/



























































































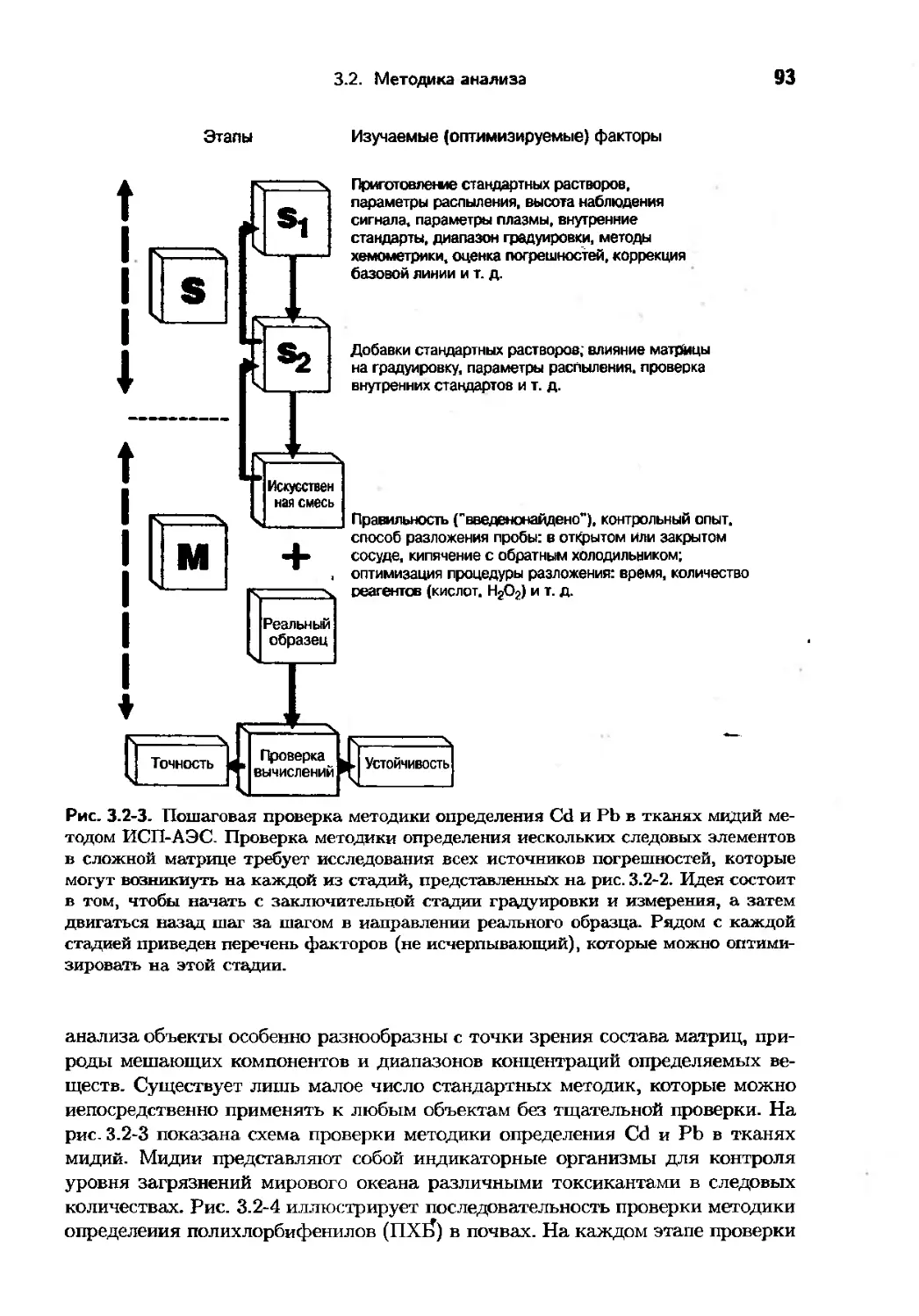

















































































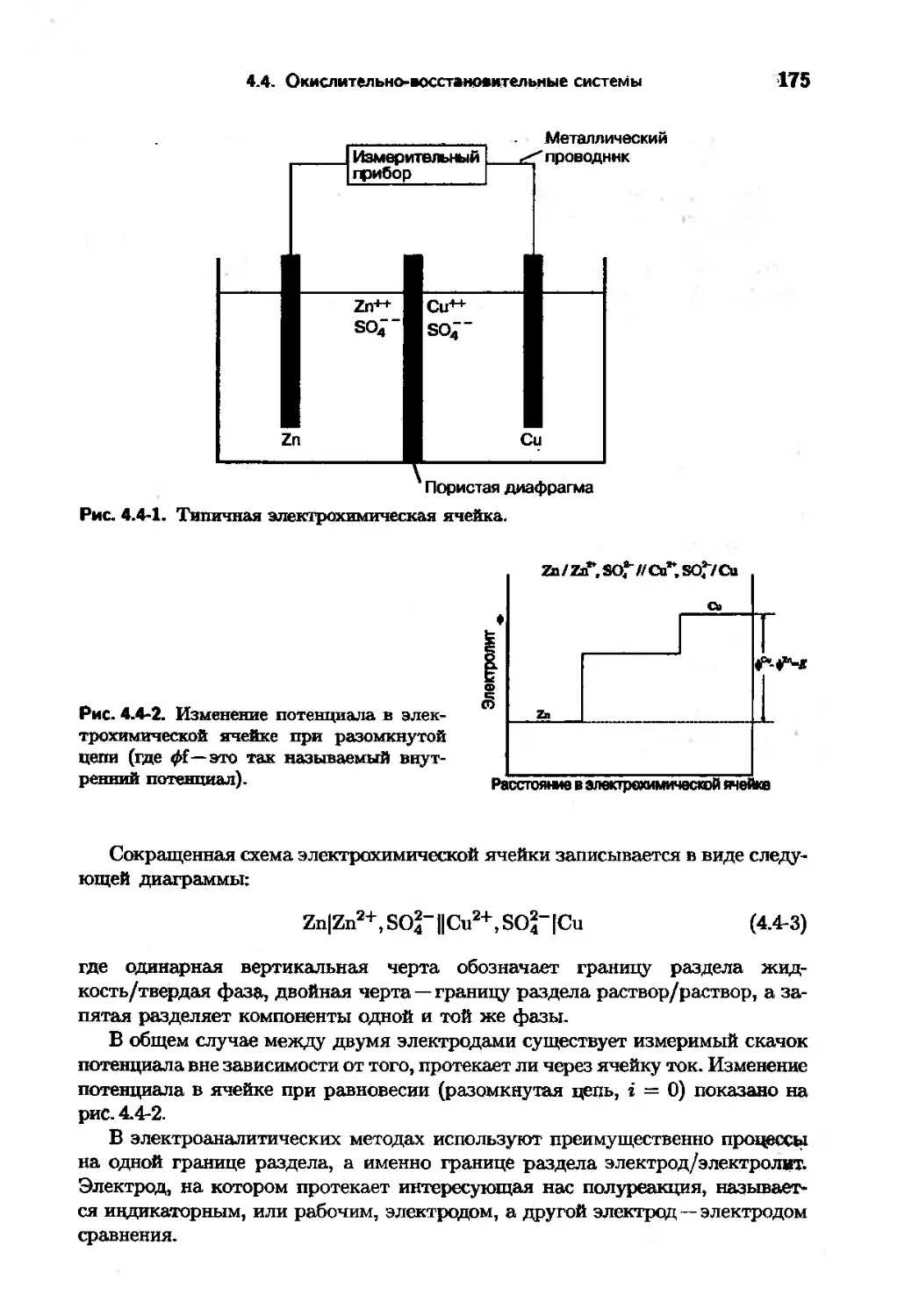














































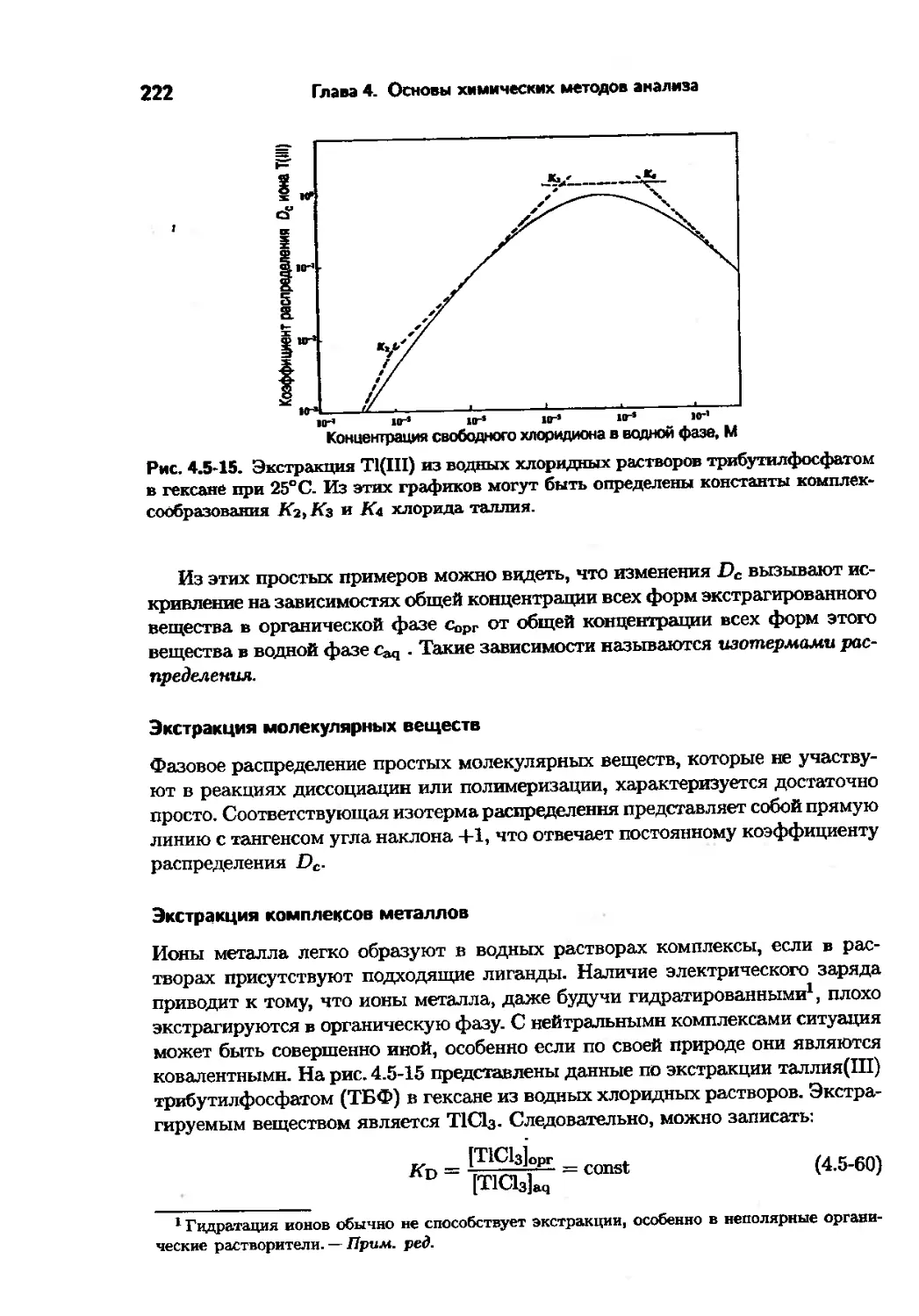













































































































































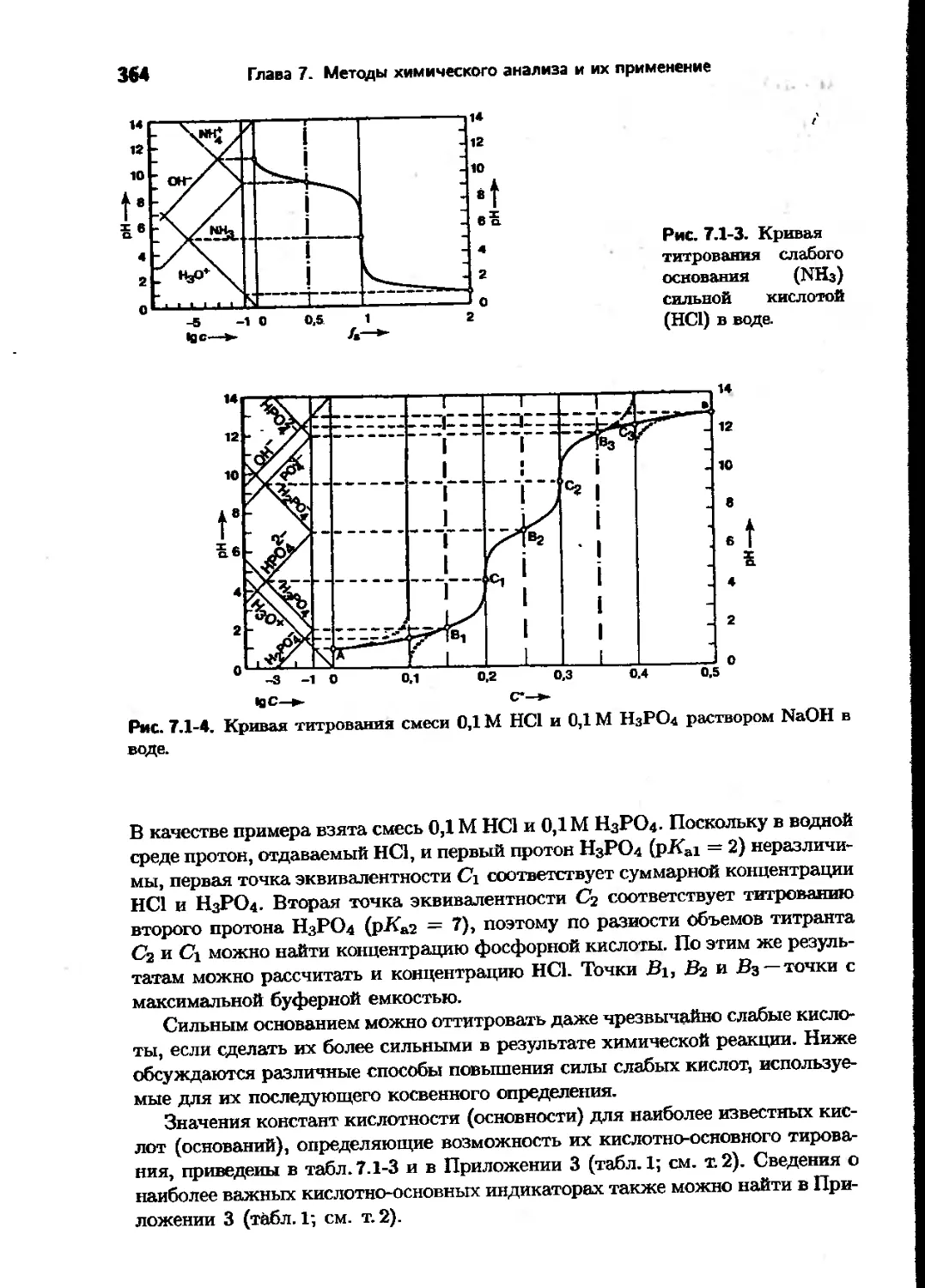























































































































































































































































Text
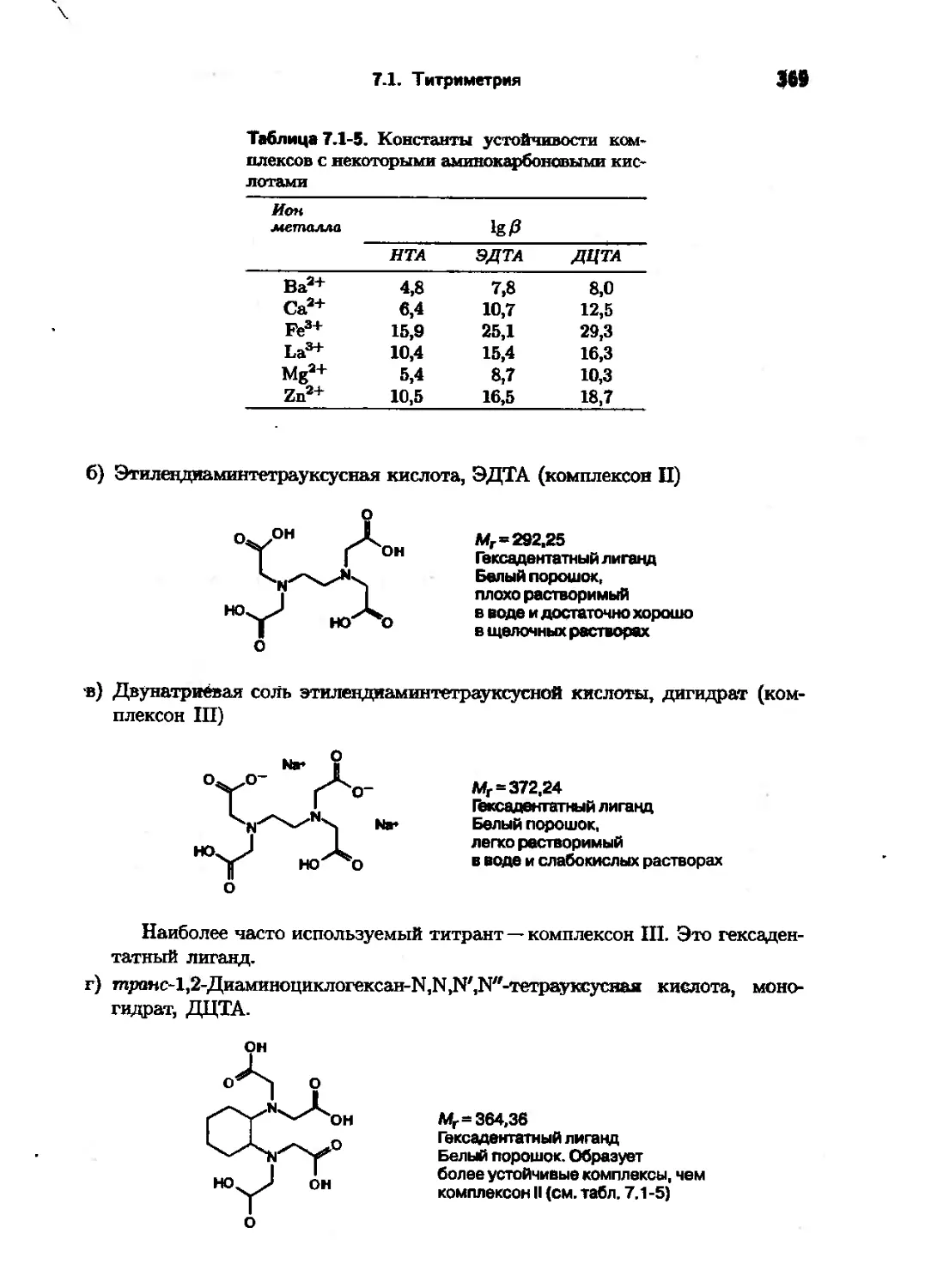
Analytical Chemistry
The Approved Text to the FECS Curriculum ’ Analytical Chemistry
Edited by R. Kellner, J.-M. Mermet, M. Otto, H. M. Widmer
CONTRIBUTORS
KARL CAMMANN, Institute for Chemical and Biochemical, Sensor Research, Munster, Germany
(Section 1.1)
KEIICHIRO FUWA, Tokyo, Japan (Section 1.2)
JEANETTE G. GRASSELU, Ohio University, Department of Chemistry, USA (Section 1.3)
D. BERNARD GRIEPINK, CEC, Brussels, Belgium (Chapter 3)
ELIZABETH A. H. HALL, Institute of Biotechnology, University of Cambridge, Cambridge, United
Kingdom (Sections 7.8, 7.9)
ELO H. HANSEN, Departroent of Chemistry, Technical University of Denmark, Lengby, Denmark
(Section 7.4)
ROBERT A KELLNER, Institute Of Analytical Chemistry, Technical University of Vienna, Vienna, Austria (Sections 1.1, 7.1, 7.2, 7.6)
WILLEM E. VAN DER UNDEN, Faculty of Chemical Technology, University of Twente, Enschede, The Netherlands (Sections 2.1-2.3)
EDDIE A MAIER, European Commission, Standard Measurements and Testing Programme (MO75), Brussels, Belgium (Chapter 3)
LAURI NIINISTO, Helsinki University of Technology, Laboratory of Inogranio and Analytical
Chemistry, Espoo, Hnland (Section 7.5)
MATTHIAS OTTO, Institute of Analytical Chemistry,- Freiberg University of Mining and Technology, Freiberg, Germany (Sections 5.1-5.5, 7.7)
DOLORES PfeREZ-BENDITO, Department of Analytical Chemistry, University of C<5rdoba,
Cdrdoba, Spain (Chapter 6)
KLARA TOTH, Institute of General and Analytical Chemistry, Technical University of Budapest, Hungary (Sections 4.4, 7.3)
H. MICHAEL WIOMER, Ciba-Geigy Ltd., Basel, Switzerland (Sections 4.1-4.3, 4.5, 5.8, 7.1, 7.2)
PIER GIORGIO ZAMBONIN, Department of Chemistry, Campus University, Bari, Italy (Section 2.4)
WILEY-VCH
Weinheim • New York • Chichester Brisbane • Singapore • Toronto
''"ЛУЧШИЙ"' ЗАРУБЕЖНЫЙ .УЧЕБНИК .
АНАЛИТИЧЕСКАЯ ХИМИЯ
ПРОБЛЕМЫ И ПОДХОДЫ
В двух томах
Том 1
Редакторы
Р. Кельнер, Ж.-М. Мерме, М. Отто, Г. М. Видмер
Перевод с английского канд. хим. наук А. Г. Борзенко, канд. хим. наук А. В. Гармаша, канд. хим. наук А. А. Горбатенко, канд. хим. наук М. А. Проскурнина, канд. хим. наук Г. В. Прохоровой, канд. хим. наук А. И. Элефертова
под редакцией академика Ю. А. Золотова
«Мир» «АСТ» МОСКВА 2004
УДК 548
БВК 24.4
А64
IS»N 5-17-mO6-l (♦#».)
Учей&£м^
Аналжтпеская химия. Проблемы и подходы: В 2 т: Пер. AM с Л Кедьвера. Ж.-М. Мерме,
М. dtrm.Bt. NUF ф!ир»:(Х)®%Йэдательст»о ACT»,
2004.—НРчадйЛ вйрувьмный учебник).'* • '
Т1. — 608 слил. _fi >т
ISBN 5-ОЗ-0ОЗБвО-*С#й*». ft 1) ,i*
ISBN 5-03-003550- К«Мф»): >
ISBN 3-527-28881-3 (ыгл.)
ISBN 5-17-019774-8 («ACT». T. 1)
написанное коллективом
преподавателе* и авторитетных ученых-аналитиков из ведущих университетов и научных центров Европы. рекомендована в качестве основного учебника по аналитической: химий в европейских вузах. В русском издании выходит в двух томах.
В т. 2 включены части III-V и приложения справочного характера: ч. Ш посвящена физико-химическим методам анализа; ч. IV - методам хемометрики; в ч. V рассмотрены интегрированные аналитические системы, применяемые в гибридных методах и в производственном анализе.
Для студентов университетов, Технологических и педагогических вузов.
УДК 543
ББК24.4
химии
Федеральная целевая программа «Культура России» (подпрограмма «Поддержка полиграфия книгоиздания Россия»)
Издание осуществлено прж финансовой поддержке Российского фонда фуждамвятадьпм яесаедонаиий во проекту М 00-03-46012 Издание впущено В свет при участий химического факультета Московского гвсуддр^г»еМЯогоукнн«ренгота мм. М.В. Ломоносова
Редакторы Р. Кельнер,Ж.-$4, Мерме, М. Ono, М. Видмер Зав. редакцией каяд. хим. наук Т.Й. Почкаева. Ведущий редактор И.С. Беленькая.
Художник М;М. Иванов
Оригинал-макет подготовлен В.Н. Цлаф в пакете
Подписано к печати Dfi.12.2003 г. Формат 70x100/16
Печать офсетная. Объем 22,75 бум. л. Уел. йен. л. 1Й),15. Уч.-изд. д. 45,90 Саиипрно-зоцдешюлогичжжое ваключедие М 77.9в.02.9бЗ.Д.008286.12.02 от О0.12ЖО2 г. Общероссийский классификатор продукции ОК-ООб-98, том 2; 953005 - литература учебная Изд. М 3/9887. Тираж 5 000 нкз. (1-й завод - SbOO нкз.) Заказ J4 5520
Издательства «Мир» Мднястеретеа РФ Во далям печати, телерадиовещания и средств массовых коммуникаций 107996, ГСП-6, Москва, 1-й Рижский пер., 2 ООО «Издательство АСТ», 667000, Республика Тыва, г. Кызыл, ул. Кочетова, д. 28 Диапозитивы изготовлены в издательстве «Мир»
Отпечатано с готовых диапозитивов во ФГУП ИПК «Ульяновский Дом печати». 432980, г. Ульяновск, ул. Гончарова, 14
ISBN 5-03-003560-5 («Мир*. Т. 1)
ISBN 5-03-003559-1 («Мир»)
ISBN 3-527-28881-3 (англ.)
ISBN 5-17-019774-8 («ACT*. Т. 1)
ISBN 5-17-18406-1 («АСТ»)
© 1998 WUey-VCH Verlag GmbH Originally published in the English language by Wiley-VCH Verlag GmbH, Pappelallee 8m 11-69469 Weinheim, Federal Republic of Germany, under the title «Keller, Mermet, Otto, Widmer: Analytical Chemistry»
© перевод и». русский язык, оформление «Мир», 2003
ПРЕДИСЛОВИЕ РЕДАКТОРА ПЕРЕВОДА
Бесконечные дискуссии о том, что такое аналитическая химия, хорошо известны научной общественности. Часто можно встретить утверждение, что аналитическая химия — это наука об определении состава веществ и материалов. Встречается и такое: аналитическая химия—это то, чем занимаются аналитики. Но отбросив все курьезы, аналитическую химию, строго говоря, можно определить как науку о принципах, методах и средствах определения химического состава и строения химических соединений, веществ и материалов. В книге вы найдете несколько иное определение, но это понятно: Ведь аналитическая химия не только быстро развивается, но и меняется в своих методологических основах. Куда существеннее понять и принять, что эта наука важная, живая н интересная.
Аналитическая химия как область науки имеет мощный фантастический1 по объему фундамент в виде практических работ по анализу и контролю резальных, всем нужных объектов. Анализ крови и мочи; контроль производства лекарств; контроль качества и безопасности пищевых продуктов; анализ воды, которую мы пьем и в которой купаемся; оценка степени чистоты воздуха; анализ почв; быстрое обнаружение взрывчатых веществ, ядов и наркотиков; анализ геологических объектов, например при разведке полезных ископаемых; проверка марки бензина—да где только не делаются химические анализы! Сам этот, далеко не полный перечень химикоаналитических объектов говорит многое о чрезвычайной важности аналитических служб и науки, которая эти службы обеспечивает идеями, методами, приборами, реактивами, способами обработки результатов и т. д.
Поэтому изучать аналитическую химию необходимо при подготовке самых разных специалистов и, разумеется, не только химиков. Совершенно очевидно, что и фармацевт, и агрохимик, и эколог, и военный химик, и геохимик — все эти специалисты должны понимать основы химического анализа, иметь соответствующие практические навыки. Для обучения необходимы учебники, причем не только глубоко излагающие современное состояние науки и тщательно продуманные с методической точки зрения. Учебники могут быть разнообразными в этом отношении, поскольку требования к уровню обучения для разных специальностей совершенно различаются.
Перед нами новое руководство по аналитической химии иностранных авторов; оно солидно, объемисто. История создания этого учебника необычна. Как правило, книга, призванная служить учебным пособием или учебником, созда
6
Предисловие редактора перевода
ется одним автором или же таковыми выступают два-три единомышленника; подчас это может быть результатом работы большого авторского коллектива— преподавателей н сотрудников одной кафедры или одного института и тогда отдельные главы или разделы готовят соответствующие специалисты, а книга в целом обобщает, как правило, многолетний методический опыт целого коллектива. Так, например, иаписан недавно вышедший двумя изданиями двухтомник < Основы аналитической химии», над которым работала кафедра аналитической химии МГУ им. М. В. Ломоносова1. Но перед вами сейчас учебник, авторы которого—ученые и одновременно преподаватели более чем из десяти стран! Согласимся, что такой подход—редкость. Тем неменее даже четыре редактора этого английского издания представляют четыре разные страны.
Дело в том, что Европа интегрируется; введение единой валюты Европейского Союза ярко демонстрирует эту интеграцию, и возможно это, не последний пример. Химики стран Европы тоже сотрудничают между собой, в том числе в рамках Федерации европейских химических обществ (ФЕХО), которая в числе прочих имеет Отделение аналитической химии (ОАХ). Одна из задач О АХ—совершенствование преподавания аналитической химии. Этой цели служат постоянно организуемые рабочие встречи, секции на крупных форумах, симпозиумы или специальные конференции по вопросам преподавания. В какой-то момент появилась идея разработать единую программу по аналитической химии (Eurocurriculum), которую можно было бы рекомендовать университетам разных европейских стран. Эта задача была решена. Такая весьма обширная и современная по охвату материала н глубине его изложения программа отражена в этом оригинальном учебнике (не один учебник из существовавших и применяемых в учебном процессе не годился для поставленной задачи). Создание этого учебника курировала специально сформированная группа редакторов-организаторов, которые и подобрали авторский коллектив (вполне естественно, многонациональный), выработали концепцию и дали авторам необходимые указания, а затем отредактировали полученный материал.
Когда все это было осуществлено, такой учебник — весьма нетрадиционный и почти всеохватный — появился в 1998 году. Сейчас он выходит в русском переводе. Конечно, за те четыре-пять лет, что прошли после написания книги, в аналитической химии кое-что обновилось, но учебник есть учебник, в него положено включать устоявшееся и существенное. Четыре-пять лет не столь большой срок, чтобы учебный материал нужно было «перетряхивать».
Ну а теперь собственно о содержании учебника. Прежде всего он состоит из объемных глав, что обычно не бывало в других учебниках (часто конкретные сведения излагались весьма кратко). Например, имеется глава о сенсорах, ядерном магнитном резонансе, рентгеноструктурном анализе н других методах структурных исследований, об анализе поверхности. Весьма много внимания уделено масс-спектрометрин, рентгенофлуоресцентному анализу, математическим методам (хемометрике). В той или иной степени описаны почти все недавно зародившиеся перспективные направления аналитической химии, скажем, аналитические схемы и приборы на микрочипах.
М.: Высшая школа, 1997 и 2001.
Предисловие редактора перевода 7
Материал в больший»^ случаев представлен на весьма высоком научном уровне и методически в общем и целом очень неплохо. Правда* в отдельных главах изложение излишне пространное, детализированное; иапомин&б-щее большие научные обзоры, написанные для специалисте®. Можно мягко покритиковать раздел о биосенсорах, который во многом не соответствует высокому уровню учебника. К сожалению, в таком большом труде, конечно же, не удалось избежать некоторых ошибок. Те, что были обнаружены при переводе и редактировании, исправлены, причем в очевидных случаях даже без каких-либо указаний, в иных даны объяснения в примечаниях.
Перевод выполнен следующими сотрудниками кафедры аналитической химии Московского университета им. М. В. Ломоносова: А. Г. Борзенко (разд. 4.1, 4.5, гл. 15, 16, приложение), А.В.Гармаш (предисловие; гл. 1-3, гл. 12, 13, приложение), А. А. Горбатенко (разд. 4.3, 7.4-7.Э, гл. 8, приложение), Е.В.Проскурнина (гл. 9, 10), М. А. Проскурнин (гл.6, 11), Г. В. Прохорова (разд. 4.2, 4.4, 7.1-7.3), А. И. Элефтеров (гл. 5, 14).
Ю.А: Золотов
ПРЕДИСЛОВИЕ ПРЕЗИДЕНТА ФЕДЕРАЦИИ ЕВРОПЕЙСКИХ ХИМИЧЕСКИХ ОБЩЕСТВ
Среди всех естественнонаучных дисциплин, вероятно, именно аналитическая химия претерпела самое бурное развитие в последние десятилетия, в соответствии с чем иеймоверно возросло число аналитических методов, степень их сложности и масштабы использования. Это породило проблемы в' преподавании аналитической химии: что следует включать в общий учебный курс по этому предмету и в каком объеме?
С момента своего образования четверть века назад Федерация европейских химических обществ (ФЕХО, англ. FECS) проявляла пристальный интерес к химическому образованию и преподаванию аналитической химии; обе эти области представлены отдельными подразделениями в структуре ФЕХО. После обширной подготовительной работы Отделение аналитической химии фЕХО (ОАХ ФЕХО, англ. DAC/FESC) разработало концепцию типового курса аналитической химии, которая была благожелательно встречена в Европе и Америке. Следующим шагом явилось создание данного учебника, который может служить средством распространения этого курса во всем мире. _
Как считал И. М. Кольтгоф, один из великих химиков-аналитиков первооткрывателей в аналитической химии, стеория указывает путь решения задач, а решает эксперимент». Данный учебник поможет привить студенту аналитическое мышление, для чего приводятся необходимые теоретические основы, детали эксперимента и аппаратуры н дается обзор реальных приложений аналитической химии. Но главная цель этой книги состоит в том, чтобы научить студентов применять полученные знания для решения реальных проблем.
Быстрый технологический прогресс и возрастающее внимание общества к проблемам окружающей среды дают уверенность в том, что число аналитических задач и их сложность будут, безусловно, возрастать. Позвольте выразить надежду, что эта своевременная книга внесет вклад в образование и обучение будущих химиков-аналитиков с тем, чтобы общество располагало достаточным числом квалифицированных специалистов, способных решать аналитические проблемы и достойно ответить на эти вызовы времени.
Сентябрь 1997 г.
Лаури Нийнисто
ПРЕДИСЛОВИЕ ПРЕДСЕДАТЕЛЕЙ ОТДЕЛЕНИЯ АНАЛИТИЧЕСКОЙ ХИМИИ ФЕХО
Аналитическую химию можно считать и самым старым и самым молодым направлением химической науки. Еще Аристотель употреблял термин «аналитика» в связи с задачами аргументации и доказательств с помощью силлогизмов1. Йо лишь сравнительно недавно аналитическая химия оформилась в самостоятельную научную дисциплину, основанную на теоретическом знании и накопленной информации. Процесс обособления аналитической химии в отдельное направление берет начало с работ Роберта Бойля, получил продолжение благодаря работам Лавуазье, Берцелиуса, Вёлера й Либиха, а около 100 лет назад с появлением книги Вильгельма Оствальда «Научные осцовы аналитической химии» аналитическая химия как автономная, весьма сложная и в высшей степени привлекательная область науки утвердилась окончательно. Очень быстрое развитие аналитической химии продолжается и поныне: достаточно упомянуть об огромном вкладе лишь некоторых из совре1 менных выдающихся ученых: Ярослава Гейровского (электроанализ), Ричарда Р. Эрнста (ЯМР), Герхарда Бинйнга и Генриха Рорера (сканирующая туннельная и атомно-силовая микроскопия). Многолетние исследования химиков-аналитиков привели к накоплению огромного объема’ эмпирических знаний (как фундаментального, так и практического характера) о веществе.
Химия составляет одну из основ общей человеческой Культуры, промышленности и торговли, поставляя разнообразную продукцию д ля повседневных нужд: пищу, одежду, строительные материалы, лекарства и материалы для медицины, транспорта, связи. Будучи изначально чисто эмпирической наукой, причем преимущественно «лекарского» направления, химия демонстрирует сегодня современный эксперимент, изучая превращения и свойства веществ; в основе химической науки лежат физические, химические и математические законы. В химии постепенно сформировались вполне самостоятельные, но в то же время и тесно связанные между собой направления: органическая химия, неорганическая химия, биохимия, пищевая химия, химическая технология, физическая химия и, наконец, о чем пойдет речь в этой книге, аналитическая химия.
Несомненно, что во многом благодаря современной аналитической химии с ее многочисленными высокочувствительными и селективными методами в
1 Аристотель имел в ваду не химический анализ. Но верно и то, что некоторые анализы проводили еще в глубокой древности, в том числе в античные времена. — Прим. per).
10 Предисловие-председателей Отделения амвлитической химии ФЕХО
обществе утвердилось понимание важности экологических проблем^ а .также возникли системы контроля качества на производстве, в сферах здравоохранения и охраны окружающей среды. Мировой рынок аналитических приборов непрерывно расширяется и к настоящему моменту оценивается впечатляющей цифрой порядка 1 триллиона долларов США. В современной химической промышленности в случае выхода технологического процесса за пределы оптимальных условий под «недреманным оком» управляющих программ процесс немедленно останавливается. Большинство химических фирм Европы приняли принцип «устойчивого развития» общества в качестве основополагающей философии своего бизнеса. Аналитической химии принадлежит решающая роль в обеспечении контроля за технологическим процессом и его успешного протекания, а также в сохранении глобального экологического баланса в целом.
Отделение аналитической химии (ОАХ) Федерации европейских химических обществ приняло следующее определение:
□ Аналитическая химия—это научная дисциплина, которая развивает и применяет методы, средства и общую методологию получения информации о составе и природе вещества (в пространстве и времени).
Данный учебник должен сыграть уникальную роль в университетском об- ! разовании; он отвечает именно этому определению аналитической химии, данному ОАХ: в нем изложены весомые свидетельства того факта, что аналитическая химия действительно стала самостоятельной областью науки, причем науки информационной, призванной ответить на крайне важный теоретический и практический вопрос о том, как устроен материальный мир.
Для химиков-аналитиков вопрос этот сводится обычно к определению состава пробы с помощью химических реагентов, физических методов исследования и биотестов. Именно практические соображения пронизывают все изложение. В некоторых случаях методы аналитической химии являются чисто химическими; так, кислотно-основиое титрование полностью основано на прн-йгенении химических реагентов. Есть и чисто физические методы, такие, как рентгеноспектральный анализ. Однако во многих случаях четко разграничить природу применяемого метода трудно, например в хроматографии (условно отнесенной в учебнике к «химическим методам») или сканирующей туннельной микроскопии (рассматриваемой как один из «физических методов»). В действительности в обоих методах можно найти и химические, и физические аспекты.
Аналитическая химия — междисциплинарная наука, базирующаяся на законах химии, физики, математики, информатики и биологии. Ее задача—извлечение информации путем исследования образца, причем достичь этого следует, не допуская искажений, т. е. задача—установление истины о строении материального мира. Конкретные задачи, решаемые химиками-аналитиками, могут показаться тривиальными, но это совсем не так, если учесть сложность современных объектов анализа (как промышленных, так и природных) и необходимость во многих случаях получать результаты в реальном времени н без разрушения образца (in situ). Настоятельные потребности современной мировой торговли и производства привели к появлению национальных и, что еще
Предисловие председателей Отделений аналитической химии ФЕХО 11
•более важно, международных организаций обеспечения качества EURACHEM и СПАС. Эти организации требуют, чтобы даже лаборатории е большим опытом работы доказывали уровень своей компетентности, периодически проходя процедуру аккредитации, включающую проверку оборудования и методик иа их соответствие технологическим стандартам, а также проверку квалификации персонала. Требования международной аккредитации обусловливают необходимость унификации образования химиков-аналитиков в разных странах, причем соответствующий ученый курс должен базироваться на едином научном языке и общепринятых базовых принципах. Поскольку сейчас неоспоримую роль международного языка в химии и физике играет английский в ОАХ в качестве первой попытки создания единого курса аналитической химии разработана англоязычная версия программы курса «Аналитическая химия» (основной раздел программы см. [Ляс/. Chem. 1994, 66, 98А], дополнительные разделы— [М'ез. J. Anal. Chem. 1997, 357, 197]).
Данный учебник «Аналитическая химия» написан в соответствии с программой ОАХ «Аналитическая химия» и ее ранней версии («WPAC — Eurocuricculum»)1. Главная концепция учебника—сбалансированное сочетание традиционных методов химического анализа (часть П), современных биотестов (также часть П) и физических методов (часть III), а также методов хемометрики (часть IV). Изложению конкретных методов (части с П по ГУ) предшествует вводная часть I, где обсуждаются общие вопросы: задачи аналитической химии и ее значение для общества, аналитический цикл, обеспечение и контроль качества. Книгу завершает часть V «Интегрированные аналитические системы», которая имеет производственную направленность и рассматривает достаточно сложные стороны аналитической науки: гибридные методы и производственный анализ, которые играют важную роль в современной химической технологии и значение которых еше.более возрастет в будущем. Для обеспечения высокого уровня подачи материала в качестве авторов были привлечены специалисты в соответствующих областях. В учебнике достигнуто хорошее сочетание необходимой фундаментальности при изложении основных принципов и гибкости подхода при переходе к новейшим методам анализа. Поэтому удалось охватить как классические методы (например, кислотноосновное или комплексометрическое титрование), что необходимо для понимания действия современных химических сенсоров, так н последние достижения в области физических методов, хемометрики и производственного анализа, во многом определяющих развитие аналитической химии в будущем. Даже сложные вопросы, например, атомно-силовая микроскопия или миниатюризован-ные интегрированные аналитические системы, изложены вполне доступно (на уровне основ). Возможная схематичность отдельных глав обусловлена решением редакторов ограничить объем книги. Для более детального ознакомления с соответствующими вопросами можно использовать множество специальных руководств, в чем также имеются рекомендации в соответствии с программой ОАХ.
1 С сентября 1996 г. WPAC (Working Party on Analytical Chemistry) переименован в DAC — Отделение аналитической химии (ОАХ ФЕХО).
12
Предисловие председателей Отделения аналитической химии ФЕХО
Университетское образование предполагает подготовку по аналитической химии, позволяющую не только работать в современной промышленности, но и выполнять фундаментальные научные исследования. В аналитической химии важное место занимает понятие «истина». Поэтому студентам следует внимательно изучить гл. 2 («Процесс анализа») и 3 («Обеспечение и контроль качества»). Кредо сэра Карла Поппера1 «Приближение к истине в принципе возможно» следует считать базовым философским положением аналитической химии как фундаментальной науки.
Что касается прикладных аспектов, то отметим лишь, что мы сейчас имеем дело с колоссальным потоком аналитических данных, возникающих в результате. невероятно большого числа анализов —10 м илллиардов в год! В рамках данного учебника по общему курсу аналитической химии редакторы постарались показать, что на современную аналитическую химию возложена как никогда большая ответственность за будущее развитие цивилизации. Например, на основе полученных аналитических данных были смоделированы изменения содержания оксидов азота в стратосфере при полетах сверхзвуковых самолетов; благодаря этому удалось добиться сокращения таких полетов, которые приводят к разрушению озонового слоя. Это лишь один пример, демонстрирующий, что аналитик обязан получать правильные результаты с тем, чтобы полученные знания можно было использовать при решении реальных проблем. Вообще познание истины составляет основу мудрости — качества, необходимого при принятии долговременных решений, в том числе политических/
Мы убеждены, что аналитическая химия, изучаемая повсеместно в-аспекте баланса между свободой и ответственностью (что как раз и предусмотрено в данном учебнике и программе ОАХ), может стать ключевой наукой для обеспечения безопасного будущего человечества!
Июнь 1997
Р. Кельнер, X. Малисса и Э. Пунгор
Председатели ОАХ ФЕХО: 1993-97,1975-81 и 1981-87.
1 Поппер Карл Раймунд (1902-1994)—философ, логик и социолог. Родился в Австрии. Примыкал к Венскому кружку. С 1945 в Великобритании. Предложил философскую концепцию критического рационализма, теорию роста научного знания построил как антитезу неопозитивизму. Выдвинул принцип фальсифицируемости (опровержимости), служащий критерием демаркации между наукой и «метафизикой». Теория «трех миров» Поппера утверждает существование физического и ментального миров, а также мира объективного знания. Работы по теории сознания, вероятностной логике и теории выводимости. Выступил с критикой марксизма и принципа историзма. В противовес иррационализму и релятивизму защищал рационализм. — Прим, перев.
БЛАГОДАРНОСТИ
Редакторы выражают глубокую признательность издателю Кристине Диллик за ее терпение, полезные советы и всестороннюю, эффективную и постоянную поддержку, без которых эта книга не могйа бы выйти в свет.
За рецензирование отдельных глав и множество ценнейших предложений мы хотим выразить глубокую благодарность группе ученых, профессоров, внесших свой вклад в этот международный проект:
F. Adams, UIA, Antwerpen, Belgium
К. Ballschmiter, University of Ulm, Germany
M. F. Camoes, University of Lisboa, Portugal
Ch. Ducauze, Mational Institute of Agronomy, Paris, France
R. R. Ernst, Swiss Federal Institute of Technology, Zurich, Switzerland
S. Gal, Technical University, Budapest, Hungary
D. J. Harrison, University of Alberta, Edmonton, Canada
J. A. de Haseth, University of Georgia, USA
J. Mink, University of Veszprem, Hungary
N. M. M. Nibbering, University of Amsterdam, The Netherlands
R. Niessner, Technical University of Munich, Germany
B. te Nijenhuis, Gist-brocades NV, The Netherlands
A. Sanz-Medel, University of Oviedo, Spain
G. R. Scollary, Charles Stuart University, Australia
G. ToTg, University of Dortmund, Germany
M. Valcarcel, University of Cdrdoba, Spain
C. Wilkins, University of California, Riverside, USA
0. S. Wolfbeis, University of Regensburg, Germany
Ю. А. Золотов, Российская академия наук, Россия
ОБОЗНАЧЕНИЯ
а выход оже-электронов
А оптическая плотность
Ьо свободный член, фоновый сигнал
61 коэффициент чувствительности
61/2 ширина на половине высоты
с концентрация
С ковариация
CV коэффициент вариации (относительное стандартное отклонение)
d расстояние, межплоскостное расстояние решетки, среднее откло-
нение
D коэффициент диффузии
е электрон
Е энергия, электродный потенциал, математическое ожидание, по-
грешность
Е(ХГ) г-й (нецентральный) момент X
£{(Х — р)г} г-й центральный момент X ~~
Еа энергия активации
Ев энергия связи
EKttH кинетическая энергия
/ частота
f(z) функция плотности стандартного нормального распределения
F коэффициент Фишера, скорость потока
F(z) функция стандартного нормального распределения
G энергия Гиббса
Н высота, эквивалентная теоретической тарелке (ВЭТТ), энталь-
пия
ДЯ изменение энтальпии
I интенсивность, ядерное спиновое число, сила тока, индекс удер-
живания Ковача, ионная сила интенсивность излучения источника
j внутреннее квантовое число
J константа расщепления
к константа скорости, коэффициент относительной чувствитель-
ности
к' коэффициент емкости
Обозначения
:Д5
К константа равновесия
Км константа скорости Михаэлиса—Ментен
I орбитальное квантовое число
L расстояние от образца до экрана
т магнитное квантовое число
М мультиплетность
Мг относительная молекулярная масса
М молярная концентрация (моль/л)
п порядок отражения, ось вращения, показатель преломления
п ось вращения с инверсией
N число импульсов, поверхностная плотность, число тарелок
NA численная апертура
Р угловой ядерный момент, вероятность
Q число падающих налетающих частиц
г коэффициент корреляции, радиус
R разрешение, сопротивление
Rj коэффициент ослабления (относительная скорость)
з стандартное отклонение (оценка)
S энтропия, мера сходства
s2 дисперсия (оценка)
t время, толщина пленки, коэффициент Стьюдента
ti/2 время полупревращения, период полураспада
Лм мертвое время
tn время удерживания
Т истинное значение измеряемой величины, пропускание-
71 время спин-решеточной релаксации
7Ь время спин-спиновой релаксации
й средняя линейная скорость молекул в подвижной фазе
U напряжение, потенциал постоянного тока
v скорость реакции, линейная скорость
v средняя линейная скорость компонента
Vo начальная скорость реакции
V дисперсия
объем подвижной фазы
Vr удерживаемый объем
w ширина пика
х скалярная переменная
х вектор из величин х
X матрица из величин х
х среднее, среднее арифметическое серии из п измерений
у переменная
Y выход продуктов распыления
z величина заряда
Z стандартное нормальное отклонение, атомный номер
16
Обозначения
а уровень значимости, коэффициент селективности, степень дис-
социации
/3 выход детектора ионов, фазовое отношение, общая константа
устойчивости
^1/2 ширина на половине высоты
X2 коэффициент распределения (хи-квадрат)
6 химический сдвиг
е молярный коэффициент поглощения
Ф плотность потока, работа выхода
А длина волны, постоянная радиоактивного распада
т] эффективность, распространенность (изотопа)
7 гиромагнитное отношение
Г гамма-функция
д генеральное среднее, магнитный момент, ионная сила
|/ число степеней свободы, частота
AQ угол захвата детектора
0 угол рассеяния (дифракции)
р плотность
о константа экранирования, генеральное стандартное отклонение
о2 генеральная дисперсия
тр длительность импульса
uj выход флуоресценции, циклотронная частота
0 двугранный угол
{ } твердая фаза
СОКРАЩЕНИЯ
AAC АВГ АЦП АЭД БРЛ ВВП вид.
ВОЗ ВЧ-Ge ВЭЖХ вэкэ вэтсх вэтт ВЭУ гжх гтх гх ДМА ДНК ДТА жжх жтх жх ИДА ик исп ИСП-АЭС
ИСП-МС их ИЮПАК
кгх кзэ
атомно-абсорбционная спектрометрия {англ. AAS) анализ выделяющихся газов (EGA) аналого-цифровой преобразователь (ADC) атомно-эмнссионный детектор (AED) безэлектродная разрядная лампа (EDL) валовый внутренний продукт (GNP) видимая область электромагнитного спектра (VIS) Всемирная организация здравоохранения (WHO) германий высокой частоты (HPGe)
высокоэффективная жидкостная хроматография (HPLC) высокоэффективный капиллярный электрофорез (НРСЕ) высокоэффективная тонкослойная хроматография (HPTLC) высота, эквивалентная теоретической тарелке (НЕРТ) вторичный электронный умножитель (SEM) газо-жидкостная хроматография (GLC) газо-твердофазная хроматография (GSC) газовая хроматография (GC) динамичный механический анализ (DMA) дезоксирибонуклеиновая кислота дефференциальный термический анализ (DTA) жидко-жидкостная хроматография (LLC) жидко-твердофазная хроматография (LSC) жидкостная хроматография (LC) иминодиуксусная кислота
инфракрасный (свет, диапазон электромагнитного спектра) (IR) индуктивно-связанная плазма (ICP)
атомно-эмиссионная спектроскопия с индуктивно-связанной плазмой (ICP-AES)
масс-спектрометрия с индуктивно-связанной плазмой (ICP-MS) ионная хроматография (IC)
Международный союз теоретической и прикладной химии (ШРАС)
капиллярная газовая хроматография (CGC) капиллярный зонный электрофорез (CZE)
18
Сокращения
КР комбинационное рассеяние (рамановская спектроскопия)
КЭ капиллярный электрофорез (СЕ)
КЭХ капиллярная электрохроматография (СЕС)
ЛПК лампа с полым катодом (HCL)
МИБК метилизобутилкетон
МСВНЧ масс-спектрометрия вторичных нейтральных частиц (SNMS)
МСД масс-спектрометрический детектор (MSD)
МЭКХ мицеллярная электрокинетическая хроматография (МЕСС)
НАА нейтронно-активационный анализ (NAA)
НТА нитрилотриуксусная кислота
НКЭ насыщенный каломельный электрод (SCE)
ОАХ Отделение аналитической химии ФЕХО (DAC, до сент. 1996 WPAC)
ОДС октадецилсиликагель (ODS)
ПАР пиридилазорезорцин (PAR)
ПАУ полициклические ароматические углеводороды (RAH)
ПВП полиэтилен высокой плотности (HPDE)
ПЗИ прибор с зарядовой инжекцией (CID)
ПЗС прибор с зарядовой связью (С CD)
ПИА проточно-инжекционный анализ (FIA)
ПИД пламенно ионизационный детектор
ПК персональный компьютер (PC)
ПКПН полый капилляр с покрытым носителем (SCOT)
ПКПС полый капилляр с покрытыми стенками (WCQI)
ППЗ прибор с переносом заряда
ППС поверхностно-пористые сорбенты (PLB)
ПФД пламенно-фотометрический детектор (FPD)
ПХБ полихлорированные бифенилы (РСВ)
ПЧД позиционно-чувствительный детектор (PSD)
РД рентгеновская дифракция (XRD)
РИМС резонансно-ионизационная масс-спектрометрия
РФС ЭД рентгенофлуоресцентная спектрометрия с дисперсией по энергиям (ED-XRF)
СВЭ стандартный водородный электрод (SHE)
СИ Международная система единиц измерения (SI)
ССИ свободный спад индукции (FID)
СФХ сверхкритическая флюидная хроматография (SFC)
ТБФ трибутилфосфат (ТВР)
ТГ термогравиметрня (TG)
ТД термодилатометрня (TD)
ТИД термононный детектор (TID)
ТМА термомеханический анализ (ТМА)
ТМС тетраметилсилан (TMS)
ТСХ тонкослойная хроматография (TLC)
Сокращения
19
УФ
ФЕХО ФПП ХИ ЦАП ЭДТА ЭЗД эоп ЭТА
2D A ALA АС АЕМ AES
AFM AFNOR AFS АОАС AL APCI API ARM ARUPS ASTM ATR BB BCR BE
BIPM BSI CAALS
CAMM CAR CEN CENELEC CFA CF-FAB
CID
ультрафиолетовый (свет, диапазон в электромагнитном спектре) (UV)
Ферерация европейских химических обществ (FECS) фракционирование в поперечном поле (FFF) химическая ионизация (CI) цифро-аналоговый преобразователь этилендиаминтетрауксусная кислота электрозахватный детектор (ECD) электроосмотический поток (EOF) электротермический атомизатор, эманационный термический анализ (ЕТА)
двумерный
Американская ассоциация аккредитации лабораторий переменный ток аналитическая электронная микроскопия атомно-эмиссионная спектрометрия, оже-электронная спектрометрия атомно-силовая микроскопия
Французская ассоциация стандартизации атомно-флуоресцентная спектрометрия Ассоциация официальных химиков-аналитиков атомный слой
химическая ионизация при атмосферном давлении ионизация при атмосферном давлении микроскопия атомного разрешения
УФ-фотоэлектронная спектрометрия с угловым разрешением Американское общество испытания материалов нарушенное полное отражение широкополосный
Бюро стандартов
обращенная геометрия (магнитный сектор + электростатический анализатор)
Международное бюро мер и весов Британский институт стандартов Консорциум автоматизированных аналитических лабораторных систем компьютерное молекулярное моделирование непрерывное добавление реагента
Европейский комитет по стандартизации
Европейский комитет по стандартизации в электротехнике непрерывный проточный анализ масс-спектрометрия с ионизацией быстрыми атомами в непрерывном потоке диссоциация при столкновении
20
Сокращения
СГГАС
COSY СРАА CRM CTD
CV
DAD DAS DBE DC DCI DCP DCPU DIN DLI DSC DTG EAL EB
EC EELS EFTA
El EL ELD
EN EPA EPXMA ERD ESA ESP ETSI
EU EXAFS FAB
FD FDA
FG FIM FIR FNAA
Международная организация по сотрудничеству в области обеспечения единства измерений (прослеживаемости) в аналитической химии корреляционная спектрометрия активационный анализ заряженными частицами (аттестованный) стандартный образец, образец сравнения прибор с переносом заряда (ППЗ) коэффициент вариации (относительное стандартное отклонение) детектирование (детектор) с помощью диодной матрицы дезаминосульф адимидин эквивалент двойной связи постоянный ток десорбционная химическая ионизация плазма постоянного тока дихлорфенилмочевина
Германский институт стандартизации прямой ввод жидкости дифференциальная сканирующая калориметрия дифференциальная термогравиметрия
Европейская кооперация по аккредитации лабораторий .
прямая геометрия (электростатический анализатор 4- магнитный сектор)
Европейская Комиссия
спектрометрия характеристических потерь энергии электронов Европейская ассоциация свободной торговли ионизация электронным ударом электролюминесценция электролюминесцентный детектор
Европейские стандарты
Агентство по охране окружающей среды (США) электронно-зондовый рентгеноспектральный микроанализ детектор упругой отдачи электростатический анализатор электрораспыление' ’
Европейский институт стандартов телекоммуникаций Европейский Союз дальняя тонкая структура рентгеновских спектров поглощения бомбардировка быстрыми атомами полевая десорбция
Управление пищевых продуктов н лекарств (США) функциональная группа полевая ионная микроскопия дальняя ИК-область активационный анализ с активацией быстрыми нейтронами
Сокращения
21
FT фурье-преобразование
FT-ICR спектрометрия ионного циклотронного резонанса с фурье-
преобразованием
FTIR инфракрасная спектрометрия с фурье-преобразованием
FT-MS масс-спектрометрия с фурье-преобразованием
FWHM ширина на половине высоты (пика)
GDL лампа тлеющего разряда
GDMS масс-спектрометрия с тлеющим разрядом
GF-AAS атомно-абсорбционная спектрометрия с графитовой печью
GLP «Хорошая лабораторная практика» (сборник нормативных до-
кументов)
GMP «Хорошая производственная практика» (сборник нормативных
документов)
GOD глюкозооксидаза
GS алгоритм Грама—Шмидта
ICTAC Международный союз по термическому анализу и калориметрии
IDF Международная молочная федерация
IDMS масс-спектрометрия с изотопным разбавлением
ILAC Международная кооперация по аккредитации лабораторий
IN А А инструментальный нейтронно-активационный анализ
IQR межквартильный размах
IRN радиоактивные индикаторы
ISO Международная организация по стандартизации
ISO/REMCO Консультационный комитет ISO по образцам сравнения
ISP ионное распыление
ISS спектрометрия рассеяния ионов
JCPDS Объединенный комитет по стандартам в порошковой дифракци-
онной спектрометрии
KRS-5 бромид-иодид таллия (кристаллический материал для нарушен-
ного полного отражения)
LAMMS лазерная микрозондовая масс-спектрометрия
LARIS резонансно-ионизационная масс-спектрометрия с лазерной ато-
LBB
LEED
LNRI SNMS
LQ
LRI SNMS
LRMA MALDI
MCA
МСТ
MEIS
мизацией
закон Бугера—Ламберта—Бера
дифракция электронов низкой энергии
масс-спектрометрия вторичных нейтральных частиц (МСВНЧ) с лазерной нерезонансной пост-ионизацией нижняя квартиль
масс-спектрометрия вторичных нейтральных частиц (МСВНЧ) с лазерной резонансной пост-ионизацией лазерный рамановский микроанализ
лазерная десорбция/ионизация в матрице
многоканальный анализатор
теллурид ртути-кадмия
спектроскопия рассеяния ионов средних энергий
22
Сокращения
MIP MIR
MS MS-MS M&T NATA NBS Nd:YAG
NEXAFS
NICI NIR NIST NMR NPD NRA OECD OIML Ox PA PAA PCDD PD
PDF PE PFIA PFK PFTBA PICI PMT ppb PPm ppt PTFE PVD Q
QA QC RBS Red REELS
REM RHEED
микроволновая плазма средняя ИК-область масс-спектрометрия
тандемная масс-спектрометрия
Программа измерений и испытаний Европейской Комиссии Национальная ассоциация по испытаниям (Австралия) Национальное бюро стандартов США (сейчас NIST) алюмо-иттриевый гранат, легированный неодимом (материал для лазерной техники)
ближняя (околопороговая) тонкая структура рентгеновских спектров поглощения
химическая ионизация отрицательно заряженными ионами ближняя ИК-область электромагнитного спектра Национальный институт стацдартов и технологий (США) ядерный магнитный резонанс
детектор для определения азота и фосфора
анализ с использованием мгновенных ядерных реакций Организация экономического сотрудничества и развития Международная организация официальной метрологии окислитель
сродство к протону
протоно-активационный анализ полихлордибензодиоксины плазменная десорбция
файл по порошковой дифракции —
фотоэлектрон
производственный проточно-инжекционный анализ перфторокеросин
перфтортрибутиламин
химическая ионизация положительно заряженными ионами фотоумножитель (ФЭУ)
часть на миллиард (млрд”1,10”9)
часть на миллион (млн-1,10-6)
часть на триллион (трлн-1,10”12) политетрафторэтилен
осаждение из паровой фазы квадрупольный фильтр обеспечение качества контроль качества
спектрометр резерфордовского рассеяния восстановитель
спектроскопия характеристических потерь энергии отраженных электронов
отражательная электронная микроскопия дифракция быстрых (отраженных) электронов
Сокращения
23
RIC RIMS
RM
RMD RNAA
RS
RSC
RSD
SCA
SDM SDS-PAGE
SEC
SEM SERS SEXAFS
SIM SIMS SIRIS
SNMS SOP SPM SRM SSMS STM
STS TC TCD TCDD TEELS
TGA TGS THEED TIC TIMS TOF TSP
UHV UPS UQR
реконструированная ионная хроматограмма резонансно-ионизационная масс-спектрометрия (РИМС) образец сравнения
относительное среднее отклонение
радиохимический нейтронно-активационный анализ
рамановская спектроскопия (спектроскопия комбинационного рассеяния)
Королевское химическое общество (Великобритания) относительное стандартное отклонение одноканальный анализатор сульфадимидин
электрофорез в полиакриламидном геле с додецилсульфонатом натрия
эксклюзионная хроматография
сканирующая электронная микроскопия
рамановское рассеяние, усиленное поверхностью тонкая структура рентгеновских спектров поглощения в приповерхностном слое
детектирование по выбранному иону масс-спектрометрия вторичных ионов резонансно-ионизационная масс-спектрометрия с распылением , (масс-спектрометрия вторичных частиц с постоянной резонансной ионизацией)
масс-спектрометрия вторичных нейтральных частиц стандартная рабочая методика сканирующая зондовая микроскопия стандартный образец сравнения искровая масс-спектрометрия
сканирующая туннельная микроскопия сканирующая туннельная спектрометрия технический комитет (CEN или ISO) катарометр
тетрахлордибензодиоксины
спектрометрия характеристических потерь энергии прошедших электронов
термогравиметрический анализ
триглицинсульфат
дифракция быстрых (прошедших) электронов хроматография с регистрацией по суммарному ионному току масс-спектрометрия с термической ионизацией времяпролетный масс-спектрометр термораспыление сверхвысокий вакуум
ультрафиолетовая фотоэлектронная спектрометрия верхняя квартиль
24 Сокращения
VIM VML VOX WD-XRF WPAC Международный словарь по метрологии Официальный словарь по метрологии летучие органические галогены рентгеиофлуоресцентиая спектрометрия с волновой дисперсией Рабочая группа по аналитической химии ФЕХО, в сентябре 1996 переименована в DAC (русск. ОАХ)
XAS XPS XRF ZAF •рентгеноабсорбционная спектрометрия рентгеновская фотоэлектронная спектрометрия рентгенофлуоресцентная спектрометрия ZAF-коррекция (введение поправки на атомный .номер, поглощение и флуоресценцию)
_________________ Часть
ОБЩИЕ ВОПРОСЫ
1
ЗАДАЧИ АНАЛИТИЧЕСКОЙ ХИМИИ
И ЕЕ ЗНАЧЕНИЕ ДЛЯ ОБЩЕСТВА
1.1. АНАЛИТИЧЕСКАЯ ХИМИЯ И ОБЩЕСТВО
1.1.1. Введение: историческая справка
Прежде чем обсудить задачи аналитической химии и ее фундаментальное значение для современного общества, кратко окинем взглядом историю общества, включая историю науки и, в частности, химии.
Согласно одной из теорий, Вселенная образовалась 15 миллиардов лет назад в результате «Большого взрыва» и с тех пор непрерывно расширяется. Материя Вселенной — в форме звезд, космической пыли, газов — состоит из химических элементов. А наша Земля принадлежит Солнечной системе, входящей в одну из галактик Вселенной. Таким образом, роль химических процессов как объективно существующих процессов превращения веществ была велика во все времена. Люди появились на Земле около 4 миллионов лет назад и с этого времени сумели выжить в борьбе за существование^в значительной мере благодаря накопленным знаниям о свойствах и превращениях веществ и способах производства различных материалов они создали цивилизацию, охватывающую различные стороны деятельности и знаний человечества, в том числе естественные науки. Современная химия состоит из множества дисциплин, одна из которых—аналитическая химия [1.1-1].
Древнегреческие философы Платон (428-347до н.э.) и Аристотель (384-322 до н.э.) полагали, что материя состоит из четырех основных элементов — земли, воды, воздуха и огня. В средние века (с конца V до середины XIV в.) химические знания обогатились в значительной мере в связи с попытками осуществить заветную мечту человечества—превратить различные металлы в золото (ввиду высокой стоимости последнего). Так возникла алхимия — средневековый этап развития химии.
На другом конце Земли, на востоке, главным образом в Китае, ряд философов подобно древним грекам, представляли себе строение материи на основе пяти элементов — земли, воды, огня, дерева и металла. Помимо превращения веществ в золото китайских алхимиков интересовало получение «эликсира жизни» — лекарства, способного победить старение и смерть. Эти исследования стимулировались тем обстоятельством, что древние китайские императоры, в частности Цинь Шихуанди (259-210 до н. э.), правитель царства Цинь, создавший единую централизованную империю Цинь (221-206 до н.э.), были
1.1. Аналитическая химия и общество
27
весьма озабочены проблемой долголетия. Впоследствии в Китае наблюдался значительный прогресс в таких областях, как производство бумаги, пиротехника и восточная медицина, однако уровень развития химии (в ее современном понимании) оставался низким. Сходная ситуация была в Японии. Современная наука, включая химию, начала развиваться там трлько после буржуазной революции Мэйджи в 1868г. До этого японцы сумели приобрести лишь весьма ограниченные знания химии и западной медицины, главным образом от голландских путешественников [1.1-2, 1.1-3].
Честь создания первой атомистической теории принадлежит Демокриту (ок. 460-370 до н.э.), греческому философу, полагавшему, что материя Вселенной состоит из неделимых и неизменных частиц, атомов. Впоследствии эта идея оказала влияние на таких великих ученых и философов, как Н. Коперник (1473-1543), Ф.Бэкон (1561-1626), Р. Бойль (1627-1691), И. Ньютон (1643-1727). Затем английский химик Дж. Дальтон (1766-1844) создал современную атомистическую теорию. Следует отметить, заслуги А. Л. Лавуазье, который ввел в химию количественные измерения, послужившие мощным импульсом развития аналитической химии.
В частности, Лавуазье провел знаменитый опыт по измерению массы оксида ртути, образующегося из ртути и воздуха (или кислорода). Он доказал, что масса оксида ртути в точности равнялась сумме масс двух его компонентов. Лавуазье, павшего жертвой Великой французской революции, всегда будут помнить как человека, который заложил основы химии в ее современном понимании [1-1-41- Для более подробного ознакомления с историей развития химии рекомендуем книгу Ф. Сабадвари1. _
1.1.2. Задачи аналитической химии
Для исследования любых проблем, касающихся как естественных, так и общественных наук, первым делом обычно используют аналитический подход. Это означает, что проблему сначала расчленяют на более простые составляющие и изучают их по отдельности. Затем, после объединения полученных таким путем отдельных порций информация, проблема может быть, наконец, постигнута в целом. Анализ вещества выполняют сходным образом. Сначала образец разделяют на химические компоненты, такие, как атомы, ионы или молекулы (см. ч.П). В то же время современные физические методы анализа часто позволяют исследовать вещество in situ в его исходном состоянии, без предварительного разделения или разрушения (см. ч.Ш). Первичная задача химического анализа—установить природу и количества химических компонентов, присутствующих в системе. После этого возможно установление состава и строения исходного исследуемого объекта в целом, как описано выше.
Во второй половине двадцатого века благодаря стремительному развитию инструментальных аналитических методов появилась целая область, называемая «компьютерно ориентированной аналитической химией» (см. ч-IV). В на
1 Есть русский перевод книги: Сабадвари Ф., Робинсон А., История аналитической химии.—Мир, М., 1984. — Прим. ред.
28 Глава 1. Задачи аналитической химии и ее значение для общества
стоящее время стало возможным получение разнообразной информации о веществе с использованием «интегрированных аналитических систем» (см. ч. V). Современное определение аналитической химии, принятое в Эдинбурге на конференции «Евроанализ» в 1993г., звучит следующим образом: «Аналитическая химия—это научная дисциплина, которая развивает и применяет методы, средства и общую методологию получения информации о составе и природе вещества (в пространстве и времени)» [1.1-5].
1.1.3. Роль аналитической химии в жизни общества
Люди окружены объектами природного и искусственного происхождения. Воздух, вода, почва, горные породы, растения и животные составляют естественную окружающую среду. Искусственная окружающая среда состоит из зданий, предметов одежды, пищевых продуктов, машин, книг и прочих продуктов, которые общество создает для жизнеобеспечения современных людей. Все эти естественные и искусственные объекты состоят из материалов, которые, в свою очередь, включают различные химические вещества. Для идентификации и оценки качества материалов требуются соответствующие методы. Помимо сведений о форме, размерах, твердости, цвете и других физических свойствах окружающих предметов человеку необходима информация и об их химическом составе и свойствах. Такую информацию может предоставить только аналитическая химия. Аналитическая химия, таким образом, — неотъемлемая составляющая жизни современного человека.
Для лучшего понимания характера отношений между аналитической химией и современным обществом рассмотрим некоторые из традиционных классификаций, принятых в аналитической химии.
а) Наиболее наглядна классификация с точки зрения природы анализируемого объекта или материала: анализ воды, минерального сырья, пищевых продуктов и т. д.
В промышленности для обеспечения качества стали—одного из основных конструкционных материалов — необходимы методы анализа сталей и сплавов. С тех самых пор, как появились первые методы «мокрого» химического анализа, во всех крупнейших промышленных компаниях мира работали специалисты-аналитики в этой области.
Клинический анализ жизненно необходим в здравоохранении. Современные больницы должны располагать средствами для проведения надежных анализов крови и мочи для достоверной диагностики заболеваний. Родственными областями являются фармакологический и биологический анализ. В последнее время широко развивается анализ объектов окружающей среды — воздуха, природных вод, почв, биологических материалов.
б) Другой тип классификации основан на химической природе определяемых компонентов. Задача полного анализа — определение всех веществ, содержащихся в пробе; в этом случае суммарная масса определяемых компонентов должна быть равна массе исходной пробы. Примером может служить полный анализ горных пород, — в частности, лунного грунта, которым занималось
1.1. Аналитическая химия и общество
29
множество ученых во всем мире. В то же время часто кеобкодимо определять только некоторые компоненты (вещества или отдельный элементы), входящие в состав пробы. Современный элементный анализ позволяет определять все элементы в неорганических и органических соединениях. При анализе объектов окружающей среды очень важно уметь определять такие специфические компоненты, как оксиды азота и серы, озон, полихлорбифенилы, диоксины. Задачу определения диоксинов осложняет то обстоятельство, что они представляют собой смесь множества изомеров, значительно различающихся ею своей токсичности. Очевидно, сколь важное значение для общества имеет определение радионуклидов, таких, как ^Sr, 137Cs, 235U, 249Pu, входящих в состав материалов атомной энергетики или продуктов их деления1.
в) В настоящее время химический анализ включает в себя качественный и количественный анализ. Задача качественного анализа—установить, присутствует ли в пробе данный компонент; количественного—определить его содержание [1.1-6]. В повседневной жизни многие явления можно описать в «количественных» терминах: расстояние между двумя точками, размеры здания, количество пищи, уровень способностей ребенка, степень серьезности преступления и т.д. Иногда результаты химического анализа не представляется возможным выразить в точных величинах (миллиграммах, миллионных долях и т.д.), но можно описать в форме «содержание вещества довольно велико» или «очень мало» (и такого рода понятия тоже часто используют в повседневной жизни). В этом случае говорят о полуколичественном анализе.
Как отмечено выше, современные методы аналитической химии (спектроскопические, в частности) получили столь широкое развитие, что их можно использовать для решения самых разнообразных задач."Это показывает, насколько велика роль аналитической химии в современном обществе. Следует ожидать, что в будущем она возрастет еще больше.
Литература
[1.1-1] MillerS. L., OrgelL.E. The Origins of Life in the Earth. New Jersey: Prentice-Hall, 1974.
[1.1-2] Radzinsky W. A History of China. Oxford: Pergamon Press, 1979.
[1.1-3] KakuK. Chemistry and Chemical Industry, 77ie Chem. Soc. of Japan, 1992, 45, 388.
[1.1-4] Petrucci R.H. General Chemistry. New York: Macmillan Publishing, 1972.
[1.1-5] Kellner R. Education of Analytical Chemistry in Europe. Anal. Chem., 1994, 66, 98A.
[1.1-6] LaitinenH. A. Analytical Chemistry, Encyclopedia of Chemistry, New York; McGraw Hill, 1992, pp. 72-74.
1 Эта классификация в более или менее полном виде может выглядеть следующим образом: изотопный анализ, элементный, структурно-групповой (например, функциональный), молекулярный, вещественный (оценка форм существования компонентов и их содержания), фазовый. — Прим. ред.
30 Глава 1- Задачи аналитической химии и ее значение для общества
1.2. ХИМИК-АНАЛИТИК В ЦЕНТРЕ РЕШЕНИЯ ПРОБЛЕМ
ОБЩЕСТВА
Цели изучения
( • Сформировать представление о химике-аналитике как о профессиональном «решателе проблем>, использующем набор различных методов, обладающем широким мышлением и руководствующемся обобщенным знанием.
• Понять специфические аспекты практической работы аналитиков в условиях промышленности, особенно с точки зрения ее влияния на окружающую среду.
• На примере задач из области экологии ознакомиться с некоторыми общими подходами к решению аналитических проблем.
1.2.1. Введение
Широко распространено мнение, что десятилетия, приходящиеся на рубеж XX-XXI веков, войдут в историю как «информационная эра». Принимая во внимание поразительные достижения последних лет в области сбора, хранения, обработки, передачи и представления информации, эту точку зрения следует считать оправданной. Высокопроизводительные компьютеры, накопители данных, спутниковая передача информации, всемирные телевизионные сети и, наконец, создание «виртуальной реальности» —вот лишь некоторые примеры, иллюстрирующие этот тезис.
□ Мы живем в «информационную эру», радикально изменявшую всю общественную жизнь и, в частности, возможности аналитической химии.
В результате этого «технологического взрыва» немало приобрела и аналитическая химия. В жизнь вошли локальные компьютерные сети, соединяющие аналитические приборы, методы, использующие фурье-преобразование спектроскопических данных, системы сравнительного поиска аналитических данных для идентификации неизвестных веществ. В то же время аналитическую химию следует по праву считать и-одним из «столпов», на которых держится вся информационная эра. Ведь прежде, чем информацию собрать, сохранить, обработать и передать, ее необходимо получить. А именно получение новой, достоверной информации и является, по существу, целью химического анализа.
Для достижения этой цели необходимо точно сформулировать задачу, привлечь для ее решения, если это необходимо, множество методов (в том числе из смежных наук) и доказать, что полученные результаты правильны и воспроизводимы. Разработкой соответствующей методологии занимается аналитическая химия как наука. Применение химико-аналитической методологии в смежных областях, таких, как биотехнология, химическая промышленность, материаловедение, наука о пище, экология, в сочетании с новейшими технологиями «информационной эры» — открывает новые перспективы.
В этом разделе приведены примеры тех достижений современной цивилизации, в основе которых лежат успехи аналитической химии. Здесь также по
1.2. Химик-аналитик в центре решения проблем общества
31
казано, каким образом научная группа может работать как единая команда, чтобы точно определить характер задачи и установить, что для ее решения действительно использованы все возможные подходы. Обсуждается и ряд других важных проблем, касающихся деятельности аналитиков,— в частности, как следует представлять сложные результаты своей работы в форме, понятной неспециалистам, например руководству или общественности.
1.2.2. Индустриальная среда
Еще раз вспомним поразительные успехи химии в создании новых материалов, так преобразивших всю нашу жизнь за последние 50 лет. Ученые и инженеры разработали много новых лекарств, искусственных волокон и пластмасс, новые прочные покрытия, сплавы для авиационной и космической техники, полупроводниковые материалы для микроэлектроники. Одновременно мы стали придавать гораздо более серьезное значение проблемам химии живой материи, охраны здоровья, исследованию окружающей среды на Земле и в космосе.
□ Промышленность широко использовала достижения аналитической химии для разработки новых материалов и технологий.
Многое из этого стало возможным лишь благодаря тому, что люди умели идентифицировать химические вещества, устанавливать их структуру, соотносить структурные особенности с физическими свойствами, детально изучать механизмы химических реакций в реальных условиях, а также применять современные точные методы для анализа объектов окружающей среды.
Аналитические методы и приборы — вот то, что необходимо для получения информации о веществе. Сейчас стало возможным анализировать дажё“мель-чайшие частицы вещества, определять содержание компонентов на уровне миллионных долей и ниже. Возможно, ничто не характеризует прогресс современной науки и широту применения новейших технологий столь ярко, как поразительные достижения аналитического приборостроения за последние одно-два десятилетия. Особо следует отметить компьютеризацию химического анализа. Она позволила не только улучшить и ускорить процесс сбора информации, но и создать множество сложных методов обработки данных, реализация которых без помощи компьютеров была бы невозможна.
Около 80% выпускников по специальности «аналитическая химия» в дальнейшем работают в промышленности. Поэтому роль аналитической химии в промышленности необходимо рассмотреть особо. Эффективное использование достижений аналитической химии есть важная составляющая деятельности любой успешно работающей заводской лаборатории. Соответственно, велика и роль химиков-аналитиков.
О Химик-аналитик, работающий в промышленности, должен быть квалифицированным «решателем проблем».
Разумеется, необходимый уровень образования и квалификации аналитиков зависит от характера тех проблем, которые им нужно решать. Слово «проблема» здесь является поистине ключевым. Современный химик-аналитик, работающий в промышленности,—это в первую очередь «решатель проблем»,
32 Глава 1. Задачи аналитической химии и ее значение для общества
несущий полную ответственность за то, чтобы его знания и навыки, а также имеющееся в его распоряжении оборудование применялись максимально эффективно для решения аналитических задач, стоящих перед производством.
Что должен знать и уметь химик-аналитик? Универсального ответа быть не может, поскольку условия современного производства весьма разнообразны. Тем не менее можно выделить некоторые общие требования.
□ Химик-анаяитик должен иметь хорошие знания в области теории аналитических методов и соответствующей аналитической техники.
1. Аналитик должен владеть техникой эксперимента и теоретическими знаниями в объеме, необходимом для того, чтобы осмысленно пользоваться разработанными методиками и получать с их помощью точные результаты. Залогом успеха в любой области аналитической химии является хорошее знание теоретических основ каждого используемого метода. Без таких знаний аналитик не может быть ученым и творчески подходить к решению проблем. Очень важно четко понимать, для каких задач тот или иной метод может быть использован с максимальной отдачей и, наоборот, каковы его принципиальные недостатки, ограничения. Владение техникой эксперимента жизненно необходимо для аналитика. Без эксперимента теория мертва. Студент должен не просто уметь работать на приборе, но и ясно понимать, достаточны ли его характеристики (например, отношение сиг-нал/шум или разрешающая способность) для решения данной конкретной задачи. Для этого равно необходимы и теоретическое образование, и лабораторная практика, одно без другого теряет смысл. Практическая работа учит студента не только самостоятельно думать, но и самостоятельно действовать. Это умение особенно ценно в условиях быстрого развития новых аналитических методов.
2. Аналитик должен постоянно практиковаться в своей области, чтобы уметь выполнять как рутинные, так и «уникальные» анализы и развивать необходимые для этого методы и методики. Только постоянная практика учит студента искать компромисс между точностью анализа и затратами времени на его выполнение, консультироваться со специалистами из других областей с тем, чтобы найти оптимальный путь решения проблемы. Аналитическая химия не сводится к применению готовых методик к серии образцов. Химик-аналитик должен быть творческой и предприимчивой личностью, постоянно готовой применить весь арсенал своих теоретических знаний.
□ Новаторство и умение работать в коллективе—важные составляющие успеха при работе в промышленности.
3. Аналитик должен уметь руководить работой своих коллег и действовать как член единой команды в рамках аналитической исследовательской программы организации, осуществлять научный поиск в неизведанных областях аналитической химии.
Умение каждого человека достигать этих целей зависит от уровня образования, тренировки по месту работы, последующего обучения и накопленного
1.2. Химик-аналитик в центре решения проблем общества
33
опыта. Очевидно, что только некоторая, ограниченная часть требуемых навыков может быть получена в университете. Конечно, эти общие соображения применимы не только к аналитику, работающему в заводской лаборатории, но и ко всем химикам-аналитикам, да и ко всем химикам вообще.
Каковы же специфические особенности работы химиков-аналитиков в условиях промышленности?
Заводские химики-аналитики вынуждены балансировать между временем, стоимостью и точностью, которые являются ключевыми элементами каждой задачи из реальной жизни (рис. 1.2-1). Задача может поступить из любой части фирмы, организации и т. д., и аналитик должен быть достаточно квалифицированным, чтобы использовать все доступные ему ресурсы для решения этой задачи.
Для химика, работающего в промышленности, с фактором времени связаны две важные проблемы. Первая из них состоит в рациональном использовании собственного времени и времени коллег. В условиях конкуренции огромную роль играет эффективное использование всех ресурсов. В лабораториях университетов или исследовательских институтов, хотя они и стали уделять больше внимания фактору стоимости, фактор времени все еще не является основной заботой, но для аналитика, работающего в промышленности, эффективное использование рабочего времени становится жизненной необходимостью, и он должен приучать себя к этому уже со студенческой скамьи.
□ Выигрыш в затратах и своевременность аналитических результатов —ключевые промышленные факторы в промышленности.
Вторая проблема времени — это требование компромисса между правильностью и быстротой анализа. Если работа завода или дорогая экспериментальная программа зависят от результатов анализов, просто хороший ответ, данный вовремя, часто оказывается значительно более ценным, чем совершенно идеальный ответ, данный слишком поздно. Это не следует понимать в том смысле, что работа в заводской аналитической лаборатории «быстрая и грязная». Некоторые весьма точные аналитические работы необходимы и проводятся в заводских лабораториях, например, в фармацевтических компаниях, проверяющих чистоту продукта перед поступлением его на рынок. Опубликованное в журнале Physics Today письмо о том, как достичь успеха в промышленных исследованиях, выражает уместное в данном случае правило точности: «Не тратьте время и усилия на проведение измерений, более точных, чем те, которые оправданы природой эксперимента. Если погрешность при одном измерении не может быть меньше 10%, не имеет смысла измерять другие параметры с точностью до 0,1%.»
2 Аналитическая химия. Том 1
34 Глава 1. Задачи аналитической химии и ее значение для общества
Заводские лаборатории расходуют на анализ большое количество денег. Промышленность меньше обеспокоена стоимостью оборудования, поскольку она может окупиться. Необходимость и значимость ответа обычно перевешивает стоимость его получения. Экономика диктует, что необходимо учитывать не только стоимость получения ответа в аналитической задаче, но также и стоимость убытков, связанных с задержкой других производственных операций или самих исследований в случае, если получение ответа вовремя невозможно.
□ Аналитики могут получить задание с ограниченной информацией об источнике пробы или ее чистоте.
В промышленных условиях аналитику постоянно приходится работать в условиях неопределенности. Например, заводские исследовательские лаборатории могут изучать составы реакционных смесей, полученных с использованием новых типов катализаторов органического синтеза. На ранних стадиях исследования составы таких смесей весьма сложны. Зачастую по новой тематике число опубликованных данных невелико, чтобы можно было предположить, какие продукты присутствуют в образце. При этом аналитик стоит перед лицом настоящей «неизвестности». В других случаях, таких, как проблемы заводского производства, «проблема» может быть равносильна болезни. Нередко эта появляющаяся на столе аналитика темная взвесь, помеченная невразумительным образом, например, «проба, отобранная из резервуара на Иэокрекере П» или «загрязнение из неизвестного источника». Сравните это с типичными условиями работы большинства академических лабораторий, анализирующих пробы, отобранные в строго контролируемых условиях!
В обоих случаях—в. академической лаборатории и на производстве—аналитик должен собрать всю химическую информацию, имеющую отношение к задаче. Затем он должен разработать законченный аналитический план для решения поставленной задачи. Напомним, что многие промышленные пробы являются действительно «неизвестными» образцами, а пробоотбор сам по себе привносит новую неопределенность. Получение представительной пробы может быть почти таким же трудным, как и сам анализ. Пробоотбор и пробо-подготовку следует считать жизненно важной частью учебного курса аналитической химии. Статистика может быть мощным инструментом, н каждый студент должен по крайней мере знать разницу между правильностью и воспроизводимостью, а также, чем отличаются анализ и определение (см. гл. 2).
□ Задачи могут меняться, но методология решения задач не изменится.
Задачи, которые встанут перед промышленностью через несколько лет, а также методы решения этих задач, которые будут доступны в то время, могут радикально отличаться от того, что мы имеем сегодня. Это результат быстрых Изменений за последнее десятилетие не только в развитии новейших аналитических методов (таких, как атомно-силовая микроскопия, к примеру), но и во всевозрастающей сложности задач, возникающих перед обществом. В течение следующего десятилетия, согласно прогнозам ученых Национального института стандартов и технологий (NIST) в США, технические возможности аналитических измерений совершат такой же скачок, какой имел место за последние
1.2. Химик-аналитик в центре решения проблем общества 35
три десятилетия. Мы должны понимать, что обучение сегодняшних студентов специфическим методам двадцать первого века невозможно. Поэтому усилия необходимо сконцентрировать на том, что имеет непреходящее значение. Даже если задачи изменятся, то общая методология решения задач не изменится. Она по-прежнему будет включать:
— понимание химических основ;
— правильные пробоотбор и пробоподготовку;
— умения использовать методы разделения;
— соответствующий уровень градуировки и выбор стандартов;
— умение выбрать наилучший метод или методы для конкретного этапа аналитических измерений.
Как сказал Филипп У. Уэст при вручении ему премии Фишера Американского химического общества в 1974 г., «Аналитическая химия—это не спектрометры, полярографы, электронные микроскопы и прочее; это экспериментирование, наблюдения, установление фактов и логические заключения» [1.2-2]. В особенности в промышленности, где оборудование легче доступно, мы должны контролировать концентрацию иа каждом этапе контроля. Целью является ответ, а не метод. Задача и ее решение — это вершина; каждый химик-аналитик должен быть способен задачу разрешить.
1.2.3. Аналитический подход
□ Химик-аналитик должен представить достаточно обоснованную информацию, на основе которой руководство предприятия могло бы принять осмысленное решение.
Если аналитик-исследователь может действительно эффективно решать проблемы общества, то это совершенно другое измерение его роли, которая должна быть признана. Удачливый химик-аналитик должен быть адептом множества методов и быть в курсе происходящего иа передовой химических знаний. Функция химика-аналитика-^ предоставить достаточно обоснованную информацию, имеющую необходимую статистическую значимость, чтобы о материалах и задачах могло быть принято осмысленное решение. Подчеркнем здесь еще раз, что химик-аналитик по сути является лицом, решающим задачу, и чтобы выполнять работу наиболее эффективно, он должен использовать всеобъемлющий аналитический подход для разрешения задачи. Этот «аналитический подход» может быть определен следующими шагами (см. также гл. 2):
— правильно определить задачу;
— убедиться, что имеющиеся пробы представительны для решения этой задачи;
— взаимодействовать с заказчиком, чтобы получить от него сведения о задаче и определить требуемое соотношение между точностью и срочностью ответа;
— разработать план анализа, предусматривающий оценку последовательности операций к выбор наилучших методов;
2*
36 Глава 1. Задачи аналитической химии и ее значение для общества
— выполнить работу с использованием накопленного опыта и хорошего знания химии;
— сообщить ответ (а не данные), включающий оценку воспроизводимости и надежности всех чисел, со всеми предостережениями или ограничениями при использовании этих данных;
— интерпретировать информацию и результаты в ясном, последовательном и осмысленном отчете, соответствующем решению данной задачи.
□ Самый ответственный этап работы химика-аналитика'—это интерпретация и представление результатов.
Таким образом, ошибочно считать «измерение» основной ролью химиков-аналитиков, не считая рутинных, повторяющихся анализов. Химик-аналитик с самого начала должен быть активным участником группы, решающей задачу. В большинстве заводских лабораторий химию-аналитик выступает в качестве вспомогательной единицы, обеспечивающей работу исследовательской или производственной группы. Важно, чтобы он был активным участником в данном проекте, а не просто послушным источником аналитических результатов.
Химик-аналитик может внести значительный вклад в исследования или производство, взаимодействуя с экспертами на этапе планирования- задачи. В этом случае аналитик полностью вникает в общую суть задачи; например, знание источников сырья, условий производства, методов извлечения и потенциально возможных промежуточных продуктов. При необходимости с этими знаниями аналитик может предложить способы пробоотборК к комбинацию аналитических методов, наиболее подходящие для правильного разрешения задачи.
Представляется нелогичным, если аналитик не вовлечен в установление наилучшего способа сбора представительных и имеющих смысл проб, а также подходящего порядка проведения анализа. Ведь аналитик в группе, решающей задачу, имеет широкий обзор методов, которые могут быть использованы. В наше время сложных, сопряженных аналитических методов сила таких комбинированных методов очевидна (см. гл. 14), так как их применение дает максимум информации.
Наиболее важный вклад химиков-аналитиков — разъяснение механизма химических процессов, что значительно важнее, чем простое определение уровней концентрации; очень важно понимать, например, как работает катализатор, обеспечивая и выход, и селективность, или какие изменения могут произойти в структуре из-за изменения последовательности добавления молекул при полимерном синтезе. В области охраны окружающей среды потенциально важным может быть разъяснение токсикологического механизма влияния химиката на жизненно важные процессы.
Обобщая (рис. 1.2-2), можно сказать, что ученый-аналитик должен профессионально взаимодействовать с заказчиком для правильного определения задачи. Аналитический подход требует также, чтобы аналитик взаимодейство-
' 1.2. Хаммк-аналмтик в центре решения проблем общества
37
Рис. 1.2-2. Взаимодействия ученых-аналитиков при решении аналитических задач.
вал с другими учеными-аналитиками и применял их экспертную оценку для использования комбинированных методов, обеспечивающих информацией для решения задачи. И наконец, ученый-аналитик должен быть в курсе прогресса аналитических методов во всех областях методологии и оборудования. Только тогда химик-аналитик является настоящим «решателем проблем».
Это предполагает, что академическое химическое обучение должно обеспечить скорее общее широкое образование, нежели избыточную специализацию, поскольку в промышленности значительно проще специализироваться, чем расширить свой аналитический образовательный уровень. Основы теоретических знаний химии и экспериментальной практики должны закладываться в университете.
Еще один взгляд на роль химика-аналитика был представлен деХасетом [1.2-3], который отмечал, что химик-аналитик должен быть экспертом и в частном, и в общем. В промышленности необходимо также, чтобы химик-аналитик был участником команды. Можно утверждать, что ни один промышленный проект не является плодом усилий одного человека. Профессор Гари Хифтье из Университета Индианы, получивший премию Американского химического общества в области аналитической химии и премию за успехи в преподавании в Индиане, признавал оба эти требования. Он писал:
«... Студент часто наталкивается на трудности, когда пытается закончить дипломный проект за разумный промежуток времени, стараясь достичь достаточной широты кругозора, чтобы стать ответственным лицом, решающим задачи. Очевидно, что если студент проводит оригинальное исследование, требуемое для дипломной работы, он должен быть сосредоточен иа своей тематике. К сожалению, эта самая сосредоточенность часто делает студента менее способным к решению проблем. Для преодоления этой проблемы в моей исследовательской группе я применил следующую тактику. У каждого должна быть широкая и достаточно разнообразная тематика. Неизбежно, что наиболее тесные контакты студента, как профессиональные, так и социальные, находятся внутри той же самой исследовательской группы. Если каждый в этой группе
38 Глава 1. Задачи аналитической химии и ее значение для общества
работает в большом числе областей науки, каждый студент впитает понемногу такой широты кругозор и станет экспертом и в других областях, помимо его собственного исследования.
Кроме того, другая тактика, которую я применял, состоит в том, что студент должен работать в исследовательской команде. При этом каждый студент Приобретает не только более широкий научный кругозор, но и учится работать в окружении, ориентированном на коллективную работу. Интересно, что любой студент может стать лидером группы и может пригласить в группу кого-нибудь еще, включая приезжих ученых и стажеров.»
□ Для химика-аналитика важно поддерживать свой опыт на современном уровне и обновлять его путем непрерывного образования.
При обсуждении образования химиков-аналитиков также уместно обсудить важность повторного обучения и обновления навыков. Краткие курсы для таких целей повсеместно предлагаются профессиональными обществами, компаниями, производящими оборудование, и академическими институтами.
1.2.4. Решение аналитических проблем в науке об окружающей среде
Как уже отмечали выше, нет сомнений в том, что возможность проанализировать структуру и состав материала имеет важное значение в промышленном секторе и в обществе. Анализ конечных продуктов и поточный анализ промежуточных продуктов прямо на месте действительно обеспечили «промышленную революцию», и они-являются -жизненно необходимыми. Успехи в биотехнологии, медицине, электронике, катализе, в производстве полимеров и новых материалов и т. д. не были бы столь эффективными без новых аналитических методов.
□ Быстрые изменения технических возможностей химиков-аналитиков, произошедшие в последние 50 лет, позволили снизить пределы обнаружения, работать с меньшими пробами и с меньшими уровнями концентраций.
Рис. 1.2-3 иллюстрирует быстрые изменения пределов обнаружения, размеров пробы и времени анализа, произошедшие за время после второй мировой войны [1.2-4]. Пределы обнаружения от микрограммового уровня дошли
Рис. 1.2-3. Технический прогресс аналитической науки за время после второй мировой войны.
1.2. Химик-анаяитик в центре решения проблем общества
39
до уровня менее пикограммов. Пробы миллиметрового размера являлись достижением в 1950 г.; сегодня мы часто оперируем с пробами размерами менее микрометра или даже с единичными атомами, наблюдаемыми в аналитический электронный микроскоп; такие методы стали едва ли не рутинными. Время анализа уменьшилось с нескольких часов, а в отдельных случаях и дней, ранее требовавшихся для полного качественного и количественного анализа, до фемтосекунд, как, например, в случае спектроскопических исследований молекулярной динамики в режиме реального времени непосредственно в процессе эксперимента.
□ Потребности контроля окружающей среды заставляют аналитиков обнаруживать и определять содержание веществ во все более низких концентрациях и в более сложных матрицах.
Обнаружение и определение следовых количеств элементов и молекул во все более сложных матрицах сегодня легко выполнимы. Именно такие задачи обычно возникают при анализе объектов окружающей среды. Более того, некоторые люди склонны рассматривать успехи аналитической химии как негативный фактор в изучении окружающей среды, поскольку способность измерять крайне малые количества приводит к новым законодательным ограничениям. Возможность «видеть» ранее невидимые компоненты имеет как «хорошую», так и «плохую» стороны. С «плохой» стороны, мы можем идентифицировать некоторые компоненты, которые действительно являются вредными, и в то же время, инициировать беспричинную «охоту на ведьм» по отношению к материалам, которые могут этого не заслуживать. Но разве не было бы лучше для промышленности и общества направить наши усилия в аналитической науке на идентификацию большего числа компонентов в объектах окружающей среды для обеспечения раннего предупреждения экологических катастроф, опасных для людей и всего живого? Аналитическая наука могла бы таким образом помочь нам смягчить изменения в окружающей среде или адаптироваться к ним.
Таким образом, мы подошли ко второй стороне вопроса, т. е. к результатам оценки риска, управлению риском и сообщениям о риске. Эти слова вместе с понятиями «предел обнаружения», «селективность», «правильность», «аналитическая вариабельность», «гарантии качества» и «управление качеством» (см. гл. 2 и 3) должны быть определены и сделаны понятными для общества, законодательных органов и промышленности. Мы больше ие можем отделять эффективность науки от нашего предназначения сообщать обществу находки и неопределенности науки.
Какую же роль играют химики-аналитики в промышленности и экологии и как сделать их работу более эффективной? Как им следует влиять на качество связи между учеными, работающими в промышленности, и законодательными интересами или интересами общества?
Как показано на рис. 1.2-4, за период с 1955 по 1990 годы были достигнуты поразительные успехи в развитии аналитической науки в следовом анализе объектов окружающей среды. Однако следует принимать во внимание и иные аспекты. Речь идет о реальных практических возможностях, не всегда адекват-
40
Глава 1. Задачи аналитической химии и ее значение для общества
Рис. 1.2-4. Изменение пределов обнаружения в рутинных оценках качества воды с 1955 по 1990 гг.
ных состоянию науки; укажем, например, на неопределенность в пробоотборе для следового анализа, а также отнюдь не прямое соотношение между тем, что может быть измерено, и тем, что пагубно для человеческого здоровья.
Токсичность '—
Большинство из нас справедливо опасается раковых заболеваний. Мы можем определять крайне низкие содержания веществ, токсичность которых установлена и допустимое содержание которых в пище регламентируется на уровне, близком к нулевому. Отмечено, что в течение 1980-1990-х годов предел обнаружения снизился с концентраций, которые вызывают заметные нарушения в организме человека, до концентраций, которые на шесть или более порядков величины ниже [1.2-5]. Коротко говоря, не существует научно достоверного способа оценить надежность экстраполяции их токсичности на столь малые концентрации.
□ Из-за биологических различий между животными и человеком нет надежного способа переноса на людей данных о токсичности веществ, полученных на животных.
Новые данные [1.2-6] вступают в противоречие с результатами тестов на животных, по которым ранее оценивали концерогениость веществ. Полагают, что прежние работы были основаны на неясных идеях о возникновении рака, и, следовательно, были сделаны необоснованные допущения о влиянии малых доз на основе данных, полученных для высоких доз веществ. В недавней работе Эймса [1.2-6] показано, что во фруктах и продуктах присутствуют природные пестициды, причем их концентрация в сто и более раз превышает концентрацию любых синтетических веществ.
1.2. Химик-анаяитик в центре решения ярсблем общества
41
Аналитические источники погрешностей
О Неопределенность результатов анализа возрастает с уменьшением концентраций.
В реальных условиях аналитики сталкиваются с различными причинами неопределенности, с разнообразными источниками погрешностей в конечных результатах. Хотя такие источники погрешностей, как пробоотбрр, загрязнение пробы и низкая селективность методик, известны, на практике избежать их крайне сложно. В результате для уровня концентраций 1 млрд-1 средняя межлабораторная погрешность для ряда методик оказалась близкой к ±50%. Более того, погрешность быстро возрастает при переходе к более низким концентрациям (см. гл. 3 и 12).
□ Трудно достигнуть согласования между результатами различных лабораторий, даже когда они используют стандартные методики.
Отсутствие контроля качества при анализе образцов окружающей среды обсуждается, например, Хертцем (Hertz) [1.2-7] и иллюстрируется в табл. 1.2-1. В этом исследовании, проводимом Национальным бюро стандартов США (NBS, ныне Национальный институт стандартов и технологий, или NIST), результаты, полученные по «круговой системе» разными лабораториями, выполняющими рутинные анализы образцов, выпадают за пределы точности, достигаемые квалифицированными химиками при хорошей лабораторной практике. Например, при определении полихлорированных бифенилов (ПХБ) в нефти один из шести образцов, отправленных на анализ по круговой системе, ие содержал добавок Arochlor (ПХБ). Анализы NBS подтвердили отсутствие ПХБ в этом образце на уровне предела обнаружения методик. Тем не менее, 9 из 18 лабораторий сообщили о наличии ПХБ во всех шести образцах! Факторы, вносящие вклад в разброс результатов, включают процедуры пробоподготов-ки, чистоту реагентов, загрязнения образца из других источников, а также отсутствие доступных стандартных образцов (см. гл. 3).
Таблица 1.2-1. Изучение межлабораторного разброса результатов.
Изучаемая система Число образцов Число лабораторий Разброс (относительное стандартное отклонение)
ПХБ в нефти 6 18 38-64%
Se в плазме крови 27 а
Дкаротин в плазме крови 4 11 100% (на один образец) 35% (генеральное среднее)
Органические загрязнители в воде Рассолы из токсичных сбросов6 5 500%
* Диапазон результатов 38-100 мкг/л.
6 Для каждого из 9 определяемых элементов результаты различались в десятки раз.
42
Глава 1. Задачи аналитической химии и ее значение для общества
Оценка риска
□ Для широких слоев населения «оценка риска» — новое или вообще незнакомое понятие.
В свете огромных современных возможностей аналитической науки в определении сверхмалых количеств веществ и в то же время из-за трудностей в надежной реализации этих возможностей, очевидно, следует искать компромисс между затратами на химико-аналитическую работу и оценкой риска д ля окружающей среды. Технологические новшества открывают более широкие возможности в детектировании и анализе и стимулируют появление совершенно новых процессов или продуктов [1.2-8]. В то же время применение новых методов анализа дает импульс совершенствованию технологии с целью уменьшения загрязнений и выбросов. Как отмечено в статье Уайта и Рода [1.2-8], «технология формирует общество, а общество создает технологию».
Приведем пример из истории исследований загрязнений свинцом. Наличие свинца в воздухе и воде было результатом сжигания угля, работы металлургической промышленности, а после второй мировой войны—также использования этилированного бензина для автомобилей. Токсическое воздействие свинца было хорошо известно задолго до 1970г., когда Акт о чистой воде в Соединенных Штатах положил начало систематическим определениям вредных уровней свинца и контролю за его выбросами. Однако для детального анализа связей между человеческой деятельностью, загрязнением окружающей среды свинцом и экологическими последствиями требовались более низкие 'пределы обнаружения, чем доступные к тому времени.
Рис. 1.2-4 показывает разительное улучшение возможностей определения свинца в воде. На повышение чувствительности методов анализа общество отвечало технологическими новациями: чтобы отказаться от Црименения свинца, стали использовать неэтилированный бензин; заменили краски на основе свинца и свинцовые водопроводные трубы; отказались от припоев, содержащих свинец (например, стали производить бесшовные алюминиевые банки). Цикл осведомленности, социального отклика и технологической инновации стал прямым следствием нашей способности определять малые количества свинца в образцах сложного состава.
1.2.5. Стандарты для процедур и измерений в аналитической лаборатории
□ Для анализа объектов окружающей среды критичным является использование стандартных образцов, содержащих следовые количества определяемых компонентов, и аттестованных методик.
Разумно управляемая и эффективно работающая аналитическая лаборатория должна использовать хорошие н общепринятые лабораторные методики. Должен присутствовать контроль качества для всех методов и методик (см. гл.З). Это предусматривает единство измерений (traceabibily, «прослеживаемость») для всех первичных и вторичных стандартов и частую градуировку. Необходима также переносимость методологии между различными лабораториями или отделами организации (например, между исследовательской и
1.2. Химик-аиалитик в центре решения проблем общества
43
Таблица 1.2-2. Системы химического анализа
Выделение определяемого вещества Разделение определяемых веществ Определение
Растворение в микроволновом поле Газовая хроматография Элементный анализ ИСП-АЭС
Плавление Жидкостная хроматография Атомная абсорбция
Растворение Модификация матрицы Элементный анализ исп-мс
Жидкостная экстракция Сверхкритическая флюидная экстракция Жидкостная экстракция Пламенно-ионизационное Спектроскопическое
Твердофазная экстракция Масс-спектрометрия
заводской лабораториями). Для увеличения производительности следует использовать автоматизированные системы [1.2-9]. Национальные лаборатории стандартов в большинстве стран призваны выполнять эти задачи. Промышленности требуются Надежные современные аналитические измерения для выполнения требований по охране природы и для улучшения качества продуктов и процессов, с тем чтобы достичь конкурентного преимущества.
Примером взаимодействия промышленности и правительства в этой области является Консорциум автоматизированных аналитических лабораторных систем (CAALS), основанный при NIST в Соединенных Штатах в 1989 г. для координации совместной работы промышленности с NIST по развитию автоматизированных аккредитованных методов химического анализа. Принятое консорциумом понятие системы химического анализа включает составляющие, показанные в табл. 1.2-2. В процессе развития аналитической системы исследователи разрабатывают: а) схему (методику) химического анализа; б) стандартные образцы и в) методологию обеспечения качества. NIST является также поставщиком стандартных образцов состава и всех градуировочных н проверочных систем в Соединенных Штатах.
Весьма важной международной организацией, систематизирующей аналитические стандарты и методики, является Международный союз теоретической и прикладной химии (ИЮПАК). Его Аналитическое отделение работает очень активно, ио и все другие отделения (физической, неорганической, органической, макромолекулярной, клинической, медицинской и прикладной химии) также имеют комиссии, работающие, помимо всего прочего, над проблемами номенклатуры н методов в электрохимии, молекулярной спектроскопии, пищевой химии, химии полимеров, биотехнологии.
1.2.6. Будущее аналитической науки
□ Новые методы постоянно увеличивают возможности аналитической химии.
Сейчас на горизонте появилось много новых технологий, которые заметно воздействуют на возможности аналитиков будущего. Волоконная оптика, лазеры, хемометрика и нанотехнология—вот лишь некоторые из них. Проблемы
44
Глава 1. Задачи аналитической химии и ее значение для общества
Таблица 1.2-3. Будущие пути развития аналитической науки
Автоматизация и робототехника
Сети приборов
Истинно интеллектуальные приборы
Более сложные методы свертывания данных
On-line сенсоры и мияиатюризованные системы
Усовершенствованные дистанционные методы
Таблица 1.2-4. Будущие потребности аналитической химии
Более высокая чувствительность/селективность
Усовершенствованные сочетания аналитических методов
Тонкий трехмерный микро-, нано- и субповерхностный анализ
Более глубокое понимание и широкое использование метрологии
Возможность выполнять анализы в более жестких ситуациях in situ
Прямое зондирование локализации энергии в молекулах, состояний переходов и реакционной динамики
Интерпретация первичных аналитических данных с помощью экспертных систем
общества сложны, как никогда ранее, особенно в областях технологии окружающей среды, биотехнологии, передовых материалов да и информационной революции в целом. Некоторые из путей перспективного развития аналитической науки представлены в табл. 1.2-3. Следуя этими путями, аналитик должен буцет обеспечивать новые потребности иауки, приведенные в табл. 1.2-4. Поскольку мы продолжаем применять аналитический подход к новым вызовам, используя новые методы и методики, аналитик, который способен решать проблемы, продолжает оставаться ключевой фигурой в промышленности, науке и правительстве. Аналитическая химия занимает центральное место по отношению ко всем другим отраслям знания.
Литература
[1.2-1] Sommer, А. И., Phys. Today, Sept. 1976, 9.
[1.2-2] West, P.W., Anal. Chem., 1974, 46, 9, 784A.
[1.2-3] deHaseth, J., Spectroscopy, 1990, 5, К* 1, 20.
[1.2-4] Grasselli, J. G., in: Analytical Applications of Spectroscopy II: Davies A.M.C., Greaser, C.S. (Eds.). Cambridge, UK: The Royal Society of Chemistry, 1991.
[1.2-5] Harris, W.E., Anal. Chem., 1992, 64, 655A.
[1.2-6] Ames, Б. N., Gold, L. S., Chem. Eng. News, 1991, Jan. 7, 28.
[1.2-7] Hertz, H.S., Anal. Chem., 1988, 60, 76A.
[1.2-8] White, R.M., Rod, S.R., Environ. Sci. Technol., 1990, 24, 460.
[1.2-9] Salit, M. L., Guenther, F. R., Kramer, G. W., Griesmeyer, J. M., Anal. Chem., 1994, 66, 361A.
1.3. Задачи аналитической химии в исследовательских лабораториях
45
1.3. ЗАДАЧИ аналитической химии применительно К ИССЛЕДОВАТЕЛЬСКИМ ЛАБОРАТОРИЯМ1
1.3.1. Введение
Как правило, задача аналитической химии состоит в нахождении ответа на поставленный вопрос или в решении некоторой проблемы (см. разд. 1.1, 1.2). Очень часто для принятия важных решений требуются непосредственно результаты химических измерений. В ряде случаев это могут быть не результаты количественного анализа в традиционном смысле, а некоторые качественные характеристики. В частности, существует множество более или менее стандартизованных процедур для определения параметров, иных, чем концентрация (таких, как кислотное число, химическое потребление кислорода, содержание органического углерода, скорость фильтрации, биотоксичность нт. д.). В судебной практике часто требуется установить наличие или отсутствие определенного вещества. В материаловедении больший интерес, чем валовое содержание компонента, может представлять его локальное или поверхностное распределение либо же его содержание в некоторых «особых» точках образца (см. гл. 10). Правильность и хорошую воспроизводимость химических измерений призвана гарантировать система обеспечения качества (см. гл.З). Результаты анализа не должны зависеть от оператора, места и времени.
В этом отношении воспроизводимость результатов гораздо важнее, чем сходимость, т. е. случайная погрешность результатов, полученных одним и тем же аналитиком в одной и той же лаборатории в серии параллельных измерений' (см. разд. 2.4). Хорошая сходимость — необходимое, но не достаточное условие для полного контроля качества работы химической лаборатория. Международные соглашения, особенно в сфере торговли, требуют, чтобы результаты анализов не содержали систематической погрешности и могли быть проверены вне зависимости от того, кем, где и когда оии были получены. Результат химического измерения должен представлять собой истинное утверждение, и должна существовать возможность доказательства его истинности (см. гл. 3).
Практически истинность результата анализа неизвестного объекта означает, что большинство экспертов согласны с этим результатом. Во многих случаях сообщество экспертов определяет национальные или международные стандарты, служащие основой для проверки результатов измерений на протяжении всей измерительной процедуры (единство измерений, прослеживаемость). Измерительные методики часто стандартизуют и представляют в виде точного описания процедуры (см. гл.З).
Существуют «первичные», абсолютные, не требующие градуировки методы количественного химического анализа. Однако в большинстве инструментальных методов требуется градуировка с использованием необходимых образцов сравнения. Любая методика анализа в принципе может быть усовершенствована, и не следует этому препятствовать. Потому очень важно, чтобы любая стандартная рабочая методика не рассматривалась как утвержденная раз и навсегда. Даже в случае самых отработанных методик тщательная экспертиза может привести к выводу о возможности или необходимости их улучшения (с
1В соответствии с документом CITAC К* 1 (обеспечение качества в химическом анализе).
46
Глава 1. Задачи аналитической химии и ее значение для общества
точки зрения экономии материалов, стоимости, сокращения объема отходов и т. д.). Однако необходимо убедительно продемонстрировать, что результаты в этом случае получаются по крайней мере не хуже, чем при использовании традиционной методики. Это призваны сделать эксперты (способные СуДйть о деталях новой методики) под руководством органов аккредитации [1.3-1,1.3-2] .
Для оптимизации контроля качества в исследовательских лабораториях целесообразно разбить всю измерительную процедуру на отдельные блоки — «единичные операции», которые объединяются при необходимости в «единичные процедуры», из которых, комбинируя их желаемым образом, можно составить всю методику. Такой подход позволяет получить большое разнообразие методик. Кроме того, если установлено, что отдельные блоки дают правильные результаты применительно к определенной задаче или определенному компоненту, то с высокой вероятностью н вся методика также будет правильной. Таким образом, процедура поверки методик в этом случае упрощается: теперь для контроля качества достаточно лишь поверить правильность комбинаций отдельных стадий в целом.
Сейчас объединенная рабочая группа CITAC/EURACHEM готовит инструкцию по контролю качества для исследовательских лабораторий иа основе последней инструкции ISO25 с учетом специфики аналитической химии—по всем возможным «единичным операциям и процедурам». Квалифицированный аналитик должен уметь разбить последовательность описанных действий на блоки. Это требует знаний и в первую очередь опыта. Чтобы избежать многочисленных проб и ошибок при поиске оптимального сочетания блоков для разработки новой методики, необходимы обширные познания в области различных методов анализа.
Стандартная рабочая методика может строиться в форме дерева, содержащего различные варианты принятия решений. При ее разработке необходимо помнить, для какой цели она разрабатывается. Одна из них—обеспечение контроля качества. Для этого необходимо, чтобы все лабораторные процедуры выполнялись как можно более точно разными людьми и в разное время. Другая задача—обеспечение надежной прослеживаемости (см. разд. 3.2.1) методики. Эксперты должны иметь возможность оценить характеристики для каждого модуля методики, как бы давно ни были получены результаты. Наконец, руководящее лицо, удостоверяющее результаты анализа, должно иметь возможность проследить весь непрерывный ход операции с пробой. Описание стандартной рабочей методики ие должно быть многословным. Оно должно содержать минимум информации, достаточной, чтобы квалифицированный аналитик смог понять и выполнить все необходимые действия.
1.3.2. Примеры построения методик анализа
Описанный выше общий подход к построению методики предусматривает ее разбиение на ряд более или менее тесно связанных этапов (блоков, модулей). Естественными разграничительными линиями между блоками могут служить этапы, на которых выполнение методики может быть прервано, а проба может храниться длительное время без последствий для результатов анализа. Как в качественном, так и в количественном анализе такое разбиение обычно проводят в соответствии с общей логикой аналитического процесса (см. гл. 2).
1,3. Задачи аналитической химии в исследовательских лабораториях 47
Последний схематически можно представить в виде описанной ниже последовательности стадий.
Постановка аналитической задачи
Точную формулировку вопроса, на который необходимо дать ответ с помощью химических измерений, следует определить путем соглашения между потребителем результатов анализа и опытным химиком-аналитиком, хорошо знакомым с различными методами анализа, их достоинствами и недостатками. Следует обращать особое внимание на то, чтобы состав пробы не изменился в ходе отбора пробы н ее хранения.
Только квалифицированный аналитик способен понять общую концепцию каждой конкретной методики, ибо только он отдает себе отчет в ее характеристиках по точности и прочих и может принять необходимые меры, чтобы избежать неверной постановки задачи. Уже на этой стадии возможно разработать предварительный план анализа в виде комбинации отдельных блоков, включая условия отбора, хранения и подготовки пробы (см. разд. 2.2). В дальнейшем должно стать ясно, какого рода методы анализа следует использовать. Это могут быть, в частности, некоторые стандартизованные процедуры, призванные определить некоторое обобщающее свойство, методы анализа поверхности, определения следовых содержаний (см. гл. 10), методы анализа объектов окружающей среды, пищевых, промышленных продуктов и т. д.
В окончательном виде аналитическая задача должна включать в себя тре-буемые характеристики правильности и воспроизводимости, а также возможность сопоставления с другими данными, что необходимо для правильной интерпретации результатов.
Необходимые требования
Для получения надежных и правильных результатов, которые возможно сопоставлять с другими данными, полученными где-либо в мире, следует четко оговорить все необходимые для этого условия и требования—это первое условие для правильной интерпретации данных (необходимый минимум точности, включая правильность и воспроизводимость, форму представления, допускающую сопоставление с другими результатами). Эти требования следует затем перевести на язык технических условий.
Аналитик должен также сформулировать требования к прослеживаемости (см. разд. 3.2.1) методики, учитывая временные и стоимостные ограничения, доступный уровень точности и возможный риск получения ошибочных результатов. На стадии планирования анализа следует оценить каждый выбранный блок методики с помощью квалифицированного специалиста с тем, чтобы избежать возможных систематических погрешностей. Необходимо с самого начала предусмотреть возможные действия в случае угрозы их возникновения.
Персонал
Химические анализы, особенно в исследовательских лабораториях, должны выполняться высококвалифицированными и опытными специалистами (или под их руководством), имеющими соответствующий и документально подтвержденный уровень профессиональной квалификации, приобретенный в ходе
48 Глава 1. Задачи аналитической химии и ее значение для общества
предыдущего обучения в вузе или на производстве (см. гл. 3)'. Соответствующее образование можно получить в результате специализации в области аналитической химии при обучении в университете. Требования к уровню необходимого для этого образования сформулированы Рабочей группой по Аналитической химии (WPAC, ныне DAC, т. е. Отделение аналитической химии, ОАХ) Федерации европейских химических обществ (ФЕХО) в 1993 г. в документе под названием WPAC EUROCURRICULUM. Ои и послужил основой для создания как данного учебника, предназначенного для обучения общему курсу, так и серии специализированных (в том числе электронных) учебников для студентов младших курсов, а также системы Eurocourse и серин пособий, доступных через Интернет, для студентов старших курсов. Все это вместе взятое, а также практический опыт работы не менее двух лет, является абсолютно необходимым для того, чтобы занять руководящую должность в аналитической лаборатории. Кроме того, полная система управления качеством должна обеспечить постоянное переобучение и повышение квалификации в условиях стремительно изменяющихся измерительных технологий.
Лабораторные условия
Система полного контроля и управления качеством предъявляет также строгие требования к условиям работы лабораторий с точки зрения чистоты, безопасности и надежности. В некоторых помещениях допускается работа только определенного круга лиц. Режим работы таких помещений должен быть соответствующим образом документирован.
Оборудование
Все оборудование, используемое в лабораториях, должно по своим характеристикам (проверенным с помощью установленных процедуру удовлетворять требованиям, необходимым для решения поставленных задач. В соответствии с предназначением оборудования следует постоянно принимать меры по уходу за ним, контролю его характеристик и периодической калибровке.
Как правило, все оборудование, используемое в химических лабораториях, делится на следующие классы:
— оборудование общего назначения;
— мерная посуда;
— стандарты для измерения химических и физических величин;
— приборы для измерения физических величин (аналитические приборы); — компьютеры и иные устройства для обработки данных.
Реактивы
Квалификация реактивов, используемых для химических анализов, должна быть достаточной для решения поставленных задач. Желательно приобретать все реактивы у тех поставщиков, которые внедрили у себя систему обеспечения качества (в США, например, иа основе стандартов ISO 9000) и способны гарантировать требуемую квалификацию. Сейчас это особенно важно, поскольку для результатов любого количественного анализа стало необходимо обеспечить полную «прослеживаемость» по отношению к единице измерения «моль». Особое внимание к качеству реактивов следует уделять при определении следовых
1.3. Задачи аналитической химии в исследовательских лабораториях
49
седержаний. Фоновый сигнал необходимо определять отдельно, а не автоматически вычитать его из показаний прибора, поскольку при наличии матричных эффектов его величина становится неопределенной.
Оптимизация стратегии пробоотбора и анализа
Общая стратегия пробоотбора и собственно анализа должна гарантировать, что конечная цель анализа будет достигнута, а полученные результаты будут правильными (см. разд. 2.2). Важность стадии пробоотбора невозможно переоценить. Если проба не является представительной, то полученные результаты нельзя ни при каких условиях отнести к исходному материалу анализа, какой бы прекрасный метод анализа не был использован и как бы тщательно анализ не был выполнен. Пробоотбор всегда является источником погрешностей, величина которых зависит главным образом от степени гомогенности исходного материала.
Оптимальную стратегию пробоотбора аналитик должен разработать совместно с заказчиком (потребителем результатов анализа). Способ отбора пробы тесно связан со способом последующего ее анализа; при его разработке очень важно творческое мышление и здравый смысл. Блок стандартной рабочей методики, описывающий стратегию пробоотбора, может представлять собой схему в виде дерева, в котором варианты принятия решений (число и размер порций пробы) должны соответствовать требуемому уровню точности результатов и степени неоднородности объекта анализа.
Чтобы избежать ошибочного обнаружения, переопределения компонента или неверной интерпретации результатов, важно разработать стратегию пробоотбора и для контрольных образцов. Последние часто бывают необходимы в целях сравнения. Это могут быть, например, образцы почв, не загрязненные определяемым компонентом, образцы материала с составом, отвечающим нормативам и т. д.
План пробоотбора должен содержать также временную схему всех сопутствующих действий: подготовки требуемой тары (включая ее очистку), подготовки реагентов, необходимых для предотвращения разложения пробы в процессе ее хранения и транспортировки в лабораторию. Следует рассчитать и подготовить необходимый запас контейнеров для сбора всего требуемого числа проб (включая контрольные) и для хранения реактивов.
Пробоотбор
Отбор пробы следует проводить в точном соответствии с разработанным планом и задачей анализа. Он должен обеспечить представительность пробы. При оценке общей погрешности результатов анализа следует учитывать и неизбежную погрешность, связанную с пробоотбором.
Любое изменение состава пробы в ходе пробоотбора должно быть документировано и удостоверено. Прн этом следует принимать во внимание специфические свойства определяемого компонента. Летучесть, чувствительность к свету, термическая нестабильность, склонность к биодеградации, высокая химическая реакционная способность—все это следует серьезно учитывать при разработке стратегии пробоотбора и выборе оптимального способа его осуществления. Все оборудование и средства, используемые для отбора пробы, ее
SO Глава 1. Задачи аналитической химии и ее значение для общества
сокращения (см. ниже) и подготовки (в частности, экстракции), должны быть задокументированы, так же, как и все результаты, касающиеся контрольных («холостых») проб.
Персонал, участвующий в отборе и подготовке проб, должен быть соответствующим образом обучен и в ходе работы строго следовать предписаниям стандартной рабочей методики.
Гомогенизация пробы
Любая проба, доставленная в лабораторию, нуждается в дополнительной гомогенизации перед ее сокращением — в противном случае ее представительность не может быть гарантирована. Если неоднородность пробы (наличие в ней различных фаз) видна на глаз, то качество гомогенизации может определять качество результатов анализа в целом. Стандартные рабочие методики, описывающие стадию подготовки пробы, должны предусматривать разнообразные способы гомогенизации. При оценке общей погрешности результатов необходимо количественно оценить погрешность, связанную со стацией сокращения пробы. Последняя погрешность определяет число сокращенных проб, которые следует проанализировать, чтобы достичь требуемой точности результатов анализа.
Сокращение пробы
Очень важной стадией анализа является сокращение (уменьшение) пробы — отбор точно известной порции общей (генеральной) пробы, доставленной в лаг бораторию. При сокращении пробы необходимо учитывать всю информацию о степени ее неоднородности.
Сокращенная проба должна быть во всех отношениях подобна всему объекту анализа в целом. Погрешность, связанную с сокращением пробы, можно оценить количественно методами статистики (см. разд. 2.4) иЯГоснове результатов анализа большого числа порций пробы.
Количество сокращенной пробы лучше всего определять гравиметрически (см. разд. 7.2). Навески анализируемого материала и контрольных проб в ходе взвешивания следует обрабатывать в точности одинаково и одновременно с сокращенными пробами. При работе с суспензиями (кровь, плазма крови) следует избегать отбора аликвот с помощью мерной посуды, поскольку содержание твердых или коллоидных частиц (таких, как макромолекулы белков) может изменяться от аликвоты к аликвоте.
Обработка сокращенной пробы
В ходе операций пробоподготовки, таких, как растворение, разложение, сожжение, плавление и т. д., следует применять все меры, исключающие внесение систематических погрешностей. Лишь очень малое число методов анализа, в частности, некоторые варианты иейтронно-активационного анализа, не требуют пробоподготовки. Стацдартные рабочие методики пробоподготовки должны содержать описание множества различных методов, а также специфических мер предосторожности применительно к каждому типу пробы и определяемого компонента. На стадии пробоподготовки возможно включение в аналитический процесс некоторых элементов системы обеспечения качества (см. разд. 2.2 н гл. 3). Наряду с анализируемой пробой через весь процесс пробоподготовки следует провести также все контрольные образцы (в том числе
L3. Задачи аналитической химии в исследовательских лабораториях 51
меченые), образцы сравнения (приготовленные в лаборатория) и стандартные образцы, используемые в ходе анализа. При этом образцы сравнения и стандартные образцы по своему матричному составу должны максимально точно соответствовать пробе.
Разделение и концентрирование (см. гл. 5)
Наилучший способ контроля качества на стадии разделения и концентрирования трудно описать в деталях, поскольку все здесь зависит от множества факторов, касающихся специфики как объекта, так и поставленной задачи. Он может быть весьма жестким, громоздким или дорогостоящим, но в любом случае он должен обеспечивать минимальную погрешность. В некоторых случаях требуется, чтобы отделение определяемого компонента от матрицы было количественным. Отметим, что общая правильность результатов часто в решающей мере определяется стадией разделения и концентрирования. Процент потерь определяемого компонента (или загрязнения им пробы) в каждом случае свой, зависящий от природы как процесса, так и матрицы. Чем меньше содержание определяемого компонента, тем выше вероятность прямого или косвенного влияния какого-либо компонента матрицы на результаты анализа. В любом случае стандартная рабочая методика разделения должна содержать всю информацию, необходимую для ее точного выполнения, а также способ определения процента извлечения определяемого компонента.
В ходе разделения также важно, чтобы все используемые контрольные об-, разцы и образцы сравнения (меченные определяемым компонентом или близким к нему по свойствам внутренним стандартом) обрабатывались в точности тем же способом, что и проба, и по возможности одновременно с ней (параллельно либо через непродолжительное время). _
Определение
Заключительная стадия химического измерения состоит в установлении наличия либо отсутствия компонента в пробе, в определении его содержания либо измерении какой-либо обобщающей характеристики пробы. Международный словарь общих терминов метрологии определяет измеряемое количество как <атрибут... вещества, который может быть определен на качественном уровне и измерен количественно». Результатом заключительного измерительного процесса обычно является значение сигнала, которое само по себе ие представляет интереса для аналитика. Поэтому измерительную систему необходимо градуировать (за исключением методов кулонометрии, разд. 7.3, титриметрии и гравиметрии, разд. 7.1 и 7.2) с тем, чтобы установить функциональное соотношение между измеряемым сигналом и концентрацией или количеством компонента в пробе. Это соотношение может быть достаточно сложным. В простейшем случае оно представляет собой линейную зависимость, но это не является необходимым (см. разд. 12.2). В ходе поверки методики необходимо показать, что градуировочная функция, вне зависимости от того, как она была построена—с учетом или без учета влияния матрицы — позволяет получать правильные результаты применительно к анализируемой пробе. Кроме того, при градуировке важно не путать общий аналитический сигнал, сигнал фона и сигнал контрольного образца. Операция градуировки должна быть сплани
52 Глава 1. Задачи аналитической химии и ее значение для общества
рована таким образом, чтобы обеспечить «прослеживаемость» измерений по отношению к общепринятым единицам измерений либо аттестованным стандартным образцам.
Ответственность за выбор и использование методов и средств измерения, адекватных поставленной задаче, всегда лежит на аналитической лаборатории — вне зависимости от того, предлагает ли лаборатория свой метод самостоятельно, согласовывает его с заказчиком или использует метод, предлагаемый заказчиком либо требуемый в соответствии с нормативами.
Перед выполнением анализа следует проверить работоспособность измерительной аппаратуры. Стандартная рабочая методика должна содержать описание краткого теста, цель которого — проверить основные характеристики прибора, в первую очередь показатели селективности, чувствительности, стабильности и правильности. Применительно к исследовательским лабораториям и вообще при решении новых аналитических задач проверка правильности особенно важна ввиду отсутствия предварительных данных, касающихся применяемой методики.
Обработка результатов
Градуировка представляет собой установление функциональной зависимости д(С) между величиной измеряемого сигнала S и концентрацией С определяемого компонента: S = д(С). Для расчета концентрации из величины сигнала следует использовать функцию, обратную к д. Качество градуиророчной функции есть непосредственный показатель того, насколько верно с химической точки зрения понята данная система. Даже автоматизированные приборы должны предоставлять пользователю возможность выбора: какую градуировочную функцию использовать, каким способом выполнять коррекцию фона, ка^ой алгоритм применять для обработки данных. Корректность процедуры обработки данных должна быть доказана. Однако в ряде случаев спектральные приборы выполняют ряд действий автоматически, без проверки их корректности, но, например, вычитание спектров корректно только в том случае, когда наклон градуировочной функции не зависит от природы матрицы.
С помощью методов хемометрики определяют все аналитические характеристики методики в целом—такие, как предел обнаружения, нижняя граница определяемых содержаний, рабочий диапазон измерений, показатели правильности, воспроизводимости и другие (см. гл. 4 и 12). Все они должны быть определены и задокументированы. Целесообразно использование контрольных карт для того, чтобы быстро обнаружить нежелательные явления и оперативно их устранить.
При расчете результатов необходимо также рассчитать или хотя бы оценить общую погрешность данных, включая погрешность, вносимую в процессе пробоотбора.
Поверка и аттестация методики
Методики, разрабатываемые самостоятельно (в исследовательской лаборатории), необходимо перед их практическим применением тщательно проверить, а результаты проверки документировать и официально заверить. Для оценки систематической погрешности следует по возможности использовать стан-
1.3. Задачи аналитической химии в исследовательских лабораториях
53
д^ртяые образцы, а при их отсутствии сравнивать полученные результаты с данными, полученными другими методами,—желательно базирующимися на иных физических принципах. Оценка погрешности—важная составная часть проверки методики, необходимая для последующего контроля качества. Рекомендации по проверке методик содержатся в соответствующем руководстве СПАС.
Профессиональный долг химиков-аналитиков—гарантировать, что получаемые ими результаты пригодны для решения тех задач, для которых они предназначены. Сотрудники аналитических лабораторий несут ответственность за то, что результаты, поставляемые ими заказчикам, являются правильными и могут быть проверены путем сопоставления с данными, полученными другими. Правильность результатов должна быть достаточной для верного принятия решений, базирующихся на их основе. В этом н состоит причина необходимости проверки методик и оценки погрешности результатов: заказчик должен знать, на какую степень достоверности результатов ои может полагаться. Поэтому величину погрешности следует рассчитывать общепринятыми единообразными способами и представлять в легко интерпретируемой форме.
Представление результатов
Результаты измерений следует представлять в таком виде, чтобы заказчик легко мог их понять и сделать интересующие его выводы. Представление результатов не должно искажать их смысла. Полная погрешность (неопределенность) результата должна быть рассчитана или хотя бы оценена и представлена вместе с самими результатами (либо отдельно по требованию заказчика).
Представляемые результаты должны включать среднюю измеренную величину и стандартное отклонение, характеризующее случайную погрешность. При представлении общей погрешности (неопределенности), включающей также и систематическую составляющую, необходимо' дополнительно указать стандартное отклонение и число параллельных измерений.
Отчет о результатах
В отчете о результатах анализа должна содержаться вся необходимая информация, позволяющая задним числом ответить на любые вопросы, касающиеся хода анализа, оценить его качество, а при необходимости и повторить от начала до конца, включая стадию пробоотбора. Все изменения методики должны быть детально описаны с тем, чтобы любой другой специалист мог воспроизвести всю процедуру, буквально следуя указаниям, содержащимся в отчете.
1.3.3. Заключение
Проведенный краткий обзор требований, предъявляемых к современному химическому анализу, призван продемонстрировать студентам, каким образом в аналитической лаборатории возможна разработка стандартной рабочей методики на основе отдельных стандартных блоков операций. Следует подчеркнуть, что, хотя каждый блок можно поверить по отдельности применительно к решению тех или иных задач, важную роль играет и поверка конечного результата в целом. Одиако, если это по тем или иным причинам (отсутствие
54 Глава 1. Задачи аналитической химии и ее значение для общества " '
стандартных образцов или образцов сравнения с подходящей матрицей",'невозможность осуществления всей комбинации отдельных стадий и т. д.)> то проверка каждого отдельного этапа является наилучшим выходом из положения. Таким образом, модульный принцип построения методик является очень эффективным, он позволяет на основании контроля качества отдельных стадий делать правдоподобные выводы о качестве методики в целом и экономит много времени и средств.
Наконец, подчеркнем, что задача данного раздела состоит ие только в том, чтобы дать представление о сложности построения современных методик анализа, разрабатываемых в исследовательских лабораториях и об абсолютной необходимости получения результатов, сопоставимых с другими данными на всемирном уровне. В сочетании с конкретными разделами курса, представленными в других главах, данный раздел призван стимулировать внимание студентов к таким аспектам каждого метода, как проблемы точности (воспроизводимости и правильности) и «прослеживаемости», играющими огромную роль в нашем обществе, основанном на высоких технологиях.
Литература
[1.3-1] Kellner, Ret al., Fresenius* J. anal. Chem. 1997, 357, 197.
[1.3-2] Kellner, R et al., Anal. Chem. 1994, 66, 98A.
2
ПРОЦЕСС АНАЛИЗА
Цели изучения
• Понять роль химического анализа в процессе решения задач, поставленных заказчиком.
• Понять важность взаимодействия аналитика и заказчика.
• Ознакомиться с различными стадиями процесса анализа.
• Сформулировать основные понятия, отражающие аналитические характеристики методики.
2.1. ВВЕДЕНИЕ
Круг объектов, изучаемых в аналитической химии, чрезвычайно широк. В частности, объектами анализа могут быть: ~
— образцы сырья и готовой продукции на производстве (в том числе химическом);
— образцы почвы для решения вопроса о необходимости применения удобрений;
— пробы природной воды для оценйи степени загрязнения;
— пробы питьевой воды для сертификации ее чистоты;
— пробы руд для оценки содержания металлов и, тем самым, их коммерческой ценности;
— образцы крови для определения содержания холестерина;
— предметы старинной живописи, содержание в которых определенных пигментов может свидетельствовать о времени их создания и происхождении. Приведенные примеры показывают, насколько разнообразны задачи, решаемые аналитической химией. Поэтому аналитикам необходим общий язык, чтобы понимать друг друга. Поясним некоторые важнейшие термины.
В аналитической химии предмет исследования называют объектом. Часть объекта, непосредственно используемая для анализа, называется образцом или пробой, а исследуемые соединения или элементы, входящие в ее состав, — компонентами или анолитами. Аналиты находятся в матрице образца, которая
56
Глава 2. Процесс анализа
сама по себе не является предметом изучения аналитика, но может повлиять на результаты анализа. Такое влияние называется матричным эффектом. Для получения достоверных данных результаты анализа необходимо сравнить с результатом контрольного опыта.
Приступая к анализу любого объекта, химик должен четко понимать цель анализа. Обычно эту цель ставят другие люди—заказчики. В приведенных примерах заказчиками являются соответственно инженер на производстве, крестьянин, служба водного контроля, металлургическое предприятие, врач, директор музея. Очень часто сначала задача бывает сформулирована не на аналитическом языке (включающем такие понятия, как общий состав материала, содержание определяемого компонента или его строение). Поэтому прежде всего аналитику надо собрать всю информацию, необходимую для того, чтобы переформулировать задачу в терминах аналитической химии. Таким образом, нужен диалог между заказчиком и аналитиком с целью согласования постановки задачи.
В настоящее время в распоряжении аналитиков имеется настолько широкий круг средств, что выбор подходящего метода анализа нередко представляет собой трудную задачу, требующую большого опыта, а часто и хорошей интуиции. Единого «наилучшего» метода анализа не существует: каждая конкретная проблема требует своего подхода. Поэтому студенту прежде всего следует научиться систематическому подходу к выбору методики, основанному на глубоком понимании сущности наиболее важных методов.
□ Пригодные результаты анализа можно получить только тогда, когда четко поставлена общая задача.
Для этого в первую очередь необходимо располагать объективными характеристиками возможностей аналитических методик. Наиболее важные из таких аналитических характеристик обсуждаются в разд. 2.3. Кроме того, следует сформулировать поставленную задачу с точки зрения требуемых аналитических характеристик.
□ Аналитические характеристики являются объективными показателями возможностей методики.
На практике важную роль могут играть и дополнительные ограничения, в частности, доступность оборудования, необходимость проведения экспертизы, наличие подготовленного персонала. Оптимальным является метод анализа, способный обеспечить необходимые аналитические характеристики с учетом возможных ограничений.
2.2. АНАЛИТИЧЕСКИЙ ЦИКЛ
Общую схему процесса анализа можно представить в виде таблицы (табл. 2.2-1), в которой подчеркнута роль взаимодействия между аналитиком и заказчиком. Все этапы, указанные в ней, влияют на конечный результат. Поэтому их целесообразно кратко обсудить в порядке следования.
2.2. Аналитический цикл
57
Таблица 2.2-1. Общая схема полного аналитического процесса
Общая постановка задачи Заказчик Оценка степени загрязнения нефтью приповерхностного слоя почвы в городском сквере
Постановка конкретной аналитической задачи Заказчик «-> аналитик Насколько обширна область загрязнения
Выбор методики Аналитик Методика экстракции, отделения и определения нефти
Пробоотбор Заказчик + аналитик Отбор проб 100г), проверка их на представительность
Пробоподготовки. Аналитик Гомогенизация, сокращение пробы, экстракция ССЦ
Измерение Аналитик Газохроматографический анализ аликвоты экстракта
Обработка результатов Аналитик Идентификация хроматографических пиков, определение содержания компонентов
Выводы Аналитик Превышают ли полученные значения предельно допустимые?
Отчет Аналитик заказчик Рекомендации дальнейших действий- для решения проблемы
2.2.1. Общая постановка задачи
Как отмечено выше, часто общая постановка задачи находится обычно вие компетенции аналитика и вообще химии. Заказчик может вообще не иметь представления о возможностях и технических аспектах химического анализа. Результатом интенсивного обсуждения задачи между заказчиком и аналитиком должно явиться ясное понимание общей основы предстоящей аналитической процедуры.
2.2.2. Постановка конкретной аналитической задачи
Клиент и аналитик должны совместно определиться с предметом исследования: что необходимо определить и что возможно определить. На этом этапе необходимо получить ответы на вопросы, подобные следующим.
— Что представляет собой объект или образец, что нужно определить: молекулярный, элементный состав, или, скажем, функциональные группы?
— Качественный или количественный анализ требуется? Какая нужна точность количественного анализа?
58
Глава 2. Процесс анализа
— Какое количество материала доступно для анализа? Каковы примерные содержания определяемого компонента?
— Что представляет собой матрица образца?
— Достаточно ли определить только один компонент или требуется многокомпонентный анализ?
— В какие сроки требуется провести анализ? Другими словами: какое максимально допустимое время может пройти от взятия пробы до выдачи готовых результатов?
— Предполагается ли повторный анализ подобных образцов и, если да, то с какой периодичностью? Или же это единичный анализ?
— Желательно ли осуществлять непрерывный контроль системы, автоматизированный анализ?
— Допустимо или нет разрушение образца в ходе анализа?
Этот перечень вопросов ни в коей мере не является исчерпывающим. Он призван лишь показать, в каком русле должен происходить диалог, чтобы получить всю необходимую информацию для правильного выбора аналитического метода и методики.
2.2.3. Выбор методики
Выбор методики имеет решающее значение. Он определяет общие затраты на выполнение анализа, включающие как стоимость оборудования, так и~ оплату труда персонала. Выбор методики неизбежно ограничен некими рамочными условиями, такими, как количество образца, допустимое время^анализа, а также, возможно, дополнительной информацией.
2.2.4. Пробоотбор
В ходе лабораторного практикума студенты, как правило, имеют дело с гомогенными образцами. Поэтому они склонны недооценивать важность процедуры пробоотборв, являющейся на самом деле ключевым звеном любой аналитической методики [2.2-1]. На практике достоверность результатов анализа часто определяется качеством пробоотбора. Иногда анализируют весь объект целиком (например, древнее украшение) с помощью неразрушающего рентгенофлуоресцентного метода. Однако в большинстве случаев (подобных, скажем, определению железа в партии руды, перевозимой по морю) пробоотбор необходим. Пробоотбор состоит из двух стадий: а) разработка плана пробоотбора и б) отбор проб как таковой. Химику никогда не следует приступать к анализу, не выяснив предысторию образца (как выполняли отбор, хранение и консервацию пробы, подвергали ли пробу предварительной обработке и т. д.), а также насколько он представителен по отношению ко всему объекту. В зависимости от способа пробоотбора, природы определяемого компонента и его содержания, состава матрицы зависят меры, которые необходимо принять, чтобы избежать какого бы то ни было изменения состава пробы.
Твердые материалы. Проба должна быть представительной, но по экономическим соображениям ее размер не должен быть больше, чем это строго
2.2. Аналитический цикл
59
необходимо. Размер пробы зависит от требуемой точности анализа, степени неоднородности материала и размера его частиц. Иногда для обеспечения представительности требуются пробы массой несколько килограммов или даже сотен килограммов. Такую большую пробу необходимо измельчить, просеять и гомогенизировать перед сокращением ее до лабораторной пробы подходящих размеров. В ходе измельчения выделяется тепло, которое может вызвать потери летучих компонентов, а контакт вновь образованных поверхностей с воздухом может привести к окислению и изменению степени окисления некоторых элементов (например, окисление Fe(II) до Fe(III)).
Жидкости и газы. Обычно жидкости и газы достаточно однородны или могут быть легко гомогенизированы, поэтому их пробы, как правило, невелики. Однако используемые для их хранения сосуды имеют малый объем и соответственно большую величину отношения поверхности стенок к объему. Это может вызвать достаточно большие потери за счет адсорбции. Поэтому необходимо добиться установления равновесия между пробой и стенками путем длительного ополаскивания сосуда (эту операцию называют уравновешиванием).
□ Взаимодействие между пробой и стенками контейнера может изменить состав пробы. Поэтому стенки контейнера необходимо предварительно обработать таким образом, чтобы исключить эту возможность.
Описание аппаратуры для пробоотбора не входит в нашу задачу. В специальной литературе можно найти подробное обсуждение конкретных способов отбора газообразных, жидких и твердых проб. Но в любом случае должно быть полностью исключено загрязнение пробы со стороны устройств для пробоотбора и сосудов для хранения пробы. Все пробы должны быть четко подписаны с указанием источника, даты и времени отбора, а также определяемых компонентов. Если существуют специальные формы заполнения данных о пробе, следует руководствоваться ими. Наконец, следует помнить, что иногда отбор проб может быть очень опасен; в этих случаях необходимо принимать соответствующие меры предосторожности, соблюдая правила техники безопасности.
2.2.5. Транспортировка и хранение проб
При транспортировке состав пробы может измениться вследствие протекания реакций, вызванных интенсивным перемешиванием. При транспортировке пробы по трубопроводам возможна диффузия газов через стенки труб и вызванные этим потери или загрязнение пробы.
Между отбором пробы и ее исследованием может пройти значительное время. Например, пробы антарктического льда могут быть проанализированы в другой части земного шара. При этом также возможны потери компонентов за счет адсорбции на стенках или загрязнение пробы.
2.2.6. Пробоподготовка
Пробоподготовка преследует несколько целей. Одна из них — перевод пробы в физическое состояние, требуемое для выбранной методики (см. рис. 2.2-1).
60
Глава 2. Процесс анализа
Еще одна задача—позаботиться о том, чтобы содержание определяемого компонента соответствовало оптимальному для данной методики диапазону. При анализе жидких образцов это обычно достигается разбавлением или, наоборот, концентрированием следовых компонентов. Для твердых проб часто требуется их перевод в жидкое состояние с помощью простого растворения или разложения. В ряде методов, предназначенных для непосредственного анализа твердых образцов, требуется тщательная очистка поверхности проб или придание им электропроводности. В последнем случае чаще всего используют смешивание пробы с графитовым порошком или напыление иа ее поверхность золота.
При определении следовых компонентов исходные концентрации часто столь низки, что непосредственное определение невозможно ни одним из Существующих методов (2.2-2]. В этих случаях необходимо концентрирование, чаще всего с помощью экстракции, сорбции или ионного обмена. Эти же методы могут быть использованы и как методы разделения, чтобы отделить мешающие компоненты. Мешающее влияние посторонних компонентов можно устранить и добавлением специальных реагентов. Этот способ называется маскированием.
□ Часто для устранения мешающего влияния посторонних компонентов необходимо их отделение.
□ Мешающее влияние бывает возможно устранить путем перевода мешающего компонента в другую форму.
Состояние пробы
Форма после пробооодготоми
Форма, модимм априбор
(жидкость) (ж.
(твердое тело) (тв.
Рис. 2.2-1. Способы пробоподготовки:"
1. Непосредственный ввод пробы в прибор.
2. Превращение газа в жидкость — конденсация или экстракция из газовой фазы.
3. Превращение газа в твердое тело—конденсация.
4. Превращение жидкости в газ — испарение.
5. Непосредственный ввод пробы в прибор или жидкостная экстракция.
6. Превращение жидкости в твердое тело—осаждение, выпаривание растворителя, лиофильное высушивание.
7. Превращение твердого тела в газ — испарение.
8 Превращение твердого тела в жидкость — растворение, мокрое разложение.
9. Непосредственный ввод пробы в прибор.
2-2. Аналитический цикл
61
2.2.7. Измерение (определение)
В основе большинства инструментальных методов анализа лежит сравнение сигнала образца с сигналами одного или нескольких образцов сравнения точно известного состава; таким образом, необходимой частью методики является градуировка. В разд. 2.4 и гл. 12 подробно рассмотрены статистические аспекты обработки результатов измерений. Пока же мы лишь отметим, что показания измерительных приборов подвержены нежелательным, но неизбежным флуктуациям, называемым шумом. Уровень шума обычно характеризуют стандартным отклонением флуктуаций сигнала (см. разд. 2.4). Для улучшения качества результатов необходимо увеличение отношения си гнал/шум. Это можно обеспечить как на стадии измерения, так и на стадии обработки данных.
2.2.8. Обработка данных
В настоящее время большинство аналитических приборов снабжено компьютерами. В результате операция преобразования данных в аналитическую форму (концентрации компонентов или их структурные параметры) стала неотъемлемой составной частью аналитической системы. Она осуществляется автоматически — аналитику нет нужды обрабатывать и даже вообще контролировать первичные данные. Поэтому очень важное значение имеет правильная работа используемого программного обеспечения.
О Решающим фактором успеха в химическом анализе является проверка как самой методики, так и программного обеспечения.
Во многих случаях, например, при идентификации веществ методами распознавания образе®, анализ вообще невозможен без помощи компьютеров. Исходным материалом для построения образов могут служить, в частности, данные инфракрасных или масс-спектров (см. разд. 9.2 и 9.3). Процедуры, основанные иа сравнении спектра неизвестного соединения со спектрами индивидуальных веществ, хранящихся в памяти компьютера, называются поиском в базах данных или библиотечным поиском (см. гл. 13).
2.2.9. Выводы и отчет
Аналитик несет полную ответственность зв результаты, которые собирается сообщить. Он должен четко указать, с какой точностью получены все данные, избежать любой возможной двусмысленности в их интерпретации. Для обеспечения качества результатов (см. гл. 3) необходимо проверять применяемые методики на аттестованных стандартных образцах (материалах известного состава, проанализированных различными методами в различных лабораториях). Если стандартные образцы недоступны, можно сравнить полученные результаты с данными, полученными с помощью независимой методики (желательно, аттестованной арбитражной методики). Аналитик должен быть уверенным в том, что полученные им данные действительно необходимы для решения общей задачи, поставленной заказчиком.
62
Глава 2. Процесс анализа
Подчеркнем, что аналитик несет ответственность не только за результаты анализа как таковые, но и за все последующие заключения, которые могут быть полученные на основе этих результатов.
□ В отчете следует четко указать, какова точность полученных данных. Должна быть исключена любая возможность, позволяющая заказчику прийти к необоснованным выводам.
2.3. АНАЛИТИЧЕСКИЕ ХАРАКТЕРИСТИКИ
Аналитическую методику можно охарактеризовать как с помощью понятий, описывающих качество получаемых результатов, так и с помощью категорий преимущественно экономического характера (последние здесь не рассматриваются):
Характеристики качества результа- Экономические характеристики
тов анализа
Чувствительность Капитальные затраты
Воспроизводимость Продолжительность анализа и связан-
ные с этим затраты на оплату труда
Правильность Особые меры безопасности '
Предел обнаружения Стоимость установки и ремонта оборудо-
вания
Нижняя граница определяемых содержа- Стоимость обучения персонала, квали-
НИЙ фикационные надбавки
Селективность Затраты, связанные с потреблением об-
разцов
Рабочий диапазон Стоимость реактивов
Помимо этого, важными характеристиками методики являются: требуемый способ пробоподготовки, возможность неразрушающего или многокомпонентного анализа н др.
К сожалению, термины, обозначающие аналитические характеристики, часто употребляются в разных значениях. В данной книге эти термины используются в строгом соответствии с рекомендациями Международной организации по стандартизации (ISO) н Международного союза теоретической н прикладной химии (ИЮПАК).
Правильность
Правильность есть характеристика близости среднего результата измеренной величины к постулируемому истинному значению (см. разд. 2.4).
Воспроизводимость
Воспроизводимость есть характеристика разброса результатов измерений относительно среднего значения (см. разд. 2.4 и рис. 2.3-1).
2.3. Аналитические характеристики
63
Результаты правильные, но плохо воспроизводимые
Результаты неправильные и плохо воспроизводимые
Рис. 2.3-1. Иллюстрация понятий правильность н воспроизводимость.
Чувствительность
Характеристикой чувствительности является коэффициент чувствительности — мера степени изменения аналитического сигнала Y прн изменении концентрации: S = dY/dc (см. разд. 12.2).
Предел обнаружения
Если аналитический сигнал лишь близок к средней величине сигнала контрольного опыта (фоновый сигнал), то возникает вопрос, обусловлено ли это превышение наличием определяемого вещества или же флуктуациями фонового сигнала, называемыми шумом. Предел обнаружения соответствует сигналу, превышающему среднее фоновое значение в к раз (т. е. отношение полезный сигнал/шум равно к). Как правило, значение к выбирают равным 3 (см. разд. 12.1). Если сигнал выше предела обнаружения, то это свидетельствует о наличии определяемого вещества, а если ниже предела обнаружения—о его отсутствии.
Нижняя граница определяемых содержаний
В отличие от качественного анализа, где требуется лишь установить факт наличия вещества, в количественном анализе необходимо определить его содержание. При этом численные величины, характеризующие это содержание, должны удовлетворять необходимым точностным требованиям, для чего подвергаются дальнейшей обработке различными методами математической статистики, например, в самом простом случае вычисляют среднее значение. Поэтому нижняя граница определяемых содержаний (характеризующая возмож
64
Глава 2. Процесс анализа
ности метода в плане количественного анализа) по абсолютному значению всегда выше, чем предел обнаружения (возможности качественного анализа).
Селективность
Селективность характеризует то, насколько сильно посторонние компоненты пробы влияют на результат анализа. В разных методах анализа используют различные способы количественной оценки селективности (см., например, разд. 5.1 и 7.3). Иногда употребляют термин «специфичность», означающий, что никакие компоненты, кроме определяемого, не влияют на величину аналитического сигнала. Однако подобное вряд ли возможно (за исключением, может быть, лишь некоторых так называемых гибридных методов), поэтому термина «специфичность» следует избегать.
□ Международная договоренность относительно использования терминологии создает основу взаимопонимания.
2.4. ПОГРЕШНОСТИ В ХИМИЧЕСКОМ АНАЛИЗЕ
Цели изучения
• Усвоить фундаментальные понятия, касающиеся типов, источников и распространения погрешностей в химическом анализе.
• Подчеркнуть значение понятий «систематическая погрешность* и «правильность* в аналитической химии.
• Разъяснить значение терминов «сходимость*, «воспроизводимость», «лабораторная погрешность*, «погрешность методики».
• Рассмотреть, какая основная информация необходима для правильного представления аналитических данных.
2.4.1. Правильность и воспроизводимость
Из практики известно, что результаты любых измерений содержат погрешности, которые могут значительно искажать экспериментальные данные. Что следует, предпринять, чтобы минимизировать погрешности, зависит от природы самой погрешности. Обычно различают три типа погрешностей: грубые, случайные н систематические [2.4-1].
Грубые погрешности возникают в результате серьезных отклонений от стандартных условий эксперимента. Их следует всячески избегать, но, к несчастью, время от времени они все-таки случаются. Причинами могут быть сбои в работе прибора, значительные загрязнения реактивов, случайные потери образца и т. д. При обнаружении источников грубых погрешностей обычно не остается ничего иного, кроме как прекратить эксперимент н начать его заново,
В дальнейшем будут обсуждаться лишь случайные (Ьклуч) и систематические (Eclttv} погрешности. Основные особенности этих двух типов погрешностей сведены и сопоставлены в табл. 2.4-1.
2.4. Погрешности в химическом анализе
65
Таблица 2.4-1. Сводка важнейших характеристик случайных и систематических погрешностей
Случайные (недетерминированные) погрешности
1. Источник — неопределенность результатов (персонального, инструментального или методического происхождения).
2. Не устранимы, но могут быть уменьшены за счет аккуратности в работе.
3. Проявляются как разброс данных относительно среднего.
4. Определяют воспроизводимость.®
5. Количественно характеризуется мерами воспроизводимости (например, стандартным отклонением).
Систематические (детерминированные) погрешности
1. Источник — смещение результатов (персонального, инструментального или методического происхождения).
2. В принципе могут быть выявлены н уменьшены (или даже полностью устранены).
3. Проявляются как расхождение между средним и истинным значением.* 6
4. Определяют правильность.®
5. Количественно характеризуются как разность между средним и истинным
значениями.”
& См. текст.
6 Если случайная погрешность не настолько велика, что маскирует систематическую. в Справедливо, если число измерений достаточно велико для надежной оценки среднего.
Пожалуй, наиболее важными характеристиками качества аналитической -методики являются правильность н воспроизводимость. Обычно сначала оценивают воспроизводимость, поскольку систематическую погрешность (определяющую правильность) можно оценить только после того, как известнавели-чина случайной погрешности.
Случайную погрешность можно оценить из серии параллельных (повторных) измерений. Например, если XitX2t...Xn — значения объемов стандартного раствора в серии п независимых повторных титрований, то эти значения обычно отличаются друг от друга вследствие погрешностей, неизбежно возникающих в ходе любой измерительной процедуры. Простейшая операция, которую можно произвести над этими данными, — усреднение, например, путем вычисления среднего арифметического:
Х = £- (2.4-1)
г
□ Среднее (синоним — «среднее арифметическое») может служить характеристикой «центральной» величины для серии п повторных измерений.
□ Во многих случаях медиана является более реалистичной характеристикой «центральной» величины, чем среднее.
Средняя величина является более хорошей оценкой истинного (неизвестного) значения измеряемой величины, чем каждый результат в отдельности. Для этой же цели можно использовать центральное значение, называемое медианой. Если все результаты расположить в порядке возрастания, то медиана
3 Аналитическая химия. Том 1
66
Глава 2. Процесс анализа
представляет собой величину, равноотстоящую от наименьшего и наибольшего значения. При нечетном числе данных медиана находится в середине этого ряда. Например, в ряду из девяти значений
10,10; 10,20; 10,40; 10,46; 10,50; 10,54; 10,60; 10,80; 10,90
медиана равна пятому по порядку значению, а именно, 10,50. В данном случае она совпала со средним арифметическим. Отметим, что для любой величины, имеющей симметричную функцию распределения (см. ниже), и только в этом случае медиана и среднее совпадают. При четном числе данных медиана равна полусумме двух расположенных посередине значений. Так, если добавить к приведенному выше ряду десятое значение 12,80, то медиана станет равна (10,50 +10,54)/2 = 10,52. Сравните это значение с новым средним арифметическим, равным 10,73. Как будет более подробно сказано в разд. 12.1, резко выпадающие значения (промахи) сильно влияют на величину среднего н незначительно—на величину медианы. Поэтому если набор данных может содержать промахи, использование медианы предпочтительнее, чем среднего.
□ Среднее, равное 10,73, больше любого из трех близко расположенных в середине ряда значений 10,46, 10,50 и 10,54. Очевидно, что оно является менее правдоподобной оценкой «центрального значения», чем медиана, равная 10,50. Причина состоит в том, что в данном случае значение 12,80 является промахом.
Значения Хг в серии п величин в большей или меньшей-степени разбросаны относительно среднего. Величина разброса определяется как качеством работы оператора, так н качеством применяемой методики. Близость друг к другу отдельных значений в серии результатов повторных (параллельных) измерений, т. е. степень разброса данных относительно среднего X, называется воспроизводимостью. В качестве меры воспроизводимости широко используются следующие величины:
выборочное стандартное отклонение
E№-x)2j1/2
(2.4-2)
относительное стандартное отклонение относительное стандартное отклонение (%) дисперсия
s
St~x
Sr% = (j)100
V = s2
(2.4-3)
(2.4-4)
(2.4-5)
Здесь п — число результатов. В уравнении (2.4-2) величина п — 1 называется числом степеней свободы. Мерой воспроизводимости могут служить также
2.4. Погрешности в химическом анализе
67
следующие величины (хотя их использование не рекомендуется):
среднее отклонение
относительное среднее отклонение
относительное среднее отклонение (%)
d=------
п
RMD = i
RMD% = (i) 100
(2.4-6)
(2.4-7)
(2.4-8)
Таким образом, фраза «результаты хорошо (плохо) воспроизводимы» означает, что данные характеризуются малым (большим) разбросом относительно среднего н соответственно низкими (высокими) значениями s и d.
D Относительное стандартное отклонение, выраженное в процентах (sr%), называют также коэффициентом вариации (CV).
D Число степеней свободы есть число независимых данных в серии. Для серии из п независимых данных оно равно п. Однако в серии п отдельных отклонений (Xi — X) содержится только п — 1 независимых данных, поскольку их сумма всегда равна нулю. Таким образом, в результате вычисления среднего одна степень свободы оказывается потерянной, и лишь п — 1 величин отклонений являются независимыми характеристиками воспроизводимости серии данных.
□ Сходимость и собственно воспроизводимость —два частных случая воспроизводимости (в широком смысле).
Опыт показывает, что воспроизводимость результатов зависит от 1£го, были они получены в разных лабораториях или в одной и той же лаборатории, но за более длительный период времени, например в течение нескольких дней, а не часов. Поэтому различают, в частности, внутрилабораторную и межлабораторную воспроизводимость, называемые соответственно сходимостью и собственно воспроизводимостью (в узком смысле слова). Согласно рекомендациям ISO, термин сходимость следует использовать для характеристики близости отдельных результатов, полученных для одного н того же образца одним н тем же методом в одних н тех же условиях (оператор, аппаратура, лаборатория) в течение короткого промежутка времени. Аналогично, воспроизводимость в узком смысле слова характеризует близость отдельных результатов, полученных для одного и того же образца одним и тем же методом, но в различных условиях (в частности, различными операторами, в разных лабораториях, на разном оборудовании, в разное время). Очевидно, что понятия «сходимость» и «воспроизводимость» имеют один и тот же общий смысл, но характеризуют соответственно строго н менее строго контролируемые условия эксперимента.
Воспроизводимость, понимаемая в широком смысле слова, состоит из двух составляющих: случайных внутри лабораторных погрешностей (сходимость) и систематических погрешностей, присущих каждой отдельной лаборатории. В общем случае систематическая погрешность каждой лаборатории заранее не известна, а распределение этих погрешностей близко к нормальному. Таким образом, сходимость всегда лучше, чем воспроизводимость (в узком смысле).
з*
68
Глава 2. Процесс анализа
2.4.2. Смещение и точность
Обозначим истинное (как правило, неизвестное) значение измеряемой величины как Т. Тогда разность
Ei = Xi - Т (2.4-9)
есть погрешность величины Хг. При достаточно большом числе параллельных измерений среднее значение X относительно «устойчиво». Оно является оценкой величины р — среднего значения, которое могло бы быть получено при бесконечном числе параллельных измерений. В этом случае разность X — Т называется смещением или систематической погрешностью Ест:т. Подчеркнем, что рассчитанная таким образом величина смещения является лишь оценочным значением для истинной величины смещения, поскольку используемая для ее расчета величина X сама по себе является оценкой.
