или с катализатора. Поэтому все окислительно-восстановительные реакции катализируются в основном металлами (Fe, Ni, Pt и др.), а также некоторыми полупроводниками (V2O5, Сг2О3 и др.).
В кислотно-щелочных реакциях катализ осуществляется переходом протонов. В качестве катализаторов в этом случае могут выступать различные оксиды (TiO2, А12О3 и др.), сильные кислоты или основания.
В ряде работ экспериментально доказано, что удельная поверхностная активность катализатора в значительной степени зависит от его химического состава. Химический состав катализатора определяет скорость образования и разрушения поверхностных соединений и, следовательно, удельную поверхностную активность.
В работах Н. Н. Семенова гетерогенные каталитические реакции рассматриваются каю поверхностные радикальные цепные реакции, в которых катализатор, обладая свободными валентностями, может действовать как свободный радикал, возбуждая образование цепей. Так, реакция окисления СО кислородом
2СО4-О2=2СО2 в значительной степени ускоряется в присутствии паров воды, что вызвано развитием цепей с участием свободных радикалов *ОН и *Н.
— 201 —
Схематически это можно представить следующим образом: .он + со==со2+ - н
н + о2= ОН 4- - О СО+ . о =со2
С точки зрения электронной теории может быть объясним и органический катализ, однако в виду своей сложности он далеко выходит за пределы настоящего руководства.
§ 61. Ферменты как катализаторы
В процессе жизнедеятельности в любом живом организме совершаются сложнейшие и многообразные превращения химических веществ различной природы. Подавляющее большинство, а по некоторым данным, даже все химические'реакции в живых организмах протекают с участием биологических катализаторов — ферментов. Этим и объясняется легкость прохождения этих реакций.
Ферментативный катализ существенно отличается от химического катализа. Эти отличия сводятся к следующему.
Каталитическая активность. По своей активности биологические катализаторы в миллионы раз превосходят активность химических катализаторов. Даже лучший из неорганических катализаторов — атомная платина уступает, например, ферменту каталазе по своей активности в расчете на 1 активный центр в тысячи раз. О скорости ферментативных реакций можно судить по следующему примеру: 1 моль фермента сахарозы способен расщепить в 1 cjk 1000 моль свекловичного сахара.
Присутствие ферментов в ничтожно малых количествах способно расщеплять огромные количества реагирующих веществ. Так, 1г кристаллического пепсина расщепляет 50 кг коагулированного яичного белка, а 1 г кристаллического ренина свертывает 72 т молока. Фермент пероксидаза, который ускоряет окисление субстрата за счет перекиси водорода, проявляет свою активность при разбавлении 1 вес. ч. фермента в 500000 000 вес. ч. воды.
Согласно рекомендации международной комиссии по номенклатуре ферментов каталитическая активность фермента может быть охарактеризована его «молекулярной активностью», под которой следует понимать число молекул данного субстрата или эквивалентов затронутых групп, превращаемых за 1 мин одной молекулой фермента при оптимальной концентрации субстрата.
Высокая химическая специфичность. В отличие от химических катализаторов ферменты обладают значительно большей специфичностью: каждый из них действует лишь на строго определенную реакцию или группу реакций, протекающих в организме. Предполагается, что в организме человека одновременно функционирует около 1000 различных ферментов. При этом они образуют сложные ферментативные системы, которые обеспечивают в живой клетке протекание целого ряда строго последовательных и согласованных между собой реакций. Если бы ферменты не обладали столь высокой специфич-
202 —-
ностью, это привело бы к быстрому распаду всех веществ в клетках и к гибели всего организма.
Специфичность ферментов подразделяется на абсолютную (или химическую) и стереохимическую.
Абсолютная специфичность — это действие каждого фермента на вещество строго определенного химического-состава. Например, фермент уреаза катализирует только гидролиз мочевины, фермент пепсин — только расщепление белков, каталаза действует лишь на перекись водорода.
Стереохимическая специфичность заключается в том, что ферменты действуют только на определенные стереоизомеры органических соединений. В качестве примера подобной специфичности можно указать на действие двух ферментов: а и р-глюкозидазы. Фермент а-глю-козидаза действует только на а-глюкозиды, а р-глюкозидаза — на р-глюкозиды, что хорошо видно на приведенной схеме:
Место действия а - глюкозидазы
I---Т----
н—с—о-сн3
Н—С—он
I О
но—с—н
Н—С— он
н-с--------
СН2ОН
Место действия 3 - глюкозидазы
I I------
сн3—О—С—н
н—с—он I О но—С—н
Н—С— он
I СН2ОН
Причины столь высокой специфичности ферментов еще до конца не изучены. Существует целый ряд теорий, объясняющих механизм действия ферментов.
Так, немецкий химик Фишер для объяснения специфичности фермента по отношению к данному субстрату в свое время предложил гипотезу замка и ключа. Согласно этой гипотезе молекула субстрата точно соответствует по своей форме некоторому участку на молекуле фермента. Фишер полагал, что ключ — субстрат точно подходит к ферменту-замку, без какого бы то ни было нарушения формы обеих молекул. Однако, как показали исследования, в ряде случаев гипотеза Фишера не может объяснить некоторые факты.
Для того чтобы привести эту теорию в соответствие с опытными данными, К о ш л а н д несколько видоизменил модель «ключ—замок». Согласно его гипотезе субстрат, присоединяясь к активному центру, изменяет его форму, обеспечивая таким образом идеальное их соответствие. Иными словами, функциональные группы в активном центре принимают специфическую пространственную конфигурацию . только тогда, когда их вынуждает к этому присутствие субстрата.
Так, образование фермент-субстратного комплекса может происходить за счет электрически заряженных группировок как на фермен-
— 203 —
те, так и на субстрате. Такими группировками могут быть
О
R—'С—-о- или R—NHJ и др.
В результате подобного взаимодействия в субстрате могут происходить определенные химические изменения, выражающиеся в образовании новых функциональных групп с совершенно иными полярными свойствами. После реакции фермент и субстрат как бы отталкиваются друг от друга, и фермент вновь готов вступить во взаимодействие с другой молекулой субстрата. Химически измененный субстрат отщепляет продукт реакции.
Таким образом, специфичность фермента обусловливается его конфигурацией, строением и электрическими свойствами активной группы фермента.
Инактивация. В процессе протекания каталитической реакции фермент постепенно разрушается и утрачивает свою активность. Это явление получило название инактивации. Опыт показывает: чем большей активностью обладает фермент, тем он сильнее разрушается в процессе катализа. Этим свойством ферменты существенно отличаются от неорганических катализаторов, которые, как уже отмечалось, остаются без изменения в продуктах реакции.
Строение ферментов. По сравнению с неорганическими катализаторами ферменты имеют значительно более сложное строение. Каждый фермент содержит белок, которым и обусловлена высокая специфичность биологических катализаторов. Па своему строению ферменты подразделяются на два больших класса: однокомпонентные и . двухкомпонентные. К однокомпонентным относятся ферменты, состоящие только из белковых тел, которые обладают каталитическими свойствами. У этих ферментов роль активных групп выполняют определенные химические группировки, входящие в состав белковой молекулы и получившие название активных центров.
В настоящее время свыше 100 известных однокомпонентных ферментов получено в кристаллическом виде.
К двухкомпонентным относятся такие ферменты, которые состоят из белковой и небелковой части, называемой простетической группой. Было предложено активную простетическую группу называть агон, а белковый носитель — ферон или иначе апофермент. Исследования показали, что белковая часть двухкомпонентного фермента (ферон) оказывает решающее влияние на специфичность его действия. Вместе с тем соединение активной группы с белком приводит к огромному возрастанию ее каталитической активности.
Было также показано, что прочность связи агона и ферона у разных ферментов различна. У некоторых ферментов, например, дегидрогеназ, катализирующих окисление различных субстратов путем отнятия водорода (дегидрирование), эта связь является непрочной. Такие ферменты легко диссоциируют и распадаются на агон и ферон. По предложению выдающегося французского биохимика Г. Бертрана, агоны, легко отделяющиеся от белковой части фермента, обычно называют коферментами.
— 204 —
В качестве примера двухкомпонентного фермента можно назвать фермент пируватдекарбоксйлаза, который расщепляет пировиноградную кислоту на уксусный альдегид и двуокись углерода
снзС0С00н-та^тдекарбоксилаза ->сн3сон+со2.
Химическая природа активной группы пируватдекарбоксилазы в настоящее время полностью выяснена. Она представляет собой соединение молекулы витамина Вх и двух остатков фосфорной кислоты. Пируватдекарбоксилаза является примером фермента, активная группа которого содержит витамин. Как показали исследования, витамины являются неотъемлемой составной частью целого ряда важнейших ферментов (каталаза, пероксидаза и др.).
Влияние внешних условий. По своей природе ферменты значительно более чувствительны к изменению внешних условий по сравнению с неорганическими катализаторами. В частности, ферменты «работают» в значительно более узком диапазоне температур. Температурный оптимум большинства растительных ферментов 40—60° С, животных ферментов 40—50° С. Если температура превысит эти пределы, активность фермента очень быстро падает, а при 70—80° С происходит их необратимое разрушение, обусловленное денатурацией белка. Лишь очень немногие ферменты способны в определенных условиях выдержать нагревание до 100° С без потери активности.
Неорганические катализаторы, как показывает опыт, могут отлично работать и при более высоких температурах — до нескольких сот градусов.
В отличие от неорганических катализаторов ферменты проявляют свою активность в строго определенном диапазоне значений pH среды. В табл. 43 приведены значения pH, при которых различные ферменты проявляют свою максимальную активность.
Таблица 43
Оптимальные значения pH для некоторых ферментов
Фермент
Субстрат
Р-Фруктофуранозидаза
Уреаза
Попаин
Пепсин
Аргиназа
Сахароза Мочевина Белок Белок Аргинин
рн
6,6
2,0
9,9
Как видно из этой таблицы, диапазон значений pH весьма широк для активности различных ферментов. Влияние pH на активность ферментов объясняется изменением состояния ионизации не только фермента и субстрата в отдельности, но и фермент-субстратного комплекса.
— 205 —
Неорганические катализаторы практически не зависят от реакции среды. -
Таковы основные отличия биологических катализаторов — ферментов — от неорганических.
В настоящее время известно свыше 800 ферментов. В зависимости от типа катализируемых реакций все ферменты подразделяются на шесть основных групп: 1) оксидоредуктазы (окислительно-восстановительные ферменты); 2) трансферазы (ферменты переноса); 3) гидролазы (ферменты, осуществляющие гидролитическое расщепление субстрата); 4) лиазы (отщепление от субстрата отдельных групп с образованием двойных связей или присоединение групп к двойным связям); 5) изомеразы (ферменты, осуществляющие изомеризацию); 6) лигазы (ферменты, осуществляющие синтез).
Ферменты широко используются в технологии производства пищевой промышленности. Так, протеиназы применяются в хлебопечении. Эти ферменты осуществляют расщепление крахмала й гидролиз белковых веществ. За счет этих процессов не только происходит улучшение структуры хлебного мякиша, но и увеличивается объем выпеченного изделия. Фермент инвертаза используется в кондитерской промышленности: прибавка его к конфетной массе предохраняет продукцию от черствения. Если прибавить пектиназу к винограду или другим фруктам во время их прессования, получается повышенный выход сока. Виноградные вина, обработанные этими ферментами, обладают хорошим вкусом и цветом, становятся более прозрачными. При получении глюкозы из крахмала используется фермент глюкозидаза. Производство сыров, пива, получение многих лекарственных препаратов и многих других полезных для человека продуктов не обходится без применения ферментов.
Не менее важное значение имеет знание биологии ферментов и для агронома в целях получения наиболее высоких и устойчивых урожаев сельскохозяйственных культур. Исследования, проведенное в последние десятилетия, показали, что имеется непосредственная связь между питанием растения и направлением обмена веществ, совершаемым в нем под влиянием многочисленных ферментов.
При недостатке влаги и при сильном повышении температуры в растении гидролитические процессы распада начинают преобладать над синтетическими, что может в конечном итоге привести организм к гибели. Условия минерального питания растений также оказывают большое влияние на направление и скорость ферментативных процессов в них. В частности, недостаток в почве калия приводит к усилению распада сахарозы и к накоплению в растении большого количества моносахаридов. Внесение калийных удобрений устраняет эти нежелательные явления и приводит к усилению процессов синтеза белков. Таким образом, калий повышает активность ферментов, катализирующих образование и расщепление белков.
Внесение фосфорных удобрений в первую очередь сказывается на увеличении интенсивности синтеза крахмала и сахарозы, причем, скорость распада их при этом значительно ослабляется. Образование белков при этом увеличивается, но не в такой степени, как в случае сахарозы и крахмала. При хорошем обеспечении растения фосфором в нем содержится, как правило, больше сахарозы и крахмала, но несколько меньше белков. При недостатке фосфора наблюдается обратная картина.
Не менее важное значение по степени влияния на деятельность ферментов имеют также и азотные удобрения. Так, избыток азота в растении приводит к значительному снижению содержания в нем сахарозы и крахмала. Недостаток же азота, наоборот, стимулирует активность ферментов., катализирующих образование этих веществ.
Все эти примеры свидетельствуют о том, что ферментативный аппарат растения есть не только сложный, но и чрезвычайно тонкий и чувствительный механизм, который определяет весь ход обмена веществ всего растительного организма. Агроном, сознательно изменяя условия выращивания и, главным образом, питания растений, может менять не только интенсивность, но и направленность действия биохимических процессов и таким образом формировать урожай.
— 206 -
§ 62. Скорость гетерогенных химических процессов
Гетерогенными процессами называются такие процессы, в которых взаимодействие реагирующих веществ происходит на поверхности раздела фаз, т. е. взаимодействие между веществами, находящимися в различных соприкасающихся фазах. Такие реакции чрезвычайно широко распространены в природе и в различных областях производства. Можно привести много примеров гетерогенных реакций, которые имеют большое значение в сельскохозяйственном производстве, например, процессы, протекающие в почве с удобрениями, процессы корневого питания растений, растворение газов в почвенном растворе, действие различных ядохимикатов и т. д.
Отличительной чертой всех гетерогенных процессов является не только их сложность, но и многостадийность. Как правило, любая гетерогенная реакция состоит по меньшей мере из трех стадий. Первая стадия — это перенос реагирующих веществ к поверхности раздела фаз, т. е. к зоне реакции. Второй стадией является собственно сама, химическая реакция. Третья заключается в отводе продуктов реакции из зоны, где эта реакция происходит.
Таким образом, скорость любой гетерогенной реакции слагается из скорости самой реакции и скоростей подвода реагирующих веществ к границе раздела и отвода их от нее, главным образом, путем диффузии.
Опыт показывает, что если скорость одной из последовательных стадий гетерогенного процесса значительно меньше других, то суммарная скорость будет определяться скоростью этой наиболее медленной стадии. Если же скорости отдельных стадий сравнимы между собой, суммарная скорость гетерогенной реакции не обязательно должна быть равна скорости самой медленной стадии, так как все стадии взаимно связаны между собой. Более быстропротекающие стадии могут оказать влияние и на скорость самой медленной из них.
Поскольку в гетерогенных процессах взаимодействие происходит или начинается на поверхности раздела фаз, для таких процессов большое значение имеет не только состояние, но и размеры, т. е. площадь соприкасающихся фаз. Иными словами, скорость гетерогенной реакции прямо пропорциональна степени дисперсности реагирующего вещества, Кроме того, скорость этих реакций также находится в прямой зависимости от скорости диффузии, т. е. от скорости, с которой молекулы газа или растворенного вещества поступают к зоне реакции. *
Скорость диффузии измеряется количеством молей dn диффундирующего вещества через единицу площади за бесконечно малый отрезок времени dt. Количественная сторона этого явления выражается законами Фика. В частности, первый закон Фика имеет следующее математическое выражение:
dn
7t
— DS
dC
dx
(V, 34)
т. e. количество диффундирующего вещества в направлении от большей концентрации к меньшей за время dt пропорционально площади S се-
— 207 -
чения, через которое происходитдиффузия, и градиенту концентрации d&dx (т. е. изменению концентрации dC на отрезке длины dx). D — коэффициент пропорциональности, называемый коэффициентом диффузии. Знак минус указывает, что концентрация в направлении диффузии убывает.
В стационарном диффузионном потоке градиент концентрации dC?dx будет величиной постоянной и его можно заменить соотношением
» гДе б — толщина диффузионного слоя, через который идет диффузия, Со и С — концентрации по обеим границам этого слоя. Скорость диффузии, отнесенная к единице площади S, равна:
dC 1 dn
~dt~= S ' ИГ'
Учитывая уравнение (V,34), можем написать:
(V.36)
Таким образом, скорость диффузии пропорциональна разности концентраций и обратно пропорциональна толщине слоя. Коэффициент диффузии D зависит от природы диффундирующего вещества и среды, в которой происходит диффузия, а также от температуры и в меньшей степени от концентрации и давления. Он имеет размерность — пло-щадь/время.
В качестве примера гетерогенных реакций можно рассмотреть процесс растворения твердых тел в жидкости. При этом ограничимся рассмотрением наиболее простого случая, когда процесс растворения протекает в две стадии. Первая стадия — это когда взаимодействие твердого тела с растворителем завершается образованием вокруг поверхности растворяемого вещества насыщенного раствора. Вторая стадия растворения завершается отводом растворенного вещества в объем раствора путем диффузии.
Математическое выражение скорости растворения твердых тел в жидкости, предложенное в свое время Н. А. Щ у к а р е в ы м (1896), имеет следующий вид:
dC
— = ^(Снас~-С), (V,37)
где dC/dt — скорость нарастания концентрации растворенного вещества в объеме раствора, S — площадь поверхности растворяемой твердой фазы, К — коэффициент растворения, Снас — концентрация насыщенного раствора на поверхности растворяемого вещества, С — концентрация растворенного вещества в объеме раствора.
Из сравнения уравнений (V,36) и (V,37) видно, что коэффициент растворения связан с толщиной диффузионного слоя и коэффициентом диффузии следующим образом:
(V,38)
208
Из вышеизложенного можно сделать вывод о том, что скорость растворения, так же как и скорость любого гетерогенного процесса, определяется процессом диффузии и зависит от площади соприкасающихся фаз.
Многие агротехнические и агрохимические мероприятия в земледелии основаны на учете скорости протекания гетерогенных реакций. Так, применение в качестве фосфорного удобрения гранулированного суперфосфата на почвах, богатых полуторными оксидами, оказывается более эффективным, , чем применение порошковидного суперфосфата. Объясняется это тем, что гранулированный суперфосфат по сравнению с обычным имеет значительно меньшую поверхность соприкосновения с почвой и почвенным раствором, в результате чего скорость свя-зывания^ фосфат-ионов в труднорастворимые и практически недоступные для растений формы в виде фосфатов железа и алюминия значительно меньше. Гранула в почве растворяется медленнее, постепенно, и потому корневая система растения оказывается более обеспеченной подвижными формами фосфора.
Рядковое внесение фосфорных удобрений также преследует эту цель. При . таком внесении растение, с одной стороны, имеет фосфорные удобрения в непосредственной близости от корневой зоны, что облегчает питание на более ранней стадии развития, с другой — локализованное внесение суперфосфата замедляет процесс связывания его в труднорастворимые формы.
Для большей эффективности специалисты сельского хозяйства стремятся как можно больше измельчить удобрение и по возможности лучше перемешивать его с почвой. Например, эффективность таких агромероприятий, как известкование кислых почв и гипсование солонцовых, в значительной мере зависит от степени дисперсности вносимых в почву веществ и от качества перемешивания их с почвой. В самом деле, проку от извести или гипса (СаСО3 и CaSO4), внесенных в почву в виде крупных агрегатов, будет очень мало, так как площадь соприкосновения их с почвой и почвенным раствором мала, следовательно, и скорость взаимодействия с почвенным поглощающимся комплексом также окажется мала. Вот почему перед внесением в почву извести и гипса их рекомендуется хорошо измельчить. Подобные примеры можно легко продолжить.
Зная физико-химические свойства почв, а также гетерогенные процессы, в ней протекающие, агроном может сознательно регулировать скорость этих процессов путем использования научно обоснованных агротехнических приемов.
§ 63. Фотохимические реакции
Химические реакции, протекающие под воздействием света, называются фотохимическими, а сам раздел физической химии, занимающийся их изучением, получил название фотохимии. Примеров фотохимических реакций можно привести очень много. Так, смесь газов водорода и фтора на свету взрывается, аммиак разлагается на водород и азот, бромид серебра разлагается с выделением металлического серебра, что широко используется в фотографии, процесс отбелки тканей кислородсодержащими соединениями хлора также протекает под воздействием света и т. д. К числу фотохимических процессов относятся и реакции фотосинтеза, в результате которых в зеленых растениях из двуокиси углерода и воды образуются различные органические соединения, главным образом углеводы.
Все фотохимические процессы подчиняются так называемому закону Гроттуса: химическое превращение вещества может вызвать только тот свет, который этим веществом поглощается. Отраженные веществом лучи, а также прошедшие сквозь него не вызывают никаких химических превращений.
209
Опбт показывает, что количество вещества, прореагировавшего под действием поглощенного света, зависит от мощности света и от времени, в течение которого вещество было им облучено. Напомним, что мощность светового потока — это количество энергии, которое переносит световой поток через определенную площадь сечения в единицу времени. Обычно эта мощность выражается в эрг!сек. Количественно фотохимический процесс выражается уравнением
m=Kco/, (V,39)
где т — количество прореагировавшего вещества, г, со — мощность поглощенного веществом света, эрг!сек, t — время освещения, сек, К — коэффициент пропорциональности, зависящий от природы происходящей фотохимической реакции. Этот коэффициент численно равен массе прореагировавшего вещества, которая приходится на единицу поглощенной световой энергии.
Однако следует учесть, что уравнение (V,39) справедливо только для так называемых первичных фотохимических процессов. Во многих случаях под влиянием фотохимической реакции могут возникнуть другие реакции, уже не зависящие от действия света. Поэтому общая масса прореагировавшего вещества может оказаться значительно больше, чем следует из уравнения (V,39).
Из курса физики известно, что свет обладает двойственной природой: волновой и корпускулярной. Такие явления, как дифракция света, интерференция, свидетельствуют о его волновой природе. Явление фотоэффекта (отрыв ^уг поверхности вещества электронов под воздействием света) дает представление о его корпускулярной природе.
Луч света, по современным представлениям, представляет собой электромагнитное излучение, которое характеризуется следующими параметрами: длиной волны X, частотой v, массой и энергией фотона е. Возникновение его обусловлено переходом электронов в атоме с орбиталей более удаленных от ядра на орбитали, расположенные ближе к ядру. Этот перескок электронов сопровождается уменьшением энергии на некоторую величину, т. е. ее излучением. Энергия, потерянная атомом, и есть энергия электромагнитных колебаний. Испускание атомом электромагнитных колебаний, так же как и их поглощение, происходит не непрерывно, а целыми неделимыми порциями — квантами. Величина кванта света, или, как его еще называют, фотона выражается следующим равенством:
s=hv, * (V,40)
где е — энергия фотона, h — коэффициент пропорциональности, называемый постоянной Планка, равный 6,62-10-27 эрг/сек, v — частота колебаний.
Кванты света — фотоны — являются материальными частицами, несущими энергию света. Каждому кванту (фотону) соответствует световая волна частоты v. Частота колебаний v связана с длиной волны X соотношением:
v = 4~. (V.41)
Л
— 210 — 1
где с — скорость света, равная З-Ю10 см!сек. Как следует из уравнения (V,41), частота колебаний имеет размерность сект1.
Чем короче длина световой волны, т. е. чем больше ее частота, тем более мощные кванты составляют световой луч. В табл. 44 приведены некоторые данные, характеризующие различные области общего спектра электромагнитных излучений.
Таблица 44
Характеристика различных областей электромагнитного излучения
Излучение X, см V, сек 1 Е = Л V Масса фотона, г
. эрг Эв
Инфракрасное IO"2 7,6.10“! 3-1 о12 3,9-101* 1,99-10-** 2,71-Ю-*2 1,24-Ю-2 1,63 2,1-10-35 2,8-10~33
Видимое 7,6-10"! 4-10-5 3,9-10** 7,58-10м 2,71-Ю-*2 4,96- Ю-*2 1,63 3,12 2,8-10-33 5,5-10-33
Ультрафиолетовое Ю’ t-' 1 1 О О —ч < чК сч 7,58.10** 1,5-10*? 4,96-10-*2 9,9-10-*° 3,12 619,6 5,5-10-33 1,1- ю-30
Рентгеновское 2.10-7 6.10-10 1,5-1017 4,8-1018 9,9-10~10 ЗЮ’7 619,6 1,97.10s 1,1 -10—30 з-ю-28
Т-излучение* * В радиоакт] пия, чел показано 6-io-10 7,4-10"12 явных процесса в таблице. 4,8-10*» 4,1-102* X возможны и 3.10’7 2,7-10-5 5олее высокие 5 1,97-105 1,67-107 значения эне{ з-ю-29 2,8-10-2в >гий у-излуче-
Из этой таблицы видно, что энергия фотона быстро возрастает с уменьшением длины волны излучения (и соответственно с увеличением частоты), В видимой части спектра наиболее мощными квантами обладают фиолетовые лучи, наименее мощными — красные. Еще более мощные кванты несут рентгеновские, а затем гамма-лучи радиоактивных веществ.
'При попадании световой частицы в атом, фотон полностью передает свою энергию атому. В ряде случаев этой энергии бывает достаточно, чтобы расколоть молекулу какого-либо вещества на отдельные атомы. При этом может произойти соответствующая перегруппировка атомов и образоваться новое химическое соединение.
Как показывает опыт, энергия обычной химической связи в большинстве случаев составляет 30—100 ккал/моль, что соответствует примерно 1 —10 эв на одну связь. Таким образом, на химическую реакцию может оказать влияние только излучение, имеющее энергию фотонов не ниже указанных величин. Фотоны, энергия которых лежит в пределах этого интервала, носят название фотохимических.
Между количеством лучистой энергии, поглощенной молекулами данного вещества, и количеством фотохимически прореагировавших атомов или молекул химического соединения существует определенное
- 211 —
соотношение, выражаемое законом фотохимической эквивалентности Эйнштейна. Этот закон имеет следующую формулировку: число атомов или молекул, подвергшихся первичному фотохимическому превращению, равно числу поглощенных ими квантов света.
Для суждения о том, применим ли к данной фотохимической реакции закон Эйнштейна, пользуются понятием о квантовом выходе реакции.
Квантовым выходом реакции называется отношение числа молекул или атомов, прореагировавших в результате совокупности всех предшествующих реакций, к числу поглощенных квантов (фотонов), т. е.
число прореагировавших молекул число поглощенных фотонов
В табл. 45 приведены квантовые выходы некоторых фотохимических реакций.
(V,42)
Таблица 45 f
Квантовые выходы некоторых фотохимических реакций
Исходные Еещесп а Продукты реакции Длина еолны, см Квантовый выход
ci2, н2 Вг2, Н2 ^(»Hj2, Вг2 СН3Вг, NO СН3СОСН3 НС1 НВг СвНцВг, НВг CH3NO, Вг2 С2Н0, СО, СН4 3,03-10~6—5-Ю"6 5-10-8—5,78-10-» 4,7-Ю-6 2,537-10-5 3,13-10-5 1* и-—- О Of tO 1 1 I —* •— to о 05
Из этой таблицы видно, что квантовый выход не для всех фотохимических реакций равен единице. Объясняется это тем, что в ряде случаев вслед за собственно фотохимической реакцией происходят вторичные так называемые темновые реакции, в результате чего на один поглощенный фэтон приходится в конечном итоге не одна, а несколько молекул продукта реакции. Например, в указанной в табл. 45 реакции взаимодействия водорода и хлора на один поглощенный фотон приходится до 100 000 прореагировавших молекул, что объясняется в данном случае наличием цепной реакции.
Наряду с реакциями, квантовый выход которых больше единицы, известны фотохимические реакции с квантовым выходом меньше единицы (табл. 45). Причины этого явления могут заключаться в том, что часть фотонов поглощается посторонними веществами, находящимися в смеси с реагирующими веществами. В некоторых случаях пониженный квантовый выход обусловливается обратимостью химической реакции, а также передачей некоторыми молекулами, поглотившими фотоны, энергии другим молекулам в процессе взаимных столкновений.
Фотохимические реакции имеют много общего с обычными химическими реакциями. Для них, так же как и для «темновых» реакций, требуется определенное предварительное возбуждение (активация)
— 212 —
молекул, после чего последние вступают в химическое взаимодействие. Принципиальное отличие состоит в том, что в фотохимических реакциях молекулы возбуждаются за счет лучистой энергии, а в обычных «темновых» реакциях — за счет других источников, например нагревания.
Опыт показывает, что иногда фотохимические процессы осуществляются под действием излучения, хотя оно совершенно не поглощается реагирующими веществами. Казалось бы в данном случае имеет место отступление от закона Гроттуса. Однако исследования показали, что эти реакции происходят только тогда, когда к реагирующим веществам примешиваются некоторые посторонние примеси, которые, поглощая световую энергию, передают ее затем реагирующим веществам. Эти примесные вещества получили название сенсибилизаторов. Механизм действия сенсибилизаторов состоит в том, что молекула сенсибилизатора при поглощении фотона переходит в возбужденное состояние, а затем, столкнувшись с молекулой реагирующего вещества, передает ей избыток своей энергии, вызывая тем самым химическое превращение. Примеров сенсибилизированных реакций можно привести очень много. Так, путем добавления к фотоэмульсии некоторых веществ, выполняющих роль сенсибилизатора, можно значительно повысить ее чувствительность к красным лучам света. Известный всем хлорофилл также является сенсибилизатором фотохимических реакций образования органических веществ в зеленых растениях.
§ 64. Фотосинтез в растениях
Из всех известных в природе фотохимических процессов наии большее значение имеет фотосинтез. Основоположником учения о фото* синтезе является К. А. Тимирязев. Фотосинтез является основой существования всего живого на земле. Фотосинтез зеленых растений — это единственный первоисточник накопления органического вещества на Земле, которое служит для питания человека и животных. Вся растительность земного шара создает ежегодно около 120 млрд, тонн органического вещества, из них примерно 10 млрд, тонн производит человек, выращивая на площади около 2,5 млрд, гектаров пищевые и кормовые растения. •
Кислород, содержащийся в атмосфере Земли, имеет биогенное происхождение, т. е. также является продуктом фотосинтеза зеленых растёний. Расчет показывает, что растениями ежегодно возвращается в атмосферу около 4,7 -1011 тонн газообразного кислорода.
Сущность процесса фотосинтеза. В зеленом листе растения под воздействием солнечной радиации протекает целый комплекс фотохимических процессов, в результате которых из воды, углекислого газа и минеральных солей образуются крахмал, клетчатка, белки, жиры и другие сложные органические вещества. Процесс фотосинтеза очень сложен. Он осуществляется при непосредственном участии важнейшего природного фотокатализатора — хлорофилла и сопровождается целым циклом химических превращений, не зависящих от солнечной радиации. В этих превращениях участвует большое количество разно
— 213 —
образных биокатализаторов — ферментов. Суммарное уравнение фотосинтеза обычно выражают в виде реакции превращения двуокиси углерода и воды в гексозу
6СО2 4“ 6Н2О — 4~ 6()2
Однако это уравнение, как и большинство суммарных уравнений в биологии, не выражает основных особенностей процесса.
Рис. 77. Разложение светом двуокиси углерода
В изучении фотосинтеза, как уже упоминалось, большую роль сыграли работы К. А. Тимирязева. Важнейшей его заслугой является материалистическое научное обоснование фотосинтеза. Тимирязев впервые доказал, что фотосинтез подчиняется закону сохранения и превращения энергии. Таким образом был нанесен сокрушительный удар по идеалистическим воззрениям на процесс фотосинтеза, объяснявшим его действием какой-то мифической и нематериальной «жизненной силы».
Не менее важной заслугой Тимирязева является открытие роли хлорофилла как сенсибилизатора фотохимических реакций, происходящих Яри фотосинтезе. Он экспериментально установил, что фотосинтез осуществляется преимущественно в красных и синих лучах видимого спектра. Тимирязев провел следующий опыт. Ряд стеклянных трубочек, наполненных смесью воздуха и двуокиси углерода и содер
жащих по одному одинаковому зеленому листу, был выставлен на разложенный с помощью трехгранной призмы солнечный свет так, что в каждой части солнечного спектра находилась одна трубочка. Через каждые несколько часов определялось содержание двуокиси углёрода в трубочках. Оказалось, то усвоение СО2 происходит только в тех лучах, которые поглощаются хлорофиллом, т, е. в красных, оранжевых и желтых частях спектра. Некоторые результаты опыта представлены на рис. 77 в виде графика, на котором по оси ординат отложены количества поглощенной СО2 в каждой из трубочек.
Таким образом, Тимирязев показал, что именно хлорофилл являет
ся поглотителем света в зеленых растениях и что этот пигмент, поглощая кванты света, обладает способностью передавать их далее молекулам веществ, являющихся исходными при фотосинтезе, При этих реак
— 214 —
циях хлорофилл испытывает обратимое окислительно-восстановительное превращение. Структура молекулы хлорофилла показана ниже.
В основе этой структуры лежит порфириновое ядро (I), называемое хлорином. Оно состоит из четырех соединенных СН-мостика^и остатков пиррола, которые связаны двумя основными и двумя координационными связями с центральным атомом магния. Кроме того, в молекулу хлорофилла входит остаток молекулы высокомолекулярного непредельного спирта фитола (II). В настоящее время известно не менее пяти видов хлорофилла, которые отличаются друг от друга строением молекулы.
Помимо хлорофилла, который является основным видом фотосинтетических пигментов, в зеленом листе (в так называемых хлоропластах, представляющих собой сложные специализированные биологические структуры) содержатся и другие пигменты — каротиноиды и фикобелины, которые обычно называют вспомогательными. Эти пигменты, по современным представлениям,-принимают известное участие в фотосинтезе, а также защищают хлорофилл от фотоокисления. Помимо пигментов, основными компонентами хлоропластов, в которых собственно и осуществляется весь процесс фотосинтеза, являются липоидные вещества и белки, которые содержат большое количество ферментов, необходимых для осуществления последующих стадий фотосинтеза, не связанных с воздействием солнечной радиации.
Многие вопросы фотосинтеза, несмотря на бурное развитие науки, остаются мало изученными и до настоящего времени. Как уже упоминалось ранее, процесс фотосинтеза состоит из двух стадий — световой и темновой, причем обе эти стадии тесно связаны между собой.
Поскольку исходным процессом фотосинтеза является поглощение света хлорофиллом, приближенно фотосинтез можно представить в виде следующей схемы,
— 215 —
* j
В световой стадии хлорофилл, поглотив квант света, переходит в возбужденное состояние и в таком виде через ряд промежуточных процессов вызывает разложение молекулы воды на атом водорода Н и радикал ОН по схеме:
X4-/iv=X*
Х* + Н2О=Х + Н-ЬОН
где символом X условно обозначена молекула хлорофилла, X* — та же молекула в активном состоянии.
Далее молекула хлорофилла, присоединяя атом водорода, восстанавливается. Радикалы ОН, соединяясь попарно, образуют молекулу перекиси водорода Н2О2, которая как непрочное соединение распадается на воду и кислород
Х4-Н-ХН 4ОН = 2Н2О2 2Н2О2 = 2Н2О 4-О2.
После завершения этих реакций наступает темновая стадия процесса фотосинтеза, сущность которой состоит в передаче водорода молекулой восстановленного хлорофилла молекуле СО2 с образованием органических соединений типа углеводов. Этот процесс совершается под действием соответствующих ферментов по схеме
4Н 4-СО2—СН2О-|- Н2О
В итоге за счет полимеризации получается конечный продукт фотосинтеза— гексоза С6Н12О6.
То, что выделяемый в процессе фотосинтеза кислород принадлежит воде, а не двуокиси углерода, было доказано А. П. Виноградовым (1946) с помощью метода меченых атомов. Так, при использовании воды Н218О весь ее кислород 18О был найден после осуществления фотосинтез?! в свободном молекулярном кислороде, а при работе с 18СО2 и H21GO выделялся свободный кислород 16О, тогда как кислород 18О входил в состав органических соединений. Установление этого факта имело существенное значение для теории фотосинтеза, так как ранее в течение более 100 лет предполагалось, что молекулярный кислород образуется путем светового разложения или фотолиза СО2.
Приведенная выше схема фотосинтеза является лишь приближенной и не отражает всех деталей этого чрезвычайно сложного явления, В последние годы было установлено, что на восстановление одной молекулы СО2 до углерода затрачивается не один, а 8—12 квантов энергии. Это свидетельствует о том, что в процессе фотосинтеза происходит по крайней мере восемь первичных фотохимических реакций, которые совершаются в определенном порядке с другими (не фотохимическими) реакциями.
Известно, что далеко не каждая молекула хлорофилла или другого пигмента, поглотившая свет и сохранившая достаточное количество энергии для фотохимической реакции, является центром подобной
— 216 -
реакции. На самом деле фотохимическая активность, т. е. непосредственная связь с фотохимической реакцией, осуществляется лишь примерно одной молекулой из 200—250 молекул хлорофилла. Об этом явлении А. Г. Пасынский пишет: «... Могло бы создаться неправильное представление, что основная масса хлорофилла является фотохимически неактивной и играет в листе роль запасного вещества, как иногда предполагалось в литературе.
В действительности такое положение является необходимым следствием квантовой природы действующего света. Поглощение света данной молекулой хлорофилла не происходит непрерывным потоком; кванты света, падающие подобно каплям дождя, поглощаются все время разными молекулами хлорофилла.
По данным Рабиновича, даже на прямом солнечном свету каждая молекула хлорофилла поглощает квант света всего один раз за0,1сек, а при менее благоприятных условиях — гораздо реже. Между тем скорость последующих ферментативных реакций является чрезвычайно высокой. Если бы в этих условиях каждая молекула хлорофилла была самостоятельным центром фотохимической реакции, связанным с необходимыми вспомогательными ферментами, то такое устройство было бы столь же нецелесообразно, как если бы каждый участок крыши, на который падает отдельная капля дождя, был оборудован самостоятельным водостоком. В листе для подобного устройства просто не хватило бы места, не говоря уже о том, что оно могло бы использоваться лишь незначительную часть времени.
Напротив, соединение большой группы (200—250) молекул хлорофилла с одним центром фотохимической реакции обеспечивает его непрерывную работу, подобно тому, как присоединение одного водостока к достаточно значительной поверхности крыши позволяет получить из отдельных капель непрерывный поток воды. Ясно, что при этом вся масса молекул хлорофилла активно участвует в полезном процессе, хотя она связана лишь с одним центром превращения поглощенной лучистой энергии в химическую»*.
Все это лишний раз подтверждает чрезвычайную сложность процесса фотосинтеза, каждая ступень которого требует не только определенных условий среды, но и очень сложной системы вспомогательных веществ, а также строго определенной внутренней структуры внутриклеточного содержимого. На важность структурных факторов указывает то, что зеленый лист, подвергшийся механическому воздействию (например, если прокатать его на стекле толстой стеклянной палочкой), теряет способность к фотосинтезу.
Изучение процессов фотосинтеза очень важно не только с чисто теоретической точки зрения, но и с точки зрения получения высоких и устойчивых урожаев. Познать эти процессы, научиться управлять ими — вот те задачи, на решение которых направлены в настоящее время усилия целой армии отечественных и зарубежных ученых.
*А. Г. Пасынский. Биофизическая химия. «Высшая школа», 1968.
217 **
§ 65. Фотосинтез и урожай сельскохозяйственных культур
t
Основная задача, стоящая при изучении природы и механизма фотосинтеза, — это разработка путей и способов повышения продуктивности растений. В ходе формирования урожая любых сельскохозяйственных культур ^фотосинтезу принадлежит ведущая роль. Это утверждение можно иллюстрировать следующими примерами. В процессе фотосинтеза растения весь углерод усваивают из внешней среды, углерод же в урожае составляет примерно 42—45% веса сухой массы. В период наиболее интенсивного роста суточный привес сухой биомассы на гектар посевов составляет примерно 80—150 кг, а иногда и 300—500 кг. При этом в течение дня растения усваивают из воздуха через листья 150—300 и даже 1000 кг углекислого газа. Это количество углекислого газа соответствует его содержанию над гектаром площади в слое воздуха высотой 30—200 м.
При оценке урожаев также хорошо видна ведущая роль фотосинтеза. Например, сахарная свекла при урожае в 250—300 ц[га за вегетационный период усваивает около 100—150 кг азота, 25—30 кг фосфора, ПО—160 кг калия и около 4200 кг углерода. Причем в процессе фотосинтеза растения усваивают около 20m углекислого газа, что соответствует содержанию СО2 в слое воздуха высотой в 4 км над гектаром.
Решающая роль фотосинтеза в формировании урожая ни в коей мере не умаляет значения и других видов питания растений: азотного, фосфорного, калийного. Однако при формировании урожая все виды питания в конечном итоге реализуются через основную функцию растений — фотосинтез. Вот почему усилия агронома должны быть направлены на то, чтобы суммарная работа фотосинтетического аппарата растений была наиболее продуктивной.
Опыт показывает, что зависимость между деятельностью фотосинтетического аппарата растений и урожаями очень сложна. При этом необходимо учитывать • целый ряд факторов и условий, в которых протекает фотосинтез: площадь листовой поверхности изучаемой культуры, интенсивность и продолжительность работы фотосинтетического аппарата, отношение между процессами новообразования и расходом органического вещества и др.
Для оценки работы фотосинтетического аппарата пользуются понятием коэффициента эффективности фотосинтеза (КЭф)* С его помощью можно высчитать количество сухой биомассы, которое образуется в течение суток растением при усвоении в течение дня 1 кг СО2. При благоприятных условиях КЭф может приближаться к 0,5, а в неблагоприятные периоды падать до нуля или принимать отрицательные значения. Если умножить этот коэффициент на количество усвоенного 1 м2 листовой поверхности углекислого газа, а также на показатель площади листьев, можно получить размеры биологического урожая за сутки. Суммируя же эти^пок азател и за количество дней вегетационного периода, можно вычислить биологический урожай, т. е. вес общей сухой биомассы.
Такова зависимость биологического урожая от размеров и работы фотосинтетического аппарата растений. Однако важно создание благоприятных условий не только для формирования биологического, но и хозяйственного урожая культур. Для характеристики этих условий пользуются понятием так называемого коэффициента хозяйственной эффективности фотосинтеза (КХоз)- Путем умножения величины биологического урожая на этот коэффициент можно вычислить*, размер хозяйственного урожая.
Опыт показывает, что все агротехнические приемы — применение удобрений, поливов и прочие мероприятия—должны быть направлены на достижение наивысших значений показателей и Кхоз. Одно из важнейших условий высокой продуктивности посевов растений заключается в том, чтобы они поглощали как можно больше солнечной энергии. Это связано с размером площади листьев в понёвах. Однако увеличение листовой поверхности сверх определенного предела приводит к снижению коэффициента усвоения солнечной радиации и уже не дает эффективной прибавки урожая.
Таким образом, одна из главных задач, которую должен решать агроном, состоит в том, чтобы получать культуры с оптимальным развитием площади листьев. При этом они должны обладать наиболее высокими показателями про
— 218 —
дуктивности фотосинтеза. Опыт показывает, что площадь листьев около 30— 40 тыс. мЧга является оптимальной для получения высоких урожаев с достаточно высокими коэффициентами использования энергии солнечной радиации. Если площадь листьев в посевах составляет лишь 10—15 тыс. мЧга, то ее нужно увеличить путем внедрения более высокой агротехники. В тех же случаях, когда исходный агротехнический уровень высок и площадь листьев в посевах достигает около 30—40 тыс. мЧга, дальнейшее повышение урожая сопряжено с решением более трудных задач, в частности, связанных с пространственным расположением листового аппарата растений. Вопрос о посевах как целостной оптической системе (об оптимальной их структуре)—новый и очень важный; его изучением занимаются многие ученые. Опыт показывает, что идеальным по структуре может быть посев, в котором листья верхних горизонтов имеют вертикальное или близкое к нему расположение и достаточно хорошо пропускают свет в толщу травостоя, где пространственная ориентировка листьев приближается к горизонтальной. В решении вопроса о листовой структуре свое слово должны сказать и селекционеры при выведении новых сортов сельскохозяйственных культур.
В качестве примера недоучета структуры листового аппарата можно указать на неудачную попытку ввести в культуру каучуконосные одуванчики кок-сагыз, крым-сагыз, которые имели плотно распластанную на земле розетку листьев. В результате этого даже в сильно загущенных посевах площадь листовой поверхности этих растений не превышала 10—15 тыс. м2!га и урожаи получались очень низкими. В данном случае ни минеральное питание, ни высокая агротехника не могли оказать сколько-нибудь существенного влияния на урожай, так как фотосинтетический аппарат этих растений имел свой предел.
»
Глава VI
ХИМИЧЕСКОЕ РАВНОВЕСИЕ
§ 66. Понятие о химическом равновесии. Закон действующих масс •аг
Многие химические реакции протекают так, что взятые вещества целиком превращаются при данных условиях в конечные продук-ты реакции, т. е. процесс идет до конца. Эти реакции называются необратимыми. Примером такой реакции может служить взаимодействие металлического цинка с раствором серной кислоты
Zn -И H2SO4 = ZnSO4 4- Н21
Однако при очень многих химических превращениях процессы могут протекать как в прямом, так и в обратном направлениях. Такие реакции называются обратимыми. Так, если в замкнутом сосуде при температуре 500° С и давлении в 300 атм поместить эквивалентные количества азота и водорода, то между ними возникнет реакция
N24-3H2->2NH3
По мере накопления аммиака он будет разлагаться по уравнению 2NH3-N2+3H2 *
Таким образом, реакция между азотом и водородом протекает обратимо, поэтому при записи ее в виде уравнения вместо знака равенства обычно ставят знак обратимости
N2 + 3H2z± 2NH3
Реакция, идущая слева направо, называется прямой, а идущая справа налево — обратной.
Если по ходу реакции измерять концентрации участвующих в ней веществ, можно выявить следующие закономерности. В начале реакции концентрация азота и водорода уменьшается, а концентрация аммиака возрастает, т. е. идет прямая реакция. По мере же увеличения количества аммиака начинает преобладать обратная реакция, т. е. его разложение до исходных продуктов — азота и водорода. В начальный момент реакции, когда концентрации исходных веществ велики, скорость прямой реакции будет наибольшей, затем она постепенно уменьшается. По мере увеличения концентрации продуктов реакции скорость обратного процесса будет пропорционально увеличиваться. Наконец, через некоторое время наступит такое состояние, когда скорости прямой и обратной реакции станут равными. В этом случае в сосуде будут находиться в определенном количественном соотношении все три компонента рассматриваемой химической системы. И сколько бы мы ни выдерживали данную систему при вышеуказанных условиях, концентрация аммиака, водорода и азота останется неизменной, причем на долю аммиака будет приходиться 26,4 об. %,
— 220 —
а на долю водорода и азота — 73,6 об. %. Иными словами, система при данных условиях будет находиться в состоянии равновесия.
Необходимо отметить, что химическое равновесие следует понимать не как состояние покоя, а как процесс, идущий беспрерывно с одинаковой скоростью в обоих направлениях. При этом концентрации веществ не меняются, что и создает видимость покоя; Такое состояние называется химическим динамическим равновесием.
Обратимые химические реакции были изучены русским ученым Н. Н4 Б е к ето в ы м (1865), который установил влияние концентрации реагирующих веществ на направление и скорость химического процесса* В частности, наблюдая действие газообразного водорода на соли и оксиды некоторых металлов, он пришел к выводу, что вытесняющее и восстанавливающее действие водорода зависит от давления, под которым находится газ, т. е. от массы водорода. Таким образом, Бекетов вплотную подошел к формулировке закона действующих масс.
Эта формулировка в более общей форме была дана позднее Гульдбергом и Вааге (1867): скорость химической реакции пропорциональна действующим массам. Под действующими массами понимаются мольные концентрации веществ, участвующих в реакциях*
Если обозначить концентрации веществ А и В через [А] и [В], то скорость химической реакции согласно закону действующих масс можно записать уравнением
г = /([А][В], (VI,!)
где К — константа скорости химической реакции, - показывающая, какая доля исходных веществ реагирует в единицу времени.
Очень часто встречаются такие реакции, в которых в элементарном акте участвуют несколько молекул одного и того же вещества, например
/пА 4- лВ = рАВ.
В этом случае скорость реакции выразится общей формулой
v«=KfA]/7I[Bp. - (VI, 2)
Как видим, в приведенном варианте концентрация входит в уравнение скорости в степени, равной числовому коэффициенту химического уравнения реакции. Таким образом, уравнения (VI,1) и (VI,2) являются - математическим выражением закона действующих масс*
Количественная характеристика состояния динамического химического равновесия может быть выражена через так называемую константу химического равновесия, которая легко может быть выведена из следующих рассуждений. Для обратимой химической реакции типа mA 4- пВ рС -J- qD
скорость прямой реакции согласно закону действующих масс равна ^1=К1 [А]т [В]«» (VI', 3)
а скорость обратной реакции составляет
P2=K2[C]p(Df. (VI, 4)
— 221 —
В момент химического равновесия = vs,
т. е
Ki [АГ [В]«=к2 [Cf[Df.
(VI, 5)
Преобразовав уравнение (VI,5), можно написать для любой химической реакции, протекающей в растворах или в газообразной среде, выражение константы равновесия
i.EW., (V,.6)
^2 [А]"2 [В]л
Таким образом, константа химического равновесия К есть величина, численно равная отношению произведения действующих масс продуктов реакции к произведению действующих масс исходных реагирующих веществ. Причем стехиометрические коэффициенты являются знаком степени при соответствующих действующих массах.
Константа химического равновесия является характерной для каждой химической реакции величиной. Она, как показывает опыт, не зависит от концентрации реагирующих веществ, но изменяется с температурой.
Поскольку константа химического равновесия, как следует из уравнения (VI,6), равна отношению констант скоростей прямой и обратной реакций, она показывает, во сколько раз прямая реакция идет быстрее обратной при данных условиях и при данном произведении концентраций реагирующих веществ, равном единице.. Если К > 1, то быстрее идет прямая реакция, и, наоборот, если К < 1» быстрее идет обратная реакция.
Так, если величина Д равна 107, это означает, что при данных условиях прямая реакция идет в 10 миллионов раз быстрее, чем обратная, если же Д равно 10~5, обратная реакция идет в 100 тысяч раз быстрее, чем прямая, и т. д. Ниже приводятся константы некоторых равновесных систем
СО + Н2О СО2 + Н2 К == I (/ = 850°С) [COJ [Н2О] ' '
N2 + 3H2i±2NH3 К = , ,1N,^3J2 „ = 0,507 (/ = 400° С) 1^21 1^2]
Н2 + 12 2HI к =—ЩЦ— = 50,04 (/ = 448° С)
Если в реакции участвуют газообразные вещества, в выражение константы равновесия (VI,6) можно ввести вместо концентраций парциальные давления. Так, для гомогенной газовой химической реакции типа
mA 4- лВ рС qD,
где т, /?, р, q — стехиометрические коэффициенты, можно записать в следующем виде:
к Р&Р1
РаП
уравнение (VI,6)
‘ (VI, 7)
222
где Р — парциальное давление вещества, /<р — константа равновесия, выраженная через парциальные давления.
Знание величины константы химического равновесия позволяет производить очень важные расчеты: по известным концентрациям взятых веществ можно вычислить равновесные концентрации продуктов реакции и, таким образом, судить об эффективности процесса при данных условиях.
§ 67. Смещение химического равновесия. Синтез аммиака и получение азотных удобрений
Химическое равновесие является динамическим, т. е. подвижным. При постоянных внешних условиях — концентрация, давление, температура — химическое равновесие сохраняется как угодно долго. Изменение хотя бы одного из указанных факторов немедленно ведет к нарушению равновесия, смещая его в ту или иную сторону.
Влияние различных факторов на смещение химического равновесия отражено принципом Ле Шателье (1884). Этот принцип указывает направление смещения равновесия: при изменении в равновесной системе одного из параметров состояния (Р, Т, С) происходит сдвиг равновесия в направлении процесса, ведущего к ослаблению произведенного воздействия.
В качестве примера рассмотрим, как влияет изменение концентрации, температуры и давления на реакцию синтеза аммиака
N2 + ЗН2 2NH3 4- 92,048 кдж
Влияние изменения концентрации. Согласно принципу Ле Шателье увеличение концентрации одного из компонентов равновесной химической реакции приводит к сдвигу равновесия в сторону усиления той реакции, при которой происходит химическая переработка этого компонента. И, наоборот, уменьшение концентрации одного из компонентов приводит к сдвигу равновесия в сторону образования этого компонента.
В рассматриваемом случае увеличение количеств азота или водорода в системе усиливает прямую реакцию, т. е. равновесие смещается вправо, в сторону образования аммиака. С другой стороны, уменьшение, например, азота в этой системе вызовет смещение равновесия в сторону диссоциации аммиака.
Влияние температуры. Общее правило, определяющее влияние температуры на химическое равновесие, имеет следующую формулировку: повышение температуры способствует сдвигу равновесия в сторону эндотермической реакции и, наоборот, понижение температуры сдвигает равновесие в сторону экзотермической реакции.
Поскольку процесс синтеза аммиака сопровождается выделением тепла, при повышении температуры равновесие сдвигается в направлении справа налево и выход аммиака — конечного продукта реакции — уменьшается. Вот почему в заводских условиях синтез аммиака из водорода и азота стараются вести при наиболее низких температурах.
1 ~
223 —
Реакции, протекающие без тепловых эффектов, не смещают своегя равновесия при изменении температуры. Повышение температур» в этом случае приводит лишь к более быстрому установлению равно! весия, которое было бы достигнуто в данной системе и без нагревания! но за более длительное время. 1
Влияние давления. Как показывает опыт, давление оказывает! заметное влияние на смещение только тех равновесных реакций, в ко! торых участвуют газообразные вещества. При увеличении давлении равновесие смещается в сторону той реакции, которая сопровождается! образованием меньшего количества молей газообразных веществ, а при! понижении давления — в сторону образования большего количества! молей газообразных веществ. 1
Как видно из уравнения, реакция, идущая в направлении образо-] вания аммиака, сопровождается уменьшением объема, так как вместо] четырех молей исходных продуктов образуется только два моля аммиа-1 ка. Таким образом, увеличение давления способствует образованию! аммиака, а уменьшение давления способствует его разложению. I
Приложение принципа' Ле Шателье к технологическому процессу] синтеза аммиака дает возможность выбрать наиболее благоприятные* условия осуществления этого процесса, т. е. применение высоких дав-j лений при оптимальных температурах. В табл. 46 показано влияние^ давления на процентное содержание аммиака в газовой смеси различных температурах.
при
Таблица
Процентное содержание аммиака в равновесной смеси с азотом и водородом (при N2!H2 = 1 : 3)
46
Содержание аммиака в смеси, %, при давлении, атм
1000
100
300
300 —— 52,0 71,0 92,0
400 0,44 25,1 47,0 79,8
450 0,23 16,4 35,8 69,8
500 0,13 10,6 26,4 57,5
550 0,08 6,8 19,1 41,2
600 0,05 4,5 13,8 31,4
Из этой таблицы видно, что наиболее благоприятными условиями для выхода аммиака является давление 1000 атм и температура 300° С.
Синтез аммиака в заводских условиях осуществляется путем связывания азота воздуха с использованием катализаторов, в качестве которых берется смесь Fe и Fe2O3 с небольшой добавкой А12О3 и КОН, и является очень важным методом промышленного получения азотной кислоты и других азотных соединений.
Аммиак хорошо растворяется в воде и в таком виде (аммиачная вода) в настоящее время широко используется в сельском хозяйстве в качестве жидкого азотного удобрения. Промышленность выпускает
— 224 —
аммиачную воду двух сортов: первый содержит 25% аммиака (20,5% азота), второй—20% (16% азота).
Из аммиака в промышленности получают не только азотную кислоту и ее соли, но и другие соединения азота, которые являются ценными удобрениями. Окисление аммиака в заводских условиях осуществляется в специальных установках с применением в качестве катализатора сплава платины с 5—10% родия. Катализатор изготовляется обычно в виде тонкой сетки, сквозь которую продувается смесь аммиака с воздухом, содержащая примерно 12 об. % аммиака. При этом имеет место следующая химическая реакция
4NH3 -Ь 5О2 = 4NO + 6Н2О
Поскольку реакция окисления аммиака является экзотермической, смесь быстро нагревается. Максимальный выход окиси азота составляет около 98% от теоретического. Получение азотной кислоты из окиси азота в дальнейшем сводится к окислению NO до NO2 и растворению-последней в воде. Схему процесса можно представить в виде следующих уравнений
2NO+O2=2NO2
2NO2 + Н2О = HNO3 + HNO2
HNO2 -b NO2 = HNO3 + NO
Из азотной кислоты производят наиболее ценное азотное удобрение — аммиачную селитру NH4NO3, содержащую 34—35% азота. В заводских условиях ее получают путем нейтрализации 45—58%-ной азотной кислоты газообразным аммиаком
NH3 4-HNO3 = NH4NO3
с последующим упариванием раствора до состояния плава (94,5— 98,5% NH4NO3). Далее в зависимости от получаемой марки удобрения соль из плава кристаллизуют или гранулируют.
Из аммиака и азотной кислоты получают и другие азотсодержащие удобрения—мочевину СО (NH2)2, сульфат аммония (NH4)2SO4, хлорид аммония NH4C1, кальциевую селитру Са (NO3)2.
Применение азотных удобрений не только имеет решающее значение в повышении урожаев сельскохозяйственных культур на большинстве почв, но и улучшает его качество, увеличивает содержание белка в зерне и кормовых продуктах. Азотные удобрения вносят под все культуры. Особенно эффективно применение азотных удобрений в сочетании с другими минеральными удобрениями — фосфорными и калийными.
§ 68. Равновесие в гетерогенных системах.
Правило фаз
Принцип Ле Шателье применим не только к гомогенным, но и к гетерогенным системам. В качестве примера рассмотрим гетерогенную химическую реакцию восстановления двуокиси углерода
С СО2 2СО—41,22 ккал
8 Зак. 560
— 225 —
Эта система содержит твердую фазу (уголь) и газообразную (смесь паров СО2 и СО). Поскольку процесс восстановления СО2 идет с поглощением тепла, т. е. является эндотермическим, то согласно принципу Ле Шателье нагревание системы сместит равновесие в сторону увеличения выхода СО, а ее охлаждение будет сдвигать равновесие влево. Изменение давления также окажет существенное влияние на ход этой реакции, так как она в направлении слева направо сопровождается увеличением числа молей газообразной фазы. Повышение давления будет препятствовать протеканию прямого процесса, а уменьшение давления — способствовать ему.
Опыт показывает, что если гетерогенная реакция протекает без изменения числа молей газообразной фазы, то изменение общего давления не оказывает влияния на состояние равновесия реакции. В качестве примера можно указать такие реакции
Fo.0 4- СО Fe 4- СО2 МпО 4- СО Мп 4- СО3
Закон действующих масс применим к гетерогенным системам лишь с определенными допущениями. Область его приложения ограничена однородными частями равновесной системы. Равновесие же между неоднородными частями системы не подчиняется закону действующих масс.
Рассмотрим в качестве примера гетерогенную реакцию диссоциации карбоната кальция при высокой температуре в замкнутом сосуде
. СаСО3 СаО 4- СО2
*
В этой системе СаСО3 и СаО находятся в твердом состоянии, а СО2 — в газообразном. Если бы вышеуказанная реакция протекала как гомогенная реакция в газовой фазе (что можно допустить), можно было бы написать:
РСаО рсо2 ЛзаСО,
(VI, 8)
где Рсасо3, Рсо2, Рсао — упругости паров соответственно СаСО3, СО2 и СаО.
Однако упругость паров твердых СаСО3 и СаО является для данной температуры величинами постоянными, поэтому:
гСаСО3
Подставив это выражение в уравнение (VI, 8), получим:
\=¥со., (VI, Ю)
откуда
PCoJ=4L= const (VI, 11)
Ai
Равенство (VI, 11) показывает, что давление Рсо2 может иметь только одно определенное значение при данной температуре. Это дав-
— 226 —
называется давлением диссоциации СаСО3 или упругостью диссоциации. Оно не зависит от концентрации СаСО3 и СаО в данной системе.^ показывает> что давление диссоциации для реакции СаСО3^± —* СаО + СО2 достигает 760 мм рт. ст., т. е. 1 атм при температуре 880° С в то время как при 500° С это давление составляет всего лишь 0 11 мм рт. ст. Следовательно, при температуре 880° С начинается интенсивное разложение СаСО3.
Качественная характеристика гетерогенных равновесных систем, в которых не происходит химического взаимодействия, а наблюдается лишь переход составных частей системы из одного агрегатного состояния в другое, дается правилом Гиббса. Это правило основано на втором законе термодинамики и относится к системам, находящимся в состоянии истинного равновесия.
Введем основные понятия этого правила — фазы, компонента и степени свободы.
Фаза — совокупность частей гетерогенной системы, обладающих одинаковым составом и свойствами и отделенных от других составных частей ограничивающими поверхностями. При переходе через поверхности происходит скачкообразное изменение свойств веществ, образующих данную гетерогенную систему. Фаза может быть простой и смешанной.
Простая фаза состоит из одного химически индивидуального вещества (например, бензол в виде эмульсии в воде образует чистую фазу).
Смешанная фаза состоит из двух или более химически индивидуальных веществ (например, смеси газов, жидкие и твердые растворы).
Любая фаза может представлять собой сплошную массу (например, вода в суспензии глины в воде) или же совокупность более или менее крупных частиц (капельки воды, образующие туман в воздухе, составляют одну фазу).
Вещества, которые входят в состав системы и которые могут быть извлечены из системы и существовать вне ее, называются составляющими веществами. Например, водный раствор сахара образован из двух составляющих: воды и сахара.
В химических системах некоторые составляющие вещества могут образоваться в результате химического процесса, протекающего в системе. Такие составляющие вещества называются зависимыми. Первоначально же взятые составляющие систему вещества называются независимыми.
Компонентами называются независимые составляющие, наименьшего числа которых вполне достаточно, чтобы построить любую фазу в системе, находящейся в равновесии. В физических системах число компонентов равно числу составляющих систему веществ, так как последние не вступают между собой в химическое взаимодействие. В химических же системах число компонентов меньше числа составляющих веществ на число химических уравнений, по которым вещества, образующие систему, обратимо реагируют между собой при данных условиях существования системы.
8* — 227 —
Рассмотрим подсчет числа компонентов на конкретных примерах. Возьмем равновесную гетерогенную систему
СаСО3 СаО + СО2
Система химическая, составляющих веществ три. Число уравнений химических реакций в системе равно одному. Число компонентов в системе равно 3—1 = 2. Значит, система эта двухкомпонентная.
Другой пример. Рассмотрим систему из трех веществ, вг которой между составляющими протекает реакция
NH4C1 NH3 + HCI
т. е. реакция разложения NH4C1. Оба продукта реакции — NHS и НС1 находятся в газообразной фазе и равенство числа образовавшихся молей приводит к уравнению равенства концентраций (в моль!л)
[HC1] = [NH3]
В этом случае мы имеем дело с двумя уравнениями, которыми связаны составляющие вещества
NH4C1^±NH34-HC1
[HC1] = [NH3]
Таким образом, число компонентов в этой системе будет равно 3—2 = 1, где 3 — число составляющих веществ, 2 — число уравнений их связывающих. Как видим, данная система будет однокомпонентной.
Число степеней свободы характеризует вариантность системы, т. е. число независимых переменных (давление, температура и концентрация компонентов), которые можно произвольно изменять в некоторых пределах так, что число равновесных фаз в системе остается неизменным.
Например, состояние идеального газа характеризуется тремя параметрами: Ру V, Т. Из них любая пара является независимыми переменными, характеризующими термодинамическое состояние системы, которое можно изменить произвольно, без нарушения вида и числа фаз в системе. Иными словами, число степеней свободы для идеального газа равно двум, что соответствует двум произвольно заданным параметрам Р и Т; Р и С или С и Ту а третий будет определен из уравнения состояния.
Другой пример. Равновесная система лед вода пар существует при строго определенных параметрах: t° = 0,0098° С и Р == = 4,579 мм рт, ст. Эта система состоит из трех фаз и одного компонента (вода). Незначительное изменение хотя бы одного из параметров приводит к смещению равновесия в сторону изменения числа фаз системы. Так, повышение температуры будет способствовать переходу воды в пар, в результате чего число фаз уменьшится. К аналогичному нарушению равновесия системы приведет и изменение давления. Таким образом, для рассматриваемой системы степень свободы равна нулю, т. е. нет таких параметров, изменение которых не нарушило бы термодинамического равновесия данной системы.
— 228 —
Гиббс (1878) вывел следующее уравнение, выражающее условия фазового равновесия:
С=/( + 2-. Ф, (VI, 12)
4
пр с____число степеней свободы, К — число компонентов, Ф — число
фаз в системе.
Это соотношение известно как закон равновесия фаз и называется правилом фаз. Правило фаз имеет следующую формулировку: в равновесной многофазной системе число степеней свободы равно числу компонентов плюс два, минус число фаз.
В табл. 47 указано число степеней свободы различных систем при различных числах фаз и компонентов.
Таблица 47
Примеры фазовых равновесных систем
Число
Система « степеней
фаз компонентов свободы
Ненасыщенный раствор двух солей %
(раствор—пар) 2 3 3
Насыщенный раствор двух солей • 1
(раствор—осадок—пар) 4 3 1
Пересыщенный раствор трех солей (раствор—пар) 2 4 4
Раствор спирта и NaCI в воде (раствор—пар) 2 3 3
Смесь вода—а н ил и н —бензол—с п и рт (жидкость—пар) 4 4 2
Из уравнения (VI, 12) и данных табл. 47 можно сделать следующие выводы:
1) чем больше компонентов содержит система (при данном числе фаз), тем больше степень ее свободы;
2) чем большее число фаз находится в равновесии между собой в системе (при данном числе компонентов), тем меньшее число степеней свободы имеет система.
Лишенную степеней свободы систему (например, лед — вода — пар) предложено называть инвариантной. Если число степеней свободы С = 1, такую систему называют моновариантной, если С = 2, то би-вариантной, и т. д.
В настоящее время правило фаз является критерием равновесного состояния систем и помогает в решении ряда производственных задач, связанных с процессами в химических многофазных системах. Это правило широко применяется в различных областях химии и химической технологии, особенно в металлургии, в галлургии, в производстве различных стройматериалов, пластмасс.
Большой вклад в развитие учения о фазовых превращениях был внесен советскими учеными Н, С. Курнаковым, А. И. Цветковым и др.
— 229 —
§ 69. Применение закона действующих масс к равновесным системам «раствор — осадок».
Правило произведения растворимости |
I
Системы, содержащие жидкую и твердую фазы — «раствор — I осадок», имеют исключительно важное значение в аналитической 1 химии, почвоведении и геологии. Раствор, концентрация которого при 1 соприкосновении его с твердой фазой увеличивается, т. е. скорость I растворения превышает скорость кристаллизации, а состояние динамического равновесия еще не наступило, Является ненасыщенным. Если раствор находится в равновесии с твердой фазой растворенного 1 вещества, он является насыщенным. 1
Таким образом, между раствором и осадком в состоянии насыщения имеет место динамическое равновесие: в единицу времени в раствор переходит такое же количество вещества, какое выделяется из раствора.
Предположим, имеется насыщенный раствор малорастворимой соли КАн, которая в растворе полностью распадается на ионы. В ре- .] зультате между твердой фазой (КАн) и ионами (К+ и Ан-) устанавливается равновесие i
КАн К++Ан~* I
твердая фаза раствор
Обозначим через число катионов (К+), через п2 — число анионов (Ан-), приходящихся на единицу поверхности твердой фазы. Ско- ’ рость vly с которой катионы будут переходить в раствор, пропорциональна числу катионов, находящихся на поверхности осадка, т. е. j
Ч = К1П1. (VI, 13) j
где /G — коэффициент, постоянный при данной температуре.
В это же время будет идти обратный процесс — осаждение катионов на поверхности твердой фазы. Чем больше анионов на единице поверхности твердой фазы, тем больше будет осаждаться катионов. Количество осажденных катионов будет зависеть также и от концентрации их в растворе. Таким образом, скорость осаждения катионов (г2) прямо пропорциональна числу анионов, находящихся на этой поверхности, и активности катионов (пк+) в растворе, т. е, 1
^=К2и2ок+, (VI, 14)
где К2 — коэффициент, постоянный при данной температуре.
При растворении твердой фазы число катионов, переходящих в раствор, постепенно уменьшается, а число катионов, отлагающихся на поверхности твердой фазы, увеличивается. Наконец, наступает такой момент, когда скорость перехода катионов в раствор равна скорости осаждения их на поверхности твердой фазы, т, е. I
^1 = ^21
следовательно,
(VI, 15)
— 230 —
Рассуждая так, получим следующие выражения скоростей подобных процессов по отношению к анионам: скорость перехода анионов в раствор
(V 16)
скорость осаждения анионов из раствора на поверхности твердой фазы
(VI, 17)
Так как при наступлении равновесия КАн К+ + Ан’
v3=^c>4, (VI, 18)
то
^з«2= e. (VI,' 19)
ан
Перемножив левые и правые части равенств (VI, 15) и (VI, 19) между собой, получим:
Ki щ K3n2=K2 «2йк+ (VI, 20)
откуда
Ку К*
°к+ °ЛВ- к2 ка
= ПР.
(VI, 21)
Произведение концентраций, точнее активностей, ионов труднорастворимого электролита в его насыщенном растворе при постоянной температуре есть величина постоянная.
Это произведение активностей, ионов называется произведением растворимости (ПР), а сформулированное выше свойство насыщенного раствора труднорастворимого электролита — правилом произведения растворимости.
Например, в насыщенном растворе сульфата бария наблюдается следующее равновесие
BaSO4 Ва2+ + SO2- •
твердая раствор фаза
Произведение растворимости BaSO4 будет равно
Cl у i £Z 9
Ва2+ SO4
= ПР
BaSO4
1,08.Ю”1».
Если какой-либо труднорастворимый электролит диссоциирует с образованием двух или нескольких одинаковых ионов, например:
Кт Анп тКа+ 4- пАн6~",
где тип — соответственно число катионов и анионов, образованных из одной частицы электролита, а+ и Ь~ — валентности катиона и ^аниона, то выражение произведения активностей ионов примет вид
ПР — пт пп
Ка+ Ан^Г“*
(VI, 22)
— 231 —
Иными словами, при вычислении величины произведения растворимости активность каждого из ионов должна быть возведена в степень, равную числу одинаковых ионов, образующихся при распаде одной частицы. В табл. 48 приведены произведения растворимости некоторых малорастворимых соединений.
Таблица 48 «
Растворимость и произведение растворимости некоторых веществ (t = 25° С)
Формула вещества Произведение концентраций ионов Произведение растворимости Растворимость, моль/л
AgCl [Ag+] [C1-] l,56-10-10 1,2-10-5
HgCla (Hgi+nci-p 1.1-10-18 6,5-10-?
FeS [Fl2+] [S2-] 4,0-Ю-20 2-10-10
MnS я [Mn2+] [S2~] • 1,4-Ю-Ч 3,1.10-8
CaSO4-2H2O [Ca2+] [SO2-] 6,0-10-s 7,8-Ю-3
BaSO4 [Ba2+][SO2-] 1,08-10~10 1,0-10-5
CaCO3 ' [Ca2 + ][CO2-] 4,8-10-» 6,9.10-5
MgCO8 [Mg2 + ][CO|-] 1,0-10-5 3,2-10-3
FeCO8 [Fe2 + ][CO|_] 2,5-lQ-11 1 5,0-IO-6
MnCO, [Mn2 + ][CO|~] ( 8,8-10-11 9,3.10-в
Необходимо отметить, что в случае образования твердой фазы ее концентрация в выражении закона действующих масс принимается равной единице. Так, для гомогенной реакции какого-либо неэлектролита
A +Br±C + D
константа равновесия равна
„ °с «о
К=------. _ (VI, 23)
аА'«В
Если А и В представляют собой твердые вещества, то „ • .-И»
аС °D
Л = -^-=«с«о=ПР. (VI, 24)
Таким образом, в случае гетерогенных равновесных систем типа «осадок —раствор» произведение растворимости труднорастворимого соединения численно равно его константе химического равновесия.
Правило произведения растворимости имеет очень большое значение в химии. Используя его, можно заранее предвидеть направление реакции и возможность перехода того или иного иона из раствора в осадок или, наоборот, из осадка в раствор.
— 232 —
Если в растворе произведение активностей ионов труднорастворимого электролита меньше величины его произведения растворимости,
т. е.
а . а _ < ПР й , к+ Ан КАн
то в этом растворе может идти процесс растворения электролита и он не будет осаждаться. Если в растворе произведение а^+ адн“ равно произведению растворимости (ПРкан). такой раствор является насыщенным. Выпадение электролита в осадок начнется тогда, когда произведение активностей его ионов, находящихся в растворе, превысит величину произведения растворимости данного вещества, т. е.
а . а > ПР
К+ Ан КАн
-Произведение растворимости изменяется с изменением температуры. В табл. 49 показано изменение произведения растворимости для насыщенного раствора AgCl с повышением температуры.
Таблица' 49
Изменение произведения растворимости AgCl при повышении температуры
t, °C npAgCl t, °c npAgCl
5 10 25 0,21 10-10 0,37.10-10 1,56 10-10 50 100 13,2-10-10 21,5-10"10
Зная произведение растворимости какого-либо электролита, можно найти его растворимость не только в воде, но и в растворе*содержащем одноименный ион.
Одним из способов, повышающих полноту осаждения иона, является прибавление некоторого избытка осаждающего реактива. Поскольку произведение активностей иона в насыщенном растворе малорастворимого электролита есть величина постоянная, увеличение концентрации одного из ионов приводит к более полному удалению из раствора в осадок другого иона.
Например, прибавление ионов Ва2+ к насыщенному раствору BaSO4 приводит к уменьшению концентрации другого иона SO2-, т. е. к образованию твердой фазы. Количество BaSO4 в растворе уменьшится. Однако произведение активностей ионов (йва2+aso2-), находящихся в растворе, останется неизменным и будет равно произведению растворимости сульфата бария.
§ 70. Связь константы химического равновесия с максимальной работой реакции
Химическая система в состоянии равновесия отвечает наименьшему запасу свободной энергии. Любое отклонение от равновесного состояния сопровождается возрастанием свободной энергии в системе.
— 233 —
В этом случае, согласно второму началу термодинамики, возникает процесс самопроизвольного перехода данной системы в сторону уменьшения свободной энергии, за счет выделения которой возможно совершение полезной работы.
Максимальная работа изотермической реакции при постоянном давлении и постоянной температуре определяется изменением так называемого изобарного потенциала — AZ. Эта величина характеризует максимальную полезную работу реакции и указывает на направление протекания реакции,
Рис. 78. Схема «ящика» Вант-Гоффа
Максимальная работа реакции связана с константой равновесия простым соотношением, которое впервые было выведено Вант-Гоф-фом в 1885 г. Он доказал, что между константой химического равновесия (или произведением растворимости ПР) и разностью приращения свободных энергий существует логарифмическая зависимость: — AZ = — RT In Д', где R — газовая постоянная, Т — абсолютная температура по Кельвину.
Перейдем теперь к непосредственному доказательству связи между константой равновесия и максимальной полезной работой (убылью свободной энергии).
Пусть реакция N2 + 3H2Z^2 NH3 проводится при постоянной температуре в камере (ящик Вант-Гоффа) с тремя окошками, каждое из которых снабжено специальной полупроницаемой перегородкой, пропускающей только один из трех газов (рис. 78). При этом заслонка / может пропускать только азот, заслонка 2 — только водород, а заслонка —только аммиак. В самой камере находится равновесная смесь газов с равновесными давлениями Рн2, Рм2 и Pnh3. В цилиндрах, соединенных с окошками, под поршнями помещены индивидуальные газы N2, Н2 и NH3, находящиеся под давлениями соответственно Ръ Р2 и Р3, превышающими равновесные. Реакция образования аммиака проводится путем одновременного введения в камеру реагентов и вывода из камеры продукта при давлениях газов, равных (бесконечно близких) их равновесным парциальным давлениям» Для этого движением поршней в цилиндрах исходные давления газов Н2 и N2 предварительно и давление NH3 после вывода из камеры доводятся до равновесных. Размеры камеры принимаются столь большими,
— 234 —
что при введении в нее 1 моль азота, 3 моль водорода и удалении 2 моль аммиака равновесная их концентрация не изменится.
Работа Л, выполняемая идеальным газом при изотермическом его расширении, связана с изменением давления следующим соотношением:
Рл
Д = In—1, (VI, 25)
•2
где Рг и Р2— соответственно начальное и конечное давления газа. Согласно уравнению (VI,25) работа по переводу газа от давления
Рг к равновесному давлению Pn, выразится уравнением:
Л1 = /?Т1п-А-. , (VI, 26)
Аналогично этому работа по переводу трех молей водорода от давления Р2 к равновесному давлению Рн2 выразится уравнением:
A2~3RT . (VI, 27)
rN2
Удаление из камеры через заслонку 3 двух образовавшихся молей аммиака и перевод их от равновесного давления Pnh3 к давлению Р3 будет сопровождаться работой, равной
Д3 = 2RT In . (VI, 28)
Р 3
Учитывая, что введение реагентов и извлечение продукта под равными давлениями газов как вне, так и внутри камеры работой не сопровождается, а также суммируя максимальные работы А х и А 2, совершаемые при обратимом изотермическом расширении азота и водорода в цилиндрах до величины парциальных равновесных их давлений в камере, и затраченную работу А3 на сжатие двух молей аммиака после вывода его из камеры, получим величину максимальной работы всего процесса:
Яобщ= Л + Аг + А3=RT In + 3RT 1П -р- + 2RT In ,
p рз p p3
Лбщ = ^1п-^--/?Т1п -N* н*. (VI,29)
pl Pnh,
Так как реагирующие смеси во время операций находятся в равное веском состоянии, можно записать:
PNH
ЯР = р рз “> (VI, 30)
rN2rH2
где — константа равновесия. С учетом этого выражеция уравнение (VI, 29) примет следующий вид:
Ao6l4=RT}nKv-RT]n-^r=-^Z. (VI, 31)
Р1Р 2
— 235 —
Если состав реакционной смеси определять через концентрацию, для максимальной работы реакции при постоянном объеме получим:
2
Лобщ = /?Т1пКь—/?Т In' = - AZ. • (VI, 32)
сиЛн2
Соотношения (VI, 31) и (VI, 32) носят название уравнений изотерм реакции. Как видно из этих уравнений, максимальная работа реакции зависит от первоначального состава реакционной смеси и связана с константой равновесия. Для стандартных условий, за которые прини--маются парциальные давления или концентрации (активности) компонентов реакционной системы, равные единице (например, Рг = = Р2 =ж Рз = I атм или - Cn2 = Сн2 = Cnh3 = 1 моль!л), получается выражение для стандартной максимальной работы изотермической реакции:
Л0бщ=—= (VI, 33)
В этом уравнении константа равновесия может быть выражена различными способами (через парциальные давления^концентрации, сте-. пень диссоциации и т. д.).
Для изотермических реакций,. протекающих при постоянном давлении, максимальная полезная работа реакции определяется изменением изобарного потенциала (ЛОбщ = — AZ) и для стандартных условий (парциальные давления компонентов равны единице) носит название стандартного изобарного потенциала (AZ°):
AZ° = —ЯТ1п/<. (VI, 34)
Стандартные изобарные потенциалы образования (AZ°) для различных соединений вычислены и сведены в соответствующие таблицы. Зная эти потенциалы, можно определить не только величину и изменения свободной энергии любой реакции, но и константу химического равновесия, или произведение растворимости. Для условий Т = 273,16 + + 25° С после перевода натуральных логарифмов в десятичные уравнение (VI,34) можно записать:
AZ°=—1,364 IgK, откуда
'гк=-^а- ^35)
Если конечные продукты представляют собой твердую фазу, то
AZ°
ignp=———. (VI, 36)
1,364
§71. Применение закона действующих масс к растворам слабых электролитов
К растворам слабых электролитов, как к равновесным гомогенным системам, приложим закон действующих масс, Например, слабая уксусная кислота диссоциирует по уравнению
СНзСООН # СН3СОО - + Н+
— 236 —
В соответствии с законом действующих масс константа равновесия для этой реакции будет равна:
[СН3СОО-][Н+] [СНзСООН]
(VI, 37)
где в квадратных скобках указаны равновесные концентрации*.
Константа равновесия для случаев диссоциации молекул получила * название константы электролитической диссоциации. Эта константа у слабых электролитов не зависит от концентрации раствора (она зависит только от температуры). Так, константа диссоциации уксусной кислоты -при 25° С равна К = 1,85-10“5. Малая величина константы диссоциации свидетельствует о том, что равновесие в процессе диссоциации уксусной кислоты в растворе устанавливается при большом избытке недиссоциированных молекул. Независимость этой константы от разбавления хорошо подтверждается следующими данными:
Разбавление, л/моль . . . Константа диссоциации К ;
13,57 868,4 3474,0
1,845-IO-5 1,850.10-5 1,855-10-5
Константа электролитической диссоциации является характеристикой силы электролита. Чем константа диссоциации кислоты или основания больше, тем сильнее данный электролит. Например, уксусная кислота (К = 1,85* 10~б) в.о много раз сильнее цианистоводородной, у которой К = 4,8 ИО"10, и примерно в 10 раз слабее муравьиной, у которой К = 1,8«10”4. В качестве примера в табл. 50 приведены константы диссоциации некоторых слабых электролитов.
Таблица 50
Константы диссоциации некоторых слабых кислот и оснований при 25° С
Название электролита
Название электролита
К
Муравьиная кислота .
Уксусная кислота . . Пропионовая кислота Масляная кислота . . Угольная кислота .
Угольная кислота К2 • Азотистая кислота . .
1,8-10“4 • 1,85-10-5 1,34.10-5 1,48-10-5 3,5-10~7 5,0.10"11 4,00-10-4
Гидроокись аммония Анилин
Метиламин.........
Гидразин .........
Гидроокись свинца .
Фенол.............
Пиперидин.........
1,8-10-5 4,6-10-10
4,0-10"4 1,7.10-в
9,6.10-4 1,0-ю-10
1,6.10-3
Константа диссоциации имеет постоянное значение (при данной температуре).только для слабых электролитов. Подобного постоянства для сильных электролитов не наблюдается. -
Основываясь на законе действующих масс, можно вывести уравнение, которое связывает константу электролитической диссоциации К
* Концентрации ионов измеряются в г-ион!л, а концентрации недиссоциированных молекул — в моль!л раствора.
— 237 —
электролита со степенью его диссоциации и с концентрацией раство-
ра. В качестве примера возьмем какой-либо бинарный электролит 1 с концентрацией С. Степень электролитической диссоциации его обо- | значим через а. Тогда концентрация каждого из ионов в растворе будет I равна аС, а концентрация недиссоциированных молекул С — аС ~ 1
= С (1 —ос). Подставляя эти значения в уравнение (VI,37), получим: ]
к (аС)2 • к
1\ = ---=
(VI, 38)
1 — а
Выведенная формула является аналитическим выражением так называемого закона разведения. Этот закон впервые был выведен В. Оствальдом. Он применим только к слабым одно-одновалентным электролитам. Поскольку концентрация есть величина, обратная разбавлению, т. е. С = 1/V, где V — объем раствора в литрах, содержащий 1 г-же электролита, то с учетом разбавления (разведения) уравнение (VI, 38) примет следующий вид:
(VI, 39)
а2
17_________
(l-a)V’
Величина а у слабых кислот очень мала, поэтому ею в знаменателе формулы (VI, 38) можно пренебречь, считая, что 1 — а = 1. С учетом этого уравнение (VI, 38) примет более простой вид:
К = а2 С и а =
(VI, 40)
Формула (VI, 40) позволяет вычислить степень электролитической диссоциации слабой кислоты или щелочи, когда константа К известна.
По величине константы диссоциации слабые электролиты делятся на умеренно слабые (Д' = 10'2 — 10~4), слабые (К = 10"5— 10-9) и очень слабые (/< = 1О"10 и меньше).
Опыт показывает, что для сильных растворов закон разведения Оствальда не применим, так как для них величина К с увеличением концентрации непрерывно возрастает.
§ 72. Протолитическая теория кислот и оснований
Понятия «кислота» и «основание» имеют свою продолжительную историю идо сих пор не получили достаточно удовлетворительного определения. Многие реакции нейтрализации способны протекать в неводных растворах, при этом не происходит образования воды в результате соединения ионов Н+ и ОН“, хотя соли и образуются. Например, в реакции взаимодействия хлористого водорода с аммиаком, протекающей в бензоле, кислота теряет свои свойства, в результате чего образуется соль
При взаимодействии карбонатов с кислотами образуются соль и вода, т. е. результат получается такой же, как и при нейтрализации кислоты щелочью, однако ионы ОН“ в этих реакциях не участвуют, например
К2СО3 4- 2HNO3=2KNO3 + Н2О + СО*
— 238 —
или в ионной форме
СО*- + 2Н+= Н2СО3 = Н2О-J-CO2
На основании этих реакций были предложены новые определения кислот и оснований. Кислотой стали называть вещество, способное отщеплять протоны, а основанием — вещество, способное присоединять протоны.
Как известно, положительно заряженный ион водорода (или протон) обладает по сравнению с другими ионами особыми свойствами. В силу того, что он имеет очень малые размеры (диаметр протона примерно в 105 раз меньше диаметра других ионов), заряд его, концентрируясь в очень малом объеме иона, создает вокруг него сильное электрическое поле. Поэтому ионы водорода не способны сколько-нибудь долго существовать в растворе в свободном виде. Они очень быстро вступают во взаимодействие с молекулами воды, образуя так называемые ионы гидроксония
н+ + н2о^±н3о+
В силу своих свойств ион водорода обладает по сравнению с другими ионами наибольшей подвижностью.
Ион гидроксония следует рассматривать как кислоту, так как он способен отдавать протон, а воду — как основание. В общем виде подобные реакции можно представить следующим уравнением:
Кислота Основание 4- Н+
Кислоты и основания каждой такой простой пары называются српряженными. В качестве примера можно привести реакции электролитической диссоциации различных кислот, иона аммония и др.
NH+ # NH3+ Н + HSO^- #SO*- -I- H+ CH3COOH CH3COO- 4-H+
В более общем случае для того, чтобы кислота реагировала как кислота, необходимо наличие основания, способного присоединить протон, т. е. будет иметь место двойное равновесие типа:
Кислота! 4- Основание^ Кислота2 4- Основаниех
В качестве примера можно указать следующие реакции
СН3СООН 4- Н2О Н3О+ 4- СН3СОО-H2O4-NH3^±NH4+4'OH~
NH+ + Н2О^±Н3О+ 4-NH3
HCN4-H2O^tH3O+4-CN- и т. д.
Рассмотренные примеры подтверждают относительность понятий кислоты и основания. Одно и то же вещество в различных реакциях может выступать в роли кислоты и основания. В приведенных примерах в подобной роли выступает вода. В одном случае она присоединяет протоны, в другом — отдает их. Опыт показывает, что провести резкую грань между кислотой и основанием не всегда возможно. Более того, в ряде случаев в одном и том же веществе могут быть заложены два противоположных начала, так что эти вещества могут выступать и в качестве кислоты и в качестве основания.
Таким образом, реакции взаимодействия кислот с основаниями с образованием новых кислот и оснований называются протолитическими.
В системе из двух способных взаимодействовать с протоном веществ основанием всегда является то, которое прочнее его связывает, т. е. характеризуется большим протонным сродством. Например, в ряду NH3 — Н2О — HF протонное сродство убывает, и потому в смеси с аммиаком вода будет функционировать как кислота, а в смеси с HF — как основание.
