/












































































































































































































































































































Text
Л. М. ЯКИМЕНКО
ПРОИЗВОДСТВО
ХЛОРА,
КАУСТИЧ ЕСКО Й СОДЫ
И НЕОРГАНИЧЕСКИХ
ХЛОР ПРОДУКТ© В
МОСКВА - ХИМИЯ • 1974
' УДК 661.4 + 661.332.1
Я 45
Якименко Л. М.
Производство хлора, каустической соды и 4 неорганиче-
ских хлорпродуктов. М. «Химия», 1974 г. 600 с., 107 табл.,
221 рис., список литературы 1440 ссылок.
Книга представляет собой инженерную монографию,
в которой освещен весь комплекс производства хлора,
каустической соды, важнейших неорганических хлорпро-
дуктов (хлоратов и перхлоратов, жидкого хлора, хлорной
и соляной кислот, хлористого водорода, хлоридов и т. д.)
и систематизированы достижения последних лет в этой
области химической технологии.
1515
Книга предназначена для инженерно-технических ра-
ботников химических предприятий, научно-исследователь-
ских и проектных организаций. Она будет полезна также
преподавателям, аспирантам и студентам вузов, специали-
зирующимся в области прикладной электрохимии.
31403-158
050 (О1)-74
4
© Издательство «Химия», 1974
t
> f
f
- СОДЕРЖАНИЕ
Предисловие .............................. . ...................... 7
Глава 1. Развитие производства и потребления хлора, каустической соды
и неорганических хлорпродуктов ................................... 9
л
Литература................................................. 23
V л
Глава 2. Теоретические основы производства хлора и каустической соды
электролизом растворов хлоридов щелочных металлов .... 25
Свойства хлора, едких щелочей и водорода......................... 25
Хлор .............................................. • . . 25
Каустическая сода и едкое кали . . ......................... 29
- Водород .................................................. 31
Теоретические основы электролиза водных растворов хлоридов щелоч-
ных металлов . .............................................. 33
Процесс электролиза водных растворов хлоридов щелочных ме-
таллов в электролизерах с ртутным катодом и с диафрагмой . . 33
Разделение продуктов, получающихся на электродах.............41
Аноды . ............................................... . 57
Баланс напряжения и расход электроэнергии на электролиз . . 82
Выход по току ........................................ 100
Материальный баланс электролизера........................ 108
Тепловой баланс электролизера . . . . ... . ................ ИЗ
Использование восстановительной способности амальгам щелоч-
ных металлов . . . ........................ . . ......... 117
Литература ................................................ 118
Глава 3. Конструкции электролизеров для получения хлора и каустиче-
[ Ской соды . . . . ...................................... 125
Электролизеры с твердым катодом . . ............................ 125
I ’ Электролизеры БГК-13 ................................... 126
I Электролизеры БГК-17 и БГК-50..............-............... 129
[ Электролизеры типа ДА.................................... 138
Электролизеры типа Хукер ................................ 139
| Электролизеры типа Даймонд . . . . ....................... 145
5 '
1*
3
4
Электролизеры с биполярным включением электродов . ... . . . .
Биполярный электролизер большой мощности.................
Электролизеры с малоизнашивающимися анодами .............
Основные показатели электролизеров с твердым катодом . . . .
Электролизеры с ртутным катодом . .......... . . ............
Общие принципы устройства электролизеров с ртутным катодом
Конструкции электролизеров ................С............... .
Электролизеры с вертикальным расположением катода . . . . .
Электролизеры типа Р-101 ................................
Электролизер Р-20 ............................... . . .
Мощные конструкции отечественных электролизеров..........
Электролизеры фирмы «Кребса» ........................ .
Электролизеры фирмы «Сольве»...................... . . .
Электролизеры фирмы «Де-Нора»...............
Электролизеры фирмы «Олин-Матисон» ............
Электролизеры фирмы «Куреха» ............................
Электролизеры фирмы «Уде» . ..............• . ........
Электролизеры типа СУ, QA и СДМ ..............
Электролизер фирмы «Асахи»...............................
Биполярные электролизеры с ртутным катодом .........
Новые направления в развитии конструкции электролизеров
с ртутным катодом . .....................
Электролизеры с малоизнашивающимися анода
Основные показатели электролизеров с ртутным катодом . .
Литература ....................................л. . .
iflis
Глава 4. Основные стадии производства хлора и каустической соды . .
/ 4
Технологические схемы производства хлора и каустической соды электро-
лизом водных растворов хлоридов щелочных металлов.................
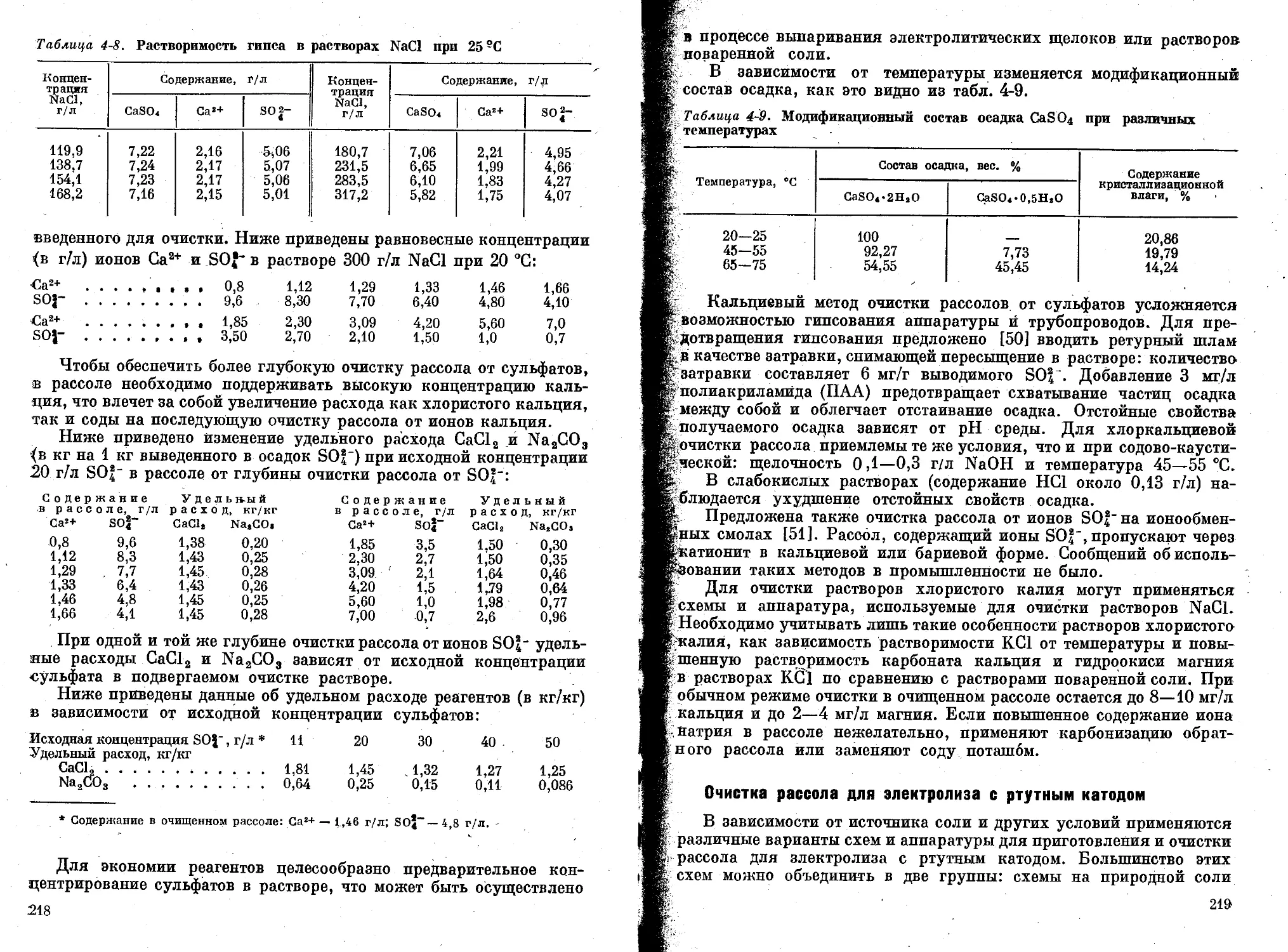
Приготовление и очистка рассола.............................. . .
Приготовление рассола .............................. . . . .
Очистка рассола для электролиза с диафрагмой ................
Очистка рассола для электролиза с ртутным катодом ......
148
151
153
156
156
156
170
170
171
174
176
176
178
179
180
181
184
184
185
185
186
187
187
193
193
197
197
206
219
Охлаждение, осушка и перекачка хлора и водорода ..................' 229
Охлаждение, осушка и перекачка хлора ...................... 229
Охлаждение, очистка и осушка водорода ...................... 239
Эксплуатация цехов электролиза '..................... 241
Объединение электролизеров в серии ......................... 241
Размещение электролизеров................................ 245
Обслуживание цехов злектролиза............................. 247
Выпарка и плавка каустической соды , ............................ 249
Выпарка электролитических щелоков . ...................... 249
Вывод сульфатов .............z............................ 261
Очистка каустической соды, получаемой электролизом с диаф-
рагмой ................................................. 264
Плавка каустической соды . . .............................. 267
4
Регенерация ртути из шламов и очистка сточных вод от ртути ... . . . 270
Литература . ............................................... 275
V ь
Глава 5. Другие способы получения хлора............t ....... . 280
Электролиз водных растворов щелочных металлов с получением кальци-
нированной соды . ................................................
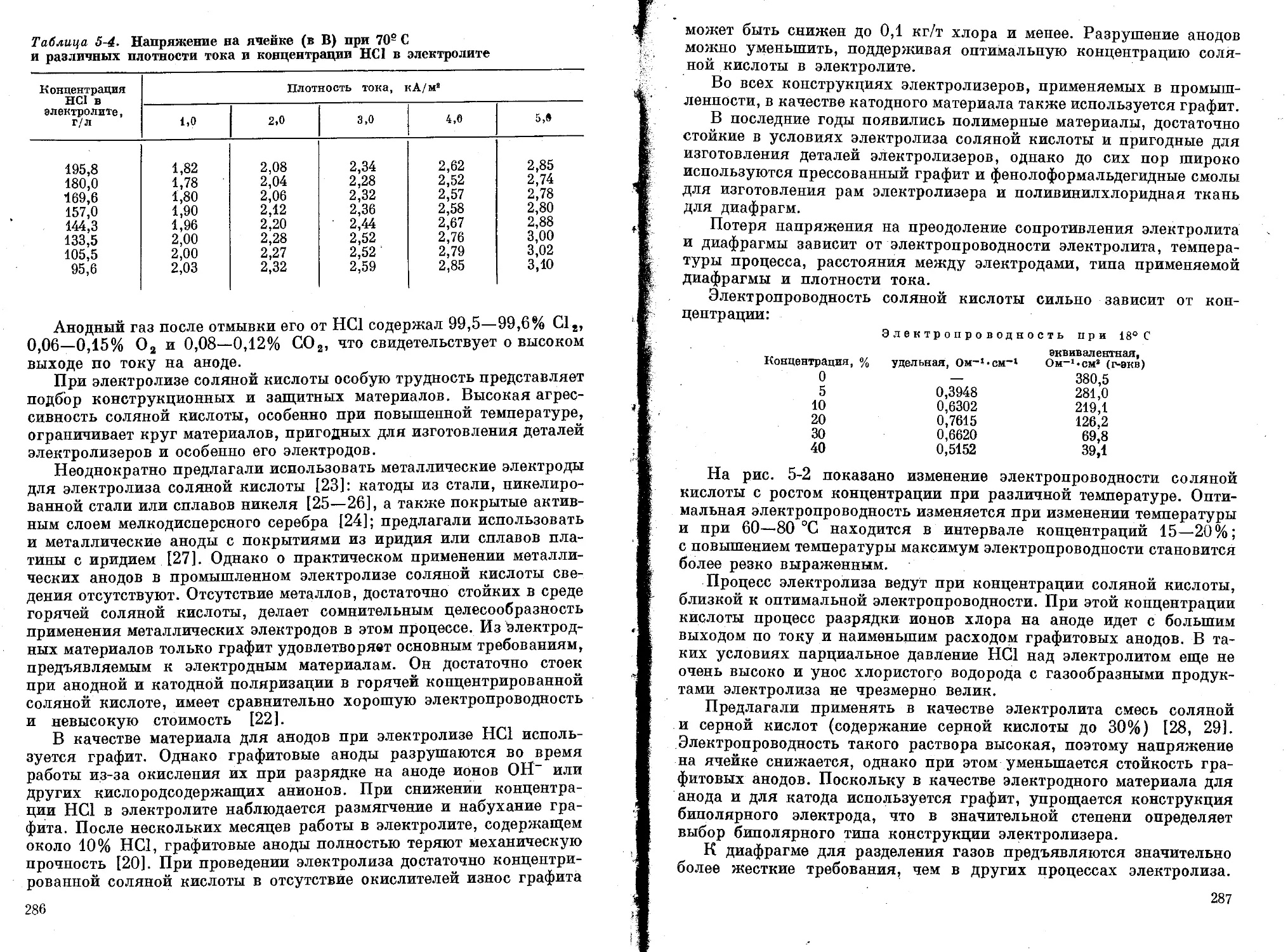
Электролиз соляной кислоты ............................
Прямые методы электролиза соляной кислоты .
Косвенные методы'электролиза соляной кислоты.................. \
Хи
wis
ческиё методы получения хлора
Литература
281
282
285
297
303
306
Глава £.
Жидкий хлор
жидкого хлора ......
производства жидкого хлора
Свойства
Развитие
Термодинамические основы процесса сжижения хлора ........
Технологические схемы производства...............................
Извлечение хлора из его смесей с воздухом или инертными газами . . .
Аппаратура установок для сжижения хлора . .............. . . , .
Компримирование хлора ,............................... . .
Конденсация хлора ................................... . .
Холодильные установки......................................
Испарение жидкого хлора............................... .
• Хранение и транспортирование жидкого хлора.....................
Автоматизация контроля и управления процессом сжижения хлора . .
Техника безопасности ................................... . . . .
Литература ................................................
309
309
312
316
327
339
340
340
346
349
351
352
360
362
363
Глава 7- Хлораты щелочных и щелочноземельных металлов
366
Производство хлоратой щелочных и щелочноземельных металлов . . . 366
Производство хлората натрия..................................... 368
Технологические схемы производства ................... . . . . . 384
Периодические схемы производства......................... 384
Непрерывная схема с выпаркой .......................... . 385
Конструкции электролизеров ...................................... 395
а Производство хлората калия.................................... 407
Производство хлоратов кальция и магния . . . . ,................ 412
Хлорат кальция ........................................... 413
Хлорат магния ч.............. . . ....................... 414
Литература ............ . .................... 415
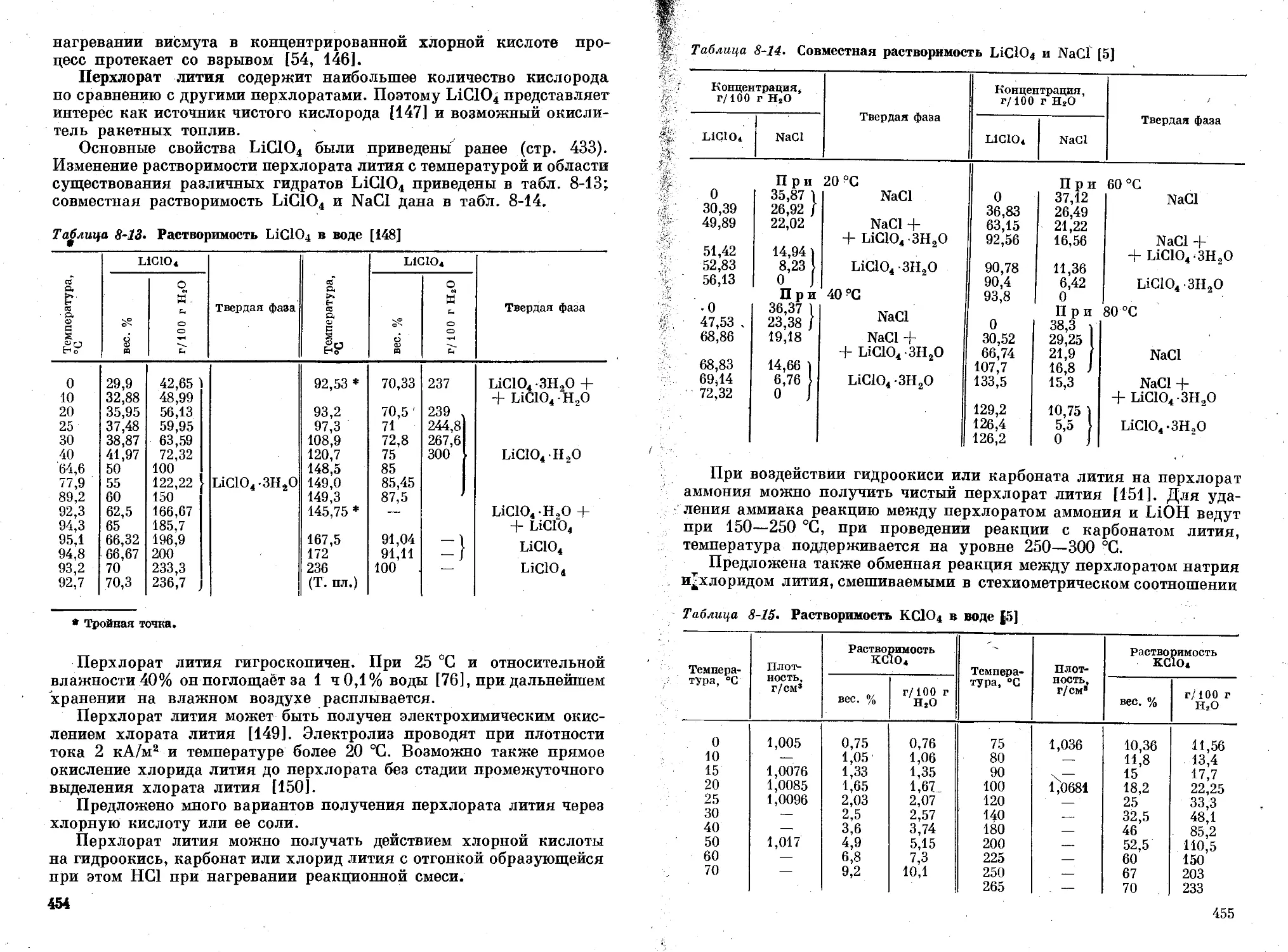
Глава 8. Хлорная кислота и перхлораты ............................ 420
‘ Хлорная кислота .....................г..................... , . . 421
Свойства хлорной кислоты......................... : л . . 421
Способы получения хлорной кислоты........................... 427
Перхлораты металлов.............................................. 432
5
*4
н
I
Свойства перхлоратов ................................... . 432
Способы получения перхлоратов металлов . . . ........... . 434
Перхлорат натрия..............;........................... 436
•Перхлорат аммония . ............................... . . . 448
Перхлораты других металлов ............................... 453
Перхлораты неметаллов ....................................... . . 457
Литература ............................................... 460
. *
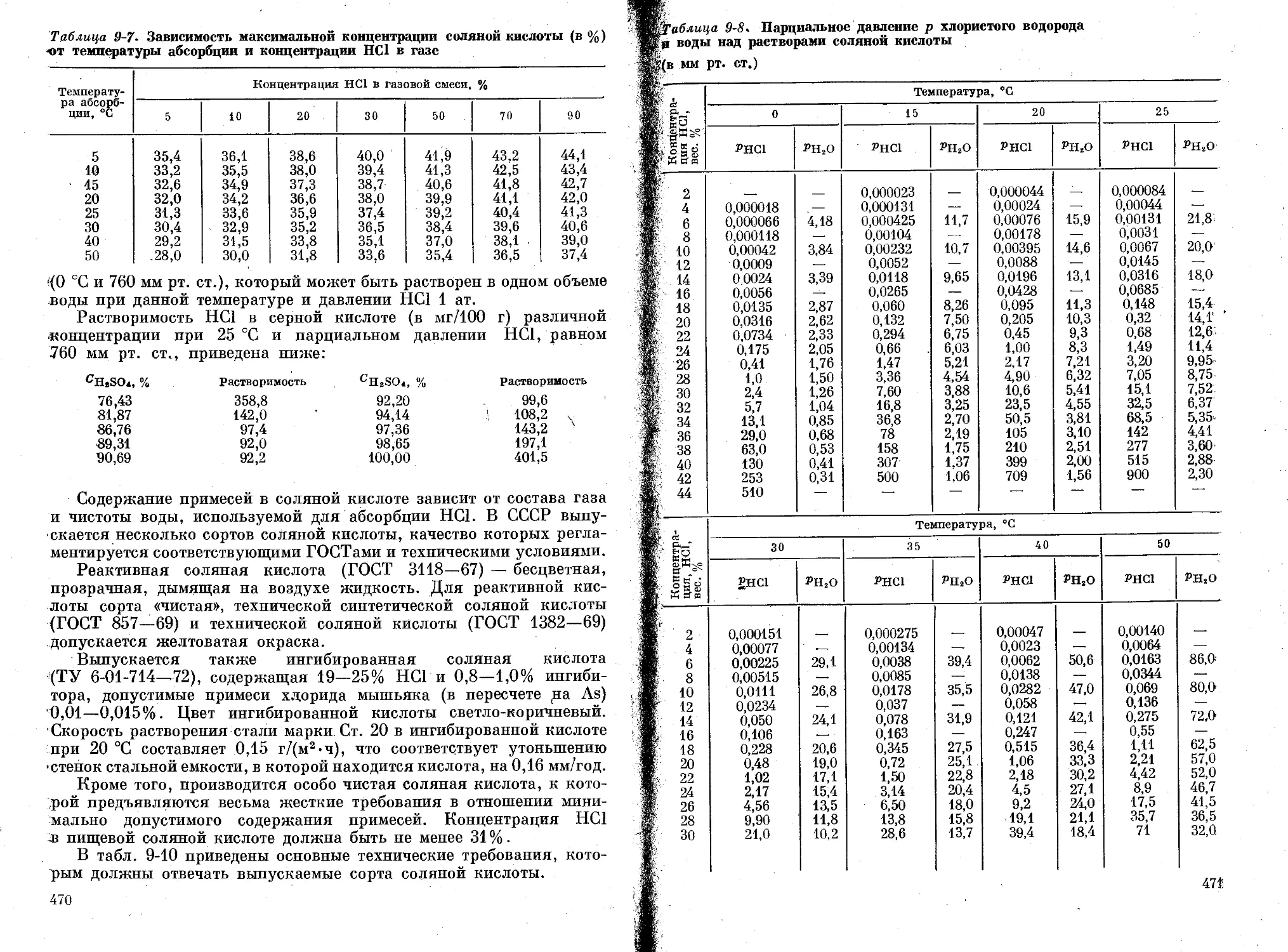
Глава Р. Хлористый водород и соляная кислота..................... 464
Свойства хлористого водорода.................................... 464
Свойства соляной кислоты.................... . . ............. . 469
Производство хлористого водорода и соляной кислоты . . .......... 479
Способы получения хлористого водорода........................... 480
Сульфатный метод получения хлористого водорода ............ 480
Синтез хлористого водорода из элементов . ’............... 483
Попутный или абгазный хлористый водород ................... 487
Способы получения соляной кислоты .............................. 492
Изотермическая абсорбция................................... 493
Адиабатическая абсорбция.................................. 495
Получение 100%-ного хлористого водорода путем стриппинг-процесса 502
Переработка абгазного хлористого водорода, сильно разбавленного па-
рами воды..................................................... 505
Получение жидкого хлористого водорода............................ 510
Материалы, стойкие в среде хлористого водорода и соляной кислоты . . 511
Литература .............................................. 512
*
Глава 10. Производство хлоридов. Канд. техн, наук Фурман А. А. . . 515
Хлористый алюминий ............................................. 515
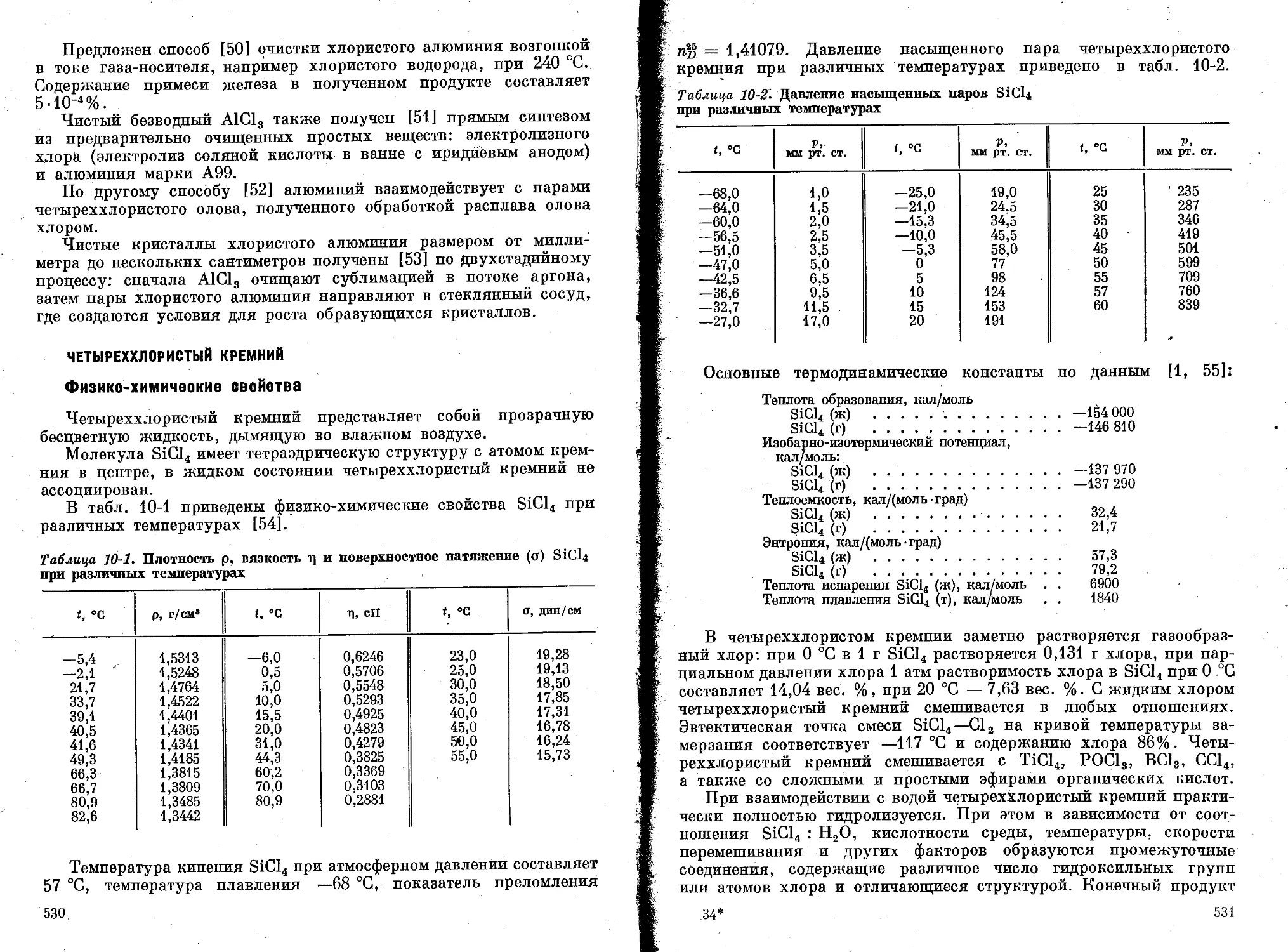
Четыреххлористый кремний ....................................... 530
Четыреххлористый титан ....................................... 543
Хлориды фосфора............................................... . 559
Хлорное железо ....................................... 567
Хлористый цинк ....................................... 572
Литература .......................................... 576
Предметный указатель . . . ............................... 583
ПРЕДИСЛОВИЕ
Быстрое развитие хлорной промышленности связано в основном
с расширением производства хлорорганических продуктов — ви-
нилхлорида, хлорорганических растворителей, инсектицидов и др.
Хотя доля неорганических хлорпродуктов в общем потреблении хлора
сравнительно невелика, их значение для промышленности и народ-
ного хозяйства трудно переоценить.
В Советском Союзе за последние десятилетия создано и продол-
жает развиваться производство многих неорганических хлорпро-
дуктов.
Увеличивается производство жидкого хлора, хлоридов алюми-
ния, кремния, титана, железа, цинка и хлоридов других металлов,
применяемых в менее широких масштабах. Развивается производство
хлоратов натрия, магния и калия, вырабатываются в значительных
количествах хлораты , кальция и перхлораты щелочных металлов
и аммония.
Серьезные технические и экономические проблемы возникают
в связи со значительным увеличением количества хлористого водо-
рода, получающегося в качестве отхода в ряде производств органи-
ческих и неорганических хлорпродуктов. Заслуживает большого
внимания проблема рационального использования абгазного хло-
ристого водорода, в частности получение из концентрированных
и разбавленных растворов соляной кислоты чистого 100%-ного НС1
для применения его в ряде процессов органического синтеза и окси-
хлорирования.
Литературы по производству неорганических хлорпродуктов
крайне мало. В последние годы издано несколько инженерных моно-
графий, посвященных производству хлора, каустической соды и не-
которых неорганических хлорпродуктов. Так, с участием автора
и под его редакцией вышли книги по производству хлора и каусти-
ческой соды Методом электролиза с диафрагмой, а также с ртутным
катодом, по подготовке и очистке рассола для электролиза, по химии
и технологии получения безводных хлоридов металлов, методам по-
лучения жидкого хлора. Однако по многим производствам — хло-
ристого водорода и соляной кислоты, хлоратов натрия, калия,
кальция, магния, перхлоратов и хлорной кислоты, водных раство-
ров хлоридов железа, алюминия и некоторых других продуктов —
I
нет литературы, в которой были бы систематизированы последние
достижения в области их технологии. Кроме того, монографии по
отдельным видам технологии производства хлора, каустической
соды и хлорпродуктов не могут заменить книгу, охватывающую весь
комплекс этих производств. *
Такие монографии по технологии хлора, каустической соды и
хлорпродуктов были изданье в основном в 30-х года! и, естественно,
в значительной степени устарели.
1 Предлагаемая книга является попыткой восполнить существу-
ющий пробел. Она рассчитана на широкий круг инженерно-техни-
ческого персонала промышленных предприятий, работников научно-
исследовательских и проектных институтов, а также работников
системы управления промышленностью. Как нам кажется, книга
может быть полезна студентам и преподавателям вузов и техникумов,
специализирующимся в области химической технологии.
Глава 10, посвященная изложению технологии получения хлори-
дов металлов, написана канд. техн, наук А. А. Фурманом.
Автор приносит глубокую благодарность И. Д. Модылевской
' за большую помощь в написании книги, полезные советы и участие
в обсуждении многих вопросов, возникающих при работе над кни-
гой. ‘
Естественно, что не все вопросы, поднятые в книге, освещены
одинаково полно.
Все критические замечания и указания о недостатках будут при-
няты автором с благодарностью.
Автор
4
ГЛ А В A 1
РАЗВИТИЕ ПРОИЗВОДСТВА И ПОТРЕБЛЕНИЯ ХЛОРА,
КАУСТИЧЕСКОЙ СОДЫ
И НЕОРГАНИЧЕСКИХ ХЛОРПРОДУКТОВ
Если щелочи давно были известны человечеству, то элементарный
хлор люди стали сознательно применять сравнительно недавно,
хотя многие соединения хлора ими уже давно, и широко использо-
вались.
Первое практическое применение хлора связано с отбеливающим
его действием. Для целей отбелки и дезинфекции нашли применение
водные растворы гипохлорита калия — КОС], получившие назва-
ние «жавелевой воды», и водные растворы гипохлорита натрия.
5 В тот же период была приготовлена хлорная известь, производство
которой начало широко развиваться в XIX и первой полЬвине
XX века в ряде стран. Хлорная известь использовалась не только
для нужд дезинфекции и санитарии, но и для хлорирования воды
и отбеливания в целлюлозной и текстильной промышленности.
Во второй половине XX века в связи с применением жидкого хлора
и других химикатов для отбеливания и обработки питьевой воды
4 и канализационных стоков во многих странах производство хлорной
извести резко сократилось.
По мере развития химической промышленности расширяется
ассортимент хлорпродуктов, разрабатываются способы получения
и организуется производство большого числа неорганических и ор-
ганических хлорсодержащих веществ: гипохлоритов кальция, на-
трия и лития, соляной кислоты, хлоратов и перхлоратов, хлоридов
алюминия, цинка, железа, титана, кремния, фосфора и других эле-
ментов, используемых в качестве катализаторов в химических син-
тезах, как полупродукты в производстве ряда химических товаров,
как коагулянты при очистке питьевой воды и канализационных
стоков. *
, г Большое значение приобрело производство хлорсодержащих
полимерных продуктов, в частности поливинилхлоридных смол и
хлоропренового каучука, а также хлорсодержащих растворителей
(дихлорэтана, перхлоруглеродов, трихлорэтилена и продуктов хло-
рирования метана — четыреххлористого углерода, хлороформ^, хло-
ристого метилена и хлористого метила). Значительная масса хлора
расходуется на получение хлорпроизводных бензола, многочислен-
ных средств защиты растений от вредителей и болезней и других
хлорпродуктов.
*
9
7
А
Гэды
Рис. 1-1. Мировое производство
хлора и каустической соды:
1 — хлор; 2 — каустическая сода.
Хлор в больших количествах используется также при производ-
стве некоторых продуктов, не содержащих его в окончательном
виде. В производствах сульфонола, этиленгликоля, глицерина
в качестве промежуточных продуктов применяются такие клорсо-
держащие соединения, как хлоркеросин, хлористый аллил. При
получении конечного продукта хлор выводится из системы в виде
хлоридов металлов (NaCl, СаС12) или хлористого водорода. В по-
следнее время наблюдается тенденция к вытеснению хлорных мето-
дов получения этих продуктов и замене их бесхлорными, так как
при этом резко уменьшается коли-
чество сточных вод.
В конце XIX в. были разработаны
и предложены промышленные ме-
тоды получения хлора и каустиче-
ской соды электролизом водных рас-
творов хлоридов щелочных метал-
лов по способу как с твердым като-
дом и диафрагмой, так и с ртутным
катодом. Первые промышленные
установки электролиза водных рас-
творов хлорида натрия возникли в
Европе и Северной Америке в 1892 г.
Хлорная промышленность за по-
следние десятилетия во многих
индустриальных странах мира раз-
вивалась более быстрыми темпами,
чем вся промышленность в целом. Среднегодовые темпы роста (в %)
общепромышленного производства продукции химической и хлор-
ной промышленности за период 1950—1970 гг, для некоторых стран >
приведены ниже: >
Общепро- Химическая Хлорная
мышленное промышлен- промышлен-
производство . ность ность
США ...................................... 4,1 7,3 8,25
ФРГ ...................................... 7,2 11,7 10,9
Франция ............................... 5,9 9,1 13,5
Италия . . ............................ 8,0 11,3 12,9
Это объясняется тесной связью хлорной промышленности с нефте-
химической, с производством полимерных материалов и другими
отраслями новой техники. Динамика роста мирового производства
хлора и каустической соды приведена на рис. 1-1 [1—3]. С 1962
по 1970 г. общее мировое производство хлора удвоилось и к 1980 г.
ожидается дальнейшее удвоение выработки хлора |4].
По объему производства хлор занимает 5-е место среди основных -
неорганических продуктов, уступая по масштабам производства
только серной кислоте, аммиаку, кальцинированной и каустической
соде.
10
В табл. 1-1 и 1-2 приведены данные о развитии производства хлора
и каустической соды в основных капиталистических странах за
последние 20 лет [3—21].
I
Таблица 1-1. Мировое производство хлора и производство его
в крупнейших капиталистических странах
Производство, тыс. т.
Страна 1950 1960 1965 и 1970 1971
Мировое производство . . . .
США .....................
Япония....................
ФРГ ......................
Англия .............. . . .
Франция ..................
Италия ...................
Канада ...........* . . . .
3200 8400 14 000 20 000
1891 4207 5 878 8 850 8474
17 617 1 246 2 380
219 658 1 081 1 725
310 563 792 - । ' -
82 331 588 1 027
45 292 578 — 1»
131 291 520 774 781
Таблица 1-2. Мировое производство каустической соды и производство ее
в ^крупнейших капиталистических странах
Производство, тыс. т
1965 1966 г 1967 1968 1969 1970
1971
Страна
1960
Мир 11 200 15 700 17 100 18 400 19 900 21 200 22 600
- США 4 510 6 200 6 900 7 188 7 983 8 726 9 070 10 207
Япония 840 1 304 1 398 1 648 1 911 2 272 2 674 2 889
ФРГ 776 1 178 1 303 1 424 1 503 1 541 1 725 2 000
Англия .... 840 870 810 830 820 860 1 000
Франция . ... 597 672 762 876 968 1 042 1 094 1 178
Италия .... 426 702 713 801 832 870 950 1 022
Канада .... 338 583 650 731 844 856 951|
Нижё показана динамика роста производства каустической и
кальцинированной соды (в тыс. т.) в нашей стране [22, 23]:
Год
1960
1965
1966
1967
1968
1969
1970
Na2COs 95%-ная
1887
2871
2963
3169
3292
3392 (100%)
3489 (100%)
NaOH 92%-ная]
765
1303
1392
1524
1599
1669 (100%у
1783 (100%:
Уровень развития хлорной промышленности характеризуется
производством хлора и каустической соды на душу населения страны.
11
г. приведены ниже:
Примерные данные о производстве хлора и каустической соды в мире
(в кг) на душу населения в 1966
Хлор
Мировое производ-
ств©
США
Канада
ФРГ
31,4
21,4
Каустическая
сода
Япония
5,15
34,7
35,6
Нидерланды
Франция
Италия . .
Хлор
. . 15,0
. . 14,5
. . 14,5
. . 13,5
. . 13,1
Каустическая
сода
14,9
15,8
16,0
15,9
16,0
I
хлорорганических продуктов, получаемых
При производстве
заместительным хлорированием углеводородов, не более 50% хлора,
затраченного на производство, входит в состав конечного продукта.
Остальное количество хлора выделяется в виде так называемого
абгазного, или попутного, хлористого водорода. При производстве
такого рода органических хлорпродуктов образуются большие
количества абгазной соляной кислоты и возникает проблема рацио-
нального ее использования. Большие количества абгазного хлори-
стого водорода выделяются также при дегидрохлорировании (на-
пример, при получении винилхлорида из дихлорэтана, трихлор-
этилена из тетрахлорэтана и др.).
Некоторое количество абгазной соляной кислоты непосредственно
после ее получения или очистки потребляется народным хозяйством,
однако основная проблема заключается в переработке избыточной
соляной кислоты в концентрированный хлористый водород и исполь-
зовании последнего для целей гидрохлорирования в производствах
хлористого винила, хлористых этила и метила, хлоропренового
каучука и других продуктов, а также для процессов окислительного
хлорирования, например этилена, пропилена или метана.
Переработка хлористого водорода в хлор разнообразными хими-
ческими методами или электролизом соляной кислоты частично
используется в ряде стран, однако не находит широкого применения
в промышленности из-за экономических соображений. Химические
методы регенерапии хлора и электролиз соляной кислоты приме-
няются в промышленности там, где по местным условиям не могут
быть использованы другие, более экономичные методы переработки
абгазной соляной кислоты. Более подробно вопросы рационального
использования абгазного хлористого водорода будут рассмотрены
ниже.
Химические методы получения хлора из соляной кислоты или
хлоридов (NaCl, NH4C1 и др.) в ближайшей перспективе не имеют
шансов на большое развитие. Однако использование хлористого
водорода для процессов гидрохлорирования и получения дихлор-
этана и других органических хлорпродуктов в процессах оксихлори-
рования этилена, метана и других углеводородов может существенно
сократить потребность в хлоре и обеспечить рациональное использо-
вание побочного хлористого водорода, получаемого в ряде процес-
12
сов, связанных с выделением больших количеств попутного хлори-
стого водорода.
Хлорная промышленность выпускает обширный ассортимент про-
дуктов с самыми разнообразными свойствами и использует много
различных технологических приемов; предприятия хлорной промыш-
ленности обычно объединяют комплекс из большого числа сложных
и разнообразных производств. Поэтому при строительстве крупного
хлорного комбината на базе использования нефтехимического сырья
стоимость цехов, непосредственно относящихся к производству
хлора и каустической соды, обычно не превышает 10—15% общей
суммы затрат на строительство такого комбината.
Основные количества хлора потребляются внутри химической
промышленности для выработки разнообразных хлорпродуктов.
В разных странах для этих целей расходуется от 75 до 90% выраба-
тываемого хлора. Точных данных о распределении хлора и каусти-
ческой соды между отдельными отраслями промышленности и от-
дельными потребителями в различных странах в литературе не опу-
бликовано. Имеющиеся сообщения дают заметный разброс и не-
сколько противоречивы.
Ниже приведены примерные данные о распределении хлора
(в %) между отдельными потребителями в СССР (на 1967 г.):
Химическая промышленность ....
Целлюлозно-бумажная промышленность
Коммунальное хозяйство ............
Цветная металлургия...............
Легкая промышленность . . . . . .
Здравоохранение ...................
Экспорт .......................
Прочие ........................
86,5
5,9
2,7
2,2
0,1
0,2
1,4
1,0
Всего . . . 100,0
В отличие от хлора доля каустической соды, потребляемая в хими-
ческой промышленности для производства различного рода химика-
тов, значительно меньше и составляет обычно в различных странах
от 30 до 40% ее общей выработки. Остальное количество каустиче-
ской соды расходуется в различных отраслях народного хозяйства.
Ниже приведены данные, характеризующие распределение каусти-
ческой соды (в %) между различными потребителями в нашей стране
(в 1968 г.):
тяш
Химическая промышленность .................... . .
в том числе
хлорная промышленность...................... . . .
промышленность красителей и органических продуктов
промышленность химических волокон . . . . . . . .
основная химическая промышленность ........
горнохимическая промышленность . ................
продукция прочих отраслей химической промышленности
Нефтеперерабатывающая и нефтехимическая промышленность
Целлюлозно-бумажная промышленность ..................
Цветная металлургия . . . /.........................
Черная металлургия ... . / . . . ... .... . . .
41,9
7,5
6,2
17,9
0,5
0,1
9,7
12,0
5,8
4,3
2,2
Пищевая промышленность . .
Легкая промышленность . . .
Прочие отрасли промышленности
Очистка воды на электростанциях
Ветеринарные нужды .....
Экспорт ......................
5,7
3,2
18,0
2,8
1,8
2,3
В настоящее время преобладающая роль в производстве хлора
и каустической соды принадлежит электрохимическим методам их
получения по способу электролиза водных растворов поваренной
соли. ' . '
Небольшие количества хлора выделяются в качестве побочного
пРОДукта при электролизе расплавов хлоридов металлов в произ-
водстве магния, натрия, кальция, лития и некоторых других Метал-
лов. Однако масштабы этого производства хлора невелики и целиком
определяются потребностью в перечисленных выше металлах.
В 1968 г. примерно 96,3% хлора производилось электролизом
водных растворов хлоридов щелочных металлов, 0,4% — электроли-
зом соляной кислоты, 2,1% — электролизом расплавов и 1,2% —
химическими методами.
Электрохимическое производство хлора с момента его зарождения
развивалось по двум методам: электролиза с твердым катодом и диа-
фрагмой и электролиза с ртутным катодом. Оба метода имеют свои
преимущества и недостатки. В различные периоды развития хлор-
ной промышленности менялась доля каждого из методов в производ-
стве хлора как в отдельных странах, так/и в мировой хлорной про-
мышленности .
Основным преимуществом метода электролиза с ртутным катодом
является возможность получения каустической соды или едкого_
кали (при электролизе водных растворов КС1) высокой степени чи-
стоты.Если проводить разложение амальгамы очень чистой водой
й предусмотреть хорошую отмывку амальгамы, поступающей из
электролизера в разлагатель, от увлекаемого амальгамой анолита,
то в разлагателе можно получать щелочи реактивной чистоты и даже
особо чистые. Для этого необходимо предохранить получаемую
лочь от загрязнения продуктами коррозии аппаратуры, трубопрово-
дов, емкостей и тары для хранения.
При производстве хлора по методу электролиза с диафрагмой
получается техническая щелочь, содержащая в зависимости от обра-
ботки ее на стадии выпарки 1,5—2,5% хлоридов, примеси сульфатов,
хлоратов, железа и другие загрязнения.
Из различных методов очистки каустической соды, получаемой
по методу электролиза с диафрагмой, промышленное применение
нашел только метод экстракции примесей из щелочного раствора
жидким аммиаком. Очищенная этим способом каустическая сода
по качеству приближается к полученной электролизом с ртутным
катодом, однако несколько уступает ей по чистоте.
Основными преимуществами метода производства электролизом
с диафрагмой является снижение удельных капиталовложений,
и
14
необходимых для организации производства, и более низкая себе-
стоимость получаемой каустической соды.
В качестве сырья в процессе электролиза с диафрагмой могут
быть использованы дешевые естественные или искусственные рас-
солы, получаемые подземным растворением соли. Для электролиза
с ртутным катодом необходима твердая соль для донасыщения обед-
ненного анолита, поэтому затраты на сырье по этому методу значи-
тельно выше, чем по методу электролиза с диафрагмой.
Серьезной народнохозяйственной проблемой является использо-
вание для производства хлора и каустической соды огромных коли-
честд^попутной поваренной соли, выделяемой при производстве
калийных удобрений, при комплексной переработке рассолов соля-
ных озер и в других производствах [24].J
В последнее время привлекает внимание подземное донасыщение
анолита для электролизеров с ртутным катодом и возможность упо-
требления для донасыщения анолита обратной соли диафрагмейного
электролиза. И то, и другое решение может обеспечить электролиз
с ртутным катодом более дешевым исходным сырьем — солью.
Однако эти варианты не нашли еще широкого применения в промыш-
ленности.
При проведении электролиза с ртутным катодом расход электро-
энергии выше, а расход пара ниже, чем при электролизе с диафраг-
мой, так как в последнем случае большое количество пара затра-
чивается на выпарку электролитических щелоков с целью получе-
ния товарной каустической соды. Поэтому районы с дешевой элек-
трической энергией и дорогим паром наиболее выгодны для метода
электролиза с ртутным катодом и, наоборот, в районах с дорогой
электроэнергией и дешевым паром целесообразно развивать электро-
лиз с диафрагмой. Если соль, необходимую для донасыщения ано-
лита в электролизе с ртутным катодом, получают выпаркой рассо-
лов, расход пара на производство приближается к расходу приэлек-
тролизе с диафрагмой.
Серьезным недостатком метода электролиза с ртутным катодом
является необходимость применения больших количеств ртути и
значительные потери ее в производстве, приводящие к загрязнению
атмосферы и сточных вод. Для уменьшения масштабов загрязнения
прдроды ртутными выбросами применяются меры [25], однако они
связаны со значительными материальными затратами [26].
Оба метода электролиза зародились и получили применение
в промышленности практически одновременно, тем не’менее длитель-
ное время развивался преимущественно метод электролиза с диафраг-
мой. С расширением промышленности искусственных и синтетических
волокон возникла потребность в значительных количествах чистой
каустической соды. Для удовлетворения этой потребности стал уси-
ленно разрабатываться метод электролиза с ртутным катодом, пре-
имущественное развитие он получил в последние 15—20 лет.
В настоящее время доля ртутного метода в мировом производ-
стве составляет 53,8%, а метода с^диафрагмой — 42%. Изменение
15
Таблица 7-Л. Соотношение различных методой производства
каустической соды в СССР (ориентировочно)
•и
Годы
Объем
продукции,т
В том числе
Соотношение методов, %
в электролизе
в каустификации
каустифи-
кация, %
известко-
вый
с диа-
фрагмой
электро-
лиз, %
С катода™ Ферритный
1950 1960 1965 1970 1972 320 367 745 864 1199 000 1 783 000 1 898 400 73,0 79,5 85,7 89,0 89,2 27,0 20,5 14,4 11,0 10,8 1 98,8 82,8 79 . 67,5 65,2 1,2 17,2 21 32,5 34,8 ч 77,5 77,3 41,0 40,2 1 22,5 22,7 59,0 59,8
4 доли ртутного метода в общем производстве хлора приведено н
В последнее время доля метода с ртутным катодом в общем произ-
водстве хлора и каустической соды возрастает. При общем большом ^
росте производства по методу электролиза с диафрагмой доля егхи
несколько снизилась. В табл. 1-3 при-
ведены данныеоб- изменении соотно-
шения методов производства каустиче-
ской соды (каустификация и электро-
лиз) и соотношения ртутного и диафраг-
менного методов электролиза.
Затраты, необходимые на строитель-
ство и эксплуатацию установок по
очистке каустической соды, получен-
ной электролизом с диафрагмой, на-
столько ее удорожают, что она ста-
новится неконкурентной с чистой
каустической содой, полученной элек-
тролизом с ртутным катодом, как по
размеру капиталовложений, так и по
продукта. Поэтому очистка каустиче-
не получила
Годы
Рис. 1-2.
Доля метода
с ртут-
ным катодом в общем произ-
водстве хлора:
1 — мировое производство; 2 ~—
производство в СССР.
себестоимости получаемого
ской соды, получаемой электролизом с диафрагмой
широкого применения в промышленности.
Быстрому развитию метода электролиза с ртутным катодом в по-
следние десятилетия способствовали также сильные изменения в тех-
нике производства в направлении интенсификации процесса, укруп-
нения единичной мощности и усовершенствования конструкции
электролизеров, улучшения показателей их работы, снижения удель-
ной загрузки ртути и уменьшения ее, безвозвратных потерь.
Особое значение приобрело применение так называемых метал-
лических анодов. Все эти новые технические решения существенно
16
\ улучшили технико-экономические показатели метода электролиза
с ртутным катодом.
Хотя техника электролиза с диафрагмой также развивалась
и совершенствовалась, однако в последние 20—30 лет в этой области
не было такого прогресса, какой наблюдался в методе электролиза
с ртутным катодом. Поэтому технико-зкономические показатели
обоих методов производства несколько сблизились.
Однако по-прежнему удельные капиталовложения и себестоимость
каустической соды и хлора для метода электролиза с ртутным като-
дом значительно выше, чем для метода электролиза с диа-
фрагмой.
5 Ограниченность ресурсов ртути, а также наличие потребителей, •
не предъявляющих высоких требований к чистоте каустической соды,
обусловили значительное развитие производства хлора и каусти-
ческой соды на ближайший период по методу электролиза с диа-
фрагмой.
-Масштаб производства хлора и каустической соды методом элек-
тролиза с ртутным катодом определяется потребностью народного
хозяйства в чистой каустической соде. Остальное количество хлора
й каустической соды целесообразно получать по методу электролиза
с диафрагмой.
Сейчас, в связи с применением металлических анодов в электро-
лизерах с диафрагмой, открылись широкие возможности конструиро-
вания мощных фильтр-прессных электролизеров с диафрагмой и би-
полярным включением электродов. Первые сообщения [27—29]
о промышленных конструкциях электролизеров такого типа произ-
водительностью по хлору 30—70 т/сут свидетельствуют о том, что
в ближайшие годы можно ожидать новых технических решений
в области промышленного электролиза с диафрагмой и резкого
улучшения технико-экономических показателей этого метода произ-
водства.
Диафрагменный метод производства хлора является доминиру-
ющим в двух крупнейших промышленных странах мира СССР и США.
Доля его в этих странах составляет 65—80%.
В настоящее время Около 7,5 млн. т хлора производится на заво-
дах, использующих метод производства с диафрагмой. В ряде стран,
прежде всего в СССР, США, Японии [30] и некоторых странах на-
родной демократии намечаются к строительству и строятся новые
крупные цеха для производства хлора и каустической соды по ме-
тоду электролиза с диафрагмой. В последние годы диафрагменный
метод производства получил некоторое распространение даже в та-
ких странах как ФРГ, где до последнего времени преимущественно
применялся метдд электролиза с ртутным катодом. Это объясняется
в значительной степени повышением требований органов санитар-
ного надзора по ограничению выбросов ртути в атмосферу и водные
бассейны.
Наиболее экономичным является комбинирование обоих методов
производства с подземным выщелачиванием соли и использованием
2 Заказ 843
17
обратной соли, получаемой при выпарке электролитических
и
щелоков
диафрагменного способа в ртутном способе производства.
При комбинировании обоих методов производства возникают
трудности с очисткой обратной соли от амальгамных ядов. Однако
эта задача в настоящее время успешно решается.
В предприятиях, строящихся по комбинированному методу,
целесообразно в первую очередь вводить производство по методу
электролиза с диафрагмой с выпуском продукции в объеме 55—60%
общей производительности, а во вторую очередь — производство
по методу электролиза с ртутным катодом, используя для донасыще-
ния анолита обратную соль после электролиза с диафрагмой. Про-
дукция, полученная при электролизе с ртутным катодом, должна
составлять не более 40—45% общей производительности.
Метод электролиза расплавленной поваренной соли на движу-
щемся свинцовом катоде с получением хлора и каустической соды
не доведен до промышленной реализации и не имеет на это перспек-
тив в ближайшее время.
На заре развития хлорной промышленности, когда потребность
в хлоре была ограничена, основным продуктом являлась каустиче-
ская сода. Ограниченность сбыта и потребления хлора сдерживала
возможное развитие электрохимического способа производства. Од-
нако в связи с организацией производства большого ассортимента
разнообразных хлорпродуктов открылись возможности для при-
менения огромного количества хлора. Основа развития хлорной
промышленности — все растущий спрос многих отраслей промышлен-
ности и народного хозяйства на хлор и различные хлорсодержащие
продукты.
Производство хлора и каустической соды развивается в основном
по пути электролиза водных растворов NaGl. Электролиз раство-
ров КС1 используется весьма ограниченно, что объясняется неболь-
шой потребностью промышленности и народного хозяйства в едком
кали.
Получаемый в качестве побочного продукта водород частично
цспользуется на хлорных^заводах для синтеза|хлористого водорода
и некоторых^других продуктов. Однако при использовании водорода
для. каталитического гидрирования и других процессов необходима
его тщательная очистка, особенно при производстве водорода элек-
тролизом с ртутным катодом.
Производство каустической соды химическими методами непре-
рывно сокращается и в настоящее время в большинстве стран уже
не имеет серьезного значения. Потребность в хлоре растет'быстрее;
чем в каустической соде [31], и в ближайшие годы можно ожидать
полного вытеснения химических методов производства каустической
соды.
Имеются работы по электролизу растворов поваренной соли
с получением кальцинированной соды вместо каустической [32],
приведены также отдельные примеры реализации этого процесса
в промышленности.
18
. .b
'• 4.
ill
• В литературе было опубликовано много сообщений об ожидаемом
избытке каустической соды и необходимости переработки ее в каль-
цинированную соду [31, 33] и о разработке методов производства
хлора, не связанных с одновременным получением каустической соды.
Однако эти предположения пока не находят подтверждения в прак-
тике. В настоящее время на рынке химических продуктов не ощу-
щается избытка каустической соды, а наоборот, сообщается о сокра-
щении ее запасов до минимума за последние годы и значительном
< росте цен на каустическую соду [30, 34].
В литературе описано много работ и патентов в области электро-
лиза с ионообменными диафрагмами с получением чистой и концен-
трированной каустической соды без применения ртутного катода
[35, 36]. Однако эти работы не доведены до разработки про-
мышленной конструкции электролизера и внедрения в промыш-
; ленность. Имеются лишь сообщения о строительстве в Японии
опытной установки с ионообменными. мембранами для получения
> хлора и чистого едкого натрия производительностью по хлору
4400 т/год [37].
Процессы электролиза растворов хлоридов щелочных металлов
< с ионообменными диафрагмами достаточно хорошо изучены в лабора-
тории и дальнейшее развитие этого метода в настоящее время лими-
тируется отсутствием диафрагм, пригодных для создания крупных
промышленных электролизеров. Применяемые для этой цели ионо-
обменные мембраны не обладают 100%-ной селективностью, что
не позволяет получать столь же чистую каустическую соду, как
и по методу электролиза с ртутным катодом. Без разработки мембран
с достаточно высокой селективностью нельзя рассчитывать на успеш-
ное использование этого метода.
Перспективы применения в промышленности процесса электро-
- лиза с ионообменными диафрагмами будут ограничены более сложной
конструкцией электролизеров, повышенным расходом электроэнер-
гии на проведение процесса и более низким качеством каустической
соды по сравнению с ртутным методом [38].
В связи с тем, что спрос на хлор и хлорпродукты растет быстрее,
чем на каустическую соду, в последнее время вновь возник интерес
к разработке и реализации в промышленности способов получения
хлора, не связанных с одновременным получением каустической
соды. Разрабатываются различные химические методы получения
хлора окислением хлористого водорода, регенерацией хлора из
хлористого аммония, электролизом соляной кислоты.
За последние 5—10 лет в промышленности получили практическое
применение два новых метода производства хлора — электролизом
соляной кислоты [39, 40] и из хлористого аммония (нашатыря) [41].
Эти методы получения хлора не связаны с одновременным выделе-
нием каустической соды, в качестве сырья в них используются трудно
реализуемые отходы производства хлорорганических продуктов
и кальцинированной соды. Однако эти методы имеют небольшой
удельный вес в общем производстве хлора.
s*
I '
19
i
Для электролиза соляной кислоты разработаны конструкции
биполярных электролизеров фидьтр-прессного типа [42] на нагрузку
до 10—12 кА с числом ячеек до 40 [43]. Установки для электролиза
соляной кислоты оборудованы в ряде стран [44]. Для снижения
напряжения при электролизе предложено добавлять к электролиту
соли палладия [45], а также соли меди и железа с деполяризацией
катода путем подачи кислорода [46]. Разрабатывается также электро-
лиз НС1 в расплаве смеси хлоридов щелочных и щелочноземельных
металлов [47, 48] с целью снижения напряжения на ячейке примерно
до 1,45 В против 1,8—2,0 В, необходимых при электролизе водных
растворов. Электролиз соляной кислоты для регенерации хлора ив
попутного хлористого водорода находит применение в ФРГ, США,
Японии и других странах. Однако даже в такой стране как ФРГ,
где электролиз соляной кислоты нашел наибольшее применение/
доля его в общем производстве хлора составляет около 4% [4].
Серьезным конкурентом электрохимических методов производства
хлора в ближайшей перспективе могут явиться методы окислитель-
ного гидрохлорирования различных органических продуктов с по-
мощью хлористого водорода, подучаемого в качестве побочного про-
дукта. Этот процесс широко используется за рубежом, в настоящее
время он разрабатывается в нашей стране и позволит заменить хлор
в ряде промышленных синтезов.
Тем не менее в ближайшее десятилетие электрохимические методы
производства хлора и каустической соды, по-видимому, сохранят
главную роль [49].
Быстрый рост хлорной промышленности сопровождался развитием
техники производства хлора и каустической соды на всех стадиях
производственного процесс^.
Основные принципы конструирования электролизеров с твердым
и ртутным катодами, разработанные ранее, широко применяются
и сейчас в промышленной практике, но* в уровне техники процесса
электролиза водных растворов поваренной соли и в аппаратурном
оформлении этого процесса в последнее время произошли большие
изменения. Они заключаются в интенсификации процесса эдектро-
лизА за счет повышения электродной плотности тока, укрупнении
размеров электролизеров и другого оборудования, в повышении
компактности, надежности и устойчивости их в работе за счет ис-
пользования новых типов конструкций аппаратов, новых электрод-
ных и коррозионно-стойких конструкционных материалов, разра-
ботки методов оптимизации условий проведения процесса. -
Интенсификация процесса электролиза за счет повышения элек-
тродной плотности тока экономически целесообразна, если она
не сопровождается значительным повышением напряжения на
электролизере и соответственно увеличением расхода электро-
энергии.
Из всех составляющих напряжения на электролизере при повы-
шении плотности тока в наибольшей степени возрастает потеря
напряжения на преодоление сопротивления электролита, поэтому
I •• '
разработка способов снижения этих потерь во многом определяла
степень интенсификации, достигаемую в практической работе.
В методе электролиза с ртутным катодом при горизонтальном
расположении анодов были разработаны способы улучшения отвода
пузырьков выделяющегося хлора из зоны прохождения тока, а также
технически удобные приемы регулирования расстояния между элек-
тродами за счет опускания анодов по мере их срабатывания в про-
цессе электролиза* Это позволило в течение последних 10—15 лет
перейти к использованию в электролизерах с графитовыми анодами
плотности тока 7—10 кА/м2 вместо 2—5 кА/м2 без существенного
увеличения напряжения на электролизере.
Для метода электролиза с твердым катодом и диафрагмой, где
применяется вертикальное расположение анодов, нет рациональных
методов регулирования межэлектродного расстояния. Это обстоя-
тельство, а также отсутствие диафрагмы, приспособленной для ра-
боты с высокими плотностями тока, ограничивало интенсификацию
процесса в электролизерах с твердым катодом. В промышленных
конструкциях электролизеров <с твердым катодом плотность тока
не превышает 1,3—1,5 кА/м2.
Тенденция к укрупнению единичной мощности аппаратов в силь-
ной степени проявляется также и в производстве хлора и каустиче-
ской соды. В электролизерах с ртутным катодом нагрузка выросла
от 15—30 кА в сороковых годах, до 100—150 кА в шестидесятые
годы [50] и в настоящее время достигла 300—500 кА в конструкции
последних электролизеров [51, 52]. Благодаря применению повы-
шенных плотностей тока и разработке новых технических решений
сильно сократилось количество ртути для первоначального запол-
нения электролизеров.
Вместо горизонтальных широко применяются вертикальные кон-
струкции раздагателей, что сокращает необходимый для производ-
ства запас ртути, уменьшает необходимую производственную пло-
щадь и улучшает работу электролизера.
При электролизе с твердым катодом увеличение нагрузки на
электролизере не было таким большим, как в способе с ртутным
катодом, однако за последние 10—15 лет она возросла на электро-
лизерах этого типа от 25—30 до 50—60 кА. До последнего времени
в процессах электролиза с диафрагмой использовались электроли-
зеры типа БГК (в нашей стране) и электролизеры Хукер и Даймонд
(за рубежом). *
Работа по укрупнению электролизеров с диафрагмой продол-
жается. Для создания очень мощных электролизеров такого типа
используются принципы секционных и биполярных конструкций [2].
Свойства графитовых анодов во многом определяли конструкцию
применяемых электролизеров, режим их работы и ряд технических
решений по производству хлора в целом. Поэтому внимание исследо-
вателей и инженеров было направлено на разработку более стойких
с хорошими конструктивными свойствами анодных материалов.
С развитием промышленного производства титана работы по
IE
21
созданию малоизнашивающихся анодов нашли свое практическое
разрешение.
Из многочисленных предложений и вариантов по созданию ано-
дов на титановой основе с нанесенным на нее активным слоем наи-
больший интерес представляют аноды с активным слоем из смешан-
ных окислов рутения и титана, условно называемые металличе-
скими анодами. Такие аноды в последние годы находят большое
применение в промышленности как для метода электролиза с ртут-
ным катодом, так и в электролизерах с диафрагмой [53]. В электро-
лизерах с ртутным катодом такие аноды позволяют увеличить плот-
ность тока до 12—15 кА/м2 без повышения напряжения и даже
с некоторым снижением его против практически имеющегося на
электролизерах с графитовыми анодами. Помимо этого, стабильные
во времени размеры анодов исключают необходимость периоди-
ческого опускания анодов, что позволяет упростить конструкцию
электролизеров.
В электролизерах с диафрагмой применение металлических
анодов позволяет повысить плотность тока до 2—3 кА/м2, обеспечить
стабильный во времени энергетический и температурный режимы
работы электролизера и снизить затраты электроэнергии на произ-
водство при одновременной его интенсификации. Применение метал-
лических анодов облегчает решение конструкции биполярного элек-
тролизера с диафрагмой, открывает новые пути развития электро-
химического метода получения хлора и каустической соды как по
методу с ртутным катодом, так и по способу электролиза с диафраг-
мой.
За последние годы техника подготовительных отделений хлор-
ного производства, а также отделений первичной переработки про-
дуктов электролиза претерпела большие изменения;
Хлорная промышленность все в большей степени переходит к ис-
пользованию дешевого сырья в виде естественных рассолов и рассо-
лов, получаемых подземным растворением соли. Операции подго-
товки и очистки рассола практически на всех крупных заводах пере-
ведены на непрерывный процесс с осветлением растворов в осветли-
телях различных типов. Широкое применение получают осветлители
со шламовым фильтром. Для интенсификации процесса осветления
применяют флокулянты, например гидролизованный полиакриламид.
Для фильтрования рассола используются автоматические насыпные
фильтры или фильтры Келли [54].
Для охлаждения хлора применяются холодильники смешения
или поверхностные титановые холодильники; для перекачки и ком-
примирования — турбокомпрессоры и винтовые компрессоры на
3,5 и 12 кгс/см2. Для очистки от загрязняющих аэрозолей влажный
и осушенный хлор фильтруют через фильтры из стекловолокна,
а также электрофильтры. Остаточное содержание влаги после осушки
составляет 60—100 мг/м3, что снижает скорость коррозионных
процессов на стадии компримирования, транспортирования и исполь-
зования хлора.
22
Применяются схемы двухступенчатого сжижения хлора с коэффи-
циентом сжижения, близким к 0,99.
Разработаны и используются в промышленности мощные системы
выпарки электролитических щелоков для получения товарной каусти-
ческой соды, а также растворов поваренной соли с-целью получения
твердой NaCl для нужд электролиза с ртутным катодом.
Широко применяется автоматизация контроля и управления
технологическим процессом на отдельных стадиях производственной
схемы. От автоматизации отдельных стадий и узлов производства
хлорная промышленность переходит к комплексной автоматизации
контроля и управления цехами и комплексами цехов хлорных
заводов [55]. Есть сведения об использовании аналоговых и цифро-
вых вычислительных машин для управления отдельными стадиями
производственного процесса [56] и работой комплекса^производств
хлора и продуктов на его основе [55].
ЛИТЕРАТУ|РА
1. Якименко Л. М., Хим. наука и пром., 3, № 4, 424 (1958); 16, № 6
691 (1971).
2. Я к и м е н к о Л. М. Электролизеры с твердым катодом. М., «Химия»,
1966.
3. Кореньков Л. Г. и др., Основные направления и пропорции развития
неорганических и органических химических производств в капиталисти-
ческих странах. СЭВ. Информация о научно-техническом сотрудничестве.
Приложение. Т. 19. М., 1970. См. с. 91.
4. Н a s s К., Chem. Ing. Techn., 43, № 4, 149 (1971).
5. OPDR (Oil Paint and Drug. Rep.), 196, № 11, 4, 27 (1969); 197, № 6,' 4,
17 (1970).
6. Chemicals, 14, № 2, 5, 17 (1967); 15, № 2, 11; № 3, 7, 12 (1968).
7. Химическая промышленность за рубежом, НИИТЭХИМ, № 8, 40, 75 (1967);
№ 6, 72, 76; № 7, 88; № 12, 28 (1968); № 2 , 86 (1970).
8. Chem. Eng. News, 44, № 49, 30 (1966); 45, № 1, 69; № 2, 20 (1967); 46,
№ 37, 94 (1968); 47, № 52, 98 (1969).
9. Chem. Ind., 19, № 6, 377 (1967); 20, № 6, 400 (1968); 21, № 8, 529 (1969);
22, № 4, 235 (1970).
10. Chem. Age, 97, № 2482, 9; № 2488, 18; № 2506, 23 (1967); 98, № 2569,
25 (1968).
11. M i 1 n e r G. R., J ef.f ery- T. C., J. Electrochem, Soc., 117, Яг 10,
353 (1970).
12. M isui waniec M., Nowak H., Chimik, 23, Яг 10, 375 (1970).
13. Can. Chem. Proc., 52, Яг 9, 9 (1968); 53, Яг 9, 43 (1969).
14. Chem. Market Abstr., 59, Яг 6, 437, 530, 545, 560 (1967); 60, Яг 5, 510, 630,
642 (1968).
15. Chim. et ind., 97, Яг 9, 1415 (1967); 103, Яг 7, 753 (1970).
16. Inform, chim., Яг 62, 32, 80 (1968); Яг 71, 157 (1969).
17. Chem. Ind. Intern., №1,1 (1969).
18. Rev. prod, chim., 70, № 1353, 34; № 1358, 308 (1967); 71, № 1371,313 (1968).
19. Europ. Chem., № 7, 5 (1967).
20. Иоффе Б. С., С а с c-T и с о в с к и й Б. А., Якименко Л. М.,
Вестн. техн, и эконом, информ., НИИТЭХИМ, Я^ 3, 63 (1962).
21. Пасманик М. И., С а с с-Т исовскии Б. А., Якимен-
ко Л. М., Производство хлора и каустической соды. Справочник. М.,
«Химия», 1966.
22. Промышленность СССР. М., «Статистика», 1964.
23. СССР в цифрах в 1967. М., «Статистика», 1968.
23
24. Резник Б. Г;, Экономика химической промышленности, НИИТЭХИМ,
№ 9, 50 (1970).
25. R о s е n z*w е i g N. D., Chem. Eng., 78, № 5, 70 (1971); Gardi-
ner W. С., M u n о z F., Chem. Eng., 78, № 19, 57 (1971); Chem. Ind.,
2, 75 (1972). ,
26. Chem. Week, 108, № 8, 75, 76, 78 (1971); Chem. Eng. News, 50, № 7, 15
(1972); Europ. Chem. News, 22, № 514, 27; № 515, 26; № 516, 30 (1972). -
27. Chem. Week, 106, № 14, 45 (1970).
28. Chem. Eng., 77, № 7, 56 (1970).
29. OPDR (Oil Paint and Drug. Rep.), 197, № 17, 26 (1970); Chem. Eng. News,
48, № 47, 32 (1970).
30. Chem. Econom. Eng. Rev., 3, № 5, 28 (1971); Japan Chem. Week, 12, № 594,
4; № 595, 1; № 597, 1 (1971); 14, № 667, 21 (1973); Chem. Week, 109, № 4,
22, 24 (1971); Jeffery T. C., Fiihrm eist er L. C.,J. Electrochem.
Soc., 118, № 10, 265 (1971).
31. Chem. Eng., 74, № 23, 128 (1967).
32. Бельг, пат. 737255 (1970); пат. США 3431193 (1969); канад. пат. 842774
(1970); франц, пат. 2102742 (1972).
33. Japan Chem. Week, 10, № 453, 5; № 456, 2; № 461, 2 (1969).
34. Europ. Chem. News, 17, № 415r 10; № 416, 6 (1970); 19, № 470, 4 (1971);
Chem. Econom. Eng. Rev., 4, № 2, 33 (1972).
35. К a d e n H., S c h w a 1 e K., Chem. Techn., 19, № 2, 87 (1967).
36. Англ. пат. 961199 (1964); 1199952 (1970); 1270375 (1972); пат. США
2967807, 3017338 (1961); 3135673 (1964); 3496077 (1970); 3654104 (1972);
Йанц. пат. 2035402 (1971).
em. Week, 110, № 24, 39 (1972); Chem. Eng., 79, № 14, 51 (1972).
38. Minagawa M.t J. Electrochem. Soc. Japan, 36, № 1, 60 (1968).
39. Wihnaker K*, Kiichler L., Chem. Techn., Bd. I, Anorganische
Technologie, Munchen, 1970, p. 228.
40. Hass K., Chem. Ing. Techn., 40, № 12, 557 (1968).
41. Japan Chem. Week, 6, № 273, 2 (1965); 10, № 461, 3; 10, № 456, 2 (1969);
пат. США 3627471 (1971).
42. Пат. ГДР 52350 (1966); пат. ФРГ 1085575 (1967).
43. Chem. Ind:, 21, № 6, 399 (1969).
44. Japan Chem. Week, 6, № 255, 3 (1965).
45. Пат. США 3336209 (1967).
46. Англ. пат. 1140481 (1969); пат.-США 3486334 (1969); франц, пат. 1556981
(1969); Hihe F., Jamakawa К., J. Electrochem. Soc., 116, № 3,
105 (1969).
47. Япон. пат. 7006685 (1970).
48. Jochizawa S., J. Electrochem. Soc. Japan, 36, №1, 56 (1968); J o-
chizawa S., Denki Kogaku (News Letters), 2, № 1, 3 (1968).
49. Trans. Inst. Chem. Eng., 46, № 2, 38, 54 (1968).
50. Волков Г. И. Производство хлора й каустической соды методом электро-
лиза с ртутным катодом. М., «Химия», 1968. '
51. Sommers Н. S., Chem. Eng. Progr., 61, № 3, 94 (1965); Electrochem.
Techn., 6, № 3—4, 124 (1968); Chem. Age India, 21, № 11, 1013 (1970).
52. D e Nora O., Chim. e. ind., 50, № 6, 624 (1968).
53. De Nora O., Inform, chim., № 68, 67, 69 (1968); №77, 28 (1969);
Chem. Ing. Techn., 42, № 4, 222 (1970).
54. Фурман А. А., Ш p а й б м а н С. С. Приготовление и очистка рас-
сола. М., «Химия», 1966.
55. Ломакин И. Л., Радун Д. В., Ловачев А. Л., Бала-
шов Л. Н., Автоматизация хлорных производств. М., «Химия», 1967.
56. Busing W., Chem. Ing. Techn., 43, № 4, 177 (1971).
Г Л А В A 2
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ ПРОИЗВОДСТВА ХЛОРА
И КАУСТИЧЕСКОЙ СОДЫ ЭЛЕКТРОЛИЗОМ
РАСТВОРОВ ХЛОРИДОВ ЩЕЛОЧНЫХ МЕТАЛЛОВ
СВОЙСТВА ХЛОРА, ЕДКИХ ЩЕЛОЧЕЙ И ВОДОРОДА
Хлор
Хлор входит в VII группу периодической системы элементов»
ат. в. 35,453, мол. в. 70,906, атомный номер 17.
При нормальных условиях свободный хлор — зеленовато-жел-
тый газ с характерным резким и раздражающим запахом. Он сжи-
жается при —34,05 °C, образуя прозрачную жидкость янтарного
цвета, затвердевающую при —101,6 °C и давлении 1 атм. Обычно
хлор представляет собой смесь 75,53% 35С1 и 24,47% 37С1.
Ниже приведены основные физико-химические и термодинамиче-
ские свойства хлора:
Температура, °C
плавления ....................... . .,............ . “101,6
кипения (сжижения) при 1 атм .................. —34,05
Критические константы
температура, °C . . ........................... 144
давление, атм.................................... 76,1
плотность, г/см3.............................. 0,573
Удельный объем, см3/г ............................. 1,745
Плотность, г/л
- сухого газа (при 0 °C и 1 атм)............... . 3,209
насыщенного пара (при 0 °C и 3,617 атм)........ 12,08
жидкого хлора (при 0 °C и 3,617 атм) .......... 1470,6
» » (при 15,6 °C и 6 атм) .......... . 1422,3
твердого хлора при —102 °C, г/см3........... . 1,9
Удельный объем, м3/кг
сухого газа (при 0 °C и 1 атм).................... 0,3116
насыщенного пара (при 0 °C и, 3,617 атм)....... 0,0828
жидкого хлора (при 0 °C и 3,617 атм) .......... 0,00068
Давление даров при 0 °C, атм ..................... 3,617
Вязкость при 20 °C, сПз
газа . ; .......... ......................... 0,0140
жидкого хлора.................................. 0,35
Теплота, кал/г
плавления твердого хлора (при темп, пл.) ..... 22,9
парообразования (при темп, кип.)............. 67,4
Теплопроводность, ккал/(м-ч-°C)
газа при 0 °C ................................. 0,0208
» при 55,5 °C . . . ..........0,0242
жидкого хлора при 30 °C........... . . -. . . ,. . 0,533
25,
Энтальпия, ккал/кг
сухого газа .............................
насыщенного пара
жидкого хлора ..
Энтропия, ккал/(кг-°С)
сухого газа . ........................
насыщенного'пара .....................
жидкого хлора ........................
Показатель преломления при 14 °C .........
Удельная электропроводность при —70 °C,
Ом"1-см"1 ........ f . ...................
Статическая диэлектрическая проницаемость
при —33,2 °C . . .................
при 10 °C ... ........................
Дипольный момент жидкого хлора (от —65 до
18 °C) в . . .........................
129,4
128,7
64,7
0,329
0,312
0,208
1,367
1-10"16
2,048
1,970
0,23
На рис. 2-1 приведена диаграмма Молье для хлора. Примеры
пользования диаграммой Молье:
Is. Жидкий хлор и его насыщенный пар находятся при температуре 10 °C,
парос оде ржание х = 0,2. Состояние этой системы характеризуется точкой А.
В точке А абсолютное давление Р = 5,124 ат, энтальпия i ~ 114,7 ккал/кг,
энтропия S = 1,054 ккал/(кг - 9С), удельный объем V ~ 12,2 л/кг.
2. Состряние хлора, находящегося при 80 9С и абсолютном давлении 1 ат,
характеризуется точкой В. В этих условиях хлор находится в состоянии пере-
гретого пара. В точке В энтальпия I = 174 ккал/кг, энтропия S —
1,302 ккал/(кг'9С), удельный объем V ~ 415 л/кг.
Давление пара хлора при различных температурах приведено
на рис. 2-2. Растворимость хлора в воде и растворах поваренной
соли приведена на рис. 2-3. При охлаждении раствора хлора в воде
до температуры ниже 9,6 °C выпадают желтые октаромбические кри-
сталлы гидрата хлора С12-пН2О (п = 12, 10, 8, 7, 4; по последним
данным, п — 6).
Разложение гидрата хлора происходит по реакции
[С12 • 6Н2О]тв С12газ+ 6Н20жидк
Парциальное Давление хлора над его гидратом возрастает
с по-
вышением температуры:
°C 0 2 4 6 8 9 9,6 10 12 14 16
мм рт. ст. , , . 249 320 398 496 620 701 760 797 992 1246 1520
Предельная температура существования твердого гидрата хлора
при атмосферном давлении равна 9,6 °C. При —0,24 °C и парциаль-
ном давлении хлора 244 мм рт. ст. одновременно существуют четыре
фазы: твердый гидрат, лед, вода, насыщенная хлором, и газовая
смесь (хлор + пары воды). При температуре ниже —0,24
жение гидрата хлора протекает по реакции
°C разло-
[С12*6Н2О]тв С12газ + 6Н20тв
При этом парциальное давление хлора составляет:
/, °C ............... —0,24 —1 —2 —3 —4 —6 —8 —10
Pclj, мм рт. ст.......... 244 234 223 213 203 185 169 156
26
?
&
Абсолютное даб ленив Z7, ат
жл
jS®' /
Рис. 2-1. Диаграмма Молье i — IgP для
хлора
Растворимость гидрата хлора в воде приведена ниже:
Температура, °C
Растворимость,
г/100 г Н2О
Температура, °C
Растворимость,
г/100 г Н2О
—0,24
0
3
6
7
' 0,490
0,507
0,615
0,714
0,869
9
12,5
20
27
28,5
0,908
1,112
1,854
3,690
3,627
В сильно разбавленных водных растворах гидрат хлора гидро-
лизуется с образованием НС1 и HOG1. <
Рис. 2-2. Зависимость
давления паров жидкого
хлора от температуры.
Температура, °C
Рис. 2-3. Растворимость хлора
в воде и растворах хлористого нат-
рия при различных температурах.
При введении соли аммония в водный раствор хлора образуются
треххлористый азот и НС1. Треххлористый азот NG13 образуется
при взаимодействии аммиака или солей аммония с хлором или хлор-
новатистой кислотой:
NH3 + 3C12 —> NC13 + 3HC1 (2.1)
NH4G14-3C12 —> NG134-4HC1 (2.2)
NH4C1+3HOC1—> NC13 + 3H2O + HC1 (2.3)
При взаимодействии хлористого аммония с хлорноватистой
кислотой при pH = 9,5 образуется монохлорамин, при pH — 4,5 и
температуре 32 °C и выше — треххлористый азот, при температуре
ниже 0 °C NG13 не образуется.
Треххлористый азот представляет собой ярко-желтую, масло-
образную жидкость с сильным запахом, пары которой раздражают
слизистую оболочку глаз.
Плотность NG13 равна 1,653 г/см3, мол. в. 120, 366, температура
плавления —40 °C, температура кипения 71 °C. Треххлористый
азот нерастворим в воде, но растворим в бензоле, сероуглероде:
хлороформе, двуххлористой сере, медленно разлагается на рассеян-
ном свету. При хранении под холодной водой разлагается в течение
28
24 ч на азотную и соляную кислоты. Перегоняется без разложения
в воздухе, в среде водорода, кислорода, этилена, но взрывается в среде
-озона, а также при соприкосновении с жирами. При работе с трех-
хлористым азотом необходимо соблюдать соответствующие меры
безопасности (надевать перчатки, защищать лицо щитом).
Если в рассоле, поступающем на электролиз, или в воде, приме-
няемой для охлаждения хлора в холодильниках смешения, присут-
ствует аммиак, соли аммония или амины, NC13 может появляться
как примесь к хлору. При сжижении такого хлора треххлористый
азот практически полностью переходит в состав жидкого хлора.
При испарении жидкого хлора NC13, вследствие высокой темпе-
ратуры его кипения, концентрируется в остатке жидкого хлора и
может послужить причиной сильных взрывов. При полном испаре-
нии жидкого хлора в проточном испарителе опасность концентри-
рования NC13 исключается.
Частые взрывы в аппаратах для очистки ацетилена объясняются
-образованием в них NC13 из хлорной извести и примесей аммиака,
присутствующего в ацетилене.
Газообразный хлор, получаемый электролизом водных растворов
щелочных металлов, должен содержать не менее 96% хлора и не
более 2% СО2 и 1% Н2. Хлор, полученный методом электролиза
с диафрагмой, должен содержать не более 0,5% Н2. Содержание
влаги после осущки не должно превышать 0,04 вес. %.
В последнее время требование к качеству газообразного хлора,
применяемого в синтезе ряда органических хлорпродуктов, сильно
возросли. Содержание влаги в хлоргазе ограничивается 40—100 мг/м3,
снижается допустимое содержание брома, сое^нений серы и других
примесей.
Каустическая сода и едкое кали
Требования к различным сортам технического едкого натра при-
ведены в табл. 2-1 (примеси в жидком продукте даны в пересчете
на 100%-ный продукт) [5].
Выпускаются также две марки улучшенного едкого натра, полу-
чаемого по методу электролиза с ртутным катодом [6], которые
должны удовлетворять приведенным ниже требованиям:
Марка II
Содержание NaOH, %, не менее . .
Содержание примесей, %, не более
Na2CO3 .......................
NaCl . . . . *................
железо (в пересчете на Fe2O3) .
SO4...........................
Са ...........................
Si02 . . . ...................
хлораты (в пересчете на NaCl О 3)
алюминий (в пересчете на А12О3)
Си
Ni
Марка I
45
0,3
0,02
0,001
0,02
0,0014
0,008
0,01
0,003
0,00003
0,00002
0,6
0,05
0,001
0,02
0,0014
0,008
0,01
0,01
0,00005
0,00002
ГЛ51Ш
29
Марка I Марка II
Мп . . . ’........................ 0,00005. 0,00005
Ва ............................... 0,0001 0,0001
Mg ............................... 0,0001 0,0001
РЬ . . . ......................... 0,0002 0,0002
К . . . ......................... 0,03 0,1
Hg .............................. 0,0001 0,0001
Коэффициент светопропускания, % , не
ниже .............. 90 80
Требования к качеству технического едкого
в табл. 2-2 [7].
Таблица 2~1. Требования к техническому едкому натру
кали приведены
Содержание, % Марка А, ртутный Твердый Жидкий
сорт марка Б, диафраг- менный химический, марка марка В; диафраг- менный
1 2
Г Д
NaOH, % , не менее ....
Примеси, %, не более
Na2CO3 ..............
NaCl ................
Fe2O3 ...............
окислы Fe, Al и Мп . .
-SO4 ..............
Са............
металлы группы H2S .
SiO2.................
NaClO3 ..............
алюминий в пересчете на
А12О3
42 0,6 96 2,0 96 3,0 92 2,5 43 2,0 2,5 42 " 2,0
0,05 1,0 1,5 3,5 1,0 2,0 2,2
0,0015 - - — . — -
V 0,03 Не мир вор- уется 0,05 Не нор- мируется
0,02 —* — — 1
0,006 1 — —
0,003 1 — . — -
0,008 “ — ——'— > м
0,01 — « —
0,01 — * в-
техническому едкому кали
Таблица. 2-2? Требования к
Содержание, %
Твердый Жидкий
-2^ Б В Г
Едкие щелочи в пересчете на КОН, не
менее ...........................
Примеси, не более ...............
хлориды в пересчете на С1 . . . .
SO4 .........................
Fe...................... .
NaOH ..............
КС103 ........... ...........
Si ..... ....................
Са . ........................
Al ............... .........
нитраты и нитриты в пересчете на азот
95,0
1,0
0,7
0,05
0,03
2,0
0,2
0,02
0,01
0,005
0,003
93,0
2,0
0,9
0,1
0,05
3,0
0,2
52,0
0,6
0,7
0,03
0,007
2,0
0,2
0,02
0,01
0,005
0,003
50,0
1,0
0,9
0,1
0,01
3,0
0,3
30
Концентрация N аОН, вес. %
Рис. 2-4. Политерма раство-
римости NaOH в воде.
Концентрация КОН , вес. %
Рис. 2-5. Политерма рас-
творимости КОН в воде.
Выпускаются также реактивные и особо чистые едкий натр и едкое
кали.
Политермы растворимости едкого натра и едкого кали приведены
на рис. 2-4 и 2-5.
Водород
Основные физико-химические свойства водорода приведены ниже:
ISI5
Молекулярный вес ......................
Мольный объем (при О °C и 760 мм рт. ст), л
Температура, °C
к
ения
2,016
22,43
—252,8
—259,4
плавления ........................................
Критические константы
температура, °C ........................................—239,9
давление, атм . ..................................... 12,8
плотность, кг/л .....................................0,031
Плотность
при 0 °C и 760 мм рт. ст., кг/м3 ..................... 0,0899
при темп, кип., кг/л .............................. 0,0709
относительная (по воздуху) . ..................... 0,0695
Удельная газовая постоянная, ккал/(кг °C) .......... . 986,96
* Теплота, ккал/кг
плавления .......................................... 14,0
парообразования (при 760 мм рт. ст.) ............ . 108,5
Объем жидкости, образующейся из 1 м3 газа (при 15 °C и
760 мм рт. ст.), л................................... 1,166
Удельная теплоемкость (при 20 °C и 760 мм рт. ст.),
ккал/(кг-°С)
Сп ..................................................3,408
cv ........................ . .................... 2,42
Cp/cv ••••,•........................................, 1,407
Теплопроводность при 0 °C и 760 мм рт. ст., ккал/(м-ч °C) 0,140
Вязкость при 0 °C и 760 мм рт. ст., сП..................0,0085
31
Таблица 2-3* Пределы воспламеняемости
Смесь водорода •ь Предельное содер- жание Н2, % Температура самовоспла- менения, °C Максималь- ная скорость распростра- ' нения пла- мени, см/с
нижнее верхнее
С кислородом . 4,5 95 450 890
с воздухом 4,1 74,2 510 267 X
Пределы воспламеняемости смесей водорода с кислородом и воз-
духом приведены в табл. 2-3.
Скорость распространения взрывной волны [8] при протекании
реакции 2Н2 + О2 = 2Н2О (в м/с):
По опытным данным ........... 2821
По расчетным данным . . . . . 2864
Рис. 2-6. Пределы взрываемости смесей хлора,
водорода и воздуха.
Теплота сгорания водорода с кислородом с образованием воды:
Н2+1/2О2Н20жидк + 68 520 ккал/моль Н2
Н2 + 1/2О2 = Н2Ожидк+3055 ккал/м3 Н2 (при норм, усл.)
Н2 + 1/2О2 = Н2О жидк + 33 990 ккал/кг Н2
и соответственно при образовании пара
Н2-Н/2О2 = Н20парЧ-57 840 ккал/моль Н2
Н2 + 1/2О2^Н2Опар + 2575 ккал/м3 Н2 (при норм, усл.)
Н2 + 1/2О2 — Н2Опар + 28 630 ккал /кг Н2
32
1^4
J '• •-
Водород в смеси с хлором и воздухом также образует взрыво-
опасную смесь. Пределы взрываемости смесей приведены на
рис. 2-6 [9].
J!
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ ЭЛЕКТРОЛИЗА
ВОДНЫХ РАСТВОРОВ
ХЛОРИДОВ ЩЕЛОЧНЫХ МЕТАЛЛОВ
ч
Процесс электролиза водных раотворов хлоридов
щелочных металлов в электролизерах о ртутным катодом
и диафрагмой
При электролизе водных растворов поваренной соли или хлори-
стого калия как в способе с диафрагмой, так и с ртутным катодом
основным процессом на аноде является разряд ионов хлора с выде-
лением газообразного хлора.
На катоде в электролизерах с ртутным катодом происходит раз-
ряд ионов щелочного металла с образованием амальгамы натрия
или калия. При разложении этой амальгамы водой в разлагателях
получают чистые растворы гидроокисей натрия или калия и водород.
В электролизерах с твердым катодом на катоде происходит разряд
молекул воды с выделением газообразного водорода и образованием
соответствующей щелочи. При этом получают смешанные растворы
щелочей и хлоридов. Суммарный процесс для электролиза с ртут-
ным катодом \
11
NaCl + *Hg —> NaHgx + i/2Cl2 (2.4)
• /
и для электролиза с диафрагмой
NaCl + H2O—> i/2H2 + i/2Cl2 + NaOH (2.5)
F ^Теоретическое напряжение разложения Е$ можно определить
исходя из максимальной работы реакции (2.4) и (2.5):
(2.6)
или из теплового эффекта этих же реакций с учетом коэффициента
температурной зависимости \
*
$
*
J
1
Е° nF -0,239 +Т dT <2’7)
где А и Q — максимальная работа и тепловой эффект реакции; п и F — число
г-экв вещества, участвующего в реакции, и число Фарадея. •
Величина Е$ зависит от температуры и при 25 °C теоретическое
напряжение составляет около 2,17 В для электролиза с диафрагмой
и 3,16 В — для злектролиза с ртутным катодом.
Одновременно с _ основными электродными процессами может
протекать ряд побочных и вторичных реакций, приводящих к сни-
жению коэффициента полезного использования тока. При проведе-
нии электролиза водных растворов хлоридов щелочных металлов
3 Заказ 843 33
/
A
одновременно с разрядом ионов СГ” на аноде и выделением газооб-
разного хлора
С1- —► 1/гС12+е
(2-9)
(2.10)
может выделяться также кислород
20Н- > 1/2О2+Н2О + 2е
или
Н20 > 1/2О2 + 2Н+ + 2е
Стандартный потенциал реакции (2.10) равен 1,2296, а реак-
ции (2.8) — 1,358—1,3595 В [10]. Однако перенапряжение выделе-
ния кислорода на обычно применяемых анодных материалах выше,
Рис. 2-7.. Зависимость выхода кислорода по
току от pH для различных типов анодов:
7 — ПТА, 80 °C, 2 кА/м2; 2 — RuO2,80 °C, 10 кА/м2;
3 _ рьОо, 80 °C, 2 кА/м2; 4 — МпО2, 90 VC, 1 кА/м2;
5 — МпО2, 90 °C, 2 кА/м2.
чем у хлора. Поэтому в
условиях электролиза слЦ
/бокислых концентрирован-\
; ных растворов хлоридов 1
I щелочных металлов про- j
исходит преимуществен- ?
ное выделение хлора. При J
электролизе на платино-;
вых анодах соотношение»
потенциалов выделения
хлора и кислорода таково,
что получают практически
чистый хлор [11]. При
электролизе с графито-
выми анодами одновре-
менно с хлором разря-
жается также и кислород.
Количество тока, расходу-
емого на разряд кисло-
рода, зависит от применя-
емого анодного материала и условий проведения анодного процесса.
Обычно оно колеблется от 0,5 до 3,0%.
Исследованы образование ионов СЮз и потери выхода по току
на выделение кислорода в процессе электролиза с диафрагмой на
окисно-рутениевых анодах [11а]. При проведении электролиза с ма-
лой плотностью тока (когда перенапряжение невелико), доля тока,
затрачиваемая на выделение кислорода, возрастает.
Потенциал реакции (2.10) зависит от pH среды, в нейтральном
растворе он снижается до 0,815 В [10], поэтому при повышении
концентрации ионов ОН” или снижении концентрации ионов С1"
изменяется соотношение скоростей их разряда и возрастает доля
тока, затрачиваемая на выделение кислорода. При наличии в элек-
тролите примесей таких ионов, как ОН", СЮ", ClOg, SO4", HSOJ и др.,
возможны значительные потери выхода по току. Особенно сильно
влияют на выход по току примеси ОН" и С1О“.
На рис. 2-7 приведена зависимость выхода кислорода по току
при электролизе концентрированных растворов поваренной соли
(300 г/л) при 90 °C от pH электролита для различных анодов.
34
При использовании графитовых анодов происходит окисление
материала анода выделяющимся кислородом с образованием дву-
окйси^л^ода. Условия, благоприятствующие анодному выделению
кислорода, способствуют усиленному разрушению графитовых ано-
дов. При промышленном электролизе водных растворов хлоридов
(елочных металлов при низком значении pH выделяющийся кисло-
род расходуется в основном на окисление графита. В сильно щелоч-
ных электролитах окисление графитовых анодов почти не происхо-
дит ' [12].
На твердом катоде в щелочном растворе происходит разряд
молекулы воды [13—16]
JI
II
Н2О+е = ОН“ + Н ад (2.11)
Атомы водорода после рекомбинации выделяются в виде молеку-
лярного водорода
2Н ад «—► Н2
(2.12)
Остающиеся в растворе ионы ОН" образуют с ионами щелочного
металла гидроокись этого металла. Разряд ионов щелочных металлов
калия или натрия из водных растворов на твердом катоде невозмо-
жен из-за более высокого значения потенциала их разряда.
Катионы, разряжающиеся на катоде при более положительном
потенциале, чем Н+, обычно присутствуют в католите в ничтожных
количествах (ионы железа, меди и др.) и их можно практически не
учитывать.
Одновременно с разрядом ионов Н* на катоде может происходить
восстановление растворенного в жидкости хлора* а также анио-
нов СЮ" и C1OJ
СЮ- + Н20 + 2е= С1- + 20 н-
(2.13)
C10j+3H20 + 6e==Cl- + 60H- (2.14)
Выделяющийся на аноде хлор частично растворяется в анолите.
С повышением температуры раствора и концентрации в нем поварен-
ной соли растворимость хлора снижается. При растворении хлора
происходит его гидролиз с образованием хлорноватистой и соляной
кислот
С12 + Н2о НС10 + НС1 (2.15)
или
С12 + 0Н- НС10 + С1- (2.16)
S
Константа равновесия этого процесса
[НС10ЯС1-]
[С12][0Н] (2Л/)
или
К1Д [НСЮИС1-ЦНЧ (218)
[С12]
При 25 °C константа Кг = 3,9-10“4.
3* 35
(2.19)
С повышением концентрации ионов С1” и Н+ равновесная кон-
центрация хлорноватистой кислоты снижается.
Хлорноватистая кислота мало диссоциирована. Константа диссо-
циации ее при 25 °C равна
Кг= ^нсю)' =3-7-10~8
Концентрация ионов СЮ", образующихся в анолите вследствие
диссоциации хлорноватистой кислоты, незначительна и не вызывает ;
существенного изменения хода анодного процесса. При попадании
ионов ОН” в анодное пространство происходит нейтрализация хлор-
новатистой кислоты с образованием хорошо диссоциирующего
гипохлорита натрия или калия
нею+он-
(2.20)
или
С124-2ОН- С1О"+СГ + Н2О
(2.21)
*
При этом нейтрализация хлорноватистой кислоты сдвигает
реакцию (2.16) вправо, и новые порции хлора будут подвергаться
гидролизу с образованием хлорноватистой кислоты.
6 процессе электролиза с ртутным катодом попадание ионов ОН“
в анодное пространство возможно при разрядке молекул воды на
к атоде и ли при ^частичном разложении амальгамыв электролизере. "
При электролизе с твердым катодом это происходит вследствие пере-
мешивания анолита с католитом за счет диффузии или электроми-
> грации ионов ОН" из катодного пространства в анодное.
Если все количество щелочи, образующейся на катоде, будет
поступать к аноду, то процесс может быть проведен без выделения
газообразного хлора с получением гипохлоритных растворов. По-
тенциал разряда ионов G1O" более отрицателен, чем ионов С1’г,
поэтому будет происходить совместный разряд ионов С1О“ и OF"
на аноде. В результате образуются ионы хлорноватой кислоты и вы-
деляется кислород
•ч • "
6С10- + ЗН20 = 2С10з+4С1- + 11/202+6Н+ + 6е (2.22)
Образование солей хлорноватой кислоты может происходить
также и химическим путем по реакции:
2HC1O+C1O- = C1OJ + 2C1" + 2H+ (2.23)
Если процесс электролиза проводить без разделения электродных
продуктов, то в зависимости от условий его проведения конечным
продуктом электролиза может быть соль хлорноватой кислоты или
растворы гипохлорита щелочного металла. Процесс электролиза
концентрированных растворов поваренной соли без разделения
электродных продуктов широко используется в промышленности
с целью получения хлората шицхия.