Выражение для Ег можно переписать в виде
Ei = (Xi - X) + (X - Т) =
— Погрешность случайная — Смещение, т. е. Еслуч — Есист (2.4-10)
Источниками систематических погрешностей может быть лабораторная погрешность н/или погрешность методики. Последняя является систематической погрешностью, присущей данной методике. Она вносит вклад в, общую погрешность результатов, получаемых в любой лаборатории, использующих эту методику. Поэтому систематическая погрешность, оцененная в ходе межлабораторного эксперимента, идентична погрешности методики. (Межлабораторный эксперимент заключается в том, что пробы одного идого же объекта анализируют во множестве случайно выбранных лабораторий, используя одну и ту же методику). Если в выражении (2.4-10) X представляет собой среднее из результатов X», полученных в разных лабораториях (рис. 2.4-1), то Есист является погрешностью методики. Для отдельной лаборатории систематическая погрешность представляет собой сумму систематической погрешности методики и систематической погрешности, присущей данной лаборатории. Эта величина является мерой точности результатов. Точность — понятие не вполне однозначное: под точностью можно понимать как разность X—Т (абсолютная точность среднего), так и Хг- — Т (абсолютная точность единичного результата). В первом случае точность есть синоним систематической погрешности (сумма погрешности методики и лаборатории), а во втором — представляет собой комбинацию систематической и случайной погрешностей.
Таким образом, представляется разумным оставить термин «точность» лишь для разговорной речи, а для строгого изложения использовать взамен только однозначные понятия, такие, как лабораторная систематическая погрешность, систематическая погрешность методики и общая погрешность (комбинация систематической погрешности методики и случайной погрешности).
□ Смещение есть постоянная (систематическая) погрешность, проявляющаяся как постоянное отклонение среднего от (постулируемого) истинного значения в большую или меньшую сторону.
2.4. Погрешности в химическом анализе
69
Отдельная лаборатория
Систематическая погрешность методики
Систематическая погрешность лаборатории
Сходимость нормально распределенная внутрилабораторная случайная погрешность
Истинное значение
У
Нормально распределенные систематические погрешности лабораторий Нормально распределенные внугрилабораторные случайные погрешности
Зистемагическая погрешность методики Случайная погрешность (воспроизводимость)
Межлабораторный эксперимент
Рис. 2.4-1. Схема разложения погрешности на составляющие. Общая погрешность величины Xij (результат г-го измерения, выполненного в у-й лаборатории) есть сумма внутрилабораторной случайной погрешности, лабораторной систематической погрешности н систематической погрешности методики. Применительно к отдельной лаборатории правильность характеризуется общей систематической погрешностью — суммой двух последних составляющих. Внутрилабораторная случайная погрешность характеризует лабораторную воспроизводимость (сходимость). Лабораторная систематическая погрешность, которая применительно к данной лаборатории есть компонент общей систематической погрешности, в условиях межлабораторного эксперимента обычно считается нормально распределенной величиной н, таким образом, вносит вклад в межлабораторную воспроизводимость (другим ее компонентом служит внутрилабораторная сходимость) Xj означает среднее из всех результатов для у-й лаборатории, а X — общее среднее из всех результатов.
D Погрешность состоит из систематической и случайной составляющей. Для единичного измерения можно оценить только общую погрешность Е: Е = X-Т
D Малость смещения называется правильностью.
Малость любой погрешности называется точностью.
D Результаты называются точными, если они хорошо воспроизводимы и в то же время не смещены.
D Методы непараметрической статистики не базируются ни на каких предпосылках относительно закона распределения данных. 14х чаще используют в тестах значимости (подробнее см. разд. 12.2), чем для оценивания.
D Медиана есть устойчивая оценка среднего значения набора данных.
то Глава 2. Процесс анализа
2.4.3. Представление аналитических данных
Как мы уже подчеркивали, результат эксперимента, не сопровождаемый оценкой экспериментальной погрешности, лишен смысла. Наилучшим н постоянно рекомендуемым способом такой оценки служит представление результата в виде среднего (X) и его доверительных границ (X ±ts/у/п) для заданной (например, 95%) вероятности (подробнее см. разд. 12.1). Заметим, однако, что такой сйособ расчета предполагает нормальный закон распределения исходных данных (см. разд. 12.1). При этом предварительно, до вычисления выборочных среднего и стандартного отклонения, используемых в дальнейших расчетах, следует с помощью соответствующих тестов выявить все возможные промахи н исключить их из серии данных.
Часто распределение результатов анализа лишь приближенно соответствует нормальному закону (например, оно, как и нормальное, может быть симметричным и унимодальным, однако обладать «хвостами», значительно более выраженными по сравнению с нормальным распределением), а серии данных могут содержать промахи. В подобных случаях целесообразно использовать устойчивые (робастные) статистические методы и представлять данные графически в форме, показанной на рис. 2.4-2. Обычно в робастных методах вместо среднего используют медиану, а вместо выборочного стандартного отклонения — в частности, межквартильный размах. Поясним значение последней характеристики. Мы определили медиану как среднее по порядку значение серин результатов. Аналогично, можно определить величины, являющиеся средними по порядку между наименьшим значением и медианой и между' медианой и наибольшим значением. Они называются, соответственно, нижней (LQ) и верхней (UQ) квартилью. Межквартильный размах (IQR) есть разность UQ-LQ. Кроме того, данные можно представить просто в виде среднего (X) с указанием числа результатов п, из которых оно рассчитано, и стандартного от-
Рнс. 2.4-2. Форма представления серии результатов измерений (на примере серии 10,10; 10,20; 10,40; 10,46; 10,50; 10,54; 10,60; 10,80; 10,90; 12,80). Прямоугольник включает в себя 50% всех данных (например, находящихся в пределах межк-вартилыного размаха IQR), медиана представлена горизонтальным отрезком. Вертикальные отрезки, выходящие за границы прямоугольника, ограничивают диапазон данных, считаемый приемлемым, например в пределах от LQ-1.5IQR до UQ+1,5 IQR, где LQ и UQ — соответственно нижняя и верхняя квартиль. Все значения, лежащие вне этих пределов, считаются выпадающими и изображаются в виде отдельных точек.
2.4. Погрешности в химическом анализе
71
клонения s как меры воспроизводимости. Например, для серии из 5 параллельных значений объема (мл), измеренных 50-мл бюреткой с ценой деления 0,1 мл
19,14; 19,13; 19,09; 19,21; 19,08
следует записать X ± s — 19,11 ± 0,02 (п — 5). Менее удовлетворительным, но весьма распространенным способом характеризовать погрешности результатов является соглашение о значащих цифрах. В соответствии с ним число цифр, используемых для записи результата, уже само по себе является показателем его точности. В записи результата принято приводить только значащие цифры, т. е. все цифры, известные точно, и одну сомнительную. Таким образом, в приведенном выше примере результаты правильно указаны с четырьмя значащими цифрами.
Для более подробного пояснения понятия «значащая цифра» рассмотрим следующие величины: 1537; 153,7; 15,37 и 1,537. Все они содержат по 4 значащие цифры. Величина 1,537 - 103 также содержит 4 значащие цифры. И все следующие величины — 0,001075, 0,01075; 0,1075; 1,075; 10,75; 107,5; 1075; 1,075 • 10-3 н 10,75 -10-2 — тоже содержат по 4 значащие цифры. Таким образом:
— все нули, предшествующие первой ненулевой цифре, являются незначащими;
— все нули между ненулевыми цифрами являются значащими.
А вот нули в конце записи числа могут быть как значащими, так и незначащими. Рассмотрим результат измерения разности потенциалов, записанный как 1750 мВ. В этом случае ситуация неоднозначная, поскольку нельзя сказать, представляет ли собой эта запись величину 1,75 103 мВ (3 значащие цифры) нли 1,750 103мВ (4 значащие цифры; в этом случае погрешность вольтметра составляет порядка единиц милливольт).
В качестве еще одного примера рассмотрим различные формы записи объема мерной колбы. Пусть номинальный объем колбы равен одному литру; в этом случае погрешность обычно составляет около ±0,4 мл. Запись значения объема как 1,0 л (2 значащие цифры) означает, что объем известен с точностью до нескольких десятых литра, что неверно. С другой стороны, запись 1000 мл снова неоднозначна, поскольку из нее невозможно определить (не располагая дополнительной информацией), сколько же из указанных нулей являются значащими. Лучший способ избежать двусмысленности — использовать экспоненциальную форму записи 1,0 • 103мл, которая правильно показывает нам, что объем известен с точностью до нескольких десятых миллилитра. Таким образом, можно сделать следующие выводы:
— заключительные нули значимы, только если они находятся после десятичной запятой;
— везде, где это возможно, следует представлять результат в виде величины, содержащей необходимое число значащих цифр, умноженной на степень десяти.
72
Глава 2. Процесс анализа
Следует быть внимательным при выборе необходимого числа значащих цифр для величин, представляющих собой результаты расчетов. Например, для приведенных выше данных среднее значение объема следует записать как 19,11мл, поскольку, как уже было сказано, неопределенность содержится в цифре сотых долей миллилитра (на что указывает величина стандартного отклонения). Однако при записи данных, которые предполагается использовать для дальнейших вычислений, лучше избегать слишком грубого округления. В этих случаях во избежание возможной потери точности можно после последней значащей цифры указать еще одну, записывая результат в виде X ± s — 19,112 ± 0,026 (п — 5) и проводя необходимое округление только на последней стадии расчетов.
Приведенный пример поучителен еще с одной точки зрения. В соответствии с «соглашением о значащих цифрах» среднее значение объема следует записать как X — 19,11. Часто такую форму записи ошибочно истолковывают в том смысле, что погрешность в последней цифре равна ±1. Конечно же, это не так! Подчеркнем, что «соглашение о значащих цифрах» позволяет указать лишь непосредственно первую неопределенную цифру, но отнюдь не величину этой неопределенности. Для указания последней необходимо рассчитать какую-либо характеристику точности, например выборочное стандартное отклонение (s — 0,02g в данном случае).
Для величин, получаемых в результате расчетов, существуют следующие практические правила определения числа значащих цифр.
• При сложении и вычитании результат имеет не большё“3начащих цифр после десятичной запятой, чем наименьшее число таких цифр среди всех исходных значений.
Пример
Рассчитаем молекулярную массу HNO.3- Атомные массы элементов равны: Н 1,00797; N 14,0067; О 15,9994.
Мол. масса = 1,00797 + 14,0067 + 47,9982 = 63,01287 « 63,0129г/моль
• При умножении и делении общее число значащих цифр результата равно наименьшему общему числу значащих цифр среди всех исходных данных.
Пример
Рассчитаем молярную концентрацию (М) 70%(масс.) раствора HNO3, имеющего плотность 1.413 кг/л.
М=1,413кг - л-1 - 0,70 г г-1 1000 г • кг”1/63,0129 г моль”1 = 15,6967 ~ 16моль/л
2.4. Погрешности в химическом анализе
73
• При логарифмировании число цифр мантиссы логарифма равно числу значащих цифр исходной величины.
Пример
Чему равно значение pH для раствора 1,9 10~2 М HNO3?
pH = - lg[H+] = - 1g 1,9 • ИГ2 = 1,7212 » 1,72
• При потенцировании (вычислении антилогарифмов) число значащих цифр результата равно числу десятичных цифр мантиссы исходной величины.
Пример
Чему равна концентрация Н+ для раствора с pH 4,75?
[Н+] = КГ4,75 = 1,7782 10-Б » 1,8 • 10-5 моль/л
2.4.4. Распространение погрешностей
Часто интересующая нас величина представляет собой результат вычисления, полученный из нескольких независимо измеренных величин. Каждая из них содержит погрешность, которая вносит свой вклад в общую погрешность результата. Это явление называется распространением погрешностей. Конкретный способ распространения погрешностей определяется видом соотношения между исходными и вычисленным значениями. При этом для Вычисления случайных (табл. 2.4-2) и систематических (табл. 2.4-3) погрешностей используют разные формулы, что обусловлено различной природой этих двух составляющих погрешности как таковых.
Как видно из табл. 2.4-2, для линейной функции дисперсия является аддитивной величиной. Вследствие этого наибольшее стандартное отклонение обычно вносит преобладающий вклад в общую величину стандартного отклонения (Ту. Поэтому попытки уменьшить значение <ту за счет уменьшения малых составляющих погрешностей исходных данных обычно не приводят к успеху. Обратите внимание, что при вычитании исходных величин их дисперсии все равно складываются, поэтому малые относительные погрешности исходных данных в результате вычитания могут быть многократно усилены. Предположим, например, что для смеси трех компонентов X, У и Z двумя независимыми методами найдены содержания (%) компонентов X и Z. Они оказались равными соответственно 80% и 15% с относительной систематической погрешностью +2%. Легко можно показать, что при расчете содержания У по разности (У — 100-X-Z) его относительная систематическая погрешность составит около —38%.
74
Глава 2. Процесс анализа
Таблица 2.4-2. Распространение случайных погрешностей
Случай Способ расчета Случайная погрешность
1 у = К + КаА + КвВ +... Су = (Кдгтд)2 4- (Квсгв)2 + ...
2* у = KAB/CD (<т,/»)2=(^л/Л)2+(<гв/В)2+(<7с/С)2+(<7е/В)2
з6 у = №) (Ту = ахЛу/6х
А, В, С, D — величины, измеряемые по отдельности и независимые друг от друга. Kt К а, Кв,... — константы, значения которых предполагаются точно известными. Величину у можно вычислять и из оценок всех указанных величин. В этом случае следует везде заменить А, В, С,... на их оценки A, В, С,.., и стандартные отклонения trv,tTA, (тв,... —также их оценками (выборочными стандартными отклонениями) SfhsA,SB,--
а Заметьте, что при возведении в степень (например, у = Л2) погрешность нельзя рассчитать как для операции умножения (у = А - А), поскольку исходные величины не являются независимыми. Для общего случая у = КАГ можно показать, что (ту/у - та а/А.
6 Для более общего случая у = f(x,z,t,...) существует формула
4 = W/dxfri + (»//а2)2^ + (0//at)2<7? +...
из которой можно получить все выражения, приведенные для случаев 1-3. Для важного случая у = Klnx (или у = К 1g ж) справедливо выражение
ау = Ках[х
(соответственно, ov —
Таблица 2.4-3. Распространение систематических погрешностей
Случай Способ расчета Систематическая погрешность
1 У = К + КаА + КвВ+ ... Др = Ка ДА 4- КвЬВ + ...
2 у = К АВ/CD Др/р = ДА/А + АВ/В + ДС/С + ДР/D
3* У = /(*) Др = Axdp/dx
А, В, С, D — величины, измеряемые по отдельности и независимые друг от друга. К, К а, Кв, - .. — константы, значения которых предполагаются точно известными. ДА, Д В, Д С, ДР—систематические погрешности A,B,C,D. Если знаки систематических погрешностей известны, их следует учитывать при расчетах. В некоторых случаях общая систематическая погрешность может оказаться равной нулю.
а Например, для логарифмической функции у — k 1g х имеем
Др = 0,434/с(Дх/х) н Др/р = 0,434Ax/(xIgx)
Пример
Студент получил задание приготовить 1л раствора NaCl (мол. масса 58,443) с концентрацией приблизительно 0,1 М. Он отвесил (по разности между массами тары с навеской и пустой тары) 5,8970 г вещества. Известно, что стандартные отклонения показаний весов и объема мерной колбы составляют соответственно 0,0001 г и 0,4 мл. Каково относительное стандартное отклонение концентрации полученного раствора?
2.4. Погрешности в химическом анализе
Рассчитаем концентрацию раствора (М) как
М = m[{Mg V) — 0,10090 моль • л'1
Здесь т—масса навески NaCl, Мг — молекулярная масса NaCl (которую будем считать точной величиной) н V — объем колбы. В соответствии с п.2 табл. 2.4-2 относительное стандартное отклонение величины М составляет
ам/М = {(<7m/m)2 + (<7v/V)2}1/2
Поскольку значение массы NaCl найдено по разности, ат следует рассчитать в. соответствии с п. 1 табл. 2.4-2:
о,„ = (ff,? + ff2)1/2 = {(I • 10“4)2 + (1 • 10“4)2}1/2 = 1,4 • 10“* г
Отсюда = 1,4 • 10-4/5,8970 = 2,3т 10“5.
Относительное стандартное отклонение объема полученного раствора составляет
o-y/V = 0,4/1000,0 = 4 • МГ4 (или 0,04%)
Отсюда им/М = {(2,3? • 10-6)2 + (4 10 4)2}1/2 = 4,1 -10-4.
Окончательно получаем:
ам = 4,1 10-4 • 0,10090 = 4,0 10-5
Таким образом, неопределенность результата содержится в пятой десятичной цифре. Студенту следует записать значение концентрации как (1,0090 ± 0,0004) -10-1 М. Как видно, случайная погрешность концентрации стандартного раствора' весьма мала. Из сравнения величин ат[тп. и ov/V следует, что основным источи ником погрешности является погрешность градуировки колбы. Поэтому использование более точных (и более дорогих!) весов бессмысленно, поскольку это не приведет к существенному улучшению точности окончательного результата.
Пример
Концентрация Н+ в растворе составляет (1,5 ± 0,1) 10-3 М. Чему равно стандартное отклонение величины pH, рассчитанной из этого значения?
pH = - lg[H+] = - 1g 1,5 • 10"3 = 2,82
В соответствии с правилами распространения погрешностей (табл. 2.4-2):
(Трн = 0,434Кс[н+]/(Н+] = 0,434 • (0,1 • 10-3)/1,5 1(Г3 = 0,03
Обратите внимание, что в данном случае К — — 1, однако знак этого коэффициента при расчетах был опущен, поскольку стандартное отклонение —всегда положительная величина. Обратный способ вычислений позволяет оценить стандартное отклонение [Н+] из значения (и стандартного отклонения) pH. Так, если pH раствора составляет 4,70±0,05, то
[н+] = Ю-4’70 = 1,7 • 10-5 и
ff|H+| = 2,303<грн[Н+] = 2,0 • КГ6
отсюда
[Н+] =(1,7±0,2)10-еМ
76
Глава 2. Процесс анализа
Пример
Пропускание светопоглощающего раствора 7 при некоторой длине волны равно 0,565. Произведение длины оптического пути Ь на молярный коэффициент поглощения е равно 2,06 -104 л/моль (примем, что это значение точное). Погрешность фотометра составляет 0,003 единицы пропускания. Оцените погрешность рассчитанного значения концентрации этого раствора.
Из закона Ламберта—Бера получаем
-Ig7 = Д = еЬС
Отсюда
С = (-еЬ)-1 IgT = 1,2 кг5 * М
ас = 0,434Кат/Т = 0,434 (-2,06 104)-* 0,003/0,565 = 1,1 10-7 га 1 • 10'7
(знак минус опущен!) Таким образом,
С= (1,20 ±0,01)- 10-5М
Относительное стандартное отклонение составляет стс/С = 0,9%.
Литература
[2.2-1] Gy, Р.М., Sampling of Heterogeneous and Dynamic Material Systems, Data Handling in Science and Technology, Vol. 10, Amsterdam: Elsevier, 1992.
[2.2-2] Dunemann, 1., Begerow, J., Bucholski, A., Sample Preparation for Trace Analysis, Ullmann’s Encyclopedia of Industrial Chemistry, Vol. B5, 65-93. Weinheim: VCH Publishing Group, 1994.
12.4-1] Massart, D.L., Vandeginste, B.G.M., Deming, S.N., Michotte, Y., Kaufmann, I., Chemometrics: a textbook. Amsterdam: Elsevier, 1988.
Литература общего содержания
Anderson R.L., Practical statistics for analytical chemistry. Van Nostrand Reinhold, New York, 1987.
Barnett V., Lewis T., Outliers in statistical data. Wiley, New York, 2nd edn., 1984.
Conover W.J., Practical non parametric statistics. 2nd Edition, Wiley, New York, 1980.
Green J.R., Margerison D., Statistical treatment of experimental data. Elsevier Sci. Publishing Co., Amsterdam 1978.
Hawkins D.M., Identification of outliers. Chapman&Hall, London, 1980.
Kratochvil B., Taylor J.K., Sampling for chemical analysis. Anal. Chem., 53 (1981) 924A.
Lindey D.V-, Scott W.F., New Cambridge elementary statistical tables. Cambridge University Press, Cambridge, 1984.
Liteanu C., Rica I., Statistical theory and methodology of trace analysis. Ellis Ног wood Chichester 1980,
Massart D.L., Dijkstra A., Kaufman L., Evaluation and optimization of laboratoiy methods and analytical procedures. Elsevier Sci. Publishing Co., Amsterdam 1978.
McCormick D., Roach A., Chapman N.B., Measurement, statistics and computation. John Wiley & Sons Chichester 1987.
Miller J.C., Miller J.N. Basic statistical methods for analytical Chemistry. Part I. Statistics of repeated measurements. A review. Analyst 113 (1988) 1351.
2.4. Погрешности в химическом анализе 77
Miller, J.C., Miller J.N. Statistics for analytical chemistry. Second Edition. Ellis Horwood Chichester, 1988.
Miller J.N., Outliers in experimental data and their treatment. Analyst, 118 (199) 455.
Neave H.R., Elementary statistics tables. George Allen & Unwin, London, 1981.
Warnimont G.T., Spendley W., Use of statistics to develop and evaluate analytical methods. AO AC, Arlington VA, USA, 1985.
Woodget B.W. Cooper D., Chapman N.B. Samples and standards. John Wiley & Sons, Chichester, 1987.
Youden W.J., Steiner E.H., Statistical mannal of the Association of Official Analytical Chemists. AOAC, Arlington VA, USA, 1975.
Вопросы и задачи
1. Чем различаются случайная и систематическая погрешности?
2. Никель в нержавеющей стали можно определить гравиметрическим методом путем осаждения диметилглиоксимом в слабощелочной среде. Для маскирования главного мешающего компонента—Fe(III)— используют винную кислоту. Методика включает следующие стадии.
— Взятие навески пробы, содержащей никель, ее растворение в достаточном объеме б М НС1, добавление б М HNO3, кипячение.
— Добавление необходимого количества маскирующего агента—водного раствора винной кислоты.
— Растворение навески диметил глиоксима (осадитель) в этаноле, прибавление' необходимого количества этого раствора к раствору пробы, добавление б М NH3 до слабощелочной реакции среды.
— Кипячение раствора и его последующее охлаждение до комнатной температуры- _
— Фильтрование осадка через фильтрующий тигель (предварительно доведенный до постоянной массы при 110°С) и его промывание до отсутствия С1~ в промывных водах.
— Высушивание тигля с осадком до постоянной массы при 110°С. Расчет содержания никеля в образце.
Попробуйте установить, погрешности какого типа с наибольшей вероятностью связаны с каждой из указанных стадий. Используя свой опыт (теоретический и, желательно, также практический) в области гравиметрии, попытайтесь оценить, которая из этих стадий вносит в результаты наибольшую погрешность.
3. Что означают термины устойчивые (робастные) и непараметрические статистические методы?
4. При спектрофотометрическом определении железа получена следующая серия параллельных значений оптической плотности*
0,390; 0,380; 0,385; 0,381; 0,380; 0,370; 0,375
Для этой серии данных найдите:
— медиану;
- среднее;
— стандартное отклонение;
- относительное стандартное отклонение (%);
— абсолютную и относительную (в тысячных долях) погрешность в предположении, что истинное значение оптической плотности равно 0,370.
78
Глава 2. Процесс анализа
5. Объясните значение следующих терминов: смещение, правильность, точность, систематическая погрешность методики, систематическая погрешность лаборатории.
6. Что такое сходимость и воспроизводимость?
7. Что такое межлабораторный эксперимент? Для чего его проводят?
8. Выполните предлагаемые вычисления, представьте результат с необходимым числом значащих цифр.
— Рассчитайте молярную концентрацию 37% (масс.) раствора НС1 (мол. масса 36,441) с плотностью 1,201 кг/л.
- Рассчитайте pH 2,5 10~3 М НС1.
— Рассчитайте концентрацию Н+ в растворе с pH 2,58.
9. Рассчитайте относительное стандартное отклонение молярной концентрации раствора NaaCOs (мол. масса 105,99 г/моль), полученного растворением 5,3870 г вещества в мерной колбе объемом 1 л. Примите, что навеска была взята по разности, а стандартные отклонения показаний аналитических весов и градуировки колбы составляют соответственно 0,00012 г и 0,5 мл.
10. Рассчитайте относительное стандартное отклонение (%) концентрации вещества, раствор которого имеет оптическую плотность 0,248 в предположении, что стандартное отклонение пропускания составляет
а) 0,003 единицы пропускания;
б) 0,010 единицы пропускания.
11. Рассчитайте стандартное отклонение величины pH для (1,2 ±0,1) • 10~2 М НС1.
12. Рассчитайте относительную систематическую погрешность концентрации Й+ для раствора, измеренное значение pH которого составляет 3,02, если показания pH-метра имеют положительную систематическую погрешность 0,08 единицы pH.
3
ОБЕСПЕЧЕНИЕ И КОНТРОЛЬ КАЧЕСТВА
Цели изучения
• Дать представление о доступных аналитику средствах обеспечения службы качества.
• Рассмотреть практические аспекты обеспечения и контроля воспроизводимости и правильности результатов, проверки (аттестации) надежности, те. методик (см. гл.2).
• Обсудить законодательные и правовые аспекты обеспечения и контроля качества.
• Подчеркнуть экономическое значение обеспечения качества химического анализа.
3.1. КАЧЕСТВО АНАЛИЗА И ЗАДАЧИ АНАЛИТИЧЕСКОЙ ХИМИИ
3 .1.1. Определения и общие понятия
Химический анализ играет важную роль в решении множества проблем:
— оценка состава и свойств и, следовательно, качества материалов н промышленной продукции;
— контроль производственных процессов;
— оценка степени воздействия промышленного производства на окружающую среду;
— исследовательская и проектная работа.
Клинический анализ вносит все возрастающий вклад в развитие средств медицинской диагностики. Анализ объектов окружающей среды необходим для принятия решений по ее защите.
Реальная роль химических измерений в различных областях экономической и социальной деятельности едва ли поддается точной оценке. Тем не менее ряд авторов попытались охарактеризовать эту роль количественно. Сотрудники американского Национального бюро стандартов (NBS, ныне Национального
80 Глава 3. Обеспечение и контроль качества
института стандартов и технологий, NIST) Уриано и Грават подцеркиваютвййл чительный вклад химических измерений в валовой внутренний продукт (ВВП) США [3.1-1]. По оценке Герца [3.1-2], в 1988 г. в США ежедневно выполнялось около 250 миллионов химических анализов. Порядка 10% из них (т. е. 25 миллионов ежедневно!) оказывались низкого качества и нуждались в повторном выполнении, что в годовом исчислении составило 5 млрд. долл, дополнительных затрат. В некоторых отраслях (например, электронной промышленности), где требования к составу продукции особенно жесткие, нуждаются в повторных анализах до 30% образцов. Куин своими недавними оценками, полученными б рамках общеевропейской программы по испытаниям и измерениям, предпринятой организацией BCR (Bureau Communitaire de Reference) [3.1-3], подтвердил эти данные; по его мнению, ие менее 5% в ВВП развитых стран Запада составляют измерения. Согласно оценке Тёльга, в 1982 г. на повторение химических анализов неудовлетворительного качества было затрачено до 12 миллиардов немецких марок.
Но даже эти данные не отражают разнообразных побочных экономических н социальных последствий неправильных измерений. Их невозможно непосредственно охарактеризовать в виде бюджетных цифр. Однако их масштаб становится понятным, если учесть, сколь серьезные законодательные и экономические решения основываются на результатах измерений. К ним, в частности, относятся:
— закрытие предприятий;
— мероприятия по охране труда;
— меры по утилизации отходов;
— выбраковка продукции; ~
— гуманитарные последствия, возникающие в результате техногенных аварий.
□ Неправильные измерения приносят ежегодно убытки в несколько миллиардов долларов США.
Все сказанное свидетельствует не только о важности химических измерений самих по себе, но и о необходимости улучшения их качества с тем, чтобы гарантировать доведение высококачественных результатов до потребителя. Слово «потребитель» здесь рассматривается в весьма широком смысле. В роли потребителя может выступать:
— заказчик аналитических исследований;
- коллега по лаборатории или производству;
— врач, нуждающийся в результатах медицинских анализов;
- органы власти;
— органы правосудия;
— таможенные органы,
— короче, все, кому необходимо получить ответы на вопросы, на которые может ответить химический анализ.
В этой главе обсуждаются доступные аналитикам средства обеспечения качества. Использование этих средств должно помочь избежать ненужных потерь времени и денежных средств из-за неверных результатов, приводящих к
3.1. Качество анализа и задачи аналитической химии
81
ошибочным выводам и влекущим за собой значительные убытки и снижение благосостояния людей.
То, что называется обеспечением качества, включает все действия, предпринимаемые для правильного планирования химического анализа. Приведем официальные определения этого и ряда родственных понятий, предложенные Международной организацией по стандартизации (ISO).
Обеспечение качества — все планируемые и систематически предпринимаемые действия, которые необходимы для гарантии того, что продукт, процесс или служба соответствуют заданным требованиям качества [3.1-5].
Качество — совокупность свойств и характеристик продукта (или службы), определяющая способность продукта (или службы) удовлетворять тем или иным заданным или подразумеваемым нуждам [3.1-5].
Качество не приходит само собой. Оно достигается только при проведении большого числа заранее запланированных мероприятий.
Контроль качества—технические и организационные средства, используемые для достижения требований качества [3.1-5].
Обеспечение и контроль качества являются составными частями системы качества [3.1-6].
Система качества — организационные структуры, обязанности, процедуры и ресурсы для осуществления управления качеством [3.1-5].
Управление качеством — составная часть общего управленческого процесса, которая определяет и проводит в жизнь политику качества [3.1-5].
Политика качества — совокупность официально установленных руководством направлений организационной работы, касаюшихся качества [3.1-5].
3 .1.2. Качество результатов анализа и необходимая информация
□ Если потребитель действительно желает получить ответ на поставленный вопрос, то аналитик должен играть ключевую роль на протяжении всего процесса анализа. Общее качество аналитической службы достигается, если представляя разультаты анализа, аналитик учитывает запросы потребителя.
Для достижения требуемого качества аналитик должен быть вовлечен в исследовательский процесс на всем его протяжении, начиная от формулировки задачи потребителем и кончая представлением заключительного отчета. Это означает, что заказчик должен советоваться с аналитиком и по вопросу выбора образца для анализа, и по поводу определяемых параметров, и относительно уровня точности, необходимого для правильного ответа на поставленные вопросы. Это позволит аналитику выбрать для решения задачи метод, обоснованный как с научной, так и с экономической точек зрения. Слишком сложная и дорогостоящая методика не нужна для определения небольшого числа простых характеристик. В то же время, если методика недостаточно точна, то ее нельзя использовать для изучения долговременных, медленно протекающих процессов — таких, как природное самоочищение загрязненных территорий или загрязнение грунтовых вод вследствие просачивания стоков, а также для оценки качества высокотехнологичной продукции. Всегда, когда аналитик
82
Глава 3. Обеспечение и контроль качества
Рис. 3.1-1. Спираль «обеспечение качества» — «контроль качества». Отдельные этапы обеспечения и контроля качества работы аналитика можно представить в виде круга. Когда на всех этих этапах получаются адекватные результаты, то окончательная обработка данных и их соответствующее представление обеспечивают обоснованный ответ на первоначальный вопрос, поставленный потребителем. Если же такого ответа не получено, аналитик и потребитель могут совместно уточнить задачу, для решения которой служит следующий «круг качества». При этом вероятность реше-
ния задачи возрастает с каждым последующим кругом.
планирует и выполняет работу, его главной целью должен быть ответ на первоначальный вопрос, поставленный потребителем.
На рис. 3.1-1 представлены основные этапы того, что характеризует «подход с точки зрения качества». Система обеспечения качества призвана гарантировать, что приняты все необходимые меры по контролю качества и, тем самым, что весь «цикл качества» находится под контролем. Если на заключительной стадии выясняется, что удовлетворительное решение не может быть получено из-за непредвиденных обстоятельств, то весь цикл следует повторить. При этом в большинстве случаев удается подойти ближе к правильному ответу за счет использования информации, полученной в результате первой попытки. На рис. 3.1-1 это изображено в виде второго, меньшего, круга «спирали качества» [3.1-7]. В некоторых наиболее сложных ситуациях для достижения удовлетворительных результатов требуется повторение нескольких таких кругов. Однако по экономическим соображениям аналитик должен стремиться к тому, чтобы их было как можно меньше. Это достигается только тесным сотрудничеством между потребителем и аналитиком как при планировании работы, так и в ходе ее.
Эта концепция всеобщего обеспечения качества, охватывающая все действия от постановки задачи до получения решения, находит среди аналитиков все большее понимание [3.1-8, 3.1-9]. Можно сделать вывод, что роль аналитика вовсе не ограничивается лабораторными измерениями, но также крайне важна при постановке задачи и формулировании выводов.
Требования потребителей к производителям относительно качества продукции и услуг весьма высоки. Разрабатываются и требуют контроля множество
3.1. Качество анализа и задачи аналитической химии
83
Рис. 3.1-2. Факторы, воздействующие нв «спираль качества». Возможности обеспечения и контроля качества зависят от ряда факторов, которые следует учитывать при планировании работы. Среди них могут быть внутренние ограничения, обусловленные, в частности, квалификацией персонала, характером инфраструктуры и управления, необходимостью аккредитации лабораторий и аттестации методик. Возможны и ограничения, связанные с требованиями законодательства или экономическими аспектами.
Рис. 3.1-3. Средства, необходимые для организации «спирали качества». В лаборатории имеется множество средств для организации необходимой системы качества. Они применяются на разных уровнях лабораторной структуры, по отношению к персоналу или в ходе контроля качества аналитической работы как таковой.
параметров, характеризующих экологическую безопасность продукции и производств (отсутствие токсичных веществ, возможность полной утилизации). Контроль качества должен гарантировать, что необходимые требования будут достигнуты. И именно аналитик есть тот человек, который может разработать средства обеспечения качества. На практике для создания необходимой системы качества требуется выполнение ряда условий. На организацию и реализацию этой системы влияют различные объективные и субъективные факторы, представленные на рис. 3.1-2. Рис. 3.1-3 иллюстрирует различные имеющиеся
84
Глава 3. Обеспечение и контроль качества
в распоряжении аналитика средства для организации и претворения в жизнь системы качества.
3.2. МЕТОДИКА АНАЛИЗА
Методика анализа —это последовательность действий, с помощью которых аналитик получает необходимую информацию. Методика должна быть полностью адаптирована к поставленной задаче. Аналитик должен выбрать подходящую методику и выявить все возможные источники погрешностей. Такое исследование называется проверкой (и нередко аттестацией) методики анализа.
3.2.1. Выбор метода (методики)
После того, как аналитик правильно уяснил себе суть проблемы, он должен выбрать соответствующие метод и методику. Если подобные анализы никогда раньше не выполнялись, разработка методики обычно начинается с изучения научной литературы. Немаловажную роль играет и практический опыт самого аналитика и его коллег в сходных областях деятельности. В этой книге рассмотрены наиболее важные методы химического анализа и примеры их использования; читатель может обратиться непосредственно к соответствующим главам.
Любая аналитическая методика включает следующие операции (см. гл. 2):
— отбор и хранение представительной пробы; _
— пробоподготовку;
— непосредственно определение;
— расчеты и представление результатов.
Возможности выбора метода н методики могут быть ограничены доступностью необходимого оборудования и опытом работы персонала. Ключевой этап методики — перевод образца в форму, обеспечивающую получение правильных результатов и совместимую с выбранным способом измерения. Разработка методики включает в себя выбор отдельных взаимно согласованных операций и разработку средств контроля, позволяющих убедиться, что при последовательном выполнении этих операций вся методика в целом дает надежные результаты. Результаты анализа называются достоверными, если они правильны и хорошо воспроизводимы. Хорошая воспроизводимость достигается путем минимизации случайных погрешностей, а правильность — путем устранения систематических погрешностей.
Определения
Для правильного понимания сущности проверки методики анализа, описанной далее в разд. 3.2.2, необходимо определить ряд понятий. Интерпретация этих базовых понятий с точки зрения статистики изложена в разд. 12.1. Приведенные ниже определения взяты из Международного словаря общих и фун
3.2. Методика анализа
85
даментальных терминов метрологии (3.2-1), нормативного документа ISO 3534 «Статистика: словарь и символы» (3.2-2) и Словаря официальной метрологии (3.2-3).
Случайная погрешность — составляющая погрешности измерения, которая в процессе повторных измерений одной и той же величины изменяется непредсказуемым образом (3.2-1).
Систематическая погрешность — составляющая погрешности измерения, которая в процессе повторных измерений одной и той же величины остается постоянной или изменяется предсказуемым образом (3.2-1).
Воспроизводимость — близость друг к другу результатов, полученных с помощью одной и той же экспериментальной процедуры несколько раз в контролируемых условиях (3.2-2).
Правильность — близость измеренной величины к ее «истинному значению» (3.2-2). Методика, обладающая как правильностью, так и хорошей воспроизводимостью, называется точной.
Истинное значение (измеряемой величины)—значение, являющееся подлинной характеристикой данной величины в тех условиях, при которых данная величина рассматривается (3.2-1). Словарь официальной метрологии приводит следующее определение этого понятия применительно к процессу измерения: «Истинное значение—это значение, которое можно было бы получить при устранении всех несовершенств измерительной процедуры» (3.2-2].
Помимо требований правильности и воспроизводимости, выбор методики ‘ анализа может быть ограничен требованиями законодательства или специфическими запросами потребителя. В речи аналитиков часто встречаются следующие термины, характеризующие методику с точки зрения ее назначения или официального статуса (3.2-4). ~
Официальная методика (3.2-4) —методика, предписанная к употреблению законодательными актами или иными нормативными документами официальных организаций (таких, как американские Агентство по охране окружающей среды (ЕРА) или Управление пищевых продуктов и лекарств (FDA), Европейская директива и др.).
Стандартная общепринятая методика — методика, разработанная официальной организацией (Международная организация по стандартизации (ISO), Европейский комитет по стандартизации (CEN), Германский институт стандартов (DIN), Британский институт стандартов (BSI), Французская ассоциация стандартизации (AFNOR) и т. д.) с использованием межлабораторной проверки. Характеристики правильности и воспроизводимости таких методик надежно установлены (3.2-4).
Модифицированная методика — стандартная методика, видоизмененная или упрощенная с целью приспособить ее для анализа объектов другого типа [3.2-4].
Экспрессная методика — методика для быстрого анализа большого числа образцов (3.2-4).
Рутинная методика — методика, используемая в повседневной аналитической практике. Это может быть, в частности, официальная или стандартная методика (3.2-4).
86
Глава 3. Обеспечение и контроль качества
Автоматизированная методика — методика, использующая автоматическое оборудование [3.2-4).
В научной литературе (например, в руководстве ISO X135)-существует также термин «арбитражная методика».
- Арбитражная методика — методика, обладающая хорошими аналитическими характеристиками и используемая в лаборатории, высокая квалификация которой официально установлена [3.2-5). Это означает, что для арбитражной методики как систематическая, так и случайная погрешности должны быть пренебрежимо малы по сравнению с требуемыми характеристиками правильности и воспроизводимости результатов. В качестве арбитражного можно использовать, например, метод масс-спектрометрии с изотопным разбавлением прн условии выполнения анализа в соответствующей лаборатории (например, в лаборатории по определению состава ядерного топлива).
Классификация методов анализа
В руководстве ISO X132, посвященного градуировке и использованию стандартных образцов в химическом анализе [3.2-6], методы анализа подразделяются на три класса с точки зрения специфики процедуры градуировки. Под градуировкой понимается набор операций, проводимых в определенных условиях и призванных установить соответствие между показаниями измерительного прибора либо измерительной системы и соответствующими им известными значениями измеряемого параметра [3.2-1).
Расчетные (абсолютные) методы
Расчетный метод —это метод, в котором конечный результат находят из результатов измерений (таких величин, как масса образца, объем раствора титранта, масса осадка), полученных в процессе анализа, путем вычислений, основанных на фундаментальных физических или химических законах [3.2-6). При использовании расчетных методов от аналитика требуется лишь измерить все величины, необходимые для получения конечного результата, провести необходимые расчеты и оценить погрешности данных. Примерами расчетных методов химического анализа являются титриметрический, гравиметрический и кулонометрический методы (см. разд. 7.1, 7.2 и 7.3).
Относительные методы
Относительный метод—это метод, основанный на сравнении результатов измерений для анализируемого образца и серии образцов сравнения известного состава при использовании системы определения, для которой зависимость сигнала (отклика) от содержания (в идеальном случае линейная) в соответствующем рабочем диапазоне определяется экспериментально и которую не требуется рассчитывать теоретически. Содержание определяемого компонента в пробе находят с помощью интерполяции зависимости отклика детектора от содержания, полученной с помощью образцов сравнения [3.2-6).
3.2. Методика анализа
87
Для относительных методов предполагается, что различия в валовом составе пробы и образцов сравнения оказывают пренебрежимо малое (по сравнению с погрешностями измерений) влияние на величину сигнала. Поэтому их использование часто требует предварительной пробоподготовки с целью устранения мешающих эффектов. Примерами относительных методов могут служить многие современные спектроскопические и хроматографические методы.
Сравнительные методы
Сравнительный метод—это метод, основанный на сравнении сигналов для анализируемого образца и серии образцов сравнения при использовании системы определения, чувствительной не только к содержанию определяемого компонента, но и к различиям в составе матрицы [3.2-6). В этом случае неучет любых различий в составах матриц приводит к ошибочным результатам. Поэтому для градуировки следует использовать образцы сравнения (в том числе стандартные образцы), состав матрицы которых известен и близок к составу матрицы пробы. Такие методы достаточно экспрессны и часто используются для контроля производственных процессов (например, рентгенофлуоресцентный метод анализа с волновой дисперсией — в металлургии, производстве керамических и порошковых материалов) и для определения физических параметров (вязкости, распределения частиц по размерам и т. д.).
Рассмотренные три категории методов различаются с точки зрения спосо- _ ба нахождения концентрации определяемого компонента, т. е. характера связи между сигналами пробы и сигналами образцов сравнения (практические примеры градуировки применительно к конкретным методам можно найти в различных главах этой книги). Для расчетных (абсолютных) и относительных методов эта связь устанавливается с помощью образцов сравнения, приготовленных на основе известного количества чистого вещества стехиометрического состава (при условии строгого контроля всех этапов методики), а для сравнительных методов — с помощью образцов сравнения (стандартных образцов) с известным составом матрицы, для которых содержание определяемого компонента надежно установлено. Такая связь, установленная в результате непрерывной цепочки действий с использованием стандартных измерительных процедур, называется прослеживаемостью.
□ Химические измерения обычно сопровождаются разрушением образца ввиду необходимости его перевода в иное физическое состояние. Превращения, претерпеваемые образцом в ходе анализа, не должны повлиять на его результаты. Возможность проследить весь ход процесса шаг за шагом называется прослеживаемостью.
Прослеживаемость
Главная задача проверки методики (см. разд. 3.2.2) состоит в слежении за поведением образца сравнения (стандартного образца) на всем протяжении анализа.
Прослеживаемость — это возможность документирование проследить всю историю, местонахождение некоторого объекта или характер применяемых
88
Глава 3. Обеспечение и контроль качества
действий либо аналогичных объектов или действий [3.1-5]. Применительно к химическому анализу это означает, что все стадии методики необходимо, выполнять и документировать таким образом, чтобы иметь возможность получать всю необходимую информацию без искажений. Иными словами, все этапы методики должны быть связаны в единую цепь, на протяжении которой (а не только на заключительной стадии измерения как такового) следует применять процедуры сравнения с использованием соответствующих измерительных стандартов, как правило, международных или национальных [3.2-5[. Ими служат, в частности, основные единицы СИ, фундаментальные константы, стандартные образцы. Связь результатов измерения с этими стандартами должна быть четко показана.
Выводы
В зависимости от типа используемого метода анализа и определяемой характеристики для демонстрации связи между сигналом прибора, измеряемым на заключительной стадии, и составом образца, к которому он относится, необходимо использовать различные средства. В общем случае качество методики анализа описывается с помощью аналитических характеристик, относящихся к двум группам.
1. Основные характеристики:
— воспроизводимость (сходимость и воспроизводимость в узком смысле);
— правильность;
— чувствительность.
2. Вторичные характеристики:
— селективность;
— диапазон линейности сигнала (рабочий диапазон);
— устойчивость.
Некоторые из этих характеристик обсуждаются в разделе, посвященном проверке методик.
Селективность методики призвана гарантировать, что величина сигнала действительно определяется именно содержанием интересующего нас вещества. При недостаточной селективности любые посторонние компоненты влияют на величину сигнала, как, например, в хроматографическом анализе с использованием неселективных детекторов (пламенно-ионизационного, электронозахватного). В каждой главе данной книги, посвященной описанию того или иного метода, обязательно рассматриваются вопросы, связанные с его селективностью.
Чувствительность — главный ограничивающий фактор при определении следовых содержаний. Очевидно, что любая методика должна быть достаточно чувствительной для определения компонента в соответствующем концентрационном диапазоне. Если чувствительность измерительного устройства оказывается недостаточной, аналитик может предпринять различные действия для ее повышения. При этом, однако, необходимо изменить также выбор, способ оптимизации и проверки других стадий методики. Аналитик, в частности, может:
3.2. Методика анализа
89
— изменить способ регистрации сигнала (разумеется, при наличии соответствующей аппаратуры);
— увеличить размер пробы, соответственно изменив стадию пробоподготовки;
— ввести дополнительную стадию концентрирования определяемого компонента.
Для грубой оценки рабочего диапазона концентраций данной методики можно использовать модельные чистые растворы определяемых компонентов.
3.2.2. Проверка методики
Суть проверки методики анализа понятна из следующего определения:
Проверка методики анализа — это процедура, позволяющая продемонстрировать, являются ли результаты достоверными и воспроизводимыми, а сама методика пригодной для решения поставленной задачи.
Для проверки методики аналитик должен установить н устранить все возможные источники погрешностей. При этом он должен показать, что результаты анализа полностью прослеживаемы по отношению к используемым стандартам. Решение этой задачи возможно только в рамках соответствующих лабораторных структур, при наличии подготовленного и заинтересованного персонала и подходящего, поддерживаемого в рабочем состоянии оборудования.
Проверка оборудования, компьютеров и программного обеспечения
Проверке методики предшествует проверка оборудования, а также компьютеров и программного обеспечения (при использовании автоматизированных методик). Проверка оборудования и компьютеров обеспечивается силами поставщиков. Хотя все оборудование обязательно проверяется производителями, однако с учетом того, что аналитик несет полную ответственность за свои результаты, он должен все равно проверить все спецификации производителя с точки зрения чувствительности, стабильности показаний, диапазона линейности отклика и других характеристик.
□ Хороший аналитик проверяет все приборы перед их использованием.
Международный консорциум пользователей оборудования, включающий организации SIREP, WIB и EXERA1, испытал 126 приборов различных типов и пришел к выводу, что 75% из них не отвечают заявленным характеристикам [3.2-7]. На рис. 3.2-1 показаны основные типы испытанного оборудования и классификация отмеченных недостатков. Основная причина расхождений между заявленными и реальными характеристиками состоит в том, что производители и, в частности, их торговые подразделения склонны преувеличивать возможности своей продукции, особенно в отношении новых типов приборов. В
1 SIREP, WIB и EXERA — национальные организации (Великобритании, Нидерландов и Франции соответственно), входящие в Международный консорциум пользователей оборудования (International Instrument Users’ Association). Например, EXERA — это Association des Exploitants d’Equipements de Mesure, de Regulation et d’Automatisme. SIREP ныне называется El — Evaluation International
90
Глава 3. Обеспечение и контроль качества
Типы испытанного оборудования 1989-1992 гг.
б Сводка результатов исследования
126 приборов за период 1989-1992 гг.
Категория
Изначально неудовлетворительная работа
Несоответствующая спецификациям
- в стандартных условиях
- в нестандартных условиях
Выход из строя
Характеристики не соответствуют
заявленным в документации
Необходимость модификации после поставки
Даля приборов
49% приборов не отвечают характеристикам, — заявленным производителем
75% приборов не отвечают характеристикам, заявленным производителем или ожидаемым потребителем 25% отвечают всем харектернстикам
Рис. 3.2-1. Соответствие оборудования заявленным характеристикам (отчет EXERA, 1993). о — распределение оборудования, испытанного в период 1989-1992 г., по типам; б — результаты испытаний показывают, что 75% приборов не отвечают характеристикам, заявленным производителями или ожидаемых потребителями, в том числе 49%—характеристикам, заявленным производителями. Это свидетельствует о том, что аналитику необходимо самому тщательно поверять новое оборудование. (Авторы благодарят организации EXERA, WIB и SIREP и в особенности г-на М. Дежардена за поддержку и согласие на публикацию результатов их исследований.)
любом случае перед практическим использованием нового оборудования аналитику следует самому проверить все его спецификации, причем не только на растворах чистых веществ, но и на реальных образцах.
Проверка собственно методики анализа
В химическом анализе интересующая аналитика величина (как правило, это содержание того или иного компонента) почти никогда не измеряется непо-
3.2. Методика анализа
91
Анализируемый объект
Отбор пробы (стратегия и техника)
п проб анализируемого материала
Предварительная подготовка пробы (гомогенизация), сокращение
Анализируемая проба
Пробоподготовка(разложение, экстракция, гранулирование, облучение и т. д.)
Гомогенная проба (жидкая, газообразная, сверхкритический флюид)
Очистка (выделение компонентов, фильтрация и т. д.)
Очищенная проба
Разделение (хроматография, электрофорез и т. д.)
Чистое вещество
Измерение: градуировка, измерение сигнала
Результат
Рис. 3.2-2. Последовательность стадий методики анализа. Методика анализа (соот* ветствует «Полевой и лабораторной работе» в спирали «Обеспечение и контроль качества» на рис. 3.1-1) состоит из различных этапов. После отбора пробы ее разделяют на отдельные порции, которые, возможно, требуют предварительной химической и (или) физической обработки. Во многих методиках необходимо удаление матрицы, а затем, возможно, и выделение определяемого компонента в чистом виде. Для некоторых методик отдельные стадии не требуются, например пробогюдготОВка для инструментального нейтронно-активационного анатаза (стрелка 1), выделение компонентов для многоэлементного атомно-эмиссионного анализа с ИСП (стрелка 2). Для определения следовых количеств органических веществ обычно требуются все указанные стадии.
средственно (в отличие от измерений физических величин — массы, длины, времени и т. д.). Перед измерением должна быть отобрана представительная генеральная проба, аналитик должен сократить ее до размеров лабораторной пробы и выделить из нее определяемый компонент или преобразовать его в форму, совместимую с выбранным способом регистрации. Это означает, что может потребоваться изменение физического или химического состояния пробы. Это изменение необходимо проводить под строгим контролем для обеспечения прослеживаемости процесса измерения по отношению к используемым стандартам. Обычно методика включает в себя следующие стадии: пробопод-готовку (разложение, экстракцию, очистку) и (или) разделение компонентов; градуировку (для относительных и сравнительных методов); заключительную стадию измерения и регистрации сигнала. Эти традиционные этапы изображены на рис. 3.2-2.
В ряде методик одну или несколько стадий можно исключить—например, удаление матрицы в инструментальном нейтронно-активационном анализе. Но
92
Глава 3. Обеспечение и контроль качества
в любом случае задача состоит в последовательном превращении пробы в то состояние, которое необходимо для измерения сигнала. При этом состав образца (первоначально—сложный) все более упрощается до тех дор, пока ие обеспечивается совместимость с системой регистрации. Любое действие на каждой из указанных стадий является потенциальным источником погрешности, вносящей вклад в общую неопределенность результата. Аналитик должен выявить и по возможности минимизировать все такие источники погрешностей. Для этого необходимо разработать стратегию исследования для каждой из стадий анализа. Один из подходов состоит в последовательном изучении стадий методики, представленных на рис. 3.2-2, снизу вверх — по мере усложнения системы. Начать следует с самого простого — образцов сравнения, используемых для градуировки. Исследовав стадию градуировки и измерения сигнала, аналитик шаг за шагом движется вверх, в направлении реального образца. На рнс. 3.2-3 и 3.2-4 показана последовательность таких действий применительно к сравнительным методам на примерах определения следовых количеств неорганических и органических веществ.
При проверке каждой новой стадии аналитик должен убедиться в том, что выводы, полученные на предыдущей стадии, остаются справедливыми. Если это не так, он должен вернуться назад и выбрать другие условия. Случайные погрешности можно оценить и минимизировать в ходе внутрилабораторных исследований. Для выявления н устранения систематических погрешностей необходимы внешние средства — например, в форме стандартных образцов или меж лабораторного исследования.
Помимо оценки достоверности результатов, полученных самим аналитиком, следует оценить, как поведет себя методика в реальных условиях повседневных анализов в руках лаборантов и при наличии ограничений экономического порядка; иными словами, аналитик должен оценить устойчивость методики.
О Перед тем, как перейти к следующей стадии проверки методики, убедитесь, что вы полностью контролируете предыдущую стадию. Постарайтесь заранее предусмотреть проблемы, которые могут возникнуть на следующей стадии.
Устойчивость методики — это ее относительная нечувствительность к возможным незначительным изменениям порядка ее проведения, квалификации реактивов и окружающих условий [3.2-8].
Как правило, стандартные методики описаны достаточно подробно, чтобы их проверка не вызвала затруднений. Применительно к отдельной лаборатории такая проверка обычно сводится лишь к оценке воспроизводимости, чувствительности, устойчивости и проверке правильности. Для методики же, разработанной самостоятельно, аналитик должен сам выполнить всю проверку целиком. Для этого необходимо разбить всю методику на отдельные этапы, подобные описанным выше, н доказать, что все возможные источники систематических погрешностей устранены, а также оценить вклад каждой отдельной стадии в общую погрешность.
Для более детального пояснения пошаговой стратегии проверки методики обратимся к примерам определения следов органических н неорганических веществ в объектах окружающей среды. В этой области химического
3.2. Методика анализа
93
Этапы Изучаемые (оптимизируемые) факторы
Приготовление стандартных растворов, параметры распыления, высота наблюдения сигнала, параметры плазмы, внутренние стандарты, диапазон градуировки, методы хемометрики, оценка погрешностей, коррекция базовой линии и т. д.
Добавки стандартных растворов; влияние матрицы на градуировку, параметры распыления, проверка внутренних стандартов и т. д.
Реальный образец
Проверка вычислений
Правильность ("введенонайдено”), контрольный опыт, способ разложения пробы: в открытом или закрытом сосуде, кипячение с обратным холодильником; оптимизация процедуры разложения: время, количество реагентов (кислот. Н2О2) и т. д.
Устойчивость
Рис. 3.2-3. Пошаговая проверка методики определения Cd и РЬ в тканях мидий методом ИСП-АЭС. Проверка методики определения иескольких следовых элементов в сложной матрице требует исследования всех источников погрешностей, которые могут возникнуть на каждой из стадий, представленных на рис. 3.2-2. Идея состоит в том, чтобы начать с заключительной стадии градуировки и измерения, а затем двигаться назад шаг за шагом в направлении реального образца. Рядом с каждой стадией приведен перечень факторов (не исчерпывающий), которые можно оптимизировать на этой стадии.
анализа объекты особенно разнообразны с точки зрения состава матриц, природы мешающих компонентов и диапазонов концентраций определяемых веществ. Существует лишь малое число стандартных методик, которые можно непосредственно применять к любым объектам без тщательной проверки. На рис. 3.2-3 показана схема проверки методики определения Cd и РЬ в тканях мидий. Мидии представляют собой индикаторные организмы для контроля уровня загрязнений мирового океана различными токсикантами в следовых количествах. Рис. 3.2-4 иллюстрирует последовательность проверки методики определения полихлорбифенилов (ГЕХЕт) в почвах. На каждом этапе проверки
94
Глава 3. Обеспечение и контроль качества
Этапы Изучаемые (оптимизируемые) факторы
Приготовление стандартных растворов чистых веществ и их смесей для градуировки, методы хемометрики, выбор растворителя и колонки, условий хроматографирования, внутренних стандартов, пределы обнаружения (нижние границы определяемых содержаний), диапазон линейности детектора, эффекты памяти и т. д.
Растворы, содержащие возможные мешающие компоненты. Повторное изучение и оптимизация перечисленных выше факторов.
Экстракт незагрязненной почвы с добавками стандартных растворов ПХБ: поведение внутреннего стандарта, условия газокроматографического разделения и детектирования: при возникновении трудностей вернуться на шаг назад.
Оптимизация операции очистки на экстрактах реальных образцов: емкость колонки, проблемы коррекции базовой линии, отрицательных пиков, выделения отдельных фракций, возможные потери, эффекты памяти, полнота извлечения внутреннего стандарта.
Проблемы, связанные с экстракцией: выбор растворителя (растворителей), избирательность, выбор внутренних стандартов, полнота их извлечения, влияние влажности, необходимость предварительной пробоподготовки (высушивание, обработка кислотами), матричные эффекты, проведение контрольного опыта
Рис. 3.2-4, Пошаговая проверка методики определения полихлорбифенилов (ПХБ) в почве методом ГЖХ. Применительно к определению следовых количеств органических веществ в объектах окружающей среды также справедливы принципы, проиллюстрированные на рис. 3.2-3. Однако здесь методика обычно включает больше стадий, нуждающихся в проверке, чем при определении следов неорганических веществ. Одним из наиболее сложных этапов является проверка стадии экстракции компонентов из твердой матрицы.
необходимо получить всю информацию, требуемую для надежного выполнения следующего этапа.
Изучение литературы
В научной литературе почти невозможно найти настолько подробные описания методик н их проверки, чтобы их можно было без риска непосредственно применять для решения практических задач. Вопросы, касающиеся контроля качества, обычно не обсуждаются вообще. Поэтому аналитик должен рассмат
3.2. Методика анализа
95
ривать опубликованные данные в качестве не более чем общих указаний, на основе которых ему предстоит разработать свою собственную методику и оценить ее характеристики применительно к реальным объектам. Даже стандартные методики нельзя механически переносить на объект с другой матрицей без полной проверки. В частности, стандартная методика, разработанная для питьевой воды, может оказаться неприменимой для природных и тем более сточных вод. Аналогично, методика анализа донных отложений не предназначена для анализа почв или бытовых отходов. Поиск в литературе может подсказать лишь общие черты методики, оптимальной для решения данной задачи, но не более того. Применительно к рассмотренным примерам можно сказать, что опубликованные данные способны лишь в самом общем виде указать на характер пробоподготовки (обработка кислотами, растворители для экстракции), очистки пробы и способ регистрации сигнала.
□ Международная система единиц измерения (СИ) введена для избежания путаницы в терминологии. Поэтому к системе СИ следует относиться с уважением и применять ее в полном объеме.
Проверка заключительной стадии регистрации сигнала (стадия S на рис. 3.2-3 и 3-2.4)
Следует отметить, что в ходе градуировки очень часто возникают ошибки: . этот вывод следует из результатов дискуссии по материалам межлабораторного эксперимента, проводившегося в течение нескольких лет под эгидой BCR. Вот основные типы таких ошибок:
- арифметические ошибки в вычислениях (в частности, при расчетах концентраций);
— ошибки, связанные с разбавлением;
- ошибки, вызванные потерями летучих растворителей;
— использование первичных стандартов нестехиометрического состава или недостаточной чистоты без проверки их качества;
— различные загрязнения, помехи, неподходящие внутренние стандарты либо неправильный способ их добавления к образцам сравнения и анализируемым пробам;
- неправильная коррекция фона н способ учета результатов контрольного опыта;
— неправильное использование единиц измерения (таких, как частей на тысячу, миллион, миллиард; необоснованное применение соотношения 1г = 1 мл к органическим растворителям или концентрированным кислотам при приготовлении стандартных растворов);
- несоответствие матриц пробы н образцов сравнения и другие ошибки.
Изучение системы с использованием растворов чистых веществ необходимо для выявления, оценки и решения всех проблем, связанных с регистрацией сигнала: селективности, диапазона линейности отклика (для некоторых типов
96
Глава 3. Обеспечение и контроль качества
датчиков, — например, электронозахватного детектора или датчиков, используемых в атомно-абсорбционном анализе—он достаточно ограничен), прослеживаемости по отношению к соответствующим чистым веществам, чувствительности, правильности и воспроизводимости градуировки. На этом же этапе необходимо исследовать, оценить масштаб и по возможности решить все проблемы, связанные с взаимным влиянием, хроматографическим разделением, выбором внутреннего стандарта для количественного анализа, применением средств хемометрики для правильной градуировки (см. гл. 12). Для обеспечения всего этого аналитик должен приготовить запасные стандартные растворы веществ известной степени чистоты н стехиометрического состава. Вопросы выбора исходных веществ, приготовления, хранения и работы с такими растворами подробно описаны Д. Э. Уэллсом с соавторами [3.2-9] применительно к определению ПХБ и других органических веществ и Дж. Р. Муди с соавторами [3.2-10] применительно к неорганическому анализу. Можно выделить следующие основные рекомендации.
Исходные вещества и их запасные растворы следует хранить в надежном месте (в сейфе, холодильнике, морозильнике), доступ к ним следует контролировать, должно быть выделено лицо из числа старшего персонала, ответственное за их приготовление и распределение. Органические вещества, как правило, растворяют в летучих растворителях; в этих случаях предпочтительнее использование герметичных сосудов, например ампул. Необходимо обеспечить защиту от яркого света (сосуды из темного стекла) и повышенных темйератур (холодильник, морозильник). Перед каждым использованием следует осматривать стандартные растворы на предмет образования осадков или хлопьев. До и после отбора аликвоты сосуд со стандартным раствором необходимо взвешивать для обнаружения возможных потерь за счет испарения или утечки. Запасные растворы следует заменять с периодичностью, определяемой устойчивостью веществ. Ошибки, допущенные в ходе градуировки, могут вызвать различия в результатах, полученных в разных лабораториях, на несколько порядков величин [3.1-7].
Влияние матрицы
При переходе к изучению экстрактов или продуктов разложения пробы все выводы (в частности, относительно параметров градуировки, диапазона линейности, условий хроматографирования, внутренних стандартов н т. д.), полученные на первой стадии проверки, следует проверить заново применительно к новой ситуации. Матрица н ее компоненты могут стать источниками новых помех (матричные эффекты). Прн определении следов органических загрязнителей стадия изучения влияния матрицы исключительно важна, поскольку в этом случае для регистрации сигнала часто используют неселективные детекторы (например, электронного захвата, пламенно-ионизационный, ультрафиолетовый)- Таким образом, правильность величины сигнала определяется эффективностью хроматографического разделения.
Часто необходимо, чтобы растворы, используемые для градуировки (в частности, способом добавок), имели тот же состав матрицы, что и проба. По
3.2. Методика анализа
97
грешности, вызванные неучетом матричных эффектовгиллюстрируют данные табл. 3.2-1. Они получены в ходе межлабораторной аттестации стандартных образцов и показывают важность соответствия составов матриц [3.2-11]. Для всех элементов (за исключением кобальта, для которого не устранены дополнительные источники погрешностей) при соответствии составов матриц пробы и образцов сравнения получены результаты, значительно более близкие к аттестованным данным, а согласование результатов разных лабораторий существенно лучше.
Таблица 3.2-1. Иллюстрация влияния соответствия составов матриц пробы н образцов сравнения на результаты определения следовых содержаний (массовые доли, мг/кг) металлов в легких песчаных почвах (стандартный образец BCR 142R [3.2-11)) методом ИСП-АЭС&.
Элемент Результаты при несоответствии составов матриц Результаты при соответствии составов матриц Аттестованное содержание
Со общ. 6,0±0,2 7,9±0,5 5,61±0,31
Со к.В. 4,6±0,5 6,1±0,5 н/а
Си общ. 6О7±9 667±18 696±12
Си к.В. 655±4 745±12 707±9
Мп общ. 139±2 151±5 15б±4
Ми к.в. 122±2 151±4 н/а
Ni общ. 216±5 249±8 247±7
Ni к.в. 207±3 26б±5 251±6
Zn общ. 1826±34 2072±47 2122±23
Zn к.в. 1856±15 2238±26 2137±50
“Результаты анализа указаны как среднее (из 5 параллельных) ± стандартное отклонение, аттестованные содержания — с доверительным интервалом для доверительной вероятности 95%.
и/а — не аттестовано.
к.в. — кислотная вытяжка, (царская водка).
Для всех элементов, кроме Со и Си, выявлены н устранены все источники погрешностей, кроме тех, что вызваны несоответствием составов матриц.
При определении следов органических веществ перед хроматографированием экстракта его обычно необходимо очистить от таких совместно экстрагирующихся компонентов матрицы, как лнпиды, серосодержащие вещества, пигменты н другие потенциально мешающие компоненты. Эта операция позволяет также предохранить от выхода из строя хроматографическую колонку и детекторы (электронного захвата, источник ионизации при масс-спектрометрическом детектировании). Важно также выявить возможные источники потерь в ходе очистки. Для этого можно использовать введение добавок определяемого компонента в экстракт. Применение добавок позволяет также оценить емкость системы очистки и тем самым установить примерное число определений без ее регенерации в ходе рутинных анализов. Учитывая
4 Аналитическая химия. Том 1
98
Глава 3. Обеспечение и контроль качества
возможность потерь определяемого компонента, целесообразно использовать подходящий внутренний стандарт.
Твердая проба (стадия М на рис. 3.2-3 и 3.2-4)
На этой стадии проводится оптимизация процедуры извлечения компонентов из пробы или разложения пробы и оценивается ее эффективность. Для неорганического анализа стадию разложения можно проверить с помощью метода, позволяющего определять элементы непосредственно в твердой матрице (например, рентгенофлуоресцентного или инструментального нейтронноактивационного). В органическом анализе проверка стадии экстракции крайне затруднительна и часто не дает удовлетворительных результатов. Обычно практически невозможно доказать, что извлечение протекает количественно. В этом случае возможны два подхода. Один из них состоит в выполнении последовательных извлечений (экстракций) и определении остаточных содержаний ПХБ в полученной серии экстрактов. Если ПХБ больше невозможно про-экстрагировать ни одним из известных способов, это может указывать (но не доказывать!) на то, что достигнута полнота извлечения. Вторая возможность состоит во введении в пробу добавок во все возрастающих концентрациях [3.2-12J. После экстракции следует убедиться в том, что найдено все количество добавленного вещества- Этот метод работает только в том случае, если добавка контактирует с пробой в течение времени, достаточного для ее перехода в то же физическое состояние, что н определяемый компонент в матрице. Повторяя эту процедуру с переменными количествами добавки, можно, наряду с полнотой экстракции, оценить и ее воспроизводимость. Наконец, когда все стадии методики отработаны, оптимизированы и проверены, следует выполнить весь анализ от начала до конца и разработать систему показателей; позволяющих контролировать качество результатов в ходе рутинных анализов.
Устойчивость методики
Методику можно использовать для повседневных анализов только в том случае, если она достаточно устойчива. В этом случае на результаты анализа не влияют такие факторы, как
— незначительные изменения температуры в ходе пробоподготовки, старение хроматографических колонок (ухудшающее их разделяющую способность), переменная остаточная влажность образцов после высушивания н т. д.;
— замена частей оборудования (аппарата Сокслета, бомбы для сожжения и т. д.);
— выполнение анализа другим лаборантом;
— небольшие изменения в составе матрицы, диапазоне концентраций определяемого вещества (в частности, двух одинаковых образцов почв просто не бывает) и т. д.;
— изменения параметров окружающей среды — температуры, влажности, атмосферного давления.
Необходимо исследовать, в какой мере повлияет на результаты изменение всех параметров, которые в ходе разработки н проверки методики поддержи
3.2. Методика анализа
99
вались постоянными. Для этого соответствующие параметры, в особенности критически важные, следует варьировать. Затем необходимо оценить устойчивость методики в целом, в том числе и с точки зрения выполнения ее различными лаборантами. Методика, слишком чувствительная к изменениям параметров, может оказаться непригодной. При разработке стандартной методики проверка устойчивости с помощью межлабораторных испытаний является необходимой стадией. Для оценки устойчивости методик предложены приемы хемометрики (3.2-8] и экспертные системы (3.2-13].
Контрольные точки
После полной проверки всех стадий методики целесообразно разработать процедуру контроля, позволяющую быстро устанавливать источник погрешностей, которые могут возникнуть при ее повседневном использовании. В первую очередь следует контролировать те параметры, которые в ходе проверки оказались критически важными. В табл. 3.2-2 приведен примерный перечень таких источников применительно к определению следов элементов в растительных материалах. В идеальном случае система контроля должна быть органично встроена в методику, а соответствующие предупреждения — выдаваться автоматически. Помимо указанных параметров, можно контролировать чувствительность определения (по наклону градуировочной прямой), эффективность хроматографической системы (используя два вещества, специально добавляемые к пробе и образцам сравнения; они же могут служить и внутренними, стандартами). Результаты контроля должны быть включены в стандартную форму отчета о результатах анализа, чтобы можно было быстро принять меры. В каждом конкретном случае требуется своя собственная система контрольных точек. Некоторые стандартные методики, а также методики, рекомендованные Ассоциацией официальных химиков-аналитиков (АОАС) [3.2-14], включают в себя такую систему контрольных точек. Все больше стандартов, разработанных для решения сложных аналитических задач, содержат критерии и характеристики, применимые для контроля различных этапов методик.
Контрольные карты
Для полностью проверенной методики существует возможность статистического контроля ее характеристик с помощью контрольных карт. Подобные карты используют при периодическом анализе стандартных образцов для контроля их представительности, однородности н устойчивости во времени (подробнее требования к стандартным образцам обсуждаются в разд. 3.3.4).
Если качество работы лаборатории стабильно высокое, колебания получаемых результатов обусловлены лишь малыми случайными погрешностями и обычно невелики. Только в этом случае методику можно считать полностью проверенной и использовать статистический контроль с помощью контрольных карт. Такой контроль позволяет установить появление в ходе анализов нового источника систематических погрешностей, а также в ряде случаев следить за воспроизводимостью результатов (при выполнении параллельных измерений). Для этого аналитик периодически анализирует один и тот же образец и представляет результаты графически. Предварительно необходимо выполнить се-
Таблица 3.2-2. Возможные источники погрешностей и способы их устранения при определении следов элементов в растительных
материалах
Источник системати-
Этап методики ческой погрешности Знак ° Способ устранения
А. Методы, требующие разрушения объекта (деструктивные) Предварительные операции Взвешивание +/—
Приготовление растворов, +/-измерение объема
Влажность образца Адсорбция/десорбция воды +/-
Разложение, окисление Улетучивание -
Адсорбция/десорбция +/-
Неполное разложение -
Загрязнение реактивами +
Загрязнения, вызванные по- + судой, аппаратурой
Загрязнения из атмосферы + Пробоподготовка, разделе- Адсорбция, необратимое -ние, очистка, концентрнро- осаждение вание
Загрязнения
Неполнота реакции
Градуировка
Регулировка весов
Использование калиброванной мерной посуды, контроль температуры
Приведение к массе сухого образца
Для летучих элементов (As, Se,...) проводить операции в герметичной аппаратуре
Использование сосудов из тугоплавкого стекла, обработанного кислотами, либо покрытых пластиком
Использование кислот-окислителей при повышенном давлении; проверка остатка на полноту разложения
Использование реактивов необходимой чистоты; контрольный опыт
Если возможно — промывка кислотами; при содержаниях определяемого компонента менее 1 мкг/г — обработка паром ^контрольный опыт
Работа в «чистой комнате» или герметичном боксе; контрольный опыт Контроль pH, добавление комплексообразующих реагентов
Б. Инструментальные активационные методы
Подсчет числа импульсов Перекрывание пиков, внут- +/-реннее облучение
Высокий фон +/-
Неподходящая геометрия +/— источника
Облучение Сам о экранирование +/-
Изменение потока нейтро- +/— нов
Градуировка +/-
см. Разложение, окисление
Добавление избытка реагента; предварительные теоретические расчеты Образцы сравнения на основе чистых веществ стехиометрического состава, при необходимости контроль их чистоты и стехиометричности; при возможности применение различных способов градуировки; построение и анализ градуировочных графиков; соответствие составов матриц образцов сравнения и пробы; способ добавок
Разделение пиков; выбор времени затухания; радиохимический активационный анализ как альтернатива
Переход к радиохимическому активационному анализу
Образцы сравнения и проба должны быть в одной и той же форме (те и другие —в виде растворов или порошков), в идентичных сосудах, на одинаковом расстоянии от детектора и т. д.
Проверка отсутствия экранирования в данном концентрационном диапазоне Контроль интенсивности облучения
Те же меры, что и для деструктивных методов; особое внимание к стабильности параметров облучения образцов сравнения.
+ завышение содержания; — занижение содержания.
3.2. Методика анализа
101
Рис. 3.2-5. Пример Х-карты. Каждая точка представляет результаты анализов одного и того же объекта. Среднее (тп) и стандартное отклонение (з) рассчитывают из серии предварительных параллельных определений (не менее 10), проведенных в течение нескольких дней. Границы предупреждения (т ± 2з) и тревоги (т ± Зз)
Рис. 3.2-6. Пример R-карты. Каждая точка представляет разность между результатами двух параллельных анализов одного и того же объекта. Положения границ предупреждения н тревоги рассчитывают так же, как и для Х-карт (см. рис. 3.2-5 и текст). R-карты используют для контроля сходимости.
рию параллельных определений н оценить среднее и стандартное отклонение, характеризующее воспроизводимость методики. Данные по воспроизводимости служат основой для расчета местоположения границ «предупреждения» н «тревоги» (см. рис. 3.2-5 и 3.2-6).
1W
Глава 3. Обеспечение и контроль качества
Исходно контрольные карты были разработаны в качестве одного из средств статистического контроля производства для непрерывного слежения за качеством продукции. Затем их стали использовать и для статистического контроля качества лабораторных анатазов, позволяющего оперативно предупреждать о выходе методики из-под контроля и необходимости ответных действий со стороны аналитика. Если хотя бы один результат выходит за пределы границ тревоги, качество анализа уже де гарантировано. Однократное превышение предела предупреждения сигнализирует аналитику о возможных проблемах в будущем, хотя н не требует немедленного принятия мер. Однако если предел предупреждения превышен два раза подряд, необходимо установить причину этого. Если результаты неоднократно, например, 10 раз подряд, оказываются по одну и ту же сторону от средней линии, это может свидетельствовать о появлении источника систематической погрешности. Когда качество результатов выходит из-под контроля, от аналитика требуются действия аналогичные тем, которые он предпринял бы при видоизменении методики. В самом крайнем случае может понадобиться полная повторная проверка.
Помимо карт рассматриваемого вида, называемых картами Шьюарта, в химическом анализе применяют так называемые Х-карты, на которых откладывают средние из нескольких параллельных определений, и R-карты, на которых откладывают разности из даух параллельных результатов. Такие карты используют для контроля воспроизводимости методики. Для своевременного обнаружения медленных изменений, связанных, например, со старением аппаратуры и стандартных растворов, смещением шкалы длин волн и т.-д., можно использовать карты накопленных сумм. На них откладывают накопленные (кумулятивные) суммы разностей между результатами анализа образца и действительным содержанием компонента. Если это содержЯйие аттестовано (например, при использовании стандартных образцов), то карты накопленных сумм позволяют также контролировать правильность. Дополнительные сведения об использовании контрольных карт (периодичность измерений, положения границ предупреждения н тревоги) содержатся в гл. 12.
Средства хемометрики
Описанию методов хемометрики посвящена гл. 12. Эти методы очень полезны как для разработки методики, так и при проверке ее различных стадий. Их можно применять в процессе градуировки, для оптимизации методики, для обработки данных, а также для статистического контроля методики в течение времени.
3.3. КАК ДОСТИЧЬ ПРАВИЛЬНОСТИ АНАЛИЗА
После оптимизации воспроизводимости методики следует оценить ее правильность. Это можно сделать путем сравнения результатов анализа одного н того же образца, полученных в разных лабораториях или путем анализа стандартного образца подходящего состава. Возможно и участие в межлабораторном эксперименте с использованием различных методов анализа. Аналитик должен хорошо понимать, что правильность оценить невозможно, не прибегая к срав-
3.3. Как достичь правильности анализа
103
Рис. 3.3-1. Результаты межлабораторного эксперимента по определению кадмия в тканях мидий, проведенного под эгидой BCR. Межлабораторное среднее ± стандартное отклонение составило 0,35 ± 0,04 мг/кг. Три лаборатории (отмечены кружками) использовали метод масс-спектрометрии с изотопным разбавлением (ввиду высокой воспроизводимости этот метод позволяет обнаружить даже очень незначительные систематические погрешности) и получили различные результаты. Этот пример показывает, что даже так называемые арбитражные методы не гарантируют отсутствия систематических погрешностей. Не в меньшей мере, чем от других факторов, праг вильность результатов зависит от организации лабораторной системы обеспечения и контроля качества.
нению с независимыми данными. При этом даже так называемые арбитражные методы могут содержать систематические погрешности. Подобный случай иллюстрирует рис. 3.3-1, где представлены результаты определения кадмия в тканях мидий. Три лаборатории использовали масс-спектрометрию с изотопным разбавлением и получили результаты, различающиеся настолько, что это свидетельствует о наличии систематической погрешности по крайней мере в одной из них.
□ Правильность обеспечивает возможность сопоставления результатов, полученных в разных лабораториях (см. разд. 3.3.1).
3.3.1. Зачем нужна правильность
Согласно новому определению ISO (см. разд. 3.2.1), точность включает в себя правильность и воспроизводимость. Воспроизводимость результатов обеспечивается использованием системы обеспечения качества (см. выше) и проверенных методик анализа. Для оценки правильности необходимо использовать внешние средства. Правильность является базой для сопоставления результатов, полученных в разное время н в разных лабораториях. Типичным случаем, когда необходимы правильные (н точные в целом) результаты, является мониторинг состояния окружающей среды. Например, постановления о сокращении вредных выбросов могут иметь измеримые последствия толь
194 Глава З. Обеспечение и контроль качества
ко по прошествии значительного времени. Чтобы их обнаружить, требуется применение очень точных методик в течение длительного периода. Многие исследователи считали, что для обнаружения столь медленных тенденций и, тем самым, демонстрации эффективности принятых мер, достаточно лишь хорошей воспроизводимости. Однако они не учли, что оборудование н методология измерений постоянно совершенствуются, а для обеспечения высокой воспроизводимости на протяжении длительного времени требуется постоянство системы измерений. Кроме того, если результаты недостаточно точны, невозможно сравнивать данные, полученные различными исследователями мира. Сходные проблемы возникают и в сфере торговли — например, когда необходимо удостоверить качество экспортируемой продукции путем измерения-их характеристик. Различия в данных, полученных производителями и потребителями, могут вызвать серьезные коммерческие конфликты. И в медицине, где результаты клинических анализов являются основой правильного диагноза, помимо долговременной воспроизводимости, требуется и правильность—особенно если пациент меняет лечебное учреждение.
Существуют три способа установления правильности результатов:
— сравнение с данными, полученными другим методом;
— сравнение с данными‘другой лаборатории;
— анализ стацдартньрс образцов.
3.3.2. Сравнение с данными другого метода
Для каждого метода характерны свои типичные источники погрешностей. Например, для спектроскопических методов элементного анализа— ААС, ИСП-АЭС, ИСП-МС—источником погрешности может быть операция , кислотного разложения (окисления) матрицы образца. Однако в инструментальном нейтронно-активационном анализе эта операция отсутствует, поэтому этот метод можно использовать для контроля методик, включающих разложение пробы, — например, ИСП-АЭС.
Если данные, полученные двумя методами, хорошо согласуются, это может свидетельствовать о правильности обоих результатов. При этом чем сильнее различаются между собой’отдельные стадии соответствующих методик, тем больше оснований для такого вывода. Если же методики на какой-то стадии схожи, то можно не заметить одинаковой систематической погрешности, обусловленной этой общей стадией. Результаты сравнения можно считать достоверными только тогда, когда данные получены с помощью двух совершенно различных методик под одинаково строгим лабораторным контролем. Если квалификация лаборантов в области метода, используемого для сравнения, недостаточна, то это может привести к неверным выводам или даже послужить источником дополнительных погрешностей. Поэтому для проверки правильно^ сти сравнительный анализ двумя методами трудно провести силами одной лаборатории; это делается редко. Эффективным средством может служить лишь сравнение с данными другой лаборатории — в том числе и для долговременного контроля характеристик методики, а также при постановке в лаборатории но
3.3. Как достичь правильности анализа
105
вого метода. В последнем случае для заключительной проверки правильности необходимо либо использование стандартных образцов, либо межлабораторный эксперимент.
3.3.3. Сравнение с данными другой лаборатории
□ Можно учиться не только на собственных ошибках, но и на чужих успехах.
Простейший способ улучшенйя качества работы лаборатории-—участие в межлабораторном эксперименте. Межлабораторный эксперимент состоит из серии практических работ, в ходе которых все участвующие лаборатории должны определить один или несколько компонентов в одном и том же образце, приготовленном организаторами.По окончании эксперимента его результаты предоставляются всем участникам. Межлабораторные эксперименты проводят для разных целей: оценки характеристик методики (например, для ее последующей стандартизации), изучения новой или трудной области химического анализа, аттестации стандартных образцов. В.Хорвитц в докладе IUPAC [3.3-1] дает определение межлабораторного эксперимента и отмечает его разновидности.
Межлабораторный эксперимент—это исследование, в ходе которого в нескольких лабораториях анализируют один или несколько идентичных однородных образцов в определенных условиях. Результаты исследования обобщаются в едином отчете.
Оценка характеристик лаборатории—это межлабораторный эксперимент, включающий один или несколько анализов одного или нескольких образцов, выполняемых в ряде лабораторий по одной и той же методике, предложенной или используемой в одной из участвующих лабораторий. Результаты, полученные в этой лаборатории, сопоставляются с результатами, полученными в других лабораториях, или с известным (действительным) содержанием. Целью исследования, как правило, служит оценка или улучшение аналитических характеристик лаборатории. Для исследования, посвященного оценке квалификации лаборатории (или отдельного аналитика), используют также термин «квалификационные испытания» (например, в руководстве ISO/IEC №25 [3.3-2] и стандарте EN 45001 [3.3-3]).
Оценка характеристик методики — это межлабораторный эксперимент, в ходе которого каждая лаборатория анализирует серию идентичных образцов с помощью одной н той же методики по единой схеме. Результаты используются для оценки характеристик методики — в первую очередь внутри- й межлабораторной воспроизводимости, а при необходимости н других: систематической погрешности, чувствительности, предела обнаружения, а также других параметров, используемых для внутрилабораторного контроля качества.
Исследование с целью аттестации образца—это межлабораторный эксперимент, результатом которого является действительное («истинное») значение концентрации компонента в исследуемом образце (или другая его характеристика), приводимое, как правило, с оценкой его неопределенности.
Межлабораторный эксперимент по оценке характеристик лаборатории позволяет его участникам оценить характеристики своих лабораторий путем сравнения собственных результатов с результатами, полученными другими, возможно более квалифицированными, аналитиками. Часто различные участии-
106
Глава 3. Обеспечение и контроль качества
ки используют разные методы. Это позволяет выявить в собственной методике неизвестные источники систематических погрешностей. Соответствующим образом организованный эксперимент позволяет продемонстрировать правильность результатов, получаемых в лаборатории. Возможность этого зависит от трех основных факторов:
— заинтересованности сотрудников и организации самой лаборатории;
— заинтересованности других участников эксперимента;
— организации эксперимента в целом.
Последнее обстоятельство наиболее важно и требует от устроителей широкой эрудиции во многих областях. Они должны показать свою способность организовать эксперимент так, чтобы все участники были уверены в том, что получат интересующую их информацию, т. е. точностные характеристики. Недавно АОАС и ISO/REMCO опубликовали согласованные принципы организации квалификационных испытаний [3.3-4].
Основные требования к экспериментам по оценке характеристик лабораторий в равной мере относятся и ко всем прочим видам межлабораторных экспериментов [3.3-5].
3.3.4. Стандартные образцы
Приведем следующие определения.
Образец сравнения — это однородный материал или вещество, одна ‘или несколько характеристик которого установлены достаточно надежно для того, чтобы использовать их для градуировки, контроля измерительных процедур и определения характеристик других материалов [3.3-6].
Стандартный образец —это образец сравнения, для которого одна или несколько характеристик удостоверены в соответствии с официальной процедурой, о чем имеется соответствующий документ (аттестат). Процедура аттестации обеспечивает прослеживаемость образца в единицах СИ (применительно к составу—в молях вещества), в которых выражена соответствующая характеристика. Каждое аттестованное значение сопровождается указанием его неопределенности при указанной доверительной вероятности [3.3-6].
Классификация стандартных образцов
Анализ стандартных образцов является простейшим способом достижения н доказательства правильности. Стандартные образцы, поставляемые надежными производителями (см. разд. 3.4.2), служат средством, связывающим воедино результаты аналитиков всего мира. С помощью стандартных образцов можно оценить показатели качества аналитической лаборатории в любой момент времени. В этом состоит большое преимущество использования стандартных образцов перед постановкой межлабораторного эксперимента. Стандартные образцы отвечают всем требованиям точности на различных этапах проверки методики. По своей природе стандартные образцы разделяют на следующие разновидности:
— чистые вещества и их растворы, используемые для градуировки и (или) идентификации;
33. Как достичь правильности анализа
107
— материалы с матрицей известного состава, используемые.для градуировки в сравнительных методах анализа, (разд. 3.2.1);
— материалы, матрица которых максимально точно воспроизводит матрицу реального объекта, с удостоверенным содержанием определенных компонентов (используются при проверке методик);
— материалы, параметры которых определяются применительно к заданной аналитической процедуре, например: содержание элемента в водной или кислотной вытяжке либо в биологически доступной форме, содержание пестицидов, экстрагируемых хлороформом и т. д.; в этом случае аттестованное значение находят в соответствии с выбранной процедурой, выполняемой в строго контролируемых условиях.
□ Состав стандартных образцов гарантируется ведущими аналитиками.
Стандартные образцы первых двух типов представляют особый интерес, поскольку они специально изготавливаются и их состав известен полностью.
Прн аттестации чистых веществ обычно указывают максимально возможное содержание (в массовых долях) различных примесей. Для образцов металлов достаточно привести массовые доли всех примесных элементов, определить которые позволяет чувствительность доступных методов анализа. Аттестация неорганических солей или оксидов более трудна, поскольку здесь требуется определить н стехиомегричность состава—в частности, содержание кристаллизационной воды. Определение же чистоты и стехиометричности органичен ских веществ — самая трудная задача, так как здесь не всегда возможно даже установить природу всех примесей. Для определения примесей следует использовать методы, основанные на измерении массы— такие, как хромато-масс-спектрометрия. Стандартные растворы следует готовить по навеске- в лабораториях, имеющих подготовленный и специально обученный персонал. При этом следует применять метрологически аттестованные методики взвешивания и контролировать чистоту н стехиомегричность исходных веществ.
Приготовление материалов заданного состава (металлов, сплавов, порошкообразных оксидов и имитаторов руд, образцов цемента, например, для рентгенофлуоресцентного анализа с волновой дисперсией) требует специальных знаний. Его можно осуществить по навескам металлов н других веществ известной чистоты.
Для третьего из рассмотренных типа стандартных образцов состав матрицы неизвестен или известен лишь частично. В этом случае аттестовать содержание компонентов можно лишь с помощью сложных аналитических процедур, точностные характеристики которых хуже, чем для измерения массы. Обычно аналитики предпочитают природные, не обогащенные определяемым компонентом образцы, состав которых как можно ближе к составу реальных объектов. При использовании же добавок, специально вводимых в образец, их содержание нельзя определять по навеске, поскольку—в частности, для органических микрокомпонентов — они могут быть лишь частично поглощены матрицей из-за потерь вследствие адсорбции на стенках сосудов, испарения или разрушения в процессе гомогенизации лабо стабилизации. Следует использовать матрицу, не содержащую вводимого компонента вообще, или удостовериться, что его исходное содержание пренебрежимо мало. Кроме
108
Глава 3. Обеспечение и контроль качества
того, в твердых материалах введенная и исходно содержащаяся в матрице части одного и того же микрокомпоиента обычно различаются по своему поведению. Это может быть обусловлено различиями в физическом или химическом состоянии и может исказить результаты пробоподготовки (разложения, экстракции). Тем не менее в некоторых случаях, когда устойчивость или однородность природного образца недостижима, введение в него добавок является единственной возможностью. Это может иметь место, например, для образцов вод. При оценке точностных характеристик методики ход анализа природных вод с добавками и полностью искусственных образцов, имитирующих природные, практически одинаков.
К последнему типу относятся образцы, параметры которых аттестуются в соответствии с определенной аналитической процедурой. Эта процедура может представлять собой как некоторую методику целиком (в частности, это может быть стандартная методика, например, при аттестации содержания летучих органических галогенов), так и ее отдельный этап. В последнем случае аттестуемый параметр характеризует содержание некоторой фракции определяемого компонента, обладающей определенным свойством, например, заданным содержанием элементов или соединений в подвижной фракции почв либо отложений.
□ Аттестация стандартных образцов предполагает, что она выполняется наиболее квалифицированными аналитиками, использующими наилучшие на данный момент методики. Любая ошибка, допущенная в ходе аттестации, вводит в заблуждение все мировое сообщество аналитиков.
В течение нескольких последних лет под эгидой BCR был проведен крупномасштабный межлабораторный эксперимент по определению водоизвлекаемой фракции следовых элементов в пахотных почвах и отложениях. В результате был выработан ряд мер по контролю качества подобных анализов [3.3-7]. Исследования, проведенные приблизительно в 20 лабораториях, показали, что при строгом соблюдении предписанной процедуры содержание водоизвлекаемой фракции ряда элементов не зависит от завершающего метода определения н может быть аттестовано. Соответствующие стандартные образцы сейчас находятся в стадии изготовления (в рамках программы M&T-BCR [3.3-3]). Сходные идеи лежат в основе оценки таких параметров, как содержание биологически доступной формы токсичных или питательных веществ в пищевых продуктах, извлекаемой формы токсикантов в стоках и др.
Процедуры аттестации стандартных образцов с реальной матрицей подробно описаны в руководстве ISO №35 [3.2-5]. Конкрентые примеры аттестации в соответствии с изложенными в них принципами можно найти в [3.3-9, 3.3-10].
Применение стандартных образцов
Градуировка, прослеживаемость, оценка точностных характеристик
Главная сфера применения стандартных образцов—доказательство того, что результаты, получаемые с помощью некоторой методики в данной лаборатории, правильны и, таким образом, могут быть использованы повсеместно. Иными словами, стандартные образцы позволяют прослеживать результаты анализа в первую очередь по отношению к самим образцам и, следовательно, к
3.4. Правовые аспекты обеспечения и контроля качества 149
тем единицам, в Которых выражены их аттестованные характеристики. Ряд стандартных- образцов (аттестованные растворьц сплавы, чистые металлы и др.) можно непосредственно использовать для градуировки.
Стандартные образцы с реальной матрицей следует оставить для проверки правильности методики и демонстрации ее прослеживаемости по отношению к основным единицам СИ (см. приложение'fe "конце т.2)". В случае относительных методов для проверки правильности следует выбрать стандартный образец с матрицей^максимально подобной матрице реального объекта, для которого методика предназначена. Использование стандартных образцов с реальной матрицей для проверки правильности относительных методов описано в руководстве ISO К*33 [3.3-11]. Стандартные образцы являются наилучшим средством для решения этой задачи.
□ Используя стандартные образцы, вы заранее знаете правильный результат.
Оценка характеристик лабораторных образцов сравнения
Обычно стандартные образцы выпускаются в количествах, недостаточных для повседневного лабораторного контроля качества (например, с помощью контрольных карт). Задача изготовления (или приобретения) образцов сравнен ния для таких нужд лежит на самом аналитике. Аналитик может сопоставить образцы сравнения с подобными ему стандартными образцами и с помощью последних оценить точностные характеристики н продемонстрировать просле-1 живаемость методики. Однако в рамках одной лаборатории установить прослеживаемость образцов сравнения по отношению к стандартным образцам достаточно трудно. Желательно, чтобы образцы сравнения анализировали в нескольких лабораториях, причем по возможности различными методами.
3.4. ПРАВОВЫЕ АСПЕКТЫ ОБЕСПЕЧЕНИЯ
И КОНТРОЛЯ КАЧЕСТВА
Химический анализ играет все возрастающую роль в механизме принятия решений на официальном, законодательном и частном уровнях. Поэтому всеми, кому необходимы результаты анализов—в том числе н на политическом, и на коммерческом уровне,—давно признанр, что качество предоставляемых результатов необходимо гарантировать. Ряд случаев с драматическими последствиями, имевших место в фармацевтической промышленности и вызванных неудовлетворительной достоверностью или .плохой прослеживаемостью результатов анализа, заставил власти принять меры к организации соответствующей контролирующей системы.
Впервые строгие международные правила контроля качества выполнения анализов (такие, как GLP — «Хорошая лабораторная практика») были введены в фармацевтической промышленности и на производстве токсичных веществ (а постепенно н в других отраслях химической промышленности). В большинстве развитых стран (в некоторых, как, например, Австралии, очень давно) по отношению к аналитическим лабораториям были введены так называемые системы аккредитации. Сейчас наметилась тенденция объединить требования стандартов EN 45000-ISO/IEC и GLP в единый всемирно применяемый стандарт, сопровождаемый общими руководящими указаниями, обя
110
Глава 3. Обеспечение и контроль качества
зательными для всех лабораторий. Это позволит упростить систему оценки лабораторий и в некоторых случаях избежать повторного аудита.
3.4.1. Оценка квалификации лабораторий
«Хорошая лабораторная практика» (GLP)
□ Нормативы «Хорошая лабораторная практика» (GLP) представляют собой первую попытку всеобъемлющего законодателъного.контроля производства в области химической промышленности. Нормы GLP предназначены главным образом для проверки прослеживаемости информации о практической деятельности.
Впервые нормативы GLP были разработаны в 1978 г. американским Управлением пищевых продуктов и лекарств (FDA). Затем американское Агентство по охране окружающей среде (ЕРА) в 1983 г. издало аналогичные нормативы, касающиеся производства токсичных веществ для промышленности и сельского хозяйства. После этого, следуя также пионерской работе Всемирной организации здравоохранения, принципы GLP приняла своим решением С81/30 от 1981 г. Организация экономического сотрудничества и развития (OECD) [3.4-1]. Впоследствии они стали обязательными для всех производителей стран—членов OECD в области контроля качества в фармацевтической промышленности и на производстве токсичных веществ (например, пестицидов). Совет Европейского Союза принял ряд руководящих документов, касающихся согласования (87/18/ЕЕС), проведения инспекций н проверки соблюдения правил GLP (88/320/ЕЕС и 90/18/ЕЕС).
В сфере своей компетенции нормы GLP касаются организационного процесса и общих условий планирования, выполнения и контроля'лабораторной работы, ведения документации и отчетности [3.4-1]. Их задача—гарантировать, что любые действия, предпринимаемые в ходе разработки продукции, исследований ее безопасности и токсичности, прослеживаемы и их ход может быть полностью восстановлен в любой момент. В. Мертц с corp. [3.4-2] характеризует задачи GLP следующим образом: «GLP направлена лишь на то, чтобы обеспечить получение всеобъемлющей информации об общих целях, планировании, выполнении, оценке и представлении результатов исследований. Не больше и не меньше. GLP не касаются ни конкретные требования, предъявляемые к выполнению анализов, ни их конкретные задачи, ни интерпретация результатов...» Иными словами, выполнение правил GLP само по себе не гарантирует требуемой точности результатов. Для этого существуют другие средства—стандартные образцы и участие в квалификационных испытаниях. GLP, так же, как и системы аккредитации, позволяют лишь улучшить общие условия работы и предложить меры, позволяющие достичь необходимой точности.
Системы аккредитации
□ Прн аккредитации проверяется соответствие лаборатории общим требованиям, необходимым для ее работы на должном уровне.
Нормативы GLP были разработаны применительно к исследованиям токсичности новых химических или лекарственных веществ. Постепенно в систему
3.4. Правовые аспекты обеспечения и контроля качества
111
GLP вовлекались все новые отрасли науки и промышленности, особенно в том, что касается требований безопасности. Однако аналитические лаборатории общего профиля остались не охваченными, и нормы GLP к ним, как правило, не применялись. Для таких лабораторий, а также лабораторий, относящихся к другим отраслям промышленности, были разработаны иные системы оценки, и некоторые из них существуют в течение долгого времени. В зависимости от сферы деятельности (анализы продуктов питания, объектов окружающей среды, биомедицинские анализы и т. д.) система контроля применялась в форме административных инспекций, квалификационных испытаний или того и другого одновременно. В некоторых странах в основе системы контроля аналитических лабораторий лежит руководство ISO/IEC j 25 от 1978 г. [3.3-2]. В Европейском Союзе (EU) 21 декабря 1989 г, была принята резолюция Совета Министров 90/С/10/01 о «всеобъемлющем подходе к аттестации соответствия», содержащая руководящие принципы, приведенные в сериях европейских стандартов EN 45000 [3.3-3] применительно к аналитическим лабораториям и EN 29000 (непосредственно включающих положения ISO/IEC 9000) [3.1-6] применительно к производственной деятельности. Система аккредитации аналитических лабораторий включает в себя семь стандартов:
• EN 45001 — общие критерии деятельности аналитических лабораторий;
• EN 45002 — общие критерии аттестации аналитических лабораторий;
• EN 45003—общие критерии для органов по аккредитации лабораторий;
• EN 45011 — общие критерии для органов по аттестации продукции;
• EN 45012 — общие критерии для органов по аттестации систем качества;
• EN 45013—общие критерии для органов по аттестации персонала;
• EN 45014—общие критерии для деклараций поставщиков о соответствии.
Резолюция Совета Министров EU предусматривает взаимное признание принципов деятельности систем аккредитации, принятых в странах-участницах. Стандарт EN 45001 приводит перечень требований и рекомендаций по обеспечению и контролю качества работы аналитических лабораторий. Они включают в себя рекомендации относительно системы управления, инфраструктуры, квалификации персонала, оборудования, применяемых методик и особенно подробно—вопросы, связанные с градуировкой и применением стандартных образцов. Отметим, что требование обязательного участия лаборатории в квалификационных испытаниях в этом стандарте отсутствует— организация таких испытаний отнесена к компетенции органов аккредитации. При этом в стандарте EN 45002 ясно указывается, что документы об аккредитации лаборатории не могут быть автоматически выданы или подтверждены лишь на основании результатов, показанных лабораторией в ходе квалификационных испытаний. Эту систему нормативов приняли и некоторые страны, не входящие в EU, но ратифицировавшие Европейское соглашение о свободной торговле. Все западноевропейские органы аккредитации сотрудничают с Европейской кооперацией по аккредитации лабораторий (EAL) с целью совместного внедрения и развития своих систем аккредитации. В неевропейских странах также существуют сходные системы аккредитации, например NATA в Австралии, AALA в США. На всемирном уровне органы аккредитации сотрудничают в рамках Международной кооперации по аккредитации лабораторий (ILAC). Ряд международных химических обществ — такие, как IUPAC и АОАС, — так
112
Глава 3. Обеспечение и контроль качества
же вносят свой вклад вразвитие общих подходов^ терминологии, единых принципов «и средств улучшения качества работы аналитических лабораторий.
Стандарты ISO 9000/EN 29000
D tSO 9000 представляет собой серию основных стандартов, касающихся систем качества Применительно к производству и сфере услуг.
Требования GLP направлены лишь на обеспечение безопасности химической продукции. Серия стандартов ISO 9000 [3.1-6] имеет своей задачей обеспечение качества любой продукции или деятельности в сфере услуг. В 1987 г. Европейский коминет по стандартизации (CEN) включил положения ISO 9000 в соответствующие стандарты серий EN 29000, утвержденные упоминавшейся резолюцией Совета Министров СЕ 90/С/10/01 от 21 декабря 1989 г. Серия ISO 9000/EN 29000 включает пять стандартов:
• ISO 9000/EN 29000—общие принципы и определения;
• ISO 9001/EN 29001 — показатели систем качества и требования при разработке продукции и услуг;
• ISO 9002/EN 29002 — показатели систем качества и требования при производстве и поставках продукции;
• ISO 9003/EN 29003—показатели систем качества и требования при заключительном инспектировании и испытаниях;
• ISO 9004/EN 29004— руководство по управлению качеством.
В стандартах ISO 9001, 9002 и 9003 и их аналогах серии EN .29000 рассматриваются измерения и требования к их качеству. Таким образом, деятельность аналитиков, которые работают в лабораториях, относящихся к сфере промышленности или услуг, и стремятся получить аттестацию в соответствии с иормами ЕМ 29000, будет находиться под контролем и аудитом.
3;4;2. Аккредитация производителей образцов сравнения и Стандартных образцов
Образцы, сравнения и стандартные образцы являются важным средством проверки того, что результаты анализа достоверны и сравнимы ^ежду собой. Поэтому качество образцов сравнения и стандартных образцов, а также научный и технический уровень их производителей должны быть аттестованы. За последние годы число таких производителей, особенно образцов сравнения, возросло, но качество производимых материалов не всегда было должным образом удостоверено. Чтобы помочь производителям в решении этой задачи, а потребителям в оценке компетентности производителей, Международная организация по стандартизации (ISO) начала разработку системы показателей качества для производства образцов сравнения и стандартных образцов. В основу положены нормы серии стандартов ISO 9000 и руководства ISO №5. Выпущен проект руководства ISO, содержащий интерпретацию положений стандартов ISO 9000 и руководства ISO №25. В нем предложен механизм, позволяющий установить, что производство образцов сравнения и стандартных образцов выполняется в соответствии с требованиями упомянутых документов. Этот проект охватывает следующие положения.
3.4. Правовые аспекты обеспечения и контроля качества
113
— Организационные требования: система управления, политика качества, система качества, квалификация и обучение персонала, сотрудничество с партнерами, хранение и долговременный контроль продукции, система документации и отчетности, послепродажное обслуживание.
— Контроль производства: планирование, приготовление материалов, прослеживаемость, градуировка, измерительные приборы и методы измерений, проверка однородности и стабильности образцов, обработка данных и их аттестация.
Технические требования приведены в руководствах ISO ЖИ [3.4-3] и J035 [3.2-5]. В случае успеха этого проекта ISO будет введена система аттестации производителей образцов сравнения и стандартных образцов. В свою очередь это поможет потребителям образцов сравнения и стандартных образцов правильно выбирать деловых партнеров.
3.4.3. Аттестация персонала
Первый официальный документ, который получает любой химик, —это диплом об окончании учебного заведения. После этого аналитик должен поддерживать и повышать свой профессиональный уровень путем регулярного чтения научной литературы, участия в научных мероприятиях, а также в силу необходимости во внутрилабораторной квалификационной аттестации. Участие в межлабораторных экспериментах (см. разд. 3.3.3) открывает дополнительные возможности оценить свой уровень и выявить необходимость дополнительного обучения. Для повышения своей квалификации аналитик должен использовать любые средства в пределах лаборатории: осваивать иовые-ирибо-ры и методы, учиться определять новые аналитические параметры, овладевать новыми сферами анализа. Аккредитация лабораторий, в частности, требует, чтобы в лаборатории была предусмотрена и реализована программа непрерывного повышения квалификации сотрудников.
Только по окончании вуза и получении ученой степени химик может рассчитывать на признание со стороны руководства. Очень немногие официальные структуры, занимающиеся последипломным образованием, выдают документы общепризнанного образца, но почти во всех западных странах химические общества организуют программы непрерывного последипломного обучения и выдают свидетельства об участии в них; ряд университетов проводит курсы по изучению и преподаванию отдельных разделов аналитической химии. Несколько лет назад в Великобритании Королевское химическое общество учредило Регистр «продвинутых» химиков-аналитиков. В него могут быть внесены только члены общества при выполнении ряда требований — в частности, наличия трех письменных рекомендаций. Подобные регистры ведут и другие химические общества (например, общество медицинской химии), а Федерация европейских химических обществ учредила звание «Европейский химик» [3.3-4].
В рамках общих требований к аккредитации лабораторий европейский стандарт EN 45013 [3.3-3] предусматривает критерии для органов, занимающихся аттестацией персонала. Эти аттестационные требования касаются не только химиков-аналитиков, но и руководящих работников, а также сотрудников службы обеспечения качества.
114
Глава 3. Обеспечение ^контроль качества
3.4.4. Стандартизация
Официальные стандарты
□ Официальные стандарты, или нормативы, служат средством законодательного закрепления сложных технических понятий.
Официальные стандарты (нормативы) предназначены для установления и поддержания требований к качеству продукции, а также для улучшения сопоставимости результатов анализов. Они также являются первым этапом на пути законодательного закрепления этих требований. В нормативных документах как законодательного, так и коммерческого уровня часто встречаются ссылки на стандарты как основание для применения тех или иных требований. В аналитической химии официальные стандарты доказали свою важность во многих сферах, таких, как стратегия и методика пробоотбора, определение ряда обобщающих параметров (общее содержание органического углерода или органических галогенов, различные фракции органических веществ — экстрагируемая, водоизвлекаемая, биологически доступная) и их измерения.
В ряде других областей аналитической химии аналитики часто испытывают неудобства от отсутствия официальных стандартов, особенно в том, что касается законодательных аспектов. Положение усугубляется тем, что многие стандартные методики к настоящему времени устарели, однако остаются обязательными, поскольку законодательство давно не менялось. Таким образом, несмотря иа появление методик более высокого качества, аналитик вынужден пользоваться старыми ввиду требований закона. С подобными ситуациями часто сталкиваются при анализе медицинской, пищевой продукции и при контроле окружающей среды.
Применение стандартной методики не гарантирует отсутствие погрешностей. В ходе экспериментов под эгидой BCR наблюдались значительные расхождения в результатах лабораторий, использующих одни и те же стандартные методики [3.4-5]. Причиной могут быть систематические погрешности, ненадлежащее выполнение методики оператором, а также неоднозначности в формулировках самих стандартов. Кроме того, стандартные методики часто разрабатывают высококвалифицированные аналитики, которые могут забыть о необходимости адаптировать методику к повседневным измерениям, выполняемым менее квалифицированным персоналом. Формулировки стандартов также часто принадлежат химикам высшей квалификации, поэтому нет гарантии, что они будут понятны любому, кто пожелает применить эту методику. Таким образом, каждую методику, подлежащую стандартизации, следует обязательно проверять на устойчивость.
Органы стандартизации признали важность стандартных методик для достижения прогресса в химическом анализе, а также трудности, связанные с применением стандартов. В новейших стандартах указываются лишь общие принципы выполнения методики, а конкретные требования к точностным и другим аналитическим характеристикам оставлены на усмотрение специальных стандартов. Помимо всего прочего, такие специальные стандарты призваны продемонстрировать, что соответствующие аналитические характеристики реально достижимы (например, при анализе стандартных образцов).
3.5. Выводы
115
Органы стандартизации
Органы стандартизации существуют на региональном, национальном и международном уровне. Они могут полностью или частично подчиняться органам государственной власти или учреждаться в рамках профессиональных либо коммерческих организаций. Для европейских стран — членов Европейского Союза и Европейской ассоциации свободной торговли—поставщиками стандартов служат организации CEN/CENELEC/ETSI; соответствующие общеевропейские стандарты постепенно вытесняют национальные. Более ста стран участвуют в работе ISO. Между ISO и CEN в 1991 г. было заключено соглашение с целью избежать дублирования в работе. В соответствии с этим соглашением CEN использует стандарты, разработанные ISO, если они отвечают потребностям CEN. Направления деятельности ISO и CEN весьма разнообразны. В их рамках существуют сотни технических комитетов, каждый из которых руководит неколькими рабочими группами. Процедура утверждения каждого нового, особенно международного, стандарта, очень длительна и обычно занимает несколько лет. Стоимость разработки стандартов также очень велика. Обычно ее финансируют промышленные предприятия, общественные организации, национальные органы стандартизации, а при разработке стандартов на уровне Европейского Союза—Европейская Комиссия.
Профессиональные организации также разрабатывают свои официальные стандарты, которые затем могут быть утверждены ISO или CEN. Типичным примером может служить деятельность Международной молочной федерации (IDF), разрабатывающей стандарты для молока и молочных продуктов, утверждаемые ISO.
3.5. ВЫВОДЫ
□ «Дешевле —это нв всегда лучше, но лучше —это всегда дешевле» (Деминг).
Обеспечение и контроль качества необходимы аналитикам для того, чтобы они могли представлять потребителям осмысленные и достоверные результаты. Выполнение анализов без использования средств обеспечения и контроля качества следует считать непродуктивной деятельностью: «лучше не иметь никаких результатов, чем иметь нетравильные». Американскому основоположнику в области обеспечения и контроля качества на производстве У. Э. Демингу принадлежит также следующее высказывание: лдешевле — это не всегда лучше, но лучше — это всегда дешевле*. Как следует из материала данной главы, для достижения качества аналитик должен хорошо знать фундаментальные науки — химию, математику, статистику, физику, биохимию, в некоторых случаях — биологию, быть сведущим в области аппаратуры, а также в основах управления и экономики. Какой замечательный выбор!
Литература
[3.1-1] Uriano, G. A., Gravatt, С. С., The role of reference materials and reference methods in chemical analysis. CRC Critical Review in Analytical Chemistry, October 1977.
[3.1-2] Hertz, H.S., Anal, chem 1988, 60(2), 75A-80A.
116
Глава 3. Обеспечение и контроль качества
[3.1-3] Quinn, T.J., Bankvall, С., Harrington, М. G., Machado Jorge, H., Repussard, J., Reuter, H.W., Evaluation of the BCR Programme 1988-1992, Measurement and Testing in Europe, Report EUR 15041 EN, Commission of the European Communities, Luxembourg, 1992.
[3.1-4] Tolg, G-, private communication, 1982.
[3.1-5] ISO, Quality Vocabulary (ISO/IEC Standard 8402), International Organization for Standardization, Geneva, CH, 1986.
[3.1-6] ISO, Quality management and Quality Assurance Standards. Guidelines for Selection and- .Use (ISO/IEC Standard 9000), International Organization for Standardization, Geneva, CH, 1987.
[3.1-7] Broderick, В. E-, Cofino, W.P., Cornells, R, Heydorn, K., Horwitz W., Hunt D.N.E., Hutton, R. C. Kingston, H.M. Muntau H., Baudo R., Rossi D-, van Raaphorst, J. G., Lub, T. T., Schramel, P., Smyth, F. T., Wells, D. E., Kelly, Д. G., Mikrochim. Acta (Wien), II (1-6), 523-42, 1991-
[3.1-8] Valcarcel, M. Rios, A. Anal. Chem 1993, 65(18), 78A-787A.
[3.1-9] Cofino W. P., in: Environment Analysis Techniques, Applications and Quality Assurance — Techniques and Instrumentation in Analytical Chemistry, Barcelo, D. (Ed.) Amsterdam: Elsevier Science Publischers, 1993; Vol. 13, pp. 79-105.
[3.2-1] BIPM/IEC/ISO/OIML, International Vocabulary of Basic and General Terms in Metrology, International Organization of Standardization, Geneva, CH, 1984.
[3.2-2] ISO (1977), Statistics — Vocabulary and Symbols — Part 1: Probability and General Statistical Terms Revision of ISO 3534', International Organization for Standardization, Geneva, CH 1977.
[3.2-3] OIML, Vocabulary of Legal Metrology, International Organisation fot Legal Metrology, Paris, R., 1978.
[3.2-4] Garfield F. M., Quality Assurance Principles for Analytical Laboratories. 2 nd ed. AOAC International Ed., Arlington VA, USA, 1991.
[3.2-5] ISO, Certification of Reference Materials — General and Statistical. Principles, ISO Guide 35-1985 E, International Organization for Standardization, Geneva, CH 1985.
[3-2-6] Marschal A., Calibration of Chenical Analyses and'Use of Certified Reference Materials, Draft ISO Guide 32, ISO/REMCO №62 Rev. August 1993, International Organization for Standardization, Geneva, CH, 1993.
[3.2-7] EXERA, SIREP and WIB, Internal ionol Instrument Users’ Associations, Studies on the compliance of analytical instruments, EXERA— Parc Technologique ALATA, BP 2, F-60550 Verneuil en Halatte, F, 1993.
[3.2-8] Massart D. L. Vandeginste B. G.in., Deming S. N-, Michotte Y., Kaufmann L-, Chemometrics: a textbook. Data Handling in science and technology—Vol. 2, Amsterdam: Elsevier Science Publischers В. V., 1988.
[3.2-9] Wells, D. E., Maier E. A., Griepink B., Intern. J. Environ. Anal. Chem 1992, 46, 255-64.
[3.2-10] Moody J. R. Greenberg, R. R., Pratt K. W., Pains T. C., Anal. Chem 1988, 60(21), 1203A-18A.
[3.2-11] Maier E. A. Griepink B., Muntau H., Vercoutere K., Certification of the total content (mass fractions) of Cd, Co, Си, Mn, Pb, Ni and Zn and the aqua regia soluble contents (mass fractions) of Cd, Pb, Ni and Zn in a light sandy soil (CRM 142R). Report EUR 15283 EN, Commission of the European Communities, Luxembourg, 1993.
[3.2-12] Wells D. E., in: Environment Analysis Techniques, Applications and Quality Assurance — Techniques and Instrumentation in Analytical Chemistry, Barcelo, D. (Ed.). Amsterdam: Elsevier Science Publishers В. V., 1993, Vol. 13, pp. 79-105.
3.5- Выводы
117
[3.2-13] Mullholland М., Walker N., van Leuven, J. A., Buydens L., Maris F-, Hindriks H-, Schoenmakers P. J. Mikrochim. Acta (Wien), 1991, 11(1-6), 493-503.
[3.2-14] AOAC (1990), Official Metholds of Analysis. 15 th ed. Association of Official Analytical Chemists, Arlington, VA, USA.
[3.3-1] Horwitz W., Nomenclature for interlaboratory studies, 4th draft, IUPAC, Analytical Chemistry Division, Commission Vl, project 27/87.
[3.3-2] ISO, Giadelines for Assessing the Technical Competence of Testing Laboratories, ISO/IEC Guide 25-1978 E, International Organization for Standardization, Geneva, CH, 1978.
[3.3-3] CEN, General Criteria for the Operation of Testing Laboratories (European Standard 45001), CEN/CENELEC, Brussels, B, 1989.
[3.3-4] Thompson, M., Wood R., The international harmonised protocol for the proficiency testing of (chemical) analytical laboratories, IUPAC/ISO/AO AC, Pure & Appl. Chem 1993, 65(9), 2123-44.
[3.3-5] Maier E. A., Quevauviller Ph., Griepink B., Rymen T., van der Jagt H., van Rooij, M. A. F. P. The role of interlaboratory studies in the improvement of the quality of chemical measurements, first draft, IUPAC, Analytical Chemistry Division, Commission V2, project QA 2/91, 1992.
[3.3-6] ISO, Terms and definitions used in connection with reference materials, ISO Guide 30 revised version 1991, International Organization for Standardization, Geneva, CH, 1991.
[3.3-7] Griepink B., Intern. J. Environ. Anal. Chem 1993, 51, 123-128.
[3.3-8] Ure, A., Quevauviller Ph., Muntau H., Griepink B., Improvement in the determination of extractable contents of trace metals in soil and sediments prior to certification, Report EUR 14763 EN, Commission of the European Communities, Luzembourg, 1993.
[3.3-9] Maier E. A., in: Environment Analysis Techniques, Applications and Quality Assurance — Techniques and Instrumentation in Analytical Chemistry, Barcelo D. (Ed.) Amsterdam: Elsevier Science Publischers, 1993, Vol. 13, pp 383-401-
[3.3-10] Wise S- A., (1993), in: Environment Analysis Tehcniques, Applications and Quality and Quality Assurance — Techniques and Instrumentation in Analytical Chemistry, Barcelo D. (Ed.). Amsterdam: Elsevier Science Publishers В. V., 1993; Vol. 13, pp. 403-446.
[3.3-11] ISO, Uses of certified materials, ISO Guide 33, International Organization for Standardization, Geneva, CH, 1989.
[3.4-1] OECD, Decision of the Council: C81/30 (final) Annex 2, OECD Guidelines for Testing Chemicals, *OECD Principles of Good Laboratory Practice», OECD Paris, F, 1981.
[3.4-2] Merz, W., Weberruss U., Wittlinger R., Fres. J. Anal. Chem 1992, 342, 779-782.
[3.4-3] ISO, Contents of certificates of reference materials, ISO Guide 31, International Organization for Standardization, Geneva, CH, 1981.
[3.4-4] Thomas, J. D. R., Fres. J. Anal. Chem 1993, 347, 25-28.
[3.4-5] Marchandise H., Fres. Z. Anal. Chem 1987, 326, 613-16.
Вопросы и задачи
1. В чем заключается роль химика-аналитика?
2. Кто определяет стратегию анализа?
3. Что такое обеспечение качества и контроль качества?
4. Возможен ли (целесообразен ли, необходим ли) контроль качества без обеспечения качества?
118
Глава 3. Обеспечение и контроль качества
5- На ком лежит ответственность за проверку оборудования?
6. Зачем и когда необходимо проверять оборудование?
7. Каковы основные и вторичные характеристики качества аналитической методики? Приведите их определения.
8. Что включает в себя проверка методики анализа?
9- Зачем и когда необходима проверка методики?
10. Как проверить стадию предварительной обработки пробы?
11. Как проверить стадию градуировки?
12. На что следует обращать внимание в ходе градуировки?
13. В чем разница между сходимостью и воспроизводимостью?
14. Что такое устойчивость методики? Почему эта характеристика очень важна?
15. Несет ли потребитель ответственность за результаты, полученные им от аналитика?
16. Что такое межлабораторный эксперимент? Какие задачи он решает?
17. Что такое образцы сравнения? Для чего их применяют?
18. Каковы основные характеристики и свойства образцов сравнения? Как их готовят?
19. Как удостовериться в однородности и стабильности образца сравнения?
20. Приведите определения стандартного образца. Д ля чего используют стандартные образцы?
21. Что означают следующие термины: GLP, аккредитация лабораторий, аттестация лабораторий? К какому роду деятельности относится каждый из них? *
22. Относится ли термин «аккредитация» только к аналитическим лабораториям?
23. Что такое контрольные карты и контрольные точки? В чем их роль в рамках общего процесса проверки методики?
24. Как добиться точности результатов анализа?
25. Необходимы ли правильность, воспроизводимость и, следовательно, точность для того, чтобы можно было сопоставлять результаты из разных лабораторий?
26. Обеспечение качества—это роскошь или экономическая необходимость?
27. Могут ли официальные стандарты гарантировать качество результатов и, если да, то всегда ли?
28. Что следует делать, если в ходе межлабораторного эксперимента лаборатория получила неудовлетворительные результаты?
29. Как связаны аккредитация лаборатории и ее квалификационные испытания?
30. Кто организует процесс аккредитации и несет за него ответственность?
31. Для того чтобы потребитель смог сделать осмысленные выводы из результатов анализа, достаточно ли, чтобы они были правильными?
32. А достаточно ли в этом случае хорошей воспроизводимости?
33. Можете ли вы указать общественные и экономические ситуации, в которых для решения поставленных задач необходима высокая точность результатов анализа?
34. На какие основные классы (с точки зрения особенностей измерительной процедуры) делятся методы анализа?
35. Что означает термин «прослеживаемость результатов анализа»?
36. Как достичь прослеживаемости? По отношению к чему прослеживают результаты анализа?
Часть I
ХИМИЧЕСКИЕ МЕТОДЫ АНАЛИЗА
В этой части книги мы рассмотрим химические основы аналитической химии и те аналитические методы, которые основаны на непосредственном химическом взаимодействии определяемого вещества с аналитическими реагентами. Причиной, объясняющей такое внимание к химическим реакциям определяемых компонентов, является широкое использование подобных реакций в методах качественного (идентификация) или количественного анализа. Кроме того, во многих методах разделения часто используют вспомогательные химические реакции, поэтому развитие теоретических основ этого подхода предопределяет дальнейший прогресс в этой области.
Как правило, для корректного решения поставленных перед аналитиком задач он должен уверенно оперировать познаниями в области общей, физической, неорганической и органической химии. Однако в современной аналитической химии существует тенденция к расширению требуемой области знаний и часто необходимо использовать те знания, которые находятся за границами классической химии. В этом случае аналитическая химия представляется междисциплинарной наукой, включающей еще и знания по биологии, медицине, материаловедению и микротехнологии.
□ Анализ химическими методами—та часть аналитической химии, в которой анализируемую пробу подвергают химическому взаимодействию с аналитическим реагентом.
ОСНОВЫ ХИМИЧЕСКИХ
МЕТОДОВ АНАЛИЗА . ч
~ -5 * - * i- »
Основы химических методов анализа базируются на закономерностях химических реакций, используемых для аналитических определений. Это могут быть реакции подготовки пробы на отдельной стадии анализа, позволяющие исключить влияние матрицы на результаты определений (например, осаждение, пре-и постколоиочная дериватизация в хроматографии, см. гл. 5, и др.).
При описании основ этих методов наиболее важны равновесия в гомогенных и гетерогенных системах, условия равновесия.
4.1. РАВНОВЕСИЕ В ГОМОГЕННЫХ СИСТЕМАХ
Цели изучения
• Описать равновеское состояние и механизмы, приводящие к нему.
• Определить различия между гомогенным и гетерогенным равновесиями, а также между однокомпонентной и многокомпонентной системами.
• Выявить роль, которую играют химические и физические равновесия в аналитической химии. Отметить различие между равновесием и стационарным состоянием, а также обсудить соотношения между константой равновесия и термодинамическими свойствами.
4.1.1. Введение
Большинство аналитических методов, используемых для количественного определения химических веществ, основываются на реакциях, протекающих в гомогенных растворах. Химические равновесия, устанавливающиеся при этом, локализованы в одной фазе и называются гомогенными равновесиями. В противоположность этому, в большинстве методов разделения используют реакции, в которых образуется вторая фаза (например, осадок) или химические вещества распределяются между двумя или более сосуществующими фазами. Устанавливающиеся равновесия называются гетерогенными равновесиями. Гомогенные и гетерогенные системы могут быть образованы отдельным веществом, но могут состоять и из нескольких компонентов- Такие системы называются соответственно одно- и многокомпонентными.
4.1. Равновесие в гомогенных системах
121
В разд. 4.1 обсуждаются гомогенные равновесия и связанные с ними вопросы аналитической химии, а также примеры гомогенных равновесий, представляющие интерес для химика-аналитика. Гетерогенные же равновесия и связанные с ними собственно аналитические вопросы рассматриваются в разд. 4.5. Гомогенная фаза является единственной формой состояния и отделяется от своего окружения определенной фазовой границей.
□ Термины гомогенные и гетерогенные (системы) — антонимы.
4.1.2. Состояние равновесия
В системе, в которой большое число веществ взаимодействую? друг с другом, происходят химические изменения до тех пор, пока не достигается некоторое устойчивое состояние. Это конечное состояние называется состоянием равновесия системы.
Любое состояние химического равновесия может быть описано константой равновесия К. Это очень полезный параметр, и он может быть использован для описания большого ряда аналитически важных химических процессов, таких, как кислотно-основные взаимодействия, комплексообразование, а также окислительно-восстановительные (редокс) реакции. Гетерогенные равновесия играют важную роль в современных аналитических методах разделения, таких, как экстракция и хроматография. Константа равновесия — это средство, с помощью которого могут быть представлены и подробно описаны химические равновесия всех типов. Введение этой характеристики системы позволяет объяснять и моделировать особенности химических процессов в сложных системах.
При описании всех видов химических равновесий ключевое значение имеет понимание ряда термодинамических понятий. Эти параметры чрезвычайно полезны при описании не только модельных равновесий, но и равновесий, устанавливающихся в реальных системах. Константа равновесия связана с такими термодинамическими параметрами системы, как свободная энергия, энтальпия (тепловое содержание) н энтропия:
AG°
(4.1-1) ill
и
AG0 = ДЯ° - TAS0 (4.1-2)
где К —константа равновесия, AG0, АЯС и Д5° — стандартная свободная энергия, стандартная энтальпия и стандартная энтропия реакций соответственно, Т—температура в градусах Кельвина, R—газовая постоянная 8,314 Дж/ (моль-К).
4.1.3. Равновесные системы
Если химические реагенты смешать вместе, реакции могут происходить до тех пор, пока не будет достигнуто состояние равновесия. С этого момента на макроскопическом уровне не наблюдается никаких дальнейших изменений. Изме
122
Глава 4. Основы химических методов анализа
нения могут быть химической или физической природы, а фазовые переходы могут быть сложными. Движение к равновесию лучше всего рассмотреть на примере системы, в которой изменяются лишь физические параметры. Рассмотрим два одинаково больших объема в некотором сосуде, разделенных общей стенкой и заполненных одинаковым количеством разных идеальных газов при одинаковом давлении (например, А и В в объемах I и П соответственно). Благодаря тепловому движению молекулы газа постоянно сталкиваются друг с другом и со стенками сосуда.
При открывании отверстия в разделяющей стенке молекулы газов могут свободно двигаться внутрь соседних объемов (рис. 4.1-1,а-г). Непосредственно после открывания поток газа А движется в объем П, а В — в объем I. С течением времени при постоянно открытом отверстии левая часть сосуда будет все еще обогащена А, а правая — в основном молекулами В (рис. 4.1-1,б-в). В конце концов два газа полностью смешиваются, и любая взятая из сосуда проба содержит А и В в соотношении 1:1 (рис. 4.1-1,г).
□ В химическом анализе «равновесия» описывают не состояние покоя, а устойчивое динамическое состояние, при котором обе реакции (прямая и обратная) протекают с одной и той же скоростью.
В конечном, равновесном, состоянии тепловое движение молекул продолжается как и прежде, но одинаковые количества газа А движутся во всех направлениях. Эти передвижения являются беспорядочными и нейтрализуют друг друга. Следовательно, на макроскопическом уровне кажется, что система находится в покое. Про такую систему говорят, что она находится в динамическом равновесии. В состоянии равновесия нет потока энергии или массы внутри или через границы системы. Все компоненты остаются"» системе и характеризуются временем пребывания (или временем жизни) тд, равным бесконечности. Таким образом, данная равновесная система является закрытой системой во время-инвариантном (не зависящем от времени) состоянии. Все химические и физические равновесия представляют собой состояния динамического равновесия закрытых систем. Этот факт можно проиллюстрировать следующим примером. При растворении диоксида углерода в воде образуется угольная кислота:
CO2(aq) + Н2О -> Н2СОз (4.1-За)
Символ (aq) свидетельствует о том, что в реакции принимает участие растворенный, а не газообразный СО2. Чем больше растворяется СО2, тем больше образуется Н2СОз. Скорость образования угольной кислоты прямо пропорциональна концентрации CO2(aqj:
d|H^°-=*:i|c°2^)1 С4-1-4)
где ki = 0,03 с-1 при 25°С (константа скорости прямой реакции). Эта реакция сопровождается структурными изменениями. Молекула СО2 имеет линейное строение:
:о;;с::о; или о=с=о (4.1-5)
< ачии<т^м168ВввЕвввйввйввы
AAAAAJXAAAAZtAAAAAAi^AAAAntiMKniWglABBBBBABBBBBBBBBl . «AAAAAAAAAAAAAAhu.ААаАВВААВАВВВВВВВВВБВВВВДДЭВв)
ААААААААААААААВАВАВАВААВААВВАВВАВАВВВВВВВВВВВВВВ ААААДАААААААААААААВАААВВАВВАВАВВВБВВВВВВВЕВВБВВВ
ААААААААААААААВААВААААВВААВВВВВАВАВВВБВВВВВБВВВВ аааааааалааааааавававаававававвавввввввввввввввв
б
в
ЛА.ЧАЛ V- кЬАЧ..hAhAKV- А^В^-САВАВАВАВАВВВАВАВВАВВВВ.
АААААААЬАНАН.WuVdl Л & К\ВДЬ. \ВЛВЛНАВ. \В>«АВВВАВВ < АААААВААВАВАВАВАВАВАВАВАВАВАВАВАААВАВВААВВВВВВВВ АААААААААВАВАВАВАВВААВАВАВАВАВААВААВВАВВБВВВВВВВ ААААААВАААВАВАВАВААВВААВАВАВАВАВАВВАВВАВАВВВВВВВ aaaaaaabaaarararabbararararararararraaarrbhrhrbr ААААААААВАВАВАВАВАВВАВАВААВААВАВАВВВААВАВВБВВВВВ ааааааваавааваавваававввававававввааававвавввввв аааааааававававааввавававававававвававававвввввв АААААААВАААВАВАВВААВАВАВАВАВАВААВАВАВАВВВВВВВВВВ аааааааававававааававававававававававваввввввввв АААААААААВАВАААВАВААВВАВАВАВАВАВАВАВВВАВВВВВВВВВ
BABAABABBABABAABABBABABABABAABBABABABABAABBBAAAB АВАВААААВАВВААВБВАВАВАВАВВААВАВАВВАВАВААВАВАВАВВ ВАВАВААВВАВААВАВАВАВВАВААВАВАВАВАВВААВАВАВВАВАВА ЛВАВАВВААВАВАВААВААВВАВАВВАВААВВАВВАВАВАВАВАВАВА АВВАВААВАВАВАВАВААВАВВААВВВАВАВАВАВААВВАВАВАВАВА ВАВАВАВАВВАВАВААВАВАВВАВАВААВАВАВАВАВАВААВАВААВВ АВАВВАВАААВАВАВАВААВАВАВАВАВАВВАВАВАВАВАВВАВАВАВ ААВАВВАВАВАВАВАВВАВАВАВВАВАВАВАВАВАВАВАВАВААВААВ вававававававававававвававаававававававваававава ABABABABABAAABBABABABABABBABABABABABBABABAABABAB BABABABABAAABABABABBABABABABABABABABAABABABBABAB ВАВААВААВАВВВАВАВВАВААВАВАВАВАВАВЁАВАВАВАВАВАВАВ ^ababababababbababaababababababbabababababbaba
Рис. 4.1-1. Иллюстрация состояния равновесия, а—газа А и В равного объема и при равном давлении разделены общей стенкой; б — разделяющая стенка убрана, и газы распространяются по всему объему; смешивание газов только началось; в — газы постепенно смешиваются при диффузии; в областях, удаленных от центра, они полностью перемешаются позднее, чем в центре; г — полное смешение; диффузия все еще продолжается, однако она незаметна на макроскопическом уровне.
124
Глава 4. Основы химических методов анализа
в то время как угольная кислота является плоской молекулой с новыми связями:
1Г
ov НС)
^С=О или 'С (4.1-6)
.0 но
н
Таким образом, совершенно очевидно, что CO2(aq) и Н2СО3 являются химически различными веществами, хотя аналитическими методами различить их трудно. Различие, однако, может быть установлено кинетическими исследованиями.
При исследовании реакции 4.1-За было установлено, что реакция не идет до конца и COafag) существует в растворе совместно с Н2СО3. Взаимные превращения заканчиваются, когда концентрации CO2(aq) и Н2СОз удовлетворяют определенному соотношению, не зависящему от времени при постоянной температуре:
^Й = 670 при 25°С (4.1-7)
[H2CO3J
Таким образом, менее 0,2% растворенного СО2 вступает в реакцию гидратации (4.1-За). Это можно объяснить только тем обстоятельством, что одновременно с реакцией 4.1-За протекает обратная реакция:
Н2СО3 - CO2(aq) + Н2О (4.1-36)
В соответствии с этой реакцией скорость образования СОг^утаожет быть выражена следующим образом:
dtCOitai)] ^[ИгСОэ] (4.1-8)
at
где к2 — 20с-1 при 25°С (константа скорости обратной реакция). Эта реакция идет почти до конца. Поскольку реакции 4.1-За и 4.1-36 протекают одновременно, в системе устанавливается равновесие:
CO2(aq) + Н2О й Н2СОз (4.1-Зв)
*3
При таком описании равновесия в форме уравнений 4.1-За-4.1-Зв, вещества, расположенные справа, считают продуктами реакции, а слева—реагентами. Для равновесной системы скорость образования продукта должна быть равна скорости, с которой из продуктов вновь образуются реагенты. Отсюда следует, что:
ар^Оз] = = к1 [СО2м] = Мл2СОз] (41.9)
Перегруппировка позволяет получить:
= V = = 20С-~Т = 670 п₽в 25°С (4.1-Юа)
[Н2СО3] к} К 0,03с-1 ' '
4.1. Равновесие в гомогенных системах
125
Полученное уравнение демонстрирует характерную связь между константой равновесия и константами скоростей прямой и обратной реакций. Эта взаимосвязь может быть представлена в общем виде обратимой реакцией
аА 4- ЬВ 4- - • • сС + dD 4-
(4.1-11)
где а, Ь, с, d—стехиометрические коэффициенты реагентов А, В и продуктов С, D. Скорость прямой реакции Vf определяется выражением
vt = Л1[А]“[В]Ь-•• (4.1-12)
а скорость обратной реакции иг:
«r=fe[CnD]‘‘.--
При равновесии Vf и vr равны и, следовательно, А:1[А]‘‘[В]1'-- = МС]с[В]“---
Отсюда константа равновесия К равна:
к2
(4.1-13)
(4.1-14)
(4.1-15)
Кинетический подход также демонстрирует, что величина константы равновесия изменяется прн изменении температуры. Скорость теплового движения молекул уменьшается при понижении температуры. Следовательно, реакционная способность реагирующих веществ и продуктов уменьшается и константы ki и к2 принимают меньшие значения. Эти эффекты температуры неодинаковы для прямой и обратной реакций, но зависят от различия энергий активации
этих двух процессов. Так, при 0°С —
к2 = 2,3с-1 (4.1-16)
И
fci = 0,0024с-1 (4.1-17)
что приводит к равновесному отношению
[СО2(ас|)| 1
^-^ = - = 950 при ОС
(4.1-106)
□ Константа равновесия обратимой реакции К = где к+—константа скорости прямой реакции, — константа скорости обратной реакции.
4.1.4. Стационарное состояние
Стационарное состояние — это не зависящее от времени (время-инва-риантное) состояние открытой системы, в которой имеются сбалансированные потоки массы и/или энергии через границы системы. Для того чтобы достичь стационарного состояния, вход и выход энергии и/или массы должны быть сбалансированы и оставаться постоянными. Это условие удовлетворяется, если имеется связанный со стационарной системой источник потока энергии
126
Глава 4. Основы химических методов анализа
и/или массы, обладающий большой емкостью. Компоненты системы, пересекающие ее границы, характеризуются конечным временем пребывания в системе. Стационарные свойства3 проявляемые системой, иногда можно сравнивать с определенными свойствами, которыми обладает система, находящаяся в состоянии равновесия (такими, как например, константа равновесия). В связи с этим в некоторых случаях стационарное состояние может быть и аппроксимировано равновесной моделью. Так, большинство водных систем в природе являются стационарными, а не равновесными системами. В морской воде компоненты возникают с той же скоростью, с которой и удаляются, например, по реакциям осаждения. Благодаря относительно большим временам пребывания компонентов морской воды в системе (порядка 103 лет), для описания процессов, происходящих в ней, можно использовать равновесные модели.
Многие природные циклы, такие, как углеродный и гидрологический (ответственный за ресурсы пресной воды на планете), представляют собой стационарные системы, зависящие от подвода солнечной энергии и ее отвода от земной поверхности (обычно в виде тепла). В этом смысле Земля — это энтропийный насос.
Пример стационарного состояния системы
Стационарное состояние и его квазиравновесные свойства хорошо иллюстрируются схемой радиоактивного распада, в которой исходное вещество играет роль источника энергии и массы. В природе существуют три радиоактивных семейства—урановое, ториевое и актиниевое, с исходными веществами U288, Th232 и Ц235 соответственно. Они характеризуются чрезвычайно большими константами распада, сравнимыми с возрастом Земли.
□ Радиоактивный распад — пример стационарного состояния.
Уран238, исходное вещество уранового радиоактивного семейства (с массовым числом п + 2), нестабилен и распадается, давая нестабильное дочернее вещество Th234, которое в свою очередь распадается до следующих дочерних веществ (рис. 4.1-2). Радиоактивные семейства могут включать большое число различных дочерних веществ, что иногда приводит к разным изотопам одного и того же элемента (с массовыми числами, отличающимися на 4 единицы), таким, как Th234 и Th230. Все элементы с атомным номером больше 81 (таллий) имеют природные радиоизотопы, относящиеся к трем радиоактивным семействам. Некоторые дочерние вещества характеризуются очень коротким периодом полураспада (например, At216 имеет период полураспада ti/г = 30 мкс). Период полураспада и время жизни в таких реакционных семействах связаны количественно. Время жизни tr представляет собой среднее время, в течение которого отдельная химическая или физическая единица остается в системе и ее состояние не меняется. Период полураспада представляет собой время, за которое точно половина исходного материала прореагировала с образованием другой физической или химической единицы. Поскольку
*1/2 = ^ И 7Я=| (4Л’18)
4.1. Равновесие в гомогенных системах
127
Элемент и атомный номер (число протонов'
Уран 92 238 4,49-1 и 0е лет /9-распад 2 ^2,4 ми • 10® лет
Протактиний 91 а-распад 234Ра^ 1,18 мин /3-распад о-распад
Торий 90 234 24,1 Th сут 230 8-10 Th 4 лет
Актиний 89 а-распад
Радий 88 22 1,62- Ra 103 лет
Рис. 4.1-2. Радиоактивный распад 238 как пример стационарного состояния.
где к — константа скорости реакции первого порядка (в случае радиоактивных семейств она называется константой распада Л), дальнейшие преобразования дают следующее соотношение:
т« = (Йз = 1’443^ ^-19)
Первые стадии распада уранового радиоактивного семейства представлены на рис. 4.1-2. Видно, что в стационарном состояний всегда имеется одинаковое число отдельных дочерних веществ на единицу массы исходного вещества.
4.1.5. Взаимодействия растворитель—растворенное вещество
Растворенное в воде вещество, как правило, ионизировано (по крайней мере в некоторой степени). В этом случае о растворе говорят как о растворе электролита. Однако ионы обычно не «голые», а существуют в виде продуктов специфических взаимодействий. Эти взаимодействия, в соответствии с их физической природой, могут быть классифицированы как ион-ионные, ион-дипольные или ковалентные (рис. 4.1-3). По химической природе взаимодействия можно классифицировать как ионную ассоциацию, гидратацию (или сольватацию) и комплексообразование (рис. 4.1-4).
Эти две классификации дополняют друг друга. Комплекс обычно не является продуктом чисто ковалентного взаимодействия: во многих комплексах ионный вклад может быть значительным или даже превосходить ковалентный вклад. Примером комплекса, который удерживается в основном ионными связями, является фторид алюминия AIF3.
128
Глава 4. Основы химических методов анализа
Иондипольное взаимодействие
Ковалентное взаимодействие
Растворенное вещество
Ионионное взаимодействие
Рис. 4.1-3. Основные реакции в растворе, классифицированные в соответствии с природой взаимодействия.
Гидратация или сольватация
Комплекс© образование
Растворенное вещество
Ионная ассоциация
Рис. 4.1-4. Основные реакции в растворе, классифицированные в соответствии с типом образующихся продуктов.
Между процессами комплексообразования, гидратации, сольватации иконной ассоциации не существует абсолютного различия. Это может быть продемонстрировано на примере соединений, присутствующих в водной фтороводородной кислоте. Образование иона HF2 [4-1-1] (который относится к той же категории, что и Н(Н2РО4)2 [4-1-2], Н(Юз)з [4-1-3] и Н(ОАс)2 [4.1-4]) в основном обусловлено электростатическими взаимодействиями, и эта частица может рассматриваться как ионный ассоциат. В то же время, HF2 может рассматриваться и как Комплекс, в котором ион водорода выступает как центральный атом, а фторид-ионы—как лиганды. Более того, HF2 может рассматриваться как сольват. В жидкой фтороводородной кислоте, характеризуемой самодис-социацией:
HF^H++F~ (4.1-20)
формальные ионы Н+ и F не существуют в виде «голых» частиц. Они сольватируются с образованием H2F+, HF2 или высших сольватов. Если имеет
4.1. Равновесие в гомогенных системах
129
Рис. 4.1-5- Ион-дипольные взаимодействия при гидратации аниона А- и катиона В+.
место перенос протона, то реакция может рассматриваться как протолиз, а H2F+, HFg —как продукты этого процесса.
Этот пример демонстрирует невозможность проведения четких различий между гидратом (или сольватом), комплексом и ионным ассоциатом. В этом случае использование формальной классификация (рис.4.1-3 и 4.1-4) может оказаться весьма полезным.
Ионная гидратация
□ Химические реакции в полярных растворах. Электролиты
Образование гидратной оболочки вокруг иона можно рассматривать как ион-дипольное взаимодействие, иллюстрируемое рис. 4.1-5. При этЬм энергия гидратации может быть рассчитана в предположении о кулоновском взаимодействии между точечным зарядом и диполем (сумма взаимодействий меж-, ду ионом и двумя точечными зарядами диполя). Оцененные величины для многих электролитов находятся в согласии с экспериментально измеренными теплотами гидратации- Лучше всего это проявляется для ионов, имеющих электронную конфигурацию инертного газа. Очевидно также, что благодари большему ион-дапольному взаимодействию небольшой ион будет сильнее гидратироваться; таким образом, многозарядные ионы гидратируются сильнее, чем однозарядные.
Однако эта модель не всегда дает хорошее согласие с экспериментальными данными. Например, установлено, что теплота гидратации иона серебра Ag+ существенно больше, чем ионов натрия или калия, хотя величина кристаллографического радиуса иона серебра лежит между соответствующими величинами для этих ионов щелочных металлов. Существуют и другие факты, указывающие на то, что гидратация ионов тяжелых металлов связана с процессами комплексообразования с ковалентным характером связи. В качестве примера можно отметить изменение окраски при растворении некоторых соединений переходных металлов, например, Сг(Ш) или Ni(II), и низкую скорость процесса их растворения в воде. Ковалентное взаимодействие возможно только с катионами, поскольку только кислородный атом молекулы воды может участвовать в образовании ковалентных связей (ковалентно связанный атом водорода не может образовать вторую ковалентную связь).
Гидратация анионов, схематически представленная на рис. 4.1-5, реально не наблюдается. Малые ионы, такие, как F", С1“ и т. д-, взаимодействуют с молекулами воды через водородные связи, как это продемонстрировано на
5 Аналитическая химия. Том 1
130
Глава 4. Основы химических методов анализа
Рис. 4.1-6. Гидратация аниона, основанная на водородных связях, а — небольшой высокоэлектроотрицательный анион; б — большой комплексный анион с электроотрицательными внешними группами (оксианион).
рис. 4.1-6,в. Эверет и Коулсон (4.1-5] установили, что данная структурная форма, получающаяся при образовании водородных связей (рис. 4.1-6,а), характеризуется более низкой энергией, чем в случае ион-дипольного взаимодействия. Большие анионы, такие, как перренат-ион ReO^, стабилизируют структуру путем образования дополнительных водородных связей между молекулой воды и электроотрицательным атомом аниона (рис. 4.1-6,6).
Как видно, за гидратацию ионов отвечают электростатические и ковалентные связи в комбинации с водородными. Следует рассмотреть еще один тип гидратации, который не ограничивается ионными биполярными частицами. Многие газы, особенно те, которые имеют электронную конфигурацию инертных газов, способны образовывать так называемые клатраты (например, хлор образует клатрат со стехиометрическим составом бС12-4бН2О)..При этом растворенное вещество входит в полости, образующиеся в пространственной структуре воды. Тот факт, что даже благородные газы способны образовывать клатраты, свидетельствует об отсутствии химического взаимодействия между ними и растворителем- Очевидно, что эффективно поглощаться могут только те вещества, диаметр молекул которых меньше размера полости в пространственной структуре растворителя.
4.1.6. Теория электролитической диссоциации Аррениуса
Шведский химик Сванте А. Аррениус (1859-1927) описал диссоциацию электролитов следующим образом. Кислота, основание или соль, растворенные в воде, диссоциируют на ионы, н устанавливается равновесие:
МтАо тМ*'++аА/’-
(4.1-21)
Равновесие описывается соответствующей константой диссоциации Кдисс:
“дисс — - -
(4.1-22)
Ионы М"+ и А^_ могут в растворе двигаться независимо. Присутствие ионов делает раствор электропроводным.
Если имеется полная диссоциация, равновесие 4.1-21 сдвигается вправо и основными частицами в растворе являются ионы. Каждый моль соли дает определенное количество молей ионов, в соответствии со стехиометрическим составом. Например, 1 моль {NaCl} дает 1 моль Na+ и 1 моль С1~, в то время
4.1. Равновесие в гомогенных системах
131
как 1 моль {NaaSCU} дает 2 моля Na+ и 1 моль SOJ-, в результате получаются соответственно 2 М и 3 М ионные растворы. (Необходимо отметить, что заключение формулы вещества в фигурные скобки {} означает, что данное вещество находится в твердом состоянии, а не рассматривается как мономерная частица в растворе; фактически, {АВ} означает А», В со.)
Электропроводность полностью диссоциированного раствора электролита прямо пропорциональна концентрации ионов и, следовательно, концентрации электролита. Это объясняется тем, что ионы являются носителями тока в растворе. Поэтому, если концентрация ионов удваивается или утраивается, проводимость изменяется.
При неполной диссоциации основной частицей растворенного вещества является молекулярное соединение МтАа. Можно рассчитать равновесную концентрацию ионов [М1'+] и [А^“] из константы равновесия КдИСС. Таким образом:
= Кдж^А.] (4.1-23)
но, поскольку [М*'+]/т — [А^Ч/а, можно записать:
(-)“[М-+Г+“ = Кдвсс|МтАо] (4.1-24)
\7П/
Аррениус установил, что электролиты полностью диссоциированы только в очень разбавленных растворах. В связи с этим им было введено новое понятие — степень диссоциации а. Эта величина определяется как доля Диссоциированных молекул раствора. Следовательно, а принимает значения от нуля (неэлектролиты) до 1,0 (сильные электролиты).
В случае полной диссоциации вещество MWAO диссоциирует на"т нонов М*'+ н а ионов АР~. В этом процессе каждая молекула растворенного вещества дает д ионов, где:
д = т + а (4.1-25)
Однако, если диссоциация неполная, то п молекул MmAn дают п(1 — а) недиссоциированных частиц и пар, ионов. Тогда общее число частиц:
п(1 — а 4- ар) (4.1-26)
Если число частиц, образованных из одной молекулы растворенного вещества, обозначить как г, где i — (1 — а 4- ар), то получается следующее соотношение:
Величина а может быть определена экспериментально, поскольку значение i получают из измерений осмотического давления, повышения температуры кипения или понижения температуры плавления, а величина р может быть рассчитана из стехиометрии растворенного вещества.
5*
m
Глава 4. Основы химических методов анализа
□ Сильные и слабые электролиты: Аррениус
На основании величин а Аррениус установил следующие различия между сильными и слабыми электролитами:
— Для слабых электролитов величины а существенно зависят от концентрации электролита. При высоких значениях концентрации а стремнтбя к нулю. В разбавленных растворах эта величина меньше единицы. К этому классу относится большинство органических кислот и оснований, а также соединений многих тяжелых металлов.
- Для сильных электролитов величина а практически не зависит от концентрации электролита и всегда близка к 1,0. Сильные электролиты обеспечивают высокую электропроводность растворов. К этому классу относятся многие соли, а также сильные минеральные кислоты и основания.
Необходимо иметь в виду, что эта классификация относительная и применима к водным средам. Электролит, сильный в водном растворе, может оказаться слабым в органическом растворителе с небольшим дипольным моментом или низкой диэлектрической проницаемостью.
4.1.7. Ион-ион ные взаимодействия и ионная ассоциация
В растворе электролита взаимодействия между ионами осуществляются через электростатическое притяжение и отталкивание (кулоновские силы) и, следовательно, зависят как от заряда ионов, так и от среднего расстояния между взаимодействующими ионами.
Кулоновские силы определяются уравнением —
(4.1-28)
Dr2
где е, и е^ —заряды взаимодействующих ионов i и у, D — диэлектрическая проницаемость среды, г —расстояние между ионами.
Кулоновские силы притяжения являются причиной образования ионных пар и высших ассоциатов. Однако в водных растворах эффекты образования ионных пар становятся значительными только при достаточно высоких концентрациях иоиов (поскольку при этом уменьшается среднее расстояние между ионами). В органических растворителях, диэлектрическая проницаемость которых значительно, меньше, процесс образования ионных пар становится существенным и при относительно низких концентрациях. Образование ионных пар часто наблюдается при жидкостной экстракции, что используют для увеличения переноса вещества в органическую фазу.
4.1.0. Ионная активность
Как было показано выше, растворенное вещество может существовать в растворе в нескольким формах. Рассмотрим 0,5 М раствор НС1, в котором растворено несколько граммов соли железа(Ш). В этот раствор добавлен некоторый реагент Х3~, который осаждает железо в виде FeX. Поскольку в исходном
4.1. Равновесие в гомогенных системах
133
растворе присутствуют разные формы железа(Ш), процесс осаждения можно записать в виде следующей схемы:
Fe3++X3-
Fe(H2O)3+ + Х3-
FeCl2++X3-Fe(H2O)5CI2+ + X3-
{FeX} {FeX}+6H2O {FeX} + Cl-
{FeX} + Cl" + 5H2O
(4.1-29а) (4.1-296) (4.1-29в) (4.1-29г)
Эти реакции не единственные, которые описывают суммарный процесс осаждения. Возникает вполне резонный вопрос о том, какие из приведенных выше реакций протекают в реальной системе. С точки зрения только термодинамики однозначно ответить на этот вопрос нельзя, поскольку необходимо одновременно рассматривать и кинетические ограничения соответствующих реакций. При установлении равновесия не имеет значения выбор конкретного пути реакции, так как для всех этих реакций достигается одно и то же равновесное состояние.
Учитывая это обстоятельство, можно прийти к выводу о том, что рассмотрение всего комплекса возможных реакций, в том числе и с участием промежуточных частиц, не является необходимым. После установления равновесия достаточно знать исходный и конечный состав системы. Однако при этом следует быть осторожным при обращении с таким термодинамическим понятием как концентрация, широко используемым в выражениях закона действующих масс. Например, для процесса (4.1-29г) можно записать:
[С1-][Н2О15 [Fe(H2O)sCI2+][X3~]
Как правило, точные концентрации частиц, представленные в выражении, ие известны. Более того, возникает ряд вопросов:
(4.1-30)
— гидратируется ли хлорид-ион и следует ли учитывать концентрацию гидрата;
— известно ли для него число гидратации;
— как рассчитать концентрацию Fe(H2O)sCI2+;
— чему равна концентрация воды [НаО]?
Для решения подобных проблем и для упрощения расчетов вводят понятие активности, которая заменяет концентрацию, но связана с ней посредством так называемого коэффициента активности.
Понятие активности очень удобно, хотя может создавать некоторые проблемы для неопытного химика. В силу того, что существуют различные концентрационные шкалы (например, молярная концентрация с, моляльная концентрация т и мольная доля п), существуют и различные шкалы коэффициентов активности. Однако в любой системе активность не должна зависеть от выбора концентрационной шкалы. Следовательно, для различных коэффициентов активности необходимо использовать и различные символы:
• fi — коэффициент активности, относящийся к шкале мольных долей;
• 7f — коэффициент активности, относящийся к моляльной концентрационной шкале;
• yi — коэффициент активности, относящийся к молярной концентрационной шкале.
134
Глава 4. Основы химических методов анализа
Коэффициент активности необходимо определять по отношению к стандартному состоянию, для которого условно полагают, что этот коэффициент равен 1. Наиболее удобным состоянием, очевидно, является раствор, в котором нет процессов гидратации, сольватации, комплексообразования, ионной ассоциации. Можно представить, что в растворе присутствует только «голое» растворенное вещество. Однако такое стандартное состояние, конечно же, является гипотетическим.
Полностью устранить взаимодействия между растворителем и растворенным веществом невозможно, но можно попытаться исключить процессы комплексообразования (поддерживая концентрацию любого потенциального лиганда равной нулю) и ионной ассоциации (удаляя ноны противоположного заряда на бесконечное расстояние друг от друга). Подобная ситуация существует в бесконечно разбавленных растворах н приближается к ней в очень разбавленных. Хотя бесконечно разбавленный раствор, с физической точки зрения, и является идеальным случаем, реализовать его практически очень трудно.
Исходя из практических соображений, Силлен (Lars G.Sillen) предложил принять стандартное состояние в постоянной ионной среде. Это система, в которой взаимодействие между средой и растворенным веществом (сольватация и комплексообразование), хотя и не полностью отсутствует, по крайней мере является постоянным. Применительно к системе уравнений реакций, рассмотренной выше, это означает, что отношения концентраций «голого» иона железа (4.1-29а), гексааква-иона железа (4.1-296), а также железо-хлоридных комплексов (4.1-29в и 4.1-29г) остаются постоянными, так что константы равновесия не изменяются с концентрацией, и таким образом, выполняется идея постоянной ионной среды. Однако значения констант равновесия, полученные для этой системы, строго говоря, верны только для этой среды.—
Поскольку отношения активностей являются гораздо более простыми в постоянной ионной среде, можно рассмотреть их до изложения теории Дебая— Хюккеля (1923), но необходимо отметить, что они получили полное признание только после того, как были полностью поняты ограничения приближения Дебая—Хюккеля.
Активность в постоянной ионной среде
Льюисом и Рэндаллом было установлено, что величина константы равновесия (полученная при подстановке концентраций ионов в закон действующих масс) для растворов с постоянным составом ионной среды не изменяется.
□ Зависимость концентраций от активности: мечта и реальность. Введение к дальнейшему изучению теории Дебая—Хюккеля
Выше указывалось, что рост концентрации электролита приводит к увеличению степени ионной ассоциации и понижению «активной концентрации» свободных иоиов в растворе. Это означает, что более концентрированные растворы ведут себя менее идеально, чем разбавленные. Однако, как отметил Льюис, та же степень неидеальиости наблюдается и при постоянстве ионной силы. Он определил ионную силу р в соответствии с уравнением
А‘=|^(с<г.?) (4.1-31)
4.1. Равновесие в гомогенных системах
135
Таблица 4.1-1. Ионная сила для ряда 1,0 М растворов
Электролит Формула Thn Ci 22 Ж = 5 £(<**?) i
NaCl 1:1 [Na+] = 1,0 1,0 1,0
[СГ] = 1,0 1,0
СаСЬ 2:1 [Ca2+] = 1,0 4,0 3,0
[СГ] = 2,0 1,0
Na2&04 1:2 [Na+] = 2,0 l,o 3,0
[SOJ-] = 1,0 4,0
Mgso4 2:2 [Mg2+] = 1,0 4,0 4,0
[SO2*] = 1,0 4,0
LaCl3 3:1 [La3+] = 1,0 9,0 6,0
[C17J = 3,o 1,0
Na3PO4 1:3 [Na+j = 3,0 1,0 6,0
poj-] = i,o 9,0
K4Fe(CN)fi 1:4 [K+] = 4,0 1,0 10,0
[Fe(CN)M = 1,0 16,0
Ре3(РО4)г 2:3 [Fe2+] = 3,0 4,0 15,0
[PO?-] = 2,0 9,0
Mg2[Fe(CN)e] 2:4 [Mg2+j = 2,0 4,0 12,0
[Fe(CN)g-] = 1,0 16,0
LaaPO4 3:3 [Las+] = 1,0 9,0 9,0
[PO]3’ =1,0 9,0
Ионные взаимодействия в растворе, основанные на электростатических эффектах, в основном определяются не специфическими свойствами взаимодействующих ионов, а их числом (концентрация с^) и зарядом (формальный заряд на ионе z$). Льюис установил, что чем выше заряд иона, тем больше выражена неидеальность раствора. Так, если для раствора однозарядных ионов данной концентрации наблюдается определенная величина отдельного «ион-независимого» свойства, тот же эффект будет наблюдаться в растворе многозарядных ионов при существенно меньшей концентрации. Поскольку кулоновские силы зависят от произведения зарядов ионов, в рамках обсуждаемой модели влияние среды также должно квадратично зависеть от заряда.
В уравнении 4.1-31 Ci означает молярную концентрацию иона i, a Zj—заряд этого иона. Таким образом, двухзарядный ион оказывает четырехкратно, а трехзарядный—девятикратно более сильное влияние на ионную силу, чем однозарядный ион. При расчете ионной силы предполагается полная диссоциация электролита. В табл. 4.1-1 представлен расчет ионной силы ряда электролитов.
Льюис установил, что коэффициенты активности ионов сохраняются постоянными в растворах с одинаковой ионной силой, но имеющих разный состав (например, при замене иона Na+ на Н+). Это выполняется в тех случаях, когда ионы ие вступают в реакцию комплексообразования с фоновым электролитом. Следовательно, имеется возможность изменять концентрации ионов без изменения их коэффициентов активности. Как следует из экспериментальных
136
Глава 4. Основы химических методов анализа
данных, коэффициент активности остается близким к единице при низкой концентрации реагирующего иона (не превышает 10% от концентрации инертного электролита). На основе этого подхода можно изменять pH среды с постоянной ионной силой без изменения коэффициентов активности изучаемых ионов. В таком случае активность ряда ионов (включая и ион водорода) может быть определена потенциометрическими методами (например, с помощью стеклянного или металлического электродов).
В качестве инертных электролитов наиболее часто используются перхлораты и нитраты. Обычно они имеют исключительно слабую тенденцию вступать в реакции комплексообразования. Их соли, имеющие в качестве катионов ионы щелочных металлов, хорошо растворимы в воде (кроме КСЮ4). На основе этих солей могут быть приготовлены растворы с ионной силой > 3,0 моль/л.
Активность при бесконечном разбавлении (теория Дебая—Хюккеля)
Петер Дебай (1884-1966) и Эрих Хюккель (1896-1980) рассчитали потенциальную энергию взаимного притяжения ионов в растворе. Расчет основывался на модели, учитывающей ионную ассоциацию в рамках модели Бьеррума и имеющей определенные ограничения:
— Силы межионного взаимодействия являются исключительно электростатическими; всеми другими взаимодействиями пренебрегают.
— В расчетах используется диэлектрическая проницаемость чистого растворителя; все структурные изменения из-за присутствия растворенных электролитов пренебрежимо малы (это не относится к концентрированным растворам, в которых диэлектрическая проницаемость чистого растворителя уменьшается из-за диэлектрического насыщения).
-'-'Все AOR& рассматриваются как бесструктурные точечные неполяризуемые заряды (строго говоря, это не совсем верно, поскольку-при приближении ионов друг к другу возникает поляризация).
Электрический потенциал, возникающий в результате межионного притяжения, является небольшим по сравнению с кинетической энергией теплового движения ионов (это верно лишь, если среднее межионное расстояние больше, чём бьеррумовская критическая величина с).
— Сильные электролиты рассматриваются как полностью диссоциированные при всех концентрациях (в очень концентрированных растворах это не так).
Поскольку идеальное поведение наблюдается только при бесконечном разбавлении, стандартным состоянием является бесконечно разбавленная система, в которой все коэффициенты активности равны единице. При любой конечной концентрация коэффициенты активности, рассчитанные по теории Дебая—Хюккеля, меньше единицы.
Для расчета среднего коэффициента активности Дебаем и Хюккелем было предложено следующее уравнение:
lg!/± = -Az+z-yfil
(4.1-32)
4-.1. Равновесие в гомогенных системах
137
где z+ и — зарады катиона и аниона, д—ионная сила раствора, а А —константа, коэффициент Дебая—Хюккеля, полученный из расчета Дебая—Хюк-келя:
А = 1,842 10'/(ОТ372)
где D—диэлектрическая проницаемость чистого растворителя, 7—абсолютная температура. Поскольку D в воде равно 78,54, в чистой воде при 25°С А = 0,5091.
Уравнение (4.1-32) известно как предельное уравнение Дебая—Хюккеля. В зависимости от рассматриваемых зарядов уравнение выполняется для концентраций электролита не более 10_3~10-2 моль/л.