Протолитическая теория кислот и оснований впервые была предложена Бренстедом (1923). Для кислот протонная трактовка совпадает с обычной и лишь расширяет ее на неводные растворы. В случае же оснований подход уже
— 239 —
будет совершенно иной. Например, едкий натр NaOH считается основанием с протолитической точки зрения не потому, что он способен отщеплять гидроксидион ОН~, а потому что этот ион способен присоединять протон (с образованием молекулы Н2О). Таким образом, в данном случае основанием является не NaOH, а именно ион ОН“.
Всякий растворитель, в состав молекулы которого входит протон, способный участвовать в.обменной реакции, имеет амфотерные свойства, проявляющиеся в собственной диссоциации. Собственная диссоциация характеризуется тем, что часть молекул растворителя ведет себя как кислота, а часть — как основание. Например:
Кислотах 4- Основание2 Кислота2 4- Основание!
Н2ОН2ОНЭО+ -f-ОН-
Так как в данном случае имеет место химическое равновесие, всякое изменение концентрации одного из двух ионов должно сопровождаться эквивалентным изменением концентрации другого. В водных растворах оба подхода — и классический, и протонный — практически приводят к одним и тем же результатам.
§ 73. Ионное произведение воды. Понятие о pH как показателе реакции среды
Вода является средой, в которой протекают самые разнообразные химические процессы. Она обладает хорошей растворяющей способностью и вызывает электролитическую диссоциацию многих растворенных в ней веществ. Более того, химически чистая вода сама является слабым электролитом и подобно кислотам, основаниям и солям частично диссоциирует на ионы
Н2О;±Н+ 4-Он-
Ионы водорода легко гидролизуются,. образуя ионы гидроксония Н3О+, которые в- дальнейшем изложении мы ради простоты будем обозначать как Н+.
Тщательно очищенная вода, как показал Кольрауш, обладает очень незначительной электропроводностью. Константа электролитической диссоциации воды, выражаемая согласно закону действующих
масс уравнением
[Н+][ОН~] [Н2О]
(VI, 41)
при 22° С составляет 1,8«10”16, а степень диссоциации 1,7-10~9. Учитывая столь незначительную степень диссоциации воды (из 555 млн. молекул ее только одна диссоциирует на ионы), концентрацию недис-социированных молекул можно приравнять к общему количеству воды и считать ее постоянной величиной, т. е. [Н2О] = const.
В связи с этим уравнение (VI.41) можно представить в виде
[Н+][ОН-]=К[Н2О]==КВ,
(VI, 42)
где Къ — ионное произведение воды.
Поскольку в литре воды находится 1000/18 — 55,6 моль Н2О, то, подставляя это значение, а также численное значение константы диссоциации воды в уравнение (VI,42), получим:
(НгО1 = 1,8.10-ie-55,6 = 10-14 (при22°С). (VI, 43)
— 240 —
Таким образом, в любом водном растворе, при постоянной температуре произведение концентраций (точнее, активностей) ионов водорода и гидроксид-ионов сохраняет вполне определенное, постоянное значение, равное ионному произведению воды.
Здесь необходимо еще раз напомнить, что вышеприведенная закономерность справедлива для очень разбавленных растворов, для которых активности практически равны аналитическим концентрациям. Для растворов электролитов обычных концентраций только величина произведения активностей ионов ан+ «он*, а не произведение концентраций [Н+] [ОН*] является постоянной величиной, т. е.
ан+%н-=Нн+]/[°н-1=/21н+]10Н-]-Кв, (VI, 44)
п 1 о м
где f — коэффициент активности.
В уравнении ионного произведения воды [Н+]или [ОН*] никогда не могут быть равными нулю, так как любая величина, умноженная на ноль, дает ноль. С другой стороны, если концентрация (активность) одного иона стремится к нулю, т. е. становится бесконечно малой величиной, то в этом случае концентрация (активность) другого иона должна быть бесконечно большой величиной, что не имеет никакого физического смысла. Однако если в воде будет растворена кислота и тем самым повысится содержание Н+-ионов, концентрация гид-роксид-ионов должна понизиться так, чтобы сохранилось постоянное значение 7(в, равное при 22° С всегда 10“и. Совершенно аналогичное действие произойдет при введении в раствор щелочи. От избытка гид-роксид-ионов значение /<в должно остаться прежним. Иными словами, численное значение Кв. не зависит от природы растворенного вещества, поскольку [Н+] и [ОН*] являются величинами сопряженными.
Опыт показывает, что диссоциация воды представляет собой эндотермический процесс, поэтому в соответствии с принципом Ле Щателье при повышении температуры равновесие будет смещаться в сторону образования ионов, что повлечет за собой увеличение /<в:
при 0°С Кв= 0,13 -10—14 = 10 —14 >8 9, Ю°с Кв = 0,36-10-14 = 10-14. 18° С Кв= 0,74-Ю“14 = 10“14»13, 22° С Кв = 1,00-10~14 = 10-14, 25° С Кв= 1,27-10~14 = 10—13 >90, ’
30°С Кв= 1,89-10-11= 10-73» 53° С Кв= 5,66- 10-и = 10-13.25, 80° С Кв = 34,00-10~14 = 10~12,4 7, 100° С Кв = 74,00-10-14 = 10-12>13.
Из курса неорганической химии известно, что все свойства, характерные для кислот, зависят от наличия в растворе ионов водорода Н+ (точнее, ионов гидроксония Н3О+), а свойства, характерные для щелочей,— от гидроксид-ионов ОН*. Поэтому растворы, в'которых [Н+] = [ОН*], называются нейтральными; если [Н + ] > [ОН ], растворы называются кислыми, и если [Нч ] < [ОН*], то раствор щелочной.
— 241 —
Чистая вода имеет нейтральную реакцию, потому что в ней-[Н+] = [ОН"] = 10"14 = 10-7 г-ион/л (при 22°С). Концентрации Н+,
с которыми приходится иметь дело на практике, обычно выражаются весьма малыми величинами. Так желудочный сок, являющийся самой кислой жидкостью организма человека, имеет концентрацию , Н+-ионов около 10"1 г-ион/л, концентрация Н+-ионов в слюне — око-ло 10-2 г-ион!л и т. д.
Как видим, характеризовать кислотность или щелочность раство- Ж ров числами с отрицательными показателями степени (10“5, 10-12) ®
практически неудобно. Поэтому реакцию водных растворов, показывающую степень их кислотности или щелочности, принято выражать не концентрацией (или активностью) ионов Н+ или ОН", а так называе- М мым водородным показателем pH. Ж
Показатель водородных ионов (pH) впервые был предложен как W условное обозначение в 1909 г. Зеренсеном (р — первая буква слова ж potenz); он определяется общей формулой
pH = -lgaH+, (VI, 46) В
а для разбавленных растворов я
рН=—Ig[H+]. (VI, 46)
Следовательно, водородным показателем называют величину, числен- Н но равную отрицательному десятичному логарифму концентрации (или активности) водородных ионов, выраженной в грамм-ионах на литр.
Для абсолютно чистой воды при 22° С pH = — 1g 10"7 = 7.
Для кислых жидкостей pH < 7, а для щелочных pH > 7. #
Логарифмируя ионное произведение воды, т. е. уравнение (VI, 42), ft получим I
[Н+][Он-]==Кв = 10-“ , 1
Ig[H+]4-lg[OH-]=lg Ю-i4.
Изменив знаки на обратные, запишем: — 1g [Н+ ] — 1g [ОН~] = = —lg 10"14. Учитывая, что — 1g [Н+] = pH, а — 1g [ОН“| = рОН, будем иметь:
pH 4-рОН =1g Кв = — 1g 10-14 = 14,00 (при 22^ С).
Если —1g Кв как. постоянную величину обозначить через р/<в, то выражение примет вид
pH + РОн = рКв = 14,00. (VI, 47)
Зависимость активности ионов Н+ от активности ионов ОН" и pH от рОН видна из табл. 51.
Как видно из этой таблицы, растворы кислот с ан+ больше 1 г-ион/л имеют pH больше нуля, а растворы щелочей с аон" больше 1 г-ион/л имеют pH больше 14. Практически можно считать, что pH изменяется в пределах от 0 до 14. Из данных таблицы видно, что при увеличении Пн+ в 10 раз pH уменьшается на единицу, увеличение ян+ в 100 раз влечет за собой уменьшение pH на 2 единицы, в 1000 раз — на 3 единицы и т. д.
— 242 —г
Таблица 51
Активность водородных и гидроксид-ионов и величины ’pH и рОН при 22° С
°Н + °он- Кв pH рОН рН + рОН
4,0 0,25-10~14 Ю-14 —0,6 14,6 14
1,0 10-14 10-14 о,о 14,0 14
10-1 Ю~1з 10-14 1,0 13,0 14
10~2 Ю-12 Ю-14 2,0 12,0 14
IQ,"8 ю-11 Ю-u 3,0 11,0 14
ю-4 Ю-ю Ю-14 4,0 10,0 14
10-5 10-в 10-U 5,0 9,0 14
ю-° ю-8 10-14 6,0 8,0 14
10"7 10~’ Ю“14 7,0 7,0 14
ю-° ю-° Ю-14 8,0 6,0 14
10-’ 10-5 Ю-14 9,0- 5,0 14
Ю-io io-4 Ю-14 10,0 4,0 14
10-11 10-а Ю-u 11,0 3,0 14
10-12 10~2 Ю-14 12,0 2,0 14
10-!3 ю-1 Ю-14 13,0 1,0 14
ю-14 1,0 Ю-14 14,0 0,0 14
0,25-10—14 4,0 Ю-14 14,6 -0,6 14
Зная концентрацию (активность) водородных ионов Н+ в растворе, можно определить водородный показатель (pH) и, наоборот, можно определить концентрацию Н+-ионов по известному значению pH. Для этой цели при отсутствии таблицы логарифмов можно пользоваться специально составленной таблицей, в которой промежуточные числа, не указанные в таблице, устанавливаются путем интерполяции (табл. 52),
Таблица 52
Пересчет числовых значений активности ионов водорода ан+ в единицы pH
ан+ pH ан + pH
1,12-10“<"+Ч л-^0,95 3,55-10“<"+Ч и 4-0,45
1,26-10—<"+ ’> /г чЬ 0,90 3,98-10“<"+Ч п 4 0,40
1,41-10“<"+Ч п 4>0,85 4,47-10“ <"+ ° п + 0,35
1,59-10“ <"+ ° п ф 0,80 5,01-10“<"+Ч п + 0,30
1,78-10“ 1"+° п 4-0,75 5,63-10“("+Ч п -^0,25
2,00-10“<" + Ч п + 0,70 6,31-10“<"+Ч п + 0,20
2,24-10“1"+Ч п + 0,65 7,08-10“1”+Ч п4-0,15
2,51-ю-1"+ч п + 0,60 7,94-10“<"+О п 4-0,10
2,82-10“<"+Ч п 4s 0,55 8,91-10-1'’+» п 4- 0,05
3,16-10-1"+° п 4х 0,50 — 1
— 243 —
2,24 • 10-6 г-ион!л. Вычислить pH. Для решения ноль-
Разберем правило пользования этой таблицей на конкретных примерах.
Примеры: 1. а
зусмся табл. 52.
pH=/z 4-0,65 = (6 —1)4-0,65 = 5,65.
’ = 2,63 • 10~10 г-ион!л. Вычислить pH. В данном случае пользуем-
2. а
ся методом интерполяции
pH = п + о,55 4- (2’ ^20О-2’ °-5- = П + 0,55 + 0,03 = (10-1) + 0,58 = 9,58. Л
2,82 — 2,51
1
3. Определить «н+, если pH = 8,8.
В данном случае п = 8, следовательно, ан+=1,59-10-<8+1) = 1,59.10~9 г-ион/л. Я
4. Определить «н+, если рН = 4,27. Вычисление pH проводим следующим ж образом:
(5,63—5,01)0,02
а , =5,63---—----- ------10-<4+1> = (5,63—0,25).10-5=5,38 г-ион/л.
н-г 0,05
Так же легко вычисляется pH раствора, если известна концентрация (или активность) гидроксид-ионов в этом растворе. Например, [ОН“] = 0,00001 г-ион!л. Вычислить pH раствора. Для вычисления используем уравнение (VI, 46):
pH = 14,0—рОН = 14,0—(—1g 10-4 = 14,0—5,0=9,0. &
* , J
§ 74. Роль концентрации водородных ионов Ц
в биологических процессах I
Как известно, каждый ион играет особую роль в различных биологических процессах, однако водородные ионы занимают особое положение среди других. Концентрация водородных ионов является одной из важных констант внутренней среды организмов. Так, pH крови человека равняется 7,36. Малейшее отклонение от этого значения ведет к серьезным нарушениям жизнедеятельности. Активность различных ферментов, а также специфика происходящих в тка-нях биохимических процессов тесно связаны с определенными довольно узкими интервалами pH. Например, пепсин желудочного сока активен при pH = 1,5— 2,0; содержащийся в слюне птиалин, ускоряющий процесс осахаривания крахмала, наиболее активен при pH — 6, 7, т. е. почти в нейтральной среде. В зависимости от pH среды ферменты могут катализировать совершенно различные реакции. Так, тканевые катепсины при реакции среды, близкой к нейтральной, катализируют синтез белка, а при кислой реакции — его расщепление. При отклонении величины pH от оптимальных значений активность ферментов, как показывает опыт, сильно снижается или даже вовсе прекращается, что в конечном итоге приводит организм к гибели.
Концентрация водородных ионов имеет большое значение в жизнедеятельности различных микроорганизмов. Например, сугубо бактериальный процесс усвоения атмосферного азота клубеньковыми бактериями на корнях бобовых растений идет при определенном значении pH почвы, равном примерно 7,2. Дифтерийный микроб развивается при pH в пределах 7,3—7,6, а кишечный микроб — при pH 6—7 и т. д.
— 244 —
Использование растениями различных питательных элементов из почвы также в значительной мере зависит от pH среды. Так, исследованиями Д. Н. Прянишникова было установлено, что нитратный азот (NO3) лучше усваивается растениями в слабо кислой среде при pH, равром примерно 5, а азот в аммиачной форме (NH^) лучше усваивается в нейтральной среде при pH ~ 7. Отношение I высших растений к pH почвы также различно. Для каждого растения установлена наиболее благоприятная, оптимальная реакция среды. В табл. 53 приведены оптимальные значения pH для различных сельскохозяйственных культур.
Таблица 53
Оптимальные значения pH для развития растений по данным Д. Н. Прянишникова
Растения Оптимальное значение pH Допустимое значение pH
Люпин Л—5 4—6
Картофель .... 5 4—8
Овес 5-6 4—8
Рожь О 1 4—7
Лен 5—6 4—7
Растения Оптимальное значение pH Допустимое значе ние pH
Пшеница .... 6—7 5-8
Горох 6—7 5 8
Клевер 6-6,5 5—8
Свекла . -. . . . 7 6-8
Кукуруза .... 7 00 1
Люцерна 7—8 00 1 о
Как видно из этой таблицы, сельскохозяйственные растения меньше страдают от кислотности, чем от щелочности почвы.
Нейтральной реакцией с небольшими отклонениями в кислую или щелочную сторону обладают черноземы;подзолистые, дерново-подзолистые и болотные почвы имеют, как правило, кислую реакцию среды; засоленные почвы, наоборот, отличаются щелочной реакцией и т. д. Необходимо отметить, что по видовому составу луговой растительности можно приблизительно определять pH почвы. Так, присутствие растения «щучки» (Deschampsia flexuosa) указывает на то, что pH этой почвы находится в пределах 3,5—3,9. Преобладание осоки (Carex) указывает на pH почвы в пределах 4,5—4,9; а преобладание «мать-и-мачехи» (Tussilago farfa-ra) — на pH 7,5—7,9 и т. д. ,
Многие агрохимические приемы направлены на создание в почве наиболее благоприятной для растений и почвенной микрофлоры реакции среды. Например, путем внесения в кислые почвы извести (СаСО3) добиваются устранения избыточной кислотности, вредной для растений и некоторых полезных в земледелии микроорганизмов.
Внесение в почву гипса (CaSO4) и железного купороса (FeSO4) способствует устранению избыточной щелочности почвенного раствора.
Не менее важное значение имеет реакция среды и в области технической биохимии. Например, контроль технологического процесса и качества готовой продукции по величине pH имеет большое значение во многих отраслях пищевой, мясной и молочной промышленности, в частности в хлебопечении, сыроварении, пивоварении, переработке молочных продуктов, фруктов, овощей, изготовлении кож, табака и во многих других отраслях народного хозяйства. Величина pH влияет на протекание важных ферментативных процессов в гидролизной промышленности, крахмало-паточном производстве, виноделии.
Не менее важное значение имеет водородный показатель в химической технологии. В частности, под влиянием pH могут изменяться растворимость, фильтрация, вязкость, поверхностное натяжение, осмотическое давление, набухание и другие свойства. Вот почему определение концентрации водородных ионов нашло применение во всех областях не только биологии, но и химии, агрохимии, почвоведения, физиологии, бактериологии, медицины и в других областях науки и практики. 4 *
— 245 —
§ 75. Активная и общая киздотность растворов. Кислотность и щелочность почв
Любая кислота или щеЛ)чь содержит в свободном состоянии определенное количество ионов Н+ (точнее, Н3О+) или ОН~. При нейтрализации кислоты щелочью ионы связываются в молекулу воды. При этом все катионы водорода, содержащиеся в растворе, участвуют в процессе нейтрализации.
Таким образом, общее количество катионов водорода, содержащееся в единице объема раствора, носит название общей или титруемой кислотности. Количество же свободных гидратированных ионов водорода составляет так называемую активную кислотность раствора.
И та и другая кислотность выражаются обычно в г-ион!л или, что то же самое, в г/л. В табл. 54 сопоставлены титруемая (общая) и активная кислотность для 1 н. растворов некоторых кислот.
Таблица 54
Активная и общая кислотность некоторых кислот
Кислоты Степень диссоциации в 1н. растворе Общая кислотность, г/л Активная кислот ность, г/л
НС1 0,79 1 0,79
HNO3 0,82 1 0,82
HCN 0,000036 1 0,000036
CH3GOOH . . 0,0034 1 0,0034
Из данных этой таблицы видно, что различные кислоты при одинаковой общей кислотности довольно резко отличаются по активной кислотности.
Концентрация ионов Н+ (в г-ион/л), а также pH для одноосновных слабых кислот и оснований могут быть вычислены теоретическим путем» Покажем это на конкретном примере с уксусной кислотой, которая диссоциирует по уравнению
СН3СООН^СН3СОО- +Н+
Применяя к этой реакции закон действующих масс, получим: [Н+] [СНзСОО-]
[СНзСООН] ’ (VI»48)
где /< — константа электролитической диссоциации уксусной кислоты. Из уравнения диссоциации уксусной кислоты имеем
[Н+] = [СН3СОО-].
Стало быть, уравнение (VI, 48) можно написать так
„ [Н+]2
“ [СНзСООН]* (V!, 49)
Если обозначить через а степень диссоциации уксусной кислоты,
— 246 —
а через С аналитическую концентрацию ее, получим: (Н+]=[СН8СОО-]=Са, 1СН3СООН]=С— Са= (1—а) С.
Уравнение (VI,49) можно представить в следующем виде:
откуда
[Н+]=/КС (1-а). (VI, 51)
Учитывая, что для слабых кислот а 1, можно без большой погрешности принять (1 —а) « 1, тогда
[Н+]=/КС. (VI, 52)
Прологарифмировав это выражение и поменяв знаки на обратные, получим окончательную формулу для вычисления pH слабых одноосновных кислот
lgIH+] = 4 igK +4 lgС, (VI, 53)
откуда
рН=—^-IgK—-у IgC.
Обозначая — 1g К через р/<, получим:
PH = 4-p/(-4*lgC- № 54)
Совершенно аналогичная формула получается и в случае одноосновного слабого основания, например
NH4OH#NHJ+ОН-
В данном случае согласно уравнению (VI,54) получим: РОН=4-рК-4 lgС, (VI, 55)
где д — константа диссоциации слабого основания, С — концентрация этого основания. С учетом уравнения (VI,47) окончательная формула для вычисления pH одноосновных слабых оснований будет иметь следующий вид:
рН=14 — 4-pK + 4-lgC. (VI 56)
Пример. Вычислить pH 0,1 н. раствора уксусной кислоты. Поскольку константа диссоциации уксусной кислоты К = 1,85 • 10~5, для решения воспользуемся формулой (VI, 53):
рН = -4“ lg (1,85-IO-®)—ig 0,1 =4-4,73 + 0,5 = 2,86.
В настоящее время советские и зарубежные ученые получили много новых данных о природе почвенной кислотности и путях ее устранения. При агрохимической характеристике почв различают три вида почвенной кислотности: актив-
— 247 —
ную (или актуальную), обменную и гидролитическую. Последние два вида объединяются общим названием потенциальной кислотности.
Активная кислотность. Под активной кислотностью понимается концентрация (точнее активность) водородных ионов в почвенном растворе или в суспензии. Суспензия готовится путем обработки почвы пятикратным количеством дистиллированной воды. После пятиминутного взбалтывания почва отделяется от раствора путем центрифугирования и уже потом в этом растворе определяется pH либо колориметрическим, либо потенциометрическим методами. Ввиду исключительно большого значения реакции для плодородия почв, почти каждое агрохимическое и почвенное исследование включает в обязательном порядке определение pH водной вытяжки.
Обменная кислотность. Эта кислотность обнаруживается в результате обмена поглощенных почвенными коллоидами (высокодисперсной частью почвы) ионов водорода или алюминия на катионы солевого раствора, которым обрабатывается почва.
Если привести почву во взаимодействие с раствором нейтральной соли, например с хлоридом калия, произойдет вытеснение ионов водорода в раствор. При этом образуется соляная кислота НС1, а эквивалентное количество калия поглотится почвой. Эту реакцию можно представить в виде следующей схемы:
[ПК] Н 4- КС1 [ПК] К 4- НС1
где [ПК] — почвенный поглощающий комплекс.
Нетрудно догадаться, что чем больше ионов водорода будет вытеснено из почвенного поглощающего комплекса, тем больше будет обменная кислотность почвы. То же самое произойдет с почвами, содержащими обменный алюминий. Эту реакцию можно представить следующим образом:
[ПК] А14-ЗКС1 ;± [ПК] К3 4-А1С13
А1С13 + ЗН2О А1 (ОН)3 4- ЗНС1
Для определения обменной кислотности почву обычно обрабатывают 1 н. раствором хлорида калия. В центрифугате (или фильтрате) определяют величину pH, . т. е. концентрацию водородных ионов и общее количество образовавшейся кислоты, путем титрования ее щелочьючв присутствии фенолфталеина в качестве индикатора. Обычно обменная кислотность почвы бывает активной, а солевая вытяжка имеет более низкое значение pH/чем почвенный раствор. В качестве примера в табл. 55 приведены результаты измерения pH различных почв.
Таблица 55
Значение pH некоторых типов почв
(по Н. И. Горбунову)
ПОЧБЫ Глубина взятия образца, см. pH вытяжки
водной солевой
Дерново-подзолистая 5—11 16—24 58—68 90-100 5,0 5,3 5,0 5,2 3,8 4,2 3,6 3,4
Чернозем обыкновенный О ООО 1 1 1 ь—1 СП ►—‘ ООО о 7,0 7,1 7,4 6,1 6,2 6,3
Солонец » 0—20 20—40 40—60 80—100 7,5 7,7 7,6 8,2 6,6 6,8 6,5
Краснозем 0—10 40—50 155—160 4,9 5,0 5,0 со со —< । ТГ Tj" ^7
— 248 — »
В почвах, имеющих щелочную реакцию водной суспензии (pH>7), обменную кислотность искать не следует: ее там нет, но в слабокислых и близких к нейтральным почвам незначительная обменная кислотность иногда обнаруживается.
Гидролитическая кислотность. Эта кислотность образуется в результате обработки почв раствором соли сильного основания и слабой кислоты, например, апетата натрия. Такая соль называется гидролитически щелочной, так как в водном растворе она имеет щелочную реакцию:
CHgCOONa + Н2О СН3СООН + NaOH
При взаимодействии ацетата натрия с почвой могут происходить различные реакции в зависимости от содержания в ней поглощенных ионов алюминия и водорода. Реакция взаимодействия ацетата натрия с почвой протекает по следующей схеме:
[ПК] Н 4- CH3COONa [ПК] Na 4- СН3СООН
или по схеме:
[ПК] А1 4- 3CH3COONa [ПК] Na3 4- Al (CH3COO)g Al (СН3СОО)з4-ЗН2О^ А1 (OH)34- ЗСН3СООН
Количество образующейся кислоты соответствует величине гидролитической кислотности почвы.
Гидролитическая кислотность обычно бывает больше, чем обменная. По мнению Л. Н. Прянишникова, нейтральная соль КС1 (в случае определения обменной кислотности) вытесняет лишь часть поглощенного водорода (или алюминия), а щелочная соль CH3COONa вытесняет почти весь поглощенный водород (алюминий), именно поэтому гидролитическая кислотность бывает больше обменной.
Щелочность почвенного раствора. Если водная суспензия почвы имеет pH больше 7, в ней присутствуют соли щелочных металлов. В зависимости от того, какими солями вызывается щелочность, различают несколько ее видов: щелочность от присутствия карбонатов щелочных металлов (Na2CO3, -К2СО3), щелочность от их бикарбонатов (NaHCO3, КНСО3) и щелочность от бикарбонатов щелочноземельных металлов [Са(НСО3)2, Mg(HCO3)2].
Общая щелочность вызывается всеми упомянутыми солями. На практике ее определяют обычно путем титрования водной вытяжки или почвенного раствора кислотой в присутствии различных индикаторов.
X
Таблица 56
Щелочность почв и значение pH (по Н. И. Горбунову)
Почва Глубина взятия образца, см pH водной вытяжки Щелочность, мг-экв на 100 г почвы
от карбонатов общая
0—5 8,3 0,1 0,62
Серо-коричневая 10—20 8,3 0,2 0,57
30—40 8,3 Нет 0,60
' 90—100 8,4 » 1,00
0—10 8,2 0,2 1,00
Серозем 15—25 8,3 0,1 0,56
50—60 8,2 0,1 0,46
2—10 8,5 0,2 1,00
Солончак 12—25 25—30 9,0 8,5 0,3 0,02 1,20 0,80
40—60 8,4 0,1 0,62
— 249 —
Так, по количеству кислоты, пошедшей на нейтрализацию, определяют: а) щелочность от карбонатов щелочей — по изменению окраски фенолфталеина от розового к бесцветному; б) щелочность общую — по изменению цвета метилового оранжевого от желтого к розовому и т. д. В табл. 56 приведены величины щелочности некоторых типов почв.
§ 76. Реакция среды в растворах солей, гидролиз
Опыт показывает, что реакция среды в водных растворах зависит не только от наличия в них кислот или оснований, но также и от присутствия некоторых солей. Многие соли, растворяясь в воде, способны смещать реакцию среды в ту или иную сторону. При этом происходит химическое взаимодействие между ионами соли и ионами Н+ и ОН- воды, сопровождающееся образованием слабых кислот или слабых оснований. Эта реакция получила название гидролиза соли.
Таким образом, гидролиз — это процесс взаимодействия ионов соли с ионами воды. В результате гидролиза растворы большинства солей имеют кислую или щелочную реакцию. В качестве иллюстрации приведем три наиболее типичных случая реакции гидролиза солей.
1. Соль, образованная сильным основанием и слабой кислотой. В водном растворе она подвергается гидролизу, в результате чего образуется слабая кислота и сильное основание, и реакция среды становится щелочной. Примером может служить гидролиз ацетата натрия, протекающий по схеме
CH3COONa СНзСОО- + Na
Н2о#н+ 4-ОН-П сНзСоон
В данном случае гидролиз ведет к увеличению концентрации ионов ОН- в растворе. Например, pH 0,1 н. раствора ацетата натрия равен 9,9.
2. Соль, образованная слабым основанием и сильной кислотой. Примером может служить хлорид аммония NH4C1. При гидролизе его равновесие между недиссоцииро-ванными молекулами воды и ионами (Н2О Н++ ОН~), как и в предыдущем случае, тоже нарушается. В результате гидролиза образуется слабое основание и избыток ионов водорода. Гидролиз хлорида аммония можно изобразить следующей схемой:
nh4ci#nh+ 4-С1-
Н2О^ОН~ + Н+
Н г.
nh4oh
Связываясь с гидроксид-ионами воды, ионы NH+ образуют слабодис-социирующую гидроокись аммония, в растворе появляется избыток ионов водорода, в результате чего реакция смещается в кислую сто-рону« Так, pH 0,1 н. раствора NH4C1 равен 5,3.
3. Соль, образованная слабой кислотой и слабым основанием. Этот вид гидролиза можно рассмотреть
— 250 —
на примере ацетата аммония CH3COONH4, диссоциирующего на ионы NH+ и СН3СОО“. В данном случае с ионами Н+ и ОН“ воды реагируют и катионы и анионы соли
CH3COONH4 # NH+ + СН3СОО’
Н2О^±ОН“4-Н +
NH4OH СНзСООН
Как видим, в результате гидролиза ацетата аммония происходит образование двух слабых электролитов: гидроокиси аммония и уксусной кислоты. Так как константы диссоциации уксусной кислоты и гидроокиси аммония очень близки по величине
^сн8соон —1 >75-10 6 И'KNH*OH—1,79-10 5 *
(при 25° С), то и образующиеся в результате гидролиза кислота и ос нование практически равны по силе. Раствор такой соли оказывается нейтральным. Так, например pH 0,01 н. раствора CH3COONH4. равен 7.
В заключение отметим, что. соли, образованные сильным основанием и сильной кислотой, гидролизу не подвергаются, Ионы таких солей не образуют с ионами Н+ и ОН~ воды слабодиссоциирующих или труднорастворимых соединений, В этом случае равновесие между недиссоциированными молекулами и ионами воды не нарушается и растворы их остаются нейтральными (pH практически равен 7). К этой группе относятся соли: NaCl, КС1, NaNO3, ВаС12 и дре Соли, отнесен-
Таблица 57
Характер среды в растворах различных солей
Сила основания или кислоты, образовавших соль Происходит ли гидролиз Реакция раствора соли pH раствора СОЛ!
Сильное Сильная Нет Нейтраль- ная 7
KCl, NaNOg, KCIO4, ВаС12, Na2SO4, КМпО4 и др.
Слабое Слабая Да, в сильной степени Близка к нейтральной «7
(NH4)2S, AI2S3, (NН4)2СО3 и др.
Сильное Слабая Да Щелочная ^>7
KCN, CH3COONa, К2СО3идр-
Слабое Сильная » Кислая 7
NH4CI, Fe(NO3)3, A12(SO4)3 и др-
*— 251 —
ные к группе (3), как уже отмечалось, в водных растворах также имеют pH, близкое к 7. Однако они резко отличаются от солей, образованных сильным основанием и сильной кислотой. Последние гидролизу не подвергаются и потому pH раствора не меняют. Соли группы (3) сильно гидролизуются, образуя в водных растворах определенные и очень устойчивые pH, близкие к 7.
В табл. 57 показан характер среды в растворах различных солей.
Рассмотрим, чему равняется pH гидролитически щелочных или гидролитически кислых солей. В качестве примера возьмем гидролиз ацетата натрия
CH3COONa 4- Н2О СН3СООН 4- NaOH
Ионное уравнение гидролиза этой соли будет иметь вид
СН3СОО- 4- Н2О СН3СООН 4- ОН“
Следует принять во внимание, что соль и основание как сильные электролиты полностью диссоциированы, а слабая кислота практически* недиссоциирована.
Применяя закон действующих масс к данной равновесной реакции, получим
[СНзСООНПОН-]
[СН3СОО-][Н2О] ’ ' }
Поскольку концентрация воды практически остается величиной постоянной, ее можно объединить с константой равновесия, т, е.
КГ=К[Н2О] =
[СНзСООН] [ОН-j [СН3СОО-1
(VI, 58)
где Kv— константа гидролиза, равная К [Н2О].
Из уравнения диссоциации слабой уксусной кислоты
CHsCOOH сн3соо- + н+
на основании закона действующих масс можем записать
_ [Н+][СН3СОО-]
, к [СНзСООН]
откуда
|СН,СООН1= .
Ан
Подставляя это выражение в уравнение (VI,58), получим:
_ [Н + ][ОН~][СН3СОО-]
г КГ[СН3СОО-]
(VI, 59)
(VI, 60)
Произведя сокращения и учитывая, что [Н+] (ОН-] == Кв, уравнение (VI,60) примет следующий вид:
Кв
Кг=~Г‘ (VI, 61)
Ак
— 253 —
Таким о разом, константа гидролиза гидролитически щелочной соли равна отношению ионного произведения воды к константе диссоциации слабой кислоты, образованной в результате реакции гидролиза. С учетом этого уравнение (VI,58) можно представить в таком виде?
[ОН-] [СНзСООН] Кв ~ [СН3СОО-] “ Кк •
(VI, 62)
Поскольку при гидролизе ацетата натрия образуется равное количество молекул уксусной кислоты и ионов ОН-, т. е. [СН3СООН] = [ОН-], уравнение (VI,62) можно переписать так:
[ОН-р _ Кв
[СН3СОО~] Кк’
(VI, ТЗ)
Так как по сравнению с общей концентрацией С соли ацетата нат
рия гидролизу подвергается лишь незначительная часть анионов СН3СОО“, без большой погрешности можно принять, что в растворе [СН3 СОО-] = С, где С — аналитическая концентрация CH3COONa. Тогда уравнение (VI,63) примет следующее выражение:
Кв _ [ОН-]2
Кк С
(VI, 64)
Из уравнения ионного произведения воды можем записать:
>он-'-]й+
Подставляем это выражение в уравнение (VI, 64):
Кв Кв
Ки“ С [Н+р ’
откуда [Н+]
(VI, 65)
I
Логарифмируя это выражение и меняя знаки, получим окончательное уравнение для вычисления pH гидролитически щелочной соли:
-lg[H+] = —J-lgKB — -i-K„ + 4 IgC,
или, учитывая, что при 22° С Кв = 10-14 и — lg/CK = р/f, узнаем
pH = 7+-^- РК + 4- lg С. (VI, 66)
& £
Например, нужно вычислить pH 0,1 н. раствора CH3COONa.
Для решения пользуемся формулой (VI,66). Поскольку константа диссоциации уксусной кислоты Кк — 1,85 • 10~“б, а концентрация С ~ 0,1 н., подставляя еги данные в формулу, получим:
рН=7+~( — 1g 1,85.10-»)+-|- 1g 0,1=7+2,36—0,5=8,86. £ £
Совершенно аналогичный вывод формулы для вычисления pH можно сделать и для гидролитически кислой соли. Формула будет
— 253 —
такая же, только поменяются знаки на обратные:
pH =7—у рК—i-lgc, (VI, 67)
где p^ — отрицательный логарифм константы диссоциации слабого основания, С — концентрация гидролитически кислой соли»
§ 77. Буферные растворы и буферное действие
Как показывает опыт, разбавленные растворы сильных кислот и оснований, обладающие слабокислой или слабощелочной реакцией, характеризуются непостоянством pH. Однако смесь, например, уксусной кислоты и ее соли CH3COONa обладает способностью сохранять постоянство pH. Можно к этой смеси добавить небольшое количество кислоты или щелочи, а также разбавить ее, но pH раствора при этом почти не изменится. Такое свойство растворов сохранят.ь~определенное_ значение pH называется буферным действием 1А растворы, обладающие буферным действием, получили название буферных растворов или буферных смесей.
Буферные растворы по своему составу бывают в основном двух типов. Они могут состоять/? из слабой кислоты и ее гидролитически щелочной соли неслабого основания и гидролитически кислой соли этого основания/
В качестве иллюстрации можно привести следующие буферные смеси:
СН3СООН 4- CHgCOONa—ацетатный буфер;
Н2СО3 4- NaHCO3—бикарбонатный буфер;
INH4QH 4- NH4CI — аммиачный буфер;
NaH2PO4 4- Na2HPO4—фосфатный буфер.
Последний буфер, как видим, состоит из смеси двух солей, одна из которых — эднэзамещенная, . вторая — двухзамещенная соль фосфорной кислоты. Причем первая соль (NaH2PO4) играет роль слабой кислоты.
Буферным действием могут обладать растворы, состоящие из анионов разных слабых кислот, таких, как фосфат-цитратный буфер
Na2HPO44- С6Н8О7
Сущность буферного действия смеси слабой кислоты с ее солью можно рассмотреть на примере ацетатного буферного раствора. В этом растворе происходят следующие реакции электролитической диссоциации
СН3СООН^±СН3СОО-4-Н+ | .
CH3COONa #СН3СОО- 4- Na+ J (а)
Поскольку степень диссоциации кислоты очень мала, в растворе преобладают ее недиссоциированные молекулы. Ацетат натрия, являясь сильным электролитом, диссоциирует полностью на ионы СН3СОО“ и ионы Na+4 Таким образом, в ацетатной буферной смеси
— 254 —
присутствие в большом количестве анионов СН3СОО“ смещает равновесие при диссоциации уксусной кислоты в сторону образования ее молекул. Причем, диссоциация уксусной кислоты может быть настолько подавленной, что кислоту можно считать практически недиссоци-ированной. В результате этого активная кислотность смеси очень мала. Добавление кислоты или щелочи к ацетатной смеси не вызывает существенного изменения концентрации водородных ионов в растворе. Так, при добавлении соляной кислоты к ацетатному буферу происходит реакция обменного разложения с одним из компонентов смеси (CH3COONa)
CH3COONa + НС1 -> NaCl -f- CH3COOH (6)
Na+" Cl-
Как видим, сильная кислота в результате этой реакции заменяется * эквивалентным количеством слабой кислоты. В соответствии с законом разбавления Оствальда (VI,38) увеличение концентрации уксусной кислоты понижает степень ее диссоциации, в результате чего концентрация ионов водорода в буферном растворе увеличивается очень незначительно.
Так же незначительно изменяется pH буферного раствора при добавлении к нему небольшого количества щелочи. При этом щелочь реагирует с уксусной кислотой (реакция нейтрализации), в результате чего гидроксид-ионы связываются с ионами водорода с образованием молекул воды
СНзСООН -Ь NaOH -> Н2О +£H3COONa (в)
CH3COO-+Na+
В конечном итоге этой реакции добавляемая щелочь заменяется эквивалентным количеством слабоосновной соли, которая влияет на реакцию среды в значительно меньшей степени, чем NaOH, Поскольку в результате этой реакции уксусная кислота расходуется, можно было бы ожидать значительного снижения содержания ионов Н+в Однако вместо прореагировавших ионов кислоты Н+ и СН3СОО- за счет потенциальной кислотности образуются новые ионы Н+ и СН3СОО~, и активная кислотность смеси (pH) почти не изменяется.
Как показывает опыт, каждая из буферных смесей характеризуется определенной концентрацией водородных ионов, которую буферная система и стремится сохранять при добавлении к ней кислоты или щелочи. Рассмотрим на примере ацетатной буферной смеси, что же определяет ее pH.
В соответствии с законом действующих масс константа диссоциации уксусной кислоты равна:
[Н + ][СН3СОО~~]
[СНзСООН]
откуда
[Н+]=К
[СНзСООН]
[СНзСОО-] •
(VI, 68)
255 —
Это равенство справедливо для раствора, в котором содержится только одна уксусная кислота. Как уже отмечалось, добавление к ра-створу уксусной кислоты ацетата натрия подавляет ее диссоциа-1 цию, в результате чего концентрацию молекул недиссоциированно^ СН3СООН можно без больших погрешностей принять равной общей концентрации кислоты, т. е.
[СН3СООН] = [кислота].
(VI, 69)
Учитывая, что соль CH3COONa как сильный электролит в водном-растворе диссоциирована полностью, можно принять, что общая кон-центрация аниона СН3СОО~ практически равна аналитической концентрации соли в данной буферной смеси, т. е.
[СН3СОО-]=[соль]. (VI,70)
Подставляя значения (VI,69) и (VI,70) в уравнение константы диссоциации (VI,68), получим:
„[кислота]
[Н + ]=К -7---А (VI, 71)
[соль]
Логарифмируя это выражение и меняя знаки, запишем:
— 1g [Н + ] — —1g К + 1g [соль] — 1g [кислота]
или
pH = рК 4-1g [соль] —1g [кислота], • (VI, 72)
где рК — отрицательный логарифм константы диссоциации уксусной кислоты.
Отметим, что уравнение (VI,72) справедливо и для буферных растворов, состоящих из смеси слабого основания и гидролитически кислой соли. В этом случае уравнение будет иметь вид:
рН = рК 4- 1g [соль] —1g [основание], (VI, 73)
где рК — отрицательный логарифм константы диссоциации слабого основания. Из приведенных уравнений следует, что pH буферного раствора зависит от величины константы диссоциации слабой кислоты или слабого основания, а также от соотношения концентраций компонентов буферных смесей.
Таким образом, для приготовления буферных смесей с желаемым значением pH необходимо взять слабые кислоты или основания с соответствующими значениями констант диссоциации, а также подбирать определенные соотношения компонентов. Влияние этих факторов на pH буферного раствора показано в табл. 58.
Из таблицы видно, что в зависимости от соотношения компонентов, входящих в состав ацетатного буфера, можно приготовить растворы со значениями pH в пределах от 3,7 до 5,6, в случае фосфатного буфера — от 5,9 до 7,7 и у аммиачного — от 8,4 до 10,3. -
Кроме того, из этой же таблицы видно, что для разных буферов при одном и том же соотношении компонентов значение pH будет различно. Так, при соотношении компонентов 9 : 1 у ацетатного, фосфатного и аммиачного буферов значение pH будет соответственно
— 256 —
Таблица 58
Буферные ряды
Соотношение компонентов в буферной смеси
ацетатный
pH буферных систем
фосфатный
аммиачный
9:1 8:2 7:3 6:4
5:5 4:6 3:7 2:8
1:9
3,72 4,05 4,27 4,45 4,63 4,80 4,99
5,23 5,57
5,91
6,24
6,47
6,64
6,81
6,98
7,17
7,38
7,73
10,28 9,95 9,73 9,55 9,37 9,20 9,01 8,77 8,43
3,72; 5,91 и 10,28. Это различие обусловлено разной величиной констант диссоциации кислот и оснований, входящих в эти растворы (табл, 50),
На практике обычно пользуются готовыми таблицами, в которых указано, в каких отношениях должны быть взяты компоненты буферных смесей для получения буферных растворов с желаемым значением pH. Так, в табл> 59 приведены соотношения компонентов для ацетатного буферного раствора.
Поскольку константа электролитической диссоциации К при данных условиях постоянна, pH буферного раствора будет зависеть только от отношения концентраций кислоты (или основания) и соли, взятых для приготовления буферной смеси, и не зависит от абсолютного значения этих концентраций. Поэтому при разбавлении буферных растворов концентрация водородных ионов (pH) должна оставаться неизменной. Опыт показывает, что даже значительное разбавление буферных растворов в 10—20 раз и более мало отражается на их pH (табл. 60).
Как видно из данных табл. 60, величина pH буфера изменяется незначительно, несмотря на сильное разбавление раствора. Для срав-' нения укажем, что в незабуференном растворе, например, сильной кислоты или сильного основания при разбавлении в 10 раз концентра-
W
Таблица 59
Соотношение компонентов в ацетатной буферной смеси
pH 0,2 н. раствор CHsCOONa, мл 0,2 и. раствор сн3соон, мл pH 0,2 н. раствор CH3COONa, МЛ 0,2 н. раствор СН3СООН, МЛ
3,6 ' 1.5 18,5 4,8 12,0 8,0
3,8 2,4 17,6 5,0 14,1 5,9
4,0 3,6 16,4 _ 5.2 15,8 4,2
4,4 7,4 12,6 5,4 17,1 2,9
4.6 9,8 10,2 5,6 18,1 1,9
*♦
9 Зак. 560
— 257 —
Таблица 60
Влияние разбавления ацетатной буферной смеси
Показатели буфера
Исходная концентрация (0,1 к.)
Разбавление в 10 раз
(0,01 н.)
Разбавление в 100 раз (0,001 н.)
pH н+] кажущаяся степень диссоциации CH3COONa
4,62
2,36-10“£
0,79
4,67
2,14-10““-0,87
4,74 l,8-i0-2
1,0
ция водородных ионов изменяется во столько же раз, Так 0,01 н. раствор НС1 имеет pH = 2, а при разбавлении его в 100 раз — pH = = 4 и т. д.
Наблюдаемое незначительное повышение pH буферной смеси при ее сильном разбавлении обусловлено увеличением кажущейся степени диссоциации ее соли.
Способность буферных растворов противодействовать резкому изменению pH при прибавлении к ним кислоты или щелочи является ограниченной. Буферная смесь поддерживает pH постоянным только при условии, что количество прибавляемых к раствору сильной кислоты или щелочи не превышает определенной величины. Превышение этого количества вызывает резкое изменение pH, т. е.. буферное действие раствора прекращается.
Предел, в котором проявляется буферное действие, называется буферной емкостью. Буферную емкость выражают количеством грамм-эквивалентов сильной кислоты или основания, которое следует добавить к 1 л буферного раствора, чтобы сместить pH на единицу, т, е.
С pHj-pHo’
(VL 74)
где В — буферная емкость, С — количество сильной кислоты или ос*-нования, г-экв, рН0 — водородный показатель до добавления сильней кислоты или основания, рЩ — водородный показатель после добавления кислоты или щелочи.
Величина буферной емкости зависит от концентрации компонентов буферной смеси и отношения между этими концентрациями,
Зная сущность механизма действия буферных систем, нетрудно догадаться, что наибольшей буферной емкостью обладают растворы, содержащие большие концентрации входящих в состав буфера компонентов, и растворы, составленные из компонентов, взятых в равных количествах. Влияние величины соотношения компонентов буферных смесей на их емкость связано с тем, что при равных величинах числителя и знаменателя величина дроби наиболее устойчива к изменению своего числового значения. Поэтому и величина соотношения компонентов, входящих в состав буфера, будет меньше подвержена изменениям. В табл. 61 приведены данные, показывающие, как изменяется pH
х
— 258 —
Таблица 61
I
1 Изменение pH ацетатной буферной смеси в зависимости от величины соотношения кислоты и соли (в мг-экв) при добавлении 5 мг-экв НС1 или NaOH
СНзСООН CHjCOONa Величина соотношения Исходное значение pH Добавлено 5 мг-экв CH3COOH Измененное значение pH Величина смещения pH (Д)
CH3COONa
94/6 15,7 3,53 НС1 99/1 2,73 0,80
90/10 9,0 3,78 НС1 95/5 3,43 0,33
85/15 5,67 3,98 НС1 90/10 3,78 0,20
75/25 3,0 4,25 НС1 80/20 4,13 0,12
50/50 1,0 4,74 НС1 55/45 4,66 0,08
50/50 1,0 4,74 NaOH 45/55 4,82 0,08
25/75 0,33 5,21 NaOH 20/80 5,33 0,12
15/85 0,18 5,48 NaOH 10/90 5,68 0,20
10/90 0,11 5,68 NaOH 5/95 6,01 0,33
6/94 0,063 5,93 NaOH 1/99 6,73 0,80
ацетатной буферной смеси в зависимости от соотношения и абсолютной концентрации входящих в ее состав компонентов.
ТаКим образом, буферные растворы обладают следующими свойствами:
1) концентрация водородных ионов буферных смесей мало зависит от разбавления;
2) добавление к буферным смесям небольших количеств (в пределах буферной емкости растворов) кислоты или щелочи мало изменяет pH;
3) величина буферной емкости зависит от концентрации компонентов буферной смеси и от отношения между этими компонентами.
§ 78. Биологическое значение буферных систем. Буферность почв и почвенного раствора
Явление буферности имеет чрезвычайно большое биологическое значение, так как обусловливает постоянство pH в различных тканях животного и растительного организма.
Буферные системы в живых организмах поддерживают постоянство pH в крови и тканях. Как показали специальные исследования, в живом организме в процессе обмена образуются большие количества кислых продуктов. Так, в организме человека за сутки образуется такое количество различных кислот, которое эквивалентно 20—30 л однонормальной сильной кислоты. Сохранение постоянства реакции внутри организма обеспечивается наличием в нем мощных буферных систем.
В организме человека особенно большую роль играют белковый, бикарбо-натный и фосфатный буферы.
Буферной смесью крови является карбонатная смесь, состоящая из NaHCO3 и СО2, а также фосфатные смеси, состоящие из одно- и двузамещенного фосфата и двузамещенной соли этой же кислоты: NaH2PO4 + Na2HPO4. Бикарбонатный буфер присутствует в крови также в довольно большой концентрации. Зная коли-
9*
— 259 —
чества бикарбонатов и свободно растворенной СО2, можно определить pH крови йо следующей формуле:
,, . [бикарбонат] __
pH«6,11 4- 1g-———-------, (VI, 75)
где 6,11 — показатель константы, характерной для крови и близкой по величине к кажущейся константе диссоциации угольной кислоты; [бикарбонат] — концентрация бикарбоната в объемных процентах; [СО2] — концентрация свободно растворенной угольной кислоты.
Этой формулой широко пользуются на практике при различных медикобиологических исследованиях.
Однако наиболее мощными буферными системами крови являются так называемые гемоглобиновые буферы, которые составляют примерно 75% всей буферной емкости крови. Сущность действия этих буферных систем заключается в следующем. Кислые продукты обмена веществ взаимодействуют с калиевой солью гемоглобина с образованием эквивалентного количества их калиевых солей и свободного гемоглобина, обладающего свойствами слабой органической кислоты. Таким образом, подкисления крови не происходит.
Углекислота, связанная с гемоглобином, в конечном итоге выделяется в воздух через легкие, однако сдвига pH крови в щелочную сторону не происходит, так как образующийся при этом оксигемоглобин значительно кислее гемоглобина.
Большое значение в поддержании постоянного pH внутри живых клеток имеет так называемый белковый буфер. Этот буфер состоит из протеина (Pt) и его соли, образованной сильным основанием Na или К. Компоненты этого буфера можно представить как Pt — СООН — слабо диссоциирующая кислота и ее соль Pt — COONa
Pt—СООН Pt — COO - 4- H+
Pt—COONa Pt —COO- 4- Na+
Эта буферная система будет действовать аналогично буферным смесям, рассмотренным ранее.
При увеличении концентрации ионов водорода соль белка будет реагировать с кислотами, образуя весьма слабо диссоциирующую кислоту и нейтральную соль по уравнению
2Pt — COONa 4- H2SO2 Na2SO4 + 2Pt—СООН
При взаимодействии же с щелочами в реакцию вступает кислота и вместо сильного основания образуется слабоосновная соль белка
Pt—СООН 4- NaOH Pt — COONa 4- Н2О
В силу амфотерности даже отдельная белковая молекула обладает буферным действием, т. е. обладает способностью связывать кислоты и щелочи с образованием солей
СООН СООН
Pt^ 4- НС1 Pt(^
NH2 NH3C1
Как видим, при добавлении сильной кислоты образуется слабо кислая соль белка (так называемый солянокислый протеин).
Прибавление щелочи приводит к образованию слабо основной соли белка (протеината натрия)
СООН COONa
Pt^ 4-NaOH->Pt\ 4-Н2О
nh£ nh2
— 260 —
Отметим что i/ce растительные соки также представляют собой буферные растворы Почвенные растворы аналогично растворам биологического проис-
^п^прния обладают определенной буферностью, что хорошо видно из следующего опыта Если взять две навески почв с нейтральной реакцией (одну суглинистую, ovrvio __песчаную) и к обеим навескам добавить одинаковое количество кисло-
ты 0 05 н концентрации, то после взбалтывания pH равновесных растворов суспензий окажется неодинаковой. В суспензии песчаной почвы pH будет значительно ниже, чем в суглинистой.
Таким образом, суглинистая почва препятствует сильному подкислению, обладает гораздо большей буферностью по сравнению с песчаной.
Если эти почвы довести до нор-
мальной влажности, а затем из них по соответствующей методике отжать путем прессования почвенные растворы, то они также будут обладать различной буферностью. Для того чтобы изменить реакцию почвенного раствора на единицу, в случае суглинистой почвы необходимо добавить значительно больше сильной кислоты (или сильного основания), чем в случае песчаной.
Опыт показывает, что буферность почвы зависит от составами свойств твердой фазы почвы, а также от состава почвенного раствора. В частности, высокая буферность суглинистой почвы обусловливается наличием в ней большого количества илистых и коллоидных частиц, которые содержат поглощенные катионы — кальций, магний и др. Если в такую почву внести кислоту, подкисления не произойдет в силу следующей обменной реакции
[ПК]$, +
4- 4НС1 [ПК] Hd + СаС12 4- MgCla
Рис. 79. Кривые титрования черноземной (/) и дерново-подзолистой (2) почв
В результате этой реакции в растворе вместо ионов водорода появляются нейтральные соли GaCla, MgCl2.
В песчаной же почве, бедной коллоидами, поглощенных оснований содер-
жится очень мало, поэтому ионы водорода кислоты практически не поглотятся и останутся в растворе.
Буферность твердой фазы почвы обусловливается в основном двумя факторами: количеством почвенных коллоидов и составом поглощенных катионов. Большое значение имеет также энергия поглощения водородных ионов почвенными коллоидами и степень диссоциации последних. Поскольку органические вещества почвы преимущественно состоят из слабых кислот (т. е. кислот, имеющих очень малую константу диссоциации), они в значительной степени будут связывать поступающие в почвенный раствор ионы водорода и тем самым оказывать буферное действие против подкисления почвы. Опыт показывает: чем больше данная почва содержит органического вещества, тем выше ее буферное действие.
Почвенный раствор обладает буферностью в том случае, если в нем присутствуют соли сильных оснований и слабой кислоты. К сильным основаниям относятся натрий, калий, к более слабым — кальций и магний. Из слабых кислот в почвах встречаются гуминовые и фульвокислоты, щавелевая и др. Из сильных кислот в почве встречаются серная и азотная. Эти кислоты попадают в почву с удобрениями или освобождаются при поглощений растениями питательных элементов из физиологически кислых удобрений, например аммония из (NH4)2SO4 и т. д. Чем выше содержание в почвенном растворе этих солей, тем выше его буферная способность.
- 261 —
Практика сельского хозяйства показывает, что в слабобуферных почвах реакция среды может довольно резко изменяться от внесения физиологически кислых или щелочных удобрений. В почвах, обладающих хорошей буферностью, этого не происходит.
Определение буферности почв по отношению, например, к кислотам производят путем титрования, добавляя к почвенной суспензии небольшими порциями кислоту и измеряя реакцию суспензии (pH). Если при этом реакция изменяется очень заметно, почва обладает сравнительно малой буферностью. Результаты определения буферности почвы можно изобразить графически в виде соответствующих кривых титрования. На рис. 79 изображены подобные кривые титрования. Из рисунка видно, что в буферной почве значение pH изменяется гораздо меньше, чем в небуферной, при добавлении одного и того же количества кислоты.
Путем внесения органических и минеральных коллоидов (например, ила или глин) буферность почвы можно значительно улучшить. В кислых почвах буферность по отношению к кислотам можно повысить путем внесения в почву извести. На почвах с малой буферностью рекомендуется вносить минеральные удобрения в несколько приемов уменьшенными дозами, чтобы предотвратить резкое смещение реакции среды.
§ 79. Индикаторы и их применение
Величина pH имеет большое значение, поэтому методам ее определения уделяется самое серьезное внимание. Методы определения pH могут быть различными. В настоящее время наиболее широкое распространение получили электрометрический и колориметрический методы (буферный и безбуферный).
. Первый метод является наиболее точным, хотя и более сложным. Колориметрические методы более просты, но менее точны, чем электрометрические.
Колориметрические методы определения pH основаны на принципе изменения цвета кислотно-основных индикаторов.
Кислотно-основные индикаторы в большинстве представляют собой довольно сложные органические вещества, которые проявляют слабокислотные или слабощелочные свойства, и которые в растворах меняют свою окраску в зависимости от pH среды.
В настоящее время считают, что изменение цвета индикатора связано с изменением строения его молекулы. Появление же окраски у молекул органического соединения часто связано с образованием в их структуре так называемых хромофорных групп, содержащих двойные связи, например: — N = О; — N = N — ; С = О и др. Очень важными хромофорами являются более сложные группы, в структуре которых имеется цепь сопряженных двойных связей. В качестве примера можно назвать хиноидную группировку, которая имеет строение типа = / “ ’ ^пыт показывает, что увеличение числа
4 хромофорных групп углубляет окраску, в то время как наличия в молекуле вещества одной хромофорной группы (одна двойная связь) бывает недостаточно для появления окраски.
Индикаторы, которые имеют различную окраску в кислой и щелочной средах, получили название двухцветных (метиловый красный, лакмус и др.), а индикаторы, которые имеют одну окраску — одноцветных (фенолфталеин и др.).
— 262 —
Рассмотрим механизм действия индикаторов на примере одноцветного индикатора фенолфталеина. Этот индикатор, как известно, в кислой среде бесцветен, в щелочном же растворе приобретает розовую окраску. Она получается благодаря тому, что в щелочной среде образуется соль фенолфталевой кислоты, которая имеет в своей структуре уже не фенольную группу, а хинонную хромофорную группу.
Таким образом, фенолфталеин в щелочном растворе ведет себя как слабая кислота. В общем виде реакцию диссоциации кислотного индикатора можно представить в виде следующего уравнения
Hind #Н+ + Ind~ окраска 1 окраска 2
где Hind и Ind-—соответственно недиссоциированная и диссоциированная формы индикатора, имеющие различные окраски.
Диссоциация слабых кислот или оснований зависит от реакции среды, поэтому при разных значениях pH в растворах будет находиться различное количество молекул и ионов индикатора. Иными словами, окраска раствора будет определяться соотношением этих разно окрашенных соединений.
Учитывая, что все индикаторы представляют собой слабые электролиты, для определения константы их диссоциации используем закон действующих масс. Так, для равновесной реакции (VI,76) будем иметь
„ [Н + ] [Ind-] [Н + ] [окраска 2]
Л —---------------------------------
[Hind] [окраска 1]
(VI, 77)
где К — константа диссоциации кислотного индикатора [ ] —
концентрации соответствующих компонентов в растворе.
Уравнение (VI,77) можно переписать в следующем виде:
[Ind-] К
[Hind] “ [Н+|
или (окраска 2] = [окраска 1] [Н+]
(VI, 78)
Нетрудно догадаться, анализируя уравнение (VI,78), что окраска раствора зависит от концентрации Н+-ионов в растворе. Точка перехода окраски индикатора зависит от величины его константы диссоциации. В самом деле, из уравнения (VI,78) следует, если
[окраска 2]
н---------77 = 1, ТО К = [Н+].
[окраска 1]
(VI, 79)
Несложный анализ этих соотношений показывает, что чем большее численное значение будет иметь константа диссоциации индикатора, тем при более низком значении pH будет находиться точка (область) пере ода его окраски, т. е. когда [окраска 1] = [окраска 2L
Из уравнения (VI,78) можем записать:
[Н+]=к [окраска 1]..
[окраска 2]
(VI, 80)
Логарифмируя это уравнение и меняя знаки на обратные, получим
lg [Н + ] = 1g /<+ 1g [окраска 1] — 1g [окраска 2],
— 263 —
или
откуда
— lg [Н+]= — lg 7<4-1g [окраска 2] —1g [окраска 1],
тт „ , [окраска 2] , „
рН = рК + 1g 4—(VI, 81) [окраска 1]
где рК — отрицательный логарифм К индикатора.
В этом уравнении рК есть не что иное, как точка перехода.
Таким образом, точкой перехода (p/Q индикатора называется то значение pH, при котором количество непродиссоциировавших молекул индикатора равно количеству молекул, подвергшихся диссоциации.
Сдвиг реакции среды в ту или иную сторону от точки перехода (р/С) сопровождается нарушением соотношения окрашенных форм индикатора и, следовательно, изменением цвета:
1) рН<рК; 2)рН —РД; 3) pH > рК; (Ind-) < (Hind) (Ind-) = (HInd) (Ind-) > (Hind).
Зона перемены.
кислошкраски* ‘ * основной окраски * '
Рис. 80. Изменение окраски индикатора метилового красного в зависимости от величин^! pH
Человеческий глаз улавливает окраску раствора в том случае, когда количество окрашенного иона индикатора будет составлять не менее 10% от общей концентрации индикатора.
Если, обращаясь к уравнению (VI,81), принять, что [окраска 2] 9%, [окраска 1] 91%, то
получим:
pH — рК 4- 1S ~ = рК 4- 1g 0,1,
откуда
рН = рК—1.
(VI, 82)
С другой стороны, если принять, что [окраска 1] = 9%, а [окраска 2] = 91%, то согласно уравнению (VI,81) будем иметь:
pH = pK4-lg — = рК 4- 1g 10, у
откуда
рН-=рК4-1. (VI, 83)
Таким образом, изменение цвета индикатора происходит приблизительно в интервале двух единиц pH, так как р/С — величина всегда постоянная (рис. 80). Интервал индикатора — величина постоянная и в значительной степени зависит от способности исследователя улавливать изменение окраски индикатора. В табл. 62 показаны интервалы
— 264 —
LJ
Таблица 62
Серия индикаторов по Кларку и Лабсу
Назедкие индикатора
Тимоловый СИНИИ . . Бромфенол синий . . Метиловый красный . Бромкрезол пурпурный Бромтимол синий . . . Феноловый красный. . Крезоловый красный . Тимоловый синий . *
р.г< Зона перехода окраски (pH) Переход окраски
1,7 1,2-2,8 Красно-желтый
4,1 3,0—4,6 Желто-си ний
5,1 4,4—6,0 Красно-желтый
6,3 5,2—6,8 Желто-пурпурный
7,1 6,0—7,6 Желто-синий
7,7 6,8—8,4 Желто-красный
8,1 7,2—8,8 Желто-красный
8,8 7,8—9,8 Желто-синий
перехода окраски некоторых индикаторов, наиболее часто применяемых для опредёления pH, а также для титрования растворов.
Все сказанное в отношении точки перехода и зон изменения окраски индикатора может относиться не только к двухцветным, но и к одноцветным индикаторам.
§ 80. Принципы колориметрического определения pH
Существует несколько методов определения концентрации водородных ионов (pH). Из них наиболее простым является колориметрический, т. е. индикаторный метод. Этот метод измерения концентрации бесцветных водородных ионов возможен только в присутствии индикаторов. Сущность его заключается в том, что изменение окраски различных индикаторов происходит при разных концентрациях водородных ионов. Все колориметрические методы определения pH основаны на законе Ламберта — Бугера—Бера, согласно которому для двух растворов, одинаково поглощающих свет, произведение концентрации С на толщину слоя раствора h есть величина постоянная:
Ch = Clhl = const. (VI, 84)
Из этого закона следует, что при совпадающей интенсивности окраски двух растворов, из которых концентрация одного является величиной известной (стандарт), можно узнать концентрацию другого раствора.
Имея набор стандартных окрашенных растворов с различной концентрацией водородных ионов (pH), выбирают тот из них, который по интенсивности окраски ближе всего подходит к исследуемому раствору. В этом случае, при условии одинаковой толщины слоя обоих растворов, pH исследуемого раствора равно pH стандартного раствора.
Для сравнения интенсивности цвета стандартных растворов с окраской исследуемого раствора пользуются различными приборами: фотометрами, колориметрами, компараторами и специальными штативами (рис. 81 и 82).
Компаратор представляет собой деревянную колодку с шестью гнездами, расположенными в два ряда. В нижней части компаратора высверлено три круглых отверстия, которые с задней стороны закрываются матовым или синим стеклами для создания однородного фона.
Существуют два основных колориметрических метода: буферный и безбу-ферный.
Сущность буферного метода заключается в следующем: приготовляют ряд стандартных буферных растворов с постепенно возрастающими значениями pH и добавляют к каждому из них несколько капель соответствующего индикатора. Таким способом, используя несколько индикаторов с различными точками
1 I
- 265 —
перехода, получают цОтную шкалу. Затем такое же количество капель индикатора добавляют к исследуемому раствору и сравнивают, например, в компараторе его окраску с окраской стандартных растворов. Совпадение окраски растворов свидетельствует о совпадении и pH исследуемого раствора с pH стандарта.
Для приготовления буферной шкалы сравнения пользуются формулой (VI, 72) или табл. 59 и 60.
Поскольку буферная шкала обычно приготовляется с несколькими индикаторами (например, в диапазоне pH от 4,2 до 8,4 берут три индикатора: метиловый красный, бромтимоловый синий и феноловый красный), то для правильного выбора индикатора сначала грубо определяют pH исследуемого раствора с помощью так называемого универсального индикатора. Универсальный индикатор пред
Рис. 81. Компаратор: а — вид спереди; б — вид сзади
ставляет собой смесь индикаторов с зонами перехода, последовательно охватывающими широкую область pH от сильно кислых до щелочных значений. Ниже приведен состав (в г) универсального индикатора с зоной перехода pH 1 — 10.
Тимоловый синий ... 1 Метиловый красный ... 0,4
Бромтимоловый синий . . 0.8 Фенолфталеин.................0,2
Диметиламиноазобензол 0,6
Указанные выше индикаторы растворяются в 1 л этилового спирта, затем в этот раствор добавляется несколько капель 0,1 н. NaOH до появления желтой окраски. Ниже указана окраска универсального индикатора при различных значениях pH раствора. I
pH Окраска
3 Красная
4 Оранжевая
5 Оранжево-желтая
6 Желто-оранжевая
7 Зеленая
8 Тем но-зелен а я
10 Синяя
Добавляя к исследуемому раствору несколько капель универсального индикатора, по цветной его шкале производят грубое определение значения pH. Зная примерный pH, выбирают такой индикатор, который имеет точку перехода в этой области pH. Окрасив исследуемый раствор этим индикатором, сравнивают его окраску с окраской буферной шкалы сравнения, по которой и определяют более точное значение pH.
Сущность безбуферного определения pH (так называемый метод Михаэлиса) заключается в использовании стандартных рядов, полученных с одно
— 266 —
цветными индикаторами группы нитрофенола в 0,001 н. растворах NaOIli 1) В-линитпоФенол с диапазоном pH от 2,8 до 4,5; 2) у-Х нитрофенол с диапазоном pH отТо до 5,5; 3) «-нитрофенол - от 5,2 до 7,0 pH и 4) .и-нитрофе-Н°Л Таки^’образо’м по методу Михаэлиса может быть определено pH растворов в довольно широком диапазоне — от 2,8 до 8,4. Предварительно надо определить приблизительное значение pH с помощью универсального индикатора. Затем производят выбор индикатора и уже с этим индикатором производят более точное определение pH.
Рис. 82. Штатив для сравнения цвета индуктора при буферном методе определе-. ния pH
Колориметрические методы определения pH имеют целый ряд недостатков. Они недостаточно точны: pH измеряется с точностью до 0,2. Индикатор сам является слабой кислотой или слабым основанием, и прибавление его к исследуемому раствору вызывает смещение равновесия»
-ь „ [Hind] |Н’|-КЫ’
т. е. [Н+] после смешивания индикатора с раствором будет выражать концентрацию Н+, обусловленную не только испытуемым раствором, но и индикатором одновременно. Щелочная или кислотная ошибка индикатора особенно сильно сказывается на точности определения pH в случае растворов, обладающих малой буферностью.
Кроме того, в присутствии многих нейтральных солей и белков область перехода окраски индикатора меняется, что приводит к большому искажению результатов. Эта солевая ошибка обусловлена высокой концентрацией солей в исследуемом растворе, изменяющей растворимость и степень диссоциации самого индикатора.
Белковая ошибка индикатора обусловливается наличием в растворах белковых веществ, которые, обладая амфотерными свойствами, могут взаимодействовать с кислотными и основными индикаторами, а также адсорбировать индикатор, изменяя его общую концентрацию в растворе.
Большие искажения результатов получаются и при изменении температуры. С повышением температуры изменяется константа диссоциации индикатора. Так, для n-нитрофенола при 0° С р/< = 7,30, а при 50° С р/< — 6,81. С изменением температуры изменяется и pH стандартных растворов.
При пользовании колориметрическими методами определения pH большие ошибки могут возникать в случае, если исследуемые растворы мутны или окрашены. Вот почему в окрашенных и мутных растворах, а также в растворах различных биологических жидкостей, содержащих значительные количества солей и белков, определение pH следует проводить более точными, хотя и более сложными электрометрическими методами.
ЭЛЕКТРОХИМИЯ
Учение об электродвижущих силах гальванических элементов является одним из основных разделов электрохимии. Начало изучению электродвижущих сил было положено еще М. В. Ломоносовым (1750), который в своих работах отмечал связь между химическими и электрическими явлениями. Позднее наблюдения итальянского физиолога Гальвани (1780) и обширные работы итальянского физика Вольта (1780) привели к открытию гальванических элементов.
В 1800 г. Вольта изобрел первый химический источник тока, так называемый вольтов столб, который был собран из пластинок различных металлов, разделенных прослойками ткани, смоченной электролитом. Исследования привели Вольта к открытию контактной разности потенциалов, возникающей при соприкосновении металлов различной природы. В первых исследованиях в качестве чувствительного прибора для обнаружения малой разности потенциалов ученый использовал свежеанатомированные мышцы лягушки. Этот случай является наглядным примером того, как биологические методы исследования нередко могут способствовать успешному развитию физики и других точных наук.
Открытие химических источников тока и контактной разности потенциалов оказало большое влияние на все последующее развитие электрохимических явлений. В настоящее время методы электрохимии получили широкое распространение в агрохимии, физиологии растений, в биологии, почвоведении, а также во многих других смежных дисциплинах.
§ 81. Электродный потенциал. Уравнение Нернста
Если в чистую воду погрузить пластинку какого-либо металла, то согласно гидратной теории Д. И. Менделеева ионы металла будут взаимодействовать с полярными молекулами воды. Иными словами, поверхностно расположенные катионы этого металла будут гидратироваться молекулами воды и переходить в окружающий раствор, заряжая его положительно, т. е. металл будет как бы поверхностно растворяться (рис. 83).
Однако электроны, в избытке остающиеся в металле, заряжают его поверхностный слой отрицательно. В результате этого между ионами металла, перешедшими в раствор, и поверхностью металлической пластинки возникают силы электростатического притяжения, в силу чего ионы, окружающие пластинку, образуют так называемый двойной электрический слой, схема которого приведена на рис. 84. Этот слой препятствует дальнейшему растворению металла и в системе устанавливается подвижное равновесие, которое характеризуется равными скоростями как растворения, так и обратного осаждения ионов из раствора на поверхности металлической пластинки.
— 2G8 —
Первоначально считали, что двойной электрический слой имеет плоское строение. Он уподоблялся конденсатору,* одна из обкладок которого расположена на поверхности металла другая — в слое, прилегающем к электроду жидкости. Расстояние между обкладками равнялось молекулярному диаметру. Согласно этой теории, которую обычно связывают с именем Гельмгольца (1879), учитывалось только лишь проявление электростатических сил взаимодействия между зарядами противоположного знака. Основным недостатком теории Гельмгольца явилось то, что она не учитывала изменение свойств двойного электрического слоя с изменением концентрации электролита и его температуры.
Рис. 83. Гидратация поверхностно расположенных • катионов металла в воде — поверхностная растворимость металла (схема):
тп — поверхность раздела твердой и жидкой фаз
Рис. 84. Двойной электрический слой на границе металл — жидкость (диффузное строение слоя)
В разработке современной теории строения двойного электрического слоя на границе твердая фаза — жидкость и методов его исследования ведущая роль принадлежит А. Н. Фрумкину и его школе.
Работы А. Н. Фрумкина и его учеников установили, что слой ионов, располагающийся в жидкости, благодаря действию двух противоположно направленных сил (электростатического притяжения и теплового движения) имеет диффузное строение, т, е. он проникает в жидкость на некоторую глубину (рис, 84).
Определенная часть ионов удерживается вблизи поверхности раздела металл — электролит, образуя обкладку двойного слоя с толщиной, отвечающей среднему радиусу ионов электролита. Остальные ионы, входящие в состав двойного слоя, распределяются диффузно, с постепенно убывающей плотностью заряда.
Таким образом, при соприкосновении металла с водой ионы его находятся под действием двух конкурирующих сил: электростати-
— 269 —
J .
ческого притяжения, возникающего между ионами металла и молекулами воды (явление гидратации), и электростатического притяжения со стороны электронного газа, определяющего прочность кристаллической решетки. |
Вполне понятно, что чем прочнее кристаллическая решетка металла, тем труднее иону металла перейти в раствор. Чем выше величина энергии гидратации, тем с большей жадностью молекулы воды взаимодействуют с этими ионами, и тем легче им выделиться в раствор.
В результате взаимодействия двух указанных взаимно противоположных сил растворение металла в воде приобретает характер только поверхностного процесса и охватывает лишь очень узкую область на границе металл — жидкость. В этом поверхностном слое концентрация ионов металла, несмотря на его чрезвычайно малую растворимость, может быть довольно значительной. Кроме того, в поверхностном растворе гидратированные катионы в силу электростатических сил притяжения со стороны электронов кристаллической решетки металла совершают лишь ограниченное кинетическое движение в виде.так называемых «пристенных» скачков. Они прочно связаны с жестким каркасом кристаллической решетки металла.
Таким образом, в системе металл—вода на границе раздела фаз возникает двойной электрический слой, блокирующий поверхность металла. Образовавшаяся пограничная разность потенциалов получила название электродного потенциала (лат. potentia — возможность, мощь).
Если жидкая среда — чистая вода, для всех металлов картина в качественном отношении будет однозначной: металл заряжается отрицательно, прилегающий слой жидкости — положительно. Однако количественно для разных металлов будут наблюдаться существенные различия, что объясняется не только неодинаковой энергией связи катионов этих металлов в кристаллической решетке, но и неодинаковой гидратируемостыо самих катионов.
Несколько иная картина будет наблюдаться в том случае, если металлическую пластинку погрузить не в чистую воду, а в раствор его соли. При этом могут иметь место три случая.
1. Исходная концентрация ионов данного металла ц растворе С меньше концентрации Со, соответствующей равновесному состоянию ионов после погружения в раствор металлической пластинки, т. е. C<ZC0. При этом будут происходить оба процесса — переход металла в раствор
(a)
и выделение металла из раствора
Ме+4-^±Ме, (б)
Поскольку С < С0, преобладающим будем процесс (а), т, е. выделение электронов превысит их поглощение, В результате этого поверхность металлической пластинки зарядится отрицательно, а прилегающий слой раствора — положительно.
— 270 —
2. Концентрация ионов металла С больше равновесной концентрации с о, т. е. С > Со. В этом случае наблюдается обратное явление: ионы металла из раствора выделяются на поверхности металлической пластинки. Чтобы ионы металла могли выделиться, они должны присоединить электроны согласно уравнению (б). Поскольку источника электронов в системе нет, выделение металла на поверхности пластинки происходит в виде ионов. В результате поверхность приобретает положительный заряд.
3. При условии С = Со, вся система будет находиться в состоянии подвижного равновесия, разность потенциалов между жидкостью и металлом равна нулю. В этом случае из раствора осаждается на единицу поверхности металла столько же катионов, сколько их выходит в двойной электрический слой.
Принимая это во внимание, нетрудно найти математическую зависимость между величиной скачка потенциала на границе соприкосновения металла и раствора и концентрацией (точнее, активностью) ионов этого металла в растворе.
Поскольку переход ионов из металла в раствор есть процесс обратимый и совершается он без изменения температуры, то при этих условиях система совершает максимально полезную работу. Напомним, что способность системы произвести максимально полезную работу называется свободной энергией, обозначаемой буквой А.
Электрическая работа перевода 1 г-иона из металла в раствор или обратно согласно реакции Men+ + -> Me равна:
А — пРг, • (VII, 1)
где л — заряд иона, 8 — потенциал электрода (в вольтах), F — число Фарадея.
По уравнению изотермы химической реакции
( а1Ле \
A = RT In К —In ——-— , (VII, 2)
\ Me"+ /
где R — универсальная газовая постоянная, Т — температура, при которой протекает реакция, К — константа равновесия данной реакции, «ме — активность металла, аме п+ — активность ионов металла в растворе.
Раскрыв скобки и заменив натуральные логарифмы на десятичные, получим:
Л=2,303/?7 1g К—2.303RT 1g Ме----. (VII, 3)
Ме'!+
Подставляя в уравнение (VII,3) значение А из (VII,!), получим:
лГе=2,303ЯТ lg A — 2,303/?Tlg —----,
°Меп+
откуда
2,303/? 7" , „ 2.3O3RT , °ме
е=---------1g К —--------1g -------.
nF nF а{Леп+
(VII, 4)
(VII, 5)
— 271 —
В уравнении изотермы К — константа равновесия — при данной температуре величина постоянная. Поэтому и все выражение 2>зуГ- 1g также, „постоянная величина, характеризующая электрохимическую природу электрода. Обозначив ее через е0, получим:
е—е0 —
2,3037?Т , °мс
---------1g------- nF ь
(VII, 6)
Так как активность металла принимается равной единице (аме = 1), то с учетом этого уравнение (VI 1,6) будет иметь следующее выражение:
2,3037? Т
е = е0 +—nF-----lg0Men-b- (VII, 7)
Уравнение (VI 1,7) называется уравнением Нернста.
Для расчетов удобнее предварительно произвести вычисление значения ~ 2,303 при какой-либо температуре. Например, при 18° С (Т — 291° К) это число будет равно 0,0577. Следовательно, для температуры 18° С уравнение Нернста будет иметь вид:
0,0577
е = (VII, 8)
RT
Обозначим численное значение выражения у 2,303 буквой Зависимость этого числа от температуры выразится следующей формулой:
0,05774- 0,0002 (/ — 18), (VII, 9)
где t — любая температура, при которой производится измерение разности потенциалов.
С учетом этого обозначения уравнение электродного потенциала Нернста будет иметь следующее выражение:
e = e«+TlgaMen+- <VI1’I0)
Разберем теперь вопрос, каков же физический смысл е0 в уравнении электродного потенциала Нернста.
Несложный математический анализ этого уравнения показывает, RT
что при С == 1 г-ион/л все выражение -у 2,303 1g Яме п+ обращается в нуль, и тогда:
е = е0. (VII, 11)
Таким образом, стандартным (нормальным) потенциалом называется такой потенциал, который возникает на металлической пластинке, находящейся в контакте с одноименными ионами в растворе, с концентрацией С = 1 г-ион/л.
За нулевую точку измерения потенциалов условно принят нормальный потенциал водородного электрода. В настоящее время наука
— 272 —
еще не располагает методами, позволяющими измерять абсолютное значение электродных потенциалов. Мы можем (и всегда измеряем) только разность потенциалов. Вот почему и понадобилось какой-то потенциал условно принимать равным нулю. Таким потенциалом является нормальный потенциал водородного электрода. Для изготовления его используют способность платины растворять газообразный водород. Платиновая проволока или пластинка, содержащая растворенный водород, играет роль «водородной пластинки», а функции «раствора солей» может выполнять любой водный раствор, в котором всегда присутствуют ионы водорода Н+. Причем, нормальный потенциал водородного электрода будет равен нулю при условии, что давление молекулярного водорода на пластинке равно 1 атм и Сн+ = == 1 г-ион/л.
Если нормальный потенциал какого-либо металла больше водородного, его принято считать положительным, если меньше — отрицательным.
Если все металлы расположить последовательно по возрастающей величине их нормальных электродных потенциалов, получится ряд напряжений. В табл. 63 приведены стандартные потенциалы некоторых металлов.
В этой таблице каждый электрод обозначен символом элемента, из которого он состоит, и соответствующего иона, а вертикальная линейка изображает поверхность раздела двух фаз, где имеет место скачок потенциала,
ч
Таблица 63
Стандартные потенциалы некоторых электродов при 25° С (ряд напряжений)
Электрод Электродный процесс Стандартный электродный потенциал, в
К|К+ —2,92
Ва | Ва2+ Ba^±Ba2+<- • —2,92
Mg | Mg2+ Mg^ztMg2+^(?e —2,38
Zn | Zn2+ Zn^z>Zn2+ —0,76
Fe | Fe2+ Ге;т±Ре2+ф2 —0,44
Cd | Cd2+ Cd^+Cd2+ + 2 e“ —0,40
Co | Co2+ Co^ztCo2+ ф 2 «?"' —0,27
Ni | Ni2+ Ni^±Ni2+^2 e- —0,23
Pb 1 Pb2+ РЬ^2РЬ2+-ф.2 r —0,13
(Pt)H21 2H+ Н2Г*2 Н+-ф2 e* —0,00
Си 1 Cu2+ Cu^ztCu2+ ф 2 e* +0,34
Ag | Ag+ Ag^+Ag++e- +0,80
Au | Au+ Ап~2 Аи+чЬе~ +1,70
— 273 —
Представленным в табл. 63 рядом напряжений широко пользуются в практике при составлении так называемых гальванических элементов, а также при изучении взаимодействия между металлами и кислотами, между солями тл-металлами. Зная ряд напряжений, можно предвидеть направление реакции вытеснения одних элементов другими. Так металлы, стоящие в ряде напряжений после водорода, неспособны вытеснять водород из кислот. Вытеснение металла из солей другим металлом осуществляется только в том случае, если вытесняющий металл расположен в ряду напряжений до вытесняемого.
Например, при составлении гальванического элемента из цинка и свинца в качестве положительного электрода следует взять свинцовый (е0 = —0,13 в), а в качестве отрицательного — цинковый (е0 = — 0,76 в).
Электроды подразделяются на электроды первого и второго рода* Электроды- первого рода — это электроды из металла, погруженного в раствор, содержащий ионы того же металла (например, Cu|Cu2+, Zn|Zn2+). Эти электроды обратимо обменивают катионы Metz;Men+ 4- пеу где п — число теряемых (или приобретаемых) электронов (е~).
Электроды второго рода состоят из металла, покрытого слоем труднорастворимой соли и погруженного в раствор какой-либо легко растворимой соли с тем же анионом. Такие электроды обратимы относительно этого аниона.
Для электродов второго рода выражение электродного потенциала:
е = Е0 — —1g [анионы]. (VII, 12)
п
§ 82. Гальванические элементы и их электродвижущая сила
Устройство, в котором энергия химической реакции непосредственно превращается в электрическую энергию, называется гальваническим элементом.
Гальванический элемент состоит из двух соприкасающихся друг с другом растворов электролитов, в которые погружены металлические пластинки — электроды, соединенные между собой внешним проводником. Гальванический элемент, дающий электрический ток, находится в неравновесном состоянии. С уменьшением силы тока разность потенциалов между электродами возрастает^ Если сила тока бесконечно мала и система практически находится в состоянии равновесия, элемент этот работает обратимо. Максимальная разность потенциалов, достигаемая при обратимой работе гальванического элемента, называется его электродвижущей силой (э. д. с.).
Элемент называется необратимым, если в системе хотя бы один из процессов является термодинамически необратимым.
В качестве обратимого гальванического элемента рассмотрим элемент Якоби —Даниеля, который состоит из медного и цинкового электродов, погруженных соответственно в растворы CuSO4 и ZnSO4.
— 274 —
Схематически этот элемент изображается следующим образом:
Си | CuSO41| ZnSO41 Zn eCu eZn
Рис. 85. Гальванический элемент Якоби—Даниеля
Здесь, как и в табл. 63, поверхность раздела двух фаз, между которыми имеет место скачок потенциала, обозначена вертикальной лиг нейкой. Двойная линейка означает, что в месте соприкосновения двух растворов скачок потенциала, обусловленный различной скоростью диффузии ионов (так называемый диффузионный потенциал), снят и его можно не учитывать при вычислении э. д. с. этого элемента. Металл с большей величиной стандартного потенциала (положительный электрод) принято писать слева, ас меньшей величиной (отрицательный электрод) — справа.
Общий вид гальванического элемента Якоби — Даниеля показан на рис. 85. В пористом сосуде 1 находится раствор CuSO4, в который погружен медный электрод. Этот сосуд помещен в стеклянную банку 2, содержащую цинковый электрод, находящийся в растворе ZnSO4.
На границе раздела фаз металл — жидкость образуется двойной электрический слой, при этом поверхность металла заряжается отрицательно, прилегающий слой — положительно. Однако при разомкнутой цепи процесс растворения цинка быстро достигает равновесного состояния и приостанавливает
ся. Если цинковый электрод соединить каким-либо проводником с медным электродом, будет наблюдаться совершенно иная картина (рис. 86).
В этом случае избыточные электроны с цинкового электрода потекут на медный, и во внешней цепи возникнет электрический ток, который можно измерить с помощью гальванометра. Электроны, перешедшие на медь, нейтрализуют осадившиеся на ней из раствора CuSO4 ионы Си2+, превращая их в электронейтральные атомы меди.
Остающиеся свободными сульфат-ионы через пористые стенки сосуда / проникают во внешнюю жидкость и, соединяясь с катионами Zn2+ металлического цинка, дают ZnSO4. С другой стороны, катионы цинка в процессе работы гальванического элемента также диффундируют из сосуда 2 через пористую перегородку в сосуд /, замещая там перешедшие на медную пластинку катионы Си2+. В результате этого раствор CuSO4 в сосуде 1 постепенно превращается в ZnSO4.
По мере удаления по внешней цепи с цинковой пластинки избытка электронов все новые количества катионов Zn2+ будут переходить в раствор. Гальванический элемент будет работать до тех пор, пока весь цинковый электрод не растворится, т. е. перейдет в состояние катионов Zn24\ После этого электрический ток прекращается.
— 275 —
Таким образом при работе гальванического элемента происходит одновременный перенос электричества по двум цепям: внешней (поток электронов по проволоке) и внутренней (поток катионов в жидкой фазе элемента). Как видно из рис. 86, цинк для внешней цепи играет роль катода- (посылает во внешнюю цепь отрицательно заряженные электроны), а для внутренней цепи — анода (посылаёт во внутреннюю цепь положительно заряженные катионы). Медь для внешней цепи играет роль анода (акцептор электронов), а для внутренней — роль катода (акцептор электронов).
Поток электронов
--- — ------ ----- _ -------- --- — _— —
01
0)-Катионмеди (CvH) ft-Катион цинка (Zn?)
®-Анион Sot' 9-Электрон
Рис. 86 Возникновение э. д. с. в элементе Якоби — Даниеля? пгт пористая диафрагма, проницаемая для ионов
Гальваническая цепь может быть составлена из пар самых разнообразных металлов, из которых каждый погружен в раствор своей соли. Например:
Кадмиево-цинковая цепь Cd | Cd2+ |i Zn2+ | Zn; eCd „ eZn
Серебряно-медная цепь Ag | Ag+ || Cu2+ | Си;
- Л eAg еСи
Водородно-цинковая цепь (Pt) Н21 2Н+ || Zn2+ | Zn 8н eZn
Э. д. с. всех гальванических элементов слагается из величин потенциалов, возникающих на всех границах раздела. Без учета диффузион-
— 276 —
кого потенциала основное уравнение э. д. с. будет иметь вид: <
£ = Е1-е2, (VII, 13)
где Е — э. д. с. гальванического элемента, 8т и е2 — электродные потенциалы.
Таким образом, э. д. с. любого гальванического элемента равна разности его электродных потенциалов. Уравнение (VII, 13) широко используется в электрохимии, и на нем основаны все расчеты, связанные с работой гальванических элементов.
В качестве примера рассмотрим, чему будет равняться э. д. с. только что рассмотренного гальванического элемента Якоби—Даниеля, если концентрации (активности) ионов цинка и меди равны между собой, т. е. aZn2+ = aCu2+. Для наглядности расчета запишем эту цепь:
Си | Си2+ || Zn2+ | Zn.
8Си 8Zn
На основании уравнения (VII, 13) э. д. с. этой цепи будет равна;
£ = 8cu~- 8zn • (VII. 14)
Используя уравнение Нернста (VI 1,7), можем записать:
eCu‘=-yIgaCu2++eou’ 8zn='Tfgazns++ eon- (VH. 15)
G учетом этого уравнение (VII, 14) может быть представлено так: £ = TlgaCu2++e0U+JFlgaZn2 + -80n’
или
М
E=eCu—ezn^—(iga —jga ). (VII, 16)
U v VuU 411
Поскольку по условию задачи aCu2+ = aZn2+ уравнение (VI1,16) еще более упростится:
E=ecu_gzn. (VI1, 17)
Таким образом, э. д. с. гальванического элемента, составленного из двух разных электродов, но с одинаковой концентрацией (активностью) их солей, равна разности стандартных потенциалов этих элементов.