При проведении элеЙТ^Злиза водных растворов хлоридов щелоч-
ных металлов на ртутном катоде происходит разряд ионов натрия
36 .
J
или калия вследствие высокого перенапряжения водорода на амаль-
С гамном катоде и снижения потенциала выделения ионов щелочных
'' | металлов* обусловленного деполяризую действием в результате
образования амальгам этих металлов. Параллельно на амальгамном
\ катоде протекает также процесс выделения водорода. Потенциал
< амальгамного электрода зависит от активности щелочного металла
Й в амальгаме и его ионов в растворе и может быть рассчитан из вы-
ражения
W ' Ф = Фо-^1п-^ф^ (2.24)
°ме*ме
где Фо — стандартный потенциал амальгамного электрода; Сме+, /ме+, Сме,
/Ме — соответственно концентрация и коэффициент активности щелочного
металла в растворе и в амальгаме.
При 25 °C значение <р0 можно принять равным для натрия
—1,8490 В [17], для калия—1,8592 В и для лития —2,0441 В [18].
Значения равновесного потенциала амальгамы натрия в усло-
виях, близких к промышленным, для различных концентраций ще-
лочного металла в амальгаме приведены в табл. 2-4 [1,19].
Таблица 2-4. Равновесные потенциалы амальгамы натрия при 80 °C (в В)
Концентрация,
Nad, л/г
Концентрация натрия в амальгаме, вес. %
0,489 0,398 0,296 0,237 0,203 0,148 0,099 0,051 0,010
300
250
200
150
1,844
1,854
1,867
1,878
1,828
1,840
1,851
1,871
1,805
1,818
1,826
1,844
1,800
1,809
1,822
1,832
1,795
1,806
1,818
1,829
1,774
1,784
1,796
1,808
1,752
1,762
1,773
1,785
1,731
1,743
1,758
1,769
1,680
1,684
1,697
1,710
Перенапряжение водорода на ртутном катоде можно определить
по уравнению Тафеля
r) = a + blni (2.25)
* 41
Значение коэффициента Ъ при 25 °C составляет 0,118, значение а,
рассчитанное по [20], составляет около 1,8 В.
Перенапряжение водорода на амальгамном катоде выше, чем
на ртутном [21, 22]. Точное определение перенапряжения выделения
водорода на амальгамном катоде затруднено сильным влиянием
ряда примесей на процесс выделения водорода. В нейтральном насы-
щенном растворе поваренной соли перенапряжение водорода для
амальгамы концентрацией натрия 0,055 вес. % составляет около
2,1 В, а для амальгамы промышленной концентрации — 2,15 В [23].
При очень малых плотностях тока должен происходить преиму-
щественно процесс выделения водорода^ с повышением плотности
тока и ростов перенапряжения водорода доля тока, расходуемого
на выделение водорода, снижается, поэтому основное значение
37
приобретает процесс разрядки ионов щелочного металла, который про-
ходит практически без перенапряжения. Зная для каждого процесса
зависимость между потенциалом и плотностью тока, можно рассчи-
тать распределение тока между этими процессами. Согласно [23],
при плотности тока 5 кА/м2 и проведении электролиза в чистых усло-
виях доля тока, расходуемая на выделение водорода, в зависимости
от pH электролита должна составлять от 0,01 до 0,04%.
При наличии даже небольших примесей, так называемых амаль-
гамных ядов, доля тока, расходуемая на выделение водорода, в про-
изводственных условиях часто возрастает на один-два порядка.
Действие амальгамных ядов объясняют восстановлением их до
металла и образованием на поверхности амальгамы мест с низким
перенапряжением водорода. К амальгамным ядам относятся металлы
с низким перенапряжением водорода, нерастворимые или мало-
растворимые в ртути и плохо смачиваемые амальгамой. Наибольшим
действием из практически встречающихся ядов обладают ванадий,
хром, германий и молибден [24—31]. В меньшей мере в качестве
катализаторов разложения выступают такие примеси; как железо,
никель, кобальт, вольфрам. Малое влияние на процесс разложения
оказывают примеси кальция, бария, магния и алюминия [32].
Считается, что примеси серебра, свинца, цинка, марганца и меди
не влияют на скорость реакции разложения амальгамы, а примеси
бора, кремния, фосфора и олова могут действовать как ингибиторы
разложения [33, 34].
Добавки некоторых металлов к амальгаме снижают вредное влия-
ние амальгамных ядов. Действие таких добавок, как Zn, Sn, Pb
и др., обусловлено образованием интерметаллических соединений,
связывающих металлы, которые относятся к амальгамным ядам [35].
Отмечены случаи взаимного стимулирования или ингибированного
действия примесей при совместном присутствии нескольких доба-
вок [36, 37]. Наиболее часто в практике встречается ванадий, попа-
дающий в раствор при выщелачивании из графитовых анодов в про-
цессе электролиза, и хром, присутствующий в растворе вследствие
коррозии аппаратуры.
Влияние амальгамных ядов, находящихся в виде примеси в рас-
соле, по-разному сказывается при электролизе растворов NaCl
и КС1, причем в последнем случае влияние амальгамных ядов осо-
бенно сильно. Например, в процессе электролиза растворов КС1
хром влияет на процесс уже в том случае, когда его концентрация
на порядок ниже, чем при электролизе растворов NaCl. Поэтому
при электролизе растворов КС1 содержание водорода в хлоре выше,
чем при электролизе растворов NaCl.
Возможно взаимодействие между амальгамой натрия и хлором,
растворенным в анолите или находящемся в нем в виде мелких пу-
зырьков. По мере сближения анода с катодом одновременно со сниже-
нием потерь напряжения на преодолепйеомического сопротивления
электролита должны возрастать потери выхода по току вследствие
катодного восстановления активного хлора на амальгаме натрия.
38
Зависимость напряжения на электролизере и потерь выхода
nov току от межэлектродного расстояния при различных плотностях
тока была объектом тщательного изучения [38]*. При увеличении
плотности тока можно „уменьшить межэлектродное расстояние до
определенного предела без того, чтобы при этом заметно не снизился
< выход по току из-за процессов катодного восстановления активного
хлора. При применении вертикальных или очень наклонных катодов
процессы катодного восстановления могут сильно возрастать, так
как на поверхности раздела амальгамы и раствора электролита воз-
никает интенсивная турбулизация потока.
Получаемая_jb электролизере, амальгама отводится для разло-
жения в специальный аппарат — разлагатель. Разложение амальгамы
надо рассматривать как электрохимический процесс, в котором на
аноде происходит процесс ионизации натрия Na = Na+ ~h е, а на
катоде разряд молекулы воды с выделением газообразного водорода
Н2О + е - ОН’ + V2H2.
Процесс ионизации натрия протекает без перенапряжения, его .
скорость ограничена только подводом щелочного металла к анодно
раббтайщей поверхности амальгамы. Процесс выделения водорода
на ртути й амальгаме натрия протекает с высоким перенапряжением.
Поэтому скорость разложения водой чистой амальгамы натрия
очень низка [39—45].
Для ускорения процесса разложения щелочных амальгам необ-
ходимо снизить перенапряжение выделения водорода. Это дости-
гается обычно созданием контакта проводника первого рода, име-
ющего низкое перенапряжение для выделения водорода, с амальга-
мой и раствором. Образующийся короткозамкнутый элемент имеет
в качестве анода амальгаму натрия, а в качестве катода — провод-
ник первого рода с низким перенапряжением выделения водорода.
Для того чтобы обеспечить устойчивую длительную работу элемента,
материал катода не должен смачиваться амальгамой натрия. Кроме
того, материал катода не должен в заметном количестве раство-
ряться в ртути и должен быть коррозионностойким в условиях ра-
боты разлагателей промышленных электролизеров. Из большого
числа опробованных материалов только графит нашел применение
в промышленности, хотя поиски других материалов (карбиды титана
и др.) продолжаются. В качестве насадки разлагателя предложен,
например, карбид вольфрама [45а].
Имеются предложения по интенсификации разложения амаль-
гамы путем осаждения на графите осадков металлов, плохо смачи-
ваемых амальгамой и имеющих низкое перенапряжение выделения
водорода [46]. В последнее время в качестве такой добавки был
испытан молибден [47]. Активирование насадки в горизонтальных
разлагателях сопряжено с трудностями, связанными с амальгамиро-
ванием осадков металлов на насадке разлагателя. В разлагателях
вертикального типа условия амальгамирования насадки иные,
поэтому ее удается активировать на 1—2 года пропиткой солями
железа и до 4 лет — солями молибдена [47].
39
Для процесса разложения амальгамы большое значейие имеет
сопротивление в месте контакта графитовой насадки разлагателя
с амальгамой, поэтому в горизонтальных типах разлагателей важна
глубина погружения графитовой насадки в амальгаму: с увеличе-
нием глубины погружения заметно возрастает скорость разложения
амальгамы. При увеличении глубины погружения насадки в амаль-
гаму до 20 мм происходит снижение омического сопротивления кон-
тактов [48]. Закономерности работы разлагателя амальгамы рас-
смотрены в работе [48а].
Первоначально для разложения амальгамы применялись гори-
зонтальные разлагатели, которые состояли из горизонтального
желоба, расположенного с уклоном для обеспечения движения
ртути. В желобе располагалась графитовая насадка. В разлагателях
такого типа скорость разложения амальгамы пропорциональна пери-
метру контакта трех фаз: графитовой насадки, амальгамы и раствора
щелочи. Для улучшения разложения насадку разлагателя выпол-
няют в виде плит из графита, перфорированных продольными па-
зами. По мере интенсификации процесса электролиза возникает
необходимость обеспечить разложение увеличивающегося количе-
ства амальгамы натрия. Эти обстоятельства способствовали созда-
нию и применению вертикальных разлагателей амальгамы.
Если при конструировании горизонтальных разлагателей в те-
чение длительного времени исходили из эмпирических данных, то
для вертикальных типов разлагателей возможно применение общих
закономерностей по аналогии с распространенными в технике наса-
дочными реакционными колоннами [49]. Преимуществом вертикаль-
ных разлагателей является их компактность, обеспечение практи-
чески полного разложения амальгамы и, при правильном инженер-
ном решении, уменьшение удельных загрузок ртути.
Электродвижущая сила амальгамного элемента может быть опре-
делена из выражения:
ill
f
RT ,
—ln
*
aNaOH
aNa
(2.26)
где «NaOH и еда “ активности едкого натра в растворе и натрия в амальгаме;
1,021 — стандартное значение э. д. с. амальгамного элемента, равное разности
стандартных потенциалов амальгамного и водородного электродов, В.
Значения э. д. с. амальгамного элемента при 70 °C и концентра-
ции натрия в амальгаме 0,2 вес. % для различных концентраций
щелочи "приведены ниже 150]:
J.
Концентрация щелочи, % ......... 10 20 30 40 50 60 70
Э. д. с., В ........ 1,00 0,90 0,80 0,70 0,62 0,57 0,53
С ростом концентрации щелочного металла в амальгаме увеличи-
вается э. д. с. элемента и облегчается подвод натрия к поверхности
раздела фаз, что способствует увеличению тока разложения амаль-
гамы.
40
1
II
II
При повышении концентрации щелочи происходит сильное сни-
жение э. д. с. В зоне низких концентраций щелочи это явление ча-
стично компенсируется увеличением электропроводности раствора
с ростом концентрации. Однако при концентрации NaOH выше
400 г/л и температуре около 100 °C одновременно с понижением
э. д. с. элемента наблюдается повышение электрического сопротивле-
ния и вязкости раствора, что приводит к значительному уменьшению
скорости разложения.
Повышение температуры мало сказывается на значении э. д. с.
элемента, но способствует увеличению электропроводности раствора,
снижению перенапряжения выделения водорода и вязкости раствораг,
что в результате приводит к ускорению процесса разложения амаль-
гамы,’ скорости которого зависит также от конструктивного оформле-
ния разлагателя.
Многократные попытки [51—54] использовать энергию амаль-
гамного элемента для снижения расхода электроэнергии в процессе
электролиза не нашли реализации в промышленности. Это объяст
няется трудностями, обусловленными разницей между выходами
по току в катодном и анодном процессах в амальгамном элементе.
Поэтому для предотвращения анодного растворения ртути в амаль-
гамном элементе необходимо регулировать силу тока, проходящего
через элемент.
Кроме того, превращение разлагателя в амальгамный элемент
связано с возникновением сопротивления во внешней цепи элемента,
снижением плотности тока разложения и. температуры процесса
(за счет полезного использования части энергии разложения амаль-
гамы, которая ранее целиком превращалась в тепло). Все эти фак-
торы приводят к необходимости увеличения размеров разлагателя
и требуемого количества ртути. Продолжаются поиски путей исполь-
зования энергии разложения амальгамы с применением элементов,
в "которых используются катоды с кислородной деполяризацией
процесса выделения водорода [55—58].
Предложен в качестве катода скелетный катализатор из активи-
рованного никеля [59—61]. Известно о проведении испытания на
сравнительно небольших опытных установках. На установке с на-
грузкой 3,5 кА достигнуто использование энергии амальгамы на
33—35%. Считается возможным при плотности тока 2—4 кА/м2
получить напряжение на ячейке 1,2—1,5 В [55—61].
Разделение продуктов, получающихся на электродах
В процессе электролиза с целью получения хлора и каустической,
соды необходимо разделять продукты, получающиеся на электродах.
При электролизе с ртутным катодом разделение анодных и катодных
продуктов осуществляется благодаря тому, что разложение амаль-
гамы и получение каустической соды и водорода проводятся
дельном аппарате — разлагателе. При электролизе с твердым
дом необходимы специальные меры для разделения катодных и
ных продуктов.
В ОТ'
като-
анод-
41
Пористая перегородка, погруженная в жидкость, обеспечивает
разделение образующихся в процессе электролиза водорода и хлора,
предотвращает возможность механического перемешивания като-
лита и анолита за счет конвекционных потоков и в сильной степени
снижает попадание щелочи в анодное пространство вследствие диф-^
фузии. Однако такая диафрагма не может исключить иди уменьшить
попадание щелочи в анодное пространство за счет участия ионов ОН”
в переносе тока. Перенос щелочи в анодное пространство при не-
подвижном электролите резко возрастает, а выход по току сни-
жается с ростом концентрации щелочи в катодном пространстве.
При повышении концентрации щелочи до 45—50 г/л выход по току
падает до 60—70%.
Пористые диафрагмы для электролиза с неподвижным электро-
литом применялись в нескольких конструкциях электролизеров
в конце прошлого и в первые десятилетия XX столетия.
Предотвращение потерь выхода по току за счет электролитиче-
ского переноса ионов ОН“ к аноду не могло быть обеспечено без
создания принципиально нового способа разделения продуктов
электролиза.
Если обеспечить равномерное по всему сечению электролизера
движение электролита от анода к катоду со скоростью, равной или
большей скорости электролитического переноса ионов ОН", то будут
устранены попадание ионов ОН^ в анодное пространство (за счет
их участия в переносе тока) и связанные с этим снижение выхода
по току и ускоренный износ графитовых анодов.
Чтобы избежать смешения газов, выделяющихся на электродах,
а также механического перемешивания анолита с католитом, прин-
цип противотока следует сочетать с применением пористой диа-
фрагмы или другими средствами предотвращения перемешивания
жидкостей и газов.
Различают четыре способа использования принципа противотока
электролита для разделения электродных продуктов: а) с колоколом;
б) с газозащитными оболочками на катоде^ в) с одной фильтрующей
диафрагмой; г) с двумя фильтрующими диафрагмами.
В способе с колоколом [62] достигается расслоение католита
и анолита вследствие большей плотности католита по сравнению
с анолитом. Анод располагается внутри колокола, а катоды — сна-
ружи за его стенками. При подаче рассола в анодное пространство
осуществляется постоянное движение электролита от анода к ка-
тоду и на некотором расстоянии под анодами устанавливается гра-
ница раздела щелочного католита и кислого анолита.
Разделение электродных продуктов этим способом имеет ряд не-
достатков. Небольшая нагрузка на один колокол ограничивает воз-
можность увеличения мощности электролизера. Несмотря на при-
менение малой плотности тока (около 0,3 кА/м2) из-за большого
расстояния между электродами и низкой рабочей температуры на-
пряжение на электролизере сравнительно велико и составляет
3,7—4,2 В. Разделение электродных продуктов с помощью колокола
42
JC
и противотока электролита было использовано в нескольких типах
электролизеров, однако упомянутые недостатки способствовали
вытеснению его другими методами.
В способе с газозащитными оболочками на катоде сохранен ч
в основном принцип разделения, применяемый в способе с колоколом,
однако катоды располагаются непосредственно под анодами, что(7
позволяет избежать ряд неудобств, связанных с размещением като-
дов на внешней стороне колокола. Для предотвращения перемеши-
вания католита с анолитом пузырьками поднимающегося водорода
катоды помещают в оболочки или желоба с наклоном в одном напра-
влении от 1 : 50 до 1 : 25 для отвода собирающегося там водорода.
В электролизерах с газозащитными оболочками плотность тока
достигала 0,5 кА/м2 и нагрузка на электролизер — 3,6 кА. Этот
способ был использован в электролизерах Биллитер-Лейкам [63]
и Песталоцци. Однако электролизерам с газозащитными оболочками
присущи, хотя и в меньшей степени, недостатки электролизеров
с колоколом, поэтому в настоящее время они не имеют промышленного
применения.
Способ с одной фильтрующей диафрагмой. Радикальное решение
вопроса о разделении электродных продуктов при электролизе вод-
ных растворов хлоридов щелочных металлов на твердом катоде
было получено при применении способа с фильтрующей диафрагмой.
Этот способ разделения электродных пространств электролизера
в настоящее время является практически единственным, применя-
емым в промышленности.
К проточным диафрагмам предъявляются требования достаточ-
ной механической прочности, химической стойкости к продуктам
электролиза, равномерности толщины, плотности и протекаемости
по всей площади диафрагмы, малого значения электролитического
сопротивления, доступности и дешевизны. Необходимо, чтобы проте-
каемость диафрагмы сохранялась стабильной или, если это невоз-
можно, менялась незначительно в течение длительного времени
работы.
В промышленности применяются асбестовые диафрагмы, проте-
каемость и электрическая проводимость которых зависит от их пори-
стости. Однако при одной и той же пористости диафрагма может
обладать различной протекаемостью и удельным электрическим Д
сопротивлением в зависимости от размера и характера пор. Р
Свойства диафрагмы зависят от диаметра и длины пор. Поскольку
асбестовые диафрагмы не имеют регулярной структуры, размеры
пор диафрагмы изменяются в широких пределах и приходится рас-
сматривать их среднее значение.
Пористость диафрагмы d можно рассчитать, зная истинную ри
и кажущуюся рк плотности образца асбестовой диафрагмы
d = l—-рк/Ри (2.27)
Средняя длина пор Z, средний их радиус г (в см) и общее сечение
пор q (в см2) можно определить из результатов электроосмотических
43
измерений, скорости протекания жидкости через диафрагму и дан-
ных по электрическому сопротивлению диафрагмы и пропитываю-
щего ее раствора.
Гидростатическое давление Р, уравновешивающее электроосмо-
; j тический перенос раствора плотностью р че^рез диафрагму, опреде-
•U ляется из выражения
р_
г2л'
Скорость v протекания раствора (в см3/с) через диафрагму при
том же приложенном напряжении будет равна
(2.28)
(2.29)
(2.30)
V = -7
где £ — потенциал границы раздела, В; Е — приложенное напряжение, В;
е — диэлектрическая постоянная раствора; ц — вязкость раствора, сП.
Из уравнений (2. 28) и (2. 29) получаем:
Коэффициент увеличения электрического сопротивления диа-
фрагмы К по сравнению с сопротивлением чистого раствора может
быть определен экспериментально. Он связан со средней
пор I и общим их сечением q выражением
K-ljq
Из выражения (2.30) с учетом уравнения (2.31) получаем
длиной
(2.31)
Масса раствора g, пропитывающего диафрагму, равна
Из уравнений (2.31) и (2.33) получаем
(2.33)
(2.35)
Вместо длины пор иногда применяют коэффициент извилистости
пор р, представляющий собой отношение длины пор I к толщине
диафрагмы:
Р = 1/Ь (2.36) -
Протекаемость диафрагмы может быть определена иэ выражения
(2.37)
’ г
44
, где F—объем протекающей через диафрагму жидкости, см3; F— площадь
: диафрагмы, дм2; Н— гидростатическое давление жидкости, см вод. ст.; t —
1 время, ч; ц — вязкость раствора, сП; К — коэффициент протекаемости.
Коэффициент протекаемости диафрагмы численно равен количе-
J ству жидкости (в см3), прошедшей через диафрагму площадью 1 дм2
W и толщиной 1 см за 1 ч при гидростатическом напоре 1 см вод. ст.
Ж ^и вязкости жидкости 1 сП.
Электрическое сопротивление диафрагмы равно сопротивлению
электролита, заполняющего ее поры, и для 1 см2 площади диафрагмы
определяется из выражения
Рд= (2:з8)
О
где р — удельное электрическое сопротивление электролита, заполняющего
поры диафрагмы, Ом • см.
Диффузия продуктов электролиза через диафрагму определяется
из выражения
Fd(ct — c2)Dt
g d₽2
(2.39)
где F — площадь диафрагмы, см2; сх — с2 — разность концентраций вещества
по обе стороны диафрагмы, г-экв/л; t — время, сут; D — коэффициент диффу-
зия, см2/сут.
Коэффициент диффузии для хлоридов и гидроокисей К и Na
приведен в табл. 2-5.
Таблица 2-5* Коэффициент диффузии D при 18 °C
Концентрация,
г-экв/л
NaCl
NaOH
КС1
КОН НС1
0,5
1,0
1,077
1,070
1,310
1,330
’1,841
1,885
2,217
диффузии при других температу-
Для определения коэффициента
рах можно воспользоваться выражением
В процессе электролиза большое значение
протекаемости диафрагмы по всей ее. площади,
диафрагмы с неравномерной плотностью по
(2.40)
и
L
имеет равномерность \ ।
В случае применения
ее площади, скорость
движения электролита будет в разных частях поверхности диафрагмы
различна. В местах повышенной плотное™ диафрагмы протекаё-
мость ее будет ниже средней величины. Если на каком-либо участке
диафрагмы скорость движения электролита будет меньше скорости 4
движения ионов ОНГ к аноду, в этих местах возникнут условия для
переноса ионов ОН?“ в анодное пространство. Последнее вызовет
снижение выхода по току и ускоренное разрушение графитовых
анодов.
Применяемые на практике асбестовые диафрагмы не отличаются
идеальной равномерностью протекания по всей площади. Поэтому
практически достигаемые выходы по току ниже теоретически воз-
можных.
Скорость протекания электролита через диафрагму зависит
от давления фильтрации. При горизонтальном расположении диа-
фрагмы давление при фильтровании жидкости через диафрагму будет
одинаковым по всей площади диафрагмы.
При вертикальном расположении диафрагмы в случае незаполнен-
ного катодного пространства давление фильтрации увеличивается
от верхних точек диафрагмы к нижним почти линейно с высотой.
Некоторое отступление от линейности обусловлено неодинаковым
газонаполнением анолита по высоте. В электролизерах с заполненным
катодным пространством зависимость давления фильтрации от вы-
соты более сложная.
В процессе работы протекают сложные физико-химические
процессы взаимодействия асбестовой диафрагмы с электролитом,
происходит набухание волокон асбеста, сжатие и деформация под
влиянием давления. Кроме того, на диафрагме могут отлагаться
твердые частички графита, гидроокиси магния и железа, карбоната
кальция, а также продукты хлорирования масла, используемого
для пропитки анодов. Эти процессы приводят к изменению свойств
диафрагмы [64—66].
Протекаемость диафрагмы изменяется особенно сильно в первое
время после начала фильтрования. В зависимости от условий в период
от нескольких дней до 1—2 нед. .происходит так называемое форми-
рование диафрагмы, после чего ее свойства в течение длительного вре-
мени остаются примерно постоянными. При этом вследствие набухания
волокон свойства диафрагмы медленно меняются [67]. В результате
протекания процессов изменения структуры асбестовой диафрагмы
и постепенного загрязнения ее графитовым шламом, гидроокисями
магния, железа, солями кальция и другими примесями протекаемость
диафрагмы уменьшается и по истечении определенного времени
становится недостаточной для поддержания концентрации щелочи
в допустимых пределах. Чтобы избежать резкого снижения выхода
по току, необходимо произвести смену или регенерацию диафрагмы.
Для регенерации диафрагму промывают водой. Предложена
также промывка ее кислотой [68], однако при этом возможно раз-
рушение диафрагмы и коррозия стальных деталей электролизеров.
Для увеличения срока службы и восстановления оптимальной про-
текаемости диафрагм предложено обрабатывать их оксикарбоновыми
кислотами [69], а также добавлять к рассолу поверхностно-активные
добавки, например изопропиловый спирт, дизтиленгликоль, зтил-
целлозольв и др. [70].
Несмотря на длительное использование асбестовых диафрагм
в практике промышленного производства хлора и каустической
соды, процессы, происходящие при длительной эксплуатации таких
диафрагм, изучены еще недостаточно.
46
Протекаемость жестких диафрагм увеличивается с ростом давления
фильтрации в очень широких пределах.
Исследования, проведенные Лифатовой, Кришталиком и др,
[64—66], показали, что протекаемость свежей асбестовой диафрагмы
после ее формирования увеличивается с ростом давления фильтра-
ции только в пределах изменения давления от 0 до примерно 300 мм
жидкостного столба. При дальнейшем повышении давления протекае-
мость практически более не растет.
При увеличении давления на уже сформировавшуюся диафрагму
ее протекаемость на короткий срок возрастает, а затем постепенно
снижается и принимает значение, нормальное для увеличенного
давления. Если диафрагма сформировалась прц определенном давле-
нии и затем переведена на работу при более низком давлении, ее
протекаемость оказывается ненормально низкой. С другой стороны,
при изменении направления фильтрации жидкости через диафрагму
ее фильтрующая способность увеличивается.
Специфическая зависимость протекаемости асбестовых диафрагм
от давления связана с деформацией волокон асбеста под влиянием
давления фильтрации, что приводит к снижению пористости диа-
фрагмы, уменьшению среднего диаметра пор и изменению коэффи-
циента протекаемости. Деформация волокон асбестовой диафрагмы под
влиянием давления фильтрации в основном имеет неупругий харак-
тер. Это подтверждается зависимостью протекаемости диафрагмы
от максимального давления фильтрации, приложенного ранее к диа-
фрагме. При упругих деформациях протекаемость диафрагмы целиком
э определялась бы приложенным в данный момент давлением фильтра-
ции и не зависела' бы от ранее испытанного диафрагмой давления.
Если в диафрагме волокна асбеста жестко связаны друг с другом,
как, например, в абсолатексной диафрагме, деформация асбестовых
волокон затруднена и протекаемость этих диафрагм зависит, так же
как и жестких, от давления фильтрации в широком интервале давле-
ний.
При изменении плотности тока протекаемость уже сформировав-
шейся диафрагмы меняется. Строгой зависимости между этими вели-
чинами не наблюдается, поэтому изменение плотности тока приводит
к изменению концентрации щелочи в католите [71]. Еслиплотность
тока меняется по высоте диафрагмы, то влияние плотности тока
на протекаемость в различных точках диафрагмы будет проявляться
по-разному. При этом протекаемость диафрагмы может отличаться
от ее протекаемости при равномерном распределении плотности тока.
При вертикальном расположении асбестовой диафрагмы неравно-
мерность протекания рассола по высоте может наблюдаться в верхней
части диафрагмы как в случае работы с пустым, так и с заполнен-
ным катодным пространством.
В электролизерах с незаполненным катодным пространством
протекаемость диафрагмы, а соответственно и концентрация щелочи
в католите по высоте катода, будет изменяться только в верхней
части катода, где давление фильтрации на диафрагму возрастает
47
от 0 до 300 мм столба жидкости. В нижней части катода протекае-
мость диафрагмы и концентрация щелочи в католите не изменяются
с высотой.
Схема изменения давлешш фильтрации и концентрации щелочи
в католите по высоте катода для электролизера с вертикальной диа-
к
фрагмой и незаполненным катодным пространством приведена на
рис. 2-8, а. Для упрощения принято, что вся диафрагма по высоте ч
Давление фильтрации, мм жидкостного столба *
Концентрация щелочи в католите, г/л
Рис. 2-8. Изменение" давления фильтрации и концент-
х рации щелочи в католите по высоте катода:
а — незаполненное катодное пространство; б — полностью
заполненное катодное пространство; в — не полностью заполо-
ненное катодное пространство; 7 — давление фильтрации;
2 — концентрация щелочи в католите.
разделена на две резко отличающиеся друг от друга зоны; зону
с линейной зависимостью протекаемости от давления и зону, где
протекаемость практически не зависит от давления фильтрации.
В действительности зависимость протекаемости от давления
более сложная и переход от одной зоны к другой происходит посте-
пенно, с отклонениями от линейной зависимости.
С учетом принятого упрощения концентрация щелочи С на раз-
личной высоте диафрагмы в зоне переменной протекаемости может
быть определена из выражения
С = С0 + К1
(2Л1)
где CQ — концентрация щелочи в зоне постоянной протекаемости, г/л; Z —
расстояние точки определения от нижней точки зоны переменной протека-
емости, см.
1 >
Коэффициент К зависит от условий опыта, в среднем его величина
может быть принята в пределах 5—6 г/(л-см).
Накопление в диафрагме большого количества осадка гидро-
окиси магния, солей кальция и других прймесей может существенно
изменять ее механические свойства и зависимость протекания от да-
вления. По своим свойствам такая диафрагма приближается к жест-
ким диафрагмам. Распределение концентрации щелочи по высоте
• ъ
SI
катода на такой диафрагме может быть очень неравномерным, что
приводит к снижению выхода по току. При промывке асбестовой
диафрагмы водой восстанавливаются ее обычные свойства.
' В верхней части диафрагмы протекаемость понижена, а концен-
трация щелочи повышена. Здесь скорость фильтрации рассола может
быть ниже скорости электролитического переноса ионов ОН“.
Процесс электролиза на этой части катодной поверхности будет
идти с пониженным выходом по току, что снизит общий средний
выход по току для электролизера в целом.
\ ' Неравномерность протекаемости диафрагмы по высоте и связан-
ное с этим изменение концентрации щелочи могут быть устранены,
если поддерживать уровень анолита над верхним краем катода не
ниже 300 мм. При этом вся Поверхность диафрагмы будет работать под
давлением фильтрации более, высоким, чем 300 мм столба жидкости,
т. е. в режиме, при котором протекаемость диафрагмы более не зави-
сит от давления фильтрации. ,
Равномерная протекаемость рассола по всей высоте вертикально
расположенной диафрагмы может создаваться при условии полностью
, заполненного жидкостью катодного пространства. При этом в пер-
вом приближении можно считать, что давление фильтрации по всей
высоте диафрагмы одинаково и равно высоте столба анолита над
верхним краем катода или над уровнем католита, если последний
находится выше верхнего края катода. Тогда протекаемость всей
площади диафрагмы может легко регулироваться в определенных
Ч1 пределах изменением давления фильтрации за счет превышения
уровня анолита над католитом от Одо 300 мм столба жидкости. В дей-
ствительности равенство давления фильтрации по высоте диафрагмы
может быть нарушено из-за различия плотностей анолита и католита.
Плотность католита выше, чем анолита, поэтому в электролизе-
рах с заполненным катодным пространством давление фильтрации
может снижаться от верхних к нижним частям диафрагмы. При
большой высоте электродов, высокой концентрации щелочи в католите
и малом давлении фильтрации в верхней части диафрагмы возможно
снижение давления фильтрации в нижней части диафрагмы до нуля
и даже изменение направления давления, т. е. будет происходить
фильтрация католита в анодное пространство.
Соотношение плотностей анолита и католита зависит от их газо-
наполнения.
Газонаполнение жидкости в анодном пространстве электроли-
зера целесообразно снижать, чтобы избежать излишних потерь
напряжения на преодоление омического сопротивления анолита.
Газонаполнение католита может быть увеличено без опасения вы-
звать при этом дополнительный рост напряжения на электролизере.
Повышая с помощью различных конструктивных особенностей
катода газонаполнение католита, можно уравнять плотности газо-
наполненных жидкостей в анодном и катодном пространствах элек-
тролизера и избежать изменения давления фильтрации по высоте
диафрагмы. Это обстоятельство имеет особое значение при большой
А
( 4 Заказ 843 49
высоте электродов и работе с высокими концентрациями щелочи
в католите.
Однако в большинстве конструкций электролизеров, рассчитан-
ных на работу с заполненным катодным пространством, последнее
заполнено не полностью для облегчения отвода водорода. Уровень
католита в катодном пространстве обычно ниже верхнего края катода
и диафрагмы на 40—100 мм. При этом выше уровня католита диа-
фрагма работает как в электролизере с незаполненным катодным
пространством. В этой части диафрагмы давление фильтрации и
щелочность католита изменяются с высотой и становятся постоян-
ными только для той части диафрагмы, которая находится нижо
уровня жидкости в катодном пространстве.
Схема распределения давления фильтрации и концентрации
щелочи по высоте диафрагмы для электролизера с заполненным
катодным пространством приведена на рис. 2-8, б и в. Принято,
что вследствие различного газонаполнения жидкостей в катодном
и анодном пространстве их кажущиеся плотности равны между
собой.
При работе электролизера с заполненным катодным пространством
зона переменной протекаемости и соответственно переменной щелоч-
ности католита меньше по сравнению с электролизером с незаполнен-
ным катодным пространством. Если для последнего эта зона соста-
вляет около 300 мм (по высоте), то в электролизерах с заполненным
катодным пространством она равна разности между верхним уровнем
катода и уровнем католита. Это . позволяет работать с более высоким
средним выходом по току при одинаковой концентрации щелочи
в катодных щелоках.
Применение заполненного катодного пространства позволяет
регулировать протекаемость диафрагмы по мере ее старения путем
изменения уровня анолита и иметь более концентрированные катод-
ные щелока.
Наиболее благоприятные условия работы создаются для диа-
фрагмы при полностью заполненном катодном пространстве. Если
разница между верхним уровнем катода и уровнем католита дости-
гает 300 мм, электролизер работает как с незаполненным катодным
пространством.
Электролизеры с фильтрующей диафрагмой чувствительны к пере-
рывам тока. Помимо изменения протекаемости, выключения тока
и перерывы в работе могут приводить к ржавлению стального катода.
При последующем включении электролизера продукты коррозии,
пропитывающие диафрагму, могут восстанавливаться до металли-
ческого железа, проникающего от катода через диафрагму в анодное
пространство. Металлизированная диафрагма начинает работать
как катод, что приводит к загрязнению хлора водородом и попаданию
щелочи в анодное пространство. Аналогичные процессы протекают
при нарушении целостности диафрагмы. При длительных остановках
электролизеров катодное пространство для предотвращения коррозии
заполняют щелочным раствором.
t-
50
При горизонтальном расположении электродов и диафрагмы
в качестве фильтрующего слоя могут быть использованы порошко-
образные материалы. При вертикальном расположении диафрагмы
такая возможность исключается и применяются асбестовые диа-
фрагмы, сформированные тем или иным методом. Горизонтальные
диафрагмы применялись в широко распространенных в 20—40-х гг.
электролизерах Сименс-Биллитера [72].
Горизонтальная диафрагма в некоторых пределах автоматически
регулирует свою протекаемость. По мере формирования, забивки
пор и увеличения сопротивления диафрагмы прохождению через
нее жидкости происходит повышение уровня жидкости в анодном
пространстве до тех пор, пока давление фильтрации не будет доста-
точным для обеспечения нужной протекаемости диафрагмы. После
повышения уровня анолита выше предельно допустимого следует
поменять диафрагму.
При вертикальном расположении электродов и диафрагмы при-
меняют диафрагму из асбестовой бумаги или картона, либо так
называемую осажденную диафрагму, формируемую непосредственно
на катоде под вакуумом из взвеси асбестовых волокон в соляно-
щелочном растворе. Для электролизеров с малоизнашивающимися
анодами предложена диафрагма из волокнистого материала, заклю-
ченного между двумя сетками [73], а также насосная диафрагма
с добавками латекса [73а].
Для изготовления асбестовых диафрагм обычно применяются
щелочестойкие сорта хризотилового асбеста состава 3MgO*2SiO2*
-2Н2О. G целью повышения кислотостойкое™ диафрагмы при ее
приготовлении предложено вносить добавку антифилитового асбе-
ста [74]. Обычно в составе асбеста имеются примеси железа и некото-
рых других элементов. Для изготовления асбестовых тканей приме-
няются длинноволокнистые сорта, для асбестовой диафрагменной
бумаги и осажденных диафрагм — коротковолокнистые сорта.
Технология изготовления асбестовой диафрагменной бумаги ана-
логична технологии бумаги и картона. После сухого размола, уда-
ления пыли и загрязняющих его пород, асбест подвергается мокрой
обработке в роллах. Взвесь асбестовых волокон с добавками клеющих
веществ (обычно крахмала) подается на бумажные машины с сетча-
тыми барабанами, на которых под вакуумом формируется асбестовая
диафрагменная бумага. Затем образующаяся бумага подвергается
сушке. Свойства асбестовой бумаги зависят от степени разработки
волокон асбеста и режима работы бумажной машины.
Для электролиза растворов поваренной соли применяется асбе-
стовая диафрагменная бумага со следующей характеристикой:
нии
Толщина, мм............................. .
Масса 1 м2, г ...............................
Влажность, %, не более ................ . . .
Разрывная длина в сухом состоянии, м, не менее
вдоль ... ...................................
поперек .................................
0,65 + 0,04
450—550
3
360
180
4*
51
Коэффициент протекаемости . . . ............... . 0,016—0,028
Средний диаметр пор, мм....................... . 0,001
Коэффициент извилистости пор .................... 1,45—1,50
Объем пор, % ................................. —60
При применении диафрагмы из асбестовой бумаги используются
катоды сравнительно простых форм, позволяющих наносить на катод-
ную поверхность листовую асбестовую диафрагму. Наиболее удобно
производить монтаж и уплотнение краев диафрагмы на цилиндри-
ческой поверхности катода. При этом необходимо уплотнять только
верхний и нижний края диафрагмы. При обкладке диафрагмы
V- и W-образных катодов возникает необходимость уплотнения
Сжатый воздух
Рис. 2-9. Принципиальная схема установки для насасы-
вания диафрагмы:
1 — ролл; 2 — фильтр; 3 — бак для приготовления асбестовой суспен-
зии; 4 — .катод на поддоне; 5 — сушильная камера; 6 — ресиверы;
7 — вакуум-насос; 8 — напорные баки длн асбестовой суспензии; 9 —
калорифер длн подогрева воздуха; 10 — вентилятор.
диафрагмы на торцах катодной поверхности. При применении
конструкции катода с кривизной в двух направлениях использование
диафрагмы из асбестовой бумаги становится затруднительным,
а для сложных поверхностей катода — невозможным.
В современных конструкциях электролизеров с твердым катодом
как в нашей стране, так и за рубежом применяет практически
только осажденную диафрагму. Технология ее получения и свойства
диафрагмы исследованы 3. И. Лифатовой, Л. И. Кришталиком
и др. Осажденная диафрагма наносится непосредственно на катод
при просасывании пульпы асбестового волокна в соляно-щелочном
растворе через поверхность катода, представляющую собой "метал-
лическую сетку, на которую откладываются асбестовые волокна.
Такой способ образования диафрагмы позволяет применять конструк-
ции катодов с очень развитой и сложной по конфигурации поверх**
- ностью. Схема установки для осаждения диафрагмы приведена
на рис. 2-9.
В^нашей стране для приготовления осажденной диафрагмы при-
меняется асбестовое волокно мягкой или полужесткой текстуры
III и IV сортов: М-3-55 и М-4-10 или П-3-50 и П-4-20. Асбестовое
волокно III и IV сортов берется в соотношении от 3 : 2 до 1:1.
При мокрой обработке в роллах происходит расщепление асбе-
стового волокна на более тонкие волокна. При этом частично может
происходить также дробление волокон. "В зависимости от марки
применяемого асбестового волокна, сетки катода и требуемой проте-
каемости диафрагмы устанавливается определенное число оборотов
ролла, время обработки волокна и присадка ножей.
При употреблении асбестового волокна полужесткой текстуры
проводится более длительная обработка в ролле, чем при исполь-
зовании волокна мягкой текстуры. Обычно применяется ролл,
в котором емкость ванны составляет 2 м3, длина барабана 825 мм
и мощность привода 15 кВт. Число ножей барабана — 54, а число
ножей планки корпуса — 24. Обычная частота вращения ролла —
238 об/мин, при этом линейная скорость Движения ножей барабана
составляет около 10 м/с. После разработки взвесь асбестового
волокна в солянощелочном растворе должна отстаиваться не болен
чём на 2% в час.
После ролла асбестовое волокно отфильтровывается от воды
и
и используется для получения суспензии для насасывания диафрагмы.
С целью получения устойчивой суспензии ее готовят в соляно-
мелочном растворе. Для этой цели применяют электролитическую
щелочь. Суспензия должна содержать 50—60 г/л NaOH и 13—15 г/л
асбестового волокна.
Для равномерного осаждения асбестовой диафрагмы необходимо
. поддерживать определенную концентрацию асбестовых водакон в.СУ.С7^^
пензии. При более высокой концентрации асбеста могут получаться
Диафрагмы неоднородной толщины по всей поверхности.
При получении диафрагмы катодное пространство корпуса
г катода соединяют с вакуумной линией и погружают катод в бак
с асбестовой суспензией. При просасывании жидкости через сетча-
. тую поверхность катода на ней откладывается слой асбестовых воло-
кон. Обычно корпус катода устанавливают на специальный поддон
и при насасывании заполняют корпус катода асбестовой суспензией.
При этом отпадает необходимость в специальном баке и исключается
смачивание тепловой изоляции на наружных стенках катодов.
Во время насасывания диафрагмы следят, чтобы уровень суспензии
был на 70—100 мм выше верхнего слоя катода. После окончания
насасывания катод вынимают иэ бака с асбестовой суспензией^
а при насасывании на поддоне — отсасывают избыток суспензии
в вакуум-сборники через-специальный штуцер в поддоне.
Для получения более равномерного слоя диафрагмы и уменьше-
ния расхода суспензии процесс насасывания обычно ведут с плавным
постепенным повышением вакуума в катодном пространстве до
400 мм рт. ст. Разработано и применяется на практике автоматическое
регулирование процесса насасывания диафрагмы по заранее
53
заданной программе. Такой режим насасывания позволяет получать
более однородные диафрагмы. Примерная схема изменения вакуума
во время насасывания диафрагмы приведена ниже:
Время насасывания, мин . . 1
Вакуум, мм рт. ст. ...........10
Время насасывания, мин . »
Вакуум, мм рт. ст. » . . . .
2 3 4 5 6 7 8
10 10 10 20 70 120 180
9 10 11 12 13 14 15
230 290 340 390 400 400 400
При максимальном вакууме около 400 мм рт. ст. насасывание
продолжается около 3 мин, затем прекращается и при тех же условиях
через диафрагму просасывается воздух для ее уплотнения и подсушки
в течение примерно 10 мин. Применяют также повторное насасывание
в течение 5—10 мин при максимальном вакууме. Расход суспензии
на образование 1 м2 диафрагмы составляет обычно 220—250 л.
Приготовленная диафрагма сушится при 100—110 °C воздухом,
просасываемым через нее. За рубежом иногда отказываются от сушки
диафрагмы, но при этом диафрагма не должна долго храниться во
влажном состоянии. При сборке электролизеров возможность повре-
ждения диафрагмы сильно возрастает при применении влажных,
не высушенных диафрагм.
Осажденная асбестовая диафрагма характеризуется следующими
средними показателями:
Толщина диафрагмы, мм ..................... . . .
Масса 1 м3 диафрагмы, кг .........................
Разрывная длина в сухом состоянии, м, не^иенее
Влажность до сушки, % .................... . »
Относительное электрическое сопротивление, кратность
Коэффициент протекаемости...............
2,5—3,0
2,2—2,4
400
55,0—60,0
4 2__1 5
0,023—0*025
Поведение осажденной диафрагмы в процессе электролиза ана-
логично диафрагме из асбестовой бумаги.
Обязательным условием получения качественной и равномерной
по толщине диафрагмы является соответствующая подготовка поверх-
ности катода перед насасыванием. Сетка катода должна быть равно-
мерной; отверстия, существенно большие ячейки сетки и сплошные
места (наплывы сварки, остатки старой диафрагмы и т. п.) на поверх-
ности катода не допускаются. При образовании осажденных диа-
фрагм нельзя допускать острых углов на покрываемых диафрагмой
поверхностях. Для получения равномерной диафрагмы должен
быть обеспечен свободный подвод асбестовой пульпы ко всем частям
покрываемого катода. Необходимо избегать в конструкции катода
длинных и узких щелей. В таких щелях возможно неравномерное
на площади отложение диафрагмы.
Применение электродов большой высоты требует соблюдения
на всей площади диафрагмы оптимальных соотношений между скоро-
стью протекания электролита через диафрагму и плотностью тока
на ней. Оптимальные условия создаются при таком соотношении ука-
занных величин, когда степень превращения хлорида в гидроокись
составляет 50—55%. Оптимальные условия для процесса электро-
54
лиза при концентрации NaCl в исходном рассоле 310 г/л создаются
при соотношении:
Л = ^-=12,3-4-13,5 мл/(А-ч) (2.42)
£
где i — плотность тока на диафрагме, А/см2; В — протекаемость раствора
через диафрагму, мл/(см2*ч).
Если значение Л ниже указанных пределов, происходит резкое
уменьшение выхода по току и увеличение износа графитовых
ацодов из-за попадания ионов ОН“ в анодное пространство. При
возрастании значения А выше указанных пределов концентрация
щелочи в католите снижается против оптимальной и несколько умень-
и
шается выход по току из-за увеличения количества хлора, попадаю-
щего в катодное пространство вместе с электролитом, протекающим
через диафрагму.
При вертикальном расположении электродов и диафрагмы
(особенно при применении электродов большой высоты) могут наблю-
даться весьма значительные колебания значения Л по высоте диа-
фрагмы как в результате изменения условий работы диафрагмы и
скорости протекания электролита через диафрагму по ее высоте,
так и вследствие неравномерного распределения плотности тока
по высоте электродов.
Применяя полностью заполненное катодное пространство и выби-
рая рациональные конструктивные формы катода, можно обеспечить
равномерность давления фильтрации и равную скорость противо-
тока электролита по всей высоте диафрагмы и при большой высоте'
электродных элементов электролизеров.
Неравномерность в распределении плотности тока по высоте
электродов обусловлена главным образом двумя факторами; наибо-
лее важным из них является значительная потеря напряжения на
преодоление омических сопротивлений графитовых электродов.
Обычно потеря напряжений в теле катода не превышает нескольких
десятков мВ и не может быть определяющей при рассмотрении
причин, вызывающих неравномерность в работе электродов по высоте.
Потеря напряжения в теле графитового анода обычно во много раз
больше и является в большинстве случаев основной причиной нерав-
номерного распределения плотности тока по высоте электродов [75].
Падение напряжения в теле анода возрастает по мере уменьшения
сечения анода вследствие его разрушения в процессе работы, а также
из-за повышения удельного сопротивления графита, сопровождаю-
щего износ графитовых анодов. Для электролизера БГК-17 с нижним
подводом тока к анодам, работающего при низкой плотности тока
(около 520 А/м2), за период работы анодов падение напряжения
в теле анода возрастает с 0,18—0,20 В в начальный период работы
до 1,2—1,4 В к концу тура работы анодов. С увеличением высоты
анодов потери напряжения на преодоление омического сопротивления
соответственно возрастают. Потери напряжения в графитовом аноде
могут быть снижены уменьшением длины пути тока по телу электрода
за счет применения двойного подвода тока к аноду: комбинирования
нижнего и верхнего подводов или применения бокового токонодвода.
. Вторая причина, вызывающая неравномерность распределения
плотности тока по высоте электродов заключается в изменении удель-
ного сопротивления электролита вследствие различного его газо-
наполнения по высоте электролизера.
В зависимости от способа подвода тока к электродам влияние
обоих этих факторов может или суммироваться, или частично ком-
пенсировать друг друга. При нижнем токоподводе к анодам влияние
Рис: 2-10. Схема электролизера с одной (я) и с двумя (б) фильтру-
ющими диафрагмами:
1 — катод; 2 — катодная диафрагма; 3 — анод; 4 — анодная диафрагма,
обоих факторов складывается, и неравномерность распределения
плотности тока по высоте электродов будет максимальной. Нижние
части поверхности электродов будут работать с максимальной плот-
ностью тока, верхние — с минимальной. При верхнем токоподводе
к анодам увеличение газопаполнения анолита в верхней части
электролизера будет частично компенсировать влияние сопротивле-
ния анода. При комбинированном подводе тока для нижней части
анода влияние этих факторов будет суммироваться, а для верхней
части частично компенсировать друг друга.
Способ с двумя фильтрующими диафрагмами. На рис. 2-10 приве-
дены схемы электролизера с одной и двумя фильтрующими диафраг-
мами. В электролизере с двумя фильтрующими диафрагмами рассол
подается в среднее пространство между диафрагмами и фильтруется
как в катодное, так и в анодное пространство электролизера. При
соблюдении необходимой скорости протекания рассола через катод-
ную диафрагму представляется возможность предотвратить попада-
ние щелочи в среднее пространство за счет участия ионов ОН-
в переносе тока подобно тому, как это имеет место в способе с одной
фильтрующей диафрагмой. При этом, в отличие от метода с одной
фильтрующей диафрагмой, к катоду поступает раствор хлорида
щелочного металла, не содержащий растворенного хлора, что исклю-
чает потери выхода по току за счет попадания кислого анолита к ка-
тоду.
56 - '
IE
При движении рассола через диафрагму в анодное пространство
можно поддерживать концентрацию хлорида щелочного металла
в анолите на желаемом уровне. Кроме того, движение потока рас-
сола через анодную диафрагму навстречу электролитическому
переносу ионов водорода позволяет предотвратить или уменьшить
попадание Н+ в среднее пространство электролизера. Поэтому при
применении двух фильтрующих диафрагм можно теоретически полу-
чить более высокий выход по току по сравнению с методом с одной
фильтрующей диафрагмой. Практически реализация такого способа
разделения электродных продуктов связана с усложнением процесса
и аппаратуры.
В качестве катодной диафрагмы может быть использована обыч-
ная асбестовая диафрагма. Применялась ткань из белого канадского
асбеста. Для анодной диафрагмы, омываемой с анодной стороны
кислым анолитом, необходимо использовать кислотостойкие диа-
рагмы (ткань из кислотостойкого голубого капского асбеста).
При работе с двумя диафрагмами значительно труднее осущест-.
вить регулирование скорости протекания рассола через катодную и
анодную диафрагмы, что, естественно, усложняет конструкцию са-
мого электролизера. Введение второй диафрагмы и дополнительного
среднего пространства приводит к увеличению расстояния между
электродами, потерь напряжения на преодоление омического сопро-
тивления электролита и диафрагм и увеличению общего напряжения
на электролизёре. Наконец, серьезным затруднением является
необходимость донасыщения кислого анолита, вытекающего из анод-
ного пространства, подобно тому, как это делается в методе электро-
лиза с ртутным катодом.
Способ разделения с двумя фильтрующими диафрагмами исполь-
зовался в электролизерах Гауса, Циба — Монтей [76], Финлей [77]
и др., однако теоретически возможные преимущества этого способа
разделения электродных продуктов не удалось получить ни в одном
из известных вариантов таких конструкций.
Аноды
До последнего времени в промышленности в качестве материала
для изготовления анодов электролизеров применялись преимущест-
венно графитированные материалы.
В начале развития электрохимического метода производства
хлора и каустической соды, когда технология получения искусствен-
ного графита еще не была реализована в промышленности, в качестве
анодного материала использовались угольные блоки и в меньшей
степени — отливки из магнетита. Значительное применение в каче-
стве анодного материала находила также платина как в чистом виде,
так и в виде платиноиридиевого сплава.
Однако угольные аноды малостойки в условиях электролиза
водных растворов хлоридов щелочных металлов, магнетитовые аноды
обладают повышенным потенциалом выделения хлора и низкой
электропроводностью, недостаточно стойки при электролизе и
57
1 т хлора при правильном
неудобны в обработке. Электроды из платины и сплавов платины
с иридием имели высокую стойкость в условиях электролиза, что со-
ответствовало требованиям, предъявляемым к малоизнашивающимся
анодам. Однако конструктивно эти аноды были сложны, а высокая
стоимость дефицитных металлов платиновой группы делала зти
аноды малоэкономичными. Поэтому платиновые аноды, так же как
угольные и магнетитовые, в производстве хлора и каустической соды
были полностью вытеснены графитовыми анодами.
Графитовые аноды имеют серьезные недостатки, осложняющие
проведение процесса электролиза. Графитовые вноды подвергаются
в процессе электролиза разрушению. В электролизерах с твердым
катодом и диафрагмой расход анодов на 1 т хлора при правильном
ведении процесса составляет 3,5—6,0 кг [78] и в методе с ртутным
катодом соответственно 2—3 кг [23]. Вследствие износа анодов
электролизеры с твердыми катодами и диафрагмой работают с изме-
няющимся в течение тура работы напряжением и в переменном
температурном режиме. В электролизерах с ртутным катодом-тре-
буется частое регулирование межэлектродного расстояния по мере
износа анодов.
Для замены изношенных анодов в электролизерах требуется
затрата большого количества труда и материалов для ремонта. Про-
дукты разрушения графита загрязняют хлор и каустическую соду,
ускоряют забивку пор диафрагмы и приводят к повышенному выде-
лению водорода в электролизерах с ртутным катодом.
Работы по улучшению качества графитовых анодов, разработке
способов их импрегнирования и оптимизации процесса электролиза
дали возможность только несколько снизить удельные расходы гра-
фитовых анодов, однако не смогли устранить перечисленные выше
недостатки.
Поэтому в последние годы вновь возрос интерес к замене графита
различными малоизнашивающимися анодами для электролиза хло-
ридов щелочных металлов с целью получения хлора и каустической
соды. Основой для этого послужили успехи в производствах таких
металлов, как титан или тантал, которые могут служить в качестве
тгокоподводящей основы для активного покрытия, например, из
металлов или окислов металлов платиновой группы, не подверга-
ющихся коррозионному разрушению при анодной поляризации
в растворах хлоридов щелочных металлов [79]. Это позволяет исполь-
зовать в качестве анода тонкий слой активного покрытия, галь-
ванически (или каким-либо другим путем) осажденный на титановом
или танталовом электроде. Более подробно о малоизнашивающихся
анодах будет изложено ниже.
Технология изготовления угольных и графитовых анодов описана
в литературе [80—82]. Искусственный графит обладает свойствами,
которые делают этот материал пригодным для использования в ка-
честве анодного материала в ряде злектрохимических процессов.
Графитовые аноды обладают удовлетворительной химической стой-
костью, сравнительно хорошей электропроводностью и высокой
58
Рис. 2-11. Зависимость из-
носа анодов от кислотности
анолита:
1 — новые аноды; 2 — аноды,
бывшие в эксплуатации.
механической прочностью. Графит отличается от угольных электро-
дов высокой степенью чистоты, значительно меньшим содержанием
золы и кристаллической структурой. Большинство примесей улету-
чивается в процессе графитирования при температуре около 2200 °C.
Искусственный графит поддается механической обработке, изде-
лиям из графита можно придать геометрическую форму, необходимую
и удобную для конструирования анодного блока электролизера.
Перенапряжение выделения хлора на графитовых анодах сравнил
тельно невелико. В условиях работы промышленных хлорных'
электролизеров оно практически мало отличается от перенапряжения
выделения хлора на платиновых анодах (если процесс на платиновых
анодах вести при низких значениях pH) и
существенно ниже (на 0,3—0,5 В), чем на
магнетитовых анодах.
В зависимости от условий ведения про-
цесса электролиза (pH анолита, концент-
рация щелочи, концентрация хлористого
натрия и сульфатов в анолите, темпе-
ратура электролиза, качество анодов
и др.) графитовые аноды подвергаются
значительному разрушению [83—97], их
расход колеблется в пределах от 2 до
10—12 кг графита на 1 т хлора.
Разрушение графитовых анодов опре-
деляется в основном скоростью окисле-
ния графита кислородом, выделяющимся
на аноде одновременно с хлором, а также
при воздействии гипохлорита и хлорно-
ватистой кислоты, образующихся при
гидролизе хлора, на материал анода.
Окисление графита с образованием двуокиси углерода и неболь-
ших количеств окиси углерода приводит также к нарушению связи
4 между отдельными зернами графита и механическому осыпанию
частиц анода.
Исследования [90] показали, что скорость износа графита резко
уменьшается при повышении кислотности анолита от нуля до
0,01 г-экв/л и при дальнейшем возрастании содержания кислоты
остается практически неизменной. Полученная зависимость приве-
дена на рис. 2-11.
Кислотность анолита определяет условия разрядки кислорода
при неизменной концентрации хлорида. При содержании кислоты
более 0,01 г-экв/л выделение кислорода связано с разрядом молекул
воды и практически не усиливается с изменением кислотности. Выска-
зано предположение, что при разрядке молекул воды происходит
интенсивное сгорание графита, тогда как при разрядке ионов ОН”
преимущественно выделяется элементарный кислород [91].
В промышленном электролизере кислотность анолита опреде-
ляется совместным протеканием нескольких процессов. Одни из
. 59
этих процессов увеличивают, а другие, наоборот, снижают кислот-
ность анолита. Основным процессом, приводящим к повышению
кислотности анолита, является разряд молекул воды с выделением
кислорода, т. е. процесс, приводящий к потере выхода по току
и окислению анодов
. Н2О > 1/2О2 + 2Н+ + 2е (2.10)
Такое же положение наблюдается при разряде ионов СЮ" и
окислении их до 010 3" в соответствии с выражением (2.22).
Увеличению кислотности способствуют также процессы хлориро-
вания органических веществ, присутствующих в графите и приме-
няемых для пропитки анодов, или поступающих
примесей
с рассолом в виде
(2.43)
удельного расхода
г/л легко окисля-
RH+C12 = RC1 + HC1
На этом основаны предложения по снижению
графита за счет добавления к рассолу 0,05—2,2
ющихся органических соединений с целью понижения pH электро-
лита. Однако органические примеси не должны содержать соедине-
ний азота во избежание образования NC13 [98].
Наконец, при гидролизе хлора, растворенного в анолите, происхо-
дит повышение кислотности рассола за счет образования соляной
и хлорноватистой кислот.
Все процессы, приводящие к повышению концентрации ионов Н+,
связаны с эквивалентным снижением выхода хлора по току. В даль-
нейшем, при поступлении анолита в катодное пространство, происхо-
дит также эквивалентное снижение выхода щелочи по току.
Кислотность анолита может также поддерживаться за счет подачи
на питание электролизера кислого рассола. Такой способ регулиро-
вания кислотности применяется при электролизе с ртутным катодом
и неоднократно предлагался и проверялся как в опытных, так и
в производственных условиях для электролиза с твердым катодом
и диафрагмой. При питании электролизеров с твердым катодом
кислым рассолом столкнулись с явлениями усиленного разрушения
асбестовых волокон диафрагмы, выщелачивания из них магния
и осаждения гидроокиси магния в толще диафрагмы. Пока, насколько
известно, проводившиеся в этом направлении опыты не позволили
добиться увеличения выхода по току и снижения удельных-затрат
графита. Однако работы в этом направлении не прекращаются.
Предложено проводить электролиз с добавлением к анолиту
до 20% HG1 (в расчете на выделяющийся хлор) [99]. При этом необ-
ходимо также применять кислотостойкую диафрагму.
Расход кислоты в анодном пространстве электролизеров с твердым
катодом определяется прежде всего затратами ее на нейтрализацию
ионов ОН", поступающих в анодное пространство.
. В электролизерах с ртутным катодом количество ионов ОН",
попадающих в электролит, определяется разрядкой молекул воды
на катоде или разложением амальгамы в электролизере. При загряз-
нении электролита амальгамными ядами эти процессы могут акти-
визироваться.
•60
~ В электролизерах с твердым катодом попадание щелочи в анодное
пространство может происходить главным образом за счет участия
ионов ОН“ в переносе тока при повышении степени конверсии NaCl
более 50—55%.
При питании электролизера щелочным рассолом значительное
количество кислоты расходуется на нейтрализацию едкого натра
и соды, содержащихся в рассоле. В балансе кислоты в анодном
пространстве существенное значение имеет ее вынос с электролитом
естественно, возрастают
Потеря массы, г
Рис. 2-12. Влияние выхода
по току на потери в массе
анода.
из анодного пространства, а в электролизерах с твердым катодом —
также за счет участия ионов Н+ в переносе тока. Обе эти статьи
расхода кислоты из анодного пространства,
с увеличением кислотности анолита и
в электролизерах с твердым катодом при-
водят к соответствующему расходу щелочи
на нейтрализацию кислоты в катодном
пространстве.
Кислотность анолита устанавливается
в электролизере в результате одновремен-
ного протекания перечисленных выше
процессов. В электролизере, работающем
с высоким выходом по току, всегда уста-
навливается более низкое значение pH,
чем в электролизере с пониженным вы-
ходом по току.
При проведении электролиза с плати-
новыми анодами или анодами, содержа-
щими активное покрытие из металлов
платиновой группы, вследствие большого
перенапряжения выделения кислорода по
сравнению с графитовым анодом реакция
(2.10) протекает с очень малой относитель-
ной скоростью. В анодном пространстве устанавливается значительно
более высокое значение pH, чем при тех же условиях в элек-
тролизере с графитовыми анодами.
Таким образом, показатель кислотности анолита отражает сово-
купность процессов, протекающих в электролизере и влияющих
на выход по току и скорость разрушения графитовых анодов.
Для электролизеров с твердым катодом зависимость износа
I
анодов от концентрации щелочи в католите и выхода по току одина-
кова по характеру. Данные о зависимости износа анодов от выхода
по току [83], степени, превращения хлорида в гидроокись и концен-
трации NaCl в анолите [84] приведены на рис. 2-12 и 2-13.
При абсолютном увеличении износа (за единицу времени) удель-
ный расход графита с ростом плотности тока снижается, что обусло-
влено увеличением потенциала анода с ростом плотности тока и
изменением относительных скоростей разряда ОН" и С1" . При очень
высоких плотностях тока наблюдается явление распыления гра-
Примеси сульфатов снижают стойкость графитовых анодов [94].
При концентрации SO4_ более 5 г/л износ увеличивается примерно
на 2,5 кг графита на 1 т щелочи на каждые 10 г/л Na2SO4 [87, 95].
Для объяснения механизма влияния сульфатов на износ анода
высказано предположение [92]^
что за счет абсорбции ионов SO4"
Концентрация NaCl, г/л
Плотность тока 7A/cmz
2
Рис. 2-13. Зависимость износа графитовых анодов от
степени превращения хлорида в гидроокись при 80 РС
(а), от концентрации хлорида натрия в анолите при
80 ?С (б), от температуры (в) и от плотности тока при
80?С (г):
1 — г/1000 (А-ч); 2 — степень разрушения за 510 ч испыта-
ния, %.
на поверхности графита тормозится процесс разрядки ионов С1“
и ОН" и создаются условия для разрядки молекул воды и выделения
кислорода с относительно более высокой скоростью.
Вследствие пористости графита процесс электролиза проходит
не только на наружной поверхности электрода, но и частично на
поверхности пор в глубине электрода [88]. Потенциал и соответ-
ственно плотность тока в порах анода быстро снижаются от наружной
62
поверхности в глубину электрода. Это приводит к тому, что на поверх-
ности пор, расположенных в глубине электрода, изменяется соотно-
и
ение скоростей электрохимического выделения хлора и кислорода
в пользу последнего.
Для снижения износа графитовых анодов стремятся избегать
попадания ионов ОН" из катодного пространства в анодное, не
допускать повышения pH анолита и концентрации ионов SO|“
в электролите, а также работать при возможно высокой концентрации
хлористого натрия в анолите. В электролизерах с ртутным катодом
стремятся также работать на рассоле, не содержащем амальгамных
ядов.
Увеличение стойкости графитовых анодов достигается за счет
пропитки их различными материалами, например льняным или
тунговым маслом. В последние годы для пропитки используют 15—
25%-ный раствор масла в легколетучем растворителе, например
четыреххлористом углероде. При такой пропитке износ графита
сокращается примерно в 1,4 раза [100, 101]. Предложены также
и другие пропитывающие материалы.
Фирма «Ниппон Карбон» сообщила о новом способе пропитки
графитовых анодов определенными полимерами винила [102], что
позволяет сократить износ графита в 2 раза по сравнению с непро-
питанным при плотностях тока 8,6—10 кА/м3. Однако, по многим
наблюдениям, с повышением плотности тока эффект пропитки графита
снижается, что объясняют вытеснением процесса электролиза из
мелких пор на поверхность графитового анода [103].
Недостатки пропитки электродов заключаются в некотором
повышении потенциала анода (при плотности тока около 1 кА/м3
на 50—100 мВ) и выделении в процессе электролиза продуктов
хлорирования масла, которые, осаждаясь на диафрагме, изменяют
ее’протекаемость и сокращают срок службы. Промывка такой диа-
фрагмы, как правило, не эффективна, поэтому для восстановления
нормальной работы электролизера необходима замена удиафрагмы.
Действие пропитки можно объяснить тем, что пропитывающий
материал, образуя пленку, защищает места контакта зерен материала
графитового анода, снижая при этом скорость механического разру-
шения анодов. Однако сокращение работающей поверхности анода
из-за образования пленки приводит к росту действительной плот-
ности тока и повышению потенциала графитового анода. Последнее,
особенно существенно в условиях работы при высоких плотностях
тока, как это имеет место при электролизе с ртутным катодом.
Исследования показали, что при применении пропитанных анодов
изменяется соотношение выделяющихся на аноде хлора и кислорода
в пользу хлора. Выход хлора по току возрастает (затраты тока
на выделение кислорода снижаются). Несколько увеличивается содер-
жание свободного кислорода в хлоргазе за счет уменьшения коли-
чества образующейся СО2.
В соответствии со сказанным о влиянии пропитки на стойкость
анодов, импрегнирование электродов приводит к сокращению,
63
главным образом, механического износа анода за счет уменьшения
осыпания зерен графита.
Если скорость химического износа в результате пропитки сокра-
щается в 1,2—1,4 раза, то скорость механического износа для хорошо
пропитанных электродов снижается в 1,8—2,4 раза. Соотношение
химического и механического износа для непропитанных электродов
составляет обычно около 1,2, а для хорошо пропитанных электродов
оно увеличивается до 1,8—2,0.
Пропитка анодов приводит к уменьшению активной поверхности
графитового анода [104] и некоторому увеличению потенциала
выделения хлора. Поэтому при высоких плотностях тока на про-
питанных электродах легче достигается критическое значение потен-
циала, при котором наступает сильное увеличение износа графито-
вого анода. Поэтому пропитка анодов применяется только для диа-
фрагменного метода электролиза, но не для ртутного [105, 106].
Прй применении пропитанных анодов в современных конструк-
циях диафрагменных электролизеров расход графита при правильной
эксплуатации составляет 3,5—6,0 кг на 1 т хлора, в электролизерах
с ртутным катодом расход графита обычно ниже 2—3 кг/т хлора.
К графитовым анодам, применяемым в современных электроли-
зерах с твердым катодом, после пропитки их раствором льняного
масла в GG14, предъявляются следующие требования:
9,5
0,3
95
1,67
210
Удельное сопротивление, Ом*мм2/м, не более .............
Содержание золы, %, не более ...........................
Износ анодов при испытании в НС1 (5 г/л), г/(1000А*ч),
не более ......... .....................................
Плотность, г/см3, не менее , . /........................
Прочность на сжатие, кгс/см2, не менее .................
п
В электродах, используемых в электролизе с ртутным катодом,
дополнительно должны отсутствовать примеси, способствующие
разложению амальгамы щелочных металлов. Электроды, предназна-
ченные для ртутного электролиза, должны содержать не более 0,2%
золы и не более 20 частей на миллион ванадия. Графитовые плиты
для ртутного электролиза применяются без пропитки, поэтому
допускается их износ до 130 г/(1000 А-ч). Удельное сопротивление
должно быть в пределах от 8 до 13 Ом*мм2/м.
В процессе электролиза удельное сопротивление графита воз-
растает по сравнению с исходным его значением за счет нарушения
контактов между отдельными зернами материала [106]. К концу
, тура работы графитового непропитанного анода удельное сопроти-
вление его может увеличиться примерно в два раза; для пропитанных
электродов оно возрастает в меньшей степени. Так, для пропитанных
анодов удельное сопротивление увеличивается до 10—Д2,9 Ом*мм2/м,
а для цепропитанных — до 18,8—22,0 Ом*мм2/м (по сравнению
с 8 Ом-мм2/м для новых анодов).
В зависимости от конструктивного оформления электролизера
применяются графитовые аноды различной формы: в виде плит,
' призм или стержнейжруглого или квадратного сечения. В современ-
64 ’
ных конструкциях электролизеров большой мощности с ртутным
катодом и с диафрагмой применяются графитовые плиты различных
размеров. В зависимости от расположения электродов, плотности
тока и способа подвода тока размеры графитовых плит колеблются
по длине от 300 до 1200 мм, по ширине от 100 до 250 мм и по толщине
от 30 до 90 мм. Наиболее распространенные размеры плит и стержней,
выпускаемых отечественной промышленностью для электролиза
растворов хлоридов щелочных металлов, приведены в табл. 2-6.
Таблица 2-6- Размеры плит и стержней (в мм)
Аноды графитовые
Длина
Ширина
Толщина
Для электролизеров с твердым катодом
Для электролизеров с ртутным катодов
Стержни
До 1100
До 1100
До 1100
550—700
210—680
155—290
51
180
175
51
• 50
50
90/70
90
Диаметр
65; 68; 70;
70/65
*
•Л!
1?
Я
I
л
В последнее время наблюдается стремление к увеличению толщины
графитовых плит для ртутного метода электролиза до 90 мм и более
с целью лучшего использования графита. Для диафрагменного *
электролиза, наоборот, наблюдается тенденция к уменьшению
толщины анодов до 35—40 мм.
При использовании более толстых плит доля неиспользованной
части графитового анода, выбрасываемой при ремонте, снижается
й за счет этого сокращаются удельные затраты графитовых анодов.
Увеличение толщины плит влияет также благоприятно на уменьше-
ние омических потерь напряжения в аноде. Необходимо учитывать,
что увеличение начальной толщины графитовых плит в случае
применения горизонтальных анодов может привести к необходи-
мости увеличить высоту электролизера или к сокращению газового
объема электролизера и ухудшить сепарацию брызг электролита,
уносимого с хлором. '
В электролизерах с ртутным катодом применяется горизонтальное
расположение анодов; в электролизерах с диафрагмой — преиму-
щественно вертикальное.
При горизонтальном расположении анодов выделяющийся на
аноде хлор в виде пузырьков собирается под электродом и, если его
не отводить, газ может экранировать поверхность анода. При этом
значительная часть поверхности электродов может оказаться выклю-
ченной из работы, а остальные части поверхности электродов будут
работать при повышенной плотности тока. Также неравномерно
может распределяться плотность тока и по слою электролита между
электродами. В местах скопления газовых пузырьков под анодом
5 Заказ 843 65
плотность тока по электролиту будет близка к нулю, а в местах,
свободных от газовых скоплений, — завышенной. Если возрастание
действительной плотности тока на электродах вследствие экрани- ,
рования части поверхности анодов газовыми скоплениями приводит
к сравнительно небольшому увеличению перенапряжения на элек-
тродах, то повышение плотности тока в" электролите вызывает проб-
но рциональный рост величины потерь напряжения на преодоление
омического сопротивления
электролита. С увеличением плотности
ГН"111! 1
1 х 11 1 * ‘
мм
ООО о о
о о о о
о о о <о о
О О О' о
0 0 0-0 0
о о о о
о о о о о
о о о о о
о о о о
О О О р О
о о о' о
о о О о о
о о о о
о о о о о
Рис. 2-14. Формы перфорации анодов из графитовых
плит. !
л
тока влияние газонаполнения электролита и экранирования газо-
выми скоплениями поверхности горизонтально расположенных элек-
тродов сильно возрастает, так как при этом пропорционально повы-
ению плотности тока растет количество газа, выделяющегося
на единице поверхности анода.
Для облегчения отвода выделяющегося хлора графитовые плиты
перфорированы — снабжены отверстиями или прорезями различной
конфигурации. При определении оптимальной перфорации графи-
товых плит необходимо учитывать разрушение материала анода
во время работы. С целью создания наиболее благоприятных условий
для отвода газовых пузырьков графитовую плиту целесообразно
снабдить достаточно частой перфорацией с тем, чтобы размеры гори-
зонтальных площадок на работающей поверхности анода были
сведены к минимальным и путь газового пузырька от места его
образования на нижней поверхности анода до ближайшего отверстия
перфорации или ближайшей прорези в аноде был минимальным.
i Однако слишком частые прорези или отверстия в материале анода
сильно, уменьшают его механическую прочность и могут привести
к разрушению и осыпанию анода в процессе работы. Различные формы
перфорации графитовых плит, применяемых в электролизерах с гори-
зонтальным расположением анодов, показаны на рис. 2-14.
7 Наиболее целесообразна конструкция плиты, изображенная на
рис. 2-14, г и д. Конструкция плиты по образцу а неудобна, так
£
л;
и
4 как для нее характерно значительное ослабление механической
прочности по сравнению с г и д.
В электролизерах с вертикальным расположением анодов пу-
зырьки хлора, выделяющегося на аноде, свободно поднимаются
Зверх в жидкости, поэтому в конструкции графитового электрода
нет необходимости предусматривать специальные устройства для
облегчения отвода газа.
При использовании металлических анодов и, повышенных плот-
ностей тока применяют так называемые проницаемые аноды с отво-
\ дом выделяющегося хлора на обратную сторону анода через отвер-
стия перфорации. ,
В зависимости от материала анода различны также приемы под-
вода тока к работающей поверхности электрода.
В электролизерах для получения хлора и каустической соды,
' применяющих графитовые аноды, подвод и распределение тока
по работающей поверхности анода осуществляется обычно с помощью
материала самого электрода. В конструкциях электролизеров с ртут-
ным катодом используются также металлические проводники, поме-
ченные внутри графитовых стержней или защищенные от действия
хлора и анолита защитными чехлами или втулками из фарфора либо
полимерных материалов.
В последние годы в связи с развитием производства титана появи-
лась возможность использовать титановые токоподводы для графи-
товых электродов. Очевидно, что при этом должны быть соблюдены
условия, предотвращающие возможность образования окисных(пле-
нок с большим переходным сопротивлением на поверхности контакта
графитового электрода с титановым токоподводом [79, 107]. Такие
условия могут быть созданы пропиткой графитового электрода
в месте контакта. Предложено использовать корзины из титановой
решетки или, сетки, заполненные кусками графита, выполняющими
роль анода [108].
Новые возможности в конструировании токоподвода к графитовым
электродам открываются при соединении графита с титаном сваркой.
Однако применение титановых токоподводов к графитовым анодам
пока еще не вышло из стадии опытной проверки на нескольких
электролизерах промышленного размера. При использовании окисно-
рутениевых или платинотитановых анодов подвод тока к работающей
поверхности анода осуществляется с помощью титановых или биме-
таллических проводников.
В электролизерах с ртутным катодом ^горизонтальным располо-
жением анодбв подвод тока осуществляется через специальные токо-
подводящие стержни. Наиболее часто подвод тока к графитовой
анодной плите производится графитовым токоподводящим стержнем,
контакт между токонодводящим стержнем й плитой осуществляется
с помощью резьбы, соединения на конусе или запрессовки (рис. 2-15).
Соединение плиты со стержнем на конусе или методом запрессовки
представляется более целесообразным, так как требует меньшей
затраты труда и обеспечивает надежный контакт с малым переходным
5* 67
сопротивлением. Контакт с помощью конуса или запрессовки требует
большей точности при проведении работ.
Наиболее широко распространен подвод тока к плите одним
графитовым токоподводящим стержнем. При увеличении плотности
тока, особенно в электролизерах с ртутным катодом, потребовалось
усиление токоподвода
к плите. Были сделаны попытки применить
а б в г
Рис. 2-15. Типы соединения анодной плиты с токо-
подводящим стержнем:
а — на резьбе; б — на конусе; в — с запрессовкой; г — с за-
щитной трубкой.
II
два стержня на одну плиту (рис. 2-16, б). Однако такое решение
не нашло широкого применения.
В промышленности предпочитают осуществлять подвод тока по
способу а, что объясняется трудностью точного центрирования двух
токоподводящих стержней. При неточности в установке и закрепле-
нии токоподводящих стержней усложняется установка анода в крышке'
Рис. 2-16. Подвод тока к плите одним (а) или
двумя (б) токоподводящими стержнями:
1 — плита; 2 — токоподводящий стержень.
электролизера, а при работе и регулировании межэлектродного
расстояния наблюдаются случаи обрыва плиты от токоподводящего
стержня. Поэтому подвод тока к плите по двум токоподводящим
стержням применяют только при использовании гибких резиновых
крышек или эластичных уплотнений стержней в крышке. Обычно же
при конструировании предпочитают уменьшение размеров плиты
с сохранением токоподвода одним стержнем.
В большинстве случаев для подвода тока к плите используются
токоподводящие стержни диаметром до 100 мм. Стержни диаме-
тром 64—65 мм применялись в электролизерах Сименс—Биллитер
?и в ряде конструкций ртутных электролизеров, В электролизерах
68
с ртутным катодом фирм «Матисон» и «Де Нора» применяются стержни
диаметром до 100 мм.
Для уменьшения электрического сопротивления токоподводящего
графитового стержня применяют металлические (медные, латунные
или стальные) токоподводящие вставки к графитовым стержням.
Одновременно такие металлические проводники служат для подвода
Рис. 2-17, Способы подвода тока к графитовым стержням:
а — впайка гибкого проводника в графитовый стержень; б — впайка металлического стержня
и подсоединение гибкого проводника на конусной клемме; в — резьбовой контакт металли-
ческого стержня с графитовым и подсоединение гибкого проводника хомутом; г — скользя-
щий контакт графитовых втулки и стержня; д — контакт чугунный колпак —г графитовый
стержень» .
I
тока от наружной токоподводящей шины к графитовому токоподво-
дящему стержню.
На рис. 2-17 приведены различные варианты установки токопод-
водящих металлических проводников, а также устройств для подвода
тока к графитовым стержням при отсутствии металлических вставок.
Для обеспечения электрического контакта металлические вставки
подвергают лужению и после установки в отверстие графитового
стержня заливают свинцовооловянным сплавом. Применяются также
металлические сердечники, ввинчиваемые в тело графитового стержня.
Предложен токоподвод к плите графитовым стержнем с конусной
резьбой и металлическим штырем внутри стержня или непосред-
ственно контактирующим с плитой [109]. К вертикальным графито-
вым стержням и плитам анодный контакт осуществляется непосред-
ственно с помощью хомутов или накладок.
Контакты защищают от действия анолита путем специальной
пропитки токоподводящих стержней или так называемых головок
электродов. Для этой цели применяется льняное масло (но не раствор
льняного масла в СС14), парафин, горный воск и др. Основная цель
.пропитки — обеспечение полной непроницаемости графита для ано-
лита или хлора.
69
В электролизерах с вертикальным расположением анодов подвод
тока может быть осуществлен в виде верхнего, нижнего или бокового
токоподвода (рис. 2-18).
При верхнем токоподводе верхний конец электрода обычно
проходит через крышку электролизера и служит для присоединения
к нему токоподводящей шины. При этом значительная часть элек-
трода по его длине не используется и при-ремонте электролизера
выбрасывается. Чтобы уменьшить расход графита, стремятся снизить
и
Рис. 2-18. Схемы подвода тока к вертикальным анодам:
а — верхний; б — нижний; в — боковой подвод тока; I — графитовый
анод; 2 — место подвода тока к аноду; 3 — крышка электролизера;
4 — днище электролизера; 5 — боковая стенка; 6 — защита анодного
контакта; 7 — уровень электролита.
расстояние от верха катода до крышки электролизера. Это ограни-
чивает возможность регулирования протекаемости диафрагмы за
счет изменения уровня анолита и ухудшает сепарацию брызг жидко-
сти от хлора. При применении верхнего токоподвода ц§рбходимо
также обеспечить уплотнение мест прохода электродов через крышку.
В настоящее время особенно широко применяется нижний подвод
тока к графитовым анодам. G нижним токоподводом к анодам рабо-
тают наиболее мощные и современные электролизеры БГК-17,
БГК-50, Хукер, Даймонд. При нижнем подводе тока к анодам
крышка электролизера имеет минимальное количество отверстий,
поэтому легко достигается герметичность электролизера. При таком
подводе тока неработающая часть анода значительно меньше. В зави-
симости от конструкции токоподвода и способа его защиты от дей-
ствия анолита высота неиспользованной части анода может меняться.
Нижний подвод тока в электролизерах на большую нагрузку
осуществляется двумя методами: с помощью механического контакта
и заливкой свинцом. В СССР принят первый метод подвода тока.
Подвод тока от анодной шины к графитовым плитам в электролизе-
рах БГК-17 и БГК-50 осуществляется через стальное днище, которое
служит токопроводником, обеспечивающим с помощью специального
устройства надежный контакт анодов с токонесущим днищем. При
таком подводе тока исключается применение свинца, обеспечивается
70
и
- Wfc
: 'i
удобство и безопасность монтажа и Демонтажа анодного блока элек-
тролизера. конструкция токоподвода позволяет точно регулировать
и фиксировать положение анодных плит и обеспечивает точное со-
блюдение межэлектродного расстояния при сборке электролизера.
Для защиты от коррозии контактную часть анодов заливают
<$лоем защитной массы на основе битумных материалов. Композиция
^защитной массы подбирается таким образом, чтобы получаемый слой
был стоек к воздействию кислого
4^
* •*' ’ --
^ анолита и растворенного в нем хлора.
й
/•
Рис. 2-19. Схёма двойного токо-
подвода к анодам:
1 — верхняя анодная шина; 2 — уро-
вень электролита; 5 — графитовый
анод; 4 — токоподводящее днище;
5 — подвод тока к днищу; « — за-
щита нижнего контакта; 7 — нижний
анодный контакт; 8 — крышка элек-
тролизера; 9 — верхний анодный
контакт.
И Для надежной защиты контактов
S' масса при рабочей температуре элек-
тролизера должна быть пластичной,
при этом обеспечивается самоуплот-
^цение в местах возможных наруше-
|иий плотности. Для удобства про-
ведения ремонтных работ приме-
| няется/защитная масса, которая при
| комнатной температуре достаточно
К хрупка и легко удаляется обыч-
Кдыми приемами механизации ремонт-
R ных работ. G целью дополнительной ;
Л1защиты от действия растворенного ;
анолите хлора на защитную массу
^Враносится тонкий слой бетона.
|||| Для сокращения потерь дапря-
ж?^жения на преодоление омического
^{сопротивления графитовых анодов и
^обеспечения более равномерного
^распределения плотности тока по ра-
Вбочей поверхности анодов можно
^Применять двойной подвод тока:
|р;К верхней и нижней части анода.
||?Схема электролизера С двойным под-
водом тока приведена на рис. 2-19.
Для верхнего и нижнего подвода
тока могут быть использованы опи-
санные ранее конструкции контакта
и способы токоподвода.
Применение двойного токоподвода
высоту электродов и мощность электролизера при той же производ-
ственной площади. При этом, однако, . возникает необходимость
увеличения высоты производственного здания и усложняется обслу-
живание и ремонт электролизеров. Для обеспечения удобства обслу-
живания контактов на крышке электролизера и его верхней частц
требуется двухэтажное расположение цеха электролиза или устрой-
ство дополнительных площадок.
При боковом вводе анодов возникают затруднения в уплотнении
мест прохода графитового электрода или токоподвода к нему через
71
л’^
у
$
Я
л:
ч
У
il
Ji
•V?
.А/
с*
til-
ЛГ-
1Й
№с*:
Т, •
8ь
£
Г
ЙР^’
№
ij?-
II
позволяет увеличить рабочую
стенку электролизера. Трудности усугубляются тем, что место
прохода должно находиться под напором столба анодной жидкости.
Принцип бокового подвода тока к графитовым анодам открывает
возможность конструирования электролизеров с большой рабочей
высотой электродов без увеличения потерь напряжения на преодоле-
ние омического сопротивления электродов?
В энергетическом балансе электролизера потери напряжения
на преодоление омического сопротивления электролита имеют суще-
ственное значение. Значение этих потерь возрастает с ростом плот-
ности тока на электродах и соответственно в электролите.
В электролизерах всех конструкций стремятся сократить потери
напряжения на^ преодоление омического сопротивления, электролита.
С этой целью пытаются достичь максимального значения удельной
электропроводности электролита за счет повышения концентрации
хлорида в растворе и его температуры. В ряде конструкций электро-
лизеров предусматриваются меры, облегчающие выделение и отвод
газообразных продуктов (хлора) из межэлектродного пространства
с целью уменьшения газонаподнения и связанного с ним увеличения
сопротивления газонаполненного электролита.
При прочих равных условиях падение напряжения на преодоле-
ние сопротивления электролита пропорционально расстоянию между
электродами. В ходе электролиза по мере разрушения Графитовых
анодов изменяются условия проведения процесса. Напряжение
на электролизере возрастает как за счет увеличения омического
сопротивления анодов по мере их износа и диафрагмы при ее старении
и забивке пор, так и вследствие повышения потерь напряжения на
преодоление омического сопротивления электролита при увеличении
расстояния между электродами. По мере роста напряжения изме-
няется также тепловой баланс электролизера.
В конструкциях электролизеров с горизонтальным расположе-
нием электродов применяются устройства для опускания анодов
по мере их износа. В горизонтальных электролизерах с ртутным
катодом, работающих с высокими плотностями тока, применяются
устройства для опускания анодов во время работы без перерыва тока.
Некоторые конструкции таких устройств схематически показаны
на рис. 2-20.
Регулирование межэлектродного расстояния указанными выше
способами возможно в электролизерах с односторонней работой
анодов. В большинстве конструкций электролизеров с диафрагмой
и вертикальным расположением анодов используется двусторонняя
работа анодов. Для этих конструкций электролизеров нет достовер-
ных данных о практически применяемых способах восстановления
межэлектродного расстояния по мере износа графитового анода
Имелись лишь указания о регулировании межэлектродного рас-
стояния на дисковых электролизерах с ртутным катодом.
Регулирование межэлектродного расстояния при двусторонней
работе анодов может быть осуществлено путем применения клино-
видных анодов [110], как это показано на рис. 2-21.
72 ' '
Перемещая анод в вертикальном направлении, можно менять
межэлектродное расстояние.
В электролизерах с твердым катодом типа БКГ иногда в середине
тура работы анодов заменяют катодный блок другим с более толстыми
катодными пальцами. При этом достигается сближение электродов
Рис. 2-20. Устройства для опускания анодов
при горизонтальном расположении электро-
дов:
а — стационарное устройство для индивидуального
опускания; б — переносное устройство для индиви-
дуального опускания; в — устройство для групйо-
вого опускания; 1 — токоподводящий стержень;
2 — крышка электролизера; з — подвод тока; 4 —
сальниковое уплотнение; 5 — эластичное уплотне-
ние; 6 — регулировочное устройство.
во второй половине тура
работы анодов.
Практическая разра-
ботка способов регулиро-
вания межэлектродного
расстояния в современных
электролизерах с твердым
катодом и вертикальным
расположением электродов
представляет большой ин-
терес.
Рис. 2-21. Схема устройства
для восстановления межэлек-
тродного расстояния при дву-
сторонней работе анодов:
1 — анод; 2 — катод с диафраг-
мой; з — токоподводящий стер-
жень; 4 — устройство для, опуска-
ния анодов; 5 — крышка электро-
лизера.
В течение всей истории развития электрохимического метода
пройзводства хлора и каустической соды проводились исследования
с целью разработки анодов, мало изнашивающихся в процессе
электролиза. Различные варианты конструкций анодов из платино-
вой или платино-иридиевой проволоки или фольги не могли конкури-
ровать с графитовыми анодами из-за сложности конструкции и
дороговизны платиновых материалов.
Исследования электрохимического поведения титана в условиях
его анодной поляризации в водных растворах хлоридов щелочных
металлов [79] заложили основу для создания мадоизнашивающихся
анодов (МИА), в которых механической основой, по которой подво-
дится ток к работающей поверхности анода, служит титан иди
73
II
биметаллическая композиция меди, стали или алюминия с титаном.
На титановую поверхность анода наносят слой активной массы,
которая создает анодно работающую поверхность электрода, опре-
деляя величину потенциала анода и его электрохимические показа-
тели в процессе электролиза. При электролизе на поверхности ти-
тана, не покрытой активной массой, образуется запорный слой
о кисетов титана, предотвращающий коррозию титана й снижаю
плотность тока на открытых поверхностях титана до очень малой
величины.
Напряжение пробоя этого защитного слоя в растворах хлоридов
щелочных металлов в зависимости от условий лежит между 6 и 14 В,
т. е. значительно выше, чем это имеет место при обычном электролизе
водных растворов хлоридов. Поэтому при нанесении активного слоя
на титановую основу электрода не обязательно добиваться сплош-
ного, беспористого покрытия титана. Это позволило применить \
покрытия, которые ранее многократно, но безуспешно пытались
использовать при нанесении на такие обычные конструкционные
материалы, как сталь, медь и др.
В качестве активного слоя, помимо металлов платиновой группы,
могут быть применены окислы металлов высшей валентности, стой-
кие в условиях анодной поляризации и обладающие достаточной
электропроводностью. Этим требованиям в некоторой степени отве-
чают Fe3O4 [111], РЬО2 [112], MriO2 [113-118] и Co2O3 [119].
В последнее время предложено использовать смешанные окислы
металлов платиновой группы и некоторых других, в частности
изоморфные окислы TiO2 h,RuO2 [120—124].
Поведение титановой основы таких электродов при анодной
поляризации было изучено на примере платинотитанового анода
(ПТА). В растворах, содержащих 300 г/л NaCl, плотность тока
на титановой поверхности в 104—105 раз меньше, чем на платине
при том же потенциале. Ориентировочно скорость окисления поверх-
ности титана в ПТА [79, 125] составила 0,2—0,4 мкм/год, что близко
к скорости окисления титана в анолите хлорного электролизера
без анодной поляризации [126]. Изучение кинетики анодного окисле-
ния титана продолжается [127]. Если активный слой, наносимый
на титан, вступает во взаимодействие с основой электрода, необхо-
димо предпринимать специальные меры для предотвращения образо-
вания переходного сопротивления между титаном и активным покры-
тием электрода [128—130].
Предлагалось также в качестве основы анода при нанесении
активных покрытий вместо титана использовать графит [131].
При этом снижается первоначальная стоимость электродов, так как
цена титана сравнительно велика.
Было предложено наносить на, графит плотный тонкий слой
титана или тантала и затем покрывать анод активным слоем [132].
При использовании графита в качестве токонесущей основы анода
необходимо на графит наносить плотное покрытие, чтобы предотвра-
тить разрушение графитовой основы электрода в процессе электролиза.
74
>v
•J .
1?
?»й
M'.V
.^. Л-.. I .
Л Г!
д?Лл
?!Ч's*/
i •
<Ь
,V''*
I r ,• 1_
4?л?
'Av
:«
Ofe
^|||
Ж.и
®.
jf.:
Зж
.s® К;...
Ж
•wfe
ТД|Д&?''
й5ЙЖ?й?
МИА обладают большими преимуществами перед графитовыми
анодами, так как позволяют иметь постоянное расстояние между
Электродами в течение всего времени работы электролизера. Это
Обеспечивает постоянство напряжения на электролизере и стабиль-
ный температурный режим работы электролизера. В электролизерах
<; ртутным катодом отпадает необходимость в опускании анодов
и, процессе работы, что резко экономит трудовые затраты,
й При использовании МИА достигается снижение напряжения на
ячейке и может быть получена экономия электроэнергии. Эти преиму-
щества обусловлены применением активного покрытия с более низким
^ Яйачением потенциала выделения хлора, чем на графите, и созданием
й конструкции анода с облегченным отводом газовых. пузырьков из
4^;;доны прохождения тока. Такой анод исключает загрязнение хлора *
^продуктами разрушения графита и позволяет снизить требования
К качеству применяемого рассола по содержанию примеси сульфатов.
Наибольшее внимание привлекает идея разработки малоизнаши-
®^Йающихся анодов без использования дорогих и дефицитных металлов
эд^Йлатиновой группы. Известно, что для производства хлората натрия
Ш&.разработаны и широко используются в промышленности пере-
^ркисно-свинцовые аноды на титановой или Графитовой основе
134]. Однако для электролиза водных растворов хлоридов
‘^йЩелочных металлов с целью получения хлора и каустической соды
'пока не удалось разработать подобные аноды. Работы в этом направле-
'^’мии продолжаются, в качестве активного покрытия используются
^композиции на основе окислов свинца, марганца, железа, кобальта "
и 'других металлов.
В настоящее время практическое развитие получили работы по
^ созданию МЦА на основе металлов платиновой группы и в первую
|Дочередь платины и рутения.
Первым шагом в создании малоизнашивающихея анодов (МИА)
были разработка и испытание в процессе электролиза хлоридов
лочных металлов, и промышленное использование в катодной защите
и в некоторых электрохимических процессах анодов из титана,
покрытого активным слоем металлов платиновой группы или их
сплавов (ПТА). Хотя после появления окиснорутениевых анодов
интерес к ПТА снизился, однако и в последнее время продолжается
интенсивная работа по усовершенствованию этого типа электродов.
В последнее время опубликовано много предложений по применению
в качестве анода в электролизерах для получения хлора и каусти-
ческой соды титана, покрытого слоем платины или Других металлов
платиновой группы или их сплавов [135—141].
Предложено активное покрытие йз сплава рутения с платиной
или родием, а также сплавы титана с марганцем и железом, в которых
часть железа может быть заменена металлами платиновой группы
или такими металлами, как Ni, Со, Мп, Сг, V, Мо [138]. Предложены
различные формы электродов — сетчатые, пластинчатые, перфори-
рованные [142—144]. Сообщается, что при нанесении слоя из метал-
лов платиновой группы на основу из пористого титана или тантала
л
А
п
£
К'
в#
Ж/
«
s$‘-
получают электроды, стойкие в условиях периодического контакта
с амальгамой натрия [145].
Для улучшения адгезии металла платиновой группы к титану
и снижения потенциала выделения хлора на электроде предложено
предварительно обрабатывать титан в расплаве хлоридов [146]
натрия, калия или их смесей, покрывать титан слоем платины/ ।
содержащей 0,01—0,1% висмута [147] и другие примеси [148], либо |
двумя слоями платины с различными добавками к электролиту [1491 i
с последующей термообработкой активного слоя после его нанесения
на титановую основу [150]. ,
Было предложено наносить на титан тонкий слой платины в ва-
кууме с последующим гальваническим покрытием электрода плати-
ной или другими металлами платиновой группы [151].
Возможность повышения плотности тока и меньшая толщина ПТ А
по сравнению с графитовыми анодами позволяют создавать более
компактные электролизеры.
Много работ посвящено изучению стойкости платины и других
металлов платиновой группы при анодной поляризации их в раство-
рах хлоридов. Исследовалось электрохимическое поведение титана,
покрытого платиной, родием, иридием [152, 153], а также сплавами
платины с иридием [154] и сплавами с палладием [155, 156]. Сплавы
платины с иридием отличаются от чистой платины значительно
большей стойкостью при электролизе. Так, при электролизе 32 %-ной
соляной кислоты доля тока, расходуемая на растворение платинового
анода, составляет около 5%, а при применении сплава из платины/
с 10% иридия эта доля снижается до 0,9% [157].
Установлено сильное влияние на износ платины добавок к элек-
тролиту органических веществ [158—160]. Скорость коррозии пла-
тины уменьшается при длительном ведении процесса гэлектролиза,
и, наоборот, коррозионное разрушение увеличивается при перерывах
тока [111, 161—163].
В условиях проведения электролиза водного раствора NaCl
(270 г/л) при 80 °C и плотности тока 0,1 А/см2 скорость растворения
платины составляет 2—5-10"9 А/см2 [161]. Очень высокая стойкость
платины и ее сплавов с иридием затрудняет точное определение
скорости анодного растворения активного покрытия. Исследование
с применением радиоактивных изотопов платины [125, 161, 164] Г
позволило установить скорость растворения платины в условиях
анодной поляризации и влияние на нее длительности процесса
электролиза, перерывов тока, значения анодного потенциала и
других факторов. При удовлетворительной устойчивости платино-
вого и особенно платиноиридиевого покрытия титана в условиях
анодного выделения хлора отмечалась очень малая устойчивость
таких покрытий к действию амальгамы [165]. Для защиты активного
покрытия из металлов платиновой группы от разрушения при кон-
такте с амальгамой предложено наносить на анод пористый защитный
слой, например, из магнетита, титана, сульфата магния [166] или
применять анод из пористого титана с нанесением активного покры-
76
п
ПТА
8000
6000 -
3 ависимость потен-
платинотитанового
Рис. 2-22.
циала
и платинового анодов qt анодной
плотности тока и значения pH'
анолита при 80 РС:
1 — значения потенциала анода на мо-
дельном электролизере; 2 — резуль-
таты лабораторных измерений.
Л 4ооо
2000
тия из металлов платиновой группы на обратную сторону анода,
не обращенную к слою амальгамы [167].
