/












































































































































































































































































































































































































































































































































































































































Author: Кристиан Г.
Tags: аналитическая химия физическая химия химическая физика химия
ISBN: 978-5-94774-389-0
Year: 2009




Text
ЛУЧШИЙ ЗАРУБЕЖНЫЙ УЧЕБНИК
АНАЛИТИЧЕСКАЯ
ХИМИЯ
ANALYTICAL CHEMISTRY
Sixth Edition
Gary D. Christian
University of Washington
JOHN WILEY I SONS. INC.
ЛУЧШИЙ ЗАРУБЕЖНЫЙ УЧЕБНИК
Г. КРИСТИАН
АНАЛИТИЧЕСКАЯ
ХИМИЯ
В двух томах
1
Перевод с английского
канд. хим. наук А. В. Гармаша,
канд. хим. наукН. В. Колычевой,
канд. хим. наук Г. В. Прохоровой
Вступительная статья
академика РАН Ю. А. Золотова
Москва
БИНОМ. Лаборатория знаний
2009
УДК 543
ББК 24.5я73
К82
Кристиан Г.
К82 Аналитическая химия : в 2 томах. / Г. Кристиан ; пер. с
англ. — М. : БИНОМ. Лаборатория знаний, 2009. — (Лучший
зарубежный учебник).
ISBN 978-5-94774-389-0 (БИНОМ. ЛЗ)
ISBN 0-471-21472-8 (англ.)
Т. 1. — 623 с.: ил.
ISBN 978-5-94774-390-6
В учебном издании, написанном американским ученым и талант-
ливым преподавателем, методически выдержанно изложен универси-
тетский учебный курс по аналитической химии, причем рассмотрены
новейшие методы анализа, а также анализ важнейших для современ-
ной аналитической практики объектов (клинический и экологический
анализ). В заключительном разделе приведено 40 лабораторных работ.
В русском издании выходит в двух томах. В т. 1 вошли гЛ. 1-15.
Для студентов вузов, изучающих аналитическую химию, аспирантов
и преподавателей.
УДК 543
ББК 24.5я73
Первый тираж издания осуществлен при финансовой поддержке
Российского фонда фундаментальных исследований по проекту № 06-03-46018
Учебное издание
Серия: «Лучший зарубежный учебник»
Кристиан Гэри
АНАЛИТИЧЕСКАЯ ХИМИЯ
В двух томах
Том 1
Ведущий редактор канд. хим. наук Т. И. Почкаева
Редактор канд. хим. наук Д. К. Новикова
Художник С. Инфантпэ, Н. А. Новак. Художественный редактор О. Г. Лапко
Компьютерная верстка: Л. В. Катуркина
Подписано в печать 01.12.08. Формат 70x100/16.
Усл. печ. л. 50.70. Тираж 1700 экз. Заказ3771.
Издательство «БИНОМ. Лаборатория знаний»
125167, Москва, проезд Аэропорта, д. 3
Телефон: (499) 157-5272, e-mail: binom@Lbz.ru, http://www.Lbz.ru
ISBN 978-5-94774-390-6 (Т. 1)
ISBN 978-5-94774-389-0 (БИНОМ. ЛЗ)
ISBN 0-471-21472-8 (англ.)
Copyright © 2004 John Wiley & Sons, Inc.
All rights reserved.
Authorized Translation from
the English language edition
published by
John Wiley & Sons, Inc.
© Перевод, оформление,
БИНОМ, Лаборатория
знаний, 2008
Об авторе
Профессор Гэри Кристиан — известный американский ученый-аналитик и вы-
дающийся педагог. Он — автор многих научных публикаций по проточно-ин-
жекционному анализу, спектроскопическим и электрохимическим методам. Его
научные заслуги отмечены многими почетными наградами, в том числе высшей
наградой Американского химического общества (ACS) в области аналитиче-
ской химии — премией Фишера, премией отделения аналитической химии ACS
за выдающиеся достижения в области преподавания, золотой медалью журнала
Taianta и др. Кристиан избирался председателем отделения аналитической хи-
мии ACS, главным редактором международного журнала по аналитической хи-
мии Taianta, членом редколлегии многих других журналов.
Гэри Кристиан — автор нескольких учебников. Наибольшее признание из
них получил учебник «Аналитическая химия», который выдержал в США уже
шесть переизданий, начиная с 1971 г. В американских университетах этот учеб-
ник используют чаще, чем другие учебники по аналитической химии. Рецензии
на него были опубликованы в ряде журналов — все положительные.
Все это дало основание осуществить перевод книги на русский язык. Разу-
меется, было использовано последнее, шестое издание. Хотя после выхода аме-
риканского учебника прошло несколько лет, я уверен, что для хорошего
учебника по фундаментальной учебной дисциплине этот срок некритичен.
Содержание учебника, я думаю, смогут оценить сами читатели, прежде все-
го преподаватели. Конечно, как и в любом другом переводном учебнике приве-
денный в этой книге материал не вполне соответствует принятым в наших
высших учебных заведениях программам курса аналитической химии. Зато
учебник Кристиана чрезвычайно современен. В учебнике активно используются
компьютерные технологии, описаны методы работы в современной аналитиче-
ской лаборатории, приведены методики анализа важнейших объектов, включа-
ющих и биологические жидкости, и объекты экоанализа.
Переводные книги всегда способствуют повышению уровня преподавания
в нашей стране: они используются преподавателями для актуализации учебной
программы и методики преподавания, отдельные их разделы изучаются студен-
тами, аспирантами и молодыми учеными для знакомства с новыми методами и,
конечно же, они очень интересны для отечественных авторов учебных изданий
как путеводитель в их творческой работе. Опытные преподаватели вузов по-
мнят, что в 1970-е годы было переведено сразу несколько учебников и учебных
пособий. Наиболее известные из них — это двухтомник «Основы аналитиче-
ской химии» (авторы Скуг Д., Уэст Д. — М.: Мир, 1979) и «Количественный
6
ОБ АВТОРЕ
анализ» (авторы Фритц Дж., Шенк Г. — М.: Мир, 1978). Даже в наши трудные
времена удалось издать европейский учебник по аналитической химии (Пер. с
англ.; в 2-х т.; под ред. Р. Кельнера и др. — М.: Мир, 2005). Вероятно, подобным
образом будет «работать» и книга Г. Кристиана.
Я с большим удовольствием представляю этот перевод российским читате-
лям. Мне особенно приятно это сделать, поскольку я давно знаю автора лично,
выполнял с ним совместный проект по проточному анализу, бывал в его лабора-
тории в Сиэтле, да и в его доме; мы многократно встречались на международных
конференциях. Профессор Кристиан рад был узнать о подготовке русского изда-
ния его популярного учебника.
Мне также приятно осознавать, что перевод этой всеобъемлющей книги
был осуществлен большей частью сотрудниками моей кафедры: канд. хим. наук
А. В. Гармашом (введение, гл. 1-5, 9-11, 18, 23, практические работы, приложе-
ния), совместно канд. хим. наук Н. В. Колычевой и А. В. Гармашом (гл. 6-8),
канд. хим. наук Г. В. Прохоровой (гл. 12-15), канд. хим. наук А. В. Ивановым
(гл. 19-22). Другие главы перевели канд. хим. наук Е. Э. Григорьева (гл. 16-17) и
канд. хим. наук Т. П. Мосолова (гл. 24-26).
Я уверен, что эта книга, содержащая очень важный и современный матери-
ал по аналитической химии, методические качества которой проверены много-
кратными ее переизданиями за рубежом, обогатит библиотеку учебной
литературы университетов и высших специальных профессиональных заведе-
ний и будет востребована преподавателями и студентами.
академик Ю. А. Золотов
Сью — за десятилетия радости
и за поразительный талант быть бабушкой
Тане и Тэффи — за солнечный свет,
которым они освещают нашу жизнь
Предисловие
Учитель открывает дверь, но войдешь ли ты в нее, зависит от тебя.
Задача аналитической химии — охарактеризовать материю с химической точки
зрения, как качественно, так и количественно. Эта задача важна применительно
едва ли не к каждому аспекту нашей жизни, поскольку все, чем мы пользуемся,
представляет собой химические вещества. Чарльз Н. Рейли в конце своей жизни
сказал: «Аналитическая химия — это то, чем занимаются химики-аналитики».
Чем же они занимаются, мы и рассмотрим в данной книге.
Книга предназначена для студентов вузов, специализирующихся в химии и
областях, связанных с ней. В ней рассматриваются теоретические основы и мето-
ды количественного химического анализа, ознакомившись с которыми можно по-
добрать способ для определения количества того или иного вещества в пробе. Вы
научитесь разрабатывать методики анализа, узнаете, какая информация для этого
необходима, как отбирать лабораторную пробу, чтобы ее состав был представи-
тельным (т. е. соответствовал составу всего образца в целом), как подготовить
пробу для анализа, какие средства измерений есть в распоряжении химика-анали-
тика, как оценивать статистическую значимость результатов анализа. Гл. 24-26
иллюстрируют применение полученных вами знаний в сферах клинического ана-
лиза, геномики и протеомики, отбора проб и анализа объектов окружающей среды.
Рассмотренные в книге примеры применения методов аналитической хи-
мии не случайно относятся к наукам о живом, клинической химии, проблемам
загрязнения воды и воздуха, промышленному анализу. Значение аналитической
химии становится особенно понятным лишь тогда, когда приходит осознание
того, что неправильный анализ крови может угрожать жизни пациента, а ошибки
при контроле качества продукции могут обернуться серьезными финансовыми
потерями для производителя. Выполнение непрерывных автоматизированных
анализов позволяет химической промышленности экономить миллионы долла-
ров и обеспечивать максимальную эффективность производства.
Для кого предназначена данная книга?
Данная книга содержит общий курс количественного анализа. Объем приведен-
ного в ней материала заведомо больше того, что можно пройти за один семестр.
Поэтому преподаватель вправе самостоятельно выбирать те темы, которые он
8
предисловие
считает наиболее важными. Остальные разделы могут в этом случае рассматри-
ваться как дополнительный материал. Книгу можно использовать независимо от
того, в какой последовательности излагаются химические и инструментальные
методы анализа. В любом случае я надеюсь, что вы найдете время, чтобы прочи-
тать и те разделы, которые интересны сами по себе, хотя, возможно, и не являются
обязательными. Впоследствии их можно будет использовать в качестве спра-
вочной информации.
Что осталось неизменным?
Шестое издание книги существенно переработано и дополнено новыми источ-
никами (главным образом более поздними), но многие особенности предыду-
щих изданий сохранены. Каждой главе предшествует небольшой абзац,
раскрывающий общее содержание главы. Ключевые слова выделены жирным
шрифтом, а основные уравнения заключены в рамку . Это поможет читателю
при повторении материала. Чтобы обратить внимание на наиболее важные поня-
тия (и также помочь при повторении материала), некоторые предложения выде-
лены курсивом.
На протяжении всей книги внимание читателя постоянно обращается на
анализ размерностей физических величин. Это позволит лучше прочувствовать
постановку задачи. В книге используются размерности величин в системе СИ и
их общепринятые обозначения (л, мл, моль, сит. д.). Понятия «нормальность» и
«эквивалент» в книге также рассматриваются, однако основное внимание все же
уделено использованию понятий «молярность» и «моль». Вопросы, связанные с
«нормальностью», изложены таким образом, что преподаватель может не исполь-
зовать это понятие, если сочтет это не нужным.
Для облегчения работы задачи и рекомендуемая литература сгруппированы
по темам. Литературные источники значительно обновлены, добавлено много
новых задач. Всего в книге содержится 673 вопроса и задачи (а также руководст-
во по их решению).
В предыдущее издание было добавлено много новых тем, большинство из
которых сохранились в тексте данного издания, так как они важны для изучения
материала, например:
• статистика малых выборок;
• статистика пробоотбора;
• систематический подход к расчетам химических равновесий, основанный
на законах сохранения массы и заряда;
• гетерогенные равновесия;
• использование логарифмических диаграмм для представления сложных
равновесий (добавлен раздел по построению таких диаграмм при помощи
электронных таблиц);
• спектрометры с матрицами диодов;
• инфракрасная спектроскопия с преобразованием Фурье;
• ИК-спектроскопия в ближней области;
ПРВДИСЛОВИЕ
9
• оптоволоконные сенсоры;
• газовая хромато-масс-спектрометрия.
Что изменилось?
Изменено расположение некоторых глав для того, чтобы логически связать их
содержание с другими главами. По многочисленным просьбам читателей глава,
посвященная базовым средствам и операциям в аналитической химии, помещена
в начало книги (гл. 2). Возможно изучение отдельных разделов этой главы, необ-
ходимых для выполнения лабораторного практикума. Гл. 10, посвященная гра-
виметрическому методу анализа и равновесным процессам образования осадка,
в настоящем издании предшествует главе, описывающей реакции осаждения и
осадительное титрование. Такое расположение глав позволяет упорядочить из-
ложение близких по смыслу вопросов.
Основной метод анализа смесей веществ — хроматография — в последнее
время переживает бурное развитие, ее возможности непрерывно расширяются.
В связи с этим описание хроматографических методов расширено и дополнено:
целых три главы (19-21) посвящены основам хроматографии, газовой хроматог-
рафии и жидкостной хроматографии. Устаревшие методы, такие как бумажная
хроматография, не рассматриваются.
Что нового?
С момента выхода предыдущего, пятого, издания книги прошло почти десять
лет (вот результат моей слишком долгой работы в качестве декана!). За это вре-
мя многое изменилось. В шестом издании представлен ряд новых тем и глав, из-
менен характер изложения. Основные добавления состоят в следующем:
• для проведения вычислений, статистического анализа и построения графи-
ков на протяжении всей книги используются электронные таблицы Excel.
При помощи электронных таблиц построены многие кривые титрования,
рассчитаны значения молярных долей а и построены распределительные диа-
граммы а—pH, а также концентрационно-логарифмические диаграммы. Инст-
рукции по использованию электронных таблиц изложены в доступной форме,
так что можно легко понять их алгоритм. Обычно в таких инструкциях просто
указано, какую формулу необходимо ввести в определенную ячейку. Например,
если ячейка В11 должна содержать частное от деления чисел, находящихся в
ячейках А2 и АЗ, то в тексте приводится формула (=А2/А3). Результат появляет-
ся в ячейке Bl 1, в которую эта формула была введена. Номера ячеек, содержа-
щих формулы, в тексте выделены жирным шрифтом. Анализируя электронные
таблицы, часто приходится разбираться, что означает та или иная формула, на-
пример, значениями каких величин являются числа, частное которых вычисля-
ется в приведенном выше примере. Со сложными формулами и выражениями и
такая работа может быть весьма утомительной. Поэтому в инструкциях к элект-
ронным таблицам приведены не только формулы, непосредственно вводимые в
10
ПРВДИСЛОВИЕ
ячейки, но и исходные выражения. Читатель лучше оценит удобство такой фор-
мы представления инструкций, когда непосредственно начнет работать с приве-
денными в книге примерами для электронных таблиц.
• Добавлена новая глава «Хорошая лабораторная практика» — сборник нор-
мативных документов по обеспечению качества результатов химического
анализа.
Вопросы практического применения аналитической химии приобрели осо-
бую важность, когда административно были введены достаточно сложные и
строгие обязательные нормативы, призванные обеспечить правильность резуль-
татов химического анализа. Каждому аналитику, стремящемуся сделать карьеру,
известно, что знание этих нормативов будет весьма благоприятно воспринято ра-
ботодателем. Данная глава может служить введением к изучению этих вопро-
сов, а также кратким справочником. Она включает в себя следующие разделы:
• поверка (валидация) методики анализа;
• обеспечение качества результатов;
• аккредитация лабораторий;
• электронная документация и электронные подписи (новые нормы 21
CRF, часть 11).
• Еще одна новая глава посвящена геномике и протеомике.
Аналитическая химия сыграла решающую роль в успешном завершении ис-
торического проекта «Геном человека». Читатель, вероятно, знает об этом про-
екте. Разработанные для него методики стали рутинными для установления
последовательности ДНК сложных организмов, в судебной экспертизе и других
областях. В настоящее время начинается развитие протеомики, где аналитиче-
ская химия будет решать чрезвычайно важные и неосвоенные проблемы, связан-
ные с картированием белков; введение в эти методы также здесь описано. Глава
рассматривает следующие вопросы:
• проект «Геном человека»;
• секвенирование генов;
• полимеразная цепная реакция (ПЦР);
• ДНК-чипы;
• двумерный гель-электрофорез и времяпролетная масс-спектрометрия
с ионизацией методом лазерной десорбции из матрицы-мишени
(MALDI-TOF).
Кроме перечисленных выше книга содержит следующие новые темы:
• калибровка мерной посуды;
• ускоренная и микроволновая экстракция и вскрытие проб;
• ионселективные полевые транзисторы;
• спектральные базы данных в Интернете — коммерческие и бесплатные;
• твердофазная экстракция (сорбция) — раздел расширен; твердофазная мик-
роэкстракция;
ПРЕДИСЛОВИЕ
11
• номенклатура в хроматографии; рекомендованные ИЮПАК термины и
обозначения;
• теория эффективности хроматографических колонок — раздел расши-
рен;
• программное обеспечение для моделирования хроматографических про-
цессов и их использование для разработки методик;
• колонки для капиллярной газовой хроматографии — раздел расширен и об-
новлен;
• дискретный, непрерывный проточный пробоотбор и термодесорбция в га-
зовой хроматографии;
• быстрая газовая и жидкостная хроматография;
• жидкостная хромато-масс-спектрометрия (ЖХ-МС);
• масс-анализаторы для ГХ-МС и ЖХ-МС;
• неподвижные фазы для ВЭЖХ — раздел расширен; применение капилляр-
ных колонок малого диаметра для повышения чувствительности;
• капиллярный электрофорез — раздел расширен; капиллярная электрохро-
матография.
Практические работы
В книге описано 40 практических работ, иллюстрирующих большинство мето-
дов анализа. Работы сгруппированы по темам. Для каждой работы приведены
методика проведения эксперимента и соответствующие уравнения реакций, по-
этому читатель может сразу понять, что именно анализируется и каким образом.
Перечислены все реагенты и растворы, которые необходимо приготовить перед
выполнением работы, что позволяет выполнить работу быстро и эффективно.
Эксперименты, по возможности, продуманы таким образом, чтобы, в соответст-
вии с требованиями техники безопасности, избежать использования асбеста,
хлороформа, тетрахлорида углерода и бензола. Все работы, особенно связанные
с титрованием, спланированы так, чтобы уменьшить расход реагентов за счет
приготовления минимально возможных объемов реагентов (титрантов).
Исключены три работы, в которых используется ртуть. Удалена также
работа по бумажной хроматографии, поскольку в настоящее время бумажная
хроматография практически полностью вытеснена тонкослойной хроматогра-
фией. Для экономии места описание неферментативных кинетических методов
и соответствующая работа заменены работой по ферментативным методам. По
тем же причинам исключена работа по хроматографическому разделению коба-
льта и никеля на анионообменной колонке.
Добавлена новая работа (практическая работа 18) по определению жестко-
сти воды методом микротитрования (описание работы предоставил профессор
Джон Ричардсон из Шиппенсбургского университета). Аппаратура и техника
этой работы могут при желании быть использованы и в других работах по титри-
метрии. Если ваш преподаватель разработает и опробует такую работу вместе
12
ПРВДИСЛОВИЕ
с вами, я могу включить ее описание в следующее издание книги. Добавлены
две групповые работы (39 и 40), иллюстрирующие принципы статистической
поверки, которые описаны в гл. 4. Одна из них посвящена поверке методики и
контролю качества результатов; при этом отдельные члены рабочей группы вы-
полняют разные этапы поверки в рамках единого эксперимента. Вторая работа
моделирует межлабораторный эксперимент. В этой работе необходимо рассчи-
тать нормированные величины (z) для результатов, полученных каждым студен-
том в ходе одинакового для всех эксперимента (или нескольких таких
экспериментов). Зная z, студент может оценить качество своей работы.
Электронные таблицы. Мы настоятельно рекомендуем вам в ходе вы-
полнения практических работ использовать электронные таблицы для постро-
ения градуировочных графиков и статистического анализа результатов
эксперимента.
Компакт-диск
К книге прилагается компакт-диск, содержащий полезный материал, дополняю-
щий текст книги*. При чтении диска в первую очередь щелкните кнопкой мыши
на словах «Read те», чтобы увидеть подробное содержание диска. Файлы сгруп-
пированы в разделы («Вспомогательные данные», «Числовые таблицы», «Тексто-
вые таблицы», «Лабораторное оборудование», «Задачи для электронных таблиц»
и «Интернет-ссылки»). Там же содержатся примеры, которые могут служить хо-
рошим подспорьем при работе с электронными таблицами.
Несколько слов об Интернет-сайтах
В книге дано более 100 ссылок на Интернет-сайты, содержащие полезные до-
полнительные материалы. Чтобы не набирать адреса сайтов вручную, на ком-
пакт-диске приведены все имеющиеся в книге ссылки в порядке их появления,
глава за главой. Этот же список имеется и на сайте издательства John Wiley &
Sons, Inc. на странице, посвященной данной книге.
Чтобы открыть сайт, достаточно щелкнуть кнопкой мыши по соответствую-
щей ссылке. Если это не приводит к успеху, скопируйте ссылку, вставьте ее в
адресное окошко браузера и выполните команду Go (Перейти) или просто на-
жмите клавишу Enter (Ввод) на клавиатуре. Иногда страница может не откры-
ться. В этих случаях можно ввести адрес домашней страницы сайта и затем
перейти по ссылке к нужной странице. На сайтах фирм часто бывает необходи-
мо перейти к странице Product (Продукция). Можно воспользоваться и сред-
ствами поиска, имеющимися на многих сайтах. Адреса некоторых страниц со
временем могут быть изменены или вообще ликвидированы. При возникнове-
нии проблем с открытием сайтов пользуйтесь средствами поиска Интернета
(такими, как Google), осуществляя поиск по названию фирмы или ключевым
словам интересующей вас темы.
Имеется ввиду оригинальное издание на английском. В русской версии книги компакт-диск отсутствует.
Читатели могут обратиться на сайт J. Wiley & Sons, Inc.
ПРЕДИСЛОВИЕ
13
Домашняя страница книги в Интернете
У издательства John Wiley & Sons, Inc. есть страница в Интернете, посвященная
данной книге. Она содержит дополнительные материалы, которые могут время
от времени пополняться или обновляться. На этот сайт следует сообщать обо
всех ошибках, обнаруженных в книге. Дополнительные материалы к отдельным
главам книги представлены в виде краткого текста, если по нему щелкнуть кноп-
кой мыши, он развернется в полный текст. На сайте имеются также главы «Ме-
тодология поиска научной литературы» и «Решение аналитических проблем:
выбор метода анализа» из издания The Encyclopedia of Analytical Chemistry. Ин-
тернет-ссылки, приведенные в книге, также собраны на этом сайте; со временем
их адреса могут изменяться. Кроме того, на сайте имеются все рисунки и таб-
лицы, содержащиеся в книге; их можно скачивать и использовать для
подготовки демонстрационных материалов. Сайт находится по адресу:
www.wiley.com/college/christian.
Благодарности
При подготовке данной книги большую роль сыграли помощь и замечания
многих специалистов. Я особенно благодарен читателям предыдущих изданий
за их комментарии и предложения по доработке материала; я всегда приветст-
вую такие предложения. Мои коллеги оказали мне неоценимую помощь в ка-
честве критиков. Разумеется, иногда они высказывали противоположные
взгляды на содержание и расположение отдельных глав или разделов, но в ко-
нечном результате получился вариант, близкий к оптимальному, и который,
как я надеюсь, покажется читателям легким и приятным для чтения и изуче-
ния. Хочу особо поблагодарить профессоров Дениса Анджо (Университет
штата Калифорния, Лонг-Бич), Кевина Чемблисса (Университет Бейлор), Май-
кла Дегранпра (Университет Монтаны), Цзиньмо Хуана (Колледж Нью-Джер-
си), Айру Кралл (Северо-восточный университет), Гэри Лонга и Харольда
Макнейра (Технический университет Вирджинии), Джоди Редпеннинга (Уни-
верситет Небраски), Джона Ричардсона и Томаса Шредера (Шиппенсбургский
университет), Бенджамина Русилоски (Университет штата Делавэр), Джеймса
Рыбарчика (Болльский университет), Шерил Клейн Стивенс (Университет
Ксавье, Луизиана) и Филиппа Фегеля (Университет Среднего Запада). Профес-
сор Норманн Доричи (Университет штата Вашингтон) внес ценный вклад в
подготовку гл. 25, посвященной геномам и протеомике. Я благодарю также
Мэка Картера за его высококвалифицированную помощь, связанную с работой
на компьютере, и Шейлу Паркер за помощь в преодолении многих случайных
трудностей.
Высоким уровнем качества данное издание обязано профессионалам из из-
дательства John Wiley & Sons. Редакторы Дэвид Харрис и Дебора Бреннан контро-
лировали издательский процесс от начала до конца. Их ассистент Кэти Донован
осуществляла весь процесс редактирования и в связи с этим решала множество
вопросов. Производственный редактор Элизабет Суэйн отвечала за подготовку
14
предисловие
печатных форм текста. Сандра Ригби работала в качестве редактора иллюстра-
ций, ей вы обязаны рисунками, имеющимися в книге. Эрнестина Франко (Рет
Editorial Services) тщательно выполнила окончательную корректуру при подго-
товке книги к печати. Работать с этими людьми в ходе долгого, но приносящего
удовлетворение процесса создания книги было настоящим удовольствием.
На протяжении всех двух лет работы над книгой самой надежной опорой
была моя жена и лучший друг Сью. Она старательно расшифровывала и перепе-
чатывала рукописный текст. Спасибо, спасибо, спасибо!
Пособие по решению задач
В распоряжении преподавателей и студентов имеется всеобъемлющее пособие
по решению задач, в котором приведены полные решения всех задач и ответы на
все вопросы, содержащиеся в книге. Решения задач, требующих использования
электронных таблиц, приведены в виде таблиц на компакт-диске. Решения задач
с четными номерами приведены в Приложении F.
Гари Д. Кристиан
Январь 2003 г.
Сиэтл
Преподавать значит дважды учиться.
Жозеф Жубер
Список электронных таблиц, использованных в тексте
Во многих главах книги содержатся указания
к использованию электронных таблиц для по-
строения графиков и проведения вычислений.
Ниже приведен список приложений Microsoft
Excel, для удобства использования сгруппи-
рованных по темам. Все соответствующие
таблицы содержатся на компакт-диске (CD).
Таблицы, предназначенные для решения за-
дач, есть только на диске. Остальные таблицы
имеются и в тексте, и на диске. Прежде чем
обращаться к диску, всегда следует постарать-
ся составить необходимую таблицу самостоя-
тельно. Вы можете копировать таблицы с
компакт-диска на свой компьютер для их по-
следующего использования.
Работа с электронными таблицами
(разд. 3.8)
Заполнение ячеек: с. 127(1)
Сохранение таблицы: с. 128(1)
Печать таблицы: с. 129(1)
Абсолютные и относительные адреса ячеек:
с. 129(1)
Использование статистических функций Excel:
с. 131(1)
Полезные функции Excel: LOG 10, PRODUCT
(ПРОИЗВЕЛ), POWER (СТЕПЕНЬ), SQRT
(КОРЕНЬ), AVERAGE (СРЗНАЧ), MEDIAN
(МЕДИАНА), STDEV (СТАНДОТКЛОН),
TTEST (ТТЕСТ), VAR (ДИСПА) — с. 132(1)
Статистические расчеты
Стандартное отклонение: гл. 3, задачи 14, 15,
16, 20,21
Среднее (средневзвешенное) стандартное от-
клонение: гл. 3, задача 27
F-тест: гл. 3, задачи 25, 26, 28
/-Тест: гл. 3, задачи 29, 30
/-Тест для нескольких выборок: гл. 3, задача 42
Наложение погрешностей: гл. 3, задача 17 (сло-
жение-вычитание), 18 (умножение-деление)
Использование электронных таблиц
для построения градуировочных
графиков (разд. 3.18, рис. 3.9)
Мастер диаграмм
Линия тренда
Регрессионное уравнение по МНК, г2
Нахождение тангенса угла наклона, свободно-
го члена, коэффициента детерминации (без по-
строения графика): разд. 3.19, задачи 37,40,41
Программа ЛИНЕЙН (LINEST) для выполне-
ния дополнительных статистических вычисле-
ний (разд. 3.20, рис. 3.10, примеры 3.21, 3.22);
десять статистических функций: вычисление
углового коэффициента, его стандартного от-
клонения, г2, F-статистики, суммы квадратов
отклонений, свободного члена, его стандарт-
ного отклонения, стандартного отклонения
оценки (предсказания значений у), числа степе-
ней свободы, остаточной суммы квадратов.
Расчет неизвестной концентрации по градуиро-
вочной зависимости: с. 36(2); гл. 3, задача 38
Расчет стандартного отклонения неизвестного
содержания по градуировочной зависимости:
с. 37(2)
Расчет концентрации и ее стандартного откло-
нения методом добавок: с. 105(2)
Расчет концентрации и стандартного отклонения
методом внутреннего стандарта: с. 177(2);
гл. 20, задача 18
Построение распределительных
диаграмм а—pH (рис. 7.1, Н3РО4):
с. 345(1)
16
СПИСОК ЭЛЕКТРОННЫХ ТАБЛИЦ, ИСПОЛЬЗОВАННЫХ В ТЕКСТЕ
Построение концентрационно-лога-
рифмических диаграмм: с. 359(1);
гл. 7, задача 61 (СН3СООН)
Использование а-коэффициентов,
с. 365(1): гл. 7, задача 64 (яблочная кис-
лота, Н2А); гл. 7, задача 68 (рис. 7.4,
Н3РО4, Н3А)
Построение кривых титрования
НС1 раствором NaOH (рис. 8.1): с. 375(1);
СН3СООН раствором NaOH: с. 377(1);
Н2СгО4 раствором NaOH (титрование двухос-
новной кислоты): CD, вспомогательные дан-
ные, гл. 8; с. 396(1);
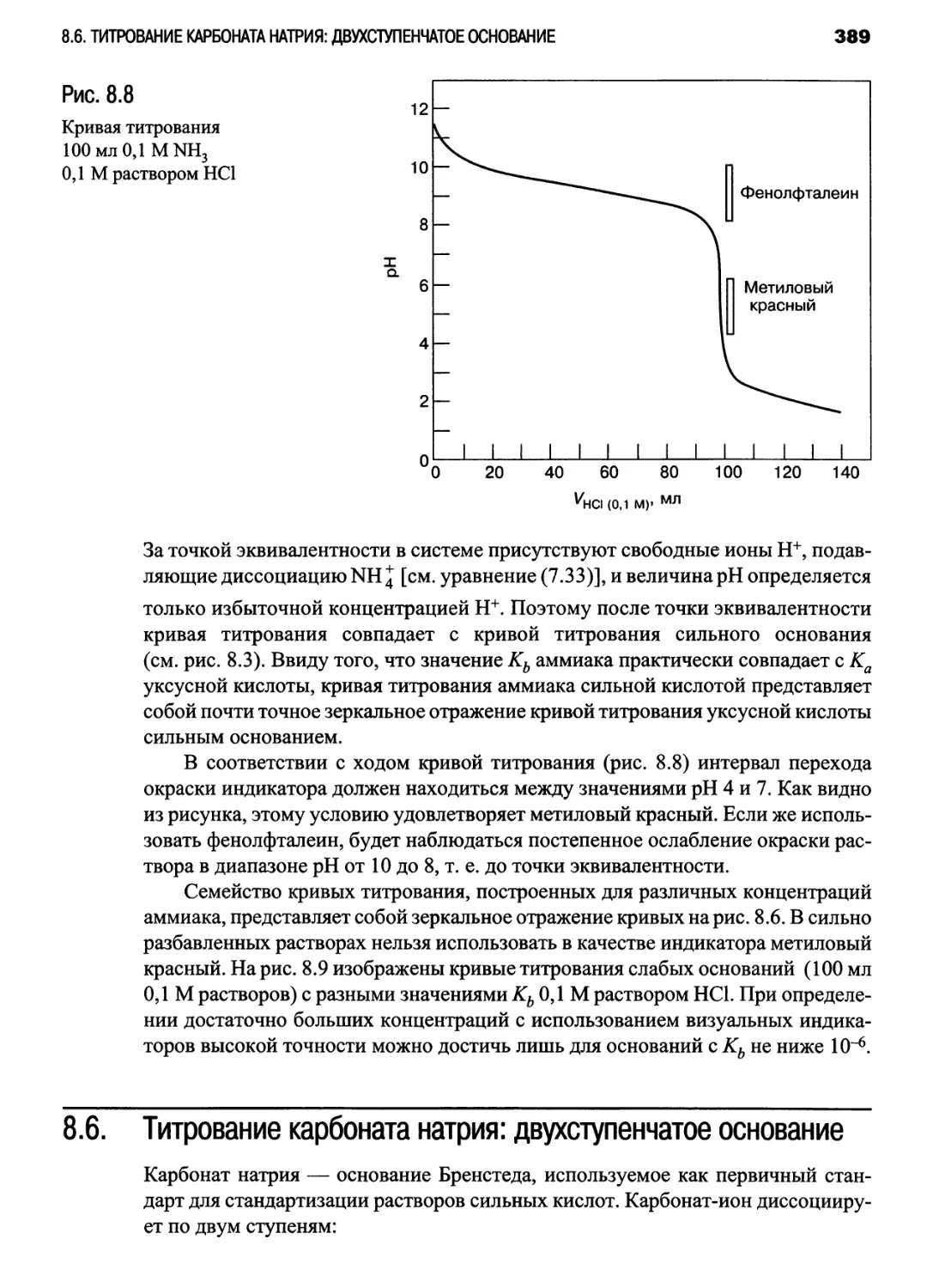
NH3 раствором НС1 (рис. 8.8): гл. 8, задача 35;
Са2+ раствором ЭДТА: гл. 9, задача 21;
СГ раствором AgNO3 (рис. 11.1): гл. 11, задача 12;
Fe2^ раствором Се4+ (рис. 14.1): гл. 14, задача 21
Построение первой и второй производных кри-
вой титрования (рис. 14.2 и 14.3): с. 589(1)
Построение зависимостей lg K'f от pH
(рис. 9.2): гл. 9, задача 20
Построение зависимостей
p-коэффициентов от концентрации
лиганда (зависимость р-коэффициентов
для системы Ад+— NH3: гл. 9, задача 22
Различные расчеты и построения
Калибровка мерной посуды (табл. 2.4): с. 72(1)
Приведение к весу в вакууме с учетом плотно-
сти образца (CD, гл. 2)
Расчеты активностей, коэффициентов активно-
сти: гл. 6, задачи 22, 23, 24, 26
Расчеты в гравиметрии: стр. 460(1); гл. 10, за-
дача 40 (пример 10.2, расчет массовой доли
р205)
Зависимость растворимости BaSO4 от [Ва2+]
(рис. 10.5): гл. 10, задача 41
Зависимость растворимости от ионной силы
(рис. 10.6): гл. 10, задача 42
Зависимость Ван Деемтера: гл. 19, задача 11
Нахождение скорости реакции: гл. 22, пример 22.2
Построение зависимости Лайнуивера-Берка
(пример 22.1): гл. 22, задача 17
Стандартные вычисления в Excel
Решение квадратного уравнения при расчетах
равновесных концентраций (пример 6.1): гл. 6,
задача 25; гл. 7, пример 7.19 (CD)
Расчет растворимости из произведения раство-
римости: с. 460(1); гл. 10, задача 43 (пример
10.9)
Решение систем уравнений (пример 16.5, спек-
трофотометрический анализ смесей)
Глава 1
ЗАДАЧИ АНАЛИТИЧЕСКОЙ ХИМИИ,
ИЛИ ЧЕМ ЗАНИМАЮТСЯ ХИМИКИ-АНАЛИТИКИ
Пока наше знание не измерено
и не выражено в цифрах,
оно немногого стоит.
Лорд Кельвин
Задача аналитической химии — охарактеризовать материю с химической точки
зрения и ответить на два важных вопроса: «что?» (качественный анализ) и
«сколько?» (количественный анализ). Все, что мы используем и потребляем, со-
стоит из химических веществ (химических соединений), и знание химического
состава многих веществ важно для повседневной жизни. Аналитическая химия
играет важную роль практически во всех областях химии, например в сельскохо-
зяйственной, клинической, судебной, фармацевтической химии, химии окружа-
ющей среды, химическом производстве и металлургии. Содержание азота в
удобрении определяет его ценность. Пищевые продукты необходимо анализиро-
вать на содержание как токсичных (в частности, остаточных количеств пестици-
дов), так и полезных веществ (витаминов). В воздухе больших городов необходимо
определять содержание монооксида углерода. Больным диабетом необходимо по-
стоянно контролировать содержание глюкозы в крови (в действительности боль-
шинство заболеваний диагностируется на основании результатов химических
анализов). Наличие на руке человека, защищавшегося от нападавшего, следов
элементов, содержащихся в порохе, доказывает, что из оружия был произведен
выстрел. Качество промышленной продукции часто зависит от соотношения в ней
химических веществ, поэтому определение их содержания — необходимая опера-
ция в ходе контроля качества. Так, содержание углерода в стали определяет ее
качество, а чистота лекарственных препаратов — их эффективность.
1.1. Что такое аналитическая наука?
Из приведенных примеров уже, в общем-то, понятно, что же представляет собой
дисциплина аналитическая химия. Было предпринято много попыток опреде-
лить ее содержание более строго и детально. В конце жизни Чарльз Н. Рейли ска-
зал: «Аналитическая химия — это то, чем занимаются химики-аналитики» [2].
Эта дисциплина расширила свои границы за пределы собственно химии, и мно-
гие ученые являются сторонниками использования более широкого понятия —
аналитическая наука. Этот термин используется в докладе Национального на-
учного фонда США, озаглавленном «Развитие курса аналитических наук». Но
18
ЗАДАЧИ АНАЛИТИЧЕСКОЙ ХИМИИ, ИЛИ ЧЕМ ЗАНИМАЮТСЯ ХИМИКИ-АНАЛИТИКИ
даже этот термин не в полной мере отражает ту роль, которую в современной
аналитической химии играют развитие инструментальных методов и практиче-
ские приложения. В связи с этим одно из предложений состоит в том, чтобы ис-
пользовать термин аналитическая наука и технология [3].
В 1992 г. Федерация европейских химических обществ объявила конкурс на
лучшее определение аналитической химии. Было выбрано следующее определе-
ние, данное К. Камманом [Fresenius' J. Anal. Chem., 343 (1992) 812-813]:
Аналитическая химия предоставляет методы и средства, необходимые
для того, чтобы заглянуть в материальный мир... с целью ответа на
основные вопросы, касающиеся материала образца:
• Что?
• Где?
• Сколько?
• Каковы расположение, структура, форма?
Отделение аналитической химии Американского химического общества пред-
лагает всеобъемлющее определение аналитической химии, которое можно найти
на его сайте (www.acs-analytical.duq.edn/whatisanalyticalchem.html). Ниже приведе-
на его основная часть:
Аналитическая химия занимается поиском новых, улучшенных средств
определения химического состава природных и искусственных материа-
лов. Техника и методы этой науки используются для идентификации ве-
ществ, которые могут присутствовать в материале, и для определения
точных количеств идентифицированных веществ.
Химики-аналитики работают над повышением надежности существу-
ющих технологий, чтобы удовлетворять все возрастающим требованиям
к качеству измерений, постоянно возникающим в нашем обществе. Они
адаптируют проверенную методологию к новым типам материалов или к
тому, чтобы ответить на новые вопросы об их составе и реакционной спо-
собности. Они выполняют исследования, чтобы разработать совершенно
новые принципы измерений, и находятся на переднем крае использова-
ния в практических целях главных открытий и изобретений — таких, как
лазеры или микрочипы. Их усилия направлены на удовлетворение нужд
многих наук.
• В медицине аналитическая химия составляет основу клинических
лабораторных тестов, помогающих врачам диагностировать забо-
левания и добиваться успехов в их лечении.
• В промышленности аналитическая химия предоставляет средства
для испытаний сырья и гарантии качества готовой продукции, хи-
мический состав которой крайне важен. Многие продукты, исполь-
зуемые в быту, топливо, красители, лекарства и другие вещества
перед тем, как поступить в продажу, анализируют с помощью мето-
дик, разработанных химиками-аналитиками.
• Степень загрязнения окружающей среды часто оценивают при по-
мощи тестов на предполагаемые загрязнители, используя средства
и методы аналитической химии.
1.2. КАЧЕСТВЕННЫЙ И КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
19
• Питательную ценность продуктов питания определяют при помощи
химических анализов на содержание основных компонентов — таких
как белки или углеводы, а также микрокомпонентов — таких, как
витамины или минеральные вещества. Даже калорийность пищи ча-
сто вычисляют на основании результатов химических анализов.
Кроме того, химики-аналитики вносят существенный вклад в самые раз-
нообразные области, например криминалистику, археологию и науку о
космосе.
1.2. Качественный и количественный анализ:
какую информацию сообщает нам каждый из них?
Дисциплина аналитическая химия состоит из качественного анализа и коли-
чественного анализа. Задача первого — идентификация элементов, ионов или
соединений, присутствующих в образце (при этом может представлять интерес
и наличие лишь одного определенного вещества), задача количественного ана-
лиза — определение того, сколько данного компонента (одного или нескольких)
содержится в образце. При этом образец может представлять собой твердое
тело, жидкость, газ или смесь. Наличие остатков пороха на руке человека обыч-
но представляет интерес лишь с качественной точки зрения (его количество не-
существенно), но цена угля определяется количественным содержанием в нем
примеси серы.
Количественный анализ
Качественный анализ
Печатается с любезного разрешения Merck KGaA.
20
ЗАДАЧИ АНАЛИТИЧЕСКОЙ ХИМИИ, ИЛИ ЧЕМ ЗАНИМАЮТСЯ ХИМИКИ-АНАЛИТИКИ
Зарождение аналитической химии
Фактически, химические измерения и средства, необходимые для их проведения, упоми-
наются в самых ранних письменных источниках. Испытания золота огнем описаны в
библейской книге Пророка Захарии (13:9), а царь Вавилона жаловался египетскому фа-
раону Амменофису IV (1375-1350 гг. до н. э.) на то, что золото, которое тот ему послал,
«теряло в весе» после прокаливания в печи. Возможно, необходимость установления ис-
тинной ценности золота, возможно, была главным стимулом к получению знаний об ана-
лизе. Архимед (287-212 гг. до н. э.) выполнил неразрушающий анализ золотой короны
царя Герона. Он поместил корону, а также куски золота и серебра, равные ей по весу, в
сосуды с водой и измерил объемы воды, вытесненные каждым из трех объектов. Объем
воды, вытесненный короной, находился между объемами, вытесненными золотом и се-
ребром. Это доказывало, что корона состояла не из чистого золота!
Весы появились так давно, что в самых ранних из найденных документов это при-
писывается богам. Вавилоняне создали стандартные весы около 2600 г. до н. э. Они
считали весы настолько важным инструментом, что их использование контролировали
жрецы.
Алхимики собрали воедино химические познания, которые легли в основу качест-
венного анализа в его современном понимании. Роберт Бойль в книге «Химик-скеп-
тик» (1661 г.) использовал термин аналитик. Антуана Лавуазье считают «отцом
аналитической химии» благодаря тщательным количественным экспериментам, кото-
рые он провел (с использованием аналитических весов) для доказательства закона со-
хранения массы. По роду основной деятельности Лавуазье был сборщиком налогов, а
занятия наукой он рассматривал как хобби. Именно из-за своей деятельности по сбору
налогов он был казнен на гильотине 8 мая 1793 г. во время Великой французской рево-
люции.
Гравиметрия получила развитие в XVII веке, а титриметрия — в XVIII и XIX веках.
В 1829 г. Гей-Люссак при титриметрическом определении серебра достиг точности в
0,05% относительной погрешности!
Учебники по аналитической химии стали появляться в 1800-х гг. В 1845 г. в Герма-
нии Карл Фрезениус опубликовал «Руководство по количественному химическому ана-
лизу», а в 1894 г. вышла книга Вильгельма Оствальда «Научные основы аналитической
химии». Эта книга содержала теоретические объяснения многих явлений в аналитиче-
ской химии при помощи констант химических равновесий и оказала большое влияние на
развитие химии.
В XX веке наблюдалось развитие инструментальных средств анализа. Второе изда-
ние книги Стивена Попова «Количественный анализ», вышедшее в 1927 г., включало
электрохимические методы анализа, кондуктометрическое титрование, колориметриче-
ские методы. К настоящему времени был совершен большой прогресс в развитии техно-
логии химического анализа, которая включает сложные и высокопроизводительные
приборы, управляемые компьютерами и позволяющие проводить весьма сложные ана-
лизы и измерения при очень низких концентрациях веществ.
Данная книга научит читателя основам аналитической химии и вооружит средст-
вами для решения большинства аналитических задач. Более подробно об истории разви-
тия аналитической химии можно узнать из работы [7].
1.2. КАЧЕСТВЕННЫЙ И КОЛИЧЕСТВЕННЫЙ АНАЛИЗ
21
Качественный анализ можно проводить при помощи селективных химиче-
ских реакций или с использованием аппаратуры. Образование белого осадка при
добавлении раствора нитрата серебра к раствору пробы свидетельствует о нали-
чии хлорид-ионов. В результате некоторых химических реакций возникает ха-
рактерное окрашивание, свидетельствующее о наличии тех или иных классов
органических соединений, например, кетонов. Инфракрасные спектры позволя-
ют получить «отпечатки пальцев», уникальные для определенных органических
соединений или их функциональных групп.
Необходимо четко различать термины селективный и специфичный.
• Селективная реакция или тест протекает с несколькими или многими веще-
ствами, но предпочтительно — с одним определенным веществом.
• Специфичная реакция или тест протекает только с одним веществом.
К сожалению, очень мало реакций являются специфичными, но многие из них
обладают селективностью. Селективности можно достичь при помощи различ-
ных приемов, например:
• Специальная пробоподготовка (в частности, экстракция, осаждение).
• Использование селективных детекторов в измерительных приборах.
• Направленное химическое превращение (дериватизация) определяемого
компонента — например, дериватизация определенных функциональных
групп при помощи детектирующих реагентов.
• Использование хроматографии — высокопроизводительного метода разде-
ления.
Таким образом, лишь немногие методики анализа селективны сами по себе.
Обычно селективность достигается при помощи подходящей подготовки пробы
и выбора условий измерения.
Для количественного анализа данные о качественном составе образца часто
бывают известны заранее (например, известно, что кровь содержит глюкозу).
В противном случае аналитику необходимо выполнить качественные испыта-
ния прежде, чем заняться значительно более трудным количественным анализом.
Современные химические измерительные системы часто обладают достаточно
высокой селективностью, поэтому результаты количественных измерений од-
новременно являются и доказательствами качественного состава образца. Тем
не менее простые качественные тесты часто выполняются значительно быстрее,
чем процедуры количественного анализа. Качественный анализ делится на две
области: неорганический и органический. Неорганический анализ обычно пре-
подается во вводных курсах химических дисциплин, органический лучше пре-
подавать лишь после того, как студент освоит курс органической химии.
Для сравнения качественного и количественного анализа рассмотрим, на-
пример, последовательность аналитических процедур при тестировании проб на
запрещенные вещества во время Олимпийских игр. Список запрещенных препа-
ратов включает около 500 физиологически активных веществ различных классов:
стимуляторов, стероидов, бета-блокаторов, диуретиков, наркотиков, анальгети-
22
ЗАДАЧИ АНАЛИТИЧЕСКОЙ ХИМИИ, ИЛИ ЧЕМ ЗАНИМАЮТСЯ ХИ МИКИ-АНАЛИТИКИ
ков, местных анестетиков и седативных средств. Некоторые из них можно обнару-
жить только по их метаболитам. Необходимо быстро протестировать множество
спортсменов, поэтому нерационально подвергать каждую пробу детальной про-
цедуре количественного анализа. Анализ включает в себя три стадии: стадия бы-
строго скрининга, стадия идентификации и, возможно, стадия количественного
анализа. На стадии быстрого скрининга пробы мочи в течение короткого време-
ни тестируют на наличие в них целых классов соединений, позволяющих отличить
их от «нормальных» проб. Для этого используют различные методы, включая
иммунный анализ, газовую и жидкостную хроматографию. Приблизительно в
5% случаев пробы показывают наличие веществ, которые могут быть или не
быть в числе запрещенных, однако нуждаются в идентификации. Пробы, вы-
звавшие подозрения в ходе скрининга, заново подвергают циклу пробоподготовки
(возможно, включающей гидролиз, экстракцию, дериватизацию) в зависимости от
природы соединений, которые надо идентифицировать. Для их идентификации
используют высокоселективный метод, сочетающий газовую хроматографию и
масс-спектрометрию (ГХ-МС). В этом методе сложные смеси разделяют при по-
мощи газовой хроматографии, а затем идентифицируют отдельные компоненты
при помощи масс-спектрометрии, позволяющей получать информацию о струк-
туре молекул веществ. Сочетание масс-спектрометрических и собственно хрома-
тографических (времена удерживания) данных позволяет с высокой вероятностью
сделать заключение о присутствии того или иного вещества. Анализ методом
ГХ-МС является длительным и дорогостоящим, поэтому его используют только
при крайней необходимости. После идентификации для некоторых веществ не-
обходимо точно определить их содержание, поскольку они могут присутство-
вать и в нормальных пробах (например, поступая из пищи, из лекарственных
средств или выделяясь в ходе жизнедеятельности организма — эндогенные стеро-
иды), но в низких концентрациях. Таким образом, необходимо подтвердить, что
их концентрации превышают норму. Для этого используют методы количественно-
го анализа — например, спектрофотометрию или ту же газовую хроматографию.
В данной книге рассматриваются главным образом методы количественно-
го анализа. При рассмотрении практического применения этих методов приве-
дены примеры из различных областей: наук о живом, клинической химии,
химии окружающей среды, техники безопасности и охраны здоровья, производ-
ственного анализа.
Давайте сначала кратко ознакомимся с процессом анализа в целом, а затем
перейдем к более подробному изучению методов (в последующих главах).
1.3. Мы начинаем процесс анализа
Общая схема процесса анализа приведена на рис. 1.1. Химик-аналитик должен
осуществлять все его стадии, он является ключевым звеном рабочей группы и
определяет, что, почему и как следует делать. Ниже подробно рассмотрены
стандартные операции химического анализа, общие для большинства типов ана-
лизов.
1.3. МЫ НАЧИНАЕМ ПРОЦЕСС АНАЛИЗА
23
Постановка задачи: что необходимо узнать (не обязательно все!)
Прежде чем планировать ход анализа, аналитик должен знать, какую информа-
цию требуется получить, для кого, с какой целью и какого рода пробу предстоит
проанализировать. Кроме того, он должен уметь устанавливать хорошие отно-
шения с заказчиком и это, возможно, окажется решающим моментом. В качестве
заказчика может выступать кто угодно — американское Агентство по охране окру-
жающей среды (Environmental Protection Agency, ЕРА), потребитель промышлен-
ной продукции, инженер, родственники — и у каждого будут свои критерии и
требования, свое понимание того, что включает в себя химический анализ и что
он означает. Поэтому важно, чтобы общение с заказчиком шло на языке, понят-
ном обеим сторонам. Если кто-нибудь поставит вам на стол бутылку с напитком и
спросит: «Что в ней находится?» или «Безопасно ли это в употреблении?», вам,
возможно, потребуется объяснить, что всего известно уже около 10 миллионов
различных химических соединений. Заказчик, который скажет: «Я хочу узнать,
какие элементы здесь содержатся», должен понять, что при средней стоимости
определения одного элемента порядка 20 долларов общая стоимость определения
85 элементов составит 1700 долларов, в то время как, возможно, содержание лишь
некоторых элементов представляет для него интерес.
Ко мне часто приходят дилетанты с образцами косметики, мечтающие вы-
пускать подделки и сделать состояние на их распространении. Но когда они узна-
ют, что определение необходимых ингредиентов в этой продукции при помощи
ряда сложных методов анализа может им обойтись очень дорого, они всегда
предпочитают заново обдумать свои намерения.
Такие понятия, как «безопасно», «отсутствует» или «нулевое содержание»,
не имеют ни строгого определения, ни смысла. Фразы типа «эта вода безопасна в
употреблении» — совсем не то, что должен говорить аналитик. Все, что он мо-
жет сделать, — это представить результаты анализа, охарактеризовав при этом
их точность, указав доверительные границы. Заказчик сам должен решить (воз-
можно, посоветовавшись с другими специалистами), можно ли эту воду исполь-
зовать для питья без угрозы для здоровья. Аналогично, не правильно указывать
в качестве результата «нуль», вместо этого должно быть: «ниже предела обнару-
жения» (который определяется характером измерений и типом измерительного
прибора). В подобных случаях необходимо говорить, что используемые методики
и средства измерений имеют ограниченные возможности. В то же время некото-
рые современные приборы позволяют измерять чрезвычайно малые содержа-
ния — например, на уровне нескольких частей на триллион (ppt), и это тоже
представляет собой проблему (по своей природе чаще всего политическую) при
разработке официальных нормативов. Например, может быть принят закон, тре-
бующий, чтобы в воде «отсутствовало» то или иное вещество. На практике в подоб-
ных случаях устанавливают предельно допустимые концентрации, определяемые
лишь возможностями анализа — тем, насколько низкие содержания можно об-
наружить. Но такие концентрации могут оказаться намного ниже природного
содержания этого вещества в воде или уровня, до которого его содержание мож-
но снизить. Химики-аналитики должны уметь эффективно доводить до сведе-
ния общественности, что представляют собой результаты анализов.
24
ЗАДАЧИ АНАЛИТИЧЕСКОЙ ХИМИИ, ИЛИ ЧЕМ ЗАНИМАЮТСЯ ХИМИКИ-АНАЛИТИКИ
Постановка задачи
Необходимо учитывать:
• В чем состоит задача: что необходимо узнать? Нужен
качественный анализ или количественный?
• Для чего будет использована полученная информация?
Кто будет ее использовать?
• Когда она будет нужна?
• Насколько точными (правильными и воспроизводимыми)
должны быть результаты?
• Сколько средств отпущено на анализ?
• Аналитик должен посоветоваться с заказчиком, чтобы
составить осмысленный и эффективный план анализа,
включая получение пробы
Выбор метода и методики
Необходимо учитывать:
• Тип пробы
• Размер пробы
• Необходимую пробоподготовку
• Требуемую чувствительность (в зависимости от
диапазона концентраций определяемого вещества)
• Требуемую селективность, возможные мешающие
влияния
• Требуемую правильность и воспроизводимость
• Доступность аппаратуры
• Опыт работы, необходимость привлечения других
специалистов
• Стоимость анализа
• Быстроту выполнения анализа
• Должен ли анализ быть автоматизированным?
• Имеются ли в химической литературе необходимые
методики?
• Имеются ли стандартные методики?
Получение представительной пробы
Необходимо учитывать:
• Тип пробы, ее размер, степень гомогенности
• Погрешности пробоотбора
Рис. 1.1. Отдельные стадии химического анализа
Постановка задачи будет определять способ пробоподготовки, необходи-
мое количество пробы, требуемую чувствительность метода, его точностные ха-
рактеристики — правильность и воспроизводимость*, и то, какие операции
Правильность — это мера близости результата измерения к истинному значению измеряемой величины.
Воспроизводимость (прецизионность) — это мера близости результатов повторных измерений между со-
бой. Хорошая воспроизводимость не означает хорошую правильность. Более подробно эти понятия об-
суждаются в гл. 3.
1.3. МЫ НАЧИНАЕМ ПРОЦЕСС АНАЛИЗА
25
разделения могут потребоваться для устранения мешающих влияний. Как пра-
вило, определение микросодержания компонентов не требует такой высокой
точности, как определение основных компонентов, но в этом случае следует
уделять больше внимания предотвращению загрязнения пробы в ходе анализа.
Как только выбран метод анализа (принцип измерения), необходимо вы-
брать конкретную методику. При этом следует руководствоваться целым рядом
соображений, в том числе квалификацией аналитика, его опытом работы с теми
или иными приборами, доступностью необходимой аппаратуры, требуемой чув-
ствительностью и точностью, стоимость анализа и объемом средств, отпущен-
ных для его выполнения, временем выполнения анализа и тем, насколько быстро
должны быть получены результаты. Часто в справочниках описана одна или бо-
лее стандартные методики для определения данного аналита (компонента, ко-
торый необходимо определить) в пробе данного типа. Это вовсе не означает,
что методика может быть автоматически использована для проб другого типа.
Например, стандартная методика американского Агентства по охране окружаю-
щей среды для анализа почвенных вод может дать неверные результаты примени-
тельно к сточным водам. В химической литературе (журналах) содержится
множество описаний конкретных видов анализа. Целесообразно начать поиск ли-
тературы с реферативного журнала Chemical Abstracts (http://info.cas.org), изда-
ваемого Американским химическим обществом. Он содержит рефераты всех
статей, публикуемых в основных химических журналах мира. В журнале име-
ются годовые и сводные указатели, а многие библиотеки оснащены компьютер-
ными средствами поиска. Можно непосредственно изучать основные журналы
по аналитической химии, например, Analytica Chimica Acta, Analytical Chemistry,
Analytical Letters, Analyst, Applied Spectroscopy, Clinica Chimica Acta, Clinical
Chemistry, Journal of the Association of Official Analytical Chemists, Journal of
Chromatography, Spectrochimica Acta, Taianta. Если необходимая методика не
описана в литературе, аналитик часто может использовать данные литературы
об определяемом веществе, чтобы самостоятельно разработать подходящую
схему анализа. Наконец, аналитик может разработать свою собственную мето-
дику анализа данной пробы, опираясь на свои знания и опыт. Описания разнооб-
разных методик определения большого числа веществ содержатся в источниках,
приведенных в Приложении А.
Примеры описаний выполнения анализа разных типов проб содержатся в
гл. 24-26. В них рассмотрены некоторые широко используемые клинические,
биохимические и экологические анализы. Для выполнения отдельных анализов
применяют различные методы, разобранные в предыдущих главах. Поэтому
гл. 24-26, посвященные практическому применению аналитической химии, по-
лезно прочитать дважды: сейчас и после того, как будет изучена основная часть
книги. В этом случае можно в полной мере почувствовать, в чем состоит анализ
реальных проб и как выполняются такие анализы.
После того как задача поставлена, наступает очередь следующих этапов
анализа.
26
ЗАДАЧИ АНАЛИТИЧЕСКОЙ ХИМИИ, ИЛИ ЧЕМ ЗАНИМАЮТСЯ ХИМИКИ-АНАЛИТИКИ
Получение представительной пробы:
невозможно анализировать объект целиком!
Обычно для химического анализа используют лишь небольшую часть того объ-
екта, который необходимо охарактеризовать. Но если размер объекта очень мал,
а его вещество не требуется использовать в дальнейшем, объект можно анализи-
ровать и целиком. Примером может служить определение следов пороха на
коже руки человека. Чаще всего исследуемый материал представляет собой
определенную ценность, и поэтому в результате отбора пробы он должен изме-
ниться как можно меньше.
Материал, от которого отбирается проба, может быть твердым, жидким или
газообразным. По своему строению он может быть гомогенным или гетероген-
ным. В первом случае для анализа может быть использована любая произвольно
отобранная проба. Во втором случае может представлять интерес пространст-
венное изменение состава пробы, тогда необходимо отобрать несколько проб.
Если же требуется установить среднее содержание компонента, используют
специальную технику пробоотбора, чтобы полученная проба была представите-
льной. Например, для определения среднего содержания белка в партии зерна
можно взять небольшую пробу из каждого мешка (для крупных партий — из
каждого десятого мешка), объединить их и тем самым получить генеральную
пробу. Технически отбор пробы легче всего осуществить, если исследуемый ма-
териал движется перед вами. Чем больше размер частиц материала, тем больше
должна быть и его генеральная проба. Генеральную пробу следует уменьшить,
чтобы получить лабораторную пробу массой несколько граммов или десятков
граммов. Из нее отбирают анализируемую пробу массой от нескольких мил-
лиграммов до нескольких граммов. При сокращении размера пробы может потре-
боваться многократный отбор порций (например, по две из четырех четвертей) и
их перемешивание, а также измельчение и просеивание для того, чтобы в конце
концов получить однородный порошок, пригодный для анализа. Таким образом,
генеральная проба имеет размер, достаточный для того, чтобы отобрать из нее
для анализа много порций. Лабораторная проба — это небольшая гомогенизи-
рованная порция генеральной пробы. Анализируемую пробу непосредственно
подвергают анализу. Отдельные способы отбора проб твердых, жидких и газо-
образных материалов рассмотрены в гл. 2.
При отборе проб биологических жидкостей могут играть роль условия, в ко-
торых они взяты, — например, принимал ли пациент перед этим пищу. Состав
крови до и после приема пищи значительно отличается, и для многих анализов
необходимо брать кровь натощак. После взятия образцов крови к ним бывает не-
обходимо добавить консерванты, например фторид натрия при определении
глюкозы, антикоагулянты. Их наличие может повлиять на результаты анализа.
Пробы крови можно, в зависимости от конкретных требований к анализу,
анализировать в виде цельной крови или предварительно разделить и анализи-
ровать плазму либо сыворотку. В большинстве случаев важным физиологиче-
ским показателем является концентрация вещества вне красных кровяных клеток,
поэтому для анализа используют сыворотку или плазму.
1.3. МЫ НАЧИНАЕМ ПРОЦЕСС АНАЛИЗА
27
Если порцию цельной крови оставить на несколько минут, то содержа-
щийся в ней растворимый белок фибриноген в результате сложной последова-
тельности химических реакций (с участием ионов кальция) превращается в
нерастворимый белок фибрин, составляющий основу геля — сгустка крови.
Поры пространственной сетки, образованной молекулами фибрина, захватыва-
ют красные и белые кровяные клетки. Таким образом, они входят в состав сгуст-
ка, хотя и не участвуют непосредственно в его образовании. Затем сгусток
сжимается и из него выделяется соломенно-желтая жидкость, сыворотка кро-
ви. Она не свертывается и остается в жидком состоянии неограниченно долго.
Процесс свертывания крови можно предотвратить при помощи добавок ве-
ществ-антикоагулянтов, например, гепарина или цитрат-ионов (образующих
комплексы с ионами кальция). После добавления антикоагулянтов для анализа
можно использовать аликвоту цельной крови либо отделить красные кровяные
клетки центрифугированием и анализировать оставшуюся розоватую жидкость,
называемую плазмой. То есть, сыворотка — это жидкость, получаемая путем
отделения от нее свернувшейся крови. Плазма — это жидкость, получаемая пу-
тем отделения несвернувшихся красных кровяных клеток. По своему химическо-
му составу плазма и сыворотка почти идентичны. Основное различие состоит в
том, что сыворотка не содержит фибриногена.
Подробности отбора проб различных материалов можно прочитать в спра-
вочниках по отдельным аспектам химического анализа. Можете посмотреть, в
частности, некоторые ссылки в конце данной главы.
Следует принимать специальные меры предосторожности при обработке и
хранении проб, чтобы предотвратить или минимизировать их загрязнение, по-
тери или изменение состава матрицы. Основными источниками загрязнений и
изменений пробы могут быть: 1) посуда, 2) атмосфера и 3) воздействие света.
Если результаты анализов предполагается использовать в качестве судебных до-
казательств, то необходимы специальные меры по охране проб. В ходе нашумев-
шего дела по обвинению известного американского спортсмена О. Дж. Симпсона
в убийстве в телевизионных новостях показывали кадры, на которых, в частно-
сти, люди помещали пробы, находящиеся без всякой охраны, в кузов автомоби-
ля, где температура воздуха была повышенной. Хотя это обстоятельство могло и
не повлиять на правильность результатов, оно дало защите повод дискредитиро-
вать результаты анализов.
Может потребоваться защита проб от действия атмосферы или света. На-
пример, если пробы представляют собой щелочные растворы, то они будут взаи-
модействовать с углекислым газом воздуха. При анализе образцов крови на
содержание СО2 их также следует защитить от действия атмосферы.
Необходимо принимать во внимание, что проба может быть неустойчивой.
В частности, растворы глюкозы неустойчивы, поэтому к пробам крови для опре-
деления глюкозы необходимо добавлять стабилизатор — фторид натрия. Разу-
меется, присутствие стабилизатора не должно искажать результаты анализа.
Белки и ферменты имеют склонность с течением времени денатурировать, поэ-
тому их надо определять немедленно. Следовые компоненты при хранении мо-
гут быть утрачены вследствие адсорбции на стенках сосуда.
28
ЗАДАЧИ АНАЛИТИЧЕСКОЙ ХИМИИ, ИЛИ ЧЕМ ЗАНИМАЮТСЯ ХИ МИКИ-АНАЛИТИКИ
Пробы мочи неустойчивы, из них со временем осаждается фосфат кальция,
захватывая ионы металлов и другие вещества, которые могут представлять ин-
терес для анализа. Осаждение фосфата кальция можно предотвратить, подкис-
ляя пробу до pH 4,5 — обычно добавлением 1-2 мл ледяной уксусной кислоты
на 100 мл пробы. Пробы лучше хранить в холодильнике. Пробы мочи, а также
цельной крови, плазмы, сыворотки и тканей, можно хранить в замороженном
виде в течение долгого времени. Пробы крови, из которых удалены белки, более
устойчивы, чем пробы, не подвергшиеся обработке.
Пробы, которые содержат газы, вызывающие коррозию, часто взаимо-
действуют с материалом сосуда. Это касается, в частности, диоксида серы. Кро-
ме того, SO2, содержащийся в газах автомобильных выхлопов, часто теряется
из-за растворения в воде, образующейся в результате конденсации содержащей-
ся в пробе влаги. В подобных случаях предпочтительнее анализировать газ в по-
токе.
Подготовка пробы к анализу:
возможно, ее состав потребуется изменить
Первый шаг собственно анализа — измерение количества анализируемой пробы
(например, ее массы или объема). Это потребуется для расчета процентного со-
держания определяемого компонента. Количество анализируемой пробы дол-
жно быть измерено с той точностью, которая в данном случае необходима. Для
измерения массы обычно используют аналитические весы с погрешностью из-
мерения порядка 0,1 мг. В твердых пробах часто анализируют сухой остаток, по-
этому их следует предварительно высушить при температуре 110-120 °C (если
проба, разумеется, устойчива при этих температурах) и охладить в эксикаторе
перед взвешиванием. В некоторых случаях вследствие высокой гигроскопично-
сти проб могут потребоваться более высокие температуры и более продолжитель-
ное нагревание (например, в течение ночи). Требуемое количество анализируемой
пробы зависит от содержания в ней определяемого компонента и того, сколько
вещества необходимо для измерения сигнала. Для определения главных компо-
нентов может быть достаточно лишь 100-200 мг пробы, в то время, как для опре-
деления следовых компонентов может потребоваться несколько граммов. Как
правило, для анализа отбирают несколько параллельных проб, чтобы набрать
данные для статистической оценки точности анализа и получить более досто-
верные результаты.
По своей природе метод анализа может быть неразрушающим (недеструк-
тивным) — как например, при определении свинца в образцах красок рентге-
нофлуоресцентным методом, основанным на облучении пробы потоком
рентгеновских лучей и измерении интенсивности возникающего при этом ха-
рактеристического вторичного рентгеновского излучения. Однако гораздо
чаще твердые образцы перед анализом необходимо перевести в раствор. Неор-
ганические вещества можно растворить в различных кислотах и других средах,
обладающих окислительно-восстановительными или комплексообразующими
свойствами. Для растворения веществ, нерастворимых в кислотах, может потре-
1.3. МЫ НАЧИНАЕМ ПРОЦЕСС АНАЛИЗА
29
боваться сплавление с кислыми или щелочными плавнями, после чего эти веще-
ства становятся растворимыми в разбавленных кислотах или воде. Например,
сплавление с карбонатом натрия приводит к образованию карбонатов, раствори-
мых в кислотах.
Органические вещества, которые необходимо проанализировать на содер-
жание неорганических веществ, например, следов металлов, можно разрушить
посредством сухого озоления. Озолением называется разложение органических
веществ путем сжигания. В этом случае пробу медленно сжигают в печи при
температуре 400-700 °C, после чего получается неорганический остаток, рас-
творимый в разбавленных кислотах. С другой стороны, органические вещества
можно разрушить путем мокрого сожжения (минерализации) при нагревании с
кислотами-окислителями. Минерализация — это разложение органических ве-
ществ путем окисления в растворе. Обычно используют смесь серной и азотной
кислот. Биологические жидкости иногда можно анализировать непосредствен-
но. Однако часто определению мешают белки, поэтому их необходимо удалить.
Это можно сделать как посредством сухого озоления, так и мокрого сожжения.
Белки можно также осадить различными реагентами, после чего отфильтровать
или отцентрифугировать и получить безбелковый фильтрат.
Если определяемый компонент представляет собой органическое вещество,
методы пробоподготовки, использующие окисление, неприменимы. В этих слу-
чаях аналит можно отделить экстракцией или диализом, либо растворить пробу
в подходящем растворителе. Иногда и в этом случае возможно недеструктивное
определение. Примером может служить непосредственное определение белка в
пищевых продуктах при помощи ИК-спектроскопии в ближней области.
После перевода пробы в раствор следует создать определенные условия
среды, необходимые для осуществления следующего этапа анализа (разделения
или измерения). Например, часто необходимо установить определенное значе-
ние pH анализируемого раствора или добавить специальные реагенты, взаимо-
действующие с мешающими примесями и тем самым маскирующие их. Кроме
того, может потребоваться проведение реакции с участием самого аналита, чтобы
перевести его в форму, необходимую для разделения или измерения. Например, для
спектрофотометрического определения необходимо получить окрашенный про-
дукт реакции, а для определения методом газовой хроматографии — продукт,
обладающий летучестью. Для гравиметрического определения железа в виде
Fe2O3 требуется, чтобы все железо находилось в виде Fe(III) — обычной форме
существования этого элемента. Напротив, для титриметрического определения
с бихромат-ионами железо необходимо полностью перевести в Fe(II) до прове-
дения титрования. В этом случае необходимой стадией пробоподготовки явля-
ется восстановление.
Все растворители и реагенты, используемые для растворения пробы и дру-
гих операций пробоподготовки, должны иметь высокую чистоту (их квалифика-
ция должна быть не ниже ч. д. а.). Но даже такие реагенты могут содержать
следовые количества определяемого компонента. Поэтому необходимо подго-
товить и провести несколько параллельных контрольных опытов. Это особен-
но важно при определении следов веществ. Всегда проводите контрольные
30
ЗАДАЧИ АНАЛИТИЧЕСКОЙ ХИМИИ, ИЛИ ЧЕМ ЗАНИМАЮТСЯ ХИ МИКИ-АНАЛИТИКИ
опыты\ В идеальном случае образец контрольного опыта содержит все компо-
ненты реальной пробы, кроме определяемого вещества. Его проводят через все
стадии анализа, используя такие же количества всех реагентов (включая воду).
Результат измерения, полученный в контрольном опыте, вычитают из результа-
тов, полученных для реальных проб. Тем самым находят чистое содержание ана-
лита в пробе. Если сигнал контрольного опыта имеет значительную величину,
это может привести к потере смысла результатов анализа. В то же время провес-
ти «идеальный» контрольный опыт часто оказывается невозможно.
Выполнение операций разделения
Чтобы устранить мешающие влияния и добиться требуемой селективности из-
мерения, а также чтобы сконцентрировать аналит для обеспечения более высо-
кой чувствительности или точности, часто бывает необходимо выполнить одну
или несколько последовательных операций разделения компонентов. Предпочти-
тельнее отделять аналит от матрицы, а не наоборот: в этом случае меньше вероят-
ность потерь аналита. Операции разделения могут представлять собой осаждение,
экстракцию несмешивающимся с водой растворителем, хроматографический
процесс, диализ или отгонку.
Измерение аналитического сигнала: вы выбираете метод!
Выбор метода измерения количества аналита зависит от множества факторов.
Не последнюю роль играет содержание аналита в пробе и требуемая точность
результатов. Множество существующих методов анализа различаются уровнем
селективности, чувствительности, точности (правильности и воспроизводимо-
сти), стоимости и скорости выполнения анализа. Гравиметрический метод
обычно включает в себя селективное отделение аналита в виде осадка с последу-
ющим крайне неселективным способом измерения — измерением его массы.
Титриметрический метод основан на взаимодействии аналита с измеренным
объемом раствора реагента известной концентрации в ходе процесса, называе-
мого титрованием. Свидетельством завершения реакции служит резкое изме-
нение какого-либо физического или химического свойства системы. Методы
гравиметрии и титриметрии обладают очень высокой точностью — относитель-
ная погрешность может составлять десятые доли процента и даже менее. Но они
позволяют определять только относительно большие количества аналита (поряд-
ка миллимолей или миллиграммов) и пригодны лишь для определения основных
компонентов. Титриметрический анализ выполняется быстрее гравиметриче-
ского и потому, при наличии выбора, он предпочтительнее.
Инструментальная техника используется во многих методах анализа. Ее
изучают в рамках отдельной дисциплины — инструментальных методах ана-
лиза. Инструментальные методы основаны на измерении физического свойства
пробы, например, электрических свойств или поглощения электромагнитного
излучения. Примерами инструментальных методов анализа могут служить спек-
трофотометрия (в ультрафиолетовой, видимой, инфракрасной области спектра),
флуориметрия, атомная спектроскопия (абсорбционная и эмиссионная), масс-спек-
1.3. МЫ НАЧИНАЕМ ПРОЦЕСС АНАЛИЗА
31
трометрия, спектрометрия ядерного магнитного резонанса (ЯМР), рентгенов-
ская спектроскопия (абсорбционная, флуоресцентная), электрохимические
методы анализа (потенциометрия, вольтамперометрия, кулонометрия), хрома-
тография (газовая, жидкостная) и радиохимические методы. В целом инструмента-
льные методы более чувствительны и селективны, чем классические (гравиметрия
и титриметрия), но они менее точны: относительная погрешность составляет
1-5% или более. Кроме того, инструментальные методы обычно более дорого-
стоящие, капиталоемкие. Однако с точки зрения стоимости одного определения
они могут быть дешевле, если учитывать и затраты рабочей силы. Инструмента-
льные анализы, как правило, выполняются быстрее, их можно автоматизиро-
вать, с их помощью можно определять содержание нескольких компонентов
одновременно. Особенно большими возможностями для анализа сложных сме-
сей обладают хроматографические методы. Они позволяют осуществлять разде-
ление и измерение одновременно. Разделение достигается за счет того, что
компоненты смеси имеют различное сродство к материалу хроматографической
колонки и при пропускании смеси через колонку выходят из нее (элюируются) в
течение разного времени. Для определения используется подходящий детектор,
реагирующий на каждое выходящее из колонки вещество. В результате форми-
руется сигнал в виде пика, площадь которого пропорциональна количеству ана-
лита.
В табл. 1.1. приведены характеристики различных методов анализа, описан-
ных в данной книге: чувствительность, воспроизводимость, селективность, ско-
рость и стоимость выполнения анализа. Данные, приведенные в этой таблице,
типичны для большинства приложений разных методов. Эти методы могут быть
использованы и для других целей, и в отдельных случаях характеристики могут
оказаться лучше указанных. Для титриметрического определения более низких
концентраций необходимо детектировать конец титрования инструментальны-
ми средствами. Если для решения задачи можно применить несколько методов,
то выбор определяется наличием необходимого оборудования и квалифициро-
ванного персонала, а также личными предпочтениями аналитика. Например, для
определения нитратов в речной воде на уровне 1 части на миллион (1 ppm или
1,6 • 10~5 М) можно использовать метод спектрофотометрии после проведения
реакции диазотирования с участием нитрат-ионов и образованием окрашенного
продукта. Фториды в зубной пасте можно определить потенциометрически при
помощи фторидселективного электрода. Сложные смеси углеводородов бензи-
на можно разделить и проанализировать при помощи газовой хроматографии.
Глюкозу в крови можно определить кинетическим методом по скорости фер-
ментативной реакции между глюкозой и кислородом, катализируемой фермен-
том глюкозооксидазой. При этом можно измерять как скорость расходования
кислорода, так и скорость образования продукта реакции — пероксида водоро-
да. Степень чистоты серебряного слитка можно определить гравиметрически,
растворив небольшую пробу объекта в азотной кислоте, осадив хлорид серебра
действием хлорид-ионов и измерив массу осадка.
Все методы анализа можно разделить на абсолютные и относительные.
В основе абсолютных методов лежит расчет содержания аналита с использова-
Таблица 1.1.
Сравнение различных методов анализа
Метод Ориентировоч- ный диапазон концентраций (моль/л) Ориентиро- вочная точ- ность (%) Селективность Скорость выполнения анализа Стоимость Объекты
Гравиметрия lO-'-lO-2 од низкая-средняя малая низкая неорг.
Титриметрия КН-ЦН 0,1-1 низкая-средняя средняя низкая неорг., орг.
Потенциометрия lO-’-lO-6 2 высокая высокая низкая неорг.
Электрогравиметрия, кулонометрия 0,01-2 средняя низкая-средняя средняя неорг., орг.
Вольтамперометрия 1о-мо-10 2-5 высокая средняя средняя неорг., орг.
Спектрофотометрия 10-3-10-6' 2 высокая-средняя высокая-средняя низкая-средняя неорг., орг.
Флуориметрия 10-6-10-9 2-5 средняя средняя средняя орг.
Атомная спектроскопия 10-3-10-9 2-10 высокая высокая средняя-высокая неорг. (многоэлементный)
Хроматография 10-3-10-9 2-5 высокая высокая-средняя средняя-высокая орг. (многокомпонентный)
Кинетические методы 10-2-10-10 2-10 высокая-средняя высокая-средняя средняя неорг., орг., ферменты
1.3. МЫ НАЧИНАЕМ ПРОЦЕСС АНАЛИЗА
33
нием фундаментальных констант, например, атомных масс. Так, в гравиметри-
ческом анализе аналит превращают в нерастворимое производное известного
химического состава и взвешивают его. Например, для определения хлорид-
ионов получают хлорид серебра AgCl. Массовая доля аналита в осадке известна,
в рассмотренном примере массовая доля хлорид-ионов составляет
оГ1_ = Мг п/Мг АаГ1 = 35,453/143,32 = 0,24737
СД Г к_/1 Г /\gVzi
(где Мг — относительная молекулярная масса). Таким образом легко рассчи-
тать содержание хлорид-ионов в полученном осадке. Однако большинство мето-
дов являются относительными, т. е. требуют сравнения результатов с данными,
полученными для раствора известной концентрации. Например, в титриметри-
ческом анализе используют знание стехиометрии реакции аналита с реаген-
том. Соляная кислота реагирует с гидроксидом натрия в соотношении 1 : 1
согласно уравнению
НС1 + NaOH -> NaCl + Н2О
Объем раствора гидроксида натрия, необходимый для полного протекания
этой реакции, можно измерить. Если молярная концентрация гидроксида натрия
в растворе известна, то можно рассчитать количество NaOH (в молях), добавлен-
ного к пробе (как произведение концентрации на объем). Так можно узнать и ко-
личество НС1 (в молях) в пробе. Но для этого необходимо в первую очередь
приготовить раствор реагента (гидроксида натрия) точно известной концентра-
ции. Таким образом, титриметричекий метод является относительным.
Большинство инструментальных методов анализа также являются относи-
тельными. Прибор регистрирует аналитический сигнал, обусловленный какими-
либо физическими свойствами пробы. Например, спектрофотометры измеряют
долю электромагнитного излучения источника света, поглощаемую раствором
пробы. Эту величину можно соотнести с концентрацией определяемого компо-
нента только посредством сравнения с такой же величиной, измеренной для рас-
твора с известной концентрацией аналита. Иными словами, в инструментальных
методах анализа требуется градуировка при помощи образцов сравнения.
Аналитический сигнал может быть связан с концентрацией определяемого
компонента линейно или нелинейно. Градуировка заключается в приготовлении
серии стандартных растворов аналита с известной концентрацией (как правило,
затем их подвергают той же обработке, что и анализируемую пробу), измерении
аналитического сигнала (величины отклика прибора) для каждого из них и по-
строении градуировочной кривой — зависимости аналитического сигнала от
концентрации аналита. При помощи этой зависимости определяют неизвестное
содержание аналита по величине его сигнала. Современные приборы, управляе-
мые компьютерами, часто позволяют проводить эту операцию автоматически и
выдают конечный результат в виде значения концентрации.
На величину аналитического сигнала может оказывать влияние состав мат-
рицы образца. В подобных случаях для того, чтобы устранить матричные эффек-
ты пробы, градуировку следует осуществлять по методу стандартных добавок.
34
ЗАДАЧИ АНАЛИТИЧЕСКОЙ ХИМИИ, ИЛИ ЧЕМ ЗАНИМАЮТСЯ ХИМИКИ-АНАЛИТИКИ
При этом к отдельным порциям раствора пробы добавляют известные количест-
ва стандартного раствора аналита и измеряют увеличение сигнала, обусловленное
добавкой. При использовании метода добавок стандартный раствор находится в
той же среде, что и аналит. Этот способ градуировки будет обсуждаться подроб-
нее при рассмотрении отдельных методов анализа.
Вычисление результатов и предоставление отчета:
характеристика объекта в целом
После определения концентрации аналита в пробе полученные результаты ис-
пользуют для расчета количества аналита в исходном образце. Результат может
быть выражен как в абсолютных, так и в относительных величинах. Как прави-
ло, приводят относительное содержание, например, в виде массовой доли
(в процентах, частях на миллион или других величинах). Оно характеризует
правильность результатов. Можно выполнить серию параллельных определений
(три или более) и привести в отчете данные о воспроизводимости результатов —
например, величину стандартного отклонения. Данные о воспроизводимости пред-
ставляют важность, поскольку они позволяют охарактеризовать неопределен-
ность результата (см. гл. 3). Аналитик должен критически оценить, насколько
разумны полученные результаты в свете постановки исходной задачи. Помните,
что потребитель часто не имеет научной подготовки и может воспринимать
ваши цифры как истину в последней инстанции. Только вы, как аналитик, може-
те объяснить смысл этих цифр. Аналитик должен уметь дать квалифицирован-
ный совет по поводу всего, что касается смысла результата анализа. Поэтому
важно, чтобы вы нашли общий язык с «потребителем» ваших данных во всем,
что касается интерпретации результатов.
1.4. Поверка методики: вы должны доказать, что она работает!
Проблеме правильности результатов анализа необходимо уделять большое вни-
мание. Существуют два типа погрешностей измерений: случайные и система-
тические. Результат любого измерения содержит некоторую неопределенность.
Это приводит к тому, что результаты параллельных измерений распределены
случайным образом, например, в соответствии с законом нормального (гауссов-
ского) распределения. Эксперимент можно и нужно планировать таким образом,
чтобы уменьшить этот разброс, но его никогда нельзя устранить полностью. Сис-
тематическая погрешность приводит к смещению всех результатов измерений в
одном направлении. Это может происходить, например, вследствие того, что
матрица пробы уменьшает аналитический сигнал. Возможно, что аналитиче-
ские весы неправильно откалиброваны и все значения масс оказываются зани-
женными или завышенными. Из образца может быть не полностью удалена
влага — это тоже приводит к систематическим погрешностям.
1.4. ПОВЕРКА МЕТОДИКИ: ВЫ ДОЛЖНЫ ДОКАЗАТЬ, ЧТО ОНА РАБОТАЕТ!
35
Для обеспечения правильности результатов необходимо прежде всего пра-
вильно откалибровать измерительные приборы. Но это лишь первый шаг. В ходе
отработки методики следует проанализировать образцы, в которые добавлено
известное содержание аналита (в количествах как выше, так и ниже его пример-
ного содержания в пробе). Полученные результаты (после вычитания найденно-
го содержания аналита в самой пробе, проанализированной по той же методике)
должны находиться в пределах требуемой точности анализа. Для поверки новой
методики можно также сравнить результаты, полученные с ее помощью, с резу-
льтатами анализа той же пробы при помощи другой, общепринятой, методики.
Кроме того, существует множество стандартных образцов известного состава;
анализ стандартных образцов также позволяет убедиться в правильности испо-
льзуемой методики. Самый лучший способ проверить методику состоит в том,
чтобы проанализировать стандартный образец известного состава. В прода-
же имеются, например, стандартные образцы вод с известным содержанием
пестицидов или почв с известным содержанием приоритетных загрязнителей,
используемые для контроля окружающей среды. Американский Национальный
институт стандартов и технологий (National Institute of Standards and Technology,
NIST) изготавливает стандартные образцы объектов с матрицами различной
природы (сталей, опавших листьев и т. д.), содержание ряда аналитов в которых
тщательно определено, по крайней мере, двумя независимыми методами и офи-
циально удостоверено. Для всех содержаний указаны доверительные интервалы.
Различные агентства и промышленные концерны могут предоставить образцы
для межлабораторных экспериментов, в ходе которых образцы рассылают лабо-
раториям-участникам случайным образом. До выполнения анализов составы об-
разцов участникам не известны.
При использовании стандартных образцов их следует анализировать впере-
мешку с реальными пробами. Контрольный стандартный образец необходимо
анализировать хотя бы раз в день. Зависимость результатов его анализа от вре-
мени откладывают на карте контроля качества (контрольной карте). На ней же
откладывают известное значение стандартного отклонения методики. Если до-
пустить, что среднее значение измеряемой величины не изменяется со време-
нем, а ее распределение отвечает нормальному закону, то результат измерения
должен отличаться от истинного значения более чем на удвоенное стандартное
отклонение лишь в 1 случае из 20, а на 2,5 стандартных отклонения — в 1 случае
из 100. Если отклонения встречаются чаще, это указывает на наличие неском-
пенсированных погрешностей, которые могут быть вызваны нарушениями рабо-
ты аппаратуры, разрушением реагентов, неправильной градуировкой и другими
причинами.
Выполнение всех анализов, результаты которых предполагается использо-
вать в официальных целях (в том числе в суде), необходимо тщательно протоко-
лировать, а соответствующие методики — поверять в соответствии с нормами,
регламентируемыми государством. Для обеспечения поверки результатов ана-
лизов разработаны нормативные документы, называемые «Хорошей лаборатор-
ной практикой» (Good laboratory practice, GLP). Подробно эти правила
рассмотрены в гл. 4.
36
ЗАДАЧИ АНАЛИТИЧЕСКОЙ ХИМИИ, ИЛИ ЧЕМ ЗАНИМАЮТСЯ ХИМИКИ-АНАЛИТИКИ
1.5. Как определить размер пробы?
Виды анализа часто классифицируют в зависимости от требуемого размера про-
бы. Такая классификация условна, а границы классов нечетки. В соответствии с
ней анализ можно разделить на следующие виды: мезо-, полумикро-, микро- и
ультрамикроанализ. Последние две категории часто объединяют под названи-
ем «анализ следовых количеств». В табл. 1.2 приведена приблизительная клас-
сификация методов в зависимости от массы или объема пробы. Классификация
по объему обычно применяется в клинических лабораториях. Для операций с
микро- и ультрамикроколичествами вещества необходима специальная аппара-
тура и микровесы.
Таблица 1.2
Классификация методов анализа по размеру пробы
Название Масса пробы (мг) Объем пробы (мкл)
Мезоанализ >100 >100
Полумикроанализ 10-100 50-100
Микроанализ 1-10 <50
Ультрамикроанализ <1
«Анализировать» и «определять» — различные понятия
Термины «анализировать» и «определять» различаются по значению. Говорит-
ся, что пробу анализируют на содержание в ней одного или нескольких компо-
нентов. Компоненты, содержание которых измеряют, называются аналитами.
Процесс измерения содержания аналита называется определением. Например,
в ходе анализа крови на содержание хлоридов мы определяем концентрацию
хлорид-ионов. Таким образом, пробу анализируют, чтобы определить в нем со-
держание компонента.
Компоненты пробы можно разделить на главные (содержание более 1%),
сопутствующие (0,1-1%) и следовые (менее 0,1%). Компоненты с содержани-
ем порядка частей на миллион и менее можно назвать ультраследовыми.
Анализ может быть полным или частичным в зависимости от того, опре-
деляют ли все компоненты пробы или только некоторые из них. Чаще всего в
ходе анализа необходимо определить содержание одного конкретного компо-
нента или класса веществ.
1.6. Некоторые полезные Интернет-сайты
Кроме многочисленных книг и других источников, отмеченных в основном тек-
сте и Приложении А (литература по аналитической химии), существует большое
1.6. НЕКОТОРЫЕ ПОЛЕЗНЫЕ ИНТЕРНЕТ-САЙТЫ
37
число Интернет-страниц, которые могут быть полезны химикам-аналитикам в
качестве дополнительных ресурсов. Разумеется, они часто претерпевают изме-
нения. Появляются и новые страницы. Однако те из них, что перечислены ниже,
в любом случае могут служить хорошей отправной точкой для получения мно-
гочисленной полезной информации.
Химия в целом
1. www.arc.org — домашняя страница Американского химического общества.
Имеется информация о журналах, конференциях, химические новости, по-
исковые базы данных (включая Chemical Abstracts) и многое другое.
2. www.chemweb.com — виртуальный клуб химиков. На сайте есть базы дан-
ных и сводные таблицы химического содержания. Действуют дискуссион-
ные форумы, ориентированные на различные химические дисциплины,
в частности, аналитическую химию. Для вступления в клуб необходимо за-
регистрироваться, регистрация бесплатная.
3- www.chemsoc.org — сеть сайтов химических обществ, поддерживаемая
британским Королевским химическим обществом. Информацию на сайт
предоставляют около 30 национальных химических обществ. Имеется вир-
туальная Периодическая таблица элементов, которую вы можете загрузить
в качестве экранной заставки компьютера.
4- http://micro.magnet.fsu.edu/primer/java/scienceopticsu/powersoflO/index.html
— посмотрите на это наглядное представление масштабов различных объек-
тов — начиная от протона и кончая Млечным путем, находящимся в
10 миллионах световых лет от Земли.
Аналитическая химия
1. www.acs-analytical.duq.edu — домашняя страница Отделения аналитиче-
ской химии Американского химического общества. На сайте имеется мно-
жество ссылок и других ресурсов, позволяющих переходить к различным
страницам, связанным с аналитической химией, по всему Интернету.
2. www.chem-uni.potsdam.de/linkcenter/analchem.html — коллекция ссылок
по аналитической химии, ведущих к университетским и другим сайтам все-
го мира.
3. www.cstl.nist.gov/nist839 — страница Отделения аналитической химии
Национального института стандартов и технологий США (NIST). Содер-
жит информацию о каждом из пяти своих направлений: спектрохимиче-
ских методов, методов органического анализа, газовой метрологии и
классических методов, молекулярной спектроскопии и микрожидкостных
методов, ядерно-физических методов анализа.
4. www.rsc.org/lap/rsccom/dab/analdiv.htm — сайт аналитического отделения
Королевского химического общества — британского аналога отделения
аналитической химии Американского химического общества.
38
ЗДЦАЧИ АНАЛИТИЧЕСКОЙ ХИМИИ, ИЛИ ЧЕМ ЗАНИМАЮТСЯ ХИМИКИ-АНАЛИТИКИ
5. http://chemweb.chem.uconn.edu/Microchem/Index.html — сайт Американ-
ского микрохимического общества. Содержит ссылки на страницы науч-
ных сообществ по аналитической химии, конференций и личные страницы
отдельных ученых.
6. http://analytical.chemweb.com/home — форум по аналитической химии
сайта Chemweb, упомянутого выше.
7. www.asdlib.org — цифровая лаборатория по аналитическим наукам (Ana-
lytical Sciences Digital Laboratory, ASDL). Находится в начальной стадии
развития, но обещает стать ключевым источником ссылок на рецензируе-
мые сайты, посвященные преподаванию и технике аналитической химии.
Дополнительную информацию об этом сайте можно получить на тексто-
вом сайте, упомянутом ранее.
Что мы узнали из этой главы?
• Задача аналитической науки — охарактеризовать материю с химической
точки зрения: что и сколько? — стр. 17
• Аналитик должен знать, какого рода информацию о веществе ему надо по-
лучить, и уметь приготовить представительную пробу — стр. 23, 26
• Лишь в редких случаях химические измерения специфичны, поэтому для
обеспечения высокой селективности необходимы специальные дополните-
льные операции — стр. 28
• Вы должны уметь выбрать подходящий метод анализа — стр. 30
• Методики необходимо поверять — стр. 34
• Существует много Интернет-страниц, посвященных аналитической хи-
мии — стр. 36
Вопросы
1. Что такое аналитическая химия?
2. В чем разница между качественным и количественным анализом?
3. Перечислите основные этапы методики анализа. Кратко охарактеризуйте
каждый этап.
4. В чем разница между понятиями «анализировать» и «определять», «проба»
и «аналит»?
5. Что такое контрольный опыт?
6. Перечислите основные способы измерений, применяемые в химическом
анализе.
7 Перечислите основные методы разделения, применяемые в химическом
анализе.
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
39
8. Дайте определение инструментальных методов анализа.
9. Что такое градуировочный график?
10. В чем различие между специфичной реакцией и селективной реакцией?
11. Пользуясь табл. 1.1, предложите методы для решения следующих задач:
а) определение степени чистоты NaCl в поваренной соли; б) определение
содержания уксусной кислоты в уксусе; в) определение pH воды плаватель-
ного бассейна.
Рекомендуемая литература
Общая
1. J. Tyson, Analysis. What Analytical Chemists Do. London: Royal Society of
Chemistry, 1988. Небольшая книга, в которой в весьма сжатой форме об-
суждается, чем и как занимаются химики-аналитики.
2. R. W. Murray, «Analytical Chemistry Is What Analytical Chemists Do», Edito-
rial, Anal. Chem., 66 (1994) 682A.
3. G. M. Hieftje, «But Is It Analytical Chemistry?», Am. Lab., October (1993) 53.
4. T. Kuwana, Chair, Curricular Developments in the Analytical Sciences. Отчет о со-
вещаниях 1996 и 1997 г., финансируемых национальным научным фондом
(National Science Foundation) (kuwana@sunglower.com).
5. R. A. DePalma and A. H. Ullman, «Professional Analytical Chemistry in Indust-
ry. A Short Course to Encourage Students to Attend Graduate School», J. Chem.
Ed., 68 (1991) 383. Описание краткого курса производственной аналитиче-
ской химии, преподаваемого по заявкам в университетах и колледжах спе-
циалистами компании Proctor & Gamble, с целью разъяснить, чем
занимаются химики-аналитики в промышленности. Основной акцент сде-
лан на тезисе: химик-аналитик — человек, решающий проблемы. С вопро-
сами обращаться по адресу parry. d@ pg.com.
6. С. A. Lucy, «How to Succeed in Analytical Chemistry: A Bibliography of Reso-
urces from the Literature», Taianta, 51 (2000) 1125. Обзор литературы на
темы: как приобрести оборудование, как написать рукопись и как получить
работу в области аналитической химии.
7. G. D. Christian, «Evolution and Revolution in Quantitative Analysis», Anal.
Chem., 67 (1995) 532А. История аналитической химии с момента возникно-
вения человечества.
Энциклопедии и справочники
8. R. A. Meyers, editor-in-chief, Encyclopedia of Analytical chemistry. Chichester:
Wiley, 2000. 15-томное издание, в котором 10 томов посвящены приложе-
ниям, а 5 томов — теории и аппаратуре.
40
ЗАДАЧИ АНАЛИТИЧЕСКОЙ ХИМИИ, ИЛИ ЧЕМ ЗАНИМАЮТСЯ ХИ МИКИ-АНАЛИТИКИ
9. A. Townsend, editor-in-chief, Encyclopedia of Analytical Science. London: Aca-
demic, 1995. 10-томное издание, охватывающее все практические аспекты
аналитических наук. Описаны все методы определения отдельных элемен-
тов, соединений и классов соединений в любых физических и биологиче-
ских матрицах.
10. J. A. Dean, Analytical Chemistry Handbook. New York: McGraw Hill, 1995.
Подробное описание классических и инструментальных методов анализа,
предварительных аналитических операций, включая методы разделения,
статистических методов в аналитической химии.
Пробоотбор
11. F. F. Pitard, Pierre Gy’s Sampling Theory and Sampling Practice. Vol. 1: Hete-
rogeneity and Sampling. Vol. II: Sampling Correctness and Sampling Practice.
Boca Raton, FL: CRC Press, 1989.
12. P. M. Gy and A. G. Boyle, Sampling for Analytical Purposes. Chichester: Wiley,
1998. Введение в теорию пробоотбора, написанное Пьером Ги и разъясня-
ющее выведенные им расчетные формулы — первоначально применитель-
но к отбору твердых проб, но справедливые и для жидких и многофазных
проб.
13. Р. М. Gy, Sampling of Heterogeneous and Dynamic Material Systems. Amster-
dam: Elsevier, 1992.
14. G. E. Baiulescu, P. Dumitrescu, and P. Gh. Zugravescu, Sampling. New York:
Ellis Horwood, 1991.
15. N. T. Crosby, ed., General Principles of Good Sampling Practice. London: Roy-
al Society of Chemistry, 1995.
16. P. M. Gy, «Introduction to Theory of Sampling I. Heterogeneity of a Population
of Uncorrelated Units», Trends Anal. Chem., 14(2) (1995) 67.
17. P. M. Gy, «Tutorial: Sampling or Gambling?». Process Control and Quality, 6
(1994) 97.
18. B. Kratochvil and J. K. Taylor, «Sampling for Chemical Analysic», Anal. Chem.,
53 (1981) 924A.
19. B. Kratochvil, «Sampling for Microanalysis: Theory and Strategies», Fresenius
J. Anal. Chem., 337 (1990) 808.
20. J. F. Vicard and D. Fraisse, «Sampling Issues», Fresenius’ J. Anal. Chem., 348
(1994) 101.
21. J. M. Hungerford and G. D. Christian, «Statistical Sampling Errors as Intrinsic
Limits on Detection in Dilute Solutions», Anal. Chem., 58 (1986) 2567.
Мешающие влияния
22. W. E. Van der Linden, «Definition and Classification of Interference in Analyti-
cal Procedures», Pure & Appl. Chem., 61 (1989) 91.
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
41
Стандартные растворы
23. В. W. Smith and М. L. Parsons, «Preparation and Standard Solutions. Critically
Selected Compounds», J. Chem. Ed., 50 (1973) 679. Описано приготовление
стандартных растворов для 72 элементов — металлов и неметаллов.
24. М. Н. Gabb and W. Е. Latchem, A Handbook of Laboratory Solutions. New
York: Chemical Publishing, 1968.
25. http://pubs.acs.org/reagents/committee.html— страница комитета по ана-
литическим реагентам Американского химического общества.
Гоадуировка
26. Н. Mark, Principles and Practice of Spectroscopic Calibration. New York: Wi-
ley, 1991.
Стандартные образцы
27. H. Klich and R. Walker, «COMAR—The International Database for Certified
Reference Materials», Fresenius' J. Anal. Chem., 345 (1993) 104.
28. Office of Standard Reference Materials, Room B311, Chemistry Building, Nati-
onal Institute of Standards and Technology, Gaithersburg, Maryland 20899.
29. National Research Council of Canada, Division of Chemistry, Ottawa KIA OR6,
Canada.
30. R. Alverez, S. D. Rasberry and G. A. Uriano, «NBS Standard Reference Materi-
als: Update 1982», Anal. Chem., 54 (1982) 1239A.
31. R. W. Seward and R. Mavrodineanu, «Standard Reference Materials: Summary
of the Clinical Laboratory Standards Issued by the National Bureau of Stan-
dards», NBS (NIST) Special Publications 260-271, Washington, DC, 1981.
32. Стандартные образцы углей. Распространяются департаментом по энерге-
тике США (U.S. Department of Energy, Pittsburgh Mining Technology Center,
P. O. Box 10940, Pittsburgh, PA 15236). Образцы охарактеризованы по 14 па-
раметрам, включая содержание главных и следовых элементов.
Глава 2
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ
ХИМИЧЕСКОГО АНАЛИЗА
Сначала получи результат;
потом можешь делать с ним что угодно.
Марк Твен
Прежде, чем приступать к эксперименту, прочтите данную главу. В химическом
анализе для получения информации нужно провести измерения, порой специ-
фичные. Аналитическое оборудование и аппаратура, необходимые для этого,
рассматриваются в соответствующих главах книги. В то же время существует и
ряд общих для большинства методов положений, связанных с выполнением экс-
перимента. Эти вопросы обсуждаются в данной главе. К универсальному анали-
тическому оборудованию относятся аналитические весы, мерная посуда, а также
сушильные шкафы и фильтры. Некоторые тонкости, связанные с использовани-
ем этого оборудования, лучше всего объяснит ваш преподаватель непосредствен-
но в ходе выполнения лабораторных работ, тем более, что инструментальный
аппарат в различных лабораториях может несколько отличаться. Здесь же мы от-
метим лишь наиболее общие приемы при проведении анализа, которыми дол-
жен владеть квалифицированный аналитик.
Рисунки с изображением наиболее часто используемой лабораторной посу-
ды и другого оборудования приведены на компакт-диске.
2.1. Лабораторный журнал - самый главный документ
Прежде всего вам необходимо усвоить, что в аналитической лаборатории, более
чем где бы то ни было еще, имеют значение чистота и аккуратность. Это отно-
сится и к ведению лабораторного журнала. Все результаты необходимо записы-
вать в журнал чернилами непосредственно тогда, когда они получены. Работая в
аналитической лаборатории, вы быстро поймете, что выполнение этого требова-
ния — в ваших же интересах. Во-первых, вы сэкономите время на переписывании
данных. Кроме того, приучив себя записывать результаты сразу и в обязательном
порядке, вы станете более организованными и в самой работе. Это будет допол-
нительным выигрышем во времени. Во-вторых, если вы будете вести записи не-
посредственно в процессе эксперимента, то сможете обнаружить ошибки в
результатах измерений или расчетах. Наконец, записывая результаты сразу в
журнале (а не на черновиках), вы обезопасите себя от возможной потери или ис-
2.1. ЛАБОРАТОРНЫЙ ЖУРНАЛ - САМЫЙ ГЛАВНЫЙ ДОКУМЕНТ
43
кажения данных. Хорошее ведение лабораторного журнала — залог получения
достоверных результатов.
Для профессиональных химиков-аналитиков требование записывать на-
блюдения и результаты измерений непосредственно в журнал имеет особенно
большое значение. Полнота документации необходима в криминалистических,
производственных лабораториях, для совершения юридических действий, пода-
чи патентных заявок. В производственных исследовательских лабораториях,
страницы журнала должны быть подписаны (заверены) с указанием даты другим
лицом, знакомым с сутью работы. Это может понадобиться для установления
приоритета при подаче заявки на патент.
Приведенное ниже описание титриметрического определения карбоната
натрия в образце технической соды можно считать хорошим примером того, как
следует вести лабораторный журнал.
Дата: 7 сентября 2003 г.
Определение карбоната натрия в образце технической соды.
Суть анализа. Навеску соды растворяют в воде и титруют стандартным раствором соля-
ной кислоты с индикатором бромкрезоловым зеленым. Соляную кислоту предварите-
льно стандартизуют по первичному стандарту — карбонату натрия. Массы навесок
карбоната натрия и технической соды рассчитывают по разности.
Источник', описание практической работы 7.
Реакция титрования'. СО|~ + 2Н+ = Н2СО3
Стандартизация НС1
"гНа2С0/МГ)
| 105,99(мг/ммоль)- Инс1 (мл)
^а2СО3<МГ)
СНС1 (мг/ммоль) =
I WrNa2CO3 (МГ/ ММОЛЬ)- ИНС1 (мл)
Взятие навески Na2CO3 № опыта 1 2 3
Масса, г:
бюкса с веществом 24,2689 24,0522 23,8597
бюкса с остатками вещества 24,0522 23,8597 23,6269
Na^jCC^ 0,2167 0,1925 0,2328
Масса Na2CO3, мг Титрование Na2CO3 Показания бюретки, мл: 216,7 192,5 232,8
конечное 40,26 35,68 43,29
начальное 0,03 0,00 0,02
Объем НС1, мл 40,23 35,68 43,27
Концентрация НС1, М: 0,10164 О,1О18о 0,10152
Среднее: 0,10165М
Стандартное отклонение: 1,6 • 10~3
Размах: 2,8 • 10~3
(См. продолжение)
44
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
Анализ образца соды (Продолжение)
сна (ммоль// мл) * ^нс1 (мл) ®№,со, - л/1навсски •1Ч-№,СО,(МГ/ММОЛЬ) (мг) •100% =
0,10165 (ммоль/ мл) • FHC1 (мл) • ~ • 105,99(мг/ ммоль) $
тнавески(МГ) Взятие навески технической соды
№ опыта Масса, г: 1 2 3
бюкса с веществом 25,6728 25,4673 25,2371
бюкса с остатками вещества 25,4673 25,2371 25,0027
навески 0,2055 0,2302 0,2344
Масса навески, мг Титрование технической соды Показания бюретки, мл: 205,5 230,2 234,4
конечное 35,67 40,00 40,70
начальное 0,00 0,01 0,05
Объем НС1, мл 35,67 39,99 40,65
Массовая доля Na2CO3, % Среднее: Стандартное отклонение: Размах: 93,50 93,50% 0,9 • 10“3 1,7 • 10~3 93,58 93,42
Приведенный пример — это лишь сокращенный вариант, в котором опуще-
ны все численные расчеты. В полной версии журнал должен содержать все вычис-
ления. В этом случае при необходимости ход вычислений можно впоследствии
проверить и найти ошибку.
Рекомендуется не заполнять все страницы журнала подряд, а оставлять сво-
бодные места: например, все левые страницы отводить под черновые вычисления,
а правые страницы—для обобщающих данных. Важно также, чтобы все данные
были записаны с правильным числом значащих цифр. Верное число значащих
цифр в результатах измерений и вычислений имеет решающее значение для
правильной интерпретации результатов анализа. Данная проблема обсуждает-
ся в гл. 3. Прежде, чем приступать к работе в лаборатории, этот материал следует
повторить.
2.2. ЛАБОРАТОРНЫЕ МАТЕРИАЛЫ И РЕАГЕНТЫ
45
Рекомендации по ведению лабораторного журнала
Лабораторный журнал — это хроника всей вашей работы как химика-аналитика. В нем
задокументировано все, что вы делаете. Это источник информации для отчетов, публи-
каций и официальных представлений. Успех или неудача деятельности вашей компании
может зависеть от того, насколько хорошо вы ведете эту документацию. Лабораторный
журнал является официальным документом при подаче заявки на патент, разработке
нормативных актов (при поверке методик, инспекциях и иных юридических действиях).
Запомните: то, что не записано, то не сделано! Именно ваш лабораторный журнал явля-
ется тем местом, где вы фиксируете новые идеи, которые в будущем могут стать основой
патента, поэтому важно записывать, в чем эти идеи заключаются и когда они появились.
Особенности ведения лабораторного журнала определяются личными предпочте-
ниями. Но существует ряд общих правил, выполнение которых необходимо для хороше-
го ведения журнала:
• Используйте тетрадь в твердом переплете (отсутствует угроза потери листов).
• Последовательно нумеруйте все страницы.
• Никогда не вырывайте листы. Если текст, написанный на странице, более не нужен,
просто перечеркните его.
• На каждой странице указывайте дату и подписывайте ее. Кроме того, пусть страницы
вашего журнала подписывает и датирует (возможно, спустя короткое время) кто-либо
еще, указывая: «С содержанием ознакомился такой-то».
• При описании экспериментов указывайте название работы и ее цели, ссылки на источ-
ники.
• Записывайте все данные в тот день, когда вы их получили.
Несколько слов об электронных журналах. Программное обеспечение со-
временных приборов часто позволяет собирать, хранить и обрабатывать данные,
поступающие непосредственно от прибора, используя в расчетах результаты
градуировки. В этом случае важно, чтобы программное обеспечение и градуи-
ровка прибора, так же, как и все стадии собственно методики анализа, были по-
верены. Это одно из требований «хорошей лабораторной практики» (гл. 4).
2.2. Лабораторные материалы и реагенты
В табл. 2.1 указаны свойства ряда материалов, часто применяемых в производст-
ве лабораторной аппаратуры. Для изготовления лабораторной посуды (стака-
нов, колб, пипеток, бюреток) чаще всего используется боросиликатное стекло
(торговых марок Pyrex, Kimax). Оно устойчиво к действию горячих растворов и
резким изменениям температуры. Для специальной посуды, используемой в
Таблица 2.1
Свойства некоторых материалов, используемых для изготовления лабораторной посуды
Материал Максимальная рабочая температура (°C) Устойчивость к резким изменениям температуры Химическая устойчивость Примечания
Боросиликатное стекло 200 Выдерживает резкое изменение на 150 °C Слабо подвергается действию горячих растворов щелочей Торговые марки: Pyrex (Coming Glass Works), Kimax (Owens-Illinois)
Мягкое стекло Низкая Разрушается растворами щелочей He содержит бора. Торговая марка Coming
Щелочеустойчивое стекло Более чувствительно, чем боросиликатное
Плавленый кварц 1050 Высокая Устойчив к действию большинства кислот, галогенов В кварцевых тиглях сплавляют различные вещества
Кварцевое стекло 1000 Высокая Более устойчиво к действию щелочей, чем боросиликатное стекло Используется так же, как плавленый кварц. Торговая марка Vycor (Coming)
Фарфор 1100 (глазированный) 1400 (неглазированный) Средняя Высокая
Материал Максимальная рабочая температура (°C) Устойчивость к резким изменениям температуры Химическая устойчивость Примечания
Платина Около 1500 Устойчива к действию большинства кислот и расплавов солей. Разрушается царской водкой, расплавами нитратов, цианидов, хлоридов при температуры >1000 °C. Используют также в виде сплавов с золотом, серебром и другими металлами Для повышения твердости обычно используют в виде сплавов с иридием или родием. В платиновых тиглях проводят сплавление и обработку фтористоводородной кислотой
Никель и железо При сплавлении пробы загрязняются материалом тигля Никелевые и железные тигли используют для проведения сплавления с пероксидами
Нержавеющая сталь 400-500 Высокая Не разрушается щелочами и кислотами, кроме конц. НС1, разб. H2SO4 и кипящей конц. HNO3
Полиэтилен 115 Не разрушается растворами щелочей и HF. Разрушается под действием многих органических растворителей (к действию ацетона и этанола устойчив) Пластичный материал
Полистирол 70 Не разрушается под действием HF. Разрушается многими органическими растворителями Достаточно хрупкий
Тефлон 250 Устойчив по отношению к большинству химических веществ Тефлоновая посуда используется для хранения растворов и реагентов при определении следов металлов
48
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
особых целях, могут применяться другие материалы, например, более устойчи-
вые к действию химических реактивов, высоких температур и т. д.
На форзаце одного из томов приведены различные квалификации чистоты
химических реактивов в соответствии с классификацией Американского хими-
ческого общества. В аналитической лаборатории следует использовать только
те реактивы, которые имеют квалификацию не ниже, чем «чистый для анализа»
(ч. д. a., A.C.S. reagent-grade) или «первичный стандарт» (primary standard). Для
выполнения анализов почти всегда используют реагенты квалификации ч. д. а.
Для приготовления стандартных растворов в титриметрии используют пер-
вичные стандарты.
Американское химическое общество публикует сводки критериев оценки
чистоты и качества основных лабораторных реактивов. Квалификация реакти-
вов, не указанных в этих списках, устанавливается непосредственно производи-
телями в соответствии с их собственными критериями.
Для реактивов квалификации ч. д. а., удовлетворяющих общим требовани-
ям чистоты, часто дополнительно указывают (на этикетке тары) результаты ана-
лизов на содержание отдельных примесей. Реактивы квалификации «первичный
стандарт», как правило, имеют содержание основного вещества не ниже 99,95%.
Они более дорогие, чем реактивы ч. д. а., и используются только для приготов-
ления стандартных растворов или для стандартизации растворов других ве-
ществ посредством проведения химических реакций (титрования). Не все
реактивы производятся в квалификации «первичный стандарт». Существуют
особые квалификации чистоты растворителей, применяемых для специальных
целей, например, «спектрально чистый» или «чистый для хроматографии». Та-
кие растворители подвергают специальной очистке, чтобы удалить примеси, ме-
шающие при выполнении конкретных задач. Аналогично этому, для определения
следовых количеств металлов выпускаются особо чистые кислоты. Их очищают
специальными приемами и более тщательно анализируют на содержание следов
различных элементов (обычно оно должно составлять не более нескольких час-
тей на миллиард).
Помимо коммерческих производителей, реактивы квалификации «первичный
стандарт» выпускает Национальный институт стандартов и технологий США.
В каталоге NIST Special Publication 260 перечислены выпускаемые институтом
стандартные образцы (см. страницу http://ts.nist.gov/ts/htdocs/230/232.htm, со-
держащую информацию о программе разработки стандартных образцов и их пе-
речень). Стандартный образец — это вещество сложного состава (например,
сплав), содержание компонентов которого точно установлено в результате ана-
лизов и официально удостоверено. Стандартные образцы используют для про-
верки и градуировки методик анализа.
На форзаце одного из томов приведены концентрации некоторых коммер-
ческих препаратов кислот и щелочей.
2.3. АНАЛИТИЧЕСКИЕ ВЕСЫ - НЕОБХОДИМЫЙ ИНСТРУМЕНТ
49
2.3. Аналитические весы - необходимый инструмент
Взвешивание — неотъемлемая операция почти любого анализа, которую испо-
льзуют для измерения массы пробы, для приготовления стандартных растворов.
В химическом анализе мы имеем дело с достаточно малыми массами, порядка
нескольких граммов, нескольких миллиграммов или даже меньше. Как правило,
массу необходимо измерять не менее чем с тремя или четырьмя значащими циф-
рами, поэтому лабораторные весы должны быть одновременно и чувствитель-
ными, и точными. Существует много сложных способов достижения этого
сочетания. Однако чаще всего для взвешивания используют полезный и универ-
сальный инструмент — аналитические весы.
Большинство аналитических весов, применяемых в настоящее время, явля-
ются электронными. В то же время до сих пор продолжают использовать меха-
нические одночашечные весы, поэтому мы опишем операции и с такими весами.
В основе действия весов обоих типов лежит сравнение веса одного тела с весом
другого (у электронных весов это осуществляется в ходе калибровки), на их ра-
боту оказывают влияние одни и те же факторы, например, дрейф нуля или вытал-
кивающая сила воздуха. Отметим, что на самом деле мы оперируем величинами
масс, а не весов. Вес тела — это сила тяжести, действующая на него. Она разная
в различных точках земного шара. Масса определяется только количеством ве-
щества, из которого состоит тело. Она не зависит от местоположения объекта.
Электронные весы — это очень удобно
Современные электронные весы удобны в работе, их показания более точны, а
устройство более надежно, чем у механических весов. При работе с ними не тре-
буется таких характерных для механических весов операций, как считывание
масс разновесов, вращение микрометрических винтов и считывание их показа-
ний, арретирование чашек. За счет этого измерения значительно ускоряются.
Общий вид электронных весов с цифровым табло показан на рис. 2.1, а принцип
их работы схематически представлен на рис. 2.2. В отличие от механических ве-
сов, в них отсутствуют разновесы и призмы. Чашка весов соединена с подвиж-
ным рычагом (рис. 2.2, 2), вся подвижная система уравновешена при помощи
электромагнита. Положение рычага контролируется специальным устройст-
вом — электрическим сканером (рис. 2.2, 7). Он также управляет электромагни-
том, поддерживая силу тока в его обмотке таким образом, чтобы чашка весов
при любой нагрузке находилась в исходном положении. Сила тока, протекаю-
щего через обмотку электромагнита, пропорциональна весу предмета, находя-
щегося на чашке весов. Эта величина преобразуется в цифровую форму и
передается микропроцессору, в свою очередь преобразующему сигнал в соот-
ветствующее значение веса. Оно отображается на цифровом табло. При этом
возможно автоматическое вычитание веса тары.
Таким образом, в основе действия электронных весов лежит принцип ком-
пенсации под действием электромагнитной силы, впервые описанный Ангстре-
мом в 1895 г. Но в процессе их работы происходит сравнение веса одного тела с
50
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
Рис. 2.1
Общий вид электронных
аналитических весов. С любезного
разрешения Denver Instrument Со.
Denver Instrument Company —
собственник всех рисунков
весом другого. В начале работы весы необходимо «обнулить», т. е. откалибро-
вать при помощи тела известного веса. Когда на чашку весов помещают ка-
кой-либо предмет, его вес при помощи электронных устройств сравнивают с
известным весом. Это один из вариантов автоматической калибровки. Совре-
менные весы обладают целым рядом дополнительных возможностей, например,
позволяют осуществлять автоматическую компенсацию колебаний нулевого поло-
жения или флуктуаций результатов вследствие колебаний здания.
Электронные весы имеют единственную управляющую клавишу, при помо-
щи которой можно включать и выключать весы, обнулять табло и автоматически
вычитать вес тары. Поскольку результаты взвешивания являются электрическим
сигналом, их легко обрабатывать и хранить при помощи персонального компью-
тера. Можно автоматически рассчитывать статистические характеристики про-
цесса взвешивания.
Рис. 2.2
Принцип работы электронных весов:
1 — электронные разновесы; 2 — рычаг;
3 — обмотка электромагнита; 4 — датчик
температуры [К. М. Lang, American
Laboratory, March, 1983, p. 72]. Публикуется
с разрешения American Laboratory, Inc.
2.3. АНАЛИТИЧЕСКИЕ ВЕСЫ - НЕОБХОДИМЫЙ ИНСТРУМЕНТ
51
Электронные аналитические весы выпускаются для разных диапазонов масс
и с разной ценой деления. Макровесы позволяют взвешивать объекты массой до
160 г при цене деления 0,1 мг, а полумикровесы — до 30 г при цене деления
0,01 мг. Микровесы позволяют взвешивать с точностью до 1 мкг, а ультрамикро-
весы — от 0,1 мкг и ниже.
На пьезоэлектрических кварцевых весах можно взвешивать объекты массой
до 100 мкг. Они позволяют зафиксировать изменения массы порядка 1 нг (Г10-9 г)!
Чувствительным элементом весов является тонкий диск из кристаллического
кварца, на который посредством осаждения из пара нанесена тончайшая пленка
золота. Диск совершает колебания при строго определенной частоте, например,
10 МГц. Эта частота изменяется при малейшем изменении массы диска. Измене-
ние частоты колебаний измеряют и преобразуют в величину массы. Таким спосо-
бом можно зафиксировать очень малые изменения массы — порядка нескольких
процентов от массы моноатомнот^о или мономолекулярного слоя покрытия зо-
лотой подложки. Можно следить за изменением массы объекта во времени.
Одночашечные механические весы
Несмотря на то что механические весы постепенно вытесняются электронными,
они до сих пор находят широкое применение. Поэтому было бы неправильным
оставить механические весы без рассмотрения.
Взвешивание свиньи по методу Бернса:
1) Возьми совершенно прямую доску и уравновесь ее, положив на лесопильные
козлы.
2) Положи свинью на один конец доски.
3) Клади булыжники на другой конец доски до тех пор, пока она не придет в равно-
весие.
4) После этого тебе осталось узнать общий вес булыжников.
Механические аналитические весы представляют собой по существу рычаг
первого рода, используемый для сравнения двух масс. Рис. 2.3 иллюстрирует
принцип их действия. Точка опоры Л находится между точками приложения сил
В и С. Символом Мх обозначена неизвестная, а М2 — известная масса. Принцип
действия весов основан на том, что в условиях равновесия MXLX = М2Ь2. Если
сконструировать рычаг так, чтобы Lx = £2, то при равновесии Мх = М2. На коро-
мысле весов помещена указательная стрелка, позволяющая судить о наличии
равновесия. В ходе взвешивания подбирают значение М2 тех пор, пока стрел-
ка не возвратится в исходное положение, т. е. положение при отсутствии нагруз-
ки на чашке весов. На самом деле при помощи весов измеряют не массу, а вес.
Однако значения весов при заданных условиях взвешивания пропорциональны
значениям масс. Поэтому в обиходе термины «масса» и «вес» используют как
синонимы, а операцию измерения массы называют «взвешиванием». Гирьки из-
вестной массы называются разновесами.
52
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
Рис. 2.3
Принцип действия механических
аналитических весов
В L2 С
В большинстве случаев механические аналитические весы являются одно-
чашечными. Типичная схема устройства таких весов изображена на рис. 2.4, а
внешний вид стандартных одночашечных весов — на рис. 2.5.
В отличие от рассмотренного выше случая, рычаг, используемый в одноча-
шечных весах, является несимметричным. Он опирается на ребро призмы, а вес
чашки весов, на которую помещают объект, действует на один из концов коро-
мысла рычага. Когда чашка весов не нагружена, к этому концу приложена сила
тяжести набора разновесов общей массой 160-200 г. На другом конце рычага
чашка отсутствует. Вместо нее там находится постоянный груз-противовес,
уравновешивающий чашку и разновесы. Он же является частью поршня-амор-
тизатора. Когда на чашку весов помещают взвешиваемый объект, часть разнове-
сов необходимо снять, чтобы восстановить равновесие. Это осуществляется
при помощи ручек, расположенных на передней панели весов. При их вращении
определенные разновесы (или их сочетания) поднимаются вверх и снимаются с
коромысла. Таким образом, манипуляции с разновесами никогда не производят-
Арретирование
и освобождение
чашки
Ручки управления
разновесами <
РИС. 2.4. Типичная схема устройства одночашечных весов
2.3. АНАЛИТИЧЕСКИЕ ВЕСЫ - НЕОБХОДИМЫЙ ИНСТРУМЕНТ
53
ся руками. Суммарный вес разновесов, снятых с коромысла, равен весу предме-
та, находящегося на чашке весов.
Обычно массы разновесов кратны 1 г или 0,1 г в зависимости от типа весов.
Поэтому нельзя добиться полного уравновешивания коромысла путем снятия
разновесов. Остаточный дисбаланс автоматически регистрируют при помощи
оптической системы. На один из концов коромысла нанесена оптическая шкала,
деления которой освещаются лампой и проецируются на экран со шкалой-нони-
усом. Последние десятичные цифры значения массы (до 0,1 мг) считывают с этой
шкалы и прибавляют к массе снятых разновесов. Величина остаточного дисбалан-
са может отображаться и на цифровом табло.
Исходное положение ненагруженной чашки весов и соответствующее ему
положение шкалы называются нулевой точкой весов, а положение под нагруз-
кой — точкой покоя. При взвешивании точку покоя устанавливают так, чтобы
она совпала с нулевой точкой. Обычно положение нулевой точки предваритель-
но устанавливают так, чтобы показание шкалы было нулевым. Для этого переме-
щают нониус при помощи специальной вращающейся ручки. Перед взвешиванием
всегда проверяйте положение нулевой точки.
В состоянии равновесия одночашечные весы находятся под постоянной на-
грузкой 160-200 г, независимо от массы взвешиваемого предмета. Однако поло-
жение центра тяжести коромысла изменяется в зависимости от веса предмета.
Соответственно, несколько изменяется и чувствительность весов.
Все разновесы одночашечных весов находятся внутри корпуса. Их положе-
ние управляется только ручками, расположенными на передней панели. Одна
ручка предназначена для манипуляций с разновесами массой в десятки граммов
(т. е. от 10 до 90 г), другая — единицы граммов (от 1 до 9 г), третья (если имеет-
ся) — сотни миллиграммов (от 0,1 до 0,9 г). Масса разновесов, снятых с коро-
мысла, отображается при помощи счетчиков, также расположенных на передней
панели. Равновесие коромысла устанавливается достаточно быстро благодаря
наличию воздушного поршневого демпфера.
С весами следует обращаться осторожно, чтобы не повредить ребра призм в
те моменты времени, когда весы не находятся в работе, а также когда объекты
помещают на чашку или снимают с нее. Для защиты призм и коромысла пред-
назначена рукоятка переключателя арретира, имеющая три положения. В сред-
нем положении как коромысло, так и чашка арретированы. В одном из двух
других положений чашка весов частично освобождается. Это положение исполь-
зуют тогда, когда приближенно определяют вес объекта. В последнем положе-
нии чашка полностью освобождается, чтобы весы могли прийти в равновесие.
При использовании одночашечных весов взвешивание может быть выполнено
менее чем за минуту.
Полумикро- и микровесы
В предыдущих разделах основное внимание уделялось обычным макровесам или
аналитическим весам. Они позволяют проводить измерения масс до 100-200 г с
точностью до 0,1 мг. Для решения большинства повседневных аналитических
54
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
Рис. 2.5
Внешний вид стандартных одночашечных весов.
(С любезного разрешения Arthur Н. Thomas Company.)
задач этого достаточно. В то же время для весов всех рассмотренных типов су-
ществует возможность увеличения чувствительности посредством изменения
влияющих на нее параметров: уменьшения массы коромысла и чашки, увеличе-
ния длины коромысла и изменения положения его центра тяжести. Для изготов-
ления коромысла можно использовать более легкие материалы, поскольку
ввиду уменьшения общей нагрузки (по сравнению с обычными макровесами)
снижаются требования к прочности конструкционных материалов.
Полумикровесы позволяют взвешивать с точностью до 0,01 мг, ъ микровесы —
до 0,001 мг (1 мкг). Предельные нагрузки для таких весов соответственно ниже, чем
для обычных весов, а при работе с ними требуется повышенная аккуратность.
Дрейф нулевой точки
Нулевая точка весов не есть нечто постоянное, что может быть определено и
установлено раз и навсегда. Ее положение изменяется под воздействием множе-
ства факторов, в том числе изменений температуры и влажности окружающей
среды, а также статического электричества. Поэтому положение нулевой точки
следует проверять во время работы с весами, по крайней мере, каждые полчаса.
Вес в вакууме — наиболее точная величина
Результаты взвешиваний отражают, разумеется, вес тела в воздухе. Однако любое
тело теряет в весе столько, сколько весит вытесненный им воздух. В этом состоит
закон Архимеда (см. материал о возникновении аналитической химии, приведен-
ный в гл. 1). Плотность воздуха составляет 0,0012 г/см3 (1,2 мг/см3). То есть на
тело объемом 1 мл действует выталкивающая сила воздуха, равная 1,2 мг! Если
плотности объекта и разновесов равны, то одинаковы и их потери в весе, и изме-
ренное значение в точности соответствует весу объекта в вакууме, где выталкива-
ющая сила не действует. Но если эти плотности сильно различаются, различия в
2.3. АНАЛИТИЧЕСКИЕ ВЕСЫ - НЕОБХОДИМЫЙ ИНСТРУМЕНТ
55
величинах выталкивающих сил приведут к небольшой погрешности взвешива-
ния. Такая ситуация возникает при взвешивании объектов как с очень высокой
плотностью (например, платиновых сосудов — плотность платины составляет
21,4 г/см3, или ртути с плотностью 13,6 г/см3), так и с низкой плотностью (напри-
мер, воды, плотность ~1 г/см3). Для сравнения: плотность разновесов, обычно
применяемых для взвешивания, составляет около 8 г/см3. При очень тщательной
работе эту погрешность следует учитывать в виде соответствующей поправки.
Вычисление поправок на выталкивающую силу воздуха для одночашечных весов
описано в работе [14], а калибровка разновесов на одночашечных весах — в рабо-
те [10]. Выталкивающую силу, действующую натару для взвешивания, не следует
принимать во внимание, поскольку при вычислении массы объекта по разности
ее величина сокращается. Поправку на выталкивающую силу часто необходимо
учитывать при калибровке мерной посуды. Одни и те же поправки на выталкива-
ющую силу применимы как к механическим, так и к электронным весам (калибру-
емым при помощи разновесов из материала известной плотности).
В действительности в использовании поправок на выталкивающую силу,
как правило, нет необходимости, поскольку соответствующие погрешности час-
то сокращаются, например, при вычислении массовой доли определяемого ком-
понента. В этом случае одна и та же погрешность присутствует как в числителе
(концентрация стандартного раствора или масса гравиметрической формы), так
и в знаменателе (масса навески) алгебраического выражения. Разумеется, чтобы
эта погрешность оставалась действительно постоянной, все взвешивания необ-
ходимо производить в таре, изготовленной из одного и того же материала (оди-
наковой плотности).
Например, при калибровке мерной посуды вводить поправку на приведение
веса к значению в вакууме действительно необходимо. Для этого измеряют мас-
су воды или ртути, которая содержится в сосуде либо вытекает из него. Зная
плотность при соответствующей температуре, можно из массы рассчитать объ-
ем жидкости. Но даже в этом случае погрешность определения составляет по-
рядка одной десятой процента. В большинстве случаев такой погрешностью
можно пренебречь. Для точных расчетов существуют специальные таблицы пе-
ревода веса воды или ртути в воздухе к весу в вакууме при различных темпера-
турах.
Вес объекта в воздухе можно привести к весу в вакууме по следующей фор-
муле:
w =W- + W-
rr vac air "air
0,0012 0,0012
Do Dw
(2.1)
где FTvac — вес в вакууме, г; FTair — измеренный вес в воздухе, г; Do — плотность
объекта, г/см3; Dw — плотность материала разновесов, г/см3; 0,0012 (г/см3) —
плотность воздуха.
56
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
Плотности наиболее распространенных материалов разновесов — латуни и не-
ржавеющей стали — составляют 8,4 и 7,8 г/см3 соответственно. Пример расчета
веса воды должен убедить вас в том, что даже в этом случае величина поправки
составляет порядка одной части на тысячу.
Пример 2.1
Удобный способ калибровки пипетки состоит во взвешивании воды, вытекшей
из нее. Объем воды может быть рассчитан из точных значений ее массы и плот-
ности при данной температуре. Предположим, что надо градуировать пипетку с
номинальным объемом 20 мл. Для взвешивания воды будем использовать колбу
с притертой пробкой. Пусть измеренное значение веса пустой колбы составляет
29,278 г, а колбы с водой — 49,272 г (используются разновесы из латуни). Како-
во значение веса содержащейся в колбе воды, приведенное к значению веса в ва-
кууме?
Решение
Вес воды на воздухе равен увеличению веса колбы и составляет
49,272-29,278 = 19,994 г
Плотность воды составляет 1,0 г/см3 (более точные значения в интервале темпе-
ратур от 10 до 30 °C см. в табл. 2.4). Отсюда
Wvac = 19,994 +19,994|
I 1,0
0,0012
8,4
= 20,015 г
Пример 2.2
Выполните расчеты с данными из примера 2.1 при условии, что разновесы изго-
товлены из нержавеющей стали (плотность 7,8 г/см3).
Решение
При расчетах избегайте округлений вплоть до получения конечного результата.
Вы получите ту же самую величину:
jyvac=19,994 + 19,994|^^-^^ | = 20,015 г
I 1,0 7,8 )
Таким образом, к весам любых типов можно применять одни и те же величины
поправок на выталкивающую силу (табл. 2.4 на стр. 72).
2.3. АНАЛИТИЧЕСКИЕ ВЕСЫ - НЕОБХОДИМЫЙ ИНСТРУМЕНТ
57
Источники погрешностей при взвешивании
Некоторые источники погрешностей при взвешивании — дрейф нулевой точки,
разновесы, выталкивающая сила воздуха — уже упоминались. Источником наибо-
лее серьезных погрешностей являются изменения температуры окружающей среды
или объекта. Они вызывают конвекционные потоки воздуха, которые обусловлива-
ют смещение нулевой точки или точки покоя. Горячие или слишком холодные
предметы перед взвешиванием необходимо привести к температуре окружающей
среды. Гигроскопичные материалы могут поглощать влагу, поэтому следует по
возможности избегать взвешивания в условиях высокой влажности или длитель-
ного нахождения образца на воздухе перед взвешиванием и в ходе его.
Общие правила взвешивания
Как мы видели, существует несколько типов весов. Порядок операций при рабо-
те с ними может быть различным для весов, изготовленных разными производите-
лями. Конкретный порядок работы с вашими весами вам объяснит преподаватель.
Но в любом случае важно: 1) предохранять призмы от повреждений; 2) предо-
хранять все детали весов от пыли и коррозии; 3) избегать загрязнений объекта
(как самой пробы, так и тары) или иных изменений его веса в ходе взвешивания
и 4) избегать грубых ошибок, вызванных конвекционными потоками воздуха.
Ниже приведены общие правила, которые вы должны усвоить до того, как при-
ступать к работе с любыми аналитическими весами. Выучите эти правила!
1 Никогда не берите взвешиваемый объект пальцами. Переносите объект,
обвернув его полоской чистой бумаги или при помощи щипцов.
2. Взвешивайте при комнатной температуре, чтобы избежать возникнове-
ния конвекционных потоков воздуха.
3- Никогда не кладите химические вещества непосредственно на чашку ве-
сов. Для взвешивания используйте тару (бюксы, стаканчики, часовые
стекла) или полоску кальки. Если вы все же просыпали вещество, немед-
ленно сметите его мягкой кисточкой.
4. Перед взвешиванием всегда закрывайте створки камеры весов. В про-
тивном случае вы можете получить неверные результаты, вызванные по-
токами воздуха.
5. При работе с механическими весами никогда не помещайте на чашку и
не убирайте с нее объект или разновесы, не убедившись, что коромысло
весов и чашка арретированы.
Взвешивание твердых веществ
Твердые вещества обычно взвешивают (а неметаллические — и высушивают) в
весовых бюксах (рис. 2.6). Они снабжены стандартными притертыми стеклян-
58
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
Рис. 2.6. Бюксы для взвешивания
Рис. 2.7. Блюдечко для взвешивания
ными пробками. Гигроскопичные, поглощающие влагу из воздуха вещества не-
обходимо взвешивать в сосудах с хорошо притертой пробкой (крышкой). После-
довательное взятие нескольких навесок одного и того же объекта (а также
гигроскопичных веществ) удобнее всего выполнять по разности. В этом случае
время работы сокращается, особенно при использовании механических весов. Для
электронных весов экономия времени не столь велика. Идея такого взвешивания
состоит в том, что сначала взвешивают бюкс со всем содержащимся в нем вещест-
вом, затем порцию вещества отбирают (например, отсыпают), количественно
перенося ее в сосуд для растворения. Бюкс с оставшимся веществом снова взве-
шивают и рассчитывают массу отобранного вещества по разности. Затем точно
так же отбирают следующую порцию вещества и снова взвешивают бюкс, и так
далее. Например, см. записи в лабораторном журнале на с. 43-44 (определение
карбоната натрия в образце технической соды).
Очевидно, что при таком способе работы на каждую навеску приходится
одно взвешивание (и еще одно в самом начале). При этом масса каждой навески
представляет собой разность двух результатов взвешивания, а общая погреш-
ность значения массы складывается из погрешностей каждого из этих результатов.
В ходе взвешивания по разности необходимо держать бюкс плотно закрытым в
тех случаях, когда вещество гигроскопично или по каким-либо иным причинам
не может перед взвешиванием долго находиться в контакте с атмосферным воз-
духом. Если же опасность воздействия атмосферы отсутствует, бюкс закрывать
не обязательно.
Для непосредственного (прямого) взвешивания в качестве тары исполь-
зуют блюдечки (рис. 2.7) или лодочки для взвешивания, а также часовые стекла
и специальную бумагу (кальку). Сначала взвешивают пустую тару, а затем тару
с навеской. В этом случае для каждой навески необходимы два взвешивания.
После взвешивания навеску пересыпают в сосуд для растворения; остатки про-
бы со стенок тары переносят при помощи кисточки. Прямое взвешивание воз-
можно только в том случае, если проба не гигроскопична.
При очень точных (до десятых долей миллиграмма) взвешиваниях необхо-
димо предохранять тару от загрязнения посторонними веществами, которое мо-
жет вызвать изменение ее веса. В частности, не следует брать тару руками,
поскольку даже выделения пота могут исказить результат. Прежде чем брать
2.3. АНАЛИТИЧЕСКИЕ ВЕСЫ - НЕОБХОДИМЫЙ ИНСТРУМЕНТ
59
тару, ее следует обернуть полоской бумаги. Твердые пробы перед взятием наве-
ски часто бывает необходимо довести до постоянной массы. Во всех случаях
перед взвешиванием проба должна приобрести температуру окружающей среды
(весовой комнаты).
Взвешивание жидкостей
Жидкости обычно взвешивают методом прямого взвешивания. Жидкость поме-
щают в предварительно взвешенный сосуд (например, бюкс) и плотно закрывают,
чтобы предотвратить возможное испарение жидкости во время взвешивания, за-
тем взвешивают сосуд с жидкостью. Если массу жидкости определяют по разно-
сти, отбирая ее порцию пипеткой, необходимо несколько раз промыть пипетку,
собирая промывные воды в тот же сосуд, в который была перенесена основная
порция. При отборе порции необходимо следить, чтобы не потерять каплю жид-
кости из носика пипетки.
Взвешивание: какая точность вам необходима?
В химическом анализе существуют два типа взвешивания — ориентировочное
и точное. Ориентировочное взвешивание (с двумя или тремя значащими цифра-
ми) обычно проводят в тех случаях, когда достаточно знать массу вещества с
точностью, не превышающей нескольких процентов, например, при приготов-
лении растворов реагентов, которые предстоит стандартизовать при помощи
стандартных растворов точно известной концентрации, или при отборе проб ве-
ществ, которые необходимо высушить и впоследствии взвесить точно либо до-
бавить в раствор для создания определенной среды. Таким образом, для всех
веществ, количества которых не используются для расчета результата анализа,
достаточно ориентировочного взвешивания. Для ориентировочного взвешива-
ния не требуются аналитические весы, вполне достаточно технических весов
любой конструкции.
Точное взвешивание необходимо при взятии навески анализируемой про-
бы, взвешивании сухой гравиметрической формы в ходе гравиметрического
анализа или высушенного реагента, используемого для приготовления первич-
ного стандартного раствора. Во всех этих случаях значение массы объекта непо-
средственно используется для расчета результатов анализа. Это значение должно
быть известно не менее чем с четырьмя значащими цифрами. В этом случае
взвешивание осуществляется только на аналитических весах, как правило,
с точностью до 0,1 мг. Лишь в редких случаях берут навеску вещества, масса
которой точно известна заранее (например, 0,5000 г). Как правило, берут при-
мерное количество вещества (в данном случае около 0,5 г), но взвешивают его с
требуемой точностью (например, 0,5129 г). На аналитических весах необходимо
проводить лишь те взвешивания, результаты которых будут использоваться в
количественных расчетах. Некоторые вещества вообще никогда не взвешивают
на аналитических весах. Например, таблетированный гидроксид натрия столь
гигроскопичен, что непрерывно поглощает воду из атмосферы. Поэтому резуль-
таты взвешивания гидроксида натрия невоспроизводимы, а степень его чистоты
60
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
неизвестна. Чтобы приготовить раствор гидроксида натрия точно известной
концентрации, порцию вещества взвешивают ориентировочно на технических
весах, растворяют в воде, а затем полученный раствор стандартизуют при помо-
щи стандартного раствора кислоты.
2.4. Мерная посуда
Пожалуй, единственный метод анализа, где не требуются точные измерения
объема, — это гравиметрия. Во всех остальных методах, в которых используют-
ся растворы, такие измерения необходимы.
Мерные колбы
Мерные колбы используют для того, чтобы разбавить раствор
до точно известного объема. Вместимость мерных колб мо- ZX
жет изменяться в широких пределах — от 2 л и более до 1 мл. ( )
Стандартная мерная колба изображена на рис. 2.8. Мерные
колбы всегда отградуированы на заполнение (англ, «to conta-
in»). Это означает, что при определенной температуре (20 или
25 °C) объем жидкости, содержащейся в колбе, точно равен но-
минальному при условии, что нижний край мениска жидкости
(ее поверхности, искривленной вследствие капиллярного эф-
фекта — рис. 2.14) касается метки — горизонтальной линии, ) I
нанесенной на горлышко колбы. Температурный коэффици- / \
ент расширения стекла, из которого изготавливают мерные / \
колбы, достаточно мал, поэтому можно считать, что неболь- / \
шие колебания комнатной температуры не влияют на значе- у J
ние объема. Мерные колбы (как и другие мерные сосуды,
характеризующиеся меньшей точностью, например, мерные
цилиндры) часто маркируют ТС — «to contain». Кроме того, рИд 2.8
на стенке колбы, наряду с номинальным значением объема,
многие производители указывают и максимальную погреш-
ность этого значения. Например, на мерной колбе вместимостью 250 мл может
быть указано «±0,24 мл», что соответствует приблизительно 0,1% относитель-
ной погрешности. Вместимость мерных колб должна быть точно известна.
При работе с мерной колбой в нее сначала помещают небольшое количество
растворителя, обычно дистиллированной воды. Реактивы не следует помещать в
сухие колбы, поскольку поверхность стекла обладает сильным адсорбирующим
действием. Раствор в мерной колбе следует готовить поэтапно. Сначала в колбу
вносят тот реагент (твердый или жидкий), который необходимо растворить, а за-
тем добавляют растворитель в количестве не более двух третей объема колбы.
При этом все частицы реагента, прилипшие к стенкам горлышка колбы, следует
смыть на дно колбы. После этого реагент полностью растворяют и лишь затем
2.4. МЕРНАЯ ПОСУДА
61
добавляют остальную часть растворителя так, чтобы нижний край мениска ка-
сался метки колбы (метка при этом должна быть расположена на уровне глаз).
Если после этого на горлышке колбы выше метки остаются капельки воды, их
следует удалить фильтровальной бумагой. Необходимо протереть досуха шли-
фы колбы и притертой пробки.
Наконец, раствор в колбе перемешивают. Для этого колбу переворачивают,
плотно придерживая пробку, и интенсивно вращают или встряхивают ее в тече-
ние 5-10 с. После этого колбу снова переворачивают дном вниз и оставляют в та-
ком положении до тех пор, пока вся жидкость не стечет со стенок горлышка. Эту
процедуру повторяют 5-10 раз.
Примечание. Если в ходе разбавления вы перелили жидкость выше метки,
то и в этом случае имеется возможность сохранить и использовать раствор. Для
этого наклейте вдоль горлышка колбы тонкую полоску бумаги и отметьте на ней
остро заточенным карандашом, избегая перекосов, положение нижнего края ме-
ниска. Раствор тщательно перемешайте и перенесите в сухой сосуд. Затем запол-
ните колбу водой до фабричной метки. Наконец, при помощи бюретки (или
градуированной пипетки небольшого объема) добавляйте в колбу воду до тех
пор, пока нижний край мениска не коснется отметки, сделанной вами на бумаге.
Запишите значение объема воды, который вам пришлось для этого добавить,
прибавьте его к номинальному объему колбы и пересчитайте концентрацию рас-
твора, используя полученное значение объема.
Пипетки
Пипетки предназначены для того, чтобы отмерить и перенести определенный
объем жидкости. В частности, пипетки часто используют для отбора аликвоты
(определенной порции) раствора. Чтобы доля, которую составляет аликвота от
общего объема раствора, была известна, необходимо, чтобы был известен и об-
щий первоначальный объем раствора. Можно отобрать несколько последовате-
льных аликвот одного и того же раствора, но в этом случае важно, чтобы между
отборами раствор не испарялся и не разбавлялся. Существуют два основных
типа пипеток — мерные и градуированные (рис. 2.9 и 2.10). Некоторые разно-
видности градуированных пипеток называют клиническими или серологиче-
скими пипетками.
В отличие от мерных колб, пипетки отградуированы на вытекание (англ,
to deliver) определенного объема жидкости при заданной температуре и часто
помечаются TD. Как и в случае мерных колб, небольшие колебания температу-
ры не влияют на объем пипеток. При калибровке пипеток учитывают, что неко-
торая часть жидкости остается внутри в виде тонкой пленки на стенках, причем
толщина этой пленки в некоторой степени зависит от скорости опорожнения пи-
петки. Как правило, пипетки градуируют в расчете на свободное вытекание под
действием силы тяжести. При работе с пипетками необходимо следить, чтобы
скорость вытекания жидкости всегда была одной и той же.
Мерные пипетки отградуированы только на одно определенное значение
объема. Их используют в тех случаях, когда требуется отобрать объем жидкости
62
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
РИС. 2.9. Мерные пипетки
РИС. 2.10. Градуированные пипетки
с наибольшей точностью. Как правило, такими пипетками можно дозировать
объемы с точностью до четырех (в случае необходимости, при условии соответ-
ствующей градуировки — пяти) значащих цифр. Допустимые погрешности объ-
емов мерных пипеток класса А приведены в таблице на переднем форзаце
данного тома. Градуированные пипетки представляют собой цилиндрические
трубки, на поверхность которых нанесены значения объема с определенным ша-
гом. Они не настолько точны, как мерные пипетки, поскольку их внутренний
диаметр все же не вполне постоянен по высоте. В этом случае искажения формы
пипеток сильнее сказываются на величинах объемов, чем в случае мерных пипе-
ток с характерным утолщением по середине. Кроме того, толщина остаточной
пленки жидкости на стенках пипетки изменяется в зависимости от отмеренного
объема. При помощи градуированных пипеток можно достичь точности объема
в лучшем случае с тремя значащими цифрами, если только не откалибровать пи-
петку специально для определенного значения объема.
2.4. МЕРНАЯ ПОСУДА
63
Большинство градуированных пипеток калибруют на свободное вытекание,
т. е. с учетом того, что небольшой объем жидкости остается в носике. Его не сле-
дует вытряхивать или выдувать. Пипетку при опорожнении держат вертикаль-
но, касаясь кончиком стенки сосуда. Жидкость при этом вытекает плавно, без
разбрызгивания, а величина объема капли, остающейся в носике, хорошо вос-
производится. Жидкость из носика пипетки частично вытягивается под действи-
ем сил притяжения к стенкам сосуда.
Иногда пипетки (в том числе градуированные, если они отградуированы в
пределах всего внутреннего объема) калибруют на выдувание. При использова-
нии таких пипеток для того, чтобы отмерить номинальное значение объема, сле-
дует выдуть из носика остатки жидкости. Пипетки такого типа легко распознать,
поскольку в верхней части они всегда помечены одним или двумя кольцами
шероховатого стекла. Их не надо путать с цветными кольцами, используемыми
для маркировки объема пипетки. Из таких пипеток остаток жидкости следует
выдувать только после свободного вытекания основного объема под действием
силы тяжести. Преждевременное выдувание жидкости увеличивает скорость ее
истечения и приводит к изменению толщины остаточной пленки на стенках.
Мерные пипетки выпускают объемом от 100 до 0,5 мл и менее. Объемы гра-
дуированных и серологических пипеток могут составлять от 25 до 0,1 мл. Их
можно использовать и для высокоточной дозировки (особенно небольших объе-
мов), если специально откалибровать применительно к конкретному значению
объема. Мерные пипетки большого объема обычно характеризуются слишком бо-
льшой скоростью истечения — более высокой, чем скорость стекания пленки
жидкости со стенок. Кроме того, их диаметр слишком велик, из-за чего снижает-
ся точность считывания показаний.
Необходимо всегда обтирать наружную поверхность пипетки после заполне-
ния. При использовании неводных растворителей, а также растворов повышенной
вязкости пипетку следует заново откалибровать при помощи соответствующего
растворителя или раствора, чтобы учесть изменение скорости истекания.
Для заполнения пипетки жидкостью используют резиновые груши, по-
ршневые или иные пипетаторы. Прежде, чем ими пользоваться, поупражняйтесь
в заполнении пипетки водой с их помощью. Запрещается засасывать жидкости
ртом.
Шприцевые пипетки
Пипетки этого типа можно использовать для дозирования как больших, так и ма-
лых объемов. Положение метки на шприцевых пипетках может быть несколько
смещено относительно номинального объема, но воспроизводимость дозировки
может быть очень высокой — особенно если пипетка снабжена автоматическим
поршнем. При этом исключаются погрешности объема, обусловленные наличи-
ем пленки жидкости на внутренних стенках, поскольку вся жидкость выталкива-
ется поршнем. Значение объема вытекающей жидкости можно откалибровать с
высокой точностью. Шприцевые пипетки удобно использовать для дозирования
микролитровых объемов. Шприцевые пипетки объемом несколько микролитров
64
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
Рис. 2.11
Микролитровая пипетка фирмы Hamilton
используют для ввода проб в газовые хроматографы. Пипетка такого типа, снаб-
женная иглой, изображена на рис. 2.11. Погрешности объемов таких пипеток не
выше, чем у других микропипеток. Дополнительным достоинством шприцевых
пипеток является возможность дозирования любого объема в пределах общего
объема пипетки.
Использование шприцевых пипеток очень удобно для точного дозирования
вязких или летучих жидкостей. При использовании обычных пипеток значения
объема будут искажены, поскольку для таких жидкостей толщина остаточной
пленки на стенках иная, чем для воды. Шприцевые пипетки удобно использо-
вать также для быстрого дозирования жидкостей. При последовательном дозиро-
вании нескольких жидкостей они хорошо перемешиваются, вследствие быстрого
вытекания.
Еще один тип шприцевых пипеток представлен на рис. 2.12. Такие пипетки
позволяют работать одной рукой, быстро отмерять постоянные объемы жидко-
Рис. 2.12
Одноканальная и
многоканальная пипетка
со съемными
наконечниками и
планшеты для
клинических анализов.
С любезного разрешения
Thermo Labsystems
2.4. МЕРНАЯ ПОСУДА
65
стей и широко применяются в клинических лабораториях. К пипеткам прилагается
набор сменных наконечников из пластмассы, например, на основе полипропилена.
Материал наконечников не смачивается водой. Поэтому искажения объемов,
обусловленные наличием пленки жидкости на стенках, отсутствуют. Кроме
того, поверхность таких материалов мало загрязняется. В пипетке содержится
распираемый пружиной поршень, который управляется кнопкой. Поршень име-
ет два положения упора: верхнее — дня набора жидкости, нижнее — для полно-
го опорожнения пипетки. В процессе работы жидкость никогда не контактирует
с самим поршнем, она наполняет только наконечник. Объемы пипеток могут со-
ставлять от 1 до 1000 мкл. Воспроизводимость дозирования зависит от величи-
ны объема и в лучшем случае составляет 1-2%.
Во многих случаях нет необходимости знать реальное значение объема, до-
зируемого микропипетками рассматриваемого и других типов, поскольку эти
пипетки используются для относительных измерений. Например, одну и ту же
пипетку — а следовательно, одно и то же значение объема — можно использо-
вать для дозирования аликвоты анализируемой пробы и стандартных растворов
с целью построения градуировочной зависимости. В подобных случаях воспро-
изводимость объема играет более важную роль, чем его абсолютное значение.
Стандартом для калибровки пипеток в Европе (где производится большая часть
пипеток такого типа) служит немецкий стандарт DIN 126650 (и соответствую-
щий ему международный стандарт ISO 8655). Калибровка пипеток производит-
ся гравиметрическим методом (путем взвешивания воды). В стандарте DIN не
указываются отдельные пределы для правильности и воспроизводимости, а ис-
пользуется значение суммарной погрешности, рассчитываемой как сумма пре-
дельно допустимой систематической погрешности и удвоенного стандартного
отклонения. Полученная величина представляет собой интервал, в котором с
95%-й вероятностью находится значение объема, дозируемого пипеткой (о стан-
дартных отклонениях и доверительных интервалах см. гл. 3). В табл. 2.2 приве-
дены предельно допустимые согласно стандарту DIN погрешности объемов для
одноканальных пипеток, а в табл. 2.3 — раздельные характеристики правильно-
сти и воспроизводимости для многоканальных пипеток.
Помимо шприцевых пипеток с ручным управлением, существуют механи-
зированные шприцевые пипетки, а также пипетки с электронным управлением и
перестраиваемым значением объема. С их помощью можно многократно дози-
ровать различные объемы жидкостей в автоматическом режиме. Выпускаются
также пипетки с несколькими каналами для одновременного дозирования не-
скольких, например, 12 или 16, аликвот. Их используют для добавления растворов в
ячейки специальных планшетов, применяемых в биотехнологии и клинических ла-
бораториях, где часто требуется обрабатывать тысячи проб (рис. 2.12). Дополните-
льную информацию о пипетках со сменными наконечниками можно получить
непосредственно у представителей их производителей, например, на сайтах
www.finnpipette.com и www.eppendorf.com.
66
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
Таблица 2.2
Предельные погрешности для одноканальных шприцевых пипеток
согласно стандарту DIN 12650*
Номинальный объем, (мкл) Максимальная погрешность, (мкл) Относительная погрешность, (%)
1 ±0,15 ±15,0
2 ±0,20 ±10,0
5 ±0,30 ±6,0
10 ±0,30 ±3,0
20 ±0,40 ±2,0
50 ±0,80 ±1,6
100 ±1,50 ±1,5
200 ±2,00 ±1,0
500 ±5,00 ±1,0
1000 ±10,00 ±1,0
2000 ±20,00 ±1,0
5000 ±50,00 ±1,0
10000 ±100,00 ±1,0
* Указаны пределы в случае испытания пипеток производителем в строго контролируемых условиях. Если ис-
пытания проводит потребитель в обычных условиях, предельные величины погрешностей следует удвоить.
С любезного разрешения Thermo Labsystems Оу, Финляндия.
Таблица 2.3
Правильность и воспроизводимость одноканальных цифровых шприцевых пипеток
Finnpipettes
Диапазон (мкл) Шаг (мкл) Объем (мкл) Правильность Воспроизводимость*
(мкл) (%) ст. откл. (мкл) отн. ст. откл. (%)
0,2-2 0,01 2 ±0,050 ±2,5 0,040 2,0
0,2 ±0,024 ±12,0 0,020 10,0
0,5-10 0,1 10 ±0,100 ±1,0 0,050 0,5
1 ±0,025 ±2,5 0,020 2,0
0,5-10 0,1 10 ±0,100 ±1,0 0,080 0,8
1 ±0,035 ±3,5 0,030 3,0
2-20 0,1 20 ±0,200 ±1,0 0,080 0,4
2 ±0,060 ±3,0 0,030 1,5
5-40 0,5 40 ±0,240 ±0,6 0,120 0,3
5 ±0,100 ±2,0 0,100 2,0
10-100 1,0 100 ±0,80 ±0,8 0,20 0,2
10 ±0,30 ±3,0 0,10 1,0
20-200 1,0 200 ±1,20 ±0,6 0,40 0,2
20 ±0,36 ±1,8 0,14 0,7
200-1000 5,0 1000 ±6,00 ±0,6 2,00 0,2
200 ±1,80 ±0,9 0,60 0,3
100-1000 5,0 1000 ±6,00 ±0,6 2,00 0,2
100 ±1,00 ±1,0 0,60 0,6
* ст. откл. — стандартное отклонение, отн. ст. откл. — относительное стандартное отклонение.
С любезного разрешения Thermo Labsystems Оу, Финляндия.
2.4. МЕРНАЯ ПОСУДА
67
Бюретки
Бюретки применяются для точного дозирования переменных
объемов растворов. Их главная сфера применения — титрова-
ние, в ходе которого к раствору пробы непрерывно добавляют
стандартный раствор реагента до тех пор, пока не будет достиг-
нута конечная точка титрования, характеризующая заверше-
ние реакции. Обычные бюретки для титрования имеют шкалу
объемов, оцифрованную от 0 до 50 мл с ценой деления 0,1 мл.
Такая бюретка изображена на рис. 2.13. Пользуясь шкалой,
можно считывать показания до сотых миллилитра по положе-
нию мениска между делениями (с точностью ±0,02-0,03 мл).
Выпускаются также бюретки объемом 10,25 и 100 мл и микро-
бюретки объемом до 2 мл. Цена деления таких микробюреток
составляет 0,01 мл, что позволяет считывать показания до тысяч-
ных долей миллилитра. Ультрамикробюретки объемом 0,1 мл
имеют цену деления 0,001 мл (1 мкл); их используют для титро-
вания микролитровых объемов растворов.
Как и для пипеток, для бюреток обычных типов толщина
пленки жидкости на стенках влияет на точность дозировки.
Этот фактор может вносить существенные погрешности, если
скорость истечения жидкости не постоянна. Рекомендуется до-
бавлять жидкость по каплям с достаточно низкой скоростью,
15-20 мл в минуту, а по завершении титрования подождать не-
сколько секунд для полного стекания жидкости со стенок. На
практике вблизи конечной точки титрования скорость состав-
ляет всего несколько капель в минуту, поэтому специально
ждать, пока жидкость стечет, нет необходимости. В непосред-
ственной близости конечной точки можно добавлять жидкость
даже меньшими порциями, чем одна капля, если слегка открыть
и тут же закрыть дозирующий кран, после чего прикоснуться кончиком бюретки
к стенке сосуда. Порцию жидкости, приставшую к стенке, следует смыть в тит-
руемый раствор дистиллированной водой.
Работа с мерной посудой и уход за ней
Мы уже упоминали некоторые меры предосторожности при работе с мерными
колбами, пипетками и бюретками. Более подробно об этом расскажет ваш пре-
подаватель. В данном разделе обсуждим лишь общие правила работы с мерной
посудой.
Важнейшее значение имеет чистота внутренней поверхности посуды. Если
внутренняя поверхность покрыта слоем грязи или жира, жидкость не будет сте-
кать равномерно, в результате чего образуются отдельные капельки на стенках.
В этом случае калибровка посуды будет неправильной. Для начала следует не-
сколько раз промыть посуду изнутри водой. Затем можно промыть ее разбавлен-
ной азотной кислотой и снова несколько раз водой. Горлышки мерных колб
Рис. 2.13
Стандартная
бюретка
68
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
удобно мыть при помощи ершика, но при этом необходимо следить за тем, что-
бы не поцарапать внутренние стенки. Пипетки можно промывать небольшими
порциями раствора моющего средства, поворачивая их, чтобы омыть всю поверх-
ность. В продаже имеются специальные высокоэффективные моющие растворы.
Пипетки и бюретки следует как минимум дважды ополоснуть тем раство-
ром, которым они будут заполнены. Если пипетка или бюретка мокрая, ее следу-
ет сначала ополоснуть дистиллированной водой, а затем как минимум трижды
используемым раствором. Объем, идущий на каждое ополаскивание, должен со-
ставлять приблизительно одну пятую часть объема пипетки или бюретки. Не забы-
вайте предварительно ополаскивать пипетки и бюретки раствором, которым
будете их заполнять! Мерные колбы следует отмыть от следов предыдущего
раствора двумя-тремя порциями дистиллированной воды. Поскольку в дальней-
шем мерная колба будет заполнена водой до метки, высушивать ее нет необхо-
димости.
Обратите внимание, что с аналитической мерной посудой нельзя поступать
так, как часто обращаются с посудой в лабораториях по органическому синтезу:
высушивать в сушильном шкафу (это может привести к изменению ее объема),
при помощи полотенца, ополаскивать летучим органическим растворителем,
например, ацетоном (все это может вызвать загрязнение). Как правило, и не тре-
буется, чтобы мерная посуда была сухой. Вместо высушивания следует просто
ополоснуть посуду тем раствором, который будет в нее помещен.
При считывании показаний объемов следует избегать погрешностей парал-
лакса, связанных с неправильным взаимным расположением глаза, мениска и
шкалы. Это относится к считыванию показаний любой шкалы, в частности,
стрелочного указателя на аналитических весах. Правильное расположение гла-
за — на уровне мениска. Если глаз расположен выше мениска, считанное значе-
ние будет меньше истинного, и наоборот.
После работы с мерной посудой ее следует немедленно промыть дистилли-
рованной водой. Сухую посуду необходимо сначала промыть раствором моюще-
го средства. Мерные колбы при хранении закрывают пробками (рекомендуется
предварительно заполнить их дистиллированной водой). Бюретки перед хране-
нием заполняют дистиллированной водой и закрывают резиновыми пробками.
Как получить правильные и хорошо воспроизводимые
результаты титрования: общие указания
Если кран бюретки изготовлен из тефлона, смазывать его не нужно. Убедитесь,
что он вставлен достаточно плотно, для того чтобы раствор не подтекал, но не
настолько плотно, чтобы его было трудно поворачивать. Если кран изготовлен
из притертого стекла, его возможно придется смазать. Для этого поверхность
крана покрывают тонким слоем специальной смазки (не силиконовой). Вблизи
отверстия этот слой должен быть особенно тонким; необходимо следить, чтобы
смазка не попала в отверстие. После смазывания кран вставляют и вращают не-
сколько раз. Слой смазки должен быть прозрачным, а раствор не должен подте-
кать. Если слой смазки слишком толстый, смазка может попасть внутрь бюретки
2.4. МЕРНАЯ ПОСУДА
69
или закупорить отверстие крана. Из кончика бюретки или отверстия крана лиш-
нюю смазку удаляют при помощи тонкой проволоки. Повторим, что тефлоно-
вые краны не нуждаются в смазке. После того как кран подготовлен к работе,
бюретку заполняют выше нулевой отметки и открывают кран, чтобы заполнить
раствором носик. Проверьте, чтобы в носике бюретки не было пузырей воздуха.
В процессе титрования они могут выйти наружу и вызвать погрешность результа-
тов. Если пузыри есть, их надо выпустить под напором струи раствора, несколько
раз резко открывая и закрывая кран. Не должно быть пузырей и в самом объеме
бюретки. Если они все же есть, это означает, что бюретка скорее всего грязная.
Для установления начального уровня жидкости следует медленно спустить
раствор до нулевой отметки. Затем необходимо подождать и убедиться, что вся
жидкость стекла со стенок. Начальное показание, если оно отлично от нуля, сле-
дует считать с точностью до 0,02 мл (для бюреток объемом 50 мл). Для удобства
считывания показаний следует поместить темный предмет (можно даже палец)
за бюреткой на уровне мениска либо воспользоваться специальным осветителем
мениска (рис. 2.14). Осветитель мениска состоит из белого (сверху) и черного
(снизу) полей. Их границу необходимо расположить точно на уровне мениска.
При считывании показаний избегайте погрешностей параллакса.
Титруемый раствор помещают в коническую колбу. Колбу ставят на поверх-
ность белого цвета, а бюретку располагают над горлышком колбы. Раствор пе-
ремешивают, вращая колбу правой рукой, а краном управляют левой рукой
(рис. 2.15). При обхвате крана, показанном на рисунке, создается некоторое дав-
ление и предотвращается подтекание раствора. Для более эффективного переме-
шивания раствора можно использовать магнитную мешалку.
В ходе титрования индикатор сначала изменяет окраску в непосредствен-
ной близости от места падения капли титранта, поскольку там создается его ло-
кальная избыточная концентрация. При перемешивании титрант равномерно
РИС. 2.14. Осветитель мениска для бюреток Рис. 2.15. Правильное выполнение титрования
70
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
распределяется по всему объему колбы и взаимодействует с титруемым веще-
ством, поэтому исходная окраска индикатора быстро восстанавливается. По
мере приближения к конечной точке это восстановление окраски протекает все
медленнее, и раствор необходимо перемешивать все более тщательно. Перед
самой конечной точкой титрование прекращают и смывают раствор со стенок
колбы дистиллированной водой. Как уже говорилось, показания объема счи-
тывают с точностью до 0,02 мл, но объем капли может быть больше (от 0,02 до
0,05 мл). Поэтому перед самым концом титрования капли следует добавлять по
частям. Для этого кран очень медленно поворачивают до тех пор, пока из носи-
ка не появится часть капли, после чего кран закрывают. Затем прикасаются но-
сиком к стенке колбы и смывают титрант в колбу дистиллированной водой.
Можно также перенести часть капли из носика бюретки в колбу при помощи
стеклянной палочки с оттянутым концом. Признаком окончания титрования
является резкое и устойчивое (в течение по меньшей мере 30 с) изменение
окраски раствора.
Как правило, титрование повторяют три раза. Предварительно можно вы-
полнить ориентировочное титрования для приближенной оценки требуемого
объема титранта. Знание этой величины позволит ускорить процесс последую-
щего титрования.
По завершении титрования остатки титранта следует вылить. Возвращать
их обратно в сосуд с раствором титранта нельзя.
Если для индикации конечной точки титрования измеряют какое-либо фи-
зическое свойство, например, потенциал, то титруемый раствор перемешивают
магнитной мешалкой.
Точность и допустимые погрешности объема мерной посуды
Американским Национальным институтом стандартов и технологий (NIST)
установлены максимально допустимые абсолютные значения погрешностей
объемов для различных типов мерной посуды. Некоторые из них приведены на
переднем форзаце данного тома. Для посуды объемом около 25 мл относитель-
ные погрешности составляют порядка одной части на тысячу (0,1%), для посу-
ды меньшего объема погрешности выше. Буква «А» на мерной колбе, пипетке
или бюретке, свидетельствует о том, что посуда удовлетворяет классу точно-
сти А. Однако это ничего не говорит о реальной точности дозировки объема. В
продаже имеется специальная посуда, удовлетворяющая требованиям точности
NIST или сертифицированная NIST, но она значительно дороже, чем несертифи-
цированная. Более дешевая посуда может характеризоваться погрешностями
вдвое большими, чем установлено требованиями NIST. В то же время не со-
ставляет большого труда самостоятельно откалибровать такую посуду. При
этом будет достигнута точность дозировки объема, сопоставимая с требовани-
ями NIST или даже превышающая ее (см. практическую работу 2). Для боль-
шинства анализов достаточно использовать посуду класса точности А. Ее
можно дополнительно градуировать так, чтобы точность удовлетворяла
требованиям NIST.
2.4. МЕРНАЯ ПОСУДА
71
Для бюреток объемом 50 мл точность считывания показаний составляет
±0,02 мл. Поскольку показания бюретки всегда считывают дважды, общая абсо-
лютная неопределенность результата может достигать ±0,04 мл. Относительная
неопределенность (погрешность) в этом случае обратно пропорциональна вели-
чине объема. Очевидно, что при использовании 50-мл бюретки для снижения от-
носительной погрешности до 0,1% необходимо, чтобы объем титранта составил
не менее 40 мл. Если объем титранта меньше, следует использовать бюретки ме-
ньшего объема. Пипетки также характеризуются определенной погрешностью
считывания показаний. В отличие от бюреток, показания пипеток считывают то-
лько один раз. Погрешности, связанные с каждым считыванием показаний, на-
капливаются. См. гл. 3. «Наложение погрешностей».
Калибровка мерной посуды:
как достигнуть максимальной точности
Пример 2.1 иллюстрирует, как можно использовать уравнение (2.1) для калиб-
ровки мерной посуды, чтобы ввести поправку на выталкивающую силу воздуха,
т. е. привести массу воды, используемой для калибровки, к ее массе в вакууме.
Для нахождения объема посуды следует разделить массу содержащейся в ней
воды (в вакууме) на плотность воды при соответствующей температуре.
В табл. 2.4 приведены значения массы в вакууме для количества воды, масса
которой составляет один грамм в воздухе при атмосферном давлении и разных
температурах. Поправки рассчитаны для стальных разновесов плотностью
7,8 г/см3. Указанные значения используют для расчета объема мерной посуды
при ее калибровке из результатов взвешивания воды, содержащейся в ней или
вытекающей из нее. При использовании латунных разновесов плотностью
8,4 г/см3 значения мало отличаются от приведенных в таблице (см. пример 2.2).
В табл. 2.4 приведены также небольшие поправки на температурное расширение
или сжатие материала посуды — боросиликатного стекла (Pyrex или Kimax). Та-
кие стекла характеризуются объемным коэффициентом расширения порядка
0,000025 на градус Цельсия при температурах вблизи стандартной (20 °C). Это
приводит к изменению объема примерно на 0,0025% при изменении температу-
ры на один градус. Например, для объема 1 мл изменение составит 0,000025 мл.
Для воды изменение объема вблизи 20 °C составляет порядка 0,02% на один гра-
дус. Для пересчета значений объемов (и концентраций) с учетом расширения
(сжатия) воды можно использовать значения плотности воды при разных темпе-
ратурах, также приведенные в табл. 2.4.
Табл. 2.4 с введенными в нее формулами есть на прилагаемом к книге ком-
пакт-диске. Вы можете заменить значение в ячейке столбца В на реальный вес
воды (в колбе, пипетке или бюретке), который вы измерили при соответствую-
щей температуре, и автоматически рассчитать объем посуды при той же темпе-
ратуре и при 20 °C. Порядок работы с электронными таблицами описан в гл. 3.
На компакт-диске находятся также таблица и рисунок, изображающие зависи-
мость относительной погрешности взвешивания (по отношению к массе в ваку-
уме) от плотности объекта.
72
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
Таблица 2.4
Данные для калибровки мерной посуды
А В _ С 0 Е F G н
1 Данные для калибровки мерной посуды
2 Вес в вакууме рассчитан применительно к стальным разновесам плотностью 7,8 г/см3.
3 Температурный коэффициент расширения боросиликатного стекла составляет 0,000025 мл/мл/°С.
4 I
5 Скопируйте эту таблицу, находящуюся на компакт-диске (глава 2), и используйте ее для расчетов объемов
6 мерной посуды по результатам калибровки. Для этого в ячейку столбца В, соответствующую заданной
7 температуре Т (столбец А), занесите вес воды, содержащейся в мерной посуде и измеренный на воздухе
8 при этой температуре. После этого в той же строке будут автоматически рассчитаны значения веса воды
9 в вакууме при температуре Т (столбец D) и при 20 °C (столбец F). Округлите рассчитанные значения до
10 необходимого числа значащих цифр (обычно четырех или пяти).
11
12 Т, °C Вес воды Вес в Объем Расширение стекла Объем при 20 °C, Плотность воды,
13 на воздухе, г вакууме, г при Т, мл по отн. к 20 °C, мл мл г/мл
14 10 1,0000 1,0010 1,0013 -0,000250 1,0016 0,9997026
15 11 1,0000 1,0010 1,0014 -0,000225 1,0017 0,9996081
16 12 1,0000 1,0010 1,0015 -0,000200 1,0017 0,9995004
17 13 1,0000 1,0010 1,0017 -0,000175 1,0018 0,9993801
18 14 1,0000 1,0010 1,0018 -0,000150 1,0020 0,9992474
19 15 1,0000 1,0010 1,0019 -0,000125 1,0021 0,9991026
20 16 1,0000 1,0010 1,0021 -0,000100 1,0022 0,9989460
21 17 1,0000 1,0010 1,0023 -0,000075 1,0023 0,9987779
22 18 1,0000 1,0010 1,0025 -0,000050 1,0025 0,9985896
23 19 1,0000 1,0010 1,0026 -0,000025 1,0027 0,9984082
24 20 1,0000 1,0010 1,0028 0,000000 1,0028 0,9982071
25 21 1,0000 1,0010 1,0031 0,000025 1,0030 0,9979955
26 22 1,0000 1,0010 1,0033 0,000050 1,0032 0,9977735
27 23 1,0000 1,0010 1,0035 0,000075 1,0034 0,9975415
28 24 1,0000 1,0010 1,0038 0,000100 1,0037 0,9972995
29 25 1,0000 1,0010 1,0040 0,000125 1,0039 0,9970479
30 26 1,0000 1,0010 1,0043 0,000150 1,0041 0,9967867
31 27 1,0000 1,0010 1,0045 0,000175 1,0044 0,9965162
32 28 1,0000 1,0010 1,0048 0,000200 1,0046 0,9962365
33 29 1,0000 1,0010 1,0051 0,000225 1,0049 0,9959478
34 30 1,0000 1,0010 1,0054 0,000250 1,0052 0,9956502
35
36 В ячейки, выделенные жирным шрифтом, введены формулы, приведенные ниже. Они скопированы вниз для
37 всех температур. Об установке таблицы см. главу 3.
38 Ячейка С14: Wvac = Wair + Wair(0,0012/Do - 0,0012/Dw) = Wair(0,0012/1,0 + 0,0012/7,8)
39 L = В14+В14*(0,0012/1,0-0,0012/7,8) скопирована вниз
40 Ячейка D14: \/т(мл) = WvacT(r)/DT(r/M л)
41 C14/G14 С скопирована вниз
42 Ячейка Е14: Расш. стекла = (Т - 20) (град.) • 0,000025 (мл/мл/град.) • Wajr(r)
43 1 | (А14-20)*0,000025*В14 скопирована вниз
44 Ячейка F14: V20rc=VT - Glassexp= D14-E14 скопирована вниз
2.4. МЕРНАЯ ПОСУДА
73
Пример 2.3
а) Пользуясь табл. 2.4, рассчитайте объем пипетки (номинальное значение
20 мл) по результатам взвешивания воды в воздухе из данных примера 2.1. При-
мите, что взвешивание проводили при 23 °C. б) Приведите полученный объем к
20 °C (изменения обусловлены сжатием стекла), в) Сравните значения объемов,
рассчитанные из величины веса на воздухе и из величин веса в вакууме и плот-
ности воды.
Решение
а) Из данных табл. 2.4 при 23 °C объем воды весом один грамм (на воздухе) со-
ставляет 1,0035 мл. Объем колбы составит
19,994 г • 1,0035 мл/г = 20,064 мл
б) Уменьшение объема за счет сжатия стекла при изменении температуры от
23 °C до 20 °C оставляет 0,0015 мл (0,000025 мл/мл/°С • 20 мл • 3 °C), поэтому
объем пипетки при 20 °C составит 20,062 мл.
в) Плотность воды при 23 °C равна 0,99754 г/мл, поэтому из величины веса в ва-
кууме
20,015 г / 0,99754 г/мл = 20,064 мл
получено то же значение, что и в пункте (а).
Пример 2.4
Вы приготовили раствор соляной кислоты и стандартизировали его титровани-
ем первичного стандарта — раствора карбоната натрия. В процессе титрования
температура воздуха в помещении составляла 23 °C. Найденное значение кон-
центрации — 0,11272 М. После этого в лаборатории вышла из строя отопитель-
ная система, и, когда вы использовали эту кислоту для титрования, температура
упала до 18 °C. Какой при этом стала концентрация титранта?
Решение
М 18 °с = м23 °с • (А8 °с /7?23 сс) = о,11272 • (0,99859/0,99754) = 0,11284 М
(о значащих цифрах и о том, что означает цифра нижнего индекса результата,
см. гл. 3).
74
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
Техника калибровки мерной посуды
Как правило, результаты калибровки мерной посуды представляют с пятью зна-
чащими цифрами, что соответствует максимально возможной точности дози-
ровки объемов. Следовательно, чистая масса воды также должна быть измерена
не менее, чем с пятью значащими цифрами. Это означает, что, если объем посу-
ды превышает 10 мл, то достаточно взвешивания с точностью до 1 мг. Такое
взвешивание можно легко и быстро осуществить на технических весах, не при-
бегая к использованию более точных аналитических весов. Иногда бывает до-
статочно и четырех значащих цифр (о значащих цифрах см. гл. 3). Например,
калиброванный объем пипетки с номинальным объемом 10 мл может составлять
9,997 мл. Это значение столь же точное, как и 10,003 мл.
1. Калибровка мерных колб. Взвешивают чистую и сухую колбу вместе с
пробкой. Затем колбу заполняют до метки дистиллированной водой. На гор-
лышке колбы не должно быть капель. Если они есть, их удаляют фильтроваль-
ной бумагой. Температура колбы с водой должна равняться комнатной. После
этого взвешивают колбу с водой и измеряют температуру воды с точностью до
0,1 °C. Разность результатов взвешивания равна весу на воздухе воды, содержа-
щейся в колбе.