Позднее Дебай и Хюккель предложили расширенное уравнение Дебая— Хюккеля. Оно расширяет концентрационный диапазон применимости уравнения 4.1-33 до концентраций КГМО"1 моль/л:
где а—ионный размерный параметр (ионный радиус), выраженный в А, В— второй коэффициент Дебая—Хюккеля, рассчитанный из соотношения
50,29 (DT)1/2
(4.1-34)
В равно 0,32865 в чистой воде при 25°С-
Можно видеть, что расширенное уравнение Дебая—Хюккеля ограничивается предельной формой, когда 1 Ва^/р. Для большинства ионов а ж ЗА, так что произведение Ва близко к 1,0. Следовательно, предельное уравнение заменяет его расширенную форму при д < 0,01 моль/л.
Выло предпринято много попыток расширить концентрационный диапазон при расчетах активности. Однако рассмотрение других уравнений, таких, как например, уравнение Гюнтельберга, находятся вне рамок данной книги.
Отклонение от идеальности для систем, в которых экспериментально измеренные в разбавленных растворах коэффициенты активности уменьшаются с ростом концентрации электролита, обусловлено ион-ионными взаимодействиями (образование «ионных облаков»). Иногда эти взаимодействия называют дальнодействующими или просто дальними. Существуют короткодействующие взаимодействия (с малым радиусом действия), причем наиболее важным из них является гидратация. В разбавленных системах влияние гидратации постоянно, поскольку сама активность воды не изменяется. Следовательно, коэффициенты активности в разбавленных системах не зависят от гидратации ионов, что учтено выбором стандартного состояния. Таким образом, необходимо понимать, что понятие коэффициента активности относится к гидратированному нону. При возрастании концентрации электролита активность растворителя должна понижаться, влияя тем самым на положение равновесия сольватации. Равновесие сольватации при этом сдвигается в сторону менее сольватированных или «голых» иоиов. Следовательно, коэффициенты активности «голых» ионов возрастают. Это наиболее справедливо для небольших ионов,
138
Глава 4. Основы химических
таких, как Н+, Li+ и Ве2+. Например, в минеральных кислотах коэффициенты активности могут принимать значения вплоть до 103 или даже больше в концентрированных растворах. Для этих систем зависимость коэффициентов активности от концентрации выглядит следующим образом. Коэффициенты активности отклоняются от 1,0 (значение для бесконечно разбавленных растворов) при концентрациях вблизи 10“4-10-3моль/л. Обычно минимум наблюдается для сильно гидратированных ионов при концентрации 0,1-0,5 моль/л. Затем коэффициенты активности вновь возрастают и принимают значения около 1,0 при концентрациях между 1,0 и 2,0моль/л.
Эти эффекты не могут быть объяснены в рамках модели Дебая—Хюккеля, поскольку специфическое взаимодействие ион-растворитель, характеризующее гидратацию ионов, отличается от ион-ионного взаимодействия. Однако с целью расширения концентрационного диапазона был предложен ряд поправок и эмпирических модификаций уравнения Дебая—Хюккеля. В общем случае получено следующее уравнение:
= (4Л-35)
В этом уравнении С — эмпирический коэффициент, определяемый экспериментально.
Робинсон и Стокс предложили еще одно уравнение:
= (4Л-36)
Это выражение является наиболее точным из имеющихся и позволяет рассчитать коэффициенты активности в растворах по крайней мере до концентрации электролитов 2-4моль/л. Однако для расчета необходимы числа гидратации ионов Н, которые почти всегда неизвестны.
Существует другая проблема, связанная с использованием коэффициентов активности. Невозможно экспериментально определить коэффициент активности отдельного иона. Это обусловлено тем, что каждый ион всегда сосуществует с другим, так называемым противоионом. Коэффициент активности индивидуального иона, следовательно, является гипотетической величиной. Экспериментально находят средний коэффициент активности у±, для которого выполняется следующее соотношение:
0.0, = (4.1-37)
где di и aj— активности отдельных ионов, с^ и cj — концентрации ионов (в молярной шкале), у, и у$ — гипотетические коэффициенты активности катиона г и аниона у; yj-— средний коэффициент активности. Таким образом, можно заключить, что:
Й = 7? = (ад) (4.1-38)
Поскольку концентрация электролита рассчитывается из общего количества исходно растворенного вещества, а не из реальных ионных концентраций, активность электролита должна определяться с учетом этого обстоятельства.
4.1. Равновесие в гомогенных системах
139
Например, 1,0 М раствор НС1 дает 1 моль/л Н+ и 1 моль/л CI . В этом случае коэффициент активности определяется следующим образом:
Vi = 2521 = (Va+Va-)1'2 (4.1-39)
CHG1
При этих условиях найденный коэффициент активности равен 0,812. Следовательно:
внс1 = (oh+^ci-)1^2 = 0,812
где сна = 1,0.
Этот расчет может быть проиллюстрирован еще одним примером. Рассмотрим раствор, приготовленный из 0,2 молей HNO3 и 0,3 молей NaCl. Объем раствора 1 л. Он содержит:
[Н+] = 0,2 моль/л
[СГ] = 0,3 моль/л
Следовательно, можно рассчитать формальную концентрацию HCI:
ода = (сн+с01- )1/2 = (0,2 - 0,3)1/2 = х/адё = 0,245 моль/л
в то время как ионная сила д равна 0,5 моль/л.
Литература
[4.1-1] Broene Н. Н., De Vries Т. J. Amer. Chem. Soc. 69 (1947) 1644.
[4.1-2] Selvaratnam M., Spiro M. Trans. Faraday Soc. 61 (1965) 360.
[4.1-3] Pethybridge A. D., Prue J. E. Trans. Faraday Soc. 63 (1967) 2019.
[4.1-4] Martin D. L., Rossotti F. J. C. Proc. Chem. Soc. 1959, 60. —
[4.1-5] Everett D. H., Coulson C. A. Trans.Faraday Soc. 36 (1940) 633.
Вопросы и задачи
1. Рассчитайте ионную силу модельного образца морской воды, содержащего следующие компоненты в литре раствора:
0,478 моль Na+ 0,550 моль СГ
0,064 моль Mg2+ 0.028 моль SO2~
Ответ'. Ионная сила:
д = 1 (0,478 + 4 - 0,064 + 0,550 + 4 0,028) = 0,698 моль/л.
2. Какой фундаментальный закон описывает химическое равновесие?
3. В каких случаях можно наблюдать гетерогенное, а в каких—гомогенное равновесие?
4. При каких условиях слабый электролит может стать сильным?
5. Концентрация является результатом многих аналитических измерений. Между тем в законах физической химии фигурирует величина активности. При каких экспериментальных условиях можно устранить это несоответствие и перейти от активностей к концентрациям?
140
Глава 4. Основы химических методов анализа
4.2. КИСЛОТНО-ОСНОВНОЕ РАВНОВЕСИЕ
Цели изучения
• Ознакомиться с различными концепциями кислот и оснований, с реакциями переноса протонов.
• Ознакомиться с концепцией pH, понять значение pH индикаторов и буферов.
• Научиться рассчитывать концентрации кислот и оснований, а также значение pH растворов и буферов по данным концентрациям.
4.2.1. Введение
Образная теория кислот и оснований была предложена аптекарем из Парижа Николя Лемери (1645-1715). В своем «Курсе химии» (1675) он попытался объяснить физические и химические свойства веществ на языке их формы и структуры. В этом отношении его попытка выглядит авангардистской. Согласно представлениям Лемери кислоты на своей поверхности имеют острые шипы, вызывающие на коже колющие ощущения. Основания, названные им щелочами, состоят из пористых тел. «Шипы» кислот проникают в «поры», при этом они ломаются или притупляются, и кислоты превращаются в нейтральные соли.
По мере открытия и изучения химических элементов (что стало возможным только после развития атомной и молекулярной теории) концепция кислот и оснований изменяется, и делаются попытки приписать кислотные свойства отдельным элементам или молекулярным группам. Антуан Л. Лавуазье (1743-1794) назвал кислотами комбинация радикалов и кислорода, а основаниями — комбинации металлов и кислорода. Его идеи получили признание несмотря на то, что уже давно была известна бескислородная «муриевая» (соляная, хлороводородная) кислота. Однако в то время полагали, что хлор—это оксид, а не элемент. В 1809 г. Жозеф Л. Гей-Люссак (1778-1850) в содружестве с Луи Ж. Тенаром (1777-1857) обнаружили, что хлориды, называвшиеся тогда му-риатами, кислорода не содержат. Однако оба они были настолько сильными приверженцами идей Лавуазье, что посчитали свои данные скорее ошибочными, чем пригодными для создания новой концепция. Такую концепцию предложил английский химик Хэмфри Дэви (1778-1829), доказавший, что хлор — это элемент и что хлороводородная кислота не содержит кислорода. Он считал, что кислотные свойства веществ обусловлены наличием водорода (1810), и эта теория вскоре стала общепринятой.
Современный подход к решению проблемы стал возможным только после развития Аррениусом теории электролитической диссоциации, включающей концепцию сильных электролитов (солей, кислот и оснований). Он высказал свои идеи Шведской академии наук в 1883 г., а в 1887 г. опубликовал свою теорию. Согласно Аррениусу, при диссоциации кислоты образуется ион водорода и анион, а при диссоциации основания—гидроксид-иои и соответствующий катион. Теория Аррениуса смогла объяснить и различия в силе кислот, связанные с различиями в степени их диссоциации.
4.2. Кислотноосновное равновесие
141
В дальнейшем трудности возникли при попытке объяснения явлений кислотности и основности в неводных растворах. Так, раствор- аммиака в эфире проявляет основные свойства, хотя гидроксид-иона в этом растворе нет. Артур Лэпуорт (1872-1941), профессор университета в Манчестере, преподававший органическую, а также физическую и неорганическую химию, рассматривал кислоты как доноры ионов водорода. Одним из его учеников был Томас М. Лоури (1874-1936). Независимо друг от друга он и датский профессор Йоханнес Н. Брёнстед (1879-1947) на основе вдей Лэпуорта создали теорию кислот и оснований, называемую теперь теорией Брёнстеда—Лоури (1923).
4.2.2. Современная концепция кислот и оснований.
Теория Брёнстеда—Лоури
□ Наиболее научно обоснованная теория кислот и оснований.
Согласно теории Врёнстеда—Лоури, кислота—донор протонов, основание— акцептор протонов. Каждой кислоте соответствует сопряженное основание и каждому основанию—сопряженная кислота
Кислота Основание Ч-Протон (4-2-1)
Поэтому основание, сопряженное с сильной кислотой, является слабым, а основание, сопряженное со слабой кислотой, — сильным. Вместе они образуют сопряженную кислотно-основную пару, и кислота без сопряженного основания— понятие бессмысленное. В водных растворах протон Н+, имеющий чрезвычайно маленький ионный радиус, в свободном виде не существует, а в результате взаимодействия с водой превращается в Н3О+ и более высокогидратирован-ные ионы. Кислотно-основное равновесие, таким образом, не является просто процессом диссоциации, а представляет собой протолиз, т. е. реакцию переноса протона, в которой участвуют две сопряженные кислотно-основные пары:
НХ + Н2О <=* Н3О+ + Х“ (4.2-2)
Эту реакцию формально можно представить в виде двух полуреакций
НХ й Н+ + X-Кислота 1 Протон Основание 1
И
Н+ + Н2О Н3О+
Протон Основание 2 Кислота 2
Суммарное равновесие описывается константой
[Н,О+](Х-]
[НХ][Н2О]
(4.2-2а)
(4.2-26)
(4.2-3)
Концентрация мономерной формы воды неизвестна, ио в разбавленных водных растворах она. по крайней мере, постоянна, и согласно определению стандартного состояния системы ее можно включить в значение константы. В окончательном виде выражение для константы равновесия (рассчитанной для рас
142
Глава 4. Основы химических методов анализа
твора с постоянной ионной силой или для бесконечно разбавленного раствора) можно записать так:
КЧНО1-/Г [НзО+][Х‘]
— Ла —-----[н5с]-- (4.2-За)
Здесь КА —константа кислотной диссоциации (или константа кислотности). Строго говоря, Кл характеризует не только силу кислоты НХ, но и силу основания Н2О. Вот почему константы диссоциации одной н той же кислоты в разных растворителях различаются. Следует иметь в виду, что фактическая концентрация НзО+ неизвестна, а величина (НзО+] в действительности представляет собой сумму концентраций всех гидратироватаых форм протона. Это означает, что с тем же успехом вместо [НзО+] можно писать [Н+]. Для полностью продиссоциировавшей одноосновной кислоты равновесная концентрация Н+ равна ее общей концентрации.
В отличие от теории Аррениуса и более ранних теорий кислот и оснований, концепция Брёнстеда—Лоури применима и для таких видов кислотноосновного равновесия, которые даже формально нельзя представить в виде процессов диссоциации. Известны соединения, не содержащие атомов водорода или гидроксидных групп, но дающие кислые или щелочные растворы. Это объясняется тем, что в результате взаимодействия с растворителем (водой) они образуют продукты с кислотно-основными свойствами. Приведем несколько примеров:
БОз +2Н2О Н3О+ + HSO4 (4.2-4)
Триоксид серы Гидросульфат-иои
NH3 +Н2О^ NH4 +ОН- ~ (4.2-5)
Аммиак Ион аммония
{LiH} +Н2О Li+ + О1Г + Н2 (4.2-6)
Гидрид лития
{NaNH2} +Н2О <=? Na+ + NH3 +ОН“ (4.2-7)
Амид натрия Аммиак
В дальнейшем мы будем использовать концепцию Брёнстеда—Лоури, хотя есть и другие теории кислот и оснований. В основе концепции Брёнстеда—Лоури лежит реакция переноса протона, в которой участвуют две сопряженные кислотно-основные пары. Константа кислотности К& поэтому не является однозначной характеристикой силы кислоты. Вследствие нивелирующего эффекта воды относительную силу сильных кислот в водных растворах охарактеризовать нельзя. Для решения этой проблемы были предложены так называемые функции кислотности. Наиболее известной из них является функция кислотности Гаммета. Однако прн обсуждении реакций, в которых фигурирует вода (как фаза), мы ограничимся концепцией Брёнстеда—Лоури.
Для полноты картины следует упомянуть концепцию Гилберта Н. Льюиса (1875-1946), предложенную им в 1916 г. Согласно Льюису, кислота является акцептором электронной пары, а основание—донором электронной пары. Следовательно, к кислотно-основным относятся не только реакции переноса протона. В рамках этой теории протон не играет никакой особой роли, а кислотно
4.2. Кислотно-основное равновесие
143
основные , реакции рассматриваются с позиций координационной химии. Кислотами Льюиса являются такие соединения, как BF3, А1С1з, РеВгз н т. п., а NH3, все амины и многие кислородсодержащие соединения (эфиры, спирты, кетоны) —это основания Льюиса.
Протоноакцепторные и протонодонорные свойства в водных растворах
В водных растворах, содержащих донор протонов (кислоту), образуется ион гидроксония (или гидрония) НзО+, и в этом процессе вода и сопряженное с кислотой основание конкурируют за обладание протоном согласно уравнению
НХ + Н2О Н3О+ + Х“ (4.2-2)
Кислота 1 Основание 2 Кислота 2 Основание 1
В зависимости от относительной силы кислот (или оснований), участвующих в этом процессе, равновесие слабо смещено в ту или другую сторону, либо все участники процесса сосуществуют в соизмеримых концентрациях. Вода является относительно слабой кислотой и для сильных доноров протонов, таких, как минеральные кислоты (НСЮ4, НС1, НВг и т. д.) и некоторые металлокомплексные кислоты (HMnO4, HFeCU и т. п.), равновесие реакции практически нацело смещено вправо. Эти сильные кислоты диссоциируют практически полностью. Анионы, подобные FeCl^ , являются настолько слабыми основаниями, что их я не рассматривают как основания. Кроме того, FeCl^ вряд ли существует в водных растворах, поскольку для этого необходима очень высокая концентрация СГ"-ионов, но в органических растворителях (метилнзобу-тилкетон, эфир и т. п.) HFeCl4 существует в виде сольватированной ионной пары.
Молекулярные газы, как и электролиты, в воде могут образовывать кислые растворы. Например, газообразный НС1—соединение молекулярное, но, попав в воду, он гидратируется с выделением значительного количества тепла (сравнимого с выделяющимся при взаимодействии сильной кислоты с основанием средней силы, таким, как NH3). Это еще раз подтверждает тот факт, что вода является основанием. Аналогичные реакции протекают при растворении в воде SO3. Сначала газообразный SO3 гидратируется, затем образовавшаяся кислота диссоциирует по стадиям:
SO3 + Н2О H2SO4 Первая стадия: гидратация (4.2-8)
H2SO4 + НгО НзО+ + HSO4 Вторая стадия: диссоциация (4.2-9)
HSO^ 4- Н2О <=± Н3О+ + SO|~ Третья стадия: диссоциация (4.2-10)
Все эти равновесия смещены вправо н правильнее было бы написать:
SO3 + Н2О —> H2SO4 Полная гидратация (4.2-8)
H2SO4 + НгО —» Н3О+ + HSO4 Полная диссоциация (4.2-9)
HSO4 + Н2О —* HgO+ + SOj" Почти полная диссоциация (4.2-10)
144
Глава 4. Основы химических методов анализа
Но так бывает не всегда. Известны оксиды, для-которых равновесия гидратации и диссоциации смещены лишь частично:
SO2 + Н2О *— H2SO3 Неполная гидратация (4.2-11)
H2SO3 + Н2О -> Н3О+ + HSO3 Почти полная диссоциация (4.2-12)
HSOg + Н2О <— НзО+ + SOf” Неполная диссоциация (4.2-13)
. Оксид углерода гидратируется, но преобладающей формой в водных растворах является CO2(aq), а не Н2СОз (см. разд. 4.1). Угольная кислота Н2СО3 — кислота средней силы, но поскольку она сосуществует в равновесии не только с диссоциированной формой НСО3, но и с гидратированным диоксидом углерода CO2(aq) (или СО2- Н20), то фактически это кислота слабая, т. е. диссоциирует не полностью.
□ Из-за неполной диссоциации Н2СО3 ведет себя как слабая кислота. При разбавлении диссоциация слабых электролитов увеличивается (закон разбавления Оствальда).
СО2 + Н2О->СО2-Н2О СО2-Н2О<-Н2СО3 Н2СО3 + Н2О 4- Н3О+ + НСОз НСОз + Н2О 4- н3о+ + СО1'
Полная гидратация (4.2-14)
Молекулярная перегруппировка (4.2-15)
Диссоциация (неполная) (4.2-16)
Неполная диссоциация (4.2-17)
Константа равновесия второй реакции порядка 1,5 10^, поэтому [СО2-Н2О]/[Н2СОз] = 670 н константа кислотности
_ [Н3ОЧ[НСОз] _ 6>37
в [Н2С03]общ
(4.2-18)
Символом [Н2С0з]общ обозначена суммарная концентрация [СО2-Н2О]+ |Н2СОз]. Фактически константа кислотности Н2СОз должна быть почти в 670 раз больше. Следовательно, эта кислота достаточно сильная, сравнимая с HSO^ н гораздо более сильная, чем уксусная.
Есть оксиды неметаллов, не растворяющиеся в воде н не взаимодействующие с ней, поэтому они не проявляют ни кислотных, ни основных свойств. Примерами могут служить NO2 и NO, используемые из-за инертности к воде в аэрозольных консервантах в пищевой промышленности.
Некоторые жидкие и твердые молекулярные вещества реагируют с водой с образованием кислых растворов. Серная кислота H2SO4 существует в молекулярной форме. Она имеет низкую электрическую проводимость н становится сильным электролитом только прн добавлении воды. Поскольку при этом выделяется значительное количество тепла, можно полагать, что протекает кислотно-основная реакция, в которой вода является основанием. Известно также, что при очень высоких концентрациях хлорная кислота существует в
4.2. Кислотно-основное равновесие
145
виде бимолекулярных частиц. Наиболее вероятно сосуществование следующих равновесий
Н3О+ + С1О4 й НзО+С1Од (4.2-19)
Ионная пара
НзО+СЮд + СЮ4 Н3О(С1О4)2 , или Н(С1О4)2 (4.2-20)
Тройной ионный ассоциат
НзО(СЮ4)2 + Н3О+ (H3O+C1OJ)2, или H3O+H(C1O4)J (4.2-21) Бимолекулярные Ассоциат из
частицы четырех ионов
В этих уравнениях НзО+С1О^ и НзО(С1О<1)2 [или Н(СЮ^д ] соответственно означают ионную пару и обе формы тройного ионного ассоциата. В случае хлорной кислоты ионная пара н тройной ионный ассоциат теряют гидратную воду гораздо легче, чем свободный ион гидроксония. Тройные ионные ассоциаты обычно существуют в средах с низкой диэлектрической проницаемостью, т. е. в растворителях, применяемых в экстракции.
Слабые кислоты и кислоты средней силы, такие, как уксусная, диссоциируют лишь на доли процента
СНзСООН + Н2О Н3О+ + СНзСОО- (4.2-22)
Для галогенидов неметаллов в водных растворах характерны реакции обмена с образованием гидратированных катионов, обычно являющихся сильными донорами протонов:
РС13 + ЗН2О Р(ОН2)1+ + ЗСГ (4.2-23)
Р(ОН2)1+ + ЗН2О^ЗН3О++НзРОз ^4.2-24)
Н3РО3 + Н2О Н3О++ Н2РО3 (4.2-25)
H2POJ + Н2О Н3О+ + HPOi~ (4.2-26)
НРО1-+Н2О«-НзО+ + РО|- (4.2-27)
Первое из этих равновесий похоже на процесс диссоциации большинства галогенидов тяжелых металлов:
{CuCl2} + 4Н2О 7S Си(Н2О)4+ + 2СГ (4.2-28)
н
SnCl4 + 6Н2О Sn(H2O)g+ + 4СГ (4.2-29)
Но для галогенидов металлов вторая стадия протекает значительно менее полно:
Cu(H2O)4+ + Н2О [Cu(OH)(H2O)J] + Н3О+ (4.2-30)
Sn(H2O)g+ + 2Н2О {Sn(OH)4} + 4НзО+ (4.2-31)
В водных растворах акцепторов протонов образуются гидроксид-ионы, потому что вода является кислотой более сильной, чем протонированное основание и происходит перенос протона от молекулы воды к основанию. Поэтому вода проявляет нивелирующий эффект относительно сильных оснований.
146
Глава 4. Основы химических методов анвлиза
Равновесная концентрация гидроксид-иона в одномолярном растворе сильного основания равна [ОН“] — 1,0 М. Такой раствор можно получить из амида натрия NaNH2 или гидроксида лития LiOH.
Способность воды выступать в роли кислоты (донора протонов) можно продемонстрировать на большом числе примеров. Наиболее известной, вероятно, является реакция с аммиаком:
NH3 + Н2О — NHJ + ОН" (4.2-32)
Как следует из записи уравнения реакции, с водой реагирует лишь малая часть аммиака и более чем 99% его существует в виде NH3- Однако образовавшихся ОН"-ионов достаточно для того, чтобы водный раствор аммиака имел рН> 7. Следует заметить, что растворенный аммиак существует не в виде молекул NH4OH, а в виде гидрата NH3-H2O.
Щелочные растворы образуются н при растворении в воде оксидов металлов. Этот процесс обычно сопровождается выделением значительных количеств тепла
{Na2O} + Н2О -> 2Na+ + 2ОН“ (4.2-33)
В некоторых случаях образуется малорастворнмый в воде гидроксид
{СаО} + Н2О -> {Са(ОН)2} <=t Са2+ + 2ОН~ (4.2-34)
Некоторые анионы, присоединяющие протон от воды или другого растворителя н образующие в водных растворах гидроксид-ионы и соответствующие катионы, как н упоминавшиеся выше оксиды, являются сильными основаниями. К ним относятся также пероксиды, сульфиды, фосфаты, карбонаты н фториды: _
F” + Н2О ОН" + HF (4.2-35)
О2" + Н2О <=! 2ОН" (4.2-36)
S2-+ Н2О ОН- I HS“ (4.2-37)
В кислых растворах эти реакции протекают по второй стадии
HS" + Н3О+ Н2О + H2S (4.2-38)
СОУ + Н2О О1Г + НСОз (4.2-39)
Для последнего равновесия в кислых растворах существует еще одна стадия:
НСОз + Н3О+ 2Н2О + СОг(ач) (4.2-40)
Из этих реакций следует, что ионы S2" и СО2- существуют только в щелочных растворах, поэтому в заметных количествах NH4 не может сосуществовать с СО|" или S2".
Особый интерес представляет фосфат-нон, реагирующий с донором протонов в три стадии:
POj" + Н2О НРО^" + ОН" (4.2-41)
НРО^~ + Н2О Н2РО7 + ОН" (в нейтральном растворе) (4.2-42) Н2РО7 + Н3О+ Н3РО4 + Н2О (в кислом растворе) (4.2-43)
4.-2. Кислотно-основное равновесие
147
Заметим., что в результате всех этих реакций pH раствора повышается. Очевидно, что ионы водорода и гидроксид-ионы в водных растворах находятся в равновесии с растворителем:
ОН"+Н+^Н2О (4.2-44)
что можно записать как реакцию автопротолиза воды
ОН“ + Н3О+ 2Н2О
(4.2-44)
□ Ионное произведение воды /Cw и его зависимость от температуры
Равновесие 4.2-44 описывает самоионизацию воды и может быть охарактеризовано так называемым ионным произведением воды Ку,.
Ку, = [Н+][ОН~] или Ку, = [Н3О+][ОН~] (4.2-45)
№ — ионное произведение прн бесконечном разбавлении (стандартное состояние) — определяется как
№ = ан+ • аон- = 7’[Н3О+][ОН-] (4.2-46)
Значения К® в зависимости от температуры даны в табл. 4.2-1.
Таблица 4.2-1. Зависимость ионного произведения воды от температуры
т, ° с к°,м2 т,°с к°,м2
0 КГ14’96 30 £q-13,83
10 10“14*63 40 jq-13,53
20 50 Ю-13.26
25 10-14ю 60 Ю-13,02
Подобные реакции переноса протонов протекают во всех растворителях, обладающих способностью отдавать н присоединять протон. Реакция', переноса протона протекает чрезвычайно быстро, что делает ее пригодной для аналитических целей, и поэтому кислотно-основные реакции используют в титри-метрии и других методах анализа.
4.2.3. Показатель кислотности pH
Вода обладает способностью протонировать илн депротонировать растворенные в ней вещества, что делает водные растворы либо щелочными, либо кислыми. Это можно было бы охарактеризовать через парциальное давление протонов (если бы свободные протоны существовали в растворе), либо через энергию переноса протона от кислоты к воде (соответственно от воды к основанию). Более удобно для описания этого явления использовать величину pH:
pH = -lg[H+] (4.2-47а)
148
Глава 4. Основы химических методов анализа
такое определение впервые введено Сёренсеном в 1909г. В этом выражении [Н+] представляет суммарную концентрацию всех гидратированных форм протона, т. е. НзО+, Н9О4 и т. п.1
Общепринято использовать символ «р» для обозначения отрицательного десятичного логарифма какой-либо величины. Следовательно, рОН и рКл есть соответственно
рОН = - = 14 - pH (4.2-48)
и
pKa = -lgKa (4.2-49)
Для уксусной кислоты при 25° С
745'10-5 = Ю-4’756 (4.2-50)
[1ЮАс]
н РКа = 4,756.
В водных растворах не могут быть достигнуты значения pH ниже —1 н выше 15. Б других растворителях интервал pH значительно отличается от указанного для воды, поэтому кнслотно-основные реакции в неводных средах могут быть полезными для специального применения. На рис. 4.2-1 приведены ионные произведения некоторых растворителей в сравнении с ионным произведением воды-
Измерить величину pH можно сделать различными способами; это одна из наиболее распространенных процедур в химии. Для ориентировочной оценки pH можно использовать изменение окраски веществ, называемых индикаторами, но более точные результаты дают электрохимические методы. Существует ряд электродов, потенциал которых строго зависит от активности нонов водорода. Наиболее известным и часто применяемым является стеклянный электрод, но есть и другие, например, водородный электрод (платиновый электрод в атмосфере Н2), хингидронный электрод и другие, используемые в случаях, когда стеклянный оказывается непригодным.
При 25°С чистая вода имеет pH 7,00, а концентрации ионов водорода и гидроксида равны ([НзО+] = [ОН~] = 1 • 10-7М). При 0°С pH чистой воды равен 7,48, а при 60°С — 6,51, но концентрации ионов водорода и гидроксида при этом также равны. Такие растворы называют нейтральными.
□ Величина pH — мера кислотности или основности водных, а также и неводных растворов (см. рис. 4.2-1).
Водный раствор с концентрацией ионов водорода> превышающей концентрацию гидроксид-ионов, называют кислым; его pH при 25°Q меньше 7,00. Водный раствор, в котором концентрация гидроксид-ионов выше концентрации нонов водорода, называется щелочным; при 25°С его pH больше 7,00. Понятно, что существует непрерывный переход от кислых к нейтральным и щелочным растворам.
1 Величина pH, строго говоря, есть обратный логарифм не концентрации, а активности иона водорода (см. об этом ниже). — Прим. ред.
4.2. Кислотноосновное равновесие
149
I f°H1 - иг1* Вода । [CHjOHj*] (CHjOl - кг’б Метанол .
(СдНдОН*] • - кг»
Этанол
[СН3СООН2*] |СН3СОО1 - КГ13
Уксусная кислота
I
(HCOOHj*] [HCOO-J - Ю-Ю
Муравьиная кислота
(NKj4] [NH2-j - IO*33
Аммиак
[H3SO4*J • (HSO4-] - кН Серная кислота
(C2H5OC2ftf] [CjHjOCjM^] - ?
Диэтиловый эфир
-10 О 10 20
Соответствующий pH в воде
Рис. 4.2-1. Константы автопротолиза различных растворителей и соответствующие диапазоны pH.
Если концентрация и активность иона (в достаточно концентрированных растворах) значительно различаются, величину pH следует выражать через активность инов водорода, поэтому
— В растворах постоянного ионного состава, когда значения коэффициентов активности принимают постоянными (фактически равными 1,0), возможно использование первоначальной концепции Сёренсена. В этих случаях pH исследуемого раствора можно легко найти из сравнения с величиной pH, измеренной для известного стандартного раствора с той же ионной силой. Например, для 0,1 М раствора сильной кислоты (НС1, HCIO4 и др.) при ион
ISO
Глава 4. Основы химических методов анализа
ной силе 1,0 М можно принять pH — 1,0. Любую величину pH, измеренную стеклянным электродом или каким-то другим способом, можно отнести к этой стандартной величине и найти, таким образом, концентрацию ионов водорода в анализируемом растворе.
— Для бесконечно разбавленных растворов величину pH можно выражать через активность нонов водорода:
ран = - 1g ац+ = - lg[H+) - 1g ун+ (4.2-476)
Но практическое значение такого подхода невелико, так как известно, что коэффициент активности индивидуального нона измерить нельзя. Этот подход, следовательно, полезен лишь для низких концентраций, когда у#+ сушествен-но не отличается от единицы.
Указанные трудности были разрешены после принятия определения ИЮПАК, основанного на рекомендациях Роджера Г. Бейтса. Согласно этим рекомендациям, pH определяют относительно стандартного буфера, для которого pH рассчитывают как для бесконечно разбавленного на основе измерений в ячейке с жидкостным соединением (стеклянный электрод в паре с каломельным). Измеренное значение pH не строго идентично ран, поскольку потенциал жидкостного соединения и активность индивидуального иона нельзя оценить, не прибегая к нестрогим допущениям.
4.2.4. Кислотно-основные индикаторы
Кислотно-основными индикаторами являются соединения с кислотно-основными свойствами, протонированная и депротонированная формы которых имеют различную окраску. Обычно кислотную форму обозначают символом Inda, а основную — Indb- Обе формы образуют кислотно-основную сопряженную пару
= (4.2-51)
Заметим, что заряды для простоты опущены. Возможно, что либо кислотная, либо основная форма является положительно заряженной (катионы, такие, как RaNH+ или Ее(Н2О)бОН2+), нейтральной (например, вода, которая является н кислотой, н основанием) или отрицательно заряженной (анионы, такие, как HSO4 , являющиеся н кислотами, и основаниями).
Преобразуя уравнение 4.2-51:
[h+)=k-S (4-2-51а)
получаем
pH = - lg|H+] = рКм + 1g (4.2-52)
[inaaj
В индикаторной системе Inda н Indb имеют разную окраску. Обычно изменение окраски индикатора наблюдается в интервале 1-2 ед. pH относительно величины pKind- Это легко понять из приведенных ниже расчетов:
4.2. Кислотно-основное равновесие
151
— . Если pH = pKind» то Igflndb] — Igflnd*]. Обе формы присутствуют в равных концентрациях, и наблюдается смешанная окраска кислотной и основной форм.
- Если pH = рКыа -1, то lg[Indb] = lg[Inda] -1 н [Inda]/[Indb] “ 10, т. е. 90,9% индикатора находится в кислотной форме. Доминирует окраска кислотной формы индикатора.
- Если pH = pKind + 1, то lg[Indb] = lg[Inda] + 1 и [Inda]/[Indb] ~ 0,1, т. е. 90,9% индикатора находится в виде основной формы и только 9,1% в виде кислотной. Доминирует окраска основной формы.
Индикаторы, являющиеся многоосновными кислотами, имеют несколько переходов окраски (табл. 4.2-2).
Таблица 4.2-2. Характеристики наиболее часто используемых индикаторов
Индикатор Цвет pH перехода окраски P-Alnd
Inda Indb
Метиловый фиолетовый Желтый Синий 0,0-1,6 1,0
Крезоловый красный* Желтый Красный 0,4-1,8 1,0
Тимоловый синийа Красный Желтый 1,2-2,8 2,0
Бромфеноловый синий Бесцветный Желтый 2,8-4,0 3,3
Конго красный Синий Красный 3,0-5,0 4,0
Метиловый оранжевый Красный Желтый 3,1-4,4 3,6
Бромкрезоловый зеленый Желтый Синий 3,8-5,4 4,5
Метиловый красный Красный Желтый 4,4-6,1 5,0
Ализарин* Бесцветный Желтый 5,5-6,6 6,0
Ализарин6 Желтый Красный 5,7-7,3 6,0-7,6 6,5
Бромтимоловый синий Желтый Синий 6,8
Феноловый красный Желтый Красный 6,6-8,0 7,3
Нейтральный красный Красный Янтарный 6,8-8,0 7,4
Крезоловый Красный6 Красный Желтый 7,0-8,8 7,9
Тимоловый Синий6 Желтый Синий 8,0-9,6 8,7
Фенолфталеин Бесцветный Розовый 8,2-10,0 9,1
Тимолфталеин Бесцветный Синий 9,3-10,6 10,0
л Индикатор с несколькими переходами окраски, первое кислотно-основное равновесие.
6 Индикатор с несколькими переходами окраски, второе кислотно-основное равновесие.
Если известны молярные коэффициенты поглощения обеих форм индикатора, то по отношению их концентраций можно определить значение pH раствора. При pH = pKind половина индикатора существует в кислотной форме, половина—в основной. Но это не обязательно означает, что окраска раствора промежуточная; окраска раствора определяется смесью цветовых оттенков обеих форм лишь прн близких молярных коэффициентах поглощения.
Прн точных измерениях pH возможности использования индикаторов ограничиваются тем, что они сами обладают кислотно-основными свойствами н по
Глава 4. Основы химических методов анализа
152
этому могУт присоединять или отщеплять протоны. Вызванная этим погрешность может быть значительной при микротитровании.
4.2.5. Буферы
Наиболее важное применение кислотно-основных систем обусловлено их буферизующей способностью. Многие химические реакции сопровождаются выделением протонов (в водных растворах — ионов гидроксония) или гидроксид-ионов. Если они остаются в сфере протекания реакции, наблюдается соответствующее изменение pH. Но если в растворе присутствует буфер, то его компоненты реагируют с выделяющимися ионами водорода или гидроксид-ионами и в результате pH изменяется незначительно. Буферы состоят из смеси слабой кислоты и сопряженного основания.
□ pH-буфер состоит из смеси слабой кислоты и сопряженного основания.
Пример
Буферное действие лучше всего продемонстрировать на практическом примере. Равновесия в растворе многоосновной фосфорной кислоты характеризуются приведенными ниже константами:
Н3РО4 Н2РО4 + Н+, рКп « 2 (4.2-53)
Н2РО4 НРОГ + Н+, рК.2 ге 7 (4.2-54)
НРОГ й РО.- + Н+, рК»з ге 12,5 (4.2-55)
Запишем выражение для константы второго равновесия (рК»2 ~ 7)
К.2 = = 1,0 Ю-7 — (4.2-56)
[Н2РО4 j
Приготовим буферный раствор, растворив в 1 л воды КГ*1 моль Na2HPO4 и 10-1моль NaH2PO4. Полученный раствор содержит 0,1М Н2РО4 и 0,1М НРО4“, а общая концентрация фосфата 0,2 М.
Добавим к этому раствору 10~Змоль НС1. Если это количество НС1 добавить к 1л чистой воды, то концентрация ионов водорода станет равной 10~3М, а pH 3,0. В фосфатном буферном растворе свободные ионы водорода прореагируют с НРО4- и в результате образуются ионы Н2РО4
НРОГ + Н+ -> Н2РО? (4.2-57)
Эта реакция демонстрирует принцип Ле Шателье. Положим, что в результате реакции расходуется 10“ 3 моль Н+, поэтому
[НРО4-] = 0,100 - 0,001 = 0,099 М
[Н2РО7] = 0,100 + 0,001 = 0,101 М
и [Н+] = 1 • Ю-7§^ = 1,0202 -10~7 М.
Из добавленных Ю-3 моль НС1 не прореагировало только 2,02 • 10~9 моль и после добавления кислоты в результате установился pH 6,99. Видим, что 0,2 М фосфатный буфер отлично сопротивляется добавленному количеству кислоты. Аналогичные расчеты pH после добавления 10~3 моль NaOH дали бы значение pH 7,01.
4.2. Кислотно-основное равновесие
153
Теперь приготовим раствор, содержащей 0^01 МНРОд- и 0,01 М И2РО4, и добавим к нему то же количество (10“3 моль) НС1. Исходное значение pH, как и в первом случае, равно 7,00. Пусть в реакции вновь расходуется 10~3 моль НС1:
[НРОГ ] = о, 010 - о, ай = 0,009 м
[Н2РО4] = 0,010 + 0,001 = 0,011 м
и [Н+] = 1 - lO-’Sgl = 1,222 • Ю-’М.
Из 10“3 моль НС1 не прореагировало только 2,22 10-вмоль, поэтому пред-положение вновь корректно. Конечный pH 6,92.
Вновь можно видеть, что 0,02 М фосфатная смесь —также хороший буфер, хотя и послабее описанного в первом случае. Буферная емкость второго раствора ниже емкости первого.
Приготовим раствор, содержащий 10-3М НРО3- и 10-3М Н2РО7 и вновь добавим к нему 10-3 моль НС1. Если предположить, что с НРО3- прореагировала вся добавленная кислота, то в растворе не останется НРО^" -
Получился раствор, содержащий 2 -10-3 М NaHaPO* и 1 • 10-3 М NaCl, т. е. в нем находится
3,0-10"3М Na+
2,0-10-3М Н2РО7
1,0-КГ3 М СГ
Часть Н2РО4 продиссоциирует с образованием равных количеств НзО’ НРО^~, поэтому можно написать
[Н2РО4] =2,0-10"3М
[Н+] = [НРО4"] < 2,0 10~3 М _
Используя выражение для константы второго равновесия, получим
1»1=1.10-М
или
[П+]|НРС>Г] = 2,0 • 10 10 и [Н+] = 1,414 • ИГ5 М
Следовательно, полученный раствор имеет pH 4,85.
0,002 М фосфатный буферный раствор не является столь же эффективным, как ранее описанные, и его емкость мала. Но если результат сравнить с полученным после добавления 10-3 моль НС1 к 1 л чистой воды (pH 3,00), то буфериру-ющий эффект еше ощущается.
Наконец, приготовим смесь 10“4М НРО3- и 10~4М Н2РО7 и добавим к ней 10-3 моль/л НС1. При взаимодействии с НРС>4~ расходуется 10-4 моль НС1 и остается 9 • 10~4 моль НС1. Это не совсем так, поскольку Н2РО4 —тоже основание и может реагировать с Н+ с образованием Н3РО4. Но это кислота сильная, поэтому относительно малого количества НС1 явно недостаточно для полного протонирования Н2РО4 . Итак, имеем смесь, содержащую
3 • 10-4 М Na+
2-10“4М Н2РО7
9-10-4М Н+
10 • 10"4 М СГ
154
Глава 4. Основы химических методов анализа
Из этой смеси х моль Н+ удаляется в виде Н3РО4 и остается 2 • 10 4 — х моль Н2РО4 - Принимая рКа1 = 2,0, находим
„ , [Н+ИНаРО7] (6 IO”4 - ^) (2 -10-4 - а:)
= - [H,POJ "------------------х
ИЛИ
18 • 10*’ - 9 • 10~4x - 2 • 10*4х + х2 - 10*2х = 0
и хг-1,11- 1<Г2х + 1,8- КГ7 = 0.
Решая уравнение, находим
[Н3РО4] = х = 1.624 • 10*6 М
|Н+] = 8,838 10*4М [Н2РО4|= 1,838-10*4М
и, следовательно, pH 3,05. Этот раствор не является буферным.
Как было показано ранее, pH буферного раствора определяется уравнением
pH = pKa + lg^J (4.2-58)
где [НА] и [А-] — концентрации кислоты и сопряженного основания в буферной смеси. Это уравнение иногда называют уравнением Гендерсона—Хассельбаха. Можно показать, что
pOH = pA'b + lgB^ ~ (4.2-59)
[А 1
где Къ — константа основности сопряженного основания (Kw/Ка), также характеризующая кислотно-основное равновесие
|Н[М 1 (4-2‘60)
КЬ = Е® (4.2-61)
[А ]
Наиболее эффективный буфер состоит из смеси слабой кислоты и сопряженного основания с соотношением 1:1, т. е. с pH = рКа. Это не всегда достижимо, но если нужно приготовить буфер с определенным pH, следует взять кислоту с рАа, близким к требуемому значению pH. Если рКа и pH не совпадают, отношение [А-[/[НА] надо сделать таким, чтобы (рКач- Ig[A“] — lg[HA]) было равно нужному значению pH.
Буферный раствор способен сохранять неизменное значение pH при добавлении сильной кислоты или сильного основания. В зависимости от соотношения концентраций кислоты и сопряженного основания система может сопротивляться добавлению того или иного количества сильной кислоты или основания. Это свойство буфера, называемое буферной емкостью, определяется количеством молей сильной кислоты или основания, которое необходимо для
4.2. Кислотноосновное равновесие
155
изменения pH 1л буферного раствора на 1 единицу pH. Можно вцдеть, что в рассмотренном примере буферная емкость первого раствора в 10 раз больше буферной емкости второго раствора:
Емкость буфера 1 = 10”3моль/л/0,0087 = 1,15 10"1 М
Емкость буфера 2 = 10"3моль/л/0,087 = 1,15 • 10“2 М
Можно показать, что максимальной буферной емкостью обладают буферы с соотношением концентраций кислоты и сопряженного основания 1:1.
Растворы с высокими или низкими pH (растворы сильных оснований или кислот) также обладают большой буферной емкостью, хотя содержат только одну из форм сопряженной кислотно-основной пары, например растворы NaOH или НС1.
Стандартные буферные растворы
Национальным бюро стандартов CIIIA(NBS), ныне Национальным институтом стандартов и технологий (NIST), рекомендован ряд стандартных буферов с точно известными значениями pH. Наиболее важные из них приведены в табл. 4.2-3.
Таблице 4.2-3. Стандартные буферные растворы
Состав раствора pH при
10°С 25° С 38° С
0,05 М тетраоксалат К 1,67 1,68 1,69
0,05 М цитрат КН2 3,820 3,776 3,775
0,05 М фталат КН 3,998 4,008 4,030
0,025 М КН2РО4, 0,0025 М Na2HPO4 6,923 6,865 6,840
0,01 М Na2B4O7 9,332 9,180 9,081
0,025 М NaHCO3, 0,0025 М Na2CO3 10,179 10,012 9,903
Насыщ. р-р (25°С) Са(ОН)2 13,00 12,45 12,04
Рекомендуемая литература
Бейтс Р. Определение pH. Теория и практика: Пер. с англ.— Л.: Химия. 1968.
Hammett L.P. Physical Organic Chemistry (1940).
Харнед Г., Оуэн Б. Физическая химия растворов электролитов: Пер. с англ. — М.:
ИЛ, 1962.
Lowry Т. М. Historical Introduction to Chemistry (1936).
Робинсон P., Стокс P. Растворы электролитов: Пер. с англ. — М.: ИЛ, 1963.
StellmanJ. М. The Story of Early Chemistry (1924).
Вопросы и задачи
1. Дайте определение понятия pH.
2. Что такое кислотно-основные индикаторы?
3. Как можно приготовить буферный раствор? Приведите пример.
4. Опишите зависимость ионного произведения воды от температуры.
156
Глава 4. Основы химических методов анализа
4.3. КОМПЛЕКСООБРАЗОВАНИЕ
Цели изучения
• Сделать обзор координационной химии и ее влияния на аналитическую химию.
• Выяснить, что такое комплексное соединение, координирующая способность ионов металлов, ступенчатый процесс комплексообразования, а также охарактеризовать хелаты и полиядерные комплексы.
• Ознакомиться с важной ролью координационной химий в аналитической химии и с введением в комплексометрические методы.
4.3.1. Введение
Теория валентных состояний, в значительной степени основанная на органической химии, была разработана в XIX в. Результатом этой теории стало представление о валентности, в соответствии с которой бинарные соединения, такие, как Nad, СаС12, СгС1з и PtCU, считают находящимися в насыщенном состоянии. Таким образом, можно считать, что устойчивый продукт любой реакции описывается стехиометрическим отношением, полученным из отношений валентных состояний (степеней окисления) различных реагирующих веществ, например:
N2 + 3H2^2NH3 (4.3-1)
Однако в конце XIX в. были найдены вещества, которые можно классифицировать только как соединения более высокого порядка. Они образуются за счет соединения обычных бинарных веществ, например:
СгС13 + 6NH3 СгС13 • 6NH3 (4.3-2)
PtCl4 + 2КС1 PtCh - 2KC1 (4.3-3)
PtCl4 + nNH3 PtCl4 • nNH3 (4.3-4)
где n — 2,3,4,5,6.
Эти соединения были названы комплексами. Согласно первому определению, комплексы рассматривали как соединения, в которых сочетание реагирующих веществ выражено более высокой стехиометрией, чем это следует из формальных валентных состояний реагирующих веществ. Особый интерес представляли соединения переходных металлов, такие, как
[Co(NH3)nC16-n]Cln_3, п = 3,4,5,6 (4.3-5)
в которых Со(Ш) может быть заменен на Rh(III), 1г(Ш) и т. д.
Подобные соединения были синтезированы Йоргенсеном и Бернером (1893), которые описали эти соединения, в частности их следующие свойства:
— стехиометрический состав;
— окраска и кристаллические формы;
— поведение в реакциях осаждения;
— электролитические свойства.
4.3- Комплексообразование
157
Альфред Вернер (1866-1919) показал, что такие соединения имеют различную окраску, проявляют различия в электропроводности и по-разному ведут себя в реакциях осаждения.
□ В комплексах атомы одного и того же элемента могут иметь различные химические свойства, как, например, С1 в [Со(ИНз)е]С1з и [Со(КНз)бС1]С12.
Желтое соединение, позднее описанное как (Со(ЫНз)б]С1з, проявляет свойства электролита состава 3:1 (сравните с FeCla), а с нитратом серебра дает осадок, соответствующий отношению Со:С1, равному 1:3.
Пурпурное соединение, позднее описанное как [Со(МНз)5С1]С12, ведет себя как электролит состава 2:1 (сравните с FeCla), и только два из трех атомов хлора осаждаются нитратом серебра.
Фиолетовое соединение, позднее описанное как [Со(ЫНз)зС1з] ведет себя как неэлектролит, и не дает осадка с нитратом серебра.
В рамках координационной теории был введен ряд определений:
• Координационный центр, или центральный атом: обычно ион металла.
• Лиганды: группы, которые окружают центральный атом; обычно анионы или нейтральные основные группы.
• Координационное число: число лигандов, находящихся в ближайшем окружении координационного центра1 * *.
• Структурное расположение лигандов вокруг координационного центров, с разными координационными числами (КЧ) например,
КЧ 6 - октаэдрическое расположение;
КЧ 4—тетраэдрическое или плоскоквадратное расположение;
КЧ 3—плоскотреугольное расположение; —
КЧ 2— линейное расположение.
• Квадратные скобки [] используют для описания той части соединения, которая не диссоциирует на более мелкие части, т. е. собственно комплекс.
Вернер первым подтвердил, что модель, основанная на этих понятиях, согласуется с наблюдаемыми свойствами этих соединений. Он сумел показать, что анионы вне-скобок в упомянутых выше соединениях могут легко замещаться другими анионами. Было синтезировано следующее желтое соединение с рядом различных анионов {Со(ИН3)6)Хз, где X — F", С1“, Вг“, I-, NO3 , С1О7, 1/2 C2Oj~, 1/2 SO*' и т. д.
Однако наибольший успех и признание теория Вернера получила благодаря открытиям, демонстрирующим ее правильность более непосредственным образом. Вернер, получивший в 1913 г. Нобелевскую премию, синтезировал цис- и транс-изомеры |Co(NH3)4C12}4“ (рис. 4.3-1, в) и оптические изомеры комплекса трис(этилендиамино)кобальта, что подтвердило, таким образом, октаэдрическую структуру комплексов кобальта (рис. 4.3-1,б).
Представления Вернера не утратили актуальности н по сей день. Однако здесь мы не делаем различий между комплексными и бинарными соедине-
1 Это не совсем точно: речь должна идти о числе атомов лигандов. Лиганды бывают поли-
дентатными, т. е. один лиганд может занимать несколько координационных мест. — Прим,
ред.
158
Глава 4. Основы химических методов анализа
Рис. 4.3-1. Комплексные соединения кабальта, подтвердившие правильность модели Вернера для координационных соединений, а — цис- и тпранс-изомеры [Со(КНз)4С1г]+; б —оптические изомеры комплекса трис(этилендиамино)кобальта, подтверждающие октаэдрическую структуру этого соединения.
ниями. Нет необходимости вводить отдельную классификацию для бинарных соединений и соединений высшего порядка, поскольку в большинстве случаев их структурные аспекты одинаковы. Простые бинарные соединения, например СгС1з, тоже являются комплексами, и такая химическая формула имеет смысл только в том случае, если структура неизвестна. В твердой фазе такие соединения, как {СгС1д} или {FeCh}, в действительности не характеризуются координационным числом 3, а в водном растворе молекулярная частица ЕеС1з имеет в первой координационной сфере молекулы воды. Следовательно, будет логичным считать все члены ступенчатого комплексообразования (на-пример, Fe3+ = Fe(H2O)|+, FeCl2+ = Fe(H2O)5Cl2+, FeClJ = Fe(H2O)4Clf) и соответствующие частицы твердой фазы комплексами. Любую реакцию катионов (или кислот Льюиса) с устойчивыми молекулярными или анионными частицами, содержащими свободную электронную пару (основания Льюиса), называют комплексообразованием, независимо от того, каков основной тип вклада в координационную связь, ковалентный или электровалентный.
4.3.2. Образование ковалентной связи
Хотя ковалентная связь играет важную роль в образовании комплексов, следует помнить, что комплексообразование может также включать электростатические взаимодействия. Но сейчас мы сосредоточим внимание на описании и характеристиках ковалентной связи.
Электронная конфигурация инертных газов (электронный октет) энергетически предпочтительна для большинства легких элементов. Не все элементы могут достичь такой конфигурации за счет прямого переноса электронов. Для
4.3. Комплексообразование
159
многих элементов потенциал ионизации слишком велик, чтобы удалить все избыточные электроны, а энергия сродства слишком мала, чтобы присоединить недостающие электроны.
□ В соответствии с современными взглядами, каждое соединение или ион, состоящие из центрального атома и лигандов, можно рассматривать как комплекс (например. 5Од“—тетраоксосера(6+)-комплекс).
Конфигурация октета часто может быть достигнута, когда реагирующие партнеры обобществляют электронные пары. Такой тип связи обычен в органической химии, однако он свойствен не только химии углерода и неметаллов. Обобществление электронных пар характерно для образования ковалентной связи: каждый атом стремится к конфигурации электронного октета на своей внешней электронной оболочке. Исключением является водород, поскольку его внешняя электронная оболочка может содержать лишь два электрона. Однако правило октета соблюдается не всегда. Например, в SFg сера имеет двенадцать электронов на внешней оболочке.
Одинарная ковалентная связь подразумевает наличие одной обобществленной электронной пары (дублета), двойная связь—двух пар ит. д. Связывающие дублеты могут образоваться различными путями. Либо каждый атом предоставляет но одному электрону, либо электронную пару обеспечивает только один из партнеров. В последнем случае мы имеем кислотно-основную реакцию Льюиса (донор и акцептор электронной пары) и, следовательно, образующаяся координационная связь будет полярной. Другими словами, образуется электрический диполь, и создаются соответствующие электростатические силы. Примером такой кислотно-основной реакции Льюиса (нейтрализации) может служить образование соединения между трихлоридом бора и трихлор-идом фосфора.
Подобные полярные связи образуются и в том случае, когда для образования связывающего дублета электроны предоставляет каждый из атомов, но только тогда, когда из-за структурного расположения и неодинакового притяжения электронного дублета различными атомными ядрами связывающая электронная пара смещена от центра связи к одному из ядер. Такая связь образуется в молекуле воды, в которой предполагается значительная степень ионного взаимодействия. Полярные связи образуются при связывании элементов, сильно различающихся по электроотрицательности. Это неудивительно, особенно для связей, которые образует кислород или фтор с некоторыми ионами металлов (щелочные и щелочноземельные металлы).
Молекулярные орбитали, описывающие ковалентное связывание, могут образовываться за счет перекрывания соответствующих атомных орбиталей. Ковалентные связи проявляют направленность, чего нет в случае ионных связей.
Для ковалентных соединений следует отметить некоторые особенности: величины энтальпии и энтропии у них обычно отрицательны, а теплоемкость имеет слабую зависимость от температуры. Эти свойства ковалентных соеди-
160
Глава 4. Основы химических методов анализа
Таблица 4.3-1- Свойства ковалентных и ионных соединений
Свойство Ионное соединение Ковалентное соединение
Температура плавления Летучесть Растворимость в воде Растворимость в неполярных растворителях Электропроводность Энтальпия Н° Энтропия S0 ая°/ат Направленность связи Окраска Высокая Низкая Хорошая Низкая Проводящие растворы Положительная Положительная или отрицательная Положительная Ненаправленная Составная из окраски отдельных ионов Низкая Высокая Низкая или нерастворимо Хорошая Непроводящие Отрицательная Отрицательная Близка к нулю Направленная Любая
нений вместе с другими характеристиками представлены в табл. 4.3-1, где проведено их сравнение со свойствами ионных соединений.
4.3.3. Координационные свойства ионоа металлов и лигандов
В координационной химии было предпринято много попыток классификации обширных данных, собранных поколениями ученых. Важно принять такую классификацию, которая помогла бы выявить важнейшие общие принципы в этом огромном количестве информации.
Мы примем классификацию Герольда Шварценбаха (1904-1978), профессора неорганической химии Швейцарского федерального института технологии в Цюрихе. Он разделил комплексообразующие агенты на две группы, назвав их общими и селективными. Все комплексообразующие агенты классифицированы в соответствии с координирующимся атомом лиганда, а свойства более сложных систем связаны и сравниваются со свойствами намного более простых бинарных комплексов, образуемых тем же металлом и одноатомным анионом или его протонированной формой в качестве лиганда. Таким образом, к гидроксокомплексам относят все кислородсодержащие комплексы, а к сульфидным (тио-) комплексам — все серосодержащие комплексы.
Кислородсодержащие лиганды и фторид-ион относятся к группе общих комплексообразующих агентов. Почти все ионы металлов образуют комплексы с этими лигандами. Благодаря высокой электроотрицательности атомов лиганда следует ожидать значительного ионного вклада в координационную связь.
Другие галогенид-ионы проявляют селективные свойства, образуя слабые комплексы с некоторыми ионами металлов и исключительно прочные—с другими. Селективность возрастает с ростом атомного номера галогена, но, в общем, она меньше, чем селективность некоторых других лигандов, таких, как цианид-ион и фосфор- и серосодержащие лиганды. Следует отметить, что электроотрицательность этих лигандных атомов значительно меньше, чем фтора и кислорода.
4.3. Комплексообразование
161
Ральф Г. Пирсон, профессор химии в Северо-Западном университете в Эванстоне, Иллинойс, разделил атомы лигацдов и ионы металлов на мягкие и жесткие кислоты и основания в соответствии с их электроотрицательностью и поляризуемостью. Весьма привлекательно рассматривать комплексообразование как частный случай концепции кислота-основание, или, наоборот, рассматривать протонную кислоту (кислота Брёнстеда—Лоури) как водородный комплекс соответствующего основания. Ионы металлов действуют как кислота Льюиса, а большинство лигандов действительно является основаниями Льюиса.
В разд. 4.3.2 мы кратко отметили устойчивость конфигурации электронного октета, который соответствует конфигурациии инертных газов. В этой конфигурации внешние з- и р-электронные оболочки заполнены.
Существуют и другие электронные конфигурации, также обеспечивающие устойчивость. Так, иная устойчивая конфигурация найдена для некоторых металлов, а именно Ni°, Pd° и Pt°. Здесь в дополнение к заполненным s- и р-оболочкам имеется также заполненная d-оболочка (10 электронов). Внутренние (s- и р-) электроны образуют сферически симметричную оболочку инертного газа (с нулевыми магнитным и электрическим моментами). Индивидуальные орбитали d-электронов асимметричны, и в процессе заполнения d-оболочки (1,2,..., 9,10 электронами) магнитный и электрический дипольные моменты могут накладываться друг на друга. Хотя полная d-оболочка (10 электронов) также характеризуется сферической симметрией и, следовательно, имеет нулевой магнитный момент, электронная оболочка может быть легко поляризована при наложении электрического или магнитного поля.
Похожее устойчивое состояние наблюдается для некоторых тяжелых металлов, что иллюстрируется примером электронной структуры ртути. Здесь в дополнение к полностью заполненной d-оболочке заполнена также s-оболочка следующего уровня. Такая ситуация отмечается для электронных конфигураций [Ni°]ns2, [Pd°Jns2 и [Pt°]ns2. Наблюдаемую дополнительную устойчивость объясняют с помощью так называемого эффекта инертной пары, который также отвечает за неожиданную устойчивость Hg°, Т1+, РЬ2+ и Bi3+. Внешняя оболочка имеет в данном случае электронную конфигурацию гелия.
Традиционно металлы подразделяют на А- и В-металлы, между которыми в периодической таблице расположены переходные металлы (Т-металлы). Ионы A-металлов (или А-катионы) имеют электронную конфигурацию инерт-•ного газа. Частью этой группы являются ионы редкоземельных металлов М3+. А-катионы имеют сферическую симметрию и их магнитный и электрический дипольные моменты равны нулю. Они трудно поляризуемы, и отнесение их Пирсоном к жестким кислотам вполне справедливо. Очевидно, играет важную роль ионный радиус, определяя степень комплексообразования для ионов этих металлов. Более устойчивые комплексу образуются между ионами с меньшим ионным радиусом и бблыпим электрическим зарядом (кулоновское взаимодействие). Например, комплексы между Ве2+ и F- или ОН“ более устойчивы, чем между Mg2+ и теми же анионами, а А13+ образует более устойчивые фторидные и гидроксокомплексы по сравнению с Ве2+ и катионами щелочных металлов. Комплексы, образуемые жесткими кислотами и жесткими основаниями, характеризуются преобладающим ионным вкладом в связывание. В
в Аналитическая химия. Том 1
162
Глава 4. Основы химических методов анализа
водном растворе А-катионы предпочитают в качестве лигандов фгорид-ионы и образуют более устойчивые комплексы с HjO, ОН" и О2-, чем с другими основаниями. Действительно, многие А-катионы образуют нерастворимые фториды и гидроксиды (их можно рассматривать как полиядерные комплексы)1. Основные лиганды, такие, как NH3, HS", CN" и даже S2", неспособны эффективно конкурировать с более электроотрицательными кислородным и фторидным лигандами. Анионы, которые образуют нерастворимые осадки, такие, как СО;,-, С2О^ (оксалат), SOj", РО|~ и CrOj", всегда содержат кислород в качестве координирующего атома лиганда. Органические растворители, которые могут сольватировать эти катионы, также обычно содержат кислород (спирты, кетоны и т. д.). Для галогенидных комплексов установлен следующий ряд устойчивости:
F" » СГ > Br" > I"
Устойчивость комплексов для данного А-катиона обычно возрастает с увеличением электроотрицательности атома лиганда.
Ионы В-металлов (В-катионы) имеют электронную структуру Ni°, Pd° н Pt°, т. е. у них 10 d-электронов на внешней электронной оболочке в дополнение к конфигурации инертного газа. Мы должны также включить в эту группу катионы с электронной конфигурацией инертной пары s-электронов ([Ni°]ns2, [Pd°]ns2 н |Pt°]ns2).
В-катионы также сферически симметричны, но легче поляризуются, чем А-катионы, н классификация Пирсона исходит из этого. Ионные радиусы В-катионов нечетко выражены. В-катионы образуют комплексы с кислородными лигандами (гидратированные ионы действуют как кислоты и теряют протоны при относительно низких значениях pH, приводя к соответствующим гидроксо-комплексам). Однако такие лиганды, как NH3, SH", S2", CN" и более тяжелые Галогенид-ионы, способны успешно конкурировать с кислородными атомами лигандов. Фторид-ион играет лишь незначительную роль в комплексообразовании этих ионов. Действительно, устойчивость комплексов увеличивается при уменьшении электроотрицательности координированного лигандного атома:
F>O>N»Cl>Br>I«C»S
электроотрицательность: ----------——------—»
уменьшается
„ F<O<NssCl<Br<I<C<S
устойчивость комплекса: ----——— ----------►
возрастает
Похожая обратная тенденция устойчивости комплексов В-металлов (по сравнению с комплексами A-металлов) наблюдается и при рассмотрении связи между устойчивостью комплексов и зарядом иона. Это очевидно дз сравнения устойчивости комплексов изоэлектронных ионов металлов. В ряду Ag+, Cd2+, In3+ устойчивость комплексов уменьшается при возрастании положительного заряда иона металла; это указывает на возрастание ковалентного вклада в связывание, что подтверждают отрицательные значения энтальпий н энтропий образования. Однако необходимо сознавать, что А- или В-характер
1 Растворимость соединения отнюдь не характеризует его устойчивость. Многие мало-
устойчивые комплексы плохо растворимы. — Прим. ред.
4.3. Комплексообразование
163
(Или, используя терминологию Пирсона, жесткость или мягкость) Ж является Количественным параметром, и у одного и того же иона мбТ&ЛХЖ Могут проявляться оба свойства. Например, ион РЬ2+ проявляет типичный жесткий характер по отношению к электроотрицательным лигандам F” и кислороду, тогда как окраска осадка иодида и нерастворимость сульфида показывают, что, по крайней мере, для более тяжелых галогенидов (Вг~ н 1~), В-характер перевешивает A-характер, и в соединениях, содержащих менее электроотрицательные лиганды, образуются ковалентные связи. В целом, можно отметить, что ионы металлов с внешней электронной конфигурацией инертной пары обладают склонностью быть жесткой кислотой.
Ионы переходных металлов (Т-катионы) занимают в этой классификации промежуточное положение между А- и В-металлами. Их свойства часто представляют компромисс между мягким и жестким поведением. Т-катионы имеют частично заполненные d-оболочки (1,2,..., 9 электронов). Мы можем рассматривать их как сферическое ядро, окруженное мягкой и легко деформируемой внешней электронной оболочкой. Электронная плотность вокруг ядра распределена несимметрично. Магнитный и электрический дипольные моменты могут принимать различные значения в зависимости от числа электронов в d-оболочке.
Фторидные комплексы в этой группе металлов обычно слабые; наиболее устойчивые из них — с Fe3+ и Сг3+ — могут легко превращаться в полиядер-ные гидроксокомплексы. Однако в комплексах ионов этих двух металлов NH3 не может замещать гидроксолнганды, тогда как ионы Со(П), Ni(II) и Си(П) успешно образуют более устойчивые комплексы с NH3, демонстрируй, что В-характер усиливается по мере роста числа d-электронов. Fe3+ и Сг3+ не образуют сульфидных комплексов в водных растворах, a Mn2+, Fe2+, Со2+, Ni2+ и Си2+ образуют нерастворимые сульфиды. Ni2+ и Со2+ образуют очень устойчивые цианокомплексы, Ni(CN)2- и Co(CN)|-, aFe(II), Fe(III), Со(Ш) и Сг(1П) образуют инертные гексацианокомплексы M(CN)g 6. Ионы металлов, близкие к В-металлам (Pd2+, Pt2+, Pt4+ и Au3+), отдают сильное предпочтение лигандам, содержащим серу и фосфор, по сравнению с лигандами, содержащими кислород и фтор.
Из этого краткого обзора можно заключить, что при прочих равных условиях ионы с более низкими зарядами имеют более выраженный В-характер (мягкость). С другой стороны, высокий заряд усиливает поведение иона как жесткой кислоты. Ионы с пятью или более d-электронами мягче, чем ионы с меньшим числом электронов (Ti3+, V3+ и Мп3+ образуют гидратированные ионы, которые ведут себя как протонные кислоты умеренной силы).
Гарри Ирвинг и Р. Дж. П. Уильямс показали, что комплексы двухвалентных металлов первого переходного ряда с отдельными лигандами располагаются в следующий ряд устойчивости (ряд Ирвинга-Уильямса) [4.3-1]:
Mn2+ < Fe2+ < Со2+ < Ni2+ < Cu2+ < Zn2+
□ Мягкие и жесткие центральные атомы и теория поля лигандов.
Эту последовательность можно объяснить, исходя из теории кристаллического поля и теории поля лигандов. Кристаллическое поле вокруг центрального иона металла создается электрическим полем лигандов, выстроенных во-в*
164
Глава 4. Основы химических методов анализа
круг иона металла. Это поле воздействует на энергию d-орбиталей и вызывает их расщепление. Если d-электроны могут занять более низкие энергетические уровни, то комплекс становится более устойчивым (стабилизация полем лигандов). Мп2+ (5 электронов) и Zn2+ (10 электронов) не могут получить преимущества от такой стабилизации, поскольку они сферически симметричны. Каждая орбиталь однократно или дважды занята в Мп24" или Zn£+ соответственно.
В итоге мы отметим следующее:
• Ионы A-металлов и первые несколько членов группы переходных металлов (например, Cr3+, Mn3+, Fe3+ и Со3+), а также UO2+ и VO2+ и такие частицы, как ВЁз, SO3, СО2, RCO+ и R3C4* (R —алкильная группа), являются жесткими кислотами. Они мало поляризуемы и сферически симметричны. Их сродство к лигандам выстраивается в следующий ряд:
N»P,O»S,F »СГ >Вг >Г,РО1~ »SO1~ »ClOJ,COi“ »NOJ
Это показывает, что нитраты и перхлораты щелочных металлов лучше всего подходят на роль инертных электролитов для поддержания постоянной ионной силы.
• Ионы из группы В-металлов и последние члены группы переходных металлов (Pd2+, Pt2+, Pt4*, Ап34" и т. д.) являются мягкими кислотами. Они характеризуются низкой электроотрицательностью и высокой поляризуемостью. Соответствующий ряд устойчивости их комплексов выглядит так:
Р » N,S » O,S2~ > CN" > Г > Br” > СГ « N > О > F
4.3.4. Ступенчатое комплексообразование
Обсудим образование комплексов в свете изложенного в предыдущем разделе.
Химическое равновесие в растворе комплексного соединения включает непрерывные образование и распад комплекса:
к1
М + L—ML (4.3-6)
k-i
Если в раствор иона металла вводят подходящий лиганд, можно ожидать образования комплексов, в которых ион металла играет роль координационного центра, собирающего молекулы лиганда в своей координационной сфере. Число и тип координируемых лигандов влияют на структуру комплекса. Места в координационной сфере могут быть заняты одной молекулой лиганда с несколькими координируемыми атомами (полидентатные лиганды, образующие хелаты), а атомы лиганда могут также координироваться с более чем одним центром (образуя полиядерные комплексы). Эти аспекты будут рассмотрены в данном разделе.
Обсуждая комплексообразование, следует помнить, что ионы металла в растворе имеют присущую им координационную оболочку. Реагирующие лиганды не просто включаются в пустую координационную оболочку, а замещают другие лиганды, которые уже занимают координационные места. Это
4.3. Комплексообразование
165
замещение происходит ступенчатым образом. Мы знаем, что ионы металла существуют в растворе в виде гидратированных частиц, в которых роль лиганда играет вода (кислород является координируемым атомом). Поэтому замещение лигандных молекул воды должно зависеть от концентрации частиц свободного введенного лиганда, которые будут координированы. Эта концентрация лиганда («лигандное давление») определяет, как много молекул воды может быть замещено лигандом, причем замещение протекает ступенчато: по всему ансамблю ионов в растворе сначала ионы металла меняют в своей внутренней координационной сфере одну молекулу воды на лиганд, прежде чем начнется вторая ступень замещения и следующий лиганд будет включен в координационную оболочку металла. Другими словами, не происходит полного замещения внутренней сферы отдельного нона металла, пока не прошла предыдущая стадия замещения лигандов по всему ансамблю ионов металла в растворе.
Удобно описывать комплексообразование как присоединение лигандов к свободному иону металла, хотя на самом деле происходит ступенчатое замещение в гидратированном ноне металла.
МХ(‘'-*)+ + Xх" <=! МХ^"2х)+ мх(„+1-ад+ + XJ£_ мх(—ад+
Процесс замещения лигандов записывается так:
М(Н2О)^+ + Xх” <=> М(Н2О)п_>Х<‘'-’£)+ + ун2о
М(Н2О)„_3Х‘,'-*)+ + Xх" М(Н2О)„_„х','-ад+ + (у - »Н2О
M(H2O)„_sX?"t1-w+ + Xх- МХ(‘"'х)+ + (n - s)H2O
Как упомянуто выше, это замещение может включать замену того же или большего числа молекул воды, чем число молекул лиганда, входящих в координационную сферу.
Отрицательно заряженные лиганды (анноны) обычно образуют анионы с более низкими координационными числами (например, FeCl^, КЧ 4), чем в нейтральных частицах (например, Ее(Н2О)2С1з, КЧ 5) или в положительно заряженных комплексах (например, Fe(H2 O^Cl^ и Fe(H2O)sCl2+, КЧ 6).
Это позволяет нам записать образование комплексов как ступенчатое присоединение лигандов, а не как замещение лигандов, поскольку высвобождаемую воду можно исключить из соответствующего выражения закона действующих масс. Дело в том, что в обычных условиях работы с растворами постоянной нонной силы активность воды в растворе не меняется. Тогда формальную реакцию комплексообразования выражают с помощью ступенчатой константы комплексообразования Кг и соответствующих общих констант или /3^. Используя эти константы, следует применять те же понятия активности, которые мы’обсуждали в связи с подходом Силлена для среды с постоянной ионной силой, теорией Дебая—Хюккеля для бесконечно разбавленных систем и рассмотрением кислотно-основных равновесий:
[MX,-] = [MXJ _ [MX,]
’ [MX.-JKXT’ [М][Х]‘Р-’’' [MXj][xp-j
(4.3-7)
166
Глава 4. Основы химических методов анализа
Рис. 4.3-2. Ступенчатое комплексообразование бромида таллия, рассчитанное из констант устойчивости, приведенных в тексте.
' Электрические заряды различных комплексов, лигандов и ионов металла для простоты опущены. В качестве практического примера приведены константы комплексообразования или устойчивости для бромидных комплексов
таллия(Ш): к’ = Явп = 108'16 (4,3'8) ^=[т1в™;[вг-гioe,is (4-з-9) k3=(tS-]=104,36 (43-10) ^-щвХг 1O2'SS (4-3-П)
□ Комплексообразование является ступенчатым процессом (см. рис. 4.3-2).
Общепринято иллюстрировать ступенчатое комплексообразование логарифмическим графиком зависимости числа комплексных частиц от концентрации свободного лиганда (рис. 4.3-2).
Комплексообразование можно также описать с помощью общих констант:
ft = К1 = 108’15 (4.3-8)
= = 1014,30 (43'12)
4.3. Комплексообразование
167
A = K1K2K3=p^^Zp=10,e-“ (4.3-13)
-ft = KlKiK3Ki = = io*.*’ (4.3-14)
ft,2 = Ki = IO6’15 (4.3-15)
^=« = {пЙт = 10ВД (43-16)
ftx = K2K9K4 = _^g_ = 10i3.36 (4.3.17)
ft.3 = Кз = Ю4-36 (4.3-18)
107,21 (4-319)
Если комплексное соединение в твердой фазе (что обозначается скобками {}), внести в водный раствор в отсутствие свободного лиганда, следует ожидать распада комплекса. Известно, что многие формы комплексов существуют в твердом виде (например, {Иаз[Т1Вгб]}), но распадаются в водном растворе (поскольку комплексный анион Т1Вг|~ не может существовать сам по себе в отсутствие свободного Вг”). Замещение лигандов теперь протекает в обратном направлении, и молекулы воды заменяют и высвобождают лиганд из координационной оболочки иона металла (подобно диссоциации кислоты). Этот процесс называют выравниванием комплекса. Соответствующую константу равновесия называют константой неустойчивости. Она представляет собой просто обратную величину соответствующей константы устойчивости.
Комплексообразование:
Т13+ + Вг- —> Т1Вг2+, Ку— константа устойчивости (4.3-20)
Выравнивание:
Т1Вг2+ — Т13+ + Вг-, Ка- константа неустойчивости (4.3-21)
В последующем обсуждении константы неустойчивости не рассматриваются.
Комплексообразование протекает в несколько отчетливых этапов, которые, обычно не накладываются друг на друга. Другими словами, значения различных Ki образуют последовательность:
Ki>K2>K3>Ki ' (4.3-22)
Как правило, отношение меньше, чем рассчитанное из статистиче-
ского рассмотрения. Нам следует помнить, что в случае анионных лигандов последовательно образующиеся комплексы имеют различный электрический заряд, так что каждый дополнительный лиганд испытывает все меньшее кулоновское притяжение или в случае комплексного аниона должен преодолевать большее кулоновское отталкивание. Это означает, что К\/К2 больше, чем К2/К3 и т. д. Существуют и другие эффекты, связанные со структурными изменениями (изменение координационного числа) комплекса в процессе
168
Глава 4. Основы химических методов анализа
ступенчатого комплексообразования. Это может происходить и с образованием ковалентной связи. Для образования такой связи должно произойти перекрывание орбитали иона металла с орбиталями лигандов. Это перекрывание определяет валентный угол н структуру комплекса. Например, ТШг^ имеет тетраэдрическое строение. Если образуется комплекс более высокого порядка, координационное число должно увеличиться от четырех до пяти или шести (как в Т1Вг^“), что сопровождается соответствующим изменением структуры. Мы видели уже, что FeCl^ имеет координационное число шесть (октаэдр), тогда как FeCl3 и FeCl^ имеют координационные числа пять н четыре. Наиболее часто встречающиеся структуры суммированы в табл. 4.3-2.
Таблица 4.3-2. Наиболее часто встречающиеся структуры комплексных частиц
Координа- Типичный
ционное Гибридные ион
число орбитали Структура металла Примеры
6
sp dsp2 или sp2d sp3 Линейная Плоскоквадратная Тетраэдрическая Н+ (для сравнения) Ag+ Hg2+ Au+ Pd2+ Pt2+ Ni2+ Fe" T13+ Co2+ Li+ Be2+ B(III) C(IV)
dPsp3 или sp3dP Октаэдри- ческая Вольшинст ионов металлов
Водородная связь, H2F+
{Ag2O}, Ag(CN)2 , Ag(SH)" HgCl2, HgBr2, Hgl2, HgS2’ AuC12
Pd(H2O)l+. Pd(NH3)J+
ptcij-
Ni(CN)|“, М(диметилглиоксим)
FeCl^, FeBr^
HCI4 , TlBr<
CoCll-, Co(NCS)?-
Li(H2O)+
Be(H2O)2+
B(OH)J
Органические соединения
Co(H2O)2+, Fe(H2O)f+, Fe(H2O)f+, Cr(H2O)f+, Na(H2O)g, большинство комплексов ЭДТА
Замещение лигандов может протекать быстро или медленно. Скорость комплексообразования лучше всего описывается средним временем жизни отдельного лиганда в координационной сфере иона металла. Хорошей иллюстрацией этого служит скорость обмена воды для различных ионов металлов. А13+ занимает положение в середине ряда ионов металлов, представленных на рнс. 4.3-3. Среднее время жизни аквалиганда в его координационной сфере для обмена воды составляет величину порядка 1 с. Ва2+ и Hg2+ обменивают свою координационную воду исключительно быстро (10~8 с), тогда как Сг3+ и Ru3+ — крайне медленно (время полупревращения для обмена ~106 с).
4.3. Комплексообразование
169
Се3* РЗЭ Tut 4-
Ct* N*+ К* U* Ва2* Се2* Mg2* Ba2* AI3*
s«3+
Cr2* Ма2* Со2* V2* Сг3*
Со2* Fe2* М2* Rn2*
Kg2* Cd2* In2* G*3* Fe3* *Ru3*
Zu2* Co3* •Co3*
I I I I I I I I I I
10-10 10-e I0-« 10-4 10-2 iqO l02 iq4 l06 l08
Время, c
Рис. 4.3-3. Среднее время жизни молекул воды в координационной сфере различных ионов металлов (аквакомплексы). РЗЭ — ионы редкоземельных элементов.
Время полупревращения реакции определяют как время, необходимое для того, чтобы половина исходных веществ превратилась в продукты. Для реакции первого порядка уравнение скорости записывается в виде
= Л[М(Н2О)6(Н2О)*] (4.3-23)
где частица MfHaOJsfHgO)* обменивает одну отдельную молекулу воды (помеченную звездочкой), а к — константа скорости реакции первого порядка (с-1). При интегрировании получим:
[М(Н2О)С(Н2О)‘ ], = i [M(H20)s(H20)*]oe~fct (4.3-24)
Если t — ti/2, то по определению:
[М(Н2О)Б(Н2О)‘], = |[М(Н2О)5(Н2О)-]0
Тогда
«1/2 = (4.3-25)
На константу скорости обмена лигандов обычно оказывает влияние как природа иона металла, так и природа лиганда. Однако существуют общие свойства, которые можно связать с природой иона конкретного металла. Например, все реакции с участием Сг(Ш), Co(III), Ni(III) и Pt(IV) идут относительно медленно по сравнению с реакциями, в которых участвуют другие ионы металлов в тех же степенях окисления.
4.3.5. Хелаты и полиядерные комплексы
В состав одной молекулы лиганда может входить несколько координируемых атомов и лиганды такого типа могут образовывать несколько связей с центральным атомом металла. Если все связи образуются с одним и тем же центром, тогда комплекс называют хелатом, а про лиганд говорят, что он является полидентатным. Мы различаем моно-, би-, три-, тетра, пента- и гекса-дентатные лиганды:
монодентатные лиганды: Cl~, F~, NH3, Н2О н т. д.;
170
Глава 4. Основы химических методов анализа
бидентатные лиганды: оксалат, этилендиамин (H2N—СН2—СН2—NH2), глицин;
тридентатные лиганды: иминодиуксусная кислота (ИДА);
тетрадентатные лиганды: нитрилотриуксусная кислота (НТА);
гексадентатные лиганды: этилевдиаминтетрауксусная кислота (ЭДТА).
На рис. 4.3-4 показаны структуры ИДА, НТА и ЭДТА.
Г" №
CHj-COQH ИДА
CHj-CQQH
CHj-COQH НТА
ЭДТА
Рис. 4.3-4. Структуры распространенных комплексообразующих агентов.
□ Хелатные комплексы более устойчивы, чем обычные, потому что энтропия системы при образовании хелата возрастает.
Комплексообразование между ионом металла и полидентатным лигандом называют хелатообразованием или хелатированием. Наиболее очевидным признаком хелатирования центрального атома является наличие циклической системы, как, например, при образовании комплекса между Ni2+ и димети-лглиоксимом. Диметилглиоксим образует пятичленный цикл с ионом никеля (рис. 4.3-5). Соответствующий хелат является нейтральным веществом, и он образует нерастворимый осадок.
Все представляющие интерес хелаты содержат пяти- или шестичленные циклы1. Меньшие или более крупные циклы создавали бы слишком большое искажение валентных углов. Хелаты с циклами большего размера могут быть получены только для Ag+, Hg2+ и Au+, так как ионы этих металлов предпочитают линейную координацию (валентный угол 180°) и, таким образом, стабилизируются при образовании хелатов, включающих шести-, семи- и восьмичленные циклы.
Полидентатные лиганды, образующие заряженные хелаты, используют в титриметрических методах, поскольку эти хелаты водорастворимы. Нейтральные хелаты часто нерастворимы в воде, и поэтому их широко используют в гравиметрии и жидкостной Экстракции-
Комплексы, образуемые монодентатными лигандами, обычно менее устойчивы, чем хелаты. Например, если раствор Си(КНз)2+ разбавляют водой, то наблюдается замещение аммиака на воду. Такое явление намного более выражено для этого аммиачного комплекса, чем для соответствующего хелата, в котором аминогруппы связаны этиленовыми группами (например, в H2N-CH2-CH2-NH-CH2-CH2-NH2 или H2N-CH2-CH2-NH2).
1 Четырехленные циклы имеются, например, в дитиокарбаматах, играющих большую роль в химическом анализе. — Прим. ред.
4.3. Комплексообразование
171
о-—но
II I
CHj-ON-OH H3C-C-N^ I*-C-CH3
Ni2* + 2 | -» | | + 2H*
CHj-ON-OH H3C-C-N'' ^N-C-CH3
I I
OH-..-o
Диметил гл оксим Ni( Диметил гл оксим)2
Рис. 4.3-5. Никель с диметилглиоксимом образует нейтральный комплекс, содержащий пятичленные циклы; комплекс дополнительно стабилизирован водородными мостиками.
Рис. 43-6. Полиядерные комплексы, присутствующие в равновесных системах хромат — дихромат и дихромат — трихромат.
Рис. 4.3-7. Полиядерная структура с гидроксигруппами в качестве мостиковых лигандов.
Такая стабилизация полидентатных комплексов по сравнению с моноден-татными лигандами называется хелатным эффектом. Он играет важную роль в природных водных системах. Концентрация свободных монодентатных лигандов в таких системах обычно невелика, и образования соответствующих комплексов не наблюдается. Хелаты же имеют столь высокую устойчивость, что их можно обраружить даже в природных водах.
Атомы лиганда могут участвовать в комплексообразовании более чем с одним координационным центром. В этом случае образуются полиядерные комплексы, а лиганд выполняет роль мостика. Кислород является наиболее важным мостиковым лигандом. Примерами таких полиядерных комплексов служат равновесия хромат-дихромат и дихромат-лрихромат (рис. 4.3-6).
Оксо- и гидроксокомплексы могут связываться через мостики сходным образом, образуя полиядерные структуры характерного строения. Низкомолекулярные полимеры (олигомеры) обычно растворимы в водном растворе, но по мере увеличения степени полимеризации частицы становятся менее растворимыми. На рис. 4.3-7 приведены типичные структуры гидроксокомплексов.
172
Глава 4. Основы химических методов анализа
4.3.6. Кинетика комплексообразования
Константа равновесия комплексообразования связана с константами скоростей
прямой и обратной реакции:
M + L=* ML к-1 (4.3-26а)
При установлении равновесия:
*i[M][L] = (4.3-266)
[ML) _ [М][Ц kl - ГС П-К1 (4.3-26в)
□ Быстро образующиеся водорастворимые хелаты играют важную роль в современных методах анализа воды (например, при определении жесткости воды).
Очевидно, что величина константы равновесия зависит как от к±, так и от fc-i, и сами константы скоростей дают значительно больше информации о механизмах комплексообразования, чем Кг, которая может, в принципе, являться результатом бесконечно большого числа комбинаций различных величин ку и k-i.
Как мы видели, лабильность комплексов и ионов металлов (оцениваемая по относительным скоростям обмена растворителя, рис. 4.3-3) значительно различается. Из того, что сказано выше, ясно, что термодинамическая устойчивость (представленная Ку) не будет простой функцией кинетической лабильности (представленной константами скоростей ki и fc-i).
Наше современное понимание механизмов неорганических реакций основано на анализе данных о комплексообразовании Cr(III), Rh(III) и других ионов, образующих кинетически инертные комплексы, но комплексные соединения, содержащиеся в природных водных системах, обычно значительно более лабильны.
Следует также отметить, что аква-ион в водной среде создает три области: внутреннюю сферу, где растворитель и молекулы других лигандов соединены с координационным центром в определенном стереохимическом порядке; вторую, сравнительно неупорядоченную область, где молекулы воды образуют сандвичи между стереохимически жесткими сферами; и третью область — относительно упорядоченный растворитель в общем объеме.
Некоторые реакции комплексообразования протекают очень медленно, ио в аналитических процессах, таких, как комплексометрическое титрование, используют быстрые реакции комплексообразования. Реакции ионов металлов с такими хелатообразующими реагентами, как НТА и ЭДТА, протекают достаточно быстро для таких приложений.
Литература
[4.3-1] Irving, H.R., Williams, R.J.P.. Nature 162, 746 (1948).
4.4. Окислительно-восстановительные системы
173
4.4. ОКИСЛИТЕЛЬНО-ВОССТАНОВИТЕЛЬНЫЕ СИСТЕМЫ
Цели изучения
• Объяснить химические равновесия, включающие перенос электронов от одного химического вещества к другому.
• Описать электрохимическую ячейку.
• Объяснить электрохимические основы потенциометрии, вольтамперометрии и кулонометрии.
4.4.1. Основы электроаналитических методов
Электрохимические методы важны для химического анализа на основе окислительно-восстановительных реакций. В них имеют дело с электрохимическим равновесием и измеряют электрические параметры (потенциал Е, ток г, заряд Q и сопротивление Л), связанные с электрохимическими процессами в электрохимической ячейке. Электрические свойства раствора зависят как от природы компонентов раствора, так и от их концентрации, и позволяют разрабатывать качественные и количественные методы анализа. Каждый из электрических параметров может быть измерен сам по себе, либо в сочетании с другими, либо как функция времени t или объема реагента Vr, и это создает большое разнообразие электроаналитических методов (табл. 4.4-1).
Таблица 4.4-1. Основные электроаналитические методы
Метод Измеряемый электрический параметр Контролируемая переменная
Прямая потенциометрия Потенциометрическое титрование Вольтамперометрия Кулонометрия Прямая кондуктометрия Кондуктометрическое титрование Е Е как функция Vk i как функция Е или t Q 1/R 1/R как функция Vk t = 0 £ = 0 Етл массоперенос Е или i и массоперенос Е, и массоперенос
4.4.2. Окислительно-восстановительные реакции
Окислительно-восстановительные реакции, также называемые редокс-реакциями,—это реакции обмена электронами между реагентами. Мы уже научились описывать давление протонов, связанное с активностью протонов в растворе, через pH, хотя мы и знаем, что в растворе отсутствуют несвязанные протоны. Подобным же образом можно описать окислительно-восстановительное состояние раствора через гипотетическое электронное давление рг, связанное с активностью электронов.
174
Глава 4. Основы химических методов анализа
□ Окисление всегда сопровождается восстановлением и, наоборот, восстановление всегда сопровождается окислением.
Поскольку в растворе нет свободных электронов, они должны восприниматься в связи с их носителем, донором электронов. Электрон может покинуть донор (восстановитель), только перейдя к акцептору электронов (окислителю). Таким образом, окисление всегда сопровождается восстановлением н наоборот. Для реакции восстановления
О2 + 4Н+ + 4е* -> 2Н2О (4.4-1а)
в которой расходуются четыре электрона, нужна реакция окисления, поставляющая четыре электрона, например,
4Fe2+ -> 4Fe3+ + 4е" (4.4-16)
Из сочетания уравнений 4.4-1а и 4.4-16 получаем суммарную окислительно-восстановительную реакцию
O2+4H++4Fe2+F±4Fe3+4-2H2O (4.4-1в)
описываемую константой равновесия
_ [Fe3+]4[H2O]2
р“" “ [Fe2+]4[H+]4[O2] ' ’
Заметим, что мы выбрали обе полуреакции таким образом, чтобы в полной реакции электроны отсутствовали. Однако мы можем ввести гипотетическую активность электронов ае и получить из нее ре, аналогично ац+ и pH.
Уравнения 4.4-1а и 4.4-16 представляют собой полуреакции, а уравнение 4.4-Ib— полную окислительно-восстановительную реакцию. В этой системе ионы железа существуют в различных степенях окисления, где у Fe3^ более высокая степень окисления, чем у Fe2+.
Шкала ре
Существует множество окислительно-восстановительных реакций, и каждая из них характеризуется своим значением ре точно так же, как каждое кислотно-основное равновесие связано с конкретным значением pH. Для сильных кислот характерны низкие pH, для сильных оснований — высокие pH. Аналогично, сильные окислители характеризуются низкими или отрицательными значениями ре, а сильные восстановители — высокими ре. В дальнейшем мы покажем, как связан ре с удобной измеряемой величиной, окислительно-восстановительным потенциалом Е. Для описания окислительно-восстановительного состояния системы удобно измерять окислительно-восстановительный потенциал в электрохимической ячейке.
4.4.3. Электрохимические ячейки
Электрохимическая ячейка в общем случае состоит из двух электродов, чаще всего электронных проводников, разделенных не менее чем одним электролитом, т. е. ионным проводником (рис. 4.4-1).
4.4. Окислительно-восстановительные системы
175
Рис. 4.4-1. Типичная электрохимическая ячейка.
Рис. 4.4-2. Изменение потенциала в электрохимической ячейке при разомкнутой цепи (где ф1— это так называемый внутренний потенциал).
Сокращенная схема электрохимической ячейки записывается в виде следующей диаграммы:
Zn|Zn2+,SOj-||Cu2+,SOl-|Cu (4.4-3)
где одинарная вертикальная черта обозначает границу раздела жид-кость/твердая фаза, двойная черта — границу раздела раствор/раствор, а запятая разделяет компоненты одной и той же фазы.
В общем случае между двумя электродами существует измеримый скачок потенциала вне зависимости от того, протекает ли через ячейку ток. Изменение потенциала в ячейке при равновесии (разомкнутая цепь, г — 0) показано на рис. 4.4-2.
В электроаналитических методах используют преимущественно процессы на одной границе раздела, а именно границе раздела электрод/электролит. Электрод, на котором протекает интересующая нас полуреакция, называется индикаторным, или рабочим, электродом, а другой электрод — электродом сравнения.
Глава 4. Основы химических методов анализа
176
Для решения задач в электроаналитической химии можно измерять равновесный потенциал (потенциометрия), контролировать потенциал ячейки (вольтамперометрия, кулонометрия) или измерять сопротивление раствора (кондуктометрия). В зависимости от типа прибора во внешней цепи за окислительно-восстановительным процессом в электрохимической ячейке можно наблюдать или же контролировать его протекание (рис. 4.4-1).
В реакции
Zn + Cu2+^Zn2+ + Cu (4.4-4)
если реагенты и обе полуреакции пространственно разделены, электроны могут переноситься с анода на Катод по внешнему проводнику.
Если прибор во внешней цепи «пассивен», ои либо пропускает, либо не пропускает электроны во внешнюю цепь, но если используют «активный» прибор, то ои контролирует направление потока электронов. Таким образом, изменяя условия эксперимента, можно заставить электроны двигаться в самопроизвольном или противоположном направлении между окислителем и восстановителем. Особенностью экспериментальной электрохимии является то, что направление и глубину протекания реакции в ячейке можно контролировать прибором во внешней цепи.
□ Электрохимическая ячейка (такая, как на рис. 4.4-3) может работать как гальванический элемент за счет самопроизвольной химической реакции, протекающей до установления равновесного потенциала (согласно уравнению Нернста 4.4-12), или как электролитическая ячейка за счет приложенной извне разности потенциалов.
Электрохимическая ячейка, в которой реакция протекает самопроизвольно, называется гальваническим элементом-, в электролитической ячейке реакция протекает в обратном направлении (рис. 4.4-3). Иначе говоря, электрохимические ячейки, вырабатывающие и потребляющие электрическую энергию, называются соответственно гальваническими элементами и электролитическими ячейками.
Катод Cu2++2a“-*Cu
б Электролитическая ячейка
Анод
1гО—*>/iO2 + 2Н+ + 2а"
Рис. 4.4-3. Электрохимическая ячейка, а—гальванический элемент; б — электролитическая ячейка. [Bard, A. J., Faulkner, L. R. (1980), Electrochemical Methods, New York, Wiley, p. 15].
4Л. Окислмтеяьно-восстанмительмые системы
177
В гальваническом элементе протекают реакции
• на аноде (окисление):
Zn S=t Zn2+ + 2е~ ' (4.4-5)
• на катоде (восстановление):
Cu2+ + 2е" Си (4.4-6)
В гальваническом элементе разность потенциалов ДЕ, измеренная при <разомкнутой цепи», связана со свободной энергией Д(? реакции в ячейке:
Д£ = -^ (4.4-7)
где знак <минус> означает, что реакция протекает самопроизвольно (т. е. идет слева направо), п—число электронов, участвующих в реакции, F—постоянная Фарадея (9648бКл/моль, заряд одного моля электронов).
Суммарная реакция в ячейке протекает справа налево, если разность потенциалов ячейки ДЕ отрицательна, и слева направо, если она положительна. При ДЕ = 0 реакция в ячейке находится в состоянии равновесия, скорость достижения которого зависит от природы системы.
Потенциал ячейки равен разности потенциалов правого [на диаграмме ячейки, см. ур. (4.4-3) ] и левого электродов ячейки:
Еяч = ЕПрМ — Елея = Ек — Е& (4.4-8)
а его знак определяется в соответствии со Стокгольмским соглашением ИЮПАК, если в гальваническом элементе левый электрод является анодом, а правый—катодом.
□ Окислительно-восстановительные реакции могут протекать вне электрохимической ячейки при растворении или в титриметрии (см. разд. 7.2), например, Ce4+-f-Fe2+ Ce3+ + Fe3+. Система с более высоким положительным потен-
циалом Е° (Се4+/Се3+ ... 1,71 В) является окислителем относительно другой системы с более низким потенциалом (Fe^/Fe24...0,77В).
Потенциал электрода
Измерить можно только разность потенциалов, но не потенциал изолированного электрода. Поэтому электродный потенциал измеряют относительно стандартного водородного электрода (СВЭ) в электрохимической ячейке с жидкостным соединением в отсутствие тока во внешней цепи (г = 0):
Pt, Н2(1 атм) |Н+(а = 1)|| Мп+(о = 1)|М (4.4-9)
СВЭ Индикаторный электрод
Стандартный водородный электрод—это платиновый электрод (платинированная пластинка), насыщенный водородом при продувании газа над электродом при постоянном давлении 1 атм и погруженный в водный раствор кислоты с известной постоянной активностью ионов водорода (ан+ = 1). Платина не принимает участия в электрохимической реакции и служит только переносчиком электронов. В основе работы СВЭ лежит полуреакция
2H+(aq) -Ь 2е~ Н2(газ) (4.4-10)
178
Глава 4. Основы химических методов анализа
Потенциал СВЭ принят равным нулю вольт при любой температуре. СВЭ может быть катодом или анодом, в зависимости от второй полуреакции в ячейке.
□ «Электрохимический ряд» —это набор полуреакций в порядке возрастания значений их _Е°, измеренных в гальваническом элементе относительно СВЭ (см. приложение в т.2).
Электродный потенциал — это потенциал ячейки из данного электрода в качестве катода и СВЭ в качестве анода. Соответствующая полуреакция на индикаторном электроде имеет вид
Мп+4-пе-^М (4.4-11)
(4.4-12)
(4.4-13)
Потенциал Е электрода для указанной окислительно-восстановительной системы определяется уравнением Нернста:
nF nF
где Е° — стандартный электродный потенциал, R — универсальная газовая постоянная, Т- абсолютная температура, F — постоянная Фарадея, п — число электронов, участвующих в электродной реакции, а аМп+ — активность иона металла Мп+.
Если «м**4 — 1, то Е = Е° — стандартный потенциал полуреакции. При изменении ам~+ на порядок значение Е изменяется на 59,16 мВ/n при 25°С (298 К). Окислительно-восстановительное состояние системы при равновесии принято описывать окислительно-восстановительным потенциалом. Значения стандартных потенциалов полуреакций приводятся в справочниках.
Теперь мы можем связать ре с Е:
EF ₽£ _ 2.3026ЛТ
Поскольку на практике с СВЭ работать достаточно сложно, обычно используют другие электроды сравнения. Чаще всего такими вторичными электродами сравнения служат хлоридсеребряный электрод (Ag|AgCl, КС1) и каломельный электрод (Hg|Hg2C12, КС1). Их потенциалы относительно СВЭ определены очень точно (ур. 4.4-14). Например, если ячейка состоит из насыщенного каломельного электрода (НКЭ) в паре с СВЭ, э.д.с ячейки равна потенциалу НКЭ:
Pt,H2|H+(n = 1)||КС1(нас.),Не2С12(нас.)|Нё (4.4-14)
-Енкэ = 4-241 мВ при 25°С. Электродный потенциал любой полуреакции определяется как потенциал ячейки, состоящей из исследуемого электрода и СВЭ.
Соотношение ток — потенциал
Ток в электролитической ячейке переносится электронами в фазе электрода и во внешних проводниках и ионами в растворе электролита. Для возникновения тока необходимы реакции переноса заряда (окисления-восстановления) на границе раздела электрод | электролит.
4.4. Окислительно-восстановительные системы
179
□ Электролитическая ячейка в химическом анализе.
Потенциал электрода в электролитической ячейке можно изменять с помощью внешнего источника (рис. 4.4-4). Если приложенный извне потенциал Евн более отрицателен, чем равновесный или же потенциал разомкнутой цепи в электрохимической ячейке (Евн < ЕРавн)> иа катоде происходит восстановление, и протекает катодный ток. Если потенциал электрода становится более положительным, энергия электронов в фазе электрода снижается, электроны поступают из электролита в фазу электрода, и в результате электрохимического окисления протекает анодный ток.
Если через электрохимическую ячейку протекает макроскопический электрический ток, то на обоих электродах происходят фарадеевские процессы. В соответствии с законом Фарадея на окисление или восстановление 1.моля ионов требуется nF кулонов (1 кулон —это электрический заряд, переносимый током в один ампер за одну секунду). Соответственно скорость электродной реакции определяется уравнением
t
!)(моль/с) = [idt (4.4-15)
at nr nr J
о
где Q — количество электричества, равное интегралу тока г по времени; N — число молей вещества, прореагировавших на электроде, ап — число электронов, участвовавших в электродной реакции.
Отсюда следует, что ток г является мерой скорости электрохимической реакции, но из этого правила есть исключения. На границе раздела электрод | раствор реакции переноса заряда могут не происходить в некотором диапазоне потенциалов из-за термодинамических или кинетических затруднений. В этих условиях могут протекать адсорбция и десорбция, и структура границы раздела электрод | раствор может меняться с изменением потенциала или состава раствора. Эти нефарадеевские (емкостные) процессы вызывают протекание во внешней цепи тока, называемого током заряжения или емкостным током. При протекании электродной реакции одновременно идут как фарадеевские, так и нефарадеевские процессы.
Источник питания
Рис. 4.4-4. Типичная электролитическая ячейка, присоединенная к внешнему источнику тока. [Bard, A.J., Faulkner, L.R. (1980), Electrochemical Methods, New York, Wiley, p. 18].
Q Cu | Cd | Cd(NO3)2 (1 Jlfl IIKCI (нас.)|Н02С121 Hfl | Cu (+)
Cd2+ + 2o” = Cd £° = -0,403 В отн. СВЭ
HggClj + 20_ = 2Hg + 2СГ £° - 0.241 В отн. СВЭ
110
Глава 4. Основы химический методов анализа
Рис. 4.4-5. Кривые ток— потенциал, в—идеально поляризуемый электрод; б — идеально иеполяризуемый электрод. Пунктирная линия показывает поведение реального электрода, приближающееся к идеальному в ограниченном интервале токов или потенциалов.
Электрод, на котором отсутствует перенос заряда через границу электрод | раствор вне зависимости от приложенного внешнего потенциала, называется поляризуемым электродом (рис. 4.4-5,а). Например, ртутный электрод в растворе хлорида калия в отсутствие растворенного кислорода является идеально поляризуемым электродом. Интервал его поляризации ограничивается
в анодной области потенциалом окисления Hg:
Hg + СГ - |Hg2CI2 + е~ (при ~ +0,25 В отн. СВЭ), (4.4-16)
а в катодной области потенциалом восстановления К+:
К+ + Hg + е~ -> K(Hg) (при ~ -2,1 В отн. СВЭ) (4.4-17)
В интервале потенциалов, ограниченном этими реакциями, реакции переноса заряда отсутствуют. Величина нефарадеевского тока (тока заряжения), протекающего в ячейке, зависит от величины поляризующего потенциала. Этот ток необходим для заряжения границы раздела электрод | раствор, которая ведет себя как емкость, отвечающая заданному значению потенциала.
Примерами идеально неполяризуемого электрода являются платиновый электрод или медный электрод, погруженный в хорошо перемешиваемый раствор с изменяющейся концентрацией ионов меди(П). Тока нет, пока приложенный извне потенциал не превышает потенциала восстановления ионов меди до металлической меди. При более высоком значении протекает реакция
Си2+ + 2е“ ₽ Си (Е° = 0,337 В) (4.4-18)
Поверхность электрода покрывается медью, электрод деполяризуется, а его потенциал определяется уравнением Нернста (25°С):
£ = Д° +°'°^916 lg[Cu2+] (4.4-19)
4.4. Окислительно-восстановительные системы
181
Если электрод деполяризован, в ячейке протекает ток, ие вызывающий отклонения потенциала от равновесного (рис. 4.4-5,б).
□ Поляризация: нарушение линейной зависимости г от Е согласно закону Ома (Е = iR).
Разность между приложенным извне потенциалом, при котором деполяризуется поляризуемый электрод, и равновесным потенциалом называется перенапряжением (поляризацией) тр Перенапряжение характерно и для катодных, и для анодных процессов, на которые влияют следующие факторы:
- массоперенос;
- перенос электронов (перенос заряда) через границу раздела электрод j раствор;
— химические реакции, предшествующие или следующие за реакцией переноса электрона.
Величина тока в электрохимической ячейке часто лимитируется самой медленной стадией электродной реакции. При заданной плотности тока величина перенапряжения определяется скоростью различных стадий реакции: т)с (перенапряжение массопереноса, или концентрационная поляризация), тд (перенапряжение переноса заряда, или кинетическая поляризация), (перенапряжение или поляризация, связанные с предшествующей реакцией). Электродную реакцию можно представить сопротивлением R, состоящим из набора сопротивлений, в различной степени замедляющих электродный процесс: Rc, Rt и т. д. Выстрой реакции соответствует небольшое сопротивление, медленной реакции — высокое.
□ Перенапряжение растет с повышением плотности тока (А/с|Ла). Микроэлектроды поляризуются легче, чем большие электроды.
4.4.4. Реакции, контролируемые переносом заряда
Рассмотрим процесс восстановления, при котором электроны переходят от восстановителя (Red) к окислителю (Ох):
Ох + пе~ =*==*= Red (4.4-20)
где kf—константа скорости прямой реакции, кг — константа скорости обратной реакции.
В процессе электрохимического восстановления, т. е. при Евн, значительно более отрицательном, чем равновесный, скорость контролируется такими процессами, как перенос электронов через границу раздела электрод | раствор или массоперенос окисленной формы из объема раствора к поверхности электрода, и устанавливается новое равновесие:
п СО(а:=0)
где Со^^о) и cR(®=o)— концентрации окисленной и восстановленной форм на поверхности электрода (ж — 0).
182
Глава 4. Основы химических методов анализа
Величина результирующего тока определяется уравнением
г = nFA(k{Co — кгс&)
где А — площадь поверхности электрода. Константы скорости реакции переноса заряда зависят от £вн:
kf — kf exp
кг — к° exp
onKEBH1 ЯГ ]
(l-a)nFEm
RT
(4.4-22)
(4.4-23)
(4.4-24)
где ка — константа скорости, измеренная при Ев„ = 0, т. е. константа скорости соответствующей химической реакции, а а—коэффициент, отвечающий доле электродного потенциала (потенциала на границе раздела), дающей вклад в константу скорости соответствующей химической реакции.
Из этих выражений видно, что электродный потенциал влияет на константы скорости соответствующих химических реакций через изменение энергий активации. Например, если потенциал катода отрицательнее равновесного потенциала, это облегчает перенос восстановителя, поскольку его потенциальная энергия увеличивается на a.nFEy что сказывается в уменьшении энергии активации катодной реакции. И наоборот, более отрицательный электродный потенциал уменьшает скорость анодной реакции в соответствии с выражением
i = nF A {eoAf exp - erf exp [(1~ j^"1] } (4-4-25)
anF(EB„
Яравн )] _ .о [(l-o)nF(E»H-Еравк)
== - сцКг ехр ———--------------
Прн равновесии Евн = 0, i = ik — ia - «о» kf — kr = к и zq = nFkAc, где zq -tok обмена.
Таким образом, прн Явн:
(44-26) где Ев„ — Ер&вя ~ у, у — перенапряжение переноса заряда.
Влияние плотности тока на форму кривой ток-потенциал показано на рис. 4.4-6.
4.4.5. Реакции, контролируемые массопереносом
Если скорость электролрой реакции н соответствующей химической реакции велика по сравнению со скоростью массопереноса, то скорость электродного процесса лимитируется скоростью массопереноса vMn, с которой электроактив-ное вещество доставляется к поверхности электрода. Массоперенос, т. е. дви-жение вещества из объема раствора к поверхности электрода, осуществляется за счет миграции заряженных частиц в электрическом поле, механической конвекции вследствие перемешивания раствора или вращения электрода или диффузии под влиянием градиента концентрации.
□ В количественном полярографическом анализе диффузия—основной механизм массопереноса к электроду в отличие от миграции и конвекции.
4.4. Окислительно-восстановительные системы
183
Рис. 4.4-6. Влияние плотности тока на форму кривой зависимости тока от потенциала. [Bard, A. J., Faulkner, L.R. (1980), Electrochemical Methods, New York, Wiley, p. 110]. Реакция: О + ne R, a — 0,5, n = 1. T = 298 K.
Массоперенос вещества к поверхности электрода вследствие миграции, конвекции и диффузии в направлении оси х можно описать уравнением Нернста— Планка:
= + (4.4-27)
ОХ л.1 ох
где Ji (ж)— поток частиц г (моль-с“1-см-2) на расстоянии х от поверхности электрода', — коэффициент диффузии (см2/с); 8а{х)/8х—градиент концентрации на расстоянии х\ 8ф[х)/8х— градиент потенциала; Zi н а — заряд и концентрация частиц г соответственно; v — скорость потока (мл/с).
В вольтамперометрии первостепенное значение имеет массоперенос вещества под действием градиента химического потенциала, т. е. градиента концентрации.
Диффузионный массоперенос (в отсутствие миграции и перемешивания раствора) электроактивного вещества можно описать законами Фика:
где х — расстояние от поверхности электрода.
Приближенно это можно записать так:
^мп = то[со ~ со(х = 0)] (4.4-29)
где со — концентрация электроактивного вещества О в объеме раствора; со(т — 0) —его концентрация у поверхности электрода; то — коэффициент массопереноса (то соответствует Dq/8o, где $о~ толщина гипотетического неподвижного диффузионного слоя на поверхности электрода).
184
Глава 4. Основы химических методов анализа
Рис. 4.4-7. Изменение концентрации у поверхности электрода при >
Ервля. в —на ртутном капающем электроде; 6 — на твердых электродах.
Изменение концентрации электроактивяого вещества у поверхности {х = 0) электродов разной природы в различные моменты времени после включения внешнего источника напряжения Ет показано на рнс. 4.4-7.
Максимальная скорость массопереноса О наблюдается при cq{x = 0) = 0. В этих условиях величина тока равна так называемому предельному диффузионному току id, где
id — nFAmoCo
(4.4-30)
Вопросы и задачи
1. Как узнать о наличии комплексов и комплексных ионов?
2. Что такое мягкий и жесткий центральные ионы?
3- Что такое хелаты и почему они устойчивее, чем обычные комплексы?
4. Опишите две основные области применения хелатов в аналитической химии!
5. Нарисуйте схемы двух основных типов электрохимических ячеек (гальванический элемент и электролитическая ячейка) и опишите их применение в аналитической химии на двух конкретных примерах (например, для измерения pH и определения содержания кислорода в речной воде).
6. Что такое стандартный водородный электрод (СВЭ) и какова его роль в теории?
7. Что такре поляризуемый электрод?
8. Почему в полярографическом анализе важно, чтобы основным источником массопереноса к электроду была диффузия, а не миграция или конвекция?
4.5. Гетерогенные равновесия 185
4.5. ГЕТЕРОГЕННЫЕ РАВНОВЕСИЯ
Цели изучения
• Дать описание состояния равновесия систем, состоящих более чем из одной фазы, и обсудить роль таких систем в аналитической химии.
• Обсудить теоретические основы гетерогенных равновесий, на которых базируется большинство методов разделения. Закономерности, имеющие место в таких системах, служат базой для понимания процессов аналитического разделения, хотя в большинстве случаев равновесие (в термодинамическом смысле) не достигается, поскольку время установления равновесия в реальной системе сопоставимо с временем, затрачиваемым на всю аналитическую процедуру.
• Проиллюстрировать тот факт, что формальное теоретическое рассмотрение гетерогенных равновесий, как и в случае с гомогенными равновесиями, важно для адекватного описания соответствующих аналитических процессов.
4.5.1. Термодинамическое рассмотрение
Введение
Равновесие, в котором находится система, состоящая более чем из одной фазы, называется гетерогенным равновесием. Принципы многофазного равновесия широко применяются для описания процессов и методов разделения. В качестве примеров таких методов можно привести кристаллизацию, экстракцию, хроматографию и зонную очистку.
Состояния гетерогенного равновесия и связанные с ними кинетические аспекты играют важную роль в химии окружающей среды, поскольку основой многих природных химических циклов являются многофазные системы. Так, например, качество пресноводной воды определяется в основном присутствием растворенных веществ, привнесенных из других фаз. Такими веществами могут быть растворенные газы из атмосферы, следовые количества соединений металлов и других загрязняющих агентов естественного и техногенного происхождения. Поэтому адекватное описание гетерогенных равновесий важно при изучении таких систем.
Принципы, лежащие в основе описания гетерогенных равновесий, были сформулированы Дж. Уиллардом Гиббсом (1839-1903) в 1876 г. До ознакомления с правилом фаз Гиббса необходимо ввести некоторые параметры и расширить некоторые идеи, упомянутые в разд. 4.1.
Считается, что система приходит в состояние термодинамического равновесия при установлении термического, механического и химического равновесий между фазами, входящими в состав данной системы. Это можно записать следующим образом:
dG = VdP - SAT + 52 = ° (4.5-1)
где объем V, энтропия S и число молей П£ компонента г — экстенсивные (зависящие от количества вещества) переменные, а температура Т, давление Р и химический потенциал — интенсивные (не зависящие от количества веще
186
Глава 4. Основы химических методов анализа
ства). Переменные V, S и п* являются аддитивными величинами, н для полной системы их можно получить в виде суммы объемов, энтропий и количества молей различных фаз А, В н С. Например,
N
V = VA+ VB+ VC = '£v (4.5-2)
1
где VA, VB и Vе — объемы фаз А, В и С.
Аналогично можно записать:
N
s = Xs (4.5-3)
1
н
N
ni — пх (4.5-4)
1
Многофазная система может быть отделена границами, не позволяющими ей обмениваться теплом, работой и веществом с внешним окружением. Такая система называется термодинамически закрытой. Однако это не относится к границам между отдельными фазами внутри самой системы. Это означает, что если система выведена из состояния равновесия, то обмен теплом, работой н веществом между фазами возможен вплоть до возвращения в исходное равновесное состояние. Возникает вопрос, могут ли фазы обмениваться теплом, работой и веществом в том случае, если равновесие уже установилось. Рассмотрим, что представляют собой термическое, механическое и химическое равновесия.
Прежде всего рассмотрим условия реализации механического равновесия. Для простоты выберем двухфазную систему с фазами А и В н предположим, что давление РА в фазе А больше, чем давление Рв в фазе В. Следовательно, при расширении одной из фаз и соответственно сжатии другой в системе совершается работа бы. При совершении этой работы в условиях равновесия процесс можно считать обратимым (бы = Ладг).
Таким образом можно рассчитать изменения объема в каждой из фаз:
= (4.5-5)
SVB = (4.5-6)
Поскольку энтропия и состав системы не изменяется, общее изменение объема 6V должно быть равно нулю:
SV = <5Va + <5VB = 0 (4.5-7)
Следовательно:
бы бы
рА + рв - 0 (4.5-8)
Полагая, что в условиях механического равновесия давление во всех фазах должно быть одинаковым, для данной двухфазной системы можно записать:
РА = Рв (4.5-9)
4.5. Гетерогенные равновесия
187
и, следовательно, в равновесном состоянии индивидуальные фазы не изменяются в объеме не происходит никакого обмена работы между фазами системы.
Аналогичным образом можно рассмотреть н термическое равновесие. В этом случае экстенсивную переменную V, используемую в изложенных выше рассуждениях, следует заменить на S, а интенсивную переменную F-на Т. Повторив ход рассуждений, можно сделать вывод о необходимости равенства температур во всех фазах прн термическом равновесии. Для рассматриваемой двухфазной системы имеем:
ТА = Тъ (4.5-10)
В равновесном состоянии индивидуальные фазы обмениваются теплом, а энтропия остается постоянной.
Наконец, рассмотрим химическое равновесие, вводя химический потенциал pi в качестве интенсивной переменной и число молей п, компонента г в качестве экстенсивной переменной. Прн химическом равновесии отсутствует направленный перенос вещества через фазовые границы, а химический потенциал для любого компонента системы должен быть одинаковым во всех фазах. Для рассматриваемой двухфазной системы получим:
дА = Hi (4.5-11)
Правило фаз Гиббса
Состояние системы, состоящей из р фаз и с компонентов описывается однозначно, если для каждой фазы известны температура, давление и состав. (Символ р означает число фаз, а не давление Р.) Если система находится в равновесии, то для описания системы требуется лишь ограниченное число переменных. Это число независимых интенсивных переменных называется числом степеней свободы F. В дальнейшем мы получим значение F для гетерогенной равновесной системы с с компонентами и р фазами. Компонент г в фазе А характеризуется мольной долей ХА, определяемой как
ХА =
En*
(4.5-12)
где
(4.5-13)
Чтобы описать состав фазы А, необходимо знать значения мольных долей только с—1 компонентов, поскольку мольная доля последнего компонента может быть рассчитана по уравнению (4.5-13). Следовательно, каждая фаза может быть описана 2 4- (с — 1) переменными. Поскольку при равновесии значения температуры н давления во всех фазах одинаковы, суммарное число переменных, описывающих систему в целом, равно 2 + (с — 1)р. Однако некоторая часть н этих переменных функционально связана друг с другом. Мы уже показали, что химический потенциал каждого компонента одинаков для всех фаз, находящихся в равновесии. Следовательно, количество независимых
18S
Глава 4. Основы химических методов анализа
переменных сокращается на р — 1 для каждого компонента или на с(р — 1) для всей системы. Отсюда можно определить общее число независимых переменных, или степеней свободы, как
F=c-p+2 (4.5-14)
Это уравнение называется правилом фаз Гиббса н обычно применяется для систем, находящихся в состоянии равновесия.
□ Правило фаз Гиббса показывает степень свободы (число независимых переменных) в гетерогенной системе с с компонентами и р фазами.
Фазовые равновесия и фазовые диаграммы
Интерес химиков-аналитиков к теоретическому описанию гетерогенных систем продиктован главным образом необходимостью дальнейшего совершенствования методов разделения. Мы подходим к термодинамике гетерогенных систем с этой точки зрения, н для удобства различаем фазовое равновесие и распределительное равновесие (равновесие распределения).
При фазовом равновесном разделении фазы представляют собой смеси, в которых изучаемое вещество является главным компонентом. Следовательно, состав фаз удобно выражать в мольных долях.
В распределительном равновесии фазы включают помимо интересующего нас вещества и иные компоненты. Исследуемое вещество не является основным компонентом, и фазовый состав удобнее выражать через концентрацию пробы, например моль/л.
Однокомпонентные системы
Для однокомпонентных систем правило фаз показывает, что возможны трн различных случая. Соотношение
F = 3 — р
позволяет различать бивариантные однофазные, моновариантные двухфазные и инвариантные трехфазные системы.
В однофазной системе максимальное число степеней свободы равно двум, поэтому наиболее удобным способом описания системы является двумерная фазовая диаграмма. Обычно в качестве интенсивных переменных выбирают Т и Р. На рнс. 4.5-1 приведена фазовая диаграмма воды. Анализ этой диаграммы приводит к следующим выводам:
— Давление пара льда при температуре ниже 0°С меньше, чем давление пара жидкой воды, и, следовательно, лед является более стабильной формой существования воды в этой области. Переход через кривую «Давление пара НаО(ж)», разделяющую на диаграмме области твердой фазы—льда и паровой фазы, отражает сублимацию или обратный процесс, конденсацию. Ниже давления 4,58 мм рт. ст. лед не может превратиться в воду, н единственный возможный путь превращения — сублимация.
— Кривая, разделяющая области существования воды и пара, приведена для интервала температур от 0 до критической точки. В открытой системе эти две фазы сосуществуют только в области существования жидкой воды, т. е.
4.5. Гетерогенные равноеееия
189
от 0 до 100°С. Однако в закрытой системе эти две фазы продолжают сосуществовать до тех пор, пока не будет достигнута критическая точка. Эта точка характеризуется критическим давлением (Рс — 218,3 атм) н критической температурой (Тс = 374,1°С).
— Вдоль кривых, разделяющих две сосуществующие фазы, степень свободы сокращается до F — 1; при этом условии любому выбранному давлению однозначно соответствует фиксированная температура, и наоборот.
— За пределами Рс н Тс существует только одна фаза, сверхкритический флюид. Его свойства отличаются от свойств жидкой воды и водяного пара. Сверхкритическая область имеет две степени свободы, т. е. к системе может быть приложено любое давление при фиксированной температуре, а при данном давлении существует свободный выбор температуры, поскольку Т > Тс и Р > Рс-
— В тройной точке сосуществуют все три фазы воды н, следовательно, отсутствует степень свободы; температура 273,16°С и давление 4,58 мм рт. ст. являются фиксированными.
— Температура плавления льда слабо зависит от давления, уменьшаясь с ростом последнего (в этом проявляется аномальное поведение воды по сравнению с другими веществами).
190
Глава 4. Основы химических методов анализа
Сверхкритические флюиды
□ Сверхкритический флюид—интересное в аналитическом плане состояние вещества.
Кратко остановимся на свойствах сверхкритических флюидов. В табл. 4.5-1 суммированы критические давление, температура н плотность некоторых сверхкритических флюидов. Подобные флюиды используют в сверхкритической флюидной экстракции (СФЭ) н сверхкритической флюидной хроматографии (СФХ).
Таблица 4.5-1. Критические параметры веществ
Соединение Тс,°С Рс,атм Ос,г/см?
Ацетонитрил 274,7 47,7 0,24
Аммиак 132,5 112,5 0,24
Аргон -122,3 48,0
н-Бутан 152 37,5 0,23
Вода 374,1 218,30 0,34
Гелий -267,9 2,26
Диоксид азота (N2O4) 157,8 100
Диоксид углерода 31,0 72,9 0,47
Диэтиловый эфир 192,6 35,6 0,27
Гемиоксид азота (N2O) 36,5 71,7 0,43
Криптон - 63,8 54,3
Метан - 82,1 45,8
Метанол 240,0 78 0,27
Неон -228,7 26,9
1-Пентен 191 39,9 0,24
н-Пропан 96 42 0,22
Сероуглерод 279 78
Этан 32,2 48,2 0,20
На рис. 4.5-2 представлена фазовая диаграмма СОг- Поскольку нас интересует плотность жидкой фазы в сверхкритической области,то диаграмма представлена в виде зависимости давления от плотности. Как видно из рисунка, значительные изменения плотности флюида (0,25-0,95 г/см3) могут быть достигнуты выбором соответствующих давления и температуры.
Плотность сверхкритического флюида может изменяться в широком диапазоне. Это весьма важно, поскольку чем выше плотность, тем лучше проявляются сольватирующие свойства флюида по отношению к растворенным в нем веществам. Растворимость веществ в сверхкритических флюидах часто превышает растворимость в обычных жидкостях. Поэтому сверхкритические флюиды весьма удобны для использования в экстракции, свойства их можно легко изменять применительно к решению конкретной аналитической задачи.
В отличие от обычных жидкостей, сверхкритические флюиды сжимаемы и, следовательно, изменяя плотность флюида (что достигается соответствующим изменением давления н температуры), можно управлять его сольватационными свойствами и растворимостью в нем различных веществ.
4.5. Гетерогенные равновесия
191
4.5.2. Системы газ —жидкость
Введение
Газы в определенной степени растворимы в жидкостях. Равновесный процесс перехода из одной фазы в другую осуществляется на границе раздела между растворителем (например, водным раствором) н атмосферой (паровой фазой). Растворимость газов в жидкостях может быть рассчитана по закону Генри.
Переход вещества из жидкого в газообразное состояние называется испарением, а обратный процесс — конденсацией. Сочетание испарения и последующей конденсации химического вещества называется перегонкой или дистилляцией, а продукт дистилляции — дистиллятом. Процесс дистилляции особенно важен для разделения больших объемов жидких смесей. Чтобы повысить эффективность разделения, этот процесс часто проводят в специальных ап-
1В2 Глава 4. Основы химических методов анализа
каратах—дистилляторах, работающих в режиме многократного чередования процессов испарения и ковденсации.
Процессы дистилляции остаются очень популярными для разделения сложных жидких смесей. В промышленном процессе переработки нефти, например, миллионы тонн сырой нефти фракционируют на коммерческие продукты, такие, как бензин, топливо для котлов и газ. Дистилляционные процессы регулярно используют также в научных лабораториях доя разделения и очистки синтезированных соединений от побочных продуктов.
В последние несколько десятилетий развиты более эффективные методы разделения, и аналитические методы разделения, основанные на дистилляции, часто заменялись новыми методами. Однако дистилляция все еще играет роль в препаративных разделениях, включая операции очистки пробы.
В промышленности широко применяется адсорбция нежелательных газообразных компонентов жидкостями (процессы промывки). Промывку используют в некоторых методиках очистки отходящих газов, образующихся в мусоросжигающих печах.
Чрезвычайно эффективное разделение обеспечивает газовая хроматография, где имеет место распределение газообразных компонентов, например между газом и жидкой фазой, нанесенной иа твердую подложку (например, капилляры со специальным покрытием на стенках в капиллярной газовой хроматографии или специальный набивочный материал в газовой хроматографии с набивными колонками).
Термины газ и пар часто используются взаимозаменяемо. Для систем, в которых соединение существует при комнатной температуре в жидком или твердом ваде, чаще используется термин «пар», в то время как термин «газ» используется в том случае, когда вещество находится при комнатной температуре в газообразном состоянии, т. е. выше температуры кипения.
Давление пара
В открытой системе жидкость находится в равновесии со своим паром и наг блюдаемое равновесное давление пара зависит от температуры и природы жидкости. Давление пара достигает атмосферного давления в точке кипения. На рис. 4.5-3 представлены зависимости давления паров этанола, воды и н-октана от температуры.
Бинарные смеси
Температура кипения бинарной смеси двух полностью смешивающихся жидкостей зависит от состава жидкой фазы. Определим отношение концентраций компонентов в жидкой фазе как
Vs- (4.5-15)
Ав
а отношение концентраций в газовой фазе как
(4.5-16)
4.5. Гетерогенные равновесия
193
Рис. 4.5*3. Зависимости давления паров этанола, воды и н-октана от температуры.
где Ха, Хв, Ya и Yb — мольные доли А и В в жидкой и газовой фазе соответственно.
Теперь мы можем рассчитать относительную летучесть а следующим образом:
Ya_ Ха
Yb ^Хв
(4.5-17)
или
а =
YaXb
YbXa
(4.5-17а)
а — мера обогащения паровой фазы веществом В.
Заметим, что А и В выбираются таким образом, что а > 1. Обычно наблюдаемые на практике значения а находятся в двапазоне от 1,0 до 5,0. Коэффициент а можно определить и как отношение равновесных давлений паров веществ А и В при данной температуре:
Й.
Рв
(4.5-18)
7 Аналитическая химия. Том 1
184 Глава 4. Основы химических методов анализа
Из данных, представленных на рис. 4.5-3, мы можем непосредственно определить значения а бинарных смесей вода-этанол и этаиол-х-октан. Для двух температур 70 и 80°С получаем следующие значения:
этанол—вода а = 2,2 и 2,3 соответственно
этанол — н-октан а = 4,4 и 4,5 соответственно
Если пар в ходе дистилляционного процесса конденсируется, то получается жидкая фаза с более высокой концентрацией летучего компонента по сравнению с исходной смесью. Это прекрасно видно на диаграмме, демонстрирующей составы жидкой и паровой фаз смеси (рис. 4.5-4).
Рассмотрим гипотетическую бинарную смесь, состоящую из компонентов А и В. В чистом виде А и В имеют точки кипения при 105 и 80°С соответственно. Следовательно, летучесть компонента В выше. В соответствии с диаграммой на рис. 4.5-4 температура кипения смеси состава 10% В —90% А равна 99,5°С (точка 1). Паровая фаза при этой температуре имеет следующий состав: 36,5% В —63,5% А (точка 2). Если эта паровая фаза конденсируется, то температура кипения образующейся жидкой фазы составляет 92°С (точка 3). При последующем испарении этой жидкой фазы состав образующейся паровой
Состав фаз
Рис. 4.5-4. Фазовая диаграмма с составом жидкости и пара; а—температура кипения чистого А; b—температура кипения чистого В.
4.5. Гетерогенные равновесия
195
фазы будет: 30% А — 70% В (точка 4). При осуществлении нового цикла конденсации паровой фазы данного состава получится жвдкость с температурой кипения, равной 85°С (точка 5), последующее испарение которой приведет к образованию пара, имеющего состав 90% А—10% В (точка 6).
Приведенные выше рассуждения справедливы в том случае, если объем исходной смеси существенно превышает объем собранного дистиллята. Обычно при нагревании исходная смесь меняет свой состав в сторону низких значений концентрации более летучего компонента. При этом наблюдается непрерывный сдвиг температуры кипения смеси в сторону более высоких значений, указывающий на то, что концентрация летучего компонента в дистилляте уменьшается. Обогащение исходной модельной смеси компонентом В (от 10 до 90%) может быть реализовано путем проведения трех последовательных операций дистилляции. Модельная система, используемая для объяснения процесса дистилляции, была выбрана произвольно. В принципе, диаграммы, отражающие зависимость состава жидкой и паровой фаз от температуры, по внешнему виду могут отличаться от диаграммы, приведенной на рис. 4.5-4. На рис. 4.5-5 представлена фазовая диаграмма бинарной смеси вода-этанол. Эта система является
° м «о во м «о Вода, % (масс.)
100 во *о «о м о Этанол, % (масс.)
Рис. 4.5-5. Фазовая диаграмма бинарной смеси вода-этанол.
196
Глава 4. Основы химических методов анализа
Рис. 4.5-6. Простейшая лабораторная установка для дистилляции.
примером того, что не всегда в процессе дистилляции можно получить чистые жидкости. Состав паровой и жидкой фаз для смеси, содержащей 96% этанола и 4% воды, одинаков, а температура кипения такой смеси принимает наименьшее из всех возможных значений. В этом случае говорят, что этанол и вода образуют азеотропную смесь, разделить которую дистилляцией невозможно.
Этот факт может быть использован для приготовления вполне определенных растворе®. Например, хлороводородная кислота и вода образуют раствор, называемый постоянно кипящей хлороводородной кислотой. Ее легко приготовить путем дистилляции в простом аппарате (рис. 4.5-6) и можно использовать в качестве первичного стандарта для кислотно-основного титрования. Она может долго храниться без изменения состава. Постоянно кипящую НС1 готовят дистилляцией НС1 с плотностью 1,18 (~ 38% НС1) при скорости 34 мл/мин, отбраковывая первые 75% и последние 5-10% дистиллята. В табл. 4.5-2 перечислены свойства постоянно кипящей НС1.
Теоретические тарелки
В нашем теоретическом эксперименте по дистилляции (рис-4.5-4) стадии, Включающие испарение жидкой фазы и последующую конденсацию пара, выполнялись В виде последовательных шагов. В сложном дистилляционном оборудовании такие стадии частично перекрываются и при этом улучшается эф-фективность разделения- Воображаемые индивидуальные стадии называются теоретическими тарелками. Теоретическая тарелка может быть определена Как воображаемое устройство, которое дает то же различие в составе, что су-
4.5. Гетерогенные равновесия
197
Таблица 4.5-2. Постоянно кипящая НО
Давление в ходе дистилляции, мм рт. ст. НС1 в дистилляте, % (масс.) Количество граммов дистиллята, содержащее 1 моль НС1
600 20,638 176,55
640 20,507 177,68
680 20,413 178,50
700 20,360 178,96
730 20,293 179,55
740 20,296 179,77
760 20,221 180,19
770 20,197 180,41
780 20,173 180,62
800 20,125 181,05
ществует при равновесии между жидкой смесью и ее паром. Любая часть колонны, которая дает обогащение в составе от точки 1 х 2, от 3 к 4 и от 5 к 6 (рис. 4.5-4), является теоретической тарелкой. Эффективность процесса разделения может быть улучшена путем разработки специальных дистилляторе®. Идея состоит в том, чтобы поместить воображаемые тарелки в аппарат, в котором дистиллят находится в равновесии со своим паром. Для того чтобы обогатить летучий компонент В от 10 до 90% потребуются три стадии, и, следовательно, можно встроить воображаемые тарелки в дистилляционную колонну, где жидкие смеси с 36,5, 70,0 и 90% летучего компонента В находятся в равновесии со своими жидкими фазами. Хотя на самом деле концентрация более летучего компонента изменяется непрерывно вдоль всей длины дистилляционной колонны.
Эффективность тарелки определяется отношением числа реальных теоретических тарелок к числу теоретических. На практике эффективность тарелок лежит в интервале 50-75%. Эффективность дистилляционной колонии определяется ее длиной и числом реальных тарелок. Мерой эффективности разделения служит высота, эквивалентная одной теоретической тарелке (ВЭТТ). В условиях суммарного оттока, в зависимости от конструкции дистиллятора достигаются величины ВЭТТ 1-60 см. Типичный дистиллятор высотой 50 см может создавать 10 теоретических тарелок и обычно имеет величину ВЭТТ 5 см.
Растворимость газов
Газы могут растворяться в жидкостях. Большинство животных, обитающих в океанах или пресных водоемах, для дыхания используют кислород, растворенный в воде.
Газированная вода—это питьевая вода, в которой растворен СОг при. давлении несколько более высоком, чем атмосферное. При атмосферном давлении газ выделяется в виде пузырьков. Из этих наблюдений мы можем извлечь наиболее важные представления о поведении газа, связанном с его раствори-
мостью.
198
Глава 4. Основы химических методов анализа
Растворимость газов в растворителях может быть рассчитана по закону Генри. Уильям Генри (1774-1836), английский физик и химик, установил, что количество газа, абсорбированного жидкостью при данной температуре, пропорционально парциальному давлению газа над жидкостью:
[А]=ра№ (4.5-15а)
где [А] — концентрация газа А, рд — парциальное давление газа, Кд — коэффициент пропорциональности, называемый константой Генри (моль-л-1-атм-1).
4.5.3. Системы твердое вещество-жидкость
Равновесие осаждения
Растворимость твердых веществ в воде и других растворителях—это видимое проявление конкуренции двух различных процессов. Первый, называемый кристаллизацией, — результат связывающих взаимодействий в твердом теле. В ходе кристаллизации происходит рост кристалла в среде, содержащей такие же ионы или молекулы, как и его собственные. Второй процесс, называемый растворением, является результатом взаимодействий молекул растворителя с молекулами или ионами на поверхности кристалла. Это приводит к разрушению кристаллической решетки и образованию все более и более концентрированного раствора до тех пор, пока либо не растворится вся твердая фаза, либо не установится равновесие между конкурентными процессами, твердая фаза насыщенный раствор.
Одиночный, «голый», ион или молекула в растворе находятся не в наиниз-шем энергетическом состоянии и, следовательно, стремятся минимизировать свою энергию путем агрегирования или сольватирования. Их поверхность не насыщена, и ряд потенциальных мест связи вакантны. Под действием межионных или межмолекулярных сил они вступают в реакции с соседними иоиами или молекулами в растворе и на поверхности твердой фазы. Наш интерес сфокусирован на двух процессах, связанных соответственно с кристаллизацией и растворением.
— Образование связей между частицами растворенного вещества. Это приводит к образованию агрегатов и, что более важно, осадка. В этом процессе энергия решетки возрастает. Чем выше энергия решетки, тем больше частиц растворенного вещества удаляются из раствора. Следует иметь в виду, что поверхность осадка остается «ненасыщенной», т. е. ионы или молекулы, занимающие поверхностные места, используют не все связи, которые потенциально могут образовывать со своим окружением (рис. 4.5-7). Следовательно, иа поверхности могут наблюдаться особые эффекты. К ним можно отнести либо адсорбцию и другие поверхностные явления, либо процессы, в которых, молекулы растворителя взаимодействуют с частицами вещества, расположенными на поверхности и переносят их от поверхности в фазу раствора.
— Образование связей между гипотетическими «голыми» частицами растворенного вещества и молекулами воды или молекулами растворите-
4.5. Гетерогенные равновесия
Рис. 4Д5-Т. Модель, демонстрирующая меньшее число связей поверхностных групп по сравнению с группами в объеме твердого тела. Точками обозначены места с ненасыщенными связями на поверхности твердого тела.
199
•А-В-А-В-А-В-А-В-А-В-А-В-А-В-
•В-А-В-А-В-А-В-А-В-А-В-А-В-А-
•А-В-А-В-А-В-А-В-А-В-А-В-А-В-
ля. Это приводит к образованию гидратов или сольватов и сопровождается выделением энергии. Подобные взаимодействия могут простираться на несколько гидратных оболочек, благодаря чему, в частности, небольшие ионы могут внедряться в структуру растворителя.
В растворе существует конкуренция между процессами кристаллизации и сольватации. В большинстве случаев гидратационные или сольватационные связи слабее, чем взаимодействие твердое вещество—твердое вещество, но обычно число таких связей больше числа связей молекулярной или ионной структурной единицы в твердой фазе. Процесс растворения может быть экзо-и эндотермическим. Последнее возможно, так как в процессе растворения энтропия возрастает. Возрастание энтропии можно объяснить тем, что раствор представляет более беспорядочно распределенную систему в отличие от высоко упорядоченного твердого состояния. В процессе растворения природа растворителя играет важную роль. Вода может оказывать большое влияние на силы межионного взаимодействия ионной твердой фазы, поскольку в среде с высокой диэлектрической проницаемостью кулоновские силы между ионами значительно ослаблены.
Оба конкурирующих процесса определяют термодинамическое равновесие, которое сдвигается в сторону образования продукта с более низкой энергией. Однако имеет место постоянный обмен частиц растворенного вещества на поверхности твердого вещества. Это может быть продемонстрировано с помощью радиохимического метода. Когда достигается состояние равновесия, можно применить закон действующих масс и охарактеризовать равновесие константой равновесия. Эта константа должна быть связана с растворимостью твердого вещества в жидкой фазе. Вальтер Нернст (1864-1941) в 1889 г. впервые определил таким образом константу равновесия. Он назвал ее произведением растворимости К8р.
Перед детальным рассмотрением этой величины необходимо рассмотреть общее влияние ионных и ковалентных связей на растворимость. В ионной кристаллической решетке составляющие ее ионы противоположно заряжены и удерживаются вместе электростатическими силами. Для таких ионных кристаллов характерны хорошая растворимость в воде и слабая растворимость в неполярных растворителях. Такое поведение называется солеподобным или солевым. Твердые вещества с преимущественно ионными взаимодействиями называются солями. Таким образом, растворимость многих солей уменьшается при добавлении органического растворителя к водному раствору- Этот факт часто используется в гравиметрическом анализе. Как правило, все соли явля
200
Глава 4. Основы химических методов анализа
ются сильными электролитами, и окраска твердого вещества или его водных растворов является окраской индивидуальных ионов.
Твердые решетки, образованные ковалентными силами, обычно проявляют слабую растворимость в воде. Решетку составляют молекулярные группы или комплексные, а не простые ионы. Эти соединения часто являются слабыми электролитами, и имеют тенденцию подвергаться реакциям комплексообразования в водных растворах. Нередко окраска твердого вещества отличается от окраски раствора. Подобное поведение демонстрируют большинство сульфидов тяжелых металлов, например, {HgS} — красный или черный; {CdS} — желтый; {HgI2J — красный.
В следующем разделе будет обсуждена связь между растворимостью и произведением растворимости. Мы увидим, что растворимость ие всегда может быть рассчитана из произведения растворимости. Пернет изначально определил произведение растворимости с некоторыми оговорками, указывая на то, что растворимость, рассчитанная с помощью произведения растворимости, поддается интерпретации только в том случае, если раствор, находящийся в равновесии с твердой фазой, содержит составные части твердой фазы исключительно в форме свободных ионов. Сванте Аррениус к тому моменту уже разработал теорию сильных и слабых электролитов и показал, что даже слабые электролиты полностью диссоциируют в разбавленном растворе. Вскоре после того, как Нернст опубликовал свои идеи, Нильс Бьеррум ввел концепцию полной диссоциации для ионных соединений (солей) в растворах электролитов и необходимость в оговорках Нериста отпала. В течение последующих лет различие между ионными и ковалентными соединениями не воспринималось всерьез. Полагали, что вещества с низкой растворимостью (ковалентные вещества) всегда полностью диссоциированы в растворе, так как благодаря их низкой растворимости их концентрация в растворе очень низка. Следовательно, их также рассматривали как соли. Как мы увидим позже, это совершенно неверно, и это неправильное представление часто вызывает некоторую путаницу, особенно для неопытного студента.
Знание характеристик растворителя и растворенного вещества позволяет химику устанавливать ряд правил, которые полезны при использовании равновесий осаждения. Так, полезно знать, что все нитраты, большинство перхлоратов ({КСЮ4} и {NH4CIO4} —исключения) и почти все соли щелочных металлов очень хорошо растворимы в воде.
Растворимость и произведение растворимости
Растворимость s вещества в воде определяется как масса растворенного твердого вещества, находящегося в равновесии с избытком твердой фазы1. Говорят, что такой раствор является насыщенным. Растворимость обычно зависит от температуры. Необходимо иметь в виду, что насыщенные и концентрированные растворы ие одно и то же. Например, слабая растворимость хлорида се
1 Речь идет о массе растворенного твердого вещества, отнесенной к определенному объему раствора. — Прим. ред.
4.5. Гетерогенные равновесия 201
ребра в воде приводит к насыщенному раствору с концентрацией AgCl порядка 10"5 моль/л (при 25°С), который, очевидно, не является концентрированным..
Растворимость может быть выражена разным образом. Для химика-аналитика наиболее удобна молярная шкала, т. е. моли иа литр, но другие единицы, такие, как грамм иа 100 мл растворителя (например, в Handbook of Chemistry and Physics использована эта шкала) тоже часто используются. Если мы предположим, что растворенное вещество полностью диссоциировано (сильные электролиты, соли и т. д.), то двухфазная система может быть описана следующим равновесием, в котором хлорид натрия взят как пример сильного электролита:
{NaCl} Na+ + СГ (4.5-19а)
Соответствующая константа равновесия определяется следующим выражением:
[Na+][C1~] [{NaCl}J
При записи выражения для константы равновесия используются те же понятия активности, что и введенные в разд. 4.1. Активность чистой твердой фазы всегда равна единице по определению. В результате этого упрощения получаем соотношение, известное как произведение растворимости К8р:
Квр = [Na+ЦСГ] (4.5-21а)
Если заряды ионов неодинаковы, соответствующее уравнения становится более сложным. Для соли общего состава МаВь, диссоциирующей в водном растворе на а молей ионов Mv+ и b молей ионов В^~, при осаждении устанавливается следующее равновесие:
{МоВь}^аМ*'++ЬВ'3- равновесие осаждения (4.5-196)
а произведение растворимости определяется как
к,р = [М*'+]“[В^“]Ь (4.5-216)
В условиях полной диссоциации и при отсутствии в исходной водной фазе катионов М*'+ и анионов В^3- каждый моль растворенной соли дает а молей катионов и b молей анионов. Растворимость s, выраженная в молях твердой фазы иа литр раствора (а ие в терминах концентраций индивидуальных ионов), связана с концентрацией ионов Mv+ и В^3-:
[M"+]=as (4.5-22)
и
[В^“] = bs (4.5-23)
где s выражено в моль/л. Произведение растворимости может быть определено как
Квр « [М‘/+]°[В^-]Ь = (as)a(bs)b = a°bbsa+b (4.5-21в)
202
Глава 4. Основы химических методов анализа
откуда получаем
□ Произведение растворимости—особая форма закона действующих масс, использование которой позволяет рассчитать растворимость данного соединения в воде при определенных условиях.
Собственно говоря, это отношение связывает растворимость и произведение растворимости только при определенных условиях (оговорка Нериста; см. выше) и только если учтены следующие соображения:
— Связь «растворимость — произведение растворимости» имеет силу, только если растворенное соединение полностью диссоциировано. Если в растворе существуют недиссоциированное вещество или любая его ассоциированная форма либо если катионы и анионы, получившиеся при растворении, образуют комплексы, то растворимость больше, чем величина, рассчитанная из произведения растворимости.
— Если исходная водная фаза содержит любой из ионов, получающийся при растворении твердого вещества (общие ионы), то растворимость меньше, чем величина, рассчитанная из произведения растворимости. Однако закон постоянства ионного произведения (произведение растворимости — особый случай ионного произведения) сохраняется, и, таким образом, концентрация общего иона становится больше, а концентрация его противоиона—меньше, чем значения, рассчитанные иа основе предположения о том, что никакого эффекта общего иона не существует.
- Изменение коэффициентов активности влияет на растворимость электролита. Обычно коэффициенты активности уменьшаются с ростом общей концентрации инертного электролита (ионной силы) и, соответственно, растворимость становится больше.
Растворимость з не является термодинамической величиной, однако произведение растворимости — термодинамическая характеристика гетерогенного равновесия при условии, что используются надлежащие концепции активности (теория Дебая—Хюккеля, постоянная ионная сила и т. д.).
Необходимо отметить, что произведение растворимости Кйр не является безразмерной величиной. Для 1:1 электролита она имеет размерность моль2-л“2, для 1:2 электролита —моль3-л-3 и т. д. Следовательно, бессмысленно сравнивать произведения растворимости электролитов с различной стехиометрией (или различными процессами диссоциации).
На растворимость вещества влияют различные факторы, которые будут обсуждаться в последующих разделах.
Влияние температуры
На растворимость и произведение растворимости твердого вещества влияют изменения температуры. Связи в кристаллической решетке твердого вещества,
4.5. Гетерогенные равновесия
Рис. 4.5-8. Зависимость диэлектрической проницаемости воды от температуры при атмосферном давлении.
как и связи и структура водного раствора, ослабевают при повышении температуры. Также с ростом температуры уменьшается диэлектрическая проницаемость воды (рис. 4.5-8). Все эти явления влияют на растворимость солей и являются причиной уменьшения растворимости многих сульфатов (сульфатов редкоземельных металлов, кальция и лития), ацетатов (ацетата кальция) и карбонатов (карбоната лития). Однако для большинства систем действие этого эффекта компенсируется процессом гидратации. Концентрация свободной «мономерной» воды (активность воды) возрастает с разрушением структуры воды, вызванным повышением температуры. Следовательно, в горячих и холодных растворах возможно существование различных гидратов. Вследствие этого растворимость может сильно изменяться с изменением температуры. Такой случай проиллюстрирован на рис. 4.5-9 на примере NaaSO.^. Большинство солеподобных электролитов проявляют в горячих растворах более высокую растворимость, чем в холодных растворах. Это противоположно поведению газов при растворении. Некоторые соли демонстрируют чрезвычайно высокий температурный эффект. В ряде случаев в горячей воде (при температуре, близкой к температуре кипения) растворяется почти в 50 раз больше соли, чем в
204
Глава 4. Основы химических методов анализа
Температура, °C
Рис. 4.5-9. Зависимости растворимости NaaSOa, NaaSCU - ЮН3О, NaCl, MgS04 и MgCl2 от температуры.
холодной (при температуре, близкой к температуре замерзания). Такой большой температурный эффект имеет бура (ПагВдОт-ЮНгО), компонент многих моющих средств. Однако есть и другие вещества, обладающие низкой растворимостью в растворах при повышенной температуре и демонстрирующие поведение, аналогичное поведению газов. Интересно отметить, что хлорид натрия имеет приблизительно одинаковую растворимость во всем температурном интервале 0 — 100°С. Различное влияние температуры на электролиты может быть использовано в ряде синтезов и аналитических процессов.
Влияние размеров частицы на растворимость
Небольшие частицы твердого вещества находятся в менее выгодном энергетическом состоянии, чем крупные. Это обусловлено тем, что их поверхность относительно велика при их небольшом объеме. На рис. 4.5-7 показано, что поверхность твердого тела находится в химически ненасыщенном, и, следовательно, энергетически менее выгодном состоянии по сравнению с насыщенной внутренней частью кристалла. В крупных телах только относительно небольшая доля атомов или молекул занимают места на поверхности. В отличие от этого, тела с малыми размерами обладают значительной долей атомов или молекул, находящихся на поверхности. Следовательно, в этом случае вклад энергии, приходящейся на поверхностные атомы или молекулы, в общую энергию тела оказывается больше. Поскольку энергии тел с разными геометрическими. размерами различны, указанные тела должны по-разному вести себя и при растворении. Необходимо провести различие между так называемой макро- и микрорастворимостью (рис. 4.5-10). Как правило, неизменная обычная (мак-ро)растворимость наблюдается в том случае, если размер частицы превышает в диаметре ~ 10“3 мм, в то время как растворимость для меньших кристаллов
4.5. Гетерогенные равновесия
205
Рис. 4.5-10. Микро- и макрорастворимость кристаллического вещества.
Микрораст-
воримость
Макрорвстворимость
Размер частицы
зависит от размера частицы. Так, хромат свинца РЬСгО4 слабо растворим в воде. Его растворимость (макрорастворимость) —1,24 • 10-4 моль/л. Это значение найдено для осадка со средним размером частиц 3,0 • 10~2 мм. Однако, осадок со средним размером частиц ~ 9,0 «10-4 мм характеризуется величиной растворимости 2,1 • 10~4 моль/л. Отношение микро- и макрорастворимости:
«МИКрО _ 2,1 • 10 4 _ । gg
«макро 1,24 • 10“4 " ’
Аналогичный расчет может быть выполнен для произведения растворимости:
Квр = s2
Для больших частиц
К,р = (1,24 10-4)2 = 1,54 • 10“6 моль2 • л“2 а для частиц с диаметром 9 - 10-4 мм:
К,р = (2,10 10-4)2 = 4,41 • 10-8моль2 • л-2
Кяр,микрО _4,41 • 10 8 _
“ 1,54-10-8- ’
Эти большие различия должны представлять интерес для химика-аналитика, применяющего гравиметрические методы. Следовательно, важно изучить условия получения макроскопических осадков. Из насыщенных и пересыщенных растворов образуются кристаллы различных размеров.
Системы с низкой растворимостью при выпадении осадка могут образовывать большое число центров кристаллизации. В результате получается мелкокристаллический осадок, поскольку каждая образующаяся частица твердой фазы имеет в близком соседстве много твердых частиц й только небольшой объем раствора, из которого она извлекает ионы для своего роста. Если растворимость высокая, каждый из небольшого числа вновь образовавшихся кристаллов может расти до значительных размеров. Вещества с низким произведением растворимости, такие, как AgCl, BaSC>4, используются в гравиметрических методах. Поэтому необходимо обратить внимание иа процедуру, приводящую к образованию макроскопических осадков. Обычно следует учитывать, что большие кристаллы получаются из растворов, которые:
— не сильно пересыщенные (избегать высоких концентраций);
— разбавленные;
- горячие;
— медленно охлаждаются.
206
Глава 4. Основы химических методов анализа
Эффект общего иона
До сих пор мы рассматривали растворимость электролита, в котором твердая фаза находится в равновесии с чистой водой. Необходимо помнить, что не имеет значения, как достигается состояние равновесия. Один и тот же результат получается и в случае, когда твердое тело находится в равновесии с определенным водным раствором, и когда твердое тело сформировано в реакции осаждения при добавлении химических веществ. На практике осаждение вызывается растворами, содержащими в избытке осаждающий реагент.
Ясно, что растворимость твердого вещества в насыщенном растворе равна нулю, и что она мала в растворе, который содержит ионы этого вещества (общие ионы), но еще не насыщен. Аналогично этому мы обычно ожидаем уменьшения растворимости в системах, в которых только один из составляющих ионов присутствует в исходном растворе. Этот результат может быть получен путем применения принципа Ле Шателъе к следующему равновесию:
{МаВь} оМ|/+ + ЬВ0~ (4.5-196)
В соответствии с принципом Ле Шателъе, когда М*'+ или В^- находятся в избытке в растворе, равновесие сдвигается влево. Это приводит к уменьшению растворимости по сравнению с системой, в которой М*'+ или В&~ отсутствуют в исходном растворе.
Основываясь на этих рассуждениях, можно сделать заключение, что эффект общего иона приводит к уменьшению растворимости. Однако этот качественный вывод должен быть подкреплен количественными соотношениями, касающимися растворимости систем с общим ионом и полученными на основе выражения для произведения растворимости. В математическом плане эта задача весьма сложна (за исключением систем, содержащих 1:1, 2:2 или 3:3 электролиты), поскольку требует использования уравнений высокого порядка. Обозначим концентрацию общего иона в исходном растворе как с. Тогда растворимость s может быть рассчитана с помощью выражения для произведения растворимости. Рассмотрим бинарный электролит, содержащий катион и анион в соотношении 1:1. Не имеет значения, какой ион мы выберем в качестве общего. Допустим, что это анион. Концентрации катиона в равновесной системе, следовательно, равна з, а концентрация аниона с 4- в. Таким образом,
К8р = [Mv+] [В^~] = s(s 4- с) = з2 4- зс ’ (4.5-21г)
Для п:п-электролита (где п = 1,2,3, и т.д.) и = /3. Перегруппировывая, получаем:
в2 4- зс — Кер = 0
или
—с±у/с2 4-4KsP
(4.5-246)
2
4.5. Гетерогенные равновесия
207
Расчеты растворимости
Произведение растворимости хлорида серебра {AgCl} равно
Кар = 1,0 • 1О"10 моль2 • л"2 (4.5-25)
В чистой воде это дает растворимость 1,0 - 10~5 моль/л.
Рассмотрим расчет растворимости хлорида серебра в растворе, исходно содержащем 10~3, 10~5 и 1(Г7 моль/л хлорид-ионов, введенных в виде {NaC!}. Можно предположить, что небольшие концентрации электролита не влияют на коэффициенты активности хлорида серебра и что они остаются в основном постоянными.
Случай 1
[СГ] = 10“3 моль/л = с
Расчет дает:
К8р = 1(T10 = s(s 4- с) = s2 + sc
_ -10-3 ± х/10“6+4>°-10-10 _ (1,00020-1,00000) 10“3 _
®“ 2 2
= 1,0 • 10 ~7 моль/л.
Случай 2
[СГ] = 10-5моль/л = с
Расчет дает:
-10"6 ± 0О-10 + 4,0 • Ю-10 (2,2361 - 1,00000) • 10"5 _
S~ 2 2
= 6,1805 • 10~6моль/л.
Случай 3
[СГ ] = 10-7моль/л = с
Расчет дает:
__ -10’7 ± ^/10"14 4-4,0-10”10 (2,000025 - 0,0100000) • 10"Б _
®“ 2 ” 2
= 9,95 • 10-6моль/л
Влияние побочных равновесий а растворе: гидролиз и комплексообразование
□ Гидролмз и комплексообразование могут вносить большой вклад в реальную растворимость веществ.
Растворимость твердого вещества больше, чем растворимость, рассчитанная из выражения для произведения растворимости, если составляющие его ионы (один тип или более) участвуют в двухфазном распределении или в побочных равновесных процессах в водной фазе — реакциях гидролиза и комплексообразования.
2ОТ
Глава 4. Основы химических методов анализа
Рассмотрим, например, сульфид таллия {TfeS}, для которого '
Квр = 7,0 - 10-23моль3 • л-3 при 25°С (4.5-26)
Из этой величины можно рассчитать растворимость соли в чистой воде: з — 2,60 • 10“8 моль/л. Одиако наблюдаемая в действительности растворимость существенно выше. Одновременно с растворением соли наблюдается увеличение pH. Очевидно, приведенный расчет не вполне корректен, поскольку основывается на предположении о существовании в нейтральном водном растворе только первичных ионов Т1+ и S2-. Это предположение, действительно, неверно, так как сульфид-ион взаимодействует с растворителем:
S2“ + Н2О <=? HS" + ОН" (4.5-27)
HS + Н2О H2S 4- ОН“ (4.5-28)
Эти равновесия описываются соответствующими константами равновесия Kj и К2:
[HS-HOH-] “ [S2-] (4.5-29)
[H2S][OH~] (4.5-30)
[HS ]
Учитывая, что
= [Н+][ОН~] = 10“14 (4.2-45)
получим константы кислотности Кд.1 и Кад:
[Н2&] (4.5-31)
[H+][S2-] м Ка’2- [HS-] 10 (4.5-32)
Отсюда:
Ki = = 1,00 ^А,2 (4.5-29а)
и к к2=^- = 1о-7 Кд.1 (4.5-30а)
Выражение для Ki показывает, что при образовании гидросульфид-ионов (HS") получается равное количество гидроксид-ноиов (ОН~), сдвигающих pH
в сторону более высоких значений. В области pH 9-12 анионы HS преоблада-
ют, что позволяет пренебречь образованием H2S. Можно записать:
[HS-]»[S2-] и [HS-] = 1[Т1+] = [ОН-]
Следовательно,
+ 2-10-»
Р 1 - [т1+]
4.5. Гетерогенные равновесия •
209
Подстановка в выражение для Кд,2 дает-
[HS-] 14 = [Т1+ЦТ1+1 = [Т1+]2
[S2]{H+1 4-10-14[S2-] 4 1O-14[S2-]
Таким образом,
[S2-] = j[Tl+]2
и
[T1+]2[S2-] = К.„ = 70 10~24
ИЛИ
j[Tl+)4 = 7,0-10-23
Окончательно имеем:
[Т1+] = 4,09 - 10-6моль/л
[ОН-] = [SH-] = 2,05 • 10-6моль/л
[S2-] = 4,18 - 10-12моль/л [Н+] = 4,88 - 10-9моль/л
□ Вблизи pH 9 эффективная растворимость TlaS приблизительно в 100 раз больше, чем величина, рассчитанная из произведения растворимости в пренебрежении гидролизом.
Эффективная растворимость (s — 2,05-10 6 моль/л) приблизительно в 100 раз выше растворимости, рассчитанной из произведения растворимости, если пренебречь взаимодействием с растворителем. В этом расчете продемонстрирован тот факт, что реальная равновесная концентрация свободного сульфид-иона [S2-] ниже ожидаемой величины, поскольку этот ион выводится из раствора за счет реакции с растворителем. Концентрация иона таллия в растворе при этом существенно возрастает.
Равновесие, описываемое уравнением 4.5-196, всегда сдвигается вправо, если в водном растворе ионы М"+ или В^— вступают в реакции с растворителем (гидролиз), противоионом (ассоциация ионов) или с посторонними веществами, присутствующими в системе (комплексообразование). Если ионы М1'4' или В^~ являются акцепторами или донорами протонов, растворимость становится зависимой от величины pH раствора. Очевидно, что растворимость соли должна расти с уменьшением pH, если ее ион обладает основными свойствами (акцептор протонов). Карбонаты, фосфаты, бораты, оксалаты, сульфиты, сульфиды, фториды, ацетаты и хроматы демонстрируют повышенную растворимость в растворах с низким pH, поскольку анионы СО|", РО4-, В (ОН)4 или BOJ, С2О2-, SO2", S2~, F-, О Ас- и СгО2" проявляют основные свойства. Следует отметить, что хромат-иои не образует форму гидрохромата, однако для дихромат-иона должна быть рассмотрена кислая форма хромата в соответствии с уравнениями:
2СгО2" + 2Н+^ 2НСгО4 Сг2О2" + Н2О (4.5-33)
210
Глава 4. Основы химических методов анализа
Большинство трехзарядных катионов, таких, как Fe3+ и А13+, взаимодействуя с растворителем, проявляют протонодоиорные свойства. Более наглядно это проявляется, если записать эти катионы в виде Fe(H2O)|+ и А1(Н2О)|+. При отщеплении иона водорода образуются соответствующие гидроксбкомплексы. Следовательно, растворимость должна возрастать с ростом pH.
M3+ + iH2O₽M(OH)?-i + iH+ (4.5-34а)
что можно записать как
М3+ + ЮН* М(ОН)3-‘ (4.5-346)
Ионы Сг(Ш), Fe(III), А1(Ш), Bi(III) и Sb(III) осаждаются в виде гидроксидов при значениях pH выше 5 ([ОН-] > Ю-9 моль/л). Однако растворимость Сг(ОН)з, А1(ОН)з, Sb(OH)s, а также Zn(OH)2 увеличивается при высоких значениях pH. Объяснить этот факт можно на примере гидроксида цинка протеканием в растворе следующей реакции:
{Zn(OH)2} +2ОН“ Zn(OH)3“ (4.5-35)
Осадок Растворенная
форма
При образовании некоторых гидроксокомплексов происходит выделение воды. В этом случае выявление зависимости растворимости от значения pH раствора, очевидно, осложнено:
Сг(ОН)7 СгО(ОН)" 4- Н2О (4.5-36)
А1(ОН) “ А1О2 4- 2Н2О (4.5-37)
Растворимость осадка также повышается, если один из составляющих ионов подвергается комплексообразованию в присутствии добавленного реагента. Так, хлорид серебра растворяется в растворе, содержащем аммиак или избыток цианид- или тиосульфат-ионов:
{AgCl} 4- 2NH3 г± Ag(NH3)} 4- СГ (4.5-38)
{AgCl} 4- 2CN" Г Ag(CN)a 4- СГ (4.5-39)
{AgCl} 4- 2S2O1- Ag(S2O3)|- 4- СГ (4.5-40)
Во всех этих системах концентрация свободных ионов серебра [Ag+] существенно уменьшается, так что произведение равновесных концентраций ионов [Ag+][C1“] становится меньше, чем произведение растворимости Кар. Рассмотрим одну систему более детально.
Пример
К ~ 10“4 моль твердого хлорида серебра (около 15 мг) добавляют 100 мл 0,5 М раствора аммиака. При этом образуется комплекс Ag(NH3)£. Константа образования этого комплекса:
А = = (4М1)
4.5. Гетерогенные равновесия
211
Установлено, что осадок растворяется полностью, и практически все растворенное серебро присутствует в растворе в виде комплекса Ag(NHa)J (10~3 моль/л). Можно рассчитать концентрацию свободного иона серебра следующим образом:
ft - .. Д7ЙА2 = 16 •10? = 107 20 [Ag+](0,5)2
Отсюда
'А*+1 = 0,25Tb-10^ 2-510~И = 10'В’6°моль/л
Концентрация свободных хлорид-ионов составляет 10"3 моль/л, а произведение равновесных концентраций (Ag+][C1~J — величину 2,5 • 10"13 моль2-л-2, которая значительно ниже произведения растворимости (1О“10 моль2-л"2). Следовательно, весь хлорид серебра находится в растворе, и никакой твердой фазы не остается.