Подставляя из табл. 63 численные значения s^u и 8z0n, получим:
£== +0,34 —(—0,76) = 1,1в.
Опыт показывает, что эта величина очень хорошо совпадает с экспериментально найденной э. д. с. медно-цинкового элемента.
§ 83. Диффузионный потенциал. Биологическое значение диффузионных и мембранных потенциалов
Говоря о гальваническом элементе, мы рассматривали только границу раздела металл — раствор его соли. Теперь обратимся к границе раздела между растворами двух различных электродов. В гальванических элементах на границах соприкосновения растворов могут возникать так называемые диффузионные
— 277 —
\
1
потенциалы. Они возникают не только на границе раздела между растворами различных электролитов, но и на границе между растворами одного и того же электролита в том случае, когда концентрация растворов неодинакова. Причина возникновения потенциала в подобных случаях заключается в неодинаковой подвижности ионов в растворе.
Представим себе два соприкасающихся раствора соляной кислоты различной концентрации (рис. 87). Пусть концентрация левого раствора Cj больше концентрации правого С2. В этом случае НС1 диффундирует в направлении, показанном стрелками, т. е. слева направо. Поскольку ионы водорода обладают примерно в пять раз большей подвижностью, чем ионы хлора, они при своем передвижении будут опережать ионы хлора. За то время, в течение которого через границу раздела двух растворов в область меньшей концентрации перейдет, скажем, 50 ионов
на । на на | ка
io,5h. 1 а «г— о,1 н. ।
I I
г
I I
Рис. 87. Схема возникновения диффузионного потенциала
водорода, ионов хлора успеет перейти только 10.3а счет этого явления в области меньших концентраций окажется избыточный положительный заряд, а в области меньших концентраций — отрицательный. Наличие такого двойного электрического слоя и является причиной скачка потенциала.
^Скачок потенциала на границе между неодинаковыми по составу или по концентрации растворами называется диффузионным потенциалом.
Величина диффузионного потенциала зависит, как показывает опыт, от различия подвижностей ионов, а также от различия концентраций соприкасающихся растворов. В табл. 67 приведены значения диффузионных потенциалов для целого ряда растворов, концентрации которых находятся приблизительно в отношении 1:10.
Таблица 64
Величина диффузионных потенциалов
Соприкасающиеся растворы электролитов 1 Диффузионный потенциал, б‘ Соприкасающиеся растворы электролитов Диффузионный потенциал, в
КС1|НС1 KCl|NaCl NH4Cl|NaCl KC1|HNO3 —0,0357 4-0,0011 4-0,0098 —0,0378 NaCl NaCl NaOH NaOH HC1|HC1 КСЦКС1 —0,0187 —0,0346 4-0,0378 —0,0004
Диффузионный потенциал можно определить экспериментально, а также вычислить. Так, величина диффузионного потенциала (ед), возникающего при соприкосновении растворов различной концентрации одного и того же электролита, дающего однозарядные ионы, вычисляется по формуле
ед = ^---у 0,0577 1g —, (VII. 18)
/к + /в °2
где ZK и 4» — соответственно подвижности катиона и аниона; а± и а2 — активности соприкасающихся электролитов, причем ах > а2. Если бинарный электролит дает два двухзарядных иона, правая часть выражения (VII, 18) должна быть разделена на 2.
Если соприкасаются между собой два бинарных электролита, дающие однозарядные ионы и имеющие равные концентрации, диффузионный потенциал вычисляется по следующей формуле;
ед = 0,0577 1g
(VII, 19)
где ZK и Za — подвижности ионов одного электролита: Zk и l'a — подвижности
ионов другого электролита.
При точных вычислениях э. д. с. гальванических цепей обязательно должна вводиться поправка на величину диффузионного потенциала. Однако на практи-
ке чаще всего устраняют влияние диффузионного потенциала, включая между растворами электролитов насыщенный раствор хлорида калия. Так как подвижность ионов калия и хлора примерно одинакова (Z + = 64,4 и Z =65,5cw1-c.4t2),
то диффузионный потенциал, вызываемый таким электролитом, будет равен
нулю.
Обычно пользуются насыщенным раствором КО, который помещают в специальную U-образную трубку (сифон). Для того чтобы раствор КС1 не выливался из сифона, хлорид калия предварительно растворяют в горячем 1%-ном растворе агар-агара и уже потом этим раствором заливают сифон. После охлаждения раствор агар-агара застывает в виде студня, который обладает почти такой же электропроводностью, как и чистый раствор КС1.
Соединение двух полуэлементов (электродов) с помощью агар-агарового сифона или, как его еще называют, электролитического ключа, показано на рис. 88. С учетом устранения диффузионного потенциала с помощью насыщенного раствора КС1 гальванический элемент Якоби —Даниеля изображают следующей схемой:
Си | Си2+ | КС1 | Zn2+ | Zn
8Си 8Zn
Аналогично изображают и другие гальванические цепи, например водородно цинковую цепь:
(Pt) Н2 | 2Н+ | КО | Zn2+ | Zn и т. д.
8н 8Zn
Диффузионные потенциалы могут возникать и в биологических объектах при повреждении, например, оболочек клеток. При этом нарушается избирательность их проницаемости и электролиты начинают диффундировать в клетку или из нее— в зависимости от разности концентраций. В результате диффузии электролитов возникает так называемый потенциал повреждения, который может достичь величин порядка 30—40 милливольт. Причем поврежденная ткань заряжается отрицательно по отношению к неповрежденной.
Диффузионный потенциал может сильно возрасти, если растворы электролитов различных концентраций разделить специальной мембраной, проницаемой только для ионов какого-нибудь знака. На рис. 89 приведено схематическое изображение мембраны, проницаемой только для катионов. Это свойство мембраны обусловлено тем, что имеющиеся на ее поверхности отрицательно заряженные
279 —
свободные карбоксильные группы притягивают и пропускают только катионы; анионы как одноименно заряженные частицы отталкиваются. Существуют мембраны, которые пропускают через себя только анионы. Примером подобных мембран являются, например, оболочки эритроцитов, в которых избирательность мембраны обусловливается положительно заряженными аминогруппами.
В ряде случаев возникновение мембранного потенциала связано с тем, что поры мембраны не соответствуют размерам ионов определенного знака. Мембранные потенциалы весьма стойки и могут без изменения сохраняться долгое время В* тканях растительных и животных организмов, даже внутри одной клетки/ ^имеются мембранные и диффузионные потенциалы, обусловленные химической 'и морфологической неоднородностью внутриклеточного содержимого.
Рис. 88. Схема электролитического ключа: Рис. 89. Мембранный потенциал,
1 — агар-агаровый сифон с раствором КС1; 2 и обусловленный отрицательным 33-
з —электроды ' рядом мембраны
Различные причины, изменяющие свойства микроструктур клетки, приводят к освобождению и диффузии ионов, т. е. к появлению различных биопотенциалов и биотоков. Роль этих биотоков в настоящее время еще до конца не изучена, но имеющиеся экспериментальные данные свидетельствуют об их важном значении в процессах саморегуляции
§ 84. Концентрационные цепи
Известны гальванические элементы, в которых электрическая энергия образуется не за счет химической реакции, а за счет разницы концентраций растворов, в которые опущены электроды из одного и того же металла. Такие гальванические элементы называются концентрационными. В качестве примера можно назвать цепь, составленную из двух цинковых электродов, погруженных в растворы ZnSO4 различной концентрации (рис. 90):
Zn | Zn2+ (С\) |j Zn2+ (СЁ) I Zn
Si e2
В этой схеме Сг и С2 — концентрации электролитов, причем Сг > Са, металл обоих электродов один и тот же, стандартные потенциалы их (ezon) также одинаковы. Однако из-за различия концентрации катионов металла равновесие
Zn Zn2+ ч-2е-
— 280 —
в растворе в обоих полуэлементах неодинаково. В полуэлементе с менее концентрированным раствором (С2) равновесие несколько сдвинуто вправо, т. е.
Zn Zn2+ 4-
В этом случае цинк посылает в раствор больше катионов, что приводит к возникновению на электроде некоторого избытка электронов. По внешней цепи они будут перемещаться ко второму электроду, погруженному в более концентрированный раствор сульфата цинка ZnSO4.
Таким образом, положительный заряд будет иметь электрод, погруженный в раствор большей концентрации (Сх), а отрицательно зарядится электрод, погруженный в раствор меньшей концентрации.
В процессе работы гальванического элемента концентрация Сг постепенно уменьшается, концентрация С2 — увеличивается. Элемент работает до тех пор, пока сравняются концентрации у анода и катода.
Вычисление э. д. с. концентрационных элементов рассмотрим на примере цинкового
Рис. 90. Цинковая концентрационная цепь:
М -* агар-агаровый сифон, содержащий хлорид калия
концентрационного элемента.
Допустим, что концентрация Сг = 1 г-ион!л, а С2 — 0,01 г-ион/л. Коэффициенты активности Zn2+ в растворах этих концентраций соответственно равны: fi = 0,061, a f2 = 0,53. Для вычисления э. д. с-цепи воспользуемся уравнением (VII, 13). На основании уравнения Нернста (VI 1,8) можем написать:
Zn 0,0577
Zn °»°577т„ п гл 1g я2,
(VII, 20)
(VII, 21)
где а г и а2 — активности катионов Zn2+ в первом и во втором полуэлементах. Подставляя эти значения ех и е2 в уравнение (VII,13), получим:
0,0577, Zn 0,0577
—— lg0i-egn--——
или
0,0577 , ч
—-— (Ig «1—1g «2)«
(VII, 22)
(VII, 23)
Учитывая, что
а2=f2C2~ 0,53-0,01 = 0,0053;
281 —
получим:
О 0577
Е = -^— (1g 0,061—1g 0,0053).
(VII, 24)
1g 0,061 =2,785=—1,215;
1g 0,0053 = 3,724 = —2,276;
Е = 0,0288 (2,276— 1,215) =0,0288-1,061 = 0,030в.
Таким образом, э. д. с. цинковой концентрационной цепи равна 0,030 в. Общее уравнение для вычисления э. д. с. подобных цепей имеет вид:
п а2
(VII, 25)
Из уравнения (VII, 25) видно, что концентрацию ионов в данном растворе можно легко вычислить, если составить цепь, один из электродов которой будет опущен в исследуемый раствор, а другой — в раствор с известной активностью тех же ионов. Для этой цели’ необходимо только измерить э. д. с. составленной цепи» что может быть легко сделано
с помощью соответствующей установки. Концентрационные цепи широко используются в практике для определения pH растворов,
произведения растворимости труднорастворимых соединений, а также для определения валентности ионов и констант нестойкости в случае комплексообразования.
§ 85. Электроды сравнения
Как уже отмечалось, потенциалы различных электродов измеряются по отношению к потенциалу нормального водородного электрода. Наряду с водородным в электрохимии в настоящее время широко применяется другой электрод сравнения — так называемый каломельный электрод, который, как показал опыт, обладает постоянным и хорошо воспроизводимым потенциалом.
Водородный электрод. Благородные металлы, например золото, платина и некоторые другие, обладают прочной кристаллической решеткой, и их катионы не переходят в раствор из металла. Следовательно, такие металлы не имеют на границе металл — раствор своего характерного скачка потенциала. Однако если на поверхности этих металлов адсорбируются вещества, которые способны окисляться или восстанавливаться, эти металлы с адсорбированными веществами уже представляют собой системы, находящиеся в равновесии с раствором. Если веществом, адсорбирующимся на поверхности благородного металла, является газ, электрод называется газовым.
Газовым электродом является так называемый водородный электрод. В нем в качестве инертного металла используется платина, на которой адсорбируется газообразный водород. Для увеличения адсорбирующей поверхности платину электролитически покрывают слоем высокодисперсной платины (платиновой чернью). Такие электроды
— 282 —
называются платинированными. Водородный электрод обратим относительно иона водорода и, следовательно, является электродом первого рода.
Таким образом, платиновая пластинка или проволока, поглотившая молекулярный водород и опущенная в раствор, содержащий ионы водорода, представляет собой водородный электрод. Поскольку сама платина не участвует в электродной реакции (ее роль сводится лишь к тому, что она поглощает водород и, будучи проводником, делает
Рис. 91. Разновидность водородного электрода: а — грушевидный: 1 — платиновый электрод; 2 стеклянная трубка; 3— краны; 4 — трубка для подачи водорода; 5 — гидравлический затвор; 6 — сифон; б — колоколообразный: 1 платиновая пластинка, 2 — стеклянная трубка; 3 — ртуть
возможным перемещение электронов от одного электрода к другому), химический символ платины в схеме водородного электрода обычно заключают в скобки:
(Pt) Н2|2Н+ /
Существуют различные конструкции сосудов для водородного электрода, две из которых показаны на рис, 91.
На поверхности водородного электрода устанавливается равновесие
Н2^±2Н^±2Н+ + 2е~
В результате этих процессов на границе между платиной и раствором ионов водорода образуется двойной электрический слой, обусловливающий скачок потенциала. Величина этого потенциала при данной температуре зависит от активности водородных ионов в растворе и от
— 283 —
количества поглощенного платиной газообразного водорода, которое пропорционально его давлению: 1
ен = —^-lgPH2+MlgaH + , (VII, 26)
где пн+ — активность водородных ионов в растворе, Рн2 — давление, под которым поступает для насыщения электрода газообразный водород. Опыт показывает: чем больше давление для насыщения платины водородом, тем более отрицательное значение принимает потенциал водородного электрода.
Электрод, состоящий из платины, насыщенной водородом под давлением в 1 атм и погруженной в водный раствор с активностью ионов водорода, равной единице, называется нормальным водородным электродом. По международному соглашению потенциал нормального водородного электрода условно принят равным нулю, с этим электродом сопоставляют потенциалы всех других электродов.
В самом деле, при Рн2 = 1 атм выражение для потенциала водородного электрода будет иметь вид:
ен — MlgaH+ или 8Н == Ж 1g [Н+]. (VII, 27)
Уравнение (VII, 27) справедливо для разбавленных растворов.
Таким образом, при насыщении водородного электрода водородом под давлением в 1 атм (Рн2 = 1 атм) потенциал его зависит только от концентрации (активности) водородных ионов в растворе. В связи с этим водородный электрод может применяться на практике не только как электрод сравнения, но и как индикаторный электрод, потенциал которого находится в прямой зависимости от присутствия Н+-ионов в растворе.
Практически приготовление водородного электрода представляет значительные трудности. Нелегко добиться, чтобы давление газообразного водорода при насыщении платины равнялось точно 1 атм. Кроме того, газообразный водород должен поступать для насыщения со строго постоянной скоростью, к тому же для насыщения необходимо применять совершенно чистый водород, так как уже весьма малые количества некоторых примесей, особенно H2S и H3As, «отравляют» поверхность платины и тем самым препятствуют установлению равновесия Н2 2Н+ + 2е~. Получение же водорода высокой степени чистоты связано со значительным усложнением аппаратуры и самого процесса работы. Поэтому на практике чаще применяется более простой каломельный электрод, обладающий устойчивым и отлично воспроизводимым потенциалом.
Каломельный электрод. Неудобства, связанные с практическим применением водородного электрода сравнения, привели к необходимости создания других, более удобных, электродов сравнения, одним из которых является каломельный электрод.
Существуют различные конструкции сосудов каломельного электрода (рис. 92). Для приготовления каломельного электрода на дно сосуда наливают тщательно очищенную ртуть. Последнюю сверху покрывают пастой, которая получается растиранием каломели Hg2CI2
— 284 —
с несколькими каплями чистой dtv™ с
калия KCI. Поверх пасты наливают раство1ГкГ|ИИ раствора хлоРВДа мелью. Металлическая ртуть добя и пят.,оВОР ’ насыш£нныи кало-
окисления каломели до ртути НрС1 рмая в пастУ. предохраняет от проволоку, от которой Уже плат мапио ртуть погРУ>кают платиновую мельный электрод схематически запиеываетсТсХ“ КЛемм% Кало' иисывается следующим образом:
Hg | Hg2 С1г, КС1
Запятая между Нр„С1 и ктч '
нет поверхности раздела так как "0™ Л° Между этими веществами и дела, так как они находятся в одном растворе.
Рис. 92. Каломельные электроды:
/ — ртуть; 2 —платиновая проволока; 3 — паста из ртути, каломели и раствора хлорида калия; 4 —отросток для соединения каломельного электрода с другими электродами; 5 — раствор хлорида калия
Рассмотрим, как работает каломельный электрод. Каломель, растворяясь в воде, диссоциирует с образованием ионов Hg+ и С1~
HgaCh^2Hg+ Ч-2СН
В присутствии хлорида калия, содержащего одноименный с каломелью ион хлора, растворимость каломели снижается. Таким образом, при данной концентрации КС1 и данной температуре концентрация ионов Hg+ будет постоянной, чем собственно и обеспечивается необходимая устойчивость потенциала каломельного электрода.
Потенциал (ек) в каломельном электроде возникает на поверхности соприкосновения металлической ртути с раствором ее ионов и может быть выражен следующим уравнением:
= + Л 1g lHg+1.
(VII, 28)
285 —
Ввиду малой способности ртути переходить в раствор потенциал имеет положительный знак по отношению к потенциалу нормального водо родного электрода. v ]
Каломель Hg2Cl2— вещество, трудно растворимое в воде. Прг 25° С ее произведение растворимости равно:
FIP = [Hg+] [С1~] — 2»10“18. (VII, 29)
Так как ПР при постоянной температуре есть величина постоянная, увеличение концентрации иона хлора может оказать существенное влияние на концентрацию ионов ртути, а следовательно, и на потенциал каломельного электрода.
Из уравнения (VI 1,29):
ПР
[Hg+] = ——; • (VII, 30)
Подставляя это выражение в уравнение (VII, 28), получим:
ПР
eK = e^ + ^lg—— (VII, 31)
[Cl J
или
ек = eHg + Ж 1g (ПР) —Ж 1g [Cl- ]. (VII, 32)
Объединяя постоянные при данной температуре величины во и Ж 1g (ПР) в одну величину и обозначая ее через е£, получим уравнение потенциала каломельного электрода:
= 2/<lg[Cl-]. (VII, 33)
Таким образом, потенциал каломельного электрода в конечном итоге зависит от концентрации (активности) ионов хлора в растворе, находящемся над слоем каломели Hg2Cl2, поэтому каломельный элек- -грод может быть отнесен к электродам второго рода. j
В насыщенном растворе КС! при 18° С потенциал каломельного электрода 8К = 0,2503 в\ в случае 1 н. раствора КС! 8К = 0,2864 в, в 0,1 н. КС1 8К — 0,3380 в. В практике чаще всего применяют каломельные электроды двух типов — с однонормальным раствором КС! и с насыщенным раствором этой соли.
Пользуясь каломельным электродом, можно опытным путем определить потенциал любого электрода. Так, для определения потенциала цинкового электрода составляют гальваническую цепь из цинка, погруженного в раствор ZnSO4, и каломельного электрода
Hg | Hg2 Cl2, КС11 KCI | ZnSO41 Zn eZn
Допустим, что экспериментально определенная э. д. с. этой цепи дает величину Е = 1,0103 в. Потенциал каломельного электрода 8К =» — 0,2503 в. Потенциал цинкового электрода будет равен:
£===tK~eZn»
откуда
eZn=£K-^
— 286 —
1
пли
eZn = 0,2503 — 1,0103 = — 0,76e.
Рис. 93. Хлорсеребряный электрод:
1 ~ серебряная проволока; 2 — слой AgC ; 3 — раствор НС1 или КС
Заменяя в данном элементе цинковый электрод медным, можно определить потенциал меди и т. д. Таким образом можно определить потенциалы почти всех электродов.
Хлорсеребряный электрод. Помимо каломельного электрода, в лабораторной практике в качестве электрода сравнения широкое распространение получил также хлорсеребряный электрод. Этот электрод представляет собой серебряную про' волоку или пластинку 7, припаянную к медной проволоке и впаянную в стеклянную трубку. Серебро электролитически покрывают слоем хлорида серебра 2 и помещают в раствор КС1 или НО. На рис. 93 показана одна из конструкций этого электрода.
Потенциал хлорсеребряного электрода, так же как и каломельного, зависит от концентрации (активности) ионов хлора в растворе и выражается уравнением:
8xc = ^-^lg(Cl*], (VII, 34)
где вхс — потенциал хлорсеребряного электрода, е*с — нормальный потенциал хлорсеребряного электрода. Схематически хлорсеребряный электрод записывается следующим образом:
Ag | AgCl, КС1
8ХС —
Потенциал этого электрода возникает на границе раздела Ag|Ag+. При этом имеет место следующая
электродная реакция
AgCl + е~
Ag Ц- С]
Ввиду чрезвычайно малой растворимости AgCJ потенциал хлорсеребряного электрода имеет положительный знак по отношению к нормальному водородному электроду.
В 1 н. растворе КО потенциал хлорсеребряного электрода по водородной шкале при 25° С равен 0,2381 в, а в 0,1 н. растворе ехс == — 0,2900 в и т. д. По сравнению с каломельным электродом хлорсеребряный электрод имеет значительно меньший температурный коэффициент, т. е. его потенциал в меньшей степени изменяется с температурой.
§ 86. Индикаторные электроды
Для определения концентрации (активности) различных ионов в растворе электрометрическим методом на практике используются гальванические элементы, составленные из двух электродов — электрода сравнения с устойчивым и хорошо известным потенциалом и индикаторного, потенциал которого зависит от концентрации (активности)
—. 287 —
определяемого иона в растворе. В качестве электродов сравнения наи!’ более часто применяют каломельный и хлорсеребряный электроды^ Водородный электрод для этой цели в силу его громоздкости употреб! ляют значительно реже. Гораздо чаще этот электрод используют в качестве индикаторного электрода при определении активности водород! ных ионов в исследуемых растворах (pH). 1 Остановимся на характеристике индика! торных электродов, получивших за послед! ние годы наиболее широкое распространение! в различных областях народного хозяйства!
Хингидронный электрод. Одним из ши-1 роко распространенных в практике элект! родов, потенциал которых зависит от актив! ности водородных ионов в растворе, являет! ся так называемый хингидронный электрод! Этот электрод весьма выгодно отличается от’ водородного электрода своей простотой и удобством в работе. Он представляет собой; платиновую проволоку /, опущенную в сосуд с исследуемым раствором 2, в котором, предварительно растворяют избыточное коли-' чество порошка хингидрона 3 (рис. 94).
Хингидрон представляет собой эквимолекулярное соединение двух органических соединений — хинона С6Н4О2 и гидрохинона С6Н4(ОН)2, кристаллизующихся в виде мелких темно-зеленых с металлическим блеском а гидрохинон — двухатомным
Рис. 94. Хингидронный электрод:
—» платиновая проволока; — исследуемый раствор, котором растворен, хин-3 —. избыток хин-
2 в гидрон;
гидрона; 4 — ртуть; 5— мед-
ная проволока
игл. Хинон является дикетоном, спиртом
С=0 С—ОН
/\ /\ НС С НС СН
II II II II
НС СН НС СН
^сСо ^С^-ОН
хинон гидрохинон
В состав хингидрона входит одна молекула хинона и одна молекула гидрохинона С0Н4О2 -С6Н4(ОН)2. При приготовлении хингидронного электрода хингидрон всегда берут в количестве, гарантирующем на- I сыщенность им раствора, т. е. он должен оставаться частично не растворившимся в осадке. Необходимо отметить, что насыщенный раствор получается при внесении очень маленькой щепотки хингидрона, так как его растворимость в воде составляет всего около 0,005 моль на 1 л воды.
Рассмотрим теорию хингидронного электрода. При растворении в воде происходят следующие процессы: хингидрон распадается на хинон и гидрохинон
С6Н4О.2.СбН4(ОН)2#С6НаО2 + С0Н4(ОН)а (а)
— 288 —
Гидрохинон, являясь слабой кислотой, в незначительной степени дис-еоциирует на ионы по уравнению
СвН, (ОН)8#СвН4О|-+2Н+ (б)
В свою очередь образовавшийся ион хинона может окисляться в хинон при условии отвода электронов
С6 Н4 Оо"~ С6 Н4 О2 4- 2е~ (в)
Суммарная реакция, протекающая на катоде, будет иметь следующий вид
С6 Н4 (ОН)2 Z=±-Ce Н4 о2 ч- 2Н+ 4- 2е-
Константа равновесия этой реакции равна: [С6Н4О2][Н+р
(г)
: (VI 1,35)
К ’ [С6 н4 (ОН)2]
Благодаря тому, что в растворе, насыщенном хингидроном, концентрации хинона и гидрохинона равны, концентрация водородного иона будет величиной постоянной. •
Хингидронный электрод можно рассматривать как водородный рри очень малом давлении водорода (приблизительно Ю~24 атм). Предполагают, что в этом случае вблизи электрода протекает следующая реакция
Сс Н4 (ОН)2 Сб Н4 О2 4- Н2
Образующийся газообразный водород насыщает под таким давлением платиновую проволоку или пластинку, опущенную в раствор.
»' Электроны, образующиеся согласно реакции (г), переходят на платину, в силу чего возникает разность потенциалов между платиной и прилегающим раствором. Таким образом, потенциал данной системы зависит от соотношения концентраций окисленной и восстановленной форм и от концентрации ионов водорода в растворе. С учетом этого уравнение электродного потенциала хингидронного электрода имеет вид:
[Сб н4 o2j [Н+р [СбН4(ОН)2] ’
(VI 1,36)
где —• стандартный потенциал хингидронного электрода.
Так как в насыщенном растворе хингидрона отношение
[С6н4О2] [CeH^OHh]” *
то уравнение (VI 1,36) примет вид:
8xr=^r4-MlgrH+]. (VII,37)
Из формулы (VI 1,37) видно, что потенциал хингидронного электрода находится в прямой зависимости от концентрации (точнее, от активности) водородных ионов в растворе.
10 Зак. 560
— 289 —
В результате практических измерений было установлено, что нор* мальный потенциал хингидронного электрода == 1) равен 0,7044 в при 18° С. Поэтому, подставляя в уравнение (VII,37) вместо е*г и Ж их численные значения, получим окончательное уравнение потенциала хингидронного электрода:
8ХГ «= 0.7044 4 0,0677 1 g (Н+]. (V11,38)
Таким образом, потенциал хингидронного электрода, так же как и водородного, зависит при данной температуре только от концентрации (активности) водородных ионов в растворе.
Сурьмяный электрод. Электроды, составленные, из некоторых металлов и их труднорастворимых оксидов, носят название металлооксидных электродов. Будучи погруженными в раствор, они также могут играть роль своеобразного водородного электрода. При этом потенциал металлооксидных электродов определяется равновесием!
w Me 4 пНг О Mem О„ 4- 2яН4 4- 2 л/? ~
Схематически эти электроды можно представить так;
Me I Mem Оп | H+
Для получения металлооксидных электродов можно использовать сурьму, вольфрам, ртуть, свинец, серебро и другие металлы.
Наиболее широкое распространение в практике получил так называемый сурьмяный электрод:
I Sb|SbjOs|H^
который представляет собой брусок сурьмы, отлитый на воздухе. Как известно, сурьма па воздухе покрывается слоем оксида, который при соприкосновении с водой образует гидрат. На электроде устанавливается следующее равновесие
2Sb 4 Зну О Sb? 4 6Н+ 4 6е-
или
-|-Sb +~-НгОг± Д-Sb, °з + Н++ е-И Z о
Сурьма и ее окись рассматриваются как твердые вещества, находящиеся в стандартных состояниях, когда активности равны единице. Поскольку активность воды в разбавленных растворах приближенно равна единице, потенциал сурьмяного электрода (eSb) должен быть функцией концентрации (активности) ионов водорода в растворе:
= 1g 1Н+], (VI j ,39)
где es£ — стандартный (нормальный) потенциал сурьмяного электрода. Опыт показывает, что сурьмяный электрод не обеспечиваем высокую точность измерения pH. Состояние металла (плавленный или электролитически осажденный, полированный или травленый) и окиси сурьмы оказывает определенное влияние на поведение электрода. Точность измерения pH с помощью сурьмяного электрода
— 290 —
может быть значительно повышена (до ± 0,1 pH) путем тщательного калибрования его потенциала в серии буферных растворов с известным значением pH. Дальнейшее определение pH исследуемых раство-' ров проводят уже по этой калибровочной кривой или прямо по шкале потенциометра, градуированной в единицах pH»
Стеклянный электрод. Этот электрод в настоящее время получил самое широкое распространение.
Для изготовления стеклянного электрода применяют стекло определенного химического состава»
А Б В
Рис. 95. Виды стеклянных электродов:
Л — стеклянный электрод Мак—Иннеса; Б — шариковый электрод; В — электрод с защитной муфтой: Г — электрод с вогнутой мембраной; 1 — стеклянная мембрана; 2 — 0,1 н, раствор НС1; 3 — хлорсеребряный электрод; 4 — наружная (утолщенная) часть электрода; 5 — защитная муфта
Одной из наиболее часто употребляющихся форм стеклянного электрода является стеклянная трубка, заканчивающаяся тонкостенным шариком. Шарик заполняют раствором НС1 с определенной концентрацией ионов Н+, в который погружен вспомогательный электрод (например, хлорсеребряный). Иногда стеклянные электроды изготовляют в виде тонкостенной мембраны из стекла, обладающего водородной функцией. Мембрана припаивается к концу стеклянной трубки. Для микроанализа используют также стеклянные электроды с вогнутой во внутрь стеклянного шарика мембраной (рис. 95).
Стеклянный электрод отличается от уже рассмотренных электродов тем, что в соответствующей ему электродной реакции не участвуют электроны. Наружная поверхность стеклянной мембраны служит источником водородных ионов и обменивается ими с раствором подоб
10*
— 291 —
но водородному электроду. Иными словами, электродная реакция сводится здесь к обмену ионами водорода между двумя фазами — раствором и стеклом:
Н+ = Нст
Поскольку заряд водородного иона соответствует элементарному положительному количеству электричества и переход иона водорода из одной фазы в другую эквивалентен перемещению единичного заряда (п = 1), потенциал стеклянного электрода (ест) может быть выражен следующим уравнением:
ест = е^ + Ж 1g _Hi. , Г1ст
(VI 1,40)
где е” — стандартный потенциал стеклянного электрода.
Как показали исследования, в реакцию обмена, помимо ионов водорода, вовлекаются также входящие в состав стекла ионы щелочного металла. При этом они частично заменяются на ионы водорода, а сами переходят в раствор. Между поверхностным слоем стекла и раствором устанавливается равновесие ионообменного процесса:
Нет “Ь М+ { * Н+ Мет
где М+ в зависимости от сорта стекла могут быть ионами лития, натрия или другого щелочного металла.
Условие равновесия этой реакции выражается законом действующих масс:
Кобм —
н * мст
анс+ ам+
(VII,41)
Величина этой константы обмена зависит от свойств стекла, из которого изготовлен электрод, а также от температуры. Для исследованных до настоящего времени стекол значение К колеблется от 10-1 до 10-15.
Исходя из предположения, что в стекле данного сорта сумма активностей ионов водорода и ионов щелочного металла постоянна, т. е*
+ + — а*
V» —* *
(VI 1,42)
уравнение константы обмена (VII,41) можно переписать в следующем виде:
Л ОбМ —
(VI 1,43)
Решая это уравнение относительно ан+/ан + , получим ан+ _ °н++^обм °м+ внс+ ~ а
(VII,44)
— 292 —
Замена а +7ан + в УРавнении электродного потенциала стекла (VII 40) его значением из уравнения (VI 1,44) приводит к следующему выражению:
(VI Г. 45)
где постоянная Ж Iga включена в стандартный потенциал стеклянного электрода &от-
Таким образом, в общем случае потенциал любого стеклянного э ектрода обусловливается двумя величинами: активностью ионов водорода и активностью щелочного металла,
Если в растворе ан+ > /<обм ам, то
е0т = 8Г+ ^'gaH+=8ST—^Рн‘
(VI 1,46)
т. е. электрод обладает водородной функцией и потому может служить индикаторным электродом при "Определении pH.
Если в растворе ан+ < Кобм ам+, то
ест = еМ = 8$Т + Ж ‘g Кобм + ж 'g ам+
или
(VH.47)
т. е. электрод обладает металлической функцией. В этом уравнении в величину е™ входит слагаемое, содержащее константу обмена, т. е, e^==8c0T + Mig/<o6M. (VII.48)
Стеклянный электрод с металлической функцией может использоваться в качестве индикаторного электрода для определения активности ионов соответствующего щелочного металла.
Таким образом, в зависимости от сорта стекла (точнее, от величины константы обмена) стеклянный электрод может обладать водородной и металлической функцией.
Изложенные представления о стеклянном электроде лежат в основе термодинамической теории стеклянного электрода, разработанной Б. П. Никольским (1937) и основанной на представлении о существовании обмена ионами между стеклом и раствором.
Схематически стеклянный электрод с водородной функцией можно записать так:
Ag | Ag Cl | HC11 стекло | H+ (раствор) exc eo ест
В качестве внутреннего электрода здесь взят хлорсеребряный электрод.
Ввиду того, что в уравнении стеклянного электрода (VII,46) величина Ж на практике получается несколько меньше теоретической и е£т зависит от сорта стекла и даже от способа приготовления электрода (т. е. является неустойчивой величиной), стеклянный электрод (так же как и сурьмяный) перед определением pH исследуемого раствора
— 293 —
I
предварительно калибрируют по стандартным буферным растворам, pH которых точно известен.
На рис. 96 показан калибровочный график стеклянного электрода. На оси абсцисс откладывают величины pH буферных растворов, а на оси ординат — значения э. д. с. гальванической цепи, составленной
из стеклянного электрода и электрода сравнения.
Преимущество стеклянного электрода перед водородным и хингидронным электродами заключается в том, что он позволяет определять
Рис. 97. Натрий-стеклянный электрод:
1 — стеклянный тонкостенный шарик; 2 — нерастворимая соль HgaSO^ 3 — 1 н. раствор Na2SO<;
4 — платиновая проволока; 5 — стеклянная трубка; 6 — оправа и ручка для крепления электрода; 7 <— экранированный провод; 8 — муфта для присоединения к потенциометру
Рис. 96. Кривая ка-1 либрования стеклянного электрода
pH раствора любого химического соединения в достаточно широком диапазоне значений. К недостаткам стеклянного электрода следует прежде всего отнести его хрупкость и большое внутреннее сопротивление. Обычно для изготовления стеклянного электрода используют стеклянные мембраны с толщиной стенок от 0,01 мм и меньше. Так как стеклянный электрод имеет высокое сопротивление (порядка нескольких десятков мегом) и проводит очень малый ток (10~7 — 10~8‘я), измерение э. д. с. гальванических элементов, составленных с его участием, возможно только с помощью усилительной схемы — электронным ламповым потенциометром. В целях предупреждения утечки тока необходимо использовать экранированные провода с хорошей изоляцией.
Натрий-стеклянный электрод. За последние 10—15 лет в практике лабораторных исследований находят все более широкое применение стеклянные электроды
— 294 —
металлической функцией. Они обратимо (в соответствии с уравнением электродного потенциала Нернста) отвечают изменением своего потенциала на изменение концентрации (активности) ионов металла в растворе [уравнение (VII, 47)].
Наибольшее практическое применение в лабораторной практике за послед-' ние годы нашел так называемый натрий-стеклянный электрод.
Его обычно изготовляют в форме трубочки с выдутым на конце тонкостенным шариком. Внутреннее заполнение натрий-стеклянного электрода отличается от стеклянного электрода с водородной функцией. Шарик электрода заполняют 1 н. раствором Na2SO4, в который добавляют сульфат ртути (I) Hg2SO4 так, чтобы на дне шарика оказался небольшой ее нерастворившийся осадок. Для отвода потенциала во внутрь натрий-стеклянного электрода вставляется амальгамированный платиновый контакт (рис. 97).
Таким образом, схематически натрий-стеклянный электрод можно представить так:
(Pt) Hg | Hg2 SO4, Na2 SO41 стекло | Na+ (раствор) eHg e0 eNa
При погружении такого электрода в раствор, содержащий ионы натрия, на внешней поверхности стеклянной мембраны возникает потенциал (eNa), величина которого может быть выражена уравнением:
i
' eNa«e0Na+MIgaNa+. . ; .(VII ,49)
Таким образом, потенциал натрий-стеклянного электрода находится в прямой зависимости от концентрации (активности) ионов натрия в растворе. Опыт показал, чтот этот электрод может обладать очень высокой специфичностью: посторонние катионы и анионы, находящиеся в растворе, оказывают сравнительно малое влияние на величину его потенциала.
Ввиду неустойчивого значения на практике производят калибрование натрий-стеклянного электрода по, стандартным растворам натрийсодержащих солей. По этой калибровочной кривой и проводят в дальнейшем определение концентрации ионов натрия в исследуемых растворах.
Помимо натриевой функции стеклянные электроды, приготовленные из специальных сортов стекла, могут обладать, например, калиевой функцией. Однако эти электроды не нашли широкого применения в практике и потому здесь не рассматриваются. •‘а
§ 87. Методы измерения электродвижущих сил
Электродвижущая сила любого гальванического элемента может быть измерена либо включением в цепь чувствительного вольтметра, либо компенсационным методом. В первом случае через цепь обязательно протекает электрический ток, во втором — э. д. с. измеряется при отсутствии тока. В практике методом непосредственного измерения э. д. с. не пользуются. И вот почему.
Обозначим внутреннее сопротивление элемента /?2; сопротивление самого вольтметра 7?х; истинную э. д. с. измеряемого гальванического элемента Е; силу тока в цепи /; на основании закона Ома получим:
ИЛИ £ = //?! + //?2=£1+£2-
(VI 1,50)
В этом случае стрелка вольтметра, включенного в цепь, будет показывать только падение напряжения Еъ которое носит название
- 295 —
напряжения на клеммах и представляет собой только часть истинной э9 д. с.:
Z?i==E—Е2=Е-----— . (VII.51)
1+-^-Ri
Из этого уравнения видно, что показания вольтметра могут приближаться к истинному значению э. д. с. элемента только в том случае, если /?2 (что не всегда практически можно выполнить). При работе гальванического элемента в нем происходят различные изменения концентрационного и химического характера, которые
Рис. 98. Нормальный кадмиевый элемент:
/ — ртуть; 2 — паста HgaSO4 и CdSO4; 3 — амальгама кадмия; 4 •— насыщенный раствор и кристаллы CdSO4
ведут к непрерывному уменьшению э. д. с. (так называемое явление поляризации). В этом легко убедиться, если в течение сравнительно длительного промежутка времени проследить за показаниями стрелки вольтметра, включенного в цепь: показания будут непрерывно уменьшаться. Таким образом, измерить истинное значение э. д. с. гальванического элемента с помощью чувствительного вольтметра даже при кратковременном включении его в цепь не представляется возможным.
Компенсационный метод измерения э. д. с. гальванических элементов лишен всех перечисленных
выше недостатков. Однако прежде чем перейти к изложению его сущности, остановимся на работе стандартного элемента, который употребляется в компенсационной установке. •
такие гальванические элементы, кото-для измерения э. д. с. Эти элементы требованиям: строгой воспроизводимо-
Нормальными называются рые могут служить эталоном должны отвечать следующим
стью; минимальным температурным коэффициентом.
Опыт показал, что так называемый элемент Вестона лучше, чем другие элементы, отвечает этим условиям. В элементе Вестона электро
дами являются металлическая ртуть и амальгама кадмия, электролитом — раствор, насыщенный по отношению к сульфатам обоих
металлов.
На рис. 98 показан нормальный элемент Вестона. Он представляет собой Н-образный стеклянный сосуд, в дно которого впаяны платиновые проволочки, служащие полюсами элемента. На дне одного колена находится амальгама кадмия (12,5% кадмия и 87,5% ртути), на дне другого — ртуть, покрытая слоем пасты из сульфата ртути (I) и сульфата кадмия. Пасту получают путем растирания ртути с твердыми
— 296 —
— Н20 в присутствии очень небольшого количества
H^gSO4 И CdSO4 • 2 Н 2^ В Прису ICltSUrl U4CHD nCUUJIDUJUlU ЛМЛНЧСС 1 DO насыщенного раствора CdSO4. Оба колена содержат мелкие кристаллы CdSO4 • 8/3 Н2О (примерно до половины сосуда) и дальше заполнены насыщенным раствором CdSO4. Сверху оба сосуда запаиваются или закрываются парафинированными пробками. Необходимо следить за тем, чтобы между поверхностью раствора и пробкой (или запаянными концами) имелся небольшой пузырек воздуха, так как в противном случае при повышении температуры сосуд может лопнуть.
т
Рис. 99. Схема установки для определения э. д. с.:
/ — аккумулятор; 2 — исследуемый гальванический элемент; 3 — гальванометр; 4 — нормальный элемент; 5 — выключатель (телеграфный ключ); 6 и 7 — переключатели; 8 — подвижный контакт; ас — реохорд •
Схематически нормальный электрод можно представить в следу ющем виде:
Pt, Hg | Hg2 SO41 Cd SO41 Cd + амальгама 1 Pt
Электрическая энергия в нормальном элементе получается за счет химического процесса
Cd + Hg|+ —>Cd2+ + 2Hg
Положительным электродом в элементе служит ртуть, отрицатель ным — амальгама кадмия. Э. д. с. элемента точно измерена и при 20° С равна 1,0183 в. Зависимость э. д. с. от температуры выражается уравнением:
£„ = 1,01830 + 0,000046 (/ — 20). (V11,52)
Э. д. с. нормального элемента при работе не изменяется, так как раствор является насыщенным по отношению к обеим солям.
Теперь приступим к изложению принципа компенсационного метода измерения э. д. с. гальванических элементов.
Сущность метода компенсации состоит в том, что э. д. с. исс ду -мого элемента уравновешивается разностью потенциалов котор получается на части реохорда компенсационной установ (р
— 297 —
Иными словами, в методе компенсации э. д. с. исследуемого элемента сравнивается с известной э. д. с. другого элемента. Для питания реохорда к концам его обычно присоединяют двухвольтовый аккумулятор 1. Проволока реохорда обязательно должна быть однородной по толщине, так как только при этом условии по всей ее длине будет происходить равномерное изменение потенциала.
Для определения э. д. с. исследуемого элемента его подсоединяют в боковую цепь так, чтобы направление тока элемента было противоположно току аккумулятора (т. е. оба элемента присоединяются одноименными полюсами друг к другу). Передвигая ползунок реохорда, можно найти такое положение, при котором гальванометр покажет полное отсутствие тока в боковой цепи. В этом э. д. с. падения напряжения аккумулятора на участке реохорда ab равно э. д. с. исследуемого элемента. Искомая электродвижущая сила будет равна:
Я" . . -
: ЕХ = Е&К~ , , (VII ,53)
ао
где Ех и Еак — соответственно э. д. с. исследуемого элемента и аккумулятора; abx—отрезок проволоки, на котором падение напряжения аккумулятора равно э. д, с. исследуемого элемента.
Если скользящий контакт реохорда сдвинуть с его нулевого значения влево, будет работать исследуемый элемент, если подвижной контакт сдвинуть вправо, через элемент начнет протекать ток от аккумулятора.
Поскольку э. д. с/ аккумулятора изменяется, то непосредственно из уравнения (VI 1,53) э. д. с. исследуемого элемента вычислена быть не может. Чтобы устранить это затруднение, в боковую цепь вместо исследуемого элемента включают нормальный элемент Вестона, э. д. с, которого, как уже отмечалось, точно известна (Еа), После компенсации э, д. с, элемента получим:
иЬ-н 4
£н = еак—г- (VII,54)
ао
где abH — отрезок реохорда, на котором падение напряжения аккумулятора равно э. д. с. нормального элемента, Разделив уравнение (VII,53) на уравнение (VII,68), получим: i
Ех abx
Еи aba
откуда
ЕХ=ЕН--^ . (VII,55)
Таким образом, по уравнению (VII,55) можно вычислить непосредственно величину электродвижущей силы любого гальванического элемента.
— 298 —
§ 88. Электрометрический (потенциометрический) метод определения pH
Концентрацию водородных ионов в растворах наиболее точно можно определять электрометрическим методом. Для этой цели нужно составить гальваническую цепь так, чтобы потенциал одного из электродов находился в зависимости от концентрации ионов Н+. Такими электродами являются рассмотренные ранее водородный, хингидрон-ный, сурьмяный и стеклянный электроды.
Рис 100. Водородно-водородная гальваническая концентрационная цепь
Рассмотрим наиболее употребительные в практике определения pH гальванические цепи.
Водородная цепь. В основу этого метода определения положен принцип измерения э. д. с. в концентрационном элементе, составленном из двух водородных электродов, один из которых погружен в исследуемый раствор, другой, служащий электродом сравнения, — в буферный раствор, pH которого точно известен. На рис. 100 показана водородно-водородная цепь. Схема этой цепи следующая:
(Pt) Н21 Н+ (ст) I КС11Н+ | Н2 (Pt).
еН (ст) еН
Э. д. с. ее равна
(ст)—ен-
В этой цепи более положительный потенциал имеет водородный электрод, имеющий точно известную концентрацию водородных ионов.
— 299 —
Исходя из уравнения электродного потенциала водородного электрода (VII, 27), можем написать:
ен (ст)=^ lg [H+lci и eH=Mlg[H+]. (VII,57)
Подставляя эти значения в уравнение (VI 1,56), получим:
E=^lg[H+lCT—^lg[H+] или Е=Ж(рН —рНст).
откуда легко найти pH исследуемого раствора:
Е Е
Рн==-^-+Рнст или Рн=^^ + Рнст- (VII,58)
В уравнении (VII,58) Е — э. д. с. водородно-водородной цепи» рНст — pH буферного стандартного раствора.
Рис. 101. Каломельно-водородная гальваническая цепь
Если в качестве электрода сравнения взять нормальный водородный электрод, у которого [Н+1ст=1 г-ион/лу уравнение (VII,58) примет еще более простой вид:
РН=^7- (VI,’59)
Каломельно-водородная цепь. В этой цепи водородный электрод является индикаторным электродом, каломельный — электродом сравнения (рис. 101). Испытуемый раствор, находящийся в стаканчике водородного электрода /, соединен с каломельным электродом через солевой мостик 2 с насыщенным раствором КС1. Схематически цепь изображается так:
Hg | Hg Cl2, КС11 KC11H+1 Ha (Pt)
8k 3H
— 300 —
(VI1,60)
н-
a
потенциал
В этой цепи положительным электродом является каломельный элект-под 3' так как концентрация водородных ионов в исследуемом растворе меньше единицы, потенциал водородного электрода — меньше нуля, Э и еет отрицательный знак, Таким образом, э. д. с. каломельноводородной цепи равна.
Е = ек-в
Если потенциал каломельного электрода известен, водородного электрода равен i
ен =//< lg (Н+), нетрудно найти э. д. с. каломельно-водородной цепи:
£==ек — Ж lg [H+J, или — lg [Н+ ]=—тт
zr\
откуда
£ —ен pH , или рр| _
(VII .61)
£—0,2503 0,0577
где 0,2503 — потенциал каломельного (насыщенного) электрода при
Хингидронно-каломельная цепь. Для составления хингидроннокаломельной цепи к исследуемому раствору прибавляют на кончике перочинного ножа хингидрон, размешивают и опускают в раствор неплатинированный платиновый электрод. Исследуемый раствор посредством солевого мостика с насыщенным раствором КО соединяют с каломельным электродом (рис. 102). Схематически эта цепь обозначается следующим образом:
(Pt) Н2 | хингидрон, ехг
Н+1КСЦКС1, Hg.2Cl2|Hg. ек
В ней положительным является хингидронный электрод, а отрицательным — каломельный. Э. д. с. цепи будет равна:
£ = ехг—8К. (V11.62)
Потенциал каломельного электрода известен, потенциал хингидронного электрода равен:
exr = ^r + ?/(lg[H+J. (VII,63)
Подставляя значения потенциалов в уравнение (VI 1,62), получим:
£ =е*г -Ь/К 1g [Н+| —зк, или — 1g [Н-ь] =»
8лГ — £к— £ е0Г — еК— £
= ------, или Рн = -2—-------------------. (VI 1,64)
/7\ /Д
Подставляя вместо е*г, sK и Ж их численные значения, получим: 0,7044 — 0,2503—£
— 301 —
или
0,4541-Е
₽ 0,0577
.» I
(VIL65)
Двойная хингидронная цепь. Для измерения pH в практике часто применяется двойная хингидронная цепь, т, е. цепь, составленная из двух хингидронных электродов. Эта цепь составляется следующим образом.
Рис. 102. Схема хингидронно-каломельной гальванической цепи:
1 — хингидронный электрод; 2 — каломельный электрод; $ —стакан с испытуемым раствором; 4 — стакан с насыщенным раствором КС1; 5—агаровый сифон с хлоридом калия
В один стакан наливают раствор, pH которого известен. Обычно в качестве стандартного раствора берут буферную смесь, состоящую из одного объема 0,1 н. НС1 и 9 объемов 0,1 н. KCL Такой раствор, именуемый раствором Вейбеля, имеет pH 2,04. В другой стакан наливают исследуемый раствор, pH которого необходимо определить, В оба стакана добавляют в избытке хингидрон и вставляют платиновые электроды. В целях устранения диффузионного потенциала цепь соединяется через агаровый сифон с насыщенным раствором КС1. Схематически двойную хингидронную цепь можно записать так:
(Pt) Н2 | хингидрон, Нст|КС1|Н+, хингидрон | Н2 (Pt)
Знаки электродов в данной схеме указаны для случая, если [Н+]ст больше [Н+] исследуемого раствора. Если это условие не выполняется, знаки заряда электродов в цепи будут обратными. Э. д. с. двойной хингидронной цепи равна:
Е ~ ехг (СТ ) — ехг • (VII, 66)
Подставляя в это уравнение значения потенциалов хингидронных электродов, получим:
Е = е5г + Ж lg [Н+]ст—ejr—Ж 1g [Н+] или pH =-^-+ рнст. (VII,67)
Учитывая, что в качестве стандартного раствора берется оаствов
Вейбеля, уравнение (VII, 67) примет следующий вид: ₽
Р"=М577 + 2’04- (VI 1,68)
Рис. 103. Иономер ИМ-2М
Хлорсеребряно-сурьмяная цепь. Наша промышленность выпускает портативные приборы для измерения pH с помощью хлорсеребряно-каломельной цепи. Схематически эту цепь можно изобразить следующим образом:
Ag | Ag Cl, КС1 | КС] | Н+ | Sb2 O31 Sb exc ecyp
На рис. 103 показана схема иономера ИМ-2М, предназначенного для измерения pH с помощью сурьмяного электрода, взятого в паре с хлорсеребряным электродом. Этот прибор состоит из измерительного устройства (гальванометра), шкала которого градуирована в единицах pH, сурьмяного электрода /, выполненного в виде чашки из сурьмы в пластмассовом корпусе и хлорсеребряного электрода сравнения 2.
Определение pH с помощью ионометра ИМ-2М проводят следующим образом. Сначала прибор настраивают по стандартному буферному раствору. Для этого в подготовленную чашку сурьмяного электрода наливают буферный раствор, например, с pH 6, 8, погружают в него хлорсеребряный электрод. Если при включении прибора стрелка отклонится от значения pH 6,8 по шкале, то необходимо специальной ручкой так называемого корректора стрелку прибора повернуть так, чтобы она остановилась на делении шкалы, равном 6,8. После предварительной настройки проводят измерение pH исследуемого раствора. Для этого стандартный раствор в чашке сурьмяного электрода заменяют на исследуемый и снимают показания pH по шкале прибора.
— 303 —
Рис. 104. Простейшая схема элемента со стеклянным электродом:
1 —- парафиновый блок; 2 — каломельный электрод; 3 — солевой мостик; 4 — крышка (канифоль с парафином); 5 — стеклянный электрод
Рис. 105. Принципиальная схема лампового потенциометра:
/ — переключатель на три положения; 2 — лампа тетрод 6V6-C (6П2); 3 —• сопротивление 1400 ом; 4 — переменное сопротивление 0—150 ом; 5 — переменное сопротивление 0—300 ом; 6 — гальванометр; 7 — сопротивление шунт-гальванометра (0—100 ом); 8 — сопротивление 300 ом; 9 — выключатель питания прибора; 10 — батарея питания прибора 12 в; // — контакт для подключения общего провода (нормального элемента и стеклянного электрода); 12— стеклянный электрод; 13 — нормальный элемент; 14 — сопротивление 1,5 ком; 15 — мостик; 16 -• двухвольтовый аккумулятор
Каломельно-стеклянная цепь, В этой цепи стеклянный электрод с водородной функцией является индикаторным электродом, а каломельный — электродом сравнения. На рис, 104 показана простейшая
Рис. 106. Схема включения в цепь лампового потенциометра, реохорда, испытуемого и нормального элементов;
/ — аккумуляторы; 2, 4 и 10 — сопротивления;
и 9 — выключатели; 5 — реохорд; — аккумулятор на 2 в; 7 — гальванометр; 8 — тройной переключатель (СЭ — стеклянный электрод; НЭ — нормальный элемент); // — нормальный элемент;
12 — испытуемый элемент
схема элемента со стеклянным электродом, которая условно записывается так:
Ag | AgCl, НС1 | стекло | Н+ | КС1 | КС1, Hg2Cl2 | Hg.
Вхс 60 8ст
Прежде чем приступить к измерению pH исследуемого раствора, проводят калибрование стеклянного электрода по серии буферных растворов с известным значением pH, и по полученным данным строят калибровочный график (см. рис, 96). Далее уже по значению э, д. с# с помощью графика определяют pH исследуемого раствора. ч
Работая со стеклянным электродом, необходимо иметь в виду, что из-за большого внутреннего сопротивления стеклянного электрода
— 305 —
сила тока, протекающего через элемент со стеклянным электродом, очень мала.Так как обычные гальванометры (стрелочный или зеркальный) не могут быть использованы в компенсационной установке в качестве нуль-инструмента, для измерения э. д. с. элементов с большим внутренним сопротивлением применяют такие потенциометры, которые практически не потребляют тока исследуемого элемента.
Этому условию отвечают ламповые потенциометры. Принципиальная схема одного из ламповых потенциометров и способ подключения к нему гальванометра, нормального элемента, реохорда и измерительной ячейки показаны на рис. 105 и 106. Потенциометр собран на лампе металлической серии типа 6V6-C (6П2).
Наша промышленность выпускает большое число различных ламповых pH-метров. В этих приборах шкалы измерительного устройства градуированы в единицах pH, так что необходимость строить градуировочный график отпадает. Наиболее широко распространенными являются приборы марок ЛП-58, ЛПУ-0,01, ППП-58 и др.
§ 89. Преимущества и недостатки различных потенциометрических методов определения pH
Не все методы, рассмотренные в предыдущем параграфе, являются в одинаковой мере удобными и точными. Так, применение водородного электрода не практично из-за громоздкости и сложности изготовления, а также потому, что электрод не работает в агрессивных средах, содержащих, например, сильные окислители или вещества, «отравляющие» платину, — мышьяк и др. Не пригоден водородный электрод и для измерения pH биологических жидкостей, так как различные органические вещества, например белки, осаждаются на поверхности платиновой пластинки, в результате чего показания водородного электрода искажаются.
Применение хингидронного электрода также ограничено целым рядом недостатков. Этот электрод не может применяться для измерения pH жидкостей, в которых его значение выше 8, так как в щелочной среде хингидрон ведет себя как кислота и взаимодействует с гидроксид-ионами (реакция нейтрализации). Хингидронный электрод обладает так называемой солевой ошибкой, т. е. его показания сильно изменяются в присутствии значительного количества различных солей. Неустойчивость показаний его обнаруживается также в случае присутствия в измеряемом растворе сильной окислительно-восстановительной системы.
Сурьмяный электрод имеет определенные преимущества перед водородным и хингидронным электродами, однако значительно уступает им в точности определения. Применяется электрод там, где не требуется высокая точность.
Стеклянный электрод одинаково хорошо работает в любых агрессивных средах как в кислой, так и в щелочной областях pH. Потенциал его устанавливается быстро и обладает очень хорошей воспроизводимостью. С помощью стеклянного электрода можно измерять pH с точностью до тысячных долей. Единственное неудобство при работе со стеклянным электродом — необходимость использования для измерения э. д. с. специальных потенциометров, снабженных усилительными устройствами. Однако это препятствие легко устранимо, так как наша промышленность ежегодно выпускает огромное количество установок подобного типа. В частности, освоен выпуск специальных стеклянных электродов в виде игл, которые позволяют определять pH, например, непосредственно в почвенной суспензии или внутри растения и т. д. Большим преимуществом стеклянного, а также сурьмяного электродов является то, что они не загрязняют раствор и поэтому могут широко применяться в пищевой промышленности при автоматическом контроле различных технологических процессов
— 306 —
§ 90. Определение в почвах поглощенного натрия с помощью натрий-стеклянного электрода
Стеклянные электроды с металлической функцией можно использовать для непосредственного определения натрия, находящегося в почвенном растворе и в поглощенном состоянии. Это имеет большое практическое значение. Почвоведы и агрохимики давно установили, что наличие иона натрия в виде солей (солончаковые почвы) распыляет почву, делает ее бесструктурной и тем самым снижает или совсем лишает ее плодородия. Требуется количественное определение ионов натрия в почвах.
К сожалению, ионы натрия на дают труднорастворимых, малоионизирован-ных или окрашенных соединений с анионами легко доступных реактивов и потому не могут быть количественно определены весовым методом в первом случае, ’ объемным — во втором и колориметрическим — в третьем.
Необходимо было найти способ определения натрия, основанный на других его свойствах. В последнее время появились методы, удовлетворительно разрешающие задачу количественного определения ионов натрия в растворах. К таким методам относятся пламенно-фотометрический (см. гл. VIII) и потенциометрический с применением натрий-стеклянного электрода.
При потенциометрическом определении концентрации ионов натрия с помощью натрий-стеклянного электрода в качестве электрода сравнения обычно берется насыщенный каломельный или хлорсеребряный электрод:
(Pt) Hg I Hg2SO4, Na2SO4 I стекло | Na+ | KCI | KC1, Hg2CI2 | Hg
eHg e0 eNa . , eK
Э. д. с. этой цепи равна
£=eNa-eK. (VII,69)
Потенциал натрий-стеклянного электрода имеет следующую зависимость от кон-, цёнтрации ионов натрия в растворе:
8Na = 8oa+Mlg[Na+]. (VI 1,70)
Строго говоря, приведенная зависимость eNa от [Na+] справедлива лишь для очень разбавленных растворов, по мере же увеличения концентрации ионов натрия в растворе потенциал натрий-стеклянного электрода изменяется несколько инаг че, чем это следует из уравнения (VII, 70). В этом случае вместо концентрации в формулу (VII, 70) необходимо пбдставить активность ионов натрия aNa, которая выражается уравнением;
°Na+ = ^Na+ , (VII ,71)
где /j — коэффициент активности.
Для того чтобы по уравнению (VII, 70) определить аналитическую концентрацию ионов натрия в растворе, необходимо знать численное значение его коэффициента активности. В почвенном растворе, где помимо натрия присутствуют и другие ионы в самых разнообразных соотношениях, определение этого коэффициента представляет значительные трудности. Для преодоления их было предложено выравнивать коэффициенты активности в стандартных и испытуемых растворах путем создания избытка индифферентной соли, которая нивелирует разницу в ионной силе растворов. Так, коэффициенты активности ионов натрия остаются практически постоянными, если раствор содержит СаС1а или ВаС12 в концентрации от 0,8 до 1,0 М. Это позволяет определять содержание натрия в засоленных почвах, пользуясь следующим методом.
Навеску почвы обрабатывают 1 н. раствором BaClg, затем почву отфильтровывают и в фильтрате измеряют э. д. с. стеклянно-каломельной цепью. Содержание натрия находят по градуировочному графику, который строят в координатах э. д. с. — lg [Na+] по серии стандартных растворов, содержащих точно известное количество натрия на фоне 1 н. ВаС12.
Содержание натрия этим методом можно определить так же легко, как и pH раствора потенциометрическим методом.
— 307 —
§ 91. Окислительно-восстановительные потенциалы
Окислительно-восстановительными называются такие реакции, при которых происходит взаимное окисление и восстановление различных веществ. Окисление вещества сопряжено с удалением электронов из составляющих его частиц, а восстановление — с присоединением их к частицам. Иными словами, окислительно-восстановительные реакции происходят с передачей электронов от восстановителя к окислителю.
Примером окислительно-восстановительной реакции может служить восстановление хлорида железа (III) хлоридом олова в растворе
Рис. 107. Схема окислительно-восстановительной цепи
2FeCl3 + ZnCl2 2FeCl2 + SnCl.
или в ионной форме
2Fe3+ +Sn2+ 2Fe2+ 4-Sn4+
В этой реакции электроны от ионов олова (II) переходят к ионам железа (III).
Ионная окислительно-восстановительная реакция может быть осуществлена в гальваническом элементе с двумя окислительно-восстановительными электродами. Напомним, что окислительно-восстановительный электрод представляет собой пластинку инертного металла (платины, золота), опущенную в раствор, содержащий ионы различной зарядности.
Если платиновую пластинку опу-
стить, например, в раствор хлорида железа (III), а вторую такую же — в раствор соли олова (II), разделить растворы электролитов пористой перегородкой (или соединить их с помощью агарового сифона), а затем соединить платино
вые пластинки между собой с помощью металлического проводника, в цепи возникает электрический ток. Обнаружить его можно с по-
мощью гальванометра, включенного в цепь этого элемента (рис. 107). Электрическая энергия в данном элементе возникает за счет окис
ления ионов олова
Sn2-!’ — 2е~ -> Sn4+
и восстановления ионов железа
Fe3+ 4-е~ Fe2+
Ионы олова (II), отдавая электроны металлу, сообщают электроду положительный заряд. В то же время ионы железа (III) стремятся присоединить электроны, принадлежащие металлу, сообщая электроду положительный заряд. В данном случае инертный металл (платина) играет роль передатчика электронов и не претерпевает в процессе реакции никаких химических превращений. В этом и заключается
— 308 —
отличие окислительно-восстановительных элементов от других гальванических элементов, в которых хотя и происходят реакции окисления — восстановления, электроды в процессе реакций химически изменяются (например, растворение цинка в медно-цинковом элементе Якоби — Даниеля).
Таким образом, к окислительно-восстановительным гальваническим элементам относятся такие элементы, которые состоят из двух окислительно-восстановительных электродов. Причем каждый из электродов представляет собой пластинку из благородного металла, опущенную в раствор разнозаряженных ионов. Потенциал, возникающий на границе соприкосновения пластинки с раствором, называется окислительно-восстановительным потенциалом.
Рассмотрим теперь, как зависит величина и знак этого потенциала от соотношения концентраций (активностей) ионов разной зарядности в растворе. Допустим, что мы имеем водный раствор солей железа (III) и (II), в который погружена платиновая пластинка. Поскольку эти ионы оказываются разнозарядными, между ними должно существовать следующее равновесие:
Fe2+ ^z± Fe3+
Как и во всех нейтральных реакциях, здесь можно определить кон
станту равновесия:
[Fe3+] [е~]
[Fe2+]
(VI 1,72)
Введение в данную систему дополнительного количества электронов повлечет за собой уменьшение концентрации окисленной и повышение содержания восстановленной форм. Уменьшение же концентрации электронов будет способствовать увеличению окисленной и понижению восстановленной форм*
Таким образом, характер и направление процесса в окислительно-восстановительной реакции зависит от того, насколько легко восстановитель отдает свои электроны и насколько прочно эти электроны связывает окислитель. Иными словами, окислительные свойства той или иной ОВ-системы определяются прочностью связи электронов с окисленной и восстановленной формами ее компонентов. Это позволяет выражать окислительно-восстановительные свойства любых систем при помощи так называемого ОВ-потенциала, возникающего на поверхности платиновой пластинки при погружении ее в эти системы. Величина потенциала, обозначаемого eft, зависит от того, насколько легко система отдает свои электроны. Можно считать, что потенциал окислительно-восстановительного электрода будет равняться:
ь , RT , рм] 8/i==eo4-~ ,n:—г» nF
(VII, 73)
где [ем 1 — концентрация электронов (или упругость электронного газа) в металле; [е~1 —та же величина для окислительно-восстановительной системы; п — разность между зарядностями ионов (для рассматриваемой системы Fe3+ Fe2+ п = 1),
— 309 —
Из уравнения (VII, 72) определим величину [е*"]:
(VII,74)
Подставляя это значение в уравнение (VII,73),
/ RT
еЛ = ео + — 1 п
k7][Fe3+l К [Fe2+]
Вследствие большой [ем | можно считать К в одну постоянную
емкости металлического
постоя иной. Объед и н я я
получим:
(VI 1,75)
электрода величину величины [ем|, ее и
в 0, получим:
RT
[Fe3+]
[Fe2+] ’
или
еЛ = е© 1g
(VII, 76)
Таким образом, для любой окислительно-восстановительной реакции типа:
Red Ox
где Ох — окислитель, Red — восстановитель, е~ — электрон, потенциал окислительно-восстановительного электрода выразится уравнением:
еЛ = е0+— 1g
п
[Ох]
[Red] ’
(VI 1,77)
Более высокий потенциал показывает, что реакция
Red Ох-]-е~
сдвинута вправо. При низком потенциале реакция будет сдвинута влево.
Все это приводит к выводу о том, что величина окислительно-восстановительного потенциала служит мерой интенсивности процессов окисления — восстановления, протекающих в данной системе, и зависит от соотношения в ней концентраций (активностей) окисленной и восстановленной форм ионов, образующих данную систему. Величина е0 в уравнениях (VII,76) и (VII,77) называется нормальным потенциалом окислительно-восстановительного электрода. Она определяется по отношению к нормальному водородному электроду и равна потенциалу данного окислительно-восстановительного электрода при условии, что активные концентрации ионов обеих зарядностей равны между собой, т. е. [Ох] = [Red]. В качестве иллюстрации в табл. 65 приведены некоторые величины нормальных потенциалов для различных окислительно-восстановительных электродов.
— 310 —
Таблица 65
Нормальные окислительно-восстановительные потенциалы s0 (в вольтах) в воде при 25 °C
Электрод Элект родным процесс е.
(Pt) | Сг2+, Сг3+ Сг3+4>е" —0,41
(Pt) | Sn2+, Sn4+ 8п4+ф2 £ZtSn2+ +0,153
(Pt) | Cu+, Cu2+ Си2+ 4* z±Cu+ 4-0,167
(Pt) | Fe2+, Fe3+ Fe3+^>^”< Z±Fe2+ 4-0,771
- (Pt)|Tl+, Tl3+ е- 7±Т1+ 4-1,25
(Pt)|Mn2+, Mn3+ Мп3+ 4* Z±Mn2+ 4-1.51
(Pt) PbSO4| PbO2 1 H2SO4 РЬО2ф4 Н+ + 2 -±РЬ2+ф2 Н2О 4-1,685
(Pt) | Co2+, Co3+ Со3+4>е"<- 2tCo2+ 4-1,840
; При сочетании какого-либо электрода в гальваническом элементе с одним из нижестоящих в этой таблице электродов, имеющим более положительный потенциал, протекает реакция окисления. И наоборот* реакция восстановления будет протекать при сочетании электрода с вышестоящими более отрицательными электродами.
Нормальные окислительно-восстановительные потенциалы характеризуют окислительно-восстановительную способность веществ по отношению друг к другу, так как они являются мерой изменения изобарного потенциала AZ (максимальной работы) при окислительно-восстановительном взаимодействии веществ. Нормальный потенциал любой окислительно-восстановительной системы может быть вычислен по следующей формуле:
А^реалции
Ео== 23,06п ’
(VII ,78)
где AZ—стандартное изменение свободной энергии, отнесенное к 1 моль или 1 г-ион, п — число валентных электронов, участвующих в реакции, 23,06 — число Фарадея, ккал/в, отнесенное к 1 моль или к 1 г-ион.
Так, для рассмотренной выше системы Fe2+ Fe3+ + е~, потенциал которой выражается уравнением (VII,76), можно записать:
А^реакции AZpe3-|----AZpe2-f
23,06л-= 23,06
(VI 1,79)
Подставляя в это уравнение численные значения AZ°Fe3 + и AZ°Fe24 из специально составленных таблиц, получим:
(-3,00)-(~20,30) п _ о — — —" —(I ti а
— 311 —
С учетом этого уравнение окислительно-восстановительного потенциала для системы Fe8+ —Fe24 примет следующий вид:
£^ = 0,77 4-м 1g
(VI 1.80)
где [Fe8+] и [Fe2+] — активные концентрации данных ионов в растворе.
Все рассматриваемые уравнения, выражающие величину ОВ-по-тенциала, были выведены для простейших реакций, когда, кроме окислителя и восстановителя, в реакции не участвуют другие компоненты. В наиболее общем случае под знаком логарифма должны находиться равновесные концентрации (активности) всех участвующих в реакции соединений. Иными словами, в уравнении (VII,77) под окисленной формой следует понимать все ионы раствора в правой стороне уравнения реакции Red 7^ Ох + е~, а под восстановленной — все ионы раствора слева от знака равенства.
Так, для какой-либо реакции типа
Am + B4-Nv^z± An4-D4-Cx4-(n—m) е~
окислительно-восстановительный потенциал будет равен:
eft = е0 4-— lg-----------,
п а л a Am * Nv
(VII,81)
где А" и Аш — ионы соответственно окисленной и восстановленной форм, В и D — твердые фазы, Nv и Сх — ионы раствора, не участвующие в окислительно-восстановительных реакциях.
Часто, особенно при реакциях органических соединений, окисление и восстановление происходит с участием водородных ионов. Роль их во всех окислительно-восстановительных процессах следует оценивать исходя из конкретных условий реакции. При этом возможны три случая: 1) водородные ионы вовсе не участвуют в ОВ-реакции; 2) водородные ионы непосредственно участвуют в окислительных процессах; 3) косвенное влияние водородных ионов.
Первый случай. Ионы водорода не принимают участия в окислительно-восстановительных реакциях простых ионов металлов и ряда анионов. Например:
a) Fe3+ 4~е“ Г"* Fe2+ ео = О,77 в\
б) Си+ ео = О,17 в;
в) Fe (CN)|~ + e~^± Fe (CN)^~ eo = O,36 в.
Во всех этих подобных реакциях di не зависит от pH, если оба компонента смеси одинаково хорошо растворимы.
Второй случай. Реакции многих органических соединений, а также некоторых кислородсодержащих анионов часто сопровождаются образованием воды или слабых кислот. В этих реакциях непосредственное участие принимают ионы водорода, потому и величина e/i таких окислительно-восстановительных систем зависит от pH.
— 312 —
Примером подобных реакций может служить рассмотренный нами ранее хингидронный электрод, а также реакции типа:
а) 4-2Н+ 4—2. 2HgO g0=if77 e*t
б) NO7 + &?- + 10Н+ NHt + ЗН2О е0 = 0,87 в,
или в общей форме
Ох + пе~ + wH+ Red.
Уравнение ей для таких систем с участием т ионов водорода записывается так:
Ж [Ох] [Н+Г Ж. [Ох] тЖ. гтт^, „утт onv еЛ«=е0-Ь--1g-----------«=e0 + — ^ТоТи4'------[Н+]. (VII,82)
п [Red] п [Red] п
Из этого уравнения следует, что по мере подкисления раствора ей систем увеличивается. Величина сдвига ОВ-потенциала в общем случае зависит от числа электронов и числа ионов водорода, участвующих в реакции, и определяется коэффициентом уравнения (VII, 82): тЖ 2t303RTm т
п nF п
Третий случай. В реакциях этого типа водородные ионы оказывают на ей косвенное влияние в результате изменения раствори-
мости компонентов системы при подкислении или подщелачивании раствора. В качестве примера можно рассмотреть равновесие в системе Fe2+ — Fe8+. Допустим, что одновременно имеется 0,1 н. раствор Fe2+ и Fe8+ железа. Величина ей определяется уравнением (при условии, что коэффициенты активности близки к единице):
ей = е0-|-0,0577 1g
[Fe3+]
[Fe2+]
При pH, например, равном 6,0, количество ионов железа в растворе можно вычислить из произведений растворимости:
[Fe2+] [ОН^]2 = 4,8.К)-16 и [Fe3+] [ОН“]3 = 4,5.10~40в
откуда
4 Ю“4о 4 R. 1Л-40
[Fe’+J = =4’5'I0'W М> <V1I,83)
[Fea+]=i±12zle=^^=4i8 м. (VI1>84) 1 1 [ОН-]2 ю~1в
В этих уравнениях значения [ОН“] были найдены из уравнения ионного произведения воды. Для pH = 6,0 [ОН-] = 10~8 * г-ион/л.
Из полученных равенств видно, что ионы Fe2+ находятся в растворе, тогда как ионы Fe3+ находятся в осадке. Следовательно, при pH = 6,0 потенциал окислительно-восстановительной системы определяется только [Fe2+ ] при условии, что в растворе нет каких-либо органических веществ, сохраняющих подвижность [Fe8+].
Если этот раствор подкислить до pH = 1,0, количество ионов IFe2+] в растворе не изменится, а гидроксид железа (III) полностью
— 313 —
растворится. В результате [Fe2+] = [Fe3+], a e/i = e0. Рассмотрим несколько подробнее, как же зависит растворимость Fe (ОН)3 от pH среды. Из уравнения (VII, 83) имеем:
[Fe3+] =
4,5-10~4° [ОН-]3
Заменяя значение [ОН+] на [Н+] из ионного произведения воды,
получим:
- 10~14
[Н+][ОН-] = 10-14, откуда [ОН-]=——.
С учетом этого
[Fe3+] = 4,5.102 [Н+]3.
Логарифмируя выражение (VII, 85), получим:
(VI 1,85)
|g[Fe3+] = 2,65 + 3 lg [Н+] йли lg [Fe3+] = 2,65—ЗрН. (VII,86)
Таким образом, концентрация (активность) иона Fe3+ в растворе . контролируется только величиной pH. Задаваясь произвольными значениями pH, можно составить таблицу активности иона Fe3+ в растворе (табл. 66):
Таблица 66
. Активность иона Fe3+ в зависимости от величины pH
pH
п_. 3+ г- ион ге
а 3+ ?/
Fe
I
I
I ]
I
I
I j
I
I ll
I
I
I
j
I I
J
)
I
1,0
2,0
3,0
4,0
5,0
6,0
7,0
8,0
9,0
10,0
— 0,35' — 3,35 — 6,35 — 9,35 —13,35
—15,35
—18,36
—21,35 —24,35
—27,35
4,47-10“1 4,47-10”4 4,47-10-7
4,47-10-10 4,47-10""13 4,47-10-16 4,47-10“19 4,47-Ю-22 4,47-10“25 4,47-IO’28
2,5-10+l 2,5-Ю-2
2,5-10-5
2,5-10“8
2,5-10 —11 2,5-10-14
2,5-10~17 2,5-10“2° 2,5-10“23
2,5-10“26
Как видно из таблицы, растворимость гидроксида железа (III)
при pH = 1,0 практически безгранична. При повышении величины pH на единицу растворимость гидроксида железа (III) уменьшается в 1000 раз и уже при pH = 3,0 активность иона Fe3+ измеряется стотысячными долями грамма на 1 л воды.
Таким образом, в подобных случаях pH, влияя на растворимость одного из компонентов окислительно-восстановительной системы, оказывает существенное влияние на ОВ-потенциал этой системы.
§ 92. Окислительно-восстановительные реакции и потенциалы в почвах
Окислительно-восстановительные реакции играют важную роль в процес-/ сах почвообразования, на что впервые указал В. Р. Вильямс. Установлено, что в почве широко развиты различные окислительно-восстановительные процессы, в связи с чем ее можно рассматривать как сложную окислительно-восстанови-
— 314 —
тельную систему. Нормальный рост и развитие растений возможны только при определенном окислительно-восстановительном состоянии почвы.
Характерной особенностью почвенных условий является необратимость большинства реакций окисления и восстановления, протекающих в почве. Обратимые реакции, которые полностью подчиняются всем разобранным выше теоретическим положениям, свойственны только некоторым окислительно-восстановительным системам — обратимому окислению и восстановлению железа (Fe3+
Fe2+), марганца (Мп4+ Мп2+), азота (N5+ N3+) и др. С другой стороны,
^Гпочве протекает большое число окислительно-восстановительных реакций биохимической природы.
Важнейшим и наиболее сильно действующим окислителем в почве является молекулярный кислород, содержащийся в почвенном воздухе и почвенном растворе. Поэтому направление и развитие окислительно-восстановительных процессов в почве тесно связано с условиями ее аэрации и, следовательно, зависит от всех свойств почвы, влияющих на ее газообмен — структуры, плотности, механического состава, а также влажности. Ухудшение аэрации в результате повышения влажности почвы, ее уплотнение, образование так называемой корки на ее поверхности и целый ряд других причин приводят к снижению окислительно-восстановительного потенциала почвы.
Опыт показывает, что в нормально аэрируемых почвах ОВ-потенциал варьирует в пределах 300—650 мв. Заболачивание и оглеение снижают его до величины, меньшей 200 мв. Бурное развитие биологических процессов также способствует снижению еА почвы вследствие потребления кислорода микроорганизмами и корнями растений.
От окислительно-восстановительных условий в почве (т. е. от величины еА) j зависит подвижность, а следовательно, и доступность растениям таких элементов, ' как железо, марганец, азот и др. Например, увеличение содержания в почве’ (точнее в почвенном растворе) веществ с высоким окислительно-восстановительным потенциалом отрицательно сказывается на процессах фиксации атмосферного азота микроорганизмами (так называемыми азотобактерами, в частности azoto-bacter chlorococcum). Процессы нитрификации, денитрификации и аммонификаД ции также идут при определенных окислительно-восстановительных условиях, j т. е. в определенном интервале значений еА.
Превращение соединений серы (аналогично остальным микробиологическим 1 реакциям) находится в зависимости от ОВ-состояния в почве. Так, сульфиды могут окисляться серобактериями, а сульфаты восстанавливаться сульфатреду пирующими бактериями по следующей схеме SO|~ S4O|~ ^±S20f“ S SH“. При разложении органических веществ в условиях высоких значений ОВ-потен-циала в почве образуются преимущественно сульфаты, а при низких значениях ОВ-потенциала, т. е. в анаэробных условиях, — сульфиды.
В настоящее время накоплены данные о содержании в различных почвах соединений железа, марганца и нитратов, которые позволяют сделать определенные обобщения и указать примерные граничные окислительно-восстановительные условия (т. е. интервал оптимальных значений еА и pH нормального усвоения высшими растениями этих элементов. На рис. 108 представлены некоторые из этих данных.
На рис. 108 прямоугольник ABCD охватывает окислительно-восстановительные условия, характерные для большинства почв нашей страны. Прямая ограничивает область нормального снабжения культурных растений марганцем, прямая KL — область нормального обеспечения железом. Проведенная примерно на уровне значений еА = 340 мв линия MN отделяет область усиленной денитрификации, т. е. область, в которой идут микробиологические процессы восстановления нитратов до нитритов и свободного азота. Наиболее благоприятные условия для питания высших растений марганцем и железом определяются заштрихованной площадью, а с учетом денитрификационных процессов— площадью с двойной штриховкой.
Опыт показывает, что для различных почв по-разному изменяется еА в зависимости от изменения внешних условий. Для одних почв изменения значений еА происходят резко, для других — не столь резко, поскольку окислительно-восстановительные системы почв обладают различной загруженностью (емкостью) и податливостью к изменениям. Рассмотрим это положение более подробно.
— 315 —
Рис. 108. Граничные условия нормального питания растений (по Сердобольскому):
косая штриховка — область нормального снабжения железом и марганцем; двойная штриховка —- область
нормального снабжения железом, марганцем и нитратами
Окислительно-восстановительный потенциал среды определяется в общем
виде следующим уравнением:
[Ох] [Red]
п
Обозначим концентрацию окисленной формы данной системы V, а общую концентрацию окислителя и восстановителя В. Тогда ОВ-потенциал системы можно выразить уравнением:
(VII ,87)
Если в рассматриваемую систему прибавить небольшое количество сильного окислителя dVt то e/i этой системы также увеличится до Eh 4- dshf т. е.
eh + deh = — 1g ———. п В — V
Продифференцировав уравнение (VII, 87), получим:
dth Ж / 1 1 \ dvh Ж В
-- __ - ---- 4_ -- ИЛИ ----- — --------- dV п \ V B—VJ dV п V (В — V)
(VI 1,88)
(VI 1,89)
Это уравнение характеризует степень изменения окислительно-восстановительного потенциала в зависимости от изменения концентрации окисленной формы. Степень изменения Eh с изменением V зависит от двух переменных (В и V), т. е. от общей концентрации участвующих в данной системе компонентов и от концентрации окисленной формы.
Поскольку в уравнении (VII, 89) величина V составляет лишь часть величины В (если В условно принять за единицу), V будет изменяться от нуля до еди-
— 316 —
ницы. Нетрудно догадаться, что ч^м ближе У к нулю или единице, тем больше степень изменения. Из этого можно сделать вывод о том, что при малых количест-
IX окисленной формы по сравнению с восстановленной (либо при малых количествах восстановленной формы по сравнению с окисленной) достаточно добавить небольшое количество окисленной формы (или соответственно восстановленной), ЧТОбЫ СИЛЬНО ...... ------ ----
ОВ-системы:
Если У —
сдвинуть окислительно-восстановительныи потенциал данной
0,5В (т. е. В = 1, а V deh . . dV n
1/2), то
(VI 1,90)
В этом случае
В этом случае степень изменения eZi с небольшими изменениями У будет очень мала. Таким образом, eh той или иной системы в случае, когда окисленная форма равна восстановленной, очень слабо реагирует на небольшие изменения У.
Рис. 109. Изменение степени загруженности окислительно-восстановительной системы с изменением восстановленной формы (V) и общей концентрации окислительно-восстановительной пары (В)
Величину deh/dV называют податливостью окислительно-восстановительной системы. Опыт показывает, что на практике гораздо удобнее пользоваться не понятием податливости, а понятием загруженности окислительно-восстановительной системы. Загруженность представляет собой величину, обратную податливости, и обозначается обычно символом р. Таким образом, загруженность ОВ-системы может быть выражена следующим уравнением
Ж (В-У) У п В
(VII,91)
Использование понятия загруженности удобнее в том смысле, что оно приближается к понятию забуференности, применимому по отношению к pH.
Из уравнения загруженности ОВ-системы (VII, 91) видно, что загруженность системы зависит от параметров п, В и У. Чем больше число валентных электронов, участвующих в реакции, тем выше загруженность окислительно-восстановительной реакции. Влияние параметров В и У на загруженность хорошо иллюстрирует рис. 109. Он подтверждает, что с увеличением концентрации участвующих компонентов (В) загруженность системы увеличивается. Влияет на загруженность также и соотношение окисленной и восстановленной форм компонентов, входящих в состав данной ОВ-системы.
Максимальная загруженность или стабильность окислительно-восстановительной системы наступает в том случае, если окисленная и восстановленная формы этой системы находятся примерно в одинаковой концентрации (т. е. У = 50%), причем абсолютная концентрация всех компонентов этой системы (В) также должна быть больше всех остальных ОВ-систем, находящихся в растворе.
— 317 —
4
В почве отношение окислителя к восстановителю редко может быть равно еди* лице, а концентрация ОВ-системы в целом низкая. Этим и объясняется чрезвычайная податливость почвы к изменению и снижению его величины при увлажнении. Опыт показывает, что повышение содержания органического вещества ' в почве повышает ее буферность (загруженность). Вот почему все мероприятия, » направленные на повышение содержания гумуса в почве, положительно сказываются на ее ОВ-состоянии.
Поскольку в почве всегда имеется большое количество окислительно-восстановительных систем, главной или, как ее еще называют, потенциалопределя-ющей системой всегда будет та, которая имеет максимальную загруженность по сравнению со всеми остальными. Иными словами, окислительно-восстановительный потенциал любой среды, в том числе и почвы, определяется максимально загруженной системой, которая потому и называется потенциалопределяющей. В зависимости от конкретных условий эта система может быть химической (реакции окисления — восстановления) и биологической^на -которой работают микроорганизмы).
Окислительно-восстановительный потенциал ей, а также загруженность буферной системы являются важными параметрами для характеристики почвы
ферной системы являются важными параметрами для характеристики с точки зрения ее плодородия. ' •-
§ 93. Экспериментальное определение окислительно-восстановительного потенциала в почве
Для измерения окислительно-восстановительного потенциала в (или в каких-либо других объектах исследования) обычно используются
почвах платиновые электроды. Поверхность электрода должна быть очень гладкой. Платиновая пластинка или проволока диаметром около 0,5 мм впаивается в стеклянную трубку и в таком виде погружается в почвенный образец. В качестве электрода сравнения обычно берут насыщенный каломельный электрод. При пблевых определениях окислительно-восстановительного потенциала Хорошо зарекомендовали ^себя так называемые платинированные стеклянные электроды. На изготовление их расходуется очень небольшое количество Платины, что позволяет значительно экономить дорогостоящий металл. Платинированные стеклянные, электроды, предложенные советскими учеными М. С. Захарьевским и М. С. Рабиновичем, вместо платиновой проволоки (или пластинки) имеют тонкий налет металлической платины на конце электрода. Из одного грамма платины можно изготовить до 200 электродов. Йа рис. ПО показаны индикаторные платиновые электроды различной конструкции.
В зависимости от цели исследования определение окислительно-восстановительных потенциалов почвы проводят непосредственно в поле или в лаборатории. Для характеристики ОВ-условий самой почвы в естественном ее состоянии следует проводить полевые измерения, так как во взятых из почв образцах при транспортировке и хранении их происходят нередко значительные изменения величины потенциала, главным образом в связи с.переменой в условиях аэрации.
В полевых условиях применяются каломельные электроды игольчатой формы системы Крюкова. В этих электродах (рис. 111) контакт с почвой осуществляется через асбестовую нить, впаянную в конец стеклянной трубки. Игольчатые каломельные электроды имеют сравнительно небольшое омическое сопротивление и легко погружаются в почву.
Оба электрода (платиновый и каломельный) подключают к потенциометру, на котором и измеряют возникшую э. д. с. в милливольтах. Так как величину е/г
принято выражать всегда по водородному электроду, к полученному при измерении числу милливольт прибавляют число милливольт, равное потенциалу каломельного электрода. Измерение ей проводят технически так же, как определение pH с хингидронным электродом, но без добавки в раствор или почву хингидрона. Измерения ей рекомендуется проводить на ламповых потенциометрах, при которых поляризационные токи, мешающие определению, крайне ничтожны и практически не обнаруживаются.
Полевые измерения могут быть разовые и стационарные при изучении, например, динамики ОВ-условий за тот или иной промежуток времени. При р«$р4
— 318
ВЫХ измерениях определение еА можно пвоволмтк »
него разреза, по отдельным генетическим гоо^зонтяЗ Л® свежвв«РЫТ0™ почвен-мерное расположение электродов в почвенном пя1па^Н®РИС- 112 показано при-установки электродов в стенку разреза в ней ппДТ?630 При измеРениях eft. Для палочкой небольшие углубления в котопыа варительно делают заостренной мом. Платиновые электроды необходимодогружать •ПеКТрОДЫ ЛеГКИМ наж-
^вес^ое0сос^оаяЛнаие.3иезРмееНрИениеТШ^яЫзаВтельноМпроОЧВа~ЭЛе^Р°1ле^^Ла°вЧало^в рав-
среднее „„
<1
Рис. ПО. Индикаторный платиновый электрод:
<1 — обычного типа; б — платинированный электрод За харь-ейского, / — платина; 2 ртуть; 3 — медная проволока! 4 — слой платины; 5 — менделеевская замазка
Рис. 111. Каломельный электрод Крюкова:
I — каломель; 2 — ртуть; 3 — платиновый контакт; 4 —• ватная пробка; 5 — насыщенный раствор КС!; 6 — пробка; 7 — кристаллы КС!; 8 — асбестовый фильтр; 9 — асбестовая нить
При полевом стационарном изучении, например, динамики ОВ-услови1) электроды в почву устанавливаются один раз на все время наблюдений на разную (в зависимости от цели исследования) глубину. Причем на каждую глубину помещают не менее трех электродов, располагая их на расстоянии 10—20 см друг от Друга. Сроки замеров ей устанавливаются в зависимости от задач исследования.