2. Калибровка пипеток. Взвешивают сухую коническую колбу, закрытую ре-
зиновой пробкой, или бюкс, закрытый стеклянной пробкой (выбор сосуда зави-
сит от объема воды, который предстоит взвесить). Пипетку заполняют водой
(температура которой известна), опорожняют в предварительно взвешенный со-
суд, используя стандартную технику пипетирования, и быстро закрывают сосуд,
чтобы избежать потерь воды вследствие испарения. Сосуд с водой взвешивают и
рассчитывают значение веса на воздухе воды, отмеренной пипеткой.
3. Калибровка бюреток. Бюретки калибруют аналогично пипеткам, но в этом
случае взвешивают несколько порций воды. Внутренняя поверхность бюретки
может не быть строго цилиндрической, поэтому дозируемый объем может отли-
чаться в ту или иную сторону от номинального. Это различие может быть раз-
ным для разных объемов. Поэтому рекомендуется откалибровать бюретку на
несколько значений объемов с шагом 20% от общего объема (например, 10 мл
для бюретки объемом 50 мл). Рекомендуется каждый раз заполнять бюретку во-
дой до нулевой отметки и отмерять очередную порцию воды в сухую колбу для
взвешивания. Это позволяет предотвратить потери воды вследствие испарения.
Можно заполнить бюретку один раз и отмерять последовательные порции в
одну и ту же колбу, но в этом случае все операции надо проводить быстро. Объ-
ем воды для взвешивания должен быть точно известен и близок к номинально-
му, но не обязательно в точности равен ему. Поэтому опорожнять бюретку
можно быстро, но после дозирования каждой порции следует подождать 10-20 с
для полного стекания воды со стенок и записать точное значение объема. По-
правки к значениям объема откладывают в виде зависимости от самого значения
2.4. МЕРНАЯ ПОСУДА
75
объема и соединяют полученные точки ломаной линией. Из полученного графи-
ка интерполяцией можно найти поправку для любого значения объема. Как пра-
вило, для пипеток объемом 50 мл поправки к номинальным значениям находятся
в пределах ±0,05 мл.
Пример 2.5
Бюретку объемом 50 мл калибруют, отбирая воду порциями по 10 мл. Бюретку
каждый раз заполняют водой до нулевой отметки. Результаты калибровки при-
ведены ниже.
Показания бюретки (мл) Вес порции воды (г)
10,02 10,03
20,08 20,03
29,99 29,85
40,06 39,90
49,98 49,86
Постройте зависимость поправки к объему от величины объема порции. Темпе-
ратура воды 20 °C, при взвешивании использованы стальные разновесы.
Решение
Из табл. 2.4 (или электронной таблицы 2.4 на компакт-диске) находим:
Wvac = 10,03 + 10,03 (0,00105) = 10,03 + 0,01 = 10,04 г
VT= 10,04 г / 0,9982 г/мл = 10,06 мл
Аналогично рассчитываем другие значения. Получаем данные для построения
требуемой зависимости:
Номинальный объем (мл) 10,02 20,08 29,99 40,06 49,98 Реальный объем (мл) Поправка (мл) 10,06 +0,04 20,09 +0,01 29,93 -0,04 40,01 -0,05 50,00 0,00
Эти данные откладывают в виде графика (по оси х — номинальные значения
объема, по оси у—поправки). Номинальные значения можно принять округлен-
но равными 10, 20, 30, 40 и 50 мл.
76
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
Выбор мерной посуды: насколько точный объем должен быть?
Как и при взвешивании, при измерении объема возникают ситуации, когда его
значение должно быть известно точно, и ситуации, когда требуется лишь прибли-
женное значение. Например, приготовить стандартный раствор 0,1 М НС1 невоз-
можно путем разбавления до определенного значения точно отмеренного объема
концентрированной соляной кислоты, поскольку концентрация соляной кислоты
в исходном растворе не известна с требуемой точностью. Полученный разбавлен-
ный раствор все равно необходимо будет стандартизовать. Поэтому в данном слу-
чае объем концентрированной кислоты можно отмерить приближенно (и затем
провести стандартизацию разбавленного раствора). Из таблицы, приведенной на
переднем форзаце данного тома, видно, что концентрация коммерческих препара-
тов концентрированной соляной кислоты составляет около 12,4 М. Чтобы приго-
товить 1 л раствора с концентрацией около 0,1 М, необходимо отмерить примерно
8,1 мл концентрированной кислоты и разбавить до 1 л. Точное измерение объема
этой порции (а также объема воды, используемой для разбавления) было бы в
этом случае пустой тратой времени. Для измерения объема кислоты вполне доста-
точно использовать градуированный мерный цилиндр объемом 10 мл (или граду-
ированную пипетку того же объема), а разбавлять кислоту можно в любом, даже
неградуированном, сосуде объемом 1 л. С другой стороны, если требуется точно
разбавить исходный стандартный раствор, его объем необходимо отмерить мер-
ной пипеткой, а разбавление проводить в мерной колбе. Любые измерения объе-
мов, значения которых используются для расчетов содержаний веществ, следует
проводить с определенной для каждого конкретного случая точностью. Как пра-
вило, эта точность составляет четыре значащие цифры. Чтобы достичь такой
точности, необходимо использовать мерные пипетки и мерные колбы. Высокая
точность требуется и при взятии навесок (пробы и реактивов для приготовления
стандартных растворов), а также при разбавлении. Для точной дозировки пере-
менных объемов, как и при титровании, используют бюретки. Приготовление
растворов реактивов, необходимых лишь для создания определенных условий
(например, буферов для контроля pH), не требует высокой точности. Для этого
можно использовать мерную посуду меныпей точности, например, мерные ци-
линдры. С высокой точностью необходимо измерять только те значения объе-
мов, которые будут использованы для расчета содержания вещества.
2.5. Приготовление стандартных растворов щелочей
В качестве щелочного титранта обычно используют гидроксид натрия. Он все-
гда содержит значительные количества воды и карбоната натрия и, следователь-
но, не может являться первичным стандартом. Для выполнения точных работ
NaOH необходимо освободить от карбоната, поскольку в ходе титрования кар-
бонат натрия создает буферную систему. В результате этого переход окраски
индикатора в конечной точке становится менее четким. Кроме того, при исполь-
зовании индикатора фенолфталеина титрование раствором NaOH, содержащим
2.6. ПРИГОТОВЛЕНИЕ СТАНДАРТНЫХ РАСТВОРОВ КИСЛОТ
77
карбонат, приводит к систематической погрешности, поскольку в этих условиях
ионы СО3“ титруются только до НСО3 (поэтому NaOH предпочитают титровать
с метиловым оранжевым: при этом ионы СО3” титруются до СО2). Иными сло-
вами, кажущаяся молярная концентрация раствора щелочи увеличивается из-за
того, что образующиеся ионы НСО3 также способны титроваться.
Карбонат натрия практически нерастворим в концентрированных, близких
к насыщенным, растворах гидроксида натрия. Для удаления карбоната натрия
навеску NaOH растворяют в объеме воды, численно равном (в мл) массе NaOH в
граммах. Раствор оставляют на несколько дней для осаждения Na2CO3 и акку-
ратно декантируют* прозрачный маточный раствор. Маточный раствор можно
также отфильтровать через фильтрующий тигель Гуча с асбестовой прокладкой
(нельзя промывать фильтр после фильтрования). Однако асбест является канце-
рогенным материалом, поэтому первый способ предпочтительнее. Таким спосо-
бом нельзя освободить от карбонатов растворы КОН, так как К2СО3 растворим в
концентрированных растворах КОН.
Вода также содержит растворенный СО2, поглощенный из воздуха. Как пра-
вило, в ходе серийных анализов, не требующих высокой точности, погрешности,
вызываемые примесями карбонатов или СО2 в воде, можно считать пренебрежимо
малыми. Если же требуется высокая точность, СО2 необходимо удалить из всей
воды, используемой для приготовления растворов (особенно щелочных) для
кислотно-основного титрования. Это проще всего сделать при помощи кипяче-
ния с последующим охлаждением под струей воды.
Растворы гидроксида натрия обычно стандартизуют титрованием точных
навесок первичного стандарта — гидрофталата калия (КНР). Гидрофталат-ион
является слабой кислотой (Ка = 4 • 10-6), по силе сопоставимой с уксусной. Для
индикации конечной точки используют фенолфталеин. Растворы гидроксида
натрия хранят в закрытых пластмассовых сосудах для предотвращения погло-
щения СО2 из воздуха. Если же сосуд должен быть открытым (например, бу-
тыль, снабженная сифоном), ее предохраняют от контакта с воздухом при помо-
щи трубочки, заполненной аскаритом (асбестом, пропитанным NaOH). О приго-
товлении и стандартизации раствора NaOH см. практическую работу 6.
2.6. Приготовление стандартных растворов кислот
Для титрования оснований обычно применяют растворы соляной кислоты. Бо-
льшинство хлоридов растворимы в воде, при использовании этого титранта по-
бочные реакции протекают редко. С ним удобно работать. Как правило, соляная
кислота не может являться первичным стандартом (хотя иногда в качестве пер-
вичного стандарта используют азеотропную смесь НС1 и воды, кипящую при
* Раствор следует хранить в пробирке или другом подходящем сосуде, закрытой пробкой для предотвраще-
ния поглощения углекислоты воздуха. В противном случае углекислый газ будет продолжать реагировать
с гидроксидом натрия, образуя карбонат. Нельзя использовать стеклянные пробки: они «схватятся» кон-
центрированным раствором щелочи. Можно использовать резиновые пробки.
78
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
постоянной температуре). Поэтому готовят растворы соляной кислоты прибли-
зительной концентрации простым разбавлением концентрированной НС1. Для бо-
лее точных работ воду, используемую для разбавления, следует прокипятить,
хотя в данном случае это не имеет столь большого значения, как при приготов-
лении растворов NaOH. Кроме того, СО2 плохо растворим в сильнокислых рас-
творах и улетучивается из них при перемешивании.
Для стандартизации растворов НС1 обычно используют растворы первично-
го стандарта карбоната натрия. Его недостатком является нечеткий переход
окраски при использовании большинства индикаторов (метилового красного,
метилового пурпурного и т. д.). Чтобы переход стал более резким, вблизи конеч-
ной точки раствор нагревают до кипения. При использовании модифицированно-
го метилового оранжевого нагревания можно избежать, но переход недостаточно
резкий и в этом случае. Еще один недостаток карбоната натрия — малая моляр-
ная масса эквивалента Na2CO3. Другим первичным стандартом может служить
трис(гидроксиметил)аминометан (THAM) (HOCH2)3CNH2, он негигроскопичен
и более удобен в работе. Однако это вещество представляет собой достаточно
слабое основание (Кь =1,3-10-6) и также имеет малую молярную массу. При его
использовании индикация конечной точки не осложняется выделением СО2.
Этот первичный стандарт рекомендуется использовать всегда, кроме тех случа-
ев, когда раствор НС1 будет затем использован для титриметрического определе-
ния карбонатов. О приготовлении и стандартизации растворов соляной кислоты
см. практическую работу 7.
Если имеется стандартизованный раствор NaOH, можно стандартизировать
НС1 путем титрования ее аликвоты этим раствором. Переход окраски индикато-
ра в конечной точке очень резкий, титрование в этом случае можно выполнить
быстрее. Но раствор NaOH является вторичным стандартом. Концентрация
вторичных стандартов известна менее точно, чем первичных. Любая погреш-
ность, допущенная в ходе его стандартизации, скажется и на значении концент-
рации НС1. Следует титровать НС1 раствором щелочи, а не наоборот, для того,
чтобы уменьшить возможное поглощение СО2 раствором в колбе для титрова-
ния. При использовании NaOH индикаторами могут служить фенолфталеин или
бромтимоловый синий.
2.7. Другая аналитическая посуда и аппаратура:
подготовка и обработка проб
Помимо устройств и посуды для измерения массы и объема, при выполнении
анализов широко используют и другое оборудование.
Отбор проб крови
Для отбора проб крови используют шприцы.* Стеклянный или пластмассовый
шприц обычно снабжен иглой из нержавеющей стали или алюминия. При отбо-
* Вы не имеете права самостоятельно отбирать пробы крови, если не обучены этому. Этим занимается спе-
циально обученный персонал.
2.7. ДРУГАЯ АНАЛИТИЧЕСКАЯ ПОСУДА И АППАРАТУРА: ПОДГОТОВКА И ОБРАБОТКА ПРОБ
79
ре проб крови проблем, связанных с их загрязнением, как правило, не возникает.
Лишь в некоторых случаях при определении следовых количеств элементов не-
обходимо принимать специальные меры предосторожности. Вместо шприцов
часто используют специальные вакуумированные пробирки, закрытые резино-
вой пробкой. При их использовании один конец иглы погружают в вену, а другим
протыкают пробку. При этом порция крови втягивается в вакуумированное про-
странство. Если надо проанализировать цельную кровь или плазму крови, то в
пробирке может находиться антикоагулянт для предотвращения свертывания.
Если для анализа требуются небольшие количества крови, то кровь берут не
из вены, а из пальца. Из пальца можно выдавить до 0,5 мл крови или немного бо-
льше. Кровь собирают в маленькую пробирку, проколов палец стерилизован-
ным острым предметом.
Эксикаторы
Эксикаторы предназначены для хранения проб в сухой атмосфере в процессе
их охлаждения и перед взвешиванием. Иногда эксикаторы используют для вы-
сушивания влажных проб. В эксикаторах охлаждают образцы и посуду, которые
подвергались нагреванию (прокаливанию, высушиванию). Если анализируемые
пробы или реагенты подвергались нагреванию, то перед взвешиванием их необ-
ходимо охладить в эксикаторе. Стандартный вид стеклянного эксикатора изоб-
ражен на рис. 2.16. Эксикатор представляет собой герметично закрытый сосуд, в
котором поддерживается атмосфера с низкой влажностью. На дно эксикатора
помещают осушитель (например, хлорид кальция), поглощающий влагу. Осу-
шитель необходимо периодически менять, так как он со временем приходит в
негодность. Признаком этого служит видимое слеживание или расплывание
осушителя. Внутри эксикатора находится фарфоровая подставка с отверстиями,
в которые помещают бюксы, тигли и другую посуду перед взвешиванием. Герме-
тичность эксикатора обеспечивают при помощи стеклянной крышки с притертыми
краями, которые смазывают специальной смазкой. В вакуумных эксикаторах в
крышку вставлена трубка с краном для откачивания воздуха. В таких эксикато-
рах содержимое хранится в вакууме.
Крышку эксикатора следует открывать лишь на непродолжительное время
и только при необходимости. При каждом открывании крышки внутрь эксикато-
ра попадает атмосферный воздух, удаление влаги из которого происходит доста-
Рис. 2.16
Эксикатор и подставка
для него
80
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
точно медленно. Если эксикатор долго держать открытым, осушитель быстро
придет в негодность. Тигли и другую посуду, нагретую докрасна, перед помеще-
нием в эксикатор необходимо охладить на воздухе около 60 с. В противном слу-
чае воздух внутри эксикатора сильно нагреется еще до того, как крышка будет
закрыта, и при последующем его охлаждении внутри эксикатора образуется раз-
режение. Это, в свою очередь, приведет к тому, что при снятии крышки эксика-
тора атмосферный воздух резко проникнет внутрь, в результате чего частицы
пробы могут рассыпаться, потеряться. Нагретые бюксы следует хранить в экси-
каторе, не закрывая их крышками, иначе при охлаждении разрежение создастся
в бюксе, и крышку бюкса невозможно будет открыть. Крышку бюкса охлажда-
ют в эксикаторе рядом с самим бюксом.
В табл. 2.5 приведены наиболее распространенные осушители и их свойст-
ва. Отработанные оксид алюминия, перхлорат магния, оксид кальция, хлорид
кальция и силикагель можно регенерировать, нагревая их до 150,240, 500,275 и
150 °C соответственно.
Печи и сушильные шкафы
Муфельные печи (рис. 2.17) предназначены для прокаливания проб при высо-
ких температурах для перевода осаждаемых форм в гравиметрические и для
сжигания органической матрицы перед определением неорганических веществ.
Муфельные печи должны быть снабжены средствами регулирования температу-
ры, поскольку при температурах выше 500 °C возможны потери некоторых эле-
ментов. Муфельные печи позволяют создавать температуры до 1200 °C.
Сушильные шкафы используют для высушивания образцов перед взве-
шиванием. Стандартный вид сушильного шкафа показан на рис. 2.18. Для обес-
печения равномерного нагревания сушильные шкафы снабжены эффективной
Таблица 2.5
Некоторые распространенные осушители
Осушитель Емкость Расплывается* Торговое наименование
СаС12 (безводный) Высокая Да
CaSO4 Умеренная Нет Drierite (W. A. Hammond Drierite Со.)
СаО Умеренная Нет
MgClO4 (безводный) Высокая Да Anhydrone (J. Т. Baker Chemical Co.); Dehydrite (Arthur H. Thomas Co.)
Силикагель Низкая Нет
А12О3 Низкая Нет
р2о5 Низкая Да
При поглощении влаги превращается в жидкость
2.7. ДРУГАЯ АНАЛИТИЧЕСКАЯ ПОСУДА И АППАРАТУРА: ПОДГОТОВКА И ОБРАБОТКА ПРОБ
81
РИС. 2.17. Муфельная печь. С любезного разре-
шения Arthur Н. Thomas Company
РИС. 2.18. Сушильный шкаф. С любезного
разрешения Arthur Н. Thomas Company
системой вентиляции. Рабочая температура, как правило, составляет 110 °C, но
возможно повышение до 200-300 °C.
Вытяжные шкафы и камеры
В вытяжных шкафах проводят выпаривание растворов. Выпаривание хлорной
кислоты или кислых растворов, содержащих перхлораты, проводят в шкафах,
снабженных системой улавливания паров либо специально предназначенных
для работы с хлорной кислотой (т. е. элементы его конструкции должны быть
устойчивы к действию хлорной кислоты).
При определении следовых содержаний, например, следов металлов, необ-
ходимо принимать меры предосторожности во избежание загрязнения образцов.
Однако обычный вытяжной шкаф — это едва ли не самое «грязное» место лабо-
ратории, поскольку в него втягивается весь окружающий воздух, обтекая при
этом пробу. Для создания очень чистых рабочих зон служат ламинарные каме-
ры. Воздух в них подается после предварительной фильтрации, а внутри камеры
создается некоторое избыточное давление, чтобы в нее не проникал нефильтро-
ванный воздух извне. Схематическое устройство стандартной ламинарной каме-
ры изображено на рис. 2.19. В таких камерах используют высокоэффективные
фильтры, задерживающие все аэрозольные частицы размером более 0,3 мкм.
Если в ходе работы выделяются пары, то предпочтительнее работать в камерах с
вертикальным ламинарным потоком. В этом случае пары уносятся током возду-
82
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
Рис. 2.19
Схема ламинарной камеры. С любезного
разрешения Dexion, Inc., 344 Beltine
Boulevard, Minneapolis, MN
Высокоэффективный
воздушный фильтр
П редварител ьн ы й
воздушный фильтр
ха и не попадают на оператора. Существуют специальные средства для поглоще-
ния ядовитых паров.
Промывал ки
Промывалки того или иного типа должны быть в любой аналитической лабора-
тории. Их используют для количественного перенесения твердых веществ или
растворов из сосуда в сосуд, для промывания осадков. В продаже имеются про-
мывалки, разнообразные по типу и размеру. Некоторые из них изображены на
рис. 2.20. Можно самостоятельно сделать промывалку из плоскодонной круглой
колбы и стеклянных трубок (рис. 2.20, б).
РИС. 2.20. Промывалки: а — полиэтиленовая (на сжатие); б — стеклянная (на выдувание)
2.7. ДРУГАЯ АНАЛИТИЧЕСКАЯ ПОСУДА И АППАРАТУРА: ПОДГОТОВКА И ОБРАБОТКА ПРОБ
83
Центрифуги и фильтры
Центрифуги находят множество разнообразных применений. В частности, в
клинических лабораториях с их помощью разделяют кровь на фракции (напри-
мер, отделяют сыворотку). Белки отделяют осаждением, а затем также центри-
фугируют.
Существуют различные типы фильтров для отделения осадков (например, в
гравиметрическом анализе). На рис. 2.21 изображены тигель Гуча, стеклянный и
фарфоровый фильтрующие тигли. Тигель Гуча изготовлен из фарфора; в его
донышке имеются отверстия. На дно такого тигля помещают фильтрующий
диск из пористого стекла. Ранее в тиглях Гуча чаще всего использовали асбесто-
вые фильтры. Но работа с ними была неудобной (такие фильтры прочно удержива-
ют мелкие частицы осадков) и потенциально опасной (сухие асбестовые волокна
обладают канцерогенными свойствами). Дно стеклянного фильтрующего тиг-
ля изготовлено из пористого стекла. Размер пор обозначают буквами F (англ,
fine, мелкопористый), М (англ, medium, среднепористый) или С (англ, coarse,
крупнопористый). Дно фарфорового фильтрующего тигля сделано из порис-
того неглазированного фарфора. Не рекомендуется использовать стеклянные
фильтры при работе с концентрированными щелочными растворами, поскольку
в такой среде они могут разрушаться. Максимальные рабочие температуры для
различных материалов тиглей приведены в табл. 2.1.
Студенистые осадки (такие, как водный оксид железа) нельзя фильтровать
через фильтрующие тигли, поскольку эти осадки забивают их поры. Фильтрова-
ние таких осадков идет медленно даже при использовании бумажных фильтров.
Для фильтрования тигли помещают в специальные держатели, которые
вставляют в горлышко колбы для фильтрования (рис. 2.22). Между колбой для
фильтрования и насосом помещают предохранительную колбу.
Беззольные бумажные фильтры широко используют для количественно-
го получения осадка во всех случаях, когда есть возможность полностью сжечь
фильтр, осадок при этом переходит в форму, пригодную для взвешивания
(см. гл. 10). Существует множество разновидностей фильтровальной бумаги для
работы с осадками разных типов. Виды бумаги, производимые фирмами Whatman
(http://www.whatman.plc.uk) и Schliecher and Schuell (www.s-und-s.de/english-
index.html), перечислены в табл. 2.6.
РИС. 2.21. Фильтрующие тигли: а — Гуча; б — стеклянный; в — фарфоровый
84
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
Таблица 2.6
Виды фильтровальной бумаги
Частицы осадка Whatman Schliecher and Schnell
Очень мелкие (например, BaSO4) № 42 (2,5 мкм) № 589/2 или 5, синяя или красная лента (2-4 мкм)
Средние (например, AgCl) № 40 (8 мкм) № 589/2, белая лента (4-12 мкм)
Студенистые и крупнокристаллические осадки (например, Fe2O3 • хН2О) № 41 (20-25 мкм) № 589/1, черная лента (>12-25 мкм)
РИС. 2.22. Применение держателей для фильтрующих тиглей
Техника фильтрования
Скорость фильтрования можно увеличить за счет выбора подходящего типа фи-
льтровальной бумаги. На рис. 2.23 изображен правильно сложенный фильтр.
Фильтр складывают так, чтобы его дольки, составляющие по фильтра, налага-
лись друг на друга не полностью (зазор должен составлять приблизительно
3 мм). От фильтра с той стороны, которая будет прилегать к воронке, отрывают
уголок размером 5-6 мм. Это обеспечивает плотное прилегание фильтра без пу-
зырьков воздуха. Сложенный фильтр помещают в воронку, расправляют и сма-
чивают водой. Носик воронки заполняют водой и прижимают фильтр к стенкам.
Если все сделано правильно, носик воронки остается заполненным водой, пузы-
рьков не образуется. В этом случае давление столба воды обеспечивает высокую
скорость фильтрования. После подготовки фильтра необходимо немедленно
2.7. ДРУГАЯ АНАЛИТИЧЕСКАЯ ПОСУДА И АППАРАТУРА: ПОДГОТОВКА И ОБРАБОТКА ПРОБ
85
Рис. 2.23
Правильно сложенный бумажный фильтр
приступать к фильтрованию. В ходе фильтрования осадок должен заполнять по-
верхность фильтра не более чем на треть-половину, поскольку частицы осадка
часто имеют склонность «ползти» вверх по фильтру. Нельзя также допускать,
чтобы уровень жидкости поднимался над краем фильтра.
Перед фильтрованием осадок должен осесть на дно стакана. Дайте осадку
отстояться. В этом случае основную массу прозрачного раствора можно пере-
нести на фильтр декантированием. Эта порция отфильтруется очень быстро, по-
скольку поры фильтра еще не забиты частицами осадка.
В ходе декантации и особенно перенесения на фильтр основной массы осад-
ка следите за тем, чтобы не потерять часть осадка. Этого легко избежать, если
переносить осадок при помощи стеклянной палочки и промывалки, как показа-
но на рис. 2.24. В ходе декантации следует сливать раствор по палочке, которая
направляет его непосредственно в фильтр, предотвращая разбрызгивание. Оса-
док легче промывать, пока он еще находится в стакане. Для этого после деканта-
ции маточного раствора обмойте стенки стакана несколькими миллилитрами
промывной жидкости и снова дайте осадку отстояться. Затем декантируйте про-
мывную жидкость и повторите промывание еще два-три раза. После этого пере-
несите на фильтр основную массу осадка, держа стеклянную палочку и стакан
одной рукой, как показано на рис. 2.24, и смывая осадок на фильтр струей про-
мывной жидкости из промывалки.
Если осадок необходимо собрать количественно (например, в ходе грави-
метрического анализа), то последние порции осадка снимают со стенок стакана
при помощи резинового наконечника (рис. 2.25). Остатки осадка со стенок и с
наконечника смывают струей промывной жидкости. Вместо резинового нако-
нечника осадок можно собирать на фильтровальной бумаге. Для этого стенки
стакана протирают небольшим кусочком беззольной фильтровальной бумаги
(удерживая его пинцетом) и переносят его на фильтр.
После того, как весь осадок перенесен на фильтр, его промывают пятью-
шестью небольшими порциями промывной жидкости. Это значительно эффек-
тивнее, чем добавление одного большого объема сразу. Струю жидкости следу-
ет направлять вращательным движением, чтобы по возможности собрать осадок
в углу фильтра. Прежде, чем добавлять следующую порцию, дождитесь полного
стекания предыдущей. Не забудьте проверить полноту осаждения. Проверьте
осадок на полноту промывания при помощи качественной реакции на присутст-
вие осадителя в последних нескольких каплях фильтрата.
86
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
РИС. 2.24. Правильная техника переноса осадка на фильтр
Рис. 2.25. Палочка с резиновым наконечником
2.8. ПРОКАЛИВАНИЕ ОСАДКОВ В ГРАВИМЕТРИЧЕСКОМ АНАЛИЗЕ
87
2.8. Прокаливание осадков в гравиметрическом анализе
Если осадок прокаливают в фарфоровом фильтрующем тигле, из него сначала
следует удалить влагу на малом огне. Прокаливание можно проводить в муфель-
ной печи или на пламени горелки. При использовании горелки фильтрующий
тигель следует поместить в обычный тигель (фарфоровый или платиновый).
В противном случае восстановительные газы пламени будут проникать сквозь
поры фильтра.
Если фильтрование проводили через бумажный фильтр, фильтр с осадком
вынимают из воронки, аккуратно сворачивают и помещают в тигель так, чтобы
часть фильтра, содержащая основную массу осадка, была обращена ко дну
тигля. После этого фильтр необходимо высушить и озолить. Для этого тигель
устанавливают наклонно на угол фарфорового треугольника, слегка приоткрыв
крышку (рис. 2.26). Сначала на малом пламени горелки подсушивают содержи-
мое тигля, следя за тем, чтобы оно не рассыпалось. Затем интенсивность нагре-
вания постепенно увеличивают по мере того, как влага испаряется, а фильтр
обугливается. Следует избегать попадания пламени (обладающее восстанови-
тельными свойствами!) внутрь тигля. Если внезапно появляется большое коли-
чество дыма, это свидетельствует о том, что бумага может загореться. В этом
случае горелку необходимо убрать. Если возгорание все-таки произошло, пламя
следует немедленно погасить, накрыв тигель крышкой. Конечно, при этом на
крышке останутся частички угля, однако впоследствии они выгорят. Наконец,
когда выделение дыма прекратилось, обугленную бумагу полностью сжигают,
постепенно повышая температуру пламени. При этом частицы угля в тигле рас-
каляются, но они не должны давать пламени. Нагревание продолжают до тех
пор, пока полностью не сгорит весь уголь, а также частицы смолы на стенках и
крышке тигля. После этого тигель с осадком можно начинать прокаливать. Да-
льнейшее прокаливание можно также проводить на горелке (при максимально
высокой температуре пламени) либо в муфельной печи. Прокаливание тигля на-
чинайте постепенно.
Прежде чем помещать в тигель осадок или фильтр с осадком, тигель надо
довести до постоянной массы. Тигель высушивают (первый раз нагревая его в
течение часа, последующие разы — в течение получаса), если осадок нужно бу-
дет высушить, и прокаливают, если осадок нужно будет прокалить. Масса счи-
тается постоянной, если последовательные результаты взвешивания тигля на
учебных аналитических весах различаются не более чем на 0,3-0,4 мг. Таким же
образом доводят до постоянной массы тигель с осадком. В этом случае последу-
ющие нагревания также проводят в течение вдвое меньших промежутков време-
ни, чем первое. Перед каждым взвешиванием тигель необходимо охлаждать в
эксикаторе не менее получаса. Если тигель раскалился докрасна, то перед тем,
как помещать в эксикатор, его сначала следует охладить до отсутствия каления.
Тигель берут специальными тигельными щипцами — обычно из нержавеющей
стали или никелированными (во избежание загрязнений ржавчиной). Прежде
чем взвешивать тигель, на всякий случай поднесите к нему руку (не касаясь тиг-
ля!), чтобы убедиться, что от него не исходит тепло.
88
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
Рис. 2.26
Тигель с крышкой на фарфоровом треугольнике
в процессе обугливания фильтра
2.9. Отбор пробы: твердой, жидкой, газообразной
Отбор представительной пробы — это аспект аналитической химии, с которым
начинающий студент часто не сталкивается, поскольку предполагается, что про-
бы, которые ему дают для практических занятий, являются гомогенными и пред-
ставительными. Однако именно эта операция может оказать решающее влияние
на весь ход анализа. Процесс пробоотбора может ограничивать точность и
важность результатов анализа в целом. Потенциально это самое слабое звено во
всей цепочке аналитических операций. От правильного обращения с пробой
крови во время ее отбора и после него иногда может зависеть жизнь пациента.
Если аналитик просто получил пробу, не участвуя сам в процессе ее отбора, то
результаты анализа могут быть квалифицированы лишь как результаты анализа
пробы «какой она была предоставлена». Если необходимо получить что-то бо-
льшее, то вся последовательность операций с пробой с самого начала должна
быть задокументирована.
Многие профессиональные сообщества разработали точные инструкции
для отбора проб различных веществ, в частности, Американское общество по те-
стированию и материалам (ASTM, http://www.astm.org), Международная ассоциа-
ция официальных химиков-аналитиков (АОAC International, http://www.aoac.org),
Американская ассоциация общественного здоровья (АРНА, http://www.apha.org).
При наличии необходимого опыта и применении статистических методов про-
боотбор можно выполнять с той же точностью, что и собственно определение.
О важных статистических аспектах пробоотбора см. гл. 3. К сожалению, ана-
литики часто оставляют эту проблему без должного внимания. В то же время,
степень простоты или сложности пробоотбора зависит от природы пробы.
В первую очередь нужно получить пробу, представительную по отношению
ко всему целому. Такая проба называется генеральной. В зависимости от харак-
тера исходного материала ее размер может варьироваться от нескольких граммов
(или даже менее) до килограммов. После того, как получена представительная ге-
неральная проба, может возникнуть необходимость ее уменьшения до таких раз-
меров, чтобы с ней было удобно работать. Такая уменьшенная проба и называется
собственно пробой (или лабораторной пробой). Для выполнения анализа из нее
берут отдельную порцию — аликвоту. Она называется анализируемой пробой.
2.9. ОТБОР ПРОБЫ: ТВЕРДОЙ, ЖИДКОЙ, ГАЗООБРАЗНОЙ
89
Отбирая отдельные аликвоты, можно выполнить несколько параллельных ана-
лизов одной и той же пробы.
В клинических лабораториях генеральные пробы можно, как правило, непо-
средственно использовать в качестве лабораторных, поскольку генеральные
пробы обычно невелики и однородны (например, пробы мочи или крови). Раз-
мер анализируемой пробы в этом случае составляет от нескольких миллилитров
до менее чем одной капли (нескольких микролитров).
Ниже мы рассмотрим некоторые проблемы, касающиеся отбора генераль-
ных проб твердых, жидких и газообразных веществ.
1. Твердые вещества. Отбор твердых проб — задача наиболее трудная из-за не-
однородностей вещества, различий в размерах его частиц и даже внутренних
различий в пределах одной частицы. Простейший, но, как правило, наименее на-
дежный способ отбора твердых проб — отбор наугад, когда случайным образом
берется проба и предполагается, что она представительна. Такой способ приво-
дит к удовлетворительным результатам только тогда, когда исходный материал
гомогенный. Для большей надежности в качестве генеральной пробы лучше
взять 1/100-1/50 часть вещества (если только оно не является однородным). Чем
больше размер частиц вещества, тем большей должна быть генеральная проба.
Очень простой и в то же время наиболее надежный способ отбора больших
проб твердых веществ — отбирать тогда, когда вещество находится в движении.
В этом случае в дело идет любая отобранная порция. Такой отбор можно
проводить систематически, отбирая отдельные «аликвоты», в сумме представ-
ляющие весь исходный материал. Приведем некоторые примеры.
В ходе погрузки или разгрузки мешков с цементом представительную про-
бу можно получить, отбирая, например, каждый пятидесятый мешок (или порцию
из каждого такого мешка). При транспортировке зерна на тачках можно отби-
рать по лопате зерна из каждой тачки. Все полученные таким путем порции объ-
единяют и получают генеральную пробу.
2. Жидкости. Жидкости, как правило, гомогенны, поэтому отбор жидких проб
проводить намного легче. В результате диффузии жидкость перемешивается
очень медленно. Поэтому, если жидкость негомогенна, перед отбором пробы ее
следует хорошо перемешать. Если жидкий объект гомогенен изначально, доста-
Отбор пробы. Из журнальной коллекции Chemical
Heritage Foundations' Othmer Library
90
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
точно лишь произвольным образом отобрать от него единственную пробу. При
отборе проб крови этот способ пригоден для решения любых практических за-
дач. В то же время перед отбором проб мочи рекомендуется собирать ее в течение
суток. В этом случае результаты будут более надежными, чем при однократном
взятии пробы.
Что действительно имеет очень важное значение при отборе проб биологи-
ческих жидкостей, так это момент отбора. Состав крови до и после еды значите-
льно различается. Во многих случаях кровь следует брать натощак. Иногда к
пробам крови добавляют консерванты, например, фторид натрия (для консерва-
ции глюкозы) или антикоагулянты.
Пробы крови можно, в зависимости от конкретных требований к анализу,
или анализировать в виде цельной крови, или предварительно разделять и анали-
зировать плазму или сыворотку. В большинстве случаев важным физиологиче-
ским показателем является концентрация вещества вне красных кровяных клеток,
поэтому для анализа используют сыворотку или плазму. Подробнее об отборе
проб биологических жидкостей см. гл. 24.
Если жидкие образцы негомогенны, а объемы достаточно малы, их следует
интенсивно перемешать и немедленно отобрать пробу. В частности, это касается
суспензий, частицы которых имеют склонность оседать. Из больших объемов
(например, из жидкости, текущей по трубам) пробы лучше всего отбирать после
пропускания жидкости через насос, поскольку при этом жидкость интенсивно
перемешивается. Пробы воды из больших резервуаров отбирают с помощью
специального заборника, позволяющего отбирать пробы с разных уровней. Про-
бы с разной глубины лучше отбирать по диагонали, а не по вертикали. Получен-
ные пробы жидкости можно проанализировать по отдельности и усреднить
результаты либо объединить пробы в одну генеральную пробу и выполнить не-
сколько параллельных анализов. Второй способ предпочтительнее, поскольку
он позволяет охарактеризовать точность результатов.
3. Газы. Наиболее распространенный способ отбора газов основан на вытесне-
нии газом жидкости. Жидкость выбирают так, чтобы газ был в ней малораство-
рим и не взаимодействовал с ней химически. Чаще всего используют ртуть. В
сосуд со ртутью газ подают сверху, ртуть при этом свободно вытекает снизу.
При таком способе на собирание средней пробы уходит много времени. В неко-
торых случаях достаточно отобрать одну произвольную пробу, например, пробу
выдыхаемого воздуха. Пациент при этом выдыхает в вакуумированный сосуд.
Выхлопные газы автомобильных двигателей также собирают в большие вакуу-
мированные сосуды из пластмассы. Подробнее об отборе проб объектов окружа-
ющей среды см. гл. 26.
Знание точного объема генеральной пробы газа не всегда бывает необходи-
мо. В пробах часто измеряют концентрации, а не количества тех или иных ком-
понентов. Если же знание объема газа необходимо, нужно учитывать, что на его
величину, а следовательно, и на величины концентраций оказывают влияние
температура и давление. Поэтому эти параметры имеют важное значение для
анализа газов.
2.10. ВЫСУШИВАНИЕ И РАСТВОРЕНИЕ ПРОБ
91
Все сказанное не относится к определению газообразных компонентов, рас-
творенных в жидкостях, например, СО2 или О2 в крови. В подобных случаях ру-
ководствуются правилами отбора жидких проб. Затем пробы соответствующим
образом обрабатывают, чтобы измерить содержание газа непосредственно в
жидкости либо выделить растворенный газ и затем проанализировать.
2.10. Высушивание и растворение проб
После отбора пробы для дальнейшего анализа ее, как правило, необходимо пере-
вести в раствор. Предварительно может потребоваться высушивание пробы,
взвешивание или измерение ее объема. Если проба уже представляет собой рас-
твор (пробы сыворотки крови, мочи, воды), могут потребоваться операции
осаждения, экстракции, концентрирования аналита. Разумеется, это может по-
требоваться и при анализе других проб.
В данном разделе описаны общие приемы перевода в раствор проб как неор-
< ганических, так и органических веществ. К таким приемам относятся растворе-
ние металлов и других неорганических веществ в кислотах или при помощи
сплавления со щелочными плавнями, разложение органических и биологиче-
ских материалов для определения неорганических компонентов при помощи
мокрого сжигания или сухого озоления, а также удаление из биологических
проб белков, мешающих определению неорганических или органических ком-
понентов.
Высушивание проб
Пробы твердых веществ часто содержат переменные количества адсорбирован-
ной воды. Неорганические вещества перед взвешиванием, как правило, высуши-
вают. Для этого пробу выдерживают 1-2 ч в сушильном шкафу при 105-110 °C.
Чтобы удалить более прочно связанную воду — например, кристаллизацион-
ную — может потребоваться более высокая температура.
Следует иметь в виду, что в ходе высушивания возможно разложение про-
бы или протекание побочных реакций. Вещества, которые неустойчивы при по-
вышенных температурах, высушивают в эксикаторе. Использование вакуумных
эксикаторов позволяет ускорить процесс высушивания. Если навеску берут без
предварительного высушивания, то результаты анализа можно отнести только к
пробе «как она есть»; это необходимо указать в отчете.
Образцы биологических тканей (растительных и животных) обычно высу-
шивают при повышенных температурах. Для таких образцов существуют раз-
ные формы представления результатов анализа — в расчете на массу влажного,
сухого образца или золы, см. гл. 1.
Растворение проб
Перед определением содержания аналита, как правило, необходимо провести
некоторые операции с пробой, чтобы перевести определяемое вещество в рас-
92
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
твор или, в случае биологических проб, избавиться от мешающих органических
веществ, например, белков. Способы пробоподготовки делятся на два основных
типаки: деструктивные (с полным разрушением матрицы) и недеструктивные
(либо частично деструктивные). Способы первого типа можно использовать
лишь тогда, когда аналит является неорганическим веществом либо в ходе анали-
за его переводят в неорганическое вещество (например, в ион аммония при опре-
делении органического азота по методу Кьельдаля — см. ниже). Если же аналит
представляет собой органическое вещество, применяют способы второго типа.
Растворение неорганических проб
Для многих неорганических веществ хорошими растворителями являются силь-
ные неорганические кислоты. Для растворения металлов, находящихся в ряду
напряжений левее водорода, чаще всего применяют соляную кислоту. Азотная
кислота, будучи сильным окислителем, растворяет большинство металлов, спла-
вы цветных металлов и сульфиды, нерастворимые в других кислотах.
Хлорная кислота в безводном состоянии (при нагревании до удаления вла-
ги) является очень сильным и высокоэффективным окислителем. Она растворя-
ет большинство металлов, а также разрушает примеси органических веществ.
Но ее следует применять с большой осторожностью, поскольку со многими лег-
ко окисляющимися веществами, в первую очередь органическими, она реагиру-
ет со взрывом.
Ряд неорганических веществ нерастворим в кислотах. Для перевода таких
веществ в раствор необходимо применить сплавление с плавнями — кислыми
или щелочными. Пробу смешивают с плавнем в соотношении 1:10-1:20 и нагре-
вают в тигле из подходящего материала до расплавления смеси. Сплавление за-
канчивают, когда плав станет прозрачным (обычно через 30 мин). Охлажденный
затвердевший плав растворяют в разбавленной кислоте или воде. В ходе сплав-
ления нерастворимые вещества взаимодействуют с плавнем, образуя раствори-
мые продукты. Один из наиболее распространенных щелочных плавней —
карбонат натрия, образующий растворимые в кислотах карбонаты.
Разложение органических веществ
для проведения неорганического анализа:
сжигание и растворение в кислотах-окислителях
Растительные и животные ткани, компоненты биологических жидкостей и дру-
гие органические вещества обычно разлагают посредством мокрого сожжения
под действием кипящих кислот-окислителей или смесей кислот либо путем
сухого озоления в муфельной печи при высоких температурах (400-700 °C).
В ходе мокрого сожжения органические вещества окисляются кислотами до ди-
оксида углерода, воды, других летучих продуктов и удаляются, а остаток содер-
жит лишь неорганические компоненты в виде солей или кислот. При сухом
озолении окислителем является атмосферный кислород. Органическое вещест-
во при этом сгорает, а остаток представляет собой неорганические вещества.
При сухом озолении можно использовать и дополнительные окислители.
2.10. ВЫСУШИВАНИЕ И РАСТВОРЕНИЕ ПРОБ
93
1. Сухое озоление. При анализе органических и биологических материалов час-
то используют различные виды сухого озоления, мокрого сжигания, а также их
сочетания. Но простое сухое озоление в отсутствие дополнительных реагентов
все же наиболее распространенный способ. В ходе сухого озоления органическое
вещество сгорает. При этом почти полностью извлекаются следы таких эле-
ментов, как свинец, цинк, кобальт, сурьма, хром, молибден, стронций и железо, а
их потери за счет улетучивания или связывания невелики. Обычно используют
фарфоровые тигли. Но если озоление проводится при температурах выше 500 °C,
в этом случае могут наблюдаться потери свинца за счет улетучивания (особенно
в присутствии хлоридов, как, например, в пробах крови или мочи). Потери свин-
ца за счет связывания меньше при использовании платиновых тиглей.
При добавлении окислителя к пробе эффективность озоления возрастает.
Один из лучших окислителей — нитрат магния. С его помощью можно количе-
ственно извлекать, кроме вышеперечисленных элементов, также мышьяк, медь
и серебро.
Биологические жидкости и влажные ткани перед помещением в муфельную
» печь выпаривают досуха на водяной бане или при осторожном нагревании.
В ходе озоления температуру печи следует повышать постепенно во избежание
слишком быстрого сгорания и задымления.
По завершении озоления остаток обычно смывают 1-2 мл горячей концент-
рированной или 6 М соляной кислотой и переносят в колбу или стакан для даль-
нейшей обработки.
Другой способ сухого озоления называется низкотемпературным. В этом
случае применяют радиочастотный разряд, вызывающий образование активиро-
ванных радикалов кислорода. Они очень реакционноспособны и разрушают ор-
ганическое вещество при низких температурах. Озоление можно проводить при
температурах ниже 100 °C, вследствие чего потери за счет улетучивания мини-
мальны. Кроме того, становится меньше загрязнений, поступающих из атмосферы
и со стенок тигля, а также уменьшаются потери на стенках тигля. Исследования с
использованием радионуклидов показали, что после полного окисления органи-
ческого вещества 17 представительных элементов извлекаются количественно.
Элементный анализ на содержание основных элементов органических ве-
ществ (например, углерода или водорода) обычно проводят путем сжигания
пробы в токе кислорода с последующим поглощением продуктов сгорания.
Пробу в платиновой лодочке помещают в трубку, нагревают и пропускают кис-
лород. Углерод при этом количественно окисляется до СО2, а водород —
до Н2О. Эти газы пропускают через систему предварительно взвешенных пат-
рончиков, заполненных соответствующими поглотителями. Например, для по-
глощения СО2 применяют аскарит (асбест, пропитанный гидроксидом натрия),
а для поглощения Н2О используют дегидрит (перхлорат магния). Привес погло-
тительного патрончика.равен массе, соответственно, СО2 и Н2О, выделившихся
из пробы. В этом методе анализа большое значение имеют конкретные детали,
поэтому, если вам придется его использовать, обратитесь к специальной литера-
туре по органическому элементному анализу.
94
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
2. Мокрое сожжение. В ходе мокрого сожжения органическое вещество окис-
ляется под действием кислот-окислителей. Распространенным способом окис-
ления органических веществ является мокрое сожжение под действием смеси
азотной и серной кислот. Серную кислоту обычно берут в небольшом объеме
(например, 5 мл), а азотную — в большем (20-30 мл). Мокрое сожжение, как
правило, проводят в колбе Кьельдаля (рис. 2.28). Азотная кислота разрушает
основную часть органического вещества, но не способна разрушить его остатки.
В ходе процесса азотная кислота испаряется, и остается одна серная кислота.
Об этом свидетельствует появление густого белого дыма SO3, циркулирующего
в колбе. К этому моменту раствор становится очень горячим. При такой темпе-
ратуре серная кислота растворяет остатки органического вещества. Если орга-
нического вещества слишком много или оно особо устойчиво к разложению, то
под действием серной кислоты может наблюдаться обугливание. В этом случае
следует добавить еще азотной кислоты. Сжигание продолжают до тех пор, пока
раствор не станет прозрачным. Всю процедуру следует проводит в вытяжном
шкафу. ,
Значительно большей эффективностью обладает смесь азотной, хлорной и
серной кислот в соотношении приблизительно 3:1:1 по объему. Десяти милли-
литров такой смеси обычно достаточно для полного разложения 10 г свежих
биологических тканей или крови. Горячая безводная хлорная кислота — очень
сильный окислитель. Она достаточно легко разлагает даже самые последние
остатки органического вещества. Нагревание ведут до тех пор, пока не испарит-
ся вся азотная кислота и не появятся пары хлорной кислоты. Они обладают ме-
ньшей плотностью, чем SO3, и поэтому заполняют весь объем колбы. Можно
вести нагревание и дальше, до полного испарения хлорной кислоты и появления
паров SO3, легко отличимых на глаз. Кислоты можно добавлять и постепенно.
В начале разложения в колбе должно быть достаточно много азотной кислоты
для разложения основной части органического вещества. Обязательно должна
присутствовать и серная кислота, чтобы проба не выпарилась досуха, а также во
избежание угрозы взрыва хлорной кислоты. Все работы, связанные с исполь-
зованием хлорной кислоты, следует проводить в вытяжном шкафу, специально
предназначенном для этого.
Указанная смесь становится еще более эффективной, если к ней добавить
небольшое количество Mo(VI), играющего роль катализатора. В этом случае по-
сле испарения воды и азотной кислоты окисление происходит очень бурно с силь-
ным выделением дыма и процесс завершается за считанные секунды. В результате
общее время разложения сильно сокращается.
Часто используют и смесь азотной и хлорной кислот. С хлорной кислотой
следует обращаться очень осторожно! Азотная кислота испаряется первой,,и
необходимо следить, чтобы хлорная кислота после этого не испарилась почти
досуха: в этом случае возможен сильный взрыв. Не рекомендуется применять
эту смесь до тех пор, пока вы не приобретете достаточный опыт в области мок-
рого сжигания. Хлорную кислоту нельзя добавлять непосредственно к орга-
ническим или биологическим материалам: всегда сначала добавляйте избыток
азотной кислоты. При использовании хлорной кислоты взрывы, как правило,
2.10. ВЫСУШИВАНИЕ И РАСТВОРЕНИЕ ПРОБ
95
вызваны образованием пероксидов, перед самым взрывом жидкость темнеет
(например, становится желто-коричневой). Некоторые органические вещест-
ва — такие как этанол, целлюлоза и многоатомные спирты — могут вызвать
внезапный взрыв горячей концентрированной хлорной кислоты, вероятно, из-за
образования этилперхлората.
При помощи смеси азотной, хлорной и серной кислот можно количественно
извлекать цинк, селен, мышьяк, медь, кобальт, серебро, кадмий, сурьму, хром,
молибден, стронций и железо. При использовании серной кислоты свинец часто
теряется. Для извлечения свинца следует использовать смесь азотной и хлорной
кислот; при этом извлекаются и все перечисленные элементы. Для предотвраще-
ния потерь селена в смеси должна присутствовать хлорная кислота. Она поддер-
живает сильно окислительную среду и предотвращает обугливание, в ходе
которого могут образоваться летучие соединения селена в низших степенях
окисления. Пробы, содержащие ртуть, нельзя разлагать методом сухого озоле-
ния, а мокрое сжигание следует проводить в приборе с обратным холодильни-
ком, поскольку ртуть и многие ее соединения летучи. Частичное разложение
органического вещества при этом часто предпочитают проводить при комнат-
ной температуре или'даже на холоду. Например, при анализе мочи, содержащей
существенно меньше органических веществ по сравнению с кровью, можно вос-
становить ртуть до металла при помощи Cu(I) и гидрохлорида гидроксиламина,
а органическое вещество разрушить перманганатом калия при комнатной тем-
пературе. Затем ртуть можно растворить и определить ее содержание.
Многие азотсодержащие соединения можно определять по методу Кьель-
даля, превращая азот в сульфат аммония. В ходе разложения по Къелъдалю азот
превращают в ион аммония, который затем отгоняют в виде аммиака и
оттитровывают. Смесь для разложения состоит из серной кислоты и сульфата ка-
лия (для повышения температуры кипения кислоты и, следовательно, эффектив-
ности разложения). Добавляют также катализатор, в качестве которого могут
выступать соединения меди или селена. После разложения органического веще-
ства смесь подщелачивают гидроксидом натрия и отгоняют аммиак в стандарт-
ный раствор соляной кислоты, взятый в избытке. Непрореагировавшую кислоту
оттитровывают стандартным раствором гидроксида натрия и по разности опре-
деляют количество поглощенного аммиака. Из найденного количества аммиака
и известного содержания азота в определяемом соединении можно рассчитать
содержание интересующего соединения. В частности, метод Кьельдаля является
самым точным методом определения белка. Более подробно об этом см. в гл. 8.
Как мокрое сожжение, так и сухое озоление имеют свои достоинства и огра-
ничения применения. Сравнительные достоинства различных методов разложе-
ния органических веществ подробно изучены. Тем не менее до сих пор нет
единого мнения о том, какому методу отдать предпочтение. Сухое озоление
привлекательно благодаря своей простоте и отсутствию серьезного завышения
результатов ввиду загрязнения пробы, поскольку в этом методе используют малые
количества реактивов (или вообще их не используют). Потенциальные источники
погрешностей при сухом озолении — потери элементов из-за улетучивания и
удерживания на стенках тигля. Эти адсорбированные элементы, в свою очередь,
96
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
могут загрязнять последующие пробы. Мокрое сожжение может быть предпоч-
тительнее благодаря быстроте (однако здесь требуется повышенное внимание со
стороны оператора), менее высоким рабочим температурам и отсутствию потерь
на стенках сосуда. Главный источник погрешностей при мокром сожжении —
загрязнения, вносимые реактивами. Остроту этой проблемы можно уменьшить,
используя особо чистые кислоты, которые сейчас широко доступны. Время, тре-
буемое для разложения одним или другим методом, зависит от состава пробы и
технических особенностей конкретного варианта метода. Обычно для сухого
разложения необходимо 2-4 ч, а для мокрого сожжения — 0,5-1 ч.
Микроволновая пробоподготовка
В настоящее время микроволновые печи широко используют для быстрого и эф-
фективного высушивания проб и их разложения кислотами. Сначала для этого
использовали бытовые печи, однако их использование было ограничено рядом
причин. Сейчас разработаны и используются специальные лабораторные печи;
именно они и будут рассмотрены ниже. Микроволновая пробоподготовка позво-
ляет уменьшить время, необходимое для разложения пробы, с нескольких часов
до нескольких минут, а также снизить фоновый сигнал благодаря использова-
нию меньших количеств реактивов.
1. Как происходит нагревание в микроволновых печах? Микроволновое из-
лучение занимает в электромагнитном спектре диапазон между инфракрасным
и радиочастотным излучением с длиной волны в области от 1000 мкм и выше
(300-300000 МГц или 3 • 108—3 • 1011 Гц, см. рис. 16.2). Оно состоит из двух со-
ставляющих — электрической и магнитной, векторы которых взаимно перпен-
дикулярны. Электрическая составляющая обусловливает перенос энергии от
источника излучения к облучаемому объекту. Воздействие микроволнового из-
лучения на частицы вещества проявляется в виде двух эффектов: вращения ди-
полей и ионной проводимости. Первый из них играет более значительную роль.
Когда микроволновое излучение проходит через вещество, молекулы, обладаю-
щие дипольным моментом, стремятся ориентироваться так, чтобы он был распо-
ложен по направлению вектора электрического поля. Чем полярнее молекула,
тем сильнее она взаимодействует с излучением. Это вызывает вращение моле-
кул и приводит к нагреванию вещества. Степень поглощения микроволновой
энергии зависит от величины дипольного момента и диэлектрической проницае-
мости вещества. Энергия поглощается сильнее всего, если время релаксации мо-
лекул совпадает с частотой микроволнового излучения. Для больших молекул,
например, молекул полимеров, время релаксации слишком велико, но по мере
нагревания вещества время релаксации уменьшается, а поглощение энергии воз-
растает. Наоборот, маленькие молекулы — такие как молекулы воды — релак-
сируют с частотой, более высокой, чем частота излучения. По мере нагревания
частота релаксации еще более увеличивается, а поглощение энергии в этом слу-
чае уменьшается.
Явление ионной проводимости состоит в том, что ионы, присутствующие в
веществе, под действием переменного электрического поля начинают попере-
2.10. ВЫСУШИВАНИЕ И РАСТВОРЕНИЕ ПРОБ
97
менно двигаться в противоположных направлениях. Это также приводит к на-
греванию объекта. По мере роста температуры ионная проводимость возрастает.
Поэтому вещества, содержащие ионы, с ростом температуры поглощают микро-
волновое излучение все сильнее. Деионизированная вода в микроволновом поле
нагревается медленно, но солевые растворы нагреваются очень быстро. Раство-
ры кислот тоже являются хорошими проводниками тока и нагреваются быстро.
Таким образом, нагревание вещества в микроволновом поле происходит за
счет вращения молекулярных диполей и движения ионов. Взаимодействие мик-
роволнового излучения с различными веществами может происходить по-раз-
ному. Материалы, хорошо отражающие микроволновое излучения — такие как
металлы — обладают высокой теплопроводностью. В микроволновом поле они
не нагреваются, а отражают падающее излучение. Вещества-изоляторы про-
зрачны для микроволнового излучение и также не нагреваются. Вещества, со-
стоящие из полярных молекул и ионов, хорошо поглощают микроволновое
излучения и нагреваются в микроволновом поле. Энергия микроволнового излу-
чения слишком мала для того, чтобы разорвать химические связи (хотя под дей-
ствием микроволнового излучения многие химические реакции ускоряются; это
обстоятельство вызвало повышенный интерес к использованию микроволново-
го излучения в химическом синтезе). Отмеченные свойства отражающих и изо-
лирующих материалов нашли применение при изготовлении микроволновых
устройств для разложения проб.
2. Конструкция лабораторной микроволновой печи. Для лабораторных це-
лей сначала использовали бытовые микроволновые печи. Однако быстро выяс-
нилось, что они нуждаются в модификации. Бытовые микроволновые печи
непригодны для нагревания проб малого размера. Лабораторные пробы обычно
значительно меньше по размерам, чем порции пищи, поэтому они поглощают
лишь незначительную часть энергии, генерируемой магнетроном. Непоглощен-
ное излучение возвращается к магнетрону, вызывая его перегрев и быстрый выход
из строя. Может возникнуть даже короткое замыкание. Поэтому лабораторные
печи конструируют так, чтобы защитить магнетрон от излучения. Основными
узлами таких печей (рис. 2.27) являются магнетрон, нагрузка, волновод, микро-
волновая камера и гомогенизатор микроволнового потока. Микроволновое из-
лучение, генерируемое магнетроном, по волноводу направляется в камеру.
Гомогенизатор перемешивает потоки микроволновой энергии и распространяет
ее в разных направлениях. Нагрузка изготавливается из ферромагнитного мате-
риала и размещается между магнетроном и волноводом. Она отклоняет микро-
волновое излучение, возвращающееся из камеры, к массивному керамическому
блоку, охлаждаемому вентилятором и, таким образом, защищает магнетрон.
Стандартная частота излучения 2450 МГц, используемая для приготовле-
ния пищи, оказалась удобной и для лабораторной работы. Мощность излучения
лабораторных печей обычно составляет 1200 Вт.
3. Кислотное разложение в микроволновых печах. Как правило, для предо-
хранения печи от паров кислот разложение проводят в автоклавах — закрытых
98
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
Рис. 2.27. Схематическое устройство лабораторной микроволновой печи [G. Le Blanc, LC/GC
Suppl., 17(6S) (1999) S30]. С любезного разрешения LC/GCMagazine
пластмассовых сосудах из тефлона PFA (перфторалкоксиэтилена) или поликарбо-
ната. Кроме того, использование автоклавов имеет дополнительные преимущества.
Внутри сосуда создается повышенное давление, в результате чего температуры ки-
пения кислот повышаются. В этих условиях разложение происходит быстрее.
Нет и потерь летучих элементов. Современные печи снабжены устройствами
контроля давления и температуры (оптоволоконные температурные зонды из
материалов, прозрачных для микроволнового излучения). Использование тем-
пературного контроля позволяет осуществлять процессы экстракции, ускоряемые
под действием микроволнового поля: в этом случае необходимо поддерживать
относительно низкие температуры во избежание химического разложения ве-
ществ.
Пробоподготовка без разрушения или с частичным
разрушением матрицы
Очевидно, что для определения органических веществ можно использовать толь-
ко недеструктивные методы пробоподготовки. Иногда и при определении метал-
лов не возникает необходимости разрушать молекулярную структуру образца, в
частности, при анализе биологических жидкостей. Например, ряд металлов в
сыворотке крови или моче можно определять атомно-абсорбционным методом
путем прямого распыления пробы (при необходимости разбавленной) в пламя
горелки. Компоненты твердых веществ, в частности, почв, также иногда опреде-
ляют без разрушения матрицы, посредством экстракции подходящими реагента-
ми. Перед экстракцией образец необходимо тщательно измельчить и перемешать,
2.10. ВЫСУШИВАНИЕ И РАСТВОРЕНИЕ ПРОБ
99
а экстракцию проводить при кипячении с обратным холодильником. Следы
многих металлов можно экстрагировать из почв 1 М растворами хлорида аммо-
ния или уксусной кислоты. Некоторые элеме'нты отгоняют в виде летучих про-
изводных, например, селен в виде хлорида или бромида.
Безбелковые фильтраты
Белки, содержащиеся в биологических жидкостях, мешают при многих опреде-
лениях. Их следует предварительно отделить, не разрушая матрицу образца. Для
этого используют ряд реагентов, вызывающих коагуляцию и осаждение белков,
в частности, трихлоруксусную кислоту, вольфрамовую кислоту (смесь вольфра-
мата натрия и серной кислоты) или «нейтральную смесь» гидроксида бария и суль-
фата цинка. К отмеренному объему пробы (например, сыворотки крови) добавляют
отмеренный объем раствора реагента. После осаждения белка (приблизительно
через 10 мин) раствор фильтруют через сухой бумажный фильтр (не промывая
его) либо центрифугируют, после чего анализируют аликвоту полученного безбел-
кового фильтрата. Более подробно о приготовлении различных видов таких
фильтратов см. в гл. 24 (разд. «Отбор и хранение проб») и в описаниях соот-
ветствующих лабораторных работ.
Техника работы при высушивании й растворении проб
При высушивании и растворении проб следует соблюдать осторожность.
Если твердую пробу высушивают в бюксе, то бюкс следует открыть и поместить
вместе с крышкой в стакан во избежание распыления. Стакан подписывают и
прикрывают часовым стеклом.
Растворение навески проводят в стакане или конической колбе. Если при
этом возможно вспенивание, стакан прикрывают часовым стеклом. После пол-
ного растворения пробы стенки сосуда обмывают дистиллированной водой. Ча-
совое стекло также обмывают водой, собирая смывную жидкость в стакан.
После этого может потребоваться упарить раствор для уменьшения его объема.
Это удобнее всего делать в стакане, неплотно прикрытом часовым стеклом. Рас-
твор нагревают медленно, не доводя до кипения. Для нагревания используют во-
дяную баню или электроплитку с регулятором температуры.
Растворение в колбе Кьельдаля удобно тем, что при этом не происходит
разбрызгивания раствора. Колбы Кьельдаля (названные так потому, что перво-
начально их применяли только для определения азота по Кьельдалю) также
удобно использовать для всех видов мокрого сжигания органических проб и рас-
творения металлических образцов в кислотах. Колбы Кьельдаля выпускаются
различных объемов от 10 до 800 мл. Некоторые из них изображены на рис. 2.28.
Пробу и необходимую кислоту помещают на дно колбы. Колбу нагревают в на-
клонном положении. В этом случае даже при кипении раствора не происходит
его разбрызгивания, поскольку кислота конденсируется на стенках горлышка
колбы. Нагревать колбу можно на горелке или при помощи специального элект-
ронагреваемого держателя, позволяющего оперировать с несколькими колбами
одновременно.
100
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
Рис. 2.28
Колбы Кьельдаля
2.11. Безопасность в лаборатории
До того, как проводить эксперименты, вам необходимо ознакомиться с правила-
ми техники безопасности в лаборатории! Прочтите приложение D и материалы,
которые предоставит вам ваш преподаватель. Ваш преподаватель дополнитель-
но ознакомит вас со специфическими правилами и нормами работы в вашей ла-
боратории, а также с местонахождением реактивов. Более подробно вопросы
лабораторной техники безопасности обсуждаются в руководстве Safety in Acade-
mic Chemistry Laboratories, изданном Американским химическим обществом
[32]. В нем рассматриваются вопросы защиты персонала и ведения лаборатор-
ной документации, описаны рекомендованные приемы экспериментальной рабо-
ты, дана характеристика опасности различных химических веществ, приведены
рекомендации по изучению паспортов безопасности химических веществ и ма-
териалов (Material Safety Data Sheets, MSDS), описано защитное оборудование и
правила действий в чрезвычайных ситуациях. Приведены также правила утили-
зации отходов, терминология, касающаяся классификации отходов. В руковод-
стве содержатся также нормативы, регламентирующие контакт с опасными
химикатами, выработанные американским Управлением по технике безопасно-
сти и здоровью (Occupational Safety and Health Administration, OSHA) и требования
американского Агентства по защите окружающей среды. Подробно рассмотрены
правила обращения с неорганическими и органическими пероксидами, приве-
ден обширный список несовместимых химических веществ и нормы хранения
горючих и легковоспламеняющихся жидкостей. Этот буклет рекомендуется
прочитать всем студентам и преподавателям. Он бесплатно распространяется
(по одному экземпляру) Американским химическим обществом (The American
Chemical Society, Washington, DC, 1-800-227-5558).
Еще одно руководство, изданное Американским химическим обществом,
The Waste Management Manual for Laboratory Personnel [33] содержит обзор офи-
циальных нормативных актов по утилизации лабораторных отходов.
ВОПРОСЫ
101
Всегда носи в лаборатории защитные очки! Публикуется с любезного разрешения Merck KGaA
Что мы узнали из этой главы?
• Аккуратно заполняйте свой лабораторный журнал — стр. 42
• Используйте реактивы квалификации ч. д. а. — стр. 48
• Как пользоваться аналитическими весами — стр. 49
• Мерная посуда и правила обращения с ней — стр. 60, 67
• Как откалибровать мерную посуду — стр. 71
• Как приготовить стандартные растворы кислоты и щелочи — стр. 76, 77
• Распространенное лабораторное оборудование для подготовки и обработки
проб — стр. 78
• Как отфильтровать и подготовить осадок для гравиметрического определе-
ния — стр. 87
• Как отбирать пробы твердых, жидких и газообразных веществ — стр. 88
• Как приготовить раствор, содержащий определяемый компонент—стр. 91
Вопросы
1 - Опишите основные типы посуды, используемой для измерения объемов. Ука-
жите, какие из них отградуированы на заполнение, какие — на вытекание.
2. Опишите принцип действия и порядок работы с аналитическими весами.
102
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
3. Почему микровесы более чувствительны, чем обычные аналитические весы?
4. Что означает маркировка «TD» и «ТС» на мерной посуде?
5. Как проводится взятие навесок по разности?
6. В чем разница между нулевой точкой и точкой покоя весов?
7. Назовите основные правила работы с весами.
8. Опишите приготовление стандартных растворов НС1 и NaOH.
9. Опишите принципы, лежащие в основе сухого озоления и мокрого сжига-
ния органических и биологических материалов. Перечислите преимущест-
ва каждого метода.
10. Назовите два основных способа перевода в раствор неорганических веществ?
11. Что такое безбелковый фильтрат? Как его приготовить?
12. Какого рода ситуаций необходимо тщательно избегать, чтобы безопасно
использовать хлорную кислоту для разложения органических веществ?
13. Что такое генеральная, лабораторная, анализируемая проба? А что такое
случайная проба?
14. Какие процессы обусловливают нагревание вещества в микроволновом поле?
Задачи
Градуировка мерной посуды, температурные поправки
15. Вы калибруете мерную колбу номинального объема 25 мл, заполняя ее до
метки дистиллированной водой (температура 22 °C). Сухая закрытая проб-
кой колба весит 27,278 г, а заполненная закрытая колба — 52,127 г. Для
взвешивания использованы стальные разновесы. Чему равен объем колбы
при данной температуре? Чему он равен при 20 °C? Для самопроверки вве-
дите значение веса на воздухе при 22 °C в соответствующую ячейку элект-
ронной таблицы 2.4 (на компакт-диске) и сравните ответ с полученным
вами значением.
16. Вы калибруете пипетку номинального объема 25 мл при 25 °C, используя
стальные разновесы. Вес воды, вытекшей из пипетки, равен 24,971 г. Чему
равны объемы пипетки при 25 и 20 °C?
17. Вы откалибровали бюретку объемом 50 мл при 20 °C и получили следую-
щие величины поправок объемов:
Показания бюретки (мл) Поправка (мл)
10 +0,02
20 +0,03
30 0,00
40 -0,04
50 -0,02
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
103
После этого вы стали работать с бюреткой жарким летним днем при 30 °C.
Чему теперь равны значения поправок?
18. Вы приготовили стандартный раствор при 21 °C, а работаете с ним при
29 °C. Исходная концентрация раствора составляла 0,05129 М, а какой она
стала к моменту работы с ним?
Рекомендуемая литература
Реактивы и стандарты
1. Reagent Chemicals, American Chemical Society Specifications, 9th ed., Washin-
gton, DC: American Chemical Society, 2000.
2. Dictionary of Analytical Reagents, London: Chapman & Hall, 1993. Данные no
более чем 10000 реактивам.
3. J. R. Moody and E. S. Beary, «Purified Reagents for Trace Metal Analysis», Ta-
lanta, 29 (1982), 1003. Описано приготовление особо чистых кислот пере-
гонкой при пониженном давлении.
4. R. С. Richter, D. Link, and Н. М. Kingston, «On-Demand Production of High-
Purity Acids in the Analytical Laboratory», Spectroscopy, 15(1) (2000) 38. Опи-
сано приготовление особо чистых кислот при помощи коммерческой уста-
новки для перегонки при пониженном давлении.
5. www.thomsmithlabs.com — сайт компании «Thom Smith Laboratories», по-
ставляющей готовые комплекты реактивов (стандарты и образцы для ана-
лиза) для студенческих лабораторных работ по аналитической химии;
www.sigma-aldrich.com/analytical— сайт компании-поставщика стандарт-
ных растворов.
Аналитические весы
6. D. F. Rohrbach and М. Pickering, «А Comparison of Mechanical and Electronic
Balances», J. Chem. Ed., 59 (1982) 418.
7. R. M. Schoonover, «А Look at the Electronic Analytical Balance», Anal. Chem.,
54(1982) 973A.
8. К. M. Lang, «Time-Saving Applications of Electronic Analytical Balances», Am.
Lab., March (1983) 72.
Калибровка весов
9. W. D. Abele, «Laboratory Note: Time-Saving Applications of Electronic Balances»,
Am. Lab., 13 (1981) 154. Обсуждается калибровка весов при помощи стандар-
тов массы, откалиброванных Американским национальным институтом
стандартов и технологий.
104
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
10. D. F. Swinehart, «Calibration of Weights in a One-Pan Balance», Anal. Lett.,
10(1977) 1123.
Калибровка мерной посуды
11. G. D. Christian, «Coulometric Calibration of Micropipets», Microchem. J.,
9(1965) 16.
12. M. R. Masson, «Calibration of Plastic Laboratory Ware», Taianta, 28 (1981)
781. Таблицы для калибровки полипропиленовых сосудов.
13. W. Ryan, «Titrimetric and Gravimetric Calibration of Pipettors: A Survey»,
Am. J. Med. Technol., 48 (1982) 763. Калибровка пипетаторов объемом от 1
до 500 мкл.
14. М. R. Winward, Е. М. Woolley, and Е. A. Butler, «Air Buoyancy Corrections for
Single-Pan Balances», Anal. Chem., 49 (1977) 2126.
15. R. M. Schoonover and F. E. Jones, «Air Buoyancy Corrections in High-Accuracy
Weighing on Analytical Balances», Anal. Chem., 53 (1981) 900.
«Чистые» лаборатории
16. J. R. Moody, «The NBS Clean Laboratories for Trace Element Analysis», Anal.
Chem.,54(\<№!} 1358A.
Пробоотбор
17. J. A. Bishop, «An Experiment in Sampling», J. Chem. Ed., 35 (1958) 31.
18. J. R. Moody, «The Sampling, Handling and Storage of Materials for Trace Ana-
lysis», Phil. Trans. Roy. Soc. London, Ser. A, 305 (1982) 669.
19. G. E. Baiulescu, P. Dumitrescu, and P. Gh. Zugravescu, Sampling, Chichester:
Ellis Horwood, 1991.
Подготовка и растворение проб
20. G. D. Christian, «Medicine, Trace Elements, and Atomic Absorption Spectro-
scopy», Anal. Chem.,M\Y) (1969) 24A. Приготовление растворов биологиче-
ских жидкостей и тканей.
21. G. D. Christian, Е. С. Knoblock, and W. С. Purdy, «Polarographic Determinati-
on of Selenium in Biological Materials», J. Assoc. Ojfic. Agric. Chemists,
48 (1965) 877; R. K. Simon, G. D. Christian, and W. C. Purdy, «Coulometric De-
termination of Arsenic in Urine», Am. J. Clin. Pathol, 49 (1968) 207. Использо-
вание Mo(VI) в качестве катализатора при мокром сжигании.
22. Т. Т. Gorsuch, «Radiochemical Investigations on the Recovery for Analysis of
Trace Elements in Organic and Biological Materials», Analyst, 84 (1959) 135.
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
105
23- S. Nobel and D. Nobel, «Determination of Mercury in Urine», Clin. Chem.,
4 (1958) 150. Мокрое сжигание при комнатной температуре.
24- G. Knapp, «Mechanical Techniques for Sample Decomposition and Element
Preconcentration», Mikrochim. Acta, II (1991) 445. Смеси кислот и их исполь-
зование для мокрого сжигания и других способов разложения.
Разложение в микроволновом поле
25- Н. М. Kingston and L. В. Jassie, eds., Introduction to Microwave Sample Prepa-
ration: Theory and Practice. Washington, D.C.: American Chemical Society,
1988.
26. H. M. Kingston and S. J. Haswell, eds., Microwave-Enhanced Chemistry: Fun-
damentals, Sample Preparation, and Applications, Washington, DC: American
Chemical Society, 1997.
27. H. M. Kingston and L. B. Jassie, «Microwave Energy for Acid Decomposition at
Elevated Temperatures and Pressures Using Biological and Botanical Samples»,
Anal. C/zem.,58(1986) 2534.
28. R. A. Nadkami, «Applications of Microwave Oven Sample Dissolution in Analy-
sis», Anal. Chem., 56 (1984) 2233.
29. B. D. Zehr, «Development of Inorganic Microwave Dissolutions», Am. Lab.,De-
cember (1992) 24. Также приведены свойства смесей кислот, используемых
для разложения.
30. J. К. Poudrier, «The Micro wave Oven as a Specialized Instrument», Today's
Chemist at Work (American Chemical Society), July/August (1995) 29.
31. www.cemx.com — сайт компании СЕМ — производителя лабораторных
микроволновых печей.
Безопасность в лаборатории
32. Safety in Academic Chemistry Laboratories, 6th ed., American Chemical Society,
Committee on Chemical Safety. Washington, D.C.: American Chemical Society,
1995.
33- The Waste Management Manual for Laboratory Personnel, Task Force on
RCRA, American Chemical Society, Department of Government Relations and
Science Policy. Washington, DC: American Chemical Society, 1990.
34. A. K. Furr, ed., CRC Handbook of Laboratory Safety, 4th ed. Boca Raton, FL:
CRC Press, 1993.
35- M.-A. Armour, Hazardous Laboratory Chemicals Disposal Guide, Boca Raton,
FL: CRC Press, 1990.
36. R. J. Alaimo, ed., Handbook of Chemical Health and Safety, Washington, DC:
American Chemical Society, 2001.
106
ОСНОВНЫЕ ИНСТРУМЕНТЫ И ОПЕРАЦИИ ХИМИЧЕСКОГО АНАЛИЗА
37. М. S. Lesny, «It's a Bad, Bad, Bad, Bad World», Today's Chemist at Work (Ame-
rican Chemical Society), December (2000) 26. Перечислены и обсуждаются
многочисленные правительственные, общественные и университетские
сайты, посвященные проблемам производственной безопасности и охраны
здоровья.
Паспорта безопасности химических веществ и материалов (MSDS)
38. http://siri.org/msds — поисковая база данных в режиме online, ссылки на
другие сайты, посвященные тем же проблемам.
39. www.env-sol.com — сайт Solutions Software Corporation. Перечни MSDS
доступны на DVD или CD-дисках.
Глава 3
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
В АНАЛИТИЧЕСКОЙ ХИМИИ
Факты упрямы, но статистика куда сговорчивее.
Марк Твен
43,8% всех статистических данных бесполезны.
Мудрость аналитика
Несмотря на то, что обработка данных производится после их получения, мы
считаем целесообразным обсудить вопросы обработки данных в начале книги,
поскольку знание основ статистического анализа важно при выполнении экс-
периментов. Оно необходимо для оценки значимости полученных данных, а
следовательно, для контроля каждой стадии анализа. Качество планирования
эксперимента (включая оценку требуемого размера пробы, необходимой точ-
ности измерений и числа параллельных определений) определяется тем, на-
сколько верно вы понимаете, что будут представлять собой получаемые вами
данные.
Применение электронных таблиц для обработки данных сильно облегчило
статистические и прочие вычисления. Сначала вы ознакомитесь со всеми дета-
лями вычислений на бумаге вручную, а затем с использованием электронных таб-
лиц, чтобы продемонстрировать преимущества таких расчетов при выполнении
рутинных вычислений. В этой главе мы узнаем, как можно пользоваться
таблицами Excel для решения задач аналитической химии.
3.1. Правильность и воспроизводимость - разные вещи
Понятие правильности характеризует степень близости измеренного значения
некоторой величины и ее истинного значения. Истинное значение в строгом
смысле слова, как правило, никогда не известно. Поэтому более реалистическим
определением правильности была бы близость между измеренным значением и
значением, принимаемым за истинное.
При условии хорошей техники эксперимента, например, при сравнении по-
лученных данных с результатами анализа стандартного образца сходного соста-
ва, можно сделать разумные предположения о правильности методики — в
пределах, ограниченных нашими знаниями о составе стандартного образца и
измерительной процедуре. Конечно, точность, с которой известен состав стан-
дартного образца, тоже не абсолютна, поскольку также определяется характе-
ристиками некоего измерительного процесса.
108
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
РИС. 3.1. Иллюстрация понятий «правильность» и «воспроизводимость».
Воспроизводимость по определению есть степень близости между резуль-
татами повторных измерений одной и той же величины. Мерой воспроизводимости
может служить стандартное отклонение, относительное стандартное отклонение,
размах данных или доверительный интервал (например, соответствующий 95%-й
доверительной вероятности) вокруг среднего. Хорошая воспроизводимость еще
не гарантирует высокой правильности: результаты анализа могут содержать си-
стематическую погрешность. Например, измеренные значения масс нескольких
одинаковых проб могут содержать одну и ту же погрешность. Эта погрешность
не влияет на воспроизводимость, но влияет на правильность. И наоборот, вос-
производимость может быть относительно низкой, а правильность, по крайней
мере, за счет случайных причин, оказаться высокой. Поскольку в реальной жиз-
ни истинные значения результатов анализа никогда не известны заранее, то чем
выше воспроизводимость, тем больше вероятность получить правильный резу-
льтат. Бесполезно надеяться, что результат случайно окажется правильным при
плохой воспроизводимости. Поэтому аналитик всегда стремится в первую оче-
редь добиться получения хорошо воспроизводимых результатов.
Если хочешь попасть в цель, стреляй куда угодно,
а потом то, во что ты попал, назови «целью».
Эшли Бриллиант
Понятия «правильность» и «воспроизводимость» можно проиллюстрировать
при помощи мишени (рис. 3.1). Предположим, в ходе упражнений по стрельбе
все пули попали в яблочко (мишень слева). В этом случае результаты правильны
и хорошо воспроизводимы. Мишень посередине демонстрирует результаты хо-
рошо воспроизводимые (рука твердая), но неправильные. Возможно, у оружия
сбит прицел. Результаты стрельбы по мишени справа плохо воспроизводимы, и
потому средний результат, скорее всего, неправильный. Таким образом, мы ви-
дим, что хорошая воспроизводимость необходима для достижения правильности,
но она отнюдь не гарантирует ее.
Как мы увидим вскоре, чем больше параллельных результатов имеется, тем
более надежным окажется средний результат. Число требуемых параллельных
измерений определяется требуемой правильностью и известной воспроизводи-
мостью методики.
3.2. ДЕТЕРМИНИРОВАННЫЕ (СИСТЕМАТИЧЕСКИЕ) ПОГРЕШНОСТИ
109
3.2. Детерминированные (систематические) погрешности
На показатели правильности и воспроизводимости влияют погрешности двух
типов. Детерминированные погрешности — это погрешности, величина кото-
рых может быть определена (как следует из их названия), которых в принципе
можно избежать или которые можно скорректировать. Они могут быть постоян-
ными по величине, например, если все взвешивания проводятся на одних и тех
же неоткалиброванных весах. Они могут быть и переменными по величине, од-
нако в силу их природы систематические погрешности можно вычислить и скор-
ректировать, как в случае бюретки, погрешности показаний которой составляют
разные значения для разных объемов.
Величина детерминированной погрешности может быть пропорциональна
размеру пробы или зависеть от нее более сложным образом. Чаще всего такие
погрешности имеют один и тот же знак, например, погрешности, обусловленные
потерями осадка за счет растворимости (отрицательные). Иногда их знак может
изменяться в зависимости от условий. Примером могут служить изменения объ-
ема раствора и его концентрации при изменении температуры. В этом случае их
можно скорректировать, если измерить температуру раствора. Такие детерми-
нированные погрешности, поддающиеся измерению, называются системати-
ческими. Детерминированные, или систематические, погрешности имеют не
случайный характер.
К наиболее распространенным систематическим погрешностям относятся:
1. Инструментальные погрешности. Возникают из-за неверных показа-
ний приборов, при использовании неоткалиброванных весов или мерной
посуды.
2. Субъективные погрешности. Это погрешности, обусловленные ошиб-
ками в работе аналитиков. Они уменьшаются с накоплением опыта рабо-
ты и при повышении внимания ко всем выполняемым руками операциям.
К типичным операциями, вызывающим такие погрешности, относятся
дозирование и перенос растворов, растворение пробы при нагревании
(за счет вспенивания, кипения раствора), высушивание пробы (если оно
проводится не до конца) и т. д. Такие погрешности трудно скорректиро-
вать. Другие источники субъективных погрешностей — арифметические
ошибки в ходе вычислений и предвзятость при считывании показаний
приборов.
3. Методические погрешности. Эти погрешности являются наиболее серь-
езными. Большинство погрешностей, упомянутых выше, можно миними-
зировать или скорректировать, но погрешностей, внутренне присущих
методике, избежать невозможно до тех пор, пока не будут изменены
условия определения. К источникам погрешностей методики относятся
110
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
соосаждение примесей и потери осадка за счет растворения, побочные
реакции, неполнота протекания реакций, загрязнения реактивов. Иногда
погрешности можно достаточно легко скомпенсировать, в частности,
путем проведения контрольного опыта. Контрольный опыт — это про-
ведение через все этапы методики пробы, не содержащей определяемого
компонента. Проведение контрольного опыта и последующее вычита-
ние полученных результатов из результатов анализа реального образца
является общепринятой практикой. Если, несмотря на все принятые
меры, погрешности методики остаются слишком большими, необходимо
выбрать другой подход к решению задачи. Впрочем, иногда приходится
пользоваться данной методикой за неимением лучшей...
Систематические погрешности могут быть аддитивными или мультипли-
кативными в зависимости от характера их влияния на результаты анализа. Для
обнаружения систематических погрешностей повсеместно принято добавлять к
пробе известное количество определяемого компонента и рассчитывать меру
правильности — отношение добавленного количества к найденному (см. гл. 1,
поверка методики). Анализ образцов известного состава (в первую очередь под-
ходящих стандартных образцов) также полезен для контроля погрешностей ме-
тодики и инструментальных погрешностей.
3.3. Недетерминированные (случайные) погрешности
Другой тип погрешностей — это недетерминированные погрешности, часто
называемые случайными. Они отражают неопределенность результата, прису-
щую любому измерению. Случайные погрешности проявляются в виде неболь-
ших различий между результатами последовательных измерений одной и той же
величины, проводимых одним и тем же аналитиком в одних и тех же условиях.
Конкретное значение каждой такой погрешности невозможно предсказать и
оценить заранее. Массив значений случайных погрешностей подчиняется неко-
торому закону случайного распределения. Поэтому для того, чтобы из серии экспе-
риментальных данных оценить наиболее вероятный результат, можно применять
методы теории вероятностей.
Детальное рассмотрение математических проблем теории вероятностей вы-
ходит за рамки данной книги. Отметим лишь, что случайные погрешности хими-
ческого анализа обычно подчиняются нормальному, или гауссовому, закону
распределения. График функции распределения Гаусса изображен на рис. 3.2.
Параметр о представляет собой стандартное отклонение для бесконечной се-
рии результатов (генеральной совокупности) и характеризует их воспроизводи-
мость, т. е. степень разброса данных. Из рис. 3.2. видно, что чем больше
3.4. ЗНАЧАЩИЕ ЦИФРЫ: СКОЛЬКО ТРЕБУЕТСЯ ЦИФР?
111
Рис. 3.2
Кривая нормального
распределения
погрешностей
Отклонения от среднего (в единицах стандартного отклонения)
погрешность по абсолютной величине, тем реже она встречается, а кроме того,
частоты появления положительных и отрицательных погрешностей одинаковы.
Погрешности, которые нами не замечены,
бесконечны и очень разнообразны.
В отличие от них, замеченные нами погрешности
конечны по определению.
Том Гибб
По сути недетерминированные погрешности обусловлены ограниченностью воз-
можностей аналитика контролировать или корректировать внешние условия
либо невозможностью распознать действие факторов, влияющих на результат
измерения. Некоторые случайные погрешности вызваны статистическим харак-
тером явлений, например, погрешности, связанные с подсчетом элементарных
частиц. Иногда при изменении условий некоторые погрешности неизвестной
природы исчезают. Но, разумеется, полностью исключить в ходе эксперимента
все мыслимые источники случайных погрешностей нельзя. Аналитик должен
лишь стремиться уменьшить их до допустимого или пренебрежимо малого
уровня. Недетерминированные погрешности имеют случайную природу. Избе-
жать их нельзя.
3.4. Значащие цифры: сколько требуется цифр?
Самое слабое звено во всей цепи операций любого анализа — то измерение, ко-
торое выполняется с наименьшей точностью. Бессмысленно стремиться прово-
дить другие измерения, с большей точностью, чем лимитирующее. Число
значащих цифр, необходимое для представления результата измерения с соот-
ветствующей точностью, называется числом значащих цифр. Поскольку неоп-
ределенность (неточность) любого измерения составляет по меньшей мере ±1 в
последней значащей цифре, следует оставлять все цифры, которые известны
точно, плюс одну недостоверную. Последняя цифра результата измерения имеет
неопределенное значение. Не следует писать после нее дополнительные цифры.
112
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
Каждая цифра характеризует число единиц в том десятичном разряде, который
она представляет. Например, в числе 237 содержатся 2 сотни, 3 десятка и 7 еди-
ниц.
Цифра 0 может быть либо значащей, либо просто указывать на положение
запятой. Однако число значащих цифр результата измерения не зависит от поло-
жения запятой. Рассмотрим число 92067. Это число имеет пять значащих цифр
независимо от того, где стоит запятая. Например, это может быть 92067 мкм,
9,2067 см, 0,92067 дм или 0,092067 м — все эти величины имеют одно и то же
число значащих цифр. Они просто представляют разные способы выражения од-
ного и того же результата измерения (в разных единицах). В последнем случае
нуль между запятой и цифрой 9 лишь указывает место расположения запятой.
Если нуль расположен после запятой, то всегда понятно, является ли он знача-
щей цифрой или просто указывает на положение запятой. Например, в числе
727,0 нуль не является указателем положения запятой и представляет собой зна-
чащую часть числа. Но если нуль находится перед запятой, то интерпретация его
роли может быть неоднозначной. Нуль, предшествующий запятой, всегда знача-
щий, если он расположен между двумя ненулевыми цифрами (как в приведен-
ном примере с числом 92067). Однако для числа, записанного как 936600,
невозможно определить, является ли один из нулей, или оба, или ни один из них
простым указателем положения (предполагаемой) дробной части либо пред-
ставляет собой значащую составную часть результата измерения. Поэтому в по-
добных случаях лучше всего записывать число в экспоненциальной форме,
указывая перед показателем степени только значащие цифры. Например, число,
записанное как 9,3660 • 105 имеет пять значащих цифр. В этом случае число
936600 следует считать имеющим шесть значащих цифр.
Пример 3.1
Укажите, сколько значащих цифр содержат числа, записанные в приведенной
ниже форме. Укажите в них нули, являющиеся значащими.
0,216; 90,7; 800,0; 0,0670
Решение
0,216 три значащие цифры
90,7 три значащие цифры, нуль значащий
800,0 четыре значащие цифры, все нули значащие
0,0670 три значащие цифры, только последний нуль является значащим
Значимость последней цифры результата измерения можно проиллюстри-
ровать следующим образом. Предположим, каждый студент в группе измеряет
длину стержня, используя одну и ту же линейку. Линейка отградуирована с це-
3.4. ЗНАЧАЩИЕ ЦИФРЫ: СКОЛЬКО ТРЕБУЕТСЯ ЦИФР?
113
ной деления 1 мм. Можно выполнить измерение, округляя результат до 0,1 мм
(считывая показание по положению конца стержня между делениями), но по-
следняя цифра оценочная и потому неточная. Может быть получена, например,
следующая серия результатов:
36,4 мм; 36,8 мм; 36,0 мм; 37,0 мм; среднее — 36,6 мм.
Умножение и деление: мыслите относительными
категориями
Для многих измерений в запись результата включают одну цифру, которая явля-
ется оценочной и потому недостоверна (в последнем примере это десятые доли
миллиметра). Эта цифра представляет собой последнюю значащую цифру чис-
ла. Все цифры, следующие за ней, лишены смысла. При умножении и делении
неопределенность этой цифры переносится на конечный результат, ограничивая
тем самым число его достоверных цифр. Результат умножения или деления со-
держит, по крайней мере, такую же относительную неопределенность, как и тот
сомножитель (делимое, делитель), который обладает наибольшей относитель-
ной неопределенностью значения, т. е. одно из чисел с наименьшим числом знача-
щих цифр. Результат умножения или деления не может быть точнее наименее
точного числа участвующего в операции. Будем считать, что это число лимити-
рует точность результата. Если есть несколько чисел, участвующих в арифмети-
ческих действиях и имеющих одинаковое и наименьшее число значащих цифр,
то лимитирует точность то из них, у которого мантисса (число, состоящее из
всех выписанных подряд значащих цифр) является наименьшей по абсолютной
величине. Например, из двух чисел, имеющих по 3 значащих цифры — 0,0344 и
5,39, точность лимитирует первое (мантисса 344 меньше, чем 539).
Пример 3.2
В каждой следующей паре чисел найдите то, которое лимитирует точность при
операциях умножения и деления: а) 42,67 или 0,0967; б) 100,0 или 0,4570;
в) 0,10 или 0,0067.
Решение
а) 0,0967 (оно имеет три значащие цифры, а 42,67 — четыре).
б) 100,0. Оба числа имеют по четыре значащие цифры, но мантисса первого рав-
на 1000, а второго 4570. Соответственно, у первого числа неопределенность
значения — 1 часть на 1000, а у второго — 1 часть на 4570.
в) 0,10. Оба числа имеют по две значащие цифры, но неопределенность значе-
ния первого — 1 часть на 10 (10%), а второго — приблизительно 1 часть
на 70.
114
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
Пример 3.3
Приведите результат следующих вычислений с наибольшим возможным чис-
лом значащих цифр и укажите, какой из участников арифметических действий
лимитирует точность результата.
35,63 -0,5481 -0,05300
1,1689
•100% = 88,5470578%
Решение
Точность лимитирует число 35,63. Поэтому результат надо представить как
88,55%, а производить вычисления более, чем с пятью значащими цифрами
(одну цифру оставляем в качестве запасной и затем округляем), смысла не име-
ет. Обратите внимание, что число 100% служит только для перемещения пози-
ции запятой и, следовательно, является точной величиной (можно сказать, что
оно содержит «бесконечно много» значащих цифр). Заметьте также, что число,
лимитирующее точность, характеризуется относительной неопределенностью
не менее чем 1 часть на 3600, поэтому и результат будет иметь относительную
неопределенность в лучшем случае 1 часть на 3600, или порядка 2,5 на 8900. Та-
ким образом, при выполнении вычислений следует не только правильно пред-
ставить результат (с точностью до последней значащей цифры), но и оценить ве-
личину его неопределенности. Конечный результат должен состоять только
из значащих цифр. При выполнении измерений следует стремиться к тому, чтобы
все результаты, используемые для расчетов, характеризовались приблизительно
одной и той же относительной неопределенностью значений.
Если мантисса конечного результата по абсолютной величине меньше ман-
тиссы числа, лимитирующего точность вычислений, то к результату вычислений
следует дописать еще одну цифру для того, чтобы не снизить неопределенность
его значения. Эту цифру пишут в виде нижнего индекса, чтобы подчеркнуть, что
она менее надежна.
Пример 3.4
Приведите результат следующих вычислений с максимальным числом знача-
щих цифр. Укажите число, лимитирующее точность вычислений.
_4W891 =
132,6-0,5247
Решение
Лимитирует точность число 891. Поскольку мантисса результата меньше, чем
мантисса этого числа, представим результат как 546,6. Последняя цифра записа-
ЗА ЗНАЧАЩИЕ ЦИФРЫ: СКОЛЬКО ТРЕБУЕТСЯ ЦИФР?
115
на в виде нижнего индекса, чтобы показать, что она менее надежна. Лимитирую-
щее точность число имеет относительную неопределенность порядка 1 части на
900, а результат — не менее 6 частей на 5500 (0,6 части на 550).
В ходе операций умножения и деления допускается округлять результат до
соответствующего числа значащих цифр на каждом этапе вычислений. Однако
для промежуточных результатов все же лучше сохранять по одной запасной
цифре и затем округлить конечный результат до нужного числа значащих цифр.
Сложение и вычитание: мыслите абсолютными категориями
При сложении и вычитании с цифрами обращаются иначе, чем при умножении и
делении. Здесь мы имеем дело не с относительными, а с абсолютными неопреде-
ленностями. В этом случае число значащих цифр результата зависит от того, где
расположены десятичные запятые в записи исходных величин. Число десяти-
чных знаков результата сложения или вычитания равно наименьшему их числу
среди участников этих арифметических действий. Предположим, мы рассчи-
тываем молярную массу вещества Ag2MoO4 из относительных атомных масс со-
ставляющих его элементов:
Ag 107,87 0
Ag 107,87 0
Mo 95,94
О 15,99 94
О 15,99 94
О 15,99 94
О 15,99 94
375,67 76
Атомная масса молибдена известна лишь с точностью до 0,01 а.е.м. Поэто-
му молярная масса любого соединения, содержащего молибден, не может быть
известна с более высокой точностью. Следовательно, максимальная точность, с
которой мы можем представить значение молярной массы Ag2MoO4, — 375,68.
Перед сложением или вычитанием все слагаемые (уменьшаемое, вычитаемое)
можно округлить в соответствии с числом десятичных знаков наименее точно
известной величины. Но, как и в случае умножения или деления, лучше оставить
по одной запасной цифре и по окончании вычислений округлить ее.
Подводя итог обсуждению значащих цифр, отметим два момента. Первый: с
какой точностью вам нужно знать конечный результат? Если, к примеру, вам не-
обходимо знать только, содержит проба: 12 или же 13% определяемого компо-
нента, то все требуемые измерения (и вычисления) достаточно проводить с
двумя значащими цифрами. Если масса навески составляет около 2 г, нет необ-
ходимости взвешивать ее точнее, чем до 0,1 г. И второй: с какой точностью вы
116
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
можете проводить измерения? Очевидно, что, если вы можете измерить значе-
ния оптической плотности окрашенного раствора лишь с тремя значащими циф-
рами (например, значение А = 0,447), то нет смысла измерять массу навески
точнее, чем до трех значащих цифр (например, 6,67 г).
Если мантисса какой-либо измеряемой величины меньше мантисс результа-
тов других измерений, то имеет смысл представлять результаты измерений с од-
ной дополнительной значащей цифрой. Поясним это на следующем примере.
Предположим, вы взвешиваете два объекта с весьма близкими массами (порядка
100 мг) и хотите получить значения их масс с одинаковой точностью, например,
до 0,1 мг или 1 части на тысячу. Масса первого объекта составляет 99,8 мг, а вто-
рого — 100,1 мг. Вы взвесили оба объекта с одинаковой точностью, но второе
значение приводите с одной дополнительной значащей цифрой. Здесь видна
аналогия с тем, как мы добавляем одну цифру к результату умножения или деле-
ния, если его мантисса меньше, чем мантисса числа, лимитирующего точность
вычислений.
Если известно, какая величина лимитирует точность в ходе расчетов, то при
необходимости точность результата можно повысить, либо сделав эту величину
больше (например, увеличив размер навески), либо повысив точность ее измере-
ния (например, проводя взвешивание на более точных весах с дополнительной
значащей цифрой). Кроме того, желательно увеличивать точность измерения
любых величин, мантисса которых мала по сравнению с мантиссами других ве-
личин, для того, чтобы неопределенности значений разных величин стали близ-
ки друг к другу.
В ходе анализа разные величины следует измерять с одинаковой абсолют-
ной неопределенностью, если их предстоит складывать или вычитать, и с одина-
ковой относительной неопределенностью, если их предстоит перемножать или
делить.
Прежде чем сообщить полученный тобою результат
кому бы то ни было, проверь его еще раз.
Эдмунд Ч. Беркли
Если вычисления включают в себя как операции сложения-вычитания, так и
умножения-деления, каждый этап необходимо контролировать отдельно. Реко-
мендуется на всякий случай сохранять одну дополнительную цифру в промежу-
точных результатах, а затем округлить ее в окончательном результате. При
использовании калькулятора можно сохранять все цифры промежуточных резу-
льтатов. Однако не следует думать, что все числа, выдаваемые калькулятором,
верны. Всегда старайтесь сначала оценить примерный порядок ожидаемого ре-
зультата. Если он должен быть около 2%, а калькулятор показывает 0,02, то, ве-
роятно, вы забыли умножить на 100. А если вы ожидаете получить результат
около 20%, а видите 4,3, то, скорее всего, вы допустили ошибку в расчетах, а воз-
можно, в измерениях.
3.4. ЗНАЧАЩИЕ ЦИФРЫ: СКОЛЬКО ТРЕБУЕТСЯ ЦИФР?
117
Пример 3.5
Приведите результат следующих вычислений с максимально возможным чис-
лом значащих цифр:
97 7
-100,0 + 36,04
32,42 )
687
Решение
301 36 +36,04 337 4
---= 0,0491!
687 687 1
В первой серии действий (деление и умножение) точность лимитирует число
97,7. Результат этих действий запишем как 301,36. Перед тем, как произвести
сложение, мы добавили к результату дополнительную (пятую) цифру. Результат
сложения мы округлим до четырех цифр, поскольку, при делении делитель бу-
дет иметь только три значащие цифры. В ходе заключительной операции деления
точность лимитирует делитель — число 687. Но результат по своей мантиссе мень-
ше, чем делитель, поэтому мы запишем его с одной дополнительной цифрой. От-
метим, что, если бы на первом этапе мы округлили результат до 301,4, то
числитель дроби стал бы равным 337,5, а конечный результат—0,4913 (впрочем,
он и в этом случае остается соизмеримым с экспериментальной погрешностью).
Логарифмирование: мыслите категориями мантисс
При переходе от логарифмов к антилогарифмам и наоборот логарифмируемое
число и мантисса логарифма имеют одно и то же число значащих цифр. Напом-
ним, что для логарифмической величины мантиссой называется его дробная
часть (см. приложение В, обзор правил действий с логарифмами). Все десяти-
чные знаки мантиссы логарифма, в том числе нули, являются значащими. Рас-
считаем значение pH 2,0 • 10-3 М раствора НС1, так как pH = -lg[H+]:
pH = -1g 2,0 • IO"3 = (-3 + 0,30) = 2,70
В этом примере -3 является характеристикой логарифма — точной величи-
ной, показателем степени 10 в числе 10-3; 0,30 является мантиссой, логарифмом
величины 2,0, имеющей две значащие цифры. Поэтому и мантисса представлена
с двумя значащими цифрами. Таким образом, здесь запись логарифмической ве-
личины содержит три цифры, несмотря на то, что величина концентрации приве-
дена с двумя значащими цифрами. В свою очередь, при взятии антилогарифмов
результат содержит столько значащих цифр, сколько десятичных знаков имеет
мантисса логарифма. Антилогарифм величины 0,072, мантисса которого содер-
жит три цифры, будет записан как 1,18, а логарифм 12,1 —как 1,083 (1 —харак-
теристика, 0,083 — мантисса, имеющая три значащие цифры).
118
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
3.5. Правила округления
Если цифра, следующая за последней значащей цифрой, больше 5, округление
ведут в большую сторону, а если меньше 5, то в меньшую сторону:
9,47 * 9,5
9,43 « 9,4
Если отбрасываемая цифра равна 5, последнюю значащую цифру округляют до
ближайшей четной:
8,65 « 8,6
8,75 « 8,8
8,55 « 8,6
Это правило основано на допущении, что последняя значащая цифра с равной
вероятностью может оказаться как нечетной, так и четной. Поэтому, если мас-
сив чисел достаточно велик, то в нем будет примерно одинаковое число четных
и нечетных цифр, предшествующих цифре 5. Отметим, что правило округления
до четного действует только тогда, когда необходимо отбросить только одну
цифру 5 (а не две цифры, например 51).
3.6. Способы выражения правильности
Для характеристики правильности результата можно использовать различные
численные величины. В любом случае для оценки правильности необходимо
иметь для сравнения значение измеряемой величины, принимаемое за истинное.
Абсолютная погрешность
Разность между измеренным и истинным значением с учетом знака называется
абсолютной погрешностью. Она имеет ту же размерность и выражена в тех же
единицах, что и измеряемая величина. Если проба содержит 2,62 г определяемо-
го вещества, а по результатам анализа найдено 2,52 г, то абсолютная погреш-
ность составляет-0,10 г. Если измеряемая величина представляет собой среднее
из нескольких значений, то погрешность называется средней. Среднюю по-
грешность можно вычислить и как среднее значение (с учетом знаков) индивиду-
альных погрешностей отдельных результатов.
Относительная погрешность
Отношение абсолютной или средней погрешности к истинному значению на-
зывается относительной погрешностью. Ее обычно выражают в процентах.
Так, в приведенном выше примере относительная погрешность составляет
(-0,10/2,62) • 100% = -3,8%. Относительной мерой правильности называется
отношение измеренного значения к истинному (также выраженное в процен-
3.7. СТАНДАРТНОЕ ОТКЛОНЕНИЕ - ВАЖНЕЙШАЯ СТАТИСТИЧЕСКАЯ ВЕЛИЧИНА
119
тах). В том же примере мера правильности составляет (2,52/2,62) • 100% = 96,2%.
Подчеркнем, что истинное значение никогда не бывает известно, поэтому для
вычисления как относительной погрешности, так и относительной меры правиль-
ности используют данные двух измерений.
Помимо процентов, относительную погрешность выражают и в других еди-
ницах. При очень точных измерениях относительная погрешность может со-
ставлять менее 1%, поэтому удобнее использовать более мелкие единицы. 1%
означает одну часть на 100, или 10 частей на 1000. Малые погрешности обычно
выражают в частях на тысячу (англ, part per thousand, ppt). Так, число 23 по от-
ношению к числу 6725 составляет 3,4 ppt. Воспроизводимость результатов изме-
рений также часто выражают в частях на тысячу.
Пример 3.6
Результат анализа составляет 36,97 г, а истинное значение принимается равным
37,06 г. Чему равна относительная погрешность (в частях на тысячу)?
Решение
Абсолютная погрешность = 36,97 г - 37,06 г = -0,09 г
Относительная погрешность = ‘ Ю00%о = -2,4 ppt
Для обозначения частей на тысячу используют символ %о подобно тому, как для
обозначения частей на сто — символ %.
3.7. Стандартное отклонение - важнейшая статистическая
величина
При проблемах с воспроизводимостью
проведи измерение только один раз.
Мудрость аналитика
Любой набор результатов анализа должен сопровождаться характеристикой их
воспроизводимости. Существуют различные способы выражения воспроизво-
димости.
Для воображаемого бесконечного набора результатов измерений введем ве-
личину, называемую стандартным отклонением и обозначаемую су:
-ц)2
НЧ- (ЗЛ)
120
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
где N — число измерений, х. — результат отдельного измерения, ар — среднее
из бесконечного набора результатов, т. е. предельное значение среднего при
7V —> оо. В действительности мы всегда располагаем лишь конечным набором ре-
зультатов. Среднее конечного набора данных обозначается х и вычисляется как
^(xf /7V), х —> ц с увеличением N.
Величину х можно использовать вместо ц при вычислении стандартного от-
клонения. Но в этом случае сумма квадратов отклонений единичных значений
от среднего [числитель дроби в выражении (3.1)] содержит не N, а только (N- 1)
независимых слагаемых (поскольку, зная (N- 1) единичных результатов и сред-
нее, всегда можно вычислить оставшийся единичный результат). Как говорят
статистики, в этом случае из исходных N степеней свободы одна степень свободы
использована для вычисления среднего, а для вычисления стандартного отклоне-
ния остается (N- 1) степеней свободы.
Можно показать, что для конечного числа данных (особенно при N < 30) бо-
лее точное приближение для о (оценка) получатся в случае, если в знаменателе
выражения (3.1) вместо общего числа данных N использовать число степеней
свободы (N- 1) (для компенсации возможных различий между х и ц). Тогда
стандартное отклонение, рассчитанное из конечного числа данных, будет выра-
жаться следующей формулой:
(3.2)
Величина s представляет собой лишь приближенную оценку и — тем более точ-
ную, чем больше число данных, из которых она рассчитана. Поскольку число ре-
зультатов, получаемых при химическом анализе, невелико, характеристикой
воспроизводимости всегда служит s. Для малого числа данных (четыре и менее)
существует другой способ оценки s — см. разд. 3.15 и уравнение (3.17).
Пример 3.7
Рассчитайте среднее и стандартное отклонение для следующей серии результа-
тов анализа: 15,67, 15,69 и 16,03 г.
Решение
xi х(-х (х,-х)2
15,67 0,13 0,0169
15,69 0,11 0,0121
16,03 0,23 0,0529
S 47,39 S 0,47 S 0,0819
3.7. СТАНДАРТНОЕ ОТКЛОНЕНИЕ - ВАЖНЕЙШАЯ СТАТИСТИЧЕСКАЯ ВЕЛИЧИНА
121
УX/ 47 39
х —2-—= 15,80
N 3
10,0819
V 3-1
= 0,20 г
Стандартное отклонение можно рассчитать и по следующей формуле, экви-
валентной выражению (3.2):
'IX2-(£*• )2Л
#-i
(3.3)
Эту формулу удобно использовать при вычислениях на калькуляторе. Отметим,
что многие калькуляторы снабжены встроенными программами для автомати-
ческого вычисления стандартного отклонения после ввода данных.
Пример 3.8
Рассчитайте стандартное отклонение для данных, приведенных в примере 3.7,
пользуясь уравнением (3.3).
Решение
X,- X,2
15,67 245,55
15,69 246,18
16,03 256,96
S 47,39 S 748,69
, (748,69-(47,39)г/3 =
\ 3-1
Различие в 0,01 г по сравнению с результатом расчета в примере 3.7 стати-
стически незначимо, поскольку колебания самих данных составляют по мень-
шей мере ±0,2 г. Пользуясь формулой (3.3), следует в промежуточных вычислениях
(х?) сохранять одну или даже две дополнительные цифры.
Стандартное отклонение, рассчитанное описанным выше способом, являет-
ся оценкой возможной погрешности результата единичного измерения. Сред-
ний результат из N измерений характеризуется меньшим возможным разбросом
значений по сравнению с единичными результатами. Чем больше N, тем меньше
этот разброс. При очень большом N среднее практически не отличается от ц, и
122
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
разброс его значений стремится к нулю. Можно показать, что среднее из /^резуль-
татов в W раз более достоверно, чем единичное значение. Так, случайная погреш-
ность среднего из четырех результатов измерений вдвое меньше, чем погрешность
результата единичного измерения. Воспроизводимость среднего значения возрас-
тает пропорционально корню квадратному из числа измерений.Таким образом,
Стандартное отклонение среднего = 5mean
(3.4)
Стандартное отклонение среднего иногда называют стандартной погрешно-
стью.
Часто стандартное отклонение выражают в относительной форме — в виде
относительного стандартного отклонения, представляющего собой отноше-
ние стандартного отклонения к среднему. Обычно его выражают в процентах и
в этом случае иногда называют коэффициентом вариации.
Пример 3.9
При выполнении параллельных взвешиваний получены четыре результата: 29,8,
30,2,28,6 и 29,7 мг. Рассчитайте стандартное отклонение единичного результата
и среднего значения. Выразите их в абсолютных единицах измерения и в отно-
сительных величинах (%).
Решение
xi xt-x (х,.-х)2
29,8 0,2 0,04
30,2 0,6 0,36
28,6 1,0 1,00
29,7 0,1 0,01
118,3 S 1,9 S 1,41
- 118’3 ЭОА
х =—— =29,6
4
141
s = Л —— = 0,69 мг (абсолютное стандартное отклонение)
V 4-1
• 100% = 2,3% (относительное стандартное отклонение)
0,69
5mean = ~~i=‘= ^34 мг (абсолютная стандартная погрешность)
л/4
034
• 100% = 1,1% (относительная стандартная погрешность)
3.7. СТАНДАРТНОЕ ОТКЛОНЕНИЕ - ВАЖНЕЙШАЯ СТАТИСТИЧЕСКАЯ ВЕЛИЧИНА
123
Воспроизводимость результатов измерений можно повысить, увеличив чис-
ло данных. Однако, как видно из уравнения (3.4), стандартное отклонение сред-
него обратно пропорционально не числу измерений, а лишь корню квадратному
из этого числа. Рано или поздно даже незначительное улучшение воспроизводи-
мости потребует слишком большого числа дополнительных данных. А чтобы
уменьшить стандартное отклонение, например, в 10 раз, число измерений необ-
ходимо увеличить в 100 раз.
Фактор случайности необходим для того,
чтобы результаты статистических расчетов
получились правильными.
Мудрость аналитика
Числом повторных измерений, достаточным для практических целей, мож-
но считать такое, при котором стандартное отклонение среднего, характеризую-
щее случайную погрешность, станет сравнимым по величине с систематической
погрешностью (если последнюю, разумеется, невозможно выявить и скорректи-
ровать). Это связано с тем, что систематические погрешности невозможно устра-
нить посредством повторных измерений.
Из величины стандартного отклонения можно рассчитать интервал, в кото-
рый результат измерения попадает с той или иной вероятностью. На рис. 3.2
изображены такие интервалы применительно к нормальному распределению.
Пользуясь уравнением кривой нормального распределения, можно показать ма-
тематически, что 68% всех величин отклонений единичных данных от среднего
попадает в интервал с полушириной s (точнее, а, поскольку этот вывод, строго
говоря, справедлив лишь для бесконечной серии данных). Аналогично, 95% от-
клонений попадают в интервал с полушириной 25, а 99% — в интервал с полуши-
риной 2,55. Таким образом, 68% результатов единичных измерений находятся в
интервале х ± 5, 95% — в интервале х ± 25, 99% — в интервале х ± 2,55 и т. д.
Повторим, что величины этих интервалов рассчитаны в предположении,
что серия результатов измерений бесконечна (см. ниже о понятии «доверитель-
ный интервал» а также пример 3.15). В действительности нельзя утверждать, что
истинное значение с вероятностью 95% попадает в интервал х ± 25. На то есть, по
крайней мере, две причины. Во-первых, это могло бы быть справедливо только
при отсутствии систематических погрешностей. Систематические погрешности
приводят к смещению всей кривой нормального распределения относительно
истинного значения. Во-вторых, число результатов всегда конечно. При этом,
чем оно меньше, тем менее достоверны значениях и 5. В то же время существует
способ расчета доверительных интервалов (см. далее) из величин х и 5, который
учитывает это обстоятельство.
Понятно, что данные по воспроизводимости результатов можно предста-
вить разными способами. Если результат представлен в виде х ± 8, всегда следу-
ет указывать, к каким условиям это относится и что именно означает 8.
В частности, это может быть 5,25, стандартное отклонение среднего или относи-
тельное стандартное отклонение.
124
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
В статистике часто используют величину, называемую дисперсией. Она
представляет собой квадрат стандартного отклонения s2. Мы будем пользовать-
ся величинами дисперсий при рассмотрении F-теста Фишера и наложения по-
грешностей (разд. 3.13).
3.8. Электронные таблицы
Электронные таблицы — это эффективные вычислительные программы, кото-
рые можно использовать в различных целях, в частности, для анализа данных и
построения графиков. Ими удобно пользоваться в ходе обработки данных, при
выполнении повторных измерений, отображении результатов в графической
или табличной форме. Электронные таблицы имеют встроенные функции, на-
пример, для вычисления стандартных отклонений и других статистических вели-
чин, выполнения расчетов по формулам и с данными, вводимыми пользователем.
К широко используемым электронным таблицам относятся Microsoft Excel, Lo-
tus 1-2-3 и Quattro Pro. В основе они все сходны, небольшие отличия есть в конк-
ретных командах и синтаксисе. В наших примерах мы будем использовать Excel
как наиболее распространенную и доступную программу.
Возможно, вы уже пользовались электронными таблицами ранее и знакомы
с их основными функциями. Здесь мы обобщенно рассмотрим те аспекты, кото-
рые представляют наибольший интерес для решения прикладных задач аналитиче-
ской химии. За более подробной информацией следует обратиться к руководствам
по работе с таблицами или справочной системе самой программы Excel Help, на-
жав соответствующую кнопку на панели инструментов.
На странице http://science.csustan.edu/titorial/Exel/index.htm находится пре-
восходное руководство по пользованию таблицами Excel, составленное препо-
давателями Университета штата Калифорния (округ Станислаус). В нем описа-
ны основные рабочие функции этих таблиц, включая ввод данных и формул,
форматирование ячеек, построение графиков и регрессионный анализ. Обязатель-
но прочтите его перед тем, как продолжать работу, — вы найдете это руководство
очень полезным. Сайт Университета западного Кентукки www.wku.edu/-conteed/
СНЕМЗЗО содержит сводку инструкций по построению графиков при помощи
таблиц Microsoft Excel и Lotus 1-2-3. Посетите также сайты, на которые ссыла-
ются страницы excelhandout.html и lotushandout.html.
Лист электронной таблицы состоит из ячеек, расположенных по столб-
цам (обозначенным А, В, С, ...) и строкам (1, 2, 3, ...). Отдельная ячейка обо-
значается комбинацией буквы столбца и номера строки, например, ВЗ. Это
обозначение ячейки называется также ее адресом или ссылкой. Для иллюстра-
ции на рис. 3.3 в некоторых ячейках приведены их обозначения-адреса. Если
навести указатель мыши (в форме крестика) на любую ячейку и щелкнуть левой
кнопкой мыши, то соответствующая ячейка становится активной (ее границы
выделяются жирными линиями), обозначение активной ячейки отображается в
верху экрана слева от окна формул, а ее содержимое — правее, в окне формул
после знака равенства.
3.8. ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
125
Заполнение ячеек
В каждую ячейку можно ввести текст, число или формулу. Именно благодаря
использованию формул электронные таблицы так удобны в работе. Они позво-
ляют проводить одни и те же вычисления с разными числами. Правила пользова-
ния формулами мы проиллюстрируем на примере расчета значений веса воды,
вытекающей из двух различных пипеток номинального объема 20 мл, по разно-
сти между весом мерной колбы, наполненной водой, и весом пустой колбы.
Каждый шаг вычислений сверяйте с рис. 3.4.
Откройте программу Excel, щелкнув кнопкой мыши по ее ярлыку (или вы-
звав ее из раздела Программы стартового меню). Чтобы ввести в ячейку текст,
число или формулу, дважды щелкните по ней. В перечисленные ниже ячейки вве-
дите следующее (по завершении ввода данных в ячейку нажимайте клавишу Enter):
Ячейка А1: Чистый вес
Ячейка АЗ: Пипетка
Ячейка А4: Колба + вода, г
Ячейка А5: Колба, г
Ячейка А6: Вода, г
Если вы хотите внести исправления в содержимое ячейки, дважды щелкни-
те по ней, после чего редактируйте текст. Текст можно редактировать и в окне
формул. Если вы щелкнете один раз, вводимый текст заменит прежний. Чтобы
полностью отобразить достаточно длинный текст в ячейках столбца А, увеличь-
те ширину этого столбца. Для этого установите указатель мыши точно на линию,
разделяющую столбцы А и В, в верхней части таблицы (в строке, содержащей
обозначения столбцов), и, оставляя кнопку мыши нажатой, перетаскивайте указа-
тель вправо до тех пор, пока ширина столбца не станет достаточной для отображе-
ния всего текста. При этом сместятся и все ячейки, расположенные правее.
Рис. 3.3
Ячейки электронной таблицы
А В С D Е
1 А1 В1 С1 ит. д.
2 А2 В2 С2 ит. д.
3 АЗ ВЗ СЗ ит. д.
4
5
Рис. 3.4
Заполнение ячеек
А В С D
1 Чистый вес
2
3 Пипетка 1 2
4 Колба+вода, г 47,702 49,239
5 Колба, г 27,687 29,199
6 Вода, г 20,015 20,040
7
8 ячейка В6=В4-В5
9 ячейка С6=С4-С5
10
126
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
Продолжите ввод.
Ячейка ВЗ: 1
Ячейка СЗ: 2
Ячейка В4: 47,702
Ячейка С4: 49,239
Ячейка В5: 27,687
Ячейка С5: 29,199
Ячейка В6: =В4-В5
Формулу в ячейку В6 можно ввести и так: ввести символ «=», затем щелк-
нуть на ячейке В4, затем ввести символ «-», затем щелкнуть на ячейке В5. Все
ячейки, составляющие прямоугольный блок В4-С6, необходимо предваритель-
но отформатировать, чтобы числовые данные содержали три знака после запя-
той. Для этого сначала выделите этот блок (щелкните на ячейке в одном его углу
и перетащите указатель мыши к ячейке в противоположном углу). Затем в строке
Меню последовательно нажмите Формат: Ячейки: Число (Format:
Cells: Number). В появившееся окошко Число десятичных знаков
(Decimal places) введите 3 и нажмите клавишу ОК.
В ячейку С6 также необходимо ввести формулу. Вы, конечно, можете ее
просто набрать точно так же, как и при вводе в ячейку В6. Но можно поступить
проще, скопировав ее из ячейки В6. Подведите указатель мыши к правому ниж-
нему углу ячейки В6 и перетащите его в ячейку С6. При этом в ячейке С6 ока-
жется нужная формула. Точно так же можно поступить и при заполнении всех
ячеек, расположенных справа от С6, если вы калибруете еще другие пипетки.
Чтобы заполнить ячейку формулой, находящейся в соседней ячейке (сверху или
слева), можно поступить и так: выделить заполняемую ячейку, а затем последо-
вательно нажать Правка: Заполнить : Вниз (Edit: Fill: Down) (или
Вправо (Right), в зависимости от того, где расположена ячейка с копируемой
формулой).
Дважды щелкните на ячейке В6. При этом отобразится формула, содержа-
щаяся в этой ячейке, а также высветятся все другие ячейки, задействованные в
формуле. Эту же операцию проделайте и с ячейкой С6. Обратите внимание, что
каждый раз, когда вы щелкаете кнопкой мыши (один или два раза) на ячейке, со-
держащей формулу, то эта формула появляется в окне формул.
Сохранение электронной таблицы
Чтобы сохранить только что созданную вами электронную таблицу, последова-
тельно нажмите Файл: Сохранить как (File: Save As). Я предпочитаю
сначала сохранять все документы на рабочем столе. Оттуда их можно перемес-
тить в любую папку, например, Мои документы (Му Documents). В этом слу-
чае они не будут потеряны. Так что в верхнем из появившихся окон выберите
Рабочий стол (Desktop). В нижнее окно введите имя документа, например,
Pipet Calibration. После этого нажмите клавишу Сохранить (Save). Если вы за-
тем захотите переместить документ в какую-либо папку какого-либо диска, пе-
ретащите его ярлык с рабочего стола в соответствующую открытую папку.
3.8. ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
127
Печать электронной таблицы
Последовательно нажмите Файл: Параметры страницы (File: Page
Setup). По умолчанию лист печатается на бумаге книжного формата (Portrait),
т. е. 81 х 11 дюймов, ориентированного вертикально. Если вы выводите на пе-
чать большое число столбцов, может быть целесообразно ориентировать лист
бумаги горизонтально (Landscape). Если вы хотите, чтобы таблица была напеча-
тана с разделительными линиями сетки, нажмите Лист: Сетка (Sheet:
Gridlines). После этого все готово к печати. Нажмите Печать: ОК
(Print: ОК). На печать будет выведена только рабочая область листа, без
обозначений столбцов и строк.
Абсолютные и относительные адреса ячеек
В приведенном выше примере в ходе копирования формулы мы использовали
относительные адреса ячеек. Формула, введенная в ячейку В6, по существу,
означает следующее: «Из числа, расположенного на две ячейки выше (относите-
льно данной, т. е. В6), вычесть число, расположенное на одну ячейку выше».
Формула, скопированная в ячейку С6, содержит те же инструкции, но уже при-
менительно к ячейке С6.
Однако часто требуется, чтобы в каждом вычислении участвовала величи-
на, находящаяся в какой-то конкретной, одной и той же, ячейке — например, не-
кая константа. В этом случае мы должны задать в формуле адрес этой ячейки как
абсолютный адрес. Абсолютный адрес записывается при помощи знака «$», по-
мещаемого как перед обозначением столбца, так и перед номером строки, на-
пример, $В$2. В этом случае как бы мы ни перемещали формулу по строкам и
столбцам таблицы, адрес останется одним и тем же.
Проиллюстрируем сказанное на примере создания таблицы для расчета
средних значений из нескольких серий данных. Заполните лист таблицы следу-
ющим образом, сверяясь с рис. 3.5:
А1: Средний объем титранта
АЗ: Порядковый номер
ВЗ: Серия А, мл
СЗ: Серия В, мл
В4: 39,27
В5: 39,18
В6: 39,30
В7: 39,22
С4: 45,59
С5: 45,55
С6: 45,63
С7: 45,66
А4: 1
Порядковые номера титрований (1^1) можно ввести вручную. Однако су-
ществуют и автоматические способы ввода в соседние ячейки чисел, возраста-
ющих с определенным шагом. После ввода числа 1 в ячейку А4 нажмите
128
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
Правка: Заполнить : Прогрессия (Edit: Fill: Series). В открыв-
шемся окне выберите опции По столбцам (Columns) и Арифметическая
(Linear), установите величину шага (Step Value) равной 1, Предельное
значение (Stop Value) равным 4 и нажмите ОК. Числа от 2 до 4 будут введе-
ны в ячейки А5-А7. Можно также предварительно выделить диапазон ячеек
(начиная с исходной А4), которые вы хотите заполнить. В этом случае нет не-
обходимости вводить предельное значение. Кроме того, вы можете осуществить
ввод при помощи формул. В ячейку А5 введите «=А4+1» (в этой формуле исполь-
зован относительный адрес}. Затем протяните эту формулу вниз, выделив ячей-
ку А5 и нажав Правка: Заполнить : Вниз (Edit: Fill: Down). Наконец,
после ввода формулы можно выделить ячейку А5, установить указатель мыши на ее
нижний правый угол и при нажатой кнопке мыши перетащить указатель до ячейки
А7. При этом формула автоматически скопируется во все ячейки диапазона.
Теперь в ячейку В8 введем формулу для расчета среднего. Оно равно сумме
значений, разделенной на число значений (оно находится в ячейке А7).
В8: =СУММ(В4:В7)/$А$7
Адрес делителя содержит символы «$», поскольку это абсолютный адрес
(далее мы скопируем эту формулу вправо, в ячейку С8, и при этом адрес делите-
ля не должен измениться). Помещая символ $ перед обозначением столбца и пе-
ред номером строки, мы гарантируем, что этот адрес останется неизменным при
копировании формулы как по горизонтали, так и по вертикали. Выражение
sum(B4:B7) — это синтаксическое выражение программы, предназначенное
для суммирования массива чисел — в данном случае начиная с ячейки В4 и за-
канчивая В7. При вводе этого выражения сами адреса ячеек набирать не обязатель-
но. Достаточно ввести «=СУММ(» , затем щелкнуть на ячейке В4, перетащить
указатель мыши до ячейки В7 и в заключение ввести «)».
Таким образом, мы рассчитали среднее для серии А и хотим сделать то же
самое для серии В. Для этого выделим ячейку В8, наведем курсор на ее правый
А I' В С D
1 Средний объем титранта
2
3 Порядковый номер Серия А, мл Серия В, мл
4 1 39,27 45,59
5 2 39,18 45,55
6 3 39,30 45,63
7 4 39,22 45,66
8 Среднее: 39,24 45,61
9 Ст. отклонение 0,053150729 0,047871355
10
11 Ячейка В8= СУММ(В4:В7)/$А$7 Скопировать вправо в ячейку С8
12 Ячейка В9= СТАНДОТКЛОН(В4:В7) Скопировать вправо в ячейку С9
13 Жирным шрифтом выделены те ячейки, в которые были введены формулы
Рис. 3.5. Относительные и абсолютные адреса ячеек
3.8. ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
129
нижний угол и при нажатой кнопке мыши перетащим его в ячейку С8. В ней не-
медленно появится значение среднего для второй серии! Щелкните дважды по
ячейке С8, и вы увидите содержащуюся в ней формулу. Обратите внимание, что
делитель имеет тот же адрес, что и в ячейке В8 (поскольку он абсолютный), а ад-
реса суммируемых значений изменились (они относительные). Если бы мы не
сопроводили адрес делителя символами «$», то программа считала бы его отно-
сительным, и в ячейке С8 он был бы не А7, а В7.
Использование статистических функций Excel
Программа Excel содержит множество математических и статистических функ-
ций. Их можно использовать в расчетах в готовом виде вместо того, чтобы писать
формулы. Попробуем использовать одну из таких функций для автоматического
вычисления среднего. Выделите пустую ячейку и щелкните мышкой на символе
«Д», расположенном на панели инструментов. При этом откроется окно Мас-
тер функций (Paste Function). В меню Категория (Function Cate-
gory) выберите Статистические (Statistical). После этого окно
приобретет следующий вид:
В меню Функция (Function Name) выберите СРЗНАЧ (AVERAGE) и нажмите
ОК. Далее в окне Число (Number) введите В4:В7 и снова нажмите ОК. Будет
вычислено то же самое значение среднего, которое вы получили при помощи соб-
ственной формулы. Можно также активировать ячейку и вручную ввести в нее
формулу =СРЗНАЧ(В4:В7) (=average(B4:B7)). Попробуйте и этот способ.
Рассчитаем теперь стандартное отклонение результатов. Выделите ячейку В9.
В меню Функция (Function Name)окна Статистические (Statisti-
cal Functions) выберите СТАНДОТКЛОН (STDEV) (или непосредственно
введите =СТАНДОТКЛОН(В4:В9) (=stdev(B4:B9)) в ячейку В9). Скопируйте
формулу из ячейки В9 в ячейку С9. Рассчитайте стандартные отклонения по
формуле (3.2) и сравните их с результатам расчетов программы Excel. Рассчи-
танные значения стандартных отклонений следует округлить, например, для се-
рии А до 0,05 мл.
130
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
Полезные синтаксические выражения
Excel содержит множество математических и статистических функций (синтак-
сических выражений), использование которых упрощает ход вычислений. Вни-
мательно изучите списки функций [меню Вставка (Paste) Функция
(Function Name)] в категориях функций Математические (Math &
Trig) и Статистические (Statistical Function) окна Мастер фун-
кций (Paste Function) (открывается нажатием на кнопку с символом «£»
на панели инструментов). Ниже приведены некоторые функции, которые пона-
добятся при изучении этой книги:
Математические и тригонометрические функции
LOG 10 ПРОИЗВЕЛ (PRODUCT) СТЕПЕНЬ (POWER) КОРЕНЬ (SQRT) десятичный логарифм числа произведение массива данных возведение в степень квадратный корень
Статистические функции СРЗНАЧ (AVERAGE) МЕДИАНА (MEDIAN) СТАНДОТКЛОН (STDEV) ТТЕСТ (TTEST) среднее из серии данных медиана серии данных стандартное отклонение серии данных доверительная вероятность, соответствующая статистике теста Стьюдента
ДИСПА (VAR) дисперсия серии данных
Эти синтаксические выражения можно вводить вручную (как мы делали ра-
нее), указывая в скобках после имени функции диапазон ячеек, содержащих ар-
гументы.
Это краткое руководство послужит вам основой для более полного изуче-
ния возможностей электронных таблиц. Любую формулу, приведенную в книге,
вы можете ввести в активную ячейку и подставить в нее соответствующие дан-
ные для расчетов. Кроме того, электронные таблицы снабжены разнообразными
средствами для анализа данных. Вы можете строить графики и диаграммы, на-
пример, градуировочные зависимости (графики зависимости сигнала отклика
прибора от концентрации) и проводить статистический анализ полученных
данных. Некоторые примеры приведены далее в этой главе.
3.9. Наложение погрешностей: это не простое суммирование
При рассмотрении значащих цифр мы отметили, что для результата умножения
или деления относительная неопределенность не может быть выше, чем для
участвующей в операции величины, которая характеризуется наибольшей
относительной неопределенностью. Аналогично обстоит дело с абсолютными
неопределенностями значений при сложении или вычитании. До сих пор мы не
3.9. НАЛОЖЕНИЕ ПОГРЕШНОСТЕЙ: ЭТО НЕ ПРОСТОЕ СУММИРОВАНИЕ
131
рассматривали конкретные численные значения этих неопределенностей, а про-
сто предполагали, что неопределенность составляет по меньшей мере плюс-ми-
нус одну единицу в последней цифре каждого числа.
Если же неопределенность каждого значения известна, то существует воз-
можность оценить численное значение неопределенности результата вычислений.
В ходе последовательности вычислений неопределенность каждого исходного дан-
ного вносит свой вклад в общую неопределенность результата. Как говорят,
происходит наложение неопределенностей — относительных или абсолютных в
зависимости от характера арифметических действий (умножение-деление или
сложение-вычитание).
Сложение и вычитание:
мыслите категориями абсолютных дисперсий
Рассмотрим наложение неопределенностей при сложении и вычитании на сле-
дующем примере:
(65,06 ± 0,07) + (16,13 ± 0,01) - (22,68 ± 0,02) = 58,51 (±?)
Неопределенности, приведенные в этом примере, характеризуют случайные
(недетерминированные) погрешности, присущие соответствующим величинам и
выраженные в виде их стандартных отклонений. Максимально возможная по-
грешность суммы составила бы ±0,10. Это произошло бы в случае, если погрешно-
сти аргументов, во-первых, все одновременно приняли бы свои максимальные
значения, а, во-вторых, имели бы одинаковые знаки. Минимально возможная по-
грешность составила бы 0,00. Это возможно, если погрешности аргументов случай-
ным образом скомпенсируют друг друга. Оба эти крайних случая маловероятны;
наиболее вероятное значение погрешности находится где-то посередине. Мож-
но показать, что наиболее вероятное значение погрешности суммы или разности
равно квадратному корню из суммы абсолютных дисперсий аргументов (напо-
минаем, что при сложении и вычитании суммируются абсолютные неопреде-
ленности). Иными словами, абсолютная дисперсия суммы (разности) равна
сумме абсолютных дисперсий аргументов. Если а = b + с - то
= ^ь +S2C +s2d
sa = ^sb + Sc + sd
(3.5)
(3-6)
В приведенном примере
sa =7(±o>°7)2 +(±0,01)2 +(±0,02)2 =
= 7(±49-10“4) + (±1-10-4) + (±4-10“4) =
= V±54-10-4 =±7,3-IO-2
132
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
Таким образом, результат составляет 58,51 ±0,07. Число ±0,07 характеризует
абсолютную неопределенность. Если мы хотим оценить относительную неопре-
деленность, то она составит
=2i2Z.ioo%=±од,%
58,51 2
Пример 3.10
Вы получили три равные по весу партии урановой руды. По данным анализа со-
держание урана в них составило 3,978 ± 0,004%, 2,536 ± 0,003% и 3,680 ± 0,003%
соответственно. Каково среднее содержание урана и чему равны абсолютная и
относительная неопределенности этой величины?
Решение
_ _ (3,978 ± 0,004%) + (2,536 ± 0,003%) + (3,680 ± 0,003%)
3
Неопределенность суммы равна
sa =7(±0,004)2 + (±0,003)2 +(±0,003)2 =
= 7(±16 • 10’6) + (±9 • 10’6) + (±9 • 10"6) =
= 7±34-1О~6 = ±5,8 • 10’3% U
Отсюда абсолютная неопределенность результата равна
10194
± 0,006% = 3,398 ± 0,006% U
3
Обратите внимание, что число 3 в знаменателе этой дроби точное и неопреде-
ленности не содержит. Поэтому относительная неопределенность содержания
урана составляет
5,8 10’3%U . 1П_3
----------= 2-10 или 0,2%
3,398% U
3.9. НАЛОЖЕНИЕ ПОГРЕШНОСТЕЙ: ЭТО НЕ ПРОСТОЕ СУММИРОВАНИЕ
133
Умножение и деление:
мыслите категориями относительных дисперсий
Рассмотрим следующую последовательность действий:
(13,67 ±0,02)-(120,4 ±0,2) = 356 0 (± ?)
4,623 ±0,006
В этом случае (при умножении и делении) складываются относительные неоп-
ределенности, а наиболее вероятная погрешность выражается корнем квадратным
из суммы относительных дисперсий. Таким образом, относительная дисперсия ре-
зультата равна сумме относительных дисперсий аргументов.
Если а = bc/d, то
(Sa ) rel (sb ) rel + (Sc ) rel (Sd ) rel
(3.7)
(Sa ) rel
) rel + (Sc ) rel + (Sd ) rel
(3.8)
В приведенном примере
(^)rei =^^ = ±0,0015
* rel 13,67
+02
fe)rel = —^— = ±0,0017
e 120,4
, . ±0,006 xnnniQ
(^)rel =---—--- = ±0,0013
d rel 4,623
±0,0015)2 +(±0,0017)2 +(±0,0013)2
) rel
= д/(±2,2 • 10-6 ) + (±2,9 • 10-6 ) + (±1,7 • 10-6 ) =
= д/(±6,8 • 10—6) = ±2,6 • 10“3
Абсолютная неопределенность рассчитывается как
sa = а • (sa ) rel = 356,0 • (±2,6 • 10’3) = ±0,93
Таким образом, результат следует представить как 356,0 ± 0,9.
Пример 3.11
Рассчитайте неопределенность величины количества хлорид-ионов (в миллимо-
лях), содержащихся в 250,0 мл анализируемого раствора, если на титрование
трех аликвот по 25,00 мл было израсходовано 36,78, 36,82 и 36,75 мл раствора
нитрата серебра. Концентрация раствора AgNO3 равна 0,1167 ± 0,0002 М.
134
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
Решение
Среднее значение объема титранта равно
36,78+36,82 + 36,75
—----------------— = 36,78 мл
3
Неопределенность этой величины выразим в виде стандартного отклонения:
xi Xj-X (Х;-Х)2
36,78 0,00 0,0000
36,82 0,04 0,0016
36,75 0,03 0,0009
S 0,0025
/0,0025
V 3-1
= 0,035. С учетом неопределенности среднее значение объема
составляет 36,78 ± 0,04 мл.
Количество хлорид-ионов в аликвоте составляет
(0,1167 ± 0,0002 ммоль/мл) • (36,78 ± 0,04 мл) = 4,292 (±?)
_ ±0,0002
<Л+е1 - ОД167
= ±0,0017
_ ±0,035
Sc reI 36,78
= ±0,00095
(sa ) reI = д/ (±0,0017)2 + (±0,00095)2 =
= д/(±2,9 • 10’6 ) + (±0,90 • IO-6) =
= 7+3,8-IO"6 =±1,9-IO-3
Абсолютная неопределенность содержания Cl- (в миллимолях) составляет
4,292 • (±0,0019) = ±0,0082 ммоль
ммоль С1_ в 25 мл = 4,292 ± 0,0082 ммоль
ммоль С1~в 250 мл = 10(4,292 ± 0,0082) = 42,92 ± 0,08 ммоль
В ходе расчетов мы сохраняли по одной дополнительной цифре во всех проме-
жуточных результатах. Обратите внимание, что абсолютная неопределенность
содержания вещества пропорциональна размеру пробы. Она не остается посто-
янной, например, при увеличении в два раз массы или объема пробы.
Если расчетная формула содержит как операции умножения-деления, так и
сложения-вычитания, вычисление общей неопределенности следует проводить
поэтапно.
3.9. НАЛОЖЕНИЕ ПОГРЕШНОСТЕЙ: ЭТО НЕ ПРОСТОЕ СУММИРОВАНИЕ
135
Пример 3.12
Вы получили три партии железной руды весом 2852,1578 и 1877 фунтов (1b). Не-
определенности этих величин составляют ±5 1b. По результатам анализа содержа-
ние железа в этих партиях составляет 36,28 ± 0,04%, 22,68 ± 0,03% и 49,23 ± 0,06%
соответственно. Стоимость руды составляет 15 центов в расчете на один фунт
железа. Сколько вам предстоит заплатить за руду и какова неопределенность
этой величины?
Решение
Следует для каждой партии руды рассчитать массу железа и ее неопределен-
ность, а затем сложить значения масс и рассчитать неопределенность получен-
ного результата. Относительные неопределенности значений масс железа равны
= ±0,0017 — = ±0,0032 — = ±0,0027
2852 1578 1877
Относительные неопределенности результатов анализа равны
^^ = ±0,0011 =^ = ±0,0013 =^ = ±0,0012
36,28 22,68 49,23
Значения масс железа, содержащегося в партиях руды, равны
(2852±51b)-(36,28±0,04Q/o) ре
100
(sa ) ге1 - д/(±0,0017)2 +(±0,0011)2 = ±0,0020
sa =1034,7 • (±0,0020) = ±2,11b
lb Fe= 1034,7 ±2,1
(Везде сохраняем по одной дополнительной цифре!)
(1578±51ЬИ22,68±0,03°/о) ре
100
(sa ) ге1 = д/(±0,0032)2 + (±0,0013) 2 = ±0,0034
sa = 357,89 • (±0,0034) = ±1,2 1b
lb Fe= 357,9 ±1,2 lb
(1877±51b)-(49,23±0,06Q/o)= +
100
(sa ) rel = -J(±0,0027)2 + (±0,0012) 2 = ±0,0030
sa = 924,05 • (±0,0030) = ±2,8 lb
lb Fe = 924,0 ±2,8 lb
136
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
Всего железа = (1034,7 ±2,11b) + (357,9 ± 1,21b) + (924,0 ± 2,81b) =
= 2316,6 (±?) 1Ь
sa = 7(±2,1)2 + (±1,2)2 +(±2,8)2 = ±3,71b
Общая масса железа = 2317 ± 41b
Стоимость составляет (2316,6 ±3,7 1b) ($0,15/1Ь) = $347,49 ± 0,56
Таким образом, вам предстоит заплатить 347,50 ± 0,60 долл.
Пример 3.13
Вы определяете содержание уксусной кислоты в уксусе путем титрования
стандартным раствором гидроксида натрия с индикатором фенолфталеином.
Порцию уксуса объемом около 5 мл взвесили в бюксе на аналитических весах.
Масса навески, рассчитанная по разности между массами заполненного и пустого
бюкса, составила 5,0268 г. Неопределенность результата отдельного взвешивания
составляет ±0,2 мг. Для определения точной концентрации раствора гидроксида на-
трия его стандартизовали, титруя точные навески особо чистого гидрофталата
калия. Концентрации, рассчитанные по результатам трех титрований, составили
0,1167, 0,1163 и 0,1164 М. На титрование пробы уксуса израсходовано 36,78 мл
стандартного раствора гидроксида натрия. Неопределенность результата считы-
вания показаний бюретки составляет ±0,02 мл. Какова массовая доля (%) уксус-
ной кислоты в уксусе и какова неопределенность этого значения?
Решение
Для нахождения массы навески необходимы два взвешивания — пустого и запол-
ненного бюкса. Каждый результат характеризуется неопределенностью ±0,2 мг,
поэтому неопределенность значения чистой массы навески (разности двух зна-
чений масс) составляет
5wt = (±0,2)2 + (±0,2)2 = ±0,3 мг
Среднее значение молярной концент-
рации раствора гидроксида натрия
равно 0,1165 М, а его стандартное от-
клонение (принимаемое в качестве
меры неопределенности) — 0,0002 М
(его можно рассчитать при помощи
Excel, см. фрагмент таблицы).
А В
1 N1 0,1167
2 N2 0,1163
3 N3 0,1164
4 Ст. отклон.: 0,000208
5
6 Ячейка В4 = СТАНДОТКЛОН(В1:ВЗ)
3.9. НАЛОЖЕНИЕ ПОГРЕШНОСТЕЙ: ЭТО НЕ ПРОСТОЕ СУММИРОВАНИЕ
137
Для нахождения объема раствора титранта необходимо двукратное считыва-
ние показаний бюретки (начального и конечного), поэтому неопределенность
объема титранта рассчитываем точно так же, как и неопределенность массы
навески:
svol = д/±0,022 + ±0,022 = ±0,03 мл
Число молей уксусной кислоты в пробе равно числу молей прореагировавшего с
ней гидроксида натрия. Поэтому массовая доля уксусной кислоты составляет
%НОАс=
(0,1165 ± 0,0002) ммоль/мл • (36,78 ± 0,03) мл • 60,05(мг/ммоль НО Ас)
=----------------------------------------------------------• 1U0/о
(5026,8 ±0,3)
= 5,119±?%
Неопределенность значения молярной массы уксусной кислоты будем считать
пренебрежимо малой (ее можно рассчитать с шестью значащими цифрами, об-
щий результат от этого не изменится).
(5м)ге1
±0,0002
0,1165
= ±0,0017
(*voi)rei = ^ = ±0,00082
эо, / о
+03
(5wt)rel = ^£- = ±0’00°060
d 5()26>8
Неопределенность результата анализа составляет
(stotal)rel = 7(±0,0017)2 +(±0,00082)2 +(±0,00060)2 = ±0,0020
5totai= 5,119- 0,0020 = ±0,010% уксусной кислоты
Таким образом, содержание уксусной кислоты составляет 5,119 ± 0,01%. Отно-
сительная неопределенность равна 0,2%.
Наибольший вклад в неопределенность результата внесла неопределен-
ность значения молярной концентрации раствора гидроксида натрия. Это еще
раз показывает, насколько важно тщательно стандартизовать растворы (а также
выполнять любые другие операции калибровки и градуировки, см. гл. 2).
138
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
3.10. Наложение погрешностей и значащие цифры
Ранее мы отмечали, что неопределенность результата вычислений характери-
зует, насколько точно известен этот результат. Иными словами, величина не-
определенности определяет число значащих цифр. Рассмотрим следующий
пример:
(73,1 ± 0,2) • (2,245 ± 0,008) = 164,1 ± 0,7
Значение неопределенности указывает нам, что результат следует привести с че-
тырьмя десятичными цифрами несмотря даже на то, что сомножитель, формаль-
но лимитирующий точность вычислений (73,1), имеет три значащие цифры
(здесь, кстати, нам не нужно приписывать к результату дополнительную цифру
в виде нижнего индекса, поскольку мы указываем реальное значение его неоп-
ределенности). Но это противоречие — кажущееся. Среди сомножителей наи-
большая величина относительной неопределенности составляет 0,0036, а
относительная неопределенность результата равна 0,0043. Таким образом, вслед-
ствие наложение погрешностей неопределенность несколько возросла. Если из-
вестны конкретные значения неопределенностей аргументов вычислений, то
точность не обязательно лимитирует тот из них, у которого наименьшее число
значащих цифр. Например, относительная неопределенность числа 78,1 ± 0,02
составляет 0,003, а числа 11,21 ± 0,08 — 0,007. Число значащих цифр результа-
та вычислений в общем случае определяется величиной его неопределенности,
обусловленной наложением погрешностей.
Рассмотрим еще один вычислительный пример:
(73,1 ± 0,9) • (2,245 ± 0,008) = 164,1 ± 2,1 = 164 ± 2
Неопределенность результата составляет несколько единиц, поэтому в его запи-
си все цифры, следующие после запятой, не имеют смысла. В этом примере отно-
сительные неопределенности аргумента, лимитирующего точность, и конечного
результата примерно одинаковы (±0,012), поскольку относительная неопреде-
ленность другого аргумента существенно меньше.
Пример 3.14
Приведите результаты следующих вычислений с необходимым числом знача-
щих цифр:
(а) (38,68 ± 0,07) - (6,16 ± 0,09) = 32,52
(12,18 ±0,08)-(23,04 ±0,07) = 8(_
1 ’ 3,247 ±0,006
3.11. КОНТРОЛЬНЫЕ КАРТЫ
139
Решение
(а) Рассчитанное значение абсолютной неопределенности результата равно ±0,11.
Поэтому результат следует представить как 32,5 ±0,1.
(б) Рассчитанное значение относительной неопределенности результата равно
0,0075, а абсолютной — 0,0075 • 86,43 = 0,65. Поэтому результат следует за-
писать как 86,4 ± 0,6, несмотря на то, что все числа, использованные в расче-
тах, содержат по четыре значащие цифры. Дело в том, что неопределенность
в четвертой цифре каждого из них достаточно велика (существенно превы-
шает одну единицу в соответствующем знаке); вследствие этого и неопреде-
ленность результата оказывается выше, чем это могло бы показаться на
первый взгляд. Относительная неопределенность результата вычислений со-
ставляет 0,0075, а наибольшая из относительных неопределенностей аргу-
ментов — 0,0066, очень близкое значение.
3.11. Контрольные карты
Контрольная карта качества — это график зависимости результата измерений
одной и той же величины от времени. Он строится в предположении, что измеря-
емая величина постоянна во времени и имеет гауссову функцию распределения.
Контрольные карты используют для проверки того, что результаты измерений
не выходят за рамки статистически допустимых отклонений. На контрольную
карту наносят среднюю горизонтальную прямую, представляющую истинное
(или принимаемое за таковое) значение измеряемой величины, а также одну или
две пары симметрично расположенных параллельных граничных линий — внут-
ренние и внешние контрольные границы. Положение этих границ рассчитыва-
ют из значения стандартного отклонения методики измерения, которое обычно
известно достаточно надежно и может считаться хорошим приближением для
величины о.
На контрольную карту периодически наносят результаты анализа одного
и того же образца с известным содержанием определяемого компонента. Пример
контрольной карты изображен на рис. 3.6. На ней отмечены результаты ежеднев-
ных определений общего содержания кальция в контрольных пробах сыворотки
крови. В ходе анализов эти контрольные пробы с известным содержанием каль-
ция случайным образом перемежают с реальными образцами. Внутренние конт-
рольные границы, как правило, проводят на расстоянии 2о от средней линии. При
этом вероятность выхода результата за границы, обусловленного чисто случайны-
ми факторами, составляет лишь 1 случай из 20. Внутренние границы называют еще
«границами предупреждения». Внешние контрольные границы откладывают на
расстоянии 2,5о либо Зег от средней линии. В этом случае вероятность случайно-
го (т. е. при отсутствии систематических погрешностей) выхода результата за
границы составляет 1 случай из 100 либо 500 соответственно. Обычно анализи-
руют одну контрольную пробу на одну партию реальных проб (например,
20 проб). Поэтому в течение дня можно получить несколько (7V) контрольных
140
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
+2,5ст 5 2
Контрольная карта результатов определения кальция за октябрь 2007
-2,5ст |__L J. J_ J_ J__l_l_l_l_L_L_L_L_L_L J_ J_ J_ J__l_l_l_L_L_L_L_L_L_L_L_l
ОКТ. 1 2 3 4 5 6 7 8 9 10 11 12 13141516171819202122232425262728293031
Дата
Рис. 3.6. Типичная контрольная карта
значений, а на карте откладывать среднее из них. Но в этом случае случайный
разброс данных должен быть в VjV раз меньше по сравнению с разбросом еди-
ничных результатов.
Если на карте обнаруживается систематический дрейф в одном направле-
нии (точек с одной стороны от средней линии становится существенно больше,
чем с другой), на это следует обратить особое внимание. Это может означать,
что либо эксперимент выходит из-под контроля, либо наблюдаются системати-
ческие погрешности анализа. Тенденция к выходу точек за контрольные границы
указывает на наличие одного или нескольких источников детерминированных
погрешностей. Аналитик должен проверить, не испортились ли реактивы, пра-
вильно ли работают приборы, нет ли помех за счет внешних условий и других
факторов. Появление дрейфов может также указывать на загрязнение реактивов,
ошибки в градуировке или приготовлении стандартов, а также на изменение со-
става контрольных образцов.
3.12. Доверительный интервал: насколько надежно значение?
Стандартное отклонение, рассчитанное из серии экспериментальных данных,
обобщенно характеризует воспроизводимость, присущую данной методике ана-
лиза. Но эта величина сама по себе еще не содержит информации о том, насколь-
ко близко может находиться среднее значение х от истинного значения. Здесь и
далее будем считать, что систематические погрешности отсутствуют; в этом
случае истинное значение равно ц. Однако теория статистики позволяет, исходя
из экспериментальных значений среднего и стандартного отклонения, рассчитать
интервал, внутри которого может с заданной вероятностью находиться величи-
на ц. Этот интервал называется доверительным интервалом, его границы —
доверительными границами, а соответствующая вероятность — доверитель-
ной вероятностью (ее обычно выражают в процентах).
3.12. ДОВЕРИТЕЛЬНЫЙ ИНТЕРВАЛ: НАСКОЛЬКО НАДЕЖНО ЗНАЧЕНИЕ?
141
Положение доверительных границ для среднего значения рассчитывают по
формуле:
тт - , ts
Доверительные границы = х ± ——
(3.9)
Здесь t—статистический коэффициент, величина которого зависит от заданной
доверительной вероятности и числа степеней свободы (напомним, что оно равно
числу результатов измерений минус единица). Значения t для различных дове-
рительных вероятностей и значений числа степеней свободы v приведены в
табл. 3.1. Заметьте, что полуширина доверительного интервала — это произве-
дение коэффициента t на стандартное отклонение среднего (s/ Jn). Аналогично
можно рассчитать доверительный интервал и для единичного значения х (в этом
случае #= 1). Доверительный интервал для единичного значения равен х ± ts.
Он в V7V раз шире, чем для среднего. При этом число степеней свободы ко-
эффициента t должно быть равно числу степеней свободы стандартного от-
клонения з (число данных, из которых оно рассчитано, минус единица).
Таблица 3.1
Значения коэффициентов t для v степеней свободы
при различных доверительных вероятностях*
V Доверительная вероятность
90% 95% 99% 99,5%
1 6,314 12,706 63,657 127,32
2 2,920 4,303 9,925 4,089
3 2,353 3,182 5,841 7,453
4 2,132 2,776 4,604 5,598
5 2,015 2,571 4,032 4,773
6 1,943 2,447 3,707 4,317
7 1,895 2,365 3,500 4,029
8 1,860 2,306 3,355 3,832
9 1,833 2,262 3,250 3,690
10 1,812 2,228 3,169 3,581
15 1,753 2,131 2,947 3,252
20 1,725 2,086 2,845 3,153
25 1,708 2,060 2,787 3,078
00 1,645 1,960 2,576 2,807
* v = N- 1 — число степеней свободы.
142
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
Пример 3.15
Образец технической соды проанализировали на содержание Na^C^ титрова-
нием стандартным раствором соляной кислоты. Для трех параллельных титро-
ваний получены значения 93,50, 93,58 и 93,43% Na2CO3. В каком интервале
значений с 95%-й вероятностью при отсутствии систематических погрешностей
находится истинное содержание Na2CO3?
Решение
При слишком большой доверительной вероятности доверительный интервал
оказывается слишком широким и может захватывать значения, содержащие сис-
тематическую погрешность. При слишком малой доверительной вероятности
доверительный интервал оказывается слишком узким, в него могут не попасть
значения, не содержащие систематической погрешности. Обычно оптимальны-
ми считаются доверительные вероятности 90-95%.
Среднее значение составляет 93,50%. Абсолютное стандартное отклонение
(рассчитайте его при помощи электронной таблицы!) составляет 0,075%. Для
95%-й доверительной вероятности и двух степеней свободы t = 4,303, и довери-
тельный интервал составляет
тт - - . ts 4,303-0,075 оасп,п1п0/
Доверительный интервал = х ± —— = 93,50 ±--—----= 93,50 ± 0,19%
4N V3
Таким образом, при отсутствии систематических погрешностей истинная вели-
чина с вероятностью 95% находится между 93,31 и 93,69%. Этот интервал явля-
ется доверительным интервалом для среднего результата измерений. В то же
время мы видели, что для единичного результата при бесконечном числе изме-
рений истинное значение в 95% случаев попадает в интервал шириной в два
стандартных отклонения (рис. 3.2). Действительно, для v = оо величины t равна
1,96 (табл. 3.1).
Вспомним также (разд. 3.7, рис. 3.2), что единичный результат измерения в
68% случаев попадает в интервал ±1о, в 95% случаев — в интервал ±2о и в 99%
случаев — в интервал ±2,5о. Поэтому возможно оценить величину стандартного
отклонения, зная величину доверительного интервала, и наоборот. Если для еди-
ничного значения для 95%-й доверительной вероятности доверительный интер-
вал составляет 27,37 ± 0,06 г, то стандартное отклонение примерно в два раза
меньше 0,06, т. е. 0,03 г. Эта величина одновременно является полушириной до-
верительного интервала для 68%-й доверительной вероятности. Строго говоря,
эти выводы справедливы лишь для бесконечной серии измерений. При малом
числе измерений значения коэффициентов t увеличиваются, и пропорционально
им увеличивается ширина доверительного интервала.
3.13. ТЕСТЫ СТАТИСТИЧЕСКОЙ ЗНАЧИМОСТИ: ЕСТЬ ЛИ РАЗЛИЧИЕ МЕЖДУ ДАННЫМИ?
143
Для среднего значения ширина доверительного интервала непрерывно уме-
ньшается с ростом N, поскольку при этом уменьшаются t и s/VtV. Поэтому чем
больше измерений вы проведете, тем более уверенно можете гарантировать, что
истинное значение при отсутствии систематических погрешностей будет нахо-
диться в заданных границах или, наоборот, что эти границы будут более узкими
при заданной доверительной вероятности. В то же время с некоторого N значе-
ния t (как и s/Jn) начинают убывать очень медленно (табл. 3.1). Поэтому суще-
ствует разумный предел, выше которого нецелесообразно увеличивать число
параллельных измерений для увеличения точности.
3.13. Тесты статистической значимости:
есть ли различие между данными?
В ходе разработки новой методики анализа часто возникает необходимость
сравнить ее результаты с результатами общепринятой (возможно — стандарт-
ной) методики. Но как можно выяснить, есть ли значимое различие между резу-
льтатами, полученными при помощи двух методик? Здесь мы опять должны
прибегнуть к помощи статистики.
Решение вопроса о том, значимо ли один набор данных отличается от друго-
го, зависит не только от степени различия средних значений, но также от количе-
ства данных и степени их разброса. Существуют специальные статистические
таблицы, с помощью которых можно оценить, насколько велико должно быть
различие результатов, чтобы его нельзя было считать случайным. Для оценки
степени различия между разбросами серий данных используют F-тест, а для
оценки различия между средними значениями — Z-тест.
F-тест
F-тест показывает нам, имеется ли значимое различие между стандартными от-
клонениями двух серий данных. Для этого вычисляют величину F, представляю-
щую собой отношение дисперсий (т. е. квадратов стандартных отклонений)
сравниваемых серий:
(3.10)
При этом необходимо, чтобы sf было больше s|. Величина F характеризуется
двумя значениями числа степеней свободы Vj и v2, каждое из которых равно чис-
лу данных в соответствующей серии минус единица.
Если величина F, рассчитанная по уравнению (3.10), превосходит соответ-
ствующую табличную величину F для выбранной доверительной вероятности,
144
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
Таблица 3.2
Коэффициенты F для 95%-й доверительной вероятности
\v' V2\ 2 3 4 5 6 7 8 9 10 15 20 30
2 19,0 19,2 19,2 19,3 19,3 19,4 19,4 19,4 19,4 19,4 19,4 19,5
3 9,55 9,28 9,12 9,01 8,94 8,89 8,85 8,81 8,79 8,70 8,66 8,62
4 6,94 6,59 6,39 6,26 6,16 6,09 6,04 6,00 5,96 5,86 5,80 5,75
5 5,79 5,41 5,19 5,05 4,95 4,88 4,82 4,77 4,74 4,62 4,56 4,50
6 5,14 4,76 4,53 4,39 4,28 4,21 4,15 4,10 4,06 3,94 3,87 3,81
7 4,74 4,35 4,12 3,97 3,87 3,79 3,73 3,68 3,64 3,51 3,44 3,38
8 4,46 4,07 3,84 3,69 3,58 3,50 3,44 3,39 3,35 3,22 3,15 3,08
9 4,26 3,86 3,63 3,48 3,37 3,29 3,23 3,18 3,14 3,01 2,94 2,86
ю 4,10 3,71 3,48 3,33 3,22 3,14 3,07 3,02 2,98 2,85 2,77 2,70
15 3,68 3,29 3,06 2,90 2,79 2,71 2,64 2,59 2,54 2,40 2,33 2,25
20 3,49 3,10 2,87 2,71 2,60 2,51 2,45 2,39 2,35 2,20 2,12 2,04
30 3,32 2,92 2,69 2,53 2,42 2,33 2,27 2,21 2,16 2,01 1,93 1,84
то различие между дисперсиями значимо. Табличные величины F для 95%-й до-
верительной вероятности приведены в табл. 3.2.
Пример 3.16
Вы разрабатываете новую фотометрическую методику определения глюкозы в сы-
воротке крови и используете для сравнения стандартную методику Фолинь—By.
Пользуясь приведенными ниже результатами параллельных анализов одной и
той же пробы, установите, существует ли значимое различие в воспроизводи-
мости двух методик.
Ваша методика (мг/дл) 127 125 123 130 131 126 129 среднее (xj) 127 Методика Фолинь—By (мг/дл) 130 128 131 129 127 125 среднее (х2) 128
3.13. ТЕСТЫ СТАТИСТИЧЕСКОЙ ЗНАЧИМОСТИ: ЕСТЬ ЛИ РАЗЛИЧИЕ МЕЖДУ ДАННЫМИ?
145
Решение
2 _ E(x/i ~xi)2 _ 50
51 Nt-1 7-1
2_Z(x«-2-x2)2 _ 24 _
.Vo
2 W2-l 6-1
Отношение дисперсий рассчитывают таким образом, чтобы оно было больше
единицы. Табличное значение F для Vj = 6 и v2 = 5 составляет 4,95. Поскольку
рассчитанное значение меньше табличного, мы приходим к выводу, что значи-
мого различия в воспроизводимостях методик нет, т. е. их случайные погрешно-
сти — одного порядка.
t-Тест Стьюдента:
есть ли различие в результатах анализа по разным методикам?
Очень часто возникает необходимость установить, существует ли статистиче-
ски значимое различие между результатами, полученными при помощи двух
разных методик. Иными словами: можно ли считать результаты этих методик
одной и той же величиной? Для решения таких задач используют Z-тест Стью-
дента. Z-Тест используют для решения вопроса о том, является ли статистически
значимым различие между средними двух серий данных.
Будем называть одну из методик проверяемой, а другую — проверочной. Из
результатов этих методик специальным образом рассчитывают эксперимента-
льную величину t и сравнивают ее с табличным значением (табл. 3.1) для выбран-
ной доверительной вероятности и соответствующего числа степеней свободы.
Если рассчитанное значение t превосходит табличное, то при заданной довери-
тельной вероятности различие между результатами двух методик следует счи-
тать значимым. Если же оно не превосходит табличную величину, то у нас нет
никаких оснований считать различие данных значимым. Разумеется, это не
означает также, что полученные результаты идентичны.
Ниже описаны три различных варианта использования Л-теста. Один из них
применяют тогда, когда известно (из других результатов измерений) значение ц
с точностью, достаточной для того, чтобы его можно было считать истинным.
Если такое значение не известно, можно проанализировать одну и ту же пробу
по двум методикам несколько раз. Можно также проанализировать серию раз-
ных проб по двум методикам. При этом одна из используемых методик не дол-
жна содержать систематической погрешности.
146
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
1. Использование /-теста при известном значении измеряемой величины.
Заметьте, что уравнение (3.9) дает интервал значений измеряемой величины при
отсутствии систематических погрешностей. Поэтому можно записать, что
(3.11)
Из этого следует, что
±г = (х_ц)_
(3.12)
Если величина ц является достаточно надежной оценкой истинного результата
анализа, полученной по данным других измерений (например, она может быть
указана в паспорте стандартного образца, выпускаемого американским Нацио-
нальным институтом стандартов и технологий — NIST, либо представлять собой
значение атомной массы — основы химического анализа), то можно использовать
уравнение (3.12) для того, чтобы решить, являются ли результат измерений и его
истинное значение статистически эквивалентными.
Пример 3.17
Вы разрабатываете методику определения следов меди в биологических матери-
алах, основанную на мокром сжигании пробы с последующим атомно-абсорбцион-
ным определением. Для проверки правильности методики вы проанализировали
стандартный образец листьев яблони, предоставленный NIST. В результате от-
бора и анализа пяти проб получено среднее значение 10,8 ррш и стандартное от-
клонение 0,7 ppm. Паспортное значение содержания меди равно 11,7 ppm.
Можно ли считать результаты методики статистически правильными при 95%-й
доверительной вероятности?
Решение
д/ДГ д/5
+/= (х-ц)—_ = (10,8-11,7)^ =2,9
s 0,7
Поскольку произведено пять измерений, число степеней свободы (N-V) равно 4.
Соответствующее табличное значение t (табл. 3.1) для 95%-й доверительной ве-
роятности равно 2,776. Это меньше рассчитанного значения. Следовательно,
разработанная вами методика содержит систематическую погрешность. Иными
словами, с вероятностью не менее 95% различие между полученным результа-
том и паспортным значением не является случайным.
3.13. ТЕСТЫ СТАТИСТИЧЕСКОЙ ЗНАЧИМОСТИ: ЕСТЬ ЛИ РАЗЛИЧИЕ МЕЖДУ ДАННЫМИ?
147
Из уравнения (3.12) следует, что чем выше воспроизводимость результатов,
т. е. чем меньше величина s, тем больше рассчитанное значение t. В этом случае
возрастает вероятность, что табличное значение t окажется меньше рассчитан-
ного. Таким образом, чем выше воспроизводимость результатов, тем легче выя-
вить неслучайные различия в данных. Кроме того, рассчитанные значения t
возрастают и с ростом числа измерений [множитель VV в формуле (3.12)]. Поэ-
тому результаты сравнения очень больших серий данных часто оказываются
статистически значимыми. Однако различия в данных, выявляемые в подобных
случаях, бывают очень тонкими и не имеют практического значения.
2. Сравнение средних двух серий данных. Z-Тест можно применять и для срав-
нения двух средних значений. В этом случае уравнение (3.12) несколько видоиз-
меняется: вместо ц в него подставляют среднее для второй серии, а вместо
величины, обратной стандартному отклонению среднего (VVIs), выражение:
N^2 Z
Nl+N2/ Р
где sp — средневзвешенное стандартное отклонение двух серий. Таким образом,
формула (3.12) преобразуется в
±f_*l ~Х2 N1N2
Sp \n1+n2
(3.13)
Средневзвешенное стандартное отклонение (его определение см. ниже) — это
величина, иногда используемая как улучшенная оценка воспроизводимости ме-
тодики. Она же используется для расчета воспроизводимости по двум сериям
данных в парном Z-тесте (см. ниже). Во всех подобных случаях воспроизводи-
мость оценивают из нескольких серий результатов анализов, например, данных,
полученных в разные дни или для образцов, несколько различающихся по соста-
ву. Это иногда бывает предпочтительнее, чем оценивать воспроизводимость из
единственной серии данных. Предпосылкой для таких оценок является допуще-
ние, что каждая серия данных характеризуется одной и той же величиной случай-
ной погрешности. В общем случае средневзвешенное стандартное отклонение
sp рассчитывают по формуле:
£(Л1 -*1) + £(*Z2-*2)2+- + £(x,* ~хк)2
N-k
(3.14)
где х!, х2, ..., хк — средние для 1 -й, 2-й, ..., £-й серии данных, axiX, xi2, ...,xik —
отдельные значения из соответствующих серий. N— общее число данных, рав-
148
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
ное сумме + N2 +...+ Nk. Например, если имеется 5 серий по 20 данных в каж-
дой (хотя в общем случае число данных в каждой серии может быть разным),
то к= 5, a N = 100. Число степеней свободы составляет (N - к), что равно
(N} - l) + (N2 -1) +...+ (Nk - 1). Таким образом, уравнение (3.14) представляет со-
бой комбинацию выражений для расчета стандартного отклонения каждой серии.
При использовании /-теста для сравнения двух средних предполагается, что
обе серии данных характеризуются приблизительно одинаковыми стандартны-
ми отклонениями, т. е. каждое значение s является приближенным значением
одной и той же величины и. Это предположение можно проверить при помощи
описанного выше F-теста.
Пример 3.18
Разработана новая методика гравиметрического определения Fe(III), в ходе ко-
торой железо осаждают в кристаллической форме в виде соединения включения с
борорганическим реагентом. Правильность методики проверили путем сравнения
результатов анализа железной руды с результатами, полученными при помощи
стандартной методики, включающей осаждение гидроксида железа аммиаком и
взвешивание Fe2O3. Результаты анализов (массовая доля железа, %) приведены
ниже.
Проверяемая методика
20,10%
20,50
18,65
19,25
19,40
19,99
х, = 19,65%
Стандартная методика
18,89%
19,20
19,00
19,70
19,40
х2 = 19,24%
Есть ли значимое различие между результатами двух методик?
Решение
х,-1 xn-xi (xn-Xi)2 х/2 хг2-х2 (Х,.2-Х2):
20,10 0,45 0,202 18,89 0,35 0,122
20,50 0,85 0,722 19,20 0,04 0,002
18,65 1,00 1,000 19,00 0,24 0,058
19,25 0,40 0,160 19,70 0,46 0,212
19,40 0,25 0,062 19,40 0,16 0,026
19,99 0,34 0,116 ~ ^г)2 = 0,420
SC^-Xj)2 = 2,262
3.13. ТЕСТЫ СТАТИСТИЧЕСКОЙ ЗНАЧИМОСТИ: ЕСТЬ ЛИ РАЗЛИЧИЕ МЕЖДУ ДАННЫМИ?
149
F _Д|2_ 0,262/5 _П1
-/ 0,420/4 ’
Эта величина меньше табличной (6,26). Поэтому серии данных имеют сопоста-
4 вимые величины стандартных отклонений, и мы можем применить /-тест для
сравнения средних:
'£(*,1 ~*1)2 +Z(x.-2 ~хг)2 _ 12,262 + 0,420
N{+N2-2 У 6 + 5-2
.19,65-19,24 6-5 _
ZL Г — л I — -э
0,546 V 6 + 5 3
Табличное значение t для девяти степеней свободы (N} + N2 - 2 = 6 + 5 - 2 = 9) при
95%-й доверительной вероятности равно 2,262. Таким образом, между результа-
тами, полученными при помощи двух методик, нет статистически значимого
различия.
Точно так же, как результаты анализа одной пробы по двум методикам,
можно сравнивать и результаты анализа двух проб при помощи одной мето-
дики.
3- Парный /-тест. В клинических лабораториях новую методику часто проверя-
ют при помощи стандартной методики, анализируя серию проб, несколько раз-
личающихся по составу (в пределах физиологических состояний). Для этого
случая экспериментальное значение t вычисляют несколько иначе. Сначала для
каждой z-й пробы рассчитывают разность D. между результатами анализа двух
методик (с учетом знака). Затем рассчитывают среднее значение разности, от-
клонения (Z) -D), и из них — стандартное отклонение sd. Значение t находят по
формуле:
(3.15)
(3.16)
150
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
Пример 3.19
Вы разрабатываете новую методику определения азота мочевины крови и хоти-
те узнать, значимо ли отличаются ее результаты от результатов анализа при помо-
щи стандартной методики в диапазоне значений концентраций, которых можно
ожидать для большинства реальных проб. Предварительно было установлено,
что воспроизводимости обеих методик значимо не различаются. При анализе се-
рии проб получены следующие результаты:
Проба А Ваша методика (мг/дл) 10,2 Стандартная методика (мг/дл) 10,5 А -0,3 Di-D -0,6 (D-D? 0,36
В 12,7 11,9 0,8 0,5 0,25
С 8,6 8,7 -0,1 -0,4 0,16
D 17,5 16,9 0,6 0,3 0,09
Е 11,2 10,9 0,3 0,0 0,00
F 11,5 И,1 0,4 S 1,7 D = 0,28 0,1 0,01 S 0,87
Решение
Sd= ^1=0,42
d V6-1
0,28
0,42
•л/б =1,63
Табличное значение t для 95%-й доверительной вероятности и пяти степеней
свободы составляет 2,571. Рассчитанное значение t меньше табличного, следо-
вательно, при выбранной доверительной вероятности значимое различие между
результатами двух методик отсутствует.
Вывод о значимости различия двух величин, полученный при 95%-й дове-
рительной вероятности, обычно считается достаточно достоверным. Если такое
различие наблюдается даже при доверительной вероятности 99%, то этот вывод
еще более достоверный. Напротив, если выполнять Z-тест при слишком малой
доверительной вероятности (например, 80%), то можно ошибочно заключить,
что между значениями наблюдается различие, когда на самом деле его нет. Эта
ситуация называется ошибкой I рода. С другой стороны, при слишком высоких
доверительных вероятностях можно не заметить различия, когда оно есть
(ошибка II рода). В целом чем меньше рассчитанное значение /, тем более на-
дежным становится вывод об отсутствии значимого различия между сравнивае-
мыми значениями. Если при 95%-й доверительной вероятности рассчитанное и
3.14. ОТБРОС ПРОМАХОВ: Q-TECT
151
табличное значения t оказываются близки, то для окончательного решения во-
проса о различии величин обычно проводят дополнительные исследования.
3.14. Отброс промахов: Q-тест
Третий закон Финегла: в любом массиве данных величина, наиболее очевидным
образом представляющаяся правильной, ошибочна.
Часто в серии результатов параллельных анализов один из результатов за-
метно отличается от остальных. В таких случаях необходимо принять решение
исключить такой результат из серии или оставить его. К сожалению, критерия,
позволяющего однозначно решить, является ли результат грубым промахом или
обусловлен «обычной» случайной погрешностью, не существует. Разумеется,
всегда есть соблазн удалить вообще все крайние значения, поскольку они могут
существенно ухудшить общую статистику — увеличить значения стандартного
отклонения и дисперсии, а также значительно изменить величину среднего. Но
единственным надежным основанием для удаления результата может служить
установленный факт грубой ошибки, допущенной при его получении. Сохра-
нять такой результат в массиве данных нельзя.
На практике для решения вопроса о сохранении или удалении результата
следует руководствоваться опытом и здравым смыслом в не меньшей степени,
чем результатами каких-либо статистических тестов. Часто опытный аналитик
уже заранее примерно представляет себе, какую воспроизводимость следует
ожидать от методики, и легко распознает подозрительные данные. Если ожидае-
мое стандартное отклонение известно, то можно, например, удалять все резуль-
таты, отстоящие от среднего более чем на 2s или 2,5s: такие результаты могут
встретиться лишь в одном случае из 20 или 100 соответственно.
Тем не менее предложено множество тестов, позволяющих решать вопрос
об удалении результата чисто статистическими методами. Все они так или иначе
основаны на оценке величины некоторого интервала, внутри которого должны
находиться результаты, имеющие статистический смысл. Проблема состоит в
том, как же именно оценить такой интервал. Если он окажется слишком узким,
то вполне «хорошие» результаты могут быть отброшены. Если слишком широ-
ким — в серии слишком часто будут оставаться ошибочные данные. Среди ряда
тестов одним из наиболее статистически корректных (в применении к сериям
малого размера) является 0-тест. Именно его рекомендуется использовать для
выявления промахов. 0-тест состоит в следующем. Сначала все данные распо-
лагают в порядке возрастания или убывания. Затем вычисляют разность между
подозрительным значением и значением, ближайшим к нему, и делят эту раз-
ность (а) на размах всей серии (w) — разность между наибольшим и наимень-
шим значением: Q = a/w. Полученное отношение сравнивают с табличным
значением Q. Если рассчитанное значение Q равно табличному или превышает
его, подозрительное значение следует отбросить. Табличные значения Q для
90-, 95- и 99%-й доверительной вероятности и разного количества данных в се-
рии приведены в табл. 3.3.
152
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
2-тест используют для того, чтобы решить, обусловлено
ли выпадающее значение систематической
погрешностью. Если нет, то оно вызвано лишь обычной
случайной погрешностью и его надо оставить в серии
данных.
Таблица 3.3
Значения Q для разных значений доверительной вероятности*
Число данных в серии Доверительная вероятность
£?90 295 299
3 0,941 0,970 0,994
4 0,765 0,829 0,926
5 0,642 0,710 0,821
6 0,560 0,625 0,740
7 0,507 0,568 0,680
8 0,468 0,526 0,634
9 0,437 0,493 0,598
10 0,412 0,466 0,568
15 0,338 0,384 0,475
20 0,300 0,342 0,425
25 0,277 0,317 0,393
30 0,260 0,298 0,372
*По данным D. В. Rorabacher, Anal. Chem., 63 (1991) 139.
Пример 3.20
При определении хлоридов в отдельных аликвотах объединенной пробы сыво-
ротки крови получены следующие результаты (ммоль экв./л): 103, 106, 107 и
114. Одно из значений вызывает подозрения. Установите, можно ли при 95%-й
доверительной вероятности считать, что оно обусловлено лишь случайными по-
грешностями.
Решение
Подозрительным значением является 114 ммоль экв./л. Разность между ним и
ближайшим соседним значением (107 ммоль экв./л) составляет 7 ммоль экв./л.
Размах всей серии составляет 114-103 = 11 ммоль экв./л. Таким образом, рас-
считанное значение Q составляет 7/11 = 0,64. Табличное значение для серии из
четырех результатов составляет 0,829. Поскольку рассчитанное значение Q
меньше табличного, можно считать, что подозрительное значение обусловлено
лишь случайными погрешностями. Его не следует исключать из серии данных.
3.14. ОТБРОС ПРОМАХОВ: Q-TECT
153
Для малых серий данных (от трех до пяти значений) g-тест может выявить
промах только в том случае, если его отличие от остальных значений весьма ве-
лико. При этом вероятность, что промах останется в серии, высока, следовательно,
значение среднего арифметического окажется сильно искаженным. В подобных
случаях рекомендуется в качестве результата анализа указывать не среднее
арифметическое, а медиану. Для серии данных, упорядоченных по возрастанию,
медианой называется значение, находящееся в центре этой серии (если число
данных нечетное), либо полусумма двух центральных значений (если число дан-
ных четное). Перед средним арифметическим медиана имеет то преимущество,
что ее величина не подвержена столь сильному влиянию промахов. В приведен-
ном выше примере медиана составляет (округленно) 106 (среднее из двух центра-
льных значений, (106 + 107)/2), а среднее — 108. В данном случае эти величины
оказались близки.
Для интерпретации малых (три-пять значений) серий данных, когда одно из
значений заметно отличается от остальных, а оценка воспроизводимости суще-
ственно хуже ожидаемой, можно предложить следующий способ.
1 - Оцените случайную погрешность, которую разумно ожидать от исполь-
зуемой методики, и решите, действительно ли отклонение подозритель-
ного значения столь велико. Имейте в виду, что для серии из трех
значений, когда два из них очень близки, применение g-теста часто при-
водит к неверным выводам (см. ниже).
2- Проверьте все данные, на основании которых было рассчитано подозри-
тельное значение, и подумайте, не были ли допущены ошибки в ходе по-
лучения этих данных.
3. Если нет возможности получить дополнительные данные, примените
g-тест.
4. Если результат g-теста свидетельствует о том, что значение следует
оставить, то для малых наборов данных в качестве конечного результата
используйте медиану, а не среднее арифметическое.
5. В крайнем случае повторите анализ еще раз. Если новый результат хоро-
шо согласуется с теми данными, которые можно считать заведомо верны-
ми, то это дополнительный аргумент в пользу того, что подозрительное
значение следует отбросить. В то же время не следует повторять анали-
зы, добиваясь получения «верного», на ваш взгляд, результата.
g-тест бессмысленно применять к серии из трех значений, два из которых
совпадают: тест всегда укажет на необходимость исключения третьего значения,
как бы мало оно ни отличалось от двух других. Действительно, в этом случае
а = w, и рассчитанное значение g всегда равно 1. То же самое, очевидно, относит-
ся к серии из четырех результатов, три из которых совпадают, и так далее.