Если эксперимент выполняется с бромидом серебра, значение произведения равновесных концентраций ионов (10~12,60 моль2 л"2) является величиной того же порядка, что и произведение растворимости:
К.р = [Ag+HBr“l = 10-12’3°моль2 • л-2 (4.5-42)
Следовательно, осадок слаборастворим. Однако можно видеть, что бромид серебра легко растворяется в концентрированном аммиаке (около 15 моль/л). Для этой концентрации аммиака концентрация ионов серебра рассчитывается в соответствии с :
'А*+1 = (18,0W(P = 22ГТГЖ = 2'77 •10'13 = 10”2’“
и
[Ag+]Br“] = 2,77 • 10“16 = 10-“-“ моль2 • л~2
Полученное значение существенно ниже величины произведения растворимости. Если концентрированным аммиаком обработать иодид серебра, то твердая фаза не растворится. Произведение растворимости иодида серебра:
к.„ = [Ag+J[I“] = 10-“'3“ (4.5-43)
Это значение существенно меньше рассчитанного выше произведения равновесных концентраций ([Ag+J[I-J = ю-1Б,Б6 моль2-л"2).
Этот вид комплексообразования может использоваться в гравиметрических методах. Рассмотрим присутствующую в растворе смесь хлоридов, бромидов и иодидов, которые должны быть отделены друг от друга. Галогениды легко выделяются при осаждении нитратом серебра. При обработке осадка разбавленным раствором аммиака удаляется только хлорид серебра, в то время как бромид и иодид остаются в твердой фазе. После отделения раствора от осадка бромидов и иодидов и удаления аммиака выпариванием можно вновь осадить хлорид серебра (если выпаривание осуществляется медленно и осторожно, то хлорид серебра выпадает в мелкокристаллической форме). При последующей обработке первого осадка, содержащего бромид и иодид серебра, концентрированным аммиаком растворяется бромид серебра и таким образом отделяется от
Глава 4. Основы химических методов анализа
212
иодида серебра. Бромид также может быть получен в твердой форме при выпаривании аммиака. Итак, можно достичь количественного разделения всех трех галогенидов.
Процессы комплексообразования, приводящие к увеличению растворимости, не ограничиваются взаимодействием только одного из компонентов твердого вещества. В конце концов, те же силы, которые обеспечивают нерастворимость осадков (энергия кристаллической решетки, т. е. сильные связи в твердом веществе, которое можно рассматривать как полиядерный комплекс), могут действовать в противоположном направлении, способствуя образованию растворимых бинарных комплексов или высших гомологов (заряженные комплексные ионы). Следующий пример иллюстрирует это утверждение.
Пример
□ Кривая растворимости как функция от р[С1~] для AgCI имеет сложный вид (см. рис. 43-11).
Для хлорида серебра известны следующие константы комплексообразования. Они относятся к гомогенному равновесному состоянию (т. е. AgCI представляет собой мономолекулярное соединение, которое не следует путать с твердофазным
Рис. 4.5-11. Логарифмический график общей растворимости хлорида серебра в зависимости от концентрации хлорид-ионов, контактирующих с твердым {AgCI}. Прямые линии представляют вклад каждого из комплексе» в общую растворимость.
4.5. Гетерогенные равновесия
213
веществом {AgCI}).
Ag++ СГ AgCI, Кг [Д-]-103,00 (4.5-44)
AgCI + СГ = AgCI 7, Кз = 1О2’30 .(4.5-45)
AgClJ + СГ AgCl|“, Кз = 10°’“ (4.5-46)
AgCg^ +Cl- г: AgCI3-, Kt = 10-°“ (4.5-47)
Эти константы равновесия описывают гомогенные равновесия, однако нас интересуют гетерогенные системы, содержащие твердую фазу AgCI. С помощью Ki можно рассчитать концентрацию молекулярного AgCI, используя значение произведения растворимости AgCI, определенное для равновесия твердого {AgCI} с водным раствором:
где
[Ag+ЦСГ] = JO-10,0
Следовательно,
[AgCI] - 1О3’00 - Ю"10,0 - 1О~7,0
Последняя величина — термодинамическая константа, как и Кяр> и не зависит от концентрации хлорид-иона! Другими словами, если твердый хлорид находится в равновесии с водной фазой, мы всегда имеем в растворе 10—7 моль/л молекулярного AgCI. Теперь мы можем рассчитать Кв для гетерогенных равновесий,
устанавливающихся в растворе:
{AgCI} Ag+ + СГ (4.5-19в)
Последнее равновесие описывается произведением растворимости:
К.„ = [Ag+ЦСГ] = (4.5-25)
Равновесие {AgCI} Г* AgCI описывается константой K„r.
KBi = [AgCI] = ИГ7-0 (4.5-48)
Равновесие {AgCI} + СГ AgCl^ описывается константой Кд:
К.з = 1^?1 = K2[Agd] = K2KS1 = IO”4’70 (4.5-49)
М J
Равновесие {AgCI} 4- 2СГ AgCI7 описывается константой Каз:
Мз = = К2Кз[А8СЧ = К-2Кз = 10“3’85 (4-5-50)
Равновесие {AgCI} + ЗСГ AgCI3- описывается константой
К.4 = ^СП3 1 = К2 Кз K1!AgC4 = К‘зК1 = 10-4,50 (4.5-51)
214
Глава 4. Основы химических методов анализа
Теперь можно рассчитать концентрацию всех комплексов, находящихся в водном растворе и равновесии с твердым {AgCl}. Каждый комплекс вносит определенный вклад в общую растворимость. Каждый из этих вкладов описывается прямой линией на соответствующем логарифмическом графике (рис. 4.5-11). Мы цолучаем:
- ^0~1°^°<llg^Ag+^-
1 1 [СГ| dlg[CTJ
[AgCla-] = 10^"|C1-]djgffi~J;
[Agcirj=io-^madyff -[Agci’-| = lo-^cr^W?^!,
-- -1 (наклон1 = —1) (4.5-52)
= 0 (наклон — 0) (4.5-53)
= 1 (наклон = 1) (4.5*54)
= 2 (наклон = 2) (4.5-55)
= 3 (наклон = 3) (4.5-56)
Этот пример иллюстрирует тот факт, что знание значения только произведения растворимости не всегда дает возможность правильно рассчитать растворимость твердой фазы. Значительное увеличение растворимости за пределами определенной концентрации лиганда (в рассматриваемом случае приблизительно [СГ] = Ю 4 моль/л) вызвана комплексообразованием. Этот эффект проявляется особенно сильно для веществ, образующих ковалентные связи, и, следовательно, наблюдается для веществ, получающихся в результате взаимодействия мягкой кислоты и мягкого основания. Эти вещества характеризуются небольшим произведением растворимости. Значительное увеличение растворимости ожидается для сульфидов, селенитов и теллуритов ионов тяжелых металлов.
Безусловно, растворимость {HgS} обусловлена такими комплексами, как Hg(SH)2, HgSSH” и HgS|“, и в широкой области pH растворимость более чем в 1О20 раз больше, чем рассчитанная из произведения растворимости (включая влияние гидролиза S2-). Действительно, сульфид ртути растворим в щелочных сульфидных растворах в макроскопических количествах в форме HgS*-
Известно также, что сульфид серебра образует комплексы в сероводородных и сульфидных растворах. В этих растворах находятся такие формы, как AgSH, Ag(SH)2 и биядерный Ag2S(SH)2-. На рис. 4.5-12 представлена растворимость серебра в 0,02 моль/л сульфидном растворе как функция pH. Это очень сложная система, так как гидросульфид-ион HS“ и сульфид-ион S2- являются лигандами, участвующими в процессах комплексообразования. Концентрация этих лигандов зависит от pH, поскольку они представляют кислотную (HS-) и основную (S2-) формы участника одного и того же кислотноосновного равновесия. Из рис. 4.5-12 видно, что при pH 0, 7 и 13 общая растворимость серебра приблизительно в 104,1014 и 1016 раз выше, чем концентрация
Точнее, тангенс угла наклона. — Прим, перев.
4.5. Гетерогенные равновесия
215
Рис. 4.5-12. Зависимость растворимости {Ag2S} в 0,02 моль/л растворах сульфида от pH. Растворимость не определяется ионами [Ag+], которые преобладают только в нереализуемой области pH < —2. Од нако это результат присутствия трех комплексов серебра: AgSH, Ag(SH)" н HSAgSAgSH2-.
216 Глава 4. Основы химических аталиаа
ионов серебра. На самом деле свободные ионы серебра никогда не существуют как преобладающая форма серебра в практической области pH. Сплошная линия на рис. 4.5-12 представляет молярную растворимость s твердой фазы {Ag2S} и, следовательно, является суммой половинных значений концентраций Ag+, AgSH и Ag(SH)2 и полной Концентрации Ag2S(SH)2--
Известно, что последовательность осаждения сульфидов тяжелых металлов из горячих природных сульфидно- и гидросульфидных вод (серных источников) не отвечает последовательности соответствующих произведений растворимости. Этот фацт может быть объяснен только в предположении об образовании растворимых комплексов, рассмотренных выше. Такие комплексы известны для сульфидов Hg, Ag, Au, Си, Pb, Zn, Cd, In, Ni, Fe, As и Sb.
Ионный обмен
Растворимость твердого вещества в растворителе — вполне определенное свойство всех кристаллических тел. В водных растворах большинство твердых веществ дисссоциируют и существуют в виде катионов и анионов, что описывается ур. 4.5-20.
Если раствор не содержит больше никакого электролита, анионы и катионы присутствуют в растворе в стехиометрическом отношении и растворимость может быть выражена с использованием Клр (ур. 4.5-21). Однако эти закономерности не всегда соблюдаются. Так, многие неорганические вещества ведут себя иначе. Часто анион (например, силикат-анион) является частью нерастворимой кристаллической структуры (матрицы), а катионы компенсируют избыток отрицательного заряда жестко фиксированных анионов. В этом случае катионы удерживаются в кристаллической решетке чисто электростатическими силами. В процессе растворения такие полярные связи могут разрушаться под действием дипольных молекул воды, в то время как ковалентные связи остаются достаточно устойчивыми к этим взаимодействиям.
Поскольку катионы удерживаются в матрице относительно слабыми электростатическими силами, то они занимают в ней дыры, щели или полости и могут легко замещаться другими катионами аналогичного заряда и размера. Однако катионы с большим зарядом и меньшим ионным радиусом удерживаются на твердой поверхности гораздо сильнее, чем катионы с небольшим зарядом и большим ионным радиусом.
Глины относятся к классу минералов. В этом случае стехиометрия соединений не фиксируется и не может быть выражена простыми целыми числами. Эти материалы плохо растворимы, ио могут обменивать определенные катионы в своей решетке на катионы, присутствующие в растворе (например, в морской воде, в которой содержание электролитов составляет ~ 0,7моль/л). Такое замещение называется процессом ионного обмена и может быть охарактеризовано следующим равновесием:
R .. ,Х+ + Y+ ВТ .. .Y+ +Х+
(4.5-57)
4.5. Гетерогенные равновесия
217
и соответствующей константой равновесия Kz
где Yr и Хд —доля мест, занимаемых Y+ й Х+ соответственно.
В современной аналитической практике вместо природных ионообменйиков (минералов типа глины и цеолитов) используются синтетические органические продукты, так называемые ионообменные смолы. Коммерчески доступными являются анионо- и катионообменные смолы различных размеров (ситовое число) и различных торговых марок (Дауэкс, Амберлит и другие). Матрицу этих смол обычно получают полимеризацией стирола и дивинилбензола, формирующих трехмерную структуру, к которой присоединены такие ионные группы, как SO3 (сульфогруппа) или третичные амины (CH2-NR3, где R может быть, например, СН3). Если матрица отрицательно заряжена в растворе, юна ведет себя как катионообменник, если положительно—как анионообменник.
В случае анионообменной смолы положительный заряд встраивается в матрицу и должен компенсироваться отрицательно заряженными ионами, которые легко обмениваются с другими анионами раствора, контактирующего со смолой. Ионообменные смолы используются в разнообразных аналитических методиках. Довольно часто их применяют в хроматографических методах, например в ионной хроматографии в подавляющей колонке.
Если используют смесь катионообменной смолы в Н+-форме (отрицательный заряд матрицы компенсируется ионами водорода) и анионообменник в ОН“-форме (положительный заряд матрицы компенсируется гидроксидионами), все катионы удаляются из раствора и замещаютей ионами Н+, а всё анионы—ионами ОН~. Однако ионы водорода в’растворе взаимодействуют с гидроксид-ионами:
Н+ + ОН- Н2О (4.2-44)
Это равновесие описывается ионным произведением воды:
Kw = [Н+][ОН“] = 10“14 при 25°С (4.2-45)
Поскольку число положительных и отрицательных зарядов в растворе должно быть одинаково (электронейтральность), можно записать:
[Н+] = [ОН-] = 10~7 при 25°С
Следовательно, из раствора могут быть удалены все электролиты, и получающаяся вода будет чрезвычайно чистой. Данный процесс имеет практическое значение, и вода, полученная этим способом, называется деионизованной. Она характеризуется низкой электропроводностью и часто используется предпен чтительнее, чем дистиллированная вода, которая всегда содержит небольшое количество электролитов. В то же время нейтральные вещества, такие, как сахар и т. д., не могут быть удалены с помощью подобного ионообменного процесса.
218 Глава 4. Основы химических методов анализа
4.5.4. Системы жидкость-жидкость
Введение
Методы, основанные на распределении растворенного вещества между несме-шивающимися жидкими фазами, широко используются в аналитических, препаративных промышленных и лабораторных процессах.
В двухфазной системе, состоящей из даух несмепшвающихся жидких фаз, химическое вещество распределяется в соответствии с его относительной растворимостью в индивидуальных фазах. Распределение этого компонента между двумя фазами часто используют для концентрирования или разделения веществ или групп веществ друг от друга или от матрицы. Этот процесс называется жидко-жидкостной экстракцией или просто жидкостной экстракцией.
□ Жидкостная экстракция является эффективным инструментом для концентрирования следовых количеств ионов металлов иэ водных растворов в органические растворители (например. ССЦ) с помощью органических лигандов НА, образующих гидрофобные хелаты металлов МАП.
Препаративный вариант жидкостной экстракции все еще очень распространен в лабораториях органического синтеза. В промышленности активно совершенствуются непрерывные экстракционные процессы и аппаратура контроля за этими процессами. Использование на практике динамических свойств экстракционных систем привело к развитию метода противоточной экстракции.
Во время и после второй мировой войны для разделения редкоземельных металлов и соединений трансурановых элементов были разработаны специальные методы экстракции селективными растворителями. Жидкостная экстракция была самым популярным методом разделения до того, как в арсенале химиков-аналитиков появились более эффективные методы, такие, как хроматография (1950-е-1960-е годы).
Жидкостная экстракция играла важную роль в Манхеттенском проекте, гигантском научно-исследовательском проекте получения расщепляющихся материалов, используемых при создании атомной бомбы. В то время был доступен только один метод получения 239Ри в ядерном реакторе-размножителе. Необходимость производства значительных количеств этого металла потребовало развития химических методов извлечения, работающих в экстремальных условиях.
Сегодня роль жидкостной экстракции среди аналитических методов менее значима, поскольку для реализации этой процедуры требуется достаточно продолжительное время. На смену ей приходят более современные и эффективные методы (см. гл. 5).
Реальная и термодинамическая константы распределения
Распределение растворенного вещества S, находящегося в равновесии между водной фазой и органическим растворителем, может быть описано уравнением
S(aq) S(opr) (4.5-59а)
4.5. Гетерогенные равновесия
219
Совсем не обязательно выбирать воду в качестве одной из фаз. В принципе, можно описать распределение растворенного вещества S между любой парой несмешивающихся растворителей (жидкостью 1 и жидкостью 2):
Si^S2 (4.5-596)
Такие системы описываются константой равновесия:
(4-5'6о)
PJ«q PJ1
где Ко — реальная константа распределения. Определение термодинамической константы распределения требует знания соответствующих коэффициентов активности. Однако они обычно неизвестны, и в качестве первого приближения можно использовать значения равновесной концентрации в соответствующих фазах. Следовательно, полученные величины зависят от выбранных условий и, собственно говоря, не являются постоянными. Константа распределения Kd связана С термодинамической константой распределения следующим образом:
= (4.5-61)
2/aq
где з/ОрГ и т/aq — коэффициенты активности в органической и водной фазах соответственно.
Коэффициент распределения
Используемые на практике экстракционные системы обычно не являются идеальными в термодинамическом смысле. Например, при рассмотрении экстракции кислоты НА из водной в органическую фазу необходимо учитывать существование кислоты в водной фазе в двух формах: диссоциированной и недиссо-циированной. Реальная константа распределения определяется в соответствии с выражением
Kd = <4-5-62)
Одиако информация о формах нахождения вещества вг контактирующих фа-здх обычно отсутствует. Например, в водной фазе кислота НА может сосуществовать с ее диссоциированной формой, а в органической фазе могут образовываться ионные ассоциаты или продукты полимеризации экстрагированного вещества. В практике используют отношение общих концентраций растворенного вещества в каждой из фаз распределительного равновесия, называемое коэффициентом распределения Dc:
& _ [общая концентрация всех форм НА]орг _ [НА]общ, орг 5-63)
[общая концентрация всех форм HA]aq [Н A]O6m,aq
Формы, в которых может находиться распределяющееся между двумя фазами вещество, различаются, и их соотношение часто определяется концентрацией вещества. Поэтому, как правило, величина Dc также оказывается зависимой от концентрации.
220
Глава 4. Основы химических методов анализа
Рассмотр ской фазами водной фазе Для опис нием конста] им- проблему распределения кислоты между водной и органиче-более подробно, предположив, что кислота умеренно сильная в и практически не диссоциирована в органической. алия равновесия в водной фазе можно воспользоваться выражены кислотности Ад: (4-2-18)
В соответств как: ии с ур. 4.5-63 коэффициент распределения может быть выражен Dc = JHAhr-, (4.5-64) [HAJaq + [A Jaq
Записывая в ыражение реальной константы распределения кислоты:
можно объцд (инить ур. 4.5-64 и ур. 4.5-60а с тем, чтобы получить выражение [НА]орГ Кд (НА]орг (4'5 65) Ко Ко [Н~%
которое при упрощении дает (415'65а) [Н jaq + Кд
□ В данной жидкостной экстракционной системе с константой распределения Kd коэффициент распределения Dc существенно зависит от pH водной фазы (для pH > рКд).
На графике зависимости lg Dc от pH, представленном на рис. 4.5-13, имеются два прямолинейных участка:
При [H+]aq > Ка справедливо соотношение Dc « Kd, а при Кд > [H+]aq
Ос = (4.5-656)
Еще один интересный случай наблюдается, когда вещество полимеризуется в органической фазе. Например, при экстракции уксусной кислоты в неполярный органический растворитель, например бензол, вещество в органической фазе может димеризоваться. В этом случае имеются два связанных друг с другом равновесных состояния. При значениях pH водной фазы ниже 2 преобладающим веществом в ней является недиссоциированная уксусная кислота.
Константа распределения КЬ описывает распределение молекулярного вещества в органической и водной фазах:
HACaq - НАсорг тг — [НАс]орг D [HAcU
(4.5-60а)
4.5. Гетерогенные равновесия
221
Рис. 4.5-13. Зависимость коэффициента распределения от pH при экстракции гипотетической карбоновой кислоты (рКл ~ 5) органическим растворителем из водного буферного раствора (Kd = 104).
Рис. 4.5-14. Гипотетическая экстракция уксусной кислоты из водной фазы в органический растворитель, в котором кислота образует димеры, (а—угол наклона)
а Ь"2,оРг описывает процесс димеризации в органической фазе:
2НАсорг [HAcjj.opr
^2,орг —
[(HA^alop. [ПАс]2р,
График зависимости [НАс]орг от [HAcjaq приведен на рис. 4.5-14.
(4.6-80)
222
Глава 4. Основы химических методов анализа
Рис. 4.5-15. Экстракция Т1(Ш) из водных хлоридных растворов трибутилфосфатом в гексане при 25°С. Из этих графиков могут быть определены константы комплексообразования К2, Кз и Кл хлорида таллия.
Из этих простых примеров можно видеть, что изменения Dc вызывают искривление на зависимостях общей концентрации всех форм экстрагированного вещества в органической фазе Сорг от общей концентрации всех форм этого вещества в водной фазе Caq . Такие зависимости называются изотермами распределения.
Экстракция молекулярных веществ
Фазовое распределение простых молекулярных веществ, которые не участвуют в реакциях диссоциации или полимеризации, характеризуется достаточно просто. Соответствующая изотерма распределения представляет собой прямую линию с тангенсом угла наклона 4-1, что отвечает постоянному коэффициенту распределения Dc.
Экстракция комплексов металлов
Ионы металла легко образуют в водных растворах комплексы, если в растворах присутствуют подходящие лиганды. Наличие электрического заряда приводит к тому, что ионы металла, даже будучи гидратированными1 *, плохо экстрагируются в органическую фазу. С нейтральными комплексами ситуация может быть совершенно иной, особенно если по своей природе они являются ковалентными. На рис. 4.5-15 представлены данные по экстракции таллия(Ш) трибутилфосфатом (ТБФ) в гексане из водных хлоридных растворов. Экстрагируемым веществом является TIQ3. Следовательно, можно записать:
Ко = №1Г|3^РГ = const (4.5-60)
1 Гидратация ионов обычно не способствует экстракции, особенно в неполярные органи-
ческие растворители. — Прим. ред.
4.5. Гетерогенные равновесия
223
В предположении о том, что Т1С12+, TIClJ, TICI3 и Т1С14 являются основными формами в водной фазе, полученное выражение можно переписать в следующем виде:
[Т1С1з]орг = const[TlCI2+]„[Cr]^ где [TlCl2+]aq может быть установлена равной [ТЦШЭДа^овщ:
[т1С13]Орг = constpic^j^ia-u
где {TlClJjaq может быть установлена равной (П(П1)]ач,общ:
[Т1С1з]орг = const [TlCl3]aq
где [TlClsJaq может быть установлена равной [Т1(Ш)]аЧ1общ и:
[Т1С1з]ерг = constfTlCl^UCrt1
(4.5-67а)
(4.5-676)
(4.5-67в)
(4.5-67г)
где [TlCl^Jaq может быть установлена равной [Т1(Ш)]ад,Общ-
Из этих соотношений могут быть получены константы комплексообразования в следующем виде:
К* [ст]
(4.5-68а)
(в точке пересечения касательных с тангенсами угла наклона, равными 4-2 и 4-1);
(4.5-686)
(в точке пересечения касательных с тангенсами угла наклона, равными 4-1 и 0);
(4.5-68в)
(в точке пересечения касательных с тангенсами угла наклона, равными 0 и —1).
В случае определенных органических растворителей некоторые ионы металлов экстрагируются в форме анионного комплекса MX”, где г = v 4-1 (и— заряд на свободном незакомплексованном ноне металла). Это особенно важно для тетраэдрических комплексов, таких, как Т1(Ш)Х7, Fe(III)X4 , ReO4 .и СЮ7, которые хорошо экстрагируются в виде кислот некоторыми органическими растворителями. В зависимости от природы органического растворителя экстрагируемое вещество существует в широкой области в виде свободных анионов сильных кислот НМеХ4. На рис. 4.5-16 представлены изотермы распределения железа(Ш) между водным раствором НС1 (2,0 н 1,0 моль/л) и ме-тилизобутилкетоном (гексон, МИБК). Это прекрасный пример для иллюстрации многих процессов, рассматриваемых в аналитической химии растворов, включая процессы комплексообразования, гидратации/сольватации и ионной ассоциации.
На рис. 4.5-16,а имеются три прямолинейных участка на каждой из изотерм. При низких значениях существует линейная зависимость между концентрациями железа(Ш) в водной и органической фазах (прямая с тангенсом угла наклона, равным 4-1). При концентрациях железа(Ш) в органической фазе
Рис. 4.5'16. Экстракция железа(Ш) из 1,0 н 2,0 моль/л водной НС1 метилизобутилке-тоном при 20°С. а — эксперимент (данные распределения); б — интерпретация: концентрационный график.
4.5. Гетерогенные равновесия
225
более 10~5 моль/л (пересечение I) тангенс угла наклона линейной зависимости становится равным 4-0,5, И при концентрациях более 2'10”3 моль/л вновь становится равным 4-1(пересечение П). Необходимо отметить, что в обеих системах точка пересечения II соответствует одним и тем же концентрациям железв(Ш) в органической фазе.
Пересечение I на графиках отражает проявление общего ионного эффекта. Так, при экстракции 2,0 моль/л НС1 в отсутствие железа НС1 экстрагируется в органическую фазу, достигая концентрации 8 • 10~4 моль/л и 2,5 • 10“Б моль/л для ^диссоциированной и диссоциированной форм соответственно (рКд.орг = 6,1). При экстракции железа(1П) концентрации ионов водорода и хлорид-ионов не могут быть одинаковыми, как только концентрация аниона FeCl^ принимает значения того же порядка. При этом эффект общего ионна становится значительным, н равновесные концентрации [Н+]о₽г и [С1—]о₽г начинают различаться, однако ионное произведение [Н+|орг[С1~]орг остается постоянным. Проявление этого эффекта выражается в изменении наклона изотермы распределения от 4-1 до 4-0,5, указывающем на то, что FeC17iOpr является доминирующей формой железа(Ш) в органической фазе, сосуществующей с тем же количеством противоионов Н+,- Поскольку из 1 моль/л НС1 экстрагируется меньшее количество хлороводородной кислоты ([НС1]орг = 4 • 10 3 и [Н+]Орг — [С1~]орг = 1,8 • Ю 5 моль/л), точка перегиба оказывается при другой (1,8 • 10~5 моль/л) концентрация железа(Ш) в органической фазе.
Пересечение II наблюдается на графиках при одной и той же концентрации железа(Ш) в органической фазе, указывая на то, что имеет место реакция ассоциации. В органической фазе появляются две дополнительные формы железа. С одной стороны, ионная пара или недиссоциированная HFeCU.opr становится основной формой, концентрация которой больше равновесной концентрации [FeCl^]орг- С другой стороны, в органической фазе также существенно увеличивается концентрация иона ЩРеСЦ)^ • Наклон 4-1 для кривых, соответствующих этой форме, на рис. 4.5-16,6 при более низкой концентрации увеличивается до 4-1,5. Это можно также объяснить постоянством произведения [H+]opr[H(FeC14)2 ]о₽г- Необходимо отметить, что Н(РеСЦ)з следует рассматривать как димерную форму, один моль которой соответствует двум молям железа(Ш). Это обстоятельство должно учитываться при определении общей концентрации железа(Ш) в органической фазе.
Представленные экстракционные данные не дают информации о гидратации и сольватации экстрагированной формы. Однако в серии дополнительных экспериментов может быть показано, что свободные ионы водорода в органической фазе гидратируются четырьмя молекулами воды (Н9О4 ) и кроме того—сольватируются тремя молекулами МИБК. Также очевидно, что числа сольватации для иона водорода в ионе H(FeC14)2 должны быть другие. Это прекрасный пример, показывающий, что большинство экстракционных явлений могут быть объяснены, когда есть все необходимые аналитические данные.
8 Аналитическая хжмим. Том 1
226
Глава 4. Основы химических методов анализа
Ион-парные реагенты
В экстракционных системах с органическими растворителями, характеризующимися низкой диэлектрической проницаемостью, часто наблюдается явление образования ионных пар. Примером тому является рассмотренная выше экстракция железа(Ш) из водного раствора хлороводородной кислоты в МИБК.
В качестве положительно заряженных реагентов часто используются тетра-алкиламмониевые соединения RiN+ (например, хлорид тетр абутиламмония). Алкилсульфаты (ROSO3) используются в качестве отрицательно заряженных нон-парных реагентов. На процессы образования ионных пар большое влияние оказывают температура и длина алкильной группы в реагенте.
4.5.5. Системы газ — твердое вещество
Как известно, твердые поверхности связывают молекулы газов, с которыми они контактируют. Этот процесс называется адсорбцией, твердое вещество, используемое для реализации этого процесса, — адсорбентом, а адсорбированные вещества — адсорбатами. Адсорбция происходит на внешней поверхности твердого вещества и/или на внутренней, т. е. в порах, щелях или капиллярных каналах. Обратный процесс называется десорбцией.
В общем случае в основе явления лежат силы физического притяжения, например вандерваальсовы. Если в процессе участвуют химические силы, имеет место химическая адсорбция, называемая хемосорбцией. Хемосорбция обычно происходит при повышенной температуре, поскольку имеет большую энергию активации, и обычно протекает медленнее, чем физическая адсорбция.
Изотермы адсорбции
Количество адсорбированного газа выражается как объем адсорбированного газа при стандартных температуре и давлении, а распределение химического вещества между твердой н газообразной фазами описывается изотермами адсорбции (при постоянной температуре), представляющими собой график зависимости объема адсорбированного газа от его парциального давления. Увеличение давления способствует росту объема адсорбированного газа, а повышение температуры действует в обратном направлении. На рис. 4.5-17 приведены различные типы изотерм адсорбции. Рис. 4.5-17, в отражает идеальную систему, хотя и в реальных системах некоторый участок изотермы адсорбции может быть линейным. Рис. 4.5-17,6 описывает адсорбционное насыщение, возникающее в системе. Этот случай реализуется в системах, имеющих ограниченное число адсорбционных мест. Рис. 4.5-17, в иллюстрирует тот случай, когда парциальное давление адсорбируемого газа достигает критического значения, выше которого происходит конденсация газа (процесс, обратный кипению). Кривая на рис. 4.5-17,г представляет собой комбинацию только что описанных изотерм адсорбции, состоящую из линейной области при низких парциальных давлениях адсорбата, насыщение прн умеренных давлениях и достижение критического значения, после которого происходит конденсация.
4.5. Гетерогенные равновесия
227
____________ § ®-
Рис. 4.5-17. Изотермы адсорбции.
Адсорбция газов находит широкое применение в технике; так, адсорбция газов на активированном угле обычно используется для очистки загрязненного воздуха, и играет важную роль в удалении токсичных компонентов из атмосферы (например, в противогазах), для очистки отходящих газов мусоросжигающих печей и т. д.
Адсорбция газов на твердых поверхностях интересна в общехимическом плане, но некоторые ее приложения играют особую роль в аналитической химии. Адсорбция газов на твердых носителях используется при аналитическом определении загрязняющих и опасных веществ в воздухе (промышленная гигиена) и для пробоотбора в системах очистки н фильтрации. В данном случае газы адсорбируются на специально подготовленных материалах (например, на силикагеле, глиноземе, пористых полимерах, в том числе на пенополиуретане), которые используются для концентрирования газообразных компонентов на адсорбенте до их термической десорбции и ввода, например, в газовый хроматограф для последующего разделения и количественного определения.
Адсорбирующие материалы
В большинстве аналитических приложений процессы адсорбции и десорбции должны быть обратимыми. Реже используются процессы хемосорбции, хотя даже при физической адсорбции процессы могут быть не полностью обратимыми и характеризоваться гистерезисным поведением. Очевидно, что получение материалов-адсорбентов с контролируемыми свойствами является актуальной задачей, на решение которой направлены усилия многих исследователей.
Вид (пористая/непористая) и площадь поверхности (м2/г) адсорбента определяют его свойства. Полезные материалы имеют хорошо развитую площадь поверхности (около 100 м2/г и более) с порами контролируемого размера. Ве
8*
228
Глава 4. Основы химических методов анализа
щества с большой площадью поверхности являются также эффективными катализаторами.
Активированный уголь существует во многих формах, различающихся по площади поверхности, пористости и поверхностной активности. Поверхность обычно модифицируют различными химическими или физическими способами, чтобы сделать ее менее химически активной. С целью получения многоцелевых адсорбентов на основе силикагелей и глинозема разработаны сложные методики модифицирования свойств исходных материалов.
□ Материалы, обратимо адсорбирующие газы (активированный уголь, молекулярные сита, пористые полимеры и т. д.), используются в современном следовом газовом анализе для концентрирования веществ.
Некоторые вещества, такие, как природные цеолиты, имеют вполне определенные структуры с порами молекулярных размеров, и называются молекулярными ситами. Молекулы газов с размерами меньшими, чем размеры полостей молекулярных сит, могут проникать во внутреннюю часть пористой структуры и эффективно улавливаться. Большие молекулы не проникают внутрь пористой структуры и проходят через колоночный наполнитель с молекулярными ситами без существенной задержки. Размеры полостей можно регулировать специальными приемами подготовки.
Некоторые из адсорбирующих материалов получают синтетически. Например, пористые полимеры получаются путем полимеризации стирола, сшитого с дивинилбензолом. Продукт имеет открытую структуру, аналогичную молекулярным ситам. Обычно доступны для приобретения пористые полимеры различных типов, такие, как Тепах, Porapax, Chromosorb и XAD. Они особенно полезны в системах очистки и фильтрации для концентрирования следовых количеств определяемых компонентов. Определение проводится после термической десорбции.
Во многих методиках пробоподготовки используются концентрирующие системы, содержащие адсорбирующие материалы. Они используются либо в пассивном варианте, когда доставка определяемого компонента осуществляется за счет диффузии, либо в активном, когда осуществляется принудительная прокачка анализируемой пробы.
□ Носители, пропитанные специальными химическими веществами, меняющими окраску в зависимости от концентрации газа, используются для скрининга токсичных газов в промышленности и окружающей среде.
Возможна адсорбция некоторых соединений на носителях, пропитанных химическими веществами (индикаторами), которые меняют цвет при достижении некоторого критического количества адсорбированного газа или пара. Немецкая компания Drager разработала ряд приборов для определения в воздухе токсинных компонентой. Эти приборы в основном предназначены для решения задач промышленной гигиены.
4.5. Гетерогенные равновесия
229
Вопросы и задачи
1. Напишите правило фаз Гиббса и объясните его смысл.
2- Используя фазовую диаграмму воды, объясните поведение системы в сверхкри-тичвсгсой области.
3. При каких условиях корректный р&счет растворимости соли в воде можно проводить, используя только значение произведения растворимости? Рассмотрите случаи AgCl и СаСОз-
4. Как системы газ — твердое вещество можно использовать для быстрых аналитических измерений?
5. Какие побочные реакции могут значительно увеличить растворимость соли в воде а) В области pH 1-14; б) в присутствии посторонних ионов; в) в присутствии общего иона?
6. Запишите 'равновесия, реализующиеся при экстракционном концентрировании следов ионов металла из водных растворов в органические растворители с использованием органического лиганда НА.
5
ХРОМАТОГРАФИЯ
Цели изучения
• Понять основные принципы разделения веществ за счет распределения их между неподвижной и подвижной фазами.
• Изучить наиболее важные хроматографические методы, такие, как газовая хроматография (ГХ), высокоэффективная жидкостная хроматография (ВЭЖХ), тонкослойная хроматография (ТСХ) и ионная хроматография (ИХ).
• Получить представление о новых методах—сверхкритической флюидной хроматографии, капиллярном электрофорезе и проточном фракционировании в поперечном поле.
5.1. ПРИНЦИПЫ ХРОМАТОГРАФИЧЕСКОГО РАЗДЕЛЕНИЯ
Эффективное разделение химически подобных соединений возможно лишь в случае повторяющихся актов разделения. Однако последовательное использование актов разделения (например, согласно распределению Крейга) на практике пригодно лишь для препаративных целей.
Эффективное многократное разделение может быть осуществлено с использованием хроматографических методов. По современным оценкам приблизительно 60% всех анализов в мире выполняется хроматографически.
Русский ботаник Михаил Цвет предложил метод колоночной хроматографии в 1903 году. Используя колонку, содержащую мелко размолотый карбонат кальция, он смог разделить экстракты пигментов из листьев, такие, как хлорофиллы, а также ксантофилл н спириллоксантин. При этом он наблюдал внутри колонки цветные зоны, что побудило его предложить термин хроматография (от греческих слов, означающих «цвет» и «пишу»).
5.1. Принципы хроматографического разделения 231.
5.1.1. Обзор хроматографических методов
Хроматография основана на прохождении разделяемых компонентов через систему подвижной и неподвижной фаз. Для этого пробу растворяют в подвижной фазе, которая может представлять собой жидкость, газ или сверхкритический флюид, и пропускают ее через неподвижную фазу, находящуюся либо в колонке, либо на твердой поверхности. Благодаря взаимодействиям компонентов пробы с неподвижной фазой они через некоторое время заметно разделяются.
Б зависимости от способа получения хроматограмм различают внутренние и внешние хроматограммы. В случае внутренней хроматограммы разделяемые компоненты пробы проходят разное расстояние за одинаковое время. После разделения они все еще находятся на неподвижной фазе и там же детектируются. Этот вид хроматограмм типичен для плоскостных вариантов, таких, как бумажная или тонкослойная хроматография (разд. 5.3.4). Неподвижная фаза расположена на пластинке, а подвижная движется через неподвижную за счет капиллярных сил или под влиянием гравитации.
Внешние хроматограммы получают в колоночной хроматографии — в газовой, высокоэффективной жидкостной или флюидной хроматографии. В этом случае все компоненты проходят одинаковый путь по колонке, и благодаря специфическим взаимодействиям с неподвижной фазой они через различное время выходят из колонки и затем могут быть детектированы.
Классификация методов колоночной хроматографии по природе используемых подвижной и неподвижной фаз представлена в табл. 5.1-1. Два основных механизма хроматографического разделения — это распределение н адсорбция. Адсорбционная хроматография основана на непосредственном взаимодействии разделяемого вещества с поверхностью неподвижной фазы, например, в ГТХ или ЖТХ. Распределительная хроматография связана с наличием иммобилизованной жидкой неподвижной фазы (ГЖХ, ЖЖХ).
Для жидкостной хроматографии возможны колоночный и плоскостной варианты. Основные физико-химические принципы Для обоих вариантов одни и те же. Газовая же хроматография ограничена колоночным вариантом.
В дальнейшем теоретические принципы хроматографии будут освещены на примере колоночной хроматографии.
Таблица 5.1-1. Классификация методов колоночной хроматографии по природе подвижной и неподвижной фаз
Неподвижная фаза Подвижная фаза
Газ Флюид Жидкости
Твердая ГТХ (газо-твердо-фазная хроматография) СФХ (сверхкритическая флюидная хроматография) ЖТХ (жидко- твердофазная хроматография)
Жидкая ГЖХ (газо-жидкостная хроматография) ЖЖХ (жидкожидкостная хроматография)
232 Глава 5. Хроматография
5.1.2. Получение хроматограмм
Основной способ получения хроматограмм в колоночной хроматографии — элюентная хроматография. В этом варианте проба, растворенная в подвижной фазе, вводится в верхнюю часть колонки. Затем с использованием подвижной фазы осуществляется элюирование разделяемых веществ до тех пор, пока они не будут детектированы в конце колонки. Рис. 5.1-1 объясняет принцип элю-ентной хроматографии на примере разделения веществ А и В.
□ Другие варианты получения хроматограмм, такие, как фронтальная и заместительная хроматография, являются дополнением к элюентной хроматографии. Во фронтальном варианте проба непрерывно подается в систему до наступления «проскока» разделяемой смеси. Это может быть использовано в целях очистки, например, для удаления жесткости из воды или осушки растворителей.
После того как проба введена в колонку, ее компонента распределяются между подвижной и неподвижной фазами. Если подвижная фаза после этого непрерывно поступает в форме элюента, вещества распределяются вдоль колонки между новыми порциями подвижной и новыми частями неподвижной фаз. Для соединений, удерживающихся сильнее на неподвижной фазе, требу-
Проба
Подвижная фаза
* *АГГ Д,
-й
<4* ’V Неподвижная
Дни
v- at-
б
Сигнал детектора
Время
Рис. 5.1-1. Разделение двух веществ А и В с использованием элюентной хроматографии. а — развитие внутренней хроматограммы на неподвижной фазе; б — внешняя хроматограмма, регистрируемая с помощью детектора.
5.1. Принципы хроматографической) разделения
233
ется больше времени для разделения, чем для веществ, взаимодействующих слабее. В идеале вещества разделяются после соответствующего времени элюирования и индивидуально детектируются в конце колонки.
□ Термин элюент является синонимом «подвижной фазы».
В колоночной хроматографии запись сигнала детектора как функции времени элюирования или объема элюента называется хроматограммой. Если наблюдать за перемещением зон веществ вдоль колонки, можно отметить два эффекта (см. рис. 5.1-1). Расстояние между зонами возрастает. Однако, в то же время, зоны становятся шире, вызывал некоторое ухудшение разделения. Следовательно, разделение компонентов на хроматограмме может быть улучшено, если:
а) либо скорости движения веществ селективно изменять,
б) либо размывание пиков поддерживать иа минимально возможном уровне.
Величины, влияющие на скорости движения и размывание пиков, рассматриваются в последующих разделах.
5.1.3. Параметры хроматограмм
Начнем описание параметров хроматограмм со скорости движения, с которой частицы движутся по колонке. В простейшем случае прохождение веществ между подвижной (М) и неподвижной (S) фазами определяется равновесием распределения. Коэффициент распределения К для этого равновесия можно получить с использованием концентрации вещества в неподвижной фазе (cs) и подвижной фазе (см):
К = — (5.1-1)
См
□ Коэффициент распределения описывает константу равновесия для разделяемого вещества между двумя несмешивающимися фазами. Его также называют распределительным отношением.
Коэффициент распределения не может быть напрямую определен из хроматограммы. Общее время удерживания, обозначаемое tR, непосредственно получается из хроматограммы. Мы уже использовали этот параметр без описания на рис. 5.1-1,б. Ддя разъяснения рассмотрим хроматограмму еще раз на рис. 5.1-2.
Маленький пик со временем удерживания 4м соответствует соединению, которое не удерживается. При этом символ М отражает принадлежность к подвижной фазе, т. е. время удерживания £м (время подвижной фазы или лучше мертвое время) в точности соответствует времени, за которое молекулы подвижной фазы проходят через колонку. Часто в эту величину неверно определяют как общее время от момента ввода пробы до прохождения через детектор.
□ Разность между общим временем удерживания и мертвым временем называется исправленным временем удерживания, т. е. Ir = tn — tM.
234
Глава 5. Хроматография
Рис. 5.1-2. Элюентная хроматограмма для пробы из двух компонентов. Первый пик в левой части хроматограммы со временем удврживания tw (Мертвое время) соответствует неудерживаемому компоненту.
Зная время удерживания, можно рассчитать линейную скорость движения определяемого вещества v и молекул подвижной фазы и:.
(5-1-2)
(5.1-3)
гдв L—длина слоя сорбента в колонке.
Как же можно установить зависимость между временем удерживания и коэффициентом распределения? Для этого снова обратимся к элюированию веществ в колонке. Поскольку вещество перемещается по колонке, находясь в подвижной фазе, то его удерживание зависит от времени его пребывания в подвижной фазе, а именно удерживание тем больше, чем меньше время пребывания в подвижной фазе. Неудерживаемые вещества все время проводят в подвижной фазе, а вещества, которые взаимодействуют с неподвижной фазой, находятся в подвижной фазе лишь некоторую часть времени. Эта часть времени может быть описана с использованием соотношения между массой определяемого вещества в подвижной фазе и его общей массой в колонке:
_ cmVm 1
V — U..т------— = U-----ГТ-
СМ Ии + CS Vs г + CS Vs
CMVM
(5-1-4)
где массы выражены произведением концентраций определяемого вещества На объем подвижной или неподвижной фазы.
□ В ур. 5.1-4 скорость движения определяемого вещества выражается как относительная скорость движения подвижной фазы.
Если в ур. 5.1-4 ввести коэффициент распределения К, получится зависимость скорости движения вещества от скорости подвижной фазы:
v = и
(5.1-5)
Объемы неподвижной и подвижной фаз можно получить непосредственно из экспериментальных данных.
5.1. Принципы хроматографического разделения 235
Для второго слагаемого в знаменателе ур. 5.1-5 в хроматографии вводится новая величина, коэффициент емкости к':
к' = К^-=Л (5.1-6)
ПИ р
гдв Ум/Уз^-так называемое фазовое отношение /3.
□ В последнее время коэффициент емкости в литературе также называют фактором удерживания и обозначают к.
Используя коэффициент емкости к', мы также можем установить связь между временами удерживания. Подставляя ур. 5.1-6 в уравнение для скорости движения 5.1-5, получаем:
* = (51-7)
Если скорости движения заменим соответствующими выражениями 5.1-2 и 5.1-3, то получим:
L L 1
*r *м 1 + к'
После преобразования получим следующее уравнение для связи между коэффициентом емкости как параметром, описывающим коэффициент распределения, и исправленным временем удерживания:
к’ = tR-tu = «к (5 д.9)
tM tM
Таким образом, коэффициент емкости может быть установлен непосредственно из хроматограммы на основании общего времени удерживания интересующего компонента и мертвого времени удерживания (рис. 5.1-2). Значение коэффициента емкости обычно находится в диапазоне от 1 до 5. Если коэффициент емкости значительно меньше единицы, значит соединение элюируется слишком быстро, так что его время удерживания трудно отличить от времени удерживания подвижной фазы. Если коэффициент емкости превышает 20, то на практике это приводит к неприемлемо большим временам удерживания.
Коэффициент селективности, также называемый фактором разделения, является мерой разделения двух веществ и в хроматографии обозначается а. Коэффициент селективности для разделения двух веществ, А и В, определяется выражением
а=^ (5.1-10)
Кд
гДе К — коэффициенты распределения веществ А или В соответственно. Поскольку по хроматограмме мы можем определить только коэффициенты емкости, удобнее выразить коэффициент селективности, используя ур. 5.1-6:
(5.1-11)
236
Глава 5. Хроматография
Определение а из данных хроматограммы возможно заменой в ур. 5.1-11 коэффициента емкости его выражением 5.1-9:
Величины, получаемые для определения разрешения хроматограмм с использованием коэффициентов емкости и селективности, будут использованы в последующих разделах.
5.1.4. Теория хроматографии
Рассмотрим еще один эффект—размывание пика при прохождении по колонке. Ширина пика находится в прямой зависимости от эффективности разделения, т. е. от эффективности колонки. Эта зависимость получается из классической теории хроматографии.
□ Эффективность колонки описывает степень размывания хроматографического пика.
Классическая теория
Классическая теория хроматографии рассматривает процесс хроматографического разделения как результат совокупности дискретных актов распределения в колонке в целом. Мартин и Синг (Нобелевские лауреаты 1952 г.) ввели понятия высоты тарелки (т. ё. высоты, эквивалентной теоретической тарелке, ВЭТТ) и числа теоретических тарелок. Предполагается, что на каждой теоретической тарелке устанавливается равновесие для вещества между подвижной и неподвижной фазами. Если вещество движется по колонке, это означает, что происходит последовательный переход от одного акта разделения или одного равновесия к другому.
Число теоретических тарелок N рассчитывается как отношение общей длины колонки L к высоте, эквивалентной теоретической тарелке, Н:
(5.1-12) п
Связь между дисперсией пика и пройденным веществом расстоянием (длина колонки L) или временем удерживания, £r, определяется как высота, эквивалентная теоретической тарелке, Н, или, короче, — высота тарелки-.
Н=а1 (5.1-13)
= Ф (5-1-14)
er
Дисперсия а2, относящаяся к длине колонки, измеряется в см2, а дисперсия ст2, относящаяся к времени удерживания,—в с2. Если длина колонки измеряется в см, то и размерность Н в обоих уравнениях—также в см.
□ Дисперсия определяется как квадрат стандартного отклонения а в распределении Гаусса.
5.1. Принципы хроматографического разделения
Й7
Рис. 5.1-3. Определение стандартного отклонения о по хроматограмме через ширину ника у основания w — или полуширину Ь1/з (т- е. ширину пика на половине высоты /г).
Меньшая высота тарелки Н приводит к большей эффективности колонки, а следовательно, и к лучшему разрешению.
На практике непосредственно используют данные хроматограммы для определения числа теоретических тарелок. Для этого аппроксимируют ширину пика на половине высоты, по величине ширины пика у его основания, w. Ширина у основания может быть определена по пересечению касательных, проведенных в точке перегиба, с базовой линией гауссовой кривой (рис. 5.1-3).
□ Хроматографические пики также называют хроматографическими полосами или зонами.
Из параметров гауссова распределения известно, что внутри стандартного отклонения величиной ±2<rt находится примерно 96% площади под гауссовой кривой. Измеренные как времена удерживания, результаты для стандартного отклонения на оси времени соответствуют ±2<it или w — 4at для ширины у основания. Если подставить эту величину в уравнение для высоты тарелки (5.1-14), то получается:
Переходя к числу тарелок, получим
N = 16 (^) (5.1-16)
□ Высота тарелки и число теоретических тарелок характеризуют эффективность колонки.
Таким образом, число теоретических тарелок может быть определено из хроматограммы измерением времени удерживания и ширины пика у основания. Более правильные результаты обычно получаются при расчете по модифицированному уравнению с использованием ширины на полувысоте 5^2 (см-рис. 5.1-3):
N = 5,54 (^-) (5.1-17)
Выражения д ля высоты тарелки и числа тарелок обычно используются для оценки хроматографического разделения. Однако теория тарелок лишь представляет аппроксимацию процессов, на самом деле происходящих в колонке.
238
Глава 5. Хроматография
Так, идея повторяющегося установления равновесия при разделении нереалистична, поскольку равновесие трудно достижимо из-за движения подвижной фазы. А при сравнении колонок на основе теории тарелок в числа тарелок следует всегда использовать одно и то же вещество в пробе.
Кинетическая теория
Согласно этой теории, размывание пиков происходит благодаря кинетическим эффектам. Эти эффекты возникают из-за конечной скорости процессов массопереноса, который протекает при движении вещества по колонке. Действие этих эффектов зависит от длины возможных переходов между подвижной и неподвижной фазами и, следовательно, непосредственно пропорционально скорости потока подвижной фазы.
Для описания этих явлений следует изучить зависимость высоты тарелки от линейной скорости потока или проще линейной скорости й (см/с). Рис. 5.1-4 иллюстрирует эти зависимости для жидкостной и газовой хроматографии. Важные независимые переменные для описания эффективности колонки описаны в табл. 5.1-2.
Таблица 5.1-2. Переменные, влияющие на эффективность колонки в хроматографии
Переменная Обозначение Размерность
Линейная скорость подвижной фазы й см/с
Коэффициент диффузии в под вижной фазе Ом см2/с
Коэффициент диффузии в неподвижной фазе Ds см2/с
Диаметр зерна сорбента dp CM
Толщина жидкого слоя неподвижной фазы dt CM
Скорость десорбции вещества td c
Диаметр колонки dc CM
Как можно заключить из рис. 5.1-4, минимум для высоты теоретической тарелки в жидкостной хроматографии (ЖХ) находится при более низких скоростях потока, чем в газовой хроматографии (ГХ). Следует отметить, что этот
а Жидкостная хроматография б Газовая хроматография
Рис. 5.1-4. Зависимость числа теоретических тарелок Н от линейной скорости потока и в подвижной фазе для жидкостной (а) и газовой (б) хроматографии.
5.1. Принципы хроматографического разделения
239
минимум часто вообще не проявляется для обычных скоростей в ЖХ. Согласно рисунку, в ЖХ могут быть в принципе достигнуты меньшие высоты тарелки, чем в ГХ. Это преимущество, однако, теряется из-за длин колонок, которые могут быть достигнуты на практике. Если в ЖХ из-за создаваемого давления можно работать с колонками длиной максимум 25-50 см, то в ГХ могут быть использованы колонки длиной до 100 м. ‘ ”J
Для математической аппроксимации функции К(й) на рис. 5-1-4 было предложено несколько моделей. Изначально это было уравнение, взятое из инженерной области и перенесенное на хроматографический процесс, — уравнение ван Деемтера:
Н = А + ^ + Сй (5.1-18)
и
Здесь высота тарелки зависит от линейной скорости подвижной фазы и описывается коэффициентами А д ля диффузии Эдди, В для продольной диффузии и С для массопереноса между подвижной и неподвижной фазами. Коэффициент А описывает расстояние, проходимое потоком подвижной фазы до того, как его скорость значимо изменяется под действием сорбента. Это изменение скорости называется диффузией Эдди (или вихревой диффузией.—Пер.).
Следующее уравнение более подробно описывает модель:
Н = А + ^+Свй + Сый (5.1-19)
и
Коэффициенты В, См и Cs соответствуют продольной диффузии, коэффициенту массопереноса в подвижной фазе и коэффициенту массопереноса в неподвижной фазе. Вклад отдельных слагаемых в ур. 5.1-19 объясняется более подробно в табл. 5.1-3 и на рис. 5.1-5. В капиллярной хроматографии коэффициент А равен нулю, поскольку отсутствует поперечный поток.
Таблица 5.1-3. Кинетические факторы размывания пиков (описание символов см. табл. 5.1-2)а
Фактор Слагаемое в ур. 5.1-19
Продольная диффузия В 2kr>DjA й й
Массоперенос из жидкого слоя неподвижной фазы и внутрь него qk'dju Cs (l + k'^Ds
Массоперенос из твердого слоя неподвижной фазы и внутрь него 2idfc'u Cs“ (1 + к')г
Массоперенос в подвижной фазе „ _ М.<й)д CMU
* Лр, q —постоянные, / характеризует функциональную зависимость.
Так же, как и уравнение ван Деемтера (5.1-18), ур. 5.1-19 должно приводить к минимальной высоте тарелки при оптимальной скорости потока. Коэффициент В описывает влияние продольной диффузии. Это влияние создается диффузией частиц от центра пика по направлению потока подвижной
240
Глава 5. Хроматография
Рис. 5.1-5. Вклад диффузионной составляющей В/П и массопереноса Смй н С$и в высоту тарелки Н.
фазы или против него. Коэффициент В не зависит от размера частиц неподвижной фазы. Он прямо пропорционален коэффициенту диффузии вещества в подвижной фазе и, следовательно, увеличивается при уменьшении относительной молекулярной массы. Коэффициент В не имеет большого значения при скоростях потока, используемых в жидкостной хроматографии. Однако в газовой хроматографии он очень важен.
□ Константа равновесия для распределения вещества между двумя несмеШива-ющимися растворителями называется константой распределения.
Влияние продольной диффузии на высоту тарелки обратно пропорционально линейной скорости, поэтому можно сказать, что диффузия уменьшается с увеличением скорости потока подвижной фазы. Напротив, высота тарелки уменьшается при увеличении линейной скорости из-за эффектов массопереноса. Этот эффект возникает, потому что для массопереноса между фазами требуется конечное время. Часто в проточной системе недостаточно времени для достижения состояния равновесия, так что уменьшение массопереноса становится более очевидным при увеличении скорости потока.
Слагаемое массопереноса в подвижной фазе, Смй, представляет конвекти-ный компонент дисперсии потока. При некоторых ограниченных условиях он соответствует диффузии Эдди в уравнении ван Деемтера. Это слагаемое массопереноса обратно пропорционально коэффициенту диффузии в подвижной фазе и напрямую зависит от диаметра частиц сорбента dp, как и от диаметра колонки dc (табл. 5.1-3).
Для слагаемого С$й, которое описывает массоперенос внутрь и из неподвижной фазы, должны быть рассмотрены два случая, в зависимости от того, имеем мы дело с жидкой иммобилизованной или твердой неподвижной фазой.
В случае жидкой неподвижной фазы доминирует равновесие распределения. Эффект возрастает при увеличении толщины жидкой пленки и при уменьшении коэффициента диффузии в неподвижной фазе. Утоньшение пленки и уменьшение коэффициента диффузии замедляют подход вещества к границе раздела для массопереноса.
Если неподвижная фаза состоит из твердого вещества, то коэффициент массопереноса С$ зависит от скорости адсорбционных и десорбционных процессов.
5.1. Принцип^ хроматографического разделения
241
Вообще, цель хроматографии — эффективное разделение за более короткое время. Исходя из теоретических рассуждений, это-означает, что следует достичь низкой высоты тарелки (минимум Я), и что функция Я(й) должна быть как можно более ровной, чтобы высокие Скорости разделения могли бы быть обеспечены без потери эффективности. В принципе низкие значения Щтп и ровные кривые для функции Щи) могут быть достигнуты следующими приемами:
а) малый размер твердых частиц или малая толщина слоя жидкого покрытия неподвижной фазы;
б) гомогенная упаковка неподвижной фазы с использованием сорбента с максимально близким размером частиц;
в) малый диаметр колонки;
г) большие коэффициенты диффузия в неподвижной фазе и малые коэффициенты диффузии в подвижной фазе. В ГХ коэффициенты диффузии в подвижной фазе могут быть значительно уменьшены снижением температуры.
Поскольку коэффициенты диффузии для различных размеров молекул различаются, размывание пиков также зависит от относительной молекулярной массы. Малые молекулярные массы благоприятствуют повышению эффективности колонки. Одиако молекулярные массы определяются разделяемыми веществами и поэтому не могут быть свободно выбраны.
5.1.5. Разрешение Rs как характеристика разделения пиков
Мы уже познакомились с коэффициентом селективности (ур. 5.1-11) для определения селективности разделения двух компонентов в хроматографии. Однако коэффициент селективности как отношение коэффициентов распределения или коэффициентов емкости отражает только селективность используемой фазовой системы. Из теории хроматографии мы знаем, что эффективность колонки также определяется числом тарелок N и величиной коэффициента емкости.
□ Разрешение характеризует возможность хроматографической колонки разделить два вещества.
Таким образом, мы используем хроматографическое разрешение Rs как характеристику всей системы. Для разделения двух гауссовых пиков А и В разрешение рассчитывается с использованием ширин пиков у основания (рис. 5.1-6):
= Ж = (51'20)
считая, что w и wa ~ wb-
242
Глава 5. Хроматография
Рис. 5.1-6. Хроматограмма смеси веществ А и В. Хроматографическое разрешение Rs может быть определено по ур. 5.1-20 из определенных значений.
□ Пики часто оказываются асимметричными из-за размытого заднего фронта (а) или из-за перегрузки (б):
Подставив ур. 5.1-16 для числа теоретических тарелок, получим:
Или, используя коэффициент емкости из ур. 5.1-9:
= (51'22)
Путем введения коэффициента селективности из ур. 5.1-11 и преобразований, получим выражение для хроматографического разрешения:
Выражение для разрешения часто записывается в упрощенной форме в случае, когда коэффициенты емкости очень близки, т. е. = к'в = к':
./дг F
j?s = ^(a_l)_£_ (5.1-24)
Если известно разрешение, можно рассчитать число теоретических тарелок, зная коэффициент селективности и коэффициент емкости для компонента В:
jv=^fe)2(i^)2 (51-25)
Основываясь на том, что разрешение зависит от а, к' и N (или Н), можно оптимизировать разделение. Индивидуальные величины можно варьировать независимо друг от друга. Коэффициент селективности а может быть модифицирован путем выбора типа молекулярных взаимодействий, т. е. сменой неподвижной фазы. Коэффициент емкости А/ может быть изменен варьированием температуры в газовой хроматографии или составом подвижной фазы в жидкостной хроматографии. Число теоретических тарелок N можно изменять
5.1. Принципы хроматографического разделения
243
через длину колонки или через оптимизацию высоты тарелки Н. Напомним, что высота тарелки может зависеть от скорости потока, размера частиц сорбента, вязкости фаз (и, следовательно, коэффициентов диффузии Ds и 7?м) или от толщины пленки иммобилизованной жидкости на неподвижной фазе (табл. 5.1-3).
В случае симметричных пиков одинаковой интенсивности можно Считать, что Rs = 1 для разделения вплоть до базовой линии или 4(т-разделении. Упрощенно можно предполагать, что в случае асимметричных пиков или для пиков с сильно отличающимися интенсивностями для полного разделения требуется более высокое разрешение.
5.1.6. Качественная информация
Хроматографические методы могут быть использованы как для качественного, так и для количественного анализа, а также для препаративных целей. Мы не будем останавливаться подробно на особенностях препаративной хроматографии.
В случае внутренней хроматограммы качественная информация заложена в положении вещества в неподвижной фазе, а во внешней хроматограмме—во времени удерживания или в объеме удерживания. Несмотря на то что воспроизводимость данных об удерживании, как правило, значительно ниже, чем, например, точность длин волн в спектроскопии, измерив удерживание вещества относительно стандартных веществ, можно достаточно установить присутствие или отсутствие вещества. Сочетание хроматографического разделения со спектроскопическим детектированием повышает точность идентификации вещества. Так, в газовой хроматографии (ГХ) используют масс-спектрометрические детекторы, а в высокоэффективной жидкостной хроматографии (ВЭЖХ)-УФ-детекторы с диодной матрицей.
В качественном хроматографическом анализе многокомпонентных смесей следует всегда представлять себе, что пиковая емкость колонки ограничена. Пиковая емкость отражает число пиков, которые могут быть разрешены друг за другом за определенный промежуток времени (рис. 5.1-7). Согласно Гид-динсу, в элюентной хроматографии пиковую емкость п можно приблизительно рассчитать по следующему уравнению:
n=l+—1п^ (5.1-26)
Рис. 5.1-7. Представление пиковой емкости колонки.
Объем удерживания
244
Глава 5. Хроматография
Таблица 5.1-4. Типичные значения пиковой емкости при различных числах теоретических тарелок в ГХ, ЖХ и гель-хроматографии (по Гиддингсу)
Число теоретических тарелок N Пиковая емкость п
Гем ГХ ЖХ
100 3 11 7
400 5 21 13
1000 7 33 20
2500 11 51 31
10000 21 101 61
где Ir и Vp - объемы удерживания пиков, элюирующихся первым и последним соответственно.
Если число компонентов, присутствующих в пробе, превышает пиковую емкость, то неизбежно перекрывание пиков, и в одном пике будут элюироваться два или большее число компонентов. Таблица 5.1-4 показывает типичные значения пиковой емкости для газовой хроматографии (ГХ), жидкостной хроматографии (ЖХ) и гель-фильтрации (см. также рис. 5.1-7).
5.1.7. Количественный анализ
Основа количественного анализа в колоночной хроматографии — определение высоты или площади пика. В случае внутренних хроматограмм может быть измерена полная интенсивность пятна вещества, например в тонкослойной хроматографии (ТСХ). Хроматографические методы являются методами относительными, т. е. можно сказать, что градуировка проводится путем определения стандартных веществ. При этом можно использовать как внутренние, так и внешние стандарты.
При определении высоты пика должна быть соблюдена неизменность формы пика при изменении условий хроматографирования, иначе высота пика не будет прямо пропорциональна концентрации. Параметры хроматографической системы, такие, как температура колонки, скорость потока и объем вводимой пробы, должны быть точно контролируемы. Кроме того, должна быть исключена перегрузка колонки.
Размывание пика не имеет значения, когда определяется площадь пика. Для этого используется численное интегрирование при помощи компьютера. Площади пиков могут быть приблизительно определены вручную перемножением высоты и полуширины пика (площадь = Л.61/2, см. рис. 5.1-3). Однако в случае очень узких пиков при определении площадей пика могут возникнуть проблемы.
□ Проблемы с численным интегрированием возникают при определении точного положения начала и конца пика. Это вызвано шумом базовой линии (см. разд. 12.3).
5.1- Принципы хроматографического разделения
245
Пример. Хроматографические параметры
Рассчитайте длину хроматографической колонки, обеспечивающей удерживание двух соединений А и В с коэффициентом селективности a — 1,05 при разрешении Rs — 1; коэффициент емкости для второго пика к'в = 2,0; высота тарелки Н = 1мм.
Из ур. 5.1-12 известно, как рассчитать длину колонки исходя из высоты тарелки и числа теоретических тарелок. Преобразование этого уравнения дает:
L = N-H
В этом уравнении неизвестно число теоретических тарелок N. Однако его можно следующим образом рассчитать, зная коэффициент селективности, разрешение и ширину пика. Высота тарелки зависит от среднего времени удерживания двух пиков и их ширины (ур. 5.1-16):
Средняя ширина пиков определяется по ур. 5.1-20:
Неизвестное время удерживания первого пика А можно получить из известного коэффициента емкости и фактора селективности а согласно ур. 5.1-11:
Q
Поскольку мертвое время неизвестно, отношение коэффициентов емкости непосредственно соответствует отношению исправленных времен удерживания, т. е.
a _ кв — tjA
кд — tjA
в случае tM = 0 или по крайней мере tM « <r- Тогда в приведенном выше уравнении мы можем использовать коэффициенты емкости вместо исправленных времен удерживания Численные расчеты дадут:
Ц = = 1.9°
a 1,05
ГО=*Ы1 = 2.00-1,90 Rs 1
L = N Н = 6084 -1 мм = 6084 мм или 6,084 м
Результат можно округлить н считать, что требуемая длина колонки составляет около 6 м.
Вопросы и задачи
1. Какие функции разделения важны для хроматографических процессов, н какие фундаментальные физические и химические эффекты лежат в основе этих процессов?
246
Глава 5. Хроматография
2. Объясните эффекты, вызывающие размывание пиков в хроматографии.
3. Опишите взаимосвязь между высотой тарелки и скоростью подвижной фазы в хроматографической колонке.
4. Охарактеризуйте взаимосвязь между временем удерживания и текущим равновесием, длиной колонки и скоростью потока.
5. С помощью каких параметров можно улучшить разрешение хроматографического разделения и как они могут быть определены из хроматограммы?
5.2. ГАЗОВАЯ ХРОМАТОГРАФИЯ
В газовой хроматографии определяемые соединения испаряются и элюируются с колонки при помощи газа в качестве подвижной фазы. Используемая подвижная фаза является просто газом-носителем, так что взаимодействия подвижной фазы с определяемым веществом практически нет. Неподвижной фазой может служить твердое вещество, на котором разделяемые компоненты адсорбируются. На практике газо-твердофазная хроматография (ГТХ), или адсорбционная хроматография, особенно важна при анализе газов воздуха и будет рассмотрена в конце раздела. Жидкость в качестве неподвижной фазы применяют в основном для анализа органических соединений. Этот вид называется газо-жидкостной хроматографией (ГЖХ) или просто газовой хроматографией. Преобладающий принцип разделения при этом—распределение веществ между жидкой неподвижной фазой и газообразной подвижной фазой.
5.2.1. Параметры удерживания и коэффициент распределения
□ В качестве параметра удерживания на практике чаще используется время удерживания.
Прежде чем обсудить возможности разделения веществ в газовой фазе, рассмотрим факторы, определяющие удерживание в газовой хроматографии. Чтобы учесть влияние давления и температуры, вместо времени удерживания используют объем удерживания. Общий объем удерживания Vr и объем подвижной фазы Vm получают умножением соответствующих времен удерживания на объемную скорость потока газа-носителя F (в мл газа-носителя в минуту):
VR = F-tR (5.2-1)
VM = F-tM (5-2-2)
Пик воздуха или метана может быть использован для определения времени удерживания подвижной фазы. Чтобы правильно описать удерживание компонентов в неподвижной фазе, объем удерживания подвижной фазы следует вычесть из общего объема удерживания. Эта разница называется исправленным объемом удерживания:
V£ = Vr-Vm
(5.2-3)
5.2. Газовая хроматография
247
Исправленный объем удерживания—характеристика вещества; через объем неподвижной фазы Vs он может быть непосредственно связан с коэффициентом распределения К:
Vr = KVS (5.2-4)
Из-за сопротйвления разделяющей колонки давление газа на входе в колонку больше, чем на выходе из нее. Значит, вдоль колонки существует градиент давления. Благодаря сжимаемости газа-носителя скорость потока газа возрастает с повышением разности давления. Чтобы описывать объем удерживания независимо от падения давления, исправленный объем удерживания корректируют коэффициентом j (коэффициент Мартина) и истинным объемом удерживания Vfj:
VN=j-V^ (5.2-5)
где j = 3[(pi/po)2 — l]/2[(pj/po)3 — 1], a Pi и po—давление на входе колонки и на выходе из нее, соответственно.
Удельный объем удерживания V& который зависит от истинного объема удерживания Vn, массы неподвижной фазы Ws (г) и температуры колонки Т (К), может быть получен из истинного объема удерживания:
Удельный объем удерживания менее всего зависим от условий анализа. Однако имеется немало трудностей для определения этой величины, поэтому на практике для сравнительных определений предпочитают использовать исправленный объем удерживания или другие относительные величины удерживания.
еь’
5.2.2. Разделение в газовой фазе i
Чтобы определить эффективность разделения различных колонок, используют число теоретических тарелок N и высоту тарелки Н из кинетической теории.
□ Коэффициенты диффузии для газов и жидкостей: см. табл. 5.4-1.
Продольная диффузия (величина В в ур. 5.1-19) особенно значима в газовой хроматографии по сравнению с другими хроматографическими процессами. Причина этого—высокие коэффициенты диффузии в газах, примерно в 104 раз больше, чем в жидкостях. Поэтому минимум функции Я (и) значительно более широкий и расположен при более высоких значениях линейной скорости потока подвижной фазы.
Однако параметры теории тарелок и кинетические модели в хроматографии недостаточны для установления соответствия, т. е. выбора разрешающей способности колонки для конкретной задачи анализа. Для этого нужно также включить в качестве дополнительного фактора эффективность разделения
248
Глава 5. Хроматография
колонки. В газовой хроматографии она зависит от давления пара соединения и степени взаимодействия с неподвижной фазой.
Эффективность разделения может быть получена из законов Генри и Рауля; получаемое соотношение называется формулой Херингтона:
lg%=lepl+le^ (Б'2‘7)
где V& и VSi — удельные объемы удерживания для компонентов 2 и,1 соответственно. Тогда для времен удерживания двух разделяемых компонентов применимо следующее соотношение:
или + Л (5.2-8)
*Ri Р2У2 Р2 Т2
Отношение истинных объемов удерживания соединений 1 и 2 пропорционально отношению давлений пара чистых компонентов р® и и отношению коэффициентов активности компонентов 7® и 7° при бесконечном разбавлении.
Согласно этому, разделение двух компонентов определяется их относительной летучестью. Естественно, давление пара зависит от температуры. В то же время, коэффициенты активности — характеристика взаимодействий разделяемых компонентов с неподвижной фазой. Значит, они определяют селективность неподвижной фазы, а именно одной из десятка тысяч имеющихся жидких фаз.
Различия между давлениями пара формируют основу разделения химически близких соединений, таких, как, например, члены гомологических рядов. Вещества с одинаковыми температурами кипения могут быть разделены на основании различий в коэффициентах активности. Нежелательные дистилля-ционые азеотропы в данном случае не имеют значения.
5.2.3. Составны^ части газового хроматографа
Модули газового хроматографа изображены на рис. 5.2-1. Различие газохроматографических систем состоит в типе используемого газа-носителя, в системе ввода пробы, а также в используемых колонках и детекторах.
Газы-носители
В качестве газов-носителей используются инертные газы (гелий, аргон), а также азот, диоксид углерода и водород. Выбор газа-носителя отчасти определяется детектором. Газ иногда пропускают через молекулярные сита для удаления следов воды. Поток газа обеспечивается избыточным давлением газового баллона, поэтому можно работать без насоса. Чтобы получать воспроизводимые результаты измерений поток носителя следует поддерживать неизменным.
При работе с колонкой в изотермическом режиме достаточно установить давление на колонке с помощью даухступенчатого крана-редуктора. При программировании температуры или при использовании переключения колонок в дополнение к учету изменения текучести необходимо использовать регулятор потока. Для измерения скорости потока на входе в колонку может быть
5.2. Газовая хроматография
249
использован ротаметр, а на выходе—мыльно-пузырьковый измеритель потока. Для набивных колонок расход варьируют между 25 и 150мл/мин, а для капиллярных —в пределах 1-25 мл/мин.
Система ввода пробы
При анализе газообразных веществ проба может быть введена непосредственно в поток газа-носителя (объем пробы до 20 мкл). Жидкие и твердые пробы предварительно испаряют в инжекционном испарителе. Систему ввода пробы можно нагревать; она соединяет поток газа-иосителя с колонкой (см1, рис. 5.2-1). Она герметично закрывается диафрагмой из силиконовой резины, называемой септа.
Пробу вводят в систему, протыкая септу. Ввод следует осуществить так, чтобы получалась пробка пара. Медленный ввод пробы приводит к широким пикам и трудностям в количественном обсчете хроматограмм. При использовании набивных колонок обычно работают с объемами вводимой пробы 0,5-20 мкл. В капиллярной ГХ объемы до 0,001 мкл могут вводиться в колонку с использованием разделения газового потока в делительных системах ввода пробы. Системы ввода пробы, работающие автоматически, обеспечивают во время ввода пробы воспроизводимость до относительной погрешности 0,5%.
Температура испарителя обычно задается примерно на 50°С выше температуры кипения наименее летучего компонента вводимой смеси.
Заполнение колонок и термостаты колонок
Разделяющие колонки можно изготавливать из трубок, материалом которых служит нержавеющая сталь, стекло или плавленый кварц. В настоящее время плавленый кварц приобретает все большее распространение в качестве материала колонок. Прочность колонок поддерживается полиимидным покрытием. Колонки для предварительного нагрева помещают в термостат. Существует различие между набивными и капиллярными колонками. В набивных колон
Глава 5. Хроматография
250
ках неподвижная фаза нанесена на гранулированный материал-носитель. Этот наполнитель помещается в колонку с внутренним диаметром от 3 до 8 мм и длиной от 1 до Зм. Капиллярные колонки не содержат материала-носителя. В этом случае жидкая неподвижная фаза распределена на стенках капилляров. Например, жидкость попадает в колонку при пропускании через колонку концентрированного раствора неподвижной фазы. Другой путь состоит в выпаривании (с использованием вакуумного насоса) растворителя из колонки, полностью заполненной сильно разбавленным раствором. Капилляры могут быть длиной до 100 м и внутренним диаметром 0,15-1 мм.
Короткие набивные колонки устанавливаются внутри термостата колонки выпрямленными или в U-образной форме. Более длинные набивные колонки и капилляры монтируют в термостатах в виде спиралей диаметром от 10 до 30 см.
Перед использованием колонки должны быть хорошо прогреты в потоке газа-носителя для удаления остатков растворителя или для активации силикагеля или молекулярных сит.
Детекторы
Наиболее универсальными детекторами в газовой хроматографии являются катарометр и пламенно-ионизационный детектор (ПИД). Для специфического детектирования соединений все шире применяется масс-спектрометрический детектор (ГХ-МС). Кроме того, используют и другие принципы детектирования, обеспечивающие селективность или высокую чувствительность. Наиболее важные способы детектирования описаны ниже.
Катарометр
В этом детекторе теплопроводность газа-носителя—гелия или водорода—снижается в присутствии определяемого вещества. Теплопроводность гелия и водорода примерно в 6-10 раз выше, чем у органических соединений. Другие газы-носители не могут использоваться в этом детекторе, поскольку отличие их теплопроводности от теплопроводности детектируемых веществ слишком мало. Теплопроводность определяют, измеряя сопротивление нагретой металлической нити (рис. 5.2-2). Измеряемое сопротивление и сопротивление сравнения включены в электрический мост. Катарометр работает пропорционально концентрации. Поскольку катарометр — неизбярательный детектор, его мож-
т
Газ
Рис. 5.2-2. Катарометр.
5.2. Газовая хроматография
251
Таблица 5.2-1. Детекторы для ГХ
Детектор Детектируемые компоненты Предел обнаружения Линейный диапазон
Катарометр Неизбирателен 10“8 г/мл 104
пид Соединения, содержащие СН 1(Г13 г/с 107
ЭЗД Электроотрицательные группы 5 10“14 г/с 5 ТО4
тид Р 10-1' г/с 10'
N 10“14 г/с
пфд Р 3 10“13 г/с 10'
S 2 10“" г/с
ПИД — пламенно-ионизационный детектор, ЭЗД—электронозахватный детектор, ТИД — термоионный детектор, ПФД — пламеннофотометрический детектор
но использовать для детектирования как органических, так и неорганических соединений.
Как можно видеть в табл. 5.2-1, предел обнаружения и линейный (динамический) диапазон у катарометра хуже, чем у других детекторов. Он не годится для капиллярной ГХ из-за его низкой чуствительности. Преимуществом же катарометра является его недеструктивный характер действия на определяемые компоненты.
Пламенно-ионизационный детектор (ПИД)
В настоящее время ПИД—наиболее широко используемый детектор. Принцип детектирования основывается на изменении электрической проводимости водородного пламени в электрическом поле при попадании в пламя органических веществ. Органические соединения, выходящие из колонки, подвергаются пиролизу и, следовательно, распадаются на фрагменты. При последующем окислении кислородом, попадающим в пламя, образуются ионы:
СН* + О СНО+ + е“ (5.2-9)
Поток ионов регистрируется как падение напряжения на собирающем электроде (рис. 5.2-3)
ПИД чувствителен ко всем соединениям, содержащим связи С~С или С-Н. Именно поэтому он и получил широкое распространение. Относительно низкая чувствительность вплоть до полной нечувствительности может наблюдаться по отношению к таким функциональным группам, как карбонильная, спиртовая, галогенная или аминогруппа. Детектор также нечувствителен к негорючим газам: Н20, С02, S02, NO-c-
Поскольку детектор реагирует на число атомов углерода в единицу времени, его сигнал пропорционален массе детектируемого вещества. Следовательно, изменения в скорости потока подвижной фазы оказывают лишь незначительное влияние на сигнал детектора. ПИД характеризуется очень низким
252
Глава 5. Хроматография
Рис. 5.2’3. Принципиальная схема пламенноионизационного детектора.
пределом обнаружения и большим линейным диапазоном (табл. 5.2-1). Однако компоненты пробы разрушаются при детектировании.
Электронозахватный детектор (ЭЗД)
Принцип работы электронозахватного детектора (ЭЗД) близок к принципу действия пропорционального счетчика для измерения рентгеновского излучения. Под действием /^излучателей, таких, как 63Ni или тритий, в потоке газа-иосителя происходит ионизация и появляются электроны. В отсутствие детектируемых соединений ток, протекающий через ячейку, остается постоянным. Уровень тока уменьшается в присутствии органических соединений, особенно если эти соединения могут захватывать электроны. Среди них такие соединения, которые содержат электрофильные (электроотрицательные) группы—галогены, пероксиды, хиноны, фталаты или нитрогруппы. Таким образом, ЭЗД можно использовать, например, в качестве высокочувствительного специального детектора для хлорсодержащих инсектицидов.
ЭЗД нечувствителен к аминам, спиртам и углеводам. Другие аналитические характеристики представлены в табл. 5.2-1.
Специальные детекторы
Термоионный детектор (ТИД). ТИД используется как высокоспецифичный детектор для соединений, содержащих азот и фосфор (табл. 5.2-1). Его чувствительность к этим элементам примерно в 10000 раз выше, чем к углероду. ТИД—пламенный детектор с безводородной газовой смесью. Между горелкой и собирающим электродом на платиновой проволоке закреплена стеклившая частица, содержащая рубидий. Вокруг нее формируется плазма, в которой со-
5.2. Газовая хроматография
253
• Р=Е Q=F=S Рис. 5.2-4. Радикалы фосфора в термоионном детектировании.
единения, содержащие N и Р, образуют радикалы, например:
^C-N^ —-C=N|
(5.2-10)
Эти радикалы реагируют с паром рубидия вокруг стеклянной частицы, давая ион рубидия:
•C=N| + Rb*~* |C=N|“ +Rb+ (5.2-11)
Катион рубидия захватывается отрицательно заряженной стеклянной частицей, в то время как цианид-ион либо движется непосредственно к собирающему электроду, либо сгорает под действием электронов.
На рис. 5.2-4 представлены радикалы, участвующие в детектировании фосфора.
Спектроскопические детекторы
Пламенно-фотометрический детектор (ПФД). ПФД—простейший фотометрический детектор для селективного детектирования фософра и серы. Органические соединения частично сгорают в водородно-воздушном пламени, при этом с помощью фотоумножителя измеряется эмиссия при 526 нм для фосфора и при 394 нм для серы.
Атомно-эмиссионный детектор (АЭД). АЭД также работает с использованием эмиссионных эффектов. Это элемент-специфичный детектор, основанный на атомной эмиссии элементов, таких, как N, Р, S, С, Si, Hg, Вт, Cl, Н, D, F или О. Атомизация и испускание света проходит в гелиевой микроволновой плазме (см. разд. 8.1). Детектирование эмиссии света проводится с использованием фотометра с диодной матрицей в диапазоне длин волн от 170 до 780 нм.
Точность определения элементов в АЭД не очень высокая, относительное стандартное отклонение от 2 до 20%. Несмотря на это, АЭД может быть использован для получения данных об относительном элементом составе. Чувствительность детектора особенно высока для углерода, фосфора и серы. Динамический диапазон значительно меньше, чем, например, для ПИД.
Элемент-специфичное детектирование особенно важно при анализе сложных смесей. Так, например, спирты можно селективно детектировать в бензиновой смеси, измеряя линии атомной эмиссии кислорода.
Масс-спектрометрический детектор (МСД). Прямое сочетание масс-спектрометрии с колонкой возможно в капиллярной ГХ благодаря низким скоростям потока газа-носителя (от 1 до 25мл/мин). В случае набивных колонок поток газа-носителя должен быть разделен с помощью делителя потока.
Более детально возможности сочетания ГХ с МС, так же, как с ИК-спектроскопией, описаны в гл, 14.
254
Глава 5. Хроматография
5.2.4. Неподвижные фазы для газо-жидкостной хроматографии
Для создания эффективных колонок необходимо добиться высокого числа тео-ретичесих тарелок, хорошей селективности неподвижной фазы и высокой степени емкости колонки по жидкой фазе, что обеспечивается высоким уровнем покрытия иммобилизованной жидкостью.
Различие между набивными и капиллярными колонками для газовой хроматографии уже было описано. Сначала в ГХ использовали исключительно набивные колонки. В них на носитель нанесена неподвижная фаза. Как правило, набивные колонки в длину составляют менее 5 м. Абсолютный максимальный предел —20 м, при этом колонка получается очень громоздкой, а падение давления, которое пропорционально плотности набивки, становится очень большим. Таким образом, для набивной колонки число теоретических тарелок менее 10000.
С применением капиллярных колонок Голеем (1958) эти ограничения были преодолены. Поскольку неподвижная фаза наносится иа внутренние стенки капилляров, поток газа-иосителя не затруднен. Поэтому длины колонок могут достигать 100 м, а число теоретических тарелок —100 000 и более. Кроме того, при этом объем подвижной фазы значительно больше, чем неподвижной.
Материалы носителя для набивных колонок
Идеальный материал носителя неподвижной фазы состоит из маленьких гомогенных сферических частиц, которые химически инертны, термически и механически устойчивы и имеют удельную площадь поверхности от 0,5 до 4 м2/г. Размер частиц носителей составляет около 150-250мкм (что соответствует американским мерам измерения 100-60 меш).
О Диатомовые земли (диатомит) состоят из кремневых скелетов ископаемых диатомовых (кремневых) водорослей.
Диатомит — наиболее часто используемый материал носителя. Он содержит до 90% аморфной кремневой кислоты и представляет собой скелетные отложения диатомовых водорослей ископаемого происхождения. В этих водорослях транспорт питательных веществ происходил через поры посредством молекулярной диффузии. Диатомит отлично подходит в качестве носителя для ГХ, поскольку обеспечивает тот же тип молекулярной диффузии. В зависимости от удельной площади поверхности оптимум емкости по непожвижной жидкой фазе достигается при ее содержании от 5 до 30%.
Коммерчески доступны также другие материалы для носителей на основе силикагеля (см. разд. 5.3.1), пористых стеклянных шариков или полимеров (см. табл. 5.2-4).
Капиллярные колонки
Иммобилизировать жидкую неподвижную фазу в капилляре можно двумя способами. Неподвижная фаза может наноситься либо на внутреннюю стенку капилляра в виде пленки (полый капилляр с покрытыми стенками —ПКПС), либо на пористую подложку толщиной примерно 30 мкм (полый капилляр с
5.2. Газовая хроматография
255
покрытым Носителем—ПКПН) (рис. 5.2-5). В последнем случае также может быть использован диатомит. ТЬикослойные капилляры отличаются от тонкопленочных более высокой нагрузочной емкостью, поскольку содержат значительно большее количество неподвижной фазы. Однако эффективность тонкослойных капилляров ниже и находится между эффективностью набивных и тонкопленочных колонок.
Рис. 5.2-5. Колонки в газо-жидкостной хроматографии.
Основной материал колонок — плавленый кварц, который производят из вы-сокоочищенного кварца с очень низким содержанием оксидов металлов.
Сравнительная характеристика набивных и капиллярных колонок представлена в табл. 5.2-2.
Таблица 5.2-2. Характеристики набивных и капиллярных колонок для газовой хроматографии
Параметр Набивные ПКПС ПКПН
Длина, м 1-5 10-100 10-100
Внутренний диаметр, мм 2-4 0,1-0,75 0,5
Эффективность, число тарелок на метр 500-1000 1000-4000 600-1200
Количество вводимой пробы, нг 10-106 10-1000 10-1000
Относительное давление Высокое Низкое Низкое
Инактивация поверхности
Необработанные специальным образом носители или капиллярные колонки представляют проблему для пользователя, поскольку все еще содержат на поверхности силикагеля остаточные силанольные группы (рис. 5.2-6), что приводит к физической адсорбции полярных соединений, таких, как спирты или ароматические углеводороды. Это затрудняет десорбцию и приводит к размыванию пиков на хроматограмме и появлению пиков с размытым задним фронтом.
Для предотвращения нежелательных взаимодействий остаточные силанольные группы инактивируют диметилсилильными или триметилсилильны-
он ОН он он
— I I -о I ,о I Si Si Si Si
Рис. 5.2-6. Поверхность полностью гидролизованного силикагеля.
256
Глава 5. Хроматография
ми группами. При этом- формируется гидрофобная поверхность. Такаягинакти-вация включает реакцию силикагеля с дихлорсиланом и доследующую стадию промывки метанолом:
Z Vя- / V*1
—si-он +а-я-а —► —я-о—s-a +на
XI 4 си.
сн, с“>
/Г» /Г’
—s-о—я-а ♦tUj-он—» —s-o—s-oCHj+na
4 сн, 4 сн,
Остаточные центры активности появляются из-за загрязнения оксидами металлов. Этой проблемы нет в капиллярах из плавленого кварца, если они сделаны из высокоочищенных материалов.
Неподвижные фазы
Жидкости, используемые в качестве неподвижных фаз в ГХ, должны быть термически и химически устойчивыми. Они должны быть малолетучими, чтобы ие происходила их десорбция с колонки. Температура кипения неподвижной фазы должна быть примерно на 100°С выше требуемой температуры колонки. Разделяющая жидкость должна обладать определенной селективностью, т. е. должна обеспечивать различные коэффициенты распределения для определяемых веществ. Однако коэффициенты распределения не должны быть ни слишком большими, ни слишком малыми, иначе удерживание соединений будет слишком сильным или настолько слабым, что не будет разделения.
О Для повышения термостабильности колонок проводят сшивку неподвижной фазы непосредственно внутри колонки после ее покрытия полимером.
При выборе разделяющей жидкости применимо правило <Подобное растворяется в подобном».
Типичные полярные фазы содержат функциональные группы —CN, — С=О, —ОН, —CF3 или полиэфирные. Они обладают отличной селективностью по отношению к спиртам, органическим кислотам и аминам. Неполярные фазы содержат углеводороды или силоксаны, которые подходят для разделения насыщенных или галогенированных углеводородов. Вещества средней полярности, такие, как простые эфиры, кетоны или альдегиды, следует разделять на соответствующим образом модифицированных фазах. В табл. 5.2-3 указаны температурные диапазоны и полярности основных разновидностей разделяющих жидкостей. Структурные формулы некоторых из них представлены на рис. 5.2-7.
Химически связанные фазы занимают особое место среди неподвижных фаз. Они обладают более высокой термостабильностью и не десорбируются с колонки, поскольку разделяющая жидкость химической реакцией ковалентно связывается с поверхностью материала носителя (носитель или внутренняя стенка капилляра).
5.2. Газовая хроматография
257
Таблица 5.2-3. Жидкие неподвижные фазы в ГХ (некоторые формулы см. рис. 5.2-7)
Разделяемые соединения Фаза ТЪыперо-тпура, °C Полярность
Углеводороды Сквалан 20...150 Неполярная
Аполан-87 50...300 Неполярная
Полигликоли Полиэтиленгликоль (CARBOWAX) 50...225 Полярная
Сложные Этиленгликоль сукцинат 100...200 Сильнополярная
эфиры Д ии.чл пени няни п ат 20... 125 Среднеполярная
Соединения, содержащие азот 1,2,3-Трис- (2-цианоэтокси)пропан (CYANO В) 110...200 Полярная
Силиконы Метилсилоксаны (OV-1, SE-30) 20...300 Неполярная
Фенилсилоксаны (OV-22) Средяетюлярная
Нитрилосилоксаны (ОЕ-4178) Сильнополярная
В принципе, разделяющие фазы могут быть получены также с использованием подходящего модифицирования, в результате чего возможно решение большинства хроматографических задач (см. табл. 5.2-3).
Разнообразие взаимодействий разделяемых веществ с неподвижной фазой затрудняет выбор подходящей фазы для конкретной задачи анализа. При этом
/С. и. Н ед I /Ч8**37
37 18 ^CH-fCH^jC—Сн^-сн «37°* СЛ S^37
Аполан-87
ИО-СН—CHj-to-CHj-CHj-bOH
Пояиэтмленгяиколь
О
О
HO-CHj-CHp О-С-СНрСН—С—O-CHf
он
Полизтиленгликольсукцивэт
CH—O-CHj-CHj-CN CH—O-CHj-CHj-CN CHj-O-CHj-CHj-CN
1,2,3-Трис-(2-цианозтокси)пропан
Рис. 5.2-7. Структурные формулы некоторых жидких фаз.
CHj—-Si—О—
—Si—О-I _сн,
Si—СИ, СИ,
Полмдиметилсилоксаны
9 АдАлт 7—» Том 1
258
Глава 5. Хроматография
следует учитывать следующие типичные взаимодействия между выделяемым веществом и неподвижной фазой:
— неспецифические дисперсионные силы (силы Лондона), типичные для алканов и бензола;
— ориентационные силы (силы Кессома) между постоянными диполями, например, в водородных связях;
— индукционные силы (силы Дебая) между постоянными и наведенными диполями;
— силы химических связей в виде комплексов с переносом заряда, например, между ароматическим углеводородом и ионом металла в хиральной фазе.
О Хирэльные фазы используют для разделения оптических изомеров (энантиомеров). Их получают, например, из оптически активных аминокислот.
Поэтому Роршнайдер и позже Мак-Рейнольдс разработали характеристические параметры, позволяющие характеризовать типичные взаимодействия со стандартными веществами (бензол, этанол, бутанон, нитрометан, пиридин) и иа основании этих данных выбирать селективные фазы. Принцип определения таких данных заключается в аддитивном накоплении индексов удерживания для веществ с прибавляющимися функциональными группами и типами СВЯЗИ.
5.2.5. Применение газо-жидкостной хроматографии
Газовую хроматографию можно использовать просто для разделения соединений, например при исследовании чистоты вещества или препаративном выделении вещества из смеси. Однако химик-аналитик в первую очередь заинтересован в использовании ГХ для качественного и количественного анализа, что и будет освещено ниже.
Качественный анализ
На хроматограмме качественная информация содержится в данных об удерживании, или, точнее, в исправленном объеме удерживания или в истинном времени удерживания (см. разд. 5.2.1). Если есть подходящее стандартное соединение для сопоставления с исследуемым, то это неизвестное соединение может быть идентифицировано по его расположению на хроматограмме.
При этом, однако, простой перенос абсолютных данных по удерживанию на другие хроматографические условия недопустим. Как уже отмечалось, удерживание сильно зависит от следующих экспериментальных условий:
— температура колонки;
— скорость газа-носителя;
— тип газа-носителя;
— падение давления в колонке;
— тип и количество неподвижной фазы;
— размеры колонки (длина и диаметр).
В принципе можно исключить некоторые влияния, если данные удерживания исправить на удельный объем удерживания (ур. 5.2-6). Однако на практике
5.2. Газовая хроматография
259
это выполнить слишком сложно. Поэтому прилагают усилия, чтобы либо определить относительные параметры удерживания, либо гарантировать идентификацию с использованием спектроскопических измерений. Для последнего обычно служит масс-спектрометрия.
Индексы удерживания Ковача
Относительные данные удерживания определяют по методу Ковача (1958), основанному на том, что внутри гомологического ряда н-алканов существует линейная зависимость между логарифмом исправленного времени удерживания и числом атомов углерода в соединении. Удерживание изучаемого соединения связано с удерживанием н-алканов и может быть определено следующим образом:
Индекс удерживания I (индекс Ковача) соединения равен 100-кратному числу атомов углерода гипотетического н-алкана с таким же временем удерживания, как и у определяемого соединения.
Согласно этому определению я-алканы имеют индекс, равный 100-кратному числу атомов углерода в молекуле при любых температурах на всех разделяющих колонках, например 600 для я-гексана, 800 для н-октана.
Для определения индекса удерживания определяемое вещество хроматографируют в смеси, которая содержит по крайней мере два н-алкана. При этом время удерживания определяемого вещества должно находиться между временами н-алканов.
Расчет индекса проводится по следующей формуле:
I = 100у ( \ +1002 (5,2.12)
где йи, — время удерживания определяемого вещества х, н-алкана с числом углеродных атомов z и я-алкана с числом углеродных атомов z + у, при этом у—добавочное число атомов углерода по сравнению с z.
Индексы удерживания определены для многих соединений и доступны в виде табличных данных для большинства обычных неподвижных фаз. Они относительно независимы от температуры.
Пример
На рис. 5.2-8 изображена газовая хроматограмма для определения индекса Ковача бензола. В этом примере бензол хроматографируют вместе с я-пеитаном и н-гептаном. С учетом времени удерживания н-пентана (z = 5) tes = 2,0 мин, н-гептана (z+y = 5+2) £r(e+2) = 2,8 мин и £rx = 2,56 мин для бензола получается следующий индекс удерживания:
/=w02(^i^)+100-5 = 640
На рис. 5.2-9 изображена процедура определения индекса Ковача бензола на колонке со скваланом при 60° С.
Количественный анализ
Мы ввели высоту и площадь пика в качестве меры концентрации соединений иа хроматограмме (разд. 5.1). Для надежного анализа прежде всего необходимо:
260
Глава 5. Хроматография
Рис, 5.2-8. Газовая хроматограмма для определения индекса удерживания Ковача.
Рис, 5.2-9. Определение индекса Ковача бензола с использованием н-алканов на колонке со скваланом при 60° С.
— полное и воспроизводимое испарение пробы;
— достаточное разделение и правильная идентификация определяемого вещества.
Кроме того, если детектор реагирует линейным образом, то и высота, и площадь пика находятся в линейной зависимости от массы определяемого вещества (или, в случае детектора по теплопроводности, — от его концентрации).
Изотермическая ГХ и ГХ с программируемой температурой
Коэффициент распределения, описываемый в газо-жидкостной хроматографии через объемы (ур. 5.2-4), зависит от температуры, как и любая константа равновесия. Объемы удерживания также зависят от давления пара соединений (ур. 5.2-8). Повышение температуры вызывает увеличение давления пара и, следовательно, ведет к более высокой скорости элюирования. Эта корреляция определяется зависимостью Клаузиуса—Клайперона. В интегральной форме она представляется следующим образом:
lgP=2^T+COnSt (5-2'13)
где AVH—молярная энтальпия испарения чистого вещества. Согласно этому, давление пара уменьшается и, как может быть показано, время удерживания также увеличивается логарифмически при уменьшении температуры.
5.2. Газовая хроматография
261
□ Программирование температуры аналогично градиентному элюированию в ЖХ и программированию давления в СФХ.
В изотермической ГХ температура в колонке поддерживается постоянной. Этот метод приемлем в ограниченном диапазоне температур кипения разделяемых соединений. Однако для смесей с большим диапазоном температур кипения (> 100°С) возникают проблемы. Если выбирается слишком высокая температура, то пики появляются на хроматограмме слишком быстро и они неполностью разделены. При слишком низких температурах увеличивается время анализа, а соединения с высокой температурой кипения появляются в виде размытых пиков в конце хроматограммы, и их сложно обрабатывать.
Эти недостатки можно преодолеть с использованием ГХ с программируемой температурой. Для этого температуру во время анализа поднимают, как правило, непрерывно или через равные промежутки ступенчатым образом. Начальная температура выбирается при этом таким образом, чтобы наиболее легколетучие компоненты могли бы быть оптимально разделены. Соединения с более высокими температурами кипения сначала удерживаются в начале колонки и начинают двигаться по ней лишь при повышении температуры.
Сравнение изотермической ГХ и ГХ с программируемой температурой для разделения смеси спиртов представлено на рис. 5.2-10.
Таким образом, с использованием программирования температуры могут быть разделены удобным для анализа образом смеси с широким диапазоном температур кипения. Дополнительным преимуществом для количественных определений является то, что пределы обнаружения и точность обработки пиков могут оставаться при этом постоянными на всей хроматограмме.
5.2.6. Адсорбционная хроматография
Исторически именно адсорбционная хроматография была использована первой. Основа газо-твердофазной хроматографии—адсорбирующая среда в качестве неподвижной фазы. Разделение происходит за счет процессов сорб-ции/десорбции. Оно может проводиться как в набивных, так и в капиллярных колонках. В принципе, в качестве полой капиллярной колонки может быть использован капилляр, внутренняя стенка которого активирована. Более типична иммобилизация адсорбента на внутренней стенке капилляра. По аналогии с колонками ПКПН такие колонки называются тонкослойными капиллярами или полыми капиллярами с пористым слоем (рис. 5.2-11).
По сравнению с распределительной хроматографией адсорбционная хроматография обладает следующими преимуществами:
— широкий температурный диапазон;
— хорошая стабильность базовой линии, что особенно важно для ГХ с программируемой температурой и для сочетания ГХ с МС;
— быстрое установление равновесия (экспресс-анализы).
К сожалению, имеются и существенные недостатки:
— асимметричные пики из-за малого линейного интервала изотермы адсорбции;
— высокие энтальпии адсорбции, приводящие к большому времени анализа;
Глава 5. Хроматография
1 I I ।-------1--------1----
48 05 142 190 237 285
Температура, °C
Рис. 5.2-10. Разделение смеси спиртов с использованием изотермической ГХ при 175° С (а) и ГХ с программируемой температурой (б).
Рис. 5.2-11. Колонки в адсорбционной хроматографии.
- гетерогенные поверхности и каталитическая активность многих адсорбентов;
- небольшое количество адсорбирующих сред, которые к тому же трудно стандартизовать.
В качестве неподвижной фазы используют неорганические адсорбенты, такие, как молекулярные сита (силикаты алюминия) или графитизированная сажа, а также пористые полимеры, такие, как сополимеры стирола и дивинил-
5.2. Газовая хроматография
263
бензола. Удельные поверхности адсорбционных фаз значительно больше, чем в распределительной хроматографии (см. табл. 5.2-4).
Таблица 5.2-4. Твердые наполнители колонок
ТЪп Торговал марка ЛГаксимоль-нал рабочая температура, °C Удельная площадь поверхности, л?/г Использование
Диатомит CHROMOSORBA GASCHROM 400 0,5-4 Носитель для ГЖХ
Силикагель PORASIL 400 1,5-500 Все хроматографические задачи
Активирован- 400 1300 Неорганические
ный уголь газы
Сополимеры CHROMOSORBB 275 50-800 Низкомолекулярные,
полистирола PORAPAK Р, Q, Т 250 100-600 полярные вещества
Тефлон CHROMOSORB Т 250 7-8 Чрезвычайно полярные вещества
Адсорбционная хроматография имеет особое значение для разделения газов— водорода, азота, кислорода, метана, диоксида углерода и инертных газов, а также легких углеводородов.
Вопросы и задачи
1. На набивной колонке со скваланом в качестве жидкой неподвижной фазы хлороформ (Ткип 61°С) элюируется перед четыреххлористым углеродом (Ткип 77°С). При использовании в качестве неподвижной фазы нитрилосилоксана порядок удерживания обращается. Объясните различный порядок элюирования на основании уравнения Херингтона.
2. Сравните фазовое отношение для двух капилляров с внутренним диаметром 0,24 мм и 0,36 мм и одинаковой толщиной пленочного слоя dp = 0,2 мкм. Какая из колонок больше подходит для анализа смеси низкокипящих соединений, а какая — для анализа смеси высококипящих или термолабильных соединений?
3. Как влияет на высоту тарелки скорость подачи газа-носителя?
4. Какой из вариантов газовой хроматографии лучше использовать для:
а) анализа многокомпонентной пробы с широким диапазоном температур кипения;
б) определения остаточных мономеров в полимерной пленке;
в) анализа смеси аминокислот;
г) разделения изомеров?
5. Почему масс-спектрометр особенно подходит в качестве детектора в ГХ? Какие инструментальные проблемы могут возникать при использовании различных вариантов ГХ?
6. Опишите кратко методы идентификации неизвестных соединений в ГХ.
7. В чем разница между различными видами открытых капиллярных колонок, используемых в ГХ?
Глава 5. Хроматография
264
5.3. ЖИДКОСТНАЯ ХРОМАТОГРАФИЯ
Особенность жидкостной хроматографии (ЖХ) — жидкая подвижная фаза. В классической жидкостной хроматографии, предложенной М. С. Цветом в начале XX века (1903г.), использовали стеклянные колонки, имевшие внутренний диаметр от 1 до 5 см и длину от 50 до 500 см. Для обеспечения достаточных скоростей потока (до 1 мл/мин) использовали частицы размером 150-200 мкм. При этом, однако, разделение часто занимало много времени.
Попытки увеличить скорость потока использованием вакуума или насосов не позволяли добиться существенного улучшения, поскольку, как уже известно из теории хроматографии, простое увеличение линейной скорости потока подвижной фазы приводит к снижению эффективности колонки. Достаточно быстро пришли к заключению, что более высокая эффективность может быть достигнута лишь уменьшением размера частиц сорбента. Также было понятно, что требуемое для малых размеров частиц давление не может быть достигнуто с помощью обычного оборудования д ля ЖХ.
Кардинальные изменения произошли в конце 1960-х годов, когда были введены в употребление частицы диметром от 3 до 10 мкм и новое поколение оборудования. Классическая жидкостная хроматография используется в настоящее время в препаративных целях, а также для демонстрационных экспериментов. В этом разделе будут рассмотрены наиболее важные элементы скоростной жидкостной хроматографии— высокоэффективной жидкостной хроматографии (ВЭЖХ).
5.3.1. Высокоэффективная жидкостная хроматография (ВЭЖХ)
Принципы разделения и эффективность
Разделение с использованием жидкой подвижной фазы могут быть основаны на четырех принципах:
— адсорбция;
— распределение;
— ионный обмен;
— эксклюзия.
□ Аффинная хроматография может рассматриваться как пятый принцип разделения а ЖХ. Этот аид разделения основан на специфических взаимодействиях между молекулами отделяемого вещества и молекулами, закрепленными на неподвижной фазе. Например, антитела, иммобилизованные на неподвижной фазе, специфически взаимодействуют с определенным белком из пробы.
Исторически первым использованным принципом разделения была адсорбция в форме бюидко-твердофазной хроматографии. Она служит для разделения неполярных веществ, изомеров и соединений таких классов, как, например, алифатические углеводороды и алифатические спирты. Принцип распределения используется в Жидко-жидкостной хроматографии. К этому методу относится нормалъно^фазовая и обращенно-фазовая хроматография. Последний
5.3. Жидкостная хроматография
Рис. 5.3-1. Влияние размера частиц сорбента в ЖХ на высоту тарелки Н.
метод в настоящее время наиболее широко используется в анализе. Хроматография на основе ионного обмена подразумевает перенос в колоночную хроматографию классического ионного обмена, проводимого в статическом режиме. Высокоэффективный вариант ионообменной хроматографии называется ионной хроматографией. Принцип эксклюзии основан на эффекте молекулярного сита: соединения определенного молекулярного размера не могут проникать в поры используемого сорбента в отличие от молекул меньшего размера. Соответствующий метод называется гелъ-хроматографией или эксклюзионной хроматографией.
Как правило, в хроматографии редко осуществляется какой-то один механизм разделения — адсорбция, распределение, ионный обмен или эксклюзия; чаще в разделении в большей или меньшей степени участвуют несколько механизмов одновременно.
Размер частиц сорбента
Из динамической теории хроматографии можно сделать вывод, что высота тарелки непосредственно связана с размером частиц сорбента через коэффициент массопереноса См (табл. 5.1-3). Следовательно, уменьшение размера частиц снижает высоту тарелки и повышает эффективность разделении колонки.
Рис. 5.3-1 показывает изменение высоты тарелки в зависимости от линейной скорости потока подвижной фазы для разных размере» частиц сорбента. Как уже отмечалось, минимум функции Н(й) в ЖХ может наблюдаться, однако, лишь при очень малых, не используемых на практике скоростях потока.
Аппаратура
Классическое устройство для жидкостной хроматографии предусматривает наличие следующих модулей:
— емкость для элюента, содержащую подвижную фазу. В самом простом случае это может быть капельная воронка;
— разделяющая колонка, сделанная из стекла, обычно с внутренним диаметром около 1 см н длиной около 30 см; сорбент удерживается в колонке стеклянным пористым фильтром или стекловатой (см. рис. 5.1-1);
— шприц или инжекционный кран для подачи раствора пробы;
2U
Глава 5. Хроматография
Рис. 5.3-2. Схема устройства для ВЭЖХ с предколонкой.
- коллектор фракций, с помощью которого вручную или автоматически собираются фракции по нескольку миллилитров; фотометр с проточной ячейкой служит для непрерывного детектирования в элюенте.
При использовании частиц с размером 3-10 мкм колонки и соединительные трубопроводы должны поддерживать скорость потока около 1 мл/мин, что соответствует давлению до 15 МПа. В этом случае не может быть использовано классическое устройство для жидкостной хроматографии. Модули устройства для ВЭЖХ представлены на рис. 5.3-2. Устройство состоит из емкостей для растворителей, являющимися компонентами подвижной фазы, насоса, системы подачи пробы, или инжектора, предколонки (необязательно), разделяющей колонки и детектора. Для уменьшения размывания пиков мертвый объем системы, особенно системы ввода пробы и детектора, должен быть минимальным.
Растворители
Растворители, используемые в качестве подвижной фазы, хранятся в емкостях, которыми служат бутыли из стекла или нержавеющей стали. Растворенные газы, которые могут приводить к образованию пузырьков и мешать детектированию, а также нерастворенные вещества должны быть удалены из растворителей. Простейший способ очистки растворителя — пропускание его
5.3. Жидкостная хроматография
267
Рис. 5.3-3. Форма градиента для бинарного элюента, состоящего из метанола и воды.
через мембранный фильтр под вакуумом. Растворенные газы, такие, как азот и кислород, могут также быть удалены пропусканием инертного газа (например, гелия) или выдерживанием в ультразвуковой бане.
Существуют два вида элюирования — изократическое и градиентное. При изократическом методе работают с одним растворителем постоянного состава, например, 30% метанола и 70% воды. Лучшее разделение достигается с использованием градиентного элюирования. При этом состав элюента непрерывно изменяется по определенной программе. Обычно используют растворители различной полярности. Например, в водно-метанольном элюенте доля метанола увеличивается с 30 до 70% (об.) линейно или по другой программе (см. рис. 5.3-3). Эффект градиента растворителя сравним с эффектом температурного градиента, известного в ГХ. Достигается сокращение времени анализа, а пики можно обрабатывать с одинаковой точностью по всей хроматограмме.
Насосные системы
К насосам для ВЭЖХ предъявляются следующие требования:
— генерация давления до 15 МПа;
— слабые остаточные пульсации;
— химическая стойкость;
— обеспечение скорости потока от 0,1 до 10 мл/мин;
— воспроизводимость и контроль скорости потока с относительной погрешностью не более 0,5%.
Для изготовления насосов применяются нержавеющая сталь, тефлон, керамика. Стойкость к давлению обеспечивается сапфировыми клапанами. Существуют вытеснительные и возвратно-поступательные насосы. Вытеснительные насосы работают по принципу шприца, поэтому называются также шприцевыми. Около 200 мл подвижной фазы всасывается внутрь и выдавливается без пульсаций в хроматографическую систему. Такие насосы, однако, имеют серьезный недостаток — прерывание подачи подвижной фазы для заполнении цилиндра и промывки поршня. Вытеснительные насосы и сегодня используются в микроколоночной ВЭЖХ, поскольку в этом виде хроматографии требуются небольшие расходы подвижной фазы, но в основном в настоящее время ис-
268
Глава 5. Хроматография
пользуются возвратно-поступательные насосы (рис. 5.3-4). Как правило, они работают как двухплунжерные (двухпоршневые) насосы с фазовым сдвигом движений на 180° для уменьшения пульсаций давления. Такие насосы также называют обратноколебательными вытеснительными насосами. Для предотвращения прямого контакта растворителя с плунжером насоса возможен вариант плунжерно-диафрагменных насосов. В этом случае движение плунжера передается на диафрагму с использованием гидравлики.
Преимуществами короткопоршневых насосов являются малые внутренние объемы (от 40 до 400мкл), высокое давление на выходе (до 60МПа), а также постоянный поток, не зависящий от давления, создаваемого колонкой, и вязкости растворителя.
Градиент может создаваться либо со стороны насоса, где имеется низкое давление, либо с другой стороны, с высоким давлением. Если два или три растворителя для получения элюента смешиваются со стороны всасывания насоса, речь идет о градиенте низкого давления.
Для создания градиента высокого давления требуется два насоса. Один компонент или, в случае тройной смеси, два компонента присутствуют в постоянном отношении. Третий компонент смешивается с выходной стороны насоса согласно запрограммированному градиенту.
Градиенты высокого давления обеспечивают более точный Состав элюента, чем градиенты низкого давления, поскольку объемное сжатие при смешении различных растворителей более существенно в варианте градиентов низкого давления. Для предотвращения повреждения насоса и загрязнения колонки компоненты элюентов должны быть предварительно профильтрованы через одномикронный фильтр.
Система ввода пробы
Система ввода пробы должна обеспечивать введение объемов от 5 до 500 мкл. Кроме того, она должна выдерживать давление в системе. Поэтому используют систему ввода пробы в виде петлевого дозатора. С помощью микролитре-
5.3. Жидкостная хроматография
2W
Рис. 5.3-5. Кран ввода пробы для ВЭЖХ. а—заполнение дозирующей петли; б — ввод пробы в систему
кого шприца раствор пробы через иглу поступает во входное отверстие петли 6-ходового крана (рис. 5.3-5). При этом элюент после насоса направляется непосредственно в колонку. Прн переключении крана определенный объем пробы поступает в систему под действием протекающего через петлю элюента. Затем кран переводится в прежнее положение.
Для высокоточного ввода пробы предпочтительны системы, работающие в автоматическом режиме. Они основаны на том же принципе петлевого дозатора и переключаются с помощью сжатого воздуха (или электричества. — Иерее.).
Сорбенты
Внутренняя сторона колонки обычно представляет собой полированную поверхность из нержавеющей стали. Шероховатая поверхность приводила бы к уменьшению числа теоретических тарелок, т. е. к уменьшению эффективности колонки. Кроме того, используют колонки из специальных сортов стекла (дюран, пирекс). Такие колонки вставляются в систему в виде картриджей (рис. 5.3-2). Соединения с трубопроводами из нержавеющей стали делают с Помощью фиттингов (конические металлические запирающие кольца). Стандартная колонка изготавливается длиной 250 мм й внутренним диаметром 4,6 мм, заполнена она частицами размером 5 или Юмкм. При этом обычно достигается эффективность около 50 000 теоретических тарелок на метр.
Для сокращения расхода растворителей, которые для целей ВЭЖХ должны быть очень чистыми, все чаще используют микроколонки с внутренним диаметром до 1мм и длиной от 30 до 75 мм (микроколоночная ВЭЖХ). Использование частиц размером 3 мкм может повысить эффективность колонок до 100000 теоретических тарелок на метр.
Для зашиты разделяющей колонки или для предварительного разделения часто применяют короткие предколонки. Их могут заполнять более крупным сорбентом, чтобы не использовать слишком высокие давления.
Глава 5. Хроматография
270
Заполнение колонок частицами размером менее 20 мкм является отдельной проблемой. Высокая поверхностная энергия сорбента и связанный с ней электростатический заряд не позволяют проводить сухое заполнение. При заполнении колонок с помощью жидкости следует избегать градиента размера частиц в колонке за счет седиментации.
Для заполнения колонки сорбент суспендируется в жидкости. Для заполнения колонок сорбентами с обращенными фазами может быть использован метанол. Еще лучших результатов можно достичь с использованием плавающих суспензий. При этом разница в плотностях твердой фазы и жидкости компенсируется специфически выбираемой диспергирующей жидкостью, такой, как дибромметан.
□ Плавающие суспензии — особый аид суспензии твердого вещества а жидкости (например, «жидкая» глина).
Детекторы
Для детектирования в ВЭЖХ существуют два варианта:
1. Измерение общей характеристики подвижной фазы, например показателя преломления или электропроводности. В этом случае детектируемое вещество измеряется косвенно по изменению характеристики подвижной фазы.
2. Измерение характеристики самого детектируемого вещества, например его УФ-поглощения, флуоресценции или диффузионного тока на электроде.
□ Детекторы в ВЭЖХ могут быть разделены на детекторы общих свойств, например, рефрактометрический детектор, и детекторы свойств определяемого соединения, например, УФ-детектор.
УФ-детектор наиболее часто используется, детектирование с его помощью составляет свыше 70% случаев.
Абсорбционные детекторы
Чтобы измерить светопоглощение элюента на выходе из колонки, используют проточную ячейку. Устройство жидкостной проточной ячейки Z-образной формы изображено на рис. 5.3-6. Для уменьшения размывания пиков размер ячейки делают минимальным — от 1 до 10 мкл. Длина оптического пути колеблется от 2 до 10 мм. Для измерений в УФ-диапазоне оптические окна ячейки должны быть сделаны из кварца.
Наиболее простой вариант УФ-детекторов — фильтровые фотометры. В этом случае из света ртутной лампы выделяется эмиссионная линия и она используется для детектирования. Обычно используют линии 254 нм, поскольку все ароматические соединения имеют максимум поглощения при этой длине волны и большинство органических, а также неорганических веществ поглощают в этой спектральной области.
Детектор с изменяемой длиной волны может работать и при одной длине волны, которая задается при помощи решеточного или призменного монохроматора аналогично сканирующему фотометру.
5.3. Жидкостная хроматография
271
Рис. 5.3-6. Жидкостная проточная ячкйка для УФ-детектирования в ВЭЖХ.
и», (н Поглощение
Время, мин
Рис. 5.3-7. Трехмерная диаграмма сигнала детектора с фотодиодной матрицей при определении фенантрена.
Спектрофотометры с фотодиодной матрицей используются для получения полного УФ-спектра. Информация представляется в виде трехмерной, или контурной, диаграммы, где поглощение отображается в зависимости от времени удерживания и длины волны (рис. 5.3-7). По контурной диаграмме очень легко выбрать длину волны, наиболее подходящую для отдельного вещества.
Пределы пропускания растворителей для ВЭЖХ в УФ-области спектра даны в табл. 5.3-2.
В принципе, в качестве детекторов в ВЭЖХ можно использовать фотометры и в ИК-области спектра. Однако в настоящее время их применение ограничено лишь специальными случаями из-за сильного поглощения типичных элюентов, таких, как вода и метанол.
Глава-5. Хроматография
2Т2
Флуоресцентные детекторы
На флуоресцентных детекторах достигается чувствительность примерно в 1000 раз выше по сравнению с УФ-детекторами. Для возбуждения в флуоресцентных детекторах наиболее часто используется ртутная лампа. Ксеноновые лампы высокого давления также применяются, но для особых задач. Кроме того, длины волн возбуждения и испускания можно выбирать при помощи монохроматоров, или в качестве детектора можно использовать флуоресцентный спектрометр. Для анализа лекарств или природных соединений часто используется собственная флуоресценция веществ. Для детектирования нефлуоресцирующих соединений следует получить их производные (см. рис. 5.3-12).
Рефрактометрический детектор (рефрактометр)
Измерение коэффициента преломления подвижной фазы является универсальным способом детектирования, поскольку этот показатель совершенно неизбирателен и реагирует практически на все соединения. В основе метода лежит различие между коэффициентами преломления чистого элюента и элюента, содержащего компоненты пробы.
В принципе, для измерений можно использовать как свет, отраженный призмой, так и отклоненный. На рис. 5.3-8 изображено устройство рефрактометрического детектора, работающего на принципе отражения. В этом случае отраженная часть света не измеряется непосредственно, иначе угол обзора будет непрерывно меняться. Поэтому свет измеряется после прохождения через элюент и отражения от стальной пластины. Одновременно стальная пластина играет роль термостата.
Рефрактометры состоят из измерительной ячейки н ячейки сравнения (дифференциальный рефрактометр). Рефрактометрический детектор значительно менее чувствителен, чем, например, УФ-детектор. Его высокая температурная чувствительность также является существенным недостатком, поскольку детектор должен очень точно термостатироваться,—если возможно, до ±0,001°С. Этот детектор не подходит для градиентного элюирования. Он используется для соединений, которые не поглощают свет в УФ-области, например для сахаров.
К детектору
Отраженный луч
Рис. 5.3-8. Принцип действия рефрактометрического детектора, измеряющего отраженный свет после прохождения его через элюент.
\
\ 5.3. Жидкостная хроматография 273
\
Электрохимические детекторы
Для Электрохимического детектирования можно использовать вольтамперометрию, амперометрию, кулонометрию и кондуктометрию.
Кондуктометрическое детектирование широко распространено в ионной хроматЬграфии (разд. 5.3.2), где работают с проточными кондуктометрами.
Кулонометрическое детектирование возможно, если определяемое вещество может быть трансформировано кулонометрически генерируемым реагентом, например, галогенид е кулонометрически получаемыми ионами Ag+. Кулонометрические детекторы применяются достаточно редко.
Вольтамперометрическое детектирование подразумевает запись характеристики ток-потенциал с помощью динамического (например, ртутного капающего) или стационарного электрода. Чтобы не останавливать поток элюента, изменение потенциала на рабочем электроде должно быть достаточно быстрым по сравнению со скоростью потока подвижной фазы, а иначе будет невозможно измерить диффузионный ток. Для этого требуется скорость сканирования потенциала 1 В/с. Кроме того, требуется использование полностью обратимых окислительно-восстановительных реакций. Поэтому детектирование на основе вольтамперометрии остается лишь для специальных исследований'.
Амперометрическое детектирование более предпочтительно на практике. К рабочему электроду, изготовленному из стеклоуглерода, золота или платины, прикладывается постоянный потенциал, и предельный диффузионный ток при данном потенциале измеряется относительно электрода сравнения. Чтобы не загружать электрод сравнения, подключают вспомогательный электрод (рис. 5.3-9).
Амперометрический детектор подходит для всех соединений, которые способны восстанавливаться или окисляться в диапазоне установленного рабочего электрода. Чтобы не допускать поглощения кислорода из элюента, окислительные процессы необходимо тщательно регулировать. Селективное и достоверное детектирование биохимических веществ, таких, как гормоны стресса адреналин н норадреналин (катехоламины), в пикомолярном диапазоне подтверждает важность использования амперометрического детектора.
Рис. 5.3-9. Электрохимический детектор со стеклоуглеродным рабочим электродом.
274
Глава 5. Хроматограф*»
Недостатком амперометрических детекторов является опасность отравления поверхности электрода в случае проб, содержащих поверхностно-активные вещества, например белки в сыворотке крови или детергенты в сточных водах.
Спектроскопические детекторы
Мы уже познакомились с одним спектроскопическим детектором — детектором с фотодиодной матрицей. Другие возможности, используемые на практике,—это комбинации ВЭЖХ с масс-спектрометрией для идентификации органических соединений и с атомно-абсорбционной спектрометрией или с атомно-эмиссионной спектрометрией для определения элементов. Особенности, связанные с этими моментами, освещены в главе о сопряженных методах (гл. 14).
Распределительная хроматография
Жидкостная хроматография, основанная на распределении,—наиболее широко распространенный метод по сравнению с методами, использующими адсорбцию, ионный обмен и молекулярную эксклюзию. Методом распределительной хроматографии обычно определяют незаряженные полярные соединения с относительной молярной массой до 3000. Современные варианты позволяют определять также высокомолекулярные соединения.
Неподвижные фазы
В качестве неподвижных фаз в жидкостной хроматографии могут служить жидкости, иммобилизованные на носителе. В других случаях носитель может быть модифицирован с помощью химической реакции, при этом получаются химически закрепленные фазы.
Иммобилизованные жидкости
В классической ЖХ работают с жидкостями, которые удерживаются на носителе за счет физической адсорбции. Типичным носителем является силикагель с площадью поверхности 10-500 м^г или оксид алюминия с площадью поверхности 60-200м2/г. Неподвижными фазами служат полярные жидкости, такие, как вода и триэтиленгликоль. Подвижная фаза—неполярная, как, например, гексан или диизопропиловый эфир.
Хроматография, в которой используется полярная неподвижная фаза и менее полярная подвижная фаза, называется нормально-фазовой хроматографией. Хроматография с противоположным расположением фаз (относитель
\
\ 5.3. Жидкостная хроматография 275
но ^полярные неподвижные фазы, например, углеводороды, н полярные подвижные фазы, например, вода или метанол) называется обращенно-фазовой хроматографией.
ЖХ с физически иммобилизованными жидкостями в качестве неподвижной фазы сохранили свое значение лишь в классической нормально-фазовой хроматографии. Из-за непрочной связи жидкости с носителем должно быть предусмотрено его насыщение жидкостью из подвижной фазы. Перед разделением неподвижная и подвижная фазы насыщаются соответствующим растворителем или же жидкая неподвижная фаза подбирается как часть подвижной фазы (фазы с контролируемым растворителем). Даже прн таких предосторожностях при применении жидких неподвижных фаз нельзя использовать градиентное элюирование.
В связи с этим более важными с практической точки зрения являются фазы с химически (ковалентно) закрепленными жидкостями.
Химически закрепленные фазы
Неподвижные фазы этого типа могут быть приготовлены н для нормально-фазовой и для обращенно-фазовой хроматографии. В настоящее время хроматография на обращенных фазах составляет около 75% всего использования ВЭЖХ.
Обращенные фазы
В качестве материала носителя наиболее часто применяется силикагель- кроме того, используются оксид алюминия или полимерные ионообменники. В отличие от классических полимерных ионообменных материалов, силикагель является жестким н ненабухающим материалом, т. е. поглощение гелем воды не изменяет его объем.
Силикагель используют в виде однородного, пористого (объем пор около 1,2 мл/г), механически стойкого материала с размером частиц 3, 5 или Юмкм. Сферические частицы силикагеля предпочтительнее 'частиц неправильной формы.
Поверхность полностью гидролизованного силикагеля состоит из силанольных (гидроксильных) групп, как уже было показано выше (рис. 5.2-5). Их количество на поверхности составляет около 8 мкмоль/м2. Для гидрофобизации силикагель подвергают обработке алкилхлоросиланами, при этом в качестве химически привитых фаз образуются силоксаны (группы Si—О—Si).
Наиболее часто используются алкильные группы Cie (н-октадецил), а также Се (н-октил). Длинные углеводородные группы располагаются параллельно друг другу н перпендикулярно поверхности частиц. Они образуют щеточную поверхность (рис. 5.3-10).
Поверхность обращенной фазы можно рассматривать как жидкость, хотя до настоящего времени не совсем понятно, является лн удерживание веществ результатом их физической адсорбции на поверхности или поверхность ведет себя как жидкая углеводородная среда.
276
Глава 5. Хроматография
Фаза С8
Рис. 5.3-10. Часть поверхности Св (октильной)-обращенной фазы. Остаточные силанольные группы, выступающие с поверхности, частично связаны водородными связями, а также кислородными атомами в силоксановые связи.
Время удерживания, мин
0 4 в 12 16
Время удерживания, мин
Рис. 5.3-11. Сравнение удерживания с использованием силоксановых обращенных фаз с алкильными группами метил (Ci) н октил (Cg)-1 —урацил, 2 — фенол, 3 — ацетофенон, 4—нитробензол, 5 — метилбензоат, 6 - толуол. Подвижная фаза —50/50 (об.) метанол/вода; скорость потока 1 мл/мин.
а о
Удлинение алкильной цепи ведет к увеличению удерживания соединений (рис. 5.3-11). Кроме того, максимально допустимое количество пробы зависит от типа углеводорода. Так, количество пробы примерно удваивается при переходе от фазы С4 к фазе Cis-
Когда силикагель силанизируют, поверхность может быть покрыта силанольными группами не более, чем на 50%, что соответствует примерно 4мкмоль/м2. Поверхностные силанольные группы являются сильно полярными и могут приводить к адсорбции полярных соединений (рнс. 5.3-10). Это заметно на хроматограммах — видны пики с размытым задним фронтом. Поэтому остаточные силанольные группы стараются инактивировать, насколько это возможно. Для этого их покрывают трнметилхлорсиланом и метилсилок-саном (см. разд. 5.2).
Силоксановые фазы устойчивы в полярных подвижных фазах, содержащих воду, метанол или ацетонитрил прн pH от 2 до 8. Прн pH выше 8 они гидролизуются, прн этом происходит разрушение и реорганизация сорбента.
Нормальные фазы
Химически привитые фазы важны и в нормально-фазовой хроматографии. В табл. 5.3-1 приведены примеры полярных функциональных групп химически
5.3. Жидкостная хроматография
277
привитых фаз для этого вида хроматографии. Функциональные группы приведены в порядке уменьшения полярности.
Таблица 5.3-1. Полярные функциональные группы в качестве химически привитых фаз в нормально-фазовой хроматографии
Группа Функциональная группа
Диол -(СН2)3ОСН2СН(ОН)СН2ОН
Циано -(CH2)3C=N
Амино ~(CH2)nNH2 с п — 3 или 4
Диметиламино -(CH2)3N(CH2)2
Диамино ~(CH2)3NH(CH2)2NH2
Материалы носителя
Для предколонок в качестве носителя обычно используют частицы из непористого стекла или полимера диаметром около 30мкм. Поверхность таких материалов покрывают пористым слоем силикагеля, оксида алюминия или полимерного ионообменника (поверхностно-пористые сорбенты).
Жидкость может адсорбироваться на этих слоях в качестве неподвижной фазы или закрепляется на поверхности за счет химической модификации.
©
Силикагель
Подвижные фазы
В отличие от ГХ, где подвижной фазой служит инертный газ, в ЖХ наблюдаются значительные взаимодействия между разделяемыми веществами и жидкой подвижной фазой. Поэтому при разработке метода выбор подвижной фазы в ЖХ является важным фактором.
Из теории хроматографии известно, что удерживание и появление на хроматограмме компонентов пробы согласно ур. 5.1-23 зависит от числа теоретических тарелок N, коэффициента емкости к' и коэффициента селективности а.
Значения кг я а можно варьировать, изменяя свойства подвижной фазы путем смены растворителей. При этом наиболее важной характеристикой является полярность.
В принципе, сначала выбирают неподвижную фазу, которая имеет полярность, близкую к полярности разделяемых компонентов. Затем выбирается подвижная фаза—таким образом, чтобы по возможности к' оказался в интервале от 2 до 5. Если полярность подвижной фазы слишком близка к полярности
278
Глава 5. Хроматография
неподвижной, получаются слишком малые времена удерживания, а если полярности подвижной и неподвижной фаз различаются очень сильно, времена удерживания получаются слишком большими.
Элюотропные ряды
Для определения полярности растворителей были созданы элюотропные ряды. Обычно используют индекс полярности Снайдера, который выделил «сильные* полярные и слабые (слабополярные или неполярные) растворители. Основа шкалы полярности—измерение растворимости в диоксане, нитрометане и этаноле. В табл. 5.3-2 указаны индексы полярности Р' некоторых растворителей для ЖХ. Полярность увеличивается от неполярных, «слабых» алканов к наиболее полярному из всех растворителей — воде.
Таблица 5.3-2. Элюотропный ряд растворителей для жидкостной хроматографии в порядке увеличения полярности
Растворитель Индекс полярности Р' Элюирукицал сила (SiOz) УФ-прозрачность, нм
Фторалкан < -2 -0,2 200
Циклогексан 0,04 0,03 200
н-Гексан 0,1 0,01 195
Тетрахлорметан 1,6 0,11 265
Диизопропиловый эфир 2,4 0,22 220
Толуол 2,4 0,22 285
Диэтиловый эфир 2,8 3,1 0,38 215
Дихлорметан 0,34 230
Тетрагидрофуран 4,0 0,35 210
Хлороформ 4,1 4,3 0,26 235
Этанол 0,68 205
Уксусная кислота 4,4 0,38 255
Диоксан 4,8 0,49 215
Метанол 5,1 0,73 205
Ацетонитрил 5,8 0,50 190
Нитрометан 6,0 0,49 380
Вода 10,2 Высокая 170
Для расчета полярности смеси растворителей индексы полярности индивидуальных растворителей суммируются. Например, расчет полярности водно-метанольной смеси с объемным отношением 70/30 будет выглядеть так:
7*4 ета нол /в ода — 0,ЗРметанол + 0,7Рвода = 1,53 + 7,14 = 8,67
или в общем виде для смеси т растворителей:
(5.3-1)
где Ф, — объемная доля растворителя г.
5.3. Жидкостная хроматография
279
Элюирующая сила
Данные по элюирующей силе, или силе растворителей, eo(Si02), которые относятся к силикагелю в качестве неподвижной фазы, представлены в табл. 5.3-2. Понятно, что подобные значения зависят от выбранной неподвижной фазы, что однако не исключает возможности относительной оценки растворителей с использованием индексов полярности.
Следовательно, данные по элюирующей силе, приведенные в табл. 5.3-2, в первую очередь специфично применимы к полярным неподвижным фазам в адсорбционной хроматографии на силикагеле или, после деления на 0,8 — на оксиде алюминия. Если используют неполярные неподвижные фазы, элюо-тропный ряд обращается. Например, вода как сильно полярный расворитель обладает невысокой элюирующей силой на углеводородных фазах по сравнению с неполярным гексаном. Полярность и элюирующая сила должны, следовательно, рассматриваться по отдельности, несмотря на то что для некоторых комбинаций фаз они могут быть идентичны.
В заключение следует рассмотреть удерживание в нормально- и обращенно-фазовой хроматографии. В нормально-фазовой ЖХ, где полярность неподвижной фазы больше полярности подвижной фазы, полярные соединения элюируются последними. Их время удерживания тем выше, чем более неполярная подвижная фаза используется. Напротив, в обращенно-фазовой хроматографии полярные соединения элюируются первыми, а чем более полярная подвижная фаза используется, тем сильнее удерживаются неполярные соединения.
Выбор подвижной фазы
Наиболее рационально подвижную фазу можно было бы выбирать на основании действующего механизма разделения. Однако часто механизмы удерживания перекрываются или они не могут быть заново изучены для каждой хроматографической задачи. Поэтому состав подвижной фазы часто выбирают путем испытаний различных смесей растворителей, взятых на основе имеющегося у оператора опыта, или путем систематической процедуры согласно принципам многопараметрической оптимизации (см. разд. 12.4).
Установление характеристик удерживания, т е., например к' разделяемых соединений, автоматически не обеспечивает реального разделения, которое определяется коэффициентом селективности а. Смена разделяющей колонки часто означает значительное изменение коэффициента селективности. Далее, для улучшения коэффициента селективности и вообще селективности может также быть оптимизирован состав подвижной фазы.
В обращенно-фазовой хроматографии для такой оптимизации используют, например, смесь метанола, ацетонитрила и тетрагидрофурана. Воду используют для установления значений к'.
В случае смешанных растворителей можно последовательно настолько точно регулировать элюирующую силу, что возможно получать хроматограммы с наилучшим разрешением; этот прием позволяет решать большое число хроматографических задач.
280
Глава 5. Хроматография
В нормально-фазовой хроматографии аналогичным образом используют смеси диэтилового эфира, дихлорметана и хлороформа. Элюирующая сила модифицируется н-гексаном.
Применение распределительной хроматографии
Если предстоит решать новую аналитическую задачу, прежде всего, как правило, выбирают ВЭЖХ на привитых обращенно-фазовых сорбентах. В отличие от большого числа неподвижных фаз в газовой хроматографии, неподвижные фазы в ВЭЖХ ограничены использованием практически единственной С18-обращенной фазы. При этом регулирование удерживания или селективности достигается варьированием состава подвижной фазы.
Обращенно-фазовую ВЭЖХ используют практически во всех областях, где необходимо отделять полярные соединения. Среди них фармацевтический, биохимический, криминалистический, клинический, промышленный анализы, а также проверка продуктов питания или определение вредных соединений, например пестицидов, полициклических ароматических углеводородов (ПАУ), полихлорированных бифенилов (ПХБ).
Если при изократическом элюировании не удается достичь требуемой селективности, обычно прибегают к использованию градиента растворителей. Кроме того, элюирующая сила подвижной фазы может быть изменена варьированием величины pH или добавлением ион-парных реагентов. В обоих случаях целью является получение в подвижной фазе нейтральных частиц, которые могут взаимодействовать с неполярной обращенной фазой. В случае ион-парной хроматографии определяемое соединение ионного характера реагирует с соответствующим противоионом. Примеры определений в присутствии нон-парных реагентов представлены в табл. 5.3-3.
Таблица 5.3-3. Ион-парные реагенты для обращенно-фазовой ВЭЖХ
Определяемое соединение Противоион Подвижная фаза
Амины ск>7 0,1 м НСЮ-л/вода/ацетонитрил
Карбоновые кислоты (C4H9)4N+ pH 7,4
Сульфоновые кислоты ((C16H33)CH3)3N+ Вода, пропанол
Селективным регулированием уровня pH можно достичь преобладания той кислотной или основной равновесной формы, которая требуется в данном случае.
Еще один способ изменить свойства системы в целом для решения аналитической задачи — дериватизация определяемых веществ. Таким образом может быть изменена полярность соединения, увеличена чувствительность его детектирования, а отклик может стать более селективным. Например, аминокислоты дериватизуют в гидролизатах белка реакцией с дансилхлоридом (1-диметиламинонафталин-5-сульфонилхлоридом), при этом получаются флуоресцирующие соединения (рис. 5.3-12).
5.3. Жидкостная хроматография
281
Рис. 5.3-12. Дериватизация аминогруппы аминокислоты или пептидного остатка (R-NHa) с использованием дансилхлорида с образованием флуоресцирующего производного.
Рис. 5.3-13. Образование комплекса хираль-ной фазы, полученной из оптически активного комплекса L-пролина с медью(П), для разделения энантиомеров аминокислот.
Энантиомеры разделяют с использованием хиралъных фаз. Используемые в этом случае сорбенты состоят из силикагеля в качестве носителя и полимерного покрытия, к которому химически прикреплены оптически активные молекулы. Взаимодействие с хиральной фазой может быть основано, например, на комплексообразовании, как показано на рис. 5.3-13 для определения оптически активной аминокислоты с ипользованием комплекса L-пролина с медью(П).
Особое использование нормально-фазовой хроматографии—разделение ПАУ с дополнительными алкильными цепями. Здесь важная цель —определять следы потенциально канцерогенных и мутагенных соединений в воздухе, сточных водах и промышленных технологических растворах. Хотя незамещенные ПАУ можно разделить на обращенных фазах, их алкилированные аналоги невозможно разделить по размеру кольца. В этом случае в качестве нормальной неподвижной фазы может быть использована диаминовая фаза, а в качестве элюента—гептан.
Другие примеры разделений на нормальных фазах—групповые разделения алканов или липидов, а также разделение стероидов, сахаров и жирорастворимых витаминов.
282
Глава 5. Хроматография
Адсорбционная хроматография
Адсорбционная, или жидко-твердофазная, хроматография — старейший метод, впервые примененный М. С. Цветом. В качестве неподвижной фазы используют силикагель и оксид алюминия. Силикагель более приемлем, поскольку он имеет более высокую нагрузочную емкость и доступен в различных вариантах исполнения.
Основой удерживания соединений являются различные адсорбционные процессы на твердом адсорбенте, когда молекулы подвижной фазы конкурируют с молекулами определяемого вещества. Адсорбция энергетически более выгодна по сравнению с распределением между двумя жидкостями. Более сильные взаимодействия и диффузия в неподвижную фазу обусловливают меньшие времена удерживания по сравнению с распределительной хроматографией. Однако изотермы адсорбции линейны лишь в небольшом диапазоне концентраций, поэтому легко происходит перегрузка колонки.
Адсорбция локализована на адсорбционных центрах поверхности. Сильно полярные молекулы, такие, как вода, могут инактивировать поверхность, поэтому содержание воды в используемом растворителе является важной характеристикой в адсорбционной хроматографии.
Поскольку неподвижная фаза более полярна, чем подвижная, которая состоит, к примеру, из дйхлорметана н изооктана, адсорбционная хроматография не вполне правильно считается нормально-фазовой хроматографией.
Для оценки элюирующей силы растворителей предпочтительнее вместо индекса полярности использовать элюирующую силуг (табл. 5.3-2). Элюирующая сила—это величина энергии адсорбции растворителя на единицу площади поверхности. Для перевода значений элюирующей силы системы на силикагеле к значениям для систем на оксиде алюминия приведенные величины следует разделить на 0,8. Практически нет различий в удерживании на силикагеле и оксиде алюминия в качестве неподвижных фаз. Времена удерживания увеличиваются в общем в такой последовательности:
Алкены < ароматические углеводороды < галогенированные соединения и сульфиды < простые эфиры < нитросоединения < сложные эфиры ~ спирты амины < сульфоны < сульфоксиды < амиды < карбоновые кислоты.
Адсорбционная хроматография особенно подходит для разделения неполярных соединений, которые малорастворимы в воде и поэтому не могут быть разделены с использованием распределительной хроматографии. Как и в случае распределительной хроматографии, достаточно просто разделяются вещества с различающимися функциональными группами.
С помощью адсорбционной хроматографии также могут быть эффективно разделены позиционные изомеры и стереоизомеры. На рнс. 5.3-14 изображено два примера таких разделений.
Так же, как и в распределительной хроматографии, может быть проведена систематическая оптимизация состава подвижной фазы. В некоторых случаях для оценочной проверки дополнительно используют тонкослойную хроматографию, которая может быть отнесена к плоскостному варианту адсорбционной хроматографии.
5.3. Жидкостная хроматография
283
Рис. 5.3-14. Варианты разделения различно замещенных молекул с использован нием адсорбционной хроматографии, а — стереоизомерные формы цис- и транс-пиразолинов; б—позиционные изомеры азапроизводных фенантрена.
5.3.2. Ионная хроматография как классический и высокоэффективный метод
Высокоэффективный вариант ионообменной хроматографии называется ионной хроматографией. Ионная хроматография служит для разделения и определения ионов с использованием ионообменников. В особенности это касается неорганических анионов.
Динамический метод в ионном обмене основан на принципе элюентной хроматографии (рнс. 5.1-1).
Классическая ионообменная хроматография
Классическая ионообменная хроматография проводится на пористых нонооб-менниках, синтезированных из сополимера стирола и дивинилбензола. Изначально она была разработана для разделения химически очень близких редкоземельных элементов с помощью катионообменников. Определение ионов, собранных по фракциям, осуществляли титриметрическими методами.
О Кроме ионообменной хроматографии ионообменные смолы также используются в статических условиях.
В качестве примера можно рассмотреть разделение ионов металлов в виде хлоридных комплексов наснльноосновном анионообменнике. Через ионообмен-ник пропускается 12 М соляная кислота, а затем вводятся ионы металлов в соляной кислоте такой же концентрации. Никель сразу же выходит из колонки. Это связано с тем, что очень слабые хлоридные комплексы никеля не удерживаются на колонке. Другие металлы элюируются с колонки ступенчатым градиентом при более низких концентрациях соляной кислоты. Последова-
284
Глава 5. Хроматография
Рис. 5.3-151 Разделение ионов металлов в виде хлоридных комплексов на сильно основном анионообменник^ методом ступенчатого градиентного элюирования соляной кислотой.
телыюсть соответствует устойчивости хлоридных комплексов. Например, для ионов меди комплексообразование протекает по следующей реакции:
Си2+ + 4СГ [СиСЦ]2' (5.3-2)
На рис. 5.3-15 изображена ионообменная хроматограмма разделения Ni2+, Мю2+, Со2+, Cu2+, Fe3+ и Zn2+.
Ионы металлов, собранные при различных концентрациях соляной кислоты, могут быть определены индивидуально титриметрически либо фотометрически.
Ионная хроматография
Современная ионная хроматография, разработанная в середине 1970-х годов, использует обычные хроматографические блоки для ВЭЖХ. Необходимо было решить две проблемы. Во-первых, нужно было усовершенствовать классические ионообменные полимерные сорбенты. Они не подходили для использования в высокоэффективном варианте из-за их сжимаемости под действием давления, набухания в растворах и медленной диффузии разделяемых молекул в поры сорбента. Во-вторых, не существовало универсального детектора для детектирования неорганических ионов.
Сорбенты
Классические ионообменные полимерные сорбенты были заменены в иониой хроматографии покрытыми материалами, в которых поверхность непористого стекла или полимерные частицы покрывали слоем ионообменника. Такие сорбенты имели в диаметре 30-40 мкм (пелликулярные ионообменники). Во втором варианте использовали пористый силикагель, аккуратно покрытый жидкими ионообменниками, по аналогии с адсорбционной хроматографией.
В обоих случаях диффузионные процессы заметно ускорялись по сравнению с классическими ионообменниками. Однако емкость новых ионообменных материалов получалась низкой.
5.3. Жидкостная хроматография
285
Кондуктометрический детектор
В качестве универсального принципа детектирования неорганических ионов используется измерение электропроводности. В водных растворах ионы обладают проводимостью, пропорциональной их концентрации. Прямое детектирование разделяемых компонентов в элюенте, как это делается в ЖХ, однако, было невозможно. Концентрация электролитов в элюентах, обычных для ЖХ, была слишком высокой, чтобы можно было распознать очень низкие сигналы элюированных ионов на фоне высокой фоновой электропроводности.
Ионная хроматография с подавляющей колонкой
Проблема детектирования была решена соединением аналитической колонки с подавляющей колонкой и путем селективного выбора подвижных фаз.
При определении анионов в качестве подавляющей колонки используют катионообменник в кислотной форме. Элюент состоит из смеси карбоната и гидрокарбоната натрия. После того, как анионы, например, хлорид н нитрат, разделяются на аналитической колонке, заполненной анионообменником, элюент реагирует в подавляющей колонке согласно следующим уравнениям:
Na+ + НСО3 + Н + <=» Na+ + Н2СО3 (5.3-3)
2Na+ + СО^“ + 2Н + 2Na+ + Н2СО3 (5.3-4)
Черта над символом иона обозначает, что данный ион находится в фазе ноно-обменника. Такой перевод ионов подвижной фазы в непроводящую недиссо-циированную угольную кислоту подавляет электропроводность элюента. В то же время определяемые ноны, например, хлорид н нитрат, не подвергаются превращению в подавляющей колонке и их электропроводность может быть детектирована с очень высокой чувствительностью.
На рис. 5.3-16 изображена хроматограмма определения восьми анионов.
Для определения катионов в аналитических колонках используют кати-онообменники. Подвижной фазой служит соляная кислота, а подавляющая колонка должна содержать анионообменник в ОН-форме.
Прн этом в элюенте происходит реакция нейтрализации соляной кислоты согласно уравнению:
Н+ + СГ + ОН СГ + Н2О (5.3-5)
Снова детектируемые ионы остаются в растворе единственными проводящими соединениями, например катионы Na+ или Mg2+.
Недостатком подавляющей колонки является то, что ее следует регенерировать через определенные промежутки времени (каждые 10 часов), поскольку емкость ее ограничена. В связи с этим в современном оборудовании подавляющие колонки заменяют непрерывно действующим мембранным подавителем. На рис. 5.3-17 изображено устройство мембранной подавляющей системы. Поток элюента окружен двумя ионообменными мембранами, снабжающими элюент ионами Н+ или ОН- в зависимости от типа определения. Мембрана непрерывно обновляется потоком регенерирующего раствора кислоты или основания, направленным навстречу потоку элюента.
286
Глава 5. Хроматография
Рис. 5.3-16. Ионохромвтографический анализ смеси анионов с использованием элюента 2,8 мМ ИаНСОз/2,ЗмМ Na2C03-
Поток регенерирующего Элюент
Поток = : регенерирующего раствора
Рис. 5.3-17. Микромембранный подавитель. Элюент отделен от регенерирующего раствора, текущего в противоположную сторону, ионообменными мембранами.
Вместо слоистого устройства потоков элюента и регенерирующего раствора, как это изображено на рис. 5.3-17, возможно кольцевое устройство с использованием подавителей с полыми волокнами.
Одноколоночная ионная хроматография
Использования подавляющей колонки можно избежать, если электропроводность подвижной фазы поддерживать очень низкой. Для этого необходимо работать с ионообменниками низкой емкости н элюентами с низкой электропроводностью. В качестве элюентов широко используются слабые органические кислоты, такие, как, например, изофталевая, бензойная или салициловая. Уровень pH должен строго контролироваться, чтобы поддерживать ионную силу и фоновую электропроводность элюента. Существующая остаточная электропроводность элюента подавляется в данном случае не ионообменным подави-
телем, а с помощью электроники.
соон
соон
Изофталевая кислота
'Органические ноны, такие, как аминокислоты, также могут быть проанализированы в одноколоночном варианте. Такие определения можно проводить с обычным оборудованием для ВЭЖХ. Однако возможности детектирования в таком случае ниже, чем в варианте с подавлением.
5.3. Жидкостная хроматография
287
Подавляющая колонка также не требуется, если можно использовать фотометрический детектор. Мы уже говорили о косвенном фотометрическом детектировании, когда такое вещество, как изофталевая кислота, поглощающее свет в УФ-области спектра, обеспечивает постоянное фоновое светопо-глощение. Ионы, элюируемые с колонки, измеряют косвенно по замещению изофталевой кислоты.
Прямое фотометрическое детектирование возможно при использовании послеколоночной реакция. Для такого варианта определения ионов металлов также можно использовать мембранную систему (см. рис. 5.3-17). Комплексообразующий реагент, например ПАР (пиридилазорезорцин), вводят в поток элюента через полупроницаемую мембрану. Элюент остается практически бесцветным при протекании чистой подвижной фазы. Если же в элюенте оказываются ионы металла, происходит комплексообразование и развивается интенсивное окрашивание на бесцветном фоне.
5.3.3. Гель-хроматография
Гель-хроматография — особая форма жидкостной хроматографии. Она основана на разделении молекул по их различному размеру (рис. 5.3-18). Все молекулы, имеющие размеры выше определенного, исключаются из силикагеля или полимерного сорбента с определенным размером пор {эксклюзионная хроматография). Молекулы с молекулярной массой ниже эксклюзионного предела сорбента соответствующим образом удерживаются. В отличие от всех методов ЖХ, обсуждавшихся до этого, гель-хроматография не основана ни на каком химическом или физическом взаимодействии с неподвижной фазой.
Гель-хроматография важна для анализа смесей высокомолекулярных соединений, таких, как белки или полимеры.
□ В гель-хроматографии распределение молекул между неподвижной и подвижной фазами основано на размере молекул и частично на их форме и полярности.
Во взаимодействиях молекул с перистой неподвижной фазой можно различить два крайних случая. Молекулы с диаметром, большим, чем средний диаметр пор, полностью исключаются из сорбента и элюируются вместе с подвижной фазой первыми. Молекулы с диаметром, значительно меньшим, чем поры сорбента, могут свободно проникать через него н остаются в неподвижной фазе наибольшее время, поэтому элюируются последними.
*---Подвижная фаза
Гель (неподвижная фаза)
Рис. 5.3-18. Принцип гель-хроматографии.
288
Глава 5. Хроматография
Молекулы среднего диаметра проникают в поры сорбента в зависимости от размера и отчасти в соответствии с формой молекул. Они элюируются с различными временами удерживания между пиками двух крайних случаев.
Обычно в гель-хроматографии для описания удерживания используют объем удерживания, который является произведением скорости потока подвижной фазы и времени удерживания, получаемых из хроматограммы (см. уравнения 5.2-1 и 5.2-2).
Общий объем удерживания К>бщ для колонки, заполненной силикагелем или полимером, равен
Уобщ = V0 + Ур + Кель (5.3-6)
где Vq — мертвый объем, a объем пор. В гель-хроматографии мертвый объем связан с незаполненным объемом и соответствует теоретическому объему для прохождения через колонку полностью исключенных молекул. Для молекул, которые могут свободно проникать в поры, объем может быть определен из общих объемов за вычетом мертвого объема н объема пор, т. е. максимально достижимый объем равен Vo + Vp (рнс. 5.3-19).
Элюируемый объем Ve молекулы, которая находится некоторое время в неподвижной фазе, зависит от мертвого объема и некоторой доли К от объема пор следующим образом:
VE = Vo + KVv (5.3-7)
Коэффициент К соответствует коэффициенту распределения, который в гель-хроматографии может принимать значение от 0 до 1:
в случае, когда молекула полностью исключается, К = О
для молекул, свободно проникающих в поры частиц, К = 1
для частично исключаемых молекул 0 < К < 1
Этот простой подход хорошо описывает только те случаи, когда нет дополнительных взаимодействий, например адсорбционных, возникающих между элюируемыми молекулами и поверхностью геля. Если взаимодействия существуют, коэффициент К может быть больше единицы. В таком случае могут наблюдаться пики, соответствующие объему удерживания Vr, обычно используемому в жидкостной хроматографии (рис. 5.3-19,а), который может находиться вне эксклюзионного диапазона.
Преобразование ур. 5.3-7 позволяет записать коэффициент распределения в обычном виде, аналогичном ур. 5.1-1:
К Ve - Vo = cs (5,3_8)
*р См
Коэффициент распределения является важной характеристикой для сравнения различных сорбентов. Волее того, на основании этой величины все фундаментальные теоретические предпосылки, описанные в разд. 5.1, могут быть перенесены на гель-хроматографию.
5.3. Жидкостная хроматография
289
Рис. 5.3-19. Удерживание и градуировка в гель-хроматографии. а — удерживание стандартных соединений в диапазоне молекулярной эксклюзии с величнами К от О до 1 и относительными молекулярными массами от 10е до 103. Последний (пятый) пик соответствует соединению, имеющему химические взаимодействия с сорбентом; б — логарифмическая зависимость между молекулярной массой и объемом удерживания (элюирования) стандартных соединений; в — определение молекулярной массы на основании хроматограммы неизвестной пробы. Для этого элюирование должно проводиться в тех же условиях, что и градуировка (объем вводимой пробы, скорость потока).
10 Аяалхтячесхм химия. Том 1
290
Глава 5. Хроматография
Неподвижная фаза
В качестве сорбентов используют пористые стекла, частицы силикагеля или полимеры, а также полисахариды. Размер частиц сорбентов 5-10 мкм.
Пористые стекла и силикагели имеют множество преимуществ благодаря быстрому установлению равновесия диффузии элюируемых молекул и молекул растворителя в поры, благодаря высокой устойчивости этих материалов даже при высоких температурах, а также благодаря простоте производства сорбентов. Для них достигаются размеры пор от 40 до 2500 А. Недостатком таких материалов, могут оказаться адсорбционные эффекты, поэтому их поверхность обычно инактивируют силанизацией.
Изначально в качестве полимерных сорбентов использовали лишь сополимеры стирола и дивинилбензола. Размер пор таких смол контролируется количеством дивинилбензола, повышающего степень сшивки. Поскольку данные материалы являются гидрофобными, их можно использовать лишь в комбинации с неполярными подвижными фазами.
В настоящее время доступны также гидрофильные гели, производимые из сульфированного дивинилбензола или полиакриламидных смол.
Размер пор типичных сорбентов представлен в табл. 5.3-4. Он напрямую связан с эксклюзионным пределом данного сорбента. Эксклюзионный предел — это такой размер молекул, выше которого не наблюдается удерживания. Эксклюзионный предел указывается как относительная молекулярная масса (Мг).
Таблица 5.3-4. Типичные сорбенты для гель-хроматографии
Средний размер пор, А Эксклюзионный предел (относительная молекулярная масса)
Сополимер стирола и дивинилбензола
ю2 700
10“ 1-Ю4...20-Ю4
10е 5 10® ...10 • 10®
Силикагель
125 0,2 104.. .5 • 104
500 0,05-10®. ..5 105
1000 5-10®. ..20-10®
Следует отметить, что информация о молекулярных массах, приведенная в табл. 5.3-4, в конечном счете не имеет решающего значения для хроматографического поведения, а скорее описывает объем, занимаемый свернутой сольватированной молекулой в элюенте. Этот объем называется гидродинамическим объемом.
Подвижная фаза
Выбор подвижной фазы зависит от типа используемой неподвижной фазы. Гель-хроматографические системы могут быть разделены на гельфильтрационную и гель-проникающую хроматографию.
5.3. Жидкостная хроматография
291
В гель-фильтрационной хроматографии проводят разделение водорастворимых веществ на гидрофильных сорбентах. При этом используются водные элюенты, содержащие, как правило, буфер для контроля уровня pH. В гель-проникающей хроматографии применяются гидрофобные сорбенты и соответственно неполярные органические растворители, такие, как тетрагидрофуран, дихлорметан или толуол. С помощью гель-проникающей хроматографии определяют соединения, малорастворимые в воде.
Детекторы
Детектирование в гель-хроматографин можно осуществлять с помощью детекторов, с откликом, пропорциональным концентрации (например, дифференциальный рефрактометрический или фотометрический детекторы в УФ-или ИК- области спектра).
Проточный вискозиметр обычно используют в качестве специального детектора для высокомолекулярных веществ. При этом измеряется фоновая вязкость элюента. При прохождении через детектор определяемого высокомолекулярного соединения вязкость возрастает и результирующий сигнал регистрируется в виде пика.
Применение
Гель-хроматография служит для определения веществ с относительной молекулярной массой более 2000, причем гель-фильтрационная хроматография применяется для разделения водорастворимых соединений, а гель-проникающая — для разделения водонерастворимых соединений.
В случае гель-фильтрации отделяют, например, природные высокомолекулярные вещества от более низкомолекулярных соединений или от солей. Таким образом белки могут быть отделены от аминокислот или более низкомолекулярных пептидов, если эксклюзионный предел сорбента составляет несколько тысяч дальтон (см. табл. 5.3-4).
Гель-проникающая хроматография служит для разделения гомологов и олигомеров, например, жирных кислот с относительной молекулярной массой между 100 и 350. Для этого выбирают смолы с эксклюзионным пределом около 1000.
Оба вида гель-хроматографии могут быть использованы для нахождения молекулярных масс или определения распределения по молекулярным массам природных соединений или полимеров.
Молекулярные массы компонентов пробы находят на основе стандартных соединений, имеющих характеристики, близкие к характеристикам определяемых компонентов. Основой градуировки является логарифмическая зависимость между объемом элюирования и относительной молекулярной массой МТ (см. рис. 5.3-19,6). Эта зависимость может быть аппроксимирована в средней части диапазона прямой линией:
1g Мг = Ь0 -MgHs (5.3-9)
где Ьо и 61 — регрессионные параметры, а Ге — объем элюирования (удерживания).
10*
292
Глава 5. Хроматография
Водорастворимыми стандартами могут служить декстран, полиэтиленгликоль, сульфированные полистиролы или белки. Полистирол, по-ли(тетрагидрофуран) и полиизопрен—типичные водонерастворимые стандарты.
Принцип определения молекулярных масс компонентов пробы объясняется на рис. 5.3-19,в.
Преимущества
Точно определенный диапазон эксклюзии между мертвым объемом Vo и суммой мертвого объема и объема пор Vq + Vp обеспечивает малые времена удерживания, определяемые коэффициентом распределения К. Получаются узкие пики, что позволяет проводить их правильную количественную обработку. Поскольку, как правило, проба не вступает ни в какие химические или физические взаимодействия, ее компоненты элюируются без потерь. Следовательно, данный метод пригоден для препаративного разделения. В то же время, проба не влияет на разделяющую колонку, как это происходит в других вадах жидкостной хроматографии.
Недостатки
Из-за узкого диапазона эксклюзии (значение К от 0 до 1) пиковая емкость в гель-хроматографии ограничена (см. табл. 5.1-4). Более того, не могут быть разделены вещества близкого размера, например изомеры. Считается, что разделение на основании молекулярных масс может быть успешным только при различии масс как минимум на 10%.
5.3.4. Тонкослойная хроматография
В предыдущих разделах, посвященных ЖХ, обсуждалась лишь колоночная элюентная хроматография. В этом разделе рассмотрены плоскостные варианты ЖХ, в которых фиксируется внутренняя хроматограмма (см. разд. 5.1).
Плоский слой материала, служащего неподвижной фазой, может сам по себе выступать в роли носителя (например, в бумажной хроматографии), или в роли неподвижной фазы, когда в ваде слоя наносится на подложку из стекла, синтетического материала или металла. Подвижная фаза движется по слою за счет капиллярных сил. Она также может двигаться под действием гравитации или электрического потенциала.
□ Бумажная и тонкослойная хроматография, а также электрохроматография, относятся к плоскостным методам.
Наше рассмотрение будет ограничено наиболее широко распространенным плоскостным методом — тонкослойной хроматографией (ТСХ). ТСХ является модифицированной формой ЖХ. Метод часто применяют как предварительный этап колоночного разделения, поскольку его легче осуществить и разделяющая система может быть опробована быстрее.
ТСХ составляет основу скрининговых тестов в химических, промышленных, клинических, фармацевтических, биохимических и биологических лабораториях.
5.3. Жидкостная хроматография
293
Неподвижные и подвижные фазы
Неподвижными фазами, применяемыми в ТСХ, служат те же материалы, что и в ВЭЖХ для разделений, основанных на адсорбции, распределении (нормально или обращенно-фазовом), ионном обмене и эксклюзии. Эти материалы в виде мелко размолотых частиц наносятся на плоские пластины размером 5 х 10,10 х 20 или 20 х 20 см. При размере частиц 20 мкм достигается толщина слоя от 200 до 250 мкм. Во время развития хроматограммы при ее длине 12 см в течение 25 мин достигается около 200 стадий разделения.
Пластины усовершенствованы для ВЭТСХ (высокоэффективной тонкослойной хроматографии). В этом случае работают со слоями толщиной 100 мкм с размером частиц 5 мкм и меньше. Разделение получается более отчетливое и осуществляется оно за меньший промежуток времени, около 10 мин. Достигается эффективность до 4000 теоретических тарелок при длине Зсм. Однако недостатком высокоэффективных пластин является значительно меньшая нагрузочная емкость.
Нанесение пробы и получение хроматограммы
Нанесение пробы, которая должна содержать 0,01-0,1% определяемого вещества, наиболее просто осуществляется с помощью капилляра. Обычно объем пробы составляет от 0,5 до 5 мл. Каплю пробы наносят на расстоянии 1-2 см от края пластины. Для качественного анализа диаметр полученного пятна должен составлять около 5 мм. Для количественного определения необходим еще меньший диаметр. В ВЭТСХ обычно используют платино-иридиевые капилляры, с их помощью на пластину наносится 100-200 нл. Кроме того, существуют автоматические раздаточные системы, позволяющие отмерять раствор пробы с высокой точностью. Перед началом хроматографического процесса растворитель пробы выпаривается.
Для получения хроматограмм подвижная фаза должна двигаться вдоль пластины по неподвижной фазе. Для этого пластину помещают в закрытую камеру (рнс. 5.3-20). Камера насыщается подвижной фазой. Следует быть осторожным, чтобы пятно пробы не опускалось в растворитель. Растворитель за счет капиллярных сил поднимается по пластине, правда, с неодинаковой скоростью. Скорость миграции фронта растворителя является убывающей гиперболической функцией от длины пути.
Рис. 5.3-20. Камера для проведения тонкослойной хроматографии.
Пластинканоситель
Тонкий слои
Растворитель
294
Глава 5. Хроматография
Когда зона растворителя пройдет две трети пластины, пластину вынимают из растворителя и заканчивают процесс. Растворителю дают испариться, после чего пластина готова к обработке пятен.