Если рабочей поверхностью ПТА являются платина, металлы
платиновой группы или их сплавы, возможны два варианта ее соче-
тания с титановым токоподводом. Для процессов со сравнительно
высокими удельными расходами платины (требуется большая тол-
щина платинового покрытия — 10 мкм и выше) может применяться
наварка металлической фольги на титановую основу анода. При
этом, в зависимости от условий,
фольгой может быть покрыта вся
поверхность анода или только ее
j часть.
Вторым вариантом является при-
менение гальванического покрытия
титановой основы анода платиной
или металлами платиновой группы.
Такие электроды удобны для исполь-
зования в хлорной промышленности
и в других электрохимических про-
цессах, где требуемая толщина пла-
тинового покрытия не превышает 2—
10 мкм. При электролизе растворов
хлоридов потенциал таких анодов
определяется потенциалом платино-
вого покрытия и, в зависимости от
плотности тока, pH среды и других
условий колеблется в пределах от
1,4 до 2,0 В (против НВЭ). В этих
условиях незащищенные поверхности
титана платинотитановых электродов
покрываются окисной пленкой, за-
щищающей анод от дальнейшего
окисления, и работают при не-
большой плотности тока (около
1 А/м2).
Изучена зависимость стойкости ПТА от толщины слоя платино-
вого покрытия [168].
Уже при толщине платинового слоя около 1 мкм потенциал
выделения хлора на ПТА не отличается от его потенциала на сплош-
ном .платиновом электроде.
На рис. 2-22 даны значения потенциалов платиновых и ПТА
при различной плотности тока, снятых в лабораторных условиях
и на модельном электролизере при 80 СирН = 1 иЗ. Там же приве-
дены значения потенциала ПТА, снятые на промышленной модели
электролизера.
В зависимости от pH электролита значение потенциалов на плати-
новом и платйнотитановом аноде может сильно изменяться. При
повышении pH потенциал выделения хлора может возрастать на 0,3—
77
0,5 В. При одном и том же pH значение потенциала на платиновом
аноде и ПТ А одинаково при равных плотностях тока.
В табл. 2-7 приведены результаты длительного испытания ПТА
в электролизере БГК-17 при плотности тока 1000 А/м2 и pH около 3
[125].
Таблица 2~7. Результаты испытания ПТА
Показатели
Продолжительность, сутки
0 6 22 34 37 93 109
Температура, °C.......
Напряжение, В
между катодом и ПТА
катодом и графитовым
анодом
Анодный потенциал на
ПТА, В
94
2,9
3,4
96
2,9
3,4
и;
96
3,0
3,5
1,45
96
3,0
3,7
1,53
96
2,9
3,6
1,44
97
3,0
1,47
100
3,0
4,2
Анодный потенциал, измеренный на графитовом
анодный потенциал, измеренный на графитовом аноде в равных
условиях, составляет 1,43—1,54 В, т. е. практически не отличается
от потенциала на ПТА. При проведении электролиза при более
высоком значении pH потенциал ПТА при плотности тока до 2000 А/м2
может возрастать до 1,8—2,0 В [125]. Ряд исследователей отмечали
явление пассивирования платиновых анодов при электролизе рас-
твора NaC] в определенных условиях. Для активации платинового
анода в этих условиях помимо ведения процесса при низком значе-
нии pH [125] предложено применять пульсацию тока [169] либо
использовать в качестве активного покрытия сплавы платины с ири-
дием [170]. Однако для получения длительного эффекта необходимо
увеличить содержание иридия до 20—30%.
Несмотря на одинаковое значение потенциала ПТА,и графитового
анода при применении ПТА достигается снижение напряжения на
электролизере и уменьшение удельного расхода электроэнергии.
Снижение напряжения можно получить за счет сокращения потерь
напряжения на преодоление сопротивления электролита и тела
электрода. Особенно сильно это сказывается к концу тура работы
электролизеров с графитовыми анодами. В начале испытания напря-
жение на ячейке с ПТА на 0,5 В, а в конце на 1,2 В ниже по сравнению
с ячейкой с графитовыми анодами.
На электролизерах БГК-17 расход платины на 1 т хлора соста-
вляет около 0,5 г [125]. Этот расход увеличивается при перерывах
процесса электролиза, и, наоборот, значительно сокращается при
длительной работе без перерывов. Применение сплавов платины
с иридием позволяет сократить удельные расходы платины. Вслед-
ствие высокой стойкости платины в ЦТА применяются платиновые
покрытия малой толщины — 2—3 мкм. Такие электроды работают
II
II
11
11
в промышленных образцах электролизеров с диафрагмой более 4 лет
при плотности тока 1200—2000 А/м2.
Из-за дороговизны и дефицитности платины ПТА не находят
в настоящее время применения в хлорной промышленности, но
ироко используются в ряде электрохимических процессов вместо
платиновых анодов.
Практическое использование в хлорной промышленности МИА
получили после разработки окиснорутениевых анодов [171, 172],
в которых основой электрода служит титан. Возможно также приме-
нение тантала, ниобия, циркония или их сплавов, однако из-за
высокой стоимости этих металлов нашел применение только титан.
На титановую основу электрода различными способами наносится
смесь окислов рутения и некоторых неблагородных металлов (Ti,
Fe, Pb, Со, Мо и др.) [120-124].
Из металлов платиновой группы наиболее доступны для промыш-
ленного использования палладий и рутенйй — спутники платины.
Однако металлы палладий и рутений нестойки при анодной поля-
ризации в условиях электролиза водных растворов хлоридов щелоч-
ных металлов, а также в щелочных и окислительных средах [154,
172L Поэтому аноды, полученные покрытием титана слоем метал-
лического рутения, не пригодны для электролиза водных растворов
хлоридов щелочных металлов.
Стойкость покрытия из окислов рутения и смеси окислов рутения
с окислами других металлов при анодной поляризации в растворах
хлоридов щелочных металлов сильно возрастает.
Окиснорутениевые аноды, разработанные фирмой «Де-Нора»
совместно с фирмой «Даймонд», достаточно устойчивы для использо-
вания в промышленности. Окисно рутениевые аноды имеют низкое
перенапряжение выделения хлора [173] — менее 50 мВ при плот-
ности тока 10 кА/м2. Меньшая плотность рутения по сравнению
с плотностью платины позволяет получать покрытия одинаковой
толщины, но с пониженной затратой металла.
На рис. 2-23 приведены поляризационные кривые для анодов,
где в качестве активно работающей поверхности использовали галь-
ванически осажденный слой платины (ПТА), слой РЪО2, осажденный
из азотнокислого раствора и слой МпО2, нанесенные на титан элек-
трохимическим и термохимическим способами, слой магнетита, полу-
ченный при окислении железа, а также смесь окислов рутения с оки-
слами других металлов на титановой основе. Кривые сняты в ра-
створах NaGl при 80 °C и в интервале плотностей тока от 500
до-10 000 А/м2 [174, 175].
При равном значении плотностц тока аноды из двуокиси рутения
имеют наиболее низкий потенциал выделения хлора. Низкое пере-
напряжение для выделения хлора на таких анодах и возможность
создания конструкции электрода, проницаемого для газообразного
хлора, дают возможность повышать плотность тока до 10 кА/м2
и более, сохраняя напряжение на электролизере, равное 3,7—3,9 В.
Окиснорутениевые аноды позволяют без существенного ухудшения
. ' 79
процесса электролиза повысить степень использования NaCl в элек-
тролите и снизить требования к очистке рассола от ионов SO|~
[173, 176].
При применении таких анодов в электролизерах с диафрагмой
возрастает плотность тока до 2—3 кА/м2, увеличивается компакт-
ность электролизера при сохранении постоянного электрического
и теплового режима в течение тура работы электролизера, а также
возрастает срок службы Диафрагмы, улучшается качество хлора
и каустической соды и снижаются затраты электрической энергии
на производство при одновременной его интенсификации.
Плотность тока, А /м 2
Рис. 2-23. Поляризационные кривые различных анодов
в растворе NaCl концентрацией 300 г/л при 80 ?С:
1 — RuO2; 2 — графит; 3 — МпО2; 4 — РЪО2; 5 — ПТА; 6 — маг-
нетит.
Использование окиснорутенйевых анодов (ОРА) в электролизерах
с ртутным катодом позволяет значительно увеличить плотность тока
по сравнению с электролизерами с графитовыми анодами. Указы-
вается [176, 177], что электролизеры с ОРА при плотности тока
13 кА/м2 имеют напряжение 3,95 В против 4,3 В на электролизерах
с графитовыми анодами при плотности тока 9,3 кА/м2. Снижение
напряжения достигается за счет создания рациональной конструкции
проницаемых анодов, обеспечивающих легкий отвод пузырьков
хлора из зоны прохождения тока. Помимо этого, ОРА имеют низкое
значение потенциала выделения хлора. На электролизерах с ОРА
при повышении плотности на 1 кА/м2 напряжение возрастает на 100 мВ
против 200 мВ на электролизерах с графитовыми анЬдами [176].
При замене графитовых анодов на окиснорутениевые в электроли-
зерах с ртутным катодом также исключается загрязнение хлора
двуокисью углерода, достигается более глубокое разложение рассола
и снижаются требования к очистке рассола от сульфатов. Поскольку
отпадает необходимость регулирования положения анодов, сильна
сокращаются трудовые затраты. Экономия электроэнергии при
применении ОРА может достигать 20% [177].
Применение ОРА в электролизерах с ртутным катодом снимает
также необходимость периодического регулирования анодов, что
80
упрощает конструкцию электролизеров и позволяет исключить
устройства для опускания анодов по мере их износа.
Использование малоизнашивающихся анодов упрощает конструк-
цию биполярного электролизера с диафрагмой и открывает пути
создания биполярного электролизера с ртутным катодом. Работы
в этом направлении намечают новые пути развития электрохими-
ческого способа получения хлора и каустической соды как по методу
с диафрагмой, так и с ртутным катодом.
Технология приготовления ОРА не
освещена в литературе. Есть указания
о получении ОРА термохимическим спо-
собом: нанесением на титановый анод
смеси солей рутения и титана или других
добавок с последующей термообработкой
для получения активного слоя, содер-
жащего окислы рутения [123]. Предложен
также способ изготовления таких анодов
осаждением слоя металла платиновой
группы или, сплава этих металлов с по-
следующим окислением этого слоя в раз-
личных условиях.
Ведутся поцски новых анодных ма-
териалов и композиций, не содержащих
металлов платиновой группы [178, а],
используя, например, для образования
активного слоя окислы марганца [116],
железа и его сплавов [179]. Предложены
различные варианты образования актив-
ного слоя анода из металлов платиновой
группы в сочетании с разнообразными
пленкообразующими металлами [180].
В случае применения малоизнашивающихся анодов в электро-
лизерах с ртутным катодом и горизонтальным расположением
электродов необходимо предусматривать отвод выделяющегося
на аноде хлора из зоны прохождения тока. Для этой цели разрабо-
таны различные конструкции пластинчатых электродов, а также
электроды из перфорированных листов. Вопрос об оптимальной
перфорации такого анода был изучен [181] на модели электролизера
с ртутным катодом, работающей на водном растворе NaOH;.
Зависимость напряжения на электролизере от степени перфора-
ции для анода с отверстиями диаметром 6—8 мм показана на рис. 2-24.
Минимальные значения напряжения отмечены при степени перфо-
рации 35—40% при всех плотностях тока для анодов как толщи-
ной 10 мм, перфорированных отверстиями диаметром 8 мм, так и
толщиной 3 мм с отверстиями диаметром 6 мм.
Увеличение степени перфорации анода облегчает выход газа
из межэлектродного пространства. Хотя при этом удаляется часть
лобовой поверхности анода, однако его работающая поверхность
6 Заказ 843 81ч
ЗП\---1--!---1---1——1
'10 20 30 М 50 60
Степень перфорации °/о
Рис. 2-24. Зависимость на
пряжения на электролизере
от степени перфорации
анода при различной плот-
ности тока [толщина анода
и диаметр отверстий пер- .
форации
(в мм: ।
и 6)]:
1 — 2 кА/м2; 2 — 4 кА/м2; 3 —
6 кА/м2; 4 — 8 кА/м2. '
соответственно
a — 10 и 8; 6 — 3
возрастает из-за развития поверхности стенок отверстий перфорации
и уменьшения экранирующего действия газа. При этом эффективное
сопротивление слоя электролита уменьшается и снижается напря-
жение на электролизере. С ростом степени перфорации уменьшение
напряжения будет продолжаться до тех пор J пока не достигнет
некоторого оптимального значения. При дальнейшем увеличении
степени перфорации эффект отвода газа не будет покрывать эффекта
от потери лобовой поверхности анода, обращенной к катоду, и эффек-
тивное сопротивление электролита вновь начнет возрастать, вызывая
увеличение напряжения на электролизере. Кроме того, с увеличе-
нием степени перфорации анода возрастают потери напряжения
на преодоление омического сопротивления внутри материала анода
за счет уменьшения сечения для прохождения тока вдоль анода.
Баланс напряжения и расхсд электроэнергии
на электролиз
*
Стоимость электроэнергии, расходуемой на электролиз, соста-
вляет значительную часть общих затрат на производство, расход
электроэнергии постоянного тока на единицу продукции прямо
пропорционален напряжению на электролизере и обратно пропор-
' ционален выходу продукта по току.
Ддя определения удельного расхода электроэнергии постоянного
тока (в кВт*ч/т) можно пользоваться выражением:
И7_ 1000
т т) 4
где т — электрохимический эквивалент, г/(А-ч).
*
Значение К составляет:
(2.44)
хлора ..............................
100%-ной NaOH ......................
92% -ной NaOH ......................
100%-ной КОН . .................
расход электроэнергии постоянного
755
670
616
477
тока
зависи-
расчете
для
ДЛЯ
ДЛЯ
для
Удельный
мости от напряжения на электролизере и выхода по току в расчете
на хлор и 100%-ную каустическую соду приведен в табл. 2-8 и 2-9.
В современных конструкциях электролизеров выход продуктов
по току мало зависит от типа электролизера и при правильном режиме
эксплуатации колеблется от 94 до 97%. Поэтому значение удельных
затрат электроэнергии постоянного тока определяется в основном
напряжением на электролизере.
Удельный расход электроэнергии переменного тока зависит
также от коэффициента полезного действия преобразовательной
подстанции:
^пер^^/4! (2.45)
где И^пер — удельный расход электроэнергии переменного тока на единицу
продукции, кВт • ч; 41 — общий коэффициент полезного действия преобразо-
вательной подстанции.
*
у
Таблица 2-8. Расход электроэнергии постоянного тока на 1 т хлора (вкВт-ч)
W
г'М;
W
’АВ.:
Ъ:;-
Выход по току, %
90
92
94
98
96
Напряжение на
электролизере, В
3,00 2519,27 2464,50 * 2412,07 ’ 2361,82 2313,61
3,20 2687,22 2628,80 2572,87 2519,27 2467,85
3,40 2855,17 2793,10 2733,68 2676,72 "2622,10
3,60 3023,12 2957,40 2894,48 2834,18 2776,34
3,80 3191,07 3121,70 3055,28 2991,63 2930,58
4,00 3359,03 3286,00 3216,09 3149,09 3084,82
4,20 3526,98 3450,30 3376,89 • 3306,54 3239,06
4,40 3694,93 3614,60 3537,70 3463,99 3393,30
4,60 3862,88 3778,90 3698,50 3621,45 3547,54
4,80 4030,83 3943,20 3859,31 3778,90 3701,78
'5,00 4198,36 4107,51 4020,11 3936,36 3856,03
5,20 4366,73 4271,81 4180,92 4093,81 4010,27
5,40 4534,69 4436,11 4341,72 х. 4251,27 4164,51
Таблица 2-9. РасхЬд электроэнергии постоянного тока на 1 т
100%-ного NaOH (в кВт-ч)
Напряжение на
электролизере, В
Выход по току, %
к* • •
90
92
94
96
98
3,0
3,6
5,0
2231,1
2378,5
2532,6
2680,0
2827,4
2974,8
3128,9
3276,3
3571,1
3718,5
3872,6
4020,0
2184,2
2331,6
2472,3
2619,7
2767,1
2914,5
3055,2
3202,6
3350,0
3497,4
3638,1
3785,5
3932,9
2137,3
2278,0
2425,4
2566,1
2706,8
2854,2
2994,9
3135,6
3276,3
3423,7
- 3564,4
3705,1
2090,4
2231,1
2371,8
2512,5
2653,2'
2793,9
2927,9
3068,6
3209,3
3350,0
3490,7
3631,4
3765,4
2050,2
2184,0
2324,9
2458,9
2599,6
2735,6
2867,6
3008,3
3142,3
3283,0
3417,0
3557,7
3691,7
t:
4,0
Общее напряжение на электролизере Е зависит от ряда факторов
и может быть представлено как сумма отдельных составляющих,
определяющих значения скачков потенциала на электродах и .потерю
напряжения на отдельных участках пути прохождения тока в элек-
тролизере.
Общее напряжение на электролизере Е может быть записано
для электролизера с твердым катодом
Е = фа “Fфк4~*Па“Ь *]r< “F Еэл “Ь-^Диаф “Н^диф’Ь ^ан “Ь-^катп-^конт (2.46)
для электролизера с ртутным катодом
Е “фа 4~фк + + #эл+ #ан + #k+#КОНТ
(2.47)
83
где фа» фк “ равновесные потенциалы анода и катода; ца, т}к — перенапряже-
ние на аноде и катоде; £Эл — падение напряжения на преодоление электриче-
ского сопротивления электролита; £диаф — падение напряжения в диафрагме;
Ядиф — диффузионный потенциал на границе кислого анолита и щелочного
католита; £ан — падение напряжения на аноде; £кат — падение напряжения
на катоде; £конт — падение напряжения в контактах, подводящих ток шин
с катодом и анодом.
Общее напряжение на электролизере в большой степени зависит
от применяемой плотности тока, рабочей температуры электролиза
состава электролита, материала электродов и конструкции электро-
лизера.
Конструкция электролизера определяет расстояние между рабо-
тающими поверхностями электродов, условия отвода образующихся
в процессе электролиза газов, а также геометрические формы и раз-
меры электродов, от которых зависят потери напряжения на преодо-
ление электрического сопротивления электродов и токопроводящих
деталей электролизера.
Ниже будут рассмотрены факторы, влияющие на величину отдель-
ных составляющих напряжения на электролизере.
'V
Равновесные потенциалы анода и катода
В условиях работы промышленных электролизеров с твердым
катодом на аноде происходит разряд ионов хлора из растворов NaCl
концентрацией около 265 г/л, или 4,53 моль/л, а на катоде разряд
ионов водорода из растворов, содержащих примерно 120 г/л, или
3 г-экв/л, NaOH, и 190 г/л, или 3,35 г-экв, NaCl.
Нормальный потенциал выделения хлора фа по реакции
ч С1“ ~ 1/ 2OI2 4” &
составляет при комнатной температуре фа = 1,359 В.
Равновесный потенциал выделения хлора зависит от активности
хлор-ионов в растворе и составляет
фа —Фа (^Cl-/ci”) (2.48)
• где Cci-концентрация ионов хлора, г-ион/л; /с1—- коэффициент актив-
ности ионов хлора.
.л
Принимая /ci- = 0,810, получим значение равновесного потен-
циала анода при комнатной температуре:
фа = 1,359 - 0,0001987 1g (4,53 - 0,810) = 1,326 В
Нормальный потенциал выделения водорода на катоде
1/2Н2“^=Н+
равен при комнатной температуре нулю (ф° = 0,Q В).
Равновесный потенциал выделения водорода на катоде из раствора
католита составляет г
^4-^lg(<WH+) (2.49)
где Сц+ , /н+ — концентрация и активность ионов водорода в католите.
J.
84
При комнатной температуре активность ионов ОН в католите,
содержащем 3 г-экв/л NaOH и 3,35 г-экв/л NaCl, можно принять
равной /он- = 0,785 и К - [Н+Ь [ОН~] = 1,05-10^14. Тогда
Фк=^+0;0001987Че = -0,846В
О,U • U, /оЭ
Теоретическое напряжение разложения растворов поваренной
соли при комнатной температуре составит
Ео = Фа — фк = 1,326 — (-0,846) = 2,172 В.
Температурный коэффициент зависимости э. д. с. равен
^/^ = 0,0004 В/9С.
I - . •
Поэтому теоретическое напряжение разложения при 95 °C соста-
вит
Я0 = фа — фк = 2,172 —0,0004(95 —25) = 2,144В
При электролизе с ртутным катодом равновесный потенциал
анода можно определить по выражению (2.48), а равновесный потен-
циал амальгамного катода может быть рассчитан по уравнению
0 2,ЗЯТ ^Na+/Na+
Фак = ф£к--Z~Fr~ 1g 7;—7--- (2.50)
GNa*Na
гДе Фак — стандартный потенциал амальгамы натрия можно принять равным
—1,849В [17,23]; CNa+; /Na+ и ^Na,/Na— концентрация и коэффициент ак-
тивности ионов натрия в растворе и натрия в амальгаме.
•I *
Теоретическое напряжение разложения при электролизе с ртутным
катодом составляет около 3,1 В и практически мало меняется в усло-
виях промышленного электролиза [182].
Перенапряжение на аноде и катоде
Процесс разряда ионов хлора и водорода всегда происходит при
потенциале более высоком, чем термодинамически обратимый, т. е.
наблюдается явление перенапряжения. Перенапряжение зависит
от материала электрода, состояния его поверхности, плотности тока,
температуры процесса, состава электролита, продолжительности
процесса электролиза и других причин.
На платинированной платине перенапряжение выделения водо-
рода и хлора невелико, однако на других электродных материалах
оно составляет значительную часть общего напряжения на ячейке.
Равновесный потенциал разряда на аноде молекул воды с выделе-
нием газообразного кислорода ниже равновесного потенциала выде-
ления хлора, поэтому получение практически чистого хлора при
электролизе водных растворов хлоридов щелочных металлов ста-
новится возможным из-за большего (по сравнению с хлором) пере-
напряжения выделения кислорода на применяемых в практике
анодных материалах: графите, платине, окислах рутения или магне-
тите.
85
Таблица 2Л0. Перенапряжение Хлора и Кислорода (в мЙ)
Перенапряжение связано с замедлением скорости одной из стадий
электродного процесса. Перенапряжение выделения водорода было
подробно исследовано и достаточно освещено в литературе [13, 16].
Перенапряжение выделения хлора в различных условиях изучено
менее подробно. Однако определению значения потенциала выделе-
ния хлора на графитовых анодах в условиях работы промышленных
электролизеров посвящено значительное количество работ как в на-
шей стране [183—185], так и за рубежом [186—190].
\ Перенапряжение выделения хлора и кислорода на платине,
графите и магнетите приведено в табл. 2-10.
С увеличением плотности тока растет разница между значениями
перенапряжения для кислорода и хлора. Особенно велика зта раз-
ница на платиновых электродах. Поэтому при применении плати-
новых анодов анодный процесс идет с очень высоким выходом хлора
по току; происходит незначительный разряд кислорода на платино-
вом аноде из концентрированных растворов NaCl.
Невелико перенапряжение выделения хлора также на окисно-
рутениевом аноде. Приводятся данные [173], что перенапряжение
выделения хлора на окиснорутениевом аноде не превышает 50 мВ
при плотности тока 10 кА/м2.
С ростом температуры потенциал выделения хлора несколько
снижается. В табл. 2-11 приведены значения потенциалов выделения
хлора на разный материалах при различных температурах.
Плотность
тока,
А/м»
Таблица 2-11- Потенциал выделения хлора из 22—24%-ных растворов NaCl
Температура, °C
16,5 ' 50 ч_ 75 25 50 * 75 25' 50 75
М агнетит
1,85 1,75 1,74
1,90 1,80 1,79
1,95 1,84 1,83
1,98 1,87 1,87
2,01 1,90 1,90
2,04 1,92 1,91
2,10 1,94 1,92
2,12 1,96 1,93
1,99 1,94
— 2,01 1,95
П л а т и/н и р о - г р а ф и т
ванная
- платина
100 1,36 1,34 1,34 1,43 1,38 1,33
200 ' 1,37 1,35 1,35 1,49 1,44 1,35
300 1,37 1,35 1,35 4,52 ' 1,47 1,38
400 1,38 1,36 1,35 1,55 1,48 1,40
500 1^,38 1,36 1,35 1,58 1,50 1,42
600 1,39 1,36 1,36 1,61 1,51 1,44
700 1,39 1,37 1,37 1,64 1,52 1,45
800 1,40 1,38 1,66 1,53 1,46
900 1,40 1,38 * 1,68 1,54 1,47
1000 1,41 1,39 1 1,70 1,55 1,48
Значение перенапряжения выделения хлора зависит также от
сорта углеграфитового материала.
Потенциал выделения хлора на одном и том же сорте графита
может существенно изменяться в зависимости от предварительной
проработки анода.
87
f
В табл. 2-12 приведены значения потенциала выделения хлора
на графите из раствора 300 г/л NaCl при pH = 4,0—4,5 и различный
плотностях тока и температуре в зависимости от предварительной
проработки4 анодов.
Таблица 2-12. Потенциалы выделения хлора из раствора NaCl
(предварительная проработка анода при плотности тока 1,25; 2,00
и 3,00 А/см2) [78]
)СТЬ Мем2 Температура, °C *
Плотнс тока, j 37 49 58 । 68 ! 37 ч 49 58 63 37 49 68 *
Предварительная проработка
0,045 * При 1,25 А/см2 При 2,0 А/см' 2 При 3,0 А/см2
0,045 1,460 1,423 1,392 1,363 1,448 1,413 1,382 1,362 1,436 1,400 1,354
0,055 1,466 1,428 1,403 1,368 — 1 >
0,065 1,477 1,436 1,408 1,372 1,467 1,426 1,395 1,370 1,458 1,416 1,369
0,075 1,484 1,441 1,413 1,382 11 » — ——
0,085 1,492 1,448 1,419 1,388 1,484 1,442 1,407 1,382 1,462 1,432 1,380
0,100 1,500 1,457 1,428 1,397 — * — —
0,125 1,517 1,466 1,438 1,407 1,507 1,456 1,423 1,394 1,482 1,447 1,392
0,150 1,528 1,477 1,449 1,413 1,516 1,468 1,432 1,403 1,493 1,456 1,396
0,200 1,547 1,493 1,462 1,426 — - — — —
0,250 1,558 1,509 1,472 1,436 1,544 1,495 1,453 1,421 1,518 1,482 1,417
0,300 1,574 .1,521 1,482 1,447 < 1 — * —— i —
0,350 1,584 1,525 1,488 1,455 1,560 1,515 1,465 1,432 — -
0,400 1,600 1,540 1,498 1,462 1,467 — * - — *- ™
0,450 1,610 1,548 1,508 1,575 1,527 1,481 1,439 1,550 1,510 1,434
0,500 1,618 1,558 1,512 1,472 1,582 1,536 1,486 1,443 1,556 1,515 1,439
0,600 1,638 1,570 1,524 1,480 — ! —
0,700 1,654 1,582 1,534 1,487 1,605 1,552 1,500 1 1,455 1,575 1,530 1,452
0,800 1,663 1,596 1,545 1,498 - 1 у ——
0,900 1,674 1,605 1,554 1,501 1,623 1,565 1,509 1,463 1,587 1,543 1,547 1,459
1,000 1,687 1,615 1,560 1,508 1,631 1,570 1,512 1,466 1,595 1,467
1,100 1,697 1,625 1,567 1,515 *' — — 1 1—
1,250 1,708 1,635 1,577 1,527 1,646 1,584 1,528 М76 1,612 1,554 1,471
1,400 1,654 1,592 1,533 1,480 1,618 1,568 1,474
1,500 * м 1,661 1,597 1,538 1,487 1,625 1,574 1,478
1,750 — — 1,670 1,604 1,546 1,494 1,634 1,582 1,486
2,000 \ 1 1,679 1,612 1,553 1,506 1,643 1,590 1,496
2,250 — 1 — — J — — — ' 1,653 1,597 1,500
2,500 — — — > — 1,658 1,604 1,505
2,750 — 1 U- —— . — t 1,664 1,610 1,513
3,000 — — —- — — — — 1 —11 1,672 1,614 ;1,518
На рис. 2-25 даны [23] значения потенциала выделения хлора
на графитовом электроде в широком интервале плотностей тока
и температур. Перенапряжение выделения водорода в зависимости
от материала твердого катода приведено в табл. 2-13.
Предложено много способов снижения перенапряжения выделе-
ния водорода путем подбора материала катода или покрытия катода
слоем материала с более низким перенапряжением [191—197].
88
Материал
Таблица 2-13. Перенапряжение выделения водорода из 16%-ного раствора
NaOH при 80 °C
Плотность тока, А/м2
100 500 юоо 2000
Никель катайый .............
Железо
никелированное . ... . .
обработанное струей песка
Сталь с присадкой 5% Ni . .
Железо, покрытое NiS . . . .
Платинированная платина . .
Однако при проверке этих
•. • • • 360 390 420 470
♦ • • • 160 240 260 300
« • » 120 180 220 270
» • * • 110 150 180 230
' • » 20 60 80 100
• • ♦ • 10 30 45 55
предложений в производственных уело
катодного потенциала
виях не удалось воспроизвести снижение
и напряжения на электролизере, полу-
ченного в лабораторных условиях.
Исследования, проведенные Кохановым
и др. [198, 199], показали, что на потен-
циал катода, устанавливающийся при дли-
тельной работе хлорного электролизера,
большое влияние оказывает присутствие
небольших количеств иона СЮ" в като-
лите. В присутствии этих ионов пред-
отвращается рост во времени катодного
потенциала на стальном катоде, наблю-
даемый обычно при проведении опытов
в «чистых» условиях, т. е. в отсутствие
ионов С1О"\
При температурах, близких к обыч-
ным рабочим температурам промышлен-
ного электролиза, в присутствии СЮ"
в католите устанавливается потенциал
катода значительно более низкий по сра-
внению с потенциалом стального катода
в чистых условиях. Это явление можно
объяснить тем, что в присутствии гипо-
хлорита возможно протекание локаль-
ных процессов растворения и после-
дующего осаждения железа катода, при-
водящих к развитию поверхности катода
и снижению перенапряжения выделения
водорода.
Разряд ионов натрия на амальгам-
ном катоде проходит практически без
Рис. 2-25. Зависимость по-
тенциала выделения хлора
на графитовом электроде
от плотности тока при раз-
личных температурах:
1 — при 40 °C; 2 — при 50 °С^
3 при 60 °C; 4 — при 70 °С^
5 — при 80 °C; 6 — при 85 °C.
перенапряжения. Потенциал ртутного катода определяется концен-
трацией щелочного металла в поверхностном слое амальгамы.
89
Перенапряжение выделения водорода на ртутном и амальгамном
катоде высоко, что обеспечивает возможность проведения процесса
электролиза с образованием амальгамы
и
(елочных металлов с высо-
ким выходом по току. При попадании амальгамных ядов на поверх-
ность амальгамы образуются местные включения с низким пере-
напряжением выделения водорода и наблюдается увеличение расхода
тока на разряд водорода.
Паден ие и а п р я жен и я в электролите
Для наиболее распространенного в практике электролиза случая,
когда плоские электродные поверхности расположены параллельно
Друг другу, падение напряжения в электролите (в В) может быть
определено из выражения:
= (2.51)
где i — плотность тока в электролите, А/см2; р — удельное сопротивление
электролита, равное 1/х, Ом • см; х — удельная электропроводность электро-
лита, Ом"1 •см*1; Ъ — расстояние между электродами, см; К — коэффициент
увеличения удельного сопротивления электролита за счет газовых пузырьков.
Если работающие поверхности анодов и катодов отличаются
друг от друга, для расчета i можно принять усредненное значение
рабочей поверхности электродов.
Удельндя электропроводность растворов NaCl возрастает с повы-
шением концентрации и температуры раствора. Значение электро-
Таблица 2~14. Удельная электропроводность растворов NaCl при
различных температурах
G °с
Ом-^’см**1
т-----------
Ом"1 .см-1
X,
Ом-1, см'1
д
3,74
4,29
4,76
5,26
анные Ангела^
[200]
. 18,0
31,2
51,2
69,8
70,0
18,0
30,1
50,2
70,5
18,0
30,2
50,1
70,5
18,0
йЗО,1
*50,3
70,5
Данные Гантмана с
с о т р. [201]
0,1933
0,2501
0,3419
0,4298
0,4328
0,2048
0,2603
0,3599
0,4615
0,2098
0,2707
0,3757
0,4850
0,2150
0,2760
0,3855
0,4997
4,2
4,3
4,4
4,5
4,6
80
90
95
80
90
95
80
90
95
80
90
95
80
90
95
0,511
0,565
0,593
0,516
0,567
0,595
0,522
0,571
0,596
0,523
0,575
0,600
0,527
0,579
0,606
4,7
№
4,8
4,9
5,0
5,05
80
90
95
80
90
95
80
90
95
80
90
95
80
90
95
0,530
0,585
0,612
0,537
0,590
0,617
0,541
0,596
0,622
0,548
0,605
0,628
0^49
0,606
0,629
90
г
Таблица 2-15. Удельная электропроводность растворов Na€l и NaOH
(в Ом"1 • см’1)
Состав раствора при 20 °C, г/л Т е м п е р а ту р а, °C и
NaCl NaOH и 25 50 60 70 80
о
50
60
70
80
90
100
0
50
60
70
80
90
166
0
50
60
70
80
90
100
310
155
124
93
62
31
о
300
150
120
90
60
30
0
290
145
116
87
58
29
0
о
106,0
127,2
148,4
169,6
190,8
212
О
102,6
123,1
143,6
164,2
184,7
205,2
О
99,2
119,0
138,9
158,7
178,5
198,4
0,2502
0,3178
0,3310
0,3437
0,3570
0,3705
0,3906
0,2483
0,3181
0,3324
0,3483
0,3639
0,3798
0,3981
0,2454
0,3205
0,3368
0,3527
0,3697
0,3864
0,4040
проводности концентрированнь^
температурах приведены в табл.
Удельная электропроводность
0,3828
0,5184
0,5446
0,5738
0,6060
0,6386
0,6710
0,3793
0,5184
0,5468
0,5770
0,6082
0,6415
0,6749
0,3728
0,5158
0,5467
0,5795
0,6119
0,6460
0,6789
0,4380
0,6000
0,6384
0,6779
0,7168
0,7558
0,7968
0,4341
0,6028
0,6418
0,6799
0,7197
0,7586
0,7988
0,4268
0,6029
0,6419
0,6800
0,7197
0,7600
0,8014
0,4962
0,6968
0,7399
0,7853
0,8309
0,8780
0,9290
0,4909
0,6875
0,7322
0,7772
0,8249
0,8775
0,9283
0,4824
0,6914
0,7365
0,7812
0,8274
0,8771
0,9278
0,5523
0,7876
0,8412
0,8952
0,9507
' 1,0072
1,0656
0,5462
0,770
0,8334
0,8882
0,9456
1,0021
1,0585
0,5368
0,7711
0,8230
0,8781
0,9357
0,9954
1,0548
растворов NaCl при различных
2-14.
электролитических щелоков при
различных степени превращения хлорида в гидроокись и исходном
содержании поваренной соли в рассоле приведена в табл. 2-15 и 2-16»
Таблица 2-16 • Удельная электропроводность электролитических щелоков
При 95 °C
40
50
55-
60
70
При 80 °с
При 90 °с
[Степень превра-
щения, %
2,42 3,63w 0,7445 2,49 3,75 0,8555 2,60 3,87 0,920
3,09 3,12 0,7915 3,20 3,23 0,9025 3,39 3,36 0,985
. 3,48 2,84 0,8156 3,62 2,97 " 3,83 3,15 0,972
3,84 2,58 0,8410 4,07 2,72 0,9920 4,29 2,88 1,050
4,65 2,00 0,8955 4,95 2,12 1,045 5,40 2,35 1,091
91
Газонаполнение^ %
Рис. 2-26. Увеличение
удельного сопротивле-
ния электролита в за-
висимости от газон апо л-
нения:
1 — измеренное; 2 — рас-
считанное.
Газонаполнение электролита определяется соотношением ско-
рости образования газовых пузырьков в электродном пространстве
ячейки в процессе электролиза и скорости удаления их из электро-
лита. Газонаполнение электролита возрастает с увеличением плот-
ности тока, высоты электродов и вязкости электролита. Оно повы-
шается также при уменьшении межэлектродного расстояния и объема
электродного пространства.
При повышении температуры снижается вязкость электролита
и облегчается подъем пузырьков газа в электролите, но увеличи-
вается объем газов. При температуре выше 70—80 °C йроисходит
быстрый рост парциального давления паров
воды, насыщающих газ в пузырьках, увели-
чивается объем влажного газа, выделя-
ющегося в электродном пространстве, и как
следствие этого, возрастает газонаполнение
электролита [202].
Зависимость удельного сопротивления
электролита от газонаполнения [203] пока-
зана на рис. 2-26. Для расчета электропро-
водности электролитов при различном газо-
наполнении предложен [204—206] ряд урав-
нений, расчет по которым дает результаты,
близкие к данным, приведенным на рис. 2-26.
На основании экспериментальных дан-
ных подсчитано [207], что для электроли-
зера с диафрагмой, работающего при плот-
ности тока 715 А/м2, межэлектродном рас-
стоянии 17 мм и высоте электродов 1,4 м,
удельная электропроводность электролита
в верхней части электролизера снижается вследствие газонаполнения
в 1,2—1,3 раза при рабочей температуре 90 °C и в 1,5 раза при 100 °C.
Газонаполнение электролита зависит также от условий отвода из
него газовых пузырьков, в частности от их размеров и наличия
организованной циркуляции электролита, способствующей подъему
и удалению пузырьков газа. Снижение газонаполнения анолита
достигается за счет создания естественной циркуляции внутри элек-
тролизера. С этой целью в конструкции электролизеров предусматри-
ваются так называемые циркуляционные вертикальные каналы,
свободные от электродов и4 соединенные тем или иным способом
с верхней и нижней частями электродных пространств всех ячеек
электролизера.
При этом в электродных пространствах ячеек газонаполненный
электролит поднимается вдоль электродов вверх, а в циркуляцион-
ном канале электролит, освобожденный от газовых пузырьков,
опускается вниз и вновь поступает в нижнюю часть электродных
пространств ячеек электролизера. 4
Движущая сила циркуляции электролита Д77 (в мм вод. ст.)
обусловливается различием удельных весов у х — чистого и у 2 — газо-
наполненного электролита и для ячейки высотой I мм может быть
^айдена из выражения
ДЯ^ЦУх-уз) (2.52)
Интенсивность циркуляции электролита зависит от газонаполне-
ния, высоты электролитической ячейки, полноты выделения газовых
пузырьков из электролита перед поступлением его в циркуляционный
канал и гидравлического сопротивления на пути движения цирку-
лирующего электролита.
Предложены также конструкции электролизеров с организован-
ной циркуляцией электролита через наружный сепаратор для выде-
ления из него хлора [208]. Такой сепаратор может быть использован
также для донасыщения твердой солью циркулирующего анолита
[209].
Помимо электропроводности газонаполнение анолита и католита
оказывает влияние также на давление фильтрации жидкости через
диафрагму. В зависимости от газонаполнения в анодной и катодной
частях ячейки давление фильтрации может изменяться по высоте
ячейки. Газонаполнение анолита всегда стремятся свести к минимуму,
организуя с этой целью циркуляцию электррлита.
Газонаполнение в катодной части ячейки можно регулировать
в определенных пределах путем изменения конструктивных пара-
метров катодного элемента: с уменьшением толщины катодной ячейки
и объема катодного пространства газонаполнение католита воз-
растает. Изменяя толщину катодного пальца, можно регулировать
газонаполнение католита и изменять его плотность.
На рис. 2-27 нанесены кривые, характеризующие изменение по
высоте диафрагмы гидростатического давления со стороны анолита
(кривая 1) и со стороны католита при различных значениях толщины
катодной ячейки (кривые 2—5), при межэлектродном расстоянии
18 мм, температуре 90 °C и концентрации щелочи в католите 130—
140 г/л. Как видно из рисунка, для катодной ячейки толщиной 40 мм
в нижней части диафрагмы давление фильтрации меняет свой знак.
При толщине катодной ячейки менее 10 мм (или толщине катодного
пальца при двусторонней работе катода менее 20 мм) создаются
условия для поддержания давления фильтрации постоянным по
высоте диафрагмы.
Для различных точек по высоте электролитической ячейки по-
тери напряжения в аноде Д2?а и в электролите АЕЭл существенно
меняются, что приводит к изменению плотности тока и к соответ-
ствующему изменению электродных потенциалов и потерь напря-
жения в диафрагме.
Потеря напряжения на катоде и металлических проводниках
ДЯК также может изменяться по высоте электролитической ячейки,
однако абсолютное значение этой величины невелико и ее измене-
ния не оказывают существенного влияния на баланс напряжения.
В работе [210], приняв линейную зависимость потенциала элек-
трода от плотности тока, выведено выражение для расчета распреде-
93
ления плотности тока по высоте электрода. Результаты расчетов,
проведенных на аналоговой электронной вычислительной ма
ине
применительно к электролизеру с нижним подводом тока, показаны
на рис. 2-28. Распределение плотности тока по высоте электрода
отклоняется от линейной зависимости. Неравномерность в распреде-
лении плотности тока по высоте электрода сильно возрастает к концу
тура работы электродов. По-видимому, кривые на рис. 2-28 показы-
вают картину распределения плотности тока по высоте электрода,
близкую к действительности. Изучение распределения плотности
тока в электролизере с диафрагмой при высоте электродов, равной
1100 мм, указывает [211] на то, что при низких плотностях тока
(г—500 А/м2) и для новых анодов неравномерность плотности тока
Высота у мм
Рис. 2-27. Изменение гидростатического давления
анолита и католита по высоте диафрагмы при раз-
личной толщине катодной ячейки:
1 — давление анолита; 2 — давление католита при толщине
катоДной ячейки 40 мм; 3 — то же, при s20 мм; 4—то же, при
10 мм; 5 — то же, при 5 мм.
по высоте электрода невелика, однако она возрастает при увеличе-
нии плотности тока и уменьшении межэлектродного расстояния и
снижается с повы
лита.
и
сением температуры и скорости подачи электро-
!Г/-*
!'3*
.If
*
if-
Г ‘3-
ж
^ Увеличение удельного сопротивления электролита за счет газо-
llнаполнения приобретает большое значение с возрастанием плотности
Рис. 2-28. Соотношение действительной и средней плотности тока на различной
высоте электрода.
При плотности тока 0,1 А/см2: 1 — в начале тура; 2 — в середине тура; з — в конце тура
работы анодов.
При плотности тока 0,05 А/см2: 4 — в начале тура; 5 — в середине тура; 6 — в конце тура
работы анодов.
I*
f
f
тока и при горизонтальном расположении электродов, как это на-
блюдается, например, при электролизе с ртутным катодом.
Исследование^ газонаполнения и разработка различных спосо-
бов его снижения в последние годы являются предметом многих
Г
95
работ [212]. Основным методом снижения газонаполнения при гори-
зонтальном расположении анодов является перфорация анодов.
Для уменьшения газонаполнения анолита предложено также распо-
лагать аноды с уклоном или смывать пузырьки газа струей жидкости,
быстро протекающей по поверхности анода [213—217]. Для электро-
лизеров с ртутным катодом предложена с целью увеличения ско-
рости движения электролита рециркуляция части анолита через
электролизер без его донасыщения [218]. Специальной перфорацией
анодов стараются создать условия для непрерывной циркуляции
электролита у рабочей поверхности анода с целью быстрого отвода
пузырьков газа с рабочей поверхности анода и из межэлектродного
пространства ячейки [219, 220]. Для этой цели предложено снаб-
жать графитовые плиты мелкими отверстиями для подъема пузырьков
газа и крупными для опускания электролита после отделения его
от газа. Таким образом обеспечивается внутренняя замкнутая цир-
куляция электролита.
Влияние степени перфорации графитовых анодов на потерю на-
пряжения в электролизере с ртутным катодом вследствие изменения
газонаполнения электролита видно из данных* приведенных
в табл. 2-17 [36].
Таблица 2-17. Зависимость потерь напряжения за счет газонаполнения
электролита от размеров анодного элемента (в В)
Размеры анодного
элемента, мм
Плотность тока, кА/м2
1 2 а 3 5 7
51 X 51
24 X 24
12 X 12
6X6
0,14
0,07
0,03
0,02
0,28
0,14
0,06
,0,02
0,42
0,21
0,09
0,03
0,70
0,35
0,15
0,05
0,98
0,49
0,21
0,07
1,40
0,70
0,30
0,10
При высоких плотностях тока облегчение удаления пузырьков
хлора из-под анода за счет перфорации приобретает большое зна-
чение .
В электролизерах с твердым катодом, работающих при сравни-
тельно невысоких плотностях тока (0,10—0,15 А/см2),
коэ
ициент
увеличения сопротивления электролита за счет газонаполнения
обычно не превышает 1,2—1,3. При начальном межэлектродном рас-
стоянии 1,1—1,3 см средние потери напряжения на преодоление
омического сопротивления электролита и диафрагмы составляют
при плотности тока около 1 кА/м2 около 0,5 В. По мере износа гра-
фитовых анодов межэлектродное расстояние возрастает, а следова-
тельно, увеличиваются и потери напряжения в электролите.
В электролизерах с ртутным катодом, при межэлектродном рас-
стоянии 3 мм, плотности тока 10 кА/м2 и интесивной перфорации
анода потеря напряжения в электролите составляет около 1,1 В,
в том числе около 0,6 В за счет газонаполнения.
96
W'
В электролизерах с твердым катодом состав электролита в порах ,
диафрагмы может быть неодинаковым по ее толщине. В слое диа-
рагмы, непосредственно примыкающей к катоду, сортав слоя анало-
гичен составу католита. Состав электролита с обратной стороны
диафрагмы зависит от режима работы электролизера. При низкой
концентрации щелочи в католите, когда скорость противотока суще-
ственно выше скорости движения ионов'QH", поры диафрагмы запол-
нены в основном кислым хлорсодержащим анолитом. Когда ско-
рость противотока электролита равна или близка к скорости движе-
ния ионов ОН- , граница между кислым анолитом и щелочным като-
литом проходит в толще диафрагмы. При очень высокой концентрации
щелочи в католите (более 155 г/л), когда скорость противотока
меньше скорости движения ионов ОН", диафрагма полностью про-
питана католитом, и граница между кислым анолитом и католитом
проходит по жидкости с анодной стороны диафрагмы.
Для расчетов работы электролизера в оптимальных условиях
можно принимать состав электролита в порах диафрагмы аналогич-
ным составу католита.
Падение напряжения на преодоление электролитического сопро-
тивления диафрагмы может быть определено из выражения
£диаф = -^р- (2.53)
II
где i — плотность тока в диафрагме, А/см2; р — удельное сопротивление элект-
ролита, заполняющего поры диафрагмы, - Ом • см; b — толщина диафрагмы, см;
d — объемная пористость, доли; р — коэффициент извилистости, пор, характе-
ризующий отношение длины пор к толщине диафрагмы.
Диффузионный потенциал £ДИф на границе между кислым ано-
литом и щелочным католитом невелик и в практических расчетах
и
его можно не учитывать. .
Потери напряжения на преодоление омического сопротивления
анода и катода Еа и Ек рассчитываются по закону Ома, как для про-
водников первого рода. Необходимо учитывать, что при применении
графитовых анодов по мере их износа сопротивление анодов возра-
стет как за счет уменьшения их сечения, так и в результате увели-
чения удельного сопротивления графита, обусловленного ухудше-
нием электрического контакта между отдельными зернами графита
в оставшейся неразрушенной части графитового анода.
i'4'V''
$1 римерные балансы напряжения
.ЭД. л е к т р о л и з ер о в
^Напряжение на электролизере зависит от многих факторов.
Н$|более сильное влияние оказывают применяемая плотность тока;
РЭДЙая температура электролиза, состав электролита, материал
элв^родов и конструкция электролизера, определяющая расстояние
«Заказ 843 ' 97
между электродами, а также способ отвода образующегося в про-
цессе электролиза хлора.
С повышением плотности тока увеличивается напряжение на
электролизере и расход электроэнергии, однако снижаются капита-
ловложения и удельные затраты труда на единицу продукции.
Экономически выгодную плотность тока рассчитывают для конкрет-
ных условий производства, она зависит прежде всего от стоимости
электроэнергии [219—222]. В настоя
и
(ее время для таких расчетов
используются электронно-вычислительные машины.
При электролизе с твердым катодом напряжение возрастает во
время тура работы электродов вследствие износа электродов и за-
бивки диафрагмы. При электролизе с ртутным катодом напряжение
в большой степени зависит от перфорации анода и способа регули-
рования межэлектродного расстояния по мере износа анодов.
В табл. 2-18 ^приведен [78, 223] баланс напряжения на электро-
лизерах с твердым катодом Б ГК-17 при различной плотности тока.
Таблица 2-18* Баланс напряжения на электролизерах БГК-17
при различной плотности тока (в В)
Плотность тока, А/м2
Показатели
f
520
730
910
Температура электролита, °C .........
Общее напряжение на электролизере..............
Потенциал
анода......................................
катода....................................
Падение напряжения, В
в электролите и диафрагме ........
» катоде .....................................
» аноде ............................. . .
» анодном днище . ........................
» анодной шине ...........................
» катодной шине ..........................
» ДРУГИХ прово,
ПИН
ках и контактах
Сумма составляющих
Расхождения
90
3,305
1,525
0,962
0,316
0,061
0,312
0,061
0,061
0,014
0,046
3,358
0,053
1,6
92
3,456
1,576
0,972
0,476
0,055
0,332
0,046
0,070
0,016
0,056
3,599
0,143
4,0
94
3,542
1,583
0,975
0,516
0,047
0,346
0,039
0,065
0,010
0,063
3,611
0,102
2,8
В
% .
среднему за тур
Баланс составлен для напряжения, близкого к
работы электролизера, рабочей температуры, установившейся при
соответствующем напряжении на электролизере, если питание его
производится рассолом при температуре около 80 °C.
В табл. 2-19 ^приведены [23] расчетные значения составляющих
баланса для ртутных электролизеров при межэлектродном расстоя-
нии 3 мм и такой^перфорации электрода, когда отдельные элементы
анода составляют 15 мм. Каждой плотности тока отвечает рабочая
температура электролита, установившаяся в процессе работы.
98
Таблица 2-19. Баланс напряжения на электролизере с ртутным катодом
при различной плотности тока (в В)
Плотность тока, кА/ма
Показатели
1
ю
Температура электролиза, РС . . .
Равновесный потенциал
катода............................
анода................'. . - . .
Перенапряжение
на катоде .... ...................
на аноде .....................
Падение напряжения в электролите
без учета газонаполнения . . .
за счет газонаполнения ......
/ Средний прирост напряжения за счет
износа анодов.................. .
Потеря напряжения в проводниках пер-
вого рода.........................
Общее напряжение ................
45
1,72
1,32
0,04
0,10
0,09
0,06
0,02
0,2
3,50
60
1,77
1,33
0,07
0,12
0,14
0,12
0,04
0,2
3,70
70
1,78
1,32
0,06
0,11
0,18
0,18
0,06
0,2
3,80
80
1,79
1,31
0,05
0,12
0,27
0,30
0,10
0,2
4,10
85
1,81
1,31
0,04
0,12
0,35
0,42
0,14
0,2
4,30
85
1,82
1,31
0,04
0,14
0,51
0,60
0,20
0,2
4,80
IlSISIfi
Значения напряжения на электролизере, приведенные
в табл. 2-18 и 2-19, достигаются при строгом соблюдении требуемых
условий эксплуатации. Фактическое значение напряжения в произ-
водственных условиях обычно бывает выше.
Для электролизеров с ртутным катодом напряжение на электро-
лизере может быть определено^ удовлетворительной точностью при
различных плотностях тока по уравнению
2? —3,2 +5г (2.54)
где i — плотность тока, кА/м2; Ь — коэффициент, зависящий от конструкции
электролизера и колеблющийся от 1,5 до 0,3.
Для расчета напряжения на электролизере с ртутным катодом
в зависимости'от расстояния между электродами (а, мм), темпера-
туры процесса (£, °C) и плотности тока (г, кА/м2) предложено выра-
ч жение [224]:
г
я=3,1+(0,06 +0.0175а) (1,73 —0,0108г) i (2.55)
Приведенные в уравнении (2.55) коэффициенты пригодны для
определенной конструкции анода и при изменении ее должны быть
скорректированы. Так, для пластинчатого металлического анода
(ПТА) предложено выражение [224].
0,017 + 0,00315 + 0,01а .
’ + 0,11 + 0,0054? *г
где Ь — ширина электродного элемента, мм.
7* 99
(
йыход по току
При полном использовании тока на каждые 1000 А-ч электри-
чества, прошедшие через электролизер, на айоде должно выделяться
1,323 кг хлора, а на катоде 0,0376 кг водорода и образоваться
1,492 кг NaOH (при электролизе растворов NaCl) или 2,094 кг КОН
(для растворов КС1). При электролизе с ртутным катодом на катоде
должно выделиться 0,858 кг натрия. Если количество выделяющихся
газов выразить в объемах сухих газов при 0 *С и 760 мм рт. ст., то
при прохождении 1000 А-ч на электродах должно выделиться по
0,419 м3 хлора и водорода.
В действительности полезное использование тока в целевом про-
цессе никогда не достигает 100%. Вследствие протекания на элек-
тродах побочных электродных реакций, не связанных с образованием
целевых продуктов, и наличия вторичных процессов, приводящих
к потере части уже выработанных продуктов, количество последних
всегда меньше, чем следует из расчета по закону Фарадея. Для учета
суммарных потерь продукта вводится понятие выхода по току, под
которым понимают отношение фактически полученного продукта
к его количеству, которое должно быть получено, исходя из закона
Фарадея, без протекания побочных или вторичных процессов и без
потерь уже полученного продукта.
При электролизе водных растворов хлоридов щелочных метал-
лов образуются три продукта: газообразный хлор, раствор щелочи
и водород. Выходы по току каждого из них могут несколько раз-
личаться между собой. Обычно выход по току определяют по щелочи,
так как учет получаемых при электролизе электролитических ще-
локов может быть произведен с большей точностью, чем количества
газообразных хлора и водорода.
Если теоретически рассчитанное количество продукта электро-
лиза g, а практически полученное количество gx, то<выход по току ц
определится:
или в %
11
II
(2.56)
1]=gl/g
n = ^/^.100 (2.57)
Понижение^выхода по току вызывается двумя группами причин.
1. Протекание на электродах параллельно с основным процес-
сом, приводящим к получению нужного продукта, других побочных
электродных процессов. К ним можно отнести разрядку на аноде
ионов ОН" или других кислородсодержащих ионов с выделением
кислорода вместо хлора, окисление ионов СЮ" до хлората, раз-
рядка ионов Вг", если они присутствуют в рассоле. К этой же группе
относятся процессы хлорирования различных органических соеди-
нений, содержащихся в графите анодов, материалах, применяемых
для их пропитки, или в рассоле, поступающем на электролиз.
На катоде могут протекать процессы катодного восстановления
растворенного в электролите хлора, гипохлорита, а также частично
хлората с потерей выхода щелочи по току в электролизерах с ртут-
г ’ *
100
катодом и водорода в электролизерах и твердым катодом. При
^^электролизе с ртутным катодом разрядка водорода приводит к сни-
► женило выхода щелочи пр току.
: 2. Вторичные процессы, связанные со смешением полученных на
электродах продуктов — хлора и каустической соды и последующим
их взаимодействием между собой. Характер и значение этих про-
цессов меняются в зависимости от применяемого способа электро-
Лиэа, метода разделения электродных пространств и режима работы
электролизера.
Приведенные выше две группы процессов, приводящих к сниже- f
нйю выхода по току, взаимосвязаны, так как при смешении продук-
ч , тов электролиза образуются ионы СЮ- и СЮ3“, которые могут уча-
сФвовать в катодном и анодном электродных процессах, способству-
ющих снижению выхода по току.
Основным фактором, определяющим выход по току в электро-
£8
Ц * I
pa
|." А>.|;
• -‘W'Ab-
II
III
5^4
. ...
-Ж лйзерах с твердым катодом и проточной диафрагмой, является ско-
Ж
Л
г:
-
V-.
Л •
£
i 11
W. i ।
f
жк
Ж';
Ж*?
gfe
Ж,
Жа
я
;’.ЭД№^
ИВ
Яр
S';
рость протока электролита и связанная с ней концентрация щелочи
в католите или степень конверсии хлорида в гидроокись. В электро-
лизерах с горизонтальным расположением электродов и диафрагмы
протекаемость последней и щелочность католита по всей площади
диафрагмы одинаковы. В электролизерах с вертикальным располо-
жением диафрагмы концентрация щелочи в католите на различной
высоте диафрагмы может быть разной. Поэтому на различных участ-
ках диафрагмы по высоте выход по току может быть различным.
Вначале рассмотрим наиболее простой случай, когда плотность
тока и скорость фильтрации электролита по всей площади диафрагмы
одинаковы. Близкие к зтому условия наблюдаются, например,
в современных конструкциях электролизеров с вертикальным рас-
положением электродов и полностью заполненным катодным про-
странством.
До тех пор, пока скорость движения электролита от анода к ка-
тоду равна или больше скорости электромиграции ионов ОН“,
обеспечивается возможность проведения электролиза с высокими
выходами по току. При этом потери выходов по току могут быть обу-
словлены помимо выделения на аноде небольших количеств кисло-
рода переносом хлора с анолитом в катодное пространство, участием
ионов Й4 в переносе тока и явлениями диффузии.
Растворенный в анолите хлор, поступая в катодное пространство,
будет реагировать со щелочью, снижая выход по току хлора и
лочи. ’ При последующем катодном восстановлении образовавше-
гося гипохлорита
II
II
II
ч.
СЮ--ЬН2О + 2в = С1’+2ОН- J
вновь образуется щелочь, но снижается выход водорода но току.
Снижение выхода по току будет также вызываться явлениями
диффузии растворов через диафрагму, а также участием ионов Н+
в переносе тока. Перенос ионов Н+ в катодное пространство приводит
к соответствующему снижению выхода щелочи по току и к росту pH
101
1.
анолита, что создает условия для электролитического выделения
кислорода на аноде.
В табл. 2-20 приведены данные о возможном снижении выхода
по току за счет переноса в катодное пространство хлора, растворен-
ного в анолите.
Таблица 2.20* Снижение выхода по току (в %) за счет хлора,
поступающего в катодное пространство с анолитом [111]
Концентрация, вес. % Температура электролиза, %
NaCl в анолите . NaOH в католите 20 40 . 60 80
20 6 5,45 3,45 1,78 0,78
8 4,09 2,59 1,34 0,59
- 10 3,27 2,07 1,07 0,39
12 2,72' . 1,72 0,89 0,39
25 6 3,48 1,97 1,01 0,47
8 2,61 1,47 0,76 0,35
10 2,06 1,18 0,61 0,28
12 1,74 0,93 0,51 0,23
Указанные потери резко снижаются с ростом рабочей темпера-
туры электролиза и повышением концентрации поваренной соли
в анолите и щелочи в католите. В современных промышленных элек-
тролизерах, работающих при высокой температуре, эти потери не
превышают 1%.
При очень высокой скорости противотока, электролита, когда
концентрация
щелочи в католите
низка, потеря выхода по току
и
из-за поступления в катодное пространство растворенного в ано-
лите хлора увеличиваются. При уменьшении скорости противотока
возрастает концентрация щелочи в католите и снижаются потери
выхода по току. Это наблюдается до тех пор, пока скорость фильтра-
ции электролита не станет равной или близкой к скорости электро-
миграции ионов ОН". При дальнейшем снижении скорости фильт-
рации электролита ионы ОН“ будут попадать из католита в анод-
ное пространство тем в большей степени, чем больше разница между
скоростью движения ионов ОН и скоростью фильтрации,электро-
лита через диафрагму. Снижение выхода по току будет расти при
уменьшении скорости противотока. Таким образом, выход по току
будет зависеть от скорости противотока анолита или, в конечном
счете, от концентрации щелочи в катодных щелоках.
Сказанное выше иллюстрируется данными Ангела [200], приве-
денными на рис. 2-29, о зависимости выхода по току при электролизе
раствора поваренной соли с концентрацией 5,45 г-экв/л в электро-
лизерах с горизонтальной диафрагмой от концентрации получае-
мой щелочи. С уменьшением скорости противотока и ростом концент-
102
О 7 2 3 4 5 в
Концентрация NaOH
в католите, н.
Рис. 2-29. Зависимость выхода по
току от концентрации щелочи в ка-
толите при электролизе 6,45 н. рас-
твора поваренной соли с горизон-
тальной диафрагмой:
7— выход по току; 2 — содержание ги-
похлорита; 3 — содержание хлората.
рации щелочи выход по току медленно увеличивается и достигает
максимального значения при концентрации щелочи около 3,8 г-экв/л.
В этих условиях скорость противотока близка к скорости элек-
тромиграциц ионов ОН~. При дальнейшем снижении скорости проти-
вотока и росте концентрации ще-
, лочи выше 3,8 г-экв/л выход по
току резко падает за счет попада-
ния ионов ОН" в анодное про-
4 *странство. При этом в католите
возрастает концентрация хлората
вследствие взаимодействия щелочи
и хлора в анолите.
В этих рассуждениях концен-
трацию щелочи можно заменить
степенью превращения ап хлорида
в гидроокись. Тогда максималь-
ный выход по току достигается
при ссп = 0,50 — 0,55.
Если электролиз проводился
при повышенных температурах
(90—100 вС), то испарение влаги
и унос паров воды с хлором и водородом может существенно изменять
концентрацию щелочи и хлорида в католите и искажать характер зави-
симости выхода по току от коццентрации щелочи. При использова-
нии степени превращения хлорида в гидроокись изменение концен-
трации солей в католите за счет испарения влаги не отражается на
зависимости, определяющей выход по току.
Рис. 2-30. Зависимость выхода по току от концентрации щелочи в католите
в электролизерах с вертикальной диафрагмой:
< 7 — электролизеры с обычной асбестовой диафрагмой; 2 — электролизеры с асболатексной
диафрагмой.
Зависимость выхода по току от концентрации щелочи в католите
подтверждена и уточнена результатами большого числа опреде-
лений этой величины при различной концентрации щелочи. На
рис. 2-30 показана зависимость выхода по току от концентрации
щелочи в католите в электролизерах типа БГК [78, 225, 226].
При изменении концентрации щелочи от 70—80 г/л примерно
до 155 г/л (около 3,8 г-экв/л) выход по току возрастает в незначи-
103
Уровень
г анолита
Уровень верха катода
Рис. 2-31. Схема распределе-
ния концентрации щелочи по
высоте диафрагмы.
CQ 755
Концентрация NaOH , г/л
тельной степени. Для практических целей в этом интервале концент-
рации щелочи можно принять выход по току величиной постоянной
и равной примерно 0,98. При концентрации щелочи выще 155 г/л
выход по току снижается.
Разброс экспериментальных данных затрудняет установление точ-
ного характера зависимости выхода по току от концентрации щелочи
для этого участка кривой. Для практических условий при концент-
рации щелочи до 200 г/л с достаточной точностью можно принять
линейную зависимость выхода по току (выраженного в долях еди-
ницы) от концентрации щелочи с угловым коэффициентом —0,29 • 10“2.
Исходя из принятых выше допуще-
ний выход по току для концентраций
щелочи ниже 155 г/л будет равен 0,98,
а для более высоких концентраций бу-
дет определяться из выражения
ц = 0,98—0,29 (С —155) 10“2 (2.58)
где С — концентрация щелочи в католите, г/л. .
Для зоны переменной концентрации
щелочи, как это было рассмотрено ранее.
С —C0-|-6Z (2.41)
где Cq—концентрация щелочи в зоне по-
стоянной протекаемости диафрагмы, г/л; I —
расстояние от нижней точки зоны переменной
протекаемости диафрагмы, см.
Рассмотрим сначала процесс в элек-
тролизере с незаполненным катодным
пространством.
Распределение концентрации щелочи в католите по высоте диа-
фрагмы показано на рис. 2-31. На участке /0 по высоте диафрагмы
концентрация щелочи Со постоянна и менее 155 г/л. На участке
v 155 —Со
I = ----~_ концентрация щелочи меняется в соответствии с ура-
внением (2.41) от Со до 155 г/л: На участке диафрагмы (Zo + Г)
выход по току можно принять равным 0,98, поскольку концентра-
ция щелочи на всех участках диафрагмы находится в пределах от
CQ до 155 г/л.
На участке диафрагмы 1п концентрация щелочи в католите воз-
растает от 155 г/л до значения, равного
tl
<7 = 1554-62"
11
(2.59)
и выход по току по высоте будет непрерывно меняться в соответствии
с изменением концентрации щелочи в католите.
Для каждого элемента dl высоты диафрагмы на участке Г выход
по току будет определяться значением концентрации щелочи в като-
лите в этом элементе.
104
11
-P\ ’ * I . • .* :
'•<•' л :
Средний выход по току на участке Г можнб найти интегрирова-
Жнием в пределах от 0 до 1п выражения
ЙГЖ. * •
-* г •
^cp, i" -2
(2.60)
'• Подставляя в уравнение (2.60) значения т] и С из выражении
4 ;:’ (2.58) и (2.59) и интегрируя его, получаем значение среднего выхода
току на участке диафрагмы
.. Ж*
(2.61)
W?
Ж
.'В
! ЛйЖ
w
Ж’.
г?/ &••• •••
*>•
> i'.t J
№
'чму.'
T|CD «=0,98 —0,87-10’2Г
•up, 4
Средний выход по току для всего электролизера можно опреде-
лить из выражения
0,98 (lQ + l')+ (0,98-0,87.10'2/*) I" /О
Л1ср —
Ж Выход по току на электролизере тем выше, чем меньше относи-
|<тельная длина участка
< Если максимальная концентрация щелочи в католите в верхней
точке катода не превышает 155 г/л, выражение (2.62) приобретает
вид ц == 0,98 и все участки диафрагмы работают с максимально воз-
можным выходом по току.
Ж > При CQ ~ 155 г/л участок Г = 0, Г = Z, и выражение (2.62)
примет вид
tl
O,98Zo + (0,98 — 0,87 • 10~2Z) Z
Пср =----------у—rr-----------
ч
IK
,в.
^.V
£&':
S!
tag г, -
Kh'i
И*',!
p
Ж-
Если Со больше 155 г/л, путем аналогичных рассуждений полу-
чим выражение
А
[0,98—0,29 - 10-2 (Со —155)] ZQ + [0,98 — 0,29 10'2 (Со—155)—0,87 • 10'2/] Z
Лер — --- ! 7_ : ?
*.
Л
¥
w
1
(2.64)
Для электролизеров с заполненным катодным пространством вы-
ражения (2.59) — (2.64) сохраняются в силе, причем в данном слу-
чае I — расстояние от верха катода до уровня католита.
В приведенных выше рассуждениях исходили из равномерности
протекания электролита и равенства плотности тока по всей поверх-
ности диафрагмы, В действительной процессе электролиза это усло-
вие может нарушаться. Например, плотность тока существенно из-
меняется на различных участках диафрагмы, особенно к концу тура
работы электролизера при изношенных анодах.
Неравномерность протекания электролита через диафрагму
может быть вызвана различной плотностью диафрагмы на разных ее
участках и наличием дефектов диафрагмы. Кроме того, протекание
жидкости по порам диафрагмы происходит с различной скоростью.
105
У
3
В крупных порах можно ожидать движения жидкости с большей
скоростью, чем в малых порах. Движение жидкости у стенок будет
более медленным, чем в средней части пор.
Поэтому практически при равенстве средней скорости противо-
тока и скорости электромиграции ионов ОН“ не исключена возмож-
ность переноса ионов ОН’ в анодное пространство. Для получения
максимально возможного выхода по току необходимо вести процесс
электролиза при средней скорости противотока, превышающей ско-
рость электромиграции ионов ОН" .
При практическом осуществлении процесса электролиза на луч-
ших современных конструкциях электролизеров с диафрагмой
удается получать выхода по току обычно не более 96—97%. Такое
снижение выхода по току в значительной степени объясняется пере-
носом ионов ОН’ из-за неравномерности протекания электролита
по сечению всех пор диафрагмы.
Приближенно выход по току в данный момент может быть опре-
делен на основе анализа анодного газа.
При этом исходят из того, что основные потери выхода по току
обусловлены выделением кислорода на аноде и образованием экви-
валентного количества двуокиси углерода. Выделение свободного
кислорода и образование СО очень невелико и их можно не учиты-
вать. Для образования одного объёма СО2 необходимо затратить
такое же количество электрического тока, как и для получения двух
объемов С12.
Если содержание хлора в анодном газе составляет А %, а Дву-
окиси углерода Л'%, то выход по току будет равен
Т1= d А., (2.65)
Выражение (2.65) дает несколько завышенные значения выхода
по току, так как в нем не учитывается снижение выхода по току из-за
растворения хлора в аналите й связанное с этим образование гипо-
хлоритов и хлоратов. Это обстоятельство в какой-то мере компенси-
руется тем, что помимо окисления графита дополнительным источ-
ником двуокиси углерода в анодном газе может быть реакция ней-
трализации соды, содержащейся в свежем рассоле. Это приводит
к занижению определяемой величины.
Такой приближенный метод определения выхода по току при-
годен только в случае применения графитовых анодов. При исполь-
зовании малоизнашивающихся анодов содержание двуокиси угле-
рода в анодном газе обусловлено только протеканием реакции ней-
трализации соды в анодном пространстве. В этом случае для при-
ближенного расчета выхода по току вместо содержания двуокиси
углерода подставляется содержание кислорода в анодном газе.
Следует также учитывать возможность подсоса кислорода с возду-
хом через неплотности аппаратуры и трубопроводов.
Предложено [227] выражение для более точного определения вы-
хода по току на основе анализа газа и электролита (анолита и ка-
106
.4
’ толита), учитывающее потери выхода по току в связи с растворением
хлора в анолите и образованием гипохлоритов и хлоратов.
Однако для практических целей оно неудобно, так как требует
проведения большого количества анализов.
При проведении электролиза с ртутным катодом в «чистых»
условиях потери тока вследствие разряда водорода на катоде могут
быть очень невелики 123], и содержание водорода в хлоре не будет
превышать сотых долей процента. Но за счет присутствия различных
примесей и других причин в промышленных электролизерах содер-
жание водорода в хлоре составляет от 0,1—0,2 до 1,0—1,5%.
При загрязнении амальгамными ядами электролита или воды, пода-
ваемой на разложение амальгамы, или при других отклонениях
- процесса, содержание водорода в хлоре может превышать указанные
пределы.
Скорость разряда водорода на катоде мало зависит от обычных
показателей режима электролиза — концентрации натрия в амальгаме
и NaCl в рассоле, температуры процесса электролиза — до тех пор,
пока эти показатели находятся в пределах, применяемых в промк
in
ленном электролизе. В основном на скорость разряда водорода
влияют наличие загрязнений амальгамными ядами, а также нару-
шения нормального функционирования разлагателя или системы
циркуляции ртути в электролизере.
Потери тока на катоде связаны также с восстановлением рас-
творенного в анолите хлора и ионов СИУ и СЮ" по реакциям (2.13)
и (2.14). Эти потери зависят от концентрации активного хлора в ано-
лите и гидродинамических условий (скорости движения амальгам-
ного катода и электролита), определяющих скорость подвода актив-
ного хлора к поверхности катода [228, 229Г.
Концентрация активного хлора зависит не только от факторов,
определяющих растворимость хлора в рассоле, но и от pH анолита.
Значительное выделение водорода на катоде приводит к образованию
в анолите гидроксильных ионов и, соответственно, повышению кон-
центрации С1О~ и СЮ~.
Особенно возрастают потери тока на восстановление активного
хлора при больших уклонах ртутного катода вследствие турбулиза-
ции электролита на поверхности раздела с амальгамой [230—232].
В электролизерах с ртутным вертикальным катодом для снижения
потерь на восстановление активного хлора применяют диафрагму.
Плотность тока, расходуемого на процессы восстановления ак-
тивного хлора, определяется условиями подвода восстанавливае-
мого вещества к катоду; она практически не зависит от общей плот-
ности тока и для обычных конструкций ртутных горизонтальных
электролизеров составляет примерно 100—200 А/м2 [23]. Поэтому
с повышением плотности тока относительные потери тока снижаются,
и выход по току возрастает.
Помимо выхода по току полезно исследовать выход по энергии
для процессов электролиза. Эти процессы в промышленности обычно
107
протекают в необратимых условиях, поэтому напряжение на ячейке
электролизера превышает теоретическое напряжение разложения.
Это связано с наличием перенапряжения на электродах и потерями
напряжения на преодоление омических сопротивлений электролита
и проводников первого рода. Часть затрачиваемой электрической
энергии теряется также потому, что выход по току ниже единицы.
Обычно под выходом по энергии понимают отношение химиче-
ской энергии продуктов электролиза к общему количеству электри-
ческой энергии, затраченной на их получение. При этом под хими-
ческой энергией продуктов электролиза подразумевают энергию,
которая может быть получена при осуществлении реакции, обрат-
ной процессу электролиза. ?
Разница между общим количеством электрической энергии, за-
траченной на электролиз, и химической энергией полученных при
этом продуктов, характеризует количество энергии, превращающейся
в процессе электролиза в тепло. Иногда в качестве меры полезной
затраты энергии принимают не тепловой эффект обратной реакции
соединение продуктов электролиза, а максимальную работу этой
реакции.
В соответствии с изложенным для определения выхода по энергии
может быть использовано выражение
Л=-^-г] (2.66)
-^яч
где Еа и ЕяЧ — теоретическое напряжение разложения и практическое напря-
жение на ячейке.
электролизера
Материальный баланс
В процессе электролиза происходят изменения концентрации
электролита, которые обусловлены участием ионов в переносе тока,
химическими процессами, протекающими на электродах и в объеме
электролита, испарением влаги и уносом ее с газообразными
продуктами электролиза — хлором и водородом.
В электролизерах с твердым катодом объем католита VK
меньше объема поступающего на электролиз рассола Уо. Уменьше-
ние объема связано с превращением хлорида натрия в гидроокись,
имеющую меньший молекулярный объем, расходом воды на хими-
ческий процесс и испарение и унос в виде паров с выделяющимися
газами. Степень изменения объема католита
Со
V
1
(2.67)
где С0 и Ск — мольная концентрация хлорида щелочного металла в исходном
растворе и католите; Сх — мольная концентрация гидроокиси щелочного ме-
талла в католите.
1 - г *
✓
"Ч
Ниже.приведен расчет концентрации соли в анолите, а также соли
и щелочи в католите с учётом испарения и уноса влаги с газами.
108 ‘ .
^Незначительный расход воды, связанный с протеканием побочных
реакций, не учитывается. Для упрощения принято, что выход по
| току, температура и давление газов для анодного й катодных продук-
&\:тов одинаковы.
/При подаче в анодное пространство рассола, моляльная концент-
^| рй(ция которого тт10, на 1000 г воды (55,5 моль) поступит тп0 молей
При степени разложения соли а через ячейку должна быть
'Щйгйропущено ат0/т} фарадеев электричества и на аноде должно выде-
СТ « ТП V ПППО
to
• fw И . I .* .1.' k
/ЖЧ-.'
w
w
* rfe ;
‘•и
to
to
W’ .
.to-. -
^ пропущено атп0/т| фарадеев электричества и на аноде должно выде-
литься ат0 г-экв хлора.
; Если процесс электролиза проводить в условиях, исключающих
^^электролитический перенос ионов ОН" иэ катодного пространства
в анодное, то количество ионов хлора, Перенесенное током в анод-
ное пространство из катодного, составит паатп0/ц г-ионов, где па —
число переноса хлор-иона.
* Общее количество г-ионов хлора, находящееся в рассматриваемом
объеме анолита и поступающее затем в катодное пространство при
- Стационарном процессе, составит .
MaOtW0 rim i*) АЯЛ
- У)-----(^.ио)
Afa = mo
или после преобразования
(2.69)
(2.70)
Ж
г
; to
даг
.®to
Ж
’йил**' ’
Ж-/.
eto
K'
Afa = m0H^—[1 —na —(1—Г])] •
Учитывая, что число переноса катиона пк = 1 — па, получим:
1
Ma — 1п
Если для упрощения принять, что выход по току ц = 1, выра-
жение (2,70) приобретает вид:
Л/а = Wg (1 —CZZ2K) (2.71)
На аноде выделяется cwn072 молей газообразного хлора. Если
пренебречь объемом СО2, О2 и подсасываемого к хлору воздуха, то
количество молей влаги, уносимой с хлор-газом в виде паров, со-
ставит
__ тОа
?С1,—9-
(2.72)
кд
to
ц f!
tfl
ПА
где Р “ общее давление влажного газа; р' — парциальное давление паров
цоды в хлоре над анолитом.
Моляльность анолита можно определить из выражения
я _ 55,5
та~М* 55,5-gcl,
(2.73)
1:А'
или
та •=•
.ат$
m0 И
— [пк — (1 — Ц)] •
ч
(2.74)
109
При ц — 1
-----—-------«о (1 — )
55,5—ДЛ • -^-Р1-г
На катоде образуется ат0 молей гидроокиси натрия и выделяется
а/п0/2 молей газообразного водорода; при этом на химическую ре-
акцию расходуется ат молей воды.
Количество воды, уносимой с водородом в виде паров воды,
составит
ш0
ft
~fi
(2.76)
где р"— парциальное давление паров воды в водороде над католитом.
Содержание поваренной соли в католите определится по раз-
ности !между поступившим и разложившимся количествами
ЛГк — т$ (1 — а)
Содержание воды в католите составит
(2.78)
Моляльность католита (по NaCl) составит
лг0 (1 —а) 55,5
mK =
0,5p'
(2.79)
ft
a no NaOH
mKa
woa • 55,5
55,5-am0
г/
“77
(2.80)
Суммарная моляльность католита по NaCl и NaOH
ra=m^b
Степень изменения количества воды в электролите в
электролиза
(2.81)
процессе
fl
55,5 —
Чтобы перевести единицы концентрации из моляльности в г/л
(С), можно воспользоваться выражением:
lOOOwp
m + 1000/M
(2.83)
где т — моляльность раствора; р — плотность раствора; М — молекулярный
вес растворенной соли.
110
Ж*
' ,X?i. • Гм.- <
(2.84)
(2.85)
водяного пара над электро-
опубликовано. Для практи-
водяного пара над смешан-
Учитывая уравнение (2.82), моляльность католита по поварен-
ной соли и каустической соде составит
лгк=лг0(1—:0с) Ь
Данных о парциальном давлении
литическими щелоками в литературе не
ческих расчетов парциальное давление
ными растворами NaCl и NaOH можно определить по принципу
аддитивности. Ниже в табл. 2-21 и 2-22 приведено давление водяного
пара над растворами NaCl и NaOH.
Таблица 2-21. Давление^паров воды над растворами NaCl
(в мм рт. ст.)
и
растворами гидроокиси натрия
воды над
Таблица 2-22. Давление паров
(в мм. рт. ст.)
Снижение парциального давления паров воды
ческими щелоками лк может быть приближенно
над электролити-
принято равным
111
,
сумме снижения парциального давления над соответствую
щми рас-
и
творами ГИДРООКИСИ натрця ЛкаОН и хлористого натрия ЛхаС1
I* 1
— nNaOH +‘JlNaCl
(2.86)
III
При таком подсчете парциальное давление паров воды над като-
литом мало отличается от* парциального давления над насыщенным
раствором поваренной соли при той же температуре.
Если принять, что pf = р" = р, выражение (2.84) примет вид
55,5
/ А . Р
(2,87)
d —
ч
Ниже приведены значения 6, рассчитанные для электролиза
раствора поваренной соли (т = 6,0) при степени превращения хло-
рида в гидроокись а = 0,5, общем давлении газов 760 мм рт. ст.
и различных значениях парциального давления паров воды р:
р, мм рт. ст. Ь
р, ъш рт, ст. Ь
р. мм рт. ст, b р, мм рт. ст. Ъ
О 1,055
100 1,062
200 1,078
300 1,100
400 1,130
500 1,185
550 1,240
600 1,345
650 1,600
700 3,180
710 56,600
719 оо
III
При парциальном давлении паров воды над электролитом выше
400—500 мм рт. ст. унос паров воды резко возрастает. При парци-
альном давлении паров около 720 мм рт. ст. теоретически с газами
должна быть унесена вся вода из раствора. Поэтому при сильном по-
вышении температуры электролиза происходит интенсивное испа-
рение влаги, пересыщение раствора и выделение кристаллов соли,
которые забивают поры диафрагмы и приводят к нарушению нор-
мального процесса электролиза.
Материальный баланс электролизера осложняется наличием при-
месей, например соды, щелочи и сульфатов, в питающем электроли-
зер рассоле, протеканием процессов выделения на аноде кислорода
и окисления графитовых анодов с образованием в основном дву-
окиси углерода, а также вторичных процессов растворения и гидро-
лиза хлора в анолите и последующих реакций между растворенным
хлором и ионами ОН" с образованием гипохлорита и хлората. Од-
нако для практических целей приведенная выше приближенная схема
расчета материального баланса обычно дает достаточно точные
результаты.
Образовавшийся в электролизере гипохлорит практически пол-
ностью восстанавливается на катоде с образованием исходного хло-
рида натрия. Количество хлората натрия, уходящего с катодными
щелоками, не превышает обычно десятых долей Процента от образо-
вавшейся каустической соды. Поэтому в практических балансах элек-
тролизера эти процессы могут не учитываться. Расход воды и обра-
зование двуокиси углерода за счет сгорания анодов в связи с выде-
112
ж
г-^
с
лением кислорода можно учесть приближенно, приняв, что снижение
выхода по току связано лишь с разрядом ионов ОН“ на аноде.
Расход воды на разложение составит 9 (1 — ц), а количество дву-
окиси углерода, образовавшегося от сгорания анодов, равно
11 (1 — ц). При температуре 90—95 °C, поддерживаемой в совре-
менных электролизерах, потери воды на побочные процессы не пре-
вышают 0,5—1,0% общего расхода воды на химические процессы
и испарение.
При электролизе с ртутным катодом выражения для расчета
Материального баланса электролизера имеют более простую форму.
При сохранении тех же обозначений получаем
Ма~т^(1 — а) (2.88)
Количество влаги, уносимой с хлором, можно определить по урав-
нению (2.72) и
'w
ат0
то (1 — а)
(2.8?)
•t
л
г
Ж V
ft.
Материальный баланс разлагателя должен учитывать расход воды
на химическую реакцию, на испарение и унос паров с водородом.
В мощных электролизерах с ртутным катодом унос воды в виде па-
ров с водородом составляет значительную величину, поэтому прак-
тикуют установку на разлагателе холодильников водорода для кон-
денсации основного количества паров воды и ртути и возвращения
конденсата в разлагатель.
Ж,
i
м-
I
$
Тепловой балано электролизера
Повышение температуры в электролизере приводит к сниже-
нию напряжения и увеличению выхода по току. Поэтому, несмотря
на возрастание скорости коррозионных процессов и, в частности,
усиление износа графитовых анодов, стараются работать при повы-
шенной температуре. Однако увеличение температуры в электроли-
зере ограничено определенным пределом. При приближении рабо-
чей температуры к температуре кипения электролита резко повы-
шается парциальное давление паров воды, возрастает объем влаж-
ных газов, выделяющихся на электродах, что приводит к росту
газонаполнения и соответственно увеличению напряжения на ячейке.
При чрезмерном повышении температуры электролит «вскипает»,
увеличивается унос электролита с газами. Такое явление наблю-
дается при температуре выше 100—105 °C.
Ж В электролизерах с ртутным катодом температуру в электроли-
зере стараются не поднимать выше 80—90 °C из-за отсутствия гум-
х^гровочных материалов, обладающих достаточной стойкостью при
более высокой температуре.
f В современных конструкциях мощных электролизеров удельный
. вес потерь тепла наружными стенками невелик. В электролизерах
8 Заказ 843
113
с твердым катодом и диафрагмой эти потери тепла снижают путем
применения тепловой изоляции.
Температура электролизера зависит в основном от напряжения
на нем и температуры подаваемого на питание рассола.
В результате износа графитовых анодов во время работы электро-
лизера изменяется напряжение на нем и повышается его рабочая
температура.
При высоких плотностях тока к концу тура работы анодов тем-
пература в электролизере может подняться выше допустимой. Это
приобретает особое значение для электролизеров с твердым катодом,
где обычно нет возможности регулирования межэлектродного рас-
стояния по мере износа анодов. В таких случаях рабочую темпера-
туру электролизера можно снизить питанием электролизера хо-
лодным рассолом. Холодильные элементы для охлаждения электро-
лита в современных конструкциях электролизеров не применяются.
Приход энергии в электролизере составляется из физического
тепла, приносимого с питающим электролизер рассолом, и электри-
ческой энергии постоянного тока, затрачиваемого на электролиз.
Некоторое количество тепловой энергии выделяется также при
сгорании графитовых анодов до СО2.
Тепловой эффект образования гипохлорита при реакции хлора
со щелочью и последующего восстановления его до хлористого нат-
рия можно не учитывать, так как в сумме последовательных реак-
ций получается исходный продукт — хлористый натрий. Можно
учитывать тепловой эффект образования хлората, но его количество,
образующееся при электролизе, невелико и поправка незначительна.
Все побочные процессы, снижающие выход по току в энергети-
ческом балансе, в практических расчетах можно учесть, приравняв
их к процессу электролиза воды с выделением кислорода и водорода.
Расход энергии в балансе электролизера будет включать тепло-
вой эффект реакции разложения хлорида натрия и воды на хлор,
водород и гидроокись натрия, а также реакции разложения воды
на водород и кислород, физическое тепло, уносимое из электроли-
зера с катодными щелоками, хлором и водородом, энтальпию паров
воды, уносимых из электролизера с газообразными продуктами элек-
тролиза, и потери тепла стенками аппарата в окружающую среду.
Некоторые из показателей технологического режима работы
электролизеров мало изменяются в процессе работы и поэтому прак-
тически не влияют на изменение материального и теплового балан-
сов электролизера. К таким показателям можно отнести концентра-
цию хлорида в исходном рассоле, выход по току. Другие показа-
тели, наоборот, значительно изменяются за тур работы электроли-
зера. К ним относятся прежде всего напряжение на электролизере
и степень превращения хлориду в гидроокись. Изменение напря-
жения на электролизере влечет за собой изменение температуры
электролиза и связанное с этим увеличение количества испаренной
в электролизере воды, а следовательно, концентрации NaCl и NaOH
в католите.
114
1
t
I
Испарение влаги и унос её с газообразными продуктами электро-
лиза автоматически приводят к установлению нового теплового ра-
вновесия электролизера на новом температурном уровне.
Для электролизеров с твердым катодом предложена [233] тепло-
вая диаграмма (рис. 2-32), связывающая температуру подогрева
рассола, напряжение на электролизере, степень превращения хло-
рида в гидроокись и удельные тепловые потери электролизера через
наружные стенки с температурой процесса, количеством испаренной
в электролизере воды и концентрацией едкого натра и поваренной
\ соли в католите.
В верхнем левом поле диаграммы нанесены линии равных кон-
центраций едкого натра и поваренной соли в католите, равных плот-
ностей католита, линии равных температур электролиза и количе-
ства испаренной воды, а также линии выпадения кристаллов поварен-
ной соли при 20 и 100 °C. В верхнем правом поле нанесены линии
. равных напряжений на электролизере.
На нижних правом и левом полях нанесены линии равных тем-
ператур подогрева питающего рассола и равных значений величины
g' из выражения
(2.90)
где g' — коэффициент, учитывающий потери тепла через стенки электролизера
и влияние температуры; q — удельные потери тепла через стенки аппарата
при температуре помещения 28 °C, ккал/ч на 1 кг NaOH; t2 — температура
электролиза, °C; р — теплота испарения воды при температуре электролиза,
ккал/кг.
Диаграмма составлена при питании электролизера рассолом кон-
центрацией 312 г/л NaCl.
С помощью диаграммы можно с достаточной для практических
целей точностью рассчитывать показатели теплового режима работы
электролизера,
л
Для иллюстрации приведем пример пользования диаграммой.
Электролизер работает при V ~ 3,3 В, температуре питающего рассола
t — 70 °C, степени превращения хлорида в гидроокись t, = 0,5 и величине
q = 0,3 кг/кг.
Для определения неизвестных показателей работы электролизера проводим
через точку £ = 0,5 на шкале степени превращения вертикальную линию.
Через точку Ь± пересечения этой линии с линией, отвечающей температуре пи-
тающего рассола t = 70 9С, в левом нижнем поле диаграммы проводим гори-
зонтальную линию до точки Ci — заданного значения q = 0,3. Из этой точки
проводим вертикаль до точки dr пересечения с линией V ~ 3,3 В. Горизонтальная
линия из точки dt пересекается с первоначально проведенной вертикальной
линией. Точка пересечения характеризует тепловой режим работы электро-
лизера.
Значения интересующих нас показателей определяются по близлежащим
кривым разных величин. Так, в нашем примере температура электролиза =
= 93,5 °C; количество испаренной воды S = 0,75 кг/кг NaOH; концентрация
NaOH в католите 127 г/л; концентрация NaCl в католите 187,5 г/л; плотность
католита ук — 1,224 кг/л.
Аналогично можно решать различные задачи теплового баланса электро-
лизера.
8* > 115
Рис. 2-32. Тепловая диаграмма диафрагменного электролизера:
А — граница области выпадения соли из католита при 20 °C; В — то же, при 100 °C.
1
i*-?
ь"*1 L ••
«Лж&4злЬ ,:u„. •
&
ЧЯЪ / •
и
-^1
гЙ?>
О;
I
В
•••
Ш-
fv '
'W-
fe-
В электролизерах с ртутным катодом необходимо рассчитывать
тепловой баланс отдельно для электролизера и разлагателя, хотя
они связаны между собой циркуляцией ртути. В электролизере при-
ход энергии составляется из физического тепла, приносимого рас-
солом и ртутью, поступающей из разлагателя, и электрической
энергии, затрачиваемой на электролиз.
Расход энергии включает тепловой эффект процесса разложения
хлорида натрия с выделением хлора и образованием амальгамы нат-
рия, и тепло, уносимое с амальгамой, анолитом, хлором и насыщаю-
щими его парами воды и теряемое через стенки в окружающую
среду.
Основным способом регулирования температуры является под-
держание заданного напряжения на электролизере путем опускания
анодов по мере их срабатывания и изменение температуры рассода,
подаваемого на питание. ;
В процессе разложения амальгамы выделяется большое коли-
чество тепла, повышается температура. Основная доля потерь тепла
в разлагателе приходится на испарение воды. В, мощных агрегатах
потери тепла за счет теплоотдачи через стенки невелики, но про-
исходит большой унос паров воды и ртути с водородом из разлага-
теля. Чтобы уменьшить унос ртути и стабилизировать работу разла-
гателей, их снабжают индивидуальными обратными холодильни-
ками для водорода.
w
Использование восстановительной способности амальгам
щелочных металлов
>•
l5'
В производстве хлора и каустической соды по методу электро-
лиза с ртутным катодом амальгама натрия используется только для
получения гидроокиси щелочного металла и водорода. При этом
водород часто не находит полезного применения, особенно в связи
с загрязнением газа парами ртути.
Энергия, выделяемая при разложении амальгамы, фактически
расходуется лишь на повышение температуры в разлагателе. Это
позволяет интенсифицировать процесс разложения и уменьшить
размеры разлагателя и загрузку ртути.
Вопрос об использовании энергии разложения амальгамы
с целью получения электрической энергии или снижения затрат
электрической энергии на процесс электролиза рассматривался
ранее.
Восстановительная способность амальгам щелочных металлов
может быть использована для проведения реакций восстановления,
причем возможности зтого процесса велики. Высокое значение пе-
ренапряжения выделения водорода на ртути и амальгамах щелоч-
ных металлов позволяет проводить с помощью амальгам процессы,
окислительно-восстановительный потенциал которых ниже, чем
воды.
ii
117
Разработано много процессов, основанных на использовании
восстановительной способности амальгам для получения как неор-
ганических [234 — 236], так и многих органических продуктов [234,
236, 237].
Многие из этих процессов нашли применение в промышленности,
однако ни один из них не может быть сравним по масштабам с про-
цессом получения каустической соды электролизом с ртутным ка-
тодом.
Ниже приведены некоторые из таких процессов.
Получаемый продукт
Реакция получения
Сульфид натрия
Дитионит натрия
Хлорит натрия
Перекись натрия
Азобензол
Диметилгидразин
Пинакон
d-сорбит
Na2S4 + 6Na^ 4Na2S
2SO2 +4Na^ Na2S2O4
C1O2 + 2Na^ XaC102
O2 + 2Na-> Na2O2
C6H5NO2 -> C6H5N3
(CH3)2N-NO-> (CH3)2NHNH2
(CH3)2CO^ (CH3)2COH-COH—COH
Восстановление а-глюкозы
СОН—(CH3)2
Некоторые из этих и аналогичных процессов применяются лишь
сравнительно в небольшом масштабе в промышленности. Предста-
вляет большой интерес использование амальгамы для получения круп-
нотоннажных продуктов, так как это могло бы существенно улучшить
экономические показатели процесса электролиза с ртутным катодом.
Неоднократно рассматривался вопрос о получении металличе-
ского натрия, используя амальгаму в качестве исходного сырья.
Предлагалось получать натрий разделением ртути и натрия пу-
тем разгонки амальгамы [238], а также электролизом в неводных
электролитах, в котором амальгама натрия из электролизеров с ртут-
ным катодом должна служить анодом. В качестве электролита мо-
гут применяться смеси солей с низкой температурой плавления [239,
240], а также алюминийалкидные или боралкидные соединения
[241]. Однако сообщений о промышленном применении этих спосо-
бов переработки амальгамы натрия на металлический натрий не
было опубликовано.
ЛИТЕРАТУРА
*1.
*2.
3.
4.
5.
6.
7.
8.
9.
Пасманик М. И., С а с с-Т исовский Б. А., Якимен-
ко Л. М. Производство хлора и каустической соды. Справочник. М.,
«Химия», 1966.
Справочник химика. Т. I, II. М., Госхимиздат, 1963; т. III, 1964.
Справочник по содовой промышленности (Сода когё бокецу то букку).
Изд. фирмы «Ниппон Сода», Токио, 1960.
Gmelins, Handbuch der anorganischen Chemie, № 34, 8 AufL, Berlin,
1962. .
Натр едкий технический. ГОСТ 2263—59.
Натр едкий улучшенный. ГОСТ 11078—71.
Едкое кали техническое. ГОСТ 9285—69.
Хютте. Справочник. Т. I. М., Госхимиздат, 1934.
Suzuki О., Fukunaga Т., J. Electrochem. Sос. Japan,
104 (1956).
№ 3
24,
118
10. Г-лессман С. Введение в электрохимию. М., Издатинлит, 1951; Ла-
тимер В., Окислительные состояния элементов и их потенциалы в вод-
ных растворах. М., Издатинлит, 1964.
И. Веселовская И. Е., Ходкевич С. Д., Якименко Л. М.,
Электрбхимия, 5, № 8, 906 (1969).
1. 1а.- Perret J., Parker J., J. Electrochem. S ос., 119, № 3, 107 (1972).
12. К о x а н б в Г. H., M и л о в а Н. Г., Электрохимия, 6, № 1, 73 (1970).
13. Фрумкин А. Н., Багоцкий В. С., Иофа 3. А., Каба-
нов Б. Н. Кинетика электродных процессов. М., Изд. МГУ, 1952; Фрум-
кин А. Н., Л у к о в ц е в П. Д., Левина С. Д., Acta Phisi-
cochim., 11, 21 (1969).
14. Ф р у м к и н А. Н. Труды совещания по электрохимии. М., Изд. АН СССР,
1953. См. с. 21—46.
15. Ф р у м к и н А. Н., Z. Electrochem., 59, 807 (1965).
16. Антропов Л. И., ЖФХ, 26, 1688 (1952).
17. A 11m and A. J., Pollak W. G., J. Chem. Soc., 115, 1120(1919).
18. К а п ц а н О. Л., Кинетика разложения амальгам. Канд, дисс., МГУ,
1950.
19. S u g i п о Т., А о k i К., J. Electrochem. Soc. Japan, Overseas Ed., № 1 —
3, 15 (1959).
20. Багоцкий В. С., Яблокова И. Е., ЖФХ, 23, 413 (1949).
21. Коршунов В. Н., Иофа В. А., ДАН СССР, 141, № 1,143 (1961).
22. Фрумкин А. Н., Коршунов В. Н., Иофа 3. А., ДАН
СССР, 141, № 2, 443 (1961)
23. В о л к о в Г. И. Производство хлора и каустической соды методом
электролиза с ртутным катодом. М., «Химия», 1968.
^24. Волков Г. И., ЖПХ, 27, 681 (1954).
25. X о м я к о в В. Г., Зубова И. Е. Труды МХТИ. Вып. 18. М., 1954,
См. с. 84. *
26. Angel J., Briinland R., J. Electrochem. Soc., 99, 443 (1952); 100,
39 (1953); Angel J. et al., J. Electrochem. Soc., 104, 167 (1957).
27. Antweiler H., Карре у H. J., Meihold G., Chem. Ing.
Techn., 34, № 5, 387 (1962).
28. Antweiler H., Schaefer J. P., Chem. Ing. Techn., 43, № 4,
180 (1971).
29. В a 1 e j J., P a s e k a J., Chem. Ing. Techn., 39, № 12, 725 (1967).
30. Hauck G., Durr W., Chem. Ing. Techn., 34, № 5, 369 (1962); 39,
720 (1967).
31. Stokmann V., Landsberg R., Chem. Techn., 17, 148 (1965).
32. Коршунов В. H., Гладких И. П., Волков Г. И., Электро-
химия, 6, № 1, 117 (1970). *
33. Конончук -Т. И. и др., Хим. пром., № 41, 599 (1965).