154
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
3.15. Статистика малых массивов данных
Статистические методы, разработанные для больших массивов данных, не все-
гда хорошо работают для малых. В предыдущих разделах мы обсудили возможные
способы оценки центрального значения (среднее арифметическое, х), разброса дан-
ных (стандартное отклонение, s) и границ доверительного интервала (при помощи
f-теста) для серии данных, имеющих нормальное распределение. Эти подходы хо-
рошо работают только в случае, когда массив данных достаточно велик. Но в хи-
мическом анализе мы, как правило, имеем дело с массивами данных малого
размера—менее 10, иногда всего 2-3. В этих случаях более подходящими могут
оказаться другие способы оценки.
Одним из методов, специально предназначенных для малых массивов дан-
ных, является рассмотренный выше 2-тест и основанные на нем правила интер-
претации подозрительных результатов.
Медиана может оказаться лучшей оценкой, чем среднее
В качестве оценки центрального значения серии данных может быть использо-
вана медиана М. Ее преимущество в том, что она не столь подвержена влиянию
экстремальных значений (выбросов), как среднее арифметическое х. Относите-
льную (по сравнению со средним арифметическим) эффективность использова-
ния медианы как оценки «истинного» среднего (т. е. среднего, рассчитанного
для бесконечной совокупности данных) можно охарактеризовать в виде показа-
теля эффективности Ем— отношения дисперсий медианы и среднего арифмети-
ческого при заданном размере серии. Величины Ем приведены в табл. 3.4. Они
изменяются от 1 для серии из двух значений (в этом случае медиана всегда сов-
падает со средним) до 0,64 для достаточно больших серий данных. Это означает,
что медиана, вычисленная из серии, содержащей 100 данных (в этом случае Ем
можно принять равным 0,64), является столь же надежной оценкой «истинного»
среднего значения, как и среднее арифметическое, рассчитанное из серии
100 • 0,64 = 64 данных. Аналогично медиана для 10 данных (Ем =0,71) содержит
столько же информации, как и среднее для 10 • 0,71 = 7 данных. При использова-
нии медианы отпадает необходимость решать вопрос о том, содержит ли серия
грубые промахи, и, следовательно, применять 2-тест. Показано, что для серии
из трех значений нормально распределенной величины медиана в любом случае
является лучшей оценкой, чем среднее из двух «лучших» (ближе всего располо-
женных друг к другу) значений.
Использование размаха вместо стандартного отклонения
Для небольших массивов данных величина размаха R является очень эффектив-
ной оценкой разброса результатов. Показатели эффективности для размаха ER,
приведенные в табл. 3.4, свидетельствуют о том, что для серий из четырех или
менее значений размах является практически столь же надежной оценкой, как и
стандартное отклонение. Причина заключается в том, что для малых серий дан-
ных стандартное отклонение само по себе является достаточно плохой оценкой
3.15. СТАТИСТИКА МАЛЫХ МАССИВОВ ДАННЫХ
155
Таблица 3.4
Показатели эффективности и коэффициенты пересчета
статистических характеристик для серий из 2-10 значений*
Число данных Показатель эффективности _ Коэффициент отклонения KR Коэффициент для расчета довери- тельного интервала по размаху
для медианы Ем для размаха ER {г, 0,95 {Г, 0,99
2 1,00 1,00 0,89 6,4 31,83
3 0,74 0,99 0,59 1,3 3,01
4 0,84 0,98 0,49 0,72 1,32
5 0,69 0,96 0,43 0,51 0,84
6 0,78 0,93 0,40 0,40 0,63
7 0,67 0,91 0,37 0,33 0,51
8 0,74 0,89 0,35 0,29 0,43
9 0,65 0,87 0,34 0,26 0,37
10 0,71 0,85 0,33 0,23 0,33
00 0,64 0,00 0,00 0,00 0,00
*По данным R. В. Dean, W. J. Dixon, Anal. Chem., 23 (1951) 636.
разброса (хотя и самой лучшей из всех возможных!). Из величины размаха для
серии данного размера можно оценить стандартное отклонение для бесконечной
совокупности данных, т. е. получить оценку величины о. Для этого значение
размаха необходимо умножить на коэффициент отклонения KR, значения ко-
торого также приведены в табл. 3.4.
s =RKr (3.17)
Для серии из четырех данных, приведенных в примере 3.9, стандартное от-
клонение составляет 0,69 мг, а размах — 1,6 мг. Умножив последнее значение на
величину KR для N= 4, получим s =1,6 мг • 0,49 = 0,78 мг, что близко к величине
стандартного отклонения. С ростом N эффективность размаха как оценки раз-
броса данных уменьшается по сравнению со стандартным отклонением.
При вычислении стандартного отклонения вместо среднего арифметиче-
ского можно использовать медиану. В этом случае влияние резко выделяющих-
ся данных также уменьшается. Еще раз воспользуемся данными примера 3.9:
для расчета стандартного отклонения по формуле (3.2) используем медиану
(29,8) вместо среднего. В этом случае стандартное отклонение окажется равным
0,73 вместо 0,69.
Расчет доверительных интервалов из величин размахов
Значения s, рассчитанные из величин размахов, можно использовать для расчета
границ доверительных интервалов. Эти расчеты производятся по аналогии с
обычной формулой (3.9), но значения коэффициентов пропорциональности надо
брать другие:
156
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
Доверительный интервал = х ± Rtr
(3.18)
Вместо коэффициентов t из табл. 3.1 используют специальные коэффициенты tr,
приведенные в табл. 3.4 для 95- и 99%-й доверительной вероятности. Так, для
данных, приведенных в примере 3.15, доверительный интервал, рассчитанный
по уравнению (3.18) для 95%-й доверительной вероятности, составляет
93,50 ± 0,19 • 1,3 = 93,50 ± 0,25% Na2CO3.
3.16. Линейный метод наименьших квадратов:
как провести прямую?
Если требуется линейная зависимость, получи только два результата.
Мудрость аналитика
Аналитику часто приходится строить линейные зависимости, например, градуи-
ровочные прямые. Для обеспечения правильности результатов построение гра-
дуировочных зависимостей имеет решающее значение — ведь именно с их
помощью мы находим содержания веществ в неизвестных образцах! Однако все
результаты измерений характеризуются некоторой неопределенностью, и дан-
ные, используемые для построения градуировочных зависимостей, не составля-
ют исключения. Они неизбежно имеют разброс относительно прямой, и часто
прямую в таких случаях проводят интуитивно, на глаз, просто приложив линей-
ку так, чтобы точки были разбросаны относительно прямой более-менее равно-
мерно. Естественно, более надежным было бы использование статистических
методов, позволяющих определить наиболее вероятное расположение прямой.
Электронные таблицы содержат специальные статистические функции, позво-
ляющие без труда рассчитывать параметры линейных и даже нелинейных зави-
симостей, наилучшим образом соответствующих экспериментальным данным.
Но сначала мы рассмотрим, каким же образом программа рассчитывает эти па-
раметры и осуществляет их статистическую обработку.
Если предполагается, что зависимость между переменными х и у линейна,
то данные должны удовлетворять уравнению
у = тх + b (3.19)
Символом у обозначена зависимая переменная, символом х — независимая
переменная, а параметры т и b называются, соответственно, угловым коэффици-
ентом (тангенсом угла наклона) и свободным членом. Последний представляет
собой длину отрезка, отсекаемого прямой на оси ординату. Как правило, у явля-
ется результатом какого-либо измерения, соответствующего переменной вели-
чине х (рис. 3.7).Так, при построении градуировочного графика в спектро-
фотометрическом анализе у — это измеренное значение оптической плотности,
соответствующее концентрации стандартного раствора х. Нашей задачей явля-
ется нахождение параметров т и Ь.
3.16. ЛИНЕЙНЫЙ МЕТОД НАИМЕНЬШИХ КВДЦРАТОВ: КАК ПРОВЕСТИ ПРЯМУЮ?
157
х (независимая переменная)
РИС. 3.7. Прямая, построенная по методу наименьших квадратов
Метод наименьших квадратов
Статистическими методами можно показать, что экспериментальный набор дан-
ных наилучшим образом описывает та прямая, для которой сумма квадратов
отклонений экспериментальных значений от рассчитанных (эти отклонения
называются остатками) минимальна. Способ построения таких прямых назы-
вается методом наименьших квадратов. Если х — заданная величина (напри-
мер, концентрация), а у — измеряемая величина (оптическая плотность при
спектрофотометрических измерениях, площадь пика при хроматографических
измерениях и т. д.), то отклонения рассчитывают вдоль вертикальной оси у.
В этом случае предполагают, что значения независимой переменной xz не содер-
жат погрешностей. Обозначим экспериментальное значение у, соответствую-
щее заданному значению х-, как у., а рассчитанное значение у (т. е. лежащее на
прямой) — как j>z. Очевидно, величина^ в точности равна mxt + b. Тогда сумма
квадратов отклонений S равна:
s = Y(yi-yl)2=^yi-(mxi+b^2 (3.20)
Наилучшей прямой является та, для которой величина S минимальна. Для
нахождения соответствующих параметров следует продифференцировать выра-
жение для S по т и Ь, приравнять производные нулю и решить полученную сис-
тему из двух уравнений относительно т и Ь. Решениями являются:
£(*,- -х)(у,- - у)
£(*,• -к)2
Ъ- у — тх
(3.21)
(3.22)
где х — среднее из всех значений х/5 а у — среднее из всех значений у.. Выраже-
ние (3.21) можно преобразовать в форму, более удобную для вычислений (осо-
бенно на калькуляторе):
158
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
-[(Zx«)2 /"]
(3.23)
где п — общее число точек (пар значений х/5 уг).
Значения углового коэффициента и свободного члена, рассчитанные по ме-
тоду наименьших квадратов, характеризуют наиболее вероятное прохождение
прямолинейной зависимости.
Пример 3.21
В образце зерна определяли рибофлавин (витамин В2) путем измерения интен-
сивности флуоресценции вытяжки в 5%-й уксусной кислоте. Для построения
градуировочной зависимости были измерены интенсивности флуоресценции се-
рии стандартных растворов рибофлавина возрастающей концентрации. По по-
лученным данным при помощи метода наименьших квадратов рассчитайте
параметры наилучшей прямолинейной градуировочной зависимости и опреде-
лите концентрацию рибофлавина в анализируемом растворе (интенсивность его
флуоресценции равна 15,4).
Концентрация Интенсивность
рибофлавина, мкг/мл = ррт (х;) флуоресценции, усл. ед. (у,) X,2 Х1У,
0,000 0,00 0,0000 0,00
0,100 5,8 0,0100 0,58
0,200 12,2 0,0400 2,44
0,400 22,3 0,16Оо 8,92
0,800 43,3 О,64Оо 34,64
£х,- =1,500 Хл =83,6 £>• = О,85Оо =46>58
(£х,.)2 =2,250 п = 5 х = У х,- =0,3000 п y = S2± = 1б,72 п
Решение
При помощи уравнений (3.23) и (3.22) получаем:
46,5 8 - [(1,500 - 83,6)/5]
О,85Оо -2,250/5
= 53,75 (усл. ед./ррш)
b = 16,72 - (53,75 • О,ЗООо) = О,6о (усл. ед.)
3.16. ЛИНЕЙНЫЙ МЕТОД НАИМЕНЬШИХ КВАДРАТОВ: КАК ПРОВЕСТИ ПРЯМУЮ?
159
РИС. 3.8. Градуировочный график, построенный методом наименьших квадратов по данным при-
мера 3.21
В ходе вычислений мы сохраняли максимальное число значащих цифр. Посколь-
ку экспериментальные данные (у) получены с точностью до одного знака после
запятой, величины т и b также округлим до одного знака после запятой. Таким
образом, уравнение градуировочной прямой имеет вид (ppm = мкг/мл):
у (усл. ед.) = 53,8 (усл. ед./ррт) • х (ррт) + 0,6 (усл. ед.)
Рассчитаем концентрацию в анализируемой пробе:
15,4 = 53,8х + 0,6
х = 0,275 мкг/мл
Для построения градуировочного графика достаточно взять два произвольных
значения х, отстоящих далеко друг от друга, рассчитать соответствующие им
значения у (или наоборот) и провести прямую через эти точки. На рис. 3.8
изображены экспериментальные точки и градуировочный график. Этот рису-
нок был построен при помощи программы Excel, позволяющей также вывести
уравнение прямой линии и значение квадрата коэффициента корреляции. Оно
является мерой близости экспериментальных и рассчитанных значений (см. да-
лее). Приведенные значения имеют дополнительные десятичные цифры, посколь-
ку их число определяется программой автоматически.
160
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
Стандартные отклонения углового коэффициента и свободного
члена: они характеризуют неопределенность результата
Будем исходить из допущения, что для каждой точки градуировочного графика
значение ^-представляет собой случайную величину, распределенную по норма-
льному (гауссовому) закону. В соответствии с уравнением (3.20) отклонение ор-
динаты точки от градуировочной прямой равно угу1 = у - (тх + Ь). Из этих
значений можно рассчитать стандартное отклонение величин у. Расчет прово-
дится по аналогии с уравнением (3.2) за исключением того, что число степеней
свободы равно N - 2 (две степени свободы затрачены на нахождение двух пара-
метров — углового коэффициента и свободного члена):
/[£ У- - (Е У,- )2/N] - m2[^i - )2>]
N-2 N N-2
Эта величина называется стандартным отклонением относительно линии рег-
рессии. Неопределенности (стандартные отклонения) параметров т и b опреде-
ляются величиной s и могут быть из нее рассчитаны. Стандартное отклонение
углового коэффициента т равно
s2
s = _-У_
т Ш^)2
I
fc2-(&)2>
(3.25)
Здесь х, как и ранее, есть среднее из всех значений xz.
Стандартное отклонение свободного члена равно
Из стандартных отклонений, характеризующих неопределенности значений}/, т
и £, можно рассчитать стандартное отклонение содержания вещества в анализи-
руемой пробе х, пользуясь уравнением (3.19) и известным нам законом наложе-
ния (распространения) погрешностей*.
Указанный способ оценки неопределенности значения х достаточно приближенный. Закон наложения по-
грешностей в той его форме, которая приведена в книге, справедлив только в том случае, если все аргументы
взаимно некоррелированы. В то же время параметры градуировочной зависимости т и Ь, как правило, связа-
ны между собой отрицательной корреляцией. Это обстоятельство можно учесть и получить более реалистич-
ную оценку стандартного отклонения х. См., например, [Аналитическая химия. Проблемы и подходы. T. 2 /
Под ред. Р. Кельнера и др. М.: Мир, ООО «Издательство АСТ», 2004, с. 470]. — Прим, перев.
3.16. ЛИНЕЙНЫЙ МЕТОД НАИМЕНЬШИХ КВДЦРАТОВ: КАК ПРОВЕСТИ ПРЯМУЮ?
161
Пример 3.22
Оцените неопределенности значений углового коэффициента, свободного члена
и величину для данных из примера 3.21, а также неопределенность найденного
значения концентрации рибофлавина.
Решение
Чтобы рассчитать все требуемые величины неопределенностей, необходимы
значения ^yz2, (^yz2 )2, )2 и т2. Для данных, приведенных в при-
мере 3.21, (^у. )2 = (83,6)2 = 6989,0, (£х2)2 = O,85Oo, QV )2 = 2,250 и
т2 = (53,75)2 = 2,889 • 103. Значения (у,)2 равны 0,0, 33,6, 148,8, 497,3 и 1874,9.
Отсюда Ху- = 2554,6 (значение приводим с запасными цифрами). Из уравне-
ния (3.24):
_ /(2554,6-6989,0/5) - (53,75)2(О,85Оо -2,250/5) _+()б
sy А s ’ з Усл’ ед-
Из уравнения (3.25):
Sm
' (0,63)2
О,85Оо -2,250/5
= ±1,0 усл. ед./ррш
Из уравнения (3.26):
_ | 0,850о л А
sh =0,6 о --------------= ±0,4, усл. ед.
ь у 5(О,85Оо)-2,250 1
Отсюда т = 53,8 ± 1,0 и b = 0,6 ± 0,4.
Концентрацию рибофлавина в пробе рассчитаем как
(у ± sy)-(b ± sb )_ (15,4 ±0,6)-(0,6 + 0,4) _ПТ7 . 9
w±jm 53,8+1,0
Используя общие принципы наложения погрешностей (при сложении- вы-
читании суммируются абсолютные дисперсии, при умножении-делении — от-
носительные), получим оценку неопределенности величины х:
х = 0,215 ± 0,014 мкг/мл (ppm).
О расчетах стандартного отклонения относительно линии регрессии и стандарт-
ного отклонения х при помощи электронных таблиц см. в гл. 16.
162
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
3,17. Коэффициент корреляции и коэффициент детерминации
Коэффициент корреляции служит мерой корреляции (взаимной зависимости)
между двумя величинами.* Если связь между переменными х и у не является
строго функциональной, то нет смысла говорить о «наилучшем» значении у, со-
ответствующем данному х. Можно говорить лишь о «наиболее вероятном» зна-
чении у. Чем ближе экспериментальные значения к наиболее вероятным, тем
более определенной является зависимость между х и у. Это положение служит
основой для вычисления различных величин, характеризующих степень корре-
ляции между переменными.
Среди таких величин одна из самых простых по способу вычисления —
коэффициент корреляции Пирсона г. Он рассчитывается по формуле:
у (*,- -х)(^- - у)
nsxsy
(3.27)
где п — число данных (пар значений х,у), sx — стандартное отклонение х, sy —
стандартное отклонение у, хги — отдельные значения х и соответствующие им
значения у, х и у— средние значениях и у. Уравнение (3.27) можно записать в
другой форме:
^х^-пху
7(£Х2 ~ wx2xZ^2 -«х2)
^In^xf -(.^хУ ][n^yj ~(^уУ]
(3.28)
Несмотря на устрашающий внешний вид, формула (3.28) обычно оказывается
более удобной для вычисления г, чем формула (3.27), особенно при использова-
нии калькулятора.
Наибольшее возможное значение г равно 1. В этом случае между двумя пе-
ременными имеется строгая линейная зависимость. Если значение г равно нулю,
переменные можно считать полностью независимыми друг от друга. Наимень-
шее возможное значение г равно -1. В этом случае между переменными также
наблюдается строгая линейная зависимость. Отрицательное значение коэффи-
циента корреляции указывает на то, что с возрастанием одной переменной (напри-
мер, х) другая переменная (у) убывает. В этом случае коэффициент корреляции
между величинами х и -у по абсолютной величине тот же, что и между хи у, только
положительный.
Более строго — мерой близости этой зависимости к линейной. — Прим, перев.
3.17. КОЭФФИЦИЕНТ КОРРЕЛЯЦИИ И КОЭФФИЦИЕНТ ДЕТЕРМИНАЦИИ
163
Пример 3.23
Рассчитайте коэффициент корреляции для данных из примера 3.19, приняв зах
результаты, полученные при помощи вашей методики, а за у — при помощи
стандартной методики.
Решение
Рассчитаем значения ^xz2 = 903,2, у? = 855,2, х = 12,0, у = 11,7и ]>\z- у{ - 878,5.
Отсюда по уравнению (3.28):
878,5 -6-12,0-11,7
________________________________= 0,991
7(903,2 - 6 • 12,02 )(855,2 - 6 • 11,72 )
Коэффициент корреляции можно рассчитать для данных градуировочного
графика. Он показывает, насколько близка к линейной связь между измеренны-
ми значениями аналитического сигнала и концентрациями стандартных растворов.
Как правило, если значение г находится в диапазоне 0,90-0,95, это свидетельствует
об удовлетворительном качестве градуировки, если в диапазоне 0,95-0,99 —
о хорошем, а если превышает 0,99 — об отличном качестве. При тщательной ра-
боте иногда можно получить значения г, превышающим 0,999.
При расчете коэффициента корреляции обе переменные рассматриваются
как равноправные. В действительности одна из них обычно является независи-
мой, а другая — зависимой. Численные значения г часто сбивают с толку. Вели-
чина коэффициента корреляции, равная 0,98, кажется весьма близкой к единице,
но соответствующий градуировочный график может быть довольно плохим.
Если значения г, меньше 0,9, это в любом случае свидетельствует о плохом каче-
стве градуировочной зависимости.
В этом отношении более наглядной оценкой соответствия двух серий дан-
ных служит квадрат коэффициента корреляции г2. Именно его вычисляет боль-
шинство стандартных статистических программ (включая Excel — см. рис. 3.8).
Величина г, равная 0,90, соответствует величине г2, равной лишь 0,81, а для
г = 0,95 г2 = 0,90. О близости серий данных можно судить по числу девяток в
значении г. Три девятки и более (г > 0,999) свидетельствуют об отличном соот-
ветствии. В дальнейшем в качестве меры близости серий данных мы будем ис-
пользовать величину г2. Ее часто называют коэффициентом детерминации.
Необходимо подчеркнуть, что коэффициент корреляции характеризует лишь
степень взаимозависимости между двумя массивами переменных в целом и со-
вершенно не свидетельствует о численной близости их значений. Вполне возмож-
на ситуация, когда между результатами анализов, полученными при помощи двух
методик, наблюдается высокая корреляция (значение г2 близко к единице), но
164
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
Z-тест показывает значимое различие между их средними значениями. Это мо-
жет быть, например, в случае, если одна из методик содержит постоянную сис-
тематическую погрешность. Если результаты анализов, полученные для разных
проб при помощи двух методик, представить в виде точек на плоскости (ось х —
данные одной методики, ось у — другой методики), то близость величины г2 к
единице говорит лишь о том, что точки хорошо ложатся на некоторую прямую
линию. Однако угловой коэффициент этой прямой может сколь угодно отличать-
ся от единицы, а свободный член — от нуля. В подобных случаях можно при по-
мощи эмпирических поправочных коэффициентов привести результаты обеих
методик в соответствие друг с другом во всем исследуемом диапазоне концент-
раций.
3.18. Использование электронных таблиц для построения
градуировочных графиков
Использование электронных таблиц позволяет избежать расчетов параметров
линейных зависимостей и их построения на листе миллиметровой бумаги вруч-
ную. Используем данные примера 3.21 для построения графика, изображенного
на рис. 3.8, при помощи Excel.
Откройте новый файл Excel и введите в указанные ячейки следующие дан-
ные.
Ячейка А1: Концентрация рибофлавина, мкг/мл (подберите необходимую
ширину столбца)
Ячейка В1: Интенсивность флуоресценции
Ячейка АЗ: 0,000
Ячейка А4: 0,100
Ячейка А5: 0,200
Ячейка А6: 0,400
Ячейка А7: 0,800
Ячейка ВЗ: 0,0
Ячейка В4: 5,8
Ячейка В5: 12,2
Ячейка В6: 22,3
Ячейка В7: 43,3
Отформатируйте ячейки так, чтобы числа в столбце А содержали три, а в столб-
це В — одну цифру после запятой.
Нажмите на кнопку Мастер диаграмм (Chart Wizard), расположен-
ную на панели инструментов. На экране появится окно Шаг 1 из 4 : тип
диаграммы (Step 1-Chart Type).
3.18. ИСПОЛЬЗОВАНИЕ ЭЛЕКТРОННЫХ ТАБЛИЦ ДЛЯ ПОСТРОЕНИЯ ГРАДУИРОВОЧНЫХ ГРАФИКОВ
165
Выберите тип Точечная (Scatter) и далее Точечный (Scatter) (по
line), после чего нажмите клавишу Далее (Next).
В появившемся окне в строке Диапазон данных (Data Range) введите
АЗ:В7 [или откройте вкладку Ряд (Series), после чего введите диапазоны ад-
ресов переменных X и Y в отдельных строках и затем откройте вкладку Диапа-
зон данных (Data Range)].
Отметьте позицию В столбцах (Columns) и нажмите клавишу Далее
(Next).
В появившемся окне, содержащем несколько строк, введите текст:
• в строку Название диаграммы (Chart Title) — «Градуировочный
график»
• в строку Ось X (Value (X) axis) — «Концентрация рибофлавина»
• в строку Ось Y (Value (Y) axis) — «Интенсивность флуоресценции»
Откройте вкладку Линии сетки (Gridlines) и снимите отметку
Основные линии (Maj or gridlines).
Откройте вкладку Легенда (Legend) и снимите отметку Добавить ле-
генду (Show legend).
Откройте вкладку Подписи данных (Data labels) и отключите все
позиции (ради интереса можете попробовать проставлять отдельные галочки и
посмотреть, что получится).
Нажмите клавишу Далее (Next).
166
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
В появившемся окне отметьте позицию На отдельном листе (As New
sheet); в соответствующей строке при этом будет текст Диаграмма 1
(Chart 1).
Нажмите клавишу Готово (Finish). После этого градуировочный график
появится на отдельном листе файла Excel.
Теперь отобразим уравнение, полученное методом наименьших квадратов, и
величину г2. Выделите область диграммы. В строке Меню откройте меню Диа-
грамма (Chart) и выберите пункт Добавить линию тренда (Add Trend-
line). Откроется новое окно, в котором выберите пункт Линейная (Linear).
Далее откройте вкладку Параметры (Options) и отметьте позиции:
• Показывать уравнение на диаграмме (Display equation on
chart)
• Поместить на диаграмму величину достоверности аппрок-
симации (R2) (Display R-squared value on chart).
Нажмите клавишу OK.
Теперь взгляните на диаграмму. Щелкните по ней клавишей мыши, чтобы
убрать маркеры концов отрезка. Окошко с текстом, содержащим уравнение пря-
мой, можно переместить влево и увеличить. Для этого щелкните клавишей
мыши на уравнении. Оно окажется в прямоугольном окошке с небольшими
квадратиками по периметру. Щелкните по левому нижнему квадратику и пере-
тащите окошко влево вниз. Чтобы увеличить размер шрифта, в меню Формат
(Format) выберите Выделенные подписи данных ... : Шрифт (Se-
lect data labels : Font). Выберите размер шрифта 14 и нажмите ОК. Пе-
ретащите уравнение ближе к прямой. Вы можете увеличить и размер шрифта
подписей осей. Для этого щелкните по соответствующей подписи и далее посту-
пайте, как описано выше.
Область построения диаграммы имеет серый фон. Давайте от него избавим-
ся. Щелкните в произвольной точке фона, затем в меню Формат (Format) выбе-
рите Выделенная область построения ... (Select Plot Area).
Щелкните на квадратике, окрашенном в белый цвет, и нажмите ОК. После всего
этого ваша диаграмма будет иметь вид, подобный рис. 3.8.
При построении диаграммы вы можете в самом начале выделить ячейки, со-
держащие данные (АЗ:В7). В этом случае их адреса будут автоматически поме-
щены в соответствующую строку окна Диапазон данных (Data Range).
Кроме того, вы можете разместить диаграмму не на отдельном листе, а на том
же, куда вводили данные. Для этого вместо На отдельном листе (As New
sheet) вам следует отметить позицию На имеющемся (As object in); в соот-
ветствующей строке будет текст Лист 1 (Sheet 1). Рисунок, появившийся на ли-
сте с данными, вы можете переместить в любое место листа. Для этого щелкните
по рисунку и перетащите его за уголок. На рис. 3.9 изображен график, размещен-
ный на листе с данными. Попробуйте создать его. Если график размещен на листе
с данными, то при изменении данных в ячейках, использованных для построения,
график будет автоматически изменяться. Попробуйте проделать и это, пред-
варительно сохранив файл, а затем переименовав его. Если вы хотите вывести на
печать только график, то выделите его, щелкнув по нему кнопкой мыши.
3.19. РАСЧЕТ УГЛОВОГО КОЭФФИЦИЕНТА, СВОБОДНОГО ЧЛЕНА И КОЭФФИЦИЕНТА ДЕТЕРМИНАЦИИ
167
3.19. Расчет углового коэффициента,
свободного члена и коэффициента детерминации
при помощи электронных таблиц
Использование статистических функций Excel позволяет рассчитать величины
углового коэффициента, свободного члена и коэффициента детерминации ли-
нейной зависимости без построения графика. Откройте новый файл и введите
данные из примера 3.21 в ячейки АЗ:В7, как показано на рис. 3.9. В ячейку А9
введите текст «Свободный член», в ячейку А10 — «Угловой коэффициент», в
ячейку All — «R2». Выделите ячейку В9, нажмите fx: Статистические
(Statistical) и прокрутите меню с именами функций вниз до появления
строки ОТРЕЗОК (INTERCEPT). Выделите эту строку и нажмите ОК. В строке
Известные_значения_х (Known_x ’ s) введите диапазон адресов АЗ:А7, а в
строке Известные_значения_у (Known_y ’ s) — ВЗ:В7. Нажмите ОК, и ве-
личина свободного члена появится в ячейке В9. Аналогично поступите с ячей-
кой В10, выделив ее и выбрав в качестве имени функции SLOPE (НАКЛОН).
Значение углового коэффициента появится в ячейке В10. Наконец, выделите
ячейку Bl 1 и выберите имя функции КВ ПИРСОН (RSQ). В ячейке Bl 1 появится
значение г2. Сравните полученные значения с приведенными на рис. 3.9.
РИС. 3.9. Градуировочный график, вставленный в рабочий лист файла Excel (Лист 1)
168
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
3.20. Программа ЛИНЕЙН (UNEST) для выполнения различных
статистических расчетов
Электронные таблицы Excel снабжены программой ЛИНЕЙН (LINEST), позво-
ляющей быстро рассчитывать различные статистические характеристики для
наборов данных, — например, величины углового коэффициента, свободного
члена и их стандартные отклонения, коэффициент детерминации, стандартное
отклонение предсказания, а также многие другие, которые мы здесь обсуждать
не будем. Программа ЛИНЕЙН (LINEST) позволяет рассчитать одновременно 10
статистических величин, располагаемых в двух столбцах рабочего листа.
Откройте новый файл Excel и введите в ячейки АЗ : В7 данные градуировоч-
ного графика из примера 3.21 так же, как вы делали ранее (сверяйтесь с рис. 3.10).
Статистические характеристики будут помещены в’ 10 ячеек, поэтому давайте
сначала пометим каждое из них. Используем для рассчитанных величин ячейки
В9 : С13. Введите подписи:
Ячейка А9: угловой коэффициент
Ячейка А10: станд. откл. углового коэффициента
Ячейка All: R2
Ячейка А12: F
Ячейка А13: сумма квадратов отклонений
Ячейка D9: свободный член
Ячейка D10: станд. откл. свободного члена
Ячейка DI 1: станд. откл. оценки
Ячейка D12: число ст. своб.
Ячейка D13: остаточная сумма квадратов
А В С D Е
1 Концентрация рибофлавина, мкг/мл Интенсивность флуоресценции
2
3 0,000 0,0
4 0,100 5,8
5 0,200 12,2
6 0,400 22,3
1 0,800 43,3
8
9 угловой коэффициент 53,75 0,595 свободный член
10 станд. откл. углового коэффициента 1,017759 0,419633 станд. откл. свободного члена
11 R2 0,998926 0,643687 станд. откл. оценки
12 F 2789,119 3 число ст. своб.
РИС. 3.10. Использование программы ЛИНЕЙН (LINEST) для статистических расчетов
3.21. ПАКЕТЫ ПРИКЛАДНЫХ СТАТИСТИЧЕСКИХ ПРОГРАММ
169
Выделите ячейки В9:С13 и нажмите fx. В меню Статистические (Statis-
tical) выберите ЛИНЕЙН (LINEST) и нажмите ОК. В строке Изв_знач_у
(Known_y's) введите диапазон адресов ВЗ:В7, а в строке Изв_знач_х
(Known_x ’ s) — АЗ: А7. Затем в каждой из строк Константа (Const) и Ста-
тистика (Stats) наберите Истина («true»). Для продолжения вычислений
нажмите на клавиатуре комбинацию клавиш Shift, Ctrl и Enter и отпустите их.
Рассчитанные статистические величины появятся в выделенных ячейках. Ука-
занное сочетание клавиш необходимо нажимать всегда при использовании фун-
кций, возвращающих массив значений в диапазон ячеек. Значение углового
коэффициента появится в ячейке В9, а его стандартное отклонение — в ячейке
В10. Величина свободного члена и его стандартное отклонение появятся в ячей-
ках С9 и СЮ соответственно. Сравните величины стандартных отклонений с
рассчитанными в примере 3.22, а сами значения углового коэффициента, сво-
бодного члена, а также г2 — с рассчитанными в примере 3.22 или приведенными
на рис. 3.8.
Ячейка С11 содержит величину, названную «стандартное отклонение
оценки». Это синоним термина «стандартное отклонение относительно линии
регрессии» — уже известной нам величины, являющейся мерой погрешности
при расчете значений у. Чем эта величина меньше, тем ближе эксперименталь-
ные данные к рассчитанным. Ячейка В12 содержит экспериментальную величи-
ну F, ячейка С12 — число степеней свободы, использованное для ее расчета,
ячейка В13 — сумму квадратов, обусловленную регрессией, ячейка С13 — оста-
точную сумму квадратов.
Сколько значащих цифр необходимо указывать в записи статистических па-
раметров прямой? Ответ дают величины стандартных отклонений. Угловой коэф-
фициент характеризуется стандартным отклонением 1,0, поэтому его значение
лучше всего указать как 53,8 ± 1,0. Стандартное отклонение свободного члена
равно 0,42, поэтому значение свободного члена запишем в виде 0,6 ± 0,4.
См. также пример 3.22.
3.21. Пакеты прикладных статистических программ
Программа Excel содержит множество статистических функций, перечислен-
ных в меню Сервис (Tools). Для доступа к ним в меню Надстройки. . .
(Add-Ins) отметьте Analysis ToolPak, нажмите ОК и вернитесь к работе с
файлом. После этого в меню Сервис (Tools) появится пункт Анализ данных
(Data Analysis). В нем вы найдете 19 различных статистических программ.
Попробуйте поупражняться с ними: некоторые программы вы найдете очень по-
лезными. Одной из них, безусловно, является Поиск решения (Solver) —
программа для решения сложных уравнений. Как пользоваться этой программой,
описано в гл. 6. Посмотрите также список коммерческих программных продук-
тов для выполнения как простейших, так и более сложных статистических рас-
четов, приведенный на сайте www.wiley.com/college/christian.
170
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
6 делений
Рис. 3.11. Один из способов оценки предела обнаружения. При непрерывной регистрации сигна-
ла видны как флуктуации фона, так и пик, обусловленный наличием аналита. Пик аналита можно
считать надежно детектируемым, если его высота вдвое превосходит размах колебаний фона (т. е.
в данном случае составляет 12 делений шкалы, считая от среднего уровня фона)
3.22. Предел обнаружения:
в аналитической химии нет понятия «нуль»!
До сих пор мы обсуждали применение статистических методов для вероятност-
ной оценки надежности конкретных численных значений, характеризующих ре-
зультаты анализа. В то же время, поскольку каждый инструментальный метод
характеризуется определенным уровнем шумов, связанным со спецификой из-
мерительного процесса, всегда существует предел содержаний, ниже которого
вещество вообще не может быть надежно обнаружено (детектировано). Шум ха-
рактеризует воспроизводимость фонового сигнала, т. е. сигнала в отсутствие
определяемого компонента. Он существует всегда, даже когда сама по себе ве-
личина фонового сигнала пренебрежимо мала. Шум может быть обусловлен
флуктуациями темнового тока фотоумножителя, колебаниями пламени горелки
атомно-абсорбционного спектрометра и множеством других факторов.
Минимальная концентрация вещества, при которой сигнал аналита статисти-
чески значимо отличается от фонового, называется пределом обнаружения. Су-
ществует множество способов оценки предела обнаружения. Например, пределом
обнаружения можно считать концентрацию, при которой сигнал вдвое превышает
размах колебаний фона (рис. 3.11). Однако общепринято считать пределом обна-
ружения такую концентрацию, при которой сигнал превышает фоновый на вели-
чину, равную утроенному стандартному отклонению фонового сигнала.
Пример 3.24
В ходе определения чистоты аспирина спектрофотометрическим методом полу-
чили ряд значений оптической плотности фона (раствора сравнения). Эти вели-
чины составили 0,002, 0,000, 0,008, 0,006 и 0,003. Стандартный раствор аспирина
с концентрацией 1 мкг/мл имеет оптическую плотность 0,051. Чему равен пре-
дел обнаружения аспирина?
3.22. ПРЕДЕЛ ОБНАРУЖЕНИЯ: В АНАЛИТИЧЕСКОЙ ХИМИИ НЕТ ПОНЯТИЯ «НУЛЬ»!
171
Решение
Стандартное отклонение фонового сигнала составляет 0,0032 единиц оптиче-
ской плотности, а среднее значение фонового сигнала — 0,004 единицы. Предел
обнаружения соответствует концентрации аналита, для которой сигнал превы-
шает уровень фона на 3 • 0,0032 = 0,0096 единиц оптической плотности. Значение
сигнала стандартного раствора за вычетом фона составляет 0,051 - 0,004 = 0,047.
Предел обнаружения равен 1 мкг/мл • (0,0096/0,047) = 0,2 мкг/мл, а соответству-
ющее ему значение аналитического сигнала — 0,0096 + 0,004 = 0,014.
Точность определения концентрации на пределе обнаружения, в соответствии с
определением этой величины, составляет 33%. Для надежных количественных
измерений концентрация должна быть как минимум в 10 раз выше (2 мкг/мл в
приведенном примере).
Предпринимались многочисленные попытки более строгого статистическо-
го обоснования понятия «предел обнаружения». На Международной конферен-
ции по гармонизации технических требований для регистрации лекарственных
средств, используемых человеком (ICH, см. гл. 4) разработан ряд руководств по
проверке методик анализа [18]. В руководстве ICH Q2B, посвященном проверке
методологии, предлагается рассчитывать предел обнаружения на основе вели-
чин стандартного отклонения сигнала s и углового коэффициента градуировоч-
ной зависимости S (коэффициента чувствительности). При этом обе величины
должны быть определены в концентрационном диапазоне, близком к пределу
обнаружения. Для расчета предела обнаружения (limit of detection, LOD) пред-
ложена формула
LOD = 3,3 (s/S) (3.29)
а для расчета нижней границы определяемых содержаний (limit of quantitation,
LOQ) — формула
LOQ=10(s/5) (3.30)
В качестве стандартного отклонения сигнала можно использовать как не-
посредственно значение стандартного отклонения фона, так и стандартное от-
клонение относительно линии регрессии (остаточное стандартное отклонение)
или стандартное отклонение свободного члена уравнения линии регрессии.
Две последние величины можно рассчитать при помощи статистических функ-
ций Excel.
Международный союз по теоретической и прикладной химии (IUPAC) ре-
комендует использовать для расчета предела обнаружения коэффициент 3 в
уравнении (3.29), а за величину s принимать стандартное отклонение фонового
сигнала. При разумном числе измерений это обеспечивает доверительную веро-
ятность около 95%. Для определения величины s следует провести 7-10 измере-
172
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
ний. В любом случае следует иметь в виду, что предел обнаружения является
сугубо оценочной величиной. Не стоит предпринимать усилий для слишком
точного ее определения.
3.23. Статистика пробоотбора:
сколько брать проб, какого размера?
Возможно, именно отбор представительной, неискаженной пробы является наи-
более важной стадией любого анализа. Физические принципы отбора проб мате-
риалов разного типа (твердых, жидких, газообразных) были рассмотрены ранее
в гл. 2. Теперь мы рассмотрим процесс пробоотбора с точки зрения статистики.
Пробоотбор — ключевой этап в обеспечении точности
результатов
В большинстве случаев правильность и воспроизводимость результатов анализа
лимитируются не стадией измерения сигнала, а стадией пробоотбора. Общая
дисперсия результатов анализа s2 складывается из дисперсий, обусловленных
отбором пробы ($£) и прочими аналитическими операциями (s2):
s2=s2+s2
(3.31)
Составляющую дисперсии, обусловленную пробоотбором, можно найти,
например, путем отбора ряд проб и их анализа при помощи высокоточной мето-
дики. Нет смысла стремиться уменьшить величину sa менее чем до | — выиг-
рыш в точности будет несущественным. Например, если абсолютное стандартное
отклонение, связанное с пробоотбором, составляет 3,0%, а с измерением и дру-
гими стадиями анализа — 1,0%, то общая дисперсия s2 = (3,0)2 + (1,0)2 = 10,0,
или s0 = 3,2%. Таким образом, 94% общей погрешности обусловлены пробоотбо-
ром и только 6% — измерением (за счет измерения величина s0 возрастает с 3,0%
до 3,2%). Если погрешность, обусловленная пробоотбором, велика, то лучше ис-
пользовать пусть недостаточно точный, но быстрый метод анализа и за одно и то
же время проанализировать больше проб.
Следует учесть, что на разброс результатов анализа влияют не только по-
грешности анализа, но и изменчивость содержания аналита в самом объекте.
Именно этот фактор представляет интерес для аналитика. Если охарактеризо-
вать эту изменчивость при помощи дисперсии s2, то общий разброс результатов
анализа составит 5^otal = s2 + s2 +s2. Для надежной интерпретации результатов
химического анализа сумма s2 + s2 не должна превышать 20% от . Более по-
дробно об этом см. в работе [М. Н. Ramsey, «Appropriate Precision: Matching Analyti-
cal Precision Specifications to the Particular Application», Anal. Ргос.,Ш (1993) 110].
3.23. СТАТИСТИКА ПРОБООТБОРА: СКОЛЬКО БРАТЬ ПРОБ, КАКОГО РАЗМЕРА?
173
Оценка содержания вещества в объекте
По результатам анализа отдельной пробы можно оценить содержание вещества
во всем объекте в целом. Это делается при помощи ^-теста для выбранного зна-
чения доверительной вероятности по уравнению (3.11). В качестве х используют
среднее значение результатов анализа пробы, а в качестве s — стандартное от-
клонение, рассчитанное из результатов предыдущих анализов серий аналогич-
ных проб. Можно рассчитать s и из результатов анализа данного объекта, если
проанализировано достаточное число проб.
Минимальный размер пробы
Для обеспечения правильного отбора проб гетерогенных материалов разработан
ряд статистических правил, основанных на величине дисперсии пробоотбора.
Для хорошо перемешанного материала, состоящего из отдельных частиц раз-
личной природы, минимальный размер отдельной порции пробы можно оценить
из величины константы пробоотбора Ингамелла Ks по уравнению
wR2 = Ks
(3.32)
где w — масса анализируемой пробы, а А — относительное стандартное откло-
нение, характеризующее неоднородность состава материала и выраженное в
процентах. Ks численно равна массе пробы, характеризующейся неопределенно-
стью состава на уровне 1% при 68%-й доверительной вероятности. Ее можно
рассчитать из ряда значений стандартных отклонений результатов анализа проб
разной массы. Из уравнения (3.32) следует, что дисперсия пробоотбора обратно
пропорциональна массе пробы. Чем больше размер пробы, тем меньше диспер-
сия результатов ее анализа.
Пример 3.25
Применительно к методике определения содержания азота в пшеничном зерне кон-
станта пробоотбора Ингамелла составляет 0,50 г. Пробу какой массы следует взять,
чтобы относительное стандартное отклонение пробоотбора составило 0,2%?
Решение
w (0,2)2 = 0,50 г
w = 12,5 г
Полученный результат не означает, что необходимо анализировать всю эту про-
бу. 12,5 г — это масса генеральной пробы. Ее необходимо тонко измельчить, пе-
ремешать и лишь несколько сотен миллиграммов полученного гомогенного
материала использовать для анализа. Но если пробу не подвергать гомогениза-
ции, тогда следует анализировать ее целиком.
174
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
Минимальное число порций
для составления генеральной пробы
Число отдельных порций анализируемого материала, необходимое для дости-
жения требуемой степени надежности результатов анализа, можно оценить по
следующему уравнению:
(3.33)
где t — коэффициент Стьюдента для выбранной доверительной вероятности,
sj —дисперсия пробоотбора, г—желаемое значение относительного стандарт-
ного отклонения среднего результата анализах. Величина^ представляет собой
абсолютное стандартное отклонение, выраженное в тех же единицах, что и х.
Таким образом, п оказывается безразмерной величиной. Значения х и s можно
получить из результатов предыдущих анализов или оценить априори. Посколь-
ку г равно sx /х, формулу (3.33) можно переписать в виде
(3.34)
В этом случае sx и можно выразить либо в абсолютных, либо в относительных
единицах, но обе величины — одним и тем же способом. Величина t зависит от
и, значение которого неизвестно. Поэтому сначала задают произвольное значе-
ние t и используют для расчета «-итерационную процедуру.
Пример 3.26
Содержание железа в объединенной партии руды составляет около 5% (масс.), а
относительное стандартное отклонение пробоотбора — 0,021 (2,1%). Сколько
проб нужно отобрать, чтобы обеспечить относительное стандартное отклонение
результатов анализа г, равное 0,016 (1,6%; соответственно, абсолютное стандар-
тное отклонение составит 0,016 • 5% (масс.) = 0,08% (масс.)) при 95%-й довери-
тельной вероятности?
Решение
Для расчетов можно использовать как уравнение (3.33), так и (3.34). Мы исполь-
зуем второе. В качестве начального приближения для t возьмем значение 1,96
(для п = оо при 95%-й доверительной вероятности, см. табл. 3.1) и рассчитаем
первое приближение для п. Выберем новое значение /, соответствующее рассчи-
танному и, и будем повторять расчеты до тех пор, пока не получим постоянное
значение п.
3.23. СТАТИСТИКА ПРОБООТБОРА: СКОЛЬКО БРАТЬ ПРОБ, КАКОГО РАЗМЕРА?
175
Для п = 7, t = 2,365.
Для п = 10, t = 2,23.
1,962 -0,0212
п =---------------= 6,6
0,0162
2,3652-0,0212
п =---------------= 9,6
0,0162
2,232 -0,0212
0,0162
Выполните аналогичные расчеты, используя уравнение (3.33), и сравните полу-
ченные значения п.
Уравнение (3.33) справедливо для случая, когда распределение концентра-
ции аналита в массе пробы является нормальным. Как мы неоднократно отме-
чали, в этом случае 68% значений величины находятся в пределах одного, а
95% — в пределах двух стандартных отклонений относительно среднего. Для
нормального распределения величина генеральной дисперсии а2 обычно суще-
ственно меньше, чем среднее значение. Однако в ряде случаев распределение
концентраций может подчиняться закону Пуассона. Распределение Пуассона
похоже на нормальное, но для него дисперсия sj равна среднему значению х.
В этом случае уравнение (3.33) упрощается:
(335)
Обратите внимание на то, что, поскольку численно равна х, эти величины со-
кращаются, однако их единицы измерения различны. Ввиду того, что дисперсия
равна среднему, разброс данных в этом случае шире, и для получения предста-
вительной пробы требуется гораздо больше порций материала.
Если материал настолько неоднороден, что в нем можно выделить отдель-
ные комья или клочья, стратегия пробоотбора усложняется. Каждый комок мож-
но рассматривать как отдельную фракцию и отбирать из него пробу отдельно.
Если же материал имеет отчетливо выраженную структуру, например, состоит
из отдельных слоев, а цель анализа — получение данных о его среднем составе,
то число порций, отобранных из каждой фракции, должно быть пропорциональ-
но размеру фракции.
176
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
Что мы узнали из этой главы?
• Правильность и воспроизводимость — стр. 107
• Классификация погрешностей измерений — стр. 109
• Значащие цифры в результатах измерений и вычислений — стр. 111, 138
• Стандартное отклонение — стр. 119
• Как пользоваться электронными таблицами — стр. 124
• Наложение погрешностей — стр. 130, 138
• Контрольные карты — стр. 139
• Некоторые статистические понятия и тесты: доверительный интервал,
/-тест, F-тест— стр. 140, 143, 145
• Правила отброса промахов — стр. 151
• Построение прямых методом наименьших квадратов, коэффициент детер-
минации— стр. 156, 163
• Применение электронных таблиц для построения градуировочных графи-
ков— стр.164
• Предел обнаружения — стр. 170
• Статистика пробоотбора — стр. 172
Вопросы
1 - В чем разница между правильностью и воспроизводимостью?
2. Что такое детерминированная (систематическая) и недетерминированная
(случайная) погрешность?
3- Среди приведенных ниже распространенных причин, вызывающих по-
грешности результатов анализа, укажите, в каких случаях погрешность бу-
дет систематической, в каких — случайной. Систематические погрешности
подразделите далее на инструментальные, субъективные и методические,
а) Взвешиваемая проба гигроскопична, б) При газохроматографическом
анализе один из компонентов пробы химически взаимодействует с материа-
лом колонки, в) При анализе радиоактивной пробы в одних и тех же усло-
виях каждый раз регистрируемые скорости счета импульсов несколько раз-
личаются. г) Кончик пипетки, используемой в ходе анализа, разбит, д) При
измерении высоты одного и того же хроматографического пика два лабо-
ранта сообщают различные результаты.
Задачи
При решении статистических задач сначала выполните вычисления вручную, а
затем при помощи статистических функций Excel и сопоставьте результаты. См.
также компакт-диск, задачи 14-18, 20, 21, 25-30 и 37^40.
3.23. СТАТИСТИКА ПРОБООТБОРА: СКОЛЬКО БРАТЬ ПРОБ, КАКОГО РАЗМЕРА?
177
Значащие цифры
4- Сколько значащих цифр содержит запись каждого из приведенных ниже
чисел?
а) 200,06, б) 6,030 • 10-4, в) 7,80 • 1010.
5- Сколько значащих цифр содержит каждое из представленных чисел?
а) 0,02670, 6)328,0, в) 7000,0, г) 0,00200.
6- Рассчитайте молярную массу LiNO3 и приведите ее с необходимым числом
значащих цифр.
7- Рассчитайте молярную массу PdCl2 и приведите ее с необходимым числом
значащих цифр.
8. Приведите результат следующего арифметического выражения с макси-
мально возможным числом значащих цифр: 50,00 • 27,8 • 0,1167.
9- Приведите результат следующего арифметического выражения с максима-
льно возможным числом значащих цифр:
(2,776 • 0,0050) - 6,7 • 10~3 + (0,036 • 0,0271).
10- Аналитик планирует определить содержание меди в бронзе спектрофото-
метрическим методом. С какой точностью следует брать навеску, если ее
масса составляет около 5 г, а значения оптической плотности (А) измеряют
с точностью до 0,001 единицы? Перед измерением раствор разбавляют таким
образом, чтобы оптическая плотность находилась в диапазоне 0,1 < А < 1,
в котором погрешность ее измерения минимальна.
Представление результатов
11. Стандартный образец плазмы крови с содержанием хлорид-ионов
102 ммоль экв./л проанализировали с помощью кулонометрического тит-
рования ионами серебра. Результаты повторных анализов составили 101 и
98 ммоль экв./л. Рассчитайте: а) среднее значение, б) абсолютную погреш-
ность среднего, в) относительную погрешность среднего (%).
12- Партия таблеток с ядерным топливом была взвешена для того, чтобы уста-
новить, насколько их масса соответствует номинальной. Массы таблеток
составили 127,2,128,4,127,1,129,0 и 128,1 г. Рассчитайте: а) среднее значе-
ние, б) медиану и в) величину размаха.
13- Рассчитайте абсолютные и относительные (в % и частях на тысячу) по-
грешности следующих результатов измерений:
Измеренное значение Аттестованное значение
а) 22,62 г 22,57 г
б) 45,02 мл 45,31 мл
в) 2,68% 2,71%
г) 85,6 см 85,0 см
178
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
Стандартное отклонение
14- В образце бронзы определяли содержания олова и цинка. Получили следу-
ющие результаты: Zn — 33,27, 33,37 и 33,34%, Sn — 0,022, 0,025 и 0,026%.
Рассчитайте абсолютное и относительное стандартное отклонение для
каждой серии результатов.
15- Результаты параллельных определений жесткости воды составили 102,2,
102,8,103,1 и 102,3 ppm СаСО3. Для этой серии данных рассчитайте: а) стан-
дартное отклонение, б) относительное стандартное отклонение, в) стандарт-
ное отклонение среднего, г) относительное стандартное отклонение среднего.
16- Параллельные анализы серебряного сплава на содержание серебра дали
следующие результаты: 95,67, 95,61, 95,71 и 95,60% Ag. Рассчитайте:
а) стандартное отклонение, б) стандартное отклонение среднего, в) относи-
тельное стандартное отклонение среднего (в %)
Наложение погрешностей
17- Оцените неопределенность результатов следующих вычислений:
а) (128 ± 2) + (1025 ± 8) - (636 ± 4);
б) (16,25 ± 0,06) - (9,43 ± 0,03);
в) (46,1 ± 0,4) + (935 ± 1).
18 - Оцените абсолютную неопределенность результатов следующих вычислений:
а) (2,78 ± 0,04) • (0,00506 ± 0,00006);
б) (36,2±0,4)/(27,1 ±0,6);
в) (50,23 ± 0,07) • (27,86 ± 0,05)/(0,1167 ± 0,0003).
19- Оцените абсолютную неопределенность результата следующего вычисле-
ния: [(25,0 ± 0,1) • (0,0215 ± 0,0003) - (1,02 ± 0,01) • (0,112 ± 0,001)] • (17,0 ±
± 0,2)/(5,87 ± 0,01).
Доверительный интервал
20- В результате повторных экспериментов по стандартизации раствора по-
лучены следующие значения его молярной концентрации: 0,5026, 0,5029,
0,5023, 0,5031, 0,5025, 0,5032, 0,5027 и 0,5026 М. В предположении отсут-
ствия систематических погрешностей рассчитайте интервал, в котором с
95%-й вероятностью находится истинное значение средней концентрации
раствора.
21 - При определении содержания натрия в отдельных порциях пробы крови
при помощи ионселективного электрода получены следующие значения:
139,2,139,8,140,1 и 139,4 ммоль экв./л. В каких пределах находится истин-
ное значение (считая, что систематические погрешности отсутствуют):
а) с 90%-й вероятностью; б) с 95%-й вероятностью; в) с 99%-й вероятностью?
22- Содержание свинца на поверхности листьев деревьев, растущих вблизи
шоссе, определяли спектрофотометрическим методом с дитизоном. Стан-
3.23. СТАТИСТИКА ПРОБООТБОРА: СКОЛЬКО БРАТЬ ПРОБ, КАКОГО РАЗМЕРА?
179
дартное отклонение, рассчитанное для трех параллельных определений,
составило 2,3 ppm. Рассчитайте ширину доверительного интервала для
90%-й доверительной вероятности.
23. Для методики определения хлоридов в крови при помощи кулонометриче-
ского титрования найдено значение стандартного отклонения, равное
0,5 ммоль экв./л. Какова ширина доверительного интервала для среднего из
трех параллельных определений при 95%-й доверительной вероятности?
24. Для данных по стандартизации раствора из задачи 31 рассчитайте диапа-
зон, в котором с 90%-й вероятностью может (при отсутствии системати-
ческих погрешностей) находиться истинное значение концентрации
раствора.
Статистические тесты значимости
25. Было проведено исследование, имевшее целью установить, есть ли корре-
ляция между содержанием хрома в крови и определенным заболеванием.
Для этого проанализировали ряд проб крови больных людей и сравнили их
с результатами анализов контрольной группы здоровых людей. Установи-
те, можно ли различия в результатах анализов этих двух групп отнести на
счет случайных факторов или они свидетельствуют о предполагаемой кор-
реляции. Содержания хрома (ppb) для контрольной группы составляют 15,
23,12,18,9,28,11,10, а для группы больных людей — 25,20,35,32,15,40,
16, 10, 22, 18.
26. Для проверки результатов определения этанола в вине ферментативным
методом использованы данные газохроматографического анализа. При мно-
гократном анализе одной и той же пробы двумя методами получены следу-
ющие данные: ферментативным методом — 13,1, 12,7, 12,6, 13,3, 13,3%
этанола; газохроматографическим методом— 13,5,13,3,13,0,12,9% этано-
ла. Можно ли считать, что для 95%-й доверительной вероятности фермен-
тативный метод дает те же результаты, что и газохроматографический?
27. В лаборатории проводят оценку воспроизводимости определения креати-
нина в сыворотке крови фотометрическим методом по реакции со щелоч-
ным раствором пикрата с образованием окрашенного продукта. Чтобы
получить наиболее надежную оценку, провели несколько серий анализов
различных проб в течение нескольких дней. Из приведенных ниже величин
оптической плотности рассчитайте средневзвешенное стандартное откло-
нение результатов.
День 1, проба А День 2, проба В День 3, проба С
0,826 0,682 0,751
0,810 0,655 0,702
0,880 0,661 0,699
0,865 0,724
хА = 0,845 хв = 0,666 хс = 0,719
180
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
28. Ниже приведены результаты параллельных определений кальция в пробе
крови методами атомно-абсорбционной спектроскопии (ААС) и фотомет-
рии (по вновь разработанной методике). Есть ли значимое различие в вос-
производимостях этих методик?
Данные
ААС
(мг/дл)
10,9
10,1
10,6
11,2
9,7
10,0
Среднее 10,4
Данные
фотометрии
(мг/дл)
9,2
10,5
9,7
11,5
11,6
9,3
10,1
11,2
Среднее 10,4
29. Дихромат калия — окислитель, используемый для титриметрического
определения железа в форме Fe(II). Дихромат калия можно получить в
чистом виде и непосредственно приготовить из него стандартный раствор
известной концентрации. Однако часто раствор дихромата калия тем не
менее стандартизуют, титруя им раствор с известным содержанием Fe(II)
(приготовленный из железной проволоки особой чистоты или электроли-
тического железа) по той же методике, что используется для анализа про-
бы. Это делают потому, что в ходе титрования ионы Fe(II) окисляются до
ионов Fe(III), окраска которых может накладываться на окраску индика-
тора, используемого для установления конечной точки титрования, и тем
самым вызывать некоторую погрешность. Концентрация раствора дихро-
мата калия, рассчитанная по навеске, составила 0,1012 М, а по данным
стандартизации — 0,1017, 0,1019, 0,1016, 0,1015 М. Верно ли предполо-
жение о том, что концентрация дихромата калия, найденная в результате
титриметрической стандартизации, статистически значимо отличается от
истинной?
30- В атомной промышленности ведется строгий учет полученного, перевезен-
ного и использованного количества плутония. Каждую получаемую партию
плутониевых таблеток тщательно анализируют, чтобы проверить, насколько
чистота плутония, а следовательно, и его количество соответствуют дан-
ным поставщика. Данные по содержанию плутония в одной из партий
составили 99,93, 99,87, 99,91 и 99,86%. По данным поставщика, содержание
плутония составляет 99,95%. Можно ли считать эту информацию соответст-
вующей действительности?
3.23. СТАТИСТИКА ПРОБООТБОРА: СКОЛЬКО БРАТЬ ПРОБ, КАКОГО РАЗМЕРА?
181
Q-тест
31 - При стандартизации раствора получен следующий ряд значений его концен-
трации (М): 0,1067,0,1071,0,1066 и 0,1050. Следует ли один из этих результа-
тов (при 95%-й доверительной вероятности) считать промахом и отбросить?
32. Содержит ли промахи (при 95%-й доверительной вероятности) серия дан-
ных, приведенных в задаче 14?
33. Для оценки воспроизводимости методики проведен ряд повторных анали-
зов одного и того же объекта. Получены следующие значения содержания в
нем определяемого компонента (%): 22,23,22,18, 22,25, 22,09 и 22,17. Сле-
дует ли считать, что значение 22,09% не является промахом при 95%-й дове-
рительной вероятности?
Статистика малых серий данных
34. Для данных, приведенных в задаче 15, оцените стандартное отклонение по
величине размаха. Сравните полученную оценку с результатом непосред-
ственного расчета стандартного отклонения, проведенного в ходе выпол-
нения задачи 15.
35 - Для данных из задачи 20 оцените границы доверительного интервала по ве-
личине размаха при 95%-й доверительной вероятности. Сравните эту оцен-
ку с полученной в задаче 20 из величины стандартного отклонения.
36. Для данных из задачи 21 оцените границы доверительного интервала по ве-
личине размаха для 95- и 99%-й доверительной вероятности. Сравните эти
оценки с полученными в задаче 21 из величины стандартного отклонения.
Метод наименьших квадратов
37. При помощи уравнения (3.22) рассчитайте тангенс угла наклона линейной
зависимости, описывающей данные из примера 3.21. Сравните результат с
величиной, рассчитанной по уравнению (3.23).
38. Для построения градуировочного графика для фотометрического опреде-
ления фосфора в моче провели реакцию ряда аликвот стандартного раствора
фосфатов с Mo(VI), восстановили образовавшуюся фосфорномолибдено-
вую кислоту до «синей» формы и измерили оптическую плотность (А) по-
лученных растворов. Из приведенных ниже данных методом наименьших
квадратов рассчитайте параметры градуировочной прямой и содержание
фосфора в пробе мочи.
Концентрация фосфора, мкг/мл А
1,00 0,205
2,00 0,410
3,00 0,615
4,00 0,820
Проба мочи 0,625
182
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
39- Оцените неопределенности значений углового коэффициента и свободного
члена линейной зависимости, рассчитанной в задаче 38, а также неопреде-
ленность содержания фосфора в анализируемой пробе.
Коэффициент корреляции
40- Из приведенных ниже данных рассчитайте коэффициент корреляции меж-
ду содержанием токсина, вырабатываемого культурой грибов, и содержа-
нием экстракта дрожжей в питательной среде.
Образец Содержание экстракта дрожжей, % Содержание токсина, мг
(а) 1,000 0,487
(б) 0,200 0,260
(в) 0,100 0,195
(г) 0,010 0,007
(д) 0,001 0,002
41 - Культуры, описанные в задаче 40, характеризуются следующими количе-
ствами сухой массы грибов (мг): (а) 116, (б) 53, (в) 37, (г) 8 и (д) 1. Рассчи-
тайте коэффициент корреляции между сухой массой грибов и количеством
выработанного токсина.
42- Новая методика определения холестерина в сыворотке крови основана на
измерении скорости потребления кислорода в ходе реакции окисления хо-
лестерина кислородом, катализируемой ферментом холестериноксидазой.
Несколько образцов проанализировали как с использованием этой методи-
ки, так и по стандартной фотометрической методике Либермана. Определи-
те из приведенных данных при помощи /-теста, существует ли статистически
значимое различие между результатами двух методик, а также рассчитайте
коэффициент корреляции между ними. Примите, что методики характери-
зуются одинаковой воспроизводимостью.
Проба Ферментативная методика, мг/дл Фотометрическая методика, мг/дл
1 305 300
2 385 392
3 193 185
4 162 152
5 478 480
6 455 461
7 238 232
8 298 290
9 408 401
10 323 315
3.23. СТАТИСТИКА ПРОБООТБОРА: СКОЛЬКО БРАТЬ ПРОБ, КАКОГО РАЗМЕРА?
183
Предел обнаружения
43 - Вы определяете алюминий в образцах растений при помощи флуориметри-
ческой методики. Для семи проб контрольного опыта получены значения
интенсивности флуоресценции, равные 0,12, 0,18, 0,25, 0,11, 0,16, 0,26 и
0,16 единиц. Стандартный раствор, содержащий 1,0 ppm алюминия, дает
сигнал 1,25 единицы. Чему равен предел обнаружения? Какое значение
сигнала ему соответствует?
Статистика пробоотбора
44- В результате анализа высокоточным методом (относительное стандартное
отклонение менее 1%) серии проб краски массой 0,4 г на содержание свинца
относительное стандартное отклонение пробоотбора оказалось равным 5%.
Какой массы следует брать пробы, чтобы снизить эту величину до 2,5%?
45- Содержание меди в руде составляет около 3% (масс.). Сколько порций
руды следует отобрать, чтобы относительное стандартное отклонение ре-
зультата анализа при 95%-й доверительной вероятности составило 5%? Аб-
солютная погрешность пробоотбора составляет 0,15% (масс.).
Рекомендуемая литература
Общие вопросы статистики
1. Р. С. Meier and R. Е. Zund, Statistical Methods in Analytical Chemistry, 2nd ed.
New York: Wiley, 2000.
2. J. C. Miller and J. N. Miller, Statistics and Chemometrics for Analytical Chemist-
ry, 4th ed. Englewood Cliffs, NJ: Prentice Hall, 2000.
3. J. C Miller and J. N. Miller, «Basic Statistical Methods for Analytical Chemistry.
A Review. Part 1. Statistics of Repeated Measurements». «Part 2. Calibration
and Regression Methods», Analyst, 113 (1988) 1351; 116 (1991)3.
4. M. Otto, Chemometrics: Statistics and Computer Application in Analytical Che-
mistry. London: Wiley, 1999.
Q-тест
5- R. B. Dean and W. J. Dixon, «Simplified Statistics for Small Numbers of Obser-
vations», Anal. Chem., 23 (1951) 636.
6- W. J. Blaedel, V. W. Meloche, and J. A. Ramsay, «А Comparison of Critiera for
the Rejection of Measurements», J. Chem. Educ., 28 (1951) 643.
7- D. B. Rorabacher, «Statistical Treatment for Rejection of Deviant Values; Criti-
cal Values of Dixon’s ’Q' Parameter and Related Subrange Ratios at the 95%
Confidence Level», Anal. Chem., 63 (1991) 139.
8. С. E. Efstathiou, «А Test for the Simultaneous Detection of Two Outliers Among
Extreme Values of Small Data Sets», Anal. Lett, 26 (1993) 379.
184
ОБРАБОТКА ДАННЫХ И ЭЛЕКТРОННЫЕ ТАБЛИЦЫ
Контроль качества результатов анализа
9- J. К. Taylor, «Quality Assurance of Chemical Measurements», Anal. Chem., 53
(1981) 1588A.
10- J. K. Taylor, Quality Assurance of Chemical Measurements. Boca Raton, FL:
CRC Press/Lewis, 1987.
11. J. K. Taylor, «Validation of Analytical Methods», Anal. Chem., 55 (1983) 600A.
12- J. O. Westgard, P. L. Barry, and M. R. Hunt, «А Multi-Rule Shewhart Chart for
Quality Control in Clinical Chemistry», Clin. Chem., 27 (1981) 493.
Метод наименьших квадратов
13- P. Galadi and B. R. Kowalski, «Partial Least Squares Regression (PLS): A Tuto-
rial», Anal. Chim. Acta, 185 (1986) 1.
Предел обнаружения
14- G. L. Long and J. D. Winefordner, «Limit of Detection. A Closer Look at the
ШРАС Definition», Anal. Chem., 55 (1983) 712A.
15- J. P. Foley and J. G. Dorsey, «Clarification of the Limit of Detection in Chroma-
tography», Chromatographia, 18 (1984) 503.
16- J. E. Knoll, «Estimation of the Limit of Detection in Chromatography», J. Chro-
matogr. ScL, 23 (1985) 422.
17- Analytical Methods Committee, «Recommendations for the Definition, Estima-
tion and Use of the Detection Limit», Analyst, 112 (1987) 199.
18- 1CH-Q2B Validation of Analytical Procedures: Methodology (International
Conference on Harmonization of Technical Requirements for Registration of
Pharmaceuticals for Human Use, Geneva, Switzerland, November 1996).
Статистика пробоотбора
19. В. Kratochvil and J. K. Taylor, «Sampling for Chemical Analysis», Anal. Chem.,
53 (1981) 924A.
20. M. H. Ramsey, «Sampling as a Source of Measurement Uncertainty: Techniques
for Quantification and Comparison with Analytical Sources», J. Anal. Atomic
Spectrosc., 13 (1998) 97.
21. G. Brands, «Theory of Sampling. I. Uniform Inhomogeneous Material», Frese-
nius' Z. Anal. Chem., 314 (1983) 6; II. «Sampling from Segregated Material»,
Z. Anal. Chem., 314 (1983) 646.
22. N. T. Crosby and I. Patel, eds., General Principles of Good Sampling Practice.
Cambridge, UK: Royal Society of Chemistry, 1995.
23. S. K. Thompson, Sampling, New York: Wiley, 1992.
3.23. СТАТИСТИКА ПРОБООТБОРА: СКОЛЬКО БРАТЬ ПРОБ, КАКОГО РАЗМЕРА?
185
Использование электронных таблиц
24- D. Diamond and V. С. A. Hanratty, Spreadsheet Applications in Chemistry
Using Microsoft Excel. New York: Wiley, 1997.
25. H. Freiser, Concepts and Calculations in Analytical Chemistry: A Spreadsheet
Approach. Boca Rato, FL: CRC Press, 1992.
26. R. De Levie, How to Use Excel in Analytical Chemistry and in General Scientific
Data Analysis. Cambridge, UK: Cambridge University Press, 2001.
Глава 4
«ХОРОШАЯ ЛАБОРАТОРНАЯ ПРАКТИКА»:
ОБЕСПЕЧЕНИЕ КАЧЕСТВА РЕЗУЛЬТАТОВ АНАЛИЗА
Мы можем преодолеть гравитацию,
но иногда падаем под тяжестью бумажной работы.
Вернер фон Браун
В гл. 1 мы рассмотрели основные принципы выполнения количественного ана-
лиза, а в гл. 2 и 3 обсудили вопросы, касающиеся методологии и статистики от-
бора проб, а также основные приемы обработки и анализа данных. Соблюдение
этих общих правил в основном достаточно для того, чтобы правильно осуществ-
лять измерительные процедуры, а при использовании хорошо отработанных ме-
тодик высока вероятность получения приемлемых, правильных результатов.
Однако в зависимости от цели, для которой предназначаются результаты анали-
за, всего этого может оказаться недостаточно для того, чтобы удовлетворить за-
просы заказчика. Сказанное в особенности касается случаев, когда анализ
проводится в соответствии с требованиями законодательства, в целях кримина-
листической экспертизы и вообще во всех ситуациях, когда результаты анализа
предстоит защищать в суде. Чтобы гарантировать, насколько это возможно, что
представленные результаты анализа являются правильными (в заранее предпи-
санных или документированных пределах), применительно к аналитическим ла-
бораториям были разработаны концепции «хорошей лабораторной практики»
(англ, good laboratory practice, GLP), поверки (аттестации, валидации) методик
и обеспечения качества результатов анализа. Целый ряд правительственных ор-
ганизаций (американские Агентство по защите окружающей среды — ЕРА и
Управление по пищевым продуктам и лекарственным средствам — FDA) и обще-
ственных организаций (Международная ассоциация официальных химиков-ана-
литиков — АОАС International, Американское общество по тестированию и
материалам — ASTM и другие) выпустили свои собственные нормативы, каса-
ющиеся GLP, поверки методик и обеспечения качества. Все эти нормативы име-
ют ряд общих черт. Краткое ознакомление некоторых из нормативов мы начнем
с рассмотрения основ GLP.
При выборе средств обеспечения качества результатов анализа руководство
лабораторий и сами аналитики должны руководствоваться как минимум здра-
вым смыслом, принимая во внимание цели анализа, опыт работы, доступность
методик, ограничения, обусловленные временем и стоимостью анализов, и тому
подобные соображения. Но чем строже вы будете придерживаться официально
установленных норм, тем в большей мере вы (и не только вы) будете уверены в
правильности результатов. Необходимо запомнить, что правильное выполнение
«ХОРОШАЯ ЛАБОРАТОРНАЯ ПРАКТИКА»: ОБЕСПЕЧЕНИЕ КАЧЕСТВА РЕЗУЛЬТАТОВ АНАЛИЗА
187
Зачем нужна «хорошая лабораторная практика»?
Ответ на этот вопрос может показаться очевидным. Тем не менее мы проиллюстрируем
его рассказом о том, в какое замешательство была однажды повергнута одна из ведущих
аналитических лабораторий мира — лаборатория Федерального бюро расследований
(ФБР). В 1995 г. лаборатория ФБР участвовала в расследовании дела особой важности —
взрыва Федерального здания Альфреда П. Мурры в Оклахома-сити. В результате этого
взрыва часть здания была разрушена, 168 человек погибли и сотни получили ранения. Ла-
боратория ФБР провела определение взрывчатых веществ в пробах, взятых на месте пре-
ступления. Результаты их анализа стали важнейшими судебными доказательствами. Жюри
присяжных признало Тимоти Маквея виновным по всем пунктам обвинения, включая уча-
стие в заговоре, терроризм и тяжкое убийство первой степени. Однако адвокаты Маквея
нашли в обвинительном заключении слабые места и представили суду опубликованный
незадолго до того 157-страничный доклад Департамента юстиции по поводу деятельности
лаборатории ФБР. В нем были собраны многочисленные данные, свидетельствующие о
недостатках в организации и проведении работ в этой лаборатории (только три страницы
из этого доклада были приняты судом). Доклад явился результатом 18-месячного рассле-
дования, инициированного одним из сотрудников лаборатории. Этот сотрудник выдвинул
против своей лаборатории множество обвинений, в том числе указал на сотни случаев,
когда, по его мнению, имели место загрязнения проб в подразделении, занимающемся
определением взрывчатых веществ. Этот сотрудник даже свидетельствовал в суде в поль-
зу защиты! В результате, правда, Департамент юстиции не нашел доказательств загрязне-
ния проб, результаты анализа которых были представлены в деле, и большая часть
обвинений «доброжелателя» не была принята во внимание. Но в то же время были обнару-
жены недостатки в ведении лабораторной документации, касающиеся регистрации резу-
льтатов анализов, представления отчетов, а также общих вопросов делопроизводства.
Департамент юстиции признал, что руководство лаборатории не смогло обеспечить испо-
льзование официально утвержденных процедур и форм отчетности. Результатом этого
расследования явилось около сорока предписаний общего характера, направленных на ис-
правление недостатков и улучшение практики работы лаборатории, включая требование
обязательной аккредитации в Американском обществе директоров криминалистических
лабораторий Коллегии по аккредитации лабораторий (ASCLD/LAB). Вот некоторые из
этих предписаний.
• Каждый сотрудник, выполняющий анализ вещественного доказательства, должен со-
ставить и подписать отдельный отчет.
• Каждая папка, заведенная по конкретному делу, должна содержать все текстовые доку-
менты, распечатки, графические и прочие данные, которые могут потребоваться для
принятия решения.
• Лаборатория должна разработать специальную систему хранения и поиска данных.
• Следует обновить письменные инструкции, касающиеся работы с вещественными до-
казательствами и мер по предотвращению их загрязнения.
Конечно, многие из этих требований обусловлены спецификой работы лаборатории ФБР и
вряд ли могут относиться к большинству других лабораторий. Тем не менее эта история
иллюстрирует, насколько важным может быть соблюдение норм «хорошей лабораторной
практики». Если бы в лаборатории ФБР строго придерживались этих норм, ее сотрудники,
вероятно, не имели бы столько неприятностей в ходе описанного расследования.
188
«ХОРОШАЯ ЛАБОРАТОРНАЯ ПРАКТИКА»: ОБЕСПЕЧЕНИЕ КАЧЕСТВА РЕЗУЛЬТАТОВ АНАЛИЗА
анализа не сводится лишь к получению пробы и проведению с ней необходимых
процедур. Ваши действия должны быть правильным образом задокументирова-
ны. В противном случае они могут оказаться пустой тратой усилий, времени и
средств.
4.1. Что такое «хорошая лабораторная практика»?
Точное определение понятия «хорошая лабораторная практика» зависит от того,
кто и с какой целью дает это определение. В самом широком смысле слова GLP
включает в себя вопросы, связанные с общей организацией работы лаборатории,
управлением, персоналом, возможностями лаборатории, ее оборудованием, вы-
полняемыми операциями, поверкой методик, обеспечением качества работы и
ведением документации. Цель всех этих мероприятий — удостоверить, что каж-
дый этап анализа выполняется правильно. Конкретные аспекты достижения
этой цели могут быть различными для разных лабораторий.
Нормы «хорошей лабораторной практики» разработаны всемирными орга-
низациями, такими как Организация по экономическому сотрудничеству и раз-
витию (ОЭСР, OECD) или Международная организация по стандартизации
(ISO). Национальные правительственные организации приняли эти нормы для
своих нужд в качестве правил, которым должны следовать все лаборатории, вы-
полняющие анализы законодательно регламентированных веществ. Примерами
могут служить фармацевтические рецептуры, пищевые продукты и загрязните-
ли окружающей среды.
GLP обеспечивает правильность представляемых результатов. GLP
можно определить как «свод правил, рабочих прописей и практических реко-
мендаций, которые устанавливает данная организация и которые являются
обязательными для обеспечения качества и правильности результатов анализов,
выполняемых в лаборатории» [М. Valcarcel, Principles of Analytical Chemistry.
Berlin: Springer, 2000. P. 323]. Нормы GLP всегда содержат два раздела: стандар-
тные рабочие процедуры (англ. Standard Operating Procedures, SOP) и блок обес-
печения качества (англ. Quality Assurance Unit, QAU). Раздел стандартные
рабочие процедуры содержит подробное описание всех видов деятельности,
выполняемых в лаборатории. Примерами могут служить процедуры доставки
проб, пробоподготовки, выполнения анализов, поддержки работы оборудова-
ния, хранения документации и тому подобное. В лаборатории должны иметься
прописи стандартных рабочих процедур для каждой используемой методики.
Подробные описания методик анализа различных проб доводятся до каждого
аналитика и лаборанта. Все сотрудники обязаны следовать этим предписаниям.
Как правило, в таких прописях методики описываются более подробно, чем в на-
учных публикациях, посвященных разработке методики. Высококвалифициро-
ванные химики-аналитики могут и не нуждаться в столь подробных указаниях,
но уровень подготовки и опыт работы персонала могут быть разными в разных
лабораториях.
4.2. ПОВЕРКА МЕТОДИКИ АНАЛИЗА
189
Содержание блока обеспечения качества, как правило, лишено специфи-
ки, присущей данной лаборатории. Оно составляется в соответствии с интереса-
ми руководства организации, к которой лаборатория принадлежит. Этот блок
призван гарантировать внедрение мер обеспечения качества и их непрерывное
использование. В частности, эти меры включают периодический аудит лабора-
тории. Правила, составляющие блок обеспечения качества, призваны гарантиро-
вать, что лаборатория придерживается «хорошей лабораторной практики».
Каждый, кто работает в лаборатории, обязан выполнять эти правила.
4.2. Поверка методики анализа
Поверка (валидация) методики — это процесс, призванный документально до-
казать, что результаты, получаемые при помощи методики, приемлемы с точки
зрения цели их использования.
В целом поверка методики всегда охватывает два аспекта:
• суть проблемы и требования к данным;
• суть методики и ее возможности.
Сначала уясните себе суть задачи и необходимые требования, а затем выбери-
те методику, отвечающую этим требованиям. Как мы уже отмечали в гл. 1, ве-
сьма желательно, чтобы аналитик был вовлечен в процесс постановки задачи,
чтобы гарантировать правильную формулировку требований. Если требования,
предъявляемые к результатам анализа, нечетко сформулированы или нереали-
стичны, стоимость анализа может оказаться неоправданно высокой из-за исполь-
зования методики более точной, чем это необходимо. Если же методика менее
точна, чем требуется, результаты могут оказаться неадекватны поставленной за-
даче. Наконец, если точностные характеристики методики неизвестны, то неиз-
вестна и ценность полученных результатов. В ходе разработки и поверки любой
методики на первом этапе устанавливается минимум требований, представляю-
щихся разумными как с точки зрения общих возможностей самой методики, так
и предполагаемой сферы ее применения. Насколько правильной и воспроизво-
димой должна быть методика? Какие примерно концентрации предполагается
определять с ее помощью?
Иерархия методологических понятий
В гл. 1 мы описали в общих чертах, из чего складывается процесс химического
анализа. Иерархию методологических понятий, связанных с химическим анали-
зом (табл. 4.1), можно вкратце представить следующим образом:
Метод методика пропись инструкция
190
«ХОРОШАЯ ЛАБОРАТОРНАЯ ПРАКТИКА»: ОБЕСПЕЧЕНИЕ КАЧЕСТВА РЕЗУЛЬТАТОВ АНАЛИЗА
Таблица 4.1
Иерархия методологических понятий химического анализа*
Понятие Определение Пример
Метод Физический принцип, используемый для получения информации о составе вещества Спектрофотометрия
Методика Конкретное применение метода для решения определенных аналитических задач Методика определения диоксида серы с парарозанилином
Пропись Письменные указания, необходимые для выполнения методики Стандартная методика испытаний Веста-Гаеке для определения диоксида серы в атмосферном воздухе ASTM D2914
Инструкция Последовательность однозначных указаний, которым без исключений необходимо следовать для того, чтобы результаты анализа были приняты для решения данной задачи Стандартная методика ЕРА определения диоксида серы в атмосферном воздухе с парарозанилином
* Перепечатано из [J. К. Taylor, Anal. Chem., 55 (1983) 600А]. Опубликовано American Chemical Society в 1983 г.
Именно эти стадии аналитик в ходе разработки каждой методики должен выпол-
нить. Глубина проработки методики зависит от задачи, для решения которой ме-
тодика предназначается. При выполнении всех этих этапов методика будет
аттестована. При этом необходимо определить все ее аналитические характери-
стики, перечисленные ниже.
Понятие метод анализа означает физический принцип, используемый для
получения информации о составе вещества. Примером может служить метод
спектрофотометрии, при помощи которого концентрацию находят по величине
светопоглощения соответствующим образом подготовленного раствора пробы.
Методика включает в себя адаптацию метода (при помощи необходимых хими-
ческих операций), чтобы от стал селективным по отношению к данному аналиту.
Пропись представляет собой письменные указания, необходимые для выпол-
нения методики. Именно здесь начинают действовать нормы GLP. При этом на-
личие прописи еще не означает, что методика является стандартной. Наконец,
инструкция — это последовательность специальных предписаний, которым не-
обходимо следовать без исключений, чтобы результаты анализа можно было ис-
пользовать для определенной цели — например, для издания нормативных
актов ЕРА или выполнения тех или иных правовых действий. Такая методика
должна быть аттестована на правильность результатов при определении данно-
го аналита в данной матрице. В этом случае методика является стандартной.
4.2. ПОВЕРКА МЕТОДИКИ АНАЛИЗА
191
Процесс аттестации методики
Вопрос о необходимости аттестации методики и о процедурах, которые для это-
го следует выполнить, решает сам аналитик. В то же время существуют хорошо
разработанные руководства и правила, которые могут помочь в решении этого
вопроса.
Правительственными и международными организациями издан ряд доку-
ментов, регламентирующих процесс аттестации методик, в особенности тех, кото-
рые будут использованы в качестве официальных. В целом аттестация включает в
себя изучение следующих характеристик:
• селективность
• линейность градуировочной
зависимости
• правильность
• воспроизводимость
• рабочий диапазон
• предел обнаружения
• нижняя граница определяемых
содержаний
• устойчивость
• чувствительность
Все это лучше всего сделать непосредственно в ходе разработки методики (если
окажется, например, что чувствительность методики недостаточна, зачем ее раз-
рабатывать дальше?). На рис. 4.1 схематически представлен общий ход процесса
аттестации методики. Его отдельные аспекты рассматриваются в следующих
разделах.
Селективность
Селективность характеризует возможность определения того или иного аналита
без помех со стороны матрицы образца (включая другие аналиты). Влияние мат-
рицы может приводить как к завышению, так и к занижению результатов. Для
оценки селективности следует сравнить аналитический сигнал аналита в при-
сутствии возможных компонентов матрицы с сигналом раствора, содержащего
только аналит. При этом решающее значение имеет выбор общей методологии
измерительного процесса. Даже методика, ранее аттестованная в целом, может
оказаться непригодной при определенном составе матрицы.
Линейность градуировочной зависимости
При аттестации методики необходимо установить, что аналитический сигнал
линейно зависит от концентрации аналита в некотором диапазоне концентра-
ций. Для этого, как правило, необходимо использовать пять стандартных раство-
192
(ХОРОШАЯ ЛАБОРАТОРНАЯ ПРАКТИКА»: ОБЕСПЕЧЕНИЕ КАЧЕСТВА РЕЗУЛЬТАТОВ АНАЛИЗА
Рис. 4.1. Общая схема процесса оценки характеристик и аттестации методики анализа [J. К. Taylor,
Anal. Chem., 55 (1983), 600А]. Опубликовано American Chemical Society в 1983 г.
ров с различными концентрациями в диапазоне от 50 до 150% от предполагаемой
концентрации аналита. В этом случае искривление градуировочной зависимо-
сти легко заметить. Сигнал каждого стандартного раствора необходимо изме-
рить по крайней мере трижды.
О близости градуировочной зависимости к прямо пропорциональной часто
судят по величинам коэффициента детерминации (г2) и свободного члена урав-
нения линейной регрессии (фонового сигнала). Обычно значение г2 > 0,998 прини-
мается достаточным для того, чтобы зависимость можно было считать линейной.
Величина свободного члена (фонового сигнала) должна быть достаточно мала
по сравнению с сигналом, соответствующим средней концентрации аналита, на-
пример, составлять не более 2% от него. Следует иметь в виду, что все эти реко-
мендации носят сугубо практический характер и еще не гарантируют линейности.
Помимо этого, градуировочную зависимость всегда необходимо исследовать
визуально. Часто отклонения от линейности (пусть небольшие) наблюдаются в
областях высоких или низких концентраций. По этой причине для построения
градуировочных графиков может оказаться предпочтительнее взвешенный ме-
тод наименьших квадратов, основанный на том, что чем меньше отклонения то-
чек от расчетной зависимости, тем больший вклад они вносят в оценку ее
параметров. Один из способов оценки линейности состоит в построении зависи-
4.2. ПОВЕРКА МЕТОДИКИ АНАЛИЗА
193
мости так называемого фактора отклика (англ, response factor, RF) от концент-
рации. Фактор отклика определяется как
Фактор отклика = (сигнал - фон)/концентрация (4.1)
Если указанная зависимость является горизонтальной прямой, то это свидетель-
ствует о линейности градуировки в соответствующем концентрационном диапа-
зоне. При хорошей линейности отношение аналитического сигнала (за вычетом
фона) к концентрации должно быть приблизительно постоянным. Допустимы
колебания фактора отклика в пределах 2-3% от значения, соответствующего
средней концентрации. Рассмотрим градуировочный график, приведенный на
рис. 3.8. Для него величина свободного члена равна 0,595. Соответствующая
зависимость фактора отклика от концентрации приведена на рис. 4.2. Угловой
коэффициент описывающей ее линии регрессии составляет -1,48 единицы RF на
1 ppm. Это соответствует изменению величины RF на 1,0 единицы в изучаемом
диапазоне концентраций (0,1-0,8 ppm), т. е. 1,8% от среднего значения RF, рав-
ного 54,4. Таким образом, градуировочный график можно считать линейным.
Если градуировочная зависимость значительно отклоняется от линейной в
диапазоне 50-150% от выбранной концентрации аналита, можно попробовать
выбрать более узкий диапазон концентраций, например, 80-120%. В этом слу-
чае степень линейности графика может оказаться выше.
Правильность
Напомним, что правильностью называется близость результата анализа к истин-
ному содержанию аналита в пробе. Пожалуй, среди всех характеристик методики
Концентрация, ppm
Рис. 4.2. Зависимость фактора отклика от концентрации для данных, приведенных на рис. 3.8.
Фактор отклика рассчитан как разность между интенсивностью флуоресценции при данной кон-
центрации (0,1, 0,2, 0,4 и 0,8 ppm) и свободным членом уравнения регрессии, деленная на соответ-
ствующее значение концентрации
194
«ХОРОШАЯ ЛАБОРАТОРНАЯ ПРАКТИКА»: ОБЕСПЕЧЕНИЕ КАЧЕСТВА РЕЗУЛЬТАТОВ АНАЛИЗА
именно правильность оценить труднее всего. При оценке правильности следует
принимать во внимание не только стадию измерения сигнала, но и отбор пробы,
и пробоподготовку. Существуют три основных способа проверки правильности
методики. В порядке возрастания их доказательной силы это:
• метод «введено-найдено»;
• сравнение с результатами другой методики, относительно которой извест-
но, что она правильная;
• анализ стандартных образцов.
Сущность метода «введено-найдено» состоит в том, что в пробу вводят из-
вестное количество аналита. Его можно добавлять как к «чистой» матрице (в ко-
торой содержание аналита пренебрежимо мало), так и к пробе, содержащей
некоторое количество аналита. В последнем случае по одной и той же методике
анализируют как пробу с добавкой, так и пробу без добавки, и полученные резу-
льтаты вычитают. Следует использовать как минимум три разных содержания
добавки — наибольшее, наименьшее и среднее в пределах выбранного диапазо-
на. Пробу каждого состава необходимо приготовить и проанализировать не ме-
нее трех раз.
Более надежным способом проверки правильности является анализ пробы
при помощи другой независимой методики. Относительно нее должно быть из-
вестно, что она дает правильные результаты применительно к матрицам соот-
ветствующего типа. Независимость методики означает, что она должна иметь
как можно меньше общих стадий с проверяемой методикой. В идеале даже про-
боподготовка должна производиться по-другому. Выбрать методику для сравнения
можно при помощи научной литературы (журналов, справочников, сборников
стандартных методик) — там могут быть приведены методики, применимые к
объектам интересующего вас типа (однако, возможно, вы не сможете использо-
вать ее для анализов из-за ее высокой стоимости, недоступности оборудования
и т. д.). Если ничего подходящего для вашего конкретного случая найти не уда-
ется, можно использовать более общую методику, о которой известно, что она
правильна. В этом случае хорошее совпадение результатов, полученных при по-
мощи проверяемой и проверяющей методики, с высокой вероятностью свидете-
льствует о том, что и та, и другая применима к образцам изучаемого типа. Если
же результаты расходятся, то сделать какие-либо определенные выводы сложно,
поскольку каждая из методик может оказаться неприменимой к исследуемым
образцам (хотя более вероятно, что неправильные результаты дает все-таки но-
вая методика).
Наилучшим способом проверки правильности является анализ стандарт-
ного образца, состав которого по возможности должен быть идентичен составу
анализируемого образца. Национальный институт стандартов и технологий
США (NIST) решает задачу обеспечения правильных и сопоставимых результа-
тов измерений, разрабатывая, аттестуя и распространяя аттестованные стан-
дартные образцы. Программа NIST охватывает более 1000 таких образцов,
применимых: (1) для фундаментальных измерений в научных исследованиях и
4.2. ПОВЕРКА МЕТОДИКИ АНАЛИЗА
195
метрологии; (2) анализах объектов окружающей среды; (3) анализах, связанных
с охраной здоровья и (4) анализах промышленных материалов и готовой продук-
ции. У NIST есть стандартные образцы для определения химического состава,
физических свойств, стандартные образцы конструкционных материалов и
многие другие (см. http://ts. mst.gov/ts/htdocs/230/232/info/index, htm). Эта ор-
ганизация является главным координирующим центром при взаимодействии с
частными, государственными и международными организациями, занимающи-
мися аналогичной деятельностью. Среди таких организаций — Американское
общество по тестированию и материалам (ASTM), Американская ассоциация
клинической химии (ААСС), Международный союз теоретической и приклад-
ной химии (ГОРАС), Международная организация по стандартизации (ISO) и
Европейский Союз (EU). Соответствующую информацию можно найти на их
сайтах.
В паспорте стандартного образца указываются содержания его компонен-
тов и их статистические характеристики (стандартные отклонения). Если резу-
льтат вашего анализа отличается от паспортного значения не более, чем на
удвоенное стандартное отклонение, то с 95%-й вероятностью расхождение в
результатах связано с чисто случайными факторами. Вы можете установить
для себя и другие допустимые пределы расхождений в зависимости от содер-
жания аналита — например, 2% или даже — при следовых содержани-
ях— 10%.
Стандартный образец, идентичный по составу вашей пробе, может отсутст-
вовать, но может иметься подобный ей. Результаты проверки правильности с ис-
пользованием таких стандартных образцов также достаточно надежны.
Чтобы результаты, полученные при анализе стандартного образца или при
сравнении с независимыми данными, были достаточно надежны, необходимо
провести не менее семи параллельных анализов (число степеней свободы дол-
жно быть не менее шести).
Воспроизводимость
Воспроизводимость методики анализа можно охарактеризовать при помощи
многократных анализов одной и той же гомогенизированной пробы. Можно
оценить воспроизводимость методики в целом, включая пробоподготовку, или
отдельных ее стадий. Воспроизводимость, охарактеризованная в максимально
строго контролируемых условиях (анализ отдельных аликвот одной и той же
гомогенизированной пробы в одной лаборатории в течение одного дня), назы-
вается сходимостью. Если требуется, можно оценить и межлабораторную вос-
производимость (например, в ходе оценки устойчивости методики, см. далее).
Оценка воспроизводимости отдельных стадий анализа может включать в
себя, например, оценку воспроизводимости ввода пробы в газовый хроматограф
(при помощи многократного ввода одного и того же анализируемого раствора).
В этих случаях каждую исследуемую операцию также необходимо повторить не
менее семи раз.
196
ХОРОШАЯ ЛАБОРАТОРНАЯ ПРАКТИКА»: ОБЕСПЕЧЕНИЕ КАЧЕСТВА РЕЗУЛЬТАТОВ АНАЛИЗА
Чувствительность
Под чувствительностью методики понимают возможность различить с ее помо-
щью две близкие концентрации аналита.*
Ее характеристикой служит наклон градуировочной зависимости, называе-
мый также коэффициентом чувствительности. В различных концентрационных
диапазонах он может быть разным. В этом случае коэффициент чувствительности
следует рассчитать отдельно в области низких, средних и высоких концентраций.
Чувствительность, а также воспроизводимость методики определяют, сколько
значащих цифр следует приводить в записи результатов анализа. Бессмысленно
записывать результат как 11,25%, если методика позволяет различать содержа-
ния лишь на уровне 0,1 абсолютного процента (или, в данном случае, 1% отн.).
Рабочий диапазон
Рабочим диапазоном методики называется область концентраций, в которой ре-
зультаты анализа обладают приемлемой правильностью и воспроизводимостью.
Как правило, он включает в себя диапазон линейности градуировочной зависимости.
Критерии «приемлемости» определяются характером задачи, для решения которой
разрабатывается методика. Разумеется, воспроизводимость результатов зависит от
концентрации. Она всегда ухудшается в области низких концентраций (рис. 4.3),
а иногда и в области высоких концентраций, например, в спектрофотометрии.
Предел обнаружения
Предел обнаружения рассчитывают в соответствии с определением этой вели-
чины, данным в гл. 3. Как правило, для этого проводят серию параллельных анали-
зов матрицы и рассчитывают среднее значение фонового сигнала и его стандартное
отклонение. Затем в матрицу вводят аналит в количестве, близком к предполага-
емому пределу обнаружения (например, в таком, чтобы сигнал превышал фоно-
Рис. 4.3
Зависимость относительного
стандартного отклонения
от концентрации
На практике под «чувствительностью» обычно понимают несколько другое, а именно, способность опре-
делять (или обнаруживать) малые содержания вещества («методика высокочувствительна» — значит, она
позволяет определять очень малые концентрации). Это можно рассматривать как частный случай приве-
денной в тексте трактовки, а именно — как способность различить нулевую и ненулевую концентра-
цию. — Прим, перев.
4.2. ПОВЕРКА МЕТОДИКИ АНАЛИЗА
197
вое значение на величину, равную 10 стандартным отклонениям фона), и снова
измеряют сигнал. Из полученных данных рассчитывают концентрацию, при кото-
рой аналитический сигнал превышает фоновое значение на величину, равную ут-
роенному стандартному отклонению фона. Это и будет пределом обнаружения.
Нижняя граница определяемых содержаний
Это наименьшая концентрация, которую можно определить с приемлемой пра-
вильностью и воспроизводимостью. В зависимости от концентрационного диапа-
зона и характера поставленной задачи «приемлемым» можно считать значение
относительного стандартного отклонения 10-20% или какое-либо иное. Если
требуемая точность не оговорена, то за нижнюю границу определяемых концен-
траций принимают концентрацию, при которой сигнал превышает фоновое зна-
чение на величину, равную 10 стандартным отклонениям сигнала фона.
Устойчивость методики
Ранее рассмотренное нами понятие «воспроизводимость» является весьма об-
щим. Применительно к разным условиям и ситуациям оно может приобретать
тот или иной конкретный смысл. Например, можно оценить воспроизводимость
в строго контролируемых условиях (выполнение анализов в одной лаборатории
на одном и том же оборудовании в течение одного дня). Как отмечено ранее, в
этом случае воспроизводимость называется сходимостью (повторяемостью).
Воспроизводимость можно охарактеризовать и применительно к более широко
варьируемым условиям, когда анализы выполняются в одной лаборатории, но в
течение нескольких дней, на разном оборудовании (например, на разных хрома-
тографических колонках), с использованием реактивов из разных источников
и т. д. Такая воспроизводимость называется внутрилабораторной воспроизво-
димостью или промежуточной прецизионностью. Исследование внутрилабора-
торной воспроизводимости позволяет выявить факторы, которые могут повлиять
на разброс результатов анализа и, следовательно, по возможности должны под-
держиваться постоянными. Существует и такое понятие, кж устойчивость мето-
дики. Оно показывает, насколько чувствительны результаты анализа к небольшим
неконтролируемым изменениям различных параметров — размера пробы, тем-
пературы, pH раствора, концентрации реактивов, времени проведения реакции и
других. При изучении устойчивости методики необходимо, в частности, оце-
нить стабильность реактивов, стандартных растворов и самих проб во времени.
Влияние каждого параметра необходимо или исследовать отдельно, или исполь-
зовать достаточно сложные статистические методы факторного планирования
эксперимента с одновременным варьированием нескольких параметров (кото-
рые мы здесь рассматривать не будем).
Наконец, межлабораторная воспроизводимость оценивается на основании
результатов анализов порций одной и той же гомогенизированной пробы в раз-
ных лабораториях (среди которых одна обычно выбирается как основная для
сравнения). Согласно современным рекомендациям, именно межлабораторную
воспроизводимость предложено называть просто воспроизводимостью (а вое-
198
ХОРОШАЯ ЛАБОРАТОРНАЯ ПРАКТИКА»: ОБЕСПЕЧЕНИЕ КАЧЕСТВА РЕЗУЛЬТАТОВ АНАЛИЗА
производимость в широком смысле слова называть прецизионностью). При изу-
чении межлабораторной воспроизводимости основной акцент часто делается не
столько на изучении разброса результатов самого по себе, сколько на оценке
расхождений между результатами, полученными в разных лабораториях (так
называемое «лабораторное смещение»). Конечно, каждая лаборатория стремит-
ся к тому, чтобы ее «смещение» (по отношению к основной лаборатории) не пре-
вышало допустимого уровня.
Прежде, чем переходить к следующему разделу, изучите еще раз рис. 4.1,
иллюстрирующий различные этапы аттестации методики и их взаимосвязь. Да-
лее мы рассмотрим проблему контроля качества результатов анализа в контек-
сте общих проблем обеспечения качества.
Изучение межлабораторной воспроизводимости в реальных условиях
Является ли вклад, вносимый межлабораторным разбросом данных, существенным? От-
личается ли межлабораторная воспроизводимость от внутрилабораторной? Ответ на оба
вопроса положительный. Кроме того, межлабораторная воспроизводимость зависит от
концентрации аналита. Уильям Хорвитц с сотрудниками оценили межлабораторную
воспроизводимость данных, полученных в течение двух десятилетий, изучив более
10000 массивов данных различных лабораторий (см. R. Н. Albert, Chemical & Enginee-
ring News, September 13, 1999, p. 2). Они вывели выражение зависимости абсолютного
межлабораторного стандартного отклонения результатов анализа 5 от концентрации С
(выраженной в виде безразмерной массовой доли; например, для содержания 1 мг/кг
С ~ 1 • 106). Из этой зависимости следует, что 5 = 0,02С°’85 (для межлабораторного от-
носительного стандартного отклонения sr (%) = 2С-0’15). Если начать отсчет с чистого
вещества (С= 1), для которого sr = 2%, то межлабораторное относительное стандартное
отклонение увеличивается вдвое при уменьшении концентрации на два порядка. Это
правило соблюдается всегда, независимо от природы аналита, матрицы, метода и даты
анализа. Оказалось, что воспроизводимость результатов анализов проб любого типа —
сельскохозяйственной продукции, геологических или фармацевтических образцов —
так, по существу, и не изменилась за полвека, несмотря на использование современных
инструментальных методов! В соответствии с приведенным выражением можно ожи-
дать, что, например, для результатов определения пестицида, содержание которого со-
ставляет порядка 1 ppm (1 мг/кг, С = 1 • 10"6), относительное стандартное отклонение
составит 16%. Проверьте это, выполнив соответствующие расчеты по любой из приве-
денных формул. Для облегчения расчетов используйте электронные таблицы Excel.
Приведенные эмпирические соотношения были подтверждены и более поздними
исследованиями (М. Thompson, Р. J. Lowthian, [J. АОАС Int., 80 (1997) 6786]). Кроме
того, в них было показано, что межлабораторный разброс данных примерно вдвое пре-
вышает внутрилабораторный. Несколько улучшить межлабораторную воспроизводи-
мость позволило лишь применение чрезвычайных мер по контролю качества в ходе
межлабораторного исследования, проведенного ЕРА (см. [./. АОАСInt., 79 (1996) 589]).
Однако в этом случае стоимость каждого анализа составила порядка 1000 долларов; кро-
ме того, резко возросло время выполнения анализов. Это пример явного несоответствия
между дополнительными затратами средств и времени и выигрышем в качестве.
4.3. ОБЕСПЕЧЕНИЕ КАЧЕСТВА РЕЗУЛЬТАТОВ АНАЛИЗА
199
4.3. Обеспечение качества результатов анализа:
действительно ли методика по-прежнему
дает правильные результаты?
После того, как методика аттестована, необходимо гарантировать, что она дает
правильные результаты в процессе применения. Для этого существует система
обеспечения качества — комплекс мер, призванных обеспечить и документа-
льно подтвердить, что характеристики методики по-прежнему отвечают необхо-
димым требованиям. Эти меры входят в упоминавшийся ранее блок обеспечения
качества. В целом этот блок включает письменную документацию, касающуюся
результатов аттестации методики, последующих мероприятий и протоколы всех
действий, совершаемых с пробами. В нем предусмотрены многочисленные меры
по контролю качества, основанные на результатах количественных измерений.
Основные меры по контролю качества рассмотрены далее.
Контрольные карты
Для каждой методики, применяемой в лаборатории, нужно непрерывно вести
карту контроля качества (см. рис. 3.6). На эту карту наносят результаты анализа
стандартных образцов с известным содержанием аналита, которые выполняют
каждый день (а еще лучше — в ходе анализа каждой новой партии образцов).
Стандартные образцы шифруют и случайным образом размещают среди проб.
Если измеренная величина выходит за заранее установленные границы (опреде-
ляемые стандартным отклонением методики), необходимо выяснить, не содер-
жит ли результат систематической погрешности, например, из-за загрязнения
реагентов или дрейфа показаний прибора. В последнем случае требуется прове-
сти повторную градуировку.
Ведение и хранение документации
Это нудная, отнимающая много времени, но крайне важная часть мер по обеспе-
чению качества. Все действия, выполняемые в лаборатории с целью обеспечения
качества, должны быть письменно задокументированы. Должна быть документаль-
но зафиксирована вся цепочка действий, выполняемых с пробой, операции гра-
дуировки и оценки характеристик приборов, выполнение стандартных рабочих
процедур, исходные результаты измерений, результаты расчетов и итоговые от-
четы. Все документы должны быть подписаны и датированы сотрудниками, ко-
торые их составляли или ответственны за их содержание.
Квалификационные испытания
Одним из способов документально удостоверить квалификацию лаборатории
является ее участие в совместных межлабораторных экспериментах. Такие экс-
перименты проводят официальные органы. Они распространяют порции одного
и того же гомогенизированного материала между несколькими лабораториями
для анализа. Затем результаты, полученные разными лабораториями, а также не-
200
«ХОРОШАЯ ЛАБОРАТОРНАЯ ПРАКТИКА»: ОБЕСПЕЧЕНИЕ КАЧЕСТВА РЕЗУЛЬТАТОВ АНАЛИЗА
определенности этих результатов сопоставляют между собой. При этом истин-
ное значение содержания определяемого компонента может быть неизвестно; в
этом случае сопоставление проводят относительно среднего из всех полученных
результатов. Однако лучше, если для анализа используют аттестованный стан-
дартный образец, состав которого известен (но не участвующим лабораториям!).
Этот вариант особенно предпочтителен, в частности, когда разные лаборатории
используют разные методы анализа.
Один из способов представления результатов межлабораторного экспери-
мента состоит в расчете так называемых величин z для каждой лаборатории. Эта
величина служит мерой отклонения результата, полученного в данной лаборато-
рии, от значения, принимаемого за истинное:
где X • — среднее из i параллельных результатов, полученных в данной лабора-
А
тории, X — значение, принимаемое за истинное (аттестованное или общее
А
среднее), s — стандартное отклонение величины X.
Пример 4.1
Представьте, что вы участвуете в межлабораторном эксперименте по определе-
нию кальция в сыворотке крови. Содержание кальция в пробе составляет
5,2 ммоль экв./дл, стандартное отклонение этой величины — 0,2 ммоль экв./дл.
Пробы разосланы в 10 лабораторий для анализа атомно-абсорбционным мето-
дом. Вы получили три параллельных результата: 5,0, 4,7 и 4,8 ммоль экв./дл.
Чему равна величина z для вашей лаборатории? Что это значение характеризует?
Решение
Средний результат, полученный в вашей лаборатории, равен 4,85 ммоль экв./дл
(стандартное отклонение 0,15 ммоль экв./дл). Величина z равна
4,85-5,2 17.
z -------— = -1,75
0,2
Это означает, что ваш результат занижен по сравнению с истинным значением
на величину, равную двум стандартным отклонениям принятого значения со-
держания кальция. Таким образом, с 95%-й вероятностью это различие обуслов-
лено систематическими, а не случайными, погрешностями.
4.5. ЭЛЕКТРОННАЯ ДОКУМЕНТАЦИЯ И ЭЛЕКТРОННЫЕ ПОДПИСИ: НОРМЫ 21 CFR, ЧАСТЬ 11
201
Рис. 4.4
Типичное распределение z-величин при
межлабораторных квалификационных
испытаниях
На рис. 4.4 представлены результаты одного из представительных межлабора-
торных квалификационных испытаний. Как правило, результаты нескольких ла-
бораторий не укладываются в допустимые границы.
4.4. Аккредитация лабораторий
Другой формой сторонней оценки квалификации лаборатории является аккре-
дитация лаборатории, выполняемая уполномоченными на то государственными
или иными службами. Как правило, аккредитация лаборатории — дело доброволь-
ное, но она может оказаться обязательной, если лаборатория планирует выпол-
нять официальные анализы. Аккредитация — это процедура, по результатам
которой уполномоченный орган официально признает лабораторию компетент-
ной в сфере выполнения тех или иных задач. Процедура аккредитации может
иметь форму инспекторской проверки (на качественном уровне) работы лаборато-
рии, чтобы удостовериться в выполнении лабораторией всех правил «хорошей ла-
бораторной практики» (правильное ведение и хранение документации, проверка
методик, проведение квалификационных испытаний и т. д.). Аккредитация может
быть проведена и по-другому—например, на основании результатов анализов пре-
доставленных стандартных образцов. Но в любом случае аккредитованную лабора-
торию будут периодически проверять, возможно, без предупреждения.
4.5. Электронная документация и электронные подписи:
нормы 21 CFR, часть 11
В лаборатории многие операции выполняются при помощи компьютеров, начи-
ная от ввода кода пробы до составления итогового отчета. Традиционный спо-
соб составления документации для проверок или иных официальных действий
состоял в распечатывании бумажных копий всех документов, которые затем
подписывали, представляли в соответствующие организации и хранили. Однако
эта процедура занимает много времени, требует места для хранения бумаг, да и
просто сводит на нет преимущества использования компьютеров. Гораздо эф-
202
«ХОРОШАЯ ЛАБОРАТОРНАЯ ПРАКТИКА»: ОБЕСПЕЧЕНИЕ КАЧЕСТВА РЕЗУЛЬТАТОВ АНАЛИЗА
фективнее было бы хранить и подписывать документы непосредственно в элект-
ронном виде. Это обеспечивало бы более быстрый доступ к ним, возможность
поиска по базам данных и одновременного изучения данных из нескольких ис-
точников, чтобы выявить в них общие закономерности или тенденции. В тече-
ние 6 лет Управление по пищевым продуктам и лекарственным средствам США
(FDA) разрабатывало процедуры, призванные обеспечить ведение безбумажной
документации в рамках существующих нормативов, известных как «хорошая
производственная практика» (англ, good manufacturing practices, GMP). Нако-
нец, в 1997 г. FDA выпустило Заключительные правила, касающиеся электронной
документации, электронных подписей и представлений электронных документов,
известные как 21-й Кодекс федеральных установлений, часть 11 (21 CFR, part 11;
см. [«Electronic Records; Electronic Signatures», Fed. Reg., 62 (1997) 1000, 13230;
64 (1999) 41442]). Их можно найти на сайте www.fda.gov/ora в разделе Compliance
References. Главной проблемой при разработке этих правил был тот факт, что
электронные документы слишком просто исказить или сфальсифицировать,
случайно либо намеренно. Заключительные правила устанавливают критерии, в
соответствии с которыми FDA может считать электронные документы имеющи-
ми такую же силу, как и бумажные, а электронную подпись — такую же силу,
что и собственноручная, а также гарантировать полноту, правильность и аутен-
тичность информации, хранимой в электронном виде.
Электронная документация
Для ведения электронной документации на компьютере устанавливается элект-
ронная система контроля за полнотой данных, операциями их сохранения, вос-
становления и архивирования, а также использованием электронных подписей.
Эта система должна быть проверена; она должна осуществлять свою работу в те-
чение всего периода работы программного обеспечения. При изменении или об-
новлении рабочих программ необходимо обеспечить совместимость прежних
данных с новой версией.
Серьезной проблемой является то, что форма баз данных, хранящихся в
электронных документах, часто является динамической, т. е. их содержимое из-
меняется всякий раз при обновлении информации. Хуже того, данные можно
изменить или уничтожить, не оставив никаких следов от первоначальной ин-
формации. Поэтому доступ к системе должны иметь только пользователи, име-
ющие на это право. Необходимы периодические проверки операционной
системы. Система контроля должна обеспечивать регистрацию даты и времени
каждого действия с данными. Если в базе данных производятся изменения, сис-
тема фиксирует, кто произвел эти изменения, когда, что представляют собой
прежние и новые данные и по какой причине данные были изменены.
Электронные подписи
Как уже отмечалось, доступ в систему должен быть лишь у лиц, имеющих на это
право. Степень безопасности доступа зависит от того, открыта или закрыта сис-
тема. Технологии электронных подписей могут включать как обычные способы
4.5. ЭЛЕКТРОННАЯ ДОКУМЕНТАЦИЯ И ЭЛЕКТРОННЫЕ ПОДПИСИ: НОРМЫ 21 CFR, ЧАСТЬ 11
203
идентификации (ввод имени пользователя и пароля), так и более изощренные
системы биометрической идентификации, основанные на определении физиче-
ских характеристик (сканирование отпечатков ладоней или пальцев, радужной
оболочки или сетчатки глаза). Системы биометрической идентификации доро-
гостоящи и используются редко, особенно если доступ к компьютеру имеют не-
сколько пользователей. Имя пользователя и пароль являются уникальными, они
не могут быть изменены пользователем. В то же время пароль периодически
изменяется самой системой.
Документ 21 CRF, часть И, разрешает использование электронной доку-
ментации и электронных подписей, но не предписывает этого. Но чем больше
компьютерных систем будет проверено и адаптировано для ведения электрон-
ной документации, чем больше производителей будет включать в свои приборы
такие компьютеры, тем все более обычным станет ведение документации в элек-
тронном виде. Вероятно, что вслед за FDA подобные стандарты введут у себя и
другие организации.
Нормы CROMERRR Агентства по защите окружающей среды
Бюро информации по вопросам окружающей среды (OEI) американского Агентст-
ва по защите окружающей среды (ЕРА) разработало систему правил CROMERRR
(Cross Media Electronic Reporting and Record-Keeping Rule) с целью устранить сущест-
вующие законодательные препятствия к ведению и хранению электронной докумен-
тации в рамках широкого спектра программ ЕРА. Этот 80-страничный документ
можно найти на сайте www.epa/gov/cdx/cromerrr rule.pdf(www.epa.dov/cdx —
головной сайт ЕРА, занимающийся распространением официальной информа-
ции). Критерии, разработанные для ведения электронной документации в рам-
ках CROMERRR, согласуются с нормами 21 CFR, часть 11.
О затратах
Поскольку даже системы обеспечения качества еще не гарантируют получения правиль-
ных результатов (см. на стр. 198 «Изучение межлабораторной воспроизводимости в ре-
альных условиях»), необходимо, по крайней мере, тщательно фиксировать показатели
правильности, выявлять возможные причины искажения результатов и столь же акку-
ратно регистрировать все действия, направленные на их устранение. Разумеется, все это
требует немалых затрат. Введение в действие системы обеспечения качества не обходит-
ся без существенных первоначальных расходов как средств, так и времени. Подсчитано,
что постоянно действующая система обеспечения качества отнимает от 20 до 30% бюд-
жета лаборатории. Поэтому важно, чтобы эта система работала должным образом, на-
столько эффективно, насколько это возможно (а это, в свою очередь, означает, что
руководитель лаборатории должен ясно понимать, что для этого требуется; можете ли
вы быть таким руководителем?) и чтобы к ней относились всерьез все сотрудники лабо-
ратории (а вот это уже точно касается вас!).
204
«ХОРОШАЯ ЛАБОРАТОРНАЯ ПРАКТИКА»: ОБЕСПЕЧЕНИЕ КАЧЕСТВА РЕЗУЛЬТАТОВ АНАЛИЗА
4.6. Некоторые официальные организации
Многие государственные, негосударственные национальные и международные
организации разработали свои собственные правила аттестации методик и «хо-
рошей лабораторной практики». В основе большинства из них лежат принципы,
являющиеся плодом совместной работы международных организаций. Некото-
рые из важнейших таких организаций перечислены ниже. Более подробные све-
дения о каждой из них можно получить на соответствующем сайте. Хотя бы
бегло просмотрите их. Вы можете получить более обширную (и намного!) ин-
формацию, переходя по ссылкам, расположенным на этих сайтах. Ведь сфера
стандартизации и правового регулирования — это целый особый мир, и вы, бе-
зусловно, ощутите его неповторимый аромат.
Международная организация по стандартизации (ISO): www.iso.ch
Международная конференция по гармонизации (ICH): www.ich.com
Организация экономического сотрудничества и развития (ОЭСР, OECD):
www.oecd.org
Управление по пищевым продуктам и лекарственным средствам США (FDA):
www. fda. gov /cder
Агентство по защите окружающей среды США (ЕРА): www.epa.gov
Бюро по принудительным мерам и согласованиям, подразделение целост-
ности лабораторных данных: http://es.epa.gov/labdata.html
Бюро по твердым отходам: www.epa.gov/osw
US-ЕРА, регион 4, подразделение по науке и поддержке экосистем:
www. ера. gov / region4/sesd
Американская ассоциация клинической химии (ААСС): www.aacc.org
Американская ассоциация химии зерна (ААСС): www.scisoc.org/aacc
Американская ассоциация нефтехимиков (AOCS): www.aocs.org
Общество обеспечения качества (SQA): www.sqa.prg
Американское общество по тестированию и материалам (ASTM): www.astm.org
Международная ассоциация официальных химиков-аналитиков (АОАС Interna-
tional): www.aoac.org
Национальный институт стандартов и технологий США (NIST): www.nist.gov
Практические упражнения по использованию процедур GLP
Практическая работа 39 позволяет попрактиковаться в аттестации методики и
контроле качества, а практическая работа 40 имитирует пример квалификацион-
ных испытаний. Эти работы предназначены для группового выполнения в ауди-
тории. Прочтите их описания, даже если их выполнение не предусмотрено
вашим учебным планом.
ВОПРОСЫ
205
Что мы узнали из этой главы?
• «Хорошая лабораторная практика» (GLP) — что это такое, как это приме-
нять— стр. 188
• Как аттестовать методику: оценка селективности, линейности, правильно-
сти, воспроизводимости, чувствительности, границ рабочего диапазона,
предела обнаружения, нижней границы определяемых концентраций,
устойчивости — стр. 191
• Обеспечение качества: контрольные карты, ведение документации, квали-
фикационные испытания — стр. 199
• Электронная документация — стр. 201
• Официальные организации, предоставляющие информацию о GLP —
стр. 204
Вопросы
«Хорошая лабораторная практика»
1 . Что такое «хорошая лабораторная практика» (GLP)?
2. Для чего применяют GLP?
3. Что такое стандартные рабочие процедуры?
4- Каковы характерные особенности блока обеспечения качества?
Аттестация методик
5- Что представляют собой два главных аспекта аттестации методики?
6- В чем состоит первый этап разработки методики?
7- В чем разница между методом, методикой, прописью и инструкцией?
8. Назовите основные особенности процесса аттестации любой методики?
9- Что такое фактор отклика?
10 - Какими способами можно добиться линейности градуировочного графика?
11 - Каковы основные способы достижения правильности методики?
12. Сколько измерений необходимо провести для получения статистически до-
стоверных данных в ходе аттестации методики?
13. В чем разница между сходимостью, внутрилабораторной и межлаборатор-
ной воспроизводимостью и устойчивостью методики?
14. В чем состоят основные требования к ведению электронной документации
и к электронным подписям?
206
«ХОРОШАЯ ЛАБОРАТОРНАЯ ПРАКТИКА»: ОБЕСПЕЧЕНИЕ КАЧЕСТВА РЕЗУЛЬТАТОВ АНАЛИЗА
Обеспечение качества
15. Что такое обеспечение качества и контроль качества?
16. В чем заключаются основные процедуры контроля качества?
17- Что такое нормированная величина z?
18- Что такое аккредитация лаборатории?
Задачи
Аттестация методик
19- Ниже приведены данные для построения градуировочной зависимости для
газо-хроматографического определения этанола в крови.
Концентрация этанола, % (масса : объем) Площадь пика, усл. ед.
О 0,0
0,020 43
0,040 80
0,080 155
0,120 253
0,160 302
0,200 425
При помощи электронных таблиц Excel постройте градуировочный график
с линейной зависимостью, полученную методом наименьших квадратов.
Рассчитайте угловой коэффициент и свободный член. Рассчитайте значе-
ния фактора отклика и тангенс угла наклона его зависимости от концентра-
ции. На сколько процентов изменяется фактор отклика относительно
среднего значения в этом концентрационном диапазоне?
20- Подтвердите, что при определении пестицида в концентрации 1 ppm отно-
сительное стандартное отклонение, характеризующее межлабораторную
воспроизводимость, составляет 16%. Расчеты по каждой из формул, приве-
денных в разд. «Изучение межлабораторной воспроизводимости в реальных
условиях», проведите с использованием Excel.
Обеспечение качества
21- Предположим, что вы участвуете в межлабораторном эксперименте по
определению содержания свинца в листьях. Всем участвующим лаборато-
риям предоставлен гомогенизированный стандартный образец опавших
листьев с паспортным содержанием свинца 10,3 ± 0,5 ppm. Для анализа вы
используете кислотное разложение с последующим атомно-абсорбцион-
ным определением. По вашим данным, полученным для семи параллель-
ных аликвот, содержание свинца составляет 9,8 ± 0,3 ppm. Чему равна
величина z для вашей лаборатории?
РЕКОМЕНДУЕМАЯ литература
207
22. Посетите сайты по меньшей мере трех правительственных организаций
или профессиональных сообществ, перечисленных в разд. 4.6, и откройте
на них страницы, посвященные аттестации методик. Запишите, что общего
и чем отличаются документы, представленные на этих страницах.
Рекомендуемая литература
Интернет-сайты
1. www.labcompliance.com/index.htm — содержит обширную информацию,
касающуюся согласования лабораторных нормативов. Посетите страницу
по ссылке Regulations, на которой приведен список определений испо-
льзуемых терминов. Отличный учебник по GLP.
2. http://21 cfrpart 1 l.com — коммерческий сайт, посвященный вопросам согла-
сования лабораторных нормативов. Полезные ссылки по этой теме.
3. www/PDA.org — on-line конференция по вопросам, касающимся аттестации
компьютеров и нормативов 21 CFR, часть 11.
4. www.iscric.nl/GLP.htm — международный информационно-справочный
центр по вопросам почвоведения. Содержит руководство по управлению
качеством в лабораториях, исследующих почву и растения.
5. www.waters.com — сайт компании Waters. В разд. Applications есть
ссылка на страницу Introduction to Validation (введение в атте-
стацию методик), где приведен хороший обзор руководств по аттестации
методик Фармакопеи Соединенных Штатов (USP) и Международной кон-
ференции по гармонизации (ICH).
«Хорошая лабораторная практика»
6. J. М. Miller and J. В. Crowther, eds., Analytical Chemistry in a GMP Environ-
ment: A Practical Guide. New York: Wiley, 2000.
7. J. Kenkel, A Primer on Quality in the Analytical Laboratory. Boca Raton, FL:
CRC Press, 2000.
8. W. Gamer, M. S. Barge, and P. Ussary, eds., Good Laboratory Practice Stan-
dards. Washington, DC: American Chemical Society (Oxford), 1992.
Обеспечение и контроль качества
9- Н. Marchandise, «Quality and Accuracy in Analytical Chemistry», Fresenius' J.
Anal. Chem., 345 (1993) 82.
10- H. Y Aboul-Enein, R-I. Stefan, and G-E. Baiulescu, Quality and Reliability in
Analytical Chemistry. Boca Raton, FL: CRC Press, 2000.
11 - F. E. Prichard, Quality in the Analytical Chemistry Laboratory. New York: Wi-
ley, 1999.
208
«ХОРОШАЯ ЛАБОРАТОРНАЯ ПРАКТИКА»: ОБЕСПЕЧЕНИЕ КАЧЕСТВА РЕЗУЛЬТАТОВ АНАЛИЗА
12 - М. Sargent and G. MacKay, eds., Guidelines for Achieving Quality in Trace Ana-
lysis. Cambridge, UK: Royal Society of Chemists, 1995. (Available from the
American Chemical Society). Информация от Американского химического
общества
13 - J. М. Green, «А Practical Guide to Analytical Method Validation», Anal. Chem.,
68(1996) 305A.
14- M. Swartz and I. S. Krull, Analytical Method Development and Validation. New
York: Marcel Dekker, 1997.
15- L. Huber, Validation and Qualification in Analytical Laboratories. Buffalo Gro-
ve, IL: Interpharm, 1999.
16- M. Stoeppler, W. R. Wolf, and R. S. Jenks, eds., Reference Materials for Chemi-
cal Analysis: Certification, Availability and Proper Usage. New York: Wiley,
2001.
17- D. G. Rhoads, Lab Statistics Fun and Easy: A Practical Approach to Method Va-
lidation. Washington, DC: AACC (Американская ассоциация клинической
химии), 1999.
18- D. A. Sanders, Passing Your ISO 9000/QS-9000 Audit: A Step-by-Step Guide.
Washington, DC: AACC, 1996.
19- R. D. McDowall. Validation of Spectrometry Software. Part II: Roles of Valida-
tion Plan and User Requirement Specifications. Spectroscopy, 16 (7) (2001), 30,
www.spectroscopyonline.com. Рассмотрены технические аспекты и необхо-
димые составляющие части плана аттестации.
20- J. Kenkel, A Primer on Quality in the Analytical Laboratory. Boca Raton, FL:
Lewis, 1999.
Глава 5
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ -
ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
В химическом анализе приходится иметь дело с измерениями количеств твер-
дых веществ и концентраций растворов. Из этих данных рассчитывают массы
веществ. Для градуировки измерительных приборов или для титрования необхо-
димо готовить растворы известной концентрации. Массу аналита в растворе вы-
числяют как произведение концентрации раствора на его объем. Зная массу
исходных веществ, находят массу предполагаемых продуктов реакции. Для вы-
полнения всех подобных вычислений необходимо знание стехиометрии, т. е.
количественных соотношений, в которых взаимодействуют химические вещест-
ва. Из стехиометрических соотношений вычисляют и коэффициенты пересчета,
позволяющие выражать результаты анализа в тех или иных величинах.
В этой главе мы разберем такие фундаментальные понятия, как масса, коли-
чество вещества, моль, эквивалент. Мы рассмотрим также различные формы
представления результатов анализа применительно к твердым веществам и рас-
творам. Наконец, мы познакомимся с основами титриметрического метода ана-
лиза и использованием стехиометрических соотношений для расчета массы
аналита в титриметрии.
5.1. Обзор основных понятий
В основе всего количественного анализа лежит небольшое число фундаменталь-
ных понятий атомно-молекулярной теории, которых мы коснемся ниже. Без со-
мнения, вы уже знакомы с ними по курсу общей химии, но мы все же вкратце
напомним о них еще раз, поскольку они играют очень важную роль в ходе вы-
числений в аналитической химии.
Атомная, молекулярная и формульная масса*
Грамм-атомная (или просто атомная) масса простого вещества есть общая
масса определенного числа его атомов, одного и того же для любого элемента.
* В настоящее время вместо понятий «грамм-атомная», «грамм-молекулярная» и «грамм-формульная мас-
са», приведенных ниже, используется единое понятие «молярная масса», т. е. масса одного моля частиц
любой природы, в том числе воображаемых (эквивалентов). — Прим, перев.
210
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
Это число выбрано таким образом, чтобы грамм-атомная масса изотопно чис-
того простого вещества углерода-12 составила точно 12 г. Оно называется чис-
лом Авогадро и равно 6,022 • 1023 атомов на грамм-атомную массу любого
элемента.
Поскольку в природе элементы встречаются в виде смесей изотопов, испо-
льзуемые в химии атомные массы являются средними из атомных масс изотопов
элементов, рассчитываемыми с учетом природной распространенности каждого
изотопа. Поэтому ни для одного элемента атомная масса не является целым чис-
лом.* Например, у брома два природных изотопа, 79Вг с атомной массой 78,981338
и 81Вг с атомной массой 80,9162921. Если относительную распространенность
первого изотопа принять за 100,0, то для второго она составит 97,28 (т. е. 50,69 и
49,31%, соответственно). Среднее из этих атомных масс составит 79,904 — чис-
ло, которое мы используем в расчетах. В химии используются также грамм-мо-
лекулярные массы, определяемые как сумма атомных масс атомов, из которых
состоит молекула вещества. Применительно к веществам, состоящим не из отде-
льных молекул, а, например, из ионов (таких как сильные электролиты — кислоты,
основания, соли), правильнее было бы использовать термин грамм-формульная
масса. Ее обычно называют молярной массой. Именно этот термин мы и будем
использовать в дальнейшем. Молярная (грамм-формульная) масса характеризу-
ет массу (в г) одного моля вещества.
Что такое дальтон?
Для выражения масс таких объектов белковой природы, как хромосомы, рибо-
сомы, вирусы, митохондрии, понятие «молекулярная масса», строго говоря, не-
применимо. В этих случаях биологи и биохимики часто используют термин
дальтон. Масса одного атома 12С принимается равной 12 дальтонам. Таким об-
разом, 1 дальтон равен 1,661 • 10-24 г — величине, обратной числу Авогадро.
Масса одной молекулы, выраженная в дальтонах, численно равна ее молеку-
лярной массе, выраженной в граммах. Строго говоря, использовать дальтон в
качестве единицы измерения массы молекул было бы некорректно; эту едини-
цу следовало бы оставить лишь для объектов, подобных перечисленным
выше. Например, масса клетки бактерии Escherichia coli составляет 1 • 10-12 г,
т. е. 6 • 1011 дальтон.
Моль: основная единица измерения при работе
с химическими уравнениями
Химикам хорошо известно, что атомы и молекулы взаимодействуют в опреде-
ленных пропорциях. Но непосредственно подсчитывать число атомов или мо-
лекул, участвующих в реакции, было бы неудобно. Однако вместо числа
Даже для изотопно чистых простых веществ атомные массы не являются целыми числами (как это следует
из приведенного ниже примера) вследствие известного ядерно-физического явления, называемого дефек-
том масс. — Прим, перев.
5.1. ОБЗОР ОСНОВНЫХ ПОНЯТИЙ
211
реагирующих частиц можно использовать их относительные массы. Например,
для реакции
Ag+ + Cl--> AgCl
известно, что один ион серебра соединяется с одним хлорид-ионом. Известно
также, что атомная масса серебра составляет 107,870, а атомная масса хлора —
35,453. Таким образом, 107,870 единиц массы серебра взаимодействуют с 35,453
единицами массы хлора. Для упрощения расчетов в химии было введено поня-
тие моль. Один моль атомов, молекул, ионов или других частиц содержит
количество частиц, равное числу Авогадро (6,022 • 1023). Масса одного моля ча-
стиц численно равна их атомной, молекулярной либо формульной массе, выра-
женной в граммах.*
Итак, поскольку моль любого вещества содержит столько же атомов или
молекул, сколько моль любого другого вещества, то атомы реагируют в таких же
мольных соотношениях, каковы их атомные соотношения в соответствии с
уравнением реакции. Так, в приведенном выше примере один ион серебра реаги-
рует с одним хлорид-ионом. Следовательно, один моль ионов серебра (107,87 г)
реагирует с одним молем хлорид-ионов (35,453 г).
Пример 5.1
Рассчитайте массу (в граммах) одного моля CaSO4 • 7Н2О.
Решение
Рассчитаем молярную массу вещества. Это и будет масса одного моля (в г).
Са 40,08
S 32,06
НО 176,00
14 Н 14,11
262,25 г/моль
Число молей вещества рассчитывают как
Число молей = Масса (г) / Молярная масса (г/моль)
(5.1)
Под молярной массой следует понимать атомную или молекулярную массу ве-
щества. Так,
Число молей Na2SO4 = Масса (г) /124,04 (г/моль)
Число молей Ag+ = Масса (г) / 107,870 (г/моль)
Строго говоря, термин «грамм-атомная масса» следовало бы употреблять только применительно к атомам,
«грамм-формульная масса» — к ионным веществам, а «грамм-молекулярная масса» — к молекулярным
соединениям. Чтобы охватить вещества любой природы, мы будем использовать понятие «моль».
212
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
В экспериментах часто имеют дело с достаточно малыми количествами вещест-
ва, для которых удобнее использовать единицу измерения миллимоль. Для рас-
чета числа миллимолей используют формулу
Число миллимолей = Масса (мг) / Молярная масса (мг/ммоль)
(5.2)
Подобно тому, как число молей вещества рассчитывают из его массы, можно
рассчитать массу вещества (в г) из числа его молей:
Масса Na2SO4 (г) = Число молей • Молярная масса = Число молей • 142,04 г/моль
Масса Ag (г) = Число молей • Молярная масса = Число молей • 107,870 г/моль
Химики часто имеют дело с количеством вещества, выраженным в миллимолях.
В этом случае
Масса (мг) = Число миллимолей • Молярная масса (мг/ммоль) (5.3)
Обратите внимание, что единица г/моль — это то же самое, что мг/ммоль,
г/л — то же самое, что мг/мл, а моль/л — то же самое, что ммоль/мл. Т. е. чис-
ло г/моль = числу мг/ммоль = молярной массе; число г/л = числу мг/мл; число
моль/л = числу ммоль/мл = молярности.
Пример 5.2
Рассчитайте число молей Na2WO4 (вольфрамат натрия), содержащееся в 500 мг
этого вещества.
Решение
500 мг / 293,8 мг/ммоль • 0,001 моль/ммоль = 0,00170 моль
Пример 5.3
Чему равна масса (мг) 0,250 ммоль Fe2O3 (оксид железа)?
Решение
0,250 ммоль • 159,7 мг/ммоль = 39,9 мг
5.2. КАК ВЫРАЖАЮТ КОНЦЕНТРАЦИИ РАСТВОРОВ?
213
5.2. Как выражают концентрации растворов?
Концентрации растворов выражают множеством способов. Для количествен-
ных расчетов некоторые из них более предпочтительны, чем другие. Ниже мы
рассмотрим общепринятые в химии единицы измерения концентрации и более
подробно обсудим их использование для количественных расчетов в титриметрии.
Молярность — самая распространенная единица
измерения концентрации
Понятие моля используют для выражения концентрации растворов, особенно в
аналитической химии, где требуется знание сотношений объемов растворов реа-
гирующих веществ. По определению одномолярным раствором называется та-
кой раствор, в одном литре которого содержится один моль вещества. Его
можно приготовить, растворив один моль вещества в некотором объеме раство-
рителя и разбавив в мерной колбе до конечного объема, равного одному литру.
Можно также растворить меньшее или большее количество вещества, разбавив
до соответствующего значения объема — например, 0,01 моль в 10 мл. В более
общем случае молярностью раствора называется его концентрация, выражен-
ная в виде количества молей в литре или миллимолей в миллилитре. Молярность
сокращенно обозначается М; как правило, в аналитической химии под словом
«концентрация» понимается именно молярность. Одномолярный раствор ни-
трата серебра и одномолярный раствор хлорида натрия взаимодействуют в рав-
ных объемах в соотношении (1:1) в соответствии с уравнением реакции:
Ag+ + Cl- -» AgCl. В общем случае число молей вещества, содержащееся в про-
извольном объеме его раствора, рассчитывают как
Число молей = Молярность (моль/л) • Объем (л) (5.4)
Применительно к небольшим объемам, используемым в титриметрии, литр —
неудобная единица измерения. Удобнее работать с миллилитрами — единица-
ми, в которых отградуирована бюретка. В этом случае
Число миллимолей = Молярность (ммоль/мл) • Объем (мл) (5.5)
В аналитической химии часто выражают количество вещества в миллимолях.
Поэтому запомните эту формулу!
Пример 5.4
Раствор приготовлен растворением 1,26 г AgNO3 в мерной колбе объемом 250 мл
с последующим разбавлением до метки. Рассчитайте молярность полученного
раствора нитрата серебра. Сколько миллимолей AgNO3 содержится в растворе?
214
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
Решение
М = г/моль _ q Q297 моль/л (или 0,0297 ммоль/мл)
1,250 л
Отсюда
Число миллимолей = 0,0297 ммоль/мл • 250 мл = 7,42 ммоль
Всегда помните, что в числовых выражениях единицы измерения сокраща-
ются, образуя соответствующую единицу измерения конечного результата.
Так, в приведенном выше примере граммы сокращаются, остается моль/л (мо-
лярность). Выполнение вычислений вместе с единицами измерений с целью
проверки правильности единиц измерения результата называется анализом
размерностей. Тщательное проведение анализа размерностей важно для правиль-
ного проведения вычислительного процесса. Если вы хотите правильно выпол-
нять вычисления, всегда проводите анализ размерностей. Не ограничивайтесь
простым заучиванием формулы.
Пример 5.5
Сколько граммов NaCl содержится в 1 мл 0,250 М раствора?
Решение
0,250 моль/л = 0,250 ммоль/мл
0,250 ммоль/мл • 58,4 мг/ммоль • 0,001 г/мг = 0,0146 г/мл
Пример 5.6
Какую навеску Na^C^ следует взять для приготовления 500 мл 0,100 М раствора?
Решение
500 мл 0,100 ммоль/мл = 50,0 ммоль
50,0 ммоль • 142 мг/ммоль • 0,001 г/мг = 7,10 г
5.2. КАК ВЫРАЖАЮТ КОНЦЕНТРАЦИИ РАСТВОРОВ?
215
Пример 5.7
Рассчитайте концентрацию ионов калия (г/л) в растворе, полученном смешени-
ем 100 мл 0,250 М КС1 и 200 мл 0,100 М K2SO4.
Решение
ммоль К+ = ммоль КС1 + 2 • ммоль K2SO4 =
= 100 мл • 0,250 ммоль/мл + 2 • 200 мл 0,100 ммоль/мл = 65,0 ммоль в 300 мл
65,0 ммоль -39,1 мг/ммоль -0,001 г/мг -1000 мл/л о .
—-------------------------------------= 8,47 г/л
300 мл
Нормальность
Выражение концентрации в виде молярности широко используется в химии.
Тем не менее ряд химиков использует в количественном анализе выражение
концентрации посредством нормальности (N). Однонормальным называется
раствор, содержащий 1 моль эквивалентов вещества в литре. Эквивалентом на-
зывается реальная или воображаемая частица, взаимодействующая с одной «ре-
агирующей единицей» — одним протоном (в кислотно-основных реакциях) или
электроном [в окислительно-восстановительных; см. разд. 5.6, уравнения (5.40)
и (5.41)]. Таким образом, моль эквивалентов — это количество вещества, содер-
жащее число «реагирующих единиц», равное числу Авогадро. Число молей эк-
вивалентов равно числу молей вещества, умноженному на число «реагирующих
единиц», приходящихся на одну молекулу или атом, а эквивалентная масса —
это молярная масса, деленная на число «реагирующих единиц». В табл. 5.1 при-
ведены «реагирующие единицы» для различных типов реакций. Для кислот и
оснований число «реагирующих единиц» равно числу протонов (ионов водоро-
да), которое молекула кислоты отщепляет либо молекула основания присоединя-
ет. Для окислительно-восстановительных реакций оно равно числу электронов,
которое принимает окислитель либо отдает восстановитель. Например, серная
кислота H2SO4 содержит две «реагирующие единицы» — два протона. Поэтому
один моль серной кислоты содержит два моля эквивалентов.
Таблица 5.1
«Реагирующие единицы» для различных типов реакций
Тип реакции «Реагирующая единица»
Кислотно-основные Н+
Окислительно-восстановительные Электрон
216
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
Отсюда ее эквивалентная масса равна
_ 98,08 г/моль ,
Эквивалентная масса =---------—-----= 49,04 г/моль экв.
2 моль экв./ моль
Эквивалентная масса (как и число «реагирующих единиц») зависит от химиче-
ской реакции. Она может принимать разные значения — чаще всего в окислите-
льно-восстановительных реакциях, для которых возможны разные продукты.
Таким образом, нормальность раствора серной кислоты вдвое выше, чем
его молярность. Число молей эквивалентов рассчитывают как
Масса (г)
Число молей эквивалентов =-------------------—----------=
Эквивалентная масса (г/моль экв.)
= Нормальность (моль экв./л) • Объем (л)
Число г/моль экв. = Число мг/ммоль экв. = Эквивалентная масса
Число моль экв./л = Число ммоль экв./мл = Нормальность
(5.6)
Если использовать количества вещества, выраженные в миллимолях, то, как и
для молярности,
тт w Масса (мг)
Число миллимолеи эквивалентов =-------------------—-------------
Эквивалентная масса (мг/ммоль экв.)
= Нормальность (ммоль экв./мл) • объем (мл)
(5.7)
В клинической химии эквивалент часто определяют, используя величину
заряда иона вместо числа «реагирующих единиц». Так, эквивалентная масса
иона Са2+ равна половине его атомной массы, а число молей эквивалентов равно
удвоенному числу молей ионов. Такое соглашение удобно использовать при
расчете баланса зарядов (электронейтральности). Более подробно понятие «эк-
вивалент» рассматривается в разд. 5.3.
В научной литературе можно встретить понятие «нормальность» , посколь-
ку его широко употребляли ранее. Сейчас же его используют не столь часто, как
понятие «молярность». В разд. 5.6 мы обсудим, в каких случаях использование
нормальности действительно обладает преимуществами. Однако в основном мы
будем использовать понятия «моль» и «молярность», поскольку при этом не
возникнет неоднозначного понимания величины концентрации. В то же время
расчеты с использованием молярностей требуют знания стехиометрии реакций,
т. е. соотношений, в которых вещества реагируют между собой. В журнале Ana-
lytical Chemistry сейчас уже не разрешается использовать в статьях понятие
«нормальность» (в то время как в некоторых других журналах это по-прежнему
разрешено).
5.2. КАК ВЫРАЖАЮТ КОНЦЕНТРАЦИИ РАСТВОРОВ?
217
Формальность вместо молярности
В химии для обозначения концентрации растворов ионных веществ, не сущест-
вующих в молекулярной форме ни в твердом виде, ни в растворах, иногда испо-
льзуют термин формальность. Такая концентрация называется формальной и
обозначается F. При вычислениях «формальность» идентична молярности. Ино-
гда понятие «формальность» используют для обозначения общих (аналитиче-
ских) концентраций веществ в растворах, а «молярность» — для обозначения
равновесных концентраций. Во избежание путаницы мы будем во всех случаях
использовать исключительно термин «молярность», что является общеприня-
той практикой.
Моляльность — концентрация, не зависящая от температуры
Помимо молярности и нормальности, существует еще одна удобная величина
для выражения концентрации — моляльность (т). Одномоляльным называется
раствор, содержащий один моль вещества в 1000 г растворителя. Моляльные
концентрации удобно использовать в ходе физико-химических измерений кол-
лигативных свойств растворов (понижения температуры замерзания и давления
насыщенного пара, осмотического давления и др.), поскольку эти свойства зави-
сят только от числа частиц растворенного вещества, приходящихся на один
моль растворителя. Моляльные концентрации не зависят от температуры рас-
твора, в то время как молярные и нормальные — зависят (так как с изменением
температуры изменяется объем раствора).
Как пересчитать плотность раствора в молярность?
Для многих достаточно концентрированных коммерческих растворов кислот и
щелочей в качестве концентрации указывается массовая доля растворенного ве-
щества (в процентах). Часто возникает необходимость приготовить из таких
препаратов растворы заданной приблизительной молярности. Чтобы выполнить
соответствующие расчеты, необходимо знать плотность исходного раствора.
Напомним, что плотностью называется масса единицы объема вещества при за-
данной температуре (обычно 20 °C), выражаемая, как правило, в г/мл (г/см3).
Иногда для вещества указывают не плотность, а удельный вес. Удельный
вес — это отношение массы физического тела (например, раствора), находяще-
гося при 20 °C, к массе равного объема воды, находящегося при 4 °C (иногда
20 °C). Таким образом, удельный вес — это отношение плотностей двух ве-
ществ, т. е. величина безразмерная. Поскольку плотность воды при 4 °C состав-
ляет 1,00000 г/мл, то плотность вещества равна его удельному весу, отнесенному
к воде при 4 °C. Если удельный вес относят к воде при 20 °C, то плотность равна
удельному весу, умноженному на 0,99821 (плотность воды при 20 °C равна
0,99821 г/мл).
218
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
При 4 °C:
х w Плотность раствора, г/мл (t °C)
Удельный вес =-------------------'-------- =
Плотность Н2О (4 °C)
= 1,0000 при t - 4 °C (плотность разбавленных растворов близка
к плотности воды, т.е.РН2О = 1,0000 г/мл) =
= 0,99820 при t = 20 °C (DHp = 0,99820 г/мл)
При 20 °C:
ж т „ Плотность раствора, г/ мл (t °C)
Удельный вес =---------------------------- =
Плотность Н2О (20 °C)
= 1,0000 при t = 20 °C (для разбавленных растворов
Г>нр = 0,99820г/мл) = 1,0018 при Z = 4°C(DH2O = 1,0000г/мл)
Пример 5.8
Сколько миллилитров концентрированной 94% (г/100 г раствора) серной кисло-
ты с плотностью 1,831 г/см3 необходимо взять для приготовления 1 л 0,100 М
раствора?
Решение
Концентрированная кислота содержит 0,940 г H2SO4 в одном грамме раствора, а
один миллилитр раствора весит 1,831 г. Произведение этих двух чисел равно
массе H2SO4 (г) в одном миллилитре раствора. Из этой величины можно рассчи-
таль молярность исходного раствора:
м= -ЮООмл/л =
98,1 г/моль
= 17,5 моль Н28О4/л раствора
Этот раствор необходимо разбавить так, чтобы получить 1 литр 0,100 М раство-
ра. Следовательно, необходимо взять столько миллимолей H2SO4, сколько их
5.2. КАК ВЫРАЖАЮТ КОНЦЕНТРАЦИИ РАСТВОРОВ?
219
содержится во всем объеме конечного раствора. Рассчитаем требуемый для это-
го объем исходного раствора (х):
0,1000 М • 1000 мл = 17,5 М • х мл
Отсюда х = 5,71 мл. Такой объем концентрированной серной кислоты следует
разбавить до 1000 мл.
Способы выражения концентрации через молярность и нормальность наи-
более удобны для количественного анализа. Расчеты в титриметрии с использо-
ванием этих величин будут рассмотрены далее (см. разд. 5.5 и 5.6).
Аналитические и равновесные концентрации:
это не одно и то же
Аналитическая концентрация — это общая концентрация растворенного веще-
ства, т. е. сумма концентраций всех его химических форм, присутствующих в
растворе. В растворе с известной аналитической концентрацией вещество мо-
жет полностью или частично диссоциировать, переходя в различные химические
формы. Концентрация каждой такой формы называется равновесной. Например,
слабая уксусная кислота в растворе диссоциирует на несколько процентов (сте-
пень диссоциации зависит от концентрации и увеличивается с разбавлением
раствора):
НОАс Н+ + ОАс-
В результате образуется равновесная смесь, содержащая протоны и ацетат-ионы
в определенной концентрации. Равновесные концентрации (обычно также выра-
жаемые в виде молярности) часто бывают необходимы для расчетов с использо-
ванием констант равновесий (гл. 6). Аналитическая концентрация веществах
обозначается Сх, а равновесная концентрация частицы X — [X]. Для раствора
хлорида кальция с аналитической концентрацией 1 М равновесные концентра-
ции частиц составляют: для СаС12 — ОМ, для Са2+ — 1 М и для С1_ — 2 М. Г ово-
рят, что концентрация такого раствора составляет 1 М по Са2+.
Расчет разбавления для приготовлений раствора
нужной концентрации
Часто возникает необходимость приготовить разбавленный раствор из более
концентрированного исходного раствора. Например, может потребоваться при-
готовить из концентрированной НС1 разбавленный раствор (и затем стандартизо-
вать его) для использования в титровании. Или из одного исходного стандартного
раствора может потребоваться приготовить серию более разбавленных стандарт-
ных растворов. Во всех подобных случаях количество вещества (ммоль) в порции
исходного раствора, взятой для разбавления, должно быть равно общему количе-
220
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
ству вещества во всем объеме разбавленного раствора, т. е. СисхКисх = СразбКразб
(индексы «исх» и «разб» относятся к исходному и разбавленному раствору).
Пример 5.9
Необходимо построить градуировочный график для спектрофотометрического
определения перманганат-ионов. Имеется исходный 0,100 М раствор КМпО4 и
несколько мерных колб объемом 100 мл. Какие объемы исходного раствора не-
обходимо точно отмерить и перенести в колбы, чтобы приготовить стандартные
растворы КМпО4 с концентрациями 1,00 • 10-3; 2,00 • 10-3; 5,00 • 10-3 и 10,0 • 10-3 М?
Решение
100 мл 1,00 • 10-3 М раствора КМпО4 содержат
100 мл • 1,00 • 10-3 ммоль/мл = 0,100 ммоль КМпО4
Объем х исходного раствора, содержащий такое количество КМпО4, найдем из
следующего соотношения:
0,100 ммоль/мл • х мл = 0,100 ммоль
х = 1,00 мл
Аналогично, для приготовления остальных растворов потребуется взять 2,00;
5,00 и 10,0 мл исходного раствора и разбавить до 100 мл.
Пример 5.10
Предположим, что вы определяете содержание марганца в руде. Для этого вы
растворяете навеску руды, окисляете марганец до перманганат-ионов и опреде-
ляете их спектрофотометрическим методом. Содержание марганца в руде со-
ставляет около 5%. Навеску массой около 5 г растворяют и разбавляют до 100 мл,
а затем окисляют. Во сколько раз надо разбавить раствор перед измерением, что-
бы его концентрация оказалась в пределах градуировочного графика, описанно-
го в примере 5.9: например, составила около 3 • 10 3 М перманганат-ионов?
Решение
Раствор пробы содержит 0,05 • 5 г = 0,25 г Мп. Это соответствует мольной кон-
центрации марганца, равной 0,25 г/(55 г/моль Мп)/0,1 л = 4,5 • 10-2 М МпО4. Что-
бы получить раствор с концентрацией З Ю-3 М, его необходимо разбавить в
4,5 • 10~2/3 • 10-3 = 15 раз. Если разбавление проводить в мерной колбе объемом
100 мл, то для этого надо взять объем раствора х, равный
4,5 • 10-2 М • х мл = 3 • 10-3 М • 100 мл; отсюда х = 6,7 мл
5.2. КАК ВЫРАЖАЮТ КОНЦЕНТРАЦИИ РАСТВОРОВ?
221
Поскольку объем нужно отмерить точно, то удобно воспользоваться пипеткой
объемом 10 мл. В этом случае концентрация перманганат-ионов в фотометриру-
емом растворе составит около 4,5 • 10-3 М.
Еще расчеты при разбавлениях
Запомните: количество вещества (ммоль) до и после разбавления должно быть
одинаковым! Соотношение концентрация (М) • объем (мл) = количество вещества
(ммоль) можно использовать для расчета разбавления, требуемого приготовле-
ния раствора известной концентрации из более концентрированного. Например,
если мы хотим приготовить 500 мл 0,100 М раствора путем разбавления более
концентрированного, то мы можем рассчитать количество миллимолей вещест-
ва, необходимое для этого. Из полученного значения найдем объем концентри-
рованного раствора, который следует разбавить до 500 мл.
Пример 5.11
Требуется приготовить 500 мл 0,100 М раствора К2Сг2О7 из 0,250 М раствора.
Какой объем этого раствора необходимо разбавить до 500 мл?
Решение
Мразб ^разб— МИсх ^исх
0,100 ммоль/мл • 500 мл = 0,250 ммоль/мл • Кисх
Кисх = 200 мл
Пример 5.12
Какой объем 0,40 М раствора Ва(ОН)2 следует добавить к 50 мл 0,30 М NaOH,
чтобы получить раствор с концентрацией 0,50 М по ОН-?
Решение
Пусть х — объем раствора Ва(ОН)2 (мл). Конечный объем раствора составит
(50 + х) мл. Количество миллимолей ОН- (п) составит
ПОН- = nNaOH + 2иВа(ОН)2
222 СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
Отсюда
0,50 М • (50 + х) (мл) = 0,30 М (NaOH) • 50 (мл) + 2 • 0,40 М (Ва(ОН)2) • х (мл)
х = 33 мл (Ва(ОН)2)
Часто аналитику требуется провести серию последовательных разбавлений
раствора пробы или стандартного раствора. В этом случае расчеты выполняются
точно так же, однако здесь необходимо проконтролировать всю цепочку вычис-
лений.
Пример 5.13
Вы определяете железо спектрофотометрическим методом по реакции образо-
вания оранжевого продукта реакции Fe2+ с 1,10-фенантролином. Вам необхо-
димо построить градуировочный график, а для этого — приготовить серию
стандартных растворов для сравнения интенсивности окраски. Из сульфата же-
леза(П)-аммония приготовлен исходный стандартный раствор с концентрацией
железа 1,000 • 10-3 М. Рабочие стандартные растворы А и В приготовлены раз-
бавлением, соответственно, 2,000 и 1,000 мл исходного раствора в мерных кол-
бах объемом 100 мл. Рабочие стандартные растворы С, D и Е приготовлены
разбавлением, соответственно, 20,00, 10,00 и 5,000 мл рабочего раствора А в
мерных колбах объемом 100 мл. Каковы концентрации приготовленных рабо-
чих растворов?
Решение
Раствор А. Мисх Кисх — Мд Рд
(1,000 • 10-3 М) • (2,000 мл) = Мд • 100,0 мл
МА = 2,000 • 10-5 М
Раствор В: (1,000 • 10~3 М) • (1,000 мл) = Мв • 100,0 мл
Мв= 1,000- 10-5М
Раствор С: МА - КА = Мс • Кс
(2,000 • 10-5 М) • (20,00 мл) = Мс • 100,0 мл
Мс = 4,000 • 10-* М
Раствор D: (2,000 • 10~5 М) • (10,00 мл) = MD • 100,0 мл
MD = 2,000 • 10-* М
Раствор Е: (2,000 • 10~5 М) • (5,000 мл) = МЕ • 100,0 мл
МЕ= 1,000- 10-* м
Описанные выше расчеты применимы к химическим реакциям любых типов,
включая реакции нейтрализации, окислительно-восстановительные, осаждения и
комплексообразования. Главное требование состоит в том, что для выполнения
5.3. ВЫРАЖЕНИЕ РЕЗУЛЬТАТОВ АНАЛИЗА
223
расчетов необходимо знать количественные соотношения, в которых взаимо-
действуют вещества в соответствии с уравнением реакции.
В химической литературе существуют различные способы описания приго-
товления растворов разбавлением. Так, для приготовления разбавленной серной
кислоты из концентрированной может быть указано: «разбавление 1 + 9», что
означает «смешать 1 объем кислоты с 9 объемами растворителя (воды)». Либо
разбавление может быть указано как 1:10, что означает «разбавить 1 объем кис-
лоты до 10-кратного». Очевидно, что в обоих случаях концентрация исходной
кислоты уменьшается приблизительно в десять раз. Однако в первом случае раз-
бавление не будет точно 10-кратным, поскольку объемы жидкостей при их сме-
шении не являются строго аддитивными. Во втором же случае разбавление
будет в точности 10-кратным; для этого можно, например, отобрать пипеткой
ровно 10 мл кислоты, перенести в мерную колбу объемом 100 мл и разбавить во-
дой до метки (колбу следует частично заполнить водой прежде, чем вносить в нее
концентрированную серную кислоту!). Разбавлять растворы, смешивая опреде-
ленные объемы исходного растора и растворителя, можно только тогда, ког-
да не требуется знать точную концентрацию разбавленного раствора.
5.3. Выражение результатов анализа
Результаты анализа можно представить множеством способов. Даже начинаю-
щий аналитик должен быть знаком с наиболее распространенными из них, а
также с соответствующими единицами измерения. Почти всегда результаты
представляют в виде концентрации или других относительных величин на
основе массы или объема — например, отношения количества аналита к массе
или объему пробы. При этом единицы измерения количества аналита могут
быть разными.
Рассмотрим сначала основные единицы массы и объема в метрической сис-
теме единиц, а также основанные на них способы выражения результатов анализа.
Основной единицей массы является грамм (г); она чаще всего используется в мак-
роанализе. Для проб малого размера, а также для малых содержаний аналита ис-
пользуют меньшие единицы — миллиграмм (мг, 10-3 г), микрограмм (мкг, 10-6 г),
нанограмм (нг, 10-9 г). Основной единицей объема является литр (л). Миллилитр
(мл, 10-3 л) чаще всего используют в титриметрии. Применяют также еще мень-
шие единицы — микролитр (мкл, 10-6 л) и нанолитр (нл, 10“9 л). Для еще более
мелких единиц используют приставки «пико» (10-12) и «фемто» (10-15).
Y = иотта = 1024 М = мега = 106 н = нано = 10-9
Z = зетта = 1021 к = кило = 103 п = ПИКО = 10-12
Е = экса = 1018 д = деци = 10"1 ф = фемто = 10-15
Р = пета= 1015 с = санти = 10-2 а = атто = 10-18
Т = тера = 1012 м = милли = 10-3 z = зепто = 10-21
Г = гига = 109 мк = микро = 10-6 у = иотто = 10~24
224
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
Твердые пробы
Расчеты результатов анализа для твердых проб основаны на величинах их масс.*
Содержание макрокомпонентов чаще всего представляют в виде массовой доли,
отношения массы аналита к массе пробы, выраженной в процентах. При этом
единицы измерения массы аналита и пробы одни и те же. Например, если в об-
разце известняка массой 1,267 г содержится 0,3684 г железа, то содержание же-
леза составляет
0,3684 г • 100% = 29,08% Fe
1,267 г
Общая формула расчета массовой доли (со) в процентах (т. е. частях на 100) вы-
глядит следующим образом:
со (% масс.) = масса аналита (г) масса пробы (г) •102 (5.8)
Принято считать, что в подобных выражениях «граммы» в числителе и знамена-
теле не сокращаются — результат в квадратных скобках представляет собой
число граммов аналита на грамм пробы. В результате умножения его на 100 мы
получаем число граммов аналита на 100 г пробы. Вместо граммов можно испо-
льзовать любые другие единицы измерения массы, если для величин в числите-
ле и знаменателе они одни и те же.
Содержания следовых компонентов обычно выражают в более мелких еди-
ницах, таких как часть на тысячу (англ, part per thousand, ppt, %o), часть на
миллион (англ, part per million, ppm) или часть на миллиард (англ, part per billi-
on, ppb). Они вычисляются аналогично частям на 100 (%):
cd (ppt масс.) = масса аналита (г) масса пробы (г) •103 (5.9)
cd (ppm масс.) = масса аналита (г) масса пробы (г) •106 (5.10)
cd (ppb масс.) = масса аналита (г) масса пробы (г) •109 (5.И)
1 ppt (часть на тысячу) = 1000 ppm = 1 000 000 ppb
1 ppm = 1000 ppb = 1 000 000 ppt (часть на триллион)
Вместо термина «масса» в просторечии часто употребляют «вес». Различие понятий «масса» и «вес» см. гл. 2
5.3. ВЫРАЖЕНИЕ РЕЗУЛЬТАТОВ АНАЛИЗА
225
В данном случае тоже можно выражать величины в числителе и в знаменателе в
любых единицах массы, но в одних и тех же. Существует единица измерения
часть на триллион (одна часть на 1012 частей), которая тоже сокращенно обо-
значается как ppt, поэтому при использовании этого обозначения надо быть вни-
мательным и четко оговорить, что вы имеете в виду. В приведенном выше
примере содержание железа в пробе составляет 29,08 частей на сто, или 290,8 ча-
стей на тысячу, или 2908 частей на десять тысяч, 29080 частей на сто тысяч,
290800 частей на миллион (т. е. 290800 г железа на 1 миллион граммов пробы,
290800 фунтов железа на 1 миллион фунтов пробы и т. д.). Аналогично, 1 ppm
соответствует 0,0001 части на сто, или 10^%. В табл. 5.2 приведены соотноше-
ния единиц ppm и ppb с другими единицами концентрации. Обратите внимание,
что ppm — это мг/кг или мкг/г, a ppb — мкг/кг или нг/г.
Пример 5.14
В образце растительных тканей массой 2,6 г в результате анализа найдено
3,6 мкг Zn. Чему равно содержание цинка в образце в ppm? В ppb?
Решение
3,6 мкг . л , . л
------= 1,4 мкг/г = 1,4 ppm
2,6 г
—--------= 1,4 -10 нг/г = 1400 ppb
2,6 г
1 ppm = 1000 ppb, 1 ppb = 10-7%.
Таблица 5.2
Соотношения между различными единицами выражения следовых концентраций
Единица Сокращенное обозначение масса: масса масса: объем объем : объем
Часть на миллион (1 ppm = 10^%) ppm мг/кг, мкг/г мг/л, мкг/мл мкл/л, нл/мл
Часть на миллиард (1 ppb = 10-7% = 10~3 ppm) ppb мкг/кг, нг/г мкг/л, нг/мл нл/л, пл/мл
Миллиграмм-процент мг% мг/100 г мг/100 мл
226
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
В клинической химии иногда предпочитают для малых концентраций вмес-
то ppm использовать единицу миллиграмм-процент (мг%) — один милли-
грамм аналита в 100 г пробы. Для случая, рассмотренного в примере 5.14, содер-
жание цинка составит (3,6 • 10~3 мг/2,6 г) • 100 мг% = 0,14 мг%.
Жидкие пробы
Для жидких проб результаты анализа тоже можно представлять в виде отношения
масс. Однако чаще их представляют в виде отношения массы к объему, по край-
ней мере, в клиническом анализе. Все вычисления при этом аналогичны описан-
ным выше: процентное содержание (по массе к объему) означает количество
граммов аналита в 100 мл пробы, миллиграмм-процентное — количество
миллиграммов аналита в 100 мл пробы. Последнюю единицу особенно часто ис-
пользуют в клиническом анализе применительно к биологическим жидкостям.
Чтобы отличить эту единицу от массовых миллиграмм-процентов, ее называют
миллиграммы на децилитр (мг/дл). Децилитр равен 0,1 л или 100 мл. Если же кон-
центрация выражена в процентах, то во избежание путаницы рекомендуется ука-
зывать, что означают эти проценты — отношение массы к массе или массы к
объему. При выражении концентрации как отношения массы к объему также
используют единицы «часть на миллион» (ppm, мг/л или мкг/мл), «часть на мил-
лиард» (ppb, мкг/л или нг/мл) и «часть на триллион» (ppt, нг/л или пг/мл). Для рас-
чета концентраций в этих единицах можно использовать следующие формулы:
со (% масса : объем) = масса аналита (г) объем пробы (мл) •102 (5.12)
со (ppm, масса : объем) = масса аналита (г) объем пробы (мл) •106 (5-13)
cd (ppb, масса : объем) = масса аналита (г) объем пробы (мл) •109 (5-14)
со (ppt, масса : объем) = масса аналита (г) объем пробы (мл) •1012 (5.15)
Обратите внимание, что концентрация, выраженная в процентах по массе к объ-
ему, не означает число фунтов растворенного вещества на 100 галлонов раство-
ра: масса растворенного вещества должна быть выражена в граммах, а объем
раствора — в миллилитрах.
5.3. ВЫРАЖЕНИЕ РЕЗУЛЬТАТОВ АНАЛИЗА
227
Пример 5.15
В пробе сыворотки крови объемом 25,0 мкл найдено 26,7 мкг глюкозы. Рассчи-
тайте концентрацию глюкозы в ppm и мг/дл.
Решение
25,0 мкл —1 МЛ = 2,50 • 10-2 мл
1000 мкл
26,7 мкг (грамм) —— = 2,67 • 10-5 г
105 мкг
2,67 10~5 г глюкозы 1А6 / 1 ап m3 /
ppm = — ---—--------------10 мкг/г = 1,07 10 мкг/мл
2,50 • 10-2 мл сыворотки
или
26,7 мкг глюкозы . 1Пз ,
-------------------= 1,07-10 мкг/мл = ppm
0,0250 мл сыворотки
, 26,7 мг глюкозы • 10-3 мг/мкг л . А_ ,
мг/дл =--------------------------100 мл/дл =107 мг/дл
0,025 мл сыворотки
Обратите внимание на соотношение значений концентраций, выраженных в
ppm (масса : объем) и мг/дл.
Сколько молей в литре соответствует 1 ppm? Это зависит от молярной массы
вещества. Ориентировочное соотношение между концентрациями, выраженны-
ми в частях на миллион (или миллиард) и в молях в литре, можно получить, приняв
молярную массу аналита равной 100. В этом случае, поскольку 1 ppm = 1 • 10-3 г/л,
то молярная концентрация составит (1 • 10-3г/л)/(1 • 102 г/моль) =1 • 10-5 моль/л.
Аналогично, 1 ppb ориентировочно соответствует 1 • 10—8 моль/л. Заметьте, что
последняя величина даже меньше концентрации ионов водорода в чистой воде
(1 • 10-7М)! Разумеется, приведенные соотношения — ориентировочные, зави-
сящие от молярной массы вещества. Например, молярные концентрации рас-
творов цинка и меди с массовыми концентрациями 1 ppm различаются.
Аналогично, растворы различных веществ одинаковых молярных концентраций
будут иметь различные массовые концентрации (за исключением случаев, когда
их молярные массы равны). В основе величины молярной концентрации лежит
число частиц в единице объема, а в основе массовой — масса растворенного ве-
щества в единице объема.
Выполним некоторые расчеты с использованием реальных молярных
масс. Пусть массовая концентрация раствора бензола составляет 2,5 ppm. Мо-
лярная масса бензола (С6Н6) равна 78,1. Молярная концентрация составит
228
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
(2,5 ’ 10-3 г/л)/(78 г/моль) = 3,8 • 10-5 моль/л. Далее, пусть имеется раствор с кон-
центрацией свинца 5,8 • 10-8 М. Его массовая концентрация в г/л составит
(5,8 • 10-8 моль/л) • (207 г/моль) = 1,20 • 10-5 г/л, а в единицах ppb (мкг/л) —
(1,20- 10"5 г/л)/(1 • 106 мкг/г) = 12 ppb. Для питьевой воды, содержащей
350 пг/л тетрахлорида углерода, концентрация в ppt (нг/л) составит
(350 • 10-12 г/л) • (1 • 10-9 нг/г) = 350 • 10-3 нг/л = 0,35 ppt, а молярная концентра-
ция — (350 • 10-12 г/л)/(154 г/моль) = 2,3 • 10-12 моль/л. Для обработанной хло-
ром воды, которая может содержать следы хлорированных углеводородов, это
очень малая величина.
Основное, что следует запомнить: растворы разных веществ равных массо-
вых концентраций (в единицах масса : масса или масса : объем) в общем случае
содержат разное число молекул или других реагирующих частиц, а растворы
равных молярных концентраций — одинаковое.
Пример 5.16
(а) Рассчитайте молярные концентрации 1,00 ppm растворов Li+ и РЬ2+.
(б) Чему равна масса Pb(NO3)2, необходимая для приготовления 1 л 100 ppm рас-
твора РЬ2+?
Решение
(а) Концентрации Li и РЬ составляют 1,00 ppm = 1,00 мг/л. Отсюда
__ 1,00 MrLi/л 10-3 г/мг . 1/ч-4 / т-
Мт i = ---------------— = 1,44 • 10 моль/л Li
6,94 г Li/моль
1,00 мгРЬ/л-10"3 г/мг 6 ,
Mph = —----------------— = 4,83 -10 моль/л РЬ
207 гРЬ/моль
Поскольку свинец существенно тяжелее лития, при равных значениях массы
число молей свинца меньше, чем лития, и мольная концентрация раствора свин-
ца меньше.
(б) 100 ppm Pb2+ =100 мг/л = 0,100 г/л
0,100 г РЬ . оа 1П-4
—---------= 4,83 • 10 моль РЬ
207 г/ моль
Таким образом, в растворе должно содержаться 4,83 • 10 моль Pb(NO3)2, что
соответствует массе
4,83 • 10^ моль • 283,2 г РЬ(ЬЮ3)2/моль = 0,137 г Pb(NO3)2
5.3. ВЫРАЖЕНИЕ РЕЗУЛЬТАТОВ АНАЛИЗА
229
Единицы концентраций, выраженные в форме отношений масса : масса и
масса : объем, связаны между собой через плотность раствора. Для разбавлен-
ных водных растворов, плотность которых близка к 1 г/мл, они численно почти
равны друг другу. Дла разбавленных водных растворов концентрация, отнесен-
ная к единице массы раствора (7 г), приблизительно равна концентрации, отне-
сенной к единице его объема (7 мл).
Если аналит представляет собой жидкость, растворенную в другой жидко-
сти, то результаты анализа можно представить в виде отношения объема анали-
та к объему раствора, но такой способ представления результатов используется
редко. Вычисления в этом случае проводятся так же, как описано выше, с испо-
льзованием одних и тех же единиц объема для аналита и для раствора пробы. Ре-
зультаты определения газов можно представить в виде отношения массы к
массе, массы к объему или объема к объему.
Всегда лучше в явном виде указывать, какую форму представления резуль-
татов вы имеете в виду. По умолчанию обычно предполагается, что для твердых
проб результаты представляют как отношение массы к массе, для газов — объе-
ма к объему, а для жидкостей — массы к массе (например, для концентрированных
растворов кислот и щелочей), массы к объему (для большинства разбавленных во-
дных растворов) или объема к объему (как это принято в производстве алкоголя
в США). Содержание алкоголя в спиртных напитках принято выражать в виде
отношения объемов (1 градус = 1% об.). Поскольку удельный вес этилового
спирта равен 0,8, концентрация алкоголя, выраженная в виде отношения мас-
са : объем, равна концентрации в градусах, умноженной на 0,8.
В клинической практике для выражения содержания основных электроли-
тов (ионов Na+, К+, Са2+, Mg2+, Cl“, Н2РО4 и т. д.) в биологических жидкостях час-
то используют иные величины, а не единицы массы. Наиболее распространенной
величиной является число миллимолей эквивалентов (ммоль экв.), в данном
случае равное числу миллимолей иона, умноженному на его заряд. Результаты ана-
лиза обычно выражают в виде ммоль экв./л. Эта единица удобна для представления
общего баланса ионов: взглянув на результаты анализа, представленные в виде
ммоль экв./л, врач сразу же может оценить, насколько увеличилась или умень-
шилась общая концентрация электролитов. Очевидно, что общее число милли-
молей эквивалентов катионов должно быть равно общему числу миллимолей
эквивалентов анионов: так, один моль однозарядного (+1) катиона (1 моль экв.)
и половина моля двухзарядного (-2) аниона имеют одинаковый суммарный за-
ряд. В качестве примера баланса зарядов в табл. 5.3 приведены содержания
основных ионов в концентрациях, в норме присутствующих в плазме человече-
ской крови (а также в моче). В гл. 24 обсуждаются пределы изменчивости содер-
жания некоторых химических компонентов крови человека, а также
физиологические причины такой изменчивости.
Для любого раствора суммарные концентрации катионов и анионов, выра-
женные в ммоль экв./л, должны быть равны.
230
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
Таблица 5.3
Содержание (в норме) основных ионов в плазме крови человека*
Катион Концентрация, ммоль экв/л Анион Концентрация, ммоль экв/л
Na+ 143 С1- 104
К+ 4,5 НСО3- 29
Са2+ 5 Белок 16
Mg2+ 2,5 Н2ро; 2
SC>2- 1
Органические кислоты 3
Всего 155 Всего 155
* Воспроизведено из книги Joseph S. Annino. Clinical Chemistry, 3rd ed., Boston: Little, Brown, 1964
Число миллимолей эквивалентов вещества по его массе (в мг) рассчитыва-
ется по следующей формуле (аналогично расчету числа миллимолей):
тт Масса (мг)
Число ммоль экв =-------------------—------------=
Эквивалентная масса (мг/ммоль экв.)
_ Масса (мг)
Молярная масса (мг/ммоль)/и (ммоль экв./ммоль)
где п — заряд иона.
Эквивалентная масса Na+ равна 23,0 (мг/ммоль)/1 (ммоль экв./ммоль) =
= 23,0 мг/ммоль экв.
Эквивалентная масса Са2+ равна 40,1 (мг/ммоль)/2 (ммоль экв./ммоль) =
= 20,0 мг/ммоль экв.
Пример 5.17
Концентрация иона цинка в сыворотке крови составляет около 1 ppm. Выразите
ее в ммоль экв./л.
Решение
1 ppm = 1 мкг/мл = 1 мг/л
5.3. ВЫРАЖЕНИЕ РЕЗУЛЬТАТОВ АНАЛИЗА
231
Эквивалентная масса Zn2+ равна 65,4 (мг/ммоль) / 2 (ммоль экв./ммоль) =
= 32,7 мг/ммоль экв. Отсюда
1 (MrZn/л) ОПГ Ш? I 7
------------—------= 3,06 • 10-2 ммоль экв./л Zn
32,7 (мг/ммоль экв.)
Эту единицу измерения чаще применяют для выражения содержания главных, а
не следовых (как в данном примере) компонентов электролита.
Выражение концентраций в пересчете на различные
химические формы
До сих пор мы полагали, что аналит определяют в той самой форме, в которой
он существует в растворе (или в которой мы хотим выразить результаты). Од-
нако это не всегда так. Концентрацию можно выражать для любой химиче-
ской формы аналита. Например, при определении содержания железа в руде
мы можем определять железо в форме Fe2O3, а результаты представлять в виде
массовой доли Fe (%). Либо мы можем определять железо в форме Fe2+ (напри-
мер, титриметрическим методом), а результаты представлять в виде массовой
доли Fe2O3 (%). Это возможно при условии, что известны количественные со-
отношения между формой, содержание которой мы измеряем, и формой, в
виде которой представляем результаты. Поэтому мы можем, например, при
определении содержания кальция в воде представить результаты в виде кон-
центрации (ppm, мг/л) СаСО3 (это обычный способ представления результатов
при определении жесткости воды). Известно, что один грамм Са2+ эквивален-
тен массе СаСО3, равной отношению молярных масс СаСО3 и Са2+. Поэтому,
умножив найденную массу Са2+ на величину, равную 100,09/40,08, мы полу-
чим массу СаСО3. Конечно, это не означает, что кальций существует в раство-
ре именно в этой форме (мы можем вообще не знать, в какой форме он там
существует). Мы просто рассчитываем ту массу, которая соответствовала бы
указанной форме, и соответствующим образом представляем результат. Необ-
ходимые операции, требуемые для расчета массы аналита в желаемой форме,
будут описаны ниже.
Здесь мы заодно упомянем о различных способах выражения массы пробы
для представления результатов анализа биологических тканей и других твердых
образцов. Проба может быть взвешена в одном из трех состояний: влажном, су-
хом или озоленном. Это может относиться и к жидким пробам, хотя в этих случа-
ях состав обычно характеризуют в пересчете на единицу объема. Для нахождения
массы влажной пробы образец взвешивают непосредственно, без какой-либо об-
работки. Массу сухой пробы определяют после высушивания — при нагревании,
в эксикаторе или сублимационной сушкой. Если проба термически неустойчива,
ее нельзя высушивать при нагревании. Массу зольного остатка находят после
232
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
полного сжигания органического вещества пробы. Очевидно, такой способ вы-
ражения массы пробы можно применять только при определении неорганиче-
ских веществ.
5.4. Титриметрический анализ:
как проводить стехиометрические расчеты?
Титриметрический метод анализа — один из самых распространенных и точных
методов, особенно для определения миллимольных количеств аналита. Титри-
метрический анализ выполняется быстро, его можно автоматизировать, а также
использовать для определения достаточно малых содержаний аналита, приме-
няя чувствительные инструментальные методы индикации точки завершения
титриметрической реакции, например, посредством измерения pH. Ручное тит-
рование сейчас используют главным образом тогда, когда требуется высокая
точность, а число проб относительно невелико, например, для проверки рутин-
ных инструментальных методов. При анализе большого числа проб рациональнее
автоматическое титрование. Для автоматизации титрования можно применять, в
частности, измерение интенсивности окраски или pH раствора в сочетании с
управляющей системой, которая в необходимый момент останавливает подачу
титранта. Значение объема может быть выведено на цифровой индикатор (при-
мер см. в гл. 14). В этом разделе описаны различные виды титрования, а также
общие принципы титриметрии, включая требования, предъявляемые к стандарт-
ным растворам. Для определения содержания титруемого аналита можно испо-
льзовать общие положения о соотношении объемов растворов реагирующих
веществ, описанные в этой главе ранее. В разд. 5.5-5.7 расчеты в титриметрии
описаны более подробно. Рассматриваются, в частности, вычисления с исполь-
зованием величин молярности, нормальности и титра, а также расчеты в обрат-
ном титровании.
Требования к титриметрическим реакциям
В ходе титрования определяемое вещество (аналит), помещенный в сосуд,
взаимодействует с реагентом, добавлемым из бюретки в виде раствора извест-
ной концентрации. Такой раствор реагента называется стандартным раство-
ром, а сам реагент — титрантом. В процессе титрования измеряют объем
раствора титранта, требуемый для полного взаимодействия с аналитом. Поско-
льку концентрация раствора титранта и природа реакции, протекающей между
аналитом и титрантом, известны, то из полученных данных можно рассчитать
содержание аналита. Количество оттитрованного аналита (в молях) рассчи-
тывают из количества добавленного титранта (в молях) с учетом стехио-
метрических соотношений, в которых они взаимодействуют. Требования к
титриметрическим реакциям состоят в следующем.
5.4. ТИТРИМЕТРИЧЕСКИЙ АНАЛИЗ: КАК ПРОВОДИТЬ СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ?
233
1 . Реакция должна быть стехиометричной. Это означает, что взаимодей-
ствие аналита с титрантом должно протекать в соответствии с вполне
определенным и известным уравнением реакции. Например, в ходе
определения уксусной кислоты в уксусе титрованием раствором гидро-
ксида натрия протекает вполне определенная реакция:
НС2Н3О2 + NaOH -> NaC2H3O2 + Н2О
2. Реакция должна быть быстрой. Большинство реакций между ионами
(в частности, приведенная выше) протекает очень быстро.
3. Не должно протекать никаких побочных реакций, а титриметрическая ре-
акция должна быть специфичной. Если присутствуют мешающие веще-
ства, их необходимо удалить. В приведенном выше примере в образце не
должно присутствовать никаких других кислот.
4. В момент завершения реакции в системе должно наблюдаться резкое из-
менение какого-либо свойства. Это может быть изменение окраски раство-
ра, электрических или других физических параметров. При титровании
уксусной кислоты гидроксидом натрия после завершения реакции на-
блюдается значительное увеличение pH раствора. Изменение окраски
раствора обычно наблюдается при добавлении к раствору индикатора —
вещества, окраска которого зависит от состава и свойств раствора, на-
пример, величины pH.