Детектирование
При скрининговом тестировании обходятся без инструментального детектирования, ограничиваясь определением расположения пятен и идентификацией веществ таким образом. Чтобы определить расположение пятен на пластине, применяют следующие методы:
а) Использование хемилюминесцентных характеристик определяемого вещества. В случае органических веществ это, как правило, флуоресценция, а для неорганических — фосфоресценция.
□ Визуализация — метод определения расположения определяемого вещества на тонкослойной пластине.
б) Пропитка слоя сорбента флуоресцентным индикатором. При облучении УФ-светом разделившиеся вещества проявляются как тусклые нефлуоресцирующие пятна. В качестве таких индикаторов служат производные пирена, флуоресцеин, морин или родамин В.
в) Опрыскивание слоя неизбирательным сильным окислителем, например HNO3, КМпО4 или H2SO4. При окислении органических соединений на пластине образуются черные пятна.
г) Опрыскивание растворами групповых или специфических реагентов, таких, как нингидрин для визуализации аминогрупп (рис. 5.3-21), хлорид же-леза(Ш) для фенолов, анилинфталат для восстанавливающих сахаров или комплексообразующие реагенты для визуализации ненов металлов.
Вещество может быть очень легко идентифицировано по пятну в случае, если на той же пластине хроматографируется эталонное вещество и сравниваются реакция или цвет пятна неизвестного и эталонного веществ. Идентификация по характеристикам удерживания освещена ниже в разд. «Применение тех».
Важнейший инструментальный метод детектирования — локально разрешенное измерение диффузного светоотражения в УФ- и видимом диапазоне
r-nh2
Нингидрин Синий продукт реакции
Рис. 5.3-21. Образование синего продукта реакции нингидрина в присутствии аминогрупп.
5.3. Жидкостная хроматография
295
спектра с использованием денситометра, называемого также сканером. Может быть также использовано сочетание ТСХ, например с ИК-спектроскопией, в рабочем режиме (on-line).
Сочетание ТСХ в режиме off-line возможно со всеми хорошо известными спектроскопическими методами. Для этого пятна разделенных веществ извлекают с пластины и используют для дальнейшего анализа.
Величина Rf и коэффициент емкости
Отличительной особенностью ТСХ является обработка хроматограмм, которые представляют собой внутренние хроматограммы. В качестве величины удерживания в ТСХ используется относительная скорость Rf. Она определяется как отношение расстояния, пройденного определяемым веществом, zr, к расстоянию, пройденному подвижной фазой, zm (рис. 5.3-22):
Rf = ^ (5.3-10)
zm
Для симметричных пятен определяют их середину, а для асимметричных — используют максимум интенсивности.
Использование основ хроматографии базируется на связи между величиной Rf и коэффициентом емкости.
Промежуток времени, когда определяемое вещество остается в подвижной фазе, можно оценить из пройденного расстояния zr, деленного на линейную скорость потока подвижной фазы и:
(м = (5-3-11)
и
За время <м подвижная фаза проходит расстояние 2м- Тогда время удерживания, соответствующее времени удерживания для внешней хроматограммы, выражается как
«м = (5.3-12)
и
Рис. 5.3-22. Обработка хроматограммы в ТСХ.
296
Глава 5. Хроматография
Если подставить ур. 5.3-11 и 5.3-12 в выражение для коэффициента емкости (5.1-9), получается:
(5.3-13)
или, используя величину Rf.
(5.3-14)
Соотношение между коэффициентом распределения К и величиной Rf может быть получено преобразованием ур. 5.3-14 с использованием фазового отношения (ур. 5.1-6):
1 + Л/ i + рк
(5.3-15)
Пройденное расстояние также может быть использовано для расчета числа теоретических тарелок и высоты тарелки. Для тонкослойной хроматографии они рассчитываются следующим образом:
Число теоретических тарелок: N = 16 (5.3-16)
Высота тарелки: Н — — (5.3-17)
Применение ТСХ
Ранее уже упоминалось о многочисленных применениях ТСХ в качестве легко реализуемого метода скрининга. Важной предпосылкой для этого является возможность параллельного анализа нескольких проб на одной пластине.
□ Селективность разделения в ТСХ может быть улучшена получением хроматограммы в двух направлениях с использованием разных растворителей в каждом направлении:
I ш I I I
Растворитель А—*
В принципе качественный анализ возможен на основании данных удерживания, а также инструментального детектирования.
Однако для этого определение абсолютного значения Rf ничего ие дает, поскольку оно сильно зависит от экспериментальных условий. Среди них толщина слоя, влажность подвижной и неподвижной фаз, температура, степень насыщения паров подвижной фазой в хроматографической камере, размер пробы и пятна. Следовательно, лучше использовать относительный коэффициент
5.3. Жидкостная хроматография 297
удерживания Нотн, определяемый для вещества i по сравнению со стандартным веществом (станд):
Я»™ = „Я™ (5.3-18)
*ч(станд)
Для инструментального качественного и количественного анализа используется фотометрическое определение с помощью сканера. Преобразования нелинейной зависимости сигнала от концентрации осуществляется согласно функции Кубелки—Мунка. Для количественного определения особенно важна коррекция фонового сигнала, который сильно зависит от качества материала слоя.
По сравнению с элюентным вариантом в ВЭЖХ классическая ТСХ имеет ряд серьезных недостатков:
1. Скорость движения фронта растворителя непостоянна. Ее можно изменять, лишь меняя размер частиц материала слоя или тип используемого растворителя. Снижение скорости движения растворителя во время получения хроматограммы приводит к размыванию пятен. Поэтому ограничены длина пройденного расстояния и, следовательно, достигаемое число теоретических тарелок.
2. Благодаря дополнительным взаимодействиям через газовую фазу, а также при движении растворителя по слою состав растворителя во время разделения может изменяться. Это часто приводит к трудно воспроизводимым коэффициентам емкости и, следовательно, к плохой воспроизводимости разделения.
Эти недостатки могут быть частично преодолены получением хроматограмм с принудительным движением растворителя, как, например; в случае ТСХ при повышенном давлении или во вращательной плоскостной хроматографии.
Вопросы и задачи
1. Перечислите свойства растворителей, делающие их подходящими в качестве подвижной фазы в ЖХ.
2. Перечислите типичные детекторы в ВЭЖХ. Какие из них являются неизбирательными, а какие более селективными?
3. В чем различие между адсорбцией растворенных веществ на твердой поверхности и ионообменным процессом?
4. Как можно разделить электролиты и неэлектролиты?
5. Какие методы могут быть использованы для идентификации определяемых веществ в ВЭЖХ?
6. Что делает ТСХ отличным методом для скрининга?
7. Объясните, почему в классической ионной хроматографии необходимо использование подавляющей колонки. Какие возможности существуют для реализации ионной хроматографии в одноколоночном варианте?
298
Глава 5. Хроматография
5.4. СВЕРХКРИТИЧЕСКАЯ ФЛЮИДНАЯ ХРОМАТОГРАФИЯ
В сверхкритической флюидной хроматографии (СФХ) подвижной фазой служит сверхкритический флюид. СФХ объединила важные преимущества газовой и жидкостной хроматографии. Она особенно полезна для определения соединений, которые не определяются ни газовой, ни жидкостной хроматографией. Она применима ко всем веществам, которые, с одной стороны, нелетучи или не могут испаряться без разложения, а следовательно, не могут быть определены в ГХ. С другой стороны, это метод для соединений, которые напрямую нельзя определить и с помощью ЖХ, поскольку они не содержат функциональных групп и поэтому не могут давать сигнал в обычных спектроскопических или электрохимических детекторах для ЖХ.
Число определений веществ, которые не могут быть переведены в газовую фазу или не содержат никаких функциональных групп, составляет 25% от всех хроматографических задач. Такие задачи обычно связаны с анализом природных веществ, фармацевтически активных агентов, продуктов питания, пестицидов, полимеров, неочищенной нефти.
Из физической химии известно, что существует критическая температура вещества, выше которой оно не может быть переведено в жидкую фазу, даже если прикладывается повышенной давление. Давление, соответствующее критической точке, называется критическим давлением. Флюид находится в сверхкритическом состоянии, близком к критической точке, и имеет характеристики, промежуточные между характеристиками газов и жидкостей.
Наиболее важными параметрами для хроматографии в районе критической точки являются плотность, вязкость и коэффициент диффузии. В табл. 5-4-1 сопоставляются эти параметры для газов, сверхкритических флюидов и жидкостей. Необычно высокая плотность сверхкритических флюидов обусловливает чрезвычайно хорошую растворимость в них большого числа нелетучих веществ. Так, диоксид углерода в сверхкритическом состоянии растворяет н-алканы с числом атомов углерода от 5 до 40, а также полициклические ароматические углеводороды (ПАУ).
Таблица 5.4-1. Важные характеристики газов, сверхкритических флюидов и жидкостей
Характеристика Газ Сверхкритический флюид Жидкость
Плотность, г/см3 0,6 10-3 — 2 10~3 Вязкость, г/см- с 1 10“4 — 3 10-4 Коэффициент 1.10-i_4.10-i диффузии, см /с 0,2-0,5 0,6-2 1 10-4 - 3 10-4 0,2* 10~2 — 3-10-2 10“*-10*3 0,2-10-’-2-КГ’
Критические данные для некоторых веществ представлены в табл. 5.4-2. Все эти вещества можно использовать в качестве подвижных фаз в СФХ. Особенно подходящими для СФХ являются температуры около 1,2 Тс м давления от 1 до 3 рс.
Как можно заключить из данных табл. 5.4-2, и критические температуры, и критические давления находятся в пределах условий, обычных для ВЭЖХ
5.4. Сверхкритическая флюидная хроматография
299
Таблица 5.4-2. Критические величины для подвижных фаз в СФХ
Флюид Температура Тс, °C Давление рс, Па Плотность dc, г/см2
со2 31,3 7,39 0,468
n2o 36,5 7,27 0,457
NH3 132,5 11,40 0,235
Метанол 239,4 8,10 0,272
н-Бутан 152,0 3,80 0,228
Ди фтордихлорометан 111,8 4,12 0,558
Диэтиловый эфир 195,6 3,64 0,265
и ГХ. Следовательно, хроматограф для СФХ может состоять из блоков, аналогичных используемым в высокоэффективных ЖХ и ГХ.
Специфические характеристики сверхкритических флюидов могут также быть использованы при сверхкритической флюидной экстракции в процессе пробоподготовки. В промышленности такая экстракция может проводиться для извлечения кофеина из кофе или никотина из табака (получение сигарет с низким содержанием никотина).
Аппаратура
Как уже было упомянуто выше, оборудование для СФХ может быть смонтировано из модулей, используемых в ВЭЖХ и ГХ. Кроме того, в колонке должна быть точная установка температуры. Это достигается использованием колоночных термостатов, типичных для ГХ. Еще одна особенность заключается в том, что прибор должен поддерживать давление в колонке, но в то же время в конце колонки сверхкритический флюид должен детектироваться в виде газа. Для этого используются капилляры длиной 2-1Qcm, которые примерно в 10 раз тоньше (внутр, диаметр 5 или Юмкм) разделяющего капилляра (50-100 мкм). Они называются устройствами поддержания давления или рестрикторами. Простейший способ создания рестриктора—оттягивание разделяющего капилляра в конце колонки (рис. 5.4-1).
Давление в системе нужно точно контролировать, поскольку плотность сверхкритического флюида зависит от давления и изменения давления приводят к изменению коэффициентов емкости. Более высокое давление обеспечивает ббльшую плотность. Это позволяет повышать элюирующую силу подвижной фазы и получать меньшие времена удерживания. Например, увеличение давления диоксида углерода с 7 до 9 МПа приводит к уменьшению времен удерживания с 25 до 5 мин. Значит, в СФХ можно использовать программирование давления в форме градиента, аналогично тому, как программируется температура в ГХ и состав подвижной фазы в ЖХ.
Полиимидное покрытие
Рис. 5.4-1. Оттянутый капилляр в качестве рестриктора в капиллярной СФХ.
300
Глава 5. Хроматография
Неподвижные и подвижные фазы
Неподвижные фазы в СФХ могут реализовываться в форме набивных колонок или капилляров- Набивные колонки похожи иа колонки, используемые в ВЭЖХ для разделений на основе равновесия распределения, т. е. разделяющие колонки имеют внутренний диаметр от 0,5 до 4,6 мм и до 25 см в длину, а заполнены частицами диаметром 3-10 мкм.
В капиллярах из плавленого кварца в качестве неподвижной фазы используются силоксаны, жидкие или химически привитые на внутренних стенках. Обычные характеристики разделяющих капилляров: длина 10-20 м, внутренний диаметр 0,05-1 мм, толщина слоя неподвижной фазы 0,05-1 мкм.
Как указывалось выше, эффективные характеристики разделяющих колонок в СФХ сравнимы с характеристиками в ВЭЖХ или капиллярной ГХ.
Наиболее часто в качестве подвижной фазы используют диоксид углерода. С ним легко обращаться, он достаточно дешевый, неядовитый, без запаха, не поглощает в УФ-диапазоне вплоть до 190 нм. Критические параметры диоксида углерода таковы, что температура и давление могут варьироваться в относительно широких пределах с использованием оборудования, сходного с оборудованием для ВЭЖХ. Иногда к подвижной фазе добавляют органический модификатор, например метанол или диоксан. Другие подвижные фазы для СФХ перечислены в табл. 5.4-2.
Детекторы
Газ, выходящий из рестриктора, может быть неизбирательно продетектирован с помощью пламенно-ионизационного детектора (ПИД), использующегося в ГХ. Предпосылкой для этого является низкий фоновый сигнал, создаваемый подвижной фазой, содержащей СО?, NH3 или N2O-
В случае СФХ значительно проще, чем в ЖХ, осуществить сочетание с масс-спектрометрией, поэтому оно используется достаточно часто. Кроме того, в качестве других детекторов можно применять УФ-, ИК-флуоресцентный или пламенно-фотометрический детекторы, а также катарометр или ЭЗД (см. разд. 5.2).
Эксплуатационные характеристики СФХ
Из сравнения характеристик сверхкритических флюидов с характеристиками газов и жидкостей (табл. 5.4-1) можно сделать два важных заключения:
1. Благодаря низкой вязкости сверхкритических флюидов по сравнению с жидкостями (см. рис. 5.4-2) могут достигаться высокие скорости потоков. Поэтому разделения в СФХ осуществляются быстрее, чем в ЖХ и сравнимы по скорости с разделениями в ГХ.
2. Скорость диффузии сверхкритических жидкостей является промежуточной между скоростями диффузии в ГХ и ЖХ. В связи с этим размывание пиков больше, чем в ГХ, но меньше, чем в ЖХ.
Для объяснения этих зависимостей сравним функцию Н(«) при разделении на колонке с октадецилсиликагелем для СФХ и ВЭЖХ (рис. 5.4-2). В случае СФХ при линейной скорости потока 0,6 см/с достигается высота тарелки
5.4. Сверхкритическая флюидная хроматография
301
Рис. 5.4-2. Сравнение функции К(й) при разделении методом ВЭЖХ на октадецилсиликагеле (ОДС) и методом СФХ со сверхкритическим диоксидом углерода в качестве неподвижной фазы.
0,13 мм. При разделении методом ВЭЖХ при той же линейной скорости высота тарелки составляет 0,39 мм, что в 3 раза больше. Согласно ур. 5.1-17 на практике это означает, что пики при разделении методом СФХ в 3 раза уже, чем в ВЭЖХ.
Если сравнить на рис. 5.4-2 линейные скорости потока, соответствующие высоте тарелки 0,13 мм, т. е. минимуму функции # (и) для жидкостной хроматографии, то получится, что это 0,15 см/с для ВЭЖХ и 0,6 см/с для СФХ. Значит, в СФХ линейная скорость больше в 4 раза.
Подвижная фаза в СФХ не является инертной транспортной средой, как в ГХ. Взаимодействия выделяемого вещества с подвижной фазой сверхкритического флюида очень важны, как и в ЖХ, и их можно использовать для селективного изменения коэффициента селективности а.
Двойственный характер СФХ проявляется также в парциальном давлении выделяемого вещества. Растворение последнего в сверхкритическом флюиде происходит в условиях, очень близких к испарению, но при значительно более низкой температуре. Благодаря этому парциальное давление растворенных веществ при определенном давлении в сверхкритическом флюиде на несколько порядков больше, чем в газах. Этот факт создает основу для элюирования высокомолекулярных соединений (полимеров, крупных биомолекул) или термически неустойчивых веществ.
С помощью СФХ можно определять соединения со значительно более высокими молекулярными массами, чем в ГХ. Молекулярные массы могут быть такими же, как и в ЖХ, то есть до 105. Соединение с более высокими молекулярными массами, до 107, могут быть разделены только гель-хроматографией.
Применение
Известны многочисленные примеры применения СФХ для определения нелетучих соединений с относительно высокой молекулярной массой. С помощью СФХ анализируют природные продукты, лекарства, продукты питания, поверхностно-активные вещества, полимеры, сырую нефть и взрывчатые вещества1.
Из ур. 5.1-15 вытекает, что пики уже в 9 раз. — Прим, перев.
302
Глава 5. Хроматография
Время удерживания, мин
Рис. 5.4-3. СФХ-анализ смеси олигомерных полиэтиленов со средней молекулярной массой 740. Разделяющая колонка: 10x0,01 см набивная нормально-фазовая колонка, заполненная оксидом алюминия с частицами 5 мкм; подвижная фаза: СОг; давление 10 МПа 7 мин, затем повышение давления до 36 МПа за 25 мин, далее изобарически 36 МПа; температура термостата 100° С; детектор: ПИД.
На рис. 5.4-3 представлена хроматограмма анализа олигомерных полиэтиленов. Следует отметить, что в данном случае применялась набивная колонка. Обычно в СФХ используют градиент давления. Начинают с постоянного давления, затем повышают его линейным образом в течение определенного времени до максимального, а затем хроматографируют при постоянном давлении до конца анализа.
5.5. ЭЛЕКТРОФОРЕЗ
Электрофоретические методы основаны на миграции ионов в электрическом Иаяе. Для разделения используют различия в скоростях миграции частиц и ццдходящим образом определяют разделяющий эффект.
Большинство электрофоретических методов не являются истинно хроматографическими методами, поскольку в них отсутствует распределение определяемого вещества между подвижной и неподвижной фазами. При этом аппаратурное оформление классического бумажного электрофореза и современного электрофореза очень похоже на оформление плоскостных хроматографических методов и капиллярной колоночной хроматографии соответственно, поэтому имеющийся опыт можно перенести на электрофоретические методы.
Кроме миграции низкомолекулярных ионов можно также использовать движение коллоидных частиц, макромолекул, вирусов и целых клеток.
Классический электрофорез
Классическую форму электрофореза используют в исследованиях по биохимии и молекулярной биологии для разделения, выделения и определения белков, полинуклеотидов и других биополимеров. Различают электрофорез с носителем и без него.
5.5. Электрофорез
303
пробы в буфере
делением.
Крышка Фильтровальная
-к бумага/
й=%.
Электрод -ВЦКш
Диафрагма Буферные растворы
Рис. 5.5-2. Электрофореграмма сывороточных белков с фотометрическим опре-
Сигнал детектора
4-
Расстояние
Электрофорез без носителя, предложенный А. Тизелиусом (Нобелевская премия 1948 г.), основан на миграции ионов в буферном растворе, покрывающем раствор пробы. Буферный раствор, помещаемый в U-образную трубку, должен быть легче раствора пробы (рис. 5.5-1,о). После приложения постоянного напряжения к платиновым электродам в растворе пробы происходит частичное разделение и ионы концентрируются на границе между раствором пробы и буферным раствором (электрофорез на границе раздела фаз). Однако при этом нет полного разделения.
Электрофорез с носителем практикуется чаще. В этом случае ионы мигрируют по бумажному носителю или по гелю, такому, как агар, полимер или силикагель. Носитель насыщен раствором буферного электролита и его концы опущены в буферный раствор с электродами (рис. 5.5-1,£). К фильтровальной бумаге, пропитанной раствором электролита, прикладывается постоянное напряжение в несколько киловольт или больше.
Пятна пробы наносятся на линию на одном из концов носителя, и при разделении компонентов пробы образуются мигрирующие зоны (зонный электрофорез).
Для обработки электрофореграммы можно использовать окрашивание или применить фотометрический метод. На рис. 5.5-2 изображено классическое разделение сывороточных белков.
304
Глава 5-- Хроматография
Дальнейшее развитие
□ Амфипротные вещества, или амфолиты. — вещества, которые могут отдавать или принимать протоны.
Для определения амфолитов, таких, как аминокислоты и пептиды, используют изоэлектрическое фокусирование. В этом методе буферной смесью создается градиент pH. Из теории амфолитов известно, что при значении pH, соответствующем изоэлектрической точке, вещества в электрическом поле не движутся. Следовательно, при изоэлектрическом фокусировании можно достичь особых эффектов разделения для амфолитов, поскольку только они мигрируют к изоэлектрической точке и концентрируются в ней.
Метод широко используется в аналитической химии белков. Предпосылкой для этого является устойчивый, неподвижный градиент pH. Он достигается применением буферных растворов низкомолекулярных амфолитов, которые обеспечивают высокую буферную емкость н разнообразные изоэлектрические точки. Они фокусируются в определенном месте в электрическом поле и передают свое значение pH окружению.
Столь же важным развитием классического электрофореза стал изотахофорез (по-гречески изо означает «равный», а тахос — «скорость»): частицы мигрируют в электрическом поле с одинаковой скоростью. В других методах электрофореза частицы движутся с разными скоростями в постоянном электрическом поле, т. е. скорость частицы г определяется как
Vi = Ещ (5.5-1)
Поскольку при одинаковой скорости миграции ионов подвижность иона щ различна, в изотахофорезе электрическое поле в зонах должно неизбежно различаться:
v = Е1Щ = E2U2 =... Епип (5.5-2)
Иными словами, чем менее подвижна частица, тем больше интенсивность поля в соответствующей зоне:
«1 > U2 > - - > Un
Ei< Е? < ... < Еп
Внутри зон электрическое поле одинаковое. Значительные скачки возникают на краю зоны. Ионы, способные двигаться слишком быстро, замедляются в предыдущей зоне, а ионы, способные двигаться слишком медленно, ускоряются благодаря более Высокой интенсивности поля в последующей зоне. Происходит динамическая интенсификация зон.
Как же практически равная скорость частиц достигается на практике? В изотахофорезе для разделения используется, например, тефлоновый капилляр и два иона электролита различной подвижности. Лидирующий ион, который имеет бблыпую подвижность, вводится в капилляр с одного конца, а на другом помещается замыкающий ион с меньшей подвижностью.
6-Аминокарпонат в качестве Морфолинэтансульфокислота в качестве
замыкающего иона
лидирующего иона
5.5. Электрофорез
305
\Проба помещается между двумя электролитами. Ионы упорядочиваются в соответствии с уменьшающейся подвижностью и таким образом разделяются. Кроме того, для создания желаемого уровня pH для обоих электролитов нужно использовать буферный противоион.
Например, иммуноглобулины могут быть определены методом изотахофореза с морфолинэтансульфонатом (pH 9) в качестве лидирующего иона и ами-нокапронатом (pH 10,8) в качестве замыкающего иона. Изотахофорез становится очень распространенным в капиллярном варианте.
В принципе, использование капиллярного варианта позволяет преодолеть серьезные недостатки электрофореза. Эти недостатки связаны с сильно мешающей тепловой конвекцией при электрофорезе, которая приводит к гетерогенности и, следовательно, к размыванию пиков. Еще один недостаток классического электрофореза — длительная обработка электрофореграмм, которую отчасти сложно воспроизвести.
Капиллярный электрофорез
Перенос ряда элементов капиллярной ГХ в электрофорез привел к возрождению метода. Такое сочетание называют капиллярным электрофорезом (КЭ), капиллярным зонным электрофорезом (КЗЭ) или высокоэффективным капиллярным электрофорезом (ВЭКЭ).
Вообще, все варианты электрофореза могут быть перенесены в капиллир. Однако здесь миниатюризация способствует не сокращению диффузионных путей, как в ГХ или в СФХ, а предотвращению теплового влияния. Низкие токи в КЭ приводят к значительно меньшему уровню джоулевой теплоты. Кроме того, теплота, генерированная электрически, рассеивается гораздо проще в окружении капилляров небольшого диаметра из плавленого кварца.
Аппаратура
Блок-схема системы для КЭ представлена на рис. 5.5-3. Она состоит из двух буферных растворов, капилляра с устройством охлаждения, высоковольтного источника питания, детектора и системы сбора данных.
Рис. 5.5-3. Система для капиллярного электрофореза (КЭ).
306 Глава 5- Хроматография
Обычные капилляры, имеющие 10-100 см в длину и внутренний диаметр 25-100 мкм, состоят из немодифицированного плавленого кварца либо капилляры модифицируют химически или адсорбционно. В капилляре находится либо буферный раствор, либо гель или раствор полимера.
Для получения электрического поля 30 кВ к платиновым электродам прикладывают высокое напряжение.
Ввод пробы можно осуществлять за счет гравитационных сил. Для этого положительный конец капилляра опускают в раствор пробы и поднимают контейнер примерно на 10 см выше уровня буферного раствора на отрицательном конце. Другой метод ввода пробы—электрокинетический. Определенный объем пробы (от 5 до 50 мл) вводится в капилляр за счет электроосмотического потока, создаваемого коротким импульсом в 5 кВ в течение нескольких секунд. В автоматических системах капилляр прокалывает пробку контейнера с раствором пробы и проба вводится в капилляр.
Компоненты движутся по направлению к противоположному концу капилляра, охлаждаемого воздушной или жидкой средой. За счет очень высокого сопротивления образуется лишь небольшое количество тепла. Более того, большое отношение поверхности к объему обеспечиват хорошее рассеяние тепла, выделяемого за счет прохождения электричества, поэтому размывание пиков отсутствует. Часто размывание пиков соответствует только теоретически рассчитанному для продольной диффузии. Эффективность может достигать 500 000 и даже миллиона теоретических тарелок.
Для детектирования ионов непосредственно на колонке используются УФ-детекторы, включая детекторы с фотодиодной матрицей, а также флуоресцентные детекторы. Кроме того, существуют миниатюрные электрохимические детекторы.
Принципы разделения
Как уже упоминалось ранее, принципы электрофоретических методов на макроуровне могут быть перенесены в капилляр. Особенностью капиллярного электрофореза является внутренний электроосмотический поток (ЭОП), часто называемый просто электроосмотическим потоком. Он возникает из-за образования двойного слоя между раствором и внутренней стенкой капилляра. Внутренняя стенка плавленого кварца отрицательно заряжена за счет присутствия диссоциированных силанольных групп.
Этот отрицательно заряженный слой притягивает положительные ионы из раствора, в результате чего образуется положительно заряженное кольцо жидкости. Это кольцо движется в направлении отрицательно заряженного катода (рис. 5.5-4). Электроосмотический поток сильно зависит от pH. Он присутствует только при значениях pH более 4. Чем выше значение pH, тем более четко выражен эффект. ЭОП может быть уменьшен в результате химической модификации или покрытия внутренней стенки капилляра.
Профиль электроосмотического потока очень плоский (рис. 5.5-4) по сравнению с гидродинамическим потоком, профиль которого параболический. В результате электроосмотический поток не вызывает размывания пиков.
5-5. Электрофорез
307
Рис. 5.5*4. Электроосмотический поток (ЭОП) как результат образования двойного слоя между раствором и поверхностью капилляра.
Когда прикладывается электрическое поле, положительно заряженные ионы мигрируют к катоду, а отрицательные—к аноду. ЭОП не влияет на движение положительно заряженных ионов к катоду, поскольку они движутся быстрее ЭОП. Отрицательно заряженные ионы, однако, притягиваются положительным слоем кольца жидкости. При этом движение анионов к аноду может быть электроосмотическим потоком обращено, т. е. анионы могут медленно двигаться по направлению к катоду. Нейтральные частицы движутся вместе с ЭОП. В результате детектор может последовательно фиксировать сначала положительно заряженные, затем нейтральные и наконец отрицательно заряженные частицы.
О Додецилсульфат
Однако таким образом нельзя разделять незаряженные частицы. Для этого добавляют мицеллообразователь — мицеллярная электрокинетическая хроматография (МЭКХ). Мицеллообразователями служат поверхностно активные вещества, например додецилсульфат. Образующиеся мицеллы достаточно гидрофобны изнутри и могут включать в себя нейтральные молекулы. Заряженные мицеллы, содержащие внутри нейтральные соединения, могут перемещаться в электрическом поле и таким образом могут быть разделены, как и другие ионные частицы (рис. 5.5-4).
Применение
Электрофорез на макроуровне является частью рутинного анализа в биологии или биохимии, поскольку большинство интересующих молекул заряжены. Как правило, в этих случаях используют электрофорез с носителем на полимерах. В то же время недостатки тепловой конвекции как результата электрического нагревания сводятся к минимуму, насколько это возможно. Однако этот вариант трудоемок и требует больших затрат времени, поскольку автоматизирован может быть лишь отчасти.
Самые важные проблемы (тепловая конвекция и сложность детектирования) были преодолены с введением капиллярного электрофореза. Как высокоэффективный метод КЭ обеспечивает основу для большинства анализов смесей аминокислот, пептидов, белков, нуклеиновых кислот и других биополимеров.
308
Глава 5. Хроматография
Рис. 5.5-5. Капиллярный электрофорез основных белков на немодифицированном капилляре (а) и на капилляре, покрытом поливиниловым спиртом (б). 1 —цитохром С, 2 — лизоцим, 3 — трипсин, 4 — трипсиноген, 5 — химотрипсин; капилляр: полная длина 70 см, эффективная длина 57 см, внутренний диаметр 50 мкм; буфер: 150 мМ (а) или 50 мМ (б) фосфата натрия, pH 3. Напряжение: 30 кВ; ввод пробы: 15 кВ, 5 с; детектирование: 214 нм.
Так, фрагменты ДНК (дезоксирибонуклеиновой кислоты) могут быть определены за 10 мин на капилляре 35 см х 50 мкм с помощью боратного буфера при напряжении 8 кВ и с фотометрическим детектированием при 260 нм. В традиционном полиамидном гель-электрофорезе этот же анализ занимает 3 ч.
□ Капиллярная электрохроматография (КЭХ) —сочетание КЭ и ЖХ обещает быть мощным методом разделения.
На рис. 5.5-5 изображены две капиллярные электрофореграммы для определения основных белков. На немодифицированных капиллярах (рис. 5.5-5,а) нужно использовать относительно высокие концентрации буфера для уменьшения эффектов адсорбции на внутренних стенках. Однако при внимательном рассмотрении все равно можно разглядеть отчетливое размывание заднего фронта пика. Если капилляр покрыт поливиниловым спиртом (рис. 5.5-5,б), получаются симметричные пики. При этом для разделения на капилляре длиной 57 см можно достичь эффективности 1000 000 тарелок.
Кроме того, можно определять катионы и анионы в биологических жидкостях. При этом динамический концентрационный диапазон составляет 3 порядка.
Поскольку мицеллообразование в МЭКХ обеспечивало возможность разделения и определения нейтральных молекул, стали осуществимыми соответствующие анализы объектов окружающей среды, например определение фенолов.
5.6. Фракционирование ® поперечном поле
309
Вопросы и задачи
1. Назовите свойства сверхкритического флюида^явии^Л® для использования его в качестве подвижной фазы в хроматографии.
2. В чем разница между оборудованием для СФХ и для ГХ и ВЭЖХ?
3. Объясните важность давления в СФХ.
4. Как в электрофорезе используется изоэлектрическая точка?
5. В чем основное преимущество мицеллярной электрокинетической капиллярной хроматографии перед жцдостной хроматографией?
5.6. ФРАКЦИОНИРОВАНИЕ В ПОПЕРЕЧНОМ ПОЛЕ
5.6.1. Введение
Концепция фракционирования в поперечном поле (ФПП) была разработана в середине 1960-х годов Дж. Кельвином Гиддингсом, профессором химии университета Юты в Солт-Лейк-Сити. С тех пор рамки метода расширились, охватывая диапазон от малых макромолекул с молекулярной массой в несколько сотен дальтон до коллоидных частиц всевозможных типов и в особых случаях до 100 мкм диаметром.
ФПП является элюентным методом, как и хроматография, однако, строго говоря, не является хроматографией. Если в хроматографии разделение является результатом различного распределения компонентов пробы между подвижной и неподвижной фазами, то разделение в ФПП достигается за счет различия в скоростях компонентов в потоке под влиянием приложенного поля. Это поле удерживает частицы более мягко, и его легче контролировать по сравнению с межмолекулярными силами, используемыми для разделения в хроматографии. Методы ФПП поэтому особенно полезны для изучения макромолекул и коллоидных частиц, поскольку такие объекты анализа на активной границе раздела фаз часто подвергаются неблагоприятным или необратимым превращениям или разлагаются при прохождении через набивные хроматографические колонки.
Диффузия как основа разделения
В обоих методах, хроматографии и ФПП, важную роль играет диффузия молекул или частиц в жидкой среде. В хроматографии это движущая сила, с помощью которой молекулы достигают границы раздела фаз, где происходит распределение. В ФПП диффузия уравновешивает силы поля и определяет положение равновесного слоя каждого компонента.
Диффузия в качестве основы методов разделения привлекает внимание физиков, физикохимиков и химиков более, чем полвека, и она нашла применение во всех видах разделения. В 1920-е годы У. Уивер интересовался диффузией и установлением свойств малых частиц в жидких системах, а в 1938 г. Клаус Класиус (Мюнхенский, затем Цюрихский университеты) опубликовал большое число статей, описывающих разделение смесей газов и изотопов на основе принципа тепловой диффузии. Он заметил, что при диффузии в трубке, вдоль которой приложен градиент температуры, происходит разделение. Этот подход стал важным инструментом для разделения изотопов.
310
Глава 5. Хроматография
Преимущества методов ФПП
Основным преимуществом ФПП перед другими методами разделения является теоретическая простота. В канале в форме ленты создается ламинарный поток, профиль которого в обычных условиях параболический. Поскольку приложенное поле действует однородно в направлении, перпендикулярном потоку, на частицы пробы воздействуют силы, которые достаточно легко рассчитываются (рис. 5.6-1).
Это означает, что частицы элюируются со временами удерживания, которые также могут быть рассчитаны и, следовательно, предсказаны заранее. Существование этой твердой теоретической базы исключает необходимость проведения сложной, требующей времени процедуры градуировки и значительно упрощает разработку метода для новых проб. Оптимальные параметры системы могут быть рассчитаны для любой пробы с известными свойствами еще до первого ввода пробы, что позволяет сэкономить время и материалы.
5.6.2. Физические компоненты ФПП
Несмотря на то что каждому из вариантов ФПП соответствуют свои требования и ограничения в плане технического исполнения, принципиальное устройство более или менее неизменно. Прибор состоит из самого ФПП-канала, компонента, создающего поле или градиент, насосной системы для подачи раствора-носителя, соответствующего детектора и записывающего устройства, устройства для измерения потока и, если необходимо, коллектора фракций. Тенденции развития следуют за жидкостной хроматографией: приборы для ФПП все больше контролируются компьютером, который также служит для сбора и обработки данных. Как показано на рнс. 5.6-1, чаще используют канал в виде ленты, а не трубчатый. Такой канал вырезается из пластикового или
Рис. 5.6-1. Схематическое изображение канала разделения в ФШ1 (наверху) и профиля потока в этом устройстве (внизу).
5-6. Фракционирование в поперечном поле
311
стального листа толщиной от 0,05 до 0,5 мм, чтобы получился длинный и узкий спейсер, обычно длиной 100 см и шириной 2-3 см.
Стенки канала делают таким образом, чтобы они обеспечивали передачу движущей силы (поля), поэтому от системы к системе они отличаются.
Обычно метод ФПП не предъявляет особых требований ни к насосной, ни к детектирующей системе. Хотя ранние работы выполнялись практически исключительно с помощью перистальтического насоса для подачи носителя, в настоящее время стараются избегать использования пульсирующих насосов, так как незаполненные каналы ФПП плохо сглаживают пульсации потока. Однако открытый канал оказывает относительно низкое сопротивление потоку, поэтому пригодны насосы почти любого типа, обеспечивающие желаемый диапазон скоростей потока.
Выбор детектора сильно зависит от свойств разделяемых частиц. Детекторы для ВЭЖХ обычно подходят и для ФПП.
Объем пробы в ФПП составляет от 10 до 200 мкл при концентрации ниже 1%. Небольшие количества пробы обеспечивают улучшенное удерживание и лучшее разделение, и они предпочтительнее (если только позволяют пределы обнаружения). Умеренная перегрузка пробой проявляется в более высоких, чем предсказывалось, объемах удерживания и в излишнем размывании пиков; серьезная перегрузка приводит к искаженным или артефактным пикам. Во время разделения, однако, проба многократно разбавляется и детектирующая способность может ухудшиться.
Программируемое уменьшение величины поля могло бы быть использовано для увеличения скорости и эффективности разделения сложных смесей в широком диапазоне молекулярных масс (рис. 5.6-2). При этом сила, действующая на частицы, снижается со временем, что приводит к устойчивому повыше-
Рис. 5.6-2. Программируемое тепловое ФПП: разделение частиц с размером частиц в широком диапазоне молекулярных масс [Giddings J. С. et al., Anal. Chem., 48 (1976) 1587].
312
Глава 5. Хроматография
нию скорости движения частиц через канал. В настоящее время использование компьютеров для контроля за уровнем силы поля позволяет легко и точно варьировать его во время анализа.
Многосторонность ФПП отражает широкое разнообразие проб, к которым его можно применить благодаря ряду контролируемых параметров, влияющих на разрешение и скорость разделения. Тип поля, величина поля и способ его программирования, свойства и скорость подачи носителя и даже геометрия канала—все это может влиять различным образом и использоваться для обеспечения оптимальной селективности, скорости и точности.
Поскольку приложенное поле приводит к разделению на основании физических свойств частиц пробы (например, размера, плотности, коэффициента диффузии, электрического заряда или молекулярной массы), это определяет, какой из вариантов ФПП будет наиболее подходящим в каждом случае. Влияние поля на разделяемые компоненты пробы должно быть достаточно сильным, но различным.
5.6.3. Различные варианты ФПП
Для проведения разделения в ФПП могут применяться разнообразные поля или градиенты:
— седиментационные силы;
— тепловой градиент;
- пересечение потоков;
— электрические поля;
— магнитные поля.
Возможность дальнейшего расширения диапазона применений заключается в комбинации этих движущих сил с различными режимами работы, которые проявляются в различных способах распределения компонентов пробы по дифференциальному профилю потока в канале. Это распределение может быть диффузионно (нормальное ФПП) или гидродинамически (стерическое ФПП) контролируемо и может зависеть от коэффициентов перемещения. Оно может регулироваться наложением двух противоположных полей, приводящим к фокусированию пробы в тонком слое между стенками (гиперслойное ФПП). Наиболее широко распространены нормальный и стерический режимы.
Процесс элюирования
Проба, введенная в канал ФПП, образует зону, в которой концентрация макромолекул постоянна на протяжении всей ширины канала. Сразу же после этого поток останавливают на время, достаточное, чтобы частицы достигли равновесия под влиянием внешнего поля. Этот промежуток времени, называемый временем релаксации, изменяется в зависимости от пробы, типа н величины силы внешнего поля, а также от толщины канала и обычно составляет от нескольких часов до часа. Оно может быть значительно сокращено или вообще исключено при помощи входного делителя потока. Во время релаксации
5.6. Фракционирование в поперечном поле
313
поле вызывает движущую силу F, действующую на компоненты пробы и вынуждающую их двигаться по направлению к одной из стенок незаполненного канала—к аккумулирующей стенке. Выражение скорости этого движения U может быть записано как
где f — коэффициент трения, R — универсальная газовая постоянная, Т — абсолютная температура, D — коэффициент диффузии частиц или молекул. В наиболее общем случае, в нормальном варианте, этот процесс продолжается до достижения устойчивого состояния, когда накопление компонентов у стенки компенсируется их диффузией наружу из области высокой концентрации. Это состояние равновесия характеризуется образованием дискретных узких слоев частиц, параллельных стенке, каждая из которых имеет экспоненциальное распределение концентраций с максимальной концентрацией в непосредственной близи от стенки. Концентрационный профиль соответствует толщине I:
1 = ^ (5.6-2)
которая также является средним расстоянием от частиц до стенки. Толщина слоя наиболее подходяще представляется как доля толщины канала, w:
Х = - (5.6-3)
W
где А—безразмерный параметр удерживания. Комбинирование этих трех уравнений дает
RT1
Л = - (5.6-4)
Ясно, чтобы сделать А меньше, необходимо увеличить приложенную силу настолько, что бы энергия взаимодействий Fw достигала уровня тепловой энергии RT.
Ламинарный поток ньютоновой жидкости по любому тонкослойному каналу с умеренной скоростью приводит к параболическому профилю скорости потока со скоростью, стремящейся к нулю у стенок из-за силы трения. Именно так получается, когда жидкость движется между параллельными пластинами канала ФПП. Образующийся дифференциальный поток усиливает разделение, которое уже произошло на предыдущем этапе. Дискретные зоны, формирующиеся во время процесса релаксации, движутся по направлению к выходу потока носителя с различными скоростями, зависящими от того, насколько быстро каждый слой попадает в быстрейшую зону потока, что приводит к разделению за счет различия во времени удерживания.
Как и в хроматографии, относительное удерживание г в ФПП определяется отношением
V°
т = -- (5.6-5)
314
Глава 5. Хроматография
где У0 —мертвый объем канала, а V—объем удерживания определяемого компонента. Открытые каналы, используемые в ФПП, позволяют определить мертвый объем либо прямым расчетом из размеров канала, либо осторожным определением удерживания неудерживаемого маркера, вводимого в систему.
Теоретические времена удерживания можно рассчитать по уравнению г = 6А Jcth
Для хорошо удерживаемых компонентов (А < 0,1) это уравнение преобразуется в простую сокращенную форму
т = 6А - (5.6-7)
(5.6-6)
что приводит к общему уравнению для времени удерживания в диффузионно контролируемом ФПП:
(5.6-8)
- tOFw tr ~ 6RT
Частицы пробы размером более 1 мкм удерживаются по механизму, который отличается от механизма, характерного для описанного выше диффузионно контролируемого режима. Изменение режима иа стерический характеризуется обращением порядка элюирования, т. е. чем больше частицы, подвергаемые стерическому ФПП, тем раньше они элюируются. Когда эти большие частицы, броуновским движением которых можно пренебречь, подвергаются действию поля, они останавливаются у аккумулирующей стенки. Эта тенденция противоположна существованию гидродинамических подъемных сил, которые увлекают частицы вверх и вдаль от стенки в условиях высокой скорости. Несмотря на то, что теория такого процесса удерживания до настоящего времени не полностью разработана, понятно, что между приложенным полем и этими подъемными силами, индуцированными потоком, должен быть достигнут очень тонкий баланс. Если скорость потока мала по сравнению с приложенным полем, частицы могут адсорбироваться на стенках и элюироваться непредсказуемо долго или не элюироваться вовсе. Если скорость потока слишком велика, чтобы эффективно компенсироваться полем, подъемные силы приведут к существенному ухудшению разрешения. Если же необходимый баланс достигается, инициация потока вдоль канала после релаксации вызовет движение частиц по потоку со скоростями, определяемыми степенью, с которой они выходят в поток: равновесное расстояние от центра тяжести частиц до стенки будет примерно равно радиусу частиц. Уравнение удерживания для этого гидродинамического режима работы в таком случае может быть выражено следующим образом:
(5.6-9)
г &уг
где г — радиус частиц, а у — безразмерный фактор, примерно равный единице.
Эта чрезвычайная близость частиц к стенке обычно имеет свои недостатки и, конечно, она показывает, что структура поверхности и свойства аккумулирующей стенки особенно важны для успешного разделения в этом режиме.
5.6. Фракционирование в поперечном поле
315
Тем не менее, на примере стандартов полистирола показано, что этот метод разделения чрезвычайно успешен.
Седиментационное ФПП
Из всех вариантов ФПП до настоящего времени наиболее подробно были изучены тепловое и седиментационное ФПП. Разработано и выпускается соответствующее оборудование.
Частицы размером около 1 мкм или больше и достаточной плотности могут разделяться в седиментационном ФПП даже под действием земной гравитационной силы. Однако сила, создаваемая вращением модифицированной центрифуги, вокруг оси которой направлен канал, значительно более мощная и легче контролируемая. Такая конфигурация, изображенная на рис. 5.6-3, существенно расширяет дипазон использования метода. Выпускаемое оборудование позволяет создавать поле свыше 30000g, делая возможным удерживание частиц до 0,01 мкм. В седиментационном ФПП плотность носителя оказывает большое влияние на удерживание и должна тщательно контролироваться, чтобы обеспечить высокую точность работы. Регулирование плотности носителя путем добавления модификаторов плотности может быть использовано для индуцирования сдвига удерживания, однако технические ограничения обычно требуют, чтобы носителем были водные растворы.
После завершения релаксационного процесса поток носителя возобновляется и частицы движутся по каналу, элюируясь в порядке, определяемом их размером или плотностью. Уравнение для параметра удерживания в седиментационном ФПП следующее:
А = —-sGw
(5.6-10)
316
Глава 5. Хроматография
где s—коэффициент седиментации, a G—ускорение. Проводя разделения с носителями различной плотности, возможно одновременно определить и размер частиц, и их плотность. В общем, для каждой лаборатории, планирующей проводить высокоточные работы в области ФПП, необходим хороший денситометр.
Седиментационное ФПП используют для фракционирования образцов латекса различной полидисперсности и для определения характеристик агрегированных проб полиметилметакрилата. Метод служит для определения молекулярной массы некоторых вирусов, для фракционирования полимеризованных микросфер из сывороточного альбумина и некоторых вОдно-масляных эмульсий. Метод пригоден для разделения и изучения промышленных, биологических и экологических проб всевозможных типов — от липосом до ДНК, от клеток человека и животных до природных вод, от растворов золота и серебра до хрящевых протеогликанов. Одиако седиментационное ФПП на практике имеет ограничения по размеру частиц. Полимерные частицы, особенно те, которые слишком малы для адекватного влияния на них гравитационных сил в седиментационном ФПП, могут быть успешно изучены с помощью теплового ФПП.
Термическое ФПП
Термическое ФПП было первым вариантом ФПП, который был успешно применен в качестве инструмента макромолекулярного анализа. Стенки канала изготавливаются из теплопроводного материала (например, из меди), одна из них содержит нагревательный элемент, а внутри другой циркулирует охлаждающий агент. Регулируя температуру горячей стенки, возможно регулировать скачок температуры в тонком канале. При этом протекает термоднффузия, за счет которой молекулы в растворе при температурном градиенте подвергаются миграции относительно растворителя. Однако лишь некоторые пробы подвергаются термоднффузии в водных носителях, поэтому большинство экспериментов до настоящего времени сосредоточено на разделении синтетических полимеров в органических растворителях. Эти полимеры имеют тенденцию аккумулироваться на холодной стенке под действием температурного градиента, причем полимеры с самой высокой молекулярной массой движутся ближе всего к стенке и, следовательно, удерживаются дольше всего. Показано, что Удерживание в термическом ФПП зависит и от молекулярного размера и от состава. Толщина слоя может быть представлена следующим образом:
(5.6-11)
где DT — коэффициент термодиффузии. Разница в размерах частиц полимеров отражена коэффициентом Z), в то время как разница в химических свойствах находит отражение в DT. Полимеры с высокой молекулярной массой движутся ближе к аккумулирующей стенке за счет их низких коэффициентов диффузии, поэтому удерживаются дольше. Биологические пробы и пробы, требую
5.6. Фракционирование в поперечном поле
щие водных носителей, естественно требуют использования других методов; их анализируют с помощью проточного ФПП.
Проточное ФПП
Проточное ФПП особенно интересно, поскольку в принципе является универсальным в плане использования, а диапазон селективности в этом случае перекрывает диапазон для термического ФПП. Лимитирующим фактором является обычно состав аккумулирующей стенки, которая представляет собой полупроницаемую мембрану, соответствующей молекулярной массы, предотвращающей прохождение через нее компонентов пробы. Активное поле в этом случае пересекает поток носителя, проникающий через мембрану, и побуждает компоненты пробы дифференцированно распределяться по толщине канала согласно лишь их коэффициентам диффузии. Поскольку практически все частицы подвержены действию этого типа сил, ограничение связано лишь с проницаемостью мембраны. С помощью мембраны с ограничением молекулярной массы 10000 могут изучаться гуминовые и фульвокислоты с Мг до 300. Недавно благодаря использованию асимметричной системы, в которой растворитель, являющийся носителем, удаляется на аккумулирующей стенке полупроницаемой мембраной, а на противоположной стенке вход отсутствует, были решены некоторые технические проблемы, связанные с проточным ФПП. Такая асимметричная система ФПП нашла широкое применение для биологических проб.
Литература
Altruia, К. D., Capillary Electrophoresis Guidebook, Chapman and Hall, Van Nostrand Reinbold, 1995.
Foret F., Krivankov L., Bocek P., Capillary Zone Electrophoredid, VCH, New York, 1994.
Giddings J. C. Unified Separation Science, Wiley, New York, 1991.
Guichon G., Guillemin L.,- Quantitative Gas Chromatography, Elsevier, Amsterdam, . 1988.
Haddad P. R., Jackson P. E., Ion Chromatography: Principles and Applications, Elsevier, New York, 1990.
Hamilton R., Hamilton S., Thin Layer Chromatography, Wiley, New York;* 1987.
Hunt B. J. Holding S. R., Eds., Size Exclusion Chromatography, Chapman and Hall, New York, 1989.
Jennings W., Analytical Gas Chromatography, Academic Press, Orlando FL, 1987. Lindsey S., High-Performance Liquid Chromatography, Wiley, New York, 1988.
Sewell P, Clarke B., Chromatographic Separations, Wiley, New York, 1988.
Small H., Ion Chromatography, Plenum Press, New York, 1989.
Snyder L. R., Glaijch, J. L. Kirkland, J. J., Practical HPLC Method Development, Wiley, New York, 1988.
White С. M. Modem Supercritical Fluid Chromatography, Huethig Verlag, Heidelberg, 1988.
6
КИНЕТИКА И КАТАЛИЗ
Цели изучения
• Ознакомиться со значительным достижением аналитической методологии — использованием химической кинетики; эта методология основана на прямых измерениях скорости реакций, а также на исследовании явлений, отражающих две основные составляющие современной аналитической химии — физическую и химическую динамику процессов.
• Ознакомиться с измерениями, проводимыми для химических систем, изменяющихся во времени, и научиться по этой дополнительной информации выяснять механизм химических реакций, а также оценивать сложность процессов, сопутствующих аналитической реакции до момента достижения равновесия.
6.1. ВВЕДЕНИЕ
Термодинамика отвечает иа вопросы о том, почему происходит реакция и как в ходе реакции изменяется полная энергия системы. При этом рассматриваются только характеристики исходных веществ и конечных продуктов. Классическая методология аналитической химии опирается на термодинамические константы, которые определяют, измерив некоторое свойство реакционной системы в равновесном состоянии. Кинетика же отвечает на такие вопросы, на которые термодинамика не дает убедительного ответа, например, какой механизм лежит в основе превращения реагирующих веществ в конечные продукты, сколько времени занимает этот процесс, какими свойствами обладают промежуточные соединения, образующиеся и расходующиеся в ходе реакции, какова энергия их образования и разрушения. Огромное число методов современной аналитической химии базируется на кинетических параметрах, получаемых из расчетов скорости реакции как функции изменений концентраций каких-либо реагентов или продуктов во времени. Кроме того, знание кинетических констант позволяет выяснять механизмы реакций и использовать их в дальнейшем для учета нестехиометрических взаимодействий. Однако только совокупность
6.2. Скорость химических реакций
319
Рис. 6.1-1. Изменение концентрации исходного вещества (а) и продукта (б) химической реакции во времени. Показаны кинетическая и равновесная области [P6rez-BenditoD., Silva М., Kinetic Methods in Analytical Chemistry, Chichester, Ellis Horwood, 1988].
как термодинамических, так и кинетических параметров необходима для понимания причин протекания реакций и их характеристик.
Вкратце можно сказать, что любой процесс, независимо от его природы, протекает с конечной скоростью и стремится достичь равновесного состояния. В результате, протекание процесса включает в себя две различающиеся области: кинетическую (динамическую), в которой реакция приближается к равновесному состоянию, и равновесную (статическую), в которой все стадии процесса достигли равновесного состояния (рис. 6.1-1). Статические измерения возможны не для всех реакций: в некоторых случаях протекают побочные процессы, в других—системы достигают равновесия крайне медленно или выход продуктов реакции не является количественным. В таких неблагоприятных случаях равновесные методы не могут конкурировать с кинетическими и последние оказываются более предпочтительными.
Использование катализаторов для ускорения медленных процессов явилось следствием их сильного влияния на кинетику химических реакций и стимулировало развитие кинетических методов аналитической химии. Поэтому в данной главе кинетические и каталитические методы рассмотрены совместно.
Проведение аналитических измерений в неравновесных условиях в значительной мере стало возможным благодаря использованию современных методов сбора и обработки данных, применению мощных приборов и компьютеров, сделавших кинетические методы столь же точными и надежными, как н методы, основанные на равновесных измерениях.
Вследствие всех рассмотренных, выше обстоятельств химическая кинетика по праву занимает место в программах по аналитической химии, что уже иашло свое отражение в учебниках по аналитической химии и монографиях, посвященных кинетическим методам анализа [6.1-1, 6.1-2].
6.2. СКОРОСТЬ ХИМИЧЕСКИХ РЕАКЦИЙ
Скорость реакции определяют как число молей вещества, израсходованного или образовавшегося в ходе реакции в единице объема за единицу времени
320
Глава б. Кинетика и катализ
(кривые а и б на рис. 6.1-1)
dc*
Скорость = ±-тг (6.2-1)
at
где —концентрация г-го исходного компонента или продукта в зависимости от знака (отрицательный для исходных веществ н положительный для продуктов). За время протекания процесса концентрации продуктов и исходных веществ меняются и постепенно приближаются к постоянным значениям по мере завершения реакции. Скорость изменения этих концентраций также меняется (и становится близкой нулю вблизи состояния равновесия). Основная задача химической кинетики заключается в изучении взаимосвязи между концентрациями и скоростями их изменения. Следовательно, время всегда играет главнейшую роль в кинетических измерениях, поэтому следует особенно тщательно и точно воспроизводить условия проведения реакций.
□ Кинетическое уравнение описывает зависимость скорости реакции от концентраций.
Кинетический закон, или кинетическое уравнение, реакции связывает скорость реакции в любой момент времени с концентрациями всех веществ, от которых она зависит. Для прямой реакции вида
А + В—»Р (I)
где Р обозначает продукты реакции, кинетическое уравнение записывается следующим образом:
Скорость = -^ = -® = ®=МА][В] (6.2-2)
где коэффициент пропорциональности к — константа скорости, которая зависит только от давления и температуры и представляет собой скорость реакции при концентрациях компонентов, равных единице.
□ Константа скорости представляет собой скорость реакции при концентрациях реагирующих компонентов, равных единице.
Таким образом, скорость реакции—это положительная величина, значение которой в любой момент времени определяется уравнением (6.2-2), устанавливающим взаимосвязь между скоростью реакции и концентрациями веществ, от которых она зависит. Скорость реакции можно рассчитать для любого момента времени, если известна константа скорости.
□ Реакция, проходящая в прямом направлении в одну стадию, называется элементарной реакцией.
Скорость одностадийной реакции вида
В —* Р (П)
которая называется элементарной стадией, или элементарной реакцией, выражается уравнением
Скорость = — —— = —— = fc[B] (6.2-3)
at at
6.2. Скорость химических реакций
321
Скорость обратного процесса предполагается при этом незначимой.
□ Молекулярность элементарной реакции — это число частиц, участвующих в этой реакции.
Молекулярность элементарной реакции (т. е. реакции, протекающей в одну стадию) определяется числом частиц, вступающих во взаимодействие. В элементарной реакции может участвовать один ион (молекула) реагента или более (две или цри частицы). В зависимости от этого реакцию называют мо-номолекулярной, бимолекулярной или тримолекулярной, при этом скорость реакции зависит от стехиометрии. Более всего в природе распространены бимолекулярные элементарные стадии, так как столкновения двух одинаковых или различных частиц происходят с большой вероятностью. Напротив, тримо-лекулярные реакции встречаются гораздо реже, так как одновременные столкновения трех частиц сравнительно маловероятны, а элементарные стадии более высокого порядка вообще на сегодняшний день не известны. Многие реакции являются результатом последовательных элементарных стадий с низкой молекулярностью .
□ Самая медленная стадия определяет общую скорость реакции и называется скоростьопределяющей или лимитирующей.
Если какая-то из элементарных стадий протекает намного медленнее остальных, то она называется скоростьопределяющей или лимитирующей стадией реакции, при этом скорость реакции в целом определяется скоростью этой стадии.
□ Время полупревращения реакции —это время, за которое концентрация реагирующего вещества уменьшается вдвое.
Другим важным параметром для описания кинетики реакции является время полураспада или период полупревращения Этот параметр характеризует время, необходимое для уменьшения концентрации реагирующих веществ вдвое. Вид математического выражения для этого параметра зависит от порядка реакции (см. разд. 6.2.1).
Все изложенные соображения основаны на предположении о необратимости реакций, т. е. незначимости протекания обратных процессов. Однако большинство химических реакций обратимы, и в ходе реакции скорость прямого процесса уменьшается, а скорость обратного возрастает до момента достижения равновесия в системе. В равновесии суммарная скорость процесса равна нулю. В результате обработка кинетических данных усложняется. Описание еще более затрудняется при наличии последовательных (А —> В —* Р), конкурирующих (Ач-В—*РиАч-С—>Р7)и цепных реакций. Гетерогенные реакции, протекающие в системах, состоящих из двух или более фаз, также весьма трудно описать при помощи простых математических выражений. Рассмотрение всех этих сложных типов реакций выходит за рамки этой главы. Далее речь пойдет в основном о необратимых гомогенных бимолекулярных реакциях.
11 Аналитическая химия. Том 1
322
Глава 6. Кинетика и катализ
6.2.1. Кинетическое уравнение и порядок реакции
□ Общий порядок реакции определяется суммой показателей степени при концентрациях, входящих в кинетическое уравнение. Частный порядок реакции по компоненту определяется показателем степени при концентрации данного компонента. ,
Общий порядок реакции представляет собой сумму показателей степени при концентрациях, входящих в кинетическое уравнение. Так, порядок реакции (I), выражение для скорости которой дается уравнением 0:2-2, равен двум, и данная реакции является реакцией второго порядка. Кроме того, можно говорить о частном порядке реакции по отношению к какому-либо ее компоненту. Частный порядок определяется показателем степени при соответствующей концентрации в кинетическом уравнении. Известны реакции шестого или даже седьмого порядка. Константа скорости реакции n-го порядка называется константой скорости n-го порядка.
Порядок реакции — это эмпирический параметр: он может быть дробным и необязательно постоянным. Его находят экспериментально для конкретных условий. Следует четко различать порядок и молекулярность реакции. Скорость элементарной реакции определяется стехиометрией данной реакции, и для элементарной реакции вида
«А + ЬВ + • Л сС + <ГО + • (III)
кинетическое уравнение записывается следующим образом:
='"=МА1“[в,ь (б-2-4)
Частные порядки сложных реакций, проходящих более чем в одну стадию, могут не совпадать со стехиометрическими коэффициентами. В качестве примера можно привести хорошо известную реакцию окисления бромида бромат-ионом по реакции
ВгОз + 5Вг~ + 6Н+ ЗВг2 + ЗН2О (IV)
кинетическое уравнение которой имеет вид
(6.2-5)
Данная реакция в целом является реакцией четвертого порядка, первого порядка по бромид- и бромат-ионам н второго порядка по ионам водорода. Очевидно, что из общего уравнения реакции нельзя делать предположения о порядках или кинетическом уравнении этой реакции. Эти величины должны быть определены экспериментально.
□ Кинетика псевдопервого порядка наблюдается, если один из компонентов присутствует в большом избытке.
Если один из реагентов, участвующих в реакции (I) (например, В), находится в большом избытке, его концентрация за время реакции меняется пренебрежимо мало, и ее можно включить в значение константы скорости к в
6.2. Скорость химических реакций
323
Рис. 6.2-1. Изменение концентрации реагента А во времени для реакций различного порядка по А. 1 — нулевой порядок; 2—первый порядок; 3 —второй порядок.
уравнении 6.2-2. В этом случае говорят, что имеет место реакция псевдопервого порядка по реагенту А или псевдонулевого порядка по реагенту В. Это можно выразить так:
-^ = fc'[A] (6.2-6)
at
В этом случае У — А: [В] является константой скорости реакции псевдопервого порядка по А, что иногда обозначают как Лд. Если кинетическое уравнение имеет вид —d[A]/di = А/ или -d[A]/di = А/[А]2, то говорят, что данная реакция — псевдонулевого или псевдовторого порядка по А соответственно.
На рис. 6.2-1 показано изменение концентрации компонента А во времени для реакций разного порядка по этому компоненту. Зависимости представляют собой убывающие экспоненциальные кривые (тангенс угла наклона касательных к которым в каждой точке представляет собой скорость реакции), за исключением зависимости, соответствующей реакции нулевого порядка по А, которая является линейной. Ее наклон (d[A]/dt) не изменяется в ходе реакции при уменьшении концентрации А. По мере уменьшения концентрации А н завершения реакции, наклоны касательных к кривым реакций псевдопервого и псевдовторого порядка по А стремятся к нулю. Ход этих кривых неодинаков (ср. кривые 2 и 3 на рис. 6.2-1), потому что константы скорости псевдопервого и псевдовторого порядка различаются, и даже при равенстве начальных концентраций этн различия определить достаточно трудно.
Как альтернативный вариант можно построить зависимость концентрации продукта Р от времени. Эта величина связана с концентрацией А выражением [А] — [А]о — [Р], где [А]о — исходная концентрация А. В результате получится возрастающая экспоненциальная кривая.
В табл. 6.2-1 приведены кинетические уравнения для прямых необратимых реакций псевдонулевого, псевдопервого, псевдовторого н псевдо-п-го порядков, а также время полупревращения и размерности констант скорости. Уравнения представлены как в дифференциальной, так и интегральной форме.
и*
Таблица 6.2*1. Кинетические уравнения, соответствующие простым необратимым реакциям, v = fc[A]e[В]ь; (п = а + Ь)а
Порядок nab Дифференциальная форма Интегральная форма Время полупревращения Единицы размерности k
ООО = * [А]о-[А)(-М [A]o/2t маль-л-1-с-1
1 1 0 -^ = *[А] 1п^ = И6 1п2/* с"1
2 1 1 -^£1 = fc[A][B] „l. 1 гХГ In MPfe = kt 1 In (2- л моль-' с-1 dt i и j |в]о - [Ajo [B]o[A]t fc({B]o-[A]o) \ [B]o J
2 2 0 -^ = *iAl2 rarijfe'*4 l/*{A]o Л.МОЛВ-.С-’
-1 -1 0 -^ = k[A]-1 1([A|§ - [A]? = kt 3[A]g/8A моль’ л-’-с-1
dt n-lVfA]?*1 [A}„-1J (n- 1)*(A|S"‘ "0Л1
* По данным Рёгег-BenditoD., Silva М., Kinetic Methods in Analytical Chemistry, Chichester, Ellis Horwood, 1988.
5 Или, в экспоненциальной форме [A}t == [AJoexp(fct)
6.2. Скорость химических реакций
325
Размерность константы скорости зависит от порядка реакции. Размерность определяют, решая кинетическое уравнение относительно к и подставляя размерности всех переменных. Поскольку концентрации выражают в моль/л, а время — в секундах, для реакций нулевого, первого н второго порядков по А величину к выражают в моль/(л-с), 1/с или л/(моль-с) соответственно (размерности константы к для реакций других порядков приведены в табл. 6.2-1). Константа скорости реакции псевдо-п-го порядка имеет ту же размерность, что и константа скорости реакции n-го порядка.
Реакции первого порядка представляют наибольший интерес для аналитической химии н используются наиболее широко. Их легко осуществить, используя избытки всех реагентов за исключением одного, чтобы скорость реакции определялась этим реагентом. Относительно этого реагента реакция будет иметь псевдопервый порядок. Концентрации, входящие в уравнение 6.2-6, можно заменить любыми измеряемыми параметрами, прямо пропорциональными концентрации. Таким образом, за изменениями концентрации реагента или продукта можно следить при помощи аналитических методов.
Для определения кинетического уравнения исследуемой системы необходимо знать частные порядки реакция по всем компонентам, участвующим в процессе. Частные порядки можно определить двумя способами в зависимости от того, используется ли интегральная или дифференциальная форма кинетического уравнения. Как правило, для определения частных порядков используют дифференциальные методы, основанные на измерении начальной скорости реакции (tga). Интегральные методы в основном применяют для определения констант скорости.
□ Частные порядки реакций можно рассчитать из дифференциальной формы кинетического уравнения.
Частный порядок п в дифференциальной форме кинетического уравнения псевдопервого порядка вида
Скорость — tgo — ~ ^а[А]п (6.2-7)
где А — вещество, порядок по которому необходимо определить, и /сд — константа скорости реакции псевдо-п-го порядка для данного вещества, можно определить, логарифмируя уравнение 6.2-7:
lg(tg а) = 1g fcA + п lg[A] (6.2-8)
Данная зависимость представляет собой прямую линию с наклоном п н отрезком, отсекаемым на оси ординат, равным lgfcA, т- е. обе эти величины можно найти из графика зависимости Ig(tga) от lg[A] для разных начальных концентраций реагента А. Если эта зависимость линейна, то искомый частный порядок равен единице, что легко видеть из уравнения 6.2-7.
На практике определение частных порядков по каждому из компонентов, участвующих в реакции, основано на измерении концентрации этого компонента как функции времени при условии, что концентрации всех других веществ постоянны и существенно превосходят концентрацию этого компонента.
326
Глава 6. Кинетика и катализ
Далее строят графики зависимости выбранного свойства системы от времени при разных исходных концентрациях компонента и определяют начальные скорости реакции (тангенсы углов наклона начальных прямых участков кинетических кривых). В этих условиях изменения выбранного параметра определяются исключительно исследуемым компонентом, что позволяет определить соответствующий частный порядок реакции.
После выявления влияния каждого компонента записывают кинетическое уравнение в общей форме:
« = -^ = 4Ai“(fl|‘(C|c (6.2-9)
at
Это уравнение используется очень широко. В качестве переменных оно может включать концентрации катализаторов, ионов водорода н растворителей.
Использование и интерпретация кинетического уравнения: механизмы реакций
Кинетическое уравнение можно использовать для прогнозирования скорости реакции при данных условиях илн времени, необходимого для завершения реакции до определенной степени. Кроме этого, с помощью кинетического уравнения, а именно с помощью информации о лимитирующих и быстрых стадиях, можно определить элементарные стадии, через которые проходит химическая реакция. Это в свою очередь позволяет понять, как н почему данная реакция протекает и почему именно так, а не иначе.
□ Активированный комплекс может распадаться с образованием либо продуктов, либо исходных компонентов.
Кинетическое уравнение можно интерпретировать, рассматривая механизм реакции, который представляет собой последовательность химических стадий, составляющих реакцию и имеющих химический смысл. Действительно, механизм, соответствующий общему уравнению реакции и кинетическому уравнению, включает несколько элементарных стадий, каждая из которых согласуется с химическими и структурными принципами. Одной из основополагающих концепций, касающихся механизмов реакций, является теория активированного комплекса—некоторого нестабильного соединения, которое разрушается с образованием либо продуктов, либо исходных соединений. Кинетическое уравнение дает информацию о стадии, которая ведет к образованию активированного комплекса, который также называют «переходным состоянием». Каждая элементарная реакция проходит через состояние критического комплекса, соответствующего максимуму потенциальной энергии в ходе реакции. Здесь следует подчеркнуть различие между промежуточным соединением реакции и активированным комплексом. Первый из них обладает минимумом потенциальной энергии, н для превращения этого промежуточного соединения в активированный комплекс, приводящий к образованию продуктов реакции, необходима дополнительная активация (вследствие внутримолекулярных искажений, перегруппировки или последующей бимолекулярной реакции с другим химическим компонентом).
6.2. Скорость химических реакций
327
В рамках теории активированного комплекса рассмотрено большое число различных реакций. Основное исходное положение этой теории заключается в том, что реагирующие вещества находятся в некоем квазиравновесии с активированным комплексом и обладают характеристической частотой перехода через максимум потенциальной энергии или переходное состояние. Получающаяся в результате схема реакции отражена в уравнениях (V) н (VI):
А + В^(АВ)* (V)
(АВ)* Л С + D (VI)
где (АВ)*—активированный комплекс. Стадия (V), записанная в виде обычного равновесия, характеризуется константой равновесия К и связанной с ней термодинамической функцией.
6.2.2. Интегральные кинетические уравнения
Обычные кинетические уравнения можно интегрировать для получения функций, более сложных, чем обычные зависимости концентрации от времени. Интегрирование кинетического уравнения для реакции первого порядка по А (уравнение 6.2-6) приводит к
ln[A]f =ln[A]0-fcAt (6.2-10)
или, в форме десятичных логарифмов:
lg[A]f = lg[Aj0 - 0,4343fcAt (6.2-11)
где [А]о и [А]е — исходная концентрация соединения А и его концентрация в момент времени t соответственно, /сА — константа скорости реакции.
Уравнение 6.2-10 можно преобразовать в экспоненциальную форму:
[А], = [А]ое-‘Л[ (6.2-12)
Если наблюдение ведется за концентрацией продукта, а не исходного реагента, уравнения 6.2-10 и 6.2-11 слегка изменяются. Так, уравнение 6.2-10 преобразуется в уравнение 6.2-13
1п([Р]оо - [Р]0 = 1п[Р]оо - fcAt (6.2-13)
где [P]t н [Р]то — концентрации продукта, образовавшегося в момент времени t н оо (время, достаточное дЛя того, чтобы весь реагент А превратился в продукт Р, иными словами [А] о — [Р]оо).
Интегрирование кинетического уравнения для простейшего возможного случая реакции второго порядка по исследуемому реагенту
-^ = ЫА]2 (6.2-14)
приводит к
г —Ь
[A]t [А]о
(6.2-15)
328
Глава 6. Кинетика и катализ
Время, с
Рис. 6.2г2. Графики зависимости 1п[А] (а) и 1/[А] (£>) от времени для реакций различного порядка по А. 1 — нулевой порядок; 2 — первый порядок; 3 — второй порядок.
□ Частные порядки реакций можно также рассчитать из интегральной формы кинетического уравнения.
Уравнения типа 6.2-10 (или 6.2-13) н 6.2-15 называют «интегральными кинетическими уравнениями». Можно найти порядок по веществу А, определив, какая зависимость будет линейна. В соответствии с этими уравнениями, зависимость ln[A]t или l/[A]t от времени должна быть линейной, если реакция подчиняется первому илн второму порядку по А соответственно. Графики, выражающие эти зависимости, приведены на рис. 6.2-2. По ним достаточно легко определить порядок реакции по А. Действительно, если кривая отклоняется вниз от линейной зависимости (рис. 6.2-2,а), то реакция подчиняется нулевому порядку по исследуемому реагенту; напротив, отклонение кривой вверх свидетельствует о втором или более высоком порядке реакции по А. Интегральное кинетическое уравнение для реакции нулевого порядка по А приведено в табл. 6.2-1. Вид этих кривых очень легко различить. Как видно из рис. 6.2-1, если бы первый график (рнс. 6.2-2,а) отражал зависимость [А] от времени и не являлся бы прямой, то дальнейшее рассмотрение вопроса оказалось бы довольно сложным.
Определение частных порядков реакции при помощи интегральных методов заключается в построении графика кинетического уравнения в интегральной форме для предполагаемого порядка реакции. Если в результате мы получаем систему параллельных прямых, соответствующих различным начальным концентрациям А, то частный порядок реакции, принятый или рассчитанный из дифференциального кинетического уравнения, подтверждается. Констан-
6.2. Скорость химических реакций
329
Рис. 6.2-3. Соотношение между временем полупревращения реакции и долей непрореагировавшего реагента для реакций нулевого (1), первого (Й) и второго (5) порядков по реагенту А.
Число периодов полупревращения реагента
ту скорости реакции псевдо-п-го порядка легко рассчитать как тангенс угла наклона полученных прямых к оси абсцисс.
□ Время по лу л реа ращения реакции можно рассчитать из логарифмической формы кинетического уравнения.
Время полупревращения для реакции первого или псевдопервого порядка не зависит от концентрации реагента. Это легко продемонстрировать, решив уравнение 6.2-10 относительно t и приняв [A]t = [А]о/2:
1п[А]0 - 1п[А], Ыо/2? _ 1п2 _ 0,693
*1/2 =------(6 6)
Следовательно, как на расходование половины исходной концентрации реагента, так и на расходование от половины до трех четвертей исходного количества реагента требуется одно и то же время. Из рнс. 6.2-3 видно, что половина исходной концентрации или количества реагента исчезает за время, равное времени полупревращения. Половина оставшейся половины расходуется за еще один период полупревращения ti/2- Половина оставшейся четверти, остающейся после 2<1/2> расходуется еще за один период tjy2, т- е- спустя 3ti/2 остается только одна восьмая исходной концентрации и т. д. В случае реакции нулевого порядка по А все количество реагента будет израсходовано за 2ii/2. В случае реакции второго порядка по этому реагенту после 2ti/2 в реакционной смеси останется треть его исходного количества, четверть — после 3ti/2 н одна пятая—после 4ti/2, как легко определить из интегрального кинетического уравнения н соответствующих значений для fy2, представленных в табл. 6.2-1.
Уравнения, приведенные в табл. 6.2-1, можно использовать для расчета времени, необходимого для уменьшения концентрации А до одной десятой исходной величины. Эта величина равна l,8ti/2, 3,32ti/2 и 9^1/2 Для реакций нулевого, первого н второго порядков по А соответственно. Полученные величины можно использовать для определения порядка по данному реагенту посредством простой оценки их значений в условиях псевдо-п-го порядка. Тем не
330
Глава 6. Кинетика и катализ
менее для определения порядка предпочтительнее описанные выше дифференциальный и (или) интегральный подходы.
6.2.3. Факторы, влияющие на скорость реакции
Некоторые из реакций протекают так быстро, что любое столкновение между двумя реагирующими частицами приводит к образованию продуктов. В этом случае говорят, что реакция протекает в диффузионной области. Другие реакции протекают значительно медленнее вследствие того, что активированный комплекс слишком нестабилен и, следовательно, образуется только при столкновении частиц с чрезвычайно высокими кинетическими энергиями. К первому типу реакций принадлежит реакция между нонами водорода и гидроксидионами. Кинетическое уравнение имеет вид d[H+]/dt = — fc[H+][OH~] и константа скорости к составляет примерно 10п л/моль-с (т. е. она настолько велика, что лишь 7 10-12 с требуется для того, чтобы израсходовалась половина количества ионов водорода, добавленного к раствору, содержащему 1М гидроксид-ионов). Таким образом, с точки зрения химической кинетики существуют две большие категории реакций, а именно медленные (время полупревращения 10 с н более) и быстрые (с временем полупревращения менее Юс).
Скорость, с которой происходят столкновения между* частицами реагента, зависит от ряда факторов. К ним относятся, безусловно, концентрации реагентов. Поскольку это влияние в явном виде учитывается кинетическим уравнением, целесообразнее уделить внимание другим важным факторам — температуре, диэлектрической проницаемости растворителя и ионной силе электролитов, присутствующих в реакционной среде. Скорость реакции может меняться (увеличиваться или уменьшаться) в присутствии веществ, ускоряющих (катализаторы) или замедляющих (ингибиторы) реакцию. Катализ представляет собой явление, требующее отдельного подробного рассмотрения (разд. 6.3).
Влияние температуры
Температура оказывает влияние на большинство систем, использующихся в кинетической аналитической химии. Поэтому при аналитических измерениях ее необходимо строго контролировать. Повышение температуры приводит к росту кинетической энергии частиц реагирующих соединений, частицы движутся быстрее и сталкиваются чаще в растворе. Дополнительная кинетическая энергия повышает энергию молекул до величины энергии активации, необходимой для образования активированного комплекса.
□ Уравнение Аррениуса выражает зависимость энергии активации от температуры.
Константа скорости зависит от энергии активации. Действительно, рост энергии активации уменьшает долю от общего числа столкновений, для которых достигается требуемое значение кинетической энергии. В результате
6.2. Скорость химических реакций
331
уменьшается частота столкновений, приводящих к реакции, что находит свое отражение в уменьшении константы скорости, которая зависит от частоты столкновений н от геометрии системы. Простейшее описание этих эффектов дается уравнением Аррениуса:
d(bfe) ,к917,
•~dT~ = № (62’17)
которое можно представить в интегральной форме:
k = Ae~E‘IRT (6.2-18)
где Ea~ энергия активации (Дж/моль), R— универсальная газовая постоянная (8,3143 Дж/К-моль) и Т—абсолютная температура (К). Параметр А представляет собой произведение двух факторов, а именно а) частотного множителя Z, равного общему числу столкновений, происходящих за 1 с в объеме 1 л раствора, содержащего 1 моль каждого реагента; этот фактор обычно принимает значения около 1011 л/моль-с (кроме реакций между многозарядиыми ионами); б) стерического фактора р, представляющего собой долю столкновений, в которых реагенты пространственно расположены удобно для образования активированного комплекса. Стерический фактор зависит от структуры реагентов и изменяется в диапазоне от 0 до 1.
Для бимолекулярной индивидуальной стадии ср=1и£а = 0 константа скорости равна Z (« 1011 л/моль-с) и будет уменьшаться с ростом Ел. Таким образом, энергии активации, равные 20, 40 и ЮОкДж/моль, приведут к значениям к, равным 3 107, 1 104 и 3 10“7 л/моль-с соответственно. Поэтому, если энергия активации очень велика, лимитирующая стадия может быть очень медленной.
Влияние температуры на константу скорости проще всего описать уравнением 6.2-18 в логарифмической форме:
In к — In А — ~-RT
(6.2-19)
(6.2-20)
или
'В к 'В А 2,303ЯТ
□ Энергию активации реакции можно рассчитать из интегральной формы уравнения Аррениуса.
Для большинства реакций стерический и частотный факторы (оба учтены в параметре Л), а также энергия активации практически не зависят от температуры. Таким образом, зависимость In к (или любой другой пропорциональной величины, скажем, натурального логарифма начальной скорости реакции) от величины, обратной абсолютной температуре (1/Т), обычно представляет собой прямую линию. Из наклона соответствующего графика (—Ea/R) можно легко рассчитать энергию активации. Определив ее значение, можно рассчитать к для любой желаемой температуры по уравнению 6.2-19.
332
Глава б. Кинетика и катализ
□ Величина влияния температуры на константу скорости зависит от энергии активации.
Константа скорости многих гомогенных реакций увеличивается в 2-3 раза при повышении температуры на 10°С (при температурах, близких к комнатной). Это справедливо для реакций с энергиями активации в диапазоне от 50 до 90 кДж/моль (ниже и выше указанного диапазона данный фактор становится меньше 2 и много больше 3 соответственно). Это является результатом того, что температура по-разному влияет на скорости различных процессов в зависимости от их энергии активации. Так, из уравнения 6.2-21
Infer,= Infer.(6.2-21)
полученного вычитанием уравнения 6.2-19 для двух разных температур, следует, что нагревание реакционной смеси от 298 К (25° С) до 308 К (35° С) приводит к росту константы fcr2 всего лишь на 14% при Еа = 10 кДж/моль. При Еа — 100 кДж/моль константа увеличивается уже примерно в четыре раза, а при Еа — 300 кДж/моль — в 50 раз. В соответствии с этим, на медленные реакции (т. е. с малыми константами скорости) рост температуры будет оказывать более значимое влияние, чем на быстрые реакции (т. е. имеющие высокие константы скорости). В целом, можно сказать, что количественное влияние температуры на константу скорости зависит от соответствующей энергии активации. Реакции, протекающие быстро при комнатной температуре, можно замедлить охлаждением; медленные реакции можно ускорить нагреванием.
6.2.4. Скорости реакций: аналитическое применение
Для определения соединений при помощи кинетических методов (рис. 6.2-1) требуется прямо или косвенно измерить скорость реакции, которая связана с концентрациями реагентов (ур. 6.2-6). Для этого измеряют изменение концентрации реагента или продукта как функцию времени в экспериментальных условиях, соответствующих реакции псевдопервого порядка. В итоге строят соответствующий график этой зависимости, представляющий собой возрастающую (для продукта реакции) или убывающую (для реагента) кинетическую кривую.
Методы определения, основанные на реакциях псевдопервого порядка
Некоторые кинетические методы позволяют проводить как определение индивидуальных веществ, так н одновременное определение двух или более соединений в смесях. Эти методы основаны на применении дифференциальных или интегральных уравнений реакций псевдопервого порядка, хотя в каждом конкретном случае методология определения может меняться.
Определение индивидуальных веществ
В наиболее распространенном варианте кинетических методов используют начальный участок кинетической кривой (реакция протекает на 1-3%). Этот участок обычно линеен и его наклон прямо пропорционален концентрации измеря
6.2. Скорость химических реакций
333
емого вещества. В этих условиях реакцию можно рассматривать как реакцию псевдонулевого порядка, и уравнение 6.2-6 можно выразить в виде конечных приращений:
и=-^г = ^г=*'|А1о (6-2~22)
□ Метод тангенсов (метод начальных скоростей) позволяет получать точные данные о кинетике реакций, которые нельзя определить другими методами.
Из уравнения легко рассчитать начальную концентрацию [А]о вещества А. Этот дифференциальный метод называется методом тангенсов (или методом начальных скоростей) и обладает несколькими важными достоинствами: а) ои применим к большому числу химических реакций, которые нельзя исследовать равновесными методами вследствие их малой скорости или иеколичественного характера; б) можно избежать конкурирующих реакций, возникающих в момент, когда реакция близится к завершению; в) измерения начальной скорости реакции, сделанные в начальный момент развития процесса, являются более точными, так как наклон кинетических кривых и отношение сигнала к шуму максимальны.
□ Метод фиксированного времени и метод фиксированной концентрации являются альтернативой методу тангенсов.
Кроме метода тангенсов, широко распространены методы фиксированного времени и фиксированной концентрации. Оба метода базируются на уравнении 6.2-22 и используют либо постоянство времени At, либо постоянство А [А] (или А[Р]). Иными словами, метод фиксированного времени основан на определении концентрации реагента или продукта в заданный момент времени от начала реакции. Метод фиксированной концентрации основан на измерения времени, необходимого для того, чтобы концентрация реагента или продукта достигла заданной величины. Таким образом, А [А] (или А[Р]) и величина 1/At, обратная At, будут прямо пропорциональны [А]о, что позволяет построить соответствующий градуировочный график.
Определение одного вещества можно также осуществить при помощи интегрального метода, основанного иа интегральном уравнении псевдопервого порядка (ур. 6.2-10 или 6.2-13). Методика включает в себя построение функции ln[A]t или ln([P]TO — (P]t) от времени. Исходную концентрацию вещества А ([А]о) легко рассчитать из отрезка, отсекаемого данной прямой на осн ординат. Можно использовать и интегральные методы фиксированного времени и фиксированной концентрации, при этом уравнение 6.2-10 приводится к виду
In £11 = kA At (6.2-23)
1AJ2 подстановкой tj = 0 (при котором [A]i — [А]о); при этом либо At, либо А [А] являются постоянными.
Одновременное определение нескольких соединений
Измерение скоростей реакции позволяет эффективно решать извечную аналитическую задачу анализа смесей компонентов, близких по химическим свой
334
Глава б. Кинетика и катализ
ствам. Кинетические методы одновременного определения соединений в смесях основываются на различии скоростей, с которыми соединения вступают в реакцию с аналитическим реагентом. Поэтому можно определить состав смеси без ее -предварительного разделения. Эти методы часто называют дифференциальными кинетическими методами.
□ Кинетические методы анализа многокомпонентных систем применимы к реакциям, не являющимся реакциями псевдопервого порядка.
Несмотря на то что существует несколько методов исследования реакций высшего (не псевдопервого) порядка, чаще всего используют дифференциальные кинетические методы.
Пусть А и В—два компонента смеси, которые реагируют с реагентом R с образованием продуктов Р и Р'. Эти продукты, различные по своей природе, весьма сходны в отношении аналитического свойства, которое будет служить аналитическим сигналом. Если скорости соответствующих реакций существенно отличаются и не зависят друг от друга (т. е. отсутствуют взаимные кинетические и синергические эффекты), а процесс характеризуется первым порядком по каждому из компонентов смеси (т. е. [R]o [А]о и [R]o >> [В]о), тогда сумма концентраций А и В в момент времени t будет равна
Ct = [A]f + [B]f = (Р]то - (P]t = [А]о ехр(-kAt) + [В]о exp(-fcBt) (6.2-24)
где кА и кв — константы скорости псевдопервого порядка для компонентов А и В соответственно.
Уравнение 6.2-24 является основным для многих успешно используемых дифференциальных кинетических методов, таких, как метод логарифмической экстраполяции и метод пропорциональных уравнений. Применимость этих, а также других дифференциальных методов в огромной степени зависит от относительных величин констант скорости исследуемых реакций, а также соотношения концентраций компонентов смеси, т. е. от соотношений кА/кв и [А]о/[В]о.
Метод логарифмической экстраполяции основан на логарифмировании уравнения 6.2-24 и построении графика зависимости In Ct или ^([Р]^ — [Р]*) от времени. В случае кА кв получим нелинейную зависимость, а в случае равенства констант —линейную. Если вещество А расходуется со скоростью, большей, чем вещество В (т. е. кА > feB), тогда [A]t —» 0 и кривая в конечном итоге становится прямой (рис. 6.2-4). Уравнение этой кривой в логарифмической форме выгладит следующим образом:
In Ct = InQP]- - [Р]t) = ln[B]0 - kBt (6.2-25)
□ Метод логарифмической экстраполяции не требует предварительной информации о константах скорости.
Отрезок, отсекаемый данной прямой на осн ординат, позволяет определить исходную концентрацию более медленно реагирующего компонента [В]о, а исходную концентрацию быстрее реагирующего компонента находят по разности
6.2. Скорость химических реакций
335
Рис. 6.2-4. Применение метода логарифмической экстраполяции для реакций первого порядка [Рёгег-Bendito D-, SilvaМ., Kinetic Methods in Analytical Chemistry, Chichester, Ellis Horwood, 1988}.
[A]o — [Р]то- [B]o. В случае фотометрического определения Ct значения [Р]то и [pjt можно заменить экспериментальными значениями оптической плотности.
Метод логарифмической экстраполяции не требует предварительной информации о /са или кв- Напротив, кв можно непосредственно получить из наклона линейного участка кривой. Однако для получения надежных значений константы скорости данным методом необходимо, чтобы по крайней мере 99% более реакционноспособного компонента было израсходовано на момент измерений. Кроме того, начальные концентрации компонентов А и В должны быть точно известны.
□ Метод пропорциональных уравнений требует предварительной информации о константах скорости.
Метод пропорциональных уравнений для анализа двухкомпонентных смесей основан на измерении Ct в два момента времени и построении двух функций типа уравнения 6.2-24. Из этой системы уравнений можно определить исходные концентрации каждого из компонентов, если заранее известны соответствующие константы скорости. Следует отметить, что для этой цели можно использовать не только время, но и любую другую экспериментальную переменную, например температуру или какое-либо физико-химическое свойство реагирующих веществ. Система уравнений в этом случае имеет вид
fi = кд! [А] о + &Bi [В]о Л = &Аг[А]о + кв2 [В]о
(6.2-26)
где fi и /а обозначают выбранный измеряемый параметр, определенный для двух различных совокупностей экспериментальных условий, а кдг, кдз, кв1 и кв2 — константы пропорциональности, полученные отдельно для каждого компонента в различных условиях.
- С практической точки зрения, отношение констант скорости кд/кв должно быть на уровне 3-4 или выше. Тем не менее для смесей с отношением кон
336
Глава 6. Кинетика и катализ
стант около 1,5-2 можно использовать современные математические методы (например, аппроксимацию кривых по массиву данных), которые позволяют использовать весь объем информации, содержащийся в кинетической кривой (например, при помощи фильтра Кальмана).
6.2.5. Кинетические методы определения, основанные на некаталитических реакциях
Можно выделить два случая, когда применение кинетической методологии к иекаталитическим реакциям, описанным в предыдущих разделах, однозначно приводит к лучшим результатам, чем анализ равновесными методами. Первый случай: исследуемый процесс протекает крайне медленно или не до конца (и вблизи равновесного состояния проявляются конкурирующие реакция). Второй случай: соответствующий равновесный метод весьма трудоемок или требует длительного времени. В иных ситуациях вряд ли следует ожидать роста чувствительности или улучшения правильности определения кинетическими методами по сравнению с некинетическими. Но в любом случае метод, основанный на кинетических измерениях, часто обеспечивает лучшую селективность измерений-
Некаталитические реакции имеют особое значение в случае анализа смесей компонентов, имеющих сходные химические свойства. Для таких систем разработаны разнообразные дифференциальные методы. Их широко применяют в органическом анализе и анализе смесей ионов металлов, основанном иа различии скоростей реакций обмена лигандов в некоторых комплексах.
Основанные на некаталитических реакциях кинетические методы определения органических соединений, как индивидуальных, так и в смесях, базируются на целом ряде реакций, таких, как а) окисление определяемых соединений, б) бромирование ароматических соединений, в) конденсация или присоединение аминов н-карбонильных соединений, г) гидролиз.
Периодат-ион—один из самых широко распространенных реагентов для окисления по реакции Малапрада органических соединений, имеющих гидроксигруппу, таких, как фенолы, хлорфенолы или вицинальные гликоли. Фенолы и их производные можно определять в диапазоне концентраций от 50 до 500 мкг/мл методом фиксированного времени, измеряя оптическую плотность при 340 нм. В этой области находится максимум поглощения образующихся хинолов и хинонов [6.2-1, 6.2-2]. Некоторые органические соединения, имеющие фармакологическое значение, такие, как витамины Bi и С, также можно определить при помощи реакции окисления — восстановления. Тиамин окисляется Hg(II) до тиохрома—флуоресцирующего соединения, которое является индикаторным веществом в этом определении [6.2-3]. В данном случае кинетический метод является весьма чувствительным (предел обнаружения 2 • 10-8М). В настоящее время его используют для определения тиамина в различных лекарственных препаратах (смесях микроэлементов и поливитаминов). Катехоламины окисляются до о-бензохинонов гексахлоридом иридия и до аминохро-мов периодат-ионами [6.2-4], что дает возможность определять адреналин и
б.З. Явление катализа
337
леводопу на уровне концентраций от 0,2 до 2,0 мМ методом остановленного потока. В качестве окислителя можно также использовать перйодат.
В кинетических методах, основанных на реакциях бромирования, бром образуется in situ по реакции между бромг£т- и бромид-ионами в кислых растворах. В качестве индикаторного вещества используют метиловый оранжевый, степень обесцвечивания которого пропорциональна концентрации определяемого соединения в растворе. В качестве примеров соединений, которые могут быть определены данным методом (в варианте метода фиксированной концентрации) можно привести крезолы, ксилолы, этил- и фенилфенолы, парацетамол и салициловую кислоту [6.1-1].
Некаталитические реакции все более широко применяются для определения индивидуальных соединений и анализа смесей благодаря доступности автоматических приборов для кинетических измерений. Это в огромной степени расширило число методик, которые можно использовать для рутинного анализа (см. табл. 6.4-1), в котором до последнего времени применялись в основном каталитические кинетические методы анализа.
6.3. ЯВЛЕНИЕ КАТАЛИЗА
6.3.1. Общие положения
□ Катализ может быть гомогенным или гетерогенным.
Одним из путей ускорения медленной реакции является добавление к смеси незначительного количества катализатора. Катализаторы существенно различаются по химической природе. В частности, каталитическими эффектами обладают различные металлы, неметаллы и органические соединения (включая ферменты), которые увеличивают скорость некоторых реакций, находясь в водной среде. Это явление известно как гомогенный катализ. Если же в системе присутствует твердая фаза (например, электрод) или в общем случае имеется двухфазная система, то описываемое явление называют гетерогенным катализом. Далее мы не будем рассматривать этот случай.
Современная тенденции использования мицеллярных сред в экспериментах, связанных с измерением скоростей реакции, привела к совершенно новой возможности ускорения некоторых каталитических и некаталитических реакций. Данное физическое явление известно как мицеллярный катализ. Его можно рассматривать как вариант гомогенного катализа, несмотря на присутствие псевдофаз в системе.
Широкое применение химических катализаторов в кинетических методах в настоящее время, приведшее к их значительному развитию, основано на том, что скорость каталитической реакции прямо пропорциональна концентрации катализатора. Поэтому становится возможным разрабатывать высокочувствительные методы определения металлов и неметаллов, обладающих каталитическими свойствами. Высокая чувствительность этих методов обусловлена тем, что катализатор не расходуется в процессе реакции, а принимает в ней участие циклическим образом. Другие катализаторы (например, ферменты) обладают высокой чувствительностью по своей природе.
338
Глава б. Кинетика и катализ
□ Катализатор изменяет скорость реакции, не влияя при этом на полное изменение свободной энергии Гиббса в ходе реакции.
Катализатор можно определить как вещество, которое ускоряет химическую реакцию, не влияя на полное изменение энергии Гиббса реакции, а само явление ускорения называется «катализом» [6.3-1]. Катализатор обладает следующими свойствами: а) он не меняется с химической точки зрения в конце реакции; б) небольших его количеств обычно достаточно для того, чтобы скорость реакции изменилась существенно; в) наличие катализатора не влияет на положение равновесия обратимой реакции.
В каталитическом процессе энергия активации снижается в результате того, что катализатор непрерывно регенерируется (каталитический цикл) на одной из стадий реакции, что обеспечивает постоянство его концентрации во время процесса. Поскольку константа равновесия для данной реакции К равна ki/k-i (fcj и k-i—константы скорости прямой и обратной реакций соответственно), катализатор влияет на скорости как прямой, так и обратной реакций в одинаковой степени, т. е. ускоряет достижение равновесия, но не меняет его положение. Катализатор также увеличивает скорость несамопроизвольных реакций. Таким образом, термин «отрицательный катализ», который используют по отношению к веществам, повышающим свободную энергию активации, представляется некорректным.
Реакция, которая катализируется определяемым веществом, носит название «индикаторной реакции» и «некаталитической реакции»; для аналитического использования ее скорость должна быть крайне мала или пренебрежимо мала по (равнению с каталитической реакцией.
Скорость химической реакции может также меняться (увеличиваться или уменьшаться) в присутствии определенных веществ, известных как активаторы и ингибиторы соответственно. Явление ингибирования обычно связывают с каталитическими эффектами в ферментативных и неферментативных реакциях, хотя оно наблюдается и для других реакций (например, фотохимических или электрохимических). Явление активирования однозначно связано с каталитическими реакциями. Активатором можно называть вещество, принимающее участие на некоторой стадии реакции, для которой энергия активации ниже, чем для реакции с одним катализатором: т. е. активатор проявляет свои свойства только в присутствии катализатора. Этот благоприятный эффект приводит к значительному увеличению чувствительности определения катализатора, а также дает возможность определять сам активатор.
Как ингибирующие, так и активирующие эффекты имеют аналитическое значение [6.1-1], потому что они пропорциональны количеству ингибитора или активатора, присутствующего в растворе. Эффекты обоих типов уже нашли широкое применение в ферментативных и неферментативных реакциях. В ферментативных реакциях активирование или ингибирование обусловлено присутствием ионов металлов, тогда как в неферментативных реакциях активирующие и ингибирующие эффекты вызваны действием лигандов (например, гидрокси- или полиаминокарбоксисоединений) на ионы металлов.
6.3. Явление катализа
339
6.3.2. Кинетические уравнения и механизмы реакций
Для каталитической реакции
A + B-S.P + Y (VII)
где С—катализатор, А и В — реагирующие вещества, Р и Y—продукты, кинетическое уравнение можно составить на основе механизма каталитической реакции, включающего образование промежуточного комплекса по схеме
c+b=₽==>=cb+y (vui)
СВ + А^Р + С (IX)
где вещество В, находящееся в избытке, образует промежуточный комплекс СВ с катализатором, а А — компонент, по которому ведется наблюдение за реакцией. Следует рассмотреть два различных случая в зависимости от того, какая из стадий — VIII или IX — является лимитирующей. Общая скорость процесса зависит от соотношения величин A?i, fc-i и /с2-
□ Понятие предравновесного механизма применимо, если скорость диссоциации комплекса намного больше, чем скорость реакции между комплексом и реагентом.
Если лимитирующей стадией является реакция IX ( &2 fc-i), то реакция диссоциации комплекса будет много быстрее, чем реакция между комплексом и реагентом, — процесс достиг предравновесного состояния. Если равновесное состояние реакции VIII, выражаемое константой
*! _[CB][Y]
Кс-м--[сдвГ (63-1)
достигается первым, то общая скорость реакции будет совпадать со скоростью лимитирующей стадии, т. е. стадии IX:
= мсвнА| (6'3‘2)
Замена {В] и [С] в уравнении 6.3-1 на [В] о — [СВ] и [С] о — [СВ] соответственно ([В]о и [С]о — исходные концентрации соединений В и С) приводит к уравнению1
[CB][Y] ,6,31
С [В]„([С]0-[СВ]) (" )
Решая данное уравнение относительно [СВ] и подставляя его в ур. 6.3-2, получаем общее (полное) кинетическое уравнение для процесса в предравновесном состоянии
(в.3-4) at -KcpSJo + L *J
где [A] — концентрация реагента А в момент времени t.
1 При выводе данного уравнения также использовано опущенное авторами в тексте допущение [В]о — [СВ] « [В]о, что справедливо, так как [В]о >> [С]о, [С], [СВ]. — Прим, перев.
340
Глава 6. Кинетика и катализ
D Механизм стационарного состояния применим, если реакция промежуточного комплекса проходит намного быстрее, чем реакции образования илн диссоциации комплекса.
Если же лимитирующей является стадия VIII (fe k-i и fe fe), тогда реакции промежуточного комплекса с реагентом А будет много быстрее реакции образования и диссоциации комплекса. В такой ситуации концентрация промежуточного комплекса СВ в системе будет всегда постоянной и очень низкой — процесс достиг стационарного состояния:
ЁИ = о = [С][В] - к_1 [CB][Y] - k2[CB][A] (6.3-5)
Решая это уравнение относительно [СВ] и заменяя [С] и [В] на значения, приведенные выше, мы можем определить концентрацию комплекса [СВ]:
[гга________________fci[C]o[B(o ___________
11 МЧо + [В]о-[СВ]*)+ *_,[¥)+ fa[A] 1 '
Пренебрегая членом [СВ]2, который мал в сравнении с суммой [С]о 4- [В]о, в знаменателе данной дроби, и учитывая, что в соответствии с условиями стационарного состояния
= МОД - = fe[CB][A] (6.3-7)
подстановкой величины [СВ] (уравнение 6.3-6) в правую часть уравнения 6.3-7 получаем выражение
_d[A] ___________А1*2[С]о[В]о[А]_________
dt ki([C]o + lB]o-[CB]2) + fc_1[Y] + fc2[A] 1 1
которое представляет собой общее кинетическое уравнение для процесса в стационарном состоянии.
Общие уравнения 6.3-4 и 6.3-8 можно упростить с практической точки зрения, заменяя [А] на [А]о — [Р] ([Р] является уменьшением концентрации реагента А или увеличением концентрации продукта [Р], измеренным во время реакции), так как за большинством аналитических каталитических реакций следят именно по изменению концентраций продуктов реакции;
® = Л«ЛС]0([А]с - [Р]) (6.3-9)
где ас представляет собой функцию, содержащую все концентрационные члены уравнения 6.3-8 за исключением [С]о, [А]о и [Р], а К—член, включающий все константы скорости и константы равновесия.
С другой стороны, если некаталитическая реакция протекает с достаточной скоростью, что отражается в значимом значении константы fe, общее кинетическое уравнение будет выглядеть так:
(Ф) = Ка°[С]о([А]о - М) + Л3[В]о([А]о - [Р]) (6.3-10)
\ / набл
Вблизи начала реакции можно пренебречь [Р] по сравнению с [А]о, и, таким образом, уравнение 6.3-10 приводится к виду
ад = ) = К'[С]о + К! (6.3-10а)
6.3. Явление катализа
341
где К' и К{ — константы, если [А]о и [В]о много больше, чем [С]о, и [В]о [А]о. Для того чтобы эти условия выполнялись, реакцию необходимо проводить в условиях псевдопервого порядка по реагенту, по которому ведется наблюдение, так как концентрация катализатора, по определению, не меняется в процессе реакции.
Для каталитического цикла (определяемого реакциями VIII и IX), являющегося основой каталитических определений, весьма важны реакции образования хелатов и других комплексов катализатора и реагента. Многие реакции, включая ферментативные, ведут себя в соответствии с предравновесным механизмом. Ферменты образуют ассоциаты с некоторым реагентом (субстратом). Субстрат высвобождается из комплекса фермент — субстрат во время лимитирующей стадии и может катализировать разложение другой молекулы субстрата, вновь начиная каталитический цикл.
Еще один путь, по которому катализатор может взаимодействовать с компонентами реакционной смеси, включает его реакцию с А, приводящую к образованию продукта Р и активированной формы катализатора С*, которая затем реагирует с В, образуя второй продукт реакции Y по следующей схеме:
А + С —Р + СГ (X)
C»+B6“^Yf-C (XI)
Стадия X является лимитирующей. К механизму этого типа принадлежат многие окислительно-восстановительные каталитические реакции, причем образование формы С* часто сопровождается изменением степени окисления катализатора. Для обеспечения данной схемы взаимодействия исследуемая система должна соответствовать двум важнейшим условиям: а) окислительно-восстановительный потенциал каталитической системы Ес должен быть более положительным, чем потенциал пары P/А, и более отрицательным, чем потенциал пары B/Y (т. е. Ед/y > Ес > £?р/а)> 6) прямое взаимодействие между А и В, хотя и разрешенное с термодинамической точки зрения, должно быть запрещено кинетически. Кроме того, реакция между катализатором и соединением В должна быть очень быстрой.
Все эти общие механизмы оказываются не столь простыми при переходе к реальным системам. В действительности каждая стадия может включать более чем одну реакцию, каждая из которых и может быть настоящим лимитирующим процессом.
Каталитические неферментативные реакции
Кинетические уравнения, применимые к реакциям, катализируемым соединениями, не являющимися ферментами,—это упрощенные уравнения 6.3-9 и 6.3-10. Класс подобных реакций включает обычные каталитические реакции и реакции Ландольта. Реакции первого типа включают самые разнообразные взаимодействия, в том числе окислительно-восстановительные, кислотноосновные, хемилюминесцентные реакции и реакции комплексообразования.
342
Глава б. Кинетика и катализ
□ В кинетических методах анализа широко распространены окислительно-восстановительные каталитические реакции.
В настоящее время наиболее распространенным типом индикаторных реакций в кинетических методах анализа являются окислительно-восстановительные реакции. Обычно используются такие окислители, как пероксид водорода, кислород воздуха, бромат-, перйодат-, иодат- и персульфат-ионы, Fe(III) и Ce(IV), и такие неорганические восстановители, как олово(П), железо(II), мьппьяк(1П), иодид- и тиосульфат-ионы, а также большое число органических восстановителей (амины, фенолы и азокрасители). Катализаторами обычно служат многовалентные ионы металлов, имеющие вакантные d-орбитали и образующие комплексы с каким-либо компонентом реакции (например, органическим восстановителем). Чаще всего это металлы с валентностью четыре (Zr, Hf, Th), пять (V, Та, Nb), шесть (Mo, W), а также два и три (Fe, Мп, Си, Со) или благородные металлы (Ag, Os, Pd, Ru, Pt, Rh, Ir). Напротив, только немногие неорганические анионы обладают каталитической активностью, среди них наиболее часто используют иодид-, нитрит-, сульфид- и бромид-ионы. Некоторые ионы металлов обладают преимущественным ускоряющим действием на окисление неорганических или органических соединений определенным окислителем (например, марганец действует практически исключительно на реакции с периодат-ионами, тогда как ванадий проявляет себя обычно в присутствии бромат-ионов).
□ Реакция между церием и арсенитом представляет собой типичный случай окислительно-восстановительной каталитической реакции.
Приведем некоторые из наиболее типичных примеров каталитических окислительно-восстановительных систем. Это система Ce(IY)-As(III), катализируемая иодидом (реакция Сэндела-—Кольтгофа), система иод-азид натрия, катализируемая серосодержащими соединениями, разложение пероксида водорода Си(П), Со(П), Мп(П) и Fe(III), система пероксид водорода-иодид, катализируемая Fe(III), Mo(VI), W(VI), Zr(lV) и Hf(IV), система пероксид водородагидрохинон, катализируемая Cu(II), система периодат-малахитовый зеленый, катализируемая Мп(П).
Реакцию Сэндела—Кольтгофа проводят в сернокислой среде, несмотря на то, что, по имеющимся данным, каталитическая активность иодида в азотнокислой среде выше в 20 раз. Хлорид-ионы ингибируют данную каталитическую реакцию при концентрациях выше примерно 0,34 М, но ускоряют ее при низких концентрациях. Кинетика и механизм данной реакции подробно изучены [6.3-2]. Предложенный механизм, согласующийся с экспериментальными данными, выглядит следующим образом:
I” + Ce(IV) Се(Ш) +1 (XII)
I + As (III) 2 ? Промежуточное соединение (Х1П)
Промежуточное соединение 4- Ce(IV) As(V) 4- Се(Ш) 4-I” (XIV)
I + Ce(IV) -S Се(Ш) + Г (XV)
6.3. Явление катализа
343
l+ + As(III) =р=* As(V) + Г (XVI)
Следует подчеркнуть, что реакции XII—XIV, с одной стороны, и XII, ХШ и XVI, с другой стороны, представляют собой два различных каталитических цикла, каждый из которых по отдельности не согласуется с экспериментальными фактами.
Взаимодействие между катализатором и органическим соединением (например, амином или фенолом) обычно включает предварительное образование комплекса с переносом заряда между ними, устойчивость которого напрямую зависит от pH среды. Следовательно, данный параметр имеет очень заметное влияние на каталитическую активность иона металла. В то же время неорганические субстраты взаимодействуют с катализатором посредством переноса электронов и могут привести к образованию нестабильных промежуточных соединений на других стадиях, аналогично приведенному выше примеру системы Ce(IV)-As(III).
□ Хемилюминесцентные реакции часто включают окислительно-восстановительные процессы.
Хемилюминесцентные реакции, которые ускоряются следовыми количествами ионов металлов, обычно включают процесс окисления. Наиболее хорошо изученная хемилюминесцентная реакции—это окисление люминола (5-амино-2,3-дигидро-1,4-фталазиндиона) пероксидом водорода. Чрезвычайно малые количества таких ионе® металлов, как Со(П), Cu(II), Сг(Ш), Мп(П), Fe(III) и Ni(II), при pH 10-11 увеличивают квантовый выход хемилюминесценции. Люминол превращается в двухзарядный анион, который затем окисляется до возбужденного синглетного состояния, которое испускает излучение при разложении до аминофталат-ионов [6.3-3] согласно следующей схеме:
(XVII)
Другими пригодными окислителями являются кислород воздуха и персульфат-ионы. В качестве субстратов в реакциях данного типа используются также люцигенин и лофин, хотя и гораздо реже, чем люминол.
□ Большинство реакций Ландольта включают окислительно-восстановительные процессы, в основе некоторых лежат процессы комплексообразования или кислотно-основного взаимодействия.
Реакции Ландольта в большинстве случаев также включают окислительно-восстановительный процесс (кислотно-основную реакцию или реакцию комплексообразования). Этот тип реакций основан на эффекте Ландольта [6.3-4],
344
Глава 6. Кинетика и катализ
Рис. 6.3-1. Кинетические кривые для реакций Ландольта полученных в отсутствие (I) и в присутствии (2) катализатора. Кривая 3 соответствует индикаторной реакции (А + В —» Р) [Pftez-Bendito D., Silva М., Kinetic Methods in Analytical Chemistry, Chichester, Ellis Horwood, 1988].
который заключается в наличии некоторого индукционного периода в случаях, когда медленная реакция связана с быстрой через продукт восстановления первой в соответствии со схемой
А + В--------->Р
медленно
P+L *? >Y быстро
(XVIII)
(XIX)
Поскольку вторая реакция быстрее, чем первая, продукт (Р) может быть определен после того, как соединение L (реагент Ландольта) будет полностью из-расходованово второй реакция. Если реакция XVIII ускоряется катализатором (рис. 6.3-1), время, прошедшее от начала процесса до момента, когда появляется продукт реакции, будет являться мерой концентрации катализатора, которая в свою очередь непосредственно связана с индукционным периодом t{ согласно эмпирическому уравнению [С]о = Ki/ti или [С]о = Ki/t% в зависимости от рассматриваемой системы. Одной из типичных реакций Ландольта является окисление иодид-ионами Н2О2, ВгОз или SzOg-, катализируемое молибденом, ванадием и медью в присутствии аскорбиновой кислоты или тиосульфат-ионов в качестве реагентов Ландольта.
Ферментативные каталитические реакции
Ферментативные реакции представляют собой особый тип каталитических реакций. Реакция между единственным субстратом и ферментом может быть записана следующим простым образом:
E + S=f=*ES «2
ES-^E+P
(XX)
(XXI)
где Е, S, ES и Р — фермент, субстрат, комплекс фермент-субстрат и продукт соответственно. Однако кинетика ферментативных каталитических реакций может значительно осложняться наличием нескольких комплексов ES. В при-
6.3. Явление катализа
345
Рис. 6.3*2. Влияние концентрации субстрата на скорость ферментативной каталитической реакции с единственным субстратом.
ближении [Е] С [S] и предположении, что стадия XXI является лимитирующей, Михаэлис и Ментен вывели кинетическое уравнение данной реакции:
® = VO = fc3[ES] (6.3-11)
UE
Вследствие того, что предыдущая реакция (XX) является равновесной, т. е.
,, fa [ESI fa [E][S]
и принимая во внимание, что [ES] = [Е]о — [Е] ([Е]о—общая равновесная концентрация фермента), заменяем [Е] в уравнении 6.3-12 и решаем его относительно [ES]. Подставляем полученное значение в уравнение (6.3-11) и в результате получаем
fc3K[E]0[S] fc3[E]0[S]
vo= i+k[sr = KfTM ( }
где Ks — 1/K.
Существуют два возможных крайних случая. Если концентрация субстрата [S] столь мала, что K[S] С 1, скорость реакции приблизительно определяется выражением
ц0 и fc3K[E]0[S] (6.3-14)
что согласуется с экспериментальным фактом, согласно которому ферментативные реакции имеют первый порядок по S, 'когда концентрация [S] мала (рис. 6.3-2). С другой стороны, когда [8]столь велика, что [S] 1,
г?о « ^з[Е]о (6.3-15)
Экспериментально подтверждено, что когда концентрация субстрата достаточно велика реакция подчиняется нулевому порядку по S. (рис. 6.3-2). Относительная величина [S], естественно, зависит от величины К.
Если предположить, что /с3 не обязательно должна быть много меньше ^2, подход, основанный на рассмотрении стационарного состояния, позволяет объяснить поведение многих ферментативных реакций. В рамках этого подхода
346
Глава 6. Кинетика и катализ
концентрация фермент-субстратиого комплекса [ES] остается постоянной, т. е. d[ES]/di = 0 и
кг [Е] [S] = *2 + fc3[ES] (6.3-16)
Исходя из тех же предпосылок, которые использовались при объяснении механизма предравновесия, можно вывести выражение типа уравнения 6.3-13.
= =fc3[E]0is]
k2 + k3 ! [S] Кы + [S]
которое называется уравнением Михаэлиса—Ментен. Член Км — (&2 + кз)/к^ носит название «константы Михаэлиса—Ментен».
□ Константу Михаэлиса—Ментен можно использовать для оценки сродства субстрата и фермента.
Для случая кз С fa Км = Ks в уравнении 6.3-13, которое, таким образом, становится особым случаем уравнения 6.3-17. Следовательно, при кз « fa константа Км представляет собой всего лишь меру сродства фермента к субстрату, так как только в этих условиях она становится равной Ks. С другой стороны, Км представляет собой эмпирическую константу, совпадающую с концентрацией субстрата, когда скорость реакции достигает половины своей максимальной величины vmax (рис. 6.3-2). Если Км = [S], то поскольку fa[E]o = «шах> уравнение 6.3-17 упрощается:
_ t?n..lx[Sj _ k'max V ~ 2[S] ~ 2
Концентрация фермента в ферментативной каталитической реакции столь мала, что предравновесие достигается практически только за счет избытка субстрата. Это позволяет определить, участвует реагент в предравиовесии или в лимитирующей стадии.
Ферментативные каталитические реакции не только высокочувствительны, но и чрезвычайно селективны. Ферменты представляют собой белки-катализаторы, которые воздействуют только на определенные субстраты. Например, среди всех биохимических реакций, протекающих в человеческом организме, гидролиз мочевины является единственным процессом, на скорость которого оказывает влияние присутствие фермента, называемого уреазой. Уреаза проявляет каталитический эффект на превращение мочевины в аммиак и карбонат-ионы:
CO(NH2)2 + 2H2O—'^2NH+ + HCOs (XXII)
Поскольку уреаза не расходуется в этом процессе, весьма небольшое количество этого фермента может эффективно ускорить гидролиз очень больших количеств мочевины. Сама по себе эта реакция очень медленная (время полупревращения при температуре человеческого тела 37°С или 310 К составляет примерно 105лет), но существенно ускоряется уреазой.
6.3. Явление катализа
347
Глюкозооксцдаза—это еще один высокоспецифичный фермент. Он катализирует окисление Д-Л-глюкозы до глюконовой кислоты в присутствии ряда других сахаров, многие из которых также реагируют с этим ферментом, но с существенно меньшей скоростью.
Активность ферментов характеризуют международными единицами активности. Единица активности фермента определяется как количество фермента, превращающего 1 мкмоль субстрата при 25°С за 1 мии при оптимальных экспериментальных условиях, включающих pH, ионную силу и концентрацию субстрата.
Мицеллярным катализ
Мицеллярный катализ представляет собой особый тип катализа. Известно, что мицеллы, образованные ПАВ при достаточно высоких концентрациях в водном растворе, способны изменять скорость химических реакций в результате того, что реагирующие компоненты притягиваются к поверхности мицелл, и вызванный этим эффект концентрирования приводит к более быстрой реакции. Реагенты могут взаимодействовать с мицеллами различными путями: находиться внутри их гидрофобной оболочки, адсорбироваться в поверхностном слое за счет электростатических взаимодействий и т. д. Несмотря на то что концентрация мицелл не меняется в ходе реакции, мицеллы не являются катализаторами в строгом смысле, так как они не участвуют ни в какой стадии реакции и каталитических циклах. Таким образом, данный эффект имеет скорее физическую, а не химическую природу.
□ Для случая мицеллярного катализа применима псевдофазовая кинетическая модель.
Рисунок 6.3-3 иллюстрирует наиболее вероятные взаимодействия на примере бимолекулярной реакции между соединениями А и В в присутствии мицелл [6.3-5]. Здесь кт и kv,— константы скорости реакции в мицеллярной и водной фазах соответственно, а к'т и — константы скорости в случаях, когда только одно из реагирующих соединений взаимодействует с мицеллами.
Мйцелла
Продукты
!tk«
** км
В '4 ' * Продукты
+ _
А 4„—г Продукты
Водный раствор
Рис. б.3-3. Взаимодействия в бимолекулярной реакции в присутствии мицелл [Рёгег-BenditoD., RubioS., Trends Anal. Chem., 1993, V. 12, Р.9].
348
Глава 6. Кинетика и катализ
На основе псевдофазовой кинетической модели можно записать константу скорости реакции второго порядка данной бимолекулярной реакции как
(l + fcAC)(l+fcBC)
где подстрочные индексы m, w, А и В относятся к фазе мицелл, водной фазе и реагентам, Рд и Рв являются коэффициентами распределения реагентов (т. е. Ра — [A]m/[A]W); С —молярная концентрация ПАВ минус критическая концентрация мицеллообразования, V — молярный объем ПАВ (иными словами, факторы CV и 1 — CV отражают объемные доли мицеллярной и водной фаз соответственно), кд и кв — константы связывания реагентов, кд — (Рд — 1)У и кв = (Рв - 1)V.
Эффективность мицеллярного катализа оценивают как соотношение между скоростью реакции в присутствии и в отсутствие мицелл, зависящее от двух факторов согласно следующему выражению:
кжсп _ кт кдкв
Ъ ~ k„v(y/kX+Vkiy 1 ’
Первый множитель, km/kw, отвечает за изменения собственной реакционной способности реагентов в присутствии мицеллярной среды, тогда как второй множитель кдкв/\Г(у/кд + \/^в)2 зависит от того, насколько эффективно реагирующие вещества концентрируются у поверхности мицелл, что в свою очередь является функцией констант связывания реагентов мицеллами (кд и кв) и молярного объема ПАВ (V). Эксперименты с различными реакциями и моделями показали, что именно концентрация реагентов в малом объеме мицеллярной псевдофазы определяет наблюдающийся рост скорости реакции. Этот факт имеет большое аналитическое значение, так как он является основой улучшения чувствительности и (или) селективности кинетических определений, основанных как на некаталитических, так и каталитических реакциях.
6.3.3. Аналитическое применение каталитических реакций
Вне зависимости от механизма реакции уравнения 6.3-9 и 6.3-10 из разд. 6.3.2 в одинаковой степени пригодны для кинетического определения катализаторов. Для этого требуется запись (обычно в автоматическом и полуавтоматическом режиме) кинетических кривых, т. е. изменений концентрации индикаторного вещества во времени. Кривая будет иметь убывающий или возрастающий характер в зависимости от того, является индикаторное вещество исходным соединением или продуктом реакции (см. рис. 6.1-1). Концентрации в данных уравнениях можно заменить любыми экспериментально измеряемыми параметрами, пропорциональными концентрации. Для слежения за реакциями обычно используют физические методы определения. При этом обеспечиваются непрерывные измерения аналитического сигнала (оптической плотности, потенциала, температуры, люминесценции или проводимости).
Методы определения катализаторов в основном идентичны методам, используемым в случае некаталитических реакций. Методы, основанные на из-
6.3. Явление катализа
349
Рис. 6.3-4. Применение метода начальных скоростей (метода тангенсов) [PCrez-BenditoD., Silva М-, Kinetic Methods tn Analytical Chemistry, Chichester, Ellis Horwood, 1988].
мерении индукционного периода, описаны выше при рассмотрении реакций Ландольта. Метод начальных скоростей (метод тангенсов) используют в предположении, что [Р] незначимо по сравнению с [А]. Последняя величина может считаться постоянной в уравнении 6.3-9, что дает возможность упростить его до вида 6.3-11. График данного уравнения, представляющий собой изменение [Р] (через измеряемый параметр) в зависимости от времени будет, следовательно, иметь прямой участок (рис. 6.3-4,а), наклон которото определяется уравнением
«о= ® = tg “ = = fc'|C]o + к'1 (62'21)
Данное выражение представляет собой вариант уравнения 6.3-11. Оно непосредственно связано с концентрацией катализатора и является основой применения данного метода определения. При построении зависимости г>о (tg«) от концентрации катализатора [С]о для нескольких образцов с известной концентрацией катализатора (рис. 6.3-4,б) подучают прямолинейную зависимость, проходящую через начало координат (А) или не проходящую через начало координат (Б) в зависимости соответственно от того, можно пренебречь некаталитической реакцией или нельзя.
Дифференциальный метод фиксированного времени основан на измерении концентрации исходного вещества или продукта в заданное время от начала реакции. Он основан на уравнении 6.3-21, решенном относительно А[Р]
Д [Р] = fc'[C]0At + At (6.3-22)
□ Кинетические методы можно использовать для определения катализаторов.
Поскольку величина At постоянна, график зависимости Д[Р] от [С] о будет прямой линией с тангенсом угла наклона кЧМ и отрезком, отсекаемым на оси
350
Глава 6- Кинетика и катализ
Рис. 6.3*5. Применение метода фиксированного времени [P£rez-BenditoD., Silva М., Kinetic Methods in Analytical Chemistry, Chichester, Ellis Horwood, 1988].
Рис. 6.3-6. Применение метода фиксированной концентрации [P6rez-Bendito, D..
Silva, M, Kinetic Methods in Analytical Chemistry, Chichester, Ellis Horwood, 1988].
ординат, равным k^At. Данный метод иллюстрируется рис. 6.3-5 для случая fcj = 0.
Дифференциальный метод фиксированной концентрации основан на измерении времени, необходимого для того, чтобы в реакционной среде прошли заданные изменения. 'Решая уравнение 6.3-22 относительно 1/АС, получим
1 _ У[С]0 + At Д[Р]
Поскольку величина Д[Р] постоянна, график зависимости l/At от [С]о будет прямой линией с наклоном А//Д[Р] и отрезком, отсекаемым на оси ординат, равным &£/Д[Р]. Данный метод иллюстрируется рис. 6.3-6 для случая к{ = 0.
Если в уравнении 6.3-9 нельзя пренебречь величиной [Р] по сравнению с [А]о, то это уравнение можно проинтегрировать по конечному, но не обязательно короткому, временному интервалу. Это служит основой интегральных методов обработки кинетических данных (методы тангенсов, фиксированного времени и фиксированной концентрации), которые реже встречаются в аналитической практике, чем дифференциальные методы.
Три описанных метода используют для определения ионов металлов и различных соединений (за исключением ферментов), например неорганических
6-3. Явление катализа
351
анионов. Изредка их также применяют для определения реагентов (например, Н2О2), участвующих в индикаторной реакции. В любом случае основным приложением данных методов является высокочувствительное определение катализаторов (пределы обнаружения обычно лежат в диапазоне 10”9-10~7 М, но иногда достигают значений 10”12-10-1°М). Столь высокая чувствительность может быть дополнительно улучшена при использовании активатора, что часто применяется в практике кинетических аналитических определений.
Другой путь увеличения чувствительности существующих каталитических методов определения — использование мицеллярных сред. Мицеллярный (имеющий физическую природу) и химический катализ сочетают в целом ряде методов, чувствительность которых повышается практически на порядок по сравнению с определением в водной среде. Данный эффект объясняется различными механизмами, в частности, концентрированием катализатора на поверхности мицелл при образовании комплекса с одним из реагентов, участвующих в индикаторной реакции [6.3-5].
□ Кинетические методы можно использовать для определения субстрата в ферментативной реакции.
Ферментативные каталитические реакции применяют для определения субстратов чаще, чем для определения самих ферментов (их активности). Помимо высокой чувствительности ферментативного определения субстратов (а также других веществ при их косвенном определении) подходящий фермент обеспечивает высокую селективность определения. Определение субстратов базируется на использовании уравнения 6.3-17, но при этом величина [S] должна быть существенно ниже, чем Км- Тогда уравнение упрощается:
4) = ^“ps] = ^ = vps] (6.3-24)
-ОМ ЛМ
Полученное выражение является уравнением реакции первого порядка (рис. 6.3-2). Из ур. 6.3-24 непосредственно вытекает, что скорость ферментативной реакции пропорциональна концентрации субстрата при данной неизменной концентрации фермента, а значит, можно успешно использовать описанные выше методы начальных скоростей (тангенсов), фиксированного времени и фиксированной концентрации. Для выполнения этого условия на практике отношение [S]/Km не должно превышать 0,2, а еще лучше, чтобы оно было ниже 0,05. При постоянной концентрации субстрата скорость реакции будет пропорциональна концентрации фермента, как показывает уравнение 6.3-24.
Кинетические определения металлов-катализаторов (и других веществ), соединений и ферментов столь многочисленны, что здесь невозможно привести даже их краткий обзор. Мы отсылаем читателя, интересующегося определением металлов-катализаторов, к монографиям, приведенным в разд. 6.1, и нескольким обзорам [6.3-7, 6.3-8]. Литература о ферментативных реакциях весьма многочисленна и хорошо известна.
352
Глава 6. Кинетика и катализ
6.4. ИЗМЕРЕНИЕ АНАЛИТИЧЕСКОГО СИГНАЛА В КИНЕТИЧЕСКИХ И КАТАЛИТИЧЕСКИХ МЕТОДАХ
Быстрое развитие методов, основанных на измерении скоростей реакций (т. е. в кинетической области, см. рис. 6.1-1) в основном является результатом крупных технических достижений в области аналитического приборостроения за последние годы. В частности, это связано с появлением все более мощных компьютеров, позволяющих автоматизировать аналитический процесс. Развитие инструментария позволило решить все проблемы, связанные с традиционными измерениями времени, и привело к улучшению правильности и воспроизводимости аналитических определений. В настоящее время все большее число серийно выпускаемых приборов снабжаются специальными устройствами для проведения кинетических измерений.
Вместо абсолютных значений измеряемого параметра (оптической плотности, флуоресценции или потенциала), в кинетических методах измеряют изменение этого параметра в ходе реакции как функцию времени. Таким образом, статические сигналы, вызванные, к примеру, фоновым поглощением образца, не вносят погрешности. Это является одним из основных преимуществ кинетических методов перед статическими измерениями. В то же время кинетические методы требуют строгого контроля измерений времени и температуры. Преобразованный для обработки сигнал должен иметь максимально возможную точность по шкале времени. Температуру тоже следует контролировать достаточно строго (колебания ее не должны превышать ±0,01—0,1°С), так как она оказывает значимое влияние на скорость реакции (см. разд. 6.2.3).
Выбор техники измерений скорости реакции определяется ее временем полупревращения. Приборное обеспечение для измерения медленных реакций (с временем полупревращения более 10 с) проще, чем применяемое для быстрых реакций (с временем полупревращения менее 10 с). Точно такие же требования предъявляются и к технике смешивания реагентов, а также к измерению и обработке аналитических сигналов.
□ В большинстве случаев методики кинетического определения включают три основные стадии.
Аналитическая методика расчета скоростей реакции включает в себя три стадии: а) подготовка, предварительные измерения, отбор аликвотных частей и смешивание реагирующих компонентов (т. е. исследуемого образца и реагентов); б) детектирование и преобразование сигнала при постоянной температуре, осуществляемое при помощи измерительной аппаратуры (например, фотометра или потенциометра), настроенной таким образом, чтобы следить за изменением какого-либо свойства исходного вещества или продукта в ходе реакции; в) сбор полученных данных для одновременной или последовательной обработки вручную или при помощи компьютера. Полностью автоматизированные системы выполняют все три стадии, а частично автоматизированные системы используются только на стадиях (б) и (в).
На первой стадии смешивание исследуемого образца и реагентов можно проводить в открытой илн закрытой системе в зависимости от того, постоянны внешние экспериментальные условия или нет. Закрытые системы пригод
6.4. Измерение аналитического сигнала 353
ны для медленных реакций (например, каталитических), тогда как открытые системы лучше работают в случае реакций с быстрой кинетикой. Их также можно успешно использовать для медленных реакций при автоматизированном массовом анализе.
6.4.1. Закрытые и открытые системы
Кинетические методы требуют тщательного смешивания исследуемого образца и реагентов при постоянной температуре перед переносом смеси в детектирующую систему для проведения измерений. Это может быть выполнено вручную (что и делается обычно в случае закрытых систем) или автоматически.
Смешивание вручную проводят просто гомогенизацией реагентов в реакционном сосуде, которым может служить фотометрическая кювета или потенциометрическая ячейка. Момент, в который добавлен последний реагент (обычно при помощи шприца), считается началом реакции (t = 0) и, следовательно, должен быть точно известен. Как правило, реагенты перед смешиванием тер-мостатируют, иапример в кюветном отделении, для того, чтобы реакционная смесь достигла необходимой температуры за очень короткое время. В другом варианте исследуемый образец и реагенты можно смешать в мерной колбе и довести объем раствора до метки, после чего аликвоту разбавленной смеси переносят в детектирующую ячейку. Реагенты можно доставлять в измерительную систему автоматически, что увеличивает воспроизводимость измерений. В частности, это относится к управляемому компьютером анализатору Technicon RA 1000 и центрифужным анализаторам, входящим в комплект фотометрических детекторов, широко распространеных в клинических лабораториях.
Автоматическое смешивание реагентов в случае быстрых реакций требует специальных механических устройств, поскольку их необходимо детектировать в открытых системах. Данные устройства могут быть непрерывными проточными анализаторами или статического типа. Метод остановленного потока является наиболее широко применяется при использовании первого типа систем (для быстрых реакций), тогда как <стат»-методы и непрерывного добавления реагента наиболее распространены при работе со статическими устройствами. Они описаны в следующем разделе.
6.4.2. Автоматизированные кинетические методы анализа
□ «Стат»-методы можно использовать для слежения за каталитическими реакциями в открытых системах.
«Стат»-методы являются наиболее часто используемым вариантом прн детектировании каталитических реакций в открытых системах. В этом случае реагент добавляют в реакционный сосуд с такой скоростью, чтобы характеристическое свойство исследуемой реакции оставалось постоянным. В начале процесса добавляют небольшое количество необходимого реагента до тех пор, пока не достигается желаемого значения детектируемого параметра (pH, оптическая плотность). Любое отклонение от этого состояния в результате дальнейшего развития реакции немедленно компенсируется автоматическим добавлением аналитического реагента. При этом скорость достижения постоянного сигна-
12 Аналитическая химия. Том 1
354
Глава 6. Кинетика и катализ
Рис. 6.4-1. Экспериментальная установка, использованная для реализации метода непрерывного добавления реагента [Marquez М., SilvaM., Рёгег-BenditoD., Anal. Chim. Acta, 1990, V. 237, P. 353].
ла пропорциональна концентрации катализатора. Этот подход использовали для кинетического определения иодид-иона по его каталитическому эффекту в реакции между Ce(IV) и As(III) при фотометрическом детектировании ионов церия(ГУ), который служил титрантом, добавляемым с переменной скоростью.
□ Методику непрерывного добавления реагента можно использовать для получения кинетической кривой.
Методика непрерывного добавления реагента с точки зрения техники измерений подобна описанной выше методике добавления реагента порциями. Эта методика основана на непрерывном добавлении реагента в реакционную смесь с постоянной скоростью. При этом получают кинетическую кривую, наклон прямого участка которой прямо пропорционален концентрации анализируемого компонента смеси. За скоростью реакции следят при помощи спектрофотометрии, флуориметрии или даже при помощи измерений хемилюминесценции. На рис. 6.4-1 показана принципиальная схема прибора, реализующего данный метод (6.4-1]. Он состоит из автоматической бюретки (дополнительный блок), фотометрического или флуориметрического детектора, включающего зонд для измерения оптического сигнала, погруженный в исследуемый раствор, и компьютер. Эта методика, позволяющая работать в открытых статических системах, была специально разработана для быстрых некаталитических реакций и представляет собой выгодную альтернативу методике остановленного потока с точки зрения простоты аппаратурного оформления и техники измерений. Метод непрерывного добавления реагента позволяет напрямую, без применения дифференциальйых кинетических методов (см. разд. 6.2-4), анализировать смеси, состоящие из двух или более компонентов. График изменения сигнала во времени для двухкомпонентной смеси обычно имеет два следующих друг за другом линейных участка с различным наклоном. Из этих данных можно определить скорости реакции и, затем, определить концентрации каждого их компонентов.
6.4. Измерение аналитического сигнала
355
Рис. 6.4-2. Блок-схема спектрофотометра (спектрофлу ори метра), работающего в режиме остановленного потока [P6rez-BenditoD., Silva М., Kinetic Methods in Analytical Chemistry, Chichester, Ellis Horwood, 1988].
□ Методику остановленного потока обычно используют для быстрых реакций.
Методика смешивания реагентов в режиме остановленного потока обычно включает смешивание исследуемого раствора и растворов реагентов при помощи двух шприцев, автоматически управляемых пневматическим устройством, в проточной кювете или смесителе, который служит также и ячейкой для наблюдений. Поток затем резко останавливают при помощи третьего шприца, и регистрируют аналитический сигнал как функцию времени. Этот подход обычно требуется для быстрых реакций (с временем полупревращения порядка нескольких миллисекунд или секунд). Производительность данного подхода очень сильно зависит от мертвого времени прибора (определяемого временами смешивания и временами прокачки и останова), которое должно быть меньше, чем время полупревращения реакции примерно на два порядка. На рис. 6.4-2 изображен спектрофотометр (или флуориметр) с устройством, позволяющим работать в режиме Остановленного потока, связанный с автоматическим пробоотборником и управляемым компьютером в режиме реального времени. Это экспериментальное устройство может быть использовано для исследования механизмов реакций, включающих быстрые стадии.
Описанные выше кинетические методы нашли широкое применение при определении большого числа неорганических и органических соединений. Автоматизированные системы особенно важны для аналитического контроля параметров в массовом анализе, включающем как некаталитические, так и каталитические реакции [6.4-2]. В табл. 6.4-1 перечислены некоторые приложения кинетических неферментативных методов в различных аналитически важных областях (например, анализ объектов окружающей среды, клиническая химия и фармацевтика).
12*
Таблица 6.4-1. Некоторые примеры использования кинетических неферментативных методов в анализе объектов окружающей среды, клиническом и фармацевтическом анализе
Объект Определяемое вещество Примечания
Питьевая вода Марганец - Автоматический фотометрический метод
Ванадий Цинк, магний, медь и никель6 - Метод стандартных добавок с флуориметри-ческим детектированием* - Непрерывный проточный анализ (ЗОпроб/ч) Предварительное ионообменное разделение Анализ смесей
Водопроводная Иодид Фосфат, силикат Кобальт* Автоматический фотометрический мониторинг Использование центрифужного микроанализатора Удаление мешающих элементов, Fe и Mg
вода Речная вода Нитрит Гипохлорит" Медь Проточный анализ в режиме остановленного потока (Звбпроб/ч) Проточный анализ’в режиме остановленного потока Сравнение с непламенной ААС
Морская вода Железо Иодат, иодид Карбарил + 1-нафтол* Кальций + магний6 Также использован для морской и водопровод-. ной воды Использование автоматического анализатора Technicon I AutoAnalyzer Проточный анализ в режиме остановленного потока, также Применен для анализа овощей Проточный анализ смесей в режиме останов-
Сточные воды Хлорсодержащие соединения" Кадмий + марганец6 ленного потока Также применен для других типов вод Проточный анализ в режиме остановленного
Воздух Цианид Бензальдегид, формальдегид* Сульфид" потока Сточные воды после процесса электролитического покрытия Использован в производстве синтетических волокон Проточный анализ в режиме остановленного
Почвы Оксиды азота* Карбофуран* потока/мнкродистилляция Определение смесей NO-NOj Проточный анализ в режиме остановленного
Сыворотка крови Медь + железо потока Метод непрерывного добавления реагента
Моча Мочевая кислота*, креатинин*, мочевина* Иодид Проточный анализ в режиме остановленного потока Использование автоматического анализатора
Лекарственные Мочевая кислоте, сульфонамиды* Эпинефрин + норэпинефрин* Ртуть Парацетамол Technicon I AutoAnalyzer Проточный анализ в режиме остановленного потока Проточный анализ в режиме остановленного потока Метод непрерывного добавления реагента Проточный анализ в режиме остановленного
Тиамин* Тз и Т4, теофиллин*, прокаин Хлоропромазин + перфеназин Сульфонамиды*, витамин В12* потока с флуориметрическим детектированием Окисление до тиохрома Проточный анализ й режиме остановленного потока Проточный анализ в режиме остановленного потока Метод непрерывного добавления реагента
Типы реакций: 4 некаталитические; 6 обмена лигандов; в хемилюминесцентные.
6.4. Измерение аналитического сигнала
357
Литература
[6.1-1] Рёгег-BenditoD., Silva М., Kinetic Methods in Analytical Chemistry, Chichester: Elies Horwood, 1988 (Перес-БендитоД, Сильва M., Кинетические методы в аналитической химии. —М.: Мир, 1991).
[6.1-2] MottolaН. A., Kinetic Aspects of Analytical Chemistry, New York: John Wiley & Sons, 1988.
[6.2-1] Buckman N.G., Magee R. X, Hill J. O. Anal Chem. Acta, 1983, V. 153, P. 285.
[6.2-2] Sherman L.R., Trust V.L., Hoang H., Taianta, 1981, V. 28, P. 408.
[6.2-3] Ryan M. A., Ingle J. D. Jr., Anal Chem., 1980, V. 52, P. 2177.
[6.2-4] Pelizzetti E., MentastiE., Anal. Chim., Acta, 1976, V. 85, P. 161.
[6.3-1] IUPAC, Nomenclature of Kinetic Methods of Analysis, Pure & Appl. Chem 1993, V. 65, P. 2291.
[6.3-2] Rodriguez P. A., Pardue H.L., Anal. Chem., 1969, V. 41, P. 1369.
[6.3-3] Guibault G. G., Practical Fluorescence: Theory, Methods and Techniques, Chapter 9, New York: Dekker, 1973.
[6.3-4] LandoHH., Ber. Deut. Chem. Ges., 1986, V. 19, P. 1317.
[6.3-5] Рёгег-BenditoD., RubioS., Trends Anal. Chem. 1993, V. 12, P. 9.
[6.3-6] Berezin I. V., MartinekK., Yatsimirskii A.K., Russ. Chem. Rev. 1973, 42, 787 (БерезинИ. В., МартинекК. ЯцимирскийА. К. Усп. Химии. 1973, Т. 48, С. 787).
[6.3-7] MottolaН. А., Рёгег-BenditoD., MarkH. В. Jr., Anal. Chem., V. 62, Р. 411 R.
[6.3-8] MottolaН. А., Рёгег-BenditaD., Anal. Chem., 1992, V. 65, P. 407R.
[6.4-1] Marquez M., Silva M., Рёгег-BenditaD., Anal. Chim. Acta, 1990, V. 237, P. 353.
[6.4-2] Рёгег-BenditoD., SilvaM., Gomez-Hens A., Trends Anal. Chem., 1989, V- 8, P. 302.
Вопросы и задачи
1. Реакция А + В С + D подчиняется кинетическому уравнению d[A]/di = — fc[A] с константой к = 1,0-103 с-1 при 297К. Каково время полупревращения для этой реакции при 297 К? = 7 КГ4 5 с.)
2. Константы скорости для реакции равны 8,93 10-7 и 8,12 10* й л/моль-с при 650 н 750 К соответственно. Какова энергия активации данной реакции? (Ел = 182,8 кДж/моль)
3. Реакция Co(OH2)4C1J + ОН" Со(ОН2)4СЮН+ + С1- подчиняется уравнению скорости d[Co(OH2)4ClJ]/dt = —fc[Co(OH2)4ClJ]. Предложите вероятный механизм реакции
/ Co(OHa)4ClJ Со(ОНз)4С1+ + С1 Лимитирующая стадия \ Со(ОН2)4С1+ + ОН- Со(ОН2)4С1ОН+ Быстрая стадия
4. Константа скорости для реакции первого порядка удваивается при повышении температуры от 20° С до 30° С. а) Как изменится константа скорости при повышении температуры от 85° С до 95°С? б) Какова энергия активации данной реакции? [а) Повысится в 1,66 раза; б) Ел — 52,38 кДж/моль.]
5. Разложение 1,1-дигидроксиэтана описывается реакцией СНзСН(ОН)2 —» СНзСНО + Н2О. Кинетическое уравнение данной реакции в водном растворе, содержащем 0,1 М уксусной кислоты, имеет вид d[CH3CH(OH)2]/dt = —&[СНзСН(ОН)2], при этом константа скорости первого порядка равна 0,32 с-1.
358 Глава 6. Кинетика и катализ у? .
Какое врем# требуется для того, чтобы 90% 1,1-дигидроксиэтана прорй^гиро-1»алО в слу^е^чогда его исходная концентрация равна 1 • 10“3М, а исходная концентрация уксусной кислоты—0,1 М? (t = 72 с.)
6. Кинетическое уравнение для реакции А+В C+D имеет вид d[A]/dt — —А;[А][В], константа скорости второго порядка равна 2,22 -10-3 л/моль-с. а) Какое время требуется для того, чтобы в растворе, изначально содержащем 1 • 10”4 М вещества А и 0,01 М вещества В, прореагировало 90% А? б) Какова будет концентрация А в этом растворе спустя 48 ч после начала реакции? [a) t = 28,8 ч; б) 2,16 10”6 М.]
7. Реакция второго порядка по отношению к одному из ее составляющих завершилась на 75% за 92 мин в случае, когда исходная концентрация этого компонента составляла 0,240 М. Какое время потребуется для того, чтобы концентрация этого вещества в растворе составила 0,043 М, если исходная концентрация равна 0,148 М? (t = 121 мин.)
8. Разложение оксида азота(У) по реакции 2N2O5 ₽ 2N2O4 + О2 подчиняется кинетическому уравнению первого порядка, имеющему вид dfNsOsj/dt = —fcp^Og]. В некотором эксперименте исследовали раствор N2O5 в тетрахлориде углерода, н было найдено, что его исходная концентрация снижается вдвое за 900 с. Какова величина константы скорости этой реакции в данных экспериментальных условиях? (fc = 7,7 10”4с-1)
9- При исследовании реакции (СНз^СВг^)+Н2О(Ж.) (СНз)3СОН(ад)+Н++Вг~
при 298 К получены следующие данные:
t, 103 с 5 20 60 100 150
[(СНз)зСВг], 10”2 моль/л 3,80 3,08 2,33 1,76 0,502
а) Каков порядок реакции по отношению к (СНз)зСВг?
б) Каково значение константы скорости n-го порядка при 298 К?
в) Какова исходная концентрация (СНз)зСВг?
[а) первый; б) — к = 1,5- Ю^с”1; в) 0,041 М.]
10. Какое значение должно иметь отношение констант скоростей реакции (Аа/Лв) для смеси двух компонентов А и В, чтобы в момент, когда прореагировало 99,9% А, степень завершенности реакции по В составляла 0,1%? = 6900.)
11. Скорость окисления глюкозы (G) кислородом при помощи глюкозооксидазы (GO) при pH 5,6 н 0°С в присутствии фермента с концентрацией 1,17 • 10~Б М описывается уравнением
1 _ к2 + кл 1 1
v к2к< + fcx[G] + fc3[O2]
а) Выведите выражение, связывающее скорость реакции и концентрацию глюкозы при концентрации кислорода 1 • 10~3 М.
б) Если считать, что скорость реакции пропорциональна концентрации фермента, то как выглядит кинетическое уравнение, учитывающее концентрации фер-ыента н глюкозы?
Константы fci, к2, кз н равны 2,1 103моль“1-с"1, 650 с”1, 1,3 10е моль~1-с”1 и 370 с”1, соответственно.
[a) v = 2,1 • 103[G]/(l + 10,5[G]); б) v = 1,8 • 106[G][GO]/(1 + 10,5[G])J
7
МЕТОДЫ ХИМИЧЕСКОГО АНАЛИЗА И ИХ ПРИМЕНЕНИЕ
В этой главе рассматривается применение теоретических основ химического анализа к решению практических проблем, при этом включен весь набор методов анализа от волюмометрии до гравиметрии и от электроанализа до анализа в потоке н химических сенсоров. В настоящее время химики-аналитики решают сложнейшие проблемы в биотехнологии и биологии. Поэтому существенное внимание уделено анализу биохимических систем и применению биосенсоров.
7.1. ТИТРИМЕТРИЯ
Цели изучения
• Понять суть титриметрических методов количественного анализа.
• Уяснить общие требования к реакциям в титриметрических методах— реакция должна протекать быстро, стехиометрично и не осложняться другими компонентами анализируемого раствора. Важным требованием является также легкость установления конечной точки титрования.
в Понять важность требования — изменение стандартной свободной энергии реакции (AG°) должно быть большим, чтобы реакция протекала количественно.
• Понлть, что этим требованиям удовлетворяют не все реакции; в гомогенных системах для титриметрии с успехом можно использовать кислотноосновные, окислительно-восстановительные реакции и некоторые реакции комплексообразования.
7.1.1. Кислотно-основное титрование
Введение
Реакции переноса протона обычно протекают чрезвычайно быстро (хотя известны и медленные кислотно-основные реакции, например, депротонирование CO2(aq)), и поэтому они очень удобны для титриметрии. Немаловажно и то, что в результате реакции образуются продукты известной стехиометрии, и то, что имеется много химических индикаторов и инструментальных способов для обнаружения конечной точки.
360 Глава 7. Методы химического анализа и'их применение
D Большинство реакций переноса протона пригодны’ для титриметрнй. -й к л ’ ? * * - > ?А- - < . -‘т, <
В основе инструментальной индикации конечной точки, как правило, лежит измерение pH, но используют и другие методы, например кондуктометрию. Единственным ограничением является автопротолиз растворителя (воды или другого амфотерного растворителя), в результате которого образуются Н3О+ и ОН-. Нивелирующий эффект воды по отношению к сильным кислотам и сильным основаниям обсуждался в разд. 4.2. Он ограничивает интервал pH для кислотно-основного титрования значениями 0 и 14. Поэтому иногда используют неводные растворители, обладающие другим или более широким интервалом pH. На рис. 4.2-1 приведены рабочие диапазоны pH ряда известных растворителей в сравнении со шкалой pH для воды.
Методы кислотно-основного титрования основаны на измерении pH. С помощью химических индикаторов измерения можно провести с точностью до одной единицы pH. Для более точных измерений применяют электроды, реагирующие на изменение концентрации ионов водорода или, лучше, их активности. Такие электроды известны; они Подробно представлены в разд. 4.4.
Наиболее широко применяется стеклянный электрод, но в растворах с высоким pH или с высокой ионной силой его показания могут отличаться от теоретически предсказанных. Типичный стеклянный электрод обладает идеальным откликом на pH в интервале pH от 1 до 12. Изменение потенциала (мВ) на единицу pH можно рассчитать по уравнению
Е = Е2 - Ei = 2,3°У—(lg[H+j2 - Igp+h)
где R—газовая постоянная (8,3143 Дж-К-1-моль-1); Т—температура (К); F— постоянная Фарадея (966485,0 Кл-моль-1); Е — разность потенциалов, наблюдаемая для растворов 1 и 2 с концентрациями ионов водорода [H+]i и [Н+]2-
Рассчитанные'значения (мВ):
0°С: 54,20 мВ 10°С : 56,18 мВ 20°С : 58,16 мВ 25° С : 59,16 мВ
30° С : 60,14 мВ
40° С : 62,13 мВ
50°С : 64,12 мВ
60°С : 66,11 мВ
Кривая титрования
□ Кривая титрования — удобный способ представления процесса титрования.
Процесс титрования очень удобно представить графически. В дальнейшем будем рассматривать только титрование щелочью, но поскольку подобные результаты получаются и при титровании кислотой, выводы применимы к обоим типам титрования.
Прежде чем перейти к обсуждению получаемых кривых титрования, необходимо ввести ряд допущений и обозначений;
1. Все теоретические положения рассматриваем для случаев, когда титрантом служит NaOH.
7.1. Титриметрия 361
2. Концентрация титранта (NaOH) достаточно высока (> 1,0М) и можно не учитывать изменение объема в процессе титрования.
3. Концентрацию титруемой кислоты обозначим Са. Если кислота (протонированная форма) условно обозначается формулой НВ, сопряженным основанием является однозарядный анион В-.
Св = [НВ] + [В~] (7.М)
□ В кислотно-основном титровании вместо объема титранта можно использовать степень оттитрованное™ (см. ур. 7.1-2).
Величина /в, называемая степенью оттитрованности, определяется соотношением
Число молей добавленного основания .
js — тт , тттэ (7.1-2)
Число молей исходной кислоты НВ
Величина степени протонирования определяется как
а.= И (7-1-3)
поэтому [НВ] = СЕая я [В-] = С,(1 — а,).
Следующие пять уравнений необходимы для расчета пяти неизвестных величин — [Na+], [Н+], [НВ], [В'], [ОН-]:
[Na+]=/,C, (7.1-4)
С, = [НВ] + [В-] (7.1-5)
Ги+1ГТ>—1
Константа кислотности К& == —rrrrTi— (7.1-6)
[HBj
Константа автопротолиза Kw — [Н^ОН-] — 10"14 (7.1-7)
Уравнение электронейтралъности [Na+] + (Н+] = [ОН-] + [В-] (7.1-8)
Рассмотрим три различных случая титрования.
Титрование сильной кислоты (НО) сильным основанием (NaOH)
Результаты расчета для Св = 0,01М приведены в табл. 7.1-1, а их графическое представление в координатах pH —степень оттитрованности дано на рис. 7.1-1. Такая кривая характерна для титрования любой одноосновной сильной кислоты сильным основанием. Вместо степени оттитрованности по оси аб-цисс можно отложить соответствующий объем добавленного основания.
В точке /в = 1,0 (конечная точка титрования) величина dpH/d/B, характеризующая наклон кривой титрования, достигает максимума. Конечную точку титрования легко обнаружить по резкому скачку pH или с помощью любого индикатора с pKj =4-10. Это означает, что выбор индикатора ие затруднителен. Минимальные значения dpH/d/B в начале и конце кривой титрования указывают на то, что растворы сильных кислот и сильных оснований обладают значительной буферной емкостью.
362
Глава 7. Методы химического анализа и их применение
Рис. 7.1-1. Кривая титрования сильной кислоты (НС1) сильным основанием (NaOH) в воде. Левая часть рисунка—логарифмическая диаграмма Хэгга.
Таблица 7.1-1. Концентрации ионов водорода и гидроксида при различных степенях оттитрованности
/. [Н+1 [ОН“1 рн
0,0 10“3 10”13 2,00
0,1 9,0-Ю-3 1,1-10*13 2,05
0,5 5,0-Ю-3 2,0-10”13 2,30
0,9 10-3 10”11 3,00
0,99 ю-4 Ю-w 4,00
0,999 10-Е 10“в 5,00
1,0 10-7 10”’ 7,00
1,001 10”9 Ю-5 9,00
1,01 10“10 10”4 10,00
1,1 10-11 10”3 11,00
1,5 2,0-10“13 5,0-Ю-3 11,70
1,9 1,1 иг13 9,0-Ю-3 11,95
2,0 10“13 10-2 12,00
□ Выбрать индикатор для титрования сильной кислоты сильным основанием несложно.
Титрование слабой кислоты (СН3СООН) сильным основанием (NaOH)
Расчетные данные для Са — 0,01 М приведены в табл. 7.1-2. Для облегчения расчетов введены некоторые упрощения. Константа кислотности
== 1,74 • 10"Б (рКа = 4,76).
Из кривой титрования (рис. 7.1-2) видно, что dpH/d/8 достигает минимальной и максимальной величины в точках /, = 0,5 и /, = 1,0 соответственно. При fa —0,5 система обладает максимальной буферной емкостью, так как [НВ] — [В~] и поэтому наблюдается минимальное изменение pH при добавлении титранта. Точку минимального наклона можно использовать для нахождения рКв неизвестной слабой кислоты, так как в этой точке pH = рКв. Конечную точку можно обнаружить по скачку pH, но он значительно меньше, чем при титровании сильной кислоты. Для обнаружения конечной точки тит-
7.1. Титриметрии
363
Таблица 7.1-2. Титрование слабой кислоты (уксусная кислота, рК& = 4,76) сильным основанием (NaOH) . .
/. [Na+ [Н+| (В-| (НВ] рн Уроненное соотношение ,
0,0 0.00 4,20-10“* 4,20 10”* 9,58 - 10”’ 3,38 (Н+] = [В“)
од 1,0-10“’ 1,35-10”* 1,14 • 10“3 8,86-10“’ 3,87 |Na+) + [H-4 = [£>')
0,2 2, ОНГ3 6,76 -10“6 2,06-10”’ 7,94-10“’ 4,17 [Na+] + [Н+] = [В”)
0,3 3,0- 10“3 4,00 ЦТ5 3,04-10“’ 6,96-10“’ 4,40 [N»4 + [н+] =-[В“]
0,4 4,0-10“’ 2,58-10”® 4,03-10“’ 5,97-10“’ 4,59 [Na+] + [Н+] = [В“]
0,5 5,0-10“’ 1,74-10”® 5,02-10”’ 4,98-10“’ 4,76 [Na+J + [Н+] = [В”]
0,6 6,0-10”’ 1,15-10”® 6,01-10“’ 3,99-10”’ 4,94 [Na+J + [H+l = [В-]
0,7 7,0 -10“’ 7,14 - 10”® 7,01 10“’ 2,99-10“’ 5,13 [Na+J + [Н+] = [В-]
0,8 8,0-10“’ 5,40 10“® 8,00-10“’ 2,00-10”’ 5,26 [Na+J = [B”J
0,9 9,0-10"’ 1,93-10”® 9,00-10”’ 1,00-10“’ 5,72 [Na+J = [B“]
0,99 9,9-10“’ 1,76-10”’ 9,90-10”’ 1,00-10“* 6,76 [Na+] = [B“]
1,0 1,0-КГ8 4,16-10”® 1,00-10“’ 2,40 10“6 8,38 [Na+] = [OH“]
1,1 1,1-10-’ 1,00 10“11 1,00-10“’ 5,76-10“® 11,00 [Na+J - [B”J = [OH-]
1,5 1,5-10“’ 2,00-10-” 1,00 10”’ 1,15 • 10"® 11,70 [Na+] - [B“] = [OH“]
1,9 1,9-10“’ 1,11-10“” 1,00-10”2 6,46-10“10 11,95 [Na+J - [B“] = [OH“J
2,0 2,0-10“’ 1,00-10“” 1,00-10”’ 5,76-10”® 12,00 [Na+] — [B“] = [OH”J
Рис. 7.1-2. Кривая титрования слабой кислоты (СНзСООН) сильным основанием (NaOH) в воде.
рования пригодно гораздо меньшее число индикаторов: только индикаторы с рК, от 7 др 10 (см. табл. 4.2-2).
□ Для титрования слабых кислот сильными основаниями пригодны индикаторы с рК; в интервале 7-10.
Титрование слабого основания (NH)3 сильной кислотой (НО)
Кривая титрования NH3 (С = 0,1М) раствором НС1 представлена на рис. 7.1-3. Видно, что в конечной точке наклон кривой максимален. Скачок pH меньше, чем в случае титрования сильной кислоты сильным основанием и сравним со скачком на кривой титрования слабой кислоты, но смещен к более низким значениям pH.
Как видно из кривой титрования на рис. 7.1-4, сильная кислота в присутствии слабой может быть оттитрована, если сила кислот заметно различается.
364
Глава 7. Методы химического анализа и их применение
Рис. 7.1-3. Кривая титрования слабого основания (NH3) сильной кислотой (НС1) в воде.
Рис. 7.1-4. Кривая титрования смеси 0,1 М НС1 и 0,1 М Н3РО4 раствором NaOH в
воде.
В качестве примера взята смесь 0,1 М НС1 и 0,1 М Н3РО4. Поскольку в водной среде протон, отдаваемый НС1, и первый протон Н3РО4 (pKai = 2) неразличимы, первая точка эквивалентности Ci соответствует суммарной концентрации НС1 и Н3РО4. Вторая точка эквивалентности С2 соответствует титрованию второго протона Н3РО4 (рКаз = 7), поэтому по разности объемов титранта С2 и Ci можно найти концентрацию фосфорной кислоты. По этим же результатам можно рассчитать и концентрацию НС1. Точки Bi, В2 и Вз~точки с максимальной буферной емкостью.
Сильным основанием можно оттитровать даже чрезвычайно слабые кислоты, если сделать их более сильными в результате химической реакции. Ниже обсуждаются различные способы повышения силы слабых кислот, используемые для их последующего косвенного определения.
Значения констант кислотности (основности) для наиболее известных кислот (оснований), определяющие возможность их кислотно-основного тирова-ния, приведены в табл. 7.1-3 и в Приложении 3 (табл. 1; см. т. 2). Сведения о наиболее важных кислотно-основных индикаторах также можно найти в Приложении 3 (табл. 1; см. т. 2).
Таблица 7.1-3. Сравнительная сила кислот и оснований
рк. Кислоты Основания ~рКъ
Очень сильные Чрезвычайно слабые
< —5 СЮ<Н Ei H++C1O7 ~23
< -5 IH у» h++i- ~23
~-3,5 ВгН у» H+ + Вг- ~20
~-2 С1Н т-» Н++СГ *-17.
SO4H2 Ei Н+ + SO4H- ~ 17
-1,74 ОН+ Н++ОН2 15,74
-1,32 NO3H у* H+ + NO£ 15,32
0 С1О3Н Ei Н++С1О- 14
0 ВгОзН pt Н+ + ВгОз 14
Сильные Очень слабые
1,42 С2О4Н2 у» Н+ + С2О4Н- 12,58
1,64 Ю3Н Ei Н++1О, 12,36
1,92 SO3 + ОН2 H++SO3H- 12,03
1,92 SO4H- Ei Н+ +SO2 12,08
1,96 РО4Н2 Ei Н+ + РО4Н2 12,04
2 S2O3H- y> H++S2O^- 12
2,22 Fe(OH,)J+ Ei H++Rs(OH)(OH2)|+ 11,78
2,32 AsO4H4 у» н+ + А»О4н5+ 11,68
3,14 FH Ei н+ +F+ 10,86
3,35 NO3H r-r H++NO3 10,65
3,7 нсоон Ei н+ + нсоо- 10,3
4 NCSH ri H++NCS- 10
4,21 С2О4Н- Ei H++C2oJ_ 9,79
Слабые Слабые
4,74 СН3СООН Ei Н+ + CH3CO2 9,25
4,85 А1(ОН2)5+ Ei н+ + А1(ОН)(ОН2)£+ 9,15
6,52 C62 + 0H2 Ei н+ + СОзН- 7,48
6,92 SH3 Ei н+ + SH2 7,03
7 ASO4H3 Ei H+ + AsO4H2- 7
7 SO3H- Ei H++SO2- 7
7,12 PO.HJ Ei H+ + FO.H2- 6,38
7,25 СЮН Ei н+ + СЮ" 6,75
8,69 BrOH Ei н+ + НгСГ 5,31
9,24 во3н2 у» Н+ + ВО3Н- 4,76
9,22 ASO3H3 Ei н+ + АаОзН, 4,78
9,25 nhJ Ei Н+ + NH3 4,75
Очень слабые Сильные
9,40 NCH Ei H++NC- 4,60
9,61 Zn(OH2)2+ z± H+ +Zn(OH)(OH2)£ 4,39
10 S1O4H4 Ei н+ + S1O4H3 4
10,6 IOH si н+ + Ю- 3,4
10,4 CO3H- T~> н+ + СО," 3,6
11,62 O2H2 Ei h+ + o,h- 3,38
12 S1O4HJ Ei н+ +S.O.H5- 2
12,32 PO4H2- Ei H+ + POJ- 1,58
13 AsO.H2- Ei н+ + AsO2- 1
13 SH“ Ei н+ + S2- 1
13,52 ASO3H7 Ei H++AsO3H3- 0,48
Чрезвычайно слабые Очень сильные
15,74 OH2 Ei н+ + ОН- -1,74
~23 NH3 Г» н+ + NH7 г» -9
~24 OH- r~» н+ + о2- ~ -10
~34 CH4 Г-* н+ + сн3 ~ -20
38,6 H2 zi н+ +Н- -24,6
366
Глава 7. Методы химического анализа и их применение
□ Возможно последовательное титрование сильной и слабой кислот (сильного и слабого основания) в смеси. Даже и для таких сложных систем на ©сновании диаграммы Хэгга можно построить кривую титрования (см. рис. 7.1-4).
Способы повышения силы слабых кислот
В водных растворах очень слабые кислоты определять прямым титрованием основанием нельзя. Однако есть ряд химических реакций, позволяющих заменить- слабую кислоту эквивалентным количеством более сильной кислоты, пригодной для прямого титрования основанием.
а) Осаждение аниона слабой кислоты. Для ряда анионов осадителем может служить ион бария.
2НВ + Ва2+ -» {ВаВ2} + 2Н+
Здесь {ВаВг} —осадок соли бария, например,
2HPOJ+ + ЗВаС12 — {Ваз(РО4)2} + 2Н+ + 6СГ
б) Окисление слабой кислоты до кислоты с более высоким содержанием кислорода.