Опыт показывает, что величина &h не остается постоя иной за весь период измерений: она изменяется в зависимости от аэрации, температуры, увлажнения, выпадения атмосферных осадков, норм и сроков полива и т. д.
Приемы измерения ей в лабораторной обстановке остаются теми же, какие описаны выше для полевых измерений. Лабораторные опыты по изучению
-319 —
ОВ-условий в зависимости от микробиологической деятельности, разложения орга-нического вещества почвы, перехода различных элементов почвы из одного окислительного состояния в другое ставят тац^ чтобы платиновые электроды в течение всего опыта находились в исследуемой почве. Опыт показывает, что даже кратковременное извлечение электродов серьезно нарушает равновесие, установившееся на поверхности электрода, и вносит ошибки в результаты наблюдений.
§ 94. Потенциометрическое титрование
Потенциометрическое титрование, как и определение активных концентраций ионов, является важнейшим приложением измерений э. д. с. При потенциометрическом титровании эквивалентная точка определяется не по изменению цвета инди-
Рис. 112. Установка электродов в почвенном разрезе для измерения ОВП:
1 — платиновые электроды; 2 — каломельный электрод Крюкова; 3 — потенциометр
катора, как это делается при обычном объемном методе титрования, а по изменению скачка потенциала индикаторного электрода. Особую ценность приобретает потенциометрическое титрование в том случае, когда нельзя использовать обычные индикаторы, например, при титровании окрашенных или мутных растворов. Кроме прямого аналитического назначения, потенциометрическое титрование часто используют для определения свойств титруемых соединений — константы диссоциации слабых
электролитов, количества активных групп и некоторых других.
Рассмотрим процесс титрования какой-либо сильной кислоты
щелочью с использованием в качестве индикатора водородного электрода. Для сравнения берем каломельный электрод. Схема этой установки показана на рис. 113. Титруемый раствор (в нашем случае кислоту) строго определенного объема наливают в стакан, погружают в него водородный электрод и агар-агаровый ключ (сифон) каломельного электрода и включают мешалку. Далее из бюретки небольшими порциями добавляют раствор щелочи, титр которой точно известен. После каждого прибавления щелочи производят определение pH с помощью потенциометра, к которому подсоединены соответствующим образом водородный и каломельный электроды.
В начале титрования концентрация водородных ионов (а следовательно, pH среды) будет уменьшаться постепенно. Однако вблизи эквивалентной точки это уменьшение будет значительным даже от прибавления весьма малых количеств щелочи. После прохождения эквивалентной точки изменение pH раствора вновь становится небольшим. Соотношения между изменением концентрации ионов Н+ и потенциалом водородного электрода (£) в случае титрования 0,1 н. раствора соляной кислоты 0,1 н. раствором едкой щелочи показаны в табл. 67,
— 320 —
Таблица 67
*
Изменение [Н+], pH и Е при нейтрализации °»1 н. раствора НС1 0,1 н. раствором NaoH
Рализовано кислоты, % н+ pH Е, в
0 ю-1 1,0 —0,058
90,0 io-2 2,0 —0,116
99,0 ю-3 3,0 —0,174
- 99,9 ]0-4 4,0 —0,232
п я 10°.° Прибавлен избыток NaOH % 10-’ 7,0 -0,406
0,1 10-10 10,0 —0,580
1 ,0 ю-11 11,0 —0,638
10,0 ю-12 12,0 —0,696
Как видно из табл. 67, на нейтрализацию последней части кислоты (0,1%) при добавлении такого же избытка едкого натра (0,1%) приходится изменение концентрации водородных ионов от 10“4 до
Рис. 113. Схема гальванической цепи при потенциометрическом титровании:
I =- каломельный электрод; 2 — солевой мостик; 3 « водородный электрод; 4 — мешалка; 5 — бюретка
10~10, что соответствует изменению pH на шесть единиц. Именно по этой причине кривая титрования сильной кислоты сильной щелочью, изображенная на рис. 114, около самой точки эквивалентности практически перпендикулярна осп абсцисс. Это резкое изменение pH около
11 Зак. 060
321
Рис.
114. Кривая титрования соляной кислоты едким натром
точки эквивалентности называется скачком титрования (нейтрал и з а -ц и и). Как видно из рис. 114, точка Л, соответствующая моменту нейтрализации раствора (pH 7,0), находится как раз посередине скачка нейтрализации, а потенциал водородного электрода при этом равен
E = 0,0581g 10-7 = —0,058-7 = = •— 0,406 в.
В практике иногда используют упрощенные варианты схем, в которых вместо съемки полных кривых ограничиваются титрованием до какого-либо наперед заданного значения pH. В качестве индикаторных
родной ряный
электродов при потенциометрическом титровании в практике наиболее часто применяются стеклянные электроды с водо-и натриевой функциями, хингидронный электрод, хлорсереб-электрод — при определении хлорид-иона, платиновый
электрод — при титровании окислительно-восстановительных систем и др.
В последние годы широко применяется потенциометрическое титрование в неводных средах. Оно позволяет значительно расширить класс исследуемых органических и неорганических веществ, нерастворимых в воде. Используя различные растворители, удается отти-тровывать смеси веществ, выявлять присутствие незначительных примесей, четко определять константы многоосновных кислот. Неводное титрование находит широкое применение в органической и полимерной химии, фармацевтической, парфюмерной и пищевой промышленности. В ряде случаев чувствительность потенциометрического метода превышает чувствительность обычного объемного метода, а также метола
кондуктометрического титрования.
§ 95. Свинцовый аккумулятор
На процессах окисления—восстановления основана работа широко распространенного химического источника электрического тока — свинцового аккумулятора. Это также гальванический элемент, но материалы в нем подобраны с таким расчетом, чтобы была возможна максимальная обратимость процесса, иными словами, чтобы многократное повторение циклов зарядки и разрядки совершалось без необходимости добавления участвующих в его работе веществ.
В настоящее время аккумуляторы получили широкое и разнообразное применение в различных областях народного хозяйства. Они являются необходимой принадлежностью буквально всех машин, на которых установлены двигатели
— 322 —
мгтоеннего сгорания. Шахтные электровозы, грузовые электрокары, подводные лки также работают на использовании свинцовых аккумуляторов. Не менее 71 ипокое распространение имеет свинцовый аккумулятор и в повседневной лабораторной практике, так как является дешевым и удобным источником постоянного
тока.
Свинцовый аккумулятор состоит из трех свинцовых пластин в виде решеток или рам с ребристой поверхностью (рис. 115). Отверстия в этих пластинах замаза-
Рис. 115. Свинцовый аккумулятор:
J — общий вид; 2 — решетчатая свинцовая пластинка (отдельно)
ны тестом, состоящим из окиси свинца и воды. Пластины погружены в 22— 28%-ный раствор серной кислоты. При взаимодействии окиси свинца с серной кислотой на поверхности пластин образуется тонкий слой сульфата свинца
РЬО 4- H2SO4 = PbSO4 + Н2О
Рассмотрим реакции, проходящие в свинцовом аккумуляторе. На отрицательном полюсе идет реакция окисления свинца
РЬ — 2е*“=РЬ2+
Образовавшиеся в результате этой реакции ионы свинца соединяются с анионами серной кислоты, образуя нерастворимую в воде соль PbSO4
Pb2+ + SO2-=PbSO4
Электроны, отдаваемые атомами свинца, движутся по проводнику к положитель-I ому полюсу, где они восстанавливают четырехзарядный свинец до двухзарядного, который дает с серной кислотой нерастворимый сульфат
РЬО2 + 2е“ + 4Н+ + so|“=PbSO4 + 2Н2О
Если через свинцовый аккумулятор пропускать постоянный ток, предварительно соединив положительный полюс аккумулятора с положительным полюсом источника постоянного тока, а отрицательный полюс аккумулятора — с отрицательным полюсом источника тока, реакции, происходящие при разрядке аккумулятора, будут идти в обратном направлении.
Электроны, посылаемые внешним источником тока, входят в отрицательный полюс аккумулятора и восстанавливают двухзарядные ионы свинца до металлического свинца
PbSO4 + 2е~ =РЬ 4- SQ2—
11*
— 323 —
На положительном полюсе аккумулятора идет реакция окисления двухзарядного свинца до четырехзарядного в результате потери двух электронов
PbSOa—2е- + Н2О с=РЬО2 + 4Н+ + SO^~
Оба процесса — разрядка и зарядка аккумулятора — можно изобразить одним суммарным уравнением
разрядка
Pb+PbO-> + 4H++SO^—;--------2РЬ5Ол4-2Нс.О
зарядка
После долговременного пропускания тока одна пластина заполняется губчатым свинцом, другая — пористым слоем двуокиси свинца РЬО2. По окончании зарядки начинается электролиз воды: у катода выделяется газообразный водород, а у анода — кислород. В результате реакции жидкость в растворе как бы закипает, что является внешним признаком окончания процесса зарядки аккумулятора.
"Э. д. с. свинцового аккумулятора долго держится около 2 в, затем, по прошествии определенного времени работы аккумулятора, она начинает падать. Как только э. д. с. снизится примерно до 1,8 в, дальнейшую разрядку аккумулятора необходимо прекратить во избежание его порчи.
Одним из недостатков свинцового аккумулятора является его относительно большая тяжесть. Поэтому в ряде случаев используют более легкие аккумуляторы, например железо-никелевые, которые относятся к щелочным (электролитом является раствор щелочи, обычно КОН). Щелочные аккумуляторы в отличие от свинцовых не боятся толчков и встряхиваний, хорошо переносят длительное пребывание в разряженном состоянии. Однако щелочные аккумуляторы обладают и некоторыми недостатками: у них меньший коэффициент полезного действия по сравнению со свинцовым, меньшая величина э. д. с., а также меньшая емкость. Напомним, что емкость аккумулятора выражается в ампер-часах и определяется тем наибольшим количеством электричества, которое можно получить от заряженного аккумулятора.
«Истинный химик должен быть теоретиком и практиком»
Л1. В. Ломоносов
Глава VIII
ФИЗИКО-ХИМИЧЕСКИЕ МЕТОДЫ, ПРИМЕНЯЕМЫЕ В ПРАКТИКЕ АГРОХИМИЧЕСКИХ И ПОЧВЕННЫХ ЛАБОРАТОРИЙ
При изучении структуры и свойств веществ в настоящее время наряду с химическими методами исследования приобретают большое значение физико-химические методы. Последние обладают целым рядом преимуществ, так как являются более быстрыми и более надежными. В большинстве случаев они не вызывают каких-либо изменений в исследуемых объектах, а для проведения их требуется значительно меньше вещества, чем при химическом анализе. Физико-химические методы оказываются единственно возможными, когда изучаются тонкие различия в структуре молекул различных веществ.
В настоящее время физико-химические методы анализа находят широкое применение в агрохимических лабораториях: с помощью термического, рентгеноструктурного, инфракрасно-спектроскопического, спектрофотометрического и многих других методов исследования получены ценные данные о важных физико-химических процессах, протекающих в почвах, а также в растительных и животных организмах.
Современный ученый-агроном должен иметь достаточно полные представления о физико-химических методах исследования, что позволит ему не только яснее представлять процессы, протекающие в почве и растениях, но и свободно читать научную литературу и разбираться в результатах научных исследований, выполненных на молекулярном уровне, самому ставить и разрешать отдельные вопросы в повышении плодородия почв.
§ 96. Термографический метод
Термографический анализ получил развитие и широкое распространение в различных областях народного хозяйства, в частности в почвоведении. С его помощью можно решать различные задачи минералогии почв. Термографический анализ основан на изучении тепловых эффектов, происходящих в результате физико-химических превращений веществ при изменении их температуры.
При нагревании минералов происходят различные физико-химические процессы: удаление рыхлосвязанной и конституционной воды, разрушение кристаллической решетки и образование новых минералов. Эти реакции сопровождаются поглощением или выделением теплоты. Все изменения температуры фиксируются при помощи термопар и записываются на фотобумаге лучом света, отраженным от чувствительного зеркального гальванометра, к которому подключены термопары. Полученные кривые называют термограммами или кривыми нагревания. Прибор Для термоанализа называется пирометром. Наиболее употребительны такие марки пирометров: ПК-55, ФПК-59, УТА-1 и др.
Термограммы получают следующим образом. В одно из отделений двухкамерного тигля набивают навеску почвы или какого-то другого исследуемого материала, а во второе отделение — эталонное вещество (А12О3, MgO и др.), которое не претерпевает никаких изменений при нагревании. Один спай термопары (обычно платиновой и платинородиевой) помещается в центр исследуемого образца. Другой — в центр эталонного вещества. Схема соединений этих термопар с гальванометрами, а также принципиальная схема пирометра показаны на рис. 116.
Тигель с термопарами 1 помещают в печь 2 и нагревают с равномерной скоростью до 1200° С. С ростом температуры э. д. с. термопары увеличивается и соот
— 325 —
ветственно отклоняется зеркальце гальванометра 3. Луч света, отраженный зеркальцем, фиксируется на фотобумаге 6.
Как видно из рис. 116, обе термопары соединены таким образом, что одна измеряет температуру нагрева (нормальная термопара), другая — разность температур между образцом и эталонным веществом (дифференциальная термопара). При отсутствии различий в температурах образца и эталона тока в цепи термопар не будет, так как оба спая нагреты одинаково, и возникающие термотоки гасят друг друга. Самописец зарегистрирует прямую линию, практически параллельную оси абсцисс. При наличии в испытуемом веществе тепловой реакции возникает разность температур между образцом и эталоном. В цепи термопар появится ток, и самописец зарегистрирует дифференциальную кривую. Причем отклонение на термограмме дифференциальной кривой вниз от нулевого положения соответствует эндотермическому эффекту, а вверх — экзотермическому.
Рис. 116. Схема устройства аппарата для термического анализа (пирометра) :
/ — тигель с термопарами; 2 — печь; 3 — гальванометры; 4 — образец; 5 — эталон; 6 — самописец; 7 — холодные спаи; 8 — сосуд Дьюара; 9 — выключатель гальванометров; 10 — мотор; // — осветители; 12 — общий автоматический выключатель; /<3 — выключатель освештелей и мотора; 14 — трансформатор осветителей и мотора
В некоторых пирометрах наряду с записью термограммы исследуемого вещества одновременно происходит и запись потери веса его при нагревании.
Каждый из минералов обладает характерной термограммой (рис. 117). Для примера приведем описание термограмм нескольких минералов, наиболее часто встречающихся в почвах. Так, каолинит имеет два термических эффекта (рис. 117, /): эндотермический при 590° С, и экзотермический при 975° С. Первый обусловлен разрушением кристаллической решетки и удалением конституционной воды. Экзотермический эффект обусловлен образованием нового кристаллического вещества из продуктов разрушения каолинита.
Термограмма галлуазита (кривая 2) отличается от термограммы каолинита наличием эндотермического эффекта при 100° С, обусловленного выделением адсорбированной воды, которой нет у каолинита.
На термограмме монтмориллонита (кривая 3) наблюдаются три эндотермических эффекта: соответственно при 115°, 650° и 825° С — а также один экзотермический — в интервале температур 925—1055° С. Первый эффект (иногда раз-
— 326 —
пвоенный) обусловлен выделением рых-лосвязанной и гигроскопической воды, второй и третий —разрушением кристаллической решетки минерала. Экзотермический эффект обусловлен, как и в случае с каолинитом, образованием нового кристаллического вещества.
На кривых нагревания гидрослюди-стых минералов также наблюдаются три эндотермические и одна экзотермическая реакции (рис. 117). Первая низкотемпературная эндотермическая реакция с максимумом при 120—160° С обусловлена потерей межслойной воды; вторая начинается в интервале 450—500° С и имеет максимум в пределах 550—600° С. При этом происходит потеря ОН-групп кристаллической решетки и начинает частично изменяться * структура минерала. Третья эндотермическая реакция наблюдается в интервале между 850—900° С. При этом происходит полное разрушение структуры гидрослюды. Экзотермическая реакция между 900 и 1000° С связана с перекристаллизацией аморфных продуктов разрушения гидрослюды.
Таким образом, сравнивая термо-
Рис. 117. Термограммы некоторых минералов:
1 — каолинит; 2 — галлуазит;
3 — монтмориллонит; 4 — вермикулит
грамму почвы или какого-то другого мине-
рального объекта с термограммами чистых минералов, можно определить, какие минералы содержатся в исследуемой пробе.
§ 97. Электронная микроскопия
За последние годы электронный микроскоп получил широкое применение в различных исследованиях в области биологии, вирусологии, а также в почвоведении при минералогических анализах глин и почв.
Как известно, современный световой микроскоп позволяет рассматривать объекты размером не менее одного микрона, так как разрешающая способность микроскопа невелика. Напомним, что разрешающая способность (d) микроскопа определяется формулой:
, 0,61Х
<7=-------,
п sin а
(VIII,1)
где d — наименьшее расстояние между двумя предметами, при котором они еще видны раздельно, X — длина волны света, п — коэффициент преломления света в данной среде, а — апертурный угол, равный половине угла вершины конуса лучей, попадающих в объектив микроскопа. Попутно отметим, что чем больше апертурный угол, тем больше разрешающая способность микроскопа. В современных микроскопах этот угол равен 70°.
В табл. 68 приведены средние значения длин волн (см) различных электромагнитных колебаний.
Как видно из табл. 68, длина волны видимого света не менее 4,0 • 10-5 см (4000 А). Апертура лучших световых микроскопов равна 70°. Коэффициент преломления среды можно повысить, поместив между предметом и линзой жидкость С высоким показателем преломления, напри*мер кедровое масло, до п = 1,5. Подставив в формулу (VIII, 1) численные значения всех рассмотренных величин, определим наименьшее разрешающее расстояние. Оно равно примерно 0,2 мк. Практически такого разрешения добиться очень трудно, и частицы коллоидных размеров, т. е. меньше 0,2 мк, в световом микроскопе различить не удается.
— 327 —
Таблица 68
Средние значения длин волн электромагнитных колебаний
Вид излучения
Длина волн, см
Инфракрасное излучение Белый (видимый) свет . Ультрафиолетовые лучи Рентгеновские лучи . . Волны потока электронов
1,2.1g-1—7,6-10-5
7,6 • 10“5—4,0-10“5 4,0-10-5—1,5.10“7
1,5-10-7—6,0-10-10 5,0-10~10
В электронном микроскопе вместо света на объектив направляется пучок электронов, длина волны которого в 50—100 тысяч раз меньше длины волны света. Роль оптической системы здесь играют электрические или магнитные поля.
Разрешающая способность электронного микроскопа в 50—100 раз больше таковой светового микроскопа. В электронный ми-
кроскоп можно наблюдать объекты размером 10—15 А, а в отдельных случаях 6—8 А.
В электронном микроскопе можно выделить следующие основные узлы:
1) осветительная система: электронная пушка и конденсорная линза. Источником электронов является вольфрамовая нить, накаленная до 2400° С;
Рис. 118. Схема и ход лучей: а - в оптическом микроскопе; б — в электронном магнитном микроскопе
Рис. 119. Электронный ми-, кроскоп УЭМБ-100
- .328 —
2) камера для образцов с предметным столиком, куда помещают исследу-е‘ 3) линза-объектив, дающая первичное изображение объекта;
4) одна или несколько проекционных линз для вторичного увеличения;
5) фотокамера с флуоресцирующим экраном и фотокассетой для наблюде-ия И фотографирования исследуемого объекта.
ни Чтобы исключить столкновение электронов с молекулами воздуха, в приборе поддерживается высокий вакуум порядка 10“4 — 10~5 мм рт. ст.
Ход электронных и световых лучей и схема образования изображения электронном и в световом микроскопе очень сходны (рис. 118). Схемы обоих приборов имеют аналогичные элементы: источник освещения и линзы — конден-
Рис. 120. Электронномикроскопические снимки глинистых минералов:
а — монтмориллонит; б —каолинит; в — галлуазит
сорную, объективную и проекционную. Принципиальные отличия заключаются в том, что в электронном микроскопе источником освещения является электронная пушка, линзами служат магнитные или электрические поля, а для того, чтобы полученное изображение сделать видимым, применяют специальные экраны, покрытые светящимся составом.
Внешний вид электронного микроскопа марки УЭМБ-100 представлен на рис. 119. Это один из лучших отечественных микроскопов.
В настоящее время исследования с помощью электронного микроскопа широко проводятся в почвоведении. Кристаллы различных минералов можно отличить по форме, сличая полученные на электронном микроскопе снимки. а рис. 120 приведены снимки кристаллов некоторых высокодисперсных минералов, которые наиболее часто встречаются в почвах: монтмориллонита, каолинита, галлуазита.
С помощью электронного микроскопа можно также решать такие вопросы в области почвоведения, как природа связи минеральных и органических коллои-
— 329 —
дов между собой. Непосредственное наблюдение расположения органических коллоидов и бактерий на поверхности минералов стало доступно только при таком увеличении, какое может дать электронный микроскоп.
§ 98. Рентгенографический метод
Рис. 121, Рентгеновский аппарат
УРС-55А
Рентгенографические исследования в последние годы получили широкое распространение в почвоведении. Определение содержания высокодисперсных минералов в почвах или в каких-либо других объектах методом рентгенографии основано на явлении дифракции рентгеновских лучей от плоскостей кристаллов и интерференции. Лучи, отраженные от исследуемого объекта, попадают на фотобумагу, где образуют симметрично расположенные линии дугообразной формы различной степени интенсивности.
Атомы, молекулы и ионы, образующие кристалл, располагаются в нем в правильном порядке, образуя стр у к-. туру. Характер этого порядка может быть разным, но для данного вида кристалла всегда одинаков.
В качестве примера может служить хлорид натрия (см. рис. 12). Его кристаллы построены из ионов натрия и хлора, которые, чередуясь, располагаются правильными рядами, объединяющимися в плоскостную решетку. В узлах решетки в шахматном порядке находятся ионы Na+ и С1". Плоскости располагаются в пространстве параллельно одна другой (смещаясь на одно звено) и образуют пространственную кристаллическую решетку*.
Решетка имеет три измерения и ее
можно представить в виде примыкающих друг к другу параллелепипедов, каждый из которых можно назвать элементарной ячейкой. В пространственной решетке можно выделить узловые прямые и узловые плоскости, которые находятся друг от друга на определенном расстоянии d, которое является характерным для каждой системы плоскостей данного кристалла.
Длины волн рентгеновских лучей имеют тот же порядок, что и расстояние между атомами или ионами в кристаллах (10“8слг). Именно благодаря этому при дифракции рентгеновских лучей от граней кристалла и получается рентгенограмма, которая дает возможность, в частности, определить расстояние между узловыми плоскостями, а также между частицами в кристалле. Каждая линия на рентгенограмме соответствует отражению рентгеновских лучей от определенных узловых плоскостей. При расшифровке рентгенограмм сначала делают точные промеры расстояний между симметрично расположенными линиями (дугами). Зная
эти величины, можно сравнительно легко рассчитать межплоскостные расстояния в кристаллах, являющиеся характерными для каждого минерала.
При расчете пользуются формулой Брегга—Вульфа:
nX = 2dsinO, (VIII,2)
в
где п — целое число или порядок отражения, от разных параллельно расположенных плоскостей в кристалле, X — длина волны падающего на объект рентге-
* Расположение частиц в пространстве может быть иным и более сложным, чем у хлорида натрия. В таком случае усложняется геометрическая форма самих кристаллов.
— 330 —
невского луча, dрасстояние между сосрппн.,., .
скольжения, т. е. угол между падающим лучом ^Ип^ЗЛаМИ ₽ешетки> 6 — угол
Угол 0 равен k2s, где k - постоянная величина Х?'0'
изводится рентгеносъемка, 2s — расстояние п ДЛЯ камеРьЬ в которой про-
Для рентгенографического анализа приме^яю^ИМеТ₽аХ На Ре«тгенограмме. струкции. На рис. 121 показан аппарат марки Ж «а.ппа₽аты различной кон-роко применяется в минералогических и почвентХ / КОТОРЫЙ Довольно ши-
Каждыи из высокодисперсных минепяп^о Х лабораториях.
нограмму (рис.122). Так, на рентгенограмме каочи™™ /л хаРактеРнУЮ рентге-ся расстояние d = 7,15 А, а также d = 3 58- 9 59 9 чо характеРньш являет-т" Д„ •„в„
Рис. 122. Рентгенограммы некоторых глинистых минералов: / —каолинит; 2 — галлуазит; 3 — галлуазит с несовершенной структурой; 4— диккит
,ся межплоскостное расстояние 14—16 А. Кроме того, используются и другие линии рентгенограммы: d = 4,46 — 4,48; 2,55—2,59; 1,49 А. При нагревании, а также при насыщении глинистых минералов органическими наполнителями (глицерин, этиленгликоль) происходят заметные изменения в расположении линий на рентгенограммах. Эти приемы широко применяются при минералогических исследованиях таких сложных полиминеральных систем, какими являются почвы. Метод рентгенографии особенно хорошо себя зарекомендовал при минералогическом анализе высокодисперсной (менее 0,001 мм). части почвы, в состав которой входят глинистые минералы.
Сравнивая рентгенограммы высокодисперсных частиц почвы с рентгенограммами известных глинистых минералов, можно определить, из каких минералов состоят частицы.
§ 99. Метод инфракрасной спектроскопии
Инфракрасная спектроскопия (ИК-спектроскопия) стала одним из основных методов исследования состава и строения различных химических соединений, сажными преимуществами этого метода являются его быстрота, наглядность поручаемых результатов, а также возможность проведения анализа с весьма малыми количествами (несколько миллиграммов) исследуемого вещества.
г- 331 —
Инфракрасный спектр любого химического соединения — одна из наиболее важных его характеристик и может использоваться для идентификации вещества. При сравнении двух соединений идентичность их инфракрасных спектров указывает (за очень редким исключением) на идентичность самих соединений. Инфракрасные спектры любых веществ дают ценные сведения об их внутренней структуре. Для расшифровки ИК-спектра нужно найти соответствие между положением полос поглощения и наличием отдельных структурных элементов в молекуле, а уже затем по ИК-спектру определить присутствие тех или иных атомных групп и их взаимное расположение.
AXz-HOMitUWte АХг-веерное /U2“ крутильное
UPu МШсшнде иливнеплоскостное -или внеллоскостное >
АХ2-маятникоВое АХ^-симметричное АХ5~ асимметричное
А Х2~симметричное АХ5-симметричное
АХ^-асимметричное АХ3-асимметричное
Рис. 123. Валентные колебания молекул типа АХ2 и АХ3
Метод инфракрасной спектроскопии в последние годы стал особенно широко применяться в органической и неорганической химии, биохимии, биологии, почвоведении, минералогии и во многих других областях науки и народного хозяйства.
Что же собой представляет инфракрасный спектр любого химического соединения? Известно, что все молекулы состоят из атомов, соединенных между собой химическими связями. Атомы в молекуле находятся в постоянном колебательном движении. В простейшем случае, когда молекула вещества состоит только из двух атомов А и Б, происходит единственный вид колебаний — периодическое растяжение и сжатие по линии связи А — Б. Движение химически связанных атомов в молекуле можно сравнить с непрерывным колебанием систем шаров, которые связаны между собой пружинами. Оба вида колебательного движения хорошо описываются математически при помощи закона Гука. Колебательная частота в случае двухатомной молекулы определяется уравнением:
(VIII,3)
— 332 —
где v — колебательная частота, см*1, с — скорость света, jj — силовая постоянная связи и р — приведенная масса, определяемая уравнением:
тАтБ и =---;--,
(VIII,4)
где и тБ — массы отдельных атомов А и Б.
Аналогичным образом могут рассматриватьсячвалентные колебания и в более сложных молекулах, хотя здесь становятся возможными и другие виды колебаний, в частности деформационные колебания. При валентных колебаниях изменяются в основном длины связей, углы между ними остаются приближенно неизменными. В случае деформационных колебаний изменяются главным образом углы между связями. Валентные колебания молекул типа АХ§ и АХ3 схематично представлены на рис. 123, где стрелки показывают направление периодического смещения атомов, а знаки плюс и минус соответствуют периодическим смещениям перед плоскостью или за плоскостью бумаги.
Инфракрасное излучение принято характеризовать волновым числом у, которое выражается в обратных сантиметрах (сж^1) и называется обычно частотой, хотя на самом деле единица частоты у имеет размерность обратной секунды (сек"1). Для характеристики излучения используется также длина волны %, измеряемая в микронах. Волновое число у связано с длиной волы % следующей простой зависимостью:
10*
Таким образом, частота у представляет собой величину, равную числу длин волн, выраженных в микронах, уложенных на расстоянии 1 см. Например, интервал от 2,5 до 40,0 мк соответствует интервалу от 4000 до 250 см-1.
Как видно из уравнений (VIII, 3) и (VIII, 4), частоты колебаний зависят не только от самой природы отдельных связей, но и от всей молекулы и ее окружения. В случае системы шариков, связанных пружинками, происходит совершенно аналогичное явление — на колебание одной пружинки оказывает влияние вся система в целом. Если по такой системе произвести удар, амплитуды колебаний шариков возрастут. Аналогичное явление происходит и в молекуле вещества при воздействии на нее электромагнитных волн (инфракрасных лучей): амплитуды колебаний отдельных связей и вместе с ними колебаний электрических зарядов будут увеличиваться.
Полной аналогии между системой шаров на пружинах и молекулой какого-либо химического вещества быть не может: колебательные энергетические уровни молекулы изменяются скачкообразно. В системе же шаров на пружинах амплитуды колебаний изменяются непрерывно. Следовательно, молекула поглощает только те частоты инфракрасного излучения, энергия которых точно соответствует разностям между двумя уровнями энергии связи, т. е. амплитуды колебаний возрастают скачкообразно.
Таким образом, при взаимодействии молекулы какого-либо вещества с электромагнитным полем (инфракрасным излучением) может произойти передача энергии от поля молекуле, в результате чего молекула перейдет из одного квантового состояния в другое (возбужденное). Приобретенная молекулой величина энергии Е связана с частотой поглощенного излучения v простым соотношением:
AE = Av, (VIII,5)
где h — постоянная Планка (А «= 1,623 • 10"2- эрг-сек).
При возвращении молекулы в исходное состояние она испускает такой же квант энергии ДЕ частоты у. Итак, если исследуемое вещество облучать инфракрасным светом с непрерывно меняющейся частотой, определенные участки спектра излучения будут поглощаться молекулой, в результате чего луч, проходящий через вещество, в области поглощения будет ослабляться. Регистрируя интенсивность прошедшего через данное вещество инфракрасного излучения в зависимости от длин волн или от волновых чисел, можно получить кривую, на которой
— 333 —
будут видны полосы поглощения. Эта кривая и называется инфракрасным спектром поглощения.
Инфракрасные спектры могут дать весьма богатую информацию, поскольку каждая молекула имеет только ей присущий набор колебаний, который зависит от массы и взаимного расположения атомов в данной молекуле, межатомных расстояний, типов меж- и внутримолекулярных взаимодействий. Даже самые незначительные изменения в структуре молекул отражаются на инфракрасных спектрах поглощения, поэтому инфракрасный спектр поглощения может являться однозначной характеристикой вещества, образно говоря его «паспортом».
Рис. 124. Принципиальная схема однолучевого инфракрасного спектрометра:
/ « источник инфракрасных лучей; II — монохроматор; 111 — усилитель термотока; IV « регистрирующее устройство; 1 — источник излучения (штифт Нернста или глобар); 2 — исследуемое вещество; 3 — входная щель монохроматора; 4 — выходная щель монохроматора; 5-—отсчетное устройство; 6 — приемник излучения (термоэлемент)
Метод инфракрасной спектроскопии может применяться для идентификации различных химических соединений. Поскольку полосы поглощения непосредственно отражают состояние связей и наличие в молекуле различных атомных груп-пироврк, можно получать весьма ценную информацию о структуре данного вещества. Интенсивность полосы поглощения в ИК-спектре любого химического соединения пропорциональна количеству исследуемого вещества, и это обстоятельство лежит в основе еще одного важного направления применения инфракрасной спектроскопии — количественного анализа.
Прибор, в котором получают запись инфракрасного спектра поглощения, горит название инфракрасного спектрофотометра (или спектрометра). Принцип действия инфракрасных спектрометров в общем довольно прост, сложны лишь их механические и электрические устройства, необходимые для воспроизведения малейших изменений поглощаемой энергии в виде точно зарегистрированного спектра. Поскольку обычное стекло не прозрачно для инфракрасных лучей, в инфракрасных спектрометрах оптика изготовляется из кристаллов галидов (NaCl, КВг, LiF), которые не поглощают инфракрасные излучения.
Принцип работы инфракрасного спектрометра заключается в следующем. Инфракрасное излучение, получаемое от специального источника, проходит
— 334 —
через исследуемое вещество и далее разлагается в спектр с помощью моно-кроматора. Отдельные лучи этого спектра последовательно направляются на приемник излучения и затем, после усиления, фиксируются соответствующим записывающим устройством. Принципиальная схема однолучевого прибора представлена на рис. 124. Имеются более сложные двухлучевые инфракрасные спектрометры, в которых световое излучение от источника разделяется
на два потока: один из них проходит через кювету с исследуемым веществом, другой — через аналогичную кювету, не содержащую этого вещества.
Наша промышленность серийно выпускает однолучевые инфракрасные спектрометры моделей ИКС-9, ИКС-11, ИКС-12, а также двухлучевой прибор марки И КС-14.
На рис. 125 приведены инфракрасные спектры поглощения некоторых высокодисперсных глинистых минералов, наиболее часто встречающихся в почвах.
Полосы поглощения ПК-спектров любых веществ, в том числе и минералов, обусловлены колебаниями определенных атомных групп и их связей. Гак, полосы, обусловленные Si — О колебаниями, в спектрах диккита и каолинита наблюдаются при v — 435, 470, 685, 1010, 1030 и 1110 сл/"1. Полосы поглощения, приписываемые Si — О — А1 колебаниями, проявляются при v= =750 и 790 см"1, полоса при v = = 910 см"1 обусловлена колебаниями связи Н — О — А1. Все рассмотренные полосы поглощения характерны для обоих минералов. Отличие в этой области спектра каолинита и диккита заключается в том, что диккит имеет в своем ПК-спектре две полосы поглощения: слабую при v = 645 см-1 и очень интенсивную при v=1435 слт"1. В ПК-спектре каолинита эти полосы отсутствуют.
Однако главные отличия в спектрах минералов каолинитовой группы наблюдаются в коротковолновой области. Колебания в этом участке спектра обусловлены О — Н-группами, входящими в состав исследуемых минералов. Например, каолинит имеет очень узкую и интенсивную полосу поглощения при V —3700 слг"1 и менее интенсивную при v = 3630 см-1. . Полосы поглощения при v = 3675 и 3660 см-1 у него очень слабые. У диккита только три очень интенсивных полосы поглощения с максимумами при v = 3630, 3660 и 3710 см-1.
Минералы группы галлуазита содержат в своих ПК-спектрах те же полисы поглощения, что и минералы группы каолинита. Отличие наблюдается
Рис. 125. ПК-спектрограммы различных глинистых минералов: а — каолинит; б — галлуазит; в монтмориллонит; г — вермикулит
335 —
в коротковолновой области спектра. Так, галлуазит характеризуется лишь двумя узкими полосами поглощения при V —3640 и 3700 см~\ причем полоса 3640 см"1 значительно интенсивнее аналогичной полосы поглощения при v = =3630 сл/”1 каолинита. Для галлуазита характерно обратное (чем у каолинита) соотношение интенсивностей этих полос.
Из этого примера явствует: даже близкие по структуре глинистые минералы можно сравнительно легко идентифицировать по их инфракрасным спектрам поглощения. Напомним, что идентификация этих минералов с помощью других физико-химических методов (термический анализ, рентгенография) очень затруднена.
Метод инфракрасной спектроскопии широко применяется во многих областях сельского хозяйства. В частности, с его помощью изучают гуминовые соединения почвы, а также проводят анализ растений на содержание различных органических соединений (аминокислотный состав, сахара, кислоты жирного ряда и целый ряд других жизненно важных веществ).
§ 100. Фотометрический анализ
Фотометрический метод количественного анализа основан на измерении све-топоглощения растворами окрашенных соединений. Через слой окрашенного вещества свет частично проходит, частично отражается, частично поглощается (рис. 126).
Интенсивность* падающего светового потока при прохождении через поглощающий раствор разлагается на составляющие:
/о = 7 4- /п 4- /от >
где / — интенсивность светового потока, прошедшего через слой вещества, /от — интенсивность отраженного светового потока, /п — интенсивность светового потока, поглощенного окрашенным веществом.
Интенсивность падающего светового потока /0 и прошедшего через раствор / можно определять экспериментально. Эти две величины связаны между собой соотношением (закон Бугера—Ламберта):
l = (VIII,6)
где е — основание натуральных логарифмов, а — коэффициент поглощения, I — толщина поглощающего слоя. Для различных фотометрических исследований наиболее удобно выражать интенсивность светопоглощения уравнением:
D=lgy-. (VIII,7)
Эта величина называется оптической плотностью. Связь между концентрацией поглощающего раствора и его оптической плотностью выражается законом Бера:
D=lg-y = *iC, (VIII,8)
где k± — коэффициент пропорциональности, С — концентрация растворенного вещества.
Зависимость интенсивности прошедшего через раствор монохроматического светового потока от интенсивности падающего потока света, концентрации окрашенного вещества и толщины просвечиваемого слоя выражается уравнением:
/ = /0.10“^. (VIII,9)
* Под интенсивностью света понимают энергию светового потока, испускаемого источником света в 1 сек внутри телесного угла, равного единице. Единица световой энергии (люмен-секунда) — это энергия светового потока в 1 ллг, которая расходуется в течение 1 сек,
— 336 —
Где £ — коэффициент погашения, который зависит от природы растворенного вещества, температуры, растворителя и длины волны падающего светового потока. Это соотношение носит название основного закона светопоглощения или объединенного закона Бугера—Ламберта—Бера. Оно лежит в основе большинства фотометрических методов анализа.
Рис. 126 Прохождение светового потока через окрашенный раствор
Если концентрация С выражена в моль/л, а толщина слоя I в см, то k становится молярным коэффициентом погашения и обозначается Уравнение (VIII,9) в этом случае будет иметь вид:
7 = /0.Ю“е’-сг- (VIII ,10)
Если раствор подчиняется закону Бугера—Ламберта—Бера, то оптическая плотность раствора прямо пропорциональна концентрации поглощающего вещества, толщине слоя раствора и молярному коэффициенту погашения:
£) = ехС/. (VIII,11)
Различают в основном три группы способов измерений концентрации окрашенного соединения в растворе: визуальное сравнение, фотоколориметрирование и спектрофотометрирование. Остановимся на краткой их характеристике.
Визуальный метод сравнения. Как известно, визуально можно довольно точно устанавливать равенство интенсивности окрасок двух растворов. Визуальный колориметрический метод отличается быстротой и простотой. В силу этого он получил довольно широкое распространение в количественном агрохимическом анализе. Он принципиально основан на том, что согласно закону Бугера— Ламберта—Бера при определенных условиях существует прямая пропорциональ-• ная зависимость между интенсивностью окраски раствора и концентрацией окрашенного соединения. Окраски стандартного и испытуемого растворов сравнивают в приборе, который носит название колориметра. Уравнивания окрасок обоих растворов добиваются изменением толщины слоев окрашенных растворов, что осуществляется при помощи погружателей, которые представляют собой призмы, связанные с отсчетной шкалой и изготовленные из оптического стекла.
Наиболее распространенным прибором является колориметр марки КОЛ-1М, общий вид которого показан на рис. 127.
В момент равенства окрасок стандартного и испытуемого растворов^ оптические плотности их одинаковы. Таким образом, отношение концентраций растворов обратно пропорционально толщине их поглощающих слоев:
с I 1
* ст
откуда
(VIII, 12)
- 337 —
где Сх и Сст—соответственно концентрации исследуемого и стандартного растворов, а 1Х и /ст — толщина слоев этих растворов. Визуальный метод колориметрии имеет существенные недостатки. Он является очень субъективным: точность анализа во многом зависит от индивидуальных особенностей зрения исследователя. Кроме того, при длительной и непрерывной работе глаза сравнительно быстро устают, что отрицательно сказывается на точности измерений.
Фотоколориметрический анализ. В фотоколориметрии степень поглощения света окрашенными растворами определяется не визуально, а при помощи специальных приборов с фотоэлементом. Такие приборы называются фотоэлектроколориметрами (ФЭК}. Фотоэлемент преобразует световую энергию, проходящую через окрашенный раствор, в электрическую. Поскольку сила возникающего фототока прямо пропорциональна интенсивности падающего на фотоэлемент света, с помощью ФЭК можно непосредственно измерить ослабле-
Рис. 127. Внешний вид концентрационного калориметра К0Л-1М:
1 — окуляр; 2 —- диск со светофильтрами; 5 — лупа нониуса отсчетной. шкалы; 4 — рукоятка перемещения погружателя; 5 — трансформатор; 6 — патрон для лампы; 7 =₽ кожух; <3-—погру-жатель; 9 — кювета
ние интенсивности первоначального светового потока. Нет необходимости каждый раз готовить стандартные растворы. При работе с ФЭК обычно строят 'градуировочный график по серии растворов с известной концентрацией окрашенного соединения. Такой график особенно удобен при массовых однотипных анализах.
В настоящее время существует много фотоэлектроколориметров различных конструкций. Все они имеют обязательно следующие элементы схемы: 1) осветитель, 2) светофильтры, 3) кюветы, 4) фотоэлементы, 5) система регулируемых сопротивлений, 6) гальванометры.
Различают два типа фотоэлектроколориметров: однолучевые, или приборы с одним фотоэлементом, и двухлучевые, или приборы с двумя фотоэлементами В настоящее время однолучевые фотоэлектроколориметры практически не применяются. В двухлучевом приборе оба фотоэлемента соединены по принципу противотока, гальванометр показывает разницу фототоков между исследуемым раствором и чистым растворителем, так что небольшие колебания света осветителя практически не отражаются на точности определений.
Современные фотоэлектроколориметры ФЭК-М, ФЭК-Н-52, 54, 57, ФЭК-56, которые наиболее широко распространены, являются двухлучевыми приборами и имеют одинаковые оптические схемы. На рис. 128 показан внешний вид и прин-
— 338 -
иальная схема фотоэлектроколориметра ФЭК-М. Как видно из рисунка, один ЦИфотоэлементов находится в «контрольном» световом потоке, что позволяет ИЗт матически компенсировать колебания тока в цепи осветителя. Для уравни-аВ ия двух световых потоков в ФЭК-М применена щелевая диафрагма. Для rto-ва Вения точности измерений в прибор вмонтировано два набора из четырех Фетофильтров: нейтрального, синего, зеленого и красного. Оптическая характе-п стика этих светофильтров представлена на рис. 129. От визуальных приборов фотоэлектроколориметры отличаются значительно более высокой точностью.
Рис. 128. Принципиальная схема (а) и внешний вид (б) фотоколориметра ФЭК-М:
1— лампа; 2 и 2' —• конденсаторы; 3 и 3' — зеркала; 4 и 4'— светофильтры; 5 и 5, 7 и 7' — линзы; 6 и 6' — кюветы с растворами; 8 и 8' — призмы; 9 и 9' — фотоэлементы;
10 и //—’фотометрические клинья; 12 — щелевая диафрагма; 13 — отсчетный oapaoanj 14 — гальванометр; 15 — шкала отсчетных барабанов; 16 — шторка, перекрывающая световые потоки; /7— арретир гальванометра; 18 — механический корректор; /9 — пере* ключатель чувствительности гальванометра; 20 — кюветодержатель; 21 — переключатель светофильтров
Рис. 129. Спектральная характеристика светофильтров:
1 «=» синий; 2 — зеленый, 3 красный
Спектрофотометрический анализ. Наиболее совершенным и сложным фотометрическим прибором является спектрофотометр. Ослабление интенсивности светового потока в спектрофотометре измеряется с помощью фотоэлементов. Однако в отличие от фотоэлектроколориметров спектрофотометры дают возможность применять строго монохроматический свет для проведения фотометрических измерений. Достигается это с помощью специальной призмы, которая разлагает «белый» свет в спектр, и щелевого устройства. Все это позволяет выделить очень узкий участок спектра с определенной длиной волны. Измерение светопоглоще-ния в узком участке спектра дает более строгую пропорциональность между концентрацией исследуемого окрашенного соединения и численным отклонением показания прибора. Рассмотрим это положение на конкретном примере.
Допустим, что исследуемое вещество имеет спектр поглощения, представленный на рис. 130, а. Максимума поглощение достигает при длине волны X = 550 нм, а минимума — при \ = 640 нм. Спектры поглощения построены для трех различных концентраций, причем Ci < С2 < С3. На рис. 130, б показана зависимость оптической плотности при Хмакс 11 *мин от концентрации раствора определяемого вещества. Нз него видно, что при изменении концентрации в интервале от Q до С2 (ДС) соответствующее ему изменение оптической плотности Д£) бу-
4
339
дет значительно больше при Хмакс, чем при Хмин. Поскольку практическая ошибка измерения оптической плотности раствора приблизительно одинакова во всем диапазоне измерений, при большем изменении AD (ошибка измерения) соответствующее ему изменение концентрации раствора АС (ошибка определения) будет гораздо больше при Хмин, чем при Хмакс. Иными словами, точность опреде ления будет тем выше, чем ближе длина волны поглощаемого света к Хмакс.
Рис. 130. Сравнение точности измерения оптической плотности раствора и фотометрического определения при разных длинах волн поглощаемого света:
а — спектры поглощения растворов; б — зависимость D от С при Хмакс И Хмин
К совершенно аналогичному выводу можно прийти при анализе математического выражения закона Бугера—Ламберта—Бера (VIII, 10):
Cl.
Продифференцировав зто выражение по С, получим
dD • AD
т. е. изменение оптической плотности раствора AD с изменением концентрации АС тем больше, чем больше молярный коэффициент погашения е^. Этот коэффициент всегда является наибольшей величиной при Хмакс.
Преимущества применения монохроматического света при фотометрических измерениях заключаются и в том, что в ряде случаев можно проводить измерения в присутствии посторонних веществ, поглощающих.свет в близких к максимуму поглощения определяемого вещества областях спектра. При этом влияние посторонних веществ уменьшается или вовсе устраняется. Таким образом, применение монохроматического света увеличивает специфичность фотометрических методов анализа. В этом собственно и заключается главное отличие фотоколориметрии от спектрофотометрии. В спектрофотометре есть возможность измерять оптическую плотность исследуемого раствора при любой длине волны поглощаемого света, что невозможно при работе с фотоэлектроколориметром.
В настоящее время промышленность СССР выпускает целый ряд спектрофотометрических приборов: СФ-4, СФ-4А, СФД-2 и др., а также саморегистрирующие спектрофотометры: СФ-10, СФ-8, СФ-9.
На рис. 131 показан общий вид и принципиальная оптическая схема широко распространенного спектрофотометра СФ-4А. рабочий диапазон этого прибора находится в пределах длин волн 220—1100 нм. Более сложный по устройству спектрофотометр СФ-9 дает возможность автоматически регистрировать спектр пропускания и излучения в области 186—2500 нм. Внешний вид прибора показан на рис. 132.
340 —
15 17 15 74 12 1
Рис. 131. Спектрофотометр СФ-4А:
а — общий вид: 1 — осветитель; 2 — рукоятка передвижения фотоэлементов; 3 — кюветное отделение; 4 — рукоятка каретки с кюветами; 5 — переключатель чувствительности; 6 — потенциометр чувствительности; 7 — рукоятка установки длин волн; 3 —переключатель пределов измерения; 9 и 10 — рукоятки для компенсации темнового тока; 11 — рукоятка шторки фотоэлемента; 12 — рукоятка для изменения ширины щели; 13 — рукоятка установки светофильтра; 14 — шкала ширины щели; 15 — шкала длин волн; 16 — шкала темнового Тока; 17 — шкала отсчетного потенциометра; 18 — осушительный патрон; 19 — отделение для установки фотоэлементов; б — оптическая схема: / — осветитель; 2— вогнутое зеркало; 3 — плоское зеркало; 4 — входная щель; 5 — защитная кварцевая пластинка; 6 — сферическое зеркало; 7 — диспергирующая призма; 8 — выходная щель; 9 — линза; 10 — светофильтр; 11 — кювета;
12 — защитная пластинка; 13 — фотоэлемент
Все фотометрические приборы (колориметры, фотоэлектроколориметры и спектрофотометры) широко применяются в агрохимическом анализе. Определение азота, фосфора, железа, марганца, меди и других элементов в почвах и растениях, изучение качественного и количественного состава органического вещества почвы, определение красящих и дубильных веществ в плодах, винах и виноматериалах — таков далеко не полный перечень вопросов, которые можно решать с помощью методов фотометрии.
§ 101. Методы анализа по фотометрии пламени
Метод «фотометрии пламени» или пламенная фотометрия за последнее время получил широкое распространение в агрохимическом анализе. Этот метод позволяет быстро определять элементы с точностью от 2 до 4%, а иногда до 0,5—1%, что вполне достаточно для практических целей. Известно, что в пламени сначала
происходит поглощение энергии атомами элемента в связи с переходом некоторых
Рис. 133. Внешний вид портативного пламенного фотометра ППФ-УНИИЗ
Электронов на более удаленные от ядра орбиты. Затем совершается обратный процесс — переход электронов на более близкие к ядру орбиты, идущий с выделением энергии в виде лучей с определенной длиной волны.
Принцип метода пламенной фотометрии чрезвычайно прост: исследуемый раствор в виде аэрозоля (мелких брызг) вводят посредством специального распылителя в пламя горелки, работающей на каком-нибудь горючем газе. Возникающее в пламени излучение определяемого элемента отделяется при помощи светофильтра или монохроматора от излучения других элементов и, попадая на фотоэлемент, вызывает фототок, который измеряется специальным высокочувствительным
гальванометром.
Вследствие невысокой температуры пламени, используемого в качестве источника возбуждения, возникающий спектр излучения несложен и содержит лишь легко возбудимые элементы. Простота спектров дает возможность применять для выделения искомой спектральной линии светофильтры или монохроматоры с малой дисперсией. Благодаря быстроте и простоте анализа, несложности аппаратуры и достаточно высокой точности метод пламенной фотометрии успешно применяется для определения щелочных и щелочноземельных металлов, которые очень трудно определять химическими методами.
Работа на пламенном фотометре сводится к следующему. По серии заранее приготовленных стандартных растворов, содержащих определяемый элемент, строят калибровочную прямую, откладывая по одной оси концентрации элемента, а по другой—показания гальванометра. Далее снимают показания гальваномет-
ра при введении в пламя горелки исследуемых растворов и по графику определяют концентрацию. Метод отличается высокой производительностью. За рабочий день можно проанализировать не одну сотню проб. Неслучайно он нашел широкое применение в агрохимии для определения обменных оснований в почвах и для анализа удобрений и растительных материалов.
На рис. 133 показан общий вид портативного пламенного фотометра марки ППФ-УНИИЗ, который предназначен для определения калия, натрия и кальция в почвенных вытяжках и других растворах.
— 342 —
§ 102. Оптические методы анализа
К оптическим методам анализа относятся рефрактометрия и поляриметрия. Эти методы получили широкое распространение в агрохимическом анализе.
Рефрактометрия. Рефрактометрические методы анализа основаны на определении коэффициента преломления исследуемого вещества. Впервые рефрактометрия была применена для исследования состава жидкостей еще в середине XVIII в. М. В. Ломоносовым.
При прохождении луча света из среды с одной плотностью в среду с другой плотностью меняется его направление — он испытывает преломление или рефракцию. Это явление вызвано тем, что скорость света зависит от оптической плотности среды: чем плотнее среда, тем скорость меньше. С изменением же скорости света на границе раздела двух сред с различной плотностью связано изменение направления падающего луча.
Рис. 134. Преломление света и полное внутреннее отражение
Отношение синуса угла падения луча к синусу угла его преломления называется коэффициентом или показателем преломления (рис. 134, а):
sin а sin 0
(VIII,14)
где а — угол падения, а 0 — угол преломления.
Угол падения и угол преломления — это углы, образованные лучом и перпендикулярами, восставленными из точки падения луча на поверхность раздела двух сред.
Если луч света переходит из более плотной среды в среду с меньшей плотностью (например, из стекла в жидкость или воздух), наступает момент, когда угол преломления становится равным 90°, и луч, не попадая в другую среду, будет скользить по поверхности раздела. Лучи с еще большим углом падения отражаются от поверхности. Угол падения, при котором угол преломления достигает 90° и луч скользит по поверхности раздела двух сред, называется углом полного внутреннего отражения (рис. 134, б).
Из уравнения (VIII, 14) при условии sin 0 = 1 получаем:
ci и Г
n = —р—= sina', (VIII ,15)
где а' —угол падения, при котором наблюдается полное внутреннее отражение. Следовательно, коэффициент преломления равен синусу угла полного внутреннего отражения. Этот способ определения показателя преломления вещества и ле
— 343 —
жит в основе многих конструкций рефрактометров. Следует отметить, что для особо точных измерений коэффициента преломления используется другой — интерферометрический принцип определения, который здесь не рассматривается.
Коэффициент преломления вещества зависит от ряда факторов и прежде всего от длины волны падающего света. Поэтому для коэффициентов преломления всегда указывают, какой длине волны они соответствуют. Обычно коэффициент преломления определяется для следующих длин волн: желтая линия натрия (линия D) — X = 5893 А; красная линия водорода (линия С) — к — 6562, 8 А; синяя линия водорода (линия F) — Х = 4861,3 А; фиолетовая линия водорода (линия G) — % = 4340,5 А. Поэтому при особо точных измерениях в рефрактометрах пользуются монохроматическим светом с указанными длинами волн.
Коэффициент преломления зависит и от температуры окружающей среды, поэтому при рефрактометрических измерениях постоянство температуры играет большую роль. Для этой цели наиболее сложные рефрактометры оснащаются специальными термостатическими устройствами.
Рис. 135. Оптическая схема рефрактометра:
1 — осветительная призма; 2 — измерительная призма; 3 — компенсатор; 4 — зазор, куда помещается несколько капель исследуемой жидкости
Принципиальная оптическая схема обычного рефрактометра изображена на рис. 135. Луч А при отражении от зеркала попадает через узкое окно на осветительную призму, в ней А преломляется и проходит через слой исследуемой жидкости. Далее луч попадает в измерительную призму, снова преломляется и попадает в зрительную трубу. Так проходят все лучи, угол падения которых на поверхности жидкости меньше предельного угла. Эти лучи освещают часть выходного окна, видимого в зрительной трубе. Лучи, падающие на поверхность исследуемой жидкости под углом, равным предельному или даже больше его (лучи В и С на схеме), в зрительную трубу не попадут, в результате чего часть выходного окна измерительной призмы не будет освещена, и в трубе мы увидим границу светотени. '
Вращением призмы относительно трубы добиваются совмещения светотени с крестом нитей окуляра зрительной трубы, а связанный с призмами указатель показывает по шкале коэффициент преломления исследуемого раствора или же концентрацию растворенного вещества (например, сахарозы).
Компенсатор 3, состоящий из двух призм (из которых одна закреплена неподвижно, а вторая может передвигаться относительно первой), предназначен для компенсации дисперсии света и для создания резкой границы темнового поля в окуляре.
Рефрактометры довольно широко применяются в агрохимических лабораториях, в основном для определения концентрации сахара в растворах. Наша промышленность выпускает специальные рефрактометры-сахариметры. На рис. 136 показан общий вид наиболее распространенного рефрактометра-сахариметра марки РПЛ (рефрактометр прецизионный лабораторный), который выпускается Киевским заводом контрольно-измерительных приборов.
Отличие сахариметра от обычного рефрактометра состоит в том, что в сахариметре измерительная призма неподвижна, а граница светотени получается на шкале показателей преломления с помощью специального зеркальца, вращением которого она может перемещаться по шкале. Шкала проградуирована в весовых
— 344 —
процентах сахарозы. При измерениях перекрестие нитей окуляра совмещают с границей светотени и делают отсчет по шкале. Промышленность выпускает также портативные сахариметры (например, марки РП), которые предназначены для определения сахара в плодах непосредственно в полевых условиях. При помощи п штативного ручного пресса получают сок исследуемого плода и в нем определяют содержание сахара.
Лабораторные сахариметры широко используются также для определения свободной и связанной воды в растениях по изменению концентрации стандартного раствора сахарозы после взаимодействия его с исследуемым объектом.
Поляриметрия. Этот метод определения концентрации оптически активных веществ основан на измерении угла вращения поляризованного света. Как следует из электромагнитной теории света, колебания световых волн происходят
1 Плоскость колебаний
Плоскость поляризации.
I
Рис. 137. Естественный и поляризованные лучи
Рис. 136. Рефрактометр-сахариметр РПЛ: / — камера с измерительной призмой; 2 — камера с осветительной призмой; 3 — компенсатор; 4— отсчетный барабан; 5 — окуляр с диафрагмой; 6 —зажим; 7— колонка с шарниром; 8—массивная подставка
в плоскости, перпендикулярной к направлению луча. У обычного естественного луча эти колебания происходят во всех плоскостях, которые перпендикулярны к направлению луча (рис. 137).
Кристаллические решетки некоторых кристаллов обладают способностью пропускать через себя свет только с определенным направлением колебаний. Свет, выходя из такого кристалла, имеет колебания только в одной плоскости. Такой свет называется поляризованным. Плоскость, в которой происходят колебания его, называется плоскостью колебаний поляризованного света, а перпендикулярная к ней плоскость — плоскостью поляризации (рис. 137).
Все вещества в кристаллическом состоянии, а также растворы различных веществ могут быть разделены на две категории, в зависимости от их отношения к поляризованному свету.
Вещества, способные изменять (вращать) плоскость поляризации света, называются оптически активными; вещества, не способные вращать плоскость поляризации, — оптически неактивными. Оптическая активность, как известно, может быть обусловлена в основном двумя причинами: 1) особенностями строения кристаллической решетки вещества (SiO2, NaC103) или 2) особенностями строения молекулы вещества; оптическая активность этих веществ обусловлена наличием в молекуле асимметричных атомов углерода, серы, азота и других элементов. Так, глюкоза C6H12Of{ имеет две различные оптически активные формы: а-
— 345 —
f-глюкоза
глюкозу и f-глюкозу, которые отличаются расположением атомов в молекуле: сс-глюкоза
.//°
/
и— С—ОН
НО—С—н
он
он
Этот тип оптически активных веществ проявляет свою активность только в растворенном или расплавленном состоянии. Если через слой оптически активного вещества проходит поляризованный луч, плоскость поляризации луча оказывается повернутой на некоторый угол, который называется углом поворота плоскости поляризации.
Рис. 138. Схема поляриметрического исследования
Поляриметрические измерения проводят при помощи двух специальных призм — николей, изготовленных из прозрачной разновидности исландского шпата СаСО3 специальным способом. Один из николей играет роль поляризатора, другой — анализатора. Оба николя обладают свойством пропускать через себя лучи света, колебания которых лежат строго в одной плоскости (поляризованный луч). - I
На рис. 138 показана схема поляриметрического исследования. Если поляризатор и анализатор установлены параллельно (рис. 138, п), то свет пройдет через оба николя. Если анализатор повернуть на 90°, лучи света не пройдут через анализатор (рис. 138, б), потому что плоскости колебания лучей, которые пропускают в данном случае оба николя, взаимно перпендикулярны. Света за анализатором не будет: николи, как говорят, установлены на темноту. Если между Николями поместить раствор оптически активного вещества, в анализаторе вновь появится свет (рис. 138, в).
- 346 —
Рис 139. Поляриметр-глюкозиметр ПГ: I — поляризатор, состоящий из двух призм николя; 2 — анализатор; 3 — камера для помещения исследуемого раствора; 4 — траверза, на которой смонтированы основные узлы поляриметра; 5 — колонка; 6 — массивное основание; 7 —• диск; 8 — гильза, снимающаяся при установке поля зрения на одноцветность; окулятор для отсчета показания шкалы; 10 — нониус, служащий для установки шкалы на нуль; 11 — кремальерная подача для передвижения клина и шкалы; 12 — экран для установки источника освещения
Это объясняется тем, что вышедший из раствора луч света имеет колебания же в какой-то другой плоскости MN и может быть разложен по правилу параллелограмма на два луча (рис. 138, г). Один из этих лучей будет иметь колебания в плоскости анализатора и потому может пройти через него. Чтобы вновь установить николи на темноту, необходимо анализатор повернуть на некоторый угол 6 так, чтобы его плоскость стала перпендикулярной плоскости колебаний луча, т. е. к плоскости MN, Угол Р называется углом поворота плоскости поляризации. Он зависит от толщины слоя оптически активного вещества, его концентрации в растворе и индивидуальных свойств.
Одни оптически активные вещества вращают плоскость поляризации по часовой стрелке (правое, плюсовое вращение), другие — против часовой (левое, минусовое вращение). Угол вращения плоскости поляризации р связан с концентрацией оптически активного вещества в растворе и толщиной слоя этого раствора довольно простым соотношением:
р = ±[сс] CZ, (VIII, 16) где С — концентрация, г! мл, I — толщина слоя, дм.
Величина [а], называемая удельным вращением, — это угол вращения плоскости поляризации раствором, содержащим 1 а/мл оптически активного вещества при толщине слоя I = 1 дм. Знаки плюс и минус отвечают правому и левому вращению.
Удельное вращение плоскости поляризации зависит от природы вещества, длины волны поляризованного света и температуры. Поэтому все исследования вращения плоскости поляризации должны проводиться при определенных зна
чениях длины волны и температуры. Обычно вращение относится к температуре 20° С и желтой линии натрия [ос]о°.
На основании рассмотренного выше соотношения (VII 1,16) можно определить концентрацию оптически активного вещества в растворе. Для этого необходимо измерить угол вращения плоскости поляризации с известной толщиной слоя раствора и удельным вращением. Измерение углов вращения плоскости поляризации производят в специальных приборах, называемых поляриметрами.
Поляриметр простейшего типа состоит из поляризатора, трубки, в которой находится исследуемый раствор, и анализатора. При работе с поляризатором этого типа анализатор устанавливают «на темноту», а затем вводят трубку с раствором. При этом поле светлеет за счет вращения плоскости поляризации оптически активным раствором. Поворотом анализатора можно добиться нового потемнения поля, при этом угол поворота анализатора соответствует углу пращения плоскости поляризации раствора, его отсчитывают непосредственно по шкале.
На несколько другом принципе основано устройство так называемых полу-тенсвых поляриметров, в которых угол вращения определяется не по наибольшей освещенности или затененности оптического поля, а по достижению равномерной слабой освещенности — полутени.
~ 347 —
На рис. 139 показан общий вид полутеневого поляриметра — глюкозиметра марки ПГ, который в настоящее время выпускается Киевским заводом контрольно-измерительных приборов.
Поляриметрический анализ широко распространен в агрохимическом анализе при определении концентрации оптически активных веществ. Особенно большое значение он имеет в сахарной промышленности, где применяется для определения содержания сахаристых веществ, а также в масляной промышленности, где используется совместно с рефрактометрическим методом для идентификации масел.
§ 103. Хроматографические методы анализа
Хроматографические методы анализа за последнее время нашли самое широкое применение в решении важнейших вопросов не только сельского хозяйства, но и химии, биологии, атомной техники, медицины, а также в нефтеперерабатывающей, нефтехимической, химической и газовой промышленности. Эти методы успешно применяются для автоматизации различных технологических процессов.
Обладая высокой чувствительностью, методы хроматографии дают возможность разделять и выделять в чистом виде различные вещества из сложных смесей близких по своей природе химических соединений. Именно благодаря хроматографическому анализу появилась возможность разделения столь близких* по свойствам соединений, как стереоизомеры, а также разнообразные вещества, входящие в состав растений, — аминокислоты, органические кислоты, различные углеводы, ферменты, пептиды, неорганические вещества и многие другие. Попутно отметим, что разделение веществ с применением физических и химических методов является сложным и трудоемким процессом, который требует значительно больше времени.
Наши знания об антибиотиках, витаминах, алкалоидах, а также о динамике обмена веществ в растениях почти целиком получены методами хроматографии. Для хроматографического анализа требуются незначительные количества исследуемого вещества — десятые доли миллиграмма, а иногда микрограммы и даже доли микрограмма.
Впервые хроматографический метод анализа был открыт русским ботаником М. С. Цветом в 1903 г. Он использовал для целей разделения и очистки веществ их неодинаковую адсорбируемость на разных адсорбентах в присутствии различных жидких фаз. Характеризуя основной принцип своего метода, М. С. Цвет писал: «При фильтрации смешанного раствора через столб адсорбента пигменты... расслаиваются в виде отдельных, различно окрашенных зон. Подобно световым лучам в спектре, различные компоненты сложного пигмента закономерно распределяются друг за другом в столбе адсорбента и становятся доступными количественному определению. Такой расцветочный препарат я назвал хроматограммой, а соответствующий метод анализа — хроматографическим методом».
Существуют четыре вида хроматографического анализа: адсорбционная хроматография, осадочная хроматография, распределительная хроматография и газо-жидкостная хроматография. В зависимости от механизма адсорбции растворенного вещества адсорбционная хроматография может быть разделена на два подвида: молекулярная хроматография и ионообменная хроматография. С помощью молекулярной хроматографии разделяют неэлектролиты в неводных растворах. Ионообменная хроматография используется для разделения ионов.
Молекулярная адсорбционная хроматография. Молекулярная адсорбция основана на том, что поверхность различных адсорбентов обладает определенным количеством свободной потенциальной энергии, мерой которой является энергия единицы поверхности, называемая капиллярной постоянной. Поскольку согласно второму закону термодинамики процессы протекают в сторону уменьшения свободной энергии, поверхностная потенциальная энергия всегда стремится к минимальным значениям за счет накопления на поверхности адсорбента веществ с меньшей капиллярной постоянной, т. е. за счет адсорбции.
— 348 —
Адсорбент, т. е. материал, субстрат, на котором адсорбируются (накапливаются) другие вещества, должен обладать как можно большей поверхностью и большей, чему этих веществ, капиллярной постоянной. Чем меньше капиллярная постоянная адсорбируемого вещества, тем оно более способно снизить уровень потенциальной энергии тел с большей капиллярной постоянной (в данном случае адсорбента) и вступить с ним в более прочную связь.
В качестве адсорбентов применяют обычно тонко измельченные карбонат кальция, крахмал, окись алюминия, сахар, инулин и другие вещества. Адсорбентом равномерно заполняют стеклянную трубку (колонку) и через нее фильтруют раствор, содержащий несколько веществ, подлежащих разделению. Поскольку разделяемые вещества обладают различной капиллярной активностью по отношению к адсорбенту, они адсорбируются в колонке в порядке убывания адсорбционной активности, т. е. в верхней части колонки накапливается наиболее капиллярно активное вещество, в нижней — наименее капиллярно активное.
При промывании колонки каким-либо растворителем адсорбированные вещества передвигаются вниз по колонке с определенной скоростью, образуя отдельные зоны, каждая из которых содержит только одну составную часть анализируемой смеси-. Фильтрат можно собирать раздельно или, разделив колонку на части, из каждой выделять адсорбированное вещество. С этой целью хроматографические колонки иногда изготовляют разборными, состоящими из ряда отдельных хорошо пришлифованных друг к другу колец.
В качестве примера на рис. 140 изображена хроматограмма хлорофилла, которая впервые была получена Цветом в результате пропускания через хроматографическую колонку с адсорбентом петролейно-эфирного экстракта исвежих тканей зеленых растений и промывания колонки бензолом. Описание этой хромо-тограммы приведено в табл. 69. Помимо веществ, указанных в таблице, сквозь адсорбент проходил, не поглощаясь, желтый (в петролейном эфире) каротин.
Таблица'69
Описание хроматограммы по Цвету
Зона
Окраска
Название пигментов
1
п HI IV
V
VI VII (VIII)
Бесцветная
Желтая Желто-зеленая Зелено-синяя Желтая
ъ
»
(Стальная серая)
Коллоидальные спутники хлорофилла
Ксантофилл (3
Хлорофиллин (3 Хлорофиллин а Ксантофилл а"-Ксантофилл а' Ксантофилл а (Хлорофиллан)
М. С. Цвет, применив свой метод, впервые показал, что зеленый пигмент листьев состоит из четырех главных компонентов, которые теперь различают как хлорофиллы а и Ь, ксантофилл и каротин.
Итак, разделение веществ методом молекулярно-адсорбционной хроматографии основано на различной адсорбируемости разделяемых веществ и различной скорости передвижения отдельных зон при промывании хроматографической колонки неводным растворителем.
Ионообменная хроматография. Ионообменная адсорбционная хроматография в отличие от молекулярной протекает под действием сил электровалентном (химической) связи. Она основана на обратимом стехиометрическом обмене содержащихся в хроматографируемом растворе ионов на подвижные ионы адсорбента, который носит название ионита или ионообменника. Если в молекулярной хроматографии вымывание адсорбированных веществ производят чистым раствори
— 349 —
телем, то в ионообменной в качестве вымывающего вещества применяют растворы электролитов.
По знаку заряда обменивающихся ионов иониты подразделяются на катиониты, или катионообменники, и аниониты, или ионообменники. Амфотерные иониты, которые способны к обмену как катионов, так и анионов, называются амфолитами.
Среди ионитов наибольшее практическое применение получили синтетические иониты, получаемые на основе синтетических смол, которые обладают большой адсорбирующей способностью. Ионообменные смолы являются типичными гелями. Их каркас состоит из полимерной углеводородной сетки, называемой матрицей, в которой фиксированы группы, несущие заряд. У катионитов это чаще всего группы—SO^, —СОО“, —РО|~, —AsO^; у анионитов группы— NH3, = = NH2, = N+, sS+. На рис. 141 схематически показана структура синтетической
Рис. 140. Хро-мато грамм а хлорофилла
У/Матрица с фикси-® - Противоионы рытыми ионами Q_KouoHbi
Рис. 141. Схематическое изображение структуры синтетической ионообменной смолы
ионообменной смолы. Заряд каркаса компенсируется зарядом ионов противоположного знака, так что в целом ионит электронейтрален. Эти компенсирующие ионы называются противоионами.
В порах ионита содержатся не только противоионы, но и молекулы растворителя и другие растворенные вещества, в числе которых могут быть и ионы того же знака, что и заряд каркаса. Такие ионы носят название коионов. Таким образом, содержание противоионов в ионите определяется двумя факторами — величиной заряда каркаса и содержанием коионов.
При соприкосновении ионита, содержащего противоионы одного вида, с раствором, в котором находятся ионы другого вида, происходит ионообменный процесс, который можно представить в виде следующих реакций:
катионный обмен
RAn -Н+ + Na+ 4- Cl - ;z± RAn “Na+ 4- Н+ 4- Cl -анионный обмен
RKt+ ОН ~ 4* Na+ 4- Cl - ^z± RKt+Cl “ 4- Na+ 4- ОН ~
Здесь R — полимерный радикал, образующий вместе с ионогенной группой кар-I ас ионита, а Ап~ и Kt+ — ионогенная группа или фиксированный ион, обусловливающий заряд каркаса.
Поскольку свойства ионитов в значительной степени зависят от природы находящихся в них противоионов, при характеристике ионита всегда указывают, какой именно ион является противоионом. Так, если говорят, что катионит находится в водородной форме (Н-форме), значит противоионами этого катионита
350 —
1яются ионы Н+; аналогично, если говорят, что данный анионит находится хлоридной форме (Cl-форме), это значит, что противоионами в этом анионите являются хлорид-ионы.
Сама матрица ионита гидрофобна, но фиксированные ионы гидрофильны, поэтому матрица обладает способностью к набуханию, и смола превращается в полиэлектролит. Синтетические ионообменные смолы являются гелями полиэлектролитов, способными к набуханию. Однако набухаемость их ограничена в силу того, что в полимерной молекуле каркаса имеются поперечные связи.
Свойства синтетических ионитов целиком зависят от числа и типа фиксированных ионов, а также от строения матрицы, точнее от количества поперечных связей в ней. Важнейшим условием успешного разделения веществ при помощи ионообменной хроматографии является правильный выбор ионообменника, его подготовка, а также определение условий проведения анализа, особенно размеров колонки.
В качестве колонок применяют стеклянные трубки с краном внизу. Колонку заполняют ионитом и обычно промывают сверху, хотя есть колонки и с восходящим током жидкости. На рис. 142 и 143 показаны наиболее простые цилиндрические хроматографические колонки, которые широко применяются в лабораторной практике.
Наша промышленность выпускает большой ассортимент ионообменных смол, которые полностью отвечают всем требованиям практики.
Ионообменная хроматография широко применяется в агрохимическом анализе. С ее помощью в лабораторных условиях получают дистиллированную воду высокой степени чистоты, проводят разделение аминокислот, сахаров, карбоновых кислот: ее также используют для изучения явлений поглощения в почвах, процессов питания растений и запасов питательных веществ, находящихся в усвояемом для сельскохозяйственных культур состоянии в почве.
За последнее время в агрохимии нашел большое применение ионитный метод извлечения из почвы усвояемой фосфорной кислоты и калия, а также других питательных элементов.
Осадочная хроматография. Метод осадочной хроматографии также служит для разделения ионов. Он разработан в 1948 г. Е. Н. Талоном.
В осадочной хроматографии разделение веществ осуществляется в результате образования фильтрации исследуемого раствора через колонку и образования и растворения при этом осадков. Этот процесс повторяется многократно, т. е. является динамическим процессом. В результате многократного повторения элементарных актов образования и закрепления осадка в определенном месте колонки, а также его растворения при различной растворимости получающихся осадков и происходит разделение смеси веществ методом осадочной хроматографии.
Адсорбент (например, безводная окись алюминия, карбонат кальция) тщательно смешивается с веществом-осадителем, с которым ионы, подлежащие разделению, дают трудно растворимые осадки. Затем этим составом набивают хроматографическую колонку и пропускают исследуемый раствор. По мере продвижения раствора вдоль колонки образуются зоны, содержащие разделяемые ионы в виде соответствующих осадков.
Метод осадочной хроматографии используется для аналитического разделения катионов и анионов.
Распределительная хроматография. Распределительная хроматография основана на различном распределении разделяемых веществ между двумя несмеши-вающимися жидкостями.
Распределительная хроматография делится на колоночную и бумажную. В колоночной хроматографии твердый носитель (пористое вещество) прочно удерживает на своей поверхности одну из жидкостей, которая служит неподвижным растворителем. Вторая жидкость, не смешивающаяся с первой, служит подвижным растворителем. Она пропускается через колонку с небольшой скоростью. В бумажной хроматографии в качестве носителя применяется бумага, которая обладает способностью удерживать в своих порах значительные количества жидкости, играющей роль неподвижного растворителя.
Поскольку сущность распределительной хроматографии заключается в использовании различия в коэффициентах распределения компонентов разделяемой смеси между двумя несмешивающимися жидкостями, этот вид хроматогра-
— 351 —
фии еще называют жидкостно-жидкостной распределительной хроматографией.
Из указанных выше двух вариантов жидкостно-жидкостной распределительной хроматографии наибольшее распространение получила бумажная, поэтому рассмотрим ее несколько подробнее. Носителем для неподвижного растворителя (например, воды) в бумажной хроматографии является фильтровальная бумага специального изготовления. Подвижной фазой служит какой-то органический растворитель. Последний, протекая через участок бумаги, содержащий исследуе
Рис. 142. Цилиндрические хроматографические колонки:
а — с нисходящим током жидкости; б — с восходящим током жидкости; / — напорный сосуд; 2 — стеклянная вата; 3 — сорбент; 4 — патрубок для выхода жидкости
Рис. 143. Хроматографические колонки, работающие при повышенном (д) и пониженном (б) давлении:
1 — 01 баллона с сжатым газом, 2— к вакуумному насосу
а
мое вещество, частично его растворяет и уносит с собой. Достигнув участка бумаги, не содержащего данного вещества, растворитель отдает определенную ею долю водной фазе. Следующие порции подвижной жидкости (растворителя) должны растворять остаток исследуемого вещества, переносить его еще дальше и тоже передавать водной фазе. Переходя то к органическому подвижному растворителю, то к водной фазе, исследуемое вещество перемешается вместе с потоком жидкости.
Движение зон компонентов разделяемой смеси может быть количественно охарактеризовано величиной которая называется подвижностью. Эта величина равна отношению скорости зоны исследуемого вещества к скорости движения
— 352 —
фронта растворителя. Так как от скоростей зависят и пути, пройденные компонентами разделяемой смеси от своего начального положения (х) и фронта растворителя (ху), то
(VIII, 17)
где к — путь, пройденный тем или иным компонентом разделяемой смеси от своего начального положения, х/ — путь, пройденный фронтом растворителя от TOiO же положения. На рис» 144 дано пояснение этого соотношения. Величина легко может быть определена на основании экспериментальных данных, так как ж и х/ доступны непосредственному измерению на хроматограмме.
Фронт растворителя
Начальная линия
Рис. 144. Схема определения Rf компонентов смеси по результатам хроматографии на бумаге:
Xi, х2, Xf — путь, пройденный соответственно первым и вторым компонентами смеси и растворителем
Рис. 145. Камера для получения нисходящей хроматограммы:
1 — цилиндр; 2 — корковая пробка; 3 — ванночка с подвижным растворителем; 4 — Г-образная стеклянная полочка; 5 —• полоска бумаги; 6 -*• груз; 7 — бюкс с неподвижным растворителем
Техника бумажной хроматографии не сложна. Хроматографирование обычно проводят в герметически закрытых сосудах или камерах, где поддерживается насыщенная парами растворителя атмосфера. Это предотвращает испарение растворителя с листов фильтровальной бумаги. Капля исследуемого раствора наносится на некотором расстоянии от края бумажной полосы. Подвижный растворитель наливают в ванночку, куда опускают конец бумажной полосы. Бумагу закрепляют так, чтобы она свободно провисала. Сосуд, в котором происходит хроматографирование, плотно закрывают и помещают в термостат на все время опыта. Колебания температуры на протяжении всего опыта не должны превышать ±1,5° С.
Под действием капиллярных сил подвижная жидкость передвигается по бумаге и разделяет составные части исследуемой смеси на отдельные зоны, которые при различии Rf разделяемых веществ передвигаются по слою бумаги с неодинаковыми скоростями. Опыт считают законченным, когда фронт подвижной жидкости достигнет противоположного края полосы бумаги. После этого бумагу вы
12 Зак. 560
— 353 —
нимают из сосуда, высушивают и проявляют опрыскиванием соответствующим раствором, дающим окрашенные соединения с компонентами разделяемой смеси. При одновременном хроматографировании ряда растворов берут более широкую полосу бумаги и вдоль ее края на так называемую стартовую линию наносят капли исследуемых растворов с интервалами в 2—3 см.
Бумажная хроматография обычно выполняется в двух вариантах: восходящим и нисходящим движением растворителя. На рис. 145 показан общий вид простейшей камеры для получения нисходящей хроматограммы.
Если при помощи одного растворителя разделить сложную смесь не удается, можно последовательно применять два растворителя, которые обладают различными коэффициентами распределения. Такую хроматограмму называют двумерной.
Рис. 146. Схема получения двумерной хроматограммы (А— место нанесения исследуемого раствора):
а — хрсматограмма, подученная после хроматографирования первым растворителем; смесь, состоящая из шести компонентов, не разделилась; б — та же хроматограмма перед опусканием бумаги во второй растворитель; в — хроматограмма, полученная после хроматографирования во втором растворителе и проявления; смесь разделилась. Стрелкой указано направление движения фронта подвижной фазы
Для получения двумерной хроматограммы применяют квадратные листы бумаги. Каплю исследуемого раствора вначале наносят на бумагу в ее левом углу (рис. 146, а), а затем пятно высушивают. Далее нижний край бумаги опускают в один из растворителей и хроматографируют по восходящему методу. Как только фронт растворителя достигнет верхнего края бумаги, хроматографирование прекращают, бумагу высушивают и поворачивают на 90° (рис. 146, б). В таком положении бумагу помещают в другой сосуд для хроматографирования, опускают ее нижний край во второй растворитель и хроматографируют по восходящему методу, как и в первом случае. После высушивания хроматограммы ее проявляют заранее выбранным проявителем (рис. 146, в).
Иногда бумажную хроматографию объединяют с электрофорезом: на разделяемую смесь одновременно или последовательно с обычной хроматографией воздействуют электрическим полем постоянного тока. Такой вид хроматографии получил название электрофоретической хроматографии. Он более сложен, широко применяется для разделения различных белков и им подобных соединений.
Помимо качественного обнаружения разделяемых веществ на хроматограммах, метод бумажной хроматографии может быть использован и для количественных определений. Существуют две группы методов количественного бумажного хроматографического анализа: методы, основанные на вымывании анализируемых веществ, и методы, не требующие удаления анализируемых веществ.
Сущность первой группы методов заключается в том, что пятна анализируемых веществ вырезают из хроматограммы и экстрагируют вещество из бумаги, а затем уже в экстракте определяют концентрацию его любым из доступных способов. Во второй группе методов концентрацию разделяемого вещества определяют по интенсивности окраски пятна. Это измерение проводят с помощью специальных фотометрических приборов — денситометров. Концентрация вещества в пятне (и в исследуемом растворе) и интенсивность окраски пятна связаны между собой линейно.
— 354 —
но недавно (1952), но за последнее время уже получил довольно широкое НИс пр остр а нение в биологии, химии, а также во многих важных отраслях народ-ного хозяйства.
носитель
Рис. 147. Схема хроматографа:
1 *— дозатор; 2 — детектор; 3 — приемник;
4 — регистратор
разделении смеси отношение носит коэффициента распре-
газо-жидкостной заключается в
пы бумажной хроматографии в настоящее время вошли в практику аг-Методь улабораторий. С ее помощью определяют аминокислотный состав рохимически объектов. Белки, сахара, карбоновые кислоты и другие важные биологичес живОтных вещества могут быть определены с помощью этих медля растеми
Т0Д°г зо-жидкостная хроматография. Этот вид хроматографии был открыт срав-1 а о I едавно (1952), но за последнее время уже получил довольно широкое цитель нение в биологии, химии, а также во многих важных отраслях народ-РЗСПРхозяй*ства. В газо-жидкостной хроматографии происходит распределение Н0Г° онентов исследуемой смеси между газообразной и жидкой фазами, из кото-К°МПпоследняя является неподвижной. Отношение концентрации анализируемо-г вещества в жидкой неподвижной фазе к его концентрации в газовой фазе играет первостепенную роль в веществ. Это название i деления.
Сущность хроматографии следующем. Проба исследуемого вещества сложного состава вместе с газом-носителем (аргон, водород, углекислый газ и др.) проходит через специальные колонки, наполненные инертным твердым сорбентом, на который предварительно нанесена жидкая фаза. Проходя через колонку, исследуемые вещества разделяются (хроматографируются) и на выходе появляются порознь в определенном порядке.
Прибор для проведения газовой хроматографии носит название хроматографа. Принципиальная схема хроматографа представлена на рис. 147. Устройство 1 — дозатор — служит для ввода в хроматографическую колонку газовой, жидкой или твердой пробы исследуемого вещества. Пробу можно ввести либо непосредственно в поток газа-носителя (например, шприцем), либо в ограниченный объем, из которого оно транспортируется газовым потоком'в колонку 2, где и происходит разделение.
Поскольку основным способом определения состава анализируемой смеси в газовой хроматографии является метод выходной кривой, после колонки устанавливают специальный детектор 3, который фиксирует изменение состава выходящей из колонки газовой смеси. Кривые, записанные на ленте специального регистратора 4, и есть хроматограммы. Наша промышленность в настоящее время серийно выпускает различные марки хроматографов, из которых наиболее распространенными являются ХПА-2, ХПА-4, ХТП-63, УХ-1 и многие другие.
С помощью газо-жидкостной хроматографии проводят качественный и количественный анализ практически любых органических соединений, в том числе и таких, которые являются жизненно важными для культурных растений.
* * *
Перечисленные физико-химические методы далеко не полностью исчерпывают все возможности физической химии. Многие методы, открытые буквально в последние годы, уже прочно входят в обиход биологических наук. Это в полной /алл °тносится к “ методам исследования, как ядерный магнитный резонанс (ЯМР), электронный парамагнитный резонанс (ЭПР), масс-спектроскопия и мно-тие другие, с помощью которых уже сейчас решаются сложные биологические проблемы. И нет сомнения, что овладев в должной мере физической химией и се методами исследования, можно успешно решать проблемы, которые ставит перед нашим сельским хозяйством Коммунистическая партия Советского Союза.
—J355J—
Часть вторая КОЛЛОИДНАЯ ХИМИЯ
ВВЕДЕНИЕ
Предмет коллоидной химии и ее значение для промышленности и сельского хозяйства. Коллоидная химия изучает физико-химические свойства гетерогенных высокодисперсных систем и высокомолекулярных соединений. Коллоидная химия —важный самостоятельный раздел физической химии, изучающий физико-химические свойства высокомолекулярных и высокополимерных соединений в твердом состоянии и в растворах. Коллоидная химия уделяет особое внимание роли поверхностных явлений на границе раздела фаз.'
В настоящее время коллоидная химия занимается уже не только химическим строением и химическими реакциями, протекающими в коллоидных системах, но и физической структурой, физическими (и даже механическими) свойствами и физико-химическими процессами, характерными для высокодисперсных и высокомолекулярных систем. Вот почему правильнее было бы называть этот раздел науки более общим термином — физическая химия дисперсных систем.
Из курса физической химии известно, что если одно вещество в более или мелко раздробленном (дисперсном) состоянии равномерно распределено в массе другого вещества, то систему называют дисперсной. Раздробленное вещество в этом случае называют дисперсной фазой, а среду, в которой оно распределено — дисперсионной средой. Так, система, представляющая собой взмученную в воде глину, состоит из взвешенных мелких частиц глины — дисперсной фазы и воды — дисперсионной среды.
Для характеристики и классификации различных дисперсных систем в практике широко пользуются понятием степень дисперсности D, которая определяется как величина, обратная величине размера (диаметра) дисперсной частицы а, если ее размер выразить в сантиметрах: D — 1/а см'1. Отсюда следует, что степень дисперсности есть величина, показывающая, какое число частиц можно уложить вплотную в 1 см.
Иногда применяется и другая характеристика степени дисперсности — так называемая удельная поверхность, которая представляет собой отношение поверхности S данного тела*к занимаемому объему V, т. е. 5уд = S/V, где 5УД — удельная поверхность. С повышением степени дисперсности величина удельной поверхности быстро растет.
— 356 —
Рее дисперсные системы по величине частиц дисперсной фазы о степени дисперсности можно разделить условно на три группы: " Пбодисперсные, коллоидно-дисперсные и молекулярно (ионно)-дисперсные (табл. 70).
Таблица 70
Классификация дисперсных систем по степени дисперсности
Группы D, см~Г а, см Характеристика дисперсных частиц
I Грубодисперсные 105 io-5 Не проходят через тонкие
(простые дисперсии, суспензии) (>100 ммк) бумажные фильтры; сравнительно быстро оседают (или всплывают); не диализируют и не диффундируют; видимы в обычный микроскоп
Н. Коллоидно-дис- 105—107 10-S—Ю-’ Проходят через самые тонкие
персные (дисперсоиды) (100—1 ммк) фильтры, но задерживаются в ультрафильтрах; заметно не оседают; не диализируют и очень слабо диффундируют; невидимы в обычный микроскоп, но обнаруживаются при помощи ультрамикроскопа
III. Молекулярно 107 10-’ Проходят через все фильтры;
(ионно)-дисперсные (дисперсиды) (<1 ммк) не оседают; хорошо диализируют и диффундируют; не обнаруживаются и в ультрамик-
' 1 роскопе
Дисперсные системы третьей группы, известные под общим названием истинных, или молекулярных растворов, всесторонне исследуются в физической химии. Эти системы являются наиболее изученными, так как сравнительно просты по составу и структуре (дискретными единицами в них являются либо простые молекулы, либо ионы), а поведение определяется простыми и четкими закономерностями. Молекулярно- и ионно-дисперсные системы могут образоваться самопроизвольно, они являются системами равновесными и термодинамически устойчивыми, подчиняющимися правилу фаз.