5. Точка, в которой к титруемому веществу добавлено эквивалентное, т. е.
стехиометрическое, количество титранта, называется точкой эквива-
лентности. Точка, в которой титрование прекращают, считая реакцию
завершенной на основании наблюдаемого изменения какого-либо свой-
ства раствора, называется конечной точкой титрования. Объемы тит-
ранта в конечной точке титрования и точке эквивалентности должны
совпадать в пределах требуемой точности результатов, а если они разли-
чаются, то это различие должно быть воспроизводимым.
6. Реакция должна протекать количественно, т. е. равновесие реакции
должно быть сильно смещено вправо. Это условие необходимо для того,
чтобы изменение свойств системы вблизи конечной точки титрования
было достаточно резким для обеспечения требуемой точности результа-
тов. Если равновесие недостаточно смещено вправо, изменение свойства
системы (например, величины pH) будет плавным, что затруднит точ-
ную индикацию конечной точки титрования.
234
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
Стандартные растворы: они бывают разные
Стандартный раствор, приготавливаемый растворением точной навески хими-
чески чистого вещества с последующим разбавлением в мерной колбе до точного
значения объема, называется первичным стандартом. Если же чистота вещества
недостаточна, готовят его раствор с концентрацией, приблизительно соответст-
вующей желаемой, а затем его стандартизируют — устанавливают точное зна-
чение концентрации, например, титрованием раствора первичного стандарта.
Так, гидроксид натрия невозможно получить достаточно чистым для того, что-
бы непосредственно приготовить раствор первичного стандарта. Поэтому его
раствор стандартизируют, титруя раствор первичного стандарта — например,
гидрофталата калия. Гидрофталат калия является твердым веществом, которое
можно получить в химически чистом виде и точно отвесить. Раствор, концентра-
цию которого определяют посредством титрования раствора первичного стан-
дарта, называется вторичным стандартом. Из-за погрешности титрования его
концентрация известна менее точно, чем концентрация первичного стандарта.
Расчеты, используемые в ходе стандартизации, рассмотрены ниже.
Чтобы вещество могло служить первичным стандартом, оно должно отве-
чать следующим требованиям.
1. Чистота вещества должна составлять практически 100,00%. Допустимо
содержание примесей не более 0,02% при условии, что оно точно известно.
2 Вещество должно быть устойчивым при высушивании и тем более — при
комнатной температуре. Первичные стандарты перед взвешиванием все-
гда высушивают или прокаливают.*
3- Вещество должно быть легко доступным и недорогим.
4- Желательно (хотя и не обязательно), чтобы значение его эквивалентной
массы было большим. В этом случае масса навески, необходимой для
приготовления раствора, будет достаточно большой, а относительная
погрешность взвешивания — маленькой.
5- В отношении первичного стандарта, используемого для титрования, со-
храняют силу все требования к титриметрическим реакциям,, перечис-
ленные выше. В частности, равновесие его реакции с титрантом должно
быть сильно смещено вправо, чтобы изменение свойств системы в ко-
нечной точке титрования было достаточно резким.
Некоторые исключения возможны в отношении первичных стандартов, являющимхся кристаллогидратами.
5.4. ТИТРИМЕТРИЧЕСКИЙ АНАЛИЗ: КАК ПРОВОДИТЬ СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ?
235
Классификация титриметрических методов
Обычно выделяют четыре класса титриметрических методов.
1 - Кислотно-основное титрование. Многие вещества, как неорганические, так и
органические, представляют собой кислоты или основания и могут быть от-
титрованы стандартным раствором сильного основания либо сильной кисло-
ты. В этом случае конечную точку титрования легко детектировать — либо
при помощи индикаторов, либо контролируя изменения pH при помощи
pH-метра. Кислотность или основность многих органических кислот и основа-
ний можно повысить, проводя титрование в неводных растворителях. Это по-
зволяет титровать даже достаточно слабые кислоты и основания, поскольку
изменение pH вблизи конечной точки титрования становится более резким.
2. Осадительное титрование. Этот вид титрования основан на том, что тит-
рант образует с аналитом малорастворимое соединение. Примером может
служить титрование хлорид-ионов раствором нитрата серебра с образова-
нием осадка хлорида серебра. Для индикации конечной точки титрования
можно использовать индикаторы либо контролировать потенциал индика-
торного электрода.
3. Комплексометрическое титрование. В комплексометрическом титрова-
нии титрант представляет собой вещество, образующее водорастворимый
комплекс с аналитом, ионом металла. Обычно титрант является хелатооб-
разующим лигандом*. Часто используют и обратное комплексометриче-
ское титрование. Одним из наиболее распространенных хелатообразующих
реагентов в комплексометрическом титровании является этилендиамин-
тетрауксусная кислота (ЭДТА). Она взаимодействует со многими ионами;
селективного протекания реакции можно добиться, в частности, с помощью
контроля pH раствора. Для индикации конечной точки титрования исполь-
зуют индикаторы, образующие с ионом металла интенсивно окрашенные
комплексы.
4. Окислительно-восстановительное титрование. В этом случае окислитель
титруют раствором восстановителя или наоборот. Напомним, что окисли-
телем называется вещество, которое в ходе химической реакции присоеди-
няет электроны, а восстановителем — которое отдает. Чтобы реакция
протекала полностью и конечная точка титрования фиксировалась отчет-
ливо, различие в окислительно-восстановительных свойствах реагирующих
веществ должно быть достаточно велико. В идеальном случае окислитель и
восстановитель должны быть достаточно сильными. В этом виде титрова-
ния также можно использовать соответствующие индикаторы либо конт-
ролировать протекание реакции электрохимическими способами.
Перечисленные виды титрования, а также средства, используемые для ин-
дикации конечной точки, будут рассмотрены далее в соответствующих главах.
Хелатообразующим (от греч. %т|Хг| — клешня) называется лиганд, содержащий два или более атомов, спо-
собных одновременно связаться с ионом металла. ЭДТА содержит шесть таких атомов.
236
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
5.5. Расчеты в титриметрии: использование молярностей
Для расчетов в титриметрии мы будем использовать главным образом величины
молярностей. Другим способом выражения концентраций, часто используемым
в титриметрии, является нормальность, в основе которой лежат понятия экви-
валент и эквивалентная масса (вместо молярной). Величина нормальности,
или эквивалентной концентрации, зависит от конкретной реакции, которая в
этом случае должна быть явно указана. Ввиду того, что ряд преподавателей
предпочитают использовать понятие нормальности, а студенты могут встретить
это понятие в научной литературе, мы в следующем разделе рассмотрим расче-
ты с использованием понятий эквивалента и нормальности.
Различные способы выражения концентраций были рассмотрены в этой
главе ранее. Ниже приведены основные соотношения.
тт w Масса (г) Число молей = — Молярная масса (г/моль) тт w Масса (мг) ЧЬТРТТЛ АЛТЛ ТТ ТТ1ТА4ГТ TTPLT — (5.17)
T.KLUJ1V IV111J1J1111V1 VJlVKL Молярная масса (мг/ммоль)
Число молей Число миллимолей (5.18)
Молярная концентрация (М) — — Объем (л) Объем (мл)
Хорошо запомните эти соотношения. Они лежат в основе всех расчетов,
используемых в титриметрии, для приготовления растворов и их разбавления.
Не забывайте о единицах измерения!
Преобразовав эти равенства, получим выражения для расчета других вели-
чин:
Молярность (М, моль/л) • Объем (л) = Число молей
Молярность (М, ммоль/мл) • Объем (мл) = Число миллимолей
Масса (г) = Число молей • Молярная масса (г/моль)
Масса (мг) = Число миллимолей • Молярная масса (мг/ммоль)
Масса (г) = Молярность (М, моль/л) • Объем (л) • Молярная масса (г/моль)
Масса (мг) = Молярность (М, ммоль/мл) • Объем (мл) • Молярная масса (мг/ммоль)
(5-19)
(5.20)
(5.21)
В титриметрии обычно используют количества вещества порядка миллимолей и
объемы порядка миллилитров. Обратите внимание, что во всех этих выражениях
5.5. РАСЧЕТЫ В ТИТРИМЕТРИИ: ИСПОЛЬЗОВАНИЕ МОЛЯРНОСТЕЙ
237
используется одно и то же численное значение молярной массы независимо от
того, выражено оно в г/моль или мг/ммоль. В то же время при использовании
всех величин с приставкой «милли» (миллимоль, миллиграмм, миллилитр) сле-
дует быть очень внимательными. Некорректное использование этих величин по-
влечет за собой 1000-кратную ошибку в результате вычислений.
Предположим, что для титрования раствора пробы, содержащей хлорид-
ион (NaCl), требуется 25,0 мл 0,100 М раствора AgNO3. В соответствии с уравне-
нием реакции
Cl~ + Ag+^ AgCl
Ag+ и С1~ взаимодействуют в соотношении 1:1, поэтому число миллимолей С1“
равно числу миллимолей Ag+, необходимых для титрования. Массу NaCl в рас-
творе (мг) можно рассчитать следующим образом:
wNaCi= ^AgNO3 (мл)' cAgNO3 (ммоль/мл) = 25,0 мл *0,100 ммоль/мл = 2,50 ммоль
wNaCi= wNaCi (ммоль) • A/NaC1 (мг/ммоль) = 2,50 ммоль • 58,44 мг/ммоль = 146 мг
Массовую долю (%) аналита А, взаимодействующего с титрантом в мольном со-
отношении 1:1, можно рассчитать по следующей общей формуле:
Ф (%) = ^аналита (мг) 1QQO/O _ ^аналита (ММОЛЬ) • -^аналита (мг/мМОЛЬ) j QQO^ _
™ пробы (Мг) ™ пробы (Мг)
^титранта (мМОЛь/мл) • ^титранта (мл) • /^аналита (мг/мМОЛЬ)
---------------------------------------------------• 1U U /о
(5.22)
т пробы (МГ)
где т — масса, п — количество вещества, М— молярная масса, с — концентра-
ция, V— объем. Не забывайте о единицах измерения!
Эта формула представляет собой комбинацию элементарных соотношений
между различными величинами с использованием соответствующих единиц изме-
рения. С этой точки зрения ее полезно проанализировать, а не просто запомнить.
Пример 5.18
Навеску образца массой 0,4671 г, содержащего бикарбонат натрия, растворили и
оттитровали 40,72 мл 0,1067 М стандартного раствора соляной кислоты. Реак-
ция протекает по уравнению
НСО' +Н+^Н2О + СО2
Рассчитайте массовую долю (%) бикарбоната натрия в образце.
238
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
Решение
Поскольку реакция протекает в мольном соотношении 1:1, число миллимолей
бикарбоната натрия равно числу миллимолей соляной кислоты:
иНС1 (ммоль) = 0,1067 ммоль/мл • 40,72 мл = 4,3448 ммоль НС1 = wNaHco3 (мм°ль)
(мы сохраняем в промежуточных результатах запасные цифры; в этом случае
получится тот же самый ответ, что и при выполнении всех вычислений в один
прием).
wNaHco3 = 4,3448 ммоль • 84,01 мг/ммоль = 365,0! мг
365,0, MrNaHCO3 л лл/
®NaHCO3 Я 100/о - 78,14/о.
3 467,1 мг пробы
Или, объединяя все стадии расчетов в одну,
chci (М) • Кнс1 (мл) • ^NaHCO3 1 ппо/
°>NaHCO3 =--------------’100%
«пробы (мг)
0,1067 ммоль/мл -40,72 мл -84,01 мг/ммоль 1ПП0/ ПО1Л0/
—-----------------------------------------• 100/о — 7 о,14/о
467,1 мг
Некоторые соображения, полезные при расчетах
с использованием молярностей
Многие вещества реагируют между собой в мольном отношении, отличном от
1:1. Поэтому простые расчеты, приведенные в предыдущем примере, не могут
быть применены ко всем реакциям. В то же время можно записать общую рас-
четную формулу, применимую к любой реакции, если использовать отношение
стехиометрических коэффициентов.
Рассмотрим в общем виде реакцию
аА + ГГ->Р (5.23)
где А — аналит, Т — титрант, взаимодействующие в мольном отношении alt с
образованием продуктов Р. Из этого уравнения можно заключить, что
иА(ммоль) = (ммоль) • —
иА(ммоль) = ст (ммоль/мл) • Кт (мл) • —
(5.24)
(5.25)
5.5. РАСЧЕТЫ В ТИТРИМЕГРИИ: ИСПОЛЬЗОВАНИЕ МОЛЯРНОСТЕЙ
239
тА (мг) = пА (ммоль) • МА (мг/ммоль)
тА (мг) = сТ (ммоль/мл) • Кт (мл) • — * МА (мг/ммоль)
(5.26)
(5.27)
Обратите внимание, что для уравнивания числа молей аналита и титранта испо-
льзуется множитель alt. Во избежание ошибок полезно запомнить мнемониче-
ское правило: чтобы рассчитать количество аналита, количество титранта надо
умножить на отношение alt (а записываем первым, в числителе). И наоборот,
если необходимо рассчитать количество титранта по известному количеству
титруемого вещества (аналита), последнее необходимо умножить на t/a (t запи-
сываем первым). Но, разумеется, наилучшим способом избежания ошибок явля-
ется анализ размерностей.
Можно получить и общее выражение для расчета массовой доли аналита А
в образце (по результатам титрования раствора его навески стандартным раство-
ром титранта Т) аналогично тому, как было получено уравнение (5.22):
Ф (%) = ^аналита (мг) 1QQO/O _ ^аналита (ММОЛЬ) • а/1 * А/аналита (мг/мМОЛЬ) jqqo^
А т пробы (мг) т пробы (мг)
_ ^титранта (мМОЛь/мл) • Титранта (мл) • a/t • Л7аналита (мг/мМОЛЬ) qqo/
«пробы (МГ)
(5.28)
И в этом случае во избежание ошибок необходимо тщательно проводить
анализ размерностей на каждой стадии вычислений. В соответствии с уравнени-
ем реакции именно величина alt является тем множителем, который переводит
число миллимолей титранта в эквивалентное число миллимолей аналита.
Пример 5.19
Для определения содержания карбоната натрия в технической соде ее навеску
массой 0,2638 г оттитровали 0,1288 М стандартным раствором соляной кислоты.
На титрование израсходовано 38,27 мл этого раствора. Уравнение реакции име-
ет вид
СО|~ + 2Н+ -> Н2О + СО2
Рассчитайте массовую долю (%) карбоната натрия в образце.
240
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
Решение
Число миллимолей карбоната натрия равно половине числа миллимолей кисло-
ты использованной для титрования (поскольку эти вещества взаимодействуют в
соотношении 1:2, a/t = |). Отсюда
иНс1= 0,1288 ммоль/мл • 38,27 мл = 4,929 ммоль
«Na2co3 = 4,929 ммоль • | = 2,4645 ммоль
^Na2co3 = 2,4645 ммоль • 105,99 мг/ммоль Na2CO3 = 261,2j мг
О^СОз
261,2, MrNa^COo
---?-----------3- 100% = 99,02% Na,CO3
263,8 мг пробы
Или, выполнив все действия в один прием,
сНс1 (ммоль/мл) • КНС1 (мл) • | со (мг/ммоль)
®Na2CO3 =----------------------—------—------------100%
теобразца(МГ)
0,1288 ммоль/мл-38,27 мл-105,99мг/ммоль
------------- ------------2----------------100% = 99,02%
263,8 мг
Пример 5.20
Какой объем (мл) 0,25 М раствора H2SO4 понадобится для взаимодействия с
10 мл 0,25 М раствора NaOH?
Решение
Уравнение реакции имеет вид
H2SO4 + 2NaOH -> Na2SO4 + 2Н2О
Число миллимолей H2SO4, вступившей в реакцию, вдвое меньше числа милли-
молей NaOH. Поэтому
CH2SO4 * ^H2SO4 “ CNaOH ’ ^NaOH ’
Отсюда
5.5. РАСЧЕТЫ В ТИТРИМЕТРИИ: ИСПОЛЬЗОВАНИЕ МОЛЯРНОСТЕЙ
241
0,25 ммоль / мл NaOH • 10 мл NaOH • 1
И„ „„ =--------------------------------------= 5,0 мл
Н2Ь°4 0,25 ммоль /мл H2SO4
Обратите внимание, что в этом примере мы умножили количество титранта на alt.
Пример 5.21
Чистоту препарата салициловой кислоты С6Н4(ОН)СООН (в условиях титрова-
ния она отщепляет один протон) определяют титриметрическим методом. Какую
навеску салициловой кислоты следует взять, чтобы массовая доля основного ве-
щества, выраженная в процентах, была в пять раз больше, чем объем (мл) 0,0500 М
раствора NaOH, израсходованный на титрование?
Решение
Пусть объем раствора NaOH (мл) равен х. Тогда массовая доля салициловой кис-
лоты НА (%) составит со = 5х. Отсюда
100%,
^навески (мг)
ИЛИ
0,0500 ммоль/мл • х мл NaOH • 1 • 138 мг НА/ммоль 4 ,
5х =-------------------------------------------100%
^навески (^г)
Отсюда ^навески= 138 мг.
cNaOH (ммоль/мл) • KNa0H (мл) • 1 ммоль НА/ммоль NaOH • М(мг/ммоль)
Приведенные примеры можно использовать для расчетов результатов тит-
рования, описанных в гл. 8.
Расчеты результатов стандартизации и титриметрического
определения: они взаимно обратны
Если вещество для приготовления раствора титранта невозможно получить в
чистом виде (или хотя бы известной степени чистоты), готовят раствор титранта
приблизительной концентрации, а затем определяют ее точно. Эта операция на-
зывается стандартизацией. Обычно ее проводят посредством титрования точно
известного количества раствора первичного стандарта, приготовленного по на-
веске. Из объема титранта, израсходованного на титрование первичного стандар-
242
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
та, можно рассчитать концентрацию раствора титранта. При стандартизации
концентрация раствора титранта неизвестна, а количество титруемого ве-
щества (первичного стандарта) известно.
Пусть в уравнении (5.23) титруемое вещество (аналит, А) представляет со-
бой первичный стандарт. Тогда
"станлапта (ММОЛЬ) = ^станлапта (МГ) / (МГ/ММОЛЬ)
СТаНДарТа z 1ДаНДар1а v z 1ДаНДар1а v z
«титпанта (ММОЛЬ) = Сти_анта (ММОЛЬ/МЛ) • К.—.,— (мл) = Пстанлапта (ММОЛЬ) • t/d.
ТИграНТа 4 z IMlpdrild v z IHlpdriLd v z С1апДар1а z
Отсюда
^титранта (ММОЛЬ/МЛ) =
^стандарта (ММОЛЬ) • t/ а
^титранта (мл)
Или, объединив приведенные выше выражения,
/«станпапта (МГ)/^стянпапта (мг/мМОЛЬ) • t!а стип,анта (ММОЛЬ/МЛ) = стандартаV стаНДарта^ титранта у , . г титранта V / (5.29)
Пример 5.22
Раствор соляной кислоты приблизительной концентрации 0,1 М приготовлен
разбавлением концентрированной НС1 в 120 раз. Его отстандартизовали титро-
ванием навески 0,1876 г первичного стандарта — прокаленного карбоната на-
трия:
СО^ + 2Н+ -» Н2О + СО2
На титрование израсходовано 35,86 мл раствора кислоты. Рассчитайте моляр-
ную концентрацию этого раствора.
Решение
Число миллимолей соляной кислоты равно удвоенному числу миллимолей от-
титрованного карбоната натрия.
nNa2co3 = 187,6 мг / 105,99 мг/ммоль = 1,77Оо ммоль «на = снс1 (ммоль/мл) • 35,86 мл = 1,77Оо ммоль • 2
Отсюда 1,770п ммоль -2 л к сНГ| = 5 = 0,09872 М 35,86 мл
5.5. РАСЧЕТЫ В ТИТРИМЕТРИИ: ИСПОЛЬЗОВАНИЕ МОЛЯРНОСТЕЙ
243
Или, объединив расчеты в одну формулу,
_ WNa2CO3 (МГ)/ MNa2CO3 (мг/мМОЛЬ) • | _
СНС1 “ Г \ “
^НС1 (МЛ)
187,6 мг/105,99 мг/ммоль -2 n х#
=--------------------------= 0,09872 М
35,86 мл
Обратите внимание, что количество титруемого вещества (аналита) Na2CO3 ум-
ножается на величину t/a (отношение числа миллимолей титранта к числу милли-
молей аналита). Заметьте также, что, хотя все результаты измерений приведены с
четырьмя значащими цифрами, в расчетах было использовано значение моляр-
ной массы Na2CO3 с пятью значащими цифрами. Если бы это значение округлили
до четырех цифр, то именно эта величина лимитировала бы точность вычислений
(неопределенность ее значения составила бы порядка одной части на тысячу, а
неопределенность значения массы 187,6 мг — примерно вдвое меньше). Сове-
туем вообще взять за правило использовать в расчетах значения молярных
масс с одной запасной цифрой — в частности, при расчетах на калькуляторе.
Для иллюстрации расчетов в титриметрии применительно к разным типам
реакций и различной стехиометрии приведем еще несколько примеров.
Пример 5.23
Железо(П) титруют в слабокислом растворе 0,0206 М раствором перманганата
калия. Реакция протекает по уравнению
5Fe2+ + MgO; + 8Н+ -> 5Fe3+ 4- Mn2+ 4- 4Н2О
На титрование израсходовано 40,2 мл раствора титранта. Какова масса железа (мг)
в титруемом растворе?
Решение
Число миллимолей железа в 5 раз больше числа миллимолей перманганата, поэ-
тому
nFs (ммоль) = mFe (мг) / MFe (мг/ммоль) = СкМп04 (ммоль/мл) • (мл) • f
mFe (мг) = 0,0206 ммоль/мл • 40,2 мл • 5 • 55,8 мг/ммоль = 231
244
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
Подобные расчеты описаны и в гл. 14 (применительно к окислительно-вос-
становительному титрованию).
Ниже приведены примеры реакций осадительного и комплексометрическо-
го титрования и соответствующие выражения для расчета массы аналита (мг) из
количества титранта (ммоль). В последнем уравнении H2Y2- означает анион
ЭДТА.
С1~ + Ag+-> AgCl
2С1- + Pb2+ -» РЬС12
PO4~ + 3Ag+—> Ag3PO4
2CN- +Ag+-> Ag(CH)~
2CN- + 2Ag+ -> Ag[Ag(CN)2]
Ва2++ SO4“ -> BaSO4
Wcl- (мг) = CAg+(ммоль/мл) • Кдё+(мл) • 1 • Mcl- (мг/ммоль)
7WC1- (мг) = Cpb2+ (ммоль/мл) • Kpb2+ (мл) • 2 Мс1- (мг/ммоль)
/ироз_ (мг) = сAg+(ммоль/мл) • KAg+(Mn) • | • Мроз_ (мг/ммоль)
тг„_ (мг) = с. + (ммоль/мл) • К +(мл) • 2 • (мг/ммоль)
(мг) = с. + (ммоль/мл) • V. „+(мл) • 1 • Мгм_ (мг/ммоль)
TWBa2+ (мг) = CS02- (ммоль/мл) • KS02- (мл) • 1 • /ИВа2+ (мг/ммоль)
Са2+ + H2Y2" -> CaY2~ + 2Н+ тСа2+ (мг) = СэдТА (ммоль/мл) • ГЭДТА (мл) • 1 • (мг/ммоль)
Эти формулы удобно использовать при расчетах результатов осадительного и
комплексометрического титрования (гл. 8 и 11).
Пример 5.24
Алюминий определяют титрованием раствором ЭДТА по уравнению
А13+ + H2Y2- -> A1Y- + 2Н+
На титрование раствора навески пробы массой 1,00 г израсходовано 20,5 мл рас-
твора ЭДТА. Раствор ЭДТА стандартизовали титрованием 25,0 мл 0,100 М рас-
твора СаС12. На титрование израсходовано 30,0 мл раствора ЭДТА. Рассчитайте
массовую долю (%) А12О3 в образце.
Решение
Са2+ и ЭДТА взаимодействуют в мольном соотношении 1:1, поэтому
0,100 ммоль/мл СаС19 -25,0 мл СаС19 л .
------------------?-----------= 0,0833 ммоль/мл
30,0 мл ЭДТА
5.5. РАСЧЕТЫ В ТИТРИМЕТРИИ: ИСПОЛЬЗОВАНИЕ МОЛЯРНОСТЕЙ
245
Число миллимолей А13+ равно числу миллимолей ЭДТА, израсходованного на
титрование. Число миллимолей А12О3 вдвое меньше (поскольку 1А13+ соответст-
вует 1А12О3). Поэтому
‘ЭДТА ' ^ЭДТА ' | '^А1,О3
®ai2o3 =-------------------—-100% =
пробы
0,0833 ммоль/мл -20,5 мл -i -101,96 мг/ммоль
=-------------------------------------------100% = 8,71%
1000 мг
Как быть, если аналит и титрант могут реагировать
несколькими способами?
Как вам известно из курса общей химии, ряд веществ могут взаимодействовать
друг с другом с образованием различных продуктов. В этом случае коэффици-
ент перевода количества титранта в количество аналита зависит от конкретной
реакции. Например, карбонат-ион может реагировать как двухпротонное или
однопротонное основание:
СО^- + 2Н+ -> Н2О + СО2
ИЛИ
со^+н+->нсо3
В первом случае число миллимолей Na2CO3 равно | числа миллимолей од-
неосновной кислоты, во втором случае оно равно числу миллимолей кислоты.
Аналогично, фосфорную кислоту можно титровать как одноосновную
Н3РО2 + он- -> н2ро; + Н2О
или двухосновную
Н3РО4 4- 2ОН- -> НРО; + 2Н2О
Пример 5.25
В кислой среде перманганат калия взаимодействует с Н2О2 с образованием Мп2+:
5Н2О2 + 2МпО; + 6Н+ 5О2 + 2Мп2+ + 8Н2О
В нейтральной среде он взаимодействует с MnSO4 с образованием МпО2:
3Mn2+ + 2MnO; + 4ОН- -> 5О2 + 5МпО2 + 2Н2О
246
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
Рассчитайте объем (мл) 0,100 М КМпО4, который пойдет на взаимодействие с
50,0 мл 0,200 М Н2О2 и с 50,0 мл 0,200 М MnSO4.
Решение
Будьте внимательны при расчете числа миллимолей! Число миллимолей МпО4
равно двум пятым числа миллимолей Н2О2, вступившей в реакцию:
СМпО4 ’ ^МпО4 ” СН2О2 ’ ^Н2О2 ’ 5
0,200 ммоль/мл Н7О9 -50,0млН7О7 |
К, п_ = ---------------2-^--------= 40,0 мл
п 4 0,100 ммоль/мл МпО4
Число миллимолей МпО4, вступившего в реакцию с Мп2+, равно двум тре-
тям числа миллимолей Мп2+:
с -V = с .V .2
MnO; MnO; Mg2+ Mg2+ з
0,200 ммоль/мл Мп2+ -50,0 мл Мп2+ -2
К, п_ =------------------------------------= 66,7 мл
МпО4 - . ~ ~ , ------ ’
0,100 ммоль/мл МпО4
Пример 5.26
Щавелевая кислота Н2С2О4 является восстановителем, взаимодействующим с
КМпО4 следующим образом:
5Н2С2О4 + 2МпО; + 6Н+ -> 10СО2 4- 2Мп2+ 4- 8Н2О
Кроме того, щавелевую кислоту можно оттитровать раствором щелочи как
двухосновную. Сколько миллилитров 0,100 М NaOH и 0,100 М КМпО4 вступит
в реакцию с 500 мг Н2С2О4?
Решение
1) WNaOH = 2«н2с2о4
500 мг НоСоОя
0,100 ммоль/мл NaOH • х мл NaOH =---2—2 4 • 2
90,0 мг/ммоль
Отсюда х = 111 мл NaOH
2) ПКМпО4 = 5ПН2С2О4
0,100 ммоль/мл КМпО4 • х мл КМпО4 = мг_^2^"2^4 .1
90,0 мг/ ммоль 5
Отсюда х = 22,2 мл КМпО4
5.5. РАСЧЕТЫ В ТИТРИМЕТРИИ: ИСПОЛЬЗОВАНИЕ МОЛЯРНОСТЕЙ
247
Пример 5.27
Чистые Na2C2O4 (Na2A) и КНС2О4 • Н2С2О4 (КН3А2, при титровании отщепля-
ются все три протона) смешаны в таком соотношении, что на титрование одной
и той же массы смеси расходуются одинаковые объемы 0,100 М КМпО4 и
0,100 М NaOH. В каком соотношении смешаны вещества?
Решение
Предположим, что на титрование расходуется 10,0 мл раствора титранта, что со-
ответствует 1,00 ммоль NaOH или КМпО4. Щелочью титруется только
КНС2О4 • Н2С2О4 (КН3А2), поэтому
ПКН3А2 = nNaOH ’ 3
1,00 ммоль NaOH • | = 0,333 ммоль КН3А2
В то же время (см. пример 5.26) один миллимоль Na2C2O4 (Na2A) взаимодейст-
вует с | миллимолями КМпО4. Отсюда
ПКМпО4 = WNa2A * 5 + ПКН3А2 ’ 5
1,00 ммоль KMnO4 = nNa2A * | + 0,333 ммоль КН3А2 • |
Отсюда nNazA = 1,83 ммоль.
Вещества находятся в мольном отношении 1,83 ммоль Na^ / 0,333 ммоль КН3А2,
или 5,50 : 1. Массовое соотношение составляет
5,50 ммоль Na2A-134мг/ммоль
---------------------------3,38 : 1
1 ммольКН3А2 -218мг/ммоль
Если реакция протекает медленно, используйте обратное
титрование
Иногда взаимодействие аналита с титрантом происходит медленно, и конечная
точка титрования не может быть четко зафиксирована. Примером может слу-
жить анализ таблеток антацида (суспензия, содержащая гидроксиды алюминия
и магния. — Прим, перев.) путем титрования сильной кислотой, например, НС1.
В подобных случаях целесообразно использовать обратное титрование. Для
этого к пробе добавляют в небольшом избытке точно известное количество реа-
гента, который мог бы служить титрантом. После того, как аналит полностью
прореагирует (в присутствии избытка реагента скорость реакции увеличивается),
оставшееся непрореагировавшее количество реагента определяют при помощи тит-
рования стандартным раствором другого вещества. Зная исходное количество
реагента и определив его непрореагировавшее количество, можно рассчитать
количество аналита, вступившего в реакцию с реагентом:
248
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
Количество прореагировавшего реагента (ммоль) =
Исходное количество реагента (ммоль) - Оттитрованное количество реагента (ммоль)
Масса аналита (мг) = Количество прореагировавшего реагента (ммоль) х
Отношение стехиометрических коэффициентов х Молярная масса аналита (мг/ммоль)
Пример 5.28
Хром(Ш) взаимодействует с ЭДТА (H4Y) медленно, поэтому его определяют
обратным титрованием. Для анализа лекарственного препарата, содержащего
Сг(Ш), навеску массой 2,63 г растворили и добавили 5,00 мл 0,0103 М ЭДТА. По
завершении реакции непрореагировавшее количество ЭДТА оттитровали 1,32 мл
0,0122 М раствора Zn2+. Какова массовая доля (%) хлорида хрома в препарате?
Решение
Как Сг3+, так и Zn2+ взаимодействуют с ЭДТА в мольном соотношении 1:1:
Cr3+ + H4Y -> CrY" + 4Н+
Zn2+ + H4Y -» ZnY2" + 4H+
Исходное количество ЭДТА равно
0,0103 ммоль/мл ЭДТА • 5,00 мл = 0,0515 ммоль
Количество непрореагировавшего ЭДТА равно
0,0112 ммоль/мл Zn2+ • 1,32 мл = 0,0148 ммоль
Количество прореагировавшего ЭДТА равно
0,0515 ммоль (исходно) - 0,0148 ммоль (осталось) =
= 0,0367 ммоль ЭДТА = количеству ммоль Сг3+
Масса оттитрованного СгС13 равна
0,0367 ммоль СгС13 • 158,4 мг/ммоль = 5,81 мг
®СгС13
5,81 мг СгС13
2630 мг пробы
•100% = 0,221 %
Или, выполнив все вычисления в одну стадию,
_ (^ЭДТА ’ КЭДТА “ cZn2+ ’ *Zn2+ '1 ‘ МСгС13 ) _
®СгС13------------------------------------100 /о-
пробы
(0,0103 ммоль/ мл • 5,00 мл - 0,0112 ммоль/ мл • 1,32 мл ¥ 1 • 15 8,4 мг/ ммоль
---------------------------------------------------------------100%=0,221%
2630 мг
5.5. РАСЧЕТЫ В ТИТРИМЕТРИИ: ИСПОЛЬЗОВАНИЕ МОЛЯРНОСТЕЙ
249
Пример 5.29
В навеске пиролюзита массой 0,200 г определили содержание марганца следую-
щим образом.* К пробе добавили 50,0 мл 0,100 М раствора сульфата желе-
за(П)-аммония для восстановления МпО2 до Мп2+. По завершении реакции из-
быток Fe2+ оттитровали в кислой среде 0,0200 М раствором КМпО4. На титрова-
ние израсходовано 15,0 мл раствора. Рассчитайте содержание марганца в образце,
выразив его в виде массовой доли Мп3О4 (%). Разумеется, в действительности
марганец в пробе может присутствовать в других формах, однако в ходе вычис-
лений мы будем предполагать, что он присутствует именно в виде Мп3О4.
Решение
Fe2+ взаимодействует с МпО4 по уравнению
5Fe2+ 4- MnO; + 8Н+ -> 5Fe3+ 4- Мп2+ 4- 4Н2О
Поэтому число миллимолей непрореагировавшего Fe2+ в пять раз больше числа
миллимолей МпО4, израсходованного на титрование. Не забывайте, что реа-
гент может взаимодействовать с анолитом и с титрантом в разных мольных
соотношениях.
С МпО2 ион Fe2+ взаимодействует по уравнению
МпО2 4- 2Fe2+ 4- 4Н+ -» Мп2+ 4- 2Fe3+ 4- 2Н2О
Число миллимолей МпО2 в два раза меньше числа миллимолей Fe2+, прореаги-
ровавшего с ним. Кроме того, число миллимолей Мп3О4 составляет | от числа
миллимолей МпО2 (поскольку 1 МпО2 соответствует | Мп3О4). Отсюда число
миллимолей Fe2+, вступившего в реакцию, равно
nFe2+(ммоль) = 0,100 ммоль/мл Fe2+ • 50,0 мл Fe2+ -
- 0,0200 ммоль/мл МпО4 • 15,0 мл МпО4 • 5 = 3,5 ммоль
С0Мп3О4
«Мпо2 (ммоль)= 3,5 ммоль Fe2+ • | = 1,75 ммоль
пмп3о4 (ммоль) = 1J5 ммоль МпО2 • | = 0,5 83 ммоль
0,58, ммоль Мп тОл -228,8 мг/ммоль Мп ЛЬ
соМп о = ---------—-------------’100% = 66,7%
Мпз°4 200 мг пробы
Или, объединяя все вычисления в одну стадию,
_ ^Fe2* ’ ~ СМпО4 ’ ^МпО4 ‘ ’ 2 ‘ 3 ‘ ^Мп3О4 _
' '-------------------------------------------1UU 70 —
(ОД 00 • 50,0 - 0,0200 • 15,0 • 5) • Д • | • 228,8
- 21 -100% = 66,7%
200
Указанным способом можно определить лишь содержание марганца в степени окисления +4. —Прим, перев.
250
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
5.6. Использование нормальностей -
другой способ вычислений в титриметрии *
Часто вещества взаимодействуют в мольном соотношении, отличном от 1 : 1.
Соответственно их растворы с равными молярными концентрациями взаимо-
действуют в объемном соотношении, также отличном от 1 : 1. Однако, исполь-
зуя понятия «эквивалент» и «нормальность», можно и в этом случае вести
расчеты точно так же, как и для веществ, взаимодействующих в мольном соот-
ношении 1:1. Для этого вводят специальный способ выражения концентраций,
традиционно называемый нормальностью. Концентрации, выраженные в еди-
ницах нормальности, обозначают символом N подобно тому, как концентрации,
выраженные в единицах молярности, — символом М. Нормальность раствора
равна числу молей эквивалентов** вещества в литре раствора:
Нормальность (N) = моль экв./л = ммоль экв./мл
(5.30)
Под эквивалентом понимается частица (возможно, воображаемая), взаимо-
действующая с одной «реагирующей единицей» — например, протоном в кис-
лотно-основных реакциях. Так, одна частица НС1 в ходе кислотной диссоциации
отщепляет один протон. Поэтому эквивалентом хлористого водорода является
одна частица НС1, а молем эквивалентов — один моль НС1. С другой стороны,
H2SO4 отщепляет два протона. Поэтому один моль H2SO4 равен двум молям эк-
вивалентов H2SO4 (а сам эквивалент в этом случае представляет собой вообра-
жаемую частицу «| H2SO4»). Число молей эквивалентов можно рассчитать из
числа молей вещества следующим образом:
Число молей эквивалентов = Число молей вещества х
х Число «реагирующих единиц», приходящихся на одну молекулу
Понятия «эквивалент» и «нормальность», ранее применявшиеся широко, сейчас используются для расче-
тов не столь часто, как понятие «молярность». Если ваш преподаватель предложит вам изучить этот раз-
дел, сделайте это; в противном случае можете его пропустить. — Прим, автора.
Столь резкое противопоставление понятий «нормальность» и «молярность» представляется спорным. Ав-
тор прав в том, что термин «нормальность», как и символ N для ее выражения, в настоящее время действи-
тельно употребляются редко и не рекомендованы к использованию. Однако само понятие, которое автор
этим термином обозначает, есть лишь частный случай мольной концентрации, а именно — мольная кон-
центрация эквивалентов вещества («эквивалентная концентрация»). Например, если, эквивалент КМпО4
составляет у КМпО4, то такие обозначения, как 0,05000 моль экв./л КМпО4 или 0,05000 М | КМпО4 (вместо
действительно устаревшего 0,05000 N КМпО4) вполне правомерны и широко распространены и в современ-
ной литературе. Именно такие обозначения использованы в переводе. — Прим, перев.
В современной трактовке «эквивалент» есть «условная частица, в целое число раз меньшая (или равная)
соответствующей ей формульной единицы — атома, молекулы, иона, радикала и др.» [Б. Д. Степин. Хими-
ческая энциклопедия. Т. 5. С. 405. М.: БРЭ, 1998]. Автор же придерживается устаревшей трактовки, пони-
мая под эквивалентом моль таких частиц. В переводе понятие «эквивалент» и все, что с ним связано,
изложено с современной точки зрения. — Прим, перев.
5.6. ИСПОЛЬЗОВАНИЕ НОРМАЛЬНОСТЕЙ - ДРУГОЙ СПОСОБ ВЫЧИСЛЕНИЙ В ТИТРИМЕГРИИ
251
Число миллимолей эквивалентов = Число миллимолей вещества х
х Число «реагирующих единиц», приходящихся на одну молекулу
Число «реагирующих единиц» зависит от природы реакции и для одного и
того же вещества может быть различным. Особенно часто это наблюдается в
окислительно-восстановительных реакциях.
Что такое «эквивалентная масса»?
Эквивалентная масса — это сокращенное название молярной массы эквива-
лента, т. е. масса (в граммах) такого количества вещества, которое взаимодейст-
вует с одним молем «реагирующих единиц». Так, для НС1 эквивалентная масса
Мэкв равна молярной массе М. В то же время для H2SO4 количество вещества,
содержащее один моль протонов, равно половине моля, поэтому эквивалентная
масса H2SO4 составляет половину молярной массы:
_ (г/моль)
Экв. масса НО =---—-----------
1 (моль экв./моль)
Экв. масса H2SO4 =
2 (моль экв./моль)
Пример 5.30
Рассчитайте эквивалентные массы следующих веществ: a) NH3, б) Н2С2О4 (при-
менительно к реакции с NaOH), в) КМпО4 (применительно к реакции восстанов-
ления Mn(VII) до Мп2+).
Решение
а) Одна частица NH3 представляет собой один эквивалент NH3. Поэтому
„ кт„ Mnh (г/моль) 17оз
Экв. масса =----------------=------= 17,03 г/моль экв.
3 1 1
б) Одна частица Н2С2О4 содержит два эквивалента,
„ Мн2с2о4(г'/моль) 90,04 ЛСА_ .
Экв. масса Н9С9О4 =---------------=-----= 45,02 г/моль экв.
2 2 4 2 2
в) В ходе указанной реакции атом Мп изменяет свою степень окисления с +7 до
+2, присоединяя пять электронов:
МпО; + 8Н+ + 5е" = Мп2+ + 4Н2О
Поэтому
С» ™ ГА MKMnO4 (г/моль) 15804
Экв. масса КМпО4 =--------------=--------= 31,608 г/моль экв.
5 5
252
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
Подобно тому, как число молей вещества можно рассчитать делением его
массы (г) на молярную массу (г/моль), можно рассчитать и число молей эквива-
лентов путем деления массы вещества на его эквивалентную массу:
Число молей экв. = Масса (г) / Экв. масса (г/моль экв.)
Число миллимолей экв. = Масса (мг) / Экв. масса (мг/ммоль экв.)
(5.31)
Нормальность (эквивалентная концентрация) равна частному от деления числа
молей эквивалентов на объем раствора:
, , п моль экв. т (г)/М 9КВ (г/моль экв.) с (моль экв/л, N) = = —— L Г(л) И(л) . . п ммоль экв. ?п(мг)/М,кв (мг/ммоль экв.) с (ммоль экв/мл, N) = = —-—-— - Г (мл) У (мл) (5.32)
где Л/экв — молярная масса эквивалента.
Пример 5.31
Рассчитайте эквивалентную концентрацию растворов следующего состава:
a) Na2CO3 5,300 г/л (применительно к реакции взаимодействия СО3“ с двумя
протонами); б) 5,267 г/л К2Сг2О7 (применительно к реакции восстановления
хрома до Сг3+).
Решение
а) Поскольку СО3" взаимодействует с 2Н+ (с образованием Н2СО3), то
с =-------5,300 Г;/л------= 0,1000 моль экв./л (0,1000 N)
(105,99/2) г/моль экв.
б) Каждый атом Cr(VI) восстанавливается до Сг(Ш), поэтому одна частица
К2Сг2О7 взаимодействует с шестью электронами:
Сг2О2" + 14Н+ + бе- = 2Сг3+ + 7Н2О
с =-------5,26? г/л------= 74 модь экв /л z0 1074 N)
(294,19/6) г/моль экв.
5.6. ИСПОЛЬЗОВАНИЕ НОРМАЛЬНОСТЕЙ - ДРУГОЙ СПОСОБ ВЫЧИСЛЕНИЙ В ТИТРИМЕГРИИ
253
Для нахождения числа молей эквивалентов не всегда необходимо в явном
виде рассчитывать молярную массу эквивалента. Можно просто использовать
стехиометрический фактор т* для перевода числа молей вещества в число мо-
лей эквивалентов. Стехиометрический фактор равен числу эквивалентов, со-
ставляющих одну частицу вещества.
Число молей эквивалентов = Число молей вещества • т
Эквивалентная концентрация (моль экв./л, N) =
= Молярная концентрация (моль/л, М) • т
Эквивалентная масса (г/моль экв.) = Молярная масса (г/моль) / т
Для окислительно-восстановительных реакций в ходе расчетов, связанных
с эквивалентами, нельзя просто полагаться на величину изменения степени
окисления элемента. Необходимо использовать стехиометрическое уравнение
полуреакции. Например, когда дихромат-ион восстанавливается до Сг3+, урав-
нение полуреакции имеет вид
Сг2О2’ + 14Н+ + бе- 2Сг3+ + 7Н2О
Отсюда т = 6 (шесть электронов, приходящихся на одну частицу Сг2О2“). Поэтому
эквивалентная концентрация 0,1 М раствора К2Сг2О7 составляет 0,6 моль экв./л
(0,6 N).
Преимущество выражения концентраций посредством нормальности, а
количества вещества — при помощи эквивалентов состоит в том, что один
моль эквивалентов вещества А всегда взаимодействует с одним молем эквива-
лентов вещества В. Так, один моль эквивалентов NaOH (= 1 моль NaOH) взаи-
модействует как с одним молем эквивалентов НС1 (= 1 моль НС1), так и с одним
молем эквивалентов H2SO4 (= | моль H2SO4). Поэтому можно рассчитывать мас-
су аналита непосредственно из числа молей эквивалентов титранта, так как оно
равно числу молей эквивалентов аналита. Все вычисления с использованием эк-
вивалентных концентраций (нормальностей) выполняются точно так же, как вы-
числения с использованием молярностей для реакций, протекающих в мольном
соотношении 1:1.
Следует подчеркнуть, что число молей эквивалентов и величина эквивалент-
ной концентрации зависят от природы конкретной реакции. Например, Na^C^
может взаимодействовать как с одним протоном (СО3“ + Н+ -> НСО3), так и с
двумя протонами (СО3“ + 2Н+ -> Н2СО3). В первом случае одному иону СО3" со-
ответствует одна «реагирующая единица», во втором — две. Поэтому использо-
вание величин эквивалентных концентраций, количества молей эквивалентов и
эквивалентной массы связано с определенной опасностью: все эти величины
имеют смысл только применительно к конкретной реакции, и эта реакция дол-
жна быть явно указана. Например, если концентрация раствора КМпО4 записана
просто как 0,1000 N без указания, что за реакция имеется в виду, то это может
Сейчас чаще используют обратную величину f=\hn, называемую фактором эквивалентности. —Прим, перев.
254
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
Число миллимолей эквивалентов А (иэкв А) = Число миллимолей эквивалентов Т (иэкв т)
«экв А (ММОЛЬ ЭКВ.) = —---(М£)---------- = (ммоль экв,/л> tf) . (МЛ)
Мэкв А (мг/ммоль экв.)
тА (мг) = иэкв т (ммоль экв.) • Мэкв А (мг/ммоль экв.)
тА (мг) = сэквТ (ммоль экв./л, N) • Кт (мл) • Мэкв А (мг/ммоль экв.)
быть как реакция восстановления перманганат-иона до Мп2+ (обычно протекаю-
щая в кислой среде), так и реакция восстановления до МпО2 (в нейтральной сре-
де). В первом случае число «реагирующих единиц» (электронов), приходящихся
на один перманганат-ион, равно пяти, во втором — трем. Если же указана моль-
ная концентрация вещества (молярность), то это величина однозначная. Раствор
1 М КМпО4 может быть 5 N или 3 N раствором в засисимости от того, в какой ре-
акции он используется.
Итак, запомните, что число «реагирующих единиц», а следовательно, и чис-
ло эквивалентов, приходящихся на одну частицу вещества, зависит от того, в ка-
кой реакции это вещество участвует. Однако в любом случае для аналита А и
титранта Т в точке эквивалентности:
(5.33)
(5-34)
(5.35)
(5.36)
Эквивалентную массу А рассчитывают точно так же, как и эквивалентную
массу Т, т. е. с использованием числа «реагирующих единиц», взаимодействую-
щих с одной молекулой А либо отщепляемых ею.
Общее выражение для расчета массовой доли определяемого компонента в
пробе с использованием эквивалентных концентраций имеет вид, аналогичный
уравнению (5.22):
титранта (ММОЛЬ ЭКВ./мЛ, N) • Ктитранта (мл) • 1 • Мэкв анадита (мг/мМОЛЬ ЭКВ.)
т пробы (мг)
(5.37)
Единица в этом выражении означает отношение числа миллимолей эквивален-
тов А к числу миллимолей эквивалентов Т. Оно всегда равно единице.
Пример 5.32
Пробу массой 0,4671 г, содержащую бикарбонат натрия (основание, присоеди-
няющее один протон), растворили и оттитровали 40,72 мл стандартного раство-
ра соляной кислоты. Соляную кислоту стандартизовали титрованием навески
0,1876 г карбоната натрия; на титрование израсходовано 37,86 мл кислоты
5.6. ИСПОЛЬЗОВАНИЕ НОРМАЛЬНОСТЕЙ - ДРУГОЙ СПОСОБ ВЫЧИСЛЕНИЙ В ТИТРИМЕТРИИ
255
(уравнение реакции см. в примере 5.18). Рассчитайте массовую долю (%) бикар-
боната натрия в пробе.
Решение
Na2CO3 — основание, в ходе титрования присоединяющее два протона. Поэтому
z . . «экв№2со3 (ммоль экв.) (Mr)/[MN со (мг/ммоль)/2]
сэкв НС1 (ммоль экв-/л) =---,z , .------=-----—-----------—-------------— =
КНС1 (мл)
>НС1 (мл)
187,6 мгNa9ССК/(105,99/2мг/ммоль экв.) л
=----------?---------—----------------- = 0,09350 ммоль экв./мл
37,86 мл НС1
сэквНС1 ’^НС1 ’[^NaHCOj^]
®NaHCO3 -------------------------100% =
пробы
0,09340 ммоль экв./мл НС1 • 40,72 мл НС1 • 84,01/1 мг/ммоль экв. NaHCO3 _
476,1 мг пробы
= 67,18%.
Еще раз повторим, что один моль эквивалентов любого вещества всегда
взаимодействует с одним молем эквивалентов другого вещества:
л (А) = л (В)
(5.38)
Отсюда следует соотношение, позволяющее рассчитать соотношение объемов
растворов реагирующих веществ:
сэкв а (ммоль экв./мл, N) • КА (мл) = сэкв т (ммоль экв./мл, N) • Кт (мл) (5.39)
Пример 5.33
Сколько миллилитров 0,25 М раствора H2SO4 необходимо для реакции с 10 мл
0,25 М раствора NaOH?
Решение
Поскольку одна частица H2SO4 содержит две «реагирующие единицы», то экви-
валентная концентрация раствора H2SO4 вдвое выше, чем молярная:
2NaOH + H2SO4 -> Na^O.^ + 2Н2О
Сэкв H2SO4 = 2 моль экв./моль • 0,25 моль/л = 0,50 моль экв./л
256
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
Эквивалентная концентрация (нормальность) раствора NaOH равна его моляр-
ной концентрации, поскольку одна частица NaOH взаимодействует с одной «ре-
агирующей единицей»:
Сэкв NaOH = °>25 М0ЛЬ Экв-/л
сэкв H2SO4 (ммоль экв./мл) • (мл) = сэкв NaOH (ммоль экв./мл) • KNaOH (мл)
0,50 ММОЛЬ экв./мл ’ ^н28О4 (МЛ) ~ 0’25 ммоль экв./мл • 10 мл
^H2SO4 = 5>° мл
Расчет разбавления, необходимого для приготовления раствора заданной
эквивалентной концентрации из более концентрированного, выполняется точно
так же, как и для молярных концентраций (см. уравнение (5.29) и пример 5.22).
В табл. 5.4 приведены основные соотношения между величинами, связан-
ными с числом молей вещества и числом молей эквивалентов вещества.
«Реагирующие единицы» (протоны и электроны)
в расчетах эквивалентных концентраций
1. Кислотно-основные реакции. Как было отмечено ранее, для кислот и осно-
ваний «реагирующей единицей» является протон Н+. Если вещество реагирует
как кислота, необходимо определить число реакционноспособных протонов, со-
держащихся в одной молекуле вещества. Если вещество реагирует как основа-
ние, следует найти число протонов, взаимодействующих с одной его молекулой.
В обоих случаях
Mjkb
м
Число протонов
(5.40)
Таблица 5.4
Сопоставление величин, основанных на числе молей вещества и числе молей
эквивалентов вещества
п (моль) • т (моль экв./моль) = иэкв (моль) п (ммоль) • т (ммоль экв./ммоль) = иэкв (ммоль)
с (моль/л, N) • т (моль экв./моль) = = сэкв (моль экв./л, N) с (ммоль/мл, N) • т (ммоль экв./ммоль) = = сэкв (ммоль экв./мл, N)
М (г/моль): т (моль экв./моль) = = Мэкв (г/моль экв.) М (мг/ммоль): т (ммоль экв./ммоль) = = Л/экв (мг/ммоль экв.)
5.6. ИСПОЛЬЗОВАНИЕ НОРМАЛЬНОСТЕЙ - ДРУГОЙ СПОСОБ ВЫЧИСЛЕНИЙ В ТИТРИМЕТРИИ
257
Число протонов зависит от конкретной реакции, в которой участвует вещест-
во. Так, H2SO4 диссоциирует нацело по обеим ступеням, поэтому на один моль
H2SO4 приходится два моля эквивалентов «реагирующих частиц». В ходе титрова-
ния оба протона вступают у реакцию одновременно, поэтому для H2SO4 существу-
ет единственная конечная точка титрования, соответствующая оттитровыванию
двух протонов. Н3РО4 по первой ступени диссоциирует в значительной мере, по
второй — слабо, а по третьей — настолько слабо, что третий протон оттитровать
невозможно. Поэтому у фосфорной кислоты можно оттитровать по ступеням
один или два протона, наблюдая две отдельные конечные точки. Конечная точка,
соответствующая оттитровыванию третьего протона, не наблюдается. Если тит-
рование проводят так, чтобы оттитровать только первый протон, то в данной конк-
ретной реакции на одну молекулу Н3РО4 приходится лишь одна «реагирующая
единица». Число молей эквивалентов в этом случае равно числу молей Н3РО4. Если
же оттитровывают два протона (в этом случае объем титранта будет вдвое больше,
что позволит уменьшить относительную систематическую погрешность результа-
та), то число молей эквивалентов будет вдвое больше, чем число молей Н3РО4.
Na^O-j — достаточно сильное основание, способное взаимодействовать с
кислотами с образованием NaHCO3. NaHCO3 — слабое основание, которое тем
не менее можно оттитровать по одной ступени до угольной кислоты:
СО^“ + Н+ НСОз
НСО3 + Н+ Н2О + со2
При титровании Na^C^ мы также можем наблюдать две отдельные конечные
точки, а число молей эквивалентов (и молярная масса эквивалента) Na^C^ зави-
сит от того, какая реакция используется для титрования. Поэтому, прежде чем
приступать к каким-либо расчетам с использованием понятия «эквивалент», не-
обходимо точно установить, какая реакция имеется в виду.
Пример 5.34
Раствор карбоната натрия приготовлен растворением 0,212 rNa2CO3 с последу-
ющим разбавлением до 100 мл. Рассчитайте эквивалентную концентрацию
(нормальность) этого раствора, если карбонат натрия используется как: а) одно-
ступенчатое; б) двухступенчатое основание.
э) ^экв
б) £*ЭКВ
Решение
mNa2CO3 (Mr)/[^Na2CO3212 МГ/106,0 МГ/ММОЛЬ ЭКВ. _ ____ .
=-----— ----------—-— =------------------------= 0,0200 ммоль экв./мл
К(мл) 100 мг
mNa2co3 (Mr)/[^Na2co3 212 мг/0,5 -106,0 мг/ммоль экв. _ л .
=----— ----------—-— =----------------------------= 0,0400 ммоль экв./мл
К(мл) 100 мг
258
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
2. Окислительно-восстановительные реакции. В окислительно-восстанови-
тельных реакциях «реагирующей единицей» является электрон. Восстанови-
тель отдает электроны и, таким образом, окисляется, а окислитель принимает
электроны и вследствие этого восстанавливается. Например, в реакции
5Fe2+ + MnO; + 8Н+ 5Fe3+ + Мп2+ + 4Н2О
каждый ион Fe2+ (восстановитель) теряет один электрон, а каждый ион МпО^
(окислитель) приобретает пять электронов, восстанавливаясь из Mn(VII) до
Мп(П). Расчеты с использованием числа «реагирующих единиц» выполняются
точно так же, как и для кислотно-основных реакций:
Число приобретенных или отданных электронов
(5.41)
В приведенном примере для железа число молей эквивалентов равно числу мо-
лей Fe2+, а эквивалентная масса равна атомной массе железа. В то же время для
перманганат-иона число молей эквивалентов в пять раз больше числа молей
МпО;, а эквивалентная масса равна одной пятой молярной массы МпО^. Но, как
и для кислотно-основных реакций, один моль эквивалентов восстановителя все-
гда взаимодействует с одним молем эквивалентов окислителя.
Пример 5.35
Иод (12) является окислителем, который при взаимодействии с восстановителя-
ми восстанавливается до иодид-ионов (I-). Какую навеску (г) иода следует взять
для приготовления 100 мл 0,100 N раствора 12?
Решение
В ходе реакции каждая молекула 12 приобретает два электрона:
12 + 2е~ —> 2I-
Поэтому эквивалентная масса 12 составляет половину его молярной массы:
А/, (г/моль)
М ЭКВ 12 ~ / 7
2 2 (моль экв./моль)
тх (мг) тх (мг)
сэкв (ммоль экв./мл, N) • И(мл) = иэкв (ммоль экв.) = —г--= — -
^эквЬ
19 L2,
5.6. ИСПОЛЬЗОВАНИЕ НОРМАЛЬНОСТЕЙ - ДРУГОЙ СПОСОБ ВЫЧИСЛЕНИЙ В ТИТРИМЕТРИИ
259
т1 (мг)
0,100 ммоль экв./мл • 100 мл =-----------------
254/2 мг/ ммоль экв.
wl2 = 0,100 ммоль экв./мл • 100 мл • (254/2 мг/ммоль экв.) = 1270 мг
Таким образом, следует взять навеску массой 1,27 г.
Пример 5.36
Рассчитайте эквивалентную концентрацию (нормальность) раствора Н2С2О4
0,25 г/л в зависимости от того, чем это соединение является: кислотой или
восстановителем.
Решение
При расчете эквивалентной концентрации раствора необходимо учитывать, яв-
ляется Н2С2О4 кислотой или восстановителем. Эквивалентная масса Н2С2О4,
выступающей в роли кислоты, равна половине ее молярной массы. Отсюда
250 мг/(90,04/2 мг/ммоль экв.) л ,
=------——-— ------------------ = 0,00555 ммоль экв./мл
1000 мл
В ходе окисления каждый оксалат-ион отдает два электрона, превращаясь в СО2
(каждый атом углерода переходит из степени окисления +3 в +4). Поэтому и
здесь эквивалентная масса равна половине молярной, а эквивалентная концент-
рация Н2С2О4 как восстановителя такая же, как кислоты.
Формулы для расчетов с использованием нормальностей
Приведем формулы, наиболее часто употребляемые в титриметрических расче-
тах при использовании понятия эквивалентной концентрации (нормальности).
, . т (мг)
иэкв (ммоль экв.) =-------i— --------
А/экв (мг/ ммоль экв.)
. , хтч «чкв(ммоль экв.)
б? (ммоль экв./мл, N) = ------------
экв К(мл)
(5.42)
(5.43)
260
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
Для расчетов результатов титрования вещества А раствором титранта Т обычно
используют одну из двух следующих формул (помните о единицах измерения!):
сэкв т (ммоль экв./мл, N) • Кт (мл) • Л/экв А (мг/ммоль экв.) = (мг) (5.44)
сэквТ (ммоль ЭКВ./МЛ, N) • Гт (мл) • Мэкв А (мг/ммоль экв.) / тпро5и (мг) • 100% = <оА (%) (5.45)
5.7. Титр - способ быстрого выполнения серийных вычислений
Для серийных (рутинных) вычислений в титриметрии часто бывает удобным
рассчитать титр раствора титранта. Титр — это масса аналита (выражаемая
обычно в миллиграммах), химически эквивалентная 1 мл раствора титранта. На-
пример, если указано, что титр раствора дихромата калия равен 1,267 мг Fe, это
означает, что один миллилитр такого раствора взаимодействует с 1,267 мг желе-
за. Массу оттитрованного железа находят умножением израсходованного объе-
ма титранта на его титр. При желании титр может быть выражен в виде массы
любой формы аналита, например, FeO или Fe2O3.
Пример 5.37
Концентрация стандартного раствора дихромата калия составляет 5,442 г/л.
Чему равен его титр, выраженный в виде массы Fe3O4 (мг)?
Решение
Железо титруется в виде Fe2+, а один ион Сг2О7“ взаимодействует с шестью
ионами Fe2+, что соответствует содержанию железа в 2Fe3O4:
6Fe2+ + Cr2O^ + 14Н+ -> 6Fe3+ + 2Cr3+ + 7H2O
Молярность раствора К2Сг2О7 равна
5 442 г/ л
cn n2_ (М) = ’ v = 0,01850 моль/л
Сг2°7 294,19 г/моль
Отсюда титр равен
. / 2 ммоль Fe4O, мгРеаО4
0,01850 ммоль К2Сг2О7/мл •---------3 — -231,54-----—-— =
1 ммоль К2Сг2О7 ммоль Fe3O4
= 8,567 мг Fe3O4/ мл К2Сг2О7
5.8. МАССОВЫЕ СООТНОШЕНИЯ - ОНИ НЕОБХОДИМЫ ДЛЯ РАСЧЕТОВ В ГРАВИМЕТРИИ
261
5.8. Массовые соотношения - они необходимы
для расчетов в гравиметрии
В ходе гравиметрического анализа (гл. 7) аналит превращают в малораствори-
мое соединение, а затем взвешивают. Из величины массы осадка, используя мас-
совые соотношения между аналитом и осадком, можно рассчитать массу аналита.
Рассмотрим главные положения, лежащие в основе таких расчетов.
Почти всегда химическая форма аналита, в которой его взвешивают (грави-
метрическая форма), отличается от той формы, в виде которой должен быть
представлен результат анализа. Поэтому необходимо рассчитать массу желае-
мой формы, соответствующую данной массе гравиметрической формы. Это
можно сделать, используя простую пропорцию. Например, если необходимо
определить массовую долю хлорид-ионов в обазце, а гравиметрическая форма
представляет собой AgCl, то можно записать:
С1~ _оса-д^ь> AgCl
Из одного моля С1“ образуется один моль AgCl, поэтому \
^сг _ Ал (г Cl/моль Cl) 1 моль С1
mAgC1 (г) AfAgC1 (г AgCl/моль AgCl) 1 моль AgCl
или
wcl_ (г) = mAgC| (г) • (г Cl/г AgCl)
^AgCl
Иными словами, масса С1, содержащегося в определенном количестве AgCl, рав-
на массе AgCl, умноженной на массовую долю, которую составляет С1 в AgCl.
В гравиметрическом анализе число молей аналита рассчитывают из числа мо-
лей осадка с учетом стехиометрических соотношений между ними.
Если требуется выразить результаты анализа в виде массы С12, то схема рас-
четов выглядит следующим образом:
С^ос адитель>
Из одного моля С12 образуются два моля AgCl, поэтому
™С12(Г) МС12(гС12/мольС12) 1 моль С12
mAgCi (г) МAgCl (г AgCl/моль AgCl) 2 моль AgCl
или
Ма
та2 (г) = mAgci (г) • - ~ (г С12/г AgCl)
2^AgCl
262
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
то есть
70,906 г С12/моль С12
(г) = ^доГ1 (г)--------------------—---------------------
С12 AgC1 (2 моль AgCl/моль С12) (143,32 г AgCl/моль AgCl)
Следите за единицами измерения!
Это выражение можно записать и так:
lмoльAgCl 1мольС12 79,906 г С12
тС] (г) = т^сх (г)-----------------------------
С12 AgC1 143,32 г AgCl 2 моль AgCl 1мольС12
Отношение молярной массы искомой формы аналита к молярной массе гра-
виметрической формы, умноженное на отношение числа молей, называется
гравиметрическим фактором (ГФ):
ГФ
(гравиметрический
фактор)
Молярная масса
искомой формы
Молярная масса
гравиметрической формы
а
~Ъ
Моль искомой формы
Моль гравиметрической
формы
(5.46)
Здесь а и b — целые числа, уравнивающие числа атомов «ключевого» (т. е. со-
держащегося в обеих формах) элемента искомой и гравиметрической форм. Для
примеров, приведенных выше, гравиметрические факторы равны [МС1 ] • 1/1
и [А/С12 ^AgCi ] ’ 1/2. Обратите внимание, что в общем случае одну или обе вели-
чины молярных масс следует умножить на целое число, чтобы уравнять число
атомов «ключевого» элемента.
Гравиметрический фактор равен массе аналита, соответствующей единице
массы осадка. Массу искомой формы вещества рассчитывают умножением мас-
сы осадка (точнее, гравиметрической формы) на гравиметрический фактор:
т (искомой = т (гравиметри- М (искомой формы)_______ а
формы, г) ческой формы, г) А/(гравиметрической формы)
(5.47)
Заметьте, что в этих выражениях правильность записи единиц измерения и ве-
ществ, к которым они относятся, можно проверить при помощи анализа размер-
ностей (при правильной записи единицы измерения и вещества сократятся).
Например:
j22^p-AgCTf- 1/2 (кхол1ге1^Лиоль^А§СГ) •
Л/С12 (г С12 /мож~СГ2)
MAgC1 (tAgCI/MOflbTtgCi)
= т(г С12)
5.8. МАССОВЫЕ СООТНОШЕНИЯ - ОНИ НЕОБХОДИМЫ ДЛЯ РАСЧЕТОВ В ГРАВИМЕТРИИ
263
Результат этого расчета представляет собой массу газа С12, который можно
было бы получить из образца. Если хлориды осадить и взвесить в виде РЬС12, то
расчеты будут выглядеть следующим образом:
2СГ осадитель> РЬС12 или С12->РЬС12,
отсюда
тС1_ (г) = тиРЬС| (г) • 2Мс1 (г СУ г РЬС12) = mpbC1 (г) • ГФ
^PbCl2
или
МГ1
"Ъ2 (0 = «рьс12 <г) • 77^ (г С1г/Г РЬС1г) = ™Pbci2 (0 • ГФ
^РЬС12
При расчете массы одного вещества из эквивалентной ей массы другого ве-
щества полезно провести анализ размерностей всех единиц измерения с тем,
чтобы в итоге получить правильное значение массы желаемой химической фор-
мы. Один из этапов таких вычислений — расчет гравиметрического фактора.
Особенно полезно вычислить его значение при выполнении массовых, рутин-
ных расчетов. Если гравиметрический фактор известен, то для нахождения массы
аналита в соответствующей форме достаточно просто умножить массу гравимет-
рической формы на гравиметрический фактор.
Пример 5.38
Рассчитайте массу бария и хлора, содержащихся в 25,0 г ВаС12.
Решение
тлВа = 25,0 г ВаС12 • 1 (моль Ba/моль ВаС12) •
137,3 (г Ва/моль Ва) Л . _
-----------------------= 16,5 г
208,2 (г ВаС12/моль ВаС12)
тс, = 25,0 г ВаС12 • (моль Cl/моль ВаС12) • -35,45 (Г С1/ моль С1)-= 8,51 г
1 2 208,2 (г ВаС12/моль ВаС12)
Пример 5.39
Для определения алюминия образец руды перевели в раствор, осадили алюми-
ний основанием в виде А1(ОН)3, а затем осадок прокалили до А12О3 и взвесили.
Чему равна масса алюминия в образце, если масса прокаленного осадка состав-
ляет 0,2385 г?
264
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
Решение
/ч /4 7/ Л1/ А1ГЧЧ Мд. (г Al/моль А1)
™ai (г) = о (г) • | (моль Al/моль А12О3) • —-А‘ -------———
Ма1о3(гА12Оз/МОЛьА12°з)
= 0,2385 (г А12О3) • j •-26,982 (гА1/мольА1)-= о, 12623
1 101,96 (г А12О3/моль А12О3)
Гравиметрический фактор равен
2Мм (г Al/г А12О3) = 2<26^982гА1/моль А1) = 0 27 А1/г А1 0
Ммп 101,96 г А1,ОЧ/моль А17О,
/112^3 Z Э / Z J
Используя эту величину, находим:
тлА1 = 0,2385 (г А12О3) • 0,52927 (г Al/г А12О3) = 0,12623 г
Ниже приведено еще несколько примеров расчета значений гравиметриче-
ского фактора.
Искомая форма Гравиметрическая форма Гравиметрический фактор
so3 Ва$О4 MSOj /MBaSo4
Fe3°4 Fe2°3 ^РезОд /^F^Oj
Fe Ре2^3 ^Fe/^FejOj
MgO Mg2P2O7 2A^Mgo/WMg2P2O7
P2°5 Mg2P2O7 /^Mg2P2O7
Другие примеры расчетов в гравиметрии приведены в гл. 7. Операции, вы-
полняемые в гравиметрическом анализе, подробно описаны в гл. 10.
Что мы узнали из этой главы?
• Как рассчитать число молей и молярность [основные уравнения (5.4), (5.5)] —
стр. 213
• В какой форме представляют результаты анализа — стр. 223
• Как рассчитать массу и массовую долю аналита из величины молярности,
объема и стехиометрических коэффициентов [основные уравнения (5.5),
(5.17)-(5.22), (5.27)] — стр. 236
ЗАДАЧИ
265
• Нормальность как еще один способ выражения концентраций [основные
уравнения (5.30)-(5.32), (5.36)] — стр. 250
• Массовые соотношения в гравиметрическом анализе [основное уравнение
(5.47)] —стр. 261
Вопросы
1. Объясните, в чем разница между способами выражения концентраций в
виде отношения масса : масса, масса : объем и объем : объем.
2 Объясните, что представляют собой единицы измерения ppm и ppb, выражен-
ные посредством отношений масса : масса, масса : объем и объем : объем.
3. Что означает термин «эквивалентная масса», используемый в клиническом
анализе? Для чего его используют?
4. Перечислите требования к титриметрической реакции. Назовите четыре
основных класса титриметрических методов.
5 - Что такое точка эквивалентности в титровании? Конечная точка титрования?
6- Что такое стандартный раствор? Как его готовят?
7- Какие требования предъявляются к первичным стандартам?
8. Почему желательно, чтобы первичный стандарт имел большую молярную
массу?
Задачи
Расчет массы и количества вещества
9- Рассчитайте массы веществ (г), требуемых для приготовления следующих
растворов: (а) 250 мл 5,00% (масса : объем) NaNO3; (б) 500 мл 1,00% (мас-
са : объем) NH4NO3; (в) 1000 мл 10,0% (масса : объем) AgNO3.
10- Чему равна процентная концентрация (в единицах масса : объем) следующих
растворов: (a) Na^C^ 52,3 г/л; (б) КВг 275 г/500 мл; (в) SO2 3,65 г/200 мл?
11. Рассчитайте молярные массы следующих веществ: (а) ВаС12 • 2Н2О;
(б) КНС2О4 • Н2С2О4; (в) Ag2Cr2O7; (г) Са3(РО4)2.
12. Рассчитайте количество вещества (ммоль), содержащегося в 500 мг следу-
ющих веществ: (а)ВаСгО4; (б) СНС13; (в) КЮ3 • НЮ3; (г) MgNH4PO4;
(д) Mg2P2O7; (е) FeSO4 • C2H4(NH3)2SO4 • 4Н2О.
13. Рассчитайте массу (г) каждого из веществ, перечисленных в задаче 12, тре-
буемую для приготовления 100 мл 0,200 М раствора.
14 Рассчитайте массу (мг) веществ, требуемую для приготовления следующих
растворов: (а) 1,00 л 1,00 М NaCl; (б) 0,500 л 0,200 М сахарозы (С12Н22ОИ);
(в) 10,0 мл 0,500 М сахарозы; (г) 0,0100 л 0,200 М Na2SO4; (д) 250 мл
0,500 М КОН; (е) 250 мл 0,900% (г/100 мл раствора) NaCl.
266
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
15 - В лаборатории имеются исходные стандартные растворы следующего соста-
ва: 0,100 М НС1, 0,0200 М NaOH, 0,0500 М КОН, 10,0% (масса : объем) НВг
и 5,00% (масса: объем) Na2CO3. В каких объемах этих растворов содержат-
ся следующие количества растворенных веществ: (а) 0,0500 моль НС1;
(б) 0,0100 моль NaOH; (в) 0,100 моль КОН; (г) 5,00 г НВг; (д) 4,00 rNa2CO3;
(е) 1,00 моль НВг; (ж) 0,500 моль Na2CO3?
Расчеты с использованием молярных концентраций
16 - Рассчитайте молярные концентрации каждого катиона и аниона в растворе,
приготовленного смешением порций по 10,0 мл следующих растворов:
0,100 М Mn(NO3)2, 0,100 М KNO3 и 0,100 М K2SO4.
17. Раствор, содержащий 10,0 ммоль СаС12, разбавили до 1 л. Рассчитайте мас-
совую концентрацию (г/мл) СаС12 • 2Н2О в полученном растворе.
18- Рассчитайте молярную концентрацию каждого из следующих растворов:
(a) H2SO4 (10,0 г в 250 мл раствора); (б) NaOH (6,00 г в 500 мл); (в) AgNO3
(25,0 г в 1,00 л).
19- Рассчитайте массу (г) каждого из следующих веществ, содержащуюся в
500 мл раствора: (а) 0,100 М Na2SO4; (б) 0,250 М Fe(NH4)2(SO4)2 • 6Н2О;
(в) 0,667 М Ca(C9H6ON)2.
20- Рассчитайте массу (г) каждого из следующих веществ, необходимую для
приготовления растворов: (а) 250 мл 0,100 М КОН; (б) 1,00 л 0,0275 М
К2Сг2О7; (в) 500 мл 0,0500 М CuSO4.
21 - Какой объем (мл) концентрированной соляной кислоты (38,0% масс., плот-
ность 1,19) следует взять для приготовления 1 л 0,100 М раствора?
22. Рассчитайте молярную концентрацию каждого из следующих коммерче-
ских растворов кислот и оснований: (а) 70,0% НС1О4 (плотность 1,668);
(6)69,0% HNO3 (плотность 1,409); (в) 85,0% Н3РО4 (плотность 1,689);
(г) 99,5% СН3СООН (плотность 1,051); (д) 28,0% NH3 (плотность 0,898).
Расчеты с использованием единиц ppm
23. Раствор содержит 6,0 мкмоль Na2SO4 в 250 мл. Какова концентрация на-
трия в этом растворе в ppm? Сульфат-иона?
24- В 100 мл раствора с концентрацией К+, равной 325 ppm, определяют калий
следующим образом. Калий осаждают в виде тетрафенилбората К(С6Н5)4В,
растворяют осадок в ацетоне и определяют концентрацию тетрафенилбо-
рат-иона (С6Н5)4В~ в полученном растворе. Чему равна концентрация тет-
рафенилборат-иона в растворе в ppm, если объем раствора равен 250 мл?
25. Рассчитайте молярные концентрации растворов следующих веществ, кон-
центрация каждого из которых составляет 1 ppm: (a) AgNO3, (б) A12(SO4)3,
(в) СО2, (г) (NH4)4Ce(SO4)4 • 2Н2О, (д) НС1, (е) НС1О4.
ЗАДАЧИ
267
26. Рассчитайте концентрации в ppm каждого из 2,50 • 10-4 М растворов:
(а) Са2+, (б) СаС12, (в) HNO3, (г) KCN, (д) Мп2+, (е) МпО;.
27. Вам необходимо приготовить 1 л раствора, содержащего 1 ppm Fe2+. Какую
навеску (г) сульфата железа-аммония FeSO4 • (NH4)2SO4 • 6Н2О следует
растворить в воде и разбавить до 1 л? Какова молярная концентрация полу-
ченного раствора?
28. В пробе руды массой 0,456 г содержание Сг2О3 составляет 0,560 мг. Выра-
зите содержание Сг2О3 в руде в виде массовой доли: (а) в процентах,
(б) в частях на тысячу, (в) в частях на миллион.
29. Какую навеску (г) NaCl следует взять для приготовления 1 л раствора с кон-
центрацией 100 ppm: (а) по Na+, (б) по С1“?
30. Имеется раствор КС1 с концентрацией 250 ppm К+. Из него необходимо
приготовить 1 л 0,00100 М раствора С1_. Какой объем раствора (мл) следует
взять для разбавления?
31. Какова масса К+ (г) в одном литре 500 ppm раствора КС1О3?
Расчеты разбавлений
32. 12,5 мл раствора разбавили до 500 мл. Концентрация разбавленного рас-
твора составила 0,125 М. Какова молярная концентрация исходного рас-
твора.
33. Какой объем 0,50 М H2SO4 следует добавить к 65 мл 0,20 М H2SO4, чтобы
получить 0,35 М раствор? Допущение: суммарный объем при смешении не
изменяется.
34. Какой объем (мл) 0,10 М H2SO4 неохбодимо добавить к 50 мл 0,10 М NaOH,
чтобы получить раствор с концентрацией 0,050 М по H2SO4? Допущение:
суммарный объем при смешении не изменяется.
35. Необходимо приготовить рабочие стандартные растворы глюкозы с кон-
центрацией 1,00 • 10~5, 2,00 • 105, 5,00 • 10-5 и 1,00 • 10-4 М из исходного
0,100 М раствора. В вашем распоряжении имеются мерные колбы объемом
100 мл и пипетки объемом 1,00, 2,00, 5,00 и 10,00 мл. Опишите схему при-
готовления рабочих растворов.
36. Для спектрофотометрического определения марганца пробу массой 0,500 г
растворили в кислоте и разбавили в мерной колбе до 250 мл. Из полученно-
го раствора отобрали три аликвоты по 50 мл в конические колбы объемом
500 мл и добавили окислитель пероксодисульфат калия для окисления мар-
ганца до перманганата. По завершении реакции каждую смесь количест-
венно перенесли в мерную колбу объемом 250 мл, разбавили до метки и
измерили светопоглощение, обусловленное перманганат-ионами. Среднее
значение концентрации, определенное при помощи градуировочного гра-
фика, составило 1,25 • 10 5 М. Какова массовая доля (%) марганца в пробе?
268
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
Расчеты при стандартизации растворов
37. Известно, что препарат технической соды содержит 98,6% Na^O^ На тит-
рование 0,678 г этого препарата требуется 36,8 мл раствора серной кисло-
ты. Какова мольная концентрация кислоты в этом растворе?
38. Для стандартизации раствора гидроксида натрия приблизительной концен-
трации 0,1 М используют первичный стандарт — сульфаминовую кислоту
(NH2SO3H). Какую навеску сульфаминовой кислоты следует взять, чтобы
на ее титрование пошло около 40 мл раствора NaOH?
Расчет результатов анализа
39. Степень чистоты образца особо чистой лимонной кислоты (Н3С6Н5О7, три
титруемых протона) определяли титрованием раствором NaOH. На титро-
вание навески массой 0,2678 г израсходовано 38,31 мл 0,1087 М раствора
NaOH. Каково содержание основного вещества в препарате (для особо чис-
той лимонной кислоты требуется не менее 99,5%)?
40. На титрование кальция в пробе сыворотки крови объемом 200 мкл потребо-
валось 2,47 мл 1,87 • 10-4 М раствора ЭДТА. Какова концентрация (в мг/дл)
кальция в крови?
41. На титрование 0,372 г загрязненного ВаС12 • 2Н2О потребовалось 27,2 мл
0,100 М AgNO3. Рассчитайте: (а) массовую долю (%) С1 в образце и (б) сте-
пень чистоты вещества (%).
42. Для определения содержания железа в руде образец растворили в кислоте,
восстановили все железо до Fe2+ и оттитровали 0,0150 М стандартным рас-
твором К2Сг2О7. На титрование железа, содержащегося в пробе массой
1,68 г, потребовалось 35,6 мл раствора. Какова массовая доля железа (%) в
образце в пересчете на Fe2O3? Уравнение реакции титрования см. в приме-
ре 5.37.
43. Для определения кальция пробу массой 2,00 г растворили, осадили кальций
в виде СаС2О4, осадок растворили в кислоте и оттитровали оксалат-ионы
35,6 мл 0,0200 М КМпО4. Какова массовая доля кальция в пробе (%) в пере-
счете на СаО? При титровании протекает реакция
5Н2С2О4 + 2МпО; + 6Н+ -> 10СО2 + 2Мп2+ + 8Н2О.
44. Раствор перманганата калия приготовили растворением 4,68 г КМпО4 в
воде с последующим разбавлением до 500 мл. Какой объем (мл) этого рас-
твора потребуется для взаимодействия с железом, находящимся в 0,500 г
руды с содержанием 35,6% Fe2O3? Уравнение реакции титрования см. в
примере 5.29.
45. Образец содержит ВаС12 и инертное вещество. Какую навеску образца сле-
дует взять, чтобы объем 0,100 М AgNO3 (в мл), израсходованный на титро-
вание, был численно равен массовой доле (%) ВаС12 в образце?
ЗАДАЧИ
269
46- На титрование 0,250 г загрязненного А1С13 израсходовано 48,6 мл 0,100 М
раствора AgNO3. Какой объем 0,100 М раствора ЭДТА понадобится для
титрования 0,350 г этого препарата? ЭДТА взаимодействует с А13+ в соот-
ношении 1:1.
47- Навеску чистой одноосновной органической кислоты массой 425,2 мг от-
титровали 28,78 мл 0,1027 М раствора NaOH. Чему равна молярная масса
кислоты?
48- Степень чистоты Zn(OH)2 определили при помощи титрования соляной
кислотой. На титрование навески массой 0,287 г израсходовано 37,8 мл рас-
твора НС1, который стандартизовали гравиметрически, осадив хлорид-
ионы в виде AgCl. Из аликвоты НС1 объемом 25,0 мл получено 0,462 г
AgCl. Какова степень чистоты Zn(OH)2?
49- На титрование навески чистого КНС2О4 • Н2С2О4 • 2Н2О (содержит три тит-
руемых протона) требуется 46,2 мл 0,100 М NaOH. Какой объем (мл)
0,100 М КМпО4 требуется на титрование такой же навески? Уравнение ре-
акции титрования см. в условии задачи 43.
Обратное титрование
50- Образец содержит Na2CO3 и инертное вещество. К навеске образца массой
0,500 г добавили 50,0 мл 0,100 М НС1, прокипятили для удаления СО2 и от-
титровали избыток кислоты 0,100 М раствором NaOH. На титрование из-
расходовано 5,6 мл раствора. Какова массовая доля (%) Na^Oj в образце?
51 - Для определения пероксида водорода к навеске анализируемого раствора
массой 0,587 г добавили 25,0 мл стандартного раствора 0,0215 М КМпО4,
взятого в небольшом избытке, непрореагировавший КМпО4 оттитровали
5,10 мл стандартного раствора 0,112 М Fe2+. Какова массовая доля (%) Н2О2
в образце? Уравнения протекающих в ходе определения реакций см. в при-
мерах 5.25 и 5.29.
52. Определение серы в стали основано на превращении серы в H2S, поглощении
сероводорода стандартным раствором иода (10,0 мл 0,00500 М 12) и титро-
вании избытка иода раствором тиосульфата натрия. На титрование израс-
ходовано 2,6 мл 0,00200 М раствора Na2S2O3. Сколько миллиграммов серы
содержалось в образце? В ходе определения протекают следующие реакции:
H2S +12 -+ S + 21- + 2Н+
I2 + 2S2O^~ -> 21- + S4O^‘
Титр
53- Выразите титр 0,100 М раствора ЭДТА в виде мг ВаО/мл.
54- Выразите титр 0,0500 М раствора КМпО4 в виде мг Ре2О3/мл.
55- Титр раствора нитрата серебра составляет 22,7 мг Cl/мл. Чему равен его
титр, выраженный в виде мг Вг/мл?
270
СТЕХИОМЕТРИЧЕСКИЕ РАСЧЕТЫ - ОСНОВНОЙ РАБОЧИЙ ИНСТРУМЕНТ АНАЛИТИКА
Расчеты эквивалентных масс
56- Рассчитайте эквивалентные массы следующих веществ, реагирующих как
кислоты или основания: (а) НС1, (б) Ва(ОН)2, (в) КН(Ю3)2, (г) H2SO3,
(д) НС2Н3О2 (уксусная кислота).
57- Рассчитайте значения мольных концентраций (молярностей) 0,250 N рас-
творов всех кислот и оснований, упомянутых в условии задачи 56.
Расчеты с использованием эквивалентной массы
58. Рассчитайте значения эквивалентной массы КНС2О4, реагирующего: (а) как
кислота, (б) как восстановитель в реакции с МпО4 в соответствии с уравне-
нием 5НС2О; + 2МпО; + 11Н+ -> 10СО2 + 2Мп2+ + 8Н2О.
59. Оксид ртути HgO можно определить при помощи реакции с иодид-ионами
(HgO + 41" + Н2О -+ Hgl4" + 2ОН) и последующего титрования кислотой.
Чему в этом случае равна эквивалентная масса HgO?
60. Рассчитайте массу одного моля эквивалентов (г) следующих веществ при-
менительно к указанным реакциям: (a) FeSO4 (Fe2+ -+ Fe3+), (б) H2S (—> S°),
(в) H2o2 Н О2), (г) Н2О2 (->Н2О).
61. Для титрования Ag+ (с образованием AgCl) можно использовать раствор
ВаС12 • 2Н2О. Сколько миллимолей эквивалентов содержится в 0,5000 г
ВаС12 • 2Н2О?
Нормальность (эквивалентная концентрация)
62. В воде растворили 7,82 г NaOH и 9,26 г Ва(ОН)2 и разбавили полученный рас-
твор до 500 мл. Какова нормальность этого раствора как раствора основания?
63. Какая масса триоксида мышьяка As2O3 необходима для приготовления 1 л
0,1000 N раствора As(III), окисляющегося в ходе окислительно-восстано-
вительной реакции до As (V)?
64. 2,73 г КНС2О4 • Н2С2О4 (три диссоциирующих протона), содержащего
2,0% инертных загрязнений, и 1,68 г КНС8Н4О4 (один диссоциирующий
протон) растворили в воде и разбавили раствор до 250 мл. Какова нормаль-
ность этого раствора как кислоты в предположении, что все диссоциирую-
щие протоны взаимодействуют с основанием?
65. Раствор КНС2О4 • Н2С2О4 • 2Н2О (три диссоциирующих протона) имеет
концентрацию 0,200 N как кислота. Какова нормальность этого раствора
как восстановителя, реагирующего в соответствии с уравнением, приве-
денным в условии задачи 43?
66. Na2C2O4 и КНС2О4 • Н2С2О4 смешали в таком соотношении, что нормаль-
ность полученного раствора как восстановителя (уравнение реакции см. в
условии задачи 43) в 3,62 раза больше его нормальности как кислоты. Како-
во массовое соотношение компонентов в смеси?
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
271
67. Какую навеску К2Сг2О7 следует взять для приготовления 1,000 л его 0,1000 N
раствора применительно к реакции Сг2О7“ + 14Н+ + бе -» 2Сг3+ + 7Н2О ?
Расчеты эквивалентных концентраций ионов
68. Концентрация хлорида в растворе составляет 300 мг/дл. Чему равна его
концентрация, выраженная в ммоль экв./л?
69. Концентрация кальция в растворе составляет 5,00 ммоль экв./л. Чему равна
его концентрация, выраженная в мг/дл?
70. Содержание хлоридов в пробе мочи составляет 150 ммоль экв./л. Если
предположить, что хлориды присутствуют в моче в виде хлорида натрия,
какова будет концентрация NaCl в этой пробе, выраженная в г/л?
Расчеты в гравиметрии
71. Чему равна масса марганца, содержащегося в 2,58 г Мп3О4?
72. Для определения цинка его осадили и взвесили в виде Zn2Fe(CN)6.
(а) Какова масса цинка в пробе, из которой получено 0,348 г осадка?
(б) Какую массу осадка можно получить из 0,500 г цинка?
73. Рассчитайте величину гравиметрического фактора для следующих систем:
Искомое вещество Гравиметрическая форма
Мп Мп3О4
Mn2O3 Мп3О4
Ag2S BaSO4
CuCl2 AgCl
Mgl2 Pbl2
Рекомендуемая литература
1. Т. R. Hadjiioannou, G. D. Christian, С. Е. Efstathiou, and D. Nikolelis, Problem
Solving in Analytical Chemistry. Oxford: Pergamon, 1988.
2. Q. Fernando and M. D. Ryan, Calculations in Analytical Chemistry. New York:
Harcourt Brace Jovanovich, 1982.
3. M. R. F. Ashworth, Titrimetric Organic Analysis. New York: Interscience, 1964.
Глава 6
ОСНОВНЫЕ ПОЛОЖЕНИЯ ТЕОРИИ ХИМИЧЕСКОГО
РАВНОВЕСИЯ
Худшая форма неравности есть
попытка сделать неравные вещи равными.
Аристотель
Химические реакции никогда не идут только в одном направлении, даже если
они могут протекать почти до конца. Другими словами, между компонентами
реакции устанавливается равновесие, при котором скорости реакций, идущих в
прямом и обратном направлении, равны. В этой главе мы рассмотрим понятия
«равновесие» и «константа равновесия», а также изучим общие подходы к рас-
четам с использованием констант равновесия. Мы обсудим активность ионных
частиц и научимся вычислять коэффициенты активности. Эти величины нужны
для расчетов с использованием термодинамических констант устойчивости, в
частности, при рассмотрении влияния посторонних ионов, описанного в конце
этой главы. Также их используют для расчетов в потенциометрии (гл. 13).
6.1. Химические реакции: понятие скорости реакции
В 1863 г. Н. Гульдберг и П. Вааге сформулировали то, что мы теперь называем
законом действующих масс, согласно которому скорость химической реакции
пропорциональна «действующим массам» реагирующих веществ. Гульдберг и
Вааге вывели константу равновесия, определив равновесие как условие, при ко-
тором скорости прямой и обратной реакций равны. Рассмотрим химическую ре-
акцию
аА + 6В ^=г сС + dD
(6.1)
Согласно Гульдбергу и Вааге, скорость прямой реакции равна константе, умно-
женной на концентрацию каждой частицы в степени, равной числу молекул,
участвующих в реакции*:
7=^[А?[Вр
(6.2)
Квадратные скобки [ ] обозначают в данном случае эффективную концентрацию, выраженную в единицах
моль/литр. Эффективная концентрация будет обсуждаться при рассмотрении активностей в разделе о вли-
янии посторонних ионов.
6.1. ХИМИЧЕСКИЕ РЕАКЦИИ: ПОНЯТИЕ СКОРОСТИ РЕАКЦИИ
273
где ту — скорость прямой реакции, а к?- константа скорости, которая зависит
от таких факторов, как температура и присутствие катализаторов. [А] и [В] обо-
значают молярную концентрацию реагентов А и В, выраженную в единицах
моль/литр. Подобным же образом Гульдберг и Вааге записали уравнение для
обратной реакции:
rh = ^[CF[D]^ (6.3)
Для равновесной системы скорости прямой и обратной реакций равны:
^[ahbWJCHdH (6.4)
Преобразуя эти уравнения, получаем молярную константу равновесия реак-
ции К (справедлива только для разбавленных растворов):
[C]C[D]J _kf _К
[A]a[B]h kb
(6-5)
Выражение (6.5) представляет собой правильное выражение константы равно-
весия, но способ получения этого выражения не всегда применим. Это связано с
тем, что в действительности скорость реакции зависит от механизма реакции,
определяемого числом сталкивающихся частиц, тогда как выражение константы
равновесия зависит только от стехиометрии химической реакции. Сумма показа-
телей степени в выражении для скорости реакции указывает на порядок реакции
и его величина может отличаться от стехиометрических коэффициентов (см. гл. 22).
Примером может служить реакция восстановления тиосульфата иодидом:
sp*’ + 31--> 2SO^ +I3
В действительности скорость реакции равна ^[S2O][I-] (реакция второго по-
рядка), а не ^[S2Og~][I-]3, как можно было бы ожидать из уравнения химической
реакции (реакция имела бы четвертый порядок). Единственное разумное обоснова-
ние выражения константы равновесия дает термодинамический подход, в осно-
ве которого лежит очень важная характеристика процесса — энергия Гиббса.
В разд. 6.3 рассмотрено ее использование для расчета значений термодинамиче-
ских констант равновесия.
Величину К можно рассчитать, измерив концентрации А, В, С и D в состоя-
нии равновесия. Заметим, что чем выше скорость прямой реакции по сравнению
со скоростью обратной реакции, тем больше величина константы равновесия и
тем сильнее будет смещено равновесие реакции вправо.
В начальный период реакции между А и В скорость прямой реакции боль-
шая, так как концентрации А и В еще высоки; при этом обратная реакция идет
медленно вследствие очень малых значений С и D (скорость этой реакции пер-
воначально равна нулю). По мере протекания реакции концентрации А и В уме-
ньшаются, а С и D — увеличиваются, что приводит к снижению скорости
274
ОСНОВНЫЕ ПОЛОЖЕНИЯ ТЕОРИИ ХИМИЧЕСКОГО РАВНОВЕСИЯ
§ g Изменение Равновесие
РИС. 6.1. Протекание химической реакции
прямой реакции и повышению скорости обратной реакции (рис. 6.1). В конеч-
ном счете, скорости обеих реакций становятся равными и система достигает со-
стояния равновесия. В этой точке концентрации каждого из компонентов А, В,
С и D остаются постоянными (относительные значения концентраций будут за-
висеть от стехиометрии реакции и от того, насколько сильно реакция смещена
вправо). Однако система остается в динамическом равновесии, при котором
прямая и обратная реакции протекают с одинаковой скоростью.
Выражение для константы равновесия представляет собой отношение, в ко-
тором концентрации продуктов реакции находятся в числителе, а концентрации
исходных веществ — в знаменателе. Такая запись является условной, но по до-
говоренности широко используется. Большая величина константы равновесия
указывает на то, что равновесие сильно смещено вправо.
Следует отметить, что конкретная реакция с большой константой равнове-
сия может протекать справа налево, если в начальный момент концентрация
продуктов реакции достаточно велика.
Кроме того, величина константы равновесия ничего не говорит о том, на-
сколько быстро реакция достигнет равновесия. Большая величина константы
равновесия не гарантирует того, что реакция будет протекать с заметной
скоростью. Некоторые реакции в действительности протекают настолько мед-
ленно, что это невозможно зафиксировать. Значение константы равновесия про-
сто говорит о возможности и направлении протекания реакции, а не о том,
достаточно ли реакция быстрая, чтобы ее можно было применить на практике.
(См. гл. 22, посвященную кинетическим методам анализа для измерения скоро-
стей реакций и их применения для анализа.)
Для реакции (6.1) равновесие будет достигнуто, скорее всего, при разных
скоростях прямой и обратной реакций. Например, если сначала смешать С и D,
то равновесие может быть достигнуто медленнее или быстрее, чем для обратной
реакции.
6.2. ТИПЫ РАВНОВЕСИЙ
275
6.2. Типы равновесий
Мы можем записать выражение константы равновесия для многих химических
процессов. Примеры различных типов равновесных процессов приведены в
табл. 6.1. К равновесным процессам относятся диссоциация (кислот/основа-
ний, растворение), образование комплексов, окислительно-восстановитель-
ные реакции, процессы распределения вещества между двумя фазами (вода и
неводный растворитель — жидкостная экстракция; адсорбция из воды на поверх-
ность, как в хроматографии, и другие). Некоторые из этих равновесных процес-
сов мы обсудим ниже и в следующих главах.
6.3. Свободная энергия Гиббса и константа равновесия
Возможность протекания реакции термодинамически определяется по измене-
нию ее энтальпии (AZ7) и энтропии (AS). Энтальпия — это теплота, погло-
щенная в результате эндотермической реакции, протекающей при постоянном
давлении. При выделении теплоты (экзотермическая реакция), ДЯ имеет отри-
цательное значение. Энтропия — это мера беспорядка, или хаотичности, веще-
ства или системы.
Система всегда стремится к снижению энергии и увеличению степени хао-
тичности, т. е. к уменьшению энтальпии и увеличению энтропии. Например,
камень, лежащий на вершине холма самопроизвольно скатится вниз (состояние
с более низкой энергией), а шарики, уложенные в коробку по цвету, при встря-
хивании расположатся в хаотическом порядке. Суммарное влияние энтальпии и
энтропии выражается свободной энергией Гиббса, G:
G = H-TS
(6.6)
Таблица 6.1
Типы равновесий
Равновесий процесс Реакция Константа равновесия
Диссоциация кислот и оснований НА + Н2О Н3О+ + А- Ка, константа кислотности
Растворение осадка МА Мл+ + Ал“ ПР, произведение растворимости
Комплексообразование Мл+ + aL’’- ML("-afc)+ /Суст, константа устойчивости комплекса
Окислительно-восстановительная реакция А . + В ^=7 А + В j red ox ox red -Кравн» константа равновесия реакции
Распределение между фазами АщО Т A-organic KD, коэффициент распределения
276
ОСНОВНЫЕ ПОЛОЖЕНИЯ ТЕОРИИ ХИМИЧЕСКОГО РАВНОВЕСИЯ
где Т — абсолютная температура в градусах Кельвина; G — мера энергии систе-
мы, а система самопроизвольно стремится к состоянию с более низкой энергией.
Изменение энергии системы при постоянной температуре можно выразить урав-
нением:
АС = А#-ТА5 (6.7)
Таким образом, процесс будет самопроизвольным, если значение AG отри-
цательное, обратный процесс будет самопроизвольным, если значение AG по-
ложительное, равновесие установится при AG равном нулю. Следовательно,
протеканию реакции благоприятствует выделение теплоты (АН имеет отрица-
тельное значение), как в экзотермических реакциях, и увеличение энтропии
(положительное значение AS). АН и AS могут быть и положительными, и отрица-
тельными, но от относительного вклада каждой величины и от температуры за-
висит, будет ли AG отрицательным и, соответственно, будет ли реакция
самопроизвольной.
Стандартная энтальпия Н°, стандартная энтропия S° и стандартная свобод-
ная энергия G° представляют собой термодинамические величины, выраженные
для одного моля вещества, находящегося в стандартных условиях (Р = 1 атм,
Т= 298 К, концентрация равна единице). Тогда,
AG° =AH°-TAS° (6.8)
Взаимосвязь AG° с константой равновесия реакции отражается следующим
уравнением:
K=e~^G°/RT (6.9)
ИЛИ
AG° = -RT\n К = -2,303RT \gK (6.10)
где R — газовая постоянная (8,314 Дж К-1моль-1).
Следовательно, если известна стандартная свободная энергия реакции, мож-
но рассчитать константу равновесия. Очевидно, что чем более отрицательно зна-
чение AG°, тем больше будете. Отметим, что AG° и AG несут информацию о том,
может ли реакция протекать самопроизвольно, но не несут никакой информации
о скорости протекания этой реакции.
6.4. Прицип ЛеШателье
Равновесные концентрации реагирующих веществ и продуктов реакции можно
изменить, оказав на систему определенное воздействие, например, изменив тем-
пературу, давление или концентрацию одного из компонентов системы. Послед-
ствия таких изменений могут быть предсказаны на основании принципа
Ле Шателье: если на систему, находящуюся в состоянии химического равнове-
6.7. ВЛИЯНИЕ КОНЦЕНТРАЦИИ НА ПОЛОЖЕНИЕ РАВНОВЕСИЯ
277
сия, оказать воздействие, равновесие сместится таким образом, чтобы умень-
шить или нейтрализовать эффект от воздействия. Рассмотрим подробно
влияние температуры, давления и концентрации на химическое равновесие.
6.5. Влияние температуры на константу равновесия
Как было отмечено, температура влияет на константу скорости прямой и обрат-
ной реакций, и, следовательно, на константу равновесия [или, что более коррек-
тно, температура влияет на свободную энергию — см. уравнение (6.10)].
Повышение температуры сместит равновесие в направлении, в котором проис-
ходит поглощение теплотыа, в результате произойдет устранение эффекта ока-
занного воздействия. Так, в случае прямой эндотермической реакции (протекает с
поглощением теплоты) равновесие сместится вправо, что приведет к увеличению
константы равновесия. Обратное утверждение справедливо, если прямая реакция
является экзотермической. Степень смещения зависит от количества теплоты.
Кроме того, что температура влияет на положение равновесия, она также
довольно сильно влияет на скорости прямой и обратной реакций, и поэтому тем-
пература влияет на скорость, с которой устанавливается равновесие. Это объяс-
няется тем, что число и энергия столкновений между реагирующими частицами
увеличиваются с повышением температуры. Скорость многих эндотермических
реакций возрастает в два-три раза при повышении температуры на каждые 10 °C.
6.6. Влияние давления на положение равновесия
Давление сильно влияет на положение химического равновесия для реакций,
происходящих в газовой фазе. Увеличение давления способствует смещению
равновесия в направлении уменьшения объема системы.
Но для реакций, протекающих в растворах, изменение давления незначитель-
но влияет на положение равновесия, так как жидкости нельзя сжать так сильно,
как газы.
6.7. Влияние концентрации на положение равновесия
Концентрация исходных веществ и продуктов реакции не влияет на величину
константы равновесия, но оказывает влияние на положение равновесия. Направ-
ление смещения равновесия легко предсказать, исходя из принципа Ле Шателье.
Рассмотрим реакцию железа(Ш) с иодид-ионом:
31- + 2Ре3+^1з +2Fe2+
Если система находится в состоянии равновесия, определяемом константой рав-
новесия, то добавление или удаление одного из компонентов приведет к нару-
278
ОСНОВНЫЕ ПОЛОЖЕНИЯ ТЕОРИИ ХИМИЧЕСКОГО РАВНОВЕСИЯ
шению и затем повторному установлению равновесия. Например, мы увеличиваем
концентрацию ионов железа(П). Исходя из принципаЛе Шателье, равновесие смес-
тится влево, чтобы снизить эффект воздействия (увеличения концентрации про-
дукта реакции). В конечном счете, равновесие восстановится и его положение
будет определяться той же величиной константы равновесия.
6.8. Катализаторы
Катализаторы повышают либо снижают скорость, с которой устанавливается
равновесие, влияя на скорости как прямой, так и обратной реакций. Но катали-
заторы в одинаковой степени влияют на скорости обеих реакций и поэтому их
применение не приводит к изменению величины константы равновесия.
Катализаторы имеют большое значение для аналитической химии, так как
используются для ускорения некоторых важных аналитических реакций, иду-
щих слишком медленно при нормальных условиях. (Об использовании в анали-
тических целях ферментов см. гл. 22.) Примером может служить применение
тетраоксида осмия для ускорения реакции титрования мышьяка(Ш) церием(1У);
константа равновесия этой реакции достаточно велика, но скорость в обычных
условиях слишком мала, чтобы реакцию можно было использовать для титрова-
ния. Измерение увеличения скорости медленной реакции в присутствии катали-
затора можно использовать для определения концентрации катализатора.
6.9. Полнота протекания реакций
Если равновесие реакции смещено вправо настолько, что оставшееся количест-
во определяемых (исходных) веществ слишком мало, чтобы его можно было
определить используемым методом анализа, говорят, что реакция прошла до
конца. Реакцию,равновесие которой смещено вправо на 75%, считают прошед-
шей до конца. Но для проведения точных измерений в количественном анализе
равновесие реакции должно быть смещено вправо как минимум на 99,9°/о. Если
же равновесие не так сильно смещено вправо, можно применить принцип
Ле Шателье. Можно либо увеличить концентрацию реагирующего вещества,
либо уменьшить концентрацию продукта реакции. Получение большего количе-
ства продукта реакции может быть достигнуто следующими способами: (1) уда-
лением газообразного продукта реакции, (2) осаждением продукта реакции,
(3) путем образования в растворе устойчивого комплекса продукта реакции,
(4) с помощью избирательной экстракции.
Из сказанного следует, что принцип Ле Шателье лежит в основе управления
химическими реакциями. Это особенно важно для биохимических реакций, на
равновесие которых могут оказывать значительное влияние такие внешние фак-
торы как температура. Катализаторы (ферменты), как будет показано в гл. 22,
играют ключевую роль во многих биологических и физиологических процессах.
6.10. КОНСТАНТЫ РАВНОВЕСИЯ ПРОЦЕССОВ ДИССОЦИАЦИИ И АССОЦИАЦИИ
279
6.10. Константы равновесия процессов диссоциации
и ассоциации: слабые электролиты и осадки
При растворении в воде вещество в большинстве случаев частично или полностью
диссоциировано, или ионизировано. Частично диссоциированные электролиты на-
зывают слабыми электролитами, а полностью диссоциированные электролиты —
сильными электролитами. Например, уксусная кислота только частично иони-
зируется в воде и поэтому является слабым электролитом. Хлороводородная
кислота полностью ионизируется и поэтому является сильным электролитом.
(Реакции диссоциации кислот в воде в действительности представляют собой
реакции переноса протона НО Ас + Н2О ±=? Н3О+ + О Ас-.) Некоторые вещества
полностью ионизируются в воде, но имеют ограниченную растворимость; их на-
зывают малорастворимыми. Вещества могут соединяться в растворе с образо-
ванием диссоциирующего продукта, например, комплекса. Примером может
служить реакция меди(П) с аммиаком, продуктом которой являются частицы
Cu(NH3)*+.
Слабый электролит диссоциирует только частично. Малорастворимое ве-
щество — это сильный электролит, так как та его часть, которая растворяется,
диссоциирует полностью. Диссоциацию слабых электролитов или раствори-
мость малорастворимых веществ можно количественно описать с помощью
констант равновесия. Константы равновесия для полностью растворимых и
полностью диссоциированных электролитов представляют собой бесконечно
большие величины. Константа равновесия является конечным числом, если дис-
социация протекает менее чем на 100%. Рассмотрим диссоциацию частицы АВ:
АВ^А + В (6.11)
Константу равновесия можно записать в общем виде так:
[А][В]_ [АВ] равн (6.12)
Чем больше величина тем в большей степени пройдет диссоциация. Напри-
мер, чем больше будет константа равновесия для кислоты, тем сильнее кислота.
Некоторые частицы диссоциируют ступенчато, и для каждой ступени дис-
социации можно записать выражение константы равновесия. Соединение А2В
диссоциирует следующим образом:
А2В^А + АВ Кх = -[~][АВ] [А2В] (6.13)
АВ^А + В К [АВ] (6.14)
280
ОСНОВНЫЕ ПОЛОЖЕНИЯ ТЕОРИИ ХИМИЧЕСКОГО РАВНОВЕСИЯ
Диссоциация соединения представляет собой сумму этих двух процессов:
А2В 2А + В
_ [А]2[АВ]
равн [А2В]
(6.15)
Общая константа равновесия может быть получена перемножением уравнений
(6.13) и (6.14):
г г г - - ^[АВ]
Аравн А1А2 га г га
(6.16)
Если частицы вещества диссоциируют ступенчато, как в этом примере, как пра-
вило, значения последовательных констант равновесия постепенно уменьшают-
ся. Следует отметить, что при расчетах равновесий в качестве единицы измерения
концентрации вещества в растворе всегда используется моль/л.
Если реакция записана в обратной форме, равновесие устанавливается то
же, но выражение константы равновесия будет обратное. Так, для приведенного
выше примера, А + АВ, Яравн (обр) = [АВ]/([А][В]) = 1Жравн (прям). Если £равн
для прямой реакции равна 105, то Х’ц для обратной реакции будет 10-5.
Эти положения применимы и к процессам ассоциации частиц, только
обычно в этом случае величина константы равновесия будет скорее больше
единицы, чем меньше, так как такие реакции смещены в сторону образования
продукта (например, комплекса). Константы кислотно-основного равновесия,
комплексообразования и растворения осадков рассматриваются в соответству-
ющих главах.
6.11. Расчеты с использованием констант равновесия:
каковы равновесные концентрации?
Константы равновесия используют для расчета равновесных концентраций раз-
личных частиц, например, для нахождения концентрации ионов водорода при
диссоциации слабой кислоты. В данном разделе мы рассмотрим общий подход к
расчету с использованием констант равновесия. Приемы, используемые для рас-
чета специфических равновесий, обсуждаются в соответствующих главах.
Химические реакции
Иногда возникает необходимость в знании равновесных концентраций реаген-
тов и продуктов химической реакции. Это, например, может понадобиться для
построения кривой титрования или для расчета потенциала электрода в растворе.
6.11. РАСЧЕТЫ С ИСПОЛЬЗОВАНИЕМ КОНСТАНТ РАВНОВЕСИЯ
281
Пример 6.1
Вещества А и В реагируют с образованием веществ С и D:
A + B^C + D =
[А][В]
Константа равновесия К равна 0,30. Предположим, что реакция протекает меж-
ду 0,20 моль А и 0,50 моль В, растворенных в 1 л. Рассчитайте равновесные кон-
центрации реагентов и продуктов реакции.
Решение
Исходные концентрации А и В составляют 0,20 М и 0,50 М соответственно, тог-
да как исходные концентрации С и D равны нулю. К моменту наступления рав-
новесия концентрации А и В уменьшатся, а концентрации С и D увеличатся.
Пусть х — равновесная концентрация С или количество прореагировавших
А и В в моль/л. Поскольку при образовании каждого моля С образуется один
моль D, то равновесная концентрация D тоже будет х. Исходные концентрации
А и В можно рассматривать как аналитические концентрации и обозначить их
как СА и Св. Равновесные концентрации этих компонентов обозначаются
[А] и [В]. Концентрации А и В уменьшатся на величину х, следовательно
[А] = СА - х и [В] = Св - х. Равновесную концентрацию получают вычитанием
количества вещества, выраженного в единицах концентрации, прореагировав-
шего из исходной (аналитической) концентрации. Тогда равновесные концент-
рации можно выразить следующим образом:
[А] [В] [С] [D]
Исходная концентрация 0,20 0,50 0 0
Изменение (х, ммоль/мл) -х —х +х +х
Равновесная концентрация 0,20-х 0,50 -х X X
Можно подставить эти значения в выражение для константы равновесия и
решить полученное уравнение относительно х:
-------—--------= 0,30
(0,20-х)(0,50-х)
х2 = (0,10-0,70х+ х2)0,30
0,70х2 +0,21х-0,03 =0
282
ОСНОВНЫЕ ПОЛОЖЕНИЯ ТЕОРИИ ХИМИЧЕСКОГО РАВНОВЕСИЯ
Это квадратное уравнение можно решить относительно х, используя формулу
для нахождения корней квадратного уравнения, приведенную в Приложении В:
-b±^Jb2 -4ас
х =--------------=
2а
_-0,21±70,212 - 4 0,70 ОДЗО _
2 0,70
-0,21 ± JO,044 + 0,084 П1, „
1,40
[А] = 0,20 - х = 0,09 М
[В] = 0,50-х = 0,39 М
[С] = [D] =х = 0,11 М
Другим способом нахождения х является метод последовательных при-
ближений. При использовании этого метода сначала следует пренебречь вели-
чиной х по сравнению с исходными концентрациями для упрощения расчетов
(принять аналитическую концентрацию за равновесную) и рассчитать исходное
значение х. Затем вычесть полученное значение х из СА и Св, чтобы оценить рав-
новесные концентрации А и В, и рассчитать новое значение х.
Расчет повторяют до тех пор, пока значение х не станет постоянным.
Первый расчет —— = 0,30
0,20 0,50
х=0,173
Расчеты сходятся гораздо быстрее, если все время сохранять дополнитель-
-----------—-------------= 0,30
(0,20-0,173)-(0,50-0,173)
х= 0,051
-----------—-------------= 0,30
(ОДО-0,051) (0,50-0,051)
х=0,142
-----------—-------------= 0,30
(0,20-0,142)-(0,50-0,142)
х = 0,079
-----------—-------------=0,30
(ОДО - 0,079) • (0,50 - 0,079)
х =0,124
ную значащую цифру.
Второй расчет
Третий расчет
Четвертый расчет
Пятый расчет
6.11. РАСЧЕТЫ С ИСПОЛЬЗОВАНИЕМ КОНСТАНТ РАВНОВЕСИЯ
283
Шестой расчет — = 0,30 (0,20-0,124) (0,50-0,124) х= 0,093
Седьмой расчет — = 0,30 (0,20 - 0,093) • (0,50 - 0,093) х=0,114
Восьмой расчет — = 0,30 (0,20-0,114) (0,50-0,114) х = 0,104
Девятый расчет — = 0,30 (0,20-0,100) (0,50-0,100) х = 0,110
Будем считать равновесное значение х равным 0,11, так как это значение совпа-
дает с результатом седьмого расчета. Следует отметить, что в этих итерациях
значения х колеблются относительно равновесной величины. Приближение
значения х к значению С приведет к необходимости выполнения большого ко-
личества итераций, обусловленной колебанием искомой переменной (рассмот-
ренный пример не самый лучший для использования этого метода). Существует
более эффективный путь завершения итерации. Возьмите среднее арифметиче-
ское от первой и второй итерации для проведения третьей, которая дает значе-
ние близкое к окончательному (в данном случае, 0,112). Еще одна или две
итерации покажут, что мы достигли равновесной величины. Попробуйте этот
способ!
В примере 6.1 к моменту наступления равновесия осталось значительное
количество вещества А даже при избытке В; это связано с тем, что величина кон-
станты равновесия не очень большая. В большинстве реакций, имеющих значе-
ние для аналитической химии, константы равновесия имеют большие величины,
и равновесие очень сильно смещено вправо. В этих случаях равновесные кон-
центрации реагентов, если их берут не в избытке, обычно очень малы по сравне-
нию с концентрациями других веществ. Это упрощает расчеты.
Excel, Поиск решения: решение квадратного уравнения
Электронные таблицы Microsoft Excel содержат очень полезную программу для
решения уравнений — Поиск решения (Solver). Чтобы использовать эту про-
грамму, следует сначала переписать уравнение в виде формулы, при вычисле-
нии дающей известную величину. Поиск решения выполняет вычисления по
284
ОСНОВНЫЕ ПОЛОЖЕНИЯ ТЕОРИИ ХИМИЧЕСКОГО РАВНОВЕСИЯ
этой формуле в обратном порядке, с помощью итерационного метода подбирая
одно или несколько входящих в формулу значений переменных так, чтобы в ре-
зультате вычислений получился заданный ответ. Подобные программы есть и в
других электронных таблицах.
Итак, для работы с программой Поиск решения следует записать уравнение
в виде формулы, правая часть которой представляет собой постоянное значение,
а левая — выражение, включающее неизвестные, искомые, величины. Это мо-
жет быть выражение, которое требуется минимизировать (как в методе наимень-
ших квадратов), максимизировать, приравнять нулю или любой другой
постоянной величине. В примере 6.1. мы уже переписали выражение константы
равновесия именно таким образом — в виде формулы, в левой части которой
стоит алгебраическое выражение, а в правой — нуль: 0,70х2 + 0,21х - 0,30 = 0.
Она содержит единственную переменную — х.
Как правило, вызов программы Поиск решения находится в меню Сервис
(Tools) программы Excel. Если его там нет, он может находиться в меню Над-
стройки (Add-In). При вызове программы Поиск решения открывается
следующее диалоговое окно для задачи, приведенной в примере 6.1:
Диалоговое окно содержит три окошка ввода параметров. Одно из них озаглав-
лено как Установить целевую ячeйкy(Set Target Cell). В это окошко
6.11. РАСЧЕТЫ С ИСПОЛЬЗОВАНИЕМ КОНСТАНТ РАВНОВЕСИЯ
285
следует ввести адрес ячейки, содержащей расчетную формулу (символы $ в ад-
ресе можно не указывать). Если выделить мышью соответствующую ячейку (до
или после вызова Поиска решения), ее адрес будет введен автоматически. Вто-
рое окошко озаглавлено значению (Equal То). В него необходимо ввести то
значение, которое должно получиться в результате вычисления по этой формуле
(в данном случае это 0). Третье окошко озаглавлено Изменяя ячейки (By
Changing Cells). В него вводят адреса ячеек, в которых находятся величины
искомых переменных (в данном случае х). Если переменных более одной, адреса
соответствующих ячеек разделяют запятыми. Поскольку значение х заранее не-
известно, соответствующую ячейку можно оставить пустой или ввести в нее
произвольное значение — оценку искомой величины.
Для решения поставленной задачи создайте электронную таблицу, содер-
жащую в соответствующих ячейках все значения, которые будут использованы
в расчетах (коэффициенты квадратного уравнения а, b и с, равные соответствен-
но 0,70,0,21 и -0,03). Выберите ячейку для величины х и введите в одну из ячеек
расчетную формулу в следующем виде:
=[0,70]*[х]л2+[0,21]*[х]+[-0,030]
Здесь под [...] понимается адрес ячейки, содержащей соответствующее значе-
ние: например, [х] — это адрес ячейки, содержащей значение х. В примере, приве-
денном на рисунке, константы находятся в ячейках ВЗ, В4 и В5, а величина х —
в ячейке С7, поэтому формула будет выглядеть так: =ВЗ*С7Л2+В4*С7+В5. Для
ввода самой этой формулы использована ячейка Е5. Обратите внимание, что
сразу же после ввода формулы в ней появилось значение -0,03 (соответствую-
щее результату вычислений при х = 0), поскульку ячейка для х была оставлена
пустой.
Вызовите программу Поиск решения. При появлении диалогового окна
установите курсор в окошко Установить целевую ячейку и щелкните
кнопкой мыши. Окошко должно быть пустым; если там уже указан адрес ка-
кой-либо ячейки, удалите его. Затем щелкните мышью на ячейке Е5 или вручную
введите в окошко «Е5». Затем таким же образом введите в окошко Изменяя
ячейки адрес С7. Наконец, в окошке Равной выберите опцию значению и
введите 0. Теперь все готово для решения уравнения. Однако рекомендуем
предварительно открыть окошко Параметры (Options) в меню Сервис (To-
ols) и установить требуемую точность вычислений (относительная погреш-
ность), например, 0,000001 (1 • 10-6). Эта величина должна быть по крайней мере
в 100 раз меньше, чем любая из констант (а, b и с), используемых в расчетах, а
также чем предполагаемое значение х. В данном случае точности 1 • 10-6 доста-
точно, но если в иной ситуации вычисление окажется невозможным, следует в
первую очередь увеличить точность вычислений (уменьшить допустимую по-
грешность на несколько порядков) и повторно выполнить расчет с помощью По-
иска решения.
Вернитесь в окошко Поиск решения и нажмите на клавишу Выполнить
(Solve). Вы получите сообщение Решение найдено (Solver found а
solution) и увидите в ячейке для х его значение, равное 0,10565:
286
ОСНОВНЫЕ ПОЛОЖЕНИЯ ТЕОРИИ ХИМИЧЕСКОГО РАВНОВЕСИЯ
А В С D : \ ,Е I F . | . G | н
1 Использование программы Поиск решения (Solver) для решения квадратного уравнения, Пример 6.1
2 I I I
3 а= 0,7 формула =ахл2 +Ьх-с = 0 (значение 0 не вводят)
4 Ь= 0,21 Поиск решения:
5 с= -0,3 Результат вычислений по формуле: -8Е-08 Е5 = формула для вычислений
6 Результат должен быть равен 0
7 0,10565^ I формула в ячейке Е5: =ВЗ*С7Л2+В4*С7+В5
8 Поиск решения: С7 = изменяемое значение I I
Соответствующее значение в правой части уравнения (результат вычислений по
формуле с полученным значением х) равен -8Е-08 — очень малой величине, дей-
ствительно близкой к нулю.
Квадратное уравнение может иметь и отрицательный корень. В данном слу-
чае, правда, он нас не интересует. Но если возникнет задача найти и это значение
х, то следует использовать соответствующие опции в окошке Ограничения
(Constraints). Нажмите на клавишу Добавить, установите в окошко Ссылка
на ячейку и щелкните кнопкой мыши в ячейке С7. Затем подведите указатель
к символу <= (меньше или равно) и введите в диалоговом окне Ограничение
значение 0. Нажмите на клавиши ОК и затем Поиск решения. Вы получите
значение х = -0,40565 (а результат вычислений по формуле, равный -9Е-07).
При изменении некоторых опций в диалоговом окне Solve можно полу-
чить несколько другое значение ответа. В дальнейшем мы будем использовать
Поиск решения для решения особенно трудоемких математических задач.
Готовая электронная таблица, созданная для решения задачи в соответст-
вии с условиями примера 6.1, находится на компакт-диске. Вы можете исполь-
зовать ее для решения квадратных уравнений с другими значениями параметров,
вводя в соответствующие ячейки новые значения а, b и с и вызывая Поиск
решения. Разумеется, вы можете также по своему усмотрению изменять саму
расчетную формулу.
Пример 6.2
Предположим, что константа равновесия в примере 6.1 равна 2,0 • 1016. Рассчи-
тайте равновесные концентрации А, В, С и D.
Решение
Поскольку значение К очень велико, равновесие реакции между А и В будет
практически полностью смещено вправо и к моменту наступления останутся толь-
ко следовые количества А. Обозначим х равновесную концентрацию А. В реак-
цию вступят равные количества А и В и образуются эквивалентные количества
С и D (около 0,20 М для каждого). Мы можем выразить равновесные концентра-
ции веществ следующим образом:
6.11. РАСЧЕТЫ С ИСПОЛЬЗОВАНИЕМ КОНСТАНТ РАВНОВЕСИЯ
287
[А]=х
[В] = (0,50 - 0,20) + х = 0,30 + х
[С] = 0,20-х
[D] = 0,20-x
или записать в виде равновесной реакции:
А + В С + D
х 0,30 + х 0,20-х 0,20-х
В целом, мы можем сказать, что количество образовавшихся С и D равно
количеству прореагировавшего вещества А за вычетом х, имеющем очень мале-
нькое значение. Поскольку х очень мало по сравнению со значениями 0,20 и
0,30, то им можно пренебречь. Таким образом
[А]=х
[В]« 0,30
[С]» 0,20
[D] « 0,20
Неизвестна концентрация [А]. Подставив полученные нами числа в уравнение
константы равновесия, мы получим
вдр.сд0 0 10,6
х-0,30
х = [А] = 6,7 • 10-18 М (аналитически определить невозможно).
В этом случае расчет значительно упростился, так как мы не учитывали ве-
личину х из-за ее малого значения по сравнению с другими значениями концент-
рации. Если бы х была сравнимо с другими значениями, тогда задачу следует
решать по-другому, используя квадратное уравнение или метод последователь-
ных приближений, начиная с первого рассчитанного значения х. В целом, если
величина х составляет меньше 5% от предполагаемой концентрации, то ей
можно пренебречь. В этом случае погрешность самой величины х составляет
обычно 5% или меньше. Это допущение возможно, когда концентрация продукта
реакции составляет менее 1% от константы равновесия, т. е. С < 0,01 Кравн.
Пример 6.3
А и В взаимодействуют следующим образом:
А + 2В^2С
[А][В]2
288
ОСНОВНЫЕ ПОЛОЖЕНИЯ ТЕОРИИ ХИМИЧЕСКОГО РАВНОВЕСИЯ
Предположим, что 0,10 моль А реагирует с 0,20 моль В в объеме 1000 мл;
К= 1,0 • 1010. Каковы равновесные концентрации А, В и С?
Решение
А и В участвуют в реакции в стехиометрических количествах, поэтому оба реа-
гента практически полностью израсходованы, остаются лишь следовые количе-
ства. Пусть х — равновесная концентрация А. Тогда при равновесии:
А + 2В 2С
х 2х 0,20 - 2х » 0,20
На каждый моль А, который расходуется или образуется, образуется или
расходуется два моля С и расходуется или образуется два моля В. Подставляем в
выражение константы равновесия:
-^^- = 1,0-1 о10
х -2х2
= 1,0-1010
4х3
х = [А] = з|4’0'10 = ^1,0-10-12 = 1,0 • КГ* М
V 4,0-1О10
[В] = 2х =2,0Ю~4М
(Результатом является аналитически определяемая, но не очень значимая вели-
чина по сравнению с исходной концентрацией.)
Равновесия диссоциации
Расчеты, связанные с процессами диссоциации, не очень отличаются от приме-
ров, приведенных выше для химических реакций.
Пример 6.4
Рассчитайте равновесные концентрации А и В в 0,10 М растворе слабого элект-
ролита АВ с константой равновесия 3,0 • 10-6.
Решение
АВ А + В
к =
равн [АВ]
6.12. ВЛИЯНИЕ ОДНОИМЕННЫХ ИОНОВ: СМЕЩЕНИЕ РАВНОВЕСИЯ
289
Равновесные концентрации [А] и [В] неизвестны и равны. Обозначим их х. Рав-
новесная концентрация [АВ] равна ее исходной аналитической концентрации
за вычетом х.
АВ — А + В
0,10-х х х
При диссоциации величиной х можно пренебречь по сравнению с аналитиче-
ской концентрацией С, если С > 100 АГравн.
Величина достаточно мала, поэтому мы можем пренебречь величиной
х по сравнению с числом 0,10. В противном случае мы должны будем решать
квадратное уравнение. Подставив х в выражение константы равновесия АГравн,
получаем:
— = 3,0-10“6
0,10
х = [А] = [В] = 7з,О-1О“7 = 5,5 • Ю^М
6.12. Влияние одноименных ионов: смещение равновесия
Согласно принципу Ле Шателье на положение равновесия можно влиять, из-
меняя концентрацию одного или более участников реакции. Пример 6.5 иллюст-
рирует это.
Пример 6.5
Пересчитайте концентрацию А в примере 6.4, считая, что раствор содержит еще
0,20 М компонента В.
Решение
Мы можем выразить равновесные концентрации следующим образом:
[АВ] [А] [В]
Исходная концентрация 0,10 0 0,20
Изменение (х — количество —х +х +х
диссоциированного АВ, ммоль/мл)
Равновесная концентрация
х
0,10-х»0,10
0,20+ х« 0,20
290
ОСНОВНЫЕ ПОЛОЖЕНИЯ ТЕОРИИ ХИМИЧЕСКОГО РАВНОВЕСИЯ
Величина х теперь будет меньше, чем в предыдущем примере, из-за обыч-
ного ионного влияния В, поэтому мы можем ей пренебречь. Подставив полу-
ченные значения в выражение константы равновесия АГравн получаем:
*'0,20 = 3,0 ю-6
0,10
х = 1,5-10“6М
Концентрация А уменьшилась почти в 400 раз.
Влияние одноименных ионов может быть использовано для смещения ана-
литических реакций в нужную сторону или для их применения в количествен-
ном анализе. Часто для смещения равновесия изменяют кислотность растворов.
Регулирование pH— это общепринятый способ смещения равновесия.
Например, протеканию реакции титрования бихроматом калия способству-
ет повышенная кислотность среды, так как в реакции участвуют протоны. Титро-
вание иодом, слабым окислителем, обычно проводят в слабощелочных растворах,
для того чтобы сместить равновесие до полного завершения реакции, как, на-
пример, при титровании мышьяка(Ш):
Н3AsO3 + I2 + Н2О H3AsO4 + 21- + 2Н+
6.13. Общий подход к расчетам химических равновесий:
как решить любую задачу
После краткого знакомства с некоторыми задачами, связанными с химическими
равновесиями, рассмотрим общий подход к расчетам равновесных концентра-
ций в системах любой сложности. Он состоит в выявлении неизвестных концент-
раций всех участвующих в равновесии частиц и составлении системы уравнений,
число которых должно быть равно числу неизвестных. Чтобы упростить систе-
му, можно принять некоторые допущения, пренебрегая концентрациями одних
частиц по сравнению с другими (подобно тому, как мы это делали ранее). В об-
щем случае система уравнений включает выражения материального баланса
для ряда веществ и одно уравнение электронейтральности. Рассмотрим снача-
ла, как составлять такие выражения.
Уравнения материального баланса
Принцип материального баланса вытекает из закона сохранения массы. Он
гласит, что в химических системах (в которых отсутствуют ядерные превраще-
ния) число атомов каждого элемента в ходе реакции остается постоянным. На
практике вместо числа атомов используют мольные концентрации частиц.
6.13. ОБЩИЙ ПОДХОД К РАСЧЕТАМ ХИМИЧЕСКИХ РАВНОВЕСИЙ: КАК РЕШИТЬ ЛЮБУЮ ЗАДАЧУ
291
Взаимосвязь между этими концентрациями легко установить из уравнений со-
ответствующих реакций.
Пример 6.6
Запишите уравнения материального баланса для 0,100 М раствора уксусной кис-
лоты.
Решение
В системе существуют равновесия
НОАс Н+ + ОАс-
Н2О н+ + он-
Общая (аналитическая) концентрация уксусной кислоты равна сумме кон-
центраций всех ее форм:
сноас = [НОАс] + [ОАс-] = 0,100 М
Еще одно уравнение материального баланса может быть записано для рав-
новесной концентрации Н+ — частицы, образующейся как из НОАс, так и из
Н2О. При этом одна частица Н+ приходится на одну частицу ОАс- и на одну ОН-,
следовательно,
[Н+] = [ОАс-] + [ОН-]
В уравнении материального баланса аналитическая концентрация вещества
равна сумме равновесных концентраций всех его форм (с учетом стехиометри-
ческих коэффициентов).
Пример 6.7
Запишите уравнения материального баланса для 1,00 • 10-5 М раствора
[Ag(NH3)2]Cl.
Решение
В системе существуют следующие равновесия:
[Ag(NH3)]2Cl -> Ag(NH3)2 + Cl-
Ag(NH3)2 Ag(NH3)+ + NH3
Ag(NH3)+^ Ag+ + NH3
NH3 + H2O = NHj + OH-
H2O H+ + он-
292
ОСНОВНЫЕ ПОЛОЖЕНИЯ ТЕОРИИ ХИМИЧЕСКОГО РАВНОВЕСИЯ
Концентрация ионов С1~ равна общей концентрации растворенного вещест-
ва, т. е. 1,00-10-5 М. Этой же величине равна и сумма концентраций всех частиц,
содержащих серебро:
CAg = [Ag+] + [Ag(NH3)+] + [Ag(NH3)+] = [C1-] = 1,00 • 10-5 м
В системе присутствуют следующие частицы, содержащие азот:
NH* NH3 Ag(NH3)+ Ag(NH3) +
Концентрация азота в составе Ag(NH3)2 вдвое больше, чем концентрация
Ag(NH3) 2. Общая концентрация азота также вдвое больше общей концентрации
исходного вещества, поскольку его молекула содержит два атома азота. Поэто-
му можно записать:
CNH3 = [NHJ] + [NH3] + [Ag(NH3)+] + 2[Ag(NH3)^] = 2,00 • 10-5 M
Наконец, можно записать также
[ОН-] = [NHJ] + [Н+]
Некоторыми из приведенных равновесий и концентрациями соответствую-
щих частиц можно пренебречь по сравнению с концентрациями других частиц и
тем самым упростить расчеты. В частности, можно не учитывать последнее
уравнение материального баланса.
Таким образом, для одной системы можно записать несколько уравнений
материального баланса. Некоторые из них дублируют друг друга и вообще не
требуются для расчетов (иначе число уравнений превысит число неизвестных).
Другие можно упростить или вообще исключить, поскольку концентрации уча-
ствующих в них частиц могут быть малы по сравнению с концентрациями дру-
гих частиц. Все это станет более понятным после рассмотрения еще нескольких
примеров.
Уравнения электронейтральности
В соответствии с законом сохранения заряда все растворы электрически ней-
тральны. Это означает, что ни положительно, ни отрицательно заряженные час-
тицы не находятся в избытке, а суммарный заряд катионов равен суммарному
заряду анионов. Таким образом, для любой равновесной системы можно запи-
сать лишь одно уравнение электронейтральности.
6.13. ОБЩИЙ ПОДХОД К РАСЧЕТАМ ХИМИЧЕСКИХ РАВНОВЕСИЙ: КАК РЕШИТЬ ЛЮБУЮ ЗАДАЧУ
293
Пример 6.8
Запишите уравнение электронейтральности для раствора H2S.
Решение
В системе существуют равновесия:
H2S^H+ + HS“
HS" Н+ + S2"
Н2О Н+ + ОН"
В результате диссоциации H2S образуется катион Н+ и два аниона — HS" и
S2-, а в результате диссоциации воды — Н+ и ОН-. Количество ионов Н+, образо-
вавшихся в результате диссоциации H2S по двум ступеням, равно удвоенному
количеству ионов S2-, а образовавшихся в результате диссоциации по первой
ступени — количеству ионов HS~: на каждый ион S2" образуется по два иона Н+,
на каждый HS" — по одному Н+ (и на каждый ОН" — также по одному Н+). Для
всех однозарядных частиц концентрация их зарядов равна концентрации самих
частиц. Но для ионов S2" концентрация зарядов вдвое больше концентрации са-
мих частиц, поэтому в данном случае для нахождения концентрации зарядов
следует концентрацию S2" умножить на два. В соответствии с принципом элект-
ронейтральности суммарная концентрация положительных зарядов должна быть
равна суммарной концентрации отрицательных зарядов. Отсюда получаем:
[Н+] = 2[S2~] + [HS"] + [ОН"]
Обратите внимание, что общая концентрация заряда всегда равна равновес-
ной концентрации частицы, умноженной на величину ее заряда, даже если час-
тица (в данном случае Н+) может образуется из разных источников.
Пример 6.9
Запишите уравнение электронейтральности для раствора, содержащего КС1,
A12(SO4)3 и KNO3, пренебрегая диссоциацией воды.
Решение
[К+] + 3[А13+] = [С1-] + 2[SO 4~] + [NO3]
294
ОСНОВНЫЕ ПОЛОЖЕНИЯ ТЕОРИИ ХИМИЧЕСКОГО РАВНОВЕСИЯ
Пример 6.10
Запишите уравнение электронейтральности для насыщенного раствора CdS.
CdS^Cd2+ + S2-
S2- + H2O^HS+OH-
Решение
В растворе существуют равновесия
hs- + h2o^h2s + oh-
Н2О Н+ + ОН-
И в этом случае концентрации зарядов для однозарядных частиц (Н+, ОН-,
HS-) равны равновесным концентрациям соответствующих частиц. Но для Cd2+
и S2- концентрации зарядов вдвое больше концентраций частиц. Условие равен-
ства суммарных концентраций положительных и отрицательных зарядов запи-
шется так:
2[Cd2+] + [Н+] = 2[S2-] + [HS-] + [ОН-]
Пример 6.11
Запишите уравнение электронейтральности для системы, рассмотренной в при-
мере 6.7.
Решение
[Ag+] + [Ag(NH3)+] + [Ag(NH3)2] + [NHj] + [Н+] = [Cl-] + [ОН-]
В этом случае все частицы — однозарядные, поэтому концентрации зарядов
равны концентрациям соответствующих частиц.
Общий подход к расчетам равновесий
В общем случае для систем, включающих множество химических равновесий,
для расчета равновесных концентраций частиц нужно выполнить следующие
шаги:
6.13. ОБЩИЙ ПОДХОД К РАСЧЕТАМ ХИМИЧЕСКИХ РАВНОВЕСИЙ: КАК РЕШИТЬ ЛЮБУЮ ЗАДАЧУ
295
1 - Записать уравнения всех реакций, протекающих в системе.
2. Для каждой реакции записать выражение константы равновесия.
3. Составить все уравнения материальных балансов.
4. Составить уравнение электронейтральности.
5 - Сравнить общее число различных частиц, концентрации которых входят
в полученную систему уравнений (составленную на основании шагов 2,
3 и 4), и число независимых уравнений в системе. Если число независи-
мых уравнений равно числу частиц, система имеет решение. Теперь за-
дачу в принципе можно решить чисто математически.
6- Принять разумные допущения, основываясь на сравнительных величи-
нах концентраций частиц. Это позволит упростить математику, но для
этого необходимо мыслить как химик!
7. Решить систему и найти неизвестные концентрации.
8. Проверить обоснованность сделанных допущений!
Рассмотрим с этих позиций один из ранее решенных примеров.
Пример 6.12
Решить задачу, приведенную в условии примера 6.4, на основе общего подхода к
расчетам химических равновесий.
Уравнения реакции
АВ=А+В
Выражение константы равновесия
К
равн
_ [А][В] _ 30.10-б
[АВ]
(1)
Уравнения материальных балансов
Сдв = [АВ] + [А] = 0,10 М (2)
[А] = [В]
(3)
Напомним, что символом С обозначают общие (аналитические) концентрации.
Уравнение электронейтральности
Записать нельзя, поскольку в системе нет заряженных частиц.
296
ОСНОВНЫЕ ПОЛОЖЕНИЯ ТЕОРИИ ХИМИЧЕСКОГО РАВНОВЕСИЯ
Сравнение числа неизвестных и числа уравнений
Система содержит три неизвестных ([АВ], [А] и [В]) и состоит из трех урав-
нений (выражение константы равновесия и два уравнения материального ба-
ланса).
Упрощающие допущения
Требуется рассчитать равновесные концентрации А, В и АВ. Поскольку ве-
личина К мала, можно ожидать, что диссоциация АВ будет протекать в малой
степени, поэтому из (2) получаем:
[АВ] = Сдв - [А] = 0,10 - [А] « 0,10 М
При введении допущений используйте те же простые правила, что и ранее
(СА > 100А7равн для равновесий диссоциации, СА < 0,01 А7равн для равновесий ассо-
циации).
Решение системы уравнений
[АВ] — найдено ранее;
[А] можно найти из (1) и (3):
^1Е!=з,о1(г6
0,10
[А] = -До-Ю"7 = 5,5 Ю^М
[В] можно найти из (3):
[В] = [А] = 5,5-10"4М
Проверка допущений
[АВ] = 0,10 - 5,5 • 1(Н = 0,10 М (в пределах значащих цифр)
Получен тот же самый результат, что и при интуитивном подходе, описан-
ном в примере 6.4. Может показаться, что общий подход слишком сложен и
формален. Применительно к рассмотренному крайне простому случаю это,
возможно, и так. Однако следует понимать, что общий подход применим для
расчетов любых равновесных систем, вне зависимости от степени сложности за-
дачи. Вы можете столкнуться с задачами, связанными с таким большим количе-
ством частиц и, соответственно, равновесий, что с точки зрения интуитивного
подхода они покажутся безнадежно сложными. В то же время хорошее интуи-
тивное «чутье» при решении равновесных задач — это исключительно ценное
качество. Большой опыт в решении разнообразных задач позволяет развить это
чутье. По мере накопления опыта вам будет все легче делать разумные допуще-
ния, сокращая тем самым формальные моменты, заложенные в общем подходе.
6.13. ОБЩИЙ ПОДХОД К РАСЧЕТАМ ХИМИЧЕСКИХ РАВНОВЕСИЙ: КАК РЕШИТЬ ЛЮБУЮ ЗАДАЧУ
297
Пример 6.13
Решите задачу, описанную в условии примера 6.5, основываясь на общем подхо-
де. Считайте, что заряд А равен +1, заряд В -1, а дополнительное количество
В (0,20 М) внесено в виде полностью диссоциирующего соединения МВ.
Решение
Уравнения реакций
АВ = А+ + В-
МВ -> М+ + в-
Выражение константы равновесия
^равн = [А+][В ] = 3,0 10"6 (1)
р [АВ]
Уравнения материального баланса
САВ = [ АВ] + [А+] = 0,10 М (2)
[В"] = [А+] + [М+] = [А+] + 0,20 М (3)
Уравнение электронейтральности
[А+] + [М+] = [В-] (4)
Отметим, что уравнение электронейтральности часто не требуется.
Сравнение числа неизвестных и числа уравнений
Система содержит три неизвестных ([АВ], [А+] и [В-]; концентрация М+ из-
вестна и равна 0,20 М) и состоит из трех независимых уравнений (выражение
константы равновесия и два уравнения материального баланса; уравнение элек-
тронейтральности тождественно второму уравнению материального баланса).
Упрощающие допущения
1) Поскольку величина £ мала, АВ диссоциирует незначительно. Отсюда
из (2)
[АВ] = 0,10 - [А+] ~ ОДОМ
2) [А] « [М], поэтому из (3) и (4)
[В-] = 0,20 + [А+] « 0,20 М
Решение системы уравнений
Теперь [А] находим из (1):
[AJ^2O= ю-6
0,10
[А+] = 1,5-10’6М
298
ОСНОВНЫЕ ПОЛОЖЕНИЯ ТЕОРИИ ХИМИЧЕСКОГО РАВНОВЕСИЯ
Проверка допущений
1) [АВ] = 0,10-1,5 • 10"6 = 0,10М
2) [В] = 0,20 + 1,5 • 10-6 = 0,20 М
Допущения, приведенные в разд. 6.10 и 6.11, подходят для многих случаев,
рассматриваемых в рамках общего подхода. Использование общего подхода для
систем, включающих большое число равновесий, также рассмотрено в гл. 8.
Перечислим некоторые общие правила для приближенного решения задач,
связанных с расчетом химических равновесий. Эти правила применимы к кис-
лотно-основным равновесиям, равновесиям комплексообразования, окисления-
восстановления и др. Таким образом, все равновесия можно рассматривать еди-
нообразно.
1. Записать уравнения всех реакций, протекающих в системе.
2. Для каждой реакции записать выражение константы равновесия с испо-
льзованием численных значений.
3. Основываясь на химической специфике процессов, выбрать (обозначив
через х) равновесную концентрацию одной из частиц, которую можно
считать достаточно малой по сравнению с равновесными концентрация-
ми других частиц.
4. Выразить через х равновесные концентрации всех остальных частиц,
при необходимости посредством вычитания или прибавления этой вели-
чины к известным значениям аналитических концентраций.
5 - Принять разумные допущения, пренебрегая величиной х по сравнению с
аналитическими концентрациями в тех случаях, когда это возможно. Как
правило, это допустимо, если аналитическая концентрация в 100 или бо-
лее раз превышает соответствующую константу равновесия, либо если х
составляет не более 5% от величины аналитической концентрации.
6- Подставить все приближенные выражения равновесных концентраций
частиц в уравнения системы и решить ее относительно х.
7> Если допущения, сделанные на шаге 5, оказались неоправданными, ре-
шить систему более точно — с использованием формулы полного квад-
ратного уравнения или метода последовательных приближений.
Применение этих правил станет более понятным из примеров, приведенных
в последующих главах, посвященных отдельным типам равновесий.
6.15. АКТИВНОСТИ И КОЭФФИЦИЕНТЫ АКТИВНОСТИ: КОНЦЕНТРАЦИИ - ЭТО ЕЩЕ НЕ ВСЕ!
299
6.14. Гетерогенные равновесия:
концентрации твердых веществ не записываем
По сравнению с равновесиями в растворах существующие в гетерогенных сис-
темах равновесия имеют ряд особенностей. Равновесия, все участники которых
находятся в растворе, обычно устанавливаются достаточно быстро. Но если в
равновесии участвуют две фазы, скорость достижения равновесия, как правило,
существенно меньше. Примером может служить равновесие при распределении
аналита между неподвижной (например, твердой) и подвижной фазами в хрома-
тографии. Строго говоря, полное время установления равновесия бесконечно.
Только если скорость пропускания подвижной фазы через хроматографическую
колонку достаточно мала, равновесие можно считать достигнутым. Растворение
или образование осадка тоже происходит не мгновенно.
Еще одно отличие гетерогенных равновесий от гомогенных состоит в ха-
рактере влияния количеств участвующих в них веществ на положение равновесия.
Гульдберг и Вааге экспериментально установили, что если одним из компонентов
обратимого химического процесса является твердое вещество, его «действую-
щая масса» постоянна и не зависит от того, сколько этого вещества присутствует
в системе. Таким образом, добавление дополнительного количества твердого ве-
щества не смещает равновесия. Поэтому в выражения констант таких равнове-
сий нет необходимости включать члены, содержащие концентрации твердых
веществ: «концентрацию» чистого твердого вещества в стандартном состоянии
можно принять равной единице. Например, для равновесия
CaF2 Са2+ + 2F"
выражение константы выглядит как
Л7равн = [Ca2-][F-p
Сказанное справедливо и для чистых жидкостей (недиссоциирующих), если
они участвуют в равновесии (например, для ртути). Кроме того, для реакций,
протекающих в разбавленных водных растворах, стандартная концентрация
воды также принята равной единице. Поэтому в этих случаях концентрацию
воды не следует записывать в выражения констант равновесий.
6.15. Активности и коэффициенты активности:
концентрации - это еще не все!
Как правило, присутствие посторонних индифферентных электролитов (не со-
держащих ионов, которые участвуют в рассматриваемом равновесии) влияет на
положение равновесия — усиливает диссоциацию слабого электролита или уве-
личивает растворимость осадка. Причина состоит в том, что катионы электроли-
та притягивают анионы аналита и в то же время эффективно экранируют их, тем
самым снижая их эффективную концентрацию и смещая равновесие. Это явле-
300
ОСНОВНЫЕ ПОЛОЖЕНИЯ ТЕОРИИ ХИМИЧЕСКОГО РАВНОВЕСИЯ
ние называется солевым эффектом или влиянием посторонних ионов; с увеличе-
нием зарядов ионов (как постороннего электролита, так и ионов, участвующих в
равновесии) солевой эффект проявляется все сильнее. Строго говоря, солевой
эффект не вытекает непосредственно из принципа Ле Шателье. Однако, если
рассуждать, используя понятия «эффективных концентраций», то он оказывает-
ся в чем-то аналогичным влиянию одноименного иона.
«Эффективная концентрация» иона, присутствующего в растворе, называ-
ется его активностью. При помощи величин активностей солевой эффект мож-
но описать количественно (см. ниже). Величины активностей играют важную
роль в потенциометрии (см. гл. 13). В данном разделе мы покажем, как можно
оценить величину активности.
Коэффициенты активности
Активность иона at определяется выражением
ai Cifi
(6.17)
где Cz — равновесная концентрация иона i, а величина f. называется коэффици-
ентом активности. Активность выражают в тех же единицах, что и концентрацию
(обычно моль/л). Коэффициент активности — величина безразмерная, однако его
численное значение зависит от способа выбора стандартного состояния. Вели-
чины коэффициентов активности зависят от общего содержания ионов в растворе
и их зарядов. Коэффициенты активности играют роль поправок на межионные
взаимодействия. В очень разбавленных растворах с общей концентрацией ионов,
не превышающей IO-4 М, коэффициенты активностей электролитов практиче-
ски равны единице, а активности приблизительно равны концентрациям. С ростом
концентрации ионов — как посторонних, так и участвующих в рассматриваемом
равновесии — коэффициенты активности обычно убывают, а величины актив-
ностей становятся меньше величин концентраций.
Ионная сила
Из всего сказанного следует, что коэффициенты активности зависят от общего
содержания ионов в растворе. Обобщенной мерой электростатических взаимо-
действий в растворе может служить ионная сила ц. По определению она равна
(6.18)
где Zz — заряд иона определенного вида. Для расчета ионной силы необходимо
учитывать концентрации всех ионов, присутствующих в растворе, — как катио-
нов, так и анионов. При значении ионной силы менее 1(Н коэффициенты актив-
ности можно считать равными единице.
6.15. АКТИВНОСТИ И КОЭФФИЦИЕНТЫ АКТИВНОСТИ: КОНЦЕНТРАЦИИ - ЭТО ЕЩЕ НЕ ВСЕ!
301
Пример 6.14
Рассчитайте ионную силу 0,2 М раствора KNO3 и 0,2 М раствора K2SO4.
Решение
Для раствора KNO3 С 71 +Г 72 еК+^К+ ^^NO3 NO3 И= 2“ [К+] = 0,2 М [NOj] = 0,2 М 0,2• I2 4-0,2• I2 . ц = — = 0,2 моль/л 2
Для раствора K2SO4 С 72 + С 72 Ьк+Лк+ -t- <-so2- ^SO2- 2 [К+] = 0,4 М [SO2-] = 0,2 М 0,412+0,2-22 . LL = = 0,6 МОЛЫЛ 2
Обратите внимание, что ионная сила раствора K2SO4, содержащего двухза-
рядный ион SO4“, втрое больше ионной силы раствора KNO3 той же концентра-
ции. Многозарядные ионы вносят больший вклад в ионную силу.
Если в растворе присутствует несколько электролитов, для расчета ионной
силы следует учитывать суммарные концентрации и заряды каждого из ионов.
Для раствора любого электролита заданного состава ионная сила пропорциона-
льна концентрации. Для сильных кислот, которые можно считать полностью
диссоциированными, расчет ионной силы ничем не отличается от такого же рас-
чета для солей. Для частично диссоциированных кислот следует сначала оценить
концентрации их ионизированных форм. Очень слабые кислоты, как правило,
можно считать недиссоциированными и пренебречь их вкладом в ионную силу.
Пример 6.15
Рассчитать ионную силу раствора, содержащего 0,30 М NaCl и 0,20 М Na^O^
CNa+ZNa+ + Ссг Zcr + CsoT Zl(%- 0,70 I2 + 0,30 • I2 + 0,20 • 22 Л
ц =--------------------------— -----------------------= 0,90 моль/л
2 2
302
ОСНОВНЫЕ ПОЛОЖЕНИЯ ТЕОРИИ ХИМИЧЕСКОГО РАВНОВЕСИЯ
Расчет коэффициентов активности
В 1923 г. Дебай и Хюккель вывели теоретическое выражение для расчета коэф-
фициентов активности. Это выражение, известное как уравнение Дебая-Хюк-
келя, выглядит следующим образом:
0,51 7;2Тц
1 + 0,33 а /Л/ц
(6.19)
Величины 0,51 и 0,33 относятся к водным растворам при 25 °C (первая из
них включает произведение диэлектрической проницаемости и абсолютной
температуры в степени -3/2 каждая). Величина az — это параметр размера
иона, равный эффективному диаметру гидратированного иона в ангстремах
(А; 1 А = 100 пм (пикометров) = 1 • Ю-10 м). Точность, с которой известна вели-
чина az, ограничивает точность расчетов по уравнению Дебая-Хюккеля. Для бо-
льшинства однозарядных ионов az можно принять равным 3 А, поэтому для
практических целей уравнение Дебая-Хюккеля обычно используют в упрощен-
ном виде:
(6.20)
Для многих многозарядных ионов значение az может достигать 11 А. Одна-
ко для значения ионной силы, не превышающих 0,01, второй член в знаменателе
выражения (6.20) мал по сравнению с единицей. Поэтому неточности в значени-
ях az становятся несущественными, и уравнение (6.20) можно использовать при
значении ионной силы 0,01 и ниже. Более точное уравнение (6.19) можно испо-
льзовать вплоть до значения ионной силы, равного 0,2. В работе [9] (см. в конце
главы) приведены значения az для различных ионов, а также рассчитанные для
них по уравнению (6.19) коэффициенты активности для значений ионной силы
от 0,0005 до 0,1 моль/л. На компакт-диске (гл. 6) содержится взятый из этой ра-
боты список значений параметров размера некоторых ионов.
Пример 6.16
Рассчитайте коэффициенты активности для ионов К+ и БОд- в 0,0020 М раство-
ре сульфата калия.
6.15. АКТИВНОСТИ И КОЭФФИЦИЕНТЫ АКТИВНОСТИ: КОНЦЕНТРАЦИИ - ЭТО ЕЩЕ НЕ ВСЕ!
303
Решение
Ионная сила раствора равна 0,0060, поэтому можно применить уравнение (6.20):
1g ОД.Р^бббО.О^
1+7о,ообо
/„+ =10~°’037 =10-1 -1О0,963 =0,918
IV
, _ 0,51-227о,0060 п1.
-|8Л“- ° i+^ooso =0Л4?
f =ю"°’147 =10-1 -Ю0’85’ = 0,71,
SO4
Пример 6.17
Рассчитайте коэффициенты активности для ионов К+ и SO4“ в 0,020 М растворе
сульфата калия.
Решение
Ионная сила раствора равна 0,060, поэтому следует использовать уравнение
(6.19). Из данных работы [9] находим, что а к+ =3 Аиа^. = 4 А. Для К+можно
по-прежнему использовать уравнение (6.20):
, . 0,5112д/а060 П1_
- 1g /к* =---г-—"• = °’1О1
1 + 70,060
fv+ =1О~°’101 =10-1 -1О0’899 =0,794
J К 7 ч
Для SOд используем уравнение (6.19):
_ 0,51-22д/^060
lg/gO2" — I----— 037 g
4 1 + 0,33-4,070060
/SO2- =Ю-1 -10°’622 =о,419
При использовании уравнения (6.20) в этом случае получилось бы значение
0,396. Обратите внимание, что значения коэффициентов активности меньше,
чем для 0,002 М K2SO4, особенно для сульфат-иона.
304
ОСНОВНЫЕ ПОЛОЖЕНИЯ ТЕОРИИ ХИМИЧЕСКОГО РАВНОВЕСИЯ
На компакт-диске (гл. 6) находятся электронные таблицы для расчета коэф-
фициентов активности по уравнениям (6.19) и (6.20).
Для более высоких значений ионной силы предложено множество эмпириче-
ских уравнений. Чаще всего используется уравнение в модификации Дэвиса [8]:
-1g/,. =0,51 Z,1 2
П=-0,Зц
(6.21)
которое справедливо до значений ионной силы порядка 0,5 моль/л. Используйте
это уравнение для ц, лежащих в интервале 0,2-0,5 моль/л. Коэффициенты актив-
ности, рассчитанные по этому уравнению, выше, чем по уравнению Дебая-Хюк-
келя.
При очень высоких концентрациях электролитов коэффициенты активно-
сти возрастают и могут превышать единицу. (Активности ионов в 0,01 М раство-
ре НС1 в 8 М NaCl почти в 100 раз выше, чем в таком же растворе в чистой воде!
Значение pH этого раствора составляет около 0,0. См. [F. Е. Critchfield, J. В. Joh-
nson, Anal. Chem., 30 (1958) 1247 и G. D. Christian, CRC Crit. Rev. in Anal. Chem.,
5(2) (1975) 119-153].) Причина состоит в снижении активности растворителя
(воды), в результате чего ионы частично десольватируются. При этом возраста-
ет их реакционная способность и, следовательно, активность. Обратите внима-
ние, что по сравнению с уравнением Дебая-Хюккеля в уравнении (6.21) как раз
и содержится «поправочный» член, увеличивающий рассчитанное значение ft
при высоких значениях ионной силы.
Приведем некоторые выводы, касающиеся оценки коэффициентов актив-
ности.
1 Коэффициенты активности всех ионов одинакового заряда при данной
ионной силе примерно одинаковы. Они не зависят от природы и концент-
рации индивидуальных ионов, создающих ионную силу. Чем выше заряд
посторонних ионов, тем сильнее их влияние на величины активностей.
2. С ростом заряда иона его поведение все больше отличается от идеально-
го, а рассчитанные коэффициенты активности—от экспериментальных.
3. Для растворов электролитов сложного состава рассчитанные коэффици-
енты активности менее точны, чем для растворов индивидуальных элек-
тролитов.
4. Коэффициенты активности неэлектролитов (т. е. нейтральных молекул),
как правило, можно принимать равными единице вплоть до значений
ионной силы, равных 0,1 моль/л. Даже вплоть до значений порядка 1 от-
клонения коэффициентов активности от единицы невелики. Сказанное
относится, в частности, к молекулам слабых кислот НА — для них коэф-
фициенты активности почти всегда считают равными 1.
6.16. ВЛИЯНИЕ ПОСТОРОННИХ ионов
305
6.16. Влияние посторонних ионов: термодинамическая константа
равновесия и коэффициенты активности
В начале предыдущего раздела мы упоминали, что введение постороннего ин-
дифферентного электролита обычно увеличивает степень диссоциации слабых
кислот из-за эффекта экранирования ионов (т. е. уменьшения их активности),
образующихся при диссоциации. Использование понятия активности позволяет
количественно оценить влияние этого эффекта на положение равновесия.
До сих пор, оперируя с константами равновесий, мы полагали, что солевой
эффект отсутствует. Это допущение справедливо лишь при ионной силе, близкой
к нулю, когда коэффициенты активности практически равны единице. Однако
более строгим было бы использование в выражениях констант равновесий не
концентраций, а активностей. Константы равновесий, записанные в такой фор-
ме, называются термодинамическими (а с использованием равновесных кон-
центраций, как это мы делали до сих пор, — реальными). Термодинамические
константы можно рассматривать как предельные значения реальных констант
при экстраполяции на бесконечное разбавление раствора. Так, для равновесия
диссоциации вещества АВ термодинамическая константа К равн записывается так
° _ ’ аВ _ [М/д ' [®1/в
равн ~ ~
(6.22)
аАВ [АВ]/ав
Поскольку реальная константа равновесия записывается в этом случае
как£равн = [А][В]/[АВ],то
(6.23)
ИЛИ
= К =
равн равн
(6-24)
Численное значение А?равн остается постоянным при любой ионной силе.
При нулевой ионной силе А?равн = А?равн, но при больших величинах ионной силы
реальную константу следует рассчитывать по уравнению (6.24) для каждого
конкретного значения ионной силы. Все константы равновесий, приведенные в
Приложении С, относятся к нулевой ионной силе и, таким образом, являются
термодинамическими. Для многих равновесий экспериментально определены и
значения реальных констант при различных ионных силах. Их можно непосред-
306
ОСНОВНЫЕ ПОЛОЖЕНИЯ ТЕОРИИ ХИМИЧЕСКОГО РАВНОВЕСИЯ
ственно использовать в расчетах равновесий при соответствующем значении
ионной силы раствора, не прибегая к расчету коэффициентов активностей.
Пример 6.18
Термодинамическая константа диссоциации слабого электролита АВ на ионы
А+ и В /С°авн = 2 • 10"8. Рассчитайте: (а) значение реальной константы этого рав-
новесия в присутствии индифферентного электролита, создающего ионную
силу 0,1 моль/л, принимая коэффициенты активности А+ и В- равными 0,6 и 0,7
соответственно; (б) степень диссоциации АВ в 1,0 • 10^ М растворе в чистой
воде и в присутствии индифферентного электролита в указанных условиях.
Решение
а) АВ А + В~
[А+][В]
[АВ]
^•ав- _ [а+]/аИВ-]/в-
адв [АВ]/^
Коэффициент активности незаряженной частицы принимаем равным 1, поэ-
К
равн
К
равн
тому о к равн _ [А+][В+] , , „ f f [АВ] лравн /д+ “'В-
2-Ю-8 8 равн х $ |0 — о
равн "/А+-/в- 0,6-0,7 ’
б) АВ — А++ В- 1 • Ю^-х х х
В чистой воде /А+ = » 1 (так как ц < 1 • 10-4 моль/л), х « 1 • 10-4 и
0^=2.10-
[АВ]
=2-10-8
1-Ю*4
х= 1,4-10-6
1 10 —м
Степень диссоциации составляет —----100% = 1,. %
1,0-ю-м
ЗАДАЧИ
307
В 0,1 М растворе соли
[АЖ]=5.ю-
[АВ]
*'х— = 5 10~8
1,0-10^
х = 2,2-ю-6
2„-10-6
Степень диссоциации составляет —--- • 100% = 2,2 %
1,0 Ю"4
Таким образом, степень диссоциации возросла на 57%.
В гл. 7 приведены примеры расчетов, учитывающих солевой эффект, для
кислотно-основных равновесий, а в гл. 10 — для равновесий осадок-раствор.
Однако в иллюстративных целях мы во всех остальных случаях на протяжении
всей книги будем пренебрегать влиянием посторонних ионов. В большинстве
ситуаций аналитика интересуют лишь относительные изменения равновесных
концентраций частиц, и пренебрежение коэффициентами активности, как пра-
вило, не меняет общей картины.
Что мы узнали из этой главы?
• Понятие константы равновесия [основные уравнения (6.12), (6.15)] —
стр. 279
• Расчеты с использованием констант равновесий — стр. 280
• Использование Поиска решений (Excel) для решения квадратных уравне-
ний — стр. 283
• Общий подход к расчетам равновесных концентраций; уравнения матери-
ального баланса и электронейтральности — стр. 290
• Активности и коэффициенты активности [основное уравнение (6.19)] —
стр. 299
• Термодинамические константы равновесия [основное уравнение (6.22)] —
стр. 305
Задачи
Расчеты химических равновесий
1 - Вещества А и В взаимодействуют по уравнению А + В С + D. Константа
равновесия равна 2,0 • 103. Если исходные концентрации А и В составляют
0,30 и 0,80 М соответственно, чему будут равны концентрации А, В, С и D
в условиях равновесия?
308
ОСНОВНЫЕ ПОЛОЖЕНИЯ ТЕОРИИ ХИМИЧЕСКОГО РАВНОВЕСИЯ
2- А и В взаимодействуют по уравнению А + В 2С. Константа равновесия
равна 5,0 • 106. Если исходные концентрации А и В составляют 0,40 и
0,70 М соответственно, чему будут равны концентрации А, В и С в услови-
ях равновесия?
3- Константа диссоциации салициловой кислоты С6Н4(ОН)СООН по первой
ступени равна 1,0- 10-3. Рассчитайте степень ее диссоциации (в %) в
1,0 • 10-3 М растворе. Диссоциацией по второй ступени пренебречь (см.
также задачу 25 ниже).
4. Константа диссоциации синильной кислоты HCN равна 7,2 • 10-10. Рассчи-
тайте степень ее диссоциации (в %) в 1,0 • 10-3 М растворе.
5- Рассчитайте степень диссоциации салициловой кислоты (см. задачу 3),
если раствор содержит также 1,0 • 10-2 М салицилата натрия.
6 - Сероводородная кислота H2S диссоциирует по ступеням. Константы диссо-
циации равны 9,1 • 10-8 и 1,2 • 10-15 соответственно. Напишите уравнение
полной диссоциации сероводородной кислоты и рассчитайте константу
этого равновесия.
7. Ионы Fe2+ и Сг2О7” взаимодействуют по уравнению 6Fe2+ + Сг2О7“ + 14Н+
6Fe3+ + 2Сг3+ + 7Н2О. Константа равновесия этой реакции равна 1 • 1057.
Рассчитайте равновесные концентрации железа и хрома во всех указанных
формах, если смешать по 10 мл 0,02 М раствора К2Сг2О7 в 1,14 М НС1 и
0,12MFeSO4B 1,14 МНС1.
Общий подход к расчетам химических равновесий
8. Запишите уравнения материального баланса: а) для насыщенного раствора
Bi2S3, б) для раствора Na2S.
9- Запишите уравнения материального баланса и электронейтральности для
0,100 М раствора [Cd(NH3)4]Cl2.
10. Проверьте, правильно ли записаны следующие выражения с точки зрения
уравнений электронейтральности и материального баланса:
a) [NO" ] = [Н+] - [ОН-] для 0,2 М раствора HNO2;
б) [СН3СООН] = 0,2 - [Н+] + [ОН-] для 0,2 М раствора СН3СООН;
в) [Н2С2О4] = 0,1- [Н+] + [ОН-] - [С2О;_] для 0,1 М раствора Н2С2О4;
г) [HCN] = [ОН-] - [Н+] для 0,1 М раствора KCN;
Д) [Н2ю;] = 1([ОН-] - [Н+] - [НРО^] - 3[Н3РО4]) для 0,1 М раствора
Na3PO4;
е) [HSO4] = 0,2 - [Н+] - [ОН-] для 0,1 М раствора H2SO4 (предполагая, что
диссоциация H2SO4 на Н+ и HSO4 протекает полностью).
11. Запишите уравнения материального баланса для насыщенного водного
раствора BaF2, содержащего F~, HF, HF2 и Ва2+.
ЗАДАЧИ
309
12. Запишите уравнение материального баланса для водного раствора Ваз(РО4)2.
13- Рассчитайте pH 0,100 М раствора уксусной кислоты, используя уравнения
материального баланса и электронейтральности.
Ионная сила
14- Рассчитайте ионную силу следующих растворов: а) 0,30 М NaCl, б) 0,30 М
Na^C^, в) 0,30 М NaCl + 0,20 М K2SO4, г) 0,20 М Al2 (SO4)3 + 0,10 М Na2SO4.
15 - Рассчитайте ионную силу следующих растворов: а) 0,20 М ZnSO4, б) 0,40 М
MgCl2, в) 0,50 М LaCl3, г) 1,0 М К2Сг2О7, д) 1,0 М T1(NO3)3 + 1,0 М Pb(NO3)2.
Активность
16 - Рассчитайте коэффициенты активности ионов Na+ и С1_ в 0,00100 М раство-
ре NaCl.
17- Рассчитайте коэффициенты активности каждого иона в растворе, содержа-
щем 0,0020 М Na2SO4 и 0,0010 М A12(SO4)3.
18. Рассчитайте активность иона NO3 в 0,0020 М растворе KNO3.
19- Рассчитайте активность иона СгО4“ в 0,020 М растворе Na^rC^.
Термодинамические константы равновесий
20- Запишите выражения термодинамических констант следующих равнове-
сий:
a) HCN Н+ + CN-
б) NH3 + Н2О NH4 + ОН-
21. Рассчитайте pH 5,0 • 10-3 М раствора бензойной кислоты: а) в воде, б) в при-
сутствии 0,05 М K2SO4.
Задачи для решения при помощи электронных таблиц
22- Не обращаясь к компакт-диску, составьте электронную таблицу с програм-
мой расчета коэффициентов активности по уравнению (6.20). Сравните ее с
приведенной на компакт-диске. Для проверки правильности выполните
вычисления, используя обе таблицы.
23- Рассчитайте коэффициенты активности ионов К+ и SO4“ для данных из
примера 6.16, пользуясь электронной таблицей с компакт-диска для расче-
тов по уравнению (6.20). Сравните полученные результаты с рассчитанны-
ми вручную и приведенными в ответе к примеру.
24- Рассчитайте коэффициенты активности для данных из примера 6.17, поль-
зуясь электронными таблицами с компакт-диска для расчетов по уравнениям
(6.19) и (6.20). Сравните полученные результаты с рассчитанными вручную и
приведенными в ответе к примеру.
310
ОСНОВНЫЕ ПОЛОЖЕНИЯ ТЕОРИИ ХИМИЧЕСКОГО РАВНОВЕСИЯ
25- Рассчитайте концентрацию х из данных задачи 3 (см. выше), используя
программу Excel Поиск решения. В задаче требуется решить квадратное
уравнение.
26- Решите задачи с 16 по 19, используя программу Excel.
Рекомендуемая литература
Химическое равновесие
1. A. J. Bard, Chemical Equilibrium. New York: Harper & Row, 1966.
2. T. R. Blackbum, Equilibrium: A Chemistry of Solutions. New York: Holt, Rine-
hart and Winston, 1969.
3- J. N. Butler. Ionic Equilibrium. A Mathematical Approach. Reading, MA: Addi-
son-Wesley, 1964.
4- G. M. Fleck, Ionic Equilibria in Solution. New York: Holt, Rinehart and Wins-
ton, 1966.
5. H. FreiserandQ. Fernando, Ionic Equilibria in Analytical Chemistry. New York:
Wiley, 1963.
6- A. E. Martell and R. J. Motekaitis, The Determination and Use of Stability Cons-
tants. New York: VCH, 1989.
Метод последовательных приближений
7- S. Brewer, Solving Problems in Analytical Chemistry. New York: Wiley, 1980.
Активность
8. C. W. Davies, Ion Association. London: Butterworth, 1962.
9- J. Kielland, «Individual Activity Coefficients of Ions in Aqueous Solutions»,
J. Am. Chem. Soc„ 59 (1937) 1675.
10. K. S. Pitzer, Activity Coefficients in Electrolyte Solutions, 2nd ed. Boca Raton,
FL: CRC Press, 1991.
11. PC. Meier, «Two-Parameter Debye-Huckel Approximation for the Evaluation
of Mean Activity Coefficients of 109 Electrolytes», Anal. Chim. Acta,
136(1982) 363.
Глава?
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
Кислота дает три побочных эффекта.
Улучшает хорошую память, сокращает короткую память,
а третий я забыл
Тимоти Лери
Кислотность или основность раствора часто является важным условием для про-
текания химической реакции. Для сохранения pH раствора на необходимом уров-
не нужно использовать соответствующие буферы. Кроме того, закономерности
установлений кислотно-основных равновесий важны для понимания процессов
кислотно-основного титрования и влияния кислотности на химические реакции,
например, на реакции комплексообразования и осаждения. В гл. 6 мы обсужда-
ли фундаментальные положения о константах равновесия. Объектами рассмот-
рения этой главы будут различные расчеты, связанные с кислотно-основными
равновесиями, включая слабые кислоты и основания, гидролиз солей слабых
кислот и оснований, буферы, полиосновные кислоты и их соли, а также физио-
логические буферные растворы. В первую очередь приведем обзор известных
кислотно-основных теорий и основных концепций pH.
7.1. Кислотно-основные теории
Для объяснения и классификации кислотных и основных свойств веществ было
предложено несколько кислотно-основных теорий. Вы, возможно, хорошо знаете
теорию Аррениуса, которая применима только к водным растворам (см. работу
[J. Am. Chem. Soc., 36 (1912) 353], в которой изложены впечатления Аррениуса о
трудностях, с которыми принимали его теорию). Другие теории являются более об-
щими и применимы к другим растворителям. Давайте обсудим наиболее распро-
страненные кислотно-основные теории.
Теория Аррениуса: Н+ и ОН~
В 1894 г. Аррениус, будучи аспирантом, предложил теорию (за что получил Нобе-
левскую премию), согласно которой кислота — это вещество, которое диссоци-
ирует в воде (частично или полностью) с образованием ионов водорода
(образующих с молекулами растворителя ионы гидрония Н3О+):
НА + Н2О Н3О+ + А-
312
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
Основание диссоциирует в воде с образованием гидр оксид-ионов. Слабые (ча-
стично диссоциированные) основания обычно диссоциируют следующим образом:
В + Н2О ВН+ + он-
а сильные основания, такие как гидроксиды металлов (например, NaOH), диссо-
циируют в соответствии с общим уравнением:
М(ОН)„ -> М"+ + юн-
Совершенно очевидно, что эта теория ограничена только теми случаями,
когда в качестве растворителя выступает вода.
Теория сольвосистем: катионы и анионы растворителя
В 1905 г. Франклин предложил концепцию кислот и оснований, основанную на
представлении об ионизации растворителя с образованием катионов и анионов,
например, 2Н2О Н3О+ + ОН" или 2NH3 *=? NH 4 + NH 2. Кислотой называется
вещество, которое способствует образованию кислотного катиона раствори-
теля, основанием — вещество, которое способствует образованию основного
аниона растворителя. Так, NH4C1 является сильной кислотой в жидком аммиа-
ке (подобно НС1 в воде: НС1 + Н2О Н3О+ + С1"), тогда как NaNH2 в аммиаке —
сильным основанием (подобно NaOH в воде); при ионизации этих соединений
образуются соответственно катионы и анионы растворителя. Этанол ионизиру-
ется следующим образом 2С2Н5ОН С2Н5ОН2 + С2Н5О". Следовательно, эток-
сид натрия NaOC2H5 является сильным основанием в этом растворителе. Теория
Франклина похожа на теорию Аррениуса, но применима также и для других спо-
собных ионизироваться растворителей, кроме воды.
Теория Бренстеда—Лоури:
присоединение и отщепление протонов
Теория сольвосистем подходит для ионогенных растворителях, но неприменима
для кислотно-основных реакций в неионогенных растворителях, таких как бен-
зол или диоксан. Бренстед и Лоури независимо друг от друга предложили тео-
рию, которая известна как теория Бренстеда—Лоури. Согласно этой теории
кислотой является вещество, способное отщеплять протон (донор протонов),
а основанием — вещество, способное присоединять протон (акцептор прото-
нов). Таким образом, мы можем записать уравнение «полуреакции»:
кислота = Н+ + основание
(7.1)
Кислота и основание, принимающие участие в этой полуреакции, составляют
сопряженную пару. Свободные протоны не существуют в растворе, поэтому в
растворе должен уже находиться акцептор протонов (основание) до того, как
7.1. КИСЛОТНО-ОСНОВНЫЕ ТЕОРИИ
313
Таблица 7.1
Реакции кислот и оснований по Бренстеду
Растворитель Кислота1 + Основание2 -> Кислота2 + Основаниеj
NH3 (жидкий) НОАс NH3 NH4 ОАс-
н2о НС1 н2о н3о+ сг
н2о NHJ Н2О н3о+ NH3
н2о н2о ОАс- НОАс ОН-
н2о НСО3 он- н2о СО*-
с2н5он NH+ с2н5о- С2Н5ОН NH3
с6н6 о о Н-пикрат CLHSNH9 О J Z c6h5nh+ пикрат-
донор протонов (кислота) освободит свой протон. Это значит, что происходя-
щие в растворе процессы можно описать комбинацией двух полуреакций. Неко-
торые кислотно-основные реакции в различных растворителях приведены в
табл. 7.1. В первом примере ацетат-ион представляет собой сопряженное осно-
вание уксусной кислоты, а ион аммония — сопряженную кислоту аммиака. Пер-
вые четыре примера иллюстрируют ионизацию кислоты или основания в
растворителе, остальные являются примерами реакций нейтрализации между
кислотой и основанием в растворителе.
Из данного выше определения ясно, что вещество не может проявлять свой-
ства кислоты, если отсутствует основание, способное принять протон. Так, кис-
лоты будут полностью или частично ионизироваться в основных растворителях
(таких как, вода, жидкий аммиак или этанол) в зависимости от основности рас-
творителя и силы кислоты. Но в нейтральных или «инертных» растворителях
степень ионизации незначительна. Однако ионизация в растворителе не являет-
ся обязательным условием для протекания кислотно-основной реакции, как сле-
дует из последнего примера, в котором пикриновая кислота взаимодействует с
анилином.
Теория Льюиса: электронная теория
Еще одна теория была предложена в 1923 г. Г. Н. Льюисом — электронная тео-
рия кислот и оснований. В теории Льюиса кислота — это вещество, которое мо-
жет принимать электронную пару, а основание — это вещество, которое может
отдавать электронную пару. Основания часто содержат атомы кислорода или
азота, являющиеся донорами электронов. Как кислоты могут рассматриваться и
вещества, не содержащие водорода. В качестве примеров кислотно-основных ре-
акций по теории Льюиса можно привести следующие:
314
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
Н+ (сольватированный) + :NH3 -> H:NH3
R
AlCl3 + :О -> Cl3Al:OR2
R
H
О: + Н+ -> Н2О:Н+
Н
Н+ + :ОН“ -> Н:ОН
Во втором примере хлорид алюминия—это кислота, а эфир—основание.
7.2. Кислотно-основные равновесия в воде
Выше мы уже обсуждали, что при растворении в воде кислоты и основания дис-
социируют, или ионизируются, причем степень зависит от их силы. «Сильный»
электролит диссоциирует полностью, тогда как «слабый» электролит диссоциирует
частично. В табл. 7.2 перечислены некоторые часто используемые электролиты,
как сильные, так и слабые. Другие слабые кислоты и основания перечислены в
Приложении С.
Хлороводородная кислота — сильная кислота, она ионизируется полно-
стью:
НС1 + Н2О Н3О+ + ci-
(7.2)
Величина константы равновесия для уравнения (7.2) равна бесконечности.
Таблица 7.2
Некоторые сильные и слабые электролиты
Сильные Слабые
НС1 НС1О4 H2SO* HNO3 NaOH NaC2H3O2 HC2H3O2 (уксусная кислота) NH3 С6Н5ОН (фенол) НСНО2 (муравьиная кислота) C6H5NH2 (анилин)
* Диссоциация по первой ступени проходит полностью в разбавленном растворе, по второй ступе-
ни — частично (К2 = I О-2).
7.2. КИСЛОТНО-ОСНОВНЫЕ РАВНОВЕСИЯ В ВОДЕ
315
Протон Н+ существует в воде в виде гидратированного иона—иона гидро-
ния Н3О+. Возможно существуют и другие гидраты, в частности, Н9Од. Ион
гидрония обозначают Н3О+ для удобства и для того, чтобы подчеркнуть поведе-
ние в рамках теории Бренстеда.
Уксусная кислота* представляет собой слабую кислоту, диссоциирующую
лишь частично (несколько процентов):
НОАс + Н2О Н3О+ + ОАс" (7.3)
Для этой реакции мы можем написать выражение константы равновесия:
< (7.4)
аНОАс ’ аН2О
где Ка — термодинамическая константа кислотности (см. разд. 6.16), а —
активность указанной частицы. Катионы и анионы солей также могут частично
взаимодействовать с водой, например, ацетат-ион полученный в результате дис-
социации соли, взаимодействует с водой с образованием уксусной кислоты
НАс.
Под активностью можно подразумевать эффективную концентрацию иона
(это обсуждалось в гл. 6). Влияние протонов на химические реакции часто опре-
деляется их активностью, и именно активность протонов измеряют широко ис-
пользуемым прибором pH-метром (гл. 13). Методы расчета коэффициентов
активности обсуждались в гл. 6.
В разбавленных растворах активность воды остается практически постоян-
ной и принята равной единице (стандартное состояние). Поэтому уравнение
(7.4) можно записать следующим образом:
<=flH3O+ flOAc- (75)
67 НОАс
Чистая вода в небольшой степени ионизируется или, иными словами, под-
вергается автопротолизу:
2Н2О Н3О+ + ОН- (7.6)
Константа равновесия для этой реакции имеет следующее выражение :
-°-- (7.7)
ан2о
------------ о
II
Мы будем использовать символ ОАс для обозначения ацетат-иона СН3—С—О .