HNO2 + ”О” — HNOg, H2SO3 + ”0” H2SO4
в) Комплексообразование. Примером может служить реакция образования эфира с многоатомными спиртами
НзВОз + 2С3Н5(ОН)3 -+ ВО4(С3Н5ОН)2 +Н30+ + 2Н2О
Аналогичный прием можно использовать для титрования NH^ (слабая кислота) сильным основанием—добавляют формальдегид, превращающий NH^ в гексаметилентетрамин (уротропин) с выделением свободных ионов водорода:
NHj + Н2О NH3 + Н3О+ SS C6Hi2N<, +Н3О+
Уротропин
7.1.2. Комплексометрическое титрование
Введение
В основе методов комплексометрического титрования лежит стехиометрическая реакция нона металла с водорастворимыми бидентатными или полиден-татными лигандами, называемыми комплексонами.
Реакция иона металла с комплексоном должна протекать быстро и количественно, образующийся продукт должен быть растворим в воде. Реакция должна протекать практически нацело, если реагирующие вещества присутствуют в стехиометрических количествах. Первое требование относительно скорости реакции обязательно для всех видов титрования. Последнее требование означает, что константа устойчивости образующегося комплекса должна быть высока.
□ Титрантами могут служить комплексоны, быстро реагирующие с определяемым ионом с образованием водорастворимого устойчивого комплекса.
7.1. Титримстрия
367
Большинство практически важных лигандов являются полидентатными хелатообразующими полиаминами и аминополикарбоновыми кислотами. Комплексы с не образующими хелатов (мондентатными) лигандами, как правило, имеют недостаточно высокие константы устойчивости. Это можно видеть на примере комплексов одного и того же иона (Си2+) с монодентатными'(аммйак)' и тетрадентатными (триаминотриэтиламин) лигандами.
а) Аммиачные комплексы меди (см. также табл. 7.1-4).
Таблица 7.1-4. Константы устойчивости комплексов меди и цинка с некоторыми полиаминами (1g/3)*
Лиганд Формула Zn Си
Аммиак NH3 2,3; 2,3 2,4; 2,1 4,1; 3,5 2,9; 2,1
Этилендиамин H2NCH2CH2NH2 5,9; 5,2 10,7; 9,3
Диэтилентриамин h2nch2ch2nhch3ch2nh2 8,9; 5,5 16,0; 5,3
1,2,3-Триаминопропан H2NCH2CHNH2CH2NH2 6,8; 4,3 11,0; 9,0
Триэтилентетраамин h2nch2ch2nhch2ch3nh-
(Триен) ch2ch2nh2
Д 01 > Д '-Триамйно-триэтиламин N(CH2CH2NH2)3 14,7 18,8
а Экспериментальные условия: 20°С, концентрация иона 0,1 М. (См. G- Schwarzenbach. Analyst. 80, 713 (1955).]
В результате взаимодействия с монодентатными лигандами образуются четыре относительно неустойчивых комплекса.
Cu2+ + NH3 Cu(NH3)2+
Cu(NH3)2+ + NH3 й Cu(NH3)|+
Cu(NH3)2+ + NH3 Cu(NH3)|+
Cu(NH3)3 + NH3 Cu(NH3)2+
lgKi = 4,l lgK2 = 3,5 lgK3 = 2,9 lgb-4 = 2,1
Cu2+
+ 4NH3 Cu(NH3)J+ lg/3d = 12,6
□ Комплексы с монодентатными лигандами в отличие от комплексов с полидентатными лигандами для титриметрии не удобны, так как недостаточно устойчивы1 (табл. 7.1-4 и рис. 7.1-5).
6) Комплекс меди с триаминотриэтиламином (см. также табл. 7.1-4)
Комплекс с тетрадентатным лигандом образуется в одну стадию. Вследствие хелатного эффекта он имеет гораздо более высокую константу устойчивости, поэтому скачок на кривой титрования выражен более четко. Хелатный эффект обусловлен повышением энтропии системы. При образовании хелата один полидентатный лиганд вытесняет из исходного аквакомплекса (например, Си24" координирует четыре молекулы воды) несколько молекул воды, сле-
1 В то же время известны очень устойчивые комплексы с монодентантными лигандами, но они образуются не в одну стадию, что и делает их неудобными для титриметрии. —- Прим,
персе.
368
Глава 7. Методы химического анализа и их применение
Рис. 7.1-5. Кривые титрования цинка полиаминами. Титранты: аммиак (7), этилендиамин (5), диэтилентриамин (3), триэтилентетрамин (триен) (4), триаминотриэтиламин (5).
довательно, число несвязанных частиц увеличивается и энтропия возрастает (рис. 7.1-5).
Cu2+ + N(CH2CH2Nh2)3 CuN(CH2CH2NH2)2+ , lg 0 = 18,8
Комплексообразующие реагенты
В продаже имеется ряд комплексообразующих реагентов, образующих водорастворимые хелаты. Их применяют главным образом для определения жесткости воды.
а) Нитрилотриуксусная кислота, НТА (комплексон I)
Мг = 191,14
Тетраде нтатный лиганд Белый порошок, легко растворимый в щелочных растворах
7.1. Титриметрия
369
Таблица 7.1-5. Константы устойчивости комплексов с некоторыми аминокарбоновыми кислотами
Ион металла lg£
HTA ЭДТА ДЦТА
Ва2+ 4,8 7,8 8,0
Са2+ 6,4 10,7 12,5
Ре3+ 15,9 25,1 29,3
La3+ 10,4 15,4 16,3
Mg2+ 5,4 8,7 10,3
Zn2+ 10,5 16,5 18,7
б) Этилендиаминтетрауксусная кислота, ЭДТА (комплексон П)
Мг = 292.25 Гексадентатный лиганд Белый порошок, плохо растворимый в воде и достаточно хорошо в щелочных растворах
в) Двунатриевая соль этилендиаминтетрауксусной кислоты, дигидрат (комплексон III)
Мг = 372,24
Гексадентатный лиганд Белый порошок, легко растворимый в воде и слабокислых растворах
Наиболее часто используемый титрант — комплексон III. Это гексадентатный лиганд.
г) транс-1,2-Диаминоциклогексан-Н^,^,Н"-тетрауксусяая кислота, моногидрат, ДЦТА.
он
Мг= 364,36
Гексадентатный лиганд
Белый порошок. Образует более устойчивые комплексы, чем комплексон II (см. табл. 7.1-5)
зге-
Гпава 7. Методы химического анализа и их применение
Рис. 7.1-6. Октаэдрическая структура хелата, образованного гексадентатным лигандом ЭДТА с двухзарядным ионом металла. Донорными являются два атома азота аминогрупп и четыре атома кислорода карбоксильных групп.
ЭДТА — часто используемый комплексон. На рис. 7.1-6 показана трехмерная структура хелата, указаны шесть донорных атомов лиганда, координируемых центральным атомом.
Равновесия, влияющие на реакцию комплексонометрического титрования
Согласно приведенной ниже схеме, реакция, лежащая в основе комплексонометрического титрования1, может осложняться конкурирующими реакциями:
[MY]-*
у ОН" и м(он);"у Образование гидроксокомплексов
Реакции с посторонними лигандами
уН+
II
Кислотно-основные реакции
L
И
MLY""X
Образование смешанолигандных комплексов
В отсутствие конкурирующих реакций (идеализированный случай) равновесие реакции можно описать реальной константой
/3 =
[MY] [M][Y]
При протекании конкурирующих реакций (одной или нескольких одновременно) необходимо ввести поправки на отклонения от идеальности с помощью
1 Титрование, при котором в качестве титрантов используют полиаминополикарбоновые кислоты (комплексоны), называют комплексонометрическим. — Прим. ред.
7.1. Титриметрии
371
Рис. 7.1-7. Зависимость условной константы устойчивости ff комплекса Zn—ЭДТА от pH при Cnh3 = 0,1М.
^-коэффициентов (а изменяется от 1 до 0) и пользоваться условной константой устойчивости /?':
отсюда р' ~ рамау.
_[М] _ [Y]
См И ~[MY]
р=-...IMYI
Ом См Оу Су
□ Наряду с реакцией комплексонометрического титрования в растворе одновременно протекает множество конкурирующих реакций, существенно влияющих на устойчивость комплекса (равновесие описывается условной константой, ft', пример на рис. 7.1-7).
□ Влияние конкурирующих реакций отражает условная константа.
Пример-, титрование Zn2+ раствором комплексона Ш (YH^-) в растворе NH3
Реакция титрования: Zn2+ 4- YH2- —» ZnY2- + 2Н+
Реакция комплексообразования: Zn2+ 4-Y4” ZnY2-, j0znY Конкурирующие реакции:
а) Кислотно-основные с участием ЭДТА
Y4- + Н+ YH3- рКм = 10,3
YH3" 4- Н+ YH2- рКа,3 = 6,2
Глава 7. Методы химического анализа и их применение
зп
б) Комплексообразование с участием ЫНз
Zn2+ +NH3 Zn(NH3)2+
Zri(NH3)2++NH3 Zn(NH3)i+
= /fenY«Zn’+QY<-
в) Образование гидроксокомплексов (pH> 10)
Zn2++ 4OH“ Zn(OH)2-
Константы устойчивости ряда комплексов приведены в табл. 7.1-5.
Установление конечной точки титрования
Копейную точку в комплексонометрическом титровании можно установить
а) используя прямую регистрацию кривой -титрования электрохимическими методами (разд. 7-3). Главными методами является амперометрия в случае, если потенциалы полуволн «свободного» иона металла и хелата различаются, или потенциометрия, если имеется ион-селектнвный электрод. За точку эквивалентности принимают точку перегиба на кривой титрования (рис. 7-1-5);
б) с помощью металлохромных индикаторов. Это комплексообразующие реагенты с различно окрашенными свободной и связанной в индикаторный комплекс формами. Комплекс определяемого иона с индикатором должен быть значительно менее устойчивым, чем комплекс иона металла с титрантом.
До начала титрования индикатор реагирует с ионом металла и образует окрашенный металл-индикаторный комплекс. В процессе титрования образуется более прочный комплекс с титрантом, поэтому вблизи точки эквивалентности появляется иначе окрашенная форма свободного индикатора. При сопоставимой интенсивности окраски свободного и закомплексованного индикатора точку эквивалентности можно обнаружить по высвобождению 50% индикатора.
Известны металлохроыные индикаторы для различных ионов. Рассмотрим некоторые из них.
Эриохромовый чёрный Т (для Mg, Pb, Cd, Мп, Zn)
Это трехосновная кислота, используемая обычно в виде однозамещенной натриевой соли.
7.1. Титриметрия
373
Индикаторная реакция: НЭХЧТ2 +Mg2+ Mg(3X4T)_ +Н+
голу6ой красный
ЭХЧТ чувствителен к окислителям, водные растворы стабилизируют аскорбиновой кислотой, а ионы Fe3+ восстанавливают гидроксиламином. Выпускаются «индикаторные буферные таблетки», представляющие смесь индикатора ЭХЧТ, красителя (переход окраски красный/серый/зеленый) н буфера для pH 10.
Полезными при комплексонометрическом титровании являются также следующие индикаторы: Калгон (для Са, Cd, Mg, Мп, Zn)
он но
HOjS-
н тайрон (для Fe3+) °»8'
Применение комплексонометрического титрования
В большинстве случаев объектом анализа является смесь ионов. Селективность определения в таких случаях обеспечивают специальными приемами (см. * также разд. 4.3). В конкретных случаях можно использовать
— выбор титранта;
— выбор индикатора;
— регулирование pH;
— маскирование мешающих нонов, образующих более устойчивые хелаты с титрантом;
- предварительное осаждение мешающих катионов.
Способы титрования
а) Прямое титрование: ЭДТА образует хелаты со многими катионами, но если константы устойчивости заметно различаются, возможно последовательное титрование. Так, Fe3+ и Са2+ можно определить при совместном присутствии, оттитровав сначала Fe3+ при pH 3, а затем Са2+ при pH 10.
6) Обратное титрование: при отсутствии подходящего индикатора к анализируемому раствору прибавляют избыток стандартного раствора ЭДТА, затем остаток его титруют раствором Mg2+ в присутствии эрнохромового черного Т.
в) Заместительное титрование: к анализируемому раствору, например Fe^, прибавляют Избыток М§ЭДТА2~ и выделившийся Mg2+ титруют ЭДТА.
г) Косвенное титрование применяют для определения анионов, например сульфат-нона. Для этого сульфат-ион осаждают избытком стандартного раствора Ва2+ и остаток Ва2+ титруют ЭДТА.
’374 Глава 7. Методы химического анализа и их применение
Пример
а) Определение жесткости воды
Жесткость воды характеризуют суммарным содержанием Са2+ и Mg2+ (мМоль/л). и определяют, титруя оба иона одновременно при pH 10 раствором ЭДТА в присутствии эриохромового черного Т. В литературе описаны маскирующие агенты для главных мешающих ионов: Fe3+ и Мп24 (гидроксиламин), А13+ (триэтаноламин), Си2+ (цианид).
б) Раздельное определение Са2+ и Mg2+
Устанавливают pH 12 с помощью NaOH и после осаждения Mg(OH)a титруют ионы Са2+ раствором ЭДТА в присутствии >алгона. Для устранения погрешности за счет-соосаждения Са2+ осадок Mg(OH)2 растворяют НС! и вновь осаждают Mg(OH)a с помощью NaOH. Точное содержание Mg2+ находят по разности результатов определения жесткости воды и Са2+.
□ Жесткость веды характеризуется суммарным содержанием кальция и магния.
7.1.3. Электрохимическое титрование
Электрохимическое титрование — классические тетраметрические методы, в которых конечную точку определяют электрохимически. Различают потенциометрическое, амперометрическое и кондуктометрическое титрование (см. ниже и, более подробно, в разд. 7.3).
Потенциометрическое титрование
Потенциометрию используют для наблюдения за процессом титрования, т. е. для установления конечной точки титрования, если имеется индикаторный электрод по крайней мере для одного из веществ, участвующих в химической реакцииЦдля титранта, титруемого вещества или для обоих). Точка перегиба на кривой титрования (т. е. с максимальным значением ДЕ’/ДГд) совпадает с точкой конца титрования, если кривая титрования симметрична. Для любой реакции со стехиометрией, отличной от 1 : 1, кривая титрования асимметрична. Погрешность титрования из-за асимметрии кривой титрования можно частично устранить, если концентрацию титранта определяют точно в тех же экспериментальных условиях, что и концентрацию определяемого вещества.
□ Электрохимическое детектирование позволяет зарегистрировать не только конечные точки титрования, но и полные кривые титрованил.
На рис. 7.1-8 показаны типичные кривые осадительного титрования, а также различные способы нахождения конечной точки титрования (непосредственно по интегральной кривой, по первой и второй производной). Необходимо помнить, что погрешность определения конечной точки влияет на погрешность потенциометрического титрования.
Потенциометрическое определение конечной точки можно также использовать в различных автоматизированных методах титрования. Но при любом способе титрования индикаторный электрод в потенциометрии служит только
7.1. Титриметрия
375
Рис. 7.1-8. Типичные кривые потенциометрического титрования хлорида ионами серебра.
для установления конечной точки титрования, а концентрацию определяемого компонента находят на основании стехиометрии химической реакции.
□ Потенциометрически со стеклянным электродом можно -зарегистрировать кривые кислотно-основного титрования, а также кривые титрования смесей (как, например, на рис. 7.1-4).
Амперометрическое титрование
Амперометрия как способ детектирования широко используется в различных проточных методах, таких, как проточно-инжекцноииые и хроматографические методы, а также в классической титриметрии и кулонометрическом титровании. В последнем из них измерение тока используют для нахождения конечной точки титрования.
□ Амперометрически можно регистрировать, например, изменение концентрации иона металла в процессе осаждения или определять остаточное содержание воды в «безводных» органических растворителях по Карлу Фишеру.
Материал рабочего электрода важнее в непрерывных проточных методах, чем в случае титрования, главным образом, из-за необходимости механической и долговременной функциональной стабильности. Разнообразные уголь
зп
Глава 7. Методы химического анализа и их применение
ные электроды, такие, как стеклоуглеродный электрод или электрод из угольной пасты (смесь графитового порошка н парафина или силиконового масла), особенно распространены в ВЭЖХ и прЬточно-инжекционном анализе. В амперометрическом титровании в качестве индикаторного (поляризуемого) электрода используют ртутный капающий электрод, платиновый или графитовый микроэлектрод. Противоэлектродом служит либо электрод сравнения (т. е. неполяризуемый электрод, потенциал которого остается постоянным при относительно небольших токах), такой, как насыщенный каломельный электрод (НКЭ),либо какой-то другой поляризуемый электрод. Варианты амперометрического титрования:
— титрование с одним поляризуемым электродом;
— титрование с двумя поляризуемыми электродами (биамперометрическое титрование).
Амперометрическое титрование с одним поляризуемым электродом
Метод основан на измерении тока в процессе титрования в присутствии фонового электролита при заранее выбранном потенциале индикаторного электрода в перемешиваемом растворе. Потенциал выбирают таким образом, чтобы было возможно электрохимическое окисление или восстановление либо определяемого вещества, либо титранта, либо того н другого1. Кривая амперометрического титрования представляет собой зависимость предельного тока от объема титранта или степени оттитрованности. Вид кривой титрования зависит от того, какой из участников реакции титрования является электроактивиым и от обратимости окислительно-восстановительной пары, участвующей в реакции титрования.
В качестве примера рассмотрим следующую реакцию титрования:
РЬ2+ + CrOj РЬСгОд (7.1-9)
Потенциал индикаторного ртутного капающего электрода поддерживают постоянным относительно электрода «равнения (НКЭ), и измеряют ток в зависимости от объема добавленного титранта. Если титруют при потенциале О В, ток остается равным нулю вплоть до конечной точки титрования, поскольку при этом потенциале восстанавливается только CrOl". После точки эквивалентности ток растет, что обусловлено избытком электроактнвного титранта. Конечную точку титрования находят экстраполяцией двух линейных участков кривой титрования* Точки на кривой вблизи конечной точки титрования не рассматривают, поскольку растворимость образовавшегося осадка искажает кривую титрования.
Если на индикаторном электроде установить потенциал —1 В, соответствующий предельному току восстановления и ионов свинца, н титранта, получается V-образная кривая титрования.
В качестве индикаторного при амперометрическом титровании можно также использовать вращающийся платиновый электрод. Более высокое значение предельного тока вследствие конвективной диффузии и низкого остаточного
1 Обычно это потенциал на участке предельного тока электроактнвного участника реакций титрования.’—Прим, перев.
7.1. Титриметрия
377
тока на электроде с постоянной площадью поверхности существенно понижают предел обнаружения. Так, амперометрическое титрование можно использовать для количественного определения веществ в интервале концентраций от 10-5 до 10“® М. Но необходимо помнить, что низкое перенапряжение водорода на платиновом электроде может оказаться серьезной проблемой при использовании его в качестве катода в кислых растворах.
Относительное стандартное отклонение в амперометрическом титровании обычно менее 0,024),03. Селективность прямого амперометрического титрования можно улучшить соответствующим подбором реакции Титрования. Так, соединения с одинаковыми Е-цъ можно оттитровать в присутствии друг друга подходящими титрантами.
Амперометрическое титрование с двумя поляризуемыми электродами
Амперометрическое титрование с использованием двух идентичных поляризуемых электродов'одинакового размера называют биамперометрическим титрованием. Ток измеряют как функцию объема Титранта при наложении на электроды относительно небольшой разности потенциалов (10-100мВ). Один электрод выступает в качестве анода, другой функционирует как катод. Форма кривой титрования сильно зависит от обратимости электродной реакции с участием определяемого вещества и титранта. Биамперометрическое титрование используют обычно для определения конечной точки окислительно-восстановительного титрования.
Рассмотрим, например, титрование 1г раствором SaOj- при небольшой разности потенциалов Д.Е, приложенной к двум платиновым электродам. Реакция титрования:
I2 + 2S2Of~ 2Г + S40j- (7.1-10)
Для качественной интерпретации кривых титрования необходимо рассмотреть окислительно-восстановительные пары 12/1~ н ^O^/S^g . Ток в электрохимической ячейке протекает, если на обоих электродах идет электродная реакция. Если приложенный потенциал мал, то ток протекает, если оба компонента обратимой окислительно-восстановительной системы присутствуют в растворе. Величина тока определяется компонентом с меньшей концентрацией.
Система 12/1~ обратима иа платиновом электроде, а система SaO^/S^g-необратима. Поэтому при небольшом приложенном потенциале протекает реакция
12 + 2е“—»2Г" (7-1-11)
а реакция
S2O|- + 2е- -> S4O^“ (7.1-12)
не протекает.
Следовательно, в начале титрования ток отсутствует, поскольку в титруемом растворе иет ионов I-; можно измерить только остаточный ток.
По мере титрования в растворе в результате окислительно-восстановительной реакции образуется I-, и протекает фарадеевский ток, обусловленный электродными реакциями
37Й
Глава 7. Методы химического анализа и их применение
на катоде: 1,1
Ь + 2е~-»2Г (7.1-13)
и на аноде:
2Г-»12 + 2е" (7-1-14)
Ток определяется компонентом окислительно-восстановительной пары с меньшей концентрацией, поэтому он достигает максимума при степени оттитро-
ваниости 50%, когда концентрации^ и 1“ равны, а затем уменьшается из-за расходования 12 в реакции титрования. Прн 100% степени оттитрованное™ и далее в растворе нет 12 и наблюдается только остаточный ток. Кривые такого типа называют кривыми титрования с ’’мертвой’1 конечной точкой, поскольку ток падает практически до нуля при достижении конечной точки титрования и далее не изменяется.
Форму кривой титрования можно предсказать, рассматривая кривые ток — потенциал для титранта н определяемого вещества н величину A-S.
Титрование воды по Карлу Фишеру
Методика пригодна д ля определения воды в очищенных растворителях, а также кристаллизационной воды. Реагент Карла Фишера, содержащий, например, иод, диоксид серы, пиридин в молярном соотношении 1 : 3 : 10 в метаноле, можно использовать для прямого титрования воды в любом растворителе, не реагирующем с диоксидом серы и/или иодом. Растворители, содержащие альдегиды и кетоны, титровать нельзя, поскольку онн связывают диоксид серы.
□ Метод Карла Фишера используют для определения воды.
Методика основана на реакции диоксида серы с иодом в присутствии воды:
SO, + ь + 2Н,О Й SO1" + 21- + 4Н+ (7.1-15)
В присутствии пиридина равновесие может сместиться вправо:
+ Н2О-----*•
О"2 * @"80, * @N
(7.1-16)
Для стандартизации реагента Карла Фишера используют метанольный раствор соединения, содержащего известное количество воды, например, СНзСООН • ЗН2О.
В процессе титрования измеряют ток как функцию известного объема стандартизированного реагента Карла Фишера, т. е. г — V (мл). Конец титрования устанавливают методом титрования до мертвой конечной точки.
7.1. Титриметрия
379
Кондуктометрическое титрование
Специфичность кондуктометрии повышается при использовании селективного реагента. На этом основано кондуктометрическое титрование, в котором измеряют изменение электропроводности раствора как функцию количества добавленного титранта Vr.
Количественное определение основано на определении объема титранта, затраченного на достижение момента стехиометричности химической реакции. Конечную точку титрования находят по пересечению прямых, проведенных через линейные участки кривой титрования.
□ В кондуктометрических методах часто требуются селективные реагенты.
Основные преимущества кондуктометрического титрования перед классическим и потенциометрическим титрованием:
— можно титровать окрашенные н мутные растворы;
— для построения кривой титрования достаточно небольшого числа точек, удаленных от точки эквивалентности.
Последнее позволяет преодолеть проблекСу медленного установления потенциала вблизи конечной точки при потенциометрическом титровании и значительно ускоряет титрование.
Определение конечной точки титрования по изменению электропроводности используется обычно в кислотно-основном и осадительном титровании. В качестве иллюстрации рассмотрим пример кислотно-основного титрования:
Н+ + А" + М+ 4- ОН* = Н2О + М+ + А" (7.1-17)
Подчеркнуты ноны, присутствующие в растворе до конечной точки титрования.
Таким образом, при добавлении титранта (МОН) ионы Н+ заменяются ионами металла, а концентрация аниона остается неизменной. До точки эквивалентности число ионных частиц неизменно, только ионы водорода замещаются на ионы металла, и поскольку подвижность ионов водорода существенно выше подвижности нонов металла, электропроводность раствора падает, пока не достигается точка эквивалентности. После точки эквивалентности электропроводность определяется избытком гидроксид-ионов и возрастает с увеличением концентрации титранта. Кривая титрования сильной кислоты сильным основанием имеет V-образную форму.
□ Кондуктометрически можно титровать смеси слабых кислот в водных окрашенных или мутных растворах.
Напротив, при титровании слабой кислоты слабым основанием, электропроводность исходного раствора мала из-за слабой диссоциации. Соль, образующаяся в результате титрования, диссоциирует полностью, что приводит к повышению электропроводности. Однако при достижении точки эквивалентности концентрация сильного электролита остается постоянной, а увеличивается только концентрация слабого основания.
зи Глава 7. Методы химического анализа и их применение
В общем случае четко выраженная конечная точка наблюдается только при титрований сильной кислоты сильным основанием или слабой кислоты слабым основанием1. г‘
Гидролиз, диссоциация н растворимость продукта реакции вызывают искривление кривой титрования вблизи конечной точки. Прн удалении от конечной точки, т. е. при степенях оттитрованности 0-50% н 150-200%, эти эффекты подавляются. Экстраполяция прямолинейных участков позволяет точно определить конечную точку титрования.
Как и во всех титриметрических методах, титрант должен быть в 10 раз концентрированнее, чем титруемый раствор, чтобы изменение объема было небольшим. При необходимости вводят поправку на разбавление, умножая электропроводность на (V + t>/V), где V —исходный объем раствора, a v — добавленный объем титранта.
Вопросы и задачи
1. Сформулируйте общие принципы титриметрических методов анализа.
2. Какие виды титриметрических методов вам известны?
3. Для титрования 0,01 М NaOH раствором 0,1 М НС1 изобразите кривую титрования и объясните различные участки кривой, а также значение pH конечной точки, используя диаграмму Хэгга.
4. Используя соответствующую диаграмму Хэгга, нарисуйте кривую титрования 0,Ш М ТЧНз раствором 0,1 М НС1 и объясните различные участки кривой, а также pH буферной точки и конечной точки!
5. При титрования Н3РО4 раствором NaOH в воде, в соответствии с рис. 7.1-4, можно определить только две точки эквивалентности (С\ и Сг) и одну буферную точку (В2). Объясните, почему другие точки на кривой не видны!
6. Как можно повысить силу слабой кислоты перед титрованием?
7. Какие лиганды можно успешно использовать в комплексоиометрии и почему?
8. Опишите принцип действия комплексонометрических индикаторов.
9. В каких экспериментальных условиях можно регистрировать кривые титрования?
7.2. ГРАВИМЕТРИЯ
Цели изучения
• Рассмотреть достоинства и недостатки гравиметрии.
• Описать условия количественного выделения осадка.
• Ознакомиться с основами электрогравиметрии.
7.2.1. Введение
В гравиметрии определяемое вещество (в большинстве случаев ион) осаждают в виде малорастворимого соединения определенной стехиометрии. После выделения и высушивания осадок взвешивают на аналитических весах н по его массе и известной стехиометрии находят количество определяемого компонента.
7.2. Гравиметрия ЗВ J
□ Гравиметрические методы чрезвычайно точны.
Гравиметрические методы широко используют для стандартизации, хотя титриметрические и инструментальные методы вытеснили «гравиметрию из большинства стандартных исследований и .работ общего -характера- Вообще говоря, гравиметрические методы чрезвычайно точны, потому чтона аналитических весах можно взвесить вещества с высокой степенью точности. Массу можно определить до пятой цифры после запятой, тогда как погрешность даже наиболее точных титриметрических и инструментальных методов редко бывает ниже 1-0,1%.
Точность (правильность н воспроизводимость) гравиметрического метода зависит от условий осаждения, а также свойств осадка. Если требуется высокая точность, осадок должен удовлетворять следующим требованиям:
— он должен иметь известную и воспроизводимую стехиометрию;
— должен мало растворяться в маточном растворе и промывной жидкости (определяемый компонент должен количественно осаждаться);
— подвергаться минимальному влиянию других элементов и компонентов системы;
— иметь малую поверхность (кристаллический осадок), чтобы адсорбция посторонних примесей была минимальной;
— быть легко фильтруемым н промываемым, т. е. легко отделяться от маточного раствора и примесей при промывании подходящим растворителем;
— быть термически устойчивым, чтобы его можно было высушить»не изменив состава;
— быть устойчивым в высушенном виде (гигроскопичность вещества создает дополнительные проблемы).
7.2.2. Гравиметрические методы
Очень важно, чтобы при осаждении получался чистый, легко фильтруемый осадок, поэтому при выполнении гравиметрических определений требуются некоторое умение и опыт.
□ Для получения правильных результатов гравиметрического определения необходимо контролировать условия осаждения.
Некоторые из вышеупомянутых свойств зависят от техники осаждения. Кристаллические вещества обычно имеют определенный стехиометрический состав, их легко отделить от маточного раствора фильтрованием и центрифугированием и. эффективно промыть минимальным объемом промывной жидкости. К тому же для кристаллических осадков характерна относительно малая величина удельной поверхности, поэтому поверхностная активность относительно мала (особенно в сравнении с аморфными осадками), и количество адсорбированных или окклюдированных примесей минимально.
Форму и размер кристаллов можно до определенной степени контролировать техникой осаждения. При быстром смешении двух концентрированных растворов реагентов образуются неудобные мелкокристаллические осадки. Если растворимость вещества существенно зависит от температуры, при резких
3t2 Глава 7. Методы химического анализа и их применение
изменениях температуры выпадают небольшие кристаллы, а растворимость понижается. Если горячий раствор охлаждается очень медленно, то у зародышей кристаллов есть возможность вырасти, прежде чем образуются новые центры кристаллизации. В пересыщенных растворах происходит спонтанная кристаллизация, в результате осадок сильно загрязняется окклюдированными примесями, содержащимися в маточном растворе. Эти примеси при промывании не удаляются.
Как правило, осадки, удобные для фильтрования, выделяют из горячих растворов. Осадитель должен быть по возможности разбавленным н добавлять его нужно по каплям при перемешивании раствора во избежание местных пересыщений.
Растворы летучих веществ нагревать нельзя, иначе могут быть значительные потери. Не рекомендуется нагревать растворы летучих кислот (H2S, НС1 и др.) или оснований (NH3 и др.). Например, при гравиметрическом определении соляной кислоты осаждением нитратом серебра раствор кислоты нельзя нагревать до добавления AgNOg. Однако после добавления AgNOg рекомендуется повысить температуру почти до точки кипения (80°С) для получения легко фильтруемого осадка. Не следует до добавления AgNOg нагревать н растворы хлоридов, поскольку осаждение проводят в кислой среде (добавление HNO3) и в растворе образуется летучий НС1.
Перед фильтрованием весь осадок должен собраться на дне сосуда, чтобы большую часть жидкости можно было декантировать.
Традиционно осадок собирают на обеззоленной фильтровальной бумаге, которую затем сжигают в платиновом тигле, чтобы остался только конечный продукт. Позднее для фильтрования стали использовать специальные керамические тигли. В любом случае для получения правильного результата важной стадией является промывание осадка. Для получения сухого вещества тигель с осадком прокаливают в печке. По разности масс пустого тигля и тигля с осадком находят массу осадка н затем рассчитывают содержание определяемого компонента.
7.2.3. Расчет результатов гравиметрического анализа
Целью гравиметрического анализа является нахождение массовой доли wa определяемого компонента в пробе в соответствии с законами стехиометрии. Формула для расчета, основанная на двух точных взвешиваниях, приведена ниже
WA = !2Д1оо = —100
е е
где wa — содержание определяемого компонента (%); тд —масса определяемого компонента (мг); е —масса навески (мг); а—масса осадка1, взвешенного после осаждения, прокаливания и т. д.; F—гравиметрический фактор.
Гравиметрическая форма. — Прим, перев.
7.2. Гравиметрия
383
Гравиметрический фактор F равен отношению молярных масс компонента А и его гравиметрической формы:
р _ Молярная масса определяемого вещества, (г/моль) Молярная масса гравиметрической формы (г/моль) - ,.1
В следующей таблице приведены примеры расчета гравиметрических факторов.
Определяемое вещество Гравиметрическая форма Гравиметрический фактор
MgO FesOd MgP2O7 РезОз 2-Af(MgO) Af(Mg2P2O7) 2M(Fe3O4) 3 Af(Fe2O3) 0,3622 0,9666
Чем меньше гравиметрический фактор F, тем выше чувствительность ме-
тода и ниже погрешность определения. Этого можно достичь, используя орга-
нические осадители с высокой молярной массой.
Определяемое Осаждаемая Гравиметри- Гравиметри-
вещество Осадитель форма ческая форма ческий фактор
Fe3+ ОН- Fe(OH)3 РезОз 0,6994
А13+ 8-Гидроксихинолин A1(HQ)3 А12Оз . 0,5293
А13+ 8-Гидроксихинолин A1(HQ)3 A1(HQ)3 0,05873
Ni2+ N« №(ДМГ)2 0,2032
Ниже приведен пример расчета прн гравиметрическом определении Ва2+ осаждением H2SO4 в виде BaS04, с последующим фильтрованием и прокаливанием в кварцевом тигле:
Масса пробы (навеска) : 0,6537г
Масса BaSO4 : 0,4288 г
= ^7’34 = 0,5884, шва = — х 100 = 38,6%
M(BaSO4) 233,40 е
7.2.4. Электрогравиметрия
В отличие от вольтамперометрии (см. разд. 7.3) электролиз можно провести в условиях, когда состав раствора меняется вследствие количественного электроокисления или электровосстановления компонентов раствора. В таких случаях электролиз проводят при большом отношении рабочей поверхности электрода А к объему раствора V и в условиях конвективно-диффузионного контроля массопереноса. Электрод с большой поверхностью дает возможность пропускать относительно высокие токи и позволяет быстро получить продукт электроокисления или электровосстановления. Основные принципы, управляющие электродными реакциями при объемном электролизе, те же, что и в условиях микроэлектролиза (вольтамперометрия).
зм Глава 7. Методы химического анализа и их применение
□ Электрогравиметрия основана на реакциях количественногоокисления и восстановления.
Методы электролиза можно классифицировать по измеряемым количествам вещества. В электрогравиметрии определяют массу вещества, выделенного на одном из электродов (рабочем электроде) в виде чистого соединения известной стехиометрии. Электрогравиметрия—это метод гравиметрического определения микроколйчеств вещества, использующий в качестве реагента электрический ток.
Электрогравиметрия относится к старейшим электроаналитическим методам, широко используемым для определения ионов металлов. В настоящее время ее применяют прежде всего для электровыделения металлов из сложных смесей.
В кулонометрических методах основой для прямого или косвенного количественного определения веществ является общее количество электричества Q, израсходованное на электролиз (см. разд. 7.3).
□ И электрогравиметрия, и кулонометрия,—высокоточные, безэталонные методы.
Принцип электролиза с контролируемым током
В процессе электролиза химическая реакция протекает в направлении, противоположном самопроизвольному, при наложении на ячейку постоянного напряжения от внешнего источника. Ячейка состоит из двух электродов, контактирующих с раствором. Если Ь’прнл < общее напряжение, наложенное на ячейку, распределяется следующим образом:
EnpM^E,-EK + iR (7.2-1)
где Ел — потенциал анода; Ек — потенциал катода; г—ток; R—сопротивление.
Потенциал каждого электрода рассматривается как сумма равновесного потенциала обратимой реакции и перенапряжения. Равновесный потенциал зависит от стандартного потенциала электрохимической полуреакции, протекающей на электроде, и активностей соответствующих веществ у поверхности электрода.
Рассмотрим следующую обратимую полуреакцию:
O + ne-^R (7.2-2)
где н О (окисленная форма) и R (восстановленная форма)1 растворимы.
Скорость электродной реакции зависит от приложенного потенциала, а соотношение концентраций О н R можно выразить уравнением Нернста
£„₽„» = + (7.2-3)
П2* CR
В процессе электролиза, т. е. в процессе осаждения металла, потенциал электрода падает при снижении концентрации со катиона металла в растворе.
В учебной литературе их обычно обозначают Ох и Red. — Прим, перед.
7.2. Гравиметрия
385
Полноту протекания электролиза можно предсказать, исходя из приложенного потенциала н уравнения Нернста. Если считать, что в начале концентрация О равна со,», Е° — стандартный электродный потенциал, V* — объем раствора, а х—доля О, восстановленного до R при потенциале электрода Е, то при равновесии число молей О составляет Цс*(1 ~ж), тогда как число молей R равно т. е.
Д.р«л = £° + ^1п^ЦЙ—^=£° + £Ln— (7.2-4)
nF V»CiX nF х ' y '
Например, при степени прохождения реакции восстановления О до R, равной 99,99% (х = 0,9999), потенциал рабочего электрода равен
= £« +0^9 /^0001) w£O+0^59 ^ (7 2 5)
т. е. стал на 236/пмВ отрицательнее, чем Е° при 25°С.
Аналогично, в условиях электрогравиметрии при осаждении ионов металла на инертном (платиновом) электроде:
0+шГ ?=!R(tb.) (7.2-6)
По уравнению Нернста
£„рнл = B° + ^ln[c,(l-i)| (7.2-7)
где с,—начальная концентрация окисленной формы, а ж—доля окисленной формы, восстановленной при электродном потенциале Е.
При учете степени электровосстановления можно рассчитать сдвиг потенциала при данной степени протекания электролиза (ур. 7.2-4).
Отсюда следует, что изменение электродного потенциала вследствие электролиза следует учитывать при использовании электролиза для селективного определения металлов.
. □ В электрогра ви метри и следует уч итыватьиэменение электродного потен цма л а
в процессе протеканий электролитической реакции.
Пусть сд и св — исходные концентрации компонентов А и В соответственно, а Ед и Е^ — окислительно-восстановительные потенциалы соответствующих полуреакций. Если Е^ > Eq, то в предположении, что обе полуреакции обратимы, условие для селективного разделения (х = 0,9999):
0,059-4 0,059
ЬА------__ = Ев + —— Св (7.2-8)
П П59
ДЕ =^^(4 +1g св) (7.2-9)
В рассмотренном случае степень протекания электродной реакции зависит от равновесных условий, но скорость электролиза мала при потенциалах, найденных по уравнению Нернста. Скорость электролиза, как и следовало ожидать, возрастает при повышении приложенного к электроду потенциала и достигает предельного значения при потенциале, соответствующем плато на кривых ток —напряжение. Поэтому потенциал электролиза обычно выбирают
18 Аналитическая химия. Том 1
386
Глава 7. Методы химического анализа и их применение
на основании экспериментальных кривых ток—напряжение для микроэлектродов (разд. 7.3), снятых в условиях, близких к условиям проведения электролиза.
Селективность методик электролитического разделения зависит от состава фонового электролита, выбранного для электролиза. Для разделения двух ркислительно-восстановительных систем с близкими значениями Е° (например, ДЕ° < 236 мВ при 25°С) можно улучшить разделение добавлением комплексообразующего лиганда, связывающего ионы одного из металлов. При введении лиганда, связывающего ион металла в комплекс, обычно наблюдается сдвиг значения Е° окислительно-восстановительной системы к отрицательным значениям. Этот подход можно использовать для разделения меди(П) и висмута^!) (££и — 0,345 В, Ejjj ~ 0,320 В). В тартратном буфере с pH 6 можно провести количественное электроразделение ионов этих двух металлов. Осаждению металлов часто мешает выделение Нг, его можно легко избежать, сделав раствор более щелочным.
На основании приведенных расчетов можно найти теоретическую разность стандартных потенциалов, необходимую для определения одного иона в присутствии другого. Она составляет 240 мВ (25°С) для однозарядных ионов и 120 мВ (25°С) для двухзарядных ионов.
Потенциал рабочего электрода можно регулировать, только изменяя внешнее приложенное напряжение. Однако, в соответствии с уравнением (7.2-4), -Е„рил влияет на значения всех потенциалов в ячейке, каждый из которых меняется по мере протекания электролиза. Поэтому единственный практический способ поддержания потенциала электрода при заданном значении заключается в непрерывном измерении потенциала катода относительно электрода сравнения. Потенциал ячейки можно поддерживать в выбранном диапазоне. Этот вид анализа называется электролизом при контролируемом потенциале катода (или анода).
Потенциал рабочего электрода относительно третьего электрода (электрода сравнения) автоматически контролируется потенциостатом.
Выход по току
Эффективность тока (выход по току) в электродной реакции (i-й реакции) равен доле суммарного тока, потраченной в i-й реакции:
Эффективность тока в t-й реакции = . и (7.2-10)
^сумгл
где icyMM ~ суммарный ток, протекающий через электролизер.
100%-ная эффективность тока (выход по току) означает, что отсутствуют побочные реакции, т. е. иа рабочем электроде протекает только один процесс.
Для макроэлектролиза выгодны большие выходы по току, поскольку при этом сокращается время анализа. В электрогравиметрии не требуется 100%-ный выход по току, если только в результате побочных реакций не образуются нерастворимые продукты, но в кулонометрическом титровании 100%-ный выход по току — обязательное условие.
□ Высокий выход по току сокращает время анализа.
7.2. Гравиметрия
387
Электролитические ячейки
Конструкции электродов для электролиза прн контролируемом потенциале показаны на рис. 7.2-1. Катодом обычно служит платиновая сетка или цилиндр из платиновой фольги диаметром 2-3 см. Преимуществом платиновых электродов является их сравнительно низкая реакционная способность и возможность их использования для осаждения ряда ионов металлов. Однако прн выделении некоторых нонов металлов, в особенности олова и цинка, требуется защитное покрытие во избежание образования сплава.
Металлы, образующие амальгамы, можно осаждать на ртутных электродах. Этот метод особенно широко используется как предварительная стадия анализа для удаления легко восстанавливающихся элементов.
Анод также можно изготовить из платины. Правильное расположение вспомогательного электрода важно для обеспечения равномерной плотности тока по поверхности рабочего электрода. Расположение электрода сравнения также важно для обеспечения длительной стабильности электрода сравнения и Ш-скачка.
Рабочий и вспомогательный электроды часто помещают в разные отделения, чтобы предотвратить смешивание электролитов. Их обычно разделяют диском из спеченного стекла или ионообменной мембраной со сравнительно низким сопротивлением, что не очень хорошо влияет на сопротивление ячейки в целом. Можно обойтись без перегородок, если правильно выбрать реакции на вспомогательном электроде, например, образование твердых (AgCI) или инертных газообразных (например, N2) продуктов.
Сопротивление ячейки и правильное расположение электрода сравнения в ячейке важны прн электролизе в неводных растворителях с низкой диэлектрической проницаемостью (например, в ацетонитриле, диметилформамиде, аммиаке).
Электрогравиметрические методы
Для электрогравиметрического анализа желательны гладкие, плотно прилипающие к поверхности электрода осадки. Качество осадка зависит и от того, в какой форме находится ион металла в растворе. Из растворов комплексных
Рис. 7.2-1. Конструкции электродов для электролиза при контролируемом потенциале.
13*
388
Глава 7. Методы химического анализа и их применение
соединений выделяются более гладкие и более плотно прилегающие осадки, чем из растворов акваионов металлов.
Время, требуемое для выполнения электрогравиметрического определения, зависит от плотности тока, но при слишком высоких плотностях тока качество осадка ухудшается. Выделение водорода во время-осаждения также ухудшает структуру осадка.
□ Электрогравиметрические методы можно использовать для количественного разделения металлов.
Чувствительность электрогравиметрии ограничивается сложностью определения небольшой разности масс чистого электрода н электрода с осадком. Обычно достаточно осадить 0,2-5 ммоль вещества. Электрогравиметрический анализ дополняется кулонометрическими методами, кроме случая, когда нельзя обеспечить 100%-ный выход по току.
Электрораэделение
В предыдущем разделе обсуждены условия для селективного количественного осаждения одного металла Mi на твердом электроде. В данном случае селек-тивндсть означает, что при данных экспериментальных условиях не происходит заметного осаждения металла М2- Таким образом, для количественного осаждении Mi (> 99,9%) в виде амальгамы ртути потенциал электрода должен быть
Е<Е?- —Впри25°С (7.2-11)
П1
(где Ef—стандартный потенциал, щ—число электронов, участвующих в электродной реакции), а для 0,1% осаждения Мг:
Е > -Е? - — В при 25°С (7.2-12)
п2
Следовательно, 99,9% отделение Mi достигается при
ДЕ = Е? - Е2 = 0,18(ni + п2) В прн 25°С (7.2-13)
или для однозарядных ионов при ДЕ — 0,36 В.
Для электроразделения обычно применяют катод из донной ртути.
Вопросы и задачи
1. Каковы экспериментальные условия достижения высокой точности гравиметрического анализа?
2. Как увеличить вероятность получения чистого осадка?
3. Сравните точность гравиметрического и титрнметрического анализа.
4. Опишите принципы электрогравиметрии и электроразделения.
5. Кыюварйзность стандартных потенциалов, необходимая для определения одного иОЩкМетадла в присутствии другого иона металла?
6. Какие типы электродов используют в электрогравиметрии?
7.3. Электроанализ
389
7.3. ЭЛЕКТРОАНАЛИЗ
Цели изучении
• Обсудить йринципы применения основных электроаналитических методов.
• Детальное описать типы и свойства электродов в потенциометрии и вольтамперометрии.
• Обсуждить возможности отдельных электроаналитических методов.
Электроаналнтические методы относятся к наиболее мощным и распространенным методам аналитической химии. Они являются результатом интенсивных исследований в области электрохимии, науки, возникшей почти сто лет назад. Электроаналнтические методы относятся к наиболее ранним инструментальным методам анализа, которые в двадцатом веке произвели переворот в аналитической химии.
К основным электроаналитическнм методам относят потенциометрию, вольтамперометрию, или полярографию, кулонометрию и кондуктометрию. Все эти Методы нашли применение в титриметрии (7.1-3). В то же время элек-троаналитические методы играют значительную роль в создании детекторов для хроматографических и родственных методов (см. гл. 5) и очень важны для разработки химических сенсоров и биосенсоров (разд. 7.7 н 7.8 соответственно).
7.3.1. Потенциометрия
Потенциометрия—один из самых простых электроаналитических методов; он используется прежде всего для измерений pH, а также для установления ионного состава биологических жидкостей, например крови и мочи, но, кроме того, и как способ преобразования селективных взаимодействий в аналитический сигнал в молекулярных сенсорных устройствах или в процессе химических реакций.
Потенциометрия основана на измерении потенциала ячейки, т. е. разности потенциалов между двумя электродами (индикаторным электродом и электродом сравнения) в отсутствие тока во внешней цепи. Это позволяет получить информацию о химическом составе раствора. Потенциал н область применения индикаторного электрода зависят от его природы и селективности. В потенциометрии используют два приема, а именно измерение потенциала электрода как функции активности (концентрации) определяемого компонента (прямая потенциометрия) и измерение потенциала как функции объема реагента, добавляемого к пробе (потенциометрическое титрование).
□ Прямая потенциометрия и потенциометрическое титрование.
Измерительный инструмент, в первую очередь, должен обеспечить минимальный отвод тока. Для этой цели пригодны потенциометр и электронный вольтметр, последний в настоящее время используют чаще.
398
Глава 7. Методы химического анализа и их применение
Потенциометрическая ячейка
В потенциометрии раствор, содержащий определяемое вещество, активность (концентрацию) которого следует установить, и индикаторный электрод образуют полуэлемент; второй полуэлемент состоит из электрода сравнения (рис. 7.3-1). Потенциал индикаторного электрода линейно зависит от логарифма активности (концентрации) определяемого иона. Электрод сравнения в растворе постоянного состава имеет фиксированный потенциал Еср1 не зависящий от состава анализируемого раствора. Оба электрода идеально неполяризуемые.
Потенциал ячейки (э.д.с.) Е описывается следующим уравнением:
Е = (Е.Ш- Вер) + Ej (7.3-1)
прав лев
где Еичд — потенциал индикаторного электрода, Еср — потенциал электрода сравнения, a Ej — потенциал жидкостного соединения.
Следовательно, э.д.с. ячейки определяется разностью потенциалов правого н левого электродов, а полуреакции на электродах определяют их окислительно-восстановительные потенциалы.
Например, в ячейке
Ag|AgCl,KCl||Ce3+,Ce4+|Pt (7.3-2)
на левом электроде (электрод сравнения) протекает полуреакция
AgCl(TB.) + е" Аё(тв.) + СГ, Е° = 0,222В (7.3-3)
а иа правом (индикаторном) электроде, т. е. на Pt, — полуреакция
Се4+ + е“ <=! Се3+, == 1,44 В (7.3-4)
Внешний электрод Индикаторный сравнения электрод
Рис. 7.3-1. Схема ячейки для потенциометрических измерений.
7.3. Электроанализ
391
Для потенциалов электродов можно записать уравнения
Ет._ = 0,222 - 0,05916 lg[Cl“ ] (7.3-5)
(Се3+1
Я,.ра.. = 1,44 - 0,059161g Ы (7.3-6)
[Се J
Потенциал (э.д.с.) ячейки при 25°С:
/ ГСе3+1 \
Е = £),р„. - Ей„. = 1,44 - 0,059161g - (0,222 - 0,05916 ЦСГ]) =
\ LCe J /
^1,218-0,059161g [Ce^.L (7.3-7)
Поскольку концентрация хлорид-ионов в электроде сравнения постоянна, потенциал ячейки зависит от соотношения [Се3+]/[Се4+].
В правильно составленной потенциометрической ячейке Еср постоянен, а Ej или постоянен или пренебрежимо мал.
□ Типы электродов в потенциометрии.
Индикаторные электроды можно классифицировать следующим образом:
а) Классические электроды
Класс 0. Инертные металлы (например, Pt, Ап) в контакте с раствором окислительно-восстановительной пары, например система Pt|Ce4+, Се3+. Идеальные инертные материалы обратимо обмениваются электронами с компонентами электролита, при этом сами не окисляются и не корродируют.
Класс 1. Обратимые: металл/нон металла (активный металл в контакте с раствором собственного иона), например Ag|Ag+ (электроды первого рода).
Класс 2. Обратимые: металл в равновесии с насыщенным раствором соли иона металла н избытком аниона X’, например, Ag|AgX,X“ (электроды второго рода).
Класс 3. Обратимые: металл в равновесии с двумя малорастворимыми солями с общим анионом (или растворимым комплексом иона второго металла) и избытком второго катиойа, например, РЬ|оксалат РЬ|оксалат Са|Са2+ (электроды третьего рода).
б) Мембранные (нон-селективные) электроды.
Электроды сравнения
Электрод сравнения, изготовленный из фаз постоянного химического состава, характеризуется известным, постоянным (не зависящим от концентрации определяемого вещества) потенциалом. Важными требованиями к электроду сравнения являются обратимость, воспроизводимость и стабильность во времени. Обратимость означает, что направление электродной реакция можно изменить, поменяв полярность электрода. Воспроизводимость выражается стандартным отклонением потенциала ячейки при последовательных измерениях
392
Глава 7. Методы химического анализа и их применение
в растворе заданной концентрации, а стабильность работы электрода оценивают по смещению отклика при измерениях в потоке и по величине стандартного отклонения для заданного раствора. Электроды сравнения, используемые в аналитической практике, являются вторичными эталонами и, как правило, электродами второго рода. Потенциалы электродов сравнения определены относительно стандартного водородного электрода (СВЭ, первичный электрод сравнения в электрохимии).
D Самым распространенным электродом сравнения является каломельный электрод.
Хлоридсеребряный электрод (Ag|AgCI, КО)
Хлоридсеребряный электрод можно приготовить, например, покрыв анод из серебряной проволоки хлоридом серебра методом электролиза 0,1 М раствора хлорида. Проволоку, покрытую хлоридом серебра, погружают в раствор КС1 с известной концентрацией, чаще всего насыщенный (3,8 М, иногда 1,0 или 0,1 М, рис. 7.3-2,а).
Потенциал хлоридсеребряного электрода возникает в результате полуреакции'
AgCl(TB.) + е“ Аё(тв.) + СГ (7.3-8)
Хлоридсеребряный электрод—самый воспроизводимый электрод сравнения с хорошей электрической н химической стабильностью при 25°С. Потенциал электрода зависит от всех компонентов раствора, влияющих на концентрацию иона серебра, поэтому его нельзя использовать без дополнительного солевого мостика в растворах, содержащих белки, бромид- или сульфид-ионы, образующие нерастворимые соединения с ионами серебра, а также в присутствии лигандов, взаимодействующих с AgCl, таких, как CN~, SCN-, и сильных окислителей или восстановителей.
Температурный коэффициент хлоридсеребряного электрода очень мал, поэтому он удобен в случаях, когда температуру невозможно поддерживать постоянной или прн температурах выше 80°С.
Каломельный электрод (Hg|Hg2CI2, KCI)
Каломельный электрод состоит из платиновой проволоки, погруженной в пасту из ртути и хлорида ртути(1) в стеклянной трубке. Внутренним раствором служит раствор хлорида калия известной концентрации, насыщенный хлоридом ртути(1) (рис. 7.3-2,в). Потенциал каломельного электрода определяется полуреакцией
Hg2Cl2(TB.) + 2е“ 2ВДж.) + 2СГ (7.3-9)
и согласно уравнению Нернста зависит от концентрации хлорид-ионов:
Е = Е°' - _'£|Н16 ДО-]2 = Е°' - 0,05916 lg[Cl“] Уравнение Нернста
(7.3-10)
7.3. Электроанализ
393
соединение (пористая керамическая пробка)
Рис. 7.3-2. Электроды сравнения.
а —хлоридсеребряный электрод; б— хлоридсе-
ребряный электрод с двойным жидкостным соединением; в — каломельный элек-
трод.
Наиболее часто используют каломельный электрод с насыщенным раствором КС1 (3,8 М при 25°С); его называют насыщенным каломельным электродом (НКЭ). Его легко приготовить, а наличие кристаллов КС1 гарантирует постоянство концентрации раствора; потенциал НКЭ равен 4-0,241В при 25°С. Каломельный электрод не рекомендуется в качестве электрода сравнения при температурах выше 75°С. Переход с одной шкалы потенциалов на другую показан на рис. 7.3-3.
Потенциал жидкостного соединения
Жидкостное соединение—это зона контакта двух растворов различного состава, образующаяся в месте соприкосновения электрода сравнения и анали-
394
Глава 7. Методы химического анализа и их применение
MJ.763 -1.00
о ЙГ” ' -0242
СВЭ 0241 0
НКЭ Лр..* лгчл . (1522 D320
£ (VU АЛ) 0.77 0.53
£ (ге /ге ) 1247 1.005
£ (11 ILT )
Е отн. СВЭ (В) Е атн. НКЭ (В)
Рис. 7.3-3. Пересчет потенциала по шкале СВЭ на шкалу НКЭ.
зируемого раствора. Эта неселективная граница раздела является источником потенциала жидкостного соединения (или диффузионного потенциала), Ej.
В потенциометрической ячейке электрод сравнения контактирует с раствором пробы через солевой мостик. Солевой мостик обеспечивает электрический контакт между двумя полуэлементами электрохимической ячейки. Смешивание двух неодинаковых фаз (различного состава) можно минимизировать, если зону соприкосновения фаз уменьшить, что обеспечивается при контакте через капилляр, диафрагму из спеченного стекла или пробку из агар-агара. Природа жидкостного соединения (анализируемый раствор | площадь контакта солевого мостика) влияет на воспроизводимость измерений потенциала ячейки. Значение потенциала жидкостного соединения в большинстве случаев неизвестно, и это лимитирует точность прямых потенциометрических измерений. В ячейке с диафрагмой жидкостное соединение обозначается двойной чертой ||.
Различные типы жидкостных соединений приведены на рис. 7.3-4. В зоне контакта (например, через диафрагму) градиент концентрации вызывает диффузию ионов. Из-за различия подвижностей ионов на границе раздела (жидкостном соединении) происходит разделение зарядов, т. е. возникает скачок потенциала, и через некоторое время ионы начинают пересекать границу с одинаковыми скоростями. Соответствующий стационарный потенциал называют потенциалом жидкостного соединения или диффузионным потенциалом. Для минимизации потенциала жидкостного соединения используют солевой мостик с насыщенным раствором, содержащим ионы с одинаковыми подвижностями, например, КС1:
Hg|Hg2Cl2|HCl(0,l М)||КС1(нас.)||КС1(0,1 M)|Hg2Cl2|Hg (7.3-11)
С повышением концентрации с раствора КС1 величина потенциала жидкостного соединения снижается, поскольку последовательные потенциалы жидкост-
7,3. Электроанализ
395
o.oi м на
од м на
олмна
олмка
o.iM на
0.05М KNO3
Рис. 7.3-4. Схематическое представление структуры различных типов жидкостного соединения [Bard, A.J., Faulkner, L.R. (1980), Electrochemical Methods, New York, Wiley, p. 63].
ного соединения становятся равными по величине и противоположными по знаку н, таким образом, компенсируют друг друга.
В продаже имеются электроды сравнения с двойным жидкостным соединением для высокоточных измерений потенциала (рнс. 7.3-2,б). Двойное жидкостное соединение не только снижает диффузионный потенциал, но и позволяет избежать загрязнения раствора пробы внутренним раствором электрода сравнения. Электроды сравнения с двойным солевым мостиком особенно удобны для точных измерений с нон-селективными электродами.
В прямой потенциометрии важно, чтобы жидкостное соединение было одинаковым при измерениях в стандартных и анализируемых растворах.
Потенциометрические индикаторные электроды
Потенциал индикаторного электрода, пропорциональный логарифму активности (концентрации) определяемого иона, можно описать уравнением Нернста или его модификациями.
Классические потенциометрические электроды
Металлические индикаторные электроды
Для электродов первого рода (например, Ag|Ag+) потенциалопределяющее равновесие на границе раздела фаз имеет вид
Мп++пе~^М (7.3-12)
При равновесии электрохимические потенциалы вещества в двух контактирующих несмешивающихся фазах, равны:
Рм»+ + пре = рм (7.3-13)
где электрохимический потенциал металла, —электрохимический
потенциал иона металла Мп+ в фазе раствора, де — электрохимический потен
396
Глава 7. Методы химического анализа и их применение
циал электронов в фазе металла, а п—число молей обменивающихся электронов в расчете на моль вещества.
Электрохимический потенциал определяется выражением
д = д + пЕф (7.3-14)
Решая уравнение для Дф, T. е. для равновесного гальвани-потетщйала (Д$- = Фыет ~ Фр-р) с учетом того, что активности электронов и атомов металла в металлической фазе равны единице, а химические потенциалы /л частиц Мп+ определяются как
~ 7^лп+ +-ЙПпом»*-»- (7.3-15)
получаем классическое уравнение Нернста для электродного потенциала:
„ 2,303йТ_ 1
E = EQ- F lg------------
nF «М"+
где E° — стандартный электродный потенциал, 2, ЗОЗЯТ/nF— так называемый коэффициент Нернста, а ам~+ — активность иона металла.
Поскольку потенциал электродов этого типа обусловлен электроннообменными равновесиями, он зависит и от других окислительно-восстановительных пар в растворе.
Электроды второго рода
Потенциал электрода второго рода (например, Ag|AgCI, (яМ)КС1) возникает также в результате окислительно-восстановительного процесса
AgCl + е~ Ag + СГ (7.3-17)
Концентрация хлорид-нона определяется произведением растворимости Кяр:
AgCl(TB.) г* Ag+ + СГ, К8Р = 1,8- 10’1О(25°С) (7.3-18)
поэтому из уравнений (7.3-17) и (7 Л-18) получаем
Е « ££g+ + 0,05916 lg Xsp -0,05916lg[Cr]
’ ' (7-3-19)
E = <g,Asci- 0,05916 lg[Cr]
Таким образом, потенциал электрода обратимо зависит от активности хлорид-иона, в идеальном случае он изменяется на 59,16 мВ (при 25°С) при изменении [СГ] на порядок.
Потенциал электрода зависит от любого вещества, влияющего на активность иона серебра: лигандов, растворяющих AgCl (таких, как CN_, SCN-), веществ, образующих менее растворимые соединения с Ag+ (таких, как S2~, Вг~, 1~), и сильных окислителей и восстановителей. В их присутствии хлоридсеребряный электрод нельзя погружать в исследуемый раствор, необходим солевой мостик.
Электрод характеризуется стабильным потенциалом только при условии, что концентрация хлорид-иона во внутреннем растворе поддерживается постоянной.
7.3. Электроанализ
397
Окислительно-восстановительные электроды
Электроды из Pt, С и Au относительно инертны и служат лишь для переноса электронов от восстановителя к окислителю в растворе. Величина потенциала платинового индикаторного электрода определяется отношением активностей окисленной (ох) и восстановленной (red) форм в растворе в соответствии с уравнением Нернста:
0,0592 ^Qred 0,0592 7red 0,0592 cfed _
Е — Е----------1g----= Л----------1g------------1g----—
П flox 71 7ox П Qw
= _ 2^®? ]g 5* (7.3-20)
71 Cqx
где E°' — формальный потенциал.
Формальный потенциал—потенциал полуреакции при общих концентрациях окисленной и восстановленной форм, равных 1М, н известной концентрации электролита.
Ион-селективные электроды
Ион-селективные электроды (ИСЭ) — полуэлементы, состоящие из ион-селективной мембраны (т. е. селективной межфазной границы), внутреннего раствора и внутреннего электрода сравнения (стандартная конструкция) (рис-7.3-5) или ион-селективной мембраны и твердофазного контакта (твердотельный электрод). Такой электрод позволяет селективно определять активности одних ионов в присутствии других; анализируемый раствор обычно является водным. Эти электроды отличаются от окислительно-восстановительных электродов (электродов нулевого, первого, второго н третьего рода), хотя они часто содержат электрод второго рода в качестве внутреннего электрода сравнения.
Вторым полуэлементом в паре с ион-селективным электродом является внешний электрод сравнения, контакт между ними осуществляется с помощью солевого моСтика. Обычно используют электроды сравнения с двойным
Рис. 7.3-5. Ион-селективные электроды, а — микроэлектрод; б — мембранный макроэлектрод.
398
Глава 7. Методы химического анализа и их применение
Анализируемый раствор
Электрод сравнения од
Ей
Мембрана
♦м
"$2
Ей = <Ф1 - Фм) + <Фм - Ф2^
Рис. 7.3-6. Изменение потенциала на границе раздела ион-се-лективная мембрана|раствор и в фазе мембраны.
J
= d
жидкостным соединением (рис. 7.3-2). Схема электрохимической ячейки с ион-селективным индикаторным электродом:
Hg|Hg2Cl2, КСЦнас.) || II проба| мембрана | внутр, р-р, AgCI | Ag
МОСТИК ИСЭ
электрод сравнения
Разность потенциалов, измеренная между двумя электродами сравнения, состоит из суммы локальных разностей потенциалов:
Е — (Д$1 4- Д02 "Г &Фз) 4* ^Фз "Г &Фм — Д^о 4* ^Фз 4" Ем (7.3-21)
гдв Д<£0 —потенциал электрода сравнения, не зависящий от концентрации определяемого иона, Д^- —- потенциал жидкостного соединения, а Д$м — мембранный потенциал Еи.
Мембранный потенциал Ем описывает поведение ион-селективного мембранного электрода (рис. 7.3-6). Для мембраны, идеально селективной по отношению к определенному иону А, равновесный мембранный потенциал определяется соотношением активностей иона А в контактирующих растворах:
(73-22)
где;Цд — активность иона А в растворе пробы, ад — активность иона А во внутреннем растворе, гд —заряд иона А.
Поскольку Пд во внутреннем растворе постоянна, уравнение для потенциала мембранного электрода имеет вид
Е = const +S 1g ад (7.3-23)
где S—угловой коэффициент градуировочного графика (крутизна электродной функции ДЕ/Д^Од), в идеальном случае равный нернстовскому, т. е. ^мВ (25°С).
На практике необходимо учитывать вклад посторонних ионов В в измеряемый потенциал. Отклик мембранного электрода в реальных условиях можно
7.3. Электроанализ
399
успешно описать уравнением Никольского—Эйзенманна:
Е = const +S 1g <4 + £ (“в)’*7'”
(7.3-24)
А#В
где Кд°2—так называемый потенциометрический коэффициент селективности, zb—заряд постороннего иона В.
Потенциометрический коэффициент селективности (Кд°в) характеризует селективность ион-селективного электрода к иону В по сравнению с основным ионом А. Иначе говоря, коэффициент селективности показывает, в какой степени электрод селективнее к нону А, чем к иону В. Например, Кд°в = Ю~Э означает, что активность иона В должна быть в 1000 раз выше активности иона А, чтобы его вклад в измеренный потенциал был тем же, что и вклад иона А. Коэффициенты селективности некоторых электродов приведены в справочниках.
D Типы ион-селективных электродов.
В зависимости от природы мембраны различают следующие типы ион-селективных мембранных электродов:
Первичные ион-селективные электроды:
• электроды со стеклянной мембраной;
• кристаллические (твердофазные) мембранные электроды;
• электроды с жидкой мембраной
— жидкие ионообменные мембранные электроды;
— электроды с жидкой мембраной с нейтральными носителями.
Сложные или многомембранные ион-селективные электроды:
• молекулярно-чувствительные устройства, такие, как газочувствительные или ферментные электроды, в которых потенциометрический детектирующий блок основан на стандартных потенциометрических электродах ранее перечисленных типов (см. разд. 7.7).
Ион-селективные полевые транзисторы ИСПТ:
• Эти типы электродов—гибриды ион-селективных электродов и полевых транзисторов из оксидов металлов МИСИТ. В ИСПТ металлический затвор МИСПТ заменен или контактирует с твердой или жидкой ион-селективной мембраной. Откликом таких миниатюрных датчиков является сила тока (разд. 7.7).
Ион-селективные электроды различных типов показаны на рнс. 7.3-7.
Стеклянные электроды
Стеклянный электрод для измерения pH *— первый ион-селективный электрод, известен с начала двадцатого века. Его широко используют для измерения pH в лабораториях и в мониторинге. Стеклянный электрод состоит из стеклянной мембраны определенного состава и внутреннего электрода сравнения, погруженного в раствор с фиксированными pH и концентрацией хлорид-иона
40ft
Глава 7. Методы химического анализа и их применение
Рис. 7-3-7. Конструкция различныхион-селективных электродов, а—стеклянный электрод; б—электрод с кристаллической мембраной; в—твёрдотельный электрод с кристаллической мембраной.
Рис. 7.3-8. Комбинированный стеклянный Электрод н стеклянная мембрана.
(рис. 7.3-7,а). Комбинированный электрод состоит из стеклянного электрода и внешнего электрода сравнения (рис. 7.3-8). Электрод сравнения контактирует с раствором пробы с неизвестным pH через жидкостное соединение.
Для получения стабильных значений потенциала стеклянный электрод необход имо некоторое время вымачивать в разбавленном буферном растворе; поверхность стекла поглощает воду н образуется слой гидратированного геля. Одновременно образуются силанольные группы, и ноны натрия в слое гидратированного геля замещаются на ионы водорода. На рис. 7.3-8 схематически изображена хорошо вымоченная стеклянная мембрана.
Возникновение мембранного потенциала обусловлено наличием сильно основных групп в решетке диоксида кремния, обладающих высоким сродством к определенным катионам, в особенности к ионам Н+, адсорбирующимся на
7.3. Электроанализ
401
отрицательно заряженных центрах:
= SiOH„o. SiO'OB + Нр.р (7.3-25)
В результате специфической адсорбции ионов водорода возникает разделение зарядов и межфазный скачок потенциала на каждой из поверхностей мембраны. Через слой сухого стекла ток переносят ионы натрия. Механизм отклика стеклянного электрода описывается так называемой моделью мембраны с фиксированным зарядом.
Разность потенциалов, измеряемая между двумя электродами сравнения, зависит от pH исследуемого раствора, поскольку все остальные межфазные потенциалы постоянны из-за постоянства состава контактирующих фаз. Изменение pH раствора на единицу в идеальном случае вызывает изменение потенциала ячейки на 59,16 мВ при 25°С.
□ Потенциал стеклянного электрода
Потенциал стеклянного электрода описывается уравнением
ан+(внутр.) ан+ (внешн.)
Е = const —0,0591g
(7.3-26)
где const — постоянная, называемая потенциалом асимметрии: представляет собой разность потенциалов между двумя одинаковыми электродами сравнения в идентичных по составу растворах по обеим сторонам мембраны.
Потенциал асимметрии обусловлен различием внешней и внутренней поверхностей стеклянной мембраны и устраняется градуировкой электрода по буферам с известным pH.
Еще в 1923 г. было показано, что стеклянный электрод, в особенности при высоких pH, реагирует на другие однозарядные катионы, такие, как ионы серебра, натрия, калия [7Л-2]. Ленгьель и Блум [7.3-3] прн систематическом изучении зависимости электродной функции от состава стекла обнаружили, что
чувствительность к ионам натрия можно повысить введением в стекло оксидов трехзарядных катионов, в основном AI2O3, В2О3 и ДР- При замене КагО на ЫгО [7.3-4] так называемую «натриевую погрешность» можно уменьшить, н мембранные электроды из такого стекла можно использовать для измерений pH вплоть до pH 13, даже при высокой концентрации натрия.
Систематическое исследование селективности стеклянного электрода показало, что структура поверхности стеклянной мембраны на границе раздела мембрана/раствор в первую очередь влияет на селективность отклика. Коэффициент селективности равен произведению константы равновесия реакции обмена на поверхности мембраны
Na+,+H+p7±H+, + Na+p
rs °Н+,пов aNa+,p-p
-Kh.Nh = ------------
GH+,p-p • ftNa+ ,пов
(7.3-27)
(7.3-28)
и отношения подвижностей ионов натрия и водорода (uNa+/«H+) в гидратированном слое стекла. Таким образом, правильно подбирая состав стеклянной мембраны, можно изменять коэффициенты селективности.
402
Глава 7. Методы химического анализа и их применение
Рис. 7-3-9. Кислотная и щелочная погрешности для ряда стеклянных электродов при 25GC [Bates, R. G. (1973), Determination of pH, 2nd ed., New York: Wiley]. Стекло: А — Корнинг 015, HjSO«; В —корнинг 015, HC1; С — корнинг 015, 1М раствор Na+; D~ бекман СР, 1М раствор Na+; Е — стекло «L and N Black», IM раствор Na+; F — бекман типа E, IM раствор Na+.
Из-за чувствительности pH-чувствительной стеклянной мембраны к ионам натрия при высоких pH (т. е. низких #н+) и высоких flNa+> измеренные значения pH ниже истинных. Погрешность, обусловленную присутствием ионов натрия, называют «щелочной» или «натриевой». Причины возникновения «кислотной» погрешности (отклонения на графике ДрН от pH в кислой среде) не ясны. На рис. 7.3-9 показано, как зависят кислотная и натриевая погрешности от состава стекла.
Оптимальный состав стеклянной мембраны, селективной к ионам Na+:
Na2O, 11% А12Оз, 18% SiO2 71% « кг3]
и мембраны, селективной к ионам К+:
Na2O, А12О3, SiO2 ВД.^10-1]
27% 5% 68%
(7.3-29)
(7.3-30)
Перед первым использованием стеклянный электрод необходимо выдерживать в разбавленных буферных растворах для образования гидратированного поверхностного слоя. Между измерениями электроды следует хранить в воде для сохранения гидратированного поверхностного слоя.
Кристаллические (твердофазные) электроды
Чувствительный элемент (материал мембраны) кристаллического (твердофазного) ион-селективного электрода состоит из ион-проводящей, умеренно растворимой в ионогенном растворителе (вода) соли, такой, как AgCI, Ag2S, LaF3 или Ag2S + CuS, Ag2S 4- PbS и Ag2S 4- CdS. Мембрана может быть гомогенной или гетерогенной. Гомогенные мембраны изготавливают из пластинки монокристалла или спрессованной таблетки соответствующей соли. В гетерогенных мембранах электродноактивное вещество включено в инертную матрицу, например в силиконовую смолу.
Электроды с кристаллическими мембранами на основе галогенидов серебра предназначены главным образом для определения анионов, но их мож-
7.3. Электроанализ
403
Рис. 7.3-10. Отклик ион-селективно-го электрода с мембраной на основе хлорида серебра на хлорид-ионы и ионы серебра.
но использовать и для определения иона серебра или его противоиона (X ) (рис. 7.3-10).
Функции отклика электрода на ионы Ag+ и X- описываются уравнениями
RT RT
Е = const -I—— 1пеДк+ — const'-— Infix' (7.3-31)
F F
и
RT
const' = const -I—~ In KsP.AgX (7.3-32)
ZiF
где Ksp.AgX — произведение растворимости соответствующего галогенида серебра.
□ Предел обнаружения ион-селективных электродов на основе AgX
Предел обнаружения ион-селективных электродов на основе галогенидов серебра зависит от природы материала мембраны и определяется минимальной активностью ионов серебра Qae+ ты. обусловленной процессами на границе раздела мембрана/раствор. Источниками поступления ионов серебра в раствор служат
1) сам материал мембраны (его растворимость определяется константой произведения растворимости галогенида серебра);
2) соосажденные и адсорбированные на мембране ионы серебра.
Соответственно, электроды с мембранами из нерастворимых солей не являются инертными, и указанные выше реакции на поверхности обусловливают предел обнаружения таких электродов порядка 10~5-10-6 М.
Фторидный электрод—один из самых важных электродов с кристаллической мембраной, поскольку существует всего несколько методов, пригодных для простого и селективного определения фторида. Электродная мембрана состоит из пластинки из монокристалла LaF3 с добавками Eu (II) для повышения электропроводности мембраны. (При введении Eu(II) происходит замещение небольшого количества La(III).] Электродная функция основана на
404 Глава 7. Методы химического анализа и их применение
селективной адсорбции фторид-ионов «а поверхности электрода, приводящей к разделению зарядов; электропроводность мембраны обусловлена только подвижностью фторид-ионов. Внутренний раствор содержит 0,1 М NaF и 0,1 М NaCl, в него погружен хлоридсеребряный электрод.
Электродная функция выполняется в диапазоне 10°-10“6 М фторид-ионов. Единственный Мешающий ион — ОН~, поскольку он может участвовать в реакции обмена на поверхности мембраны:
LaF3 + ЗОН" La(OH)3 + 3F" (7.3-33)
Поэтому при высоких pH образуется слой La(OH)3, поскольку растворимость Ьа(ОН)з й LaF3 примерно одинакова. Потенциометрический коэффициент селективности Крон =0Д, в то время как селективность электрода к фторид-иону в присутствии других анионов на несколько порядков выше.
Известно также, что ионы Н+ реагируют с фторид-ионами с образованием HF и HF7,
н+ + 2F- HF + F" HF2 (7.3-34)
не определяемых электродом. Поэтому, если понизить pH анализируемого раствора, уменьшается концентрация свободного фторид-иона и потенциал электрода повышается. Рабочий диапазон pH фторидного электрода лежит в интервале pH 5,5-6,5, создаваемом ацетатным или цитратным буфером.
Ионы А13+ и Fe3+ уменьшают содержание свободного фторид-иона в растворе вследствие комплексообразования. Если необходимо определить общее содержание фторид-иона, следует добавить лигаяд L3, образующий более устойчивые комплексы с этими катионами и высвобождающий фторид-ионы
A1F3" + L3' Al-L + 6F- (7.3-35)
Такими лигандами могут быть цитрат или этилендиаминтетрауксусная кислота (ЭДТА). Следовательно, правильный выбор условий измерения позволяет устранить влияние pH и других комплексообразователей, таких, как А13+ и Fe3+.
Электроды с жидкой мембраной
Жидкие мембраны представляют собой трех- или четырехкомпонентные мембраны с положительно или отрицательно заряженными или нейтральными подвижными носителями (переносчиками) ионов. Подвижные носители — соединения со способными связывать ионы центрами, как правило, внедренные в пластифицированные поливинилхлоридные матрицы.
Среди заряженных переносчиков наибольший интерес представляют жидкие ионообменники, используемые в электродах для определения активности ионов нитрата и кальция. Активным компонентом мембраны нитрат-селективного электрода является раствор нитрата трис(4,7-дифенил-1,10-фенантролината)?й2+ в n-нитроцимоле, а для кальциевого ион-селективного электрода ионообменником служит додецилфосфат кальция, растворенный в диоктилфенилфосфате. Потенциалопределяющая реакция, например, для от-
7.3. Электроанализ
405
Рис. 7.3-11. Химическая структура различных ионофоров, применяемых в электродах с жидкими мембранами.
клика на ион кальция, для каждой границы раздела мембрана | раствор описывается уравнением
[(RO)2PO2 ]2Са rt 2(ЯО)2РО7(пов.) + Са2+(р-р) (7.3-36)
Ионообменник облегчает транспорт ионов кальция через мембрану. Электродная функция кальциевого электрода выполняется в интервале 10-1-10-6 М, основные мешающие ионы —Н+, Zn2+, Fe2+, Mg2+ и др.
Благодаря идее Стефанака и Саймона [7.3-5] использования антибиотиков в качестве нейтральных переносчиков в электродах с жидкими мембранами, предложено огромное количество электронейтральных лигандов с различными структурами, названных ионофорами. В качестве нейтральных переносчиков использовались антибиотики, синтетические ациклические соединения, циклические полиэфиры (краун-соединения) (рис. 7.3-11) и, с недавнего времени, производные каликс(4)-аренов. Общей чертой этих ионофоров является наличие устойчивой конформации с полярными центрами (в полостях), способными захватывать катион, который оказывается окруженным липофильной оболочкой (рис. 7.3-12). Несмотря на это, ионофоры обладают достаточной гибкостью для быстрого ионного обмена.
Ионофоры, как правило, внедряют в пластифицированную поливинилхлоридную мембрану, обычно содержащую 1 масс.% ионофора, 66 масс.% неполярного растворителя (пластификатора) и 33 масс.% ПВХ. Для уменьшения сопротивления мембраны и улучшения ионной селективности в мембрану добавляют липофильную соль. Наиболее часто используемые составы для изготовления мембран приведены в табл. 7.3-1.
466
Глава 7. Методы химического анализа и их применение
Диаметр полости (А) краун-эфиров и некоторых катионов в кристаллах
Диаметр катиона LT 13 Na* 1.9 К* 2.66 О* 2.96 w 3.34
Диаметр полости краун-эфира 14-0-4 13 15-0-5 2.0±0.2 18-0-6 2.9Ю.2 21-0-7 3.8510.45
Сандвичевый комплекс состава 1:2
«Трехпалубный» сандвичевый комплекс состава 2:3
Рис. 7.3-12. Структура комплексов ионов щелочных металлов с бис-краун-эфирами.
Таблица 7.3-1. Оптимальные составы ион-селективных мембран
Состав мембраны ~ 33%ПВХ (полимерная матрица) Материал мембраны ВМ-ПВХ ВВВС-ПВХ ПВХ-СООН ПВХ-ОН ПВХ-NHa
~ 66% пластификатора Пластификатор (неполярный растворитель) НФОЭ
~ 1% ионофора (подложки для ионов) БЭГС Соевое масло ББПА ДБС БЭГФ
~ 0,5% добавки Добавки липофильной соли Na-ТФБ К-ТХФБ К-ТТФМФБ
ВМ-ПВХ— поливинилхлорид с большой молекулярной массой; ВВБС-ПВХ — сополимер винилхлорида, винилацетата и винильного спирта; НФОЭ--2-нитрофенилоктиловый эфир; БЭГС—бис(2-этил-гексил)себацинат; ББП А — бис(1-бу тилпентил)адипат; ДБС—ди-бутилсебацинат; БЭГФ — бис(2-этилгексил)фталат; Na-ТФБ—тетра-фенилборат натрия; К-ТХФБ—тетракис(4-хлорфенил)борат калия; К-ТТФМФБ—тетракис(3,5-бис(трифторметил)фенил)борат калия.
7.3. Электроанализ
407
Потенциалопределяющий процесс на межфазной границе мембра-на/раствор:
М2+ (р-р) + пБ(мембр.) MS*+ (мембр.) (7.3-37)
где Кт1П — коэффициент распределения, aS — ионофор.
Селективность мембраны зависит от койстант устойчивости и коэффициентов распределения комплексов, образованных определяемым ионом А и мешающим ионом В в процессе ионного обмена на поверхности:
В+(р-р) + А8+(мембр.) BS+(мембр.) 4- А+(р-р) (7.3-38)
Для жидких мембран с ионофорами, образующими с катионами комплексы 1:1, коэффициент селективности определяется отношением констант устойчивости (/?) комплексов:
^А°в « ~ (7-3-39)
поскольку коэффициент распределения комплексов с ионофорами не зависит от природы центрального иона.
□ Электроды с жидкими мембранами для клинического анализа.
Молекулярный дизайн ионофоров для различных катионов позволил сконструировать высокоселективные жидкостные мембранные электроды для таких биологически важных ионов щелочных и щелочноземельных металлов, как Са2+, Na+ и К+. Аналитические характеристики калиевого электрода на основе производного бис-краун-эфира показаны на рис. 7.3-13.
Важной особенностью электродов с жидкими мембранами является возможность придавать им различные формы и размеры. Калиевые электроды типа микропипеток с головкой размером в несколько микрометров очень ценны для физиологов и биологов при контроле активности иона калия in vivo, например во внеклеточных жидкостях. Селективность электродов с жидкой мембраной по отношению к ионам калия примерно в 104 раз выше, чем к ионам натрия. Срок службы электродов с жидкими мембранами в основном зависит от липофильности ионофоров, а также всех компонентов мембраны (т. е. пластификатора, солевых добавок и т. д.).
Сложные (многослойные) ион-селективные электроды
Чувствительные потенциометрические устройства состоят из основного чувствительного элемента (ион-селективного электрода) н модифицирующего мембранного слоя, который ведет себя как дополнительная селективная граница раздела. Природа модифицирующего слоя (химически активного или неактивного) повышает селективность сигнала, и чувствительность основного элемента увеличивается при определении различных неорганических газовых и органических молекул. Эти устройства рассмотрены в разд. 7.7.
408 Глава 7. Методы химического анализа и их применение
Рис. 7.3*13. Характеристики калиевого электрода на основе бис-краун-эфира; S—наклон кривой = д^-д; is кк-коэффициент селективности, найденный при активности 0,1 М методом отдельных растворов [Guibault G.G., at al (1976) Pure Appl. Chem. 48, 127].
Прямая потенциометрия
Прямая потенциометрия позволяет определять концентрацию или активность иона, если есть селективней электрод- Метод основан на сравнении потенциала индикаторного электрода (или потенциала ячейки при условии, что все потенциалы, кроме потенциала индикаторного электрода, при измерении постоянны), измеренного в растворе определяемого иона, с потенциалами, измеренными в двух или более стандартных растворах с известной концентрацией определяемого иона. Следует иметь в виду, что поведение ион-селективных электродов, в том числе и pH-чувствительного стеклянного электрода, может отклоняться от предсказываемого уравнением Нернста.
Измерение pH
Из-за невозможности измерения активности индивидуального иона, как и потенциала изолированного электрода, значение pH на основании концентрации или активности ионов водорода экспериментально определить нельзя.
7.3. Электроанализ
409
Измеряемые значения pH фактически являются «рабочими» — величинами, определяемыми на основании значений pH, постулированных для стандартных растворов:
Е1 _ Е1
pH = pH(p-p’ + 5^W (73-4<))
где рН(р-р) — постулированное значение pH стандартного раствора, Еа—соответствующий потенциал ячейки, Е — потенциал ячейки, измеренный в растворе с неизвестным pH.
Рабочая шкала pH основана на использовании общепринятых значений pH ряда стандартных растворов известного состава, измеренных в потенциометрической ячейке без жидкостного соединения, называемой ячейкой Гарнеда:
Pt|H2(l атм)Н+(он+ — 1), Cl“ |AgCl| Ag (7.3-41)
Определение понятия «рабочее значение pH» принято Национальным институтом стандартов и технологии (США), аналогичными организациями в ряде других стран и ИЮПАК.
В большинстве случаев градуирования стеклянного электрода проводят по двум точкам. Значения pH стандартных растворов должны охватывать диапазон pH исследуемых растворов и не должны отличаться друг от друга более чем на пять единиц pH (ДрН = 5), поскольку угловой коэффициент S электродной функции' непостоянен во всем диапазоне pH.
Признание понятия «рабочего» pH означает, что электрохимически измеренный pH не вполне равен значению, определяемому активностью или концентрацией иона водорода, а скорее является результатом применения общепринятой рабочей процедуры.
Определение активности (концентрации) иона (рА)
Методом прямой потенциометрии концентрацию или активность иона при наличии ион-селективного электрода можно найти по градуировочному графику. Зависимость потенциала ион-селективного электрода от активности иона может отклоняться от нернстовской, приближенно ее можно описать уравнением
Е = const ES 1g од (7.3-42)
где Е — потенциал электрода, const—постоянная, зависящая от природы мембраны, S—угловой коэффициент электродной функции, теоретически равный 59,16 мВ/рА для однозарядного нона при 25°С (положительная для катионов, отрицательная величина для анионов), а ад — активность исследуемого иона А.
Согласно уравнению (7.3-42) зависимость Е от 1g од позволяет в достаточно широком диапазоне активности получить линейный градуировочный график, использун для этого по меньшей мере два стандартных раствора.
Определение активности иона с использованием градуировочного графика Стандартные растворы готовят последовательным разбавлением исходного раствора (например, 0,1 М), приготовленного по точной навеске соли определяемого иона. Соответствующие значения активности вычисляют, используя коэффициенты активности индивидуальных ионов, определенные независимым
410
Глава 7. Методы химического анализа и их применение
методом или вычисленные по уточненному уравнению Дебая—Хюккеля (см. разд. 4.1), но следует помнить, что все методы расчета коэффициентов активности индивидуальных ионов являются приближенными.
Проблемы, связанные с определением рА по шкале активностей, те же, что и при точном измерении pH.
Точность определения рА по шкале активностей не может быть выше точности измерения для стандартных растворов. Погрешность измерения потенциала в 1 мВ соответствует относительной погрешности 4% для однозарядного иона и 8% для двухзарядного иона.
Определение концентрации методом стандартов
Основное требование к стандартным растворам заключается в том, чтобы коэффициент активности определяемого иона был одинаковым в стандартном и анализируемом растворах; оно выполняется только в случае, если ионная сила
и в стандартном, и анализируемом растворе одинакова (см. разд. 4.1).
Есть два способа приготовления таких растворов:
1) Стандартные растворы готовят тем же способом, что н анализируемый раствор. При анализе крови обычно используют сыворотку, ионный состав которой установлен независимым методом.
2) Другой способ заключается в добавлении избытка инертного электролита (не определяемого ион-селективным электродом) и к раствору пробы, и к стандартным растворам для поддержания ионной силы столь высокой, что ее изменением в различных растворах можно пренебречь (например, разбавление 1:10 раствором 5М NaNOg).
При определении таких ионов, как S2-, CN~, F", используют многоцелевые растворы. Для определения фторид-иона в пробах воды предложены буферы различного состава для регулирования ионной силы. (Например, буфер TISAB I состоит из 1М NaCl, 0,25 М уксусной кислоты, 0,75 М ацетата натрия и 0,001 М цитрата натрия.) Стандартные растворы фторида натрия и пробу воды разбавляют буфером TISAB в соотношении 1:1, чтобы сохранить значение ионной силы постоянным и установить pH к* 5,5. Цитрат необходим для высвобождения фторид-ионов из комплексов А13+, Fe3+.
Метод добавок
Метод добавок применяют для нахождения концентрации определяемого вещества в пробах со сложной или неизвестной основой. Этот метод основан на измерении изменения потенциала электрода при добавлении известного объема стандартного раствора к известному объему пробы. Объем стандартного раствора определяемого иона должен быть малым во избежание изменения ионной силы, концентрации определяемого иона и потенциала жвдкостного соединения.
7.3. Электроанализ
411
Потенциал электрода измеряют дважды: в анализируемом растворе
Ei = const +S lg(cxfxkx) (7.3-43)
и после введения добавки стандартного раствора
Е% = const 4-Slg[(c® + /^c)fxkx] (7.3-44)
где Сх — неизвестная концентрация, fx — коэффициент активности иона, кх — доля свободного определяемого иона в присутствии комплексообразующего агента, Д с—изменение концентрации за снег добавки стандартного раствора.
Совместное решение этих уравнений
Д£ = Е2 - Я1 = Sig Сд + Ас
Сх
позволяет найти искомую концентрацию
Сх = Дс (10ЛЕ/я - 1) -1 (7.3-45)
Рекомендуется добавлять определяемый ион в количестве примерно 100% от исходного. Методом множественных добавок можно не только найти концентрацию определяемого иона, но и оценить величину S для отклика электрода.
7.3.2. Вольтамперометрия
Вольтамперометрия включает методы микроэлектролиза, в которых потенциал индикаторного электрода создается внешним источником и является известной функцией времени, а получаемые зависимости ток — потенциал и ток-время служат источником информации о составе раствора. В зависимости от вида развертки потенциала и механизма массопереноса, различают вольтамперометрию с линейной разверткой потенциала (вольтамперометрию при постоянном токе), методы со ступенчатым изменением потенциала, гидродинамические методы и инверсионную вольтамперометрию.
Вольтамперометрические методы—точные методы определения многих органических и неорганических веществ (в основном, ионов металлов), которые можно электрохимически окислить или восстановить (электроактивные вещества) в следовых количествах. С помощью вольтамперометрии возможно одновременное определение нескольких веществ. Если рабочий электрод является микроэлектродом, объем анализируемого раствора сравнительно велик, а время анализа мало, концентрация определяемого вещества в объеме раствора во время анализа меняется незначительно. Вольтамперометрические методы умеренно селективны, но селективность можно значительно повысить, например сочетанием жидкостной хроматографии с электрохимическим детектированием.
Развитие вольтамперометрии ведет начало от первой работы (1922 г.) Ярослава Гейровского (вольтамперометрия при постоянном токе на ртутном капающем электроде), которому была присуждена Нобелевская премия в 1959 г.
412
Глава 7. Методы химического анализа и их применение
Рис. 7.3-14. Схема экспериментальной установки для работы при контролируемом потенциале; ИЭ— индикаторный электрод; ПЭ — противоэлектрод; ЭС—электрод сравнения (Bard, A.J. and Faulkner, L.R. (1980), Electrochemical Methods, New York, Wiley].
□ Электрохимическая вольтамперометрическая ячейка — электролитическая ячейка с одним поляризуемым электродом (ртутным капающим электродом или платиновым микроэлектродом)
Вольтамперометрическая ячейка состоит из анализируемого раствора, двух электродов разного размера и электрода сравнения. Микроэлектрод является индикаторным электродом, в то время как другой токопроводящий электрод служит вспомогательным или противоэлектродом. Потенциостат контролирует напряжение между индикаторным электродом и вспомогательным электродом, поддерживая разность потенциалов между индикаторным электродом и электродом сравнения в соответствии с заранее выбранной разверткой напряжения, обеспечиваемой программатором (или микрокомпьютером). Разность потенциалов между индикаторным электродом и электродом сравнения измеряют высокоомным контуром обратной связи (ток в нем отсутствует). Потенциостат считается «активным инструментом», который извне контролирует потенциал индикаторного электрода. На рис. 7.3-14 показана схема установки для работы при контролируемом потенциале. Для вольтамперометрического анализа водных растворов можно использовать более простую двухэлектродную схему (индикаторный электрод и электрод сравнения).
Индикаторный электрод—идеально поляризуемый электрод, т. е. электрод, характеризующийся большим сдвигом потенциала при протекании бесконечно малого тока. Поляризация электрода отвечает горизонтальному участку на кривой г~Е электрода и определяет диапазон потенциалов, пригодный для аналитических целей, поскольку в нем можно изучать процессы электрохимического окисления или восстановления определяемого вещества. Напротив, идеально неполяризуемый электрод — это электрод с фиксированным потенциалом, не изменяющимся при протекании относительно небольших токов. Неполяризуемые электроды, такие, как электроды второго рода или электрод из донной ртути с большой поверхностью, используют в вольтамперометрии в качестве электродов сравнения. Вспомогательным электродом (токопроводящим противоэлектродом) может служить, например, платиновая проволока.
7.3. Электроанализ
413
Вольтамперограммы обычно регистрируют в неперемешиваемом растворе, содержащем большой избыток инертного электролита, называемого фоновым электролитом.
Индикаторные электроды
В вольтамперометрии используют различные индикаторные электроды- наиболее часто применяемые электроды обсуждаются ниже.
Ртутный капающий электрод
В классической полярографии индикаторным электродом является ртутный капающий микроэлектрод. Ртутная капля образуется на конце стеклянного капилляра (длиной 10-20 см, внутренним диаметром 0,05 мм), соединенного гибкой трубкой с резервуаром со ртутью. Ртутные капли имеют воспроизводимый диаметр и время жизни от 2 до 6 с. Время жизни капли зависит от высоты столба ртути над капилляром, т. е. гидростатического давления ртути. Иногда используют механический молоточек, контролирующий время жизни капель. Ртутный капающий электрод обладает следующими преимуществами: 1) постоянное обновление поверхности электрода предотвращает загрязнение поверхности электрода, что выражается в высокой воспроизводимости зависимостей ток — потенциал; 2) перенапряжение водорода на ртути в водных растворах велико, поэтому можно изучать процессы восстановления элек-троактивных веществ с более отрицательными потенциалами, чем обратимый потенциал разряда ионов водорода. В кислом растворе, например, 0,1 М НС1 выделение газообразного водорода наблюдается при потенциалах отрицательнее —1,2 В; 3) ртуть образует амальгамы со многими металлами, понижая их потенциал восстановления.
Интервал поляризации ртутного электрода (отн. НКЭ) в водном растворе в отсутствие Ог~от 4-0,3 и примерно до —2,7В. В катодной области он ограничен потенциалами восстановления катионов фонового электролита (фоновый электролит добавляют для обеспечения диффузионного массопереноса). Наиболее отрицательных потенциалов можно достичь, применяя соли тетра-алкиламмония. Окисление ртути
2Hg Hg^+ + 2е~ (7.3-46)
ограничивает интервал поляризации в анодной области потенциалов. Отсюда можно заключить, что на ртутном микроэлектроде в основном можно определять восстанавливающиеся электроактивные вещества.
Статический ртутный капельный электрод
В конструкции статического электрода, как и ртутного капающего, имеется резервуар со ртутью и стеклянный капилляр, ио воспроизводимость размера капли и период капания контролируются точнее (рис. 7.3-15). Это обеспечивается с помощью соленоида, находящегося между ртутным резервуаром и капилляром, и механического молоточка у капилляра. Таким образом, правильно задав времена включения и выключения соленоида, можно регулировать время отрыва капли, и устройство можно использовать как статический электрод
414
Глава 7. Методы химического анализа и их применение
Рис. 7,3-15. Схематическое изображение коммерческого статического ртутного капельного электрода (механический молоточек для сбрасывания капли не показан) [по данным EG&G Princeton Applied Research, Princeton N. Y.]. 1 — ртуть; 2 — соленоид; 3—поршень; 4—направляющая втулка; 5 — пружина; 6 — полиуретановый наконечник; 7—затвор капилляра; 8 — держатель; 9 — манжета (на капилляре); 10 — крепление манжеты; 11 — гайка; 12 — капилляр.
(висячая капля) или капающий электрод с контролируемым периодом капаг ния.
Статический ртутный электрод обладает всеми характеристиками капающего ртутного электрода, что особенно удобно для рутинных анализов. Дополнительная особенность, связанная с постоянством поверхности капли ртути, будет обсуждена в связи с высокочувствительными импульсными методами измерения тока (см. раздел «Дифференциальная импульсная полярография», с. 428).
Твердые электроды
Для определения окисляющихся веществ обычно применяют твердые электроды. Из твердых электродов (например, Pt, Au, С), используемых в электролизе, в аналитической вольтамперометрии широкое применение нашли различного вида графитовые электроды. Это обусловлено широким диапазоном анодных потенциалов, низким электрическим сопротивлением и легкостью обновления поверхности электрода. Электроды из угольной пасты [7.3-6] и стеклоуглеродные электроды [7.3-7J используют для мониторинга электроокисле-
7.3. Электроанализ
415
Рис. 7.3-16. Тонкослойная ячейка с графитовым (стеклоуглеродным) индикаторным электродом:
ИЭ —индикаторный электрод
ВЭ — вспомогательный электрод ЭС —электрод сравнения [по данным Bioanalytical Systems Inc. West Lafayette, IN].
ВЭ
ния как в неперемешиваемом растворе, так и в потоке электролита, например, в ВЭЖХ. В последнем случае электрохимическое детектирование с использованием тонкослойной ячейки (рис. 7.3-16) позволяет определять нано- и пикограммовые количества биогенных аминов.
В анодной области интервал поляризации графитовых электродов ограничен потенциалом выделения 02(4-1,5В).
Вольтамперометрия с линейной разверткой потенциала
На ячейку подают линейную развертку постоянного напряжения (т. е. Е линейно зависит от t). Ячейка состоит из индикаторного электрода (поляризуемый микроэлектрод, например, ртутный капающий электрод, статический ртутный электрод или любой твердый .микроэлектрод), вспомогательного электрода и электрода сравнения — электроды с большой площадью поверхности и относительно неполяризуемые.
Вольтамперометрия с линейной разверткой потенциала на ртутном капающем электроде (полярография)
В полярографии ртутный капающий электрод служит индикаторным, а донная ртуть — вспомогательным электродом. При анализе водных растворов донная ртуть обычно является и электродом сравнения. Для приложенного к ячейке потенциала можно записать
Ецрмл = Е + IR = Ер&ап 4- 7) 4- iR (7.3-47)
где iR — омическое падение потенциала (£%>м)* требуемое для протекания тока через ячейку. Его значение при микроамперных токах i существенно, только если сопротивление раствора R велико. Сопротивление раствора снижают
416
Глава 7. Методы химического анализа и их применение
Рис. 7.3-17. Линейная развертка потенциала.
Рис. 7.3-1В. Полярограммы, снятые в растворе, содержащем 5-10 4 М Cd2+ и IM НС1 (I) или 0,1 М НС1 (2) [Sawyer, D.T., Roberts Jr., JX. (1980),Experimental Electrochemistry for Chemists, New York, Wiley].
добавлением инертного (фонового) электролита; -Еравн — потенциал при разомкнутой цепи, т. е. равновесный потенциал; г) — перенапряжение.
Перенапряжение является мерой энергии активации гетерогенной химической реакции (реакции переноса заряда), и оно необходимо для преодоления активационного барьера реакции. Величина перенапряжения электродной реакции зависит от материала электрода.
Линейная развертка напряжения (Е^ = Ei 4- vt, где ^ — начальный потенциал, a v — скорость поляризации (развертки)) показана на рис. 7.3-17. Ток измеряют непрерывно в течение времени жизни каждой капли (рис. 7.3-18, кривая 1). Получаемые кривые зависимости тока от потенциала (полярограммы) используют для анализа.
7.3. Электроанализ
417
Массоперенос растворенного вещества к электроду осуществляется за счет диффузии, механической конвекции и электростатического притяжения. Для полярографического анализа важно устранить две последние причины, чтобы массоперенос определяемого вещества к поверхности электрода осуществлялся только в результате диффузии. Электростатическое притяжение (или отталкивание) ионов определяемого вещества электродом становится пренебрежимо малым в присутствии фонового электролита с концентрацией в 50-100 раз выше концентрации определяемого вещества, например, 1М НС1.
Полярограммы (зависимости i от Ё)
Зависимость тока от потенциала в условиях диффузионного массопереноса называется полярограммой. На рис. 73-18 приведены полярограммы, снятые в неперемешиваемом растворе, содержащем 5 • 10“3М Cd2+ и 0,1 М НС1 (кривая 1) и 0,1 М НС1 (фоновый электролит) (кривая 2).
□ Зависимость ток — потенциал в условиях диффузионного массопереноса называется полярограммой.
Восстановление Cd2+ на капающем Hg электроде
Cd2++ 2е“^ Cd(Hg), O + ne'^R (7.3-48)
Кь
вызывает ступенчатый рост тока.
□ Классическая полярограмма состоит из трех участков: участок остаточного тока при небольших отрицательных потенциалах; участок предельного тока, где наблюдается отклонение от закона Ома из-за поляризации катода; восходящий участок, где ток контролируется переносом заряда. Последний и содержит качественную (потенциал полуволны) и количественную (диффузионный ток) информацию.
Полярограмма состоит из трех участков:
а) остаточный ток при небольших отрицательных потенциалах электрода; б) ток, контролируемый переносом заряда, при значениях электродного потенциала, соответствующих или превышающих потенциал восстановления Cd2+. На этом участке ток растет экспоненциально с увеличением потенциала (скорость реакции контролируется ее кинетикой, и стационарный ток обеспечивается диффузией);
в) предельный ток, контролируемый скоростью диффузии.
Изменение концентрации Cd2+ на поверхности электрода (ccd(xx=o)) В зависимости от приложенного потенциала схематически показано на рис. 4.4-7,а. Рост тока при потенциале около —1,2В вызван восстановлением Н+. Осцилляции тока обусловлены тем, что полярограмма записана на капающем ртутном электроде с изменяющейся площадью поверхности. Когда капля падает, ток достигает нуля.
14 Аналитическая химия. Том 1
418
Глава 7. Методы химического анализа и их применение
Остаточный ток (гг)
Остаточный ток состоит из фарадеевского тока и тока заряжения (емкостного тока). Фарадеевская компонента остаточного тока обусловлена электрохимическим восстановлением или окислением примесей из фонового электролита. Концентрация фонового электролита относительно высока по сравнению с концентрацией определяемого вещества, поэтому вклад примесей в измеряемый остаточный ток может быть значительным.
Емкостный ток—это ток, требуемый иа заряжение или разряд двойного электрического слоя (являющегося конденсатором), формируемого на границе раздела индикаторный электрод | электролит. Изменение емкостного тока в 0,1 М НС1 в зависимости от потенциала электрода показано на рис. 7.3-18, кривая 2. (Нулевое значение тока наблюдается при —0,4 В и совпадает с элек-трокапиллярным максимумом, при котором поверхность ртутной капли не заряжена.) Каждая капля заряжается или разряжается, и емкостный ток при регистрации полярограммы протекает постоянно. Его величина зависит от потенциала и площади поверхности электрода:
и д
ч = kCi аГ(Кпркл “ Ех} (7'3-49)
где А—площадь поверхности электрода, Ci — интегральная емкость двойного слоя, т — скорость вытекания ртути (мг/с), t—время жизни капли (с) и Ех — потенциал нулевого заряда (потенциал электрокапилляриого максимума).
Для капающего ртутного электрода
А 0,85m2/3t2/3 (7.3-50)
Следовательно,
^ = 0,85т»2/3Г1/3 at
и
ic = /гС,т2/3Г1/;,(Е - EJ (7.3-51)
ТЬк; контролируемый переносом заряда
Рассмотрим процесс восстановления на электроде:
O + ne-₽±R (7.3-52)
Кь
где kf—константа скорости прямой реакции, а кь — константа скорости обратной реакции.
Если Е^пркл < £?равн, т. е. Дприл отрицательнее равновесного потенциала (измеренного при г ~ 0), через ячейку протекает стационарный ток, величина которого зависит от скоростей таких процессов, как перенос электронов на электроде и массоперенос окисленной формы из объема раствора к поверхности электрода, и устанавливается новое равновесие:
Впрня = Е° - - — 1g (7.3-53)
П/ Со(®1=0)
7.3. Электроанализ
419
где cr^o) и ео(ж_о)—концентрации окисленной и восстановленной форм у поверхности электрода (ат = 0).
Величина суммарного тока определяется уравнением
г — nFA{kfCQ — кьск)
(7.3-54)
где Л—площадь поверхности электрода.
Константы скорости реакции переноса заряда зависят от Епря„-.
kf — к® exp
апГКприл1 RT J
(7.3-55)
къ = *b“exp R1 (7.3-56)
где — константа скорости, измеренная при ^прИл = 0, т. е. константа скорости соответствующей химической реакции, а — множитель, отвечающий доле электродного потенциала (межфазного потенциала), дающий вклад в соответ-
ствующую химическую реакцию.
Эти уравнения показывают, что электродный потенциал влияет на константы скорости, поскольку изменяется энергия активации. Например, смещение потенциала катода к более отрицательным значениям облегчает процесс
восстановления, поскольку потенциальная энергия увеличивается на величину anFE, что проявляется в снижении энергии активации. В то же время, сдвиг потенциала электрода к более отрицательным значениям снижает скорость анодной реакции.
Соответственно,
i = nF/{roA:°exp[-«nF7;„p„.,//IT) -cR^exp[(l - a)nFE„p„„/RT]} (7.3-57)
Из уравнения (7.3-57), уравнения Фольмера—Батлера, следует, что ток в этой области изменяется экспоненциально с изменением £прил из-за того, что скорость электродной реакции меньше скорости диффузии определяемого вещества.
Предельный ток
Скорость электродной реакции зависит от Еприл по уравнению Фольмера— Батлера (ур. 7.3-57), обсужденному выше. Для случая, когда перенос заряда и сопряженные химические реакции протекают очень быстро, скорость электродной реакции v контролируется скоростью массопереноса (цМп)
v = VM„ = —(7.3-58) nFA
Полярограммы регистрируют в иеперемешиваемых (покоящихся) растворах в присутствии фонового электролита с концентрацией в 50-100 раз выше концентрации определяемого вещества; поэтому возникающий при электролизе градиент концентрации контролирует скорость диффузии определяемого вещества к электроду:
= то [со - со(х=о)] (7.3-59)
где со — концентрация окисленной формы О в объеме раствора, Со(ж=о) — концентрация ее на поверхности электрода, а то — число переноса.
420
Глава 7. Методы химического анализа и-их применение
Значение со(ж^о) зависит от потенциала электрода. Наибольшая скорость массопереноса—при со(я=о) — 0. Ток достигает максимального значения, когда скорость диффузии окисленной формы становится постоянной; его называют предельным током (рис. 7.3-18, кривая 1).
Выражение для предельного тока выводится на основе законов диффузионной кинетики с привлечением первого и второго законов Фика. Для фарадеевского тока в момент времени t можно записать
i = nF A(7.3-60) гДе = (^)(x==ot) — поток вещества, N — количество молей.
Согласно первому закону Фика, у поверхности электрода (х = О)
Л*-»,.) = = D° (73-61)
\ dt У x—0,t \ дх Jx=O,t
Градиент концентрации у поверхности электрода как функцию времени можно найти на основании второго закона Фика:
(§)..-(₽),
Уравнение для предельного диффузионного тока и изменение Концентрации co(x,t) получают, решая уравнения для линейной диффузии при граничных условиях
COfoO) = СО> 1’пп СО(я=0) — СО, Со(0,1) — 0 (для t > 0)
Полагая, что диффузия к поверхности растущей капли ртути происходит так же, как и к плоскому электроду, Ильковйч вывел уравнение для диффузионного предельного тока:
td = knDll2comi,3tlle (7.3-63)
где к — постоянная Ильковича (fc = 706 для мгновенного и 607 для среднего диффузионного тока); D — коэффициент диффузии (см2/с), со — концентрат ция вещества в объеме раствора (М), т—скорость капания ртути (мг/с), i — период капания (с).
Среди факторов, влияющих иа величину диффузионного тока, наиболее важным является концентрация определяемого вещества: диффузионный ток пропорционален концентрации определяемого вещества в растворе. Эта четкая связь между id и концентрацией электроактивного вещества, описываемая уравнением Ильковича, является основой количественного полярографического анализа.
При определении id следует помнить, что a) id = it — ir, т. е. значение остаточного тока ir следует всегда вычитать из значения предельного тока ii; б) метод измерения id должен быть одним и тем же на протяжении всей серии опытов. Поправку иа гг можно сделать или экстраполяцией его значения на потенциал, при котором графически определяется id, или измерением в чйстом фоновом электролите.
Значения т и t зависят от геометрии капилляра ртутного капающего электрода. Произведение называют постоянной капилляра, она отражает
7-3. Электроанализ
421
влияние характеристик электрода на диффузионный ток. Постоянная капилляра и, следовательно, меняются как квадратный корень из высоты ртутного столба h. Изучая зависимость га от Л, можно показать, контролируется ли ток диффузией.
Диффузионный ток увеличивается с повышением температуры примерно на 2% на °C, поскольку с повышением температуры растет коэффициент диффузии D. Таким образом, изменение температуры может влиять на результаты анализа. Изучение влияния температуры иа диффузионный ток показало, что для определения га с погрешностью, меньшей 1%, температуру электрохимической ячейки следует поддерживать в пределах ±0,5°С.
Потенциал полуволны
Для обратимой электродной реакции в интервале потенциалов, соответствующих i > 0 и г < га, зависимость соотношения концентраций окисленной формы 00(2=0) и восстановленной формы от потенциала электрода можно описать уравнением Нериста:
Е„ркЛ = Е^-^1в^> пЕ со(х=о)
При протекании фарадеевского тока возникает градиент концентраций, и диффузия обеспечивает подвод электроакгивиых частиц к электроду.
Рассмотрим электродную реакцию:
О 4- пе" R
где R—растворимый продукт электродной реакции.
Ток восстановления на восходящем участке полярограммы определяется соотношениями
(7.3-64)
(7.3-65)
i = kD^\co - со(х=0)) (7.3-66)
i = ^/2cr(i=0) (7.3-67)
Если реакции обратимы, то уравнение, описывающее зависимость тока от потенциала на восходящем участке полярограммы, имеет вид
0,05916 i t 0,05916 D1^2 п id -i п дУ2
При i = гл/Ч, если k = k' уравнение принимает вид „ „ „ 0,05916, £>i/2
Е = Е1/2^Е° + -1-----Ig-^
(7.3-68)
(7.3-69)
и поскольку Dq = Dr, toc
Dj/2 = D'
(7.3-70)
Потенциал полуволны не зависит от концентрации и практически равен стандартному электродному потенциалу соответствующей полуреакции. Однако следует заметить, что Ец2 / D®, если продукт электродной реакции образует с ртутью амальгаму.
Поскольку потенциал полуволны не зависит от концентрации определяемого вещества в растворе, а зависит в первую очередь от его природы, его можно
422
Глава 7. Методы химического анализа и их применение
использовать для идентификации вещества, участвующего в электродной реакции. Однако это следует делать с осторожностью, поскольку, например, pH раствора, константа равновесия гомогенной реакции в растворе могут влиять на £1/2- Влияние pH заметно в реакциях электрохимического восстановления органических веществ, протекающих с участием ионов водорода:
О + пе~ + 2Н+ <=t RH2 (7.3-71)
Следовательно, £j/2 = Е° — pH. Во избежание влияния pH на Еу2 требуется буферный раствор с высокой буферной емкостью.
Факторы, влияющие на форму полярограммы
При измерении диффузионного тока id следует учитывать, что на форму полярограммы влияют
— остаточный ток;
— полярографические максимумы (максимумы тока);
— присутствие растворенного кислорода.
Полярографические максимумы различной формы появляются в области предельного тока, а также в начале плато на полярограмме. Они наблюдаются на полярограммах катионов, анионов и нейтральных молекул.
Интерпретация полярографических максимумов, т. е. причин их возникновения, еще не достаточно разработана1 *. В общем случае, их возникновение связывают с конвекцией слоя раствора у поверхности индикаторного электрода, вызванной неравномерным распределением заряда на поверхности ртутной капли. Максимумы можно подавить добавлением следовых количеств полярографически неактивных поверхностно-активных веществ (ПАВ), таких, как желатин, метиловый красный и другие красители или Тритон Х-100 (доступное ПАВ). Обычно к 10 мл раствора добавляют 0,1-0,2 мл 0,5%-ного желатина. При концентрациях выше некоторого порогового значения (> 10“4%) диффузионный ток начинает зависеть от концентрации ПАВ из-за изменения вязкости раствора при добавлении ПАВ. Следует отметить, что ПАВ необходимы только в классической полярографии при работе с ртутным капающим электродом.
Растворенный кислород в растворе, контактирующем с воздухом, восстанавливается на Hg капающем электроде с образованием двух волн одинаковой высоты:
О2 + 2Н+ + 2е~ -+ Н2О2, Е1/2 « 0,1 В(отн. НКЭ) (7.3-72)
Н2О2 + 2Н+ + 2е~ 2Н2О, £i/2 « 0,9 В(отн. НКЭ) (7.3-73)
Поскольку волны восстановления кислорода появляются в диапазоне 0-1 В, кислород мешает определению многих веществ, восстанавливающихся в этом диапазоне потенциалов. Эти помехи можно устранить, удалив О2. Из растворов с любым pH кислород удаляют продуванием азота или аргона в тече
1 С этим согласиться трудно. См., например, Я. Гейровский, Я. Кута. Основы полярогра-
фии. М.: Мир. 1965. — Прим, перев.
7.3. Электроанализ
423
ние 10 мин. В процессе измерений газ продолжают продувать над поверхностью раствора. Из щелочных растворов кислород удаляют с помощью Na2SO3 (к 10 мл раствора добавляют 0,2 мл насыщенного ИагБОз).
Полярография как метод анализа
Многие неорганические и органические соединения полярографически активны, т. е. вступают в электрохимические реакции в интервале поляризации ртутного капающего электрода. Этот интервал ограничен значениями потенциала окисления ртути (+0,3 В отн. НКЭ) и потенциалами восстановления растворителя или фонового электролита в пределах —1,8 В и —2,2 В (отн. НКЭ). Типичные полярограммы неорганических веществ показаны на рис. 7.3-19. Примеры полярографически активных функциональных групп даны в табл. 7.3-2. Наличие одной и более таких групп в молекуле и обеспечивает ее полярографическую активность.
□ Возможен анализ смесей (см. рис. 7.3-19).
Общие принципы приготовления растворов для полярографических измерений:
— раствор должен содержать полярографически неактивные фоновые электролиты, такие, как соли, кислоты или буферы;
- в растворе должно присутствовать некоторое количество поверхностноактивного вещества для подавления максимумов;
— предварительно необходимо удалить растворенный кислород.
Рис. 7.3-19. Полярограмма раствора, содержащего примерно по 0,1 ммоль/л сереб-ра(1), таллия(1), кадмия(П), никеля(П) и цинка(П) на фоне раствора 1М NH3/IM NH4CI, в присутствии Тритона Х-100 (I) и полярограмма чистого фона (2) [Meites, L. (1967), Polarographic Techniques, 2nd ed., New York: Wiley].
424
Глава 7. Методы химического анализа и их применение
Таблица 7.3-2. Некоторые полярографически активные функциональные группы
1 ФС=О ^СНО сх„ —NO2
1 х Фс=с' N- -NO
с=с- —C=N 1 сх 1 —NHOH
X 1 1 / ^С=С-С=С' х 1 1 —N=N- 1 1 —ONO
^с-с-с-о —о—о— 1 —ONO2
1 1 ос-со -s-s-X — Атом галогена -NO=N-
Для определения органических соединений часто используют неводные растворители, такие, как спирты, смесь спирта и воды, ацетонитрил, диметил-формамид, а в качестве фоновых электролитов наиболее удобны перхлорат тетраалкиламмония и перхлорат лития. Для устранения проблем, связанных с омическим падением напряжения iR, требуется трехэлектродная схема.
Полярограммы имеют две четкие характеристики, а именно Ецъ и которые и служат основой для полярографического анализа. Значение характеризует электроактивное вещество и среду, в которой проводится анализ. Таким образом, экспериментально найденное значение £^у2 можно с некоторой долей осторожности на основе табулированных значений Еуъ использовать для идентификации неизвестного вещества.
Полярография—метод количественного анализа, основанный на линейной зависимости от концентрации определяемого вещества в растворе. Для оценки концентрации можно использовать градуировочный график или метод добавок.
В условиях, контролируемых диффузией, для определения веществ при концентрации 5 -10-5-5 • 10-3 М применима классическая полярография. Предел обнаружения 1 • 10“б-5 • 10-5 М определяется значением остаточного тока.
Разрешающая способность метода зависит от разности потенциалов полуволн E\j2 электроактивных веществ А и В и соотношения их концентраций. Две полярографические волны разделяются, если Д-Ец/г > 150 — 200 мВ, а концентрации А и В примерно равны. Но если их концентрации значительно различаются, возможность одновременного определения зависит от того, является ли потенциал полуволны вещества с большей концентрацией менее или более отрицательным, чем потенциал полуволны второго определяемого вещества. •
7.3. Электроанализ
425
Влияние комплексообразующих веществ иа потенциалы полуволн восстановления ионов металлов можно использовать для разделения перекрывающихся волн. Классическим примером является разделение полярографических волн свинца и таллия на ртутном капающем электроде. В кислых и нейтральных растворах потенциалы полуволн свинца и таллия практически одинаковы и их волны перекрываются. В то же время в щелочном растворе свинец образует гидроксокомплекс, а таллий остается в виде акваиона. За счет этого между потенциалами полуволн возникает разница порядка 300 мВ, что вполне достаточно для их количественного определения при примерно одинаковых концентрациях.
Измерение диффузионных токов (id)
При измерении диффузионных токов следует
1) учесть остаточный ток tr и вычесть его значение из предельного тока ii, т. е. id = it — iti
2) использовать один и тот же метод оценки для всей серии опытов.
Для измерения id предложены различные графические методы. Все они основаны на предположении, что остаточный ток линейно растет с повышением потенциала, и, следовательно, его значение при любом потенциале можно учесть экстраполяцией.
Точность полярографического анализа
Точность полярографического определения концентрации зависит в первую очередь от воспроизводимости величины id и погрешности графического способа оценки диффузионного тока. Первая из них высока, благодаря постоянно обновляющейся поверхности электрода. Пря контролируемых условиях эксперимента погрешность определения концентрации составляет 1-5%.
Вольтамперометрия с линейной разверткой на стационарных электродах
В разное время в различных вольтамперометрических методах применяли огромное количество твердых электродов. Их возрастающую популярность можно объяснить тем, что окисление многих органических веществ нельзя изучать на ртутном электроде из-за ограниченного диапазона анодных потенциалов. Наиболее широко применяют твердые микроэлектроды из благородных металлов (платина и золото), а также угольные (графитовые) электроды. Платину и золото используют в основном в неводных средах, тогда как угольные электроды применяют в водных растворах.
На индикаторный электрод с постоянной площадью поверхности подают линейную развертку потенциала:
Et = Ei±vt (7.3-74)
где Ei—начальный потенциал, и — скорость развертки; обычно она составляет 0,001-0,1 В/с, означает направление изменения потенциала, t —время электролиза.
♦26
Глава 7. Методы химического анализа и их применение
Рис. 7.3-20. Типичная кривая ток-потенциал (вольтамйерограмма) при линейной развертке потенциала на электроде с постоянной площадью поверхности; Ei — начальный потенциал, V — скорость развертки.
Рассмотрим восстановление вещества О в растворе, содержащем избыток фонового электролита. Раствор не перемешивают, т. е. созданы условия для линейной диффузии. Значение Ei выбрано таким, чтобы электродная реакция не протекала.
Типичная зависимость ток — потенциал (вольтамперограмма с линейной разверткой потенциала) показана на рис. 7.3-20. Ее можно интерпретировать на основе кинетики диффузионного массопереноса.
На вольтамперограмме есть три четких участка. При потенциалах, более положительных, чем Е° окисления О, протекает только остаточный ток (участок А~В). При приближении потенциала электрода к Е° начинается восстановление О и возникает фарадеевский ток (участок В-С). На этом участке реакция переноса заряда протекает быстро и ток экспоненциально растет с повышением потенциала электрода. Протекание тока снижает поверхностную концентрацию О и увеличивает градиент концентрации, вызывая диффузию к поверхности электрода. При Е > Е°, поверхностная концентрация О приближается к нулю и скорость массопереноса к поверхности электрода достигает максимального значения. При дальнейшем увеличении потенциала ток снижается из-за увеличения толщины обедненного слоя (участок C-D).
Ток пика ip, измеренный в условиях диффузионного контроля, описывается уравнением Рэндлса—Шевчика.
ip = kb^Av^c (7.3-75)
где к — постоянная, называемая константой Рэндлса—Шевчика (fc = 2,7-10-2), D—коэффициент диффузии, п — число электронов, участвующих в электродной реакции, v — скорость изменения потенциала (скорость поляризации, скорость развертки), а с—концентрация электроактнвного вещества.
Из уравнения следует, что ip линейно зависит от концентрации, если A, D и v постоянны.
Электроды с постоянной площадью поверхности обладают тем преимуществом, что остаточный ток на них на порядок ниже, чем на ртутном капающем электроде. Для аналитической химии зто очень важно, поскольку предел обнаружения удается снизить на порядок. Недостатком электродов с постоянной
7.3. Электроанализ 427
площадью поверхности является то, что в некоторых случаях их поверхность может загрязняться продуктами электродной реакции, образующими на поверхности электрода нерастворимый слой. Поверхность электрода обновляют полировкой; однако обновленная и исходная поверхности могут отличаться (на 5-10%). В этом случае требуется повторная градуировка. Загрязнение поверхности в результате так называемого «пленочного эффекта» можно уменьшить, снизив концентрацию определяемого вещества.
Вольтамперометрия с треугольной разверткой поляризующего напряжения на стационарном электроде называется циклической вольтамперометрией. Этот метод позволяет изучать механизмы электродных реакций.
Методы ступенчатого наложения потенциала
Предел обнаружения в полярографии определяется величиной остаточного тока, точнее током заряжения. Различие временных характеристик емкостного тока и фарадеевского тока положено в основу новых способов снижения предела обнаружения в полярографии [7.3-8, 7.3-9}. Они основаны на измерении тока как функции времени (хроноамперометрия) после наложения импульса потенциала. Ток измеряют не непрерывно, а в течение некоторого интервала времени.
□ Для понижения предела обнаружения следует каким-либо способом снизить остаточный ток.
Так называемая стробированная постояннотоковая полярография (раньше ее называли таст-полярографией) использует тот факт, что диффузионный ток id на ртутном капающем электроде за время жизни ртутной капли растет пропорционально t1/6, в то время как емкостный ток уменьшается пропорционально t”1/3. Это позволяет, измеряя ток в конце периода жизни капли, снизить предел обнаружения на один порядок из-за улучшения соотношения id/v
При ступенчатом наложении потенциала (рис. 7.3-21) на электрод с постоянной площадью поверхности в неперемешиваемом растворе, содержащем фоновый электролит и электроактивное вещество, Еу выбирают на участке потенциалов, где фарадеевские процессы отсутствуют, а Ег — на участке восстановления О. Изменение диффузионного тока id во времени на электроде с постоянной площадью поверхности описывается уравнением Коттреля, выведенным им на основании закона линейной диффузии:
qFAD^Cg
7ft1/2
Из уравнения Коттреля следует, что фарадеевский ток уменьшается пропорционально t-1/2, поэтому правильный выбор времени измерения тока позволяет улучшить соотношение фарадеевского и емкостного токов.
Дифференциальная импульсная полярография
Развертка потенциала, налагаемого на электрохимическую ячейку в дифференциальной импульсной полярографии, показана на рис. 7.3-22. Импульсы по-
42»
Глава 7. Методы химического анализа и их применение
Рис. 7.3-21. Вольтамперометрия со ступенчатым наложением потенциала, а —развертка напряжения; б — изменение концентрации электроактивного вещества при потенциале в — результирующая г(£)-кривая.
Рис. 7.3-22. а — развертка потенциала в дифференциальной импульсной полярографии; п и 72 — времена измерения тока; б — дифференциальная импульсная полярограмма.
7.3. Электроанализ
429
тенциала ДЕ небольшой фиксированной амплитуды (10-100 мВ) периодически налагают на постоянное напряжение, линейно увеличивающееся со скоростью 0,1-0,2В/мин. Продолжительность импульсов составляет 5-100 мс.
Индикаторным электродом обычно служит ртутный капающий электрод с контролируемым временем жизни капли (статический ртутный капельный электрод). За время жизни каждой капли подают один импульс, поэтому требуется синхронизация времени жизни капли с импульсами.
Величину тока измеряют дважды за время жизни каждой капли; перед наложением импульса и затем в последний период продолжительности импульса. Прибор фиксирует разность токов Дг на каждый импульс как функцию линейно увеличивающегося потенциала. В результате получают производную кривую ток-потенциал, т. е. дифференциальную импульсную вольтамперограмму (рис. 7.3-22). Ступеньки на вольтамперограмме отвечают временам жизни капель.
Величина тока пика (максимальное значение тока) описывается уравнением
(7-3-77)
где ДЕ — амплитуда импульса, тз — продолжительность импульса, А—усредненная площадь поверхности в продолжение импульса, а со — концентрация определяемого вещества; остальные обозначения общепринятые.
Из уравнения (7.3-77) следует, что высота пика прямо пропорциональна концентрации определяемого вещества в растворе.
Ширина пика на половине высоты равна
НТ
^1/2 = 3,52— (7.3-78)
Пг
Подставляя значения постоянных в уравнение, получаем значение Wi/г, равное 90,4 мВ при п = 1 и 45,2 мВ при п = 2.
Потенциал пика зависит от ДЕ:
д г
Е„ = Е112-^ (7.3-79)
т. е. потенциал пика вещества, участвующего в электрохимическом восстановлении смещается к менее отрицательным значениям при увеличении амплитуды импульса ДЕ.
Дифференциальная импульсная полярография обладает более высокой разрешающей способностью, чем нормальная импульсная или классическая полярография. При ДЕ1/2 « 90 мВ пики на дифференциальной импульсной полярограмме, в отличие от классической полярографии, четко разделяются, и их разделение не зависит от соотношения концентраций соответствующих электроактивных веществ.
Основным достоинством дифференциальной импульсной полярографии является ее высокая чувствительность. Предел обнаружения на порядки ниже, чем в классической полярографии. Это обусловлено в первую очередь увеличением фарадеевского тока при наложении импульса потенциала (ур. 7.3-77), а также правильным выбором интервала времени для измерений.
430 Глава 7. Методы химического анализа и их применение
Суммируем преимущества дифференциальной импульсной полярографии:
— улучшенная селективность, четкое разделение пиков на полярограмме;
— низкий предел обнаружения, порядка 10“8М;
— возможность использования фонов с низкими концентрациями вплоть до 10“3 М, что уменьшает вклад примесей из фонового электролита в остаточный ток.
Инверсионные методы анализа
Основными стадиями инверсионных электрохимических методов являются
1) предварительное накопление определяемого вещества на электроде с постоянной площадью поверхности с помощью электролиза при контролируемом потенциале;
2) удаление или растворение определяемого вещества с электрода в результате вольтамперометрической или химической реакции.
□ Для понижения предела обнаружения используют предварительное электролитическое иакопление.
Общей для всех инверсионных методов является стадия предварительного накопления (осаждения), но приемы, используемые на заключительной стадии анализа, могут различаться. Если растворение осуществляется электрохимически, метод называют инверсионной вольтамперометрией (ИВА) (рис. 7.3-23); в инверсионной потенциометрии в основе стадии удаления продукта накопления лежит химическая реакция.
Поскольку инверсионные методы основаны на предварительном накоплении определяемого компонента, они пригодны для интервала концентраций 10”6-10-8 М, т. е. для определения следовых количеств металлов.
Анодная инверсионная вольтамперометрия используется в основном для определения металлов, способных образовывать амальгамы, тогда как катодная инверсионная вольтамперометрия применяется для определения веществ, образующих на поверхности электрода нерастворимые соли ртутн. В последнем методе на стадии анодного предэлектролиза образуются ионы Hg(I) и определяемые вещества (анионы) осаждаются на поверхности электрода в виде малорастворимых солей Hg(I). При катодной развертке потенциала осадок восстанавливается до иона Hg(I) и наблюдаются катодные пики анионов. Кроме ртутного в катодной инверсионной вольтамперометрии можно использовать и серебряный электрод. Методом катодной инверсионной вольтамперометрии удобно определять ионы галогенидов, сульфата, оксалата и меркаптаны.
7.3.3. Амперометрия
□ Амперометрия — частный случай постояннотоковой вольтамперометрии, основанной на измерении тока при постоянном фиксированном значении потенциала в области предельного тока.
Амперометрия основана на измерении тока при фиксированном потенциале индикаторного электрода в перемешиваемых растворах (или в потоке) или на
7.3. Электроанализ
431
\ О Развертка потенциала
Рис. 7.3-23. Анодная инверсионная вольтамперометрия, а — развертка потенциала на стадии накопления и регистрации вольтамперограммы. б- —анодная инверсионная вольтамперограмма с линейной разверткой потенциала.
вращающемся дисковом электроде. Ток возникает в результате электрохимического окисления или восстановления электроактивного вещества при наложении импульса потенциала на индикаторный электрод. Гидродинамическую вольтамперометрию, т. е. вольтамперометрию с линейной разверткой в потоке или перемешиваемом растворе, используют для выбора рабочего потенциала, соответствующего плато предельного тока на кривой ток-потенциал, полученной на электроде с постоянной площадью поверхности в растворе, содержащем электроактивное соединение и фоновый электролит.
Предельный ток, измеренный прн электрохимическом восстановлении определяемого вещества О в гидродинамических условиях, т. е. в условиях конвективной диффузии, можно выразить следующим образом:
ii — nF AttiqCq
(7.3-80)
432
Глава 7. Методы химического анализа и их применение
где то—число переноса, не зависящее от скорости потока; со — концентрация определяемого вещества.
Бели число переноса постоянно, то изменение концентрации определяемого вещества можно контролировать, измеряя силу тока. Постоянство то поддерживают, сохраняя постоянной толщину диффузионного слоя, т. ё. перемешивая раствор или позволяя ему течь, или вращая с постоянной скоростью дисковый электрод:
mo=Do/6o (7.3-81)
где Dq — коэффициент диффузии, Йо —'толщина диффузионного слоя.
Считается, что в неподвижном слое растворе (слое Прандтля) имеет место линейная диффузия. В амперометрии при движении раствора толщина диффузионного слоя остается постоянной, и, таким образом, регистрируется стационарный ток, не зависящий от времени.
Амперометрия как способ детектирования наиболее часто используется в различных проточных аналитических методах, таких, как проточноинжекционные и хроматографические методы, а также в кулонометрическом титровании и классической титриметрии. В последнем случае измерение тока применяют для фиксирования конечной точки титрования. Амперометрия нашла важное применение в качестве способа преобразования в биосенсорах.
Материал электрода более важен для непрерывных проточных методов, чем для титриметрии, поскольку необходима механическая и долговременная функциональная стабильность. Различные угольные электроды, такие, как стеклоуглеродный или угольный пастовый (смесь графитового порошка и парафинового или силиконового масла), особенно популярны в ВЭЖХ и проточно-инжекциойном анализе. В амперометрическом титровании индикаторным электродом служит либо ртутный капающий электрод, либо платиновый или графитовый микроэлектрод. Электродом сравнения может быть насыщенный каломельный электрод или другой поляризуемый электрод. Варианты амперометрического титрования:
— титрование с одним поляризуемым электродом;
- титрование с двумя поляризуемыми электродами (биамперометрическое титрование).
□ Амперометрию можно применять для определения конечной точки титрования или в качестве прямого метода определения (например, амперометряче-ский датчик для определения Os).
7.3.4. Кулонометрические методы анализа
□ В кулонометрии реагент, используемый для титрования (титрант), получается in stu в результате стехиометрической электрохимической реакции со 100%-ным выходом по току.
Кулонометрические методы основаны на измерении количества электричества, затраченного на количественное электроокисление или электровосста-
7.3. Электроанализ
433
новл<рие определяемого вещества. Количество электричества измеряют в ку-лонахлПри постоянной силе тока
\ Q = it (7.3-82)
а при изменяющейся силе тока
' Q=[‘idt (7.3-83)
Jo
Количество электричества можно выражать в фарадеях. Один фарадей составляет 96485 Кл {число Фарадея) и равен количеству электричества, необходимому для превращения одного моля эквивалентов вещества. Это позволяет рассчитать массу химического вещества, выделившегося на электроде, по количеству электричества, прошедшего через электролитическую ячейку:
t
тк = QkK = [it dt (7.3-84)
nr nr J
0
где mk — количество определяемого вещества к, образовавшегося в процессе электролиза; М—молярная масса определяемого вещества к; п—число электронов, принимающих участие в электродной реакции; F — число Фарадея, а К—константа, характеризующая определяемое вещество, участвующее в электродном процессе.
В отличие от электрогравиметрии, в кулонометрии важно, чтобы ток, протекающий через электрохимическую ячейку, расходовался только на электрохимическое восстановление или окисление определяемого вещества, поскольку его массу вычисляют по количеству электричества, затраченного при 100%-ном выходе по току. Можно полагать, что 100%-ный выход по току достигается, если отсутствуют побочные реакции. Побочными могут быть электрохимические реакции с участием растворителя, материала электрода, компонентов фонового электролита или продуктов электролиза, вступающих во вторичные электродные реакции.
В электроанализе используют амфотерные растворители, способные к электрохимическому окислению и восстановлению, поэтому для каждого растворителя существует интервал потенциалов, пригодный для аналитических целей. В водных растворах он ограничен потенциалами выделения водорода и кислорода: 0,0 В и 1,23 В при pH 0. На практике этот диапазон потенциалов всегда шире вследствие кинетических затруднений, и измерения можно проводить при потенциалах отрицательнее 1,23 В и положительнее 0 В, в зависимости от энергий активации для различных электродных материалов.
Различают
1) прямую кулонометрию, основанную на измерении количества электричества, необходимого для количественного электроокисления или электровосстановления определяемого вещества;
2) косвенную кулонометрию, основанную на применении электролитически генерированных титрантов, обычно называемую кулонометрическим титрованием.
Для достижения 100%-ного выхода по току кулонометрические измерения проводят либо при контролируемом потенциале электрода (кулонометрия при
434
Глава 7. Методы химического анализа и их применение
контролируемом потенциале, или потенциостатическая кулонометрия), либо при постоянной силе тока (гальваностатическая кулонометрия).
В потенциостатической кулонометрии, как и в электролизе при контролируемом потенциале, измерения проводят при заранее выбранном постоянном потенциале электрода. В этих условиях за протекание тока в электролитической ячейке отвечает только одна реакция. Сила тока является функцией времени. Аналитический сигнал, т. е. количество электричества, измеряют электронным интегратором. Количество электричества, израсходованного на электролиз определяемого вещества, можно измерить химическим интегратором тока (кулонометром), подсоединенным последовательно.
В кулонометрии при контролируемом потенциале массу определяемого вещества находят по количеству электричества, затраченного на электролиз. Это возможно, если:
1) известна стехиометрия электродной реакции;
2) протекает только одна электродная реакция, т. е. отсутствуют побочные реакции другой стехиометрии;
3) реакции на электроде протекает при выходе по току, близком к 100%.
Эти условия очень напоминают требования, предъявляемые к реакциям в титриметрических методах.
Изменение силы тока в потенциостатической кулонометрии регистрируют самописцем. Это позволяет оценить фоновый ток и степень завершения электролиза.
Ток в любой момент времени при электролизе в объеме раствора равен
it — nFAD^- exp (—(7.3-85) 0 \ VO J
где n—число электронов, участвующих в электродной реакции, со — исходная концентрация вещества О, V — объем раствора, 6—толщина диффузионного слоя.
В соответствии с уравнением (7.3-85) ток экспоненциально уменьшается во времени и асимптотически приближается к фоновому уровню. Таким образом, электролиз занимает 30-100 мин. Форма типичной кривой i — t показана на рис. 7.3-24.
При твердофазном электролизе, т. е. при анодном растворении металлов, ток в любой момент времени равен
it = п F A ka exp
(7.3-86)
vs (dh + O*.
где n,FA,D,S и t имеют общепринятое значение, кс и fca — гетерогенные константы скорости катодной и анодной реакций, соответственно, а /м — активность образующихся ионов металла.
Поскольку сила тока в процессе электролиза меняется, т. е. является функцией времени, количество электричества определяют интегратором тока. В соответствии с уравнением Мак-Невина и Бейкера [7.3-10], время электролиза
7.3. Электроанализ
435
Рис. 7.3-24. Зависимость ток—время в кулонометрии при контролируемом потенциале; здесь Qt — общее количество электричества (Кл), Qb —количество электричества (Кл), израсходованное на электролиз примесей в фоновом электролите, Qs— количество электричества (Кл), израсходованное на электролиз раствора пробы.
при фиксированном потенциале можно значительно сократить, если для определения Q использовать кривую i — t. Так,
Q = / itdt (7.3-87)
Jo
Из (7.3-85) при t = 0 находим
i0 = nFAD^-О
Обозначив /3 = DA/V6, получаем:
Q — г0 exp (—/0t) di = io0 (7.3-88)
Jo
Наклон (—/?/2,303) функции lg г = /(4) дает значение /0, а точка пересечения — величину io- Для определения Q, достаточно иметь 3-4 точки, по которым можно построить зависимость lg i от t и найти ф и /0. Этот подход нельзя использовать для определения Q при электролизе на ртутном электроде, поскольку поверхность электрода непостоянна.
Для определения конечной точки измерения не нужна индикаторная система; метод высокоточен и селективен.
Количество электричества можно определить химическим интегратором тока (кулонометром), например Н2 — Ы2-кулонометром, в котором происходит электролиз раствора сульфата гидразина:
N2H+ -» N2 + 2Н2 + Н+ (7.3-89)
Химические интеграторы обладают высокой точностью и позволяют непосредственно измерять количество затраченного электричества. Одним из наибо-
436 Глава 7. Методы химического анализа и их применение
лее точных является серебряный кулонометр, но для практической работы он неудобен.
Современные интеграторы тока выдают результаты непосредственно в кулонах и позволяют записывать кривые Q — t в процессе выполнения анализа.
Для кулонометрии при контролируемом потенциале требуется автоматический потенциостат. Потенциостат обычно сйабжен устройством для считывания тока, которое выдает зависимость ток-время, поступающую на электронный интегратор. С помощью источника напряжения потенциал электрода (катода или анода) устанавливают на нужном уровне и контролируют потенциометром относительно электрода сравнения- Если нуль-ииструмент зафиксирует изменение потенциала, источник напряжения восстановит заданное значение потенциала электрода. ,
Применение кулйноме^ЙМН^и код^ррлируёмом потенциале
О Кулонометрия позволяет применять химически нестабильные титранты, такие, как Bra, для титрования с высокой точностью.
В кулонометрии при контролируемом потенциале применяют многие реакции электроосаждения, используемые в электрогравиметрии, а также реакции, сопровождающиеся образованием растворимых продуктов или газов, например реакции восстановления Fe(III) до Fe(II) или окисления N2H4 до N2.
Методы при контролируемом токе
Если электролиз проводят при постоянной силе тока, количество затраченного электричества можно определить и без специальных приборов. Поскольку сила тока известна, остается только измерить время электролиза:
Q = it (7.3-90)
Условия электролиза при контролируемой силе тока можно выбрать на основании кривых ток-потенциал типа показанных на рис. 7.3-25.
На рис. 7.3-25 показан процесс анодного окисления ионов Fe(II) в серной кислоте.
При силе тока на 25 мА меньше предельного тока на аноде протекает окисление Fe(II). С увеличением времени электролиза концентрация Fe(II) в растворе уменьшается. Кривые б, в, г и д соответствуют 25, 50,75 и 100%-ной степени завершения электролиза соответственно. При концентрации Ее(П) ниже требуемой для поддержания постоянной силы тока ток может поддерживаться при заданном значении, только если потенциал анода смещается к более положительным значениям, при которых образуется Ог (кривые в, г, д):
2Н2О -> 4Н+ + О2 + 4е- (7.3-91)
С уменьшением концентрации Fe(II) все большее и большее количество электричества расходуется на окисление воды и условие 100%-ного выхода по току для Fe(II) не выполняется.
В зависимости от заданной силы тока раньше или позже по ходу электролиза начинается выделение О2- Таким образом, если определяемое вещество электроактивно, т. е. в процессе электролиза участвует в электрохимической реакции, в большинстве случаев 100%-ный выход по току получить нельзя.
7.3. Электроанализ
437
+1,5 +1,0 +0,5 0,0 Е, В
Рис. 7.3-25. Кривые ток-потенциал окисления Fe(II) в 1М H2SO4 при степенях завершенности электролиза 0 (л); 0,25 0,5 (с); 0,75 (г); 1 (й) и в присутствии
избытка Се3+ (е).
Эти сложности можно преодолеть, добавив так называемый деполяризатор. Деполяризатор может окисляться при менее положительных потенциалах, чем определяемое вещество. Так, если к раствору добавить Се(Ш), то при 1,15 В ионы Се(Ш) окисляются до Ce(IV), который затем окисляет Fe(II)—»Fe(III). Тем самым, устраняется опасность окисления воды, и Fe(II) окисляется со 100% выходом по току.
Для достижения 100%-ного выхода используют 50-10000-кратный избыток деполяризатора.
В косвенной кулонометрии определяемое вещество, как правило, не принимает участия в электрохимической реакции. Электролиз при постоянной силе тока используют для электрохимической генерации титранта или из вспомогательного реагента, или из материала рабочего электрода1. Титрант быстро и количественно реагирует с определяемым веществом. Необходимо убедиться в достижении конечной точки титрования. Наиболее часто используемыми и чувствительными методами для определения конечной точки кулонометрического титрования являются потенциометрия и амперометрия. Кулонометрическое титрование можно автоматизировать.
По аналогии с классическим титрованием, косвенную кулонометрию называют кулонометрическим титрованием. Преимущества кулонометрического титрования:
1) кулонометрически может быть генерировано огромное число титрантов;
2) при получении титранта in situ можно вычислить его точную концентрацию, если генерация протекает со 100%-ным выходом по току;
3) «добавление» титранта не вызывает разбавления анализируемого раствора;
Или из растворителя, например воды. — Прим, персе.
438
Глава 7. Методы химического анализа и их применение
4) при разумном выборе силы тока электролиза и времени электролиза можно точно контролировать количество титранта.
В настоящее время почти все необходимые титранты можно получить со 100%-ным выходом по току (табл. 7.3-3).
Таблица 7.3-3. Типичные электрогенерированные титранты и вещества, определяемые кулонометрическим титрованием [A. J. Bard, L. R. Faulkner, Electrochemical Methods, p. 384. New York: Wiley, 1980.]
Электрогене-рированный титрант Генераторный электрод и раствор Примеры определяемых веществ
Окислители Бром Иод Хлор Церий(ГУ) Марганец(Ш) Серебро(П) Pt/NaBr Pt/KI Pt/NaCl Pt/Ce2(SO4)3 Pt/MnSO4 Pt/AgNO, As(III), U(IV), NH3, олефины, фенолы, SO2, H2S, Fe(II) H2S,SO2, As(UI), вода (по методу Карла Фишера), Sb(III) As(III), Fe(II), разные органические вещества U(IV), Fe(II), Ti(III)t Г Fe(II), H2O2, Sb(III) Се(П1), V(IV), H2C2O4
Восстановители Железо(П) Титан(Ш) Олово(П) Медь(1) Уран(У), (VI) Хром(П) Pt/Fe2(S04)3 Mn(III), Cr(VI), V(V), Ce(IV), U(VI), Mo(VI) Pt/TiCU Fe(III), V(V,VI). U(VI), Rc(VIII), Ru(IV), Mo(VI) Au/SnBr4(NaBr) I2. Br2, Pt(IV), Se(IV) Р1/Сч(П)(НС1) Fe(IH), Ir(IV), Au(III), Cr(VI), 1О2 Pt/UO2SO4 Cr(VI), Fe(III) Hg/CrClafCaCh) O2Cu(II)
Осадители и комплексообразующие лиганды Серебро(1) Ртутъ(1) ЭДТА Цианид Ag/HClO» Hg/NaC104 Hg/HgNHsY2- * Pt/Ag(CN)7 Галогенид-ионы, S2-, меркаптаны Галогенид-ионы, ксантаты Ионы металлов Ni(II), Аи(ПЦ), Ag(I)
Кислоты и основания Гидроксид-ион Ион водорода Pt(-)/H2O Pt(+)/H2O Кислоты, СО2 Основания, СО2", аммиак
* Y2-эгилеидинминтетраацетат-ион.
Литература
|7-3-1] Henderson, Р. Z. Phys. Chem., 59, 118 (1908); 63, 325 (1907).
[7.3-2] Horovitz, К. Z. Phys. Chem., 15, 369 (1923).
[7.3-3] Lengyel, Б., Blum, E., Trans. Faraday Soc., 30, 461 (1934).
[7.3-4] Perley, G. A., Anal. Chem., 21, 391 (1949).
7.3. Электроанализ
439
[7.3-5] Stefanac, Z., Simon, W-, Chimia, 20, 463 (1966); Microchem. J., 12, 125 (1967).
[7-3-6] Adams, R. N., Anal Chem., 30, 1576 (1958).
[7.3-7] Yamada, S., Sato, H., Nature, 193, 261 (1962).
[7.3-8] Osteryoung, J. G., Schreiner, M. M., CRC Critical Reviews in Analytical Chemistry, 19, 1 (1988).
[7.3-9] Borman, S. A., Anal. Chem., 54, 6 (1982).
[7.3-10] MacNavin, W- M., Baker, В. B., Anal. Chem., 24, 986 (1952).
Дополнительная литература
Bard, A. J., Faulkner, L. R., Electrochemical Methods; Fundamentals and Applications, John Wiley and Sons, New York, Chichester, Brisbane, Toronto, 1980.
Bond, A. M., Modern Polarographic Methods in Analytical Chemistry, Marcel Dekker, Inc. New York and Basel, 1980. (Имеется перевод: Бонд A. M. Полярографические методы в аналитической химии. — М.: Химия, 1983.)
Borman, S. A., New Electroanalytical Pulse Techniques, Anal. Chem., 54, 698, (1982).
Freiser, H. (Ed.), Ion-Selective Electrodes in Analytical Chemistry, Vol. 1 and 2, Plenum Press, New York and London.
Hobart, H. W., Lyune, L. M., Jr., Dean, J. A., Settle, F. A., Jr., Instrumental Methods of Analysis, 2nd ed., Wadsworth Publishing Company, Belmont, California (Division of Wadsworth, Inc.), 1988.
Kissinger, P. K., Heineman, W. R. (Ed.), Laboratory Techniques in Electroanalytical Chemistry, Marcel Dekker Inc., New York and Basel, 1984.
Kolthoff, I. M., Lingane, J. J., Polarography, Vol. I., 2nd ed., Interscience Publishers, New York, London 1952.(Имеется перевод раннего издания: Кольтгоф И., Лингейн Дж. Полярография. М.-Л.: Госхимиздат, 1948.)
Meier, Р. С., Ammann, D., Osswald, Н- F., Simon, W., Ion-Selective Electrodes in Clinical Chemistry, Medical Progress through Technology, 5, 1-12 (1977).
Pungor, E., Oscillometry and Conductometry, Pergammon Press, Oxford, London, Edinburgh, New York, Paris, Frankfurt, 1965.
Wang, J. Analytical Electrochemistry, VCH, New York, 1994.
Willard, H. H., Meritt Jr., L. L., Dean, J. A., Settle Jr., F. A., Instrumental Methods of Analysis, 2nd ed., Wadsworth Publishing Company, Belmont, California (Division of Wadsworth, Inc.), 1988.
Вопросы и задачи
1. Опишите и сравните основные принципы четырех наиважнейших электроаналитических методов!
2. Почему в потенциометрии необходимо проводить измерения в отсутствие тока во внешней цепи?
3. Опишите основные типы электродов, применяемых в потенциометрии.
4. Напишите уравнение Нернста для полуреакций
a) Fe24" —» Fe3+ + е";
б) МпО7 + 8Н+ + 5е“ —» Мп2+ + 4НгО, а также проанализируйте эти уравнения в графической форме и опишите влияние pH на потенциалы полуреакций (а) и (б), используя данные приложения (см. т. 2 настоящего учебника).
5. Опишите принцип работы стеклянного рН-электрода.
6. Опишите принцип работы жидкостного мембранного калий-селективного электрода.
7. Что такое «поляризуемый электрод»?
440
Глава 7. Методы химического анализа и их применение
8- Нарисуйте классическую полярограмму и объясните наблюдающиеся на ней три области тока, а также аналитическую информацию, которую можно извлечь из полярограммы.
9. При каких условиях методом классической полярографии можно проанализировать смесь веществ?
10. Опишите принципы дифференциальной импульсной полярографии и анодной инверсионной вольтамперометрии и сравните их с классической полярографией.
11. Опишите принципы амперометрии и ее основное применение в аналитической практике.
12. Почему для применения кулонометрии важен 100% выход по току?
7.4. ПРОТОЧНО ИНЖЕКЦИОННЫЙ АНАЛИЗ
Цели изучения
• Дать введение в автоматизированный анализ.
• Описать .основные принципы ПИА.
• Продемонстрировать возможности ПИА в сравнении с периодическим и обычным непрерывным проточным анализом.
• Показать, что ПИА позволяет улучшать существующие аналитические методы.
• Показать, как ПИА реализует новые аналитические процедуры, неосуществимые обычным путем.
• Продемонстрировать возможности ПИА на примере практических приложений.
7.4.1. Периодический и непрерывный проточный анализ
Традиционно принято считать, что единственный разумный способ осуществления химического анализа—гомогенно смешать определяемый компонент и реагент (реагенты) и ждать установления химического равновесия. Целые поколения аналитиков обучены такому подходу безотносительно к тому, идет ли речь об обычном периодическом анализе или о непрерывных проточных методах, вошедших в практику в конце 1950-х годов.
□ В обычном периодическом анализе раствор неподвижен, а емкость с пробой перемещается.
В обычном периодическом анализе раствор определяемого вещества содержится внутри подходящей емкости (например, пробирки или колбы; рис. 7.4-1,л), в которую добавляют соответствующие реагенты, тщательно перемешивают, при необходимости нагревают, затем ожидают, когда будет достигнуто химическое равновесие, после чего содержимое емкости переносят в измерительную ячейку подходящего измерительного прибора, такую, как кювета спектрофотометра. Вся процедура может быть механизирована или автоматизирована, как в ряде имеющихся на рынке приборов различного типа (преимущественно клинических анализаторов). Все они, кроме центрифужных анализаторов, основаны на принципе конвейерной ленты (рис. 7.4-1,6), т.е.
ТА. Проточно-инжекционный анализ
441
Поток
Рис. 7.4-1. Сравнение ручной и автоматизированной операций, выполняемых в ходе типичного фотометрического определения [7.4-3]. а — ручная операция; б — дискретный анализатор конвейерного типа; в — непрерывно-проточный анализатор с воздушным сегментированием; отдельно показаны сегменты воздуха и жидкости, условно изображено перемешивание, приводящее к гомогенизации отдельных сегментов жидкости. Во всех этих видах анализа измерения проводят в стационарных условиях (постоянный сигнал, т. е. «плоская вершина»).
емкость продвигается через отдельные пункты, где выполняются разнообразные отдельные операции, например введение анализируемой пробы, реагентов, смешивание и нагревание, включение временнбй задержки для обеспечения завершения реакции, и, наконец, детектирование. Независимо от устройства анализаторов все они имеют общее свойство: раствор неподвижен, в то время как емкость движется, в точности имитируя процесс ручного непроточного анализа.
□ В непрерывном проточном анализе анализирующая система неподвижна, а раствор движется.
□ В ПИА стационарные условия не требуются.
В непрерывном проточном анализе используется противоположный подход: анализирующая система неподвижна, в то время как раствор перемещается по трубкам. Однако, как показало применение первой коммерческой непрерывной проточной системы AutoAnalyzer, поставляемой фирмой Technicon
442 Глава 7. Методы химического анализа и их применение
(рис. 7.4-1,в), подход к химическому анализу в этом способе не изменился — гомогенизация и достижение химического равновесия, приводящие к так называемым стационарным условиям. Чтобьгэти условия гарантировать и обеспечить индивидуальность проб, в поток вводили пузырьки воздуха. Будучи сам по себе оригинальным, этот подход все-таки требует достижения равновесных условий, поскольку наличие смеси раствора и пузырьков воздуха делает контроль времени в системе неточным. Вызов идее проведения непрерывного проточного анализа был брошен в середине 1970-х, когда в Дании [7.4-1] был разработан и описан проточно-инжекционный анализ (ПИА). Этот аналитический подход совершил революцию в выполнении химического анализа. Если раньше было необходимо работать в условиях равновесия, то для ПИА было показано, что это несущественно; новый метод позволил выполнять контролируемое смешивание пробы и реагентов в строго определенных и воспроизводимых условиях.
7.4.2. Основы метода
□ Автоматизированные измерения с гтрмощью ПИА экономичны даже для малых партий проб.
ПИА имеет ряд важных характеристик, сравнимых с характеристиками традиционных непрерывных проточных измерений: высокую производительность (обычно 100-300 пробвчас); малые времена отклика (часто менее 1 мин между инжекцией пробы и откликом детектора); значительно более быстрые нарастание и исчезновение сигнала (только несколько минут для каждой пробы) и, за исключением системы ввода, более простое и гибкое оборудование. Последние два преимущества исключительно важны, поскольку они делают возможным и экономичным автоматизированные определения для сравнительно небольшого числа проб нерутинного типа. Непрерывные проточные методы теперь уже не ограничиваются только теми случаями, когда число проб велико и аналитический метод является рутинным.
□ ПИА основан на инжекции пробы, контролируемой дисперсии и воспроизводимом контроле времени.
□ Для получения аналитического сигнала может быть использована любая точка градиента концентрации. Часто используют максимальную высоту пика.
ПИА претерпел определенные изменения, например, в последние годы он был дополнен ПосИА (последовательный инжекционный анализ [7.4-2]), но его основы сохраняют изначально определенный вид [7.4-1, 7.4-3J (рис. 7.4-2): инжекция точно измеренного объема пробы; воспроизводимый и точный контроль времени для всех манипуляций, производимых в системе с пробой от точки ввода до точки детектирования (так называемая контролируемая дисперсия); создание концентрационного градиента введенной пробы, что обеспечивает нестационарную, но строго воспроизводимую величину регистрируемого сигнала. Сочетание этих характеристик с использованием детектора, способного непрерывно регистрировать поглощение, электродный потенциал или любой другой физический параметр, меняющийся при прохождении пробы через проточную ячейку, делает ненужным достижение химического равнове-
1А. Проточноинжекционный анализ
443
КОНТРОЛИРУЕМАЯ ДИСПЕРСИЯ
ВОСПРОИЗВОДИМАЯ УСТАНОВКА ВРЕМЕНИ
Рис. 7.4-2. Дисперсная зона пробы с начальной концентрацией С°, введенной в положении S, и соответствующий выход самописца [7.4-3]. Каждой концентрации С вдоль градиента соответствует определенная величина коэффициента дисперсии D, которая может быть отнесена к фиксированному времени задержки ti, прошедшему с момента инжекции to.
сия (стационарных условий). Любая точка на пути к стационарному сигналу (рис.7.4-1,в) столь же пригодна для измерений, как и стационарное состояние, при условии, что эта точка повторимо воспроизводится, а это действительно возможно в ПИА благодаря присущей ему точной установке времени пребывания пробы. Детектирование аналитического сигнала в ПИА может быть выполнено в любой точке вдоль созданного градиента концентраций, но в большинстве случаев используется высота пика, соответствующая концентрации Стах на рис. 7.4-2, поскольку ее легко идентифицировать. Тем не менее, как показано в разд. 7.4.10, использование всех концентраций вдоль градиента создает основу для ряда абсолютно новых аналитических подходов. Однако на данный момент мы будем рассматривать в качестве аналитического сигнала максимум пика.
□ Требуется лишь несколько микролитров пробы.
□ Измерения в ПИА выполняются очень быстро.
Характеристики ПИА лучше всего, пожалуй, проиллюстрировать на практическом примере спектрофотометрического определения хлорида в одноканальной системе (см. рис. 7.4-3), основанной на последовательности реакций:
Hg(SCN)2 + 2СГ 7= HgCl2 + 2SCN”
Fe3+ + SCN" Fe(SCN)2+
Аналитическая последовательность основана на реакции хлорида с тиоцианатом ртути(П) с высвобождением тиоцианат-ионов, которые затем реагируют с железом(Ш) с образованием интенсивно окрашенного тиоцианатного комплекса железа(Ш), светопоглощение которого измеряют. Пробы с содержанием хлорида 5-75 м.д. (1м.д. — 10~4%) инжектировали (S) через дозатор объемом 30 мкл в раствор носителя, содержащий смешанные реагенты и прокачиваемый со скоростью 0,8 мл/мин. По мере распространения зоны введен-
444
Глава 7. Методы химического анализа и их применение
Рис. 7.4-3. а — схема потокораспределительной системы для спектрофотометрического определения хлорида: S — точка ввода, D — детектор, W — слив; б — аналоговый вывод сигнала при определении хлорида в диапазоне 5-75 м.д. С1 с помощью системы а. Чтобы продемонстрировать воспроизводимость измерений, каждую пробу вводили четырехкратно. Инжектируемый объем составил 30 мкл, производительность — порядка 120 проб в час. Быстрое сканирование проб 30 м.д. (R30) и 75 м.д. (R75) на правой части графика показывает степень перекрывания (менее 1%), если пробы вводят с промежутком времени 38 с (разница между Si и S2) (7.4-3].
ной пробы в потоке носителя образуется тиоцианат железа(Ш) и направляется к детектору (D) через смесительную спираль (длина 0,5 м, внутренний диаметр 0,5 мм). Последняя благодаря центробежным силам уменьшает размывание полосы (зоны пробы), что приводит к регистрации более отчетливого пика. Оптическую плотность А потока носителя непрерывно измеряли при 480нм в проточной микроячейке (объем 10мкл) и записывали (рис. 7.4-3,б). Чтобы продемонстрировать воспроизводимость аналитического сигнала, каждую пробу для каждой из семи концентраций хлорида в этом эксперименте вводили 4 раза, так что было проанализировано 28 проб. Поскольку на это потребовалось 14 мин, то средняя производительность составила 120проб/ч. Быстрое сканирование пиков проб с концентрациями 75 и 30 м.д. (показано справа на рис. 7.4-3,б) подтвердило, что к моменту времени, когда следующая проба (введенная при S2) достигает проточной ячейки, в ней остается менее 1% предыдущего раствора, и нет никакого наложения пиков, если пробы вводят с 30-секундным интервалом. Эти эксперименты отчетливо проявляют одну из ключевых черт ПИА — при прохождении через аналитический канал все пробы последовательно обрабатываются в точности тем же самым образом, или, другими словами, то, что происходит с одной из проб, точно таким же образом происходит с любой другой пробой.
7 А. Проточно-инжекционный анализ
445
7.4.3. Основное оборудование ПИА
□ Для прокачивания жидкостей используют Главным образом периста я ьтиче-ские насосы.
Наиболее часто растворы в проточно-инжекционных системах прокачивают перистальтическим насосом, в котором жидкость продавливается через пластиковую или резиновую трубку движущимися роликами. Современные насосы обычно содержат 8-10 роликов, расположенных по кругу, так что в любой момент времени каждую трубку сдавливает половина роликов. Поток управляется частично скоростью двигателя, а частично — внутренним диаметром трубки. Поскольку все трубки насоса имеют одинаковый размер стенок, так что, будучи полностью сжатыми, они заполнены в одинаковой степени, при постоянной скорости вращения перистальтического насоса скорость потока определяется внутренним диаметром трубок. Производимые трубки имеют внутренний диаметр от 0,25 до 4 мм, что позволяет менять скорость потока от 0,0005 до 40 мл/мин. Перистальтические насосы в целом удовлетворяют большинству приложений, позволяя работать не только без пульсаций, но и с несколькими трубками одновременно. В ПИА применяются насосы и других типов, такие как плунжерные, но они обычно значительно дороже (кроме того, они допускают прохождение только одного потока, что в случае многоканальной потокораспределительной системы требует нескольких насосов).
□ ПИА позволяет экономить реагенты и анализируемые пробы и сопровождается незначительным количеством отходов.
Инжектор представляет собой клапан с петлей, похожий на используемый в ВЭЖХ, т. е. оснащенный внутренней петлей для пробы. Более часто используют специально предназначенный для ПИА клапан, включающий ротор и статор с четырьмя, шестью или большим числом отдельных портов, в которых объем инжектируемой пробы, обычно 1-200 мкл (чаще всего 25 мкл) отмеряют с помощью внешней петли соответствующей длины и внутреннего диаметра. Поскольку объем пробы столь мал, для одного цикла определений много реагента не требуется. Это делает ПИА не только простым микрохимическим методом, обеспечивающим высокую производительность при минимальном расходе пробы и реагента, но н обеспечивает минимальное количество сливов. Последнее существенно, так как устранение химических отходов во многих случаях обходится дороже, чем сами реагенты.