34. DAS 1224284 (1960).
35. Пат. США 3497432 (1970); канад. пат. 833006 (1970).
36. Волков Г. И., Клица 3. Л., Сусорова Е. К., Чере-
мисина Н. В. Труды 4-го совещания по электрохимии. Изд. АН СССР,
1959. См. с. 841.
37. Angel G., L u n d е n Т. et aL, J. Electrochem. Soc., 102, 124, 246 (1955).
38. Schmidt H., HolzingerF., Chem. Ing. Techn., 35, № 1, 37 (1963).
39. Скляренко С. И., Сахаров Б. А., Ж. физ. хим., 21, 97 (1947).
40. Dunning W. G., К i 1 р a t г i k М. К., J. Phys. Chem., 42, 215 (1938).
41. Иофа 3. А., К а п ц а н О. Л., Ж. физ. хим., 26, 201 (1952).
42. Иофа 3. А., Пенк о в ска я 3. Б., ДАН СССР, 59, 265 (1948).
43. Trumpler G., Gut К., Helv. chim. acta, 33, 1922 (1950).
44. Bockris J. О. M., Watson R. G. H., J. Chim. phys., № 1, 70
(1952).
45. Волков Г. И., Ж. физ. хим., 27, 194 (1953).
45а. Пат. США 3649486 (1972).
46. Jaksic М. М., Jovanovic D. R., Chonka J. М., Electro-
chim. acta, 13, № 11, 2077 (1968).
' 119
47. Jaksic М. М., Csonka J. М., Electrochem. Techn., 4, 49 (1966).
Hund Н., Chem. Ing. Techn., 39, № 11, 702 (1967).
i. В a 1 e i J., Paseka J. Chem. Prum., 22, № 9, 458 (1972).
H i n e F., Electrochem. Techn., 2, № 3—4 (1964).
В олков Г. И., Канд, дисс., МХТИ им. Д. И. Менделеева, 1961.
Герм. пат. 73224 (1894); 88230 (1896).
Kand ler L.<, Chem. Ing. Techn., 34, № 5, 349 (1962).
Пат. США 2970095 (1961); пат. ФРГ 1050740 (1959); 1094722,
(1961),
Япон. пат. 1526, 5528, 7076, 7416, 7513 (1958).
Vielstich W., Chem. Ind. Techn., 34, 346 (1962); Schm
Vielstich W., Chem. Ing. Techn., 39, № 12, 761 (1967).
Liska S., Rev. chim., 19, № 6, 316 (1968).
Adrien J., J. Electrochem. Soc., 116, № 4, 546 (1969).
Япон. пат. 9319 (1955); англ. пат. 1185757 (1970).
Wendtland R., W in sei A., Chem. Ing. Techn., 39,
(1967).
J u s t i E., W i n s e 1 A., Kalte Verbrennung-Fuel Cells.,
Steiner, 1962.
Австр. пат. 237643 (1962).
Герм. пат. 141187 (1903).
Герм. пат. 263432 (1910).
Стендер В. В. Диафрагмы для электролиза водных растворов. М.,
Госхимиздат, 1948; Стендер В. В. и сотр. Асбестовые диафрагмы —
в кн.: Труды ГИПХ. Вып. 22. 1934.
Стендер В. В. Электрохимическое производство хлора и щелочей.
М., ОНТИ, Химтеорет, 1935.
36, № 8, 1958 (1963).
5 (1936).
49.
50.
51.
55.
56.
57.
58.
59.
60.
61.
63.
64.
65.
66.
67.
68.
69.
70.
Китаев Г. И., Асбест, № 3,
Пат. США 3467586 (1969).
Пат. США 3630863 (1971).
№
1099515
12, 756
Wiesbaden,
71.
72.
73.
в а О. Я., Смолян 3. С., Хим. пром., № 10, 788 (1966); авт. свид.
СССР 172727 (1965); Бюлл. изобр., № 14 (1965).
Haas К., Chem, Eng. Techn., 27, №5, 234 (1955).
Биллитер Ж. Новейшие достижения технической электрохимии.
М., Госхимиздат, 1934.
DAS 2134126 (1972).
74.
75.
76.
77.
78.
Л и фа т о в а 3. И., Иншакова 3. П. и др. Авт. свид. СССР
388781 (1971); Бюлл. изобр., № 29, 23 (1973).
Пат. США 3505200 (1970).
Колотухин А. Т., Мулин Е. В., Хим. пром., № 5, 395 (1960).
Англ. пат. 11872 (1913}; герм. пат. 277433 (1912); 284022 (1913).
Англ. пат. 1716 (1905); 16853, 17492 (1907).
енко Л. М. Электролизеры с твердым катодом. М., «Химия»,
1966.
79.
., Хим. пром., № 1, 49 (1962); М и ха й-
Д., Be сел ов ская И. Е.,Яки-
лова
м е н к о Л. М., Электрохимия, 9, № 6, 825 (1973).
К р ы лов В. Н. Производство угольных и графитовых электродов.
М., ГОНТИ, 1939.
Чалых Е. Ф. Производство электродов. М., Металлургиздат, 1954.
Марковский Л. Я., Оршанский Д. Я., Прянишни-
ков В. П., Химическая электротермия. М., Госхимиздат, 1952.
Jalop.et.ti Н., Rend. Accad. Sci. Fis. Mat. Sosieta Reale di Napoli,
Ser. IVa, № 12 (1941—1942).
84. J о h n s о n N., Trans. Electrochem. Soc., 86, 127 (1944).
85. V a a 1 e r L. E., Electrochem. Technol., 5, 170 (1967).
86. Jeffery T. C., Electrochem. Techn., 5, 124 (1967).
120
80
81.
83.
iv ''
I
и
r
*
*-,V'
V
•/
'Ш'
л:-
4т.
д
•J
88
89
90
01
93
94
95
96
97
98
99:
100
101
102
Potsch A., Proft R., G о t z m.a n n J., J. Chem. Techn., 19,
№ 5, 285—288’(1967).
Стендер В. В., Ксенжек О. С., ЖПХ, 31, № 1, 110 (1958).
Иоффе С. В-, ЖПХ, № И, 668 (1936); хим. пром., № 13, 784 (1963).
Кришталик Л. И., М
ЖПХ, 34, № 7, 1537 (1961).
Электрохимия, 6, № 6, 866; 6’,
Милова Н. Г., Электрохи-
ЖПХ,
№ 10, 1492 (1970); Коханов Г. Н
мия, 5, № 1, 93 (1969); Коханов Г. Н.
56, № 6, 1231 (1973).
Флисский М. М., Веселове ка
нян Р. В., ЖПХ, 33, № 8, 199 (1960).
М., ЖПХ, 31, № 12, 1832 (1958); 32, № 1, 121 (1959).
Wranglen G., Electrochem. Acta (London), 10, 43
К э й и г и X., Кэнсукэ М., Сода то энсо, 22, №1,
• 1
(1965).
С ю н д з и М.,
1 (1971).
НгоДайВ
Электрохимия, 8, № 2, 221, 225; № 3,384, 387 (1972).
Хине О, Я ,с у д а М., Denki kogaku, 39, № 6, 530 (1971).
DAS 1812724 (1969); англ. пат. 1247027 (1971); канад. пат. 863.917 (1971);
Япон. пат. 43618 (1971).
Франц, пат. 141^9612 (1965).
Кришталик Л. И., ЖПХ, 34, № 7, 1543 (1961).
Т i n n J. М., J. Electrochem. Soc., 117,
№ 2, 219 (1970).
Chem. Econom. Eng. Rev., 3, № 8, 43 (1971); 4, № 2, 36 (1972); Nagaoki
Tory. Chem. Age India, 23, № 6, 556 (1972).
Lenon P., Vaaler L., Electrochem. Techn., 1, № 5—6, 178 (1963).
5, 223 (1967).
• t
103.
104. Shobert E. J., Electrochem. Techn., ___x___
105. Sjodin B., WranglenXj., Electrochem. Acta (London), 10
(1965).
106. M улика Ф. И., Кришталик Л. И., К(
ЖПХ, 39, № 6, 1338 (1960); ЖОХ, 38, 2819 (1965).
108.
109.
110.
111.
(1959);
о x a -
Бюлл.
растворов. M:.,
применение.
Chem. Rev.,
113.
н о в Г. Н., Мулин В- Н., авт. свид. СССР 124423
изобр., № 23, 14 (1959).
Пат. США 3278410 (1966).
Пат. США 3505199 (1970).
Бельг, пат. 639014 (1964).
Биллитер Ж., Промышленный электролиз водных
Госхимиздат, 1959.
112. Ш у м а х е р И. Перхлораты: свойства, производство
М., Госхимиздат, 1963; Carr J.P., Hampson N.
72, № 6, 679 (1972).
Электрометаллургия водных растворов. Под ред. Г. Егера. М., «Метал-
лургия», 1966.
Стендер В. В., Н и к и ф о р о в А. Ф., Коновалов М. Б.,
авт. свид. СССР 195121; Бюлл. изобр., № 9 (1967).
114.
115.
116.
117.
ЖПХ, 42, № 3, 584 (1969).
нова Л. А., авт. свид. 289823 (1968); Бюлл. изобр.
Курашвили М. И., Электрохимия марганца. Т.
СССР, 1963. См. с. 375.
Пат. США 2631115 (1953).
Япон. пат. 27741 (1968).
№ 2, 11 (1971).
2. M., изд-во AH
1147442 (1969);
118.
119.
120. Англ. пат. 855107 (1960); 1111767; 1127484 (1968):
1186454, 1206863 (1970); 1260645 (1972).
121
121. Франц, пат. 1479762 (1967); 1523738 (1968); 2005403, 2020680, 2021041
(1970); 2040107, 2040116 (1971).
122. Пат. ФРГ 1115721 (1961); 1917040 (1969); пат. ГДР 72249 (1970),.
123. Бельг, пат. 710551 (196&); 725491, 725492 (1969).
124. Пат. США 3236756 (1966); 3491014 (1970); канад. пат. 860270 (1971).
125. Якименко Л. М., Джагацпанян Р. В., Веселов-
ская И. Е., Ходкевич С. Д., Хим. пром., № 10, 728 (1962).
126. G eg пег A. et al., Corrosion, 15, № 7, 6437 (1959).
127. A mm ar J. A., Kamal J., Electrochim. acta, 16, Хг 9, 1539, 1555
(1971).
128. Разина H. Ф., Электрохимия твердых и жидких систем. Алма-Ата,
изд-во АН КазССР, 1967.
129. Smyth D. М., J. electrochem. Soc., 113, № 1, 19 (1966).
130. Англ. пат. 1104078 (1968).
131. Япон. пат. 7018283 (1970).
132. Пат. ФРГ 1286513 (1969).
133. Пат. ФРГ 1182211 (1964); англ. пат. 1104078 (1968).
134. Udupa N. V. К. et. al., Indian J. Techn., 4, № 10, 305 (1966).
135. Пат. США 3055811 (1962); 3497426 (1970); 3630768 (1971).
136. Канад, пат., 616029 (1961); 833014 (1970).
137. Edwards С. Е., Denki kagaku (News Letters), 2, № 3, 4 (1968).
138. Япон. пат. 20469 (1967); 7011014, 7041201 (1970); 7102322, 7103257,
7103258 (1971).
139. Англ. пат. 1087529 (1967); 1127484, 1128136 (1968); 1244850(1971).
140. Швейц, пат. 450372 (1968).
141. Франц, пат. 1498250 (1967).
142. Пат. США 3297561 (1967); 3409533 (1968).
143. Пат. ФРГ 1240827 (1967).
144. Англ. пат. 1076973, 1087529 (1967); 1113421, 1125493, 1127484 (1968).
145. Япон. пат. 7023816 (1970).
146. Acki К., J. Electrochem. Soc. Japan, 35, № 3, 136—141 (1968).
147. Пат. США 3267009 (1966); 3293176 (1967); пат. ФРГ 1671468 (1971).
148. Англ. пат. 1111767 (1968).
149. Франц, пат. 1523738 (1968); 1594758, 1594759 (1970).
150. Англ. пат. 1113421 (1968).
151. Япон. пат. 11774 (1969); пат? США 3497426 (1970).
152. Такахаси М., Сода то энсо, 19, № 1, 9—21 (1968); J. Electrochem.
Soc. Japan, 36, № 1, 57 (1968).
153. Zirngiebl E., Chem. Ing. Techn., 39, № 12, 752 (1967).
154. Fait a G., Fiori J., Augustinski J., J. Electrochem. Soc.,
116, № 7, 928 (1969).
155. Такахаси M., Сода то энсо, 19, 52 (1968).
156. Fukunaga T., J. Electrochem. Soc. Japan, 36, № 1, 59 (1968).
157. Winnacker K., Kuchler L., Chemische Technologie, Bd. I,
Anorganische Technologie, Munchen, 1970.
158. Ф и о ш и н M. Я., Казакова Л. И., Хим. пром., 10, 760 (1964).
159. Warne М. A., Hayfield Р. С. S., Trans. Inst. Metal Finishing,
45, 83 (1967).
160. Tanaka N., Fujisawa T., Bull. Chem. Soc. Japan, 42, № 8, 2300
(1969).
161. Городецкий В. В., Дембровский М. А., Лосев В. В.,
ЖПХ, 36, 1543 (1963); Узбеков А. А., Лосев В. В., Н о с о -
в а К. И. Всесоюзная научная конференция «Пути развития и последние
достижения в области прикладной электрохимии»; ЛТИ им. Ленсовета,
1971. См. с. 132.
162. Юхниевич Р., Хейфильд П. Труды 3-го международного кон-
гресса по коррозии металлов. Т. 3. 1968. См. с. 70.
163. Mituya A., Obayshi Т., Inst. Catalysic. Hokkaido Univ., 7, 10
(1959); 8, 79 (1960).
163. Дембровский M. А., Диссертация, ФХИ им. Л. Я. Карпова, 1967.
122
165. Antler M., Butler C. A., Electrochem. Techn., 5, 126 (1967).
166. Пат. США 3103484 (1959); 3657102, 3654121 (1972); франц, пат. 1502587
(1965); нидерланд. пат. 6612095 (1965); япон. пат. 7036483 (1970);
25597 (1972); англ. пат. 1292130 (1972).
167. Нидерланд. пат. 6501716 (1964).
168. Ходкевич С. Д., Веселовская И. Е., Якименко Л. М.,
Данилова О.ХЛ., Электрохимия, 7, № 3, 357 (1971).
169. Lu Chien-Cheng et al., Denki kagaku, 38, № 3, 213 (1970); Bull.
Chem. Soc. Japan, 43, № 7,2264 (1970); J. Electrochem. Soc., 117, № 3 74
(1970).
170. Faita G., Fiori G., N i d о 1 a A., J. Electrochem. Soc., 117, № 10,
1333 (1970).
171. Chem. Week, 103, № 3, 58 (1968); 104, № 10, 15, 42; № 11, 86 (1969).
172. Chem. Eng., 77, № 19, 36 (1970).
173. De Nora O., Chim. ind., 50, № 6, 642 (1968); Chem. Ing. Techn.,
42, № 4, 222 (1970); 43, № 4, 182 (1971).
174.
Спасская E. К., Веселовская. И. E., Ходкевич С. Д.,
Ш5
Якименко Л. ЭД. Всесоюзная научная конференция «Пути развития
и последние достижения в области прикладной электрохимии». ЛТИ им.
Ленсовета, 1971. См. с. 130.
175. Якименко Л. М. Всесоюзная научная конференция «Пути разви-
тия и последние достижения в области прикладной электрохимии». ЛТИ
им. Ленсовета, 1971. См. с. 138.
176. De Nora О., Inform, chim., № 68, 67, 69 (1968); № 68, 77, 28 (1969);
J. Electrochem. Soc., 117, № 3, 74c (1970).
177. Europ. Chem. News, 14, № 339, 30 (1968); 16, № 404, 34, 36 (1969).
178. Англ. пат. 1128136 (1968).
178a. Пат. США 3649485 (1972); Канад, пат. 923069, 923072 (1973).
179. Франц, пат. 2009337 (1970); 2039723 (1971); пат. ГДР 84175 (1971).
180. Заявка на франц, пат. 2083384 (1972); франц, пат. 2093093, 2094051 (1972);
япон. пат. 7202842 (1972); пат. ФРГ 2126840 (1971); DAS 2163527 (1972);
пат. США 3632498 (1972).
1§1. Якименко Л. М., Изосенков Р. И.,- ЖПХ, 35, № 2, 342
(1962).
182. Tsukada Н. et al., J. Electrochem. Soc. Japan. Overseas Ed., 32,
№ 1, 1 (1964).
183. Мулина Ф. И.,Кришта лик Л. И., ЖПХ, 38, № 12, 2808 (1965).
184. Стендер В. В., К с е н д ж е к О. С., ЖПХ, 32, НО (1959).
185. Кришталик Л. И., Ротенберг 3. А., ЖФХ, 39, 328 (1965);
39, 907 (1965).
186. Suzuki О., Ikeda A., Abe S., J. Electrochem. Soc. Japan, 27,
№ 12, 8 (1959). '
187. D e Nora O., J. Electrochem. Soc., 97, 346 (1950).
188. Okada S. et. al., J. Electrochem. Soc. Japan, 26, № 4—6, 55 (1958).
189. Brandmair F., Chem. Rundschau, 26, № 14, 13 (1973).
190. Sugino K. et al., J. Electrochem. Soc. Japan, 27, № 1—3, 55 (1959).
191. Vielstick W., Chem. Ing. Techn., 33, № 2, 75 (1961).
192. Федотьев H. П. и др., ЖПХ, 21, № 4, 317 (1948).
193. Стендер В. В. и др., ЖПХ, 20, № 1, 36 (1947).
194. Кузьмин Л. А., П о р о й к о в а А. С., ЖПХ, 22, № 6, 572 (1949).
195. Антропов Л. И., ЖФХ, 26, 1688 (1952).
196. Бормашенко И. Б. и др., ЖПХ, 38, 12 (1'965). *
197. Воронин Н. Н. и др., Укр. хим. ж., 20, № 2, 182 (1954).
198. Коханов Г. Н. и др., ЖФХ, 36, 481 (1962); Электрохимия, 4, № 6,
685 (1968); Вести, техн, и эконом, информ., НИИТЭХИМ, № 2, 36
(1959).
199. Коханов Г. Н., Кучинский Е. М., Веселовская И. Е.
Труды IV совещания по электрохимии. М., изд-во АН СССР, 1956.
200. Ангел Г.4 Электролиз хлористых солей щелочных металлов в ваннах
с диафрагмой. Пер. с нем. М., ОНТИ, Химтеорет, 1935.
123
201. Гантман Л. В. и др. Труды по химии и химической технологии.
Вып. 2., Горький, изд. Горьков, гос. ун-та, 1958. См. с. 300.
202. Якименко Л. М., Модылевская И. Д., Ткачек 3. А.
Электролиз воды. М., «.Химия», 1970.
203. Пфлейдерер Г. Электролиз воды. Пер. с нем., М., ОНТИ, Хим-"
теорет, 1935, <
204. Коган Б. В., Софронов В. М., Хим. пром., № 2, 94 (1954).
205. М а ш о в е ц П. В., ЖПХ, 24, № 4, 353 (1951).
206. Dela Rue, Tobias С. W., J. Electrochem. Soc., 106, № 9, 827
(1959).
207. M у л ин В. E., Колотухин A. T., Хим. пром., № 8, 652 (1960);
Мул ин В. Е., ЖПХ, 43, № 12, 2638 (1970).
208. Франц, пат. 1531843 (1968).
209. Пат. США 3403083 (1968).
210. Генин Л. С., Кронгауз Е. Л., Хим. пром., № 2, 116 (1961).
211л Мина га в а М., Уэда Т., Denki kagaku, 35, 210, 268 (1967); J.
Electrochem. Soc. Japan, 35, № 1, 53; 35, № 2, 100 (1967).
212. Канад, пат. 859718 (1970); Venczel J., Electrochem. Acta, 15,
№ 12, 1909 (1970). •
213. Okada S. et al., J. Electrochem. Soc. Japan, Overseas Ed., 26, № 4—6,
E66 (1958).
214. Greens E. A., J. Electrochem. Soc., 108, № 11, 1065 (1961).
215. Gardiner W. C., Electrochem. TecJm., 1, № 3—4, 71 (1963).
216. Gardiner W, C., Sakowaki W. L., Electrochem. Techn., 1^
№ 1—2, 53 (1963).
217. T a k a t a K. et al., Denki kagaku, 32, 101, 376, 378, 429 (1964).
218. Пат. США 3627652 (1971); канад. пат. 853681 (1970).
219. Hine F., Electrochem. Techn.1, 6, № 1—2, 69 (1968).
220. S t a h 1 b e r g W., Energieanwend., 17, № 2, 33 (1968); пат. США
3507771 (1970);, япон. пат. 7014411 (1970).
221. Adam Е. et al., Chem, Ind. Techn., 37, № 6, 573 (1965).
222. Sconce J. S., Chlorine, its Manufacturing, Use and Properties, Reinhold
Publ. Co., N. Y., 1962.
223. Вой тех ев А. Г., Хим. пром., № 11, 852 (1969).
224. Волков Г. И., Кубасов В. Л., ЖПХ, 44; № 2, 451 (1971);
Волков Г. И., Лядин Ю. В., Свирская И. И., Хим. пром.,
№ 8^ 608 (1972).
225. Файн штейн С. Я. Производство хлора методом диафрагменного
электролиза. М., «Химия», 1964.
226. . Стендер В. В. Прикладная электрохимия. Харьков, изд. Харьков,
гос. ун-та 1961.
227. Сучков В. Н., Хим. пром., № 2, 49—51 (1969).
228. В о л к о в Г. И., С в и р с к а я И. И., Электрохимия, 7, № 4, 597 (1971).
229. Волков Г. И., Лядин Ю. В., Свирская И. И., Хим.
пром., № 4, 291 (1971).
230. В о л ков Г. И., К у с ь к и н а Э. И., ЖПХ, 31, 1755 (1937).
231. W a t a m a b е Т., Electrochem. Soc. Japan, Overseas Ed., 27, № 1—3,
9 (1961).
232. Sommers H. A., Chem. Eng. Progr., 53, № 10, 506 (1957); Elec-
trochem. Techn., 5, 108 (1967).
233. Сучков В. H., Хим. пром., № 8, 37 (1964).
234. Гладышев В. П., Изв. АН КазССР, 21, № 10, 31 (1965).
235. К е t е 1 а. а г J. A. A., Chem. Ing. Techn., 35, 372 (1963).
236. Mullin R. В. M., Chem. Eng. Progr., 46, 440 (1950).
237. Funke W., Chem. Ing. Techn., 35, 336 (1963).
238. Пат. ФРГ 1028348 (1958).
239. Joshizawa A., Watanabe N. et al., J. Chem. Soc. Japan, Ind
Chem. Sect., 66, 1412 (1963). ,
240. Пат. ФРГ 1200533 (1963).,
4 241. Ziegler L., Chem. Ing. Techn., 22, № 11, 229 (1950).
.-J
£
%
КОНСТРУКЦИИ ЭЛЕКТРОЛИЗЕРОВ ДЛЯ ПОЛУЧЕНИЯ ХЛОРА
И КАУСТИЧЕСКОЙ СОДЫ
1
J
r
ЭЛЕКТРОЛИЗЕРЫ С ТВЕРДЫМ КАТОДОМ
£
&
Ji'
V-
В конструкциях современных электролизеров с твердым катодом
используется проточная диафрагма. В начале развития электрохими-
ческого производства хлора применялись электролизеры с непро-
точными диафрагмами или бездиафрагменные электролизеры с про-
точным электролитом и разделением анодных и катодных продуктов
электролиза с помощью колоколов или газозащитных оболочек.
В 30-х годах был разработан способ осаждения диафрагмы на сет-
чатом катоде, и с этого времени многочисленные электролизеры с ли-
стовой или порошковой диафрагмой уступают место конструкциям
с осажденной диафрагмой.
Ниже будут описаны только последние конструкции электроли-
зеров. G более ранними конструкциями можно ознакомиться в опу-
бликованных работах [1—6].
Разработка метода получения осажденной диафрагмы открыла
новые возможности для конструирования электролизеров с сильно
развитой и сложной поверхностью катода, например гребенчатой
формы, состоящих из большого числа плоских пустотелых катодных
элементов — пальцев, расположенных в два, четыре или шесть ря-
дов. При такой форме катода отпадала ручная операция обкладки
асбестом поверхности катода, что значительно облегчало процесс
нанесения диафрагмы.
Возможность применения катодов с очень развитой поверхностью
позволила создать весьма компактные конструкции электролизе-
ров, рассчитанных на большую нагрузку. Технические и экономи-
ческие преимущества электролизеров с осажденной диафрагмой
обусловили их широкое применение во многих странах. В Советском
Союзе разработаны и применяются конструкции электролизеров
с осажденной диафрагмой — БГК-13, БГК-17 и БГК-50 [7—111.
За^рубежом распространены электролизеры с осажденной диафраг-
мой фирм «Хуцер» и «Даймонд», разработанные в США.
Все конструкции электролизеров как с диафрагмой, так и с ртут-
ным катодом предназначены для работы при атмосферном давлении.
Осуществление процесса электролиза под давлением представляет
большие трудности, но "работы в этом направлении продолжаются.
Так, предложен способ получения хлора под избыточным давлением
200 мм рт. ст. [12].
201. Гантман Л. В. и др. Труды по химии и химической технологии.
Вып. 2., Горький, изд. Горьков, гос. ун-та, 1958. См. с. 300.
202. Якименко Л. М., Моды л евская И. Д., Ткачек 3. А.
Электролиз воды. М., «Химия», 1970.
203. Пфлейдерер Г. Электролиз воды. Пер. с нем., М., ОНТИ, Хим-'
теорет, 1935.
204. Коган Б. В., Софронов В. М., Хим. пром., № 2, 94 (1954).
205. Машовец П. В., ЖПХ, 24, № 4, 353 (1951).
206. D е 1 a Rue, Tobias С. W., J. Electrochem. Soc., 106, №9, 827
(1959).
207. Мулин В. E., Колотухин A. T., Хим. пром., № 8, 652 (I960);
Мулин В. Е., ЖПХ, 43, № 12, 2638 (1970).
208. Франц, пат. 1531843 (1968).
209. Пат. США 3403083 (1968).
210. Генин Л.С., К р о н г а у з Е. Л., Хим. пром., № 2, 116 (1961).
211лМинагава М., У эд а Т., Denki kagaku, 35, 210, 268 (1967); J.
Electrochem. Soc. Japan, 35, № 1, 53; 35, № 2, 100 (1967).
212. Канад, пат. 859718 (1970); V en c z el J., Electrochem. Acta, 15,
№ 12, 1909 (1970). -
213. Okada S. et al., J. Electrochem. Soc. Japan, Overseas Ed., 26, № 4—6,
E66 (1958).
214. Greens E. A., J. Electrochem. Soc., 108, № 11, 1065 (1961).
215. Gardiner W. C., Electrochem. Tecpn., 1, № 3—4, 71 (1963).
216. Gardiner W. C., Sakowaki W. L., Electrochem. Techn., 1,
№ 1—2, 53 (1963).
217. T a k a t a K. et al., Denki kagaku, 32, 101, 376, 378, 429 (1964).
218. Пат. США 3627652 (1971); канад. пат. 853681 (1970).
219. Hine F., Electrochem. Techn.1, 6, № 1—2, 69 (1968).
220. S t ah Iberg W., Energieanwend., 17, № 2, 33 (1968); * пат. США
3507771- (1970); япон. пат. 7014411 (1970).
221. Adam Е. et al., Chem. Ind. Techn., 37, № 6, 573 (1965).
222. Sconce J. S., Chlorine, its Manufacturing, Use and Properties, Reinhold
' Publ. Co., N. Y., 1962.
223. Войтехов А. Г., Хим. пром., № И, 852 (1969).
224. Волков Г. И., Кубасов В. Л., ЖПХ, 44; № 2, 451 (1971);
Волков Г. И., Лядин Ю. В., Свирская И. И., Хим. пром.,
№ 8, 608 (1972).
225. Файнштейн С. Я. Производство хлора методом диафрагменного
электролиза. М., «Химия», 1964.
226. Стендер В. В. Прикладная электрохимия. Харьков, изд. Харьков,
гос. ун-та, 1961.
227. Сучков В. Н., Хим. пром., № 2, 49—51 (1969).
228. Волков Г. И.,Свирская И. И., Электрохимия, 7, № 4, 597 (1971).
229. Волков Г. И., Лядин Ю. В., Свирская И. И., Хим.
пром., № 4, 291 (1971).
230. Волков Г. И., Куськина Э. И., ЖПХ, 31,1755 (1937).
231. Watamabe Т., Electrochem. Soc. Japan, Overseas Ed., 27, № 1—3,
9 (1961).
232. Sommers H. A., Chem. Eng. Progr., 53, № 10, 506 (1957); Elec-
trochem. Techn., 5, 108 (1967).
233. Сучков В. H., Хим. пром., № 8, 37 (1964).
234. Гладышев В. П., Изв. АН КазССР, 21, № 10, 31 (1965).
235. Ketela.ar J. A. A., Chem. Ing. Techn., 35, 372 (1963).
236. Mullin R. В. M., Chem. Eng. Progr., 46, 440 (1950).
237. Funke W., Chem. Ing. Techn., 35, 336 (1963).
238. Пат. ФРГ 1028348 (1958).
239. J oshizawa A., Watanabe N. et al., J. Chem. Soc. Japan, Ind
Chem. Sect., 66, 1412 (1963).
240. Пат. ФРГ 1200533 (1963)..
241. Zing 1 er L., Chem. Ing. Techn., 22, № 11, 229 (1950).
Г Л Д В Д 3
КОНСТРУКЦИИ ЭЛЕКТРОЛИЗЕРОВ ДЛЯ ПОЛУЧЕНИЯ ХЛОРА
И КАУСТИЧЕСКОЙ СОДЫ
ЭЛЕКТРОЛИЗЕРЫ С ТВЕРДЫМ КАТОДОМ
" В конструкциях современных электролизеров с твердым катодом
используется проточная диафрагма. В начале развития электрохими-
ческого производства хлора применялись электролизеры с непро-
t точными диафрагмами или бездиафрагменные электролизеры с про-
точным электролитом и разделением анодных и катодных продуктов
электролиза с помощью колоколов или газозащитных оболочек.
В 30-х годах был разработан способ осаждения диафрагмы на сет-
чатом катоде, и с этого времени многочисленные электролизеры с ли-
стовой или порошковой диафрагмой уступают место конструкциям
с осажденной диафрагмой.
Ниже будут описаны только последние конструкции электроли-
зеров. G более ранними конструкциями можно ознакомиться в опу-
бликованных работах [1—6].
Разработка метода получения осажденной диафрагмы открыла
новые возможности для конструирования электролизеров с сильно
развитой и сложной поверхностью катода, например гребенчатой
формы, состоящих из большого числа плоских пустотелых катодных
элементов — пальцев, расположенных в два, четыре или шесть ря-
дов. При такой форме катода отпадала ручная операция обкладки
асбестом поверхности катода, что значительно облегчало процесс
нанесения диафрагмы.
Возможность применения катодов с очень развитой поверхностью
позволила создать весьма компактные конструкции электролизе-
ров, рассчитанных на большую нагрузку. Технические и экономи-
ческие преимущества электролизеров с осажденной диафрагмой
обусловили их широкое применение во многих странах. В Советском
Союзе разработаны и применяются конструкции электролизеров
с осажденной диафрагмой — БГК-13, БГК-17 и БГК-50 [7—11].
За^рубежом распространены электролизеры с осажденной диафраг-
мой фирм «Хуцер» и «Даймонд», разработанные в США.
Все конструкции электролизеров как с диафрагмой, так и с ртут-
ным катодом предназначены для работы при атмосферном давлении.
Осуществление
процесса электролиза под давлением представляет
большие трудности, но работы в этом направлении продолжаются.
Так, предложен способ получения хлора под избыточным давлением
200 мм рт. ст. [12].
125
J
3
W
10
12
И
13
Г iii!
l«[i
Рис, 3-1. Электролизер БГК-13:
1 — анодная шина; 2 — крышка; 3 —
аноды; 4 — бетонная футеровка; 5 —
отвод водорода; 6 — корпус электроли-
зера; 7 — катод; 3 — устройство для
регулирования уровня католита;
9 — отвод электролитической щелочи;
Ю— капельница; 11 — подвод рас-
сола; 12 — отвод хлора; 13 — катод-
ная шина.
верхним подводом тока
разработаны также кон-
электролизера БГК-13
подводом тока к анодам,
« Г* V, ° - • ч *-»
н
Электролизеры БГК-13 (3, 13, 14)
Электролизер БГК-13 выполнен в нескольких вариантах на на-
грузку от 3,5 до 5,5 кА. В начале 50-х годов наибольшее распрост-
ранение имели такие электролизеры на нагрузку 5 кА.
В электролизере БГК-13 приме-
няется цельнометаллический сварной
пальцеобразный катод из стальной
проволочной сетки, натянутой на ме-
таллический токоподводящий каркас,
со сварными контактами. Катод при-
способлен для нанесения осажден-
ной диафрагмы. Ввод анодов осу-
ществляется через крышку электро-
лизера с
к анодам.
Были
струкции
с нижним
однако они не получили применения
в промышленности.
В электролизере обеспечена воз-
можность естественной циркуляции
анолита для выравнивания его кон-
центрации и снижения степени га-
зонаполнения анолита во время
работы электролизера. Общий вид
электролизера показан на рис. 3-1.
Электролизер состоит из прямо-
угольного катодного блока и бетон-
ной крышки, через которую пропу-
щены графитовые аноды, опира-
ющиеся через специальные под-
ставки на изолированное днище
корпуса катода. Изнутри к пря-
моугольному стальному корпусу
катода (сечение 615x690 мм, вы-
сота 1140 мм) на двух противопо-
ложных сторонах корпуса приварен каркас, на который натянут сет-
чатый катод в виде гребенки с плоскими пустотелыми катодными
карманами. Карманы доходят почти до середины корпуса электро-
лизера. Между катодными карманами при сборке электролизера
устанавливают графитовые аноды. Начальное расстояние между
анодами и катодами составляет 11—13 мм. В средней части катодного
корпуса между обеими катодными гребенками остается свободное
от электродов пространство. Это облегчает естественную циркуля-
цию электролита благодаря образованию его нисхЬдящего потока
в свободном от электродов центральном пространстве.
126 I.
3.
*
Hi
' •••?
ь V,
u?
&>'
>4
У
£
г.-
E
rt«b
iE
A..'
it.'
l
Si
£*'.
Eft-
I
ft'
11
Верхний край катодной сетки с осажденной на ней диафрагмой
на 130 мм ниже верхнего края корпуса катода. При насасывании
диафрагмы на сетку асбестовая пульпа подается непосредственно
в корпус катода, а жидкость удаляется под вакуумом через штуцер
для отвода водорода. Стенки корпуса по периметру над верхним
краем катода футерованы слоем бетона, на бетонные уступы опи-
рается асбоцементная крышка плоской или куполообразной формы.
Днище корпуса катода после насасывания диафрагмы футеруется
слоем бетона, так же как и верхние борта катодного корпуса.
В электролизере установлено 14 графитовых анодов — плит тол-
щиной 50 мм, шириной 250 мм, длиной 1000 мм, расположенных
в два ряда. Аноды монтируются в крышке электролизера. Сквозь
крышку проходит четырехугольная головка анода, сечение которой
меньше сечения рабочей части анодов. В крышке электролизера
имеются также отверстия для отвода хлора и ввода рассола.
На рис. 3-1 показан электролизер с плоской крышкой. При такой
конструкции крышки облегчается ошиновка электролизера, в этом
случае медные соединительные шины имеют очень простую форму.
Для улучшения сепарации хлора от брызг анолита применяют также
крышки с куполом в средней ее части. При этом устройство шин для
подвода тока к анодам несколько усложняется.
Зазоры между крышкой и кожухом и между крышкой и анодами
уплотняются цементным раствором после установки анодов и крышки
на корпусе катода. Ток к анодам подводится с помощью медных шин,
прикрепленных болтами к головкам анодов, выступающим над
крышкой электролизера. К катоду ток подводится через стальные
шины, приваренные непосредственно к наружному корпусу. Корпус
электролизера и стальной каркас катодной сетки служат для распре-
деления тока по всей поверхности катодной сетки.
Для уменьшения утечки тока щелочь выводят из электролизера
в коллектор через капельницу. Электролизер БГК-13 может рабо-
тать при. разных уровнях католита в катодном пространстве, регу-
лируемых специальными запорными устройствами. Верхний шту-
цер для слива щелочи расположен немного ниже верхнего края
катодной сетки. Возможна работа электролизера как с заполненным,
так и незаполненным катодным пространством.
Общая рабочая поверхность анодов в электролизере БГК-13 со-
ставляет 6,15 м2, поверхность катодов 6,80 м2. При номинальной
нагрузке 5,0 кА электролизер работает при плотности тока на аноде
820 А/м2 (с новыми графитовыми электродами) и на катоде 735 А/м2.
Каждый графитовый анод в электролизере БГК-13 с трех сторон
окружен катодом, что несколько снижает рабочую плотность тока
на аноде.
Электролизеры этого типа не приспособлены для смены диа-
фрагмы в процессе работы, поэтому весь тур электролизер работает
с одной и той же диафрагмой. В течение 4—6 мес. по мере старения
диафрагмы иТзабивки ее пор осадками солей кальция, магния, же-
леза й продуктами разрушения графитовых анодов концентрация
127
щелочи в электролитических
щелоках
постепенно повышается до
130—140 г7л. Чтобы предотвратить дальнейшее увеличение концент-
рации
щелочи и снижение в связи с этим выхода по току, диафрагму
промывают водой. Это на короткий срок улучшает протекаемость
диафрагмы. Средняя продолжительность работы электролизеров
БГК-13 составляет 7—9 мес. Это обусловливает повышенный удель-
к
и
ный расход графита.
Высота верхней газовой части анодного пространства в электро-
лизере БГК-13 невелика, благодаря чему доля анодов, не исполь-
зуемая в процессе электролиза, сравнительно невелика. Вследствие
малого объема анолита над верхним краем катода электролизер
чувствителен к неравномерности питания его рассолом, при пере-
рыве или изменении скорости подачи рассода сильно колеблется
концентрация щелочи. В большей мере это обстоятельство сказы-
вается на работе электролизера с заполненным катодным простран- к
ством. Поэтому на практике предпочитают вести процесс в электро-
лизерах БГК-13 с незаполненным катодным пространством и регули-
ровать подачу рассола по постоянному уровню анолита в электро-
лизере.
Показатели работы электролизеров БГК-13 приведены ниже:
514
Нагрузка, кА................ ............. . .
Плотность тока, А/м2
на катоде . . . . . . . . . . . ... . . .
на аноде ......................
Среднее напряжение на электролизере, В . . . .
Средний выход по току, % ..................
Расход электроэнергии (постоянный ток), кВт-ч
на 1 т хлора..............
на 1 т 100%-ной NaOH ..........
Концентрация NaOH в католите, г/л ......
Концентрация С12 в хлоргазе, % . ..........
Срок службы, мес.
диафрагмы ......................
анодов ....................................
Производительность по хлору, кг/сут
электролизера . . . .......................
с 1 м2 площади пола (с учетом проходов) . .
5 ' *
735
820
3,35
93,5
2700
2400
110—125
96—98
7—9
7—9
147
105
Вследствие сравнительно небольшой мощности электролизеров
БГК-13 серии аппаратов обычно питают постоянным током парал-
лельно от общих шин преобразовательной подстанции при одинако-
вом напряжении. Поэтому нагрузка на серии электролизеров при
ее пуске обычно составляет 6,5 — 6,8 кА при напряжении на один
электролизер 3,35 В. К концу тура нагрузка на серию уменьшается;
при снижении ее до 2,5 — 3,0 кА серию электролизеров отключают
для замены анодов и диафрагм.
По компактности и малому удельному расходу электроэнергии
электролизер БГК-13 превосходит многие типы электролизеров,
применяемые в промышленности. Расход материалов на изготовле-
128
ние электролизера и требуемая производственная площадь на 1 кА
нагрузки и на мощность 1 т/сут хлора приведены ниже:
Сталь, кг ..........................
Медь, кг . . . .....................
Площадь пола (с учетом проходов), м2
на 1 кА
нагрузки
на мощность
1 т/сут хлора
1800
122
В настоящее время электролизеры БГК-13 применяются весьма
ограниченно и в ближайшие годы будут полностью заменены более
мощными современными электролизерами.
Электролизеры БГК-17 и БГК-50 (3, 13—15)
Электролизеры БГК-17 рассчитаны на номинальную нагрузку
25 кА. Они выпускаются для работы при 750 и 900 А/м2, но могут ра-
ботать и при более высокой плотности тока — до 1200 — 1300 А/м2.
Электролизеры этого типа предназначены в основном для цехов элек-
тролиза мощностью 25—100 тыс. т хлора в год.
Электролизеры БГК-50 рассчитаны на нагрузку 50 кА при плот-
ности тока около 1000 А/м2 и предназначены для цехов электролиза
мощностью 50 тыс. т хлора в год и более, но могут быть использо-
ваны и в цехах меньшей мощности, особенно если предусматривается
дальнейшее увеличение мощности производства.
В последнее время электролизеры БГК-17 и БГК-50 несколько
модернизированы. При сохранении их габаритов и плотности тока
нагрузка на электролизеры БГК-17 повышена до 32 кА, на электро-
лизеры БГК-50 — до 65 кА.
Выпускаются также электролизеры на нагрузку 15 кА при раз-
личных плотностях тока — от 500 до 1200 А/м2. Электролизеры
таких моделей целесообразно использовать в менее крупных цехах
для производства хлора и каустической соды.
В электролизерах типа БГК-17 и БГК-50 применена конструк-
ция разветвленного катода, состоящего из узких плоских катодных
пальцев, выполненных из стальной сетки и расположенных в виде
четырех или шести гребенок. Катоды электролизеров имеют сталь-
ной каркас, что обеспечивает достаточную жесткость конструкции и
позволяет распределить ток по поверхности катода. При правильном
соотношении объемов катодного и анодного пространства в этих
электролизерах можно значительно увеличить рабочую высоту
электродов без опасения снизить выход по току.
Конструкция катодного блока предусматривает повышенное га-
зонаполнение в катодном пространстве и исключает возможность
снижения давления фильтрации через диафрагму в нижней части.
В электролизерах применен нижний подвод тока к анодам. Верхняя
часть анодного пространства свободна от анодов и может быть до-
статочно развита в высоту.
9 Заказ 843
129
Электролизеры БГК47 и БГК-50 отличаются большой высотой,
что обеспечивает компактность конструкции и высокие съемы про-
дукции с единицы площади производственного здания при сравни-
тельно невысокой плотности тока, пониженные удельные расходы
электроэнергии и затраты цветных металлов по сравнению с электро-
лизерами других типов. Достигнута хорошая герметичность в местах
соединений
блока
с
12
катодного
11
Рис. 3-2. Электролизер БГК-17-25:
1 — корпус катода: 2 — подвод тока к анодному
днищу; 3 — графитовые аноды; 4 — отв£д ще-
лочи; 5 — капельница; 6 — анодное днище;, 7 —
устройство для регулирования уровня католита;
3 — расходомер рассола; 9 — крышка электроли-
зера; 10 — водородный коллектор; 11 — хлорный
коллектор; 12 — рассолопровод.
анодным комплектом и крышкой.
Схема устройства элек-
тролизера БГК-17 на на-
грузку 25 кА показана на
рис. 3-2, а внешний вид уста-
новки — на рис. 3-3.
Катодный блок предста-
вляет собой стальной корпус,
внутри которого в четыре
ряда вмонтированы гребенки
катодных пальцев, предста-
вляющих собой сплющенные
полые карманы, выполнен-
ные из металлических кар-
касов с натянутой на них
стальной проволочной сет-
кой. Толщина катодных
пальцев 20 мм. Крайние
каркасы катодных гребенок
приварены к продольным
стенкам корпуса катода, два
средние образуют двусторон-
нюю гребенку, приваренную
к торцевым стенкам кор-
пуса.
Внутреннее пространство
катодных элементов в элек-
тролизере сообщается между
собой, образуя общее катод-
ное пространство, заполнен-
ное католитом, а в верхней части — водородом. Между двумя
соседними катодными гребенками сохраняется циркуляционное про-
странство, свободное от электродов.
На сетчатую поверхность катода насасывается асбестовая диа-
фрагма. В корпусе катода предусмотрен штуцер для присоединения
к вакуумной лцнии при насасывании диафрагмы.
_ Анодный блок состоит из графитовых плит толщиной 50 мм и
шириной 250 мм, монтируемых на стальном анодном днище, которое
одновременно используется для подвода тока к анодным плитам с по-
мощью специальных контактных устройств (без применения свинца).
. ; Днище электролизера вместе с контактной частью анода для за-
щиты от действия хлорсодержащего анолита заливают битумной
130
и
л ь ' >
-..Чи ч' • b
’ • ч-
Г массой специального состава, поверх которой наносят тонкий слой
бетона. Битумная масса имеет температуру плавления, удобную для
\ее нанесения и удаления. При комнатной температуре масса доста-
точно хрупка • и легко удаляется пневматическим инструментом.
Во время работы электролизера масса размягчается и заполняет
. все пустоты, поры и возможные трещины. При этом повышается ее
^адгезия к графиту и металлу и увеличивается надежность за
деты
и
анодного контакта.
Рис. 3-3. Общий вид цеха электролиза, оборудованного электролизерами
БГК-17 на нагрузку 25 кА.
II
Ток к анодному днищу подводится с помощью контактных пла-
стин, приваренных к днищу электролизера, а ток к катоду — через
пластины, приваренные к катодному корпусу.
При установке катодного блока на анодный комплект графито-
вые плиты располагаются в промежутках между пальцами катод-
ных гребенок. При новых анодах расстояние между электродами
составляет около 12 мм. Щелочь из катодного пространства сли-
вается по нижнему штуцеру, соединенному сифонной трубой с ка-
пельницей. Уровень жидкости в катодном пространстве можно ре-
гулировать, меняя положение подвижной трубы для слива щелочи.
Водород отводится из электролизера по верхнему штуцеру катодного
блока.
Стенки корпуса катода подняты несколько выше катодных кар-
манов и образуют надкатодную камеру и раструб для установки
крышки. Для защиты от действия хлора внутренние стенки раструба
Покрывают слоем бетона.
Бетонная крышка электролизера типа БГК-17 изготавливается
в металлических формах. Для предотвращения от разрушения при
действии кислого анолита и влажного хлора крышку выполняют
из кислотобетона, стойкого в условиях работы электролизера.
При использовании таких крышек исключается загрязнение
131
анолита солями кальция и магния, как это происходит в результате
коррозионного разрушения крышки в случае применения обычного
бетона на портланд-цементе.
Крышка электролизера снабжена отверстиями для отвода хлора,
подачи свежего рассола, установки термометра, измерителя уровня
рассола и отбора проб анолита. После установки в раструбе катода
крышка уплотняется специальной замазкой.
В последнее время в электролизерах БГК-17 и БГК-50 с успехом
стали применять стальные гуммированные крышки, что облегчает
конструкцию электролизеров, их монтаж и обслуживание.
Уплотнение между анодным комплектом и катодным блоком до-
стигается за счет собственной тяжести катода с крышкой и с по-
мощью дополнительной болтовой стяжки. Особая конструкция уплот-
нительного устройства в электролизере позволяет легко и надежно
герметизировать стык между анодной и катодной частями электро-
лизера и обеспечивает точность расположения анодов между катод-
ными пальцами при сборке. Устройство для уплотнения исключает
возможность течи электролита, что позволяет поддерживать чистоту
и опрятный вид серии электролизеров во время их работы.
При применении сдвоенного сварного катода достигается макси-
мальное развитие активной катодной поверхности и интенсивная
естественная циркуляция электролита. Графитовые аноды с трех
сторон окружены катодами, что также увеличивает рабочую анодную
поверхность.
Надежный токоподвод к анодам без применения свинца, подвод
тока к катодной сетке через корпус катода и приваренный к нему
каркас обеспечивают в электролизерах БГК незначительный пере-
пад напряжения в контактах и подводе тока к электродам. Возмож-
ность при монтаже точного регулирования и фиксации положения
анодов позволяет точно устанавливать расстояние между электро-
дами и снизить напряжение на электролизере.
Конструкция электролизера дает возможность работать , при
высокой температуре анолита — до 95—100 °G, что в свою очередь
способствует снижению рабочего напряжения на электролизере
и увеличению выходов по току. Для уменьшения потерь тепла и
улучшения санитарных условий работы в цехе электролиза наруж-
ные поверхности катода электролизера покрываются слоем тепловой
изоляции. Электролизер компактен и полностью герметичен, что
устраняет утечки электролита и газов.
За счет большой высоты крышки электролизера обеспечивается
возможность изменения уровня анолита в пределах от 50 до 300—
400 мм над верхним краем катода. Поэтому электролизеры работают
с подачей постоянного количества рассола, необходимого для полу-
чения щелочи концентрацией 130—140 г/л NaOH. Контроль пита-
ния электролизера осуществляется обычно с помощью ротаметра.
На некоторых заводах подача рассола в каждый электролизер регу-
лируется по уровню анолита, который устанавливается в зависи-
мости от состояния диафрагмы и изменяется по мере ее старения.
132
Рис. 3-4. Расход электроэнергии
постоянного тока на 1 т хлора в
электролизерах БГК-17 при раз-
личной плотности тока.
Для установления требуемого уровня анализируют католит, выте-
кающий из электролизера.
Питание электролизера рассолом может осуществляться через
калиброванные отверстия диафрагмы. Работа электролизера с пода-
чей постоянного количества рассола и при одинаковой нагрузке
по току создает условия для получения максимально возможного
выхода по току при высокой концентрации щелочи.
При подаче постоянного количества рассола, необходимого для
получения щелочи концентрацией 130—140 г/л, по мере ухудшения
протекаемости диафрагмы уровень анолита в электролизере автома-
тически повышается до величины, обеспечивающей протекание пода-
ваемого рассола через диафрагму.
Когда уровень электролита достигнет
верхнего предела 300—400 мм над
уровнем католита, дальнейшее повы-
шение сопротивления диафрагмы не
может быть компенсировано увеличе-
нием высоты уровня анолита и про-
текаемость диафрагмы снижается, что
вызывает рост концентрации щелочи.
Когда концентрация щелочи превы-
сит 140—150 г/л, электролизер вы-
ключают. После замены диафрагмы
тур работы электролизера возобнов-
ляется. Иногда вместо замены диа-
фрагмы применяется ее промывка.
Промывка водой приводит к удале-
нию из диафрагмы отложений солей кальция и магния, а также
графитового шлама и восстановлению протекаемости диафрагмы.
После промывки диафрагмы уровень анолита в электролизере,
необходимый для обеспечения заданной протекаемости, снижается
и восстанавливается способность к авторегулированию величины
протекаемости. Срок работы диафрагмы после промывки меньше
срока службы новой диафрагмы.
Длительность работы электролизера без смены диафрагмы зави-
сит от плотности тока, при которой работает электролизер. В элек-
тролизерах с плотностью тока 500—700 А/м2 за время работы анодов
обычно производился дополнительно одна смена диафрагмы, при
плотности тока 900—1000 А/м2 требуются уже две дополнительных
смены диафрагмы, а при плотности тока 1200 А/м2 и выше диафрагму
необходимо менять еще чаще.
Продолжительность формирования диафрагмы, т. е. период, в те-
чение которого устанавливается ее нормальная протекаемость, за-
висит от плотности тока и колеблется в пределах от 1 до 8 сут.
Графитовые аноды служат 10^—16 мес. После износа анодов электро-
лизер поступает на перемонтаж для замены электродов и диафрагмы.
С увеличением плотности тока возрастают напряжение на элек-
тролизере и удельный расход электроэнергии. На рис. 3-4 приведена
133
зависимость удельного расхода электроэнергии постоянного тока
от плотности тока. Выход по току и удельный расход графита при
этом практически не меняются; снижается лишь длительность фор-
мирования диафрагмы.
С ростом температуры электролита при повышении плотности .
тока увеличивается унос паров воды с водородом и хлором, что при-
водит к росту концентрации щелочи в католите при неизменной сте-
пени превращения хлорида в гидроокись и при том же значении
выхода по току.
При повышении плотности тока в электролизерах можно увели-
чить их компактность, сократить затраты материалов на их изго-
товление и требуемую площадь цехов электролиза.
Электролизер БГК-50 на нагрузку 50 кА аналогичен по устрой-
ству электролизеру БГК-17. В катодном блоке оборудовано шесть
рядов катодных карманов. Схема электролизера БГК-50 дана на
Основные показатели работы электролизеров типа БГК приве-
дены в табл. 3-1.
Таблица 3-1 • Основные показатели работы электролизеров БГК-17 и БГК-50
Показатели
БГК-17
БГК-50
Сила тока, кА ....................
Катодная плотность тока, А/м2 . . . .
Напряжение, В ................ . .
Выход по току, % ..........
Расход
электроэнергии постоянного тока на
1 т хлора, кВт-я ........
графита на 1 т хлора, кг.......
Расход материалов на 1 кА нагрузки, кг
стали.............................
меди..................... . . .
Требуемая площадь на 1 кА нагрузки, м2
без учета проходов . . ........
с проходами . . ...............
Производительность, кг хлора в сутки
электролизером . .......... .
на 1 м2 площади с учетом проходов
20
570
3,32
96
2610
5—6
185
7,5
0,135
0,56
610
52,5
25
730
3,45
96
2720
5-6
148
6
0,11
0,45
765
65,4
25
925
3,60
96
2840
5-6
132
4,6
0,084
0,39
765
75,4
50
1000
3,70
96
2920
5—6
130
4,6
0,084
0,27
1530
108
время работы электролизеров происходит разрушение графи-
анодов и изменяется расстояние между работающими поверх-
Во
то вых
ностями электродов, а также возрастают потери напряжения на
преодоление сопротивления графитовых анодов, электролита и диа-
фрагмы. Увеличение сопротивления диафрагмы объясняется ее
старением и забивкой пор. Все эти процессы обусловливают повыше-
ние напряжения и увеличение количества тепла, выделяющегося
134
ifr--:
в электролизере. Поэтому энергетический и тепловой баланс элек-
тролизера непрерывно изменяются во время тура работы анодов [3].
Напряжение в конце тура работы электродов зависит от степени
срабатывания. Практические значения напряжения и рабочих
SsM I I {фа I kWt I
j
*
Q
X
У
J ,c
28 W
‘ Рис. 3-5. Электролизер БГК-50:
1— анодное днище; 2 — графитовые аноды; з — катодный кор-
пус; 4 — бетонная футеровка; 5 — крышка электролизера;
б — каркас катодных элементов; 7 — подвод тока к катоду;
8 — подвод тока к аноду; 9 — катодные пальцы.
температур в электролизере в начале и в конце работы, а также
средние за тур работы анодов при различных плотностях тока
и питании рассолом, нагретым до 80 °C, приведены в табл. 3-2.
к 135
Таблица 3-2. Изменение напряжения и температуры в электролизерах
БГК-17 [16]
•ь Плотность Тока, А/м2 Напряжение, В Температура электролита, °C
нач. конечн. сред. нач. конечн. сред.
520 3,05 3,90 3,30 84 96 90
730 3,20 4,00 3,45 86 97 92
910 *1 3,30 4,10 3,60 90 98 94
Значения напряжения на электролизере включают также потери
напряжения во внешних токоподводящих шинах (около 0,1 В).
При неизменных нагрузке и температуре подогрева питающего
рассола, рабочая температура электролита определяется в основном
Напряжение на электролизере, В
Рис. 3-6, Зависимость температуры
ацолита от напряжения на электро-
лизере при температуре питающего
рассола 82 °C.
напряжением на электролизере.
На рис. 3-6 приведены данные,
характеризующие зависимость
температуры анолита в электро-
лизерах Б ГК от напряжения на
электролизере. Эти данные полу!
чены в результате измерений
на большом числе электролизе-
ров, работающих при плотностях
тока в интервале 520—910 А/м2 [16].
Потери тепла наружными стен-
ками электролизера за счет луче-
испускания и конвекции состав-
ляют незначительную долю в об-
щем тепловом балансе электроли-
зера. Отвод основного количества
тепла из электролизера осуще-
ствляется с потоком католита и
(без учета потерь в наружной
парами воды, уносимыми с водо-
родом и хлором. Затраты тепла на
испарение влаги возрастают с по-
вышением напряжения и темпе-
ратуры электролизера. При на-
пряжении на электролизере 3,5 В
ошиновке) почти половина всего
количества тепла отводится из электролизера за счет испарения воды.
В конце тура работы анодов с ростом температуры доля затрат тепла
на испарение увеличивается еще больше.
Результаты измерения составляющих энергетического баланса
для электролизера БГК-17 приведены в табл. 3-3.
Значения электродных потенциалов мало изменяются с ростом
плотности тока и по мере износа анодов. Из остальных составляющих
наибольшую долю в энергетическом балансе электролизера соста-
136
Таблица 3-3. Баланс напряжения электролизеров БГК-25 [3]
Плотность тока, А/м®
Показатели *
520
730
910
Температура электролита, °C...........
j Общее напряжение на электролизере, В
Потенциал, В
а анода ................................
А катода .................. ............
Падение напряжения, В
в электролите и диафрагме . . - . .
в катоде................... . . .
в аноде ..........................
в анодном днище . ................
в анодной шине . .................
в катодной шине...................
в прочих проводниках и контактах .
Сумма.................. ..............
Небаланс
90
3,305
+ 1,525
-0,962
0,316
0,061
0,312
0,061
0,061
0,014
0,046
3,358
+0,053
1,6
92
3,456
+ 1,576
—0,972
0,476
0,055
0,332
0,046
0,070
0,016
0,056
3,599
0,143
4,0
94
3,542
+1,583
—0,975
0,516
0,047
0,346
0,039
0,065
0,010
0,063
3,611
0,102
2,8
* Измерения относятся к работе электролизеров с различной [плотностью тока при-
мерно в середине тура работы электродов.
вляют потери напряжения в электролите, диафрагме и аноде.
Повышение напряжения на электролизере к концу тура в основном
обусловлено ростом потерь напряжения в теле анода. На рис. 3-7
О 40 80 120 160 200 240 280 О 40 80 120 160 200 240 280
Продолжительность роботы, сутки Продолжительность работы, сутки.
Рис. 3-7. Изменение напряжения на Рис. 3-8. Падение напряжения в
электролизере БГК-17 за тур работы теле графитового анода за тур ра-
; электродов при цлотнести тока 520 А/м2. боты электродов при плотности тока
520 А/м2:
I, 2 — измерения, проведенные на двух
разных электролизерах.
приведено изменение напряжения на электролизере БГК-17 за тур
работы электродов при плотности тока 520 А/м2.
На рис. 3-8 приведены данные по изменению падения напряже-
ния в теле графитового анода на преодоление омических сопротивле-
’ ний для двух аналогичных электролизеров за тот же период. В конце
137
тура работы электродов потеря напряжения на преодоление оми-
ческого сопротивления анода резко возрастает и достигает 1,2—
1,4 В. Особенно резко увеличивается эта величина за последние
20—30 дней работы электродов.
Электролизеры объединяют в серии, которые обычно питаются
постоянным током от самостоятельной группы преобразователей
тока, причем на серии электролизеров поддерживается постоянная
во времени нагрузка. Напряжение на каждом из электролизеров
серии возрастает по мере износа графитовых анодов, однако при
правильной организации ремонта электролизеров средний возраст
электролизеров и общее напряжение в серии сохраняются в течение
работы примерно на одном уровне. - .
Для ремонта электролизеры отключаются поочередно по одному
или группами по мере старения диафрагмы или износа анодов.
Отключение отдельных электролизеров осуществляется с помощью
шунт-тележки без перерыва питания серии. Для отключения группы
электролизеров иногда устанавливают стационарные устройства.
Все операции по подготовке и сборке основных узлов электро-
лизеров производятся в специальной мастерской; в цех электро-
лиза поступают готовые катодный и анодный блоки и крышка для
сборки электролизера. При разборке электролизеров эти основные
элементы электролизера целиком передают для ремонта в мастер-
скую. Сборка электролизера в цехе электролиза заключается в уста-
новке с помощью мостового крана подготовленных заранее трех
основных узлов электролизера и присоединении электролизера к ком-
муникациям постоянного тока, рассола, щелочи, хлора и водорода.
За время тура работы анодов из-за их износа возрастает меж-
электродное расстояние. Для регулирования межэлектродного рас-
стояния в электролизерах БГК-17 и БГК-50 в середине тура работы
анодов на некоторых заводах применяют замену катодного комплекта
другим с катодными пальцами большей толщины. Это позволяет
снизить межэлектродное расстояние и напряжение на электролизере
во второй половине тура работы анодов. Замену катодных комплектов
приурочивают к смене диафрагмы у электролизеров.
Электролизеры типа ДА
В ГДР разработана конструкция электролизера ДА с вертикаль-
ным расположением электродов и диафрагмы, рассчитанного .на раз-
личные нагрузки [17]. Электролизер имеет гребенчатый катод, сталь-
ную гуммированную крышку с верхним подводом тока через головки
анодов, проходящие через крышку электролизера наружу. В элек-
тролизере исключено применение цемента и вообще силикатных
материалов, что благоприятно сказывается на сроке службы диа-
фрагмы. Схема устройства электролизера типа ДА приведена на
рис. 3-9.
Основные показатели работы электролизеров этого типа приве-
дены в табл. 3-4.
138
Таблица 3-4. Техническая характеристика электролизеров типа ДА
Модель
Показатели
ЛИ
шт-#
Ei.?'-.
Нагрузка, кА................. . . . .
Выработка хлора, кг/сут . . . . . .
Мйсса электролизера без наполнения, т
Размеры электролизера, м
длина ...........................
ширина ..........................
высота . . . ...................
ДА-16
ДА-32
ДА-60
? Ъ.:
ГяЗ" * Л 'г
И
ж
* V
s?e
С-'
№
й'н
4-/
Я,-
s:h
to
32
16
0,87
1,37
3-9. Схема устройства электролизера типа
ДА:
2,57
2,49
1,37
Рис.
1 — корпус катода; 2 — катодный палец; 3 — графитовые аноды; 4 —
крышка; 5 — днище; в — опорные лапы; 7 — устройство для слива
католита; 8— катодная шина; о— анодная шина; 10— опорный
изолятор.
При нормальном обслуживании электролизер может работать
удельным расходом электроэнергии постоянного
60
1810
*
6
ч.
тока 2520—
С
2560 кВт • ч/т 100 % -ного NaOH.
на нагрузку
дальнейшего
Электролизеры типа Хукер (18—26)
Разработка конструкции электролизера Хукер S
6,0—7,5 кА относится к началу 30-х годов. По мере
усовершенствования конструкции номинальная мощность электро-
лизера возросла до 10 кА. В электролизере Хукер S-ЗА рабочая
поверхность электродов была увеличена вдвое и нагрузка возросла
сначала до 20 [27—28], а затем до 25—30 кА [29]. Электролизеры
Хукер S-ЗВ и S-3C рассчитаны на номинальную нагрузку 30 кА.
В последнее время разработаны электролизеры Хукер S-ЗД и Хукер
8-4Д на нагрузку 40 и 55 кА.
139
Быстрое внедрение электролизеров Хукер в хлорную промышлен-
ность США, Канады, Англии и других зарубежных стран обуслов-
лено высокой мощностью этих электролизеров, их экономичностью
и улучшенными эксплуатационными свойствами по сравнению с элек-
тролизерами других типов [30—38]- Различные модели электро-
лизеров Хукер аналогичны по своему устройству. В электролизерах
Хукер применяется гребенчатый катод из стальной или медной сетки
По А -А По б‘в
3 — корпус катода; 4 — катодная шина
Рис. 3-10. Схема электрелизераТ Хукер:
1 — бетонная крышка; 2 — графитовые аноды;
предыдущего электролизера; 5 — анодный контакт; 6 — бетонное днище; 7 — защитное
покрытие; 8 — слой свинца; 9 — катодные пальцы; 10 — анодная шина соседнего электро-
лизера; 11 — катодный контакт; 12 — катодная сетка; 13 — измеритель уровня жидкости;
14 — медная анодная
иг.
с осажденной диафрагмой и нижний подвод тока к графитовым ано-
дам с помощью заливки свинцом.
Возрастание нагрузки • на электролизерах типа Хукер дости-
гается за счет увеличения рабочей поверхности электродов и повыше-
ния плотности тока на них. Рабочая поверхность электродов уве-
личена как за счет установления большего числа электродных эле-
ментов, так и вследствие увеличения их высоты. В первоначальных
моделях электролизера Хукер рабочая высота электродов составляла
140
ШК лишь около 300 мм. Затем она возросла до 450 и позднее до 600 мм.
Ж При этом выход по току не снизился, а несколько возрос. Увеличе-
нию выхода по току от 94 до 97% способствовало повышение элек-
Ж тродной плотности тока и рабочей температуры злектролиза. При-
Bi менение в электролизерах Хукер в качестве анодов графитовых
плит толщиной 32 мм ограничивает оптимальную высоту электродов
Ж в связи с большими потерями напряжения на преодоление омиче-
Ж; ского сопротивления графитового анода.
На рис. 3-10 схематично показан электролизер типа Хукер.
По форме он Приближается к кубу со стороной около 1,5 м. Электро-
лизер состоит из трех основных частей: анодного комплекта, катод-
ного блока и крышки. В бетонное днище злектролизера, имеющее
форму прямоугольного корыта, устанавливают параллельно в 30 ря-
дов графитовые плиты анодов толщиной 32 мм, шириной 150 мм
и высотой 450 мм по 3 анода в ряду. Расстояние между плитами при
Ж новых анодах 44 мм. Нижние концы графитовых анодов и медную
шину для подвода тока к анодам заливают слоем свинца толщиной
Ж 35—40 мм, обеспечивающим контакт между шиной и анодами.
Поверхность свинца защищена от воздействия анолита слоем
эластичной хлоростойкой массы, составляемой на основе природных
битумов. По периметру бетонного днища имеются бортики для
< установления катода. Катодный блок состоит из стального корпуса
и связанного с ним металлического каркаса, на котором располо-
жены два параллельных ряда'пальцеобразных катодов из стальной
или медной сетки, покрытых осажденной асбестовой диафрагмой.
Один ряд катодных пальцев сдвинут относительно другого на поло-
вину шага. При установке катодного блока на днище электролизера
пальцеобразные элементы катодов попадают в промежутки между
графитовыми анодами. Ток к катодному блоку подводится с помощью
шины, приваренной или прикрепленной к корпусу катода. Расстоя-
ние между электродами при новых анодах составляет 12 мм.
На катоде устанавливается бетонная крышка с отверстиями
для отвода хлора, подвода рассола и установления указателя уровня
рассола.
Уплотнение между анодным и катодным блоками и между катод-
ным блоком и крышкой электролизера осуществляется с помощью
J эластичных прокладок из хлоростойких материалов и замазок.
Уплотнение достигается под действием силы тяжести крышки и ка-
тодного блока. Такая система уплотнения для обеспечения герметич-
ности выдвигает высокие требования к эластичным прокладкам
и подготовке поверхности деталей под уплотнение.
Электролизер работает с заполненным катодным пространством.
Щелочь из электролизера отводится по сифонной трубе, поворотом
которой можно регулировать высоту католита в ванне. Водород
выводится по трубе из верхней части катодного блока. Соблюдение
постоянной подачи рассола и нагрузки по току обеспечивает получе-
ние электролитических щелоков, содержащих до 130г—140 г/л ще-
лочи при выходе по току 94—96%.
«<
Таблица 3-5. Примерные технологические показатели электролизеров
типа Хукер [18, 30] ' .
Показатели
Электролизе ры
Нагрузка, кА 10 15 20 25 27 * 30 *
Среднее напряжение, В 3,75 3,25 3,50 3,75 3,85 3,95
Выход по току, % 96,0 95,5 96,0 96,0 96,0 96,0
Температура католита, °C .у. . . . 94 91 95 97 98 99,5
Концентрация щелочи в католите, г/л‘ Содержание хлората в католите, части 135 133 138 138 ь 144 146
на 1000 частей Расход электроэнергии постоянного то- 1 1 1 1 I I
ка, кВт ч/т С12 2965 2535 2767 2965 3050 3120
Расход анодов, кг/т С12 ....... Средний срок службы, сут 3,4 3,9 3,5 2,6 2,6 2,6
анодов ............ 360 425 360 360 ЗЮ 280
диафрагмы . .'. . . ... . . Выработка хлора ♦, кг/сут 120 140 120 120 ^м-НЧ* —I -
электролизером . с 1м2 площади пола цеха (вклю- 302 453 605 756 815 906
чая проходы) . 4 43,98 58,67 76,73 96,27 1 ^м-НЧ* ——
Примерный состав хлоргаза [18]: 97,5% Ch; 0,2 — 0,4% Н£; 1,0 СО/, 0,5% О».
Рис. 3-11. Электролизер Хукер S-ЗА со
снятой крышкой, установленный в серии.
142
J
Таблица 3-6. Характеристика новых мощных электролизеров типа Хукер
Электролизеры
Показатели
S-ЗД 8-4Д *
Нагрузка на электролизер, кА
расчетная..............................................
максимальная ......................................
Катодная плотность тока при нагрузке, А/м2
расчетной.............................................
максимальной ......................................
Напряжение на электролизере при нагрузке, В
расчетной.......................................... . .
максимальной ......................................
Выход по току, % ........................
Расход электроэнергии постоянного тока при расчетной на-
грузке, кВт-ч/т хлора ................................
Размеры электролизера, м
ПЕПШ
длина ...............................................
высота............................................ • •
Расстояние между осями электролизеров, м ................
Площадь пола на электролизер, включая площадь проходов,
м2........................................................
Выработка хлора с 1 м2 площади пола цеха (включая про-
ходы), кг/сут ................................
Количество анодов в электролизере, шт....................
Масса анодов в электролизере, кг ........................
Рабочая масса электролизера, т ...................... .
40
1435
1579
3,96
4,08
96,2
3100
1,78
2,04
1,88
2,28
9,70
120
168
907
10,4
55
60
1287
1404
3,97 '
4,06
96,2
3120
1,78
2,04
2,11
2,28
9,70
167
168
1225
11,3 -
* Высота электродных элементов в электролизере Хукер 8-4Д увеличена.
В зависимости от плотности тока, при которой работает электро-
лизер, за период работы графитовых анодов диафрагму меняют от 2
до 4 раз. Наиболее часто производят трехкратную замену диафрагмы.
Срок работы первой диафрагмы несколько короче за счет загрязне-
ния диафрагмы продуктами хлорирования материалов, применяемых
для пропитки анодов.
Показатели работы электролизеров зависят от условий их экс-
плуатации: концентрации и чистоты рассола, плотности тока, тем-
пературы подогрева питающего рассола, своевременной замены диа-
рагмы по мере снижения ее протекаемости. Примерные показатели
работы электролизеров Хукер S, S-3A, S-ЗВ и S-3C при питании их
рассолом (около 318 г/л NaCl), подогретым до 65—70 °C, приведены
в табл. 3-5.
Характеристика электролизеров типа Хукер новейших кон-
струкций приведена в табл. 3-6 [39—41}.
На рис. 3-11 показан электролизер Хукер S-ЗА, установленный
в серии, со снятой крышкой.
По мере старения диафрагмы и увеличения ее сопротивления
протеканию рассола уровень анолита в электролизере автомати-
чески повышается от 125 до 400—430 мм над верхним краем катода
143
(насколько это позволяет высота крышки). При дальнейшем увели-
чении уровня анолита нужно менять диафрагму.
Зависимость напряжения на электролизере Хукер S-ЗА от плот-
ности тока приведена на рис. 3-12. Там же даны значения отдельных
составляющих общего напряжения на электролизере при различной
плотности тока.
Рис. 3-12. Зависимость общего
напряжения и отдельных его ча-
стей на электролизере Хукер
S-ЗА от плотности тока:
1 — общее напряжение; 2 — анодный
потенциал; 3 — катодный потенциал;
4 — потери напряжения в электролите;
5 — потери напряжения в диафрагме;
6’, 7 — потери напряжения в графито-
вых анодах и в металлических частях
электролизера.
Рис. 3-13. Напряжение на электролизере
Хукер при различных нагрузках:
1 — напряжение на новых анодах; 2 — среднее
напряжение за тур; 3 — напряжение в конце
тура работы анодов.
*
Напряжение на электролизере меняется с течением времени
в связи с износом анодов и изменением сопротивления диафрагм.
Рис. 3-14. Выход по току в электролизерах
Хукер при различной плотности тока.
На рис. 3-13 приведены
напряжения на электроли-
зере для различных сроков
работы графитовых ано-
дов , в начале и конце
тура работы электродов
и среднее за тур при раз-
ной нагрузке на электро-
лизере типа Хукер по-
следних моделей [18].
Разработана математи-
ческая модель электроли-
зера Хукер, позволяющая рассчитывать на ЭВМ напряжение на элек-
тролизере, материальный и тепловой баланс в зависимости от срока
службы, износа электродов, газонаполнения и других факторов [42].
Выход по току на электролизерах типа Хукер возрастает с повы-
шением плотности тока их при соотношении хлорида к гидроокиси
144
и
в электролитических щелоках, равном 1,4, значения выхода по току
при различных плотностях тока приведены на рис. 3-14.
z В литературе описано большое количество разнообразных ва-
риантов как конструкций электролизера типа Хукер в целом, так
и отдельных его деталей. Например, были запатентованы электро-
лизеры большой длины с установкой катодных пальцев в один ряд,
электролизеры со стальными гуммированными крышками, различные
варианты днищ электролизера, уплотнений между днищем и катод-
ным корпусом, типы катодных пальцев с установленными внутри
пружинами, предотвращающими сжатие катодных пальцев в ва-
кууме в процессе осаждения диафрагмы и др. Запатентован также
электролизер, аналогичный конструкции Хукер, но с верхним под-
водом тока к анодам. Однако все эти варианты не нашли практи-
ческого применения.
%
Электролизеры типа Даймонд
ч
Электролизеры типа Даймонд [30] применяются за рубежом
наряду с электролизерами Хукер.
Вначале в электролизерах этого типа применялась листовая
асбестовая диафрагма. Такие электролизеры были разработаны
на нагрузку 10 кА [43, 44].
При дальнейшем усовершенствовании конструкции была увели-
чена мощность электролизера и использована осажденная диа-
фрагма [44, 45]. Последний вариант конструкции электролизера
типа Даймонд [46], получивший применение в хлорной промышлен-
ности, имеет много общего с описанными ранее конструкциями
электролизеров с осажденной диафрагмой. Электролизеры типа
Даймонд выпускаются двух моделей на номинальную нагрузку 20
и 30 кА. Электролизеры имеют прямоугольную форму и размеры
1,524x1,092X1,219 м при нагрузке 20 кА и 2,134x1,092x1,737 м
при нагрузке 30 кА. Электролизер состоит из трех основных частей:
анодного блока, катода и бетонной крышки.
Анодный блок имеет форму неглубокой чугунной чаши. Аноды
из графитовых пластин высотой около 750 мм устанавливаются
в анодной чаше в вертикальном положении рядами. Подвод тока
к анодам осуществляется с помощью медных шин, вставляемых в виде
решетки в нижнюю часть анодной чаши между головками графито-
вых анодов и имеющих выводы для присоединения к шинам от катода
соседнего электролизера. Нижняя часть графитовых анодов — их
головки и токоподводящие медные шины — заливаются расплавлен-
ным свинцом, что обеспечивает электрический контакт к анодам.
Катодный комплект, аналогично электролизеру Хукер, пред-
ставляет собой прямоугольный стальной сварной корпус. Внутри
корпуса расположены катодные пальцы из металлической сетки,
на ее поверхности осаждается асбестовая диафрагма. Катодный
корпус снабжен штуцерами для отвода водорода и электролитиче-
ской щелочи.
10 Заказ 843
145
Хлор
Водород
Рассол
9
электро-
Рис. 3-15. Схема устройства
лизера Даймонд типа Д-З:
I — анодное днище; 2 — катодный корпус;
3 — крышка; 4 — медная катодная шина;
5 — анодная шина; 6 — катодные пальцы;
7 ~ графитовые аноды; 3 — отвод щелочи;
9 — указатель уровня анолита.
При установке катода на анодный блок катодные пальцы распо-
лагаются между графитовыми анодами. Внутренний объем катодных
элементов образует катодное пространство электролизера, наруж-
ный — анодное. На катодном корпусе установлена крышка из бе-
тона с отверстиями для отвода хлора и подачи рассола в электро-
лизер.
В одном из патентов фирмы «Даймонд» [46] описан электролизер,
катодные пальцы которого изготовлены из стальной сетки или, сталь-
ного перфорированного листа, расположены в один ряд и проходят
от одной внутренней катодной сетки электролизера к другой у про-
тивоположной стенки электро-
лизера. Каждый анодный блок
состоит из пяти графитовых
плит, укрепленных заливкой
свинцом в пазах медной решет-
ки, которая установлена на
металлическом днище.
Центральный циркуляцион-
ный проход между электрод-
ными элементами отсутствует.
Предполагается, что циркуля-
ция электролита осуществляется
за счет подъема его вместе с пу-
зырьками хлора вдоль верти-
кально расположенных анодов
и опускания освобожденного от
газовых пузырьков анолита
через полости в анодных про-
странствах у боковых продоль-
ных стенок корпуса электро-
лизера.
Подача рассола на питание производится через одно или два
отверстия в крышке, причем трубка, через которую подводится рас-
сол, опускается почти до дна электролизера, тогда свежий рассол
поступает в нижнюю часть электролизера. ;
Поверхность катода покрывается диафрагмой, а прилегающие
к ней внутренние поверхности корпуса электролизера, соприкаса-
ющиеся с анолитом, защищены от коррозии материалом (например,
бетоном), стойким к действию хлора и анолита.
Схема устройства электролизера Даймонд Д-З на нагрузку 30 кА
приведена на рис. 3-15. Масса такого электролизера без жидкости
составляет около 5 т, а в рабочем состоянии с электролитом — около
7 т. Электролизеры этого типа обладают достоинствами, характер-
ными для электролизеров с нижним подводом тока к анодам и оса-
жденной диафрагмой. Электролизеры приспособлены для работы
с заполненным катодным пространством при постоянных нагрузке
и питании рассолом. Детали конструкции электролизера Даймонд
до настоящего времени мало освещены в литературе.
и
146
Таблица 3-7. Технологические показатели электролизеров типа Даймонд [30]
Нагрузка, кА...............................
Плотность тока, А/м2 . . . ................
Среднее напряжение на электролизере, В . .
Температура электролита, °C .........
Выход по току, % . ............... . . .
Расход электроэнергии постоянного тока, кВт • ч/т
С12 ................................. .
Расход графита, кг/т С12 . ................
Концентрация щелочи в католите, % . . . .
Содержание хлората, части на 1000 частей NaOH
Средний срок службы, сут
анодов .......................... ...... .
_ диафрагмы........................... .
Выработка хлора, кг/сут
электролизером ............................
на 1 м2 площади пола ................ .
Номинальная нагрузка
20 кА 30 кА
20
1240
3,78
91,1
96,5
2967
3,75
10,5
0,54
230
150
612
61,6
23
х1426
3,94
93,3
96,5
3095
3,80
10,5
0,54
210
110
703
71,2
27
1164
3,68
89,4
96,5
2891
3,75
10,5
0,54
255
115
826
78,8
30
1294
3,82
91,1
96,5
3007
3,80
10,5
0,54
230
115
916
87,2
33
1420
3,94
93,9
/96,5
3091
3,75
10,5
0,54
210
110
1010
95,5
Технологические показатели электролизеров типа Даймонд при-
ведены в табл. 3-7.
Электролизеры Даймонд работают при высокой плотности тока
и отличаются сравнительно высоким удельным расходом электро-
энергии. Эти электролизеры на номинальную нагрузку 30 кА успешно
«г
Рис. 3-16. Серия электролизеров Даймонд на нагрузку 30 кА.
работали при нагрузке 34 кА. Возможно также повышение нагрузки
на электролизер до 36 кА без значительного снижения выхода по току
по сравнению с выходом при нормальной нагрузке, равным 96,5% .
10* 147
При нагрузке на электролизер 33 кА производительностью 1 т/сут
хлора требуется (включая помещение для ремонта электролизеров)
11,26 м2 производственной площади по сравнению с 17,4 м2, необхо-
димыми для оборудования цеха электролизерами при нагрузке 20 кА.
На рис. 3-16 показана серия электролизеров типа Даймонд на
нагрузку 30 кА.
Электролизеры с биполярным включением электродов
Биполярное включение электродов открывает большие возмож-
ности при конструировании мощных электролизеров. Было пред-
принято много попыток создать конструкцию такого электролизера,
однако трудности обеспечения надежной защиты от коррозии
устройств для электрического соединения между графитовой анод-
ной стороной биполярного электрода и катодной его стороной за-
держивали создание пригодных для промышленного использования
конструкций.
Несмотря, на многочисленные поцытки создать конструкции би-
полярных электролизеров для получения хлора и каустической соды
по методу электролиза с твердым катодом и диафрагмой [47—48]
до промышленного использования доведены лт
in
ь немногие.
Необходимо учитывать, что по условиям работы с проточной
диафрагмой катодное пространство электролитической ячейки необ-
ходимо располагать с обратной стороны катода. Это усложняет кон-
струкцию по сравнению с электролизерами для разложения соляной
кислоты или воды, где катодное пространство заключено между
диафрагмой и катодом.
Электролизер типа Дау является единственной конструкцией
биполярного аппарата, о промышленном использовании которой
имеются данные. Этот электролизер состоит из большого количества
ячеек, стянутых болтами аналогично секциям фильтр-пресса. Все
электроды, кроме двух крайних, работают биполярно. Ток подво-
дится к первому электроду и отводится от последнего. Аноды из
графитовых плит, графитовые стержни, подводящие к аноду ток,
и катодная сетка вместе с устройством для электрического контакта
катодной сетки с графитовыми стержнями, составляют сложный
биполярно работающий электрод. В современных конструкциях
электролизера Дау катод и анод со стороны, обращенной друг к другу,
имеют перемежающиеся выступы прямоугольной формы и одинако-
вой высоты, расположенные таким образом, что выступы одного
электрода входят во впадины другого. Это позволяет увеличить
работающую электродную поверхность и нагрузку на электро-
лизер.
Гребенчатый катод состоит из ряда катодных пальцев, изгото-
вленных из перфорированного листа или проволочной сетки с листо-
вой или осажденной асбестовой диафрагмой. Электрический контакт
между катодной и анодной сторонами биполярного электрода осу-
ществляется с помощью графитового стержня и медных токопод-
148
водов, конструкция которых допускает небольшие перекосы при
сборке ячеек.
Коллекторы для хлора и водорода проходят вдоль электролизера,
их составляют из элементов бетонных рам при сборке электроли-
зера. Стыки между отдельными ячейками заливаются уплотняющим
составом.
Электролизер сложен в изготовлении, сборке, перемонтаже и ре-
монте. Из-за трудности уплотнения электролизера его герметич-
ность достигается не всегда. Значительные недостатки этой кон-
струкции обусловили ее неконкурентоспособность по сравнению
6
Рис. 3-17. Типы устройств биполярного электродного элемента:
1 — разделительная плита из токопроводящего материала; 2 — стальная полоса, образу-
ющая раму биполярного электрода; 3 — катодная сетка; 4 — катодные кольца; 5 — стержни,
крепящие катодные пальцы к разделительной плите; 6 — графитовые аноды; 7 — медные
желоба; 8 — графитовые плиты; 9 — медная токораспределительная шина; 10 — хлоро-
стойкая масса; 11 — защитное покрытие.
с современными конструкциями электролизеров с осажденной диа-
фрагмой типа Б ГК или Хукер.
В электролизере' фильтр-прессного типа Хантер-Отис-Блю [181
к опорной электродной плите с анодной стороны прикрепляются
графитовые аноды, с катодной — с помощью медных контактов —
катоды, изготовленные из стальной сетки в виде катодных пальцев.
При сборке отдельных рам в фильтр-пресс катодные пальцы поме-
щаются в зазорах между анодами. Подача рассола и отвод продуктов
электролиза производят через коллекторы, снабженные отводами
к каждой ячейке. Фильтр-прессный электролизер может иметь до
50 ячеек, а серия из четырех электролизеров — 200 ячеек. Сообще-
ний о промышленном использовании такой конструкции в литера-
туре нет.
В последнее время проводились работы по усовершенствованию
конструкции биполярных электролизеров с целью увеличения ' их
мощности и улучшения конструкции [49]. Имеются сообщения [501
о конструкции электролизера, в котором биполярные электродные
элементы, составляющие ряд последовательно включенных ячеек,
размещаются в общем бетонном корпусе электролизера.
Варианты устройства биполярного электродного элемента пока-
заны на рис. 3-17.
149
t
Биполярный электрод монтируется на разделительной плите,
изготовленной ’ из стали или другого токопроводящего материала.
По периметру плиты приварена полоса, образующая раму биполяр-
ного электрода, вставляемую в вертикальные пазы стенки корпуса
при сборке электролизера. К краям рамы прикреплена катодная
сетка, в свою очередь к сетке присоединены катодные плоские пальцы,
// /
я Ю
"ИТПД
Рие. 3-18. -Схема устройства электролизера
Ниссо:
I — подвод тока к катоду; 2 — разделительные пе-
регородки; 3 — отвод хлора; 4 — графитовые аноды;
5— катодные элементы; 6 — отвод водорода; 7 —
подача рассола; 3 — подвод тока к аноду; о — уров-
немер; 10 — отвод электролитической щелочи; 11 —
корпус электролизера; 12 — крышка электролизера;
13 —изолятор; 14 — термометр; 15 — люк; 16 —
отверстие для подачи рассола.
rm тг пг
разделительной
и токопереда-
элементами уста-
медные про-
- закрепляемые в электрод-
ной плите на стержнях.
' Графитовые аноды рас-
полагаются с другой,
анодной стороны плиты.
Подвод тока к анодам мо-
жет осуществляться непо-
средственно от раздели-
тельной плиты (если аноды
вставляются в соответству-
ющие пазы в плите и за-
крепляются там), либо
с помощью конусных мед-
ных желобов, приварен-
ных или прикрепленных
болтами к стальной раз-
делительной плите, либо
через промежуточные гра-
фитовые литы, прикреп-
ленные болтами к разде-
лительной плите. Для
улучшения подвода тока
между^
. плитой
ющими
новлены
кладки.
На поверхность катодных пальцев и катодной сетки насасывается
диафрагма из взвеси асбестового волокна в соляно-щелочном рас-
творе. Анодная сторона разделительной плиты, анодные контакты
и поверхность рамы защищены хлоростойкой массой и слоем защит-
ного покрытия, например гуммировкой.
Внутренние полости катодных пальцев биполярного электрод-
ного элемента соединены с пространством между разделительной
плитой и катодной сеткой и составляют катодное пространство
ячейки.
Водород и электролитическая щелочь выводятся через отверстия
в раме биполярного электрода.
Концевые монополярные электроды имеют аналогичное устрой-
ство. Концевой монополярный анод выполняется так же, как и анод-
ная сторона биполярного электродного элемента; концевой моно-
полярный катод аналогичен катодной стороне биполярного электрода.
150
Много общего с описанной конструкцией имеет японский электро-
лизер типа Ниссо [51], схема устройства которого приведена на
рис. 3-18. В одном корпусе установлено 5 ячеек с биполярным вклю-
чением электродов. Нагрузка на ячейку 4 кА, плотность тока на
аноде 780 А/м2, на катоде 690 А/м2. Среднее напряжение на ячейке
3,4 В, выход по току 95%, концентрация щелочи в католите до 145 г/л.
Катодные пальцы выполнены из металлической сетки и покрыты
асбестовой бумагой. Размеры электролизера из 5 ячеек 1444 X
X 3039x900 мм; размеры анодов 203x440x25 и катодов 440 X 440 х
Х20 мм. Срок службы графитовых анодов 6—8 мес. Промежуточная
смена диафрагмы за тур работы электродов не проводится. Отме-
чается, что для электролизеров этого типа требуется меньшая пло-
щадь пола производственного помещения, они удобны в ремонте
и обслуживании и работают с малыми удельными расходами электро-
энергии.
Та же фирма разработала более мощную конструкцию электро-
лизера с биполярным включением электродов. В 1973 г. предпола-
гается пуск промышленной установки, оборудованной электроли-
зерами ВМ-50 из пяти последовательно включенных ячеек на сум-
марную нагрузку 250—300 кА [51а].
II
Биполярный электролизер большой мощности
В связи с увеличением мощности отдельных хлорных цехов
до 250—500 тыс. т хлора в год мощности диафрагменных электро-
лизеров описанных ранее конструкций недостаточны для оснащения
крупных цехов, поэтому стала неотложной задача создания новых
электролизеров с большой нагрузкой (100—200 кА и более) в одном
агрегате.
Разработка конструкций таких электролизеров на базе старых
принципов конструирования связана с большими трудностями^
так как при этом электролизеры становятся громоздкими, ослож-
няется транспортирование и ремонт деталей. С увеличением габари-
тов и массы отдельных деталей электролизеров необходимо приме-
нять краны большой грузоподъемности, увеличивать монтажные
проемы и площади для ремонта электролизеров. Преимущества^
получаемые от укрупнения электролизеров, могут быть в значитель-
ной мере обесценены этими недостатками крупных агрегатов.
Наиболее целесообразно для создания мощных электролизеров
применить принцип секционного устройства их путем набора повто-
ряющихся элементов — ячеек или секций. При параллельном со-
единении таких секций по току может быть получен мощный электро-
лизер с монополярпым включением электродов, при последователь-
ном включении — электролизер с биполярным включением электро-
дов. Биполярное решение мощного электролизера имеет свои пре-
имущества по сравнению с монополярным вариантом, так как при
этом нет необходимости в чрезмерном повышении силы тока, облег-
чается ошиновка, включение и выключение электролизера.
151
В корпусе биполярного электролизера размещены два моно-
полярных и комплект биполярных электродов. Катодная часть
биполярного электрода выполнена в виде гребенчатого сетчатого
электрода (аналогично катодам электролизера БГК-17). Катод при-
способлен для работы с осажденной диафрагмой. Анодная часть
представляет собой графитовые плиты, соединенные с катодной
частью биполярного электрода металлическими контактами. Кон-
такт между катодной и анодной частями биполярного электрода
защищен от воздействия кислого анолита слоем тугоплавкой битум-
ной массы и бетона. Уплотнением между отдельными ячейками слу-
жит слой специальной тугоплавкой массы.
В конструкции электролизера применен боковой подвод тока
к анодам, что позволяет увеличить рабочую высоту электродных
элементов без повышения напряжения за счет роста потери напря-
жения на преодоление омического сопротивления графитовых ано-
дов. При соблюдении оптимальных соотношений геометрических раз-
меров катодных элементов можно получать выход по току 96%
и выше при большой высоте рабочей части электродов. При одина-
ковой плотности тока напряжение на электролизере несколько
ниже, чем на обычных электролизерах с осажденной диафрагмой
типа Б ГК, вследствие исключения потерь напряжения в ошиновке
между электролизерами и уменьшения потерь напряжения на пре-
одоление омического сопротивления графитовых анодов.
Основные технологические показатели электролизера из трех
последовательно включенных ячеек с высотой электродных элемен-
тов более 1 м и расстоянием между электродами 15 мм, полученные
за тур работы электродов в течение 276 сут, приведены ниже:
Нагрузка на ячейку, кА .................... 15
Плотность тока, А/м2
на аноде ............................. 900
на катоде.............................. 885
Напряжение на ячейке, В .................. 3,48
Температура электролита, °C ............. 92—94
Выход по току, % 95,5
Концентрация, г/л
щелочи в католите . . . . . . , . . 134
хлоратов ........................ 0,14
Расход электроэнергии постоянного тока,
кВт -ч/т ,С12 . . ...................... 2750
Основным недостатком всех предложенных конструкций биполяр-
ных электролизеров является малый срок работы графитовых элек-
тродов и значительные трудовые затраты на сравнительно частые
переборки электролизеров для замены электродов и диафрагм.
Наиболее важной и перспективной проблемой в области электро-
лизеров с твердым катодом является разработка долгоживущих,
малоизнашивающихся электродов и диафрагмы и создание на их
основе биполярных электролизеров очень большой мощности. Тур
работы таких электролизеров между разборками для ремонта мог бы
составить 3—5 лет. Для эффективного использования малоизнаши-
152
вающихся анодов в электролизерах с твердым катодом необходима
создать новые типы диафрагм с длительным сроком работы при вы-
сокой плотности тока.
Электролизеры о малоизнашивающимиоя анодами
Для рационального и экономичного использования малоизна-
ивающихся анодов (МИА) требуются принципиально новые кон-
и
струкции электролизеров с диафрагмой. Однако применение МИА
даже в известных в настоящее время конструкциях электролизеров
типа БГК, Хукер или Даймонд, приспособленных для работы с гра-
фитовыми анодами, позволяет получать большие технические и
экономические преимущества.
Опыт длительной (в течение 4—5 лет) эксплуатации промышлен-
ных образцов электролизеров типа БГК-17 с платинотитановыми
анодами вместо графитовых [52] показал высокую надежность и
технико-экономические преимущества эксплуатации таких электро-
лизеров.
Применение МИА позволило повысить плотность тока до 2 кА/м2
(вместо 1 кА/м2 при графитовых анодах) без увеличения напряжения
на электролизере. Благодаря этому, а также вследствие уменьшения
толщины МИА до 51—-10 мм (вместо 50 мм у графитового анода) в элек-
тролизере тех же габаритов удалось увеличить нагрузку в 2,8—
3,2 раза при том же напряжении на электролизере. Это открывает
возможность для дальнейшего укрупнения монополярных электро-
лизеров и увеличения экономически выгодной нагрузки на ячейку
биполярного электролизера.
Напряжение на электролизере с МИА мало изменяется в течение
его работы. Потери напряжения на преодоление омического сопро-
тивления анода, плотность тока на аноде, анодный потенциал,
межэлектродное расстояние и потери напряжения в электролите
в течение работы электролизера с МИА остаются постоянными,
в отличие от электролизеров с графитовыми анодами, у которых
по мере износа графита все эти составляющие энергетического ба-
ланса возрастают, обусловливая непрерывный рост напряжения
на ячейке.
Некоторое незначительное повышение напряжения на электро-
лизере с МИА вызывается постоянной забивкой диафрагмы в про-
цессе работы. По мере забивки для восстановления протекаемости
диафрагмы и уменьшения ее сопротивления применяется промывка
диафрагмы горячей водой.
Практически постоянное за все время работы напряжение на
электролизере с МИА позволяет поддерживать стабильный темпе-
ратурный режим работы электролизера. Помимо этого, среднее
за тур работы электролизера напряжение существенно ниже, чем
в случае применения графитовых анодов.
При использовании МИА резко сокращаются трудовые затраты
на переборку электролизеров для смены анодов.
153
Для электролизеров с МИА не требуется тщательная очистка
рассола от SO|", так как эти примеси в рассоле не ухудшают стой- •
кость анодов, как это наблюдается для графитовых анодов. Хлор
и каустическая сода не загрязняются продуктами окисления ано-
дов и хлорирования органических веществ, применяемых для импрег-
нирования графита или содержащихся в материале графитовых ано-
дов. При применении платинотитановых анодов (ПТА) расход
платины не превышает 0,5 г/т хлора. ПТА с платиновым покрытием
толщиной 3 мкм после 4 лет эксплуатации при плотности тока 1,2—
2,0 кА/м2 оставались пригодными для дальнейшей работы и не тре-
бовали замены. Технико-экономические подсчеты показали, что
при существующих ценах на графит, титан и платину себестоимость
хлора и каустической соды при переходе на ПТА несколько сни-
жается по сравнению с работой на графитовых анодах. Однако,
несмотря на технические преимущества, использование ПТА вслед-
ствие дефицитности платины не выходило за пределы нескольких
промышленных образцов электролизеров.
Разработка способов изготовления МИА с использованием в ка-
честве активного слоя вместо платины или сплава ее с родием более
дешевого и менее дефицитного рутения создало дополнительные
стимулы для применения МИА в хлорной промышленности. На окис-
норутениевых анодах перенапряжение выделения хлора невелико,
напряжение на электролизере и удельный расход электроэнергии
снижается по сравнению с этими же показателями на графитовых •
анодах [53, 54]. г
При применении МИА, работающих с более высокими плотно-
стями тока, удельный вес потерь напряжения на преодоление оми-
ческого сопротивления электролита в общем энергетическом балансе
электролизера существенно возрастает. С увеличением плотности тока
возрастает влияние газонаполнения на повышение эффективного
сопротивления электролита. Поэтому в конструкциях электролизе-
ров с МИА предусмотрена возможность уменьшения межэлектрод-
ного расстояния и облегчения отвода пузырьков хлора, выделяю-
щихся на^ анодах. Для этой цели применяют проницаемые для газа
аноды и приближают их к диафрагме вплоть до прилегания к ней
с тем, чтобы обеспечить отвод пузырьков хлора на обратную сторону
анода.
В последнее время появилось много сообщений о разработке
конструкции диафрагменных электролизеров с металлическими ано-
дами (так часто называются малоизнашивающиеся аноды с активным
слоем из металлов платиновой группы или их окислов).
Предложены конструкции монополярных электролизеров с ано-
- дами такого типа, нижним токоподводом к ним и защитой днища
токонепроводящим слоем [55], а также с токоподводами из меди или
алюминия, защищенными от коррозии слоем титана [56].
При применении металлических анодов в электролизерах с диа-
фрагмой можно увеличить на 40—50% плотность тока и примерно
вдвое срок службы асбестовой диафрагмы [57]. По сравнению с гра-
154
и
J С
фитовыми анодами срок между переборками электролизера возра-
стает от 0,5—1 до 3—4 лет, между сменами диафрагмы от 0,25—
0,3 до 0,5—1,0 года [58].