316
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
Так как активность воды для разбавленных растворов постоянна (ее концент-
рация составляет примерно 55,3 М), то константу равновесия можно записать и так:
= ан3(У ’ аон~ (7-8)
где K°w — термодинамическая константа автопротолиза, или константа са-
моионизации воды.
Расчеты упрощаются, если пренебречь коэффициентами активности. Такое
упрощение приводит всего лишь к незначительной погрешности для разбавлен-
ных растворов, и мы будем пользоваться молярными концентрациями во всех
расчетах. Этого будет достаточно для описания рассматриваемых равновесий.
Большинство растворов, о которых будет идти речь, являются разбавленными, и
нас часто будет интересовать относительное (причем существенное) изменение
значений pH; при таких изменениях погрешность незначительна. Мы упростим
выражения, используя Н+ вместо Н3О+. Это не будет противоречием, так как мо-
лекулы воды, участвующие в сольватации других ионов или молекул (например,
ионов металлов), обычно не записывают, кроме того, в этом случае запись Н3О+
будет не вполне корректна.
Молярную концентрацию будем обозначать квадратными скобками [ ].
Для приведенных выше реакций можно записать упрощенные уравнения
HCl -> Н+ + Cl" (7.9)
НОАс^Н+ + ОАс" (7.10)
_[Н+][ОАс~]
а [НОАс]
н2о н+ + он-
Arw=[H+][OH-]
(7.П)
(7.12)
(7.13)
где Ка и Kw — молярные константы равновесия.
При 25 °C = 1,0 • 10-14. Произведение концентраций ионов водорода и
гидроксид-ионов в водных растворах всегда равно 1,0 • 10-14 при комнатной тем-
пературе:
(7.14)
[Н+][ОН-] = 1,0 • ю-14
Химикам (особенно студентам) повезло, что при комнатной температуре
численное значение Kw равно именно 1,0 • 10~14. Представьте себе, что нужно
производить расчеты pH, если значение Kw составляет, допустим, 2,39 • 10~13.
Однако вам придется иметь дело с подобным расчетом (см. разд. 7.4) для дру-
гих значений температуры.
7.3. ШКАЛА pH
317
В чистой воде концентрации ионов Н+ и ОН- равны, поскольку кроме диссо-
циирующих молекул воды нет других поставщиков этих двух частиц.
Поэтому,
[Н+] = [ОН-]
[Н+][Н+] = 1,0 • ю-14
[Н+] = 1,0- 10~7М = [ОН-]
Если в воду добавить кислоту, то можно рассчитать концентрацию гидро-
ксид-ионов при условии, что известна концентрация ионов водорода, поступив-
ших из добавленной кислоты. Вклад ионов водорода, образующих при ионизации
воды, в общую концентрацию [Н+] можно не учитывать, кроме тех случаев,
когда концентрация ионов водорода в кислоте очень мала, т. е. составляет
10~6 М и менее.
Пример 7.1
Приготовлен 1,0 • 10-3 М раствор хлороводородной кислоты. Какова концентра-
ция гидроксид-иона?
Решение
Поскольку хлороводородная кислота является сильным электролитом и ее дис-
социация протекает полностью, то концентрация Н+ равна 1,0 • 10-3 М. Таким
образом,
(1,0- Ю-3)[ОН-] = 1,0 • ю-14
[ОН-] = 1,0- ю-им
7.3. Шкала pH
Концентрации ионов водорода и гидроксид-ионов в водных растворах могут из-
меняться в очень широких пределах: от 1 М и более до 10-14 М и менее. Построить
график зависимости концентрации ионов водорода от некоторой переменной бу-
дет трудно, если изменения концентрации находятся в пределах от 10-1 М до
10-13 М. Этот диапазон обычно используется при титровании. Более удобно
сжать шкалу кислотности, если использовать логарифмы. По Серенсену pH рас-
твора выражается как
pH = -lg[H+]
(7.15)
318
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
В уравнении стоит знак минус, так как в большинстве случаев концентра-
ции, с которыми приходится иметь дело, меньше 1 М, поэтому такое выражение
позволяет получить шкалу pH в основном с положительными числами. (При бо-
лее строгом подходе pH представляет собой -1g ан+, именно эту величину измеря-
ют pH-метром со стеклянным электродом (см. гл. 13), но мы будем пользоваться
более простым уравнением (7.15). В общем показатель р какой либо величины
есть 1g этой величины, и такой способ обозначения будет использован позже
для других величин, которые могут изменяться в широких пределах, либо пред-
ставлять собой очень большие или очень малые числа (например, константы
равновесия).
Пример 7.2
Рассчитайте значение pH 2,0 • 10-3 М раствора НС1.
Решение
НС1 полностью диссоциирована, поэтому
[Н+] = 2,0 • IO’3 М
pH = -1g (2,0 • 10“3) = 3 - 1g 2,0 = 3 - 0,30 = 2,70
Похожее уравнение существует для определения концентрации гидроксид-
ионов:
pOH = -lg [ОН-]
(7-16)
Значение pH 1 М раствора НС1 равно 0, а рОН 14. Тогда как для 1 М раствора
NaOH pH 14, арОНО.
Уравнение (7.13) можно использовать для расчета концентрации гидро-
ксид-ионов, если известна концентрация ионов водорода, и наоборот. Для непо-
средственного расчета значений pH или рОН используют уравнение в
логарифмической форме
-lg Kw = -1g [Н+][ОН-] = -1g [Н+] - 1g [ОН-]
(7-17)
При 25 °C
p£w = pH + pOH
(7.18)
14,00 = pH + рОН
(7-19)
7.3. ШКАЛА PH
319
Пример 7.3
Рассчитайте значения рОН и pH 5,0 • 10-2 М раствора NaOH.
Решение
[ОН-] = 5,0 • IO"2 М
рОН = -1g (5,0 • 10-2) = 2 - 1g 5,0 = 2 - 0,70 = 1,30
pH + 1,30= 14,00
pH = 12,70
или
+ ] = lo-ioj^ = 1O-13 M
5,0 IO"2
pH = -1g (2,0 • 10-13) = 13 - 1g 2,0 = 13 - 0,30 = 12,70
Пример 7.4
Рассчитайте значение pH раствора, приготовленного смешением 2,0 мл раствора
сильной кислоты с pH 3,00 и 3,0 мл раствора сильного основания с pH 10,00.
Решение
[Н+] сильной кислоты = 1,0 • 10-3 М
ммоль Н+ = 1,0 • 10~3 М • 2,0 мл = 2,0 • 10-3 ммоль
рОН раствора основания = 14,00 - 10,00 = 4,00
[ОН-] =1,0- 10^М
ммоль ОН" = 1,0 • 10-4 М • 3,0 мл = 3,0 • 10-4 ммоль
В растворе избыток кислоты.
ммоль Н+ = 0,20020 - 0,0003 = 0,0017 ммоль
[Н+] = 0,0017 ммоль/5,0 мл = 3,4 • 10^ М
pH = -1g (3,4 • 10^) = 4 - 0,53 = 3,47
320
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
Пример 7.5
Значение pH раствора равно 9,67. Рассчитайте концентрацию ионов водорода в
растворе.
Решение
-1g [Н+] = 9,67
[Н+] = 10-9’67 = ю-10 • Ю0’33
[Н+] = 2,1 • Ю-10М
Если [Н+] = [ОН-] раствор называют нейтральным. Если [Н+] > [ОН-], рас-
твор кислый. И наконец, если [Н+] < [ОН-], раствор называется щелочным.
Концентрации ионов водорода и гидроксид-ионов в чистой воде равны 10-7 М
каждая, следовательно, значение pH воды равно 7, что соответствует нейтраль-
ному раствору. Если pH больше 7 — раствор щелочной, а если меньше 7 — кис-
лый. Обратное утверждение справедливо для рОН. В нейтральном растворе
значение рОН тоже равно 7. Помните, что при 25 °C произведение [Н+] и [ОН-]
всегда равно 1,0 • 10-14, а сумма pH и рОН — всегда равна 14. Если температура
отличается от 25 °C, то Kw не будет равно 1,0 • 10-14, и нейтральный раствор бу-
дет иметь отличные от 10-7 М концентрации ионов водорода и гидроксид-ионов
(см. ниже).
Студентам часто кажется, что значения pH не могут быть отрицательными.
У таких утверждений нет теоретического обоснования. Отрицательное значе-
ние pH указывает только на то, что концентрация ионов водорода больше 1 М.
На практике отрицательные значения pH встречаются не часто по двум причи-
нам. Даже сильная кислота в очень концентрированных растворах может находить-
ся в недиссоциированной форме. Например, 100%-ная серная кислота настолько
слабо диссоциирует, что ее даже можно хранить в железных контейнерах; в бо-
лее разбавленных растворах серная кислота диссоциирует с образованием до-
статочного количества ионов водорода для растворения железа. Вторая причина
связана с активностью. Поскольку pH в действительности представляет собой
-1g ан+, то раствор с концентрацией ионов водорода 1,1 М может в действитель-
ности иметь положительное значение pH, так как активность Н+ меньше 1,0 М*.
Это связано с тем, что при таких высоких концентрациях коэффициент активно-
сти меньше единицы (хотя при еще больших концентрациях коэффициент актив-
ности может стать больше единицы — см. гл. 6). Тем не менее с математической
точки зрения нет причин для того, чтобы значения pH (или рОН) не могли быть
Как будет показано в гл. 13, существуют трудности при определении pH растворов с отрицательными зна-
чениями pH или рОН, так как при работе с высококонцентрированными растворами кислот и оснований
появляются погрешности определения, связанные с возникновением значительного и неизвестного по ве-
личине потенциала жидкостного соединения.
7.3. ШКАЛА PH
321
отрицательными, хотя в аналитической химии это встречается редко. Значения
pH и рОН для 10 Мраствора НС1 составляют соответственно -1 и 15.
Если концентрация кислоты или основания намного меньше 10“7 М, то
вкладом такой кислотности или основности можно пренебречь по сравнению с
вкладом соответствующих ионов, образующихся в результате автопротолиза
воды. Поэтому значение pH 10-8 М раствора гидроксида натрия не будет очень
отличаться от 7. Значение pH 10~9 Мраствора НС1 не равно 9! Если концентра-
ция кислоты или основания близка к 10-7 М, то этим вкладом нельзя пренебречь
также как и вкладом соответствующих ионов воды; следовательно, в этом слу-
чае необходимо суммировать концентрации.
Пример 7.6
Рассчитайте значения pH и рОН 1,0 • 10-7 М раствора НС1.
Решение
В растворе устанавливаются следующие равновесия:
НС1 -> Н+ + С1-
н2о^н+ + он-
[Н+][ОН“] = 1,0 • ю-14
[Н ] Н2Одисс. ~ [ОН ] н2Одисс. ~~ %
Поскольку вкладом ионов водорода, образованных при диссоциации воды, не-
льзя пренебречь по сравнению с НО
[Н+] = Снс1 + [Н+]Н2Одисс
Тогда
([Н+]НС1+х) • х = 1,0 • 10“14
(1,00- 10~7 + x)-x= 1,0- 10~14
х2+1,00- 10~7х-1,0- 10-14 = 0
Решая квадратное уравнение [см. Приложение В и используйте инструмент Excel
Поиск решения (см. разд. 6.11)], получаем:
-1,00• 10-7 ±71,0-Ю"14 + 4(1,0 4 О-14)
2
322
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
Отсюда общая концентрация ионов водорода= (1,00 • 10-7 + 6,2 • 10-8) = 1,62 • 10-7 М:
pH = -1g (1,62 • 10-7) = 7 - 0,21 = 6,79
рОН= 14,00-6,79 = 7,21
или, так как [ОН-] = х,
рОН = -1g (6,2 • 10-8) = 8 - 0,79 = 7,21
Обратите внимание, что в результате добавления ионов водорода, ионизация
воды подавляется на 38% вследствие влияния одноименных ионов (принцип Ле
Шателье). При более высоких концентрациях кислоты (или основания) эффект
подавления возрастает и вклад от диссоциации воды становится незначитель-
ным. Этот вклад можно не учитывать, если концентрация протонов или гидро-
ксид-ионов от кислоты или основания будет больше или равна 10-6 М.
Расчет в данном примере является скорее формальным, так как на практике
диоксид углерода из воздуха растворяется в воде и концентрация ионов водоро-
да изменяется. Так как при растворении диоксида углерода в воде образуется
кислота, для того, чтобы получить раствор кислоты с концентрацией 10-7 М, не-
обходимо принять дополнительные меры для удаления диоксида углерода из
воды и исключения его попадания в раствор.
7.4. Значения pH при повышенных температурах: pH крови
Для студентов и химиков, имеющих дело с расчетами кислотности и шкалой pH,
оказалось очень удобным, что при комнатной температуре в водных растворах
величина pKw — целое число. При 100 °C Kw = 5,5 • 10-13, и в нейтральном рас-
творе
[Н+] = [ОН-] = д/5,5 -10-13 = 7,4 10-7 М
рН = рОН = 6,13
pKw = 12,26 = pH + рОН
У нейтрального раствора при температуре выше комнатной pH < 7.
Не все измерения проводят при комнатной температуре, поэтому иногда
необходимо учитывать зависимость величины Kw от температуры (о зависимо-
сти констант равновесия от температуры см. гл. 6). Важным примером является
измерение pH жидкостей тела человека. Значение pH крови при температуре
тела (37 °C) колеблется от 7,35 до 7,45. Это значение лишь немного выше по
сравнению с pH чистой воды при комнатной температуре. При 37 °C
7.5. СЛАБЫЕ КИСЛОТЫ И ОСНОВАНИЯ: КАК РАССЧИТАТЬ pH?
323
Kw = 2,5 • 10-14, a pKw = 13,60. Значение pH (и рОН) нейтрального раствора при
этой температуре равно 13,60/2 = 6,80. Концентрация ионов водорода (и гидро-
ксид-ионов) равна д/2,5 • 10-14 = 1,6 • 10-7М. Следовательно, нейтральный раствор
крови должен был бы иметь при 37 °C pH 6,80, а pH крови 7,4 при 37 °C указыва-
ет на то, что среда на 0,2 единицы pH более щелочная, по сравнению с той, какая
была бы при 25 °C. Такое различие важно на фоне того, что изменение pH на 0,3
единицы считается критическим.
Концентрация хлороводородной кислоты в желудке находится в пределах
от 0,1 до 0,02 М. Так как pH = - 1g [Н+], то pH 0,02 М раствора равно 1,7. Это зна-
чение будет постоянным независимо от температуры, так как концентрация
ионов водорода остается неизменной (пренебрегая изменением объема раство-
рителя). Но при 25 °C рОН = 14,0 - 1,7 = 12,3, а при 37 °C рОН =13,6-1,7=11,9.
Температура влияет не только на ионизацию воды, находящейся в теле, и
тем самым на значение pH, она также влияет на константы ионизации кислот и
оснований, которые образуют буферные системы в организме. Как будет пока-
зано далее в этой главе, это влияет на pH буферов, так что значение pH крови,
измеренное при 37 °C и равное 7,4, не будет совпадать со значением pH крови,
измеренным при комнатной температуре; а pH в желудке определяется кон-
центрацией сильной кислоты и не зависит от температуры. Именно поэтому
измерение pH крови для диагностических целей обычно выполняют при 37 °C
(см. гл. 13).
7.5. Слабые кислоты и основания: как рассчитать pH?
До сих пор мы проводили расчеты только для растворов сильных кислот и осно-
ваний, в которых, как принято считать, происходит полная ионизация.
Поскольку концентрации ионов водорода или гидроксид-ионов определяют
непосредственно из концентрации кислоты или основания, такие расчеты доста-
точно просты. Как следует из уравнения (7.3), слабые кислоты (или основания)
ионизируются лишь частично. Минеральные (неорганические) кислоты и осно-
вания, такие как НС1, НС1О4, HNO3 и NaOH, являются сильными электролитами,
которые полностью ионизируются в воде, тогда как большинство органических
кислот и оснований, нашедших клиническое применение, являются слабыми
электролитами.
Для расчета количества ионизированной формы вещества, а следовательно,
и значения pH, используют константу ионизации. Константа кислотности уксус-
ной кислоты при 25 °C равна 1,75 • 10-5:
[Н+][ОАс-]= 51о_5
[НОАс]
(7.20)
324
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
При ионизации уксусной кислоты происходит ее диссоциация с образова-
нием равных количеств ионов Н+ и ОАс-, так что левая часть уравнения (7.20)
всегда будет равна 1,75 • 10-5:
НОАс ^Н+ +ОАс- (7.21)
Если исходную концентрацию уксусной кислоты обозначить С, а концентрацию
образующихся частиц (Н+ и ОАс-) как х, можно записать равновесные концент-
рации всех частиц
НОАс ^Н+ +ОАс- (7.22)
(С - х) х х
Пример 7.7
Рассчитайте pH и рОН 1,00 • 10-3 М раствора уксусной кислоты.
Решение
НОАс^Н+ + ОАс-
Концентрации присутствующих в растворе частиц следующие:
[НОАс] [Н+] Исходные 1,00 10-3 0 [ОАс-] 0
Изменение (х = ммоль/мл ~х +х ионизированной НОАс) Равновесные 1,00 • 10-3-х х +х X
Из уравнения (7.20)
------------= 1,75 Ю-5
1,00-10-3 -х
Необходимо решить квадратное уравнение. Если в ионизированной форме нахо-
дится менее 10-15% кислоты, уравнение часто упрощают, пренебрегая величи-
ной х по сравнению с С (в этом случае 10-3 М). Это спорное (и не очень
обязательное) условие.
Упрощение возможно, если Ка меньше 0,01 С, т. е. меньше 10-4 при С=0,01 М,
10-3 при С = 0,1 М, и т. д. При таких условиях погрешность расчета составляет
5% и менее (значения получаются неколько завышенные), и обычно не превы-
шает погрешность определения константы равновесия. После упрощения полу-
чаем:
7.5. СЛАБЫЕ КИСЛОТЫ И ОСНОВАНИЯ: КАК РАССЧИТАТЬ pH?
325
—------т- = 1,75-10-15
1,00-10-3
х= 1,32- 1(НМн[Н+]
Следовательно
pH = -1g (1,32 • 1(H) = 4 - 1g 1,32 = 4 - 0,12 = 3,88
рОН= 14,00-3,88= 10,12
Сделанное упрощение в расчетах не сильно влияет на результат, потому что
константы равновесия часто определены с не очень высокой точностью (не
выше ±10 %). Так, в приведенном примере при решении квадратного уравнения
получаем [Н+] = 1,26 • 1 О*4 М (на 5% меньше) и pH 3,90. Это значение pH отлича-
ется на 0,02 единицы от значения, полученного при упрощенном расчете, что
близко к предельному значению точности, с которой проводят измерения pH
(абсолютная погрешность измерений pH не превышает 0,02 единицы — см. гл. 13.).
Почти наверняка расчет настолько точен, насколько это возможно, если учесть экс-
периментальные погрешности определения значений Ка или Кь и то, что при рас-
четах мы используем концентрации, а не активности. При расчетах мы также не
учитывали ионы водорода, образующиеся при ионизации воды (что было впол-
не оправдано); это допущение является общим за исключением очень разбав-
ленных растворов (< 10-6 М) или очень слабых кислот (Ка < 10-12).
Подобные уравнения и расчеты справедливы для слабых оснований.
Пример 7.8
Константа основности Кь аммиака равна 1,75 • 10-5 при 25 °C (совпадение с кон-
стантой кислотности уксусной кислоты всего лишь случайность). Рассчитайте
pH и рОН 1,00 • 10-3 М раствора аммиака.
Решение
NH3 + Н2О NH* + ОН-
(1,00- 10"3-х) х х
X X
^1[ОН~1=1.75.Ю"
[NH3]
326
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
Для решения применим тот же подход, что и в случае слабой кислоты. Таким об-
разом,
Х Х , = 1,75 10~5
1,00 10-3
х= 1,32 • 10-4М = [ОН-]
pOH = -lg(l,32 • 10-4) = 3,88
рН= 14,00-3,88 = 10,12
7.6. Соли слабых кислот и оснований:
их растворы не являются нейтральными
Соль слабой кислоты, например NaOAc, является сильным электролитом, почти
как все соли, и, следовательно, полностью ионизируется. Кроме того, анион
соли слабой кислоты является основанием по Бренстеду, которое будет прини-
мать протоны. Анион частично гидролизуется в воде (кислота по Бренстеду) с
образованием гидроксид-иона и соответствующей недиссоциированной кисло-
ты. Например,
ОАс- + Н2О НОАс + ОН- (7.23)
В данном случае НОАс не диссоциирует и поэтому не вносит вклад в значе-
ние pH. Такой процесс называется гидролиз иона соли. В результате гидролиза
ацетат натрия обладает свойствами слабого основания (сопряженное основание
уксусной кислоты). Чем слабее сопряженная кислота, тем сильнее сопряженное
основание, а это значит, что соль будет сильнее взаимодействовать с протоном
воды, при этом равновесие в уравнении (7.23) сместится вправо. Равновесия с
участием оснований по Бренстеду рассматривают точно также как равнове-
сия с участием слабых оснований, которые мы только что рассматривали. Мы
можем записать константу равновесия:
£н = ^ = [НОАС][ОН ] (7.24)
[ОАс ]
КИ называют константой гидролиза соли, и она равна константе основности
соли. Мы будем пользоваться обозначением Кь, чтобы подчеркнуть, что такие
соли следует рассматривать как слабые основания.
Величину Кь можно рассчитать из Ка уксусной кислоты и Kw, если умно-
жить и числитель и знаменатель на [Н+]:
к _ [НОАс]
Ь [ОАс-] [Н+]
(7.25)
7.6. СОЛИ СЛАБЫХ КИСЛОТ И ОСНОВАНИЙ: ИХ РАСТВОРЫ НЕ ЯВЛЯЮТСЯ НЕЙТРАЛЬНЫМИ
327
Произведение величин внутри пунктирной линии — это Kw, а остальная
часть уравнения равна 1/ Ка. Следовательно,
К ь= = 1,0-10 14 = 5,7 • 1О-10
Ь Ка 1,75-КГ5
(7.26)
Малая величина Кь, свидетельствует о том, что ацетат ион представляет собой
очень слабое основание, и только очень малая часть от его общего количества
подвергается гидролизу. Произведение Ка слабой кислоты и Кь ее сопряженного
основания равно Kw:
Wb = Kw (7.27)
Для соли слабой кислоты НА, которая подвергается в воде гидролизу
а-+н2о^на + он-
(7.28)
[НА][ОН-]_^ _к
[А’] Ка 1
(7.29)
Значение pH раствора такой соли (основание по Бренстеду) рассчитывают так-
же как в случае слабого основания. При гидролизе соли образуются равные ко-
личества НА и ОН-. Если исходную концентрацию А- обозначить Сд_, то
А- + Н2О НА + ОН-
(СА_ -х) хх
(7.30)
Величиной х можно пренебречь по сравнению с Сд_, если Сд_ > lOOA^, как
обычно происходит в случае для таких слабо ионизированных оснований.
Мы можем рассчитать концентрацию ОН-, используя уравнение (7.29):
(7.31)
Сравните такой ход решения с решением в примере 7.8. Они идентичны:
[ОН =
(7.32)
Это уравнение можно применять только в том случае, если СА_ > 100 2СЬ, и если
величиной х можно пренебречь по сравнению с Сд_. В противном случае необ-
ходимо решать квадратное уравнение, также как и для других оснований в по-
добной ситуации.
[OH-][OH-]_*W
328
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
Пример 7.9
Рассчитайте pH 0,1 М раствора ацетата натрия.
Решение
Запишем равновесия
NaOAc -> Na+ + О Ас- (ионизация)
ОАс- + Н2О НОАс + ОН- (гидролиз)
Запишем выражение константы равновесия
[НОАс][ОН-] Kw 1,0 10-14 ,71п-ю
[ОАс-] Ка 1,75-10-5
Обозначим через х равновесные концентрации НОАс и ОН-. Тогда, при рав-
новесии
[НОАс] = [ОН-] =х
[ОАс-]=С„. _ -х = 0,10-х
Поскольку Спд _ Kh, пренебрегаем х по сравнению с Спд _. Тогда
ОАС и ОАС
— = 5,7-1О-10
0,10
х = л/5,7-10-10-0,10 = 7,6 • 10-6 М
Сравните последнее уравнение с уравнением (7.32), а также сравните все
стадии решения этого примера с решением примера 7.8. Образующиеся молеку-
лы НОАс не диссоциируют и поэтому не влияют на значение pH:
[ОН-] = 7,6 • 10-6 М
+ = 1,0-10 14 = 1О-9М
7,6-10-6
pH = -1g (1,3 • 10-9) = 9 - 0,11 = 8,89
Подобные уравнения можно вывести для катионов солей слабых оснований
(соли полностью диссоциированы). Такие катионы являются кислотами по
Бренстеду и ионизируются (гидролизуются) в воде:
ВН+ + Н2О В + Н3О+
(7.33)
7.6. СОЛИ СЛАБЫХ КИСЛОТ И ОСНОВАНИЙ: ИХ РАСТВОРЫ НЕ ЯВЛЯЮТСЯ НЕЙТРАЛЬНЫМИ
329
Основание В не диссоциирует и не вносит вклад в значение pH. Константа
кислотности равна
Формулу для расчета константы кислотности (константы гидролиза) можно
получить, умножив числитель и знаменатель на [ОН-]:
[ВН+] [ОН"]
(7.35)
И в этом случае произведение величин, обведенное пунктирной линией — это
Kw, а остальная часть уравнения равна 1/ Кь. Следовательно,
и для иона аммония NH £
_1,0-10-14 _5? 1о_1О
Кь 1,75 10“5
(7.37)
Мы могли бы, конечно, рассчитать Ка по уравнению (7.27). Еще раз повторяем,
что численное совпадение величин Ка для иона аммония и Кь для ацетат иона
случайно.
Соли слабых оснований ионизируются с образованием равных количеств В
и Н3О+ (сокращенно обозначаемого как Н+). Поэтому мы можем определить кон-
центрацию ионов водорода (допуская, что Свн+ >100 Ка);
Опять же, это уравнение можно использовать, только если Свн+ >100 Ка. В про-
тивном случае необходимо решать квадратное уравнение.
330
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
Пример 7.10
Рассчитайте pH 0,25 М раствора хлорида аммония.
Решение
Запишем равновесия
NH4C1 -> NH4 + Cl- (ионизация)
NH4 + Н2О NH4OH + Н+ (гидролиз)
(NH4 + Н2О NH3 + Н3О+)
Выражение константы равновесия
[NH4OH][H+] _kw _ 1,0-ю-14 1П_ю
[NH4] Кь 1,75-Ю-5
Обозначим равновесные концентрации [NH4OH] и [Н+] через х. Тогда при
равновесии
[NH4OH] = [H+]=x
[NHJ] = -х = 0,25-х
Поскольку » Ка, то можно пренебречь значением х по сравнению с CNH+.
Тогда,
— = 5,7-1О-10
0,25
х = 75,7-10-10 -0,25 = 1,2 • 10-5 М
Сравните последнее уравнение с уравнением (7.39). Сравните также все ста-
дии решения этого примера с решением примера 7.7. Образующиеся молекулы
NH4OH не диссоциируют и поэтому не влияют на значение pH:
[Н+] = 1,2 • IO"5 М
pH = -1g (1,2 • 10-5) = 5 - 0,08 = 4,92
7.7. Буферные растворы: поддерживают значение pH
постоянным [или примерно постоянным]
Буферными называют растворы, которые препятствуют изменению pH при до-
бавлении к раствору небольших количеств кислоты или основания либо при раз-
бавлении раствора. Их удобно использовать для поддержания оптимального
7.7. БУФЕРНЫЕ РАСТВОРЫ
331
значения pH для протекания химической реакции. Буферные растворы состоят
из смеси слабой кислоты и сопряженного ей основания или из слабого основа-
ния и сопряженной ему кислоты, концентрации и соотношения которых заранее
определены. Таким образом, буферные растворы представляют собой смеси
слабой кислоты и ее соли или слабого основания и его соли. Рассмотрим буфер-
ный раствор уксусная кислота — ацетат (ацетатный буфер). Кислотное равнове-
сие, которое определяет эту систему, имеет вид
НОАс Н+ + ОАс-
Если добавить в систему источник ацетат-ионов (например, ацетат натрия),
концентрация ионов водорода больше не будет равна концентрации ацетат-
иона. Концентрация ионов водорода равна
+ = к [^[НОАс]
[ОАс-]
Взяв отрицательный логарифм каждой части этого уравнения, мы получим
-lg[H+]=-lg^-lg^2^ (7.41)
[ОАс ]
тт 1 [НОАс] z_
рН = р7Са-lgL J (7.42)
[ОАс ]
Если в уравнении (7.42) перевернуть дробь под знаком логарифма, то второй
член станет положительным:
рН = рАГа +lg
[ОАс~]
[НОАс]
(7.43)
Полученное уравнение называется уравнением Гендерсона—Хассельбаха.
Его используют для расчета pH растворов слабых кислот, содержащих их соли.
В общем виде уравнение можно записать для слабой кислоты НА, которая иони-
зируется с образованием аниона А~ и Н+:
НА Н+ + А" (7.44)
рН = рА? +lg^-l
[НА]
pH - рА? + 1g [сопРяженное основание]
а [кислота]
рН = р7С । 1g [акЦептоР протонов]
° [донор протонов]
(7.45)
(7.46)
(7.47)
332
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
Значение pH буферных растворов определяется соотношением концентра-
ций сопряженных пар кислота-основание.
Пример 7.11
Рассчитайте pH буферного раствора, приготовленного добавлением 10 мл 0,10 М
раствора уксусной кислоты к 20 мл 0,10 М раствора ацетата натрия.
Решение
Необходимо рассчитать концентрацию кислоты и соли в растворе. Общий объ-
ем раствора составляет 30 мл:
с,и = с2к2
Для НОАс,
0,1 ммоль/мл • 10 мл = СН0Ас • 30 мл
СН0Ас = 0,033 ммоль/мл
Для ОАс-,
0,1 ммоль/мл • 20 мл = СПА _ • 30 мл
’ ОАс
С\А _ = 0,067 ммоль/мл
Часть молекул НОАс диссоциирует на Н+ и ОАс-, и равновесная концентра-
ция НОАс будет равна концентрации добавленной кислоты (0,033 М) за выче-
том продиссоциировавшей НОАс. Однако концентрация диссоциированной
формы кислоты очень мала, особенно в присутствии добавленной соли (подав-
ление ионизации обусловлено влиянием одноименного иона), и им можно пре-
небречь. Следовательно, мы можем полагать, что равновесные концентрации
веществ равны их исходным концентрациям:
pH =-IgA: I 1g [акцептоР протонов]
а [донор протонов]
pH = - 1g (1,75 • 10-5 ) + 1g 0,067 ммоль/мл = 4,76 + 1g 2,0 = 5,06
0,033 ммоль/мл
Расчет можно упростить, сократив под знаком логарифма объемы веществ,
и использовать отношение их количеств, выраженных в ммоль:
«ноле = 0Д0 ммоль/мл • 10 мл = 1,0 ммоль
«оас- = ОДО ммоль/мл • 20 мл = 2,0 ммоль
тт Л к . 1 2,0 ммоль .
pH = 4,76 + 1g —-----= 5,06
1,0 ммоль
7.7. БУФЕРНЫЕ РАСТВОРЫ
333
Смесь слабой кислоты и ее соли можно получить добавлением избытка сла-
бой кислоты к сильному основанию (в результате их взаимодействия образуется
соль). Еще один способ заключается в добавлении избытка соли слабой кислоты
к сильной кислоте, при этом образуется слабая кислота.
Пример 7.12
Рассчитайте pH раствора, приготовленного добавлением 250 мл 0,10 М раствора
гидроксида натрия к 30 мл 0,20 М уксусной кислоты (это может наблюдаться в
ходе обычного титрования).
Решение
Выразите количества реагентов в ммоль:
«ноле= 0’20 М • 30 мл = 6,0 ммоль
«NaOH = 0,10 М • 25 мл = 2,5 ммоль
Компоненты смеси реагируют следующим образом:
НОАс + NaOH NaOAc + Н2О
После реакции
«NaOAc = 2’5 ММОЛЬ
«ноле= ^’0 - 2,5 = 3,5 ммоль
Механизм буферирования для смеси слабой кислоты и ее соли можно объ-
яснить следующим образом. Значение pH определяется отношением концентра-
ций соли и кислоты под знаком логарифма отношения:
[А”1
pH = constant + 1g i—-
[НА]
(7-48)
При разбавлении раствора соотношение компонентов буферной системы
остается постоянным, и следовательно не происходит изменения pH*.
При добавлении небольшого количества сильной кислоты она вступит в ре-
акцию с эквивалентным количеством иона А~, с образованием НА. А это значит,
В действительности значение pH немного увеличится, потому что коэффициент активности соли увели-
чится при уменьшении ионной силы раствора, а активность незаряженных молекул (например, недиссоци-
ированной кислоты) останется равной молярности (см. гл. 6), т. е. отношение увеличивается. Подробнее об
этом рассказано в конце этой главы.
334
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
что в равновесной системе НА Н+ + А-, согласно принципу Ле Шателье, до-
бавленные ионы водорода связывают ионы А-, с образованием НА, а при избыт-
ке ионов А-равновесие сильно смещено влево. Изменение отношения [А~]/[НА]
невелико и, следовательно, мало изменение pH. Если кислоту добавили бы к не
буферному раствору, а, например, к раствору NaCl, то pH значительно уменьши-
лось бы. При добавлении небольшого количества сильного основания к буферно-
му раствору оно вступит в реакцию с частью НА с образованием эквивалентного
количества иона А-. И в этом случае изменение отношения концентраций будет
небольшим.
Количество кислоты или основания, которое можно добавить не вызвав си-
льного изменения pH, определяется буферной емкостью раствора. Буферная
емкость определяется концентрациями НА и А-: чем они выше, тем больше до-
пустимое количество добавленной кислоты или основания, не приводящее к
сильному изменению pH. Буферная емкость определяется так
Р = <7СВОн /<7рН = -dCBK /JpH
(7-49)
где я?Свон и JCHA — соответственно количество (моль/л) сильного основания
или кислоты, необходимое для изменения pH на величину JpH. Буферная ем-
кость — положительная величина. Чем она больше, тем больше «сопротивле-
ние» раствора изменению pH. Для буферных растворов, состоящих из слабой
кислоты и сопряженного ей основания, с концентрацией больше 0,001 М буфер-
ную емкость можно приближенно оценить по уравнению:
2,303—наСа~
^НА +^А-
(7.50)
где СНА и С А_ — аналитическая концентрация кислоты и соли соответственно.
Так, буферная емкость смеси 0,10 моль/л уксусной кислоты и 0,10 моль/л ацета-
та натрия будет равна
Р = 2,303 0’-0- 0’10 = 0,050 М°ЛЬ (для изменения на единицу pH)
0,10 + 0,10 л
При добавлении твердого гидроксида натрия до 0,0050 моль/л, изменение
pH составит:
JpH = dCB0H/ Р = 0,0050/0,050 = 0,10 = АрН
Буферная емкость зависит не только от концентрации компонентов раство-
ра (НА и А”), но и от их соотношения. Наибольшая величина буферной емкости
получается, если это соотношение равно единице, т. е. при pH = рКа.
pH = p^ + lg| = p^ (7.51)
7.7. БУФЕРНЫЕ РАСТВОРЫ
335
Это соответствует середине кривой титрования слабой кислоты. В целом,
буферная емкость считается удовлетворительной в интервале pH, равном рКа ± 1.
Мы будем обсуждать буферную емкость на иллюстративном материале в гл. 8
(построение кривых титрования слабых кислот).
Пример 7.13
Рассчитайте изменение pH буферного 0,20 М раствора по уксусной кислоте и
ацетату натрия, после добавления к 10 мл этого раствора 1,0 мл 0,10 М раствора
хлороводородной кислоты.
Решение
В исходном буферном растворе pH равно рКа, так как отношение [ОАс-]/[НОАс]
равно единице. Значение pH составляет 4,76. Для того, чтобы рассчитать новое
значение pH после добавления НС1, необходимо определить новые концентра-
ции НОАс и ОАс-. Начнем с того, что количество ацетата в 10 мл равно
10 • 0,20 = 2,0 ммоль. Мы добавили 1,0 • 0,10 = 0,10 ммоль Н+ и в результате реак-
ции с 0,10 ммоль ОАс- образовалось такое же количество НОАс:
«ноле= 2,0 + 0,1 =2,1 ммоль
п^. _ = 2,0-0,1 = 1,9 ммоль
Такое количество уксусной кислоты и ее соли содержится в 11 мл раствора, но в
наших расчетах объемы сокращаются:
тт л , 1 1,9 ммоль/Пмл . . 1,9 ммоль
pH = 4,76 + 1g —---------= 4,76 + 1g —-----=
2,1 ммоль/Пмл 2,1 ммоль
= 4,76 + 1g 0,90 = 4,76 + (0,95 - 1) = 4,71
Изменение pH составляет -0,05 единицы. Это довольно маленькая величина,
особенно по сравнению с тем изменением, которое произошло бы, если бы то же
количество НО добавили к нейтральному, не буферному раствору; в этом слу-
чае концентрация раствора по НС1 стала бы примерно 10-2 М, а значение pH
было бы 2,0.
Следует отметить, что буферный раствор может сопротивляться изменению
pH даже в таких случаях, когда количество добавленной к нему сильной кисло-
ты или сильного основания (в моль) превышает равновесное количество ионов
Н+ или ОН- (в моль) в буферном растворе. В качестве иллюстрации можно при-
вести пример 7.13. Значение pH буферного раствора составило 4,76, следователь-
но, [Н+] =1,7 • 10-5 М, или 1,7 • 10-5 ммоль/мл, а в 10 мл раствора количество
ионов водорода, находящихся в равновесии с компонентами буферного раство-
336
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
ра, равно 1,7 • 10-4 ммоль. Мы добавили 0,10 ммоль Н+, что намного превышает
исходное количество. Тем не менее, благодаря компенсирующему действию
компонентов буферного раствора (в этом случае ОАс- реагирует с Н+), добав-
ленные ионы Н+ связываются и значение pH остается практически постоянным
до тех пор, пока количество добавленных ионов Н+ не превысит количество бу-
ферного резерва.
Подобные же расчеты применимы для смеси слабого основания и его соли.
Мы можем рассмотреть равновесие между основанием В и сопряженной кисло-
той ВН+ и записать выражение для Ка этой сопряженной кислоты (по Бренстеду):
ВН+ = В + Н+ (7.52)
[В][Н+] Х'И,
А„--------------- ( / .JJ)
[ВН+] кь
Логарифмическую форму уравнения Гендерсона—Хассельбаха можно полу-
чить так же, как это сделано выше*:
[Н+] = Ха
[ВН+]_^ [вн+]
[В] кь [В]
-ig[H+]=-ig/ca
[В] Кь [В]
(7-54)
(7.55)
pH=?ка + 1g = (?KW - PKb)+1g
[BH J [Bn J
pH = p# I lg [акцептор протонов]
a [донор протонов]
= (p^w -P^b) + lg
[акцептор протонов]
[донор протонов]
(7-56)
(7-57)
Так как рОН = pKw - pH, мы можем записать это выражение в другом виде,
заменив pH уравнением (7.56) либо (7.57) (т.е. вычесть одно из этих уравнений
изрЯ^),
ГАТТ rs 1 [ВН+] „ , [донор протонов]
рОН = рКь + lg -J = pKb + 1g
[В] [акцептор протонов]
Механизм буферного действия смеси слабого основания и его соли такой
же, как и у смеси слабой кислоты и ее соли. При добавлении сильной кислоты,
она реагирует с некоторым количеством основания В с образованием соли ВН+.
И наоборот, добавленное основание взаимодействует с ВН+, образуя слабый
электролит В.
См. уравнения (7.40)-(7.47). — Прим. пер.
7.7. БУФЕРНЫЕ РАСТВОРЫ
337
Так как изменение соотношения концентраций основания и сопряженной
кислоты будет небольшим, то и значение pH изменится незначительно. Опять
же, величина буферной емкости максимальна при pH = рКа = 14 - рКь (или при
рОН = рКь), а рабочий интервал pH таких буферных растворов составляет
р*а±1.
Пример 7.14
Рассчитайте объем концентрированного раствора аммиака и массу хлорида ам-
мония, которые необходимо взять для приготовления 100 мл буферного раство-
ра с pH 10,00, если конечная концентрация соли в буферном растворе должна
быть 0,200 М.
Решение
Нам нужно получить 100 мл 0,200 М NH4C1. Поэтому
«nh4ci = 0,200 ммоль/мл • 100 мл = 20,0 ммоль
^nh4ci = 20,0 ммоль • 53,5 мг/ммоль = 1,07 • 103 мг
Следовательно, нам необходимо взять 1,07 г NH4C1. Рассчитаем равновесную
концентрацию аммиака:
рН_+ [g [акцептор протонов] = .pj Т-щЩ
[донор протонов] ' [NH4]
10,0 = (14,00 - 4,76) + 1g-------
0,200 ммоль/мл
g 'NH-' , =0,76
0,200 ммоль/ мл
_____________= 10°’76 =5,8
0,200 ммоль/мл
[NH3] = 0,200 • 5,8 = 1,16 ммоль/мл
Молярная концентрация концентрированного раствора аммиака составляет
14,8 М. Следовательно,
100 мл • 1,16 ммоль/мл = 14,8 ммоль/мл • мл NH3
^NH3 = 7’8 МЛ
338
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
Пример 7.14
Сколько граммов хлорида аммония и сколько миллилитров 3,0 М раствора гид-
роксида натрия необходимо добавить к 200 мл воды, чтобы при разбавлении до
500 мл получить буферный раствор с pH 9,50 и концентрацией соли 0,10 М.
Решение
Нам нужно рассчитать отношение [NH3]/[NH J]. Из примера 7.14
рН = р^+1ё2Щ=^4 + 1ё™
[NH4] [NH+4]
9,50 = 9,24 + 1g
[NH3]
[NH+4]
lg№U,26
[NH+4]
[NH3]_wq,26
[NH+4]
= 1,8
Конечная концентрация NH4 равна 0,10 M, поэтому
[NH3] = 1,8 • 0,10 = 0,18 М
в конечном растворе — 0,10 М 500 МЛ — 50 ММОЛЬ
WNH3 в конечном растворе — 0,18 М 500 МЛ — 90 ММОЛЬ
Аммиак получается в количестве равном (ммоль) NH4C1, поэтому необходимо
брать суммарное количество 50 + 90 = 140 ммоль NH4C1:
wNh4ci = 140 ммоль • 53,5 мг/ммоль = 7,49 • 103 мг = 7,49 г
Необходимое количество NaOH (ммоль) равно количеству NH3 (ммоль):
3,0 М • х мл = 90 ммоль
х = 30 мл
Выше было показано, что для приготовления буферного раствора с задан-
ным значением pH выбирают слабую кислоту (или слабое основание) и ее (его)
соль с величиной рКа, близкой к нужному значению pH. Известен целый ряд та-
ких кислот и оснований, поэтому можно подобрать буферный раствор для любого
интервала pH при правильном выборе компоненты. Система из слабой кислоты и
ее соли лучше всего проявляет буферные свойства в кислом растворе, а смесь
слабого основания его соли — в щелочном.
7.8. МНОГООСНОВНЫЕ КИСЛОТЫ И ИХ СОЛИ
339
Ниже обсуждаются состав и свойства некоторых буферных систем, используе-
мых для биологических и клинических измерений. Буферные растворы, рекоменду-
емые Национальным институтом стандартов и технологии (NIST) для градуировки
pH электродов рассмотрены в гл. 13.
Вас может удивить, что в буферных смесях соль не гидролизуется водой с
образованием кислоты или основания. Это связано с тем, что гидролиз подавля-
ется в присутствии кислоты или основания. В уравнении (7.28) в присутствии
значительного количества НАс или ОН- ионизация (гидролиз) будет подавлять-
ся до незначительного уровня. В уравнении (7.33) присутствие В или Н3О+ бу-
дет также подавлять ионизацию.
7.8. Многоосновные кислоты и их соли
Многие кислоты и основания являются многоступенчатыми, т. е. молекулы этих
соединений способны последовательно ионизироваться с образованием более
одного протона или гидроксид-иона. Ионизация этих веществ проходит по ста-
диям, и для каждой стадии можно записать константу равновесия. Рассмотрим,
например, ионизацию фосфорной кислоты:
Н3РО4 н+ + н2ро; КаХ = 1,1 • 10-2 = [-^[IhP°4] (7.59)
[НЗГО4 J
Н2РО; Н+ + НРО Ка2 = 7,5-10-8 = [н ИНР04 ] (7.60)
[Н2РО4 ]
НРО?“ Н+ + РО*“ Ка3 = 4,8 • 10~13 = [Н ][Р°4 -1 (7.61)
[НРО*-]
Величины ступенчатых констант кислотности Ка многоосновных кислот
постепенно уменьшаются, так как увеличение отрицательного заряда протонсо-
держащего аниона затрудняет диссоциацию этой частицы.
В гл. 16 поясняется, что общее уравнение ионизации представляет собой
сумму уравнений для каждой ступени, а общая константа ионизации — это про-
изведение ступенчатых констант ионизации:
Н3РО4 ЗН+ + РО^
Ка = Ка1Ка2Ка3 = 4,0 • 10-22 = [^][Р°4 ] (7.62)
[H3rO4 J
Значения ступенчатых констант рА?а1, рА^2, и pX^3 равны 1,96, 7,12 и 12,32. Для
точных расчетов pH необходимо учитывать вклад протонов на каждой ступени
ионизации. Точный расчет труден и проводится по итерационной методике, по-
скольку равновесные концентрации иона водорода и анионов фосфорной кисло-
ты неизвестны. Примеры таких расчетов можно найти в работах [7, 10].
340
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
В большинстве случаев можно рассматривать каждую ступень ионизации по
отдельности. Если разница между последовательными константами ионизации со-
ставляет как минимум 104, можно с помощью титрования определить протоны, об-
разующиеся на каждой ступени ионизации, т. е. для каждого титруемого отдельно
протона (на каждой ступени) фиксировать скачок pH на кривой титрования.
(Если константа ионизации меньше 10~8, то степень ионизации будет слишком
мала, чтобы скачок pH был увиден — например, третий протон для Н3РО4. Мы
можем отдельно оттитровать первые два протона Н3РО4.) При таких условиях
расчеты упрощаются, так как систему можно рассматривать как смесь трех
слабых кислот.
Расчеты pH буферных систем,
содержащих многоосновные кислоты
Анион, образующийся на каждой ступени ионизации, входит в состав соли кис-
лоты, из которой он образуется. Так, в уравнении (7.59) Н2РО 4 — это анион соли
кислоты Н3РО4. В уравнении (7.60) НРО 4” — это анион соли Н2РО 4, а в уравне-
нии (7.61), РО4“ — анион соли кислоты НРО4~. Таким образом, каждая из пар
этих анионов представляет собой буферную систему, и буферы на основе орто-
фосфорной кослоты могут быть приготовлены в широком диапазоне pH.
Максимальная буферная емкость каждой пары соответствует значению pH, рав-
ному величине рКа частицы, играющей роль кислоты в этой паре. Можно приго-
товить фосфатные буферные растворы со значениями pH вблизи 1,96 (рА?а1),
7,12 (рЛ?а2)и 12,32 (рКа3). Пара НРО4“/ Н2РО4 является одной из буферных сис-
тем крови (см. ниже).
Пример 7.16
Значение pH крови составляет 7,40. Рассчитайте отношение [НРО4~]/ [Н2РО4] в
крови, принимая температуру равной 25 °C.
Решение
рН = рАГ । 1g [акЦепт0Р протонов]
а [донор протонов]
р^2 = 7,12
Отсюда,
pH=7,12 + lg
[HPOj-]
[н2ро;]
7.8. МНОГООСНОВНЫЕ КИСЛОТЫ И ИХ СОЛИ
341
7,40 = 7,12 4^1
^н2ро;]
[НРО2~]_1,9
[н2ро;] 1 ’
Диссоциация многоосных кислот
Поскольку для Н3РО4 величины констант ступенчатой ионизации довольно си-
льно отличаются, тоpHраствораН3РО4можнорассчитать, используя тот же
подход, как и в случае любой слабой кислоты. Ионы водорода, образующиеся на
первой ступени диссоциации, эффективно подавляют диссоциацию по двум
другим ступеням. Поэтому вклад ионов водорода от этих двух ступеней незна-
чителен по сравнению с первой ступенью.
Пример 7.17
Рассчитайте pH 0,100 М раствора Н3РО4.
Решение
н3ро4 «н+ + н2ро;
0,100-х х х
Из уравнения (7.59)
- ХХ = 1,1Ю~2
0,100-х
Фосфорную кислоту Н3РО4 можно рассматривать как одноосновную кислоту, но
величиной х пренебречь нельзя, так как она сопоставима с концентрацией С. Вели-
чиной х можно пренебречь только если С > 100АГа. В данном случае концентрация
больше Ка только в 10 раз. Поэтому необходимо решить квадратное уравнение:
х2 + 0,011х-1,1 • 10-3 = 0
_ -0,011 ± 7(0,011)2 -4(-1,110"3)
Х 2
х = [Н+] = 0,028 М
Кислота диссоциирована на 28%:
pH = -1g (2,8 • 10-2) = 2 - 0,45 = 1,55
342
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
Мы можем использовать такой подход только в том случае, если предполо-
жение, что Н3РО4 представляет собой единственный источник протонов, соот-
ветствует действительности. Анион Н2РО4 мог бы быть другим возможным
источником протонов. Из уравнения (7.60) [НРО4“] = Ка2 [Н2РО4]/ [Н+]. Если
предположить, что концентрации Н2РО4 и Н+ равны 0,028 М, тогда [НРО4“]
примерно равна Ка2 = 7,5 • 10-8 М. Это очень малая величина по сравнению с
0,028 М (концентрация Н2РО 4), поэтому другими ступенями диссоциации мож-
но пренебречь. Таким образом, мы подтвердили правомочность сделанных до-
пущений.
Доля диссоциирующих частиц при заданном значении pH.
Величина а: какова доля каждого вида частиц?
Часто требуется знать распределение разных форм многоосновных кислот в за-
висимости от pH, например, при известной концентрации ионов водорода в бу-
ферном растворе.
Рассмотрим, например, диссоциацию фосфорной кислоты. Равновесные
процессы описываются уравнениями (7.59)-(7.61). При любом заданном pH все
четыре формы фосфорной кислоты будут сосуществовать в равновесии друг с
другом, хотя концентрации некоторых из них будут очень низкими. Доля каж-
дой формы определяется значением pH. При изменении pH равновесия смещают-
ся и относительные концентрации частиц изменяются. Можно вывести общие
уравнения для расчета доли кислоты, которая существует в определенной фор-
ме, по заданной концентрации ионов водорода.
Для заданной общей аналитической концентрации фосфорной кислоты,
СНзРо4 , можно записать уравнение
Сн3РО4 = [РОд-]+ [НРОд-] + [Н2ро;] + [Н3РО4] (7.63)
где члены правой части уравнения представляют собой равновесные концент-
рации отдельных форм фосфорной кислоты. Предположим, что известна исход-
ная общая концентрация СНзРО4 и мы хотим найти доли или концентрации
отдельных форм, находящихся в равновесии.
Обозначим
[Н3РО4] ао= ? <-'Н3РО4 _[н2ро;] _[нро*-] 1А, 1 (Лэ 1 С z С СН3РО4 СН3РО4
[ро^-] а2 = — С СН3РО4 а0 + 0ц + а2 + а3 = 1
7.8. МНОГООСНОВНЫЕ КИСЛОТЫ И ИХ СОЛИ
343
где az (z = 0 4- 4)—это доля каждой формы, находящейся в равновесии. Обратите
внимание, что подстрочный индекс i обозначает число диссоциированных про-
тонов или заряд частицы. Для того, чтобы выразить общую аналитическую кон-
центрацию СНзрО4 через равновесные концентрации заданных форм, мы можем
использовать уравнение (7.63) и уравнения, определяющие константы равнове-
сия (7.59)-(7.61). Далее подставляем СНзРО4 в соответствующее уравнение, что-
бы рассчитать а через равновесную концентрацию ионов водорода и константы
равновесия. Например, для расчета а0 мы можем преобразовать уравнения
(7.59)-(7.61) относительно каждой из форм, кроме [Н3РО4], и подставить полу-
ченные выражения в уравнение (7.63):
[РОГ] = М^ (7.64)
[BPof] = S®l (7.65)
[НгРО(]Л-™ (7.66)
[Н J
Нам нужно получить уравнения, содержащие только [Н3РО4] (и [Н+], которая
является переменной величиной). Подставляем уравнение (7.66) для [Н2РО4] в
уравнение (7.65):
(7.67)
[Н+]
Далее подставляем уравнение (7.67) в уравнение (7.64) для [НРО4-]:
[Н+]
И наконец подставляем уравнения (7.66)—(7.68) в уравнение (7.63):
, _*а1Ка2*а3[Н3РО4] , *а1*а2[Н3РО4] . £а1[Н3РО4] ,
""><----------р,-]--- [Н’РО-1 (7'69>
Можно разделить каждый член этого выражения на [Н3РО4], в результате полу-
чим 1/а0. Также можно подставить это выражение в знаменатель приведенного
выше выражения для а0, при этом [Н3РО4] сократится, и мы получим выражение
для а0. В первом случае получаем
1 _KaiKa2Ka3 | *а1*а2 | ^а1 н
а 0 [Н+]3 [Н+]2 [Н+]
(7.70)
344
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
Во втором случае
(^а1£а2£а3/[Н+]3) + (ха1ха2/[н+]2) + (каХ /[Н+]) + 1
Любое из двух последних уравнений можно преобразовать в следующее вы-
ражение для а0:
_________________[Н+]3_________________
[Н+]3 + КаХ [Н+]2 + *а1^а2[Н+] +каХка2ка3
(7.72)
Используйте это уравнение для расчета доли Н3РО4 в растворе.
Подобный подход можно использовать для расчета других az (/=14-4). Для
определения, например а15 уравнения для констант равновесия надо выразить
через [Н2РО4] и подставить в уравнение (7.63), чтобы получить выражение для
СНзРо4> которое содержит только [Н2РО4] и [Н+]. Из этого выражения рассчи-
тать аР
Ниже приведены выражения для az, где /=14-4:
a = *а1[Н+]2 1 [н+]3 + каХ [Н+]2 +каХка2щ.+}+каХка2ка3 (7.73)
a = У«2[Н+] 2 [Н+]3 + Х'а1[Н+]2 +каХка2\в.+}+каХка2ка3 (7.74)
a = VA 3 [Н+]3 + КаХ [Н+]2 +каХка2щ+]+каХка2ка3 (7.75)
Обратите внимание, что уравнения (7.72)-(7.75) имеют один и тот же знамена-
тель, а сумма числителей равна знаменателю. В задаче 58 потребуется вывести
эти уравнения.
Пример 7.18
Рассчитайте равновесную концентрацию различных частиц в 0,10 М растворе
фосфорной кислоты при pH 3,00 ([Н+] = 1,0 • 10-3 М).
7.8. МНОГООСНОВНЫЕ КИСЛОТЫ И ИХ СОЛИ
345
Решение
Подставив данные в уравнение (7.72), получаем:
а =_______________________________дою-3)3____________________________=
° (1,0 • 10“3 )3 + (1,1 • 10-2 )(1,0 • КГ3 )2 + (1,1 • 10“2 )(7,5 • 1(Г8 )(1,0 Ю’3 ) +
+ (1,1 • ИГ2 )(7,5 • 10-8 )(4,8 • 10’13)
=1^124=8,з-ю-2
1,2 Ю’8
[Н3РО4] = Сн РО а0 = 0,10 • 8,3 • IO-2 = 8,3 • 10~3 М
L J Я-1 1131^4 V 7 7 7
Подобным же образом
а! = 0,92
[Н2РО;] =СНзрО4а1 = 0,10 • 0,92 = 9,2 • 10-2 М
а2 = 6,9 - 10-5
[ПРО2-] = Ся рп а2 = 0,10 6,9 • IO-5 = 6,9 • 10^ М
L я J Пзги^ Z 7 7 7
а3 = 3,3 • 10-14
[РО3“] = СНзРО4а3 = 0,10-3,3- 10~14 = 3,3- 10“15М
При pH 3 большая часть (91%) фосфорной кислоты находится в форме
Н2РО4 и 8,3% существует в форме Н3РО4. При данном pH в растворе находится
только 3,3 • 10-12 % полностью депротонированной фосфорной кислоты (ион РО 4")!
Для расчета доли каждой формы в зависимости от pH можно создать элект-
ронную таблицу. Расчетные формулы приведены непосредственно в таблице (см.
ниже), а на рис. 7.1 изображены полученные зависимости a-коэффициентов от
pH. Значения Ка следует ввести в ячейки В4, D4 и F4, а величины pH — в ячейки
столбца А. Все формулы, которые необходимо ввести в ту или иную ячейку, пере-
числены в нижней части таблицы; сначала их вводят в ячейки, выделенные жир-
ным шрифтом. Формулу для расчета концентрации ионов водорода (необходимой
для вычисления a-коэффициентов) вводят в ячейку В6, а формулу для вычисле-
ния знаменателя всех выражений, используемых для расчета а-коэффициен-
тов, — в ячейку С6. Обратите внимание, что адреса ячеек с константами кислот-
ности вводятся как абсолютные, а ячеек со значениями концентрации ионов водо-
рода— как относительные. Формулы для расчета трех a-коэффициентов вводят
в ячейки D6, Е6 и F6. Все введенные формулы следует затем скопировать вниз
вплоть до строки 34.
Построение графика осуществляется при помощи Мастера диаграмм (Chart
Wizard). Последовательно строят зависимости величин, находящихся в ячейках
D6:D34, Е6:Е34, F6:F34 и G6:G34, от величин в ячейках А6:А34. На компакт-ди-
346
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
Коэффициент а для Н3РО4
0,0 1,0 2,0 3,0 4,0 5,0 6,0 7,0 8,0 9,0 10,0 11,0 12,0 13,0 14,0
pH
Рис. 7.1 . Зависимости мольных долей форм Н3РО4 от pH. Области перекрывания кривых соответ-
ствуют диапазонам pH буферных растворов. Значения pH, при которых cq и а2 приближаются к
единице, соответствуют конечным точкам титрования Н3РО4. Относительно способа спрямления
рассматриваемых зависимостей (концентрационно-логарифмические диаграммы) см. разд. 7.12
ске (гл. 7) находится лист таблицы с исходными данными (Sheet 1) и сам график
(Chart 1). Вы можете использовать эти данные для самостоятельного воспроиз-
ведения рис. 7.1. Для этого сохраните лист с данными (Sheet 1) на своем компьюте-
ре. Откройте Мастер диаграмм и выберите опции: Тип -> Точечная диаграмма
(Scatter data) и Вид —> Плавная кривая (второй образец диаграммы в ле-
вом столбце). Затем нажмите клавишу Далее (Next) и в окошко Диапазон
(Data Range) введите строку A6:A34,D6:D34 — программа автоматически
преобразует ее к виду Sheetl!$A$6:$A$34,$D$6:$D$34. Здесь Sheetl — это на-
звание листа таблицы, содержащего исходные данные для построения диаграм-
мы, а символ «!» служит для его отделения от адресов ячеек. При раздельном
вводе данных для оси абсцисс X и ординат Y перед диапазонами адресов ячеек
следует ввести название листа с разделителем (Sheetl!) в явном виде. Нажмите
на клавишу Ряд (Series). В качестве имени первой серии введите alpha 0
(Н3РО4). Скопируйте строку Значения X в буфер обмена, чтобы использовать
ее для построения остальных серий. Нажмите на клавишу Добавить (Add) для
создания второй серии. Помните, что для этого необходимо будет ввести диа-
пазоны значений X и Y. Диапазон значений X введите в соответствующее
окошко из буфера обмена командой Вставить (Paste). Для значений Y на-
берите строку: =Sheetl !Е6:Е34 (после ввода адреса автоматически преобразу-
ются в абсолютные). Назовите эту серию alpha 1 (Н2РО4). Повторите эти
операции для столбцов F и G, чтобы создать серии 3 и 4 для а2 и а3. Нажав на
7.8. МНОГООСНОВНЫЕ КИСЛОТЫ И ИХ СОЛИ
347
А В С D Е F G
1 Расчет зависимостей величин а от pH для Н3РО4
2 Знаменатель для а,: = [Н*Р + Ка1[Н+? + К^К^Н*] + Ка1КагКаз
3 Числители: а0 = [Н+]3; а, = Ка1[Н’]2; а2 = Ка1Ка2[Н+]; а3 = Ка1Ка2Ка3
4 ка1= 1,10Е-02 Ка2= 7.50Е-08 КаЗ= 4.80Е-13
5 PH [Н*] Знаменатель а0 «1 а2 а3
6 0,0 1 1.01Е+00 9,89Е-01 1,09Е-02 8,16Е-10 3,92Е-22
7 0,5 0,316228 3.27Е-02 9.66Е-01 3.36Е-02 7.97Е-09 1,21Е-20
8 1,0 0,1 1J1E-03 9,01 Е-01 9,91 Е-02 7.43Е-08 3.57Е-19
9 1,5 0,031623 4,26Е-05 7.42Е-01 2.58Е-01 6J2E-07 9,29Е-18
10 2,0 0,01 2.10Е-06 4,76Е-01 5,24Е-01 3,93Е-06 1,89Е-16
11 2,5 0,003162 1.42Е-07 2.23Е-01 7,77Е-01 1,84Е-05 2.80Е-15
12 3,0 0,001 1.20Е-08 8,ЗЗЕ-02 9J7E-01 6,87Е-05 3,ЗОЕ-14
13 3,5 0,000316 1,1 ЗЕ-09 2,79Е-02 9.72Е-01 2,ЗОЕ-04 3.50Е-13
14 4,0 0,0001 1.11Е-10 9,00Е-03 9.90Е-01 7,43Е-04 3,56Е-12
15 4,5 3.16Е-05 1.11Е-11 2.86Е-03 9.95Е-01 2.36Е-03 3,58Е-11
16 5,0 0,00001 1,11Е-12 9.02Е-04 9,92Е-01 7,44Е-03 3.57Е-10
17 5,5 3.16Е-06 1J3E-13 2,81 Е-04 9,77Е-01 2,32Е-02 3,52Е-09
18 6,0 0,000001 1.18Е-14 8.46Е-05 9, ЗОЕ-01 6,98Е-02 3,35Е-08
19 6,5 3.16Е-07 1,36Е-15 2,32Е-05 8,08Е-01 1,92Е-01 2,91 Е-07
20 7,0 1Е-07 1,93Е-16 5,19Е-06 5,71 Е-01 4.29Е-01 2,06Е-06
21 7,5 3J6E-08 3,71Е-17 8,53Е-07 2.97Е-01 7,03Е-01 1,07Е-05
22 8,0 1Е-08 9,35Е-18 1.07Е-07 1J8E-01 8,82Е-01 4,24Е-05
23 8,5 3.16Е-09 2,72Е-18 1.16Е-08 4,05Е-02 9.59Е-01 1,46Е-04
24 9,0 1Е-09 8.36Е-19 1,20Е-09 1,32Е-02 9,86Е-01 4.73Е-04
25 9,5 3J6E-10 2.62Е-19 1,21 Е-10 4J9E-03 9,94Е-01 1,51Е-03
26 10,0 1Е-10 8, ЗОЕ-20 1.20Е-11 1.33Е-03 9,94Е-01 4.77Е-03
27 10,5 3,16Е-11 2,65Е-20 1.19Е-12 4J5E-04 9.85Е-01 1.49Е-02
28 11,0 1Е-11 8,65Е-21 1J6E-13 1,27Е-04 9.54Е-01 4.58Е-02
29 11,5 3.16Е-12 3.00Е-21 1.05Е-14 3.66Е-05 8.68Е-01 1.32Е-01
30 12,0 1Е-12 1,22Е-21 8J9E-16 9,01 Е-06 6,76Е-01 3.24Е-01
31 12,5 3.16Е-13 6,57Е-22 4,81 Е-17 1.67Е-06 3.97Е-01 6,03Е-01
32 13,0 1Е-13 4.79Е-22 2.09Е-18 2,ЗОЕ-07 1,72Е-01 8,28Е-01
33 13,5 3.16Е-14 4.22Е-22 7,49Е-20 2,61 Е-08 6J8E-02 9,38Е-01
34 14,0 1Е-14 4,04Е-22 2.47Е-21 2.72Е-09 2,04Е-02 9.80Е-01
35 В ячейки, выделенные жирным шрифтом, введены формулы:
36 Ячейка В6 = [Н+] = 10-А6
37 Ячейка С6=знаменатель B6'3+$B$4*B6'2+$B$4*$D$4*B6+$E®&$D$4*$F$4
38 Ячейка D6 - а0 - В6-3/С6
39 Ячейка Е6 = = ($В$4*В6Л2)/С6
40 Ячейка F6 = а2 = ($B$4*$D$4*B6)/C6
41 Ячейка G6 = а3 = ($B$4*$D$4*$F$4)/C6
42 Копируем каждую формулу вниз до ячейки 34 включительно
43 График А6:А34 от D6:D34, Е6:Е34, F6:F34 и G6:G34 (серии 1, 2, 3 и 4)
348
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
клавишу Параметры диаграммы (Chart options) (или пользуясь команда-
ми меню Диаграмма (Chart)) вы можете ввести название диаграммы, подпи-
си осей X и Y, основные и промежуточные линии сетки. Затем нажмите на
клавишу Готово (Finish), и вы увидите построенный график для а-коэффи-
циентов. После этого можно вносить в диаграмму дальнейшие изменения, на-
пример, изменить масштабы или числовой формат осей (для того и другого надо
навести курсор мыши на соответствующую ось, щелкнуть правой кнопкой
мыши и выбрать команду Формат оси). Можно переместить легенду с названи-
ями серий в поле диаграммы; для этого необходимо выделить легенду щелчком
мыши, а затем перетащить ее в желаемое место.
Рисунок иллюстрирует, как изменяются соотношения четырех форм фос-
форной кислоты при изменении pH, например, при титровании Н3РО4 раствором
NaOH. Несмотря на то, что концентрации некоторых форм кажутся равными
нулю выше или ниже определенных значений pH, они не равны нулю, но крайне
малы. Для иллюстрации обратимся к примеру 7.18: хотя концентрация иона РО 4“
в 0,1 М Н3РО4 при pH 3,00 и составляет всего лишь 3,3 • 10-15 М, этот ион все рав-
но участвуют в равновесии. Области pH, где две кривые накладываются друг на
друга (при значимых величинах концентраций), представляют собой интервалы
pH для приготовления буферных растворов, содержащих соответствующие две
частицы. Например, смеси Н3РО4 и Н2РО 4 можно использовать для приготовле-
ния буферных растворов с интервалом pH 2,0 ± 1, смеси Н2РО 4 и НРО 4“ — с ин-
тервалом pH 7,1 ± 1, смеси НРО 4~ и РО 4“ — с интервалом pH 12,3 ± 1. Значения
pH, при которых доля частицы равна 1,0, соответствуют конечным точкам тит-
рования фосфорной кислоты сильным основанием, а именно Н2РО 4 в первой
конечной точке (pH 4,5) и НРО4“ во второй конечной точке (pH 9,7).
Уравнение (7.69) можно было бы использовать для точного расчета концен-
трации ионов водорода, образующихся при диссоциации фосфорной кислоты в
растворе при заданной концентрации Н3РО4 (отсутствуют ионы Н+ из других ис-
точников), но этот расчет требует утомительных вычислений. При первом при-
ближении [Н+] можно было бы рассчитать из КаХ. как показано в примере 7.17,
предположив, что диссоциация фосфорной кислоты протекает только по по пер-
вой ступени. (Именно так мы и поступили в этом примере.) Первое рассчитанное
значение [Н+] можно было бы подставить в уравнение (7.69) для расчета второго
приблизительного значения [Н3РО4], нужного для второго приблизительного рас-
чета [Н+], в котором используют Ха1, и так далее, пока концентрация не стала бы
постоянной.
Соли многоосновных кислот: кислоты или основания?
Соли таких кислот, как Н3РО4, могут быть кислыми или основными. Протоносо-
держащие соли проявляют как кислотные, так и основные свойства (Н2РО4,
НРО4~), тогда как соль, не содержащая протона, представляет собой простое
основание по Бренстеду, которое подвергается гидролизу (РО4").
7.8. МНОГООСНОВНЫЕ КИСЛОТЫ И ИХ СОЛИ
349
1. Амфотерные соли. Ион Н2РО4 проявляет как кислотные, так и основные
свойства. Это значит, что он является амфотерным. Он диссоциирует как сла-
бая кислота, но он также представляет собой основание по Бренстеду, которое
подвергается гидролизу. (В конце разд. 7.12 рассмотрено, как оценить степень
протекания реакции с помощью концентрационно-логарифмических диаграмм.)
н2ро; н+ + нро^ [н2ро;] (7.76)
н2ро; + н2о н3ро4 + он- к =К^= [Н3РО4][ОН-]_ ь ка1 [Н2ро;] = 10040^ = 9Д.10_13 1,1 10’2 (7.77)
Следовательно, раствор соли может быть либо щелочным, либо кислым в
зависимости от того, какой процесс преобладает. Так как значение Ка2 для пер-
вого процесса почти в 105 больше Кь для второго процесса, то ясно, что раствор
в этом случае будет кислым.
Выражение для расчета концентрации ионов водорода в растворе, содержа-
щем ионы типа Н2РО4, можно получить следующим образом. Общая концент-
рация ионов водорода равна сумме концентраций всех форм, полученных в
результате процессов ионизации по уравнению (7.76) и ионизации воды, минус
количество ионов ОН-, полученного в результате гидролиза по уравнению (7.77).
Тогда мы можем записать
Сн+ = [Н+] общая = [Н+ ]н2О + [Н+ ]Н2ро; ~ [Н~ ]н2РО; С7’78)
ИЛИ
[Н+] = [ОН-] + [НРО 2-] - [Н3РО4] (7.79)
Мы должны учитывать вклад от ионизации воды, так как этим процессом
нельзя пренебречь, если значение pH раствора соли близко к 7 — хотя конкретно
в данном случае раствор будет кислым, поэтому ионизация воды будет незначи-
тельной.
Можно рассчитать [Н+], выразив члены в правой части уравнения (7.79) че-
рез константы равновесия по уравнениям (7.59) и (7.60) и Kw для того, чтобы
исключить все переменные, кроме [Н2РО4] и [Н+]:
_ Kw । ^2[Н2РО;] _ [н2ро;][н+]
[Н+] [Н+] ка1
350
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
Из этого уравнения (умножив каждую из сторон уравнения на [Н+], переве-
дя все члены уравнения, содержащие [Н+]2, в левую часть, и решив уравнение
относительно [Н+]2) получаем:
[П+г._^+^2[н2ро;]
!, [Н2ро;]
(7.81)
(7.82)
Это же уравнение в общем виде для НА- имеет вид:
/Ка1*„+Кд1Ка2[НА-]
У Ка1+[НА~]
В случае НА2- замените [НА-] на [НА2-], КаХ на Ка2, пКа2 наА^. Это уравне-
ние действительно для любой соли НА-, соответствующей кислоте НА (или для
НА2-, полученной из Н2А-, и так далее).
Если предположить, что равновесная концентрация [НА-] равна общей кон-
центрации соли, т. е. степень диссоциации и гидролиза аниона очень мала, тогда
эту величину вместе с константами можно использовать для расчета [Н+]. Это
предположение справедливо, если две константы равновесия (Ка1 и Кь), характе-
ризующие анион НА-, малы, и если раствор не слишком разбавлен. Во многих
случаях для слагаемых в числителе уравнения (7.83) справедливо следующее со-
отношение: Ka}Kw [НА-], поэтому первым слагаемым можно прене-
бречь. Именно такое уравнение мы бы получили, если бы не учитывали
диссоциацию воды. Более того, если [НА-] Ка1 в знаменателе уравнения
(7.83), то уравнение упрощается до
[Н+] = Х2^а3
(7.84)
В случае НА*- [Н+] = ^Ка2Ка3.
7.8. МНОГООСНОВНЫЕ КИСЛОТЫ И ИХ СОЛИ
351
Поэтому, если сделанное выше предположение справедливо, то pH раство-
ра Н2РО4 не зависит от концентрации соли! Например, в случае Н2РО4
[Н+] * ^Ка1ка2 = 71,1-Ю’2 -7,5-10"8 = 2,9 • 10“5 М
(7.85)
и pH (« 4,54) мало зависит от концентрации соли.
Анион НРОд- также является и кислотой, и основанием. В данном случае рас-
чете используются константы Ка2 и Ка3 для Н3РО4 (Н2РО 4 = Н2А и НРО 4~ = НА-).
Так как Ка2 Ка3, то pH раствора Na2HPO4 можно рассчитать по уравнению*
[Н+] « ^Ка2ка3 = 77,5-ю-8 -4,8 10"13 = 1,9 1О~10
(7.86)
и рассчитанное значение pH составляет 9,72. Поскольку значение pH растворов
амфотерных солей такого типа существенно не зависит от концентрации, то эти
соли используют для приготовления растворов с известным значением pH для
градуировки pH-метров. Например, раствор гидрофталата калия КНС8Н4О2 (КНР,
«стандартный буфер» Национального института стандартов США, гл. 13) при
25 °C имеет pH 4,0. Значение pH раствора КНР постоянно, но этот раствор не
проявляет буферных свойств. Однако растворы этих солей являются плохими
буферами по отношению к кислотам или основаниям; их pH лежит вдали от бу-
ферной области.
2. Соли, не содержащие протонов. Непротонированный фосфат-анион в рас-
творе представляет собой довольно сильное основание по Бренстеду и диссоци-
ирует следующим образом:
РО3“ + Н2О НРО2- + он- кь = —^-
КаЗ
(7.87)
Константа Ка3 очень мала, поэтому равновесие сильно смещено вправо. По-
скольку Ка3 С Ка2, дальнейший гидролиз (иона НРО 4") подавляется ионами ОН-,
образующимися на первой ступени, и pH раствора, содержащего ионы РО 4~ мож-
но рассчитать также, как и pH раствора соли одноосновной слабой кислоты. Од-
нако, поскольку Ка3 очень мала, Кь относительно велика, а количество ионов
ОН- сопоставимо с исходной концентрацией РО4“ (Св_), для расчета pH необ-
ходимо решать полное квадратное уравнение.
* В этом случае не соблюдается условие Ka^Lw < АГа2АГа3[НА2-], поэтому пренебречь диссоциацией воды в
растворах HPOj” нельзя и pH солей, содержащих этот анион, в значительной степени зависит от концент-
рации аниона в растворе. — Прим, перев.
352
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
Пример 7.19
Рассчитайте pH 0,100 М раствора Na3PO4.
Решение
х
РО + Н2О НРО + он-
0,100-х
= = = -1-0-1-0-"14- = 0,020
[РОд-] Ка3 4,8-10-13
-^=1’°-10~114 =0,020
0,100-х 4,8-10“ 3
Концентрация раствора только в пять раз больше величины Кь, поэтому решаем
квадратное уравнение:
х2 + 0,020% -2,0- 10~3 = 0
_ -0,200 ± 7(0,020)2 - 4(-2,0-10-3)
Х~ 2
х = [ОН-] = 0,036 М
pH = 12,56
Степень диссоциации (гидролиза) составляет 36% и фосфат представляет собой
довольно сильное основание. Проведите расчет с помощью программы Поиск ре-
шения, гл. 6 на компакт-диске, и сравните результаты расчета.
Пример 7.20
ЭДТА представляет собой многоосновную кислоту с четырьмя протонами (Н4У).
Рассчитайте концентрацию ионов водорода в 0,0100 М растворе Иа2ЭДТА
(Na^Y).
Решение
В растворе протекают следующие процессы:
H2Y2- ±=7 Н+ + HY3’ Ка3 = 6,9 10’7
К 10 1
и H2Y2- + Н2О Н3Y- + ОН- Kh = ^- =
К.2 2,2-10-’
7.9. ФИЗИОЛОГИЧЕСКИЕ БУФЕРНЫЕ РАСТВОРЫ
353
H2Y2- соответствует НА- в общем уравнении для расчета pH, а Н3Y- соответст-
вует Н2А. В расчете следует использовать константы равновесия Ка2 и Ка3 (пер-
вая для кислоты H3Y_, сопряженной с гидролизующейся солью). Таким образом
[Н+] = ^Ка2Ка3 = д/(2,2 10-3)(6,9-10-7) = 3,9 • 10“5 М
7.9. Физиологические буферные растворы:
они поддерживают жизнедеятельность
У здорового человека pH крови отличается удивительным постоянством и находит-
ся в интервале от 7,35 до 7,45. Это связано с тем, что кровь содержит ряд буферных
систем, которые препятствуют изменениям pH, обусловливаемым присутствием
кислых или основных метаболитов. С физиологической точки зрения изменение
pH на ±0,3 единицы считается критическим. Кислые метаболиты обычно обра-
зуются в больших количествах, чем основные, главным кислым метаболитом явля-
ется диоксид углерода. Установлено, что буферная емкость крови по отношению к
СО2 складывается из емкостей различных буферных систем следующим образом:
гемоглобин и оксигемоглобин — 62%; Н^Од/НРО^" — 22%; белок плазмы —
11%; гидрокарбонат — 5%. Белки содержат карбоксильные и аминогруппы, ко-
торые представляют собой соответственно, слабые кислоты и основания. Поэто-
му они являются эффективными буферными агентами. Клиницисты называют
комбинированную буферную емкость крови по отношению к кислотам «ще-
лочным резервом», его часто определяют в клинических лабораториях. Некото-
рые заболевания вызывают нарушения кислотного баланса организма. Например,
диабет может вызвать ацидоз, который может оказаться смертельным.
Важным диагностическим анализом является установление баланса СО2/НСО3
в крови. Это отношение связано с pH крови уравнением Гендерсона—Хассель-
баха (7.45):
рН = 6,10 + 18№211
[Н2СО,]
(7.88)
где концентрацию Н2СО3 можно считать равной концентрации растворенного
СО2 в крови; 6,10 — это рАГа1 угольной кислоты при температуре тела (37 °C).
Обычно концентрация гидрокарбоната в крови составляет около 26,0 ммоль/л,
тогда как концентрация диоксида углерода равна 1,3 ммоль/л. Следовательно,
для крови
pH = 6,10 + 1g 26 ммоль/л = 7;40
1,3 ммоль/л
354
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
Концентрацию НСО3 можно определить титриметрически (практическая
работа 8), общее содержание диоксида углерода (НСО3 + растворенный СО2)
можно определить, если подкислить раствор и измерить объем выделившегося
газа.* Если проведены оба анализа, можно рассчитать отношение СО2/ HCOj и
сделать выводы о наличии ацидоза или алкалоза у пациента. Альтернативная ме-
тодика состоит в том, что после измерения pH (при 37 °C) необходимо опреде-
лить либо только содержание НСО3, либо общиее содержание СО2, чтобы
иметь полную информацию о балансе угольной кислоты, так как соотношение
[НСОз]/[Н2СО3] можно рассчитать по уравнению (7.88).
Можно также измерить парциальное давление СО2 pCq2 (например, испо-
льзуя СО2-электрод); в этом случае [Н2СО3] « 0,30/?сс>2. Тогда нужно будет
определить только pH или [НСО3].
Обратите внимание, что уравнение (7.88) является справедливым, несмотря
на то, что в крови есть и другие буферные системы. Значение pH крови опреде-
ляется действием всех буферных систем, а отношение [НСО3 ]/[Н2СО3] устанав-
ливается в зависимости от этого pH.
Буферная система НСОз/Н2СО3 является наиболее важной при буфериро-
вании крови в легких (альвеолярная кровь). Так как кислород из вдыхаемого
воздуха соединяется с гемоглобином, окисленный гемоглобин ионизируется с
выделением протона. Образующийся избыток кислоты нейтрализуется при вза-
имодействии с НСО3:
Н++ НСО3 —> Н2СО3
Следует отметить, что отношение [НСОз]/[Н2СО3] при pH 7,4 составляет
26/1,3 = 20 : 1. Это не очень эффективное соотношение компонентов буферной
системы; так как значительные количества НСО 3 переходят в Н2СО3, то pH дол-
жно снижаться, вызывая установление нового соотношения. Но к счастью, обра-
зующаяся угольная кислота быстро разлагается на СО2 и Н2О под действием
фермента дикарбоксилазы и выводится через легкие с выдыхаемым воздухом.
Таким образом, отношение НСОз/Н2СО3 остается постоянным и сохраняет зна-
чение 20:1.
Пример 7.21
Общее содержание диоксида углерода (НСО 3 + СО2) в образце крови определи-
ли путем подкисления пробы и измерения объема выделившегося СО2 с помощью
По величине объема СО2 при определенных значениях температуры и атмосферного давления можно рас-
считать количество СО2 в миллимолях и, следовательно, его концентрацию в ммоль/л в растворе, из кото-
рого этот газ выделился. При стандартных температуре и давлении 22,4 л содержат один моль газа.
7.10. БУФЕРЫ ДЛЯ БИОЛОГИЧЕСКИХ И КЛИНИЧЕСКИХ ИЗМЕРЕНИЙ
355
манометра Ван Слайке. Оно оказалось равным 28,5 ммоль/л. Измеренное при
37 °C значение pH крови равно 7,48. Рассчитайте концентрации НСО3 и СО2 в
крови.
Решение
Рн = б,ю + 1ёП!Щ
[СО2]
7,48=6,10+lg^
[СО2]
lg^=l,38
[СО2]
[НС°3 ] = Ю1’38 = 101 • 10°>38 = 24
[СО2]
[НСО~]=24[СО2]
Но
[НСО3 ] + [СО2] = 28,5 ммоль/л
24[СО2] + [СО2] = 28,5
[СО2] = 1,14 ммоль/л
[НСО3 ] = 28,5 - 1,1 = 27,4 ммоль/л
7.10. Буферы для биологических и клинических измерений
Многие биохимические реакции, представляющие интерес для исследований,
протекают в диапазоне pH от 6 до 8. Для целого ряда реакций, особенно специ-
фических ферментативных, которые могут быть использованы в аналитических
целях (см. гл. 22), рабочий интервал pH составляет от 4 до 10 или даже выше.
При правильном выборе буферов для изучения биохимических реакций или для
использования в клинических анализах важно знать, влияют они или нет на ход
реакции. Кислотный компонент буфера должен иметь величину рАГа, близкую к
физиологическому значению pH, чтобы отношение [А_]/[НА] в уравнении Ген-
дерсона—Хассельбаха не сильно отличалось бы от единицы. Наконец, буфер
должен быть физиологически совместимым.
356
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
Фосфатные буферные растворы
К широко используемым буферным растворам относятся фосфатные. Биологи-
ческие системы обычно уже содержат некоторое количество фосфатов, и фос-
фатные буферы в большинстве случаев не оказывают мешающего влияния.
Правильно выбрав соответствующие смеси Н3РО4/Н2РО4, Н2РО4/НРО4" или
НРО4-/РО4-, можно приготовить растворы в широком диапазоне pH. Состав
ряда фосфатных буферов при постоянной ионной силе 0,2 моль/л приведен в ра-
боте [G. D. Christian, W. С. Purdy, J. Electroanal. Chem., 3 (1962) 363]. Ионная
сила является мерой общего содержания солей в растворе (см. гл. 6), она часто
влияет на протекание реакции, особенно в кинетических исследованиях. Следо-
вательно, фосфатные буферы могут быть использованы в тех случаях, когда
ионная сила должна быть постоянной. Однако буферная емкость значительно
уменьшается, как только значение pH растворов приближается к значениям pH
перечисленных выше индивидуальных солей, которые не проявляют буферных
свойств; максимальная буферная емкость соответствует интервалу pH, равному
соответствующему значению рКа ± 1, а именно 1,96 ± 1; 7,12 ± 1 и 12,32 ± 1. Фос-
фатные буферные растворы используются для поддержания определенного зна-
чения pH в тех случаях, когда к раствору нужно добавить небольшое количество
кислоты или основания либо когда кислота или основание образуются в раство-
ре. Смесь с pH 7,40 хорошо подходит для буферирования при физиологических
значениях pH; при pH 7,5 ее буферная емкость несколько снижается.
Пример 7.22
Сколько граммов NaH2PO4 и Na2HPO4 необходимо взять для приготовления 1 л
буферного раствора с pH 7,45 и ионной силой 0,100 моль/л?
Решение
Обозначим х = [Na2HPO4], а 7 = [NaH2PO4]. Имеются два неизвестных значения,
следовательно необходимы два уравнения. (Помните, что число уравнений дол-
жно соответствовать числу неизвестных, относительно которых нужно решить
задачу.) Первое уравнение — это уравнение для расчета ионной силы:
0,100 = l[Na+] • I2 + [HPOi-] • 22 + [Н2РО7] • I2
(1)
0,100 = 1[(2х+у) • 12 + х-22+у- I2]
0,100 = 3х +у
7.10. БУФЕРЫ ДЛЯ БИОЛОГИЧЕСКИХ И КЛИНИЧЕСКИХ ИЗМЕРЕНИЙ
357
Второе уравнение — это уравнение Гендерсона—Хассельбаха:
pH = pATa2 + lg
[HPOj-]
[н2ро;]
7,45 = 7,12 + 1g — (2)
У
- = 1О°’33 = 2,14
У (3)
х = 2,14У
Подставляя (3) в уравнение (1), получаем:
0,100 = 3 -2,14- у + у
у = 0,0135М = [NaH2PO4]
Подставив этот результат в уравнение (3), получаем:
х = 2,14 • 0,0135 = 0,0289 М = [Na2HPO4]
WNaH2PO4 (Г) = 0,0135 моль/л • 120 г/моль = 1,62 г/л
wNa2HPO4 (г)= 0,0289 моль/л • 142 г/моль = 4,10 г/л
Область применения фосфатных буферных растворов ограничена. Кроме
ограничений, связанных со значением буферной емкости при определенных
значениях pH, следует учитывать, что фосфат-ион образует осадки или комплек-
сы со многими многоразрядными катионами и зачастую может осаждаться в
ходе реакции или ингибировать реакцию. Фосфатный буфер не следует исполь-
зовать, например, при наличии кальция в том случае, если его осаждение по-
влияет на ход исследуемой реакции.
Трис-буферные растворы
Буферный раствор, который широко используется в клинических и биохимиче-
ских исследованиях в физиологическом диапазоне pH, готовят из дирмс(гидро-
ксиметил)аминометана [(HOCH2)3CNH2 — Tris, или ТНАМ] и сопряженной
ему кислоты (с протонированной аминогруппой). Tris может служить первич-
ным стандартом: вещество стабильно, хорошо растворимо в физиологических
жидкостях, негигроскопично, не поглощает СО2 (что важно), не осаждает соли
кальция, не оказывает ингибирующего влияния на многие ферментные системы
и совместимо с биологическими жидкостями. Для этого буфера значение рКа со-
пряженной кислоты равно 8,08 и близко к физиологическому значению pH, но, к
сожалению, его буферная емкость при pH ниже 7,5 снижается. К другим недо-
статкам можно отнести то, что первичный алифатический амин отличается значи-
тельной реакционной способностью и может взаимодействовать с льняными
волокнами, которые, например, используются в электролитическом ключе насы-
щенного каломельного электрода сравнения для измерения pH (гл. 13); для та-
358
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
ких измерений следует использовать электроды сравнения с соединениями,
сделанными из керамики, кварца или в виде втулки. Трис-буферные растворы
обычно готовят добавлением кислоты, например, хлороводородной, к раствору
Tris до установления нужного значения pH.
7.11. Влияние посторонних ионов на поведение кислот и оснований:
К°а и К°ь - изменение pH при введении солей
В гл. 6 мы рассмотрели термодинамические константы равновесия, рассчитыва-
емые на основе активностей, а не концентраций. Посторонние ионы изменяют
на величины активностей, а следовательно, влияют на степень диссоциации сла-
бых электролитов, таких как слабые кислоты или основания. Коэффициенты ак-
тивности недиссоциированных кислот и оснований, молекулы которых не имеют
заряда, практически равны единице. Тогда для кислоты НА
(7.89)
(7.90)
(7.91)
Поэтому можно ожидать увеличения Ка и усиления диссоциации с повыше-
нием ионной силы, так как коэффициенты активности в этом случае уменьшаются
(см. пример 6.18 и задачу 21 к гл. 6). Подобная закономерность характерна и для
слабых оснований (см. задачу 60 к гл. 6).
Поскольку ионная сила влияет на диссоциацию слабых кислот и оснований,
то она должна влиять и на значение pH буферных растворов. Мы можем запи-
сать уравнение Гендерсона—Хассельбаха, используя Ка или Ка:
pH = p£a+lg^J (7.92)
pH = p*fl+lg^ (7.93)
При повышении ионной силы Ка увеличивается, а рКа уменьшается. Следо-
вательно, согласно уравнению (7.92), pH уменьшается. Если буферный раствор
разбавить, его ионная сила уменьшится, что приведет к небольшому увеличению
pH, даже если отношение [А~]/[НА] остается постоянным (см. примечание на
7.12. КОНЦЕНТРАЦИОННО-ЛОГАРИМФИЧЕСКИЕ ДИАГРАММЫ
359
стр. 333 в этой главе). Для буферного раствора НРО4 /Н2РО4 отношение
^нрсЯ-^н ро- также будет уменьшаться с увеличением ионной силы, так как
влияние ионной силы на активность многозарядных ионов значительно сильнее.
7.12. Концентрационно-логарифмические диаграммы: как
изобразить изменения концентраций в широком диапазоне
Концентрационно-логарифмическая диаграмма — это график билогарифмиче-
ской зависимости, где по оси абсцисс (х) откладывается та же самая величина,
что и на распределительной диаграмме (для величин а), а по оси ординат (у) —
логарифм концентрации той или иной формы. Эту ось можно использовать для
представления концентраций любых частиц. Поэтому на концентрационно-ло-
гарифмические диаграммы в дополнение к кривым, характеризующим различные
формы кислоты, можно наносить и кривые для таких частиц, как Н+ или ОН-.
Построим, например, концентрационно-логарифмическую диаграмму для
всех частиц, присутствующих в 1,0 • 10-2 М растворе уксусной кислоты (рКа = 4,76).
Проще всего построить кривые для Н+ и ОН-. По определению, 1g [Н+] = -pH
(например, при pH 4 [Н+] = 1 • 1(Н, 1g [Н+] = -4). Зависимость для [Н+] представ-
ляет собой прямую линию с тангенсом угла наклона -1, проходящую через точ-
ку с координатами lg С = -4 и pH = 4. Аналогично, 1g [ОН-] = -рОН = pH - рАГн,.
Зависимость для [ОН-] — также прямая линия, но с тангенсом угла наклона +1.
Она проходит через точку с координатами lg С = -4 и pH = 10. Знаний угловых
коэффициентов и расположений опорной точки достаточно для изображения
этих прямых. Концентрационно-логарифмическая диаграмма, на которой пред-
ставлены только зависимости для [Н+] и [ОН-], изображена на рис. 7.2.
РИС. 7.2. Концентрационно-логарифмическая диаграмма для кислотно-основной системы. Отме-
чены опорные точки для Н+ (pH 4, lg С = - 4) и ОН- (pH 10, lg С = - 4)
360
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
Далее, необходимо построить кривые для [НОАс] и [ОАс-] (о ходе дальней-
шего обсуждения см. рис. 7.3). В сильнокислых растворах диссоциация уксус-
ной кислоты подавлена, и преобладающей формой является НОАс. Поскольку
СНОАс = 1,0 ’IO-2 М, в кислых средах величина [НОАс], будучи практически по-
стоянной, равна 1,0 • 10-2 М. Напротив, в щелочных средах величина [НОАс] ста-
новится очень малой вследствие диссоциации, и [ОАс-]« СН0Ас. Таким образом,
для щелочных растворов можно записать преобразованное выражение констан-
ты кислотно-основного равновесия:
[НОАс] = [Н ]СноАс (7.94)
Ка
Логарифмируя обе части этого выражения, получаем:
lg[HOAc] = 1g -^ЦОАс _ рН = const _ рН (7,95)
Ка
Для 1,0 • 10-2 М раствора НОАс величина константы составляет 2,76. В ще-
лочной области кривая для НОАс представляет собой прямую линию с танген-
сом угла наклона -1. При pH 6,76 величина 1g [НОАс] равна - 4,00. Эти значения
являются координатами опорной точки для проведения прямой с известным уг-
лом наклона — участка кривой для НОАс в щелочной области.
Аналогично, чтобы построить кривую для ОАс- в кислых средах, следует
учесть, что [НОАс] при этом практически постоянна и равна СН0Ас. Поэтому в
сильнокислых средах
[ОАс-] =---------- (7.96)
[Н+Кноас
1g [ОАс-] = 1g Ка • СНОАс + pH = const + pH (7.97)
Участок кривой для [ОАс-] представляет собой прямую линию с тангенсом
угла наклона+1. Для 1,0 • 10-2 М раствора НОАс величина константы составляет
-6,76, поэтому при pH, равном 2,76,1g [ОАс-] = - 4,00. По координатам этой точ-
ки и величине тангенса угла наклона, равного +1, можно построить участок кри-
вой для [ОАс-] в кислых средах.
В растворах, для которых pH » рКа, наблюдается плавный переход от одно-
го предельного случая (сильнокислые среды) к другому (сильнощелочные). При
pH = рКа имеет место равенство [НОАс] = [ОАс-]. Таким образом, при этом зна-
чении pH кривые для [НОАс] и [ОАс-] должны пересечься. В рассматриваемом
примере это происходит при концентрации каждого компонента С = 5,0 • 10-3 М
(половина от общей концентрации кислоты), или 1g С = -2,30, и pH = 4,76.
Если продлить наклонные прямолинейные участки двух кривых, то они пе-
ресекутся в точке, для которой 1g С = 1g СНОАс и pH = рЛ*а. Эту точку часто назы-
вают системной точкой (рис. 7.3). Системную точку и одну из опорных точек,
рассмотренных ранее, можно использовать для построения наклонных участков
7.12. КОНЦЕНТРАЦИОННО-ЛОГАРИМФИЧЕСКИЕ ДИАГРАММЫ
361
РИС. 7.3. Концентрационно-логарифмическая диаграмма для 0,01 М раствора уксусной кислоты
(НОАс). Опорные точки для НОАс (pH 2,76, lg С = - 4) и ОАс- (pH 6,76, lg С = - 4) отмечены круж-
ками. Чтобы построить кривые для НОАс и ОАс-, достаточно знать положение системной точки и
величины тангенсов углов наклона прямолинейных участков
кривых. В то же время при известном наклоне прямой для ее построения доста-
точно использовать лишь одну опорную точку, которой всегда может быть сис-
темная точка (хотя в действительности вблизи системной точки зависимости
несколько искривляются и непосредственно через нее не проходят). В зависимо-
сти от концентрации кислоты положение системной точки (и кривых в целом)
смещается вверх или вниз по оси lg С.
При построении концентрационно-логарифмической диаграммы в первую
очередь наносят системную точку. В дальнейшем она служит опорной точкой
для построения всех зависимостей. Если бы концентрационно-логарифмиче-
скую диаграмму требовалось построить, например, для концентрации 1 • 1(Н М,
а не 1 * 10-2 М, кривые для [ОАс-] И [НОАс] имели бы ту же самую форму, но
были бы смещены вертикально вниз на 2 логарифмические единицы концентра-
ции. В то же время зависимости для [Н+] и [ОН-] остались бы на месте.
Как и a-распределительная диаграмма, концентрационно-логарифмическая
диаграмма позволяет определить, какие формы преобладают при заданном зна-
чении pH. Но поскольку в этом случае используется не линейная, а логарифми-
ческая шкала, то можно с высокой точностью определить и концентрации тех
частиц, которые присутствуют в системе в малых количествах. Так, из диаграм-
мы видно, что при pH 2 [Н+] = 1 • 10-2 М, [НОАс] = 1 • 10-2 М, а концентрация аце-
тат-ионов [ОАс-] несколько больше, чем 1 • 10-5 М (в соответствии с уравнением
(7.97) она равна 1СИ’76). Из распределительной диаграммы (полулогарифмиче-
ской) оценить эту величину невозможно, поскольку она слишком близка к нулю.
Действительно, из выражения для константы кислотной диссоциации можно
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
рассчитать, что [ОАс-] = 1,8 • 10-5 М (что очень близко к результату нашей оцен-
ки). При общей концентрации СН0Ас, равной 0,01 М, это соответствует величине
а! = 0,0018. Такую малую величину невозможно оценить из распределительной
диаграммы.
Благодаря высокой степени детализации, достигаемой при использовании
логарифмического масштаба по оси концентраций, часто оказывается возмож-
ным получать при помощи концентрационно-логарифмических диаграмм отве-
ты (по крайней мере приближенные) на многие вопросы, связанные с расчетами
химических равновесий. Допустим, нас интересует величина pH в 1,0 • 10-2 М
растворе уксусной кислоты. Выражение электронейтральности для этого рас-
твора имеет вид
[Н+] = [ОАс-] + [ОН-]
Разумно допустить, что в растворе кислоты (кислая среда!) концентра-
ция ОН- пренебрежимо мала. Тогда выражение электронейтральности упро-
щается:
[Н+] = [ОАс-]
Иными словами, pH в таком растворе соответствует величине pH в точке пе-
ресечения кривых для [Н+] и [ОАс-]. Эта точка пересечения показана на рис. 7.3;
значение pH в этой точке равно 3,35. Также видно, что при этом значении pH
концентрация ОН- крайне мала (соответствующая точка находится ниже края
рисунка в области 10-11М). Таким образом, предположение о том, что этой вели-
чиной можно пренебречь, было справедливым. Аналогично, для 1,0 • 10-2 М рас-
твора ацетата натрия можно записать
[ОН-] = [НОАс] + [Н+] « [НОАс]
Из координат точки пересечения кривых для [ОН-] и [НОАс] можно найти
значение pH этого раствора, равное 8,35, а также убедиться в справедливости
пренебрежения величиной [Н+] (она примерно в тысячу раз меньше, чем [ОН-] и
[НОАс]).
Можно построить концентрационно-логарифмические диаграммы и для бо-
лее сложных систем. На рис. 7.4 изображена концентрационно-логарифмическая
диаграмма для фосфорной кислоты при концентрации 0,001 М (см. задачу 67
о выводе выражений для lg С применительно к разным формам кислот и зада-
чу 68 по построению концентрационно-логарифмических диаграмм на основе
a-распределительных при помощи электронных таблиц). Из нее, как и из рас-
пределительной диаграммы (см. рис. 7.1), можно сделать выводы о преобладании
при данном pH тех или иных форм. Но и в этом случае только концентрацион-
но-логарифмическая диаграмма позволяет оценить концентрации форм, содер-
жание которых мало. Так, при pH 1 концентрация формы Н3РО4 лишь немногим
меньше 1 • 10-3 М, концентрация Н2РО4 составляет 1 • 104М, а концентрации
НРО 4“ и РО 4" пренебрежимо малы. Из этих данных можно уточнить величину
7.12. КОНЦЕНТРАЦИОННО-ЛОГАРИМФИЧЕСКИЕ ДИАГРАММЫ
363
РИС. 7.4. Концентрационно-логарифмическая диаграмма для 0,001 М раствора фосфорной кислоты
концентрации Н3РО4 вычитанием концентрации Н2РО4 из 1 • 10-3 М, получив
значение ~9 • IO*4 М.
Из рис. 7.4 можно оценить значение pH растворов Н3РО4 и различных ее со-
лей. Для 1 • 10-3 М раствора Н3РО4 выражение электронейтральности имеет вид:
[н+] = [Н2РО4-] + [НРО*-] + [ро4_] + [ОН-]«[Н2ро;]
Видно, что [Н+] равна [Н2РО4] при pH 3,05. При этом значении pH концент-
рации НРО4", РО4“ и ОН- действительно пренебрежимо малы, поэтому допу-
щение [Н+] = [Н2РО4] справедливо.
Для 1 • 10-3 М раствора NaH2PO4 величину pH можно оценить из известного
соотношения [Н+] « ^К^Ка2 или pH « (ptfal + рКа2)/2 « (1,96 + 7,12)/2 = 4,54.
При этом значении pH в растворе присутствуют главным образом частицы Н2РО 4
(~ 1 • 10-3 М), Н+ (1 (У4’54 М), а также HPO 4” и Н3РО4 (приблизительно по 1 (У5>6 М).
Для 1 • 10-3MpacTBopaNa2HPO4pH«(pA:a2 + pA'a3)/2«(7,12 + 12,32)72 = 9,72.
При этом pH [Н2РО;] « РО4~ « ПУ6-55 М и [ОН-] = 1(Н-28 М.
Для 1 • 1(У3 М раствора Na3PO4 можно записать выражение материального
баланса из уравнений протонирования отдельных форм по ступеням:
[ОН-] = [нро4~] + [Н2ро;] + [Н3РО4]«[НРО*-]
Из концентрационно-логарифмических диаграмм можно оценить глубину
протекания реакций депротонирования и протонирования амфолита [см. урав-
нения (7.76) и (7.77)].
364
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
Из рис. 7.4 видно, что равенство [ОН-] = [НРО4-] достигается при pH 10,96.
При этом pH концентрации Н2РО4 и Н3РО4 много меньше, чем НРО4“, и сделан-
ные допущения можно считать справедливыми. В этом растворе [РО 4“] = 10-4»35 М
и [ОН-] = IO"3’04 М.
Упражнение для работы с электронными таблицами:
построение концентрационно-логарифмической диаграммы
для уксусной кислоты
Создадим электронную таблицу (она приведена ниже) для построения концент-
рационно-логарифмической диаграммы для 0,01 М раствора уксусной кислоты
и сравним полученный результат с рис. 7.3. Можно задать такой алгоритм вы-
числений, чтобы потом использовать эту таблицу для построения концентраци-
онно-логарифмических диаграмм и при других концентрациях уксусной кислоты
(для этого в уравнениях, включающих СН0Ас, нужно использовать эту величину
в виде ссылки на ячейку таблицы, а не в форме константы в явном виде).
Для каждой из четырех частиц — НОАс, ОАс", Н+ и ОН- — необходимо за-
дать координаты опорных точек. Относительно НОАс известно, что в кислых сре-
дах величина [НОАс] практически равна СН0Ас. Поэтому мы можем задать для
[НОАс] величину, равную СН0Ас, при pH 0 и 4 (ячейки В4 и В5; жирным шрифтом
А I В I С I D | Е I F G н
1 Концентрационно-логарифмическая диаграмма для уксусной кислоты
2 СнОАс- 0,01 ка= 1.75Е-05
3 pH [НОАс] 1д[НОАс] [ОАс] lg[OAc] |д[ня 1д[ОН]
4 0,00 0,01 -2,00 -6,76 0,00 -14,00
5 4,00 0,01 -2,00 -2,76 -4,00 -10,00
6 4,76 0,005 -2,30 0,005 -2,30 -4,76 -9,24
7 5,50 -2,74 0,01 -2,00 -5,50 -8,50
8 12,00 -9,24 0,01 -2,00 -12,00 -2,00
9 В ячейки, выделенные жирным шрифтом, введены формулы:
10 Ячейка В4: [НОАс] = СНОАс = $С$2 | (Копировать до ячейки В5)
11 Ячейка С4: lg[HOAc] = LOG10(B4) (Копировать до ячейки С6)
12 Ячейка Е4: lg[OAc ] = 1д(Ка • СН0Ас) + pH = I
13 =LOG 10($Е$2*$С$2)+А4 (Копировать до ячейки Е5)
14 Ячейка F4:1д[Н+] = -pH = (-А4) (Копировать до конца)
15 Ячейка G4:1д[ОН] = 14 - 1од[Н+] = (-14-F4) (Копировать до конца)
I6 Ячейка В6: [НОАс] = СНОАс/2 = $С$2/2
17 Ячейка D6: [ОАс] = [НОАс] = В6
18 Ячейка Е6:1д[ОАс ] = LOG10(D6)
19 Ячейка С7: lg[HOAc] = 1д(СНОАс/Ка) - pH = LOG10($C$2/$E$2)-A7 (Копировать до ячейки С8)
20 Ячейка D7: [ОАс ] = СНОАс = $С$2 (Копировать до ячейки D8)
21 График А4:А8 от С4:С8, D4:D8, Е4:Е8 и F4:F8 (серии 1, 2, 3 и 4)
22 Этот рабочий лист может быть использован и для других концентраций уксусной кислоты путем
23 изменения содержания ячейки С2. 1 I
7.12. КОНЦЕНТРАЦИОННО-ЛОГАРИМФИЧЕСКИЕ ДИАГРАММЫ
365
выделены адреса ячеек, в которые следует ввести формулу либо значение и за-
тем скопировать данные в нижележащие ячейки). При pH = рКа = 4,76 • [НОАс] =
= [ОАс-] = 1/2СН0Ас (ячейки В6 и D6). Это точка пересечения двух кривых. При
pH выше 4,76 величину lg[HOAc] рассчитывают по уравнению (7.95) — ячейки
С7 и С8.
Для ОАс- величину lg[OAc-] в кислых средах можно расрчитать по уравне-
нию (7.97) — ячейки Е4 и Е5. При pH ниже, чем рКа величину [ОАс-] можно
считать равной СН0Ас ячейки D7 и D8). Обратите внимание, что величины lg[H+]
и lg[OH-] рассчитывают непосредственно из pH при любых значениях этой ве-
личины (ячейки F4 и G4).
Заметьте, что если в ячейки Е4 и С7 ввести соответственно формулы для
расчета величин lg^*CH0Ac и lgA?a/CH0Ac посредством ссылок на ячейку, содер-
жащую значение СН0Ас (С2), а не с использованием константы (0,01 М), то табли-
ца становится пригодной для использования расчета уксусной кислоты любой
концентрации (ее значение следует лишь ввести в ячейку С2).
Электронная таблица и построенный с ее помощью график зависимости
IgC от pH приведены на компакт-диске (гл. 7). Перепишите этот файл на свой
компьютер и откройте его. На Листе 1 (Sheet 1) содержатся данные, а на от-
дельном листе Диаграмма 1 (Chart 1) — соответствующий рисунок. Сравните
его с рис. 7.3.
Рисунки построены при помощи Мастера диаграмм (Chart Wizard) с исполь-
зованием опций Тип —> Точечная диаграмма (Scatter data) и Вид —>
Плавная кривая. Чтобы посмотреть, как рисунок был построен, откройте
лист Диаграмма 1 (Chart 1), нажмите на кнопку со значком Мастер диа-
грамм (Chart Wizard) и выполните все этапы построения. Для первой серии
данных (lg[HOAc]) диапазон значений X задан выражением =Sheetl!A4:A8,
а значений Y — выражением =Sheetl!C4:C8. Для серий 2, 3 и 4 величины X
те же самые, а величины Y — Е4:Е8, F4:F8 и G41G8 (графики зависимостей для
lg[OAc-], lg[H+] и lg[OH-] соответственно). Попробуйте воспроизвести этот ри-
сунок, открыв лист с данными Sheet 1), а затем Мастер диаграмм (Chart Wizard)
нажатием соответствующей кнопки. После того как рисунок будет готов, снова
откройте лист с данными, измените значение концентрации уксусной кислоты и
посмотрите как при этом будут перемещаться вверх или вниз кривые для НОАс
и ОАс-.
Использование a-коэффициентов для построения
концентрационно-логарифмических диаграмм: это проще
Поскольку для вычисления a-коэффициентов существуют общие формулы, лег-
ко создать электронную таблицу для их расчета (подобно тому, как это сделано
для Н3РО4 — см. рис. 7.1). Чтобы использовать такую таблицу для построения
концентрационно-логарифмических диаграмм, достаточно лишь ввести в ячей-
ку какого-либо столбца формулу вычисления lg(C • cQ (для каждого а-коэффи-
циента) и скопировать ее вниз по столбцу, распространив на все строки,
366
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
содержащие величины pH. Рассмотрим случай двухосновной кислоты Н2А. Пред-
положим, что формула для расчета 04 (доля формы НА-) находится в ячейке D4,
а величина общей концентрации кислоты С—в ячейке В2. Тогда можно ввести
в ячейку Е4 следующую формулу: =LOG10($B$2*D4). В ячейке будет рассчита-
но значение lg[HA-] при соответствующем pH. Чтобы вычислить эту величину
при каждом pH, скопируйте формулу вниз вплоть до последней строки, содер-
жащей значение pH. После этого можно построить график зависимости lg[HA-]
от pH. Описанные действия следует повторить для форм Н2А (с ячейками, со-
держащими значения а0) и А2- (а2), а также заполнить столбцы с величинами
lg[H+] и lg[OH-]. Можно изменить значение общей концентрации С в ячейке В2
и посмотреть, как при этом изменится график. Часто при граничных значениях
pH рассчитанные величины концентраций оказываются очень малы — по лога-
рифмической шкале до -20 и ниже. Поэтому можно ограничить предельное зна-
чение по оси ординат (IgQ величиной, равной, например -10, и тем самым
сделать изображение основной области кривых более детальным.
Вспомогательные данные для построения такого рода диаграмм (примени-
тельно к уксусной кислоте) находятся на компакт-диске (гл. 7). См. также задачу 64
о построении аналогичной диаграммы для двухосновной яблочной кислоты. Обра-
тите внимание, что для многоосновных кислот угловые коэффициенты прямо-
линейных участков кривых не всегда равны +1 или -1.
Что мы узнали из этой главы?
• Теории кислот и оснований — стр. 311
• Кислотно-основные равновесия в воде [основные уравнения (7.11), (7.13),
(7.19)] —стр. 314
• Слабые кислоты и основания — стр. 323
• Соли слабых кислот и оснований [основные уравнения (7.27), (7.29), (7.32),
(7.36), (7.39)] —стр. 326
• Буферные растворы [основные уравнения (7.45), (7.56)] — стр. 330
• Многоосновные кислоты — величины а [основные уравнения
(7.72)-(7.75)] — стр. 339, 342
• Использование электронных таблиц для расчета а как функции pH —
стр. 345
• Соли многоосновных кислот [основные уравнения (7.83), (7.84), (7.86),
(7.87)] —стр. 348
• Логарифмические концентрационные диаграммы — стр. 359
• Использование электронных таблиц для построения графиков —
стр.364
• Использование величин а для построения графиков с помощью элект-
ронных таблиц — стр. 365
ЗАДАЧИ
367
Вопросы
1 - Объясните разницу между сильными и слабыми электролитами. «Нерас-
творимая» соль — это сильный или слабый электролит?
2. В чем суть теории кислот и оснований Бренстеда?
3. Что такое сопряженная кислота? Сопряженное основание?
4 - Напишите уравнение реакции ионизации анилина C6H5NH2 в ледяной уксус-
ной кислоте и укажите сопряженную анилину кислоту. Напишите уравнение
реакции ионизации фенола С6Н5ОН в этилендиамине NH2CH2CH2NH2 и ука-
жите сопряженное фенолу основание.
5- В чем суть теории кислот и оснований Льюиса?
Задачи
Сильные кислоты и основания
6- Рассчитайте pH и рОН следующих растворов сильных кислот:
а) 0,020 М НСЮ4, б) 1,3 • 10^ М HNO3, в) 1,2 М НС1, г) 1,2 • 10~9 М НС1,
д) 2,4 • IO’7 М HNO3.
7- Рассчитайте pH и рОН следующих растворов сильных оснований:
а) 0,050 М NaOH, б) 0,14 М Ва(ОН)2, в) 2,4 М NaOH, г) 3,0 • 10"7 М КОН,
д) 3,7 • IO"3 М КОН.
8. Рассчитайте концентрацию гидроксил-иона в следующих растворах:
а) 2,6 • 10-5 М НС1, б) 0,20 М HNO3, в) 2,7 М -10~9 М НС1О4, г) 1,9 М НС1О4.
9 - Рассчитайте концентрацию иона водорода в растворах со следующими зна-
чениями pH: а) 3,47, б) 0,20, в) 8,6, г) -0,60, д) 14,35, е) -1,25.
10- Рассчитайте pH и рОН раствора, полученного смешением равных объемов
0,10 М H2SO4 и 0,30 М NaOH.
11 - Рассчитайте pH раствора, полученного смешением равных объемов раство-
ров сильной кислоты с pH 3 и сильного основания с pH 12,00.
Влияние температуры
12. Рассчитайте концентрацию ионов водорода и pH в нейтральном растворе
при 50 °C (Kw =5,5 • 1014 при 50 °C).
13- Рассчитайте рОН образца крови, значение pH которого при 37 °C составля-
ет 7,40.
Слабые кислоты и основания
14, Значение pH раствора уксусной кислоты составляет 3,26. Какова концент-
рация уксусной кислоты и на сколько процентов кислота диссоциирована?
368
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
15- Значение pH 0,20 М раствора первичного амина RNH2 составляет 8,42.
Чему равно рКь амина?
16- Степень диссоциации одноосновной органической кислоты, Ка которой
равна 6,7 • 10-4, составляет 3,5 % при концентрации кислоты 100 г/л. Какова
молярная масса кислоты?
17- Рассчитайте pH 0,25 М раствора пропионовой кислоты.
18. Рассчитайте pH 0,10 М раствора анилина (слабое основание).
19- Рассчитайте pH 0,1 М раствора йодноватой кислоты НЮ3.
20. Серная кислота диссоциирует по первой ступени полностью, а по второй
ступени — частично, константа кислотности Ка2 составляет 1,2 • 10-2 Рас-
считайте концентрацию ионов водорода в 0,0100 М растворе H2SO4.
21 - Рассчитайте концентрацию ионов водорода в 0,100 М растворе трихлорук-
сусной кислоты.
22 - Значение рКь амина RNH2 составляет 4,20. Каково значение pH 0,20 М рас-
твора этого основания?
23 - Какова концентрация уксусной кислоты, если диссоциировано 3% кислоты?
24- Во сколько раз следует разбавить 0,100 М раствор слабой кислоты НА, чтобы
степень ее диссоциации увеличилась в два раза, если допустить, что С > 100 Ка.
Соли слабых кислот и оснований
25. Какое значение pH будет у раствора, если к 20 мл 0,25 М борной кислоты
добавить 25 мл 0,20 М раствора NaOH?
26- Рассчитайте pH 0,010 М раствора NaCN.
27. Рассчитайте pH 0,050 М раствора бензоата натрия.
28. Рассчитайте pH 0,25 М раствора хлорида пиридиния (пиридин • НС1,
C6H5NH+C1).
29. Рассчитайте pH раствора, полученного добавлением 12,0 мл 0,25 М раство-
ра H2SO4 к 6,0 мл 1,0 М раствора NH3.
30. Рассчитайте pH раствора, полученного добавлением 20 мл 0,10 М раствора
НОАс к 20 мл 0,10 М раствора NaOH.
31. Рассчитайте pH раствора, полученного добавлением к 500 мл воды по
0,10 мл гидроксиламина и хлороводородной кислоты.
32. Рассчитайте pH 0,0010 М раствора салицилата натрия С6Н4 (OH)COONa.
33- Рассчитайте pH 1.0 • 10^ М раствора NaCN.
Многоосновные кислоты и их соли
34. Рассчитайте pH 0,0100 М раствора фталевой кислоты?
35- Рассчитайте pH 0,0100 М раствора фталата калия?
36- Рассчитайте pH 0,0100 М раствора гидрофталата калия (КНР)?
ЗАДАЧИ
369
37, Рассчитайте pH 0,600 М раствора Na^.
38. Рассчитайте pH 0,500 М раствора Na3PO4.
39. Рассчитайте pH 0,250 М раствора NaHCO3.
40. Рассчитайте pH 0,600 М раствора NaHS.
41. Рассчитайте pH 0,050 М раствора тринатриевой соли ЭДТА (этиленди-
аминтетрауксусная кислота) Na3HY.
Буферные растворы
42. Рассчитайте pH раствора, представляющего собой смесь 0,050 М раствора
муравьиной кислоты и 0,1 М раствора формиата натрия.
43. Рассчитайте pH раствора, полученного смешением 5,0 мл 0,10 М раствора
NH3 и 10,0 мл 0,020 М раствора НС1.
44. Концентрация NaOAc в буферном растворе уксусная кислота — ацетат на-
трия с pH 5,00 составляет 0,100 М. Рассчитайте pH раствора, полученного
добавлением 10 мл 0,1 М раствора NaOH к 100 мл буферного раствора.
45. Буферный раствор приготовлен добавлением 20 мл 0,10 М раствора гидро-
ксида натрия к50мл0,10М раствора уксусной кислоты. Какое значение pH
будет у буферного раствора?
46. Буферный раствор приготовлен добавлением 25 мл 0,050 М раствора сер-
ной кислоты к 50 мл 0,10 М раствора аммиака. Какое значение pH будет у
буферного раствора?
47. Аспирин (ацетилсалициловая кислота) всасывается стенками желудка в
свободной (неионизированной) форме. Если пациент принимает нейтрали-
зующий кислоту препарат, который повышает pH среды желудка до 2,95, а
затем ему дают две 5-грановые таблетки аспирина (общая масса 0,65 г), то
сколько грамм аспирина могут сразу же всосаться стенками желудка при
условии его немедленного растворения в желудке? Предположите также,
что аспирин не меняет pH среды желудка. Значение рКа аспирина составля-
ет 3,50, а его молекулярная масса равна 180,2.
48. 7)шс(гидроксиметил)аминометан [(HOCH2)3CNH2 — Tris, или ТНАМ] яв-
ляется слабым основанием, которое часто используют в биохимии для при-
готовления буферных растворов. Его Кь равна 1,20 • 10^, а рКь равна 5,92.
Соответствующее значение рКа составляет 8,08, и оно близко к значению
pH физиологических буферных растворов, поэтому растворы на основе
ТНАМ при pH, близком к физиологическому, имеют хорошую буферную
емкость. Какова масса ТНАМ, необходимого для приготовления 1 л бу-
ферного раствора с pH 7,40, если к ТНАМ добавляют 100 мл 0,50 М рас-
твора НС1?
49. Рассчитайте концентрацию ионов водорода по данным задачи 21 при усло-
вии, что раствор содержит также 0,100 М трихлорацетата натрия.
370
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
Буферные растворы из многоосновных кислот
50- Какое значение pH имеет раствор, содержащий 0,10 М гидрофталата калия
(КНР) и 0,20 М фталевой кислоты (Н2Р)?
51 - Какое значение pH имеет раствор, содержащий по 0,25 М гидрофталата ка-
лия (КНР) и фталата калия (К2Р)?
52 - Общая концентрация фосфата в образце крови, определенная спектрофото-
метрическим методом, составила 3,0 • 10-3 М. Какова концентрация ионов
Н2РО; и НРО4” в образце крови при pH 7,45?
Буферная емкость
53- Буферный раствор содержит 0,10 М NaH2PO4 и 0,070 М Na^PO^ Какова
буферная емкость этого раствора, выраженная в моль/литр на единицу pH?
Как изменится pH, если к 10 мл буферного раствора добавить 10 мкл
(0,010 мл) 1,0 М раствора НС1 или 1,0 М раствора NaOH?
54- Вам нужно приготовить буферный раствор уксусной кислоты — ацетат на-
трия с pH 4,76 и буферной емкостью 1,0 М на единицу pH. Какая концент-
рация должна быть у используемых растворов уксусной кислоты и ацетата
натрия.
Буферные растворы с постоянной ионной силой
55- Какие навески Na2HPO4 и КН2РО4 необходимо взять для приготовления
200 мл буферного раствора с pH 7,40 и ионной силой 0,20 моль/л? (Об опре-
делении ионной силы см. гл. 6.)
56. Какой объем 85% (по массе) Н3РО4 (удельный вес 1,69) и какую навеску
КН2РО4 необходимо взять для приготовления 200 мл буферного раствора с
pH 3,00 и ионной силой 0,20 моль/л?
Расчет а-коэффициента
57- Рассчитайте равновесные концентрации всех частиц в 0,0100 М растворе
сернистой кислоты H2SO3 при pH 4,00 ([Н+] = 1,0 • 1(Н М).
58. Выведите уравнения (7.73), (7.74) и (7.75) для ар а2и а3 фосфорной кислоты.
Влияние посторонних ионов
59. Рассчитайте концентрацию ионов водорода для 0,0200 М раствора HCN в
0,100 М растворе NaCl (солевой эффект).
60. Выведите уравнение, аналогичное уравнению (7.91), но характеризующее
влияние посторонних ионов на незаряженное слабое основание В.
Концентрационно-логарифмические диаграммы
Для решения задачи 61 вы можете воспользоваться электронной таблицей, при-
веденной на компакт-диске для случая НОАс. Но попытайтесь также построить
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
371
кривые вручную. Для задачи 64 сами создайте электронную таблицу, используя
значения a-коэффициентов. Вспомогательные данные для построения концент-
рационно-логарифмических диаграмм для НОАс приведены на компакт-диске.
61 - Постройте концентрационно-логарифмическую диаграмму для 10-3 М рас-
твора уксусной кислоты.
62- По диаграмме из задачи 61, рассчитайте pH 10-3 М раствора уксусной кис-
лоты. Какова концентрация ацетат-ионов в этом растворе?
63 - Для условий задачи 61 выведите выражение для 1g [ОАс-] в растворе кисло-
ты и рассчитайте концентрацию ацетат-ионов при pH 2,00 в 10-3 М растворе
уксусной кислоты. Сравните эту величину с величиной, полученной по
концентрационно-логарифмической диаграмме.
64- Постройте концентрационно-логарифмическую диаграмму для 10 3 М рас-
твора яблочной кислоты, создайте для этого электронную таблицу с испо-
льзованием величин а-коэффициентов.
65- По диаграмме в задаче 64 рассчитайте pH и концентрацию каждой части-
цы в: а) 10-3 М растворе яблочной кислоты и 6)10-3 М растворе яблочнокис-
лого натрия.
66- Из условий задачи 61 выведите уравнения для кривых НА- в кислых и ще-
лочных областях.
67- Выведите уравнения функций: a) lg[H3PO4] между значениями pH, равны-
ми рЛ?а1 и рАГа2, б) lg[H2PO4] между значениями pH, равными рАГа2 и рАГа3,
в) lg[HPO 4“] между значениями pH, равными рЛГа2 и рАГа1, (г) lg[PO 4“] меж-
ду значениями pH, равными рАГа3 и рЛГа2. Проверьте правильность получен-
ных выражений по характерным точкам на кривых.
68- Постройте концентрационно-логарифмическую диаграмму для 0,001 М
раствора Н3РО4 (рис 7.4), используя выличины а. Начните с электронной
таблицы для построения графика, изображенного на рис. 7.1 (есть на ком-
пакт-диске). Сравните диаграмму с рис. 7.4. Измените концентрацию
Н3РО4 и посмотрите, как изменяются кривые.
Рекомендуемая литература
Теории кислот и оснований, буферные растворы
1 - R. G. Bates, «Concept and Determination of pH», in I. M. Kolthoff and P. J. Elving,
eds., Treatise on Analytical Chemistry, Part I, Vol. 1. New York: Wiley-Intersci-
ence, 1959, pp. 361^101.
2. N. W. Good, G. D. Winget, W. Winter, T. N. Connally, S. Izawa and
R. M. M. Singh, «Hydrogen Ion Buffers for Biological Research», Biochemistry,
5(1966) 467.
372
КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
3. D. Е. Gueffroy, ed., Л Guide for the Preparation and Use of Buffers in Biological
Systems. La Jolla, CA: Calbiochem, 1975.
4. I. M. Kolthoff, «Concepts of Acids and Bases», in I. M. Kolthoff and P. J. Elving,
eds., Treatise on Analytical Chemistry, Part I, Vol. 1. New York: Wiley-Intersci-
ence, 1959, pp. 405-420.
5. D. D. Perrin and B. Dempsey, Buffers for pH and Metal Ion Control. New Y ork:
Chapman and Hall, 1974.
Расчеты равновесий
6. S. Brewer, Solving Problems in Analytical Chemistry. New York: Wiley, 1980.
Describes iterative approach for solving equilibrium calculations.
7. J. N. Butler, Ionic Equilibria. A Mathematical Approach. Reading, MA: Addi-
son-Wesley, 1964.
8. W. B. Guenther, Unified Equilibrium Calculations. New York: Wiley, 1991.
9. D. D. DeFord, «The Reliability of Calculations Based on the Law of Chemical
Equilibrium», J. Chem. Ed., 31 (1954) 460.
10. E. R. Nightingale, «The Use of Exact Expressions in Calculating H+ Concentrati-
ons», J. Chem. Ed., 34 (1957) 277.
11. R. J. Vong and R. J. Charlson, «The Equilibrium pH of a Cloud or Raindrop:
A Computer-Based Solution for a Six-Component System», J. Chem. Ed., 62
(1985) 141.
12. R. deLevie, A Spreadsheet Workbook for Quantitative Chemical Analysis. New
York: McGraw-Hill, 1992.
13. H. Freiser, Concepts and Calculations in Analytical Chemistry. A Spreadsheet
Approach. Boca Raton, FL: CRC Press, 1992.
Виртуальный сетевой калькулятор
14. http://hamers.chem. wisc.edu/chapman/Titrator/ — виртуальный калькулятор
1,5 для расчета a-кривых на сайте Профессора Роберта Хамерса, Универ-
ситет штата Висконсин (США). В окне Virtual Titrator (виртуальный титратор)
вы можете выбрать кислоту, графики и воспользоваться, стехиометрическим
калькулятором для приготовления растворов.
Глава 8
КИСЛОТНО-ОСНОВНЫЕ ТИТРОВАНИЯ
Любезно предоставлено
Metrohm AG.
В предыдущей главе мы познакомились с основными поло-
жениями теорий кислотно-основных равновесий. Эти зна-
ния важны для построения и интерпретации кривых
титрования при использовании метода кислотно-основного
титрования. В данной главе рассмотрены различные типы
кислотно-основного титрования, включая титрование силь-
ных кислот и оснований, а также слабых кислот и оснований
(для каждого случая приводятся кривые титрования). В рам-
ках теории индикаторов обсуждается выбор подходящего
индикатора для установления конечной точки конкретных
титриметрических реакций. Рассмотрены примеры титрова-
ния слабых кислот и оснований с двумя или более титруемыми
группами, а также смесей кислот и оснований. Приведено
описание метода Кьельдаля, имеющего большое значение
при определении азота в органических соединениях и образ-
цах биологических тканей.
8.1. Сильные кислоты и сильные основания:
самый простой вариант титрования
В основе кислотно-основного титрования лежит реакция кислоты с эквивалент-
ным количеством основания. При построении кривой титрования — графика
зависимости pH раствора от добавленного объема титранта — легко понять, как
определить конечную точку титрования. Титрантом всегда является раствор
^ильной кислоты или сильного основания. Аналит может быть как сильным, так и
слабым основанием или кислотой.
374
КИСЛОТНО-ОСНОВНЫЕ ТИТРОВАНИЯ
При титровании растворов сильных кислот (сильных оснований) и титрант,
и аналит полностью ионизированы. Примером может служить титрование рас-
твора хлороводородной кислоты раствором гидроксида натрия:
Н+ + Cl- + Na+ + ОН- Н2О + Na+ + С1~ (8.1)
Ионы Н+ и ОН- взаимодействуют с образованием Н2О, а другие ионы (Na+ и С1")
не участвуют реакциях, поэтому в целом результатом реакции является превра-
щение раствора кислоты НС1 в нейтральный раствор NaCl. Кривая титрования
100 мл 0,1 М раствора НС1 0,1 М раствором NaOH приведена на рис. 8.1; для
построения этой кривой использована электронная таблица, приведенная ниже.
Чтобы построить кривую титрования, необходимо по способу, описанному
в гл. 7, рассчитать pH в зависимости от состава раствора на разных стадиях тит-
рования. При расчете концентрации частиц, присутствующих в растворе, надо
обязятельно учитывать изменение объема при титровании.
В табл. 8.1 приведены уравнения, по которым рассчитывают pH на разных
участках кривой титрования. Начальная точка кривой соответствует 0,1 М рас-
твору НС1, поэтому исходное значение pH равно 1,0. По мере титрования часть
ионов Н+ связывается с образованием Н2О. Поэтому концентрация ионов Н+ по-
стоянно уменьшается. Когда реакция проходит на 90% (добавлено 90 мл NaOH),
в растворе остается только 10% ионов Н+. Если пренебречь изменением объема,
то концентрация ионов Н+ в этой точке составит 10-2 М, а pH увеличится только
на одну единицу. (Если мы учтем изменение объема, то значение pH будет не-
много больше — см. электронную таблицу ниже.) Тем не менее, по мере прибли-
жения к точке эквивалентности (это точка, соответствующая добавлению
стехиометрического количества основания), концентрация ионов Н+ быстро
уменьшается вплоть до ее достижения, где завершается нейтрализация кислоты
и остается нейтральный раствор NaCl, а значение pH равно 7,0. Точка эквивален-
тности — это точка, в которой реакция теоретически завершается. По мере
добавления раствора NaOH концентрация ОН- быстро возрастает от 10-7 М в
точке эквивалентности до 10-2-10-1 М; на этой стадии процесса титрования в
растворе будут находиться уже NaOH и NaCl. Таким образом, вдали от точки эк-
Таблица8.1
Основные уравнения для расчета кривых титрования сильной кислоты (НХ)
или сильного основания (ВОН)
Степень оттит- рованное™ F Сильная кислота Сильное основание
Находится в растворе Уравнение Находится в растворе Уравнение
F=0 НХ [Н+] = [НХ] ВОН [ОН-] = [ВОН]
0<F< 1 НХ/Х- [Н+] =[оставшаяся НХ] вон/в+ [ОН-] = [оставшееся ВОН]
F = 1 X- [Н+] = 7x7 [ур. (7.13)] в+ [Н+] = Ддур. (7.13)]
F> 1 0Н7Х- [ОН-] = [избыток титранта] н+/в+ [Н+] = избыток титранта]
Рис. 8.1
Кривая титрования
100 мл 0,1 М НС1 раствором
0,1 М NaOH
14
ЦмаОН» мл
А в I с D I Е F G
1 Кривая тит оования 100 мл 0,1 М Н Cl 0.1 М NaOH
2 млнсг 100,00 МНСГ 0,1000
3 МмаОН= 0,1000 1^,= 1.00Е-14
4 MJ1NaOH [Н+] [ОН-] рОН pH
5 0,00 0,1 1,00
6 10,00 0,0818182 1,09
7 20,00 0,0666667 1,18
8 30,00 0,0538462 1,27
9 40,00 0,0428571 1,37
10 50,00 0,0333333 1,48
11 60,00 0,025 1,60
12 70,00 0,0176471 1,75
13 80,00 0,0111111 1,95
14 90,00 0,0052632 2,28
15 95,00 0,0025641 2,59
16 98,00 0,0010101 3,00
17 99,00 0.0005025 3,30
18 99,20 0,0004016 3,40
19 99,40 0,0003009 3,52
20 99,60 0,0002004 3,70
21 99,80 0,0001001 4,00
22 99,90 5.003Е-05 4,30
23 99,95 2,501 Е-05 4,60
24 100,00 0,0000001 7,00
25 100,05 2,5Е-05 4,60 9,40
26 100,10 5Е-05 4,30 9,70
27 100,20 1Е-04 4,00 10,00
28 100,40 0,0002 3,70 10,30
29 100,80 0,0004 3,40 10,60
30 101,00 0,0005 3,30 10,70
31 102,00 0,00099 3,00 11,00
32 105,00 0,00244 2,61 11,39
33 110,00 0,00476 2,32 11,68
34 120,00 0,00909 2,04 11,96
35 140,00 0,01667 1,78 12,22
36 В ячейки, выделенные жирным шрифтом, введены формулы:
37 Ячейка В5: [Н+] = (млНС1 х МНС1- мл№ОН х М№ОН)/(млНС1 + мл№ОН)
38 = |($B$2*$D$2-A5*$B$3)/($B$2+A5) Копировать вниз до ячейки В23
39 Ячейка Е5 = рН= [C-LOG10(B5)) Копировать вниз до ячейки Е24
40 Ячейка В24 = [Н ] = К^/2 = |SQRT(D3)
41 Ячейка С25 = [ОН-] = (млМаОН х MNa0H - млнс| х Мнс|У(млНС| + мл№0Н)
42 = | (A25*$B$3-$B$2*$D$2)/($B$2+A25) Копировать вниз
43 Ячейка D25 = рОН = -1д[ОН’] = (-LOG 10(С25)) Копировать вниз
44 Ячейка Е25 = pH = 14 - рН= 14-D25 Копировать вниз
376
КИСЛОТНО-ОСНОВНЫЕ ТИТРОВАНИЯ
вивалентности как с одной, так и с другой стороны pH меняется мало, но вблизи
точки эквивалентности происходит резкое изменение pH. Такой резкий скачок
pH позволяет определить окончание реакции измерением pH или какого-либо
параметра, который зависит от pH.
Пример 8.1
Рассчитайте значения pH в точках, соответствующих 0, 10, 90, 100 и 110% от-
титрованное™, при титровании 50,0 мл 0,100 М раствора НС1 0,100 М раство-
ром NaOH.
Решение
Следите за израсходованным и оставшимся количеством миллимолей кислоты!
При 0% оттитрованное™ pH = -1g 0,100 = 1,00. При 10% оттитрованное™ до-
бавлено 5,0 мл раствора NaOH. В исходном растворе НС1 0,100 М • 50,0 мл =
= 5,00 ммоль ионов Н+. Рассчитаем концентрацию Н+ после добавления раство-
ра NaOH:
Исходное количество Н+
Добавленное количество ОН- = 0,100 М • 5,0 мл
Оставшееся количество Н+
= 5,00 ммоль
= 0,500 ммоль
= 4,50 ммоль Н+ в 55,0 мл
[Н+] = 4,50 ммоль/55,0 мл = 0,0818 М
pH = -lg 0,0818= 1,09
При 90% оттитрованное™
Исходное количество Н+
Добавленное количество ОН" = 0,100 М • 45,0 мл
Оставшееся количество Н+
= 5,00 ммоль
= 4,500 ммоль
= 0,50 ммоль Н+ в 95,0 мл
[Н+] = 0,00526 М
pH = -lg 0,00526 = 2,28
При 100% оттитрованное™: все ионы Н+ прореагировали с ионами ОН-, и в
результате получился нейтральный 0,0500 М раствор NaCl, pH которого равен
7,00.
При 110% оттитрованное™: в растворе находится смесь NaCl и избытка
NaOH.
ммоль ОН- = 0,100 М • 5,0 мл = 0,50 ммоль ОН-в 105 мл
[ОН-] = 0,00476 М
рОН = -1g 0,00476 = 2,32; pH = 11,68
8.1. СИЛЬНЫЕ КИСЛОТЫ И СИЛЬНЫЕ ОСНОВАНИЯ: САМЫЙ ПРОСТОЙ ВАРИАНТ ТИТРОВАНИЯ
377
Обратите внимание, что до точки эквивалентности, когда в растворе нахо-
дится избыток кислоты, справедливо выражение
[Н+] = (Скислоты-
^кислоты ^основания ^основания)/ ^общий
где V — это объем. Вы легко можете использовать это уравнение для расчета
[Н+], если вам понятно решение примера 8.1. Подобным же образом, после точ-
ки эквивалентности, когда в растворе находится избыток основания, справедли-
во выражение
[Н ] (^основания ^основания ^кислоты ^кислотыУ^общий
Величина скачка на кривой титрования будет зависеть от концентрации
как кислоты, так и основания. Кривые титрования при разных концентрациях
кислоты и основания приведены на рис. 8.2. При титровании щелочи кислотой
получается зеркальное отображение этих кривых. Кривая титрования 0,1 М раство-
ра NaOH 0,1 М раствором НС1 приведена на рис. 8.3. Выбор индикаторов
проиллюстрирован на рисунках и подробнее рассматривается ниже. Выбор ин-
дикатора становится тем труднее, чем более разбавлены растворы.
Создание электронной таблицы для построения кривой
титрования раствора HCI раствором NaOH
Давайте создадим электронную таблицу, для построения кривой, изображенной
на рис. 8.1. Введите значения концентрации и объема раствора НС1 и концентра-
ции раствора NaOH и величины Kw в ячейки (В2, D2, ВЗ и D3 соответственно).
Это абсолютные значения, которые будут использованы в формулах. В элект-
Рис. 8.2
Зависимость величины скачка
вблизи конечной точки
титрования от концентрации.
Кривая/: 100 мл 0,1 МНС1
титруют 0,1 М NaOH.
Кривая 2: 100 мл 0,01 М НС1
титруют 0,01 М NaOH.
Кривая 3: 100 мл 0,001 М НС1
титруют 0,0001 М NaOH
378
КИСЛОТНО-ОСНОВНЫЕ ТИТРОВАНИЯ
Рис. 8.3
Кривая титрования
100 мл 0,1 М NaOH раствором
0,1 МНС1
^НС1 (0,1 М)’ мл
ронной таблице концентрацию ионов водорода рассчитывают из оставшегося
количества миллимолей НС1 путем деления на общий объем раствора (формула
в ячейке В5). В точке эквивалентности концентрация ионов водорода равна кор-
ню квадратному из Kw (формула в ячейке В24). А после точки эквивалентности
концентрация гидроксид-ионов равна избыточному количеству NaOH в милли-
молях, деленному на общий объем раствора (формула в ячейке С25). Обратите
внимание, что концентрация растворов НС1 и NaOH, объем раствора НС1 и зна-
чение Kw в формулах являются абсолютными значениями. Введите формулы для
расчета pH из [Н+] (ячейка Е5), рОН из [ОН-] (ячейка D25) и pH из рОН (ячей-
ка Е25).
Кривую титрования строят с помощью Мастер диаграмм (Chart Wizard) как
зависимость значений в ячейках А5:А35 (ось X) от Е5:Е35 (ось Y). Подробности
построения графика см. в электронной таблице в гл. 7 для графика зависимости
а от pH, рис. 7.1. Графическое изображение кривой титрования (Chart 1) приве-
дено на компакт-диске, гл. 8.
8.2. Установление конечной точки титрования: индикаторы
В ходе титрования необходимо установить положение точки эквивалентности.
Точка, в которой на практике завершают титрование, называется конечной
точкой титрования. Метод определения конечной точки титрования выбирают
таким образом, чтобы она была как можно ближе к точке эквивалентности. Наи-
более точный с этой точки зрения способ состоит в измерении pH в различные
8.2. УСТАНОВЛЕНИЕ КОНЕЧНОЙ ТОЧКИ ТИТРОВАНИЯ: ИНДИКАТОРЫ
379
моменты титрования и построении графика зависимости pH от объема титранта
(мл). Для этого используют pH-метр, описание которого дано в гл. 13.
Чаще применяют визуальный метод, который отличается удобством и про-
стотой. Он состоит в том, что в титруемый раствор добавляют индикатор и о
достижении конечной точки титрования судят по изменению его окраски. Ин-
дикаторами в кислотно-основном титровании служат интенсивно окрашенные
слабые кислоты или слабые основания. Окраска кислотной формы индикатора
должна заметно отличаться от окраски его основной формы. Одна форма может
быть бесцветной, но другая обязательно должна быть окрашена. Молекулы ин-
дикаторов обычно содержат большое число сопряженных органических групп,
обусловливающих окраску индикатора (см. гл. 16).
Предположим, что индикатор — это слабая кислота, обозначим ее Н1п и бу-
дем считать, что неионизированная форма окрашена в красный цвет, а ионизи-
рованная — в синий:
Н1п Н+ + 1п-
(красный) (синий)
(8-2)
Так же, как и для других слабых кислот, в данном случае применимо урав-
нение Гендерсона—Хассельбаха:
pH = p^ln + ign^J
ln [Н1п]
(8.3)
Индикатор изменяет окраску в определенном интервале pH. Ширина ин-
тервала перехода окраски индикатора зависит от способности наблюдателя раз-
личать небольшие изменения цвета. При использовании индикаторов, у которых
обе формы окрашены, один цвет обычно различим на фоне другого, когда соот-
ношение концентраций двух форм равно 10:1; при этом мы видим цвет той фор-
мы, концентрация которой выше.
На основании этой информации мы можем рассчитать интервал pH, в кото-
ром происходит переход окраски индикатора. Если видна окраска только неио-
низированной формы, то [1п_]/[Шп] = 1/10. Поэтому,
Рн=Р^ + 1ё1=Р^-1
(8.4)
Когда видна окраска только ионизированной формы, [1гг]/[Н1п] = 10/1 и
pH = рКа + lg — = рКа + 1
(8.5)
380
КИСЛОТНО-ОСНОВНЫЕ ТИТРОВАНИЯ
Таким образом, pH перехода окраски индикатора лежит в пределах от
рКа - 1 до рКа + 1. Интервал изменения pH составляет 2 единицы для боль-
шинства; в его пределах наблюдаемая окраска представляет собой смесь
двух цветов.
В середине этого интервала концентрации двух форм равны и pH = рКа.
Нужно так выбирать индикатор, чтобы его рКа было близко к значению pH в точ-
ке эквивалентности.
Подобные расчеты можно провести и для индикаторов, которые проявляют
свойства слабых оснований; в середине интервала рОН = рА^, а pH = 14 - ^Кь.
Следовательно, индикатор, который является слабым основанием, нужно выби-
рать таким образом, чтобы вблизи точки эквивалентности pH = 14 - рКь.
На рис. 8.4 приведен перечень наиболее часто используемых индикаторов с
указанием окраски и интервалов ее перехода в единицах pH.
В некоторых случаях ширина интервала может быть немного меньше: неко-
торые цвета легче различить, чем другие. Переход легче заметить, если одна из
форм бесцветная. По этой причине наиболее часто при титровании сильных кис-
лот и оснований используют, если это возможно, фенолфталеин (см. рис. 8.1, тит-
рование 0,1 М НС1). Однако при титровании разбавленных растворов интервал
перехода окраски фенолфталеина не совпадает со скачком pH на кривой титро-
вания (см. рис. 8.2), и в этом случае необходимо использовать такой индикатор,
как, например, бромтимоловый синий. Подобная же ситуация наблюдается при
Индикатор
pH
0 2 4 6 8
I---->----1-----1---1-----1----1----->----1-
10 12
---•—н- ч
Метиловый фиолетовый:_____желтый Г | фиолетовый
Кристаллический фиолетовый: _ желтый | | синий
Крезоловый красный:-----------красный | | желтый
Бромфеноловый синий:__________________желтый | | синий
Метиловый оранжевый:------------------красный | | желтый
Бромкрезоловый зеленый:--------------------желтый I I синий
Метиловый красный:-------------------------красный I | желтый
Метиловый пурпурный:-----------------------пурпурный I I зеленый
Бромтимоловый синий:-------------------------------желтый I I синий
Лакмус:---------------------------------------красный I I синий
Крезоловый красный:-----------------------------------желтый I 1 красный
Тимоловый синий:______________красный | | желтый_желтый I I синий
Фенолфталеин:--------------------------------------бесцветный I I красно-фиолетовый
Тимолфталеин:-----------------------------------------------бесцветный I I синий
Ализарин желтый R:------------------------------------------желтый I 1 красный
РИС. 8.4. Интервалы pH, соответствующие изменению окраски некоторых распространенных ин-
дикаторов
8.4. ТИТРОВАНИЕ СЛАБЫХ КИСЛОТ СИЛЬНЫМИ ОСНОВАНИЯМИ
381
титровании раствора NaOH раствором НС1 (см. рис. 8.3).* Более полный пере-
чень индикаторов приведен на заднем форзаце.
Поскольку индикатор является слабой кислотой или слабым основанием,
его необходимо добавлять в титруемый раствор в минимальном количестве,
чтобы его влияние на pH было незначительным и чтобы для изменении окраски
индикатора требовалось очень небольшое количество титранта.
Изменение окраски индикатора будет более четким, если его концентрация
будет ниже, потому что для переведения одной из его форм в другую потребует-
ся меньше кислоты или основания. Конечно, индикатор нужно добавлять в та-
ком количестве, чтобы придать раствору хорошо различимую окраску. Обычно
готовят раствор индикатора с концентрацией несколько десятых процента (мас-
са : объем) и добавляют две или три капли в титруемый раствор. Две капли
(0,1 мл) 0,01 М раствора индикатора (0,1% раствор при молярной массе
М= 100 г/моль) эквивалентны 0,01 мл 0,1 М раствора титранта.
8.3. Стандартные растворы кислот и оснований
В качестве титранта при титровании оснований обычно используют растворы
сильной хлороводородной кислоты, тогда как растворы гидроксида натрия слу-
жат обычно титрантом для титрования кислот. Большинство хлоридов раство-
римо в воде, кроме того, при использовании НС1 побочные реакции протекают
редко. С НС1 удобно работать.
Ни один из этих титрантов (растворы НС1 и NaOH) не может быть первич-
ным стандартным раствором, поэтому сначала готовят растворы с приблизите-
льной концентрацией, а затем их стандартизируют титрованием первичным
стандартным раствором кислоты или основания. При приготовлении растворов,
особенно раствора гидроксида натрия необходимо соблюдать специальные пре-
досторожности. В гл. 2 описаны методики приготовления и стандартизации рас-
творов хлороводородной кислоты и гидроксида натрия.
8.4. Титрование слабых кислот сильными основаниями:
это немного сложнее
Кривая титрования 100 мл 0,1 М раствора уксусной кислоты 0,1 М раствором
гидроксида натрия приведена на рис. 8.5. Уравнение реакции титрования следу-
ющее:
НОАс + Na+ + ОН- -> Н2О + Na+ + ОАс~ (8.6)
В действительности в данном примере можно использовать оба индикатора; индикаторная погрешность
для фенолфталеина несколько больше, но чаще всего на практике ею пренебрегают. — Прим, перев.
382
КИСЛОТНО-ОСНОВНЫЕ ТИТРОВАНИЯ
Уксусная кислота, диссоциация которой протекает в зависимости от концент-
рации всего лишь на несколько процентов, при взаимодействии с гидроксидом на-
трия образует воду и эквивалентное количество соли, ацетата натрия. Перед
началом титрования pH 0,1 М раствора НОАс можно рассчитать, как описано в
гл. 7 для слабой кислоты. В табл. 8.2 приведены уравнения, выведенные в гл. 7 и
используемые для расчета различных участков кривой титрования. В самом нача-
ле титрования часть НОАс переходит в NaOAc, и образуется буферная система.
По мере продолжения титрования pH медленно увеличивается, так как изменяет-
ся отношение [ОАс-]/[НОАс]. В точке полуоттитрованности [ОАс-] = [НОАс],
и значение pH становится равным рКа. В точке эквивалентности в растворе на-
ходится NaOAc. Так как это основание по Бренстеду, то среда в точке эквива-
лентности будет щелочной. При этом значение pH зависит от концентрации
NaOAc [см. уравнение (7.32) и рис. 8.6]: чем выше концентрация, тем выше pH.
После точки эквивалентности в растворе присутствует избыток NaOH, и диссо-
циация основания ОАс- почти полностью подавляется [см. уравнение (7.23)].
Поэтому в этих условиях значение pH определяется только избытком ОН-. Та-
ким образом, после точки эквивалентности кривая титрования совпадает с
кривой титрования сильной кислоты.
Пример 8.2
Рассчитайте pH при добавлении 0, 10,0, 25,0, 50,0 и 60,0 мл раствора титранта
(0,100 М NaOH) к 50,0 мл 0,100 М уксусной кислоты.
РИС. 8.5. Кривая титрования 100 мл 0,1 М НОАс раствором 0,1 М NaOH
8.4. ТИТРОВАНИЕ СЛАБЫХ КИСЛОТ СИЛЬНЫМИ ОСНОВАНИЯМИ
383
Таблица 8.2
Основные уравнения для расчета кривых титрования слабой кислоты (НА) или сла-
бого основания (В)
Степень оттитро- ванности F Слабая кислота Слабое основание
Присутствуют в растворе Уравнение Присутствуют в растворе Уравнение
F=0 НА [Н+]=^а-СНА (7.20) в [ОН-] = 7^-Св (пример 7.8)
0<F<l НА/А- pH = рКа + 1g (7.45) в/вн+ pH=(pKw-pKt)+lg-^B- (7.56) свн +
F= 1 А- [ОН-]=1^ СА. (7.32) V ВН+ [ОН+] = Л&СВН+ (7.39) V кь
F> 1 0Н7А- [ОН-] = [избыток титранта] Н7ВН+ [Н+] = [избыток титранта]
Рис. 8.6
Зависимость хода кривой
титрования слабой кислоты
(НОАс) от ее концентрации:
7 — 0,1 М; 2 — 0,01 М;
3 — 0,001 М. Объем титруемого
раствора 100 мл. Титрант —
NaOH, концентрация титранта
равна концентрации кислоты
Решение
В начальной точке (0 мл титранта) имеем 0,100 М раствор НОАс:
- Х'Х = 1,75-10~5
0,100-х
[Н+]=х=1,32- 10~3М
pH = 2,88
384
КИСЛОТНО-ОСНОВНЫЕ ТИТРОВАНИЯ
Рассчитаем, сколько оттитровано и сколько осталось уксусной кислоты по-
сле добавления 10,0 мл титранта. Следите за количеством оттитрованного и
оставшегося вещества! Исходное количество кислоты равно 0,100 М • 50,0 мл =
= 5,00 ммоль. Часть его прореагоровала с ОН- и превратилась в ОАс-:
Начальное количество НОАс
Добавлено ОН-: 0,100 М • 10,0 мл
Осталось НОАс
= 5,00 ммоль
= 1,00 ммоль =
= образовавшемуся количеству ОАс- в 60,0 мл
= 4,00 ммоль НОАс в 60,0 мл
Система представляет собой буферный раствор. При вычислениях величи-
ны объемов сокращаются, поэтому можно использовать непосредственно коли-
чества веществ:
pH = рк + ig J
а [НОАс]
pH = 4,76 + 1g — = 4,16
4,00
При добавлении 25,0 мл титранта половина НОАс превратилась в ОАс-, по-
этому pH = рКа:
Начальное количество НОАс
Добавлено ОН-: 0,100 М • 25,0 мл
Осталось НОАс
= 5,00 ммоль
= 2,50 ммоль =
= образовавшемуся количеству ОАс-
= 2,50 ммоль НОА
2 50
pH = 4,76 +1g ^ = 4,76
2,50
При добавлении 50,0 мл титранта вся НОАс превратилась в ОАс-. Количе-
ство ОАс- составляет 5,00 ммоль в 100 мл, а концентрация — 0,0500 М:
[ОН-]= -^[ОАс-] =
V
4,0-иг14
1,75-10-5
• 0,0500 =5,35-10"6 М
рОН = 5,27 pH = 8,73
При добавлении 60,0 мл титранта в растворе содержится NaOAc и избыток
NaOH. В присутствии ОН- влиянием ацетат-ионов можно пренебречь. Таким
образом, величина pH определяется только избыточной концентрацией ОН-:
количество ОН- = 0,100 М • 10,0 мл = 1,00 ммоль в 110 мл
[ОН-] = 0,00909 М
рОН = -2,04; pH = 11,96
8.4. ТИТРОВАНИЕ СЛАБЫХ КИСЛОТ СИЛЬНЫМИ ОСНОВАНИЯМИ
385
Участок кривой до точки эквивалентности, на котором величина pH возрас-
тает медленно, называется буферным участком. Он наиболее пологий в сред-
ней точке, т. е. там, где отношение [ОАс-]/[НОАс] равно единице (о буферных
растворах и буферной емкости см. разд. 7.7). Таким образом, буферная емкость
максимальна при значении pH, равном фКа. Буферная емкость зависит также от
абсолютных концентраций НОАс и ОАс-, возрастая с увеличением общей кон-
центрации буфера. Иными словами, с ростом суммарной концентрации НОАс и
ОАс- возрастает протяженность пологого участка кривой. При отклонении pH
от рКа в кислую область раствор может поглотить без заметного изменения pH
больше щелочи, но меньше кислоты. Однако даже при добавлении фиксирован-
ного небольшого количества щелочи изменение pH в этом случае будет больше,
чем при pH, равном рКа, поскольку кривая в этой области не такая пологая. На-
против, в более щелочной области относительно точки pH = рАГд раствор может
поглотить больше кислоты, но меньше щелочи. Подробнее о буферной емкости
см. гл. 7.
Вы могли обратить внимание, что начальные участки кривых титрования
сильных кислот и оснований (см. рис. 8.1 и 8.2) более пологие, чем у кривых тит-
рования слабых кислот. Таким образом, растворы сильных кислот и оснований
могут быть даже более устойчивы к изменениям pH при добавлении Н+ и ОН-,
чем буферные растворы. Однако область этой устойчивости ограничена очень
небольшими диапазонами pH, лежащими либо в сильнокислых, либо в сильно-
щелочных областях и наблюдается только для достаточно концентрированных
растворов кислот и щелочей. На практике редко требуется поддерживать посто-
янство значения pH в таких областях. Кроме того, pH растворов сильных кислот
и щелочей не остается постоянным при разбавлении, в отличие от pH буферных
растворов. Поэтому для поддержания постоянного значения pH в заданной об-
ласти, как правило, используют смеси слабых кислот или оснований и их солей
(сопряженных форм). Часто буферные растворы применяют просто для созда-
ния требуемого значения pH в условиях, когда к раствору не добавляют ни кис-
лоту, ни щелочь. В этих случаях удобнее использовать буферы, чем растворы
сильных кислот или щелочей.
В рассматриваемом случае титрования слабой кислоты интервал перехода
окраски индикатора должен попадать в диапазон pH приблизительно от 7 до 10
(см. рис. 8.5). Этим условиям хорошо удовлетворяет фенолфталеин. Если же ис-
пользовать метиловый красный, переход его окраски начнется вскоре после на-
чала титрования и будет медленно продолжаться вплоть до pH 6, т. е. закончится
еще до точки эквивалентности.
Зависимость хода кривой титрования и величины pH в точке эквивалентно-
сти от концентрации титруемой кислоты показана на рис. 8.6. Очевидно, что для
сильно разбавленных растворов (порядка 10-3 М) фенолфталеин оказывается не-
пригодным. При титровании слабых кислот следует тщательно подходить к
выбору индикатора. Обратите внимание, что с уменьшением концентрации сла-
бой кислоты величина pH в точке эквивалентности уменьшается (чего не проис-
ходит в случае сильных кислот).
386
КИСЛОТНО-ОСНОВНЫЕ ТИТРОВАНИЯ
А В C 0 E F g Н
1 Титрование 50,00 мл 0,05000 М НОАс 0,05000 М NaOH •v 1.00Е-14
2 V(HOAc), мл: 50,00 c(HOAc), M: 0,05000 c(NaOH), M: 0,05000 Ка: 1.75Е-05
3 V(NaOH), мл В растворе [HOAc] [OAc] [H+] [ОН] рОН pH
4 0,00 НОАс 9,35414E-04 3,028995973
5 2,00 НОАс/ОАс 0,046153846 0,001923077 3,37675071
6 5,00 НОАс/ОАс 0,040909091 0,004545455 3,802719442
7 10,00 НОАс/ОАс- 0,033333333 0,008333333 4,15490196
8 15,00 НОАс/ОАс- 0,026923077 0,011538462 4,388985166
9 20,00 НОАс/ОАс- 0,021428571 0,014285714 4,580870692
10 25,00 НОАс/ОАс- 0,016666667 0,016666667 4,756961951
11 30,00 НОАс/ОАс- 0,0125 0,01875 4,93305321
12 35,00 НОАс/ОАс- 0,008823529 0,020588235 5,124938737
13 40,00 НОАс/ОАс- 0,005555556 0,022222222 5,359021943
14 45,00 НОАс/ОАс 0,002631579 0,023684211 5,711204461
15 48,00 НОАс/ОАс- 0,001020408 0,024489796 6,137173193
16 49,00 НОАс/ОАс- 5,05051 E-04 0,024747475 6,447158031
17 50,00 НОАс/ОАс- 2.51256E-04 0,024874372 6,752597146
18 50,00 НОАс/ОАс- 2,50125E-05 0,024987494 7,75652744
19 50,00 ОАс- 3.77964Е-06 5,422549 8,57745098
20 50,00 он- 2.49875Е-05 4,602277 9,397722916
21 51,00 он- 2.48756Е-04 3,604226 10,39577395
22 51,00 он- 4.95050Е-04 3,305351 10,69464863
23 52,00 он- 9.80392Е-04 3,008600 10,99139983
24 55,00 он- 0,002380952 2,623249 11,37675071
25 60,00 он- 0,004545455 2,342422 11,65757732
26 65,00 он- 0,006521739 2,185636 11,81436342
27 70,00 он- 0,008333333 2,079181 11,92081875
28 В ячейки, выделенные жирным шрифтом, введены формулы:
29 Ячейка Е4: [Н+]=(Ка[НОАс])1/2= SQRT($H$2*$D$2)
30 Ячейка Н4: pH=-lg[H+]= (-LOG10(E4))
31 Ячейка С5: [HOAc]=(V(HOAc) • c(HOAc)-V(NaOH) c(NaOH))/(V(HOAc)+V(NaOH)
32 = ($B$2*$D$2-A5*$F$2)/($B$2+A5) (скопировать до ячейки C18)
33 Ячейка D5: [OAc-]=(V(NaOH) - c(NaOH)/(V(HOAc)+V(NaOH)) L Г
34 = (A5*$F$2)/($B$2+A5) (скопировать до ячейки D18)
35 Ячейка Н5: pH=pKa+lg([OAc]/[HOAc] г
36 S (-LOG10($H$2)+LOG10(D5/C5)) (скопировать до ячейки Н18)
37 Ячейка F19: [OH]=(Kw[OAc]/Ka)i/2
38 = SQRT($H$1 *($D$2/2)/$H$2)
39 Ячейка G19: pOH=-lg[OH]= (-LOG10(F19)) (скопировать до конца)
40 Ячейка Н19: pH = 14-pOH= 14-G19 | (скопировать до конца)
41 Ячейка F20: [OH ]=(V(NaOH) c(NaOH)-V(HOAC) • c(HOAc))/(V(HOAc)+V(NaOH))
42 = (A20*$F$2-$B$2*$D$2)/($B$2+A20) (скопировать до конца)
8.4. ТИТРОВАНИЕ СЛАБЫХ КИСЛОТ СИЛЬНЫМИ ОСНОВАНИЯМИ
387
Упражнение с электронными таблицами:
кривая титрования НОАс раствором NaOH
Давайте создадим электронную таблицу для построения кривой титрования
50 мл 0,05 М НОАс 0,05 М раствором NaOH. Сначала введем значения началь-
ного объема раствора (ячейка В2) и концентрации (ячейка D2) НОАс, концент-
рации NaOH (ячейка F2), величины Kw (ячейка G1) и Ка (ячейка Н2). Они будут
использованы в последующих расчетах зависимости pH от добавленного объема
титранта NaOH.
В начальной точке титрования значение [Н+] равно корню квадратному из
А?а[НОАс] (формула в ячейке Е4). Из этой величины рассчитывается pH (фор-
мула в ячейке Н4). С момента добавления первой порции титранта и вплоть до
точки эквивалентности система представляет собой буферный раствор (смесь
НОАс и ОАс-), поэтому необходимо рассчитать концентрацию каждого из ком-
понентов. Первая равна числу миллимолей неоттитрованной НОАс, деленному
на общий объем раствора (формула в ячейке С5). Величина [ОАс-] равна числу
миллимолей добавленного NaOH, деленному на общий объем (формула в ячей-
ке D5). Величина pH рассчитывается по уравнению Гендерсона—Хассельбаха
(формула в ячейке Н5). В точке эквивалентности рассчитывают [ОН-] из кон-
центрации ОАс- (формула в ячейке F19), а из этой величины — значения рОН
(ячейка G19) и pH (ячейка Н19). На оставшемся участке кривой pH рассчиты-
вают [ОН-] из избыточного количества NaOH и общего объема раствора (фор-
мула в ячейке F20) точно так же, как для кривой титрования НС1.
Для графического отображения кривой титрования используйте Chart Wi-
zard (Мастер диаграмм), вводя значения в диапазоны ячеек А4:А27 (ось X) и
Н4:Н27 (ось Y). Эта кривая приведена на компакт-диске (гл. 8, Chart 1).
При титровании любой слабой кислоты сильным основанием в точке экви-
валентности среда щелочная. Чем слабее кислота (чем меньше значение АГД
тем больше значение Кь сопряженного основания и тем выше pH в точке экви-
валентности.
Следует отметить, что кривые титрования, построенные по значениям pH,
которые рассчитаны по приведенным здесь достаточно простым формулам,
вблизи точки эквивалентности имеют разрывы. Причина состоит в том, что в
этих условиях допущения, сделанные при расчетах, перестают быть справедли-
выми. Это наблюдается даже при титровании сильных кислот сильными основа-
ниями в непосредственной близости от точки эквивалентности, поскольку при
этом степень диссоциации воды, не учитываемая в расчетах, становится замет-
ной. Вы можете сами убедиться во всем этом, добавляя к имеющимся в электрон-
ной таблице данным точки, соответствующие, скажем, степени оттитрованости
99,99 или 99,999% и наблюдая, как кривая титрования теряет гладкость.
На рис. 8.7 приведены кривые титрования 100 мл 0,1 М растворов слабых
кислот с разными значениями Ка 0,1 М раствором NaOH. С уменьшением Ка
уменьшается величина скачка на кривой. Как и на рис. 8.6, величина скачка уме-
ньшается также с уменьшением концентрации. Обычно считается, что для до-
статочно высоких концентраций (порядка 0,1 М) кислоты с Ка не менее 106
388
КИСЛОТНО-ОСНОВНЫЕ ТИТРОВАНИЯ
можно оттитровать с высокой точностью при использовании визуальных инди-
каторов. Используя подходящий способ сравнения окраски, можно оттитровать
с приемлемой точностью и кислоты с величинами Ка вплоть до 10-8. Повысить
точность при титровании очень слабых кислот можно путем непосредственного
построения кривых титрования при помощи рН-метра.
8.5. Титрование слабых оснований сильными кислотами
Случай титрования слабого основания сильной кислотой совершенно аналоги-
чен только что рассмотренному, но кривая титрования направлена в противопо-
ложную сторону. На рис. 8.8 изображена кривая титрования 100 мл 0,1 М
раствора аммиака 0,1 М раствором НС1. Реакция протекает по уравнению
NH3 + Н+ + Cl- -> NH+ + Cl" (8.7)
В начальной точке в растворе присутствует только NH3 (0,1 М), и pH рас-
считывают, как для растворов слабых оснований (см. гл. 7, табл. 8.2). После до-
бавления некоторого количества кислоты часть NH3 превращается в NH4, и
образуется буферная система. В точке полуоттитрованности концентрация
NH4 равна концентрации NH3, а значение pH равно (14 - рКь). В точке эквива-
лентности система представляет собой раствор NH4C1 — слабой кислоты Брен-
стеда, которая при взаимодействии с водой создает кислую среду. Как и при
титровании слабых кислот, pH в точке эквивалентности зависит от концентра-
ции титруемого вещества: чем она больше, тем pH ниже [см. уравнение (7.39)].
Рис. 8.7
Кривые титрования
100 мл 0,1 М растворов слабых
кислот с разными значениями Ка
0,1 М раствором NaOH
^NaOH (0,1 М)> мл
8.6. ТИТРОВАНИЕ КАРБОНАТА НАТРИЯ: ДВУХСТУПЕНЧАТОЕ ОСНОВАНИЕ
389
Рис. 8.8
Кривая титрования
100 мл 0,1 MNH3
0,1 М раствором НС1
За точкой эквивалентности в системе присутствуют свободные ионы Н+, подав-
ляющие диссоциацию NH д [см. уравнение (7.33)], и величина pH определяется
только избыточной концентрацией Н+. Поэтому после точки эквивалентности
кривая титрования совпадает с кривой титрования сильного основания
(см. рис. 8.3). Ввиду того, что значение Кь аммиака практически совпадает с Ка
уксусной кислоты, кривая титрования аммиака сильной кислотой представляет
собой почти точное зеркальное отражение кривой титрования уксусной кислоты
сильным основанием.
В соответствии с ходом кривой титрования (рис. 8.8) интервал перехода
окраски индикатора должен находиться между значениями pH 4 и 7. Как видно
из рисунка, этому условию удовлетворяет метиловый красный. Если же исполь-
зовать фенолфталеин, будет наблюдаться постепенное ослабление окраски рас-
твора в диапазоне pH от 10 до 8, т. е. до точки эквивалентности.
Семейство кривых титрования, построенных для различных концентраций
аммиака, представляет собой зеркальное отражение кривых на рис. 8.6. В сильно
разбавленных растворах нельзя использовать в качестве индикатора метиловый
красный. На рис. 8.9 изображены кривые титрования слабых оснований (100 мл
0,1 М растворов) с разными значениями Кь 0,1 М раствором НС1. При определе-
нии достаточно больших концентраций с использованием визуальных индика-
торов высокой точности можно достичь лишь для оснований с Кь не ниже 10"6.
8.6. Титрование карбоната натрия: двухступенчатое основание
Карбонат натрия — основание Бренстеда, используемое как первичный стан-
дарт для стандартизации растворов сильных кислот. Карбонат-ион диссоцииру-
ет по двум ступеням:
390
КИСЛОТНО-ОСНОВНЫЕ ТИТРОВАНИЯ
Рис. 8.9
Кривые титрования
100 мл 0,1 М растворов слабых
оснований с разными значениями Кь
0,1 М раствором НС1
Q.
СО?’+ Н,0 нсо: + ОН-; £Н1 = Км = —= 2,1 • 10-4 (8.8)
5 Z 5 ’ Jtll DL у ’ v
^а2
НСО" + Н2О СО2 + Н2О + ОН-; £Н2 = КЬ2 = = 2,3 • 1(Г8 (8.9)
Ка\
Здесь КаХ и Ка2 — константы кислотной диссоциации Н2СО3 по первой и второй
ступеням. Из них можно рассчитать значения Кь сопряженных оснований (СО3~
по отношению к НСО3, НСО3 — к Н2СО3) по соотношению KJCb = Kw (о вычис-
лении Кь соли слабых кислот и оснований см. гл. 7).
Карбонат натрия можно оттитровать по каждой из двух ступеней, соответ-
ствующих протонированию СО3” до НСО3 или СО2 (которая является продук-
том разложения Н2СО3 в кислых средах). Чтобы оттитровать основание по двум
ступеням, наблюдая при этом четко выраженные скачки в каждой точке эквива-
лентности, соответствующие величины Кь должны различаться как минимум в
104 раз.
Кривая титрования Na2CO3 раствором НС1 изображена на рис. 8.10
(сплошная линия). Несмотря на то что значение Кьх существенно больше 106 —
величины, достаточной для образования четко выраженного скачка в точке эк-
вивалентности, первый скачок титрования мал, что связано с образованием СО2
сразу же за точкой эквивалентности. Во второй точке эквивалентности скачок
тоже невелик, поскольку Kb2 < 10-6. К счастью, существует возможность уве-
личить этот скачок, поскольку СО2, образующийся в результате протонирова-
ния НСО3, летуч и его можно удалить кипячением раствора. Основанный на
этом способ титрования описан ниже.
8.6. ТИТРОВАНИЕ КАРБОНАТА НАТРИЯ: ДВУХСТУПЕНЧАТОЕ ОСНОВАНИЕ
391
Рис. 8.10
Кривая титрования 50 мл 0,1 М
Na2CO3 0,1 М раствором НС1.
Штриховая линия соответствует
титрованию раствора при
кипячении, в ходе которого
удаляется образующийся СО2.
Горизонтальный участок
штриховой линии соответствует
величине pH чистого раствора
IICOJ, для которого
[н+]=КХ7
В начальной точке величина pH определяется процессом диссоциации
основания Бренстеда СО 3“. После добавления титранта часть СО 3“ превращает-
ся в НСО3, и образуется буферная система СО3"/НСО3. В первой точке эквива-
лентности система представляет собой чистый раствор НСО3, для которого
[Н+] « у1К^Ка2. После первой точки эквивалентности НСО3 частично превра-
щается в Н2СО3 (т. е. СО2), и образуется другая буферная система — НСО3/СО2,
определяющая величину pH. Во второй точке эквивалентности значение pH
определяется концентрацией слабой кислоты СО2.
Для индикации первой конечной точки титрования можно использовать фе-
нолфталеин, а второй — метиловый оранжевый. Однако в обоих случаях скачок
выражен достаточно слабо. На практике фенолфталеин часто используют для
приблизительной оценки положения первой точки эквивалентности, из чего
можно оценить и положение второй точки эквивалентности (по достижении
первой точки эквивалентности фенолфталеин становится бесцветным и не ме-
шает дальнейшему титрованию). Для более точного определения используют
вторую точку эквивалентности. Однако при обычном титровании и ее положение
трудно установить с высокой точностью, поскольку переход окраски метилового
оранжевого при этом размыт вследствие буферного действия системы НСО3/СО2.
Однако если после достижения первой точки эквивалентности в ходе титро-
вания нагревать раствор для удаления СО2, то из двух компонентов буферной
системы НСО3/СО2 в растворе останется только НСО3. Гидрокарбонат-ион яв-
ляется одновременно и слабой кислотой, и слабым основанием. Величина pH
его раствора (~ 8,3) не зависит от его концентрации ([Н+] = -\1каХКа2 или
[ОН-] = см. гл. 7). В результате величина pH раствора будет практиче-
ски постоянной вплоть до второй точки эквивалентности, когда в растворе будет
392
КИСЛОТНО-ОСНОВНЫЕ ТИТРОВАНИЯ
находиться только NaCl, а значение pH будет равно 7. Ход соответствующей
кривой титрования показан штриховой линией на рис. 8.10.
Чтобы увеличить скачок вблизи второй точки эквивалентности, можно ис-
пользовать следующий прием (рис. 8.11). В качестве индикатора берут метило-
вый красный и ведут титрование до изменения окраски раствора из желтой через
оранжевую в чисто красную. Такое изменение наблюдается еще до точки экви-
валентности. Это происходит очень медленно, поскольку начинается при pH
около 6,3, задолго до точки эквивалентности. Сразу по завершении перехода
окраски титрование временно прекращают и осторожно нагревают раствор до
кипения для удаления СО2. При этом окраска раствора опять становится желтой,
соответствующей разбавленному раствору НСО3. При кипячении титруемого
раствора СО2 удаляется, а значение pH становится равным pHраствора НСО^.
Затем раствор охлаждают и продолжают титрование до повторного перехода
окраски в красную или розовую (теперь он очень резкий). Отметим, что в этом
случае pH в точке эквивалентности уже не будет равно 7 (как показано пунктир-
ной кривой на рис. 8.10), поскольку после кипячения в растворе все же остается
небольшое количество НСО3, которое по достижении точки эквивалентности
превращается в СО2.
Вместо метилового красного можно использовать бромкрезоловый зеле-
ный, изменяющий окраску в диапазоне pH от 5,4 до 3,8 с голубой через блед-
но-зеленую до желтой (см. практическую работу 7) или метиловый пурпурный
(см. практическую работу 19).
Если кипячения не проводить, то в качестве индикатора можно использо-
вать метиловый оранжевый с добавлением синего красителя ксиленцианола FF.
Такую смесь называют «модифицированным метиловым оранжевым». Си-
ний цвет является дополнительным к оранжевому, в который метиловый оран-
жевый окрашен при pH около 4. В результате вблизи точки эквивалентности
Рис. 8.11
Титрование 50 мл 0,1 М Na2CO3
0,1 М раствором НС1
с индикатором метиловым красным
8.7. ТИТРОВАНИЕ МНОГООСНОВНЫХ КИСЛОТ
393
раствор приобретает серую окраску, а интервал изменения цвета сужается по
сравнению с чистым раствором метилового оранжевого. Поэтому конечную
точку можно определить более точно. Тем не менее переход окраски все равно
менее отчетливый, чем при использовании метилового красного. При титрова-
нии с метиловым оранжевым можно для сравнения использовать раствор «сви-
детеля», содержащий индикатор в растворе гидрофталата калия; pH такого
раствора составляет около 4,0.
8.7. Титрование многоосновных кислот
Двухосновные кислоты, подобно карбонату натрия, можно оттитровать по сту-
пеням. Чтобы скачок вблизи первой точки эквивалентности был достаточно рез-
ким, необходимо, чтобы величина КаХ была по меньшей мере в 104 раз больше,
чем Ка2. Если величина Ка2 составляет не менее 10-8, наблюдается достаточно
четко выраженный скачок вблизи второй точки эквивалентности. По ступеням
можно титровать и трехосновные кислоты (например, Н3РО4), но величина Ка3
таких кислот обычно слишком мала для того, чтобы в третьей точке эквивалент-
ности наблюдался скачок. На рис. 8.12 приведен пример кривой титрования
двухосновной кислоты, а в табл. 8.3 — формулы для расчета pH на различных
участках этой кривой. В начальной точке pH рассчитывают, принимая во внима-
ние диссоциацию только по первой ступени (если раствор не слишком разбав-
лен; см. раздел гл. 7, посвященный многоосновным кислотам). Если значение
КаХ не очень велико, часто можно пренебречь количеством диссоциированной
формы по сравнению с недиссоциированной и использовать для расчета [Н+]
приближенное уравнение. В противном случае необходимо решать полное квад-
ратное уравнение (7.20) — см. пример 7.17.
РИС. 8.12. Кривая титрования двухосновной кислоты Н2А гидроксидом натрия
394
КИСЛОТНО-ОСНОВНЫЕ ТИТРОВАНИЯ
На участке до первой точки эквивалентности существует буферная система
НА7Н2А. В первой точке эквивалентности система представляет собой раствор
НА-, для которого [Н+] « ylKa}Ka2. После этой точки существует буферная сис-
тема А2-/НА; во второй точке эквивалентности величина pH определяется основ-
ной диссоциацией А2-. Если основание А2- не очень сильное, для расчета [ОН-]
можно использовать упрощенную формулу. В противном случае следует решать
полное квадратное уравнение (7.29) (см. пример 7.19).
На рис. 8.12 и в табл. 8.3 показано, какие частицы определяют значение pH на
отдельных участках кривой. Повторим, что если значение КаХ или Khx (= Kw /Ка2)
достаточно велико, то для расчета pH соответственно в начальной точке либо
во второй точке эквивалентности нельзя использовать упрощения, необходи-
мо решать полное квадратное уравнение. На практике такие случаи встречаются
редко. Если КаХ очень велика (например, для хромовой кислоты КаХ = 0,18,
Ка2 = 3,2 • 10-7), то и для первого буферного участка нельзя использовать урав-
нение Гендерсона—Хассельбаха: в этом случае концентрация ионов Н+, обра-
зующихся при диссоциации Н2А, сравнима с общей концентрацией Н2А.
В соответствии с уравнениями диссоциации
Н2А = Н+ + НА-
и
на- + н2о = н2а + он-
Видно, что равновесная концентрация Н2А связана с общей концентрацией сле-
дующим образом
[Н2А] = СНгА - [Н+] - [ОН-]
Соответственно,
[НА-^Сцд- + [Н+] - [ОН-]
Поскольку раствор кислый, величиной [ОН-] можно пренебречь:
[Н2А]=СН2А-[Н+]
[НА-]=Сна_ + [Н+]
Таким образом, для достаточно сильных кислот в выражение
КаХ = [Н+][НА-]/[Н2А]
следует подставить значения [Н2А] = СНзА - [Н+] и [НА-] = Снд + [Н+] и решить
полученное квадратное уравнение
[Н+Р + (КаХ + сНА. )[Н+] - каХс^ = о
Величины СНгА и Снд_ рассчитывают, исходя из соотношения количеств
титруемого вещества и титранта в данной точке кривой.
8.7. ТИТРОВАНИЕ МНОГООСНОВНЫХ КИСЛОТ
395
Аналогично, если величина Кьх для А2- очень велика, то в небольшой облас-
ти после второй точки эквивалентности нельзя пренебрегать количеством ионов
ОН-, образующихся в результате диссоциации А2-, по сравнению с количеством
ОН-, вносимым избытком титранта. И в этом случае тоже необходимо решить
полное квадратное уравнение после подстановки в выражение Kbx = Kw/Ka2 =
= [НА-][ОН-]/[А2-] значений [А2-] = Сд2_ - [ОН-] и [НА] = [ОН-]. Однако обыч-
но уже после добавления 0,5 мл NaOH концентрация ОН- -ионов становится до-
статочной для подавления диссоциации А2-, и значение pH можно рассчитать
непосредственно из величины избытка титранта. Наконец, для расчета [Н+] в
первой точке эквивалентности в тех случаях, когда величиной КаХ нельзя пре-
небречь по сравнению с [НА-], вместо (7.84) может потребоваться более точное
выражение (7.83) (в то же время слагаемое KaiKw в числителе расчетной форму-
лы обычно остается пренебрежимо малым). В этих случаях следует рассчиты-
вать [Н+] по формуле
[Н+]= |У*г[НА-]
Ка1+[НА-]
В непосредственной близости от точек эквивалентности могут потребовать-
ся еще более сложные выражения.