Вторая группа дисперсных систем, получивших название коллоидно-дисперсных, является основным объектом изучения коллоидной химии. Системы этой группы и получили название коллоидов, или коллоидных систем. Структурной и кинетической единицей в них являются не ион и не молекула в общем смысле, а либо комплекс (агрегат), состоящий из обычных молекул, атомов или ионов, называемых мицеллой, либо макромолекула, т. е. молекула-полимер «гигантских» размеров ~ 10~5 10~7 см, обладающая молекулярным или
частичным весом в десятки и сотни кислородных единиц. В качестве примера в табл. 71 приведены размеры молекул некоторых соединений, коллоидных частиц и некоторых клеток.
— 357 —
Таблица 71
Размеры частиц и живых микроскопических объектов
Объекты Размеры» ммк Объекты Размеры, мм к
Атом водорода Ион натрия Молекула спирта . . . Молекула гемоглобина . Молекула крахмала . . Частицы коллоидного золота 0,01 0,26 0,5 3,5 5,0 2—130 Вирусы ........ Хромосомы ...... Зерна крахмала .... Бактерии Эритроциты ...... 10—300 200—3500 7000 400—15 000 7500
С увеличением молекулярного веса в дисперсных системах второй группы можно ожидать новых качественных изменений, т. е. появления новых, более сложных свойств, которые не укладываются в закономерности более простых систем третьей группы. По мере изменения размеров частиц от наиболее крупных (грубодисперсных) к мелким и обратно соответственно изменяют кинетические, оптические, каталитические и другие свойства. Такие коллоидно-дисперсные системы занимают как бы промежуточное положение между грубыми и молекулярно-ионными системами. В табл. 72 приведены изменения некоторых свойств различных дисперсных систем.
• Таблица 72
Изменение свойств некоторых дисперсных систем
Грубые взвеси (суспензии) Коллоидные системы Молекулярно-ионные системы
Непрозрачные Не проходят через бумажный фильтр Не проходят через пергамент Гетерогенные Рассеивают свет в результате отражения и преломления Неустойчивые Со Бременем стареют Прозрачные Фильтруются Не проходят через пергамент Гетерогенные Дают конус Тиндаля Относительно устойчивы Со временем стареют Прозрачные Фильтруются Проходят через пергамент Гомогенные Оптически пусты 1 Устойчивы Не стареют
Из этой таблицы следует, что коллоидно-дисперсные системы в отличие от истинных растворов сами по себе агрегативно неустойчивы. Размеры их дисперсных частиц могут изменяться как самопроизвольно, так и под влиянием внешних факторов. Одной из причин неустойчивости коллоидных растворов является их гетерогенность. Обладая громадной суммарной поверхностью, следовательно, боль
— 358 —
шой свободной энергией, коллоидные системы в силу второго начала термодинамики стремятся к равновесному состоянию, характеризующемуся разделением системы на две фазы, имеющие минимальные межфазовые поверхности и минимальную свободную поверхностную энергию.
Агрегативная устойчивость коллоидно-дисперсных систем повышается, если на поверхности коллоидных частиц за счет свободной поверхностной энергии будут адсорбироваться молекулы (ионы) третьего компонента системы — стабилизатора. Так, если в пробирку с водой ввести небольшое количество растительного масла, при встряхивании образуется эмульсия, которая быстро расслаивается снова на два слоя — масло и воду. Неустойчивость эмульсии объясняется самопроизвольным уменьшением суммарной поверхности за счет слипания мелких капелек масла в более крупные. Однако если ввести в эту смесь небольшое количество 2%-ного раствора мыла и хорошо встряхнуть, образуется стойкая эмульсия белого цвета. Мыло в данном случае играет роль стабилизатора.
В отличие от коллоидно-дисперсных систем высокомолекулярные системы значительно более устойчивы: они дают при смешении с растворителями молекулярные растворы, подобные обычным растворам низкомолекулярных веществ, но с очень длинными цепными молекулами. Такие растворы являются гомогенными системами, они образуются самопроизвольно, потому что сам процесс растворения идет с уменьшением свободной энергии и не требует наличия стабилизатора. Растворы высокомолекулярных соединений являются термодинамически равновесными и потому обратимыми системами.
Гетерогенные коллоидно-дисперсные и гомогенные высокомолекулярные системы обладают целым рядом общих свойств, что и делает их объектом изучения коллоидной химии.
Во многом близки к коллоидно-дисперсным системам и изучаются теми же методами суспензии, эмульсии и пены (D ж 103 Ч- 105 см-1). Хотя эти системы и обладают рядом особых специфических свойств, однако их следует причислить именно к коллоидным системам.
Коллоиды очень широко распространены в природе и играют важную практическую роль, чем и определяется не только научное, но и народнохозяйственное значение коллоидной химии.
Драгоценные камни, а также другие минералы в недрах земли, пищевые продукты, одежда, обувь, дым, облака, мутная вода в природных водоемах, почва, глина — все это не что иное, как коллоидные системы. Такие биологические жидкости, как кровь, плазма, лимфа, спинно-мозговая жидкость, белки, крахмал, слизи и камеди, являются коллоидами.
Существенную роль играют коллоиды в промышленности, главным образом в таких ее отраслях, как добыча и переработка нефти, металлургическая промышленность, горнорудное дело, производство различных строительных материалов и пластмасс, синтетических волокон, синтетического каучука и резины, текстильная, лакокрасочная и пищевая промышленность, мыловаренное производство и т. п. Такие важные для промышленности технологические процессы, как обогащение
— 359 —
полезных ископаемых путем флотации, механическая и термическая обработка металлов, технология фотографических и кинематографических процессов имеют прямое отношение к коллоидно-дисперсным системам. В фармацевтической и парфюмерной промышленности многие лекарственные и бытовые препараты производятся в виде паст, кремов, мазей, тонких суспензий и эмульсий.
Исключительно важное значение имеет коллоидная химия в геологии. Представления о коллоидном состоянии вещества способствуют более углубленному пониманию процессов образования минералов, различных руд, горных пород и т. п.
В области почвоведения многие проблемы, например процессы ионного обмена, строение и свойства почвенного поглощающего комплекса, биохимия гумуса и другие также тесно связаны с коллоидной химией. Закономерности, устанавливаемые ею, дают возможность агроному не только глубже понимать процессы, протекающие в почве, но и в известной мере сознательно их изменять в желаемом направлении.
Велика роль коллоидной химии в вопросах химической защиты растений от различных вредителей и сорняков. В целях более высокой эффективности различные ядохимикаты применяются в виде суспензий, эмульсий, дымов и туманов (аэрозолей). Вот почему в системе агрономического образования коллоидной химии уделяется большое внимание. Такие важные для подготовки агронома научные дисциплины, как почвоведение, агрохимия, физиология растений и животных, метеорология, биохимия, микробиология и другие, широко пользуются основными положениями и методами коллоидной химии.
В целях более глубокого понимания предмета и задач современной коллоидной химии полезно рассмотреть ее положения в их историческом развитии.
Краткая история развития коллоидной химии. Как самостоятельная научная дисциплина коллоидная химия возникла в начале XX в., однако практические сведения о коллоидах можно найти уже в работах Аристотеля и алхимиков. Многие коллоидные системы и их свойства были хорошо известны человеку в глубокой древности и широко им использовались. Однако до середины XIX в., несмотря на успешное развитие естествознания, изучение и понимание коллоидов продвинулось очень мало. Объясняется это не только сложностью коллоидных систем, но и тем, что в XVIII и начале XIX вв. в естествознании господствовали идеалистические и вульгарно-механистические взгляды на сложные явления природы, вроде учения о «жизненной силе» и т. п.
Русские ученые внесли неоценимый вклад в будущую коллоидную химию. Так, в трудах М. В. Ломоносова (1751) четко различались явления кристаллизации и свертывания (коагуляции) растворов, описаны способы получения и свойства коллоидных растворов в воде и стекле (его знаменитые цветные стекла по существу являются твердыми растворами). Позднее, Т. Е. Ловиц (1789) впервые открыл одно из важнейших явлений, лежащее в основе коллоидной химии, — адсорбцию из растворов на твердом адсорбенте (угле). Это свойство угля Ловиц успешно использовал в практических целях для осветления сахарного сиропа и растительных масел, а также для очистки селитры, которая применялась в производстве пороха.
В 1808 г. профессор Московского университета Ф.Ф. Рейсс впервые установил факт движения частичек дисперсной фазы и дисперсионной среды под влиянием внешнего электрического поля. Эти работы легли в основу изучения влектрокинетических свойств коллоидно-дисперсных систем.
Большое значение имели работы итальянского химика С е л ь м и, который еще в 1845 г., исследуя свойства различных растворов, заметил, что биологические жидкости --сыворотка, молоко, кровь, лимфа и другие резко отличаются по
— 360 —
ойствам от обычных истинных растворов; они были им названы псевдо-своим Сельми доказал, что характерным отличием псевдорастворов (или раствор *тв0р0В от истинных растворов) является то, что образование их не ложны Р^^ сам0Пр0ИЗВ0льным раздроблением вещества на молекулы. Не сопров <ное ЗНачение имели работы Фарадея, который впервые (1857) от-МеИее явление, получившее впоследствии название «эффекта Фарадея», а также КРЬЬяпал секрет древних алхимиков — способ получения коллоидных раство-пов золота.
Р Однако началом классического периода в развитии коллоидном химии сле-т считать появление работ английского химика Грэма (1861), которого по считают «отцом» коллоидной химии. Он ввел термин и определил понятие «коллоиды». Изучая различные растворы, Грэм обнаружил, что одни вещества быстро диффундируют и проходят через растительные и животные мембраны, легко кристаллизуются. Другие обладают очень малой диффузией, не проходят через мембраны и не кристаллизуются, а образуют аморфные осадки. Так, например сравнивая время диффузии различных растворенных веществ и принимая вр^мя диффузии НС1 за единицу, Грэм получил сильно различающиеся значения:
НС1.................................... 1,00
NaCl................................... 2,33
MgSO4.................................. 7,00
Сахар.................................. 7,00
Белок.................................. 49,00
Карамель............................... 98,00
Как видим, уже в этом примере растворы можно разделить на две группы. В одну группу входят такие вещества, как НС1, NaCl, MgSO4 и сахар. Эти вещества хорошо диффундируют, легко проходят через поры животной и растительной перепонки (диализ). Во вторую группу входят белок и карамель, которые обладают ничтожно малой диффузией и не проходят через полупроницаемые перепонки.
Первые Грэм назвал кристаллоидами, вторые — коллоидами (от греческого слова kolla — клей и eidos — вид) или клееподобными веществами. Таким образом, все вещества он подразделил на два больших класса. Причем, в отличие от обычных истинных растворов, коллоидные растворы были названы Грэмом волями.
Против такого жесткого разделения химических веществ на коллоиды и кристаллоиды высказался в 60-х годах XIX в. профессор Киевского университета П. Г. Б о р щ о в, который независимо от Грэма определил сущность коллоидного раствора (золя) и коллоидной частицы. В частности, он выдвинул идею о кристаллической структуре коллоидных частиц, высказал правильное представление о коллоидной мицелле и наличии определенной связи между поверхностью коллоидных частиц с молекулами растворителя. Работы И. Г. Борщова позволяют считать его зачинателем русской коллоидной химии и одним из основоположников коллоидной химии как науки вообще.
Позднейшие исследования подтвердили правоту высказывания И. Г. Борщова о том, что правильнее говорить не о коллоидах как об особой группе веществ, а о коллоидном состоянии вещества. Такой же точки зрения придерживался и Д. И. Мен дел е^е в, который полагал возможным получение любого вещества в соответствующей среде в коллоидном состоянии. Эту идею об универсальности коллоидного состояния вещества окончательно развил и экспериментально обосновал другой русский ученый П. П. В е й м а р н, которому удалось получить в коллоидном состоянии огромное количество веществ, считавшихся до него типичными кристаллоидами. Так, раствор мыла в воде обладает свойствами коллоида, а мыло, растворенное в спирте, проявляет свойства истинных растворов. Аналогично этому типичный кристаллоид поваренная соль, растворенная в воде, дает истинный раствор, а в бензоле — коллоидный раствор и т. п. В настоящее время любое вещество можно получить в коллоидном состоянии. Поэтому нельзя говорить о коллоидных веществах, а следует говорить о коллоидно:.: состоянии тех или иных веществ.
— 361 —
Начало современного этапа развития коллоидной химии тесно связано с це-лым рядом замечательных открытий в области физики и смежных с ней наук в первые два десятилетия нашего века. За этот период произошла переоценка многих классических представлений. Разработка новых методов исследования, таких, как ультрамикроскопия (1904), рентгеноструктурный анализ (1913—1916), метод электронной микроскопии и др., позволила ученым глубже проникнуть в сущность строения коллоидов и вместе с тем далеко продвинуться в области теории. В учении о коллоидах в этот период на первый план выступает изучение поверхностно-сорбционных явлений. Эти явления были подробно исследованы русскими учеными А. А. Титовым (1910) и Н. А. Ш и л о в ы м (1916), а также зарубежными Л эн гмюром (1917) и др. Успешное применение А. В. Д у-манским центрифуги для изучения коллоидных систем послужило мощным толчком к разработке метода ультрацентрифугирования, который по существу является одним из важнейших современных методов исследования коллоидных растворов.
В решении проблемы устойчивости коллоидных систем и их коагуляции наряду с выдающимися работами таких крупных зарубежных ученых, как Дюкло, Фрейндлихи Кройт, весомый вклад внесли и наши советские ученые Н. П. Пес к о в, А. И. Р а би н о в и ч, П. А. Р е б и н д е р и многие другие.
Большую известность у нас и за рубежом приобрели работы С. М. Л и п а • това и В. А. Каргина в области исследования высокомолекулярных соединений. В частности, В. А. Каргин и его сотрудники подробно исследовали механизм процесса образования коллоидных частиц, экспериментально доказав наличие двух стадий в этом процессе.
В области исследования поверхностно-адсорбционных слоев коллоидных систем большое значение приобрели работы А. Н. Ф р у м к и н а, Б. В. Д е р я -г и н а и других советских ученых.
В нашей стране впервые были выполнены обширные исследования, касающиеся всестороннего изучения почвенных коллоидов. Выдающиеся работы академика К. К. Г е д р о й ц а явились основополагающими в учении о почвенном поглощающем комплексе. Дальнейшее развитие наука о почвенных коллоидах получила в работах В. Р. В и л ь я м с а, А. Ф. Т ю л и н а, И. Н. Антипова-Каратаева, А. Н. Соколовского, Н. П. Ремезова, С. Н. Алешина, А. В. Петербургского, Н. И. Горбунова и других советских исследователей. Из зарубежных ученых следует отметить работы Г. Вигнера и С. Маттсона.
«... вопросы коллоидной химии должно считать передовыми и могущими иметь большое значение во всей физике и химии.
(Д. И, Менделеев)
Глава IX
ОБЩАЯ ХАРАКТЕРИСТИКА КОЛЛОИДОВ И ИХ СВОЙСТВ
§ 104. Классификация дисперсных систем
Дисперсные системы (в том числе и коллоидно-дисперсные) классифицируются по следующим признакам: дисперсности, агрегатному состоянию и интенсивности взаимодействия частиц на поверхности раздела между фазами. По дисперсности системы делятся на три класса: грубые взвеси (суспензии), коллоидно-дисперсные (дисперсоиды) и молекулярно-ионные растворы (дисперсиды). Для каждого из этих классов характерна определенная степень дисперсности (см. табл. 70).
Таблица 73
Классификация дисперсных систем по агрегатному состоянию фазы и среды
Дисперсная фаза Дисперсионная среда Примеры дисперсных систем
1 вердая Газообразная Табачный дым; пыль цементная, сахарная,
» Жидкая мучная, космическая и др. Суспензия; холодное молоко, коллоидные
Т вердая растворы металлов Металлические сплавы; искусственные дра-
Жидкая Газообразная гоценные камни; цветные стекла; голубая каменная соль и др. Аэрозоли: туман, облака, распыленные ор-
Жидкая ганические вещества, газ в критическом состоянии Эмульсии: горячее молоко, маргарин, ели-
ъ Т вердая вочное масло; эмульсии масла в воде, воды в нефти, бензина в воде; некоторые кремы и мази Природные минералы с жидкими включения-
Газообразная Газообразная ми: жемчуг, опал и др. Не являются дисперсной системой, так как
Жидкая являются гомогенной смесью Различные пены: пивная, противопожарная
Твердая и др. Твердые пены; пенопласты; пемза; активи-
рованный уголь; микропористая резина; ряд ионообменных смол
— 363 —
По агрегатному состоянию обеих фаз различают следующие под, группы дисперсных систем (табл. 73). ПН
Из указанных дисперсных систем ближе всего стоят к биологи-ческим объектам и представляют наибольший интерес коллоидные ] растворы, включающие жидкую дисперсионную среду (воду) и высо-комолекулярную дисперсную фазу (белки, полисахариды, липоиды ' и др.). Коллоидные растворы с жидкой дисперсионной средой назы-вают, как уже отмечалось, @оляжГ)(от латинского слова solutus — 1 растворенный). Водные коллоидные растворы называются гидрозолями. спиртовые — алкозолями, бензоловые—бензозолями, эфирные— этерозолями и т. п. Ввиду того, что коллоидные растворы могут при известных условиях терять свою текучесть и затвердевать с образованием так называемых гелей, они называются соответственно: гидрогелями, алкогелями, бензогелями, этерогелями и т. п.
По характеру взаимодействия между частицами дисперсной фазы и дисперсной среды дисперсные системы подразделяются на лиофильные (от греческого 1уо — растворяю; philia—люблю) и лиофобные^ (phobia — страх, нелюбовь).
Лиофильные системы характеризуются интенсивным взаимодейст- j вием частиц дисперсной фазы с дисперсионной средой. Если дисперсионной средой лиофильной системы является вода, то такие системы называют гидрофильными. Например, растворы мыл, некоторых белков и т. д. 1
В лиофобных системах почти полностью отсутствует взаимодействие между молекулами среды и частицами дисперсной фазы. Лиофобные системы, в которых дисперсионной средой является вода, называются гидрофобными системами. Например, многие металлы в коллоидном состоянии, эмульсии масел в воде и др.
В настоящее время деление коллоидных систем на две основные | группы—лиофильные и лиофобные коллоиды в известной мере устарело, хотя эти термины еще широко распространены в литературе. За последние 20 лет трудами таких ученых, как В. А. Каргин, С. М. Ли- j патов и др., доказано, что системы, ранее называвшиеся лиофильны ми золями, на самом деле представляют собой не что иное, как истин- ] ные растворы высокомолекулярных соединений. В отличие от лиофобных золей эти растворы являются системами гомогенными и термодинамически равновесными. Исследования показали, что основной структурной единицей лиофильных золей является не мицелла (как у лиофобных золей), а сильно сольватированная (гидратированная) макромолекула высокомолекулярного или высокополимерного соединения. Причем для многих полярных полимеров и белков сольватация является хотя и главным, но не единственным фактором устойчивости их растворов. В значительной мере характер поведения вы- | сокомолекулярных соединений в растворах определяется свойствами их длинных цепеобразных частиц — макромолекул. Огромные размеры макромолекул, превышающие в отдельных случаях размеры коллоидных частиц, объединяют эти системы с коллоидно-дисперсными системами. Сближает их и то обстоятельство, что при концентрировании растворов высокомолекулярных соединений они обращаются
— 364 —
так называемые студни (гели), в которых решающую роль играют поверхностные (адсорбционные) явления, которые характерны для гетерогенных лиофобных коллоидов. В остальном лиофильные золи гораздо ближе к обычным молекулярным растворам.
В настоящее время признано целесообразным отказаться от терминов «лиофильные коллоиды» и «лиофильные золи», чтобы не связывать с ними представлений о мицеллярном строении этих систем, а заменить их термином «растворы высокомолекулярных соединений» (сокращенно — растворы ВМС). Мы будем следовать этому решению п лишь иногда употреблять термин «лиофильные золи» в качестве синонима нового термина. Термины «лиофильные коллоиды» и «лиофобные золи» мы оставляем исключительно в целях преемственности, поскольку в прошлом и до сего времени эти термины господствуют в учебной, научной и технической литературе.
Таким образом, по интенсивности взаимодействия частиц дисперсной фазы со средой все коллоидные системы подразделяются на лиофобные коллоиды (гидрозоли металлов, сульфидов) и высокомолекулярные соединения и их растворы (белки, полисахариды, каучук* полиамиды), именуемые по старой терминологии лиофильными коллоидами.
§ 105. Получение коллоидно-дисперсных систем
Размеры коллоидных частиц настолько велики по сравнению с молекулами дисперсионной среды, что между ними образуется поверхность раздела. Собственно коллоидные системы занимают промежуточное положение по размерам своих частиц между грубодисперсными и молекулярно-дисперсными системами.
То или иное вещество может быть получено в коллоидном состоянии при следующих условиях:
1) размеры частиц данного вещества должны быть доведены до коллоидных размеров, что осуществимо двумя методами: а) раздроблением более или менее крупных тел до частиц коллоидного размера (дисперсионные методы); б) укрупнением молекул, атомов или ионов до частиц коллоидного размера (конденсационные методы);
2) необходимо присутствие стабилизаторов (ионов электролитов, которые на поверхности коллоидной частички образуют ионно-гидратную оболочку, препятствующую слипанию частичек при их взаимном столкновении в растворе);
3) коллоидные частицы (дисперсная фаза) должны обладать плохой растворимостью в дисперсионной среде, хотя бы в момент их получения.
При соблюдении этих условий коллоидные частицы приобретают электрический заряд и гидратную оболочку, что препятствует выпадению их в осадок.
Дисперсионные методы. Механические методы. Сущность методов механического диспергирования заключается в энергичном и продолжительном растирании, размалывании и прочих механических приемах раздробления вещества. Для этих целей применяются специаль-
— 365 —
Рис. 148. Схема коллоидной мельницы
ные машины, работающие по принципу ударного размельчения и растирания диспергируемых веществ. Наиболее широкое распространение получили шаровые и коллоидные мельницы. Шаровая мельница представляет собой полый цилиндр, в котором находятся стальные или фарфоровые шарики различного диаметра. В цилиндр загружается вещество, подлежащее диспергированию, и он с помощью электромотора приводится в быстрое вращение. Измельчение вещества достигается за счет движения шаров, находящихся в цилиндре. Шаровые мельницы широко используются в технике для получения различных тонкодисперсных порошков: серы, графита, различных минеральных красок. Однако степень дисперсности, достигаемая на этих мельницах, остается сравнительно низкой — диаметр частиц составляет около 50—60 мк.
Если требуется более высокая степень дисперсности, используют специальные коллоидные мельницы. Сконструированная Плауссоном (1920) коллоидная мельница представляет собой полый цилиндр, внутри которого находится специальный ротор с лопастями, вращающийся со скоростью до 20 000 об/мин* На рис. 148 показана схема коллоидной мельницы. Измельчение вещества происходит в зазорах между лопастями ротора b и выступами а внутри корпуса в результате быстрого вращения вала. На этих мельницах может быть достигнута степень дисперсности от 0,1 до 1,0 мк.
Во всех случаях диспергирование обычно ведут, добавляя соответствующие стабилизирующие вещества, препятствующие слипанию раздробленных частиц.
Ультразвуковой метод. В последующие годы довольно широкое распространение получил метод измельчения веществ с помощью ультразвука. Механизм действия ультразвука очень сложен и в настоящее время еще сравнительно мало изучен. Можно лишь предполагать, что диспергирование взвешенных в жидкости веществ происходит под действием быстро сменяющихся сжатий и расширений системы, в результате чего появляются разрывающие силы, ведущие к раздроблению вещества. Ультразвуковые установки отличаются высокой производительностью. С их помощью можно диспергировать самые разнообразные вещества.
Метод химического диспергирования. Наиболее распространен метод пептизации. Это процесс перехода из геля в золь под влиянием диспергирующих веществ — пептизаторов. Сущность пептизации заключается в том, что к свежеполученному рыхлому осадку диспергируемого вещества прибавляют небольшое количество пептиза-
тора (чаще всего электролита), который уменьшает взаимодействие между частицами осадка и облегчает их переход в состояние золя. Пептизаторами служат различные электролиты, которые способствуют дезагрегации аморфных осадков. В качестве примера можно назвать получение золя гидроокиси Fe(OH)3 при действии на ее осадок небольшим количеством соли FeCl3, выполняющей роль пептиза-тора. Практически все рыхлые свежеобразованные осадки гидроокисей металлов, например А1(ОН)3, Zn(OH)2, подвергаются пептизации.
К химическим методам диспергирования относится и так называемый метод самопроизвольного диспергирования. Он заключается в получении коллоидных растворов веществ путем растворения их в соответствующих растворителях. Так, путем растворения в воде можно получить коллоидные растворы крахмала, желатина, агар-агара и др. Самопроизвольное диспергирование совершается без внешних механических воздействий. Этот метод широко применяется для получения растворов высокомолекулярных веществ из твердых по-t лимеров.
Образование коллоидов в природе. В природе активно протекают процессы диспергирования. Приливно-отливные явления океанов и морей, разрушающее действие прибоя, резкие колебания температур, ветер и другие явления природы развивают колоссальные силы, которые дробят горные породы вплоть до частиц коллоидных размеров. Постоянное действие ледников и рек также приводит к интенсивным процессам измельчения слагающих пород.
Мощным фактором механического диспергирования твердых горных пород является расширение воды при ее замерзании. Проникая глубоко в трещины породы и замерзая там, вода вызывает дробление породы на частицы различного (вплоть до коллоидного) размера.
Громадные массы осадочных пород, глины, лесса, которые мы встречаем в природе — все это результат диспергирования твердых горных пород, которое происходит не только под влиянием механических факторов, но и под влиянием химического воздействия (выветривание под действием углекислоты и воды), а также под влиянием биологических факторов. Животные, как и растения, так же разрыхляют горные породы и своими выделениями способствуют их изменению. Таким образом, в результате всех перечисленных выше процессов горные породы, подвергаясь глубоким физическим и химическим изменениям, могут образовать сложные коллоидные системы.
Конденсационные методы. В основе большинства конденсационных методов получения коллоидных растворов лежат различные, химические реакции: окисления, восстановления, реакции обменного разложения, гидролиза и др. В результате всех этих реакций молекулярные или ионные растворы переходят в коллоидные путем перевода растворенных веществ в нерастворимое состояние. В основе методов конденсации, помимо химических процессов, могут лежать и процессы физические, главным образом явления конденсации паров.
Рассмотрим кратко наиболее важные методы конденсации (агрегации) частиц до коллоидных размеров.
Метод окисления. Он основан на реакциях окисления, в результате которых одно из веществ может быть получено в коллоидном состоянии. Так, при окислении сероводорода кислородом воздуха или двуокисью серы можно получить золь серы
2H2S-|-О2 = 2Н2О-|-2S
2H2S + SO2 = 2Н2О 4- 3S
— 367 —
Эти реакции, как показали исследования, протекают гораздо сложнее, ? так как наряду с коллоидной серой образуется ряд тионовых кислот.
Метод восстановления. Наиболее распространенные химические £ методы получения коллоидных растворов различных металлов основаны на реакциях восстановления. Ионы, восстанавливаясь, т. е. присоединяя электроны и превращаясь в нейтральные атомы, конденсируются затем в коллоидные частицы. В "качестве примера рассмотрим реакцию получения золя золота путем восстановления перекисью водорода или формалином
2HAuC14 4- ЗН2О2
2HAuC14 + ЗНСНО 4-11КОН -
. 2Аи 4- 8НС1 4- ЗО2
2Аи 4- ЗНСООК 4- ЗКС1 4- 8Н2О
Реакцией восстановления были получены в коллоидном состоянии многие металлы Au, Ag, Pt, Rd, Ph, Os, Hg и многие другие.
Рис. 149. Схема прибора для получения коллоидов по методу электрического распыления металлов:
1 — электроды из распыленного металла;
2 — микрометрический винт; 3 сосуд с водой; 4 — штатив
Метод обменного разложения, Сущность его заключается в том, что при взаимодействии двух веществ в результате реакции обменного разложения образуется новое трудно растворимое вещество, которое при наличии определенных условий способно находиться в коллоидном состоянии. В качестве примера можно назвать реакцию получения золя сульфата бария
BaCl24-K2SO4 -> BaSO4 4-2КС1
или золя хлорида серебра AgNO3+KCI AgCl + KNO3
широко пользуются при получе-
Метод гидролиза. Этим методом
нии золей различных металлов из их солей, если в результате реакции гидролиза образуется труднорастворимая гидроокись. Так, например, труднорастворимая гидроокись железа образуется при гидролизе хлорида железа по уравнениям реакций
FeCl3 4- ЗН2О -> Fe (ОН)3 4- ЗНС1
Fe (ОН)3 4- НО - FeOCl 4-2Н2О
Образующаяся в результате этих реакций соль железа FeOCl диссоциирует частично на ионы
FeOO FeO+4-О-
Эти ионы и обеспечивают ионогенный слой вокруг частиц Fe (ОН)3, благодаря чему они удерживаются во взвешенном состоянии.
Замена растворителя. Сущность этого метода заключается в том, что при замене растворителя вещество, ранее находившееся в растворенном состоянии, выделяется из раствора в виде высокодисперсной
— 3G8 —
Фазы, нерастворимой в данном растворителе. Так, если спиртовый раствор канифоли (который представляет собой истинный раствор) небольшими порциями прибавлять в воду, образуется коллоидный раствор канифоли в воде. В данном случае спирт хорошо смешивается с водой, а канифоль очень мало в ней растворяется и поэтому выделяется в виде высокодисперсной фазы. Кроме канифоли, этим методом можно приготовлять золи серы, фосфора, мастики и т, п. также путем вливания их спиртовых растворов в воду.
Электрический метод. Этот метод, предложенный Бредигом еще в 1898 г., используемся преимущественно для приготовления коллоидных растворов благородных металлов. Сущность его заключается в получении электрической дуги между находящимися в воде электродами, состоящими из золота или платины, серебра и т. д., т. е. из металла, золь которого хотят получить. В дуге под воздействием высокой температуры металл электродов испаряется, а затем пары его конденсируются в частицы коллоидных размеров, образуя соответствующий золь. Весь процесс проводят при охлаждении. На рис. 149 показана схема прибора для получения золей металлов этим способом.
§ 106. Получение растворов высокомолекулярных веществ
Растворы высокомолекулярных соединений образуются самопроизвольно и для их устойчивости не требуется вводить стабилизирующие вещества. Все высокомолекулярные вещества состоят главным образом из цепных линейных структур, отдельные звенья которых связаны между собой прочными химическими связями, в результате чего молекулярные цепи сохраняются как в твердых полимерах, так и в растворах. Образование высокомолекулярных веществ из низкомолекулярных происходит двумя методами: полимеризацией и поликонденсацией. Полимеризацией называется соединение молекул низкомолекулярного вещества с образованием высокополимера такого же элементарного состава, как и исходное вещество. Так, при полимеризации винилхлорида получается высокомолекулярное соединение — поливинилхлорид
п (СН2=СНС1) -> (-СН2—СНС1----)п
Соединение молекул винилхлорида в данном случае происходит за счет раскрытия двойных связей. Молекулярный вес образующегося поливинилхлорида достигает 90 000 кислородных единиц.
Напомним, что молекулы низкомолекулярных веществ, образующие полимер, называются мономерами (или звеньями), а их число в макромолекуле носит название степени полимеризации.
Продукты полимеризации разнородных мономеров называются сополимерами. В качестве примера можно назвать сополимер винилхлорида и винилацетата
хСН2 = СНС1 + хСН2=СНОСОСН3 -> / — СН2 —СНС1 — сн2—сн— \
I о I
I I /
\ СОСНз/х
Поликонденсацией называется образование полимера из низкомолекулярных веществ с отщеплением от них атомов или групп атомов с образованием воды, спирта или других соединений. Элементарный состав продукта поликонденсации отличается от исходных веществ. В качестве примера можно назвать образование белков путем поликонденсации аминокислот с отщеплением воды:
2/i (NH2CHRCOOH) -> Н (—NH — CHR—СО—NH—CHR—СО—)ОНп+(2л—1) Н2О При поликонденсации одновременно с высокомолекулярным соединением образуется низкомолекулярное вещество, что позволяет считать эти реакции как бы реакциями обмена.
Типичным примером поликонденсации может служить реакция образования ортокремниевой кислоты, которая идет по схеме
ОН ОН ОН ОН
I I II
НО—Si—ОН 4- НО—Si—ОН -> НО—Si—О—Si—ОН+Н2О I I II
он он он он
Дальнейшая конденсация приводит к образованию макромолекул, имеющих следующее строение ОН ОН
I I
ОН—Si—О—[Si (ОН)2—О]х—Si—ОН
I I
он он
Опыт показывает, что растворы ортокремниевой кислоты неустойчивы во времени и имеют тенденцию образовать мицеллы или переходить в студень.
Некоторые высокомолекулярные вещества могут быть получены как полимеризацией, так и поликонденсацией. Так, полиэтиленок-сид можно получить полимеризацией окиси этилена
хСН2-СН2 -> (-СН2-СН2-О-)Х Ny/
а также поликонденсацией этиленгликоля (по Коршаку)
хНОСН2—СН2ОН -> (—СН2—СН2—О—)Х+Н2о
Высокомолекулярные системы образуются также из длинных цепных молекул (или макромолекул), которые в свою очередь были получены методами полимеризации или поликонденсации.
К высокомолекулярным системам относятся различные полимеры с линейными гибкими макромолекулами (каучук, эластомеры), линейными жесткими макромолекулами (целлюлоза и ее эфиры), спиральными макромолекулами (крахмал, гликоген) и др.
Для образования цепей полимерных соединений могут служить не только углерод или кремний, как считалось еще недавно, нои алюминий, бор, титан, фосфор, магний и многие другие элементы. Таким образом, высокомолекулярные соединения могут иметь как органическую, так и неорганическую природу.
— 370 —
Наиболее подробно разработан синтез кремнийорганических полимеров, которые обладают такими ценными свойствами, как высокая термическая устойчивость, морозоустойчивость, хорошие диэлектрические свойства, В настоящее время кремнийорганические полимеры нашли широкое применение в качестве термо- и морозостойких масел, каучуков, пластических масс, цементирующих и гидрофобилизирую-щих составов. Так, каучук, полученный на основе кремнийорганических смол, сохраняет свою эластичность при температурах от — 60 до 4- 200° С и не разрушается даже при 300° G.
Впервые синтез кремнийорганических соединений был разработан в 1936 г. советским ученым К. А. А н д р и а н о в ы м. Он первоначально получал сложные эфиры, являющиеся производными орто-кремниевой кислоты Si(OH)4. Например
СН3 О—СН3
I I
СН3—Si—О—СН3 или СН3—Si—О—СН3 I I
СН3 О—сн3
В результате гидролиза этих эфиров получаются соответствующие гидрокси лсодержащие соединения кремния, которые сразу же конденсируются с отделением молекулы воды и образованием поликонденсатов по следующей схеме
сн3 сн3 сн3
I I I
СН3—Si—ОН+НО—Si—ОН+НО—Si—СН3 I I I
СН3 СН3 СН3
СН3 СНз СНз
I I I
СН3—Si—О—Si—О—Si—СН3+2Н2О I I I
СН3 СН3 СН3 *
При небольшой степени поликонденсации (если молекулы содержат до десяти атомов кремния) получаются жидкости, применяемые в качестве смазочных масел. При более высокой степени поликонденсации получаются вещества, имеющие характер смол*
Синтетические полимеры нашли широкое применение в сельском хозяйств? для создания агрономически важной оструктуренности почвы. Искусственное оструктуривание почв осуществляется введением в почву небольших количеств структурообразующих веществ. Начиная примерно с 1950 г. в ряде стран (США, Англия, Венгрия, ГДР, СССР и др.) в качестве структурообразующих вещестз стали применять высокомолекулярные соединения — полимеры и сополимеры, главным образом состоящие из производных акриловой СН2=СН — СООН, метакриловой СН2 = С (СН8) — СООН и малеиновой СООН — СН = СН — —СООН кислот. Внесение, например, сополимера, состоящего из метакриловой кислоты (60%) и метакриламида (40%), всего лишь в количестве 0,СО 1 о с почвы существенно увеличивает водопрочность структуры. В табл. 74 приведены данные по созданию водопрочных агрегатов с помощью искусственных структура-образователей.
— 371 -«
Таблица 74
Влияние сополимера метакриловой кислоты и метакриламида на водопрочность дерново-подзолистой почвы (данные П. В. Вершинина)
Количество внесенного сополимера, % к весу почвы
Количество водопрочных агрегатов <0,25 мм, % к весу почвы
Количество внесенного сополимера, % к весу почвы
Количество водопрочных агрегатов <0,25 мм, % к весу почвы
0,1
0,05
0,01
92,0
90,2
86,0
0,005 0,001 0
56,8
51,8
18,4
Помимо синтетических высокомолекулярных соединений, широкое распространение в народном хозяйстве имеют и так называемые искусственные высокомолекулярные соединения. Это природные высокомолекулярные соединения, подвергшиеся химической обработке. Искусственные полимеры в громадных количествах получают в промышленности в виде производных целлюлозы: нитроцеллюлоза, ацетилцеллюлоза, вискоза и др. Из этих веществ получают нитролаки, искусственную кожу, бездымный порох, целлулоид, искусственный шелк, негорючую кинопленку и т. п. Продуктом химической обработки казеина является, например, искусственный роговидный материал — галалит, который широко используется в производстве предметов массового потребления.
Советские ученые создали большое число высокомолекулярных соединений, которые используются в различных областях народного хозяйства. Академик Н. Н. Семенов так определил значение полимеров в наше время: «Если девятнадцатый век часто называют веком пара и электричества, то двадцатый век делается веком атомной энергии и полимерных материалов». Без использования синтетических высокомолекулярных соединений невозможно производство радио- и электроаппаратуры, а также других машин и приборов.
§ 107. Методы очистки золей и растворов высокомолекулярных веществ
Гидрофобные золи и растворы высокомолекулярных соединений при их образовании почти всегда «загрязняются» различными примесями, чаще всего электролитами. Особенно загрязняются золи, в которые в избытке введен стабилизатор. Часто в системе присутствует исходный электролит. Для получения коллоидных растворов с наибольшей устойчивостью необходимо удалять из них примеси. Рассмотрим кратко различные методы очистки золей и растворов высокомолекулярных веществ.
Диализ. Диализ — это процесс освобождения коллоидных растворов от примесей, способных проникать через полупроницаемые мембраны. Этот метод очистки, предложенный еще Грэмом, является наиболее простым и доступным. Процесс очистки основан на способности примесных ионов и молекул малых размеров свободно проникать через полупроницаемые мембраны, тогда как крупные коллоидные частицы и молекулы высокомолекулярных соединений такой способностью не обладают.
— 372 —
Полупроницаемыми являются различные растительные, животные и искусственные мембраны; их можно приготовить из пергамента, бычьего, свиного и рыбьего пузыря, из коллодия, целлофана и т. д.
Приборы, в которых производится диализ, называются диализаторами. На рис. 150 изображен простейший диализатор Грэма. В нем очи-
щаемый золь контактирует с проточной дистиллированной водой через полупроницаемую мембрану. Чем больше разность концентраций кристаллоида по обе сторо- -ны мембраны, тем эффективнее идет диализ. Вот почему очистка золя ускоряется, если, во внешней
Рис. 150. Схема простейшего диализатора:
•пп — полупроницаемая перепонка (мембрана)
камере диализатора вода проточная или часто сменяется. Однако даже при
этих условиях диализ идет
очень медленно, длится иногда недели и даже месяцы и требует огромного количества растворителя. Для ускорения процесса диализа было предложено использовать электрический ток.
Электродиализ. Этот метод представляет собой ускоренный процесс диализа с применением электрического тока. В электролизаторах раз
Рис. 151. Электродиализатор Паули:
1 — коллоидный раствор; 2 — электроды
личных конструкций имеется три камеры (рис. 151) с внутренними стенками из полупроницаемых мембран. В среднюю камеру наливают коллоидный раствор, подлежащий очистке, а во внешние камеры растворитель — проточную воду. Во внешних камерах находятся электроды, на которые подается напряжение постоянного тока. При падении потенциала 20—50 в!см и более образуется направленное движение ионов к соответствующим электродам. Поскольку ионы свободно проходят через полупроницаемую перепонку, а коллоидно-дисперсные частицы не проходят, коллоидный раствор постепенно очищается от электролитов.
Продолжительность электродиализа в отличие от простого диализа измеряется не днями, а лишь часами и минутами, причем затрата растворителя сведена до минимума. В настоящее время широкое приме
— 373 —
нение метод электродиализа получил в биохимии и медицине, а также в народном хозяйстве.
Ультрафильтрация. Ультрафильтрацией называют фильтрование коллоидного раствора через полупроницаемые мембраны, которые укрепляются в специальных ультрафильтрах на твердой пористой подкладке.
Поскольку через поры обычной фильтровальной бумаги (от 1,5 до 5 мк) коллоидно-дисперсные частицы проходят легко, при ультрафильтрации пользуются специальными фильтрами, например целлофаном или фильтровальной бумагой, пропитанной коллодием. При уль-
Рис. 152. Ультрафильтрация под вакуумом
Рис. 153. Ультрафильтрация под давлением
трафильтрации дисперсная фаза остается на фильтре. Обычно процесс ультрафильтрации проводят под разрежением или под повышенным давлением. На рис. 152 и 153 показаны установки для обоих способов ультрафильтрации.
Методом ультрафильтрации можно производить концентрирование золей и растворов высокомолекулярных соединений, что позволяет избежать выпаривания, например, для таких соединений, которые не выдерживают высоких температур.
Применяя для ультрафильтров мембраны с определенной степенью пористости, можно в известной мере произвести разделение коллоидных частиц и одновременно приближенно определить их размеры. Этим методом впервые были определены размеры целого ряда вирусов и бактериофагов.
В настоящее время методы ультрафильтрации иногда применяются в сочетании с электродиализом. Этот комбинированный метод получил название метода электроультрафильтрации. В табл. 75 дано сопоставление относительных скоростей очистки по различным методам при сравнимых условиях.
— 374 —
Таблица 75
Относительные скорости очистки раствора
Метод Удельное . ещество
NaCl сахар
Диализ 1 0,3
•Электродиализ 163 2
Ультрафильтрация .... 14 14
Электроультрафйльтрация . 182 14
Как видим, метод электроультрафильтрации по скорости превосходит метод электродиализа.
Рис. 154. Ультрацентрифуга Сведберга:
1 — ротор; 2 — кюветы с коллоидным раствором
Ультрацентрифугирование. Идея этого метода впервые была высказана еще в 1913 г. А. В. Ду минским, который применил центрифугу для осаждения коллоидных частиц. За последние годы, с изобретением Сведбергом ультрацентрифуги, этот метод получил исключительно широкое применение в коллоидной химии. Современная ультрацентрифуга (рис. 154) представляет собой сложный аппарат, в котором ротор вращается в толстостенном металлическом корпусе в вакууме или в атмосфере водорода (для улучшения теплоотдачи) со скоростью до 60 000 об/мин и выше. Как видно из рис. 154, в роторе есть два сквозных отверстия, в которых находятся кюветы с коллоидным раствором емкостью всего 0,5 мл. По мере оседания частиц дисперсной фазы поглощение света вдоль
— 375 —
кюветы, находящейся на пути луча света, изменяется, что фиксируется на специальном экране или фотопластинке.
В ультрацентрифуге оседают не только коллоидные частицы гидрофобных коллоидов, но и молекулы белков и высокомолекулярных соединений. Помимо очистки, метод ультрацентрифугирования широко применяется в настоящее время для определения среднего радиуса коллоидных частиц, а также для вычисления молекулярного веса высокомолекулярных соединений. Работа с ультрацентрифугой очень сложна и кропотлива, так как требует тщательного учета влияния многих побочных факторов.
§ 108. Оптические свойства коллоидных растворов
По оптическим свойствам коллоидные растворы существенно от-
личаются от истинных растворов низкомолекулярных веществ, а также от грубодисперсных систем. Наиболее характерными оптическими свойствами коллоидно-дисперсных систем являются опалесценция, эффект Фарадея —Тиндаля и окраска. В основе всех этих явлении лежит рассеяние и поглощение света коллоидными частицами.^
В зависимости от длины волны видимого света и относительных размеров частиц дисперсной фазы рассеяние света принимает различный характер. Если размер стиц превышает длину товых волн, то свет от отражается по законам метрической оптики.
Рис. 155. Рассеивание света частицей, меньшей световой полуволны (схема): MN — направление освещающего луча
ча-све-них гео-При
этом часть светового излучения может проникать внутрь частиц, испытывать преломление, внутреннее отражение и поглощаться.
Если размер частиц меньше длины полуволны падающего света, наблюдается дифракционное рассеяние света; свет как бы обходит (огибает) встречающиеся на пути частицы. При этом имеет место частичное рассеяние в виде волн, расходящихся во все стороны (рис. 155). В результате рассеяния света каждая частица является
источником новых, менее интенщшщтх^волн, т. е. происходит как бы ‘самосвечеиие каждой частицы./Явление рассеяния света мельчайшими частицами получило название опалесценции. Оно свойственно преимущественно золям (жидким и твердым), наблюдается только в отраженном свете, т. е. сбоку или на темном фонц) Выражается это явление в появлении некоторой мутноватости* золя и в смене («переливах») его окраски по сравнению с окраской в проходящем свете. Окраска в отраженном свете, как правило, сдвинута в сторону большей частоты видимой части спектра. Так, белые золи (золь хлорида серебра, канифоли и др.) опалесцируют голубоватым цветом.
Эффект Фарадея—Тиндаля. Дифракционное рассеяние света впервые было замечено М. В. Ломоносовым. Позднее, в 1857 г. это явление наблюдал Фарадей в золях золота. Наиболее детально явление дифракции (опалесценции) для жидких и газовых сред было изучено Тиндалем (1868).
— 376 —
Если взять один стакан с раствором хлорида натрия, а другой — с гидрозолем яичного белка, трудно установить, где коллоидный раствор, з где истинный, так как на вид обе жидкости бесцветны и прозрачны (рис. 156). Однако эти растворы можно легко различить, проделав следующий опыт. Наденем на источник света (настольную лампу) светонепроницаемый футляр с отверстием, перед которым в целях получения более узкого и яркого пучка света поставим линзу. Если на пути луча света поставить оба стакана, в стакане с золем увидим световую дорожку (конус), в то время как в стакане с хлоридом натрия луч почти не заметен. По имени ученых, впервые наблюдавших это
Рис. 156. Эффект Тиндаля:
1 — стакан с раствором NaCl; 2 — стакан с гидрозолем яичного белка; 3— настольная лампа с светонепроницаемым футляром; 4 — оптическая линза
явление, светящийся конус в жидкости был назван конусом (или эффектом) Фарадея — Тиндаля. Этот эффект является характерным для всех коллоидных растворов.
Появление конуса Фарадея — Тиндаля объясняется явлением рассеяния света коллоидными частицами. Их размеры находятся в пределах 0,1—0,001 мк, а длина волн видимой части спектра лежит в границах 0,76—0,38 мк. Поэтому каждая коллоидная частица рассеивает падающий на нее свет. Он виден в конусе Фарадея — Тиндаля, когда луч зрения направлен под углом к проходящему через золь лучу.
Таким образом, эффект Фарадея — Тиндаля — явление, совершенно идентичное опалесценции и отличается от последней только способом коллоидного состояния, т. е. микрогетерогенности системы.
Теория рассеяния света коллоидно-дисперсными системами была разработана Рэлеем в 1871 г. Она устанавливает зависимость интенсивности (количества энергии) рассеянного света (/) при опалесценции и в конусе Фарадея — Тиндаля от внешних и внутренних факторов. Математически эта зависимость выражается в виде формулы, получившей название формулы Рэлея:
nV2
- (IX,1)
л
где / — интенсивность рассеянного света в направлении, перпендикулярном к лучу падающего света, К — константа, зависящая от
— 377 —
показателей преломления дисперсионной среды и дисперсной фазы, п — число частиц в единице объема золя, X — длина волны падающего света, V — объем каждой частицы.
Из формулы (IX, 1) следует, что рассеяние света (/) пропорционально концентрации частиц, квадрату объема частицы (или для сферических частиц — шестой степени их радиуса) и обратно пропорционально четвертой степени длины волны падающего света. Таким образом, рассеяние коротких волн происходит относительно более интенсивно. Потому бесцветные золи в проходящем свете кажутся крас-1 новатыми, в рассеянном — голубыми.
Опытная проверка формулы Рэлея показала, что применение ее^ ограничено. Во-первых, она применима только к золям, в которых г вещество дисперсной фазы не является проводником электричества и1 совершенно неприменима к металлическим золям, так как в окраске, их решающую роль играет поглощение (т. е. абсорбция) света. Во-вторых, даже для систем с частицами из непроводников это уравнение применимо только лишь для типичных золей, т. е, для частиц размером от 5 до 100 ммк.
Явлением светорассеяния Рэлей объяснял голубой цвет неба, а индийский ученый Раман — цвет морской воды. Однако рассеяние света в этих случаях происходит не за счет присутствия высокодисперсных примесей (например, пылинок, мельчайших капелек воды и т. п.). В 1907 г. Л. И. Мандельштам показал, что рассеянный свет возникает только в оптически неоднородной среде, так как в этом случае показатель преломления среды меняется от одного участка к другому. Позднее Смолуховский (1908) доказал, что такое нарушение однородности среды может возникнуть в результате теплового движения молекул как местное изменение (флуктуация) плотности, т. е. совершенно самопроизвольно на короткое время могут возникать очень малые участки, отличающиеся от соседних своей плотностью. В силу этого возникает разность показателей преломления между отдельными участками атмосферы (или морской воды) и как следствие-рассеяние света.
Явление опалесценции по своим внешним признакам сходно с явлением флуоресценции, природа которого связана с внутримолекулярным процессом. В случае флуоресценции часть падающего светового луча сначала избирательно поглощается, а затем вновь испускается (рассеивается), но уже с иной (обычно большей) длиной волны. Явление флуоресценции присуще в одинаковой мере как коллоидным, так и молекулярным растворам.
Окраска коллоидных растворов. В результате избирательного поглощения света (абсорбции) в сочетании с дифракцией образуется та или иная окраска коллоидного раствора. Опыт показывает, что большинство коллоидных (особенно металлических) растворов ярко окрашено в самые разнообразные цвета, начиная от белого и кончая совершенно черным, со всеми оттенками цветового спектра. Так, золи As2S3 имеют ярко-желтый, Sb2S3 — оранжевый, Fe (ОН)3 — красновато-коричневый, золота — ярко-красный цвет и т. п.
Один и тот же золь имеет различную окраску в зависимости оттого, в проходящем или отраженном свете она рассматривается. Многим золям присуще также явление полихромии (многоцветности), т. е. свойство золей одного и того же ве-
— 378 —
шест в а в зависимости от способа приготовления приобретать различную окраску. В данном случае окраска золей зависит от степени дисперсности частиц. Так, грубодисперсные золи золота имеют синюю окраску, большей степени дисперсности — фиолетовую; а высокодисперсные — ярко-красную. Интересно отметить, что цвет металла в недисперсном состоянии ничего не имеет общего с его цветом в коллоидном состоянии.
Необходимо отметить, что интенсивность окраски золей в десятки и сотни раз больше, чем молекулярных растворов. Так, желтая окраска золя As2Sa в слое толщиной в 1 см хорошо заметна при весовой концентрации 10~4%, а красный цвет золя золота заметен даже при концентрации 10“6%.
Красивая и яркая окраска многих драгоценных камней (рубинов, изумрудов, топазов, сапфиров) обусловлена содержанием в них ничтожных (не определимых даже на лучших аналитических весах) количеств примесей тяжелых металлов и их оксидов, находящихся в коллоидном состоянии. Так, для искусственного получения яркого рубинового стекла, употребляемого для автомобильных, велосипедных и прочих фонарей, достаточно на одну тонну стеклянной массы добавить всего лишь 100 г коллоидного золота.
Рис. 157. Схема ультрамикроскопа:
1 — кюве!а с исследуемым раствором; 2 — источник света; 3—пинты; 4 — щелевая диафрагма
Ультрамикроскоп. Явление светорассеяния в конусе Фарадея — Тиндаля лежит в основе одного из важнейших методов исследования высокодисперсных систем — с помощью ультрамикроскопа. Ультрамикроскоп был изобретен в 1903 г. Зигмонди и Зидентопфом. Отличительной особенностью ультрамикроскопа (рис. 157) является осветительная система, которая состоит из мощной вольтовой дуги 2, щелевой диафрагмы 4 и системы линз 3. Объект исследования помещают в специальную кювету /, которая крепится на предметном столике микроскопа.
В отличие от обычного микроскопа в ультрамикроскопе применяют боковое освещение. При этом свет от осветителя не попадает в объектив микроскопа и в глаз наблюдателя, поэтому фон поля зрения микроскопа темный. При рассматривании в ультрамикроскоп коллоидного раствора (например, золота) можно видеть беспрерывно движущиеся, переливающиеся всеми цветами радуги, разного размера частицы, из которых наиболее мелкие представляют собой светящиеся точки.
— 379 —
Интенсивность рассеяния света зависит от концентрации коллоидных частиц, от их размеров и формы.
Поскольку разрешающая сила ультрамикроскопа невелика (не пре-вышает таковую обычных микроскопов, имеющих увеличение в 300— 500 раз), то ни размеры, ни форма частиц в ультрамикроскоп непосредственно не различимы. И все же с помощью ультрамикроскопа можно косвенно судить о размерах и форме коллоидных частиц.
Размеры частиц можно определить приближенно путем следующего расчета.
При помощи микрометрической окулярной шкалы ультрамикроскопа выделяют определенный объем золя, в котором визуально подсчитывают число находящихся в нем коллоидных частиц. Зная общую мас-
Рис. 158. Снимки коллоидных частиц, полученные с помощью электронного микроскопа:
а—частицы золя Fe(OH)3; б — частицы золя пятиокиси ванадия; в — частицы окиси вольфрама
су диспергированного вещества и вычислив количество частиц во всем объеме, можно рассчитать массу одной частицы, и уже по ней, учитывая плотность диспергированного вещества, определить объем и размеры частицы. Пусть d — плотность частиц, С — весовая концентрация коллоидного раствора, V — выделенный оптический объем, v — число частиц в объеме V, тогда число частиц п в единице объема будет равно:
n = (IX,2)
Соответственно этому вес отдельной частицы т и ее объем w будут равны:
С т
tn ——, ш = ~с (IX,3)
п а
Принимая форму частицы за куб или сферу, можно вычислить размер коллоидной частицы по следующим формулам:
I
l=V -d' (IX-4>
— 380 —
где I — ребро куба, или
(IX,5)
где D — диаметр сферической частицы.
Ультрамикроскоп дает возможность косвенно судить о форме коллоидных частиц, хотя и весьма приближенно. Так, ровная освещенность поля зрения в ультрамикроскопе свидетельствует о том, что коллоидные частицы имеют более или менее правильную (т. е. симметрии-
Рис. 160. Прибор Доти для измерения светорассеяния: 1 — источник света; 2 — пластинка; 3 — кювета с рассеивающим раствором; 4 — пластинка из молочного стекла; 5 — фотометр Пульфриха; 6 — лимбы
Рис. 159. Молекулы нуклеиновых кислот (а) и гемоцианина (6), видимые в электронный микроскоп
ную) форму частиц (например, шар, куб, октаэдр и т.п.). «Искрящееся» или «мерцающее» поле косвенно свидетельствует о том, что коллоидные частицы имеют неправильную (несимметричную) форму частиц — палочкообразную или пластинчатую. При своем хаотическом движении эти частицы неодинаково рассеивают свет в зависимости от того, какой стороной поворачиваются они к падающему лучу. Опыт показывает: если для освещения золя в ультрамикроскопе применять не обыкновенный, а поляризованный свет, можно отличить палочкообразные частицы от пластинчатых.
Электронный микроскоп. В последние годы электронный микроскоп нашел самое широкое применение при изучении коллоидных растворов. С его помощью был полностью решен вопрос о форме коллоидных частиц и макромолекул.
Разрешающая способность лучших электронных микроскопов отечественных марок достигает 10—15 А, полное увеличение превышает 600 тысяч. С помощью электронного микроскопа были изучены размеры и форма многих лиофобных коллоидов, аэрозолей, молекул различных высокомолекулярных соединений, вирусов. На рис. 158 представлены полученные в электронном микроскопе снимки коллоидных частиц различной формы. Масштаб, указанный на этих снимках, козвдляет судить и о размерах частиц.
Возможность использования электронного микроскопа ограничена до некоторой степени необходимостью тщательно высушивать объект, так как внутри электронного микроскопа поддерживается высокий вакуум. Изучаемые образцы должны быть чрезвычайно тонкими (1 —10 мк), поскольку они сильно поглощают электроны. В настоящее время разработаны методы наблюдения в электронном микроскопе не самих образцов, а отпечатков с них (так называемых реплик), которые могут быть получены и с влажных объектов. На рис. 159 представлены полученные в электронном микроскопе снимки молекул нуклеиновых кислот и гемоцианина.
Нефелометры и нефелометрия. На явлении опалесценции и законе светорассеяния Рэлея основано действие весьма важного оптического прибора нефелометра, с помощью которого измеряют интенсивность опалесценции коллоидного раствора, а также степень мутности суспензии или эмульсии. На рис. 160 показана схема прибора Доти, предназначенного для визуального измерения светорассеяния. В этом приборе свет от источника 1 падает на рассеивающий раствор, находящийся в термостатированной кювете, й пластинкой 2 частично направляется на пластинку из молочного стекла 4, которая является стандартом «мутности». Интенсивность стандартного пучка от пластинки 4 и света, рассеянного под углом 90° в кювете 3, сравнивается в фотометре Пульфриха 5 и уравнивается с помощью лимбов 6. Отсчеты на этих лимбах А и /2 характеризуют отношение интенсивностей рассеянного света.
• В настоящее время широкое распространение получили так называемые фотоэлектрические нефелометры, в которых фотометр заменен фотоэлементами с умножителями и гальванометром, например ФЭКН-57, ФЭКН-54 и др.
Нефелометры применяются для определения степени дисперсности (размера частиц) коллоидных растворов. Этот метод основан на уравнении:
Л D2 r'l К ~D3i ~ r32 '
(IX, 6)
которое легко выводится из уравнения Рэлея (IX, 1) при постоянстве значений IQ и X для одного и того же золя с одинаковой весовой концентрацией, но с разной степенью дисперсности его частиц. Оно показывает, что интенсивность рассеяния, а следовательно, и интенсивность опалесценции и свечения конуса Фарадея — Тиндаля прямо пропорциональны кубу радиуса (размера) г частицы или обратно пропорциональны кубу степени дисперсности D. Из уравнения (IX,6) следует:
(IX,7)
Для этого метода необходимо иметь стандартный раствор (золь или суспензию) того же вещества с монодисперсными частицами, радиус которых точно известен. Это условие до некоторой степени ограничивает данный метод.
С помощью нефелометрии можно легко определить концентрацию золя. Формула для определения концентрации золя может быть легко выведена из уравнения Рэлея;
/1 _ N, V1
К N,vf
(IX,8)
' — 382 —
Лля двух золей °ДН0Г0 и того же вещества, обладающих одинаковой степенью исперсности частиц (т. е. Ух = У2) и отличающихся друг от друга только частичной концентрацией, т. е. числом частиц Д\ и N2 в известном объеме раствора, ^праведливо следующее соотношение:
Из этого соотношения выводим уравнение:
С^Сг-—, (IX,10)
*2
где Ci — искомая весовая концентрация, равная N^V^d.
Таким образом, для определения концентрации золя нефелометрическим спо-• собом необходимо иметь стандартный раствор с известной концентрацией С того же вещества, что и объект исследования.
§ 109. Молекулярно-кинетические свойства коллоидных растворов
Как показали многочисленные исследования, коллоидные системы по своим молекулярно-кинетическим свойствам принципиально ничем не отличаются от обычных (истинных) растворов, только эти свойства у золей и растворов высокомолекулярных соединений выражены значительно (в сотни и тысячи раз) слабее.
Все другие молекулярно-кинетические свойства основаны на броуновском движении.
Броуновское движение. Частицы дисперсной фазы золя под влиянием ударов молекул растворителя находятся в состоянии непрерывного хаотического движения. Так, если рассматривать какой-либо золь в ультрамикроскоп, можно заметить, что частицы золя все время беспорядочно движутся (рис. 161).
Впервые это явление было обнаружено английским ботаником Р. Броуном (1827), который подробно описал беспорядочные колебательные движения (в виде своеобразного, никогда не прекращающегося «танца»), совершаемые растительной пыльцой при рассматривании ее суспензии в воде в микроскоп. Вначале Броун считал, что эти движения присущи только живым существам, но вскоре убедился, что они свойственны любым суспензиям и эмульсиям органических и неорганических веществ при условии, что размер частиц достаточно мал (в пределах от 1 до 5 мк). Было предложено много различных гипотез для объяснения броуновского движения, но все они оказались несостоятельными.
Позднее Г у и (1888) и Э к с н е р (1900) высказали мысль о том, что броуновское движение имеет молекулярно-кинетическую природу, т. е. является следствием теплового движения. Это было впоследствии подтверждено теоретическими расчетами Эйнштейна и Смол у -ховского, а также экспериментальными работами Перрена иСведберга.
Опыты показали, что броуновское движение совершенно не зависит от природы вещества, оно изменяется в зависимости от температуры,
— 383 —
вязкости среды и размеров частиц. Под действием беспорядочных уда. ров молекул растворителя частицы дисперсной фазы также совершаю] беспорядочные движения. Перемещение в пространстве этих частщ совершается в результате усредненного действия всей совокупность ударов за время наблюдения (в 1 сек частица испытывает около 1020 уда^ ров). Число ударов, приходящихся с разных сторон, при малых раз.
Рис. 161. Броуновское дви жение частицы
мерах частиц обычно неодинаково они передвигаются в пространстве пс| сложной траектории (рис. 161). Если размеры и масса частиц дисперсной; фазы превышают определенные пределы, вероятность взаимной компенЛ сации ударов оказывается значительна но выше. Вот почему частицы разме-) ром, например, 4—5 мк совершают только небольшие колебательные движения около некоторого центра. При более крупных размерах частиц броуновское движение прекращается вовсе.
Таким образом, наблюдаемое в микроскоп смещение частицы х (рис. 161) за определенный промежуток времени является лишь статическим результатом множества смещений частицы по разным направлениям в пространстве (в их проекции в поле зрения микроскопа). Действительный путь частицы при броуновском движении (как и при молекулярном движении) проследить в ультрамикроскоп невозможно: частица за одну секунду успевает претерпеть десятки и сотни миллионов ударов молекул растворителя
и столько же раз ничтожно изменить свое направление, а человеческий глаз способен улавливать не более 10 движений в секунду и притом лишь в крупном масштабе. Это заставило в теорию броуновского движения вместо средней квадратичной скорости для газовых молекул ввести несколько иное понятие — среднее квадратичное смещение, или средний сдвиг ± Дх, как проекцию расстояния между двумя положениями частицы Л и В за время t дву х смежных наблюдений (рис. 161). Зависимость среднего смещения частицы Дх за время t от коэффициента диффузии D выражена Эйнштейном
в виде уравнения:
(IX, И)
Коэффициент диффузии для сферических частиц, значительно больших
— 384 —
по размерам, чем молекулы дисперсионной среды, выражается следующим уравнением:
(IX,12)
RT 1 D = -7----
Мп опт)/'
Уравнения (IX, 11) и (IX, 12) имеют очень большое значение в коллоидной химии, так как позволяют на основании измерений коэффициента диффузии D определить радиус взвешенных коллоидных частиц сферической формы, а также величину молекул высокомолекулярных соединений. Для частиц или макромолекул несферической формы выражение 6лт]г в уравнении (IX, 12) заменяется более сложным.
Подставляя в уравнение (IX, 11) значение D из уравнения (IX, 12), для среднего сдвига частицы получим:
(IX, 13)
Дх —
/Vo Зяту *
2RT /Vo
Изучение-броуновского движения позволило Перрену при содействии математика Ланжевена впервые экспериментально путем непосредственного подсчета в поле зрения ультрамикроскопа определить одну из наиболее важных констант — число Авогадро 2V0. Найденное им значение этого числа Л/о = 6,5 • 1026 хорошо согласуется с другими известными данными.
Таким образом, изучение броуновского движения (наряду с ультрамикроскопией) имело большое значение не только для коллоидной химии, но и для всего естествознания в целом как одно из доказательств справедливости диалектико-материалистической концепции миропонимания.
Диффузия и флуктуация. Вследствие молекулярно-кинетического движения частицы дисперсной фазы испытывают случайные смещения, например, вверх и вниз. Однако если в каком-либо растворе частицы распределены неравномерно (содержание их у дна сосуда больше, чем в верхнем слое), общее число смещений частиц снизу вверх будет больше, чем сверху вниз. При этом частицы будут передвигаться вверх до тех пор, пока не наступит выравнивание концентраций.
Самопроизвольный процесс выравнивания концентраций ионов, молекул или коллоидно-дисперсных частиц за счет их беспорядочного теплового движения (у коллоидных частиц — броуновского движения) получил название диффузии. Диффузия как самопроизвольный процесс для всех дисперсных систем подчиняется одним и тем же закономерностям, установленным Фиком для газов. Согласно первому закону Фика скорость диффузии прямо пропорциональна площади, через которую происходит диффузия, и градиенту концентрации, матически этот закон имеет следующее выражение:
dm ,, dC — = —DS—, dt dx
где dm — масса вещества (в молях или граммах), продиффундировав-шая за бесконечно малое время dt через площадь S; dC/dx — падение
13 Зак. 560
(ix,14)
— 385 —
I
(IXJ5)
концентрации на бесконечно малом отрезке диффузионного пути dx, называемое градиентом концентрации; D — коэффициент диффузии, индивидуально характеризующий диффузионную способность данной дисперсной системы. Этот коэффициент выражает количество вещества, диффундирующего в единицу времени (1 сек) через единицу площади (1 см2) при градиенте концентрации, равном единице.
С изменением концентрации в процессе диффузии величина градиента концентрации также изменяется. Поэтому необходимо знание и скорости изменения концентрации во времени, т. е. производной dCldt, что дается вторым законом Фика:
dC d2C — =D-----.
dt dx2
Основная трудность в применении обоих законов Фика до недавнего времени заключалась в определении коэффициента диффузии D. Однако трудности определения этого коэффициента для растворов и золей были преодолены после того, как Эйнштейн, изучая броуновское движение, обнаружил связь этого коэффициента со средним сдвигом Ах [уравнение (IX, 11)]. Используя закон Стокса, Эйнштейн нашел зависимость коэффициента диффузии от вязкости среды и радиуса частиц [уравнение (IX, 12)]. Диффузионный метод определения размера частиц в настоящее время дает для коллоидных растворов наиболее надежные результаты.
Изучение броуновского движения и диффузии в коллоидных системах помогло глубже вскрыть природу дисперсных систем, а также установить общность молекулярно-кинетических свойств этих систем и систем молекулярной дисперсности. Оно подтвердило реальное существование молекул и явилось убедительным обоснованием правильности материалистического мировоззрения. Дальнейшее более детальное и углубленное изучение броуновского движения привело к созданию так называемой теории флуктуаций.
Флуктуация представляет собой самопроизвольное отклонение плотности, концентрации или параметра от среднего равновесного значения в микрообъемах системы. Так, Сведберг, наблюдая явление флуктуации при подсчете числа частиц, находящихся в 1000 мк? золотого золя, нашел, что в среднем число частиц составило 1,545, но в отдельные моменты оно изменялось в пределах от 0 до 7. Отклонения можно объяснить тем, что хаотическое движение частиц приводит к случайному попаданию в выделенный микрообъем то большего, то меньшего числа частиц.
Таким образом, флуктуация представляет собой явление как бы обратное явлению диффузии, хотя оба они — результат теплового движения. Если диффузия как всякий самопроизвольный процесс должна, в соответствии со вторым началом термодинамики, идти необратимо, то флуктуация указывает на то, что второе начало термодинамики имеет статический характер, т. е. оно не применимо к отдельным» индивидуальным частицам или к малому числу их. В обоих явлениях мы видим одно из доказательств справедливости закона материалистической диалектики — единства противоположностей.
— 386 —
Осмотическое давление. Для коллоидных растворов, как и для истинных, характерно осмотическое давление. Оно подобно газовому давлению, является коллигапгивным свойством растворов, т. е. зависящим только от числа свободно движущихся коллоидных частиц.
Если учесть, что коллоидные частицы по сравнению с молекулами низкомолекулярных веществ обладают значительно большими размерами и массой, то при одной и той же весовой концентрации коллоидного и истинного растворов в единице объема золя частиц содержится значительно меньше, чем в единице объема истинного раствора. Вот почему по сравнению с последними коллоидные растворы обладают ничтожно малым осмотическим давлением.
Так, 1%-ный золь золота имеет осмотическое давление, равное 0,00045 атм, а раствор сахарозы той же концентрации и в тех же условиях 0,725 атм. Кроме того, какая-то доля измеряемого осмотического давления в коллоидных растворах (главным образом гидрофобных) обусловливается примесью электролитов.
Поскольку коллоидные растворы принципиально не отличаются от истинных растворов, к ним можно применить формулу Вант-Гоффа:
= (IX, 16)
где П — осмотическое давление золя, п — количество коллоидных частиц (в кмоль) в V л золя. Под киломолем коллоидных частиц подразумевается число Авогадро.
Если учесть, что данный золь содержит v коллоидных частиц, а 1 кмоль — Nq таких же частиц, n = vINq. С учетом этого уравнение (IX, 16) примет следующий вид:
V
ПУ = — RT,
No
откуда
Относительно малые концентрации коллоидных растворов обусловливают также ничтожно малые значения всех других величин, зависящих от числа частиц в растворе (частичной концентрации). Так, все коллоидные растворы обладают чрезвычайно малым понижением упругости пара, ничтожными (практически не поддающимися экспериментальному измерению) величинами понижения температур замерзания и повышения температур кипения. Так, понижение температуры замерзания золя золота концентрации 1 г/л при размере частиц 4 ммк равно всего 0,000004°.
Между осмотическим давлением П, числом частиц в единице объема п и средним радиусом г можно установить определенную зависимость. Как известно, весовое количество диспергированного вещества в единице объема равно 4/3irr3dn, где d — плотность раствора. Исходя из этого для'двух дисперсных систем с одинаковой дисперсионной сре-
13* . — 387 —
дои при одинаковой температуре можно записать:
4 я 4 з
— лг 1 driL = — ЛГ2 dn2 о о
.. 3 3
и г\ П1=Г2 п2,
откуда
__Г2 D1
П2 ~п2~ rf
(IX, 18)
Рис. 162. Осмометр* Геппа—Скат-чарда:
i — мембрана; 2 — коллоидный рас-шор; 3 — резервуар с растворителем;
4 — капилляр; 5 — манометр
Для отсчета берут разность
где D — степень дисперсности золя.
Таким образом, осмотическое давление коллоидных растворов обратно пропорционально кубу радиуса частиц и, следовательно, прямо пропорционально кубу степени дисперсности.
Экспериментально осмотическое давление золей измеряют в приборах осмометрах. Один из наиболее современных типов осмометров изображен на рис. 162. В этом приборе полупроницаемая мембрана из целлофана или другого материала плотно закрепляется на специальном пористом диске. Исследуемый золь (2—3 мл) находится - над мембраной, а растворитель — под мембраной, заполняя часть резервуара 3. Верхняя часть резервуара и капилляр 4 заполняются толуолом. Манометр 5 дает возможность задать практически любую наперед заданную разность давлений между коллоидным раствором и растворителем.
давлений в манометре, при которой уро-
вень толуола в капилляре остается постоянным в течение длительного промежутка времени. В этом случае осмотическое давление золя компенсируется внешним давлением. Все измерения осмотического давления необходимо производить при строго постоянной температуре. При измерении давления в манометре в сантиметрах водяного столба и при 25° С осмотическое давление вычисляется по формуле:
2,53 105С
м
(IX, 19)
где С — концентрация золя или высокомолекулярного соединения, выраженная в г на 100 мл, М. — молекулярный вес.
Так, если 1 г белка растворен в 100 мл воды (С = 1) при 25° С, молекулярный вес белка М = 10 000, то осмотическое давление П рас-
— 388 —
твора будет равно:
2,53- 1O'> • 1,0 П —----------= 2o, 3 см 11 >0.
10000
Мембранное равновесие Доннана. Более детальное изучение осмотического давления коллоидных растворов показало, что даже применение в качестве внешней жидкости ультрафильтрата этого же золя не дает результатов, которые бы точно соответствовали теоретическим. В некоторых случаях экспериментально определенное осмотическое * давление того или иного золя было больше теоретического, оно не зависело в ряде случаев от концентрации коллоида или изменялось не пропорционально концентрации его и т. п. Как показали более углубленные исследования, проведенные в свое время Доннаном, причина подобных отклонений кроется в особом равновесии электролитов, которое устанавливается в присутствии частиц (или ионов), не способных проникать через полупроницаемую мембрану.
Впоследствии само явление неравномерного распределения какого-нибудь электролита по обе стороны полупроницаемой мембраны под влиянием коллоидного электролита получило название мембранного равновесия Доннана.
Рассмотрим основные положения теории" мембранного равновесия.
Пусть имеется сосуд, разделенный на две части полупроницаемой мембраной, которая способна свободно пропускать ионы электролитов, но задерживает коллоидные частицы. В одной стороне этого сосуда помещен раствор, содержащий электролит Na+ и коллоидный анион R-, задерживаемый мембраной.
По другую сторону мембраны в этом же сосуде находится электролит NaCl, оба иона которого могут свободно проходить через мембрану. Состав растворов в сосуде в начале процесса можно представить следующей схемой:
I мембрана
R“ Na+ i ;
II
Na+Cl-
: ^2 '-'2
В этой схеме буквами С\ и С2 обозначены начальные концентрации соответствующих ионов.
По истечении некоторого промежутка времени ионы Na+ и С1~ на-. чнут свободно проходить из правой половины сосуда в левую. Причем коллоидный анион R~ все время остается в левой половине сосуда. Перемещение ионов закончится установлением динамического равновесия, т. е. количество ионов, переходящих в единицу времени в ту и другую стороны, будет одинаково. Обозначая через х количество ионов Na+ и СН, перешедших из правой половины сосуда в левую, состояние равновесия по обе стороны мембраны можно представить следующим образом:
I мембрана
Na+ Cl- : :
II Na+ Cl" (C2-x) (C2-x) — 389 -
где х не равняется 1/2С2 , как этого следовало ожидать. Распределение хлорида натрия по обе стороны мембраны не будет равномерным. Ионы, свободно диффундирующие через мембрану, должны распределиться так, чтобы произведение концентраций их в обоих отсеках сосуда было одинаковым, т. е.
(G 4-х) х = (С2—х)2-
Решая это уравнение относительно %, получим:
С22
Доля электролита NaCI, которая перешла из правой в левый, будет равна:
(IX, 20)
(IX,21)
части сосуда
(IX, 22)
Анализ уравнений (IX, 21) и (IX, 22) показывает, что если концентрация электролита велика по сравнению с концентрацией коллоида, т. е. Со Ci, то:
В этом случае электролит NaCI распределяется поровну по обе стороны мембраны. Этот вывод имеет чрезвычайно важное практическое значение, так как экспериментально определенное осмотическое давление золя наиболее близко к расчетному. Таким образом, более точные значения осмотического давления золя могут получиться при избытке электролита в ультрафильтрате.
Если концентрация электролита очень мала по сравнению с концентрацией коллоида С2 С\, то:
т. е. электролит практически не переходит из правой половины сосуда ‘в левую.
Для большей наглядности в табл. 76 приведено распределение электролита NaCI при различных соотношениях между NaCI и NaR.
Таблица 76
Распределение электролита NaCI при различных соотношениях между концентрациями NaCI и NaR
С, С£ X С2 — х X
0,01 1,00 0,497 1,01
0,10 1,00 0,476 1,1
1,00 1,00 0,380 2,0
1,00 0,10 0,083 н,о
1,00 0,01 0,10 99,0 4
~ 390 --
Из табл. 76 видно, что при С2/Сг = 100 хлорид натрия практически повну распределяется по обе стороны полупроницаемой мембраны. Если соотношение С2/С± = 0,01, то 99% хлорида натрия остается в правом отделении и только 1 % его переходит к коллоидному раствору.
Доннановское равновесие имеет очень большое значение для понимания и теоретического обоснования целого ряда явлений: осмотического давления лиофобных коллоидов и растворов высокомолекулярных соединений, отрицательной адсорбции ионов, явлений набухания, а также различных физиологических процессов.
Седиментационное равновесие. Частицы вещества, диспергированного в жидкой или газообразной среде, постоянно находятся под влиянием двух противоположно направленных сил — силы тяжести, под действием которой частицы данного вещества оседают, и сил диффузии, под влиянием которых частицы стремятся переместиться из области больших в область меньших концентраций, т.е. к равномерному распределению в объеме.
В зависимости от преобладания тех или иных сил в системе наблюдается осаждение частиц дисперсной фазы (под влиянием сил тяжести) или (в случае преобладания сил диффузии) выравнивание концентрации во всем объеме системы. Процесс оседания частиц под действием силы тяжести носит название седиментации (от лат. sedimentum — оседание). Скорость оседания частиц зависит не только от их размера, но и от разности плотностей частиц d—d0, а также от вязкости этой среды г) и математически выражается следующим уравнением:
2 г2 (d — d0) g
(IX,23)
где v — скдрость оседания частиц, г — радиус частиц, d и d0— плотности диспергированного вещества и растворителя, т] — вязкость среды, g — ускорение силы тяжести.
Из уравнения (IX, 23) видно, что с большей скоростью оседают более крупные частицы. Так, частицы серебра оседают в воде на 1 см при г = 1 • 10"2 см за 0,05 сек\ при г = I • 10~4 см — за 500 сек, а при г = 1 . Ю”6 см — за 58 суток.
Диффузия в случае более мелких частиц дисперсной фазы протекает с большей скоростью и замедляется с увеличением их размера. Если степень дисперсности вещества мала (радиус частиц больше 2 ммк), то частицы не совершают броуновского движения, следовательно, их способность к диффузии равна нулю. В данном случае сила тяжести значительно преобладает над силами диффузии.
Если в системе силы тяжести полностью уравновешены силами диффузии, наступает так называемое седиментационное равновесие, которое характеризуется равенством скоростей седиментации и диффузии. При этом через единицу поверхности сечения в единицу времени проходит вниз столько же оседающих частиц, сколько их проходит вверх с диффузионным потоком. Седиментационное равновесие наблюдается не только в коллоидных растворах, но и в молекулярно-дисперсных системах. Это раййовёсие характеризуется постепенным умень-
— 391 —
шснием концентрации частиц в направлении от нижних слоев к верхним. Распределение частиц в зависимости от высоты столба жидкости подчиняется гипсометрическому (или барометрическому) закону Лапласа в применении к золям: при увеличении высоты столба золя в арифметической прогрессии концентрация частиц убывает в геометрической прогрессии. Математически этот закон выражается в виде уравнения:
RT In —
/1=——(IX,24) Mg
где Ст — концентрация дисперсной системы на исходном уровне, С2 — концентрация этой системы на высоте ft, *714— молекулярный вес вещества, g — ускорение силы тяжести.
В табл. 77 приведены данные для системы самой разнообразной степени дисперсности.
Таблица 77
Седиментационное равновесие
Раздробленнее ьсщество
Высота, на которой частичная концентрация убыгаег в два раза, гм
Кислород 0,27
Тонкодисперсный золь зо-
лота 1,86
Золь золота средней дис-
персности 8,35
Грубодисперсный золь зо-
лота 186,0
500 000
215
2,5
2-10-5
Вначале гипсометрический закон Лапласа был выведен для молекулярно-дисперсных газообразных систем. Позднее Перрен распространил этот закон на коллоидно-дисперсные и даже на грубодисперсные системы. Работая с эмульсиями гуммигута и мастики в воде, Перрен обнаружил, что на каждые 30 мк изменения высоты столба суспензии число частиц гуммигута изменилось в два раза, т. е. точно по формуле Лапласа. Подсчитывая число частиц на разных глубинах, можно вычислить число Авогадро Мо.
В своих опытах Перрен получил значения Мо, очень близкие к полученным другими методами. Труды Перрена доказали универсальность молекулярно-кинетической теории и применимость ее к коллоиднодисперсным системам.
На определении скорости оседания частиц дисперсной фазы основаны все методы седиментационного анализа. Определив экспериментально скорость оседания частиц, можно рассчитать их размер, т. е. степень дисперсности. Размер радиуса диспер-
— 392 —
Таблица 78
Частичный (модулярный) вес белковых веществ, определенный с помощью ультрацентрифуги
Вещества Час Iочный еес Вещества Частичный I ес
Пепсин Яичный альбумин . . Гемоглобин 39 200 43 500 68 100 Фитоциан О ктоп ус - гемоцианин 279 000 2 785 000
сной частицы можно определить из уравнения (IX, 23):
d do
(IX,25)
Рис. 163. Седиментометр Фигурой-ского:
/ — кварцевый стержень; 2 — стеклянная нить; 3 — чашечка
седиментометра Фигуровского.
Поскольку оседание частиц коллоидной степени дисперсности под действием силы тяжести происходит медленно, для его ускорения применяют ультрацентрифуги, которые способны развивать центробежную силу, примерно в I 000 000 раз превосходящую силу земного тяготения. Именно при помощи ультрацентрифуги были определены размеры частиц в некоторых коллоидах, а также молекулярные веса большого числа полимерных соединений.
В табл. 78 приведены данные по определению частичного (молекулярного) веса некоторых белковых веществ, определенных с помощью метода ультрацентрифугирования.
Суспензии и эмульсии с размером частиц в интервале 1—200 мк изучаются простыми методами седиментации в так называемых седимен-тометрах. На рис. 163 показана схема
В этом приборе к упругому стеклянному (или кварцевому) стержню 1 прикреплена на стеклянной нити 2 чашечка 3, на которой по мере оседания накапливается осадок суспензии. С помощью микроскопа по специальной шкале измеряется прогиб плеча 1. В процессе оседания частиц дисперсной фазы прогиб плеча вначале увеличивается быстро,,затем все медленнее и так до полного оседания.
Результаты седиментационного анализа обычно изображают графически в виде кривой распределения, на которой по оси абсцисс наносятся значения радиуса частиц, а по оси ординат — процентное содержание частиц данной фракции, отнесенное к определенному интервалу радиусов. По данным седиментационного анализа можно, например, определять удельную поверхность порошкообразных веществ, а также наиболее вероятный радиус диспергированных частиц.
Г л а в а X
ТЕОРИЯ КОЛЛОИДНЫХ СИСТЕМ
Свойства коллоидных растворов зависят не только от степени их дисперсности, но также и от их природы. Различные коллоиды аналогично кристаллоидам могут сильно различаться по химическим свойствам. В качестве примера можно взять коллоидные растворы золота, белка и гидроксида железа (III). Как показывает опыт, химические свойства этих трех коллоидных растворов совершенно различны даже в случае одинакового размера частиц. Так, белок оседает под действием высокой температуры, но выдерживает (т. е. не выпадает в осадок) значительные концентрации электролитов.Коллоидное золото хорошо выдерживает нагревание, не осаждается кипячением, но очень чувствительно к действию электролитов. Коллоидный гидроксид железа (III), приготовленный при соблюдении определенных условий, хорошо выдерживает и нагревание, и действие электролитов.
Прежде чем приступить к изложению теории строения коллоидных частиц, необходимо отметить, что термин «коллоидная частица» носит неопределенный характер: более правильным и более точным является термин «коллоидная мицелла». Рассмотрение теории строения коллоидных мицелл рациональнее всего начать с лиофобных или (в случае водных растворов) гидрофобных коллоидных систем.
ГИДРОФОБНЫЕ КОЛЛОИДНЫЕ СИСТЕМЫ
В коллоидных растворах на границах раздела фаз возникают электрические заряды. Для понимания электрических свойств коллоидно-дисперсных систем необходимо изучение электрокинетических явлений в них, которые связаны с взаимодействием коллоидов с электролитами и наблюдаются при движении одной фазы относительно другой.
§ 110. Электрокинетические явления в коллоидных системах
Электрокинетические явления в коллоидно-дисперсных системах и связанное с этими явлениями наличие электрических зарядов у коллоидов открыл Ф. Ф. Рейсс (1808). В опытах Рейсса в кусок влажной глины 1 (рис. 164) были вставлены две стеклянные трубки 2, <3. В полученные таким способом цилиндры с глиняным дном насыпали хорошо промытый кварцевый песок 4 и налили в них до одинакового уровня воду. Опустив в цилиндры электроды от вольтова столба — одного из немногих известных в то время источников электрической энергии, включали ток. Через некоторое время под влиянием электрического поля частицы глины, отрываясь от поверхности, стали проходить сквозь песок и двигаться к положительному полюсу (аноду). Следовательно, эти частицы имели отрицательный заряд. В цилиндре с положительным электродом образовалась хорошо заметная суспен-
— 394 —
я (муть) 5. Одновременно уровень воды в цилиндре с отрицательным 1 ктродом (катодом) поднялся, а в цилиндре с положительным элек-тоодом опустился, что свидетельствовало о положительном заряде воды. р При дальнейшем изучении этих явлений обнаружилось, что они характерны для коллоидно-дисперсных систем. Движение частиц дисперсной фазы в электрическом поле к противоположно заряженному электроду получило название электрофореза.
Пытаясь определить причины поднятия уровня воды в цилиндре с отрицательно заряженным электродом, Рейсс поставил другой опыт. Он пропускал постоянный ток через прибор, состоящий из U-образной
Рис. 164. Схема установки для проведения электрофореза
Рис. 165. Схема установки для проведения электроосмоса:
7 — (J-образная трубка; 2 — кварцевый песок; 3 — вода
трубки (рис. 165), средняя часть которой была заполнена мелким кварцевым песком. В этом приборе кварцевый песок играл роль пористой диафрагмы.
После включения электрического тока уровень воды в колене с отрицательным электродом начал повышаться, а в колене с положительным электродом — понижаться. Это продолжалось до тех пор, пока разность уровней в обоих коленах не достигла определенной величины. Многочисленные опыты показали, что, как и при электрофорезе, этот процесс протекает с постоянной скоростью. Причем количество перенесенной жидкости находится в прямой зависимости от приложенной разности потенциалов и диэлектрической проницаемости и обратно пропорционально вязкости этой среды. Впоследствии явление переноса жидкости через пористые диафрагмы и узкие капилляры получило название электроосмоса.
Позднее были открыты еще два электрокинетнческих явления, противоположные (по движению фаз) электрофорезу и электроосмосу.
Так, Квинке (1859) обнаружил, что при механическом проталкивании воды через пористую диафрагму или через капилляр (что мож-
— 395 —
Рис. 166. Схема установки для обезвоживания грунтов методом электроосмоса:
1 — глубинный насос; 2 — скважина со вставленным в нее металлическим фильтром; 3 — генератор постоянного тока; 4 — металлический стержень
но осуществить, применяя давление) на противоположных сторонах этой диафрагмы (или на концах капилляра) появляется разность потенциалов, препятствующая протеканию жидкости. Это явление получило название эффекта протекания, или эффекта истечения, а возникающая разность потенциалов — потенциала протекания.