Имеются сообщения фирм «Даймонд Шемрок» и «Де-Нора» о кон-
струкции диафрагменного электролизера Элинкор 46 с металличе-
скими анодами на нагрузку 80 кА [59], о выпуске фирмой «Хукер»
электролизеров с окиснорутениевыми анодами на нагрузку до
90—100 кА. Окиснорутениевые аноды используются при рекон-
струкции действующих производств, оборудованных электролизе-
рами Хукер [60].
Фирма «Даймонд Шемрок Кемикел Компани» предлагает электро-
лизеры с диафрагмой и малоизнашивающимися анодами, основные
показатели которых приведены в табл. 3-8.
Таблица 3-8. Технологические показатели электролизеров типа ДС
Электролизеры
Показатели
ДС-31 ДС-45
ДС-85
Нагрузка, кА..........................
Плотность тока, кА/м2 ................
Напряжение на электролизере (включая
внутреннюю ошиновку), В ..............
Выход по току, % ..................
Число анодов, шт. ....................
Высота катода, м .'...................
Габариты электролизера, м
длина ................................
ширина............................
высота . .........................
Площадь пола, занимаемая электролизе-
ром с учетом проходов, м2 ............
It С
20—40
0,99—1,98
2,97—3,64
96,5
32
0,72
1,56
1,09
1,8
20,2
60—80
2,07—2,76
3,70—4,17
» 96,5
46
0,72
2,16
1,09
1,8
29,0
100—150
1,82-2,74
3,54—4,15
96,5
87
0,76
2,86
1,52
2,06
54,8
По рекламным сообщениям фирмы, малоизнашивающиеся аноды
работают не менее 4—6 лет, а срок службы диафрагмы без остановок
при хорошем качестве рассола и равномерной нагрузке равен 1—
2 годам. Для предотвращения загрязнения рассола продуктами
коррозии бетона крышки электролизеров выполнены из пластмассы
на основе полиэфирной смолы.
В наибольшей мере преимущества металлических анодов в элек-
тролизерах с диафрагмой могут проявиться при биполярном вклю-
чении электродов. Предложена конструкция биполярного электро-
лизера с титановыми электродами, покрытыми с анодной стороны
активным слоем из металлов платиновой группы [61]. Фирмы «Де-
Нора» и «Питсбург плейт гласе» разработали конструкцию биполяр-
ного электролизера с диафрагмой и металлическими анодами произ-
водительностью 30—60 т/сут хлора [62—64], что эквивалентно на-
грузке (определяемой как произведение числа последовательно вклю-
ченных ячеек электролизера на нагрузку) на электролизер от 1 до
155
2 МА. Такие электролизеры в 2—4 раза мощнее самых крупных
электролизеров с ртутным катодом, о которых имеются сообщения
в литературе.
Основные показатели электролизеров
е твердым катодом
Данные о многочисленных конструкциях электролизеров, при-
менявшихся в различное время в промышленности, приведены в ли-
тературе с различной степенью подробности. Поэтому затруднительно
составить сводную таблицу, включающую с одинаковой полнотой
основные конструктивные характеристики и технологические пока-
затели всех применявшихся в промышленности электролизеров.
В табл. 3-9 приведены такие данные лишь для основных конструкций
электролизеров, широко применявшихся в промышленности. Более
подробные данные опубликованы в работе [13].
11
ЭЛЕКТРОЛИЗЕРЫ С РТУТНЫМ КАТОДОМ
Общие принципы устройства электролизеров
е ртутным катодом
%
Электролизер с ртутным катодом состоит из электролитической
ячейки, в которой в процессе электролиза получается хлор и амаль-
гама щелочного металла, и разлагателя амальгамы. В разлагателе
образуется раствор каустической соды и водород и регенерируется
ртуть. С помощью насоса либо иного устройства обеспечивается
постоянная циркуляция амальгамы (ртути) по циклу электролити-
ческая ячейка — разлагатель. Если ртуть используется в качестве
биполярного электрода, необходимость в разлагателе амальгамы
отпадает, однако многие предположения, касающиеся разработки
схемы и конструкции электролизеров с биполярными ртутными
электродами и диафрагмой 465—68], не нашли практического при-
менения. Это объясняется конструктивными трудностями и опас-
ностью анодного растворения ртути вследствие неравенства катод-
ного и анодного выходов по току.
Предложено несколько способов создания ртутного катода.
Применение в качестве катода неподвижного зеркала ртути, поме-
[енной в корыто, связано с необходимостью периодически, по мере
образования амальгамы, заменять ее свежей ртутью. Этот способ
в промышленности не используется. Практически применяется только
движущийся ртутный катод, в котором образующаяся в процессе
электролиза амальгама непрерывно выводится из электролитической
ячейки в разлагатель и заменяется свежей ртутью. Почти во всех
применяемых на практике электролизерах движущийся ртутный
катод образуется за счет перемещения тонкого слоя ртути по пло-
скому слабо наклонному днищу электролизера. Все электролизеры
этого типа получили название электролизеров с горизонтальным
катодом.
156
11
a
£f'
if
4
Is
Многочисленные предложения по использованию в качестве
катода слоя ртути, стекающей по вертикальной или сильно наклон-
ной поверхности [71—76], не получили до настоящего времени i
рокой реализации в промышленности. При большой скорости дви-
жения амальгамы происходит интенсивное перемешивание электро-
лита в прикатодном слое, что приводит к увеличению катодного
восстановления хлора и снижению выхода по току. Кроме того,
при использовании вертикального или сильно наклонного катода
возникает необходимость в применении диафрагмы между катодом
и анодом, что сильно усложняет конструкцию электролизера.
Предложен пористый катод, через поры которого продавливается
ртуть, образуя ртутный катод [77, 78].
В конструкциях промышленных электролизеров с ртутным ка-
тодом, используемых в промышленности, ртутный катод представляет
собой поток ртути, движущийся вдоль электролизера. Предлагается
также организовать движение ртути в направлении, перпендикуляр-
ном длине электролизера, с целью уменьшения количества ртути
на днище электролизера [79]. Однако в этом случае потребуется
дополнительная загрузка ртути в устройства для распределения
и сбора потоков ртути в начале и конце ртутного катода. Выигрыш
в количестве ртути, покрывающей поверхность катода, снижается
за счет увеличения количества ртути в устройствах по распределе-
нию и сбору потока ртути на входе и выходе из электролизера.
Были сделаны попытки применения в качестве ртутного катода
движущейся металлической поверхности, смоченной ртутью [79—83].
Такой катод был использован в электролизерах, где катодом слу-
жили стальные диски, насаженные на горизонтальный вал и погру-
женные нижней своей частью в ртутную ванну. При вращении вала
на поверхности дисков образовывался постоянно меняющийся слой
амальгамы.
Другой вариант такого электрода, был использован в электроли-
зере фирмы «Асахи гласе Ко.»* [46, 84—87], где катодом служил
тонкий слой амальгамы на круглом горизонтальном вращающемся
диске. Ртуть, поступающая в центре диска, за счет центробежной
силы движется от центра к периферии, образуя ртутный катод.
Такие катоды применялись и применяются весьма ограниченно,
что можно объяснить конструктивными трудностями.
Катоды в виде падающих струй ртути [88—90] не применяются,
так как уже на небольшой высоте происходит разрыв струи ртути
на отдельные капли.
Практически в промышленности в настоящее время используются
почти исключительно электролизеры с горизонтальным катодом.
В большинстве современных конструкций по стальному днищу,
к которому подводится ток, протекает слой амальгамы. Днища
электролизеров могут покрываться гуммировкой или специальной фу-
теровкой. В этом случае подвод тока к слою амальгамы осуществляется
с помощью специальных токоподводов, утопленных несколько
ниже уровня гуммировки или футеровки днища. В электролизерах
157
Таблица 3-9* Технологические показатели различных типов электролизеров с
Электролизер
Способ разделе-
ния электродных
пространств
Анод
Катод
Корпус
Электролизеры с не
Грисгейм-
электрон
Колоколье
Биллитер-
Лейкам
Песталлоца
Нельсон
Кребс
Аллен-Мур
Гибе
Ворс
Х-2
х-з
Цементная
непроточная
диафрагма
Колокол
Газозащитные
оболочки
Газозащитные
оболочки
Угольные или
магнетитовые
плиты
Стальные Пер-
форированные
листы и. стенки
корпуса
Стальной
ящик
Электролизеры с проточным
Графитовые
плиты
То же
Графитовые
плиты
Стальные
листы
Стальные
стержни
Стальные
стержни
Бетонный
ящик
То же
Стальной
футероваг
ящик
Электролизеры с вертикальной фильтрующей
Асбестовая
листовая
диафрагма
То же
»
»
»
»
Графитовые
стержни
Графитовые
плиты
То же
V-образный
катод из пер-
форированного
стального листа
W-образный
катод из пер-
форированного
стального листа
V-образный
катод из пер-
форированного
стального листа
Стальной
ящик
То же
Цилиндрические электролизеры с вер
Один ряд
графитовых
стержней
То же
»
»
Цилиндриче-
ский из пер-
форированной
стали
То же
Стальной
цилиндри-
ческий
То же
»
2 концентри-
ческих цилин-
дрических
»
158
твердым катодом
*
Начальное расстояние между электродами, мм Размер электролизера, мм Нагрузка, кА Плотность тока, А/М2 * Среднее напряжение, В •ч Температура, °C * Концентрация щелочи, г/л Выход по току, % Расход электроэнергии, кВт>ч/т хлора
длина ширина высота
л « ь
проточной диафрагмой
100
3812
3100
электрод и том
3000
1250
870
без
2,5
200*
д и а ф р а гм ы
0,5
300
4,0
4,0
100
100 " 2650
2100
900
3,0
1,6—
1,8
500
500
3,6-
4,0
85—
95
40—
50
55-
70
60—
70
120
120
125—
140
70—
80
85
92
90
3780—
4100
3550
3350
диафрагм ой и
корытообразным катодом
40 2000 340 860 1,0 500— 600 3,8- 4,0 100— 120 90 3300
|« 1 1 —«а 2,0— 6,0 600— 650 3,6— 4,5 65— 75 100— 120 90— 95 3300- 3600
2590 378 ч 915 ч 1,5-— 1,8 450 3,8- 4,2 1 100— 125 90— 92 3300
диа
тикальной фильтрующе
V
1000
24
= 665
= 665
= 665
1,0
1000
1000
1000
1,0
1,0
1,5
ф р а г м о й
720
720
700
3,6— 3,8 60— 65 100— 120 у 92 2900— 3100 21 ♦
3,8 70— 80. 100— 120 91- 94 3100 21
3,8 70— 75 100— 110 91— 94 3100 21
3,8 75— 100— 91— 3100 31,5
/> 80 110 94
- 665
159
Продолжение табл. 3-9
to
to
to
to
Нс
Цс
1 * 1 1- 1 1 СО СО -Ьг НЬ кАь -]_ь, рь СО | СО | СО | СО |
2040 2134 1 069
1780 1092 —.1 11 । °? 1 1 1.1 1
2100 1737 1140
СП to Ci о со СП о£=! оо w ел ro> to- to- од о q О О ел 4S. -о © -о © о
1 1 to to о Sg 1 1 © оо о о й ££ о •<! о о '-Л £
СО со СО со СО со 11^^ '» Ч» Ч* I | 00 СО 00 -<l Ci со to СП О О СП
© со с, | , 1 со о © о со с 1 1 сл | | *<j 0 00 00 00 О СО СЛ СО СЛ © СП 00 | 00 | СЛ |
1 , tC^r wen ОЭ с 1 1 СЛ | СЛ а. !-* Н*- 1-^ о i-^Cp i-^Cp i-^CO ьл- 5 coo coo COO too СП | СЛ 1 СП 1 СП 1
Не
to to
Начальное расстояние
между электродами, мм
длина
ширина
высота
00
to
Нагрузка, кА
Электродная плотность
Среднее напряжение, В
Температура; °C
Концентрация щелочи
г/л
to
Выход по току, %
tO
со
tO
to
to
tO
tO
to
to
Расход электроэнергии,
кВт*ч/т хлора
Выработка хлора о Гм2
«площади пола, включая
проход, кг/сут
Продолжение табл. 3-9
d % Электролизер Способ разделе- ния электродных пространств Анод Катод Корпус
Циба-Монтей
Электролизеры с двумя
Две асбесто-
вые диафрагмы
Стальные
прутья
Бетонный
ящик
- Графитовые .
плиты
Электролизеры с б ипо ля р н ым
Дау
Мощный
биполярный
Биполярный
с металличе-
скими анодами
Асбестовая
диафрагма
Асбестовая
осажденная
диафрагма
Диафрагма
Графитовые
То же
Металлические
аноды
Металлическая
сетка
Пальцеобраз-
ные из сталь-
ной сетки
Бетонные
рамы
Стальные
защищенные
пли бетонные
рамы
НРЛП
1$
большой мощности возможны искажения потока ртути вслед-
ствие возникновения электромагнитных полей и электродина-
мических сил, возбуждаемых током большой силы. Предложено
наложение электромагнитного поля обратного направления для
устранения нарушений в гидравлике потока ртути [94].
На рис. 3-19 показаны основные типы корпуса электролизеров
с ртутным катодом. Конструкции электролизеров а использовались
в самом начале развития метода электролиза с ртутным катодом.
В последнее время в вариантах этого типа иногда применяются раз-
личные облицовочные плиты из стекла, керамики и других мате-
риалов [91, 92]. Однако неудобства, связанные с футерованием или
гуммированием стальных корпусов, привели к тому, что в боль-
инстве конструкций электролизеров гуммировка используется
и
только для защиты стенок корпуса.
Наиболее удобны типы корпуса гид, где гуммированы только
боковые стенки корпуса, а днище электролизера не защищено.
Хотя принцип конструирования электролизеров с горизонталь-
ным катодом за последние три четверти столетия мало изменился,
уровень техники электролиза значительно возрос. Плотность тока
в электролизерах с графитовыми анодами увеличилась от 1—2
до 10—12 кА/м2, а нагрузка — от 5—10 до 400—500 кА. В электро-
лизерах с металлическими анодами происходит дальнейшее увели-
162
Начальное расстояние между электродами, мм эле! л и S § З4 ширина g gg S CD w« kW высота Нагрузка, кА Электродная плотность, А/м2 ч Среднее напряжение, В Температура, °C 1 /" Концентрация щелочи, г/л Выход по току, % Расход электроэнергии, кВт’Ч/т хлора Выработка хлора с 1 м2 площади пола, включая проход, кг/сут
фильтрующими диафрагмами
3400 1600 1400 7,0
клюнением электродов
50—85 ячеек на нагрузку до 1,5 кА
4—50 ячеек на нагрузку до 25 кА
2 МА
чение плотности тока. Интенсификация процесса электролиза
с ртутным катодом экономически целесообразна при одновременной
разработке мер, предотвращающих повышение напряжения на
электролизере и увеличение расхода электроэнергии [93].
Рис. 3-19. Основные типы корпусов горизонтальных электролизеров с ртутным
катодом:
а — бетонный корпус электролизера; б — стальной корпус корытного типа с гуммирован-
ным дном; в — стальной корпус корытного .типа с голым стальным дном; г, д — стальной
корпус рамного типа с голым стальным дном.
11* .163
Увеличение плотности тока в электролизерах! с ртутным катодом
во многом связано с разработкой способов регулирования межэлек-
тродного расстояния в процессе электролиза. По мере износа гра-
фита в процессе работы и увеличения расстояния между работающей
поверхностью графитового анода и ртутным катодом необходимо
обеспечить опускание графитовых анодов.
Для опускания графитовых анодов по мере их износа предло-
жено много различных способов, которые можно разделить на само-
произвольные и принудительные.
Самопроизвольное опускание анодов может быть осуществлено
установкой между днищем электролизера и графитовыми анодами
дистанциот^ных вставок, фиксирующих межэлектродное расстоя-
ние [95]. Вставки изготавливаются из материалов, не проводящих
ток, они не должны экранировать поверхность графитовых анодов
в месте их опоры на вставки. Необходимо также эластичное уплотне-
ние в месте прохода токоподводящего стержня к аноду через крышку
электролизера, чтобы обеспечить свободное опускание электрода
по мере его износа под влиянием собственной тяжести. Подвод тока
к аноду должен быть выполнен с помощью достаточно гибкого кабеля
или пакета шин, чтобы не препятствовать опусканию анодов. Однако
при практической реализации таких предложений встретились труд-
ности, и эти способы опускания анодов применения в промышлен-
ности не нашли.
Практически во всех известных конструкциях современных элек-
тролизеров с ртутным катодом используется принудительное опу-
скание анодов по мере их износа, которое может осуществляться
как вручную, так и механически, что позволяет автоматизировать
этот процесс.
Наиболее трудной проблемой является контроль за фактическим
расстоянием между электродами. Предложено опускать аноды до
соприкосновения с амальгамным катодом (что легко контролируется
по электрическим параметрам) и затем поднимать их на заданное
расстояние [96—99]. Такой способ регулирования положения анодов
под нагрузкой связан с возникновением многочисленных кратковре-
менных коротких замыканий в электролизере, что нарушает его
нормальную эксплуатацию. Для предотвращения или ограничения
вредных последствий коротких замыканий автоматическая система
должна обеспечивать очень быстрый подъем анодов сразу же по до-
стижении их контакта с амальгамным катодом.
Опускание анодов по такому способу может быть проведено также
с выключением электролизера на время регулирования, однако это
приводит к увеличению потерь ртути за счет ее окисления и уноса
с анолитом за время выключения.
Обычно для регулирования положения анодов пользуются показа-
телями силы тока, проходящего через данный анод или группу анодов
и напряжения на электролизере или потери напряжения на одном из
элементов электролизера [100,101 ]. Для этой цели могут быть исполь-
зованы измерительные схемы с наложением переменного тока [102].
164
При опускании анодов уменьшается расстояние между электро-
дами и снижаются потери напряжения на преодоление омического
сопротивления электролита. Однако при сближении анода с като-
дом возникают условйя для увеличения потерь, обусловленных взаи-
модействием хлора с амальгамой натрия и образованием исходного
продукта — хлористого натрия. Помимо того, возникает опасность
образования эпизодических коротких замыканий между анодом
и ртутным катодом. Минимальный удельный расход электроэнергии
на производство достигается при оптимальном расстоянии между
электродами. Обычно на практике в электролизерах с ртутным
катодом стараются поддерживать межэлектродное расстояние
Поддержание оптимального межэлектродного расстояния в элек-
тролизерах с большим числом анодов представляет .технические *
трудности. Короткие замыкания на современных электролизерах
большой мощности могут приводить к нарушению целостности де-
талей токоподводящих шин и повреждению самого электроли-
зера [103]. При коротком замыкании на одном из анодов электро-
лизера общее напряжение на электролизере меняется на сравни-
тельно небольшую величину (до 100—300 мВ), так как при большом
количестве электродов многократное увеличение силы тока на одном
аноде приводит к сравнительно небольшому снижению плотности
тока на остальных анодах. Нагрузка на отдельные аноды в зависи-
мости от межэлектродного расстояния может изменяться в широких
пределах.
Для иллюстрации этого можно привести данные об изменении
силы тока на аноде электролизёра с ртутным катодом в зависимости
от его положения [104]. Если нагрузка на анод при равномерном
распределении тока между анодами электролизера составляла
1,5 кА, то при сохранении такого же напряжения на электролизере
(4,1 В) максимальная нагрузка на электрод при его максимально
возможном опускании может достичь 3,5 кА, т. е. стать в 2,3 раза
выше нормальной. При этом межэлектродное расстояние близко
К нулю, однако короткое замыкание еще не наступило. При корот-
ком замыкании на одном из анодов напряжение на электролизере
несколько снижается (до 4 В), а сила тока короткого замыкания,
проходящего через анод, возрастает примерно до 20 кА, т. е. ста-
новится в 13 раз выше нормальной. 1
При использовании графитовых анодов короткие замыкания
отдельных анодов приводят к большому увеличению силы тока
на этих анодах и к сильному перегреву токоподводящей системы
к аноду. Местные перегревы способствуют усилению процессов кор-
розии защитных покрытий деталей, уплотняющих места прохода
токоподводов через крышку, и самой крышки. Для предотвращения
вредных последствий коротких-замыканий предложено [105] уста-
навливать на токоподводах к анодам плавкие предохранители, пре-
рывающие прохождение тока при возрастании его величины выше
допустимого предела.
165
В электролизерах с металлическими анодами необходимость
в опускании анодов отпадает, так как геометрические размеры анодов
во время работы остаются неизменными. Однако требуется защищать
аноды от разрушения при возникновении коротких замыканий
между металлическими анодами и амальгамным катодом.
Эти замыкания возможны . вследствие неравномерности потока
ртути по катоду, образования амальгамных масел и пен и вслед-
ствие этого увеличения толщины амальгамного катода. Возникно-
вению коротких замыканий могут способствовать тепловые и механи-
ческие деформации днища и крышки электролизеров. Кроме того,
при применении металлических анодов следует учитывать разруше-
ние активного слоя анодов в результате восстановления амальгамой
натрия окисных соединений металлов платиновой группы и после-
дующего их растворения. В этом случае необходима малоинерцион-
ная быстро Действующая автоматическая система подъема анодов
в случае возрастания силы тока, превышающей установленные
пределы.
Автоматическое устройство для регулирования межэлектродного
расстояния может работать, если в качестве датчика используется
измеритель силы тока, проходящего через каждый анод [106]. Наи-
более эффективной для снижения напряжения на электролизере
и расхода электроэнергии на производство является автоматическая
система, предусматривающая регулирование межэлектродного рас-
стояния на каждом из анодов электролизера. При использовании
устройств, обегающих последовательно все аноды электролизера,
можно поддерживать желаемое межэлектродное расстояние на всех
анодах и минимальное напряжение на электролизере [107].
Однако в современных мощных электролизерах, у которых пло-
щадь электродов составляет до 35 м2, количество анодов очень велико,
поэтому на электролизере необходимо устанавливать десятки,
а иногда и сотни отдельных регуляторов положения анодов. Так,
на электролизере типа Уде на нагрузку 250—350 кА количество гра-
фитовых анодов составляет 216 шт. На заводе средней мощности
количество работающих электродов исчисляется десятками тысяч.
При применении металлических анодов их число на электролизере
Уде уменьшается в 4 раза, однако все же остается достаточно боль-
шим. Таким образом, осуществление автоматического регулирования
каждого из анодов связано с большими затратами на оборудование
и обслуживание многочисленных приборов и автоматических
устройств.
Намечается тенденция к объединению электродов в группы
с целью уменьшения числа точек контроля и регулирования, что
открывает практическую возможность реализации автоматического
управления межэлектродным расстоянием. Примером такого объеди-
нения является, например, последняя модель электролизера Уде,
в котором аноды закреплены в стальной защищенной крышке элек-
тролизера, последняя с помощью эластичных элементов соединяется
с его корпусом. Межэлектродное расстояние в таком электролизере
166
и
1 можно регулировать путем подъема или опускания всей крышки
Ц вместе с анодами.
При объединении анодов в группы задача регулирования облег-
J чается, так как пропорционально степени объединения анодов
|| в группы уменьшается число точек контроля и регулирования,
j Однако вследствие некоторой неравномерности установки й, в по-
Ж следующем, износа отдельных анодов фактически среднее расстоя-
Ц ние между электродами и напряжение на электролизере при группо-
ft: вом регулировании анодов несколько выше, чем при их индивидуаль-
ном регулировании. Несмотря на это, в последние годы групповое
К регулирование положения анодов широко применяется взамен инди-
В видуального.
В При установке металлических анодов необходимость в одускании
В анодов в течение работы электролизера отпадает. Применяются
В автоматические устройства, поднимающие аноды в случае коротких
В замыканий и опускающие их вновь до необходимого положения после
г устранения короткого замыкания.
% Проблема регулирования межэлектродного расстояния постоянно
а привлекает внимание инженеров и исследователей [103, 104].
Благодаря разработке удобных методов регулирования межэлек-
А.
#
тродного расстояния в процессе электролиза и установления опти-
мальных форм перфорации электродов для облегчения отвода хлора
из зоны прохождения тока, процесс электролиза был интенсифици-
рован без повышения реального напряжения на электролизере.
Увеличение плотности тока в электролизере и интенсификация раз-
ложения амальгамы обеспечили возможность создания более ком-
пактных электролизеров, рассчитанных на очень высокие нагрузки.
В практике хлорной промышленности в настоящее время исполь-
зуются все более и более крупные электролизеры. Известны электро-
лизеры с корпусом длиной 24 м (Сольве V-200) и днищем шириной
более 2 м (например, Де-Нора). При увеличении плотности тока
на графитовых анодах до 8—10 кА/м2 создание агрегата на нагрузку
500 кА не встречает больших технических трудностей, и мощность
электролизера должна определяться масштабом производства и эко-
номическими соображениями.
Для разложения амальгамы применяются как горизонтальные,
так и вертикальные разлагатели, хотя в последние годы горизон-
тальные разлагатели быстро уступают место вертикальным.
Горизонтальные разлагатели выполняются в виде герметичного
корыта, снабженного люками для загрузки и выгрузки насадки и рас-
положенного с уклоном до 20 мм/м рядом с электролизером или
под ним. При расположении разлагателя пбд электролизером сокра-
щается необходимая производственная площадь и облегчается обслу-
живание электролизера. Однако при этом ухудшается обслуживание
самого разлагателя.
Разлагатель, помимо своей основной функции, служит также для
возвращения ртути в верхнюю часть электролизера. По дну разла-
гателя течет тонким слоем амальгама, здесь же размещается насадка,
167
служащая для ускорения разложения амальгамы вследствие созда-
ния короткозамкнутых элементов амальгама натрия — раствор
NaOH — материал насадки. Насадка плавает в слое амальгамы,
часто для улучшения ее контакта с амальгамой применяются грузы
или упоры, прижимающие насадку к днищу разлагателя для лучшего
ее погружения в слой амальгамы.
Скорость разложения амальгамы пропорциональна периметру
контакта амальгама — раствор — материал насадки. Поэтому пе-
риметр контакта этих фаз должен быть как можно больше.
В качестве насадки в разлагателях такого типа применяется
преимущественно графит в виде плит. На нижней стороне графито-
вых плит прорезаются канавки шириной 4—5 мм и шагом 8—12 мм
для развития поверхности графита, увеличения периметра контакта
фаз и облегчения отвода водорода из зоны разложения амальгамы.
Возможно применение и кускового графита, однако при этом уве-
личивается количество ртути в разлагателе и затрудняется замена
насадки.
В вертикальных разлагателях в качестве насадки обычно при-
меняют кусковой графит, зажатый между решетками, что улучшает
электрический контакт между кусками графита, способствует интен-
сификации разложения амальгамы, а также предотвращает перети-
рание кусков насадки при работе разлагателя.
В последнее время предложены и другие материалы для изгото-
вления насадки, например карбиды металлов (вольфрама) или
графит, покрытый слоем карбида [108], а также комбинированная
железографитовая насадка [109]. Для интенсификации разложе-
ния [НО] предложено вводить в разлагатель растворы молибдата
и, кромё того, обматывать графитовые элементы насадки стальной
проволокой [111]. Интенсификация работы скрубберного разлага-
теля может быть достигнута наложением пульсации с помощью пуль-
сокамеры [111]. Наиболее распространены вертикальные разлага-
тели скрубберного типа, в которых сплошной фазой является рас-
твор щелочи, амальгама поступает на орошение насадки сверху про-
тивотоком водному раствору, а ртуть отводится снизу. При приме-
нении скрубберных разлагателей возрастает требуемая высота подъ-
ема ртути и повышаются требования к ртутному насосу. Необхо-
димо, чтобы скорость движения раствора была достаточной для
предотвращения образования местных зон высокой концентрации
щелочи. Образованию таких зон могут способствовать конвекцион-
ные потоки, возникающие вследствие разности плотностей пода-
ваемой на разложение воды и отводимой из разлагателя ще-
лочи.
В разлагателях с погружной насадкой вода для разложения
подается через распределитель в нижнюю часть разлагателя. Сюда же
поступает свежая амальгама. Выделяющийся в процессе разложения
амальгамы водород может быть использован для организации подъ-
ема ртути по принципу Мамут-насоса. Для обеспечения работы раз-
лагателя необходимо равномерное распределение воды по сечению
168
II
II
i;
A . j ' »
разлагателя, что затруднено при эксплуатации разлагателей боль-
шой мощности.
Длительное время применялись преимущественно горизонталь-
ные разлагатеди вследствие их простоты, надежности и устойчи-
вости в работе. После создания компактной конструкции вертикаль-
ного разлагателя и устранения истирания графитовой насадки [112]
все большее предпочтение отдается вертикальным разлагателям,
главным образом скрубберного типа. Преимуществами этих
разлагателей являются возможность полного разложения амаль-
гамы, их компактность, что резко сокращает затраты производствен-
ной площади, хороший доступ для обслуживания электролизера
и несколько уменьшенная закладка ртути по сравнению с горизон-
тальным разлагателем.
При применении вертикального скрубберного разлагателя сле-
дует принимать специальные меры ,для предотвращения истирания
кусков графита насадки во время работы. Это достигается с помощью
устройств для зажима слоя насадки, предотвращающих движение
частичек насадки относительно друг друга. Однако полностью исти-
рание насадки обычно предотвратить не удается, и растворы каусти-
ческой соды из скрубберных разлагателей загрязнены графитовой
пылью. Такие растворы обычно фильтруют через специальные
микропористые фильтры [ИЗ].
Разлагатеди с погружной насадкой, хотя и открывают принци-
пиальную возможность осуществления циркуляции ртути без спе-
циального насоса за счет водородного подъема, однако не получили
широкого распространения. Это объясняется несколько большей
закладкой ртути по сравнению со скрубберными, они неустойчивы
в работе вследствие трудности распределения сравнительно малых
количеств воды, подаваемой на разложение по сечению разлагателя.
Последний недостаток можно устранить путем циркуляции щелочи
через разлагатель.
Обычно вода и амальгама в разлагателе проходят противотоком,
образующийся раствор содержит 50—55% NaOH. Для получения
более высококонцентрированной щелочи применяют разлагатеди
с параллельным током воды и амальгамы [114]. При применении
противотока в горизонтальном разлагателе концентрация щелочи
возрастает примерно от 20% в месте ввода воды до 50—55% в месте
вывода раствора каустической соды. В вертикальных разлагателях,
вследствие интенсивного перемешивания пузырьками выделяюще-
гося водорода, концентрация щелочи в растворе, заполняющем раз-
лагатель, примерно одинакова во всем объеме и близка к концентра-
ции отбираемого раствора. С этой точки зрения условия для разло-
жения амальгамы в вертикальном разлагателе хуже, чем в горизон-
тальном. Однако турбулентный режим потоков амальгамы и раствора
и высокие температуры позволяют интенсифицировать процесс раз-
ложения амальгамы в скрубберных разлагателях.
В процессе разложения амальгамы выделяется большое количе-
ство тепла и повышается температура. Ограничением для повышения
II
II
169
температуры служит расход тепла на испарение воды из рас-
творов щелочи, заполняющей разлагатель. С водородом, уходящим
из разлагателя, уносятся большое количество паров воды и заметные
количества паров ртути. В маломощных электролизерах эти потери
не столь велики, но при укрупнении электролизеров и снижении
удельного веса теплопотерь через наружные стенки в общем тепло-
вом балансе электролизера это приводило к технологическим неудоб-
ствам. Затруднялось регулирование подачи воды в разлагатель
и возникала необходимость компенсации ртути, уносимой с водоро-
дом в виде паров.
Мощные конструкции электролизеров стали снабжаться индиви-
дуальными холодильниками для водорода, выходящего из разла-
гателей. Влага и ртуть, сконденсировавшиеся в холодильниках,
возвращались обратно в разлагатель. В таких холодильниках осу-
ществляется первичное охлаждение и грубая начальная очистка
водорода от ртути.
При применении вертикального разлагателя возникает опасность
уноса мелкораздробленной ртути с потоком щелочи. Поэтому следует
предусмотреть устройство гасителей или сепараторов для отделения
щелочи от капелек ртути [115].
Общие закономерности, определяющие зависимость закладки
ртути в электролизере от плотности тока, уклона дна электроли-
зера, соотношения между длиной и шириной электролизера, а также
зависимость содержания водорода в хлоре от закладки ртути в элек-
тролизере являются предметом исследований в нашей стране [116]
и зарубежных странах [117].
КОНСТРУКЦИИ ЭЛЕКТРОЛИЗЕРОВ
За период более 80 лет с момента создания метода электролиза
с ртутным катодом было разработано и использовалось в промышлен-
ности много различных конструкций электролизеров [1, 2, 4—6,
18, 25, 26, 118—120]. В настоящее время в промышленности исполь-
зуется много типов горизонтальных электролизеров, близких по
конструкции и описанных в периодической литературе и рекламных
материалах фирм [40, 121—128].
Ниже будет приведено описание современных советских кон-
струкций мощных электролизеров, используемых в хлорной про-
мышленности, и также наиболее распространенных зарубежных
конструкций.
Электролизеры о вертикальным расположением катода
Из электролизеров с вертикальным расположением катода можно
назвать только дисковый электролизер типа Хонзберга [129], при-
менявшийся некоторое время в промышленности. Этими электроли-
зерами была оборудована одна установка в 1940 г., она проработала
до 1969 г., и электролизеры этого типа были заменены электролизе-
170
рами с горизонтальным катодом. Электролизер типа Хонзберга
состоял из набора металлических дисков, насаженных на вал и ча-
стично погруженных в ртутную ванну. При вращении вала диски
смачивались ртутью и образовывали ртутный катод. Графитовые
аноды были размещены в промежутках между дисками. О приме-
нении в промышленности других конструкций электролизеров
с вертикальным ртутным катодом в литературе сообщений не было.
Электролизеры типа Р-101 (118, 125)
Электролизер Р-101 наиболее широко применяется в отечествен-
ной хлорной промышленности. Первоначально он был разработан
на нагрузку 100 кА при плотности тока 5,35 кА/м2. Однако затем
он был интенсифицирован, что обеспечивало возможность увеличе-
ния нагрузки за счет повышения плотности тока примерно до 8 кАЛЙЗ *
Электролизер Р-101 — горизонтальный, рамного типа с голым
незащищенным катодом, размером 13,18x1,42 м и площадью 18,7 м2,
имеет верхний входной и нижний выходной карманы и горизонталь-
ный разлагатель амальгамы, расположенный рядом с электролизе-
ром. Циркуляция ртути осуществляется с помощью конусного
ртутного насоса, размещенного в торце разлагателя. Электролизер
устанавливается с уклоном 10 мм/м. Крышка электролизера сталь-
ная гуммированная. Подвод тока к анодам осуществляется частично
через крышку, частично через специальные медные шины (чтобы
чрезмерно не утяжелять крышку, особенно при нагрузке 125—
150 кА). При работе крышка электролизера имеет анодный потен-
циал, что может усилить ее коррозионное разрушение при образова-
нии дефектов в защитной гуммировке.
Графитовые аноды размером 340x175x90 мм расположены
в электролизере в четыре ряда. Подвод тока к плитам осуществляется
через графитовые стержни и металлические шпильки, ввинчиваемые
по оси стержней. Уплотнение токоподводящих графитовых стержней
в крышке осуществляется с помощью сальников. Аноды снабжены
индивидуальными приспособлениями для регулирования межэлек-
тродного расстояния по мере износа анодов в процессе работы.
Схематически устройство электролизера Р-101 показано на рис. 3-20.
Верхний входной карман электролизера отделен от электролизера
жидкостным затвором; прдача^рассола в электролизер производится
через входной карман для ртути. Благодаря этому входной карман
хорошо промывается рассолом и всегда чист. Во входном кармане
предусмотрено устройство для отделения ртути от воды, захватывае-
мой ртутью из насоса. Этим предотвращается попадание щелочной
воды вместе с рассолом в электролизер, что могло бы привести к сни-
жению выхода по току и усиленному износу графитовых анодов.
Выходной карман расположен на 20—25 мм ниже уровня днища
электролизера, поэтому амальгама свободно стекает в него из элек-
тролизера. Для лучшего отделения амальгамы от увлекаемого ею
электролита помимо сифона для перетока ртути в разлагатель
171
в выходном кармане электролизера имеется ртутный затвор, в котором
амальгама может дополнительно промываться водой. Устройство
дополнительного ртутного затвора позволяет получать в раз л агате ле
каустическую соду высокой степени чистоты с минимальным содер-
жанием хлор-иона. Хлор и обедненный анолит отводятся вместе
через штуцер, расположенный в днище электролизера. Хлор отде-
ляется от анолита во внешнем сепараторе. Коробки верхнего и ниж-
него карманов закрыты крышками, полости карманов присоединены
Рис. 3-20. Поперечный разрез электролизера Р-101:
1 — токоподвод к аноду; 2 — сальник; з— крышка; 4 — рана;
5 — аноды; в — шунт; 7 — ртутный насос,
г
к линиям для отсоса газов с целью улучшения санитарных условий
работы в цехе.
Разлагатель амальгамы электролизера Р-101 расположен рядом
с электролизером. Корпус разлагателя— штампованный, выпол-
няется из одного листа с продольным сварным швом, расположен-
ным в верхней газовой зоне разлагателя. Разлагатель снабжен лю-
ками для загрузки и выгрузки насадки. К нижней части разлагателя
присоединена коробка — приемная камера со свободным стоком
в нее ртути из разлагателя, что способствует лучшему удалению
загрязнений и амальгамного масла из зоны, заполненной насадкой.
Из приемной камеры ртуть через ртутный затвор поступает к конус-
ному ртутному насосу и перекачивается в верхний входной карман
электролизера. Вода, подаваемая на промывку ртутного насоса,
переливается в приемную камеру разлагателя и далее поступает
на разложение амальгамы. Для уменьшения загрузки ртути разлага-
тель располагают под уклоном 18—20 мм/м.
Тепло, выделяющееся при разложении амальгамы, расходуется
на нагрев реагирующих потоков и испарение воды' Только неболь-
шая его часть теряется через стенки разлагателя в окружающую
среду. Тепловое равновесие устанавливается за счет испарения боль-
172
II
и
его или меньшего количества воды и частично ртути. Распределе-
ние температуры в разлагателе по его длине определяется количе-
ством амальгамы, разлагающейся на единицу длины разлагателя
и связанным с этим распределением по его длине концентрации ще-
лочи в растворе. Этим определяется парциальное давление паров
воды в водороде и устанавливающееся распределение температуры
по длине разлагателя.
Рис. 3-21. Поперечный разрез электролизера Р-101 с рези-
новой крышкой: х
1 — рамка подвески анодов; 2 — металлический токоподвод; з —
графитовый стержень; 4 — резиновая крышка; 5 — гибкий токо-
подвод; в— рама электролизера; 7 — аноды; 8 — шунт; 9 — пневмо-
цилиндр щунта; — катодная птина. ,
Температура водорода, выходящего из разлагателя электроли-
зера Р-101, зависит от нагрузки на электролизере, так при нагрузке
около 100—125 кА она близка к 100 °C. При этой температуре с водо-
родом уносится значительное количество паров воды и ртути. Для
стабилизации режима работы разлагателя и уменьшения уноса ртути
с водородом над разлагателями электролизеров Р-101 установлены
поверхностные кожухотрубчатые обратные холодильники для водо-
рода. Конденсирующиеся в холодильниках вода и ртуть возвра-
щаются самотеком в разлагатель.
Разработан вариант электролизера Р-101 с эластичными резино-
выми крышками (рис. 3-21). По длине электролизера расположены
четыре резиновые крышки, присоединяемые к раме электролизера
с помощью накладок. В таком электролизере принято групповое
регулирование межэлектродного расстояния. Каждая крышка имеет
две .алюминиевые рамки для подвески анодов и подвода к ним тока!
Рамки установлены на четырех опорах на раме электролизера таким
образом, чтобы была возможность одновременного группового
173
регулирования всех анодов, закрепляемых на рамке. Помимо этого,
положение каждого анода можно регулировать индивидуально.
Такая система регулирования позволяет работать с минимальным
напряжением на электролизере* Эластичная крышка электролизера
позволяет устанавливать графитовые плиты размером 680 X175 X
Х9О мм с двумя токоподводящими стержнями на плиту*
Рассматривались варианты электролизера Р-101 с нижним распо-
ложением горизонтального разлагателя, а также с вертикальным
скрубберным разлагателем, однако эти варианты не были исполь-
зованы, так как для вновь проектируемых заводов были разработаны
। и
усовершенствованные конструкции горизонтальных электролизеров
на нагрузку 200 и 300 кА с вертикальным скрубберным разлагателем.
В электролизерах Р-101 графитовые аноды имеют специальную
перфорацию, облегчающую выделение пузырьков хлора из зоны
прохождения тока. Поэтому напряжение на этих электролизерах
при работе в интенсифицированном режиме (при плотности тока
в 8—10 А/м2) не превышает 4,5—4,7 В. При нормальном обслужива-
нии и регулировании межэлектродного расстояния электролизер (
•Р-101 на нагрузку 100 кА может работать при напряжении 4,3 В
с выходом по току 96%, что соответствует расходу электроэнергии
3000 кВт-ч/т 100%-ного NaOH или 3385 кВт-ч/т хлора.
При работе с нагрузкой 100 кА и при концентрации щелочного
металла в амальгаме 0,4—0,5% в электролизер загружают 1800 кг
ртути; при концентрации щелочного металла к амальгаме 0,3%
требуемая загрузка ртути возрастает до 2100 кг. При повышении
нагрузки до 125 и 150 кА закладка ртути также несколько возрастает.
Содержание водорода в хлоре зависит от качества рассола и попада-
ния амальгамных ядов в анолит. При нормальном составе рассола
в хлоре содержится 0,3—0,6% Н2.
Для оборудования сравнительно небольших цехов электролиза
применяется аналогичный по конструкции, но меньший по мощности
электролизер Р-30. Это горизонтальный рамный электролизер с го-
лым, незащищенным днищем, стальной крышкой и сальниковым
уплотнением токоподводящих штырей в крышке. Электролизер
имеет горизонтальный разлагатель, располагаемый обычно рядом
с электролизером, и конусный ртутный насос. При нагрузке 30 кА
и нормальном обслуживании электролизер работает с плотностью
тока 5,3 кА/м2 при напряжении 4,3 В.
Электролизер допускает повышение нагрузки до 45 кА. При при-
менении анодов с интенсифицированной перфорацией плит напряже-
ние на электролизере при этой нагрузке может поддерживаться
на уровне 4,4—4,5 В.
Электролизер Р-20 (118, 131)
Электролизер Р-20 также применяется в отечественной хлорной
промышленности, но в значительно меньшем масштабе цо сравнению
с электролизерами Р-101. Электролизер горизонтальный, рамного
174
ll'
типа, с голым незащищенным стальным дном и стальной гуммиро-
ванной крышкой. Уклон катода электролизера 10 мм/м. Общая пло-
щадь катода около 19,2 м2 при длине 17 и ширине 1,125 м. Ацоды
расположены в три ряда по ширине крышки. Анодные стержни уплот-
няются с помощью сальников. Межэлектродное расстояние регули-
руется индивидуально стационарными устройствами.
Выходной карман электролизера имеет сложное устройство для
обеспечения надежного отделения амальгамы, поступающей на раз-
ложение, от увлекаемых ею примесей электролита.
Рис. 3-22. Принципиальная схема устройства электро-
лизера Р-20:
1 — входной карман электролизера; 2 — выходной карман; 3 —
скрубберный разлагатель; 4 — центробежный ртутный насос и бу-
ферная емкость.
Электролизер снабжен вертикальным скрубберным разлагателем
амальгамы с кусковой* графитовой насадкой, зажатой между двумя
решетками. Предусмотрен постоянный поджим насадки, чтобы избе-
жать усиленного истирания насадки в процессе работы. Разлагатель
диаметром около 0,5 м имеет общую высоту 4,2 м и высоту рабочего
слоя насадки 2,5 м. Разлагатель обеспечивает полное разложение
амальгамы, поэтому при работе практически не образуется легкое
амальгамное масло, что облегчает обслуживание электролизера.
После разлагателя ртуть собирается в буферной емкости и с по-
мощью погружного центробежного насоса перекачивается в верхний
карман электролизера. Высота подъема ртути составляет около 5 м.
Принципиальная схема устройства электролизера Р-20 приведена
на рис. 3-22. Электролизер первоначально был рассчитан на на-
грузку 100 кА, при которой плотность тока должна составлять около
5,0 кА/м2. При правильной эксплуатации электролизер может рабо-
тать со средним напряжением 4,3—4,4 В. Загрузка ртути составляет
2,6 т. При этом концентрация щелочного металла в амальгаме не
превышает 0,4 вес. %.
Впоследствии, без существенных изменений конструкции элек-
тролизера и разлагателя, работа их была интенсифицирована и на-
грузка на электролизере увеличилась до 150 кА. Электролизер Р-20
был также испытан при нагрузке 200 кА. Показатели работы
175
электролизера Р-20 при различной нагрузке, полученные при испы-
тании в промышленных условиях, приведены ниже:
100 кА 150 кА
Напряжение на электролизере, В .... 4,3—4,4 4,8
Расход электроэнергии . постоянного тока,
кВт*ч/т 100%-ной щелочи ...................... 3000 3350
Загрузка ртути в электролизер, т .... 2,6 2,85
Удельная загрузка ртути, кг/кА............ 26 19
Мощные конструкции отечественных электролизеров
Для строительства новых хлорных цехов в нашей стране разра-
батываются более мощные конструкции электролизеров с ртутным
катодом как с графитовыми, так и с малоизнашивающимися анодами.
Электролизёры рассчитаны на нагрузки 200—300 и 500 кА. Это
электролизеры рамного типа с голым незащищенным стальным дном
с уклоном около 10 мм/м, со стальной гуммированной крышкой и груп-
повым регулированием положения анодов. '
В электролизере с графитовыми анодами на нагрузку 200—
300 кА принята плотность тока около 8 кА/м2, в электролизерах
на нагрузку 500 кА — около 10 кА/м2. За счет специальной формы
перфораций анодов, напряжение и соответственно расход электро-
энергии невелики. Применение высокой плотности тока и значитель-
ных уклонов катода электролизера позволяют уменьшить загрузку
ртути в электролизере. В электролизерах с малоизнашивающимися
анодами принимается более высокая плотность тока.
Для разложения амальгамы применяются вертикальные скруб-
берные разлагателя с графитовой кусковой насадкой, расположенной
между двумя решетками. Перекачивание ртути из разлагателя
в верхнюю часть электролизера осуществляется центробежным
ртутным насосом.
Ниже указаны основные конструктивные и технологические пока-
затели новых мощных отечественных электролизеров с графитовыми
анодами: ?
Плотность тока, кА/м2 . . . ч . . .
Номинальное напряжение, В ....
Выход по току, % . ..............
Расход электроэнергии постоянного тока
кВт*ч/т 100%-ного NaOH . . .
Площадь катода, м2 ........... .
200 кА 300 кА 500 кА
ft
8 8 ' 10
4,5 4,5 4,7
96 96 96
3140 3140 3220
25 37,5 50
Электролизеры фирмы „Кребоа"
На рис. 3-23 и 3-24 показан электролизер фирмы «Кребс» на на-
грузку 100 кА, установленный на одном из отечественных заводов.
Корпус электролизера корытного типа, штампованный из сравни-
тельно тонкой (7 мм) стали, целиком гуммирован изнутри. Подвод
тока к ртутному катоду осуществляется с помощью 107 тарелочных
176 -
стальных контактов, расположенных в три ряда по длине электроли-
зера. Контакты утоплены на 2—3 мм ниже уровня гуммировки
днища, корпус электролизера установлен с» уклоном 6—7 мм/м.
Корпус электролизера имеет малую жесткость, поэтому равномер-
ность уклона днища и горизонтальность его установки в поперечном
направлении достигается с помощью большого числа регулируемых
опор.
Рид. 3-23. План и вид сбоку электролизера Кребс:
1 — ртутный насос; 2 — входной карман; з — анодный стержень; 4 — анодные
шины; 5 — выходной карман; в — переток амальгамы; 7 — приемная коробка
разлагателя; 8 — разлагатель; 9 — выгодной карман разлагателя; 10 — регули-
ровочные подвески; 11 — распределительная анодная шина.
Крышка электролизера стальная (толщина 6 мм) гуммированная,
для подвода тока к анодам не используется. Графитовые аноды раз-
мером 700x230x80 мм закрепляются на графитовых стержнях,
уплотняемых в крышке резиновыми кольцами — пробками. Регу-
лирование межэлектродного расстояния — индивидуальное с по-
мощью переносного устройства. Токоподводящие стержни пропи-
таны рафинированным горным воском, что обеспечивает хорощее
скольжение стержней в резиновой пробке при регулировании поло-
жения анодов.
Отвод хлора и обедненного анолита осуществляется через днище
электролизера по двум штуцерам. При этом входной карман закрыт,
по всей ширине выходного кармана имеется ртутный затвор, однако
он недостаточен для полного отделения амальгамы от увлекаемого
сю анолита.
12 Заказ 843
177
Рис. 3-24. Поперечный разрез электроли-
зера Кребс:
— токоподвод к аноду; 2 — анодная шина;, з —
крышка; 4 — шунт для выключения электроли-
зера; 5 — токоподвод к катоду; 6 — разлагатель;
7 — графитовая насадка; 8 — уплотняющая ре-
зиновая пробка.
Горизонтальный разлагатель расположен под электролизером
в нижнем этаже с уклоном 15 мм/м. Насадка из перфорированных
графитовых плит прижимается чугунными грузами. Для подъема
ртути служит центробежный
ртутный насос, помещенный
в цилиндрическом углубле-
нии в нижней части разла-
гателя.
Длина катода 14,25 м, ши-
рина 1,426 м. Загрузка ртути
в электролизер 2,9 т. При
нагрузке 100 кА электроли-
зер работает при плотности
тока 4,95 кА/м2 и при пра-
вильном обслуживании мо-
жет иметь среднее напряже-
ние 4,4 В, что при выходе по
току 96% соответствует рас-
ходу электроэнергии посто-
янного тока 3070 кВт*ч
на 1 т 100%-ного едкого
натра.
Применение гуммирован-
ного днища электролизера
усложняет конструкцию и
увеличивает загрузку ртути,
однако при нормальной экс-
хлор, содержащий 0,2—0,4%
выпускают в настоящее время
высоких плотностях тока — до
плуатации позволяет получать
водорода.
Электролизеры фирмы «Кребс»
[40, 120] для работы при более
8—10 кА/м2 и для более высоких нагрузок — 150—200 кА.
Выпускают также электролизеры, на токовую нагрузку 200 кА
с незащищенным стальным днищем для работы с графитовыми ано-
дами при плотности тока около 8 кА/м2.
Электролизеры фирмы „Сольве“ (124, 132)
Старые конструкции электролизеров фирмы «Сольве» были ши-
роко распространены и подробно описаны. При разработке новых
конструкций были предложены интересные и оригинальные техни-
* ческие решения.
Электролизер Сольве горизонтального типа с голым стальным
дном и горизонтальным разлагателем, расположенным под электро-
лизером, как это показано на рис. 3-25.
Конструкция электролизера рассчитана на замену графитовых
анодов и насадки разлагателя во время работы электролизера без
его выключения, поэтому исключена необходимость устройства
дорогостоящих шунтирующих приспособлений.
178
Электролизер закрыт крышками небольшого размера, которые
уплотняются с помощью надувного резинового шланга. Крышки
могут передвигаться вдоль электролизера. Крышки с отработанными
электродами могут быть удалены и заменены новыми без отключения
электролизера.
Рис. 3-25. Поперечный разрез элек-
тролизера Сольве V-200:
7 — площадка для обслуживания;
2 — подвод тока к анодам; 3 — аноды;
4 — катодная шина; 5 — разлагатель;
€ — графитовая насадка разлагателя.
Рис. 3-26. Схема подвода тока к анодам
в электролизере Сольве V-200:
7 — площадка для обслуживания; 2 —
устройство для регулирования анодов; 3 —
анодная шина; 4 —опорный ролик крышки
электролизера; 5 — надувная резиновая тру-
ба; 6 — резиновая прокладка; - 7 — дно элек-
тролизера; 8 — анод; 9 — крышка электроли-
зера; 10 — анодный токоподвод; 77 — медная
шайба; 12 — графитовая втулка.
Направляющие движения крышки установлены с уклоном с тем,
чтобы последний компенсировал износ анодов в процессе работы
электролизера. При передвижении элементов крышки вдоль электро-
лизера одновременно регулируется межэлектродное расстояние.
В новейших моделях электролизера предусмотрена также возмож-
ность индивидуального регулирования положения каждого анода
при сохранении указанного ранее принципа.
Пакеты графитовой насадки разлагателя также могут передви-
гаться по длине разлагателя, замена насадки возможна во время
работы электролизера.
Электролизеры могут располагаться впритык друг к другу без
проходов в здании из нескольких этажей.
Подвод тока к графитовому стержню и уплотнение осуществляется
с помощью графитовой муфты (рис. 3-26).
Электролизер V-200 на нагрузку 160—190 кА имеет катод дли-
ной 23,26 м и шириной 1,29 м, загрузка ртути 3,3 т. При нагрузке
190 кА плотность тока равна 6,33 кА/м2 и среднее напряжение 4,56 В.
Электролизеры фирмы «Де Нера»
Электролизеры фирмы «Де Нора» горизонтальные с эластичной
резиновой крышкой или крышкой из полимерных материалов и вер-
тикальным скрубберным разлагателем. Длительное время стенки
12*
179
и днища электролизеров выпускали 'с> футеровкой изнутри сиени-
том. Подвод тока к ртутному катоду осуществлялся через стальные
полосы, уложенные вдоль электролизера несколько ниже отметки
футеровки днища. Схема устройства такого электролизера дана
на ^рис. 3-27.
Рис. 3-27. Поперечный разрез электролизера Де Нора:
J — рама подвески анодов; 2 — гибкая шина; 3 — резиновая крышка; 4 — ре-
гулировочный винт; 3 — анод; 6 — корпус электролизера; 7 — шунт.
и:
и:
В последнее время эти электролизеры выпускают с незащищен-
ным стальным днищем [40, 53, 54, 124, 1251. Применение эластичной
резиновой крышки облегчает групповое регулирование анодов.
За последние 15—20 лет значительно выросла мощность электро-
лизеров этой конструкции. _С 1956 по 1970 г. их геометрические раз-
меры мало изменились, площадь катода для наиболее мощного
электролизера возросла с 30,5 до 34,2 м2. Плотность тока повышена
от 3,93 до 8,77 кА/м2, что соответствует увеличению максимальной
нагрузки от 120 до 300 кА. При применении металлических анодов
плотность тока увеличивается до 13,2 кА/м2, а максимальная на-
грузка — до 450 кА [121].
При плотности тока 8 кА/м2 удельная загрузка ртути в электро-
лизер составляет 10,5 кг/кА. За счет использования новых метал-
лических анодов предполагается в дальнейшем повысить плотность
тока и уменьшить необходимую загрузку ртути [53—54].
Разработана также конструкция электролизера Де Нора с вер-
тикальным ртутным катодом на нагрузку 40 кА с катодом в виде
проволочной сетки, орошаемой слабой амальгамой, и тканевой диа-
фрагмой. Однако в литературе не было сообщений, что эта кон-
струкция получила применение в промышленности.
Электролизеры фирмы „Олин-Матисон“
v Электролизеры этого типа выпускают на различные нагрузки.
На рис. 3-28 приведен поперечный разрез электролизера Олин*
Матисон Е-11 на нагрузку 125—135 кА. Это — рамный электроли-
180
зер с незащищенным стальным дном, скрубберным разлагателем
и групповым регулированием положения анодов. Крышка электро-
лизера стальная гуммированная; уплотнение токоподводящих анод-
ных стержней в крышке осуществляется с помощью эластичных
колпачков [40, 122, 124, 133]. х
rintl
Гг|гТ
НН
4
£
2
Г----------—
9 й 8
Ssnssa
Рис. 3-28. Поперечный разрез электролизера фирмы
«Олин-Матисон» Е-11:
1 — катодная шина; 2 — стальное дно; з — гуммировка;
4 устройство для регулирования положения анодов; 5 —
уплотняющий колпачок; 6 — опора и токоподвод к ано-
дам; 7 — ребро жесткости крышки; 8 — гибкий токоподвод;
9 — анод.
Последняя модель электролизера Олин-Матисон Е-12 с катодом
площадью 28,8 м2 на нагрузку 300 кА при плотности тока около
10 кА/м2 может работать при напряжении 4,66 В [127]. Срок службы
анодов толщиной 152 мм составляет 17«—21 мес. Сообщается о воз-
можности выпуска электролизеров с катодом площадью до 42 м2
нагрузкой при использовании графитовых анодов до 420 кА,
металлических анодов — до 630 кА.
a
Электролизеры фирмы „Куреха“
Особенностью этих электролизеров является их П-образная
форма [134, 135]. Такое решение было ранее использовано фирмой
«ИГ Фарбениндустри», но не получило широкого применения в про-
мышленности [136]. П-образная форма позволяет увеличить уклон
катода до 18 мм и уменьшить необходимую загрузку ртути за счет
исключения обратного трубопровода от разлагателя к электроли-
зеру. Электролизер снабжен скрубберным разлагателем и ковшовым
подъемником ртути (схема устройства приведена на рис. 3-29).
Переточный карман для амальгамы также может снабжаться ано-
дами и работать как часть электролизера.
Днище электролизёра стальное, незащищенное, крышка сталь-
ная гуммированная с эластичным уплотнением анодных стержней.
Электролизер на нагрузку 150 кА имеет катод шириной 1,2 м и об-
сей длиной в двух половинках 16,5 м. При этом плотность тока
II
181
доставляет 7,57 кА/м2, а напряжение на электролизере — 4,3 В. Для
увеличения’’скорости движения рассола с целью облегчения удаления
пузырьков хлора с анода применяется рециркуляция анолита в ко-
личестве до 50% общего потока. Применяются графитовые плиты
размером?Д180х287х 135 мм. Подвод тока к анодной плите осуще-
ствляется
При форсированном
1двумя графитовыми
стержнями.
Рис. 3-29. Схема устройства электролизера фирмы «Куреха»:
1 — рама электролизера; 2 — крышка; з — средняя перегородка: 4 —
вводной карман; 5 — выходной карман; 6 — разлагатель: 7 — ртутный на-
сос; 8 — отвод хлора; 9— выключатель; 10 — шины; 11 — подача воды;
12 — устройство для регулирования анодов; 13 — холодильник водорода.
режиме нагрузка на электролизер может быть увеличена до 220 кА.
При этом плотность тока возрастет до 11,6 кА/м2, а напряжение
до 4,9 В [125, 137, 138].
В последнее, время фирма предложила горизонтальные электро-
лизеры обычной конструкции со скрубберными разлагателями.
Электролизеры такого типа с катодом площадью 34,2 м2 могут ра-
ботать при нагрузке 300 кА и плотности тока 8,77 к А/м2.
Электролизеры фирмы „Уде
Фирма «Уде» разработала нормальный ряд конструкций электро-
лизеров с ртутным катодом, у которых поверхность катода составляет
соответственно 6,4, 12,8, 17,92, 26,9 и 34,56 м2. Электролизеры имеют
стальное голое днище, вертикальный разлагатель амальгамы и
182
выпускаются для работы как с графитовыми, так и с металлическими
анодами. Для облегчения переоборудования электролизеров с од-
ного типа анодов на другой для металлических анодов приняты стан-
дартные размеры 787x787 мм [139], а для графитовых анодов —
395x395 мм. Основные технические характеристики электролизеров
типа Уде приведены ниже:
Нагрузка, кА.................. . .
Плотность тока на катоде, кА/м3 . . .
Напряжение, В
без учета ошиновки .......
с учетом ошиновки . . . . . . . .
Расход электроэнергии
постоянного тока, кВт ч/т С12 . .
Удельная* загрузка ртути, кг/кА . . .
Выход по току, % ..........
С графитовыми
анодами
48—345,6
7,5—10
4,03-4,30
4,11—4,50
3210—3420
11,5-10,3
95
С металли-
ческими
анодами
64—518
10—15
3,98—4,39
4,06—4,62
3135—3460
10,3—9,2
96
Схема устройства электролизера приведена на рис. 3-30.
Электролизеры снабжены жесткой крышкой, к которой подве-
шиваются электроды. Крышка соединена со стенками корпуса элек-
тролизера и входным и выходным карманами с помощью эластичной
Рис. 3-30. Схема устройства последней модели электролизера
фирмы «Уде»:
1 — днище электролизера; 2 — анод; 3 — эластичная соединительная пленка;
4 — крышка; 5 — групповая рама для крепления анодов; 6 — соединитель-
ные шины; 7 — шунтирующий выключатель.
полимерной пленки. Межэлектродное расстояние регулируется опу-
сканием или подниманием крышки с группой анодов синхронным
электродвигателем. Управление устройством для регулирования
положения крышки и анодов может быть осуществлено со щита
по показаниям контрольно-измерительных приборов.
В ранних моделях электролизеров типа Уде применялось сталь-
ное днище, которое было составлено по длине из отдельных элемен-
тов, соединенных между собой сваркой или другим способом. При
этом была необходима термообработка днищ для снятия напряжения
183
в металле, возникающего при проведении сварочных работ. В по-
следних моделях применяется катодное днище, выполненное из одного
листа стали. Для улучшения распределения тока по всей поверх-
ности катода днище приходится делать примерно в два раза толще,
чем в предыдущих конструкциях. При увеличении расхода металла
снижаются затраты труда на изготовление, монтаж и установку
электролизеров.
raw
Электролизеры типа СУ, СА и СДМ
Помимо описанных выше применяется много других тиной гори-
збнтальных электролизеров с ртутным катодом, в которых в раз-
личных комбинациях повторяются перечисленные выше конструктив-
ные решения и самого электролизера, и разлагателя амальгамы.
В электролизерах СУ-100 (ЧССР) на нагрузку 100 кА приме-
няется голое стальное днище, эластичная резиновая крышка и гори-
зонтальный разлагатель амальгамы, располагаемый рядом с электро-
лизером. Разработан также электролизер типа СУ и на более высо-
кую нагрузку с вертикальным разлагателем амальгамы.
В электролизерах СА и СДМ (СССР и ГДР) с голым металличе-
ским днищем применяются как горизонтальные, так и вертикаль-
ные разлагатели амальгамы [141], основные показатели этих элек-
тролизеров приведены в табл. 3-10.
Таблица 3-10- Техническая характеристика электролизеров СА и СДМ
Электролизер
Показатели
СА-100
САТ-100 СДМТ-ЮО СДМТ-200
Нагрузка, кА . . . ............ . . .
Напряжение, В . Л. .................
Плотность тока катодная, кА/м2 . . .
Удельная закладка ртути, кг/кА . . . .
Разлагатель .........................
__ ( /
Площадь пола на 1 кА нагрузки, м2
100
4,4
6,675
19,5
Горизон-
тальный
0,208
100
4,4
6,675
19,0
100
4,4—4,5
7,50
16,0
200
4,4-4,5
7,50
15,0
Вертикальный
0,216 0,198 0,177
и
В электролизерах применяется групповое регулирование гра-
фитовых анодов, что снижает затраты труда на обслуживание элек-
тролизера. Подвод тока к анодам осуществляется медными шинами.
Крышка не используется для подвода тока к анодам.
г
Электролизер фирмы «Асахи» (124, 140)
, В электролизере этой конструкции вместо традиционного гори-
зонтального катода применяется катод в виде вращающегося сталь-
ного диска. За счет центробежной силы ртуть, подаваемая в центр
диска, движется от центра к периферии диска тонким слоем. Конус-
184
£
Рассол
ныи разлагатель амальгамы размещается под
диска способствует также движению раствора и
газовых пузырьков из межэлек-
тродного пространства.
Кроме того, при движении
ртути от центра к периферии тол-
щина слоя ртути уменьшается и
происходит непрерывное обновле-
ние поверхности амальгамы, что
способствует отводу выделяюще-
гося на поверхности амальгамы
натрия в объем протекающей
ртути.
Схема электролизера показана
на рис. 3-31. Электролизер на
нагрузку 30 кА имеет диск диамет-
ром 2,1 м, загрузка ртути соста-
вляет 430 кг, и при плотности
тока 9 кА/м2 он работает с напря-
жением 4,25 В [40, 84]. К недо-
статкам таких электролизеров
относится наличие крупных дви-
жущихся деталей электролизера и
затруднения, возникающие при
укрупнении электролизера.
диском. Вращение
облегчает удаление
1Ртуть
3XSX£S
Рассол
и хлор
э лек-
Ко.»:
Рис. 3-31. Схема устройства
тролизера фирмы «Асахи Гласс
1 — вращающийся катод; 2 — графито-
вый анод; 3 — крышка; 4 — подвод тока
к аноду; 5 — ртутный насос; в — вал
катода; 7 — мотор; 8 разлагатель; 9 —
выход щелочи и водорода; 10 — опорный
изолятор.
и । и i ।
Биполярные электролизеры с ртутным катодом
Обсуждалась также конструкция биполярного электролизера
с ртутным катодом, в котором отдельные ячейки расположены друг
над другом в виде многоэтажного агрегата [142]. Ячейки такого элек- .
тролизерано потоку амальгамы и рассола включались параллельно,
по электрическому току — последовательно. Все ячейки и электро-
лизер в целом имели уклон в одну сторону. Для предотвращения
сильных утечек тока подача ртути в ячейки и отвод из них амаль-
гамы в разлагатель осуществлялись через прерыватели тока.
Новые направления в развитии конструкции
электролизеров с ртутным катодом
Во многих странах ведутся поиски новых технических решений
в области электролиза с ртутным катодом. 1
Появляются новые конструкции электролизеров и всего цеха
электролиза на базе традиционной конструкции с горизонтальным
катодом. Следует упомянуть оригинальные конструкции электро-
лизеров Сольве, располагаемые в многоэтажном здании без проходов,
185
I
Kypexa, где применена П-образная конструкция электролизера,
установки электролизеров с ртутным катодом вне здания и др.
Разрабатываются также принципиально новые пути ведения про-
цесса электролиза с анодами, погруженными в ртутный катод [143—
145]. Процесс основан на том, что при погружении графитового
анода в ртуть или амальгаму при определенной плотности тока
потери напряжения в контакте графит — ртуть достигают примерно
3,1 В. При этом начинается процесс электролиза и выделяющиеся
пузырьки хлора нарушают непосредственный контакт между элек-
тродами, что обеспечивает протекание процесса электролиза с при-
емлемыми выходами по току. Здесь, вероятно, играет роль поверх-
ностное натяжение электролита на границе раздела фаз ртуть —
рассол и рассол — графитовый анод [146]. Расстояние между элек-
тродами невелико, поэтому представляется возможным вести элек-
тролиз при плотности тока около 30 кА/м2 и сравнительно неболь-
шом напряжении на ячейке (около 4,5 В).
Возможность увеличения плотности тока до величины порядка
30 кА/м2 без повышения напряжения на электролизере открывает
новые перспективы развития техники электролиза с ртутным като-
дом.
При осуществлении процесса электролиза с анодами, погружен-
ными в амальгаму, встречается ряд трудностей. Для устойчивого
протекания процесса электролиза необходимо обеспечить равно-
мерное распределение потока электролита по всей площади анода,
погруженного в амальгаму.
Электролизеры о малоизнашивающимися
анодами (МИА)
Преимущества МИА по сравнению с графитовыми анодами, ука-
занные при рассмотрении конструкции электролизеров с твердым
катодом и диафрагмой, полностью сохраняются и для электролиза
с ртутным катодом, который обычно проводится при значительно
более высокой плотности тока.
Для электролиза с ртутным катодом, помимо возможности даль-
нейшей интенсификации процесса, применение МИА позволяет зна-
чительно улучшить отвод газа из зоны прохождения тока, так как
для металлических анодов можно использовать оптимальные формы
перфорации и создать такую конструкцию проницаемого для газа
анода, которая сведет к минимуму дополнительные потери напряже-
ния, обусловленные газонаполнением электролита и экранированием
газовыми пузырьками части работающей поверхности анода. Помимо
этого, применение МИА исключает необходимость регулирования
межэлектродного расстояния в ходе работы, так как эти аноды прак-
тически не изнашиваются в процессе эксплуатации. Это значительно
упрощает конструкцию электролизера, облегчает решение вопроса
об уплотнении мест токоподвода и сокращает трудовые затраты
на обслуживание электролизеров. Для электролизеров с МИА не
186
^требуется очистка рассола от сульфатов и возможно*.более полной
использование поваренной соли в рассоле за один цикл. При этом
расход МИА, в отличие от графитовых анодов, не увеличивается.
Вследствие этого могут быть сокращены затраты на приготовление
и очистку рассола.
При установке МИА в электролизерах с ртутным катодом необ-
ходимо предотвращать возможность коротких замыканий между
ртутным катодом и МИА, поскольку даже при кратковременных
замыканиях повреждаются аноды, разрушается активное покрытие
электрода и выходят из строя МИА.
Если в качестве МИА применять платинотитановые электроды,
слой платины легко смывается с поверхности анода даже при кратко-
временных случайных коротких замыканиях. Поэтому попытки
использования ПТА в электролизерах с ртутным катодом не дали
ожидаемых положительных результатов. Для предотвращения вы-
хода из строя ПТА при случайных коротких замыканиях приходи-
лось увеличивать межэлектродное расстояние, что приводило к по-
вышению напряжения и соответственно возрастанию удедьного рас-
хода электроэнергии по сравнению с графитовыми анодами. При
установке окиснорутениевых анодов также надо предотвращать
возникновение коротких замыканий, однако восстановление окис лов
рутения, образующих активный слой МИА, амальгамой натрия
происходит не мгновенно, и, по литературным данным [53, 54],
окиснорутениевые аноды могут находиться в контакте с амальгамой
натрия в течение 20 с без опасности их повреждения.
При применении МИА необходимо поддерживать постоянный
технологический режим работы электролизера, постоянную и равно-
мерную циркуляцию ртути и питание электролизера чистым рассо-
лом, что позволит сократить до минимума образование в процессе
электролиза амальгамных масел или амальгамной пены. Поэтому
содержание кальция не должно превышать 30 мг/л, а магния — 1 мг/л
рассола [147].
В мощных электролизерах с ртутным катодом серьезная про-
блема заключается в равномерном распределении нагрузки между
электродами, особенно при применении окиснорутениевых анодов
[148, 149].
ОСНОВНЫЕ ПОКАЗАТЕЛИ ЭЛЕКТРОЛИЗЕРОВ
С РТУТНЫМ КАТОДОМ
В табл. 3-1 ^приведены краткие технические данные о некоторых
наиболее распространенных конструкциях мощных электролизеров.
Для каждого типа электролизеров приводятся преимущественно
данные по наиболее мопщой конструкции, уже применяемой в про-
мышленной практике.
187
И1ГЭХ
-BJBireisd
edaenir
-ойхяаке
Таблица 3-11. Основные показатели электролизеров с ртутным катодом
‘Аяох on
яохмя
Ю Ю Ю to
Cfr cfc о о
Ф Ф О О ffi о
188
1
ЛИТЕРАТУРА
ж
f
1. Стендер В. В. Электрохимическое производство хлора и щелочей.
М., Химтеоретиздат,1.935; Прикладная электрохимия. Изд. Харьков,
гос. ун-та, 1961.
2. Биллитер Ж. Новейшие достижения технической электрохимии. М.,
Госхимиздат, 1934; Промышленный электролиз водных растворов. Гос-
химиздат, 1959.
3. . Якименко Л. М. Электролизеры с твердым катодом. М., «Химия», 1966.
4. С а с с-Т исовский Б. А. Производство хлора. М., Госхимиздат, 1933.
5. Кузнецов В. С. Электролитическое производство хлора. М., Оборон-
гиз, 1939.
6. Хомяков В. Г., Машовец В. П., Кузьмин Л. Л. Техно-
логия электрохимических производств. М., Госхимиздат, 1949.
7. Якименко Л. М. Химическая промышленность СССР. М., Госхим-
издат, 1959. См. с. 336. -
8. Якименко Л. М., Файнштейн С. Я. Советская химическая
наука и промышленность. М., «Химия», 1967. См. с. 163.
9. 'Симон А. Г., X а и н П. Г., Якименко Л. М.., Журн. ВХО
им. Д. И. Менделеева, 6, № 1, 16 (1961).
10. Ф л е й ш м а н В. Г., Хим. пром., № 1, 9 (1959).
11. Якименко Л. М., Хим. наука и пром., 3, № 4, 424 (1958).
12. Канад, пат. 833529 (1970),
13. Файнштейн С. Я. ' Производство хлора методом диафрагменного
электролиза. М., «Химия», 1964; Генин Л. С. Электролиз растворов
поваренной соли. М., Госхимиздат, 1960.
14. Круглый С. М. Производство хлора, каустической соды и водорода.
М., «Высшая школа», 1967.
15. Электролизер БГК-17 для получения хлора и щелочей (проспект ВДНХ).
М., Госхимиздат, 1961.
16. В о й т е х о в А. Г., Хим. пром., № 11, 852 (1969); № 10, 754 (1969).
17. Greinzig G. et. al., Chem. Techn., 21, № 10, 604 (1969).
18. Sconce J. S., Chlorine, its Manufacture and Properties Uses, N. Y.,
Reinhold, Publ. Corp., 1962.
19. Пат. США 1862245, 1865152, 1866065, 1868065 (1932); 2183299 (1939);
2208778 (1940); 2330415 (1943); 2370087 (1945); 2392868, 2409911, 2409912
(1946); 2430374 (1947); 2447547 (1948).
20. Br an er A. et. al., Fortschr. Ind. Anorg. Chem. Ind. Springer, 1921,
P. 40, Bd. V, S. 2476.
21. Straw H., Chem.-Ztg., 58, 1039 (1934).
22. S t u a г t К. E., List or T. L. B., Mura у R. L., Chem. Met.
Eng., 45, № 7, 354 (1938).
23. M u г a у R. L., Trans. Am. Inst. Chem. Eng., 36, 445—462 (1940).
24. Chem. Met. Eng., 49, № 12, 114 (1942).
25. Mellor J. W., Comprehensive Treatise on Inorganic and Theoretical
Chemistry, Suppl. 2, 1, № 11, 272—322 (1956).
26. К i г k R. E. et al., Encyclopedia of Chemical Technology. VI. N. Y.,
1947.
27. Ma nt ell C. Industry Electrochemistry. N. Y., 1950.
28. H u b b a г d D. O., Chem. Eng. Progr., 46, № 9, 435 (1950); Chem. MeL
Eng., 57, № 9, 138 (1950).
29. Gardiner W. G., J. Chem. Educat., 30, № 3, 116—120 (1953).
30. International Cooperation Administration Office of Industrial Industry Ser-
vice, April 1959, IR-22275PR.
31. E h 1 e г s N. L, Hampel -С. A., J. Electrochem. Soc., 107, № 9, 791—
794 (1960).
32. M a с M u 1 1 i n R. B., Chem. Ind., 61, 41—50 (1947).
33. Canad. Chem. Proc., 37, № 1, 31 (1953); 38, № 2, 40 (1954).
34. Chem. Techn., 6, № 7 (1954).
35. Chem. Eng., 61, №2, 128—13? (1954).'
189
36. Murray R. L. et al., Trans. Electrochem. Soc., 86, № 2, 88—106 (1944).
37. Hooker Electrochem. Co., Bulletin № 20; Chem. Eng., 59, № 11, 236 (1952).
38. Industr. Chem., 28, № 333, 451 (1952); 27, № 314, 115(1951).
39. Electrochem. Techn ol., 1, № 5/6, 191—196 (1963).
40. S о m m e r s H. A., Chem. Eng. Progr., 61, № 3, 94 (1965).
41. Lewis G., Trans. Inst. Chem. Eng., 46, № 2, 38 (1968).
42. MacMullin R. B., Denki kogaku, 38, № 8, 570 (1970).
43. Chem. Eng. Progr., 44, № 12, 30 (1948); 53, № 7, 52 (1957).
44. Chem. Trade J., № 3210, 671 (1948); 140, № 3655, 1493 (1957).
45. Chem. Eng., 55, № 12, 112 (1948); 64, № 7, 160 (1957).
46. Пат. США 2666028 (1950); 2987463 (1958); 3390072 (1965).
47. Пат. США 987717 (1911); 1070454 (1913); 1365875 (1921); 1907818, 2161166,
2282058 (1939); 2920028 (1960).
48. Герм. пат. 706834 (1941).
49. Англ. пат. 1110843, 1110844 (1968).
50. Пат. США 2858263 (1962); 3337443 (1967).
51. Справочник по содовой промышленности (Сода коге бокецу то букку). Изд.
фирмы «Ниппон Сода», Токио, 1960. ,
51а. Japan Chem. Week, 13, № 652, 6 (1972).
52. Якименко Л. М., Д жа гацпанян Р. В., Веселов-
ская И. Е., Ходкевич С. Д., Хим. пром., № 10, 728 (1962).
53. De Nora О., Chemica Ind., 50, № 6, 642 (1968); Chem. Ind. Techn.,
42, № 4, 222 (1970).
54. D e Nora O., Inform. Chim., № 68, 67, 69 (1968); №77, 28 (1969);
J. Electrochem. Soc., 117, № 3, 74c (1970).
55. Бельг, пат. 739420 (1970).
56. Пат. США 3515661 (1970).
57. Н a s s К., Chem. Ing. Techn., 43, № 4, 149 (1971).
58. Strewe W., Chem. Ing. Techn., 43, № 4, 152 (1971).
59. Chem. Eng. News, 48, № 47, 32 (1970).
60. Chem. Week, 110, № 1, 32; № 14, 35 (1972).
61. Edward? G. E., Denki Kogaku (News Letters), 2, № 3, 4 (1968); ян.
пат. 20469 (1967).
62. Chem. Week, 106, № 14, 45 (1970).
63. Chem. Eng., 77, № 7, 56 (1970); 80, № 13, 62 (1973); Chem. Eng. News,
51, № 26, 12 (1973).
64. Oil Paint and Drug. Rep., 197, № 17, 26 (1970); Europ. Chem. News, 22,
№ 566, 32 (1972); 23, № 587, 30; № 590, 24 (1973).
65. Герм. пат. 70007 (1893); 73304 (1894); 106717 (1899).
66. Англ. пат. 7985, 11664 (1894); 15430 (189&); 127250 (1918).
67. Русск. пат. 2365 (1899).
68. Пат. США 676531 (1901); 692531, 692532 (1902); 1908134 (1932); 1981498
(1934); 2749301 (1956); 3332868 (1963).
69. Potter С., В is io A. L., J. Electrochem. Soc., 101, 158,637 (1954).
70. К u m e T.M Okada M., J. Chem. Soc. Japan, Ind. Chem. Sect., 58,
248 (1955).
71. Англ. пат. 490911 (1938); 1117104 (1968).
72. Герм. пат. 67851 (1893); 145794 (1904); 193768, 213808 (1906).
73. Франц, пат. 1153516 (1958); 1419637 (1965); 1494021 (1967).
74. Канад, пат. 836557 (1970); итал. пат. 590143 (1958).
75. Пат. США 2598228 (1952); 2762765 (1956).
76. Пат. ФРГ 1245931 (1967).
77. Щербак С. К., Пуставит В. Т., Ш о пирштейн И. А.,
Ха син И. А., ПриходченкоВ. Г., Волков Г. И., пат.
ФРГ 1219457 (1963).
78. Пат. ФРГ 1172239 (1962).
79. Яп. пат. 24660 (1967).
80. Пат. США 2597545 (1952).
81. Пат. ФРГ 879690 (1953); 923842 (1954).
82. Яп. пат. 1666 (1956); 2276, 2277, 7722, 7723 (1959).
190
83. Волков Г. И., авт. свид. СССР 71383 (1948); Бюлл. изобр., № 5
84. Vemura S., Kagaku Koge, Chem. Ind. (Japan), 11, № 6, 573 (1960).
85. Яп. пат. 1423 (1957); 2311, 5720 (1958); 4367, 4368, 4369, 7866 (1961).
86. Англ. пат. 852597 (1960).
87. Франц, пат. 1208271 (1960); 1325644 (1963).
88. Русск. пат. 4723 (1901).
89. Англ. пат. 15129 (1896); 25352, 25353, 25354 (1902).
90. Т oshima S., J. Electrochem. Soc. Japan, Overseas Ed., 27, № 7_______9
E153, E255 (1959); 29, № 2, E121 (1961).
91. Шведок, пат. 131224 (1950); австр. пат. 155936 (1954).
92. Яп. пат. 4201 (1962).
93. В о л к о в Г. И., Бабаян Е. Л., С и у ш е в а Э. П., Хим. пвом
№ 11, 864 (1970). ’
94. Франц, пат. 2078762 (1971); белы. пат. 763939 (1971); пат. ФРГ 2110340
(1971); пат. США 3691036 (1972).
95. Англ. пат. 627349 (1949); пат. ФРГ 931350 (1955).
96. Пат. США 3052618 (1962).
97. Бельг, пат. 705921 (1967).
98. Франц, пат. 1541435 (1968).
99. Я л о в е н к о Б. А., Грищен ко Л. 3., Хим. пром. Укр., № 1, 37
(1970).
100. Яп. пат. 3761 (1956).
101. Бельг, пат. 668236 (1965); франц, пат. 2085084 (1972).
102. Пат. ФРГ 1567948 (1970).
103. Hund Н.,Nottebohm Н., Chem. Ing. Techn., 43, № 4, 156 (1971).
104. Schafer R., Chem. Ing. Techn., 43, № 4, 160 (1971).
105. Пат. ФРГ 1173068 (1960).
106. Пат. ГДР 69604 (1969).
107. Пат. США 3531392 (1970).
108. Пат. ГДР 76010 (1970).
109. Англ. пат. 1140445 (1969); пат. ФРГ 1229996 (1970).
110. J a k s i с М. М., Csonka J. М., Electrochem. Techn., 4, № 1—2
(1966).
111. Яп. пат. 20968 (1970).
111а. Володин Н. Л., Ивенских В. Д. и др., свид. СССР 389014
(1971); Бюлл. изобр., № 29, 79 (1973).
112. Пат. США 3164477 (1962).
113. Ч в и р у к В. П., Конева Н. В. и др., авт. свид. СССР 280440
(1968); Бюлл. изобр., № 28, 20 (1970).
114. Пат. ФРГ 1017599 (1956).
115. Бельг, пат. 704247 (1968).
116. Волков Г. И., Лядин Ю. В., Свирская И. П., Хим.
пром., № 11, 838 (1971).
117. М a t u s*e k М., Frauz М., Р е s a v a Z., Chem. Prum., 21, № 12,
581 (1971).
118. В о л к о в Г. И. Производство хлора и каустической соды методом
электролиза с ртутным катодом. М., «Химия», 1968.
119. Хомяков В. Г. Краткий курс технологии хлора. М., Госхимиздат,
1933.
120. Генин Л. С. Производство хлора. М., Оборонгиз, 1938.
121. Schwab R. F., Doyle W. Н., Electrochem. Techn., 5, 228 (1967).
122. Baker I. E., Burt R. D., Chem. Eng. Progr., 63, № 12, 47 (1967).
123. Hass K. A., Chem. Ing. Techn., 39, 689 (1967).
124. Sommers H. A-, Chem. Eng. Progr., 53, № 10, 506 (1957); Electro-
chem. Techn., 5, 108 (1967); Electrochem. Techn., 6, № 3—4, 124 (1968).
125. Нодзима Сего, Кагаку коге, 19, № 7, 724 (1968).
126. Chem. Eng., 76, № 16, 143 (1969).
127. Chem. Week, 101, № 13, 89 (1967).
128- Си ба та Хироси, Никкоке гэппо, 20, № 10, 581 (1967).
191
I
Электролизер P-20. Проспект ВДНХ.
Пят СУПА 9704743 PIQ55Y
Gardiner W. C., Chem. Eng. Progr., 59, № 4, 77 (1963).
Яп. пат. 4654 (1968).
Англ. пат. 1065381 (1967).
Герм. пат. 655562Г(1932).
129. Герм. пат. 686551., 686618 (1939).
130. Электролизер P-101. Проспект ВДНХ.
131. “
. 132.
133.
134.
135.
136.
137. Tsukada Н. et al., J. Electrochem. Soc. Japan, 32, № 2, 63 (1964).
138. S h i b a t a H., Yamazaki J., Electrochem. Techn., 5, 239 (1967).
139. Stresser B., Chem. Age India, 21, № 11, 1025 (1970).
140. Mura zu mi M., Electrochem. Techn., 5, 236 (1967).
141. - - ' ** ' ----
142.
143.
144.
Chem. Age India, 19, № 6, 466 (1968). *
Пат. США 3499829 (1970); 3650919, 3650939, 3657098 (1972).
Франц, пат. 1401896 (1964).
Адаев С. П., В о л к о в Г.. И., Кубасов В. Л./ЖПХ, 12, № 1,
145.
146. А о к i К. et al., ;
147. D e N ora 0., Chem. Ing. Techn
123 (1969).
Пат. ФРГ 1146481 (1961).
J. Electrochem. Soc. Japan, 31, 817 (1962).
" _ 43, № 4, 182 (1971).
l., Chem. Age India, 21, № 11, 1013 (1970).
Г Л А В * 4
ОСНОВНЫЕ СТАДИИ ПРОИЗВОДСТВА ХЛОРА
И КАУСТИЧЕСКОЙ СОДЫ
ТЕХНОЛОГИЧЕСКИЕ СХЕМЫ ПРОИЗВОДСТВА ХЛОРА
И КАУСТИЧЕСКОЙ СОДЫ ЭЛЕКТРОЛИЗОМ
ВОДНЫХ РАСТВОРОВ ХЛОРИДОВ ЩЕЛОЧНЫХ МЕТАЛЛОВ
При электролизе водных растворов хлоридов щелочных металлов
получают хлор и растворы каустической соды. Основное количество
каустической соды обычно отгружается заводами в виде 40—50%-ных
растворов или в виде плавленого едкого натра для удовлетворения
различных нужд народного хозяйства. Хлор перерабатывается в ос-
новном на месте производства в различные хлорпродукты. Значи-
тельное количество хлора сжижается, однако основная масса жидкого
хлора потребляется на месте, количество товарного жидкого хлора,
отгружаемого различным потребителям на стороне, обычно неве-
лико.
В процессе производства хлора и каустической соды электроли-
зом водных растворов хлоридов щелочных металлов выделяется
водород, при производстве по методу электролиза с ртутным катодом
водород загрязнен парами ртути. При диафрагменном методе произ-
водства водород не содержит ртути, но может включать помимо
примесей кислорода и азота также небольшие количества хлорор-
ганических продуктов, образующихся в анодном пространстве
электролизера и поступающих затем в катодное пространство вместе
с потоком анолита.
Водород используется для получения синтетического хлористого
водорода, а также в некоторых процессах гидрирования после
необходимой очистки.
Ассортимент хлорпродуктов, получаемых на хлорных заводах,
очень велик; технологические схемы их производства весьма разно-
образны.
В этой книге рассматривается технология получения лишь основ-
ных хлорнеорганических продуктов. Настоящая глава посвящена
технологической схеме получения хлора, каустической соды и водо-
рода и первичной переработке этих продуктов с целью придания им
формы, необходимой для дальнейшего использования.
Технологическая схема охватывает процессы получения рассола
и подготовки его к электролизу, сам процесс электролиза, выпарку
и плавку каустической соды и первичную переработку хлора и водо-
рода, включающую их охлаждение, осушку и компримирование.
В зависимости от метода электролиза с твердым или с ртутным ка-
тодом, от применяемого вида соли (твердая или рассолы) и требований
13 Заказ 843 193
к каустической соде и хлору со стороны потребителей технологи-
ческие схемы различных предприятий могут несколько различаться.
На рис. 4-1 приведена принципиальная схема производства хлора
и каустической соды электролизом водного раствора поваренной
H2SO4
Рис. 4-1. Принципиальная схема производствajjxлора и каустической соды
электролизом с твердым катодом и диафрагмой.
соли по методу электролиза с твердым катодом и диафрагмой. В ка-
р
честве сырья используются рассолы, полученные подземным выще-
лачиванием поваренной соли. Состав цехов и отделений предусма-
тривает возможность производства жидкой и твердой каустической
соды и передачу потребителям и на переработку жидкого хлора,
194
сухого компримированного и испаренного хлора, хлористого водо-
рода, соляной кислоты и водорода. Процессы сжижения хлора и
производства хлористого водорода и соляной кислоты будут рас-
смотрены в следующих главах.
эис. 4-2. Принципиальная схема производства хлора и каустической соды
Рлектролизом с ртутным катодом.
При использовании в качестве сырья твердой поваренной соли
стадия подземного растворения будет исключена, однако возникает
необходимость в устройствах для хранения запаса твердой соли
и ее растворения.
На рис. 4-2 приведена принципиальная схема производства хлора
и каустической соды электролизом с ртутным катодом, причем
^3* 195
в качестве сырья применяются рассолы подземного выщелачивания
поваренной соли. Поскольку для донасыщения анолита требуется
твердая соль, в схеме предусмотрена стадия выпарки растворов для
получения кристаллической поваренной соли. Возможны варианты
схемы с донасыщением анолита природной твердой поваренной солью.
Рис. 4-3. Принципиальная схема кооперирования производства
хлора и каустической соды электролизом с диафрагмой и ртутным
катодом.
При этом отпадает необходимость в выпарке растворов поваренной
соли, но весь поток рассола после донасыщения должен быть очищен
от примесей, вносимых с природной солью. Для донасыщения анолит
можно закачивать в скважины. Если при этом проводить подземную
очистку и осветление рассола, то можно получить рассол, пригодный
для процесса электролиза с упрощением наземных устройств для
приготовления и очистки электролита.
196
Если на одном производстве используются оба метода электро-
лиза, обратная соль, выделяемая в производстве по методу с твердым
катодом и диафрагмой, может быть использована [1] для питания
цеха электролиза с ртутным катодом, как это показано на рис. 4-3.
При этом необходимо принять меры против загрязнения обратной
соли амальгамными ядами, содержащимися, например, в графито-
вых анодах или в продуктах коррозионного разрушения материалов
аппаратуры, или предусмотреть очистку получаемого после дона-
сыщения электролита от этих загрязнений. Ниже будут рассмо-
трены технологические процессы и схемы по отдельным стадиям
производственного процесса получения хлора и каустической соды.
ПРИГОТОВЛЕНИЕ И ОЧИСТИ А-РАССОЛА
и
Приготовление расоола
Основным сырьем при производстве хлора и каустической соды
служит* поваренная соль. В небольших масштабах используется
»также хлористый калий. Ниже приведены некоторые наиболее
важные физико-химические свойства поваренной соли и хлористого
калия:
NaCl KG1
2,16 1,99
1,2—1,3 —
1,25-1,45 -
801 790
11,18 —
0,204 .0,16
1,281 4,4
98,3 104,3
Плотность чистых кристаллов, г/см3
Насыпная плотность, т/м3
неслежавшейся соли.................
слежавшейся соли .......
Температура плавления, °C ....
Мольная теплоемкость, кал/(моль-град)
Удельная, теплоемкость, кал/(г-град)
Теплота растворения в 200 моль Н2О
при 18 °C, ккал/моль...............
Теплота образования, ккал/моль . .
Растворимость в системе NaCl—Н2О приведена на рис. 4-4.
Растворимость поваренной соли в воде мало зависит от температуры
и давления. В табл. 4-1 и 4-2 приве-
дена растворимость чистой поваренной
соли и КС1 в воде при различных тем-
пературах.
В присутствии других солей (хлори-
дов, сульфатов) растворимость поварен-
ной соли снижается. Природная соль
обычно поглощает влагу из воздуха
при относительной влажности выше
70—75%. Гигроскопичность природной
соли возрастает с увеличением коли-
чества загрязняющих ее примесей,
Рис. 4-4. Растворимость в системе
NaCl—Н2О.
1 - -1 h 111 I । 111 t }
О 10 20 30 4Z?
концентрация NaCl,%
197
Таблица 4-1. Растворимость NaCl в воде [3]
Температура*
°C
Концентрации NaCl
вес. %
г/л
Плотность,
г/см1
Твердая фаза
-21,2
—20
-15
—10
—5
0,15
10
20
25
30
-40
50
60
70
75
80
90
100
23,3
23,5
24,2
24,9
25,6
26,3
26,3
26,4
26,45
26,5
26,7
26,9
27,1
27,3
27,45
27,6
27,9
28,25
278
281
290
299
309
318
317
317
317
317
318
319
321
322
.323
324
327
330
1,193
1,195
1,198
1,202
1,206
1,209
1,205
1,200
1,198
1,196
1,192
1,187
1,183
1,179
1,177
1,175
1,171
1,167
Лед + NaCl 2Н3О
NaCl-2HaO
То же
»
»
NaCl-2H2O + NaCl
NaCl
То же
»
»
*
»
»
>
»
»
Таблица 4~2. Растворимость KG1 в воде [3]
ь Концентрация КС1 ..-V
Температура, Плотность, Твердая фаза ' f
•с •• Л г/см*
вес. % г/л
—10,8 19,9 . 227 1,142 Лед+КС1
—8 20,4 234 1,145 KCI
0 21,9 . 253 1,154 То же
10 23,8 277 1,165
20 25,6 301 1,174 »
25 26,45 312 1,178 »
30 27,2 322 1,182 »
40 28,7 341 1,188 »
50 30,1 359 1,194 »
60 31,4 376 1,198 »
70 32,6 392 1,202 1,203 »
75 33,2 399 »
80 33,8 407 1,205 »
90 34,9 422 1,208 »
100 35,9 434 1,210 »
особенно хлоридов кальция и магния. Вследствие гигроскопич-
ности происходит слеживание хлористого натрия, особенно при
хранении мелкой соли. Крупнозернистая соль (5 мм и вы
живается в меньшей степени [2].
еле-
и
Запасы поваренной соли в Советском Союзе очень велики. Об
балансовые запасы поваренной соли в СССР по всем категориям со-
ставляют свыше 250 млрд, т, а по категориям А + В — свыше
198
Ж*;
10 млрд. т. Запасы соли представлены залежами каменной соли,
соляными озерами с самосадочной солью и природными подземными
рассолами. В приморских районах производится также добыча
соли из морской воды бассейным или другими способами.
Запасы поваренной соли обнаружены в большинстве районов
Европейской части СССР, Сибири и Средней Азии. Не обнаружены
запасы соли (исключая морскую соль) только на Дальнем Востоке.
Химический состав каменной или самосадочной соли существенно
изменяется в зависимости от места отбора пробы или пласта. При-
мерный состав соли наиболее важных месторождений СССР приведен
в табл. 4-3.
Таблица 4~3. Примерный химический анализ поваренной соли наиболее
важных месторождении
(в % на сухое вещество) [4]
х
Месторождение
NaCl
CaSO*
CaCl,
MgSO<
MgCl»
Нераство-
римый
остаток
L Л
£
*
Каменная соль
L'
. ¥
(I
-
Артемовское
Илецкое
Солотвинское
Старобинское
Новомосковское
•..i^ras-w
ЗЙ Волгоградское
S3S *
Нахичеванское
97,5-99,0
98,0-99,0
94,0-99,0
93,0—98,0
88,0—97,0
93,0—98,0
93,2—97,6
0,3—0,4
0,8—0,9
0,1—0,3
0,05-8,4
1,0-3,0
Л;
У-г-’
’tv
Баскунчакское
Бурлинское
Туркменское
Крымское
0,2—0,3
0,05-0,1
0,03—2,0
0,01—0,06
0,05-0,3
0,02—0,05
0,032-0,05
0,03-0,1
0,05-0,3
0,2-0,5
Самосадочная соль
97,0-99,01 0,1—0,7 0,05—0,2 .0,05—0,3
98,0-99,0 0,02—0,4 0,02-0,4 0,01—0,6
97,0—98,0 0,2—1,3 - 0,1-0,6
I 98,0-99,01 0,3-0,4 |
ОД-0,4
0,1-0,6
0,6—0,7
| 0,05—0,3 |0,05—0,4 | 0,05—0,15
Г
рт’
я
Добыча соли в СССР превышает 20 млн. т в год. Около половины
ее составляет озерная соль (озеро Баскунчак), около 40% — камен-
ная соль и около 4% — бассейная и выварочная.
Вначале хлорная промышленность базировалась на твердой соли.
В настоящее время все в большей степени для производства хлора
и каустической соды начинают использовать природные и искус-
ственные рассолы, получаемые подземным растворением соли. Оте-
чественная содовая промышленность давно пользуется подземным
растворением соли и природными рассолами, состав некоторых из
них приведен в табл. 4-4 [4].
Рассолы, получаемые подземным растворением соли, обходятся
примерно 15 коп/м3, т. е. в 20 раз дешевле, чем при приготовлении
рассола из твердой привозной соли.
ИИЕИИ
1£9
I
4>
Таблице, 4-4» Состав подземных и искусственных рассолов (в г/л)
Месторождение NaCl Са2+ - Mg2+ sot
Природные рассолы
Березниковское * ......... . . .
Московское **.....................
Искусственные рассолы
Славянское ................... .
Донсода .........................
Усолье-Сибирское .................
304
312
305—310
1,52
1,67
1,2—1,6
0,38
0,20
Следы
4,38
3,89
* Содержит 0,06 г/л H2S.
** Содержит 0,4 г/л Вг~.
Подземное растворение соли в настоящее время
осуществляется
методом гидровруба [4—7]. Для обеспечения горизонтальной раз-
работки скважин под кровлей камеры создают защитную подушку
из воздуха или нефтяных фракций, подаваемых в начальный период
77777т/272Щ
Вода
Вада Рассол
Нефть ।
. Рассол
Нефть \
ZZZZZZZZ7ZZ/
77717777
777777/77Л 2
+
Рис. 4-5. Схема подземного растворения соли мето-
дом гидровруба:
а — начальный период; б — конечный период работы;
1 — породы, покрывающие и подстилающие каменную
соль; 2 — каменная соль; з — вода; 4 — тампонаж (цемент).