Таблица 8.3
Формулы для расчета кривой титрования двухосновной кислоты Н 2А
Степень оттитрованности F В растворе находится Формула
F=0(0%) н2а [H+]«^a|CHiA (пример 7.7)
[либо уравнение (7.20) — пример 7.17; полное квадратное уравнение для достаточно сильных кислот]
0 <F< 1 (0% <F< 100%) н2а/на- pH = р^, + 1g (7.45) [или (СНА_ + [Н+]) и ^н2а
(Сн А- [Н+]) для достаточно сильных кислот]
F = 1 (100%) (первая точка эквивалентности) НА- (Н+]«Хл«2 (7-84) [или (7.83) если Н2А достаточно сильная]
1 < F < 2 (100% < F < 200%) НА7А2- (j pH = рл;2 + 1g (7.45) — примеры 7.16, 7.22 Сна-
F=2(200%) (вторая точка эквивалентности) А2- [OH-]« J^-Ca2. (7.32) V а2 [либо (7.29) — пример 7.19; полное квадратное уравнение, если А2- — достаточно сильное основание]
F>2(>200%) ОН7А2- [ОН-] = [избыток титранта]
396
КИСЛОТНО-ОСНОВНЫЕ ТИТРОВАНИЯ
Используя общий подход к расчетам химических равновесий, можно прово-
дить вычисления при помощи точных уравнений материального баланса, в кото-
рых обычно не прибегают к упрощениям (за исключением того, что в кислых
средах можно пренебречь концентрацией ОН“, но не Н+, а в щелочных — Н+, но
не ОН-).
Мы не будем рассматривать примеры построения кривых титрования двух-
основных кислот, но если вы хотите выполнить хорошее упражнение для ума —
попробуйте! Попробуйте также обойтись без обычных допущений, используе-
мых для не очень разбавленных растворов слабых одноосновных кислот. На
компакт-диске (гл. 8) есть шаблон для построения при помощи электронных
таблиц кривой титрования 50,00 мл 0,1 000 М Н2СгО4 0,1000 М раствором NaOH.
Вы можете ввести в эту таблицу другие значения КаХ и Ка2 и посмотреть, как из-
менится при этом кривая. Попробуйте, например, построить кривую титрования
малеиновой кислоты. В этой таблице использованы уточненные формулы для
расчетов pH в начальной точке, первой буферной области и первой точке экви-
валентности. В то же время во второй точке эквивалентности решение полного
квадратного уравнения не используется, поскольку СгО4“ — слабое основание
(Кьх = 3,12 • 10-8). О других примерах расчетов кривых титрования см. [8].
8.8. Смеси кислот и смеси оснований
Компоненты смесей кислот (или оснований) можно оттитровать раздельно толь-
ко в том случае, если они значительно различаются по силе. Если не прибегать к
потенциометрическому титрованию (при помощи pH-метра), необходимо, что-
бы величины Ка различались по меныией мере в 104 раз. Если же одна из кислот
является сильной, то перегиб на кривой титрования в первой точке эквивалент-
ности наблюдается только в том случае, если величина Ка слабой кислоты со-
ставляет не более 1&~5 (рис. 8.13: в точке эквивалентности, соответствующей
оттитровыванию НС1, наблюдается лишь небольшой перегиб). Сначала оттит-
ровывается более сильная, затем более слабая кислота. На рис. 8.13 изображена
кривая титрования смеси соляной и уксусной кислот раствором гидроксида на-
трия. В точке эквивалентности, соответствующей оттитровыванию НС1, в рас-
творе присутствует смесь НОАс и NaCl, поэтому среда в этой точке кислая.
Затем в растворе образуется буферная система ОАс~/НОАс, что существенно
снижает величину скачка в первой точке эквивалентности. Участок кривой по-
сле оттитровывания НС1 идентичен кривой титрования НОАс (см. рис. 8.5).
При титровании смеси двух сильных кислот определить их по отдельности
невозможно: на кривой будет наблюдаться единственный скачок, соответствую-
щий оттитровыванию суммы кислот. То же само справедливо и для смеси двух
слабых кислот с близкими значениями Ка. Например, при титровании смеси ук-
сусной (Ка = 1,75 • 10~5) и пропионовой (Ка = 1,3 • Ю-5) кислот наблюдается один
скачок.
8.8. СМЕСИ КИСЛОТ И СМЕСИ ОСНОВАНИЙ
397
РИС. 8.13. Кривая титрования 50 мл смеси 0,1 М НС1 и 0,2 М НОАс 0,1 М раствором NaOH
В водном растворе молекулы H2SO4 полностью диссоциированы по первой
ступени, но для второй ступени величина Ка составляет приблизительно 102.
Тем не менее степень диссоциации по второй ступени настолько велика, что при
титровании по обеим ступеням серная кислота ведет себя как сильная. На кри-
вой титрования имеется единственный скачок. То же самое наблюдается и при
титровании смеси любой сильной кислоты и слабой кислоты с величиной Ка по-
рядка 10-2.
У сернистой кислоты H2SO3 первая константа диссоциации составляет
1,3 • 10-2, а вторая 5 • 10-6. Поэтому в смеси с НС1 первый протон сернистой кис-
лоты титруется совместно с НС1, а pH в первой точке эквивалентности определяет-
ся концентрацией образовавшихся ионов HSO 3: [Н+] = ^KaiKa2 (HSO 3 является и
кислотой, и основанием одновременно). Затем титруется второй протон серни-
стой кислоты. Объем титранта, соответствующий первой точке эквивалентно-
сти, всегда оказывается больше, чем объем, затраченный на титрование от
первой точки эквивалентности до второй, поскольку в первом случае титруются
обе кислоты. Содержание H2SO3 можно определить по объему титранта, израс-
ходованному на ее титрование по второй ступени, а содержание НС1 — по разно-
сти между объемом титранта, затраченным на на титрование обеих кислот (до
первой точки эквивалентности) и на титрование H2SO3 по второй ступени (этот
объем равен объему, требуемому для титрования H2SO3 по первой ступени).
Рассмотренный пример является иллюстративным и не имеет практического
значения: в кислой среде H2SO3 разлагается с образованием летучего SO2, что
препятствует точному определению сернистой кислоты.
398
КИСЛОТНО-ОСНОВНЫЕ ТИТРОВАНИЯ
Фосфорная кислота в смеси с сильной кислотой титруется аналогично. Сна-
чала оттитровываются совместно сильная кислота и фосфорная кислота по пер-
вой ступени, затем фосфорная кислота по второй ступени. По третьей ступени
фосфорную кислоту оттитровать невозможно, поскольку третий протон диссо-
циирован в очень малой степени.
Пример 8.3
Смесь НС1 и Н3РО4 оттитровали 0,1000 М NaOH. На титрование до первой ко-
нечной точки (по метиловому красному) затрачено 35,00 мл, а до второй (по
бромтимоловому синему) — 50,00 мл раствора титранта от начала титрования
(т. е. 15,00 мл титранта затрачено на титрование от первой точки до второй). Рас-
считайте содержание НС1 и Н3РО4 (в миллимолях) в растворе.
Решение
Вторая конечная точка соответствует оттитровыванию одного протона Н3РО4
(по второй ступени: Н2РО4 -> НРО4“). Следовательно, число миллимолей
Н3РО4 равно числу миллимолей NaOH в 15,00 мл его раствора, израсходованного
на титрование от первой конечной точки до второй:
лн3ро4 (ммоль)= cNaOH (М) ’ ^NaOH (мл) = ОД000 ммоль/мл • 15,00 мл = 1,500 ммоль
НС1 и Н3РО4 по первой ступени титруются совместно. На титрование Н3РО4
по первой ступени расходуется 15,00 мл титранта (столько же, сколько и по вто-
рой). Следовательно, на титрование НО израсходовано 20,00 мл. Отсюда
инс1 (ммоль) = 0,1000 ммоль/мл • (35,00 - 15,00) мл = 2,000 ммоль
Смеси оснований тоже возможно раститровать только тогда, когда компо-
ненты значительно различаются по силе. И в этом случае различие в величинах
констант (Кь) должно быть не менее 104, а если одно из оснований сильное, то для
другого величина Кь должна быть не более 10-5. В частности, при титровании сме-
си NaOH и Na2CO3 не наблюдается скачка в точке эквивалентности, соответству-
ющей оттитровыванию NaOH, поскольку для СО 3“ значение Kbi равно 2,11 (И.
8.9. Титрование аминокислот:
они являются и кислотами, и основаниями
Аминокислоты — соединения, играющие важную роль в фармацевтической и
биологической химии. Это амфотерные вещества (т. е. способные реагировать и
как кислоты, и как основания), содержащие кислотную карбоксильную (—СО2Н)
8.9. ТИТРОВАНИЕ АМИНОКИСЛОТ: ОНИ ЯВЛЯЮТСЯ И КИСЛОТАМИ, И ОСНОВАНИЯМИ
399
и основную аминогруппу (—NH2). В водных растворах происходит внутримоле-
кулярный перенос протона от карбоксильной группы к аминогруппе, поскольку
RNH2 — более сильное основание, чем RCO2. В результате образуется цвит-
тер-ион:
R—СП—СО2
I
NHt
Аминокислоты в силу их амфотерной природы в принципе можно титро-
вать растворами как сильных кислот, так и щелочей. Правда, многие аминокис-
лоты являются слишком слабыми для титрования в водных растворах, однако
для некоторых конечную точку можно зафиксировать достаточно отчетливо,
особенно с потенциометрической индикацией.
Цвиттер-ион можно рассматривать как основание, сопряженное двухос-
новной кислоте, диссоциирующей по ступеням:
каХ
R— CH— СО2Н Н+ + R— СП— СО2
I I
nh+3 nh;
кислота, цвиттер-ион
сопряженная
цвиттер-иону
ка2
^Н+ + R—СН—СО2
I
nh2
основание,
сопряженное
цвиттер-иону
Величины КаХ и Ка2 аминокислот часто приводят в таблицах (см. табл. С. 1 в
Приложении С). Указанные в них величины характеризуют ступенчатую диссо-
циацию протонированной формы кислоты (т. е. кислоты, сопряженной цвит-
тер-иону). При диссоциации по первой ступени образуется цвиттер-ион, а по
второй — сопряженное цвиттер-иону основание, аналогичное аниону слабой
кислоты. Таким образом, равновесия в растворе аминокислоты можно описать
аналогично равновесиям в растворе любой двухосновной кислоты. Концентра-
цию протонов в растворе цвиттер-иона рассчитывают точно так же, как и для
аниона двухосновной кислоты, диссоциированной по первой ступени, напри-
мер, НСО3 (см. гл. 7):
[W] = jKalKa2 (8.11)
Процесс титрования аминокислот аналогичен процессу титрования других
амфотерных веществ, подобных НСО3. В последнем случае титрование щело-
чью приводит к образованию СО через промежуточную область существова-
ния буферной системы СО^/НСОз, а титрование кислотой — к образованию
Н2СО3 через область существования системы НСОз/Н2СО3.
При титровании цвиттер-иона аминокислоты сильной кислотой сначала об-
разуется буферная система, состоящая из цвиттер-иона и сопряженной кислоты.
400
КИСЛОТНО-ОСНОВНЫЕ ТИТРОВАНИЯ
В точке полуоттитрованности pH = рХа1 (как и для системы НСО3/Н2СО3), а в
точке эквивалентности значение pH определяется величиной Ка сопряженной
кислоты (при титровании НСО3 — величиной КаХ для Н2СО3). Если же титро-
вать цвиттер-ион раствором щелочи, то буферная система состоит из цвит-
тер-иона и сопряженного основания. В этом случае в точке полуоттитро-
ванности pH = рАСа2 (как для системы СО3“/НСО3), а в точке эквивалентности
определяется величиной Кь сопряженного основания (равной Kw 1Ка1, как и для
СО 2-).
Аминокислоты могут содержать более одной карбоксильной и более одной
аминогруппы. В этих случаях на кривой титрования может наблюдаться
несколько скачков, как и для других многоступенчатых кислот или оснований.
Необходимое условие для этого — различие величин последовательных кон-
стант не менее чем в 104 раз.
8.10. Метод Кьельдаля: определение белков
Важным методом определения азота в белках и других азотсодержащих веществ
является метод Кьельдаля. По известной массовой доле азота в белке можно
определить и содержание самого белка. Хотя существуют и другие, более эксп-
рессные, методы определения белков, метод Кьельдаля до сих пор является стан-
дартным, с результатами которого сопоставляют результаты всех других методов.
Для определения азота пробу сначала разлагают серной кислотой. При этом
содержащийся в ней азот превращается в гидросульфат аммония:
н so
CH,N-------aCO2t +1Ж2О + cNH4HSO4
а ° с катализатор 2 2 2 ч ч
Затем раствор охлаждают, подщелачивают концентрированным раствором ще-
лочи и отгоняют аммиак, поглощая его точно отмеренным объемом стандартного
раствора кислоты, которая берется в избытке. Непрореагировавшее количество
кислоты оттитровывают стандартным раствором щелочи:
NH4HSO4^> NH3t + SO^~
NH3 + HC1 (в избытке)-> NH4C1 + HC1 (непрореагировавшее количество)
HC1 (непрореагировавшее количество) + NaOH-> Н2О + NaCl
Содержание азота находят по разности между взятым количеством кислоты и
израсходованным количеством щелочи.
Чтобы ускорить процесс разложения пробы, к ней добавляют сульфат калия
(для повышения температуры кипения раствора) и катализатор — например,
соли меди или селен. Количество азотсодержащего соединения рассчитывают
из найденной массы азота, разделив ее на массовую долю азота в веществе.
8.10. МЕТОД КЬЕЛЬДАЛЯ: ОПРЕДЕЛЕНИЕ БЕЛКОВ
401
Пример 8.4
Навеску (0,2000 г) пробы, содержащей мочевину
О
II
nh2—с—nh2
проанализировали по методу Кьельдаля. Аммиак поглотили 50,00 мл 0,05000 М
H2SO4 и оттитровали избыток кислоты 3,40 мл 0,05000 М NaOH. Рассчитайте
массовую долю (%) мочевины в пробе.
Решение
При титровании протекает реакция
H2SO4 + 2NaOH -> Na2SO4 + 2Н2О
Поэтому избыточное количество H2SO4 (ммоль) равно половине количества
NaOH, затраченного на титрование. Взаимодействие аммиака с серной кислотой
протекает по уравнению
2NH3 + H2SO4 -» (NH4)2SO4
Количество аммиака вдвое больше количества прореагировавшей с ним H2SO4.
Наконец, количество мочевины вдвое меньше количества образовавшегося из
нее аммиака. Отсюда получаем:
«мочевины.0/» = {[«H2SO4 " «№он ’ ^(ммоль Н28О4/ммоль NaOH)] X
х 2 (ммоль NH3/ммoль H2SO4) • |[ммоль ^Н2)2СО/ммоль NH3] х
Х M(NH2)2со(Мг/мМОЛЬ)/"7пробы (мг) ’ 100% =
(0,0500 ммоль/мл • 50,00 мл - 0,0500 ммоль/мл • 3,40 мл • • 2 • | • 60,05 мг/ммоль
200 мг
= 72,51%
Заметим, что количество (NH2)2CO (ммоль) равно количеству H2SO4, всту-
пившей в реакцию с аммиаком. Поэтому можно было не вычислять количество
NH3, а сразу рассчитать массу мочевины, умножив количество прореагировав-
шей H2SO4 (ммоль) на молярную массу мочевины, равную 60,05 (мг/ммоль).
402
КИСЛОТНО-ОСНОВНЫЕ ТИТРОВАНИЯ
Для очень большого числа белков массовая доля содержащегося в них азо-
та почти одинакова. Для стандартной смеси белков сыворотки крови (гло-
булинов и альбумина) фактор пересчета массы азота в массу белка равен
6,25 (т. е. эти белки содержат 16% азота). Если проба состоит практически
из одного гамма-глобулина, более точным значением является 6,24, а если
из альбумина — то 6,27.
В обычном варианте метода Кьельдаля требуются два стандартных раство-
ра — кислоты для поглощения аммиака и щелочи для обратного титрования. Но
существует разновидность метода, где требуется только один стандартный рас-
твор кислоты для прямого титрования. В этом случае аммиак поглощают раство-
ром не серной, а борной кислоты. При этом образуется эквивалентное количество
бората аммония:
NH3 + Н3ВО3 NH J + Н2ВО3 (8.12)
Борная кислота является слишком слабой для титрования, но борат-ион, об-
разующийся в эквивалентном аммиаку количестве, достаточно сильное основание
Бренстеда. Его можно оттитровать стандартным раствором кислоты с индикатором
метиловым красным. При этом присутствующий в растворе избыток очень сла-
бой борной кислоты титрованию не мешает. Поэтому нет необходимости брать
ее в точно известном количестве
Пример 8.5
Пробу пищевого продукта массой 0,300 г проанализировали на содержание белков
модифицированным методом Кьельдаля. На титрование израсходовано 25,0 мл
0,100 М НО. Какова массовая доля (%) белка в пробе?
Решение
В этом случае титрование является прямым. Число миллимолей NH3 (а следова-
тельно, и азота) равно числу миллимолей НС1, затраченной на титрование. Ум-
ножив массу азота на 6,25, найдем массу белка. Отсюда
^мочевины’ —
_ 0,100 ммоль/ мл (НС1) • 25,00 мл (НС1) 14,01 (мгТ4/ммоль НС1) • 6,25 (мгбелка/MrN)
200 мг (пробы)
х 100% = 73,0%
ВОПРОСЫ
403
Вариант метода Кьельдаля с использованием борной кислоты и прямого
титрования проще и, как правило, точнее, поскольку в этом случае требуется
стандартизация и точное измерение объема только одного раствора. Однако ска-
чок титрования в конечной точке получается не такой резкий. Поэтому для мик-
роанализа по методу Кьельдаля обычно предпочитают все же классический
вариант с обратным титрованием. Для определения белков в варианте макроана-
лиза требуется около 5 мл крови, а микроанализа — всего 0,1 мл.
До сих пор мы рассматривали определение азота лишь в тех соединениях, в
которых он находится в степени окисления -3 (как в аммиаке). Они включают в
себя соединения с амино- и амидными группами. Для анализа соединений, со-
держащих азот в более положительных степенях окисления (в частности, орга-
нических нитро- и азосоединений) их следует сначала восстановить, чтобы в
ходе последующего разложения полностью перевести азот в ион аммония. В ка-
честве восстановителей используют соединения железа(П) или тиосульфат-ион.
Что мы узнали из этой главы?
• Построение кривых кислотно-основного титрования:
• сильные кислоты, сильные основания (табл. 8.1) — стр. 373
• расчеты при помощи электронных таблиц — стр. 377
• слабые кислоты, слабые основания (табл. 8.2) — стр. 381
• расчеты при помощи электронных таблиц — стр. 387
• Кислотно-основные индикаторы [основные уравнения (8.4), (8.5)] —
стр. 378
• Титрование Na2CO3 — стр. 391
• Титрование многоосновных кислот (табл. 8.3) — стр. 393
• Титрование аминокислот — стр. 398
• Анализ азотсодержащих веществ, определение белков по методу Кьельда-
ля — стр. 400
Вопросы
1. Какова минимальная величина скачка pH, необходимая для резкого изме-
нения окраски индикатора в конечной точке титрования? Почему необхо-
дима такая величина скачка?
2. По какому критерию выбирают индикатор для кислотно-основного титро-
вания?
3- При каком значении pH буферная система обладает максимальной бу-
ферной емкостью?
4. Нейтральной, кислой или щелочной является среда в конечной точке тит-
рования слабой кислоты? Почему?
404
КИСЛОТНО-ОСНОВНЫЕ ТИТРОВАНИЯ
5- Предложите подходящий индикатор для титрования: аммиака раствором
соляной кислоты, уксусной кислоты раствором гидроксида натрия.
6 - Объясните, почему кипячение раствора карбоната натрия вблизи конечной
точки титрования позволяет увеличить скачок на кривой титрования.
7. Какой порядок величины рК самой слабой кислоты или основания, кото-
рые можно оттитровать в водном растворе?
8- Насколько должны различаться по силе две кислоты, чтобы их можно было
оттитровать раздельно при совместном присутствии?
9. ZB чем состоит различие между первичным и вторичным стандартом?
10- Что такое цвиттер-ион?
11 - Какова массовая доля азота в большинстве белков?
12- Какими кислотами предпочтительнее титровать основания? Почему?
Задачи
Расчеты при стандартизации растворов
13. Раствор соляной кислоты стандартизовали титрованием навески 0,4541 г
первичного стандарта трис(гидроксиметил)аминометана. На титрование за-
трачено 35,37 мл раствора. Какова молярная концентрация соляной кислоты?
14. Раствор соляной кислоты стандартизовали титрованием навески 0,2329 г
первичного стандарта карбоната натрия с индикатором метиловым крас-
ным, используя кипячение раствора вблизи точки эквивалентности для
удаления диоксида углерода. На титрование израсходовано 42,87 мл рас-
твора кислоты. Чему равна его концентрация (моль/л)?
15. На титрование навески 0,8592 г первичного стандарта гидрофталата калия
затрачено 32,67 мл раствора гидроксида натрия (индикатор — фенолфтале-
ин). Чему равна концентрация раствора гидроксида натрия (моль/л)?
16. К 10,00 мл раствора соляной кислоты добавили избыток нитрата серебра и
определили массу выпавшего осадка хлорида серебра. Она оказалась равной
0,1682 г. Чему равна концентрация раствора соляной кислоты (моль/л)?
Индикаторы
17. Напишите уравнение Гендерсона—Хассельбаха для индикатора В, являю-
щегося слабым основанием, и рассчитайте ширину диапазона pH, соответ-
ствующего изменению окраски индикатора. Где лежит середина этого
диапазона?
Кривые титрования
Для решения некоторых задач этого раздела можно пользоваться электронными
таблицами.
ЗАДАЧИ
405
18. Рассчитайте значения pH после добавления 0, 10,0, 25,0 и 30,0 мл раствора
титранта (0,200 М НС1) к 50,0 мл 0,100 М NaOH.
19- Рассчитайте значения pH после добавления 0, 10,0,25,0, 50,0 и 60,0 мл рас-
твора титранта (0,100 М NaOH) к 25,0 мл 0,200 М раствора кислоты НА
(^ = 2,0-10-5).
20- Рассчитайте значения pH после добавления 0, 10,0,25,0, 50,0 и 60,0 мл рас-
твора титранта (0,100 М НС1) к 50,0 мл 0,100 М раствора аммиака.
21 - Рассчитайте значения pH при значениях степени оттитрованности, равных
0, 25,0, 50,0, 75,0, 100 и 125%, в ходе титрования 100 мл 0,100 М раствора
двухосновной кислоты Н2А 0,100 М раствором NaOH по двум ступеням.
КаХ = 1,0 • 10-3, Ка2 = 1,0 • ю-7.
22 - Рассчитайте значения pH в ходе титрования 100 мл 0,100 М Na^PO.^ 0,100 М
раствором НС1 (до Н2РО4) при значениях степени оттитрованности, рав-
ных 0, 25,0, 50,0, 75,0, 100 и 150%.
Количественный анализ методом кислотно-основного титрования
23- Навеску КН2РО4 массой 0,492 г оттитровали 25,6 мл 0,112 М NaOH по урав-
нению:
н2ро; + он- нро 4’ + н2о
Каково содержание КН2РО4 (% масс.) в образце?
24- Какой объем 0,155 М H2SO4 потребуется для титрования 0,293 г LiOH (чис-
тота 90,0%)?
25. Показателем средней молярной массы жиров служит число омыления, вы-
раженное в виде массы (мг) КОН, необходимого для гидролиза (омыления)
1 г жира по уравнению
ch2co2r сн2он
I I
CHCO2R + ЗКОН СНОН + 3RCO2K
I I
ch2co2r сн2он
(природа R может быть различной). Навеску масла массой 1,10 г обработа-
ли 25,0 мл 0,250 М КОН. По завершении омыления непрореагировавшее
количество КОН оттитровали 9,26 мл 0,250 М НС1. Чему равно число омы-
ления жира и какова его средняя молярная масса (в предположении, что
проба содержит только жиры)?
26- Пробу, содержащую аминокислоту аланин CH3CH(NH2)COOH и инертные
компоненты, проанализировали по методу Кьельдаля. Навеску массой 2,00 г
разложили, аммиак отогнали и поглотили 50,0 мл 0,150 М H2SO4, избыток
которой оттитровали 9,0 мл 0,100 М NaOH. Рассчитайте массовую долю
(%) аланина в пробе.
406
КИСЛОТНО-ОСНОВНЫЕ ТИТРОВАНИЯ
27. Пробу сыворотки крови объемом 2,00 мл проанализировали на содержание
белков модифицированным методом Кьельдаля. Пробу разложили, аммиак
отогнали, поглотили разбавленным раствором борной кислоты и оттитро-
вали 15,0 мл стандартного раствора НС1. Раствор соляной кислоты стандар-
тизовали, определяя содержание азота в 0,330 г чистого (NH4)2SO4 тем же
способом. На титрование израсходовано 33,3 мл раствора кислоты. Каково
содержание белков в сыворотке крови (мг/мл)?
Количественный анализ смесей
28. Аликвоту объемом 100 мл раствора, содержащего НС1 и Н3РО4, оттитрова-
ли 0,200 М раствором NaOH. На титрование затрачено 25,0 мл по индикатору
метиловому красному и еще 10,0 мл (т. е. всего 35,0 мл) по бромтимоловому
синему. Чему равна концентрация НС1 и Н3РО4 в растворе?
29. Пробу массой 0,527 г смеси, содержащей N^CO^ NaHCO3 и инертные веще-
ства, оттитровали 0,109 М раствором НС1. На титрование израсходовано
15,7 мл с индикатором фенолфталеином и 43,8 мл (всего) с «модифициро-
ванным метиловым оранжевым». Какова массовая доля (%) Na2CO3 и
NaHCO3 в пробе?
30. С индикатором фенолфталеином гидроксид натрия и Na2CO3 по первой
ступени титруются совместно (ОН- -> Н2О, СО3“ -> НСО3). Смесь, содер-
жащую NaOH и Na^O^ оттитровали 0,250 М раствором НС1. По фенолфта-
леину затрачено 26,2 мл раствора кислоты, а по «модифицированному
метиловому оранжевому» — еще 15,2 мл. Какова масса (мг) NaOH и
Na2CO3 в пробе?
31. Карбонат натрия может сосуществовать или с NaOH, или с NaHCO3, но не с
обоими веществами одновременно, поскольку они реагируют между собой
с образованием Na2CO3. С индикатором фенолфталеином гидроксид на-
трия и Na2CO3 по первой ступени титруются совместно (ОН- -> Н2О,
СО 3" —> НСО 3). Пробу, содержащую Na2CO3 в смеси с NaOH или с NaHCO3,
оттитровали соляной кислотой. С фенолфталеином на титрование затрачено
15,0 мл, а с «модифицированным метиловым оранжевым» — всего 50,0 мл
(т. е. 35,0 мл после первой конечной точки). Раствор соляной кислоты стан-
дартизовали титрованием 0,477 rNa^C^ по «модифицированному метило-
вому оранжевому». На титрование пошло 30,0 мл раствора кислоты. Какие
вещества содержатся в пробе и в каких количествах (ммоль)?
32. Каким был бы ответ на вопрос задачи 31, если бы общий объем кислоты во
второй конечной точке составил 25,0 мл (т. е. 10,0 мл после первой конеч-
ной точки)?
33. Смесь массой 0,150 г состоит только из ВаСО3 и Li2CO3. Для титрования кар-
бонат-ионов, содержащихся в этой смеси, по второй ступени (СО 3~ -> Н2СО3)
требуется 25,0 мл 0,120 М НС1. Какова массовая доля (%) ВаСО3 в образце?
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
407
34- Образец Р2О5 содержит примесь Н3РО4. Навеску пробы массой 0,405 г рас-
творили в воде (при этом произошла реакция Р2О5 + ЗН2О -> 2Н3РО4) и от-
титровали полученный раствор 0,250 М раствором NaOH до Na2HPO4. На
титрование затрачено 42,5 мл титранта. Какова массовая доля (%) Н3РО4 в
образце?
Задачи для решения при помощи электронных таблиц
См. компакт-диск, Лист 1 (Sheet 1) для ввода данных и График 1 (Chart 1).
35. Создайте электронную таблицу для построения кривой титрования 100 мл
0,1 М NH3 0,1 М раствором НС1. Сравните построенную кривую с рис. 8.8.
Рекомендуемая литература
Индикаторы
1. G. Gorin, «Indicators and the Basis for Their Use», J. Chem. Ed, 33 (1956) 318.
2. Индикаторы / Под ред. Э. Бишопа. В 2-х томах — М.: Мир, 1976.
Кривые титрования
3. R. К. McAlpine, «Changes in pH at the Equivalence Point», J. Chem. Ed., 25
(1948) 694.
4. A. K. Covington, R. N. Goldberg, and M. Sarbar, «Computer Simulation of Tit-
ration Curves with Application to Aqueous Carbonate Solutions», Anal. Chim.
Acta, 130(1981) 103.
Титриметрические методики
5. Y.-S. Chen, S. V. Brayton, and С. C. Hach, «Accuracy in Kjeldahl Protein
Analysis», Am. Lab., June (1988) 62.
6. R. M. Archibald, «Nitrogen by the Kjeldahl Method», in D. Seligson, ed., Standard
Methods of Clinical Chemistry, Vol. 2. New York: Academic, 1958, pp. 91-99.
7- M. E. Hodes, «Carbon Dioxide Content (Titrimetric)» in M. Reiner, ed., Standard
Methods of Clinical Chemistry, Vol. 1. New York: Academic, 1953, pp. 19-22.
Виртуальный интернет-калькулятор
8. http://hamers.chem. wisc.edu/chapman/Titrator/—виртуальный калькулятор
1.5 на сайте профессора Роберта Хамерса, Университет штата Висконсин
(США). Вы можете пользоваться им для построения кривых титрования.
Можно выбирать различные типы кислот, значения концентраций и объе-
мов. Можно также строить распределительные диаграммы.
Глава 9
РЕАКЦИИ КОМПЛЕКСООБРАЗОВАНИЯ
И КОМПЛЕКСОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Простые вещи должны быть простыми.
Сложные вещи должны быть возможными.
АланКей
Многие ионы металлов образуют малодиссоциирующие комплексы с различны-
ми лигандами (комплексообразующими реагентами). Химики-аналитики широко
используют комплексы для предотвращения нежелательных реакций (маскирова-
ния мешающих компонентов). Образование комплексов можно также использо-
вать для удобного и точного титриметрического определения ионов металлов, в
ходе которого титрантом служит раствор лиганда. Этим методом, называемым
комплексометрическим титрованием, можно определять большое число метал-
лов. При этом селективность достигается главным образом за счет использования
подходящих маскирующих реагентов (обычно это другие лиганды, взаимодейству-
ющие с мешающими ионами) и контроля pH (поскольку большинство лигандов
являются слабыми основаниями, величина pH оказывает существенное влияние
на положение равновесий с их участием). В данной главе мы рассмотрим равно-
весия в растворах с участием ионов металлов и влияние pH на положение этих
равновесий. Также мы ознакомимся с процессами титрования ионов металлов
весьма распространенным комплексообразующим реагентом ЭДТА (этот вид
комплексометрического титрования называется комплексонометрическим), об-
судим факторы, влияющие на эти процессы, и индикаторы, применяемые в этих
случаях. Титриметрическое определение кальция и магния с ЭДТА обычно ис-
пользуют для определения жесткости воды. Комплексонометрическим титрова-
нием можно определять с высокой точностью почти все металлы. Помимо
перечисленного, реакции комплексообразования используют в гравиметрии,
спектрофотометрии, люминесцентном и других методах анализа.
9.1. Константа устойчивости - мера устойчивости комплекса
Комплексные соединения играют важную роль во многих химических и биохи-
мических процессах. Например, в молекулах гема (вещества, входящего в состав
крови) атомы железа прочно удерживаются ввиду того, что они связаны в комп-
лекс атомами азота. В свою очередь, эти атомы железа [в форме Fe(II)] легко свя-
зывают молекулярный кислород, разносят его от легких по всему организму, а
затем столь же легко его отщепляют. Возможность этого процесса обусловлена
9.1. КОНСТАНТА УСТОЙЧИВОСТИ - МЕРА УСТОЙЧИВОСТИ КОМПЛЕКСА
409
тем, что молекулярный кислород связывается с ионами железа слабее, чем ато-
мы азота. В то же время цианид-ион связывается намного прочнее, чем О2, и вы-
тесняет его. Поэтому цианиды являются смертельными ядами. В этом же состоит
и причина токсичности моноксида углерода, который связывается с гемом в 200
раз прочнее кислорода, образуя карбоксигемоглобин.
В растворах многие катионы образуют комплексы с разнообразными веще-
ствами, содержащими атомы с неподеленными электронными парами (обычно
это атомы N, О, S), и в силу этого способными к образованию координационных
связей с ионами металлов. Ион металла является акцептором электронной пары
(кислотой Льюиса), а лиганд, имеющий в своем составе атом-донор электрон-
ной пары, — основанием Льюиса. Число молекул лиганда, присоединяющихся к
иону металла, зависит от координационного числа металла и от числа комплек-
сообразующих групп в молекуле лиганда.
Простейшим примером может служить молекула аммиака, имеющая одну
неподеленную электронную пару и способная к образованию комплекса с ионом
меди:
Cu2+ + 4:NH3
NH3
H3N:Cu:NH3
NH3
В данном примере ион меди выступает в качестве кислоты Льюиса, а аммиак —
в качестве основания Льюиса. В результате этой реакции окраска раствора из-
меняется с бледно-голубой, присущей свободному (точнее, акватированному)
иону Си2+, на темно-синюю, характерную для аммиачных комплексов меди.
Аналогичная реакция протекает с ионами никеля; в этом случае акватированные
ионы металла окрашены в зеленый цвет, а аммиачные комплексы — в темно-си-
ний.
Аммиак образует также бесцветные комплексы с ионами серебра. Это мож-
но использовать, например, для растворения остатков хлорида серебра на филь-
трующем тигле и, таким образом, для очистки тигля. В ходе реакции один ион
Ag+ последовательно присоединяет две молекулы NH3. Каждую стадию этого
процесса характеризует своя константа равновесия, называемая ступенчатой
константой устойчивости комплекса Кр
Ag+ + NH3 Ag(NH3)+ Kfl = J = 2,5-103
[Ag+][NH3]
Ag(NH3)+ + NH3 Ag(NH3) J Kf2 = —[Ag(NH3)2]— = j 0 . j 04
[Ag(NH3)+][NH3]
410
РЕАКЦИИ КОМПЛЕКСООБРАЗОВАНИЯ И КОМПЛЕКСОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Суммарный процесс представляет собой комбинацию этих стадий. Он харак-
теризуется общей константой устойчивости, являющейся произведением ступен-
чатых констант:
Ag* + 2NH, s= Ag(NH,)t К, -К.,- К„ - ।А0"'Н1 Ь I _ 2,5 10’
7 ' f [Ag + ][NH,]2
(9.3)
Для комплексов состава 1 : 1 (М + L = ML) общая константа устойчивости, оче-
видно, равна ступенчатой: Kj = = [ML]/([M][L]). Обратите внимание, что
равновесие комплексообразования сейчас принято записывать как равновесие
ассоциации, а не диссоциации, и, соответственно, концентрация продукта реак-
ции (комплекса) записывается в числителе, а произведение концентраций ис-
ходных веществ — в знаменателе выражения константы.
Ранее равновесие комплексообразования часто записывали в обратном на-
правлении, как равновесие диссоциации комплекса. В этом случае числитель и
знаменатель выражения константы равновесия меняются местами, а значения
соответствующих констант, называемых константами нестойкости Kt или кон-
стантами диссоциации Kd, представляют собой величины, обратные констан-
там устойчивости:
Ag(NH,)t Ag+ + 2NH, Kd = — = tAgJ-№3J- = 4,0 • 10~8 (9.4)
Kf [Ag(NH3)J]
К настоящему времени константы нестойкости комплексов практически
вышли из употребления.
Пример 9.1.
Двухзарядный ион металла М2+ взаимодействует с лигандом L с образованием
комплекса состава 1:1:
M2+ + L^ML2+ Kf= [Mb2+]
[M2+][L]
Рассчитайте концентрацию иона М2+ в растворе, образованном смешением рав-
ных объемов 0,20 М растворов М2+ и L; Kj = 1,0 • 108.
Решение
М2+ и L смешаны в стехиометрически равных количествах. Комплекс достаточ-
но устойчив, поэтому реакцию можно считать протекающей практически пол-
ностью. Ввиду того, что объемы исходных растворов были равны, общие
концентрации каждого компонента уменьшились в два раза. Обозначим иско-
мую концентрацию [М2+] какх. Тогда в состоянии равновесия:
9.1. КОНСТАНТА УСТОЙЧИВОСТИ - МЕРА УСТОЙЧИВОСТИ КОМПЛЕКСА
411
М2+ + L ML2+
х х 0,10-х~0,10
Подставив эти величины в выражение для Кр получаем:
^12=1,ою8
XX
х = [М2+] = 3,2- 10-5М
Пример 9.2
Ион серебра образует устойчивый комплекс состава 1:1с триэтилентетрами-
ном (триеном, NH2(CH2)2NH(CH2)2NH(CH2)2NH2). Рассчитайте равновесную
концентрацию ионов серебра в растворе, полученном при добавлении 25 мл
0,010 М раствора нитрата серебра к 50 мл 0,015 М растовора триена; Kj= 5,0-107.
Решение
а + , а / \+ [Ag(TpneH)+]
Ag+ + триен Ag(TpneH)+ К^= - - --------—
[Ag + ] [триен]
Найдем общее количество миллимолей Ag+ и триена в растворе:
«Ag+(ммоль) = 25 мл • 0,010 ммоль/мл = 0,25 ммоль
«трИен (ммоль)= 50 мл • 0,015 ммоль/мл = 0,75 ммоль
В этом случае равновесие сильно смещено вправо, поэтому можно считать,
что практически все ионы Ag+ связываются с соответствующим количеством
(0,25 ммоль) триена, образуя 0,25 ммоль комплекса (0,50 ммоль триена остается
в избытке). Обозначим равновесную концентрацию Ag+ как х. Тогда:
[Ag+] = х = моль/л (непрореагировавший)
[триен] = 0,50 ммоль / 75 мл + х = 6,7 • 10~3 + х « 6,7 • 10-3
[Ag(rpneH)+] = 0,25 ммоль / 75 мл - х = 3,3 • 103 - х « 3,3 • 10-3
Величиной х в двух последних выражениях пренебрегаем. Подставляя по-
лученные значения, получаем:
x=[Ag+] = 9,8- 10-9М
Как видно, величиной х в расчетах действительно можно было пренебречь.
412
РЕАКЦИИ КОМПЛЕКСООБРАЗОВАНИЯ И КОМПЛЕКСОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
9.2. Хелаты. ЭДТА - самый распространенный титрант
для определения ионов металлов
Простые комплексообразующие реагенты наподобие аммиака в качестве тит-
рантов используют редко ввиду трудности индикации конечных точек титрования,
соответствующих образованию индивидуальных комплексов стехиометрического
состава. Причина этого состоит в том, что ступенчатые константы образования
отдельных комплексов обычно невелики и близки друг к другу, поэтому образо-
вание индивидуальных комплексов в чистом виде не наблюдается. В то же
время существуют реагенты, содержащие две или более комплексообразующих
групп и способные к образованию индивидуальных комплексов. Их можно ис-
пользовать в качестве титрантов. Шварценбах показал, что уже для бидентатных
лигандов (т. е. содержащих две комплексообразующие группы) наблюдается су-
щественное возрастание устойчивости комплексов. Например, комплекс меди с
этилендиамином (NH2CH2CH2NH2, еп) состава Cu(en) 2+ значительно устойчи-
вее аммиачного. Если же в молекуле этилендиамина атомы водорода при атомах
азота заместить на остатки уксусной кислоты, то полученный лиганд может об-
разовать с ионом меди только комплекс состава 1:1. Использование такого ли-
ганда для титрования не вызывает затруднений.
Чаще всего в комплексометрическом титровании используют аминополи-
карбоновые кислоты, в которых атомы азота и атома кислорода карбоксильных
групп являются донорными. Анионы этих кислот способны к протонированию;
при этом атомы азота являются более основными и протонируются легче, чем
атомы кислорода карбоксилатных групп. С подобными полидентатными лиган-
дами ионы металлов, как правило, образуют только комплексы состава 1:1 неза-
висимо от заряда иона, поскольку донорных атомов одного лиганда достаточно,
чтобы занять все места координации иона металла.
Органический реагент, имеющий две или более групп, способных к комплек-
сообразованию с ионом металла, называется хелатообразующим реагентом (хела-
тообразующим лигандом), а образующийся комплекс — хелатом. Термин «хелат»
в переводе с греческого означает «клешнеобразный». Хелатообразующие реагенты
в буквальном смысле охватывают собой ион металла, как клешнями. Титрование
хелатообразующим реагентом — разновидность комплексометрического титрова-
ния — иногда называют комплексонометрическим титрованием, так как среди
хелатообразующих реагентов для титрования чаще всего используют этилендиа-
минтетрауксусную кислоту (ЭДТА), которую называли также комплексоном:
НО2ССН2 СН2СО2Н
\ н+ н+/
nch2ch2n
-О2ССН2 СН2СО1
9.2. ХЕЛАТЫ. ЭДТА - САМЫЙ РАСПРОСТРАНЕННЫЙ ТИТРАНТ ДЛЯ ОПРЕДЕЛЕНИЯ ИОНОВ МЕТАЛЛОВ
413
Каждый из двух атомов азота и по одному атому кислорода из каждой кар-
боксильной группы, имеющие неподеленные электронные пары, способны к
образованию координационных связей с ионом металла. Таким образом, моле-
кула ЭДТА содержит шесть донорных атомов. В дальнейшем мы будем обо-
значать молекулу ЭДТА как Н4У. ЭДТА является четырехосновной кислотой.
В комплексы с металлами она входит в форме депротонированного аниона Y4-.
Таким образом, при комплексообразовании протоны ЭДТА замещаются на
ион металла.
Хелатный эффект:
чем больше комплексообразующих групп, тем лучше
Полидентатные хелатообразующие реагенты образуют более устойчивые ком-
плексы с ионами металлов, чем бидентатные и монодентатные лиганды анало-
гичного строения. Это явление имеет термодинамическую природу. Движущей
силой химических реакций может быть уменьшение энтальпии (при выделе-
нии теплоты АЯ отрицательно) или возрастание энтропии (при увеличении
беспорядка AS положительно). Вспомним уравнение (6.7) из гл. 6: из него сле-
дует, что химический процесс протекает самопроизвольно, если изменение
свободной энергии A G = АН - TAS—отрицательная величина. Что касается из-
менения энтальпии в ходе комплексообразования, то для лигандов аналогич-
ного строения оно почти одинаково. Например, координация четырех молекул
аммиака или четырех аминогрупп двух молекул этилендиамина ионом Си2+
приводит к почти одинаковому выделению теплоты. В то же время при диссоци-
ации комплекса Cu(NH3) 4+ с образованием пяти частиц увеличение энтропии
больше, чем при диссоциации комплекса Cu(H2NCH2CH2NH2)2+ с образова-
нием трех частиц. Поэтому для процесса диссоциации того комплекса, где AS
больше, AG оказывается более отрицательной величиной, а следовательно
диссоциация этого компонента должна быть более существенной. Поэтому
комплекс с монодентатными лигандами лучше диссоциирует. Комплексы с
полидентатными лигандами более устойчивы (характеризуются более высо-
кими значениями — влияет энтропийный фактор. Это явление известно как
хелатный эффект. Хелатный эффект имеет энтропийную природу. Для ли-
гандов с большим числом донорных атомов, способных, подобно ЭДТА, за-
нять все места координации иона металла, хелатный эффект выражен особенно
сильно. О дизайне хелатообразующих реагентов см. статью [С. N. Reilley,
R. W. Schmid, F. S. Sadek, «Chelon Approach to Analysis I. Survey of Theory and
Application». J. Chem. Ed., 36 (1959) 555]. Иллюстрированный эксперимент
описан во второй статье этой серии: [J. Chem. Ed., 36 (1959) 619].
414
РЕАКЦИИ КОМПЛЕКСООБРАЗОВАНИЯ И КОМПЛЕКСОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Равновесия в растворе ЭДТА
Молекулу ЭДТА можно охарактеризовать четырьмя ступенчатыми константа-
ми диссоциации кислотных протонов:*
H4Y^H+ + H3Y- 7Са1 = 1,0-10-2 = [Н+^У ] (9.5)
|H4YJ
Н,Y- — Н+ + H.Y2- Ка2 = 2,2-10-3 = [н- _ 1 (9.6)
[H3Y-]
H,Y2- — Н+ + HY3- Ка. = 6,9-10"7 = [Н ][Н? (9.7)
[H2Y2~]
HY3- Н+ + Y4" К. = 5,5 • IO"В * * 11 = SUDLJ (9.8)
[HY3"]
Прежде чем изучать этот раздел дальше, советуем повторить материал, посвя-
щенный равновесиям в растворах многоосновных кислот (см. разд. 7.8).
На рис. 9.1 изображены зависимости мольных долей каждой формы ЭДТА
от pH. Одна из этих форм, Y4-, участвует в комплексообразовании в качестве ли-
ганда. Поэтому на положение равновесия комплексообразования с участием
ЭДТА сильно влияет значение pH. Как реактив обычно используют динатриевую
соль N^^Y • 2Н2О, поскольку свободная кислота H4Y малорастворима в воде.
При растворении динатриевая соль образует анион Н2 Y2; значение pH такого
раствора находится между 4 и 5 (теоретическая величина [Н+] = у/Ка2Ка3 соот-
ветствует pH 4,4).
Константы устойчивости комплексов с ЭДТА
В качестве примера рассмотрим образование комплекса ЭДТА с Са2+. Этот про-
цесс можно представить уравнением
Са2++ Y4-CaY2- (9.9)
Выражение константы устойчивости комплекса записывается следующим
образом:
к_ [CaY2-]
f [Ca2+][Y4-]
(9-Ю)
В принципе к протонированию способны не только все четыре карбоксилатные группы, но и оба атома
азота. Это соответствует шести ступеням диссоциации и шести последовательным константам кислотно-
сти, первые две из которых равны 1,0 и 0,032. Атомы азота обладают более основными свойствами, чем
карбоксилатные атомы кислорода, и протонируются легче. Протонирование атомов азота увеличивает
растворимость ЭДТА в сильнокислых средах.
9.2. ХЕЛАТЫ. ЭДТА - САМЫЙ РАСПРОСТРАНЕННЫЙ ТИТРАНТ ДЛЯ ОПРВДЕЛЕНИЯ ИОНОВ МЕТАЛЛОВ
415
Величины констант устойчивости комплексов ЭДТА с некоторыми ионами
металлов приведены в Приложении С.
Влияние pH на равновесия с участием ЭДТА:
какова равновесная концентрация иона Y4 ?
Равновесие, представленное уравнением (9.9), с ростом концентрации Н+ сдви-
гается влево из-за конкурирующего связывания аниона ЭДТА с протонами
[с учетом этого в уравнении (9.10) можно выразить равновесную концентрацию
[Y4-] в виде а4Сн Y]« Это можно проиллюстрировать схемой
н+ н+ н+ н+
CaY2Са2+ + Y4 ^HY3’ ^H2Y2’ ^H3Y' ^H4Y
i ___________ j
Ch4y
В полном виде уравнение реакции комплексообразования можно записать в
виде
Са2+ + H4Y CaY2- + 4Н+
В соответствии с принципом Ле Шателье увеличение кислотности способ-
ствует протонированию Y4-, т. е. конкурирующему процессу. При этом в равно-
весии находятся все формы ЭДТА, хотя концентрации некоторых из них могут
быть исчезающе малыми (см. рис. 9.1). Уменьшение кислотности, напротив,
способствует образованию комплекса CaY2-.
Зная величину pH и константы соответствующих равновесий, можно по
уравнению (9.10) рассчитать концентрации свободных ионов Са2+ в различных
условиях — например, в отдельных точках кривой титрования. Равновесную
концентрацию [Y4"] при различных pH рассчитывают следующим образом
Рис. 9.1
Зависимости мольных долей
форм ЭДТА от pH
416
РЕАКЦИИ КОМПЛЕКСООБРАЗОВАНИЯ И КОМПЛЕКСОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
(см. гл. 7, раздел, посвященный многоосновным кислотам). Обозначим суммар-
ную концентрацию всех форм ЭДТА, не связанных в комплекс, как Сщу. Тогда
Ch4y = [Y4-] + [НУ3-] + [Н2У2-] + [Н3У-] + [Н4У] (9.11)
Выразив равновесные концентрации Н4У, Н3У-, Н2У2- и НУ3- через [У4-] из
уравнений (9.5)-(9.8), подставив эти выражения в уравнение (9.11) и разделив на
[У4-], получаем:
= ;h i^ [и*]1 + [н>Г . .
[Y4-] «. Х.4 к„,к„, Ка2КлК„ КлКа1Ка2Ка,
trq а4 — мольная доля формы У4-, равная [Y4-]/^ Y • Подобные выражения
можно получить и для мольных долей других форм ЭДТА — а0, ар а2 и а3
(см. гл. 7). Результаты сответствующих расчетов представлены на рис. 9.1.
Уравнение (9.12) можно использовать для расчета доли ЭДТА в виде У4-
при заданном pH. Зная же Сц4у — общую концентрацию ЭДТА, не связанного в
комплекс, — можно далее по уравнению (9.10) рассчитать концентрацию сво-
бодных ионов Са2+.
Пример 9.3
Рассчитайте долю ЭДТА в форме У4- при pH 10, а затем величину рСа в растворе,
полученном смешением 100 мл 0,100 М Са2+ и 100 мл 0,100 М ЭДТА при pH 10.
Решение
Из уравнения (9.12):
1 = 1 1,0 1О~10 (1,0-10~10) 2 (1,0-Ю"10)3
а 4 5,5-10-11 (6,9-10’7)(5,5-1041) (2,2-10"3)(6,9-10~7)(5,5-10“п)
+(1,0-1О~10)4=
(1,0 • 10-2 )(2,2 • 10"3 )(6,9 • 10-7 )(5,5 • 10-11)
= 1+ 1,82+ 2,6-10-4 + 1,2-10-11 +1,2-10~19 =2,82
а4 =0,35
Са2+ и ЭДТА находятся в стехиометрических количествах, равных
иСа2+= 0,100 М • 100 мл = 10,0 ммоль
иЭдта = 0,100 М • 100 мл = 10,0 ммоль
9.2. ХЕЛАТЫ. ЭДТА - САМЫЙ РАСПРОСТРАНЕННЫЙ ТИТРАНТ ДЛЯ ОПРВДЕЛЕНИЯ ИОНОВ МЕТАЛЛОВ
417
В результате их взаимодействия образуется эквивалентное количество
(10,0 ммоль) малодиссоциирующего комплекса CaY2-. Общий объем раствора
составляет 200,0 мл, поэтому [CaY2-] = 0,0500 М. Запишем:
Са2++ ЭДТА CaY2-
х х 0,0500 М-х®
« 0,0500 М (поскольку константа устойчивости комплекса велика)
Здесь х = Сцд — общая концентрация ЭДТА во всех формах, не связанных в
комплекс. Величина [Y4-], необходимая для расчетов по уравнению (9.10), равна
а4Сн4у. Таким образом, мы можем переписать уравнение (9.10) в виде
к_ [СаУ2~]
f [Са2+]а4Сн4у
Значение Kjравно 5,0 • Ю10 (см. Приложение С). Отсюда
5,0Ю10 = 0,0500
х 0,35 • х
х = 1,7- 10-бМ
рСа = 5,77
Условная константа устойчивости при заданном pH
Снова подставим а4Сц4у вместо [Y4-] в уравнение (9.10):
к_ [CaY2"]
f [Са2+]а4Сн4у
Перепишем это выражение в виде
(9.13)
Kf<x^ = K'f =
[CaY2~]
[Са2+]СН4у
(9.14)
Величина называется условной константой устойчивости. Она зави-
сит от а4 и, следовательно, от pH. Величина условной константы устойчивости
сохраняет постоянное значение только при фиксированном значении pH. Урав-
нение (9.14) можно использовать вместо уравнения (9.10) для расчета равновес-
ных концентраций различных частиц при заданном pH.
418
РЕАКЦИИ КОМПЛЕКСООБРАЗОВАНИЯ И КОМПЛЕКСОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Пример 9.4
Константа устойчивости CaY2- равна 5,0 • 1О10. При pH 10 рассчитанное значе-
ние а4 (пример 9.3) составляет 0,35, а условная константа устойчивости CaY2
[уравнение (9.14)] — 1,8 • Ю10. Рассчитайте рСа в растворе, полученном при до-
бавлении к 100 мл 0,100 М Са2+ при pH 10: а) 0, б) 50, в) 100, г) 150 мл 0,100 М
ЭДТА.
Решение
а) рСа = -lg [Са2+] = -1g 1,00 Ю"1 = 1,00
б) Общее количество Са2+ равно 0,100 М • 100 мл = 10,0 ммоль. Количество до-
бавленного ЭДТА равно 0,100 М • 50 мл = 5,0 ммоль. Величина условной кон-
станты устойчивости очень велика, равновесие (9.9) сильно сдвинуто вправо.
Поэтому тем количеством свободных ионов Са2+, которые образуются вслед-
ствие диссоциации CaY2-, можно пренебречь. Таким образом, количество
свободных ионов Са2+ равно общему количеству ионов Са2+, не вступивших в
реакцию:
«Са2+= 10,0 - 5,0 = 5,0 ммоль
[Са2+] = 5,0 ммоль /150 мл = 0,033 М
pCa = -lg3,3 • 10-2= 1,48
в) В точке эквивалентности почти все ионы Са2+ превращаются в CaY2-. Для
расчета равновесной концентрации Са2+ следует использовать уравнение
(9.14). Число миллимолей образовавшегося CaY2- равно исходному числу
миллимолей Са2+, а концентрация кальция равна 10,0 ммоль / 200 мл = 0,0500 М.
Из уравнения диссоциации CaY2- следует, что [Са2+] = Сщу = х, а равновес-
ная концентрация [CaY2-] = 0,0500 М -х. Однако степень диссоциации комп-
лекса мала, поэтому величиной х по сравнению с 0,0500 М можно пренебречь.
Таким образом из уравнения (9.14) следует:
Х'Х
х=1,7- lO^M^Ca2*]
pCa = -lg(l,7- 10-6) = 5,77
Сравните полученное значение с величиной, рассчитанной в примере 9.3 с
использованием К? вместо K’j.
г) Концентрация СН4у равна избыточной концентрации ЭДТА (вклад, обуслов-
ленный диссоциацией CaY2-, в этом случае еще меньше, чем в предыдущем,
поскольку теперь в системе присутствует избыток ЭДТА). Число миллимо-
лей CaY2- такое же, как и в случае в. Отсюда
[CaY2-] = 10,0 ммоль/250 мл = 0,0400 М
9.2. ХЕЛАТЫ. ЭДТА - САМЫЙ РАСПРОСТРАНЕННЫЙ ТИТРАНТ ДЛЯ ОПРВДЕЛЕНИЯ ИОНОВ МЕТАЛЛОВ 419
Избыток ЭДТА составляет 0,100 М • 150 мл - 0,100 М • 100 мл = 5,0 ммоль
Сн4у = 5,0 ммоль / 250 мл = 0,0200 М
--------------------= 1,8 -1010
[Са2+]-0,020
[Са2+] = 1,1 • 10~10М
рСа = -lg (1,1 • Ю-10) = 9,95
На рис. 9.2, подготовленном при помощи электронной таблицы (см. задачу 20),
показаны зависимости K’j для комплексов от pH ионов трех металлов с ЭДТА с
тремя ионами металлов различной устойчивости — от средней (Са2+) до высо-
кой (Hg2+). Ион кальция образует комплекс, недостаточно устойчивый для того,
чтобы этот металл можно было титровать в кислой среде (в этих условиях K’j < 1).
В то же время комплекс Hg2+ устойчив, и ртуть можно титровать в кислой среде.
При pH 13 рассчитанные значения K'j практически совпадают с поскольку
при этом pH величина а4 очень близка к единице (т. е. весь ЭДТА находится в
форме Y4-). Кривые на рис. 9.2 параллельны друг другу, поскольку для каждого
Рис. 9.2. Влияние pH на величины K'j комплексов металлов с ЭДТА
420
РЕАКЦИИ КОМПЛЕКСООБРАЗОВАНИЯ И КОМПЛЕКСОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Отметим в то же время, что влияние pH на устойчивость комплексов (т. е. на
величину АГу) может быть обусловлено изменением формы существования не
только ЭДТА, но и иона металла (расчет кривых на рис. 9.2 был произведен без
учета этого обстоятельства). В частности, возможно образование гидроксокомп-
лексов (М2+ + ОН- —> М0Н+). В этом случае ион ОН- конкурирует с анионом
ЭДТА за связывание с ионом металла подобно тому, как Н+ конкурирует с
ионом металла за связывание с Y4-.
9.3. Кривые титрования ионов металлов раствором ЭДТА
При титровании ионов металлов протекает реакция в соответствии с уравнени-
ем (9.9). На рис. 9.3 изображены кривые титрования Са2+ раствором ЭДТА при
pH 7 и 10. До точки эквивалентности концентрация свободных ионов Са2+ прак-
тически равна общей концентрации неоттитрованного кальция, поскольку дис-
социация комплекса протекает в незначительной степени (аналогично обстоит
дело и в осадительном титровании). В точке эквивалентности и после нее вели-
чину рСа рассчитывают из выражения константы устойчивости комплекса при-
менительно к заданному значению pH, как показано в примерах 9.3 и 9.4; для
расчетов можно использовать как величины Ау, так и АГу. Влияние pH видно при
сравнении двух кривых на рис. 9.3.
Расчет кривой титрования Са2+ ЭДТА при pH 10 (рис. 9.3) выполнен при по-
мощи электронной таблицы (задача 21). Как и в случае кислотно-основного тит-
рования, рассчитанные кривые имеют разрыв в непосредственной близости от
точки эквивалентности вследствие упрощений, заложенных в расчеты.
Чем устойчивее комплекс (т. е. чем больше величина АГу), тем сильнее сдви-
нуто вправо равновесие реакции (9.9) и тем больше скачок вблизи точки эквива-
лентности. Соответственно, чем устойчивее комплекс, тем ниже значения pH,
при которых возможно титрование (см. рис. 9.2). Это обстоятельство позволяет
титровать некоторые ионы металлов в присутствии других, образующих с ЭДТА
малоустойчивые комплексы и не взаимодействующих с ней в кислой среде.
Лишь немногие ионы образуют с ЭДТА настолько устойчивые комплексы, что
титрование можно вести в сильнокислой среде. Большинство ионов металлов
титруются ЭДТА только в слабокислых или щелочных растворах.
На рис. 9.4 приведены минимально возможные значения pH для титрования
раствором ЭДТА ионов различных металлов. Эти значения соответствуют кис-
лотности, при которой условная константа устойчивости Kj комплекса метал-
ла становится равной 1 • 106 (1g АГу = 6) — величине, условно выбранной в
качестве критерия наличия на кривой титрования четко выраженного скачка.
Обратите внимание, что чем меньше Ау, тем более щелочная среда (т. е. тем
большие значения а4) требуется для достижения величины АГу, равной 1 • 106.
Так Са2+, для которого значение Ауотносительно невелико (порядка 1О10) можно
титровать лишь при pH 8 и выше. Штриховые горизонтальные линии, на рисун-
ке условно делят ионы металлов на подгруппы в соответствии с величинами их
9.3. КРИВЫЕ ТИТРОВАНИЯ ИОНОВ МЕТАЛЛОВ РАСТВОРОМ ЭДТА
421
Рис. 9.3
Кривые титрования
100 мл 0,1 М раствора Са2+
0,1 М раствором Na2H2Y
при pH 7 и 10
Рис. 9.4
Минимальные значения pH,
необходимые для
титрования различных
ионов металлов раствором
ЭДТА. Воспроизведено
с разрешения С. N. Reilley,
R. W. Schmid, Anal. Chem.,
30 (1958) 947. Авторские
права принадлежат
American Chemical Society
422
РЕАКЦИИ КОМПЛЕКСООБРАЗОВАНИЯ И КОМПЛЕКСОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
констант устойчивости. В первую группу входят металлы, которые можно тит-
ровать в сильнокислой среде (pH < 4), во вторую — при pH от 4 до 7 и в третью —
только при pH > 7. При самых высоких значениях pH ионы всех металлов спо-
собны взаимодействовать с ЭДТА, но не все из них при этом можно непо-
средственно титровать ввиду образования малорастворимых гидроксидов. Напри-
мер, титрование Fe(III) или Th(IV) в щелочной среде возможно только обратным
способом либо в присутствии вспомогательных комплексообразующих реаген-
тов, предотвращающих образование гидроксидов. В промежуточной области pH
ионы третьей группы не титруются; при этих условиях возможно определение
ионов второй группы в присутствии третьей. В наиболее кислых средах титру-
ются только ионы первой группы, которые в этих условиях можно определять в
присутствии любых других ионов.
Для маскирования посторонних ионов можно использовать осаждение, комп-
лексообразование, окисление-восстановление и кинетические факторы, а также
их комбинацию. Например, Си2+ можно замаскировать восстановлением до
Cu(I) аскорбиновой кислотой или осаждением в виде Cui под действием иодид-
ионов. При определении висмута свинец можно осадить в виде сульфата. В бо-
льшинстве случаев маскирование основано на селективном образовании устой-
чивых растворимых комплексов. Гидроксид-ионы маскируют алюминий за счет
образования ионов А1(ОН)4 (или AIO2), не мешая определению кальция. Для
селективного определения Sn(II) маскируют Sn(IV) фторидами. Аммиак образу-
ет комплексы с ионами меди, вследствие чего титрование меди с индикатором
мурексидом становится невозможным. Ионы многих металлов можно титровать
в присутствии Сг(Ш) ввиду того, что комплекс Сг(1П) с ЭДТА, даже будучи весь-
ма устойчивым, образуется крайне медленно.
9.4. Индикация конечной точки.
Металлоиндикаторы - хелатообразующие реагенты
Для индикации конечной точки можно использовать потенциометрическое из-
мерение рМ при наличии подходящего электрода, например, ионселективного
(см. гл. 13). Однако проще применять химические индикаторы. Как правило, ин-
дикаторы, используемые при титровании с хелатообразующим титрантом, сами
являются хелатообразующими реагентами. Обычно это красители, относящиеся
к группе о,о’-дигидроксиазосоединений.
Типичным примером является эриохромовый черный Т. Его молекула со-
держит три протона, способных к диссоциации, поэтому его формулу можно за-
писать как Н31п. Этот индикатор можно использовать для определения магния с
ЭДТА. К раствору пробы добавляют небольшое количество индикатора, кото-
рый образует с частью ионов Mg2+ окрашенный в красный цвет комплекс (сво-
бодный индикатор имеет голубую окраску). Как только все свободные ионы
Mg2+ оттитрованы, ЭДТА вытесняет индикатор из его комплекса с магнием. При
этом наблюдается переход окраски раствора из красной в голубую:
9.4. ИНДИКАЦИЯ КОНЕЧНОЙ ТОЧКИ. МЕТАЛЛОМ ИДИ КАГОРЫ - ХЕЛАТООБРАЗУЮЩИЕ РЕАГЕНТЫ
423
Mglrr + h2y2~ - > MgY2" + Шп2- + Н+
(красный) (бесцветный) (бесцветный) (голубой)
(9.15)
Этот процесс происходит в некоторой области значений pMg. Поэтому переход
будет более резким, если брать индикатор как можно в меньшем количестве,
еще достаточном для наблюдения окраски.
Разумеется, комплекс металла с металлоиндикатором должен быть менее
устойчив, чем с ЭДТА, — в противном случае ЭДТА не сможет вытеснить инди-
катор из его комплекса с металлом. С другой стороны, комплекс с металлоиндика-
тором не должен быть слишком малоустойчивым: тогда он будет разрушаться
уже в начале титрования, а переход окраски будет размытым. Как правило, ком-
плекс иона металла с индикатором должен быть в 10-100 раз менее устойчи-
вым, чем с титрантом.
Константы устойчивости комплексов Са2+ и Mg2+ с ЭДТА достаточно близ-
ки, поэтому раздельное титрование этих ионов из одной пробы само по себе не-
возможно, даже если пытаться варьировать pH (см. рис. 9.4). Эти ионы титруются
совместно. Для индикации конечной точки можно использовать эриохромовый
черный Т (см. выше). Такое титрование используют для определения общей же-
сткости воды, т. е. суммарного содержания Са2+ и Mg2+ (см. практическую рабо-
ту 9). Жесткость воды обычно выражают в единицах ррш СаСО3, характеризуя
этим значением суммарное содержание кальция и магния. При этом эриохромо-
вый черный Т нельзя использовать для прямого титриметрического определе-
ния с ЭДТА кальция в отсутствие магния, поскольку этот индикатор образует с
кальцием слишком малоустойчивый комплекс и переход окраски выражен не-
четко. Поэтому, если проба не содержит Mg2+, для определения Са2+ к раствору
добавляют небольшое точно известное количество Mg2+ и оттитровывают сум-
му ионов, как описано выше. При этом Са2+ титруется первым, поскольку его
комплекс с ЭДТА более устойчив. Затем отдельно титруют взятое количество Mg2+
и полученный объем вычитают из объема, затраченного на титрование пробы.
Удобнее вместо Mg2+ (например, в виде MgCl2) добавлять к пробе небольшое
количество (например, 2 мл 0,005 М) раствора комплекса Mg2+ с ЭДТА. Для его
приготовления смешивают равные объемы 0,01 М растворов MgCl2 и ЭДТА и
затем точно подбирают соотношение компонентов, добавляя по каплям раствор
Mg2+ либо ЭДТА до тех пор, пока раствор (после добавления к нему буфера с
pH 10 и эриохромового черного Т) не станет фиолетовым. Окраска такого рас-
твора от одной капли 0,01 М ЭДТА должна измениться на голубую, а от одной
капли 0,01 М MgCl2 — на красную.
При добавлении раствора комплекса Mg2+ с ЭДТА к пробе, содержащей ка-
льций, ионы Са2+ вытесняют эквивалентное количество ионов Mg2+, поскольку
комплекс Са2+ с ЭДТА более устойчив. Высвободившиеся ионы Mg2+ взаимо-
действуют с индикатором. Вблизи конечной точки начинается взаимодействие
Mg2+ с ЭДТА. На него расходуется такое же количество ЭДТА, которое ранее
связалось с Са2+. Поэтому никакой поправки на содержение Mg2+ в этом случае
вносить не нужно. Данная методика описана в практической работе 9.
424
РЕАКЦИИ КОМПЛЕКСООБРАЗОВАНИЯ И КОМПЛЕКСОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Можно также добавить небольшое количество Mg2+ непосредственно в рас-
твор титранта ЭДТА. При этом соответствующее количество ЭДТА свяжется в
комплекс с магнием (а концентрация свободных ионов Mg2+ в таком растворе
будет очень мала). Разумеется, концентрация свободного ЭДТА в растворе тит-
ранта уменьшится по сравнению с исходной. Поэтому такой раствор необходи-
мо стандартизовать после добавления Mg2+ — например, титруя им раствор
первичного стандарта СаСО3 (после растворения в соляной кислоте и установ-
ления необходимого pH). При добавлении индикатора к титруемому раствору
Са2+ его окраска становится розово-красной, обусловленной образованием ком-
плекса Са2+ с эриохромовым черным Т. С началом титрования Mg2+ замещает
Са2+ в комплексе с индикатором, и окраска изменяется на винно-красную. В ко-
нечной точке она становится голубой вследствие вытеснения Mg2+ из комплекса
с индикатором. Здесь также не требуется вносить поправку на содержание
Mg2+ — оно уже было учтено в ходе стандартизации. Раствор ЭДТА, приготов-
ленный таким способом, нельзя использовать для определения ионов других ме-
таллов, кроме кальция.
Химически чистый препарат ЭДТА можно приготовить из Na2H2Y • 2Н2О
высушиванием при 80 °C в течение 2 ч. При этом кристаллизационная вода не
отщепляется. Полученный препарат можно использовать как первичный стан-
дарт для приготовления стандартных растворов ЭДТА.
Кальций и магний определяют с ЭДТА при pH 10, для создания такого pH
используют хлоридный аммиачно-аммонийный буфер. Значение pH не должно
быть слишком велико, иначе возможно осаджение гидроксидов металлов, пре-
пятствующее взаимодействию с ЭДТА или по крайней мере резко его замедляю-
щее. В то же время кальций можно титровать в присутствии магния при значениях
pH вплоть до 12 (в растворах сильных щелочей). При этом магний осаждается в
виде Mg(OH)2 и не взаимодействует с ЭДТА.
Эриохромовый черный Т, как и большинство других металлоиндикаторов,
является слабой кислотой, и его формы различной степени протонированности
окрашены по-разному, поэтому окраска его растворов зависит от pH. Например,
форма эриохромового черного Т состава Н21п_ (при pH < 6) имеет красный цвет,
HIn2- (pH 6-12) — голубой, a In3- (pH >12) — желто-оранжевый. Поэтому ме-
таллоиндикаторы можно использовать только в определенном диапазоне pH.
Изменение pH влияет также и на устойчивость комплексов ионов металлов с ин-
дикатором (так же, как и с ЭДТА).
Повторим, что в любом случае индикатор можно применять для определе-
ния только тех ионов металлов, которые при данном pH образуют с ним комплек-
сы менее устойчивые, чем с титрантом. Таким образом, при выборе индикатора
надо учитывать одновременно множество разных факторов. Тем не менее изве-
стно много подходящих индикаторов для определения большинства ионов ме-
таллов с различными хелатообразующими титрантами.
Например, при использовании кальмагита в качестве индикатора для опре-
деления Са и Mg с ЭДТА переход окраски в конечной точке более резкий, чем в
случае эриохромового черного Т. Кроме того, этот индикатор устойчивее в рас-
творах. Для определения многих ионов, образующих с ЭДТА очень устойчивые
9.4. ИНДИКАЦИЯ КОНЕЧНОЙ ТОЧКИ. МЕГАЛЛОИНДИКАГОРЫ - ХЕЛАТООБРАЗУЮЩИЕ РЕАГЕНТЫ
425
комплексы и титрующиеся при pH 1,5-3, используют ксиленоловый оранжевый.
Примерами могут служить прямое титрование тория(ГУ) и висмута(Ш), а также
обратное титрование циркония(ГУ) и железа(Ш) одним из этих ионов. Для тит-
рований с ЭДТА предложено много других индикаторов. Многочисленные при-
меры содержатся в руководстве [6] — прекрасной монографии с подробными
описаниями методик определения различных металлов.
Кроме того, в комплексонометрическом титровании используют и другие
титранты, помимо ЭДТА. Заслуживающим внимания примером является эти-
ленгликоль-бис(Р-аминоэтил)-М,М,М’,М’-тетрауксусная кислота (ЭГТА). Этот
простой эфир, по строению аналогичный ЭДТА, позволяет селективно опреде-
лять кальций в присутствии магния.
нооссн2 СН2СООН
\ н+ н+/
/ NCH2CH2OCH2CH2OCH2CH2N
ООССН2 СН2СОО-
Комплексообразующие реагенты, содержащие простые эфирные связи, вооб-
ще склонны к образованию устойчивых комплексов с ионами щелочноземельных
металлов более тяжелых, чем магний. Для комплекса ЭГТА с Са2+ величина 1g Kj
составляет 11,0, а в случае Mg2+ — лишь 5,2. О других хелатообразующих реа-
гентах и их использовании см. в работах [4] и [6].
Почти для каждого металла (кроме щелочных) существуют высокоточ-
ные методики комплексонометрического определения. Они более экспрессны и
удобны гравиметрических и к настоящему времени почти вытеснили их, за иск-
лючением нескольких случаев, где требуется — а при использовании гравимет-
рии и может быть достигнута — особо высокая точность.
В клинических лабораториях комплексонометрическое титрование приме-
няют лишь для определения веществ, присутствующих в достаточно высоких
концентрациях (поскольку титриметрия в целом не очейь чувствительный ме-
тод). Самый важный пример — определение кальция в крови [8]. Кроме того, в
медицине хелатообразующие реагенты, такие как ЭДТА, используют для лече-
ния отравлений тяжелыми металлами (например, если ребенок проглотит пред-
мет, содержащий свинцовую краску). В подобных случаях ЭДТА применяют в
виде комплексов с кальцием (например, Na2CaY), чтобы предотвратить связыва-
ние кальция, содержащегося в организме, и его вымывание из костной ткани. Тяже-
лые металлы, в частности, свинец, образуют с ЭДТА более устойчивые комплексы,
чем кальций, и вытесняют его, а затем в связанном состоянии выводятся из орга-
низма через почки.
Константы устойчивости комплексов некоторых металлов с ЭДТА приве-
дены в Приложении С.
426
РЕАКЦИИ КОМПЛЕКСООБРАЗОВАНИЯ И КОМПЛЕКСОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
9.5. Другие области применения комплексных соединений
В химическом анализе образование комплексов используют не только в титри-
метрии. Комплексообразование часто используют в экстракции, поскольку
многие хелаты металлов (в частности, с дитизоном) экстрагируются растворите-
лями, не смешивающимися с водой. Хелатные комплексы часто интенсивно
окрашены. Их образование может лежать в основе спектрофотометрического
определения ионов металлов. Существуют комплексы, обладающие флуоресцен-
тными свойствами. Даже такое свойство, как малая растворимость многих хела-
тов, может быть использовано в анализе. Примером может служить методика
гравиметрического определения никеля в виде комплекса с диметилглиоксимом.
В табл. 10.2 приведены примеры других малорастворимых хелатных комплексов.
Равновесия комплексообразования могут быть использованы в ходе хроматог-
рафических разделений. Мы уже упоминали о применении комплексообразую-
щих реагентов для маскирования, предотвращения нежелательных реакций.
Например, при экстракции ванадия в виде хелатного комплекса с 8-оксихиноли-
ном экстракцию меди подавляют введением ЭДТА, предотвращающего образо-
вание 8-оксихинолината меди.
Почти всегда на полноту протекания реакции комплексообразования влия-
ет pH раствора. Поэтому выбор и контроль значения pH (при помощи буферных
растворов) — необходимое условие оптимизации протекания желаемой реакции
либо подавления нежелательных.
9.6. Доли отдельных форм многолигандных комплексов.
p-коэффициенты: каково содержание каждой формы?
Многолигандные комплексы, такие как Ag(NH3)2, диссоциируют ступенчато
подобно многоосновным кислотам. Как и для кислот, расчеты равновесных кон-
центраций отдельных форм могут быть достаточно сложными, за исключением
случаев, когда лиганд присутствует в большом избытке. В противном случае ис-
пользуют итерационные процедуры. Если же имеется избыток лиганда, то все
расчеты выполняют аналогично расчетам для многоосновных кислот при задан-
ной концентрации ионов водорода [см. гл. 7, уравнения (7.63)-(7.75)]. Рассчита-
ем, например, равновесные концентрации отдельных форм серебра в системе,
описываемой уравнениями (9.1) и (9.2), при заданной концентрации аммиака.
Запишем выражение для мольной доли каждой формы:
R _[Ag+] R _[Ag(NH3)+]
Po“~ Pi ~
Ag Ag
^Ag
9.6. ДОЛИ ОТДЕЛЬНЫХ ФОРМ МНОГОЛИГАНДНЫХ КОМПЛЕКСОВ. р-КОЭФФИЦИЕНТЫ
427
где CAg — общая концентрация серебра во всех ионных формах. Для обозначе-
ния доли каждой комплексной формы мы будем использовать символ р, чтобы
отличить их от долей разных форм кислоты, обозначаемых а. Нижний индекс
показывает число лигандов, связанных с ионом металла. Заметим, что иногда
символ р используют для обозначения ступенчатых (а в русскоязычной литера-
туре — наоборот, общих — прим, перев.) констант устойчивости комплексов;
эти обозначения не следует путать с символами р, применяемыми в данном
случае.
Общая концентрация серебра равна сумме концентраций всех его форм:
CAg = [Ag(NH3)2] + Ag(NH3)+] + [Ag+] (9.16)
Для расчета величины ро — доли ионов Ag+ — следует выразить концентра-
ции всех других форм серебра через [Ag+] из уравнений (9.1) и (9.2) и подставить
в уравнение (9.16). Из уравнения (9.1):
[Ag(NH3)+] = ^[Ag+JtNiy (9.17)
Из уравнений (9.2) и (9.17):
[Ag(NH3)2] = ^2[Ag(NH3)+][NH3] = /^/2[Ag+][NH3]2 (9.18)
Подставляя уравнения (9.17) и (9.18) в (9.16), получаем:
CAg = ^^[Ag+JtNiy2 + ATzl[Ag+][NH3] + [Ag+] (9.19)
Подставив это выражение в качестве знаменателя в выражение для ро, получаем:
Подобные выражения можно получить и для других P-коэффициентов (Pt для
Ag(NH3)+ и Р2 для Ag(NH3)2+), подставляя соответствующие величины из урав-
нений (9.1) и (9.2). От выражения (9.20) они отличаются только числителями:
*Z1[NH3]
*Z1*/2[NH3]2
^^[NH^+^JNH^ + l
(9.21)
(9.22)
428
РЕАКЦИИ КОМПЛЕКСООБРАЗОВАНИЯ И КОМПЛЕКСОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Полезно сравнить эти выражения с уравнениями (7.72)-(7.75). Они несколь-
ко отличаются по форме, поскольку в них использованы константы ассоциации
комплексов, а не диссоциации. Если использовать константы диссоциации ком-
плексов, то выражения для ^-коэффициентов были бы идентичны выражениям
для a-коэффициентов двухосновной кислоты за исключением того, что вместо
величины [Н+] следовало бы использовать [NH3], вместо Ка—порядок сле-
дования индексов ^-коэффициентов был бы обратным (так, 0О соответствовал
бы а2) в соответствии с определениями этих величин.
Пример 9.5
Рассчитайте равновесные концентрации различных форм серебра в 0,010 М рас-
творе Ag(I) в присутствии 0,10 М NH3.
Решение
Из уравнений (9.20)-(9.22) получаем:
(2,5 • 103 )(1,0 • 104 )(0,1 0) 2 + (2,5 • 103 )(0,10) +1
и, аналогично,
Pi = 1,0- 10~3 р2=1,0
[Ag+] = CAgP0 = 0,010 • 4,0 • 10-6 = 4,0 • 10"8 M
[Ag(NH3)+] = CAgPi = 0,010 • 1,0 • 10-3 = 1,0 • 10-5 M
[Ag(NH3)2] = CAgP2 = 0,010 • 1,0 = 0,010 M
Практически все серебро находится в виде комплекса с двумя молекулами
аммиака.
При расчетах мы пренебрегли уменьшением концентрации аммиака в рас-
творе за счет связывания в комплексы. Но мы видим, что в первом приближении
в связанной форме находится 20% аммиака. Если повторить расчеты р-коэффи-
циентов для равновесной концентрации аммиака, равной 0,08 М, то величина Р2
останется равной единице. И в этом случае приходим к выводу, что почти все се-
ребро находится в форме Ag(NH3)2. Однако относительные значения Ро и рх не-
сколько изменятся. Описанная процедура лежит в основе итерационных расчетов
методом последовательных приближений. Этот метод можно использовать
во многих расчетах равновесных концентраций, когда есть возможность сделать
некоторые упрощающие допущения, в частности, для расчетов кислотно-основ-
ЗАДАЧИ
429
ных равновесий, если пренебречь концентрацией продиссоциировавшей кисло-
ты по сравнению с ее общей концентрацией (см. гл. 6). Как правило, достаточно
бывает двух-трех итераций. (См. задачу 22 по расчету P-коэффициентов для ам-
миачных комплексов серебра при помощи электронной таблицы.)
Что мы узнали из этой главы?
• Константы устойчивости комплексов — стр. 408
• Равновесия в растворах ЭДТА [основные уравнения (9.12), (9.13)] —
стр. 414
• Индикаторы для комплексонометрического титрования — стр. 422
• p-коэффициенты [основные уравнения (9.20)-(9.22)] — стр. 426
Вопросы
1 > Чем отличаются понятия «комплексообразующий реагент» и «хелатообра-
зующий реагент»?
2. На чем основано использование индикаторов в комплексометрическом
титровании?
3 - Для чего к раствору ЭДТА, используемому для титрования кальция с инди-
катором эриохромовым черным Т, добавляют небольшое количество соли
магния?
Задачи
Расчеты равновесий комплексообразования и констант
устойчивости
4. Ион кальция образует с нитрат-ионом малоустойчивый комплекс состава
1:1с константой устойчивости 2,0. Каковы равновесный концентрации
Са2+ и Ca(NO3)2+ в растворе, приготовленном смешением 10 мл 0,010 М
СаС12 и 10 мл 2,0 М NaNO3? Влиянием ионной силы можно пренебречь.
5- Константа устойчивости комплекса серебра с этилендиамином
Ag(NH2CH2CH2NH2)+ равна 5,0 • 104. Рассчитайте равновесную концентра-
цию Ag+ в 0,10 М растворе комплекса. Образованием комплексов другого
состава можно пренебречь.
6- Чему равна концентрация Ag+ в растворе его комплекса с этилендиамином
(задача 5), содержащем 0,10 М свободного этилендиамина?
7- Ион серебра образует комплексы с тиосульфат-ионом S2O3” со ступенча-
тыми константами устойчивости Kfx = 6,6 • 108 и Kf2 = 4,4 • 104. Рассчитайте
430
РЕАКЦИИ КОМПЛЕКСООБРАЗОВАНИЯ И КОМПЛЕКСОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
равновесные концентрации всех форм серебра в растворе, содержащем
0,0100 М AgNO3 и 1,00 М Na2S2O3. Влиянием ионной силы пренебречь.
Условные константы устойчивости
8- Константа устойчивости комплекса свинца с ЭДТА PbY2 равна 1,10 • 1018.
Рассчитайте его условную константу устойчивочти: а) при pH 3 и б) при pH 10.
9. Используя значения условных констант устойчивости, рассчитанные в за-
даче 8, рассчитайте величины рРЬ (т. е. -lg [Pb2+]) для 50,0 мл 0,0250 М раство-
ра РЬ2+: а) при pH 3; б) при pH 10 после добавления к нему: 1) 0 мл, 2) 50 мл,
3) 125 мл, 4) 200 мл 0,0100 М ЭДТА.
10. Условная константа устойчивости комплекса Са2+ с ЭДТА при pH 10, рас-
считанная в примере 9.4, равна 1,8 • 1010. Рассчитайте условную константу
устойчивости этого комплекса при pH 3. Сравните полученную величину с
условной константой устойчивости комплекса свинца при таком же зна-
чении pH (задача 8). Можно ли титровать свинец при pH 3 в присутствии
кальция?
Стандартные растворы
11 - Рассчитайте массу навески Na2H2Y • 2Н2О, необходимую для приготовле-
ния 500,0 мл 0,05000 М ЭДТА.
12. Для стандартизации раствора ЭДТА навеску особо чистого СаСО3 массой
0,3982 г растворили в соляной кислоте, при помощи аммиачного буфера
установили pH 10 и оттитровали раствором ЭДТА. На титрование затрачено
38,26 мл раствора. Какова молярная концентрация ЭДТА?
13- Рассчитайте титр 0,1000 М раствора ЭДТА, выразив его в мг СаСО3/мл.
14. Чему равен титр 0,01000 М раствора ЭДТА (выраженный в единицах жест-
кости воды на 1 мл), используемого для титрования 100,0 мл жесткой
воды?
Количественный анализ методом комплексонометрии
15- Пробу молочного порошка массой 1,50 г озолили, растворили и оттитрова-
ли кальций раствором ЭДТА. На титрование затрачено 12,1 мл раствора.
Титрант стандартизовали по раствору цинка, приготовленному растворе-
нием 0,632 г металлического цинка в кислоте с последующим разбавлени-
ем до 1 л. На титрование 10,0 мл этого раствора израсходовано 10,8 мл
раствора ЭДТА. Какова концентрация кальция в молочном порошке (в еди-
ницах ррш)?
16- В сыворотке крови кальций определяют микротитрованием с ЭДТА.
К 100 мкл пробы добавили две капли 2 М КОН, индикатор и оттитровали
0,00122 М ЭДТА при помощи микробюретки. На титрование затрачено
0,203 мл раствора титранта. Чему равно содержание кальция в сыворотке
крови (в мг/дл и ммоль экв./л)?
ЗАДАЧИ
431
17- В ходе титриметрического определения цианид-ионов титрованием рас-
твора серебра по Либиху сначала образуется растворимый комплекс, а в
точке эквивалентности — осадок, что свидетельствует о достижении ко-
нечной точки:
2CN- + Ag+ -> Ag(CN) 2 (в ходе титрования)
Ag(CN) 2 + Ag+ Ag[Ag(CN)2] (в конечной точке)
Навеску KCN массой 0,4723 г оттитровали 34,95 мл 0,1025 М AgNO3. Како-
во содержание основного вещества (%) в пробе?
18. Медь определяли в соленых водах вблизи пунктов сброса очистных соору-
жений следующим образом. Сначала медь отделили и сконцентрировали
экстракцией метиленхлоридом в виде дитизоната при pH 3, затем выпарили
растворитель, остаток озолили для разложения органических веществ, рас-
творили и титровали ЭДТА. Каждую из трех порций пробы объемом по 1 л
проэкстрагировали 25 мл метиленхлорида, объединенные экстракты разба-
вили в мерной колбе до 100 мл, аликвоту объемом 50 мл выпарили, озолили
и оттитровали. На титрование затрачено 2,67 мл раствора ЭДТА с титром
2,69 мг/мл по СаСО3. Какова концентрация (в ppm) меди в пробе?
19 - Хлориды в сыворотке крови определяли титрованием с Hg(NO3)2 по реакции
2С1- + Hg2+ ±=f HgCl2. Раствор Hg(NO3)2 стандартизовали, титруя 2,00 мл
0,0108 М NaCl с индикатором дифенилкарбазоном. На титрование затрачено
1,12 мл раствора Hg(NO3)2. К пробе сыворотки объемом 0,500 мл добавили
3,50 мл воды, 0,50 мл 10%-го раствора вольфрамата натрия и 0,50 мл 0,33 М
H2SO4 для осаждения белков. Затем раствор профильтровали через сухой
фильтр в сухую колбу. На титрование аликвоты фильтрата объемом 2,00 мл
затрачено 1,23 мл раствора Hg(NO3)2. Рассчитайте концентрацию хло-
рид-ионов в сыворотке крови в ммоль экв./л.
Примечание. Эта задача носит иллюстративный характер, поскольку титрование
растворами солей ртути сейчас применяют редко из-за их токсичности.
Задачи для решения при помощи электронных таблиц
См. компакт-диск, гл. 9.
20. Создайте таблицу для построения графика зависимости 1g от pH для
комплексов Са2+, РЬ2+ и Hg2+ с ЭДТА. Для этого потребуется расчет а4 для
ЭДТА и ввод величин для соответствующих комплексов. Расчет ведите
с шагом 0,5 единиц pH. Сравните полученный график с рис. 9.2.
21. Создайте электронную таблицу для построения кривой титрования 100,0 мл
0,1000 М раствора Са2+ 0,1000 М раствором ЭДТА при pH 10 в координатах
Иэдтд, мл — рСа (см. рис. 9.3). В первой точке кривой примите значение
объема титранта равным 10 мл, затем ведите расчет с шагом 20 мл, а вблизи
точки эквивалентности постепенно уменьшите шаг до 0,05 мл. Сравните
полученную кривую с результатами расчетов в примере 9.4.
432
РЕАКЦИИ КОМПЛЕКСООБРАЗОВАНИЯ И КОМПЛЕКСОМЕГРИЧЕСКОЕТИТРОВАНИЕ
22. Создайте электронную таблицу для построения графика зависимостей трех
(3-коэффициентов для системы Ag+—NH3 от значения [NH3]. Зависимость
постройте в диапазоне 0-0,01 М NH3 с начальным шагом 0,0005 М, а после
0,002 М увеличьте шаг до 0,001 М.
Рекомендуемая литература
1. Stability Constants of Metal-Ion Complexes. Part A: Inorganic Ligands. E. Hog-
feldt, ed., Part В: Organic Ligands, D. D. Perrin, ed., Oxford: Perga-mon, 1979,
1981.
2. A Martell and R. J. Motekaitis, The Determination and Use of Stability Cons-
tants. New York: VCH, 1989.
3. J. Kragten, Atlas of Metal-Ligand Equilibria in Aqueous Solution. London: Ellis
Horwood, 1978.
4. G. Schwarzenbach, Complexometric Titrations. New York: Interscience, 1957.
5. H. Flaschka, EDTA Titrations. New York: Pergamon, 1959.
6. H. A. Flaschka and A. J. Barnard, Jr., «Titrations with EDTA and Related Com-
pounds», in C. L. Wilson and D. W. Wilson, eds. Comprehensive Analytical Che-
mistry, Vol. IB. New York: Elsevier, 1960.
7. F. J. Welcher, The Analytical Uses of Ethylenediaminetetraacetic Acid. Prince-
ton: Van Nostrand, 1958.
8. F. W. Fales, «Calcium (Complexometric)», in Standard Methods of Clinical
Chemistry, Vol. 2. D. Seligson, ed. New York: Academic, 1958, pp. 1-11.
9. J. Stary, ed., Critical Evaluation of Equilibrium Constants Involving 8-Hydroxy-
quinoline and Its Metal Chelates. Oxford: Pergamon, 1979.
10. H. A. Flaschka, Chelates in Analytical Chemistry, Vols. 1-5. New York: Dekker,
1967-1976.
Глава 10
ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ И РАВНОВЕСИЕ
ОСАДОК-РАСТВОР
Некоторые нагрузки легки, некоторые тяжелы.
Есть люди, предпочитающие легкое тяжелому...
Мао Цзэдун
Гравиметрия — один из самых точных методов определения больших количеств
веществ. В ходе гравиметрического анализа определяемый компонент, как пра-
вило, селективно переводят в малорастворимую форму, т. е. осаждают. Затем
осадок отделяют, высушивают либо прокаливают (при этом, возможно, опреде-
ляемый компонент превращается в другую химическую форму) и точно взвеши-
вают. Зная массу осадка и его химический состав, можно рассчитать массу
аналита, выразив ее в желаемой форме.
Гравиметрия — не только один из самых точных, но и один из самых трудо-
емких методов анализа. Именно гравиметрию использовал Т. У. Ричардс для опре-
деления атомных масс химических элементов, за что получил в 1914 г.
Нобелевскую премию. Подробнее об этом можно узнать из его работ, посвящен-
ных изучению причин загрязнений осадков [Z. Anord. Chem., 8 (1895) 413, 419,
421]; см. также http://nobelprizes.com.
В данной главе описаны особенности отдельных этапов гравиметрического
анализа, включая подготовку раствора перед осаждением, само осаждение, при-
емы, позволяющие получать чистый осадок в удобном для фильтрования виде,
способы фильтрования и промывания осадка, прибегая к которым можно избе-
жать его потерь и загрязнения, и термообработку, проводимую с целью получе-
ния гравиметрической формы. Рассмотрены способы расчета количества аналита,
исходя из значения массы осадка, в соответствии с принципами, описанными в
гл. 5. Приведены также примеры гравиметрических определений. Глава завер-
шается рассмотрением понятия произведения растворимости и связанных с ним
вопросов, касающихся равновесий осадок-раствор.
10.1. Как успешно провести гравиметрический анализ?
Успешное проведение гравиметрического анализа зависит от правильности вы-
полнения целого ряда важных операций, специально разработанных для того,
чтобы получить чистый осадок в форме, удобной для фильтрования и взвешива-
ния. Например, при определении хлорид-ионов посредством их осаждения в
виде хлорида серебра недостаточно просто прилить к раствору пробы раствор
434
ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ И РАВНОВЕСИЕ ОСАДОК-РАСТВОР
нитрата серебра и отфильтровать осадок, — вы ошибаетесь! Чтобы достичь вы-
сокой точности в гравиметрическом анализе, требуется тщательное выполнение
всех операций, связанных с осаждением и обработкой осадка.
Этапы гравиметрического анализа
Операции, которые необходимо выполнить в ходе гравиметрического анализа
после растворения пробы, следующие.
1 - Подготовка раствора.
2. Осаждение.
3- Старение осадка.
4> Фильтрование.
5- Промывание осадка.
6- Высушивание или прокаливание.
7- Взвешивание.
8- Расчет содержания аналита.
Последовательно рассмотрим каждую из этих стадий.
Сначала подготовим раствор
Первый этап гравиметрического анализа — подготовка раствора пробы к осаж-
дению. Иногда при этом требуется предварительное отделение мешающих ком-
понентов. Кроме того, в растворе необходимо создать такие условия, которые
обеспечивают малую растворимость осадка и получение его в форме, удобной
для фильтрования, а в ряде случаев необходимо и маскирование мешающих
компонентов. Среди факторов, которые следует при этом учитывать, — объем
раствора в ходе осаждения, диапазон концентраций определяемого компонента,
наличие и состав других компонентов, температура и pH раствора.
Иногда мешающие компоненты необходимо специально предварительно
отделить. Однако в большинстве случаев осаждение аналита само по себе обес-
печивает достаточную селективность разделения. При этом важно учитывать pH
раствора, поскольку его величина часто влияет на растворимость осадков, обра-
зованных как аналитом, так и возможными мешающими компонентами. Напри-
мер, оксалат кальция практически нерастворим в щелочных средах, но при
низких pH его растворимость резко возрастает за счет связывания оксалат-ионов
с протонами с образованием слабой кислоты. 8-Оксихинолин (оксин) можно ис-
пользовать для осаждения ионов многих элементов, при этом контроль pH по-
зволяет обеспечить селективное осаждение. Например, А13+ можно осадить при
pH 4, однако для осаждения Mg2+ концентрация аниона-осадителя (8-оксихино-
лината) в этих условиях еще недостаточна. Чтобы осадить магний, необходимо
10.1. КАК УСПЕШНО ПРОВЕСТИ ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ?
435
повысить pH и тем самым сдвинуть вправо равновесие диссоциации 8-оксихи-
нолина. В то же время при слишком высоких pH может начаться осаждение гид-
роксида магния, загрязняющего осадок. Влияние других перечисленных выше
факторов станет более ясным в ходе обсуждения стадии осаждения.
| А13+ +
А1/3
Приступаем к осаждению
(при правильно выбранных условиях!)
После подготовки раствора к осаждению необходимо провести само осаждение.
При этом также следует соблюдать определенные условия. В первую очередь
необходимо, чтобы растворимость осадка была достаточно низка, т. е. чтобы
потери вещества за счет растворимости можно было считать пренебрежимо ма-
лыми. Осадок по возможности должен быть крупнокристаллическим, тогда он
легко фильтруется. Кроме того, в этом случае осадок получается более чистым:
осадки в любом случае захватывают различные частицы, присутствующие в рас-
творе, но крупнокристаллические загрязняются меньше.
Чтобы оценить всю важность выбора условий осаждения, кратко рассмот-
рим процесс осаждения в целом. При добавлении раствора осадителя (например,
AgNO3) к раствору осаждаемого вещества (например, хлоридов) образование
осадка (AgCl) протекает в несколько стадий. При образовании осадка устанав-
ливается гетерогенное равновесие (см. гл. 6 и окончание данной главы — мате-
риал о произведении растворимости), что происходит не мгновенно. Сначала
наступает состояние пересыщения, при котором фаза раствора содержит боль-
ше растворенных веществ, чем в условиях равновесия. Это состояние является
метастабильным: существует движущая сила, стремящаяся привести систему в
состояние равновесия. Переход к равновесному состоянию начинается с образо-
вания зародышей (нуклеации). Чтобы образовался зародыш осадка, некоторое
минимально необходимое число частиц растворенных веществ должно соеди-
ниться вместе и образовать микроскопическую частицу твердой фазы. Чем
выше степень пересыщения, тем выше скорость образования зародышей, тем
больше в конечном счете образуется частиц осадка и тем меньше будет их сред-
ний размер. При этом будет больше общая площадь поверхности осадка и, сле-
довательно, выше угроза его загрязнения из-за адсорбции (см. ниже).
436
ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ И РАВНОВЕСИЕ ОСДЦОК-РАСТВОР
В принципе образование зародышей может происходить и самопроизвольно.
Однако обычно оно инициируется под действием пылинок, царапин на стенках
сосуда, либо же его вызывают специально введением затравок — кристалликов
осадка (разумеется, для количественного анализа последнее неприменимо).
Исходные зародыши начинают расти за счет осаждения на их поверхности
других частиц, входящих в состав осадка, в результате могут образоваться крис-
таллы правильной геометрической формы. Этот процесс тоже протекает тем бы-
стрее, чем выше степень пересыщения. Если скорость роста кристаллов слишком
велика, возрастает вероятность появления в их структуре дефектов, а также за-
хвата примесей.
Фон Веймарн установил, что средний размер частиц осадка обратно про-
порционален величине относительного пересыщения раствора в ходе осажде-
ния, которая определяется следующим образом:
Относительное пересыщение =
где Q — концентрация частиц, образующих осадок, до начала осаждения (эта
величина определяет абсолютную степень пересыщения раствора); S — рас-
творимость осадка, т. е. концентрация составляющих его частиц в условиях
равновесия. Величина относительного пересыщения называется также отноше-
нием фон Веймарна.
Как отмечено выше, состояние пересыщения является метастабильным, и
чем выше пересыщение, тем выше скорость образования зародышей. Запомните:
Высокое относительное пересыщение —> большое число мелких частиц осадка
(большая площадь поверхности)
Низкое относительное пересыщение меньшее число более крупных частиц осадка
(малая площадь поверхности)
Очевидно, что в ходе осаждения следует поддерживать значение Q как мож-
но меньшим, а значение S — как можно большим. Поэтому обычно применяют
следующие приемы, позволяющие поддерживать оптимальные условия осаж-
дения* Все эти приемы позволяют уменьшить пересыщение и получить более
крупные частицы осадка.
В полном объеме эти приемы применимы лишь для тех осадков, которые в условиях гравиметрического
анализа можно получить в кристаллической форме (например, BaSO4). Для аморфных осадков (например,
Fe2O3 • хН2О) некоторые из этих приемов (такие как медленное ведение осаждения) могут оказаться беспо-
лезными, а некоторые — даже вредными (так, осаждение аморфных осадков из разбавленных растворов
может привести к потерям вследствие коллоидообразования). — Прим, перев.
10.1. КАК УСПЕШНО ПРОВЕСТИ ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ?
437
1. Осаждение следует проводить из разбавленных растворов, это позволя-
ет уменьшить величину Q.
2- Разбавленный раствор осадителя следует добавлять медленно, при ин-
тенсивном перемешивании. Это также позволяет поддерживать значение
Q на низком уровне, поскольку препятствует появлению локальных из-
бытков осадителя.
3 - Осаждение следует проводить из горячих растворов. При этом возраста-
ет растворимость S. В то же время растворимость не должна быть слиш-
ком большой, поскольку это может привести к неполному осаждению
(обычно осаждение считается полным, если остаточная концентрация
вещества в растворе не превышает 1 ppm). Иногда основную массу осад-
ка осаждают из горячего раствора, а затем раствор охлаждают для обес-
печения полноты осаждения.
4. Осаждение следует проводить при настолько низких значениях pH, на-
сколько это возможно для достижения полноты осаждения. Как мы виде-
ли, в кислой среде растворимость многих осадков возрастает, а скорость
осаждения, соответственно, уменьшается. Причина увеличения раство-
римости при подкислении — связывание аниона осадка с протонами.
В большинстве случаев это также способствует уменьшению степени за-
грязнения осадков. При разбавлении растворов уменьшается концентрация при-
месей, при нагревании и подкислении увеличивается их растворимость, а при
низкой скорости осаждения снижается вероятность захвата примесей частицами
осадка. Кроме того, чем больше средний размер кристаллов, тем меньше удель-
ная площадь поверхности осадка (т. е. отношение площади поверхности к массе)
и, следовательно, меньше степень адсорбции примесей. Заметим, что осадки,
растворимость которых слишком мала, в большинстве случаев трудно получить
чистыми и в легко фильтрующейся форме. Примером может служить водный
оксид (гидроксид) железа(Ш), образующий студенистый осадок с очень боль-
шой удельной площадью поверхности. Кроме того, осадки, обладающие слиш-
ком малой растворимостью, не очень удобно использовать в гравиметрическом
анализе, так как в этом случае относительное пересыщение всегда очень велико.
Осадитель необходимо добавлять в небольшом избытке для того, чтобы
уменьшить растворимость осадка за счет влияния одноименного иона и тем са-
мым обеспечить полноту осаждения. Но слишком большого избытка осадителя
следует избегать, поскольку это, помимо неоправданного расхода реагентов,
может привести к загрязнению осадка вследствие адсорбции. Если известно
примерное содержание аналита, осадитель обычно добавляют в 10%-м избытке.
Осаждение следует проверить на полноту. Для этого раствору дают отстояться и
затем добавляют к прозрачному раствору над осадком несколько капель раство-
438
ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ И РАВНОВЕСИЕ ОСАДОК-РАСТВОР
ра осадителя. Если осадок больше не образуется, осаждение считается полным.
Не забывайте проверять полноту осаждения!
Старение осадков: кристаллы становятся крупнее и чище
Известно, что очень маленькие частицы осадка с большой удельной площадью
поверхности обладают повышенной поверхностной энергией и большей раство-
римостью по сравнению с крупными кристаллами. Если осадок оставить стоять
под маточным раствором (т. е. раствором, из которого проводили осаждение),
мелкие кристаллы растворяются и оседают на крупных, а крупные, соответст-
венно, растут. Этот процесс называется старением осадка, или оствальдовским
созреванием (рис. 10.1). Оствальдовское созревание улучшает кристаллическую
структуру и увеличивает степень чистоты осадка. В ходе старения отдельные ча-
стицы могут слипаться, сначала образуя общий слой противоионов (см. ниже),
а затем цементируются, образуя между собой прочные связи. В результате об-
щая площадь поверхности может значительно уменьшиться.
Кроме того, в процессе старения уменьшается общее число дефектов крис-
таллической структуры, а адсорбированные или захваченные примеси перехо-
дят в раствор. Старение обычно проводят при повышенных температурах для
ускорения процесса, хотя в ряде случаев можно старить осадки и при комнатной
температуре. В результате старения осадки становятся и лучше фильтруемыми,
и более чистыми.*
Для многих осадков, особенно обладающих очень малой растворимостью,
низкие значения отношения фон Веймарна недостижимы. В этих случаях невоз-
можно получить осадок в кристаллической форме (т. е. в виде относительно не-
большого числа крупных частиц) и сначала образуется коллоидный раствор,
состоящий из большого числа очень мелких частиц.
Рис. 10.1
Оствальдовское созревание
При обсуждении вопросов, связанных со старением осадков, следует различать кристаллические и аморф-
ные осадки. Старение кристаллических осадков в основном протекает по механизму оствальдовского со-
зревания, и этот процесс действительно приводит к увеличению чистоты осадка. Для аморфных осадков,
как правило, преобладает процесс цементации (иначе — химического старения). Но в данном случае это
нежелательно, поскольку в результате захваченные примеси оказываются «намертво» встроенными в
структуру осадка. Поэтому аморфные осадки не рекомендуется старить более 1-2 ч. — Прим, перев.
10.1. КАК УСПЕШНО ПРОВЕСТИ ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ?
439
Первичный
адсорбционный слой
> + = Ад+
- =СГ
Cl-
Ядро коллоидной
частицы AgCI
Рис. 10.2. Схематическое изображение коллоидной частицы хлорида серебра и адсорбционного
слоя в условиях избытка хлорид-ионов
Коллоидные частицы обладают очень малым размером (от 1 до 100 мкм) и
очень большой удельной площадью поверхности и, следовательно, повышенной
склонностью к адсорбции примесей. Рассмотрим механизм коллоидообразова-
ния с общих позиций теории осаждения. В частице любого осадка образующие
его ионы расположены в определенном порядке. Например, на поверхности час-
тицы AgCI ионы Ag+ и С1_ чередуются (рис. 10.2), поэтому первоначально сум-
марный заряд поверхности равен нулю. В то же время поверхность осадка
притягивает те из образующих осадок ионов, которые присутствуют в растворе
в избытке (например, С1" в процессе осаждения хлорид-ионов нитратом сереб-
ра). При этом поверхность приобретает заряд (отметим, что для кристаллических
осадков роль этого явления относительно невелика). В результате адсорбции воз-
никает первичный адсорбционный слой ионов, адсорбированных настолько
сильно, что их можно считать неотъемлемой частью самой кристаллической ча-
стицы. Этот слой притягивает к себе ионы противоположного знака, которые об-
разуют слой противоионов (или вторичный слой); в результате суммарный
заряд частицы снова становится нулевым. Однако между первичным и вторич-
ным слоями, а также непосредственно во вторичный слой могут внедряться моле-
кулы растворителя. Если толщина вторичного слоя невелика, частицы слипаются,
образуя осадок. Этот процесс называется коагуляцией. Но если вторичный
слой достаточно рыхлый, частицы, будучи одноименно заряженными из-за пер-
вичного адсорбционного слоя, отталкиваются друг от друга, и система представ-
ляет собой коллоидный раствор.
В процессе фильтрования скоагулировавшего осадка его частицы продол-
жают удерживать адсорбированные ионы как первичного, так и вторичного
слоя, а также молекулы растворителя. Промывание такого осадка водой приво-
дит к увеличению количества растворителя (воды) между слоями и разрыхле-
440
ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ И РАВНОВЕСИЕ ОСАДОК-РАСТВОР
нию вторичного слоя. В результате частицы осадка вновь переходят в коллоидное
состояние. Этот процесс называется пептизацией; он более подробно обсужда-
ется далее, при рассмотрении операции промывания осадка. Для предотвращения
пептизации осадок промывают раствором электролита, летучего при повышен-
ных температурах. Добавление электролита приводит к сжатию вторичного
слоя и способствует коагуляции. Повышение температуры также способствует
коагуляции, поскольку при этом уменьшается адсорбция и, соответственно, эф-
фективный заряд адсорбционных слоев. Коагуляции способствует и перемеши-
вание раствора.
Если осадок склонен к образованию коллоидных растворов, это всегда вы-
зывает трудности при проведении гравиметрического анализа. Однако в некото-
рых случаях эти трудности особенно велики. Существуют два типа коллоидных
растворов — гидрофильные и гидрофобные. «Гидрофильный» буквально озна-
чает «любящий воду»; коллоиды этого типа обладают повышенным сродством к
воде. Гидрофильные коллоидные растворы обычно очень вязкие. Гидрофобные
коллоиды не обладают высоким сродством к воде, такие коллоидные растворы
называют золями.
Коагуляция гидрофобных коллоидов протекает сравнительно легко с обра-
зованием творожистых осадков. Примером может служить хлорид серебра. Коа-
гуляция гидрофильных коллоидов, таких как водный оксид железа, протекает с
большим трудом. Образующийся студенистый осадок плохо фильтруется, так
как забивает поры фильтра. Кроме того, студенистые осадки легко адсорбируют
примеси благодаря большой площади поверхности. Иногда после фильтрования
осадок необходимо переосадить, т. е. растворить и осадить повторно. При по-
вторном осаждении количество примесей в растворе значительно меньше по
сравнению с первым осаждением, следовательно, уменьшается и степень загряз-
нения осадка.
Несмотря на склонность хлорида серебра к образованию коллоидных рас-
творов, методика гравиметрического определения хлоридов относится к числу
самых точных методик анализа.
Загрязнения осадков
Осадки могут захватывать из раствора частицы посторонних веществ, хорошо
растворимых в обычных условиях. Это явление, называемое соосаждением,
приводит к загрязнению осадка. Причины соосаждения могут быть как кинетиче-
скими, так и термодинамическими. Существует множество видов соосаждения.
1. Окклюзия и инклюзия. Явление окклюзии состоит в захвате частицами
осадка посторонних веществ, не входящих в кристаллическую структуру осад-
ка. Например, кристаллы AgCI в процессе роста могут образовывать «карманы»,
в которые попадает часть раствора. При последующем прокаливании вода испа-
ряется, однако частицы растворенных в ней веществ остаются в осадке, загряз-
няя его. Инклюзией называется встраивание в кристаллическую структуру
осадка отдельных ионов, как правило, того же заряда, что и один из ионов осад-
10.1. КАК УСПЕШНО ПРОВЕСТИ ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ?
441
ка, и близких ему по размерам. Примером может служить изоморфная инклюзия
ионов К4 при осаждении NH4MgPO4. Окклюзия и инклюзия — термодинамиче-
ски невыгодные процессы.*
Окклюдированные или инклюдированные примеси трудно удалить. В неко-
торой степени может помочь старение осадка, но это не всегда оказывается эф-
фективным. Не помогает и промывание осадка. В ряде случаев для удаления
захваченных примесей необходимо растворение осадка и его повторное осаждение.
2. Поверхностная адсорбция. Как было отмечено ранее, на поверхности осадка
существует первичный адсорбционный слой из тех собственных ионов, которые
присутствуют в избытке. Это явление, называемое поверхностной адсорб-
цией, — наиболее распространенная причина загрязнения осадков. Так, при до-
стижении полноты осаждения сульфат-ионов ионами бария в растворе присут-
ствует избыток ионов Ва2+, которые притягиваются поверхностью осадка и
образуют первичный адсорбционный слой. Этот слой притягивает противо-
ионы, т. е. посторонние анионы, например, NO3 (по два на каждый адсорбиро-
ванный ион Ва2+). Таким образом, осадок сульфата бария содержит некоторое
количество нитрата бария.
Поверхностная адсорбция является термодинамически выгодным процес-
сом. Адсорбированные примеси часто можно удалить в ходе промывания — в
том числе посредством замещения на другое вещество, летучее при высоких
температурах. Работа со студенистыми осадками и в данном случае связана с осо-
быми трудностями. Старение позволяет уменьшить общую площадь поверхности
осадка и, следовательно, количество адсорбированных примесей.
3. Изоморфное замещение. Два вещества называются изоморфными, если они
обладают одинаковой стехиометрией, а также одинаковой геометрической фор-
мой кристаллов и близкими размерами кристаллических решеток. В этом случае
один ион может замещать другой в кристаллической структуре, образуя сме-
шанные кристаллы. Это явление называется изоморфным замещением. На-
пример, при осаждении Mg2+ в виде фосфата магния-аммония ион К+, имеющий
почти такой же радиус что и NH 4, может частично замещать последний, образуя
фосфат магния-калия. В тех случаях, когда изоморфное соосаждение возможно,
оно обычно представляет собой очень серьезную проблему, для борьбы с кото-
рой практически нет средств. Поэтому системы, в которых возможно изоморфное
соосаждение, редко используют в анализе. Образование смешанных кристал-
лов — процесс термодинамически выгодный, однако на него могут влиять и кине-
тические факторы.
Приведенная трактовка понятия «окклюзия» и «инклюзия» не общепринята. В отечественной литературе,
наоборот, инклюзией называют механический захват макроскопических объемов раствора, а захват инди-
видуальных ионов называют внутренней адсорбцией. Под окклюзией понимают внутреннюю адсорбцию
и инклюзию, вместе взятые. Изоморфное соосаждение к окклюзии обычно не относят, а рассматривают
как самостоятельный вид соосаждения (как это делает и автор — см. ниже). — Прим, перев.
442
ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ И РАВНОВЕСИЕ ОСДЦОК-РАСТВОР
4- Последующее осаждение. Иногда при стоянии осадка под маточным раство-
ром примесь постепенно образует с избытком осадителя свой собственный осадок.
Это явление называется последующим осаждением. Например, при осаждении
кальция в виде оксалата в присутствии ионов магния оксалат магния выпадает в
осадок не мгновенно, так как он склонен к образованию пересыщенных раство-
ров. Но если оставить раствор перед фильтрованием на слишком долгое время,
оксалат магния выпадет в осадок. Аналогично, в кислой среде в растворе, содер-
жащем ионы Си2+ и Zn2+, сначала образуется осадок сульфида меди, но со време-
нем может образоваться осадок сульфида цинка. Последующее осаждение —
процесс термодинамически выгодный, но замедленный.
Промывание и фильтрование осадка:
будьте осторожны, иначе возможны потери
Соосажденные примеси, особенно адсорбированные на поверхности, часто мож-
но удалить промыванием осадка после его фильтрования. При этом удаляются и
компоненты маточного раствора, которым пропитан осадок. Однако многие
осадки нельзя промывать чистой водой, поскольку при этом возможна пептиза-
ция. Как отмечено выше, это процесс, обратный коагуляции.
Процесс коагуляции, рассмотренный выше, является, по крайней мере, час-
тично обратимым. Как было отмечено, скоагулировавшие частицы характеризу-
ются нейтральным суммарным зарядом первичного и вторичного адсорбционных
слоев. В присутствии постороннего электролита вторичный слой противоионов
сжимается, что способствует коагуляции. Ионы электролита-коагулянта про-
должают удерживаться частицами осадка и после фильтрования. Промывание
такого осадка водой удаляет ионы коагулянта, а слой противоионов, насыщаясь
молекулами воды, снова расширяется. В результате силы отталкивания между
одноименно заряженными частицами опять могут возрасти настолько, что оса-
док частично возвращается в коллоидное состояние и проходит сквозь фильтр.
Чтобы предотвратить это явление, к промывной жидкости следует добавить не-
много индифферентного летучего электролита. Например, осадок AgCl следует
промывать разбавленным раствором HNO3 или NH4NO3 (но, не KNO3, посколь-
ку он нелетуч, — см. ниже).
Используемый электролит должен быть летучим при тех температурах, при
которых впоследствии осадок будут высушивать или прокаливать, и не должен
растворять осадок. Например, для промывания осадка хлорида серебра можно
использовать разбавленную азотную кислоту, которая замещает ионы, адсорби-
рованные на поверхности AgCl, а в ходе высушивания при 110 °C улетучивается.
Для промывания водного оксида железа используют раствор нитрата аммония.
При прокаливании осадка при высоких температурах это вещество полностью
разлагается на NH3, HNO3, N2 и оксиды азота.
При промывании осадка необходимо проверить полноту промывания. Для
этого обычно используют качественную реакцию на один из ионов осадителя,
собрав в пробирку несколько капель фильтрата после нескольких промываний
осадка маленькими порциями промывной жидкости. Например, при определе-
10.2. РАСЧЕТЫ В ГРАВИМЕТРИИ: СКОЛЬКО АНАЛИТА ПРИСУТСТВУЕТ В ПРОБЕ?
443
нии хлорид-ионов осаждением нитратом серебра фильтрат проверяют на при-
сутствие ионов Ag+ действием хлорида натрия или разбавленной НС1.0 технике
фильтрования см. гл. 2.
Высушивание или прокаливание осадка
Если осадок находится в форме, подходящей для взвешивания (т. е. в первую
очередь имеет постоянный состав), то после фильтрования и промывания его до-
статочно высушить, чтобы удалить воду и адсорбированный электролит промыв-
ной жидкости. Высушивание обычно проводят при температурах 110-120 °C в
течение 1-2 ч. Прокаливание, проводимое при существенно более высоких тем-
пературах, обычно необходимо в тех случаях, когда осадок перед взвешиванием
требуется перевести в другую химическую форму. Например, фосфат магния-
аммония MgNH4PO4 при прокаливании до 900 °C превращается в пирофосфат
магния Mg2P2O7. Водный оксид железа Fe2O3 • хН2О прокаливают до безводного
оксида. Осадки многих металлов с органическими осадителями (например, 8-ок-
сихинолином) или сульфид-ионами также прокаливают до оксидов. Техника
прокаливания также описана в гл. 2.
10.2. Расчеты в гравиметрии:
сколько аналита присутствует в пробе?
Взвешиваемый осадок по составу обычно отличается от той формы аналита, со-
держание которой требуется привести в отчете. Общие принципы пересчета
содержания одного вещества в содержание другого вещества при помощи сте-
хиометрических мольных соотношений описаны в разд. 5.8. Важным понятием
является гравиметрический фактор (ГФ), позволяющий непосредственно пере-
считать массу осадка в массу аналита. Гравиметрический фактор равен отно-
шению молярных масс аналита и осадка, умноженному на соответствующее
отношение стехиометрических коэффициентов — числа молей аналита к экви-
валентному числу молей осадка:
ГФ = ^анаДита £ (моль аналита / моль осадка)
^осадка (г/мОЛЬ) Ь
Произведение массы осадка на гравиметрический фактор равно массе ана-
лита. Например, при определении С12 после его превращения в хлорид-ион и
осаждения в виде AgCl масса С12, соответствующая 1 г AgCl, равна
Мс] (г С12 /моль С12)
т ci = -------------------------(моль С12 / моль AgCl) =
с12 AgC1MAgC! (г AgCl/моль AgCl) 2 2 ё
= wAgci • GF(r Cl2 / г AgCl) = wAgC1 • 0,24737 (г С12 / г AgCl)
444
ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ И РАВНОВЕСИЕ ОСАДОК-РАСТВОР
Пример 10.1
Рассчитайте массу аналита, соответствующую 1 г осадка, для следующих случаев:
Аналит
Осадок
Р
К2НРО4
Bi2S3
Ag3PO4
Ag3PO4
BaSO4
Решение
, М? (г[моль) ! z х
WP /т Ag3PO4 = ~Т7~~--' 1 (МОЛЬ р z МОЛЬ АёзРО4) =
^Ag3PO4 (Г/МОЛЬ)
=-----30,97(гР/мбль)----। = 0 99 г р t г А р0 = Гф
418,58 (г Ag3PO4/MOflb) 1 3
. AfK HPO (г/моль)
т к2нро4 /т Ag3po4 = -гг1—-----г • т (моль К2НРО4 / моль Ag3PO4) =
AfAg3pO4 (г/моль)
= 174)18 (г К2НР°4/моль). 1 = 0 4i6J2 г к2НРО4/г Ag3PO4 = ГФ
418,58 (г Ag3PO4/Monb) 1 3
AfBi s (г/моль)
rn Bi 2S3 /"I BaSO4 = 2 3 z ,-7 • 4 = (моль Bi2s3 / моль BaSO4) =
AfBaSO4 (г/моль)
= 514’15 (г-?125.з./моль)_ . X = 0 73429 r Bi s / r BaS0 = ГФ
233,40 (г BaSO4/моль) 3
В гравиметрическом анализе чаще всего требуется найти массовую долю
аналита <о в пробе:
zn. x масса искомого вещества (г) . .
о (%) =----------------------— • 100%
масса навески (г)
(Ю-2)
Массу искомого вещества рассчитывают по уравнению (10.1). Таким образом
z \ _ z \ ^искомого вещества (г/мОЛЬ)
^искомого вещества ™осадка ‘ z ч
^осадка (Г/МОЛЬ)
х — (моль искомого вещества/моль осадка) =
b
= тиосадка (г) • ГФ (г искомого вещества/г осадка)
(10-3)
10.2. РАСЧЕТЫ В ГРАВИМЕТРИИ: СКОЛЬКО АНАЛИТА ПРИСУТСТВУЕТ В ПРОБЕ?
445
Содержание искомого вещества А обычно выражают в процентах:
оА,% = —• 100%
навески
(Ю.4)
Таким образом, можно написать общую формулу для расчета процентного со-
держания искомого вещества (в ходе расчетов проверяйте единицы измерения!):
^искомого вещества’
^осадка (г) ’ ГФ (г искомого вещества/г осадка)
тп
'"навески
(г)
•100%
(Ю.5)
Пример 10.2
Ортофосфат-ион (РО4“) определяют взвешиванием молибдофосфата аммония
(NH4)3PO4 • 12МоО3. Рассчитайте массовую долю (%) Р и Р2О5 в пробе, если из
навески массой 0,2711 г получено 1,1682 г осадка. Выполните расчеты содержания
фосфора, используя как гравиметрический фактор, так и анализ размерностей.
Решение
тп •-----------------------
^^н4)3ро412МОо3 = 1,1682 г-(30,97/1876,5) 10()% = 7 jjр/о
^навески 0,2711 Г
®Р2О5
^Р2О5
тп •---------------—------
Af(NH4)3PQ4.i2MoO3 100о/о= 1,1682 г [141,95/(2-1876,5)] 1()()% = 1630%
^навески 0,2711 Г
Повторим расчет содержания Р, используя анализ размерностей:
1,982тЦЖ4^зРе4-12МбО'3 (30,97/1876,5) гР/г^Н4)3 Р04-^1МоОу _
0,2711г навески
= 7,111 (г Р/г навески) - 100% =7,111%
Единицы массы (NH4)3PO4 • 12МоО3 сокращаются, и в числителе остается толь-
ко единицы массы (г) Р.
Рассмотренный способ расчета принципиально ничем не отличается от рас-
чета с использованием гравиметрического фактора. Однако этот способ нагляд-
нее: в данном случае ясно видно, какие единицы измерения сокращаются.
446
ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ И РАВНОВЕСИЕ ОСАДОК-РАСТВОР
Пример 10.3
Содержание марганца в руде определяли путем превращения марганца в оксид
Мп3О4 и его взвешивания. Из навески пробы массой 1,52 г получено 0,126 г
Мп3О4. Каково содержание марганца в руде, выраженное в виде массовой доли
(%) Мп2О3? В виде массовой доли Мп?
Решение
З^Мп2О3
™Мп3О4—---------
со Мп о =------------•1 °0% = °’126г'[3'157’9/(2'228’8)]. 100% = 8,58%
ivin2u3 л ’
717 1 52 г
1,1 навески
шм„ -------------- 100% - °.126г'(3 М94/228.8) _ 5 %
т 152 г
навески 1,-^1
Следующие два примера иллюстрируют некоторые другие виды расчетов в
гравиметрическом анализе.
Пример 10.4
Какова должна быть масса навески пирита (руда, содержащая FeS2 и примеси),
взятой для анализа, чтобы масса (г) осадка BaSO4 равнялась половине массовой
доли (%) серы в образце?
Решение
Пусть руда содержит А% серы. Тогда масса BaSO4 составит j А г. Отсюда полу-
чаем:
cos, % = А =-----• 100%
™навески
«навески = 5 ' ’ 100% = 6>869 г
навески 2 233,40
10.2. РАСЧЕТЫ В ГРАВИМЕТРИИ: СКОЛЬКО АНАЛИТА ПРИСУТСТВУЕТ В ПРОБЕ?
447
Осаждение смесей: необходимо два значения массы
Пример 10.5
Масса смеси, состоящей только из FeCl3 и А1С13, составляет 5,95 г. Смесь раство-
рили, осадили металлы в виде гидроксидов и прокалили осадок до Fe2O3 и А12О3.
Масса прокаленного осадка составила 2,62 г. Рассчитайте массовые доли (%) Fe
и Al в исходной смеси.
Решение
В данном случае требуется найти две неизвестные величины, поэтому необхо-
димо составить систему из двух уравнений. Пусть масса Fe (г) равна х, а масса
Al-у. Тогда:
"lFeCl3 + wAlCl3 =5>95 (1)
^FeClj ^А1С13 , n, x + у L = 5 95 MFe MA1 (2)
162,21 133,34 55,85 26,98 (3)
2,90x + 4,94y = 5,95 (4)
'«Fe2p3 + '«A1A =2’62 (5)
^FejOj WA12O3 - _ x — 4- у — = 2,62 2MFe 2A/a1 (6)
159,69 101,96 2-55,85 2-26,98 (7)
l,43x+ 1,89^ = 2,62 (8)
Решив систему из уравнений (4) и (8), находим: х = 1,07 г,у = 0,58 г. Отсюда
®Fe = • Ю0% = 18,0%
Fe 5,95 г
®а| = ^^-100% = 9,8%
5,95 г
448
ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ И РАВНОВЕСИЕ ОСАДОК-РАСТВОР
10.3. Примеры гравиметрических определений
Как уже отмечалось, многие гравиметрические методики относятся к самым
точным методикам химического анализа. С некоторыми из них следует познако-
миться поближе. В табл. 10.1 приведены различные элементы, определяемые
гравиметрическим методом, осаждаемые и гравиметрические формы, а также
мешающие компоненты, которые должны отсутствовать при проведении анализа.
Подробнее о гравиметрическом определении элементов при помощи этих и дру-
гих методик можно узнать в специальной литературе.
Таблица 10.1
Примеры распространенных гравиметрических методик
Определяемый элемент Осаждаемая форма Гравиметрическая форма Мешающие компоненты
Fe Fe(OH)3 Fe2O3 Многие элементы: Al, Ti, Cr и др.
купферонат Fe Fe2O3 Четырехзарядные катионы
Al A1(OH)3 ai2o3 Многие элементы: Fe, Ti, Сг и др.
Al(ox)? Al(ox)3 Многие элементы. Mg в кислой
среде не мешает
Са CaC2O4 CaCO3 или CaO Все металлы, кроме щелочных и Mg
Mg MgNH4PO4 Mg2P2O7 Все металлы, кроме щелочных
Zn ZnNH4PO4 Zn2P2O7 Все металлы, кроме Mg
Ba BaCrO4 BaCrO4 Pb
SO^- BaSO4 BaSO4 NO3, РО^, СЮз
Cl- AgCl AgCl Br, Г, SCN-, CN~, S2-, S2O^’
Ag AgCl AgCl Hg(I)
po^- MgNH4PO4 Mg2p2°7 МоО^, С2О^, К+
Ni Ni(dmg)2 Ni(dmg)2 Pd
аох — анион 8-оксихинолина (однозарядный)
Mmg — анион диметилглиоксима (однозарядный)
10.4. Органические осадители
Все осадители, которые мы упоминали до сих пор (табл. 10.1, кроме 8-оксихино-
лина, купферона и диметилглиоксима), представляли собой неорганические ве-
щества. В то же время существует множество органических осадителей, широко
10.5. РАВНОВЕСИЕ ОСДЦОК-РАСТВОР: ПРОИЗВВДЕНИЕ РАСТВОРИМОСТИ
449
используемых для осаждения металлов. Некоторые из них весьма селективны, а
некоторые, наоборот, осаждают широкий круг элементов.
Достоинством многих органических осадителей является очень низкая рас-
творимость образуемых ими осадков и малая величина гравиметрического фак-
тора. Большинство таких осадителей представляет собой хелатообразующие
реагенты, которые образуют с ионами металлов малорастворимые незаряжен-
ные хелаты. Напоминаем, что хелатообразующим называется лиганд, содержа-
щий две или более функциональных групп, которые одновременно связываются
с ионом металла. Образующийся при этом комплекс называется хелатом. Более
подробно хелатные комплексы рассмотрены в гл. 9.
Большинство хелатообразующих реагентов представляет собой слабые кис-
лоты, способные образовывать комплексы с целым рядом ионов. В этих случаях
селективность обеспечивают, как правило, регулированием pH. В общем виде
реакцию комплексообразования с такими реагентами можно записать так
М”+ + иНХ^МХ„ +иН+
Молекула реагента может содержать и более одного протона, способного к от-
щеплению. Чем менее устойчив хелат, тем более высокое значение pH необхо-
димо для осаждения. Некоторые часто используемые органические осадители
перечислены в табл. 10.2. В ряде случаев образуемые осадки не имеют стехио-
метрического состава, поэтому для обеспечения точности результатов их прокали-
вают до оксидов металлов. Некоторые осадители, например, диэтилдитиокарбамат
натрия, можно использовать для групповых разделений, подобно сероводороду.
Более подробно об использовании перечисленных и других органических оса-
дителей можно узнать в специальной литературе. В частности, существует много-
томное руководство Холлингсхеда, содержащее много полезной и разнообразной
информации об использовании в химическом анализе 8-оксихинолина и его про-
изводных.
10.5. Равновесие осадок-раствор: произведение растворимости
Когда концентрация малорастворимого вещества в растворе превышает его рас-
творимость, вещество выпадает в осадок и устанавливается равновесие между
твердой фазой и составляющими ее ионами, находящимися в растворе. Таким
образом, так называемые «нерастворимые» вещества на самом деле являются не
абсолютно нерастворимыми, а малорастворимыми. Например, если твердый
хлорид серебра поместить в воду, то его небольшое количество растворяется в
соответствии с уравнением
AgCl^(AgCl)BnnH^Ag+ + Cl- (10.6)
Любой осадок при данной температуре характеризуется вполне определенной,
пусть и малой, величиной растворимости (т. е. количеством растворенного ве-
щества в единице объема насыщенного раствора), которую можно выразить
450
ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ И РАВНОВЕСИЕ ОСАДОК-РАСТВОР
Таблица 10.2
Некоторые органические осадители
Реагент Формула Осаждаемые ионы
Диметилглиоксим СН3 —C = NOH СН3 —C = NOH Ni(II) в аммиачном или ацетатном буфере; Pd(II) в среде НС1 М2+ + 2HR -> MR2 + 2Н*
а-Бензоиноксим (купрон) ОН NOH УК 1 II /к Cu(II) в аммиачно-тартратной среде; Mo(VI) и W(VI) в кислой среде (М2++ H2R —> MR + 2Н+; М2+ = Си2+, МоО2+, wol+) Прокаливание до оксидов и взвешивание
Нитрозофенилгидроксиламинат аммония (купферон) N=O y%-N-O-NH4 Fe(III), V(V), Ti(IV), Zr(IV), Sn(IV), U(IV) (M"+ + hNH4R -> MR„ + hNHJ) Прокаливание до оксидов и взвешивание
8-Оксихинолин (оксин) ОН с» Ионы многих металлов. Часто используют для А1(Ш) и Mg(II) (М^ + hHR MR„ + иН+)
Диэтилдитиокарбамат натрия S (C2H5)2N — С — S“Na+ Ионы многих металлов из кислых сред (М"+ + zzNaR MR„ + nNa+)
Тетрафенилборат натрия NaB(C6H5)4 К+, Rb+, Cs+, Tl+, Ag+, Hg(I), Cu(I), NH4, RNH3, R2NH2+, r3nh+, r4n+ из кислых сред (M++NaR —> MR+Na+)
Хлорид тетрафениларсония (C6H5)4AsC1 Cr2O2-, МпОд, ReO;, MOO4-, WO^, CIO4,13 из кислых сред (A"_ + hRCI -> R„A + иС1~)
в г/л, моль/л или любых других единицах концентрации. Обычно в водных рас-
творах в равновесии с твердой фазой находится и небольшое количество (как
правило, порядка 0,1%) вещества в недиссоциированной форме; концентрация
этой формы при данной температуре является постоянной величиной. Однако
эту величину бывает трудно оценить. Нас будет интересовать главным образом
концентрация вещества в ионизированной форме как мера его растворимости и
химической реакционной способности. В дальнейшем наличием каких-либо не-
диссоциированных форм в насыщенном растворе мы будем пренебрегать.
Для равновесия между осадком и составляющими его ионами можно запи-
сать выражение общей константы, которую можно представить как произведе-
ние констант двух последовательных равновесий с участием растворенной
10.5. РАВНОВЕСИЕ ОСАДОК-РАСТВОР: ПРОИЗВВДЕНИЕ РАСТВОРИМОСТИ
451
молекулярной формы. При перемножении ее концентрация (в рассматриваемом
примере — [AgCl]BOflH) сокращается, и выражение константы, называемой
произведением растворимости (Ks), приобретает следующий вид:
Ks = [Ag+][Cl_] (10.7)
Концентрация любого твердого вещества (в данном примере — AgCl) — это по-
стоянная величина. Ее включают в величину Ks (а не в выражение константы
равновесия!). Произведение концентраций ионов, составляющих осадок, сохра-
няет постоянное значение независимо от наличия в растворе каких-либо недис-
социированных форм. Повторим, что в качестве меры растворимости осадка мы
будем использовать концентрацию составляющих его ионов. Растворимость
можно рассчитать из величины произведения растворимости вещества при дан-
ной температуре (а на практике, наоборот, произведения растворимости рассчи-
тывают из экспериментальных данных по растворимости).
Количество малорастворимого вещества в данном объеме насыщенного
раствора не зависит от количества твердой фазы, находящейся в равновесии с
раствором (если этого количества достаточно для насыщения раствора). От объ-
ема же раствора количество растворенного вещества, напротив, зависит. Концен-
трация растворенного вещества в условиях равновесия в насыщенном растворе не
зависит от его объема, будь то объем лабораторного стакана или бассейна. Но
количество вещества, растворенного в объеме бассейна, намного больше!
Для соединений со стехиометрией, отличной от 1 : 1, например, Ag2CrO4,
выражение Ks записывается следующим образом:
Ag2CrO4^2Ag+ + CrO^ (10.8)
Ks= [Ag+]2[CrO^] (10.9)
Такого рода электролиты являются сильными и диссоциируют полностью. Поэ-
тому «ступенчатые» произведения растворимости не используются. Как и для
любой константы равновесия, величина Ks в равновесных условиях при посто-
янной температуре сохраняет постоянство. Однако для гетерогенных процессов
равновесие устанавливается более медленно, чем для гомогенных реакций в рас-
творах.
Насыщенный раствор
Пример 10.6
Для AgCl величина^ при 25 °C составляет 1,0 • 1О-10. Рассчитайте концентра-
ции Ag+ и С1~ в насыщенном растворе AgCl и растворимость AgCl (моль/л).
Решение
При растворении AgCl в соответствии с уравнением AgCl *=? Ag+ + С1“ в раствор
переходят равные количества Ag+ и С1_. Их концентрации связаны соотношени-
452
ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ И РАВНОВЕСИЕ ОСАДОК-РАСТВОР
ем Ks = [Ag+][C1-]. Обозначим растворимость AgCI (выраженную в моль/л) через S.
Поскольку из одного моля AgCI образуется по одному молю Ag+ и С1~, то
[Ag+] = [С1-] = 5
S2 = 1,0 • ю-10
5=1,0- 10-5М
Растворимость AgCI составляет 1,0 • 10~5 М.
Снижение растворимости: влияние одноименного иона
Если один из ионов, составляющих осадок, присутствует в избытке, концентра-
ция другого иона соответственно уменьшается. Следовательно, уменьшается
растворимость осадка. В этом проявляется влияние одноименного иона. Кон-
центрацию другого иона можно рассчитать из величины произведения раство-
римости.
Пример 10.7
К 10 мл 0,10 М NaCl добавили 10 мл 0,20 М AgNO3. Рассчитайте остаточную
концентрацию С1~ в растворе после установления равновесия, а также раствори-
мость AgCI в этих условиях.
Решение
Конечный объем раствора составляет 20 мл. Общее количество добавленного Ag+
равно 0,20 • 10 = 2,0 ммоль. Исходное количество С1“ равно 0,10 • 10 = 1,0 ммоль.
Из результата, полученного в примере 10.6, видно, что вклад в концентрацию
Ag+ за счет растворимости осадка невелик — порядка 10-5 ммоль/мл в отсутст-
вие избытка однименного иона. В присутствии избытка Ag+ вклад будет еще мень-
ше, так как растворимость AgCI при этом подавляется. Добавление одноименного
иона уменьшает растворимость. Поэтому количеством Ag+, перешедшим в
раствор вследствие растворения осадка, можно пренебречь по сравнению с вне-
сенным избытком Ag+. Следовательно, конечная концентрация Ag+ в растворе
составит 1,0 ммоль/20 мл = 0,050 М и
0,050 • [С1-] = 1,0 • IO"10
[С1-] = 2,0 • 10-9 М
Растворимость AgCI и в этом случае равна равновесной концентрации С1_ и со-
ставляет 2 • 10~9 М.
10.5. РАВНОВЕСИЕ ОСАДОК-РАСТВОР: ПРОИЗВВДЕНИЕ РАСТВОРИМОСТИ
453
В условиях равновесия между осадком и раствором произведение раствори-
мости всегда сохраняет постоянное значение. Следовательно, если произведение
концентраций ионов (в данном случае Ag+ и С1_) меньше или равно величине Ks,
образования осадка не происходит, В этом случае все ионы остаются в расвторе.
Для образования осадка необходимо, чтобы произведение концентраций ионов
превысило произведение растворимости.
Зависимость растворимости от стехиометрии осадка
В табл. 10.3 приведены величины произведений растворимости некоторых ма-
лорастворимых веществ, а также рассчитанные из них величины растворимости
(моль/л).* Величина растворимости не всегда прямо пропорциональна Ks, по-
скольку также зависит от стехиометрии осадка. Так, для Agl значение Ks в 5 • 1015
раз выше чем для А1(ОН)3, но рассчитанная растворимость Agl превышает эту
величину для А1(ОН)3 всего лишь вдвое. В общем случае при одинаковых значе-
ниях^ осадок состава 1 : 1 всегда обладает меньшей растворимостью, чем оса-
док другой стехиометрии. Обратите внимание на сульфид ртути, для которого
произведение растворимости составляет лишь 4 • 10“53, а рассчитанное значение
растворимости — 6 • 10-27 М! При такой концентрации средний объем раствора,
в котором находилось бы по одному иону Hg2+ и S2-, составил бы около 280 л
(попробуйте самостоятельно рассчитать это значение при помощи числа Аво-
гадро). Более полный перечень величин произведений растворимости приведен
в Приложении С.
Таблица 10.3
Произведения растворимости некоторых малорастворимых веществ
Соединение Ks Растворимость S (моль/л)
PbSO4 1,6- 10-8 1,3 • 1(H
AgCI 1,0- 10-6 1,0 • 10-5
AgBr 4 • 10-13 6 • 10~7
Agl 1 • IO"16 1 • 10-8
A1(OH)3 2 • IO’32 5 • IO’9
Fe(OH)3 4 • IO"38 2 • 10-'°
Ag2S 2 • 1(H9 4 • IO’17
HgS 4 • 10~53 6 • io-27
Приведенные в табл. 10.3 величины растворимости являются идеализированными, поскольку при их рас-
чете не учтены ни возможные побочные процессы (протонирование анионов в случае Ag2S или HgS, обра-
зование гидроксокомплексов катионов в случае А1(ОН)3, Fe(OH)3, HgS), ни сопутствующие равновесия с
участием одноименных ионов (диссоциация воды в случае А1(ОН)3 или Fe(OH)3). С учетом этих явлений
величины растворимостей в ряде случаев оказываются на много порядков выше приведенных в таблице
значений. — Прим, перев.
454
ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ И РАВНОВЕСИЕ ОСАДОК-РАСТВОР
Пример 10.8
Какую концентрацию Ag + необходимо создать в 1,0 • 10"3 М растворе NaCl, что-
бы начал выпадать осадок AgCl?
Решение
[Ag+]l,0- IO"3 = 1,0 • IO'10
[Ag+] = 1,0 • IO"7 М
Таким образом, для начала образования осадка необходимо, чтобы концентра-
ция Ag+ превысила 1 • 10"7 М.
Пример 10.9
Произведение растворимости РЫ2 составляет 7,1 - 10"9. Рассчитайте раствори-
мость РЫ2 (г/л) в чистой воде.
Решение
Равновесие осадок-раствор устанавливается в соответствии с уравнением
РЫ2 ±=? РЬ2+ + 21", для которого Ks = [Pb2+][I~]2 = 7,1 • 10"9. Обозначим раствори-
мость РЫ2 (моль/л) через 5. Тогда
[РЬ2+] = 5 и [1"] = 25
S • (2S)2 = 7,1 • 10"9
5 = з|7’Ь1° 9.=1,2-1(Г3 М
V 4
В единицах г/л растворимость составляет
1,2 • 10-3 моль/л -461,0 г/моль = 0,55 г/л
Обратите внимание, что в ходе вычислений не следует умножать концентрацию
I" на два: величина этой концентрации уже составляет 2S. Можно выполнить
расчет иначе, обозначив через S концентрацию I". Тогда величина [РЬ2+], а также
растворимость РЬ12 будут равны ±S. В этом случае рассчитанное значение S бу-
дет вдвое больше полученного предыдущим способом, но концентрации всех
частиц останутся такими же. Попробуйте убедиться в этом самостоятельно.
10.5. РАВНОВЕСИЕ ОСАДОК-РАСТВОР: ПРОИЗВВДЕНИЕ РАСТВОРИМОСТИ
455
Пример 10.10
Рассчитайте растворимость PbSO4 (моль/л) и сравните ее с растворимостью РЫ2.
Решение
PbSO4 РЬ2+ + SO^
[Pb2+][SO = 1,6 • 10-8
S-5=l,6- 10-8
S = 1,3 • 1(HM
Хотя величина Ks для РЫ2 меньше чем для PbSO4, растворимость Pbl2 выше
(см. пример 10.9) ввиду иной стехиометрии этого соединения. Если величина Ks
для осадка со стехиометрией, отличной от 1 : 1, меньше чем для осадка стехиомет-
рии 1:1, это не обязательно означает, что растворимость первого осадка меньше.
Для электролитов одинаковой стехиометрии значения растворимости рас-
положены в том же порядке, что и величины их произведений растворимости.
Однако при сопоставлении соединений разной стехиометрии этот порядок мо-
жет нарушиться. При равных значениях Ks вещество состава АВ имеет мень-
шую растворимость, чем вещество состава АС2.
Уменьшение растворимости осадка в присутствии одноименного иона исполь-
зуют в гравиметрическом анализе. Для примера рассмотрим определение суль-
фат-ионов в виде BaSO4, осаждающегося при добавлении раствора хлорида бария.
На рис. 10.3 показано влияние избытка ионов Ва2+ на растворимость BaSO4.
Пример 10.11
При каком значении pH начнется осаждение гидроксида железа(Ш) из 0,10 М
раствора FeCl3?
Решение
Fe(OH)3 Fe3+ + ЗОН~
[Fe3+][OH-]3 = 4 • IO-38
0,1 [ОН-]3 = 4- Ю-38
4-10-38
[ОН-] = з —— = 7 • 10-13 М
V 0,1
pOH = -lg (7 • 10-13) = 12,2
pH = 14,0-12,2 = 1,8
456
ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ И РАВНОВЕСИЕ ОСАДОК-РАСТВОР
Рис. 10.3
Теоретическая зависимость
растворимости BaSO4
от избыточной концентрации
Ва2+. Растворимость BaSO4
рассчитана как равновесная
концентрация SO4“
в растворе над осадком.
В отсутствие избытка SO4“
растворимость составляет
1 • IO’5 М
[Ва2+] , 104 М
Таким образом, вследствие очень малого значения Ks гидроксид железа начина-
ет осаждаться в очень кислой среде — при pH 1,8! Если попытаться растворить
FeCl3 в воде, это вещество будет медленно гидролизоваться с образованием гид-
роксида (водного оксида) железа(Ш) - студенистого ржаво-бурого осадка. Поэто-
му растворы соединений железа(Ш) необходимо стабилизировать подкислением,
например, добавляя соляную кислоту.
Пример 10.12
К 25,0 мл 0,100 М раствора AgNO3 добавили 35,0 мл 0,0500 М раствора К2СгО4.
а) Рассчитайте концентрацию каждого иона в условиях равновесия, б) Будет ли
осаждение серебра количественным? Считайте осаждение количественным, если
в осадок перешло не менее 99,9% серебра.
Решение
а) Уравнение реакции имеет вид
2Ag+ + СгО4" Ag2CrO4
Исходные количества реагирующих веществ составляют
25,0 мл 0,100 ммоль/мл = 2,50 ммоль AgNO3
35,0 мл • 0,0500 ммоль/мл = 1,75 ммоль К2СгО4
10.6. ВЛИЯНИЕ ПОСТОРОННИХ ИОНОВ НА РАСТВОРИМОСТЬ
457
С 2,50 ммоль Ag+ взаимодействуют 1,25 ммоль СгО | , т. е. в растворе оста-
нется избыток СгОд", равный 0,50 ммоль. Конечный объем раствора со-
ставляет 60,0 мл. Обозначим растворимость Ag2CrO4 (моль/л) через S.
В условиях равновесия
[СгО^] = 0,50/60,0 + S = 0,0083 + S « 0,0083 М
Ввиду того, что СгО4- находится в избытке, величиной S можно прене-
бречь по сравнению с 0,0083.
[Ag+] = 25
[К+] = 3,50/60,0 = 0,0583 М
[NO3] = 2,50/60,0 = 0,0417 М
[Ag+]2[CrO^] = 1,1- 1012
(2S)2 8,3 • 10-з = 1,1 • 10-12
5= I 1,140 12 = 5,8-КГ6
V 4 -8,3 -10-3
[Ag+] = 2(5,8 • 10-6) = 1,16 • IO-5 М
б) Доля осажденного серебра составляет
2,50 ммоль - 60 мл • 1,16 • 10~5 ммоль/мл j ° =
2,50 ммоль
Можно поступить иначе и рассчитать долю серебра, остающегося в растворе:
60,0 мл 1,1, 10~5 ммоль/мл 100% _ f) 02g%
2,50 ммоль
Таким образом, осаждение можно считать количественным.
10.6. Влияние посторонних ионов на растворимость:
термодинамическое произведение растворимости
и коэффициенты активности
В гл. 6 мы ввели понятие термодинамической константы равновесия, в выраже-
нии которой вместо концентраций используют активности с целью учета влияния
инертных электролитов на положение равновесия. В общем случае в присутствии
индифферентных электролитов растворимость осадка возрастает вследствие эк-
458
ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ И РАВНОВЕСИЕ ОСАДОК-РАСТВОР
ранирования составляющих его ионов (активность которых при этом уменьша-
ется). Для AgCl выражение термодинамического произведения растворимости
записывается в виде
K°S = • аа- = [А§+]4g+ tcr 1/сг (10.10)
Реальное произведение растворимости Ks, как мы знаем, равно [Ag+][C1~], Отсюда
s J Ag+y СГ
(10.11)
или
ks =———
f^fcr
(10.12)
Величина Ks остается постоянной при любом значении ионной силы. Ks равно
K °s при нулевой ионной силе, но при значениях ионной силы, заметно отличаю-
щихся от нуля, величины Ks необходимо рассчитывать по уравнению (10.12)
для каждого значения ионной силы. Обратите внимание, что из уравнения
(10.12) следует тот же вывод, который мы сделали ранее на качественном уров-
не: с уменьшением активности ионов величина Ks, а следовательно, и раствори-
мость, возрастают.
Пример 10.13
Рассчитайте растворимость хлорида серебра в 0,10 М NaNO3.
Решение
Величины констант равновесий, приведенные в Приложении С, соответствуют ну-
левому значению ионной силы и являются, таким образом, термодинамическими
константами.* Из табл. С.З находим, что для AgCl величина^ равна 1,0 • Ю-10.
Для расчета необходимы значения коэффициентов активности Ag+ и С1_.
Величина ионной силы составляет 0,10 моль/л. Из данных работы [9] (список ли-
тературы к гл. 6) находим, что /Ag+ = 0,75 и /сг = 0,76 [можно также использо-
вать приведенные в работе [9] величины параметра а для Ag+ и С1~ и рассчитать
коэффициенты активности по уравнению (6.19)]. Из уравнения (10.12) получаем:
В то же время для многих веществ известны экспериментальные величины Ks для различной ионной силы.
Эти величины можно непосредстванно использовать для расчетов растворимости при соответствующем
значении ионной силы, не вычисляя коэффициентов активности.
10.6. ВЛИЯНИЕ ПОСТОРОННИХ ИОНОВ НА РАСТВОРИМОСТЬ
459
Ks =
1,0-1 о-10
0,75-0,76
= 1,8 - 10-10 = [Ag+][Cl-] = 52
S = -Ю’10 = U • Ю~5 М
Эта величина на 30% выше, чем для нулевого значения ионной силы (напоми-
наем, что тогда S = 1,0 • 10-5 М). Таким образом, присутствие постороннего
электролита увеличивает растворимость осадка, особенно если он образован
многозарядными ионами.
Увеличение растворимости BaSO4 в присутствии NaNO3 вследствие влия-
ния посторонних ионов (солевой эффект) иллюстрирует рис. 10.4. Этот эффект
выражен особенно сильно, если осадок состоит из многозарядных ионов. При
очень высоких значениях ионной силы, когда коэффициенты активности могут
быть больше единицы, растворимость может, наоборот, уменьшаться. В ходе
гравиметрического анализа обычно добавляют достаточно большой избыток
осадителя. При этом влияние избытка осадителя, приводящее к уменьшению
растворимости осадка, проявляется значительно сильнее, чем влияние солевого
эффекта, приводящее к увеличению растворимости. Поэтому на практике соле-
вой эффект обычно не представляет опасности.
На растворимость осадка часто влияет и кислотность среды. Ионы Н+ с рос-
том их концентрации все более эффективно конкурируют с ионами металла за
связывание анионов осадителя (если он представляет собой анион слабой кисло-
Рис. 10.4
Теоретическая зависимость
растворимости BaSO4
от ионной силы раствора.
При нулевой ионной силе
растворимость составляет
1,0- 10-5М
460
ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ И РАВНОВЕСИЕ ОСАДОК-РАСТВОР
ты). При этом концентрация свободного аниона осадителя уменьшается, а рас-
творимость осадка—возрастает (поскольку величина/^ остается постоянной):
Мл+ + nRr MR„ (желаемый процесс)
R- + Н+ HR (конкурирующий процесс)
MR„ + иН+ Mw+ + «HR (общее уравнение реакции)
Аналогично, растворимость увеличивается при введении комплексообразу-
ющего реагента, связывающего ионы металла осадка, например, NH3 при осаж-
дении хлорида серебра:
AgCl + 2NH3 Ag(NH3)2 + Cl-
Количественное описание влияния этих факторов на растворимость рассмотре-
но в гл. И.
Упражнения с электронными таблицами
Для определения железа в руде из навески массой 2,287 г осадили Fe(OH)3, прока-
лили осадок до Fe2O3 и взвесили. Масса прокаленного осадка составила 0,8792 г.
Создайте электронную таблицу для расчета массовой доли Fe (%) в руде.
А В С D Е F G Н
1 Определение массовой доли Fe (%).
2 Масса образца, г 2,287 Масса Fe2O3, г: 0,8792
3 %Fe: 26,88797
4
5 %Fe = {[г Fe2O3 2Fe/Fe2O3 (г Fe/г Fe2O3)]/r образца] • 100%
6 = {[0,8792 г Fe2O3 • 2(55,845/159,69)г Fe/g Fe2O3]/2,287 г образца} 100%
7 В3 = (D2*2*(55,845/159,69)/В2)*100
8
9 | Ответ: 26,89% Fe.
Произведение растворимости AgCl составляет 1,0 • 1О-10. Рассчитайте раствори-
мость AgCl при помощи программы Excel Solver (Поиск решения).
А I в I с D I Е F
1 Определение растворимости AgCl с помощью Поиска решения.
2 Ks: 1.00Е-10 Формула = S2/Ks = 1-
3
4 s = 1Е-05 Формула: 1,000001
5
6 Формула в ячейке Е4: = С4Л2/В2
7 Поиск решения:
8 Е4 = Целевая ячейка
9 Установить значение 1
10 Изменяя ячейку = С4
11 Ответ: 1,0 10-5 М. I
ЗАДАЧИ
461
Что мы узнали из этой главы?
• Стадии гравиметрического анализа: осаждение, старение осадка, фильтро-
вание, промывание осадка, термообработка, взвешивание, расчеты —
стр. 433
• Расчеты в гравиметрии [основные уравнения (10.1), (10.3), (10.5)] —
стр. 443
• Произведение растворимости, влияние одноименного иона — стр. 449
• Влияние посторонних ионов на растворимость [основное уравнение
(10.10)] —стр. 457
Вопросы
1 - Опишите стандартные операции, выполняемые в ходе гравиметрического
анализа, и кратко охарактеризуйте цель каждой из них.
2. Что такое отношение фон Веймарна? Поясните смысл каждой величины,
входящей в это выражение.
3. Какую информацию относительно оптимальных условий осаждения мож-
но извлечь из формулы фон Веймарна?
4. Что такое старение осадка и для чего оно необходимо?
5- Опишите оптимальные условия осаждения, позволяющие получить чис-
тый и легко фильтрующийся осадок.
6- Что такое соосаждение? Перечислите виды соосаждения. Как можно пред-
отвратить или устранить последствия соосаждения?
7- Почему необходимо промывать осадок после фильтрования?
8. Для чего в промывную жидкость добавляют электролит? Каковы требова-
ния к этому электролиту?
9« Назовите основные преимущества органических осадителей перед неорга-
ническими.
Задачи
Гоавиметрический фактор
10- Рассчитайте массу натрия, содержащегося в 50,0 г Na^C^.
11 - Если для анализа 50,0 г использовать осаждение и взвешивание
BaSO4, какова будет масса осадка?
462
ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ И РАВНОВЕСИЕ ОСАДОК-РАСТВОР
12 - Рассчитайте значение гравиметрического фактора для следующих случаев:
Определяемое вещество Гравиметрическая форма
As2O3 Ag3AsO4
FeSO4 Fe2O3
К2О KB(C6H5)4
SiO2 KAlSi3O8
13. Какова масса оксида меди СиО, который можно получить из 1,00 г париж-
ской зелени Cu3(AsO3)2 • 2As2O3 • Cu(C2H3O2)2? Какова масса As2O3?
Расчеты в гравиметрии
14. Навеску содержащего примеси образца КВг массой 523,1 мг обработали
избытком AgNO3 и получили 814,5 мг AgBr. Какова степень чистоты об-
разца?
15- Какова масса Fe2O3, полученного из 0,4823 г железной проволоки с чисто-
той 99,89%?
16- Содержание алюминия в сплаве определяли гравиметрически осаждением
8-оксихинолином и взвешиванием осадка A1(C9H6ON)3. Из 1,021 г пробы
получено 0,1862 г осадка. Какова массовая доля (%) алюминия в сплаве?
17- Для определения железа в руде предполагается использовать метод грави-
метрии со взвешиванием Fe2O3. Желательно получить результат с четырь-
мя значащими цифрами. Какую минимальную массу навески следует взять,
чтобы получить 100,0 мг осадка, если содержание железа в пробе составля-
ет от 11 до 15%?
18. Из навески MgCl2 (степень чистоты 95%) массой 0,12 г осаждают хло-
рид-ионы в форме AgCI. Рассчитайте необходимый для осаждения объем
0,100 М AgNO3 с учетом 10%-го избытка осадителя.
19. Для определения ионов аммония их можно осадить действием H2PtCl6
в виде (NH4)2PtCl6, прокалить осадок до металлической платины
((NH4)2PtCl6 пРокали?ание>р| + 2NH4C1T + 2С12Т) и взвесить ее. Рассчитайте
массовую долю (%) ионов аммония в образце массой 1,00 г, если из него по-
лучено 0,100 г платины.
20. Определение хлорид-ионов в образце проводят посредством осаждения и
взвешивания хлорида серебра. Какую массу навески следует взять, чтобы
масса осадка численно равнялась массовой доле (%) хлорид-ионов в об-
разце?
21. Определение серы в пирите (FeS2 с примесями) проводят, превращая серу в
сульфат-ионы и осаждая их в виде BaSO4. Какую массу навески следует
взять для анализа, чтобы масса осадка (г) численно равнялась массовой
доле FeS2 (%), умноженной на 0,1000?
ЗАДАЧИ
463
22. Масса смеси, состоящей только из ВаО и СаО, составляет 2,00 г. Оксиды
бария и кальция превратили в смесь соответствующих сульфатов, масса ко-
торой составила 4,00 г. Рассчитайте массовые доли Ва и Са (%) в исходной
смеси.
23. В смеси, состоящей только из BaSO4 и CaSO4, масса бария в два раза мень-
ше массы кальция. Какова массовая доля CaSO4 (%) в смеси?
24. Масса смеси, состоящей только из AgCl и AgBr, составляет 2,000 г. Смесь
количественно превратили в металлическое серебро, масса которого соста-
вила 1,300 г. Рассчитайте массы AgCl и AgBr в исходной смеси.
Расчеты с использованием произведения растворимости
25. Напишите выражения произведения растворимости для следующих соеди-
нений: a) AgSCN, б) La(IO3)3, в) Hg2Br2, г) Ag[Ag(CN2)], д) Zn2Fe(CN)6,
е) Bi2S3.
26. Растворимость иодида висмута Bil3 составляет 7,76 мг/л. Чему равно про-
изведение растворимости?
27. Чему равны концентрации Ag+ и СгО 4” в насыщенном растворе Ag2CrO4?
28. Рассчитайте равновесную концентрацию ионов бария в растворе, получен-
ном смешением 15,0 мл 0,200 М К2СгО4 и 25,0 мл 0,100 М ВаС12.
29. При какой концентрации РО4“ начнется выпадение осадка Ag3PO4 из рас-
твора, содержащего 0,10 М AgNO3?
30. При какой концентрации Ag+ начнется выпадение осадка из раствора, со-
держащего 0,10 М РО4"? Из раствора, содержащего 0,10 М С1_?
31. При каком значении pH начнется осаждение А1(ОН)3 из 0,10 М раствора
А1С13?
32. Какая масса Ag3AsO4 может раствориться в 250 мл воды?
33. Рассчитайте растворимость Ag2CrO4 в 0,10 М К2СгО4.
34. Произведения растворимости соединений состава АВ и АС2 равны и со-
ставляют 4 • 10-18. Растворимость какого соединения (выраженная в моль/л)
выше?
35. Произведения растворимости Bi2S3 и HgS составляют 1 • 10-97 и 4 • 10~53 со-
ответственно. Какое из этих соединений менее растворимо?
36. Студент предложил определять барий гравиметрически, осаждая BaF2 при
помощи NaF. Насколько правомерно использование этой реакции в грави-
метрии, если считать, что в осадок должно перейти не менее 99,9% бария?
Примите, что содержание бария в образце составляет 200 мг, а объем рас-
твора в ходе осаждения — 100 мл.
464
ГРАВИМЕТРИЧЕСКИЙ АНАЛИЗ И РАВНОВЕСИЕ ОСАДОК-РАСТВОР
Влияние посторонних ионов на растворимость
37. Напишите выражения термодинамических произведений растворимости
для следующих систем:
a) BaSO4 ^Ba2+ + SO4~
б) Ag2CrO4 2Ag+ + СгО4"
38. Рассчитайте растворимость BaSO4 в присутствии 0,0125 М ВаС12 с учетом
влияния ионной силы.
39. Вы собираетесь определять фторид-ионы гравиметрически, осаждая их в
виде CaF2 Осадитель Ca(NO3)2 берется в таком избытке, чтобы его концен-
трация в растворе после осаждения составляла 0,015 М. Раствор содержит
также 0,25 М NaNO3. Какая масса фторид-ионов останется в растворе после
осаждения, если объем раствора составляет 250 мл?
Упражнения с электронными таблицами
40. Создайте электронную таблицу для расчета массовой доли Р2О5 в соответ-
ствии с условиями примера 10.2. Выполните с помощью электронной таб-
лицы расчет для данных, приведенных в примере. Повторите расчет для
условий: масса навески пробы составляет 0,5267 г, масса осадка—2,0267 г.
41. Создайте электронную таблицу для расчета зависимости растворимости
BaSO4 от концентрации Ва2+ в растворе (см. рис. 10.3). Постройте график
этой зависимости при помощи приложения Мастер диаграмм (Chart Wi-
zard) программы Excel и сравните его с рис. 10.3.
42. Создайте электронную таблицу для расчета зависимости растворимости
BaSO4 от величины ионной силы (см. рис. 10.4). Постройте график этой за-
висимости при помощи приложения Мастер диаграмм (Chart Wizard) Excel
и сравните его с рис. 10.4.
43. Решите пример 10.9, пользуясь для расчета растворимости приложением
Solver (Поиск решения) программы Excel.
Рекомендуемая литература
Основы гравиметрии и определение неорганических веществ
1. F. Е. Beamish and W. А. Е. McBryde, «Inorganic Gravimetric Analysis», in
C. L. Wilson and D. W. Wilson, eds., Comprehensive Analytical Chemistry,
Vol. 1A. New York: Elsevier, 1959, Chapter VI.
2. C. L. Wilson and D. W. Wilson, eds., Comprehensive Analytical Chemistry,
Vol. 1C, Classical Analysis: Gravimetric and Titrimetric Determination of the
Elements, New York: Elsevier, 1962.
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА 465
Органические реагенты
3. К. L. Cheng, К. Ueno, and Т. Imamura, eds. Handbook of Organic Analytical
Reagents, Boca Raton, FL: CRC Press, 1982.
4. R. G. W. Hollingshead, Oxine and Its Derivatives. London: Butterworth Scienti-
fic, 1954-56.
5- F. Holmes, «Organic Reagents in Inorganic Analysis», in C. L. Wilson and
D. W. Wilson, eds., Comprehensive Analytical Chemistry, Vol. 1 A. New York:
Elsevier, 1959, Chapter II.8.
Осаждение из гомогенных растворов
6- L. Gordon, M. L. Salulsky, and H. H. Willard, Precipitation from Homogeneous
Solution. New York: Wiley, 1959.
Глава 11
РЕАКЦИИ ОСАЖДЕНИЯ И ОСАДИТЕЛЬНОЕ ТИТРОВАНИЕ
Все, что не компонент раствора, —
компонент осадка.
Мудрость аналитика
Ряд анионов образует малорастворимые соединения с ионами металлов. Раство-
ры таких ионов металлов можно использовать в качестве титрантов для опреде-
ления соответствующих анионов. Например, хлорид-ионы можно титровать
растворами ионов серебра, а сульфат-ионы — растворами бария. На положение
равновесия осадок-раствор может влиять pH раствора или наличие комплексо-
образующих лигандов. Так, если анион осадка образован слабой кислотой, то в
кислых средах он связывается с протонами, что приводит к растворению осадка.
С другой стороны, если катион осадка образует растворимые комплексы с ка-
ким-либо лигандом, равновесие также сместится в сторону растворения осадка.
Например, хлорид серебра растворяется в присутствии аммиака за счет связыва-
ния ионов серебра в аммиачные комплексы.
В данной главе мы рассмотрим на количественном уровне влияние кислот-
ности и комплексообразования на положение равновесия осадок-раствор, а также
познакомимся с методом осадительного титрования (на примере таких титрантов,
как нитраты серебра и бария), включая теорию применения индикаторов различ-
ных типов. Хотя для большинства анионов, особенно неорганических, разрабо-
таны удобные методики их определения при помощи ионной хроматографии
(см. гл. 21), в случае высокого содержания более точные результаты получаются
методом осадительного титрования (там, где это возможно).
11.1. Влияние кислотности на растворимость осадков:
условное произведение растворимости
Прежде чем рассматривать осадительное титрование, рассмотрим влияние кон-
курирующих реакций на растворимость осадков. Перед изучением данной главы
следует повторить раздел, посвященный равновесиям в растворах многоосновных
кислот и расчетам мольных долей (a-коэффициентов) отдельных форм кислоты
в условиях равновесия при заданном pH (см. гл. 7).
Растворимость осадка, образованного анионом слабой кислоты, возрастает
с увеличением кислотности среды вследствие связывания аниона протонами и
11.1. ВЛИЯНИЕ КИСЛОТНОСТИ НА РАСТВОРИМОСТЬ осдцков
467
снижения равновесной концентрации в растворе. Например, для осадка состава
МА, диссоциирующего в растворе на ионы М+ и А-, устанавливаются следую-
щие равновесия:
МА^±М+ + А"
+
Н+
НА
В результате связывания аниона А- с протонами растворимость осадка увеличи-
вается. Сумма равновесных концентраций форм А- и НА составляет общую
(аналитическую) концентрацию СНА, которая (в отсутствие избытка как М+, так
и А-) равна концентрации [М+]. Используя выражения констант всех равнове-
сий, существующих в растворе, можно рассчитать растворимость осадка при
данном pH.
Рассчитаем, например, растворимость СаС2О4 в присутствии сильной кис-
лоты. В этом случае устанавливаются следующие равновесия
СаС2О4 Са2++ С2О4~ Ks= [Са2+][С2О^“] = 2,6 • 1(Г9 (11.1)
С2О 2“ + Н+ НС2О; Ка2 = [Н ][С2°4 ] = 6,1 • 10-5 (11.2)
[НС2О4 ]
нс2о;+ н+ —н2с2о4 /са1 = [Н']^С2°4] = б,5 10-2 (и.з)
LH2C2U4J
В рассматриваемом примере протоны конкурируют с ионами кальция за связы-
вание с оксалат-ионами. Растворимость СаС2О4 (5) равна концентрации [Са2+],
которая, в свою очередь, равна CH2q2q4 — суммарной концентрации всех форм,
содержащих оксалат-ион, т. е. [Н2С2О4] + [НС2О4] + [С2О4-]. В выражении произ-
ведения растворимости оксалата кальция [С2О4“] можно заменить на Сн^р о^:
^=[Са2+]СН2с2о4а2
(И.4)
где а2 — мольная доля оксалат-иона С2О 4 , равная [С2О 4 ]IC^2q2q4 . Используя
способ расчета a-коэффициентов, описанный в гл. 7 применительно к Н3РО4,
можно получить выражение
[H+]2+*ai[H+] + Va2
(И.5)
468
РЕАКЦИИ ОСАЖДЕНИЯ И ОСАДИТЕЛЬНОЕ ТИТРОВАНИЕ
Можно записать также следующее соотношение:
^=K's=[Ca2+]CH^=s2 (11.6)
а2
Величина, обозначенная как K's, называется условным произведением раство-
римости (аналогично условной константе устойчивости, рассмотренной в гл. 9).
Условное произведение растворимости сохраняет постоянное значение только
при данной величине pH раствора.
Пример 11.1
Рассчитайте растворимость СаС2О4 в растворе, содержащем 0,0010 М НС1.
Решение
„ - 6,5Ю-2 • 6Д10-5 _<7 1П-2
ci 2 —--------------------------------------------— 5,7 • 10
(1,0 10~3)2 + 6,5-10-2 • 1,0 10’3 + 6,5 Ю-2 • 6,110~5
S = ^Ks /а 2 = 72,6-10“В 9/5,7-10“2 = 2,1 • 10"1 М
Сравните эту величину с растворимостью СаС2О4 в чистой воде [которую мож-
но рассчитать непосредственно по уравнению (11.1)], составляющей 5,1 • 10-5 М.
В присутствии кислоты растворимость увеличилась в 4 раза. Подчеркнем, что
рассчитанное значение (2,1 • ПУ4 М) равно как [Са2+], так и Сн Ср . Можно рас-
считать и концентрации всех форм оксалат-ионов по отдельности, умножая по-
лученное значение на а0, а! или а2 — мольные доли (в среде 0,0010 М
НС1) Н2С2О4, НС2О4 и С2О 4“ соответственно. Мы не будем приводить выражения
для а0 и aj и ограничимся лишь конечным результатом: [С2О4“] = 1,2 • 10-5 М,
[НС2О4] = 2,0- 1СНМ, [Н2С2О4] = 3,1 • 10~6М. Попробуйте рассчитать эти значе-
ния самостоятельно. Для растворов с низкой буферной емкостью необходимо
использовать метод итераций, повторяя расчет с учетом количества протонов,
израсходованных на связывание с анионом.
В приведенных расчетах мы пренебрегли тем обстоятельством, что часть
кислоты расходуется на взаимодействие с оксалат-ионами. Но из полученных
данных следует, что около одной пятой исходного количества кислоты идет на
образование НС2О4 (количество, из которого образуется Н2С2О4, пренебрежи-
мо мало). Чтобы получить более точное решение, следует из исходного количе-
ства кислоты вычесть рассчитанное количество прореагировавшей кислоты и
повторить расчет с новым значением концентрации кислоты. Далее этот процесс
следует повторять до тех пор, пока изменения в конечном результате не окажутся
11.2. РАСЧЕТ СЛОЖНЫХ РАВНОВЕСИЙ С ИСПОЛЬЗОВАНИЕМ МАТЕРИАЛЬНОГО БАЛАНСА
469
меньше требуемой точности ответа. Повторный расчет с использованием значения
концентрации кислоты, равного 0,8 • 10-3 М, приводит к величине концентрации
кальция 1,9 • 10-4 М, что на 10% меньше первоначально рассчитанного.
Подчеркнем, что в системах, где одновременно существует множество рав-
новесий, положение каждого из них по-прежнему определяется выражением соот-
ветствующей константы. Так, в приведенном примере выражение произведения
растворимости для СаС2О4 определяет соотношение между концентрациями
Са2+ и С2О 4“ вне зависимости от того, присутствует кислота в растворе или нет.
Иными словами, произведение [Са2+][С2О4~] всегда является постоянной вели-
чиной, коль скоро в системе присутствует твердый СаС2О4. Однако количество
растворившегося СаС2О4 в присутствии кислоты возрастает, поскольку при
этом часть С2О4_ превращается в НС2О4 и Н2С2О4.
11.2. Расчет сложных равновесий с использованием
материального баланса
Для систем, в которых существует множество равновесий, можно также исполь-
зовать описанный в гл. 6 общий подход, в основе которого лежит решение сис-
темы уравнений, включающих выражения констант равновесий, уравнения
материального баланса и электронейтральности. Общий подход к расчетам хи-
мических равновесий сосбенно удобно использовать для систем с конкурирую-
щими реакциями.
Пример 11.2
Какое количество вещества МА (моль) растворится в 1 л 0,10 М НС1, если вели-
чина Ks для МА составляет 1,0- 10-8, а Ка для НА — 1,0- 10-6?
Решение
В системе протекают следующие процессы:
МА М+ + А-
а-+н+^на
н2о мг + он-
иа -> Н+ + Cl-
Константы соответствующих равновесий имеют вид
А'5=[М+][А-] = 1,0- 10-8 (1)
=10- 10-6 (2)
а [НА]
^ = [Н+][ОН-] = 1,0-10-14 (3)
470
РЕАКЦИИ ОСАЖДЕНИЯ И ОСАДИТЕЛЬНОЕ ТИТРОВАНИЕ
Уравнения материальных балансов записываются в виде
[М+] = [А-] + [НА] - Сна (4)
[Н+] = [С1-] + [ОН-] = [НА] (5)
[С1-] = 0,10М (6)
Уравнение электронейтральности имеет вид
[Н+] + [М+] = [А-] + [С1-] + [ОН-] (7)
Проверим соответствие числа уравнений числу неизвестных. Число неизве-
стных величин ([Н+], [ОН-], [С1-], [НА], [М+] и [А-]) равно шести, и число неза-
висимых уравнений также равно шести (выражение электронейтральности
представляет собой линейную комбинацию других уравнений системы, и его
можно не использовать). Число переменных величин должно быть равно числу
уравнений системы. Для упрощения расчетов можно сделать следующие допу-
щения:
1) В кислой среде диссоциация НА подавлена, поэтому можно допустить, что
[А-] [НА]. В этом случае уравнение (4) примет вид
[М+] = [А-] + [НА] « [НА]
2) В кислой среде также очень мала величина [ОН-], поэтому из (5) и (6) полу-
чаем:
[Н+] = 0,10 + [ОН-] - [НА] « 0,10 - [НА]
Расчет, Чтобы рассчитать количество МА, растворенного в одном литре рас-
твора, требуется вычислить [М+].
Из (1) получаем:
[M+]=i (8)
[А]
Из (2) получаем:
[А-] = (9)
[Н+]
Разделив (8) на (9), получаем:
[М+] = = 1,0 • 10-2 (10)
*ДНА] [НА]
В соответствии с допущением (1),
[М+]«[НА]
11.2. РАСЧЕТ СЛОЖНЫХ РАВНОВЕСИЙ С ИСПОЛЬЗОВАНИЕМ МАТЕРИАЛЬНОГО БАЛАНСА
471
В соответствии с допущением (2),
[Н+] « 1,10 - [НА] « 0,10 - [М+]
„,+1 1,0-10'2- 0,10-[М+]
[М+] =---------------------
[М+]
0,10 -[М+]
В результате решения квадратного уравнения получаем: [М] = 0,027 М.
Таким образом, в 1 л раствора указанного состава растворяется 0,027 моль МА.
Сравните эту величину с растворимостью МА в чистой воде (0,00010 моль/л). Про-
верим правомерность сделанных допущений.
Допущение (1):
[НА] « [М+] = 0,027 М
[А-] = = 1>0'1—- = 3,7 • 10-7 М
[М+] 0,027
Допущение (1) оправданно, поскольку [А-] [НА].
Допущение (2):
[Н+] « 0,10 - [М+] = 0,073 М
[ОН-] = =1,0'10 14 = 1,4 • 10-13
[Н+] 0,073
Допущение (2) также оправданно, поскольку [ОН_]<^ [О-] (или [НА]).
Пример 11.3
Рассчитайте растворимость СаС2О4 в 0,0010 М растворе соляной кислоты, исполь-
зуя общий подход.
Решение
В системе протекают следующие процессы:
СаС2О4 Са2+ + С2О^“
С2О^ + Н+^НС2О4
нс2о; + н+ н2с2о4
н2о н+ + он-
НС1 -> Н+ + С1-
472
РЕАКЦИИ ОСАЖДЕНИЯ И ОСАДИТЕЛЬНОЕ ТИТРОВАНИЕ
Константы соответствующих равновесий имеют вид
Ks= [Са2+][С2О5’] = 2,6 • 10-9 (1)
[НЧ1НСД]. [Н2С2О„] g [Н-][С2ОГ1д,6|1чо_, [нод] Kw = [Н+][ОН-] = 1,00 • ю-14 Уравнения материальных балансов записываются в виде (2) (3) (4)
[Са2+] = [С2О 24~] + [НОД] + [Н2С2О4] = СНАо4 (5)
[Н+] = [С1-] + [ОН-] - [Н(ОД] - [Н2С2О4] [С1-] = 0,0010 М (6) (7)
Уравнение электронейтральности имеет вид
[Н+] + 2[Са2+] = 2[С2О4~] + [НС2О;] + [С1_] + [ОН"] (8)
Система содержит семь неизвестных ([Н+], [ОН-], [С1-], [Са2+], [С2О 4 ], [НС2О 4 ]
и [Н2С2О4]) и семь независимых уравнений.
Допущения для упрощения расчетов:
1) Константа Ка1 достаточно велика, а Ка2 — достаточно мала, поэтому можно
предположить, что [НС2О4] >> [Н2С2О4] и [НС2О4] >> [С2О4“].
2) В кислой среде величина [ОН-] очень мала, поэтому из (6) и (7) получаем
[н+] = 0,0010 + [он-] - [н2с2о;] - [н2с2о4]« о,оою - [нс2о;] (9)
Расчеты. Чтобы рассчитать количество СаС2О4, растворенного в одном литре
раствора, требуется вычислить [Са2+].
Из (1) получаем:
[Са2+] = -Д-
[с2о*-]
(Ю)
Из (3) получаем:
[C2Oi~] =
тса2[нс2о;]
[н+]
(И)
11.3. ВЛИЯНИЕ КОМПЛЕКСООБРАЗОВАНИЯ НА РАСТВОРИМОСТЬ
473
Таким образом,
[Са2+] = —— (12)
/Са2[НС2О*-]
Из допущения (1) получаем:
[Са2+] = [НС2О;] (13)
Из допущения (2)
[Н+] «0,0010- [НС2О 4 ] « 0,0010 - [Са2+] (14)
Подставляя (13) и (14) в (12), получаем:
г„ 2+ -1 _ Ks (0,0010 - [Са2+ ]) _ 2,6 • IO"9 (0,0010 - [Са2+ ])
ТСа2[Са2 ] 6,1 -10“5[Са2+]
г_ 2+ 4,6- IO’5 (0,0010 -[Са2+])
[Са2+]
В результате решения квадратного уравнения мы находим [Са2+] = 1,9 • 1(Н М.
Этот ответ совпадает с ответом, полученным в примере 11.1 при использовании
условного произведения растворимости. Однако в том случае потребовалось
дополнительно учесть уменьшение концентрации Н+ в ходе реакции. Здесь же
мы учли это непосредственно в ходе расчетов. Обратите внимание, что рассчи-
танная концентрация НС2О4 (пример 11.1) составляет около 95% от величины
[Са2+], поэтому допущение (1) можно считать обоснованным.