Трубопроводы в потокораспределительных системах ПИА представляют собой преимущественно тонкие пластиковые трубки (из материалов типа ПВХ или ПТФЭ) с внутренним диаметром 0,5-0,8 мм. Трубки обычно свернуты спиралью или узлом для минимизации дисперсии (обеспечивают структуру вторичного потока). Как правило, длина трубок потокораспределительной системы ПИА должна быть по возможности небольшой, чтобы избежать нежелательного разбавления инжектированного раствора пробы.
Последовательный инжекционный анализ (ПосИА), являющийся в сущности разновидностью ПИА, основан на последовательном введении в удерживающую спираль точно определенных зон пробы н реагента с помощью на
446 Глава 7. Методы химического анализа и их применение
правляющего (селекторного) клапана. Затем меняют направление потока на противоположное и удержанные зоны направляют через реакционную спираль к Детектору. При этом зоны реагента и пробы проникают одна в другую и образуют область смешивания. За исключением селекторного клапана, все составные части ПосИА аналогичны используемым в ПИА. Хотя такой вариант имеет определенные преимущества в конкретных приложениях (например, незначительный расход пробы и раствора реагента), ему присущи серьезные практические ограничения (возможность работы только с одноканальной потокораспределительной системой). По этим причинам ПосИА в дальнейшем не рассматривается.
ПИА (или ПосИА) совместим с детектором практически любого типа. Одна из причин столь большого успеха ПИА как раз н заключается не только в технике управления раствором, применимой для множества задач, но и в том, что он пригоден для детекторных устройств практически любого вида. Это будет показано в последующих разделах, но основное внимание будет уделено контролируемой дисперсии, тому, как ей управлять и каким целям она служит.
7.4.4. Дисперсия в ПИА
□ Дисперсия количественно характеризуется коэффициентом дисперсии D.
Степень дисперсии, или разбавления, в ПИА характеризуется коэффициентом дисперсии D. Рассмотрим простой дисперсионный эксперимент. Раствор пробы, содержащийся в полости клапана до инжекции, однороден н имеет начальную концентрацию С°, которая, если бы ее можно было измерить с помощью детектора, дала бы сигнал прямоугольной формы с высотой, пропорциональной концентрации пробы (рис. 7.4-2). После инжектирования проба движется с потоком носителя, образуя размытую зону, форма которой зависит от геометрии канала и скорости потока. Следовательно, кривая отклика имеет форму пика, отражающую континуум концентраций (рис. 7.4-2, внизу) и образующую градиент концентраций, в котором каждый отдельный элемент жидкости имеет концентрацию, отличную от соседнего. Полезно, однако, рассматривать этот континуум концентраций состоящим из индивидуальных фрагментов жидкости, имеющих определенную концентрацию содержимого пробы С, поскольку каждый из этих фрагментов является потенциальным источником сигнала (см. также разд. 7.4.10).
□ Коэффициент дисперсии D пробы определяется как отношение концентраций содержимого пробы до и после дисперсии в том фрагменте жидкости, который дает аналитический сигнал.
Чтобы разумно сконструировать систему ПИА, важно знать, насколько исходный раствор пробы разбавляется на пути к детектору и сколько времени проходит между инжекцией пробы и регистрацией сигнала. Для этого определим коэффициент дисперсии D как отношение концентраций содержимого пробы до н после разбавления, имеющего место в таком фрагменте жидкости, который дает аналитический сигнал, т. е.:
D = C°/C
7.4. Проточно-инжекционным анализ
447
что для С — дает
П = С°/Стах (0<D<oo)
□ Коэффициент дисперсии для конкретной системы ПИ А может быть легко определен.
□ Коэффициент дисперсии есть мера степени разбавления инжектированной пробы в точке детектирования.
□ Любом пик в ПИ А есть результат двух кинетических процессов, протекающих одновременно: физического процесса дисперсии и налагающегося на него химического процесса, связанного с реакцией между пробой и реагентами.
Итак, если аналитический сигнал основан на измерении высоты максимума пика, то следует рассматривать концентрацию Стах такого воображаемого фрагмента жидкости, который соответствует максимуму на регистрируемой кривой. Таким образом, зная D, можно оценить концентрации пробы (н реагента). Определить D конкретной потокораспределительной системы ПИ А довольно легко. Простейшим способом является инжекция точно измеренного объема раствора красителя в бесцветный поток и непрерывный контроль оптической плотности дисперсной зоны красителя с помощью спектрофотометра. Чтобы получить величину Dmax, измеряют высоту (т. е. поглощение) полученного пика и затем сравнивают ее с расстоянием между нулевой линией и сигналом, полученным при заполнении ячейки неразбавленным раствором красителя. При условии соблюдения закона Ламберта—Бера (см. разд. 9.1) отношение величин соответствующих поглощений дает значение Dmaxt которое характеризует потокораспределительную систему ПИА, детектор и метод детектирования. Заметим, что такое определение коэффициента дисперсии учитывает только физический процесс дисперсии, а не протекающую химическую реакцию, поскольку D относится к концентрации содержимого пробы до и после одной лишь дисперсии. В этой связи следует подчеркнуть, что любой пик в ПИА есть результат двух кинетических процессов, которые протекают одновременно: физического процесса дисперсии (размывания) зоны н химических процессов, в основе которых лежат реакции между пробой н реагентом. Рассматриваемый физический процесс хорошо воспроизводится для каждого инжекционного цикла; и все же это не гомогенное смешивание, а дисперсия, результатом которой является градиент концентрации пробы в растворе носителя.
Дефиниция D подразумевает, что прн D, равном, например, 2, раствор пробы разбавлен потоком носителя в отношении 1:1. Параметры, управляющие коэффициентом дисперсии, являются предметом детального изучения. Вкратце можно отметить, что наибольшее влияние на D оказывают объем инжектированной пробы, физические размеры системы ПИА (длина и внутренний диаметр трубок), время пребывания пробы в системе и скорость потока. Дополнительные факторы включают возможность использования одноканальной потокораспределнтельной системы, а не разветвленной со многими трубками, а также выбор точки измерения на градиенте дисперсной зоны пробы, отличной от соответствующей максимуму пика [7.4-3].
Для удобства дисперсия пробы подразделяется на ограниченную (D = 1—2), среднюю (D = 2—10), высокую (D > 10) и пониженную (D < 1); со
448
Глава 7. Методы химического анализа и их применение
ответствующим образом сконструированные системы ПИА используют для множества аналитических задач. Ограниченную дисперсию используют, когда инжектированную пробу следует транспортировать к детектору в неразбавленном виде, т. е. система ПИА служит для аккуратного и точного транспорта пробы к детектирующему устройству (такому, как ион-селективный электрод или атомно-абсорбционный спектрофотометр). Среднюю дисперсию применяют, когда определяемый компонент должен смешиваться и реагировать с потоком носителя, образуя детектируемый продукт. Высокая дисперсия требуется только в том случае, когда проба должна быть разбавлена до величины, попадающей в измеряемый диапазон. Пониженная дисперсия подразумевает, что концентрация детектируемой пробы выше, чем концентрация инжектируемой пробы, т.е. выполняется концентрирование on-line (например, с помощью ионообменной колонки или путем соосаждения, разд. 7.4.9).
□ ПИА — широко применяемый способ работы с растворами.
□ ПИА обеспечивает выполнение таких аналитических процедур, которые сложно или невозможно выполнить по обычным схемам.
Следующие разделы дают примеры, охватывающие различные варианты дисперсии. Рассматривая обширную литературу по ПИА (к началу 1996г. число статей по ПИА превысило 7000, к ним следует добавить более 25 монографий), приходим к выводу, что здесь, конечно, можно обсудить лишь некоторые аспекты, и предлагаем читателю для дальнейшей информации обратиться к литературе [7.4-4], которая свидетельствует, что ПИА является мощным методом современной аналитической химии. Задуманный изначально как метод проведения серийного анализа, ПИА вскоре вышел за эти рамки, став широко применяемым способом работы с растворами в аналитической лаборатории, а также в промышленности, при управлении процессами. Было показано, что ои создает уникальные аналитические возможности, позволяя реализовать аналитические процедуры, которые сложно или невозможно осуществить по обычным схемам, например, такие, как определения, основанные на получении и измерении метастабильных компонентов, и исследование принципов детектирования, основанных на био- и хемилюминесценции. По этой же причине аббревиатура ПИА во многих случаях была заменена на ПИ, чтобы подчеркнуть использование проточно-инжекционных систем для работы с растворами, а не только для анализа.
7.4.5. ПИА для воспроизводимого и точного ввода пробы (ограниченная дисперсия)
Поскольку для ПИА характерен точный контроль времени, очевидно, что метод можно применять для точной и воспроизводимой транспортировки пробы к детектору, обеспечивая таким образом тщательное поддержание всех условий в течение каждого цикла измерений. Это может быть полезным, например, если детектором служит электрод или сенсор, сигнал которого контролируется диффузией, или если на поведение детекторного устройства влияет время, в течение которого образец воздействует на это устройство. Примерами детекторов первого типа служат ион-селективные электроды (ИСЭ) и биосенсо-
7.4. Проточно-инжекционным анализ
449
Рис. 7.4-4. Типичные кривые потенциал-время для определяемого иона (А) и мешающего иона (В). Если сигнал фиксируют в момент времени, обозначенный вертикальной пунктирной линией, то вклад В минимален.
ры, а второй вариант реализуется, например, в комбинации ПИА с атомноабсорбционной спектрометрией (ААС).
□ ИСЭ преимущественно работают в динамическом режиме ПИА.
□ Мешающие ионы можно дискриминировать кинетически.
Так, для потенциометрии показано, что многие нон-селектнвные электроды, работающие в динамическом режиме, способствуют получению быстрого и воспроизводимого отклика. Для ИСЭ характерны обычно довольно большие времена отклика, связанные с достижением стационарных условий, и, следовательно, трудно установить, когда именно регистрировать сигнал. В ПИА это решение целиком возлагается на систему, так как проба прибывает к детектору за время, определяемое исключительно выбором потокораспределнтель-ной системы. Но ИСЭ часто проявляют также кинетическую дискриминацию по отношению к определяемому иону и мешающим веществам (т. е. в течение экспозиции пробы с малой длительностью отклик сенсора на различные вещества может значительно изменяться, что в свою очередь можно использовать для улучшения селективности и пределов обнаружения сенсора). Это показано на рис. 7.4-4, из которого видно, что если сигнал детектируют в стационарных условиях (во время t в дальней правой части), мешающий ион В оказывает значительное влияние. Если же сигнал регистрируют в момент времени, обозначенный пунктирной вертикальной линией, то это влияние (ценой небольшого уменьшения величины сигнала первичного иона А) практически устраняется.
□ Для обеспечения линейной зависимости между концентрацией определяемого вещества и сигналом биосенсора требуется, чтобы удовлетворялось условие кинетики псевдопервого порядка. В системах ПИА такие условия легко достигаются.
Ту же концепцию управления временем экспозиции пробы можно применить для более сложных сенсоров, таких, как ферментативные. В этих сенсорах мембрану, содержащую один или несколько иммобилизованных ферментов, помещают впереди активной поверхности детекторного устройства. С помощью
16 Аналитическая химия. Том 1
450
Глава 7. Методы химического анализа и их применение
диффузии определяемое вещество транспортируется в мембрану и там разрушается ферментом, образуя продукт, который затем можно детектировать оптически или электрохимически. Условием получения линейной зависимости между концентрацией н сигналом является, однако, наличие кинетики псевдопервого порядка [7.4-5]. Другими словами, концентрация получаемого продукта, достигающего поверхности детектора, должна быть значительно меньше, чем константа Михаэлиса—Ментен. Так как эта константа для большинства ферментных систем имеет величину порядка 1 ммоль/л, а многие основы пробы (например, человеческая сыворотка или кровь) содержат значительно большие концентрации, то использование ферментных сенсоров в статических (непроточных) условиях часто осложняется необходимостью введения дополнительных мембранных слоев, чтобы ограничить диффузию определяемого вещества в ферментный слой, или в случае электрохимических детекторов — химически модифицированных электродов, в которых перенос электрона регулируется подходящим медиатором.
□ Условия для кинетики псевдопервого порядка можно создать, регулируя время, в течение которого проба действует на сенсор, т. е. регулируя скорость потока в системе ПИА.
Однако при работе в ПИА степень превращения можно просто отрегулировать с помощью времени, в течение которого определяемое вещество воздействует на ферментный слой, и, следовательно,' можно прямо управлять количеством превращенного определяемого вещества, регулируя скорость потока в системе ПИА. Этот подход показан на рис. 7.4-5,а, где изображена система для определения глюкозы с помощью амперометрического сенсора (GE), содержащего глюкозооксидазу, а на рис. 7.4-5,б приведены градуировочные графики для трех различных скоростей потока. Увеличивая скорость потока от 0,5 до 1,0 мл/мин, линейный диапазон измерений можно легко расширить от
Рис. 7.4-5. а — потокораспределительная система ПИА для определения глюкозы с амперометрическим сенсором, детектирующим ферментативно генерируемый пероксид водорода; б — градуировочные графики для глюкозы в диапазоне концентраций 0-40 ммоль/л прн трех различных скоростях потока: (А) 0,50, (А) 0,75 и (х) 1,00 мл/мин [7.4-5].
7.4. Проточно-инжекционным анализ
4S1
20 до 40 ммоль/л глюкозы, что гарантирует применимость системы для физиологических измерений. Кроме того, время воздействия можно использовать для кинетической дискриминации, используя различия в скоростях диффузии определяемого вещества н мешающих веществ в мембранном слое электрода. Таким образом, с помощью системы, показанной на рис. 7.4.-5, достигнуто полное пространственное разделение сигналов глюкозы и парацетамола, даже когда парацетамол добавляли в избытке. Помимо этого, следует обратить внимание на тот факт, что работа (био) сенсора в режиме ПИА обеспечивает постоянный контроль за самим сенсором: если регистрируемый сигнал есть мера определяемой концентрации, то сигнал нулевой линии показывает стабильность сенсора.
□ Сочетание ААС и ПИА улучшает характеристики детектирующего устройства.
Атомно-абсорбционная спектрометрия (ААС) — один из ряда аналитических методов, характеристики которого могут быть улучшены, а в некоторых случаях улучшены значительно, прн сочетании с ПИА. Так, при использовании системы, состоящей лишь из соединительной линии между на-сосом/инжектором и пламенным прибором ААС (рнс. 7.4-6), можно достичь ряда преимуществ за счет повторимого и воспроизводимого ввода пробы.
□ Короткое время экспозиции пробы устраняет или значительно уменьшает риск влияния основы/солевого загрязнения.
Как видно из рис. 7.4-6, частота ввода проб может быть существенно улучшена по сравнению с традиционным распылением растворов проб (рис. 7.4-6,о), но, что более важно, в течение времени, необходимого для распыления одной пробы, можно инжектировать две индивидуальные аликвоты пробы, что означает, что ПИА обеспечивает не только улучшение точности, но улучшает и воспроизводимость (рнс. 7.4-6,б). Более того, можно использовать как преимущество тот факт, что проба воздействует на детектор в течение очень небольшого периода времени. В оставшееся время происходит очистка детектора раствором носителя. Это означает, что отношение промывка/проба велико н, следовательно, шанс засорить горелку, например из-за высокого солевого содержания, значительно уменьшается или устраняется. Это можно видеть на рис. 7.4-6,в при сравнении градуировочных циклов для чистых водных стандартных растворов свинца и стандартных растворов, моделирующих морскую воду (3,3% NaCl); очевидно, что два градуировочных цикла идентичны. Эта особенность с еще большей очевидностью продемонстрирована Шредером с corp. [7.4-6], которые установили отсутствие какого-либо изменения регистрируемого сигнала в экспериментах, которые заключались в 160 повторяющихся инжекциях стандартного раствора меди с концентрацией 1 м.д., приготовленных на основе 30%-ного раствора NaCl.
Далее мы рассмотрим процедуры, где на динамику дисперсионных процессов накладываются химические реакции н где ПИА позволяет использовать кинетику химических процессов, т. е. кинетический эффект используют либо для повышения селективности аналитического метода, либо для того, чтобы получить информацию, которая недоступна с помощью обычных непроточных
1В*
452
Глава 7. Методы химического анализа и их применение
Рис. 7.4-6. Одноканальная потокораспределительная система ПИА для определения ионов металлов методом атомно-абсорбционной (АА) спектрометрии пламени. Записи получены при скорости потока 4,9 мл/мин и объеме инжектируемой пробы 150 мкл [7.4-3]. а — градуировочный цикл для цинка, полученный при инжектировании стандартных растворов в диапазоне 0,10-2,0 м.д.; 6 — выход самописца для стандартного раствора 1,5 м.д., полученный 1 —при инжектировании через систему ПИА и 2— при непрерывном распылении в обычном режиме(также со скоростью 4,9 мл/мин). D представляет величину коэффициента дисперсии, которая в случае 2 равна 1; в — градуировочный цикл для серии стандартных растворов свинца (2-20 м.д.), записанный без добавки (0%) и с добавкой (3,3%) хлорида натрия к стандартным растворам.
операций. Это хорошо видно на примере так называемых конверсионных методик ПИА.
7.4.6. Конверсионные методики ПИА (применения средней дисперсии)
□ Конверсионные методики ПИА — это операции, в которых недетектируемые частицы превращают в детектируемый компонент за счет кинетически контролируемой химической реакции.
Система, показанная на рис. 7.4-3, где детектирование хлорида с очевидностью требует химической реакции, является хорошим примером использования средней дисперсии. При конструировании таких систем важно помнить, что дисперсия должна быть достаточной, чтобы произошло частичное смеши-
7.4. Проточноинжекционный анализ
453
вание пробы и реагента, но не должна быть чрезмерной, чтобы не произошло нежелательного разбавления определяемого вещества и, как следствие, не ухудшился предел обнаружения.
□ Конверсионные методики дают возможность использовать кинетическую дискриминацию и кинетическое усиление.
Большинство методик ПИА основано на средней дисперсии, поскольку определяемое вещество должно быть подвергнуто какой-либо разумной «конверсии». В самом широком смысле конверсионные методики ПИА можно определить как методики, с помощью которых недетектируемые частицы превращают в детектируемый компонент за счет кинетически контролируемой химической реакции с целью проведения соответствующей пробоподготовки, генерации реагента или модификации основы пробы. При выполнении этих задач дополнительные преимущества дают кинетическая дискриминация и кинетическое усиление.
□ Кинетическая дискриминация основана на использовании различий в скоростях первичной реакции и побочных реакций.
Кинетическая дискриминация использует различие скоростей реакций реагента с определяемым и мешающими веществами. При кинетическом усилении химические реакции направляют в сторону образования определяемого вещества. В то время как в непроточном варианте процессы приводят к равновесию, так что нельзя использовать тонкие различия между скоростями реакций, в режиме ПИА малые различия в скоростях реакций с одним и тем же реагентом обеспечивают различную чувствительность измерений.
Простой пример процедуры гомогенной конверсии, использующей кинетическую дискриминацию, — использование последовательности реакций, направленной на определение хлорат-иона в технологическом растворе:
2CIO3 + 10Ti3+ + 12Н+ — 10Ti4+ + Cl2 + 6Н2О (быстро)
CI2 + LMB —> МВ (быстро)
МВ + Ti3+ -> LMB + Ti4+ (медленно)
Анализ проводят, инжектируя пробу хлората в поток носителя — подкисленного раствора титана(Ш), который затем соединяют со вторым потоком лейкометиленового синего (LMB). В то время как первые две реакции протекают очень быстро, третья реакцйя восстановления синей формы МВ идет медленно. Таким образом, концентрацию хлорат-иона можно легко определить по оптической плотности раствора МВ, образующегося во второй реакции, тогда как образование LMB имеет место уже после того, как зона пробы прошла через детектор.
□ В ПИА можно использовать промежуточные или метастабильные продукты реакций с интересными аналитическими характеристиками, такими, как высокий молярный коэффициент поглощения.
Другим примером, иллюстрирующим данный подход, служит определение тиоцианата—вещества, представляющего интерес в клинической химии, по-
454
Глава 7. Методы химического анализа и их применение
SCW+ 5-Br-PADAP + Окислитель ^ИИ*> Окрашенный продукт
(метастабилен)
Рис. 7.4-7. о —реакция тиоцианата с б-Br-PADAP. которая в присутствии окислителя (например, дихромата) в кислой среде приводит к образованию метаста-бильного продукта, характеристики которого представляют аналитический интерес; б — потокораспределительная система ПИА для определения метастабильного продукта реакция [7.4-7].
скольку обычно оно не содержится в сколько-нибудь значительных количествах в организме человека, за исключением курильщиков. Время полуразложения тиоцианата в теле составляет приблизительно 14 дней, и, следовательно, с помощью анализа жидких сред организма (слюна, кровь или моча) можно легко отличить курильщика от некурящего. Быстрый, простой и удобный ПИА-метод определения тиоцианата основан на реакции этого соединения с 2-(5-бром-2-пиридилазо)-5-диэтиламинофенолом (5-Br-PADAP) в кислой среде в присутствии дихромата в качестве окислителя (рис. 7.4-7,а), приводящей к образованию хотя и промежуточного, но интенсивно окрашенного (красноватого) продукта с высоким молярным поглощением.
Последняя особенность делает метод идеальным для определения низких содержаний тиоцианата, несмотря на то, что промежуточная природа продукта подразумевает, что он быстро образуется, а затем постепенно исчезает со временем жизни порядка 10 с. Важно, следовательно, чтобы отсчет снимали в точке, где развитие окраски максимально.
□ Воспроизводимый контроль времени в ПИА служит основой определения ме-тастабильных компонентов.
□ ПИА позволяет дискриминировать меняющийся фоновый сигнал.
Используемая система ПИА показана на рис. 7.4-7,б. Пробу (50 мкл слюны) инжектируют в поток носителя (вода), который последовательно соединяется с реагентом (5-Br-PADAP) и затем с окислителем, причем концентрация кислоты в конечной смеси составляет приблизительно 2 моль/л. Благодаря воспроизводимой установке времени можно детектировать и определять метастабиль-ный окрашенный компонент. Однако существует дополнительная проблема. Даже если тиоцианат отсутствует, 5-Br-PADAP и дихромат постепенно реагируют друг с другом, образуя вещество, которое поглощает при используемой
7.4. Протонно-икжекцмонкый анализ
455
Рис. 7.4-8. Использование ПИА для определения метастабильных частиц в системе, которая вызывает постепенное увеличение фонового сигнала. Благодаря точной и воспроизводимой установке времени в ПИА возможна регистрация сигнала в такой момент времени, который соответствует наибольшей разнице между сигналом определяемого вещества и фоновым сигналом (отмечен стрелкой и вертикальной пунктирной линией).
длине волны (570нм), т. е. пассивный фоновый сигнал. Еще больше усложняет ситуацию тот факт, что фоновый сигнал возрастает вместе со временем реак-ции. Другими словами, мы сталкиваемся с такой ситуацией, которая показана на рис. 7.4-8, где нестационарный сигнал определяемого вещества как функция времени сначала растет и затем уменьшается, в то время как фоновый сигнал постоянно растет.
При использовании ПИА путем надлежащего подбора конструкции аналитической системы можно так отрегулировать время пребывания пробы, что детектирование будет проведено в точно такое время, когда разница между сигналами достигает своего максимума (показано стрелкой и вертикальной пунктирной линией). На рис. 7.4-9 показан ряд реально регистрируемых сигналов, полученных с помощью системы ПИА, приведенной на рис. 7-4-7. Слева показаны сигналы для серии водных стандартов (в диапазоне 5-100 мкмоль/л, каждый инжектировали дважды), а справа —пять проб слюны от некурящих и затем пять проб от курильщиков (обе серии также инжектировали дважды). Как можно видеть, курящие н некурящие разделены на две отчетливые группы, но более важен с аналитической точки зрения тот факт, что хотя фоновый сигнал сравнительно велик (порядка 0,25 отн. единиц поглощения, AU), а индивидуальные сигналы находятся в диапазоне mAU, воспроизводимость параллельных измерений весьма удовлетворительна.
□ Селективность может быть увеличена при использовании набивных реакторов, включающих, например, ионообменные материалы.
Возможности увеличения селективности в ПИА становятся более впечатляющими в методиках гетерогенной конверсии, когда можно включать в потокораспределительную системы осуществление таких операций, как газовая диффузия, диализ, жидкостная экстракция или использование набивных реакторов. Так, в качестве материалов колонки можно использовать ионообмен-
456
Глава 7. Методы химического анализа и их применение
Рис. 7.4-9. Записи сигнала для тиоцианата, полученные с помощью потокораспределительной система ПИА, показанной на рис. 7.4-7. Слева: градуировочный цикл для серии стандартных растворов тиоцианата, каждую пробу инжектировали дважды. Справа: выходные сигналы для десяти проб слюны, также инжектированных дважды, из которых первые пять взяты у некурящих, а оставшиеся пять—у курильщиков. Отметим, что нулевая линия соответствует очень большому фоновому сигналу, однако сигналы весьма хорошо воспроизводимы [7.4-7].
ники, чтобы перевести компонент пробы в детектируемую форму; примером является определение различных анионов с помощью атомно-абсорбционной спектрофотометрии. Например, цианид-ион определяют после реакции на колонке, содержащей CuS, что приводит к образованию растворимого тетрацианокупрата, позволяя косвенно определять цианид методом ААС за счет стехиометрического количества меди, выделяющейся с твердой поверхности [7.4-3]. Как упомянуто ранее, ионообменники можно использовать также для предварительного концентрирования определяемого вещества, чтобы подготовить его для определенного детектирующего устройства или просто удалить нежелае-мый компонент основы, который в противном случае может оказывать мешающее влияние. Однако наиболее широко в качестве наполнителей используют иммобилизованные ферменты (разд. 7.4-7).
□ ПИА позволяет использовать реагенты в момент образования (in statu nascendi).
Чтобы завершить картину, следует заметить, что созданы реакторы, содержащие окислители или восстановители, для получения реагентов in statu nascendi (например, Ag(II), Cr(II) или V(II)); преимущество достигается за счет защитной среды системы ПИА, позволяющей получать и применять реагенты, которые из-за присущей им неустойчивости не находят практического использования при обычных аналитических условиях.
7.4. Проточно-инжекционный анализ
457
7.4.7. Системы ПИА с ферментами
□ В ПЙА выгодно использовать иммобилизованные ферменты в набивных реакторах.
Использование иммобилизованных ферментов, помещаемых в небольшие колоночные реакторы, в режиме ПИА не только повышает селективность, экономичность и стабильность за счет иммобилизации, но также обеспечивает строгую повторяемость, а следовательно, и постоянную степень преобразований от цикла к циклу. Кроме того, за счет высокой концентрации фермента, иммобилизованного в малом объеме, достигаемая высокая активность фермента облегчает быструю конверсию субстрата при минимальном разбавлении пробы, а малый коэффициент дисперсии в свою очередь приводит к более низкому пределу обнаружения. Ферменты, -необходимые для конкретного анализа, могут быть включены в один набивной реактор или в последовательно соединенные реакторы, чтобы реализовать требуемую последовательность превращений или обеспечить оптимальные условия для реакций, протекающих в индивидуальных реакторах. Для контроля за этими реакциями можно использовать как оптические, так и электрохимические детекторы. Так, для оксидаз, которые генерируют пероксид врдорода, можно использовать детектирование хемилюминесценции, основанное на реакции с люминолом.
□ Хеми-/биолюминесценция и ПИА образуют идеальное сочетание, поскольку люминесцентные реакции дают нестационарное испускание.
Хеми- и биолюминесценция особенно привлекательны как способ детектирования, в первую очередь благодаря потенциально высокой чувствительности и широкому динамическому диапазону люминесцентного метода, а также из-за простоты требуемого оборудования. Более того, люминесценция имеет дополнительное преимущество перед другими оптическими методами: поскольку свет испускается и детектируется только при наличии пробы, нет никаких проблем с контрольным опытом.
□ Использование ферментов — звено в аналитической цепи, обеспечивающее селективность. тогда как ПИА—это мощное средство количественной оценки.
В то же время люминесцентные реакции обычно дают нестационарное испускание, потому что интенсивность испускаемого света пропорциональна скорости реакции, а не концентрации вещества (рис. 7.4-10). Следовательно, излучение наиболее часто испускается в виде вспышки, интенсивность которой быстро уменьшается, и по этой причине общепринятым способом определения является интегрирование интенсивности в течение определенного периода времени и установления связи интеграла с количеством определяемого вещества. Очевидно, однако, что если интенсивность света d2?/dt можно измерить при точно определенных и воспроизводимых условиях, так что все пробы будут обработаны—физически нли химически — одинаковым образом (т. е. измерения будут выполнены при идентичных временах задержки t-t, рис. 7.4-10), то можно непосредственно связать любое значение dE/dt (предпочтительно то, которое соответствует максимальному испусканию, At) с концентрацией определяемого вещества. Это достижимо за счет ПИА, н, следовательно, сочетание
458
Глава 7. Методы химического анализа и их применение
Рис. 7.4-10. Типичный ход зависимости генерации света (hi/) от времени в био-или хемилюминесцентной реакции. Количественное определение можно выполнять либо зная зависимость между количеством определяемого вещества и выделяющейся энергией (т. е. интегрируя площадь под кривой), либо используя ПИА и определяя интенсивность dE/dt после фиксированного периода времени At, которая при соблюдении кинетики псевдопервого порядка (св 2> сд) прямо пропорциональна концентрации определяемого вещества.
мл/мин
БУФер [^(СХЦЗ- Ц
Люминол
Р1
Рйс. 7.4-11. а — потокораспределительная система для определения глюкозы с детектированием по хемилюминесценции. Пробу S инжектируют в поток буферного носителя С и продвигают к реактору ER, содержащему иммобилизованную глюко-зооксидазу, где глюкоза пробы разлагается, образуя пероксид водорода. После слияния с люминолом и гексацианоферратом(Ш) зона пробы проходит через короткий канал (2 см, что соответствует 16 мкл) в детектор света D, выход которого соединен с компьютером; б — интегрированный микроколлектор ПИА, содержащий компоненты показанной выше потокораспределительной системы а. С — поток буферного носителя, R1 —люминол, R2 — гексацианоферрат(Ш). Проточная ячейка включает два фотодиода D, заключенные в кожух Н, смонтированный на основной плате микроколлектора В [7.4-3].
7.4. Проточно-инжекционный анализ
459
люминесценции н ПИА произвело революцию в применении био- и хемилюминесценции как методов детектирования в аналитической химии.
Пример использования хемилюминесценции приведен на рис. 7.4-11, на котором показана система для определения глюкозы с помощью иммобилизованной глюкозооксидазы. Пробу инжектируют через клапан в поток буферного носителя (чтобы обеспечить постоянство pH) и затем направляют в ферментный реактор. Там глюкоза разлагается с образованием пероксида водорода, который последовательно смешивают с люминолом и гексацианоферратом (III), что приводит к возникновению хемилюминесценции, контролируемой с помощью набора фотодиодов.
Сообщается о многочисленных приложениях ферментного анализа в ПИА, преимуществом которых является тот факт, что эти компоненты составляют в аналитической цепи звено, которое обеспечивает селективность, и таким образом цепь вся становится селективной. По тем же причинам ПИА не только дополняет (био)сенсоры, но во многих случаях может быть привлекательной альтернативой [7.4-8].
7.4.8. Схемы проточно-инжекционной генерации гидридов
□ Проблемы, связанные с генерацией гидридов, в целом преодолимы при реализации этих схем в условиях ПИА.
Некоторые элементы (такие, как As, Sb, Bi, Se, Те и Ge) могут при реакции с сильным восстановителем типа тетрагидробората натрия превращаться в гидриды. Газообразные гидриды можно легко отделить от основы пробы и направить в нагреваемую кварцевую проточную ячейку прибора ААС, где они за счет нагрева атомизуются н возбуждаются излучением, так что можно селективно определить интересующий элемент. Первоначально метод генерации гидридов использовали в обычном (непроточном) варианте, но это осложнялось рядом проблем, которые иллюстрирует рис. 7.4-12.
□ Мешающие влияния за счет присутствия ионов переходных металлов, таких, как Си, Со и Ni, устраняют или кинетически дискриминируют в пользу первичной реакции образования гидрида.
Превращение определяемого вещества должно обязательно проводиться в кислой среде. Как показано на рисунке, существуют, однако, возможные побочные реакции и мешающие влияния. Сам тетрагидроборат может реагировать с кислотой, образуя водород, и в результате реагента не хватает для образования гидрида. Следовательно, необходимо готовить тетрагидроборат в слабощелочной среде и смешивать с пробой и кислотой точно тогда, когда это требуется, и жестко контролировать условия. Серьезную возможность помех представляет образование свободных металлов и осадков боридов металлов, особенно Ni, Си и Со. Если ионы этих металлов присутствуют в пробе, они восстанавливаются тетрагидроборатом, образуя коллоидные свободные металлы или бориды металлов, которые, как было показано, служат превосходными катализаторами разложения гидридов до того, как они достигнут измерительной ячейки. Однако, благодаря преобладанию в ПИА динамических условий, а также за
♦60 Глава 7. Методы химического анализа и их применение
Генерация гидрида/атомизация
ВИТ '
A»®*. Sb**, Sb**--77—» AsHz.SbHs.SnH4
КислотаСНХ) * * *
A>Hz.SbHz.SnH4 ----------» AS, Sb, Sb ♦ ПН2
Побочные/мешающие реакции
ВН«* ЗНХ ♦ Н*----------> вха* 4 Н2
t1eI*(HI.Cu,Co)---В1<* > н«° (более медленная
Кислота(НХ) реакция)
AsHz.SbHz.SbH4------——» As, Sb. Sb * ПН2
Рис. 7.4-12. Реакции, протекающие при генерации газообразных гидридов (на примере As, Sb и Sn) и возможные конкурентные побочные реакции. Последние можно существенно подавить или даже устранить за счет использования ПИА.
счет короткого времени пребывания пробы в системе, эти побочные реакции в значительной мере можно устранить или кинетически дискриминировать в пользу основной реакции. Если побочные реакции все же протекают, точный контроль времени в системе ПИА обеспечивает их протекание в одинаковой степени для всех введенных проб [7-4-3, 7.4-9J.
7.4.9. Обработка и концентрирование пробы в режиме on-line
Одной из характеристик ПИА является то, что он позволяет выполнять обработку и концентрирование пробы в режиме on-line. Эти задачи можно решать либо за счет включения подходящих колоночных реакторов (например, ионооб-менников для удержания ионных форм, инжектированных в большом объеме раствора пробы, которые затем можно элюировать малым объемом инжектированного элюента, так чтобы регистрируемый сигнал попадал в рабочий диапазон прибора [7.4-3, 7.4-9]), либо за счет конструкций самой системы ПИА. Хорошим примером здесь служит определение низких содержаний селена(1У) и мышьяка(У/Ш).
□ Коленчатый реактор является эффективным средством концентрирования за счет соосаждения.
Определение следовых количеств Se(IV) (или As) представляет особый интерес в случае водных проб, таких, как питьевая вода. Хотя сложная комбинация индуктивно-связанной атомно-эмиссионной спектрометрии (разд. 8.1) н масс-спектрометрии (ИСП-МС, разд. 8.5) позволяет определять низкие концентрации, ей присуща проблема мешающего влияния димера Аг-Аг+ при значении mlz, идентичном значению для селена, и Аг-С1+, идентичном значению для мышьяка. Следовательно, необходимо выбрать альтернативный подход, который должен включать этап предварительного концентрирования. Для
7.4. Проточно-инжекционный анализ
461
Рис. 7.4-13. Схема проточно-инжекционной атомно-абсорбционной спектрометрии с генерацией гидридов для соосаждения — растворения селена или мышьяка в режиме on-line, а — система в положении «наполнение»: пробу всасывают насосом Р1 и смешивают в режиме on-line с буфером и соосаждающим агентом La(III). Полученный осадок улавливают коленчатым реактором KR; б — система в положении «инжекция»: клапан V переключают, позволяя элюенту (НО) проходить через коленчатый реактор, растворяя и элюируя осадок, а также продвигая его к точке смешивания с восстановителем (ИаВНд), приводящим к образованию гидрида. Гидрид отделяют от жидкой основы в газо-жидкостном сепараторе SP и затем с помощью дополнительного потока аргона переносят в нагреваемую кварцевую ячейку QTA атомноабсорбционного спектрометра [7.4-11].
решения этой проблемы большие усилия были предприняты в работе Фана [7.4-9, 7.4-10], который описал оригинальный и простой способ ПИА для определения следов металлов, включающий предварительное концентрирование за счет соосаждения подходящим реагентом с последующим растворением. Развивая идею, первоначально предложенную для обычного непроточного анализа, Фан сумел включить эту процедуру on-line в ПИА, обходясь без фильтра для сбора осадка, а просто используя коленчатый реактор, сделанный из трубки Microline. Поскольку такое устройство ранее применяли для увеличения радиальной дисперсии за счет уменьшения осевой дисперсии [7.4-3], оно оказалось весьма пригодным в качестве коллектора для осадка, который просто прилипал к стенкам коленчатой трубки. В своих экспериментах Фан получал «органический» осадок, который был растворим в органической жидкости. Однако в данном случае, когда необходимо не только провести сооса-ждение для предварительного концентрирования, но и использовать преимущество генерации гидрида Se(IV) для отделения его от компонентов основы (разд. 7.4.8), крайне необходимо отыскать «неорганический» осадок, который в конечном итоге можно растворить в неорганическом растворителе. Это и было сделано при использовании La(III), который при pH порядка 9,5 осаждается в виде Ьа(ОН)з и соосаждает Se(IV) и As(III). Схема системы ПИА показана на рис. 7.4-13,а (в положении «наполнение»), где проба с помощью временной инжекции осаждается в коленчатом реакторе (KR, трубка Microline
462
Глава 7. Методы химического анализа и их применение
с длиной 100 см и внутренним диаметром 0,5 мм) за счет одновременного добавления аммиачного буфера и раствора La(III). После накопления достаточного количества осадка инжекторный клапан переключали, осадок растворяли (рис. 7.4-13,5) с помощью носителя 1,0 моль/л кислоты, затем смешивали с раствором тетрагидробората и полученный гидрид селена направляли в проточную ячейку прибора ААС с помощью дополнительного потока аргона.
С использованием этого подхода удалось снизить предел обнаружения Se(IV) до 0,006 мкг/л, относительное стандартное отклонение sr для концентрации 0,1 мкг/л составило 3%, предел обнаружения для As(V/III) был равен 0,003 мкг/л. Более того, 400-кратные количества Ni(II) и Cu(II) не оказывали сколько-нибудь существенного влияния на результаты определения.
7.4.10. Использование процесса физической дисперсии.
Градиентные методики ПИА
□ Градиентные методики ПИА основаны на использовании одной или нескольких концентраций вдоль созданного градиента концентраций.
До сих пор мы ограничивались рассмотрением максимума пика в качестве аналитического сигнала. Однако любой пик ПИА, который мы создаем, инжектируя пробу, обязательно содержит континуум концентраций, представляющих все величины между нулем и Стах (рис. 7.4-2), независимо от того, намереваемся ли мы использовать только один его компонент, такой, как максимум пика, чтобы получить наш аналитический результат.
□ Градиентные методики ПИА породили ряд новых аналитических приложений.
Однако, поскольку поток носителя несжимаем и его движением, следовательно, можно строго управлять и воспроизводить его от одного цикла измерений к другому с высокой точностью, любая часть градиента концентраций характеризуется двумя параметрами: фиксированным временем задержки Ц (рис. 7.4-2), прошедшим с момента инжекции to, и фиксированной величиной дисперсии, т. е. коэффициент дисперсии инжектированной пробы равен Ds = Cg/Cs, тогда как для реагента Dr — Cr/Cr. Следовательно, в наших силах воспроизводимо выбрать и использовать любую часть (или части), просто установив время t,, связанное с этой частью; концентрация в выбранной части будет равна С = C°/D(ti). Эта особенность создает основу для развития ряда градиентных методик ПИА (рис. 7.4-14), делая доступным ряд новых аналитических приложений. Список различных градиентных методик представлен в табл. 7.4-1 [7.4-3, 7.4-12].
□ Метод остановки потока основан на приостановке потока смеси проба-реагент.
Одним из приложений, заслуживающим отдельного рассмотрения, является метод остановки потока. Как указано в табл. 7.4-1, этот подход используют или для остановки потока смеси проба/реагент, чтобы увеличить время реакции без чрезмерного разбавления (что произошло бы, если увеличивать время
7.4. Проточно-инжекционный анализ
463
Рис. 7.4-14. Градиентное сканирование, основанное на выборе точки отсчета для разных времен задержки (каждое соответствует разному соотношению проба/реагент) [7.4-3]. В каждой точке детектор быстро сканирует диапазон длин волн (а), создавая таким образом дополнительное измерение для матрицы время—концентрация (б). Матрица состоит из ряда последовательных эмиссионных спектров, зарегистрированных на возрастающей и убывающей частях дисперсионной зоны, которая содержит Na, К и Са, инжектированные в атомно-эмиссионный спектрометр с быстрым сканирующим монохроматором.
Таблица 7.4-1. Примеры градиентных методик ПИА
Градиентное разбавление
Выбор и использование для детектирования определенной части на градиенте концентрации, причем концентрация равна С = Используют, например,
для согласования концентрации пробы с динамическим диапазоном детектора.
Градиентная градуировка
Выбор и использование нескольких частей на градиенте, концентрации в которых определяют через коэффициент дисперсии индивидуальных компонентов. Градуировка по нескольким точкам может быть проведена при одной инжекции концентрированной пробы.
Остановка потока
Увеличение чувствительности измерения за счет увеличения времени пребывания или определение концентрации пробы путем измерения скорости реакции при кинетике псевдопервого порядка, см. рис. 7.4-15.
Градиентное сканирование
Совместное использование градиентного разбавления с динамическим детектором, который для каждого уровня концентрации способен непрерывно сканировать физический параметр, например длину волны или потенциал (см. рис. 7.4-14).
Титрование
Выбор частей на возрастающей и убывающей ветвях градиента концентрации, где получена эквивалентность между титруемым веществом и титрантом, и установление взаимосвязи различия между этими частями с концентрацией инжектированного определяемого вещества.
Проникающие зоны
Использование кривых отклика градиентов концентрации, образованных при одновременной инжекции двух или более зон. Помимо экономичного способа введения растворов пробы и реагента, можно использовать для измерения коэффициентов селективности, а также для метода стандартных добавок в широком регулируемом диапазоне отношений концентраций образец сравнения/определяемое вещество.
464
Глава 7. Методы химического анализа и их применение
Рис. 7.4-15. а — простая потокораспределительная система ПИА с остановкой потока. Когда инжектируют пробу S, с помощью микропереключателя на инжекторном клапане активируется электронный таймер Т. Время от инжекции до остановки прокачки (время задержки), а также длительность периода остановки можно установить с помощью электроники; б — принцип метода ПИА с остановкой потока на примере инжектирования зоны окрашенной пробы в бесцветный поток носителя и регистрации поглощения с помощью проточной ячейки: А — непрерывная прокачка; В — прокачка в течение 9 с, остановка на 14 с, затем снова непрерывная прокачка; С — пунктирная линия изображает кривую, которую регистрировали бы, если бы в проточной ячейке в течение 14-секундного интервала остановки имела место химическая реакция нулевого или псевдонулевого порядка, т. е. наклон был бы тогда прямо пропорционален концентрации определяемого вещества [7.4-3].
реакции за счет прокачивания через длинную реакционную спираль), или чтобы контролировать реакцию в развитии (если остановка происходит внутри самого детектора), что позволяет нам видеть, как она протекает.
□ Остановку потока можно использовать или для увеличения времени реакции без избыточного разбавления, или для контроля за химической реакцией, чтобы измерить скорость реакции.
Если условия подобраны таким образом, чтобы моделировать кинетику псевдопервого порядка (избыток реагента и справедливость предположения, что концентрация пробы не изменяется за время наблюдения), скорость реак-
7.4. Проточно-инжекционный анализ
465
ции (т. е. наклон регистрируемой кривой) становится прямо пропорциональной концентрации определяемого вещества, как показано на рис. 7.4-15.
Достоинства такого подхода связаны с высокой воспроизводимостью, с которой различные сегменты градиента, образованного инжектированной пробой, могут быть, выбраны и остановлены, и из того факта, что мы можем изменить условия реакции, чтобы удовлетворить критерию реакции псевдопервого порядка, просто выбрав точку (или точки) на профиле дисперсии, которая удовлетворяет этому условию, т. е. выбрав время, когда мы производим действительную остановку зоны. Измерения скорости реакции, когда скорость образования (или расходования) определенных веществ измеряют в большом числе точек, не только улучшают воспроизводимость анализа, но и в целом обеспечивают его надежность. Дело в том, что мешающие явления, такие, как величина контрольного опыта, существование фазы запаздывания и нелинейные кривые скорости, можно легко идентифицировать и устранить. Благодаря автоматической регистрации контрольного опыта метод остановки потока привлекателен для выбора таких сфер приложения, как клиническая химия, биотехнология и контроль производства, когда часто имеют дело с основами пробы, у которых значения контрольного опыта сильно различаются.
7.4.11. Заключительные замечания
□ ПИА обеспечивает возможность разработки новых и уникальных аналитических методик.
Хотя ряд приложений ПИА был детализирован, чтобы продемонстрировать разнообразней применимость ПИА, это лишь часть приложений, которые имеются в литературе. Надеемся, что они могут послужить вдохновляющим примером для поиска новых решений. В одной из ранних работ [7.4-13] автор написал, что «возможности ПИА еще далеко не исчерпаны, потому что конечным мерилом для аналитического метода является не то, что он позволяет сделать что-то лучше, чем это можно сделать другим путем, а то, что он позволяет решить проблему, которую никаким другим способом решить нельзя». И ПИА действительно позволяет нам осуществить уникальные приложения. Единственным ограничением является только наша изобретательность.
Литература
[7.4-1] Ruzicka, J., Hansen, Е.Н., Anal. Chim. Acta 1975, 78, 145-157. [7.4-2] Ruzicka, J., Marshall, G.D., Anal. Chim. Acta 1990, 237, 329-343.
[7.4-3] Ruzicka, J., Hansen, E.H., Flow Injection Analysis. 2nd ed. New York: John Wiley & Sons, 1988.
[7.4-4] Монографии по ПИА, см. ссылку 3; Valcarcel, M., Luque de Castro, M.D., Flow Injection Analysis: Principles and Applications. Chichester: Ellis Horwood, 1987; Karlberg, B., Pacey, G.E., Flow Injection Analysis. A Practical Guide. New York: Elsevier, 1989.
(7.4-5] Petersson, B.A., Anal. Chim. Acta 1988, 209, 231-237.
4И Глава 7. Методы химического анализа tf мх применение
[7.4-6] Schrader, W., Portala, F., Weber, D., Fang, Z.L., in 5. Colloquium Atomspectrom. Spurenanalytik, Welz, B. (Ed.), Perkin-Elmer, 1989; pp. 375-383.
[7.4-7] Bendtsen, A.B., Hansen, E.H., Analyst 1991, 116, 647-651.
[7.4-8] Hansen, E.H., Taianta 1994,, 41, 939-948.
[7.4-9] Fang, Z.L., Flow-Injection Separation and Preconcentration.- Weinheim: VCH, 1993.
[7.4-10] Fang, Z.L., Flow-Injection Atomic Spectrometry. New York: John Wiley fc Sons, 1995.
[7.4-11] Nielsen, S., Sloth, J.J., Hansen, E.H., Analyst 1996, 121, 31-35.
[7.4-12] Hansen, E.H., Fees emus’ Z. Anal. Chem. 1988, 329, 656-659.
[7.4-13] Hansen, E.H., Quim. Anal. 1989, 8, 139-150.
Вопросы и задачи
1. В чем различия между обычным дискретным и непрерывным проточным анализом?
2. Каковы характерные различия между ПИА и непрерывным проточным методом?
3. Дайте определение коэффициента дисперсии D в ПИА; как его определяют для конкретной системы ПИА?
4. Дает ли коэффициент дисперсии какую-либо информацию о химической реакции, протекающей в конкретном анализе?
5. Почему выгодно работать с ион-селективными электродами и биосенсорами в режиме ПИА?
6. Многие биосенсоры основаны на использовании ферментов из-за высокой селективности последних. Чтобы получить линейную зависимость сигнала от концентрации определяемого вещества, необходимо наличие кинетики реакции псевдопервого порядка. Что это означает? И как можно достичь таких условий в системе ПИА?
7. Известно, что сочетание ПИА и ААС дает определенные преимущества. Каковы они?
8. Что означает кинетическая дискриминация и как ее можно использовать в ПИА? Приведите пример.
9. Что означает термин «реагенты in statu nascendi» и почему ПИА применим для их использования?
10. Почему привлекательно использовать ПИА в сочетании с детектированием, основанным на био- или хемилюминесценции?
11. Что подразумевает генерация гидридов? И почему выгодно использовать ПИА в этой ситуации?
12. Предварительное концентрирование пробы используют в ПИА, чтобы достичь более низких пределов обнаружения. Приведите два примера того, как можно провести концентрирование.
13. Известно, что в методе генерации гидридов некоторые переходные металлы могут оказывать мешающее влияние. Объясните, как они действуют в этом отношении.
14. Что подразумевается под градиентными методиками ПИА и на чем они основаны?
15. В ПИА используют измерения с остановкой потока? Каким целям они служат?
7.5. Термический анализ
467
7.5. ТЕРМИЧЕСКИЙ АНАЛИЗ
Цели изучения
• Описать основные принципы и приборы для главных термоаналитиче-ских методов: ТГ, ДТА, ДСК, АВГ, ТМА.и некоторых их сочетаний.
• Обсудить выбор параметров, ограничения метода и источники ошибок для ТГ и ДТА/ДСК.
• Дать руководство по использованию термоаналитических методов для решения аналитических проблем на некоторых избранных примерах исследования материалов.
7.5.1. Введение
ICTAC (Международный союз по термическому анализу и калориметрии) определяет термический анализ как «группу методов, в которых свойства пробы контролируют в зависимости от времени или температуры, когда температура пробы в специальной атмосфере программируется. Программа может включать нагревание или охлаждение с постоянной скоростью изменения температуры, либо поддержание температуры постоянной, либо любую последовательность этих процессов». Ключевыми словами здесь являются проба, программируемое нагревание или охлаждение и температура, но очевидно, что столь широкое определение включает методы, выходящие за рамки традиционной аналитической химии. Тем не менее, основные термоаналитические методы, например, термогравиметрию (ТГ) и дифференциальный термический анализ (ДТА), обычно используют для решения аналитических проблем, так как именно эти методы часто предлагают элегантный и весьма быстрый путь решения.
□ Для исследования материалов используется большое число разнообразных термоаналитических методов.
В табл. 7.5-1 перечислены основные методы, включая их утвержденные ICTAC названия и сокращения, а также свойства, контролируемые в зависимости от времени или температуры. Некоторые из этих методов существуют уже несколько сотен лет (например, ТГ, ДТА и дилатометрия), тогда как другие были разработаны сравнительно недавно для решения специальных задач исследования материалов. Термоаналитические методы весьма разнообразны; более того, эти методы можно объединять друг с другом, чтобы получить больше информации для одной пробы при одном измерении (комбинированные методы, причем наиболее частое сочетание —ТГ+ДТА). Имеется также развитая приборостроительная промышленность, предоставляющая пользователю выбор современных термоаналитических приборов с компьютерным управлением, обработкой данных и возможностью автоматизированного управления пробой.
Термоаналитическне методы в настоящее время находят широкое применение в исследовании и анализе материалов, а также в контроле качества и контроле технологических процессов. Изучаемые материалы включают полимеры, лекарственные вещества, керамику, металлы и сплавы. Их также можно
ш
Глава 7. Методы химического анализа и их применение
Таблица 7.5-1. Основные термоаналитические методы
Измеряемое свойство Метод Рекомендованное сокращение
Масса Термогравиметрия, или термогравиметрический анализ ТГ ТГА
Летучее вещество Анализ выделяющихся газов АВГ
Температура Дифференциальный термический анализ ДТА
Теплота или тепловой поток Дифференциальная сканирующая калориметрия ДСК
Механические свойства Размеры Акустические свойства Электрические свойства Магнитные свойства Оптические свойства Термомеханический анализ Динамический механический анализ Термодилатометрия Термосонометрия Термоакустометрия Термоэлектрометрия Термомагнетометрия Термооптометрия ТМА ДМА
Радиоактивный распад Эманационный термический анализ Эта
использовать, чтобы изучать прекурсоры для синтеза новых материалов и для создания оптимальных условий процесса синтеза.
В последующих разделах дан обзор основных термоаналитических методов, включая их сочетания, связанные с задачами аналитической химии.
7.5.2. Термогравиметрия (ТГ)
□ Термогравиметрия, по-видимому, наиболее широко используется как самостоятельный термоаналитический метод.
Термогравиметрия (ТГ), или термогравиметрический анализ (ТГА), — один из основных методов в термическом анализе. Прибор для ТГ—термовесы — построен на основе печи, в которой проба механически присоединена к аналитическим весам. Первоначально термовесы разработал К. Хонда в 1915 г., но с тех пор прибор был значительно усовершенствован в отношении чувствительности, автоматической записи кривой Дтп от Т, а также управления, включая скорость нагрева, атмосферу и т. д.
Термовесы
Тремя существенными составными частями современного ТГ-прибора являются весы, печь и система управления прибором и обработки данных. На рис. 7.5-1 приведено схематическое изображение типичных термовесов.
Весы
Чувствительные и надежные аналитические весы представляют центральную часть прибора ТГ, и поэтому понятно, что многие компании, выпускающие
7.5. Термический анализ
469
Рис. 7.5-1. Схематическое изображение терм о весов.
ТГ-приборы, производили или производят весы. Для весов требуются чувствительность порядка 1 мкг и максимальная нагрузка 1 г. В большинстве случаев пробы в ТГ-экспериментах фактически весят от 10 до 50 мг. Существует несколько типов механизмов для весов, включая пружинное коромысло, консоль и торсионные весы, но предпочтительнее всех взвешивающий механизм с нулевой точкой, потому что тогда проба всегда остается в одной и той же зоне нагрева печи.
Очень популярная схема весов, созданных в начале 1960-х гг., показана на рис. 7.5-2. Электромагнитные весы относительно нечувствительны к вибрациям и имеют высокую чувствительность (до 0,1 мкг) и термическую стабильность. Когда пробу помещают слева, коромысло отклоняется от равновесия. Это детектируется с помощью системы фотоячейки, которая включает вращающий электродвигатель, чтобы восстановить начальное положение коромысла. Восстанавливающая сила пропорциональна изменению веса и току, потребляемому двигателем.
Печь
Температурный диапазон печи, используемой в термовесах, зависит главным образом от материалов, из которых печь изготовлена. Если этот диапазон доходит до 1000-1100°С, то можно использовать трубки из плавленого кварца вместе с материалами нагревательного элемента типа Кантал, но температуры вплоть до 1500-1700°С требуют керамических огнеупоров, таких, как оксид
470
Глава 7. Методы химического анализа' и их применение
а б в
Горизонтальный Загрузка сверху Подвешивание
п₽оба'Л—Т । П Печь^™ 1------«--- 1^1
Рис. 7.5-3. Три основных способа размещения пробы относительно весов и печи.
алюминия или муллит. Большинство производителей термовесов предлагают приборы, которые могут достигать температуры 1500°С, но лишь немногие производят приборы для более высоких температур из-за проблем с материалами (включая нагревательные элементы), конструкцией печи, а также термопарами для измерений температуры.
Существуют в основном три способа размещения пробы относительно весов и печи (рис. 7.5-3), каждый из которых имеет свои преимущества и недостатки. Во всех случаях важно, чтобы проба находилась в однородной температурной зоне печи и чтобы механизм весов был защищен от излучательного нагрева и агрессивных газов, как выделяющихся из пробы, так и используемых в качестве реакционной атмосферы. Коммерческие термовесы предполагают использование инертной (азот или аргон) или окислительной (воздух или кислород) атмосферы, но лишь очень немногие сконструированы для работы с агрессивными или химически активными атмосферами, например хлором или диоксидом серы.
Управление прибором и обработка данных
Недавно большинство коммерческих приборов были объединены с персональным компьютером для управления циклами нагрева и охлаждения, а также для хранения и обработки данных. Он может также рассчитывать первую производную кривой Дп от Т (ТГ), которая называется кривой производной термогравиметрии (ПТГ). Кривая ПТГ может существенно помочь в интерпретации кривых ТГ за счет разрешения перекрывающихся химических реакций. Другим способом разрешения реакций и достижения термодинамического равновесия является использование изотермического нагрева или очень малой скорости нагрева. В квази-изотермической ТГ (называемой также ТГ высокого разрешения или ТГ с контролируемой скоростью) нагревание замедляется, когда начинается изменение веса. Это улучшает разрешение, но, с другой стороны, требует больше времени на ТГ-цикл. Потерю времени можно частично компенсировать, устанавливая относительно высокую скорость для тех областей, где не происходит никаких изменений.
Источники ошибок
На правильность измерения массы и температуры влияют несколько факторов. Эти факторы связаны либо с ТГ-прибором и параметрами, выбираемыми для эксперимента, либо с пробой и атмосферой. К счастью, большинством
7.5. Термический анализ
471
Таблица 7.5-2. Основные факторы, влияющие на регистрируемые массу (?п) и температуру (7) в термогравиметрии •
Плавучесть (тп)
Конденсация и реакция (тп)
Электростатический эффект (тп)
Скорость нагрева (Т)
Поток газа (7)
Держатель пробы (7)
Энтальпия реакции (7)
Размер и упаковка пробы (7)
этих факторов можно управлять или корректировать результаты с учетом их влияния.
□ Ряд факторов влияет на ТГ-кривые и может вызывать ошибки.
В табл. 7.5-2 приведены основные факторы, влияющие на термогравиметрическую кривую. Большинство из них влияет на правильность регистрации температуры, и в некоторых случаях влияние может быть драматичным. Рис. 7.5-4 дает пример того, насколько велико может быть совместное влияние скорости нагрева и размера пробы на температуру реакции. Как более малый размер пробы, так и более медленный нагрев понижают температуру, при которой протекает термическая реакция, совместный эффект может значительно превышать 100 градусов. Температурную градуировку ТГ-прибора удобно проводить со стандартами, основанными на магнитном переходе. Если, к примеру, стандарт из металлического никеля взвесить на термовесах во внешнем маг-
Рис. 7.5-4. Кривые ТГ, ПТГ и ДТА разложения Ег2(БО4)з-8Н2О на воздухе для двух различных экспериментальных условий: (I) 200 мг, 10° С/мин (сплошная линия) и (II) 20 мг, 2° С /мин (пунктирная линия).
т
Глава 7. Методы химического анализа и их применение
Температура, *С
Рис. 7.5-5. Влияние геометрии держателя пробы на разложение карбоната кальция (СаСОз).
нитном поле, он теряет ферромагнетизм в точке Кюри, Тс — 353°С. Это можно легко наблюдать как скачок на кривой Дтп от Т.
Держатель пробы также может оказывать огромное влияние, особенно когда окружающая атмосфера находится в химическом равновесии с пробой. Хорошо известным примером служит разложение карбоната кальция (рнс. 7.5-5), где открытый держатель пробы позволяет текущему газу эффективно уносить образующийся СОг- С другой стороны, лабиринтовый тигель справа мешает выходу СОг, пока его парциальное давление не превысит окружающее давление (1 атм), и, таким образом, разложение начинается при 900°С. Два держателя пробы в середине более открыты, чем лабиринтовый тигель, но и здесь влияние собственной атмосферы на температуру разложения по сравнению с открытой структурой первого держателя желобкового типа очевидно.
Применения ТГ
Одним из ранних применений ТГ было точное определение условий высушивания или прокаливания аналитических осадков. Хотя это аналитическое приложение потеряло свою значимость, все еще существует ряд проблем, для которых ТГ может дать решение. Например, она может указать содержание воды в пробе или даже отличить адсорбированную воду от конституционной, потому что вода этих типов обычно удаляется прн различных температурах.
Другим практическим примером является непосредственный анализ угля и других подобных топлив (рис. 7.5-6). Если нагревание сначала проводить в инертной атмосфере (азот), то по термограмме можно определить количества влаги и летучих веществ. Затем при фиксированной температуре термовесы автоматически переключают атмосферу на окислительную, вследствие чего углерод сгорает и его содержание, а также содержание золы можно определить по ТГ-кривой. Точность результатов, полученных на ТГ-приборе, сравнима с точностью стандартного гравиметрического метода, требующего значительно больше ручной работы.
Второй пример касается новых оксидных сверхпроводников. С момента их открытия в 1986-87гг. большинство прикладных исследований было сосредоточено на так называемом соединении 1-2-3, или УВагСидОт-а-- Содержание кислорода, определяемое величиной а?, имеет решающее значение для сверх-
7.5. Термический анализ
473
Рис. 7.5-6. Непосредственный анализ угля методом ТГ.
Температура и режим нагрева, *С
юо.в 100.6
100.4 100.2
100.0
* 99.6
I 996
99.4 99.2
99.0 988
h
700 800 900 1000
Температура, °C
Рис. 7.5-7.- Данные ТГ для УВазСизОе,»» показывающие влияние нагревания (потеря кислорода) и охлаждения (обратное присоединение кислорода вплоть дох = 6,9).
проводящих свойств. Для высокой критической температуры (90 К) х должно быть мало, т.е. содержание кислорода должно быть близко к 7. Содержанием кислорода управляют при медленном охлаждении на воздухе, и этот процесс можно контролировать и определять содержание кислорода с помощью термогравиметрии (рис. 7.5-7-).
7.5.3. Дифференциальный термический анализ (ДТА) и дифференциальная сканирующая калориметрия (ДСК)
□ ДТА и ДСК способны определять изменения энергии при нагревании или охлаждении пробы. Изучаемые явления могут иметь физическую или химическую природу.
В то время как термогравиметрия позволяет измерять изменение массы пробы при нагревании или охлаждении, методы дифференциального термического анализа (ДТА) и дифференциальной сканирующей калориметрии (ДСК) связаны с измерением изменений энергии. Оба метода тесно связаны друг с другом, давая однотипную информацию. С практической точки зрения разница заключается в принципах устройства и работы приборов: в ДТА измеряют
474
Глава 7. Методы химического анализа и их применение
разность температур между пробой и эталоном, тогда как в ДСК температуры пробы и эталона поддерживают одинаковыми и контролируют разницу в необходимой для этого мощности нагрева. Классический ДТА является наиболее. старым методом термического анализа, Ле Шателье предложил его в 1887 г. Сегодня ДТА и ДСК — самые широко используемые термоаналитические методы.
Основы методов и приборы ДТА и ДСК
ДТА
Когда пробу (S) и материал эталона (R) однородно нагревают в печи, и в пробе имеет место эндотермический эффект, ее температура Ts будет отличаться от температуры эталона Tr. Разность температур ДТ = Ts — Tr регистрируют как функцию температуры Т/?, которая практически равна температуре печи, и получают кривую ДТА (рис. 7.5-8,а). Аналогично экзотермическая реакция образует пик, но в противоположном направлении.
Материал эталона должен иметь следующие свойства. Во-первых, он не должен претерпевать термических изменений в используемом диапазоне температур. Во-вторых, он не должен реагировать с держателем пробы или с термопарой. Третье требование касается теплопроводности и теплоемкости, которые должны быть близки аналогичным характеристикам пробы во избежание смещения или искривления нулевой линии кривой ДТА. Для неорганических образцов в качестве эталонов обычно используют глинозем (AI2O3) или карбид кремния (SiC), а для органических полимеров можно использовать, например, силиконовое масло.
ДСК
В отличие от режима работы в ДТА, в ДСК разность температур между пробой и эталоном поддерживают равной нулю, т. е. ДТ = Tg — Тд = О. Это достигается с помощью независимых нагревателей, а метод называют ДСК с компенсацией мощности (рис. 7.5-8,в).
В дополнение к классическому ДТА и ДСК с компенсацией мощности существует третий вариант приборов ДТА и ДСК, а именно калориметрический
Рис. 7.5-8. Схематическая диаграмма трех основных дифференциальных термоаналитических методов, а — классический ДТА; б~ ДТА модификации Бёрсма; в-ДСК.
7.5. Термический анализ
475
ДТА или ДТА Бёрсма, который иногда также называют ДСК с тепловым потоком (рис. 7i5-8,5). В этом варианте, как и в классическом ДТА, измеряют разность температур. Пробу и эталон размещают на нагревательной пластине, которая производит управляемый тепловой поток от стенки печи к пробе и эталону. Таким образом в ДТА Бёрсма (ДСК с тепловым потоком) конструкция дает разность ДТ, пропорциональную разности теплового потока между пробой и эталоном. Все варианты ДТА и ДСК дают похожую информацию, но рабочий диапазон температур прибора ДСК с компенсацией мощности более ограничен (обычно до 700°С), чем у приборов ДТА, где высокотемпературные модели могут достигать температуры 1500°С и выше. В то же время, чувствительность приборов, работающих ниже 700°С, выше, чем у высокотемпературных, и, следовательно, проба может быть намного меньше (порядка нескольких миллиграммов); в качестве эталона достаточно иметь пустой держатель пробы.
Градуировка и интерпретация кривых ДТА и ДСК
□ Для количественных измерений прибор ДТА или ДСК должен быть проградуирован по стандартным образцам сравнения.
Для ДТА и ДСК имеется довольно обширный набор стандартных образцов сравнения (СОС). СОС выпускают в NIST (Национальный институт стандартов и технологий США) в сотрудничестве с ICTAC, и они покрывают диапазон для градуировки температур от ~-32°С до 925°С. Градуировка основана на четких переходах, которые эти материалы претерпевают при определенной температуре, например полиморфный переход а-кварца в /3-кварц при 573°С. Градуировка энтальпии (и одновременно температуры) основана на энтальпиях плавления высокочистых металлов. Металлы включают индий, олово, свинец, цинк и алюминий, что покрывает диапазон температур от 156 до 660°С или типичный рабочий диапазон прибора ДСК. Для более высоких температур можно использовать серебро (т.пл. 961°С) или золото (т.пл- 1064°С). ДСК можно использовать также для определения теплоемкости Ср пробы, но и в этом случае требуется стандартный образец, обычно сапфир (монокристалл AI2O3).
При количественном определении возникает проблема, связанная с интегрированием площади пика, которая зависит от проведения нулевой линии. Обычно компьютерные программы прибора ДТА или ДСК включают некоторые возможности для экстраполяции нулевой линии (рис. 7.5-9), но пользователь должен сделать надлежащий выбор. Измеренное изменение энтальпии ДИ (Дж/г) прямо пропорционально площади пика:
ДИ = Ак/тп (7.5-1)
где А — площадь, к — градуировочный коэффициент н т — масса пробы.
В дополнение к отчетливым переходам первого рода, таким, как плавление и полиморфные превращения, кривые ДТА и ДСК можно использовать для детектирования переходов второго рода, где ДИ = 0, но имеется изменение теплопроводности (Ср). Примеры включают стеклование полимеров и
476
Глава 7. Методы химического анализа и их применение
Рис. 7.5-9. Некоторые возможные альтернативы определения нулевой линии и площади пика в ДТА/ДСК.
Рис. 7.5-10. Температуру стеклования (Тя2) можно определить как среднюю точку между начальной (Тк1) и конечной (Тк3) температурами.
точку Кюрн ферромагнитных материалов. Процедура определения температуры стеклования показана на рис. 7.5-10.
Таблица 7.5-3. Влияние пробы и рабочих параметров на разрешение и чувствительность
Максимальное Максимальная
Параметр разрешение чувствительность
Размер пробы малый
Размер частиц малый
Упаковка пробы плотная
Скорость нагрева медленно
Теплопроводность атмосферы высокая
большой большой рыхлая быстро низкая
Как и в термогравиметрии, рабочие параметры влияют на вид кривых ДТА и ДСК. Для надлежащей интерпретации термических эффектов ключевыми факторами, которые следует учитывать, являются разрешение пиков и чувствительность. В табл. 7.5-3 дана упрощенная картина того, как различные параметры влияют на вид кривых. Помимо этого, выраженное влияние оказывает конструкция прибора. Максимальное разрешение получают, когда держатели пробы и эталона связаны вместе в блочной конструкция, тогда как изолированные держатели обеспечивают более высокую чувствительность. Чашка для пробы (открытая, закрытая или закрытая с тонким отверстием) также оказывает влияние.
7.5. Термический анализ
477
Применения ДТА и ДСК
ДТА и ДСК имеют весьма широкий диапазон применений в отношении как типа материалов, так изучаемых физических и химических явлений. В табл. 7.5-4 перечислены основные области применения, а ниже будет рассмотрено нисколько примеров. Они включают проблемы из областей как качественного, Так и количественного анализа материалов.
Таблица 7.5-4. Физические и химические явления, которые можно исследовать с помощью ДТА/ДСК
Эндотермические Экзотермические Не детектируются методом ТГ
Физические явления
Фазовые переходы X X X
Плавление X X
Кипение X
Сублимация X
Адсорбция X
Десорбция X
Абсорбция X
Магнитный переход" X
Стеклование" X
Изменение теплоемкости • X
Химические явления
Хемосорбция X
Разложение X или X
Окисление X или X
Восстановление X или X
Горение X
Полимеризация X X
Поликонденсация X
Реакции в твердой фазе X или X X
Каталитические реакции X X
а — Переходы второго рода, только изменение теплоемкости.
Определение степени чистоты
Одно из наиболее частых применений ДСК в органической и фармацевтической промышленности — это определение степени чистоты. Оно основано на уравнении Вант-Гоффа:
РТ2
Тв-Тт = ^х (7.5-2)
где То н Тт — температуры плавления чистого и загрязненного вещества соответственно, АН— энтальпия плавления чистого вещества, а ж—мольная доля примеси. Уравнение Вант-Гоффа основано иа предположении о системе с идеальной эвтектикой, образование твердого раствора или соединения между основным материалом и примесью должно быть исключено.
478
Глава 7. Методы химического анализа и их применение
ЛН ПП
Рис. 7.5-11- Чистота пробы влияет на форму и положение кривой ее плавления. Кривые плавления ДСК для проб с чистотой 97; 98,5 и 99% (слева направо).
ЮО 200 300
Температура, °C
Рис. 7.5-12. Кривые ДСК для пластмассовых отходов. ПЭНП и ПЭВП — полиэтилен низкой и высокой плотности соответственно, ПП—полипропилен, ПТФЭ—политетрафторэтилен (тефлон).
На основе единичного стандартизованного цикла ДСК компьютерная программа прибора может рассчитать степень чистоты с приемлемой точностью. На качественном уровне степень чистоты с использованием уравнения Вант-Гоффа видна по форме кривой плавления (рис. 7.5-11).
Другие применения
ДТА/ДСК может быть использован как метод «отпечатков пальцев» для качественного анализа. На рис. 7.5-12 приведены результаты анализа пластмассовых отходов, в которых можно распознать шесть различных полимеров на основе их температур плавления или перехода.
На рис. 7.5-13 приведен пример количественного анализа. Шлак доменной печи образуется в больших количествах как побочный продукт при производстве железа и стали. Для некоторых его применений необходимо знать степень кристалличности. Ее можно определить с помощью ДТА, измеряя площадь экзотермического пика выше 700°С, который связан с кристаллизацией стеклообразной части шлака (рис. 7.5-13,а). Связь между площадью пика и степенью некристалличности (стекла), определенней с помощью метода сравнения (оптическая микроскопия), имеет линейный характер (рис. 7.5-13,6).
7.5.4. Комбинированные методы
Комбинация методов, осуществляемых одновременно
□ Аналитические возможности методов термического анализа часто возрастают за счет применения не отдельного метода, а их комбинации.
Достаточно часто обнаруживают, что информации, полученной отдельным ТА методом (например, ТГ), недостаточно для того, чтобы решить конкретную
7.5. Термический анализ
479
Рис. 7.5-13. а ~ кривые ТГ и ДТА для шлака из доменной печи, показывающие только экзотермический переход, связанный с кристаллизацией выше 700°С; б—зависимость площади пика ДТА от степени некристалличности (содержания стекла).
задачу. Тогда следует добиваться дополнительной информации, осуществляя эксперимент на другом приборе (иапример, ДТА). Это требует затрат времени, а различия в конструкции приборов затрудняют сравнение двух наборов данных и получение правильных выводов.
Поскольку ТГ и ДТА (или ДСК) вместе обеспечивают взаимодополняющую информацию, преимущества проведения одновременных измерений для одной пробы очевидны. Техническая проблема объединения ТГ и ДТА была впервые решена в 1955 г. Ф. и Я. Паулик вместе с Л. Эрдеи весьма элегантно, учитывая доступные в то время электронные компоненты (рис. 7.5-14). За этой пионерской конструкцией вскоре последовал ряд других, и сегодня ряд компаний выпускает комбинированные приборы ТГ-ДТА. Обычно приборы имеют температурный диапазон до 1500°С и возможность получать и строить кривые ДТГ из ТГ-данных.
Часть ДТА в объединенном приборе ТГ-ДТА, однако, представляет собой компромисс в сравнении с прибором ДТА/ДСК, и, следовательно, объединенный прибор редко используют для истинно количественных измерений. Тем не
4И
Глава 7. Методы химического анализа м их применение
Рис. 7.5-14. Устройство комбинированного прибора ТГ-ДТА (и ПТГ) конца 1950-х гг. 7, 2—держатели пробы и эталона соответственно, 5—печь, 4 — весы, 5 — фотографическая регистрация. Отметьте образование сигнала ПТГ при движении катушки внутри постоянного магнита (6).
менее он обеспечивает дополнительную информацию, которая часто является решающей для правильной интерпретации процессов термического разложения. В объединенном приборе ТГ-ДТА следует принять во внимание те же факторы, которые влияют на отдельные кривые ТГ и ДТА, и, следовательно, очень важно должным образом знать и описывать экспериментальные условия (табл. 7.5-5).
Таблица 7.5-5. Основные пункты в рекомендациях ICTAC и ИЮПАК по представлению данных для ТГ, ДТА, ДСК и АВГ
• идентификация пробы (название, формула, состав)
• источник и история пробы (пробоподготовка и т. д.)
• атмосфера (состав, давление; статическая, динамическая или собственная)
• геометрия и материал держателя пробы
• вес пробы и ее упаковка
• тип прибора, скорость нагрева и используемая температурная программа
Пример
Неизвестное соединение (X) было проанализировано одновременно методами ТГ/ДТА в азоте. Конечный остаток, соответствующий потере веса 72,5%, рассчитанной из ТГ-кривой, был идентифицирован как СоО методом рентгеновской дифракции с использованием банка данных JCPDS. Дополнительная информация, полученная методом АВГ-МС, показала, что на первых двух этапах (1 —* * 2 и 2 —» 3) улетучивалась только вода, тогда как на этапе 3 —* 4 выделялись SO2 и SO3 в молярном отношении 1:2.
На основании этих данных можно рассчитать состав неизвестного соединения и установить механизм реакции. Во-первых, состав X можно вычислить на основе общей потери веса и информации АВГ (X должен содержать воду, серу и кислород). Достаточно близкое соответствие между рассчитанными (73,3%) и наблюдаемыми (72,5%) потерями веса указывает на состав X — C0SO4 • THsO (см. таблицу ниже). Этапы 1 -> 3 можно теперь легко идентифицировать как потерю воды в два этапа. Образование С03О4 на этапе 3 —» 4 вытекает из данных АВГ (80г:80з в отношении 1:2) и подтверждается данными потери веса.
7.5. Термический анализ
481
Другим способом одновременного измерения, часто сочетаемым с ТГ или ДТА, является анализ выделяющихся газов, который обсуждается ниже, в разд. 7.5.5. ДТА также был объединен в промышленно выпускаемом приборе с методом микроскопии с горячим предметным столиком.
На рис. 7.5-15 приведен пример комбинации ТГ-ПТГ-ДТА для нахождения оптимальных условий приготовления активированного европием оксосульфи-
Рис. 7.5-15. Одновременно регистрируемые кривые ТГ, ПТГ и ДТА для восстановления соосажденных сульфитов (Y,Eu)2(SOs)s- ЗНгО с 6%-ным содержанием Ей.
16 Аналитическая химия. Том 1
4вг
Глава 7. Методы химического анализа и их применение
да иттрия EuzYzOjS, который является красным люминофором в цветном ТВ. Синтез начинают соосаждением сульфитов Ей и Y в молярном отношении 6:100, которые затем восстанавливают монооксидом углерода. ТГ-кривая показывает дегидратацию (1) и восстановление сульфитов Ей (2) и Y (5). Как ПТГ-, так и ДТА-кривые показывают, что дегидратация проходит в два этапа, но только на кривой ДТА можно видеть фазовый переход (кристаллизацию) оксосульфида (4) при 650°С. Общая потеря веса (46,6%) меньше, чем теоретическое значение для восстановления (47,9%), что свидетельствует о протекании незначительной побочной реакции.
Комбинации с исследованием отобранных промежуточных проб (ex situ)
Для решения конкретных проблем с помощью ТА часто бывает необходимо идентифицировать промежуточные продукты и термически индуцированные процессы путем измерения ex situ, т. е. с отбором проб в середине или в конце процесса. С этой целью можно использовать все виды спектроскопических и дифракционных методов, но наиболее общепринятым и мощным методом является, пожалуй, рентгеновская дифракция порошков (см. разд. 7.5-6) и инфракрасная спектроскопия с преобразованием Фурье (ИКСПФ).
Пример использования мессбауэровской спектроскопии для идентификации промежуточных и конечных продуктов реакции приведен на рис. 7.5-16. Сульфат железа(П) получается как побочный продукт в ряде производственных процессов. Термический анализ используют для изучения его превращения в более ценные продукты. В восстановительной атмосфере монооксида углерода механизм реакции сложен, и продукты невозможно идентифицировать на основе только ТГ-кривой. На рис. 7.5-16,а показаны ТГ-кривые, записанные при двух скоростях нагрева, и точки a-d, в которых пробы были отобраны для мёссбауэровского анализа (рис. 7.5-16,б). Положение мёссбауэровских пиков и их расщепление можно использовать для однозначной идентификации различных фаз, содержащих железо. Из ТГ-кривой очевидно, что проба а представляет безводный FeSC>4, являющийся также преобладающей фазой и в пробе Ь. На следующем этапе с из мёссбауэровского спектра очевидно наличие двух ферромагнитных фаз (FeS и Ее). Конечный продукт d содержит три фазы: Ее, FeS и Fe^C. Образование цементита (ЕезС) в атмосфере СО (2 СО <=* СО2 + С) объясняет небольшое увеличение веса при более высоких температурах.
7.5.5. Анализ выделяющихся газов (АВГ)
Большинство термически индуцированных реакций при высоких температурах включают выделение из пробы летучих веществ. Поскольку ТГ дает информацию только об общей потере массы, а ДТА/ДСК — об изменении энтальпии, механизмы реакций часто бывает сложно интерпретировать. Это особенно проявляется в случае конкурирующих реакций или при образовании нескольких газообразных веществ с примерно одинаковой молекулярной массой, например HjO и NH3.
Анализ выделяющихся газов (АВГ) представляет собой метод анализа газов, который может быть использован в сочетании с ТА-измерением (обыч-
7.5. Термический анализ
483
а
Рис; 715-16. а — термическое разложение FeSCh-HsO в монооксиде углерода. Для возможных продуктов приведены рассчитанные (теоретические) весовые уровни; обозначения a-d относятся к пробам, взятым для мёссбауэровских экспериментов, показанных на рис. 6.
4М
Глава 7. Методы химического анализа и их применение
Рис. 7.5-17. Устройство реактивного сепаратора.
но с ТГ). Для АВГ можно использовать ряд методов —от простых методов детектирования газов (детектирование выделяющихся газов, ДВГ) до более сложных, основанных на газовой хроматографии (ГХ), инфракрасной спектроскопии с преобразованием Фурье (ИКСПФ) или масс-спектрометрии (МС). Методы АВГ, основанные на ГХ, были описаны уже в 1960-х гг., но из-за свойственной ей медлительности газовую хроматографию в настоящее время используют менее широко, чем ИКСПФ или МС.
□ Анализ выделяющихся газов (АВГ) методами МС или ИКСПФ бывает необходим для выяснения механизмов сложных реакций, включающих газообразные вещества.
Масс-спектрометрия в комбинации с АВГ имеет ряд преимуществ, включая помимо высокой чувствительности возможность однозначного, одновременного и быстрого детектирования нескольких газообразных веществ в рамках доступного диапазона масс прибора. Но в дополнение к проблеме представительного отбора пробы возникает существенная проблема, как сконструировать интерфейс между МС и ТА-прибором, в частности ТГ-анализатором.
Поскольку квадрупольные и времяпролетные приборы МС работают при высоком вакууме (10~8-10-1Оатм), для интерфейса ТГ-МС остается две возможности: а) использовать термовесы только под вакуумом и соединять их с МС напрямую или б) работать на термовесах при атмосферном давлении и уменьшать давление с помощью интерфейса. Очевидно, что вторая альтернатива предпочтительнее для ТА-экспериментов; имеются сообщения о нескольких конструкциях интерфейсов для сочетания атмосферная ТГ-МС. Они включают нагреваемый капилляр, двойное сопло и реактивный сепара
7.5. Термическим анализ
485
тор (рис. 7.5-17). Каждая система имеет свои достоинства и недостатки; например, нагреваемый капилляр является, вероятно, наиболее простой по конструкции системой, но капилляр легко засоряется при конденсации летучих веществ. Коммерческие приборы основаны на интерфейсе или с нагреваемым капилляром, или с двойным соплом. Что касается масс-спектрометра, основные ограничения здесь налагаются диапазоном масс, если используют прибор квадрупольного типа. Недорогой квадрупольный МС-прибор обычно покрывает диапазон масс вплоть до 200-400, что достаточно для неорганических веществ (рис. 7.5-18), но не для органических фрагментов с большими молекулярными массами.
В то же время, приборы ИКСПФ не требуют интерфейса для снижения давления, достаточно простой нагреваемой трубки для соединения газовой ячейки ИКСПФ с ТА-прибором. Трудности с ИКСПФ связаны с тем, что этим методом нельзя ни детектировать неполярные молекулы (такие, как Ог и N2), ни различать очень близкие по структуре молекулы (такие, как гомологи углеводородов).
7.5.6. Другие термоаналитические методы
Имеется большое число разнообразных свойств материалов, которые можно контролировать в зависимости от температуры (см. табл. 7.5-1). Ниже обсуждаются лишь два из наиболее часто используемых дополнительных методов и их применения.
Термомеханические методы
□ Термомеханические методы применяют для изучения полимеров и керамики.
Термомеханические методы можно разделить на два класса. Термодилатометрия (ТД) измеряет изменения размеров (термическое расширение) как функцию температуры без внешней нагрузки или давления. Термомеханический анализ (ТМА) также связан с изменением размеров, но при статической нагрузке, тогда как динамический механический анализ (ДМА) позволяет измерять различные механические параметры при динамической или пульсирующей нагрузке. Эти методы широко применяют, особенно для изучения керамики или полимеров.
Термодилатометрия
Экспериментально довольно просто измерить изменение длины в отдельном направлении как функцию температуры (рис. 7.5-19). Проблемы с точностью могут возникать только тогда, когда проба очень мала. Однако современные термодилатометры, основанные на лазерных интерферометрах, способны определять изменения размеров значительно меньше 1 мкм. Определение термического расширения в различных направлениях дает степень анизотропности н в этом смысле более информативно, чем только определение изменения объема.
Данные по термическому расширению полезны, например, для определения того, совместимы или нет подложка и тонкая пленка на ней: если различие велико, тогда пленка будет трескаться или шелушиться. Помимо этого, методом ТД можно исследовать фазовые превращения.
486
Глава 7. Методы химического анализа и их применение
Рис. 7.5-19. Схематическое изображение двух измерений ТМА в режимах сжатия (о) и расширения (б).
Термомеханический анализ (ТМА) и динамический механический анализ (ДМА)
В отличие от ТД-измерений, в ТМА и ДМА к пробе приложена значительная нагрузка. ТМА представляет собой расширение ТД-метода, в котором нагрузку можно использовать для сжатия или растяжения пробы, например. ДМА требует более сложного инструментального оформления, поскольку изменяющуюся нагрузку прикладывают периодически (синусоидально).
И ТМА, и ДМА можно использовать для изучения твердых полимеров, пленок и волокон, а также вязких жидкостей и гелей. Примеры применений включают определение температуры стеклования полимера и динамический модуль волокна (ДМА).
Высокотемпературная рентгеновская дифракция
Рентгеновская дифракция (РД) — это мощный метод идентификации кристаллических фаз и их смесей. Благодаря наличию компьютерных файлов РД-данных идентификация фаз часто очень проста, при условии что фазы кристаллические и их концентрация выше предела обнаружения метода (1-2%).
РД можно использовать как метод ex situ для идентификации промежуточных и конечных продуктов термического процесса. Такой подход весьма распространен, потому что не требует никакого специального оборудования; используют обычный порошковый дифрактометр. Для идентификации кристаллических фаз необходим файл по порошковой дифракции или на карте, или в виде компьютерного файла. Естественно, справочный файл может быть создан самим пользователем, но в большинстве случаев более удобно обращаться к банку JCPDS (Объединенного комитета по стандартам для порошковой дифракции).
□ Измерения рентгеновской дифракции можно проводить или exsitu, или in situ.
Высокотемпературная РД in situ (исследование «на месте», без отбора проб) является чисто термоаналитическим методом, поскольку изменения в кристаллической структуре пробы можно контролировать при нагревании держателя пробы с использованием температурной программы. Выпускается ряд конструкций высокотемпературных печей. Наибольшая достигаемая температу-
7.5. Термический анализ
487
Рис. 7.5-20. Запись высокотемпературной рентгеновской дифракции, показывающая как орторомбический {вверху) УВагСизОг-а; переходит в тетрагональную фазу, когда температура превышает 600°С.
ра может составлять 2500°С, но тогда прибор должен работать под вакуумом, а в качестве материалов используют только тугоплавкие металлы, такие, как молибден (т. пл. 2625°С) или вольфрам (т.пл. 3410°С). Обычно температурный диапазон доходит до 1400-1500°С, что позволяет применять платину (т. пл. 1770°С) даже в окислительной атмосфере.
Применения высокотемпературной РД включают исследование фазовых переходов, контроль твердофазных реакций и измерение термического расширения путем определения постоянной решетки как функции температуры.
Тогда как обычный детектор в порошковом дифрактометре сканирует измеряемый интервал 20, позиционно-чувствительный детектор (ПЧД) может измерять одновременно более широкий диапазон (5-120 градусов угла 20). ПЧД стали очень популярными в высокотемпературных работах, поскольку позволяют следить за быстрыми изменениями, происходящими при изменении температуры. Хорошим примером служит переход орторомбической фазы в тетрагональную в сверхпроводящем УВааСизОт-а- (так называемое соединение 1-2-3), который происходит около 600°С (рис. 7.5-20). Орторомбическая фаза обладает сверхпроводимостью с критической температурой Тс — 90 К.
Высокотемпературную РД сочетают с ДСК, и эта комбинация становится более мощным способом обнаружения и идентификации фазовых изменений, чем любой из этих методов в отдельности.
Литература
Brown, М.Е., Introduction to Thermal Analysis, Techniques and Applications, London: Chapman and Hall, 1988.
Charsley, E.L., Warrington S.B., (Eds.), Thermal Analysis — Techniques and Applications, London: Royal Society of Chemistry, 1992.
488
Глава 7. Методы химического анализа и их применение
Chang, D.D.L., De Haven, P.W., Arnold, H., Ghosh, D., X-ray Diffraction at Elevated Temperatures. A Method for in situ Process Analysis. New York: VCH Publishers, Inc. ,1993.
Gallagher, P.K., Thermoanalytical Methods, Ch.7. in Cahn, R.W., Haasen, P., Kramer, E.J., (Eds.), Materials Science and Technology — A comprehensive Treatment, Vol. 2A, Weinheim: VCH Publishers Inc., 1992.
Hatakeyama, T., Quinn, F.X., (Ed.), Thermal Analysts. Fundamentals and Applications to Polymer Science, Chichester: John Wiley & Sons, 1994.
Hemminger, W.F., Cammenga, H.K., Methoden der Termischen Analyse, Berlin: Springer-Verlag, 1989.
Hill, J.O., (Ed.), For Better Thermal Analysis. 3rd ed. London: ICTA, 1991.
Keatch, C.J., Dollimore, D., An Introduction to ThermoGravimetry. 2nd ed. London: Heyden and Sons, 1975.
Van der Plaats, G-, The Practice of Thermal Analysis. Switzerland: Mettler, 1991.
Pope, M.I., Judd, M.D., Differential Thermal Analysis . London: Heyden and Sons, 1977.
Turi, E.A., Thermal Characterization of Polymeric Materials. New York: Academic Press, 1981.
Wendlandt, W.W., Thermal Analysis. 3rd ed. New York: John Wiley & Sons, 1986.
Вопросы и задачи
1. Какие параметры оказывают влияние на ТГ-кривую и каким образом?
2. Опишите способы увеличения разрешения ТГ-данных.
3. Какие физические и химические явления можно исследовать с помощью ДТА/ДСК, но не ТГ?
4. Какой выбор параметров привод ит к максимальному разрешению в ДТА/ДСК?
5. Какой информацией рекомендуется снабжать кривые ТГ или ДТА/ДСК с целью упрощения интерпретации?
6. Как градуируется температурная шкала в цикле а) ТГ и б) ДТА/ДСК?
7. Как проявляется различие между переходами первого и второго рода при исследовании методом ДТА/ДСК? Приведите пример перехода второго рода, который можно детектировать методом ДТА/ДСК.
8. Каковы обычные комбинации ТА-методов н какие преимущества они дают по сравнению с отдельными методами?
9. Как можно выполнить АВГ? Каковы преимущества и трудности различных подходов?
10. Напишите химические уравнения термического разложения оксалата кальция, используя данные АВГ-МС, показанные на рис. 7.5-18. Рассчитайте также теоретические потери веса для каждого этапа и сравните их с наблюдаемыми потерями веса.
11. Чем в принципе различаются методы ТД, ТМА и ДМА?
12. Какими методами можно дополнительно охарактеризовать (ex situ) конечный остаток, который вы получили от ТГ-цикла, если исходная проба представляет собой а) неорганическую соль; б) органический полимер?
13. Перечислите некоторые области применения а) ТГ и б) ДТА/ДСК, в которых эти термоаналитические методы конкурируют или превосходят другие аналитические методы. Постарайтесь также объяснить, почему это так.
7.6. Элементный органический анализ
489
7.6. ЭЛЕМЕНТНЫЙ ОРГАНИЧЕСКИЙ АНАЛИЗ
7.6.1. Введение
Целью этого анализа, вообще говоря, является определение элементного состава органических соединений ,т. е. определения содержания составляющих их элементов — С, Н, N, S и О, а также галогенов, Р и металлов. Знание процентного содержания всех элементов в соединении наряду с его молекулярной массой делает возможным расчет молекулярной брутто-формулы. Поэтому методики определения процентного содержания основных составляющих органических соединений (С, Н, N и О) в миллиграммовых органических пробах были разработаны еще в начале XX в. Примечательно, что сначала для этой цели были созданы новые микровесы, чтобы обеспечить высокую чувствительность взвешивания таких проб, а именно от 0,1 до Змг пробы.
□ Чтобы определить содержание С, Н, N и О, пробу сначала необходимо подвергнуть химическому разложению.
Принцип С,Н,1Ч-метода, разработанного австрийским химиком Фрицем Преглем (Нобелевская премия 1923 г.), сохраняется и в современных инструментальных методах элементного органического анализа. Он заключается в разложении точно взвешенной навески пробы с помощью быстрого сжигания (окисления) в потоке кислорода при высокой температуре (от 1000° С до 1800°С), разделении продуктов реакции (каковы они, если образец состоит только из С, Н, N?) и последующем определении небольших количеств газов, образующихся при сгорании, а также в конечном этапе вычислений.
При данных условиях сгорания (см. ниже) органические соединения разлагаются, образуя СОг, Н2О, N2 и NO2. Кислород определяют из отдельной пробы после разложения в гелии, приводящего к образованию СО, который можно превратить в СОг и определять отдельно.
7.6.2. Методы, основанные на использовании для разделения селективной адсорбции
СН-анализатор конструкции Ф. Прегля (см. рис. 7.6-1)
Каталитическое сжигание (проба в Pt-тигле) происходит при 750°С в постоянном потоке кислорода. В качестве веществ, поддерживающих окисление, Прегль предложил CuO/PbCrO4 при 750°С и РЬОг при 190°С (почему?). Затем полученные продукты сгорания, Н2О н СОг, селективно поглощают с помощью предварительно взвешенных трубок, содержащих Mg(C104)2 и NaOH на асбесте (!) соответственно. Определение содержания С и Н в пробе основано на утомительной гравиметрической методике. Образующиеся также газы NO» и N2 в то время не определяли (почему?). Из-за экспериментальных ограничений метод был заменен современными схемами, позволяющими проводить автоматизированные определения.
490
Глава 7. Методы химического анализа и их применение
Рис. 7.6-1. Схема первого прибора, разработанного Ф. Преглем для одновременного определения С и Н.
CHN-анализатор конструкция У. Саймона (см. рис. 7.6-2)
□ Разложение в атмосфере €>2 для определения С, Н, N.
В этой первой коммерческой версии автоматического элементного органического анализатора (1963 г.) использованы в усовершенствованном варианте принципы сжигания и поглощения Прегля, но гравиметрическое определение заменено на дифференциальное измерение теплопроводности реакционных газов.
Сжигание: Приблизительно при 1000°С в кислороде органические пробы (1-3 мг) в Pt-тигле, с AgVOs/AgWC^ в качестве веществ, поддерживающих сгорание. Образующийся NOa: восстанавливают в восстановительном блоке (Си при 650°С) до N2, а избыток кислорода связывают в виде СиО. После завершения сгорания (несколько секунд) Ог выключают и включают поток носителя (Не). Галогены можно удалить адсорбцией на волокнах серебра, SO2 — окислением МпОг-
Адсорбция и определение реакционных газов: Как видно из рис. 7.6-2, газовая смесь после блоков сжигания/восстановления состоит из Н2О, СОг, N2 и Не. Последовательно удаляя НгО и затем СОг по методу Прегля, можно определить количество реакционных газов с помощью трех специальных ДТП (детекторов теплопроводности). Вся процедура занимает 14 минут.
CHNS + 0-анализатор конструкции Е. Пелла (рис. 7.6-3)
Этот современный анализатор работает с модифицированной системой сжигания и совершенно другим блоком разделения реакционных газов, основанным на газовой хроматографии.
Детектирование проводят с помощью ДТП, как в варианте Саймона. Система Пелла позволяет одновременно определять С, Н, N и S из одной микропробы и О из другой пробы при нагревании в гелии, но с использованием того же ГХ-метода разделения.
Рис. 7.6-2. Схема С, Неавтоматического анализатора, разработанного У. Саймоном.
492
Глава 7. Методы химического анализа и их применение
Сжигание: Пробу (обычно 1 мг) вводят в блок сжигания. Локальную температуру сжигания 1800° С получают за счет сильного экзотермического эффекта, когда проба в Sn-капсуле горит в кислороде при температуре печи 1000°С. Такое сжигание в динамической вспышке гарантирует полное разложение также галогенированных органических проб и других веществ, имеющих высокую термостойкость, таких, как металлоорганические и даже неорганические соединения. В том же блоке в качестве веществ, поддерживающих сгорание, используют СиО или WO3. Восстановление ГЮЖ до N2 и SO3 до SO2 проводят в восстановительной трубке с помощью меди при 650°С.
Разделение и детектирование: Реакционную газовую смесь разделяют затем с помощью газовой хроматографии, используя специально разработанную твердую фазу Poropak QS в набивной колонке. Реакционные газы элюируют в последовательности N2, СО2, Н2О и SO2 и количественно определяют с ДТП. С, Н, N, S можно определить одновременно из одной пробы массой около 1 мг приблизительно за 10 минут.
Определение кислорода
□ Разложение в атмосфере гелия для определения кислорода.
Содержание кислорода в органических пробах является важной информацией, особенно для современных топлив, в которых в качестве антидетонацион-
7.7. Химические сенсоры
493
ного агента вместо свинца используют кислородсодержащие соединения. Для таких приложений разработаны автоматические жидкостные инжекционные устройства, которые позволяют инжектировать жидкости непосредственно в пиролитическую камеру без взвешивания. Анализ на кислород проводят, нагревая органические пробы в гелии при 1120°С, при этом образуются СО, N21 Н2О, СН4 и кислотные газы (рис. 7.6-3, правая часть). Газовую смесь последовательно пропускают через содержащий никель слой углерода, являющийся эффективным катализатором восстановления (почему эффективность здесь столь важна?), и щелочной поглотитель для удаления кислотных газов. Оставшиеся газы инжектируют в ГХ-колонку, заполненную молекулярными ситами в качестве неподвижной фазы. В качестве детектора, как и прежде, используют ДТП. Зная массу пробы и определив СО, можно рассчитать содержание кислорода в пробе. Полный анализ на кислород занимает 5 минут.
Вопросы и задачи
1. Какова цель элементного органического анализа?
2. Что мы должны знать, чтобы рассчитать молекулярную брутто-формулу соединения?
3. Почему использование малых количеств пробы является преимуществом, но одновременно представляет сложность для анализа?
4. Опишите принцип элементного органического анализа Прегля.
5. Как видоизменен принцип Прегля в современных автоматических CHN-анализаторах?
6. Опишите принципы элементного органического анализатора конструкции Пелла и сравните его с анализатором конструкции Саймона.
7. При каких условиях в дополнение к С, Н и N можно определять О?
7.7. ХИМИЧЕСКИЕ СЕНСОРЫ
Цели изучения
• Обосновать необходимость и описать основные принципы работы химических сенсоров.
• Понять принципы работы электрохимических и микроэлектронных, оптических, термических и масс-чувствительных сенсоров.
• Понять принципы работы комбинации сенсоров с целью компенсации ограниченной селективности сенсоров.
7.7.1. Введение
Разработка химических сенсоров необходима для достижения ряда целей. Первой является замещение классических аналитических методов на сенсорные измерения. Типичным примером служит систематическая замена пламенных фотометров для определения компонентов электролитов крови, таких, как литий, натрий, калий и кальций, ион-селективными электродами.
494
Глава 7. Методы химического анализа и их применение
Вторая цель возникает из-за растущих требований к автоматизации производственных процессов. Измерение химических параметров необходимо для контроля и управления процессами. Так, на применении химических сенсоров основаны методы анализа on-line в контроле технологических процессов. Особые требования производства включают устойчивость к ошибкам, долговременную стабильность и температурную нечувствительность сенсоров. Дополнительные потребности возникают в связи с биотехнологическими процессами, где нужны многофункциональные биосовместимые сенсоры.
Контроль (мониторинг) химических величин также важен для охраны окружающей среды, в технике безопасности и медицине. Контроль pH и мутности воды представляет относительно простую задачу. pH можно измерять с помощью стеклянного электрода, а мутность — с помощью оптического сенсора. Предположительно, загрязнение воды тяжелыми металлами и органическими соединениями возможно непрерывно контролировать с помощью сенсоров, а ие методами off-line в лаборатории с предварительным отбором проб. Химические сенсоры нужны не только для определения индивидуальных веществ, но и для измерения суммарных параметров.
Важным требованием в области охраны окружающей среды является контроль за выбросом отходящих газов, например SO2, или за загрязнениями воздуха. В наличии уже имеются газовые сенсоры, основанные на принципах оптики или микроэлектроники. Непрерывный контроль загрязнений в почвах или твердых отходах более затруднителен; для этой цели требуются сенсоры, которые обеспечивали бы химическую информацию с помощью дистанционных измерений.
Для контроля за воздействием опасных веществ нужны недорогие сенсоры, для которых возможно массовое производство. Сенсоры этого типа, как ожидают, будут оценивать долговременную экспозицию или максимальную нагрузку. В добывающей промышленности необходимо определять взрывоопасные или ядовитые газы, для кондиционирования воздуха важно знание влажности и содержания СО.
Еще одной областью является мониторинг in situ («на месте»). Например, при операции на сердце необходимо измерять соотношение калия и натрия, поскольку это очень важный показатель состояния пациента. Другую важную область применения сенсоров в медицине представляет слежение во времени за содержанием лекарств, анестетиков и метаболитов в организме при операции. Существует растущая потребность в сенсорах, которые можно использовать в качестве неинвазивйых устройств, например для определения глюкозы в крови по ближнему ИК-спектру, регистрируемому через ноготь или кожу на пальце.
7.7.2. Характеристики и основные принципы
Химические сенсоры определяют следующим образом:
Химические сенсоры— это (небольшие) устройства, способные непрерывно определять концентрацию химических составляющих в жидкостях или
7.7. Химические сенсоры
495
газах и превращать эту информацию в режиме реального времени в электрический или оптический сигнал.
В принципе, сенсор состоит из химически чувствительного слоя, системы распознавания, преобразователя химической информации в электрический или оптический сигнал и электронного устройства для оценки данных, обычно интегрированного в сенсор. В качестве примера, для ион-селективного электрода химически чувствительным слоем служит твердая или жидкая мембрана, а преобразователь основан на электрическом потенциале, измеряемом с помощью вольтметра.
Электроника
• . \
k £ *
i
Проба с определяемым веществом
Схема сенсора
Технические характеристики сенсоров включают общие алитические критерии и специфические требования, такие, как долговременная стабильность. В табл. 7.7-1 перечислены типичные параметры, характеризующие сенсор.
Универсального основного принципа создания сенсоров не существует. Каждая практическая задача требует своего подхода, так что известны сенсоры, основанные на электрических, оптических, гравиметрических или термических принципах.
Обзор основных принципов разработки химических сенсоров дан в табл. 7.7-2. Со многими из этих принципов вы уже знакомились в разных местах этой книги. Этот раздел посвящен особым аспектам конструкции
496
Глава 7. Методы химического анализа и их применение
сенсоров, а новые принципы, такие, как принципы микроэлектронных или волоконно-оптических сенсоров, рассмотрены более детально.
Таблица 7.7-1. Типичные характеристики химических сенсоров
Предел обнаружения
Рабочий диапазон
Селективность / специфичность
Дрейф сигнала
Воспроизводимость
Миниатюризация
Механическая устойчивость
Время отклика
Долговременная стабильность
Совместимость с меняющимися условиями (давлением, температурой, взрывоопасностью, радиоактивностью, биологическими условиями, стерилизацией)
Таблица 7.7-2. Обзор схем детектирования и типов сенсоров
Схема детектирования ТЪп сенсора
Изменение проводимости Полупроводники металл-оксид Органические полупроводники
Изменение потенциала Ион-селективные электроды Твердотельные газовые сенсоры Полевые транзисторы
Изменение тока Амперометрические сенсоры (кислородные и ферментные электроды, иммуносенсоры, твердотельные газовые зонды)
Изменение резонансной частоты Пьезоэлектрические кварцевые резонаторы Сенсоры на поверхностных акустических волнах
Изменение оптических свойств Сенсоры на основе пропускания / поглощения, рассеяния, отражения, флуоресценции, преломления, измерения времени затухания, поляризации
Термические эффекты Термические/калориметрические сенсоры Пеллисторы
7.7.3. Электрохимические и микроэлектронные сенсоры
Потенциометрические сенсоры
Потенциометрические сенсоры лучше всего представлены pH стеклянным электродом и ион-селективными электродами (ИСЭ, см. разд. 7.3.1). Потенциометрические измерения можно использовать также при разработке сенсо
7.7. Химические сенсоры
497
ров для определения газов, например монооксида углерода или аммиака, и органических соединений, таких, как мочевина.
Твердотельные газовые сенсоры
Благодаря наличию ионов твердые тела с ростом температуры проявляют проводимость. Этот эффект можно использовать для создания газового сенсора. Особенно важны твердотельные электроды с проводимостью за счет оксидных ионов. Эти электроды являются редокс-электродами. Обычно материалом служит ZrO2, легированный СаО или ¥Ъ20з, в кристаллической решетке которого имеются катионные вакансии, что и объясняет ионную проводимость. Твердотельные электролитные сенсоры из ZrO2 подходят для определения кислорода в выхлопных газах или для контроля металлургических процессов, где нужно определять кислород в расплавленном железе при температуре свыше 1000°С. Ионы О2-, образующиеся в результате окислительно-восстановительной реакции
О2 + 4е" 2О2-
(7-7-1)
имеют тенденцию мигрировать в вакансии, вызывая смещение последних. Для измерений в расплавленном железе твердотельный электролит вводят в контакт с помощью платинового электрода, а результирующий потенциал измеряют вторым металлическим электродом в расплавленном железе. При определении О2 в газах твердотельный электролит внедряют в пористый платиновый электрод и измеряют электродный потенциал относительно сигнала сравнения. Обычно в качестве среды для сравнения используют воздух.
Проводящие электроды
Камера сравнения
Катод~~~
Твердотельный кислородный
сенсор для горячих газов
В соответствии с уравнением Нернста связь между потенциалом электрода Е и парциальным давлением кислорода в камерах сравнения рсравн и пробы Рпроба выглядит следующим образом:
(7.7-2)
Рпроба
Для анализа выхлопных газов автомобиля обычно используют так называемый А-зонд. Этот зонд основан на том, что для значения А = 1, при котором имеется стехиометрическое соотношение топлива и воздуха, наблюдается большой скачок парциального давления кислорода. Это изменение можно использовать для управления смесью топлива с воздухом.
498
Глава 7. Методы химического анализа и их применение
Сенсоры с газопроницаемой мембраной
Газочувствительные потенциометрические сенсоры включают электрохимическую ячейку с ион-селективным электродом и электродом сравнения. Оба они погружены в раствор внутреннего электролита. Внутренний электролит отделен от анализируемого раствора с помощью газопроницаемой мембраны (рис. 7.7-1). Микропористая или гомогенная мембрана имеет обычно толщину 0,1 мм. Микропористые мембраны изготавливают из гидрофобных полимеров, например, политетрафторэтилена (ПТФЭ) или полипропилена. В таких мембранах 70% пор имеют диаметр менее 1 мкм, так что газы могут проникать за счет эффузии, тогда как вода или ионы отталкиваются гидрофобной мембраной.
Гомогенные мембраны готовят из силиконового каучука. Газ растворяется в мембране и диффундирует сквозь нее. С целью обеспечения быстрого переноса газа эти мембраны часто значительно тоньше пористых, обычно порядка 0,02 мм толщиной.
Рассмотрим режим работы потенциометрического газового сенсора на СОг-Газ проникает через мембрану и растворяется во внутреннем электролите, который состоит из раствора NaHCOa/NaCl. Протолиз СОг протекает следующим образом:
СО2 + Н2О НСО3 + н+ (7-7-3)
Относительно высокая концентрация ионов НСОз во внутреннем растворе гарантирует прямую связь между концентрацией СО2 во внешнем растворе и активностью ионов водорода во внутреннем растворе. В качестве ион-селективного электрода на водородный ион используют обычный стеклянный электрод, а градуировка основана на уравнении Нернста. Измерение pH можно использовать и для определения других газов, таких, как SO2 или NO2. Подходящие химические реакции приведены в табл. 7.7-3.
Газопроницаемая мембрана
Рис. 7.7-1. Схема газочувствительного зонда, основанного на потенциометрических измерениях.
7.7. Химические сенсоры
499
Таблица 7.7-3. Типы коммерческих потенциометрических газовых сенсоров
Определяемое Реакция во внутреннем Детектирующий
вещество растворе электрод
со2 СО2 + 2Н2О НСОз + НзО+ pH стеклянный
no2 2NO2 + ЗН2О NO7 + NO3 + 2Н3О+ pH стеклянный или ИСЭ на NO3
so2 SO2 + 2Н2О HSO7 + НзО+ pH стеклянный
NH3 NH3 + Н2О NH+ + ОН- pH стеклянный
H2S H2S + 2Н2О <=> S2" + 2Н3О+ ИСЭ на основе Ag2S
HCN HCN + Н2О CN" + Н3О+ ИСЭ на основе AgCN
HF HF + Н2О F- + Н3О+ ИСЭ на основе LaF3
Для мембранных электродов помехи связаны с газами, которые реагируют со внутренним раствором подобно определяемым газам. Это наблюдается, например, при определении NO2 в присутствии СОг или SO2. В таких случаях стеклянный pH-электрод заменяют на ион-селективный электрод. Для определения NO2 можно использовать ИСЭ на нитрат, как можно понять из уравнения реакции в табл. 7.7-3. Эта таблица дает некоторые дополнительные примеры использования ИСЭ в качестве детектирующих. Недостатками этого вида сенсоров являются большое время отклика и низкая чувствительность.
Биокаталитические мембранные сенсоры
ИСЭ можно покрыть ферментом, который катализирует биохимическую реакцию. Например, фермент уреаза катализирует гидролиз мочевины с образованием аммиака и диоксида углерода. Продукты реакции можно определять с помощью вышеупомянутых газочувствительных ИСЭ. Комбинация подходящих ферментов и электродов позволяет определять многочисленные органические вещества. Это дает возможность создавать биосенсоры (см. разд. 7.8). Применение ферментов как биокатализаторов приводит к тому, что реакции, протекающие при умеренных значениях pH и температуры, селективны и потребляют минимальное количество субстрата.
Чтобы уменьшить расход относительно дорогих ферментов, предпочтительны устройства, в которых фермент может быть иммобилизован. Фермент иммобилизуют или непосредственно на поверхности электрода, или в твердофазном реакторе, через который пропускают пробу, а продукты детектируют на выходе из него.
Примером использования субстрат-специфичного мембранного электрода является определение мочевины с помощью чувствительного к ионам аммония стеклянного электрода в качестве ИСЭ. Ферментативная реакция основана на гидролизе мочевины в присутствии фермента уреазы:
NH2CONH2 + 2Н2О + Н+ 2NHJ + НСО3 (7.7-4)
Уреазу иммобилизуют в полиакриламидном слое, нанесенном на поверхности NH4 -стеклянного электрода. На рис. 7.7-2 показана схема устройства этого
500
Глава 7. Методы химического анализа и их применение
Защитная мембрана
ИСЭ на ионы аммония
Иммобилизованный фермент
Рис. 7.7-2. Биокаталитический мембранный сенсор на основе ИСЭ на ионы аммония и с иммобилизованным ферментом уреазой.
биосенсора. Проблемы возникают из-за ограниченной селективности этого типа аммониевого ИСЭ. В большинстве биологических проб мешающее влияние оказывают ионы натрия и калия. В качестве альтернативы можно использовать газочувствительный аммиачный электрод (табл. 7.7-3). В этом случае, однако, чувствительность ограничена, поскольку значения pH, необходимые для определения и ферментативной реакции, различаются. Максимальную чувствительность газочувствительного аммиачного сенсора получают в диапазоне pH между 8 и 9, где большая часть NHj превращается в аммиак. Для ферментативной реакции, однако, оптимальным является значение pH около 7.
Коммерческие приборы для рутинного определения мочевины работают на основе шарикового реактора с иммобилизованным ферментом. Раствор пробы пропускают через реактор. После подщелачивания раствора аммиак определяют с помощью газочувствительного аммиачного ИСЭ.
Полевые транзисторы
Рассматриваемые до сих пор потенциометрические сенсоры состоят в основном из чувствительной мембраны, соединенной с измеряющим прибором с помо-, щью твердого контакта или ион-проводящего солевого мостика. Электрическое усиление и оцифровка осуществляются отдельно в приборе, регистрирующем напряжение.
Возможна микроэлектронная интеграция сенсора и обработки сигнала, если потенциометрический сенсор основан на полевом транзисторе (ПТ). Такая микросхема может быть химически чувствительной (ХимПТ) или специфично ион-селективной (ИСПТ). Бергвельд разработал сенсоры на основе металлоксидных ПТ (МОПТ).
Структура такого транзистора приведена на рис. 7.7-3. Транзистор состоит из подложки кремния p-типа, с двумя обогащенными кремнием областями n-типа, образованными на ее поверхности. Электроды областей n-типа, исток и сток, соединены с окружением с помощью металлических контактов, таких, как напыленные алюминиевые электроды. С помощью изолятора (SiOs) крем-
7.7. Химические сенсоры
501
Рис. 7.7-3. Схематическое изображение металлоксидного полевого транзистора (МОПТ).
ниевые области почти полностью закрыты, так что соединение возможно только через электроды стока и истока. Слой нитрида кремния Si3N4 можно рассматривать в качестве дополнительного пассивирующего слоя (рис. 7.7-3).
Если напряжение (7си приложено между стоком и истоком, ток через соединительный канал не протекает, поскольку канал рп-диода заперт как между электродом истока и подложкой, так и между электродом стока и подложкой. В микросхеме есть, однако, еще один контакт, состоящий из слоя оксида металла («затвор»). Этот слой электрически отделен от подложки с помощью изолятора SiC>2-
Если приложить дополнительное напряжение между затвором и подложкой, то в n-канале между п-областями кремния образуется электрическое поле, так что между стоком и истоком протекает ток /си- Величина тока определяется напряжением. Конечно, чтобы получить ток, необходимо минимальное напряжение. Благодаря высокому сопротивлению между затвором и подложкой входной ток пренебрежимо мал. Высокое сопротивление полевого транзистора делает его подходящим входным устройством для pH- и иономеров, а также для усиления сигнала в обычных вольтметрах. Влияние напряжения сток-исток существует из-за изменений электрических характеристик транзистора (рис. 7.7-5).
Для создания ион-селективного ПТ исходный затвор следует удалить или заменить другим материалом. При удалении нижний слой SiaN4 действует как проводящий слой. При замене новым ион-селективным слоем, например мембраной из подходящего ионофора (рис. 7.7-4), он действует как ИСЭ. Электрический контакт получают с помощью электрода сравнения, как и в потенциометрических измерениях. Потенциалы, возникающие из приложенного напряжения затвора U3 и из ион-селективного слоя, складываются, и измеряв-
502
Глава 7. Методы химического анализа и их применение
Рис. 7.7-4. Сенсор, основанный на ион-селективном полевом транзисторе (ИСПТ).
мый ток /си зависит от активности потенциалобразующего иона. В результате электрическая характеристика ПТ меняется. В оптимальном случае изменение характеристики составляет 59 мВ на один электрон при изменении концентрации на один порядок величины (рис. 7.7-5).
Для практических измерений с ИСПТ ток сток-исток поддерживают на постоянном рабочем уровне. Изменение напряжения затвора от концентрации ионов компенсируют с помощью дополнительного управляющего напряжения. Это напряжение обратной связи пропорционально активности растворенных ионов, как и в случае обычных измерений, в соответствии с уравнением Нернста. Первое применение ИСПТ нашел для измерения pH, при этом наилучший нернстовский отклик получен при изготовлении затвора из ТагОб.
Другие применения ПТ для определения ионов, а также газов, ферментов и антител, указаны в табл. 7.7-4. В принципе, любые слои, применяемые в ИСЭ, можно также перенести в микроэлектронные сенсорные устройства.
Характеристики макросенсоров с ферментным покрытием детально обсуждаются в разд. 7.8.3. Иммуносенсоры, в свою очередь, основаны на принципах радиоиммунного анализа (разд. 7.8.3).
Многие методологические вариации разработки сенсоров, основанные на микролитографии, осложняются серьезными проблемами. В микроэлектрон-
7.7. Химические сенсоры
503
Рис. 7.7-5. Электрические характеристики ИСПТ: ток стока 7си зависит от суммы приложенного напряжения затвора U3 и потенциала ион-чувствительного слоя.
Таблица 7.7-4. Химически селективные полевые транзисторы
Тип
Затвор
Определяемое вещество
Газочувствительный ПТ (ГазПТ)
Ион-чувствительный ПТ (ИСПТ)
ПТ с ферментным покрытием (ФермПТ)
Иммуно-ПТ (ИМПТ)
Pd
Ион-селективные слои: Ta2Os, AI2O3, BN Газопроницаемая мембрана + AI2O3 Валиномиций
Гелевый или полимерный слой с иммобилизованным ферментом Антиген или антитело
Н2, NH3, СО
Н+
NH3, СО2
К+
Мочевина, глюкоза, пенициллин, Н2, ацетилхолин Альбумин
ных цепях участки, используемые для химического детектирования, обычно герметично закрыты в металлические корпуса или закапсулированы в пластиковые или керамические блоки. Это связано с высокой чувствительностью таких приборов к изменениям окружающей среды, таким, как изменение влажности, облучения или температуры. Эти взаимодействия основаны на силах, которыми в макродиапазоне обычно можно пренебречь, например, поверхностное натяжение, эффекты диффузии, силы Ван-дер-Ваальса или квантовые туннельные эффекты. В области микроэлектронных химических сенсоров эти взаимодействия становятся очень важными.
Ам перометрические сенсоры
Использование вольтамперометрических измерений как основы сенсора хорошо известно на примере амперометрического сенсора Кларка для определения кислорода (разд. 7.3). Рабочий электрод сенсора Кларка представляет собой платиновый электрод, связанный с серебряным анодом. Сенсор можно модифицировать, чтобы обойти необходимость регенерации серебряного электрода. Например, серебро можно использовать в качестве рабочего, а свинец—
504
Глава 7. Методы химического анализа и их применение
в качестве вспомогательного электрода. Электролитом служит гидроксид калия. Вместо восстановления кислорода на серебряном рабочем электроде, здесь окисление свинца представляет собой генерирующую электроны анодную реакцию:
РЬ + ВОН- = [РЬ(ОН)6]2- + 4е“ (7.7-5)
Поскольку гидроксид свинца растворим, то не происходит увеличения сопротивления, в отличие от электрода Кларка, где гидроксид серебра осаждается на серебряном аноде.
Амперометрические сенсоры известны и для других газов (табл. 7.7-5). Сенсоры без мембраны используют для мониторинга хлорид-иона в питьевой воде. Сенсоры иа монооксид углерода распространены в угледобывающей промышленности. Полуколичественные сенсоры этого типа дают быстрый отклик в опасных ситуациях, но не могут сравниться —в отношении разрешения сигнала—с мембранными сенсорами, такими, как электрод Кларка-
Таблица 7.7-5. Примеры амперометрических газовых сенсоров
Определяемое вещество Реакция Рабочий электрод
СО СО + ЗН2О СО2 + 2 Н3О+ + 2е_ Pt
NO2 NO2 + 3 Н2О Я NO3 + 2 НзО+ + е- Аи
SO2 SO2 + 6 Н2О Л SOJ" + 4 НзО+ + 2е_ Аи
Cl2 С12 + 2е“ 2СГ Pt
H2S H2S + 2 Ag Л AgaS + 2 НзО+ + 2е- Ag
HCN Ag + HCN + Н2О AgCN + НзО+ + Ag
Сочетание сенсора Кларка с ферментным слоем обсуждается в разд. 7.8.3 на примере определения глюкозы с глюкозооксидазой. Эту схему можно перенести на другие окислительные ферментативные реакции, например, определения галактозы с галактозооксидазой или мочевой кислоты с помощью уриказы.
Сенсоры на основе проводимости
□ Комбинацию (набор) полупроводниковых сенсоров иногда называют электронным носом. Основанные на методах распознавания образов отклики можно использовать, чтобы анализировать неизвестный газ, как качественно, так и количественно.
Еще одно свойство, которое можно использовать для газовых сенсоров,— это электропроводность. Соответствующие сенсоры состоят из оксидов металлов, таких, как SnOa, ZnO, TiO2 и ЕезОз, с проводимостью n-типа. Схема сенсора с тонким слоем SnO2 приведена на рис. 7.7-6. Кислород адсорбируется на нагреваемой поверхности этого сенсора и реагирует с определяемыми газами-восстановителями. В результате проводимость устройства изменяется и может быть измерена как неспецифический сигнал.
7.7. Химические сенсоры
505
Рис. 7.7-6. Газовый сенсор на основе алюминиевой подложки, покрытой полупроводником.
Простота устройства сенсоров на основе проводимости обеспечила широкий диапазон промышленных и бытовых их применений для обнаружения и определения Нг, РНз, NH3, SO2, СО, СН4, О2 и других газов.
Проводимость используют также в так называемых хемирезисторах. Сенсоры этого типа сенсоров изготавливают из тонкой пленки органического полупроводника, уложенного поверх пленочных электродов. В качестве материалов пленки обычно служат фталоцианины, имеющие химическую структуру, подобную структуре гемина и хлорофилла. В зависимости от центрального атома комплексообразующего агента можно приготовить сенсоры для определения СО (Zn-фталоцианин) или NO2 (РЬ-фталоцианин).
Пленочные электроды
7.7.4. Оптические сенсоры
Развитие оптических сенсоров стало возможным с появлением оптических волокон для видимого диапазона. Недавние достижения связаны с расширением спектрального диапазона, включающего волоконно-оптические сенсоры в УФ-, ближнем и среднем ИК-диапазонах. Помимо волоконно-оптических сенсоров представляют интерес также сенсоры на основе планарной оптики.
506
Глава 7. Методы химического анализа и их применение
Волновод
Устройство ввода/вывода света
Плоский волновод
В целом волоконно-оптические сенсоры генерируют оптический сигнал, пропорциональный концентрации определяемого вещества.
Измерение внутренних оптических свойств
Простейший тип волоконно-оптического сенсора включает связь световода со спектрофотометром. Используя это устройство, можно измерить интенсивность окраски или флуоресценцию растворов либо биологического вещества. Чтобы проиллюстрировать применимость волоконно-оптических сенсоров, рассмотрим некоторые основные свойства волноводов.
Волоконный кабель состоит из сердечника (волновода) и оболочки, сделанных из стекла, кварца или пластика. Диаметр кабеля составляет от 0,05 мкм до 0,6 см. Свет проходит через единичное волокно или через пучок волокон. Пучки могут быть расположены случайно или в определенных положениях на входе и выходе кабеля. В последнем случае через волоконный кабель можно передавать целую картину. Такие световоды очень важны в медицинской диагностике (эндоскопии) для обследования внутренних органов, например желудка.
Прохождение света через оптическое волокно показано на рис. 7.7-7. Когда свет достигает световода, часть его проходит, а часть — полностью отражается. Чтобы происходило полное отражение света, необходим критический угол в и показатель преломления сердечника гц должен быть выше, чем показатель преломления оболочки пг- Показатель преломления стеклянного сердечника составляет примерно 1,6, а стеклянной оболочки — около 1,5. Для измерений
преломления л, Рис. 7.7-7. Отражение света в оптическом волокне.
7.7. Химические сенсоры
507
в среднем ИК-диапазоне необходимы другие материалы, такие, как халькогенидные стекла (AsSeTe) или волокна из поликристаллических галогенидов серебра, которые прозрачны в диапазоне 2-20 мкм. Показатель преломления волокна из галогенидов серебра, состоящего из 75% AgBr и 25% AgCl, равен 2,21. Этот сердечник покрыт пластиком с показателем преломления 1,5.
Критический угол в определяется численной апертурой (NA) световода. NА зависит от показателя преломления сердечника nj и оболочки пг:
NA = sin в = у/(п^ — n|)
(7-7-6)
Чем больше численная апертура, тем больше света может световод захватить и пропустить.
Обычно используют многомодовые световоды. В этих волокнах свет проходит по световоду во многих модах. В разных модах свет ослабляется по-разному. Показатель преломления однороден и изменяется скачком на поверхности оболочки. Одномодовые световоды имеют очень малый диаметр сердечника, обычно 2-10 мкм, так что свет проходит через волокно почти линейно. Толщина оптической оболочки должна быть по меньшей мере в десять раз больше, чем диаметр сердечника. Ввод света в одномодовый световод представляет трудности, поскольку нужно добиться очень узкого угла падения. Следовательно, необходимы высокофокусированные источники света, такие, как лазеры или лазерные диоды.
Одномодовое волокно
Многомодовое волокно
Световоды соединяют со спектрофотометром как раздваивающиеся волокна (Y-кабели). На рис. 7.7-8 показано типичное устройство простого волоконного «сенсора» для измерения поглощения в жидких пробах. Свет проходит по световоду в раствор пробы и направляется с помощью отражателя в другой световод к спектрофотометру. Оптический путь света составляет удвоенное расстояние между концом волокна и отражателем.
Световод служит лишь для передачи излучения. Вместо фотометра волокно может быть связано с оптоэлектронной цепью', в этом случае источником света является светодиод (СД), а детектором — фотодиод.
Примеры применения простых волоконно-оптических сенсоров приведены в табл. 7.7-6. Если фотометрическое титрование на основе оптоэлектронного сенсора осуществить достаточно легко, то мониторинг химических процессов или грунтовых вод представляет значительно более сложную задачу. К примеру, возможно прямое детектирование органических соединений в грунтовых водах с помощью флуоресцентных измерений. Хотя нельзя определить индивидуальные вещества, качество воды можно контролировать, используя сочетание волоконной оптики, лазерного усиления и количественной спектроскопии комбинационного рассеяния. Такая система позволяет контролировать загряз-
508
Глава 7. Методы химического анализа и их применение
Рис. 7.7-8. Волоконно-оптический сенсор для измерения поглощения света в растворе пробы. Световод состоит из одного волокна или из пучка волокон.
нения на уровне миллиардных долей и на расстоянии до 1000 м при условии, что хотя бы некоторые из примесей флуоресцируют.
Таблица 7.7-6. Применения простых волоконно-оптических сенсоров
Определяемое вещество Измеряемое оптическое свойство Область применения
Ионы меди Поглощение при 930 нм Электроосаждение меди
Органические соединения Флуоресценция Мониторинг грунтовых вод
Гемоглобин Диффузное отражение при 600-750 нм Оксиметрия крови
Галотан Поглощение в ближнем ИК Анестетики
Сенсоры с системами распознавания
□ Иммобилизация может быть механической, электростатической или ковалентной.
Определение концентрации вещества в пробах, для которых нельзя использовать никакое присущее им оптическое свойство, требует системы распознавания с измеряемым изменением окраски. Для этой цели соответствующий
7.7. Химические сенсоры
509
Рис. 7.7-9. Оптический сенсор, использующий иммобилизованный реагент.
реагент часто наносят прямо на волокно (рис. 7-7-9); методы иммобилизации основаны обычно на адсорбции на (ионо)обменных смолах, включении в полимеры или ковалентной иммобилизации.
Такие сенсоры, называемые также оптродами или оптодами, разработаны для измерения pH, определения кислорода, СОг, NH3, ионов тяжелых металлов и некоторых других веществ. Соответствующий реагент иммобилизуют на торце волокна и измеряют изменение его окраски.
Чтобы получить обратимый сенсор, следует учитывать химическое равновесие между иммобилизованным реагентом и определяемым веществом в растворе (или в газовой фазе). Рассмотрим простейший случай реакции определяемого вещества А с иммобилизованным реагентом R:
А + R AR (7.7-7)
(Чтобы отличить частицы в растворе, такие, как А, от частиц на смоле, будем использовать подчеркивание для иммобилизованных частиц.)
Для равновесия между комплексом на волокне AR и иммобилизованным реагентом R справедливо выражение:
Т, [AR]
[A][R]
(7.7-8)
В случае измерения оптических свойств AR мы получаем связь сигнала с концентрацией:
[AR] = /<[A][R]
(7.7-9)
В предположении, что равновесная концентрация А почти идентична общей концентрации определяемого вещества в растворе, т. е. [А] = Сд, и что концентрация иммобилизованного реагента равна разности между общей концентрацией реагента Сц и концентрацией продукта, т. е. [R] = Cr — [AR], уравнение
510
Глава 7. Методы химического анализа и их применение
Рис. 7.7-10. Связь между концентрацией комплексообразующего реагента [ЛЯ] и концентрацией определяемого вещества Сд. Оптический сигнал обычно прямо связан с (АД]. Константа равновесия К — 1660л/моль, [Я] = 10-4моль/г.
(7.7-9) можно переписать с учетом постоянства величины К и Сд, в следующем виде:
[АВ)—
kcacr
1 + КСА
(7.7-10)
На рис. 7.7-10 показана типичная зависимость концентрации продукта реакции в иммобилизованном слое от концентрации определяемого вещества.
Градуировочные кривые линейны только при низких концентрациях определяемого вещества, т. е. для КСА С 1 или СА < 1/К. При высокой концентрации определяемого вещества график близок к асимптоте из-за насыщения реагента определяемым веществом.
Нелинейность градуировочных графиков не представляет существенной проблемы, если для обработки данных использовать микропроцессор. Недостатком остается уменьшение чувствительности с ростом концентрации. Часто, однако, сенсоры нужны только для узких диапазонов концентраций, где может быть получена линейность.
При выборе индикаторного реагента следует учитывать тот факт, что реагент должен образовывать менее устойчивые комплексы. Это гарантирует, что концентрацию реагента можно подобрать для диапазона, представляющего аналитический интерес. В этом имеется отличие от классических аналитических приложений равновесных реакций, где правилом являются высокие константы равновесия и избыток реагентов.
Химическая иммобилизация основана на ковалентном связывании реагента с материалом волокна. Иммобилизация этого типа обеспечивает весьма прочное связывание индикатора и сводит к минимуму вымывание. Необходимым условием химической иммобилизации является наличие реакционных групп у индикатора или у несущей матрицы. Как правило, эти группы получают пу-
7-7. Химические сенсоры
511
Таблица 7.7-7. Методы химической иммобилизации для оптических сенсоров н метод ы модификации поверхности
Полимер Реакционная группа Модифицируют реакцией со следующими веществами Дополнительный компонент системы
Целлюлоза Аминоэтил Вромциан, этилендиамин Карбоксилат,
Стекло, Карбоксиэтил Аминопропил Хлоруксусная кислота 7-Аминопропилтрн- сульфоновые кислоты Амины Карбоновые
силикагель Винил этоксисилан Триацетоксивинилсилан кислоты, альдегид Нуклеофилы
Полиакриламид Карбоксиэтил Сильные щелочи и кислоты Амины, белки
Таблица 7.7-8. Волоконно-оптические сенсоры с иммобилизованными реагентами
(оптоды)
Определяемое вещество Реагент/иммобилизация Принцип измерения
pH (2-5) Конго красный/ацетилцеллюлоза Отражение
pH (4-7) Флуоресцеинамин/стекло Флуоресценция
Л13+ Морин/целлюлоза Флуоресценция
к+ Валиномицин 4- MEDPIN/ПВХ Отражение
СГ Флуоресцеин/колловд серебра Флуоресценция
Влажность Coda /Со(Н2 0),гС12 / желатин Отражение
02 Ru-трис (дипиридил)/силикон Фосфоресценция
NH3 Вромтимоловый синий/силикон Затухающие волны
Альбумин Вромкрезоловый зеленый /целлофан Отражение
тем предварительной активации матрицы. Наиболее часто используемыми носителями являются целлюлоза, декстрин или агароза. Кроме того, используют синтетические полимеры, например, полиакриламид. Примеры методов химической иммобилизации приведены в табл. 7.7-7. Методы можно заимствовать из исследовательской практики в области полимеров и химии поверхности, а также модификации неподвижных фаз в хроматографии.
Применение волоконно-оптических сенсоров не ограничено измерением pH. Разработаны оптоды для катионов, анионов, газов, органических соединений и для измерения ионной силы (табл. 7.7-8). Сенсоры, основанные на поглощении и отражении, менее распространены, чем флуоресцентные сенсоры, благодаря более высокой чувствительности последних.
Перенос принципа ион-селективных электродов на оптические сенсоры привел к созданию ион-селективных оптодов. Одна из возможностей заключается в применении ионообменных равновесий между раствором и ПВХ-мембраной оптода. Определение иона калия возможно на основе равновесия:
К+ + НС+ +1 К1+ + Н+ + С (7.7-11)
где НС+ и С — протонированная и депротонированная формы соответственно хромофора, иммобилизованного на ПВХ, а I и К1+ — соответственно ионофор
512
Глава 7. Методы химического анализа и их применение
и его комплекс с ионами калия на ПВХ. Равновесие управляется условием электронейтральности.
Универсальным ионофором является валиномицин, известный по ион-селективным применениям. Хромофором служит гидрофобное соединение, например, MEDPIN (Miles(Bayer)):
Преимуществом ион-селективных оптодов является их широкий рабочий диапазон, сравнимый с диапазоном ИСЭ. Помимо этого, многие ионофоры, разработанные для ИСЭ, можно использовать также и в оптодах.
Оптоды, основанные на затухающих волнах
Измерение затухающих волн возможно в том случае, когда реагент иммобилизован на сердечнике световода (см. рис. 7.7-8). Для определения аммиака раствор бромтимолового синего в силиконе наносят на световод (рис. 7.7-11). При каждом внутреннем отражении в световоде часть луча выходит в покрытие и взаимодействует с реагентом. Такое же явление известно в ИК-спектроскопии как метод НПВО (разд. 9.2).
При определении аммиака на затухающую волну воздействует интенсивность окраски красителя, пропорциональная концентрации аммиака. Разность можно оценить, например, измеряя поглощение.
В табл. 7.7-8 приведены другие примеры разработки волоконно-оптических сенсоров.
Преимущества оптических сенсоров перед электрохимическими:
— Вся спектроскопическая информация доступна. Если необходимо, эту информацию можно контролировать в нескольких местах измерения (преимущество мультиплексности).
— Нет электрических помех, так что измерения можно проводить в сильном электрическом поле, например, при электролизе или в трансформаторах.
— Не нужен электрод сравнения.
— Фазу реагента можно сделать недорогой, так что возможно создание одноразовых сенсоров.
Ввод света
Вывод света
Краситель
Рис. 7.7-11. Оптический сенсор для измерения затухающих волн.
7.7. Химические сенсоры
513
Недостатки оптических сенсоров:
— На измерения влияет рассеянный свет. Эту помеху можно уменьшить, например, за счет импульсных источников света.
— Обратимость оптодов в жидких пробах часто весьма плохая, и регенерация сенсора становится обязательной.
— Долговременная стабильность часто ограничена из-за вымывания индикатора.
— Установление равновесия между определяемым веществом в растворе и иммобилизованным реагентом (ур. 7.7-10) приводит к относительно узкому рабочему диапазону, что не относится к ион-селективным оптодам с логарифмической зависимостью сигнал—концентрация.
7.7.5. Термические сенсоры
Измерение энтальпии реакции используют в газовых сенсорах, основанных на каталитической реакции (пеллисторах). Такие каталитические сенсоры состоят из нагреваемой проволоки, внедренной в шлаковый шарик, и каталитически активного слоя, легированного металлическими платиной или палладием (рис. 7.7-12). Проволочную спираль нагревают примерно до 550°С. Восстановительные газы, такие, как СО или СЩ, окисляются адсорбированным кислородом, а тепло реакции можно измерить по увеличению сопротивления спирали. Скорость окисления на поверхности сенсора пропорциональна концентрации определяемого газа. Точность такого неспецифичного измерения сопротивления может быть улучшена путем сравнительных измерений с использованием неактивного шлакового шарика.
Калориметрические сенсоры основаны на миниатюрных калориметрах и применяются для растворов (разд. 7.5). В простейшем случае проба проходит через реактор, на выходе которого тепло реакции измеряют термистором. Для определения субстратов (разд. 7.8) в реакторе иммобилизуют фермент. Такое устройство не является сенсором в строгом смысле нашего определения, поскольку контроль не непрерывен. Измерение теплового эффекта реакции стали практиковать, используя отдельные ферментативные реакции. Возможно определение мочевины, пенициллина, глюкозы, сахарозы, холестерина или лактата.
Рис. 7.7-12. Пеллистор, состоящий из слоя каталитически активных Pt или Pd и неактивного шлакового зерна, изготовленного из оксида.
Каталитический слой
Инертный оксидный слой
(ThO2 AI/DJ
17 Аналитическая химия. Том 1
514
Глава 7. Методы химического анализа и их применение
7.7.6. Масс-чувствительные сенсоры
□ Пьезоэлектрический кварцевый резонатор называют также кварцевыми микровесами.
Изменение массы и последующее изменение резонансной частоты в присутствии определяемого газа используют в механоакустических сенсорах. Наиболее важными из них являются пьезоэлектрические кварцевые резонаторы и сенсоры поверхностных акустических волн (ПАВ).
Поглощающий слой
Кварцевый резонатор
Рис. 7.7-13. Пьезоэлектрический сенсор на основе кварцевого резонатора.
Принцип работы кварцевого резонатора показан на рис. 7.7-13. Кварцевый резонатор, освобожденный от его оболочки, покрыт органическим слоем, поглощающим газ. Кварцевый кристалл установлен в цепи генератора в качестве элемента, задающего частоту. При осаждении газа на плоскую поверхность кристалла изменяется масса. В результате изменяется также резонансная частота. Устройство функционирует в режиме сдвига толщины.
Режим сдвига толщины
В этом режиме поверхности кристалла сдвигаются относительно друг друга. Между изменением массы Дт и изменением частоты Д/ имеет место следующая связь:
Д/ = -2,3 - 106/о2
Дт “Т
(7.7-12)
где А — площадь поверхности кварца, а /о — резонансная частота кварца без покрытия.
Частоты 10-15 МГц можно разрешить с точностью до 0,01 Гц. Таким образом, можно определять массы на уровне 10 пг. В качестве поглощающих слоев используют различные покрытия. Например, фосфорорганические соединения можно определять с помощью субстрата диаминмеди, связанного с полимерной смолой. С использованием этого материала получают обратимый сенсор, который дает отклик на диизо пропил метилфосфонат в концентрации 20 млрд-1.
7.7. Химические сенсоры
515
Масс-чувствительные сенсоры используются также для мониторинга газовых загрязнений и для определения пыли в воздухе.
□ Пьезоэлектрический кварцевый резонатор генерирует объемные акустические волны в отличие от поверхностных акустических волн, наблюдаемых в устройстве ПАВ.
Более чувствительные приборы могут быть получены с помощью сенсоров на поверхностных акустических волнах (ПАВ). Это достигается благодаря использованию более высоких частот. В соответствии с уравнением 7.7-12 обычно используют частоту 1 ГГц, что уменьшает определяемую массу до уровня фемтограммов. Принцип работы сенсора ПАВ объясняет рис. 7.7-14. На поверхность пьезоэлектрической подложки укладывается с помощью технологии интегральных схем набор электродов. После наложения на этот передатчик ВЧ-сигнала вдоль поверхности проходит сейсмическая волна Рэлея. Если подложка покрыта газочувствительным слоем, изменения в окружающем газе можно фиксировать, измеряя частоту. Селективность прибора определяется природой химического покрытия.
Радиочастотный усилитель
Сигнал
Рис. 7.7-14. Газовый сенсор на основе поверхностных акустических волн (ПАВ), например, для определения паров стирола на уровне 5 млн-1 с PtCh (этилен) (пиридин) в качестве селективного покрытия.
17*
516
Глава 7. Методы химического анализа и их применение
7.7.7. Сенсорные наборы
Большинство сенсоров не специфично, а дает отклик на несколько веществ. Чтобы скомпенсировать потерю селективности, необходимо проводить измерения с несколькими сенсорными каналами, т. е. при нескольких длинах волн, потенциалах, токах или резонансных частотах.
Многоканальные сенсоры, или сенсорные наборы, могут быть получены путем соединения нескольких отдельных сенсоров. Например, пьезоэлектрические кварцевые кристаллы могут быть связаны в такую обойму и работать одновременно. Комбинации сенсоров, основанных на полевых транзисторах, состоят из единственной цепи, где индивидуальные сенсоры образованы различными покрытиями (рис. 7.7-15). Оптические сенсоры также могут работать в многоканальном режиме, если использовать спектральный диапазон, а не одну длину волны, например, с помощью спектрофотометра с диодной матрицей. Каналом здесь будет регистрируемая длина волны.
Обработку данных для многоканальных сенсоров проводят, используя принцип одновременного многокомпонентного анализа, рассматриваемого в разд. 12.5.4.
До настоящего времени развитие сенсоров было направлено на конструирование сенсоров для определения индивидуальных химических веществ. Таким образом, основной подход, принятый в анализе off-line, был просто перенесен на разработку сенсоров. Например, оценку качества вина проводят, определяя индивидуальные компоненты с последующей оценкой данных о содержании некоторых следовых компонентов или с помощью хемометрических методов распознавания образов. Гораздо более естественным подходом было бы использование сенсора вкуса, который можно было бы использовать для оценки качества вина таким же образом, как это делают люди.
Рис. 7.7-15. Матрица ИСПТ для определения электролитов крови. рН-чувствительный слой основан на затворе из SiaN-i. Для определения натрия изолятор Sia^/SiO? легируют А1 и Na; калий определяют с помощью чувствительного слоя, содержащего валиномицин.
7.8. Биосенсоры
517
Разработка сенсоров для решения проблем такого типа все еще находится в стадии младенчества1. Рутинное использование таких сенсоров будет также зависеть от одобрения их официальными организациями.
Литература
[7.7-1] Gopel, W., Hesse, J., Zemel, J.N., (Eds.), Sensors: A Comprehensive Survey, Vol. 2, Chemical and Biochemical Sensors. Weinheim: VCH, 1991.
[7.7-2] Wolfbeis O.S. (Ed.), Fiber Optical Chemical Sensors and Biosensors, CRC Press, Boca Raton, Florida, 1991, Vols. I and II.
[7.7-3] Edmonds, T.E., Chemical Sensors, New York: Chapman & Hall, 1988.
Вопросы и задачи
1. Ионная сила оказывает существенное влияние на электрохимические измерения ионов в растворах. Какого влияния ионной силы следует ожидать в случае использования оптодов?
2. Вольтамперометрические сенсоры обычно основаны на амперометрии. Имеет ли смысл полярографический сенсор?
3. Оптоды используют изменение окраски иммобилизованного реагента. Как выглядит форма градуировочного графика?
4. Как можно скорректировать недостаточную специфичность и/или селективность сенсора?
5. Каковы различия и сходства между диодными матрицами и комбинациями сенсоров?
7.8. БИОСЕНСОРЫ
Цели изучения
• Понять, каким образом сочетать реагенты и метод определения в биоанализе и получить аналитический прибор.
• Принять решение о выборе измеряемого параметра, а значит, и типа преобразователя.
• Рассмотреть, как можно иммобилизовать реагенты вблизи преобразователя.
7.8.1. Введение
□ Биосенсоры—это аналитические устройства.
Биосенсоры... Что означает это слово? Эта книга в значительной мере посвящена детектированию реакций в растворе между пробой и добавленными реагентами с помощью аналитического метода (химического или физического) ; иногда в дополнение к собственно аналитическому этапу рассматриваются
1 За последнее время здесь достигнут заметный прогресс. Разработано несколько вариантов «электронного языка» который и представляет собой комбинацию ряда сенсоров. — Прим. ред.
518
Глава 7. Методы химического анализа и их применение
Рис. 7.8-1. Схематическое изображение биосенсора.
этапы пробоподготовки или разделения. Биосенсор — это «безреагентная» система; более корректно было бы сказать, что реагенты непосредственно интегрированы в биосенсор и, следовательно, не добавляются пользователем, что является преимуществом. Другим полезным свойством биосенсора является то, что не требуется никакой специальной подготовки пробы. Следовательно, биосенсоры, очевидно, связаны с иммобилизованными реагентами, т. е. с реакциями, инициируемыми в иммобилизованных слоях, и их исследованием. Однако эти черты уже встречались нам в главе о химических сенсорах. Так что же отличает биосенсоры?
Биосенсоры принадлежат к семейству молекулярных сенсоров и поэтому включают селективную к определяемому веществу поверхность вблизи преобразователя или интегрированную в преобразователь (рис. 7.8-1), функцией которой является передача сигнала о взаимодействии между поверхностью и определяемым веществом либо непосредственно, либо через химический медиатор. В биосенсорах специфичная к определяемому веществу поверхность использует биомолекулы, распознающие молекулярные участки или их аналоги.
Преобразователь — это детектор. Он отслеживает химическую или биохимическую реакцию, инициируемую при добавлении пробы. Природа преобразуемого параметра зависит от типа биоаналитического события, происходя-
7.8. Биосенсоры
519
щего при детектировании определяемого вещества. Однако, поскольку в ходе аналитической реакции часто могут изменяться несколько параметров, выбор устройства не обязательно ограничен единственным преобразователем. Для сенсоров можно приспособить ряд имеющихся аналитических методов. (Рассмотрите примеры, приведенные в других главах, И обдумайте, можно ли осуществить такую трансформацию.)
В этой главе обсуждаются фундаментальные принципы биосенсорного устройства и компоненты, необходимые для его построения. Необходимо помнить, что не все сенсоры имеют корни в традиционных аналитических методах. Некоторые биосенсоры не имеют аналитических аналогов, с которыми их можно было бы сравнить, они были разработаны исключительно как сенсоры.
Вероятно, любой измеримый физико-химйческий параметр можно использовать в биосенсорах, хотя делать это не всегда практично! Наиболее широко распространенные устройства используют параметры, суммированные иа рис. 7.8-2. В этой главе далее рассмотрены некоторые примеры, иллюстрирующие использование оптических и электрохимических методов. Рассмотрение других биосенсоров можно найти в книгах специалистов по биосенсорам [7.8-1—7.8-4].
Биораспознающий компонент и преобразователь
□ Биораспознающий компонент обусловливает селективность к определяемому веществу и формирует аналитический сигнал.
Биораспознающий компонент биосенсора—это белок, макромолекула или комплекс со специфической поверхностью или внутренними распознающими центрами, необходимый для распознавания определяемого вещества. Компонент обусловливает селективность по отношению к определяемому веществу и передает сигнал на преобразователь. Тип реакции, катализируемой ферментом, определяет выбор преобразователя. Определяемое вещество, а значит, и доступные методы преобразования обусловливают природу биораспознающего компонента. Рассмотрим два примера, в которых фермент используют для создания сенсора на субстрат этого фермента. На схеме 7.8-1 ферментативная реакция включает перенос электрона; таким образом, для определения холестерина можно использовать в качестве преобразователя амперометрический электрохимический сенсор. Схема 7.8-2 включает изменение [Н+]; следовательно, контроль превращения ацетилхолина возможен с помощью рН-электрода или pH-чувствительного красителя в оптическом приборе. Другие ферменты можно использовать в случае реакций гидролиза, этерификации, расщепления и т. д.; определяемое вещество обычно является субстратом фермента. (Как можно провести анализ, если вы не смогли найти подходящую ферментативную реакцию с участием определяемого вещества, но знаете, что оно является ингибитором ферментативной реакции?)
Если определяемое вещество является антигеном, оно может быть определено с помощью иммунного анализа (см. разд. 7.9). Для проведения такого анализа обычно используют метку, и выбор преобразователя зависит от природы
520
Глава 7. Методы химического анализа и их применение
Рис. 7.8-2. Параметры преобразования и тип устройства.
Схема 7.8-1. Катализируемое ферментом окисление холестерина.
Схема 7.8-2. Катализируемое ферментом превращение ацетилхолина в ацетат и холин, вызывающее увеличение |Н+].
7.8. Биосенсоры
521
Иммобилизованное антитело
Антиген пробы
Антитело
—* Метка
Рис. 7.8-3. Типы иммуносенсоров, а — Модифицированная антителами поверхность преобразователя; б — конкурентный иммунный анализ, [♦] ос l/[Ag]; в — сандвичевый иммунный анализ, [*] ос [Ag]; г —гомогенный иммунный анализ, [♦] ос l/[Ag],
метки (рис. 7.8-3). Были также предприняты попытки прослеживать комплексообразование между антигеном и антителом без метки (см. разд. «Иммунный анализ», с. 546).
Биораспознающие компоненты не ограничиваются макромолекулами. Живые клетки или их составные части могут обеспечить требуемую чувствительность к определяемому веществу, и преобразованный сигнал изменяется в соответствии с рассматриваемым клеточным процессом. Действие определяемого вещества можно контролировать по его влиянию на дыхание клетки (за которым можно следить с помощью кислородного электрода) либо на характеристики мембранного транспорта, или по усилению либо подавлению переноса заряда. Каким бы ни был параметр, клетки следует иммобилизовать на преобразователе.
Можно также использовать небиологические материалы, если возможно получение синтетических аналогов, которые отличаются всеми естественными характеристиками селективности с улучшенным преобразованием. Такие возможности выглядят очень привлекательно, но усилия по созданию селективных центров в виде, «дружественном для биосенсора», требуют больше времени и мастерства, если они столь же хороши, сколь и имитируемые природные макромолекулы.
7.8.2. Создание биологической поверхности
□ Биораспознающий компонент и другие реагенты должны быть иммобилизованы.
Независимо от метода, показанного на рис. 7.8-2, или типа биораспознавания, используемого в анализе для обеспечения селективности к определяемому веществу, существует общая черта для биосенсоров всех типов. Биораспознающий компонент иммобилизован на поверхности, обычно на поверхности преобразователя, где детектируется сигнал.
Методы иммобилизации подразделяются на несколько категорий, и выбор наилучшего метода не всегда очевиден. Участки внутри макромолекул не должны быть затронуты при иммобилизации. Однако предсказать направле
522
Глава 7. Методы химического анализа и их применение
ние реакции при иммобилизации непросто, поскольку всегда имеется несколько реакционных групп или центров, которые могут быть затронуты. Существуют два вида иммобилизации: захват и связывание.
Последний может происходить с энергией связывания от 10 до 500 к Дж/моль, так что не удивительно, что результаты могут сильно различаться. Захват часто бывает более простым, но возникает проблема вымывания реагента. Вымывание особенно велико, когда молекулы реагента меньше, чем поры захватывающей матрицы, если нет некоторого дополнительного связывания с матрицей. В таком случае иммобилизация должна сочетать связывание с захватом.
Теоретические основы планирования иммобилизации
Реакция на поверхности раздела фаз
□ Реакции на поверхности являются гетерогенными.
Помимо того, что один реагент неподвижен, а поведение другого контролируется диффузией, существует основное различие между процессом иммобилизации (хемосорбции) и бимолекулярной реакцией в гомогенной фазе. В последнем случае реакционная способность реагирующих веществ остается постоянной независимо от степени протекания реакции, пока один из реагентов не станет ее лимитировать. Реакция, таким образом, протекает с одинаковой кинетикой при t — 0 и t > 0 (рис. 7.8-4,а). При иммобилизации же отдельные адсорбционные места нельзя рассматривать как не зависящие одно от другого; адсорбция по одному из мест может значительно влиять на реакционную способность соседних мест (рис. 7.8-4,б), так что кинетика реакции изменяется во времени.
Рис. 7.8-4. Реакционная способность, а — в гомогенной фазе: кинетика не меняется при t = 0 и t > 0. Константа скорости ki одинакова при t = Ont > 0; б — в гетерогенной фазе: кинетика изменяется в соответствии с покрытием поверхности.; константы скорости: к>2 (t = 0) и кз (t > 0).
7.8. Биосенсоры
523
Рис. 7.8-5. Миграция адсорбата.
Притягивающие и отталкивающие силы между адсорбатом и адсорбентом могут простираться на 50 нм, а их величины меняются. Рассматривая начальный процесс адсорбции, следует принимать во внимание подвижность адсорбата. Адсорбированная частица может находиться в месте своей начальной адсорбции в течение всего времени или она может мигрировать от одного места к другому (рис. 7.8-5). В последнем случае необходимо преодолевать потенциальные энергетические барьеры, разделяющие соседние места. Поскольку энергия активации для миграции часто меньше энергии связывания, сделать это совсем нетрудно.
Любая поверхность имеет шероховатость (неоднородность). Если шероховатость сравнима по размеру с адсорбатом, тогда адсорбат сможет взаимодействовать с различными гранями поверхности; каждая грань определенной ориентации имеет различную реакционную способность. Следует ожидать, что эти грани будут реагировать в порядке уменьшения реакционной способности. Даже если поверхность однородна и все адсорбционные места идентичны, все равно теплота адсорбции уменьшается при увеличении степени заполнения за счет диполь-дипольного взаимодействия между адсорбированными молекулами и прибывающими молекулами. Для идеального неподвижного простого адсорбированного слоя уменьшение теплоты адсорбции линейно связано со степенью заполнения. В подвижном слое теплота адсорбции остается вначале практически постоянной, а затем резко возрастает при 50%-ном заполнении. Это свидетельствует о том, что высокая подвижность приводит к минимизации горизонтальных взаимодействий.
Диапазон сил
При присоединении биораспознающей молекулы к поверхности между ними должно произойти столкновение, после которого возможно ограниченное число процессов (рис. 7.8-6):
1. Отражение молекулы обратно в объем адсорбата с передачей (неупругое) или без передачи (упругое) энергии.
2. Передача такого количества энергии (неупругое столкновение), что молекула оказывается неспособной преодолеть потенциал у поверхности и находится в возбужденном физическом сорбированном состоянии, которое харак-
524
Глава 7. Методы химического анализа и их применение
Рис. 7.8-6. Последовательность событий на поверхности, ведущая к хемосорбции.
теризуется относительно малыми силами связывания (например, силами Ван-дер-Ваальса) и низкой энтальпией адсорбции, порядка —40 кДж/моль.
3. Последующие возможные процессы:
а) дальнейшая передача энергии поверхности в том же месте,
б) миграция молекулы по поверхности с потерей энергии в других местах, в) десорбция с приобретением энергии от адсорбента,
г) переход в хемосорбированное состояние или в начальном месте, или после миграции.
Химическая адсорбция связана с образованием химической связи с типичным изменением энтальпии порядка —400 кДж/моль.
4. После хемосорбции возможны дальнейшие процессы:
а) миграция’хемосорбированных частиц',
б) десорбция из хемосорбированного состояния-,
в) дополнительная хемосорбция, приводящая к образованию множественных соединений. .
5. Между прибывающей молекулой и другой уже адсорбированной частицей, может иметь место поверхностная реакция, не затрагивающая напрямую подложку.
Хемосорбция характеризуется большой энтальпией адсорбции, более высокой специфичностью ориентации и более стабильными слоями, чем физическая сорбция. Тем не менее на поверхности, содержащей много мест, способных к физической или химической адсорбции, будут, вероятно, накапливаться адсорбированные частицы, природа которых зависит от температуры и скорости осаждения.
После создания биораспознающей поверхности ее реакция с определяемым веществом не будет гомогенной, а будет также включать хемосорбцию.
7.8. Биосенсоры
525
Методы иммобилизации
Иммобилизация биораспознающего компонента может происходить в один или два этапа. Биораспознающая молекула может присоединиться непосредственно к поверхности преобразователя или модифицировать «несущий субстрат», который впоследствии, а иногда и одновремненно, осаждается на преобразователь.
Физическая адсорбция
□ Физическая адсорбция характеризуется низкой энергией взаимодействия.
Физическая адсорбция биораспознающей молекулы на поверхности преобразователя является простейшим из процессов иммобилизации. Одним из наиболее распространенных адсорбентов для ферментных электродов служит углерод [7.8-5-7.8-8]. Весьма подходящую поверхность для оптического преобразования в анализе растворов обеспечивает полистирол. Однако физическая адсорбция очень часто слишком слабо удерживает реагент, который в результате быстро вымывается, поэтому сенсор следует покрывать мембраной, проницаемой для определяемого вещества и способной удерживать биораспознающие молекулы иа преобразователе.
Физическая адсорбция — наиболее простой вид иммобилизации, однако вероятно, и наименее удовлетворительный, поскольку, если ие изменять саму поверхность или не влиять на активность адсорбированных частиц, контролировать места присоединения почти невозможно. Это показали Дашл и Холл [7.8-9], которые сравнили человеческий иммуноглобулин G (hlgG), адсорбированный на немодифицированной поверхности SiC>2 и на поверхности S1O2, модифицированной аминосиланом. В обоих случаях были явно похожие монослои hlgG, что подразумевало хорошие биочувствительные поверхности. Однако реакция антитела-анти-hlgG с этими слоями существенно различалась: в первом случае монослой hlgG связывал только примерно три молекулы антитела на одну молекулу адсорбированного hlgG, тогда как во втором случае— пять или шесть. Это свидетельствует о различных взаимодействиях между адсорбированным белком и поверхностью, приводящих к образованию в этих двух случаях неидентичных распознающих поверхностей.
Во всех методах сложно осуществить присоединение белка к поверхности преобразователя, поскольку оно не должно затрагивать активный участок. Если модифицированную поверхность использовать непосредственно без метки или медиатора, нужно предотвратить все иеспецифические взаимодействия, поскольку «прямые» измерения без метки, включающие обычно контроль изменения массы или показателя преломления на поверхности, не могут’различить специфические или неспецифические взаимодействия. При иммобилизации поверхности с «плотной упаковкой» неспецифическое связывание не происходит, тем не менее, иммобилизация часто бывает неэффективной для образования комплекса антитело-антиген из-за стерических препятствий. Наоборот, упаковка с большими промежутками обычно допускает неспецифические взаимодействия с расположенной ниже поверхностью, и, таким образом, можно зарегистрировать значительные ошибочные сигналы.
526
Глава 7. Методы химического анализа и их применение
Физическое удерживание в полимерных матрицах
□ Для создания полимерной матрицы, в которой удерживается реагент, можно использовать поперечно сшитые реагенты.
Распространенным методом удерживания ферментов и живых клеток является поперечное сшивание, при котором образуется гелевая матрица, где частицы рецепторов удерживаются физически. Методы получения этого «геля» многочисленны. Наиболее испытанный метод иммобилизации—это, пожалуй, поперечное сшивание с помощью глутарового альдегида, и он все еще предпочтителен для иммобилизации фазы реагента в проточно-инжекционном анализе, несмотря на проблемы с его осуществлением в большом масштабе. Во всяком случае, используются многие бифункциональные реагенты; в табл. 7.8-1 перечислены некоторые примеры наиболее часто встречающихся реагентов, которые связываются преимущественно через группы -NHa, -ОН и -SH.
Таблица 7.8-1. Бифункциональные реагенты
9» ,О
„с-<снА-<
Глутаровый альдегид
OCN-(CHJe-NCO 1СНа-СЫН-(СН^в-ЫНС-СН,1
Гаксамвпмемдимэоцианат Н,Ы'-Этилвнбис(иадацвтамид)
° 9
СНа-СН-^-СН-СНа О &
Ливинилсульфон лЛ-Дифтор-жХ-ЛИНитро-дифенилсульфон
Диаэобвнмдин Диааобенэидаи-3,3' -
дикарбоноем кислота
6CN
•<СН2)0-С
ОСНз
NCS
СН
Диметилимидаты
«'•Дииаотмоцшшто-дифвнил-2,2'-дисульфокислот а
Диааобенэыдин-2,2'-дисульфокмслота
Фенол-2,4-бис(сульфонилхлорид) 1,5-Дифтор-2,4-дкытробвнзол Трмхлортриаэми
NH^CH^R (R - -СООН; -СОИН2; л - 2-12)
Аминосоединения с длинной целью
HS(CH2)nX
Тиолы с длинной цепью
7.8. Биосенсоры
527
Все виды полимеров (например, желатин, агар, циклодекстрины, полипиррол, полианилин, поливинилхлорид, поливиниловый спирт, поливинил пиридин, полиакриламид и многие другие полимеры и гели [7.8-10-7.8-12]) тщательно изучены в качестве матриц для удерживания, но ни один не является идеальным. Некоторые прозрачны и проницаемы, так что они обладают очевидным потенциалом для создания различных типов сенсоров, но матрица для иммобилизации должна также иметь адекватные характеристики с точки зрения «хранения» иммобиизованного вещества, особенно стабилизации иммобилизованной биораспознающей молекулы. Для водных или содержащих воду растворов важна также способность к гидратации. В новом подходе используют пленкообразующие эмульсионные полимеры, для которых pH, изотоничность и гидратационные характеристики можно подобрать в зависимости от конкретной биораспознающей системы и приложения [7.8-13]. Это означает, что подобную методику иммобилизации можно использовать и для фермента, и для антитела, и для живой клетки. Эмульсию, содержащую биосистему, можно приготовить в водной среде, если даже пленка после образования становится нерастворимой.
Можно приготовить низкотемпературные золь-гель силикатные стекла [7.8-14-7.8-16], но те из них, которые переходят в «гель» в течение недель, могут быть мало пригодными для менее устойчивых биологических молекул. Многие другие низкотемпературные стекла готовятся быстрее. Для дательного удерживания небольших белковых и других молекул во многих случаях недостаточно только физического удерживания.
Модификация поверхности
□ Реагент может присоединяться с помощью ковалентной связи или прямо к преобразователю, или к слою, нанесенному на преобразователь.