разработки скважины. При этом происходит растворение пласта
соли в радиальном направлении (рис. 4-5). Производительность
скважины достигает 25—50 м3/ч, срок ее работы до 50 лет.
Можно применять также подземное донасыщение обедненного
анолита из электролизеров с ртутным катодом [8—9]. При этом
вместо воды для растворения соли подают обедненный анолит из
цеха электролиза после его тщательного обесхлоривания с целью
предотвращения коррозионного разрушения труб буровой скважины.
В последнее время делаются попытки осуществить одновременно
с растворением подземную очистку рассола, чтобы получать из сква-
жины очищенный и осветленный рассол.
200
нН С
Рассол для электролиза с диафрагмой должен быть тщательно
очищен от кальция и магния, поэтому вместе с водой для растворе-
ния подается необходимое количество щелочи и кальцинированной
соды для осаждения кальция и магния. Чтобы снизить затраты ре-
агентов, в скважину можно подавать частично карбонизованный об-
ратный рассол после выпарки каустической соды. Расход реагентов
на очистку зависит от состава примесей в пласте соли.
При высоком содержании ангидрита в пласте соли, растворяемой
в воде, часть его остается в скважине в виде нерастворимого шлама,
оседающего на дно. При подземной очистке рассола по мере осажде-
ния кальция содой возможно дальнейшее растворение новых пор-
ций ангидрита. Это способствует увеличению расхода химикатов
и содержания в очищенном рассоле сульфат-иона, если в дальнейшем
не осаждать его хлористым кальцием или барием. Для снижения
скорости растворения загрязняющих поваренную соль примесей
кальциевых соединений предложено [10—13] добавлять к воде,
подаваемой на растворение, пирофосфат, гексаметафосфат, трипо-
лифосфат или карбонат натрия. Предполагается, что при этом на
поверхности кристаллов ангидрита или других кальциевых соедине-
ний отлагается осадок малорастворимых соединений, препятству-
ющих растворению солей кальция. Насколько эти предложения
могут быть эффективными при подземном растворении соли в течение
продолжительного контакта раствора с пластом, трудно судить.
При приготовлении и очистке рассола для электролиза с ртут-
ным катодом не требуется полного удаления кальция. Поэтому при
подземной очистке рассола можно ограничиться осаждением магния.
В зависимости от соотношения Са : SO4 в пласте соли загрязнение
рассола кальцием или сульфат-ионом можно ограничить, подавая
в скважину вместе с обедненным анолитом раствор хлористого каль-
ция (при избытке ионов SO4"), или сульфата натрия (при избытке
кальция).
При подземном донасыщении анолита электролизеров с ртутным
катодом производительность скважин снижается по сравнению
с простым выщелачиванием соли водой, так как с 1 м3 рассола выво-
дится из скважины 50—60 кг соли вместо 300—310 кг при выщелачи-
вании соли водой.
Серьезной проблемой при подземном донасыщении анолита
является подбор коррозионно-стойких материалов для буровой сква-
жины. При повышенных температурах обычная черная сталь не
стойка, а проведение донасыщения при пониженных температурах
связано с большими потерями тепла и затратами на теплообменную
аппаратуру. Необходимо также принимать меры для предотвраще-
ния гипсования труб скважины, коммуникаций и теплообменной
аппаратуры в связи со снижением растворимости гипса при повыше-
нии температуры рассола.
Помимо этого, питание электролизеров с ртутным катодом рас-
солом, насыщенным по CaSO4, приводит к ухудшению их работы.
Поэтому для питания электролизеров с ртутным катодом обычно
201
II
применяют рассол, содержащий CaSO4 в количестве, меньшем чем
требуется для полного его насыщения.
Ниже приведены требования, предъявляемые к качеству соли [14],
применяемой для производства хлора и каустической соды:
Содержание, %
NaCl, не менее . .... 4 .... . 97,5
примеси в пересчете на сухое вещество,
не более
нерастворимого осадка............... 0,5
Са^+ .............................. 0,4
Mg2+ ............................ 0,05
SOt ............... . .......... 0,84
влаги............................ 5,0
К+ ................................. 0,02*
Амальгамная проба за 30 мин, мл Н2, не
более . . . '......................... 0,3 *
и
* Только для электролиза с ртутным катодом.
Твердую соль стараются перевозить водным транспортом. Если
это невозможно, используют железнодорожные или смешанные пере-
возки. При длительных перевозках в зимнее время соль смерзается
и выгрузка ее затруднена. Для механизации разгрузки соли при-
меняют саморазгружающиеся вагоны, механические лопаты или
краны [15]. Иногда при разгрузке соли в бассейны складов-раство-
рителей ее смывают из вагона струей рассола.
При применении водного транспорта в связи с сезонностью л
навигации необходимы большие запасы соли для обеспечения бес-
перебойной работы предприятия. Соль хранят на открытых бетони-
рованных площадках или в бассейнах складов-растворителей. Для
предотвращения потерь соли с рассолом, утекающим в грунт, бето-
нированные площадки или бассейны складов растворителей должны
быть выполнены тщательно и снабжены хорошей гидроизоляцией
днищ и стенок от грунта. Сообщается [16] о применении на зару-
бежных заводах бетонных бассейнов, выложенных поливинилхло-
ридом на специальном клее.
Растворение соли обычно производят в бассейнах складов-раство-
рителей или в специальных растворителях.
Скорость растворения соли определяется в основном скоростью
диффузии молекул соли из насыщенного раствора у поверхности
растворяемого кристалла соли в объем раствора [17—20]. Поэтому
скорость растворения соли возрастает с ростом температуры (рис. 4-
6) при перемешивании раствора или пропускании его через слой соли.
Скорость растворения соли и загрязняющих ее примесей раз- .
лична. Скорость растворения CaSO4 ниже, чем поваренной соли.
Поэтому при ограниченном времени контакта раствора с кристал-
лами соли можно получить рассолы с пониженным содержанием
медленно растворяющихся примесей [21]. Если в воду, подаваемую
на растворение соли, добавить соду для связывания кальция, эффект
фракционирования может быть дополнительно увеличен, по-види-
202
Температура, °C
, 4-6. Зависимость кон-
станты скорости растворения
NaCl от температуры [13].
мому, за счет осаждения на кристаллах ангидрита слоя СаСО31
затрудняющего растворение примесей.
На рис. 4-7 показан один из типов открытого бассейна-раствори-
теля поваренной соли. В складе такого типа снижаются затраты
труда на разгрузку и транспортирование соли. Отбираемый из
растворителя рассол имеет достаточную степень насыщения. Обычно
предусмотрена также возможность рециркуляции низкоконцентри-
рованного рассола, а также подачи рассола для размывки соли
из вагонов в склад.
При использовании таких растворителей происходит хорошее
усреднение соли, полученный рассол содержит примеси постоянного
состава, что облегчает регулирование
процесса очистки рассола.
Для растворения соли, хранящейся
в сухих складах, а также для дона-
сыщения анолита в электролизерах
с ртутным катодом применяются рас-
творители многих типов. Обычно это
цилиндрические или цилиндрические
с коническим дном аппараты с ложным
дном. Одна из конструкций - такого
аппарата показана на рис. 4-8. Вода
или обедненный анолит подается в ниж-
нюю часть растворителя под ложное
днище и, проходя через слой соли,
насыщается. По мере растворения соль . Рис.
дополняется сверху через загрузочный
люк. При донасыщении анолита элек-
тролизеров с ртутным катодом, содер-
жащего активный хлор, растворитель выполняют из материалов, стой-
ких в средах, содержащих активный хлор; подача соли и сам рас-
творитель должны быть герметизированы для предотвращения
загрязнения атмосферы выделяющимся хлором.
Хлористый калий также используется как сырье в хлорной про-
мышленности для получения хлора, едкого кали и водорода. Мас-
штабы производства едкого кали во много раз меньше, чем NaOH.
В природе хлористый калий встречается в виде сильвина КС1,
сильвинита — смеси сильвина с галитом, карналлита KCl*MgCl2-
KCl-MgSO4 ЗН2О, загрязненных г лини-
• оп2и, каинита
стыми веществами, карбонатами, сульфатами и другими приме-
сями.
В СССР запасы калийных солей [22] имеются на Урале (Верхне-
камское месторождение), в Белоруссии, Предкарпатье, Туркмении,
Казахстане и др. районах. Хлористый калий получают из калийных
руд галургическим и флотационным способами. Галургический спо-
соб основан на различной температурной зависимости растворимости
NaCl и КС1 в системе NaCl—КС1—Н2О: с повышением температуры
растворимость NaCl снижается, а КС1 возрастает (табл. 4-5).
203
Рассол на
очистку
Рассол
"потребителям
Рис. 4-7. Открытые склады-растворители соли:
1 — вагон; 2 — отстойник; 3 — насос.
Соль
Рис, 4-8. Растворители соли с ложным дном:
1 — штуцер для чистки растворителя; 2 — ложное дно;
3 — насос.
Таблица 4-5. Растворимость системы NaCl—KCI—Н2О
Температура,
Состав жидкой фазы,
вес. %
NaCl KCI
Твердая фаза
—20 20,6 6,0 NaCl -2Н,0 + КС1
—10 21,7 6,5 То же
-2,7 22,55 6,95 NaCl -2Н,0 + NaCl + КС1
0 22,35 7,35 NaCl + KCI
10 21,5 8,9 То же
20' 20,7 10,4 » '
30 20,1 11,85 »
40 19,6 13,25 »
50 19,1 14,7 »
60 18,6 16,15 »
80 17,55 19,05 »
100 16.8 21,7
На этом основаны галургические методы переработки сильви-
нита [2, 23, 24]. Выделяемый в виде отхода NaCl может быть также
использован для электролиза с диафрагмой.
Применении этой соли для ртутного электролиза ограничено
присутствием КС1. Из отхода NaCl не может быть получена каусти-
ческая сода марки А-2.
По флотационному способу размолотый сильвинит обрабатывают
в насыщенном растворе в присутствии флотореагентов (алифати-
ческих аминов С16—С20, например солянокислого октадециламина
C18H37NH2.HC1).
Хлористый калий получают также при переработке карналлита,
в качестве отхода в производстве металлического магния.
Технический хлористый калий первого сорта должен содержать
не менее 96% КС1 и не более 1,4% NaCl и 1% влаги [25].
Фактический состав примесей в хлористом калии, используемом
для электролиза на одном из заводов, приведен ниже [4]:
Примеси
NaCl .
Са2+ ,
Mg2+ ,
Содержание
О/
/о
. 1,1-1,4
. 0,02
. 0,01
Примеси ЦСо держание,
%
Fe3+ ....»» 0,004
SO|- ........... 0,03
Нерастворимый оса-
док .........0,09—0,11
Хлористый калий растворяют так же, как и NaCl, в раствори-
телях с ложным дном. При температуре около 40 °C можно получить
растворы, содержащие 330—340 г/л, а при 70—80 °C до 400 г/л.
Для снижения слеживаемости хлористый калий обрабатывают
октадецил амином (0,02% от массы хлористого калия).
205
Очистка рассола для электролиза с диафрагмой
К рассолу, поступающему на электролиз, предъявляются опре-
деленные v требования в отношении содержания NaCl и вредных
для процесса электролиза примесей. Требования к рассолу для
электролиза с твердым и ртутным катодами существенно различа-
ются между собой. Ниже приведены требования к очищенному
рассолу для электролизеров с твердым катодом и диафрагмой, при-
нятые на отечественных заводах:
Содержание, г/л
NaCl ..................................Не менее
310
Na2S04 ..........................Не более 6
Na2CO3.............................0,3—0,4
NaOH .............................0,05—0,10
Содержание, мг/л, не более
Са2+ ............................ 5
Mg2+ . ............ 1
Прозрачность «по кресту», мм, не менее . . 1200
Рассол, подаваемый в электролизеры с ртутным катодом, не дол-
жен содержать примесей амальгамных ядов, в первую очередь ва-
надия, хрома, молибдена и титана. Загрязнение рассола амальгам-
пыми ядами проверяется так называемой амальгамной пробой рас-
сола: объему водорода, выделяемого при разложении амальгамы
в определенных условиях [26, 27].
СГдругой сторОТыт^рассол для электролиза с ртутным катодом
не обязательно очищать от примесей кальция,, так как при исполь-
зовании графитовых анодов соли кальция- не мешают процессу элек-
тролиза . Од н^о^эти^сол^усилшвают^врецное^ действие некоторых
амальгамных^ддов.
^Специфические требования, предъявляемые к рассолу для элек-
тролиза с ртутным катодом, обусловлены значительно большим удель-
ным расходом рассола (около 35 м3 на 1 т каустической соды) по
сравнению с расходом при электролизе на твердом катоде (около
9 м3 на 1 т каустической соды), а также тем, что на стадию приго-
товления и очистки рассола при электролизе с ртутным катодом по-
ступает отработанный анолит, содержащий активный хлор и соли
ртути. Поэтому схемы приготовления и очистки рассола для ртут-
ного и диафрагменного методов электролиза различны.
На всех хлорных заводах нашей страны применяется содово-
каустический метод очистки рассола от’примесей Са и Mg, основан-
ный на образовании малорастворимых осадков СаСО3 и Mg (0Н)2.
При содово-каустической очистке протекают следующие хими-
ческие реакц
sis
СаС12 -pNagCOg — СаСО3 -р 2NaCl
CaSO4-pNa2CO3— СаСОз+ Na2SO4
Са(НСО3)2 4-2NaOH — СаСОз -f- NajCOg -p 2H2O
MgCl2+2NaOH = Mg(OH)2+2NaCl
MgSO4+2NaOH = Mg(OH)2+Na2SO4
(4.1)
206
и
*<•
$
/Л'
Для осаждения Mg обычно используется щелочь, остающаяся
в обратной соли цеха выпарки электролитических щелоков. При
небольшом содержании Mg избыток щелочи переводят в соду путем
^карбонизации обратного рассола.
При известково-содовом способе для проведения реакций (4.3)—
(4.5) вместо NaOH применяется раствор известкового молока. Изве-
стково-содовый способ очистки применяется в содовой промышлен-
ности и использование его целесообразно при высоком содержании
магния в рассоле. При кооперировании производства хлора и соды
Часто применяют рассол, полученный после известково-содовой
очистки с доочисткой его за счет подогрева до 50—60 °C и последую-
щей фильтрации.
Разрабатывался также метод очистки рассола
мощью ионообменных смол [28—30]. При этом
между ионом натрия катионита и ионами^Са2*
j2R—Na+Са«+= R2Ca + 2Na+
н
от примесей с по-
происходит обмен
(4.6)
Отработанный катионит регенерируется промывкой соляной ки-
слотой и переводится в натриевую^ форму щелочью
/ RaCa + 2HCl=2R—Н + СаС12 (4.7)
R—H + NaOH = R-Na+H2O (4 8)
(Расход реагентов на регенерацию катионита зависит от содержа-
ния в рассоле кальция и магния и степени полезного использования
обменной емкости катионита, так как он пропорционален общей
емкости ионита по Na+, Са2* и Mg2+.
Ионообменная очистка рассола не получила применения в хлор-
ной промышленности вследствие повышенного
недостаточной механической прочности смол и трудности утилиза-
ции стоков.
Растворимость образующегося при очистке карбоната кальция
снижается с повышением концентрации поваренной соли в рассоле [31 ]
и сильно зависит от избытка соды. Ниже приведены данные С. С. Шрай-
бмана о влиянии избытка соды в
кальция:
£ Избыток
СОДЫ, мг/л
100
200
300
h
Ниже
в рассоле от избытка NaOH [32]:
С о д е р ж" а н и е
при 25 °C
расхода реактивов,
рассоле на остаточное содержание
Содержание
ц и я
при 20^С
35,0
16,0
9,0
показана
мг/л
при 40 *С
20
8,0
5,0
Избыток
соды, мг/л
400
500
600
Содержание
ция,
при 20 °C
8,0
мг/л
при 40 °C
4,0
зависимость
остаточного
содержания
магния
Избыток NaOH, мг/л
О
50
100
200
магния, мг/л
при 50 °C
10,7
Не обнаружено
207
Обычно при очистке рассола содово-каустическим методом избы-
ток соды составляет 300—500 мг/л и едкого натра 50—100 мг/л;
а остаточное содержание ионов кальция и магния равно 4—5 мг/л [4].
Дальнейшее увеличение избытка соды и NaOH нерационально,
так как при этом качество очистки рассола не улучшается.
Реакция химического взаимодействия солей кальция и магния
с добавляемыми реактивами протекает практически мгновенно
после перемешивания очищаемого раствора с реактивами.
Процесс образования твердых осадков СаСО3 и Mg (ОН)2, укруп-
нения их и отстаивания зависит от многих
я
акторов и протекает
во времени. Наибольшее влияние на характер образующихся осад-
ков и скорость их отстаивания оказывает соотношение получаемых
гидроокиси магния и карбоната кальция. Рассолы с высоким отно-
шением Mg : Са при очистке образуют плохо уплотняющиеся и мед-
ленно отстаивающиеся осадки, как это можно видеть из данных,
приведенных в табл. 4-6.
Таблица 4-6- Скорость отстаивания суспензией [32]
при различном отношении Mg : Са при 20 °C
(Избыток 100 мг/л NaOH и 800 мг/л Na2CO3)
Концентрация
в суспензии, г/л
Mg Са
Скорость
отстаивания
мм/мин
Объем шлама, %
через 2 ч через 3 ч
через 24 ч
0
ОД
0,3
0,6
0,9
1,1
1,2
2
1,8
1,5
1,0
0,5
0,2
0
Карбонат кальция
0,7
25,6
15,6
5,5
3,0
2,4
1,0
62,7
15,8
47,8
11,7
35,7
1,8
2,0
4,0
10,0
12,7
26,8
способен давать перенасыщенные
0,6
1,0
1,4
2,6
4,2
5,7
10,7
растворы,
что может также затруднить процесс очистки рассола.
Избыток соды более 1,3 г/л приводит к уменьшению скорости
отстаивания осадков. Для ускорения процессов кристаллизации,
коагуляции и абсорбции, которые протекает при образовании твер-
дой фазы и укрупнении осадков, вводят контактные среды, а также
н
коагулянты и флокулянты. В качестве контактной массы в рассол
вводят осадок, полученный при предыдущих операциях очистки.
При этом наличие в рассоле сформировавшихся частиц осадка со-
здает условия для кристаллизации на их поверхности вновь обра-
зующегося осадка, более быстрого укрупнения и отстоя частиц.
Дальнейшим развитием этого принципа явилось создание аппа-
ратов для осветления рассола, в которых процессы кристаллизации,
коагуляции и образования хлопьев осадка происходят во взвешенном
слое образующегося осадка, служащего контактной средой. Осветле-
208
И
ние с применением контактной среды и взвешенного фильтра из
частиц осадка было использовано сначала для очистки воды, в насто-
ящее время оно широко используется для очистки рассола [4, 33,
34] в осветлителях типа ОВР и ЦНИИ.
В процессе очистки рассола может добавляться хлорное железо ,
для улучшения структурообразования осадка, а также коагуляции
и процессов сорбции различных примесей на образовавшемся осадке
гидроокиси железа [35]. Хлористый алюминий для этой цели не-
пригоден из-за значительной растворимости его гидроокиси в щелоч-
ных растворах. Растворимость
с ростом избытка щелочи [3]:
гидроокиси железа увеличивается
Избыток NaOH, мг/
50
150
250
400
500
Растворимость Fe8+
при 20 °C, мг/л
Не обнаружено
То ж*
0,08
Р,65
0,75
Однако при применяемом на практике избытке щелочи (50—100 мг/л)
гидроокись железа практически нерастворима.
Добавки хлорного железа широко используются в установках
водоочистки, но редко применяются при очистке рассола, так как
вследствие значительного содержания загрязняющих примесей в рас-
соле расход FeCl3 как коагулянта сильно возрастает по сравнению
с процессом водоочистки. При очистке рассола для электролиза
с ртутным катодом возможно загрязнение рассола амальгамными
ядами, вносимыми с хлорным железом.
Для ускорения агрегации частиц осадка и процессов осветления
рассола применяются различные флокулянты —высокомолекуляр-
ные полимеры с линейной или мало разветвленной цепью —
крахмал, карбоксиметилцеллюлоза, сульфатцеллюлоза, полиакрил-
амид и др. Наиболее широко применяется в качестве флокулянта
при/*очистке рассола полиакриламид
СН2— СН—"
I
со
nh2 _
п
Добавки полиакриламида при очистке рассола, получаемого
из баскунчакской или артемовской соли, а также подземного рассола
позволяют ускорять хлопьеобразование и процессы осветления
и уплотнения шлама [36]. Для повышения флокулирующего дей-
ствия полиакриламид на 20—40% гидролизуется при обработке
раствором щелочи.
Полиакриламид почти в два раза ускоряет осветление рассола
при периодическом методе. Полиакриламид широко применяется
в осветлителях с контактной массой в виде взвешенного слоя осадка.
При этом образуется осадок с хорошими физическими свойствами,
14 Закав 843 209
II
обеспечивается получение устойчивого шламового фильтра и доста-
точное осветление рассола [4]. Большой избыток полиакриламида
в очищенном рассоле может вызвать нарушения технологического
процесса на следующих стадиях производства, в частности, снизить
производительность фильтров для рассола и протекаемость диафрагм
электролизеров с твердым катодом. Поэтому количество добавляемого
полиакриламида должно быть строго дозировано.
1
21 ’
18
Соляная
кислота
Рбратныщрассол
CQ> из цеха Выпарки
(топочные
Электролит
ческцй ще-
пля
19
13 8 канализацию 15 3
Рис. 4-9. Схема очистки рассола для производства хлора и каустической соды
по методу электролиза с диафрагмой:
1 — сборник сырого рассола; 2 — подогреватель; з -- центробежные насосы; 4 — сборник
обратного карбонизованного рассола; 5 — колонна карбонизации; 6 — автоклав для гид-
ролиза полиакриламида; 7 — растворитель гидролизованного полиакриламида; 8 — бак для
раствора полиакриламида; 9 — дозатор; 10 — осветитель с-шламовым фильтром; 11 — на-
порный бак шламовой суспензии; 12 — центрифуга; 13 — приемник фильтрата; 14 — при-
емник шлама; 14 — сборник чистого рассола; 16 — фильтр для рассола; 17 — нейтрализа-
тор рассола; 18 — напорный бак соляной кислоты; 19 — сборник осветленного рассола;
20 — подогреватель рассола; 21 — напорный бак для очищенного рассола.
лок
пар
Сжатый
Воздух
I/ Сжа-
/ ¥ тыа
11 Воздух
12 алЭп
17
"Ь/б1
Очищенный,
рассол на
электролиз
Пар
20
Конденсат
Технологический процесс очистки рассола, полученного раство-
рением природной соли, состоит из осаждения ионов кальция и маг-
ния добавляемыми реактивами, осветления и фильтрования рас-
сола и нейтрализации избыточной щелочности рассола перед подачей
его на электролиз. В зависимости от типа осветлителей и фильтров,
а также местных условий технологические схемы отделений очистки
рассола могут различаться между собой. На рис. 4-9 приведена прин-
ципиальная Jтехнологическая схема непрерывной очистки рассола
для цехов электролиза с диафрагмой, включающая карбонизацию
рассола, при которой для осаждения солей кальция используется
избыточная щелочность обратного рассола из цеха выпарки.
При наличии на заводе концентрированной двуокиси углерода
Для карбонизации могут быть использованы обычные насадочные
скруббера с кольцами Рашига, а при использовании топочных газов
котельных — аппараты пенного типа. Карбонизацию рассола , сле-
дует вести таким образом, чтобы образовалось количество соды,
достаточное для осаждения кальция и создания необходимого избытка
ее в очищенном рассоле. При этом в рассоле должна оставаться
210 '
щелочь, количество которой достаточно для осаждения магния
и создания избытка щелочи в рассоле не менее 0,05 г/л. Обычно
для удобства управления процессом на- карбонизацию направляют
лишь часть обратного рассола, а остальное количество, минуя карбо-
низатор, смешивают с карбонизованным рассолом.
При отсутствии источников СО 2 применяются растворы соды.
Для этого в схему включают растворитель соды, емкости и насосы
для содового раствора. Чтобы исключить разбавление рассола при
добавлении содового раствора, последний часто готовят на обратном
рассоле. При этом концентрация Na2СО3 в растворе составляет 60—
80 г/л.
Для усреднения обычно
устанавливают (на схеме не
показаны) емкости для об-
ратного рассола из отделе-
ния выпарки каустической
соды.
Смешение рассола с рас-
творами реактивов или с об-
ратным карбонизованным
рассолом производится в сме-
сителях, где необходимо под-
держивать хорошее переме -
шивание подаваемых на
смешение жидкостей для
протекания реакции и обра-
зования хлопьев осадка.
В осветлителях с взве-
шенным шламовым фильтром
смешение производится не-
посредственно в осветлителе.
В качестве осветлителей
сола применяются аппараты Дорра и другие конструкции, в основе
которых лежит гравитационный метод [37, 38]. В последнее время
как при водоочистке, так и при очистке рассола для хлорной и содо-
вой промышленности преимущественное применение находят освет-
лители с-взвешенным фильтром осадка [4,39—43]. В нашей стране
применяются осветлители со шламовым фильтром типа ЦНИИ-1,
ЦНИИ-3, ОВР-ПШ.
На рис. 4-10 приведена схема работы осветлителя ЦНИИ-3.
Рассол вводится через сопла, расположенные тангенциально, что
обеспечивает вращательное движение рассола и хорошее перемеши-
вание его с реагентами и полиакриламидом, вводимыми непосред-
ственно в осветлитель.
* Раствор полиакриламида в чистом рассоле подвергается частич-
ному гидролизу щелочью при нагревании. Рассол, смешанный с оса-
дителями, поднимается в конической части осветлителя. При этом вра-
щательное движение рассола затухает и формируется взвешенный
14* 211
и
Шлам
tx:
{Очищенный
! рассол
в
Рассол
Шлам
Осветлитель ЦНИИ-3:
2 — окна для отвода
в шламоуплотни-
Рис. 4-10.
1 —дренажная решетка; ,
избытка шламового фильтра г -------_---
тель; з — трубопровод для подачи реагентов;
4 — распределительные сопла; 5 — донные кла-
паны; 6 — дополнительный бункер; 7 — шламо-
уплотнитель; 8 — дополнительные
отвода шлама при диаметре
7 м; 9 — труба для отвода
твора из шламоуплотяителя.
окна для
уплотнителя более
осветленного рас-
в непрерывных схемах
очистки рас-
фильтр из частиц осадка. Осветленный рассол из верхней части
осветлителя отводится через сборный желоб. Избыток шлама через
окна поступает в шламоуплотнитель и выводится из нижней части
аппарата.
На рис. 4-11 приведена схема осветлителя ОВР-ПШ, используе-
мого на некоторых отечественных заводах. Цилиндрический освет-
литель имеет механическую мешалку, вращающуюся с частотой
12—16 об/ч, что обеспечивает хорошее распределение сырого и обрат-
Рис. 4-11. Осветлитель^ О В Р-ПШЦ
1 — воздухоотделитель; ^==гптлатитготводнь1е
трубы; з — коллектор для отвода осветленного
рассола из шламоуплотнителя; 4 — мешалка
шламоуплотнителя; 5 — шламоуплотнитель;
6 — клапан для спуска отстоявшегося
шлама в шламоуплотнитель; 7 — распреде-
лители рассола; 8 — сборный желоб для
осветленного рассола; 9 — привод мешалки.
ного рассола. С помощью греб-
ков мешалка перемещает
шлам к отверстию для вывода
его из аппарата. Осветлитель
ОВР-ПШ диаметром 8 м имеет
производительность до120м3/ч.
При расходе полиакриламида
1,5—2,5 г/м3 очищенного рас-
сола получают шлам концент-
рацией твердого осадка до
400-500 г/л.
Шлам, получаемый при очи-
стке рассола, состоит из частиц
СаСО3 и Mg(OH)2. Попытки
полезного использования этого
шлама в качестве наполнителя
в резиновой и других отраслях
промышленности [44—45], для
орошения санитарных колонн
с целью улавливания хлора
вместо известкового молока [4]
не увенчались успехом и шлам
обычно сбрасывается на шла-
мовые поля. Для уменьшения
и
потерь соли
лам после освет-
лителя отделяют от рассола на
центрифугах или в барабанных вакуум-фильтрах и после репуль-
пации осадка водой откачивают в канализацию или на шламовые
поля.
На многих заводах для отстоя рассола еще используются освет-
лители Дорра (рис. 4-12). Часто корпус осветлителя выполняется
железобетонным, однако при этом требуется очень тщательное вы-
полнение работ по укладке бетона и гидроизоляции, чтобы пред-
отвратить неплотности аппарата, приводящие к потерям рассола
и быстрому разрушению осветлителя.
Распространены осветлители Дорра диаметром 18 м, высотой
цилиндрической части 6,45 м и конической 1,15 м; мешалка делает
около 5 об/ч. Осветлитель работает удовлетворительно при скорости
подъема рассола 0,5^—0,8 м/ч, что соответствует производительности
от 125 до 200 м3/ч осветленного рассола. Шлам, уплотненный до
212
содержания твердой фазы 400—500 г7л, выводится из нижней части
осветлителя.
В хлорной промышленности аппараты Дорра работают при тем-
пературе 40—50 °C. Для предотвращения конвекционных потоков,
нарушающих процесс отстаивания, отстойники необходимо разме-
щать в помещении или снабжать хорошей теплоизоляцией с тем,
чтобы колебания температуры не превышали + 2 °C. В этих условиях
Рис. 4-12. Осветлитель Дорра:
.1 — стальная чаша; 2 — кольцевой фундамент; 3 — столб; 4 — вал освет-
лителя; 5 — ферма; 6 — редуктор ; 7 — гребки мешалки; 8 — кольцевой
желоб осветленного рассола.
1
X
прозрачность осветленного рассола «по кресту» обычно достигает
400—500 мм. Вследствие громоздкости, малой производительности
и недостаточного осветления рассола аппараты Дорра уступают бо-
лее совершенным конструкциям осветлителей со взвешенным шла-
мовым фильтром.
В последнее время разработаны методы интенсификации работы
осветлителей со взвешенным шламовым фильтром осадка за счет
изменения скорости подъема раствора по высоте осветлителя: при
скорости в зоне реакции около 90 м/ч в зоне осветления и фильтрации
применяется скорость около 9 м/ч и в зоне уплотнения осадка —
около 5 м/ч [46].
Для нормальной работы электролизеров с диафрагмой и с ртут-
ным катодом большое значение имеет высокая степень осветления
рассола. Обычно после осветлителей рассол подвергают одной или
двум последовательным фильтрациям. До последнего времени двой-
ная фильтрация применялась преимущественно для растворов, по-
ступающих в электролизеры с ртутным катодом. В последнее время
на некоторых отечественных заводах начинают применять вторую
2ia
более тонкую фильтрацию рассола для питания электролизеров
с диафрагмой.
Для фильтрации рассола наиболее широко применяются насып-
ные фильтры с заполнением мраморной крошкой, антрацитом или
кварцевым песком. Однако насадка из кварцевого песка пригодна
только для фильтрации кислого рассола, так как
заметно растворяет кварцевый песок.
(елочной рассол
Для фильтрации щелочного
н
IE
Рис. 4-13. Насадочный фильтр для рас-
сола:
1 — штуцер для ввода рассола и отвода жид-
кости при промывке; 2 — люк; з — штуцер
для спуска воздуха; 4 — штуцер; 5 — штуцер
для ввода промывной жидкости и отвода рас-
сола из фильтра; 4,6 J—распределитель рассола;
7 — насадка.
Шлам
Рис. 4-14. Рамный фильтр для
рассола:
1 — рамы фильтра; 2 — крошка;
3 — зажимное устройство; 4 — фо-
нарь; 5 —гребенка.
рассола удобнее применять насадку из мраморной крошки, так как
при этом помимо фильтрации происходит снятие перенасыщения
рассола до СаСО3 за счет его кристаллизации на зернах насадки.
Ниже приведен примерный гранулометрический состав мрамор-
ной крошки для загрузки фильтров и
[4]:
IE
изменение его
за 5 мес
г
работы
Фракция, мм До 0,5
Содержание, %
до загрузки • • • » —
через 5 мес. » , , 7,2
0,5-1,0 Ji—2J
5,5 29,1
14,8 36,9
2-3
29,7
27,4
3—51 Более 5
33,1 2,6
12,4 1,3
На рис. 4-13 показано устройство фильтра насадочного типа.
Такой фильтр диаметром 3 м и с насадкой высотой до 1,2 м имеет
производительность 50—60 м3/ч. По мере забивания фильтра сопро-
тивление его возрастает, и по достижении 2,5—3,0 ат фильтр выклю-
чается на регенерацию — промывку струей воды или рассола.
Операции по переключению фильтра на промывку^могут быть арто-
214
&
Я
t
t
in
IE
IE
матизированы. Если рассол, использованный для промывки филь-
тров, возвратить вновь в цикл очистки рассола, можно предотвра-
тить потери соли на стадии регенерации фильтров.
Для хорошей работы фильтров необходимо тщательно изготавли-
вать и монтировать устройства для распределения рассола, так как
в противном случае, особенно при регенерации фильтров, насадка
частично уносится с потоком рассола, подаваемого на регенерацию,
и нарушается работа фильтра. При подаче рассола, прозрачность
«по кресту» которого в пределах 700—1000 мм, фильтр может рабо-
тать без регенерации около 10 сут.
На рис. 4-14 показана схема рамного фильтра типа Келли. Такие
ильтры широко применялись и применяются в настоящее время
на многих хлорных заводах. Рамный фильтр с поверхностью фильтра-
ции 100—НО м2 имеет производительность до 50 м3/ч рассола. Филь-
тровании обычно ведут через ткань бельтинг, покрытую сверху пала-
точной тканью. При фильтровании рассола с примесью активного
хлора применяют поливинилхлоридные фильтровальные ткани. Для
регенерации фильтровальную ткань промывают струей воды и при-
мерно раз в два месяца — ингибированной соляной кислотой. Рамные
ильтры требуют для обслуживания большой затраты рабочей
силы и труднее поддаются автоматизации, чем насыпные фильтры.
На одном из отечественных хлорных заводов с целью продления
срока службы диафрагмы электролизеров рассол после фильтро-
вания на насыпных фильтрах направляли на повторное фильтрование
через трубки из пористого фторопласта. При этом рассол практи-
чески полностью освобождался от взвешенных твердых частиц,,
а фильтры с фторопластовыми трубками не требовали слишком ча-
стой регенерации.
Для отделения рассола от твердых частиц и осветления предло-
жено использовать процесс флотации. Для этого рассол после до-
бавления NaOH и Na2CO3 смешивают с чистым рассолом, насыщен-
ным воздухом под давлением 2—3 ат. При смешивании под атмосфер-
ным давлением воздух, растворенный в рассоле, выделяется в виде
мелких пузырьков, увлекая образующийся при очистке рассола
осадок. В зависимости от состава сырого рассола соотношение рас-
сола, подвергаемого очистке, и рассола, насыщенного воздухом под
давлением, может изменяться в пределах от 2 : 1 до 5 : 1. Скорость
флотации составляет 4—6 м/ч, процесс осветления заканчивается
за 15 мин. Содержание твердого осадка в флотационной пене дости-
гает 11—13%. Указывается [471, что этот метод позволяет успешно
проводить очистку рассола при соотношении СаО : MgO = 0,5—5
в аппаратуре меньшего объема и требует меньших затрат при орга-
низации производства. После осветления рассол фильтруют на на-
сыпных фильтрах.
Осветленный рассол перед подачей на электролиз подвергается
нейтрализации до pH = 10,8 11,2 (0,05—0,1 г/л NaOH) в секци-
онном смесителе. Подача соляной кислоты в нейтрализатор может
быть автоматизирована в зависимости от pH рассола на выхода
215,
V
из нейтрализатора. Избыток соды должен регулироваться при сме-
шении^растворов, так как в условиях работы нейтрализатора (pH —
= 10,8 11,2) сода не нейтрализуется кислотой.
При электролизе с твердым катодом и диафрагмой увеличение
концентраций поваренной соли в рассоле позволяет повысить кон-
и
центрацию получаемой щелочи при сохранении того же выхода по
току, снизить удельный расход графита и несколько уменьшить на-
пряжение на электролизере за счет возрастания электропроводности.
С целью получения более концентрированных рассолов [48, 49]
очищенный рассол донасыщают чистой обратной солью из отделе-
Очищенный •
рассол
Пар
Пар л
Обратная соль из
отделениябыпарки
каустической соды
Конденсат
Рассол на
....- *
Электролиз
Конденсат
Соляная 5
кислой
та г ‘
Рис. 4-15. Схема донасыщения. рассола обрат-
ной солью из отделения выпарки:
1 — теплообменник для подогрева рассола до 70—
€0 °C; 2 — донасытитель; 3 — центробежный насос;
4 — теплообменник для подогрева рассола до
90—9В °C; 5 — нейтрализатор рассола.
/
ния выпарки электроли-
тических щелоков. Схема
установки для донасыще-
ния очищенного рассола
показана на рис. 4-15.
Очищенный рассол
после фильтров подогре-
вают до 70—80 °C и по-
дают в донасытитель. При
этом концентрация NaCl
в рассоле увеличивается
до 320—330 г/л. Этот рас-
сол подогревают во втором
теплообменнике до 90—
98 °C, чтобы исключить
кристаллизацию рассола
при частичном его охла-
ждении в трубопроводах
на пути к электролизерам. При донасыщении с обратной солью
в рассол вводится дополнительно некоторое количество щелочи,
поэтому нейтрализацию рассола соляной кислотой необходимо
проводить после донасыщения.
Такой способ донасыщения рассола связан с необходимостью
транспортировать твердую соль в донасытители. Повышение кон-
центрации соли в рассоле, поступающем на электролиз, может быть
достигнуто также за счет частичной выпарки рассола с обычной кон-
центрацией в нем поваренной соли (310—312 г/л).
Чтобы исключить выпадение кристаллов соли, должна быть
осуществлена схема автоматического регулирования количества
подводимого к рассолу тепла, обеспечивающая поддержание опре-
деленной концентрации NaOl в выходящем из выпарной установки
рассоле.
На некоторых заводах до настоящего времени используется пе-
риодическая схема очистки рассола, в которую входят те же стадии
процесса, что и в непрерывной схеме, однако смешение реагирую-
щих растворов и осветление рассола производится периодически
в реакционных баках-осветлителях. После смешения сырого рассола
с обратным карбонизованным рассолом или с раствором соды избы-
216
н
точную щелочность нейтрализуют соляной кислотой и дают осадку
отстояться. Осветление продолжается от 6 до 18 ч в зависимости
от состава примесей и других условий. Осадки от предыдущих опе-
раций используются в качестве контактной массы. После 15—20
операций баки очищают от накопившегося шлама. Периодическая
схема очистки требует очень громоздкой аппаратуры, в частности
большого числа емкостей для рассола. По подсчетам [4], для завода
производительностью 500 т хлора в сутки при применении периоди-
ческой схемы очистки рассола необходима общая емкость баков
около 19 000 м3 для хранения сырого, обратного, очищенного рас-
сола и для проведения реакции осаждения примесей и осветления
очищенного рассола. Поэтому периодическая схема очистки рас-
сола сохраняется только на старых заводах, тогда как новые и ре-
конструируемые хлорные заводы используют непрерывные схемы
очистки рассола.
Практически во всех цехах электролиза с диафрагмой отказа-
лись от очистки рассола от сульфатов с помощью ВаС12. Вывод
из рассольного цикла накапливающихся там сульфатов обычно
производится в процессе выпарки электролитических щелоков,
где на второй ступени выпарки сульфаты выпадают вместе с пова-
ренной солью. Схема вывода сульфатов из цикла при упаривании
растворов будет рассмотрена ниже.
Для очистки рассола от сульфатов может быть использован каль-
циевый метод [50]. Для осаждения ионов SOf" к очищенному рас-
солу добавляют раствор СаС12
Na8SO4+CaG]8+2H8O —GaSO4.2H2O + 2NaCl (4.9)
Растворимость сульфата кальция в растворах поваренной соли
зависит от концентрации NaCl и сильно снижается с ростом темпе-
ратуры.
В табл. 4-7 приведены данные о зависимости растворимости
сульфата кальция в растворах поваренной соли от температуры,
а в табл. 4-8 — от концентрации NaCl.
Таблица 4-7. Влияние температуры на растворимость гипса в растворе
280 г/л NaCl
Темпера-
тура,
Содержание, г/л
CaSO* Са2+ so 2- 4
Темпера-
тура,
°C
Содержание, г/л
CaS04 Са2+ SO 2- 4
5
25
50
10,35
6,53
6,45
3,05
1,93
1,90
60 6,31 1,86 4,45
70 6,16 1,81 4,35
90 5,67 1,67 4,00
С изменением pH раствора от 2 до 10 растворимость гипса в рас-
творах поваренной соли практически не меняется. Остаточное со-
держание иона SO*" зависит от избытка хлористого кальция,
217
Таблица 4-8. Растворимость гипса в растворах NaCl при 25 9С
Концен- трация NaCl, г/л Содержание, г/л Концен- трация NaCl, г/л Содержание, г/р
CaSO4 Са*+ SO 2- 4 CaSCU Са2+ so?- 4
119,9
138,7
154,1
168,2
7,22
7,24
7,23
7,16
2,16
2,17
2,17
2,15
5^06
5,07
5,06
5,01
180,7
231,5
283,5
317,2
7,06
6,65
6,10
5,82
2,21
1,99
1,83
1,75
4,95
4,66
4,27
4,07
©веденного для очистки. Ниже приведены равновесные концентрации
(в г/л) ионов Са2+ и SO** в растворе 300 г/л NaCl при 20 °C:
•Са2+
sor
£а2+
SOJ"
• • 0,8
. ♦ 9,6
, . 1,85
» t 3,50
1,12
8,30
2,30
2,70
1,29
7,70
3,09
2,10
1,33
6,40
4,20
1,50
1,46 1,66
4,80 4,10
5,60 7,0
1,0 0,7
Чтобы обеспечить более глубокую очистку рассола от сульфатов,
в рассоле необходимо поддерживать высокую концентрацию каль-
ция, что влечет за собой увеличение расхода как хлористого кальция,
так и соды на последующую очистку рассола от ионов кальция.
Ниже приведено изменение удельного расхода СаС12 й Na2CO3
(в кг на 1 кг выведенного в осадок SO*-) при исходной концентрации
20 г/л SO*' в рассоле от глубины очистки рассола от SO*':
Содержание
в рассоле, г/л
Ga2+ SOJ'
У д е л ь й
расход, кг/кг
СаС12 NasCO#
0,8 9,6
1,12 8,3
1,29
1,33
1,46
1,66
1,38
1,43
1,45
1,43
1,45
1,45
0,20
0,25
0,28
0,26
0,25
0,28
Содержание Удельный
в рассоле, г/л расход, кг/кг
Са2+ so}- СаС12 Na2CO9
1,85 3,5 1,50 0,30
2,30 2,7 1,50 0,35
3,09 ' 2,1 1,64 0,46
4,20 1,5 1,79 0,64
5,60 1,0 1,98 0,77
7,00 0,7 2,6 0,96
При одной и той же глубине очистки рассола от ионов SO*- удель-
ные расходы СаС12 и Na 2СО3 зависят от исходной концентрации
сульфата в подвергаемом очистке растворе.
Ниже приведены данные об удельном расходе реагентов (в кг/кг)
в зависимости от исходной концентрации сульфатов:
Исходная концентрация SOJ’, г/л
Удельный расход, кг/кг
СаС12.......................
Na«CO4 . ...............
11 20 30 40 - 50
J I-
1,81 1,45 1,32 1,27 1,25
0,64 0,25 0,15 0,11 0,086
* Содержание в очищенном рассоле: Са2+ — 1,46 г/л; SO}
Для экономии реагентов целесообразно предварительное кон-
центрирование сульфатов в растворе, что может быть осуществлено
218
II
1'
> процессе выпаривания электролитических щелоков или растворов
поваренной соли.
В зависимости от температуры изменяется модификационный
состав осадка, как это видно из табл. 4-9.
Таблица 4 9- Модификационный состав осадка CaSO4 при различных
температурах
Температура, °C Состав осадка, вес. % Содержание кристаллизационной влаги, %
GeSO< • 2 О CaSO4*0,5HtO
20—25 100 20,86
45—55 92,27 7,73 19,79
65—75 54,55 45,45 14,24
II
Кальциевый метод очистки рассолов от сульфатов усложняется
возможностью гипсования аппаратуры й трубопроводов. Для пре-
дотвращения гипсования предложено [50] вводить ретурный шлам
в качестве затравки, снимающей пересыщение в растворе: количество
^затравки составляет 6 мг/г выводимого SOV- Добавление 3 мг/л
полиакриламида (ПАА) предотвращает схватывание частиц осадка
между собой и облегчает отстаивание осадка. Отстойные свойства
получаемого осадка зависят от pH среды. Для хлоркальциевой
очистки рассола приемлемы те же условия, что и при содово-каусти-
ческой: щелочность ОД—0,3 г/л NaOH и температура 45—55 °C.
В слабокислых растворах (содержание НС1 около 0,13 г/л) на-
блюдается ухудшение отстойных свойств осадка.
Предложена также очистка рассола от ионов БО^на ионообмен-
ных смолах [51]. Рассол, содержащий ионы SO®', пропускают через
^катионит в кальциевой или бариевой форме. Сообщений об исполь-
зовании таких методов в промышленности не было.
Для очистки растворов хлористого калия могут применяться
схемы и аппаратура, используемые для очистки растворов NaCl.
Необходимо учитывать лишь такие особенности растворов хлористого
калия, как зависимость растворимости КС1 от температуры и повы-
енную растворимость карбоната кальция и гидроокиси магния
в растворах КС1 по сравнению с растворами поваренной соли. При
обычном режиме очистки в очищенном рассоле остается до 8—10 мг/л
кальция и до 2—4 мг/л магния. Если повышенное содержание иона
Натрия в рассоле нежелательно, применяют карбонизацию обрат-
ного рассола или заменяют соду поташом.
:4:
Очистка рассола для электролиза с ртутным катодом
В зависимости от источника соли и других условий применяются
различные варианты схем и аппаратуры для приготовления и очистки
рассола для электролиза с ртутным катодом. Большинство этих
схем можно объединить в две группы: схемы на природной соли
219
&
и на чистой соли, содержащей небольшие количества вредных при-
месей.
При использовании для донасыщения анолита твердой природ-
ной соли поток анолита загрязняется примесями, содержащимися
в соли, и подлежит очистке от них. Обычно обедненный анолит со-
держит 260—270 г/л хлорида натрия, 0,3—0,5 г/л активного хлора
и до 10—20 мг/л ртути в виде ее хлоридов. При частых остановках
электролизеров содержание ртути в анолите может возрасти до 40—
60 мг/л и более. Температура обедненного анолита обычно составляет
75—85 °C. Анолит донасыщается до концентрации 305—310 г/л NaCl
и очищается от примесей.
* Ртутный электролиз очень чувствителен даже к небольшим за-
грязнениям рассола твердыми взвесями, которые, осаждаясь на
поверхности ртути, могут усиливать выделение водорода, загрязняю-
щего хлор. Поэтому рассол перед подачей на 'электролиз подвер-
гается обязательно одной, а часто и двум последовательным фильтра-
циям. В процессе двойной фильтрации из рассола частично выводятся
также тонкие взвеси амальгамных ядов. Обычно циркулирующий
рассол надо охлаждать, так как в электролизер необходимо подавать
рассол при температуре не выше 55—65 °C.
Активный хлор, содержащийся в анолите из электролизеров,
мешает осаждению примесей в ходе очистки. Кроме того, при работе
с таким рассолом аппаратура должна быть герметична и выполнена
из коррозионно-стойких материалов. Поэтому при донасыщении
анолита твердой природной солью обычно весь его поток подвер-
гается дехлорированию.
* Количество хлора, растворенного в анолите, составляет около
2% от производимого в электролизерах. Поэтому при дехлори-
ровании стремятся возвратить хлор для полезного использо-
вания.
* Для дехлорирования анолит подкисляют до содержания 0,1 —
0,2 г/л НС1, что предотвращает гидролиз хлора, и удаляют хлор
в вакууме в насадочных колоннах при остаточном давлении 260J—
360 мм рт. ст. При этом анолит вскипает и содержание хлора в нем
снижается до 30—50 мг/л. Отсасываемый хлор вместе с парами воды
подается в общий коллектор хлора на охлаждение и сушку. С целью
дальнейшего уменьшения содержания актирного хлора анолит под-
вергается отдувке воздухом в гуммированной насадочной колонне.
Расход воздуха составляет 1—1,5 м3/м® анолита, в последнем остается
10—20 мг/л активного хлора.
Воздух от продувки, Содержащий небольшое количество хлора,
пропускают через санитарную колонну, орошаемую раствором NaOH
или известкового молока. Ёсли имеется на месте потребитель раство-
ров гипохлорита кальция, процесс обесхлоривания может быть упро-
щен. После подкисления и частичного выделения хлора анолит может
быть подан в колонну для отдувки воздухом. При этом около 1,5—
2,0% производимого хлора Хбудет перерабатываться на растворы
гипохлорита.
Ull
(4.10)
(4.Н)
(4.12)
Для окончательного обесхлоривания используются химические
методы связывания хлора. Для этой цели наиболее широко приме-
няется обработка анолита сульфидом натрия
4С12 + Na2S + 4Н2О = Na2SOi+8НС1
ЗС12 + Na2S + ЗН2О = Na2SO3 + 6НС1*
Cl2 + Na2S = 2NaCl + S
Реакция (4.10) протекает преимущественно в кислой среде в из-
бытке хлора, а реакция (4.12) — в щелочной и в избытке сульфида
натрия. »
Вместо Na2S можно также применять NaHSO3, Na2SO3, Н2О2
и другие восстановители. Практическое применение нашел способ [52,
53] разрушения остатков хлора в анолите с помощью кускового
графита или на активированном угле
С+2С12 + 2Н2О - СО2 + 4HQ. (4-13)
Обесхлоривание на активированном угле проходит быстро
и до конца. Так, для полного обесхлоривания при 60—75 °C доста-
точно нескольких секунд контакта анолита с углем. Одновременно
с обесхлориванием анолита на активированном угле происходит
сорбция примесей ртутных солей и амальгамных ядов, содержащихся
в анолите [54]. При этом ртуть и активный хлор полнее поглощаются
активированным углем с анионообменными свойствами, а примеси
пятивалентного ванадия — углем с полифункциональными кати-
онообменными свойствами.
В процессе химического дехлорирования анолита с помощью
сульфида натрия происходит осаждение ртути в виде сернистых со-
единений. Сульфид ртути можно улавливать и далее регенерировать.
Однако фильтрование коллоидного осадка сульфида ртути затрудни-
тельно. Поэтому обычно сернистую ртуть не улавливают, и часть
ртути теряется со шламами на стадии донасыщения и очистки рас-
сола. Потери ртути могут составлять 120—150 г/т NaOH. Особенно
велики потери ртути при повышении ее содержания в анолите бо-
лее 20 мг/л. Преимуществом сульфидного обесхлоривания анолита
является одновременная очистка рассола от амальгамных ядов,
значительное количество которых выводится в виде сульфидов в оса-
док.
Предлагали также проводить электрохимическое восстановление
активного хлора на катоде в диафрагменном электролизере, на
аноде которого должен был выделиться хлор [55, 56].
Принципиальная схема рассольного цикла при работе на при-
родной соли показана на рис. 4-16. Схема предусматривает трех-
ступенчатое дехлорирование анолита вакуумированием, продувкой
воздухом и обработкой сульфидом натрия. В большинстве случаев
ограничиваются двухступенчатой схемой обесхлоривания анолита:
вакуумированием или отдувкой на первой стадии и химическим
обесхлориванием — на второй. Там, где не требуется полного
221
.1*
обесхлоривания, иногда ограничиваются только первой стадией
физического обесхлоривания и на донасыщении подают анолит,;
содержащий 30—50 мг/л активного хлора.
Показанная на схеме стадия фильтрации осадка HgS мало эффек-
тивна, осадок HgS практически не отделяется на фильтре, смеши-
вается со шламами очистки и пропадает. Поэтому на многих
Рис. 4-16. Принципиальная схема рассольного цикла в производстве хлора
в электролизерах с ртутным катодом на природной соли:
1 — осушитель хлора; 2 — холодильник хлора; 3 — склад — растворитель соли; 4 — осве-
тлитель рассола; 5 — фильтр для рассола; 6 — напорный бак; 7 — аппарат для химического
обесхлоривания; 3 — аппарат для отдувки хлора воздухом; 9 — вакуумная колонна;
10 — вакуум-насос; 11 — электролизер; 12 — разлагатель; 13 — ртутный насос; 14 —
башня отмывки водорода водой; 15 — башня отмывки Н2 щелочью; 16 — башня промывки
Н4 хлорной водой; 17— аппарат для сепарации щелочи от водорода; 18 — аппарат для се-
парации анолита от хлора.
предприятиях для снижения потерь ртути отказались от химического
обесхлоривания сульфидом натрия и ограничиваются лишь вакууми-
рованием или отдувкой воздухом. При этом всю аппаратуру и ком- з
муникации отделения очистки выполняют из коррозионно-стойких 1
материалов. При работе по такой схеме потери ртути сокращаются, >
так как содержащаяся в анолите в виде сулемы ртуть проходит без |
222 f
ю
Содержание N a2SO4t г/л
4-17. Растворимость сульфата
Рис.
кальция в насыщенных растворах
NaCl в присутствии сульфата нат-
рия:
1 — при 20 °C; 2 — при 40 °C; 3 — при
60 °C; 4 — при 80 °C.
сб<Л
изменения стадию очистки рассола и возвращается вновь на элек-
тролиз.
Предложено точно дозировать сульфид натрия в количестве,
необходимом только для восстановления активного хлора без обра-
зования сульфидов ртути [56], однако такое точное дозирование
затруднительно в промышленных условиях. Полное обесхлорива-
ние анолита без осаждения ртути может быть осуществлено на акти-
вированном угле.
При работе без осаждения ртути сульфидом натрия производство
становится более чувствительным к отравлению амальгамными ядами,
так как они не выводятся с суль-
фидами.
Дальнейшее усовершенствова-
ние схемы заключается в прове-
дении частичной очистки рассола
от кальция. Поскольку присут-
£ ствие солей кальция само по себе
t не приводит к нарушению про-
цесса электролиза, возможна ра-
бота на рассоле с значительным
содержанием кальция — до 1,1 —
1,5 г/л — при обеспечении надеж-
ной очистки от магния, железа и
других примесей.
Растворимость сульфата каль-
ция в насыщенных растворах по-
; варенной соли снижается с ро-
? стом концентрации сульфата натрия, как это видно из рис. 4-17.
> Изменяя концентрацию Na2SO4 в рассоле, можна регулировать
содержание кальция в насыщенном по CaSO4 рассоле.
f Работа на рассоле, насыщенном CaSO4, позволяет предотвратить
f переход в раствор кальция из соли, если он содержится в ней в виде
V ангидрита. Если же кальций находится в поваренной соли в виде
хлорида, необходимо подавать дополнительно сульфат натрия для
связывания кальция в виде CaSO4. Если в поваренной соли имеется
избыток ионов SO*’ по сравнению с содержанием кальция, следует
добавлять СаС12 в количестве, эквивалентном избытку ионов SO*-.
Работа цеха электролиза на растворе, насыщенном CaSO4, очень
неустойчива вследствие возможного выделения кристаллов гипса
при местном повышении температуры и возрастания чувствитель-
ности процесса электролиза к влиянию примесей магния, железа
и амальгамных ядов. Поэтому на некоторых отечественных заводах
работают с неполной очисткой рассола от кальция. Перед донасыще-
нием к анолиту добавляют раствор СаС12. Тогда в насыщенном по
CaSO4 растворе, содержащем 7—8 г/л SO*-, исключена возможность
растворения примесей ангидрита, присутствующего в поваренной
соли. После донасыщения кальций частично осаждается содой,
чтобы получать рассол с насыщением по ангидриту примерно на
80—85%. При этом концентрация ионов Са2+ может поддерживаться
не более 1,0—1,2 г/л без нарушения работы электролиза [57].
При высоком содержании сульфата натрия в исходном рассоле ионы
SO^ могут частично осаждаться хлористым кальцием с последующей
очисткой всего потока рассола от Са2+ и Mg2+. Такая схема очистки
применяется на некоторых заводах в Японии [58].
* При работе электролизеров с ртутным катодом на рассоле с вы-
соким содержанием CaSO4 рекомендуется [59] перед подачей в элек-
тролизер подкислять рассол до pH = 2—З.ине допускать загрязнения
рассола Mg, Fe и другими примесями.
Если для донасыщения используется чистая соль, содержащая
малое количество Са2+, Mg2+, SO? “и других примесей, применяется
другая схема очистки рассола. Такое положение имеет место при
снабжении производства выварочной чистой солью, получаемой
выпаркой очищенных подземных или искусственных рассолов. При
этом не требуется очистка всего потока анолита после донасыщения
его чистой солью и отпадают стадии полного дехлорирования и де-
меркуризации всего потока.
При выпарке рассола с целью получения твердой соли для до-
насыщения анолита необходимо предотвращать загрязнение соли
амальгамными ядами за счет продуктов коррозии аппаратуры и ком-
муникаций выпарной установки. При применении черной стали или
стали Х18Н9Т получается соль, загрязненная амальгамными ядами
и не пригодная без очистки для донасыщения анолита. Хорошая соль
без примеси амальгамных ядов получается при изготовлении аппа-
ратуры и трубопроводов выпарки из никеля или стали ЭИ-448.
Кроме того, при получении соли необходимо ее т
(ательно промывать
и
на центрифуге от маточного раствора, который обычно содер-
жит амальгамные яды (продукты коррозии материала аппара-
туры).
Поскольку даже с чистой солью постоянно вводятся некоторые
примеси, главным образом сульфаты, а в рассольном цикле тоже
могут накапливаться загрязнения в виде продуктов коррозии ано-
дов и материалов трубопроводов и аппаратуры, для поддержания
достаточной степени чистоты рассола из цикла необходимо выводить
5—10% рассола на очистку. Доля выводимого потока зависит от
чистоты отмывки соли, подаваемой на донасыщение, и количества
загрязнений, вносимых за счет процессов коррозии.
Рассол, подаваемый на очистку, дехлорируют, очищают от ртути и
ионов SOи кальция. Принципиальная схема такой установки пока-
зана на рис. 4-18. Если установка выпарки рассола для получения
соли расположена на предприятии, загрязненный поток анолита,
отбираемого для очистки, после дехлорирования и демеркуризации
может быть направлен на смешение со свежим рассолом из скважины,
поступающим на очистку.
На рис. 4-19 показан один из вариантов схемы выпарки рассола
с целью получения соли для донасыщения анолита электролизеров
с ртутным катодом.
224
ж
/
Для вывода сульфатов из цикла обогащенные сульфатами маточ-
ники возвращаются на первую ступень очистки, где часть сульфатов
выводится в виде CaSO4. В зависимости от содержания кальция
в рассол можно добавлять раствор СаС12. Вывод сульфатов из цикла
выпарки может быть также осуществлен до схеме, применяемой
в отделениях выпарки каустической соды.
Янолит из цеха
элентррлиза
Раствор
НС1
&
о.
Ct''
Раствор
ВаС12
Раствор
ЫЭдСОд
Раствор
НС1
Раствор Раст,вор
Na2S NaOH
8
BaSO4
Очищенный
анолит ни
донасыщениё
очистну
Сжатый воздух
Рис. 4-18. Схема обесхлоривании и очистки части потока анолита от ртути
и сульфатов: .
1 — расходные баки реактивов; 2 — смеситель; 3 — отдувочная колонна; 4 — аппарат для
химического обесхлоривания; 5 — бак обесхлореяного рассола; 6 — насосы; 7 — фильтр
для отделения HgS; 8 — отстойник для вывода сульфатов; 9 — приемный бак; 10 — фильтр
для отделения сульфата бария; 11 — нейтрализатор; 12 — приемный бак нейтрализован-
ного анолита.
к
Мй
Б J
И*
Значительно реже применяется концентрирование анолита путем
упаривания вместо донасыщения твердой солью. При этом соль,
требуемая для процесса, может вводиться в систему в виде очищен-
ного рассола. При концентрировании анолита весь его поток должен
подвергаться дехлорированию и освобождению от ртути, на очистку
же должен направляться только свежий рассол. Для предотвращения
чрезмерного накопления примесей в рассольном цикле часть цирку-
лирующего рассола следует постоянно выводить для очистки по
схеме, аналогично описанной ранее.
При использовании соли, содержащей примеси амальгамных
ядов, обычные способы очистки рассола оказываются недостаточными.
При химическом обесхлоривании анолита сернистым натрием про-
исходит удаление амальгамных ядов в виде сульфидов, однако хром
не образует малорастворимых сульфидов, а сульфиды ванадия, гер-
мания, молибдена осаждаются лишь частично, поэтому в зависи-
мости от состава загрязняющих примесей необходимо каждый раз под-
бирать режим осаждения тех или иных амальгамных ядов.
Для очистки рассола от амальгамных ядов рассол может быть
обработан амальгамой натрия [60, 62]. Г. И. Волков исследовал
амальгамную очистку в аппаратах скрубберного и барботажного
типов.
1’5 Заказ 843 225
S
1
s
о
0)
S
s
s
я
s
s
юмпжодмэ en иозэо^
шт/ано ni9HHaH£t(d2D£
о
Механизм и кинетика вытеснения натрием амальгамы примесей
различных металлов из раствора были исследованы с применением
радиоактивных изотрпов [63—66]. При использовании амальгам-
ной очистки рассола происходит частичное разложение амальгамы
и подщелачивание рассола с потерей 1—2% вырабатываемой щелочи.
Предлагались также способы очистки рассола от амальгамных
ядов с помощью ионообменных смол (анионитов и катионитов) [67,
68], однако они еще не нашли применения в промышленности.
Большой интерес представляет комбинирование ртутного метода
производства хлора с диафрагменным. При этом обратная соль ста-
дии упаривания электролитических щелоков диафрагменного элек-
тролиза может быть использована для донасыщения анолита электро-
лизеров с ртутным катодом. Отпадает необходимость строительства
специальных выпарных установок для рассола, и производство элек-
тролиза с ртутным катодом обеспечивается дешевой солью [69—71].
Однако обратная соль стадии упаривания электролитических
щелоков диафрагменного злектролиза обычно загрязнена амальгам-
ными ядами, попадающими в нее вследствие коррозионного разруше-
ния аппаратуры и трубопроводов во всем цикле электролиз—выпарка,
а также из графитовых анодов электролизеров с диафрагмой, часто
содержащих примеси ванадия и других амальгамных ядов.
Попытки очистить эту соль от амальгамных ядов промывкой ее
чистым или подкисленным рассолом, или рассолом, содержащим
активный хлор, не дали положительного результата. Примеси амаль-
гамных ядов при промывке в основном остаются в соли, что позволяет
предполагать, что они содержатся внутри кристаллов соли.
Хорошие результаты были получены при растворении обратной
соли в полностью обесхлоренном щелочном анолите [72] с последую-
щей тщательной фильтрацией рассола через насыпные фильтры.
Полученный рассол выдерживал амальгамную пробу.
При донасыщении не полностью обесхлоренного анолита обрат-
ной солью стадии выпарки электролитических щелоков диафрагмен-
ного электролиза образуются рассолы, загрязненные амальгамными
ядами и не пригодные для использования в электролизе с ртутным
катодом. Можно полагать, что амальгамные яды находятся в обрат-
ной соли диафрагменного электролиза в виде окислов низшей ва-
лентности и при растворении этой соли в щелочном полностью обес-
хлоренном анолите не переходят в раствор, а отделяются при тща-
тельном фильтровании. При растворении обратной соли в анолите,
содержащем активный хлор, происходит окисление амальгамных
ядов (в основном Сг и V) с образованием растворимых соединений,
которые делают рассол непригодным для электролиза с ртутным
катодом.
Как указывалось ранее, для донасыщения анолита, использу-
емого в электролизерах с ртутным катодом, его иногда закачивают
в скважины. Можно сочетать донасыщение анолита с очисткой его
от примесей магния путем подачи в скважину подщелоченного
анолита. В зависимости от состава пластов соли, в частности от
15* . 227'
j a
31
и
соотношения Gar2*: SOV, вместе с анолитом необходимо подавать в
скважины раствор СаС1а или Na2SO4, чтобы избежать чрезмерного
загрязнения рассола ионами SOf" или Ga2+.
При такой схеме получаемый рассол будет всегда насыщенным
по CaSO4. Работа цеха электролиза с ртутным катодом на таком
рассоле затруднительна. Особенно это сказывается при применении
металлических анодов [73].
Растворы поваренной соли коррозионно-активны, причем актив-
ность растворов хлористого калия выше хлористого натрия. Корро-
дирующее действие рассолов возрастает с понижением pH и проя-
вляется в большей степени на границе раздела.фаз или при переме-
шивании растворов воздухом [74]. В щелочных средах в присутствии
0,05—0,1 г/л NaOH скорость разрушения металлов в рассолах резко
снижается [75]. Особенно агрессивны рассолы, содержащие актив-
ный хлор. Коррозия трубопроводов и аппаратуры возрастает под
влиянием токов утечки [76]. Для предотвращения коррозионного
разрушения под влиянием рассола в сочетании с токами утечки при-
нимают меры по антикоррозионной защите трубопроводов и аппа-
ратуры. Применяют гуммированные трубопроводы и арматуру
и стальные защищенные гуммировкой или футерованные плиткой
емкости.
В тех местах, где исключены токи утечки, для хранения и пере-
качки щелочных рассолов при низких температурах применяют сталь-
ную аппаратуру и трубопроводы. В этих условиях хорошей корро-
зионной стойкостью отличаются нержавеющие стали Х18Н9Т [75].
При повышенных температурах (60—80 °C) черная сталь и сталь
Х18Н9Т нестойка^ в растворах хлоридов щелочных металлов. Хо-
рошую стойкость в среде хлорсодержащего анолита имеют трубо-
проводы из титана и фторопласта-4. В последнее время для этих
сред все шире стали применять стеклопластики на основе полиэфи-
ров [77], эпоксидных смол и других полимерных материалов [78].
Цементные детали и футеровка разрушаются растворами, содер-
жащими активный хлор, При этом значительные количества кальция
и магния переходят в раствор. В анолите диафрагменных электро-
лизеров, имеющих бетонные детали, были обнаружены значитель-
ные количества кальция (50 мг/л и более) и магния (15 мг/л и более).
В процессе приготовления и очистки рассола автоматизируются
отдельные стадии контроля и управления процессами но заданиям
и программам, разрабатываемым оператором [79]. Автоматизируется
работа насосов по перекачиванию рассола, поддержание темпера-
туры рассола при подогреве, карбонизация обратного рассола из
выпарки, дозирование и поддержание соотношения реагентов, пода-
ваемых на смешение, работа насыпных фильтров, стадия ^нейтрали-
зации рассола и некоторые другие операции. Для комплексной авто-
матизации всего процесса необходимы приборы автоматического
определения состава рассола и загрязняющих его примесей. Про-
должаются работы по дальнейшему усовершенствованию процессов
и аппаратуры очистки рассола [80].
228 .
и
f
fe
г-
к
>. •
1
' t
лг
r
fi-
ОХЛАЖДЕНИЕ, ОСУШКА И ПЕРЕКАЧКА ХЛОРА И ВОДОРОДА
Хлор й водород, продуцируемые в электролизерах, загрязнены
парами воды и содержат примеси. Водород, выходящий из разл^а-
гателей электролизеров с ртутным катодом, загрязнен значитель-
ными количествами паров ртути. Первичная обработка хлора вклю-
чает охлаждение, осушку, очистку газа от загрязняющих его при-
месей и компримирование для подачи хлора потребителям по трубо-
проводам. Для уменьшения разрушения аппаратуры, трубопроводов,
арматуры и контрольно-измерительных приборов хлор должен быть
тщательно высушен. До последнего времени считалось достаточным
понижение влажности хлора до 0,04 вес. %, однако в настоящее
время требования к осушке хлора возрастают, поэтому осушка
хлора производится до остаточной влажности 100 и даже 40 мг/м3,
что соответствует содержанию влаги от 0,0031 до 0,0013 вес. %.
Водород не агрессивен с точки зрения коррозионного разрушения.
Если у потребителя нет специальных требований к качеству водо-
рода, его осушка необходима только для предотвращения замерза-
ния трубопроводов в зимнее время. Водород из электролизеров с ртут-
ным катодом следует очищать от паров ртути.
ll
Охлаждение, ооушка и перекачка хлора
Влажный хлор, выходящий из электролизеров, помимо паров
воды и двуокиси углерода содержит также некоторое количество
воздуха, поступающего извне через случайные неплотности аппара-
туры и трубопроводов, примесь водорода, окись углерода, брызги
и туман анолита. Кроме того, хлор может содержать органические
продукты: четыреххлористый углерод, хлороформ, пента- и гекса-
хлорэтан, гексахлорбензол и другие хлорированные углеводороды,
образующиеся при разрушении графитовых анодов, а также в j>e-
зультате хлорирования материалов, применяемых для импрегниро-
вания анодов и анодных токоподводов или для уплотнения или за-
щиты от коррозии деталей электролизеров.
Если в соли, применяемой при электролизе, имеются бромиды,
в хлоре также будет присутствовать бром. /
Весьма нежелательна в хлоре примесь треххлористого азота
NG13, а также моно- и дихлораминов NH2G1 и NHG12. Эти продукты
могут образоваться в анодном пространстве электролизеров при
условии, что в рассоле содержится аммиак или ионы аммония
NHS + d2 = NH2C1 + HCL
(4.14)
NHS+2С12 = NHC12 + 2HCI
(4-15)
NH3 + ЗС12 = NC13 + ЗНС1
(4.16)
?'
г
Образование треххлористого азота или хлораминов возможно так-
же при охлаждении хлора в башнях смешения и наличии в воде ио-
нов аммония. Взаимодействие между солями аммония, растворенными
229
в воде, и хлором происходит быстро и, по-видимому, лимитируется
лишь скоростью растворения хлора в воде. Скорость взаимодей-
ствия мало зависит от температуры (в интервале 20—60 °C) и не за-
висит от аниона при аммонии [81].
Образующийся треххлористый азот практически полностью отду-
вается и уносится вместе с хлором. При сжижении хлора треххло-
ристый азот растворяется в жидком хлоре и при испарении послед-
него может накапливаться в остатке неиспаренного хлора, создавая
опасность взрыва.
Коэффициент разделения NC13 и С12 при испарении жидкого
хлора равен 6—10 [81]. Предложены различные методы очистки
хлора от треххлористого азота [82], однако наиболее целесообразно
проводить процесс получения хлора в условиях, исключающих
возможность загрязнения его примесями треххлористого азота.
Это обеспечивается при использовании рассола и воды, содержащих
менее 10 мг/л ионов аммония.
Чтобы предотвратить накапливание треххлористого азота в жид-
ком хлоре и возможные в связи с этим взрывы, нельзя допускать
присутствия аммония в воде, используемой для приготовления рас-
сола и охлаждения хлора в холодильниках смешения. При наличии
солей аммония в охлаждающей воде необходимо применять для охла-
ждения хлора поверхностные холодильники или холодильники сме-
шения с замкнутым циклом охлаждающей воды.
В хлоре из электролизеров с твердым катодом и диафрагмой
содержание водорода обычно не превышает 0,2—0,5%. Только
при нарушении режима отсасывания хлора и водорода из электро-
лизеров возможно повышение содержания водорода. При электролизе
с ртутным катодом содержание водорода в хлоре должно быть не
более 1%, но иногда превышает эту величину и может быть причи-
ной взрывов в электролизерах, коллекторах и аппаратуре отделе-
ния охлаждения и сушки хлора.
Состав примесей, содержащихся в хлоре, зависит от метода элек-
тролиза, качества рассола, материалов, применяемых для пропитки
анодов, изготовления и защиты деталей электролизеров, а также
От режима работы электролизера. *
На рис. 4-20 приведена схема охлаждения, сушки и комприми-
рования хлора, применяющаяся на многих заводах. В промышлен-
ности используются и другие аналогичные схемы, отличающиеся
решением тех или иных узлов. 1
Хлор, выделяющийся в электролизерах, содержит пары воды
в количестве, соответствующем парциальному давлению их над
раствором анолита. При рабочей температуре электролиза 90—95 °C
парциальное давление паров воды над анолитом и электролитиче-
скими щелоками составляет примерно 400—500 мм рт. ст. Влаго-
содержание 1 м3 хлора и водорода * в этих условиях составляет
от 0,9 до 1,5 кг.
* Объем газа приведен к нормальным условиям.
230
г
Устройства для удаления газов электролиза и первичной их
обработки предназначены для равномерного отсоса водорода и хлора
из электролизеров, их охлаждения и конденсации основного коли-
чества паров воды, дальнейшей осушки газов и компримирования
с последующим транспортированием по трубопроводам на перера-
ботку и использование.
Для охлаждения хлора и конденсации основного количества
паров воды ранее широко применялись керамические холодильники —
целляриусы, орошаемые снаружи водопроводной водой. Применя-
лись также холодильники из стеклянных труб. Вследствие низкого
коэффициента теплопередачи, громоздкости этих холодильников,
хрупкости, чувствительности к колебаниям температуры, трудности
поддержания герметичности многочисленных соединений, холодиль-
ники такого типа уступили место холодильникам смешения, в кото-
рых охлаждение хлора осуществляется в башнях, орошаемых холод-
ной водой, как это показано на рис. 4-20. Непосредственный контакт
между хлором и охлаждающей водой позволяет создать компактные
аппараты для охлаждения хлора и полнее очистить хлор от брызг
и тумана электролита. При противотоке газа и воды экономно рас-
ходуется охлаждающая вода и достигается хорошее охлаждение
хлора с малым перепадом температур между отходящим охлажден-
ным хлором и поступающей охлаждающей водой. Сообщается [83],
что при промывке и охлаждении хлора в башнях содержание хло-
ристого натрия снижается с 30 до 10 мг/м3 хлора, а количество хлор-
органических соединений — с 40 до 30 мг/м3.
Для лучшего охлаждения хлора иногда применяют двухступен-
чатое охлаждение его в холодильнике смешения. Обычную водо-
проводную воду, используемую для первичного охлаждения, подают
не на верх аппарата, а ниже. Верхнюю же часть холодильника
смешения орошают артезианской или захоложенной водой. Более
глубокое охлаждение хлора позволяет уменьшить затраты серной
кислоты на его сушку, однако охлаждение хлора ниже 10 °C не реко-
мендуется, чтобы не допустить образования кристаллов гидрата
хлора С12'8Н2О.
При использовании поверхностных холодильников для охла-
ждения хлора конденсат, образующийся в количестве до 400—450 л/т
хлора (в зависимости от начальной температуры хлора}, насыщен
хлором. Перед спуском в канализацию такой конденсат должен быть
обезврежен (обесхлорен) во избежание потерь хлора и отравления
атмосферы при выделении хлора из канализационных вод.
, При охлаждении хлора в холодильниках смешения количество
хлорной воды увеличивается в несколько раз, так как к образующе-
муся конденсату необходимо прибавить на 1 т С12 от Д,6 до 2,5 м3
воды, используемой для охлаждения хлора (в зависимости от тем-
пературы воды). При этом потери хлора с охлаждающей водой могут
достигать нескольких процентов от общего количества вырабатыва-
емого хлора. Для максимального снижения потерь хлора необходимо
поддерживать правильный режим работы холодильника смешения,
‘232
подавая минимально необходимое количество воды и поддерживая
возможно более высокую ее температуру (до 70 °C) после холодиль-
ника смешения. Чтобы снизить потери хлора и улучшить санитарные
условия работы, принимают меры к полезному использованию
растворенного хлора и обезвреживанйю сбрасываемой в канализа-
цию воды.
"Для удаления основного количества хлора из воды после холо-
дильников смешения ее нагревают до температуры кипения и отпа-
ривают хлор, Пары воды вместе с хлором возвращаются вновь в хо-
лодильник смешения. Для более полного удаления хлора, растворен-
ного в воде, ее подкисляют соляной кислотой. При этом реакция^
гидролиза хлора С12 + H2Oz± НС1 + НСЮ сдвигается влево.
Для окончательного обезвреживания хлорной воды перед сбро-
сом в канализацию’ ее обрабатывают веществами, легко вступающими
в реакцию с хлором, или пропускают горячую воду через аппарат
с насадкой из кусков графита [84]. В качестве насадки используют
остатки отработанных графитовых электродов. Содержание раство-
ренного хлора в воде после такой очистки снижается до 0,1—0,2 г/л.
Перед сбросом в канализацию воду многократно разбавляют
холодной водой, как это показано на рис. 4-20.
Обезвреживание отработанной воды облегчается при примене-
нии замкнутого цикла охлаждающей воды в холодильнике смеше-
ния. При такой схеме работы выходящая из него вода не сбрасы-
вается в канализацию, а поступает на охлаждение в один или два
последовательно включенных теплообменника й затем возвращается
вновь в холодильник смешения для охлаждения хлора.
Количество воды, циркулирующей в замкнутом цикле, будет
возрастать за счет конденсации паров воды, поступающих с хлором.
Часть хлорной воды, которую необходимо выводить из системы
для обесхлоривания, так же как и при использовании поверхност-
ных холодильников, равна количеству образующегося конденсата.
При такой схеме объем требуемой аппаратуры и затраты энерге-
тических ресурсов и материалов, расходуемых на обесхлориванио
сточных вод, сокращаются пропорционально уменьшению количества
отводимой хлорной воды. *
Введение в схему дополнительного теплообменника для охла-
ждения циркулирующей по кругу воды ухудшает условия охлажде-
ния хлора. Температура воды^ поступающей в холодильник смеше-
ния, будет на 5—8 °C выше температуры охлаждающей воды, посту-
пающей в жидкостной теплообменник, что приводит к соответствую-
щему повышению температуры хлора после холодильника смешения.
Применение замкнутого цикла воды в холодильнике смешения повы-
шает влагосодержание хлора, поступающего на последующую ста-
дию — осушку, и приводит к увеличению удельного расхода сер-
ной кислоты на осушку. Этот недостаток может быть устранен при-
менением воды, захоложенной на специальной установке. Наиболее
целесообразный вариант схемы охлаждения хлора зависит от кон-
кретных условий производства.
233
Холодильники смешения обычно выполняются в виде стальных
гуммированных башен диаметром 1,2 м и высотой 7—8 м, заполнен-
ных кольцами Рашига 50x50 мм. Колонна охлаждает до 90 т/сут
хлора от начальной температуры 75—85 °C. Конечная температура
зависит от температуры охлаждающей воды и обычно близка к 20—
25 °C. В йормально работающей колонне температура1 отходящей
воды на 4—6 °C ниже температуры поступающего хлора, а темпе-
ратура охлажденного хлора на выходе из колонны на 5—7 °C выше
охлаждающей воды.
Для охлаждения хлорной воды, циркулирующей в системе,
и нагревания ее на стадии обесхлоривания могут применяться гра-
фитовые или титановые [85] теплообменники, а также стеклянные
( трубчатые холодильники [49], корпус и трубные решетки которых
выполнены из специальной пластмассы. Производственная площадь,
. занимаемая титановыми холодильниками, в 8 раз меньше, чем при
установке обычных холодильников. При использовании таких холо-
дильников можно применять двухступенчатое охлаждение: первая
ступень — охлаждение водопроводной водой до 30—40 °C и вторая
ступень — водой, захоложенной на специальной установке до 10—
13 °C. Необходимо предусматривать возможность ухудшения коэф-
фициента теплопередачи таких теплообменников из-за забивки их
примесями, приносимыми в холодильник с хлором. Эти хлороргани-
ческие высокомолекулярные соединения, по-видимому являются
продуктами разрушения графитовых анодов и материалов, приме-
няемых для импрегнирования графитовых электродов.
Предложено также использовать для охлаждения хлора холод-
ный рассол, подаваемый на питание электролизеров [86]. Однако
надо учитывать, что в холодильниках смешения рассол будет разба-
вляться конденсатом, образующимся при охлаждении хлора.
Транспортирование влажного хлора связано с трудностями,
обусловленными сильной коррозией аппаратуры и трубопроводов
и образованием гидратов хлора при низких температурах. Поэтому
в цехах электролиза хлор всегда подвергается сушке.
Сушка хлора производится серной кислотой в скрубберных
аппаратах. После охлаждения и отделения брызг воды хлор про-
пускают последовательно через две-три колонны с насадкой из
керамических или фарфоровых колец Рашига, орошаемых серной
кислотой. В последнюю по ходу хлора колонну подается 98%-ная
серная кислота, которая по мере разбавления в процессе сушки пере-
дается из колонны в колонну навстречу потоку хлора. Из пер-
вой по ходу хлора колонны отводится 76—80%-ная серная ки-
слота^
Ранее на первой стадии осушки хлора применялись керамические
колонны, на последующих — чугунные. В последнее время осушка
хлора обычно производится в стальных скрубберах, выложенных ч
резиной, винидуррм или опанолом и защищенных дополнительно
кислотоупорным кирпичом, диабазовой плиткой или плитками из
стекла специального сорта.
234
Для хорошей осушки хлора плотность орошения насадки колонн
поддерживают на уровне 5—10 м3/(м2*ч), т. е. близкой к захлебыва-
нию. В колоннах серная кислота циркулирует с помощью центро-
бежного насоса. В первой по ходу газа колонне, где поглощается
основная часть влаги и выделяется большое количество тепла, сер-
ную кислоту охлаждают в холодильниках. Для достижения полной
осушки циркулирующую кислоту охлаждают и в следующих по
ходу газа колоннах. Для изготовления холодильников серной кис-^
лоты применяется сталь и высококремнистый чугун.
Предложено проводить осушку хлора под давлением. Для этого
влажный хлор компримируется в ротационном компрессоре, выпол-
ненном из титана и его сплавов, и затем осушается серной кислотой
в барботажных скрубберах [87]. Такая установка более компактна
по сравнению с обычными установками для осушки хлора,
однако сведения о применении ее в промышленности пока отсут-
ствуют.
Исследование процесса сушки хлора в насадочных колоннах
с кольцами Рашига 50x50 мм показало [88], что для серной кислоты
(начальная концентрация 93%, после осушки С12—75%) число единиц
переноса, необходимое для достижения заданной степени осушки, со-
ставляет при остаточной влажности 500 мг/м3 —3,12; при 50 мг/м3
5,55 и при 10 мг/м3 — 8. При скорости газа 0,22 м/с высота единицы
переноса равна 1,62 м. До последнего времени подача свежей кислоты
и передача кислоты из одной колонны в другую выполнялась вруч-
ную. В настоящее время весь процесс сушки, включая и подачу
кислоты, начинают регулировать автоматически.
Отработанная кислота содержит до 25 г/л хлора. После отдувки
хлора кислота передается потребителям разбавленной кислоты или
поступает на концентрирование. Расход серной кислоты зависит
от температуры, до которой охлаждается хлор перед поступлением
на осушку, и составляет обычно от 15 до 30 кг/т хлора.*
В процессе осушки происходит взаимодействие между серной
кислотой и туманом и брызгами электролита в хлоре, образуются
мелко раздробленный твердый хлористый натрий и сульфат натрия,
которые могут увлекаться хлором вместе с туманом серной ки-
слоты.
Сухой хлор содержит не более 0,04% влаги (0,4 г/кг хлора). Если
далее для компримирования хлора применяются компрессоры с за-
полнением или смазкой их серной кислотой, в процессе комприми-
рования может происходить увлажнение хлора в тех случаях, когда
парциальное давление паров воды над кислотой в компрессоре при
70—80 °C выше, чем над холодной кислотой в последней по ходу газа
сушильной колонне. Поэтому на компрессоры подается 96—99%-ная
серная кислота. При обработке компримированного хлора охлажден-
ной кислотой в колоннах или аппаратах барботажного типа содержа-
ние влаги в хлоре может составлять менее 0,002—0,003% [89].
Большой интерес представляет возможность осушки хлора сер-
ной кислотой в пенных аппаратах. Такие аппараты находят
/ ’ 235
применение в промышленности для проведения различных сорбцион-
ных процессов. Они позволяют интенсифицировать процессы сушки и
сократить объем и стоимость аппаратуры и производственных зданий.
Однако следует учитывать сложности в работе пенных сорбционных
аппаратов при значительных изменениях нагрузки на аппарат,
наблюдаемых, например, при пуске цехов.
Несмотря на то что широко распространенные метод и аппаратура
для сушки хлора в колоннах с насадкой, орошаемых серной кислотой,
показали себя достаточно надежными и удобными в работе, не прекра-
щаются поиски новых аппаратурных решений. Одним из таких
направлений является проведение процесса сушки хлора распылен-
ной серной кислотой. Распыление кислоты может осуществляться
механически с помощью специальных устройств для распыления
кислоты, путем ударного слияния потоков [90] или в результате
। эффективного смешения потоков газообразного хлора и серной ки-
слоты, поступающей на осушку газа в аппаратах типа эжекто-
ров [91]. В обоик случаях сильно сокращается объем аппаратуры
для сушки. Особенно компактны аппараты типа эжектора. Однако
в них сопротивление прохождению газообразного хлора значи-
тельно выше, чем при сушке в аппаратах типа колонн. Насколько
Широко эти способы осушки найдут применение в хлорной промыш-
ленности, будет зависеть от успешного решения вопросов надёжности
л экономичности работы новой аппаратуры.
Предложена также очистка хлора от NC13 и одновременная его
осушка взаимодействием с соляной кислотой при пониженных
(до —30 °C) температурах [92].
Для перекачивания и компримирования хлора ранее применялись
компрессоры с сернокислотным заполнением типа РЖК или НЭШ;
в последнее время используются преимущественно турбокомпрессоры
большой производительности. В последнем случае необходима очи-
стка хлора от твердых и капельно-жидких примесей: тумана серной
кислоты, твердых частиц хлорида и сульфата натрия, хлоррргани-
ческих аэрозолей, хлорного железа и др. Такая очистка предотвра-
щает возможность образования отложений на поверхностях турбо-
компрессоров, трубопроводов, арматуры и контрольно-измеритель-
ных приборов и обеспечивает хорошие условия для эксплуа-
тации.
Для большого количества потребителей хлор после очистки от-
твердых и жидких загрязнений пригоден без дальнейшей его обра-
ботки.
Содержание взвешенных частиц в хлоре после сушильных башен
зависит как от условий проведения электролиза, так и от режима
охлаждения и сушки хлора. Содержание аэрозолей в осушенном
хлоре колеблется от 20 до 100 мг/м3, причем содержание тумана
серной кислоты обычно не превышает 10 мг/м3. Частицы аэрозолей
имеют диаметр менее 10 мкм, большая часть из них размером ме-
нее 0,5 мкм. Частички аэрозолей состоят в основном из хлорида
и сульфата натрия. Ниже приведен состав осадка, полученного
/
при очистке хлора фильтрованием через тонковолокнистый фильтр
(в %):
NaCl . . . 74,5
Na2SO4 - • 18,5
H2SO4 3,1
Fe ..................... 0,02
Потери при высушивании . . 2,7
Прочее 1,18
Для очистки хлора от взвесей газ пропускают через аппараты
с инертной насадкой, насадкой из железных стружек или рукавные
фильтры. Эффективность таких аппаратов невелика. В последнее
время разработаны и успешно применяются в промышленности
различные конструкции волокнистых фильтров, которые при соблю-
дении необходимых условий могут работать частично или полностью
как самоочищающиеся [93]. При применении таких фильтров для
очистки осушенного хлоргаза содержание взвесей может 'быть сни-
жено до 5 мг/м3 и менее, причем около половины остающихся в хлоре
примесей составляет туман серной кислоты.
Присутствие в виде примесей твердых частичек хлорида и суль-
фата натрия и других загрязнений приводит к постепенной забивке
фильтров, повышению их сопротивления и необходимости периоди-
ческой смены фильтрующего материала.
Чтобы обеспечить возможность самоочищения фильтра, предло-
жено проводить двукратное фильтрование хлора через волокнистые
фильтры.
Первая фильтрация проводится после охлаждения хлора. При
этом отделяется основное количество брызг и тумана электролита,
что исключает или резко сокращает возможность образования твер-
дых частичек хлорида и сульфата натрия на стадии сернокислотной
осушки хлора и обеспечивает возможность длительной работы
фильтров для отделения тумана серной кислоты от хлора без за-
бивки.
Имеется сообщения [94] о применении электростатических
фильтров для очисткй влажного хлора от капельно-жидких и твердых
загрязнений. Очистка влажного хлора от аэрозолей на электрофиль-
трах перед осушкой дает степень очистки до 99,5%, что предохраняет
насадку колонны сернокислотной сушки от забивания сульфатом
натрия и серную кислоту от загрязнения. Второй раз хлор филь-
труют после осушки от тумана серной кислоты на кассетных филь-
трах, заполненных стекловолокном. В этих условиях фильтры могут
длительное время работать без смены стекловолокна как самоочи-
щающиеся. t
Применение электрофильтров затруднено необходимостью под-
бора коррозионно-стойких материалов для изготовления фильтра
и особенно электродов. Для работы во влажном хлоре достаточной
стойкостью обладает титан.
Для некоторых потребителей хлора требуется также его очистка
от примесей газообразных хлорорганических соединений и брома.
237
Предложено обрабатывать газ жидким хлором, например промывать
в колпачковых или насадочных колоннах [95].
Примеси извлекаются жидким хлором и концентрируются в кубо-
вой жидкости. Хлор отгоняется из кубовой жидкости и возвращается
на очистку, примеси остаются в остатке.
Остаток имеет следующий примерный состав (в %):
Хлороформ ........................ 47,3
Четыреххлористый углерод . . . 14,0
Трихдорацетилхлорид ........ 12,0
Пентахлорэтан ..................... 5,0
Гексахлорэтан .................... 18,0
Хлор . . . . . . . . . . .' . . . 3,0
Прочее ............................ 1,0
Применяется комбинированная очистка хлора от твердых и
жидких загрязнений вначале на волокнистых или электростати-
ческих фильтрах, а затем на сорбентах (активированный уголь
и др.) [83, 94].
В тех случаях, когда требуется высококонцентрированный хлор
без примесей газов, его очищают от инертных газов конденсацией
и последующим испарением. В случае необходимости испаренный
хлор могут подвергать специальной очистке.
Компрессоры для отсоса хлора и водорода обычно снабжаются
регуляторами, обеспечивающими поддержание постоянного давле-
ния в коллекторах хлора и водорода в цехе электролиза. В анбдном
пространстве электролизеров обычно поддерживают небольшой
вакуум от 5 до 25 мм вод. ст. В водородном коллекторе и катодном
пространстве электролизеров рекомендуется поддерживать неболь-
шое избыточное давление, во избежание подсоса воздуха и образо-
вания взрывоопасной смеси при случайном нарушении плотности
аппаратов или трубопроводов. Однако ряд заводов длительное время
работает с небольшим вакуумом в водородном пространстве электро-
лизеров, при этом каких-либо неудобств или неприятностей не
наблюдается.
Автоматическое регулирование давления (и вакуума) в хлорном
и водородном коллекторах может осуществляться как за счет бай-
пассирования компрессора, так и дросселирования газа на входе
в компрессор. Преимущественно используются схемы автоматиче-
ского регулирования с байпассированием компрессора. Отбор’ им-
пульса для регулирования производится из места соединения трубо-
проводов от групп электролизеров в магистральный хлоропровод.
В крупных цехах электролиза, где протяженность газовых ком-
муникаций велика, необходимо учитывать сопротивление по току
газа в трубопроводах и выбирать схему и диаметры трубопроводов
для хлора и водорода с таким расчетом, чтобы не иметь чрезмерно
большой разницы давления газов в разных электролизерах.
В трубопроводах для хлора и водорода, проложенных между
электролизерами и установками для охлаждения газов, возможно
образование конденсата за счет частичного охлаждения газов.
238
|Э Поэтому при укладке трубопроводов необходимо предусматривать
J уклоны и достаточное количество дренажных устройств для отвода
ж. конденсата. Конденсат из хлорного коллектора собирается и отво-
дится на установку для обесхлоривания хлорной воды.
||| Для влажного хлора применяются фаолитовые, стеклянные,
ЦЬ керамические или асбоцементные трубы, а также стальные гумми-
!|К рованные и титановые трубопроводы, арматура и аппаратура.
II В последнее время для этой цели используются стеклопластики,
t стойкие к хлору. Для сухого хлора устанавливается аппаратура,
; трубопроводы и арматура из стали. Для увеличения коррозионной
стойкости стальных поверхностей при возможных проскоках недо-
? статочно осушенного хлора рекомендуется никелировать детали
Ш компрессоров, трубопроводов и арматуры. При толщине никелевого
слоя 30 мкм такие детали сохраняют коррозионную стойкость
Ж при 20 °C в хлоре, влажность которого до 0,3%, и при 100 °C и влаж-
ности хлора до 0,8% [96].
1 W
'«ИкК;
V Охлаждение, очнотка н ооушка водорода
w Качество водорода в большей степени, чем хлора, зависит от
Ж метода производства. При производстве по методу электролиза
W с твердым катодом и диафрагмой водород обычно, помимо паров
Ж воды, может содержать щелочной туман и воздух в результате под-
» соса последнего через неплотности электролизеров и коммуникаций,
в В водород могут иногда попадать следы хлора, если имеются повреж-
i Ж дения диафрагмы и нарушения условий отсасывания хлора и водо-
; рода из электролизеров. В некоторых случаях в водороде обнару-
» живают значительное содержание хлорорганических примесей (до
нескольких десятков мг/м3), отдуваемых водородом из электролити-
Ж ческих щелоков. Чистота водорода должна быть не ниже 98% и
Ж содержание кислорода не более 0,5%.
Ж В производстве по методу электролиза с ртутным катодом водород
в выделяется в герметических разлагателях под небольшим избыточ-
но ным'давлением; содержание Н2 должно быть не ниже 98% и* О2 —
в не более 0,4%. Температура в разлагателе электролизера с ртутным
Ж катодом из-за значительных тепловыделений при разложении амаль-
® гамы достаточно высока и в мощных электролизерах может пре-
вышать 100 °C. Поэтому с водородом уносятся из разлагателя не
Ш только большие количества паров воды, но и пары ртути.
Л В современных мощных электролизерах непосредственно на раз-
Ж. лагателях амальгамы устанавливают обратные холодильники для
Я предварительного охлаждения водорода и конденсации и возвраще-
ния в разлагатель основного количества паров воды и ртути. Но даже
ж после такого холодильника водород остается сильно загрязненным
|Ж парами ртути. При 30—40 °C на выходе из холодильника электро-
Ж лизера водород содержит от 30 до 70 мг/м3 паров ртути.
ж На рис. 4-21 показана принципиальная схема охлаждения и
Ж очистки от ртути водорода, получаемого в цехах электролиза с ртут-
Ж. 239
ным катодом. Для охлаждений служат стальные теплообменники;
сконденсировавшаяся в них ртуть отводится вместе с конденсатом
и возвращается в цикл. Дальнейшая очистка водорода от ртути
осуществляется в насадочных скрубберах, орошаемых анолитом
из электролизеров, содержащим активный хлор. При зтом пары ртути
взаимодействуют с хлором, образующаяся сулема увлекается оро-
шаемым анолитом. После этого водород промывается в щелочном
скруббере от возможных примесей хлора, увлекаемых им из ано-
лита, и в водяном скруббере отделяется от брызг щелочи. Такая
Раствор
щелочи из цеха
электролиза
Дно лит на
донасыщение
Янолит^
содержащий
хлор
Водород из
электролизеров
Отработанный
раствор щелочи
схема позволяет снизить содержание ртути в водороде примерно
до 0,1 мг/м3.
/47 И
Boon.
Рис. 4-21. Схема охлаждения, очистки от ртути и компримирования водорода
1 — холодильник водорода; 2 — отделители конденсата и ртути; 3 — турбогазодувка для
водорода; 4 — насосы; 5 —г бак для анолита; в — ^колонна для промывки водорода анолитом;
7 — бак для щелочи; 8 — колонка для промывки водорода щелочью; 9 — колонка для про-
мывки водорода водой; 10 — водородный компрессор; 11 — водоотделитель; 12 —холо-
дильник; 13 — огнепрегрйдитель; 14 — распределительная гребенка водорода.
• f
Если после такой очистки провести дополнительно адсорбцию
паров ртути из водорода на активированном угле, содержание ртути
можно снизить до 0,01 мг/м3 Н3 [97]. .
В последние годы большое внимание уделяется очистке водорода
от паров ртути. Сообщается [98] о разработке метода промывки
водорода раствором неорганических соединений до содержания
ртути 0,005 мг/м3 Н3. Предложена также схема очистки водорода
от ртути промывкой газа в насадочной колонне 20%-ной серной
кислотой, содержащей 1—2 г/л перманганата калия. Указывается,
что при этом содержание ртути становится менее 0,001 мг/м3 Н2 [99].
Наиболее эффективна очистка водорода и воздуха от ртути сорб-
ционными методами. Фирма «Монсато» предложила [100] сорбент
для ртути, снижающий ее содержание в газе до 1 части на миллиард.
Перед подачей газа на сорбцию егб предварительно фильтруют для
удаления тумана ртути.
Разработаны установки, позволяющие снизить потери ртути
с.водородом и воздухом до 45 г/сут для завода мощностью по С13
270 т/сут [101].
Возможна также очистка водорода и от ртути глубоким охлажде-
нием водорода. Для снижения содержания ртути в водороде до
санитарной нормы, равной 0,01 мг/м3, необходимо охладить водород
до —45 °C. При этой температуре парциальное давление паров ртути
соответствует примерно ее содержанию в виде паров 0,01 мг/м3 газа.
Фактическое содержание ртути может быть значительно выше, так
как в водороде помимо паров ртути будут находиться мелкие ее
капельки в виде тумана. Освобождение водорода от этого ртутного
тумана затруднительно, , поэтому вымораживание паров ртути
редко применяется в промышленности для очистки газов.