11.3. Влияние комплексообразования на растворимость:
условное произведение растворимости
Комплексообразующие реагенты могут конкурировать за связывание катиона
осадка подобно тому, как протоны кислоты — за связывание аниона. Рассмот-
рим малорастворимое соединение состава МА, диссоциирующее на ионы М+ и
А-. Пусть лиганд L образует с ионом М+ комплекс состава ML+. В этом случае в
системе существуют следующие равновесия:
МА^М+
L
н
•м
ML+J
474
РЕАКЦИИ ОСАЖДЕНИЯ И ОСАДИТЕЛЬНОЕ ТИТРОВАНИЕ
Сумма [М+] и [ML+] составляет общую (аналитическую) концентрацию См, рав-
ную [А-]. В этом случае расчеты проводят также, как при рассмотрении влияния
кислотности на растворимость осадка.
Рассмотрим, как изменяется растворимость AgBr в присутствии NH3. В сис-
теме протекают следующие процессы:
AgBr Ag+ + Вг-
(И-7)
Ag+ + NH3 Ag(NH3)+
Ag(NH3)+ + NH3 Ag(NH3)+
(П-8)
(И.9)
Растворимость AgBr (S) равна концентрации [Вг-], которая, в свою очередь,
равна CAg — суммарной концентрации всех форм, содержащих серебро, т. е.
[Ag+] + [Ag(NH3)+] + [Ag(NH3)2+]. Подобно тому, как мы делали ранее, можно
подставить в выражение произведения растворимости вместо [Ag+] величину
CAgPo> где Ро — мольная доля серебра в форме Ag+ [см. уравнение (9.20)]:
Ks = [Ag+] [Br -] = CAgp0[Br -] = 4 • 10-13
(11.10)
Отсюда
Po
= =CAg[Br-] = S2
(11-11)
Символом K's обозначено условное произведение растворимости, величина
которого в данном случае зависит от концентрации аммиака. Величина K's со-
храняет постоянное значение только при данной концентрации лиганда.
Пример 11.4
Рассчитайте растворимость бромида серебра (моль/л) в 0,10 М растворе аммиака.
Решение
Из результата примера 9.5 следует, что величина 0О при концентрации аммиака
0,1 М составляет 4,0 • 10-6.
По сравнению с величиной растворимости AgBr в чистой воде, равной 6 • 10-7 М,
это значение в 530 раз выше. Отметим, что полученная величина (3,2 • 10-4 М)
равна как [Вг-], так и CAg. Можно рассчитать и концентрации всех форм сереб-
11.4. ОСАДИТЕЛЬНОЕ ТИТРОВАНИЕ
475
ра по отдельности, умножая полученное значение на р0, или р2 — мольные
доли (в среде 0,10 М NH3) Ag+, Ag(NH3)+ и Ag(NH3) 2 соответственно. Исполь-
зуя значения P-коэффициентов из примера 9.5, получаем: [Ag+] = 1,3 • 10-9 М,
[Ag(NH3)+] = 3,2 • 10-7Ми [Ag(NH3)2] = 3,2 • 10-4 М. Обратите внимание, что
основная часть серебра находится в форме Ag(NH3)2.
Проверьте, что предположения о величине концентрации лиганда были
правильными.
При расчетах мы не учитывали количество аммиака, израсходованное на
связывание с ионами серебра. Как видим, в данном случае это количество дейст-
вительно оказалось пренебрежимо малым по сравнению с исходным: исходная
концентрация NH3 составляла 0,10 М, на образование Ag(NH3)2 затрачено
6 • 10-4 М, а на образование Ag(NH3)+ — еще меньше. Но если бы количество
связанного аммиака оказалось значительным, то для нахождения более точного
решения потребовалась бы итерационная процедура: из исходного количества
аммиака следовало бы вычесть количество аммиака, связанного в комплексы, и
рассчитать при новой концентрации аммиака новые величины Р-коэффициен-
тов и новое значение растворимости.
11.4. Осадительное титрование
Титрование растворами осадителей — важный метод определения ряда анио-
нов. Как и в любом титровании, в данном случае необходимо, чтобы равновесие
в системе устанавливалось быстро и существовал подходящий способ детекти-
рования конечной точки титрования. Глубже понять вопросы, связанные с выбо-
ром индикатора в осадительном титровании, точностью метода и титрованием
смесей, поможет рассмотрение кривых титрования.
Кривые титрования: расчет рХ
Рассмотрим процесс титрования хлорид-ионов при помощи стандартного рас-
твора AgNO3. Кривую титрования можно представить в виде зависимости рС1
(т. е. -lg[Cl-]) от объема раствора AgNO3 аналогично кривой кислотно-основно-
го титрования. Типичный вид такой кривой представлен на рис. 11.1: величина
рХ означает отрицательный десятичный логарифм концентрации галогенид-
иона. В начальной точке система представляет собой 0,1 М раствор СГ, и вели-
чина рС1 равна 1. В процессе титрования хлорид-ионы постепенно удаляются из
раствора, осаждаясь в виде AgCl, и величина рС1 определяется концентрацией
неоттитрованных хлорид-ионов. Вкладом хлорид-ионов, образовавшихся в ре-
зультате диссоциации AgCl, можно пренебречь, за исключением малой области
вблизи точки эквивалентности. В точке эквивалентности система представляет
476
РЕАКЦИИ ОСАЖДЕНИЯ И ОСАДИТЕЛЬНОЕ ТИТРОВАНИЕ
РИС. 11.1
Кривая титрования 100 мл 0,1 М
растворов хлорид-, бромид-
и иодид-ионов 0,1 М раствором
AgNO3. Среди этих галогенидов
серебра наименьшей
растворимостью характеризуется
Agl, поэтому за точкой
эквивалентности значения [Г]
меньше по сравнению
с концентрациями других
галогенид-ионов, а р!
лежит выше других рХ
собой насыщенный раствор AgCI, в котором [С1“] = ^Ks = 1 • 10-5 М (см. гл. 10) и
рС1 = 5. За точкой эквивалентности в растворе присутствует избыток Ag+, и кон-
центрацию С1_ можно рассчитать из величин [Ag+] и Ks, как показано в приме-
ре 10.7 гл. 10 ([С1-] =/Cs/[Ag+]).
Пример 11.5
Рассчитайте значения рС1 в ходе титрования 100,0 мл 0,1000 М раствора С1"
0,1000 М раствором AgNO3 при добавлении 0,00,20,00,99,00,99,50,100,00,100,50
и 110,00 мл раствора AgNO3.
Решение
Для 0,00 мл AgNO3 (начальная точка):
pCl = -lg 0,1000= 1,000
Добавлено 20,00 мл:
псх- = 100,0 мл • 0,1000 ммоль/мл = 10,00 ммоль
п . + = 20,00 мл • 0,1000 ммоль/мл = 2,000 ммоль
Ag
остаточная концентрация С1_: [С1“] = (10,00 - 2,00) ммоль /120 мл = 0,0667 М
pCl = -lg 0,0667= 1,18
11.4. ОСАДИТЕЛЬНОЕ ТИТРОВАНИЕ
477
Добавлено 99,00 мл:
п . + = 99,00 мл • 0,1000 ммоль/мл = 9,900 ммоль
остаточная концентрация Cl-: [С1_] = (10,00 - 9,90) ммоль /199 мл = 5,0 • 10-4 М
pCl = -lg (5,0- IO"4) = 3,26
Добавлено 99,50 мл:
п А + = 99,50 мл • 0,1000 ммоль/мл = 9,950 ммоль
остаточная концентрация С1~: [С1"]= (10,00-9,95) ммоль/199,5 мл = 2,5- 10-4М
pCl = -lg (2,5 • 10-4) = 3,60
Добавлено 100,0мл (точка эквивалентности):
[С1-] =7^7 = 71,0-Ю’10 =1,0 • 10-5 м
pCl = -lg (1,0 IO"5) = 5,00
Добавлено 100,50 мл:
п А + = 100,50 мл • 0,1000 ммоль/мл = 10,05 ммоль
Ag+
избыток Ag+: [Ag+] = (10,05 - 10,00) ммоль / 200,5 мл = 2,5 • 10-4 М
[С1-] = 7Cs/[Ag+] = (1,0 • 10-10)/(2,5 • IO"4) = 4,0 • 10~7 М
pCl = -lg (4,0 • 10~7) = 6,40
Добавлено 110,00 мл:
п А + = 110,00 мл • 0,1000 ммоль / мл = 11,00 ммоль
Ag
избыток Ag+: [Ag+] = (11,00 - 10,00) ммоль / 210 мл = 4,76 • 10-3 М
[С1-] = (1,0 • 10-10)/(4,76 • 10-3) = 2,1 • 10-8 М
pCl = -lg (2,1 • IO"8) = 7,67
Чем меньше величина К& тем больше скачок на кривой титрования вблизи
точки эквивалентности. Чтобы проиллюстрировать это положение, сравним
кривые титрования С1_, Вг _ и 1~ раствором Ag+ (см. рис. 11.1). Значения Ks для
AgCl, AgBr и Agl составляют 1 • 1О“10,4 • 10-13 и 1 • 10~16 соответственно. Исход-
ные концентрации всех анионов равны, поэтому вплоть до точки эквивалентности
(где ход кривых определяется только количеством неоттитрованного аниона) все
три кривые титрования совпадают. В точке эквивалентности величина [X-] тем
меньше (а рХ, соответственно, больше), чем меньше значение Ks. За точкой эк-
вивалентности [X-] также тем меньше, а рХ — тем больше, чем меньше Ks. В ре-
зультате с уменьшением растворимости продукта реакции величина скачка на
кривой титрования возрастает.
478
РЕАКЦИИ ОСАЖДЕНИЯ И ОСАДИТЕЛЬНОЕ ТИТРОВАНИЕ
Если, наоборот, титровать раствор Ag+ раствором С1”, то кривая титрова-
ния, построенная как зависимость рС1 от объема титранта, выглядела бы зеркаль-
ным отражением кривой на рис. 11.1. В этом случае концентрация хлорид-ионов
до точки эквивалентности определялась бы концентрацией неоттитрованных
ионов Ag+ и величиной Ks, а за точкой эквивалентности — только избытком тит-
ранта. Можно было бы построить кривую титрования и как зависимость pAg от
объема титранта. Такая кривая выглядела бы точно так же, как изображенная на
рис. 11.1.
Установление конечной точки:
индикаторы в осадительном титровании
В рассмотренном случае для нахождения конечной точки титрования можно из-
мерять рС1 или pAg при помощи подходящего электрода и потенциометра (см.
гл. 13). Но проще использовать индикатор. Принципы использования индикато-
ров в осадительном и кислотно-основном титровании различны: в первом слу-
чае индикатором не обязательно должно быть вещество, свойства которого
зависят от концентрации определенного иона в растворе (например, величины
рС1 или pAg).
В осадительном титровании обычно используют индикаторы двух типов.
Индикаторы первого типа при появлении в растворе избытка титранта образуют
с ним окрашенное соединение. Индикаторы второго типа называются адсорб-
ционными. При достижении точки эквивалентности они адсорбируются на по-
верхности осадка, поскольку в этой точке поверхность осадка резко меняет свои
свойства. В адсорбированном состоянии индикатор приобретает другую окрас-
ку. Рассмотрим механизмы действия индикаторов каждого типа.
1 - Индикаторы, взаимодействующие с титрантом. Можно привести несколь-
ко примеров индикаторов, которые образуют окрашенное соединение с титран-
том. Один из них используется в определении хлоридов по Мору, где титрантом
служит стандартный раствор нитрата серебра. В качестве индикатора применя-
ют растворимый хромат. Раствор хромат-ионов окрашен в желтый цвет. При до-
стижении полноты осаждения хлоридов в растворе появляются свободные ионы
Ag+, взаимодействующие с индикатором с образованием окрашенного в крас-
ный цвет осадка хромата серебра:
CrO^ + 2Ag+- » Ag2CrO4
(желтый) (красный)
(Н-12)
Важно правильно выбирать концентрацию индикатора. Начало образова-
ния осадка Ag2CrO4 должно наблюдаться непосредственно в точке эквивалентно-
сти. Рассчитанная из величины Ks концентрация Ag+ в этой точке равна 1 • 10-5 М.
Таким образом, осадок Ag2CrO4 должен начать образовываться тогда, когда
11.4. ОСАДИТЕЛЬНОЕ ТИТРОВАНИЕ
479
[Ag+] = 1 • IO'5 М. Произведение растворимости Ag2CrO4 составляет 1,1 • 10-12.
Подставляя значение [Ag+] в выражение произведения растворимости для Ag2CrO4,
находим, что для начала образования осадка концентрация СгО4~ должна со-
ставлять 0,011 М:
(1 • 10-5)2[СгО^] = 1,1 • 10-12
[СгО^] = 1,1 • 10-2 М
Если концентрация хромат-ионов превышает эту величину, осадок Ag2CrO4
начнет выпадать при концентрации Ag+ меньшей чем 1 • 10~5 М, т. е. до точки эк-
вивалентности. Если же концентрация хромат-ионов меньше этого значения, то
осадок Ag2CrO4 начнет выпадать при более высокой, чем 1 • 10 5 М, концентра-
ции Ag+, т. е. за точкой эквивалентности.
На практике создают концентрацию индикатора в пределах от 0,002 до
0,005 М. Если концентрация индикатора существенно выше, то интенсивная
желтая окраска хромат-ионов мешает увидеть появление красного осадка Ag2CrO4.
В этом случае требуется добавить значительный избыток Ag+, чтобы образова-
лось достаточно много осадка. Всегда следует проводить титрование контроль-
ного раствора, содержащего только индикатор, и затем вычесть из полученных
результатов объем титранта, затраченный на его титрование.
Титрование по Мору следует проводить при pH около 8. В слишком кислой
среде (pH < 6) часть ионов СгО 4“ переходит в форму НСгО 4, и для начала осаж-
дения Ag2CrO4 требуется больший избыток Ag+. В более щелочных средах (при
рН> 10) возможно выпадение осадка гидроксида серебра. Для поддержания
нужного значения pH к раствору обычно добавляют немного твердого карбона-
та кальция. Карбонат-ион является достаточно сильным основанием Бренстеда,
однако его концентрация в насыщенном растворе карбоната кальция невелика и
как раз создает значение pH около 8. Титрованием по Мору удобно определять
хлориды в нейтральных растворах низкой буферной емкости, например, в пить-
евой воде.
Другим примером использования индикаторов рассматриваемого типа слу-
жит методика обратного титрования по Фольгарду. Этим способом определя-
ют ряд анионов (С1-, Вг-, SCN-), образующих малорастворимые соединения с
Ag+. Титрование по Фольгарду проводят в кислой среде (в присутствии HNO3).
В ходе определения к раствору сначала добавляют в избытке точно известное
количество AgNO3 для полного осаждения аниона, а затем определяют непроре-
агировавшее количество Ag+ обратным титрованием стандартным раствором
тиоцианата калия:
X- + Ag+ —> AgX + избыток Ag+
избыток Ag+ + SCN- AgSCN
(11.13)
РЕАКЦИИ ОСАЖДЕНИЯ И ОСАДИТЕЛЬНОЕ ТИТРОВАНИЕ
Индикатором служит Fe(III) (в виде раствора железоаммонийных квасцов —
сульфата железа-аммония), образующий с избытком титранта окрашенный в
красный цвет растворимый комплекс:
Fe3++ SCN-—> Fe(SCN)2+
(И.14)
Если соединение серебра с определяемым ионом AgX менее растворимо,
чем AgSCN, отделять осадок AgX перед титрованием не требуется. Это имеет
место при титровании I- и Вг~, а также SCN-. При титровании I- индикатор сле-
дует добавлять только после полного осаждения 1~ в виде Agl, иначе иоидид-
ионы будут окисляться железом(Ш). Если же осадок более растворим, чем
AgSCN (например, AgCl), то в ходе титрования он будет взаимодействовать с
титрантом:
AgCl + SCN- -> AgSCN + С1-
(П-15)
Вследствие этого результаты титрования окажутся завышенными, а индикация
конечной точки — нечеткой. Поэтому в таких случаях осадок перед титровани-
ем следует отфильтровать.
Разумеется, все индикаторы описанного типа не должны образовывать с
титрантом соединений более устойчивых, чем продукт титриметрической реак-
ции (осадок). В противном случае изменение окраски произойдет при добавле-
нии первой же капли титранта.
2. Адсорбционные индикаторы. При использовании адсорбционных индика-
торов индикаторная реакция протекает на поверхности осадка. Адсорбционные
индикаторы представляют собой красители, существующие в растворе в виде
ионов, обычно анионов (In-). Для объяснения действия адсорбционных индика-
торов следует вспомнить механизм образования осадков (подробнее см. гл. 10).
Рассмотрим титрование ионов С1“ раствором Ag+. До точки эквивалентно-
сти в избытке находятся ионы С1, и именно эти ионы образуют первичный адсор-
бционный слой. Поэтому частицы осадка отталкивают одноименно заряженные
ионы индикатора. Вторичный слой (слой противоионов) образован катионами,
например Na+:
AgCl: Cl- :: Na+
После точки эквивалентности в избытке находятся ионы Ag+, образующие в
этих условиях первичный слой, в результате чего поверхность осадка становит-
ся положительно заряженной. Она притягивает к себе анионы индикатора, кото-
рые теперь играют роль противоионов:
AgCl: Ag+ :: In-
11.4. ОСАДИТЕЛЬНОЕ ТИТРОВАНИЕ
481
Адсорбционные индикаторы характеризуются тем, что их окраска в адсорбиро-
ванном состоянии отличается от окраски в растворе. Возможное объяснение
этого явления состоит в том, что в адсорбированном состоянии индикатор обра-
зует комплекс с ионами Ag+, который настолько малоустойчив, что не может су-
ществовать в растворе. На поверхности же осадка, т. е. как бы «в твердой фазе»
образование этого комплекса облегчается.
При использовании адсорбционных индикаторов важно учитывать pH рас-
твора. Если значение pH слишком низкое, то индикатор, который обычно явля-
ется слабой кислотой, диссоциирует в незначительной степени, и его адсорбция
в форме аниона мала. Кроме того, при данном pH индикатор не должен адсорби-
роваться слишком сильно, иначе он будет вытеснять анионы осадка (например,
Cl") из первичного слоя еще до точки эквивалентности. Разумеется, степень вли-
яния этих факторов зависит от того, насколько прочно адсорбируется анион
осадка на его поверхности, т. е. от растворимости осадка. Так, ионы Вг_ образу-
ют с ионами Ag+ менее растворимый осадок, чем ионы Cl", и соответственно,
адсорбируются сильнее. В этом случае можно использовать более сильно ад-
сорбирующийся индикатор.
Степень адсорбции индикатора можно уменьшить, увеличивая кислотность
раствора. Чем более сильной кислотой является индикатор, тем шире диапазон
pH, в котором он способен адсорбироваться. При титровании Вг _ можно исполь-
зовать индикаторы с более сильно выраженными кислотными свойствами (и си-
льнее адсорбирующиеся), чем при титровании Cl-; соответственно, титрование
можно проводить в более кислых средах. То есть чем менее растворим осадок,
тем в более кислых средах можно проводить титрование с использованием ад-
сорбционных индикаторов и тем более сильно адсорбирующиеся индикаторы
можно при этом использовать.
В табл. 11.1 перечислены некоторые адсорбционные индикаторы. Флуорес-
цеин можно использовать для определения любых галогенид-ионов при pH 7,
поскольку в этих условиях этот индикатор не вытесняет ни один из галоге-
нид-ионов. Дихлорфлуоресцеин вытесняет ионы С1_ при pH 7, но не при pH 4.
Поэтому при титровании хлорид-ионов с этим индикатором при pH 7 результа-
ты оказываются заниженными. Определение хлорид-ионов с упомянутыми инди-
каторами называют титрованием по Фаянсу. Первоначально Фаянс использовал
в качестве индикатора флуоресцеин, но сейчас чаще применяют дихлорфлуо-
ресцеин. Эозин нельзя использовать для определения хлоридов ни при каких
значениях pH, поскольку этот индикатор слишком сильно адсорбируется.
При использовании адсорбционных индикаторов конечная точка титрова-
ния, как правило, не совпадает с точкой эквивалентности. Поэтому титрант не-
обходимо стандартизовать, используя тот же индикатор, что и для анализа
пробы. В этом случае погрешности титрования практически компенсируют друг
друга, особенно если для стандартизации и для анализа расход титранта почти
одинаков.
Основной источник погрешностей при любых титриметрических определе-
ниях с участием ионов серебра — фотохимическое разложение галогенидов се-
ребра, которое катализируется адсорбционными индикаторами. Тем не менее
482
РЕАКЦИИ ОСАЖДЕНИЯ И ОСАДИТЕЛЬНОЕ ТИТРОВАНИЕ
Таблица 11.1
Некоторые адсорбционные индикаторы
Индикатор Титриметрическое определение Условия
Флуоресцеин Cl" с Ag+ pH 7-8
Дихлор флуоресцеин Cl" с Ag+ pH 4
Бромкрезоловый зеленый SCN" с Ag+ pH 4-5
Эозин Вг", Г, SCN" c Ag+ pH 2
Метиловый фиолетовый Ag+ c Cl- Кислая среда
Родамин 6Ж Ag+ c Br_ HNO3 (<0,3 M)
Торин SO^“ c Ba2+ pH 1,5-3,5
Бромфеноловый синий Hg^+ c Cl- 0,1 M
Ортохром Т Pb2+ c CrO^ Нейтральная среда, 0,02 M
при соответствующей стандартизации можно достичь точности результатов по-
рядка 0,01%.
В точке эквивалентности, когда ни один из ионов осадка не находится в из-
бытке, поверхность осадка не заряжена. Вследствие этого осадки, склонные к
коллоидообразованию (такие как хлорид серебра), в этих условиях обычно коа-
гулируют, особенно при сильном перемешивании раствора. Это весьма желатель-
но в гравиметрическом анализе, но нежелательно при осадительном титровании:
в результате коагуляции суммарная площадь поверхности осадка уменьшается,
при этом уменьшается степень адсорбции индикатора, и следовательно, рез-
кость перехода окраски. При использовании адсорбционных индикаторов (в от-
личие от гравиметрии!) желательно, чтобы осадок имел как можно большую
площадь поверхности. Коагуляцию хлорида серебра можно предотвратить, если
добавить в раствор немного декстрина.
Визуальные индикаторы удобно использовать для быстрого титриметри-
ческого анализа методом осадительного титрования с использованием ионов
серебра. В то же время широко используют и потенциометрическую индика-
цию, особенно при титровании разбавленных растворов (порядка 10~3 М —
см. гл. 14).
Определение сульфат-ионов с Ва2+
Сульфаты можно определять титрованием растворами солей бария (при этом
образуется осадок BaSO4). Как и при гравиметрическом определении сульфатов
осаждением в виде BaSO4, в этом случае могут возникать погрешности вследст-
ВОПРОСЫ
483
вие соосаждения. Такие катионы, как Na+, NH4 и, особенно, К+, склонны к соо-
саждению в виде сульфатов:
BaSO4 : SO4~ : 2М+
В результате для завершения осаждения требуется меньшее количество раство-
ра бария, и результаты оказываются заниженными. Ряд ионов металлов образует
комплексы с используемыми индикаторами и поэтому также мешают. Наконец,
некоторые анионы тоже могут соосаждаться (в виде бариевых солей), вызывая
завышение результатов. Погрешности, вносимые присутствием хлоридов, бро-
мидов и перхлоратов, обычно невелики, но нитраты, сильно искажающие резуль-
таты, должны отсутствовать.
Мешающее влияние катионов легко устранить, предварительно пропустив
анализируемый раствор через колонку с сильнокислотным катионообменником
в Н+-форме. При этом протекает реакция
2Rz-H+ + (M)2SO4 2Rz-M+ + H2SO4
В результате все катионы замещаются на протоны. Основы ионообменной хро-
матографии изложены в гл. 21.
Титрование выполняют в смешанных водно-органических средах. Присут-
ствие органического растворителя уменьшает степень диссоциации индикатора
и тем самым препятствует образованию комплексов ионов бария с индикатором.
Кроме того, в водно-органических средах осадок получается более рыхлым, что
благоприятствует адсорбции индикатора.
Что мы узнали из этой главы?
• Влияние кислотности на растворимость [основные уравнения (11.4),
(11.6)] —стр. 466
• Расчеты с использованием уравнений материального баланса — стр. 469
• Влияние комплексообразования на растворимость [основное уравнение
(11.11)] —стр. 473
• Расчет кривых осадительного титрования — стр. 475
• Индикаторы в осадительном титровании — стр. 478
Вопросы
1. Объясните суть титриметрического определения хлоридов по Фольгарду и
по Фаянсу. Какой из этих методов следует применять в кислых средах и по-
чему?
2. Объясните принцип действия адсорбционных индикаторов.
484
РЕАКЦИИ ОСАЖДЕНИЯ И ОСАДИТЕЛЬНОЕ ТИТРОВАНИЕ
Задачи
Влияние кислотности на растворимость
3. Рассчитайте растворимость AgIO3 в 0,100 М HNO3. Рассчитайте при этих
условиях равновесные концентрации IO3 и НЮ3.
4 - Рассчитайте растворимость CaF2 в 0,100 М НС1. Рассчитайте при этих усло-
виях равновесные концентрации F- и HF.
5- Рассчитайте растворимость PbS в 0,0100 М НС1. Рассчитайте при этих
условиях равновесные концентрации S2-, HS- и H2S.
Влияние комплексообразования на растворимость
6- Ионы серебра последовательно образуют с этилендиамином (еп) комплек-
сы состава 1 : 1 и 1 :2 с константами устойчивости К?х = 5,5 • 104 и
Kf2 = 1,4 • 103 соответственно. Рассчитайте растворимость хлорида серебра
в присутствии 0,100 М этилендиамина. Рассчитайте также равновесные
концентрации ионов Ag(en)+ и Ag(en) 2.
Расчеты с использованием уравнений материального баланса
При решении задач этого раздела используйте общий подход к расчетам равно-
весий на основе уравнений материального баланса. Результаты расчетов сравни-
те с полученными ранее другими способами.
7- Рассчитайте растворимость AgIO3 в 0,100 М HNO3. Результат сравните с
полученным в задаче 3.
8. Рассчитайте растворимость PbS в 0,0100 М НС1. Результат сравните с полу-
ченным в задаче 5.
9. Рассчитайте растворимость AgCl в присутствии 0,100 М растворе этиленди-
амина. Необходимые значения констант устойчивости приведены в условии
задачи 6. Результат сравните с полученным в задаче 6.
Количественный анализ методом осадительного титрования
10. в соленой воде определяли содержание хлоридов по Фольгарду. К аликво-
те пробы объемом 10,00 мл добавили 15,00 мл стандартного раствора
AgNO3 с концентрацией 0,1182 М. Непрореагировавшее количество ионов
серебра оттитровали стандартным раствором KSCN с концентрацией 0,101 М
до появления красной окраски комплекса FeSCN2+. На титрование израсхо-
довано 2,38 мл раствора. Рассчитайте концентрацию хлоридов в пробе (г/л).
11. В ходе определения хлоридов по Мору была допущена ошибка в приготов-
лении раствора индикатора. В конечной точке титрования концентрация
индикатора в титруемом растворе составила лишь 0,0011 М вместо требу-
емой 0,011 М. Какова в этом случае величина абсолютной погрешности ре-
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
485
зультатов титрования (выраженная в мл титранта), если концентрация
титранта (AgNO3) составляет 0,100 М, а объем раствора в конечной точ-
ке —100 мл? Погрешностями, связанными с собственной окраской раство-
ра индикатора, пренебрегите.
Задачи для решения при помощи электронных таблиц
12. Создайте электронную таблицу для построения кривой титрования 100 мл
0,1 М раствора С1_ 0,1 М раствором AgNO3 (см. рис. 11.1). Вы можете вос-
пользоваться шаблонами, находящимися на компакт-диске. С помощью со-
зданной таблицы постройте кривые титрования при других концентрациях
титруемого вещества и титранта (например, 0,2 М или 0,05 М; концентра-
цию титранта выбирайте равной концентрации титруемого вещества) и по-
смотрите, как при этом изменяется кривая титрования. Имейте в виду, что
при слишком низких концентрациях, особенно вблизи точки эквивалентно-
сти (99,9 или 100,1 мл титранта) применяемый способ расчетов может ока-
заться неправомерным, поскольку в таких условиях необходимо учитывать
растворимость осадка.
Рекомендуемая литература
1. О. Schales, «Chloride» in М. Reiner, ed., Standard Methods of Clinical Chemistry,
Vol. 1, New York: Academic, 1953, pp. 37-42.
Глава 12
ЭЛЕКТРОХИМИЧЕСКИЕ ЯЧЕЙКИ
И ЭЛЕКТРОДНЫЕ ПОТЕНЦИАЛЫ
Я не знаю в подлунном мире ничего,
что не было бы одним из проявлений энергии.
Эдмунд Бёрк
Одним из важнейших разделов титриметрии является окислительно-восстано-
вительное, или редокс-, титрование, основанное на взаимодействии окислителя
и восстановителя [уравнение (12.1)]. Окисление — это процесс перехода элект-
ронов к окислителю от восстановителя, который в результате окисляется, приобре-
тая более высокую степень окисления. Восстановление — процесс присоединения
окислителем отдаваемых восстановителем электронов, в результате которого
окислитель переходит в более низкую степень окисления. Чтобы понять меха-
низм окислительно-восстановительных реакций необходимо иметь представле-
ние об электрохимических ячейках и потенциалах. В данной главе помимо этих
понятий обсуждаются также уравнение Нернста (описывающее эти потенциа-
лы) и ограничения применений потенциалов. В следующей главе, посвященной
потенциометрии, рассмотрены измерения потенциалов для определения кон-
центрации, различные электроды, необходимые для этого, в том числе стеклян-
ный pH-чувствительный электрод и ионселективные электроды, а в гл. 14 —
окислительно-восстановительное титрование, а также потенциометрическое
титрование, измерение потенциала в котором используют для обнаружения ко-
нечной точки титрования. Для выполнения расчетов в титриметрии окисли-
тельно-восстановительные реакции необходимо уметь уравнивать. Правила
уравнивания будут приведены ниже и в гл. 14.
12.1. Что такое окислительно-восстановительные реакции?
Окислительно-восстановительные реакции, иногда называемые редокс-ре-
акциями, — это реакции, протекающие между восстановителем и окислителем:
Oxj + Red2 Red! + Ох2 (12.1)
В этой реакции Oxj (окислитель) восстанавливается до Red!, a Red2 (восстанови-
тель) окисляется до Ох2. Как будет показано ниже, способность вещества быть
окислителем или восстановителем зависит от окислительно-восстановительно-
го потенциала. Окислитель стремится принять электрон (электроны) и восстано-
виться до более низкой степени окисления:
12.2. ЭЛЕКТРОХИМИЧЕСКИЕ ЯЧЕЙКИ: КАКИЕ ЯЧЕЙКИ ИСПОЛЬЗУЮТ АНАЛИТИКИ-ЭЛЕКТРОХИМИКИ?
487
М*++ ие-М(™)+ (12.2)
(пример: Fe3+ + е- -> Fe2+). Восстановитель, наоборот, стремится отдать элект-
рон (электроны) и в результате окисляется:
М*+ -> М(*+")+ + пег (12.3)
(пример: 2Г-» 12 + 2е_). Если окисленная форма (ион метала) образует комплекс,
она становится более устойчивой и будет намного труднее восстанавливаться, по-
этому ее способность принимать электроны будет намного ниже, и особенно, если
восстановленная форма не связывается также легко и прочно в комплекс.
Для лучшего понимания этих процессов необходимо ознакомиться с поня-
тиями электрохимической ячейки и электродного потенциала.
12.2. Электрохимические ячейки:
какие ячейки используют аналитики-электрохимики?
Существуют два вида электрохимических ячеек — гальванические элементы и
электролитические ячейки. В гальваническом элементе самопроизвольно про-
текает химическая реакция, в результате чего он является источником электри-
ческой энергии. Типичные примеры — свинцовая батарея и обычный карманный
фонарик (электрическая энергия вырабатывается лишь при замыкании цепи, как
например, при включении карманного фонарика). Напряжение ячейки (напри-
мер, батарейки) определяется разностью потенциалов двух полуреакций. По
мере протекания реакции ячейка истощается и ее потенциал приближается к
нулю (батарейка садится). В электролитической ячейке, наоборот, электрическая
энергия внешнего источника напряжения необходима для протекания несамопро-
извольной химической реакции в направлении противоположном по сравнению с
реакцией в гальваническом элементе. Примером может служить электролиз
воды. В ячейках обоих типов электрод, на котором происходит окисление, называ-
ют анодом, а электрод, на котором идет восстановление, — катодом. Гальваниче-
ские элементы применяют в потенциометрии (это обсуждается в двух следующих
главах). Электролитические ячейки используют в таких электрохимических ме-
тодах, как вольтамперометрия. Электроактивные вещества, например, ионы ме-
талла, восстанавливаются на электроде, и при соответствующем потенциале в
результате несамопроизвольной реакции протекает ток, величина которого про-
порциональна концентрации электроактивного вещества (гл. 15).
Гальванический элемент и самопроизвольные реакции:
как измерить потенциал?
Рассмотрим окислительно-восстановительную реакцию в гальваническом эле-
менте
Fe2+ + Се4+ Fe3+ + Се3+
(12.4)
488
ЭЛЕКТРОХИМИЧЕСКИЕ ЯЧЕЙКИ И ЭЛЕКТРОДНЫЕ ПОТЕНЦИАЛЫ
Рис. 12.1
Гальванический элемент
Если раствор, содержащий Fe2+, смешать с раствором Се4+, то ионы в полу-
ченном растворе будут обмениваться электронами. Поместим Fe2+ и Се4+ в отде-
льные сосуды и соединим их солевым мостиком как показано на рис. 12.1.
(Солевой мостик позволяет переносить заряды, но предотвращает смешивание
растворов). Реакция (12.4) протекать не будет, поскольку растворы не контакти-
руют. Заметим, что солевой мостик не всегда необходим — он нужет только,
если исходные вещества или полученные на аноде или катоде продукты реагиру-
ют друг с другом и нужно предотвратить их смешивание. Поместим в каждый из
сосудов инертные платиновые проволочки и соединим их, в результате получим
гальванический элемент. С помощью микроамперметра можно обнаружить ток во
внешней цепи. На одном из платиновых электродов (на аноде) окисляется Fe2+:
Fe2+—> Fe3++ е~ (12.5)
Высвобожденные электроны по внешнему проводнику (токоотводу) переходят
на платиновый электрод (катод), находящийся в другом сосуде, на нем восста-
навливается Се4+:
Се4++ е_ —> Се3+ (12.6)
Протекающий процесс обусловлен способностью этих ионов обмениваться
электронами, суммарный результат выражается реакцией (12.4), которая проте-
кала бы и в том случае, если бы ионы Fe2+ и Се4+ находились в одном сосуде.
Платиновые проволочки служат электродами. Потенциал каждого из них обу-
словлен способностью ионов отдавать или принимать электроны и называется
электродным потенциалом. С помощью вольтметра во внешней цепи можно
измерять разность потенциалов этих двух электродов. Она тем больше, чем
выше способность Fe2+ и Се4+ реагировать между собой. Движущую силу хими-
ческой реакции (разность потенциалов) можно использовать, например, для
освещения с помощью электрической лампочки или для запуска мотора, как это
и делают с помощью аккумулятора.
12.2. ЭЛЕКТРОХИМИЧЕСКИЕ ЯЧЕЙКИ: КАКИЕ ЯЧЕЙКИ ИСПОЛЬЗУЮТ АНАЛИТИКИ-ЭЛЕКТРОХИМИКИ?
489
Полуреакции: отщепление и присоединение электронов
Уравнения (12.5) и (12.6) называют полуреакциями. Ни одна из полуреакций не
может протекать самостоятельно: обязательно должен присутствовать и донор
(восстановитель), и акцептор (окислитель) электронов. В рассматриваемом
случае восстановителем является Fe2+, а окислителем — Се4+. Каждая полуреак-
ция характеризуется определенным значением потенциала инертного электро-
да, погруженного в раствор, где эта полуреакция протекает.
Потенциал полуреакции:
относительная характеристика полуреакции
Если бы можно было измерить потенциалы отдельных полуреакций, то сравни-
вая их, можно было бы судить, какие из окислителей и восстановителей будут
реагировать друг с другом. К сожалению, способа измерения потенциала от-
дельного электрода нет, но можно измерить разность потенциалов двух элект-
родов. Потенциал полуреакции*
2Н+ + 2е-^Н2
(12.7)
произвольно принят равным 0,000 В, а электрод, в основе работы которого ле-
жит эта полуреакция, называют стандартным водородным электродом, СВЭ
(встречается и устаревшее название — нормальный водородный электрод). Его
изготавливают из платинированной платины [платиновая пластинка, электроли-
тически покрытая «платиновой чернью»; платиновая чернь катализирует полу-
реакцию (12.7)] и помещают в стеклянную трубку, через которую пропускают
газообразный водород. Относительно потенциала СВЭ измеряют потенциалы
всех других полуреакций. Разность потенциалов полуреакции (12.7) и любой
другой полуреакции измеряют с помощью гальванического элемента. В табли-
цах эти величины располагают в порядке убывания (табл. 12.1). Потенциал зависит
от концентрации, поэтому все стандартные потенциалы измерены при активностях
всех участников полуреакции, равных единице, или парциальных давлениях га-
зообразных веществ, равных 1 атм, как для газообразного водорода в случае
СВЭ. Влияние концентрации на потенциал обсуждается ниже. Более полные
сведения по стандартным потенциалам приведены в Приложении В.
Потенциалы принято приводить для полуреакций, записанных как уравнения
восстановления, поэтому их часто называют восстановительными потенциа-
лами. Мы будем использовать соглашение о знаках электродных потенциалов,
принятое на XVII Конференции ИЮПАК в 1953 г. в Стокгольме. Согласно это-
му соглашению полуреакцию записывают в форме полуреакции восстановле-
ния, и ее потенциал увеличивается по мере увеличения способности окисленной
формы полуреакции к восстановлению.
Полуреакцию можно записать и так Н+ + е ^Н2. Потенциал не зависит от способа записи полуреакции.
490
ЭЛЕКТРОХИМИЧЕСКИЕ ЯЧЕЙКИ И ЭЛЕКТРОДНЫЕ ПОТЕНЦИАЛЫ
Таблица 12.1
Стандартные потенциалы некоторых полуреакций
Полуреакция Е° (В)
Н2О2 + 2Н+ + 2е- = 2Н2О 1,77
МпО;+ 4Н+ + Зе- = МпО2 + 2Н2О 1,695
Се4+ + е- = Се3+ 1,61
МпО4 + 8Н+ + 5е- = Мп2+ + 4Н2О 1,51
Сг2О^ + 14Н+ + бе- = 2Сг3+ + 7Н2О 1,33
МпО2 + 4Н+ + 2е- = Мп2+ + 2Н2О 1,23
210 з + 12Н+ + 1 Ое- = L, + 6Н2О 1,20
Н2О2 + 2е- = гОН- 0,88
Си2 + Г + е- = Си! 0,86
Fe3+ + е- = Fe2+ 0,771
О2 + 2Н+ + 2е- = Н2О2 0,682
12(водн.) + 2е_ = 2J- 0,6197
H3AsO4 + 2Н+ + 2е- = H3AsO3 + Н2О 0,559
1з+2е- = 31- 0,5355
Sn4+ + 2e- = Sn2+ 0,154
S4O^+2e- = 2S2O^" 0,08
2Н+ + 2е' = Н2 0,000
Zn2+ + 2е_ = Zn -0,763
2Н2О + 2е- = Н2 + 2ОН- -0,828
Электродный потенциал полуреакции Sn4+ + 2е~ Sn2+ равен +0,15 В. Дру-
гими словами, потенциал этой полуреакции, измеренный относительно СВЭ, в
ячейке, подобной изображенной на рис. 12.1, должен быть равен 0,15 В. Поско-
льку эта пара имеет более высокий (более положительный) потенциал, чем СВЭ,
ион Sn4+ имеет большую склонность к восстановлению, чем ион Н+. Из сказан-
ного можно сделать следующие выводы:
1 Чем более положителен электродный потенциал полуреакции, тем легче
восстанавливается окисленная форма этой полуреакции. Иначе говоря чем
более положителен электродный потенциал, тем более сильным окис-
лителем являтся окисленная форма и тем более слабым восстановите-
лем является восстановленная форма.
12.2. ЭЛЕКТРОХИМИЧЕСКИЕ ЯЧЕЙКИ: КАКИЕ ЯЧЕЙКИ ИСПОЛЬЗУЮТ АНАЛИТИКИ-ЭЛЕКТРОХИМИКИ?
491
2 . Чем более отрицателен электродный потенциал, тем легче окисляется вос-
становленная форма полуреакции. Другими словами, чем более отрицателен
электродный потенциал полуреакции, тем более слабым окислителем яв-
ляется окисленная форма и тем более сильным восстановителем явля-
ется восстановленная форма.
Поскольку потенциал полуреакции Се4+ + е_ Се3+ является болыцой по-
ложительной величиной, следовательно Се4+ — сильный окислитель, а Се3+ —
слабый восстановитель. И наоборот, потенциал полуреакции Zn2+ + 2е_ Zn от-
рицателен, и поэтому ион Zn2+ — очень слабый окислитель, а металлический
Zn — сильный восстановитель.
Как определить направление
окислительно-восстановительной реакции?
Окисленная форма одной полуреакции способна окислить восстановлен-
ную форму другой полуреакции с более отрицательным (меньшим) потенци-
алом и, наоборот, восстановленная форма способна восстановить окисленную
в полуреакции с более положительным (большим) потенциалом.
В качестве примера рассмотрим две полуреакции
Fe3+ + e~ = Fe2+
£° = 0,771 В
(12.8)
Sn4+ + 2е" = Sn2+ Е° = 0,154 В (12.9)
Есть две возможные реакции между окисленной и восстановленной форма-
ми этих полуреакций. Их можно записать после умножения первой полуреакции
на 2 для уравнивания числа электронов и вычитания одной полуреакции из другой:
2Fe3+ + Sn2+ = 2Fe2+ + Sn4+
(12.10)
и
Sn4++ 2Fe2+ = Sn2+ + 2Fe3+ (12.11)
[Реакция не может протекать между Fe3+ и Sn4+ (оба окислители) и Fe2+ и Sn2+
(оба восстановители)]. Сравнив приведенные выше потенциалы, видим, что
протекать будет реакция (12.10), т. е. восстановленная форма полуреакции (12.9)
с менее положительным потенциалом будет реагировать с окисленной формой
полуреакции (12.8) с более высоким положительным потенциалом. Заметим, что
число отдаваемых и принимаемых электронов должно быть одинаковым в двух
полуреакциях (о способах уравнивания окислительно-восстановительных реак-
ций см. гл. 14).
492
ЭЛЕКТРОХИМИЧЕСКИЕ ЯЧЕЙКИ И ЭЛЕКТРОДНЫЕ ПОТЕНЦИАЛЫ
Пример 12.1
Расположите приведенные ниже окислители и восстановители в порядке убыва-
ния их окислительной и восстановительной способности соответственно: М11О4,
Се3+, Cr3+, IO3, Fe3+, 1~, Н+, Zn2+.
Решение
В соответствии с данными табл. 12.1 окислительная способность приведенных
окисленных форм уменьшается в ряду МПО4, IO3, Fe3+, Н+, Zn2+ (в том же ряду
уменьшается Е° соответствующих полуреакций): МпО 4 — очень сильный окис-
литель, Zn2+ — очень слабый. Для приведенных восстановленных форм восста-
новительная способность убывает в ряду: I-, Сг3+, Се3+. Из них I- — довольно
сильный восстановитель, а Се3+ — очень слабый.
Разность потенциалов, полученная вычитанием потенциала одной полуре-
акции из потенциала другой, характеризует ЭДС ячейки, возникающую при
протекании реакции в гальваническом элементе [уравнение (12.8) минус уравне-
ние (12.9) или 0,771 В - 0,154 В = +0,617 В в приведенном выше примере].* Если
рассчитанная величина положительна, реакция идет в том направлении, как
она записана. Если же получена отрицательная величина, реакция идет в про-
тивоположном направлении. Это является следствием того, что для самопроиз-
вольной реакции изменение свободной энергии отрицательно. При стандартных
условиях изменение свободной энергии
AG° = -hFAF° (12.12)
Поэтому положительная разность потенциалов обеспечивает уменьшение сво-
бодной энергии. Следовательно, сравнение стандартных потенциалов двух по-
луреакций и их знаков позволяет сказать, какая из комбинаций полуреакций
даст отрицательное изменение свободной энергии, соответствующее самопро-
извольному протеканию суммарной реакции. Например, для полуреакции
Се4+/Се3+ £° = +1,61 В (табл. 12.1), а для полуреакции Fe3+/Fe2+ £° = +0,771 В.
Поскольку величина AG° для первой полуреакции более отрицательна, чем для
второй, при вычитании полуреакции для железа из полуреакции для церия по-
лучится уравнение самопроизвольной реакции, для которой изменение сво-
бодной энергии отрицательно. Следовательно, Се4+ будет самопроизвольно
окислять Fe2+.
Далее мы будем употреблять термины «потенциал электрода» и «напряжение (ЭДС) ячейки», чтобы раз-
личать полуреакцию и суммарную окислительно-восстановительную реакцию.
12.2. ЭЛЕКТРОХИМИЧЕСКИЕ ЯЧЕЙКИ: КАКИЕ ЯЧЕЙКИ ИСПОЛЬЗУЮТ АНАЛИТИКИ-ЭЛЕКТРОХИМИКИ?
493
Анод и катод
В схеме ячейки анод принято записывать слева:
анод | раствор | катод
(12.13)
Одинарной чертой обозначают границу раздела фаз электрод—раствор или
раствор—раствор. Ячейку, изображенную на рис. 12.1, схематически можно
представить так
Pt | Fe^CCj), Fe3+(C2) || Се4+(С3), Се3+(С4) | Pt
(12.14)
где С15 С2, С3, С4 — концентрации соответствующих ионов. Двойной чертой
обозначают солевой мостик. Схема ячейки на основе полуреакций Fe3+ + е~ = Fe2+
и Sn4+ + 2е_ = Sn2+ выглядит так
Pt I Sn^Q, Sn4+(C2) II Fe3+(C3), Fe2+(C4) | Pt
(12.15)
Поскольку на аноде происходит окисление, а на катоде — восстановление,
слева записан более сильный восстановитель, справа — более сильный окисли-
тель. Для ЭДС ячейки можно написать следующее выражение
Е1 — Е1 _ Е1 — Е1 — F
^яч ^прав ^лев ^катод ^анод
(12.16)
где Е+ — более положительный электродный потенциал, Е_ — менее положи-
тельный (более отрицательный) электродный потенциал из двух рассматривае-
мых полуреакций.
Если схема записана правильно, ЭДС ячейки будет всегда положительной
и, следовательно, на катоде протекает полуреакция восстановления, на аноде —
полуреакция окисления.
Для ячейки (12.15) при стандартных условиях
^Ч=^/Fe^ - = 0,771 - 0,154 = 0,617 В
Рассмотрим несколько примеров окислительно-восстановительных реак-
ций. Так, Fe3+ не будет окислять Мп2+ и, напротив, МпО4 будет окислять Fe2+.
Окислитель средней силы 12 окисляет Sn2+, а I-, будучи достаточно сильным вос-
становителем, восстанавливает Fe3+, и т. п. Для того, чтобы реакция
протекала количественно и скачок на кривой титрования был четко выражен,
необходимо, чтобы разность потенциалов полуреакций составляла, по крайней
мере, 0,2-0,3 В.
494
ЭЛЕКТРОХИМИЧЕСКИЕ ЯЧЕЙКИ И ЭЛЕКТРОДНЫЕ ПОТЕНЦИАЛЫ
Пример 12.2
Используя величины потенциалов из табл. 12.1, напишите уравнение реакции,
включающей приведенные ниже окислительно-восстановительные полуреак-
ции, и рассчитайте напряжение соответствующей ячейки:
Fe3+ + e- = Fe2+ EQ = 0,771 В
1з +2е- = 31- Е° = 0,5355 В
Решение
Поскольку для пары Fe3+/Fe2+ стандартный потенциал значительно выше, Fe3+
более сильный окислитель, чем 1j, следовательно, Fe3+ окисляет I- и поэтому
£яч = Дсатод - £анод = “ ^Ij/r ’ Д™ написания рваКЦИИ, Протекающей В
ячейке, необходимо поступить аналогично, т. е. вычесть вторую полуреакцию
из первой (после умножения ее на 2):
2Fe3+ + 31- = 2Fe2+ + 1" и Е°ч = 0,771 - 0,536 = +0,235 В
Обратите внимание на то, что при умножении полуреакции на любой мно-
житель (который зависит от числа участвующих в полуреакциях электронов) ее
потенциал не изменяется.
12.3. Уравнение Нернста:
зависимость потенциала от концентрации
Приведенные в табл. 12.1 потенциалы даны для случая, когда активности окис-
ленной и восстановленной форм (и всех других участников) полуреакции равны
единице, и называются стандартными потенциалами, Е°. Первые таблицы эм-
пирических величин потенциалов при определенных и строго контролируемых
условиях составил Вольта. Позднее Нернст показал их практическую важность,
установив соотношение между величиной потенциала и концентрацией. Потен-
циал полуреакции, в отличие от стандартного, зависит от концентрации*, и эта
зависимость описывается уравнением Нернста:
аОх + пе~ ±=ч 6Red
„ „0 2.3026ЛГ, [Red]6
Е = Е —---------1g ----—
nF [Ох]а
(12.17)
(12.18)
Более корректно было бы использовать активности, а не концентрации, но пока мы этого делать не будем,
поскольку при титровании потенциал изменяется весьма существенно и возникающая погрешность будет
мала. Вернемся к этому в следующей главе при обсуждении вопросов, связанных с расчетом концентра-
ции по измеренной величине потенциала.
12.3. УРАВНЕНИЕ НЕРНСТА: ЗАВИСИМОСТЬ ПОТЕНЦИАЛА ОТ КОНЦЕНТРАЦИИ
495
Здесь Е—потенциал при данных значениях концентрации, п — число элек-
тронов, участвующих в полуреакции, R = 8,3143 В • Кл/(моль • К) — универсаль-
ная газовая постоянная, Т — абсолютная температура, F = 96487 Кл/экв. —
постоянная Фарадея. При 25 °C (298,16 К) величина 2,30267?77F составляет
0,05916. При произвольной температуре она равна 1,9842 • Ю-4 (°C + 273,16).
Концентрации чистых веществ, таких как твердые малорастворимые соеди-
нения или растворитель (Н2О), принимаются равными единице. Заметим так-
же, что для полуреакции, записанной в форме полуреакции восстановления, в
числителе выражения под знаком 1g записывают концентрации участников по-
луреакции, стоящих в правой части уравнения, а в знаменателе — концентра-
ции участников, стоящих в левой части.
Пример 12.3
В растворе содержится 1 • 10-3 М Сг2О 2~ и 1 10“2 М Сг3+. Рассчитайте потенциал
полуреакции при pH 2,0.
Решение
Сг2О2- + 14Н+ + бе- 2Сг3+ + 7Н2О
0,059 [Сг3+] 0,059 (110~2)2 _
Сг2О?-/сЛ 6 8|-Сг2О2-][н+]14 ’ 6 81.Ю"3 (1-ю-2)14
= l,33-^^lgl027 =l,33-27f^^ = l,06B
6 I 6 J
Рассчитанный потенциал и есть потенциал, который принимает электрод
(относительно СВЭ), погруженный в данный раствор, и характеризующий окис-
лительную или восстановительную способность раствора. Теоретически, если в
растворе совсем нет Сг3+, потенциал должен быть равен бесконечности. Однако
на практике он всегда будет иметь некоторую конечную величину, хотя ее и не-
льзя рассчитать по уравнению Нернста. Причиной этого может быть наличие
следовых количеств примесей окисленной или восстановленной формы, но бо-
лее вероятно, что в таком случае потенциал определяется какой-то другой полу-
реакцией, например, окислением или восстановлением воды.
Равновесный потенциал: после завершения реакции
Потенциал инертного электрода, погруженного в титруемый раствор, который
одержит ионы двух окислительно-восстановительных полуреакций после уста-
новления равновесия (т. е. в различные моменты титрования), можно рассчитать
496
ЭЛЕКТРОХИМИЧЕСКИЕ ЯЧЕЙКИ И ЭЛЕКТРОДНЫЕ ПОТЕНЦИАЛЫ
по уравнению Нернста (относительно СВЭ), используя данные для любой из
двух полуреакций. Это обусловлено тем, что при равновесии потенциалы обеих
полуреакций одинаковы, иначе реакция продолжалась бы. Электрод, погружен-
ный в такой раствор, приобретет равновесный потенциал. Его можно рассчи-
тать по уравнению Нернста, если известны равновесные концентрации всех
участников хотя бы одной полуреакции. Для построения кривой титрования не-
обходимо рассчитать потенциал в момент, когда добавленная порция титранта
полностью прореагировала с титруемым веществом, т. е. равновесный потенци-
ал (ЭДС ячейки равна нулю).
Пример 12.4
К 5,0 мл 0,30 М раствора Fe2+ прибавили 5,0 мл 0,10 М раствора Се4+. Рассчитай-
те потенциал платинового электрода (относительно СВЭ), погруженного в этот
раствор.
Решение
В исходном растворе было 0,30 • 5,0 =1,5 ммоль Fe2+. После прибавления
0,10 • 5,0 = 0,50 ммоль Се4+ образовалось по 0,50 ммоль Fe3+ и Се3+ и осталось
1,0 ммоль Fe2+. Если разность стандартных потенциалов полуреакций составля-
ет не менее 0,2 В, равновесие реакции смещается вправо, но при этом остается
небольшое количество ммоль Се4+ (= х) и образуется равное количество Fe2+:
Fe2+ +Се4+^ Fe3+ + Се3+
1,0 + х х 0,50-х 0,50-х
Величина х очень мала по сравнению с 0,5 или 1,0 и ею можно пренебречь.
Для расчета потенциала можно использовать любую из полуреакций, но посколь-
ку известны концентрации окисленной и восстановленной форм пары Fe3+/Fe2+,
удобно воспользоваться именно ею:
Fe3+ + е- Fe2+
0,50 1,0
£ = 0,771-0,059 1g 15^
[Fe3+]
Конечный объем раствора равен 10,0 мл, поэтому
Е = 0,771 - 0,059 1g 1,0 ммоль/10 мл = о,771 - 0,0591g 2,0 = 0,771 - 0,059 • 0,3 = 0,753 В
0,50 ммоль/10 мл
12.3. УРАВНЕНИЕ НЕРНСТА: ЗАВИСИМОСТЬ ПОТЕНЦИАЛА ОТ КОНЦЕНТРАЦИИ
497
ЭДС ячейки до начала реакции
ЭДС ячейки можно вычислить по разности потенциалов двух полуреакций, рас-
считанных по уравнению Нернста, согласно (12.16):
ЕЯЧ = Е+~Е-
(12.19)
В примере 12.2 для реакции 2Fe3+ + ЗГ~ = 2Fe2+ + Г3 получаем
г -г г -( г° 0,059, [Fe2+]2] [„о
Еяч £Fe^/F^ £i-/r £Fe^/Fe2+ 6 lg f 3+ 2 £Ц/г
\ и 1Ае J у V
0,0591п [Ре2+]2[Г]
Fe^/Fe^ Ij/г 2 8 [Fe3+ ]2[I3 ]3
0,0591£ [Г ]J =
2 [IJ] J
(12.20)
Обратите внимание на то, что для самопроизвольно протекающей реакции
под знаком 1g всегда стоит отношение концентрации (полуреакций) продуктов
реакции к концентрации (полуреакциям) исходных веществ (реагентов), как и
для полуреакции, записанной в форме восстановления. Отметим также необхо-
димость умножения полуреакции Fe3+/Fe2+ на 2 (как это уже делали при вычи-
тании одной полуреакции из другой), чтобы можно было объединить два
логарифмических слагаемых (п = 2). Идентичное выражение можно было бы по-
лучить, используя уравнение реакции, протекающей в ячейке. Заметим также,
4T0£FeW+-£Ij/r еСТЬ£яч‘
Под знаком 1g в правой части уравнения (12.20) стоит выражение констан-
ты равновесия реакции
2Fe3+ + 3I"^2Fe2+ + l3
(12.21)
По величине ЭДС ячейки можно судить о возможности протекания реакции
после соединения ее участников (подобно тому, как это происходит в эле-
менте питания), т. е. величина ЭДС определяет принципиальную возмож-
ность совершения работы/выделения энергии. При достижении равновесия
ЭДС ячейки всегда становится равной нулю и реакция завершается (ячейка
перестает быть источником энергии). Следовательно, потенциалы двух полу-
реакций при равновесии равны. Это и происходит, когда батарея «садится».
498
ЭЛЕКТРОХИМИЧЕСКИЕ ЯЧЕЙКИ И ЭЛЕКТРОДНЫЕ ПОТЕНЦИАЛЫ
Пример 12.5
В один сосуд поместили раствор, содержащий 0,0200 М КМпО4, 0,00500 М
MnSO4 и 0,500 М H2SO4, а в другой — раствор, содержащий 0,150 М FeSO4 и
0,00150 М Fe2(SO4)3. Сосуды соединили солевым мостиком и поместили в них
платиновые электроды, которые с помощью токоотводов подсоединили к вольт-
метру. Рассчитайте потенциалы каждого электрода: а) до начала реакции и
б) по завершении реакции; а также ЭДС ячейки: в) в начале реакции и г) после
установления равновесия. При расчетах полагать, что H2SO4 полностью диссо-
циирована, а объемы растворов в обоих сосудах равны.
Решение
Запишем уравнение реакции в гальваническом элементе
5Fe2+ + МпО; + 8Н+ 5Fe3+ + Мп2+ + 4Н2О
и его схему
Pt | Fe2+(0,150 М), Fe3+(0,00300 М) || МпО4 (0,0200 М), Мп2+(0,00500 М), Н+(1,00 М) | Pt
Э) £F 2+ -0,059lg= 0,771-0,059 lg 0,150 =0,671 В
Fe Fe /Fe [Fe3+] 6 0,00300
„ 0,059, [Mn2+] 0,059, 0,00500 _.„D
мп мпо;/мп2+ 5 g [Mn0-][H+]8 ’ 5 ё 0,0200 (1,00)8
б) При равновесии EFe = £Mn, поэтому для расчета можно использовать стандар-
тный потенциал любой полуреакции. Сначала рассчитаем концентрации
всех частиц при установлении равновесия. Из уравнения реакции видно, что
5 моль Fe2+ реагирует с 1 моль МпО4, поэтому Fe2+ будет в избытке. Хотя
концентрация этих ионов уменьшится на 5 • 0,0200 = 0,100 М, в растворе
останется 0,050 М Fe2+. В результате реакции образуется 0,100 М Fe3+ и его
концентрация составит 0,100 + 0,003 = 0,103 М. Поскольку фактически весь
МпО 4 восстановлен до Мп2+ (0,0200 М), то общая концентрация последнего
равна 0,0250 М. При равновесии остается небольшое количество МпО4, ко-
торое можно рассчитать из величины константы равновесия; константу,
в свою очередь, можно вычислить из условия, что в состоянии равновесия
£яч = 0 [см. уравнение (12.20) и гл. 14]. Но этого не потребуется, так как изве-
стны концентрации [Fe2+] и [Fe3-*"]:
£Мп = Еъ = 0,771 - 0,059 1g = 0,790 В
Mn Fe 6 0,103
в) £яч = £Мп - ЕГе = 1,52 - 0,671 = 0,85 В.
12.4. ФОРМАЛЬНЫЙ ПОТЕНЦИАЛ: ИСПОЛЬЗУЙТЕ ЕГО В НЕСТАНДАРТНЫХ УСЛОВИЯХ
499
г) При равновесии EMn = EFe и Еяч = О В.
Заметим, что если одна из форм полуреакции изначально отсутствует, рас-
считать исходную величину потенциала невозможно.
12.4. Формальный потенциал:
используйте его в нестандартных условиях
Величины Е° в табл. 12.1 приведены для случая, когда активности всех участни-
ков полуреакции равны 1 М, но потенциал полуреакции зависит от конкретного
состава раствора. Например, Е° для полуреакции Се4++ е~ Се3+ равен 1,61 В,
но его можно изменить, подкисляя раствор разными кислотами (см. табл. С5 в
Приложении С). Изменение потенциала обусловлено тем, что анион кислоты свя-
зывает ион Се4+ в комплекс и концентрация своюодных ионов Се4+ уменьшается.
Если состав образующегося комплекса известен, можно записать уравнение
новой полуреакции с участием аниона кислоты и рассчитать величину Е° для
этой полуреакции при активностях аниона кислоты и всех других участников
полуреакции, равных единице. Состав комплекса, однако, часто бывает неизве-
стен, и в таких случаях можно воспользоваться формальным потенциалом, Е°'.
Формальный потенциал — это потенциал полуреакции при общих концентраци-
ях окисленной и восстановленной форм полуреакции, равных 1 М, и известных
концентрациях других участников полуреакции. Например, формальный потен-
циал пары Се4+/Се3+ в 1 М НС1 равен 1,28 В. Для расчетов можно использовать
уравнение Нернста, подставляя в него величину формального потенциала вмес-
то стандартного. Величины формальных потенциалов для некоторых полуреак-
ций приведены в табл. С.5. Формальный потенциал используют если достоверно
не известно, какие именно частицы участвуют в полуреакции.
Зависимость потенциала от pH
Во многих окислительно-восстановительных полуреакциях участвуют протоны
или гидроксид-ионы, поэтому изменяя pH раствора, можно изменять величину
потенциала таких полуреакций. Рассмотрим сопряженную окислительно-вос-
становительную пару As(V)/As(III):
H3AsO4 + 2Н+ + 2е_ H3AsO3 + Н2О
Е = Е° - О’059 ig [H3AsO3]
2 [H3AsO4][H+]2
(12.22)
(12.23)
500
ЭЛЕКТРОХИМИЧЕСКИЕ ЯЧЕЙКИ И ЭЛЕКТРОДНЫЕ ПОТЕНЦИАЛЫ
После преобразования* получаем
Е = Е° + 0,059 1g [Н+] - 1g [H3AsOs]
2 6[H3AsO4]
ИЛИ
Е = Е° - 0,059 pH - 1g tH3AsO3]
2 [H3AsO4]
(12.24)
(12.25)
В последнем уравнении разность (Е° - 0,059 pH), где Е° — стандартный по-
тенциал полуреакции, можно полагать равной формальному потенциалу Е°', и
его можно рассчитать, зная pH раствора.** Так, в 0,1 М в растворе НС1 (pH 1)
Е°' = Е° - 0,059, а в нейтральной среде Е°' =Е° - 0,059 • 7 = Е° - 0,41.
В сильнокислом растворе H3AsO4 будет окислять I- до 12, но для пары
As(V)/As(III) в нейтральной среде Е°' = 0,146 В. Это меньше потенциала пары
12/1“, и поэтому реакция будет протекать в противоположном направлении, т. е.
12 будет окислять H3AsO3.
Влияние комплексообразования на величину потенциала
Если связать в комплекс один из ионов сопряженной окислительно-восстанови-
тельной пары, ее потенциал изменится за счет снижения равновесной концент-
рации этого иона. Например, для пары Fe3+/Fe2+ Е° = 0,771 В. В растворе НС1
катионы Fe3+ образуют хлоридные комплексы, в результате чего снижается кон-
центрация Fe3+ и потенциал полуреакции уменьшается. В 1 М растворе НС1 фор-
мальный потенциал этой пары равен 0,70 В. Предположим, что образуется
комплекс FeCl4, тогда полуреакцию следует записать так:
FeCl4 + е- Fe2+ + 4С1"
(12.26)
При постоянной равновесной концентрации [НС1] = 1 М
£ = 0,70-0,059 lg [Fe2+]
[FeCl4]
(12.27)
При связывании Fe3+ в комплекс, эта форма стабилизируется — ее стано-
вится труднее восстановить. Поэтому потенциал уменьшается. Если в комплекс
связать Fe2+, будет наблюдаться обратная ситуация.
* Если выразить 1g [Н+] в виде (-0,059/2)lg(l/[H+]2) = (+0,059/2) lg[H+]2, то после преобразования получим
0,059 1g [Н+].
** В данном случае дана очень приближенная оценка влияния pH. Поскольку и H3AsO4, и H3AsO3 являются
слабыми кислотами, необходимо принять во внимание константы их кислотности Ка.
ВОПРОСЫ
501
12.5. Ограничения при использовании электродных потенциалов
Величины электродных протенциалов (£° или Е°') позволяют судить о возмож-
ности протекания данной реакции, но они ничего не говорят о скорости ее про-
текания. Если полуреакция обратима (скорость переноса электронов достаточно
высока), она будет протекать со скоростью, достаточной для титриметрии. Но
если скорость переноса электронов мала, реакция будет протекать настолько
медленно, что для установления равновесия потребуется слишком много време-
ни. Такие полуреакции называют необратимыми.
Некоторые окислительно-восстановительные реакции, в которых необрати-
ма одна полуреакция, протекают быстро. Необратимое восстановление некото-
рых окислителей или окисление некоторых восстановителей, содержащих атомы
кислорода, можно ускорить с помощью подходящего катализатора. Например,
мышьяк(Ш) окисляется церием(1У) очень медленно, но реакция ускоряется в
присутствии следовых количеств тетраоксида осмия OsO4.
Итак, электродные потенциалы удобно использовать для предсказания воз-
можности протекания многих реакций, но зачастую они не позволяют судить о
том, насколько успешно можно использовать данную реакцию в титриметрии.
Что мы узнали из этой главы?
• Гальванический элемент — стр. 487
• Использование стандартных потенциалов для определения направления ре-
акции — стр. 491
• Анод, катод и напряжение ячейки [основное уравнение (12.16)] — стр. 493
• Уравнение Нернста [основное уравнение (12.18)] — стр. 494
• Вычисление потенциалов электродов до начала реакции и после ее завер-
шения — стр. 495
• Формальный потенциал — стр. 499
Вопросы
1 Что такое окислитель? Что такое восстановитель?
2. Напишите уравнение Нернста и поясните его суть.
3. Что такое стандартный потенциал? Что такое формальный потенциал?
4. Какова роль солевого мостика в электрохимической ячейке?
5. Как устроен стандартный водородный электрод?
6. Стандартный потенциал полуреакции М4+ + 2е_ М2+ равен +0,98 В.
М2+ — сильный или слабый восстановитель?
7. При какой минимальной разности потенциалов двух полуреакций скачок
на соответствующей кривой титрования выражен четко и легко определить
конечную точку титрования?
502
ЭЛЕКТРОХИМИЧЕСКИЕ ЯЧЕЙКИ И ЭЛЕКТРОДНЫЕ ПОТЕНЦИАЛЫ
8- Почему по величине стандартного или формального потенциала не всегда
можно судить о пригодности данной реакции для титриметрии?
Задачи
Окислители и восстановители
9. Расположите приведенные ниже окислители в порядке убывания их окис-
лительной способности
H2SeO3, H3AsO4, Hg2+, Cu2+, Zn2+, O3, HC1O, K+, Co2+.
10. Расположите приведенные ниже восстановители в порядке убывания их
восстановительной способности
I", V3+, Sn2+, Со2+, Cl- Ag, H2S, Ni, HF.
11. Для какой из приведенных ниже пар будет наблюдаться больший скачок на
кривой титрования одного из компонентов каждой пары другим:
(a) Fe2+—МпО; илиРе2+—Сг2О^“
(б) Fe2+—Се4+ (H2SO4) или Fe2+—Се4+(НС1О4)
(в) H3AsO3—МпО4 илиРе2+—МпО4
(г) Fe3+—Ti2+ или Sn2+—13
Гальванические элементы
12. Напишите схемы гальванических элементов для каждой из приведенных
ниже реакций, полагая концентрации всех участников равными 1 М:
(a) 6Fe2+ + Сг2О + 14Н+ 6Fe3+ + 2Сг3+ + 7Н2О
(б) Ю3 + 51- + 6Н+ 312 + ЗН2О
(в) Zn + Cu2+ Zn2+ + Си
(г) С12 + H2SeO3 + Н2О 2С1- + SeO^ + 4Н+
13. Для каждой из приведенных ниже ячеек напишите уравнение протекающей
в ней реакции:
(a) Pt | V2+, V3+1| PtCl 4“, PtCl^+, Cl-1 Pt
(6) Ag | AgCl (tb.) | Cl-1| Fe3+, Fe2+| Pt
(в) Cd | Cd2+ || СЮ3, Cl-, H+| Pt
(r) Pt | Г,I2 ||H2O2,H+|Pt
Расчет потенциала
14. Рассчитайте потенциал платинового электрода (отн. СВЭ), погруженного в
раствор, содержащий 0,50 М КВгО3 и 0,20 М Вг2 (pH 2,5).
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
503
15- Рассчитайте потенциал платинового электрода (отн. СВЭ) в растворе, по-
лученном при добавлении 90 мл 5,0 М KI к 10 мл 0,10 М раствора Н2О2, pH
которого доведено буферным раствором до 2,0.
16- В 3,0 М растворе НС1 ионы Pt4* и Pt2+ образуют хлоридные комплексы (см.
задачу 18). Рассчитайте потенциал полуреакции в растворе, содержащем
0,015 М Pt4* и 0,025 М Pt2*.
17 - Смешали равные объемы 0,100 М UO и 0,100 М V2*. Рассчитайте равно-
весный потенциал (отн. СВЭ) платинового электрода, погруженного в этот
раствор. Считать, что H2SO4 диссоциирует полностью.
Расчет ЭДС ячеек
18. Исходя из величин стандартных потенциалов полуреакций напишите урав-
нение самопроизвольно протекающей в ячейке реакции и рассчитайте ЭДС:
PtCl + 2е- PtCl + 2С1- Е° = 0,68 В
V3+ + e-^V2+ Я0 = -0,255 В
19. Рассчитайте ЭДС следующих ячеек:
(a) Pt11-(0,100М),Ц(0,0100М)|| Юз(0,100М), 12(0,0100М),Н+(0,100М) |Pt
(б) Ag | AgCl(TB.)| Cl-(0,100 М) || UO4+(0,200 М), U4+ (0,050 М), Н+ (1,00 М) | Pt
(в) Pt I Т1+ (0,100 М), Tl3+(0,0100 М) II МпО; (0,0100 М), Мп2+(0,100 М),
Н+(рН 2)1 Pt
21 - По величинам стандартных потенциалов полуреакций определите направ-
ление реакции и рассчитайте ЭДС соответствующей ячейки:
VO^ + 2Н* + еVO2* + Н2О
ио^+ + 4Н+ + 2е- и4+ + 2Н2О
Рекомендуемая литература
Уравнение Нернста
1. L. Meites, «А ‘Derivation’ of the Nemst Equation for Elementary Quantitative
Analysis», J. Chem. Ed., 29 (1952) 142.
Соглашение о знаках электродных потенциалах
2. F. С. Anson, «Electrode Sign Convention», J. Chem. Ed., 36 (1959) 394.
3. T. S. Light and A. J. de Bethune, «Recent Developments Concerning the Signs of
Electrode Conventions», J. Chem. Ed., 34 (1957) 433.
Стандартные потенцалы
4. A. J. Bard, R. Parsons, and J. Jordan, eds., Standard Potentials in Aqueous Soluti-
on. New York: Marcel Dekker, 1985.
5. W. M. Latimer, The Oxidation States of the Elements and Their Potentials in
Aqueous Solutions, 2nd ed. New York: Prentice Hall, 1952.
Глава 13
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ
И ПОТЕНЦИОМЕТРИЯ
В гл. 12 уже упоминалось об измерении потенциала полуреакции с помощью
платинового электрода. Это был частный случай; известно много электродов,
пригодных для измерения потенциалов. В данной главе рассматриваются раз-
личные виды таких электродов и способ выбора наиболее подходящего из них
для определения интересующего компонента, приборы для потенциометриче-
ских измерений и их метрологические характеристики. Описан один из важней-
ших электродов — стеклянный, а также стандартные буферные смеси для его
калибровки. Обсуждаются различные типы ионселективных электродов, при-
менение которых в потенциометрическом титровании будет рассмотрено
в гл. 14.
Отличительная особенность электродов, используемых в потенциометрии,
заключается в том, что их потенциалы являются функцией активности ионов,
а не концентрации, в связи с чем все расчеты потенциалов в данной главе выпол-
нены с использованием этой характеристики. При проведении прямых потенцио-
метрических измерений с помощью ионселективных электродов, например, при
измерении pH с помощью стеклянного электрода, очень важно понимать, что та-
кое активность и какие факторы на нее влияют. Поэтому необходимо повторить
материал гл. 6, касающийся активности и коэффициентов активности.
13.1. Металлические электроды и определение катионов
Электроды этого типа представляют собой металл (пластинка, проволока), нахо-
дящийся в контакте с раствором собственного катиона. Примером может слу-
жить металлическое серебро, погруженное в раствор нитрата серебра.
Для любого металлического электрода можно записать протекающую на
нем и обуславливающую его потенциал полуреакцию. В общем виде это можно
представить так: М | Mw+, где одинарной линией обозначена граница раздела
электрод—раствор. Для серебряного электрода можно записать
Ag | Ag
(13.1)
13.1. МЕТАЛЛИЧЕСКИЕ ЭЛЕКТРОДЫ И ОПРВДЕЛЕНИЕ КАТИОНОВ
505
и соответствующую полуреакцию
Ag+ + е~ Ag
Потенциал электрода описывается уравнением Нернста
„ _,о 2,303ЯГ, 1
y-lg —
Ag
(13.2)
(13.3)
где aAg+ — активность ионов серебра (см. гл. 6). В данной главе мы будем опе-
рировать именно этим более корректным понятием, поскольку при интерпретации
результатов прямых потенциометрических измерений возникает значительная по-
грешность из-за использования при вычислениях концентраций вместо активно-
стей.
По уравнению (13.3) рассчитывают величину потенциала относительно
стандартного водородного электрода (СВЭ — см. разд. 13.3). С повышением
концентрации (активности) Ag+ потенциал растет (справедливо для любого
электрода, пригодного для измерения концентрации катионов); т. е. для ячейки,
в которой вторым полуэлементом является СВЭ,
^изм = ^яч = ^ИЭ, отн. СВЭ = ^ИЭ “ ^СВЭ (13.4)
где £иэ — потенциал индикаторного электрода (электрода, дающего отклик
на присутствие в растворе определяемого компонента, в данном случае Ag+).
Поскольку £свэ принят за нуль, то
^яч = ^иэ (13-5)
В соответствии с записью схемы ячейки
раствор |ЕИЭ (13.6)
получаем
^яч = ^прав “ Дтев = ^ИЭ ~ ^ср = ^ИЭ “ Const (13.7)
где Еср — потенциал электрода сравнения — электрода с постоянным потенци-
алом. Наличие электрода сравнения в ячейке обязательно, но он не дает отклика
на присутствие определяемого компонента. Обычно его отделяют от анализиру-
емого раствора солевым мостиком. Заметим, что величина Еяч (или £иэ) может
быть положительной или отрицательной в зависимости от активности ионов се-
ребра или разности потенциалов двух электродов. Это противоречит ранее
сформулированному утверждению (гл. 12) в отношении гальванического эле-
мента, согласно которому его потенциал всегда положителен и тем самым ука-
зывает на протекание в ячейке самопроизвольной реакции. В потенциометрии
принято измерять потенциал при отсутствии тока во внешней цепи, так как при
этом не нарушается соотношение концентраций окисленной и восстановленной
506
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
форм у поверхности индикаторного электрода, определяющее величину потен-
циала (измерение потенциала описано ниже). Нас интересует, как изменяется
при изменении концентрации определяемого компонента потенциал индикатор-
ного электрода (зонда), измеряемый относительно электрода сравнения с прак-
тически постоянным потенциалом. Уравнение (13.7) преобразовано так, что
изменение величины Еяч адекватно отражает изменение величины £иэ, включая
его знак. Это будет рассмотрено далее при обсуждении ячеек и способов измере-
ния потенциала.
Поскольку активность металлического серебра, как и любого другого чис-
того вещества, например Си, Н2О, принята равной единице, то в уравнении
Нернста эти величины в явном виде не фигугируют, поэтому серебряный элект-
род можно применять для мониторинга активности ионов серебра в растворе.
Известно всего несколько электродов этого типа, поскольку поверхность мно-
гих металлов покрывается оксидными пленками, влияющими на их потенциалы.
13.2. Электроды металл - соль металла для определения анионов
В общем виде схему этого типа электродов можно представить следующим об-
разом: М | MX | Xw~, где MX — малорастворимая соль. Для электрода серебро -
хлорид серебра (хлорсеребряного)
Ag | AgCl(TB.) | Cl-
(13.8)
В дальнейшем будем использовать следующие обозначения: (тв.) — для ма-
лорастворимого соединения, (г) — для газообразного соединения и (ж) — для
чистого растворителя. Вертикальной линией обозначают границу раздела двух
разных твердых фаз или твердой фазы и раствора. Для соответствующей полуре-
акции
AgCl + е- Ag + С1_
(13.9)
потенциал описывается уравнением
2.303ЯГ.
^AgCl/Ag р ас\~
(13.10)
Этот электрод пригоден для измерения активности хлорид-ионов в растворе.
Обратите внимание на то, что с увеличением активности хлорид-ионов потенци-
ал уменьшается. Это справедливо для любого электрода, пригодного для опре-
деления анионов. Серебряная проволока покрыта осадком хлорида серебра [его
получают, например, при электролитическом окислении серебра в растворе, со-
держащем хлорид-ионы, в соответствии с уравнением обратимой реакции (13.9)].
Но, поскольку серебряная проволока при погружении в раствор, содержащий
хлорид-ионы, моментально покрывается тонким слоем хлорида серебра, предва-
рительной подготовки электрода обычно не требуется.
13.2. ЭЛЕКТРОДЫ МЕТАЛЛ - СОЛЬ МЕТАЛЛА ДЛЯ ОПРВДЕЛЕНИЯ АНИОНОВ
507
Заметим, что этот электрод обычно применяют для измерения ясг, но он
пригоден и для измерения aAg+, которая определяется растворимостью малора-
створимой соли. Так как ясг =KSI a Ag+, уравнение (13.10) можно преобразовать
следующим образом:
^ = ^gClAg-^^:ig— (13.И)
г а а +
Ag
^ = ^gci,Ag-^^lg^ (13.12)
5 6 F F aK +
Ag
Сравнив полученное выражение с уравнением (13.3), видим, что
= 2,303ЯТ
Е Ag+ZAg AAgCL Ag ~ [%KS
(13.13)
Фактически здесь под Ks следует понимать термодинамическое произведение
растворимости K°s (см. гл. 6), поскольку при выводе уравнения использованы
активности. Если в уравнении (13.3) заменить aAg+Ha Ks/acx-, можно получить
другую форму уравнения (13.10) — см. пример 13.1.
В растворе, содержащем смесь Ag+ и С1-, например при титровании С1- с по-
мощью Ag+, их концентрации при равновесии таковы, что потенциал серебряно-
го электрода, погруженного в этот раствор, можно рассчитать и по уравнению
(13.3), и по уравнению (13.10). Это полностью согласуется с утверждением, что
при равновесии потенциалы обеих полуреакций равны (см. гл. 12). В данном
случае полуреакции представлены уравнениями (13.2) и (13.9), и результатом
вычитания одной из них из другой будет суммарная химическая реакция'.
Ag+ + Cl-^ AgCI
(13.14)
Заметим, что при титровании С1- раствором Ag+ концентрация хлорид-ионов
уменьшается, а ионов серебра—увеличивается. Из уравнения (13.10) видно, что
потенциал увеличивается по мере уменьшения концентрации С1-, а из уравнения
(13.12) следует, что потенциал увеличивается с повышением концентрации Ag+.
Серебряный электрод можно использовать для определения и других анио-
нов, образующих с ионами Ag+ малорастворимые соединения, например, I", Вг-
и S2-. Величина Е° в каждом конкретном случае будет определяться полуреак-
цией AgX + е- Ag + X-.
Другим широко используемым электродом рассматриваемого типа является
каломельный электрод, Hg | Hg2Cl2(TB.)| Cl-. Более детально он будет описан в
разделе, посвященном электродам сравнения.
508
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
Пример 13.1
Из величин стандартных потенциалов для каломельного электрода (0,268 В) и
Hg/Hg2+-3neKTpofla (0,789 В) рассчитайте величину Ks для Hg2Cl2.
Решение
Для полуреакции Hg2++ 2е Hg
£ = 0,789-^lg^-
2 aHg^+
(1)
Для полуреакции Hg2Cl2 + 2е_ 2Hg + 2С1-
17 поде 0,05916, , ч2
Е = 0,268-------— 1g (асг у
Поскольку Ks = a Hg2+ («С1- )2, следовательно
Е = 0,268 - 0,059161g
2 aHg*
«7 пом 0,05916, „ 0,05916, 1
Е = 0,268 - —------1g Ks - —----1g-----
2 2
(2)
(3)
(4)
Из (l) и (4) получаем:
П-7СО 0,05916, „
0,789 = 0,268 - -2—-— 1g Ks
отсюда Ks = 2,44 • 10-18
13.3. Окислительно-восстановительные электроды -
инертные металлы
Окислительно-восстановительный электрод — это электрод, изготовленный
из инертного металла и погруженный в раствор, содержащий и окисленную, и
восстановленную формы полуреакции. Об этом типе электродов упоминалось
в гл. 12.
Инертным металлом обычно является платина. Потенциал инертного ме-
таллического электрода зависит от соотношения у поверхности электрода актив-
ностей восстановленной и окисленной форм полуреакции и для полуреакции
Ма++ие-^М(а-")+
(13.15)
13.3. ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ ЭЛЕКТРОДЫ - ИНЕРТНЫЕ МЕТАЛЛЫ
509
описывается уравнением Нернста
2,3037?Г.
£ ~ -----—------------- (13.16)
nF ам-
Примером может служить измерение соотношения МпОд/Мп2* с помощью
платинового электрода для полуреакции
МпО; + 8Н+ + 5е- Мп2+ + 4Н2О (13.17)
F — F° 2,303/? Г. лмп2+ (И 18^
Е - ----Т7— 1g--------------у (13.18)
5F *MnO4-
Величину pH обычно поддерживают постоянной, и поэтому в процессе
окислительно-восстановительного титрования можно измерять отношение
аМп2+^Мпо; *
Очень важным примером электрода рассматриваемого типа является водо-
родный электрод, Pt | Н2, Н+:
Н+ + е-^1Н2 (13.19)
Е = Е° 2,303ЛГ (Рн2)1/2
н+/н2 F ё а
н
Схематическое изображение водородного электрода приведено на рис. 13.1.
Слой платиновой черни электролитически осаждают на поверхности платино-
вого электрода (катода) из раствора H2PtCl6. Платиновая чернь увеличивает пло-
щадь поверхности электрода и тем самым способствует адсорбции молекул
водорода, а также катализирует процесс их окисления. Однако, если платиновой
черни слишком много, то могут адсорбироваться следовые количества других
соединений, например, органических веществ или H2S, что может привести к
возникновению погрешности.
Для газообразных веществ, в данной ситуации — водорода, в уравнении Не-
рнста вместо активности используют давление Р, выраженное в атмосферах.
При постоянном давлении газообразного водорода, равном 1 атм, для полуреак-
ции (13.19) величина Е° принята равной нулю и поэтому зависимость потенциа-
ла водородного электрода от pH описывается уравнением
2,303Р7\ 1 2,3037?Г тт
Е =--—- 1g----=----——pH
F aw F
(13.21)
510
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
Рис. 13.1
Водородный электрод
Пример 13.2
Рассчитайте pH раствора, если потенциал водородного электрода в этом раство-
ре при 25 °C, измеренный при давлении водорода 1,012 атм (с поправкой на дав-
ление паров воды при 25 °C; давление паров воды над раствором неободимо
вычесть из измереного давления газа) равен -0,324 В (относительно СВЭ).
Решение
По уравнению (13.20) находим
-0,324 = -0,059161g (1,012)1/2 = -0,05916 lg (1,012)1/2 - 0,05916 pH
pH = 5,48
Несмотря на то что водородный электрод в некоторых случаях просто необ-
ходим (например, при измерении стандартных потенциалов или pH стандарт-
ных буферных смесей — эти вопросы рассматриваются ниже), для рутинных
измерений pH его обычно не применяют. Причины заключаются в том, что элек-
трод сложен в изготовлении и эксплуатации, парциальное давление водорода
необходимо регулировать в соответствии с температурой и, наконец, раствор не
должен содержать окислители или восстановители, влияющие на измеряемый
потенциал электрода.
13.4. ГАЛЬВАНИЧЕСКИЕ ЭЛЕМЕНТЫ БЕЗ ЖИДКОСТНОГО СОЕДИНЕНИЯ
511
13.4. Гальванические элементы без жидкостного соединения -
ячейки, дающие наиболее точные результаты
Для измерения потенциала необходима «полная» ячейка, состоящая из двух по-
луэлементов (см. гл. 12). Один полуэлемент представляет собой анализируемый
раствор с погруженным в него индикаторным электродом, потенциал которо-
го зависит от концентрации определяемого компонента. Выбор другого полу-
элемента — электрода сравнения — может быть произвольным при условии,
что его потенциал не зависит от концентрации определяемого компонента. Если
его потенциал постоянен, то, во-первых, измеренную ЭДС ячейки можно счи-
тать потенциалом индикаторного электрода относительно данного электрода
сравнения и, во-вторых, любое изменение потенциала индикаторного электрода
вызовет адекватное изменение ЭДС ячейки.
Известно два вида ячеек — с солевым мостиком и без него. Последние назы-
вают ячейками без жидкостного соединения. Например:
Pt | Н2(г), НС1(р-р) | AgCl(TB.) | Ag (13.22)
Вертикальная черта обозначает границу раздела электрод-раствор. Элект-
рохимическая ячейка такого типа является гальваническим элементом, и если ее
схема записана в соответствии с принятым соглашением, т. е. для самопроизволь-
но протекающей в ней реакции, то Еяч > 0. Водородный электрод является анодом,
поскольку его потенциал меньше (см. разд. гл. 12, где обсуждается соглашение о
правилах записи схемы гальванического элемента). Такую ячейку используют
для точного измерения pH стандартных буферных смесей — см. разд. 13.12. По-
тенциал левого электрода можно рассчитать по уравнению (13.20), потенциал
правого электрода — по уравнению (13.10), а Еяч — по их разности:
р _f„o 2,303ЯГ. L» 2,303ЯТ. Он2 )'/2
Еяч -^AgCl/Ag „ ^Н*/н2 р £ (13.23)
\ г J { р ан+
о о 2303Л77 аи+ап-
^яч “-^AgCl/Ag --£н7н2 р 1g ~ й/2 (13.24)
F (Рн2)'
Уравнение протекающей в ячейке реакции можно составить, вычитая по-
луреакцию, протекающую на правом электроде, из полуреакции, протекающей
на левом электроде (в соответствии с направлением самопроизвольной реакции
и Еяч > °), т- е-
AgCl + е_ Ag + С1“
-(Н+ + е— 1Н2)
AgCl + 1Н2 Ag + С1- + Н+
(13.25)
(13.26)
(13.27)
512
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
По уравнению (13.23) можно рассчитать напряжение ячейки, в которой пра-
вый полуэлемент (электрод) служит индикаторным электродом для потенциомет-
рического определения хлорид-ионов, а левый полуэлемент является электродом
сравнения [см. уравнения (13.6) и (13.7)]. В этом случае £яч (а следовательно, и
потенциал индикаторного электрода) увеличивается с понижением концентра-
ции хлорид-ионов. Если же водородный электрод использоватьв качестве инди-
каторного электрода для измерения активности протонов или pH, то ЭДС ячейки
(и потенциал индикаторного электрода) будет увеличиваться с повышением
кислотности (с понижением pH) и согласно уравнению (13.7): £яч = £иэ - £ср
Ячейки без жидкостного соединения используют, если требуется высокая
точность результатов, поскольку в этом случае в измеренную величину потен-
циала не будет входить потенциал жидкостного соединения. Можно привести
всего несколько примеров ячеек без жидкостного соединения (иногда называе-
мых ячейками без переноса) и все они неудобны для практического применения.
Ячейки без солевого мостика на практике используют редко, поскольку на по-
тенциал электрода сравнения может влиять состав анализируемого раствора.
Поэтому обычно используют ячейки с жидкостным соединением — более удоб-
ные, хотя и дающие менее точные результаты измерений.
13.5. Гальванические элементы с жидкостным соединением:
их используют на практике
Рассмотрим пример гальванического элемента с жидкостным соединением
Hg | Hg2Cl2(TB.) | КСЦнасыщ.) || НС1(р-р), Н2(г) | Pt (13.28)
Двойной чертой обозначено жидкостное соединение двух разных по соста-
ву растворов. Обычно им служит солевой мостик, предотвращающий смешива-
ние этих растворов. В этом случае потенциал одного из электродов должен быть
постоянным и не должен зависеть от состава анализируемого раствора, а опре-
деляться только составом раствора, в котором его хранят. Левый электрод ячейки
(13.28) — насыщенный каломельный электрод — обычно служит электродом
сравнения (см. ниже). Индикаторным электродом в этой ячейке является водо-
родный электрод для измерения pH.
Потенциал жидкостного соединения:
можно ли им пренебрегать?
Недостатком ячеек рассматриваемого типа является наличие потенциала, возни-
кающего на границе раздела двух жидких фаз и называемого потенциалом жид-
костного соединения. Наличие потенциала жидкостного соединения снижает
правильность результатов потенциометрических измерений. Измеренный по-
тенциал такой ячейки описывается выражением
Е = (£ - £ ) + £ •
лЧ 4 Прав JIvb / J
(13.29)
13.5. ГАЛЬВАНИЧЕСКИЕ ЭЛЕМЕНТЫ С ЖИДКОСТНЫМ СОЕДИНЕНИЕМ: ИХ ИСПОЛЬЗУЮТ НА ПРАКТИКЕ
513
где Ej — потенциал жидкостного соединения; Ej может быть как положитель-
ным, так и отрицательным. Причиной возникновения Ej является различие ско-
рости диффузии, с которой ионы, находящиеся по обе стороны границы раздела
жидких фаз, пересекают ее. При правильном выборе раствора в солевом мостике
(или электрода сравнения с подходящим электролитом) величину Ej можно све-
сти к минимуму. В этом случае Ej можно считать практически постоянным и
учесть при построении градуировочного графика.
Рассмотрим типичный пример возникновения жидкостной границы. Допу-
стим, два различных раствора для предотвращения их смешивания разделены
мелкопористой диафрагмой из спеченного стекла. Простейший случай возник-
новения потенциала жидкостного соединения — контакт двух растворов одного
и того же электролита с разными концентрациями, например, НС1 (0,1 М) || НС1
(0,01 М) (рис. 13.2). И ионы Н+, и ионы С1“ будут с обеих сторон пересекать гра-
ницу, но суммарным итогом будет их диффузия из более концентрированного
раствора в менее концентрированный со скоростью, пропорциональной разно-
сти концентраций. Подвижность ионов Н+ примерно в 5 раз выше, чем ионов
С1“, поэтому жидкостная граница справа заряжается положительно, слева — от-
рицательно. В результате пространственного разделения зарядов и возникает
потенциал жидкостного соединения. Достаточно быстро достигается стацио-
нарное состояние, когда Н+ и С1“ будут перемещаться с одинаковыми скоростя-
ми, поскольку наличие положительных зарядов на правой стороне жидкостной
границы не позволит Н+ пересекать ее с такой же скоростью, как в момент ее воз-
никновения. В результате быстро достигается постоянная разность потенциалов.
В рассматриваемом примере Ej составляет +40 мВ и поэтому £яч = (£прав -
- £лев) + 40 мВ. Такая большая величина Ej обусловлена высокой подвижностью
ионов Н+. По мере снижения концентрации НС1 с левой стороны жидкостной
границы суммарный заряд будет уменьшаться и соответственно будет снижать-
ся потенциал жидкостного соединения, пока при выравнивании концентраций
он не станет равным нулю, поскольку в обоих направлениях будут диффундиро-
вать одинаковые количества НС1.
Н+ (0,1 М)-------► Н+ (0,01 М)
СГ (0,1 М) —► СГ (0,01 М)
Жидкостная
граница
Равновесие
+40 мВ
Рис. 13.2. Схема возникновения потенциала жидкостного соединения
514
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
Рассмотрим другой пример возникновения Ej на границе раздела 0,1 М
КС11| 0,01 М КС1. Здесь ситуация полностью аналогична рассмотренной выше,
за исключением того, что ионы К+ и С1_ пересекают границу примерно с одина-
ковыми скоростями, поскольку подвижность ионов С1~ лишь на 4% выше по-
движности ионов К+. И хотя в результате суммарный заряд на левой стороне
границы будет отрицательным, он будет достаточно малым. В данном случае Е-
отрицателен и равен -0,1 мВ. Потенциал жидкостного соединения можно свести к
минимуму, используя высококонцентрированный раствор соли, образованной
ионами с одинаковыми подвижностями, например, КС1.
Как снизить потенциал жидкостного соединения?
Выше было показано, использование электролита с примерно равными подвижнос-
тями ионов (хлорид калия) позволяет значительно снизить потенциал жидкостного
соединения. Это возможно потому, что диффузия ионов из более концентриро-
ванного раствора электролита будет значительно превышать диффузию ионов
из более разбавленного раствора и определять величину Е-. Поэтому для жидко-
стной границы КС1 (3,5 М) || H2SO4 (0,05 М) величинаEj составляет всего -А мВ,
хотя ионы водорода значительно более подвижны, чем сульфат-ионы.
В табл. 13.1 приведены величины потенциалов жидкостных соединений для
различных границ. (Обратите внимание на то, что в этой таблице слева указан
состав раствора в солевом мостике, справа — состав испытуемого раствора.
Если растворы поменять местами, изменится знак потенциала жидкостного сое-
динения.) Совершенно очевидно, что потенциал жидкостного соединения мож-
но свести к минимуму, поместив с одной стороны границы концентрированный
раствор соли, образованной ионами с близкими подвижностями, такой как КС1.
Таблица 13.1
Потенциалы жидкостных соединений при 25 °C*
Жидкостная граница E ., мВ P
0,1 MKCl||0,l MNaCl +6,4
3,5MKC1||O,1 MNaCl +0,2
3,5MKC1|| 1 MNaCl +1,9
0,01 MKC1II 0,01 MHC1 -26
0,1 MKCl||0,l MHC1 -27
3,5МКС1||0,1 MHC1 +ЗД
0,1 M KC11| 0,1 M NaOH +18,9
3,5MKCl||0,l MNaOH +2,1
3,5MKC1|| 1 MNaOH +10,5
* По данным [G. Milazzo, Electrochemie. Vienna: Springer, 1952 and
D. A. Machines and Y. L. Yeh, J. Am. Chem. Soc., 43 (1921) 2563].
13.6. ЭЛЕКТРОДЫ СРАВНЕНИЯ: НАСЫЩЕННЫЙ КАЛОМЕЛЬНЫЙ ЭЛЕКТРОД
515
Идеальный случай — если по обе стороны границы находятся растворы с одина-
ковой и высокой концентрацией такой соли. На практике это неосуществимо,
поскольку один конец солевого мостика всегда помещают в анализируемый рас-
твор. Однако, часто для снижения потенциала жидкостного соединения полу-
элемент, в котором находится другой конец солевого мостика, можно заполнить
концентрированным раствором КС1. Как уже упоминалось выше, этот полу-
элемент является в ячейке электродом сравнения. Ниже рассматривается наибо-
лее часто используемый насыщенный каломельный электрод.
Потенциал жидкостного соединения увеличивается при повышении кон-
центрации анализируемого раствора или по мере увеличения различия подвиж-
ностей ионов. В очень редких случаях потенциалом жидкостного соединения
можно пренебречь. Обычно он ниже при использовании нейтральных солей, в
случае же кислот и оснований потенциал выше вследствие высоких подвижнос-
тей ионов водорода и гидроксид-ионов. Отсюда следует, что потенциал жидко-
стного соединения будет изменяться при изменении pH раствора, и это важно
помнить при потенциометрическом измерении pH. На практике в основном ис-
пользуют солевой мостик, заполненный насыщенным или достаточно концент-
рированным раствором хлорида калия, за исключением тех случаев, когда эти
ионы определяют. Если ионы калия или хлорид-ионы мешают, можно использо-
вать хлорид аммония или нитрат калия.
Обычно солевой мостик заполняют 3%-м раствором агар-агара в насыщен-
ном растворе хлорида калия. Готовят его добавлением 100 мл холодной воды к
3 г гранулированного агар-агара и нагревают раствор на водяной бане до полу-
чения гомогенной смеси. Затем прибавляют 25 г твердого КС1 и перемешивают
до растворения. При охлаждении получается гель. Существуют различные спо-
собы создания жидкостного соединения или солевого мостика, используемые в
конструкциях электродов: стеклянные трубки со шлифами, стеклянные пробки
с впаянными в них асбестовыми волокнами, пористые стеклянные или керами-
ческие пробки, тонкие капилляры. Внутренним раствором электрода сравнения
служит насыщенный раствор КС1, который, медленно проникая через жидкост-
ное соединение, обеспечивает контакт с анализируемым раствором.
13.6. Электроды сравнения: насыщенный каломельный электрод
Важное требование к электроду сравнения состоит в том, что электрод сравне-
ния должен иметь известный и постоянный потенциал, не изменяющийся при
протекании небольших токов в процессе потенциометрических измерений
(в идеале при проведении измерений ток во внешней цепи должен быть равен
нулю, но практически всегда протекает слабый ток — см. ниже). Этому требова-
нию удовлетворяют электроды металл-соль металла. Два наиболее часто испо-
льзуемые электрода сравнения — это насыщенный каломельный Hg | Hg2Cl2 и
хлорсеребряный Ag | AgCl.
516
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
Рассмотрим насыщенный каломельный электрод (НКЭ). Термин «насы-
щенный» относится к концентрации хлорида калия. При 25 °C потенциал элект-
рода равен 0,242 В относительно СВЭ. НКЭ содержит ртуть, твердую Hg2Cl2
(каломель) и твердый КС1, смоченный насыщенным раствором КО. Смесь кон-
тактирует с насыщенным раствором КС1, содержащим немного твердого КС1
для поддержания концентрации раствора. Для обеспечения контакта со ртутью в
пасту погружают платиновый электрод, снабженный свинцовым токоотводом,
через который его присоединяют к прибору для измерения потенциала. Контакт
между раствором КС1 и анализируемым раствором осуществляется с помощью
жидкостного соединения. Обычно это асбестовая или пористая стеклянная пробка,
пропитанная насыщенным раствором КС1. Если для предотвращения загрязне-
ния анализируемого раствора необходим другой солевой мостик (НКЭ не приго-
ден для определения хлорид-ионов!), применяют электрод сравнения с двойным
жидкостным соединением, где раствор КС1 контаткирует с раствором другой
соли, который, в свою очередь, контактирует с анализируемым раствором. Ко-
нечно, при этом возникает дополнительный потенциал жидкостного соедине-
ния, но он является постоянным.
На рис. 13.3 приведена схема НКЭ. Внизу находится пробка из пористой ке-
рамики или асбестового волокна, выполняющая роль жидкостного соединения и
предотвращающая вытекание заметных количеств насыщенного раствора хло-
рида калия. Так как ртутный контакт имеет небольшую площадь, протекание то-
лько слабых токов не вызывает заметного изменения потенциала (ниже будет
показано, что при проведении потенциометрических измерений в цепи обычно
Отверстие очень
малого диаметра
обеспечения контакта
пасты с раствором KCI
Рис. 13.3
Насыщенный каломельный
электрод. С разрешения
Arthur Н. Thomas Company
волокна или пористой
керамики)
Паста из Нд,
Нд2С12 и KCI
Солевой мостик
(пробка из асбестового
Pt-проволока, впаянная
во внутреннюю трубку,
для обеспечения
контакта с пастой
Отверстие для
заполнения
раствором KCI
К потенциометру
Насыщенный
раствор KCI
13.6. ЭЛЕКТРОДЫ СРАВНЕНИЯ: НАСЫЩЕННЫЙ КАЛОМЕЛЬНЫЙ ЭЛЕКТРОД
517
протекает слабый ток). Жидкостное соединение на основе асбестового волокна
имеет высокое сопротивление порядка 2500 Ом, что ограничивает чувствитель-
ность измерений с помощью потенциометра, пригодного для измерений только
при силе тока в цепи, близкой к нулю. При увеличении сопротивления во внеш-
ней цепи сила тока в ней отклоняется от нулевой величины, и это вызывает погреш-
ность в результатах измерения потенциала. Во многих случаях (при титровании)
это не представляет собой серьезной проблемы, однако все же предпочтительно
использовать электроды с низким сопротивлением. Тем не менее насыщенный ка-
ломельный электрод с жидкостным соединением на основе асбестового волокна
вполне пригоден для измерения pH с помощью pH-метра, поскольку этот прибор
рассчитан на работу именно с электродами, имеющими высокое сопротивление.
Пример 13.3
Рассчитайте ЭДС ячейки, состоящей из серебряного электрода, погруженного в
раствор нитрата серебра с а = 0,0100 М, и НКЭ в качестве электрода сравнения.
Решение
Пренебрегая потенциалом жидкостного соединения, получаем
£_„ = £инп - £гп
мн инд ир
Ет = [£Agci/Ag - 0,0592 1g (l/aAg+)] -5нкэ = °’799 " 0,05921g (1/0,0100) -0,242 = 0,439 В
Пример 13.4
Потенциал ячейки относительно НКЭ равен -0,774 В (потенциал индикаторного
электрода более отрицательный). Рассчитайте потенциал этой ячейки относительно
хлорсеребряного (1 М КС1; £ = 0,228 В) и СВЭ электродов сравнения.
Решение
Потенциал электрода Ag I AgCl отличается от потенциала НКЭ на 0,242 - 0,228 =
= 0,014 В, поэтому относительно него потенциал ячейки равен
Яотн. Ag I AgCl = ^отн. НКЭ + 0,014 = -0,774 + 0,014 = -0,760 В
и аналогично относительно СВЭ
£отн. СВЭ = ^отн. НКЭ + 0,242 = -0,774 + 0,242 = -0,532 В
518
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
0 В (СВЭ)
+0,242 В (НКЭ)
fкч -0,760 В (отн. Ag/AgCI)
+0,228 В (Ag/AgCI)
Л -0,532 В (отн. СВЭ)
Ч-
J -0,774 В (отн. НКЭ)
РИС. 13.4. Схематическое представление пересчета величины потенциала относительно различ-
ных электродов сравнения
Расчет потенциала относительно другого электрода сравнения (переход с
одной шкалы потенциалов на другую) можно представить схематически. На
рис. 13.4 это сделано для примера 13.4. Из рисунка видно, что измеренная вели-
чина потенциала индикаторного электрода зависит от используемого при прове-
дении измерений электрода сравнения.
13.7. Измерение потенциала
Для измерения потенциала составляют гальванический элемент из подходящего
индикаторного электрода и электрода сравнения. Найденную величину ЭДС
ячейки полагают равной потенциалу индикаторного электрода относительно
данного электрода сравнения. Затем по уравнению Нернста находят искомую
активность или концентрацию определяемого компонента.
Потенциометр и рН-метр
Измерение потенциала обычно проводят с помощью двух приборов — потенцио-
метра (компенсационная схема измерения) и pH-метра (вольтметр). В настоя-
щее время в основном используют pH-метр. Определение pH по измеренному
потенциалу стеклянного (или другого) индикаторного электрода описано ниже.
Компенсационная схема пригодна для цепей с низким сопротивлением, а
pH-метр может работать в цепях и с низким, и с высоким сопротивлением, как
например, при использовании стеклянного электрода. В цепях с высоким сопро-
тивлением можно также использовать измеритель заряда — электрометр.
По своей сути рН-метр — это вольтметр, который преобразует измеряемое
напряжение в ток, и после усиления регистрирует его, т. е. это прибор с высоким
входным импедансом. (Импеданс — комплексное сопротивление в цепи перемен-
ного тока — аналог сопротивления в цепи постоянного тока. Эти приборы преоб-
разуют измеряемый сигнал в переменнотоковый сигнал для усиления). Из-за
высокого входного сопротивления таких приборов в цепи протекают очень слабые
токи (порядка 10-15—10~13 А), и химическое равновесие практически не нарушается.
Вольтметр можно использовать в случае необратимых реакций, для кото-
рых возврат в исходное состояние после смещения равновесия при протекании
тока невозможен. При измерениях в цепях с высоким входным импедансом нуж-
но использовать электроды с высоким сопротивлением (от нескольких МОм до
13.7. ИЗМЕРЕНИЕ ПОТЕНЦИАЛА
519
Ю6 Ом). Протекающий ток должен быть очень слабым, чтобы падение напря-
жения в ячейке (= iR или сила тока х сопротивление ячейки) было достаточно
мало и не вызвало заметной погрешности измерения потенциала; высокое сопро-
тивление ячейки обусловлено сопротивлением стеклянного электрода.
Существуют pH-метры с растянутой шкалой, позволяющие измерять потен-
циал с точностью до нескольких десятых долей милливольта, что почти на порядок
выше, чем при работе с обычным pH-метром. Такие приборы удобно использовать
при проведении прямых потенциометрических измерений с ионселективными
электродами.
Ячейка для измерения потенциала
Ячейка для потенциометрических измерений показана на рис. 13.5. В прямой
потенциометрии по измеренной величине потенциала индикаторного электрода
рассчитывают активность определяемых ионов. При этом необходимо, чтобы
потенциал электрода сравнения был известен или установлен эксперименталь-
но. В уравнение (13.7) для потенциала ячейки в случае использвания солевого
мостика необходимо включить потенциал жидкостного соединения, тогда
Еяч = (£иэ-£ср) + £у. (13.30)
Ej в уравнении (13.30) можно объединить с другими константами, полагая,
что величина потенциала жидкостного соединения не будет существенно менять-
ся при замене одного анализируемого раствора другим. С этим предположением
приходится соглашаться, поскольку в большинстве случаев величину £) оценить
нельзя. Итак, константа к объединяет £ср, Ej и £^э:
к = Е°иэ - Еср + Ej (13.31)
Рис 13.5. Ячейка для потенциометрических измерений
520
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
Тогда для реакции со стехиометрией 1 : 1
£яч = £-2,3037?Г1ё^-
nF «ох
(13.32)
Величину к находят, измерив потенциал стандартного раствора с известной
активностью ионов.
13.8. Определение концентрации по данным
потенциометрических измерений
Аналитику практически всегда нужно найти концентрацию определяемого ком-
понента, а не его активность. Но коэффициенты активности обычно неизвестны
и поэтому невозможно рассчитать активности ионов в растворе, используемом
для калибровки электрода.
Если ионную силу всех растворов поддерживать постоянной на одном и том
же уровне, коэффициент активности определяемого иона будет практически по-
стоянным при любой концентрации. Тогда для логарифмического слагаемого
уравнения Нернста можно записать
2,303ЯГ
nF
IgfiCi
2,303ДГ
nF
. f 2,303RT. „
nF
(13.33)
В указанных выше условиях первый член в правой части этого уравнения будет
постоянной величиной, и его можно включить в константу к, поэтому при посто-
янной ионной силе
^ох
(13.34)
Это означает, что при 10-кратном изменении концентрации окисленной или
восстановленной формы потенциал электрода изменится на ±2,3037?77(«F) В.
Лучше воспользоваться градуировочным графиком, построенным в коорди-
натах Е—lg С. Теоретически тангенс угла наклона градуировочного графика
(электродной функции) должен составлять ±2,3032?77(hF). В этом случае любое
отклонение от теоретического отклика будет учтено при построении градуиро-
вочной зависимости.
Поскольку ионная сила анализируемого раствора обычно неизвестна, для
того чтобы ее выровнять и поддерживать постоянной во все стандартные и ана-
лизируемые растворы вводят в большом избытке раствор электролита. Стандар-
13.10. ТОЧНОСТЬ РЕЗУЛЬТАТОВ ПРЯМЫХ ПОТЕНЦИОМЕТРИЧЕСКИХ ОПРВДЕЛЕНИЙ АКТИВНОСТИ
521
тные растворы должны содержать любые компоненты анализируемого раствора,
способные изменять активность определяемого компонента, например, комп-
лексообразующие соединения.
13.9. Неэлиминированный потенциал жидкостного соединения.
Почему он должен быть минимальным?
Выше мы полагали, что в уравнениях (13.32) и (13.34) величина А: одинакова при
измерениях потенциала электрода в стандартном и в анализируемом растворах.
Это справедливо лишь в случае, если потенциал жидкостного соединения на
электроде сравнения одинаков для обоих растворов. Если потенциалы жидкост-
ного соединения для стандартного и анализируемого растворов одинаковы, то Ej
не внесет погрешности в полученный результат (полагаем Ej = 0). Различие этих
двух потенциалов называют неэлиминированным потенциалом жидкостного
соединения. К сожалению, его величина неизвестна, но его вклад можно свести
к минимуму при уравнивании величин pH и ионной силы стандартного и анали-
зируемого растворов. Первое особенно важно.
13.10. Точность результатов прямых потенциометрических
определений активности: зависимость от погрешности
измерения потенциала
Точность результатов потенциометрических измерений можно оценить, исходя
из некоторой заданной погрешности измерения потенциала, например, 1 мВ
(25 °C). Для электрода, чувствительного к однозарядным ионам, например, Ag+,
справедливо выражение
Еяч = к- 0,05916 1g—!— (13.35)
aK +
Ag
И
%+=1° 0,05916 (13.36)
Погрешность измерения потенциала величиной ±1 мВ вызывает погреш-
ность в определении «Ag+ порядка ±4% независимо от величины «Ag+. Для двухза-
рядных ионов погрешность будет вдвое больше. Так, при измерении потенциала
медного металлического электрода с точностью порядка 1 мВ величина погреш-
ности определения активности ионов меди(П) составит 8%. Очевидно, что точность
результатов определения зависит от величины неэлиминированного потенциала
жидкостного соединения.
522
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
Правильность и воспроизводимость потенциометрических измерений огра-
ничивается в том числе так называемой сопротивляемостью окислительно-вос-
становительных пар (понятие, аналогичное буферной емкости кислотно-основных
буферных систем). В очень разбавленных растворах равновесие и, соответствен-
но, потенциал электрода устанавливаются медленно. В этом и заключается при-
чина, по которой потенциал электрода в разбавленных растворах измеряют с
помощью электрометров или pH-метров, способных регистрировать слабые
токи. Для устранения этой проблемы, а также для поддержания постоянного
значения ионной силы можно добавить избыток инертной соли («буфер» ион-
ной силы). Ускорить достижение равновесного потенциала помогает также пе-
ремешивание раствора.
В очень разбавленных растворах потенциал электрода зависит также и от
других электродных реакций. Так, в очень разбавленном растворе ионов серебра
-lg(l/aAg+) становится очень большой отрицательной величиной, и потенциал
электрода резко снижается. В таких условиях присутствующие в растворе окис-
лители, например кислород, могут восстанавливаться на поверхности электро-
да, и в результате образования смешанной окислительно-восстановительной
пары электрод приобретает смешанный потенциал.
Обычно нижняя граница определяемых концентраций составляет величину
порядка 1 • 10-6 - 1 • 10-5 М, но ее в каждом случае необходимо устанавливать эк-
спериментально. Следует учесть, что чем более разбавленным является ана-
лизируемый раствор, тем медленнее устанавливается равновесный потенциал.
Иная картина наблюдается при измерении pH в буферных растворах или в силь-
нокислых (сильнощелочных) растворах — из-за высокой буферной емкости по-
тенциал устанавливается быстро. При pH 10 концентрация ионов водорода
равна всего лишь 1 • 1О-10 М, но ее можно измерить с помощью стеклянного
электрода (см. разд. 13.11). В нейтральных незабуференных растворах равнове-
сие устанавливается медленно, поэтому измерять pH таких растворов сложно.
13.11 .Стеклянный электрод для измерения pH - рабочий
инструмент химика
Стеклянный электрод настолько удобен в работе, что на сегодняшний день это
наиболее часто используемый электрод для измерения pH. Окислители и восста-
новители не оказывают существенного вляния на его потенциал, и он пригоден
для измерения pH в широком интервале. Электрод обладает малым временем от-
клика и хорошо работает в физиологических системах. Ни один другой рН-чув-
ствительный электрод не обладает такими характеристиками.
Конструкция стеклянного электрода
Стандартная конструкция стеклянного электрода для измерения pH показана на
рис. 13.6. При измерении в раствор погружают только шарик. В нем находится
внутренний электрод сравнения (Ag | AgCI | С1“) и электролит для создания элек-
13.11. СТЕКЛЯННЫЙ ЭЛЕКТРОД ДЛЯ ИЗМЕРЕНИЯ pH - РАБОЧИЙ ИНСТРУМЕНТ ХИМИКА
523
Стеклянная мембрана
Рис. 13.6. Стеклянный электрод для измерения pH
трического контакта со стеклянной мембраной. Потенциал внутреннего элект-
рода сравнения обязательно должен быть постоянным, и это обеспечивается посто-
янством концентрации НС1. Схему ячейки можно представить так
внешний
электрод
сравнения
раствор
с неизвестным
pH
стеклянная
мембрана
внутренний
раствор Н+
внутренний
электрод
сравнения
Двойной чертой обозначен солевой мостик электрода сравнения. Стеклян-
ный электрод с помощью токоотвода присоединяют к выходу «индикаторный элек-
трод» на панели pH-метра, а внешний электрод сравнения (например, НКЭ) —
к выходу «электрод сравнения».
Потенциал стеклянной мембраны описывается уравнением
и 2,3037?/ 1 ^н*(внутр.)
£мемб. = const----------1g----(1337)
F А
Н(неизв.)
а напряжение ячейки — уравнением
г , 2,3037?/1
^яч j-, ё^пЧнеизв.) (13.38)
г
где к — константа, включающая потенциалы внешнего и внутреннего электро-
дов сравнения, потенциал жидкостного соединения, потенциал на внутренней
стороне стеклянной мембраны (обусловленный присутствием Н+) и еще одно
слагаемое — так называемый потенциал асимметрии.
Потенциал асимметрии — это небольшой потенциал, возникающий даже
если растворы по обе стороны мембраны идентичны. Его величина зависит от
524
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
ряда факторов, таких как неоднородность мембраны, наличие напряжения в
мембране, механическое и химическое воздействие на внешнюю поверхность
мембраны и степень ее гидратации. Потенциал асимметрии медленно изменяет-
ся во времени, особенно если мембрана высыхает, и его величина неизвестна.
Поэтому стеклянный электрод необходимо калибровать ежедневно. Для раз-
ных электродов потенциалы асимметрии отличаются из-за различий в конструк-
циях мембран.
Поскольку pH = -1g уравнение (13.38) можно представить в виде*
или
, = 2,303ЯГ
'яч * ——pH
неизв.
F
(13.39)
яч
2,3037?77F
(13.40)
Видно, что отклик стеклянного электрода при изменении pH на 1 единицу
(или на 10-кратное изменение а^) составляет 2,3032?77F. Величину к можно
оценить при градуировке электрода по стандартным буферным смесям с изве-
стным pH (см. разд. 13.12).
, „ 2,3037? Г u
* = £яч + -2-----РНСТ
F
(13.41)
При подстановке уравнения (13.41) и (13.39) получаем
F — F
— прт _|_ ^ст. яч ^неизв. яч
г1 неизв. Р^ст.
2,3037?77F
(13.42)
При измерении pH подобные вычисления не производят, поскольку шкалу
pH-метра калибруют в единицах pH (см. разд. 13.14). Заметим, что поскольку
для измерения pH применяют мембранный электрод с очень высоким сопротив-
лением (1-100 МОм ), очень важно свести к минимуму падение напряжения iR.
Для этого используют pH-метр, способный работать при протекании очень сла-
бых токов (об измерении потенциала см. выше).
В данной главе будем полагать, что pH = -1g ан+, поскольку именно ан+ измеряет стеклянный электрод.
13.11. СТЕКЛЯННЫЙ ЭЛЕКТРОД ДЛЯ ИЗМЕРЕНИЯ pH - РАБОЧИЙ ИНСТРУМЕНТ ХИМИКА
525
Пример 13.5
Потенциал стеклянного электрода (относительно СВЭ при 25 °C) в стандартном
буферном растворе с pH 4,01 равен 0,814 В. Каков будет потенциал этого элект-
рода в 1,00 • 10-3 М растворе СН3СООН? При расчете полагать а = [Н+].
Решение
В примере 6.7 в гл. 6 было найдено, что pH 1,00 • 10-3 М раствора СН3СООН рав-
но 3,88, поэтому
_ а 0,814-Е
О О _ А Г\ 1 I rlwrlOJD. Л“
откуда Енеизв яч = 0,822 В.
Обратите внимание, что потенциал увеличивается с повышением концент-
рации Н+.
Комбинированные электроды для измерения pH:
готовая ячейка
Для выполнения потенциометрических измерений необходима ячейка, состоя-
щая из двух полуэлементов — индикаторного электрода и электрода сравнения
(с солевым мостиком). Если оба эти электрода вмонтировать в один корпус, из-
мерения можно будет проводить в малых объемах растворов. Стандартная кон-
струкция такого комбинированного электрода для измерения pH показана на
рис. 13.7. Электрод состоит из двух трубок: во внутренней находится индика-
торный pH-электрод, во внешней — электрод сравнения (например, хлорсереб-
ряный электрод) и жидкостное соединение (пористая пробка). У каждого
электрода — электрода сравнения и индикаторного — имеется токоотвод. Для
того чтобы комбинированный электрод давал правильные показания важно,
чтобы цепь была замкнута, т. е. необходим контакт жидкостного соединения с
анализируемым раствором. Иногда пористым делают не все кольцо, как это по-
казано на рисунке, а только небольшую его часть. Из-за удобства при проведения
измерений в последнее время чаще pH-комбинированные электроды используют
вместо традиционной пары электродов.
От чего зависит потенциал стеклянной мембраны?
Стеклянный электрод приобретает pH-функцию в результате обмена ионов в
слое гидратированного геля на поверхности мембраны. Стеклянная мембрана
состоит из химически связанных Na^ и SiO2, и на поверхности нового стеклян-
ного электрода находятся фиксированные группировки — SiO_Na+. Известно,
526
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
Рис. 13.7
Комбинированный
электрод для
определения pH
что для приведения стеклянного электрода в рабочее состояние, его нужно неко-
торое время выдержать в воде. За это время внешний слой мембраны гидратиру-
ется. Поверхностный слой на внутренней стороне мембраны стеклянного шарика
уже гидратирован. Толщина стеклянной мембраны находится в пределах от 0,03
до 0,1 мм, а толщина гидратированного слоя — от 10“5 до 10-4 мм.
В гидратированном слое на внешней поверхности мембраны ионы Na+ за-
мещаются ионами Н+ из раствора
—SiO"Na+ + Н+ —SiO-H+ + Na+
мембрана раствор мембрана раствор
(13.43)
Другие ионы из раствора также могут обмениваться с ионами Na+ (или Н+),
но константа равновесия (13.43) очень велика из-за высокого сродства мембра-
ны к протонам. Поэтому, если электрод не погружен в сильнощелочной раствор,
в котором концентрация протонов мала, практически вся поверхность стеклян-
ной мембраны покрыта слоем кремниевой кислоты. Группировки —SiO- фикси-
рованы, но протоны могут свободно перемещаться и замещаться другими ионами.
(При изменении состава стекла для изготовления мембраны обмен протонов на
другие ионы может стать преобладающим процессом, этим пользуются при изго-
товлении стеклянных электродов, селективных по отношению к другим ионам —
см. ниже).
13.11. СТЕКЛЯННЫЙ ЭЛЕКТРОД ДЛЯ ИЗМЕРЕНИЯ pH - РАБОЧИЙ ИНСТРУМЕНТ ХИМИКА
527
Мембранный потенциал складывается из граничного и диффузионного по-
тенциалов, первый из которых зависит почти исключительно от активности
ионов водорода. Граничный потенциал возникает на поверхности стеклянной
мембраны при погружении электрода в раствор, точнее на поверхности раздела
слоя гидратированного геля и внешнего раствора из-за различия активностей
ионов водорода в растворе и на поверхности гидратированного геля. Одно из
объяснений возникновения потенциала таково: ионы водорода стремятся пере-
местится (диффундировать) в направлении участков с меньшей активностью
аналогично тому, как это происходит в жидкой среде. На поверхности мембраны
образуется заряженный микрослой, и в результате пространственного разделе-
ния зарядов возникает потенциал. Поэтому, если внешний раствор становится
более кислым (его pH уменьшается), протоны диффундируют к поверхности
геля. В результате возникновения положительного заряда потенциал электрода
в соответствии с уравнениями (13.37) и (13.38) увеличивается, а в более щелоч-
ных растворах, напротив, уменьшается.
Диффузионный потенциал возникает вследствие того, что протоны из
внутренней части слоя геля стремятся диффундировать в слой сухого стекла, со-
держащего группировки —SiO_Na+, а ионы Na+ — из слоя сухого стекла в слой
гидратированного геля. Ионы диффундируют с разной скоростью, и в результа-
те возникает своего рода потенциал жидкостного соединения. То же происходит
и по другую сторону мембраны, но ионы перемещаются в противоположном на-
правлении, поэтому диффузионные потенциалы взаимно сокращаются и мемб-
ранный потенциал в основном определяется граничными потенциалами. (Из-за
различия состояния внешней и внутренней поверхностей мембраны граничные
потенциалы могут несколько различаться — это одна из причин возникновения
потенциала асимметрии.)
Пунгор показал, что причиной возникновения потенциала является простран-
ственное разделение зарядов вследствие хемосорбции на поверхности электрода
первичных ионов (Н+) из фазы раствора. Ионы противоположного заряда (про-
тивоионы) накапливаются в растворе у поверхности электрода, в результате
пространственного разделения зарядов возникает потенциал. Это объяснение спра-
ведливо и для ионселективных электродов других типов, рассматриваемых ниже
[Е. Pungor, «The New Theory Of Ion-Selective Electrodes», Sensors, 1 (2001) 1-12 (это
электронный журнал, размещенный на странице www.mdpi.net/sensors)].
Чен К. Л. предложил применить к стеклянному электроду теорию конденса-
тора, согласно которой в щелочной среде (когда aR+ очень мала) потенциал
электрода определяют гидроксид-ионы, а не протоны [К. L. Cheng, «Capacitor
Theory for Nonfaradaic Potentiometry», Microchem. J., 42 (1990) 5. ]. Результаты
экспериментов Чена с использованием изотопов позволили ему предположить,
что общепринятая реакция обмена между ионами Н+ и Na+ не происходит и что в
щелочных растворах электрод фактически дает отклик на ионы ОН- (вспомните,
при pH 14 величина [Н+] составляет всего 1 • 10-14 М !) [С.-М. Huang et al., «Isoto-
pe Evidence Disproving Ion Exchange Reaction Between H+ and Na+ in pH Glass
Electrode», J. Electrochem. Soc., 142 (1995) Ы75]. Эта теория не является обще-
признанной, но Чен с коллегами приводят убедительные аргументы и экспери-
528
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
ментальные данные, интересные для размышления. Теория Чена имеет ряд
общих моментов с теорией двойного слоя Пунгора.
Щелочная погрешность
При измерении pH с помощью стеклянного электрода возникает два типа по-
грешностей (отклонение от теоретического отклика). Первый из них называется
щелочной погрешностью. Она обусловлена способностью мембраны реагиро-
вать наряду с протонами на присутствие других катионов. По мере снижения ак-
тивности протонов эти ионы становятся потенциалопределяющими. Хотя для
слоя гидратированного геля характерно более высокое сродство к протонам,
ионы натрия будут вытеснять оттуда протоны, если концентрация последних во
внешнем растворе станет очень мала [равновесие (13.43) смещается влево]. По-
скольку потенциал электрода зависит от соотношения а (внешн) / a Na+ (гель), элект-
род фактически становится Na-селективным.
При pH ниже 9 щелочная погрешность пренебрежимо мала, но при более
высоких значениях pH концентрация Н+ становится намного ниже по сравнению
с концентрациями других ионов и электрод дает отклик на такие ионы, как Na+,
К+. В результате электрод «чувствует» больше ионов водорода, чем их при-
сутствует в растворе, и измеренное значение pH будет заниженным. На рис. 13.8
показана зависимость щелочной погрешности от природы катиона. К сожале-
РИС. 13.8
Погрешность измерения pH
с помощью электрода,
изготовленного из стекла Coming
015, в сильнощелочных растворах,
содержащих различные катионы
[L. Meites and L. С. Thomas,
Advanced Analitical Chemistry.
Copyright® 1958, McGraw-Hill,
New York]. С разрешения
McGraw-Hill Book Company
13.11. СТЕКЛЯННЫЙ ЭЛЕКТРОД ДЛЯ ИЗМЕРЕНИЯ pH - РАБОЧИЙ ИНСТРУМЕНТ ХИМИКА
529
нию, наибольшую погрешность вызывают ионы Na+, поскольку многие анали-
зируемые растворы, особенно щелочные, содержат значительные количества
именно ионов натрия. К электродам промышленного изготовления обычно при-
лагают номограмму, позволяющую скорректировать щелочную погрешность
при известной концентрации ионов натрия. Эти электроды пригодны для прове-
дения измерений при значениях pH до ~11.
Стеклянные электроды желательно использовать для измерений при pH
ниже 11, поскольку они будут обладать меньшим временем отклика и более вы-
сокой стабильностью показаний за счет более низкого сопротивления стекла.
Изменение состава стекла мембраны может изменить ее чувствительность по от-
ношению к различным катионам и что это использовали при создании других
стеклянных ионселективных электродов.
Например, сродство стеклянного электрода к ионам натрия можно снизить,
изменив состав стекла мембраны. Так, погрешность измерения pH, обусловлен-
ная присутствием ионов натрия заметно снизится при замене в стекле Na2O на
Li2O. Такой электрод (его называют Li-стеклянным электродом) дает правильные
результаты при измерении pH в интервале от 0 до 14.
Кислотная погрешность
Второй тип погрешности определения pH с помощью стеклянного электрода —
кислотная погрешность. Она обусловлена тем, что потенциал мембраны зави-
сит от активности контактирующей с ней воды. При активности воды, равной
единицы, потенциал электрода описывается уравнением Нернста. Но в очень
кислых растворах активность воды меньше единицы (заметное количество рас-
ходуется на сольватацию протонов) и поэтому при измерении pH возникает по-
ложительная погрешность (рис. 13.9). Подобного типа погрешность возникает
также из-за уменьшения активности воды при высокой концентрации раство-
ренных солей или при добавлении неводного растворителя, например, этанола.
В этих случаях потенциал жидкостного соединения может быть достаточно вы-
соким, и это также приведет к возникновению погрешности, хотя и не очень боль-
шой при малых количествах этанола.
Рис. 13.9
Погрешность измерения pH
с помощью стеклянного электрода
в растворах НС1 [ L. Meites and
L. С. Thomas, Advanced Analitical
Chemistry. Copyright® 1958,
McGraw-Hill, New York].
С разрешения McGraw-Hill Book
Company
530
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
13.12. Стандартные буферные растворы для градуировки
pH-электродов
Национальным институтом стандартов и технологии США (NIST) разработана
серия сертифицированных стандартных буферных растворов для градуировки
шкалы измерительного прибора. Величины pH установлены при измерении с
помощью pH-чувствительного стеклянного электрода в качестве индикаторного
в паре с хлорсеребряным электродом сравнения в ячейке без жидкостного сое-
динения [см. уравнение (13.22)]. Из уравнения (13.24) видно, что для расчета по-
тенциала электрода сравнения нужно знать активность хлорид-ионов. Поскольку
ее можно рассчитать только по уравнениям Дебая—Хюккеля, это является
основной причиной, ограничивающей точность измерения pH буферов до вели-
чины порядка ±0,01 единицы pH. Парциальное давление водорода рассчитывают
по величине атмосферного давления во время проведения эксперимента, вычи-
тая давление паров воды при температуре раствора в ячейке.
Составы и величины pH стандартных буферов NIST приведены в табл. 13.2.
Хотя абсолютное значение pH нельзя измерить с погрешностью ниже 0,01 еди-
ницы, величины pH буферов, измеренные относительно друг друга, даны с точно-
стью до 0,001 pH. Это обусловлено высокой воспроизводимостью результатов
измерения потенциалов. В ряде случаев, например, при градуировке электрода,
может быть существенным различие в тысячных долях единицы pH. Влияние
температуры на pH буферов объясняется зависимостью от температуры кон-
стант кислотности и основности сопряженных пар.
Обратите внимание на то, что некоторые из приведенных в таблице раство-
ров в действительности не являются буферными. Их используют в качестве
стандартных буферов для измерения pH только потому, что в процессе градуи-
ровки электрода и при проведении pH-измерений в них не добавляют ни кисло-
ту, ни основание, и их pH не изменяется. Дело в том, что эти растворы обладают
способностью сохранять pH постоянным только при незначительном разбавле-
нии (например, [Н+ ] « y]KaiKa2). Только растворы фосфатов действительно яв-
ляются буферными растворами.
Следует заметить, что если ячейку, состоящую из стеклянного электрода и
НКЭ, калибруют по одному из стандартных буферов, а затем используют для из-
мерения pH другого раствора, полученная величина pH не будет соответство-
вать табличной величине pH второго буфера из-за потенциала жидкостного
соединения.
Для градуировки стеклянного электрода, используемой для определения pH
крови наиболее подходящей является буферная смесь КН2РО4—Na2HPO4
(pH 7,384 при 38 °C). Во многих случаях измерение pH крови выполняют при
температуре 38 °C, близкой к температуре тела человека.
О pH стандартов NIST и других способах определения pH см. [W. F. Koch,
Anal. Chem., December 1, 1997, 700A; Chem. & Eng. News, October 20, 1997, 6].
13.13. ТОЧНОСТЬ ИЗМЕРЕНИЯ pH
531
Таблица 13.2
Величины pH стандартных буферов (NIST) [R. G. Bates, J. Res. Natl. Bur. Std., A66
(1962) 179]
Буфер
Гидроксид Г (°C) Тетраоксалатный* Тартратный* Бифталатный^ Фосфатный* Фосфатный^ Боратный* кальция*
0 1,666 — 4,003 6,984 7,534 9,464 13,423
5 1,668 — 3,999 6,951 7,500 9,395 13,207
10 1,670 — 3,998 6,923 7,472 9,332 13,003
15 1,672 — 3,999 6,900 7,448 9,276 12,810
20 1,675 — 4,002 6,881 7,429 9,225 12,627
25 1,679 3,557 4,008 6,865 7,413 9,180 12,454
30 1,683 3,552 4,015 6,853 7,400 9,139 12,289
35 1,688 3,549 4,024 6,844 7,389 9,102 12,133
38 1,691 3,549 4,030 6,840 7,384 9,081 12,043
40 1,694 3,547 4,035 6,838 7,380 9,068 11,984
45 1,700 3,547 4,047 6,834 7,373 9,038 11,841
50 1,707 3,549 4,060 6,833 7,367 9,011 11,705
55 1,715 3,554 4,075 6,834 — 8,985 11,574
60 1,723 3,560 4,091 6,836 — 8,962 11,449
70 1,743 3,580 4,126 6,845 — 8,921 —
80 1,766 3,609 4,164 6,859 — 8,885 —
90 1,792 3,650 4,205 6,877 — 8,850 —
95 1,806 3,674 4,227 6,886 — 8,833 —
b — 0,05 m тетраоксалат калия (m — моляльность, но погрешность будет незначительна при использовании
молярности);
с — насыщенный (25 °C) раствор гидротартрата калия;
d — 0,5 т гидрофталат калия;
е — 0,05 т дигидрофосфат калия + 0,025 т гидрофосфат натрия;
f — 0,008695 т дигидрофосфат калия + 0,03043 т гидрофосфат натрия;
g — 0,01 т тераборат натрия;
h — насыщенный (25 °C) раствор гидроксида кальция.
Фактически буферными являются только смеси фосфатов. Величины pH изменяются при изменении темпе-
ратуры, поскольку от температуры зависят величины констант кислотности Ка.
13.13. Точность измерения pH
Точность измерения pH определяется тем, насколько достоверно известна вели-
чина активности ионов водорода в стандартном буфере. Выше упоминалось, что
точность не может быть лучше ±0,01 единицы pH, что обусловлено степенью до-
стоверности вычисления коэффициента активности индивидуального иона.
532
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
Второй причиной является наличие неэлиминированного потенциала жид-
костного соединения. Состав растворов в ячейке при стандартизации и измере-
нии pH различается. Ранее уже говорилось о том, что величину потенциала
жидкостного соединения можно свести к минимуму, поддерживая pH и состав
растворов по возможности максимально близкими. Из этого следует, что элект-
род нужно стандартизировать по буферу с pH, близким к pH анализируемого
раствора. Погрешность при стандартизации по буферу с сильно отличающимся
pH обычно составляет от 0,01 до 0,02 единиц pH, но для сильнощелочных рас-
творов может достигать 0,05 единиц pH.
Наличие неэлиминированного потенциала жидкостного соединения в соче-
тании с погрешностью в величине pH стандартных буферных растворов вызыва-
ет абсолютную погрешность измерения pH анализируемого раствора порядка
±0,02 единицы pH. Тем не менее, можно различить два близких по составу рас-
твора, pH которых отличается на ±0,004 или даже ±0,002 ед. pH, хотя точность
этих измерений и не превысит ±0,02 ед. pH. Это можно сделать потому, что для
обоих растворов величины потенциалов жидкостного соединения будут практи-
чески одинаковыми. Например, для двух образцов крови различие в величинах
их pH можно уловить, даже если оно составляет порядка всего ±0,004 ед. pH.
При относительно большом различии величин pH двух растворов увеличивается
значение неэлиминированного потенциала жидкостного соединения и провести
измерения с такой точностью уже невозможно. Если различие величин pH раство-
ров составляет порядка 0,02 единиц, заметное изменение ионной силы несущест-
венно, но при меньшем различии pH влиянием ионной силы пренебрегать нельзя.
Погрешность измерения ±0,02 единицы pH соответствует погрешности
определения а ±4,8% (±1,2 мВ)*, а погрешность в ±0,004 единицы pH—±1,0%
(±0,2 мВ). Но в целом погрешность потенциометрического измерения а^ не
превышает 5%.
Потенциал жидкостного соединения зависит от температуры, поэтому гра-
дуировка электрода и измерение pH в особо важных случаях нужно проводить
при одинаковой температуре: потенциал жидкостного соединения в интервале
от 25 до 38 °C изменяется на ±0,76 мВ при измерении pH крови и на -0,55 мВ при
измерении pH буферного раствора.
13.14. Использование pH-метра: как он работает?
Как уже было упомянуто выше, для измерения pH с помощью стеклянного элек-
трода нужен электрометр или pH-метр, поскольку у стеклянного электрода со-
противление высокое. При непосредственном измерении потенциала электрода
можно рассчитать pH по уравнениям (13.40) или (13.42). При 298,16 К (25 °C) ве-
личина 2,303RT/F составляет 0,05916; при любой другой температуре всегда
нужно вносить поправку. На сайте http://www.ph-maasurement.co.uk размеще-
Отклик электрода при 25 °C составляет 59 мВ/pH.
13.14. ИСПОЛЬЗОВАНИЕ pH-МЕТРА: КАКОЙ РАБОТАЕТ?
533
Рис. 13.10
Стандартный рН-метр
(с разрешения Denver
Instrument Company)
ны материалы, полезные для изучения основ измерения pH; в них также указаны
особенности измерения в различных средах.
На рис. 13.10 изображен цифровой pH-метр. Шкала отградуирована в еди-
ницах pH, каждая из которых соответствует 59,16 мВ при 25 °C [уравнение
(13.39)]. Прибор снабжен регулирующей кнопкой для установления величины
pH стандартного буфера. Значение pH считывают сразу же после замены стан-
дартного раствора анализируемым. Фактически с помощью этой процедуры
оценивают константу к в уравнении (13.40), включающую потенциал асиммет-
рии и другие постоянные.
У прибора есть также шкала для настройки, позволяющая регулировать вели-
чину отклика (мВ/pH) в зависимости от температуры в соответствии с коэффици-
ентом 2,3037WF. (Величина RT/nF определяет тангенс угла наклона зависимости
потенциала от pH стандартных буферов.) Например, при О °C отклик равен 54,1 мВ,
при 60 °C — 66,0 мВ. Заметим, однако, что это не может компенсировать изме-
нение pH стандартного буфера при изменении температуры, и следует использо-
вать величину pH буфера при данной температуре.
Электроды и соответствующие блоки электроники конструируют так, что-
бы в одной из точек в середине диапазона измерения активности потенциал не
зависел от температуры. Для стеклянного pH-чувствительного электрода эта
точка соответствует pH 7 (рис. 13.11). В этой точке, называемой изопотенциаль-
ной точкой, потенциал равен нулю (pH-метр фактически измеряет потенциал,
который затем преобразуется в искомую величину pH, поэтому можно регистри-
534
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
Потенциал, мВ
Рис. 13.11. Изопотенциальная точка
ровать непосредственно значение потенциала). Если потенциал при pH 7,0 не ра-
вен 0 мВ, это означает, что для данного электрода наблюдается смещение
изопотенциальной точки. Наклон градуировочной кривой зависит от температу-
ры, и новая точка пересечения даст координату фактической изопотенциальной
точки. Более детально об изопотенциальной точке и интерпретации результатов
см. [A. A. S. С. Machado, Analyst. 19 (1994) 2263].
При градуировке pH-метра электроды погружают в стандартный буфер с
pH 7,0. Измеряют температуру раствора и вводят это значение с помощью кноп-
ки для регулирования температуры. Затем с помощью кнопки «Стандартиза-
ция» или «Калибровка» показания на шкале прибора выводят на величину 7,00.
Многие современные pH-метры снабжены микропроцессорами, в которых уже за-
ложены определенные величины pH (например, 4,0, 7,0 и 10,0), поэтому про-
стым нажатием клавиши можно переключить программу с одного стандартного
буфера на другой. Затем при повторном измерении pH одного из стандартных
буферов (с pH 4,0 или 10,0, выбрав буфер, pH которого максимально близко к pH
анализируемого раствора в дальнейшем) устанавливают наклон градуировоч-
ной зависимости. Большинство современных pH-метров имеют отдельный темпе-
ратурный зонд для автоматической термокомпенсации и его можно совместить с
индикаторным рН-электродом.
Точность измерения с помощью обычного pH-метра для полной шкалы в
14 единицрН (порядка 840 мВ) составляет от±0,1 до ±0,01 pH (от±6 до ±0,6 мВ).
Результаты измерений можно регистрировать не в единицах pH, а в милливольтах
(полная шкала будет соответствовать примерно 1400 мВ). Выпускают pH-метры с
растянутой шкалой с размахом 1,4 единицы pH (140 мВ); при измерении потен-
циалов порядка 0,1 мВ достигается точность ±0,001 единицы pH.
13.15. ИЗМЕРЕНИЕ pH КРОВИ: ВАЖНО КОНТРОЛИРОВАТЬ ТЕМПЕРАТУРУ
535
При измерении pH нейтральных небуферных растворов равновесие уста-
навливается медленно, и для получения стабильных показаний требуется значите-
льное время. Раствор следует перемешивать, так как из-за растворения небольшого
количества стекла мембраны раствор на поверхности электрода может стать ще-
лочным [см. уравнение (13.43): Н2О — источник Н+ — замещается на NaOH].
13.15. Измерение pH крови: важно контролировать температуру
В гл. 7 было сказано, что величина константы равновесия буферной системы
крови изменяется при изменении температуры, и поэтому pH крови при темпе-
ратуре тела 37 °C отличается от pH, измеренного при комнатной температуре.
Следовательно, для получения результатов, соответствующих фактическому
физиологическому состоянию человека, pH необходимо измерять при 37 °C,
при этом анализируемые пробы не должны контактировать с окружающей сре-
Некоторые правила, которые следует соблюдать при измерении pH
крови:
1 • Для градуировки электрода по стандартному буферу при 37 °C тщатель-
но выберите подходящий для этой температуры буфер и с помощью со-
ответствующей кнопки pH-метра установите температуру 37 °C (отклик
61,5 мВ/pH). Хорошо использовать для градуировки два стандартных
буфера с близкими величинами pH; это гарантирует равноценность ра-
боты электродов. Перед градуировкой и проведением измерений электро-
ды следует некоторое время выдержать при 37 °C, чтобы все процессы,
протекающие на поверхности электродов, пришли в равновесие. Потен-
циал внутреннего электрода сравнения стеклянного электрода зависит
от температуры (так как температура влияет на потенциалопределяю-
щие процессы на мембране электрода), также как и потенциалы НКЭ и
жидкостного соединения. (Отметим, что в случае проведения измерений
при температуре ниже комнатной солевой мостик (электрод сравнения)
должен быть заполнен менее концентрированным раствором КС1, а не
насыщенным, так как из последнего будут осаждаться кристалы КС1, а
это увеличит сопротивление.)
2. Пробы крови необходимо содержать в аноэробной атмосфере во избежа-
ние потери или поглощения СО2. Если возможно, то измерение pH жела-
тельно провести в течение 15 мин после отбора крови, в противном случае
кровь нужно поместить в лед и измерить pH не позже, чем через 2 ч.
3. Для предотвращения пассивации электрода после каждого измерения
смывайте с него кровь солевым раствором. Оставшуюся пленку крови
можно удалить, погрузив электрод всего на несколько минут в 0,1 М
NaOH, затем в 0,1 М НО и, наконец, в воду или солевой раствор.
536
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
дой. (Вспомните, что нейтральный водный раствор при 37 °C имеет pH 6,80 и это
изменяет шкалу кислотности на 0,20 единиц pH).
Обычно пробу крови из вены берут для измерения pH, а артериальную кровь—
для специальных исследований. С 95%-й доверительной вероятностью (см. гл. 3)
можно полагать, что для артериальной крови интервал pH составляет 7,31-7,45
(среднее 7,40) для людей любого возраста и пола. Для людей пенсионного возра-
ста нормальное значение pH лежит в интервале 7,37-7,42. Различие pH венозной
и артериальной крови может достигать 0,03 единицы pH и зависеть от того, из
какого сосуда брали пробу. Величина внутриклеточного рН-эритроцита на
0,15-0,23 единицы ниже pH плазмы крови.
13.16. Измерение pH в неводных растворителях
Если перед измерением pH в неводном растворителе электрод градуировали по
стандартному водному буферному раствору, измеренная величина фактически
не характеризует активность ионов водорода, поскольку потенциал жидкостно-
го соединения может быть как угодно велик в зависимости от природы раство-
рителя. Результаты измерений, полученные таким путем, можно рассматривать
как «кажущиеся». Для неводных сред разработаны шкалы и стандарты pH, ана-
логичные используемым для измерений в водных растворах, но не вполне соот-
ветствующие друг другу. Более подробно об этом см. [3] и [М. S. Frant «How to
Measure pH in Mixed & Nonqueous Solutions», Today's Chemist at Work, American
Chemical Society, June, 1995, p. 39].
13.17. Ионселекгивные электроды
Существуют различные типы электродов, мембранный потенциал которых се-
лективно зависит от активности определенного иона (или ионов), например в
случае стеклянного электрода мембранный потенциал зависит от активности
ионов водорода. Такие электроды очень важны для определения некоторых
ионов, особенно при низких концентрациях. В отличие от других электродов их,
как правило, не «отравляют» белки, поэтому такие электроды идеально подхо-
дят для проведения измерений в биологических средах. Особенно это справед-
ливо для стеклянных ионселективных электродов.
Ни один из этих электродов не специфичен относительно данного иона, но
каждый из них обладает определенной селективностью относительно данного
иона или ионов, поэтому их и следует называть ионселективными электрода-
ми (ИСЭ). На сайте www. nico2000. net находится учебная программа (руковод-
ство для начинающих) по теории ионселективных электродов, их градуировке и
технике измерения.
13.17. ИОНСЕЛЕКГИВНЫЕ ЭЛЕКТРОДЫ
537
Электроды со стеклянными мембранами
По конструкции такие электроды не отличаются от стеклянного электрода для
измерения pH. Варьируя состав стекла мембраны, можно добиться того, что гид-
ратированный слой будет обладать более высокой чувствительностью к раз-
личным однозарядным катионам и гораздо более низкой — к протонам, чем
стеклянный электрод для измерения pH. В основе зависимости мембранного по-
тенциала таких электродов от активности этих катионов, вероятно, лежат про-
цессы ионного обмена в слое гидратированного геля, как и в случае стеклянного
pH-электрода. Поэтому возникающий граничный потенциал определяется раз-
ностью активностей катиона на поверхности геля и во внешнем (анализируе-
мом) растворе. Очевидно, что с повышением активности катиона в анализируе-
мом растворе потенциал электрода увеличивается.
Конструкция электродов аналогична показанной на рис. 13.6. Внутренний
раствор обычно содержит хлорид катиона, к которому электрод наиболее чувст-
вителен. Состав мембраны может отличаться в зависимости от фирмы-произ-
водителя, но в принципе можно выделить три основных типа стеклянных
электродов:
1 - pH-электроды. Это стеклянные электроды для измерения pH, селектив-
ность которых изменяется в ряду Н+ Na+ > К+, Rb+, Cs+ ... ^> Са2+. Как уже
говорилось выше, отклик электрода на присутствие посторонних ионов вы-
зывает щелочную погрешность. Стеклянный электрод для измерения pH —
важнейший ионселективный электрод.
2 . Катионселективные электроды. Эти электроды обладают откликом на при-
сутствие, главным образом, однозарядных катионов, и их селективность
изменяется в ряду Н+ > К+ > Na+ > NHj, Li+ ... ^> Са2+.
3 - Натрий-селективные электроды. Их селективность изменяется в ряду:
Ag+ > Н+ > Na+ » К+, Li+ ... » Са2+.
Заметим, что стеклянные электроды всех трех типов обладают откликом на
ион водорода. Поэтому, хотя чувствительность к протону электродов двух по-
следних типов гораздо ниже, для определения катионов их можно применять то-
лько при достаточно высоких значениях pH, поскольку, для того чтобы отклик
электрода зависел только от активности определяемого катиона, активность
протонов должна быть очень мала. Наименьшая допустимая величина pH зави-
сит и от типа мембраны электрода, и от природы определяемого катиона. Значе-
ние pH должно быть выше некоторой предельной величины, зависящей от
концентрации первичного (определяемого) иона. (О появлении отклика элект-
рода на ОН- при высоких pH см. теорию К. Чена в разд. «От чего зависит потен-
циал электрода со стеклянной мембраной?»)
Натрий-селективные электроды пригодны для определения Na+ в присутст-
вии значительных количеств К+, поскольку их селективность по отношению к
Na+ более чем в 3000 раз выше, чем к К+. Можно изготовить стеклянные элект-
538
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
роды, селективность которых по отношению к Ag+ будет в 1000 раз выше, чем к
Na+. Стеклянные электроды можно применять при измерениях в среде жидкого
аммиака и расплавах солей.
Электроды с твердой мембраной
Конструкция электродов данного типа показана на рис. 13.12. Наиболее извест-
ным примером служит фторид-селективный электрод, мембраной которого явля-
ется пластинка из монокристалла LaF3 с добавками европия(П) для увеличения
электропроводности. Фторид лантана очень мало растворим, и электрод облада-
ет нернстовским откликом к F- до концентрации порядка 1 • 10-5 М, а не нерн-
стовским (тангенс угла наклона градуировочного графика отличается от теоре-
тического) — даже до 1 • 10-6 М (19 ppb!). Селективность этого электрода по от-
ношению к фторид-ионам почти в 1000 раз больше, чем к хлорид-, бромид-,
иодид-, нитрат-, сульфат-, гидрофосфат- и гидрокарбонат-ионам, и в 10 раз
выше, чем к гидроксид-ионам. Определению фторида серьезно мешает только
гидроксид-ион. При использовании фторид-селективного электрода интервал
pH ограничен в кислой среде образованием фтористоводородной кислоты, а в
щелочной — откликом электрода к ионам ОН-. Это сужает рабочий диапазон
при pH от 4 до 9. Фторид-селективный электрод—один из лучших ионселектив-
ных электродов. Его с упехом применяют для определения фторидов, что чрез-
вычайно сложно сделать с помощью других методов.
Для минимизации помех при работе с фторид-селективным электродом
полезно использовать раствор, содержащий ацетатную буферную смесь с pH
5,0-5,5, 0,1 М NaCl и циклогексилендинитрилтетрауксусную кислоту (ЦДТА).
Этот раствор известен как коммерческий универсальный регулятор ионной
силы TISAB (от англ. Total Ionic-Strength Adjustment Buffer). Если этим раство-
ром разбавить в соотношении 1 : 1 стандартные и анализируемые растворы, ве-
личина ионной силы этих растворов будет практически одинаковой и большой,
а за счет этого и потенциал жидкостного соединения, и коэффициент активности
фторид-иона будут постоянными для всех растворов. Указанная буферная смесь
создает pH, при котором не образуются заметные количества HF и отсутствует
отклик электрода на гидроксид-ион. ЦДТА, подобно ЭДТА, является комплек-
Рис. 13.12
Электрод с кристаллической мембраной
Внутренний раствор
Хлорсеребряный
электрод сравнения
Синтетическая мембрана из
пластинки монокристалла
13.17. ИОНСЕЛЕКТИВНЫЕ ЭЛЕКТРОДЫ
539
сообразующим лигандом и поэтому маскирует такие многозарядные ионы, как
Al3+, Fe3+ и Si4+, реагирующие с F- и изменяющие его активность.
Другим широко используемым электродом данного типа является электрод
с мембраной из Ag2S. Этот электрод селективен и по отношению к иону Ag+, и к
иону S2- до концентрации порядка 1 • 108 М, хотя судя по величине раствори-
мости Ag2S (Ks » 10-51) можно было ожидать более низкого предела обнаруже-
ния. Скорее всего это связано с трудностями приготовления таких разбавленных
растворов, а также адсорбцией (и десорбцией) ионов на поверхности электрода
и стенках сосудов, содержащих эти растворы. Мембрана обладает хорошей ион-
ной проводимостью и низким сопротивлением, поэтому из смеси Ag2S и другой
соли серебра или сульфида другого металла, сопротивление которых может быть
достаточно высоким, можно изготовить мембрану электрода, селективного к
этим ионам. Например, электрод с мембраной из AgI/Ag2S , кроме ионов Ag+ и
S2-, обладает откликом на ион I-, а смесь CuS/Ag2S можно использовать для из-
готовления электрода, селективного и к ионам Си2+.
Главное ограничение при изготовлении электродов с поликристалличе-
скими мембранами на основе смеси солей связано с тем, что растворимость вто-
рой соли должна быть существенно выше растворимости сульфида серебра, но
все же достаточно низкой, поскольку в противном случае снизится чувствитель-
ность определения. Пока такая мембрана содержит достаточно Ag2S для обеспе-
чения электропроводности, обусловленной ионами серебра, электрод дает отклик
на ионы Ag+. В этом случае отклик на определяемый ион возникает за счет его
участия в равновесиях, аналогичных приведенным при рассмотрении электрода
металл—соль металла, таким образом концентрация ионов серебра определяет-
ся равновесием осадок—раствор. Известны и другие электроды с поликристал-
лическими мембранами, содержащими Ag2S, — это электроды для определения
С1“, Вг_, SCN , CN-, РЬ2+ и Cd2+. Определению, конечно, будут мешать ионы, ре-
агирующие с Ag+ (в случае анионселективных электродов) или с S2- (для кати-
онселективных).
Электроды с жидкой мембраной
Схематическое изображение электрода с жидкой мембраной приведено на
рис. 13.13. «Мембраной» служит слой несмешивающегося с водой жидкого
Хлорсеребряный
электрод сравнения
Внутренний водный раствор
Мембрана или солевой мостик
Жидкий ионообменник
Пористая мембрана
Рис. 13.13
Электрод с жидкой мембраной
540
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
ионообменника, удерживаемого на инертном пористом носителе (в зависимости
от производителя, носитель может быть изготовлен из синтетического эластич-
ного материала или пористого стекла), который обеспечивает контакт анализи-
руемого раствора и ионообменника при минимальном смешивании. Внутренний
электрод сравнения (Ag I AgCl) погружен в раствор, содержащий хлорид опре-
деляемого иона, например, СаС12 в Са-селективном электроде или КС1 в К-селек-
тивном электроде. Хлорид-ион стабилизирует потенциал внутреннего Ag I AgCl
электрода сравнения.
Примером электродов такого типа является кальций-селективный электрод
с мембраной на основе Са-фосфорорганического ионообменника. Чувствитель-
ность электрода определяется растворимостью ионобменника в анализируемом
растворе. Электродная функция линейна вплоть до концентрации 5 • 105 М. Се-
лективность электрода по отношению к ионам кальция в 3000 раз выше, чем к
ионам натрия или калия, в 200 раз, чем к ионам магния, и в 70 раз, чем к ионам
стронция. Рабочий интервал pH составляет от 5,5 до 11. При pH выше 11 осажда-
ется гидроксид кальция. При определении кальция нельзя использовать фосфат-
ный буферный раствор — активность ионов кальция будет уменьшаться за счет
комплексообразования или осаждения. Экспериментально установлено, что
Са-селективный и другие электроды с жидкими мембранами часто легко отрав-
ляются, например, биологическими жидкостями.
Существуют электроды, обладающие откликом не на один, а на несколько
двухзарядных ионов, например, электрод с примерно одинаковым откликом на
ионы кальция и магния. Такой электрод очень удобен для определения жестко-
сти воды. Существуют медь- и свинец-селективные электроды, а также электро-
ды с жидкими мембранами для определения нитрата, перхлората и хлорида.
Принцип их устройства тот же, за тем исключением, что вместо катионообмен-
ника используют анионообменник. В табл. 13.13 суммированы характеристики
серийно выпускаемых электродов.
Электроды с пластифицированной мембраной,
содержащей ионофоры
Надежные в практическом использовании электроды можно изготовить на осно-
ве растворенного в эластичной пластифицированной мембране нейтрального ли-
пофильного (с выраженным сродством к органическим соединениям) ионофора,
селективно связывающего в комплекс определяемый ион. Ионофор должен
быть липофильным (т. е. не гидрофильным), чтобы он не вымывался из мембра-
ны при погружении в водные растворы. Мебрану изготавливают из поливинил-
хлорида (ПВХ), обычно она содержит около 33% ПВХ, 65% пластификатора, на-
пример, о-нитрофенилокгилового эфира (о-НФОЭ), 1,5% ионофора и около
0,5% тетракис(и-хлорфенил)бората калия (КТХФБ) для повышения электропро-
водности и минимизации влияния липофильных катионов, таких как SCN-. Сам
ион ТХФБ- липофилен и отталкивает другие липофильные анионы, способные
проникать в мембрану и влиять на величину отклика электрода. Указанные ком-
поненты растворяют в тетрагидрофуране (ТГФ), полученный раствор наносят на
13.17. ИОНСЕЛЕКГИВНЫЕ ЭЛЕКТРОДЫ
541
Таблица 13.3
Характеристики некоторых ионселективных электродов
Электрод Динамический интервал концентраций Возможные мешающие ионы*
С жидкой мембраной Са2+ 10°-10-5 Zn2+(3); Fe2+(0,8); Pb2+(0,6); Mg2+(0,l); Na+(0,003)
Cl- Ю-'-Ю-5 I-(17); NOj(4); Вг(2); НСОз(0,2); SO^~, F-(0,l)
Двухвалентный 10°-10-8 Fe2+, Zn2+(3,5); Cu2+(3,1); Ni2+(1,3); Ca2+, Mg2+(1);
катион Ba2+(0,94); Sr2+(0,54); Na+(0,015)
bf; Ю-'-Ю-5 NOj(0,l); Br(0,04); OAc~ НСС>з(0,004); Cl~(0,001)
NO3 10-‘-10-5 «0^(1000); I-(20); Br(0,l); NC>2(0,04); «-(0,004);
сю; Ю-'-Ю-5 CO|”(0,0002); F-(0,00006); 80^(0,00003) I-(0,01); NO3, OH-(0,0015); Br(0,0006); F-, «-(0,0002)
К+ 10°-10~5 Cs+(1); NHj(0,03); H+(0,01); Na+(0,002); Ag+, Li+(0,001)
С твердой мембраной* F" lO’-lO-6 Максимально допустимый уровень: ОН- <0,1 F“
Ag+ или S2- 10°-10-7 Hg2+<110-7M
а Цифра в скобках характеризует относительную селективность электрода по отношению к мешающему и
определяемому ионам (о коэффициентах селективности см. ниже).
6 Указаны максимально допустимые концентрации мешающих ионов.
стеклянную поверхность и выдерживают до испарения растворителя. В резуль-
тате получается гибкая мембрана, которую можно легко вмонтировать в корпус
электрода.
Самым удачным примером электродов данного типа может служить ка-
лий-селективный электрод на основе ионофора валиномицина. Валиномицин —
это известный природный антибиотик, циклический полиэфир, размер внутрен-
ней полости которого, образованной атомами кислорода, очень близок к разме-
ру иона калия, что позволяет селективно связывать его. Электрод в 104 раз более
селективен по отношению к иону калия, чем к иону натрия.
Перспективными ионофорами для изготовления катион-селективных элек-
тродов, особенно для определения ионов щелочных и щелочноземельных ме-
таллов, являются краун-эфиры. Это нейтральные циклические эфиры, которые
можно сконструировать так, чтобы сформировать циклы нужного размера, спо-
собны образовывать комплексы с определенными катионами. Краун-эфирам
придают липофильность введением в молекулу фенильных групп или длинных уг-
леводородных цепей. В качестве примера можно привести 14-краун-4 (рис. 13.14),
селективный по отношению к иону лития в присутствии иона натрия. Цифра 4
указывает на число атомов кислорода в кольце, а 14 — характеризует размер
кольца. Размер полости 14-краун-4 и его производных подходит для связывания
542
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
Рис. 13.14
Производное 14-краун-4, селективно связывающее ион лития
в комплекс ионов лития. Введение в структуру полиэфира объемных фениль-
ных групп создает стерические препятствия для образования комплекса [кра-
ун-эфир—натрий] состава 2 : 1 и в 800 раз повышает селективность электрода к
иону лития, образующего комплекс состава 1:1. Существуют электроды на
основе краун- эфиров для определения ионов натрия, калия, кальция и других
металлов. Синтезированы ионофоры с амидными группами, селективно взаи-
модействующие с некоторыми ионами. Примеры структур ионофоров приве-
дены на рис. 13.15. За пионерские работы по краун-эфирам американский
химик Ч. Педерсен в 1987 г. был удостоен Нобелевской премии — см.
http://almaz.com/nobel/chemistry/1987c.html.
Покрытые проволочные электроды
Как сообщают Каттрал и Фрейзер, ионселективные электроды можно изгото-
вить нанесением мембраны на основе ПВХ на металлическую проволоку (на-
пример, Pt, Си, Ag), служащую токоотводом (см. Anal. Chem.. 43 (1971) 1905).
Такие электроды очень просты в изготовлении (проволоку покрывают раство-
ром ингредиентов мембраны в ТГФ и позволяют растворителю испариться),
удобны в работе и позволяют получать вполне удовлетворительные результаты.
Потенциометрические ферментные электроды:
определение субстратов
Ионселективный электрод с пленкой, содержащей иммобилизованный фермент,
пригоден для селективного определения субстрата этого фермента. Ферменты и
их свойства более подробно рассмотрены в гл. 24.
РИС. 13.15. Ионофоры для Н+, Na+ и Са2+
13.17. ИОНСЕЛЕКГИВНЫЕ ЭЛЕКТРОДЫ
543
Рассмотрим реакцию гидролиза мочевины (субстрата) в присутствии фер-
мента уреазы:
NH2CONH2 + 2Н2О + Н+ 1== 2NH4 + НСОз
Электрод для определения мочевины можно изготовить, нанеся слой геля с
иммобилизованной уреазой на поверхность катионселективного стеклянного
электрода, дающего отклик на однозарядный катион. Если такой электрод по-
грузить в раствор, содержащий мочевину, то молекулы мочевины будут диф-
фундировать в слой геля, и находящийся там фермент будет катализировать ее
гидролитическое разложение с образованием иона NH J. Образовавшиеся ионы
аммония диффундируют к поверхности электрода, и в результате на катионселек-
тивном стеклянном электроде возникает потенциал. Равновесие устанавливается
через 30-60 с, и при оптимальных условиях проведения эксперимента наблюда-
ется линейная зависимость потенциала электрода от логарифма концентрации
мочевины. При правильном выборе иммобилизованного фермента и электрода
можно изготовить и другие ферментные электроды.
Механизм возникновения потенциала
мембранного электрода
Механизм возникновения потенциала мембранных ионселективных электродов
менее изучен по сравнению со стеклянным электродом для измерения pH, и поэ-
тому гораздо меньше известно о том, за счет чего возникает потенциал и чем он
определяется. Тем не менее механизмы должны быть аналогичны. В мембране
электрода обычно содержится определяемый ион, селективно связанный реа-
гентом либо в малорастворимое соединение, либо в комплекс. Или же электрод
необходимо предварительно кондиционировать в растворе определяемого иона,
и тогда этот ион также будет селективно связан реагентом мембраны. Послед-
ний случай аналогичен образованию слоя гидратированного геля —SiO_H+ при
выдерживании стеклянного электрода в воде. При погружении ионселективного
электрода в анализируемый раствор на поверхности раздела мембрана—раствор
возникает граничный потенциал. Возможный механизм его возникновения обу-
словлен стремлением ионов диффундировать из более концентрированного рас-
твора в более разбавленный, как в случае возникновения жидкостной границы и,
соответственно, потенциала жидкостного соединения. Диффузия катионов при-
ведет к появлению положительного заряда и к увеличению потенциала электро-
да, в случае же диффузии анионов, наоборот, возникает отрицательный заряд и
потенциал уменьшается.
Из сказанного выше следует, что при изготовлении ионселективного элект-
рода очень важно подобрать материал с центрами связывания с высоким срод-
ством к определяемому иону. Так, кальций-селективный электрод с жидкой
мембраной обладает гораздо более высокой селективностью по отношению к
иону кальция по сравнению с ионами магния и калия из-за высокого сродства к
кальцию используемого органического катионообменника. На границе раздела
544
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
мембрана—раствор устанавливается равновесие с участием ионов кальция, и ве-
личина потенциала электрода определяется отношением активностей ионов ка-
льция в анализируемом растворе и фазе мембраны.
Коэффициент селективности
Если в растворе содержится только определяемый (потенциалопределяющий)
ион, то по аналогии с уравнением (13.38) для стеклянного pH-электрода для по-
тенциала ионселективного электрода можно записать
^исэ = * + -1ё«,- (13.44)
Z
где S — тангенс угла наклона электродной функции (теоретический равен
2,3037?77F), z — заряд иона с учетом знака. Часто отклик электрода оказывается
ниже теоретического (нернстовского), но для однозарядных ионов он обычно
близок к нему. Величина константы к зависит от природы внутреннего электро-
да сравнения, состава внутреннего раствора и природы мембраны. Величину
этой константы можно оценить, измерив потенциал электрода в растворе с изве-
стной активностью иона.
Пример 13.6
С помощью фторид-селективного электрода определяют содержание фторид-
ионов в воде. Стандартные и анализируемые растворы разбавили в соотношении
1:10 раствором TISAB для поддержания постоянной ионной силы. Потенциал
электрода (относительно данного электрода сравнения) в исходном 1,00 • 10-3 М
стандартном растворе равен -211,3 мВ, в 4,00 • 10-3 М стандартном растворе
-238,6 мВ, а в анализируемом растворе -226,5 мВ. Какова концентрация фтори-
да в пробе?
Решение
Поскольку после разбавления с помощью TISAB ионная сила всех растворов
одинакова и z = -1, величина отклика электрода пропорциональна lg[F_]:
£ = £ + -lg[F-] = A:-Slg[F-],
z
Сначала рассчитаем величину 5:
-211,3 = £-51g (1,00 • 10"3) (1)
-238,6 = к - S 1g (4,00 • 10-3) (2)
После вычитания уравнения (2) из уравнения (1), получаем
400 10-3
27,3 = S 1g (4,00 • 10-3) - 51g (1,00 • 10’3) = S 1g
1,00 1 о-3
13.17. ИОНСЕЛЕКГИВНЫЕ ЭЛЕКТРОДЫ
545
27,3= Sig 4,00
S = 45,3 мВ (тангенс угла наклона несколько ниже теоретического)
Теперь рассчитаем к:
-211,3 = £-45,3 1g (1,00 • 10-3)
к = -347,2 мВ
и, наконец, найдем искомую концентрацию фторид-ионов:
-226,5 =-347,2-45,3 lg [F“]
[F-] = 2,16 • 10-3М
Если электрод погрузить в раствор, содержащий смесь катионов (или анио-
нов в случае анионселективного электрода), он может реагировать на присутст-
вие посторонних катионов. Предположим, что в растворе, кроме ионов натрия,
содержатся ионы калия и электрод реагирует на присутствие и тех, и других. В
этом случае в уравнение (13.44) необходимо ввести слагаемое, учитывающее ак-
тивность ионов калия
^NaK = ^Na + 1g (а Na+ + ^NaK62 к+ ) 1
где величина £Na отражает вклад в измеренный в этом растворе потенциал ENaK
только ионов натрия. Константа A?NaK, называемая коэффициентом селектив-
ности электрода, показывает насколько электрод более селективен по отноше-
нию к иону натрия по сравнению с ионом калия. Это величина обратная ATKNa —
коэффициенту селективности электрода по отношению к иону калия по срав-
нению с натрием A?NaK = l/A?KNa. Очевидно, что величина A?NaK должна быть как
можно меньше, чтобы слагаемое A?NaKa к+ было пренебрежимо мало. Абсолютно
селективного электрода не существует. В идеальном случае можно добиться, чтобы
произведение A?NaKaK+ было пренебрежимо малым по сравнению caNa+.
Величины A?NaK и £Na можно оценить, измерив потенциал электрода в двух
различных стандартных растворах ионов калия и натрия и решив систему двух
уравнений. Можно поступить иначе: взять один из стандартных растворов, со-
держащий только ионы натрия, и найти величину £Na по уравнению (13.44).
Для смеси двух ионов с разными зарядами можно написать общее уравне-
ние, которое называется уравнением Никольского:
ЯаВ = *а+ —lg(«A + *AB«BA/ZB)
ZA
(13.46)
546
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
где zA — заряд иона А (потенциалопределяющего), zB — заряд постороннего
иона В. Так, зависимость потенциала натрий-селективного электрода от актив-
ности ионов натрия в присутствии ионов кальция описывается уравнением
£NaCa = *Na + Slg(«Na+ + ^NaCa^l) (13.47)
Поскольку все электроды в той или иной степени чувствительны к протонам,
при поведении эксперимента желательно поддерживать активность ионов водо-
рода настолько низкой, чтобы слагаемым в уравнении Никольского
можно было пренебречь. Величина A?NaK а при данном значении (?Na+ опреде-
ляет минимальную допустимую величину pH при работе с данным электродом.
Необходимо упомянуть об одной проблеме, связанной с коэффициентами
селективности. Часто коэффициенты не являются константами, потому что их
величины зависят от соотношения концентраций ионов, в связи с этим их нельзя
использовать при вычислениях в случае смеси ионов. Их используют для выбо-
ра условий измерения потенциала, при которых мешающим влиянием посторон-
них ионов, т. е. величиной А?авява/ Zb , можно пренебречь и потенциал электрода
будет определяться только величиной активности определяемого иона. Если для
нахождения концентрации используют градуировочный график, то при его по-
строении во все стандартные растворы можно ввести посторонний ион в той же
концентрации, что и в анализируемом растворе; в результате может получиться
нелинейный, но корректный градуировочный график. Этот метод применим то-
лько в том случае, когда концентрация мешающего иона практически постоянна
во всех растворах*.
Следующий пример иллюстрирует возможность использования коэффици-
ента селективности в расчетах результатов анализа.
Пример 13.7
С помощью кальций-селективного электрода определяют активность ионов
кальция в присутствии ионов натрия. Потенциал электрода (отн. НКЭ) в
0,0100 М СаС12 равен +195,5 мВ, а в растворе, содержащем 0,0100 М СаС12 и
0,0100 М NaCl, — 201,8 мВ. Какова активность ионов кальция в анализируемом
растворе, если потенциал электрода (отн. НКЭ) равен 215,6 мВ, а с помощью
натрий-селективного электрода найдено, что активность ионов натрия в нем
составляет 0,0120 М? При расчетах предположите, что электрод обладает
нернстовским откликом.
Решение
Ионная сила 0,0100 М раствора СаС12 равна 0,0300 М, а для смеси 0,0100 М
СаС12 и 0,0100 М NaCl величина ионной силы составляет 0,0400 М. По уравне-
Необходимо также, чтобы концентрация мешающего иона во всех растворах была известна. —Прим, перев.
13.17. ИОНСЕЛЕКГИВНЫЕ ЭЛЕКТРОДЫ
547
нию (6.20) находим, что коэффициент активности ионов кальция в чистом рас-
творе СаС12 равен 0,55, а в смеси коэффициенты активности ионов кальция и
натрия равны 0,51 и 0,83 соответственно. Поэтому
ЛСа = ЕСа - 29,58 1g аСа2+ = 195,5 -29,58 1g (0,55 • 0,0100) = 262,3 мВ
^CaNa = ^Са + 29,58 1g («Са2+ + ^CaNa^J
201,8 = 262,3 + 29,58 1g [0,51 • 0,0100 + £CaNa(O,83 • 0,0100)2]
*CaNa = 47
215,6 = 262,3 + 29,58 lg (aCa2+ + 47 • 0,01202)
an 2+ =0,0196 M
Ca
Обратите внимание на то, что хотя найденная величина коэффициента се-
лективности для кальция достаточно велика (электрод более селективен к иону
натрия), вклад присутствующих в смеси ионов натрия (0,0068) составляет всего
0,3 от вклада ионов кальция (0,0196), поскольку в уравнение входит активность
ионов натрия в квадрате.
Экспериментальные методы оценки
коэффициентов селективности
Приведенный выше пример иллюстрировал возможность применения уравне-
ния Никольского для идеального случая. Но, как уже упоминалось, величина ко-
эффициента селективности в действительности не может быть постоянной и
зависит от метода его оценки. Обычно используют один из двух методов — ме-
тод отдельных растворов или метод смешанных растворов. Чаще использу-
ют второй метод. Рассмотрим кратко суть обоих методов.
1. Метод отдельных растворов. Данный метод за-
ключается в построении градуировочного графика для
каждого изучаемого иона. В результате получают две
параллельные прямые, причем выше будет расположе-
на прямая для потенциалопределяющего (первичного)
иона, если это катион. Величину коэффициента селек-
тивности можно рассчитать по разности потенциалов
(см. задачу 21). Для двух однозарядных ионов А и В
-1д ХАВ = AE/S
] _ ^А *В
^АВ “
Л
(13.48)
548
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
где ЕА и Ев — потенциалы электрода при фиксированных концентрациях этих
ионов в индивидуальных растворах. Очевидно, что чем более селективен элект-
род относительно иона А, тем больше разность потенциалов (тем меньше будет
величина Ев).
2. Метод смешанных растворов. Данный метод заключается в измерении по-
тенциала электрода в растворах, содержащих оба иона. Существует много вари-
антов этого метода, но обычно используют метод постоянной концентрации
мешающего иона. Метод смешанных растворов лучше отражает условия изме-
рения потенциала в анализируемых растворах. Рассмотрим применение данного
метода на примере оценки коэффицента селективности литий-селективного элект-
рода в присутствии ионов натрия. Приготовим растворы для построения градуиро-
вочного графика для иона лития с фиксированной концентрацией ионов натрия,
например 140 мМ, как в крови. Общий вид графика показан на рис. 13.16. Верхняя
часть кривой соответствует нернстовскому отклику электрода на ион лития.
С понижением концентрации ионов лития потенциал электрода все в большей
степени определяется концентрацией ионов натрия, являющейся постоянной ве-
личиной и поэтому нижняя часть кривой соответствует смешанному отклику
электрода на оба иона. При очень низкой концентрации ионов лития электрод
реагирует исключительно на ион натрия (базовый потенциал).
Существует два способа оценки коэффициента селективности по получен-
ному графику. Первый из них основан на графическом определении точки, в кото-
рой электрод дает одинаковый отклик на оба иона. Это соответствуют значению
активности иона А, найденному экстраполяцией линейного участка кривой, при
которой потенциал определяется только ионами В (такое значение концентра-
ции А, при котором у электрода был бы тот же потенциал, что и в отсутствие
ионов В, т. е. электрод обладал бы нернстовским откликом к иону А).
Поскольку в этой точке вклад каждого иона в измеряемый потенциал одина-
ков, согласно уравнению (13.46) можно записать
РИС. 13.16
Оценка коэффициента селективности
методом смешанных растворов
аА ^АВ ав
(13.49)
13.17. ИОНСЕЛЕКГИВНЫЕ ЭЛЕКТРОДЫ
549
или
^АВ
аА
azA/zB
ив
(13.50)
где ав — фиксированная активность постороннего иона и аА — активность иона
А в точке пересечения. Например, если, в присутствии 150 мМ ионов лития ак-
тивность иона натрия в этой точке равна 1,0 мМ, то XLiNa = 1,00/150 = 6,7- 10-3 и,
следовательно, селективность электрода относительно иона лития в 150 раз
выше.
Второй способ оценки коэффициента селективности основан на использо-
вании теоретической точки, в которой вклад обоих ионов в измеряемый потен-
циал электрода одинаков. В этой точке экстраполируемый линейный верхний
участок кривой описывается уравнением (13.44), т. е.
с
^A^A + -lg(4) (13.51)
ZA
Нелинейный участок кривой описывается уравнением (13.46). Если вклад обоих
ионов в измеряемый потенциал одинаков, то аА = КАВавА^в и уравнение (13.46)
принимает вид
£ав = kA + — !g (2ga) = kA + — !g aA +---- (13.52)
ZA ZA ZA
При наличии нернстовского отклика при 25 °C
с 1 7 о
^АВ - + — 1g + — мВ (13.53)
ZA ZA
Из уравнения (13.52) следует, что значение активности иона А, вносящее такой
же вклад в измеряемый потенциал электрода, что и активность мешающего иона
В, можно определить графически. При таком значении 1g ак разность между со-
ответствующими точками экстраполированного участком и экспериментальной
кривой составляет 0,3015/zA мВ, т. е. 17,8/zA мВ. Для однозарядного иона в слу-
чае нернстовского отклика электрода это составляет 17,8 мВ и 8,9 мВ для двух-
разрядного. Далее можно опять воспользоваться уравнением (13.50).
Хотя эти два способа расчета редко дают одинаковые значения коэффици-
ентов селективности, принцип расчета один и тот же и получаемые результаты
сопоставимы.
3. Метод соответственных потенциалов. Это чисто эмпирический вариант ме-
тода постоянной концентрации мешающего иона, позволяющий аналитику по-
550
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
лучить эмпирическую величину коэффициента селективности для конкретных
условий проведения эксперимента. Допустим, что нам необходимо оценить от-
носительное мешающее влияние ионов натрия, присутствующих в крови, на ре-
зультаты определения ионов лития (порядка 1 мМ) в сыворотке крови пациента
с маниакально-депрессивным синдромом, принимавшего Li2CO3. Сначала уста-
навливают потенциал «сравнения», измерив потенциал электрода в растворе,
содержащем 140 мМ натрия (концентрация натрия в сыворотке). Затем измеря-
ют потенциал электрода в 140 мМ растворе NaCl, содержащем 1 мМ лития. На-
блюдаемое увеличение потенциала — это отклик электрода на присутствие
лития. Повышая концентрацию NaCl, определяют величину добавочной концент-
рации ионов натрия (добавки), при которой электрод обладает таким же откли-
ком, что в присутствии 1 мМ ионов лития. Отношение концентрации Li+ (1 мМ)
к найденной величине добавки Na+ является эмпирическим коэффициентом се-
лективности. Например, если электрод дает адекватный отклик при концентра-
ции натрия 240 мМ (добавка 100 мМ), то коэффициент селективности равен
1,0 • 10-2, т. е. селективность электрода по отношению к литию в 100 раз выше,
чем к натрию*. Более детально с этим методом можно познакомиться в оригина-
льных работах [16-18].