В 1878 г. Д о р н обнаружил другое электрокинетическое явление, которое заключалось в возникновении скачка потенциалов при механическом передвижении твердых частиц в жидкой фазе (в опытах Дорна—при оседании крупинок песка в воде). Указанное явление обратно электрофорезу и получило название эффекта седиментации, а возникающий скачок потенциала — потенциала седиментации.
Таким образом, приложенная извне э. д. с. вызывает движение фаз: 1) электрофорез, если в движение приходит твердая фаза по отношению к неподвижной (в целом) жидкой фазе; 2) электроосмос, если приходит в движение жидкая среда по отношению к неподвижной твердой фазе. С другой стороны, механическое передвижение фаз вызывает э. д. с. : 1) потенциал седиментации, если сообщается движение твердой дис
персной фазе по отношению к неподвижной жидкой; 2) потенциал протекания, если сообщается движение жидкой фазе по отношению к неподвижной твердой фазе (поверхности).
В настоящее время явления электрофореза и электроосмоса широко используются в технике и производстве.
Электрофорез применяется вфарфорово*м производстве для выделения из суспензий глин чистого каолина. Наиболее мелкие отрицательно заряженные частицы каолина после тщательного взмучивания в воде осаждаются на вращающемся свинцовом барабане, заряженном положительно. Построение примеси в виде положительно заряженных частиц Fe2O3, а также более крупные частицы каолина уносятся проточной водой.
С помощью электрофореза проводят покрытие различных изделий тонким стоем каучука из латекса. В этом случае отрицательно заряженные частицы латекса движутся в электрическом поле к аноду (покрываемый предмет) и осаждаются на нем. За последние годы метод электрофореза нашел широкое применение в получении оксикатодов в радиолампах.
Очень большое распространение получил метод электрофореза в борьбе с топочными дымами и производственными пылями, а также для улавливания в заводских трубах наиболее ценных отходов производства. По методу инженера Коттреля, в заводских трубах устанавливают специальные металлические стержни, на которые подается отрицательное напряжение (в десятки тысяч вольт). Частицы дыма и пыли имеют одноименный заряд со^ стержнем и отбра-
L
ываются от него с силой на внутреннюю поверхность трубы. Аналогичным способом очищаются и газы от всевозможных примесей.
Методы электроосмоса применяются в фильтрпрессах для обезвоживания различных пористых материалов, например торфа, а также для пропитки пористых материалов (например, древесины).
В последнее время электроосмос применяют для понижения уровня грунтовых вод, а также для осушения в анодной зоне грунтов, особенно глинистых. Так, он был применен при строительстве Куйбышевской гидроэлектростанции. На рис. 166 представлена схема установки для обезвоживания грунтов методом электроосмоса. Частицы коллоидно-дисперсных грунтов заряжены, как правило, отрицательно. Если в такой грунт ввести два металллических электрода, один из которых (отрицательно заряженный) опустить в специально пробуренную скважину, вода под действием электрического поля начнет перемещаться в скважину, откуда ее можно откачивать глубинным насосом.
Применяется электроосмос и в сельскохозяйственном производстве. В частности, сделана интересная попытка использовать электроосмотическую подачу воды к лемеху плуга в целях уменьшения трения между плугом и почвой при пахоте. Благодаря этому, как показали исследования, эффект уменьшения трения достиг примерно 80%.
Электроосмос применяется также при производстве кирпича для смачивания проволоки, режущей глиняные заготовки. При этом проволоку соединяют с отрицательным, а брусок глины — с положительным полюсом.
Детальное исследование электрокинетических явлений коллоиднодисперсных систем позволило сделать ряд общих выводов.
1. Все золи по знаку заряда из дисперсной фазы при явлениях электрофореза и электроосмоса могут быть разделены на положительно и отрицательно заряженные. Положительный заряд дисперсных частиц имеют гидрозоли таких гидроокисей, как Fe (ОН)3, А1 (ОН)3, а также водные растворы основных красителей (метиленовый синий, метиленовый зеленый, основной фуксин) и др. Отрицательный заряд частиц дисперсной фазы имеют гидрозоли золота, серебра, платины, а также водные растворы кислых красителей (эозин, флуоресцеин, кислый фуксин).
2. Электрофорез и электроосмос в золях не являются процессами односторонними. Оба они представляют собой единство двух противоположных процессов.
3. При наличии определенных условий во многих случаях коллоидные частицы в золях могут перезаряжаться, т. е. менять свой знак заряда на обратный.
4. Величина и знак заряда, которые несет на себе коллоидная частица, также не остаются постоянными: они меняются в зависимости от концентрации самого золя и от концентрации (а также от природы) построенных ионов, присутствующих в золях.
§ 111. Возникновение двойного электрического слоя и его строение
, На основании изучения электрокинетических явлений в коллоидных системах было установлено, что у поверхности коллоидных частиц на границе раздела фаз образуется двойной электрический слой и возникает скачок потенциала. Это обусловлено тем, что ионы одного знака необменно адсорбируются на поверхности адсорбента, а ионы противоположного знака в силу электростатического притяжения рас
— 397 —
полагаются около нее. Причем величина и знак заряда поверхности зависят от природы твердых частиц адсорбента и от природы жидкости, с которой он соприкасается.
Кроме того, заряд на поверхности частиц дисперсных систем может образоваться в результате электролитической диссоциации молекул поверхности твердой фазы, в результате чего ионы одного знака остаются в фиксированном положении на этой поверхности (т. е. кристаллической решетке), а ионы противоположного знака (противоионы) поступают в прилегающий раствор. Подобным образом возникает двойной электрический слой в растворе целого ряда высокомолекулярных соединений.
Впервые представление об образовании двойного электрического слоя было высказано Квинке (1859) и развито в работах Гельмгольца (1879). По их мнению, двойной электрический слой представляет собой подобие плоского конденсатора, одна обкладка которого находится в твердой фазе, другая — в растворе. Толщина такого конденсатора имеет порядок молекулярного радиуса. По Гельмгольцу, образование двойного электрического слоя происходит следующим образом. Сначала на поверхности частиц адсорбируется преимущественно один из ионов, который и сообщает поверхности свой знак заряда. Далее под воздействием электростатических сил притяжения противоионы (или компенсирующие ионы) стремятся расположиться возможно ближе к ионам, адсорбированным на поверхности коллоидных частиц. В результате образуются два слоя ионов, из. которых один расположен на поверхности, другой — в растворе, на расстоянии молекулярного радиуса (рис. 167, /). Такая система ионов (в целом нейтральная) получила название двойного электрического слоя по Гельмгольцу.
С развитием теории электролитической диссоциации и введением понятия об ионах появилась теория, развитая в работах Гуи (1910), согласно которой двойной электрический слой имеет диффузное строение. Дело в том, что под воздействием двух взаимно противоположных сил (электростатического притяжения и теплового движения частиц жидкости) противоионы образуют около твердой поверхности адсорбента (коллоидной частицы) диффузную ионную атмосферу (рис. 167, //). Причем концентрация противоионов, наибольшая около заряженной поверхности твердой фазы, убывает по мере увеличения расстояния от границы раздела фаз по направлению внутрь раствора.
В дальнейшем теория двойного электрического слоя получила свое развитие в работах Штерна (1924), который учел, что ионы имеют вполне определенные размеры, и центры их не могут подойти к заряженной поверхности ближе, чем на расстояние ионного радиуса. По Штерну, только часть ионов находится на молекулярном расстоянии от поверхности, образуя гельмгольцевский плоски?! конденсатор (рис. 167, III), другая часть образует диффузный двойной слой. В разбавленных растворах структура двойного электрического слоя приближается к структуре слоя Гуи, а при повышении концентрации — к слою Гельмгольца. Таким образом, структура двойного слоя зависит не от механизма возникновения зарядов на поверхности коллоидных частиц (воз-
— 398 —
пикают ли они-путем избирательной адсорбции ионов из раствора или путем диссоциации ионогенных групп молекул), а от плотности расположения зарядов на ней.
Если поверхность не проводит ток и заряды (т. е. адсорбированные ионы или ионогенные группы) расположены редко, вокруг каждого заряда (согласно теории Дебая — Гюккеля) в растворе возникает ионная атмосфера. Если же заряды на поверхности коллоидной частицы расположены плотно и тем более если поверхность проводит ток, в этих условиях за счет обобществления ионных атмосфер отдельных зарядов образуется структура двойного электрического слоя.
Рис. 167. Двойной электрический слой:
/ — по Гельмгольцу; 11— по Гуи; ///—по Штерну (вверху — расположение ионов, внизу — кривые падения потенциала)
Дальнейшее развитие диффузная теория двойного электрического слоя получила в трудах советских ученых А. Н. Фрумкина и Б. В. Дерягина.
Согласно современной теории двойного электрического слоя получили объяснение электрокинетические и электрокапиллярные явления, а также проблемы строения и устойчивости коллоидных частиц лиофобных золей. Согласно этой теории при относительном движении жидкой и твердой фаз плоскость скольжения их лежит на некотором расстоянии от твердой фазы (рис. 168, линия тп). Слой жидкой фазы толщиной в 2—3 молекулы при движении фаз остается неподвижным вместе с твердой фазой. Иными словами, непосредственно у поверхности коллоидной частицы золя образуется так называемый адсорбционный слой, который включает не только потенциалопределяющие ионы (знак которых противоположен знаку твердой фазы), но и часть противоионов, которые в обычных условиях считаются неподвижными и при движении твердой фазы перемещаются вместе с ней. Ос-
399 —
тальная
часть
противоионов составляет
диффузный слой,
в кото-
ром концентрация ионов (по мере удаления от поверхности коллоидной частицы) постепенно убывает.
Неподвижный адсорбционный слой содержит далеко не все противо-
ионы, а лишь определенную их часть, которая не в состоянии целиком компенсировать заряд твердой поверхности, а способна лишь его понизить.^ Иными словами, в адсорбционном неподвижном слое в результате взаимодействия положительных и отрицательных зарядов остается ненейтрализованным некоторый потенциал, который является частью общего потенциала поверхности твердой фазы.
Рис. 168. Схема двойного электрического слоя: а — расположение зарядов; б — кривая падения потенциала
Разность потенциалов между подвижной (диффузной) и неподвижной (адсорбционной) частью двойного электрического слоя называется электрокинетическим потенциалом. Этот потенциал обычно обозначают греческой буквой g (дзета) и потому называют дзета-потенциалом (д-потенциал).
Полное падение потенциала от его значения на поверхности MN до нулевого значения (рис. 168, 6) соответствует максимальной разности потенциалов между твердой поверхностью и всеми противоионами, вместе взятыми. Эту максимальную разность потенциалов называют термодинамическим потенциалом и обозначают греческой буквой 8 (ЭПСИЛОН).
Как видно из рис. 168, б, электрокинетический потенциал составляет лишь часть термодинамического потенциала е:
£ = еь
где ег — падение потенциала в неподвижном слое, вызываемое наличными в нем адсорбированными противоионами.
— 400
Вычисление электрокинетического потенциала и определение» его знака на практике производят, пользуясь данными электрофореза и Q электроосмоса, а также из потенциалов течения, по следующей формуле:
<x.t>
где К — постоянная, зависящая от формы коллоидно-дисперсных частиц (для малых сферических частиц К = 6, для частиц цилиндрической формы К = 4); т| — вязкость дисперсионной среды, D — диэлектрическая постоянная, 8 — градиент напряжения поля, и — средняя скорость передвижения частиц под действием электрического поля.
Рис. 169. Падение электрических потенциалов мицеллы:
О А — максимальный электродинамический потенциал на поверхности ядра мицеллы; АВ — падение потенциала в адсорбционном слое; DB — электро-кинетический потенциал (£-по-тенциал) на поверхности гранулы; ВС — падение потенциала в диффузном слое
Как видно на рис. 169, величина дзета-потенциала тесно связана с толщиной диффузного слоя противоионов. Обычно чем больше размыт этот слой, тем больше величина потенциала. Если слой противоионов предельно сжат, т. е. вовсе не размыт, дзета-потенциал равен нулю. Дзета-потенциал определяется толщиной диффузного слоя про-тивоинов, следовательно, его величина находится в обратной зависимости от концентрации электролитов, присутствующих в растворе. Увеличение концентрации электролитов влечет за собой уменьшение толщины диффузного слоя и, как следствие, уменьшение дзета-потенциала. Согласно закону действия масс повышение концентрации электролита способствует понижению концентрации противоионов в диффузном слое. При этом часть противоинов переходит из диффузного в адсорбционный слой, в результате дзета-потенциал уменьшается. Наоборот, разбавление золя способствует увеличению толщины диффузного слоя за счет перехода противоионов из адсорбционного слоя. Таким образом, дзета-потенциал очень чувствителен к посторонним электролитам. Причем, влияние на него оказывают и ионы, имеющие заряд обратного знака.
Влияние постороннего иона на величину дзета-потенциала тем сильнее, чем больше заряд иона.
Как показали многочисленные опыты, влияние природы иона на дзета-потенциал возрастает в гораздо большей степени, чем заряд ионов. Так, для ионов К+> Ва2+, А13+заряды относятся, как 1 : 2 :3, а кон
г~ 401 —
центрации, производящие одинаковое действие, относятся, как 800 : 25 : 1.
Знак электрокинетического или дзета-потенциала зависит от химической природы твердой фазы. Кислые вещества (кремниевая кислота, мастика, таннин, сульфиды металлов, сера) в водном растворе имеют, как правило, отрицательный заряд.
Особенно ясно выступает зависимость знака дзета-потенциала твердой фазы от ее химического характера при рассмотрении групп кислотного (карбоксильные) и основного (амины) характера.
§ 112. Мицеллярная теория строения коллоидной частицы
Советские ученые А. В. Думанский, Н. Н. Песков, С. М. Липатов, А. Н. Фрумкин, а также зарубежные ученые Веймарн, Паули, Фаянс, Кройт на основе теории двойного электрического слоя создали так называемую мицеллярную теорию строения коллоидных частиц. Первоначально представление о мицеллярном строении частиц распространялось на все системы, изучаемые коллоидной химией, в том числе и на лиофильные золи. Однако последующие исследования показали, что лиофильные золи, или, точнее, растворы высокомолекулярных и высокополимерных соединений, - имеют другое, весьма отличное от лиофобных золей строение. В ’настоящее врёмя мицеллярная теория строения коллоидных частиц сохраняет свое значение только для лиофобных (гидрофобных) золей.
Всякий лиофобный (гидрофобный) коллоидный раствор состоит из двух частей: мицелл и интермицеллярной жидкости. Мицеллы — это отдельные коллоидные частицы, которые в совокупности составляют дисперсную фазу золя, а интермицеллярная жидкость — это дисперсионная среда того же золя, включающая в себя, помимо среды-растворителя, все другие растворенные в ней вещества (электролиты и неэлектролиты), которые непосредственно не входят в состав мицелл.
Коллоидная мицелла имеет значительно более сложное строение, чем обычные молекулы. В ней различают две основные части: внутреннюю — нейтральную, обычно называемую ядром, и внешнюю — ионогенную, в свою очередь состоящую из двух слоев (двух ионных сфер).
Ядро составляет основную массу коллоидной мицеллы и представляет собой комплекс, состоящий из атомов (в случае гидрозолей металлов или неметаллов) или нейтральных молекул (в золях гидроксида железа (III) или иодида серебра). Общее число входящих в состав ядра атомов или молекул огромно (от нескольких сот до миллионов) и зависит от степени дисперсности золя и от размеров самих атомов или молекул.
На первом этапе развития*коллоидной химии считали, что коллоидные частицы имеют аморфную природу. Однако рентгенографические исследования доказали (как в свое время указывал И. Г. Борщев) критическое строение ядра мицеллы.
В процессе образования гидрофобного золя рост ядра в той или иной стадии может быть приостановлен созданием так называемого
- 402 —
адсорбционного слоя из ионов стабилизатора. Ионная сфера вокруг ядра коллоидной мицеллы состоит из двух слоев (или двух сфер) — , адсорбционного и диффузного. Адсорбционный слой слагается из слоя потенциалопределяющих ионов, адсорбированных на поверхности ядра и сообщающих ему свой заряд, и части противоионов, проникших за плоскость скольжения и наиболее прочно связанных электростатическими силами притяжения. Вместе с ядром эта ионная атмосфера образует как бы отдельный гигантских размеров многозарядный ион — катион или анион, называемый гранулой. Диффузный слой, расположенный за плоскостью скольжения, в отличие от адсорбционного не имеет в дисперсионной фазе резко очерченной границы. Этот слой состоит из противоинов, общее число которых равняется в среднем разности между всем числом потенциалопределяющих ионов и числом противоионов, находящихся в адсорбционном слое.
Гранула вместе с диффузным слоем противоинов составляет коллоидную частицу — мицеллу. Мицелла всегда электронейтральна. Необходимо отметить, что противоионами могут служить любые ионы тех электролитов, которые участвуют в реакциях при образовании данного золя или же присутствуют как посторонние примеси.
Для примера рассмотрим получение гидрозоля иодида серебра методом конденсации. Оно основано на химической реакции
AgNO3 + KI=AgI + KNO3
Согласно теории строения коллоидной мицеллы ядро в данном случае будет состоять из нейтральных молекул Agl. В зависимости от относительной концентрации реагирующих веществ может быть три случая.
1. Концентрация нитрата серебра больше концентрации иодида калия. При этом в системе, помимо нерастворимого комплекса Agl имеются ионы Ag+, К+ и NO з. В процессе роста ядра коллоидной мицеллы достраивание решетки Agl идет только за счет ионов Ag+, которые прочно входят в его структуру, сообщают ему электрический заряд и потому называются потенциалопределяющими. Полученный в результате адсорбции ионов Ag+ электрический заряд определяет термодинамический потенциал.
Положительно заряженное ядро притягивает оставшиеся в растворе противоионы NO3. Часть противоинов при этом входит в состав адсорбционного слоя, оставшаяся часть — в состав диффузного слоя. На рис. 170 изображена схема постепенного образования мицеллы гидрозоля Agl. z
2. Концентрация нитрата серебра меньше концентрации иодида калия. В этом случае поверхность ядра адсорбирует только анион 1~, который входит в его структуру и определяет знак заряда.
3. Концентрации нитрата серебра и иодида калия равны. В этом случае золь находится в так называемом изоэлектрическом состоянии, t. е. в состоянии, при котором электрокинетический потенциал (дзета-потенциал) равен нулю. В этом случае противоины диффузного слоя перешли в адсорбционный слой, а потому гранула лишена заряда.
- 403 -
Гис. 170. Схема постепенного образования мицеллы гидро-. золя Aql:
а — ультрамикрокристалд Agl; б — достраивание ультрамикрокристалла ядра за счет избирательной адсорбции из раствора ионов Ag+ (стадия образования адсорбционного слоя); в — образование гранулы (завершение образования адсорбционного слоя); г — образование мицеллы
Строение мицелл в коллоидной химии принято выражать особыми мицеллярными формулами. Так, в обобщенном и упрощенном виде строение любой мицеллы гидрофобного золя можно передать следующими формулами:
{т [ядро] нК+ (п—х) А~}+ хА”'
положительно заряженная мицелла,
{т [ядро] пА~ (п—х) К+}~ хК+
отрицательно заряженная мицелла,
где К+ — катионы, А“ — анионы, п — число потенциалопределяющих ионов, х — число ионов, находящихся в диффузном слое, т — число нейтральных атомов или молекул в ядре.
В этих формулах в квадратные скобки заключено ядро, а в фигурные — заряженный комплекс (гранула).
— 404 — |
Мицеллярная формула иодида серебра для первого случая (когда CAgNO3 > Cki) примет вид:
{m [Agl] nAg+ (и — х)) + xNO“
Во втором случае (когда Casno3 < Cki) формула запишется так: {m[AgI]nI~(n— х)К+}~хК+
В третьем случае, когда концентрации обоих электролитов равны, мицеллярная формула будет иметь вид:
{ т [Agl] nAg+ nNO^j0
Рассмотрим примеры образования и строения мицелл различных золей.
Подвергая гидролизу разбавленный раствор FeCl3, можно получить коллоидный раствор гидроксида железа (III). Реакция гидролиза протекает по уравнению
FeCl3 4- ЗН2О = Fe (ОН)3 4- ЗНС1
Ионным стабилизатором здесь является FeOCl, образующаяся по уравнению:
Fe (ОН)3 4- НС! = FeOCl 4- 2Н2О
Молекула стабилизатора диссоциирует по уравнению
FeOCl;z±FeO+4-Cl-
Таким образом, ядро коллоидной мицеллы гидроксида железа Fe(OH)3 состоит из большого числа молекул Fe(OH)3. Потенциалопреде-ляющим ионом является FeO+, так как ион С1“ не входит в состав ядра. Исходя из этого мицеллярная формула золя гидроксида железа (III) может быть изображена следующим образом:
(tn [Fe (ОН)3] nFeO+ (п — х) С1~} + хС1~
На рис. 171 приведено схематическое изображение мицеллы гидроксида железа (III).
Золь кремниевой кислоты относится к группе отрицательно заряженных золей. Ядро мицеллы состоит из скопления молекул кремниевой кислоты, часть которых, находящаяся на поверхности ядра, диссоциирует на ионы по уравнению
H2SiO3^±2H+4-SiO2-
причем ионы SiOi”, как прочно связанные с ядром, являются потен-циалопределяющими. Роль компенсирующих ионов (противоионов) выполняют ионы водорода, которые распределяются как в адсорбционном, так и в диффузном слоях. Схематически строение коллоидной мицеллы этого золя можно изобразить формулой:
{m[H2SiO3] nSiOj~2(n—х) Н+} ~2хН+
На рис. 172 определена схема строения мицеллы золя кремниевой кислоты.
‘— 405 —
В табл. 79 указан знак заряда мицеллярного остова (гранулы) в гидрозолях различных веществ.
Таблица 79
Знак заряда мицеллярного остова в гидрозолях различных веществ
Золи, заряженные положительно Золи, заряженные отрицательно
Гидроксиды: железа, кадмия, алюминия, хрома, церия, циркония, тория титана. Титановая кислота.
Основные красители: метиленовая синь, метиленовая фиолетовая, ночная голубая и др.
Золото, серебро, платина, сера. Сульфиды: мышьяка, меди, сурьмы, свинца, кадмия.
Кислоты: кремниевая, оловянная.
Пятиокись ванадия. Мастика, гуммиарабик. Крахмал, пектин, танин. Кислые красители: эозин, фуксин, бензопурпурин, красное конго и др.
Рассмотренная выше мицеллярная теория строения гидрофобных коллоидов дает возможность наглядно представить схему движения гранулы и противоионов при электрофорезе. На рис. 173 дана такая схема для положительно заряженных золей.
(У
Рис. 173. Схема электрофореза: а — мицелла вне электрического поля; б — мицелла в электрическом поле
Форма коллоидных частиц может быть самой разнообразной. Изучение строения и процесса образования коллоидных частиц с помощью электронного микроскопа позволило установить, что их форма во многом определяется типом кристаллической решетки ядра, которую образуют входящие в него атомы и молекулы. В зависимости от природы вещества решетка может быть как симметричной, так и асимметричной.
От формы частиц дисперсной фазы зависят некоторые свойства коллоидных растворов. Так, коллоидные системы асимметрического строения способны образовать внутреннюю сетчатую структуру; На рис. 174 и 175 приведены полученные на электронном микроскопе снимки коллоидных частиц свежеприготовленного золя сульфида мышьяка и пятиокиси ванадия. Как видно из этих рисунков, частицы пятиокиСи
l _ 407 —
Рис. 174. Коллоидные частицы свежеприготовленного золя сульфида мышьяка
Рис. 175. Иглообразные формы коллоидных частиц пятиокиси ванадия
ванадия обладают нитевидной иглообразной формой. Причем нити, переплетаясь, образуют своеобразную структуру. Именно этим и объясняется тот факт, что золь пятиокиси ванадия легко переходит в гель уже при обычной температуре.
§ 113. Электрокинетический потенциал золей и методы его измерения
Опыты показали, что в случае гидрофобных коллоидов прибавление электролита к коллоидному раствору почти не влияет на величину термодинамического потенциала, тогда как на величину электро-кинетического потенциала (дзета-потенциала) оно оказывает сильное влияние.
Благодаря чувствительности к изменению концентрации электролитов в золе электрокинетический потенциал играет большую роль для характеристики состояния различных гидрофобных систем. Во всех электрокинетических явлениях (электроосмос, электрофорез) ему принадлежит ведущая роль. Любые изменения величины и знака электро-кинетического потенциала могут привести к существенным изменениям в золях, вплоть до разрушения коллоидного раствора с выпадением дисперсной фазы в осадок (процесс коагуляции). Таким образом, дзета-потенциал является одним из важнейших факторов устойчивости гидрофобных золей. Опыт показывает, что чем больше величина этого потенциала, тем более устойчива коллоидная система.
— 408 —
I
Экспериментально определяют величину электрокинетического потенциала ,сТодом электрофореза или методом электроосмоса.
к Сущность первого метода заключается в непосредственном измерении ско-пости передвижения коллоидных частиц под действием постоянного электрического поля в специальном приборе — электрофоретической трубке. На рис. 176 показан один из наиболее простых электрофоретических приборов. Стеклянная трубка аппарата на всем протяжении и в кранах А и At имеет совершенно одинаковый просвет. Фигурно изогнутые концы трубки служат для погружения агаровых сифонов, присоединяемых к источнику электричества через промежуточные сосуды.
Исследуемый золь наливают в аппарат при открытых кранах А и Лгв таком количестве, чтобы его уровень оказался выше кранов. Затем краны закрывают и жидкость, оставшуюся выше кранов, удаляют. Далее поверх кранов (остающихся закрытыми) наливают так называемую надстилающую жидкость (обычно ультрафильтрат того же самого золя). Открыв кран В, автоматически получают одинаковый уровень жидкости в обоих коленах трубки. Перед тем как включить ток, крап В закрывают, а краны А и Аг открывают. Передвижение дисперсной фазы исследуемого золя измеряют по миллиметровой шкале, нанесенной прямо на трубке. Зная время действия и величину приложенной разности потенциалов, а также расстояние между электродами и смещение дисперсной фазы, получают скорость перемещения, рассчитанную на падение потенциала в 1 в/см.
Величину электрокинетического потенциала вычисляют по формуле (X, I). Выражая дзета-потенциал в милливольтах, е — в вольтах на 1 см, D — в милликулонах в секунду и К = 4, получим
£=1,1295 105-^. (Х,2)
Опыт показывает, что величина электрокинетического потенциала, рассчитанная по формулам (X, 1) или (X, 2), для большинства гидрофобных золей составляет около 30—40 ме. Однако имеются и высокозаряженные золи с величиной потенциала 60—70 мв. ,
Согласно уравнению (X, 1) отношение tju не зависит от размера коллоидных мицелл, однако это справедливо в том случае, если размер частиц велик по сравнению с толщиной двойного электрического слоя. Для подавляющего большинства гидрофобных золей это условие выполняется. Совершенно иную картину наблюдаем в случае высокомолекулярных соединений, например белка, радиус молекул которого сравним с толщиной двойного слоя. В этом слое электрофоретическая подвижность зависит, как показывает опыт, не только от формы, но и от размеров коллоидно-дисперсных частиц.
Метод определения величины и знака электрокинетического потенциала на основе электроосмотических явлений применяется преимущественно для исследования материалов, которые трудно (или невозможно) получить в растворенном или высокодисперсном состоянии. На рис. 177 изображен прибор Гортикова для электроосмоса, широко применяемый для определения величины и знака почвенных коллоидов. На дно пробирки помещают почву и уплотняют ее на центрифуге. Перед уплотнением в пробирку с почвой наливают воду или почвенный раствор, затем закрывают ее резиновой пробкой, через которую проходит стеклянная трубка, доходящая до самого дна пробирки. После уплотнения на центрифуге через второе отверстие в пробку вставляют еще одну трубку, конец которой находится над образцом почвы. Каждая из трубок имеет по два отростка: длинный отросток заполняется агар-агаром, приготовленным на насыщенном растворе КО; короткий представляет собой капиллярную трубку. В ней и наблюдают движение мениска жидкости, которое объясняется скольжением жидкой фазы коллоидной системы на границе коллоидной гранулы и диффузного слоя.
Длинные отростки трубок опускают вместе с электродами в стаканчики с раствором соли, затем электроды соединяют с источником постоянного тока. Чем больше заряд почвенных коллоидов, тем быстрее передвигается жидкость в капиллярных трубках. Скорость передвижения жидкости (объем жидкости,перенесенный в секунду) тем больше, чем выше величина дзета-потенциала почвенных коллои-
409 —
Рис. 176. Простейший прибор для определения скорости электрофореза
Рис. 177. Прибор для определения заряда коллоидов электроосмосом:
/ — вода или почвенный раствор; 2 — почва; 3 — агар-агар с КС!; 4 — электроды; 5 — раствор соли
дов. Связь между объемом жидкости, перенесенным в 1 сек (V), и и дзета-потен циалом выражается уравнением:
где i — сила тока (ма)\ D — диэлектрическая постоянная среды, ц — коэффициент внутреннего трения, х — удельная электропроводность, £ — электрокинетически й потенциал (мв).
Отсюда
4лТ|хУ хпУ
£-—^— = 1,1295-10^-^. (Х,4)
Dt . Di
В большинстве случаев почвенные коллоиды заряжены отрицательно, очень редко положительно. Чаще всего их дзета-потенциал составляет 30—40 мву т. е. ниже критического (критическим потенциалом называется минимальный потенциал, при котором коллоиды существуют в форме золей). Начиная с критического потенциала и при более низких его значениях, происходит коагуляция золей.
Величина дзета-потенциала является важной характеристикой почвенных коллоидов, так как имеет большое влияние на химические и физические свойства почвы. При высоком потенциале силы отталкивания между коллоидно-дисперсными частицами настолько велики, что при достаточной влажности почвенные коллоиды находятся в форме золя. При низких же значениях потенциала силы притяжения берут верх и коллоиды коагулируют, образуя гели.
Исследования дзета-потенциала на границе твердая фаза — жидкость путем электроосмоса (или потенциала протекания) позволяют сделать весьма важные вцводы о состоянии поверхности различных твердых тел. В частности, для различных грунтов и почв доказано, что характер обмена ионов на поверхности, а также ряд других важных свойств почв — способность к набуханию, свертыванию, агрегативная устойчивость и др. — тесно связаны с наличием определенного заряда на поверхности частиц, образующих коллоидную фракцию И’Очв.
410 —
§ 114. Вязкость гидрофобных золей
*
Измерение вязкости золей имеет большое значение при изучении природы коллоидных растворов. Вязкость гидрофобных золей, как показали многочисленные исследования, зависит только от концентрации и не зависит от способа их приготовления. Поскольку концентрация гидрофобных коллоидов обычно очень мала, разница между вязкостью чистой дисперсионной среды и вязкостью золя незначительна и лежит в пределах ошибки наблюдений. В тех случаях, когда есть возможность приготовить гидрофобные золи сравнительно больших концентраций, наблюдается возрастание вязкости золя с концентрацией.
Первая попытка теоретического выяснения зависимости вязкости золей от их концентрации принадлежит Эйнштейну. Рассматривая золь как суспензию, состоящую из твердых шаров, которые во взвешенном состоянии находятся в вязкой жидкости, он вывел следующую математическую зависимость:
-Л- = 1+2,5<р, (Х,5)
По
где г] — вязкость золя, — вязкость чистой дисперсионной среды, ср — отношение объема дисперсной фазы к общему объему дисперсной систем я.
Поскольку в уравнение (X, 5) не входит радиус частицы, вязкость золя должна находиться в прямой зависимости от его концентрации.
Многочисленные проверки показали, что уравнение Эйнштейна применимо в тех случаях, когда отсутствует заметное взаимодействие между мицеллами, т. е. коллоидные растворы достаточно разбавлены.
Это уравнение не применимо для золей, у которых может быть достигнута сравнительно высокая концентрация, а также для золей, у которых частицы сильно сольватированы. Сольватация способствует увеличению эффективного объема коллоидных частиц, уменьшая тем самым объем свободного растворителя. Для учета данного явления в уравнение Эйнштейна вводят величину ф — отношение объема дисперсной фазы к объему свободного растворителя, в результате чего уравнение принимает вид:
— = 14-2,5* (Х,6)
По
илч
— = 14-2,5—^—. (Х.7)
По 1~ <Р
Исследования показали, что относительная вязкость гидрофобных золей находится в прямой зависимости от величины из электрокинети-ческого потенциала.
Увеличение относительной вязкости гидрофобных золей в связи возрастанием дзета-потенциала объясняется тем, что при этом воз
— 411 —
растает концентрация противоионов, а следовательно, и сольватация (гидратация), что вызывает увеличение объема коллоидных мицелл.
Таким образом, между относительной вязкостью и радиусом коллоидных частиц существует обратная зависимость: при одинаковых концентрациях гидрофобного золя вязкость растворов, содержащих частицы меньших размеров, как правило, бывает больше, чем вязкость растворов, содержащих частицы больших размеров.
§115. Устойчивость гидрофобных золей
В одних коллоидных системах нарушение
Коллоидные растворы сравнительно мало устойчивы во времени по сравнению с молекулярными растворами. Мицелла представляет собой агрегат более или менее простых молекул, характерный для данного золя только в данный момент и для совершенно определенных условий. Под влиянием различных факторов (температуры, света, электричества, изменения концентрации, механического воздействия, присутствия ничтожно малых количеств посторонних примесей), а иногда даже и без видимых причин, в коллоидных системах протекает ряд своеобразных необратимых процессов, приводящих к изменению частиц дисперсной фазы и их выпадению в осадок. Изменение свойств коллоидной системы, происходящее в результате самопроизвольного процесса укрупнения частиц и уменьшения их числа в единице объема, называется старен нем.
устойчивости происходит сравнительно быстро, другие системы могут сохраняться годами и даже десятилетиями без видимых изменений.
Между коллоидными частицами действуют две взимно противоположные силы — притяжения и отталкивания. Под действием силы притяжения происходит слипание частиц, совершающих броуновское движение. Эти силы носят преимущественно характер молекулярного взаимодействия (так называемые ван-дер-ваальсовы силы). Силы отталкивания определяются электрическим взаимодействием между ионами двойных электрических слоев, окружающих каждую коллоидную частицу. Эти силы препятствуют сближению частиц и их соединению. В зависимости от того, какие силы преобладают в данной системе, наблюдается или коагуляция (при перевесе сил притяжения), или более высокая устойчивость (если больше силы отталкивания).
В принципе все коллоидные системы с термодинамической точки зрения являются неустойчивыми системами; практически*можно говорить об их относительной устойчивости, так как процессы, нарушающие устойчивость, мбгут в различных коллоидных системах протекать с неодинаковой скоростью.
Н. П. П е с к о в (1922) ввел в науку о коллоидах понятия кинетической и агергативной устойчивости.
, Под кинетической устойчивостью понимается способность дисперсных частиц удерживаться во взвешенном состоянии под влиянием броуновского движения. Кроме броуновского движения, факторами кинетической устойчивости являются дисперсность, вязкость дисперсионной среды, разность плотностей дисперсной фазы и дисперсионной среды и некоторые другие. Из всех перечисленных факторов наи
— 412 —
большее влияние на скорость осаждения коллоидных частиц оказывает степень дисперсности вещества. Чем меньше размер частиц, тем выше устойчивость системы. Системы, в которых скорость осаждения взвешенных частиц под влиянием силы тяжести настолько мала, что ею можно пренебречь, принято называть кинетически устойчивыми.
Под агрегативной устойчивостью понимают способность частиц дисперсной фазы оказывать сопротивление их слипанию и тем удерживать определенную степень дисперсности. Потеря агрегативной устойчивости приводит к взаимному слипанию коллоидных частиц с образованием более крупных агрегатов. Агрегативная устойчивость объяс-
няется наличием у коллоидных частиц одноименных зарядов, которые мешают им соединяться в более крупные частицы, а также наличием вокруг ядра коллоидных мицелл сольватных оболочек, состоящих из прочно связанных молекул растворителя. В настоящее время установлена прямая зависимость между толщиной (плотностью) сольватных (гидратных) оболочек и величиной дзета-потенциала и агрегативной устойчивостью данной коллоидной си-
Рис. 178. Схема уменьшения удельной поверхности в дисперсных системах:
а диссолюции; б — перекристаллизация
стемы.
Б. П. Дерягин (1945) разработал теорию устойчивости и коагуляции лиофобных (гидрофобных) золей, согласно которой сольватные (гидратные) оболочки вокруг ядра коллоидных мицелл, обусловленные сольватацией (гидратацией) ионов в диффузном слое, обладают упругими свойствами. Упругие силы жидких сольватных оболочек, препятствующие слипанию частиц, получили, по Б. П. Дерягину, название .расклинивающего давления. Это название подчеркивает, что упругие сольватные прослойки между сближенными твердыми поверхностями действуют механически, как бы расклинивая поверхности.
Таким образом, факторы агрегативной устойчивости отличны от факторов кинетической устойчивости. Это отличие состоит в том, что если повышение температуры препятствует осаждению коллоидных частиц, оно способствует агрегированию их. Если повышение интенсивности броуновского движения препятствует осаждению частиц, то слипание их происходит при столкновении в броуновском движении.
Характерной особенностью коллоидных растворов является то, что размеры их частиц могут с течением времени самопроизвольно из- ч меняться: они могут уменьшаться — процесс диссолюции (рис. 178, а) или укрупняться — процесс перекристаллизации (рис. 178, б).
Сущность процесса диссолюции состоит в неполном растворении частиц дисперсной фазы при сохранении их общего числа. Это способствует уменьшению удельной поверхности дисперсной фазы, снижению общего запаса свободной поверхностной энергии и, как результат, повышению кинетической устойчивости золя.
— 413 —
При перекристаллизации происходит укрупнение коллоидных частиц с одновременным уменьшением их числа. В качестве примера перекристаллизации может служить «созревание» фотографических эмульсий, т. е. укрупнение частиц AgBr, а также перекристаллизация суспензий BaSO4 в аналитической химии. Опыт показывает, что явление перекристаллизации в основном характерно для дисперсных систем, взвешенные частицы которых обладают способностью растворяться в дисперсионной среде.
РАСТВОРЫ ВЫСОКОМОЛЕКУЛЯРНЫХ СОЕДИНЕНИЙ
(ЛИОФИЛЬНЫЕ СИСТЕМЫ] , z
В предыдущих разделах была рассмотрена большая группа кол-
лоидных систем, которые обладают сильно развитой физической поверхностью раздела и большим избытком свободной поверхностной энергии. В этих системах, объединенных под общим названием лиофоб-
ных (гидрофобных) коллоидов, благодаря избытку поверхностей энергии образуются ионные и молекулярные адсорбционные слои, которые и сообщают агрегативную устойчивость коллоидным частицам. Стремление свободной поверхностной энергии лиофобных (гидрофобных)
коллоидов к самопроизвольному уменьшению в силу второго начала термодинамики делает их термодинамически неустойчивыми. Весьма характерным свойством этих коллоидных систем является, как известно, слабое взаимодействие между веществами дисперсной фазы и молекулами дисперсионной среды.
Однако коллоидная химия изучает и другие высокодисперсные системы—растворы высокомолекулярных соединений белков, целлюлозы, каучука, которые на заре развития коллоидной химии получили название лиофильных (гидрофильных) золей и были причислены к типичным коллоидам, так как обладают общими свойствами, характерными для коллоидных систем. К этим свойствам относятся:
1) своеобразное тепловое движение частиц растворенного вещества, аналогичное броуновскому движению;
б) очень малые, как и в типичных коллоидных растворах, скорости диффузии;
3) неспособность частиц проходить через полупроницаемые мембраны (диализ и ультрафильтрация);
4) сравнительно небольшая величина осмотического давления даже при значительной весовой концентрации;
5) более медленное протекание в растворах высокомолекулярных соединений целого ряда физических и химических процессов по сравнению с низкомолекулярными веществами;
6) повышенное стремление к образованию разнообразных молекулярных комплексов;
7) способность соединений- коагулировать и пептизироваться под влиянием внешних факторов.
Степень дисперсности частиц дисперсной фазы коллоидных и высокомолекулярных соединений также близка: для типичных золей она
— 414 —
составляет 105—107 см-1, для растворов высокомолекулярных соединений — примерно 106 —107 см-1.
Однако позднее, благодаря работам советских ученых В. А. Карги н а, С. М. Л и п а т о в а, зарубежных исследователей М е й е р а, М ар к а и др. было установлено, что лиофильные золи на самом деле не являются золями, а представляют собой истинные растворы высокомолекулярных соединений, т. е. системы гомогенные, молекулярно-или ионно-дисперсные. В растворах этих соединений взвешенными частицами являются не мицеллы (как в случае лиофобных коллоидов), а гигантских размеров макромолекулы. Вот почему термин «золь» для этих растворов является принципиально неправильным и употребляется он в настоящее время исключительно в силу традиции, поскольку термин «лиофильные золи» получил очень широкое распространение в литературе.
§ 116. Общая характеристика растворов высокомолекулярных соединений
К высокомолекулярным соединениям относится обширный класс веществ, основным признаком которых является высокий молекулярный вес (от 10000 до нескольких миллионов) и, следовательно, большой размер молекул.
Как показывает опыт, свойства высокомолекулярных соединений, а также их растворов определяются не только химическим составом, но и размерами, и формой макромолекулы. От величины и формы молекул соединений зависят прочность, гибкость, эластичность, устойчивость к многократным деформациям и ряд других важнейших технических свойств изделий, получаемых из них, при сравнительно невысокой плотности.
Все высокомолекулярные соединения делятся на две группы: природные (натуральный каучук, естественные смолы, целлюлоза, белки, крахмал, камеди) и искусственные (искусственные смолы, различные пластические массы, производные целлюлозы, синтетические каучуки). Иногда высокомолекулярные вещества подразделяются не на две, а на три группы: природные, искусственные и синтетические. В группу синтетических соединений входят все полимеры, полученные путем синтеза низкомолекулярных веществ (капрон, найлон, полиэтилен). К числу искусственных высокомолекулярных веществ относятся соединения,"получаемые в результате химической обработки природных высокополимерных соединений (в большинстве случаев это производные целлюлозы).
С геометрической точки зрения все разнообразие форм макромолекул высокомолекулярных соединений может быть сведено в основном к трем типам: линейной, двухмерной (или плоскостной) и пространственной, т. е. трехмерной.
Линейными называются такие полимеры, макромолекулы которых существуют в виде длинных неразветвленных цепей. Строение линейной макромолекулы можно схематически представить так:
____м —М —М-М-М-М—М— ...
415 —
где М — мономерная единица.
Такой тин строения макромолекул имеют многие высокомолекулярные органические соединения, в которых основной связующей единицей является четырехвалентный атом углерода.
Очень часто линейные цепи дают боковые ответвления и тогда они называются разветвленными. В этом случае макромолекулу можно отнести к типу двухмерных полимеров. Схематически разветвленную цепь макромолекул можно представить так:
м
I м I
.--м —М —М —М —М-^М —
I 1
м м
I I
м м
В более редких случаях однородные линейные макромолекулы под влиянием особых факторов (температуры, различных добавок) могут вступать друг с другом в тесную химическую связь, устанавливающуюся в отдельных участках цепи по ее длине в виде своеобразных «мостиков» и «перемычек»:
I I I I I
_м—М-М—М-М —
— м — М — М — М — м —
I I I I I
_М_М—М—М—М— I I I I I
_ М-М —М—М—М —
Такие макромолекулы получили в литературе название «сшитых» макроструктур. Поскольку «сшивание» идет в пространстве, образующиеся макромолекулы носят трехмерный характер.
В качестве примера можно указать на то, что гибкую линейную форму имеют молекулы многих синтетических и природных полимеров, натурального и некоторых видов синтетического каучука, полиэтилена, полихлорвинила, найлона, капрона, энанта. Двухмерную конфигурацию макромолекул имеют крахмал, дивиниловые каучуки, некоторые полисахариды. Трехмерной структурой макромолекул обладает эбонит, фенолоформальдегидные смолы.
Растворы высокомолекулярных соединений, несмотря на общность некоторых свойств с истинно коллоидными лиофобными растворами, имеют свои специфические особенности. Эти отличия связаны тремя общими термодинамическими признаками, характеризующими любой истинный раствор: 1) самопроизвольностью образования растворов ВМС; 2) высокой степенью их устойчивости; 3) обратимостью происходящих в них процессов.
— 416 —
Всякий лиофобный (гидрофобный) золь получается только искусственным путем за счет приложенной извне работы (химической или механической). Именно за счет этой работы золь и обладает большим избытком свободной поверхностной энергии, которая стремится к уменьшению, чем и обусловлена его агрегативная неустойчивость. I Растворы высокомолекулярных соединений могут образоваться самопроизвольно в результате неограниченного набухания, переходящего далее в обычное растворение, в результате чего происходит не увеличение, а убыль свободной энергии.
Эта важная особенность высокомолекулярных соединений объясняется весьма большой способностью молекул взаимодействовать с дисперсионной средой, что собственно и явилось причиной для употребления термина лиофильность. Именно с лиофильностью связаны и свойства большой сольватируемости и растворимости высокомолекулярных соединений по сравнению, например, с рассмотренными ранее гидрофобными коллоидами. Эта особенность и обусловливает довольно резкие различия между лиофобными золями и растворами ВМС. Если лиофобные золи могут существовать без видимых изменений только в очень незначительных концентрациях и поэтому обладают вязкостью, мало отличной от вязкости чистой дисперсионной среды, и проявляют свои диффузионные и осмотические свойства в ничтожной степени, то растворы высокомолекулярных соединений могут длительно существовать в достаточно ощутимых молярных концентрациях, следовательно, обладают заметным осмотическим давлением и повышенной вязкостью.
Лиофобные золи являются системами агрегативно неустойчивыми, растворы же высокомолекулярных соединений способны сохранять свою молярную концентрацию очень долгое время, т. е. являются системами термодинамически устойчивыми. Это экспериментально подтвердил В. А. К а р г и н, который показал, что растворы ВМС подчиняются, как и истинные растворы, правилу фаз.
И, наконец, в тесной и неразрывной связи со всем вышеизложенным находится и третий признак растворов ВМС — это обратимость всех совершающихся в них процессов с изменением температуры, давления и концентрации. Напомним, что все эти процессы являются необратимыми для лиофобных (гидрофобных) коллоидов.
Высокомолекулярные соединения способны образовывать не только истинные растворы, но и типичные лиофобные золи, если в качестве дисперсионной среды взята жидкость, по отношению к которой высокомолекулярное вещество является лиофобным. Такие коллоидные растворы отличаются ясно выраженной лиофобностью, что выражается в слабом взаимодействии вещества дисперсной фазы с дисперсной средой, требуют обязательного наличия стабилизатора для создания агрегативной устойчивости, обладают слабой диффузией и очень малым осмотическим давлением. Так же как и лиофобные золи, коллоидные растворы ВМС обладают термодинамической неустойчивостью, вызванной значительным избытком поверхностной свободной энергии.
Таким путем можно получить типичные лиофобные золи из желатины и спирта, из нитроцеллюлозы и воды.
14 Зак. 560
— 417 —
§117. Набухание и растворение высокомолекулярных соединений
Процесс растворения высокомолекулярных соединений своеобразен и отличается от растворения низкомолекулярных веществ. Растворению полимера предшествует его набухание. Оно характерно для всех высокомолекулярных соединений и никогда не наблюдается у низкомолекулярных веществ. В настоящее время благодаря работам В. А. Каргина и С. М. Липатова установлен механизм набухания. Он* сводится в основном к двум различным процессам: к процессу сольватации и процессу распределения в полимере низкомолекулярной жидкости.
Рис. 179. Схема последовательных стадий (/—4) взаимного растворения высокомолекулярного соединения и низкомолекулярной жидкости
Весь процесс растворения высокомолекулярных соединений можно условно разделить на четыре стадии. Как видно из рис. 179, в первой (начальной) стадии система состоит из двух компонентов: полимера и низкомолекулярной жидкости. Вторая стадия растворения характеризуется процессом набухания, в результате которого молекулы низкомолекулярной жидкости начинают интенсивно проникать в погруженный в нее полимер, образуя с ним прочные сольватные комплексы (студни).
Студень, по существу, — насыщенный раствор низкомолекулярной жидкости в полимере и его можно рассматривать как систему, состоящую из пространственной сетки, из частично взаимосшитых макромолекул и из молекул растворителя, которые заполняют пространства между макромолекулами. В студне дисперсной фазой служит растворитель, молекулы которого распределены в растворяемом ВМС как в дисперсионной среде.
Таким образом, набухание есть проникновение молекул растворителя в среду высокомолекулярного соединения и связанное с этим увеличение его массы и объема_£рис. 179, 2).
Третья стадия растворения ВМС характеризуется тем, что по мере дальнейшего набухания объем полимера и расстояния между макромолекулами настолько увеличиваются, что отдельные макромолекулы начинают отрываться друг от друга и переходить в слой низкомолекулярной жидкости (рис. 179, 3).
г X , ) - 418 -
В последней (четвертой) стадии растворения молекулы высокомоле-лярного вещества равномерно распределяются в силу диффузии по ' вс ему объему низкомолекулярного растворителя, образуя истинный раствор (рис. 179, 4).
Опыт показывает, что интенсивность набухания и растворения полимеров зависит от их физического состояния. Так, наиболее легко набухают и растворяются полимеры, находящиеся в высокоэластичном или вязко-текучем состояниях. Значительно медленнее и труднее растворяются полимеры, находящиеся в стеклообразном состоянии. В этом случае процесс растворения, как правило, начинается с поверхностного набухания, которое затем постепенно и очень медленно переходит в объемное набухание. Еще более трудно растворяются полимеры, находящиеся в кристаллическом состоянии. Их растворение е подавляющем большинстве случаев достигается лишь при нагревании.
Различают неограниченное и ограниченное набухание.
Неограниченное набухание — это набухание, которое в конечном итоге заканчивается растворением полимера. В качестве примера неограниченного набухания можно назвать растворение белка в воде или каучука в бензине.
Ограниченное набухание — это набухание, которое не доходит до стадии растворения. В этом случае полимер поглощает низкомолекулярную жидкость, но сам в ней не растворяется или растворяется очень мало, образуя студень. В качестве примера ограниченного набухания можно назвать набухание желатина в воде при комнатной температуре. При нагревании желатин полностью растворяется. Опыт показывает, что ограниченным набуханием обладают полимеры, которые имеют своеобразные «мостики», т. е. химические связи между макромолекулами. Такие мостики не позволяют молекулам полимера отрываться друг от друга и переходить в раствор. Кроме того, пространственная сетка, образованная такими макромолекулами, служит •своеобразной мембраной, через которую могут проникать лишь молекулы растворителя (при невозможности диффузии макромолекул). Опыт показывает, что если связь между макромолекулами у полимера прочная, полимеры, обладающие ограниченным набуханием при низких температурах, могут набухать неограниченно при высоких температурах, как, например, агар-агар или желатин.
Необходимо отметить, что до сего времени пока не удалось установить зависимость между природой растворителя и его способностью к растворению высокомолекулярного соединения. Пока пользуются правилом, установленным на практике: подобное растворяется в подобном. Так, полярные полимеры хорошо набухают в полярных жидкостях, а неполярные — в неполярных.
Скорость набухания полимеров зависит от целого ряда факторов: давления, температуры, pH среды, присутствия посторонних электролитов, степени измельчения и возраста (свежести) полимера.
В прямой зависимости находится скорость набухания и от степени измельчения. Чем выше эта степень, тем больше поверхность соприкосновения полимера с растворителем, а следовательно, и больше воз-14* — 419 —
можность проникновения молекул растворителя в данный полимер^ Опыт показывает, что чем свежее (моложе) полимер, тем больше скорость и степень его набухания.
Способность полимеров к набуханию в различных жидкостях и при различных условиях с количественной стороны может быть охарактеризована степенью набухания, под которой понимают отношение массы поглощенной низкомолекулярной жидкости к массе полимера до набухания:
где а — степень набухания; т0 — масса полимера до набухания; т — масса набухшего полимера.
Иногда степень набухания выражают в процентах, тогда уравнение (X, 8) принимает вид:
a = (Х,9>
тп
Всякий полимер, увеличиваясь в объеме при набухании, оказывает вполне определенное давление на стенки сосуда, ограничивающие полимер. Это давление набухания. В ряде случаев давление набухания достигает иногда десятков и даже сотен атмосфер. Давление набухания люди издавна использовали, в частности древние египтяне при откалывании каменных глыб при постройке знаменитых пирамид пользовались давлением набухания древесины. Для этой цели клинья из сухого дерева забивали в трещины скал и в специально проделанные отверстия, а затем поливали водой; древесина, набухая, разрывала скалу. Аналогично этому проводят свою разрушительную работу нежные корешки растений, дробя крепчайшие горные породы.
Опыт показывает, что набухание полимеров сопровождается выделением тепла. Так, при набухании 1 г сухого желатина выделяется 5,7 кал, а 1 г крахмала — 6,6 кал. Тепловой эффект, сопровождающий набухание полимера в жидкости, получил название теплоты набухания.
Различают интегральную и дифференциальную теплоту набухания.
Интегральной теплотой набухания называется то общее количество-тепла, которое выделяется при набухании 1 г сухого полимера до его полного насыщения или до какой-то определенной концентрации образовавшегося студня.
Дифференциальной теплотой набухания называется то количество тепла, которое выделяется при поглощении 1 г жидкости сухим или набухшим полимером.
Экспериментально интегральная теплота набухания определяется в специальных калориметрах, а дифференциальная теплота определяется путем соответствующего расчета из интегральных теплот.
Опыты показали, что теплота набухания зависит от природы полимера и от природы растворителя. Например, набухание 1 г ацетилцеллюлозы в трихлорэтане сопровождается выделением 11,4 кал, &
— 420 —
бензиловом спирте — лишь 8,2 кал. Определение теплоты набухания Вчеиь важно для характеристики степени сольватации (гидратации) высокомолекулярных соединений.
Набухание имеет очень большое значение не только в природе, в жизнедея-ельности человека, а также во многих производствах. Так, прорастанию зерна всегда предшествует его набухание. Целый ряд физиологических процессов, таких как сокращение мышц, образование опухолей, имеет в своей основе явления набухания. Способность кожи и волокнистых веществ растягиваться при набухании и сокращаться при высыхании широко используется в кожгалантерейном производстве при изготовлении обуви, одежды и других изделий. В производстве различного рода клеящих веществ (столярного клея, гуммиарабика, крахмального клейстера) явление набухания также играет главную роль. Кулинарная обработка большей части продуктов питания — муки, круп, овощей, мяса и т. п. сводится в основном к процессу набухания.
Не менее важное значение имеет набухание в производстве целлюлозы щелочными способами, а также в производстве пироксилиновых порохов. В качестве примера из области технологии неорганических веществ можно назвать процесс затвердевания (схватывания) Цемента. Здесь набухающим высокополимером является силикат кальция.
Начальный этап самого акта пищеварения — это тоже в известной мере процесс набухания, сопровождающийся действием механических и химических факторов, которые усиливают скорость и степень набухания.
§ 113. Свободная и связанная вода в коллоидах
Л1олекулы воды сами по себе электронейтральны. Однако стоит только поместить дипольные молекулы воды во внешнее электрическое поле, как тотчас начнет проявляться дипольный характер этих молекул. Во внешнем электрическом поле диполи воды ориентируются определенным образом в направлении электрических силовых линий.
Аналогично этому гидратация гидрофильных коллоидов обусловливается электростатическими силами, т. е. за счет электрических зарядов, возникающих вследствие ионизации. На поверхности коллоидных частиц высокомолекулярных веществ образуются оболочки, состоящие из диполей воды, ориентированных в зависимости от знака заряда ВМС своим положительным или отрицательным концом.
Те слои диполей воды, которые расположены в непосредственной близости к поверхности коллоидной частицы (макромолекуле), наиболее прочно с ней связаны и наиболее упорядоченно ориентированы.
Таким образом, в гидрофильных коллоидах, т. е..в растворах высокомолекулярных соединений, какая-то часть воды оказывается прочно связанной с коллоидными частицами и вместе с ними участвует в броуновском движении, другая же часть играет роль среды, в которой находятся коллоидные мицеллы.
В набухших полимерах (студнях) различают два вида воды: связанную (или гидратационную) и свободную (или капиллярную). Количество связанной воды в полимере зависит от его гидрофильности. Опыт показывает, что чем выше гидрофильные свойства полимера, тем больше содержит он связанной воды. Например, содержание связанной воды в желатине вдвое превышает массу сухого вещества.
Исследования многих ученых показали, что свойства связанной воды довольно резко отличаются от свойств свободной воды. По сте
14В Зак. 560
- 421
пени упорядоченности структуры связанная вода приближается к свойствам твердого тела и имеет бдльшую плотность по сравнению с водой свободной. Исследования А. Раковского (1931) показали, что плотность связанной воды на поверхности, например, набухшего крахмала колеблется в пределах 1,28—2,45. Диэлектрическая постоянная ее равна 2,2, вместо 81, что обусловливает ее пониженную способность растворять электролиты и полярные неэлектролиты. Исследования показали, что гидратационные оболочки высокомолекулярных соединений не обладают растворяющими свойствами, поэтому высокомолекулярное вещество растворяется только в свободной воде.
Используя свойство связанной воды, Гортнер и Ньютон разработали криоскопический метод определения связанной воды. Чрезвычайно простой и остроумный метод определения ее был, предложен в свое время А. В. Д у м а н с к и м, который для этой цели использовал методы рефрактометрии и поляриметрии.
* ’Х
Упорядоченность молекул воды в гидратационных оболочках, уплотненность ее обусловливает и еще одно замечательное свойство связанной воды, которое имеет большое значение для биологов и агрономов. Связанная вода при охлаждении раствора ВМС не замерзает, тогда как свободная вода подвержена замерзанию. Протоплазма животных и растительных организмов представляет собой сложнейшую систему, состоящую из высокомолекулярных соединений, поэтому вполне понятно то огромное значение, которое играет свободная и связанная вода в живой клетке.
Вопросы морозостойкости сельскохозяйственных культур тесно связаны с вопросом о связанной воде. Первоначально полагали, что причина гибели растений от пониженных температур заключается в механическом повреждении протоплазмы кристалликами образующегося льда. Однако последующие исследования показали, что механизм действия низких температур на растение гораздо сложнее: низкие температуры губительны для растения не сами по себе, а в результате их обезвоживающего действия при вымораживании воды.
Микроскопические исследования показали, что на первой стадии замораживания кристаллы льда образуются не внутри клеток, а в межклеточных пространствах. Разрастающиеся кристаллы начинают интенсивно оттягивать воду из клеток, что в конечном итоге приводит к обезвоживанию протоплазмы и резкому увеличению концентрации клеточного сока. Однако даже в полностью убитых морозом растениях клеточные стенки остаются практически неповрежденными.
Обезвоживание протоплазмы и действие повышенной концентрации электролитов клеточного сока вызывает необратимую коагуляцию протоплазмы.
Многочисленные исследования показали, что переохлаждение растений, при котором не образуются кристаллы льда, довольно легко переносится ими, причем растения выдерживают такие низкие температуры, которые их неизбежно погубили бы, если бы началось образование кристаллов льда. Однако ряд факторов способствует тому, что некоторые культурные растения сравнительно легко перезимовывают, вынося зачастую очень низкие температуры. Одним из этих факторов является, как уже отмечалось в учении о растворах, понижение точки замерзания тканевых и клеточных соков благодаря тому, что в них растворены различные электролиты и неэлектролиты. В частности, исследования показали, что в растениях под влиянием низких температур увеличивается содержание глюкозы за счет процессов гидролитического распада крахмала. Кроме того, глюкоза оказывает определенное защитное действие на клеточные белки, предохраняя их от преждевременной коагуляции.
Но самым важным фактором, защищающим культурные растения от вымораживания, является наличие в клетках связанной воды. Она прочно удерживается высокомолекулярными соединениями, в первую очередь белками. Опыт показывает, что морозоустойчивость того или иного культурного растения находится в прямой зависимости от соотношения свободной и связанной воды в нем.
— 422 —
Морозостойкость культурных растений не следует рассматривать как постоянное, раз навсегда данное, свойство. Исследования показали, что агроном в известных пределах может сознательно регулировать морозостойкость растений путем соответствующей их закалки.
Озимые злаки, выросшие в тепле, вымерзают быстрее, чем выросшие на холоде. При постоянном и постепенном снижении температуры растения все больше закаляются, приобретая высокую морозостойкость. Вот почему неожиданные ранние морозы причиняют большие повреждения озимым культурам.
Связанная вода в значительной мере лишена той подвижности, которая свойственна обычной воде. Многие белковые студни при содержании ничтожно малого количества сухого вещества имеют полутвердый характер и обладают способностью сохранять свою форму. Так, медузы, тело которых содержит всего лишь 1% сухого вещества и около 99% воды, тем не менее сохраняют и форму и достаточную жизненную стойкость.
Считается установленным, что одна из причин старения организма заключается в потере способности его тканей удерживать связанную воду на нормальном уровне. Как правило, молодые организмы содержат связанной воды значительно больше, чем старые.
Особый вид старения, например, наблюдается в процессе черствения хлеба. В свежей пшеничной муке связанной воды содержится примерно 44% от общего ее содержания, в тесте количество ее достигает уже 53%, в свежеиспеченном хлебе — 83%. Однако уже через пять суток в хлебе остается всего лишь 67% связанной воды. Таким образом, процесс черствения хлеба обусловлен потерей воды и является, по существу, необратимым процессом старения. Вот почему попытка сохранить хлеб свежим путем хранения его в герметической упаковке, например в целлофановых пакетах, не дает положительных результатов. Хлеб при этом быстро «запотевает» и покрывается плесенью, причем он все равно черствеет. Опыт показывает, что наиболее приемлемый метод сохранения хлеба свежим — хранение его при повышенной температуре (около 60° С). При этом белки значительно дольше сохраняют в себе связанную воду и хлеб остается свежим в течение шести-семи дней. На этом принципе основан старинный русский способ освежения черствого хлеба путем смачивания и последующего выдерживания в подовой печи.
§ 119. Вязкость высокомолекулярных соединений
Вязкость гидрофобных коллоидов весьма мало отличается от вязкости дисперсионной среды, причем для этих коллоидов существует пропорциональная зависимость между вязкостью и концентрацией коллоида, что математически выражается уравнением Эйнштейна (X, 5) или (X, 6). Как показали исследования, уравнение Эйнштейна оказывается совершенно непригодным для высокомолекулярных соединений, так как с увеличением концентрации вязкость их растворов непропорционально увеличивается. Причем в области небольших концентраций вязкость растворов МВС растет сначала медленно, а затем очень быстро.
Непропорциональный рост вязкости свидетельствует об увеличении объема дисперсной фазы растворов ВМС за счет гидратации. Г. Штаудингером была предложена формула, устанавливающая зависимость вязкости раствора ВМС от его концентрации и молекулярной массы частиц:
2^2-=КМ Мт, (Х,Ю)
*10
где М — молекулярная масса частиц, т — масса растворенного полимера, Км — константа (порядка 10~4).
14В*
— 423 —
В отличие от гидрофобных коллоидов вязкость растворов высоко-! молекулярных соединений зависит от их способа приготовления и меЯ няется со временем. Опыт показывает, что с течением времени обычнД относительная вязкость увеличивается. Далее вязкость растворов BM(j зависит от температуры: при повышении температуры вязкость их! быстро уменьшается. 1
Исследование высокомолекулярных соединений типа белков пока-1 зало, что минимум вязкости наблюдается в изоэлектрической точке,! в сильнокислой и сильнощелочной области. Максимум вязкости при-1 ходится на точку, соответствующую ионизации максимального числа! ионогенных групп, т. е. максимум вязкости соответствует максимуму! электропроводности растворов ВМС. !
Вязкость растворов высокополимеров зависит от присутствия по-f сторонних электролитов. При прибавлении электролитов вязкость I растворов ВМС вначале падает, затем практически не меняется.
Опыт показывает, что изменение вязкости в значительной мере за-1 висит от валентности ионов, противоположных заряду коллоидной ча- • стицы. Так, если золь агар-агара заряжен отрицательно, эффект из- менения его вязкости будет вызываться положительно заряженными ионами — катионами.
Опыт показывает, что с увеличением валентности катионов относительная вязкость агар-агара при данной концентрации электролитов уменьшается.
Помимо температуры, концентрации и побочных электролитов на величину вязкости растворов ВМС влияет также и давление. Вязкость обычных жидкостей не зависит от давления, причем истечение их начинается при любом, даже очень малом давлении. Истечение же растворов ВМС начинается лишь после того, как давление достигнет определенной величины. Объясняется это тем, что частицы ВМС, обладая, как правило, удлиненной формой, преграждают путь слоям движущейся жидкости и нарушают правильное течение их. Повышенное внешнее давление вызывает ориентацию частиц параллельно потоку, т. е. преодолевает образующиеся внутри жидкости структуры из макромолекул.
§ 120. Белки как коллоиды
Все животные и растительные ткани состоят из различных химических соединений: белков, углеводов, жиров и витаминов. И хотя все эти вещества необходимы для нормального развития организма, наибольшее значение имеют белки. Именно они служат той основной материей, из которой состоят все части отдельной клетки и целого организма. Белки являются высшей ступенью развития материи и с ними неразрывно связаны все неисчислимо многообразные проявления жизни, начиная с простейших функций самых примитивных существ и кончая сложнейшими функциями человеческой деятельности.
Строение белка. Различают белки простые и сложные. Простой белок в настоящее время рассматривается как продукт поликонденсации аминокислот, т. е. как природный полимер. Сложные белки состоят из простого белка и небелковых компонентов — углеводов, липидов, нуклеиновых кислот и других соединений.
— 424 —
Общим для всех белков является то, что их макромолекулы состоят из многих сотен звеньев, соединенных между собой так называемой пептидной связью имеющей строение
|| | или —СО—NH —
О Н
П своей природе каждое звено — остаток одной из аминокислот. Они образуют ол и пептидные цепи, из которых каждая содержит десятки и даже сотни остатков различных аминокислот.
Аминокислотами называются органические кислоты, у которых водород, входящий в состав радикала (остатка углеводорода), замещен аминогруппой NH2- Простейшей аминокислотой является гликоль, или глицин, с эмпирической формулой
(Н
I zNH2
н—С\соон
Есе другие аминокислоты, входящие в состав белка, относятся к «-аминокислотам, у которых аминогруппа NH2 связана с тем же атомом углерода, с которым связана и карбоксильная группа СООН. Все а-аминокислоты (за исключением гетероциклических) могут быть выражены общей формулой
/NH2
R<
ХСООН
где R — углеводородный радикал.
Аминокислоты отличаются друг от друга не только величиной, но и числом входящих в них групп NH2 и СООН, а также наличием в их составе атомов других элементов, таких, как S, Br, I. В настоящее время открыто около 26 различных аминокислот, входящих в состав белков. Примерно половина из них содержит лишь по одной группе NH2 и СООН и является простыми, или моно-аминокислотами.* Другие содержат две группы СООН на одну группу и обладают явно выраженными кислыми свойствами. Третья группа аминокислот обладает явно выраженными основными свойствами, так как содержит одну группу СООН на две группы NH2. Кроме того, в состав белков входят нескрлько циклических аминокислот, более сложных по составу и структуре их радикала R.
Впервые наиболее простая схема строения белка была дана Фишером (1906) в виде полипептидной цепочки.
Реакции поликонденсации протекают по следующему уравнению
HaNRiCOOH 4- H2NR2COOH H2NRiCONHR2COOH ф-Н2О
Именно с помощью полипептидной связи идет дальнейшее образование полимеров белков любой сложности. По мере увеличения числа аминокислотных звеньев в молекулах полипептидов возрастает и количество возможных изомеров. Так, английский биохимик Ричард Синдж подсчитал, что белок с молекулярным весом 3400 (сравнительно короткоцепочечный), в каждой молекуле которого содержится 288 аминокислотных остатков, а в состав входит лишь 12 аминокислот, может иметь совершенно астрономическое число изомеров — 10 300. Если бы можно было собрать воедино лишь по одной молекуле каждого из возможных изомеров этого гипотетического белка, то общий вес этих молекул составил бы Ю280 граммов. Поскольку вес нашей Земли исчисляется значительно меньшей цифрой — 1027 граммов, совершенно очевидно, что реально в природе существует лишь несколько изомеров этого белка.
Количество белков, встречающихся в природе, чрезвычайно велико. В настоящее время в чистом виде выделено несколько сот различных белков. Причем исследования показали, что общее число белков, отличающихся друг от друга по своим химическим свойствам, в любом живом организме достигает нескольких тысяч. Часто даже внешне совершенно идентичные белки из разных источников не
— 425 —
всегда оказываются одинаковыми по своему составу и структуре. Итак, все многообразие форм жизни на нашей планете обусловлено существованием нескольких миллионов отдельных белков, "каждый из которых в своем роде уникален.
Помимо полипептидной теории строения белков, существует и другая точка зрения, впервые выдвинутая в 1923 г. Н. Д. 3 е л и н с к и м и В. С. Стадниковы м. Согласно этим представлениям аминокислотные остатки в белковых молекулах соединены между собой не только в виде цепей, но и в виде колец — циклических ангидридов — дикетопиперазинов
NH
R—НС С=О
О=С СН—R
ЬШ
Белковая макромолекула представляет собой очень сложный комплекс. Повторяющимися звеньями его являются так называемые микромолекулы, в состав каждой из которых входит пептидная цепочка из 4—6 аминокислот и циклическое образование (дикетопиперазиновое кольцо).
Таким образом, белки по составу представляют собой сложные полипептиды, а по строению — высокополимеры (высокополиконденсаты).
Как показали многочисленные исследования, не только аминокислоты имеют в полипептидной цепи каждая свое место, но и сами цепи в белках расположены в строго определенном порядке. Достаточно нарушить расположение цепей в молекуле, как белок изменится и погибнет. Наряду с пептидной связью внутри белковой молекулы имеются и другие виды связи, в частности водородная. В результате этого макромолекула белка в своей основе приобретает характер очень прочного образования.
Многочисленными исследованиями было установлено, что все белки по форме их макромолекул могут быть подразделены в основном на две группы — фибриллярные и глобулярные.
Фибриллярные, или волокнистые, белки (от латинского слова fibrilla — волокно) состоят из макромолекул в виде тонких вытянутых нитей, обычно соединенных между собой. В эту группу входят белки, являющиеся составными частями кожи и сухожилий (коллаген, желатина), волоса и рога (кератин), мышц (миозины) и др. В организме они выполняют в основном механические функции, хотя некоторые из фибриллярных белков обладают и биологической активностью. Так, названный выше миозин является ферментом; он расщепляет аденазинтрифос-форную кислоту (АТФ), которая обладает большим количеством энергии, выделяемой при ее расщеплении.
Фибриллярные белки при комнатной температуре обычно не растворимы в воде, однако способны набухать в ней, что говорит об их гидрофильных свойствах.
Глобулярные (от латинского слова globula — шарик) белки состоят из макромолекул шаровидной, эллипсовидной, реже веретенообразной формы. Характерной особенностью этих белков является хорошая растворимость в воде, т.е. высокая гидрофильность. Глобулярные белкй* находятся главным образом в биологических жидкостях: в крови, лимфе, протоплазме клеток.
Белки этой группы — альбумины, а также глобулины яичного белка, молока, сыворотки крови, пепсин желудочного сока и другие выполняют в организме очень важные биологические функции.
Многочисленные эксперименты показали, что характер связей в фибриллярных и глобулярных белках одинаков. Молекулярный вес обоих основных структурных видов белка также примерно одинаков (от 30 000 до 1 000 000 и более), но форма значительно отличается. Так, у фибриллярных белков длина макроглобул в сотни и тысячи раз превышает их толщину. Например, макроглобула проколлагена с молекулярным весом 680 ООО имеет длину около 3000 А, а толщину — всего лишь несколько ангстрем.
— 426 —
।
На поверхности тех и других белков имеется большое количество гидрофильных групп, которые обусловливают создание вокруг этих макроструктур почти сплошной водной оболочки. Гидрофобные радикалы аминокислот, образующие полипептидные цепи, обращены преимущественно внутрь структуры. Несмотря па это, некоторое количество воды может быть связано и внутри белковых макроструктур. Часть гидрофильных групп может содержаться и во внутренних отделах белковых макроструктур; кроме того, некоторая часть воды может быть замкнута внутри этих структур в своеобразных «ячейках», образованных гидратированными полипептидными цепочками. И, наконец, диполи воды могут попросту вклиниваться в водородные связ ------ ---------
Принято различать интер мицеллярную ъъру, находящуюся в свободном состоянии между отдельными белковыми макромолекулами, и интрамицелляр-ную воду, находящуюся внутри белковых глобул.
Для устойчивости коллоидных частиц имеет значение только вода, создающая внешнюю водную оболочку. Именно она и препятствует столкновению и объединению белковых макромолекул.
Сложные белки имеют еще более сложное строение и состоят из макроглобулы простого белка, к которой присоединены другие компоненты: углеводы или липиды, фосфорная кислота и т. п. К этой группе относятся также и нуклеопротеиды, строенце которых особенно интенсивно изучается в последнее время. Напомним, что нуклеопротеиды состоят из белка и так называемой нуклеиновой кислоты, которая представляет собой полимер, состоящий из мононуклеотидов (мономеров), соединенных между собой сложноэфирными связями. Каждый из мономеров является одним из пуриновых
вых (тимин, цитозин или урацил) оснований, соединенных через рибозу или ди-зоксирибозу с фосфорной кислотой. Особое значение придают в биологических процессах рибонуклеиновой (РНК) и дезоксирибонуклеиновой кислотам (ДНК). Обе эти нуклеиновые кислоты встречаются в живых тканях в связанном с белком
их прочности.
4 NH3 он
6 со он
1 ь
0NH
NHJ С00’ NHt С0(Г
дсоо~
6NH, ОН
з
фсоон
рН^ИТ pH* ИГ
^СОО’
рН>ИТ
Рис. 180. Формы отдельных участков цепи макромолекул белка при различных значениях pH
(аденин или гуанин) или пиримндино-
виде.
Электрический заряд белков, помимо их своеобразного строения, является особенностью их свойств. В белковой молекуле содержится две полярные группы: основная — NH2 и кислотная—СООН, которые и сообщают макромолекуле амфотерные свойства. Белки не просто электролиты, а электролиты — амфолиты Это означает, что в водных растворах макромолекулы способны диссоциировать как кислоты, т. е. с отщеплением свободных ионов водорода
4-Н+
и как основания, т. е. с отщеплением ионов гидроксила
Макромолекулы белков имеют спиралевидную конфигурацию, которая может изменяться в зависимости от знака их заряда. Так, в нейтральном состоянии
427 —
белка противоположно заряженные ионы NH^ и СОО“ испытывают сильное притяжение друг к другу и тем самым вызывают укорачивание белковой нити и даже скручивание ее в виде спирали (рис. 180). В кислой и щелочной среде происходит отталкивание отдельных групп и растягивание цепи в целом, т. е. раскручивание спирали.
Рис. 181. Электрофоретический прибор Тизелиуса: / — U-образная трубка; 2 — электроды
Таким образом, заряд белка зависит от соотношения в его молекулах карбоксильных и аминных групп и от pH среды. Значение pH раствора белка, при котором белок становится электронейтральным, называется изоэлектрической точкой данного белка. Каждый белок имеет свое значение pH, при котором он находится в изоэлектрическом состоянии (табл. 80).
Таблица 80
Изоэлектрические точки (ИЭТ) различных белков
Белок ИЭТ
Казеин 4,6
Желатин 4,7
Альбумин яйца . . . , 4,8
Белок
Гемоглобулин
Глобулин ...«»<?»»»
Глиадин пшеницы , . * .
6,8
5,4
9,8
Опыт показывает, что в изоэлектрическом состоянии белки имеют наименьшую вязкость. Это связано с изменением формы макромолекул, так как макромолекулы в развернутом состоянии придают растворам более высокую вязкость, чем макромолекулы, свернутые в спираль или клубок.
— 428 —
Экспериментальное определение изоэлектрической точки белковых раство-п )в, как и определение изоэлектрического состояния лиофобных золей, может быть произведено прямым или косвенным методами.
Прямые методы сводятся к наблюдению за поведением частиц в электрическом поле при электрофорезе. При этом исследуемый белок подвергают электрофорезу в буферных растворах с разными значениями pH. В буферном растворе со значением pH, равным изоэлектрической точке белка, последний электронейт-рален и перемещаться в электрическом поле не будет. Наблюдения за электрофорезом приводят либо макроскопически в особых электрофоретических аппаратах, либо микроскопически в кювете ультрамикроскопа.
Наиболее совершенная техника электрофореза сложных белков (а также других веществ) была разработана Тизелиусом.
Электрофорез ведется при высоком значении разности потенциалов (8— 10 в!см) для более четкого разделения компонентов раствора по их электрофорети
ческой подвижности. На рис. 181 показана схема электрофоретического прибора Тизелиуса. Как видим, этот прибор состоит из U-образной трубки с плоскопараллельными стенками, в нижней половине которой находится испытуемый раствор, а сверху раствора находится наслаиваемый буферный раствор, заполняющий всю остальную часть прибора. Оба раствора (и исследуемый, и буферный) выравниваются по pH, по электропроводности и ионной силе.
На приборе Тизелиуса, помимо оп-
Рис. 182. Диаграммы электрофоретического анализа по Тизелиусу: а — нормальная сыворотка; б — плазма крови при множественной миэломе; в — сыворотка крови при нефрозе
ределения изоэлектрического состояния, т. е. изоэлектрической точки белков, можно проводить (для чего этот прибор главным образом и применяют) электрофоретическое разделение их по отдельным фракциям. Результаты измерений получаются в виде одного или нескольких пи-
ков на графике (рис. 182). Относительная площадь характеризует содержание компонентов в растворе. Анализ белков с помощью электрофоретического прибора Тизелиуса получил за последние годы широкое распространение при исследовании нормальных и патологических сывороток в медицине (рис. 182), нуклеопротеидов, чистых белков и их смесей.
. Помимо прибора Тизелиуса, за последние годы широкое распространение получили методы электрофореза белков на бумаге и агаровом геле. Достаточно упомянуть, что путем электрофореза в агаровом геле в нормальной сыворотке крови удается обнаружить до 20 различных белков.
Однако вернемся к обсуждению методов определения ИЭТ белков. Помимо прямых методов наблюдения изоэлектрического состояния белков, существуют и косвенные методы, которые сводятся к наблюдению максимума или минимума того или иного физического свойства, изменяющегося с изменением дзета-потенциала испытуемого раствора. Наибольшее распространение получили следующие
методы.
Определение по степени коагуляции. В пробирки наливают буферные растворы с различным значением pH, затем вносят равные количества исследуемого раствора белка и добавляют (для дегидратации) спирт. Наибольшее помутнение наблюдается в пробирке, pH которой соответствует ИЭТ.
Определение по величине набухания. Небольшие навески сухого белка насыпают в ряд пробирок, туда же приливают равные объемы буферных растворов с различным значением pH. Наименьшее набухание белка окажется в пробирке, pH которой наиболее близок к изоэлектрической точке исследуемого белка.
Определение по скорости желатинирования. В ряд пробирок, содержащих одинаковые объемы буферных растворов с различными значениями pH, добавляют концентрированный раствор белка. Быстрее всего желатинирование наступит в той пробирке, pH которой соответствует ИЭТ исследуемого белка.
Глава XI
ПОВЕРХНОСТНЫЕ ЯВЛЕНИЯ НА ГРАНИЦЕ РАЗДЕЛА ФАЗ
§ 121. Свободная энергия поверхности раздела фаз
В системах с очень развитой поверхностью раздела фаз большое значение имеют так называемые поверхностные явления. Как показали исследования, они зависят от природы вещества и величины поверхности.
Коллоидные растворы относятся к классу высокодисперсных систем и потому они обладают громадной суммарной поверхностью частиц дисперсной фазы. Для того чтобы иметь представление о поверхности коллоидов, сделаем подсчет площади всех сторон кубиков, образованных при дроблении 1 см? твердого тела на более мелкие. При дроблении суммарный объем кубиков остается тем же, а суммарная поверхность (табл. 81) быстро увеличивается.
Таблица 81
Изменение удельной и суммарной поверхности с увеличением степени дисперсности вещества
Длина ребра, см Число кубиков Суммарная поверхность, см9 X Удельная поверхность, гл”"1
1 1 ‘ 6 * 6
1 • io-1 ЫО3 6-101 6-101
1-10-а ыо6 6-102 6-10»
ыо-3 ЫО9 6-10^ 6-10«
1-ю-4 ЫО12 6-104 6-10*
1-ю-5 1 - 101э 6-10’ 6. 105
1-10-’ ыо1» 6.10е 6.10е
ЫО"7 1 - Ю21 6-107 6-Ю7
1-10-8 ЫО24 6-108 6-10®
Из таблицы видно: если 1 ои3 вещества раздробить таким образом, чтобы ребро каждого нового кубика равнялось 0,000 0001 см, то общая поверхность всех кубиков будет равна 60 000 000 см?, или 0,6 га.
Такое огромное увеличение поверхности в высокодисперсных гетерогенных системах связано с увеличением поверхностной энергии. Эту зависимость можно вывести следующим образом. Предположим, что общий запас энергии какой-то частицы равен некоторой величине Е. Эта величина состоит из двух слагаемых — энергии массы (или объема) Ет и поверхностной энергии ES'
E^Em-YEs, (XI, 1)
Энергия массы, или объема, равна Ет = Кт = KV, где т и V — соответственно масса или объем вещества. Энергия поверхности (или сво
— 430 —
бодная поверхностная энергия) выразится произведением двух сомножителей: поверхностного натяжения о как фактора интенсивности и величины общей поверхности S системы как фактора емкости, т. е.
Es = oS. (XI,2)
С учетом этого уравнение (XI, I) примет следующий вид
(XI,3)
Разделив это уравнение на V, получим выражение для общего запаса энергии, приходящейся на единицу объема (удельную поверхностную энергию) Еу
V
AV
V “ т/
oS
(XI,4)
V ‘
или
oS
(XI ,5)
Анализ этого уравнения показывает: если объем частицы V большой, то второй член очень мал и им можно пренебречь. И наоборот, если при высокой степени дисперсности объем частиц V очень мал, энергия частицы будет в основном связана с энергией поверхности. Иными словами, в грубодисперсных системах преобладает объемная энергия, т. е. энергия, связанная с массой, а в высокодисперсных системах преобладает поверхностная энергия.
Дальнейшее дробление дисперсной фазы вплоть до молекул или ионов приводит к исчезновению поверхности раздела фаз. При этом второй член уравнения (XI, 5) будет равней нулю.
Таким образом, коллоидно-дисперсные системы обладают очень большой свободной поверхностной энергией aS, так как даже при ничтожной концентрации, например в 0,001 %, суммарная поверхность раздела S в одном литре золя измеряется единицами и десятками квадратных метров.
Согласно второму принципу термодинамики, в системах, обладающих избытком свободной энергии, могут самопроизвольно протекать процессы, понижающие запас энергии. Поскольку поверхностная энергия выражается произведением двух величин а и S, уменьшение ее может идти за счет уменьшения количества свободной энергии, приходящейся на единицу поверхности, т. е. а, или за счет уменьшения величины поверхности частиц дисперсной фазы S. Уменьшение суммарной поверхности коллоидно-дисперсных частиц ведет к самоукруп-нению частиц, т. е. к коагуляции их. Уменьшение же а может происходить за счет притяжения к поверхности из дисперсионной среды молекул или атомов.
- 431 -
§ 122. Общая характеристика сорбционных явлений
Я
Опыт показывает, что чем выше степень дисперсности дянцлгл тела, тем большее количество частиц другого тела оно может поглотить Ш5ёй поверхностью. 11роцесс самопроизвольного сгущения раство-~ ренного или парообразного вещества (газа) на поверхности твердого тела или жидкости носит название сорбции. Поглощающее вещество носит название сорбента, а поглощаемое— сорбтива. Процесс, обратный сорбции, называется десорбцией. В зависимости от того, насколько глубоко проникают частицы сорбтива в сорбент, все сорбционные процессы подразделяются на адсорбцию, когда вещество поглощается на поверхности тела, и абсорбцию, когда вещество поглощается всем объемом тела. В зависимости от характера взаимодействия частиц сорбента и сорбтива сорбция бывает физическая (взаимодействие проявляется силами когезии и адгезии, т. е. силами Ван-дер-Ваальса) и химическая, или, как ее еще называют, хемосорбция (когда происходит химическое взаимодействие). В свою очередь химическая рорбция подразделяется на абсорбционную и адсорбционную.
Между физической сорбцией, хемосорбцией и типичной химической реакцией очень трудно провести четкие границы, поэтому такое деление имеет лишь условный характер.
Особое положение занимает сорбционный процесс, называемый капиллярной конденсацией. Сущность этого процесса заключается не только в поглощении, но и в конденсации твердым пористым сорбентом, например активированным углем, газов и паров. Капиллярная конденсация в первую очередь наблюдается у легко сжижаемых газов.
Из всех перечисленных выше сорбционных явлений наибольшее значение для практики имеет адсорбция, которая и будет рассмотрена наиболее подробно.
Абсорбция — это сорбция газа за счет его проникновения (диффузии) в массу сорбента. По существу она представляет собой процесс растворения одного вещества (абсорбтива) в другом (абсорбента). Распределение вещества между фазами подчиняется закону Генри, известному из физической химии. Абсорбционные процессы в настоящее время широко применяются в промышленности. Так, получение соляной кислоты в заводских условиях целиком основано на абсорбции хлористого водорода водой. На явлениях абсорбции основаны также разделение газовых смесей, очистка их от различных вредных примесей, улавливание ценной составной части газовой смеси и т. п.
Хемосорбция представляет собой наиболее глубокое взаимодействие поглощаемого вещества и поглотителя с образованием нового химического вещества. В качестве примера хемосорбции можно указать на поглощение двуокиси углерода или двуокиси серы натронной известью (смесь NaOH и Са(ОН)2), которая, поглощая их, химически взаимодействует с ними. Хемосорбцию легко можно отличить от физической абсорбции и адсорбции по значительно большему количеству выделяющейся при хемосорбции теплоты, вполне соизмеримой с теп-лотами образования химических соединений.
Капиллярная конденсация, как уже упоминалось, представляет собой процесс конденсации парообразных сорбтивов в порах твердого сорбента. Конденсация зависит от температуры, упругости пара, диаметра капилляров, а также смачиваемости поверхности твердого сорбента сорбтивом в жидком состоянии. Чем уже капилляры и чем лучше жидкость смачивает их стенки, тем при прочих равных условиях скорее происходит насыщение паров и их конденсация.
На явлении капиллярной конденсации основана так называемая рекуперация летучих растворителей при некоторых технологических процессах. Так, в процессе изготовления прорезиненных тканей на каждые 300 м ткани расходуется около 180 кг каучукового клея, который содержит около 157 кг высокосортного бензина. При высушивании ткани практически весь бензин улетучивается. При рекуперации пары бензина с воздухом отводят в адсорбер, заполненный активированным углем. В этом адсорбере и происходит адсорбция, а затем уже капиллярная конденсация паров бензина. После насыщения адсорбента бензином в адсорбер подают горячий пар, который вызывает десорбцию бензина. Далее пары воды и бензина отводятся в специальный холодильник, а уже потом в сепаратор, где сконденсированные воды и бензин отделяются друг от друга при расслаивании.
Капиллярная конденсация имеет очень большое значение для объяснения явления так называемого гистерезиса оводнения и обезвоживания в неэластичных гелях, который будет рассматриваться несколько позднее.
§ 123. Явление адсорбции I
Как известно, всякая поверхность, независимо от агрегатного состояния разделяемых ею веществ, обладает некоторым запасом свободной энергии. В силу второго начала термодинамики поверхность раздела веществ (как и любая система, обладающая запасом свободной энергии) стремится к самопроизвольному уменьшению этой энергии. Это стремление и является прямой или косвенной причиной разнообразных физических явлений, в том числе и адсорбции.
Адсорбция — это в широком смысле процесс самопроизвольного изменения концентрации вещества у поверхности раздела двух фаз, а в более узком и употребительном смысле — это повышение концентрации одного вещества у поверхности раздела двух фаз, из которых одна обычно является твердым телом. В соответствии с общей терминологией по сорбции вещество, на поверхности которого происходит накопление другого вещества, носит название адсорбента, а поглощаемое вещество — адсорбтива или адсорбата.