Для очистки водорода от паров ртути можно использовать способ
сорбции ее на активированном угле, содержащем определенное
количество адсорбированного хлора. В последнем случае хлор,
адсорбированный на активированном угле, хлорирует пары ртути
с образованием сулемы. При этом достигается достаточно полная
очистка водорода от паров ртути. Активированный уголь с анионо-
обменными свойствами может сорбировать ртути около 16 мг/г
угля [102]; для регенерации угля его обрабатывают 10%-нрй HNO3.
Применяются комбинированные схемы очистки, например охлажде-
ние и фильтрование через волокнистые фильтры с адсорбцией остав-
шегося\количества паров ртути активированным углем или другими
адсорбентами [103].
Перечисленные выше способы очистки водорода от ртути могут
быть использованы также для очистки воздуха от ртути.
Водород из злектролизеров с твердым катодом и диафрагмой
обычно охлаждают в холодильниках смешения, аналогично хлору.
Небольшие количества брызг щелочи, уносимых с водородом, спо-
собствуют выпадению солей жесткости из охлаждающей воды и за-
бивке насадки колонн, которую необходимо периодически чистить.
Для очистки водорода от примеси хлора применяют щелочную про-
мывку водорода в насадочных скрубберах аналогично тому, как
это показано на - .рис.. 4-21. Хлорорганические примеси в водороде
разлагаются в присутствии катализатора.
В большинстве цехов осушка водорода не производится. В случае
необходимости сухой водород можно получить путем сернокислотной
осушки. Возможна также осушка водорода охлаждением его в скруб-
бере, орошаемом захоложенным раствором поваренной соли, а такжо
с помощью твердых осушителей (силикагель и др.).
Для компримирования влажного водорода применяют как рота-
ционные компрессоры с жидким поршнем типа РЖК, так и турбо-
водорбдодувки. Для компримирования сухого водорода применяют
турбоводорододувки.
II
ЭКСПЛУАТАЦИЯ ЦЕХОВ ЭЛЕКТРОЛИЗА
Объединение электролизеров в оерни -
Электролизеры для питания постоянным током соединяются
последовательно в серии. В зависимости от масштаба производства
и применяемого типа электролизеров количество их в серии может
16 заказ 843 241
быть различным. Ранее, когда применялись электролизеры малой:
мощности (на нагрузку 5—10 кА или ниже), для цехов даже средней
мощности необходимо было устанавливать несколько серий электро-
лизеров.
В настоящее времц нагрузка на электролизерах с твердым Катодом
возросла до 50—60 кА, а с ртутным катодом до 300—500 кА с тенден-
цией к дальнейшему ее росту, поэтому, как правило, цехи электро-
лиза с ртутным катодом оборудуются одной серией электролизеров.
Крупные цехи электролиза по методу хс диафрагмой оснащаются
также двумя или большим числом серий. При использовании
типовых полупроводниковых выпрямителей на напряжение постоян-
ного тока 425 В цех электролиза с электролизерами БГК-50 на на-
грузку 50 кА будет иметь мощность около 60 тыс. т хлора в год,
а цех электролиза, оборудованный серией электролизеров с ртутным
катодом типа Р-200 на нагрузку 200 кА, — мощность около 200 тыс. г
хлора в год.
Ранее для питания серий электролизеров постоянным током
применялись генераторы напряжением до 250—275 В. Однако
по мере развития техники преобразования переменного тока в по-
стоянный и совершенствования конструкций электролизеров воз-
росло и напряжение постоянного тока, применяемого для питания
серий электролизеров. Увеличение напряжения постоянного тока
позволяет снизить капитальные затраты на оборудование преобразо-
вательных подстанций и при применении ртутно-выпрямительных
агрегатов повышает коэффициент полезного действия преобразова-
тельной установки. Переход на более высокое напряжение постоян-
ного тока на электролитических установках был в значительной
степени обусловлен применением ртутно-выпрямительных агрегатов.
Однако с началом широкого использования механических и
особенно полупроводниковых выпрямительных агрегатов, работа-
ющих с высоким к. п. д. даже при низких напряжениях выпрямлен-
ного тока, стали применять более низкое напряжение в процессе
электролиза растворов хлоридов. В настоящее время напряжение
на серии чаще всего составляет от 200 до 450 В. Применение более
высокого напряжения связано с неудобствами при эксплуатации
цехов электролиза [104] и требует дополнительных затрат [105]
с целью обеспечения безопасности работы обслуживающего персонала
этих цехов, снижения токов утечки и устранения связанных с этим
коррозионных явлений.
При применении повышенного напряжения постоянного тока
необходимо увеличивать рабочие проходы между рядами электро-
лизеров, а также между электролизерами и строительными кон-
струкциями или аппаратами и трубопроводами, электрически соеди-
ненными с землей, устанавливать изолированные от земли площадки
для обслуживания аппаратуры или использовать -изоляционные
материалы для устройства полов и в строительных конструкциях,
расположенных у рабочих проходов или в непосредственной бли-
зости к электролизерам.
i 242
ч. ►
/
Для предотвращения поражения персонала током при обслужи-
вании электролизеров или при проведении работ по их ремонту
необходимо надежно изолировать мостовой кран, работать в резино-
вых ботах и перчатках, периодически проверяя их на электрическое
сопротивление, применять резиновые коврики, содержать в исправ-
ности изоляцию электролизеров и коммуникаций от земли, контро-
лировать состояние изоляции и др.
С повышением напряжения увеличивается утечка тока и усили-
ваются процессы коррозии трубопроводов, аппаратуры и строитель-
ных конструкций, а также повышаются требования к прерывателям
потока рассола и щелочи. Так как обычно не удается обеспечить
полный разрыв потоков рассола, поступающего на питание в электро-
лизеры, и щелочи, вытекающей из электролизеров, и всегда наблю-
даются утечки тока по коллекторам, подводящим и отводящим рассол
и щелока, необходимо предпринимать специальные меры для защиты
от коррозии трубопроводов и оборудования (коллекторов для рассола
и щелочи и подогревателей рассола). Практикуется также изготовле-
ние трубопроводов из диэлектриков или защита их слоем непроводя-
щего ток материала.
С целью защиты аппаратуры для переработки электролитических
щелоков и подготовки рассола от коррозии вследствие утечки тока
в трубопроводах для рассола и щелоков на выходе их из цеха элек-
тролиза устанавливаются заземленные вставки. Такие вставки
снимают и отводят в землю основные токи утечки из потока раствора
и в значительной степени защищают всю аппаратуру, установленную
далее на линии рассола и щелочи, от вредного действия токов утечки.
Особенно эффективно действие таких вставок нд подогревателях
рассола, расположенных в непосредственной близости от цеха
электролиза.
Заземленные вставки в трубопроводах выполняются из мате-
риала, стойкого в условиях анодной поляризации в растворах
ренной соли, например из искусственного графита. Для этой цели
могут быть с успехом использованы также и электроды из титана или
тантала с нанесенным на них слоем платины. Для эффективной
защиты от токов утечки заземленные вставки в трубопроводах должны
иметь достаточно развитую поверхность токоснимающих электродов.
При использовании полупроводниковых выпрямителей повыше-
ние напряжения более 200—450 В уже не дает существенных преиму-
ществ в работе преобразовательных подстанций с точки зрения
коэффициента полезного действия выпрямительного агрегата, и не-
удобства, возникающие в ходе эксплуатации электролизеров при
повышенном напряжении, являются решающими. Поэтому в "случае
использования полупроводниковых или механических выпрями-
тельных агрегатов цеха электролиза растворов хлоридов обычно
работают при напряжении не выше 400—450 В.
При использовании серий сравнительно маломощных электро-
лизеров типа БГК-13 нецелесообразно осуществлять питание постоян-
ным током каждой серии от самостоятельного источника постоянного
16* 243
нова-
и
7
тока, так как мощность современных преобразователей тока больше
мощности, необходимой для питания одной серии такцх электроли-
зеров. Число агрегатов выпрямительной подстанции в таких случаях
обычно не совпадает с числом серий электролизеров, поэтому приме-
няют параллельную работу выпрямительных агрегатов на общие
шины с питанием серий электролизеров параллельно от общих
шин.
При этом распределение нагрузки между сериями электролизеров,
будет определяться сопротивлением серий, т. е.. будет зависеть от
числа электролизеров, последовательно включенных в серии, и их
состояния. При такой системе питания за время работы серии элек-
тролизеров нагрузка на нее постоянно меняется вследствие измене-
ния сопротивления электролизеров в серии, обусловленного износом
графитовых анодов и старением диафрагмы. Это затрудняет и практи-
чески делает невозможной полную автоматизацию регулирования
режима работы электролизера, в частности питания электролизеров
рассолом, в количестве, обеспечивающем оптимальную степень,
превращения хлорида в гидроокись.
При описываемом способе питания серий постоянным током
в цехах электролиза по методу с диафрагмой на режим работы элек-
тролизеров благоприятно влияют одновременное снижение проте-
каемости диафрагмы, вследствие ее старения и забивки пор, и на-
грузки на серии во время работы за счет износа графитовых анодов
и увеличения электролитического сопротивления диафрагмы. Вы-
ключение серии электролизеров для ремрнта обычно производится
после того, как нагрузка на серии падает ниже определенной вели-
чины.
При применении современных мощных конструкций электролизе-
ров, когда цеха электролиза оборудуются одной или несколькими
сериями на большую нагрузку, целесообразно иметь независимое
питание постоянным током для каждой серии электролизеров. При
этом на серии может поддерживаться постоянная нагрузка, что со-
здает благоприятные условия для работы в стабильном режиме и авто-
матизации регулирования работы серии. Хотя напряжение на каж-
дом отдельном электролизере за тур работы электродов существенно
изменяется, общее напряжение на серии обычно колеблется в неболь-
ших пределах. Выключение электролизеров для ремонта производят
по одному или группами электролизеров равномерно, в течение всего
года. В новых цехах обычно начинают выключать для ремонта
электрлизеры до окончательного износа анодов. В некоторых случаях,
особенно при строительстве цехов на новых комбинатах и при отсут-
ствии достаточного числа потребителей хлора, стараются включать
в работу серию электролизеров частями, в несколько 'сроков, что
также способствует в дальнейшем созданию равномерности в выклю-
чении электролизеров для ремонта. При правильной организации
ремонта средний срок работы электролизеров в серии, а т^кже и
среднее напряжение на электролизере практически остаются постоян-
ными во время работы серии.
244
л
ft
Л
Размещение электролизеров
/ ' • ь
В цехах электролиза с твердым катодом и диафрагмой электроли-
зеры обычно располагаются в одноэтажном здании. На рис; 4-22
по казан один из в а
БГК-17 или
в группе. В
на стойках
коллекторы;
вания электролизеров и их деталей при монтаже цеха и ремонте
электролизеров, внизу укладываются сборные коллекторы электро-
литической щелочи. Для выключения электролизеров используются
тов расположения серии электролизеров
БГК-56. Электролизеры располагаются по 12—20 шт.
узком проходе между электролизерами одной группы
монтируются рассолопровод, хлорный^и водородный
в широких проходах, используемых для транспортиро-
к'- :
:i?
ft
*
г
я .
л
it
1
И
£
Рис. 4-22. Схема расположения электролизеров
БГК-17 или БГК-50 в зале электролиза:
.1 — зал электролиза; 2 — электролизеры; з — другие
отделения цеха; 4 преобразовательная подстанция.
V
d
обычно передвижные шунт-тележки, которые подводятся к электро-
лизеру и с помощью съемных накладок присоединяются к катодной
и анодной шинам двух соседних электролизеров с одной и другой
стороны от отключаемого электролизера. После этого включается
разъединитель шунт-тележки, шунтирующий электролизер.
По мере укрупнения электролизеров с диафрагмой шунт-тележки
становятся все более тяжелыми и громоздкими. Для ^уменьшения
веса и габаритов шунтирующих устройств применяются шины
и контакты с водяным охлаждением, тем не менее шунт-тележка
для выключения электролизеров на нагрузку 25—50 кА весит бо-
лее 500 кг. Подключение шунт-тележки к электролизерам связано
с затратой физического труда и плохо поддается автоматизации.
Поэтому для серии электролизеров на 50 кА и более целесообразно
применять установку стационарных разъединителей, как это имеет
место в цехах ртутного электролиза. При этом целесообразно распо-
лагать электролизеры на перекрытии второго этажа, тогда первый
этаж используется для прокладки ошиновки, трубопроводов и уста-
новки различного вспомогательного оборудования. Такое размещение
электролизеров несколько дороже, однако более удобно при эксплуа-
тации и позволяет избежать затраты тяжелого ручного труда на
245
операции включения и выключения электролизеров в связи с ре-
монтом.
В цехе электролиза проводится только сборка электролизера
из анодного комплекта, катодного блока и крышки или его разборка
при ремонте. Детали транспортируются в отделение ремонта и по
окончании его подаются на сборку в цех электролиза с помощью
мостового электрического крана й тележек. Все операции по ремонту
электролизеров проводятся вне цеха электролиза в специально обору-
дованном отделении.
После износа графитовых анодов электролизер выключается,
разбирается и его основные узлы передаются на ремонт. На месте
удаленного монтируется новый электролизер из заранее приготовлен-
ных узлов. Полная замена всех узлов электролизера требует около
двух часов [106].
В электролизерах БГК-17 и БГК-50 за период работы анодов,
в зависимости от применяемой плотности тока, диафрагму меняют
от одного до трех раз. Для этой цели удаляют катод со старой диа-
фрагмой и на его месте устанавливают заранее подготовленный
катод с вновь осажденной диафрагмой. Анодный комплект при
этом остается на месте [107, 108].
При полной замене всех частей электролизера у старого анодного
комплекта удаляются остатки старых графитовых анодов, бетонный
и асфальтовый защитные слои и, после проверки и зачистки поверх- 4
ности днища и контактов, монтируются с помощью шаблона новые
графитовые аноды. Поверхность анодных контактов защищают
слоем асфальтовой массы и слоем бетона. Подготавливается поверх-
ность стыка анодного комплекта с катодом. Старая диафрагма смы-
вается с катода сильной струей воды. В случае необходимости катод
£о старой диафрагмой обрабатывается раствором ингибированной
соляной кислоты. Затем на катоде осаждается новая диафрагма,
после чего диафрагма подвергается сушке. В случае необходимости
изготавливаются новые крышки.
При использовании малоизнашивающихся анодов объем работ
по ремонту электролизеров значительно сокращается. Срок пробега
электродов не менее двух лет, и эа этот период необходимо проводить
только замену или регенерацию диафрагмы. Частота замены или
регенерации диафрагмы зависит от применяемой плотности тока
и качества диафрагмы.
В цехах электролиза с ртутным катодом электролизеры распо-
лагают в серии чаще всего в виде буквы П и размещают на перекры-
тиях второго этажа. В нижнем этаже прокладывают все коммуни-
кации, а также ошиновку. Там же устанавливают разлагатели амаль-
гамы скрубберного типа, сборники каустической соды и анолита,
наносы и другое вспомогательное оборудование. Имеются цехи,
в которых электролизеры типа Сольве располагаются в многоэтажном
здании без проходов между электролизерами.
Электролизеры снабжены стационарными шунтирующими разъ-
единителями, позволяющими останавливать каждый электролизер
’
Me ’ *
B&* *
I без нарушения работы всей серии. В торце цехов электролиза с ртут-
|- ным катодом на втором этаже обычно оборудуется площадка для
I ремонта электролизеров и замены графитовых анодов. Для капиталь-
| ного ремонта и смены гуммировки электролизеры вывозят из цеха
| электролиза. Зал электролиза снабжается мощным электрическим
| краном для работ по монтажу и ремонту электролизеров. Краны
I в цехах электролиза с ртутным катодом и с диафрагмой должны
| быть во взрывобезопасном исполнении и надежно изолированы
i от земли.
| С целью удешевления капитального строительства стремятся
I укрупнять производственные здания, объединяют в одном корпусе
I ряд отделений. Так, практикуется объединение в одном корпусе
преобразовательной подстанции, зала электролиза, помещений охла-
| ждения, сушки и перекачивания хлора и водорода, сжижения хлора,
I центральной станции распределения хлора и других отделений.
I При такой компановке завода сокращаются территория застройки
| и протяженность коммуникаций, удешевляется строительство хлор-
I ного завода и облегчается обслуживание и эксплуатация произ-
। водства.
I Большой интерес представляет опыт эксплуатации цехов элек-
I тролиза, расположенных вне здания. На одном из заводов в нашей
I стране, в связи с необходимостью ремонта здания цеха электролиза,
I оборудование цеха электролиза было вынесено на бетонированную
открытую площадку и успешно эксплуатировалось в течение очень
I, суровой зимы 1968—1969 г. Серии электролизеров БГК-13, распо-
ложенные на бетонированной площадке, были защищены от ветра
; легким ограждением высотой до 2 м. Несмотря на очень сильные
морозы (до —40 °C и ниже), цех работал и продолжает работать без
j серьезных затруднений. Имеются указания о строительстве цехов
[. электролиза вне здания в странах с более теплым климатом
I [109].
Обслуживание цехов электролиза
ь
ь С
В последние годы усиленно проводится автоматизация операций
контроля и регулирования процесса электролиза и вспомогательных'
процессов. Хотя еще нет примера комплексной автоматизации цехов
электролиза растворов поваренной соли, однако в настоящее время
уже технически разработана и практически осуществлена автомати-
зация подавляющего числа операций контроля и управления про-
цессом. Обслуживание цехов электролиза все в большей степени
сводится к контролю за действием приборов автоматического кон-
троля и регулирования, наладке и управлению ими.
Автоматизированы процессы перекачивания рассола в цех элек-
тролиза, подогрева, поддержания постоянства напора в рассольных
коллекторах цеха электролиза, подачи электролитических щелоков
на выпарку и растворов щелочи из цехов электролиза с ртутным
катодом на склад.
247
Осуществляется автоматическое регулирование процессов под-
кисления рассола, подщелачивания и дозирования сульфида натрия
при химическом обесхлоривании анолита электролизеров с ртутным
катодом. Автоматизировано поддержание постоянства вакуума
и давления хлора и водорода в электролизерах и сборных коллекто-
рах, а также в напорных линиях после хлорных и водородных ком-
прессоров, регулирование температуры охлажденного хлора, подачи
серной кислоты на сушку хлора и др.
В значительной степени автоматизирован аналитический контроль
производства. Однако до сего времени промышленность еще не выпу-
скает некоторых приборов, необходимых для автоматизации/прове-
дения анализов, например, электролитических щелоков.
Вновь строящиеся заводы наиболее полно оснащены средствами
автоматизации. На старых заводах средства автоматизации исполь-
зуются в меньшей мере, однако они быстро ^завоевывают себе место
и здесь, так как применение средств автоматизации позволяет не
только сократить затраты труда, но и тщательнее поддерживать
технологический режим производства и его показатели.
В цехах электролиза с диафрагмой необходимо контролировать
и регулировать питание электролизеров рассолом с тем, чтобы
количество подаваемого рассола в каждый электролизер обеспечи-
вало оптимальную конверсию хлорида натрия в гидроокись около 50%
и работу электролизеров с высоким выходом по току.
Регулирование подачи рассола может быть осуществлено по
показателям ротаметров, установленных па каждом электролизере.
Так как по мере старения диафрагмы уровень анолита возрастает,
то по его высоте при нормальной концентрации щелочи в католите
(130—145 г/л) судят о необходимости смены диафрагмы или ее про-
мывки. По напряжению на электролизере или по зависящей от
напряжения температуре в электролизере определяют степень износа
анодов и необходимость выключения электролизера для замены ано-
дов и полной переборки электролизера.
В цехах электролиза с ртутным катодом необходимо регулировать
подачу рассола на электролизеры с целью поддержания концен-
трации поваренной соли в отходящем обедненном анолите на уровне
265 г/л и подачу воды в разлагатели в количестве, необходимом для
получения щелочи с концентрацией около 650 г/л NaOH.
По концентрации щелочного металла в амальгаме на входе и
выходе ее из разлагателя контролируется скорость циркуляции
ртути и работа разлагателей амальгамы. Контроль содержания водо-
рода в хлоре и соединений активного хлора в выходящем из элект-
тролизера анолите необходим для установления нарушений в работе
электролизера и получения информации, необходимой для устране-
ния допущенных нарушений.
Чтобы обеспечить нормальную эксплуатацию электролизеров,
необходимо регулировать межэлектродное расстояние по мере износа
анодов путем их опускания, а также систематически удалять из
карманов электролизера амальгамное масло и графитовый шлам.
248
Й . ° К
!?};• •
1 * ' •
, При применении малоизнашивающихся анодов нет необходимости
к регулировать межэлектродное расстояние, но требуется автомати-
; ческая защита от коротких замыканий между анодом и катодом.
ВЫПАРКА И ПЛАВКА КАУСТИЧЕСКОЙ СОДЫ
Выпарка электролитических щелоков
Каустическая сода в промышленности и народном хозяйстве
потребляется преимущественно в виде растворов с содержанием
от 42 до 50% NaOH. За рубежом выпускается также каустическая
сода в виде 72—75%-ных растворов NaOH. Только очень ограни-
ченное количество потребителей нуждается в твердой каустической
соде, которая обычно содержит от 92 до 96% NaOH.
Хотя при перевозке товарной каустической соды в виде водных
42—50%-ных растворов на 1т NaOH приходится 1,0—1,4 т воды,
потребители предпочитают получать каустическую соду в виде
растворов, так как при этом резко снижаются затраты труда на
погрузочно-разгрузочные работы, отпадает операция растворения
щелочи и становится удобным ее транспортирование, разбавление
и дозирование.
' В производстве по методу с ртутным катодом из разлагателей
электролизеров сразу же получают каустическую соду в ее товарной
форме (42—45% NaOH).
. После охлаждения и отстаивания в баках-сборниках от увле-
каемых из разлагателей капелек ртути каустическая сода может
отгружаться потребителям. В последнее время для снижения содер-
жания ртути в товарной каустической соде ее дополнительно филь-
труют через мелкопористые фильтры.
Хорошие результаты получены при глубокой очистке каустической
соды от ртути фильтрованием через слой графитового порошка [110].
Путем фильтрования растворов каустической соды через фильтр
из смеси асбестовых волокон с графитовой пылью содержание ртути
в растворе NaOH снижается до 2,5-Ю4"5 вес. % [111]. Дополнитель-
ная очистка щелочи от ртути может быть достигнута барботажем
через подогретую щелочь газа, не взаимодействующего со щелочью
и ртутью [111, 112].
В процессе хранения и транспортирования чистой каустической
соды необходимо исключить возможность поглощения щелочью
двуокиси углерода из воздуха, а также загрязнение ее продуктами
коррозии стенок баков-хранилищ и цистерн. Для перевозки приме-
няют цистерны из нержавеющей стали цли цистерны, защищенные
гуммировкой. За границей для перевозки чистой каустической соды
используются также цистерны, выложенные изнутри листовым нике-
лем. Цистерны для перевозки каустической соды должны быть
закреплены за заводами, так как при использовании цистерн от
перевозки другой продукции остатки воды после промывки цистерн
загрязняют каустическую соду.
и
В процессе электролиза по методу с твердым катодом и диафраг-
мой образуются электролитические щелока, содержащие 100—140 г/л
NaOH и 170—200 г/л NaCl. Для получения товарной формы каусти-
ческой соды электролитические щелока следует упаривать.
Электролитические щелока ненасыщены по хлориду натрия.
В процессе испарения воды из щелоков концентрация NaOH и NaCl
возрастает и раствор становится насыщенным по NaCl. С этого
Рис. 4-23. Растворимость поваренной соли в рас-
творе NaOH различной концентрации:
1 — при 20 °C; 2 — при 60 °C; 3 — при 100 °C; 4 —
в 26 %-ном растворе NaOH; 5 — в 34 %-ном растворе NaOH;
6 — в 48 %-ном растворе NaOH*
момента в ходе дальнейшего испарения воды возрастает концентра-
ция только щелочи. Концентрация же NaCl в упариваемом растворе
понижается, так как растворимость NaC] уменьшается с ростом
концентрации NaOH (особенно в интервале от 0 до 500 г/л NaOH,
как это видно из рис. 4-23). При дальнейшем концентрировании
щелоков (содержание NaOH выше 600—650 г/л) растворимость
поваренной соли практически не меняется.
Таким образом, при упаривании электролитических щелоков
помимо испарения избыточной воды происходит также удаление
основного количества поваренной соли/ выпадающей в виде кри-
сталлов NaCl. Растворимость NaCl в растворах NaOH уменьшается
со снижением температуры, поэтому для более полного выделения
NaCl растворы каустической соды концентрируют до 600—650 г/л
NaOH и охлаждают до возможно более низкой температуры (но
не достигая температуры застывания раствора). Выпадающие при
выпаривании и охлаждении растворов кристаллы поваренной соли
отделяют от раствора фильтрованием на центрифугахf и после про-
мывки от щелочи используют для получения так называемого обрат-
ного рассола или для донасыщения анолита электролизеров с ртут-
ным катодом.
250
Количество поваренной соли, получаемой в процессе выпарки
электролитических щелок
составляет около 1,5 т/т
В рассоле, поступающем на электролиз, обычно содержится
также сульфат натрия в количестве не более 4—6 г/л. При наруше-
ниях технологического процесса содержание Na2SO4 может быть
значительно выше. При электролизе весь сульфат натрия переходит
в электролитические щелока, поэтому при их упаривании происходит
кристаллизация сульфата натрия ана-
логично NaCl. В связи со сравни-
тельно низкой исходной концентрацией
сульфата насыщение выпариваемого
раствора по Na2SO4 наступает значи-
тельно позже, чем по NaCl. При по-
следовательном концентрировании элек-
тролитических щелоков вначале выде-
ляются кристаллы чистой поваренной
соли без примеси сульфата натрия.
Насыщение по Na2SO4 обычно наступает
на последних стадиях выпаривания,
когда основная масса хлорида натрия
уже выделена из раствора и концен-
трация щелочи достигает примерно
350—400 г/л. После этого из раствора
выделяют смесь мелких плохо фильтрующихся кристаллов NaCl
и Na2SO4. Естественно, что условия, при которых начинается
совместное выделение кристаллов NaCl и Na2SO4, зависят от исход-
ной концентрации сульфата натрия в электролитических щелоках.
Ниже показано изменение растворимости сульфата натрия (в г/л)
в электролитической щелоЦи с увеличением концентрации NaOH:
зависит от состава щелоков и в среднем
ОН.
150
М
S 120
£ по
Оз
^‘цоо
О 10 20 30 W 50
^Концентрация NaOH, вес. %
Рис. 4-24. Температура кипе-
ния растворов NaOH при
атмосферном давлении:
1 — растворы NaOH; 2 — рас-
творы NaOH, насыщенные NaCl.
Концентрация Растворимость Na2SO< Концентрация Растворимость Na2SO4
NaOH, г/л при 100 °C при 20 °C NaOH, г/л при 100 °C при 20 °C
100 43 57 500 10 2,6
200 32 29 600 6,8 0,6
300 , 25 15 700 3,6 0,2
400 15 6
Выделение смеси кристаллов NaCl и Na2SO4 на последних стадиях
концентрирования электролитических щелоков используется в про-
мышленности для вывода сульфатов из цикла производства.
В ходе испарения влаги и увеличения концентрации NaOH
повышается температура кипения, раствора. На рис. 4-24 и 4-25
приведены температуры кипения растворов NaOH и КОН различной
концентрации. На рис. 4-26 приведено парциальное давление паров
воды над растворами NaOH при различных температурах.
В процессе упаривания электролитических щелоков для полу-
чения товарной щелочи необходимо испарить 7—7,5 т воды на 1 т
NaOH, при этом основную часть затрат составляет расход пара.
251
1
Помимо затрат на испарение воды тепло расходуется на подогрев
электролитических щелоков, на дегидратацию щелочи и на воспол-
нение тепловых потерь аппаратуры и трубопроводов. Теплота раство-
Концентрация КОН ,8ес.°/о
Рис. 4-25. Температура кипения
растворов КОН при различном
давлении:
1 — при 0,1 ат; 2 — при 0,5 ат; 3 —
при 1 ат; 4 — при 2 ат; 5 — при 4 ат.
Температура
Рис. 4-26. Парциальное давление водяных
паров над растворами NaOH различной
концентрации:
1 — вода; 2 — 10% NaOH; 3 — 20% NaOH;
4 — 30% NaOH; 5 — 40% NaOH; 6 — 50%
NaOH; 7 — 60% NaOH; 8 — 70% NaOH; 9 —
,75% NaOH.
Таблица 4-10.
Теплота растворения твердой
щелочи в воде
п
{в ккал/кг гидроокиси)
Концентра- ция гидро- окиси в растворе, вес. % Теплота растворения Концентра- ция гидро- окиси в растйоре, вес. % Теплота растворения
NaOH КОН л 4 NaOH КОН
5
10
15
20
26
30
35
40
45
50
298 268
296 268
294 266
288 264
280 ' 260
268 256
252 250
234
215 232
194 218
55 175 201
60 155 178
65 136 156
70 116 134
75 96 112
80 78 90
85 58 68
90 38 46
95 20 23
252
Для снижения затрат тепла применяют многоступенчатые си-
стемы выпарки с использованием сокового пара выпарных аппаратов
в качестве греющего пара последующих ступеней упаривания [113].
„При использований такой системы общая разность между темпера-
турой греющего пара, определяемой его давлением, и температурой
сокового пара в последней ступени выпарной системы распределяется
между ступенями выпарной системы [114].
Для увеличения общей разности температур повышают давление
греющего пара и снижают температуру кипения раствора в послед-
нем выпарном аппарате системы, организуя его работу под вакуумом.
Рис. 4-27. Температурная депрессия электро-
литической щелочи и раствора едкого натра
при различном абсолютном давлении (сплош-
ные линии — для электролитической щелочи;
пунктирные — для раствора NaOH).
Повышение давления греющего пара связано с необходимостью
увеличения прочности выпарных аппаратов и приводит к росту кор-
розии, обусловленному1 повышением температуры выпаривания.
Практически при выпаривании .электролитической щелочи приме-
няется пар давлением до 10—12 ат, остаточное давление в последнем
выпарном аппарате составляет 0,1—0,2 ат.
Пар, получаемый из ТЭЦ или котельных, обычно бывает пере-
гретым. Перед использованием такой пар увлажняют и превращают
в насыщенный для предотвращения снижения коэффициента тепло-
передачи и эффективности работы выпарных аппаратов. На многих
производствах в качестве греющего пара применяется пар давле-
нием 3—5 ат, что ограничивает выбор числа ступеней выпарной
системы.
Значительная часть общей разницы температур в выпарной си-
стеме расходуется на температурную депрессию при кипении раство-
ров. На рис. 4-27 показана зависимость температурной депрессии
при кипении растворов электролитической щелочи и чистых раство-
ров NaOH от концентрации при различном давлении. С увеличением
253
концентрации щелочи температурная депрессия сильно растет и
в условиях работы последнего выпарного аппарат^ (концентрация
щелочи 42—50%) может достигать 35—40 °C. Часть полезной раз-
ности температур расходуется на перегрев жидкости, что необходимо
для вынесения зоны кипения раствора из трубок греющих камер
выпарных аппаратов с целью снижения возможности выпадения
соли в трубках. Эти потери сравнительно невелики и составляют
примерно 1—2 °C на аппарат.
С ростом числа ступеней в выпарной системе почти в такой же
степени увеличивается кратность использования тепла греющего
пара и снижается его расход. При переходе от одноступенчатой
выпарки к системе из двух или большего числа ступеней полезная
разность температур в отдельном выпарном аппарате снижается,
так как общая разность температур между греющим и соковым паром
в последнем аппарате выпарной системы распределяется между не-
сколькими аппаратами. Помимо того, в каждом аппарате часть общей
разности температур теряется на температурную депрессию, перегрев
жидкости, а также в связи с падением давления сокового пара в тру-
бопроводах между аппаратами. Поэтому переход к многоступенчатой
выпарной системе приводит к необходимости увеличения размеров
выпарных аппаратов и поверхности теплопередачи в них. Получаемая
при этом экономия греющего пара полностью покрывает увеличение
затрат на аппаратуру.
Полезную разность температур в выпарнъ^х аппаратах следует
выбирать в зависимости от концентрации щелочи в упариваемом
растворе. При концентрации щелочи до 20% оптимальная разность
температур лежит в пределах 12—20 °C, при концентрации щелочи
35—45% разность температур уже составляет 25—40 °C. Чрезмерное
увеличение полезной разности температур нецелесообразно, так
как при этом усиливается выделения соли на поверхности тепло-
передачи, снижается коэффициент теплопередачи и повышается
брызгоунос с паром.
Ухудшение работы выпарных аппаратов наблюдается при отложе-
нии слоя кристаллов на теплопередающей поверхности. Для устране-
ния вредного действия инкрустации выпарные аппараты регулярно
промывают конденсатом.
На практике процесс выпаривания электролитических щелоков
ведут в выпарных аппаратах с кожухотрубчатыми подогревателями.
Греющий пар подается в межтрубное пространство, а электролити-
ческие щелока циркулируют в трубках греющей камеры. Для увели-
чения коэффициента теплопередачи и предотвращения инкрустации
греющих трубок применяются аппараты с интенсивной циркуляцией
(скорость выпариваемого раствора в трубках не ниже 1,5—2,5 м/с).
В большинстве выпарных ацпаратов поддерживается естественная
циркуляция, возникающая за счет вскипания раствора в греющей
камере. Для предотвращения вскипания раствора в самих трубках
греющих камер выпарных аппаратов и выпадения солей на поверх-
ности трубок уровень упариваемого раствора должен быть на 0,5—
254
' г
1*,0 м выше верхнего края греющих трубок. Потери полезной раз-
ности температур компенсируются увеличением надежности работы
«аппарата и возрастанием коэффициента теплопередачи.
На рис. 4-28 схематически
показан выпарной аппарат
с естественной циркуляцией
упариваемого раствора, при-
меняемый для выпарки элек-
тролитических щелоков на
первой стадии выпарки. Гре-
ющая камера аппарата диа-
метром 2000 мм с 614 труб-
ками длиной 2300 мм и диа-
метром 57x3,5 мм имеет
поверхность теплопередачи
промывки,
3000
\*2№
помещены
поверхность
составляет
по Я-Я
Греции, ай пар
в о За Зля
щелочь
К'онден сат
Д77^
опорожнения
Рис. 4-28. Выпарной аппарат с подвес-
ной греющей камерой и естественной
циркуляцией щелочи:
1 — греющая камера; 2 — сепаратор; з — подвод
воды для промывки аппарата; 4 — центробежный
брызгоотделитель; 5 — лазы.
Упаренный,
раствор с солью
В последних моделях
выпарных аппаратов диаметр
греющей камеры увеличен
до 2200 мм, высота камеры
до 2580 мм, а диаметр тру-
бок уменьшен до 38 мм. В гре-
ющую камеру
1704 трубки,
теплопередачи
450 м2. В верхней расширен-
ной части аппарата помещен
центробежный брызгоотдели-
тель для отделения капель
раствора от сокового пара.
Греющая камера лежит на
опорах в нижней части кор-
пуса. Для компенсации теп-
лового расширения трубок
греющей камеры подвод
греющего пара производится
по центральной трубе, уплот-
ненной в корпусе с помощью
сальника, образующийся кон-
денсат отводится по трубе
с компенсатором.
При разности температур 12—14 °C съем сокового пара с 1 м2
ч поверхности нагрева такого аппарата составляет 30—35 кг/ч.
На последних стадиях упаривания щелочи применяются аппараты
как с естественной, так и с принудительной циркуляцией. На стадии
окончательного упаривания вязкость растворов сильно возрастает,
поэтому целесообразно ставить аппараты с повышенной скоростью
циркуляции. В аппаратах с естественной циркуляцией для
255
увеличения циркуляции используются греющие камеры болыпрй
высоты (до 4000 мм) и устанавливаются над ними камеры вскипания,
ограничивающие объем кипящей жидкости. За счет увеличения паро-
содержания в камере вскипания возрастает циркуляционный напор.
воздух
1 Соковый
I пар
Лар
* '\кРонденсат^
'вода для
промывка
Раствор
вторичным
лар
Упаренный,
рЁРСпвдра соль
Опорожнение ^4
Рис. 4-30. Выпарной аппарат с прину-
дительной циркуляцией щелочи:
1 — греющая камера; 2 —- сепаратор; 3—
брызгоотделитель; 4 — смотровые стекла;
5 — козырек; 6 — циркуляционный насос.
Ряс. 4-29. Выпарной аппарат с есте-
ственной циркуляцией для концен-
трированных растворов каустической
соды:
1 — корпус; 2 — греющая камера; 3 —
камера вскипания; 4 — отбойник брызг
жидкости; 5 — сепаратор; 6 — труба для
стока раствора; 7 — центробежный брыз-
гоотделитель; 8 — лапы; 9 — лазы; 10 —
штуцера для термопар; 11 — смотровые
стекла.
Для уменьшения сопротивления циркуляции жидкости увеличивается
сечение по всему контуру циркуляции. На рис. 4-29 схематически
показан аппарат такого типа. Его греющая камера состоит из 492 тру-
бок диаметром 38 X 2 мм и длиной 4000 мм. Над камерой вскипания
256
установлен отбойник брызг, возникающих при интенсивном ки-
пении раствора. При полезной разности температур 25 °C съем
сокового пара с 1 м2 поверхности теплопередачи составляет 60—
80 кг/ч. *
Для окончательного упаривания каустической соды применяются
также выпарные аппараты с принудительной циркуляцией щелочи.
Устройство такого аппарата показано на рис. 4-30.
Циркуляция упариваемого раствора осуществляется с помощью
лопастного центробежного насоса, прокачивающего раствор вместе
с кристаллами соли через выносную греющую камеру. Часть раствора
в виде упаренных щелоков отводится из напорной линии насоса.
Из греющей камеры раствор поступает в сепаратор по тангенциаль-
ному вводу. Для отделения пара от брызг щелочи над уровнем жидко-
сти в сепараторе устанавливается отбойный козырек и на выходе
сокового пара из сепаратора — брызгоотделитель. Греющая камера
аппарата состоит из 127 трубок диаметром 38x2 мм и длиной 5000 мм
и имеет поверхность теплопередачи 59 м2. На аппарате устанавли-
вается циркуляционный насос производительностью 750 м3/ч при
напоре 3—4* м вод. ст. с электродвигателем мощностью 30 кВт. t
При полезной разности температур 25 °C съем сокового пара с 1 м2
поверхности нагрева составляет 70—100 кг/ч.
Выпарные аппараты с принудительной циркуляцией позволяют
иметь больший съем сокового пара с 1 м2 поверхности теплопередачи
по сравнению с аппаратами с естественной циркуляцией, однако
наибольшее распространение получают аппараты с естественной
циркуляцией, вследствие их большей мощности, отсутствия насоса
и меньших затрат на обслуживание и ремонт.
Трубки греющих камер выпарных аппаратов подвержены кор-
розионному разрушению, особенно на последних стадиях упарки
щелоков. Трубки из стали Х18Н9Т разрушаются значительно меньше,
чем трубки из черной стали. Хорошей стойкостью на всех стадиях
упарки отличаются трубки из хромистой стали Х-25 [115].
В зависимости от давления пара применяются различные схемы
цеха выпарки электролитических щелоков [116]. На рис. 4-31
показана широко применяемая на заводах схема двухстадийной
выпарки электролитических щелоков. Первая стадия выпарки
с получением средних щелоков (концентрация NaOH около 350—
400 г/л) осуществляется в трехступенчатой выпарной системе.
Вторая стадия выпарки проводится в выпарном аппарате, обогре-
ваемом соковым паром из аппаратов первой ступени первой стадии
выпарки. На схеме показана установка трех параллельно работающих
выпарных аппаратов первой ступени выпарной системы.
Электролитические щелока перед поступлением в выпарные
аппараты первой ступени последовательно проходят через тепло-
обменники, обогреваемые соответственно конденсатом греющего
пара, соковым паром второй ступени, соковым паром первой ступени
и, наконец, свежим паром и нагреваются до температуры кипения
жидкости в первой ступени выпарной системы,
р
17 Заказ 843 257
II
Выпарные аппараты первой ступени обогреваются свежим паром.
Соковый пар аппаратов первой ступени используется для обогрева
аппарата второй ступени и выпарного аппарата второй стадии
выпарки. Третья ступень первой стадии выпарки обогревается
И to JV О zfV WI £1 VVJI OU
Рис. 4-31. Принципиальная технологическая схема выпарки электролитиче-
ских щелоков:
1 — хранилище электролитических щелоков; 2 — центробежный насос для электролити-
ческих щелоков; 3 — подогреватели; 4 — выпарные аппараты первой ступени; 5 — выпар-
ной аппарат второй ступени; в — выпарной аппарат третьей ступени; 7 — бак для пульпы;
8 — центробежный насос для средних щелоков; 9 — сгустители средних щелоков — напор-
ные баки центрифуги; 10 — напорный бак средних щелоков; 11 — выпарной аппарат послед-
ней стадии выпарки; 12 — центробежные насосы для крепкого каустика; 13 — напорный
бак центрифуги; 14 — спиральный холодильник; 15 — сепараторы; 16 — водоотделители;
17 — барометрические конденсаторы; 18 —- центробежный насос для конденсата; 19 —
сборник конденсата; 20 — центрифуга; 21 — центробежные насосы для рассола; 22 —
растворитель соли; 23 — бак средних щелоков; 24 — центробежные насосы для промывных
вод; 25 — бак для промывных вод; 26 — отстойная центрифуга; 27 — бак для концентриро-
ванного каустика; 28 — вакуум-насосы; 29 — бачки вакуум-насосов; 30 — центробежный
насос для барометрической воды; 31 — барометрический ящик; 32 — хранилище крепкого
каустика.
I — электролитические щелока; II — конденсат; III — электролитические щелока и кон-
денсат для промывки соли; IV — обратный рассол; V — промывные воды на выпарку; VI —
соль с сульфатом натрия на выделение сульфатов; VII — барометрическая вода на раство-
рение соли; VIII — каустическая сода потребителю; IX — греющий пар.
соковым паром второй ступени. На первой стадии выпарки на 1 т
NaOH испаряется 5—5,5 т воды с трехкратным использованием пара,
а на второй стадии соответственно 2—2,5 т воды — с двукратным.
По мере выпаривания раствор передается из одной ступени
в Другую. На первой стадии выпарки отделение выпавших кристал-
лов соли производится только после третьей ступени, т. е. соль,
выделившаяся во второй и частично в первой ступени выпарной
258
и
системы, передается в выпарные аппараты последующих ступеней
вместе с раствором. Наличие в выпарном аппарате 12—15% кри-
сталлов поваренной соли не мешает процессу. Кристаллы соли
в упариваемом растворе являются центрами кристаллизации, что
отчасти предотвращает отложение солей на стенках трубок греющей
камеры и способствует укрупнению кристаллов.
Из третьей ступени средние щелока вместе с кристаллами соли
поступают в сгустители. Сгущенная пульпа кристаллов соли пере-
дается на центрифуги.
Для отделения соли на отечественных заводах применяют пре-
имущественно горизонтальные автоматические центрифуги АГ-1800
полунепрерывного действия производительностью около 7 т/ч соли.
В последнее время для этой цели стали использовать более компакт-
ные и высокопроизводительные непрерывноработающие центрифуги
типа НГП. После отделения от средних щелоков соль на центрифуге
промывается раствором электролитических щелоков и конденсатом.
После промывки и просушки соль срезается ножом и поступает
в растворитель для получения обратного рассола. Обратный раесол,
содержащий 305—310 г/л поваренной соли и 2—2,5 г/л щелочи,
возвращается в отделение очистки рассола.
Осветленные средние щелока поступают в выпарной аппарат
второй стадии выпаривания. На схеме показан выпарной аппарат
с естественной циркуляцией. Концентрированная каустическая сода
после выпарного аппарата второй стадии выпаривания вместе с кри-
сталлами поваренной соли и сульфата натрия поступает в сгуститель,
служащий одновременно напорным баком центрифуги.
Для снижения содержания поваренной соли в каустической соде:
ее охлаждают путем циркуляции через спиральные холодильники.
Кристаллы поваренной соли и сульфата натрия отделяют на центри-
фуге и передают на операцию вывода сульфатов из цикла.
Мелкие кристаллы смеси поваренной соли с сульфатом натрия,
образующиеся на последней стадии выпарки, плохо фильтруются
на центрифуге. Поэтому на многих предприятиях пульпу этих
кристаллов после сгустителя разбавляют пульпой от сгустителя
средних щелоков. При этом содержание сульфата натрия в соли
снижается, но облегчается работа центрифуги. Выпарные аппараты
третьей ступени и последней стадии выпарки работают под вакуумом.
Остаточное давление в них поддерживается на уровне 0,1—0,2 ат
с помощью барометрически^ конденсаторов и вакуум-насосов. Между
выпарными аппаратами и барометрическими конденсаторами уста-
навливаются сепараторы для отделения брызг щелочи, увлекаемых
соковым паром. Несмотря на это некоторое количество щелочи
попадает вместе с соковым паром в барометрические конденсаторы
и вызывает осаждение солей кальция и магния, забивающих кана-
лизационные трубы. Поэтому отвод барометрической воды стара-
ются выполнять в виде лотковой канализации, удобной для чистки.
В последнее время стали практиковать оборотные циклы для воды,
подаваемой на барометрические конденсаторы.
17* 259
I
Для технологической схемы, приведенной на рис. 4-31, необхо-
димо давление свежего пара не менее 6—7 ат. Расход пара соста-
вляет 2,7—2,9 Мкал/т 92%-ной NaOH. При более низком давлении
пара применяют на первой стадии двухступенчатую схему выпарки;
при этом затраты пара возрастают до 3,3—3,5 Мкал/т. Более эконо-
мичной по расходу пара является одностадийная схема выпарки
электролитической щелочи в три ступени. По зтой схеме все коли-
чество воды упаривается при трехкратном использовании тепла
свежего пара. Один из вариантов такой схемы показан на рис. 4-32.
Соковый пар после выпарного аппарата первой ступени, обогре-
ваемого свежим паром, поступает на обогрев аппарата второй сту-
пени. Соковый пар от второй ступени используется для обогрева
аппаратов как третьей ступени, так и окончательного упаривания.
Для работы выпарки по такой схеме необходим свежий пар с более
высоким давлением (10 ат). При этом расход пара может быть снижен
до 1,9—2,1 Мкал/т 92%тной каустической соды.
Процесс выпаривания электролитической щелочи проводится
непрерывно, его основные стадии автоматизированы [79].
Концентрация щелочи в аппарате третьей ступени выпарки
и в аппарате окончательного концентрирования второй стадии
определяется автоматически по значению депрессии в последнем.
По показаниям концентратомера автоматически производится отбор
средних щелоков из аппарата третьей ступени и готового продукта
из аппарата второй стадии выпарки. При этом уровень жидкости
в аппарате снижается и регулятор уровня в аппарате автоматически
открывает клапан подачи раствора из предыдущего аппарата или
средних щелоков в аппарат окончательного концентрирования.
Таким образом автоматически поддерживается концентрационный
режим работы выпарной установки и уровень жидкости во всех
выпарных аппаратах.
Автоматически регулируются подогрев электролитических щело-
ков, подача воды на барометрические конденсаторы, перекачивание
растворов по уровням в баках и некоторые другие операции. Отделе-
ние соли на центрифугах, промывка и подсушка соли проводятся
автоматически по заданной программе.
и
п
Вывод сульфатов
Сульфат натрия, содержащийся в исходном рассоле, в процессе
электролиза не используется и полностью переходит в электролити-
ческие щелока. При содержании Na2SO4 в рассоле, поступающем
на электролиз, 5 г/л на 1 т каустической соды на выпарку поступит
около 40 кг сульфата натрия. В процессе выпарки только небольшая
его часть уходит с каустической содой. В соответствии с раствори-
мостью Na2SO4 в концентрированной щелочи на 1 т 92 %-ной NaOH
приходится около 5 кг Na2SO4. Остальное количество сульфата на-
трия выпадает при выпаривании вместе с поваренной солью и возвра-
щается вновь в цикл производства с обратным рассолом выпйрки.
261
При питании производства солью или подземным рассолом с опре-
деленным содержанием сульфатов последние должны накапливаться
в производственном цикле до определенного предела, при котором
приход сульфатов со свежей солью или рассолом будет компенси-
роваться расходом сульфатов с каустической содой и с механиче-
скими потерями рассола в производственном цикле.
Если обозначить содержание сульфата натрия в свежей поварен-
ной соли или в свежем рассоле через Со (в %), равновесное содержа-
ние в рассоле производственного цикла через Ср (в %) и потери
рассола в производственной схеме (в долях от теоретического расхода)
через В, то при содержании сульфата натрия в товарной каустиче-
ской соде 0,5% получим уравнение материального баланса по суль-
фату натрия в производственном цикле рассола на единицу коли-
чества израсходованной на производство соли
(1+5)00-1.-^ о,5+^Ср ' <4Л7>
Равновесная концентрация сульфата натрия (в % к общему
содержанию поваренной соли в рассольном цикле) составит
CP-Cfl+C° д’342 (4.18)
При содержании сульфатов в соли или свежем рассоле ме-
нее 0,342% накопление сульфата натрия в рассоле не наблюдается.
При более высоком содержании сульфатов в соли их равновесная
концентрация в производственном цикле возрастает тем больше,
чем выше содержание сульфата натрия в исходном NaCl и чем меньше
производственные потери рассола.
В технической соли, применяемой для электролиза, допускается
содержание до 1,24% Na2SO4. При использовании такой соли содер-
жание сульфата натрия в производственном цикле должно дости-
гать 10,22% при потере рассола 10% и 5,73% — при потере 20%
рассола (или соответственно 30 и 17 г/л). Такое содержание суль-
фатов резко ухудшает показатели работы электролизеров, так как
приводит к снижению концентрации поваренной соли в рассоле
и увеличению износа графитовых анодов.
Ранее применялась очистка рассола от сульфатов путем осажде-
ния их в виде BaSO4 хлористым барием или карбонатом бария.
Такая очистка применяется иногда и в настоящее время в цехах
электролиза с ртутным катодом, работающих на твердой природной
соли, хотя все больше предпочтение отдается схеме работы с частич-
ной очисткой рассола от кальция (см. стр. 223).
В цехах электролиза с твердым катодом и диафрагмой для вывода
сульфатов иэ производственного цикла рассола используется совме-
стное выделение кристаллов сульфата натрия с кристаллами поварен-
ной соли на второй стадии выпарки электролитических щелоков.
Растворимость сульфата натрия в насыщенных растворах пова-
ренной соли сильно зависит от температуры и достигает максимума
262
при температуре около 18 °C (рис. 4-33). При обработке смеси кри-
сталлов Na2SO4 и NaCl, получаемых из выпарного аппарата послед-
ней стадии выпарки, раствором поваренной соли при 18—20 °C
можно получить растворы, содержа-
щие 90—95 г/л Na2SO4 и около
280 г/л NaCl, и вывести таким обра-
зом сульфаты из цикла. Эти растворы
могут быть затем использованы для
регенерации ионитов на очистных
установках в паро-котельных или
в содовом производстве. При отсут-
ствии потребителя из таких рас-
творов выделяют сульфат натрия,
используя зависимость его раство-
римости от температуры.
На рис. 4-34 приведена принци-
пиальная схема установки для вы-
вода твердого сульфата натрия нагре-
ванием таких растворов [117]. Смесь
кристаллов Na2SO4 и NaCl из вы-
парного аппарата второй стадии
после отделения на центрифуге и
Рис. 4-33. Совместная раствори-
мость сульфата > натрия и по-
варенной соли при различной
температуре.
промывки от щелочи обрабатывают
обедненным по сульфатам рассолом при 18—20 °C. При этом
сульфат натрия выщелачивается, а оставшаяся нерастворенной
поваренная соль отделяется на центрифуге, растворяется в воде
Соляная пульпа
из выпарного
аппарата II ступени
На выпарку
Рассол, обедненный сульфатом
Рис. 4-34. Принципиальная схема вывода сульфата натрия нагреванием:
1 — сгуститель — напорный бак центрифуги; 2 — центрифуга; з — сборник щелочи и про-
мывных вод; 4 — центробежный насос; 5 — приемник щелочи; в — бак для выщелачивания
сульфата при t = 18 — 20 °C; 7 — теплообменник; 8 — бак для рассола, обогащенного
сульфатом; 9 — напорный бак; 10 — подогреватель-испаритель; 11 — бак для рассола,
обедненного сульфатом.
263
и в виде обратного рассола, освобожденного от Na2SO4, возвращается
на очистку рассола. Обогащенный по Na2SO4 раствор поступает
в подогреватель-испаритель, где раствор нагревают до кипения
и частично упаривают для доведения его до насыщения по NaCl [118].
Выпадающие кристаллы NaaSO4 отделяют на центрифуге и в виде
товарного сульфата натрия, содержащего до 3% влаги, могут быть
отгружены потребителям. Маточник (55—60 г/л Na2SO4) возвра-
щается на выщелачивание новых порций соли из аппаратов второй
стадии выпарки.
При работе по такой схеме получают безводный Na2SO4, однако
съем сульфата натрия с 1 м3 циркулирующего рассола составляет
только около 30 кг.
Вывод сульфатов из обогащенного им рассола может быть осу-
ществлен также путем охлаждения раствора. При этом выпадает
десятиводный кристаллогидрат сульфата натрия. При охлаждении
раствора до 0—5 °C содержание сульфата натрия в растворе сни-
жается до 20—30 г/л и съем сульфата натрия с 1 м3 рассола может
составить около 60 кг, т. е. примерно в 2 раза выше, чем по методу
с нагреванием. Однако при этом выделяется Na2SO4-10H2O и для
получения товарного сульфата осадок необходимо обезвоживать.
По этой причине предпочитают осуществлять вывод сульфатов
по методу с нагреванием.
В цехах электролиза с ртутным катодом, работающих на соли,
получаемой выпаркой рассола, может применяться схема вывода
сульфатов, аналогичная описанной. Выпаривание рассола разде-
ляют на две стадии и соль второй стадии, загрязненную сульфатом
натрия, направляют на выщелачивание сульфата, который выделяют
. по схеме, аналогичной изображенной на рис. 4-34.
Может применяться также кальциевый способ вывода сульфатов,
заключающийся в обработке хлористым кальцием маточников,
обогащенных сульфатом, с выводом его иэ цикла в виде CaSO4. При
этом повышается расход соды на последующее осаждение остающе-
гося в растворе кальция.
Очистка каустической соды, получаемой 1
электролизом с диафрагмой
При электролизе с ртутным катодом получают очень чистую I
каустическую соду. Если на разложение амальгамы натрия напра- j
влять дистиллят или очищенную на ионообменных смолах воду, И
то можно получить химически чистую каустическую соду. у
Каустическая сода, полученная по методу электролиза с ртутным
катодом, всегда загрязнена ртутью. Содержание ртути зависит
от условий, при которых проводилось разложение амальгамы.
Фильтрованием через тонкодисперсные фильтры или очисткой на
осветлительных центрифугах содержание ртути может быть снижено
до 2,5-Ю"5 вес. % [111].
264
Загрязнения каустической соды попадают в нее с водой, посту-
пающей на разложение, и вносятся с амальгамой, которая также
может увлекать с собой в разлагатели из электролизера небольшие
количества электролита. Помимо того, при получении каустической
соды высокой степени чистоты необходимо учитывать возможность
загрязнения ее за счет частичного перехода в раствор продуктов
коррозионного разрушения материалов, из которых выполнены
разлагатель, его насадка, трубопроводы и емкости для хранения
растворов.
При недостаточно тщательной промывке амальгамы, поступающей
из электролизера в разлагатель, она может захватывать с собой
некоторое количество анолита и амальгамных масел. Примеси ано-
лита вносят в каустическую соду ионы СГ и SO*", а амальгамные
масла могут ее загрязнять примесями тяжелых металлов.
Если обеспечена тщательная промывка амальгамы и полное
удаление амальгамных масел, для разложения применяете^ дистил-
лированная вода особой чистоты и приняты меры, предотвращающие
загрязнение получаемой щелочи продуктами разрушения конструк-
ционных материалов (применение насадки разлагателя из особо
чистого графита, футеровка разлагателя материалами, инертными
в условиях работы разлагателя), можно получить щелочь особой
чистоты. При этом наиболее трудной задачей является подбор мате-
риалов для емкостей, трубопроводов и тары, исключающих загряз-
нение особо чистых щелочей при длительном их хранении.
Каустическая сода, получаемая выпаркой электролитических
щелоков из диафрагменного электролизера, содержит до 2% Na2CO3
и 2,2% NaCl. Такое качество каустической соды не удовлетворяет
требованиям некоторых потребителей, в частности она мало пригодна
для использования. в вискозной промышленности.
Делались попытки очистки диафрагменной каустической соды
с целью приближения ее качества к каустической соде, получаемой
электролизом с ртутным катодом, чтобы сделать ее пригодной для
наиболее квалифицированных потребителей.
Наибольшего внимания заслуживает очистка растворов каусти-
ческой соды экстракцией из нее примесей жидким аммиаком под
давлением [119]. Схема установки показана на рис. 4-35. Очистка
проводится в противоточной экстракционной колонне под давлением
около 100 ат. На верх колонны подается 50%-ный раствор каусти-
ческой соды, а в низ колонны 70—95%-ный раствор аммиака в воде.
В экстракционной колонне при 80—100 °C происходит извлечение
основных примесей из раствора каустической соды в аммиачную
фазу. Из нижней части колонны отбирается раствор чистой каусти-
ческой соды с некоторым содержанием аммиака, а из верхней части —
аммиачная фаза с экстрагированными из каустической соды приме-
сями и некоторым количеством растворенной в ней щелочи (около 5%
от поступившей на очистку).
Раствор чистой каустической соды отпаривают от остатков ам-
миака, а аммиачную фазу передают на установку регенерации
265
аммиака. После отгонки аммиака раствор
выводится из системы.
Ниже приведен состав исходной и
соды [120]:
Содержание, %
после
очистки
Состав до очистки Состав
NaOH . . 50,0 50,0 А12О3
Na2CO3 . . 0,1—0,3 0,15 SiO2
Na2SO4 . . 0,013—0,020 0,01 NH3 .
NaCl ... 1,0 0,08 Fe .
NaClO3 . . 0,05—0,1 0,0002 Ni .
CaO . . . 0,0017 0,001 Cu .
MgO . , . 0,001—0,002 0,001 Mn .
щелочи с загрязнениями
очищенной каустической
Содержание, %
после
очистки
до очистки
. . 0,0013—0,0030 0,0015
. . 0,018—0,025 0,009
— 0,0002
. . 0,0005 0,00025
0,00003 0,00001
. . 0,00003 0,00002
. . 0,00001—0,00006 0,00003
Аммиачный способ
очистки позволяет удалить основные загряз-
нения, однако получаемая при этом каустическая сода уступает
по чистоте полученной по методу электролиза с ртутным катодом.
Рис. 4-35. Очистка каустической соды путем экстракции примесей аммиаком:
1 — насос для каустической соды, поступающей на очистку; 2 — насосы для аммиачной
фазы; 3 — экстрактор; 4 — колонна для отгонки аммиака; 5 — отвод грязной щелочи; 6 —
конденсатор; 7 — сборник жидкого аммиака; 8 — промежуточный бак очищенной щелочи;
Р — кипятильник; 10 — сепаратор; >11 — холодильники; 12 — вакуум-насос; 13— скруб-
бер; 14 — выброс.
Аммиачный метод очистки каустической соды, получаемой элек-
тролизом с диафрагмой, применялся в довольно широких масштабах
в США. Однако развитие техники и улучшение технико-экономи-
ческих показателей электролиза с ртутным катодом, достигнутые
в последние десятилетия, Сделают этот способ производства чистой
каустической соды наиболее экономичным. В последнее время опу-
бликованы сообщения об усовершенствовании аммиачного способа
очистки каустической соды, и возможности проведения односта-
дийного процесса очистки не только от поваренной соли, примесей
266
W'-.-
Ш' 4
хлоратов и воды, но также и от соединений железа, никеля и дру-
гих металлов [12Q al.
Для получения чистой каустической соды использовался также
кристаллогидратный способ очистки. Каустическую соду после
концентрирования до 50% и отделения поваренной соли разбавляют
до 37% и затем охлаждают до 0 °C. При этом выделяются кристаллы
NaOH-3,5 Н2О, которые отделяются от маточника на центрифуге.
При нагревании кристаллы растворяются в кристаллизационной
воде, и полученный очищенный раствор, содержащий около 37%
NaOH, следует выпаривать для получения товарной каустической
соды. Примеси концентрируются в маточнике, который может быть
направлен на выпарку неочищенной каустической соды. Очищенная
каустическая сода содержит около 0,15% NaCl и 0,008% Na2SO4.
Предлагали очищать каустическую соду от поваренной соли
добавлением к ее 50%-ному раствору сульфата натрия в количестве,
необходимом для образования тройной соли NaCl -Na2SO4- NaOH.
Тройная соль мало растворима в концентрированных растворах
каустической соды и выпадает из них в виде осадка [121].
г" ' Возможен также электрохимический способ очистки растворов
каустической соды от загрязняющих ее примесей ионов металлов.
При этом может быть проведена очистка от катионов, разряжающихся
на катоде легче, чем водород. С этой целью предложено проводить
: электролиз на пористом катоде, через который протекает очищаемый
• раствор каустической соды [122]. Такой способ позволяет снизить
содержание примесей железа до 0,1—1; никеля — до 1,3; свинца —
до 0,4 и меди менее 0,1 частей на млн. Производительность предла-
гаемых электролизеров до 200 т очищенного рассола в сутки [123].
Плавка каустической ооды
новы
II
G
гением концентрации щелочи температура кипения рас-
творов возрастает и окончательное обезвоживание каустической
соды проводят при высокой температуре. На рис. 4-36 приведены
температуры кипения высококонцентрированных растворов каусти-
ческой соды при атмосферном и пониженном давлении.
С понижением давления температура кипения концентрирован-
ных растворов каустической соды уменьшается. При давлении 0,5 ат
90%-ные растворы NaOH застывают при температуре кипения. При
более низком давлении в значительном интервале концентраций
температура длавления раствора превышает температуру кипения.
Так, при 0,2 ат растворы, в которых концентрация NaOH составляет
от 86 до 97%, застывают при температуре, превышающей темпера-
туру кипения. Процесс обезвоживания необходимо проводить в та-
ких условиях, чтобы ни на одной стадии не образовывалась твердая
фаза. Для полного обезвоживания каустической соды при атмосфер-
ном давлении необходима температура около 500 °C. При плавке
каустической соды при пониженном давлении температура обезво-
живания может быть снижена до 350—400 °C.
267
п
1U Ранее была широко распространена плавка каустической соды
в открытых горшках при атмосферном давлении (схема установки
показана на рис. 4-37). Котлы с полусферическим днищем отливаются
из чугуна с присадкой до 3% никеля для удлинения срока службы.
Для обогрева котлов используются топочные газы. Простейшая
установка состоит из котла-подогревателя и плавильного котла.
Котел-подогреватель устанавливается выше плавителя, и подогретый
раствор самотеком поступает в
92 96 98 99 99,6 99, в
Концентрация NaOH, %
Рис. 4-36. Температура кипения вы-
сококонцентрированных растворов
каустической соды при различном
давлении:
1 — 1 ат; 2 — 0,825 ат; 3 — 0,66 ат;
4— 0,495 ат; 5 — 0,33 ат; 6 — 0,165 ат.
плавитель. Подача свежего раствора
щелочи в плавитель регулируется
по уровню в нем. Процесс плавки
ведется периодически. По оконча-
нии обезвоживания 96%-ная кау-
стическая сода с помощью верти-
кального погружного насоса пере-
качивается в железные барабаны,
В атмосферу
Рис. 4-37. Схема плавки каустической
соды в плавильных горшках:
1 — напорный бак; 2 — горшок-подогрева-
тель; 3 — плавильный горшок; 4 — топка;
5 — боров; 6 — дымовая труба; 7 — венти-
лятор; 8 — перегонная труба.
расставленные полукругом у плавильного котла. При остывании
объем плава уменьшается, поэтому расплав доливают в барабаны
после их остывания.
Общая продолжительность одной операции плавки в котлах
емкостью 16 т составляет около 4 сут. В течение 2,5 сут происходит
концентрирование каустической соды при постоянном пополнении
раствора взамен выпаренной влаги. Температура кипения повы-
шается в ходе концентрирования до 300—350 °C. Затем ее увеличи-
вают до 500—550 °C и прокаливают плав в течение 10—12 ч. После
добавления небольших количеств серы обогрев котла прекращают
и каустическую соду отстаивают в течение примерно 12 ч. При
этом плав остывает до 330—350 °C. Верхний осветленный слой плава
разливают по барабанам, а нижний слой с осадком шлама обычно
используют для специальных целей или выщелачивают и возвращают
на стадию выпарки.
п
268
Перед подачей новой партии раствора котел необходимо охйддить,
чтобы избежать бурного вскипания жидкости. Качественные хёрошо
футерованные котлы выдерживают до 100 плавок.
В процессе обезвоживания каустическая сода взаимодействует
с материалами стенки котла по реакции
2Fe + 4NaOH + Н2О = Fe2O3 + 2Na2O + ЗН3 (4.19)
Выделяющийся водород реагирует с защитной пленкой окислов
железа на стенках котла и разрушает ее, вызывая дальнейшую
коррозию. Для снижения коррозионного разрушения к плаву
добавляют селитру, которая, как сильный окислитель, обеспечивает
сохранение защитной пленки окислов железа. В последнее время
широкое применение находит катодная защита стенок котлов. Между
стенками котла, служащими катодом, и графитовым анодом про-
пускают постоянный ток 70—200 А при напряжении 6—12 В.
При плавке КОН наблюдается более сильная коррозия.
При плавке каустической соды, получаемой электролизом с ртут-
ным катодом, чтобы избежать ее загрязнения продуктами коррозии
плавильных котлов, последние выполняют из никеля или из серебра.
При этом получают едкий натр или едкое кали реактивной чистоты.
Высокую коррозионную стойкость в расплавленных щелочах имеет
никель [124].
Никелевые котлы обычно меньше чугунных. Применяют котлы
емкостью 4 м3 с электрообогревом. При огневом обогреве необходимо
применять бессернистые сорта топлива, чтобы избежать коррозион-
ного разрушения никелевых стенок котла. Для снижения коррозии
применяется также катодная защита никелевых котлов.
Для удобства потребителей некоторые заводы выпускают чешуиро-
ванную каустическую соду. При этом ее кристаллизация проводится
на вращающихся охлаждаемых барабанах, нижняя часть которых
погружена в ванну с расплавом. Слой каустической соды, кристал-
лизующийся на поверхности барабана, срезается при его вращении
ножом, образуя чешуйчатый продукт. Последний упаковывается
в барабаны, он удобен для употребления и дозировки.
В последнее время выпускается много сортов твердой каусти-
ческой соды в виде сферических частиц. Такой вид продукта удобен,
так как он свободен от мелочи и щелочной пыли, что облегчает
условия труда при его применении [125]. Насыпная плотность
этой каустической соды около 1,28 г/см3.
Периодический процесс плавки связан с рядом неудобств. Поэтому
промышленность переходит на непрерывные процессы.
При проведении плавки в открытых котлах при атмосферном
давлении для перехода к непрерывному процессу применяют каскад
котлов из нескольких подогревательных (4—6 шт.) и плавильных
(7—9 шт.) котлов, замурованных в одну общую печь с обогревом
твёрдым, жидким или газовым топливом. Непрерывный процесс
плавки осуществляется за рубежом также по схеме, показанной
на рис. 4-38 [126-128].
II
II
269
Конденсат
даутерма
Да у терм
(пары)
Рис. 4-38. Схема установки
для непрерывной плавки
каустика с даутермовым
обогревом:
1 — подогреватель; 2 — испа-
ритель; 3 — сепаратор; 4 —
барометрический конденсатор;
5 — эжектор; 6 — гидравличе-
ский затвор; 7 — сборник пла-
вленой щелочи.
Щелочь
Раствор щелочи
{50-12%)
условиях хорошая, однако
По этой схеме раствор каустической соды, содержащий 50—73 вес. %
NaOH, подается через подогреватель в прямоточный испаритель,
обогреваемый парами даутерма при 370—380 °C. Обезвоживание
проводится в вакууме. Конечная концентрация твердого вещества
•в плавленой щелочи достигает 99,5—99,9%. Смесь расплава щелочи
и водяного пара разделяется в сепараторе.
Расплавленная щелочь может быть раз-
лита в барабаны или подвергнута чешуи-
рованию или гранулированию. Испари-
тель выполняют из чистого никеля, осталь-
ную аппаратуру, соприкасающуюся с рас-
плавом щелочи — из никеля или стали,
плакированной никелем [129]. Стойкость
никеля в этих
в присутствии хлоратов резко снижается
[126, 127, 130]. Поэтому при плавке кау-
стической соды, полученной диафрагмен-
ным способом и содержащей хлораты,
вводят железную стружку или сахарозу
для их разрушения. Для обогрева испари-
теля могут применяться различные вы-
сококипящие органические соединения,
например смесь дифенила с эфиром или
низкоплавкие солевые смеси (смесь 40%
NaNO2, 7% NaNO3 и 53% KNO3)
[131, 132].
Проведение плавки каустической соды
в вакууме позволяет снизить рабочую
температуру процесса от 500—550 до
370—380 °C (без уменьшения степени обез-
воживания) и затраты тепла на плавку.
Понижение рабочей температуры повы-
шает стойкость применяемых материалов
и примерно в 5 раз увеличивает срок службы аппаратуры для
плавки.
В промышленности применялся метод плавки каустической соды
в аппаратах Фредеркинга в вакууме. Обогрев аппаратов произво-
дился с помощью стальных змеевиков, залитых в чугунных стенках
плавильных котлов. По змеевикам под высоким давлением циркули-
ровала перегретая вода.
РЕГЕНЕРАЦИЯ РТУТИ ИЗ ШЛАМОВ
И ОЧИСТКА СТОЧНЫХ ВОД ОТ РТУТИ
В производстве хлора и каустической соды электролизом с ртут-
ным катодом всегда происходит потеря ртути, величина которой
сильно зависит от состояния оборудования и общей культуры про-
изводства. На лучших предприятиях удельные затраты ртути на 1 т
270
хлора колеблются от 150 до 300 г, на других могут быть выпщЗОО г
и иногда достигают 500 г и более, \
Обычно не удается установить точный баланс потерь ртути,
так как основная доля потерь связана со случайными механическими
потерями через неплотности аппаратуры, а также при ее ремонте
и смене износившихся анодов.
Из регулярных потерь, связанных с технологическим процессом,
наибольшее значение имеют потери ртути с вытекающим из электро-
лизера анолитом. Содержание ртути в анолите нормально работа-
ющего электролизера обычно составляет 10—20 мг/л и при частых
остановках и отключениях электролизеров может возрастать до 40—•
60 мг/л [133]. Если в рассольном цикле применяется суль-
фидное обесхлоривание циркулирующего рассола, ртуть выде-
ляется в виде малорастворимого сульфида и теряется вместе со
шламами очистки рассола. Эти потери составляют 120—150г/тС12
и при частых выключениях могут превысить эту величину в 2—
3 раза.
Содержание ртути в шламах отделения очистки рассола невелико,
что затрудняет ее регенерацию.
Для сокращения потерь ртути часто работу рассольного цикла
организуют без сульфидного обесхлоривания. При этом в процессе
донасыщения и очистки рассола ртуть не выпадает в осадок и не
теряется. В этом случае потери ртути связаны с проливами и потерями
рассола, что вносит дополнительные загрязнения в сточные воды.
Сточные воды, загрязненные ртутью, образуются в основном при
промывке электролизеров и другой аппаратуры при ремонте, а также
при смывке полов. Помимо этого, загрязнены ртутью конденсат от
холодильников водорода и хлора, сточные воды из гидрозатворов
хлора и водорода. Сравнительно небольшие количества ртути те-
ряются с водородом и. вентиляционными отсосами из карманов
электролизеров. Выбросы вентиляционных отсосов в атмосферу
загрязняют ее парами ртути. Очистка водорода и газовых выбросов
от ртути была рассмотрена ранее (стр. 239).
В последнее время в США, Швеции, Японии и Канаде отмечены
случаи отравления ртутью, содержащейся в воде [134]. В Швеции
отмечено повышение содержания ртути в рыбе до 20 мг/кг, тогда
как природное содержание ее составляет 0,05—0,2 мг/кг. По неко-
торым оценкам, общее поступление ртути за год в воды морей и
океанов составляет около 104 т, в том числе около 50% за счет про-
мышленных сбросов и применения в сельском хозяйстве препаратов,
содержащих ртуть [134]. Поэтому в большинстве промышленных
стран вводятся жесткие требования в отношении содержания
в газовых и жидкостных промышленных выбросах.
В нашей стране установлен верхний предел содержания ртути
в воздухе производственных помещений, равный 0,01 мг/м3,
атмосферного воздуха населенных пунктов — 0,0003 мг/м3.
дельно допустимая концентрация ртути в воде водоемов — 5
или 5 вес. ч./млрд.
ртути
а для
Пре-
мг/м8
271
В США предельное содержание ртути в атмосфере производствен-
ных помещений в последнее время снижено с 0,1 до 0,05 мг/м3,
а ц Швеции допускается до 0,1 мг/м3 [135]. Сообщается об устано-
влении в некоторых американских штатах £!ША предельно допусти-
мой концентрации ртути в воде водоемов после разбавления на
уровне 0,5 вес. ч./млрд. [136]. В Японии для сточных вод предприя-
тий установлено предельное содержание ртути 20 вес. ч./млрд,
хотя фактическое содержание составляет 150—300 вес. ч./млрд. [137].
В последнее время появилось много публикаций о снижении
промышленных выбросов в цехах электролиза с ртутным катодом
до нескольких граммов на 1 т С12. Опубликованы сообщения об огра-
ничении в некоторых штатах США потерь ртути с промышленными
выбросами до 2,5 г/т С12 [138]. Сообщается также [136], что для
завода фирмы «Кема-Норд» в Швеции установлен верхний предел
содержания ртути в выбросах в г/т производимого хлора, не более:
С водородом .................... 0,5
С вентиляционными выбросами . . . 1,0
В сточных водах ..................0,5
।
Однако необходимо помнить, что учитываемые потери ртути
с выбросами водорода, вентиляционного воздуха и сточных вод
составляют обычно только небольшую часть общих потерь ртути.
Основную же часть составляют механические потери в виде метал-
лической ртути или ее соединений. В конечном счете эти потери
также поступают в окружающую среду и приводят к загрязнению
и водоемов, и воздушного бассейна в районе заводов.
Для снижения затрат ртути и уровня загрязнения ею окружающей
природы применяют очистку сточных вод и регенерацию ртути
из ртутьсодержащих отходов производства.
Регенерацияртутиизшламов
В производстве хлора и каустической соды методом электролиза
с ртутным катодом образуются разнообразные твердые отходы, J
содержащие ртуть. По характеру их возникновения ртутные отходы
могут быть разделены на богатые ртутью графитовые шламы, содер-
жащие до 20% ртути, и бедные ртутью отходы, в которые входят
шламы от установок очистки сточных вод от ртути, очистки рассола,
остатки отработанных графитовых анодов, графитовой насадки
разлагателей и различные загрязненные ртутью производственные ।
отходы, получаемые при5 ремонте и эксплуатации аппаратуры.
Основное количество ртутных шламов получается при чистке
входных и выходных карманов электролизеров от графитовой мелочи
и амальгамного масла. Количество шламов, выбираемых из карманов
электролизеров, зависит от многих причин и прежде всего от качества
рассола и графитовых анодов, а также от работы разлагателя амаль-
гамы. При хорошей работе разлагателя, обеспечивающего полное
разложение амальгамы, количество амальгамного масла в карманах
272
w
fc
&...•<•
©tpt*'
чЖСУ^ ’•*
ЙЖ^
ЯМрг,.т/
Ш'.
to-
Li
сильно снижается. В зависимости от этих условий количество шлама
может составлять от 0,3 до 2 кг/т каустической соды.
Содержание ртути в шламах, извлекаемых из карманов электро-
лизеров, обычно близко к 20 вес. %. Примерно 60—70% ртути может
быть выделено в виде металла из шламов в результате отстаивания
или отмывки водой и возвращено в производство. Для извлечения
остальной части ртути, а также ртути из бедных шламов необходима
термическая или химическая регенерация.
Наиболее распространена термическая регенерация ртути, заклю-
чающаяся в нагреваний ртутных шламов в различного вида печах —
ретортах до 600—800 °C и отгонке паров ртути с последующей ее
конденсацией.
Поскольку в шламах помимо металлической ртути могут при-
сутствовать также и ее соединения, в реторту обычно добавляют
около 10 вес. % обожженной извести для восстановления соединений
ртути до металлической
"f’ CaO = CaClg "f* 1 /2О2
Восстановление ртути начинается уже при температуре около
225 °C. При повышении температуры процесс ускоряется и около
600 °C протекает быстро, сопровождаясь отгонкой паров ртути.
Обычно установки термической регенерации работают периоди-
чески. Используется как огневой обогрев через стены реторты,
так и электрический с помощью наружных или внутренних нагре-
вательных элементов сопротивления или нагревателей индукцион-
ного типа. Производительность установок термической регенерации
ртути обычно лимитируется низкой теплопроводностью шламов
после отгонки из них влаги. Вследствие этого прогрев всей массы
шлама в реторте до температуры не ниже 600 °C происходит медленно
и вся операция отгонки в реторте емкостью около 0,5 т обычно зани-
мает 24—36 ч.
Для лучшего удаления паров ртути, после прогрева массы
до 600 °C, внутрь реторты подается азот для продувки.
Чтобы уменьшить опасность загрязнения воздуха парами ртути,
работают обычно при небольшом вакууме, при этом парогазовая
смесь отсасывается из реторты через конденсатор вакуум-насосом.
Для снижения температуры в парогазовую смесь перед входом в кон-
денсатор иногда вспрыскивают воду.
Если шлам в реторте прогревается во всем объеме до температуры
не ниже 600 °C, то остаточное содержание ртути в огарках после
отгонки снижается до 0,005%.
Химическая или гидрометаллургическая регенерация заклю-
чается в том, что после отстаивания и отмывки основного количества
металлической ртути ртутьсодержащие отходы подвергаются хлори-
рованию с целью превращения ртути в растворимую соль HgCl2.
После разрушения остаточного активного хлора раствор хлорной
ртути отфильтровывают от твердого остатка и ртуть осаждают из
18 Заказ 843 273
раствора в виде сульфида ртути добавлением сульфида или гидро-
сульфида натрия. Осадок сульфида ртути отделяется на фильтре.
Сульфид ртути может быть переработан в металлическую ртуть
термическим способом или переведен в раствор в виде Na2HgS2
добавлением избытка сульфида натрия. Из этих растворов ртуть
можно выделить электролизом на ртутном катоде или восстановле-
нием амальгамой натрия.
Гидрометаллургический способ извлечения ртути из ртутьсодер-
жащих растворов довольно сложен, многостадиен и связан с образо-
ванием загрязненных ртутью сточных вод. Кроме того, осадки сер-
нистой ртути плохо улавливаются фильтрами, поэтому предлагаемый
способ мало применяется в практике хлорных производств.
Очистка сточных вод от ртути
Наиболее эффективно всемерное уменьшение количества сточных
;ение их в производственный цикл. С этой целью при-
вод и возвра
и
меняют охлаждение водорода в поверхностных холодильниках,
а не в холодильниках смешения, принимают меры по предотвращению
переливов и потерь рассола, особенно в случае непредвиденных
остановок производства. Загрязненные ртутью конденсаты или сточ-
ные воды стараются возвратить в рассольный цикл для восполнения
убыли в нем воды. Однако, несмотря на эти меры, в производстве
образуются избытки загрязненных ртутью вод, которые необходимо
подвергать очистке.
Сточные воды прежде всего пропускают через отстойники — ло-
вушки ртути, где задерживается металлическая ртуть, уносимая
при промывке аппаратуры или смывке полов. Применяется также
осаждение ртути в виде сульфида с последующим длительным отста-
иванием в течение 4 сут для отделения образовавшегося осадка.
Для соосаждения и ускорения процесса осветления добавляют
хлорное железо. При этом содержание ртути в сточных водах сни-
жается до 100—300 вес. ч./млрд. [138].
Иногда сточные воды обрабатывают сульфидом и отделяют суль-
фид ртути фильтрованием, однако, как уже говорилось, эти осадки
плохо задерживаются фильтрами.
В сточных водах помимо соединений ртути может присутствовать
также и металлическая мелкодисперсная ртуть. При сульфидном
методе очистки металлическая ртуть не улавливается или улавли-
вается частично, увлекаясь образующимися осадками сульфида
ртути или гидроокиси железа при добавлении хлоридов железа.
Поэтому предложена схема очистки сточных вод и регенерации
ртути из различных шламов, предусматривающая перевод на первой
стадии очистки всей ртути в растворимое состояние обработкой
хлором [139]. После разрушения избыточного активного хлора
раствор фильтруют и извлекают из него ртуть. Извлечение может
быть проведено осаждением ртути в виде сульфидов с применением
соосадителей.
274
Предложено извлекать ртуть, находящуюся в растворе в ионном
состоянии, сорбцией на активированном угле с последующей реге-
нерацией сорбента нагреванием под вакуумом [140], а также сорб-
цией в кислой среде на анионитах с регенерацией их кислым раство-
ром поваренной соли [141]. Для извлечения ртути предложено
также фильтровать эти растворы через слой торфа, насыщенного
агентом осаждения, например сульфидом натрия. Сообщается, что
при этом можно снизить содержание ртути до 1 вес.* ч./млрд. После
насыщения торф сжигают и улавливают ртуть из продуктов сгора-
ния [142], что также не является простой задачей.
Японская фирма «Осаки-Сода» предложила двухступенчатую
очистку растворов от ртути на ионообменных смолах [139, 143].
На первой ступени на смоле марки IE содержание ртути снижается
от 3-—10 до 0,10—0,15 вес. ч./млн. Смола IE регенерируется через 1 —
7 сут. Смола IE не стойка в щелочной среде. Примеси железа, хрома,
ванадия и других тяжелых металлов не мешают извлечению ртути,
если суммарное их количество существенно меньше содержания
ртути в подвергаемой очистке воде.
Вторая ступень очистки проводится на смоле марки MR, которая
далее не регенерируется. Содержание ртути в сточных водах после
второй ступени очистки снижается до 5 вес. ч./млрд.
Предложена также очистка растворов восстановлением соедине-
ний ртути до металлической и отделением ее в циклонах и на филь-
трах [143] или испарением [144]. Для восстановления применяют
боргидрид натрия NaBO4. Если в растворе присутствуют органи-
ческие соединения ртути, их предварительно переводят в неоргани-
ческие хлорированием сточных вод [145].
ЛИТЕРАТУРА
1. Chem. Eng., 66, № 12, 118 (1959).
2. Лозин М. Е. Технология минеральных солей. М., Госхимиздат, 1961.
3. Справочник по растворимости солевых систем. Т. 3. Л., Госхимиздат, 1961.
4. Фурман А. А., Ш р а й б м а н С. С. Приготовление и очистка рас-
сола. М., «Химия», 1966.
5. К у л л е П. А. Труды ВНИИГ. Вып. 20. М., Госхимиздат, 1949.
6. Легенче н к о И. А., Хим. пром., № 8, 469 (1956).
7. Б о б к о П. С. Труды ВНИИГ. Вып. 35. М., Госхимиздат, 1959. См.
с 509.
8. Ind. Eng. Chem., 41, № 10, 2155 (1949).
9. S a nd e r s H. J., G a r d i n e г W. C., Wood J. L., Ind. Eng. Chem.,
45, № 9, 1824 (1953).
10. -Англ. пат. 1162984 (1969).
11. Пат. США 2977189 (1961).
12. Пат. ФРГ 951003 (1956).
13. Chem. Eng., 63, № 4, 110 (1956).
14. МРТУ 18/249—68. Соль поваренная для получения С12 и NaOH методом
электролиза.
15. В о р о т н и ц к и й М. К., Механизмы для добычи и погрузки соли.
М., Пищепромиздат, 1946.
16. Ind. Chim., 49, № 543, 363 (1962).
17. Здановский А. Б., Ж. физ. хим., 20, № 4—5, 379 (1946).
18* 275
18. Dav ion M., Ann. Chim., 8, № 12, 259 (1953).
19. К a r s t e n O., Z. anorg. Chem., 276, №5—6, 274 (1954).
20. Здановский А. Б. Труды ВНИИГ. Вып. 33. M., Госхимиздат, 1956.
21. Англ. пат. 1113869 (1968).
22. Калийные соли и методы их переработки. ИОНХ АН БССР. Минск, изд.
АН БССР, 1963.
23. Андреичев А. И., Нудельман Б. А. Добыча и переработка
калийных солей. М., Госхимиздат, 1960.
24. Вольфкович С. И. Общая химическая технология. Т. 1. М., Гос-
химиздат,- 1959.
25. ГОСТ 4568—49. Технический хлористый калий.
26. В о л к о в Г. И., Хим. пром., № 4, 103 (1953).
27. Волков Г. И., авт. свид. СССР 97867 (1954); Бюлл. изобр., № 4 (1954).
28. Герм. пат. 737979 (1943).
29. Апельцин А. В., Клячко В. А., Лурье Ю. Ю., Смир-
нов А. С. Иониты и их применение. М., Стандартгиз, 1949.
30. Исследования в области ионообменной, распределительной и осадочной
хроматографии. Сборник. М., Изд. АН СССР, 1959. См. с. 48.
31. Справочник по растворимости солевых систем. Т. 2. Л., Госхимиздат, 1954.
32. Борячек А. Ф,, Овечкин Е. К., У р а зовский С. С.
Труды ВИСП. Юбилейный сборник, Харьков, 1949.
33. Т е т е р к и н Е. Н., Водоснабжение и санитарная техника, № 1 (1938).
34. Кургаев Е. Ф. Методические указания по расчету осветлителей.
М., Стройиздат, 1959. .
35. Кульский Л. А., Когановский А. М., Г ороно в-
с к и й И. Г., Ш е в ч е н к о М. А. Физико-химические основы очистки
воды коагуляцией. Киев, изд. АН УССР, 1956.
36. Шрайбман С. С., Фурман А. А., Сыркина И. Г., Вестн.
техн, и эконом, информ., НИИТЭХИМ, № 10, 22 (1960).
37. Chem. Eng., 65, № 15, 56 (1958).
38. Hass R., Chem. Ind. Techn., № 5, 234 (1955).
39» Кургаев E. Ф. Осветлители типа ЦНИИ. M., Трансжелдориздат, 1954.
40. Р е з н и к Н. Ф. Теплосиловые установки. Сборник. Вып. 2. М., Транс-
желдориздат, 1958.
41. Караваев И. И., Резник Н. Ф., Водоснабжение и сантехника,
№ 9, 15 (1959).
42. Г о л ь д ш т е й н Я. Р. Труды НИОхим. Т. 13. М., Госхимиздат, 1959.
См. с. 128.
43. Гольдштейн Я. Р., ЖПХ, № 10, 953 (1947).
44. Ш о й х е т С. Н., Хим. пром., № 4, 134 (1959).
45. А й з е н б е р г В. П., Гладких И. Н. Труды НИИ соляной про-
мышленности, вып. 2 (10), 81 (1959).
46. Ф у р м а н А. А. и др., авт. свид. СССР 276002 (1969); Бюлл. изобр.,
№ 23, 27 (1970).
47. Т s ukamoto К., Хайкан тидэюцу, 13, № 2, 74 (1971).
48. Hight о wer'L, Chem. Eng., 55, № 12, 112 (1948).
49. Ind. Chemist, 27, № 314, 115 (1951); 28, № 333, 451 (1952).
50. Конончук Т. И., Бондаренко H. В., Шахновская
М. 3., Редько Л. П., Дуденкова А. Д., Хим. пром., № 11,
841 (1970).
51. Пат. США 3378336 (1968).
52. Сода то энсо, 9, № 1, 15 (1958).
53. Справочник по содовой промышленности. Токио, изд. Ниппон сода коге
тай, 1960.
54. Конончук Т. И., Пестрик ова Н. И., Тарковская И. А.
ЖПХ, 44, № 6, 1217 (1971).
55. Пат. США 3102085 (1963); англ. пат. 885818 (1961).
56. Н a v 1 i к о v a J., Chem. Prum., 10, № 9, 449 (1960).
57. Ф а йн штейн С. Я. и др., Хим. пром., № 4, 245 (1957).
276-
58. Наг a d а К., J a m а г i К., Electrochem. Techn., 5, № 3—4, 137 (1967).
59. Пат. США 2787591 (1957); 2949412 (1960).
60. Пат. ФРГ 959548 (1957).
61. Бельг, пат. 547874 (1959).
62. В о л к о в Г. И., Пыльнов В. И. и др., авт. свид. СССР 298371
(1969); Бюлл. изобр., № 11, 31 (1971).
63. Спасская Е. К., Якименко Л. М.. Электрохимия, 5, №9,
1058 (1969).
64. Якименко Л. М., Спасская Е. К., Электрохимия, 5, А 9,
1061 (1969).
65. С п а с с к а я Е. К., Я к и м е н к о Л. М. Всесоюзная конференция по
электрохимии. Тбилиси, «Мецниереба», 1969. См. с. 609.
66. Спасская Е. К., Якименко Л. М., Ткаченко В. И.,
Всесоюзная конференция по электрохимии. Тбилиси, «Мецниереба», 1969.
См. с. 611, 613.
67. Ш е й н и н а Е. А., С а л д а д з е К. М. Ионообменные сорбенты в про-
мышленности. Сборник. М., изд-во АН СССР, 1963. См. с. 153.
68. Пат. ЧССР 91138 (1958).
69. Я к и м е н к о Л. М., Хим. наука и пром., 16, №. 6, 691 (1971); Яки-
менко Л. М., Серышев Г. А., Фиошин М. Я., Хим. пром.,
№ 10, 727 (1972).
70. Gnot W., Dy lewski R., Przem. Chem., 46, № 9, 499 (1967); 47 F
№ 4, 210 (1968); Gnot W., Zesz. Nauk. Politech. Chem., № 50, 183
(1969); № 52, 3 (1970).
71. Франц, пат. 1259262 (1961).
72. Dylewski R. et al., Chem. Stosowana, Ser. A, № 3, 295 (1970).
73. D e Nora O., Chem. Ing. Techn., 43, № 4, 182 (1971).
74. Романушкина A. E., Баранник Б. П., Хим. пром., № 4,.
116 (1953).
75. Ш р а й б м а н С. С., Хим. пром., № 4, 113 (1947).
76. Г а р б е р М. Н., Коррозия и борьба с ней, 6, № 1, 32 (1940).
77. Chem. Rundshau, 23, № 10, 179 (1970).
78. S t г е w е W., Chem. Ing. Techn., 43, № 4, 152 (1971).
79. Л о м а к и н И. Л., Радун Д. В., Левачев А. Г., Вала
шов Л. Н. Автоматизация хлорных производств. М., «Химия», 1967.
80. К а н о н ч у к Т. И. и др., Хим. пром., Украины, № 1, 11 (1969). V
81. Крашенинникова А. А., Фурман А. А., Ульяно-
ва Г. С., ЖПХ, 44, 10, 2183 (1971).
82. Пат. США 2692818 (1954); 2087278, 2705219 (1955); франц, пат. 1424111
(1966); 2123800 (1972).
83. Пат. США 2910140 (1959).
84. Б а л л о Л. Г., Вести, техн, и эконом, информ., НИИТЭХИМ, № 6,
43 (1958).
85. Chem. Week, 90, № 4, 33 (1962); 88, № 12, 155 (1961).
86. DAS 1567575 (1970).
87. Франц, пат. 2017850 (1970); пат. СССР 323892 (1969); Бюлл. изобр., № 1,
219 (1972); англ. пат. 1271521 (1972); пат. ФРГ 1946096 (1972).
88. Крашенинникова А. А., Кулясова А. С., Фурман А. А.,
Юрков Л. И., Хим. пром., № 1, 73 (1971).
89. Chem. Ing. Techn., 35, № 5, 337 (1962).
90. Азизов Б. М., Лобашов А. К., Николаев А. М., авт. свид.
СССР 194773 (1967); Бюлл. изобр., № 9 (1967); Лобашов А. К.,
Гольдинов А. Л., Азизов Б. М., Развалов Г. И., Хим.
пром., № 6, 443 (1972).
91. Щербак С. К., Синайский В. С., авт. свид. СССР 173724 (1965);
Бюлл. изобр., № 16 (1965).
92. Бельг, пат. 762478 (1971).
93. Nichols J. Н., Brink I. A., Chem. Eng., 71, № 12, 221 (1964).
94. Chem. Eng., 69, № 2, 133 (1962).
95. Пат. США 2199797 (1943); 2700431 (1955); 2822889 (1958).
277
$6. М а тли с Я. В.,Чихладзе В. Д.,Утробина Ц. Т., С ыс о е/у
в а А. В., Хим. и нефт. маш., № 11, 16 (1971).
97. Sommers Н. A., Chem. Age India, 21, № 11, 1013 (1970).
98. Europ. Chem. News, 19, № 482, 30 (1971).
99. С зри дзава X., Тока К., Сода то энсо, 20, № 11, 558 (1969).
100. Chem. Eng. News, 49, № 48, 27 (1971); 50, № 7, 15 (1972).
101, OPDR (Oil Paint and Drug. Rep.), 201, № 8, 7, 11 (1972).
102. Тарковская И. А., Конончук T. И., Пестрико-
ва Н. И., ЖПХ, 43, № 12, 2623 (1970).
103. Chem. Week, 107, № 24, 50 (1970).
104. Правила и нормы техники безопасности и промышленной санитарии для
105.
106.
107.
строительства и эксплуатации производства электрохимического каустика,
хлора и водорода по диафрагменному методу. М., Госхимиздат, 1957.
Trans. Electrochem. Soc., 82, 341 (1942); 86, 127 (1944); 86, 143 (1944).
Intern. Cooper. Adm. Office. Ind. Ing. Serv., 1959, IR-22275PR.
Якименко Л. M. Электролизеры с твердым катодом. M., «Химия»,
1966.
108. Файнштейн С. Я. Производство хлора методом диафрагменного
электролиза. М., «Химия», 1964.
109. Хорьков а Н. И., Хим. пром, за рубежом, НИИТЭХИМ, № 4, 29
(1969).
110. С и р е н к о И. И., Шека И. А., Хим. технол., № 4, 20 (1971). u
111. Чвирук В.П., Конева Н.В., Укр. хим. журн., 37, № 4, 368 (1971);
авт. свид. СССР 323358 (1969); Бюлл. изобр., 1, 81 (1972); Пусто-
вит В. Т., Булавин П. Н., Хим. пром., № 1, 38 (1973). v
112. Чвирук В. П., Конева Н. В., Овруцкий М. И., Укр.
хим. ж., 38, № 3, 275 (1972).
ИЗ. Плановский А. Н., Ромм В. М., Коган С. 3. Процессы
и аппараты химической технологии. М., Госхимиздат, 1962.
114. Winkler R., Chem. Rundschau, 25, № 3, 37 (1972).
115. Орлова Ф. А., Королева Н. А., Селезнев Г. В., Б о -
н ю к П. В., авт. свид. СССР 220392 (1968); Бюлл. изобр., № 20 (1968).
116. Круглый С. М. Производство хлора, каустической соды и водорода^
М., «Высшая школа», 1967.
117. Бородулина Е. К. и др., Хим. пром., № 9, 683 (1969).
118. Бородулина Е. К., Горелова В. А., Спектор И. Э.,
Ф у р м а н А. А., Воропанов В. С., Хим. пром., № 1, 38 г
(1970); Конончук Т. И., Рогозовская М. 3., Лукья-
нова Н. К., авт. свид. СССР 363660 (1970); Бюлл. изобр., № 4, 52
(1973).
Пат. США 2373257 (1942).
119.
120. Chem. Ind., 61, № 1, 41 (1947); 63, № 8, 230 (1948).
120а. Europ. Chem. News, 22, № 566, 32 (1972); Chem. Ind.
(1973); Cor Isom G. A., Ester E. E., Jacqu
Ing. Techn., 45, № 4, 217 (1973); Feathers R. E.,
Chem. Eng., 80, № 11, 122 (1973); пат. США 3459646,
, 24, № 12, 794
ean D., Chem.
Wyche J. E.,
3647653, 3650925
(1972).
121. Герм. пат. 623876;(1936).
122. Пат. ФРГ 1592022](1971).
123. Chem. Week, 108, № 28, 43 (1971).
124. Воробьева Г. Я. Коррозионная стойкость материалов в агрессив-
ных средах химических производств. М., «Химия», 1967.
125. Can. Chem. Proc., 54, № 6, 54 (1970).
126. Chem. Eng. Progr., 46, № 10, 486 (1950).
127. Badger W. L., S t a n d i f о r d F. C., Chem. Eng., 61, № 2, 183
(1954).
128. Chem. Eng., 61, № 3, 193 (1954).
129. Chem. Proc. Eng., 38, № 1, 31 (1957).
130. Chem. Eng., 57, № 2, 215 (1950).
131. Chem. Eng., 66, № 2, 35 (1959).
278
132. Chem. Eng., 66, № 19, 98 (1959).
133. Волков Г. И. Производство хлора и каустической соды методом
электролиза с ртутным катодом. М., «Химия», 1968.
134. Chem. Ind., № 2, 75 (1972).
135. Europ. Chem. News, 22, № 544, 22 (1972).
136. Europ. Chem. News, 22, № 536, 32 (1972).
137. Japan Chem. Week, 11, № 550, 8 (1970).
138. Chem. Week, 108, № 8, 75, 76, 78 (1971).
139. Gardiner W. C., Minoz F., Chem. Eng., 78, № 19, 57 (1971);
Can. Chem. Proc., 57, № 6, 50 (1973); DAC 2230367 (1973).