В этом способе подложкой может служить либо сам преобразователь, либо полимерный материал, который будет нанесен на преобразователь в последующей операции. Не все преобразователи имеют подходящие связывающие группы на поверхности, и поэтому часто лучше обеспечить.связь с материалом, который впоследствии можно нанести на преобразователь. Большинство из этих материалов — полимеры, например сополимеры малеинового ангидрида, сополимеры метакриловой кислоты/ангидрида и их производные [7.8-17-7.8-20], или другие поверхности со свободными группами -ГШг> -SH, -СООН и т. п. Такие носители наносят на преобразователь в виде тонкого слоя. Реакционные группы, к которым будет присоединяться биораспознающая молекула, можно образовать на полимере с помощью дериватизации мономера и сополимеризации или с помощью прямой реакции с полимером. Примеры из химии связывания различных реакционных групп на биораспознающей молекуле приведены на рис. 7.8-7, ж-л.
Многие другие поверхностные группы на подложке для иммобилизации перед ковалентным связыванием должны быть функционализированы. Привлекательными реагентами для функционализации поверхностных ОН-групп являются силановые связывающие агенты типа X3S1L или XSiR^L, где X — это гидролизуемая группа, a L — лиганд (или группа, которую можно превра-
528
Глава 7. Методы химического анализа и их применение
Рис. 7.8-7. Реакция связывания различных реакционных групп с биораспознающей молекулой.
7.8. Биосенсоры
529
Схема 7.8-3. Иммобилизация на поверхность, модифицированную силаном.
Схема 7.8-4. Иммобилизация модифицированной силаном молекулы.
тить в лиганд); эти реагенты широко применяются, .особенно на кремнеземе. Термическая и гидролитическая стабильность полученных на таких носителях связей высока.
Прн проектировании желаемой модификации поверхности возможны два подхода:
а) связывающий агент присоединяется к носителю и затем реагирует с макромолекулой, которую нужно иммобилизовать (схема 7.8-3);
б) получают комплекс макромолекула-силан и затем осаждают его прямо на поверхность (схема 7.8-4).
Оба метода имеют свои преимущества и недостатки. При наличии-более одного поверхностного лиганда первый способ приводит к образованию неоднородного поверхностного продукта. Если же в макромолекуле более одной реакционной группы, то реакция с силаном приводит к получению смеси комплексов силан-макромолекула, а следовательно, и смеси продуктов иммобилизации. Первый подход, вообще говоря, предпочтительнее, поскольку набор лигандов можно легко расширить после присоединения с помощью стандартных органических реакций, которые сложно применить к самим связывающим реагентам XaSiL.
Поверхностные ОН-группы часто используются и в других реакциях связывания (рис. 7.8-7, о-е), помимо них можно использовать и другие группы, например, амидо-, -СООН н т. д, (рнс. 7.8-8). На поверхности металлов, таких, как серебро и золото, особенно эффективным способом создания полезных функциональных групп для иммобилизации стало использование самоорганизующихся слоев [7.8-21], таких, как тиолы с длинной цепью, имеющие концевую группу для связи с распознающей молекулой. В случае иммобилизации ДНК концевая КНг-группа обеспечивает связывание через тиминовое кольцо [7.8-22]. Многие другие соединения структуры Y(CH2)nX, имеющие две концевые функциональные группы, разделенные гидрофобной цепью (разделитель), самоорганизуются на поверхности в соответствии с реакционной способностью функциональных групп. Если для реакции с поверхностью выбран Y,
530
Глава 7. Методы химического анализа и их применение
Рис. 7.8-8. Химические процессы иммобилизации.
тогда упорядоченный слой, включающий разделитель, можно получить в один этап.Можно вводить различные функциональные группы, при условии что они не будут конкурировать с головной группой Y за координацию с подложкой и не будут столь велики, чтобы препятствовать плотной упаковке углеводородных цепей.
При иммобилизации распознающей системы часто бывает необходим разделитель между поверхностью и биомолекулой. В принципе, его можно присоединить как интегральную часть функционального реагента (как видно выше) или как часть биоактивной молекулы. Для более гидрофильного и более жесткого разделителя наиболее подходит олигопептид. Концевая а-аминогруппа пеп-
7.8. Биосенсоры
531
тйда может быть связана, например, с поверхностью, активированной бром-цианом, а свободный конец -СООН связанного пептида используют для соединения с биоактивной молекулой. Потенциальным недостатком такого разделителя является опасность расщепления протеазами, присутствующими в биологических пробах, но многие из процессов, упомянутых здесь, также сопровождаются таким расщеплением.
Нанесение иммобилизованного слоя
Даже успешная иммобилизация биораспознающей молекулы не всегда обеспечивает нанесение слоя на поверхность сенсора. В приведенных выше примерах реагенты для иммобилизации или полимерная матрица были нанесены точно и воспроизводимо на поверхность сенсора. Толщина и диффузионные характеристики этого слоя определяют характер получаемого отклика.
О Материалы, которые были модифицированы реагентами, следует аккуратно нанести на преобразователь.
В электронной технике нанесение и разрешение слоев осуществляют предельно тщательно. Технологии толстых и тонких пленок стали инструментом для получения миниатюрных электронных приборов. Известен целый ряд доступных методов, пригодных для нанесения на преобразователь слоев, содержащих биораспознающий компонент.
Экранную печать —метод получения толстой пленки —можно использовать для нанесения ряда электродных материалов (включая углерод и благородные металлы), а также биораспознающих слоев, медиаторов или защитных покрытий. Обычно точность нанесения поддерживается в пределах 500 мкм, хотя для определенных материалов имеются сообщения о 100 мкм.
Струйная печать может выдавать отдельные капли проводящего раствора, например, буферного раствора белка, со скоростью до 50000 капель в секунду. Размер капель можно варьировать от 0,3 до 1,5 нл, что дает разрешение 1-5 мкм в зависимости от поверхностного натяжения жидкости и гидрофобности и абсорбции подложки, на которую эту жидкость наносят.
Обычные фоторезисторы можно наносить с разрешением, достигающим 250 нм при использовании источника света 350-450 нм или менее 100 им при использовании электронного пучка. Однако при получении слоев белок/полимер реальное разрешение фотолитографии оценивают более чем в 150 мкм из-за рассеяния света, индуцированного белком.
7.8.3. Подготовка биопреобразования
Амперометрические сенсоры
Интерпретация сигнала
Стационарный ток на амперометрическом электроде при диффузионном контроле записывается в следующем виде (из разд. 7.3):
Id = (7-8-1)
а
532
Глава 7. Методы химического анализа и их применение
где d — толщина диффузионного слоя, D—коэффициент диффузии определяемого вещества в слое, [S] — концентрация субстрата.
□ Ток, измеряемый на электроде при лотенциостатическом контроле, можно связать с кинетикой ферментативной реакции.
Скорость реакции, катализируемой ферментом, равна:
Ф1 di
fe[Eo][S]
Км + [S]
где Км — константа Михаэлиса—Ментен, а [Eq] — общая концентрация фермента. Предположим, что в схеме 7.8-5 повторное окисление фермента проходит быстро и не лимитирует реакцию, тогда в простейшем случае ток на электроде прн ферментном кинетическом контроле будет приближаться к величине:
nFdfc2[E0][S] k Км + [S]
Это дает для тока максимального отклика /тах, когда [S]ZS> Км-
Лпах = 7lFd/C2[Eoj
так что для диффузионно контролируемого тока можно записать:
r _ ^[S]
А <РМЕо]
а для кинетически контролируемого тока:
= lmax[S]
k FTm + S
(7.8-3)
(7.8-4)
(7.8-5)
(7.8-6)
что говорит о том, что для данной концентрации фермента Ц уменьшается при увеличении d и увеличивается с ростом D, тогда как увеличивается при увеличении d и не зависит от D. Это предполагает, что концентрация всех частиц по всему слою одинакова и что расход любого субстрата на внешней поверхности не приводит к его исчерпанию вблизи электрода (где протекает исследуемая реакция). На практике более вероятно протекание нелинейной ферментативной реакции и следует принять во внимание точную градуировку электрода. Был разработан ряд моделей, с помощью которых можно предсказать градиенты концентраций внутри иммобилизованного слоя [7-8-23-7.8-24]. Рассмотрение градиентов концентраций и косубстратных ограничений важно, чтобы обеспечить работу биосенсора в том режиме, для которого он предназначен.
s.e
Схема 7.8-5. Кинетические этапы катализа ферментом °2 * Emd Н2О2 ♦ Еох ОКСИДазОЙ.
7.8. Биосенсоры
533
Медиаторы
□ Медиаторы можно использовать как «курьеры переноса заряда» между электродом и ферментом.
Схема 7.8-5 создает впечатление, что электрон может переноситься от восстановленной формы фермента непосредственно к электроду. На практике такое случается редко, н даже для тех ферментов, для которых это явление наблюдается, оно не может быть осуществлено с воспроизводимой эффективностью н, следовательно, не может служить идеальным источником аналитического сигнала. Поэтому электродный сигнал должен зависеть от проводника. Такой косубстратный проводник, который может вновь окислять фермент н затем сам окисляться на электроде, называется медиатором.
На схеме 7.8-6 показаны три класса ферментов, наиболее часто используемых в амперометрических ферментных электродах. Многие электроноакцепторные молекулы и комплексы рассматривались как возможные медиаторы; из схемы 7.8-6,в видно, что оксидазы могут использовать их природный медиатор— кислород, потому что продуктом взаимодействия с ферментом является пероксид водорода, который электрохимически активен. В других случаях
О FAD-оксидазы
ЕрАО + МЕОда
б NAD-дегидрогекаэы
ПХХ-дегидрогенаэы S + Epoq^-^EpqoS
Ероона ♦ Р MEDq^K, EpQQ + MEDreQ
Схема 7.8-6. Распространенные схемы анализа, восстановительные ферменты, а — FAD-оксидазы, ПХХ-дегидрогеназы (PQQ — пирролхинолинхинон).
включающие окислительно-б — NAD-дегидрогеназы, в —
534
Глава 7. Методы химического анализа и их применение
требуются синтетические медиаторы, такие, как производные ферроцена (I) (?25-бис(циклопентадиенил)железо) и многие другие [7.8-25-7.8-27].
(I)
Ферроцен
В большинстве случаев ищут медиатор, который будет завершать ферментный редокс-цикл и возвращаться в свое исходное окисленное состояние. В этой ситуации он всегда возобновляется и не должен лимитировать реакцию из-за его расхода. К сожалению, так бывает не всегда и возможны косубстратные ограничения. Можно идентифицировать два случая по их характерному от
клику.
Как обсуждалось в предыдущем разделе, биораспознающий компонент биосенсора существует в слое, следующем за преобразователем. Для амперометрического ферментного сенсора величину токового сигнала оптимизируют в соответствии с модулями Тиле, Ф^ и Ф^:
2 cffcatEo]
m. £>m[Medbl
2 = ^[Ео1
8 fs[Sb]
где [Меф,] — концентрация медиатора в объеме. Существуют модельные параметры [7.8-28], которые позволяют предсказать изменения градиента концентрации по ферментному слою. Исходя из градиента концентрации можно рассчитать ток (ср. с ур. 7-8-5).
(7.8-7)
(7.8-8)
□ Амперометрический сигнал возникает за счет электроактивных частиц, которые диффундируют к электроду, а величина сигнала модулируется свойствами диффузионного слоя.
Важной информацией, которую мы можем извлечь из этих параметров, является величина d2 (уравнения 7.8-7 и 7.8-8), показывающая, насколько чувствителен будет отклик к изменению толщины ферментного слоя. Понятно также, как важен контроль концентрации фермента [Ео] для обеспечения воспроизводимости биосенсора. Можно также предсказать, что произойдет с откликом электрода, если изменить один из параметров в модуле Тиле. Например, на рис. 7-8-9 показан отклик ферментного электрода с различной толщиной слоя, где а) медиатор регенерируется н б) медиатор расходуется. В случае более толстого слоя (а) сигнал возрастает, но линейный диапазон устройства не изменяется; в случае (б) эффект косубстратного ограничения, когда медиатор не регенерируется, с ростом толщины слоя сигнал увеличивается, но в меньшем динамическом диапазоне.
7.8. Биосенсоры
535
Рис. 7.8-9. Влияние толщины слоя на нормированный отклик медиаторного редокс-электрода, где медиатор возобновляется (а) и медиатор расходуется (б).
Оксидазные электроды
□ Ферменты оксидазы, образующие пероксид водорода, можно использовать для амперометрического определения пероксида.
Первое сообщение о биосенсоре приписывают Кларку и Лайонсу [7.8-29]; они описали «ферментный электрод», в котором использовали глюкозооксн-дазу в качестве селективной биораспознающей молекулы для определения глюкозы. Фермент удерживался в мембранном сандвиче вблизи платинового электрода. Pt-электрод, поляризованный при 4-0,6 В относительно НКЭ, давал отклик на пероксид водорода, образующийся в ферментативной реакции (схема 7.8-7). Это сообщение привело к разработке анализатора глюкозы для определения глюкозы в крови (Yellow Spring Instrument 23 YSI, 1974). Трудно отыскать более благоприятное преобразование биологической системы, чем то, которое осуществлялось в таком амперометрическом ферментном биосенсоре, т. е. перенос электрона, связанный с редокс-ферментом. Тем не менее измеряемым параметром был не прямой перенос электрона между ферментом и электродом, а реакция продукта превращения—пероксида водорода. Преобразованный электродный сигнал, таким образом, не является прямым.
Несмотря на то, что метод косвенный, он оказался вполне удовлетворительным. К сожалению, в некоторых случаях основывать определение на пероксиде водорода неудобно. Примерами служат низкие значения рОг, при которых протекание реакции ограничено, или пробы, в которых определение пероксида на электроде осложнено значительными помехами со стороны других электро-
Птокоза ♦ °2
Схема 7.8-7. Окисление глюкозы, катализируемое глюкозооксидазой.
538
Глава 7. Методы химического анализа и их применение
Схема 7.8-8. Обратимый перенос электрона к медиатору и от медиатора.
Схема 7.84). Взаимодействие редокс-медиатора с редокс-ферментом.
активных веществ (таких, как витамин С, парацетамол и др.). В этих случаях требуется медиатор, окислительные свойства которого могут конкурировать со свойствами природного окислителя фермента. Кинетика этого редокс-катализа имеет, следовательно, первостепенное значение. Как описано в разд. 7.2, графическая зависимость тока от напряжения для обратимой редокс-пары (схема 7.8-8) в растворах подчиняется уравнению Рэндлса—Шевчика:
1р = -0,4463пт(^ ' ДО1/21/1/2 (7.8-9)
В случае сопряженной реакции (например, схема7.8-9) с высокой скоростью сканирования (г/) и малой величиной кд, график ток/напряжение может не показать никаких изменений. По мере того как г/ становится сравнимой с кд, Меф,х будет удаляться из системы за счет ферментативной реакции и «кажущаяся» концентрация Medre<i будет возрастать. Прн очень низких скоростях сканирования обратный пик может исчезнуть совсем, а катодный ток достигает плато (рис. 7-8-10), не зависящего от скорости протекания, н равен:
7 = nF[Medox](£ifc6[Ered])1/2 (7.8-10)
Рис. 7.8-10. а—циклическая вольтамперограмма диаминодурола, записанная при 50 мВ/с в растворе, содержащем 0,05 мМ глюкозы при pH 7; б —то же, но в присутствии глюкозооксидазы (24мкМ).
Идентифицировать каталитический механизм можно довольно просто с помощью анализа отношения которое возрастает с ростом v. Рабочая кривая, опубликованная Николсоном н Шейном в 1964 г. [7.8-30], позволяет оце
7.8. Биосенсоры
537
нить из отношения кинетически контролируемого тока (Д) к диффузионно контролируемому току (Д)- Когда фермент насыщен субстратом ([S] Км), модель можно аппроксимировать к реакции первого порядка, посредством чего можно получить константу скорости реакции псевдопервого порядка к. Если же рассчитать к для различных концентраций [Е], можно получить константу скорости второго порядка к".
Пример
Ферроценмоиокарбоновую кислоту (FcCOOH) испытывали в качестве медиатора для глюкозооксццазы (GOD). В табл. 7.8-2 представлены данные, полученные из пика тока окисления на циклической вольтамперограмме FcCOOH в буферном растворе: 1) в отсутствие GOD (7 = Id из уравнения 7.8-7) и 2) в присутствии GOD и насыщающего количества глюкозы (0,05 М) (/ определяется каталитической реакцией по мере уменьшения v).
Для фиксированной концентрации фермента ток Ik связан с /а с помощью множителя:
(7.8-11)
Для измеренного отношения Д/Та величину у/(к/а) можно получить из рабочей кривой [7.8-30]. Когда к > а (низкая г>), то связь линейна и каталитический процесс доминирует. В этой области график зависимости (к/а) от 1/v представляет собой прямую линию с наклоном kRT/nF. Рис. 7.8-11 изображает графики к/a для различных концентраций GOD, дающие к.
к = fc"[GOD] (7.8-12)
где /с —константа скорости псевдопервого порядка, а к” — константа скорости второго порядка. На рис. 7.8-12 построен график зависимости к, взятых из рис. 7.8-11, от [GOD], чтобы получить fc". Получена константа скорости второго порядка, равная 1,9-105 л-моль-1^"1. Это сравнимо с оценкой 15-105 л-моль-1-с-г для окисления GOD кислородом, и, таким образом, показано, что производное ферроцена является хорошим медиатором.
Таблица 7.8-2. Данные из пика тока окисления ферроценмонокарбоновой кислоты в присутствии глюкозы с GOD (Ik) и без GOD (Id)
Скорость сканирования V, мВ/с (1) Id, мА л Ik, мА
5,7 мкМ [СОЛ] 12,6 мкМ [СОЛ] 16,4 мкМ [GOD] 23,7 мкМ [GOD] 32,4 мкМ [СОЛ]
100 17,0 19,4 23,4 24,6 27,8 33,4
75 14,6 17,4 21,8 23,8 27,2 32,6
50 11,8 15,4 20,4 23,9 26,6 32,0
25 8,2 13,4 19,4 22,2 25,8 31,0
10 5,0 12,8 18,8 21,6 25,0 29,8
5 3,6 12.7 18,4 21,2 24,6 23,8 28,8
2,5 2,4 12.6 18,0 20,8 28,0
538
Глава 7. Методы химического анализа и их применение
Рис. 7.8-11. Отношение к/a в зависимости от [GOD], дающее ряд значений к.
Рис. 7.8-12. График зависимости к от [GOD] для определения к".
□ Медиатор можно использовать для окисления оксидазы вместо кислорода, но он должен конкурировать с кислородом.
Оксидазный электрод, сконструированный для контроля тока окисления Medred и использующий на стадии окисления либо кислород, либо медиатор, испытывает значительное влияние кислорода на точность измерения сигнала. В этой ситуации к$ и fc4 (схемы 7.8-5 и 7.8-9, схема 7.8-6, а) конкурируют. Такой медиаторный ферментный электрод может давать заниженный сигнал, если кинетика медиатора (fc4) не превосходит окисление молекулярным кислородом (кз).
Оптимизация одного ферментативного определения может создать основу для аналогичных схем и позволяет определять другие вещества помимо первоначально интересующего определяемого вещества. Например, схема 7.8-10,а/б показывает, как можно применить систему с глюкозооксидазой в «конкурентной» реакции глюкозы с другими ферментами, использующими глюкозу в качестве субстрата, и, таким образом, определять, например, креатин, гексокиназу или креатинкиназу [7.8-31]. Если же медиатор применяют в качестве метки для антигена, комплекс антиген-медиатор можно использовать в иммуноферментном анализе (схема 7.8-11). Эта схема [7.8-32] определяется меченным ферроценом (Fc) комплексом антиген-антитело, который дезак-
7.8. Биосенсоры
539
Схема 7.8-10. Пути реакций согласно связанному с редокс-ферментом амперометрическому измерению с помощью редокс-медиатора. б — определение глюкозы; (о)+(б)—определение креатина, креатинкиназы или гексокиназы.
Схема 7.8-11. Пути реакций согласно связанному с редокс-ферментом амперометрическому измерению с помощью редокс-медиатора, для конкурентного иммунного анализа с использованием антигена, меченного ферроценом.
тивирован по отношению к переносу электрона к ферменту, а несвязанный в комплекс меченый антиген остается активным.
Такие медиаторные определения могут оказаться элегантным решением для биосенсоров. Тем не менее, оптимизация только реагента не является исчерпывающим решением. Как указывалось ранее, в биосенсоре реагент должен быть иммобилизован, чтобы взаимодействовать и с определяемым веществом, и с преобразователем как самодостаточная система.
Другие редокс-ферменты (схема 7.8-6) предполагают те же общие принципы, если их включают в биосенсор, но каждый класс имеет тонкие отличия.
Ферментные электроды на основе ИДР
□ Восстановление NAD+ на электроде при служит оптимальным (удобным) аналит»
к образованию димеров и не м сигналом.
В окислительно-восстановительных процессу большого класса ферментов вместо молекулярного кислорода используется нщвртинамиднуклеотид NAD
540
Глава 7. Методы химического анализа и их применение
или его фосфат NAD(P). Это, как правило, дегидрогеназы, участвующие в реакциях переноса водорода н описанные в схеме 7.8-6,6 Косубстратом NAD является растворимый компонент, который ассоциирован с ферментом во время реакции, но способен отсоединиться от фермента и перемещаться в объем раствора.
Существуют также многие NAD-дегидрогеназы, для которых ферментативная реакция протекает в обратном направлении, приводя, таким образом, к образованию NAD+. В случае биосенсора сигнал получали бы за счет восстановления NAD+. Восстановление NAD+ на электроде имеет тенденцию к образованию (путем радикальной полимеризации) димеров, а не восстановленного кофактора NADH. Эти димеры адсорбируются на поверхности электрода, что делает NAD+ непригодным для использования в качестве компонента биосенсора.
□ Окисление NADH на электроде обычно требует большого перенапряжения.
Окисление NADH в NAD+ можно проводить на графитовом электроде, но оно обычно идет с высоким перенапряжением или сопровождается порчей электрода [7.8-33]; следовательно, более приемлемо выполнять косвенное окисление через медиатор или сочетать окисление с другой сопряженной ферментативной реакцией, как показано на схеме 7.8-10. Эти окислительно-восстановительные агенты должны обладать теми же свойствами, что описанные в предыдущем разделе, н, хотя медиаторная кинетика не должна ограничивать реакцию, здесь нет конкуренции с молекулярным кислородом.
Ферменты пирролохинолинхинона
Для хинопротеинов молекулы хинонов играют роль кофакторов и простетнче-ских групп. В большинстве бактериальных хинопротеинов пирролохинолинхи-нон (ПХХ) связан прочно, но не ковалентно. Большинство хинопротеинов является дегидрогеназами и часто не требует растворимых кофакторов: природным электронным акцептором служит медьсодержащий белок или связанный в мембране убихинон. Эти ферменты можно использовать таким же образом, как и оксидазы. В рутинном анализе используют искусственные электронные акцепторы (схема 7.8-6,е) и подобно системам с NAD, обсуждаемым выше, эти искусственные медиаторы не подвержены мешающему влиянию кислорода, которое имеет место для оксидаз, поскольку О 2 не является косубстратом для этих ферментов [7.8-34, 7.8-35].
Клетки и клеточные компоненты
□ Живые клетки можно использовать как распознающий компонент.
Обсуждение мы до сих пор проводили, делая упор на ферменты в качестве биораспознающего компонента. Но этим компонентом не обязательно должен быть изолированный фермент, вместо этого можно использовать живую клетку [7-8-36] или компонент клетки. Описаны микробные сенсоры с некоторой специфичностью к различным определяемым веществам: от СОг до витамина В. Описано также приготовление тканей, на основе любых источников, которые
7.8. Биосенсоры
541
только можно вообразить: банановая мякоть или яблочный порошок, например, содержат высокие концентрации полифенолоксидазы, которую можно использовать для определения допамина и катехинов [7.8-37, 7-8-38]. В большинстве случаев устройства на основе этих клеток связаны с изменением давления С>2, которое контролируют с помощью амперометрического электрода, уста7 нов ленного за клеточным слоем. Это позволяет связать клеточное «дыхание» с присутствием данного определяемого вещества. Подобно описанным выше измерениям, в данном случае также осуществлй'е'гся косвенное определение.
В отличие от обсуждаемых выше систем со свободным ферментом, транспорт как определяемого вещества, так и «курьерской частицы» между электродом и бнораспознающим элементом внутри клетки может быть ограничен стенками клетки и мембранами, н химия окислительно-восстановительной молекулы (медиатора) должна быть выбрана так, чтобы обеспечить нужные свойства мембранного транспорта, а также нужную электрохимию. Такие факторы, как липофильность н поверхностные взаимодействия, играют более важную роль, чем при приготовлении ферментов.
□ Некоторые гербициды можно определять, помх ингибированию фотосинтеза.
Пример клеточного медиатора можно найти в области контроля остаточных содержаний гербицидов через механизм электронного транспорта прн фотосинтезе (ЭТФ) с использованием цианобактерий или тилакоидных мембран [7.8-39]. Многие гербициды действуют как ингибиторы ЭТФ, часто между двумя фотосистемами (PSI н РБП), как показано иа рис. 7.8-13, а результатом является уменьшение фототока.
В принципе, возможно получить один или несколько редокс-циклов между редокс-белком н электродом. Например, в описанной выше системе ЭТФ имеется NADPH-дегидрогеназа, которая расположена в плазменной мембране, связанной с выходом ЭТФ (верхняя часть рис. 7.8-13). Измерение возможно с помощью передачи через медиатор (например, феррицианид) и можно избежать таких проблем, как необходимость пересечения медиатором мембран; однако NADP проходит долгий путь «по течению» от исходной точки ингибирования, н поэтому такой косвенный путь с большей вероятностью будет подвержен мешающему влиянию других частиц «не определяемого» происхождения.
Хорошими акцепторами электронов для PS П являются п-фенилендиамины. В целом С-замещенные n-фенилендиамины способны входить в клетку (рнс. 7.8-14), восстанавливаться в системе PSII и выходить из клетки. N-Замещенные, однако, остаются в просвете после восстановления в PSII и, следовательно, не завершают медиации доставкой сообщения на электрод [7.8-40].
Потенциометрические сенсоры
□ Потенциометрические измерения связаны с уравнением Нернста.
Не все вещества можно определить с помощью окислительно-восстановительной системы; следует испробовать другие, параметры, а не только перенос электрона. Действительно, даже если для интересующего нас определя-
542
Глава 7. Методы химического анализа и их применение
Гербици. ингиби
NADPH
Рис. 7.8-13. Схематическое изображение цепи электронного транспорта фотосинтеза (ЭТФ). На входе — НгО и свет, на выходе— NADP и Ог. Белки ЭТФ представлены с помощью сокращенных названий или в виде темных кружков.
7.8. Биосенсоры
543
t шшшшш‘
700
600
500
400
300
улинурона
2001—।—।—г—j—।—т—т—।—т—।
0 60 120 160 240 300
Время, мин
Световые импульсы
Рис. 7.8-14. Диаминодурол (С-замещенный п-фенилендиамин) в качестве медиатора фототока в клетках Synechococcus. а—ток электрода как отклик на импульс света. Транспорт заряда и PSII активируется и прерывается медиатором на Q-белке (рис. 7.8-13); б—детектирование двух активных в ЭТФ гербицидов. График пикового фототока построен в зависимости от времени (ppb — миллиардные доли).
емого вещества имеется редокс-фермент, он может не всегда подходить для применения амперометрического метода. Альтернативой являются потенциометрические измерения.
Интерпретация сигнала
Измеряемый потенциал (Е) определяется уравнением Нернста (разд. 7.3), и для изменения активности иона i это уравнение имеет вид:
Е = Ё° ± -In Oi (7.8-13)
nF
так что измеряемый параметр логарифмически связан с концентрацией определяемого вещества. Как мы уже видели (разд. 7.3), хитрость с потенциометрическим измерением заключается в том, чтобы заставить его отвечать селективно только на один ион; соответственно в биосенсоре изменение ионного транспорта должно происходить в результате биораспознавания. [Н+] — главная переменная, хотя величина pH может изменяться непосредственно или через равновесие с СОг или NH3. В последнем случае можно использовать мембрану, содержащую ионофор на аммоний, или pH-чувствительную поверхность.
В случае изменения pH, связанного с ферментом, конечный отклик рН-сенсора зависит от баланса всех равновесий, включающих Н+: реакций протонирования и депротонирования продуктов ферментативной реакции, буферной емкости и т. п. Если рассматривать только прямой «выход» ферментативной реакции, без равновесий,-связанных с буферной емкостью н т. д., и предпола
d[S] di
544 Глава 7. Методы химического анализа и их применение
гать, что [Н+] непосредственно связан с расходом субстрата (S), то измеряемый потенциал будет связан с [S]. Как видно из предыдущего обсуждения, касающегося амперометрической стационарной модели, ход взаимодействия предсказывают, оценивая как диффузия в слое иммобилизованного фермента влияет на ферментативную кинетику. Здесь нет медиатора и конкуренции с молекулярным кислородом, вместо этого имеется прямое определение продукта ферментативной реакции. Используя подход, подобный описанному выше, и начиная с уравнения
D»d2[S] fc2[Eo][S] Ф/2 Km + [S]
где у— расстояние поперек мембраны, получаем тот же модуль Тиле, дающий информацию об источнике изменения величины сигнала
ф2 = ^г <7-845)
^А[Ьь]
Применение под ход ящих граничных условий приводит к значениям концентрации субстрата и/или продукта поперек ферментного слоя. Решение показывает, что когда Км [Sb], поверхностная концентрация на лежащей ниже потенциометрической мембране связана с [Sb]. Однако, когда Км [Sb], отклик будет зависеть от относительных скоростей ферментативной реакции и диффузионного массопереноса и не будет зависеть от [SbJ.
Как упомянуто выше, баланс между ферментативным и буферным равновесиями вносит вклад в измеряемый сигнал. Теоретическая кинетическая модель, учитывающая все эти связанные явления, предсказывает стационарный отклик ферментного рН-сенсора [7.8-41]; существование «нернстовского» наклона не предсказано и не имеет такого значения, как для нон-селективных электродов, выполняющих прямое измерение ионного транспорта.
Выла сделана попытка объяснить эффект, связанный с белком на границе раздела [7.8-42], и показать, как можно использовать оксидную поверхность полевого транзистора (ПТ) для наблюдения за динамическим откликом белков. На амфотерной поверхности неорганического оксида гидроксильные группы находятся в равновесии (схема 7.8-12), н, таким образом, на поверхностный потенциал Ф будет влиять значение pH в объеме ([Нь]) в зависимости от буферной емкости поверхности. Поверхности, имеющие большие количества групп OHj и О-, поддерживают значение [Н+] (Н+ в «растворе» вблизи от поверхности) постоянным в широком интервале pH, при этом Ф должен иметь нернстовский отклик на [Н£].
Когда белок также находится на поверхности, равновесие принимает более сложный вид. Белок состоит из большого числа пептидов, содержащих ионизирующиеся группы, каждая из которых имеет свое собственное равновесное значение pH (схема 7.8-13). Отношение активностей внутренних (i;n) и внешних (lex) подвижных нонов в этой «системе» дает вклад в потенциал Доннана:
7.8. Биосенсоры
545
Схема 7.8-12. Амфотерное равновесие на поверхности неорганического оксида.
равиове-
При pH, смещенном от изоэлектрической точки pl, этот потенциал соответствует разности между «внутренним pH» белка и раствором. Любые изменения aiex будут вызывать изменение -фо и заставлять белок высвобождать или принимать протон, чтобы восстановить равновесие. Во время этого восстановления непосредственное окружение белка будет испытывать временное изменение pH из-за притока/оттока протонов. Если белок иммобилизован вблизи от ион-селектнвного полевого транзистора (ИСПТ), это влияние концентрации ионов можно измерить, потому что ИСПТ очень быстро реагирует на изменения поверхностного заряда.
Ферментативный анализ
Потенциометрические измерительные устройства наиболее часто разрабатывают на основе pH-чу ветвите л ьных электродов, и в pH-ПТ используют те же аналитические реагенты, что н в ИСЭ. Можно применять любую ферментативную реакцию, которая приводит к изменению Н+ (например, схемы 7.8-2 и 7-8-14).
Использование биораспознающих молекул для анализа обеспечивает специфичность отклика биосенсоров. Однако специфичность зависит от природы бнораспознающей системы. Менее специфичная распознающая система может быть невыгодной, но в некоторых случаях, когда при этом проявляется специфичность к другим субстратам, она может оказаться полезной. Например, каталитическое расщепление связи углерод-фтор с помощью пероксидазы хрена в присутствии сенсора на фторид-йон [7.8-43] дает полезный способ детектирования фторорганических соединений.
Н2О2 + X-F„ X'Fn-i + F- + Н2О (7.8-16)
18 Аналитическая химия. Том 1
546
Глава 7. Методы химического анализа и их применение
□ Потенциометрический сигнал часто можно получить косвенным путем, вызывая изменения поверхностного заряда или селективного транспорта частиц.
В разд. 7.3 показаны другие системы потенциометрического измерения, которые могут быть пригодными для сочетания с ферментативными реакциями. Рассмотрите различные классы ферментов и определите, какие из них пригодны для потенциометрического анализа.
Иммунный анализ
Другим приложением потенциометрических сенсоров и устройств ПТ является иммунный анализ, включающий образование комплекса антитело-антиген [7.8-44]. Теоретические основы этих измерений не всегда ясны. Например, Аизава [7.8-45] приготовил и установил в потенциометрической ячейке специфичную к антителам Вассермана мембрану на основе липидного антигена из кардиолипина, фосфатидилхолина и холестерина в триацетилцеллюлозе. Предполагают, что отклик возникает за счет изменения ионообменных свойств мембраны при связывании антитела. Шасфурт и др. [7.8-46] сообщали о способе применения серии ступенчатых изменений концентрации электролита и контроля мембранного потенциала с использованием Нечувствительного ПТ с быстрым откликом (см. разд. 7.8-3) как методе иммунного анализа. Этот метод подходит для измерения любого процесса на поверхности, который включает изменение поверхностного заряда.
Оптические сенсоры
□ Оптические биосенсоры работают во внешнем или внутреннем режиме.
В предыдущих разделах об электрохимических биосенсорах для создания биосенсора манипулировали несколькими различными методами биоанализа. На примерах было показано, как можно связать биораспознающий механизм с физическим методом измерения либо напрямую, либо через медиатор. Используя тот же подход, можно разрабатывать биосенсоры на основе различных методов преобразования. В этом разделе обсуждаются особенности оптического преобразования.
Оптические биосенсоры бывают диух типов, которые отличаются по оптической конфигурации. Во внутреннем режиме падающая волна не проходит через объем пробы, а проходит в световод и взаимодействует с пробой на поверхности в затухающем поле. Во внешнем режиме падающий свет взаимодействует непосредственно с пробой, или проходя через нее или отражаясь от фазы пробы. В этом случае свет от источника не обязательно должен проходить через световод, хотя иногда бывает целесообразно передавать свет от источника к удаленной точке измерения через оптическое волокно.
Некоторые анализы биологических сред можно проводить без вмешательства биораспознающей поверхности, которая характеризует биосенсор, поскольку определяемое вещество обладает характерными ему оптическими свойствами. Например, уровни содержания кислорода in situ определяют по сдвигу поглощения между гемоглобином н оксигемоглобином. В отличие от этого, в оптическом биосенсоре определение зависит от биораспознавания в
7.8. Биосенсоры
547
иммобилизованном слое преобразователя; и подобно тому, как в электрохимических методах можно использовать косвенные измерения и медиаторы, для оптических систем можно применять метки. Однако в оптическом анализе такие метки могут быть «пассивны» или сами реагировать с определяемым веществом или продуктом биореакции. Многие из этих возможностей анализа обсуждались в предыдущих главах.
□ Оптические метки могут быть «пассивными» индикаторами или принимать участие в реакции с определяемым веществом.
Как правило, в устройствах с визуальным детектированием используются хромогенные, окрашенные реагенты-метки, тогда как красители для иммунного анализа обычно являются производными флуоресцеина. Производные флуоресцеина далеко не идеальны для сенсорных приложений, поскольку они pH-чувствительны и имеют короткую длину волны возбуждения (АВОЗб), что накладывает ограничения на используемые оптические материалы. Более предпочтительны флуорофоры с большей АаоЭб и достаточным стоксовым сдвигом (например, Техас красный). Измерять можно или непосредственно интенсивность флуоресценции, или, если протекает реакция тушения (например, тушение флуорофоров кислородом), время жизни флуоресценции. В этом случае уравнение Штерна—Фольмера принимает вид
^ = 1 + K[S] (7.8-17)
где К — константа тушения, а г — время жизни флуоресценции [7.8-47, 7.8-48]. Следует помнить, что в анализе с биораспознаванием зависимость тушения может быть подвержена серьезным помехам, поэтому следует хорошо продумать конструкцию слоев, чтобы избежать этого.
В целом, желательно использовать метки с длинноволновыми максимумами поглощения и испускания. Это предпочтительно как в случае применения световодов (поскольку наиболее общедоступные волокна плохо пропускают свет с длиной волны короче 420 нм), так и во всех других устройствах (потому что простые и наиболее дешевые источники света—светодиоды и диодные лазеры—имеют обычно длину волны больше 450нм). Работа в длинноволновой области может также иметь дополнительные преимущества благодаря более низкому флуоресцентному фону и более высокой чувствительности имеющихся фотодетекторов. Следует помнить, что не всегда удается работать в этих рамках и что некоторые из наиболее чувствительных маркирующих реагентов действуют в коротковолновой области.
Интерпретация сигнала
Рассмотрим пример, в котором метка взаимодействует с определяемым веществом. Если взаимодействие между ними можно описать просто в соответствии с равновесием
A+L^Ab (7.8-18)
так что
К =
[AL] [А][Ц
(7.8-19)
18*
548
Глава 7. Методы химического анализа и их применение
и общая концентрация метки в иммобилизованной фазе равна
[Ьобщ] — [AL] + [L]
(7.8-20)
тогда
W = Si " lALl = T^jXT <7-8-21)
|А1 = М^7ьГИ} или [А1^Мх ^lAL]} (78’22) к I 14 J к I [Ьобщ] ~ IALJ J
□ Если аналитическую реакцию можно описать как обратимое равновесие, то в результате аналитический сигнал не будет линейным.
Эти уравнения подразумевают, что отклик на определяемое вещество может не быть линейным во всем интересующем диапазоне концентраций. При анализе растворов в объеме эту проблему часто преодолевают, подбирая количество используемой метки таким образом, чтобы ее реакция с определяемым веществом попадала в диапазон, где сигнал пропорционален концентрации L или AL, в зависимости от того, какое из них вызывает оптический сигнал (например, для измерения [AL] должно быть [Ьобщ] [AL]). В противоположность этому, в иммобилизованной фазе обычно более сложно манипулировать концентрацией, чтобы попасть в этот диапазон. Вместо этого можно предложить сравнение двух длин волн, где [AL] и [L] имеют различное поглощение, дающее отношение, не зависящее от [Ьобщ]:
^ = К[А] (7.8-23)
Второй способ заключается в использовании меток как «пассивных» индикаторов. Существует несколько типов сенсоров, в которых можно использовать метки таким образом. Например, в простом конкурентном связывающем анализе в растворе отклик можно моделировать с помощью следующих двух равновесий: во-первых, между антителом или связывающим белком (Вр) н определяемым веществом (А), и, во-вторых, между антителом или связывающим белком и меченым аналогом определяемого вещества (а*):
А + Вр <=* А:Вр,
а* + Вр а* : Вр,
„ . 1А:Вр1
А [А][Вр]
[а* : Вр]
” [а*][Вр1
(7.8-24)
(7.8-25)
□ Сенсор должен быть способен различать реагенты из правой и левой частей ревновесия.
Следует ожидать, что эти равновесия будут сохраняться и при иммобилизации Вр, если константы относительного сродства Кд и KR- не зависят от концентраций и ограничений, накладываемых Иммобилизацией на реакцию с
7.8. Биосенсоры
549
Вр. Равновесие не будет зависеть от концентрации, если связывание на одном Вр не влияет на связывание на соседнем Вр. Если это справедливо и
K6„J = [а*| + |а* : Вр| (7.8-26)
[AoeJ = [А] + [А:Вр] (7.8-27)
[Вро6щ] = [Вр] + [а* : Вр) + [А:Вр] (7.8-28)
тогда будут измерять [а*] или [а*:Вр], в зависимости от того, каким образом устроен сенсор, и от их концентраций, на которые влияет отношение констант относительного сродства Кд и К*-. С помощью подстановки в приведенные уравнения получаем
[Вробщ1 = (1+уЛ|А1) (- |а>| +(7'8‘29) \ Ла* / у 1аобщ1 /
так что нормированный сигнал за счет определяется выражением
[а*] /1 + [А] X / [аобщ] — [а ] X [Вробщ) z- р
Ш = 1+1 "ХфТ? ~ fcj (7-8’30)
и
[AoeJ = [А] Ка +1] (7.8-31)
Отношение [Вробщ] : [а^щ], а также Кд : К&* являются критичными для настройки диапазона отклика и могут быть подобраны для оптимальной работы.
Из этого обсуждения можно сделать вывод, что одной из основных задач, необходимых для оптического биосенсора, независимо от того, используют «пассивную» или «активную» метку, является способность различать «свободное», или немеченое, определяемое вещество и «связанное», или меченое, определяемое вещество без проведения разделения прн анализе, как это можно сделать в классическом биоанализе, использующем растворы!
Внешние определения с меткой
Ферментативный внешний оптический сенсор
□ В оптических измерениях возможно использование пероксида водорода или других продуктов ферментативных реакций таким же образом, как в электрохимических сенсорах.
Как мы видели в предыдущей главе, многие из традиционных биоопределений основаны на оптических измерениях, но все они используют растворы без иммобилизованных реагентов н иногда с этапом разделения. Тем не менее, онн уже имеют многие из необходимых аналитических компонентов для создания биосенсора. Так как же можно осуществить этот переход? Оптические биосенсоры связаны с изучением реакций на поверхностях; первоочередной задачей, следовательно, становится перенесение реакции на поверхность. Многие наборы «домашних тестов» претерпели такой переход и сейчас выполняют оптические измерения. Рассмотрим, например, устройство для определения глюкозы, используемое диабетиками. Мы видели в предыдущем разделе, что глюкозу можно определять с помощью ферментативной реакции с глюкозооксидазой
550 Глава 7. Методы химического анализа и их применение
(схема 7.8-7). В предыдущем разделе «Оксидазные электроды» (см. с. 535) мы обсуждали амперометрическое определение глюкозы, основанное на использовании пероксида водорода либо синтетического медиатора. Ранее в этой главе биоанализ включал также применение пероксида водорода, но в этом случае пероксид определяли с помощью другой реакции, приводящей к образованию окрашенного комплекса в присутствии пероксида. «Тест-полоска» оптического биосенсора должна включать все реагенты для этого биоанализа в иммобилизованном виде. Пробу помещают на полоску и контролируют развитие окраски с помощью устройства, содержащего источник света и детектор. В этом случае образующийся хромогенный комплекс служит меткой; это «активная» метка, поскольку она участвует в реакции с пероксидом, но, в отличие от рассматриваемых выше процессов (уравнения 7.8-18-7.8-23), ее образование представляет собой быстрый необратимый процесс, протекающий наряду с основной ферментативной реакцией. Существует линейная зависимость между сигналом н концентрацией пероксида: его концентрация зависит от кинетики ферментативного процесса, а не от кинетики развития окраски.
В другом примере используют ферментативную реакцию, которая включает изменение [Н+] (см., например, схемы 7.8-2 и 7.8-14). Здесь меткой будет то вещество, оптические свойства которого изменяются вместе с pH- В этом случае этап маркирования является равновесным (уравнение 7.8-17); это значит, что линейная зависимость между определяемым веществом н сигналом возможна только при определенных условиях. На выбор рН-индикатора влияет интересующий нас диапазон pH и p/fa иммобилизованного хромофо-ра/флуорофора (который может быть иным, чем для раствора). Существует большое число таких соединений для различных диапазонов pH и с различными спектральными характеристиками, так что возможность комбинаций реакций высока. Однако, как упомянуто в предыдущем разделе, посвященном потенциометрическим измерениям, проблема, связанная с контролем реакции по изменению pH, состоит в том, чтобы характеризовать собственный pH пробы, ее буферную емкость и чувствительность равновесия к ионной силе, — каждая из этих характеристик оказывает значительное влияние на определение и должна быть принята во внимание при интерпретации результатов. Эти осложнения могут также сделать схему неподходящей.
Тем не менее, оптическую схему можно построить таким же образом, как электрохимические сенсоры, использующие отклик от косубстрата или продукта ферментативной реакции. Многие из схем анализа, обсуждаемых в предыдущих главах, подходят для такого преобразования. Например, ксантинокси-
Лени|иллин Пенициллиназа
I
Пенициллат + Н+
б
Мочевина
Уреаза
I
nh4* + нсо3”
Схема 7.8-14. Ферментативные реакции, связанные с изменением pH.
7.8. Биосенсоры
551
Люциферин
люминол* 2HJO2 * он-
Пероксмджза
I
З-Аминофталат ♦ Nj ♦ 3HgO* hv (Цзд, - 430нм)
Схема 7.8-15. Хемилюминесцентная реакция люминола с пероксидом водорода.
Люцифераза
нр
Оксилюциферин
Оксилюциферин
+Л*(1«5в2 нм)
Схеме 7.8-16. Генерация света, катализируемая люциферазой.
дазу, иммобилизованную на подходящей подложке, можно использовать для определения гипоксантина и ксантина [7.8-49]. Пероксид водорода, полученный в реакции с оксидазой (см. схему 7.8-6,а), можно определять по хемилюминесцентному механизму, основанному на реакции с люминолом (схема 7.8-15). Как можно заключить из этой схемы, для сенсора требуется совместная иммобилизация пероксидазы и подача люминола, который особенно популярен в проточно-инжекционном анализе, поскольку растворим в воде. К сожалению, в системе часто наблюдается «несовпадение» pH, потому что обычно для хемилюминесценции требуется щелочной pH порядка 10-12, тогда как многие ферменты имеют более низкое оптимальное значение pH.
□ Биолюминесценция достигает очень низких уровней определения.
Весьма привлекательно использование биолюминесцентного анализа, поскольку здесь также не требуется источник возбуждения. Биолюминесценция ассоциировлась главным образом со светлячками, у которых процесс люминесценции включает фермент люциферазу, катализирующую окисление гетероциклов люциферина (схема 7.8-16). В другой схеме (схема 7.8-17) происходит реакция бактериальной люциферазы с восстановленным кофактором флавина. Сама реакция не обеспечивает общего метода анализа, но сочетание с кофакторами, такими, как ATP, FMN или NADH, обеспечивает ее связь с другими путями метаболизма. Получены пределы обнаружения NADH на уровне 10-15 моль и линейность, составляющая пять порядков величины.
Внешний иммунный анализ с меткой
В иммунном анализе определяемое вещество не связано с продуктом каталитической реакции, а является меткой, маркирующей наличие комплексного или свободного антигена или антитела. Основы иммунного анализа обсуждаются в разд. 7.9. Так же, как в ферментативных тест-наборах (например, для определения глюкозы), на основе иммунного анализа создано большое число тест-наборов; например, в тесте на беременность — хотя здесь результат рассчитан на наблюдение пользователя за развитием окраски и не контролируется автоматически (т. е. детектором служит человеческий глаз!). На такой тест-полоске
552
Глава 7. Методы химического анализа и их применение
Схема 7.8-17. Использование бактериальных люцифераз в определении этанола для получения в возбужденном состоянии комплекса с восстановленным кофактором флавина (FMNHa)-
меченый и немеченый комплексы обычно разделяются — разделяют фазы или используют хроматографию. Затем анализ следует модели связывания белок — определяемое вещество (уравнения с 7.8-24 по 7.8-31), в которой метка наиболее часто является ферментом, а окраска развивается на последующих этапах реакции.
В принципе, можно усовершенствовать эти общие системы, чтобы получать более количественный результат в тех случаях, когда от анализа требуется больше, чем положительный или отрицательный ответ. Конфигурация сенсора зависит от того, какой вид анализа предпочтителен: конкурентный или саедцшчевый. Теоретически особенно жизненной альтернативой кажется конкурентное смещение флуоресцентной метки. Так как это равновесный процесс, то загрязнения или примеси на поверхности не должны изменять абсолютного результата, хотя вполне возможно влияние на отношение сигнал/шум [7.8-50}. Вероятным недостатком является то, что успех такого анализа со смещением очень сильно зависит от относительной кинетики связывания соединения с меткой и пробой. Для получения количественного результата они должны значительно различаться.
Общим фактором во всех этих определениях является то, что эффективность измерения зависит от этапа разделения: две формы метки должны быть отделены физически одна от другой, чтобы анализ мог их различить. Одним из способов добиться такого разделения служит проведение реакции в «мертвом объеме» сенсора, при этом измерение выполняют в определенной зоне детектирования. Такое устройство можно создать, используя ячейку с двумя отделениями: биораспознающий реагент (например, связывающий белок) иммобилизуют в одном отделении, т. е. в мертвом объеме, тогда как метка и определяемое вещество перемещаются между двумя отделениями. Определение тогда зависит от того, в правильном ли месте расположена зона детектирования.
7.8. Биосенсоры
553
□ Эффективность измерения зависит от возможности разделить связанную и свободную метки.
Естественная зона детектирования образована приемным конусом на конце световода (рис. 7.8-15,о). Типичный сенсор, таким образом, может использовать два световода, чтобы направлять свет к отдаленному концу, где происходит аналитическая химическая реакция, и обратно от него [7.8-51]. Оптические волокна имеют эффективное поле зрения, описываемое численной апертурой (NA) волокна, и, как можно видеть из рис. 7.8-15,о, чем больше NA, тем больше поле зрения. В описываемой выше конфигурации с двумя световодами неправильный выбор NA может привести к большому «мертвому объему» (рис. 7.8-15,6), который, очевидно, будет уменьшать измеряемый сигнал. Тем не менее, фокусировку поля для получения видимого и мертвого объемов можно использовать как преимущество в конкурентном анализе (рис. 7.8-16), где связывающий белок (Вр) иммобилизован в мертвом объеме, а проба (А) и меченый аналог (а*) конкурируют за места связывания, оставляя несвязанный а* диффундировать в облучаемый объем, где его и определяют.
Рис. 7.8-15. а —поле зрения в среде пробы на конце световода как функция численной апертуры (NA); б — поле детектирования в пробе при использовании двух смежных световодов для возбуждения и детектирования.
Рис. 7.8-16. Иммобилизация связывающего белка в «мертвом объеме» для конкурентного анализа.
Область наблюдения
554
Глава 7. Методы химического анализа и их применение
Пример
Конканавалин А (СопА, т. е. Вр в приведенной выше модели) был связан на полой диализной трубке, установленной на конце световода в мертвом объеме (рис. 7.8-16) |7.8-52]. Оценка величины [Вробщ], основанная на загрузке на единицу объема трубки, дает 10 мМ. Световод имел внутренний диаметр 100 мкм и численную апертуру 0,3. Для диализной трубки с внутренним диаметром 200 мкм длина мертвого объема составила ~ 0,2мм. Раствор декстрана (а*6щ), меченного FITC, заполнял световод с общей концентрацией [а*6щ] = 1,5мМ. Сенсор был, таким образом, «заряжен» для использования. Глюкозу (А) вводили за счет диффузии в диализную трубку, что приводило к конкурентному вытеснению декстрана (а*) с СопА (Кд = 3,2-102 М-1; Ка> = 7,5-104 М-1). В табл. 7.8-3 приведено значение сигнала флуоресценции, связанной с а* и измеренной в относительных единицах с помощью фотодиодного детектора. Из экспериментальной градуировки можно оценить максимальный сигнал (для [а*] = [а*6щ] = 1,5мМ) и, таким образом, получить [а*]/[а*6щ].
Таблица 7.8-3. Данные по конкурентному определению глюкозы с «мертвым объемом»
Глюкоза (А), мМ Свободный декстран (а*), отн. ед. Общий декстран (а'ов^)< отн- ед- Свободный декстран (рассн.), мМ
2,5 2,4 13 0,185 0,28
5,0 2,9 13 0,223 0,34
7,5 3,5 13 0,269 0,40
10,0 4,2 13 0,323 0,48
12,5 5,1 13 0,392 0,59
15,0 6,1 13 0,469 0,70
17,5 6,7 13 0,515 0,77
20,0 7,2 7,5 13 0,554 0,83
22,5 13 0,577 0,86
25,0 8,1 13 0,623 0,93
27,5 8,3 13 0,638 0,96
30,0 8,7 13 0,669 1,00
32,5 8,9 13 0,685 1,03
35,0 9,0 13 0,692 1,04
37,5 9,3 13 0,715 1,07
40,0 9,4 13 0,723 1,08
42,5 9,5 13 0,731 1,10
45,0 9,6 13 0,738 1,11
Построив график зависимости флуоресцентного сигнала от концентрации глюкозы (рис. 7.8-17,о), обнаружим, что при [Аобщ] > 20 мМ концентрация а* становится нечувствительной к изменению [Аобщ] , Рис. 7.8-17,е предсказывает величину сигнала на основе уравнений 7.8-30 и 7.8-31 и приведенных выше значений |а*общ], А»*, К а. и |Вробщ|- Можно видеть, что предсказанный диапазон для сенсора составляет ~ 0,5 мМ глюкозы. Ряд факторов могут давать вклад в столь значительное отклонение. Модель основана на гомогенной реакции. Ранее в разделе «Теоретические основы планирования иммобилизации» (см. с. 522) было отмечено, что гетерогенные реакции, включая взаимодействия на поверхности, имеют кинетику, которая может быть функцией заполнения поверхности, и что на Кл* и К а, вероятно, оказывает влияние общая концентрация сахара ([а*6щ] + [Аоб1Ц]). Модель также должна рас-
7.8. Биосенсоры
555
Рис. 7.8-17. Экспериментальный отклик анализа при конкурентном связывании в мертвом объеме в сравнении с точками данных моделирования, предполагающего простую кинетику конкурентного связывания, а — экспериментальные, б — расчетные, в — предсказанные точки.
Концентрация глюкозы [А], ммоль/л
сматривать концентрацию Вр по толщине диффузионного слоя в иммобилизованном слое (а не среднюю концентрацию, вычисленную исходя из общего объема). На деле получают лучшее «согласие» с экспериментальной кривой, если принять концентрацию иммобилизованного [Вр„бщ] равной 70 мМ (рис. 7.8-17,6), но действительное описание отклика должно также учитывать диффузию а* из реакционного слоя в «мертвый объем» поперек поля зрения. Сигнал не будет зависеть от диффузии только для таких концентраций, для которых время установления равновесия меньше времени диффузии.
Внутренний анализ с меткой
□ Световоды обеспечивают работу оптического сенсора во внутреннем режиме.
В этом режиме реагенты иммобилизованы на поверхности световода, который находится в контакте с фазой пробы. Свет, проходящий по световоду, способен взаимодействовать с фазой пробы (показатель преломления п?), находящейся на расстоянии 0,5-1,5 длины волны от поверхности; эта область представляет электрическое поле затухающей волны. Рассматривая лучевую модель прохождения света в световоде (показатель преломления ni) (рис. 7.8-18), можно видеть, что прохождение сопровождается серией полных
Рис. 7.8-18. Диаграмма прохождения света через «толстую» пленку (а) и «тонкую» пленку световода (6).
556
Глава 7. Методы химического анализа и их применение
(7.8-32)
внутренних отражений. Когда ni > П2, то, согласно закону Снелла, sin в ni
Sin ф П2
где # —угол падения, а ф — угол пропускания. Когда sin в > п^/п^, не существует действительного решения для ф, угол пропускания представляет собой комплексное число, и это отвечает условиям полного внутреннего отражения. Проходящая волна представляет собой затухающую волну, нормальную (ось z) к поверхности (ось х) с напряженностью электрического поля Е, которая описывается выражением
Е = Ео ехр
/ о / \2\V2
2тгП1 ( sin2#— j z
А
где остается только z-составляющая (при других условиях электрическое поле будет включать также .т-составляющую), так что поле спадает в направлении z с глубиной проникновения dp:
dp —
2тгп1
А____________
(sin2# - (n2/ni)2^
(7.8-33)
из чего следует, что ослабление сигнала максимально у поверхности и равно нулю в объеме.
На практике это означает, что имеется «зона детектирования» у поверхности и «мертвая зона» в объеме. Хотя такая конфигурация имеет то преимущество, что свет не проходит через объем пробы, она имеет и недостаток, заключающийся в том, что взаимодействие между светом и реагентами, иммобилизованными на поверхности, в обычном многомодовом световоде меньше, чем с реагентами в объеме раствора. Рассматривая лучевую модель прохождения света через световод, можно видеть, что взаимодействие с поверхностью происходит только в W «точках» полного внутреннего отражения внутри световода (см. рис. 7.8-18). 7V определяется уравнением
N = ^-ctg# 2а 6
где L — длина, 2а —толщина пленки.
(7.8-34)
□ «Зона детектирования» расположена на поверхности световода.
Тонкопленочные световоды увеличивают взаимодействие, потому что величина а меньше [7.8-52], но производить их непросто. Одним из решений служит ионный обмен на поверхности предметного стекла, дающий слой с высоким показателем преломления. Другое решение — использование низкотемпературных фосфатных стекол. Толстопленочные световоды относительно просты в изготовлении и служат недорогим материалом для сенсоров. Тем не менее, использование подходящих тонкопленочных световодов во многих случаях предпочтительнее, так как позволяет преобразовать устройства внешнего режима во внутренние оптические устройства.
7.8. Биосенсоры
557
Оптическое детектирование без метки
□ На каждой поверхности раздела свет преломляется и/или отражается, давая информацию о веществах с каждой стороны поверхности.
Как видно из уравнения 7.8-33, при данной длине волны напряженность электрического поля затухающей волны будет меняться в соответствии с отношением показателей преломления двух фаз (световода, щ, и поверхностной среды, тгг)- В биосенсорах, где поверхностный слой является местом протекания аналитической реакции, за реакцией можно следить непосредственно, если она вызывает изменение оптических характеристик слоя (подобных показателю преломления или толщине). Существует несколько способов усиления этих изменений, так что последние можно использовать в качестве аналитического сигнала.
Интерференционные методы
Когда объединяются две волны, они интерферируют друг с другом, либо конструктивно, либо деструктивно. Использование этого явления называют интерферометрией. Когда ее используют в биосенсоре, он обычно включает операцию с отражением во внутреннем режиме. Здесь мы обсудим два примера.
Модель отражения, описанная выше, рассматривает только отражение от одной границы раздела. Однако, если вместе размещено несколько слоев, тогда на каждой границе происходит и отражение, и пропускание (рис. 7.8-19). Это, в конечном итоге, приводит к «смешанному» отражению, которое является комбинацией отражений от каждой из границ раздела. Интенсивность регистрируемого отражения есть функция толщины каждого слоя и совокупного показателя преломления. Она очень чувствительна к небольшим изменениям любого параметра, и, следовательно, может быть использована для детектирования взаимодействий на поверхности многослойника. Чтобы получить максимальную чувствительность, следует тщательно выбрать диэлектрический материал (диэлектрическая проницаемость е связана с показателем преломления, п = е1'2). Непоглощающие диэлектрики (например, 8Юг) с диэлектрической проницаемостью ei = £цг (т. е. диэлектрическая проницаемость имеет только
Рис. 7.8-19. Отражение от многослойника.
Детектор
558
Глава 7. Методы химического анализа и их применение
действительный член) наносят с толщиной порядка А/4 на хорошо отражающую подложку с диэлектрической проницаемостью eq = £o,r — ifo.i (например, кремний с комплексной диэлектрической проницаемостью). Изменения в биораспознающем слое, осажденном на поверхность слоя SiCh, можно затем контролировать, измеряя и анализируя отражение, поскольку отраженные волны будут модулированы слоями. Такая многослойная система использована как преобразователь для связывания антитело-антиген [7.8-9J. Тем не менее, вообще говоря, для всех таких <безметочных» методов иммунного анализа неспецифическое связывание должно быть исключено или полностью охарактеризовано, если метод, в принципе, может быть использован в биосенсорах.
Для биосенсоров на основе интерферометрии существует важный момент — выбор точки сравнения. Необходимы два вида градуировки. Первый касается градуировки по определяемому веществу; второй требует информации о фоновом сигнале. Фон можно получить с помощью контрольного опыта; вопрос заключается в следующем: как можно включить контрольный опыт в биосенсор? В случае световодов, взаимодействующих с пробой посредством затухающего поля, это требует двух различимых поверхностей: одной — для специфических и другой — для неспецифических реакций. Одним из вариантов является конфигурация, подобная конфигурации в интерферометре Маха—Цендера (рис. 7.8-20). Сначала падающий свет разделяется,затем он проходит через све-
Вход света
Волновод
Звено сравнения
Реакционное .звено
Реакционная зона
Выход света
Рис. 7.8-20. Интерферометр Маха—Цендера.
7.8. Биосенсоры
559
товоды сравнения и пробы и впоследствии вновь объединяется. На интенсивность получаемого, многократно отраженного луча влияет длина оптического пути и отражение на границе раздела. Отражение, конечно, будет разным на поверхности, модифицированной биораспознающим компонентом, и поверхности сравнения. Когда неспецифические взаимодействия исключены, эту конфигурацию успешно используют для измерений связывания антитело-антиген.
Устройства ввода света в световод на основе решеток
□ Чтобы улучшить измерения изменений показателя преломления, можно сконструировать различные оптические схемы.
Устройство ввода на основе решетки позволяет завести свет в световод. Решетка —это периодическая структура (например, треугольная, пилообразная, синусоидальная) (рис. 7.8-21), которая нанесена на поверхность световода. Когда свет падает на решетку он возбуждает в световоде проходящую моду в зависимости от периода решетки. Чтобы усилить эффект изменения показателя преломления на границе раздела, изменения, происходящие на поверхности решетки, надо перенести на световод. Например, на рис. 7.8-21 свет, падающий вблизи устройства ввода, будет возбуждать проходящие моды, зависящие от показателя преломления окружающей среды. В этом варианте поверхность становится чувствительной к тонким изменениям показателя преломления. Этот вариант, следовательно, подходит для проведения безметочного иммунного анализа, поскольку связывание между иммобилизованным антителом и антителом пробы дает большую плотность этих молекул на расстоянии порядка длины волны вглубь от поверхности (заметьте, что глубина проникновения света составляет величину порядка длины волны, уравнение 7.8-34) и, таким образом, приводит к изменению показателя преломления. Например, конкурентный иммунный анализ можно применить для определения пестицидов, при этом на модифицированной аминосиланом поверхности устройства связи ковалентно иммобилизован триазиновый гаптен, а связывание антитела детектируют за счет измерения угла несвязывания [7.8-53, 7.8-54]. Чтобы детектировать присутствие триазиновых пестицидов, пробу следует предварительно выдержать с антителами, а затем — перенести к иммобилизованному гаптену. Очевидно, чтобы обеспечить невмешательство пользователя в эту процедуру анализа, в устройстве должен быть предусмотрен этап выдержки.
Рис. 7.8-21. Устройство связи на основе решетки.
560
Глава 7. Методы химического анализа и их применение
Поверхностный плазмонный резонанс (ППР)
В этом методе световод покрывают тонкой пленкой металла (плазмона) и измеряют изменения поля, проходящего вдоль поверхности металлической пленки. Эти изменения обусловлены тем, что воздействие внешнего электрического поля (в световоде) достаточной напряженности (см. ур. 7.8-33) передает энергию из световода и образуются осциллирующие поверхностные заряды, которые распространяются по поверхности металла. Колебания заряда должны быть связаны с Ez (полем, расширяющимся в направлении z), так что металлическая поверхность должна быть на должном расстоянии от поверхности световода в пределах глубины проникновения dp затухающего поля. Для волны поверхностного плазмона, существующей между двумя средами с диэлектрической проницаемостью Si и е? (рис. 7.8-22), волна ограничена на границе раздела волновым вектором кг в направлении распространения и kzj и кг2 в направлениях +z и —z. Условию непрерывности на границе отвечает соотношение
Mi = Мг
М М
Следовательно, к2 определяется выражением
(7.8-35)
(7.8-36)
П Поверхностный плазмонный резонанс есть колебание поверхностных зарядов (электронов в металле), распространяющееся по поверхности.
Волну поверхностного плазмона можно возбудить падающим светом той же частоты и имеющей составляющую кх. Для света, падающего на металл с диэлектрической проницаемостью ег из диэлектрического материала с ез, составляющая кх определяется выражением
М=ез^) sin2# (7.8-37)
где # —угол падения.
Плазмонный резонанс на гд/ег-границе металла, следовательно, можно возбудить, когда составляющие к^ падающей и плазмонной волн совпадают:
к2 = (= £з (^2sin20 (7.8-38)
\£1 +E2J С J \С/
Рис. 7.8-22. Конфигурации возбуждения для поверхностного плазмонного резонанса. а — конфигурация Кречмана с контактом через тонкую металлическую пленку; б — конфигурация Отто, обеспечивающая контакт сквозь воздушный зазор.
7.8. Биосенсоры
561
Чтдбы это произошло, должно выполняться условие £3 > £].
Экспериментально резонанс наблюдают как минимум отражения при угле падения G. Из уравнения 7.8-38 можно сделать вывод, что изменение £1 потребует изменения 6, чтобы сохранить условия резонанса, так что контроль положения минимума отражения очень чувствителен к реакции на поверхности металла (fj). Это является источником сигнала, измеряемого сенсором ППР, так как поверхность металла можно модифицировать с помощью биораспознающей молекулы, которая будет давать отклик на определяемое вещество, меняя на границе раздела.
Две конфигурации устройств ППР показаны на рис. 7.8.-22. В ранних сообщениях по применению ППР для детектирования связывания белков речь шла об исследованиях взаимодействий без метки, с участием, например, антител, иммобилизованных на металлических пленках [7.8-55,7.8-56]. Скоро стало ясно, что хотя можно детектировать связанный антиген, в некоторых аналитических средах часто не было возможности отличить этот сигнал от сигнала любого другого неспецифического взаимодействия с поверхностью [7.8-57]. Показатель преломления в качестве метки и другие оптические метки помогут обойти эту проблему, и, поскольку ППР столь чувствителен, достижимы низкие пределы обнаружения.
Большинство приложений этого метода лежит в области иммунного анализа или других методов со связыванием белков, а не в ферментативном анализе. Чтобы установить, возможно ли какое-либо новое приложение, необходимо выяснить, как связать измеряемый параметр с тем, что в данном методе анализа непременно должно происходить изменение £ на поверхности. Многие из определений, основанных на связывании между иммобилизованной биораспознающей молекулой и определяемым веществом, могли бы пригодиться, если разница между несвязанной и связанной с определяемым веществом распознающей молекулой достаточно велика и если реагенты можно иммобилизовать с сохранением их реакционной способности. Например, последовательность ДНК-зондов иммобилизована на модифицированной тиолом серебряной пленке, нанесенной на световод в варианте ППР [7.8-22]. Зонд проявляет характеристическое изменение в возбуждении ППР при связывании комплементарной ДНК (рис. 7.8-23). Однако, когда та же последовательность ДНК-зондов была нанесена на поверхность с помощью силана, зонд не смог связывать комплементарную ДНК и, таким образом, потерпел неудачу в качестве биосенсора. Это наблюдение заставляет нас рассматривать метод иммобилизации биораспознающего компонента как важнейший в конструировании биосенсора.
7,8.4. Заключение
Эта глава дает краткий обзор компонентов биосенсора. Она концентрирует внимание на двух основных классах биосенсоров, а именно — электрохимических и оптических, но не следует ограничиваться только этими примерами. Как сказано в начале главы, возможности конструирования биосенсоров настолько широки, насколько широко ваше воображение. Критериями успеха
562
Глава 7. Методы химического анализа и их применение
являются выбор соответствующего параметра для измерений, хороший метод иммобилизации, который можно легко приспособить для серийного производства, и пригодность вашего измерения для избранного приложения!
Литература
[7.8-1} Hall, Е.А.Н., Biosensors. Milton Keynes: Open University Press, 1990.
[7.8-2} Cass, A.E.G. (Ed.), Biosensors: A Practical Approach. Oxford: IRL Press, 1990-
[7-8-3} Gopel, W., Hesse, J., Zemel, J.N., (Eds.), Sensors: A Comprehensive Survey, Weinheim: VCH, 1991, Vols. 2 and 3.
[7-8-4} Wolfbeis O.S. ^Ed.), Fiber Optic Chemical Sensors and Biosensors, Florida: CRC Press, 1991, Vols. I and IL
[7.8-5] Ikeda, T., Hamada, H., Miki, K., Senda, M., Agric. Biol. Chem. (Japan), 1984, 49, 541-543.
[7.8-6] Randriamahazaka, H.N., Nigretto, J.M., Electroanalysis, 1993, 5, 231-241.
[7.8-7] Amine, A., Kaufman, J.M., Patriarche, G.J., Taianta, 1991, 38, 107.
[7.8-8] Wang, J., Wu, L.H., Lu, Z.L., Li, R.L., Sanches, J., Anal. Chim. Acta, 1990, 228, 251.
[7.8-9] Dushl, C., Hall, E.A.H., J. Coll. Int. Sci., 1991, 114, 368-380.
[7.8-10] Kutner, W., Storck, W.,Doblhofer, K., J. Ind. Phenom., 1992, 13, 257.
[7.8-11] Hale, P.D., Lan, H.L., Boguslavsky, L.L, Nagan, H.L, Okamoto, Y., Skotheim, T.A., Anal. Chim. Acta, 1991, 251, 121.
[7.8-12] Yon Hin, B.F.Y., Smolander, M., Compton, T., Lowe, C.R., Anal. Chem., 1993, 65, 2067.
[7.8-13] Martens, N., Hall, E.A.H., Anal. Chim. Acta, 1994, 292, 49-63.
[7-8-14] Hench, L.L., West, J.K., Chem. Rev., 1990, 90, 33-72.
[7.8-15] Braun, S., Rappoport, S-, Zusman, R., Avnir, D., Ottolenghi, M., Materials Lett., 1990, 10 (1,2), 1-5.
[7.8-16] Narang, U., Psasad, P.N., Bright, F.V., Ramanathan, K., Kumar, N.D., Malhotra, B.D., Kamalasanan, M.N., Chandra, S. Anal. Chem., 1994, 66, 3139.
[7.8-17] Hall, C.E., Hall, E.A.H., Anal. Chim. Acta, 1993, 281, 645-653.
[7.8-18] Urban, G., Jobst, G-, Kepplinger, F., Aschauer, E., Tilado, O., Fasching, R., Kohl, F., Biosensors Bioelecironics, 1992, 7, 733.
[7.8-19] Urban, G., Jobst, G., Aschauer, E., Tilado, O-, Svasek, P-, Varahram, M., Ritter, Ch., Riegebaure, J., Sensors and Actuators, 1994, 18—19, 592.
Рис. 7.8-23. Зонд на иммобилизованной ДНК, основанный на поверхностном плазмонном резонансе на серебряной пленке.
7.8. Биосенсоры
563
[7.8-20] Hall, Е.А.Н., Hall, C.E., Martens, N., Mustan, N., Datta, D., Uses of Immobilized Biological Compounds, in: ASI Series 252: Guilbault, G.G., Mascini, M., (Eds.), Kluwer Academic Publishers, 1991.
[7.8-21] Willner, I., Riklin, A., Anal. Chem., 1994, 66, 1535.
[7.8-22] Liley, M.J., Surface Plasmon Resonance for the Detection of DNA Hybridisation, PhD Thesis, University of Cambridge, 1990.
[7.8-23] Martens, N., Hindle, A., Hall, E.A.H., Biosensors Bioelectronics, 1995.
[7.8-24] Leypoldt, J.K., Gough, D.A., Anal. Chem., 1984, 56, 2896.
[7.8-25] Cass, A.E.G., Francis, G., Hill, H.A.O., Higgins, I.J., Aston, W.J., Plotkin, E.V., Scott, L.D.L., Turner, A.P.F., Anal. Chem., 1984, 56, 667.
[7.8-26] Degini, Y., Heller, A., J. Am. Chem. Soc., 1988, 110, 2615.
[7.8-27] Schlapfer, P., Mindt, W., Racine, P., Clin. Chim. Acta, 1974, 57, 283.
[7.8-28] Martens, N., Hall, E.A.H., Anal. Chem., 1994, 66, 2763-2770.
[7.8-29] Clark, L.C. Jr., Lyons, C-, Ann. N. Y. Acad. Sci., 1962, 102, 29.
[7.8-30] Nicholson, R.S., Sham, L., Anal. Chem., 1964, 36, 706.
[7.8-31] Davies, P., Green, M.J., Hill, H.A.O., Enzyme Microb. Technal., 1986, 8, 349-352.
[7.8-32] diGleria, K., Hill, H.A.O., McNiel, C.J., Green, M.J. Anal. Chem., 1986, 58, 1203.
[7.8-33] Wang, J., Chen, Q., Electroanalysis, 1994, 6, 850.
[7.8-34] Khan, G.F., Kobatake. E., Ikariyama, Y, Aizawa, M., Anal Chim. Acta, 1993, 281, 527-533.
[7.8-35] D’Costa, E.J., Higgins, I.J., Turner, A.P.F., Biosensors, 1986, 2, 71-87.
[7.8-36] Wang, J., Naser, N., Anal. Chim. Acta, 1991, 242, 259.
[7.8-37] Desphande, M.K., Hall, E.A.H., Biosensors Bioelectronics, 1990, 5, 431-448.
[7.8-38] Chen, Y, Tan, T.C., Biosensors Bioelectronics, 1994, 9, 401-410.
[7.8-39] Carpentier, R., Lemieux, S., Mimeault, M., Purcel, M., Goetze, D.C., Bioelectrochemistry and Bioenergetics, 1989, 22, 391-401.
[7.8-40] Martens, N., Hall, E.A.H., Photochem. and Photobiol., 1994, 59, 91-98.
[7.8-41] Glab, S., Koncki, R., Hulanicki, A., Electroanalysis, 1991, 3, 361-364.
[7.8-42] Bergveld, P., Stimulus Response Measurements on Protein Containing Membranes, Deposited on an ISFET Surface, in: Uses of Immobilised Biological Compounds: Guilbault, Mascini, (Eds.) Netherlands: Kluwer, 1993, pp. 289-308.
[7.8-43] Cowell, D.C., Dowman, A.A., Lewis, R.J., Pirzad, R., Watkins, S.D., Biosensors Bioelectronics, 1994, 9, 131-138.
[7.8-44] Shasfoort, R.B.M., Bergveld, P., Bomer, J., Kooyman, R.P.H., Greve, J., Sensors and Actuators, 1989, 17, 531.
[7.8-45] Aizawa, M., Kato, S., Suzuki, S., J. Membrane Sci., 1977, 2, 125-130.
(7.8-46] Shasfoort, R.B.M., Bergveld, P-, Kooyman, R.P.H., Greve, J., Biosensors Bioelectronics, 1991, 6, 477-489.
[7.8-47] Lippitsch, M.E., Pusterhofer, J., Leiner, M.J.P., Wolfbeis, O.S., Anal. Chim. Acta, 1988, 205, 1.
[7.8-48] Bannwarth, W., Schmidt, D- Stallard, R.L., Hornung, C., Knorr, R., Mueller, F., Helv. Chim. Acta, 1988, 71, 2085.
[7.8-49] Hlavay, J., Haemmerli, S.D., Guilbault, G.G., Biosensors Bioelectronics, 1994, 9, 189-195.
[7.8-50] Krull, 1989.
[7.8-51] Offenbacher, H., Wolfbeis, O.S., Fiirlinger, E., Sensors and Actuators, 1986, 9, 73-79.
[7.8-52] Mansouri, S., Schultz, J.S.. Bio/Technology, 1984, 2, 385.
[7.8-53] Nellen, P.M., Lukosz. W.. Biosensors Bioelectronics, 1991, 6, 517—525.
564 Глава 7. Методы химического анализа и их применение
[7.8-54] Bier, F.F., Schmid, R.D., Biosensore Bioelectronics, 1994,* 9, 125-130.
[7.8-55] Cullen, D.C., Brown, R.G.W., Lowe, C.R., Biosensors, 1987/88, 3, 211-225.
[7.8-56] Liedberg, B., Nylander, C., Lundstrom, I., Sensors and Actuators, 1983, 299-306.
[7.8-57] Cullen, D-C., Lowe, C.R., Sensors and Actuators, 1990, Bl, 576-579.
Вопросы и задачи
1. Какие изменения необходимо произвести, чтобы преобразовать подходящий метод биоанализа растворов в биосенсор? Какие биометоды идеально подходят для такого преобразования? С какими биометодами мы имели бы проблемы при таком преобразовании?
2. Какие типы биологических распознающих систем обычно используют в биосенсорах? Какие типы определений в каждой группе пригодны для этого? Какие идеи есть у вас относительно других биораспознающих систем, которые можно было бы использовать?
3. Редокс-ферменты часто используют в амперометрических биосенсорах .для определения субстратов фермента. Как они действуют? Каковы различные классы редокс-ферментов? Какие дополнительные реагенты требуются для амперометрического биосенсора?
4. Какое определяемое вещество измерял первый биосенсор? Как выполняли измерение?
5. Существуют различные подходы к получению слоя иммобилизованного реагента, какие именно? Когда молекула взаимодействует с поверхностью, какова последовательность событий, которые могут происходить?
6. Приведите некоторые примеры биораспознающих процессов, приводящих к изменению pH. Как можно измерить это изменение в случае биосенсора?
7. Как бы вы сконструировали оптический биосенсор для определения глюкозы и холестерина?
8. Биолюминесценция является очень чувствительным методом анализа. Опишите этапы этого анализа и укажите, как можно его встроить в другие системы для определения различных веществ.
9. Можно разработать оптический биосенсор с реагентами на поверхности световода Свет, проходящий вдоль световода, может взаимодействовать с реагентом на поверхности. Взаимодействие можно контролировать, регистрируя изменения в проходящем свете. Как это происходит? Если реагент поместить в объем раствора, а не на поверхность световода, какое влияние это оказало бы иа изменения проходящего света?
10. Почему невозможно возбудить поверхностный плазмон, облучая светом плоскую металлическую поверхность? Каковы различные конфигурации, используемые для поверхностного плазмонного резонанса?
11. Какие факторы могут влиять на точность биосенсоров? Приведите примеры со ссылкой на отдельные типы биосенсоров.
7.9. Иммунный анализ
565
7.9. ИММУННЫЙ АНАЛИЗ
Цели изучения
• Изучить равновесия антитело-аиТиген и их использование в анализе.
• Понять различные варианты иммунного анализа.
• Ознакомиться с тем, как можно проводить иммунный анализ с использованием меток.
7.9.1. Введение
□ Антитело представляет собой белок со специфичным доменом, связывающим антигены.
□ Антигены—это молекулы, способные вызывать иммунный ответ.
Иммунный анализ использует уникальную специфичность антитела, связывающего антиген, с целью селективно распознать и определить вещества, которые являются или антителами, или антигенами. Можно добиться высокой селективности иммунного анализа, потому что другие соединения в пробе распознаваться не будут. Антитела представляют собой один из главных классов белков, объединенных названием иммуноглобулины (1g). Внутри этого семейства наиболее представительными антителами являются IgG (~ 70%), Y-образный белок, состоящий из двух идентичных, связанных дисульфидными мостиками гетеродимеров с тяжелой и легкой цепями (рис. 7.9-1). Имеются два антигенсвязывающих (активных) центра, образованные вариабельными глобулярными доменами тяжелой и легкой цепей в Fab-области; специфичность этих центров к связыванию антигена определяется аминокислотами областей, определяющих комплементарность. В связывающей полости имеется максимум 17 аминокислот, так что активный центр антигена должен иметь размеры такого же порядка. Антиген не обязательно должен быть белком; это может быть любая макромолекула, которая способна индуцировать иммунный ответ и, таким образом, вызвать образования антител против нее. Молекулы с низкой молекулярной массой (такие, как гормоны или наркотики, являющиеся предметом анализа) не являются антигенными, но можно создать антитела с активными центрами, специфичными к этим молекулам, соединяя их с белковым носителем. Такие низкомолекулярные соединения известны как гаптены.
Антитела продуцируются В-лимфоцитами, причем каждая В-клетка проявляет на своей поверхности только одну Специфичность, так что инородный антиген связывают только клетки с «подходящими» активными центрами. Это связывание стимулирует деление таких клеток и образование большого количества IgG с той же специфичностью. Активный центр может быть лишь небольшой частью всей молекулы антигена, и, таким образом, у одного и того же антигена может быть несколько потенциальных активных центров. Результатом этого процесса становится образование сыворотки, содержащей неоднородную смесь частиц антител с разнообразным сродством. Такие сыворотки известны как поликлональные и, в отличие от моноклональных антител, в их кинетику распознавания вносит свой вклад каждый из активированных распознающих центров или эпитопов.
566
Глава 7. Методы химического анализа и их применение
МН2-конец цепи
NHg-конец цепи
Аитаген-
НООС СООН
-СООН-конец цепи
Рис, 7.9-1. Структура тяжелых (индекс Н) и легких (индекс L) цепей IgG, показывающая места связывания антигена.
При сближении антитела (АЬ) и антигена (Ag) первичная связывающая сила является ионной (дальнее взаимодействие), действующей на расстояниях свыше 10 нм. Медленное удаление гидратационной воды приводит к образованию водородных связей на расстояниях 0,5-0,15 нм, силы Ван-дер-Ваальса (ближнее взаимодействие) между диполями на соседних атомах становятся более значимыми и связь упрочняется. Связывание можно описать с помощью равновесия:
д _ [Ab-Ag]
Лравн - [Ab][Ag] (ЛУ 1)
где Кравн —константа равновесия, a Ab-Ag — комплекс антитело-антиген. Значения Кравн обычно изменяются в диапазоне от 106л/моль до 1012л/моль, но значения меньше, чем 108л/моль, как правило, в иммунном анализе не используют.
Определяя общую концентрацию антител [АЬобщ] как сумму концентраций связанных [Ab-Ag] и свободных [АЬ] антител:
[АЬобщ] = [АЬ] 4- [Ab-Ag] (7.9-2)
можно переписать уравнение 7.9-1:
1АЬ1 = Кр™ [А Ьобщ ] - [Ab-Ag] (7.9-3)
7.9. Иммунный анализ
567
Рис. 7.9-2. График Скэтчарда для моноклонального (о) и поликлонального (б) антитела.
На графике Скэтчарда строят зависимость отношения [Ab-Ag]/[Ag] от [Ab-Ag], где Кравн определяется наклоном, а отсекаемый на оси х отрезок — [АЬобхц].
□ Антитела могут быть моноклональными или поликлональными.
Сыворотка с единственным эпитопом специфичности представляет моноклональную систему. На соответствующем графике Скэтчарда наблюдается прямая линия (рис. 7.9-2,а). Для поликлональной системы график Скэтчарда имеет более сложный вид. Он характеризуется набором многих прямых линий (рис. 7.9-2, б).
При разработке иммунного анализа целью является проследить за равновесием между антителом и антигеном. При анализе исследуют связывание антигена с антителом и различают связанный и несвязанный антиген. Поскольку процесс имеет равновесный характер (уравнение 7.9-1), линейной зависимости от концентрации антигена нет. Следовательно, важно знать характеристический отклик связывания как функцию концентрации антигена.
Из уравнения 7.9-1 и соотношения
[А&,бщ] = [Ag] + [Ab-Ag] можем записать:
[Ab-Ag] __________1_______
[Ago6J 1 + 1/(Кравн[АЬ[) [Ab-Ag] ([Ab-Ag]/[Ag]) [AgD6„] [Ab-Ag]/[Ag] +1
Кривые, построенные исходя из этого соотношения для различных концентраций антител, показаны на рис. 7.9-3,а. Можно заметить, что для данной концентрации антител степень связывания меняется как функция концентрации антигена только в критическом диапазоне. Ниже находится область постоянного пропорционального связывания, где изменения концентрации антигена дают незначительные изменения степени связывания. Из рис. 7.9-3,б очевид-
(7.9-4)
(7.9-5)
(7.9-6)
568
Глава 7. Методы химического анализа и их применение
Концентрация антигена (ед. 1/Кравн)
Концентрация антитела, ед. 1/Крдвн
Концентрация антитела, ед. 1/Крави
Рис. 7.9-3. а—степень связывания в зависимости от концентрации антигена для различных концентраций антитела; б — степень связывания в зависимости от концентрации антитела для различных концентраций антигена; в —доля занятых центров антитела в зависимости от концентрации антитела для различных концентраций антигена.
но, что увеличение концентрации антител вызывает сдвиг области максимального изменения (т. е. потенциального динамического диапазона измерений) в область более высоких концентраций антигена.
С другой стороны, рис. 7.9-3,в показывает, что для довольно низких концентраций антител (< 0,01/Хравн) доля занятых активных центров антитела отражает концентрацию определяемого вещества и становится независимой от общей концентрации антител. Однако это не используют широко, хотя подразумевается, что изучаемые концентрации столь малы, что обеднением пробы антигеном за счет связывания можно пренебречь, при этом преимуществом является независимость от объема пробы.
Для того чтобы изучать процесс связывания независимо от уровня концентрации, обычно вводят метку, которую можно контролировать. Пути выполне
7.9. Иммунный анализ
569
ния этой операции (см. ниже) могут различаться, но при этом всегда следует учитывать ограничения, налагаемые равновесием связывания и необходимым динамическим диапазоном.
Варианты анализа
□ Иммунный анализ основан на измерении доли занятых активных центров.
Любой иммунный анализ базируется на измерениях доли занятых активных центров. Другими словами, принцип анализа основан на определении доли занятых центров, или косвенно—доли незанятых центров. Как будет показано в этом разделе, рассмотрение доли занятых активных центров антител приводит к заключению, что, если измеряют оставшиеся незанятыми центры, максимальной чувствительности достигают, когда концентрация антитела стремится к нулю; варианты, удовлетворяющие этому условию, обычно называют «конкурентными». Напротив, методы, в которых напрямую измеряют занятые центры, требуют высоких уровней антител и называются «неконкурентными». Следует, однако, сознавать, что эти термины несколько субъективны, поэтому возникает парадокс, особенно для «неконкурентного» варианта, когда анализ может быть отнесен к любой из двух категорий в зависимости от выбранного этапа. Поэтому важно помнить, что эти довольно двусмысленные термины используют применительно к этапу первичного распознавания. Из-за различных условий оптимизации конкурентный и неконкурентный иммунный анализы могут значительно отличаться в выполнении для данного диапазона концентраций: эти различия следует рассматривать при выборе оптимального варианта для конкретного приложения.
Конкурентный анализ
□ Конкурентный иммунный анализ включает конкуренцию между определяемым веществом пробы и меченым определяемым веществом за ограниченное число активных центров.
В конкурентном иммунном анализе устанавливают число центров, не занятых пробой; он может иметь различные варианты, но суть в том, что определяемое вещество пробы смешивают с меченым определяемым веществом, и они конкурируют за активные центры (рис. 7.9-4). Метод требует разделения связанного и свободного антигена или антитела с целью определения их относительных количеств. Если определяют антиген, присутствие меченого антигена позволяет оценить относительное разделение. Как можно заключить из кривых иа рис. 7.9-3, оптимальная чувствительность достигается при уменьшении концентраций меченого антигена и антитела.
Рассмотрим сигналы компонентов в конкурентном анализе: сначала обсудим условия, когда проба отсутствует и сигнал, относящийся к связанным антителу-антигену в присутствии только меченого антигена, определяется уравнением
So =
[Ab-Ag] [Ago64] ‘
(7.9-7)
570
Глава 7. Методы химического анализа и их применение
покрытая
антигеном
Свободная фракция
Рис. 7.9-4. Конкурентный анализ для тесг-определения антител.
где 5общ—сигнал, который был бы получен при прямом измерении [AgoguJ. Сигнал неспецифического связывания S„c — функция доли неспецифического связывания оставшегося свободного антигена.
[Ab-Ag]]
‘-’нс — V ГА II ‘“’общ^НС
I [Ago6ui] J
(7.9-8)
где Внс — доля неспецифически связанного антигена. Таким образом, при уменьшении концентраций меченого антигена и антитела с той целью, чтобы сместить динамический диапазон определений в область меньших концентраций, ошибки из-за неспецифического связывания начинают оказывать все большее влияние на точность определения. Каждое сочетание специфического и неспецифического связывания будет иметь свое оптимальное сочетание концентраций антитела и меченого антигена. Полное устранение неспецифического связывания или разделение связанной и свободной фаз невозможно со 100%-ной эффективностью, так что концентрацию антитела, вообще говоря, следует увеличивать, чтобы увеличить вклад специфического связывания.
В присутствии только меченого антигена So определяют по уравнению 7.9-7, так что сигнал связанного антигена, нормированный на [Ago6l4] опре
7.9. Иммунный анализ
571
деляется степенью связывания. Однако в присутствии пробы (немеченого антигена):
[Ago6ui] = [Ag*6ui] 4- [Ag*6ui] (7.9-9)
[Ab-Ag] = [Ab-Ag*] + [Ab-Ag*] (7.9-10)
[Ag] = [Ag*] + [Ag*] (7.9-11)
где Ag* — антиген пробы, a Ag* — меченый антиген.
Для данной концентрации меченого антигена сигнал связанного антигена Ag* уменьшается при небольшой добавке антигена пробы (немеченого) [AAg*].
Из кривых на рис. 7.9-5 очевидно, что изменение сигнала в результате [AAg*] является функцией концентрации антитела, используемой в анализе (рис. 7.9-5,б), и что уровень антител, дающий максимальную чувствительность, зависит от концентраций меченого и определяемого антигена. Традиционно системы иммунного анализа имеют тенденцию давать погрешность при высокой концентрации реагента со степенями связывания 0,4-0,5. Действительно, из рис. 7.9-3,а и б, видно, что связывание в этой области (0,4-0,5) слишком велико, чтобы привести к большому изменению степени связывания антигена при низкой концентрации последнего. Однако нельзя игнорировать сигнал, относящийся к неспецифическому связыванию, поэтому чувствительностью поступаются в пользу более низких помех за счет неспецифического связывания при более высоких концентрациях антител.
Хотя конкурентный вариант иммунного анализа имеет свои преимущества и очень популярен, он имеет и практические ограничения. Очевидным недостатком является то, что маркирование определяемого вещества Ag* может изменить или даже полностью устранить способность связываться с АЬ; вторая опасность особенно существенна, если маркирование включает в связы-
Рис. 7.9-5. а — кривая разбавления антитела для меченого антигена Ag* и влияние добавления небольшого количества немеченого антигена Ag*; б — изменение связывания ДЬ вследствие добавки AAg* при различных исходных степенях связывания.
572
Глава 7. Методы химического анализа и их применение
вание критический эпитоп. Если кинетика связывания меченого и немеченого антигена различается, это следует учитывать в расчетах. Иногда, особенно при использовании меченых гаптенов, метка может улучшить распознавание. Это видно, когда меченый конъюгат использует то же положение на гаптене, что и ранее используемое в белковом носителе, требуемом для получения антител.
Сандвичевый анализ
□ В Сандвичевом иммунном анализе определяемое вещество из пробы захватывается избытком антител, а количество захваченного определяемого вещества оценивают с помощью второго антитела, образующего, таким образом, сандвич.
Как упомянуто ранее, место распознавания на антигене представляет собой небольшую зону, поэтому может быть несколько различных эпитопов, которые пространственно разделены на антигене. Эту особенность можно использовать в «сандвичевом» анализе; определяемое вещество захватывается антителом АЫ, что позволяет выделить его из остальной пробы. Захваченный антиген затем выдерживают с избытком второго, меченого антитела АЬ2, которое связывается только с уже существующими комплексами антитело-антиген (рис. 7.9-6).
Меченое антитело АЬ2 должно быть способно специфически соединяться с поверхностью связанного антигена. В идеальном случае сандвичевого анализа в отсутствие меченого антитела нет никакого сигнала. На практике всегда есть некоторый сигнал, возникающий за счет неспецифического связывания АЬ2 с АЫ, так что даже в отсутствие общего сигнала:
So = _Внс5общ (7.9-12)
где Зобщ — сигнал, возникающий за счет [АЬ2ОбЩ].
В присутствии слоя АЫ связывание Ag описывается константой 7fpaBHi в соответствии с уравнениями 7.9-1-7.9-6 (где АЫ = АЬ и Кравн1 = АГравн), но затем его контролируют с помощью присоединения АЬ2 к связанному антигену Agb, описываемого константой КГравн 2-
[Aed=[Ab^7 + lAb2bl (7МЗ)
Контролируемый сигнал для связанного (АЬ2ь) и свободного (АЬ2у) меченого антитела должен различаться. Обычно измеряют связанное антитело, так что уравнение 7.9-13 означает, что будет полезным использовать избыток АЬ2, при котором соблюдаются условия
[Ab2f] » [АЬ2Ь] (7.9-14)
и уравнение 7.9-13 преобразуется в
[Agb] « [АЬ2ь] (7.9-15)
Однако существует оптимальная концентрация меченого антитела. Более высокие концентрации увеличивают неспецифическое связывание с захваченным антителом АЫ, а более низкие — больше подвержены ошибкам измерения.
7.9. Иммунный анализ
573
Твердая фаза, покрытая антителом
Свободная фракция
Рис. 7.9-6. Иммунометрическое (санд-вичевое) определение антигена.
Рис. 7.9-7. Разделение связанной и свободной фракций с использованием избытка антигена на твердой фазе.
574
Глава 7. Методы химического анализа и их применение
С другой стороны, концентрация АЫ не влияет на чувствительность до тех пор, пока его количества хватает для захвата антигена пробы (как правило, в концентрации больше чем ~ 10-9 моль/л, но обычно выбирают ~ 10-6 моль/л, чтобы попасть в область постоянной степени связывания > 90%).
7.9.2. Варианты устройства
Общее рассмотрение
В последнем разделе были очерчены основные теоретические принципы, но можно разработать почти неограниченное число вариантов связывания активных центров последовательностей и сандвичей, которые в конечном итоге достигают одной цели, а именно оценки концентрации определяемого вещества! Общими чертами классических методов иммунного анализа является то, что они определяют содержание не напрямую, а с помощью меченого аналога определяемого вещества или меченой системы распознавания определяемого вещества. Они также требуют разделения сигналов от свободного и связанного меченого маркера; это требование является первостепенным в иммунном анализе.
Разделение связанной и свободной фракций можно провести различными способами. В жидкофазном анализе растворов используют осаждение, центрифугирование и декантацию, но они дают весьма непостоянные результаты и поэтому не нашли широкого применения. Первый большой успех в этом направлении сделали Майлс и Хейлс (1968), которые предложили удалять несвязанные меченые антитела, вводя большой избыток антигена в твердой фазе (рис. 7.9-7).
Эта разработка положила начало развитию твердофазного иммунного анализа, в котором, однако, возникают дополнительные трудности, особенно в аспекте термодинамики взаимодействий. Твердофазный анализ отличается от анализа в жидкой фазе как по реакционной способности иммобилизованного антитела или антигена, так и по различию кинетики реакций в растворе и реакций, включающих твердую фазу (см. также разд. 7.8).
Эффекты поверхностной иммобилизации
□ Поверхностная иммобилизация может вызывать изменение конфигурации белка и в связи с этим изменение кинетики иммунного анализа.
В твердофазном иммунном анализе твердая фаза играет роль «несущей подложки». Ее, однако, нельзя рассматривать как пассивный компонент. Можно использовать и химическую, и физическую адсорбцию антитела или антигена, хотя большинство иммунных определений основано на физической, нековалентной адсорбции. Как обсуждалось в разд. 7.8, даже физическая адсорбция представляет собой недостаточно изученный процесс. Связывание белков, по-видимому, имеет гидрофобную природу, и все же в естественной конфигурации белков в водном растворе гидрофильные группы стремятся расположиться на поверхности, а гидрофобные группы — внутри полимера. Следовательно, связывание с гидрофобными поверхностями неизбежно вызывает изменение кон
7.9. Иммунный анализ
575
формации белка, что может привести к потере критических эпитопов, либо потому что они будут спрятаны, либо потому что сам эпитоп гидрофобен и участвует в связывании. Значит, нет ничего необычного в том, что находятся антитела, которые хорошо работают в жидкой фазе, но терпят неудачу в твердофазных системах.
Трудность разработки теоретической модели твердофазного иммунного анализа состоит также в том, что ряд основных допущений и понятий не полностью применимы. В большинстве твердофазных определений константа скорости прямой реакции между частицами в растворе и иммобилизованной распознающей фазой ограничена диффузией, а не константой сродства, относящейся к акту распознавания. Первостепенное влияние оказывают вязкость анализируемого раствора и относительное распределение активных центров, но также влияет и степень упорядочения частиц раствора при приближении к поверхности.
Поскольку биораспознающая поверхность является твердой фазой, структура пограничного слоя на ее поверхности влияет на реакцию и достижение стационарности. Хотя термин «концентрация» не очень подходит для рассмотрения фазы иммобилизованного реагента в растворе, однако в граничном слое локальная «концентрация» иммобилизованного реагента очень высока. Рассмотрим, к примеру, иммобилизованное на поверхности антитело в растворе, содержащем антиген (рис. 7.9-8). Антиген связывается с иммобилизованным антителом, вызывая обеднение антигеном слоя, ближайшего к поверхности, которое восполняется антигеном из объема раствора, причем процесс этот ограничен диффузией.
Поверхность фазы носителя рассматривают в целом как имеющую однородное распределение иммобилизованных антител, но это очевидное упрощение. Существует доказательство, что поверхность имеет фрактальную геометрию, где адсорбированные антитела собраны в островки высокой плотности, разделенные областями с малой населенностью (рис. 7.9-8,б). Обычное равновесие ассоциации/диссоциации между антителом и антигеном также имеет тенденцию к нарушению, поскольку крайне высокая «локальная» поверхностная концентрация антител в островных кластерах препятствует диссоциации антигена, и поэтому, если антиген был уже связан, процесс в значительной степени становится необратимым.
Физические методы разделения связанной и свободной метки
□ Разделение связанной и свободной метки не должно нарушать равновесия между ними.
Идеальный метод разделения не должен оказывать влияния на соотношение связанной и свободной метки и должен обеспечивать полное разделение обеих форм. Однако, как было установлено ранее, достичь этого никогда не удается. В любом случае связывание рецептор-лиганд обратимо, поэтому процедура разделения не должна изменять условия реакции, чтобы диссоциация не происходила. Большое разнообразие применяемых методов разделения отражает разнородность свойств комплекса рецептор-лиганд и требований к ме-
576
Глава 7. Методы химического анализа и их применение
Твердая фаза,
Диффузионно-контролируемый
процесс
Рис. 7.9-8. а — образование граничного слоя; б — эффекты фрактальной геометрии.
7.9. Иммунный анализ
577
году анализа. Например, основным требованием может быть скорость и простота использования, а не чувствительность или воспроизводимость. Может требоваться количественный результат, либо результаты по принципу «да-нет» Все эти критерии меняют общую схему анализа, и метод разделения связанной и свободной метки в анализе должен отражать эти потребности.
ч
Жидкая фаза
Декантация
По сути дела это простейший метод разделения; подходит и любой другой из подобных методов (процеживание, удаление жидкости и т. д.), но на практике их применение может быть связано с трудностями; для получения адекватных результатов каждый анализ требует модификации системы и методики.
Осаждение
Осаждение иммуноглобулинов легко осуществить «высаливанием» при нейтральном pH, при котором они малорастворимы, так что в случае меченого антигена связанную метку можно осадить из раствора вместе с оставшимся свободным антителом, оставляя, таким образом, свободную метку на поверхности. В качестве соли предпочитают использовать сульфат аммония, но недостатком метода в таком случае является высокий уровень неспецифического связывания.
Лучшим осадителем служит полиэтиленгликоль (ПЭГ). Его действйе подобно действию соли, отбирающей воду из структуры IgG и вызывающей ее денатурацию, в результате чего и происходит осаждение из раствора, но ПЭГ обеспечивает лучший контроль над системой. Метод широко используют в определениях ряда гормонов и стероидов различного типа, в ситуациях, когда гаптеновый антиген остается в растворе в условиях осаждения белка.
Адсорбция на твердых частицах
□ Иммунный анализ может быть гомогенным, т.е. проводиться в жидкой фазе, или гетерогенным, когда один из реагентов присоединен к твердой фазе.
Белки имеют тенденцию адсорбироваться на различных материалах, это свойство можно использовать для разделения. Целлюлоза, стекло и силикагель — все они нашли применение для адсорбции белков. Классический способ удаления растворенного вещества из раствора — использование измельченного древесного угля, но в случае белков IgG адсорбция затрудняется из-за несоответствия между большим размером молекул белка и малыми порами угля. Древесный уголь модифицируют, покрывая его декстраном и IgG, и в растворе сохраняется комплекс IgG-антиген, тогда как антиген адсорбируется на древесном угле. В случае меченого антигена этим способом удаляют из раствора свободную метку, оставляя связанную метку для определения в растворе.
В некоторых случаях адсорбция антигена на древесном угле может стать конкурентным процессом, нарушающим равновесие. В любом случае ее мож-
19 Аналитическая химия. Том 1
578
Глава 7. Методы химического анализа и их применение
но использовать лишь тогда, когда конъюгат антиген-метка достаточно мал, чтобы обеспечить эффективное разделение.
Хроматография
Во многих определениях существует значительная разница в размерах рецептора и лиганда. Антитела имеют молекулярные массы порядка 160000 и могут быть легко отделены от антигенов с молекулярной массой менее 80 000. Наиболее широко для этой цели применяется эксклюзионная гель-фильтрующая хроматография. Небольшой свободный меченый антиген удерживается на колонке, в то время как объемистый комплекс антиген-антитело элюируется. Метод обеспечивает хорошее разделение, но он дорог н требует затрат времени, так что он не подходит для рутинного, многократного использования.
Твердофазные носители
В этих методах первичная распознающая система присоединена к твердому носителю. Общим свойством этих методов является их низкое неспецифиче-ское связывание, сравнимое с жидкофазным анализом, особенно при введении этапов промывки для уменьшения неспецифичных эффектов. Выбор метода опять-таки зависит от факторов целесообразности и технических характеристик. Возможные компромиссные варианты приведены в табл. 7.9-1.
Шарики и частицы
Для твердофазной иммобилизации более всего подходят стеклянные шарики. IgG адсорбируется либо на стеклянную, либо на пластиковую поверхность, так что это метод пассивной иммобилизации, оптимальный при нейтральном pH. Высокую загрузку белка получают при таких размерах частиц, которые обеспечивают адсорбцию 10-15% белка из раствора. Используют также ковалентное присоединение, но это обычно требует большей подготовки иммобилизованного слоя (химия связывания подобна изображенной на рис. 7.8-7 и 7.8-8). Потребность в этом определяется приложением и дальнейшей обработкой иммобилизованного IgG.
Диаметр частицы не обязательно напрямую определяет емкость связывания, поскольку она зависит также от площади и природы поверхности. Однако установлено, что сефадекс и целлюлоза обычно имеют более высокую емкость связывания, чем найлон, полистирол или стекло, хотя два последних можно легко приготовить в виде более мелких частиц.
□ Твердофазными носителями могут быть частицы произвольной формы, шарики и поверхности; площадь реагирующей поверхности оказывает влияние на емкость связывания.
Среди сыпучих материалов наиболее распространен латекс (диаметр < 20 мкм) благодаря его сферической геометрии н большой удельной поверхности. Его плотность близка к плотности воды, так что на протяжении анализа он остается во взвешенном состоянии. Разделение осуществляют затем с помощью центрифугирования, декантации, микрофильтрации и т. п. Однако
Таблица 7.9-1. Системы разделения для иммунного анализа
Носитель Пример Достоинства Недостатки
Частица < 20 мкм Пористое стекло Латекс Магнитные частицы Микрокристаллическая целлюлоза Простота приготовления Высокая емкость связывания Быстрая реакция Медленное разделение (требуется центрифугирование или другой метод) Медленное магнитное осаждение
Частица < 1 мм Магнитные частицы Шарики Sepharose Шарики Сефадекс Средняя емкость связывания Быстрое магнитное осаждение Не требует центрифугирования Требуется перемешивание для удержания частиц в течение реакции во взвешенном состоянии Меньшая скорость для более крупных частиц
Частица > 1 мм Полистирол Не требует центрифугирования Низкая емкость связывания Малая скорость реакции Непостоянство в приготовлении н соединении
Мембрана Найлон Не требует центрифугирования Простота использования Низкая емкость связывания (короткий реакционный путь) Непостоянство в связывании Малая скорость
Твердая Покрытые трубки Не требует центрифугирования Высокое неспецифичное связывание
поверхность Пластинки Микротитрующие пластины Простота использования Непостоянство в связывании Малая скорость
580
Глава 7. Методы химического анализа и их применение
успешное применение этого материала для иммобилизации имело своим результатом изобретение и других оригинальных методов разделения. Например, инклюзия оксида железа (Робинсон с corp., 1973) или оксида хрома приводит к парамагнитному материалу, поэтому частицами можно управлять с помощью магнитного поля.
Для более крупных частиц и шариков, которые обычно не находятся во взвешенном состоянии, можно использовать такое парамагнитное легирование и наложение магнитного поля, чтобы поддерживать движение шариков в течение инкубационного этапа.
Мембранное фильтрование
В этом методе пробу «фильтруют» через мембрану, в которой иммобилизована захватывающая молекула (антитело или антиген). Затем в фильтрате определяют несвязанное вещество. Использование волоконных мембран увеличивает площадь поверхности, а значит, и емкость связывания; используют стекловолокно и волокно целлюлозы, ио чаще материалом мембраны служит найлон. Улучшение взаимодействия может быть получено при иммобилизации захватывающей молекулы на частицах полимера (как описано выше), которые затем улавливают в мембране.
Иммунохроматография
Дальнейшим развитием этих идей является включение захватывающей молекулы в полоску в хроматографической системе. В большинстве систем, использующих этот вариант, захватывающую молекулу (например, антитело) иммобилизуют на хроматографическом носителе (бумага, кремнезем, полимер или гель и т. д.) и фиксируют в виде полоски, так что проба (антиген), смешанная с меченым антигеном, конкурентно реагирует за захват мест распознавания, по мере того как проходит по полоске. Несвязанный материал затем можно определить, так как он мигрирует за захватывающую полоску. Другой способ— оценка количества меченого антигена, захваченного в полоске.
Разработаны также другие варианты: например, захватывающую полоску можно расширить на всю длину хроматографической системы, так что измерение основано на определении длины, на которой захватывается меченый антиген. Она связана с общей концентрацией антигена, а поскольку концентрация меченого антигена, добавленного к пробе, всегда одинакова, то для данной поверхностной плотности иммобилизованных захватывающих антител эта длина зависит от концентрации антигена в пробе.
Пластины, каналы и трубки
Пожалуй, наиболее удобной поверхностью для иммобилизации в рутинном иммунном анализе являются полистирольные микротитрующие пластины, трубки и каналы. Однако эффективность их использования в значительной мере зависит от воспроизводимости взаимодействия. Для увеличения емкости связывания и улучшения воспроизводимости разработаны способы облучения, и химической обработки поверхностей, за счет чего матрицы микротитрующих каналов массового производства улучшили воспроизводимость «идентичных»
7.9. Иммунный анализ
581
поверхностей для сравнительной иммобилизации, так что в анализе легко провести градуировку и автоматизацию, по крайней мере, частичную. Разработаны очень многие конкурентные или сандвичевые методы, использующие эти поверхности, покрытые антителом или антигеном.
Основным недостатком, особенно в случае микротитрующих пластин, является малая площадь поверхности для реакции. Пластины имеют емкость порядка 10% от емкости трубок, так что измеряемый при анализе сигнал должен быть достаточно велик, чтобы работать с этими малыми количествами. Независимо от этих ограничений, определения как с трубками, так и с пластинами проводили весьма успешно главным образом благодаря простоте их использования, а не благодаря характеристикам сигнала.
В качестве материала наиболее широко используют полистирол, а в некоторых случаях увеличивают емкость за счет высушивания на нем антитела; одним из главных достоинств является высокая степень адсорбции, несмотря на то, что площадь поверхности мала по сравнению с площадью частиц, обсуждавшихся выше. Однако это преимущество в адсорбции способствует также неспецифическому связыванию, так что определение всегда включает этап «блокировки», использующий белок, такой, как казеин, для связывания любых ненаселенных областей на поверхности полистирола после того, как была присоединена первичная распознающая молекула.
7.9.3. Метки
Радиоактивные метки
□ Наиболее широко в качестве радиоактивных меток для иммунного анализа используют 1251 и 3Н.
Обычно в качестве метки выбирают 1251, 7-изЛучатель, или 3Н (тритий), /3-излучатель, хотя используют также 57Со (для витамина В12) и 14С. Тритий дает меньшую чувствительность, чем 1251, поскольку испускаемые /3-частицы слабы {Ет^х — 18,6 кэВ; Еср = 5,5 кэВ) и легко поглощаются стенками сосуда. 7-Лучи, испускаемые 125I (Е =28-35кэВ), ие поглощаются сосудом, а эффективность счета при использовании сцинтилляционного счетчика обычно составляет около 80%.
В некоторых случаях метку можно присоединить путем простого изотопного замещения; в большинстве потенциальных определяемых веществ водород можно заместить на тритий, но природные определяемые вещества, содержащие иод (например, тироксин Т4), встречаются значительно реже. Следовательно, чтобы использовать в качестве метки 1251, обычно бывает необходимо присоединить его как «ярлык».
Белки
Простейший способ маркирования белков заключается в прямом замещении в ароматическом кольце тирозина или гистидина. Однако распад радиоактивного нуклида передает молекуле энергию, которая превышает энергию химических связей, и, следовательно, существует опасность разрыва соседних связей и нарушения молекулярной структуры. Таким образом, хотя высокий уровень
582
Глава 7. Методы химического анализа и их применение
Схема 7.9-1. Методы маркирования изотопом 1251. о —внедрение иода с помощью хлорамина-Т в качестве окислителя; б — внедрение иода с лактопероксидазой (LPO) и пероксидом водорода в качестве окислителя; в—синтез конъюгата метилового эфира тирозина: Се-кетогруппа 6-кето-17/3-эстрадиола; г — конъюгаци-онное маркирование, при котором К-гидроксисукцинимидил-3-(4-гидрокси-5-{12б1}-иодфенил)прошюнат реагирует со свободными аминогруппами.
замещения дает более высокую чувствительность, практически уровень замещения составляет порядка одного атома 1251 на молекулу белка, поскольку более высокий уровень замещения приводит к радиолизу, дезактивирующему белок.
Предложен ряд методов такого иодирования, но основные успехи в радиоиммунном анализе (РИА) получены Гинвудом и Хантером (1961; схема 7.9-1, о) при разработке метода окисления хлорамином-Т. Однако этот неспецифический окислитель подходит не для любого приложения, и растущую популярность получил ферментативный метод, использующий лактопероксидазу и пероксид водорода (Марчелонис, 1969; схема 7.9-1,б).
7.9. Иммунный анализ
583
Гаптены и полипептиды
□ Маркирование пептидов и белков обычно затрагивает ароматическое кольцо тирозина или гистидина.
Когда пептид или гаптен не содержит ароматического кольца, способного к замещению иодом (например, тирозина), обычным подходом является получение конъюгата с меченой молекулой. Очевидным выбором такой маркирующей системы является тирозиноподобный остаток, и производными, используемыми для этой цели, могут быть метиловый эфир тирозина, тирамин или гистамин и т. д. (схема 7.9-1,в). Однако доступнее другие иодированные соединения, например, N-гидроксисукцинимидный эфир З-(эт-гидроксифенил)пропионовой кислоты; схема 7.9-1,г; прямая реакция этого реагента со свободными аминогруппами приводит к присоединению через амидную связь. В целом, выбор реагента для присоединения зависит от наличия реакционных групп в пептиде или гаптене (табл. 7.9-2).
Таблица 7.9-2. Типичные реагенты, присоединяющиеся к белкам
Группа па гаптене/пептиде для присоединения
Реагент
Конъюгат
-NH2 или СООН -NH2 или СООН -NHz или -СООН Карбодиимид R1CONHR2 Изобутил хлороформиат R1CONH-R2 Изоксазолий R1-CONH-R2 RpNH NH-R2
-nh2 Глутаровый альдегид НО ОН A^NHCONH-R]
-nh2 Диизоцианаты (толуол-2,4- NHCONH-Rg дииизоцианат)
Тирозин, гистин или лизин Диазотированный Rj- N: N: N-Rp бензидин
Группа на гаптене/пептиде для активации Реагент Активируемый лиганд
-он Янтарный ангидрид 0^ у© R НО О
-он Фосген R-J-a —0—. C=NH
Соседняя -ОН BrCN о /
Кето- или альдегидная О-(карбоксиметил )-гидроксиламин r-ch=n-o-ch2cooh
584
Глава 7. Методы химического анализа и их применение
Частицы, рассеивающие свет, в качестве меток
□ Рассеяние света на частицах дает информацию о размерах частицы или агрегата, которую можно использовать для проведения иммунного анализа.
Концепция агглютинации известна с 1920-х гг. в микробиологическом анализе. Этот принцип можно положить в основу образования комплекса между антителом и антигеном, где имеется поливалентное связывание. Поливалентный белок будет реагировать со своим антителом, образуя осаждающийся комплекс, но реакция в высокой степени зависит от таких факторов, как ионная сила и ионные частицы, и осложняется изменением реакционной способности различных антигенных связывающих центров на антителе. В принципе, по мере протекания реакции образование комплекса может приводить к изменению способности среды рассеивать свет, но в идеале комплекс должен оставаться во взвешенном состоянии.
Более удобным способом является использование частиц-меток для усиления светорассеивающих свойств меченого конъюгата. Когда частицу облучают монохроматическим светом, падающее поле вызывает флуктуацию диполя и свет рассеивается перпендикулярно оси диполя в направлениях, которые дают информацию о размере частицы. Характер рассеяния зависит от соотношения между объемом частицы и длиной волны. Если длина волны намного больше, чем частица, свет одинаково рассеивается во всех направлениях, но если размер частицы увеличивается, то интерференция рассеянного света в различных точках на частице уменьшает количество света, рассеиваемого обратно в сторону источника, и образует участки высокой интенсивности (рис. 7.9-9).
При измерениях, когда частицы находятся в броуновском движении, интенсивность света, собранного под фиксированным углом, флуктуирует в соответствии с этим движением, потому что она зависит от времени, которое нужно частице для диффузии на расстояние, сравнимое с длиной волны. Рассеянный под фиксированным углом свет имеет фазовый сдвиг и вектор рассеяния К определяется выражением
K = frSin(l) (7-9-!6)
где п — показатель преломления фазы суспензии, а угол рассеяния. Интенсивность детектируемого света флуктуирует в масштабе времени, сравнимом с временем, за которое частица диффундирует на расстояние, достаточное для того, чтобы вызвать фазовый сдвиг иа тг, причем это время обратно пропорционально К.
Рис. 7.9-9. Рассеяние света в направлении, перпендикулярном оси диполя, как функция объема частицы V.
585
Наиболее распространенным металлическим <ярлыком> в этом методе является золото благодаря его высокому показателю преломления и относительной простоте приготовления. Можно получить частицы одинакового размера в диапазоне от 5 до 60 нм. Однако латексные частицы также просты в получении, а кроме того, они обеспечивают бблыпую гибкость при управлении их свойствами. Наилучшие характеристики получены при узком распределении размеров, так что суспензия в течение анализа поддерживается без излишнего перемешивания. Синтез проводят обычно одним из трех основных способов:
— полимеризация суспензии, - полимеризация эмульсии, — полимеризация набухшей эмульсии.
Процессы включают образование мицеллы поверхностно-активного вещества, которая принимает мономер и инициатор полимеризации. Средний размер частиц полученного полимера управляется концентрацией реагента, температурой и скоростью перемешивания. Мономеры с боковыми реакционными группами (например, акриловая кислота, -СООН; глицидилметилакрилат, эпоксигруппа; акриламид, -NH2) допускают последующее присоединение к антигену или антителу. Присоединение антитела к частицам показывает, что имеется оптимальная загрузка, и вне ее пределов стерические затруднения или обеднение поверхности антигеном (см. обсуждение методов поверхностной иммобилизации в разд. 7.8.2) снижает эффективность связывания. Сходные проблемы возникают также при присоединении к частицам антигена.
Эффекты неспецифического связывания могут также быть источником проблем. Изменение поверхности частиц влияет на агрегацию частиц и адсорбцию реагента, так что следует оптимизировать условия, чтобы уменьшить вандерваальсово притяжение между частицами, обеспечив достаточное кулоновское отталкивание, но не ингибируя реакцию антитело—антиген.
Флуоресцентные и хемилюминесцентные метки
□ Флуоресцентные метки обычно дают лучшую чувствительность, чем коло риметрические, потому что длины волн возбуждения и детектирования различны.
Принципы работы с этими оптическими метками во многом те же, что и с радиоактивными метками, за исключением того, что метод детектирования включает спектрофотометрическое измерение. Однако большинство прямых методов с колориметрической меткой, когда длины волн возбуждения и детектирования совпадают, редко достигают высокой чувствительности в иммунном анализе. Но можно получить улучшение на порядок величины, разделив эти длины волн. Существует дна основных класса меток, пригодных для этого; флуорофоры и хемилюминесцентные реагенты.
Флуорофоры переходят на более высокий энергетический уровень при поглощении света в одном диапазоне длин волн; в возбужденном состоянии происходит быстрый безызлучательный переход в самое низкое возбужденное состояние и затем возврат в основное состояние с испусканием фотона с большей длиной волны (рис. 7.9-10,а, б); возбужденное состояние имеет типичное вре-
586
Глава 7. Методы химического анализа и их применение
Рис. 7.9-10. Последовательность этапов, приводящая к флуоресценции.
мя жизни < 10“8с. Разница между двумя длинами волн известна как стоксов сдвиг, и чем больше сдвиг, тем проще становится отличать интенсивность испускаемой длины волны от интенсивности фона падающей длины волны. Когда квантовый выход (доля переизлученной энергии) высок, т. е. приближается к 1, метка будет наиболее чувствительной.
В течение времени жизни возбужденного состояния молекула может также участвовать в химической реакции, передавая, таким образом, энергию другим
7.9. Иммунный анализ
587
Рис. 7.9-11. Изменение интенсивности люминесценции во времени, показаны различные «окна» обработки сигнала.
Время
частицам и возвращаясь в основное состояние. Такой процесс тушения может приводить к значительной потере сигнала. Кинетика тушения флуоресценции может быть описана конкуренцией двух параллельных процессов:
Флуоресценция: М* М 4- hi/
Тушение: М' + Q М + Q’
что приводит к потере флуоресценции в соответствии с выражением
= ML’] + fcq[L’][Q] (7.9-17)
dt
и уменьшает выход флуоресценции в соответствии с соотношением
То fcf[L*] + fcq[L*][Q] 1 + MMQ1
где Io — начальная интенсивность флуоресценции, a If—конечная интенсивность после тушения.
Вслед за началом возбуждения интенсивность флуоресценции зависит от концентрации флуорофора в возбужденном состоянии и является функцией времени:
ОД = фЁИ (7.9-19)
dt
где Ф — квантовый выход. Если возбуждение происходит в режиме короткого импульса, то профиль интенсивности будет иметь форму, показанную на рис. 7.9-11, где за начальным ростом интенсивности после облучения следует затухание по мере завершения возбуждения. Этот профиль открывает несколько различных возможностей для измерения. Если изобразить зависимость интенсивности флуоресценции различных молекул от времени на одном графике, каждая из флуоресцирующих молекул показывает разный результат для указанных параметров, поскольку времена затухания и квантовые выходы индивидуальны для каждого отдельного флуорофора.
5U
Глава 7. Методы химического анализа и их применение
□ «Идеальные» флуоресцентные метки должны иметь большой стоксов сдвиг и возбуждаться с помощью недорогих источников света.
Наиболее распространенные флуорофоры, используемые напрямую в качестве меток, традиционно являются производными флуоресцеина и родамина; в первом случае при присоединении метки в качестве активного интермедиата часто используют изотиоцианатное производное. Длины волн возбужде-ния/испускания этих меток хорошо подходят для большинства флуориметров (рис. 7.9-12), хотя их положение и малый стоксов сдвиг делают затруднительным нахождение недорогих диодных источников возбуждения, которые можно было бы использовать для создания небольшого специализированного прибора.
Непрерывно разрабатываются новые флуоресцентные зонды, чтобы покрыть все более широкий спектр и приспособиться к потребностям специфических длин волн. В табл. 7.9-3 суммированы некоторые из них. Весьма чувствительными флуорофорами являются гидроксильные производные кумарина (умбеллифероны), которые открывают широкие возможности для получения различных флуоресцентных свойств. Многие из производных этого ароматического фенола, такие, как фосфаты, гликозиды и др., не флуоресцируют, ио флуоресцирующие частицы могут быть получены при их гидролизе; это свойство также можно использовать в иммунном анализе с меткой, как уже обсуждалось ранее.
Пределы обнаружения для этих флуоресцентных реагентов определяются мешающим влиянием фона и квантовой эффективностью. В первом случае собственная флуоресценция пробы, особенно если она содержит, например, сывороточные белки, NADH или билирубин, может быть очень высока. Свободный NADH, например, проявляет максимум поглощения при 340 нм и
400 500 500
Длина волны, нм
Рис. 7.9-12. Спектры возбуждения и флуоресценции белка, меченного изотиоцианатом флуоресцеина.
7.9. Иммунный анализ
589
Таблица 7.9-3. Флуоресцентные свойства некоторых флуоресцентных меток, используемых в иммунном анализе
Флуорофор ^оозбг НМ ^флуорг НЖ Время затухания, нс Молярное поглощение, л/моль Квантовый выход
Флуоресцеин 492 520 4,5 72000 0,85
Родамин С изотиоцианат Лиссамин сульфоро- 550 585 3,0 103000 0,7
дамин С хлорид Анилинонафтадин- 530; 565 595 1,0
сульфокислота 2-Метокси-2,4-дифенил- 385 471 16 0,8
3(2Н)фуранон 390 480 0,1
Метилумбеллиферон 323 386 16000
Дансил хлорид 340 480-520 14 0,3
Люцифер желтый 430 540 3,3 13000 0,2
Эритрозин 492 517 108 101000 0,01
Ru-(dipy)3 450 625 250-500 14000 0,028
Pd-Копропорфирин 396 618 700000 200000 0,17
Пирены 346 395 16000
Техас красный 596 615 80000 0,3
Эозин 520 545 101000
Флуорескамин 394 475 7,0 ОД
Хлорофилл 430-453 648-669
сильную флуоресценцию при 400 нм, которые меняют величину и положение при связывании с белком и/или подложкой. Эти мешающие соединения имеют относительно малые времена затухания, так что метод, пригодный для разделения сигналов метки и фона, должен использовать измерение флуоресценции с временным разрешением (см. рис. 7.9-11).
Для этой цели оказались полезными некоторые комплексы лантаноидов с определенными органическими лигандами, такие, как хелаты европия со стоксовым сдвигом более 200 нм и временами затухания свыше 500 ис, в частности, потому, что возможный квантовый выход составляет 30-100%. Эти хелаты использовали разными способами; например, нефлуоресцирующие лиганды, образующие комплексы с европием, можно использовать как метку, а флуоресценция, возникающая при добавлении европия, возникает по окончании имму-нокомплексообразующей стадии анализа. Ион Еп3+ в растворе гидратирован восемью или девятью молекулами воды. В этом состоянии он проявляет только слабую флуоресценцию, но удаление этих молекул воды при комплексообразовании с органическими лигандами усиливает флуоресценцию Еи3+. В течение времени жизни возбужденного состояния молекула может также принимать участие в химической реакции, передавая, таким образом, энергию другим частицам и возвращаясь в основное состояние. Такой процесс тушения вызывает значительные потери сигнала.
Этот эффект тушения может также найти полезное применение в флуоресцентном иммунном анализе с передачей возбуждения. В принципе, не тре-
590
Глава 7. Методы химического анализа и их применение
буется разделения связанной и свободной фаз. Тушитель представляет собой акцепторную молекулу, например, на антителе (рис. 7.9-13), с максимумом возбуждения, близким к длине волны испускания исходного «донорного» флуорофора, который находится на антигене. Скорость передачи обратно пропорциональна шестой степени расстояния между молекулами и зависит также от квантового выхода и «соответствия» между длинами волн донора и акцептора.
„ Ф,-Фр г~6
Фо г~е + й^6
(7.9-20)
где Ф9 и Фо — квантовые выходы донора в присутствии и в отсутствие акцептора, а Ло — расстояние, иа котором эффективность передачи составляет 50%.
Постоянно используемой парой акцептор—донор являются флуоресцеин и родамин, а получаемое 50%-ное уменьшение флуоресценции донора на ~ 50 Асравнимо с типичным внутримолекулярным расстоянием между рецепторами в IgG (порядка 100 А). Чтобы добиться максимальной близости между донором и акцептором, маркирование антитела следует проводить близко к месту связывания; однако такая направленная модификация антитела меткой затруднительна из-за недостатка уникальных групп в структуре белка. Следовательно, вводят несколько флуорофоров в случайном порядке, но слишком высокий уровень маркирования может привести к нерастворимости, поэтому типичным значением является 10-12 флуорофоров на одну молекулу IgG. По-
хожие рассуждения применяют и для донора, но в случае гаптена это соотношение может быть ограничено конъюгатом 1:1, а для белка высокая загрузка флуорофора приводит к уменьшению квантовой эффективности из-за самоту-шения. В принципе, регистрация испускания акцептора должна давать лучший сигнал, но поскольку некоторое прямое возбуждение акцептора может происходить и при длине волны возбуждения донора, это приводит к высокому фону. Выбором, следовательно, является контроль тушения флуоресценции донора.
Для того чтобы отличить сигнал от фона, а в некоторых случаях разделить сигналы связанного и несвязанного меченого антигена, можно использовать и другие молекулярные свойства. Например, когда антиген мал, меченный флуоресцеином аналог способен вращаться с высокой скоростью, а комплекс с антителом имеет значительно меньшую скорость вращения. При возбуждении популяции флуоресцентных молекул линейно поляризованным светом, вероятность возбуждения любой отдельной молекулы определяется ее ориентацией. Если «заморозить» молекулу в определенном положении, так что вращения не происходит, то в результате наблюдают флуоресценцию в плоскостях поперечного электрического (ТЕ, ±) и поперечного магнитного (ТМ, ||) полей по отдельности, а максимальное значение поляризации Р равно 0,5:
(7.9-21)
Усиление броуновского движения приводит к уменьшению поляризации. Этот принцип используют во флуоресцентной поляризации, поскольку вращение меченого флуоресцирующего антигена уменьшается при связывании с антителом (рис. 7.9-14). Таким образом, при низкой концентрации антигена в
7.9. Иммунный анализ
591
Рис. 7.9-13. Иммунный анализ путем передачи возбуждения флуоресценции.
592
Глава 7. Методы химического анализа и их применение
Флуоресцентный меченый антиген. Быстрое вращение. Низкая поляризация
Меченый антиген.
Медленное вращение Высокая поляризация
Рис. 7.9-14. Принцип иммунного анализа по поляризации флуоресценции. Связывание антитела с антигеном, меченым флуорофором, увеличивает поляризацию флуоресценции.
пробе поляризация флуоресценции максимальна, поскольку максимальное количество меченого антигена связано.
Для данного флуорофора изменение поляризации флуоресценции зависит от молекулярной массы антигена и природы комплекса с антителом; должно иметь место надлежащее изменение молекулярной массы, а этого нельзя добиться с большими антигенами. В целом, принято считать, что иммунный анализ по поляризации флуоресценции неприменим к антигенам с молекулярной массой выше 20000.
Основной проблемой гомогенного анализа, описанного выше, является влияние мешающих веществ, что не дает возможности реализовать теоретическую чувствительность. В табл. 7.9-4 проведено сравнение некоторых пределов обнаружения, полученных для отдельных вариантов с использованием флуоресцентной метки.
Таблица 7.9-4. Пределы обнаружения некоторых гаптенов, определяемых в различных вариантах иммунного анализа
Вариант Определяемое Предел
метода вещество обнаружения, моль/л
Субстратная метка Теофиллин 3 1(Г8
Тушение Гентамицин 210"®
Передача возбуждения Кодеин 1,5-10-’
Морфин 1,5 -ИГ10
Гентамицин 4,5 -ИГ®
Поляризация Фенитоин 3,5-10-®
7.9. Иммунный анализ
593
□ Хемилюминесцентные метки испускают свет за счет неустойчивого люминес-цирукяцего продукта реакции.
В отличие от флуорофоров, хемилюминесцентные реагенты не требуют для возбуждения падающего света, а испускают свет в результате химической реакции. Например, арилакридиновые эфиры, присоединенные к рецепторной молекуле через эфирную часть, могут разорвать эфирную связь за счет щелочного гидролиза на одной из стадий анализа с образованием неустойчивого люминесцирующего N-метилакридона, который разлагается с испусканием света (схема 7.9-2). Максимальная интенсивность для таких меток наблюдается обычно в течение 0,4 с с периодом полузатухания 0,9 с.
Это сравнимо с очень медленным испусканием 25 с после инициирования) производных нзолюминола. Люминол и его производные относятся к семейству циклических ацилгидразидов и испускают голубой свет (425 нм) при окислении. Изо люминол имеет более низкий квантовый выход, чем люмннол, но в отличие от люминола этот выход мало изменяется при соединении с белками через аминогруппу. Синтезированы производные изолюминола с разнообразными мостиковыми группами для присоединения к гаптенам, многие из них нацелены на пептидную связь, например, за счет реакции с карбодинмидной или сукцинимидной активацией (схема 7.9-3). Как показано в следующем разделе, хемилюминесцентный контроль значительно усовершенствован за счет
Конъюгат
Схема 7.9-3. Модифицированная карбодиимидная реакция для конъюгата изолюминола через свободную карбоксильную группу.
594
Глава 7. Методы химического анализа и их применение
идентификации ряда соединений, которые увеличивают испускание света при добавлении в реакционную среду.
Ферментные метки
Общий принцип
О Ферментные метки непосредственно не участвуют в измерении, но поскольку они вызывают превращение многих субстратных молекул на одну молекулу фермента, существует высокий потенциал усиления.
Ферменты служат, пожалуй, наиболее широко используемыми в иммунном анализе метками, однако они непосредственно не участвуют в измерении, в отличие от изотопов или флуоресцентных меток. Вместо этого, измерение может включать детектирование расхода ферментного субстрата или накопления продукта, требуя, таким образом, наличия в анализе другого реагента и привнося в метод дополнительные сложности. Тем не менее, поскольку одна молекула фермента может вызвать превращение многих молекул субстрата, имеется очень высокий потенциал усиления сигнала, и это преимущество перевешивает часто упоминаемый недостаток несовместимой оптимизации условий конечного этапа ферментативного анализа и связывания антитело—антиген.
Наиболее часто в качестве меток используют два фермента—щелочную фосфатазу (ЩФ) и пероксидазу хрена (ПХ). Выбор зависит от возможности оптического детектирования превращения субстратов этих ферментов и от легкости соединения фермента без потери активности. Щелочная фосфатаза представляет димерный гликопротеин с молекулярной массой 140 000, содержащий много свободных аминокислот для соединения. Аналогично пероксидаза хрена содержит четыре лизиновых остатка для соединения и имеет молекулярную массу 44000. В некоторых случаях для соединения можно использовать углеводные остатки.
Разработаны методы соединения фермента с антителом или антигеном в соответствии с конечным вариантом и специальными требованиями иммунного анализа. Существуют два главных подхода: гомобифункциональные агенты и гетеробифункциональные агенты. Многие из используемых реагентов те же, что и уже рассмотренные при образовании конъюгатов с радиоактивной меткой (см. табл. 7.9-2), и здесь можно применить похожее рассмотрение за исключением того, что в этом случае конъюгат образует связь с белком-меткой. Может оказаться необходимым защитить активное место фермента в процессе соединения за счет включения субстрата фермента в процессе приготовления; это особенно важно, коода происходит соединение фермента с гаптеном, поскольку гаптен может образовать связь или закрыть активное место, маскируя места распознавания и гаптена, и фермента.
Соединение с гаптеном должно, вообще говоря, использовать ту же группу, что и для образования иммуногена в первом случае, так чтобы лиганд не маскировал место распознавания. Например, в большинстве определений гидрокортизона применяют сыворотку, полученную с помощью конъюгата кортизон-3-(О-карбоксиметил)оксима, поскольку это оставляет уникальное 17 положение D-кольца доступным для распознавания, создавая, таким образом, специ
7.9. Иммунный анализ
595
фичность к кортикостероидной структуре (рис. 7.9-15). Конъюгат же с гаптеном, меченый в положении С-21, не проявляет затем сродства к антителу, так что этот анализ терпит неудачу.
□ Присоединение ферментной метки не должно блокировать место распознавания!
Точно так же, как может потребоваться разделяющая молекула между гаптеном и белком для образования иммуногена, способность меченого конъюгата к связыванию с антителом можно улучшить, если поместить подходящий разделитель между гаптеном и ферментом. Наиболее популярным гомобифунк-циональным агентом для присоединения фермента является глутаровый альдегид, но он склонен к образованию очень гетерогенных комплексов, с существенным кросс-линкингом фермент-фермент наряду с желаемым фермент-конъюгат. Гетеробифункциональные агенты позволяют обойти эту проблему с различной степенью успеха, зависящей от относительной реакционной способности разных групп. Широко используют N-гидроксисукцинимидный эфир дималеимида и карбодиимиды, но доступны многие другие соединительные агенты (см. также разд. 7.8.2).
Гомогенный ферментативный иммунный анализ
Интересным развитием идеи использовать ферментные метки является контроль соотношения «связанный свободный» путем изменения активности фермента в результате связывания в конъюгат с антителом или дополнительного связывания антиферментных антител. Такие способы анализа не нуждаются в этапе разделения, а значит, можно упростить проведение определения. Разработано несколько способов гомогенного ферментативного иммунного анализа (ФИА).
Умножающий ферментативный иммунный анализ (УФИА)
В конкурентном анализе гаптен, меченный ферментом, может конкурировать с гаптеном пробы за ограниченный запас антигаптеновых антител. Если связывание антитела с гаптеновым конъюгатом ингибирует активность фермента, то чем выше концентрация гаптена в пробе, тем меньше связывание антитела с конъюгатом, и, следовательно, тем меньше ингибирование (рис. 7.9-16). В первом варианте метода УФИА разработанном для морфина, в качестве фермента, чувствительного к ингибированию, использовали лизоцим, но было найдено, что глюкозо-6-фосфат-~ дегидрогеназа является наиболее эффективной для этого метода в отношении изменения ферментативной активности. Прн-
Рис. 7.9-15. Гидрокортизон, иллюстрирующий общую структуру стероидов, с уникальным центром «распознавания» для гидрокортизона в С-17. Соединение через С-3 не маскирует этот центр.
596
Глава 7. Методы химического анализа и их применение
Фермент, активированный связанным антителом
!y
Антитело
Гаптен, меченный ферментом
- Антиген пробы
Меченный активным ферментом гаптен определяют по окрашенному продукту
Рис. 7.9-16. Принцип умножающего ФИА-анализа. Антитело, связанное с ферментом — меткой гаптена, меняет активность фермента.
Рис. 7.9-17. Иммунный анализ с помощью фермента, меченного кофактором. Антитело, связанное с меченым гаптеном, не принимает участия в ферментативной реакции.
ложения УФИА направлены также на определяемые вещества с низкой молекулярной массой, в особенности из семейства наркотиков и терапевтических агентов. Основным недостатком этого метода является медленное установление равновесия с гаптеном в растворе, так что необходимо предварительное выдерживание пробы и антител перед добавлением ферментного конъюгата. Для определяемых веществ с большей молекулярной массой можно использовать модификации основного принципа.
Метка — кофактор фермента
Преимущество использования в качестве метки кофактора фермента, а не самого фермента, заключается в том, что его можно легко включить в схему усиления (рис. 7.9-17) н использовать в системе гомогенного анализа. Применяют конъюгаты гаптен-кофактор (например, NAD-гаптен), и в присутствии соединения фермент-NAD и субстрата конъюгат, связанный с антителом, неактивен, тогда как свободный конъюгат способен участвовать в ферментативном цикле. Каждый раз, когда конъюгат гаптен-кофактор претерпевает цикл уси-
7.9. Иммунный анализ
597
Таблица 7.9-5. Косубстраты пероксидазы для колориметрического детектирования
Косубстратп Хтах (окисленная форма), нм
2,2/-азинобис(этилбензотиазолин-6-сульфонат) (АБТС) 415 о-Фенилендиамин (ОФД) 492 (подкисленная)
3,3\ 5,5'-Тетраметилбензидин (ТМБ) 450 (подкисленная)
ления, образуется стехиометрическое количество продукта, и количество продукта накапливается по мере протекания анализа.
Образование сигнала
Образование колориметрического сигнала
□ Для проведения колориметрического иммунного анализа с ферментной меткой ферментативная реакция должна приводить к образованию окрашенного продукта.
Принципиальная идея здесь заключается в том, что ферментный субстрат следует превратить в окрашенный продукт. В случае щелочной фосфатазы обычно используют n-нитрофенилфосфат или аналогичный ароматический фосфат:
НгО
Щелочная фосфатаза
о; yszx
/О (ХлшФЮ нм)
ОН I О'-Р-ОН
но показано, что пероксидаза хрена проявляет бблыпую чувствительность. В последнем случае метка действует как косубстрат с пероксидом и донором водорода. Конечным продуктом является окисленный донор водорода, который выбирают в соответствии с его способностью поглощать в видимой области. В табл. 7.9-5 приведены распространенные косубстраты пероксидазы, имеющие подходящие абсорбционные характеристики в окисленном виде. 3,3', 5,5'-Тетраметилбензидин (ТМБ) дает наибольшее поглощение и наиболее низкий фон в сравнении с многими другими используемыми субстратами, такими, как 2,2'-азинобис(этилбензотназолин-6-сульфонат) (АБТС) или о-фенилендиамин.
Образование флуорометрического сигнала
Щелочная фосфатаза (ЩФ) больше подходит для флуоресцентных измерений, чем пероксидаза хрена (ПХ). Наиболее часто применяемым субстратом служит 4-метилумбеллиферилфосфат, который испускает свет при 448 нм при возбуждении на длине волны 365 нм. Следовательно, преимуществом ЩФ является то, что флуоресцирующее соединение можно фосфорилировать, создав, таким образом, флуорофорный субстрат для довольно неспецифнчного фермента ЩФ. В принципе, можно синтезировать широкий класс флуорофоров с различными флуоресцентными свойствами и временами затухания в качестве подходящих субстратов для ферментной метки. Такой ферментативный подход
598
Глава 7. Методы химического анализа и их применение
Субстрат, инактивированный связанным антителом, не участвует в реакции с ферментом
Гаптен, меченный субстратом
Флуорогенный продукт
Рис. 7.9-18. Флуоресцентный иммунный анализ с субстратом-меткой. Антитело, связанное с меченым гаптеном, ингибирует его как субстрат для фермента.
к флуоресцентным измерениям имеет очевидное преимущество более сильного сигнала в сравнении с обычной флуорофорной меткой, поскольку каждая ферментная метка создает много флуоресцирующих молекул.
Однако это справедливо не во всех случаях, например, в анализе, основанном на принципе УФИА, где гаптен маркирован субстратом и образует флуорогенный продукт при ферментативном катализе только тогда, когда не образуется комплекса с антителом (рис. 7.9-18). В этом случае конъюгат /3-галактозилумбеллиферона и гаптена может участвовать в качестве меченого аналога определяемого вещества в конкурентном анализе с ограниченным, количеством антител. Комплекс антитела с гаптеновым конъюгатом ингибирован как субстрат для фермента ^-галактозидазы, тогда как оставшийся свободным конъюгат гидролизуется ферментом, образуя флуорогенный продукт.
Образование хемилюминесцентного сигнала
□ Биолюминесценция предлагает очень чувствительный способ определения ферментной метки.
Щелочная фосфатаза особенно полезна при разработке способов генерации люминесцентного сигнала, поскольку многие ароматические фосфаты при отщеплении фосфатной группы образуют неустойчивый анион, который распадается с испусканием света (например, адамантил-1,2-диоксентанарилфосфат). Однако большинство выполняемых люминесцентных измерений основаны на
7-9. Иммунный анализ
Схема 7.9-4. Хемилюминесцентное окисление люминола с помощью пероксидазы хрена.
использовании пероксида. Люминол как косубстрат пероксида окисляется ПХ, образуя неустойчивый промежуточный продукт, который распадается до 3-аминофталата с испусканием света (схема 7.9-4). Хотя хемилюминесцентные реакции могут давать весьма чувствительные измерения, они, тем не менее, обычно имеют квантовый выход менее 20%.
Можно увеличить сигнал от реакции на схеме 7.9-4 на несколько порядков величины, добавляя «усилитель» (рнс. 7.9-19). Такими усилителями обычно служат фенолы и нафтолы, хотя показано, что другие молекулы тоже проявляют эффект (табл. 7.9-6). Точный механизм этого усиления до конца не ясен. Однако вероятно, что усилитель действует как переносчик радикала между ферментативными реакциями с пероксидом н люминолом, улучшая, таким образом, «медленный этап» образования люминольных радикалов.
Биолюминесценция также представляет хемилюминесцентный метод, связанный в первую очередь с механизмом, отвечающим за образование света светляками. Он включает фермент люциферазу, которая катализирует окисление гетероциклической органической молекулы (люциферина'). Люциферины имеют различную структуру, но многие из реакций, катализируемых люциферазой, включают такие кофакторы, как АТР (АТФ), FMN (флавинмононуклеотид) или NADH, так что использование этих кофакторных меток (см. рис. 7.9-17) вводит цикл в катализируемый люциферазой механизм (схема 7.9-5). С другой стороны, с помощью NAD-ферментной метки получают ферментативное усиление. Например, на рис. 7.9-20 показан ферментативный цикл, инициируемый NAD+, который приводит к люминесцентному сигналу. Такие определения NADH могут давать низкий предел обнаружения на уровне 10“15 моль NADH с линейностью свыше пяти порядков величины.
7.9.4. Мешающие влияния
Мешающие влияния — обобщенное название любого фактора, который вызывает смещение результата анализа. Большинство из наблюдаемых эффектов классифицированы как влияние основы, но это не устанавливает действитель-
600
Глава 7. Методы химического анализа и их применение
Фермент, инактивированный связанным антителом
Г1птен, меченный ферментом
Антиген пробы
Рис. 7.9-20. Включение ферментной метки в биолюминесцентный процесс.
7.9. Иммунный атшшэ
см
Люциферин
Оксилюцифарин'
Оксилюциферин +/П»
Л-562нм
АТР +РР
*°г +С02
Схема 7.9-5. АТФ в цикле генерации света люциферазой светляков.
Люцифераза
Таблица 7.9-6. Примеры хемилюминесцентных усилителей а реакции окисления люминола, катализируемой пероксидазой хрена
Производные фенола и нафтола
R- R"-
-I -Н
л-Иодфенол
-Cl -CI
2,4-Дихлорфенол
"^^соои ~н
л-П<дроксикоричная кислота
л-Фенилфенол
2-Хлор-4-фенилфенол
Производные ароматических аминов
^-снгоф-ннг
4-Бензоксианилин
N-Тетраметилбензмдин
602
Глава 7. Методы химического анализа и их применение
ную природу проблемы. Может быть несколько источников ошибок, каждый из которых может проявляться различным образом.
Эффективная концентрация определяемого вещества
□ Следует позаботиться об обеспечении оптимальных условий анализа с целью уменьшения ошибок.
В иммунном анализе определяемое вещество должно участвовать в равновесии связывания с распознающей молекулой- Если это связывание нарушено или если определяемое вещество вовлечено в связывание с другими молекулами, эффективная концентрация определяемого вещества оказывается заниженной. Во многих случаях определяемое вещество в пробе существует как в свободной,так и в комплексной формах; например, многие гормоны могут быть в равновесии с комплексом гормон-глобулин или гидрофобные стероиды могут быть распределены в липидных полостях- В первом случае эффект обычно преодолевают, добавляя блокирующий агент, тогда как липидный захват разрушают присоединением селективных поверхностно-активных веществ.
Однако, если не позаботиться о том, чтобы все определяемое вещество в пробе было в свободном виде, тогда предварительная обработка (или хранение) пробы может существенно влиять на результат. Тироксин (Т4) и многие другие малые молекулы почти полностью связаны с белком, но вытесняются с помощью неэтерифицированных жирных кислот. Поскольку эти жирные кислоты образуются при хранении пробы из-за воздействия липаз, то определение тироксина может стать скорее способом оценки возраста пробы, чем уровня определяемого вещества.
Комплексообразование может также включать ионы, а не белки или липиды, но в этом случае отсутствие комплекса может быть источником проблем, поскольку многие белковые определяемые вещества содержат центры связывания двухвалентных катионов, и антитела, выращенные на эти белки in vivo, могут распознавать конфигурацию, удерживаемую катионным комплексом (главным образом, Са2+ или Mg2+); таким образом, в отсутствие этих катионов конформация может измениться, так что распознавание с антителом становится неэффективным и анализ терпит неудачу.
Эффективность связывания антител
Для данного антитела на связывание антигена влияет наличие активного центра и его конформационная целостность. Связывание большее, чем ожидаемое, может также являться результатом неспецифического взаимодействия с определяемым веществом. В случае систем твердофазного анализа использование поверхностно-активных веществ, чтобы сделать поверхность менее гидрофобной, может уменьшить вторую проблему, но высокая концентрация поверхностно-активного вещества может привести также к потере антител, если они связаны нековалентно.
Как во всех методах анализа, всегда существует полезный рабочий диапазон, в пределах которого анализ имеет значимость. Для сандвичевых определений очень высокие концентрации определяемого вещества приводят к насы
=• «’uv»—анализ
603
щению как захватывающего Тськ и контрольного антитела, так что кажущаяся измеренная концентрация «сандвича* захватывающее антитело определяемое вещество/контрольное антитело оказывается ниже, чем ожидаемая (рис. 7.9-21). Это явление называют эффектом высокой дозы.
В конкурентном анализе наблюдается эффект низкой дозы, когда связывание меченого антигена при низкой концентрации антигена в пробе больше, чем в отсутствие определяемого вещества, из-за положительной кооперации.
Менее предсказуемыми оказываются ошибки за счет эндогенных антител, способных связывать один из аналитических реагентов. Эта проблема подобна тем, которые встречаются при эндогенном связывании антигена, обсуждаемом выше, но здесь возможные меры менее очевидны. Сандвнчевый анализ в наибольшей степени подвержен мешающему влиянию гетерофильных антител IgG и IgM в пробе, способных присоединяться к захватывающему антителу и предоставлять активный центр контрольному антителу. Существование такой проблемы неудивительно, так как антитела к животному IgG в пробе могут перекрестно реагировать с реагентными антителами, но введение сыворотки или иммуноглобулинов из тех же частиц, что и реагенты, обычно эффективно предотвращает мешающее влияние этого типа. Альтернативным подходом является использование только Fab-фрагмента, содержащего специфический активный центр в качестве контрольного антитела взамен целой молекулы IgG, что помогает избежать образования ложного сигнала.
Ошибки образования сигнала
Основным источником ошибок при образовании сигнала является какое-либо ингибирование метки. Например, флуорофор может тушиться эндогенными частицами пробы или на ферментную метку может влиять присутствие ингибиторов либо усилителей. Как было показано ранее прн обсуждении, эти
Антиген
Захватывающее антитело
Рис. 7.9-21. Эффект высокой дозы (кривая в форме «крюка»).
604
Глава 7. Методы химического анализа и их применение
эффекты можно использовать в качестве важных свойств анализа, но там, где это ие так, они могут вносить ошибки.
К сожалению, как и для многих мешающих веществ, ие всегда бывает просто предсказать или устранить проблему, так что тщательное опробывание метода анализа в той окружающей среде, в которой он будет использован, весьма существенно для полноты метода.
Литература
Bangs, L.B., Uniform latex particles. Seradyn Diagnostics Inc. Particle Technology Division 1987.
Diamandis, E.P., Clin. Biochem., 1988, 21, 139.
Hunter, W.M., Corrie, J.E.T., Immunoassay for Clinical Chemistry. Churchill Livinghstone, Edinburgh 1983.
Langone, ’ J.J., van Vunakis, H., Immunochemical Techniques, Part A. Methods in Enzymology., Vol. 70, Academic Press, New York 1988.
Langone, J.J., van Vunakis, H., Immunochemical Techniques, Part B. Methods in Enzymology., Vol. 73, Academic Press, New York 1988.
Langone, J.J., van Vunakis, H., Immunochemical Techniques, Part C. Methods in Enzymology. Vol. 74, Academic Press, New York 1988.
Langone, J.J., van Vunakis, H-, Immunochemical Techniques, Part D. Methods in Enzymology. Vol. 84, Academic Press, New York 1988.
Miles, L.E.M., Hales, C.N., Nature, 1968, 21», 186-189.
Price, C.P., Newman, D.J. (Eds.) Principles and Practice of Immunoassay. New York, Stockton Press 1991.
Robinson, P.J., Dunhill, P_, Lilly, M.D., Biotech. Bioeng., 1973, 15, 603-6.
Riibenstein, K.E., Schneider, R.S., Ullman, E.F., Biochem. Biophys. Res. Commun., 1972, 47, 846-851.
Thorell, J.L., Larson, S.M., Radioimmunoassay and related tecnhiques., Mosby, St Louis 1978.
ОГЛАВЛЕНИЕ
Предисловие редактора перевода ....................................... 5
Предисловие президента Федерации европейских химических обществ...... 8
Предисловие председателей Отделения аналитической химии ФЕХО......... 9
Благодарности ......................................................... 13
Обозначения............................................................ 14
Сокращения .......................................................... 17
Часть I. Общие вопросы
Глава 1. Задачи аналитической химии и ее значение для общества ...... 26
1.1. Аналитическая химия и общество ............................. 26
1.1.1. Введение: историческая справка......................... 26
1.1.2 Задачи аналитической химии............................. 27
1.1.3. Роль аналитической химии в жизни общества ............. 28
1.2. Химик-аналитик в центре решения проблем общества ........... 30
1.2.1. Введение.............................................. 30
1.2.2. Индустриальная среда.................................. 31
1.2.3. Аналитический подход ................................. 35
1.2.4. Решение аналитических проблем в науке об окружающей среде 38
1.2.5. Стандарты для процедур и измерений в аналитической лаборатории ........................................................ 42
1.2.6. Будущее аналитической науки .......................... 43
1.3. Задачи аналитической химии применительно к исследовательским лабораториям ........................................................ 45
1.3.1. Введение............................................... 45
1.3.2. Примеры построения методик анализа ................... 46
1.3.3. Заключение............................................ 53
Глава 2. Процесс анализа ............................................ 55
2.1. Введение ................................................... 55
2.2. Аналитический цикл ......................................... 56
2.2.1. Общая постановка задачи .............................. 57
2.2.2. Постановка конкретной аналитической задачи ........... 57
2.2.3. Выбор методики ....................................... 58
2.2.4. Пробоотбор ......................................... 58
2.2.5. Транспортировка и хранение проб ...................... 59
2-2.6. Пробоподготовка ....................................... 59
2.2.7. Измерение (определение) ....................... 61
2.2.8. Обработка данных...................................... 61
2.2.9. Выводы и отчет ..................................... 61
2.3. Аналитические характеристики ..........................-.... 62
2.4. Погрешности в химическом анализе ........................... 64
606
Оглавление
2.4.1. Правильность и воспроизвод имость ..................... 64
2.4.2. Смещение и точность.................................... 68
2.4.3. Представление аналитических данных .................... 70
2.4.4. Распространение погрешностей......................... 73
Глава 3. Обеспечение и контроль качества ............................ 79
3.1. Качество анализа и задачи аналитической химии ............. 79
3.1.1. Определения н общие понятия ........................... 79
3.1.2- Качество результатов анализа и необходимая информация .... 81
3.2. Методика анализа ........................................... 84
3.2.1. Выбор метода (методики) ............................... 84
3.2.2. Проверка методики ..................................... 89
3.3. Как достичь правильности анализа .......................... 102
3.3.1. Зачем нужна правильность.............................. 103
3.3.2. Сравнение с данными другого метода ................... 104
3.3.3. Сравнение с данными другой лаборатории ............... 105
3.3.4. Стандартные образцы .................................. 106
3.4. Правовые аспекты обеспечения н контроля качества........... 109
3.4.1. Оценка квалификации лабораторий .................... 110
3.4.2. Аккредитация производителей образцов сравнения и стандартных образцов ................................................ 112
3.4.3. Аттестация персонала ................................. 113
3.4.4. Счандартизапи я ........................-............. 114
3.5. Выводы .................................................... 115
Часть II. Химические методы анализа
Глава 4. Основы химических методов анализа ......................... 120
4.1. Равновесие в гомогенных системах ........................... 120
4.1.1. Введение........-..................................... 120
4.1.2. Состояние равновесия ................................. 121
4.1.3. Равновесные системы .................................. 121
4.1.4. Стационарное состояние ............................... 125
4.1.5. Взаимодействия растворитель—растворенное вещество .... 127
4.1.6. Теория электролитической диссоциации Аррениуса........ 130
4.1.7. Ион-ионные взаимодействия и ионная ассоциация ........ 132
4.1.8. Ионная активность .................................... 132
4.2. Кислотно-основное равновесие ............................... 140
4.2.1- Введение........................................... 140
4.2.2. Современная концепция кислот и оснований. Теория Врёнсте-да—Лоури ................................ -................. 141
4.2.3. Показатель кислотности pH ............................ 147
4.2.4. Кислотно-основные индикаторы ......................... 150
4.2.5. Буферы................................................ 152
4.3. Комплексообразование ...................................... 156
4.3.1. Введение.............................................. 156
4.3.2. Образование ковалентной связи .......-................ 158
4.3.3. Координационные свойства ионов металлов и лигавдов.... 160
4.3.4. Ступенчатое комплексообразование ..................... 164
4.3.5- Хелаты и полиядерные комплексы - -.................... 169
4.3.6. Кинетика комплексообразования ........................ 172
4.4. Окислительно-восстановительные системы ..................... 173
4.4.1. Основы электроаналитических методов .................. 173
4.4.2. Окислительно-восстановительные реакции ............. 173
4.4.3. Электрохимические ячейки.............................. 174
4.4.4. Реакции, контролируемые переносом заряда ............. 181
4.4.5. Реакции, контролируемые массопереносом ............... 182
4.5. Гетерогенные равновесия ................................... 185
4.5.1. Термодинамическое рассмотрение ..................... 185
Оглавление W7
4.5.2. Системы газ — жидкость .............................. 191
4.5.3. Системы твердое вещество-жидкость ................... 198
4.5.4. Системы жидкость-жидкость ........................... 218
4.5.5. Системы газ — твердое вещество ...................... 226
Глава 5. Хроматография ............................................. 230
5.1. Принципы хроматографического разделения....»................ 230
5.1.1. Обзор хроматографических методов .................... 231
5.1.2. Получение хроматограмм .............................. 232
5.1.3. Параметры хроматограмм .............................. 233
5.1.4. Теория хроматографии ................................ 236
5.1.5. Разрешение Rs как характеристика разделения пиков ... 241
5.1.6. Качественная информация ............................. 243
5.1.7. Количественный анализ ............................... 244
5.2. Газовая хроматография ..................................... 246
5.2.1. Параметры удерживания и коэффициент распределения ... 246
5.2.2. Разделение в газовой фазе ........................... 247
5.2.3. Составные части газового хроматографа ............... 248
5.2.4. Неподвижные фазы для газо-жидкостной хроматографии .. 254
5.2.5. Применение газо-жидкостной хроматографии........... 258
5.2.6. Адсорбционная хроматография ......................... 261
5.3. Жидкостная хроматография .................................. 264
5.3.1. Высокоэффективная жидкостная хроматография (ВЭЖХ) .... 264
5.3.2. Ионная хроматография как классический и высокоэффективный метод .................................................. 283
5.3.3. Гель-хроматография................................... 287
5.3.4. Тонкослойная хроматография .......................... 292
5.4. Сверхкритическая флюидная хроматография.................... 298
5.5. Электрофорез .............................................. 302
5.6. Фракционирование в поперечном поле ........................ 309
5.6.1. Введение............................................. 309
5.6.2. Физические компоненты ФПП............................ 310
5.6.3. Различные варианты ФПП .............................. 312
Глава 6. Кинетика и катализ ........................................ 318
6.1. Введение ............................'.................... 318
6.2. Скорость химических реакций ............................... 319
6.2.1. Кинетическое уравнение и порядок реакции ............ 322
6.2.2. Интегральные кинетические уравнения ................. 327
6.2.3. Факторы, влияющие на скорость реакции................ 330
6.2.4. Скорости реакций: аналитическое применение .......... 332
6.2.5. Кинетические методы определения, основанные на некаталитических реакциях............................................. 336
6.3. Явление катализа .......................................... 337
6.3.1. Общие положения ..................................... 337
6.3.2- Кинетические уравнения и механизмы реакций .......... 339
6.3.3. Аналитическое применение каталитических реакций...... 348
6.4. Измерение аналитического сигнала в кинетических и каталитических методах.......................................................... 352 .
6.4.1. Закрытые и открытые системы.......................... 353
6.4.2. Автоматизированные кинетические методы анализа ...... 353
Глава 7. Методы химического анализа и их применение................. 359
7.1. Титриметрии ............................................... 359
7-1-1. Кислотно-основное титрование ........................ 359
7.1.2. Комплексометрическое титрование...................... 366
7.1.3. Электрохимическое титрование ........................ 374
7.2. Гравиметрия ............................................... 380
7.2.1. Введение............................................. 380
608
Оглавление
7.2.2. Гравиметрические методы.............................. 381
7.2.3. Расчет результатов гравиметрического анализа ........ 382
7-2.4. Электрогравиметрия .................................. 383
7.3. Электроанализ ........................................... 389
7.3.1. Потенциометрия ...................................... 389
7-3.2. Вольтамперометрия ................................... 411
7-3.3. Амперометрия ........................................ 430
7.3.4. Кулонометрические методы анализа .................. 432
7.4. Проточно-инжекционный анализ ..............*............... 440
7.4.1. Периодический и непрерывный проточный анализ ........ 440
7-4.2. Основы метода ....................................... 442
7.4.3. Основное оборудование ПИА ........................... 445
7.4.4. Дисперсия в ПИА .................................. 446
7.4.5. ПИА для воспроизводимого и точного ввода пробы (ограниченная дисперсия) ............................................ 448
7.4.6. Конверсионные методики ПИА (применения средней дисперсии) 452
7.4.7. Системы ПИА с ферментами........................... 457
7.4.8. Схемы проточно-инжекциоиной генерации гидридов...... 459.
7.4.9. Обработка и концентрирование пробы в режиме on-line.. 460
7-4.10. Использование процесса физической дисперсия. Градиентные методики ПИА ........................................... 462
7.4.11. Заключительные замечания ........................... 465
7.5. Термический анализ ....................................... 467
7.5.1. Введение.......................................... 467
7.5.2. Термогравиметрия (ТГ) .............................. 468
7.5.3. Дифференциальный термический анализ (ДТА) и дифференциальная сканирующая калориметрия (ДСК) ................... 473
7.5.4. Комбинированные методы ............................. 478
7.5.5. Анализ выделяющихся газов (АВГ) .................... 482
7.5.6. Другие термоаналитические методы .................. 485
7.6. Элементный органический анализ............................. 489
7.6.1. Введение............................................ 489
7.6.2. Методы, основанные на использовании для разделения селективной адсорбции........................................... 489
7.7. Химические сенсоры ........................................ 493
7.7.1. Введение............................................. 493
7-7.2. Характеристики и основные принципы................... 494
7.7.3. Электрохимические и микроэлектронные сенсоры ........ 496
7.7.4. Оптические сенсоры ................................ 505
7.7.5. Термические сенсоры ................................ 513
7.7.6. Масс-чувствительные сенсоры ......................... 514
7-7.7. Сенсорные наборы .................................... 516
7.8. Биосенсоры ................................................ 517
7.8.1. Введение......................................... 517
7.8.2. Создание биологической поверхности .................. 521
7.8.3. Подготовка биопреобразования......................... 531
7.8.4. Заключение........................................... 561
7.9. Иммунный анализ ........................................... 565
7-9.1. Введение.......................................... 565
7.9.2. Варианты устройства.................................. 574
7.9.3. Метки ............................................... 581
7.9.4. Мешающие влияния .................................... 599