Рассмотрим пример адсорбции аммиака активированным углем. Сосуд 1 (рис. 183, а) наполнен газообразным аммиаком,и при помощи резиновых трубок 2 и 3 соединен с манометром и колбой 5, в которой находится активированный уголь. Как только установка будет собрана, откроем кран 6 и приведе^м манометрическую жидкость в обоих коленах манометра к одному уровню. Далее из колбы 5 уголь пересыпем в сосуд 1 и хорошо его встряхиваем. Спустя некоторое время увидим, что уровень в правом колене манометра сильно повысится (рис. 183, б). Это объясняется поглощением аммиака углем. Манометр 4 показывает лишь понижение давления газа в сосуде /. Однако согласно закону Бойля—Мариотта при постоянной температуре концентрация любого газа и его давление прямо пропорциональны друг другу. Так как в нашем опыте значительная часть аммиака поглотилась углем, то и давление газа в сосуде 1 в результате этого тоже уменьшилось.
- 433 —
Пусть в сосуд 1 было введено /10 молей аммиака, через некоторое время в нем осталось п молей; нетрудно догадаться, что /г0 — п составит число адсорбированных молей аммиака. Адсорбция прекращается, когда поверхность адсорбента
Рис. 183 Схема установки для иллюстрации адсорбции аммиака углем: состояние установки: а — до адсорбционного взаимодействия; б — при адсорбционном взаимодействии
г=«о п=* . (XI,6)
О
где Г — удельная адсорбция, моль/см2.
насытится газом и наступит так называемое адсорбционное равновесие, при котором скорости адсорбции ui и десорбции и2 одинаковы, т. е. ==* «2. Если для опы-по — л
та было взято tn граммов угля (адсорбента), то 1 г его адсорбирует —— молей / / V
газа.
Если общая площадь поверхности адсорбента равна S см2, то на 1 см2 поверхности адсорбируется
Количество молей поглощенного вещества на I см2 поверхности адсорбента называется удельной адсорбцией. Так как поверхность любого адсорбента практически измерить трудно, адсорбцию выражают в молях на 1 г адсорбента и обозначают буквой А: ♦
Л=—. (XI.7)
т
Связь между обоими выражениями удельной адсорбции имеет следующий вид:
Л=Г50. (XI,8)
Величина удельной адсорбции Г для данного адсорбента и данного адсорбтива зависит от двух термодинамических параметров: температуры Т и давления Р при газообразном адсорбтиве и от температуры и концентрации С при адсорбции из раствора. Причем если величина адсорбции выражается через А [уравнение (XI, 8)], она будет зависеть еще и от поверхности адсорбента. Все три величины Г, Т и С (или Р) находятся друг с другом в определенной функциональной зависимости,
• 434 —•
которая математически выражается общим термодинамическим урав
нением:
откуда
/(7, 7, С)=0 или / (7, 7, Р)=0, 7=7(7, С) или Г=/(7, 7).
(XI,9)
Из этих уравнений вытекают частные уравнения, из которых нас будет интересовать так называемое уравнение изотермы адсорбции, т, е.
Г=( (С) или Г = I (7) при 7 = const. (XI, 10)
Как показали исследования, процессы адсорбции (так же как и другие виды сорбционных процессов) обратимы. Частицы, находящиеся в адсорбционных слоях, не закреплены жестко. Некоторые из них могут выходить за пределы действия сил притяжения адсорбента, т. е. отрываться от поверхности адсорбента и уходить в окружающее пространство. Такой обратный процесс носит название десорбции. Со временем оба процесса приводят систему в состояние адсорбционного равновесия!
адсорбция десорбция,
при котором среднее число частиц, покидающих поверхностный слой, становится равным числу адсорбируемых частиц за тот же отрезок времени.
Процесс адсорбции экзотермичен, следовательно, в соответствии с принципом Ле Шателье его выгодно осуществлять при сравнительно низких температурах. Опыт показывает, что с повышением температуры в силу увеличения колебания частиц, адсорбированных поверхностью, равновесие сдвигается в сторону процесса десорбции.
Помимо обратимости и экзотермичности, адсорбция характеризуется и третьим общим признаком — чрезвычайно малой энергией активации, т. е. низким энергетическим барьером, а следовательно, большой скоростью ее протекания. Благодаря этому адсорбция с энергетической стороны имеет много сходного с обратимыми экзотермическими химическими реакциями, однако отличается от них и от хемосорбции гораздо меньшей величиной теплового эффекта.
Чаще всего адсорбция носит избирательный характер. Так, рассмотренный нами активированный уголь хорошо поглощает не только аммиак, но и хлор, однако не адсорбирует окись углерода. Поэтому нельзя пользоваться обычным противогазом при тушении пожаров, так как в зоне пожара всегда много окиси углерода.
Явление адсорбции было открыто академиком Российской академии наук Т. Е. Ловицем еще в 1786 г. Ловиц подробно описал поглощение красящих веществ углем и предложил использовать угли для промышленной очистки растворов от вредных примесей (для очистки спирта от сивушных масел, для устранения запаха питьевых вод и т. п.). Именно с работ Ловица и немецкого химика Ш е е л е началось изучение адсорбции.
Особенно интенсивно эта наука стала развиваться в начале этого столетия. Причем в ее развитии наши ученые играли и играют выдающуюся роль. Современные представления о природе адсорбционных сил были выдвинуты русскими учеными Б. И. Шишковским (1908) и Л. Г. Гуревич ем (1915).
— 435 —
Они легли в основу теории строения адсорбционных слоев Ленгмюра. Работы других наших соотечественников А, А. Т и то в а (1910), Н. Д. 3 е линек о г о в области адсорбции газов привели к созданию угольного противогаза (1915). Работы академика М. М. Д у б и н и н а и учеников его школы Н. А. Ш и-л о в а, А. В. К и се л е в а, А. Н. Фрумкина, Б. В. Д е р я г и н а, связанные с вопросами поверхностно-активных веществ и адсорбцией, получили широкое развитие в советское время. Большое значение имели исследования академика П. А. Ребиндера о влиянии адсорбционных пленок на свойства твердых и жидких поверхностей. Они легли в основу важнейшего промышленного процесса — флотации. Не меньшее значение имели эти работы и для бурения, а также резания металлов.
Немалый вклад в разработку теории и практики адсорбции внесли зарубежные ученые Гиббс, Фрейндлих, Полян и, Брунауэр.
§ 124. Адсорбция на поверхности раздела твердое вещество — газ
Адсорбция газа на твердом теле является простейшим случаем адсорбционного процесса, так как система состоит всего из двух компонентов. Конкретный пример такой адсорбции мы рассмотрели в предыдущем параграфе.
Опыт показывает, что при прочих равных условиях для твердого адсорбента и данного адсорбируемого газа количество адсорбируемого вещества будет возрастать по мере увеличения адсорбирующей поверхности. Следовательно, чтобы достигнуть большого адсорбционного эффекта, необходимо иметь как можно большую поверхность поглотителя. Способность адсорбента к поглощению газов определяется не только его пористостью, но и физическим состоянием; так, адсорбенты в аморфном состоянии лучше адсорбируют газы, чем в кристаллическом.
В качестве адсорбентов на практике применяют древесный и костяной угли, силикагель, высокодисперсные металлы, полученные восстановлением их из оксидов. Активированный уголь получают путем соответствующей активации угля-сырца твердых древесных пород. Уголь-сырец подвергают термической обработке для увеличения удельной поверхности. Активирование производят в атмосфере водяного пара или двуокиси углерода при температуре 700—900° С. При этом уголь частично реагирует с СО2 и водяным паром с образованием СО и Н2. Изменение структуры угля показано на рис. 184. Активированный уголь как адсорбент применяется в противогазах, а также для очистки воздуха на промышленных предприятиях, для осветления различных растворов и т. п. Высокая адсорбционная способность активированного угля объясняется, как это видно из рис. 184, сильно развитой поверхностью. Так, суммарная поверхность всех пор, заключающихся в 1 г такого угля, составляет от 300 до 1000 л/2. Такая огромная площадь обусловливает возникновение большого молекулярного силового поля и, стало быть, избыток поверхностной энергии на границе уголь — газ. За счет свободной поверхностной энергии и происходит адсорбция газа, т. е. повышение его концентрации в поверхностном слое угля при одновременном понижении концентрации газа в окружающем пространстве.
Как показали исследования, время пребывания молекул газа на поверхности твердого адсорбента очень мало: они удерживаются на адсорбенте всего сотые и тысячные доли секунды и, десорбируясь, замещаются на новые частицы. В конечном итоге устанавливается динамическое равновесие между свободными и адсорбированными молекулами. Скорость достижения адсорбционного равновесия для разных газов неодинакова: при адсорбции СО2 на угле равновесие наступает через 20 сек, при адсорбции О2 — через 2,5 ч, при адсорбции N2 — через 20 ч и т. п. Скорость адсорбции имеет большое значение для практического использования различных адсорбентов. Например, в широко используемом при химической защите противогазе проходящий через^ коробку воздух должен очень быстро очищаться от примесей отравляющих веществ. Это возможно лишь при высоких скоростях адсорбционных процессов.
Рис. 184. Изменение структуры угля при активировании:
1 — поры угля-сырца; 2 — поры угля-сырца в процессе активирования; 3 —- активированный уголь со свободными порами
Активированный уголь в противогазе играет роль не только адсорбента целого ряда отравляющих веществ, но и катализатора реакции разложения многих из них. В качестве примера можно указать на каталитический гидролиз фосгена
СОС12 + Н2О -> 2НС1 4- СО2
или хлорпикрина
CC13NO2 4- 2Н2О -> СО2 4- ЗНС1 4- HNO2
Опыт показывает, что адсорбция зависит не только от природы поглотителя, но и от природы поглощаемого газа (табл. 82).
Из анализа данных табл. 82 видно, что при прочих равных условиях сильнее адсорбируются те газы, которые легче конденсируются в жидкость. Следовательно, они обладают более высокой температурой кипения в сжиженном состоянии.
Для объяснения явлений адсорбции существуют различные теории. Одна из них — физическая теория, согласно которой природа адсорбционных сил чисто физическая и связана с проявлением межмолекулярных сил. Согласно химической теории, ненасыщенные силы адсорбционных поверхностных слоев являются химическими (валентными) силами.
— 437 —
Таблица 82
Адсорбция газов на угле в зависимости от температуры кипения и критической температуры адсорбируемых веществ при давлении 760 мм рт. ст.
Г аз с, м*/г КПП °C ' КРИТ °C 1 аз с, М3/с t кип °C ?крит °C
СОС12 440 — 8 4-183 СО, 48 — 78 + 31
О2 380 —10 4-137 со 9 —190 —140
NH3 181 —33 + 132 О2 8 — 182 — 119
H2S 99 —62 + 100 n2 8 —195 —147
НС1 72 —83 — 52 Н2 5 —252 —240
n2o 54 —90 + 37
Известно несколько теорий физической адсорбции, из которых интерес представляет теория мономолекулярной адсорбции Ленгмюра (1915). В построении ее ученый опирался на представление об адсорб- Я ционных силах, которые впервые были высказаны русским ученым Я Л. Г. Гуревичем. Основные положения теории Ленгмюра: Я
1. Адсорбция вызывается валентными силами или силами остаточной химической валентности. Я
2. Адсорбция происходит не на всей поверхности адсорбента,а?1ишь I на активных центрах этой поверхности. Такими центрами являются углубления и выступы, имеющиеся на любой, даже самой гладкой по- I верхности. Действие таких центров сводится к высокой ненасыщенности их силового поля, благодаря чему центры удерживают газовые молекулы. Причем активность центра тем выше, чем меньше насыщена молекула или атом адсорбента.
3. Адсорбционные силы обладают малым радиусом действия, вследствие чего каждый активный центр адсорбирует лишь одну молекулу адсорбтива, и на адсорбенте образуется мономолекулярный слой ад- ? сорбтива.
4. Адсорбированные молекулы газа не сидят прочно на поверхности адсорбента, они непрерывно обмениваются с молекулами в газовой сфере, при этом устанавливается динамическое адсорбционное равновесие. I Каждая молекула задерживается в течение короткого времени на поверхности, затем в результате флуктуации энергии молекулы отрываются от активного центра, уступая место новой молекуле.
В отличие от физической адсорбции химическая адсорбция, или хемосорбция, осуществляется при помощи химических сил. Эти виды адсорбции имеют следующие отличительные признаки: физическая адсорбция — явление обратимое, и теплота ее составляет всего 2— 8 ккал!моль, в то время как теплота химической адсорбции достигает десятков и сотен ккал!моль. С повышением температуры физическая адсорбция уменьшается, а химическая увеличивается. Объясняется это тем, что химическая адсорбция требует более значительной энергии активации (10—30 ккал!моль).
Химическая адсорбция необратима, поэтому процесс десорбции состоит не в простом отрыве адсорбированной молекулы, а в разложе-
— 438 —
НИИ поверхностного химического соединена р примера химической адсорбции можно начвятЛпкач®стве типичного на поверхности угля. Процесс хемосолбиЛ Э Ь адсоРбИию кислорода бразить следующим образом: ХеМ0с0Рбции можно схематически изо-
поверхность угля поверхность угля
до адсорбции после адсорбции
Весьма характерным является то, что при нагревании с поверхности адсорбента удаляется не кислород, а окись углерода.
Согласно современным представлениям при адсорбции проявляются все виды физических и химических сил, т. е. адсорбция по существу является физико-химическим процессом. И действительно, советские ученые Н. А. Ш и л о в, М. М. Д у б и н и н, Л. К. Л е п и н ь установили, что при различных случаях адсорбции играют роль физические и химические взаимодействия между адсорбентом и адсорбируемым веществом. Это особенно четко проявляется при адсорбции газов. Исследования показали, что при поглощении первых порций газа на чистой поверхности адсорбента чаще проявляется действие химических сил, а при последующей адсорбции газа, при повышении давления процесс переходит постепенно в чисто физический.
§ 125. Изотермы адсорбции
Как показали исследования, адсорбция увеличивается с ростом давления (концентрации) газа, однако это увеличение не беспредельно. Для каждого адсорбируемого газа (при t — const) через некоторое время над адсорбентом устанавливается предельная величина адсорбции, отвечающая равновесию между обеими фазами. Кривая зависимости адсорбции от давления (концентрации) при постоянной температуре носит название изотермы адсорбции. Она является одной из важнейших характеристик адсорбционных процессов. На рис. 185 изображены типичные изотермы адсорбции СО2 углем при различных температурах, взятые из работы А. А. Т и т о в а.
Как видно из рис. 185, повышение давления газа увеличивает адсорбируемое количество его. Однако на разных участках изотермы адсорбции это влияние сказывается неодинаково. Наиболее сильным оно оказывается в области низких давлений, где адсорбция подчиняется закону Генри для растворимости газов в жидкостях, т. е. она прямо пропорциональна давлению газа.
Дальнейшее повышение давления тоже увеличивает количество адсорбированного газа, но уже во все уменьшающейся степени. И, наконец, при достаточно высоких давлениях кривая стремится к пря
— 439 —
мой, параллельной оси аосцисс. В этом случае достигнуто полное насыщение адсорбента и повышение давления газа уже не влияет на его адсорбцию.
Таким образом, между адсорбцией и давлением (концентрацией) газа отсутствует прямая пропорциональная зависимость. Это и вызвало необходимость найти математическое выражение, которое достаточно точно описало бы экспериментальные данные. Впервые эмпириче-
Рис. 185. Адсорбция СОг при различных температурах
ское уравнение, которым пользуются и в настоящее время, было предложено Фрейндлихом. Это уравнение имеет следующий вид:
1
— = КРп, (Xl.ll)
т
где х/т — величина адсорбции на единицу веса адсорбента, Р — равновесное давление газа над поглотителем (для растворов пользуются равновесной концентрацией С), К и 1/п — константы адсорбции, характерные для данного процесса адсорбции в определенных пределах, значение которых
можно наити из опытных данных.
Рассмотрим, как определяются численные значения констант.
На рис. 186 дано графическое изображение уравнения для случая
адсорбции из жидкости. По оси абсцисс отложены равновесные концентрации С ммоль!мл, по оси ординат значения х/т моль!г. Если прологарифмировать уравнение (XI, 11), получим следующее выражение:
lg— = lgA + — igC, tn n
(XI, 12)
из которого можно найти значения постоянных /( и 1/п. Для этой цели построим график, выражающий зависимость 1g х/т — lg С. Получается прямая линия (рис. 186), отсекающая на оси ординат отрезок, равный 1g К, а тангенс угла наклона этой прямой к абсциссе дает значение 1/п.
Уравнение (XI, 12) есть уравнение прямой линии. Несмотря на то, что уравнение Фрейндлиха широко применяется на практике, оно имеет определенные недостатки. Многочисленные исследования показали, что значение адсорбции, вычисляемое на основании этого уравнения, не соответствует данным опыта в области малых и больших концентраций. Константы К и 1/п являются чисто эмпирическими и не имеют реального физического смысла.
- 440 —
Позднее (1917) Ленгмюр вывел простейшее уравнение адсорбции для случая адсорбции газа на гладкой твердой поверхности (стекло, слюда, монокристаллы), оказавшееся в дальнейшем применимым и к другим поверхностям раздела. При выводе своего уравнения Ленгмюр исходил из допущения, что адсорбционный слой мономолекуля-рен, т. е. только один слой молекул связан силами молекулярного сцепления с поверхностью. При этом указанный слой плотностью поглощает собой все адсорбционные силы поверхности адсорбента, в силу чего образование второго слоя адсорбированных молекул исключается.
Число активных мест поверхности ученый принял равным единице, а долю активных мест, связанных с адсорбированными молекулами, обозначил через х. При этом условии свободная часть поверхности рав-
Рис. 186. Изотермы адсорбции:
а —в обычных координатах; б —» в логарифмических координатах
няется 1 —х. Обозначив величину адсорбции через Г и учитывая, что при х = О Г = 0 и при к — 1 Г = Г»,, он нашел, чтох = Г/Гж, где Гзо — количество вещества, адсорбированное единицей поверхности при полном насыщении.
При постоянной температуре на твердой поверхности адсорбента устанавливается подвижное равновесие, при котором скорость адсорбции ца равна скорости десорбции цд. Скорость адсорбции (как и всякой гетерогенной реакции) пропорциональна числу возможных соударений молекул со свободной поверхностью, т. е. объемной концентрации газа С и свободной доле поверхности (1 — х):
оа — /<а С (1 х),
(XI, 13)
где /\а — константа скорости адсорбции.
В свою очередь скорость десорбции цд зависит только от степени покрытия поверхности и не зависит от концентрации газа, т. е.
^д = КдХ, (XI, 14)
где /(д — константа скорости десорбции.
15 Зак. 5G9
441 —
Из уравнения (XI, 14) следует, что чем больше степень покрытия, тем больше вероятность отрыва молекул от поверхности адсорбента. Обе константы Ка и Кд имеют различную размерность:
[/<а] = сек~1; [Дд] = моль-см~'3-сек~г.
При адсорбционном равновесии va= следовательно,
ДаС(1-х)=Дх.
Из этого уравнения находим х:
К а С Кп 4~ С
(XI, 15)
(XI, 16)
Если теперь разделить числитель и знаменатель правой части уравнения на /Са> получим следующее выражение:
Рис. 187.
Изотерма адсорбции
Лэнгмюра
(XI, 17)
В этом уравнении отношение двух постоянных величин Кд/Ка является величиной постоянной. Обозначим ее через В, а величину х заменим на Г/Гоо. Тогда уравнение (XI, 17) примет вид:
откуда
(XI, 18)
Это и есть уравнение изотермы адсорбции Ленгмюра. Оно является уравнением гиперболы (рис. 187) с асимптотой Г = Гоо.
Проанализируем уравнение. Если С -> оо, то Г = Когда С велико по сравнению с В (С В), величиной В в знаменателе уравнения (XI, 18) можно пренебречь, тогда Г = Гх. В области малых концентраций, когда С -> 0 (т. е. при С В), величиной С в знаменателе уравнения (XI, 18) также можно пренебречь, тогда
С 1
д-; Г^—Г^С. С -f- D D
Исходя из анализа уравнения коэффициент В определяется как отрезок, отсекаемый касательной, проведенной из начала координат на симптоте Г = Г^, т. е. В определяет кривизну наклона изотермы, являясь мерой адсорбционной активности вещества.
Если Г = Г/2, то Гоо/2 = • С /(С + В), откуда С + В = 2х
X С и В = С. Таким образом, константа В в уравнении Ленгмюра численно равна той концентрации, при которой одна половина актив-
— 442 —
hlix точек на поверхности адсорбента занята молекулами адеорбтива, другая остается свободной.
Опыт показывает, что уравнение изотермы адсорбции Ленгмюра сравнительно удовлетворительно дает количественную характеристику адсорбции при низких и при высоких концентрациях поглощаемого вещества. В отличие от уравнения изотермы Фрейндлиха все величины, входящие в уравнение Ленгмюра, имеют определенный физический смысл и вполне обоснованы теоретически.
Уравнение Ленгмюра исходит из расчета мономолекулярного адсорбционного слоя. Однако не все ученые разделяют эту точку зрения. По мнению П о л я н и и ряда других авторов, возможен многослойный адсорбционный слой, причем эта точка зрения имеет некоторое теоретическое и опытное обоснование.
По теории Ленгмюра, молекулы адсорбтива, притянутые к отдельным активным точкам, между собой не взаимодействуют. Однако при накоплении в адсорбционном слое молекул веществ, обладающих высоким молекулярным весом, между ними могут возникнуть значительные силы сцепления. В этом случае уравнение Ленгмюра дает неверные результаты. В ряде случаев, в частности при применении пористых адсорбентов, таких, как уголь, силикагель и др., формула Фрейндлиха дает лучшие результаты, чем уравнение Ленгмюра.
§ 126. Адсорбция на поверхности раздела жидкость — газ
Из уравнения (XI, 2) следует, что мерой свободной поверхно стной энергии единицы поверхности является поверхностное натяже ние ст, под которым следует понимать силу, действующую на единицу длины линии, ограничивающей поверхность жидкости. При обсуждении поверхностных явлений обычно говорят не о свободной поверхностной энергии, а о поверхностном натяжении, так как оно для поверхностей раздела жидкость — газ или жидкость — жидкость доступно непосредственному измерению.
В чистой жидкости состав поверхностного слоя одинаков с составом ее в объеме. Поэтому при растворении какого-либо вещества в растворителе возможны три случая: а) растворение данного вещества в растворителе не изменяет его поверхностного натяжения; б) растворение повышает поверхностное натяжение растворителя; в) растворение понижает поверхностное натяжение растворителя.
Поскольку поверхностная энергия в силу второго начала термодинамики стремится к минимуму, то в случае, когда растворение вещества понижает поверхностное натяжение растворителя, концентрация растворенного вещества в поверхностном слое должна быть больше, чем в объеме раствора. И наоборот, когда растворение повышает поверхностное натяжение, поверхностный слой раствора должен быть беднее растворенным веществом по сравнению с общим его объемом. Таким образом, на границе раздела жидкость — газ наблюдается явление уменьшения или увеличения концентрации растворенного вещества, т. е. явление а д б О р б ц ТГГГ. ~ "
15*
— 443 —
Адсорбция на границе раздела жидкость — газ отличается не- 1 которыми характерными особенностями по сравнению с процессом ад- • сорбции на границе твердое тело — газ. Во-первых, равноценность I всех участков жидкой поверхности исключает ориентацию молекул | адсорбтива в строго определенных ее местах. Во-вторых, молекулы ад- 1 сорбтива имеют возможность свободно перемещаться по поверхности j и находиться в тепловом движении. Величина адсорбирующей поверх- j ности S жидкости может быть точно измерена, чего почти нельзя сделать в отношении, например, твердых адсорбентов. Характерным 1 отличием адсорбции на границе раздела жидкость — газ является и 1 то, что изменение поверхностной энергии в процессе адсорбции может быть определено непосредственно.
Все растворимые вещества по их способности адсорбироваться на границе раздела жидкость — воздух делятся на две группы: повер- | хностно-активные и поверхностно-неактивные вещества. Первые долж- 1 ны обладать меньшим поверхностным натяжением, чем растворитель, I сравнительно малой растворимостью, а также способностью резко изменять свойства поверхности адсорбента в результате образования тонких мономолекулярных адсорбционных слоев.
Поверхностно-активные вещества по своим физико-химическим свой- | ствам делятся на три группы: молекулярные, ионогенные — анионоактивные и ионогенные — катионо-активные.
К молекулярным, или неионогенным поверхностно-активным ве- ществам относятся электрически нейтральные молекулы спиртов, карбоновых кислот, белковых веществ. Ионогенные анионо-активные по- j верхностные вещества в водной среде диссоциируют на ионы, причем поверхностно-активным будет только анион. К ним относятся мыла, | сульфокислоты, их соли и другие соединения. j
Молекулы катионо-активных поверхностно-активных веществ в водных растворах также диссоциируют на ионы, однако поверхност- j но-активным будет только катион. К ним относятся органические азотсодержащие основания и их соли.
Характерной особенностью всех поверхностно-активных веществ является то, что молекулы-их содержат две части: полярную гидрофильную группу, например ОН,СООН, NH2, и неполярный углеводородный или ароматический радикал. Полярная группа обладает значительным дипольным моментом и хорошо гидратируется. Эта группа и определяет сродство поверхностно-активных веществ к воде. В отличие от полярной группы углеводородный радикал гидрофобен, т. е. понижает растворимость поверхностно-активных веществ в воде. При взаимодействии поверхностно-активных веществ с водой молекулы их погружаются в воду своими гидрофильными группами, гидрофобная же углеводородная цепь (радикал) располагается выше уровня воды,т. е. как бы «торчит» из воды. Выталкивающее действие воды на гидрофобную часть молекул поверхностно-активных веществ способствует накоплению их в поверхностном слое жидкости. Опыт показывает: чем длиннее углеводородный радикал, тем хуже вещество растворяется в воде, тем большая доля его находится в поверхностном слое и, стало быть, выше его адсорбируемость. За счет накопления поверхностно-активных ве-
— 444 —
ществ в поверхностном слое жидкости и происходит уменьшение поверхностного натяжения раствора.
Таким образом, поверхностная активность, следовательно, и адсорбируемость вещества, зависят от природы полярной группы, строения молекулы и длины углеводородной цепи. Исследованиями установлено, что с удлинением цепи углеводородного соединения (например, кислоты) растворимость его в воде падает. Это хорошо видно из приводимых ниже данных:
Уксусная кислова Пропионовая Масляная Валериановая Капроновая
СНзСООН — растворима
СНзСН2СООН — растворима
СНзСН2СН2СООН — растворима
СНзСН2СН2СН2СООН — мало растворима
СНзСН2СН2СН2СН2СООН — не растворима
При изучении поверхностного натяжения водных растворов гомологического ряда жирных кислот было установлено, что удлинение цепи СН2 — увеличивает их способность
в жирных кислотах на радикал к адсорбции в 3,2 раза. Эта закономерность получила название правила Траубе — Дюкло. Согласно ему длина цепи жирной кислоты возрастает в арифметической прогрессии, а поверхностная активность увеличивается в геометрической прогрессии.
Правило Траубе—Дюкло применимо только для разбавленных растворов. Дело в том, что когда концентрация поверхностно-активных веществ мала, их гидрофобные части лежат на поверхности жидкости (рис. 188, а и б). При дальнейшем повышении концентрации адсорбированные молекулы заполняют все места на поверхности, образуя насыщенный слой. Как видно из рис. 188, в, такой слой представляет собой мономо-лекулярную пленку толщиной в одну молекулу адсорбированного вещества. Правило Траубе—Дюк-
fl
Рис. 188. Расположение адсорбированных молекул в поверхностном слое:
а и б — расположение молекул в ненасыщенном слое; в — расположение молекул в насыщенном слое (кружком обозначена гидрофильная часть молекул; линией — гидрофобная углеводородная часть)
ло справедливо также для температур, близких к комнатной. Опыт показывает, что с повышением температуры коэффициент 3,2 приближается к 1, так как увеличивается десорбция, и поверхностная активность падает.
Поверхностно-неактнвные вещества обладают следующими характерными особенностями: а) обладают большим по сравнению с раство-
— 445 —
ригелем поверхностным натяжением; б) имеют более высокую растворимость.
К поверхностно-неактивным веществам относятся все неорганические электролиты: кислоты, щелочи, соли, а также некоторые органические соединения, например муравьиная НСООН и аминоуксус-ная H2NCH2COOH кислоты. Поверхностно-неактивные вещества повышают поверхностное натяжение воды, так как их молекулы стремятся уйти с поверхности жидкости вглубь. Следует также упомянуть и о таких веществах, которые, будучи растворены в воде, не изменяют ее поверхностного натяжения. К числу таких веществ относится, например, сахар. Как показали исследования, молекулы этих веществ равномерно распределяются между поверхностным слоем и объемом раствора.
Математическая зависимость между поверхностным натяжением и эрг-см2, концентрацией растворенного вещества С моль! л и избытком его в поверхностном слое Г моль!см2 была установлена Г и б б с о м еще в 1876 г. и выражается следующей формулой:
С do Hr"dC’
(XI, 19)
где величина dvldC представляет собой изменение поверхностного натяжения раствора с концентрацией, R — универсальная газовая постоянная, С — молярная концентрация растворенного вещества, Т — абсолютная температура.
Из уравнения Гиббса видно, что адсорбция положительна, если da/dC z> 0, т. е. когда при адсорбции поверхностное натяжение уменьшается. Уменьшение поверхностного натяжения тем больше, чем больше концентрация растворенного вещества. Для поверхностно-неактивных веществ поверхностное натяжение, как следует из уравнения Гиббса, повышается с увеличением концентрации растворенного вещества,^ е. в данном случае адсорбция отрицательна (концентрация в поверхностном слое меньше, у А в объеме?, следовательно, — <0,
z иС/
а Г < 0. Когда — = 0, то Г — 0, т. е. адсорбции нет.
В более общем виде зависимость поверхностного натяжения от концентрации для водных растворов жирных кислот может быть выражена эмпирическим уравнением Б. И. Шишковского (1909):
До = В In (ЛС4-1),
(XI, 20)
где Ао — разность поверхностных натяжений раствора и растворителя, В и А — постоянные, величина которых может быть найдена экспериментальным путем, А — константа, изменяющаяся в гомологическом ряду в 3,2 раза для каждого последующего члена ряда в соответствии с правилом Траубе — Дюкло.
Уравнение Б. И. Шишковского может быть получено и теоретическим путем, если объединить уравнения Ленгмюра (XI, 18) и Гиббса
— 446 —
(XI, 19). Для этого надо подставить значение Г из уравнения изотермы Ленгмюра
в уравнение Гиббса]
do
dC
= —RT
do=—RTrtx
dC
C + B
и, проинтегрировав последнее выражение от С = 0 до С, получим
Q=O0—RTroo In бу+ 1).
(XI,21)
Сопоставляя это уравнение с уравнением Шишковского
о=а0—в In (/С 1),
можно сделать вывод о том, что .константа В в уравнении Шишковского имеет значение
RT
B^RTr^——, (XI ,22)
/Vq о0
где No — число Авогадро, So — площадь, приходящаяся на одну молекулу в насыщенном слое.
Таким образом, константа В в уравнении Шишковского определяется предельной адсорбцией Г^, отвечающей образованию насыщенного адсорбционного слоя, или площадью So, приходящейся на одну молекулу в насыщенном слое. Константа А в уравнении Шишковского имеет то же значение, что и константа В в уравнении Ленгмюра.
Поверхностно-активные вещества широко применяются в народном хозяйстве. Опыт показывает, что даже очень малые добавки этих веществ позволяют резко изменить условия взаимодействия соприкасающихся тел и природу их поверхности. С помощью поверхностно-активных веществ можно не только изменять условия образования различных дисперсных систем, но и управлять их устойчивостью. Добавки поверхностно-активных веществ облегчают разрушение, например, при тонком помоле веществ, а также облегчают тончайшее распыление жидкости. В ряде случаев поверхностно-активные вещества позволяют уменьшать трение между поверхностями, движущимися одна относительно другой, а также ослаблять прилипание друг к другу твердых поверхностей, например, волокон.
Добавки поверхностно-активных веществ облегчают процесс отмывания подчас самых сильных загрязнений. Так, нельзя отмыть загрязненную ткань даже горячей водой без добавления к ней мыла или какого-либо другого моющего средства.
При получении некоторых новых материалов с заданными свойствами, например резины, различных пластмасс, нельзя обойтись без поверхностно-активных веществ. При производстве синтетических материалов необходимы так называемые активные наполнители (сажа, каолин), которые резко усиливают прочность к износу, стойкость к температурам и сообщают ряд других свойств этим материалам. Добавление поверхностно-активных веществ делает наполнители еще более активными, обеспечивая хорошее смешение их с наполняемым полимером.
— 447 —.
§ 127. Адсорбция на поверхности твердое вещество — жидкость
Как показали исследования, адсорбция растворенных веществ] на твердой поверхности гораздо сложнее, чем адсорбция растворенных ’ веществ на поверхности жидкостей. Общая теория адсорбции на твер-! дой поверхности в достаточной мере еще не разработана и в настоящее ^ время. Создание общей теории этого вида адсорбции осложняется не только особым характером поверхности твердых адсорбентов, но и тем, J что при адсорбции из раствора происходит одновременная адсорбция
Рис. 189. Изотерма адсорбции из раствора
растворителя и растворенного вещества. Кроме того, необходимо учитывать взаимодействие между молекулами растворенного вещества и растворителя. Вопрос становится еще более сложным, когда растворенным веществом является сильный электролит,, и процесс адсорбции принимает ионный характер. e 1
На границе твердое тело — раствор различают два вида
адсорбции — молекулярную, или адсорбцию неэлектролитов, когда твердое тело адсорбирует молекулы адсорбтива, и адсорбцию ионную, когда адсорбент избира-
тельно поглощает из раствора один из видов ионов растворенного электролита. Рассмотрим кратко оба вида адсорбции.
Молекулярная адсорбция из растворов. При адсорбции из раствора вместе с молекулами растворенного вещества адсорбируются и молекулы растворителя. Количество тех и других молекул, адсорбируемых твердым адсорбентом, зависит от их собственной адсорбционной способности, а также от концентрации растворенного вещества. Опыт показывает, что при малых концентрациях преобладает адсорбция молекул растворенного вещества, у/ри больших — адсорбция растворителя.
•/ ( Q Q \
На рис. 189 приведена зависимость удельной адсорбции [ —----I,
где Со — концентрация растворенного вещества до адсорбции, С — концентрация вещества после адсорбции, т — масса адсорбента) от концентрации адсорбтива в растворе. Как видно из рис. 189, вначале количество адсорбированного вещества увеличивается с ростом концентрации его в растворе, затем начинает преобладать адсорбция молекул
растворителя, в результате концентрация вещества в растворе повы-шается и потому дробь — становится отрицательной, так как Со— — С < 0. Кривая опускается ниже оси абсцисс. Таким образом, более полно адсорбция происходит из растворов низкой концентрации.
П. А. Р е б и н д е р предложил правило уравновешивания полярностей, согласно которому адсорбция будет идти, если полярность ве-
— 448 —
щества С, характеризуемая диэлектрической проницаемостью ес, будет находиться между полярностью веществ А и В, т. е. при условии ея > гс > или <С &в- Так, на границе вода (в = 80) — то-
луол (в = 2,4) анилин (е — 7,3) является поверхностно-активным веществом, т. е. он хорошо адсорбируется. На границе толуол —воздух (в = 1) анилин несколько повышает поверхностное натяжение, следовательно, поверхностно-активным веществом будет являться уже толуол, растворимый в анилине.
На основании правила уравнения полярностей П. А. Ребиндера можно сделать вывод о том, что чем больше разность полярностей между растворимым веществом и раствором, т. е. чем меньше растворимость растворенного вещества, тем лучше оно будет адсорбироваться. Правило распределения полярностей разъясняет порядок ориентации молекул поверхностно-активных веществ на границе раздела твердое тело — жидкость. При этом полярная часть молекулы поверхностно-активных веществ будет обращена к полярной фазе, а неполярная часть — к неполярной. Так, при адсорбции из растворов поверхностно-активных веществ углем или силикагелем имеет место различная ориентация молекул адсорбтива. При адсорбции углем к гидрофобной его поверхности обращена неполярная гидрофобная цепь углеродных атомов, а гидрафильная часть молекулы погружена в воду (рис. 190, а). При адсорбции поверхностно-активных веществ из неполярных растворителей силикагелем адсорбируемые молекулы своей гидфрофильной частью ориентированы
к поверхности адсорбента, а своей гидрофобной углеводородной частью направлены внутрь растворителя (рис. 190, б).
Таким образом, все гидрофильные вещества (силикагель, глины) хорошо адсорбируют поверхностно-активные вещества из неполярных или слабо полярных жидкостей.* Все неполярные гидрофобные вещества (уголь, графит, тальк, парафин), наоборот, хорошо адсорбируют поверхностно-активные вещества из полярных жидкостей, например, из водных растворов.
На твердом адсорбенте возможны три случая адсорбции: 1) адсорбция положительная, если растворенное вещество адсорбируется на поверхности адсорбента в большем количестве, чем растворитель; 2) адсорбция отрицательная, когда в большем количестве адсорбируется растворитель; 3) отсутствие адсорбции, когда концентрация растворенного вещества остается одинаковой и на поверхности адсорбента и в объеме раствора. Наибольший практический интерес представляет случай положительной адсорбции.
Сама по себе скорость адсорбции вообще велика, однако в случае адсорбции на твердых адсорбентах эта скорость в какой-то мерелими-
в данном случае
Вода
дензол
5
Рис. 190. Ориентация адсорбированных молекул поверхностно-активного вещество
а -
о о
— 449 —
тируется скоростью диффузии молекул растворенного вещества. Установление адсорбционного равновесия еще более затягивается в случае мелкопористых адсорбентов, например углей. На практике для ускорения установления адсорбционного равновесия прибегают к энергичному перемешиванию и встряхиванию.
Большое влияние на адсорбируемость того или иного растворенного вещества оказывает не только его природа, но и природа адсорбента и растворителя. Этот вопрос подробно был изучен многими учеными, в частности, А. А. Титовым, Л. В. Гуревичем, П. А. Ребиндером и др. Рассмотрим более подробно зависимость адсорбции от свойств твердой поверхности и природы растворителя. В этом случае следует особо от
Рис. 191. Краевые углы смачивания. а — поверхность смачивается жидкостью; б — поверхность не смачивается жидкостью
fl
метить свойство смачивания. Если на твердую поверхность нанести капли воды, возможны три случая: 1) капля растекается по поверхности; 2) капля остается на поверхности в виде шарика; 3) капля растекается лишь частично, образуя с поверхностью некоторый так называемый краевой угол (рис. 191).
Если капля жидкости растекается по поверхности или образует с пей острый краевой угол 0, это значит, что жидкость смачивает данную поверхность. Степень смачиваемости гладких поверхностей определяется величиной угла или величиной В = cosO. Чем больше положительное значение cos0, тем больше смачиваемость (тем больше силы адгезии между молекулами жидкости и молекулами адсорбента по сравнению с силами когезии внутри жидкости). Если угол 6 тупой, т. е. если cos 0 является величиной отрицательной, смачиваемость отсутствует (силы адгезии в данном случае меньше сил когезии).
По предложению П. А. Ребиндера, твердые поверхности, хорошо смачиваемые водой, называются гидрофильными, а не смачиваемые — гидрофобными. Так как гидрофобные поверхности хорошо смачиваются неполярными органическими жидкостями (например, углеводородами),']^ называют также олеофильными поверхностями.
Таким образом, адсорбция растворенных веществ твердыми адсорбентами подчиняется одному общему правилу; чем лучше растворитель смачивает поверхность адсорбента, тем меньше адсорбция молекул растворенного вещества из данного растворителя на этой поверхности, и, наоборот, если растворитель плохо смачивает твердую поверхность, адсорбция молекул растворенного вещества на ней будет велика.
Так, если растворитель хорошо смачивает поверхность адсорбента, он сильно понижает его поверхностное натяжение, следовательно, на
— 450 —
поверхности адсорбента появляется слой адсорбируемых молекул растворителя. Для молекул растворенного вещества не остается (или остается очень мало) места на поверхности адсорбента. В том случае, когда растворитель не смачивает поверхности адсорбента, она остается свободной, и молекулы растворенного вещества адсорбируются на
поверхности.
При адсорбции жидкого вещества на твердом адсорбенте выделяется теплота. Количество теплоты, выделенное при адсорбции одним граммом порошкообразного адсорбента данной жидкости, называется теплотой смачивания. Она связана с интенсивностью адсорбции данной жидкости адсорбентом, поэтому по теплоте смачивания можно судить об адсорбционной активности поглотителя. В табл. 83 при
ведены значения теплот смачивания некоторых адсорбентов водой и бензолом.
Из данных таблицы видно, что наибольшей активностью к воде обладает силикагель, к бензолу — уголь.
Как показали исследования, природу твердой поверхности адсорбента можно изменить: гидрофильную поверхность сделать гидрофобной, а гидрофобную — гидрофильной. Для этого на твердой поверхности адсорбента создают адсорбционный слой из по-
Таблица 83
Теплота смачивания различных адсорбентов
Адсорбент Теплота смачив а-ния, кал/г
вода бензол
Силикагель . . . 19,4
Уголь s 11,6 28,3
Почва ...... 9,5 2,9
Торф 14,8 2,6
верхностно-активных веществ, на-
пример мыла, жирных кислот. Если гидрофильную поверхность обработать раствором жирной кислоты, поверхность станет гидрофобной. Молекулы кислоты, ориентируясь таким образом, что полярные группы молекул обращены к поверхности адсорбента, а углеводородные радикалы — в воздух, адсорбируются на поверхности и сообщают ей гидрофобные свойства. Капли воды, как видно из рис. 192, образуют на поверхности тупые краевые углы и уже не смачивают ее. Аналогичное явление имеет место при гидрофобизации тканей; они
становятся водонепроницаемыми в результате пропитки их соответствующими гидрофобными веществами. В качестве последних в настоящее время нашли широкое применение кремнийорганические
жидкости.
На избирательном смачивании основан процесс флотации, имеющий широкое применение при обогащении руд. Сущность этого процесса заключается в разделении смеси гидрофильного и гидрофобного порошковидных веществ на основании их избирательного смачивания различными жидкостями. Горную породу или руду перед обогащением тщательно размалывают, затем энергично размешивают в воде, к которой прибавляют небольшое количество гидрофобного вещества (масла). Примеси обычно гидрофильны, а ценная часть породы — гидрофобна. В результате этого пустая порода остается в водной фазе
— 451 —
и оседает на дно, а наиболее ценная часть, обильно смачиваемая маслом, переходит в масляную пленку, затем собирается в специальном отстойнике.
Процесс флотации возможен и без применения масла. На поверхности воды путем энеричного пропускания воздуха создают обильную пену. Гидрофобные частицы горной породы прилипают к пузырькам воздуха, затем вместе с пеной их удаляют в специальный отстойник.
Ионнообменная адсорбция. Поскольку сильные электролиты в растворах полностью или почти полностью диссоциированы на ионы, адсорбция электролитов на поверхности твердых адсорбентов в результате действия обычных адсорбционных и электрических сил имеет свои
....................... жшЫашв
а......................&
Рис. 192. Гидрофобизация поверхности: а — гидрофильная поверхность; б — поверхность, гидрофобизи-рованная поверхностно-активном веществом
специфицические особенности. Иными словами, адсорбция ионов сильных электролитов протекает под воздействием двух родов сил: молекулярно-поверхностных адсорбента и электрических сил, проявляющихся только при адсорбции ионов.
Обычно различают три основных типа адсорбции электролита: 1) эквивалентная адсорбция; 2) обменная адсорбция; 3) специфическая (избирательная) адсорбция.
При эквивалентной адсорбции происходит эквивалентное поглощение катионов и анионов электролитов, т. е. молекулы элитролитов поглощаются целиком. Сам механизм поглощения можно представить следующим образом. Лучше адсорбируемый ион данного электролита притягивает свой парный, менее адсорбируемый ион на поверхность адсорбента. При этом адсорбируемость второго иона возрастает, первого — уменьшается, так как часть его удерживается в растворе другим, хуже адсорбируемым ионом. В результате оба иона поглощаются (адсорбируются) эквивалентно, почему эквивалентную адсорбцию часто и называют молекулярной. Она характерна для слабых электролитов. При эквивалентной адсорции электронейтральность на границе фаз не нарушается.
При обменной адсорбции избирательное поглощение одного из ионов электролита, находящегося в растворе, сопровождается одновременным вытеснением другого иона того же знака из поверхности адсорбента. Обмен ионами~ттротекает~ТГ строго эквивалентных количествах, поэтому элсктронейтральность на границе раздела фаз не нарушается. Опыт показывает, что обменная адсорбция протекает более медленно, чем обычная, п се можно рассматривать как хемосорбцион-ный процесс.
— 452 —
Если при обменной адсорбции взамен поглощаемого иона нейтральной соли адсорбент отдает в раствор эквивалентное количество ионов водорода или гидроксила, такая адсорбция носит название гидролитической. Например, адсорбция на угле неогранических нейтральных солей (NaCl, КС1, KNO3) сопровождается подщелачиванием, т. е. в данном случае по преимуществу адсорбируются анионы, а в раствор поступают ионы ОН-. К гидролитической адсорбции относятся все случаи обменного выделения адсорбентом ионов Н+ или ОН- независимо от того, образовались ли эти ионы в результате тех или иных поверхностных процессов на адсорбенте или же содержались в нем заранее как составная часть молекул. Так, глинистые минералы (каолинит, монтмориллонит) могут участвовать в обменной адсорбции своими Н+-ионами.
Гидролитическая адсорбция имеет большое значение в почвенных условиях, а также в корневом питании растений. На основе представлений об обменной адсорбции К. К. Гедройц создал свое учение о почвенном поглощающем комплексе, которое имеет важное значение для разрешения проблемы повышения плодородия почв. На основании полученного им большого экспериментального материала Гедройц установил, что поглощение иона почвой из раствора сопровождается выходом из нее другого иона в строго эквивалентных количествах. По Гедройцу, носителем обменной адсорбции в почве является почвенный поглощающий комплекс, который представляет собой высокодисперсную смесь нерастворимых в воде алюмосиликатных, органических и органоминеральных соединений. Многочисленными экспериментами Гедройц доказал, что в таком обмене участвуют только катионы, причем обменная способность их тем выше, чем больше валентность (в пределах ионов одной валентности тем выше, чем больше атомный вес). В качестве примера в табл. 84 приведены данные, полученные Гедройцем при исследовании образца черноземной почвы, насыщенной барием. Почва была обработана 0,1 н. растворами хлоридов перечисленных катионов при отношении раствора к почве 10 : 1.
Поскольку вытесненные количества бария эквивалентны адсорбированным количествам катионов, ясно, что адсорбционная способность катионов возрастает при переходе от лития к железу, а для катионов одинаковой валентности возрастает с увеличением атомного веса.
Таблица 84
Вытеснение бария из почвы различными катионами (мг-экв на 100 г почвы)
Катионы Вытеснено бария, мг-экв на 100а Катионы Вытеснено бария, мг-экв на 100 г
3,8 Mg2+ 7,7
Ма+ 4,5 Са2+ 10,2
К+ 6,8 А13+ 16,7
РЬ+ 7,8 Fe3+ 18,7
453 —
Осд ановимся на конкретных примерах обменной адсорбции.
Для снижения жесткости технических вод, которая в основном обусловливается присутствием солей кальция и магния, в технике применяются либо естественные силикаты — цеолиты и глаукониты, либо искусственные алюмосиликаты щелочных металлов, называемые пермутитами, Схематически обменное действие пермутита можно представить следующим образом:
пермутит - Na
+ Ca2++SC)2-
пермутит-Са
4-2Na+4-SO2-
Применение пермутитов позволяет устранить жесткость воды, но не освобождает воду от всех катионов и анионов. Практически полное очищение воды от посторонних катионов было достигнуто лишь в недавнее время путем применения ионообменных смол. Эти смолы или иониты получают путем введения ионогенных групп (SO3H, СООН, NH2) в скелет углеводородных цепей высокополимерных соединений. Одни смолы имеют кислотный характер (поверхность их заряжена отрицательно) и потому обменно адсорбируют только катионы с заменой любого из них на ион водорода. Такие адсорбенты получили название катионитов. Другие смолы, имеющие основной характер, получили название анионитов. Эти адсорбенты адсорбируют из растворов только анионы в обмен на ионы ОН~.
Пропуская воду через особые фильтры, заполненные тонко измельченными катионитом и анионитом, ее полностью очищают от всех катионов и анионов. По чистоте очищенная таким образом вода нисколько не уступает дистиллированной воде, т. е. очищенной путем перегонки.
Очистка воды с применением ионитов может быть представлена следующей схемой:
Реакции на катионите
катионит• Н2 + Са2++2С1 катионит -Са + 2H++2CI- _
катионит - Н2 + Mg-+ + SO2- катионит • Mg + 2Н+ + SO^~
катионит-<12 + Na++CI~ катионит - Н-Na + н+ +С1-
Реакции на анионите
анионит - (ОН)2 + 2Н++ SO2-^± анионит-SO4
анионит- (ОН)2 + Н++С1-♦ анионит- ОН-Cl
При очистке воды катионит и анионит следует брать в эквивалентных соотношениях. Для регенерации, т. е. восстановления уже отра-
— 454 —
ботанных катионитов, обычно применяют 3—5%-ные растворы серной или соляной кислоты. В результате этого катиониты «заряжаются» ионами водорода - ' ' •
катионит-Са +2H+42CI"
катионит • Нг + Са2+ 4- 2С1 ~
Для восстановления анионитов применяют чаще всего 5%-ный раствор NaOH или КОН
анионит*С1
4- Na+ 4 ОН
анионит-ОН 4Na+4Cl
Возможность получать при помощи ионообменных смол очищенную воду имеет большое значение для питания котлов высокого давления, а также в ряде производств (сахарной промышленности, пивоварения, химии чистых реактивов, производстве фототоваров, лекарственных препаратов). Особенно большое значение ионообменные смолы приобрели за последнее время в винодельческой промышленности. С их помощью производят удаление излишков Fe3+, Cu2+, Са2+, вызывающих помутнение вина, а также обеспечивают сусло вина. В молочной промышленности иониты широко используются для изменения солевого состава молока. Известно, что коровье молоко богаче женского содержанием соответствующих солей и отличается характером створаживания, что зависит от соотношения кальция и казеина. Удаляя из коровьего молока с помощью ионообменных смол избыток кальция, его делают вполне пригодным для питания грудных детей.
В свое время было предложено несколько уравнений, описывающих обменную адсорбцию. Наиболее точным- оказалось теоретически выведенное Б. П. Никольским уравнение, которое имеет следующий вид:
(XI,23)
где 1\ и Г2 — количества поглощенных ионов, выраженные в мг-экв на 1 г адсорбента, и С2 — концентрации или активности соответствующих ионов в растворе, Zt и Z2 — валентности ионов, К — константа обмена.
Величина константы ионного* обмена К определяет соотношение ионов в поглощенном состоянии при заданной концентрации в растворе и должна заметно отличаться от единицы.
В практике известно немало случаев, когда на твердом адсорбенте адсорбируются избирательно преимущественно только ионы одного вида. В этом случае катионы (или анионы) поглощаются из раствора и необменно фиксируются на поверхности адсорбента, сообщая поверхности свой заряд. Такой вид адсорбции получил название специфической адсорбции, или адсорбции потенциалопределяющих ионов. Специфическая адсорбция имеет особенно большое значение в коллоидных системах при образовании так называемого двойного электрического слоя.
— 455 —
Процессы специфической адсорбции широко представлены не только в биологических объектах, но и в почвах. Согласно С. Н. А л еш и -н у, ион водорода (протон), в отличие от других катионов, может адсорбироваться многими минералами необменно, что играет большую роль в выветривании различных горных пород и образовании обменной почвенной кислотности.
Адсорбционные явления лежат в основе важнейшего метода анализа сложных смесей — хроматографии.
§ 128. Адсорбция и биологические процессы
Явления адсорбции чрезвычайно широко распространены в природе. Там, где соприкасаются газы (или пары), жидкости и твердые тела, имеют место адсорбционные процессы. Почва хорошо поглощает (адсорбирует) не только растворенные в воде органические и минеральные соединения, но и воздух, углекислоту, пары воды, аммиак. Поглощение корнями питательных элементов из почвы начинается с их адсорбции на поверхности корневых волосков и тонких неопробковевших корней. Усвоение растением углекислого газа при фотосинтезе начинается с адсорбции СО2 на внутренней поверхности листа. Превращения поглощенных солей и углекислоты связано с явлениями адсорбции и десорбции на протоплазматических структурах и поверхностях клеточных органелл, пластид, митохондрий, микросом.
В животных организмах явления адсорбции также играют очень большую роль в их жизнедеятельности. Роль адсорбции обусловлена наличием в организме огромного количества самых разнообразных поверхностей раздела — стенок сосудов, поверхности клеток, клеточных ядер и вакуолей, коллоидных частиц протоплазмы и, наконец, поверхности раздела между организмом и средой. Особенно важную функцию выполняет поверхность раздела между организмом и средой для низших организмов и организмов, живущих в воде, так как ей принадлежит существенная роль в процессах питания и обмена веществ. Исследования последних лет показали, что пищевые вещества, как правило, являются поверхностно-активными веществами, и потому первым этапом их усвоения является адсорбция, а процесс их химического превращения уже вторичен.
Чтобы более наглядно представить себе роль и значение адсорбционных процессов, протекающих в животном организме, рассмотрим адсорбционные возможности эритроцитов крови человека. Исследования показали, что эритроциты являются переносчиками различных веществ, в том числе аминокислот, которые они разносят и передают клеткам и различным тканям организма. Количество эритроцитов достигает в крови взрослого человека примерно 5 000 000 в I лш3. В среднем на 1 кг веса у здоровых мужчин приходится 450 миллиардов эритроцитов или 27 триллионов на весь организм. Учитывая, что диаметр эритроцита равен 7—8 мк, можно легко подсчитать, что общая поверхность эритроцитов всей крови человека составит примерно 3200 м2.
Большинство реакций, протекающих в организме, совершается при непосредственном участии ферментов-катализаторов.1 Исследования показали, что первые стадии действия любого фермента сводятся к адсорбции субстрата на поверхности Ьерментного комплекса, и только после этого фермент проявляет свое специфичоЛкое каталитическое действие.
Глава XU
ИЗМЕНЕНИЕ СОСТОЯНИЯ КОЛЛОИДНЫХ СИСТЕМ
Коллоидные системы обладают высокоразвитой поверхностью раздела и, следовательно, большим избытком свободной поверхностной энергии. Поэтому эти системы термодинамически неустойчивы и имеют тенденцию к самопроизвольному уменьшению межфазной энергии. Это в большинстве случаев происходит за счет уменьшения суммарной поверхности частиц дисперсной фазы золей. Если в силу создавшихся условий мицеллы золя приходят в тесное соприкосновение между собой, они соединяются в более крупные агрегаты. Этот процесс укрупнения коллоидных частиц в золях, происходящий под влиянием внешних воздействий, носит название коагуляции (от латинского слова coagula-tio — свертывание, створаживание).
Процесс осаждения укрупненных частиц твердой фазы золя называется седиментацией (от латинского sedimentation — осаждение).
Процесс коагуляции всегда связан с уменьшением степени дисперсности и обусловлен агрегативной неустойчивостью коллоидных систем. В коагуляции принято различать две стадии: скрытую коагуляцию, когда невооруженным глазом еще нельзя наблюдать какие-либо внешние изменения в золе, и явную коагуляцию, когда процесс агрегации частиц дисперсной фазы золя может быть легко обнаружен визуально.
Лиофобные золи характеризуются сравнительно короткой стадией скрытой коагуляции, для высокомолекулярных соединений период скрытой коагуляции может быть продолжительным. Часто скрытый период коагуляции в растворах высокомолекулярных соединений совсем не переходит в явную форму или заканчивается студнеобразо-ванием.
Факторы коагуляции коллоидных систем бывают весьма разнообразными. Коагуляция может быть вызвана повышением температуры, длительным диализом, добавлением электролитов, разного рода механическими воздействиями (размешиванием, встряхиванием, взбалтыванием), сильным охлаждением, ультрацентрифугированием, концентрированием, пропусканием электрического тока, а также действием на данный золь других золей. В ряде случаев коагуляция может происходить в результате химических реакций, протекающих в золях (явление старения). Поскольку главное условие уменьшения устойчивости коллоидных растворов — потеря электрического заряда, основными методами их коагулирования являются методы снятия зарядов. Чаще всего в практике для этой цели пользуются воздействием на коллоидные растворы различных электролитов.
§ 129. Коагуляция гидрофобных золей электролитами
Наиболее важным и наиболее изученным фактором коагуляции гидрофобных золей является действие электролитов. Практически все электролиты, взятые в достаточном количестве, способны вызвать коагуляцию коллоидных растворов. В частности, гидрофобные золи, ча
— 457 —
стицы которых имеют двойные электрические слои, коагулируют от прибавления сравнительно небольших количеств электролитов. Одна-ко концентрации различных электролитов по своему коагулирующему действию довольно резко различаются между собой.
Чтобы начался процесс коагуляции, нужно наличие некоторой минимальной концентрации электролита в золе. Наименьшая концентрация электролита, вызывающая коагуляцию, получила название порога коагуляции и выражается в ммоль!л. Обычно порюг коагуляции определяют по помутнению коллоидного раствора, по изменению окраски и по другим признакам.
Опытом установлено, что коагулирующее действие обычно оказывает ион, заряд которого по знаку противоположен заряду поверхности коллоидных частиц. Так, для положительно заряженных золей коагулирующим ионом электролита являются анионы, а для отрицательных — катионы. Коагулирующее действие электролита сильно возрастает с увеличением зарядности иона-коагулятора. Иными словами, ионы-коагуляторы высшей зарядности вызывают явную коагуляцию при значительно меньших концентрациях, чем ионы низшей зарядности (правило Шульце — Гарди).
Так, для катионов К+, Ва2+, А13+, отношение порогов коагуляции их хлоридов при действии на отрицательно заряженный золь As2S соответственно равно
^КС1 • ^ВаС12 • Caici., == 49,5 : 0,69 : 0,093 (ммоль/л)
или, принимая порог коагуляции иона алюминия за единицу,
Ск+:Св^:Са1з+=540 : 7>4:1-
В случае коагуляции положительно заряженного золя гидроокиси железа Fe (ОН)3 для анионов-коагуляторов С1“ и SO*- отношение порогов коагуляции калийных солей соответственно равно
С :С2. =9,0: 0,205
или округленно
Правило Шульце — Гарди носит приближенный характер, так как коагулирующее действие электролита зависит не только от зарядности его ионов. Некоторые органические однозарядные основания, например. катионы морфина, обладают более сильным коагулирующим действием, чем двухзарядные ионы. Подобное явление объясняется тем, что большие органические ионы обладают более сильной адсорбируемостью, т. е. они гораздо легче входят во внутреннюю часть двойного электрического слоя, образованного вокруг коллоидного ядра.
Пороги коагуляции ионов одного и того же знака и зарядности отличаются друг от друга. Здесь сказывается влияние не только размера ионов, но и степень их гидратации. По величине коагулирующей спо
— 458 -
собности ионы щелочных металлов можно расположить в следующий ряд
Cs+ > Rb+ > NH+ > К+ > Na+ > Li+
Анионы можно расположить в соответствующий ряд
I- >NOt> Ег->С1-
О
Такого типа ряды известны и для щелочноземельных металлов. Эти ряды ионов получили название лиотропных рядов.
Как показывает опыт, коагуляции гидрофобных золей можно вызвать и при помощи смеси электролитов. При этом возможны три случая.
1. Коагулирующее действие смешиваемых электролитов суммируется.
2. Коагулирующее действие смеси электролитов меньше, чем в случае чистых электролитов. Это явление носит название антагонизма ионов. Оно характерно для смесей ионов, имеющих различную валентность.
3. В ряде случаев имеет место взаимное усиление коагулирующего действия смешиваемых ионов. Это явление называется синергизмом ионов.
Описанные явления ни в коем случае нельзя смешивать с явлениями физиологического антагонизма ионов, под которыми обычно понимают ослабление одним катионом токсического или иного физиологического действия, вызываемого другим катионом.
Помимо электролитов, коагуляция гидрофобных коллоидов может быть вызвана смешиванием в определенных количественных соотношениях с другим гидрофобным золем, гранулы которого имеют противоположный знак. Это явление носит название взаимной коагуляции-оно может рассматриваться как частный случай электролитной коагуляции. Так, положительно заряженный золь Fe (ОН)3 может быть ско-агулирован путем прибавления отрицательно заряженного золя As2S3. Табл. 85 иллюстрирует взаимодействие этих золей. Причем в этом опыте гидрозоль Fe(OH)3 прибавляется в разных количествах к раствору, содержащему 0,56 мг гидрозоля As2S3.
Таблица 85
Взаимная коагуляция растворов F (ОН)3 и As2S3
Прибавлено Fc(OH)3, мг Результат опыта Заряд коллоидной смеси
0,8 Коагуляции нет (-)
3,2 Легкая муть (-)
4,8 Сильная муть (-)
6,1 Полная коагуляция 0
8,0 Неполная коагуляция (+)
12,8 Легкая муть (+)
20,8 Коагуляции нет (+)
— 459 —
На рис. 193 показано изменение концентрации золя сульфида мышьяка при прибавлении к нему золя гидроксида железа (III), ; Опыт показывает, что наиболее полной взаимная коагуляция стано-вится тогда, когда число разноименных электрических зарядов на > частицах обоих коллоидов будет одинаково. _ «
Явление взаимной коагуляции золей имеет чрезвычайно широкое распространение в природе и в целом ряде технологических процессов. Так, взаимная коагуляция происходит при смешении морской и речной воды. При этом ионы солей морской воды адсорбируются на заряженных коллоидных частицах речной воды, в результате чего происхо-
дит их коагуляция. По этой причине на дне постепенно скаплива- & ются большие количества ила, река мелеет, образуется множество < мелей и островков. >
С явлениями взаимной коагу- Я ляции мы часто встречаемся в быту. Чернила представляют собой 4$ коллоидные растворы различных красителей. Причем в разных чернилах коллоидные частицы заря-жены по-разному. Вот почему при
Рис. 193. Взаимная коагуляция лио- смешении разных чернил имеет ме-фобных коллоидов сто взаимная коагуляция.
На явлении взаимной коагуля-\ ции основана очистка питьевой во-
ды. Исследования показали, что при добавлении к очищаемой воде раствора А12 (SO4)3 в течение примерно первых ЗОсек в результате гидролиза коагулянта образуется коллоидная гидроокись алюминия А1(ОН)3, которая обладает громадной суммарной поверхностью, на которой происходит адсорбция коллоидных примесей очищаемой воды. Тушь при температуре ниже нуля портится — коагулирует, так как вода замерзает и коллоидные частицы сажи, лишаясь защитной гидратной оболочки, необратимо коагулируют.
Механизм электролитной коагуляции. Как известно, гидрофобные коллоиды неустойчивы в изоэлектрическом состоянии, т. е. электроней-тральные частицы коагулируют с наибольшей скоростью. На рис. 194 показана схема снятия заряда с коллоидной частицы при добавлении электролита с двухзарядными анионами. Как видим, гранула становится электронейтральной в том случае, если противоионы диффузного слоя, заряженные отрицательно, перемещаются в адсорбционный слой. Чем выше концентрация прибавляемого электролита, тем сильнее сжимается диффузный слой, тем меньше становится дзета-потенциал и, следовательно, тем быстрее начинается процесс коагуляции. При определенной концентрации электролита практически все противоионы перейдут в адсорбционный слой, заряд гранулы снизится до нуля и коагуляция пойдет с максимальной скоростью, так как отсутствие диффузного слоя обусловит значительное понижение давления рас
клинивания.
— 460 -
Коагулирующее действие электролитов не сводится только к сжатию диффузного слоя. Как показали многочисленные исследования, одновременно протекает избирательная адсорбция на коллоидной частице тех ионов электролита, которые имеют заряд, противоположный грануле. Причем чем выше заряд иона, тем интенсивнее он адсорбируется. Накопление ионов в адсорбированном слое сопровождается уменьшением не только дзета-потенциала, но и диффузного слоя (рис. 194).
Рис. 194. Снятие заряда с коллоидной частицы при добавлении электролита с двухзарядными анионами:
а — до начала коагуляции гранула заряжена положительно; б— гранула стала электронейтральной, коагуляция протекает с максимальной скоростью
При коагуляции золей электролитами имеет место также и процесс ионообменной адсорбции. При этом ионы добавляемого электролита обмениваются на одноименно заряженные противоионы адсорбционного слоя. Если ионы добавляемого электролита имеют большую зарядность по сравнению с одноименно заряженными противоионами, такая замена приводит к довольно значительному понижению дзета-потенциала.
Из всего вышесказанного не следует делать вывод о том, что основная причина коагуляции заключается в достижении некоторого постоянного для всех случаев критического дзета-потенциала. Исследования последних лет, проведенные советскими учеными В. В. Дерягиными его сотрудниками, показали, что коагулирующее действие электролитов заключается не столько в непосредственном уменьшении сил отталкивания между коллоидными частицами через понижение дзета-потенциала, сколько в том, что изменение строения двойного электрического слоя и сжатие диффузной его части, обусловленное прибавлением электролита-коагулянта, влечет за собой понижение расклинивающего действия гидратных (сольватных) оболочек диффузных ионов, разъединяющих коллоидные частицы. Иными словами,
— 461 —
необходимое для коагуляции данного золя понижение расклинивающего действия (или давления) сольватных оболочек достигается уменьшением диффузного слоя противоионов, что ведет к соответствующему понижению величины дзета-потенциала.
Для более углубленного понимания процессса коагуляции остановимся несколько подробнее на механизме действия расклинивающего давления. Согласно теоретическим воззрениям и экспериментальным данным В. В. Дерягина пленка жидкости, заключенная между двумя погруженными в нее твердыми телами, оказывает на них расклинивающее давление и тем самым препятствует их сближению. Это давле-’ ние быстро возрастает с утоныиением пленки и в большой степени снижается при прибавлении электролитов. Аналогичное явление про-
Рис 195. Схема перекрытия ионных атмосфер двух сферических частиц
исходит и ь коллоидных растворах. Как показали исследования, коллоидные частицы могут беспрепятственно приближаться друг к другу на расстояние 10-5 см. Дальнейшее сближение встречает препятствие со стороны сольватных (гидратных) оболочек, связанных с ионной атмосферой коллоидной мицеллы. Это и есть расклинивающее давление по Дерягину. Энергия, которая необходима для преодоления этого давления, быстро возрастает и достигает максимума при расстоянии между частицами примерно 10-7—10~8 см. Ван-дер-ваальсовы силы притяжения между коллоидными частицами непрерывно усиливаются, но их когезионное действие слабее расклинивающего действия тонких сольватных оболочек. Причем, электрические силы отталкивания между коллоидными мицеллами возникают лишь в том случае, если имеет место взаимное перекрытие их ионных атмосфер (рис. 195). В этом случае происходит перераспределение ионов в ионной атмосфере взаимодействующих мицелл, что приводит к появлению дополнительных сил отталкивания. В том случае, когда расстояние между мицеллами становится меньше 10~7—10~8 см, расклинивающее действие быстро уменьшается, достигает нулевого значения и наступает коагуляция.
Введение в коллоидную систему электролитов-коагулянтов вызывает уменьшение толщины ионной атмосферы, что в свою очередь сильно ослабляет ее расклинивающее действие. В результате создаются
— 462 —
более благоприятные условия для преобладания сил притяжения между коллоидными мицеллами над силами отталкивания.
В ряде случаев при добавлении к золям электролитов с многозарядными ионами, заряд которых противоположен по знаку заряду коллоидных частиц, может наблюдаться не коагуляция, а стабилизация золя и перемена знака дзета-потенциала. Это явление получило название в коллоидной химии перезарядки золей. Так, при добавлении к золю платины небольших количеств хлорида железа FeCl3 наблюдается понижение отрицательного заряда коллоидных частиц платины и их коагуляция. Дальнейшее увеличение концентрации FeCl3 приводит к перезарядке коллоидных частиц платины; они получают положительный заряд.
^Концентрация электролита-коагулятора
Рис. 197. Перемена знака потенциала
Рис. 196. Неправильные ряды при ко-
агуляции
В процессе перезарядки золь платины претерпевает следующие изменения: отрицательно заряженный золь — коагуляции нет; заряд равен нулю — коагуляция; положительно заряженный золь — коагуляции нет; заряд равен нулю — коагуляция и т. д. Процесс коагуляции золя платины раствором FeCI3 представлен в табл. 86.
Таблица 86
Зоны коагуляции золя платины хлоридом железа FeCl3
Концеш рация FeCl3, моль/л Ксагуляционный эффект Направление электрофореза Знак заряда частицы
0,0208 Коагуляции нет Перенос к аноду (-)
0,0417 То же То же (-)
0,0883 Полная коагуляция Переноса нет 0
0,1633 То же То же 0
0,2222 » » 0
0,3333 Коагуляции нет Перенос к катоду (+)
0,5567 То же То же (-)
0,8333 » i (+)
3,3330 » » (+)
6.С670 » » (+)
16,33 Полная коагуляция Переноса нет 0
33,33 То же То же 0
163,7 » » 0
— 463 —
Такое чередование состояний электронейтральности и заряжен-ности частиц называют чередованием зон коагуляции или явлением $ неправильных рядов (табл. 86). 1К|
Если данные табл. 86 представить в виде графика, где по оси абсцисс отложить концентрации хлорида железа (III), а по оси ординат — ин- ? тенсивность эффекта коагуляции, получится кривая (рис. 196).
Явление перезарядки коллоидных мицелл золя платины под влиянием FeCl3 хорошо видно на кривой изменения дзета-потенциала (рис. 197). Здесь по оси абсцисс отложены значения концентраций прибавляемого электролита-коагулятора, а по оси ординат — измененные значения дзета-потенциала. Как видим, под влиянием электролита дзета-потенциал довольно резко уменьшается по абсолютной величине, затем, переходя через нулевое значение, получает положительное значение, которое по мере дальнейшего прибавления электролита проходит через соответствующий максимум и затем уменьшается.
§ 130. Кинетика коагуляции
Поскольку коагуляция любого золя совершается не мгновенно, а требует некоторого промежутка времени, возник вопрос о с к о* М р о с т и процесса коагуляции. Протекание процесса коагуляции во времени можно наблюдать по изменению свойств коллоидного раствора, например, по изменению окраски, яркости опалесцирующего конуса Фарадея — Тиндаля, по усилению мутности.
Многочисленные исследования показали, что наиболее надежным Ji методом наблюдения процесса коагуляции во времени является метод М подсчета числа частиц за определенный промежуток времени в ультрамикроскопе. Согласно теории коагуляции золей, предложенной М. С м q л у х о в с к и м (1906), началом коагуляции считают соприкосновение двух коллоидных частиц и слипание их в один агрегат. Эти удвоенные частицы, совершая броуновское движение и встречаясь с другими такими же или одиночными частицами, способны образовать тройные, четверные и т. д. частицы — вплоть до начала седимента- I
ции. В своей теории М. Смолуховский скорость коагуляции уподобля- I
ет скорости обычных химических реакций второго порядка и на основании этого выводит соответствующее уравнение. Отличие с точки зрения кинетики заключается в том, что в случае обычной химической реакции прореагировавшие молекулы в дальнейшем не участвуют в реакции, а коллоидные частицы, слипаясь при столкновении, продолжают участвовать в процессе коагуляции, образуя все более сложные I комплексы.
В начале коагуляции образование удвоенных, утроенных и т. д. частиц протекает незаметно и более медленно; затем по мере нарастания концентрации электролита-коагулятора скорость коагуляции значительно увеличивается. Таким образом различают медленную и быструю коагуляцию. Эти понятия не следует смешивать с понятиями скрытой и явной коагуляции.
Кривая изменения скорости коагуляции золя в зависимости от концентрации электролита (рис. 198) хорошо поясняет эти понятия и их
— 464 —
Рис. 198. Кривая скорости коагуляции в зависимости от концентрации электролита (схема)
представленной на рис. 198.
связь с другими величинами, характеризующими ход коагуляции. Ня кривой OSKN отрезок OS отвечает периоду скрытой коагуляции; точка S выражает одновременно и порог коагуляции и критический потенциал, с которых начинается явная коагуляция; отрезок SK соответствует периоду медленной коагуляции, скорость которой быстро увеличивается с увеличением концентрации электролита, а затем достигает некоторой постоянной величины; точка Д' выражает не толь, ко концентрацию, вызывающую быструю коагуляцию, но и дзета-потенциал, равный нулю; отрезок KN соответствует быстрой коагуляции, независимой от концентрации прибавляемого электролита.
В табл. 87 приведены данные о продолжительности полной коагуляции гидрозоля золота при действии на него в качестве коагулятора хлорида натрия в возрастающих концентрациях.
Как видно из этой таблицы, скорость коагуляции гидрозоля золота полностью соответствует ходу кривой,
Процессы медленной коагуляции весьма слабо изучены и в настоящее время. Предполагают, что медленное протекание процесса коагуляции обусловливается тем, чго лишь очень небольшое число столкновений коллоидных частиц приводит к их слипанию (агрегации). Установлено, что слипаются лишь те частицы, у которых по какой-либо причине снизился до критического значения дзета-потенциал, или частицы, обладающие большой скоростью и при столкновении попадающие в сферу взаимного притяжения.
Процессы быстрой коагуляции изучены значительно лучше. При быстрой коагуляции каждое столкновение коллоидных частиц приводит к их слиянию (соединению). Исходя из этих представлений, Смолу-
Таблица 87
Скорость коагуляции гидрозоля Au раствором NaCl
Концентрация NaCl. ммоль! л Продолжительность коагуляции, сек Концентрация NaCl. ммоль]л Продол ж ительность коагуляции, сек
5 150 (область медленной коагуляции) 50 7,0 (область быстрой коагуляции)
10 12 • 100 7,0
20 7,2 300 500 7,5 7,0
—- 465 —
ховский и вывел уравнение, характеризующее скорость быстрой коагуляции:
(х \ т— 1
V /
Пт = По ----—- (X11 ,1)
(1+т)
где пт — число частиц, каждая из которых состоит из т частиц подвергшихся превращению за время коагуляции т, п0 — первоначальное число частиц, 0 — время, в течение которого количество частиц уменьшается вдвое против начального.
Экспериментальная проверка уравнения доказала хорошее согласие теории с опытом. Медленная коагуляция длится часы и сутки, быстрая протекает за время, исчисляемое секундами и даже долями секунды.
§ 131. Пептизация гидрофобных золей
Часто продукт коагуляции гидрофобных золей — осадок, или коагель — может быть вновь переведен во взвешенное состояние путем обработки его определенным электролитом. Так, скоагулированный золь гидроокиси железа можно вновь вернуть в исходное состояние, если осадок Fe(OH)3 обработать водным раствором хлорида железа. Процесс перехода осадка во взвешенное состояние под влиянием внешних факторов получил название пептизации. Этот процесс противоположен коагуляции, потому его называют также декоагуляцией.
Вещества, способствующие переходу коагеля в золь, называют пептизаторами. Так FeCI3, А1С13, НС1 являются пептизаторами коагеля гидроокиси железа. Обычно пептизаторами являются электролиты, вернее один из ионов электролита (так называемый ион-пептизатор). Однако в ряде случаев пептизирующим действием могут обладать и неэлектролиты, например растворитель.
Сам процесс пептизации в основном обусловливается адсорбционными явлениями, в результате которых происходит не только повышение дзета-потенциала дисперсных частиц, но и увеличение степени их сольватации (гидратации). Сообщение скоагулированным частицам дисперсной фазы золя заряда способствует, с одной стороны, общему разрыхлению осадка, с другой — переводу этих частиц во взвешенное состояние благодаря броуновскому движению. При этом происходит образование вокруг диспергируемых частиц сольватных (гидратных) оболочек, производящих свое расклинивающее действие. В табл. 88 сопоставлены процессы пептизации и коагуляции.
Как и коагуляция, пептизация гидрофобных золей не затрагивает глубинных масс коллоидного ядра. Эти процессы протекают в тончайших слоях на поверхности раздела фаз, поэтому для пептизации, как впрочем и для коагуляции, требуются незначительные количества электролитов по сравнению с количеством осадка, переводимого в состояние золя. Так, если брать одинаковое количество коагулянта и пептизировать его различным количеством пептизатора, то при малых колн-
— 466 —
Таблица 88
сопоставление процессов коагуляции и пептизации
Коагуляция
Пептизация
Заряд коллоидных частиц понижается
Структура мицеллы нарушается: диффузная ионная атмосфера исчезает, гидратация частиц резко понижается
Силы притяжения между частицами начинают превышать силы взаимного отталкивания одноименных зарядов, лежащих ниже критического потенциала коагуляции
Частицы слипаются в агрегаты, образуя хлопья: золь переходит в коагель
Броуновское движение частиц вследствие их укрупнения прекращается
Заряд коллоидных частиц повышается
Структура мицеллы восстанавливается: появляется диффузная ионная оболочка, восстанавливается гидратация частиц
Силы взаимного отталкивания частиц, одноименно заряженных выше критического потенциала пептизации, начинают превышать силы притяжения
Агрегаты частиц (хлопья коагулята) распадаются, частицы коагеля разъединяются: коагель снова переходит в золь
Броуновское движение коллоидных частиц восстанавливается
чествах происходит лишь его адсорбция без растворения осадка (кривая ОА, рис. 199), при дальнейшем повышении концентрации пептиза-тора происходит и увеличение растворимости (кривая АВ). Если и дальше увеличивать количество пептизатора, растворимость, быстро увеличиваясь, достигает определенного предела и уже не зависит от количества пептизатора (кривые ВС и CD). При большом избытке пептизатора может наступить коагуляция (кривая DE). Рассмотренная нами кривая ОЕ на рис. 199 дает типичную картину адсорбционной пептизации.
Помимо адсорбционной, различают еще диссолюционную пептизацию. Этот вид пептизации охватывает собой все случаи, когда процесс пептизации сопряжен с химической реакцией поверхностно расположенных молекул коллоидных мицелл. Он состоит из двух фаз: образования путем химической реакции растворимого электролита-пептизатора и адсорбции образовавшегося пептизатора, приводящей к образованию мицелл и пептизации коагеля. Типичным примером диссолюционной пептизации могут служить пептизация гидроокисей металлов кислотами, пептизация геля оловянной кислоты щелочами и др.
Максимальная дисперсность золей, получаемых при адсорбционной пептизации, определяется степенью дисперсности первичных частиц, образующих хлопья коагеля.
При диссолюционной пептизации граница дробления частиц может выходить из области коллоидов и достигать молекулярной и ионной степеней дисперсности. Типичный ход адсорбционной и диссолюционной пептизации представлен на рис. 199.
При постоянном количестве пептизатора и возрастающем количс-честве коагеля пептизируемость последнего сначала возрастает, дости
— 467 —
гая максимума, затем уменьшается. Эта закономерность, установленная В. Оствальдом и А. Буцагом, получила название правила осадка. Согласно ему коллоидная растворимость при пептизации зависит от количества осадка, взятого для растворения. Типичная кривая правила осадка показана на рис. 200. Таким путем протекает, например, пептизация некоторых минералов каолинитовой группы раствором гумуса. Следствием, вытекающим из правила осадка, является коагуляция золя от прибавления к нему избытка осадка, из которого он получился при пептизации. Например, золь гидроокиси алюминия
А1(ОН)3 можно коагулировать, внося в него избыток геля другого аналогичного гидрата, например Fe(OH)3.
Рис. 199. Кривая пептизации при переменном количестве пептизатора
----Количество исходного осадка
Рис. 200. Кривая «правила осадка»
Пептизация играет большую роль во многих явлениях природы и в технике. Обработка почвы раствором соли с одновалентным катионом приводит к пептизации ее коллоидной части. В этом случае имеет место не адсорбция потенциалопределяющих ионов, а обмен ионами в диффузном слое. Дело в том, что в черноземных почвах коллоидные частицы содержат в диффузном слое преимущественно ионы Са2+ и Mg2+, что обусловливает небольшую величину дзета-потенциала и слабые силы отталкивания. Почвенные коллоиды находятся в скоагулиро-ванном состоянии, поэтому они не вымываются из почвы. При обработке почвы раствором хлорида натрия ионы Са2+ и Mg2+ в диффузном слое в результате ионного обмена замещаются на ионы натрия, что приводит к пептизации почвенных коллоидов и к переходу их при достаточном увлажнении в состояние золя. Перешедшие в состояние золя коллоиды легко вымываются из верхних горизонтов почвы в нижние, в результате чего почва теряет свои ценные агрономические свойства, становится бесструктурной. Подобные процессы происходят в солонцовых почвах, содержащих значительное количество ионов натрия. Вот почему основатель учения о почвенном поглощающем комплексе К. К. Г е д р о й ц назвал кальций стражем плодородия почвы.
— 468 —
Если почву обрабатывать достаточно концентрированным раствором NaCI, ионы Са2+ в диффузном слое могут быть практически полностью замещены ионами натрия. Однако пептизации в этих условиях не будет, так как высокая концентрация электролита вызывает сжатие диффУзного слоя и преобладание сил притяжения над силами отталкивания. Последующая промывка почвы водой от избытка NaCI приводит к расширению диффузного слоя и, как результат, к пептизации коллоидов. На этом принципе основан один из методов выделения почвенных коллоидов.
П. А. Р е б и н д е р с сотрудниками изучил пептизирующее действие органических поверхностно-активных веществ на минералы, входящие в состав цементов. Адсорбируясь из раствора на поверхности зерен цемента, молекулы поверхностно-активных веществ проникают в микрощели поверхности. При этом образуются адсорбционные слои, проявляющие свое расклинивающее действие, это приводит в конечном итоге к разрыву зерен минералов на частицы коллоидных размеров.
Хорошо известное каждому моющее действие мыла тесно связано с процессом пептизации. Коллоидный ион мыла, являясь диполем, хорошо адсорбируется частичками грязи, сообщает им заряд и способствует их пептизации. Грязь в виде золя легко удаляется с моющей поверхности. Пептизация используется для увеличения прочности искусственно получаемых коллоидных систем, например, при дроблении веществ в коллоидных мельницах и т. д.
Не всякий осажденный коллоид удается снова перевести в состояние золя. Опыт показывает, что лучше всего пептизируются свежеосаж-денные рыхлые осадки, содержащие воду, например Fe(OH)s, А1(ОН)3 и др. С течением времени способность к пептизации уменьшается. Плотные осадки, полученные от гидрозолей Ag, Си, Pt, а также гидрозоли с ярко выраженной гидрофобностью практически не поддаются пептизации. Однако в ряде случаев удается пептизировать и коагулянты, не содержащие воды. Так, прокаленная окись железа пептизируется жидким стеклом; оловянная кислота, не содержащая гигроскопической воды, может быть пептизирована при кипячении ее с едкими щелочами и т. д.
§ 132. Тиксотропия
Если к золю гидроксида железа прибавить какой-нибудь коагулятор (NaCI) в количестве, недостаточном для полной коагуляции, вязкость золя начнет заметно увеличиваться. С течением времени золь может превратиться в сплошной студень (гель). При сильном встряхивании гель вновь приобретает прежнюю легкоподвижность, вязкость его уменьшается до первоначального исходного значения. Однако стоит золь на некоторое время оставить в покое, он вновь превращается в студень. Это явление изотермического обратимого перехода золь
гель получило название тиксотропии (от греч. thixis — трогать и trophos —- меняться).
469 —
Тиксотропия—^явление довольно распространенное. Оно наблюдается в золях V2O5, WO3, Fe2O3, в различная суспензиях бентонита, в растворах вируса табачной мозаики, миозина. Причем, тиксотропные гели легче всего образуются у золей, обладающих асимметричным строением частиц (например, палочкообразной формы). Тиксотроп-
ствия
между
ные структуры возникают лишь при определенных концентрациях коллоидных частиц и электролитов. Для обратимого (тиксотропного) застудевания требуется определенная величина дзета-потенциала,, лежащая выше критического. В этом случае заряд коллоидных частиц хотя и понижен, но не в такой степени, чтобы начался процесс коагуляции. В этих условиях уже становятся заметными силы взаимодей-отдельными частицами дисперсной фазы, они образуют своеобразную сетку, каркас. При сильном встряхивании связь между частицами дисперсной фазы нарушается,— тиксотропный гель переходит в золь. В состоянии покоя связи в результате соударения частиц при броуновском движении восстанавливаются, золь вновь переходит в тиксотропный гель.и т. д.
Рассмотрим, как происходит взаимодействие коллоидных частиц в зависимости от расстояния между ними. Энергия взаимодействия W двух коллоидных частиц в зависи-
мости от расстояния между их поверхностями изменяется по кривой, изображенной на рис. 201, где положительные значения W
соответствуют отталкиванию, а отрицательные — притяжению частиц. Эти кривые носят название потенциальных кривых.
Из рис. 201 видно, что на определенном расстоянии г0 существует своеобразный барьер отталкивания S, который препятствует сближению частиц. Этот барьер устраним при высокой концентрации электро-лита-коагулятора. Вследствие преобладания сил притяжения коллоидные частицы могут сблизиться при коагуляции до расстояния г2. Однако теоретические расчеты и экспериментальные данные свидетельствуют о том, что при больших расстояниях между коллоидными частицами /у (порядка нескольких толщин двойного электрического слоя) на потенциальной кривой хорошо наблюдается второй неглубокий минимум Mf который незначителен для обычных гидрофобных коллоидов и более глубок для золей, имеющих крупные асимметрические частицы. Для таких золей энергия взаимодействия в точке М может быть в несколько раз больше энергии теплового движения, что создает возможность довольно устойчивого взаимного притяжения данных частиц на больших расстояниях друг от друга.
Таким образом, наличие крупных асимметричных частиц в золе способствует образованию определенной структуры, которая придает золю гелеобразное состояние. Как видно из рис. 201, глубина максимума М бывает меньше коагуляционного минимума F, и потому
связь между коллоидными частицами на расстоянии гораздо слабее, чем при коагуляции. Подобные гелеобразные структуры могут быть легко разрушены при простом встряхивании, причем получившийся золь при стоянии вновь превращается в гель. Эти переходы могут быть повторены десятки раз.
Явление тиксотропии широко распространено в природе. Так, свойства некоторых грунтов размягчаться по$ влиянием производимого на них механического воздействия объясняется их тиксотропностью. Такие грунты называются плывунами. Плывуны, разжижаясь под действием гидростатического и гидродинамического давления грунтовой воды, затрудняют различные строительные и горные работы, заполняя выработанное пространство.
Применение глинистых растворов при производстве буровых работ также основано на явлении тиксотропии. Размешивание суспензий при накачивании в скважины делает их текучими. Попадая на стенки скважины, суспензия затвердевает, что предупреждает осыпание стенок скважины и их обвалы. Помимо укрепления стенок скважины, глинистый раствор выполняет и другие полезные функции. В частности, при временной остановке бурения глинистый раствор удерживает во взвешенном состоянии частицы выбуренных пород, что предупреждает так называемое захватывание бура, которое наблюдалось бы при оседании частиц.
Тиксотропные свойства приписывают таким сложным физиологическим структурам, как протоплазма и мускульная ткань. Раздражая иголкой тело малых лимфоцитов, Петтерфи наблюдал быстрое разжижение их протоплазмы, которая вновь быстро уплотнялась. Аналогичное явление можно наблюдать при раздражении иголкой тела мелких амеб. Явлением тиксотропии легко объясняется наблюдение К ю н е, который видел, как вдоль мышечного поперечнополосатого волоконца лягушки продвигалась нематода с такой же легкостью, как в обычной жидкости. Дело в том, что нематода при своем передвижении, механически воздействуя на тиксотропную субстанцию мускульного волокна, вызвала превращение его в золь, который после прохождения через него нематоды вновь обретал структуру.