Метод рекомендован ИЮПАК в качестве предпочтительного метода оцен-
ки коэффициентов селективности [17]. Этот эмпирический метод позволяет по-
лучить относительные отклики электрода на определяемый и мешающий ионы в
конкретных условиях проведения определения. Заряды ионов во внимание не
принимаются. Применение данного метода в отношении всех ионселективных
электродов рассмотрено в работе [18].
В какой мере применимо уравнение Никольского?
Выше уже неоднократно упоминалось о том, что величины коэффициентов се-
лективности, расчитанные по уравнению Никольского, часто отличаются в зависи-
мости от соотношения концентраций ионов и условий проведения эксперимента, и
уравнение не применимо, если отклик электрода не является нернстовским (для
мешающего иона это бывает достаточно часто). Попыткой решения этой проб-
лемы и является метод соответственных потенциалов. Бэккер с сотрудниками
предложили количественный подход, четко обосновывающий и позволяющий
интерпретировать суть метода. На основе теориии ионного обмена они вывели
уравнение, в котором правая часть выражения (13.49) представлена в виде
а величина А:дв1 названа фактором селективности [19]. Заметим, что
отношение зарядов определяемого и мешающего ионов не имеет значения. Ав-
торы обращают внимание на то, что поскольку для ионов разного заряда тангенсы
углов наклона электродных функций (градуировочных зависимостей) различны,
величины факторов селективности (и, следовательно, величин достигаемых по-
тенциалов) не будут постоянными и их трудно сравнивать, если неизвестны кон-
Подробнее об этом см. [V. Р. Y. Gadzekpo and G. D. Christian, Anal. Chim. Acta, 164 (1984) 279].
13.17. ИОНСЕЛЕКТИВНЫЕ ЭЛЕКТРОДЫ
551
кретные условия проведения эксперимента. По этой причине использование
уравнения Никольского может быть более предпочтительным. Авторы [19]
вывели также формулу для расчета величины А?ав, при которой для данного
анализируемого раствора, содержащего ионы А и В (с аА и ав соответственно)
обеспечивается максимально допустимая погрешность рАВ (%). Эта формула
применима при /?АВ менее 10%.
^ав = «а/«ваЛв (Рав/ЮО) Za/zb (13.54)
Это уравнение пригодно для оценки возможности селективного измерения с по-
мощью данного электрода.
Одна из проблем оценки истинных величин коэффициентов селективности
электродов на основе нейтральных переносчиков заряда заключается в следую-
щем. Хотя электрод и может обладать нернстовским откликом на мешающий
ион, но после выдерживания в растворе определяемого иона, он эту способность
утрачивает. Бэккер разработал способ определения неискаженных значений ко-
эффициентов селективности с использованием свежеприготовленных электродов,
проводя измерения сначала в растворах мешающего иона, а затем — определя-
емого [20, 21]. Так можно получить данные о том, какие из ионофоров действи-
тельно более селективны к определяемому иону. (См. обзор [22] данных о селек-
тивности ионселективных электродов и обзор Бэккера [14] по электродам на
основе ионофоров.)
Какова чувствительность определения с помощью
ионселективного электрода?
Пределом обнаружения с помощью ионселективного электрода считают актив-
ность (концентрацию), соответствующую точке пересечения касательных к гори-
зонтальному и восходящему участкам градуировочного графика, как показано на
рис. 13.16. Обычно это величина порядка Ю^-Ю-5 М. Для поддержания стабиль-
ного потенциала электрода во внутреннем растворе создают довольно высокую
концентрацию определяемого иона. Например, внутренний раствор кальций-се-
лективного электрода содержит 10 мМ СаС12. Бэккер с соавторами предположили,
что в результате диффузии определяемых ионов через мембрану примембранный
слой анализируемого раствора загрязняется и концентрация определяемого иона
в нем может быть порядка п • 10-6 М. Поскольку электрод реагирует на концент-
рацию определяемого иона именно в примембранном слое, предел обнаружения
определяется величиной порядка п • 10-6 М. Бэккеру удалось показать, что сни-
зив концентрацию определяемого иона во внутреннем растворе до п • 10~6 М,
можно снизить предел обнаружения в 100 и более раз и достичь порядка опреде-
ляемых величин 108 М [23, 24].
Швейцарским исследователям во главе с Претчем удалось еще больше сни-
зить предел обнаружения, создав в мембране условия для ионного обмена, исклю-
чающего вымывание определяемого иона из мембраны [25]. Это удалось сделать
552
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
путем введения во внутренний раствор другой соли (например, при определении
иона Са2+ это может быть NaCl) в относительно высокой концентрации. При
этом возникает градиент концентрации, по которому ионы Са2+ диффундируют
внутрь мембраны, а следовательно, не вымываются из мембраны. Такой прием
позволил снизить предел обнаружения до 10-12 М. Отсутствие возможности да-
льнейшего снижения предела обнаружения можно объяснить следующим образом.
Если определяемые ионы диффундируют в направлении внутреннего раствора
слишком быстро, то за счет обеднения мембраны этими ионами может возник-
нуть отрицательная погрешность. Бэккер и Претч объединились для совместно-
го изучения механизма работы ионселективных электродов и попытались
разработать стабильные системы, позволяющие получать неискаженные резу-
льтаты определения — см., например, [26, 27].
Измерения с помощью ионселективных электродов
Большинство ионселективных электродов, как и стеклянный электрод для изме-
рения pH, имеют высокое сопротивление, поэтому для проведения измерений
нужен электрометр или рН-метр. Обычно используют рН-метр с растянутой
шкалой. Часто требуется предварительная подготовка электрода — выдержива-
ние его в растворе определяемого иона.
На правильность результатов определения с помощью ионселективных
электродов влияют те же причины, которые обсуждались выше при рассмотре-
нии определения pH и других прямых потенциометрических измерений.
Для того, чтобы по градуировочному графику, построенному в координатах
Е—1g яА, найти концентрацию определяемого иона, необходимо поддерживать
в растворе постоянную ионную силу, как описано ранее [см. уравнение (13.34)].
Например, при определении концентрации несвязанного иона кальция в сыво-
ротке крови, пробу и эталонные растворы разбавляют 0,15 М NaCl. Ионселек-
тивный электрод измеряет активность только свободного иона.
Коэффициент активности иона натрия в нормальной сыворотке крови чело-
века, измеренный с помощью ионселективного электрода, оказался равным
0,780 ± 0,001, а в водной фракции сыворотки — 0,747 (в сыворотке содержится
около 96% (об.) воды). Для градуировки электродов при определении натрия и
калия в сыворотке крови используют стандартные растворы NaCl и КС1. В рас-
творах с концентрацией 1,0, 10,0 и 100,0 мМ активность иона натрия соответст-
венно равна 0,965, 9,03 и 77,8 мМ (чистый раствор NaCl), активность иона калия
составляет 0,965, 9,02 и 77,0 мМ (чистый раствор КС1).
Часто время отклика ионселективного электрода велико, и требуется дово-
льно много времени для установления стабильного потенциала электрода. Чем
ниже концентрация определяемого иона, тем медленнее он устанавливается. Но
существуют электроды с таким малым временем отклика, что их используют
при изучении кинетики реакций.
Ниже приведены достоинства и недостатки ионселективных электродов, а
также некоторые ограничения их использования.
13.17. ИОНСЕЛЕКГИВНЫЕ ЭЛЕКТРОДЫ
553
1 С помощью ионселективного электрода измеряют активность, а не кон-
центрацию иона. Это уникальная ситуация и следует помнить, что для
пересчета активностей в концентрации требуется учитывать ионную
силу раствора.
2. С помощью ионселективного электрода измеряют активность «свобод-
ных» ионов, т. е. ионов, не связанных с другими компонентами раствора.
Могут возникать погрешности за счет реакций комплексообразования,
кислотно-основных и др. видов реакций.
3. Ионселективные электроды не специфичны, они лишь обладают повышен-
ной селективностью относительно конкретного иона, в то же время потенци-
ал электрода подвержен влиянию посторонних ионов. Электроды обладают
откликом на ион водорода, поэтому рабочий интервал pH ограничен.
4- В отличие от фотометрии, с помощью ионселективных электродов мож-
но анализировать мутные и окрашенные растворы.
5 - Потенциал электрода линейно зависит от логарифма активности определяе-
мого иона, поэтому диапазон определяемых концентраций широкий, обыч-
но от 4 до 6 порядков, а погрешность определения хотя и относительно
высока, но постоянна во всем интервале выполнения электродной функции.
6- За исключением измерения потенциала в разбавленных растворах, время
отклика электрода достаточно мало и часто для выполнения измерения
требуется менее 1 мин. Существуют электроды с таким малым временем
отклика, что с их помощью можно проводить непрерывные измерения.
7- Величина отклика электрода зависит от температуры — RT/(nF).
8. Для работы в полевых условиях можно сконструировать портативные
устройства и анализировать малые объемы пробы (порядка 1 мл).
9- В процессе измерения проба не разлагается: ионометрия — недеструк-
тивный метод анализа.
10- Только с помощью лучших ионселективных электродов можно прово-
дить измерения при концентрации порядка 106 М, тогда как с помощью
других методов — порядка 10~9 М и ниже.
11 - Для получения надежных результатов электрод необходимо периодиче-
ски градуировать.
12- Известно несколько первичных стандартов активности, по сути аналогич-
ных pH-стандартам*. Но в отличие от стандартов для измерения pH они не
«забуферены» относительно определяемого иона, поэтому примеси, осо-
бенно в разбавленных стандартах, могут искажать результаты определения.
Для некоторых солей, например NaCl, стандарты активности (аналогичные pH-стандартам) выпускает На-
циональный институт стандартов и технологии США.
554
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
Несмотря на некоторые ограничения в использовании, ионселективные
электроды очень важны и полезны, поскольку они представляют собой вопло-
щение мечты химиков-аналитиков о зонде, специфичном по отношению к опре-
деляемому компоненту и поэтому не требующем сложной пробоподготовки для
жидкостей.
13.18. Ионселективные полевые транзисторы
Компактные твердотельные химические сенсоры, называемые ионселективны-
ми полевыми транзисторами (англ, ion-selective field-effect transistor, или ISFET),
можно изготовить, используя для обеспечения электрического контакта полу-
проводниковые материалы. Транзистор покрывают слоем изолятора, обычно
SiO2, и слоем Si3N4. За счет этого повышается электрическая стабильность и со-
здается слой, непроницаемый для ионов анализируемого раствора. Затем нано-
сят слой мембраны например, на основе ПВХ. При наложении потенциала
протекает слабый ток, величина которого меняется в присутствии определяемо-
го иона за счет изменения мембранного потенциала. Приложенное напряжение
регулируют так, чтобы величина силы тока вернулась к первоначальному зна-
чению и это позволяет оценить отклик электрода на определяемый ион.
На рис. 13.17 показан pH-электрод на основе полевого транзистора. В неболь-
шой зонд вмонтированы электрод сравнения и датчик температуры. Информа-
цию о таких электродах можно найти на сайтах фирм IQ Scientific Instruments,
Inc. (www.phmeters.com), Sentron, Inc. (www.sentronph.com), Jenco Instruments,
Inc. (www.servonics.com/Jenco.htm).
Сенсор на основе кремниевого чипа с быстрым откликом
Встроенный электрод сравнения и датчик температуры
Рис. 13.17. Ионселективный полевой транзистор. С разрешения IQ Scientigic Instuments, Inc.
ЧТО МЫ УЗНАЛИ ИЗ ЭТОЙ ГЛАВЫ?
555
Что мы узнали из этой главы?
• Типы электродов и расчет потенциала электрода по уравнению Нернста
[основные уравнения (13.3), (13.10), (13.16)] — стр. 504
• Жидкостное соединение и потенциал жидкостного соединения — стр. 512
• Электроды сравнения — стр. 515
• Точность потенциометрических измерений [основное уравнение (13.36)] —
стр. 521
• Стеклянный электрод для измерения pH [основное уравнение (13.42)] —
стр. 522
• Стандартные буферные смеси и точность измерения pH — стр. 530, 531
• рН-Метр — стр. 532
• Ионселективные электроды — стр. 536
• Коэффициент селективности [основное уравнение (13.46)] — стр. 544
Вопросы
1 - Что такое потенциал жидкостного соединения? Что такое неэлиминиро-
ванный потенциал жидкостного соединения? Как свести его к минимуму?
2 Объясните механизм возникновения отклика стеклянной мембраны при из-
менении pH.
3 - Какова природа возникновения щелочной и кислотной погрешности при
измерении pH с помощью стеклянного электрода?
4 . Назовите типы ионселективных электродов. Опишите их конструкцию,
различия в свойствах мембран и практическое применение.
5 - Что такое коэффициент селективности? Какая важная характеристика элект-
рода определяется его величиной? Как можно оценить величину коэффи-
циента селективности?
6 . Что такое краун-эфиры? Приведите формулу 16-краун-6.
7 . Напишите уравнение Никольского и объясните его суть.
Задачи
Стандартные потенциалы
8 - Стандартный потенциал электрода Ag I AgBr равен 0,073 В. Рассчитайте
величину произведения растворимости AgBr.
9 - Раствор тиоцианата титруют раствором ионов серебра. В конечной точке
потенциал равен 0,202 В (отн. НКЭ). Рассчитайте величину стандартного
потенциала для полуреакции Ag+ + е_ = Ag. Для тиоцианата серебра
Ks = 1,00- io-12.
556
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
Гальванические элементы
10 - Для каждой из ниже приведенных реакций: 1) напишите уравнения соот-
ветствующих полуреакций; 2) напишите схему ячейки, в которой реакция
протекает в указанном направлении; 3) вычислите стандартный потенциал
ячейки; 4) укажите полярность электродов в условиях, когда реакция будет
протекать в указанном направлении.
a) Ag + Fe3+ = Ag+ + Fe2+
6) VO 2 + V3+ = 2VO2+
в) Ce4+ + Fe2+ = Ce3+ + Fe3+
11 - Для указанных ниже ячеек напишите уравнения полуреакций, протекаю-
щих на каждом электроде, уравнения окислительно-восстановительных ре-
акций и рассчитайте ЭДС ячейки:
a) Pt, Н2(0,2 атм) I НС1(0,5 М) I С12(0,2 атм), Pt
б) Pt | Fe2+(0,005 М), Fe3+(0,05 М), НСЮ4(0,1 М) II НСЮ4(0,1 М),
VO 2 (0,001 М), V02+(0,002) М | Pt
Измерение окислительно-восстановительных потенциалов
12- Каковы будут потенциалы указанных ниже полуэлементов при стандарт-
ных условиях относительно насыщенного каломельного электрода:
a) Pt | Вг2(водн.), Вг-
б) Ag ] AgCl I Cl-
iO Pt |v3+, v2+
13 - Какова будет величина потенциала, если указанный полуэлемент включить
в цепь с насыщенным каломельным электродом? Цифры в скобках — ак-
тивности ионов.
Fe3+(0,00200 М), Fe2+(0,0500 М) | Pt
14- Раствором нитрата серебра (0,10 М) титруют 50 мл раствора, содержащего
0,10 М хлорид- и иодид-ионов. а) Рассчитайте, сколько процентов иодид-
ионов останется неосажденными в момент начала осаждения хлорида се-
ребра. б) Рассчитайте величину потенциала серебряного электрода (отн.
НКЭ) в момент начала осаждения хлорида серебра и сравните ее с теорети-
ческой величиной потенциала в конечной точке титрования иодид-иона.
в) Рассчитайте величину потенциала в конечной точке титрования хло-
рид-иона.
Расчеты проведите, используя величины концентраций (ионной силой пре-
небрегите).
ЗАДАЧИ
557
15- Зависимость потенциала электрода Hg | Hg-ЭДТА, N-ЭДТА, Nn+ зависит
от активности иона Nn+ описывается уравнением
г „о 2.303ЯГ. ^/(Hg-эдтА) 2.303ЯГ. «n-эдта 2,ЗОЗЯ7\ 1
Е = £Hg-/Hg----1g -------------------1g ----------------1g —
^/(N-ЭДТА)
Устойчивость N-ЭДТА должна быть значительно ниже устойчивости
Hg-ЭДТА (это очень устойчивый хелат, А^(Н&_эдта) = 1022). Электрод
Hg | Hg-ЭДТА можно использовать для измерения концентрации N"+ в
процессе титрования раствором ЭДТА. Выведите приведенное выше урав-
нение, исходя из потенциала электрода Hg | Hg2+. Это пример электрода
металл I хелат металла I ион металла.
16- Потенциал водородного электрода (отн. НКЭ) в кислом растворе равен
-0,465 В. Каков будет потенциал этого электрода, измеренный относитель-
но нормального каломельного электрода (1 М КС1)?
Потенциометрическое измерение pH
17- а) Какова точность измерения pH неизвестного раствора? Чем она лимити-
руется? Рассчитайте абсолютную систематическую погрешность измере-
ния потенциала в милливольтах; относительную погрешность измерения
активности ионов водорода в процентах, б) Какова воспроизводимость ре-
зультатов измерения pH анализируемого раствора? Выразите случайную
погрешность измерения потенциала в милливольтах; относительную по-
грешность измерения активности ионов водорода в процентах.
18- Измеренный потенциал стеклянного электрода (отн. НКЭ) в стандартном
буферном растворе с pH 7,00 равен 0,395 В. Рассчитайте pH неизвестных
анализируемых растворов по указанным ниже величинам потенциалов
(вспомните, что потенциал уменьшается при повышении pH):
а) 0,467 В
б) 0,209 В
в) 0,080 В
г) -0,013 В
19- Рассчитайте ЭДС ячейки, составленной из НКЭ и водородного электрода
(Рн2 = 1 атм):
а) в 0,00100 М НС1
б) в 0,00100 МСН3СООН
в) в смеси равных объемов 0,100 М СН3СООН и 0,100 М CH3COONa
При расчетах примите активности равными концентрациям.
20- Для потенциометрического измерения pH можно использовать хингидрон-
ный электрод. Для этого анализируемый раствор насыщают эквимолярной
558
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
смесью хинона и гидрохинона и измеряют pH раствора с помощью погру-
женного в этот раствор платинового электрода. Соответствующая полуре-
акция и ее Е° приведены ниже.
О ОН
|f^j + 2Н+ + 2е- = (f^| Е° = 0,699 В
О ОН
хинон гидрохинон
Каково будет значение pH раствора, насыщенного гидрохиноном, если по-
тенциал платинового электрода в этом растворе равен -0,205 В (отн. НКЭ)? По-
тенциал жидкостного соединения примите равным нулю.
Измерения с помощью ионселективных электродов
21. Из уравнений (13.44) и (13.45) следует, что для однозарядного иона
lg А?ав = (£в - EA)/S. Выведите это уравнение.
22. С помощью калий-селективного электрода определяли концентрацию ионов
калия в растворе, содержащем 6,0 • 10-3 М (активность!) цезия. Из табл. 13.3
следует, что данной электрод не различает эти ионы (KKCs =1). Если потен-
циал электрода (относительно одного и того же электрода сравнения) в
5,0 • 10-3 М КС1 равен -18,3 мВ, а в анализируемом растворе +20,9 мВ, то
какова в нем активность ионов К+? Предположите, что электрод обладает
нернстовским откликом.
23. Концентрацию нитрата в очищенных сточных вод определяют с помощью
МО3-селективного электрода. Для поддержания постоянной ионной силы
стандартные и анализируемые растворы разбавили в 20 раз 0,1 М K2SO4.
Для стандартных растворов KNO3 с концентрациями 0,0050 М и 0,0100 М
измеренные потенциалы равны -108,6 и -125,2 мВ соответственно, а для
анализируемого раствора -119,6 мВ. Какова концентрация нитрата в ана-
лизируемом растворе?
24. В пробе, содержащей 0,015 М иодида, с помощью ионселективного элект-
рода определяют концентрацию перхлорат-иона. Для поддержания посто-
янной ионной силы анализируемые и стандартные растворы разбавили
0,2 М КС1 в соотношении 1 : 10. Потенциал электрода в стандартном
0,00100 М КС1О4 равен -27,2 мВ, в стандартном 0,0100 М KI +32,8 мВ, а в
анализируемом растворе -15,5 мВ. Полагая, что электрод обладает не-
рнстовским откликом, рассчитайте концентрацию перхлорат-иона в пробе.
25. Потенциал стеклянного катионселективного электрода измеряют относи-
тельно НКЭ. В растворе NaCl с активностью иона натрия 0,100 М потенци-
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
559
ал равен 113,0 мВ, а в растворе КС1 с той же активностью иона калия —
67,0 мВ. а) Рассчитайте коэффициент селективности используя урав-
нение, выведенное в задаче 21.6) Какова ожидаемая величина потенциала
этого электрода в растворе, содержащем NaCl (а = 1,00 • 103 М) и КС1
(а = 1,00- 10’2М)?
При расчете примите, что электрод обладает нернстовским откликом,
59,2 мВ/декада.
26 Коэффициент селективности электрода для определения иона В+ относите-
льно иона А+ оценили, измеряя потенциал электрода в двух растворах с раз-
личными активностями ионов. Полученные данные: 1) 2,00 • 10-4 М А+ +
+ 1,00 • 10"3 М В+; Е = +237,8 мВ и 2) 4,00 • 10^ М А+ + 1,00 • 10’3 М В+,
£* = +248,2 мВ. Тангенс угла наклона электродной функции составляет
56,7 мВ/декада. Вычислите величину А?ав.
27. Для оценки коэффициента селективности стеклянного Na-селективного
электрода относительно калия использовали метод отдельных растворов.
Экспериментально найденные тангенсы угла наклона электродных функ-
ций составили 58,1 мВ при 10-кратном изменении активности ионов натрия
и калия, а электродная функция для иона натрия смещена на 175,5 мВ в сто-
рону положительных потенциалов. Какова величина A?NaK для этого элект-
рода?
28. Для оценки мешающего влияния натрия при определении иона калия с по-
мощью ионселективного электрода на основе валиномицина использовали
метод смешанных растворов. Все растворы для построения градуировочно-
го графика для калия содержали 140 мМ иона натрия. Координаты точки
пересечения линейных участков полученной зависимости: Е = 17,4 мВ,
концентрация иона калия 1,5 • 10-5 М. Тангенс угла наклона электродной
функции 57,8 мВ/декада. Какова величина A*NaK для этого электрода?
Рекомендуемая литература
Коэффициенты активности
1. J. Kielland, «Individual Activity Coefficients of Ions in Aqueous Solutions»,
J. Am. Chem. Soc, 59 (1937) 1675.
Потенциалы
2. W.-Y. Ng, «Conversion of Potentials in Voltammetry and Potentiometry»,
J. Chem. Ed., 65 (1988) 726.
pH-чувствительные электроды и определение pH
3. R. G. Bates, Determination of pH. New York: Wiley, 1964.
4. H. B. Kristensen, A. Salomon, and G. Kokholm, «International pH Scales and
Certification of pH», Anal. Chem., 63 (1991) 885A.
560
ЭЛЕКТРОДЫ ДЛЯ ПОТЕНЦИОМЕТРИИ И ПОТЕНЦИОМЕТРИЯ
5- J. V. Straumford, Jr., «Determination of Blood pH», in D. Seligson, ed., Standard
Methods of Clinical Chemistry, Vol. 2. New York: Academic, 1958, pp. 107-121.
6- С. C. Westcott, pH Measurements. New York: Academic, 1978.
7. H. Gdster, pH Measurement. Fundamentals, Methods, Applications, Instrumen-
tation. New York: VCH, 1991.
Ионселективные электроды и техника измерения
8- R. G. Bates, «Approach to Conventional Scales of Ionic Activity for the Standar-
dization of Ion-Selective Electrodes», PureAppl. Chem., 37 (1974) 573.
9- R. A. Durst, ed., Ion Selective Electrodes. Washington, DC: National Bureau of
Standards Special Publication 314, U.S. Government Printing Office, 1969.
10 - E. Pungor, «Theory and Applications of Anion Selective Membrane Electrodes»,
Anal. Chem., 39 (13) (1967) 28A.
11. G. A. Rechnitz, «New Directions for Ion Selective Electrodes», Anal. Chem.,
41 (12) (1969) 109A. He смотря на год издания, работа заслуживает внима-
ния из-за хорошего описания принципов.
12. G. A. Rechnitz, «Ion Selective Electrodes», Chem. Eng. News, 45 (1967) (June
12) 146. Также прекрасно изложены основы.
13. В. Fu, Е. Bakker, М. Е. Mehyerhoff, V. С Yhang, and J. Н. Yoon, «Polyion Sen-
sitive Membrane Electrodes; Principles and Biomedical Applications», Anal.
Chem., 68 (1996) 168A.
14. E. Bakker. P. Buhlmann, and E. Pretsch, «Carrier-Based Ion-Selective Electro-
des and Bulk Optodes. 1. General Characteristics», Chem. Rev., 97 (1997) 3083.
Коэффициенты селективности
15. Y. Umezawa, CRC Handbook of Ion Selective Electrodes: Selectivity Coeffici-
ents. Boca Raton, FL: CRC Press, 1990.
16. V. P. Y. Gadzekpo and G. D. Christian, «Determination of Selectivity Coeffici-
ents of Ion-Selective Electrodes by a Matched-Potential Method», Anal. Chim.
Acta, 166(1984) 279.
17. Y. Umezawa, K. Umezawa, and H. Ito, «Selectivity Coefficients for Ion-Selecti-
ve Electrodes: Recommended Methods for Reporting k fot Values», Pure Appl.
Chem., 67 (1995) 507.
18. G. Horvai, «The Matched Potential Method, a Generic Approach to Characterize
the Differential Selectivity of Chemical Sensors», Sensors and Actuators B, 43
(1997) 94. См. также G. Horvai, Trends in Anal. Chem., 16 (1997) 260.
19. E. Bakker, R. K. Meruwa, E. Pretsch, and M. E. Meyerhoff «Selectivity of Poly-
mer Membrane-Based Ion-Selective Electrodes: Self-Consistent Model Descri-
bing the Potentiometric Response in Mixed Ion Solutions of Different Charge»,
Anal. Chem., 66 (1994) 3021.
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
561
20. Е. Bakker, «Determination of Improved Selectivity Coefficients of Polymer
Membrane Ion-Electrodes by Conditioning with a Discriminating Ion», J. Elec-
trochem. Soc, 143 (4) (1996) L83.
21. E. Bakker, «Determination of Unbiased Selectivity Coefficients of Neutral Carri-
er-Based Cation-Selective Electrodes», Anal. Chem., 69 (1997) 1061.
22. E. Bakker. «Review: Selectivity of Liquid Membrane Ion-Selective Electrodes»,
Electroanalysis, 9 (1997) 7.
Ультрачувствительные ионселективные электроды
23. S. Mathison and E. Bakker, «Effect of Transmembrane Electrolyte Diffusion on
the Detection Limit of Carrier-Based Potentiometric Sensors», Anal. Chem.,
70(198) 303.
24. Y. Mi, S. Mathison, R. Goines, A. Logue, and E. Bakker, «Detection Limit of Po-
lymeric Membrane Potentiometric Ion Sensors: How Low Can We Go Down to
Trace Levels?» Anal. Chim. Acta, 397 (1999) 103.
25. T. Sokalski, A. Ceresa, T. Zwickl, and E. Pretsch, «Large Improvement of the
Lower Detection Limit of Ion-Selective Polymer Membrane Electrodes», J. Am.
Chem. 5oc, 119 (1997) 11, 347.
26. T. Sokalski, T. Zwickl, E. Bakker, and E. Pretsch, «Lowering the Detection Limit
of Solvent Polymeric Ion-Selective Electrodes. 1. Modeling the Influence of Ste-
ady-State Ion Fluxes», Anal. Chem., 71 (1999) 1204.
27. T. Sokalski, A. Cera, M. Fibbioli, T. Zwickl, E. Bakker, and E. Pretsch, «Lowe-
ring the Detection Limit of Solvent Polymeric Ion-Selective Electrodes. 2. Influ-
ence of Composition of Sample and Internal Electrolyte Solution», Anal. Chem.
71 (1999) 1210.
Глава 14
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ
И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Титрование с использванием в качестве титрантов окислителей или восстанови-
телей — очень удобный метод определения многих веществ. Конечную точку
титрования можно установить с помощью визуального индикатора или измерив
потенциал подходящего электрода и построив кривую титрования. В данной
главе обсуждается построение кривых титрования с использованием потен-
циалов полуреакций, рассматриваются наиболее важные виды окислительно-вос-
становительного титрования и способы необходимой предварительной пробопод-
готовки. Описано потроение кривых потенциометрического титрования, вклю-
чая производные кривые и графики Грана. Еще раз повторим правила уравни-
вания окислительно-восстановительных реакций, поскольку это необходимо
для обработки результатов титрования.
14.1. Уравнивание окислительно-восстановительных реакций
Для выполнения расчетов в титриметрии нужно правильно записать уравнение
окислительно-восстановительной реакции, лежащей в основе титрования. Мате-
риал, посвященный этому вопросу, находится на компакт-диске.
Существуют разные способы уравнивания окислительно-восстановитель-
ных реакций. Мы будем пользоваться методом полуреакций. Для этого реак-
цию разбивают на две полуреакции — окисления и восстановления. В любой
окислительно-восстановительной реакции реагируют окислитель и восстанови-
тель, в результате чего окислитель восстанавливается, а восстановитель окисля-
ется. Так, в реакции
Fe2+ + Се4+ -> Fe3+ + Се3+
Fe2+ — восстановитель, Се4+ — окислитель. Запишем уравнения соответствую-
щих полуреакций
Fe2+ -> Fe3+ + е_
и Се4+ + е_ Се3+
При составлении уравнения окислительно-восстановительной реакции сначала
уравнивают каждую из полуреакций. Поскольку в уравнении полной реакции
14.2. РАСЧЕТ КОНСТАНТ РАВНОВЕСИЯ РЕАКЦИЙ
563
суммарное число электронов должно быть равно нулю (для этого число отдавае-
мых и принимаемых электронов в полуреакциях должно быть одинаково), то да-
лее одну или обе полуреакции (в отличие от приведенного выше простейшего
примера) необходимо умножить на соответствующие множители, чтобы при
сложении полуреакций отдаваемые восстановителем и принимаемые окислите-
лем электроны сократились.
14.2. Расчет констант равновесия реакций: это необходимо
для вычисления потенциала в точке эквивалентности
Прежде чем перейти к обсуждению кривых окислительно-восстановительного
титрования необходимо ознакомиться с расчетом констант равновесия окисли-
тельно-восстановительных реакций из величин потенциалов полуреакций. Кон-
станту равновесия используют для расчета равновесных концентраций в точке
эквивалентности, необходимых для вычисления потенциала. Как было сказано в
гл. 12, при установлении равновесия ЭДС ячейки равна нулю, разность потенци-
алов двух полуреакций тоже равна нулю (иными словами, потенциалы полуре-
акций равны) и уравнения Нернста для обеих полуреакций можно приравнять
друг другу. Если это сделать и объединить соответствующие слагаемые, то под
знаком логарифма окажется выражение для константы равновесия реакции
[см. уравнение (12.20)], и можно найти ее численную величину. Это следует из
соотношения между свободной энергией и константой равновесия реакции. Со-
гласно уравнению (6.10) ДС?° = -RT In К. Поскольку для реакции ДС?° = -nFE°, то
-RT\nK = -nFEQ
откуда рт FF = — \nK nF (14.1)
Для самопроизвольной реакции Д(?° отрицательно и потенциал Е° положителен.
Далее величину К можно выразить через ДЕ10.
Пример 14.1
Рассчитайте потенциал (отн. СВЭ) электрода в растворе после добавления 5,0 мл
0,10 М раствора Се4+ к 5,0 мл 0,30 М раствора Fe2+, используя данные для полу-
реакции Се4+/Се3+. Результаты сравните с полученными в примере 12.4.
Решение
Это тот же случай, что и рассмотренный в примере 12.4, где расчет был сде-
лан для полуреакции Fe3+/Fe2+, так как были известны [Fe3+] и [Fe2+]. Итак, к
564
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
0,30 • 5,0 =1,5 ммоль Fe2+ добавлено 0,10 • 5,0 = 0,50 ммоль Се4+. В итоге образо-
валось по 0,50 ммоль Fe3+ и Се3+ и осталось 1,0 ммоль Fe2+:
Fe2+ + Се4+ Fe3+ + Се3+
1,0+ х х 0,50-х 0,50-х
где х — количество ммоль. Для того чтобы воспользоваться данными полуреак-
ции Се4+/Се3+ нужно найти х. Сделать это можно с помощью константы равнове-
сия, которую рассчитываем, приравнивая потенциалы обеих полуреакций. Для
полуреакции Се4+/Се3+
Се4+ + е- Се3+
Е = 1,61-0,059 1g [Се3+ ]
[Се4"]
поэтому
1,61 - 0,059 1g --- = 0,771 - 0,059 1g
[Се4"] [Fe3"]
0,84^0.059 lg^^^^-0,059
[Се ][Fe ] = 0Oj84/O,O59 = 1 Л14,2 = 1 А • 1 014 лг
[Ce4+][Fe3+] ’ равн
Большая величина АГравн указывает на то, что равновесие сильно смещено впра-
во. Поскольку в расчетной формуле объемы сокращаются, далее вместо единиц
ммоль/мл можно использовать просто ммоль.
[Се3+] = 0,50 — х « 0,50 ммоль
[Се4+] = х ммоль
[Fe3+] = 0,50 - х ~ 0,50 ммоль
[Fe-] = 1,0 + х = 1,0 ммоль
Поэтому
0,50 ммоль • 0,50 ммоль , г _ м
---------------------= 1,о ♦ 1U
х ммоль 1,0 ммоль
х = 1,6 • 10-15 ммоль (= 1,6 • 10-16 М)
Это значит, что [Се4+] очень малая, но конечная и известная величина, поэтому
по уравнению Нернста, используя количества ммоль, получаем
Е = 1,61 - 0,059 1g [Се ] = 1,61 - 0,059 1g —0-’-50ммоль = 0,75 В
[Се4+] 1,6-10 15 ммоль
Это сопоставимо с величиной 0,753 В, полученной в примере 12.4.
14.2. РАСЧЕТ КОНСТАНТ РАВНОВЕСИЯ РЕАКЦИЙ
565
Очевидно, что расчет легче выполнить для той полуреакции, для которой
известны концентрации всех ее участников. Из приведенных вычислений видно,
что при равновесии концентрации всех частиц в смеси таковы, что потенциалы
обеих полуреакций одинаковы. Заметим, что величина потенциала близка к
стандартному потенциалу (£°)* полуреакции, участник которой находится в из-
бытке, в данном случае, Fe2+.
Как правило, значения п (число электронов) в полуреакциях различаются.
В таких случаях для удобства расчетов с использованием уравнения Нернста по-
луреакции переписывают таким образом, чтобы величины п в обеих полуреак-
циях стали одинаковыми.
Если реагирующие вещества находятся в стехиометрических количествах
(как это будет в точке эквивалентности), то ни для одной из полуреакций равно-
весные конценцентрации компонентов не известны. В этом случае используют
подход, аналогичный описанному в примере 14.1.
Пример 14.2
Рассчитайте потенциал после добавления 10 мл 0,20 М раствора Fe24- к 10 мл 0,20 М
раствора Се4+.
Решение
Реагирующие вещества практически количественно превращаются в эквивален-
тные количества Fe3+ и Се3+, и их концентрации равны 0,10 М (продуктами об-
ратной реакции пренебрегаем):
Fe2+ + Се4+ Fe3+ + Се3+
0,10-х 0,10-х
х
где х—молярные концентрации Fe2+ и Се4+. Можно, как это было сделано в при-
мере 14.1, вычислить х, а затем по уравнению Нернста для любой полуреакции
рассчитать потенциал (сделайте это самостоятельно, это полезно!). Можно по-
ступить иначе.
Запишем уравнение Нернста для обеих полуреакций:
„ ° 0,059. [Fe2+]
Z7 г° ЛЛ«М X ммоль/мл
nFeE = п^Е ь 2+ - 0,0591g —------—
0,10 ммоль/мл
----lg----5Г’
«Fe [Fe3 ]
0,059 lg [Се3+].
"с* [Се4+]’
Е ~ ЕСе\Се2
77 77 ° nn,nl 0,10 ммоль/мл
«С Л = «Се^се^ Се* ~ °’059 lg--------
х ммоль/мл
Обратите внимание на то, что в обоих случаях уравнение Нернста приведено для
полуреакции, записанной в форме восстановления, хотя одно из реагирующих
Строго говоря, к формальному, Е°‘ — Прим, перев.
566
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
веществ, Fe2+, окисляется. Можно сложить эти уравнения и выразить Е — иско-
мый потенциал при равновесии:
„ „ ° ° ( х ммоль/мл
«Fe£ + «Се£ = Fe> + «Се£Се* Се* “ °’059 ------7~
re ’re се ’се I 0,10 ммоль/мл
0,10 ммоль/мл
х ммоль/мл
WFe^Fe^Fe2+ + _ 1 • 0,77 + 1 • 1,61 _ 11 g g
"Fe+^Ce 1 + 1
Рассмотренный выше прием является общим, поэтому в случае стехиомет-
рических количеств реагирующих веществ потенциал (Е в точке эквивалентно-
сти при титровании) можно рассчитать по уравнению
£_Я1£1 +И2£2
+ и2
(14.2)
где пх и Ех — число электронов и стандартный потенциал для первой полуреак-
ции, п2 и Е°2 — для второй полуреакции. Иначе говоря, Е — это средневзвешен-
ная величина из Е°. В рассмотренном выше примере Е это просто среднее из £°,
поскольку число п в обеих полуреакциях равно единице. Это уравнение справед-
ливо только для реакций, в которых не участвуют полиатомные частицы (на-
пример, Сг2О^'), и в случае отсутствия зависимости потенциала от pH (или
при pH 0). Если необходимо учесть pH или влияние концентраций в уравнение
вводят дополнительные члены (см. задачу 13). Уравнение применимо при исполь-
зовании формальных потенциалов, т. е. при конкретной кислотности раствора
(см. гл. 12).
14.3. Расчет кривых окислительно-восстановительного титрования
Используем наши представления о равновесии окислительно-восстановитель-
ных реакций для построения кривых окислительно-восстановительного титро-
вания. Форму кривой титрования можно предсказать, исходя из величин £°
полуреакций с участием титруемого вещества и титранта. В первом приближе-
нии величина скачка потенциала на кривой титрования равна разности £° полуре-
акций; до точки эквивалентности потенциал системы будет близок к Е° титруемого
вещества, за точкой эквивалентности — к Е° титранта.
Рассмотрим поцесс титрования 100 мл 0,1 М раствора Fe2+ 0,1 М раствором
Се4+ в 1 М HNO3. Каждый миллимоль Се4+ окисляет 1 миллимоль Fe2+, поэтому
14.3. РАСЧЕТ КРИВЫХ ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОГО ТИТРОВАНИЯ
567
РИС. 14.1. Кривая титрования 100 мл 0,1 М раствора Fe2+ 0,1 М раствором Се4+
конечная точка будет достигнута после добавления 100 мл титранта. Кривая тит-
рования показана на рис. 14.1. Фактически кривая представляет собой измене-
ние потенциала в процессе титрования относительно СВЭ, потенциал которого
принят равным нулю.
В гл. 13 было показано, что разность потенциалов двух полуэлементов мож-
но измерить с помощью инертных металлических (платиновых) электродов в
ячейке, подобной изображенной на рис. 12.1. Электрод, погруженный в титруе-
мый или анализируемый раствор, называют индикаторным электродом, вто-
рой электрод — электродом сравнения. Поскольку его потенциал является
постоянной величиной, будет изменяться потенциал индикаторного электрода
относительно электрода сравнения — см. рис. 14.1, на котором изменение по-
тенциала относительно СВЭ представлено в виде функции объема титранта. Эта
кривая аналогична кривой изменения pH раствора в зависимости от объема тит-
ранта в кислотно-основном титровании или изменения рМ в зависимости от
объема титранта в процессе осадительного или комплексонометрического тит-
рования. До начала титрования в растворе содержится только Fe2+ и рассчитать
величину потенциала невозможно. Как только будет добавлена первая капля
титранта, определенное количество Fe2+ превратится в Fe3+, станет известным
соотношение [Fe2+]/[Fe3+] и можно будет рассчитать величину потенциала (по
уравнению Нернста). До точки эквивалентности потенциал будет близок к вели-
чине Е°* этой пары.
Заметим, что поскольку в точке полуоттитрованности отношение
[Fe2+]/[Fe3+] = 1 и lg 1 = 0, потенциал в этой точке кривой титрования равен Е°*.
в общем случае Е°' — прим, перев.
568
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Это справедливо только для таких полуреакций, в которых не участвуют поли-
атомные частицы. Например, для полуреакции 12 + 2е_ 21~ в точке полуоттит-
рованности величина [I-] вдвое больше [12] и поэтому [1_]2/[12] = 22/1 = 4, т. е.
потенциал на (-0,059/2) 1g 4 или на -0,018 В будет меньше £°*.
Для рассматриваемого случая титрования в точке эквивалентности получаем:
Fe2+ + Ce4+^ Fe3+ + Се3+
х х С-х С-х
где С—концентрация Fe3+, которая нам известна, поскольку все Fe2+ полностью
превратилось в Fe3+ (величинах пренебрежимо мала по сравнению с Q. Неизве-
стную величину х можно вычислить, приравняв уравнения Нернста для обеих
полуреакций, как это сделано в примере 14.1, и затем рассчитать потенциал в
точке эквивалентности. Можно воспользоваться и уравнением (14.2), так как по-
лиатомные частицы в полуреакции не участвуют. За точкой эквивалентности
раствор содержит избыток Се4+ и неизвестное количество Fe2+. Теперь у нас есть
больше информации о полуреакции Се4+/Се3+, и дальнейшие расчеты легче про-
вести по уравнению Нернста для этой полуреакции. Заметим, что при наличии
избытка титранта потенциал будет близок к величине Е°* титранта. В присут-
ствии 200% избытка титранта отношение [Се4+]/[Се3+] = 1 и в этот момент
Е - Е°* этой полуреакции.
Из примера 14.3 вы увидите, что величина скачка потенциала на кривой тит-
рования зависит от разности величин Е°* полуреакций для титруемого вещества
и титранта. Для получения четкого скачка титрования эта разность должна со-
ставлять, по крайней мере 0,2 В.
Для рассматриваемого случая титрования на кривой (см. рис. 14.1) указана
точка эквивалентности. Поскольку реакция комплементарная, точка эквивалент-
ности (точка максимального наклона) лежит точно посередине восходящего
участка кривой титрования. Для некомплементарных реакций кривая не симмет-
рична относительно точки эквивалентности. Например, при титровании Fe2+
раствором МпОд точка максимального наклона смещена к верхнему перегибу
на кривой титрования, так как в реакции участвуют протоны** (см. задачу 21).
Пример 14.3
Рассчитайте потенциал как функцию объема титранта после добавления 10,0,
50,0, 100 и 200 мл 0,100 М раствора Се4+ к 100,0 мл 0,100 М раствора Fe2+.
В общем случае Е°’ — Прим, перев.
Поскольку реакция некомплементарная — в данном случае в полуреакциях участвует разное количество
электронов — 1 и 5 — Прим, перев.
14.3. РАСЧЕТ КРИВЫХ ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОГО ТИТРОВАНИЯ
569
Решение
Запишем уравнение реакции титрования
Fe2+ + Ce4+^ Fe3+ + Ce3+
10,0 мл титранта: ммоль Се4+ добавлено = 0,100 М • 10,0 мл = 1,00 ммоль
ммоль Fe2+ прореагировало = 1,00 ммоль = ммоль Fe3+ образовалось
ммоль Fe2+ осталось = 0,100 М • 100 мл - 1,00 ммоль = 9,0 ммоль Fe2+
90
Е = 0,771 - 0,059 1g — = 0,715 В
1,0
50,0 мл титранта: половина исходного количества Fe2+ окислена до Fe3+
(5,00 ммоль каждого)
Е = 0,771 - 0,059 1g ^2 = 0,771 В
5,00
100 мл титранта: ммоль Fe3+ = 10,0 -х « 10,0
ммоль Fe2+ = х
ммоль Се3+ = х = 10,0 - х ~ 10,0
ммоль Се4+ = х
Необходимо вычислить х. Поскольку расчет делается в предположении, что
равновесие достигнуто, то используя уравнение Нернста, можно записать
0,771-^lgl^= 1,61
1 [Fe3+] 1 [Се4+]
-0,84 = -0,059 1g [РеГ][СеГ] = -0,059 lg Х_авн
[Fe2+][Ce4+] Р
Хравн=1,7- 1014
Подставив в выражение для АГравн известные значения (используем ммоль, по-
скольку объемы сокращаются), находим х:
1О,О1О,ОО_1)?.1О14
X -х
х = 7,7 • 10-7 ммоль Fe2+ = ммоль Се4+
Для вычисления потенциала можно воспользоваться уравнением Нернста для
любой из полуреакций
7 7-10"7
Е = 0,771 - 0,059 1g - - = 1,19 В
10,00
Сравните полученный результат с величиной потенциала, найденной в примере
14.2. Самостоятельно рассчитайте потенциал по уравнению Нернста для полу-
570
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
реакции Се4+/Се3+. Обратите внимание на то, что найденная величина является
средним значением величин Е° двух полуреакций.
200 мл титранта', добавлен избыток титранта (Се4+) 100 мл. Теперь потен-
циал легче вычислить, используя полуреакцию Се4+/Се3+:
ммоль Се3+ = 10,0 -х « 10,0
ммоль Се4+ = 0,100 М • 100 мл + х « 10,0 ммоль
Е= 1,61-0,059 1g 1,61 В
10,0
(Потенциал в данном случае можно было рассчитать и по данным для полуреак-
ции Fe3+/Fe2+, вычислив х, как показано выше, из АГПЯВН.)
Для некомплементарных реакций необходимо принимать во внимание сте-
хиометрические коэффициенты для реагирующих веществ. Если в реакции рас-
ходуются или выделяются протоны, нужно учитывать изменение концентрации
[Н+].
Пример 14.4
Рассчитайте потенциал в точке эквивалентности при титровании 100 мл 0,100 М
раствора Fe3+ в 0,500 М H2SO4 после добавления 100 мл 0,0200 М раствора МпО 4 *.
Решение
Запишем уравнение реакции титрования (при расчете не забудьте учесть стехио-
метрию реакции: 1 ммоль Fe2+ реагирует с | ммоль МпО4):
5Fe2+ + МпО; + 8Н+ 5Fe3+ + Мп2+ + 4Н2О
х |х С-х |С-|х
ммоль Fe3+ = 0,100 М • 100 мл -х « 10,0
ммоль Fe2+ = х
ммоль Мп2+ = | • 10,0 -1 х « 2,00
ммоль МпО4 = |х
* В 0,5 М растворе H2SO4 диссоциирует по второй ступени приблизительно на 2% (рассчитайте, используя
константу кислотности, и убедитесь!), но для упрощения расчета будем полагать, что H2SO4 диссоциирует
полностью и концентрация Н+ равна 1 М.
14.4. ВИЗУАЛЬНОЕ ОБНАРУЖЕНИЕ КОНЕЧНОЙ ТОЧКИ ТИТРОВАНИЯ
571
Расчет проводим для момента достижения равновесия, поэтому для нахождения
х приравниваем уравнения Нернста, записанные для обеих полуреакций, умно-
жив полуреакцию пары Fe3+/Fe2+ на 5 для уравнивания числа электронов. Это
прозволит рассчитать величину константы равновесия реакции
[Mn2+]
0059 [Fe^= 0059
5 [Fe3+]5 5
-0)74 = °-059 lg [Mn2+][Fe3+]5
5 [MnO4 ][Fe2+]5[H+]8 5
lg + 8
[Мпо;][н+]8
0,059.
^равн
*равн = 5,o ’ 1062
paoH 7v
Исходный раствор содержал 1,00 • 100 = 100 ммоль Н+. Необходимо рассчи-
тать концентрацию Н+ по завершении реакции. Согласно уравнению реакции на
каждые 5 ммоль Fe2+ расходуется 8 ммоль Н+ (т. е. всего | • 10,0 = 16,0 ммоль),
поэтому в 200 мл осталось 84 ммоль Н+, и концентрация стала равной 0,42 М.
Для остальных участников реакции в выражения можно подставлять миллиммоли,
поскольку объемы сокращаются:
2,00 л,оо5 q62
(1х)х5(0,42)8
х = 1,1 • 10-9 ммоль Fe2+; ммоль МпОд = | (1,1 • 10-9) = 2,2 • Ю-10
Потенциал можно рассчитать по уравнению Нернста для любой из полуре-
акций
Е = 0,771 - 0,059 1g 1,1 10 = 1,359 В
10 9
Е° для пары Мп2+/МпС>4 равен 1,51 В. Обратите внимание на то, что в данном
случае полусумма Е° составляет 1,14 В. Это значит, что точка эквивалентности
(точка перегиба) для этой некомплементарной реакции смещена к потенциалу
полуреакции титранта и кривая титрования несимметрична относительно точки
эквивалентности.
14.4. Визуальное обнаружение конечной точки титрования
Очевидно, что конечную точку титрования можно обнаружить измеряя потен-
циал индикаторного электрода (гл. 13) относительно электрода сравнения и по-
строив график зависимости потенциала от объема титранта, но иногда более
удобно воспользоваться визуальным индикатором. Существует три способа ви-
зуальной индикации.
572
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ И ПОТЕНЦИОМЕГРИЧЕСКОЕТИТРОВАНИЕ
Безиндикаторное титрование
Для обнаружения конечной точки титрования можно использовать изменение
окраски титранта, если она достаточно интенсивна. Например, 0,02 М раствор
КМпО4 окрашен в глубокий красно-фиолетовый цвет, разбавленные растворы
КМпО4 — розовые. Продукт восстановления — Мп2+ — окрашен очень слабо,
почти бесцветный. При титровании восстановителей раствором КМпО4 крас-
но-фиолетовая окраска МпО4 исчезает, потому что перманганат-ион восста-
навливается до Мп2+. Как только титрант будет добавлен в необходимом для за-
вершения реакции количестве, лишняя капля МпО4 окрасит раствор в розовый
цвет и укажет на то, что титрование закончено. Совершенно ясно, что объемы
титранта в конечной точке и точке эквивалентности не совпадают, но различие
составит лишь долю капли. Эта погрешность мала и ее можно устранить, проведя
контрольное титрование, или учесть при стандартизации титранта.
Индикация конечной точки с помощью крахмала
Крахмал в качестве индикатора пригоден при титровании с участием иода.
Крахмал образует с 12 весьма устойчивый комплекс интенсивного синего цвета,
что делает его чувствительным к очень малым количествам иода. При титрова-
нии восстановителей с помощью 12 раствор будет бесцветным вплоть до точки
эквивалентности, а лишняя капля титранта придаст раствору отчетливую си-
нюю окраску.
Окислительно-восстановительные индикаторы
Рассмотренные выше способы индикации конечной точки титрования не осно-
ваны на использовании потенциалов полуреакций, хотя они определяют и полно-
ту протекания реакции титрования, и отчетливость конечной точки. Примеров
использования этих способов мало. В большинстве случаев окислительно-вос-
становительного титрования применяют окислительно-восстановительные
индикаторы. Это интенсивно окрашенные красители, являющиеся слабыми
восстановителями или окислителями, при этом их окисленная и восстановлен-
ная формы имеют разную окраску. Состояние индикатора, а следовательно, и
его окраска будут зависеть от величины потенциала в данный момент титрова-
ния. Запишем уравнение Нернста для полуреакции с участием индикатора
Охинд + не Кес1инд
Е -Е' 0.059 [RedTO]
„ [Охи„]
(14.3)
(14.4)
В процессе титрования потенциал полуреакции будет определять величину £инд
и, следовательно, отношение [RedHHJ/[OxH ], аналогично тому как в кислот-
но-основном титровании соотношение различных форм индикатора зависит от
14.4. ВИЗУАЛЬНОЕ ОБНАРУЖЕНИЕ КОНЕЧНОЙ ТОЧКИ ТИТРОВАНИЯ
573
pH раствора. Итак, в процессе титрования в зависимости от величины потенциа-
ла будет изменяться соотношение форм индикатора и его окраска. Если, как и в
случае кислотно-основного индикатора, полагать, что четкое изменение окрас-
ки можно наблюдать при изменении соотношения концентраций окисленной и
восстановленной форм индикатора в интервале от до то потенциал должен
быть равен 2 • (0,059/и) В. Если для индикатора п = 1, необходимо изменение по-
тенциала на 0,12 В. Если величина £инд близка к величине потенциала в точке
эквивалентности, потенциал быстро изменится более чем на 0,12 В, поэтому из-
менение окраски индикатора произойдет в точке эквивалентности. И вновь, по
аналогии с требованиями, предъявляемыми к кислотно-основным индикаторам,
величина рКа индикатора должна быть близка к pH в точке эквивалентности.
Если в полуреакции для индикатора [уравнение (14.3)] участвуют ионы
водорода, это нужно учесть в уравнении Нернста — в уравнении (14.4) вместо
£инд следует использовать величину формального потенциала Е°' при дан-
ном pH.
Итак, для окислительно-восстановительных индикаторов существует ин-
тервал потенциалов перехода окраски, и он должен обязательно попадать в интер-
вал скачка потенциалов на кривой титрования. Окислительно-восстановительная
полуреакция с участием индикатора должна быть быстрой или, что более точно
с точки зрения электрохимии, обратимой. Если реакция медленная или необра-
тимая (низкая скорость переноса электронов), изменение окраски происходит
постепенно (не резко) и установить конечную точку титрования трудно.
Известно много хороших окислительно-восстановительных индикаторов.
В табл. 14.1 приведены некоторые наиболее часто используемые из них (индика-
торы расположены в порядке уменьшения их стандартных потенциалов). Одним
из лучших индикаторов является ферроин [трис(1,10-фенантролин)железо(П)
сульфат], успешно применяемый во многих случаях титрования раствором це-
рия(1У). В точке эквивалентности происходит изменение красной окраски на
бледно-голубую. В табл. 14.1 приведены и другие индикаторы на основе произ-
Таблица 14.1
Окислительно-восстановительные индикаторы
Индикатор Окраска
Восстановленная форма Окисленная форма Раствор Е°,В
Нитроферроин красная бледно-голубая 1 М H2SO4 1,25
Ферроин красная бледно-голубая 1 М H2SO4 1,06
Дифениламино- бесцветная пурпурная разбавленная 0,84
сульфоновая кислота кислота
Дифениламин бесцветная фиолетовая 1 М H2SO4 0,76
Метиленовый голубой голубая бесцветная 1 М кислота 0,53
Индиготетрасульфонат бесцветная голубая 1 М кислота 0,36
574
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
водных фенантролина. Потенциал пары Сг2О 7 /Сг3+ ниже, чем пары Се4+/Се3+,
поэтому здесь нужен индикатор с более низким £°. При титровании раствором
дихромата в кислой среде в качестве индикатора используют дифениламиносуль-
фоновую кислоту. Окраска этого индикатора в конечной точке пурпурная. Удач-
ное применение индикатора может зависеть и от природы титруемого вещества,
поскольку величина скачка титрования зависит и от величины потенциала полу-
реакции с его участием.
14.5. Титрование с использованием иода -
иодиметрия и иодометрия
Окислительно-восстановительное титрование относится к наиболее важным ви-
дам титриметрии и находит широкое применение, например, для анализа пищевых
продуктов, фармацевтических препаратов и в промышленном анализе. Типичный
пример — определение сульфита в вине с помощью иода. По реакции с дихрома-
том калия можно определять спирт. Примеры из практики клинических лабора-
торий единичны, поскольку в большинстве случаев нужно определять следовые
количества, но и здесь этот вид титриметрии чрезвычайно удобен для стандарти-
зации растворов реагентов. Познакомимся с наиболее часто применямыми тит-
рантами.
Иод является окислителем и пригоден в качестве титранта для определения
достаточно сильных восстановителей. С другой стороны, иодид-ион очень мяг-
кий восстановитель и поэтому используется для определения сильных окисли-
телей.
Иодиметрия
Иод — окислитель средней силы и его используют для титрования восстанови-
телей. Этот вид титриметрии называют иодиметрией. Конец титрования уста-
навливают по появлению синей окраски иод-крахмального соединения.
Титруют обычно в нейтральной, слабощелочной (pH 8) или слабокислой среде.
В сильнощелочной среде 12 диспропорционирует с образованием гипоиодата и
иодида:
I2 + 2ОН- = IO" +1" + Н2О (14.5)
Есть три причины, не позволяющие титровать в сильнокислой среде.
Во-первых, в сильнокислой среде гидролизуется или разрушается крахмал, при-
меняемый для индикации конечной точки титрования. Во-вторых, многие вос-
становители становятся более сильными в нейтральной среде. Для примера
рассмотрим реакцию I2 с As(III):
HoAsO. + L + Н2О = H.AsO4 + 21- + 2Н+ (14.6)
•J J £ J
14.5. ТИТРОВАНИЕ С ИСПОЛЬЗОВАНИЕМ ИОДА - ИОДИМЕТРИЯ И ИОДОМЕТРИЯ
575
На равновесие этой реакции влияет концентрация ионов водорода. При низкой
концентрации ионов водорода равновесие будет смещено вправо. Мы уже виде-
ли, что согласно уравнению (12.25) в нейтральной среде потенциал пары
As(V)/As(III) настолько уменьшается, что As(III) способен восстанавливать 12.
В кислых растворах равновесие реакции смещается влево и возможно протека-
ние обратной реакции. Третьей причиной, по которой нежелательно титровать в
кислой среде, является реакция окисления I-, образовавшегося по реакции (14.6),
растворенным кислородом:
41- + О2 + 4Н+-> 212 + 2Н2О (14.7)
При титровании As(III) раствором 12 оптимальное значение pH можно создавать,
прибавляя NaHCO3. Выделяющийся при этом СО2 будет удалять растворенный
кислород, а слой СО2 над поверхностью раствора предотвратит окисление I-.
Поскольку 12 — окислитель не сильный, в качестве титранта его используют
для определения ограниченного числа восстановителей. Тем не менее ряд при-
меров можно привести (табл. 14.2), а не очень высокая окислительная способ-
ность делает 12 более селективным титрантом по сравнению с сильными
окислителями. По химическим свойствам сурьма близка к мышьяку, поэтому
при выборе pH титрования нужно учитывать те же причины. Для связывания су-
рьмы в комплекс и предотвращения гидролиза в раствор вводят тартрат.
Высокочистый 12 можно получить возгонкой, но обычно раствор иода стандар-
тизируют по первичному стандарту (восстановителю), например As2O3 (As4O6).
Оксид мышьяка не растворяется в кислотах, но растворяется в гидроксиде на-
трия. После полного растворения полученный раствор нейтрализуют. Если рас-
твор мышьяка(Ш) предстоит долго хранить, его нужно нейтрализовать или
подкислить, так как в щелочной среде мышьяк(Ш) медленно окисляется.
В воде растворимость 12 мала, но комплекс 13 очень хорошо растворим, поэто-
му раствор иода готовят, растворяя 12 в концентрированном растворе иодида калия:
12 + 1-->13 (14.8)
Поэтому фактически титрантом является 13.
Таблица 14.2
Примеры веществ, определяемых методом иодиметрии
Определяемое вещество Уравнение реакции с иодом Условия определения
H2s SO^ Sn2+ H2S + I2 -> S + 2I- + 2Н+ Кислая среда SO^~ +12 + Н2О -> SO4“ + 21- + 2Н+ Sn2+ + I2 -> Sn4+ + 21" Кислая среда
As(III) n2h4 H2AsO2 +12 + Н2О -> HAsO4“ + 21- + ЗН+ pH 8 N2H4 + 2I2 -> N2 + 4Н+ + 41-
576
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Пример 14.5
Для оценки чистоты гидразина N2H4 (сильный яд!) использовали иодиметрию.
Навеску пробы массой 1,4286 г растворили в воде и разбавили до 1 л в мерной
колбе. На титрование 50,00 мл аликвоты, отобранной пипеткой, потребовалось
42,41 мл стандартного раствора иода. Для стандартизации раствора иода 0,4123 г
первичного стандарта As2O3 растворили в небольшом количестве раствора
NaOH, установили pH 8, на титрование израсходовали 40,28 мл раствора иода.
Рассчитайте массовую долю (%) гидразина в препарате.
Решение
Согласно уравнению реакции, протекающей при стандартизации раствора иода
HjAsOj +12 + Н2О -> НАзОд- + 21- + ЗН+
из каждого моля As2O3 получается 2H2AsO 3, поэтому ммоль 12 = 2 • ммоль As2O3.
т 412,3 MrAs2O3 _ _ . А
Ст • 40,28 мл L =-------------—------• 2 ммоль L / ммоль As?Oo
2 197,85 мг As2О3/ммоль
Cl2= 0,10347 ммоль/мл
При титровании протекает реакция
N2H4 + 2I2 -» N2 + 4Н+ + 41",
поэтому
ммоль N?H4 = | • ммоль 12
масса оттитрованного N9H4 = 1,4286 г • = 0,07143 г
1000,0
g>n2h4 = [0,10347 М12 • 42,41 мл 12 • | (ммоль N2H4 / ммоль 12) х
х 32,045 мг N2H4 / ммоль] / 71,43 мг • 100% = 98,43%.
Эту задачу можно решить, и другим способом — с использованием понятий
моль эквивалентов и нормальностей (N). Молярная масса эквивалента As2O3
равна молярной массы соединения, поскольку 2 атома As в молекуле окисля-
ются от степени окисления +3 до +5. Поэтому
412,3 мг As2O3
7VT • 40,28 мл L =
2 197,85/4 (мг As 2О3/ммоль экв.)
= 0,20694 ммоль экв./мл
Каждый атом азота гидразина изменяет степень окисления от -2 до 0, т. е. расхо-
дуется 4 электрона на молекулу, поэтому молярная масса эквивалента составля-
ет молярной массы гидразина. Следовательно
14.5. ТИТРОВАНИЕ С ИСПОЛЬЗОВАНИЕМ ИОДА - ИОДИМЕТРИЯ И ИОДОМЕТРИЯ
577
_ 0,20694 N • 42,41 мл • 32,045/4 (мг N2Н4/ммоль экв.) _
СОм тт----------------------------------------------• IUUyo — У о, 43 /о
N2H« 73,43 мг
Заметим, что из-за малой молярной массы гидразина сложно взвесить требуемое
количество пробы с точностью до четвертого знака и точно отмерить аликвоту
для титрования, поэтому приходится брать большую навеску пробы.
Иодометрия
Иодид-ион является слабым восстановителем и способен реагировать с сильны-
ми окислителями. Однако в качестве титранта его не используют, главным обра-
зом, из-за отсутствия удобного способа индикации конечной точки, а также
других факторов, таких как скорость реакции.
Если в раствор окислителя ввести избыток иодида, выделившееся
количество 12 будет эквивалентно количеству прореагировавшего окислителя^
и его можно будет оттитровать раствором восстановителя. Результат будет та-
ким же, как и при прямом титровании окислителя. В качестве титранта
применяют тиосульфат натрия. Установление конечной точки титрования опре-
деляют по исчезновению окраски иод-крахмального соединения. Для расчетов
необходимо знать соотношение количества ммоль тиосульфата / ммоль опреде-
ляемого компонента. На титрование 1 ммоль выделившегося 12 требуется
2 ммоль тиосульфата.
Такой способ определения окислителей называют иодометрией. В качестве
примера рассмотрим определение дихромата:
Сг2О + 61- (добавлен в избытке) + 14Н+ 2Сг3+ +312 + 7Н2О (14.9)
I2 + 2S2O|"->2I- + S4O£- (14.10)
В результате реакции на каждый ммоль Сг2О2 выделяется 312, который затем
реагирует с 6820з", поэтому количество ммоль Сг2О2- эквивалентно | ммоль
820з“, используемого для титрования.
Йодометрически можно определять иодат:
IO3 + 51- + 6Н+ -> 312 + ЗН2О (14.11)
На каждый ммоль IO3 выделяется 312, на титрование которого расходуется
682Оз“, поэтому при расчете числа ммоль IO3 нужно умножить на | число
ммоль 82Оз“, затраченного на титрование.
578
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Пример 14.6
Медь, содержащуюся в навеске пробы массой 0,200 г, определили иодометриче-
ски, восстановив медь(П) до меди(1) иодидом:
2Cu2++ 41~ —> 2CuI +12
На титрование выделившегося иода потребовалось 20,0 мл 0,100 М раствора
Na2S2O3. Рассчитайте массовую долю (%) меди в образце.
Решение
Согласно уравнению реакции на 1 моль Си2+ выделяется 0,5 моль 12, а так как
1 моль 12 реагирует с 2 моль 820з", то 1 моль Си2+ эквивалентен 1 моль 82Оз", а
ммоль Си2+ = ммоль S2O 3~.
0,100 ммоль S9O? /мл-20 мл S9O? -мгСи/ммоль
соГ11 =---------- ------------:--------------• 100% =
200 мг пробы
0,100 ммоль/мл -20,0 мл -63,54 мг Си/ммоль 1ЛА0/ _
--------------------------------------• 100% — 63,5%
200 мг пробы
Почему окислители не титруют непосредственно раствором тиосульфата?
Сильные окислители окисляют серу в тиосульфате до более высокой степени
окисления, чем в тетратионате (например, до SO 4"), но эта реакция, как правило,
протекает нестехиометрически. Кроме того, некоторые окислители, например,
Fe3+, образуют с тиосульфатом смешанно-лигандные комплексы. При взаимо-
действии с иодидом сильные окислители количественно восстанавливаются и
при этом выделяется эквивалентное количество 12, который стехиометрически
реагирует с тиосульфатом, и для установления конца титрования есть подходя-
щий индикатор. Эту реакцию можно рассматривать как прямое титрование.
Для установления конечной точки йодометрического титрования применя-
ют крахмал — исчезновение синей окраски иод-крахмального соединения по-
зволяет зафиксировать этот момент. Не рекомендуется добавлять крахмал в
начале титрования, когда концентрация иода высока. Добавлять его нужно
вблизи конца титрования, когда при низкой концентрации иода раствор стано-
вится бледно-желтым. Существует две причины, обусловливающие такой способ
применения крахмала. Во-первых, иод-крахмальный комплекс очень медленно
диссоциирует и конец титрования установить трудно, если крахмал адсорбирует
большое количество иода. Вторая причина — в большинстве случаев йодомет-
рическое титрование проводят в сильнокислой среде, а в кислых растворах крах-
мал гидролизуется. Необходимость титрования в кислой среде вызвана тем, что
14.5. ТИТРОВАНИЕ С ИСПОЛЬЗОВАНИЕМ ИОДА - ИОДИМЕТРИЯ И ИОДОМЕТРИЯ
579
при высокой концентрации кислоты реакция между многими окислителями и
иодид-ионом ускоряется. Приведем два примера:
2МпСЦ + ЮГ + 16Н+ 512 + 2Мп2+ + 8Н2О (14.12)
Н2О2 + 2Г + 2Н+—> 12 + 2Н2О (14.13)
Титровать нужно быстро для предотвращения окисления иодида кислоро-
дом воздуха и обязательно при интенсивном перемешивании для устранения ло-
кального избытка тиосульфата, разрушающегося в кислой среде:
S2O^ + 2Н+^ H2SO3 + S (14.14)
На появление локального избытка тиосульфата указывает помутнение раствора
из-за образования коллоидной серы. В иодометрии необходим большой избыток
иодида для ускорения реакции (эффект одноименного иона). Непрореагировав-
ший иодид не мешает протеканию реакции, но он может окислиться кислородом
воздуха, если титровать не сразу после приготовления раствора, а через некото-
рое время.
Раствор тиосульфата натрия стандартизируют иодометрически, используя в
качестве первичных стандартов высокочистые К2Сг2О7, КЮ3, КВгО3 или метал-
лическую медь (при растворении образуется Си2+). При использовании К2Сг2О7
темно-зеленая окраска образующегося хрома(Ш) затрудняет установление кон-
ца титрования по исчезновению окраски иод-крахмального соединения. При
йодометрическом титровании меди(П) конечная точка титрования будет раз-
мытой, если не добавить тиоцианат-ион. Основная реакция этого процесса при-
ведена в примере 14.6. Надо учитывать, что иод адсорбируется на поверхности
осадка Cui и очень медленно реагирует с титрантом (тиосульфатом натрия). Ти-
оцианат покрывает осадок слоем CuSCN и вытесняет адсорбированный иод. До-
бавлять тиоцианат нужно вблизи конечной точки титрования, так как он
медленно окисляется иодом до сульфата. При титровании необходимо поддер-
живать pH » 3, иначе при более высоких значениях pH медь(П) выпадет в осадок
в виде гидроксида. В более кислой среде заметно ускоряется окисление иодида
растворенным кислородом, катализируемое ионами меди(П). При растворении
металлической меди в азотной кислоте образуются оксиды азота, способные
окислять иодид. Перед титрованием их нужно удалить с помощью мочевины.
Примеры йодометрических определений приведены в табл. 14.3.
Пример 14.7
При стандартизации раствора Na2S2O3 с помощью иодометрии на титрование
0,1262 г высокочистого КВгО3 израсходовали 44,97 мл раствора Na2S2O3. Рас-
считайте молярную концентрацию раствора Na2S2O3.
580
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Таблица 14.3
Примеры веществ, определяемых методом иодометрии
Определяемое вещество Уравнение реакции с иодид-ионом
Мпо; 2MnO; + 101- + 16Н+ ±=; 2Мп2+ + 512 + 8Н2О
Сг2О^- Сг2О^- + 61- + 14Н+ 2Сг3+ + 312 + 7Н2О
107 IO3 + 51- + 6Н+ 312 + ЗН2О
ВгОз ВгОз + 61- + 6Н+ Вг + 312 + ЗН2О
Се4+ 2Се4+ + 21- 2Се3+ +12
Fe3+ 2Fe3++ 2Г ±=? 2Fe2++ 12
н2о2 [катализатор — Mo(VI)] Н2О2 + 21- + 2Н+ > 2Н2О + 12
As(V) H3AsO4 + 21- + 2Н+ H3AsO3 +12 + Н2О
Cu2+ 2Cu2+ + 4I~ —2CuI+I2
hno2 2HNO2 + 21- I2 + 2NO + Н2О
SeO3" SeO3- + 41- + 6Н+ Se + 2L, + ЗН2О
O3 О3 + 21- + 2Н+ — О2 +12 + Н2О (можно определять в присутствии О2 при pH > 7)
Cl2 С12 + 21- 2С1- + 12
Br2 Вг2 + 21" 2Вг" + 12
нею НС1О + 21- + Н+ Cl" + 12 + Н2О
Решение
Из уравнений реакций, протекающих при титровании
ВгО з + 61- + 6Н+ -> Вг - + 312 + ЗН2О
312 + 6S2O 61- + 3S4O
следует, что 1 ммоль 82Оз“ = 6 • ммоль ВгО 7, поэтому
ллп-, 126,2мг КВ гО3 2 „
Со „2- • 44,97 мл =-----------------• 6 ммоль S,0 3 /ммоль ВгО 3
ЭД 167,01 мг/ммоль КВгОз 2
С„ _2_ = 0,10082 мМ
S2U3
14.6. ИСПОЛЬЗОВАНИЕ ДРУГИХ ТИТРАНТОВ-ОКИСЛИТЕЛЕЙ
581
14.6. Использование других титрантов-окислителей
Некоторые окислители, используемые в качестве титрантов, уже упоминались.
Титрант должен быть достаточно устойчив, удобен в приготовлении и хране-
нии. Если это слишком сильный окислитель, то из-за высокой реакционной спо-
собности, его устойчивость будет невысока. Так, одним из сильнейших окисли-
телей является фтор (Е° = 3,06 В), но он, конечно, не пригоден к использованию
в аналитических лабораториях. Хорошим титрантом мог бы быть хлор, если бы
он не улетучивался из водных растворов, поскольку это затрудняет приготовле-
ние и хранение его стандартных растворов.
Перманганат калия — широко применяемый титрант-окислитель. Это
очень сильный окислитель (£° = 1,51 В), и при титровании перманганатом не ну-
жен индикатор. Правильно приготовленный раствор перманганата устойчив.
Следы примесей восстановителей в свежеприготовленном растворе восстанав-
ливают небольшое количество МпО 4 до МпО2 в нейтральной среде и до Мп2+—
в кислой. Образовавшийся МпО2 катализирует дальнейшее разложение перман-
ганата, в результате чего образуется еще большее количество МпО2. Этот про-
цесс называется автокаталитическое разложение. Стабилизировать раствор
можно, удалив МпО2. Для этого перед стандартизацией раствор кипятят для
ускорения окисления примесей и оставляют стоять на ночь, затем осадок МпО2
отфильтровывают через стеклянный фильтр. Полученный раствор перманганата
можно стандартизировать по первичному стандарту Na2C2O4. Титруют образу-
ющуюся при растворении Na2C2O4 в кислой среде щавелевую кислоту:
5Н2С2О4 + 2МпО; + 6Н+ 10СО2 + 2Мп2+ + 8Н2О (14.15)
Для ускорения реакции раствор нужно нагреть. Это автокаталитическая реак-
ция, которая ускоряется продуктом — Мп2+—и протекает очень медленно, пока
не образуется некоторое его количество. В качестве первичного стандарта мож-
но также использовать чистое полученное электролитически железо. Его рас-
творяют в кислоте и перед титрованием восстанавливают до Fe2+ (см. разд. 14.8).
При титровании железа(П) перманганатом в присутствии хлорид-ионов воз-
никают осложнения. Обычно при комнатной температуре перманганат медлен-
но окисляет хлорид-ион до хлора, но в присутствии железа(П) эта реакция
ускоряется. Если металлическое железо растворили в соляной кислоте или для
восстановления до железа(П) использовали SnCl2 (см. ниже), при титровании необ-
ходимо добавить смесь Циммермана—Рейнгардта, содержащую марганец(П)
и фосфорную кислоту. Марганец(П) настолько понижает потенциал пары
МпО4/Мп2+, что перманганат уже не способен окислять хлорид-ионы— из-за
высокой концентрации Мп2+ формальный потенциал становится меньше Е°. Но
за счет этого уменьшается величина скачка потенциала на кривой титрования,
поэтому нужна Н3РО4, которая связывает в комплекс железо(Ш) и понижает по-
тенциал пары Fe3+/Fe2+. Ионы железа(П) с фосфорной кислотой не реагируют.
Иначе говоря, за счет связывания ионов железа(Ш) в комплекс равновесие реак-
582
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
ции смещается вправо и скачок титрования увеличивается, поэтому в целом ска-
чок титрования будет достаточно большим для четкого фиксирования конечной
точки титрования, но кривая при этом смещается в область более низких потен-
циалов.
Другой положительный эффект добавления фосфорной кислоты заключа-
ется в том, что смесь Циммермана—Рейнгардта предотвращает окисление С1~
перманганатом и позволяет более четко фиксировать конечную точку титрова-
ния — фосфатные комплексы железа(Ш) почти бесцветные, а хлоридные окра-
шены в интенсивный желтый цвет, и в присутствии Н3РО4 переход окраски в
конечной точке титрования становится более контрастным.
Дихромат калия К2Сг2О7 — несколько менее сильный окислитель, чем
перманганат калия. Большим преимуществом этого реагента является то, что его
можно использовать в качестве первичного стандарта и в большинстве случаев
нет необходимости стандартизировать его растворы. Раствор дихромата калия
для титрования железа(П) желательно стандартизировать по электролитически
полученному металлическому железу, но зеленая окраска ионов хрома(Ш) вы-
зывает небольшую погрешность при установлении конечной точки титрования
по индикатору дифениламиносульфонату. Стандартизация необходима только
при выполнении анализов высокой точности.
При титровании раствором дихромата калия отсутствуют проблемы, свя-
занные с окислением хлорид-ионов. Однако поскольку в 1 М НС1 формальный
потенциал пары Сг2О7“/Сг3+ снижается по сравнению со стандартным от 1,33 В
до 1,00 В, необходимо прибавлять фосфорную кислоту для снижения потенциа-
ла пары Fe3+/Fe2+. Это позволяет применять для установления конечной точки
титрования индикатор сульфонат дифениламина (EQ = 0,84 В). В отсутствие фос-
форной кислоты конечная точка будет наблюдаться намного раньше точки экви-
валентности.
Церий(ГУ) — очень сильный окислитель. Формальный потенциал церия(ГУ)
зависит от природы кислоты, используемой при приготовлении растворов: если
раствор не кислый, церий гидролизуется с образованием гидроксида. Титрова-
ние обычно проводят в среде серной или хлорной кислот. Формальный потенци-
ал соответственно равен 1,44 В и 1,70 В, т. е. в среде хлорной кислоты церий(ГУ)
является более сильным окислителем. Его можно использовать в качестве тит-
ранта вместо перманганата, что позволяет получить ряд преимуществ. Потенциал
церияЦУ) можно варьировать, изменяя природу кислоты. Скорость окисления
хлорид-ионов низка даже в присутствии железа(П), поэтому титровать можно в
присутствии умеренных количеств хлорид-ионов, не используя смесь Циммер-
мана—Рейнгардта.
Растворы церияЦУ) можно нагревать, но, во избежание окисления хлорида,
нельзя кипятить. Сернокислые растворы церияЦУ) устойчивы в течение длите-
льного времени, а растворы в азотной и хлорной кислоте хотя и разрушаются, но
очень медленно. Дополнительным преимуществом использования церия(ГУ) яв-
ляется то, что его аммонийная соль (NH4)2Ce(NO3)6 может служить первичным
стандартом и отсутствует необходимость стандартизации раствора. Основной
14.7. ИСПОЛЬЗОВАНИЕ ДРУГИХ ТИТРАНТОВ-ВОССТАНОВИТЕЛЕЙ
583
недостаток церия(1У) по сравнению с перманганатом — более высокая стои-
мость, но это не является серьезным фактором, если учесть экономию времени.
Для большинства случаев титрования в качестве индикатора используют ферроин.
Растворы церия(ГУ) можно стандартизировать, используя такие первичные
стандарты, как As2O3, Na2C2O4 или электролитически полученное железо. Реак-
ция с мышьяком(Ш) протекает медленно, и для ее ускорения вводят катализа-
тор — тетраоксид осмия (OsO4) или монохлорид иода (IC1). Индикатором служит
ферроин. Реакция с оксалатом при комнатной температуре также протекает мед-
ленно, для ее ускорения используют те же катализаторы. Однако в 2 М НС1О4
даже при комнатной температуре реакция идет быстро. В качестве индикатора
применяют нитроферроин.
Растворы церия(1У) для стандартизации обычно готовят из сульфатоцерата
аммония (NH4)2Ce(SO4)2 • 4Н2О, нитратоцерата аммония (NH4)2Ce(NO3)6 (пер-
вичный стандарт, хотя и не очень высокой степени чистоты) или оксида церия
СеО2 • 4Н2О. В качестве первичного стандарта (NH4)2Ce(NO3)6 можно использо-
вать только в том случае, когда раствор стандартизировать не нужно, так как это
очень дорогостоящий реактив.
14.7. Использование других титрантов-восстановителей
Стандартные растворы титрантов-восстановителей не применяются так широ-
ко, как растворы титрантов-окислителей, потому что большинство из них окисля-
ется растворенным кислородом. Поэтому они менее удобны для приготовления и
при хранении. Из используемых титрантов-восстановителей только тиосуль-
фат достаточно медленно окисляется кислородом, и его растворы устойчивы в
течение длительного времени. Именно это и является причиной популярности
йодометрического титрования для определения окислителей. Однако иногда не-
обходимы восстановители сильнее иодид-иона.
Железо(П) в сернокислых растворах очень медленно окисляется кислоро-
дом воздуха, поэтому его часто применяют в качестве титранта. Это не сильный
восстановитель (Е° = 0,771 В), но его можно использовать для титрования силь-
ных окислителей, таких как церий(1У), хром(У1) и ванадий(У). В первых двух
случаях в качестве индикатора пригоден ферроин, в последнем — дифенил-
аминсульфонат. Конценцентрацию раствора железа(П) нужно проверять еже-
дневно.
Хром(П) и титан(Ш) — очень сильные восстановители, но они легко окис-
ляются кислородом воздуха, и их растворы трудно хранить. Для пары Сг3+/Сг2+
стандартный потенциал равен -0,41 В, для пары TiO2+/Ti3+ — +0,04 В. Раство-
ром хрома(П) можно титровать окисленные формы меди, железа, серебра, золо-
та, висмута, урана, вольфрама и других металлов. Практически важные примеры
использования растворов титана(Ш) — титрование железа(Ш), меди(П), оло-
ва(П), хромата, ванадата и хлората.
584
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
14.8. Приготовление раствора для титрования:
предварительное восстановление или окисление аналита
После растворения пробы определяемый компонент обычно присутствует в рас-
творе либо в нескольких степенях окисления, либо в степени окисления, непод-
ходящей для титрования. Известны различные окислители и восстановители, с
помощью которых аналит можно перевести в нужную для титрования степень
окисления. Избыток реагента, используемого для предварительного окисления
или восстановления, перед титрованием необходимо удалить.
Предварительное восстановление
Используемый восстановитель не должен мешать титрованию, но если этого нельзя
избежать, то непрореагировавший остаток должен легко удаляться из раствора.
Большинство восстановителей будет реагировать с титрантом-окислителем, и
поэтому их необходимо удалять. Сульфит натрия Na2SO3 и диоксид серы в
кислых растворах являются хорошими восстановителями (Е° = 0,17 В), а их из-
быток можно легко удалить, пропуская СО2, или, в отдельных случаях, кипячени-
ем. Если SO2 в лаборатории получить сложно, можно использовать сульфит или
гидросульфит натрия в кислой среде. С их помощью таллий(Ш) можно восста-
новить до таллия(1), мышьяк(У) и сурьму(У) — до мышьяка(Ш) и сурьмы(Ш),
ванадий(У) — до ванадия(ГУ), а соединения селена и теллура — до свободного
состояния. Если реакцию проводить в присутствии катализатора тиоцианата,
можно восстановить железо(Ш) до железа(П) и медь(П) — до меди(1).
Хлорид олова SnCl2 обычно используют для восстановления железа(Ш)
при титровании раствором церия(ГУ) или дихромата. Реакция протекает быстро
в присутствии хлорид-ионов (в горячей НС1). Если для растворения образца (на-
пример, руды) используют НС1, железо частично или даже полностью будет
находиться в растворе в степени окисления +3, и его нужно восстановить:
2Fe3+ + SnCl2 + 2С1“—> 2Fe2+ + SnCl4 (14.16)
На завершение реакции укажет исчезновение желтой окраски хлоридных комп-
лексов железа(Ш). Избыток олова(П) удаляют с помощью хлорида ртути:
SnCl2 + 2HgCl2 (избыток) -» SnCl4 + Hg2Cl2 (каломель) (14.17)
Необходимо быстро при перемешивании добавить большой избыток холодного
раствора HgCl2. Если добавить слишком мало или слишком медленно, то за счет
образования локальных избытков SnCl2 некоторое количество ртути восстано-
вится до элементной ртути и выпадет серый осадок. Каломель Hg2Cl2 (молоч-
но-белый осадок) не реагирует с заметной скоростью ни с дихроматом, ни с
Ce(IV), а ртуть может реагировать. Чтобы не было большого избытка олова(П) и,
как следствие, образования металлической ртути, раствор хлорида олова при-
бавляют по каплям до исчезновения желтой окраски хлоридных комплексов желе-
14.8. ПРИГОТОВЛЕНИЕ РАСТВОРА ДЛЯ ТИТРОВАНИЯ
585
за(Ш). Если после прибавления HgCl2 появится серый осадок, раствор титровать
нельзя. Хлорид олова(П) можно также применять для восстановления As(V) до
As(III), Mo(VI) до Mo(V), а в присутствии катализатора FeCl3 можно восстано-
вить U(VI) до U(IV).
Металлы часто используют в качестве восстановителей на стадии предва-
рительной подготовки пробы. Обычно раствор пробы пропускают через колонку,
заполненную гранулированным металлом. Из колонки подготовленную пробу
медленно элюируют разбавленным раствором кислоты. Окисленная форма ме-
талла-восстановителя, попадающая в раствор из колонки, титрованию не поме-
шает, а избытка восстановителя в элюате нет, так как используемый металл в
нем не растворяется. Например, при восстановлении олова(ГУ) металлическим РЬ
РЬ + Sn4+ -> Pb2+ + Sn2+ (14.18)
элюат содержит РЬ2+ и Sn2+, а РЬ в нем нет. В табл. 14.4 перечислены наиболее
часто используемые металлы-восстановители и приведен ряд примеров их при-
менения. Восстановление с помощью металлов проводят в кислых растворах.
Поэтому металлический Zn всегда используют в виде амальгамы (раствор Zn в
металлической ртути) для предотвращения реакции Zn с кислотой:
Zn + 2Н+—> Zn2+ + Н2 (14.19)
Если восстановленный определяемый компонент быстро окисляется кисло-
родом воздуха, титровать нужно в атмосфере СО2, добавив в кислый раствор
гидрокарбонат натрия. При титровании олова(П) и титана(Ш) растворы обязате-
льно дегазируют. Некоторые вещества, легко окисляющиеся кислородом возду-
ха, элюируют из колонки, погрузив ее носик в раствор железа(Ш). При этом
железо(Ш) восстанавливается, а образовавшееся эквивалентное количество же-
леза(П) можно оттитровать дихроматом. Таким способом определяют молиб-
ден(Ш), окисляющийся железом(Ш) до молибдена(У1), и медь(1).
Таблица 14.4
Некоторые металлы-восстановители, широко используемые в пробоподготовке
Восстановитель Восстанавливаемые вещества
Zn(Hg) (редуктор Джонса) Fe(III) -> Fe(II), Cr(VI) -> Cr(II), Cr(III) -> Cr(II), Ti(IV) -> Ti(III), V(V) -> V(II), Mo(VI) -> Mo(III), Ce(IV) -> Ce(III), Cu(II) -> Cu
Ag (1МНС1) (редуктор Уолдена) А1 Fe(III) -> Fe(II), U(VI) -> U(IV), Mo(VI) -> Mo(V) (2 M HC1), Mo(VI) -> Mo(III) (4 M HC1), V(V) V(IV), Cu(II) Cu(I) Ti(IV) -> Ti(III)
РЬ Sn(IV) -> Sn(II), U(VI) -> U(IV)
Cd CIO3 -> ci-
586
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Предварительное окисление
Для окисления большинства веществ необходимы очень сильные окислители.
Один из них — безводная хлорная кислота НС1О4 — при нагревании может
окислить хром(Ш) до дихромата. Полученную смесь нужно разбавить и очень
быстро охладить для предотвращения восстановления. Разбавленная НС1О4 —
не сильный окислитель, поэтому смесь разбавляют только после окисления.
Продуктом восстановления НС1О4 является хлор, его удаляют при кипячении
разбавленного раствора. Правила техники безопасности при работе с хлорной
кислотой описаны в гл. 2. Для предварительного окисления определяемого ком-
понента перед титрованием восстановителем используют такие окислители,
избыток которых можно легко удалить.
Персульфат калия K2S2O8 — сильный окислитель, способный окислить
хром(Ш) до дихромата, ванадий(1У) до ванадия(У), церий(Ш) до церия(1У) и
марганец(П) до перманганата. Окисление проводят в горячем кислом растворе в
присутствии небольшого количества катализатора — соединений серебра(1).
Избыток персульфата разрушается при кипячении. Следует помнить, что при
кипячении также восстанавливается некоторое количество перманганата.
Бром можно применять для окисления различных элементов, например,
для окисления Т1(1) до Т1(Ш) и иодида до иода. Избыток брома удаляют с помо-
щью фенола, который легко бромируется. Хлор является несколько более силь-
ным окислителем. Перманганат окисляет V(IV) до У(У) и Сг(Ш) до Cr(VI), но
вторая реакция протекает быстро только в щелочной среде. Перманганатом
можно окислить следовые количества хрома(П1) в кислой среде, но только при
нагревании. Избыток перманганата удаляют с помощью гидразина, оставшееся
количество которого разрушается при кипячении раствора. Пероксид водорода
окисляет Fe(II) до Fe(III) и Со(П) до Со(Ш) в умеренно щелочной среде, а Сг(П)
до Сг(У1) в сильнощелочной среде.
Существует много методик окислительно-восстановительного определения
различных компонентов в разнообразных объектах. Изучение материала данной
главы должно облегчить выбор наиболее подходящей методики и понимание ее
сути.
Единственным примером использования окислительно-восстановительно-
го титрования в клинических лабораториях является определение кальция в биоло-
гических жидкостях. Кальций осаждают в виде оксалата, осадок отфильтровывают
и растворяют в кислоте. Оксалат-ион, выделившийся в количестве эквивалент-
ном содержанию кальция, титруют стандартным раствором перманганата.
В настоящее время этот метод вытесняют более удобные методы, например,
комплексонометрическое титрование с помощью ЭДТА (гл. 9) или атомно-аб-
сорбционная спектроскопия (гл. 17).
14.9. Потенциометрическое титрование (косвенная потенциометрия)
Титриметрия — удобный метод анализа при визуальном установлении конеч-
ной точки титрования. Если подходящего индикатора нет, часто конец титрова-
14.9. ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ (КОСВЕННАЯ ПОТЕНЦИОМЕТРИЯ)
587
ния можно установить потенциометрически. Потенциометрическое титрование
относится к наиболее точным методам анализа, поскольку потенциал индика-
торного электрода отражает истинное изменение активности, и поэтому конеч-
ная точка титрования часто совпадает с точкой эквивалентности. Кроме того,
как уже упоминалось выше, этот способ установления конца титрования более
чувствительный, чем визуальная индикация. Именно поэтому потенциометри-
ческое титрование часто применяют для анализа разбавленных растворов.
Техника проведения потенциометрического титрования очень проста. Уста-
новка для потенциометрического титрования представлена на рис. 13.5. После
добавления очередной порции титранта измеряют потенциал индикаторного
электрода относительно подходящего электрода сравнения и полученные дан-
ные представляют графически в виде зависимости потенциала от объема титран-
та. В точке эквивалентности наблюдается большой скачок потенциала. Поскольку
в данном случае нас интересует только изменение потенциала, нет необходимо-
сти знать точную величину потенциала индикаторного электрода. Например,
для кислотно-основного титрования не требуется калибровать стеклянный элек-
трод по стандартным буферным смесям, поскольку наклон кривой титрования
будет тот же самый, хотя кривая может быть смещена вверх или вниз по оси
потенциалов.
При титровании нас не интересуют «абсолютные» величины потенциалов,
поэтому отсутствуют проблемы, связанные с потенциалом жидкостного соеди-
нения. В процессе титрования он почти не будет изменяться, а небольшие изме-
нения можно считать незначительными по сравнению с изменением потенциала
в конечной точке титрования. Кроме того, поскольку нет необходимости точного
измерения потенциала, в качестве инструмента очень удобен обычный pH-метр с
ценой деления шкалы 10 мВ. Это позволяет оценивать величину потенциала с
точностью до единиц мВ, что приемлемо для большинства титрований.
Кислотно-основное титрование: использование рН-электрода
В гл. 8 было показано, что в процессе кислотно-основного титрования в точке
эквивалентности наблюдается резкое изменение pH. Это можно легко зафикси-
ровать с помощью стеклянного pH-электрода. Изобразив зависимость измерен-
ной величины pH от добавленного объема титранта, можно получить кривую
титрования аналогичную приведенным в гл. 8. За конечную точку титрования
принимают точку максимального наклона восходящего участка кривой тит-
рования.
Осадительное титрование:
использование серебряного электрода
В качестве индикаторного электрода в осадительном титровании используют
электрод, позволяющий проследить за изменением рМ или рА, где М — осажда-
емый катион, А—анион. Например, при титровании хлорид-ионов ионами серебра
разумно использовать уравнения (13.3) или (13.10). В первом из них lg (l/^Ag+) —
588
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
это pAg, во втором 1g ясг = —рС1, поэтому потенциал серебряного электрода бу-
дет изменяться пропорционально pAg или рС1; при 10-кратном изменении a Ag+
или ясг он изменится на 2,307?77FB (приблизительно на 59 мВ). Зависимость по-
тенциала от объема титранта имеет такой же вид, как на рис. 11.1. (Заметим, что
поскольку 0Ag+tfcl- = const, величина ясг пропорциональна 1/«А + и величина
рС1 пропорциональна -pAg, поэтому наклон кривой титрования будет один и
тот же, независимо от того что мы будем измерять или откладывать по оси орди-
нат — рС1 или pAg.)
Окислительно-восстановительное титрование:
использование платинового электрода
Широкое применение окислительно-восстановительного титрования отчасти
обусловлено тем, что в этом случае не возникает проблемы выбора индикаторного
электрода, в качестве которого используют инертные металлические индикаторные
электроды, например, Pt-электрод. Обычно и окисленная, и восстановленная фор-
мы растворимы, и в процессе титрования изменяется их соотношение. Поэтому
потенциал индикаторного электрода будет изменяться прямо пропорционально
lg (flRed/flOx), как на кривой титрования Fe2+ раствором Се4+ на рис. 14.1. Как уже
упоминалось, расчет можно провести по уравнению Нернста для любой из двух
полуреакций. Обычно при титровании pH поддерживают практически постоян-
ным, поэтому при расчете по уравнению Нернста используют величины Е°'.
Кривую потенциометрического титрования (см. рис. 14.1) можно использовать
для расчетов или выбора подходящего визуального индикатора (если Е1п « £тэ),
что особенно важно при разработке новых методик. Зная интервал скачка потен-
циала, часто можно подобрать окислительно-восстановительный индикатор так,
чтобы переход его окраски наблюдался внутри этого интервала. Или же можно
измерять потенциал в процессе титрования в присутствии визуального индика-
тора и, установив интервал изменения окраски индикатора на кривой потенцио-
метрического титрования, решить вопрос о пригодности индикатора.
Ионселективные электроды в титриметрии: измерение рМ
Ионселективные электроды можно использовать для наблюдения за изменени-
ем рМ [-1g яион, см. уравнение (13.44)] в процессе титрования. Например, кати-
онселектиный стеклянный электрод, чувствительный к ионам Ag+, пригоден для
фиксирования изменения pAg при титровании раствором нитрата серебра. Каль-
ций-селективный электрод пригоден для титрования кальция раствором ЭДТА.
Но такой электрод не должен быть чувствительным к ионам натрия, поскольку
обычно используют натриевую соль ЭДТА. Если электрод реагирует на присут-
ствующий в растворе посторонний ион, концентрация которого в процессе тит-
рования остается постоянной, из уравнения (14.46), справедливого в этом
случае, следует, что кривая титрования будет искажена. Это обусловлено тем,
что в данной ситуации потенциал определяется величиной 1g (яион + const), а не
1g яион. Если вклад постороннего иона не слишком велик, искажение кривой бу-
14.9. ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ (КОСВЕННАЯ ПОТЕНЦИОМЕТРИЯ)
589
дет незначительным и скачок потенциала вблизи конечной точки будет доста-
точным. С помощью анионселективных электродов можно титровать анионы,
например, осаждать фторид-ион лантаном(Ш) и фиксировать конечную точку
титрования с помощью фторид-селективного электрода.
Результаты потенциометрического титрования всегда более правильные,
чем данные прямых потенциометрических измерений, из-за отсутствия неопре-
деленности при измерении потенциала. В то время как в прямой потенциометрии
редко удается достичь погрешности менее нескольких процентов, погрешность в
несколько десятых долей процента обычна для потенциометрического титрова-
ния. Теперь можно сделать несколько выводов относительно потенциометриче-
ского титрования.
1. При проведении измерений в разбавленных растворах и вблизи конечной
точки потенциал обычно устанавливается (считывается) медленно, посколь-
ку равновесие в растворе устанавливается медленно.
2. На график нужно наносить величину потенциала только вблизи конечной
точки. Вблизи конечной точки титрант добавляют небльшими порциями,
например по 0,1 или 0,05 мл. Нет необходимости добавлять строго стехио-
метрическое колиичество титранта, его можно найти интерполяцией зави-
симости Е—объем титранта.
3 - Полярность индикаторного электрода относительно электрода сравнения в
процессе титрования может измениться. Так, разность потенциалов может
изменяться от одного знака, проходя через нуль, до противоположного;
в таком случае для измерения потенциала нужно изменить полярность при-
бора.
Использование производных кривых титрования
Для более точного нахождения конечной точки титрования можно использовать
первую или вторую производную кривой титрования.
1. Построение кривой первой производной. Выше уже говорилось о том, что
наклон кривой титрования максимален в конечной точке титрования, иными
словами, скорость изменения потенциала при прибавлении титранта максималь-
на в конечной точке. Поэтому, если графически отобразить функцию скорости
изменения потенциала при изменении объема титранта (ДЕ/ДР) относительно
добавленного объема титранта, получим кривую с максимумом, который будет
соответствовать объему титранта в конечной точке. Это удобно делать при до-
бавлении титранта вблизи конечной точки одинаковыми порциями. Рассмотрим
результаты титрования (см. электронную таблицу, приведенную ниже), получен-
ные вблизи конечной точки. Их можно использовать для построения интеграль-
ной и производной кривых титрования. Последние четыре столбца таблицы пока
рассматривать не будем. Для получения первой производной кривой титрова-
ния нужно построить график зависимости ДЕ/ДК от объема титранта. Такая
кривая показана на рис. 14.2. По оси абсцисс отложено среднее из двух объемов
(графа III таблицы), использованных для расчета ДЕ. Так, величине ДЕ/ДК= 0,4
590
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
РИС. 14.2. Первая производная кривой потенциометрического титрования
соответствует средний объем титранта 35,475 мл и т. д. Теоретически объем тит-
ранта в конечной точке соответствует максимуму на этой кривой, в данном слу-
чае 35,577 мл (или более корректно 35,58 мл). За максимум можно принять
точку пересечения касательных к обеим ветвям кривой. Возникающую при этом
погрешность можно существенно снизить, построив вторую производную кри-
вой титрования (см. ниже).
Заметим, что поскольку при построении кривой использовали равные объе-
мы титранта, по оси ординат можно было вместо ДЕ/Д V откладывать ДЕ. Исполь-
зование равных объемов титранта необязательно, но это упрощает вычисления.
И не старайтесь вычислять объем титранта с точностью выше 0,01 мл, хотя это
можно сделать и с точностью до 0,001 мл.
2, Построение кривой второй производной. Теоретически вторая производная
кривой титрования в конечной точке должна проходить через нуль. Последние
четыре графы в рассматриваемой таблице показывают, как можно построить
этот график. По сути вторая производная отражает скорость изменения первой
производной (графа VIII), при этом по оси абсцисс отложен средний объем тит-
ранта (графа IX). При делении данных графы VIII на данные графы IX получаем
вторую производную [Д2Е/Д V2 или Д(ДЕ/Д К^/Д К2 — графа X]. Вновь использу-
ем среднее из двух последовательных объемов титранта (графа VII), как и при
построении графика первой производной (графа III). Полученная кривая приве-
дена на рис. 14.3. Здесь также нужна экстраполяция, но получить при этом более
достоверные результаты проще, чем в случае первой производной. Кривая про-
ходит через нуль в точке при значении объема 35,579 мл, поэтому объем титран-
14.9. ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ (КОСВЕННАЯ ПОТЕНЦИОМЕТРИЯ)
591
РИС. 14.3. Вторая производная кривой потенциометрического титрования
та в конечной точке принимаем равным 35,58 мл. Как и в случае первой
производной приводим его с точностью 0,01 мл и для облегчения вычислений
оперируем равными объемами титранта. На компакт-диске (гл. 14) приведена
рассматриваемая здесь электронная таблица. Она поможет сравнить вблизи ко-
нечной точки титрования интегральную кривую и первую и вторую производ-
ные (Charts (Графики) 1, 2 и 3). Более подробно о второй производной кривой
титрования можно прочитать в работе [4].
Оба метода дают надежные результаты, если для построения графиков име-
ется достаточно большое число точек вблизи конечной точки титрования, поэто-
му титрант рекомендуется добавлять не очень большими порциями. Если они
достаточно малы, экстраполяция в случае второй производной может и не по-
требоваться, поскольку на прямолинейном участке графика, проходящем через
нуль, будет не менее двух точек. Но добавление слишком малых порций титран-
та увеличивает время выполнения анализа, и столь малые объемы трудно точно
измерить. Очевидно, что добавлять малые порции титранта требуется только
вблизи конечной точки. В ряде случаев скачок потенциала может быть настоль-
ко велик, что можно все время добавлять равные порции титранта, фиксируя ко-
нечную точку по его объему, при котором налюдается наибольшее изменение
потенциала. Иногда удобно титровать до достижения потенциала конечной точ-
ки, рассчитанного или эмпирически найденного из кривой титрования.
По поводу использования производных кривых титрования следует отме-
тить, что при каждом последующем дифференцировании уровень шумов, а сле-
довательно, и разброс точек увеличивается. Если при титровании наблюдаются
повышенный уровень шумов, возможно предпочтительнее будет использование
интегральной кривой титрования.
592
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
А В С D I LXJ . F G Н I J
1 Прямое титрование окислителя восстановителем
2 I II III IV V VI VII VIII IX X
3 Первая производная Вторая производная
4 V, мл Е Vcp1 АЕ AV, AE/AV, Vcp2 AfAE/AVJ av2 A2E/AV2
5 35,45 0,630 (AfAE/AV^/AVg)
6 35,50 0,650 35,475 0,020 0,05 0,400
7 35,55 0,680 35,525 0,030 0,05 0,600 35,500 0,20 0,050 4,00
8 35,60 0,800 35,575 0,120 0,05 2,400 35,550 1,80 0,050 36,00
9 35,65 0,860 35,625 0,060 0,05 1,200 35,600 -1,20 0,050 -24,00
10 35,70 0,890 35,675 0,030 0,05 0,600 35,650 -0,60 0,050 -12,00
11 35,75 0,910 35,725 0,020 0,05 0,400 35,700 -0,20 0,050 -4,00
12 Ячейка С6 = Vcp1 = (А5+А6)/2 Скопировать все формулы до конца
13 Ячейка D6 = ДЕ = В6-В5
14 Ячейка Е6 = AV1 = А6-А5
15 Ячейка F6 = AE/AV1 = D6/E6
16 Ячейка G7 = Vcp2 = (С6+С7)/2
17 Ячейка Н7 = AfAE/AV^ = F7-F6
18 Ячейка 17 = AV2 = С7-С6
19 Ячейка J7 = A2E/AV2 = H7/I7
20 1-я производная: График С6:С11 в зависимости от F6:F11
21 2-я производная: График G7:G11 в зависимости от J7:J11
Использование графиков Грана
для нахождения конечной точки титрования
Предположим, что вместо зависимости потенциала электрода (являющийся ло-
гарифмической функцией концентрации) от объема титранта мы построили гра-
фик, на котором по оси ординат отложена концентрация неоттитрованного
аналита в каждой точке кривой титрования. Если изменением объема раствора
пренебречь, мы должны получить прямую, отражающую уменьшение концент-
рации определяемого компонента до нуля в конечной точке титрования (при
условии, что равновесие титриметрической реакции сильно смещено вправо).
Это обусловлено тем, что если оттитровано 20%, неоттитрованным осталось
80%, если оттитровано 50% — неоттитровано 50%, если оттитровано 80% — не-
оттитровано 20% и т. д. (В практических целях при построении графика точки
откладывают только вблизи конечной точки.) За конечной точкой титрования
график также будет линейным, что обусловлено увеличением избытка добав-
ленного титранта.
Рассмотрим процесс титрования хлорид-иона раствором нитрата серебра.
В любой момент титрования, за исключением точек вблизи точки эквивалентно-
сти, когда растворимость осадка заметна по сравнению с количеством непрореа-
гировавшего хлорид-иона, можно рассчитать концентрацию хлорида, вычитая
14.9. ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ (КОСВЕННАЯ ПОТЕНЦИОМЕТРИЯ)
593
количество молей хлорида, прореагировавшего с AgNO3, из исходного его ко-
личества: CclFcl — Е'Ао |-С1-] = _С1_С1 Ag_Ag_ (14.20) ^С1 + ^Ag
Без учета коэффициента активности потенциал хлорид-селективного электрода
определяется уравнением
откуда
EH4 = A?-51g [С1-]
(14.21)
(14.22)
где S — эмпирический тангенс угла наклона в уравнении Нернста (теоретиче-
ский — 0,059 мВ), к — эмпирическая константа ячейки (теоретическая — раз-
ность величин Е° индикаторного электрода и электрода сравнения ячейки).
После подстановки уравнения (14.20) в (14.22) получаем:
1g
-cAg^Ag
, Га +>Ag
к~Еяч
(14.23)
( к-Е А
Кс| + FAg antilg —=CclKcl-CAgKAg
\ О у
(14.24)
Графически зависимость левой части уравнения (14.24) от объема титранта
(КА ) выражается прямой (в приведенных выше расчетах учтено изменение объ-
ема раствора при добавлении титранта). Этот график называется графиком
Грана [5-7]. В точке эквивалентности количества ммоль хлорид-ионов и сереб-
ра равны, т. е. левая часть уравнения (14.24) равна нулю, что проиллюстрировано
на рис. 14.4. Изгиб кривой вблизи конечной
точки объясняется тем, что хлорид серебра
имеет некоторую, отличную от нуля, рас-
творимость, в противном случае потенциал
был бы равен бесконечности. Для нахожде-
ния конечной точки через несколько пре-
дыдущих точек проводят прямую. График
Грана позволяет линеаризовать S-образ-
ную кривую титрования.
Для построения графика Грана по
уравнению (14.24) и нахождения точки пе-
ресечения прямой с осью абсцисс необхо-
димо знать константу к в уравнении
Нернста. Ее величину, а также тангенс угла
наклона электродной функции можно най-
ти, используя стандартные растворы.
Рис. 14.4
График Грана, построенный с помо-
щью уравнения (14.24)
594
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Существует несколько способов построения графиков Грана. Можно постро-
ить градуировочный график в координатах потенциал—концентрация определяе-
мого компонента и использовать его для преобразования величины измеренного
потенциала электрода непосредственно в искомую концентрацию. В этом слу-
чае точка пересечения прямой с осью абсцисс соответствует нулевому значению
концентрации по оси у. Можно использовать логарифмическую шкалу прибора
для измерения потенциала (например, pH-метра) и после градуировки по одному
или нескольким стандартам (каждые 59/и мВ будут соответствовать 10-кратному
изменению концентрации) проводить прямой отсчет искомых концентраций.
Можно поступить иначе — рассчитать антилогарифм потенциала или pH и
представить полученные значения графически в виде зависимости от объема
титранта (Е ~ lg с, antilg Е ~ с). В этом случае точка пересечения будет соответст-
вовать потенциалу при нулевой концентрации определяемого компонента.
Для построения графика Грана можно также использовать участок кривой
титрования за точкой эквивалентности. В этом случае величина слагаемого, со-
держащего антилогарифм, линейно возрастает от нулевого значения, соответству-
ющего точке эквивалентности. При таком способе построения графика потенциал,
соответствующий точке пересечения, лучше всего найти из данных титрования
контрольного образца, экстраполируя линейный участок кривой до точки пере-
сечения с осью ординат.
Величина слагаемого, содержащего антилогарифм, прямо пропорциональ-
но концентрации. При расчетах следует ввести поправку на изменение объема
раствора, умножая это слагаемое на (К+ v)/K, где V— исходный объем раствора,
v — добавленный объем титранта.
Другим достоинством использования графиков Грана, кроме их линейной
формы, является отсутствие необходимости измерения потенциала вблизи ко-
нечной точки титрования, когда из-за низкой концентрации определяемого ком-
понента потенциал устанавливается очень медленно и приходится добавлять
очень малые порции титранта. Для построения графика Грана достаточно всего
несколько точек вдали от конечной точки.
Типичный график Грана для титрования малых количеств хлорида раствором
нитрата серебра с использованием электрода Ag I Ag2S приведен на рис. 14.5, на
котором слева изображена обычная 5-образная кривая титрования. Зависимый
параметр, использованный для построения графика Грана, отложен по оси орди-
нат справа. Из-за низких концентраций реагирующих веществ наблюдается
небольшое отклонение от линейности (перегиб). Для нахождения объема тит-
ранта в конечной точке прямолинейный участок полученной кривой экстраполи-
руют до пересечения с осью абсцисс (для получения более точных результатов
проводят титрование контрольного образца и экстраполируют линейную часть
полученной кривой на нулевое значение по оси ординат). Искривление прямой
вблизи конечной точки обычно обусловлено свойствами продукта титриметриче-
ской реакции—заметной растворимостью осадка, диссоциацией комплекса и т. п.
Линейные графики обладают рядом преимуществ. Для их построения нуж-
но иметь всего лишь несколько точек, в которых титрант находится в избытке,
достаточном для подавления диссоциации продукта реакции титрования и обу-
14.9. ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ (КОСВЕННАЯ ПОТЕНЦИОМЕТРИЯ)
595
Рис. 14.5
График Грана (справа)
и кривая титрования
(слева) 100 мл 5 • 10-5 М
СГ раствором AgNO3
с использованием
электрода с мембраной
из Ag2S. С разрешения
Orion Research, Inc.
словливающем быстрый отклик электрода. Конечную точку титрования легко
найти, экстраполируя прямую до пересечения с осью абсцисс. Даже в случае
слабовыраженного перегиба на кривой титрования метод Грана позволяет за-
фиксировать конечную точку достаточно точно.
Зависимости, по форме напоминающие графики Грана, получаются при по-
строении кривых титрования в координатах ДР7ДЕ—V (т. е. с использованием
величины, обратной ДЕ7ДК, которую применяют для построения первой произ-
водной кривой титрования). В точке эквивалентности величина ДЕ/ДК мак-
симальна, а ДР7ДЕ — минимальна, вследствие чего график в координатах
ДР7ДЕ—V имеет И-образную форму. Для построения таких зависимостей, как и
в случае производных кривых титрования, по оси абсцисс откладывают среднее
из двух соседних значений объема. Чтобы получить график, более приближен-
ный к линейному, значения ДР7ДЕ следует скорректировать на изменение объе-
ма раствора.
Графики Грана удобно использовать при нахождении концентрации мето-
дом стандартных добавок. К этому методу градуировки прибегают в случаях,
если на аналитический сигнал влияет состав матрицы образца. В таких случаях
сигнал регистрируют дважды — до и после введения известного количества
стандарта, что и позволяет учесть влияние матрицы на отклик электрода. Для
большинства методов анализа характерен линейный отклик на содержание
определяемого компонента, но в потенциометрии отклик является логарифми-
ческой функцией. Метод Грана позволяет получить линейную зависимость и в
этом случае. При использовании метода добавок графики Грана строят как ли-
нейные зависимости антилогарифмического члена уравнения (14.24) от коли-
чества добавки, спрямляя их, например, методом наименьших квадратов.
596
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Рис. 14.6
Использование графика
Грана в методе
стандартных добавок.
С разрешения Orion
Research, Inc.
Содержание вещества находят экстраполяцией зависимости до пересечения с
осью абсцисс аналогично использованию метода Грана в обычном варианте
(рис. 14.6).
При использовании метода добавок очень удобно добавлять малые порции
концентрированного стандартного раствора, чтобы не вводить поправку на из-
менение объема раствора. Например, для увеличения концентрации определяе-
мого компонента на 10 мг/л к 10 мл пробы нужно добавить 100 мкл стандартного
раствора с концентрацией 1000 мг/л. Изменение объема в этом случае составит
всего 1% и, вероятно, им можно пренебречь. Желательно, чтобы концентрации
добавок были близки к концентрации определяемого компонента.
Можно использовать метод добавок и в расчетном варианте (пример 14.8).
Пример 14.8
Концентрацию кальция в сыворотке крови определяли ионометрически. По-
тенциал Са-селективного электрода в анализируемом растворе равен +217,6
мВ, а после добавления 100 мкл стандартного раствора, содержащего 2000
мкг/мл кальция к 2,00 мл анализируемого раствора он стал равным +226,8 мВ.
Тангенс угла наклона электродной функции при 10-кратном изменении актив-
ности составляет 59,2/2 мВ. Рассчитайте концентрацию кальция в образце сы-
воротки.
Решение
Поскольку использован метод добавок (матрица и ионная сила анализируемого
и стандартного растворов одинаковы), в уравнении Нернста вместо активностей
можно использовать концентрации (см. разд. 13.8) и поэтому
Е = £ + 29,6 1g [Са2+]
14.9. ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ (КОСВЕННАЯ ПОТЕНЦИОМЕТРИЯ)
597
Учтем, что при добавлении 0,100 мл стандартного раствора к 2,00 мл пробы про-
изошло разбавление в отношении приблизительно 1 : 20 и фактически добавле-
но 100 мкг/мл или, точнее, с введением поправки на 5%-ное изменение объема
С = 2000 мкг/мл • л = 95,2 мкг/мл
2,10 мл
Обозначим искомую концентрацию кальция (мкг/мл) через х, тогда
217,6 мВ = к + 29,61g х (1)
226,8 мВ = к + 29,6 1g (х + 95,2) (2)
Вычитая уравнение (2) из уравнения (1), находим х:
-9,2 мВ = 29,6 1g х - 29,6 lg (х + 95,2)
-9,2 мВ = 29,61g—-—
х + 95,2
—-— = 0,467
х + 95,2
х = 83,5 мкг/мл
Если тангенс угла наклона электродной функции отличается от теоретического
или заранее не определен, его необходимо оценить, вводя несколько добавок
определяемого компонента.
Рис. 14.7
Автоматический
потенциометрический титратор.
(С разрешения Brinkmann
Instruments, Inc.)
598
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Автоматические титраторы
Существует много автоматических титраторов, используемых для потенцио-
метрического детектирования конечной точки титрования. Эти приборы авто-
матически записывают первую или вторую производную кривой титрования и
рассчитывают положение конечной точки. Пробу помещают в сосуд для титро-
вания, а титрант, подаваемый из резервуара, находится в снабженной шприцем
бюретке (рис. 14.7). Расход титранта определяют по смещению поршня шпри-
ца приводом с электронным управлением. Титраторы можно также приме-
нять при фотометрическом детектировании изменения окраски индикатора.
Автоматические титраторы удобны, поскольку их применив ускоряет выпол-
нение анализа и повышает воспроизводимость результатов. Несмотря на мно-
гочисленные достоинства инструментальных методов анализа классическую
титриметрию до сих пор широко используют. Особенно этот метод удобен при
определении главного компонента, например, в объектах фармацевтической
промышленности.
Что мы узнали из этой главы?
• Уравнивание окислительно-восстановительных реакций — стр. 562
• Расчет констант равновесия реакций из величин стандартных потенциалов
[основное уравнение (14.1); пример (14.3)] — стр. 563
• Построение кривой окислительно-восстановительного титрования —
стр. 566
• Окислительно-восстановительные индикаторы — стр. 572
• Иодиметрия и иодометрия — стр. 574, 578
• Предварительная подготовка раствора для титрования — стр. 584
• Потенциометрическое титрование — стр. 586
• Производные кривые титрования — стр. 589
• Графики Грана — стр. 592
Вопросы
1. Назовите способы визуального детектирования конечной точки титрования.
2. В чем различие иодиметрии и иодометрии?
3. Почему иодиметрическое титрование обычно проводят в нейтральной сре-
де, а йодометрическое — в кислой?
4. Совпадают ли конечная точка титрования и точка эквивалентности в пер-
манганатометрии? Как это несоответствие можно скорректировать?
5. Объясните роль компонентов смеси Циммермана—Рейнгардта при титро-
вании железа(П) перманганатом.
ЗАДАЧИ
599
Задачи
Уравнивание окислительно-восстановительных реакций
6. Напишите уравнения следующих реакций, протекающих в водном рас-
творе:
(a) Ю3 +1- -> 12 (кислая среда)
(б) Se2Cl2 -> H2SeO3 + Se + НС1
(в) H3PO3 + HgCl2 -> Hg2Cl2 + H3PO4 + HC1
7. Напишите уравнения следующих реакций, протекающих в водном рас-
творе:
(а) МпО4" -> МпО2 + МпО4 (щелочная среда)
(б) MnO J + H2S -> Мп2+ + S
(в) SbH3 + С12О -> H4Sb2O7 + НС1
(г) FeS + NO3 —> Fe3+ + NO2 + S (кислая среда)
(Д) A1 + NO3 —> АЮ2+NH3
(е) FeAsS + С1О2 -> Fe3+ + AsO4~ + SO4" + Cl- (кислая среда)
(ж) K2NaCo(NO2)6+MnO 4 -> K+ + Na+ + Co3+ + NO 3 + Mn2+ (кислая среда)
8. Смешали равные объемы 0,10 М раствора T1NO3 и 0,20 М раствора
Co(NO3)2. Рассчитайте потенциал относительно стандартного водородного
электрода (СВЭ).
9. Рассчитайте потенциал (отн. СВЭ) при титровании 50,0 мл 0,100 М раство-
paFe2+B 1,00 МНС1О4 0,0167 Мраствором Сг2О7" после добавления 10,25,
50 и 60 мл титранта.
10 - Рассчитайте потенциал (отн. СВЭ) при титровании 100 мл 0,100 М раствора
Fe2+ в 0,500 М H2SO4 0,0200 М раствором КМпО4 после добавления 10,0,
50,0, 100 и 200 мл титранта, полагая, что H2SO4 диссоциирована полно-
стью.
Расчет потенциала в точке эквивалентности
11. Каков будет потенциал в точке эквивалентности при титровании Fe3+ рас-
твором Sn2+?
12. Уравнение (14.2) выведено для двух полуреакций с равным числом участ-
вующих электронов (и). Выведите подобное уравнение для реакции из за-
дачи 11, используя численные величины п\
2Fe3++ Sn2+^ 2Fe3+ + Sn4+
600
ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНОЕ И ПОТЕНЦИОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
13. Выведите уравнение подобное (14.2) для приведенной ниже реакции (при вы-
воде не забудьте, что в данной реакции участвуют ионы водорода):
5Fe2+ + MnO; + 8Н+^ 5Fe3+ + Мп2+ + 4Н2О
По полученному уравнению рассчитайте потенциал в конечной точке тит-
рования, рассмотренного в примере 14.4. Результат сравните с найденным
в этом примере при расчете равновесных концентраций.
14. Для приведенных ниже ячеек рассчитайте ЭДС до замыкания цепи и потен-
циал каждого каждого полуэлемента после завершения реакции. Рассчи-
тайте также константы равновесия реакций:
(a) Zn | Zn2+ (0,250 М) || Cd2+(0,0100 М) | Cd
(б) РЬ | РЬ2+(0,0100 М) || Iз (0,0100 М), I-(1,00 М) | Pt
Расчет результатов титрования
15. Селен из навески почвы массой 10 г отогнали в виде тетрабромида и собра-
ли в приемник с водой, где его гидролизовали до SeO3“. Для йодометри-
ческого титрования ЗеОз" потребовалось 4,5 мл стандартного раствора
тиосульфата. Титр раствора тиосульфата составляет 0,049 мг К2Сг2О7 /мл.
Рассчитайте содержание в ppm селена в почве.
16. Кальций, содержащийся в 5,00 мл сыворотки, осадили в виде СаС2О4 с по-
мощью оксалата аммония. Осадок отфильтровали, растворили в кислоте и
оттитровали оксалат-ион 0,00100 М КМпО4, затратив 4,94 мл раствора тит-
ранта. Рассчитайте концентрацию кальция в сыворотке, выразив результат
в ммоль экв./л.
17. Навеску массой 2,50 г, содержащую As2O5, Na2HAsO3 и инертную матрицу,
растворили и создали в растворе нейтральную среду с помощью избытка
NaHCO3. Из полученного раствора оттитровали As(III), затратив на это
11,3 мл 0,150 М раствора 12. Затем раствор подкислили НС1 (весь мышьяк в
растворе находится в степени окисления +5), добавили избыток KI и оттит-
ровали выделившийся 12, затратив 41,2 мл 0,120 М раствора Na^C^. Рассчи-
тайте массовые доли As2O5 и Na2HAsO3 в пробе.
18. Если 1,00 мл раствора КМпО4 реагирует с 0,125 г Fe2+, а на титрование
1,00 мл раствора КНС2О4 • Н2С2О4 требуется 0,175 мл раствора КМпО4, то
сколько миллилитров 0,200 М NaOH потребуется для титрования 1,00 мл
раствора тетраоксалата (титруются все 3 протона)?
19. Содержание сульфида в промышленной сточной воде (суспензии) опреде-
ляют ионометрически с помощью сульфид-селективного электрода, испо-
льзуя метод добавок. После разбавления 10,0 мл пробы водой до 25,0 мл
потенциал электрода равен -216,4 мВ, а в растворе, полученном после при-
бавления к 10,0 мл пробы 1,00 мл 0,030 М стандартного раствора сульфида
и разбавления водой до 25,0 мл, потенциал равен -224,0 мВ. Рассчитайте
концентрацию сульфида в пробе.
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
601
Графики Грана
20. Подставив в выражение для Ка слабой кислоты НА вместо [НА] и [А-] объем
титранта, покажите, что приведенное ниже уравнение справедливо вплоть
до точки эквивалентности при титровании НА сильным основанием В:
КВ[Н+] = Ка (Гтэ- Кв)= кв10-рн
где Кв — добавленный объем титранта, Ктэ — объем титранта в точке экви-
валентности. Графически зависимость Ив от Кв10_рн выражается прямой с
тангенсом угла наклона, равным -Ка, а точка пересечения прямой с осью
абсциссы соответствует точке эквивалентности.
Упражнение с электронными таблицами
21. Создайте электронную таблицу для построения кривой титрования, приве-
денной на рис. 14.1. Примите А?равн равной 1,7 • 1014 (после создания элект-
ронной таблицы см. пример на компакт-диске к гл. 14).
Рекомендуемая литература
Уравнивание окислительно-восстановительных реакций
1. С. A. Vanderhoff, «А Consistent Treatment of Oxidation-Reduction», J. Chem.
£rf.,25(1948) 547.
2. R. G. Yolman, «Writing Oxidation-Reduction Equations», J. Chem. Ed., 36
(1959)215.
Вычисление потенциала в точке эквивалентности
3. A. J. Bard and S. Н. Simpsonsen, «The General Equation for the Equivalence Po-
int Potential in Oxidation-Reduction Titrations», J. Chem. Educ.,Xl (1960) 364.
Производные кривые титрования
4. К. N. Carter and R. B. Huff, «Second Derivative Curves and End-Point Determi-
nation», J. Chem. Ed., 56 (1979) 26.
Графики Грана
5. G. Gran, «Determination of Equivalent Point in Potentiometric Titrations», Acta
Chem. Scand., 4 (1950) 559.
6. G. Gran, «Determination of the Equivalence Point in Potentiometric Titrations.
Part II», Analyst, 77 (1952) 661.
7. С. C. Westcott, «Ion-Selective Measurements by Gran Plots with a Gran Ruler»,
Anal. Chim. Acta, 86 (1976) 269.
8. H. Li, «Improvement of Gran’s Plot Method in Standard Addition and Subtracti-
on Methods by a New Plot Method», Anal. Lett., 24 (1991) 473.
Глава 15
ВОЛЬТАМПЕРОМЕТРИЯ И ЭЛЕКТРОХИМИЧЕСКИЕ
СЕНСОРЫ
Среди электрохимических методов анализа, связанных с использованием элект-
ролитических ячеек, можно найти примеры наиболее точных и чувствительных
определений. В этих методах определяемый компонент окисляется или восста-
навливается на подходящем электроде в электролитической ячейке при наложе-
нии напряжения от внешнего источника (см. гл. 12), при этом количество
электричества (или сила тока), протекающего через ячейку в процессе электро-
лиза, связано с количеством определяемого компонента. В вольтамперометрии
доля определяемого компонента, израсходованного в процессе электролиза,
может быть очень мала и фактически ею можно пренебречь — можно опреде-
лять микромолярные и даже более низкие концентрации. Поскольку потенциал
окисления или восстановления зависит от природы электроактивного вещества,
селективность методов анализа, основанных на процессе электролиза, можно
повысить при правильном выборе потенциала электролиза. Так как эти методы
анализа специфичны, часто нет необходимости предварительного разделения,
и анализ, таким образом, является весьма экспрессным.
В данной главе мы рассмотрим вольтамперометрические методы и исполь-
зуемые в них электрохимические сенсоры (датчики), включая химически модифи-
цированные электроды. В вольтамперометрических методах для осуществления
микроэлектролиза применяют микроэлектроды. Метод основан на сканирова-
нии потенциала микроэлектрода в анализируемом растворе и измерении при соот-
ветствующем значении потенциала предельного тока (в разбавленных растворах
порядка нескольких микроампер или меньше), пропорционального концентра-
ции определяемого компонента. Амперометрия — разновидность вольтамперо-
метрии, в которой при фиксированном потенциале микроэлектрода измеряют
зависимость параметров тока от концентрации определяемого компонента, из-
меняющейся, например, в процессе титрования. Этот принцип положен и в
основу работы электрохимических сенсоров.
Перед детальным изучением этих методов полезно повторить материал
гл. 13, посвященный потенциометрии.
15.1. ВОЛЬТАМПЕРОМЕТРИЯ
603
15.1. Вольтамперометрия
В основе вольтамперометрии лежит процесс микроэлектролиза на рабочем мик-
роэлектроде, изготовленном, например, из платиновой проволочки. Название
метода отражает использование зависимостей ток—потенциал. При медленной
развертке потенциала рабочего микроэлектрода регистрируют протекающий че-
рез ячейку ток как функцию наложенного напряжения. Полученная кривая на-
зывается вольтамперограммой. Если в ячейке присутствует электроактивное
вещество (способное восстанавливаться или окисляться), то при достижении
определенного отрицательного (в случае восстановления) или положительного
(в случае окисления) значения потенциала через ячейку будет протекать ток,
обусловленный электропревращением этого вещества. По договоренности катод-
ный ток (ток восстановления) считают положительным, анодный ток (ток окисле-
ния) — отрицательным. Если концентрация электроактивного вещества мала,
ток достигает предельной величины, так как вещество может подойти к электро-
ду только за счет диффузии, поэтому скорость электролиза ограничена его кон-
центрацией у поверхности электрода. Ниже будет показано, что предельный ток
пропорционален концентрации электроактивного вещества. При использовании
микроэлектрода через ячейку протекает ток порядка нескольких микроампер и
меньше, поэтому после записи вольтамперограммы концентрация вещества в
растворе практически не изменяется.
Ячейка для вольтамперометрии —
электролитическая ячейка
Ячейка для вольтамперометрических измерений состоит из рабочего микро-
электрода, вспомогательного электрода и электрода сравнения, обычно
насыщенного каломельного (НКЭ). Для контроля потенциала используют
потенциостат. Ток, протекающий через рабочий электрод, регистрируют как
функцию его потенциала, измеренного относительно электрода сравнения, а
поляризующее напряжение от внешнего источника подают в цепь, состоящую
из рабочего и вспомогательного электродов (рис. 15.1). При использовании
трехэлектродной ячейки регистрируемые кривые ток—потенциал не искажа-
ются за счет сопротивления раствора (как в неводных растворах), вызванного
омическим падением напряжения iR. Согласно закону Ома потенциал равен
произведению силы тока и сопротивления: Е = iR, где i — сила тока (А) и А —
сопротивление (Ом). При протекании тока потенциал изменяется — смещает-
ся на величину, равную z • R, где R — сопротивление раствора. Если оно доста-
точно велико, то зависимость ток—потенциал (рис. 15.2) искажается и кривая
смещается к более высоким потенциалам. В трехэлектродной ячейке регистри-
руют потенциал рабочего электрода относительно электрода сравнения в цепи,
в которой ток практически не протекает, и вольтамперная кривая не искажает-
ся за счет омического падения напряжения iR.
604
ВОЛЬТАМПЕРОМЕТРИЯ И ЭЛЕКТРОХИМИЧЕСКИЕ СЕНСОРЫ
Источник внешнего напряжения
Электрод
сравнения
отн. электрода ’
Солевой мостик
Рис. 15.1. Установка для вольтамперометрических измерений
Вольтамперные кривые — основа вольтамперометрии
В вольтамперометрии принято измерять потенциалы относительно НКЭ. Для
пересчета величин потенциалов, измеренных относительно НКЭ, на величины от-
носительно СВЭ (и наоборот) можно использовать соотношение
^отн. НКЭ ^отн. СВЭ 0,242
(15.1)
По этому уравнению можно рассчитать, при каком потенциале, наложенном от
внешнего источника напряжения, на микроэлектроде будет происходить элект-
ролиз с участием определяемого иона. (Как перейти от шкалы потенциалов от-
носительно одного электрода сравнения к шкале потенциалов относительно
другого электрода сравнения показано в примере 13.4 и на рис. 13.4.)
Вольтамперометрическая ячейка — это электролитическая ячейка, в кото-
рой при наложении потенциала от внешнего источника протекает электрохими-
ческая реакция в направлении, противоположном самопроизвольной реакции,
как это происходило бы в гальваническом элементе (см. разд. 12.2). Предполо-
жим, что в ячейку с углеродным микроэлектродом поместили 10-3 М раствор
Cd(NO3)2 и приложили от внешнего источника такую разность потенциалов
между рабочим и вспомогательным электродом, что микроэлектрод стал отри-
цательно заряженным относительно НКЭ.
15.1. ВОЛЬТАМПЕРОМЕТРИЯ
605
В этом случае на электроде протекает реакция
Cd2+ + 2е- Cd Е° = - 0,403 В (15.2)
Минимальную величину потенциала, необходимую для восстановления ионов
кадмия, можно рассчитать по уравнению Нернста (гл. 13):
00592 1
£отн нкэ = - °>403 - 1g —- 0,242 = - 0,556 В (15.3)
Этот потенциал называют потенциалом выделения. При более высоких потен-
циалах по сравнению с потенциалом выделения наблюдается линейный рост
тока согласно закону Ома
р
——— = кЕ (15.4)
цепи
Во время электролиза концентрация ионов кадмия вблизи поверхности
электрода уменьшается и возникает градиент концентраций между поверхност-
ным слоем и раствором. До тех пор пока приложенный потенциал невелик, ионы
кадмия из глубины раствора могут быстро диффундировать к поверхности элек-
трода и поддерживать ток электролиза. Но по мере увеличения потенциала сила
тока растет, что приводит к увеличению градиента концентраций, поэтому для
поддержания тока ионы должны диффундировать быстрее. Градиент концент-
раций, и, следовательно, скорость диффузии пропорциональны концентрации в
глубине раствора. Если раствор разбавленный, постепенно установится потен-
циал, при котором скорость диффузии достигнет максимума и все диффундиру-
ющие к поверхности электрода ионы будут мгновенно восстанавливаться. Это
означает, что достигнут предельный ток, i , величина которого не изменяется
при дальнейшем увеличении потенциала. Предельный ток достигается, поско-
льку определяемый ион восстанавливается быстрее, чем он диффундирует к по-
верхности электрода.
При вольтамперометрических измерениях через ячейку протекает очень
малый ток, поэтому концентрация электроактивного вещества в растворе прак-
тически не изменяется.
На рис. 15.2 показана типичная зависимость силы тока от потенциала. Если
раствор перемешивают или вращают электрод, получаются S — образная кривая
(кривая а), потому что предельный ток, достигнув некоторой величины, оста-
ется постоянным. Это объясняется тем, что диффузионный слой или градиент
концентраций, определяющий скорость диффузии определяемого компонента к
поверхности электрода, сохраняет небольшую и постоянную величину, так как
ионы непрерывно подходят к поверхности электрода в результате массоперено-
са (перемешивание). Но если электрод не вращается и находится в покоящемся
растворе, диффузионный слой будет со временем расширяться в глубь раствора
и это вызовет эскпоненциальное падение предельного тока. В результате полу-
чится «горбообразная» кривая — кривая б на рис. 15.2. Это одна из причин, по
606
ВОЛЬТАМПЕРОМЕТРИЯ И ЭЛЕКТРОХИМИЧЕСКИЕ СЕНСОРЫ
Наложенный потенциал, В
РИС. 15.2. Различные виды вольтамперограмм: а — вольтамперограмма, зарегистриванная в пе-
ремешиваемом растворе или на вращающемся электроде; б — в неперемешиваемом растворе; в —
вольтамперограмма ступенчатого восстановления (или окисления) деполяризатора или смеси двух
деполяризаторов в неперемешиваемом растворе
которым развертку потенциала на стационарный микроэлектрод подают с до-
статочно высокой скоростью, например 50 мВ/с. (В действительности даже при
перемешивании раствора имеется тенденция образования «горба».)
Потенциал выделения, необходимый для начала электрохимической реак-
ции, в некоторой степени зависит от концентрации, но потенциал, при котором
сила тока достигает половины предельной величины, от концентрации не зави-
сит и его называют потенциалом полуволны, Ет. Это константа, связанная со
стандартным или формальным потенциалом конкретной окислительно-восста-
новительной пары, поэтому вольтамперометрию используют для идентификации
восстанавливающегося или окисляющегося вещества. Потенциал, при котором
данное вещество восставливается или окисляется на электроде, зависит от приро-
ды этого вещества и, следовательно, является его характеристической величиной.
Электрод, потенциал которого изменяется в зависимости от силы протека-
ющего через него тока, называют поляризующимся электродом. Если площадь
поверхности электрода мала и на нем достигается предельный ток,.говорят, что
электрод деполяризован, а восстанавливающееся или окисляющееся на таком
микроэлектроде вещество называют деполяризатором.
Если на рабочем электроде деполяризатор восстанавливается, то протекает
катодный ток (при более отрицательных значениях потенциалах относительно
потенциала выделения), а если деполяризатор окисляется, то протекает анодный
ток при более положительных потенциалах относительно потенциала выделения.
Ступенчатое восстановление и окисление
Электроактивное вещество при определенном значении потенциала может вос-
становиться до более низкой степени окисления, а затем, при более отрицатель-
ных потенциалах, восстановиться до еще более низкой степени окисления.
Например, в аммиачном растворе медь(П) восстанавливается на графитовом
электроде до устойчивого аммиачного комплекса меди(1) при -0,2 В отн. НКЭ,
15.1. ВОЛЬТАМПЕРОМЕТРИЯ
607
а затем — при -0,5 В — до металла, при этом в каждой стадии участвует один
электрон. В таком случае на вольтамперограмме будут наблюдаться две волны,
как на кривой в на рис. 15.2. Отношение высот этих волн пропорционально отно-
шению числа электронов, участвующих в процессе восстановления или окисле-
ния. В рассмотренном примере высоты обеих волн равны.
Если в растворе находятся два или более электроактивных веществ с разли-
чающимися потенциалами восстановления, возможно их последовательное вос-
становление. Например, свинец(П) восстанавливается на графитовом электроде
при потенциалах ниже -0,4 В отн. НКЭ (РЬ2+ + 2е_ РЬ), а кадмий(И) — при по-
тенциалах ниже -0,6 В (Cd2+ + 2е~ Cd), поэтому на вольтамперограмме рас-
твора, содержащего их смесь, наблюдаются две волны: первая при -0,4 В для
свинца(П) и вторая при -0,6 В для кадмия(П). Отношение их высот пропорцио-
нально отношению концентраций веществ в растворе и числу электронов, участ-
вующих в их восстановлении или окислении.
Смесь электроактивных веществ можно проанализировать, если их волны
на вольтамперограмме четко разделяются. Для этого необходимо, чтобы раз-
ность величин Е1/2 составляла, по крайней мере, 0,2 В. Если ЕУ2 волн равны, будет
наблюдаться одна волна, высота которой равна сумме высот индивидуальных
волн. Если компонент смеси с более высокой концентрацией восстанавливается
(или окисляется) раньше, чем определяемый компонент (компоненты), его вол-
на будет маскировать все остальные волны и можно не достичь предельного
тока. В таких случаях необходимо удалить все мешающие примеси, что обычно
и делают, проводя предэлектролиз на макроэлектроде при потенциале предель-
ного тока мешающей примеси, недостаточном для электропревращения опреде-
ляемого компонента.
При ступенчатом окислении электроактивного вещества наблюдаются раз-
дельные анодные волны.
Индифферентный электролит:
необходим для проведения измерений
Во всех выше приведенных рассуждениях мы полагали, что если в покоящемся
растворе возник градиент концентраций, диффузия является единственным вари-
антом массопереноса деполяризатора к поверхности электрода. Но деполяриза-
тор может подходить к поверхности электрода и за счет электростатического
притяжения или отталкивания.
В зависимости от величины приложенного извне потенциала поверхность
электрода заряжена положительно или отрицательно, и поэтому притягивает,
или отталкивает диффундирующие к ней ионы. Возникающий в результате это-
го миграционный ток и увеличивает или уменьшает величину предельного тока.
Миграционный ток можно устранить добавлением индифферентного элект-
ролита, например KNO3, в избытке, по крайней мере в 100 раз превышающем
концентрацию определяемого вещества. Ион калия восстанавливается при более
отрицательных потенциалах и мешать не будет. При высокой концентрации ин-
608
ВОЛЬТАМПЕРОМЕТРИЯ И ЭЛЕКТРОХИМИЧЕСКИЕ СЕНСОРЫ
дифферентного электролита с заряженной поверхностью электрода взаимодейст-
вуют практически только его ионы, но в электродной реакции они не участвуют.
Индифферентный электролит — это «инертный» электролит с высокой кон-
центрацией, уменьшающий степень притяжения или отталкивания определяе-
мого иона заряженной поверхностью электрода. В результате снижается
миграционный ток.
Вторая причина необходимости использования индифферентного электро-
лита — снижение величины омического падения напряжения (iR) в ячейке.
Обычно для этого применяют растворы индифферентных электролитов с кон-
центрацией не менее 0,1 М. Это справедливо практически для всех электрохимиче-
ских методов, за исключением потенциометрии. Часто выбор индифферентного
электролита диктуется необходимостью оптимизации условий проведения ана-
лиза, например, для создания и поддержания нужной величины pH применяют
буферные смеси, а для устранения мешающего влияния некоторых прмесей ис-
пользуют селективное комплексообразование. Если ион металла склонен к ком-
плексообразованию, его электрохимическую активность можно снизить и тем
самым сместить волну его восстановления к более отрицательным потенциалам,
а иногда даже сделать этот ион электронно-неактивным. Обычно для этого при-
меняют такие лиганды, как тартрат, цитрат, цианид, аммиак и ЭДТА.
Необратимое восстановление и окисление
Если определяемое вещество восстанавливается или окисляется обратимо, вели-
чина потенциала полуволны будет близка к величине стандартного потенциала
этой окислительно-восстановительной полуреакции. Но если восстановление
или окисление протекает необратимо, механизм электродной реакции более
сложен и включает медленную стадию с высокой энергией активации, поэтому
для протекания электродной реакции с заметной скоростью необходима допол-
нительная энергия. Ее можно сообщить электроду, увеличив внешнее поляризу-
ющее напряжение. Это явление называется перенапряжением. В результате
для необратимой катодной волны значение ЕУ2 будет более отрицательным по
сравнению с величиной стандартного потенциала, а для анодной волны — более
положительным. При необратимом восстановлении наблюдается более пологая
волна, чем в случае обратимого восстановления (5-образная форма сохраняется),
но величина предельного диффузионного тока не меняется, поскольку лимити-
руется только скоростью диффузии вещества к поверхности электрода.
Рабочий интервал потенциалов:
зависимость от материала электрода
Интервал потенциалов, в котором проводят вольтамперометрические измере-
ния, зависит от материала электрода, природы растворителя и индифферентно-
го электролита, а также от кислотности раствора. При регистрации вольт-
амперограммы на платиновом электроде в водном растворе предельная величи-
на положительных потенциалов лимитируется потенциалом реакции окисления
15.1. ВОЛЬТАМПЕРОМЕТРИЯ
609
воды (Н20 —> ^02 + 2Н+ + 2е_) при условии, что индифферентный электролит не
содержит более легко окисляющегося иона, например С1“. Стандартный потен-
циал для полуреакции с участием воды равен +1,0 В отн. НКЭ, поэтому величи-
на максимального достижимого потенциала составляет в зависимости от pH
порядка+1 В отн. НКЭ. Величина максимально возможного отрицательного по-
тенциала лимитируется реакций восстановления иона водорода. Перенапряже-
ние водорода на платине низкое, поэтому проводить измерения можно только до
потенциалов порядка -0,1 В отн. НКЭ. При таких значениях потенциала кисло-
род не восстанавливается, поэтому удалять растворенный кислород не нужно,
если он не взаимодействует с компонентами анализируемого раствора.
В вольтамперометрии часто применяют графитовые электроды. Максима-
льно возможные положительные потенциалы для них примерно такие же, что и
для платинового электрода, а в катодной области с этими электродами можно
работать при несколько более отрицательных потенциалах за счет более высоко-
го перенапряжения водорода — до -1 В отн. НКЭ и даже ниже (в зависимости
от pH). Кислород при таких отрицательных значениях потенциалов электроакти-
вен, поэтому перед регистрацией вольтамперо- граммы его необходимо удалить
из раствора. Это удобно сделать, пропуская через раствор азот с помощью тон-
кого капилляра в течение 10-15 мин. Но перед этим азот пропускают через сосуд
с водой добиваясь его насыщения парами воды, чтобы не происходило испаре-
ния анализируемого раствора. По окончании удаления кислорода капилляр вы-
нимают из раствора и пропускают азот над его поверхностью во избежание
поглощения воздуха.
Важным преимуществом графитового электрода явлется то, что в отличие
от платинового электрода его поверхность не пассивируется за счет образования
оксидных пленок. Несмотря на то, что с графитовым электродом можно работать
при больших отрицательных потенциалах, чаще используют ртутный капающий
электрод (РКЭ) из-за лучшей воспроизводимости результатов. Это обусловлено
тем, что поверхность электрода постоянно обновляется — маленькие капли рту-
ти периодически вытекают из капилляра, присоединенного к резервуару со рту-
тью, и величина площади поверхности капли хорошо воспроизводится при
постоянстве высоты столба ртути над капилляром. В катодной области с ртут-
ным капающим электродом можно работать вплоть до -2В отн. НКЭ. Раздел
вольтамперометрии, в котором применяют ртутный капающий электрод назы-
вается полярографией.
Вольтамперометрию на твердых электродах широко применяют для изуче-
ния и определения веществ, окисляющихся при высоких положительных потен-
циалах, хотя эти электроды можно использовать и в случае легко окисляющихся
веществ. Однако, воспроизводимость результатов при работе с твердыми элект-
родами часто оказывается значительно ниже, чем при работе с ртутным капающим
электродом, в результате загрязнения поверхности и плохой воспроизводимости
величины ее площади.
610
ВОЛЬТАМПЕРОМЕТРИЯ И ЭЛЕКТРОХИМИЧЕСКИЕ СЕНСОРЫ
15.2. Амперометрические электроды: определение кислорода
Амперометрия — раздел вольтамперометрии, в котором при фиксированном
потенциале регистрируют протекающий через ячейку ток как функцию концен-
трации определяемого вещества. Такой принцип может быть положен в основу
работы электрохимических сенсоров, важным примером которых является кис-
лородный электрод.
Кислородный электрод состоит из тонкой полимерной пленки (например,
тефлоновой), нанесенной на поверхность платинового или золотого катода.
Пленка позволяет газам диффундировать к поверхности электрода, но она не-
проницаема для ионов раствора (рис. 15.3). Диффундирующий через мембрану
кислород восстанавливается на катоде, и через ячейку протекает ток. Необходи-
мый для протекания реакции восстановления кислорода потенциал прилагают
к цепи из индикаторного кислород-чувствительного электрода и электрода
сравнения, обычно хлорсеребряного. Между мембраной и стеклянной изоля-
цией помещают раствор или гель, содержащий электролит, для обеспечения
электрического контакта между индикаторным электродом и электродом срав-
нения.
Скорость диффузии кислорода к катоду пропорциональна парциальному
давлению кислорода в анализируемом растворе, поэтому катодный ток также за-
висит от парциального давления кислорода. Результаты считывают при атмо-
сферном давлении. Определению мешают галогены и другие газы (например,
SO2), восстанавливающиеся при заданном потенциале. Сероводород отравляет
поверхность электрода.
Для предварительной градуировки шкалы измерительного прибора исполь-
зуют образцы с известным содержанием кислорода, например, воздух, содержащий
предположительно 20,9% О2, или воду, насыщенную кислородом или воздухом.
При 37 °C на уровне моря (при РОг = 159 торр) миллилитр воды, насыщенной
воздухом, содержит 5,6 мкл О2, а миллилитр воды, насыщенной кислородом, —
Рис. 15.3
Кислородный электрод (с разрешения Arthur
Н. Thomas Company)
кольца
15.3. ЭЛЕКТРОХИМИЧЕСКИЕ СЕНСОРЫ: ХИМИЧЕСКИ МОДИФИЦИРОВАННЫЕ ЭЛЕКТРОДЫ
611
28 мкл О2. Подробно градуировка электрода, расчет Pq2 и концентрации кисло-
рода рассмотрены в [7].
Амперометрический анализатор кислорода часто применяют биохимики,
чтобы проследить за потреблением или выделением кислорода при изучении ки-
нетики биохимических и ферментативных реакций. Применяют его и в клиниче-
ских лабораториях для определения содержания ферментов или субстратов в
реакциях, протекающих с участием кислорода.
15.3. Электрохимические сенсоры:
химически модифицированные электроды
Как и потенциометрические электроды (гл. 13) амперометрические электроды
являются разновидностью электрохимических сенсоров. В последние годы много
внимания уделяют разработке электрохимических сенсоров с высокой селективно-
стью или чувствительностью. Такие характеристики присущи амперометрическим
сенсорам с химически модифицированной поверхностью электрода, которые
называют химически модифицированными электродами (ХМЭ).
Все химические сенсоры содержат трансдьюсер для преобразования от-
клика в сигнал удобный для детектирования (в амперометрических сенсорах —
ток) и химически селективный слой. Трансдьюсер может быть оптическим (на
основе оптического волокна), электрическим (потенциометрический, амперо-
метрический), термическим и др. В данной главе рассмотрены амперометриче-
ские трансдьюсеры.
Ферментные электроды для определения субстратов
Для достижения необходимой селективности при химической модификации
электродов часто применяют ферменты. В гл. 13 был приведен пример фермент-
ного потенциометрического электрода. Примером амперометрического фер-
ментного электрода является электрод для определения глюкозы, схематически
изображенный на рис. 15.4. Фермент глюкозооксидаза иммобилизован в слой
геля (например, акриламида), нанесенного на поверхность катода из платиновой
проволочки. Гель содержит также хлорид для осуществления электрического
контакта с кольцом из серебряной проволоки, покрытой хлоридом серебра, в ре-
зультате образуется электрохимическая ячейка. Фермент глюкозаооксидаза ка-
тализирует аэробное окисление глюкозы (см. гл. 22):
I ТТЛ глюкозооксидаза ТТ ГА
глюкоза + О2 + Н2О---------> глюконовая кислота + Н2О2 (15.5)
На платиновом электроде при значении потенциала порядка +0,6 В (отн. элект-
рода Ag I AgCl) протекает электрохимическое окисление Н2О2:
Н2О2 —> Н2О + Н+ + е
(15.6)
612
ВОЛЬТАМПЕРОМЕТРИЯ И ЭЛЕКТРОХИМИЧЕСКИЕ СЕНСОРЫ
РИС. 15.4. Амперометрический ферментный электрод
Глюкоза и растворенный в анализируемом растворе кислород диффундируют в
слой геля, где протекает их взаимодействие с образованием Н2О2, катализируе-
мое ферментом. Часть образовавшегося Н2О2 диффундирует к платиновому ка-
тоду, в результате окисления Н2О2 в ячейке протекает ток, при этом сила тока
пропорциональна концентрации глюкозы. В качестве альтернативного устрой-
ства можно использовать кислородный электрод Кларка, мембрана которого по-
крыта гелем, содержащим фермент глюкозооксидазу. Такой электрод применяют
для измерения расхода кислорода в процессе реакции.
Можно привести и другие примеры амперометрических ферментных элект-
родов, принцип действия которых основан на определении кислорода или пе-
роксида водорода: электрод для определения галактозы в крови (фермент
галактозаоксидаза), оксалата в моче (фермент оксалатоксидаза) и холестерина в
сыворотке крови (фермент холестеролоксидаза). Можно определять этанол по
реакции с кофактором .никотинамидадениндинуклеотидом (NAD+) в присутст-
вии фермента алкогольдегидрогеназы. В результате реакции NAD+ восстанав-
ливается до NADH, способного окисляться на электроде. Аналогичным образом
можно определять лактат в крови с помощью фермента лактатдегидрогеназы.
Каталитические электроды: редокс-медиаторы
Часто определяемый компонент восстанавливается или окисляется на электроде
необратимо (медленно), и для протекания электродной реакции необходим бо-
лее высокий потенциал по сравнению с величиной Е°. Попытки решения проб-
лемы, связанной с медленным переносом электронов, повлекли за собой
интенсивные исследования по созданию электрокатализаторов, которые мож-
но было бы ковалентно присоединить к поверхности электрода, хемосорбиро-
вать или внедрить в слой полимера. Принцип работы электрокаталитических
химически модифицированных электродов иллюстрирует рис. 15.5, где Red и
Ох — соответственно восстановленная и окисленная формы определяемого
компонента. Редокс-медиатор образует обратимую окислительно-восстановите-
льную пару и легко окисляется. Определяемый компонент быстро реагирует с
окисленной формой медиатора МОх с образованием MRed, которая тут же окис-
ляется на поверхности электрода. Слой редокс-медиатора катализирует элект-
рохимическую реакцию, снижая требуемый для ее протекания потенциал.
15.4. УЛЬТРАМИКРОЭЛЕКТРОДЫ
613
Рис. 15.5
Химически модифицированный электрод для
определения компонента, находящегося
в восстановленной форме (Red)
Электрод
Электрохимическая реакция протекает при значениях потенциалов, близких к
Е° медиатора. При небольшом приложенном извне потенциале помехи от дру-
гих электроактивных компонентов раствора, вносящих вклад в сигнал определя-
емого компонента, также будут небольшими. К электрохимическим медиаторам
относятся комплексы рутения, производные ферроцена и о-гидроксибензола.
Такие медиаторы, как метиленовый голубой, катализируют окисление Н2О2, и
электродная реакция протекает при значении потенциала порядка +0,2 В (отн.
электрода Ag I AgCl) вместо +0,6 В в отсутствие медиатора.
Иногда электроды покрывают защитным слоем для предовращения загряз-
нения поверхности электрода большими молекулами, например, белками. Так,
слой ацетата целлюлозы пропускает маленькие молекулы Н2О2, но не пропуска-
ет большие молекулы аскорбиновой кислоты, содержащиеся в биологических
жидкостях и окисляющиеся при том же потенциале. Защитный слой повышает
селективность и снижает химическое загрязнение электрода. Анионные мемб-
раны из нафиона отталкивают анионы, но позволяют катионам проникать к по-
верхности электрода.
15.4. Ультрамикроэлектроды
Амперометрические электроды диаметром порядка 5-30 мкм обладают рядом
преимуществ. Поскольку их размер меньше толщины диффузионного слоя, уве-
личивается скорость массопереноса. В итоге улучшается соотношение сиг-
нал/шум, и электрохимические измерения можно проводить в средах с высоким
сопротивлением, например, в неводных растворителях. Даже в случае непереме-
шиваемого раствора наблюдается четкая 5-образная зависимость силы тока от
потенциала, а не кривая с максимумом, поскольку исключено влияние толщины
диффузионного слоя. Для таких микроэлектродов предельный ток znp описы-
вается уравнением
znp = InFDCd
(15.7)
где п — число переносимых электронов, F— постоянная Фарадея, D — коэффи-
циент диффузии, С — концентрация и d — диаметр электрода.
Существуют различные способы конструирования ультрамикроэлектродов
[14]. Типичная конструкция показана на рис. 15.6. Электродом является микро-
диск, обычно изготовляемый из углеродного волокна. Такой электрод можно ис-
пользовать для изучения процессов окисления при положительных потенциалах
в растворах различного состава, например, для детектирования нейромедиатора
614
ВОЛЬТАМПЕРОМЕТРИЯ И ЭЛЕКТРОХИМИЧЕСКИЕ СЕНСОРЫ
Рис. 15.6
Схематичное изображение
ультрамикроэлектрода
Поперечный срез
в мозговом ликворе [см. обзор по применению микроэлектродов из углеродного
волокна — Т. Edmonds, Anal. Chim. Acta, 175 (1985) 1]. В результате электроднохи-
мических процессов на таких электродах протекают токи порядка нескольких на-
ноампер, поэтому для проведения измерений требуется потенциостат с высокой
чувствительностью.
Что мы узнали из этой главы?
• Электролитические ячейки — стр. 603
• Вольтамперные кривые — стр. 604
• Индифферентные электролиты — стр. 607
• Амперометрические электроды — стр. 610
• Химически модифицированные электроды — стр. 611
• Ультрамикроэлектроды — стр. 613
Вопросы
1 - Дайте определение следующих понятий: перенапряжение, омическое паде-
ние напряжения (iR).
2- Дайте определение следующих понятий: потенциал полуволны, деполяри-
затор, ртутный капающий электрод, предельный ток, вольтамперометрия.
ЗАДАЧИ
615
3- Назовите две причины необходимости использования индифферентного
электролита в вольтамперометрии.
4. В растворе, содержащем примерно 10-2 М Fe3+ и 10-5 М РЬ2+, требуется по-
лярографически определить содержание свинца. На ртутном капающем
электроде Fe3+ восстанавливается до Fe2+ при значениях потенциала от на-
чала поляризации электрода до —1,5 В отн. НКЭ, а при более отрицатель-
ных значениях потенциала — вместе с образовавшимся Fe2+ до Fe°. В этих
же условиях РЬ2+ восстанавливается при -0,4 В. Предложите схему поля-
рографического определения свинца.
5- Как комплексообразование влияет на вольтамперометрическое восстанов-
ление ионов металлов?
6- Что такое химически модифицированный электрод?
7- Какова роль электрокатализатора в химически модифицированном элект-
роде?
8- Назовите преимущества использования ультрамикроэлектродов.
Задачи
Вольтамперометрия/амперометрия
9- Предельный ток восстановления свинца в растворе с неизвестной концент-
рацией равен 5,60 мкА. После добавления к 10,0 мл этого раствора 1,00 мл
1,00 • 10-3 М раствора РЬ2+ ток увеличился до 12,2 мкА. Какова концентра-
ция свинца в анализируемом растворе?
10- На ртутном капающем электроде железо(Ш) при потенциалах ниже +0,4 В
отн. НКЭ восстанавливается до железа(П) и далее при -1,5 В — до металла.
Железо(П) также восстанавливается до металла. На полярограмме раство-
ра, содержащего Fe3+ и/или Fe2+, значение предельного тока при 0 В равно
12,5 мкА, а значение волны с £1/2 = -1,5 В предельного тока — 30,0 мкА.
Установите, что содержится в растворе — железо(Ш) и/или железо(П), и
рассчитайте отношение их концентраций.
Рекомендуемая литература
Общая
1 - J. J. Lingane, Electroanalytical Chemistry, 2nd ed. New York: Interscience, 1958.
Классика. Превосходное изложение основ.
2- J. Wang, Analytical Electrochemistry, 2nd ed. New York: John Wiley, 2000. Хо-
рошо использовать для ознакомления с современными электрохимически-
ми методами анализа.
3. A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Ap-
plications, 2nd ed. New York: Wiley, 2001.
616
ВОЛЬТАМПЕРОМЕТРИЯ И ЭЛЕКТРОХИМИЧЕСКИЕ СЕНСОРЫ
4- A. J. Bard and М. Stratmann, editors-in-chief, Encyclopedia of Electrochemistry,
10 volumes + index volume, New York: Wiley. Vol. 3, Instrumentation and
Electroanalytical Chemistry, P. Unwin, ed., 2003; Vol. 9, Bioelectro-chemistry,
G. S. Wilson, ed., 2002; Vol. 10, Modified Electrodes, I. Rubinstein and M. Fuji-
hira, eds., 2003.
Вольтамперометрия/амперометрия
5 - M. R. Smyth and J. G. Vos, Analytical Voltammetry. Amsterdam: Elsevier Scien-
ce, 1992.
6- R. N. Adams, Electrochemistry at Solid Electrodes, New York: Marcel Dekker,
1969. Классическое издание (уже пятое!).
7- М. A. Lessler and G. Р. Brierley, in D. Glick, ed., Methods of Biochemical Analy-
sis, Vol. 17. New York: Interscience, 1969, p. 1. Описание кислородного элек-
трода.
8. www.pineinst.com. — отличное руководство не 70 страницах по основам
вольтамперометрии и аппаратуре. Вы можете скачать пример описания
вольтамперометрического эксперимента с сайта компании Pine Instrument
Company.
Электрохимические сенсоры
9- R. W. Murray, А. Е. Ewing, and R. A. Durst, «Chemically Modified Electrodes.
Molecular Design for Electroanalysis», Anal. Chem., 59 (1987) 379A.
10- E. R. Reynolds and A. M. Yacynych, «Miniaturized Electrochemical Biosen-
sors», Am. Lab., March (1991) 19.
11 - R. I. Stefan, J. F. van Staden, and H. Y. Aboul-Enein, Electrochemical Sensors in
Bioanalysis, New York: Marcel Dekker, 2001.
12- D. Diamond, ed., Principles of Chemical and Biological Sensors, New York: Wi-
ley, 1998.
13. P. G. Edelman and J. Wang, eds., Biosensors and Chemical Sensors: Optimizing
Performance Through Polymeric Materials, ACS Symposium Series No. 487,
Washington, DC: American Chemical Society, 1992.
Ультрамикроэлектроды
14. M. Koppenol, J. B. Cooper, and A. M. Bond, «Tools and Procedures for Semiau-
tomated Production of Robust Microdisk Electrodes», A m. Lab., July (1994) 25.
Техника лабораторных работ
15- P. T. Kissinger and W. R. Heineman, Laboratory Techniques in Electroanalyti-
cal Chemistry, 2nd ed. New York: Marcel Dekker, 1996.
Оглавление
Об авторе 5
Предисловие 7
Список электронных таблиц, использованных в тексте 15
Глава 1
Задачи аналитической химии, или чем занимаются химики-аналитики 17
1.1. Что такое аналитическая наука?....................................17
1.2. Качественный и количественный анализ: какую информацию сообщает
нам каждый из них?.....................................................19
1.3. Мы начинаем процесс анализа.......................................22
1.4. Поверка методики: вы должны доказать, что она работает!...........34
1.5. Как определить размер пробы?......................................36
1.6. Некоторые полезные Интернет-страницы..............................36
Что мы узнали из этой главы? 38
Вопросы 38
Рекомендуемая литература 39
Глава 2
Основные инструменты и операции химического анализа 42
2.1. Лабораторный журнал — самый главный документ......................42
2.2. Лабораторные материалы и реагенты.................................45
2.3. Аналитические весы — необходимый инструмент.......................49
2.4. Мерная посуда.....................................................60
2.5. Приготовление стандартных растворов щелочей.......................76
2.6. Приготовление стандартных растворов кислот........................77
2.7. Другая аналитическая посуда и аппаратура: подготовка и обработка проб. ... 78
2.8. Прокаливание осадков в гравиметрическом анализе...................87
2.9. Отбор пробы: твердой, жидкой, газообразной........................88
2.10. Высушивание и растворение проб....................................91
2.11. Безопасность в лаборатории.......................................100
Что мы узнали из этой главы? 101
Вопросы 101
Задачи 102
Рекомендуемая литература 103
618
ОГЛАВЛЕНИЕ
Глава 3
Обработка данных и использование электронных таблиц в аналитической химии 107
3.1. Правильность и воспроизводимость — разные вещи...................107
3.2. Детерминированные (систематические) погрешности..................109
3.3. Недетерминированные (случайные) погрешности......................110
3.4. Значащие цифры: сколько требуется цифр?..........................111
3.5. Правила округления...............................................118
3.6. Способы выражения правильности...................................118
3.7. Стандартное отклонение — важнейшая статистическая величина.......119
3.8. Электронные таблицы..............................................124
3.9. Наложение погрешностей: это не простое суммирование..............130
3.10. Наложение погрешностей и значащие цифры..........................138
3.11. Контрольные карты................................................139
3.12. Доверительный интервал: насколько надежно значение?..............140
3.13. Тесты статистической значимости: есть ли различие между данными?.143
3.14. Отброс промахов: 0-тест..........................................151
3.15. Статистика малых массивов данных.................................154
3.16. Линейный метод наименьших квадратов: как провести прямую?........156
3.17. Коэффициент корреляции и коэффициент детерминации................162
3.18. Использование электронных таблиц для построения градуировочных
графиков...............................................................164
3.19. Расчет углового коэффициента, свободного члена и коэффициента
детерминации при помощи электронных таблиц.............................167
3.20. Программа ЛИНЕЙН (LINEST) для выполнения различных
статистических расчетов................................................168
3.21. Пакеты прикладных статистических программ........................169
3.22. Предел обнаружения: в аналитической химии нет понятия «нуль»!....170
3.23. Статистика пробоотбора: сколько брать проб, какого размера?......172
Что мы узнали из этой главы? 176
Вопросы 176
Задачи 176
Рекомендуемая литература 183
Глава 4
«Хорошая лабораторная практика»: обеспечение качества результатов анализа 186
4.1. Что такое «хорошая лабораторная практика»?.......................188
4.2. Поверка методики анализа.........................................189
4.3. Обеспечение качества результатов анализа: действительно ли методика
по-прежнему дает правильные результаты?...............................199
4.4. Аккредитация лабораторий.........................................201
4.5. Электронная документация и электронные подписи: нормы 21 CFR,
часть 11..............................................................201
ОГЛАВЛЕНИЕ 619
4.6. Некоторые официальные организации................................204
Что мы узнали из этой главы? 205
Вопросы 205
Задачи 206
Рекомендуемая литература 207
Глава 5
Стехиометрические расчеты - основной рабочий инструмент аналитика 209
5.1. Обзор основных понятий...........................................209
5.2. Как выражают концентрации растворов?.............................213
5.3. Выражение результатов анализа....................................223
5.4. Титриметрический анализ: как проводить стехиометрические расчеты?.232
5.5. Расчеты в титриметрии: использование молярностей.................236
5.6. Использование нормальностей — другой способ вычислений
в титриметрии.........................................................250
5.7. Титр — способ быстрого выполнения серийных вычислений............260
5.8. Массовые соотношения — они необходимы для расчетов в гравиметрии. ... 261
Что мы узнали из этой главы? 264
Вопросы 265
Задачи 265
Рекомендуемая литература 271
Глава 6
Основные положения теории химического равновесия 272
6.1. Химические реакции: понятие скорости реакции.....................272
6.2. Типы равновесий..................................................275
6.3. Свободная энергия Гиббса и константа равновесия..................275
6.4. Прицип Ле Шателье................................................276
6.5. Влияние температуры на константу равновесия .....................277
6.6. Влияние давления на положение равновесия.........................277
6.7. Влияние концентрации на положение равновесия.....................277
6.8. Катализаторы ....................................................278
6.9. Полнота протекания реакций.......................................278
6.10. Константы равновесия процессов диссоциации и ассоциации: слабые
электролиты и осадки...................................................279
6.11. Расчеты с использованием констант равновесия: каковы равновесные
концентрации?..........................................................280
6.12. Влияние одноименных ионов: смещение равновесия...................289
6.13. Общий подход к расчетам химических равновесий: как решить любую
задачу.................................................................290
6.14. Гетерогенные равновесия: концентрации твердых веществ
не записываем..........................................................299
620
ОГЛАВЛЕНИЕ
6.15. Активности и коэффициенты активности: концентрации —
это еще не все!.......................................................299
6.16. Влияние посторонних ионов: термодинамическая константа равновесия
и коэффициенты активности.............................................305
Что мы узнали из этой главы? 307
Задачи 307
Рекомендуемая литература 310
Глава 7
Кислотно-основное равновесие 311
7.1. Кислотно-основные теории........................................311
7.2. Кислотно-основные равновесия в воде.............................314
7.3. Шкала pH........................................................317
7.4. Значения pH при повышенных температурах: pH крови...............322
7.5. Слабые кислоты и основания: как рассчитать pH?..................323
7.6. Соли слабых кислот и оснований: их растворы не являются
нейтральными.........................................................326
7.7. Буферные растворы: поддерживают значение pH постоянным
[или примерно постоянным]............................................330
7.8. Многоосновные кислоты и их соли.................................339
7.9. Физиологические буферные растворы: они поддерживают
жизнедеятельность....................................................353
7.10. Буферы для биологических и клинических измерений.................355
7.11. Влияние посторонних ионов на поведение кислот и оснований: К° и Kf —
изменение pH при введении солей.......................................358
7.12. Концентрационно-логарифмические диаграммы: как изобразить
изменения концентраций в широком диапазоне............................359
Что мы узнали из этой главы? 366
Вопросы 367
Задачи 367
Рекомендуемая литература 371
Глава 8
Кислотно-основные титрования 373
8.1. Сильные кислоты и сильные основания: самый простой вариант
титрования............................................................373
8.2. Установление конечной точки титрования: индикаторы...............378
8.3. Стандартные растворы кислот и оснований..........................381
8.4. Титрование слабых кислот сильными основаниями: это немного сложнее... 381
8.5. Титрование слабых оснований сильными кислотами...................388
8.6. Титрование карбоната натрия: двухступенчатое основание...........389
8.7. Титрование многоосновных кислот..................................393
ОГЛАВЛЕНИЕ 621
8.8. Смеси кислот и смеси оснований...................................396
8.9. Титрование аминокислот: они являются и кислотами, и основаниями..398
8.10. Метод Кьельдаля: определение белков..............................400
Что мы узнали из этой главы? 403
Вопросы 403
Задачи 404
Рекомендуемая литература 407
Глава 9
Реакции комплексообразования и комплексометрическое титрование 408
9.1. Константа устойчивости — мера устойчивости комплекса.............408
9.2. Хелаты. ЭДТА — самый распространенный титрант для определения
ионов металлов........................................................412
9.3. Кривые титрования ионов металлов раствором ЭДТА..................420
9.4. Индикация конечной точки. Металлоиндикаторы — хелатообразующие
реагенты..............................................................422
9.5. Другие области применения комплексных соединений.................426
9.6. Доли отдельных форм многолигандных комплексов. 0-коэффициенты:
каково содержание каждой формы?.......................................426
Что мы узнали из этой главы? 429
Вопросы 429
Задачи 429
Рекомендуемая литература 432
Глава 10
Гравиметрический анализ и равновесие осадок-раствор 433
10.1. Как успешно провести гравиметрический анализ?....................433
10.2. Расчеты в гравиметрии: сколько аналита присутствует в пробе?.....443
10.3. Примеры гравиметрических определений.............................448
10.4. Органические осадители...........................................448
10.5. Равновесие осадок—раствор: произведение растворимости............449
10.6. Влияние посторонних ионов на растворимость: термодинамическое
произведение растворимости и коэффициенты активности...................457
Что мы узнали из этой главы? 461
Вопросы 461
Задачи 461
Рекомендуемая литература 464
Глава 11
Реакции осаждения и осадительное титрование 466
11.1. Влияние кислотности на растворимость осадков: условное произведение
растворимости...........................................................466
622
ОГЛАВЛЕНИЕ
11.2. Расчет сложных равновесий с использованием материального баланса.469
11.3. Влияние комплексообразования на растворимость: условное произведение
растворимости.........................................................473
11.4. Осадительное титрование.........................................475
Что мы узнали из этой главы? 483
Вопросы 483
Задачи 484
Рекомендуемая литература 485
Глава 12
Электрохимические ячейки и электродные потенциалы 486
12.1. Что такое окислительно-восстановительные реакции?...............486
12.2. Электрохимические ячейки:
какие ячейки используют аналитики-электрохимики?......................487
12.3. Уравнение Нернста: зависимость потенциала от концентрации ......494
12.4. Формальный потенциал: используйте его в нестандартных условиях...499
12.5. Ограничения при использовании электродных потенциалов...........501
Что мы узнали из этой главы? 501
Вопросы 501
Задачи 502
Рекомендуемая литература 503
Глава 13
Электроды для потенциометрии и потенциометрия 504
13.1. Металлические электроды и определение катионов .................504
13.2. Электроды металл - соль металла для определения анионов.........506
13.3. Окислительно-восстановительные электроды — инертные металлы......508
13.4. Гальванические элементы без жидкостного соединения — ячейки,
дающие наиболее точные результаты.....................................511
13.5. Гальванические элементы с жидкостным соединением: их используют
на практике...........................................................512
13.6. Электроды сравнения: насыщенный каломельный электрод............515
13.7. Измерение потенциала ...........................................518
13.8. Определение концентрации по данным потенциометрических измерений ... 520
13.9. Неэлиминированный потенциал жидкостного соединения.
Почему он должен быть минимальным?....................................521
13.10. Точность результатов прямых потенциометрических определений
активности: зависимость от погрешности измерения потенциала.............521
13.11. Стеклянный электрод для измерения pH — рабочий инструмент химика.... 522
13.12. Стандартные буферные растворы для градуировки рН-электродов......530
13.13. Точность измерения pH............................................531
13.14. Использование pH-метра: как он работает? ........................532
ОГЛАВЛЕНИЕ
623
13.15. Измерение pH крови: важно контролировать температуру..............535
13.16. Измерение pH в неводных растворителях.............................536
13.17. Ионселективные электроды..........................................536
13.18. Ионселективные полевые транзисторы................................554
Что мы узнали из этой главы? 555
Вопросы 555
Задачи 555
Рекомендуемая литература 559
Глава 14
Окислительно-восстановительное и потенциометрическое титрование 562
14.1. Уравнивание окислительно-восстановительных реакций...............562
14.2. Расчет констант равновесия реакций: это необходимо для вычисления
потенциала в точке эквивалентности.....................................563
14.3. Расчет кривых окислительно-восстановительного титрования.........566
14.4. Визуальное обнаружение конечной точки титрования.................571
14.5. Титрование с использованием иода — иодиметрия и иодометрия.......574
14.6. Использование других титрантов-окислителей ......................581
14.7. Использование других титрантов-восстановителей...................583
14.8. Приготовление раствора для титрования: предварительное
восстановление или окисление аналита...................................584
14.9. Потенциометрическое титрование (косвенная потенциометрия)........586
Что мы узнали из этой главы? 598
Вопросы 598
Задачи 599
Рекомендуемая литература 601
Глава 15
Вольтамперометрия и электрохимические сенсоры 602
15.1. Вольтамперометрия................................................603
15.2. Амперометрические электроды: определение кислорода...............610
15.3. Электрохимические сенсоры: химически модифицированные электроды.... 611
15.4. Ультрамикроэлектроды.............................................613
Что мы узнали из этой главы? 614
Вопросы 614
Задачи 615
Рекомендуемая литература 615
Кислотно-основные индикаторы
Кислотно-основной индикатор
Интервал перехода pH
Крезоловый красный розовый 0,2
л^-Крезоловый пурпурный красный 1,2
Тимоловый синий красный 1,2
и-Ксиленоловый синий красный 1,2
2,2Г,2",4,4'-Пентаметокситрифенилкарбинол красный 1,2
2,4-Динитрофенол бесцветный 2,8
4-Диметиламиноазобензол красный 2,9
Бромхлорфеноловый синий желтый 3,0
Бромфеноловый синий желтый 3,0
Метиловый оранжевый красный 3,1
Бромкрезоловый зеленый желтый 3,8
2,5-Динитрофенол бесцветный 4,0
Ализаринсульфокислота (натриевая соль) желтый 4,3
Метиловый красный красный 4,4
Метиловый красный (натриевая соль) красный 4,4
Хлорфеноловый красный желтый 4,8
Гематоксилин желтый 5,0
Лакмус экстра, чистый красный 5,0
1,8 желтый
2,8 желтый
2,8 желтый
2,8 желтый
3,2 бесцветный
4,7 желтый
4,0 желто-оранжевый
4,6 пурпурный
4,6 пурпурный
4,4 желто-оранжевый
5,4 синий
5,8 желтый
6,3 фиолетовый
6,2 желто-оранжевый
6,2 желто-оранжевый
6,4 пурпурный
7,2 фиолетовый
8,0 синий
Бромфеноловый красный оранжево-желтый 5,2
Бромкрезоловый пурпурный желтый 5,2
4-Нитрофенол бесцветный 5,4
Бромксиленовый синий желтый 5,7
Ализарин желтый 5,8
Бромтимоловый синий желтый 6,0
Феноловый красный желтый 6,4
3-Нитрофенол бесцветный 6,6
Нейтральный красный голубовато-красный 6,8
4,5,6,7-Тетрабромфенолфталеин бесцветный 7,0
Крезоловый красный оранжевый 7,0
1 -Нафтол фталеин светло-коричневый 7,1
ж-Крезоловый пурпурный желтый 7,4
Тимоловый синий желтый 8,0
и-Ксиленоловый синий желтый 8,0
Фенолфталеин бесцветный 8,2
Тимолфталеин бесцветный 9,3
Ализариновый желтый GG светло-желтый 10,2
Эпсилон синий оранжевый 11,6
6,8 пурпурный
6,8 пурпурный
7,5 желтый
7,4 синий
7,2 красный
7,6 синий
8,2 красный
8,6 желто-оранжевый
8,0 оранжево-желый
8,0 пурпурный
8,8 пурпурный
8,3 сине-зеленый
9,0 пурпурный
9,6 синий
9,6 синий
9,8 красно-фиолетовый
10,5 синий
12,1 коричневато-желтый
13,0 фиолетовый
По данным pH Indicators, Е. Merck and Со.
Квалификация химических реактивов
Квалификация Степень чистоты Примечания
Технический (technical, commercial) Неизвестна Можно использовать только для приготовления моющих растворов
Химически чистый (С.Р., chemically pure) Выше, чем у «технического», но также неизвестна
Фармакопейный США (U.S.P.) Удовлетворяет минимальным стандартам Соответствует нормам безопасности фармакопеи США с точки зрения содержания опасных для здоровья примесей
Реагент Американского химического общества (A.C.S. reagent) Высокая Соответствует минимальному набору требований, установленных комитетом по химическим реагентам Американского химического общества
Первичный стандарт (Primary standard) Наивысшая Необходимы для приготовления первичных стандартных растворов для обеспечения высокой точности титриметрического анализа
Концентрации имеющихся в продаже кислот и оснований
квалификации ч. д. а.*
Концентрация
Отн. молекулярная -------------------------------------- Плотность при
Реагент масса моль/л % масс. 20 °C (г/см3)
H2SO4 98,08 17,6 94,0 1,831
НС1О4 100,50 11,6 70,0 1,668
НС1 36,46 12,4 38,0 1,188
HNO3 63,01 15,4 69,0 1,409
Н3РО4 98,00 14,7 85,0 1,689
нс2н3о2 60,05 17,4 99,5 1,051
NH3 17,03 14,8 28,0 0,898
*У казанные концентрации — приблизительные, их нельзя использовать в расчетах при приготовлении стан-
дартных растворов.
Характеристики точности мерной посуды (класс А) согласно Национальному
институту стандартов и технологий США (NIST)*
Емкость, мл (включительно и менее) Пределы погрешности, мл
Мерные колбы Пипетки Бюретки
1000 ±0,30
500 ±0,15
100 ±0,08 ±0,08 ±0,10
50 ±0,05 ±0,05 ±0,05
25 ±0,03 ±0,03 ±0,03
10 ±0,02 ±0,02 ±0,02
5 ±0,02 ±0,01 ±0,01
2 ± 0,006
* Этим требованиям удовлетворяет продукция класса А фирм Coming Pyrex и Kimball KINAX.
Гэри Кристиан - автор более 300 научных статей и пяти книг, вклю-
чая учебники по аналитической химии. Первое издание этой книги
вышлов 1971 г Русское издание соответствует 6-му американскому
изданию, которое служит основным учебником во многих универ-
ситетах США.
Кристиан награжден премией отделения аналитической химии
Американского химического общества за выдающиеся достижения
в области преподавания, высшей наградой Американского химиче-
ского общества в области аналитической химии - премией Фише-
ра, а также другими почетными наградами.
Он возглавлял подготовку программы экзаменов по аналитической
химии в Американском химическом обществе, был составителем
программы тестирования по химии в рамках единого теста GRE,
входил в рабочую группу, составлявшую задания для Международ-
ной химической олимпиады.
Этот учебник предназначен для студентов вузов. Он послужит хоро-
шим дополнением отечественных учебных изданий.