Период застудневания при тиксотропии — величина постоянная для каждой данной системы и часто используется в качестве показателя ее устойчивости. Причем количественной оценкой тиксотропии может служить прочность образовавшегося геля и скорость отвердевания. Для определения скорости отвердевания различные образцы сравниваются по Фрейндлиху при одной и той же прочности геля. Для этой цели определяют время, необходимое для превращения в трубках стандартных размеров золя в гель такой консистенции, чтобы он не вытекал при переворачивании трубки вверх дном.
Исследования показали, что явление тиксотропии имеет место не только под влиянием механических факторов, но и при воздействиях химического и температурного порядка. Тиксочропное застудневание зависит от добавок_электролишвг,дэН и температуры. Аналогично явлению коагуляции оно ускоряется с ростом концентрации электролита. В качестве примера можно указать на гидрозоль железа [Fe(OH)3J; время отвердевания которого увеличивается примерно в 100 раз при увеличении pH на единицу. BpgM# застудневания уменьшается с повышением^температуры. Дело в том, что прИТтовышении температуры^происходит увеличение броуновского движения частиц, которое ускоряет процесс образования структуры и сокращает время ее восстановления.
471 —
§ 133. Коагуляция растворов высокомолекулярных соединений
Растворы высокомолекулярных веществ в термодинамически равновесном состоянии аналогично истинным растворам обладают абсолютной агрегативной устойчивостью. Высокая устойчивость коллоидных растворов высокомолекулярных соединений определяется двумя факторами — наличием на поверхности частиц двух оболочек: электрической и сольватной (гидратной). Для коагуляции коллоидов высокомолекулярных соединений необходимо не только нейтрализовать заряд коллоидной частицы, но и разрушить жидкостную оболочку. Выделение высокомолекулярных соединений из растворов по своему характеру отличается от коагуляции типичных гидрофобных коллоидов. Так, если для гидрофобных золей достаточно незначительных добавок электролита, чтобы вызвать коагуляцию, то для высокомолекулярных веществ этого недостаточно. Для выделения дисперсной фазы полимеров необходимы высокие (вплоть до насыщенных растворов) концентрации электролитов. Например, яичный глобулин выделяется при полу насыщении раствора сульфатом аммония, а яичный альбумин— только при полном насыщении.
Явление выделения в осадок растворенного ВМС под действием большой концентрации электролита получило название высаливания, К высаливанию неприменимо правило Шульце — Гарди, поэтому нельзя отождествлять высаливание с явлением обычной электролитной коагуляции. В отличие от гидрофобных золей явление высаливания высокомолекулярных веществ не связано с дзета-потенциалом коллоидных мицелл и заключается в нарушении сольватной (гидратной) связи между макромолекулами полимера и растворителем, т. е., иначе, в понижении растворимости полимера. При введении соли часть молекул растворителя, которая была в сольватной связи с макромолекулами ВМС, сольватирует молекулы введенной соли. Чем больше будет введено соли, тем большее число молекул растворителя покинет макромолекулы полимера и сольватирует соль. Таким образом, высаливающее действие соли заключается в ее собственной сольватации (гидратации) за счет десольватации (дегидратации) молекул высокомолекулярных веществ.
Многочисленные исследования показали, что всякое соединение, способное сольватироваться растворителем данного ВЛАС и понижать его растворимость, будет пригодным для высаливания. Так, спирт и ацетон способны отлично высаливать желатину из ее водных растворов. Аналогично происходит осаждение спиртом белка из водного раствора или осаждение ацетоном каучука из раствора бензола.
Учитывая механизм осаждающего действия электролитов и других десольватирующих веществ, К р о й т в свое время предложил общую схему осаждения высокомолекулярных веществ, которая представлена на рис. 202. Из этой схемы видно, что для осаждения макромолекул необходимо удалить водную оболочку (спиртом или другим дегидратирующим веществом) и снять заряд ее путем прибавления электролита. Причем последовательность этих операций не имеет значения. Схе
— 472 —
Спирт
Спирт
Рис. 202. Схема коагуляции (по Кройту)
ма Кройта учитывает только действие электролитов и дегидратирующих веществ, т. е. снятие заряда и водной оболочки, но совершенно не учитывает специфичности этих веществ.
В ряде случаев для осаждения многих высокомолекулярных соединений (белков, полисахаридов) снятие заряда не является обязательным условием, так как главным фактором их устойчивости служит гидратная оболочка, удерживаемая полярными, но не диссоциированными группами (эфирными и пептидными связями, спиртовыми группами). Некоторые высокомолекулярные соединения обладают высокой стойкостью к высаливанию. Например, при засолке рыбы в раствор (рассол) переходят значительные количества белковых соединений, которые остаются в нем в состоянии золя, несмотря на то, что он является почти насыщенным раствором соли. Такая высокая устойчивость к высаливанию объясняется особо сильной гидратацией белков.
Таким образом, при высаливании
высокомолекулярных веществ решающую роль играет не зарядность йонов, а их способность к гидратации и к адсорбции на коллоиднодисперсных частицах.
По своему высаливающему действию все катионы и анионы можно расположить влиотропные ряды
С2О1“ > SO*- > СН3СОО- > Cl” > NO- > I - > CNS
Li+ > Na+ > К+ > Pb+ > Cs+ > Mg2+ > Са2+ > Sn2+ > Ва2+
Расположение ионов в лиотропных рядах связано не с величиной их заряда, как в случае обычной коагуляции, а со степенью их гидратации. Чем больше ион способен связывать растворитель, тем больше его высаливающее действие. Основная роль в высаливании, как и в набухании, принадлежит анионам, катионы же оказывают уеньшее воздействие на высаливание.
Большое влияние на процесс высаливания оказывает также и степень растворимости самого полимера в данном растворителе: чем она ниже, тем полнее и быстрее происходит высаливание. В свою очередь растворимость полимерного соединения зависит от длины макромолекул и молекулярного веса полимера: чем они больше, тем меньше растворимость данного полимера, следовательно, тем легче он высаливается. На этом принципе основан метод так называемого фракционного высаливания, сущность которого заключается в последовательном высаливании из раствора все возрастающими порциями высаливателя отдельных фракций полимеров, начиная с полимеров наивысшей степени полимеризации (с наибольшим молекулярным весом). Так, на тонком сочетании действия спирта, солей и охлаждения до —5° С основаны способы детального фракционирования
16 Зак. 5G0
— 473 —
белковых смесей по Кону. Из сыворотки крови этим методом можно выделить свыше 12 различных белков.’
Для фракционирования применяют также способ постепенного понижения температуры при постоянном составе жидкости. Препаративное разделение высокомолекулярных соединений широко применяется при научных исследованиях для характеристики полидисперсности полимеров.
Как. показали „исследования, высокомолекулярные вещества, выделанные из раствора высаливанием, после отмывки их от электролитов могут быть снова переведены в раствор (явление обратимо). Коллоиды, которые при устранении фактора, вызвавшего коагуляцию, способны переходить из состояния геля в состояние золя, носят название обратимых коллоидов. Однако высокомолекулярные вещества могут при определенных условиях осаждаться и необратимо. Такое необратимое осаждение высокополимеров, в частности белков, под влиянием высокой температуры, при воздействии концентрированных кислот и щелочей, дубильных веществ, лучистой энергии называется денатурацией. При денатурации происходит не только осаждение полимеров, но и изменение их химической природы. Белки при денатурации становятся нерастворимыми и в большинстве случаев утрачивают способность к набуханию.
Высаливание имеет большое практическое значение в целом ряде технологических процессов, например, в мыловарении, в производстве красителей, канифоли и многих других искусственных волокон.
§ 134. Коацервация
В растворах высокомолекулярных соединений при изменении температуры, pH или при введении низкомолекулярных веществ иногда наблюдается явление коацервации. Внешне процесс коацервации характеризуется отделением от золя изолированных друг от друга макроскопических капель жидкости или целого жидкого слоя. Такая капля (рис. 203) содержит рой ультрамикроскопических капелек. Каждая из них состоит из нескольких первичных сольватированных частиц, сохранивших свою самостоятельность. Таким образом, от высаливания коацервация отличается тем, что вещество дисперсной фазы не отделяется от растворителя, а собирается в невидимые простым глазом жидкие капельки, которые постепенно сливаются в капельки больших размеров — вплоть до видимых невооруженным глазом, пока процесс этот не закончится полным расслоением системы на два жидких слоя. Вязкая фаза, содержащая все или почти все высокомолекулярное вещество, называется коацерватом.
Частицы высокомолекулярного соединения, входящие в состав коацерватных капель, по-видимому, отделены друг от друга тонкими гидратными оболочками. Об этом свидетельствует то, что явление коацервации обратимо. При изменении условий, вызвавших коацервацию (уменьшение концентрации электролита, изменение pH и температуры), коацерватные капли могут исчезать и система вновь переходит в однофазную. В то же время, изменяя условия в сторону усиления
— 474 —
процесса дегидратации макромолекул высокополимера, можно вызвать разрушение коацерватных капель и полное осаждение растворенного вещества.
Явление коацервации можно наблюдать, если смешивать противоположно заряженные золи, например, белка и лецитина, белка и нуклеиновых кислот. Совершенно аналогичное явление будет наблюдаться и в том случае, если к раствору белка прибавлять концентрированный раствор сульфата натрия. Коацервация, получаемая при смешении двух противоположно заряженных золей, получила название комплексной коацервации (в отличие от описанной выше простой коацервации).
оооооооооооооо ооооооооооооо оооооооооооооо ооооооооооооо
о о о о о
о о о о
И
Рис. 203. Схема коацервации:
7 — образование первичной ультрамикроскопической капельки in гидратированных макромолекул; б — вторичная капелька из «роя» первичных; в — расслоение раствора с коацерватом наверху
Явление комплексной коацервации можно наблюдать при смешивании 5%-ного раствора желатины с 5%-ным раствором картофельного крахмала. Сначала наступает микрокоацервация, а через несколько часов образуются два слоя: нижний, содержащий весь крахмал, и верхний, содержащий весь желатин. Таким образом, в данном случае происходит расслоение золя на две фазы, которые разделяются физической поверхностью раздела. Комплексную коацервацию можно наблюдать также и при взаимодействии желатина ji лецитина. При комплексной коацервации по существу происходит высаливание одного золя другим. Здесь возможны следующие наиболее типичные случаи.
1. Коллоид А во много раз гидрофильнее коллоида Б. В этом случае золь А будет оказывать высаливающее действие на коллоид Б. Причем коллоид Б выделяется из раствора в виде хлопьев.
2. Коллоид А гидрофильнее коллоида Б, но менее резко, чем в первом случае. При этом происходит явление коацервации, т. е. золь Б образует вязкий слой, содержащий гидратированные частицы коллоида Б.
3. Оба коллоида А и Б имеют примерно одинаковую гидрофильность. В данном случае золи А и Б либо смешиваются между собой во всех отношениях, либо расслаиваются с образованием двух жидких фаз.
4. Коллоид А менее гидрофилен, чем коллоид Б. Золь Б высаливает коллоид А или вызывает его коацервацию.
На рассмотренные простые примеры налагается еще влияние и других факторов, таких, как pH, концентрации электролитов или наличие каких-либо других дегидраторов.
16*
— 475 —
Коацервация, особенно комплексная, играет огромную роль в биологических процессах, совершающихся в клеточном веществе —про- 5 топлазме. По некоторым своим физико-химическим свойствам коацерваты напоминают свойства протоплазмы, поэтому считают, что именно процесс коацервации имел исключительно большое значение в истории возникновения жизни на Земле.
§ 135. Защитное действие растворов высокомолекулярных соединений
Типичные гидрофобные золи легко коагулируют при прибавлении к ним малых количеств электролитов (миллиграммы на литр). Растворы высокомолекулярных соединений, наоборот, обладают большой устойчивостью против коагулирующего действия электролитов. Многочисленными исследованиями было установлено, что растворы ВМС, будучи прибавлены к гидрофобным золям, сообщают им повышенную устойчивость к электролитам. Так, если к золю золота (гидрофобный коллоид) прибавить небольшое количество желатина, гидрозоль золота становится более устойчивым. При прибавлении электролитов даже в количествах, значительно превосходящих порог коагуляции, а также при длительном стоянии этот золь не испытывает практически никаких изменений. Если этот золь выпарить, то при смешении сухого препарата с водой вновь образуется коллоидный раствор. Та-, ким образом, типичный гидрофобный золь золота при прибавлении к нему желатина как бы приобрел свойства гидрофильного золя и стал обратимым. Подобное явление получило название защитного действия или просто защиты, а сами вещества, повышающие устойчивость гидрофобных золей, получили название защитных.
Как правило, защитным действием обладают высокомолекулярные вещества лиофильной природы (т. е. поверхностно-активные). В табл. 89 приведены важнейшие защитные вещества и указаны гидрофобные золи, которые хорошо защищаются этими веществами.
Исследования показали, что степень защитного действия растворов ВМС зависит от природы растворенного полимера и от природы защищаемого гидрофобного золя. Количественной мерой защитного действия растворов ВМС являются золотое, рубиновое и железное число. Под золотым числом подразумевают минимальное число миллиграммов защищающего высокополимера, которое является достаточным, чтобы воспрепятствовать перемене красного цвета в фиолетовый у 10 мл гидрозоля золота (0,006% концентрации, полученного по методу Зигмонди) от коагулирующего действия 1 мл 10%-ного раствора хлорида натрия.
Золотое число, - введенное в практику Зигмонди, рассчитано на самый чувствительный золь — гидрозоль золота. Позднее В. Оствальд в качестве стандарта вместо золотого числа предложил рубиновое число. Оно определяется как минимальное число миллиграммов защищающего золя, которое способно защитить 10 мл 0,01%-ного раствора красителя конго красного (конгорубина) от коагулирующего действия 1 мл 10%-ного раствора хлорида натрия.
— 476 —
Таблица 89
Важнейшие защищающие вещества
Защищающие вещества
Альбумин
Амилодекстрин Казеин Декстрин Желатин Гуммиарабик
Каучук
Защищают золи следующих веществ
Au, Pt, Ag, соли Hg, Си, As, Pb, Sn, Al, Mg, W
SiO2
Ag, Hg, Си, Hg I
Ag, Hg
Pt, Au, Ag, галиды
Платиновые металлы, Hg, HgS, Cu, WO3, S, графит, Cr2O3, MoO3
Органозоли Pb, Cu
Помимо золотого и рубинового чисел, некоторое применение получило еще более простое и легко доступное железное ч и с-л о, которое можно определить как минимальное число миллиграммов защищающего высокополимера, способного защитить Юлы золя гидроокиси железа от коагулирующего действия 1 мл 0, 005 н. раствора Na2SO4.
Сравнение золотых, рубиновых и железных чисел представлено в табл. 90.
Таблица 90 Защитное действие
Высокомолекулярное вещестьо Золотое число, мг Рубиновое число, мг Железное число, мг
Желатин 0,008 2,50 5,00
Казеинат натрия .......... 0,01 0,40 ——
Гемоглобин .... 0,25 0,80
Яичный альбумин 2,50 2,00 15,00
Гуммиарабик 0,50 — 20,00
Крахмал 25,00 20,00 20,00
Из данных таблицы видно, что желатин и казеинат натрия обладают наиболее сильным защитным действием, крахмал — наиболее слабым.
Как показали исследования, наибольшее защитное действие отмечается при одноименных зарядах высокомолекулярного соединения и коллоида, так как в противном случае они взаимно нейтрализуют заряд, и устойчивость большого объединенного комплекса, естественно, снижается.
Механизм защитного действия достаточно хорошо объясняется теорией Зигмонди, в основе которой лежит представление об адсорбционном взаимодействии между частицами защищаемого и защищающего золей. Более крупная частица гидрофобного золя адсорбирует на своей поверхности более мелкие макромолекулы ВМС с их сольват-
477 —
ними (гидратными) оболочками, и в результате этого она приобретает лиофильные (гидрофильные) свойства. В данном случае коллоидные мицеллы необратимого гидрофобного золя предохраняются от непосредственного соприкосновения друг с другом, а следовательно, и от агрегации как в случае действия на такой золь электролита-коагулятора, так и в случае концентрирования золя. На рис. 204, а показана схема подобного защитного действия. Таким образом, высокомолекулярные соединения выступают в роли стабилизатора лиофобных (гидрофобных) золей. То, что именно явление адсорбции лежит в основе защитного действия, подтверждается не только избирательным характером взаимодействия между макромолекулами ВМС и мицеллами, но и тем, что степень защитного действия увеличивается с концентрацией защищающего раствора ВМС только до полного адсорбционного насыщения поверхности мицелл защищаемого золя.
Исследования, проведенные в последние годы с применением электронного микроскопа, показали, что в случае нитевидных молекул ВМС одна макромолекула высокополимера адсорбционно взаимодействует с несколькими мицеллами (рис. 204, б). При этом мицеллы гидрофобного золя связываются в своеобразные агрегаты в виде структурных сеток, благодаря чему лишаются возможности сближаться друг с другом и коагулировать.
В некоторых случаях прибавление весьма малых количеств высокополимера к гидрофобному золю приводит к прямо противоположному результату: устойчивость золя резко понижается. Это явление называется сенсибилизацией или астабилизацией коллоидного раствора. Согласно теории П. Н. Пескова и Л. Д. Ландау астабилиза-ция происходит тогда, когда защищающий высокополимер добавляют к гидрофобному золю в таких ничтожно малых количествах, которые ниже предельного порога его защитного действия, т. е. ниже его золотого или рубинового защитного числа. Иными словами, астабилизацня наступает, когда частиц высокополимера не хватает на покрытие и защиту всей поверхности коллоидных частиц гидрофобного золя, но их достаточно для того, чтобы путем адсорбции отнять у последних стабилизирующие ионы. На рис. 204, в дана схема астабилизированной коллоидной частицы. Астабилизацня легче всего осуществляется в том случае, если оба вида частиц заряжены разноименно.
Явление коллоидной защиты играет очень важную роль в ряде физиологических процессов, совершающихся в организмах человека и животных. Так, белки крови являются защитой для жира, холестерина и ряда других гидрофобных веществ. При некоторых заболеваниях содержание защитных белков в крови уменьшается, что приводит к отложению, например, холестерина и кальция в стенках сосуда (артеросклероз и кальциноз). Понижение защитной роли белков и других стабилизирующих веществ в крови может привести к образованию камней в почках’, печени, протоках пищеварительных желез и т. п. С другой стороны, способность крови удерживать в растворенном состоянии большое количество газов (кислорода и углекислого газа) также обусловлена защитным действием белков. В данном случае белки обволакивают микропузырьки этих газов и предохраняют их от слияния»
— 478 —
Большое значение защитное действие имеет и в технологии многих производств. Так, при изготовлении ряда фармакологических препаратов, например колларгола и протаргола, используется явление защиты. Эти препараты представляют собой концентрированные золи металлического серебра, защищенного от выпадения добавкой декстрина и белковых веществ.
Путем введения в вино защитных коллоидов добиваются значительного удлинения сроков хранения вин без потери ими 'прозрачности. На явлениях защиты основано придание пенистости пиву в пивоваренном производстве, а также образование очень стойких пен в огнетушителях.
_ Лиофобная ух) _ Лиофильная
W частица частица
<
Рис. 204. Схемы защитного действия
В органическом синтезе широко используются в качестве катализаторов защитные золи таких металлов, как платина, палладий и др. Защитные коллоиды используются также при приготовлении фотографических эмульсий. В кондитерском производстве в целях предотвращения образования крупных кристаллов сахара и льда при приготовлении мороженого широко применяется желатин.
Кроме органических защитных веществ известны и неорганические. Так, получены стойкие золи Pb, Se, Fe, Au, Rh, Bi, Sb, стабилизированные оловянной или титановой кислотой. Такой краситель, как, например, кассиев пурпур является гидрозолем золота, защищенным оловянной кислотой.
§ 136. Гели, их образование, строение и свойства
Большинство растворов высокомолекулярных соединений и золи некоторых гидрофобных коллоидов способны при известных условиях переходить в особое состояние, обладающее в большей или меньшей степени свойствами твердого тела. Твердообразная текучая система, образованная коллоидными частицами или макромолекулами высокомолекулярного соединения в форме пространственного сетчатого каркаса, ячейки которого заполнены иммобилизованной жидко-
— 479 —
стыо или газом, называется гелем (от латинского слова gelatus —-замерзший). Таким образом, гели или студни представляют собой кол-лоидные системы, потерявшие текучесть в результате возникновения в них внутренних структур. Гелями являются каучук, целлулоид, клей, желатин, текстильные волокна, многие ткани растительного и животного происхождения, а также большинство продуктов пищевой промышленности—тесто, хлеб, мармелад, различные желе, а также такие минералы, как агат, опал.
По аналогии с золями, гели в зависимости от характера дисперсионной среды делятся на гидрогели, алкогели, бензогели и т. д. Бедные жидкостью или совершенно сухие студнеобразные вещества носят название ксерогелей. Примерами ксерогелей могут служить сухой листовой желатин, столярный клей (в плитках), крахмал. К типу сложных ксерогелей относят муку, сухари, печенье. Существуют студни, содержащие очень мало сухого вещества (1—2% и менее), например кисель, пищевые блюда — студень, простокваша, растворы мыл и мылообразных веществ. Такие богатые жидкостью студнеобразные системы называются лиогелями.
Особую группу гелей образуют студенистые осадки, которые получаются при коагуляции золей (гидроксида железа (III), кремниевой кислоты и т. д.), а также хлопьевидные осадки высокополимеров, образующиеся при высаливании растворов. В таких осадках связывается лишь малая часть дисперсионной среды, большая ее часть образует отдельную жидкую фазу.
Студенистые осадки, образующие отдельные фазы, называются коагелями. Бедные жидкостью хлопья и микрокристаллические порошки, образующиеся при коагуляции гидрозолей типичных гидрофобных коллоидов (Au, Ag, Pt, сульфиды), из категории гелей (коагелей) исключаются.
В зависимости от природы образующих веществ различают гели неэластичные (или хрупкие) и эластичные.
Неэластичные гели образуются коллоидными частицами SiO2, TiO2, SnO2, Fe2O3, V2O5. Эти гели впитывают всякую смачивающую их жидкость, при этом объем их почти не изменяется. Потеряв известное количество воды, гели резко меняют свои физические свойства, делаясь хрупкими. Как правило, хрупкие гели имеют сильнопористую структуру с множеством узких жестких капилляров диаметром около 20—40 А.
Эластичные гели, или студни, образуются цепными молекулами желатина, агар-агара, каучука и других полимеров и по своим свойствам отличаются от хрупких гелей. Эластичные гели поглощают не все смачивающие их жидкости, а только некоторые, которые сходны с ними по своему химическому составу или в которых вещество студня может существовать также в виде жидкого раствора. Поглощение жидкости эластичным студнем сопровождается сильным увеличением объема. Это явление, как известно, получило название набухания. По этой причине эластичные гели иначе называют набухающими гелями. Причем объем набухающего студня может в десятки раз превосходить собственный объем полимера.
— 480 —
Часто набухание студня переходит в полное его растворение (например, набухание каучука в бензине пли гуммиарабика в воде). В этом случае говорят о неограниченном набухании данного полимера. Если студень поглощает определенное количество растворителя, но не образует раствора полимера, то такое набухание называется ограниченны м. В качестве примера можно назвать набухание желатина в холодной воде, вулканизированного каучука в органических жидкостях. Иногда ограниченное набухание может переходить в неограниченное при повышении температуры или изменении
Рис. 205. Схема объединения частиц различной формы при коагуляции и желатинировании:
Ы, 2а, За — коагуляция; 16, 26, 36 — образование внутренних структур
состава среды. Так, студень желатины хорошо растворяется в воде при нагревании выше 40—42° С или при комнатной температуре при добавлении 2н. KCNS или КЕ
При обычной коагуляции коллоидный раствор разделяется на две фазы: жидкую дисперсионную среду и более или менее твердую дисперсную фазу (рис. 205, а). При гелеобразовании подобного разделения нет, вся масса раствора превращается в твердообразную нетекучую систему, во всех частях которой концентрация дисперсной фазы или высокомолекулярного соединения остается одинаковой и неизменной. При коагуляции мицеллы контактируют между собой наиболее тесно, что ведет к образованию осадка. При возникновении внутренних структур, т. е. при образовании студня, происходит объединение частиц в форме сетки или ячеек, напоминающих дзену (рис. 205, б).
На процесс гелеобразования большое влияние оказывают размеры, форма коллоидных частиц или макромолекул высокомолекулярного соединения, температура, концентрация электролитов в растворе и время. Опыт показывает, что необходимым условием геле- или студнеоб-разования является асимметричная форма коллоидных частиц или макромолекул высокополимера. Чем ярче выражена асимметричность коллоидных частиц, тем при меньшей концентрации дисперсной фазы
— 481 —
в растворе образуется гель. Наименьшее количество вещества, необходимое для построения каркаса в данном объеме, потребуется в том слу чае, если частицы имеют форму тонких палочек или нитей минимальных размеров (рис. 206, б). Несколько больше потребуется вещества при листочкообразной форме коллоидных частиц (рис. 206, а) и еще больше—при изодиаметрической форме (шар, куб) частиц (рис. 206, в).
Концы палочкообразных и игольчатых частиц и края лепестковых частиц имеют меньшую толщину сольватной (гидратной) оболочки и меньший дзета-потенциал, чем остальные части таких частиц. Поэтому в процессе гелеобразования асимметричные частицы соединяются между собой концами и краями, образуя более или менее прочный кар-
Рис. 206. Рыхлые структуры, образованные частицами различной формы
кас, охватывающий весь объем золя. Подобный каркас схематически представлен на рис. 207. На нем коллоидно-дисперсные частицы представлены в виде палочек, по концам которых расположены гидрофобные участки (зачернены). Области, защищенные сольватными (гидратными) оболочками, заштрихованы. В действительности гидрофильные и гидрофобные участки могут располагаться в макромолекулах самым разнообразным образом, чередуясь между собой.
Жидкая фаза механически включается сеткой (подобно впитыванию воды в губку), т. е. является полностью иммобилизованной, что, собственно, и приводит систему к потере легкоподвижности. Утрата золем текучести и служит внешним признаком момента образования студня.
В растворах высокомолекулярных соединений, длина макромолекул которых намного превышает их размеры в двух других направлениях, условия для образования внутренней структуры еще более благоприятные. Поэтому в растворах даже при относительно небольшой концентрации высокополимера (собенно при понижении температуры) силы притяжения между макромолекулами оказываются больше сил отталкивания, и частицы, сцепляясь друг с другом, образуют рыхлый каркас. Так, золь кремниевой кислоты образует гель при содержании
— 482 —
Рис. 207. Схема ультрами-кроскопического строения студня:
7 — гидрофобные (неполярные) участки; 2 — сольватная оболочка (лиосфера); <3 — область иммобилизованного раствори-
теля
температуры интенсивность
ее в растворе не менее 3%, а раствор агар-агара застудневает уже при содержании 0,1% в растворе.
Чем больше концентрация дисперсной фазы золя или высокомолекулярного соединения в растворе, тем с большей скоростью образуется пространственный сетчатый каркас. Например, 2%-ные и более концентрированные растворы желатина легко превращаются в студень при комнатной температуре, а 0,5%-ные и более разбавленные растворы при этой температуре студня не образуют. Влияние концентрации золя на свойство гелей можно проследить на примере только что рассмотренного типичного хрупкого геля кремниевой кислоты (табл. 91).
При значительных силах сцепления частиц в образованном каркасе, например в геле кремниевой кислоты, механическое воздействие приводит к необратимому разрушению системы. При слабых силах сцепления (гидроокись алюминия) внутреннюю структуру геля можно разрушить даже путем незначительного встряхивания или перемешивания. Однако при стоянии в этом золе внутренняя структура вновь восстанавливается, т. е. подобные гели являются тиксотропными системами.
На процессе геле-или студнеоб-разования существенное влияние оказывает температура. При повышении
теплового движения коллоидных частиц и макромолекул высокополи-мера увеличивается, поэтому связь между ними ослабляется. В резуль-те прочность пространственного сетчатого каркаса, образуемого коллоидными частицами или макромолекулами ВМС, уменьшается и гель переходит в золь. Таким образом, при повышении температуры увеличивается и минимальная концентрация дисперсной фазы или высокомолекулярного соединения в растворе, при которой возможен процесс
Таблица 91
Свойства геля кремниевой кислоты в зависимости от содержания SiO2
Содержание SiOa, %
Свойства геля
3,о
8,0
14,0
22,0
Имеет вид желе
Плотный, можно резать ножом
Вполне твердый
Д\ожно растереть в порошок
— 483 —
образования внутренней структуры. При понижении температуры концентрация уменьшается.
Как на коагуляцию, так и на процесс геле- или студнеобразования, большое влияние оказывает добавление электролитов. При этом происходит уменьшение дзета-потенциала, сжатие диффузного слоя и, как результат, уменьшение гидратной оболочки мицелл. Все это способствует образованию внутренних структур. Одни ионы ускоряют процесс гелеобразования, другие замедляют или вовсе устраняют его. На процесс геле- или студнеобразования главным образом влияют анионы, что хорошо подтверждается данными табл. 92.
Таблица 92
Влияние анионов и катионов на застудневание 5%-го раствора желатина при 15° С и pH 4,7
Электролиты" Время желатинирования, мин
Сульфат калия 25
Сульфат натрия 30
Ацетат калия . 45
Раствор желатина (5%-ный) . . без добавления электролитов 50
Хлорид натрия . 90
Хлорид калия . 85
Хлорид аммония 90
Иодид натрия . 200
Иодит калия . . 195
Роданид натрия Не желатини-
Роданид калия * рует То же
Как видно из таблицы, сульфаты ускоряют процесс застудневания, а роданиды приостанавливают его. Хлориды и иодиды замедляют процесс студнеобразования. Эти явления объясняются тем, что анионы SOI” энергично десольватируют (дегидратируют) макромолекулы желатина, что благоприятствует их взаимодействию и образованию студня. Анионы CNS” усиливают сольватацию макромолекул до такой степени, что они вообще не могут образовать пространственного каркаса, и раствор не застудневает.
Таким образом, чем больше данный ион проявляет способность гидратироваться, тем активнее в его присутствии происходит дегидратация коллоидных частиц и макромолекул ВМС. Это облегчает соединение их между собой и образование структуры. Ниже приведен ряд анионов по их действию на скорость застудневания
> СН3СОО- > Cl - > NOr
О м о
SQ2-
Ионы, стоящие в начале ряда, ускоряют застудневание, ионы, стоящие в конце ряда, замедляют его.
Геле- или студнеобразование происходит только в том случае, если к коллоидному раствору или раствору ВМС добавляется небольшое
Вг-
CNS-
— 484 —
количество электролитов, вызывающих десольватацию (дегидратацию) коллоидных частиц или макромолекул только в некоторых частях, т. е. на концах и краях. Добавление же насыщенных растворов электролитов вызывает десольватацию (дегидратацию) коллоидных частиц и макромолекул ВМС по всей их поверхности. Это в конечном результате приводит к коагуляции или высаливанию, т. е. к разделению системы на две фазы, а не к геле- или студнеобразованпю.
Золи типичных гидрофобных коллоидов (Ag, Au, Pt, As2S3) неспособны застудневать. Причина данного явления заключается в cbqco6-разном строении коллоидных частиц этих веществ, характере их гидратации и низкой концентрации золей (коллоидные растворы гидрофобных веществ не могут быть приготовлены в высокой концентрации).
Физико-химические свойства студней. Гели или студни характеризуются целым рядом свойств твердого тела. Они сохраняют форму, обладают упругими свойствами и эластичностью. Гели отличаются как от разбавленных растворов, в которых каждая коллоидная частица или макромолекула является кинетически индивидуальной, так и от компактных коагулятов или твердых полимеров. Гели по ряду свойств занимают промежуточное положение между растворами и твердыми телами.
Особый интерес представляют некоторые особенности диффузии и реакций в гелях. В водных студнях, в которых содержание воды доходит до 99% их массы, диффузия происходит почти с такой же скоростью, как.и в чистой воде. Однако явление диффузии в гелях в чистом виде наблюдается сравнительно редко. Обычно оно осложняется адсорбционными, электрическими или химическими явлениями. Рассмотрим кратко основные факторы, влияющие на скорость диффузии в гелях.
Скорость диффузии находится в обратной зависимости от концентрации геля. Чем выше эта концентрация, тем меньше скорость диффузии. Так, коэффициент диффузии электролитов снижается по сравнению с чистой водой в 10%-ном студне желатина на 50%, а в 30%-ном студне на 90%. Объясняется это тем, что в концентрированном геле резко возрастает извилистость пути, который должна совершать диффундирующая частица.
Размер растворенных молекул или ионов также оказывает огромное влияние на скорость диффузии их в гелях. Чем больше размеры растворенных частиц, тем выше задерживающее действие структурной сетки данного геля, а следовательно, тем меньше скорость диффузии. Задерживающее действие студня или геля находится в прямой зависимости от его концентрации. На этом основано использование гелей при ультрафильтрации и диализе в качестве мембран, позволяющих отделять коллоидные частицы от молекул и ионов кристаллоидов.
Не меньшее влияние на скорость диффузии оказывает и природа диффундирующего вещества. Так, например, хлориды щелочных и щелочноземельных металлов сравнительно хорошо диффундируют в гелях. Значительно хуже диффундируют сульфаты, кислоты и большинство щелочей. Влияние природы растворенного вещества на скорость диффузии связано не только с размерами молекул и ионов веществ, но так
— 485 —
же п с адсорбционным взаимодействием их с коллоидом. При этом может оказывать влияние и pH среды. Исследования показали, что вещества, ускоряющие гелеобразование, как правило, замедляют скорость диффузии в гелях, и наоборот.
Возраст студня также оказывает существенное влияние на его проницаемость. С течением времени студни уплотняются, т. е. стареют, в результате проницаемость их заметно понижается. Диффузия в гелях отличается от диффузии в жидкостях тем, что здесь отсутствует перемешивание и невозможно образование конвекционных потоков, возникающих в жидких растворах.
Гели обладают электропроводностью. Иммобилизованный растворитель в геле образует, по существу, непрерывную среду, в которой более или менее свободно могут передвигаться ионы различных электролитов. На этом явлении основано применение гелей агар-агара, приготовленных на растворе КС1 для заполнения мостиков, с помощью которых соединяют отдельные электроды в гальваническую цепь.
Совершенно по особому проходят в гелях и процессы кристаллизации. Рост кристаллов внутри студней протекает спокойно, путем медленной диффузии. Поэтому в студнях удается выращивать очень крупные кристаллы многих веществ. Так, в студне кремниевой кислоты удалось вырастить кристаллы золота (до 3 мм величиной), крупные кристаллы меди, серебра и других металлов, а также некоторых химических соединений (оксалат бария, фторосиликат бария).
В студнях, так же как и в растворах, могут протекать различные реакции. Отсутствие конвекционных потоков, а также отсутствие пе-• ремешивания придает реакциям в студнях своеобразный характер: в различных участках студня реакции могут идти независимо одна от другой. Если один из продуктов реакции твердое нерастворимое вещество, то в студне будут наблюдаться явления периодического осаждения (так называемые кольца Лизеганга) вместо образования осадка по всему объему.
Получить периодические осадки в гелях довольно просто. Надо приготовить гель на растворе соли, которая затем при взаимодействии с другой солью в процессе реакции обменного разложения приведет к образованию осадка. Например, если в гель 3,5%-ного желатина, приготовленного на растворе, содержащем 0,12 г К2Сг2О7, после застудневания его в пробирке или чашке Петри внести каплю 8,5%-ного раствора нитрата серебра, образуется ряд дисков или колец Ag2Cr2O7 (рис. 208). Сущность этих явлений заключается в том, что раствор соли нитрата серебра диффундирует во внутрь геля, где и образует осадок при взаимодействии с К2Сг2О7 по уравнению
K2Cr2O7 4-2AgNO3 -> Ag2Cr2O7 4-2KNO3
В зону выпадения осадка диффундирует К2Сг2О7 из нижележащего слоя, поэтому при дальнейшем движении AgNO3 попадает в зону с недостаточной концентрацией К2Сг2О7, и осадка не образуется. Ниже К2Сг2О7 содержится1 уже в достаточном количестве, и там появляется вторая полоса осадка. Чем дальше к периферии чашки или ближе к дну пробирки, тем чередование дисков или колец становится более
— 486 —
редким из-за постепенного уменьшения концентрации диффундирующего нитрата серебра. Реакции такого типа носят название периодических или ритмического осаждения.
Общепризнанной теории образования колец Лизеганга в настоящее время нет. В. Оствальд предложил объяснение образования периодических наслоений на основе явления пересыщения. Некоторое время нерастворимое вещество остается в ненасыщенном состоянии и под влиянием диффузии перемещается до тех пор, пока не достигнет уровня пересыщения. Тогда-то и происходит образование слоя осадка. Далее весь процесс повторяется сначала. Некоторые ученые объясняют образование слоистости явлением коагуляции: образующийся осадок движется вместе с диффундирующим веществом в виде коллоидного
Рис. 208. Периодические осадки в гелях:
a — Mg(OH)2; б — Ag2Cr2O7j в — то же, в чашке Петри
раствора; постепенно диффундирующий электролит накапливается в среде до уровня порога коагуляции, после чего происходит коагуляция коллоидного осадка с образованием хорошо заметного слоя. Затем весь процесс повторяется вновь.
Наряду с изложенными существуют и другие теории, объясняющие периодичность осаждения, на которых мы не будем останавливаться.
Слоистые структуры широко представлены в природе. В настоящее время хорошо изучены процессы образования слоистых рисунков в некоторых природных минералах (агат, яшма). Эти структуры возникают в результате ритмических реакций в гелях. Не менее часто встречаются периодические структуры и в растительных организмах.
Синерезис гелей. Как показывают многочисленные исследования, гели с течением времени меняют свои свойства, т. е. стареют. В процессе старения на их поверхности начинают появляться капельки жидкости, которые затем сливаются вместе, образуя сплошную жидкую фазу. Происходит разделение студня на две фазы—дисперсионную и дисперсную, причем это разделение не является ни коацервацией, ни коагуляцией (высаливанием). Подобный самопроизвольно возникающий процесс старения геля получил название синерезиса или отыскания.
— 487 — -
Жидкая фаза, выделяющаяся при синерезисе, является нечистым Я растворителем, а очень разбавленным раствором. Например, сыворот-- ка, образовавшаяся при «отсекании» простокваши, содержит соли и небольшие количества коллоидов. Иными словами, эта жидкость по существу является золем данного коллоида, но очень малой концентра- JJ ции. Аналогично этому выделяющаяся при явлении синерезиса дисперсная фаза представляет собой лишь более концентрированный Ш студень, так называемый «синергический сгусток», т. е. студень с еще достаточно большим количеством растворителя.
Явление синерезиса впервые наблюдалось Г рэмом еще в 60-х .
годах прошлого столетия. Более детальное и всестороннее исследование этого явления за последние 30 лет было проведено советскими учеными во главе с С. М.‘ Л и п а т о в ы м. Исследования этих ученых показали, что синерезис можно наблюдать как у типичных коллоидов, так и у студней высокомолекулярных соединений, например крахмала, W желатина, простокваши, каучука, вискозы, студней некоторых красителей. Wrf
Синерезис может протекать как самопроизвольно, так и под влиянием веществ, понижающих растворимость вещества дисперсной фазы, например электролитов. Так, студни желатина синергируют при добавлении к ним эфира, студни геранина (органический краситель) синергируют при добавлении NaCl и т. д. Наибольший интерес представляет самопроизвольный синерезис эластичных студней, являющийся не чем иным, как процессом их старения — автокоагуляции.
При синерезисе вследствие увеличения числа контактов частиц дисперсной фазы наблюдается упрочнение геля с одновременным повышением его эластичности и упругости. При этом происходит стягивание структурной сетки геля, в результате чего гель выжимает из себя зна- Я чительную часть иммобилизованной жидкости и уменьшается в объеме. Весьма характерным является то, что гель при сжатии сохраняет форму того сосуда, куда был налит золь до его застудневания, например ш форму конической колбы (рис. 209).
Таким образом, синерезис возникает вследствие изменения взаимного положения частиц дисперсной фазы геля. Отсюда становится понятным, почему электролиты-коагуляторы, увеличивающие силы притяжения между частицами, способствуют синерезису.
У студней высокомолекулярных соединений процесс синерезиса обратим. В ряде случаев достаточно простого нагревания, чтобы система, претерпевшая синерезис, вернулась в состояние исходного студня. Этим приемом широко пользуются на практике для освежения, например, каш, пюре, хлеба. В подобных случаях процесс синерезиса не сопровождается какими-либо химическими превращениями компонентов системы. Иногда же при старении коллоидов возникают раз-
личные химические процессы, в этом случае синерезис усложняется и его обратимость теряется.
Скорость синерезиса коллоидов различна. Она возрастает с повышением температуры и увеличением концентрации. Иногда увеличение концентрации геля ослабляет процесс синерезиса, что является характерным для крахмала, агар-агара, ацетилцеллюлозы и вискозы.
— 4S8 —
Рис. 209. Явление синерезиса: а — гель до синерезиса; б — разделение геля на две фазы
своих исследованиях, что при происходит уменьшение вели-
У студней, образованных белками, скорость синерезиса зависит также и от pH. Так, для желатина он активнее всего проявляется в изоэлектрической точке.
Практическое значение синерезиса велико. Чаще всего явления синерезиса в быту и промышленности нежелательны. Например, с синерезисом связано черствение хлеба и отмокание кондитерских изделий — мармелада, желе, фруктовых джемов, карамели. Борьба с черствением хлеба представляет важную народнохозяйственную проблему. Вредное действие оказывает синерезис в промышленности искусственного волокна, взрывчатых веществ, в производстве многих красителей. Примером положительного синерезиса может служить самопроизвольное отделение жидкости от творога в процессе созревания сыра и сыроварении.
Процесс синерезиса имеет важное биологическое значение. В процессе старения коллоидов происходит их уплотнение, что не может не сказаться на проницаемость клеточных мембран и цитоплазмы. Снижение проницаемости может нарушить обмен веществ между клеткой и окружающей средой. Ученые доказали в возрастных изменениях организма
чины электрического заряда и степени гидратации коллоидных частиц. В результате уменьшается способность коллоидов тканей и органов связывать воду. Более поздние исследования показали, что процессы старения белков связаны не только со структурообразованием в растворах высокополимеров, но и с явлениями медленно протекающей денатурации. Именно процессами синерезиса и дегидратации объясняется появление у тканей с увеличением возраста организма новых качеств — большей жесткости и меньшей эластичности.
Однако объяснять причину старения живого организма только старением его коллоидов нельзя. Как известно, в организме происходит непрерывный обмен веществ, процесс ассимиляции и диссимиляции, разрушение органической субстанции и образование ее. И хотя протоплазма всех организмов находится в коллоидном состоянии, причины старения их кроются не в физико-химических, а более сложных, биологических процессах. В самом деле, в любом растворе того или иного коллоида не наблюдается специфического, присущего именно живым организмам обмена веществ и энергии, явлений ассимиляции и диссимиляции. Если у коллоидов протоплазмы в процессе ее жизнедеятельности и наблюдается постепенное понижение водосвязывающей способности, уменьшение стойкости и изменение других свойств, сходных с изменениями коллоидных растворов, то они происходят в результате направленного изменения химического состава коллоидов организма, определяемых процессами обмена веществ.
— 489 —
§ 137. Почвенные коллоиды
К почвенным коллоидам относятся высокодисперсные системы, в которых дисперсионной средой служит почвенный раствор, а дисперсной фазой — частицы почвы размерами от 0,2 до 0,001 мк. В развитии учения о почвенных коллоидах, а также в выяснении их роли в создании почвенного плодородия большое значение имели работы К. К. Гедройца в первые десятилетия XX в. Они посвящены разработке вопроса о поглотительной способности почв. Под этим понятием Гедройц понимал способность почвы поглощать (задерживать) находящиеся в почвенном растворе соединения. Дело в том, что коллоиды почвы, имея огромную поверхность, обладают способностью адсорбировать из окружающей среды не только ионы электролитов, но и значительные количества газов, пароз и жидкостей.
В своих исследованиях К. К. Гедройц вскрыл закономерности обмена катионов в почвах и влияния состава обменных катионов на свойства почв, а также разработал ряд методов изучения обменного поглощения катионов. Совокупность соединений, которые обладают способностью к обменным реакциям, названа Гедройцем почвенным поглощающим комплексом. С химической точки зрения он характеризуется как комплекс нерастворимых в воде алюмосиликатных, органических и органоминеральных соединений, с физической точки зрения — как «совокупность почвенных соединений, которые находятся в почве в мелкораздробленном состоянии, это высокодисперсная часть почвы, ультрамехани-ческая часть ее, по всей вероятности, близко совпадает с коллоидной частью почвы».
Исследования академика Гедройца были продолжены и развиты В. Р. Вильямсом, Г. Вигнером, И. Н. Антиповым-Кратаевым, С. Маттсоном, А. Н. Соколовским, Н. П. Ремезовым, С. Н. Алешиным, Н. И. Горбуновым и другими исследователями.
Многочисленные исследования этих ученых показали, что адсорбционная способность почвы связана с наличием в ней коллоидных частиц, так как частицы более крупных размеров практически не участвуют в процессах поглощения и являются малоактивной с химической точки зрения ее частью.
По своей природе почвенные коллоиды делятся на минеральные, органические и комплексные, т. е. органо-минеральные. Минеральная часть (обычно преобладающая в составе почвенных коллоидов) в основном состоит из вторичных минералов, имеющих кристаллическое строение, и из аморфных веществ. В табл. 93 • приведен состав минералов высокодисперсной фракции различных почв.
Органические почвенные коллоиды в основном представлены гумусовыми веществами: гуминовыми кислотами, фульвокислотами и гумином. Органо-минеральные соединения представляют собой преимущественно соединения гумусовых веществ с глинистыми и другими вторичными минералами.
В природе почвенные коллоиды образуются не только в результате измельчения и выветривания горных пород и минералов, но и в результате различных реакций, происходящих в почвах между минеральными и органическими минералами. Как мы видели из табл. 93, качественный (а также и количественный) состав высокодисперсной части разных почв неодинаков. Так, коллоиднодисперсные частицы в тяжелых глинистых почвах составляют до 50% от веса почвы, в суглинистых — до 30%, а песчаных — до 3%.
По своему элементарному составу коллоидная фракция значительно отличается от остальной массы почвы. В почвенных коллоидах содержится меньше кремнезема и значительно больше полуторных оксидов и особенно гумусовых веществ. Одним из важнейших свойств коллоидов почвы является их высокая поглотительная способность. Они легко адсорбируют из водных растворов (точ-нее.из почвенных растворов) различные катионы: К+, Na+, Са2+, Mg2+, NH^, Н* до полного насыщения поверхности коллоидных частиц. После насыщения дальнейшая адсорбция может происходить лишь путем обмена уже адсорбированных катионов на катионы почвенного раствора. Общее количество катионов, которое могут поглотить 100 г почвы, получило название емкости поглощения или емкости обмена почвы.
Емкость поглощения принято выражать в миллиграмм-эквивалентах на 100 г почвы (мг-экв на 100 г). Для определения емкости поглощения все катионы
— 490 —
Таблица 93
Глинистые и сопутствующие им минералы по фракции <0„001 мм главнейших типов почв
(по Н. И. Горбунову)
Почвы и породы
Преобладающие или характерные для данной почвы минералы
Сопутствующие минералы
Дерново- подзол и-стые на моренных и покровных суглинках
Дер ново- подзол и-стые на массивнокристаллических или хорошо дренируемых осадочных породах
Серые лесные на покровных суглинках
Черноземы на покровных и лессовидных суглинках
Черноземы на элювии массивнокристаллических пород
Солонцы степной и полупустынной зоны на суглинках и лессах
Сероземы на лессовидных суглинках и лессах
Красноземы на элювии андезито-базаль-тов и других основных и средних пород
Красноземы на элювии гранитов и других кислых пород
Гидрослюды, вермикулиты, минералы монтмориллонитовой группы в различных соотношениях; несиликатные аморфные полуторные оксиды. Гидрослюды часто преобладают
Гидрослюды, вермикулит, каолинит, минералы монтмориллонитовой группы Распределение вторичных минералов по почвенному профилю неравномерное
Минералы групп монтмориллонита, вермикулита, гидрослюды, иногда небольшая примесь каолинита и хлорита.
Минералы монтмориллонитовой группы, вермикулит, гидрослюды, иногда небольшая примесь каолинита и хлорита
Каолинит, гидрослюды, минералы монтмориллонитовой группы, иногда небольшая примесь гетита, гиббсита
Гидрослюды, минералы монтмориллонитовой группы, иногда небольшая примесь каолинита Распределение вторичных минералов по профилю неравномерное. В верхнем слое гидрослюды преобладают над монтмориллонитом. В солонцовом горизонте соотношение обратное
Минералы гидрослюдистой и монтмориллонитовой групп в разных соотношениях
Минералы каолинитовой группы, много гетита, гиббсита; часть первичных минералов разрушена
Минералы каолинитовой группы, примесь гетита и гиббсита. Часть первичных минералов разрушена
Иногда присутствуют небольшие примеси каолинита, кварца, редко гетита, гиббсита
Амфорные вещества и кварц
Амфорные кварц
вещества и
Амфорные вещества, кварц, иногда гетит, гиббсит
Амфорные вещества и полуторные оксиды вместе с вторичными минералами образуют конкреции; кварц, иногда гетит, гиббсит
Амфорные вещества, кварц, иногда гетит, гиббсит
Амфорные вещества, полуторные оксиды
Амфорные вещества, небольшая примесь минералов монтмориллонитовой группы, кварц
— 491 —
почвы вытесняются одним (Ва2+, NH^) путем многократной обработки ее раствором соли этого катиона. Избыток соли затем отмывается, а катионвытсснителъ в свою очередь вытесняется другим ионом (например, ионом водорода из раствора соляной кислоты) и уже в растворе определяется количественно. Величина емкости поглощения любых почв зависит от целого ряда факторов: содержания высокодисперсных частиц в почве, химического и минералогического состава почвенных коллоидов, а также реакции почвы.
Так, подзолистые почвы имеют емкость поглощения в пределах от 6 до 8 мг-экв на 100 г почвы, черноземные — от 40 до 60 мг-экв, торф — от 60 до 100 мг-экв и даже более. Наибольшей емкостью обмена обладают гумусовые вещества почвы: исчисляется сотнями миллиграмм-эквивалентов на 100 г этих веществ. Вот почему наиболее богатые гумусом почвы обладают и более высокой емкостью поглощения по сравнению с малогумусовыми. В качестве примера можно назвать черноземные почвы, а также верхние горизонты почв.
В силу разнообразия природных условий и особенностей почвообразова-тельного процесса состав поглощенных катионов у различных типов почв неодинаков. Например, черноземные почвы в поглощенном состоянии содержат преимущественно кальций, а подзолистые и дерново-подзолистые, помимо кальция, содержат рбменные водород и алюминий. Солонцовые и солонцеватые почвы.содержат поглощенный натрий в различных количествах. Для красноземных почв характерно преобладание в почвенном поглощающем комплексе ионов алюминия и водорода. В табл. 94 приведены данные по составу обменных катионов и емкости поглощения в почвах различных зон пашей страны.
Согласно К. К. Гедройцу, все почвы подразделяются на насыщенные и ненасыщенные основаниями. Как видно из приведенной выше таблицы, насыщенные основаниями почвы не содержат в поглощенном состоянии ионов водорода и алюминия (черноземы обыкновенные и южные и некоторые другие). Ненасыщенные основаниями почвы, кроме кальция и магния, содержат в поглощенном состоя-
Таблица 94
Состав обменных катионов в почвах СССР (мг-экв на 100 г почвы)
Название почвы Глубина взятия образца, см Содержа -ние гумуса, % Обменные катионы Емкость поглощения Степень насыщенности основаниями, %
кальций магний водород натрий
Дерново-подзо- 0—10 ' 1,7 2,0 0,3 3,3 Нет 5,6 41
листая легсуглини- 10—20 1,6 1,7 0,4 3,3 — 5,4 10,0 39
стая 80—90 0,3 5,8 4,9 1,4 0,8 2,8 — 72
120—130 Не опр. 1,7 7,4 58,1 77
Чернозем типич- 0—10 9,6 46,0 9,1 3,0 Нет 95
ный глинистый 20—30 7,6 44,4 7,5 2,0 — 58,9 > 96
60—70 4,1 36,7 7,0 0,8 44,5 98
80—90 3,1 36,4 7,0 Нет — 43,4 100
Солонец черно- 2—8 4,6 12,8 4,4 2,9 20,1 100
земный средне- 10—16 2,8 14,1 Н,2 10,4 35,7 100
столбчатый тяже- 74—80 0,2 20,2 6,5 7,6 34,3 100
лосуглинистый 100—106 Не опр. 21,4 7,6 —— 6,4 34,9 100
Светло-каштано- 0—15 2,6 24,8 4,8 Нет 1,4 31,0 100
вйя солонцеватая 22—32 1,9 21,3 8,2 1,5 5,8 31,0 100
72—82 0,7 14,0 8,9 — 28,7 100
110—120 Не опр. 15,0 9,6 3,2 27,8 100
Краснозем су г- 3—6 3,0 1,3 0,7 26,6 Нет 28,6 7
линистый 17—25 Не опр. 1,2 1,2 44,8 1111 — 47,2 5
60—70 — 1,5 1,1 32,0 34,6 8
120—130 — 1,5 1 1,1 26,1 28,7 9
— 492 —
нии значительное количество ионов водорода и алюминия. К таким почвам относятся прежде всего красноземы, подзолистые и некоторые другие.
Как показали многочисленные исследования, важнейшие свойства почвы— водопроницаемость, влагоемкость, набухаемость, липкость, связность, структура, pH почвенного раствора — находятся в прямой зависимости от состава поглощенных катионов. Причем, адсорбированные катионы могут изменять плодородие почвы не только путем изменения ее водно-физических и физико-химических свойств, но, как впервые показал К. К. Гедройц, оказывают непосредственное влияние на рост и развитие культурных растений. Так полное насыщение почвенного поглощающего комплекса ионами Na + , К+ и Mg2+ приводит к гибели растений. Наличие этих ионов в небольшом количестве в поглощенном комплексе, наоборот, весьма благоприятно сказывается на росте и развитии растений. Насыщение почвенного поглощающего комплекса такими ионами, как Ван, Ni2+, Со2+ или Си2 + , оказалось ядовитым для всех сельскохозяйственных культур.
Катионы, адсорбированные почвой, легко вступают в обменные реакции с другими катионами почвенного раствора. Именно благодаря этому явлению состав адсорбированных почвой катионов изменяется.
Адсорбция коллоидами почв анионов носит несколько иной характер. Как показали исследования, поглощение анионов зависит от состава почвенных коллоидов, реакции среды, величины электрокинетического потенциала коллоидов, а также от особенностей самих анионов. Ряд анионов — такие, как NO^, Cl - — почвой не поглощаются. В силу этого они свободно передвигаются в почве вместе с почвенной влагой. Вообще чем выше зарядность аниона, тем больше его способность поглощаться почвой, например анион фосфорной кислоты РО| сравнительно легко поглощается почвой.
Поглощение анионов почвой в значительной степени зависит от состава почвенных коллоидов. Опыт показывает, что чем больше в почве содержится глинистых минералов и аморфных полуторных оксидов, тем больше ионов она способна поглотить.
Реакция среды также оказывает свое влияние на величину поглощения анио нов почвой. Как правило, подкисление способствует большому поглощению анионов, подщелачивание вызывает ослабление поглощения анионов. Например, поглощение фосфат-иона в подзолистой почве увеличивается в шесть раз при подкислении раствора с pH 7,3 до pH 3,5. В слабокислых, нейтральных и щелочных почвах адсорбция анионов РО*~ происходит с образованием нерастворимых или малорастворимых фосфатов кальция, железа и алюминия. Таким образом, процесс поглощения почвой анионов, в отличие от процесса поглощения катионов, происходит с образованием в ряде случаев химических соединений и потому чаще всего является необратимым.
Такое важное свойство почвы, как ее буферность, также определяется свойством ее коллоидов. Появляющиеся в почвенном растворе кислота или щелочь тотчас же вступают во взаимодействие с почвенными коллоидами как наиболее активной частью твердой фазы почвы. В результате взаимодействия часть кислоты или щелочи исчезает из раствора, т. е. нейтрализуется, следовательно, сдвиг реакции ослабляется.
Не все почвы обладают одинаковым буферным действием. Так, глинистые почвы более тяжелого механического состава, богатые коллоидами, обладают наибольшим буферным действием. Очень малым буферным действием обладают почвы легкого механического состава, содержащие к тому же мало гумусовых веществ.
§ 138. Коллоидно-химические свойства протоплазмы
Основным содержанием любой живой клетки является протоплазма — весьма сложная комплексная система, богатая водой и состоящая из ряда органических соединений. Главная роль в протоплазме принадлежит, безусловно, белкам, которые связаны с другими органическими соединениями, в первую очередь с липоидами, нуклеиновыми кислотами, гликогеном и др. Как показали многочисленные исследования, протоплазма характеризуется гомогенносз’ь.ю, нераствори-
— 493 —
Рис. 210. Молекулярная сеть (молекулярный цитоскелет гиалоплазмы): черные точки иозбражают места сцепления
гостью в воде, сократимостью, способностью к обратимым изменениям своего состава и вязкости.
По мнению ряда ученых, различные включения, встречающиеся в протоплазме живой клетки, не имеют принципиального значения для жизни и, по существу, являются специальными дифференцировками. Иными словами, в протоплазме могут быть, а могут и не быть, гранулы или вакуоли различной величины н в различном количестве. Протоплазма — бесцветное прозрачное вещество, которое получило специальное название гиалоплазмы.
Согласно современным представлениям, протоплазму следует рассматривать как сложную коллоидную систему, обладающую всеми свойствами и признаками макромолекул в растворе. Исследования, проведенные за последние годы, убедительно показали, что протоплазма построена по типу сложных коацерватов. Как уже отмечалось, белки протоплазмы представляют собой сложные соединения более простых белков с нуклеи- -новыми кислотами, углеводами, высшими жирными кислотами и т. д. Именно при соединении с белком эти вещества образуют сложные коацерваты, из которых большое значение имеют так называемые внутрикомп-лексные коацерваты.
Протоплазма живой клетки обладает еще одним важным свойством, которое сближает ее с коллоидными растворами, — это явление тиксотропии. Тиксотропные свойства протоплазмы были обнаружены при исследовании разного рода течений протоплазмы, определением вязкости, а также прямыми опытами с помощью микроманипуляций. Исследования показали, что протоплазма стоит на грани между растворимостью и нерастворимостью
в воде. В результате этого даже малейшего изменения условий внешнего или внутреннего порядка вполне достаточно, чтобы изменить эти соотношения в пользу растворимости или нерастворимости.
Таким образом, по современным представлениям протоплазма является весьма подвижным тиксотропным студнем, который легко может переходить в золь, обладающий коацерватными свойствами. В основе этих взаимных превращений лежит функциональное состояние клетки.
Однако даже в состоянии золя протоплазма сохраняет пластичность, т. е. свойства твердого тела. Об этом свидетельствуют многочисленные опыты по падению в жидкой протоплазме посторонних микроскопических частиц. Из курса физики известно, что микроскопические тела падают в жидкости с постоянной скоростью (закон Стокса). В протоплазме же подобное падение идет с задержками, толчками, с отклонениями, как будто падающие частицы на своем пути встречают невидимые препятствия. На основании этих фактов был сделан вывод о том, что в протоплазме, даже в состоянии золя, имеется тончайший цитоскелет, основой которого являются вытянутые полипептидные цепи белка. Эти цепи взаимодействуют друг с другом своими боковыми цепями, образуют тончайшую сеть, т. е. молекулярный остов протоплазмы (рис. 210).
Основным свойством цитоскелета является его подвижность. При движении протоплазмы большое число точек скрепления боковых цепей полипептидных молекул непрерывно разрывается и вновь восстанавливается. Боковые цепи полипептидных молекул белка могут взаимодействовать друг с другом в точках сцепления путем образования водородных связей или же за счет сил Ван-дер-Ваальса.
В петлях цитоскелета находятся разнообразные глобулярные белки, молекулы которых при развертывании сами могут превращаться в скелетные образо-
— 494 —
зания. Внутри цитоскелета находятся и другие органические и неорганические вещества», а также вода. Протоплазма живой клетки представляет собой пол «фазную коллоидную систему, состоящую из высокомолекулярных соединений, диспергированных в водной среде.
Однако, в отличие от тел неживой природы, характеризующихся постоянством состава и формы, протоплазма сохраняет свое постоянство в результате непрерывно идущих процессов обмена.
«Жизнь > — говорит Ф. Энгельс, — есть способ существования белковых тел, и этот способ существования состоит по своему существу в постоянном самообновлении химических составных частей этих тел»*. Это положение остается и по сей день неоспоримым, и только исходя из него можно правильно анализировать различные биологические явления.
* Ф. Энгельс. Анти-Дюринг, Политиздат, 1970, стр. 78.
I •
I
I
г
ЛИТЕРАТУРА
Агрохимия. Под ред. В. М. Клечковского, «Высшая школа», 1968.
Алешин С. Н. Руководство к практическим занятиям по физической и коллоидной химии. Изд. ТСХА, 1952.
Афонский С. В. Физическая и коллоидная химия. «Советская наука», 1954.
Балезин С. А. Практикум по физической и коллоидной химии. М., Учпедгиз, 1958.
Балезин С. А., Парфенов Г. С. Основы физической и коллоидной химии. М., Учпедгиз, 1959.
Б е р г А. Л. Введение в термографию. М., Изд-во АН СССР, 1961.
Бродский А. И. Физическая химия, т. I и И. Госхимиздат, 1948.
Б у л а н к и н И. Н. Физическая и коллоидная химия (курс лекций для биологов). Изд. Харьковского ун-та, 1957.
Булл Г. Б. Физическая биохимия. М., ИЛ, 1949.
Возбуцкая А. Е. Химия почвы. М., «Высшая школа», 1968.
Воробьевы. К. и др. Практикум по физической химии. Госхимиздат, 1950.
Горбунов Н. И. Высокодисперсные минералы и методы их исследования. М., Изд-во АН СССР, 1963.
Г е р а с и м о в Я. И. и др. Курс физической химии, т. I. М., «Химия», 1969.
Григоров О. Н. и др. Руководство к практическим занятиям по коллоидной химии. Изд. ЛГУ, 1955.
Г у л я к и н И. В. Система применения удобрений. М., «Колос», 1970.
Даниельс Ф., Альберти Р. Физическая химия. М., «Высшая школа», 1967.
Д у м а н с к и й А. В. Учение о коллоидах. М., Госхимиздат, 1970.
Жуков И. И. Коллоидная химия. Изд. ЛГУ, 1949.
Ж у к о в и ц к и й А. А., Ш в а ц м а н Л. А. Физическая химия. М., «Металлург», 1968.
3 ы р и н Н. Г., О р л о в Д. С. Физико-химические методы исследования почв. Изд. МГУ, 1964.
Измайлов Н. А. Электрохимия растворов. Изд. Харьковского ун-та, 1959.
Каблуков И. А., Г а п о н Е. Н., Г р и н д е л ь М. А. Физическая и коллоидная химия. А4., Сельхозгиз, 1949.
— 495 —
Ка п у ст и нс к и й А. Ф. Очерк по истории физической и неорганической химии в России. М., Изд-во АН СССР, 1949.
К а с а т о ч к и н В. И., Пасынский Л. Г. Физическая и коллоидная химия. М., -Медгиз, 1960.
Киреев В. А. Курс физической химии. М., Госхимиздат, 1956.
Киселева Е. В.и др. Сборник примеров и задач по физической химии. М., «Высшая школа», 1970.
Кройт Г. Р. Наука о коллоидах, т. I, ЛА., ИЛ, 1955.
К у з н е ц о в В. В. Физическая и коллоидная химия. М., «Высшая школа», 1960.
Кульман А. Г. Физическая и коллоидная химия. М., Пищепромиздат, 1963.
Леонова В. Ф. Термодинамика. М., «Высшая школа», 1967.
Липатов С. М. Физико-химия коллоидов. М., Госхимиздат, 1948.
Липатников В. Е., Казаков К. М. Физическая и коллоидная химия. М., «Высшая школа», 1968.
Марченко Р. Т. Физическая и коллоидная химия. М., «Высшая школа», 1965.
Медведев П. И. Физическая и коллоидная химия. М., Сельхозгиз, 1957.
Межени ы й П. П. Лабораторный практикум по физической и коллоидной химии. М., Сельхозгиз, 1959.
Митрофанов П. П. Физическая химия. М., «Высшая школа», 1965.
Наумов В. В. Химия коллоидов. М., 1932.
Пасынский А. Г. Коллоидная химия. М., «Высшая школа», 1968.
Песков Н. П., Александрова -П рейс Е. М. Курс коллоидной химии. М., Госхимиздат, 1948.
Петербургский А. В. Практикум по агрохимии. М., Сельхозгиз, 1963.
Плешков Б. П. Биохимия сельскохозяйственных растений. Почвоведение. По ред. И. С. К а у р и ч е в а и И. П. Гречина. М., «Колос», 1969.
Путилова И. Н. Руководство к практическим занятиям по коллоидной химии. М., «Высшая школа», 1961.
Равич-Шербо М. И., Анненков Г. А. Физическая и коллоидная химия. М., «Высшая школа», 1968.
Раковский А. В. Курс физической химии. М., Госхимиздат, 1939.
Р о у з С. Химия жизни. М., «Мир», 1969.
Ребиндер П. А. Конспект общего курса коллоидной химии (составлен К. А. Поспеловой). Изд-во МГУ, 1950.
Сердоболье к ий И. П. Химия почвы. М., Изд. АН СССР, 1953.
Соколовский А. Н. Сельскохозяйственное почвоведение. М., Сельхозгиз, 1956.
С т а й к о в Ц., К о ж у х а р о в М. Неорганична и физична коллоидна химия. София, 1962.
Т а г е р А. А. Растворы высокомолекулярных соединений. М., Госхимиздат, 1951.
Физиология сельскохозяйственных растений, т. 1—3. Изд-во МГУ, 1967.
Чухров Ф. В. Коллоиды в земной коре. М., Изд. АН СССР, 1955.
Ш а м ш и н Д. Л. Физическая и коллоидная химия. М., «Высшая школа» 1968.
Шелудко А. Коллоидная химия, пер. с болгарского. М., ИЛ, 1960.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абсорбция 432
Агрегативные состояния 10 Адсорбция 432 газов 443 изотерма 439 из растворов 448 обменная *452 отрицательная 449 положительная 449 практическое применение 454 специфическая 452 удельная 434 хроматографическая 349 эквивалентная 452
Аккумуляторы 322
Активаторы 194
Активность ионов 144
Актор 183
Акцептор 183
Аморфное состояние 30
Анализ инфракрасноспектроскопический 327 пламеннофотометрический 342 поляриметрический 345 рентгенографический 330 рефрактометрический 343 седиментационный 392 спектрофотометрический 339 термографический 325 фотоколориметрический 338 хроматографический 349 электронномикроскопический 327
Анизотропия 30
Аниониты 350, 454
Антагонизм ионов 149, 459
Антифризы 134
Ассоциация ионов 147 молекул 51, 106
Безбуферный метод определения pH 266
Белки 424
фибриллярные 426 t глобулярные 426 Броуновское движение 383 Буферная емкость 258 Буферные растворы 254 Буферный метод определения pH 265 Вещества
поверхностно-активные 53 поверхностно-неактивные 53
Вода, диаграмма состояния 131 ионное произведение 240 полярность молекул 51, 105 свободная 421 связанная 421
Водородный показатель 242 Водородный электрод 282, 284 Второй закон термодинамики 83 •<
Высокомолекулярные соединения
(ВМС) 365, 369, 415
Вязкость жидкостей 57, 411, 423
Газообразное состояние 12
Газы, адсорбция 443
идеальные 13
кинетическая теория 18
основные законы 13
парциальное давление в смесях 27
реальные 23
уравнение состояния 17, 73
Гальванический элемент 274
Вестона (нормальный 296 концентрационный 280
Якоби — Даниеля 275
Гели 480, 485
Гетерогенные процессы 207
Гетерогенный катализ 194
Гидратация ионов 101
Гидроксония ион 109, 153
Гидролиз солей 250
Гомогенный катализ 194
элемент)
Гранула 403
Давление диссоциации 227
критическое 24
набухания 420
насыщенных паров 125 осмотическое 122, 387 парциальное 27
Двойной электрический слой
Денатурация 474
Дзета-потенциал 400
399 .
Диализ 372
Диализатор Грэма 373
Дипольный момент 105
Диссолюция 413
Диссоциация электролитическая 138
Дисперсиды 363
Дисперсионная среда 356
Дисперсная фаза 356
Дисперсность системы (степень дисперсности) 356
Дисперсоиды 363
Диффузионный потенциал 277
Диффузный слой 400
Диффузия 119, 385
коэффициент 386
Диэлектрическая постоянная 106
Емкость поглощения почвы 490
Жидкости
ассоциация 51, 106
взаимная растворимость 113
вязкость 57
интермицеллярные 402, 427 интрамицеллярные 427 кристаллические 51
Ньютоновские 59
497
Загруженность окислительно-восстановительной системы 317
Закон(ы) Авогадро 16
Бойля — Мариотта 13
газовые 13
Генри 111
Гей-Люссака 14
Гесса 80
Дальтона 27
действия масс 221
Кольрауша 161
Лавуазье и Лапласа 80
Максвелла — Больцмана 190
равновесия фаз 229
разведения Оствальда 238
Рауля 126, 130
сохранения материи и движения 70
Фика 385
Защитное действие растворов ВМС476
Идеальные газы 13
Идеальные растворы 137
Изобарный потенциал 96, 234, 236,
311
Изобары идеального газа 16
Изоморфизм 41
Изотерма адсорбции 439
идеального газа 14
реального газа 24
Изотропия 31
Инактивация 205
Ингибиторы 193
Индикаторы двухцветные 262
кислотно-основные 262
одноцветные 262
универсальные 266
Индуктор 183
Ионная сила раствора 146
Ионное произведение воды 240
Ионные двойники 147
Ионные тройники 148
Ионообменные смолы 350, 454
Каломельный электрод 284, 319
Капиллярная конденсация 433
Катализ, общие понятия 192
автокаталитические реакции 193
гетерогенный 195
— мультиплетная теория 199
— теория ансамблей 200
— электронно-химическая тео-
рия 201
гомогенный 194
микрогетерогенный 196
отрицательный 193
положительный 193
Катиониты 350, 454
Коагуляция
взаимная 459
гидрофобных золей 458
растворов ВМС 472
скрытая 457
Коагуляция явная 457
Коацервация 474 Коллоиды
лиофобные (гидрофобные) 394 лиофильные (гидрофильные) 414 необратимые 474 обратимые 474
Кольца Лизеганга 486
Компонента 67, 227
Константа Ван-дер-Ваальса 27 гидролиза 252 криоскопическая 133 скорости химической реакции 176 химического равновесия 222 электролитической диссоциации 237
Коэффициент активности ионов 144 вязкости 58 диффузии газов 386 изотонический 137 поверхностного натяжения 52 полезного действия 88 термического расширения 15 электропроводности сильных электролитов 164
Кривая расслоения 115
Криоскопия 133
Кристаллические тела 30
Кристаллы жидкие 51
строение 32
Критические давления 24 объем 24 состояние 24 температура 24
Лиотропные ряды 473
Максимальная работа (полезная) 98, 234
Мицелла 357, 402
Мольная долевая концентрация 103 Моляльность 104
Молярность 103
Момент дипольный 105
Мультиплеты 199
Набухание гелей 480 неограниченное 419, 481 ограниченное 419, 481
Насыщенный пар 125
Натяжение поверхностное 51 Нефелометрия 382 Нормальность 104 Нормальный водородный электрод 284 электродный потенциал 272 элемент Вестона 296
Обратимые реакции 98, 183, 220
Объем критический 24 Опалесценция 376 Осмос 121
Осмотическое давление 122, 387
— 498
Основной термодинамический цикл
(цикл Карно) 85
Отвердевание 11
Парциальное давление 27
Парциальный объем 28
Пептизация золей 466
Первый закон термодинамики 71
Перезарядка золей 463
Перекристаллизация 117, 413
Плавление 11
Плазма
изотермическая 11
газоразрядная 11
Плазмолиз 124
Поверхностное натяжение 51
Податл и вость ок исл ител ьно- восстановительной системы 317
Подвижность ионов 161
Поликонденсация 370
Полимеризация 369
Полиморфизм 40
Полихромия золей 378
Порог коагуляции 458
Постоянная
Планка 210
сосуда 166
Потенциалы диффузионные 277
изобарные 96, 234, 236, 311
мембранные 278
нормальные 272
окисл ител ьно-восста новител ь-
ный 309
протекания 396
седиментации 396
стандартные 272
термодинамические 400
электродные 270
электрокинетические 400
Почвенный поглощающий комплекс 490
Правило
Вальдена—Писаржевского 163
Вант-Гоффа 187
произведения растворимости 231
Траубе — Дюкло 445
фаз 229
Шульце — Гарди 458
Принцип Бертло 98
Ле Шателье 223
плотнейшей упаковки 32
Промоторы 194
Простетическая группа 205
Работа максимальная 98, 234
Равновесие Доннана 389
седиментационное 391
химическое 221
Рассеяние света 377
Растворы буферные 2Е4
гипертонические 124
гипотонические 124
идеальные 119
Растворы изотонические 124 коллоидные 357
Реакции бимолекулярные 176 второго порядка 179 гетерогенные 207 мономолекулярные 176 параллельные 181 первого порядка 177 сложные 181 сопряженные 182 тримолекулярные 176 фотохимические 209 цепные 183 экзотермические 75 эндотермические 75
Решетки кристаллические атомные 34 молекулярные 34 ионные 34
Рефракция 343
Ряд напряжений 273
Свободная энергия 89, 95
Свойства систем интенсивные 67 экстенсивные 67
Связанная энергия 89
Седиментация 391, 464
. Сжижение И
Синерезис гелей 487
Системы изолированные 67 неизолированные 67
Скорость реакции влияние температуры 187 зависимость от концентрации 177г 179 истинная 175 константа 176 средняя 174
температурный коэффициент 187
Сольватация 101
Сорбент 425
Сорбтив 425
Сорбция 425
Спайность 32
Сталагмометр 55
Степень электролитической диссоциации 138
Сублимация 11
Суспензия 363
Текучесть 59
Температура абсолютная 15 замерзания 130 кипения 63 критическая 24
Теория ансамблей 200 газов, кинетическая 18 гидратная 140 мультиплетная 199 протолитическая 238 разбавленных растворов 119 сильных электролитов 143
499
Теория электролитической диссоциации 138
Теория электронно-химическая 201
Тепловая теорема Нернста 95
Тепловой эффект реакции 75
Теплоемкость 94
Теплота
гидратации 109
испарения 63
нейтрализации 78
образования 75 парообразования 11 плавления 11 приведенная 89 разложения 76 растворения 76 сгорания 79 смачивания 451 сублимации 11
Термографический анализ 325
Термостат водяной 168
Тиксотропия 469, 483
Титрование
высокочастотное 173
кондуктометрическое 172
потенциометрическое 320
Точка замерзания 433
изоэлектрическая 428
критическая 24
тройная 131
Удельнае поверхность 356
Удельная электропроводность 151
Ультамикроскопия 379
Ультрафильтрация 374
Ультрацентрифугирование 375
Универсальная газовая постоянная 17, 73
Уравнение Аррениуса 158
Ван-дер-Ваальса 26
Гиббса 446
Ленгмюра 442
Нернста 272
Никольского 455
Рэлея 377
состояния идеального газа 17, 73
Фрейндлиха 440
Шишковского 446
Устойчивость золей
агрегатная 413
кинетическая 412
Фаза 67
простая 227
смешанная 227
Ферменты 202
Флотация 451
Флуктуация 13, 92, 385
Фотосинтез 213
Фотохимические реакции 209
Хемосорбция 432
Хлорофилл 215, 350
Химическая индукция 183
Химическая кинетика 174 Химическое сродство 99 Цепи водородные 299 двойные хингидронные 302 каломельно-водородная 300 каломельно-натрий-стеклянная 307 каломельно-стеклянная 305 концентрационная 208 хингидронно-каломельная 301 хлорсеребряно-сурьмяная 303
Цикл Карно 85
Число Авогадро 16 железное 476 золотое 476 рубиновое 476 степеней свободы 228 Фарадея 159
Шульце — Гарди правило 458
Эбулиоскопическая постоянная 133
Эбулиоскопия 133
Эквивалентная электропроводность 155
Электродиализ 373
Электрод водородный 282, 284 каломельный 281, 319 натрий-стеклянный 294 платиновый 319 стеклянный 291 сурьмяный 290 хингидронный 288 хлорсеребряный 287
Электролитическая диссоциация 138 Электронно-ядерный газ II Электроосмос 395
Электропроводность при бесконечном разбавлении 156 удельная 151 эквивалентная 155
Электроультрацентрйфугирование 374
Электрофорез 395, 407 Элемент гальванический 274 нормальный (Вестона) 296 Якоби — Даниеля 275
Элементарная ячейка 32
Энергия активации 189 внутренняя 69 гидратации 109 кинетическая 68 кристаллической решетки 39 потенциальная 68 свободная 89, 95 связанная 89
Энтропия 89 стандартная 92
Эффект протекания 396 седиментации 396 тепловой 75
Фарадея — Тиндаля 376
Ядро коллоидной мицеллы 402 500 —
ОГЛАВЛЕНИЕ
Стр.
Предисловие . . , , ,..................... . ....... 3
ЧАСТЬ ПЕРВАЯ ФИЗИЧЕСКАЯ ХИМИЯ Введение . ......................................................[5
Глава I
Молекулярно-кинетическая теория трех агрегатных состояний вещества § 1. Агрегатные состояния вещества. Понятие о плазме.............10
Газообразное (парообразное) состояние вещества................. 12
§ 2. Основные газовые законы....................................13
§ 3. Молекулярно-кинетическая теория газов...................18
§ 4. Реальные газы...........................................23
§ 5. Газовые смеси. Закон Дальтона. ............................27
§ 6. Воздушный режим почвы...................................28
Твердое агрегатное состояние................................... 30
§ 7. Признаки твердого состояния.............................. 30
§ 8. Внутреннее строение кристаллов и основные типы кристаллических решеток................................................... 32
§ 9. Координационное число и энергия кристаллической решетки . . 37 § 10. Полиморфизм и изоморфизм................................. 40
§ И. Глинистые минералы, их строение, свойства и значение в почвоведении . ..................................................... 41
Жидкое агрегатное состояние .....................................50
§ 12. Характеристика жидкого состояния...........................50
§ 13. Поверхностное натяжение и поверхностная энергия............51
§ 14. Методы определения. поверхностного натяжения ... ...... 53
§ 15. Внутреннее трение (вязкость) жидкостей. ... ......... 57
§ 16. Испарение и кипение жидкостей..............................61
§ 17. Роль воды в живых организмах................................64 ,
Глава II
Основы химической термодинамики и термохимии
§ 18. Предмет термодинамики. Основные термодинамические понятия . 65
§ 19. Энергия и ее виды. Внутренняя энергия системы..............68
§ 20. Закон сохранения энергии.......................•...........70
§ 21. Первое начало термодинамики................................71
§ 22. Работа расширения газа при различных термодинамических процессах ......................................................... 7
§ 23. Тепловые эффекты химических реакций .......................75
§ 24. Основные законы термохимии и термохимические расчеты .... 80
§ 25. Второе начало термодинамики. Понятие об энтропии...........83
§ 26. Критика теории тепловой смерти Вселенной . ............... 3
— 501 —
>
§ 27. Второе начало термодинамики и живые организмы..............94
§ 28. Третье начало термодинамики. Принцип минимума свободный энергии 95
§ 29. Максимальная работа и химическое сродство..................98
Глава III
Учение о растворах
§ 30. Растворы — физико-химические системы. Концентрация растворов 100
§ 31. Сущность процесса растворения........................104
§ 32. Растворимость газов в жидкостях ... ;................110
§ 33. Взаимная растворимость жидкостей.....................113
§ 34. Растворимость твердых веществ в жидкостях............115
§ 35. Природные растворы...................................117
Разбавленные растворы ....................... 'Т
§ 36. Диффузия и осмос в растворах.........................119
§ 37. Законы осмотического давления и его биологическое значение . . 122
§ 38. Понижение давления насыщенного пара растворителя.....125
§ 39. Температуры замерзания и кипения разбавленных растворов . . . 130
§ 40. Применение методов криоскопии и эбулиоскопии.........134
Глава IV
Электропроводность растворов
§ 41. Отступление от законов Вант-Гоффа и Рауля в растворах, электролитов. Теория электролитической диссоциации........................137
§ 42. Основные положения теории сильных электролитов...............142
§ 43. Физиологическое действие ионов. Ионный антагонизм............149
§ 44. Электропроводность растворов. Удельная электропроводность . 150
§ 45. Эквивалентная электропроводность растворов ................. 154
§ 46. Связь эквивалентной электропроводности со степенью диссоциации электролита и скоростями движения ионов............................158
§ 47. Закон независимости движения ионов (закон Кольрауша) ... . 160
§ 48. Определение степени диссоциации слабых электролитов и коэффициента электропроводности сильных электролитов методом электропроводности.......................................................... 163
§ 49. Методы определения электропроводности растворов электролитов 164
§ 50. Применение методов измерения электропроводности в лабораторной практике и в агрономии.........................................163
Глава V
Химическая кинетика и катализ. Фотохимия
§ 51 Химическая кинетика реакций. Основные положения и понятия . 1741
§ 52. Классификация . гмических реакций...........................176
§ 53. Реакции первого г грядка....................................177
§ 54. Реакции второго порядка.....................................179
§ 55. Сложные реакции.............................................181
§ 56. Влияние температуры на скорость химической реакции.........187
§ 57. Влияние температуры на скорость биологических процессов . . . 191
§ 58. Зависимость скорости реакции от катализатора. Катализ гомогенный и гетерогенный....................................................192
§ 59. Основные свойства катализаторов и факторы, влияющие на катализ 196
§ 60. Теории гетерогенного катализа...............................198
§ 61. Ферменты как катализаторы .... .........202
§ 62. Скорость гетерогенных химических процессов.....207
§ 63. Фотохимические реакции......................................209
§ 64. Фотосинтез в растениях......................................213
§ 65. Фотосинтез и урожай сельскохозяйственных культур............218
— 502 —
Гл а ва VI ' Химическое равновесие
§ 66. Понятие о химическом равновесии. Закон действующих масс . . . . 220 § 67. Смещение химического равновесия. Синтез аммиака и получение азотных удобрений.................................................223
§ 68. Равновесие в гетерогенных системах. Правило фаз.............225
§ 69. Применение закона действующих масс к равновесным системам «раствор—осадок». Правило произведения растворимости..............230
§ 70. Связь константы химического равновесия с максимальной работой реакции ..... ................................................... 233
§ 71. Применение закона действующих масс к растворам слабых электролитов ............................................................236
§ 72. Протолитическая теория кислот и оснований...................238
§ 73. Ионное произведение воды. Понятие о pH как показателе реакции среды.............................................................240
§ 74. Роль концентрации водородных ионов в биологических процессах 244
§ 75. Активная и общая кислотность растворов. Кислотность и щелочность почв........................................................246
§ 76. Реакция среды в растворах солей, гидролиз...................250
§ 77. Буферные растворы и буферное действие.......................254
Биологическое значение буферных систем. Буферность гочв и почвенного раствора.................................................,259
§ 79. Индикаторы и их применение .;............................. 262
§ 80. Принципы колориметрического определения pH..................265
Глава VII Электрохимия
§ 81. Электродный потенциал. Уравнение Нернста....................
§ 82. Гальванические элементы и их электродвижущая сила...........274
§ 83. Диффузионный потенциал. Биологическое значение диффузионных и мембранных потенциалов...................................... 277
§ 84. Концентрационные цепи.......................................280
§ 85. Электроды сравнения....................................... 282
§ 86. Индикаторные электроды......................................287
§ 87. Методы измерения электродвижущих сил......................295
§ 88. Электрометрический (потенциометрический) метод определения pH 299
§ 89. Преимущества и недостатки различных потенциометрических методов определения pH....................................................306
§ 90. Определение в почвах поглощенного натрия с помощью натрий-стек-лянного электрода............................................... 307
§ 91. Окислительно-восстановительные потенциалы...................308
§ 92. Окислительно-восстановительные реакции и потенциалы в почвах 314
§ 93. Экспериментальное определение окислительно-восстановительного потенциала в почве................................................318
§ 94. Потенциометрическое титрование............................ 320
§ 95. Свинцовый! аккумулятор.................................... 322
Г л а в а VIII
Физико-химические методы, применяемые в практике агрохимических и почвенных лабораторий
§ 96. Термографический метод......................................325
§ 97. Электронная микроскопия.....................................327
§ 98. Рентгенографический метод...................................330
§ 99. Метод инфракрасной спектроскопии............................331
§ 100. Фотометрический анализ.....................................336
§ 101. Методы анализа по фотометрии пламени.......................342
§ 102. Оптические методы анализа................................ 343
§ 103. Хроматографические методы анализа....................« . . 348
— 503 —
ЧАСТЬ ВТбРАЯ
КОЛЛОИДНАЯ химия Введение ...... ..............................................356
Глава IX Общая характеристика коллоидов и их свойств
§ 104. Классификация дисперсных систем........................363
§ 105. Получение коллоидно-дисперсных систем..................365
§ 106. Получение растворов высокомолекулярных веществ.........369
§ 107. Методы очистки золей и растворов высокомолекулярных веществ . 372
§ 108. Оптические свойства коллоидных растворов . . ... ......376
§ 109. Молекулярно-кинетические свойства коллоидных растворов . . 383
Глава X
Теория коллоидных систем
Гидрофобные коллоидные системы
§ 110. Электрокинетические явления в коллоидных системах..............394
§ 111. Возникновение двойного электрического слоя и его строение . .,397
§ 112. Мицеллярная теория строения коллоидной частицы.................402
§ 113. Электрокинетический потенциал золей и методы его измерения . . 408
§ 114. Вязкость гидрофобных золей .................................. 411
§ 115. Устойчивость гидрофобных золей.................................412
Растворы высокомолекулярных соединений (лиофильные системы)...........414
§ 116. Общая характеристика растворов высокомолекулярных соединений 415
§ 117. Набухание и растворение высокомолекулярных соединений . . . 418
§ 118. Свободная и связанная вода в коллоидах.........................421
§ 119. Вязкость высокомолекулярных соединений ........................423
§ 120. Белки как коллоиды......................................... 424
Глава XI
Поверхностные явления на границе раздела фаз
§ 121. Свободная энергия поверхности раздела фаз......................430
§ 122. Общая характеристика сорбционных явлений.......................432
§ 123. Явление адсорбции............................................ 433
§ 124. Адсорбция на поверхности раздела твердое вещество — газ . . . 436
§ 125. Изотермы адсорбции..........................................439
§ 126. Адсорбция на поверхности раздела жидкость — газ.............443
§ 127. Адсорбция на поверхности твердое вещество — жидкость .... 448
§ 128. Адсорбция и биологические процессы.......................... 456
Глава XII
Изменение состояния коллоидных систем
§ 129. Коагуляция гидрофобных золей электролитами........... 4&7V
§ 130. Кинетика коагуляции...................................464
§ 131. Пептизация гидрофобных золей..........................466
§ 132. Тиксотропия.....................................♦ . . . 4®
§ 133. Коагуляция растворов высокомолекулярных • соединений .... 472
§ 134. Коацервация...........................................474
§ 135. Защитное действие растворов высокомолекулярных соединений 476
§ 136. Гели, их образование, строение и свойства.................479
§ 137. Почвенные коллоиды........................................490
§ 138. Коллоидно-химические свойства протоплазмы.................493
Литература................................................... 495
1 р. 13 к