140. Зайцев M. Г.,Белянская А. В., авт. свид. СССР 333133 (1970);
Бюлл. изобр., № 11, 89 (1972).
141. Леонтович Е. В., Постолов Л. Е., Скрынник В. А.,
Левинский М. И., авт. свид. 331038 (1969); Бюлл. изобр., № 9, 65-
(1972).
142. Can. Chem. Proc., 56, № 4, 44 (1972).
143. Rosenz weig N. D., Chem. Eng., 78, № 5, 70 (1971).
144. Europ. Chem. News, 22, № 514, 27; № 515, 26; № 516, 30 (1972); франц,
пат. 2128648 (1972); пат. ФРГ 2134716 (1972).
145. Chem. Eng., 79, № 1, 43 (1972).
ГЛ AB A 5
ДРУГИЕ СПОСОБЫ ПОЛУЧЕНИЯ ХЛОРА
Как известно, вначале для производства хлора использовались
способы окисления соляной кислоты перекисью марганца (способ
Вельдона) или воздухом в присутствии катализаторов (способ Ди-
кона). В начале XX века эти способы были полностью вытеснены
электролизом водных растворов поваренной соли. При производстве
хлора электрохимическими методами с твердым катодом и диафраг-
мой и с ртутным катодом получались одновременно эквивалентные
количества каустической соды или едкого кали при электролизе
растворов КС1. В течение длительного времени потребности народ-
ного хозяйства в каустической соде превышали потребность в хлоре
' и недостающее'количество каустической соды производилось хими-
ческим способом из кальцинированной соды. Однако применение
во многих отраслях народного хозяйства широкого ассортимента
различных хлорпродуктов привело к необходимости очень быстрого
развития производства хлора и его производных. При этом потреб-
ность в хлоре росла быстрее, чем в каустической соде [1—4], и вновь
возник интерес к химическим методам производства хлора, поскольку
они не связаны с одновременным получением каустической соды.
Во всех промышленных странах мира в последние годы в связи
с развитием производства хлора электролизом водных растворов
поваренной соли возрастает доля каустической соды, получаемой
электролитическим путем. Это происходит за счет сокращения
выпуска каустической соды методом каустификации кальцинирован-
ной соды.
При дальнейшем росте мощностей по производству хлора можно
ожидать, что в ближайшие годы химические методы производства
Каустической соды полностью потеряют свое значение и появится
необходимость искать новые области применения каустической соды.
Необходимо, однако, отметить, что, несмотря на высказываемые
юпасения о возможном перепроизводстве каустической соды [5]
и все возрастающую потребность в хлоре, каустическую соду в насто-
ящее время нельзя отнести к продуктам, которые на мировом рынке
предлагаются в количестве, превышающем спрос на них.
Учитывая перспективы развития хлорной промышленности, не-
обходимо признать целесообразным разработку различных методов
производства хлора, не связанных с одновременным получением
280
каустической соды. Все работы в этом направлении, проводившиеся
в течение последних 2—3 десятилетий, можно разбить на три группы
[5—7]:
1) видоизменение электролитического метода производства хлора
с тем, чтобы вместо каустической соды получать кальцинированную;
2) получение хлора электролизом соляной кислоты;
3) химические методы получения хлора из соляной кислоты^
и
хлоридов аммония или щелочных металлов.
Значительные количества хлора получаются в качестве побочнога
продукта при производстве щелочных и щелочноземельных металлов
электролизом расплавов их хлоридов. Наибольшее значение имеет
производство магния и натрия; в значительно меньшем масштабе
это относится к производству кальция, лития и других металлов.
Однако эти источники получения хлора целиком зависят от потреб-
ности в соответствующих металлах и трудно рассчитывать на само-
стоятельное значение их как производителей хлора. В течение по-
следних лет доля этих производств в общей мировой выработке хлора
колебалась в пределах 2—3%.
Были сделаны попытки рассматривать электролиз расплавленных
хлоридов натрия с движущимся свинцовым катодом как способ
производства хлора Q одновременным получением вместо металли-
ческого натрия его окисла или гидроокиси. Имеющиеся по этому
вопросу публикации указывали на высокую экономичность способа
по сравнению с электролизом с ртутным катодом [8—11]. Однака
детальное рассмотрение показало, что предлагаемый способ не может
конкурировать с используемыми в настоящее время способами
электролиза с ртутным катодом и с диафрагмой. Метод электролиза
расплавленной поваренной соли с применением движущегося свин-
цового катода при условии успешного решения ряда технических
вопросов и разработки промышленной технологии может быть эконо-
мичным для крупного производства металлического натрия.
Ниже мы коротко рассмотрим указанные выше три группы
способов получения хлора без одновременного получения экви-
валентного количества каустической соды.
ЭЛЕКТРОЛИЗ ВОДНЫХ РАСТВОРОВ ЩЕЛОЧНЫХ МЕТАЛЛОВ
С ПОЛУЧЕНИЕМ КАЛЬЦИНИРОВАННОЙ СОДЫ
/
Этот процесс был предложен в начале развития электрохими-
ческого метода производства хлора и каустической соды [12], ча-
стично применялся в промышленности, но не нашел широкого рас-
. пространения, так как замена каустической соды более дешевым
продуктом — кальцинированной — экономически нецелесообразна.
В последнее время в связи с предполагаемым избыточным производ-
ством каустической соды вновь обратились к этому процессу [13].
Предложено проводить карбонизацию католита в катодном простран-
стве электролизера с целью получения карбонатов. Замена ионов ОН- г
обладающих очень высокой подвижностью, менее подвижными
281
ионамй СО|~ позволяет проводить процесс электролиза с более
высокой степенью конверсии поваренной соли в щелочь без снижения
выхода по току. Конверсия поваренной соли может быть увеличена
до 70% вместо 50—55% при электролизе по методу с твердым катодом
и диафрагмой.
Исследовался также процесс электролиза с ионообменной диа-
фрагмой с получением хлора и карбоната или бикарбоната натрия [13].
При размещении производства хлора на содовом заводе электролити-
ческие щелока могут быть направлены для карбонизации на про-
изводство кальцинированной соды или для регенерации NH3 из
фильтровой жидкости [14]. Ив том и в другом случае взамен каусти-
ческой соды получают более дешевую кальцинированную соду.
Пока эти методы не нашли широкого применения в мировой промы-
шленности. В нашей стране и в ближайшие 20—30 лет вряд ли будут
созданы условия, благоприятствующие применению этих методов
в промышленности.
Имеются сообщения о применении этих методов в США, Бельгии
и Японии в ограниченных масштабах [14а]. Завод фирмы Дау во
Фрипорте мощностью 295 т/сут. был закрыт в 1970 г. вследствие
нерентабельности.
Предложено использовать для разложения амальгамы растворы
карбонатов или бикарбонатов [146], а также многочисленные ва-
рианты карбонизации каустической соды, получаемой электролизом
с ртутным катодом [14в], однако сообщений об использовании этих
процессов в промышленности нет.
ЭЛЕКТРОЛИЗ СОЛЯНОЙ кислоты
Организация производства соды по аммиачному методу Сольве
была причиной того, что в течение многих лет соляную кислоту
не получали в промышленности в виде побочного продукта и в связи
с этим не возникала проблема рационального использования хло-
ристого водорода.
Однако с развитием крупнотоннажного производства хлорорга-
нических продуктов положение изменилось. В процессах замести-
тельного хлорирования углеводородов половина расходуемого хлора
выделяется в виде хлористого водорода, более или менее загрязнен-
ного примесями органических продуктов, а также инертными газами
(в зависимости от процесса хлорирования).
Процессы, в которых используется соляная кислота, развиваются
в народном хозяйстве менее быстрыми темпами, чем процессы хлори-
рования различных органических продуктов. Поэтому в развитых
промы
н
ленных странах за последние годы сильно изменилась струк-
тура баланса производства соляной кислоты и хлористого водорода.
В табл. 5-1 приведены данные о производстве соляной кислоты
и хлористого водорода в США за последние годы.
При быстром росте общего производства соляной кислоты доля
метода получения ее из поваренной соли и серной кислоты снижается.
Количество абгазной кислоты сильно возрастает.
282
Таблица 5-1. Производство соляной кислоты в США
(в пересчете на 100% НС1)
Год Всего, тыс. т 1 В том числе (в %) Год Всего, тыс. т В том числе (в %)
ив соли и H2SO4 i из хлора и водорода из абгазов и др. р из соли и HsSO4 из хлора и водорода ‘ из абгазов и др.
1951 633 25,4 17,6 57,0 1964 1112 7,5 11,5 81,0
1952 622 23,8 20,6 55,6 1965 1190 10,8 1(1,6 77,6
1954 674 19,6 21,3 59,1 1966 1362 9,0 14,0 77,0
1956 826 15,7 19,8 64,5 1967 1453 8,7 7,1 84,2
1958 772 12,7 18,8 68,5 1968 1560 7,1 6,4 86,5
1960 880 5,0 15,9 79,1 1969 1680 7,3 6,7 86,0
1962 959 9,3 13,7 77,0 1970 1740 6,4 6,8 86,8
1963 950 11,8 12,8 75,4
Изменение структуры
приведено в табл. 5-2.
производства соляной кислоты в СССР
Таблица 5~2. Производство соляной кислоты в СССР
(в пересчете на 100% НС1)
В том числе (в %)
В том числе (в %)
1954 58,1 29,2 52,5 18,3
1956 77,1 24,5 50,3 25,2
1958 89,7 20,4 52,2 27,4
1960 102,9 18,5 47,5 34,0
1962 122,5 17,5 36,6 45,9
1963 136,6 15,4 34,6 50,0
1964 146,5 15,2 34,7 50,1
1965
1966
1967
1968
1969
1970
178,2
189,8
197,6
222,8
258,8
266,3
12,5
11,4
11,7
9,8
8,0
5,3
38,3
30,6
28,2
27,8
28,3
30,1
49,2
58,0
60,1
62,4
63,7
64,6
Большие количества хлористого водорода могут быть получены
также в качестве побочного продукта при производстве окиси магния
для промышленности огнеупоров из хлоридов магния, выделяемых
из рапы, или при производстве хлористого калия для удобрений
из смешанных солей калия и магния. На 1 т MgO, получаемой этим
методом, образуется одновременно 1,55 т хлористого водорода.
Источником хлористого водорода может быть также производство
фосфатов калия из хлористого калия и фосфорной кислоты. Разра-
батываются новые способы регенерации аммиака из хлорида аммония
283
11
в производстве кальцинированной соды с получением хлористого
водорода или хлора. В качестве-очень важной народнохозяйственной
проблемы вновь выдвигается вопрос о рациональном использовании
больших количеств отходного хлористого водорода и соляной кис-
лоты. В последние годы этой проблеме уделялось много внимания
в ряде стран. Работы велись в нескольких направлениях, из них
наиболее важные:
1) получение товарной соляной кислоты из абгазного хлористого
водорода; очистка абгазной соляной кислоты от загрязняющих ее
примесей;
2) использование абгазного хлористого водорода в хлороргани-
ческом синтезе;
3) использование абгазного хлористого водорода для окислитель-
ного хлорирования органических соединений;
4) регенерация хлора из абгазного хлористого водорода электро-
химическими методами;
5) регенерация хлора из абгазного хлористого водорода хими-
ческими методами.
Возможности использования соляной кислоты, полученной аб-
сорбцией абгазного хлористого водорода, ограничены потребностями
народного хозяйства в соляной кислоте. Загрязнения, содержащиеся
в такой кислоте, дополнительно сокращают возможности ее потреб-
ления. Потребность в соляной кислоте может существенно возрасти
при использовании ее для травления металлов взамен серной кислоты.
Разработанные в последнее время способы очистки абгазной соля-
ной кислоты от загрязняющих ее примесей и способы получения
100%-ного чистого хлористого водорода или чистой соляной кислоты
расширили возможные области использования абгазного хлористого
водорода.
Наиболее перспективно применение абгазного хлористого водо-
рода в хлорорганическом синтезе вместо применявшегося для этой
цели синтетического хлористого водорода. При производстве хло-
ристого винила, найрита, хлористого этила, хлористого метила и др.
могут потребляться многие сотни тысяч тонн, а в перспективе и мил-
лионы тонн чистого хлористого водорода. Существенным недостатком
такого использования абгазного хлористого водорода являются
ограничения при выборе сырья для соответствующих производств:
ацетилена для производства хлорвинила, метанола — для хло-
ристого метила и т. д.
Поэтому, особенно в последнее время, привлекает внимание
возможность применения хлористого водорода в смеси с воздухом
или кислородом для окислительного хлорирования таких органи-
ческих соединений, как этилен, бензол, метан и др. [15—19]. Боль-
шие экономические преимущества и широко поставленные в этом
направлении научно-исследовательские и опытные работы обеспе-
чили быстрое продвижение методов окислительного хлорирования
в промышленность.
284
ПРЯМЫЕ МЕТОДЫ ЭЛЕКТРОЛИЗА СОЛЯНОЙ КИСЛОТЫ
При электролизе водных растворов соляной кислоты на катоде
всегда выделяется водород. Процессы на аноде зависят от условий
проведения электролиза, в частности
температуры, материала анода и др.
процесса в зависимости от концен-
трации HG1 в электролите показано
[20] на рис. 5-1.
При малых концентрациях HG1
в электролите на аноде происходит
преимущественно разряд ионов ОН",
доля разряда Gif в общем токе со-
ставляет менее 50%. G увеличением <
концентрации HG1 в электролите до
8—10% процессы разрядки ОН', ,
выделения кислорода и образования
хлорной кислоты подавляются и на
аноде происходит преимущественно
разряд СР. Выход хлора на аноде
по току приближается к 100%.
Электродвижущая сила хлорно-
водородного элемента в нормальных
условиях составляет около 1,36 В.
Напряжение на ячейке при электро-
лизе содяной кислоты всегда выше
этого рачения и зависит от приме-
няемой* плотности тока, температуры
ства ячейка.
Исследован [21] баланс напряжения Электролитической
при различной концентрации соляной кислоты в электролите в ин-
тервале плотностей тока от 1 до 5 кА/м2. При расстоянии между
электродами 17 мм и для диафрагмы из фторлоновой ткани получены
данные, приведенные в табл. 5-3 и 5-4.
, от концентрации кислоты,
Изменение характера анодного
1ОО
25
В
50
в ю
Концентрация HCI, %
Рис. 5-1. Анодные процессы при
электролизе разбавленной соля-
ной кислоты:
1 — разряд гидроксил-ионов; 2 — раз-
ряд ионов хлора; з — выделение Ки-
слорода; 4 — образование
кислоты.
хлорной
состава электролита и
устрои-
ячеики
Таблица 5-3. Баланс напряжения (в В) на ячейке при 70 °C
и концентрации HCI в электролите 195,8 г/л при различной плотности тока
Показатели
Плотность тока, кА/мв
1,0 2,0 3,0 4,0
5,0
Напряжение на ячейке . . .
- Потенциал анода ............
» катода ...............
Падение напряжения
в электролите ......
в диафрагме . . . . . .
Небаланс ............... .
1,82 2,08 2,34 2,62 2,85
1,406 1,487 1,555 1,620 1,666
0,323 0,377 0,419 0,458 0,490
0,073 0,206 0,312 0,459 0,582 '
0,049 0,069 0,103 0,139 0,176
+0,03 +0,06 +0,05 +0,05 +0,06
285
Таблица 5-4. Напряжение на ячейке (в В) при 70~ С
и различных плотности тока и концентрации НС1 в электролите
К онцентрация НС1 в электролите, г/л Плотность тока, кА/м2
1,0 2,0 3,0 4,0 5 Л
195,8 1,82 2,08 2,34 2,62 2,85
180,0 1,78 2,04 2,28 2,52 2,74
169,6 1,80 2,06 2,32 2,57 2,78
157,0 1,90 2,12 2,36 2,58 2,80
144,3 1,96 2,20 2,44 2,67 2,88
133,5 2,00 2,28 2,52 2,76 3,00
105,5 2,00 2,27 2,52 2,79 3,02
95,6 2,03 2,32 2,59 2,85 3,10
Анодный газ после отмывки его от НС1 содержал 99,5—99,6% С12,
0,06—0,15% О2 и 0,08—0,12% СО2, что свидетельствует о высоком
выходе по току на аноде.
При электролизе соляной кислоты особую трудность представляет
подбор конструкционных и защитных материалов. Высокая агрес-
сивность соляной кислоты, особенно при повышенной температуре,
ограничивает круг материалов, пригодных для изготовления деталей
электролизеров и особенно его электродов.
Неоднократно предлагали использовать металлические электроды
для электролиза соляной кислоты [23]: катоды из стали, никелиро-
ванной стали или сплавов никеля [25—26], а также покрытые актив-
ным слоем мелкодисперсного серебра [24]; предлагали использовать
и металлические аноды с покрытиями из иридия или сплавов пла-
тины с иридием [27]. Однако о практическом применении металли-
ческих анодов в промышленном электролизе соляной кислоты све-
дения отсутствуют. Отсутствие металлов, достаточно стойких в среде
горячей соляной кислоты, делает сомнительным целесообразность
применения металлических электродов в этом процессе. Из электрод-
ных материалов только графит удовлетворяет основным требованиям,
предъявляемым к электродным материалам. Он достаточно стоек
при анодной и катодной поляризации в горячей концентрированной
соляной кислоте, имеет сравнительно хорошую электропроводность
и невысокую стоимость [22 ].
В качестве материала для анодов при электролизе НС1 исполь-
зуется графит. Однако графитовые аноды разрушаются во время
работы из-за окисления их при разрядке на аноде ионов ОН’ или
других кислородсодержащих анионов. При снижении концентра-
ции НС1 в электролите наблюдается размягчение и набухание гра-
фита. После нескольких месяцев работы в электролите, содержащем
около 10% HG1, графитовые аноды полностью теряют механическую
прочность [20]. При проведении электролиза достаточно концентри-
рованной соляной кислоты в отсутствие окислителей износ графита
4
может быть снижен до 0,1 кг/т хлора и менее. Разрушение анодов
можно умень
II
:ить, поддерживая
оптимальную концентрацию соля-
ной кислоты в электролите.
Во всех конструкциях электролизеров, применяемых в промыш-
ленности, в качестве катодного материала также используется графит.
В последние годы появились полимерные материалы, достаточно
стойкие в условиях электролиза соляной кислоты и пригодные для
изготовления деталей электролизеров, однако до сих пор широко
используются прессованный графит и фенолоформальдегидные смолы
для изготовления рам электролизера и поливинилхлоридная ткань
для диафрагм.
Потеря напряжения на преодоление сопротивления электролита
и диафрагмы зависит от электропроводности электролита, темпера-
туры процесса, расстояния между электродами, типа применяемой
диафрагмы и плотности тока.
Электропроводность соляной кислоты сильно зависит от кон-
центрации:
Электропроводность при 18° С
Концентрация, %
0
5
10
20
30
40
удельная, Ом-^см™1
0,3948
0,6302
0,7615
0,6620
0,5152
эквивалентная,
Ом-1*см2 (г-экв)
380,5
281,0
219,1
126,2
69,8
39,1
На рис. 5-2 показано изменение электропроводности соляной
кислоты с ростом концентрации при различной температуре. Опти-
мальная электропроводность изменяется при изменении температуры
и при 60—80 °C находится в интервале концентраций 15—20%;
с повышением температуры максимум электропроводности становится
более резко выраженным.
Процесс электролиза ведут при концентрации соляной кислоты,
близкой к оптимальной электропроводности. При этой концентрации
кислоты процесс разрядки ионов хлора на аноде идет с большим
выходом по току и наименьшим расходом графитовых анодов. В та-
ких условиях парциальное давление HG1 над электролитом еще не
очень высоко и унос хлористого водорода с газообразными продук-
тами электролиза не чрезмерно велик.
Предлагали применять в качестве электролита смесь соляной
и серной кислот (содержание серной кислоты до 30%) [28, 29].
Электропроводность такого раствора высокая, поэтому напряжение
на ячейке снижается, однако при этом уменьшается стойкость гра-
фитовых анодов. Поскольку в качестве электродного материала для
анода и для катода используется графит, упрощается конструкция
биполярного электрода, что в значительной степени определяет
выбор биполярного типа конструкции электролизера.
К диафрагме для разделения газов предъявляются значительно
более жесткие требования, чем в других процессах электролиза.
287
Это обусловлено сравнительно высокой растворимостью хлора
в соляной кислоте. При концентрации НС1 и температурах, приме-
няемых в электролизе соляной кислоты, в 1 л электролита раство-
ряется около 1,5 г хлора.
Для предотвращения снижения выхода хлора по току за счет
попадания его в катодное пространство и последующего катодного
восстановления диафрагма должна не только предотвращать про-
никновение пузырьков хлора из анодного пространства в катодное,
в
§ о Ю 20 30
Нонцентрацая HCI, %
Рис. 5-2. 3 ависимость
электропроводности со-
ляной кислоты от ее
концентрации при раз-
личных температурах:
1 — раствор НС1при 94 °C;
2 — то же, при 80 °C; 3 —
то же, при 60 °C; 4 — то же,
при 10 °C; 5 — раствор
NaCl при 60 °C.
ботке.
но и ограничивать попадание анолита в ка-
тодное пространство за счет фильтрации
или диффузии раствора через диафрагму.
Для соблюдения этого требования нужно
применять более плотную диафрагму, с 66 л ь-
шим сопротивлением диффузии по сравнению,
например, с диафрагмой при электролизе
воды. Поэтому обычно сопротивление диа-
фрагм, применяемых при электролизе со-
ляной кислоты значительно (в 5—10 раз)
выше сопротивления асбестовых диафрагм
при электролизе воды.
Для разделения получаемых при элек-
тролизе хлора и водорода применяют диа-
фрагмы из поливинилхлоридной, поливини-
лиденхлоридной или политетрафторэтиле-
новой [20] ткани или перфорированной
фольги, выдерживающих температуру до
95—100 °C [28—30]. Стойкость диафрагмы
из поливинилхлоридной ткани улучшают
введением некоторого количества фтор-
органических полимеров. По краям диа-
фрагму делают непроницаемой, что дости-
гается ее обработкой, например, поливинил-
хлоридным лаком. Края диафрагмы зажима-
ются между соседними рамами электроли-
зера. Для получения необходимой плот-
ности диафрагмы ткань подвергают соответствующей обра-
Применение политетрафторэтиленовых диафрагм позволяет ра-
ботать при более высоких рабочих температурах электролиза, что
выгодно с точки зрения снижения напряжения на ячейке и расхода
электроэнергии. ,
Диафрагма из стеклянной ткани оказалась недостаточно стойкой.
. Для изготовления других деталей электролизера, соприкасаю-
щихся с электролитом и продуктами электролиза, применяют мате-
риалы, стойкие к соляной кислоте и хлору (импрегнированный
графит, кислотостойкие пластические массы, например хавег, поли-
винилхлорид, полиэтилен, эбонит, фаолит, а также гуммированная
сталь).
288
Применяются биполярйые графитовые электроды с выступами
как с катодной, так и с анодной стороны. Был предложен и испыты-
вался длительное время насыпной анод. Однако при эксплуатации
насыпных анодов, по-видимому, возникли трудности, и в последних
моделях электролизеров применяются преимущественно сплошные
аноды. Эти трудности обусловлены необходимостью обеспечения
достаточного электрического контакта между кусками насыпного
анода и графитовой пластиной биполярного электрода во время ра-
боты электролизера. По мере разрушения кусков графита в насып-
ном аноде будет возрастать электрическое сопротивление в точках
касания кусков насадки между собой и с графитовой пластиной би-
полярного электрода. Восстановление контакта возможно только
за счет уплотнения насадки под действием силы тяжести вышележа-
щего слоя насадки анода. При плохом контакте между кусками
участие насадки в процессе электролиза может снизиться за счет
протекания анодного процесса на поверхности графитовой плиты
биполярного электрода. В этом случае насыпная насадка может
сыграть отрицательную роль, увеличивая расстояние между основ-z
ными электродами и ухудшая условия выделения газообразного
хлора и циркуляции) электролита. При больших размерах электро-
дов равномерное распределение насадки затруднительно.
Применение монолитных анодов позволяет при том же напряже-
нии на ячейке увеличить примерно вдвое плотность тока. При одном
и том же удельном расходе электроэнергии капиталовложения при
применении монолитных анодов сокращаются почти вдвое по сравне-
нию с насыпными анодами. В процессе эксплуатации монолитных
анодов происходит увеличение межэлектродного расстояния из-за
износа анодов. Однако, вследствие малого удельного износа графи-
товых анодов, увеличение напряжения на ячейке в связи с ростом
межэлектродного расстояния невелико. Дополнительный расход
на периодическую смену анодных плит с избытком компенсируется
перечисленными выше преимуществами монолитных анодов.
Процесс электролиза соляной кислоты и конструкция электро-
лизера были разработаны в 30-х годах на заводе в Биттерфельде [31,
32]. В течение длительного времени этот процесс не находил приме-
нения в промышленности. Однако исследования в этой области и
усовершенствование конструкции электролизеров продолжались
в ряде стран. Проводились работы по интенсификации процесса
электролиза за счет повышения плотности тока до 2,5 кА/м2 и выше,
снижению напряжения на ячейке электролизера, улучшению каче-
ства диафрагмы [20, 33]. Было предложено много различных вариан-
тов технического осуществления процесса электролиза соляной
кислоты и конструкции биполярных электролизеров и их отдельных
узлов [34—39], из которых только немногие получили применение
в промышленности.
Первая промышленная установка по получению хлора электро-
лизом соляной кислоты пущена в 1958 г. [29]. В последние
годы построены установки по электролизу соляной кислоты
19 Заказ 843 >89
производительностью до 110 т/сут хлора. Сообщается об эксплуата-
ции и строительстве установок'по электролизу соляной кислоты в
ФРГ, Италии, США и других странах [40—47].
Электролизеры для соляной кислоты допускают изменение на-
грузки в широких пределах. С увеличением нагрузки увеличивается
плотность тока (и соответственно напряжение на ячейке) и удельный
расход электроэнергии на 1 т хлора. Ниже приведена зависимость
напряжения на ячейке и удельного расхода электроэнергии [48]
от плотности тока:
Плотность Напряже-
тока, кА/м* ние, В
0,8 1,85
1,2 1,95
1,6 2,04
2,0 2,14
2,4 2,23
выход электро-
энергии, Плотность Напряже-
кВт-ч/т С12 тока, кА/м* ние, В
1445 2,8 2,32
1520 3,2 2,42
, 1595 3,6 2,52
1665 4,0 2,62
1740
Выход электро-
энергии,
кВт*ч/т С12
1815
1890
1965
2040
Ряд фирм поставляет установки для злектролиза соляной кис-
лоты, мало отличающиеся между собой по схеме и конструкции элек-
тролизеров.
В рекламном проспекте фирмы «Уде» приводятся следующие пока-
затели биполярного электролизера, состоящего из 30 ячеек:
Нагрузка, А
2000 3000
г
Анодная плотность тока, А/м2 ......... 1540 2300
Напряжение на ячейке, В ........ 2,0 2,3
Выход по току, % ........... 98 98
Концентрация соляной кислоты, %
поступающей в электролизер ..... 30 30
отводимой из электролизера ..... 20 20
Температура в электролизере, °C ..... . 70 70
Расход постоянного тока, кВтч/тС12 . . 1635 1850
Содержание примесей в хлоргазе, объемн. %
водорода . . . . . . . . • •. . . . . . 0,5 0,5
двуокиси углерода . . . . .... . 0,05 0,05
хлористого водорода . . . . . . . . . Следы
Водород до промывки содержит не более 2%' хлора. После про-
мывки чистота водорода возрастает до 99,5%, промытый водород
содержит только следы хлора и хлористого водорода. Общий вид
электролизера приведен на рис. 5-3.
Электролизеры для соляной кислоты выпускает также фирма
«Де-Нора» [49—51]. Промышленные электролизеры имеют 40 и более
ячеек. Рамы электролизера выполняют из фаолита, диафрагмы —
из импрегнированной поливинилхлоридной ткани, электроды —
из графита. Трубопроводы изготовляют из фаолита или поливинил-
хлорида. В электролизер подают 33%-ную охлажденную соляную
кислоту, из электролизера отводят отработанную 18%-ную НС1,
которая вместе с промывной жидкостью из колонн для хлора и во-
290
дорода поступает на донасыщение абгазным хлористым водородом
и после охлаждения вновь возвращается на электролиз.
Примеси метиленхлорида вызывают набухание [29] волокон
поливинилхлоридной диафрагмы. Высокомолекулярные органические
1
Рис. 5-3. Общий вид электролизера фирмы «Уде».
соединения мало растворимы в кислоте, легко отделяются в ре-
зультате отстаивания и, по-видимому, не вызывают затруднений
при проведении электролиза. Хлор и водород, выходящие из элек-
тролизера, промывают охлажденной
разбавленной соляной кислотой для
улавливания паров Хлористого водо-
рода и воды и затем сушат обычными
способами. Напряжение на электроли-
зере из 35 ячеек в зависимости от на-
грузки приведено на рис. 5-4.
Общий вид электролизера Де-Нора
изображен [29] на рис. 5-5. Электро-
лизер Де-Нора биполярный фильтр-
прессного типа. Комплект ячеек за-
жимается между двумя крайними стяж-
ными плитами
I
с помощью
упорных
Напряжение на электро -
лазере, В
Рис. 5-4. Зависимость напря-
жения на электролизере фир-
мы «Де Нора» из 35 ячеек от
нагрузки.
винтов. Свежая кислота подводится
в каждую ячейку через нижний рас-
пределительный канал. Газы (водород
и хлор) собираются в верхних газо-
сборных каналах и после отделения
в сепараторах от брызг кислоты отво-
дятся из злектролизеров. Подвод тока
к крайним монополярным электродам —
аноду и катоду — осуще-
ствляется через графитовые стержни, присоединяемые сверху к гра-
фитовой плите монополярного электрода.
На рис. 5-6 показано устройство ячейки в случае применения
монолитного анода, а на рис. 5-7 — насыпного кускового анода.
19*
291
0<pV
ппплгЬппл
2
Отвод
водорода
Кислота
Слив
На абсорбцию
Рис. 5-5. Схема электролизера фирмы «Де Нора» на 40 ячеек:
1 — биполярные ячейки; 2 — монополярный катодный элемент; 3— моиополярный анод-
ный элемент; 4 — подвод тока й катоду; 5—• подвод тока к аноду; 6 — стяжная плита; 7 —
упорный винт; 8 — рама для стяжки электролизера,
в 7
кислоты
Рис. 5-6. Биполярная ячейка элек-
тролизера с монолитным анодом:
1 — рама ячейки; 2—диафрагма; 3—
каНал Для подвода кислоты; 4 — катод-
ные графитовые выступы; 5 — графито-
вая разделяющая плита; 6 анодные
графитовые выступы; 7 — канал для
отвода водорода; 8 — канал для отвода
хлора.
3 2
Рис. 5-7. Биполярная ячейка электро-
лизера с кусковым насыпным ано-
дом:
1 — рама ячейки; 2 — графитовая разделя-
ющая плита; 3 — катодные графитовые вы-
ступы; 4 — насыпной кусковой анод; 5 —
канал для подвода кислоты; 8 — канал для
отвода хлора; 7 — канал для отвода водорода;
8 — диафрагма.
4
электролизеров фирмы
Ниже приведены основные показатели
«Д^-Нора»:
Нагрузка, А
1500 5000
Число ячеек . ............................. 40 50
Напряжение на электролизере, В .... 92 118
Потребляемая мощность постоянного тока,
кВт ...................................... 140 590
Номинальная поверхность электрода, м2 . 1,6 2,6
Плотность тока, А/м2 ..................... 930 1920
Общие размеры, м
длина ................................... 5,5 7,2
НГ1
высота .................................. 2,4 2,8
Расход 33% -ной соляной кислоты, л/ч . . 530 2200
Производительность электролизера
хлора, кг/сут ............................ 1770 7450
водорода, м3/сут...................... 518 2330
Требуемая площадь пола, м2................6,4 X 3,05 —
На производство 1 т С12 расходуется 1930 кВт-ч электроэнергии
постоянного тока на электролиз и 44 кВт-ч для вспомогательных
. процессов, 1,25 кг кускового графита и 40 м3 охлаждающей воды.
При работе с монолитным анодом указывается расход 0,1 кг
графита.
При включении электролизера с новыми диафрагмами напряже-
ние На 0,2—0,3 В выше указанного. По мере завершения процесса
формирования диафрагмы (примерно 1 нед.) напряжение снижается
до 2,36 В. Выход по току возрастает с увеличением плотности тока
и составляет при нагрузке 1 кА — 92%, при 1,5 кА — 94 и при
2 кА-95% [29].
Капиталовложения, необходимые для организации производства,
оцениваются в 20 000—25 000 Долл./т С12 в сутки, себестоимость
получаемого хлора около 36 долл./т.
Сообщается о выпуске фирмой «Хехст^Уде» электролизеров,
пригодных для работы при нагрузке от 2 до 10 кА [52]. Электроли-
зер (рис. 5-8) биполярного типа имеет 30 ячеек. Длина электроли-
зера 3,5 м, ширина — 2,2 м, высота — 2,2 м. Один электролизер
занимает площадь пола 4x4,8 м. Количество ячеек в электролизере
может быть увеличено до 45. Рабочая поверхность электрода 2,5 м2.
При нагрузке на электролизер 6 кА процесс электролиза проходит
при плотности тока 2,4 кА/м2 и обыщем расходе электроэнергии,
включая расход на насосы, 1800 кВт-ч/т С12. При номинальной на-
грузке 10 кА плотность тока возрастает до 4 кА/м2 и общий удельный
расход электроэнергии увеличивается до 22Q0 кВт-ч/т.
Графитовые биполярные электроды с обеих сторон снабжены
большим числом вертикальных борозд для облегчения выделения
газов и закрепляются в рамах, изготовленных из синтетических
материалов, стойких к воздействию соляной кислоты и хлора.
Диафрагмой служит ткань из поливинилхлорида. В электролизере
предусматривается раздельная циркуляция анолита и католита.
293
В электролизер поступает 23—25 % -пая и отводится примерно 18—
20%-ная НС1. Часть кислоты из цикла выводят для донасыщения
в абсорбционной колонне и возвращают вновь в цикл в виде 30 %-ной
для увеличения концентрации электролита. Рабочая температура
в| электролизере 75—80 °C.
Рис. 5-8. Общий вид биполярного электролизера
фирмы «Хехст-Уде» с 30 ячейками.
Хлор и водород охлаждаются в холодильниках смешения, здесь же
улавливаются пары НС1. Холодильники смешения орошают разба-
вленной соляной кислотой, циркулирующей в системе. Хлор сушат
серной кислотой, а водород отмывают раствором щелочи от увле-
каемого им НС1.
Если в анодном пространстве ячеек электролизера поддерживать
повышенное давление (по сравнению с катодным), часть анолита
будет проникать в катодное пространство и вывод обедненного
электролита можно проводить только из катодного цикла. При зтом
обедненная кислота, выводимая для донасыщения, не будет содер-
жать хлора. Потери тока на восстановление хлора на катоде будут
невелики и определяться количеством хлора, растворимым в объеме
анолита, который проходит через диафрагму в катодное простран-
ство.
На рис. 5-9 приведена примерная схема установки для электро-
лиза соляной кислоты. Электролизеры работают с внешней циркуля-
цией злектролита через холодильники для регулирования темпера-
туры. Разбавленная кислота из электролизеров подается на укре-
пление абгазным хлористым водородом. Хлор и водород отмываются
от паров хлористого водорода холодной водой или разбавленной
соляной кислотой. На схеме не показана система циркуляции и
г
294
охлаждения воды, поступающей на отмывку и охлаждение газов.
Хлор осушают серной кислотой
а водород промывают раствором
известкового молока или щелочи.
На рис. 5-10 показано примерное расположение установки для
электролиза соляной кислоты производительностью 21,8 т/сут хлора,
оборудованной электролизерами на нагрузку 1,6 кА. Двенадцать
о
о
Соляная кислота
после
абсорбции
Охлаждающая
бода
H2SO4
(концентри-
рованная )
16
Са (ОН)2
X СаС19
Рис. 5-9. Принципиальная схема установки для электролиза соляной кислоты:
1 — хранилище концентрированной кислоты; 2 — клапан, регулирующий подачу кислоты;
3,5 — ротаметры; 4 — смеситель; 6 — холодильник; 7 — измеритель плотности; 8 — элек-
тролизер; Р — приемный бак отработанной кислоты; 10 — регулятор давления газов; 11 —
колонна для промывки хлора; 12 — колонна для осушки хлора; 13 — колонна для отдувки
хлора из серной кислоты; 14 — колонна для отдувки хлора из разбавленной соляной кис-
лоты; 15 — колонна для промывки водорода; 16 — колонна для промывки водорода извест-
ковым молоком; 17 — холодильник; 18 — приемный бак известкового молока.
Разбавленный
хлореаз
Разбавлен- А У
ная т7
10
На абсорб-
цию
о
о
о
о
о
электролизеров по 40 ячеек в каждом для питания постоянным
током объединены в две серии по 6 электролизеров, включенных
последовательно. При полной нагрузке напряжение на серии соста-
вляет 560 В [29]. Под каждым электролизером имеется поддон из
кислотостойкого материала на случай утечки через возможные не-
плотности злектролизерал Для текущего обслуживания установки
предусматривается один аппаратчик в смену.
Разрабатываются способы снижения напряжения на ячейке
и расхода злектроэнергии на процесс электролиза. Для снижения
катодного потенциала выделения водорода предложено добавлять
к электролиту соли благородных металлов, в частности 0,2—2 г
295
f
J
соли Pd на 1 м2 активной поверхности катода, 3% солей никеля или
кобальта, что должно уменьшить напряжение разложения с 2 до
1,75 В 137, 53]; 0,5—2 моль/л хлоридов меди или железа [54];
0,0002—0,05 моль7дм2 поверхности катода солей платины [55],
покрывать катод высокодисперсным слоем серебра [24].
Для снижения перенапряжения водорода предложили выполнять
катоды из карбида вольфрама или титана, а также наносить на гра-
фитовую поверхность катрда слой карбидов этих элементов или
элементов с низким перенапряжением водорода [56, 57].
Процесс электролиза соляной кислоты и конструкция электро-
лизеров позволяют изменять в широких пределах нагрузку на элек-
тролизер без заметного ухудшения показателей работы электроли-
н2
•=—7316
•26630
-3
НС! после
абсорбции
НС! но
абсорбции)
Обедненная
кислота
Отделение
охлаждения
и промывки
газав
Отделение электролиза
Рис. 5-10. Примерное* расположение установки для электролиза соляной кис-
лоты производительностью 21,8 т хлора в сутки.
Кис-
лота
зера. Поэтому целесообразно использовать установки электролиза
соляной кислоты для потребления пиковой нагрузки энергосистем,
например в ночное время [48].
Имеются многочисленные предложения по снижению напряже-
ния на ячейке деполяризацией процесса выделения водорода за счет
применения пористого катода с подачей воздуха или кислорода
[58—61] и сочетание этого приема с добавлением солей металлов
к электролиту [54, 62—64] или каталцтически активных металлов
к материалу катода [65]. В этом случае соли металлов выполняют
роль катализаторов. '
Предложено проводить электролиз соляной кислоты в смеси
с азотной. При этом на аноде выделяется хлор, а на катоде окись
азота, регенерируемая затем в азотную кислоту с помощью газооб-
разного кислорода [66]. Имеются сведения о проведении электро-
лиза НС1 в расплаве смеси солей LiCl + КС1; СаС12 + NaCl и др.
При подаче НС1 через полый пористый катод напряжение на ячейке
ниже, чем при электролизе водного раствора хлористого водорода
296
[67—69]. Однако указывается на небольшую степень превраще-
ния НС1. Сведений об использовании в промышленности этих ме-
тодов нет.
КОСВЕННЫЕ МЕТОДЫ ЭЛЕКТРОЛИЗА СОЛЯНОЙ КИСЛОТЫ
С целью снижения удельного расхода электроэнергий и упрощения
конструкции электролизера разрабатывались косвенные методы
электролиза соляной кислоты, основанные на электролизе хлоридов
металлов. При этом на катоде не образуется водород, а происходит
восстановление металлического иона до металла, как, например,
при электролизе хлоридов никеля или ртути, или до иона с меньшей
валентностью, как при электролизе хлоридов меди или железа.
Косвенные методы электролиза соляной кислоты позволяют вести
процесс при меньшем напряжении на ячейке й меньшем удельном
расходе электроэнергии на производство хлора.
Несколько упрощается конструкция электролизера, так как
в процессе электролиза образуется только один газ и отпадает необ-
ходимость в специальных устройствах для разделения газов — элек-
тродных продуктов. Однако возникает потребность в регенерации
электролита окислением продуктов катодного восстановления или,
в случае электролиза хлоридов никеля, растворением их в соляной
кислоте. Эти стадии регенерации электролита усложняют схему
производства и требуют значительного количества дополнительной
аппаратуры. х
Ниже приведены значения нормальных потенциалов (в В) выдег
ления хлора и водорода и окислительно-восстановительных реак-
ций с ионами некоторых металлов:
Со = Со2+ + 2е
Ni = Ni2+ + 2е
Н2 = 2Н+ + 2е
Sn3+ ~ Sn4+ 4- е
Си+ — Си2+ 4- е
Си — Си2+ 4-.2е
—0,27
—0,23
0,000
0,15
0,167
0,34
Си = Си+ + е
Fe2+ = Fe3+ 4- е
2Hg = Hg2+ 4- 2е
Hg = Hg2* 4- 2e
Hgf+- 2Hg+ 4- 2e
0;52
0,771
0,798
0,854
0,910
Из приведенных данных следует, что при замене на катоде про-
цессов разрядки ионов водорода процессами восстановления ионов
двухвалентной меди до одновалентной или двухвалентной ртути
до металлической снижается значение теоретического напряжения
разложения.
Напряжение на ячейке зависит также от перенапряжения анод-
ного и катодного процессов, состава, концентрации, электропровод-
ности и температуры электролита, а также от конструкции элек-
тролизера.
Хотя нормальный потенциал разрядки ионов никеля более
электроотрицателен, чем ионов водорода, вследствие влияния пере-
численных выше факторов и, прежде всего, из-за высокого значе-
ния перенапряжения выделения водорода, процесс электролиза
f
Таблица 5-5. Значение катодного потенциала
на графитовых электродах (в мВ)
Состав электролита, г/л Плотность тока, А/м2 Увеличение потенциала при роете плотности тока от 250 до 1500 А/м2
HCI СиС12 HgCI2 250 1500
230 —840 —920 80
230 50 —270 —360 90
230 500 —50 -90 40
230 50 500 —40 —90 50
хлористого никеля можно проводить с более низким напряжением
на ячейке, чем прямой электролиз соляной кислоты.
В табл. 5-5 приведены измеренные значения катодного потен-
циала на графитовых электродах для прямого электролиза соляной
Рис. 5-11. Зависимость анодного
(£а) и катодного (£к) потенциалов
от плотности тока на графитовых
электродах:
1 — при электролизе хлорида ртути;
2 — при электролизе хлорида меди;
з — при электролизе соляной кис-
лоты.
кислоты и электролиза солянокис-
лых растворов хлоридов меди и
ртути [1].
Переход к электролизу хлоридов
меди и ртути сильно снижает дей-
ствительное значение катодного
потенциала. При электролизе хло-
ридов ртути абсолютное значение
роста катодного потенциала с повы-
шением плотности тока меньше, чем
при электролизе соляной кислоты
или хлоридов меди.
Зависимость анодного и катод-
ного потенциалов от плотности тока
при различных вариантах электро-
лиза соляной кислоты на графито-
вых электродах приведена на
рис. 5-11 [1]. Анодный потенциал
выделения хлора для прямого элек-
тролиза соляной кислоты и элек-
тролиза солянокислых растворов
меди и ртути практически одинаков.
Значения катодного потенциала существенно изменяются при пере-
ходе от одного процесса к другому.
При добавлении хлоридов металлов к раствору соляной кислоты
ее электропроводность снижается, причем для хлористой меди в боль-
шей степени, чем при добавлении хлористой ртути. Снижение удель-
ной электропроводности растворов соляной кислоты в присутствии
хлоридов металлов можно объяснить образованием комплексных
соединений. Более низкая электропроводность растворов, приме-
298
г
няемых при косвенных методах электролиза соляной кислоты,
несколько ухудйтает их энергетические показатели.
При электролизе водных растворов двухвалентной меди суммар-
ный процесс при электролизе может быть представлен в виде [70]:
2СиС12---► 2СиС1+С124-2е (5.1)
Раствор соли одновалентной меди вне электролизера окисляется
вновь до двухвалентной по реакции:
2CuCl+2НС1+1/аОа —> 2СиС1а + НаО (5.2)
При соответствующем режиме электролиза на катоде не проис-
ходит выделение водорода, что позволяет отказаться от диафрагмы
и упростить конструкцию электролизера. В этих условиях графи-
товые аноды достаточно стойки [71]. Однако при снижении содер-
жания соляной кислоты в электролите до 13% графитовые аноды
размягчаются, быстро растрескиваются и разрушаются. Присут-
ствие в электролите хлорной меди увеличивает стойкость анодов
при тех же концентрациях соляной кислоты.
Взаимодействие между хлористой медью, образующейся на
катоде, и выделяющимся на аноде хлором может быть предотвращено
путем применения фильтрующей диафрагмы. Однако подбор мате-
риала для диафрагмы затрудняется тяжелыми условиями ее работы.
Если использовать пористый полый графитовый катод, то, создавая
постоянный проток электролита через стенки пористого катода,
можно обойтись без проточной диафрагмы. Эффективность такого
способа разделения электродных продуктов зависит, от пористости
применяемого графитового катода, скорости протекания электро-
лита через стенки катода и катодной плотности тока. Для катода
с определенной пористостью с увеличением скорости протекания
электролита через стенку катода выход по току повышается до опре-
деленного оптимального значения. При дальнейшем увеличении
протекаемости выход по току снижается. Это связано с заметной
растворимостью хлора в электролите и попаданием его вместе с элек-
тролитом в катодное пространство. Повышение плотности тока при-
водит к увеличению выхода по току. При плотности тока около
4,3 кА/м2 и оптимальной протекаемости электролита получен выход
по току 90% [71].
С повышением температуры электролиза растет электропровод-
ность электролита и снижается растворимость хлора? Поэтому целе-
сообразно вести процесс электролиза при повышенной температуре.
Однако при температуре выше 80—85 °C давление паров хлористого
водорода над электролитом и унос НС1 с хлоргазом сильно возра-
стают. На опытной установке при плотности тока 1,1 кА/м2 расход
электроэнергии составил всего 880 кВт*ч/т С12, т. е. примерно в 2 раза
меньше, чем при прямом электролизе [19].
/• {
299
При проведении процесса при плотности тока около 4000 А7м2
расходные коэффициенты на 1 т хлора составляют [20]:
л Электроэнергия, кВт-ч
постоянного тока .... 1760
переменного тока .... 170
Пар, т .................... 1,25
Вода, м3 .......... 342
Хлористый водород, т .... 1,04
Хлористая медь, кг ..... . 1,8
Серная кислота, кг ..... 30
Состав электролита, поступающего в электролизер после окисле-
ния: 15% СиС12 и 20% НС1. Электролит, вытекающий из электро-
лизера, додержит 10,2% CuC12,3,7% CuCI и 20,2% НС1. Электролиз
проводится при 80—85 °C с выходом па току 82%.
На рис. 5-12 показано устройство электролизера. Для предот-
вращенйя окисления CuCI на аноде катоды выполняются из пори-
, стого графита для про-
Рис. 5-12. Схема аппарата дли электролиза
растворов CuCI:
1 — стальной корпус электролизера; 2 — битумная
масса; 3 — плита из хавега; 4 — изоляторы; 5 —
пустотелый катод из пористого графита; 6 — уро-
вень электролита; 7 — крышка; 8 — подвод тока
к аноду; 9 — подвод тока к катоду; 10 — графито-
вые аноды; 11 — медная шина.
тока электролита через
катод. Процесс электро-
лиза хлоридов меди про-
верялся на опытной уста-
новке, но не нашел приме-
нения в промышленности.
Другой метод косвен-
ного электролиза основан
на электролизе водного
раствора хлористого ни-
келя с в ыд е л ёнием н а
аноде хлора, а на катоде
металлического никеля
[72, 73]
. NiCl2 -—>Ni4-Cl2 + 2e (5.3)
Процесс проводят в двух группах электролизеров. После того
как на катоде выделилось определенное количество никеля, электро-
лизер или группа электролизеров выключается для растворения
металлического никеля в соляной кислоте [74] по реакции:
2HC14-Ni —> NiGk+ Н2 (5.4)
г
Исследования в лаборатории показали, что при электролизе
концентрированных растворов хлорида никеля при температуре
не ниже 65 °C и pH —1,3—3,5 не наблюдается выделения водорода
на катоде, поэтому диафрагма в электролизере не требуется [75].
Процесс растворения металлического никеля в соляной кислоте
проходит с недостаточной для практических нужд скоростью, однако
значительно ускоряется в растворах хлористого никеля в соляной
кислоте [75—76]. Скорость растворения металлического никеля
в кислоте за один проход соответствовала плотности тока от 269
до 323 А/м2.
300
Разработан проект установки из четырех электролизеров, в одном
из них ведется процесс электролиза при плотности тока 645 А/м2,
а в остальных трех — растворение металлического никеля, отложив-
шегося на катоде. Электролизер имеет вид прямоугольного сосуда
из хавега или другого кислотостойкого материала размером 1,2х
Х1,8x3,0 м. В вертикальных пазах продольных стенок на расстоя-
нии 6,35 мм друг от друга укреплены 239 биполярных электродов
из графитовых плит размером 1200x1500x6,35 мм. Электролизер
имеет 240 последовательно включенных ячеек и рассчитан на на-
грузку 1200 А при напряжении 440 В.
Отработанный электролит,. содержащий около 20% хлористого
никеля, обрабатывают серной кислотой для очистки от кальция
и насыщают абгазным хлористым водородом. Горячий раствор после
насыщения пропускают через электролизеры для растворения ни-
келя. Содержание хлористого никеля возрастает до 35 %, а кислот-
ность снижается примерно до 0,2%. Раствор нейтрализуют извест-
Предполагаемый расход электро-
няком и подают на электролиз»
энергии 1488 кВт*ч/т хлора.
Сообщения об использовании
в литературе отсутствуют.
В аналогичной схеме вместо
этого процесса в промышленности
никеля могут быть использованы
соли кобальта и цинка. Скорость растворения кобальта в соляной
кислоте примерно^в 25 раз выше никеля, однако кобальт дороже
и менее доступен, чем никель. При построении схемы на основе солей
цинка можно ожидать увеличения расхода электроэнергии на ста-
дии электролиза.
Процесс косвенного электролиза солянокислых растворов двух-
валентной ртути изучался в лабораторных условиях и на опытной
установке с электролизером на нагрузку 1,2жА [1].
При электролизе растворов соли двухвалентной ртути практи-
ческое значение потенциала катода на графитовом электроде зна-
чительно ниже, чем при прямом электролизе соляной кислоты.
При достаточной концентраций хлорида ртути значения катодного
потенциала на различных катодных материалах практически не
отличаются друг от друга. При понижении концентрации соли в элек-
тролите может наступить обеднение прикатодного слоя электролита
ионами Hg2+.
Процесс электролиза лимитируется скоростью подвода ионов Hg2+
к катоду и при определенной плотности тока наступает совместный
разряд ионов водорода и ртути. При этом значение катодного потен-
циала возрастает до величины, необходимой для разрядки ионов
водорода.
Для предотвращения выделения водорода на катоде и соответ-
ственно загрязнения хлора водородом необходимо процесс электро-
лиза проводить при катодной плотности тока ниже предельной плот-
ности тока для данной концентрации HgCl2 в электролите [77].
При применении ртутного катода, вследствие высокого перена-
пряжения выделения водорода на ртути, опасность разряда ионов
301
водорода на катоде уменьшается. В этом случае чистый хлор можно
получать при плотности тока на катоде до 5 кА/м2 и концентрации
HgCl2 в электролите на выходе из электролизера до 80 г/л.
Электропроводность растворов соляной кислоты снижается при
добавлении соли двухвалентной ртути. Это объясняется образова-
нием комплексных соединений, приводящих к снижению эффектив-
ной концентрации соляной кислоты в растворе. Добавление СцС12
в электролит облегчает регенерацию отработанного электролита
окислением металлической ртути соляной кислотой и кислородом.
Добавление HgGl2 и СиС12 приводит к снижению электропровод-
ности электролита и увеличению потерь напряжения на преодоление
омического сопротивления электролита в электролизере.
I Для практического осуществления процесса электролиза пред-
ложено использовать хлорный электролизер с ртутным катодом
обычной конструкции [1J. Могут быть использованы практически
все приемы, разработанные для электролиза водных растворов хло-
рида натрия, устройства для регулирования межэлектродного рас-
стояния, способы подвода и отвода электролита, циркуляции ртути
и др. Отпадает необходимость в разлагателе амальгамы, так как
продуктом электролиза является чистая металлическая ртуть.
В период подготовки к включению электролизера необходимо пред-
отвращать возможность образования каломели в результате реакции
между хлористой и металлической ртутью. На опытной установке
в процессе электролиза на образование каломели расходовалось
от 0,01 до 1,0% тока.
Для осуществления непрерывного процесса электролиза HgCl2
необходимо полученную при электролизе металлическую ртуть пере-
вести вновь в HgCl2 обработкой соляной кислотой и кислородом воз-
духа. Процесс окисления без применения катализаторов даже при
нагревании идет очень медленно. При добавлении СиС12 и других
солей скорость окисления ртути увеличивается в сотни раз.
Процессы, проходящие при этом, можно представить в виде сле-
дующих реакций, идущих параллельно или последовательно друг
за другом ;
CuCl2 + Hg—> HgCl + CuC] (5.5) ,
CuCl2 + HgCl —> HgCl2 + CuGl (5.6)
2CuCl2-f-Hg •—> HgCl2 + 2CuCl (5.7)
*• . Г
2CuC1 + 2HC1+i/2O2 •—>2CuC12 + H2O (5.8)
Как показали исследования, наиболее медленной реакцией
является окисление соли одновалентной меди кислородом воздуха.
Процесс окисления ртути проверяли в каскаде аппаратов колон-
ного типа, в которых воздух диспергировался в жидкой фазе, со-
стоящей из солянокислого раствора СиС12 и распределенной в рас-
творе металлической ртути.
Процесс проводили при новы
енной
п
температуре, его скорость зависела от развития поверхности раз-
дела фаз реагирующих веществ. На поверхности раздела капель
жидкой ртути и раствора создаются условия для образования кало-
мели. Отработанный воздух загрязнен помимо паров НС1 парами
ртути и должен подвергаться очистке.
На основе сказанного можно сделать вывод, что электролиз
растворов HgCl2, по-видимому, позволяет регенерировать хлор
из соляной кислоты с меньшими удельными затратами электроэнер-
гии, нежели при прямом электролизе соляной кислоты. Однако
необходимость применения дефицитной ртути и возможные потери
ртути в процессе производства и особенно на стадии регенерации
раствора являются серьезными недостатками этого метода,
г*
ХИМИЧЕСКИЕ МЕТОДЫ ПЬЛУЧЕНИЯ ХЛОРА
Химические методы получения хлора окислением соляной кис-
лоты использовались в начале развития хлорной промышленности.
Хлор был впервые получен Шееле окислением соляной кислоты
двуокисью марганца. Эта реакция лежит в основе метода Велдона,
предусматривавшем регенерацию двуокиси марганца. Позже был
разработан метод Дикона, основанный на окислении хлористого
водорода кислородом воздуха или чистым кислородом.
Процесс Дикона был каталитическим и непрерывным, катализа-
тором служил хлорид меди, нанесенный на дробленый кирпич или
пемзу. Реакция окисления хлористого водорода протекала с приемле-
мой скоростью при температуре около 450 °C, регулирование тем-
пературы производилось изменением скорости потокй газа через
контактную массу. На выходе из конвертора газовая смесь содер-
жала 6—8% хлора.
Окисление хлористого водорода кислородом протекает по сум-
марному уравнению реакции:
4НС1 + О2 = 2Н2О + 2С12 (5.9)
* J
Константа равновесия реакции (5.9) может быть выражена соот-
ношением:
к_ _ [flH,o]a [°С1
[аНС114 PoJ
(5.10)
где а активность соответствующего газообразного компонента,
t
Константа равновесия зависит от температуры [78—80], для
Определения ее можно пользоваться [81] следующим выражением:
еоол 7
lgg=-°° ‘-------0,930351gТ-1-1,3704• 10-«Т—1,7581 • 4,1744 (5.11)
Для расчетов, не требующих боль
II
ой точности, можно исполь-
зовать выражение:
6104 4
-----7,0994
(5.12)
303
Зависимость константы равновесия от температуры приведена
на рис. 5-13. На рис. 5-14 приведены значения равновесной концен-
трации хлора в смеси газов, образующихся по реакции (5.9), и сте-
пень конверсии НС1 в С12 при различном отношении HG1 : О2-
В качестве катализатора реакции (5.9) применяются соединения
меди, хрома и железа или композиции на их основе. Как правило,
оптимальная рабочая температура катализаторов лежит выше 350 °C,
но в этих условиях происходит довольно быстрое улетучивание актив-
ного компонента катализатора и снижение степени конверсии.
16
12
8
5с
О
250 300 400 500 1000
Температура ° К
Рис. 5-13. Зависимость 1g#
от абсолютной температу-
ры.
Рис. 5-14. Зависимость равновесной концентрации
хлора и степени конверсии НС1 от температуры:
1 — равновесная концентрация хлора при отношении
HG1 : Ог =* 4 : 1: 2 — то же, при HG1 : О4 = 2 : 1;
3 — то же, при HG1 : О, = 1 : 1; 4 — степень конверсии
HG1 при отношении HG1 : 02 — 4 : 1; 5 — то же, при
HG1 : Ог = 2 : 1; 6 — то же, при HG1: О2 — 1 : 1.
Были предприняты попытки усовершенствовать старый метод
окисления хлористого водорода по Дикону путем улучшения каче-
ства катализатора, разработки высокоактивных катализаторов, по-
зволяющих снижать температуру процесса и летучесть катализатора,
уменьшения чувствительности, катализатора к отравлению, приме-
нения вместо воздуха кислорода, проведения процесса в псевдоожи-
женном слое и др. (82—89].
Однако ни один из этих вариантов не нашел промышленного
использования, хотя опытные установки длительное время работали
как в Германии, так и в США.
Разрабатывался двухстадийный метод окисления хлористого
водорода с переносчиком хлора. Принцип метода с переносчиком
хлора состоит в расчленении процесса конверсии на несколько ста-
дий, из которых первая стадия—перевод хлористого водорода
в хлорид металла, а последняя — окисление полупродукта кисло-
родом и получение хлора. В качестве переносчиков могут приме-
няться различные поливалентные металлы, однако лучшие резуль-
таты были получены при использовании железа, меди и хрома,
304
f
t
т. e. металлов, являющихся катализаторами. По-видимому, меха-
низм действия катализаторов и переносчиков однотипен и состоит
в однократном или многократном переходе окис лов в хлориды и хло-
ридов в окислы.
При использовании в качестве переносчика соединений железа
на первой стадии процесса хлористый водород реагирует с окисью
железа
Fe2O37|-6HCF—> 2FeCl3 + 3H2O (5.13)
На второй стадии при взаимодействии хлорного железа с кисло-
родом регенерируется окись железа и образуется хлор
2FeCl3 + l,5O2 —> Fe2O3 + 3Cl2 (5.14)
1 ъ
р
Этот метод нашел применение на заводе в Брунсвике (США) [90,
91], однако в дальнейшем, производств о было ликвидировано как
нерентабельное.
В промышленности на установке производительностью 75 т/сут
хлора был внедрен метод получения хлора взаимодействием пова-
ренной соли с азотной кислотой [90, 92]. На первой стадии при обра-
ботке поваренной соли 70 %-ной азотной кислотой образуется хлор
и хлористый нитрозил
3NaCI+4HNO3 > 3NaNO3+Cl2 + NOCl + 2H2O (5.15)
Затем при обработке реакционной смеси нагретой концентриро-
ванной азотной кислотой происходит дополнительное выделение
хлора
2NOC1+4HNO3—> 6NO2 + C12 + 2H2O (5.16)
После разделения газовой смеси двуокись азота окисляют и ре-
генерируют азотную кислоту известными способами.
Предложены способы окисления НС1 и нитрозилхлорида смесью
азотной кислоты и нитратов металлов [93]. 4
Разработаны способы окисления соляной кислоты смесью азот-
ной и серной кислот в условиях, исключающих образование хло-
ристого нитрозила [90, 94—95]. Ограниченные возможности сбыта
получающегося в качестве побочного продукта нитрата натрия и тя-
желые с точки зрения коррозии условия работы аппаратуры ограни-
чивают развитие этих методов.
В последнее время появились сообщения [96] об использовании
в хлорной промышленности Японии промышленных установок по
получению хлора из хлористого аммония, однако масштабы исполь-
зования этого метода в промышленности невелики. На первой стадии
двухстадийного процесса хлористый аммоний реагирует в присут-
ствии катализатора с частично восстановленными окислами, в част-
ности железа, с выделением аммиака^ а на второй стадии при окисле-
нии кислородом выделяется хлор [971. Предложено проводить про-
цесс в кипящем слое [98].
Разработан также процесс каталитической диссоциации хлори-
стого аммония с получением аммиака и хлористого водорода [99].
20 Заказ 843 \ ЗЙ5
ЛИТЕРАТУРА
\
1. Teske W., Holleman H., Z. Electrochem., 66, № 10, 788 (1962).
2. Chem. Ing. Techn., 41, № 2, 87 (1969).
3. Chem. Ind., 21, № 2, 34 (1969).
4. Chem. Techn., 21, № 1, 55 (1969).
5. Chem. Eng., 74, № 23, 128 (1967).
6. Якименко Л. M. Электролизеры с твердым катодом. M., «Химия»,
1966.
7. Karger К., Chem. Ing. Techn., 43, № 4, 167 (1971).
8. Зарецкий С. А., Якименко Л. M. Тезисы докладов совещания
по физической химии и электрохимии расплавленных солей и шлаков.
Киев, изд-во АН УССР, 1963.
9. Алабышев А. Ф., ЗарецкийС. А., Якименко Л. М. и др.,
авт. свид. СССР 108492 (1957); Бюлл. изобр., № 4, 76 (1958).'
10. Пат. США 3104213 (1963).
11. Chem. Eng., 69, № 2, 59; № 6, 90 (1962).
12. С а с с - Т и с о в с к и й Б. А. Производство хлора. М., Госхимтехиздат,
, 1963.
13. Пат.. США 3179579, 3220941 (1965); 3431193 (1969); франц, пат. 1480430
(1967); англ. пат. 961199 (1967); 1 180448 (1970); пат. ЧССР 114352 (1965);
пат. ГДР 58497 (1966); Baley J. et al., Chem. Prum., 14, № 11, 576 (1964);
W o'lf F.* et al., Chem. Techn., 23, № 10, 607 (1971).
14. Japan Chem. Week, 10, № 453, 5; № 456, 2; № 461, 3 (1969); пат. США
3334963 (1967); 3368866 (1968); 3505009, 3514381 (1970); пат. ФРГ 12329316
(1967); 1567923, 1592018 (1970); пат. ЧССР 126347 (Г968); пат. ГДР 60294
(1968); франц, пат. 1578601 (1969); 2026416, 2028966 (1970).
14а. Chem. Age, 103, № 2713, 17 (1971); Chem. Ind., 20, № 2, 83 (1971).
146. Пат. ГДР 74027 (1970).
14в. Пат. США 3321268 (1967); 3531240 (1970); франц, пат. 1579206 (1969);
2024387 (1970).
15. Jaqueau D., Р i е s t е г L. W. et al., Chem. Ing. Techn., 43, № 4,
184 (1971).
16. Хим. пром., № 2, 16 (1958).
17. Пат. США 2636864 (1953); 2752402 (1956); 2866830 (1959); белы. пат. 643262,
643263 (1965).
18. Chem. Eng. Progr., 46, № 10, 483 (1950).
19. Oil, Paint a. Drug Rep., 82, № 13, 5 (1962).
20. Holemann H., Chem. Ing. Techn., 34, № 5, 371 (1962).
21. Л и ф а т о в a 3. И. Всесоюзная конференция по электрохимии. Тезисы
докладов. Тбилиси, «Мецниереба», 1969. См. с. 415.
22. Hass К., Trom W-, Chem. Ing. Techn., 40, № 12, 557 (1968).
23. Англ. пат. 951766 (1964).
24. Англ. пат. 1155575 (1969).
25. Нидерланд. пат. 6510493 (1966).
26. Канад, пат. 809200 (1969).
27. Австрийск. пат. 171389 (1961); франц, пат. 1273893, 1277996 (1961).
28. Пат. ФРГ 886740 (1953).
29. Sconce J. S., Chlorine its Manufacture, Properties and Uses, N. Y.,
Reinhold Publ. Corp., 1962.
30. Пат. ФРГ 1039042 (1959); 1071678 (1961).
31. Герм. пат. 173350 (1942).
32. Gardiner W. C., Chem. Eng., 54, № 1, 100 (1947); 66, № 11, 33
(1959).
33. Пат. ГДР 52350 (1966).
34. Пат. ФРГ 1112048 (1961); 1087575 (1967).
35. Япон. пат. 27102 (1964).
36. Бельг, пат. 640396 (1964).
37. Англ. пат. 1004207 (1965).
306 .
38. Пат. США 2236760, 3242065, 3236760 (1966); 3311550, 3312609, 3324023 (1967);
3415733 (1968); 3458411 (1969).
39. Франц, пат. 1265052 (1961); 1438213 (1966); нидерланд. пат. 286119 (1964).
40. Chem. Eng. News, 42, № 34, 41 (1964).
41. Japan Chem. Week, 5, № 200, 3 (1964); 6, № 255, 3 (1965).
42. Chem.-Ztg., 88, № 20, 850 (1964).
43. Chem. Eng., 67, № 24, 72 (1960); 71, № 16, 52 (1964).
44. Europ. Chem., 20, 20 (1970).
45. Doenges E., S chucker J., Chem. Ing. Techn., 37, № 5, 498 (1965);
Europ. Chem. News, 13, № 191, 49 (1965); 18, № 455, 8 (1970).
46. J an son H. C., Chem. Ing. Techn., 39, № 12, 729 (1967).
47. Galione P., Messner G., Electrochem. Techn., 3, № 11—12, 326
(1965); Chem. Ind., 21, № 6, 399 (1969).
48. Doenges E., J о n s о n H., Chem. Ing. Techn., 38, № 4, 443 (1966).
49. Ind. Chem., 33, № 396, 623 (1957).
50. Chem. Eng., 67, № 15, 63 (1960); 69, № 11, 33 (1962); 71, № 19, 172
(1964).
51. J. Electrochem. Soc., 112, № 3, 840 (1965).
52. Hydrocarbon Proc., 42, № 11, 179 (1963).
53. Пат. США 3336209 (1967); япон. пат. 8366 (1962).
54. 'Англ. пат. 1140481 (1969); франц, пат. 1556981 (1969).
55. Англ. пат. 834640 (1960).
56. Пат. ГДР 62308 (1968).
57. Berndt К., Dolle V., Kreutzberger G., Chem. Techn., 21,
.№ 10, 607 (1968).
58. Hine F., Electrochem. Techn., 4, № 11—12, 558 (1966).
59. Англ. пат. 959846 (1964); франц, пат. 1304448 (1961).
60. Т omassi W., Przem. Chem., 39, № 3, 160 (1960); 45, № 3, 131 (1966);
Bull. Sci. Acad. roy. Belg., 46, 390 (1960).
61. Tomassi W-, Jankowska H., Przem. Chem., 40, № 11, 624 (1961).
62. Hine F., J a m а к о w a K., J. Electrochem. Soc., 116, № 3, 105c (1969).
63. Пат. США 3486334 (1969).
64. Канад, пат. 840988 (1970).
65. Канад, пат. 701506 (1965).
66. Франц, пат. 1475613 (1966).
67. J о с h i z a w a S. et al., J. Elecrtochem. Soc. Japan, 36, № 1, 56 (1968).
68. J о c h i z a w a S. et al., Denki kagaku (News Letters), 2, № 1, 3 (1968).
69. Япон. пат. 7006685 (1970).
70. Пат. США 2468766 (1948); 2470073 (1948); 2666024 (1954); М е л ь ни к П. Н.,
Моргарт Р. М. и др., Хим. пром., № 7, 517 (1972).
71. Roberts S. Р., Chem. Eng. Progr., 46, № 9, 456 (1950).
72. Chem. Week, 90, № 16, 105 (1962).
73. Пат. США 3119757 (1964).
74. Chem. Eng., 66, № 17, 60 (1959); 67, № 9, 107 (1960).
75. Schroeder D, W., Ind. Eng. Chem., Proc. Design a. Developm., 1,
№ 2, 141 (1962)..
76. F r i e n d W. Z. et al., Trans. Am. Inst. Chem. Eng., 39, 731 (1943).
77. Пат. ФРГ 1131644 (1962).
78. Lem is G. N., J. Am. Chem. Soc., 28, 1318 (1906).
79. T red well W. D., Z. Electrochem., 23, 177 (1919).
80. К or ve zee A. E., Rec. trav. chim., 50, 1092 (1931).
81. A r n о 1 d C. W., К о b e K. A., Chem. Eng. Progr., 48, № 6, 293 (1952).
82. J о h n s t о n J., Chem. Eng. Progr., 44, № 9, 657 (1948).
83. Chem.' Age, 81, № 2060, 8 (1958); 86, № 2204, 551 (1961); 87, № 2239, 933
(1962).
84. Chem. Techn., 12, № 4, 188 (1960); 14, № 2, 107 (1962); Mayor V., Chem.
Rundschau, 17, № 24, 693 (1964).
85. Rev. prod, chim., 65, № 1296, 195 (1962).
86. Пат. США 2191980, 2191981, 2204733, 2204172 (1940); 2271056 (1942); 2330114
(1943); 2547928 (1951); 2602021 (1952).
20» 307
87. Англ. пат. 676667 (1952); 1192666 (1970).
88. Пат. ФРГ 857796 (1952); 857633, 873688, 873689 (1953); 1271083, 1271084
(1968).
89. Франц, пат. 1521916 (1968).
90. Chem. Eng., 68, № 9, 42 (1961).
91. Chem. Week, 80, № 24, 74 (1957).
92. Ullmann's Encyclopedic der technischen Chemie, Bd. 5, 1954. 1
93. Англ. пат. 1114430 (1968).
94. CheiA. Eng. News, 5, № 5, 14—15 (1969).
95. Пат. США 3433591 (1969).
96. Japan Chem. Week,- 6,. № 273, 3 (1965); 10, № 461, 3; №456,2 (1969).
97. Пат. ФРГ 1246690, 1246691 (1967)'; 1279004 (1968); 1592264 (1972); пат. США
3627471 (1971); япон. пат. 7201013 (1972).
98. Англ. пат. 1209044 (1970).
. 99. Канад, пат. 838596, 838597 (1970); пат. СЩА 3501431 (1970).
I ЖИДКИЙ ХЛОР '
kj * •
3b!
ALi * **
4s":
b
&:.
^7
СВОЙСТВА ЖИДКОГО ХЛОРА [(1—5)]
k . ь •
Жидкий хлор представляет собой прозрачную жидкость янтар-
ного цвета, т. кип. при атмосферном давлении —34,05 °C и т. кр. =
| = —101,6 °C. Критические константы хлора:
Температура, °C 144
Давление, атм ( . . ........ 76,1
Плотность, г/см3 ...... 0,573
Удельный объем, см3/г . . . 1,745
| Диаграмма i — lg Р для хлора приведена во 2-й главе (см. рис. 2-1).
Свойства насыщенных паров хлора приведены в табл. 6-1.
Рис. 6-1. Вязкость жидкого
хлора при различной темпег
ратуре.
Ниже приведено давление насыщенных паров над твердым хлором
при различных
Температура, °C
—171,7
-164,3
—155,7
—145,5
133,3
температурах:
Давление паров,
мм рт. ст.
10“ 5
10’ 4
10’3
10“2
10“1
Температура, °C
—118,2
—101,5
—71,9 *
—34,05 *
Давление паров
мм рт. ст.
1
10
100
760
* Над жидким хлором.
Давление насыщенных паров хлора в интервале от—154 до —103 °C
можно рассчитать по формуле
lgp^A-B/T (6.1)
где р — давление насыщенного пара, мм рт. ст.; Л = 9,950; В = 1530; Т —
абсолютная температура.
Вязкость жидкого хлора при различных температурах приведена
на рис. 6-1.
309
Таблица 6-1. Свойства насыщенных паров хлора
О
й
У дельный
объем
Энтропия,
ккал
кг* град
Энтальпия,
ккал/кг
Плотность
—70
—65
—60
—55
—50
—45
—40
—35
—30
—20
—15
—10
о
10
15
25
30
35
40
45
50
60
65
70
75
80
85
90/
95
100
105
НО
115
120
125 .
130
135
140
144
(крити-
ческая)
0,1543
0,2104
0,2823
0,3728
0,4856
0,6242
0,7925
0,9949
1,236
1,520
1,852
2,237
2,680
3,186
3,762
4,412
5,142
5,958
6,864
7,868
8,973
10,19
11,52
12,97
14,55
16,26
18,11
20,11
22,27-
24,58
27,07
29,73
32,58
35,64
38,89
42,37
46,07
50.02
54,23
58,70
63,47
68,55
73,93
78,53
0,6042
0,6088
0,6135
0,6184
0,6233
0,6284
0,6336
0,6390
0,6445
0,6502
0,6560
0,6620
0,6682
0,6746
0,6812
0,6880
0,6951
0,7024
0,7100
0,7179
0,7261
0,7346
0,7435
0,7529
0,7627
0,7729
0,7837
0,7951
0,8083
0,8201
0,8339
0,8487
0,8646
0,8820
0,9010
0,9221
0,9456
0,9725
1,0039
1,0415
1,0899
1,1541
1,2624
1,7631
1563
1174
894,4
691,6
541,8
429,7
344,9
279,6
2-29,0
189,2
157,7
132,5
112,1
95,51
81,89
70,64
61,26
53,40
46,77
41,14
36,35
32,22
28,66
25,57
22,88
20,52
18,44
16,60
14,97
13,51
12,20
11,01
9,944
8,970
8,082
7,265
6,508
5,814
5,169
4,570
4,001
3,453
2,842
1,763
73,30
72,67
72,03
71,40
70,76
70,11
69,44
68,75
68,06
67,35
66,60
65,89
65,14
64,38
63,60
62,82
62,02
61,21
60,38
59,54
58,69
57,82
56,92
56,00
55,06
54,08
53,04
51,98
50,85
49,66
48,39
47,00
45,57
43,97
42,24
40,34
38,25
35,89
33,34
30,40
27,06
23,0
17,0
0
83,86
85,01
86,15
87,28
88,41
89,55
90,69
91,85
93,00
94,16
95,35
96,49
97,66
98,83
100,0
101,16
102,33
103,49
104,66
105,83
106,98
108,14
109,31
110,48
111,65
112,83
114,05
115,26
116,50
117,77
119,08
120,47
121,87
123,40
125,00
126,68
128,47
130,43
132,47
134,78
137,36
140,2
144,2
153,41
157,16
157,68
158,18
158,68
159,17
159,66
160,13
'160,60
161,06
161,51
161,95
162,38
162,80
163,21
163,60
163,98
164,35
164,70
165,04
165,37
165,67
165,96
166,23
166,48
166,71
166,91
167,09
167,24
167,35
167,43
167,47
167,47
167,44
167,37
167,24
167,02
166,72
166,32
165,81
165,18
164,42
163,2
161,2
153,41
0,9320
0,9376
0,9430
0,9482
0,9534
0,9584
0,9634
0,9683
0,9730
0,9778
0,9824
0,9870
0,9914
0,9957
1,0000
1,0042
1,0083
1,0123
10162
1,0202
1,0240
1,0278
1,0314
1,0350
1,0385
1,0421
1,0457
1,0492
1,0527
1,0563
1,0599
1,0636
1,0668
1,0608
1,0749
1,0792
1,0837
1,0885
1,0936
1,0993
1,1053
1,1122
1,1210
1,1432
1,2928
1,2867
1,2809
1,2755
1,2705
1,2657
1,2612
1,2569
1,2529
1,2491
1,2455
1,2421
1,2389
1,2358
1,2328
1,2300
1,2273
1,2247
1,2222
1,2199
1,2176
1,2153
1,2131
1,2110
1,2089
1,2069
1,2049
1,2029
1,2009
1,1989
1,1969
1,1948
1,1923
1,1902
1,1881
1,1859
1,1835
1,1810
1,1784
1,1757
1,1724
1,1686
1,1622
1,1432
1,6552
1,6426
1,6300
1,6172
1,6043
1,5913
1,5782
1,5649
1,5518
1,5380
1,5244
1,5105
1,4965
1,4824
1,4680
1,4534
1,4387
1,4237
1,4085
1,3930
1,3773
1,3613
1,3450
1,3283
1,3112
1,2938
1,2760
1,2577
1,2388
1,2193
1,1991
1.1783
1,1566
1,1338
1,1099
1,0845
1,0575
1,0283
0,9962
0,9602
0,9183
0,8665
0,7921
0,5672
0,6398
0,8519
1,118
1,446
1,845
2,327
2,900
3,577
4,367
5,286
6,342
7,549
8,922-
10,47
12,21
14,16
16,32
18,73
21,38
24,31
27,51
31,04
34,89
39,11
43,71
48,74
54,24
60,24
66,82
74,05
82,00
90,79
100,6
111,5
123,7
137,7
153,7.
172,0
193,5
218,4
249,9
289,6
351,9
567,2
Поверхностное натяжение а на границе жидкий хлор — насы-
щенные пары хлора приведено ниже:
Темпера-
тура, °C
—72
—60
-50
—40
О, дин/см
33,0
31,2
29,2
27,3
Темпера-
тура, °C
—30
—28,7
0
а, дин/см
25,4
25,23
21,7
Темпера-
тура, СС
10
19,9
20
28
а, дин/см
20,02
18,4
18,34
16,99
Темпера-
тура, °C
49,9
50
100
(У, дин/см
13,4
13,30
4,90
Изотермический коэ
Y Y
ициент
сжимаемости 0ИЗ
жидкого хлора
при 20 °C:
Давление, атм
0,0—98,7
98,7—197,4
197,4—296
296—396
396—494
0НЗ, % на 1 атм
0,0118
0,0110
0,0102
0,00907
0,00845
Коэ
в табл.
зшг
Y Y
ициент
Р объемного расширения жидкого
хлора приведен
6-2.
Рис. 6-2. Теплоемкость жид-
кого хлора при различной
температуре.
^7г-*
Температура, °C
Удельная теплоемкость жидкого хлора~Ср
—60 °C приведена на рис. 6-2.
в интервале от 0 до
Таблица 6-2. Коэффициенты р объемного расширения хлора
1 ___________________________________________
—45
—40
—35
—30
—25
—20
-15
-10
15,1
15,3
15,5
15,8
16,2
16,5
16,9
17,5
—5
0
5
10
15
20
25
30
18,1
18,7
19,2
19,9
20,5
21,2
21,9
22,6
35
40
45
50
55
60
65
70
23,4
24,2
25,0
25,9
26,8
27,8
28,9
30,1
75
80
85
90
95
100
31,4
33,3
35,1
37,6
40,2
43,0
311
(Удельная теплоемкость твердого и жидкого
в кал/(г-град) при различных температурах:
4
хлора Ср
Твердый хлор
°C ср
—273,15 0,0045
—269,15 0,0410
—243,15 0,0990
—193,15 0,1421
—143,15 0,1719
Жидкий хлор
G°CJ ср
—93,15 0,2248
От —17,78 до +37,8 0,236
От 0 до +24 0,226
Удельная теплоемкость газообразного хлора при различном да-
влении:
Р, ат °C ср с0
1,05 10—38 0,115 0,0848
1,033 10—65,5 0,116 0,0856
7,03 10—65,5 0,128 —
Отношение : cv = 1,355.
Растворимость водорода,' кислорода
и азота в жидком хлоре
невелика. Двуокись углерода, обычно присутствующая в техниче-
ском хлоре, подвергаемом сжижению, сравнительно хорошо раство-
рима в жидком хлоре. В табл. &-3 приведена взаимная растворимость
хлора и двуокиси углерода.
1
Таблица 6-3. Взаимная растворимость хлора и двуокиси углерода
Рсо2» ат
Концентрация, мол. % i J рсо2> ат Концентрация, мол. %
со2 С12 f t со2 С12
П р и 0 °C П р и 25 °C
0,772 1,060 98,940 1,10 0,986 99,014
1,10 1,593 98,407 1,50 1,42 98,58
1,67 2,135 97,865 2,21 2,04 97,96
2,29 2,983 97,017 3,03 2,98 97,02
2,90 3,945 96,055 3,85 3,80 96,20
3,59 4,950 95,050 4,80 . . 4,74 95,26
4,10 5,640 94,360 5,55 5,46 94,54
4,78 6,582 93,418 6,54 6,43 93,47
Требования к качеству жидкого хлора в СССР определены
ГОСТом [6]. Содержание хлора должно быть не менее 99,6. вес. %.
Допускаются примеси, вес. % не более: 0,005 NC13 и 0,05 Н2О.
В жидком хлоре могут содержаться примеси хлорированных угле-
водородов, серной кислоты и продуктов коррозии стенок аппаратов
и трубопроводов, присутствие которых в ГОСТе не регламентировано.
312,
РАЗВИТИЕ ПРОИЗВОДСТВА ЖИДКОГО ХЛОРА
ч
Жидкий хло]э был впервые получен Фарадеем в 1828 г., однако
в промышленности зтот процесс был использован впервые для произ-
водства жидкого хлора как товарного продукта только в 1888 г.
Но и после этого производство жидкого хлора не получало широкого
развития вплоть до 1914 г., когда во время мировой войны сжижен-
ный хлор был применен как боевое отравляющее вещество. В даль-
нейшем хлор потерял значение как самостоятельное ОВ, но его
производство приняло крупные масштабы вследствие широкого
применения хлора в виде сжиженного газа в ряде производств и от-
раслей народного хозяйства. *
В потреблении жидкого хлора можно различать два основных
направления. Первое из них заключается в использовании жидкого
хлора вместо хлорной извести или растворов гипохлоритов натрия
и кальция для отбелки целлюлозы, тканей, а также для хлорирова-
ния воды и промышленных стоков. Применение жидкого хлора для
этих целей имеет ряд преимуществ по сравнению, например, с хлор-
ной известью, так как концентрация активного начала в жидком
хлоре значительно выше, чем в хлорной извести или в водных рас-'
творах гипохлоритов. Жидкий хлор значительно удобнее хлорной
извести при перевозке, хранении и, особенно, при дозировании.
Помимо того, в случае применения жидкого хлора, например для
хлорирования воды, последняя не загрязняется солями кальция,
как это происходит при использований хлорной извести. Поэтому
в годы после первой мировой войны в, ряде стран организуется произ-
водство жидкого хлора и одновременно с его развитием снижается
производство хлорной извести, так как жидкий хлор вытесняет
хлорную известь из основных областей ее применения.
Вытеснению хлорной извести жидким хлором способствовало
также то, что жидкий хлор поступал в продажу в различной рас-
фасовке — цистернах, контейнерах и бочках на 500 иДоОО кг и в бал-
лонах на 30—50 кг. Это создавало условия для применения жидкого
хлора не только крупными потребителями, но и в сравнительно
небольших производствах. В качестве иллюстрации этого положе-
ния ниже показано изменение размеров производства жидкого хлора
и хлорной извести в годы после первой мировой войны в США [7—12]:
я
Годы Выработка, жидкого хлора тыс. т хлорной извести Выработка, т ы с. т
Годы ЖИДКОГО хлора хлорной извести
1923 62,7 147,8 1947 520 21,8
1925 83,2 105,2 1951 1065 32,0
1927 443,2 80,4 1954 1584 22,0
1928 196,5 * 75,3 1969 4000 0
Этот процесс вытеснения хлорной извести жидким хлором харак-
терен не только для США, но и для Канады и других стран. В на-
стоящее время хдорная известь или гипохлориты для отбелки или
хлорирования воды применяются только тогда, когда потребность
в активном хлоре невелика и снабжение жидким хлором затруднено.
Электролитические способы получения водных растворов гипо-
хлоритов щелочных металлов практически полностью сошли со
-сцены. Это объясняется тем, что применение для отбелки или
хлорирования растворов гипохлорита натрия или калия стало неэко-
номичным и их заменили привозным жидким хлором.
Перечисленные обстоятельства способствовали росту потреб-
ности в жидком хлоре и увеличению его производства, однако доля
хлора, расходуемого для отбелки тканей и хлорирования воды,
в настоящее время невелика. Рост производства и потребления
жидкого хлора, особенно в последнее. время, обусловлен главным
образом развитием второго направления использования жидкого
хлора для синтеза многочисленных хлорсодержащих продуктов.
Жидкий хлор — очень удобное сырье для большого числа хлор-
потребляющих производств как на территории хлорных заводов,
так и вне ее. Для ряда предприятий особое значение имеет примене-
ние хлора высокой концентрации. Так, в процессе хлорирования
по цепному механизму примеси кислорода в хлоре затрудняют про-
текание реакции. Поэтому, несмотря на то что хлор, полученный
испарением жидкого хлора, значительно дороже хлора, непосред-
ственно получаемого из цеха электролиза, на некоторых предприя-
тиях предпочитают работать на более чистом, хотя и более дорогом'
испаренном хлоре. К числу таких производств относятся производ-
ства синтетического хлористого водорода для нужд гидро хлориро-
вания ацетилена, хлористого аллила хлорированием пропилена,
гексахлорциклогексана фотохимическим хлорированием бензола,
хлорирование полихлорвинила, полиэтилена и других продуктов.
Высокая чистота испаренного хлора обусловила проведение
процессов хлорирования с минимальным объемом абгазов и мини-
мальными потерями сырья и получаемых продуктов при сбросе инерт-
ных примесей с абгазами.
В связи с огромным ростом потребления хлора для производства
хлорорганических продуктов возникла и все более расширяется
потребность в испаренном чистом хлоре и производстве для этих
целей жидкого хлора.
Удобство транспортирования жидкого хлора способствовало
концентрации производства электролитического хлора и каусти-
ческой соды на сравнительно немногих крупных заводах и удовлетво-
рению потребности более мелких потребителей в хлоре за счет при-
возного жидкого хлора.
Заслуживает внимания предложение использовать жидкий хлор
в^качестве растворителя, в частности для С1О2 [13]. Могут быть по-
лучены безопасные в обращении растворы, содержащие до 27% 010 2.
В начале развития хлорной промышленности процесс сжижения
хлора был необходим для получения товарного жидкого хлора
с целью продажи его на сторону. Поэтому обычно сжижению под-
вергалась сравнительно небольшая часть хлора, вырабатываемого
314
| в цехах электролиза. С развитием хлорной промышленности, уве-
| личением ассортимента хлорпотребляющих производств и с появле-
I нием производств, предъявляющих высокое требование к чистоте
I хлора, цеха по получению жидкого хлора начали играть значитель-
I ную роль в системе хлорного завода. На современных хлорных за-
I водах обычно оборудуется цеха для сжижения хлора, близкие по
I мощности к общей мощности хлорного производства. При этом цеха
I сжижения ^лора приобретают функции центральной распредели-
I тельной хлорной станции завода, на которой помимо отгрузки то-
| варного жидкого хлора потребителям производится его комприми-
I рование, сжижение определенной части его, испарение части жидкого
I хлора для снабжения потребителей чистым высококонцентрирован-
I ным испаренным хлором, а также распределение электролитического
г хлора и абгазов сжижения между отдельными потребителями.
I Такая станция позволяет использовать установки сжижения
I хлора в качестве буфера, накапливающего в хранилищах жидкий
хлор в моменты, когда производство хлора на заводе превышает
его потребление и, наоборот, испаряющего жидкий хлор из запаса
в моменты недостатка хлора [14].
Развитие производства жидкого хлора и его транспортирование
на дальние расстояния открыло возможность отделить хлорпотребля-
ющие производства от хлорных заводов и обеспечивать эти произ-
водства хлором за счет перевозки его в жидком виде в цистернах.
Это приобрело особое значение для таких хлорпотребляющих
производств, которые базируются в местах расположения основного
сырья, например целлюлозно-бумажные комбинаты, производства
по переработке руд цветных металлов и другие, для которых часто
нецелесообразно организовывать свое производство хлора. Иногда
такие предприятия считают рациональным организовать свое произ-
водство хлора, как зто делают многие целлюлозно-бумажные комби-
наты.
Ниже приведены данные о росте производства жидкого хлора
в США и увеличении доли хлора, поступающего на сжижение, в по-
следние десятилетия [8—12]:
Гов Тыс. т % общего производства Год Тыс. т % общего производства
1914 0 0 1960 2172 51,5
1923 62,7 31,3 1962 2560 53,5
1938 200 43.0 1965 3100 53,2
1946' 520 49,0 1966 3410 54,2
1951 1065 46,6 1967 3595 51,7
1955 1702 55,4 1968 3890 50,8
1958 1815 55,6 1969 4000 48,0
Некоторые колебания доли хлора, подвергаемого сжижениют
можно объяснить естественными колебаниями в распределении хлора
между потребителями, а также недостаточной точностью публику-
емых данных, особенно' относящихся к внутризаводскому потребле-
нию жидкого хлора.
315
В нашей стране впервые промышленное производство жидкого
хлора было начато в* 1915 г. [7, 15]. Доля общей выработки хлора,
поступающего н^ сжижение, за последние 5 лет выросла с 41 до
45%. В ближайшие годы предусматривается увеличение производ-
ства жидкого хлора и его доли в общем производстве хлора [16].
Примерное распределение жидкого хлора между отдельными
потребителями в нашей стране за период 1965—1970 гг.:
% общего
количества
63,8—69,6 '
12,0—14,7
Отрасль
Химическая промышленность . . . .
Целлюлозно-бумажная промышленность
Цветная металлургия . ..............
Химводоочистка . . . . . ...........
Прочие потребители .................
Экспорт ............................
0,1—8,9
ТЕРМОДИНАМИЧЕСКИЕ ОСНОВЫ ПРОЦЕССА СЖИЖЕНИЯ ХЛОРА
Для сжижения хлора необходимо, чтобы-парциальное давление
его паров было равно давлению насыщенного пара хлора. Это дости-
гается снижением температуры при неизменном давлении или повы-
ением давления газообразного хлора,
или комбинированием компримирования
газа со снижением его температуры. Зави-
симость давления насыщенных паров хлора
от температуры приведена в табл. 6-1.
Для сжижения чистого хлора при
атмосферном давлении необходимо пони-
зить его температуру до —34,05 °C. Сжи-
жение чистого хлора при 0 °C Происходит
при давлении 3,762 ат.
Обычно на практике сжижению под-
вергается не чистый хлор, а газовые смеси,
состоящие из хлора и примесей, состав и
содержание которых зависят от способа
его очистки и под-
о
II
Парциальное давление> ат
Рис. 6-3. Растворимость во-
дорода, , азота и кислорода
в жидком Хлоре:
1 — кислород; 2 — азот; з —
водород (сплошная линия при
—60 °C, пунктирная — при
—30 °C).
производства хлора,
готовки к сжижению и местных условий
производства.
В качестве загрязнений в хлоре могут
присутствовать водород, кислород, азот,
двуокись углерода, треххлористый азот,
различные продукты хлорирования угле-
водородов и влага. Кроме того, в £азо-
образном хлоре могут содержаться мелкие капли серной кислоты,
увлекаемой'из башен сернокислотной сушки, а также частицы твер-
дых хлорида и сульфата натрия, образуемых в башнях сушки хлора
316
**4
при реакции серной кислоты с брызгами и туманом электролита,
увлекаемыми хлором из электролизеров.
Различные примеси ведут себя по-разному в процессе сжижения
газообразного хлора. Такие газовые примеси, как водород, кислород
и азот, имеющие очень низкую температуру сжижения, при всех
ленности способах Сжижения хлора остаются
применяемых в промыз
и
в виде несжиженных газов. Растворимость их в жидком хлоре неве-
лика, и ею обычно пренебрегают (рис. 6-3).
Наличие инертных газовых примесей в подвергаемом сжижению
газообразном хлоре определяет технологию сжижения хлора. Для
сжижения чистого хлора достаточно довести его парциальное давле-
ние до величины, отвечающей давлению насыщенных паров хлора
Содержание хлора
Содержание хлору
Рис. 6-5. Давление начала
конденсации хлора различного
состава при t — —20 9С.
Рис. 6-4. Температура конден-
сации хлора различного со-
става при Р=3 ат.
ЭШЕ
при данной температуре путем понижения температуры, повышения
давления или комбинированного действия обоих факторов. Если
обеспечить непрерывно отвод тепла, то процесс начавшейся конден-
сации будет продолжаться при постоянных температуре и давлении
до полного сжижения всего газообразного хлора. В условиях ста-
ционарных температуры и давления коэффициент сжижения может
достигать 100%. В этом случае парциальное давление хлора равно
общему давлению газа.
При сжижении хлора, загрязненного инертными газовыми при-
месями, парциальное давление хлора в смеси меньше общего давления
и составляет
Рх— ^х
(6.2)
где рх — парциальное давление хлора в смеси; Р— общее давление газовой
смеси; Сх — объемная доля хлора в смеси.
Для начала процесса конденсации хлора из газовой смеси необ-
ходимо, так же как и при сжижении чистого хлора, довести его пар-
циальное давление до величины, равной давлению насыщенного пара
хлора при данной температуре. В процессе конденсации хлора из
317
газовой смеси непрерывно происходит изменение её состава. По мере
конденсации хлора парциальное давление его в смеси снижается,
а содержание и парциальное давление несжижаемых при этих усло-
виях газовых примесей возрастает. Для продолжения конденсации
хлора необходимо снижать температуру смеси газов либо повышать
их давление.
На рис. 6-4 приведена температура конденсации чистого хлора
и его смесей с инертными газами при постоянном давлении 3 ат,
а на рис. 6-5 — давление, необходимое для осуществления этого
процесса при постоянной температуре минус 20 °C для тех же сме-
сей. Коэффициент сжижения хлора А (в долях)
А = ёж! в
(6.3)
где — количество сжиженного хлора| g общее количество хлора, содержа-
щегося в сжижаемом разе.
Если концентрацию хлора в начале сжижения обозначить через Сг
(в долях единицы) и в конце через С2, т0 коэффициент сжижения
можно представить в следующем виде:
(6.4)
Если концентрацию хлора выразить в объемн. % , выражение (6.4)
примет вид:
(01—С2)100
(100—С2)
(6.5)
Если концентрацию С % в выражении (6.5) заменить парциальным
давлением хлора по выражению (6.2), то коэффициент сжижения
можно определить по формуле:
А = 1 —
(6.6)
II
Как видно из выражений (6.4)—(6.6), коэффициент сжижения
возрастает при увеличении концентрации хлора в исходном газе,
подаваемом на сжижение, повышении общего давления газовой смеси
и при уменьшении парциального давления хлора в остатке несжи-
женного газа, т. е. при понижении температуры сжижения. Значе-
ние коэффициента сжижения хлора для различных сочетаний этих
параметров можно рассчитать по выражению (6.6).
Для определения коэффициента сжижения и других параметров
процесса сжижения можно пользоваться номограммой, приведенной
на рис. 6-6.
31S
Пример пользования номограммой. Содержание хлора в исходном газе
$3 объемн. %; абсолютное давление газовой смеси 3 ат; температура сжижения
—20 °C. Для определения коэффициента сжижения из точки на ординате, соот-
ветствующей давлению 3 ат, проводим горизонтальную линию до пересечения
Рис. 6-6. Номограмма для определения параметров процесса
сжижения хлора (в нижней части — кривые температур сжиже-
ния, в верхней — кривые коэффициентов сжижения).
с изотермой, соответствующей температуре сжижения —20 °C. Из точки пере-
сечения А восстанавливаем перпендикуляр до пересечения его с горизонтальной
линией, соответствующей исходному составу хлоргаза (93 объемн. %). Точка
пересечения В определяет значение коэффициента сжижения, равного в рас-
сматриваемом случае 87,5%. Аналогично по заданному коэффициенту сжижения
и составу хлора можно определить необходимые для этого давление и темпера-
туру сжижения.
319
Объем абхазов и содержание в них отдельных инертных примесей
можно определить по номограмме, приведенной на рис. 6-7, в зави-
, симости от степени сжижения и начального состава хлоргаза, посту-
пающего на сжижение.
По мере увеличения коэффициента сжижения содержание хлора
в остатке несжиженного газа (абгаза) уменьшается, а инертных при-
месей — возрастает.
Содержание хлора в остатке несжиженйого газа (в %) в за-
висимости от исходной его концентрации С\, выраженной в %,
Рис. 6-7. Номограмма для определения объема абгазов и со-
держания в них инертных примесей:
С — концентрация хлора в исходном газе; V — объем абгазов, м*/м* исход-
ного газа; X — кратность увеличения содержания инертных компонентов
в абгазах; А — коэффициент сжижения, %.
и коэффициента сжижения в долях может быть определена из выра-
жения:
100С1(1 — л)
2“ . 100 — АСх
* (6.7)
Содержание инертных примесей в остатке несжиженного газа
составит .
100— =
100(100—Ci)
100 — АС г
(6.8)
320
Если обозначить содержание примеси инертного газа в исходной
смеси через Сп1 (в %), то содержание ее в остатке несжиженного
газа (7п2 можно определить из выражения:
г _ IQOCni
п2 100 — А С1
(6.9)
Особое значение имеют примеси водорода в поступающей на сжи-
жение газовой смеси, так как повышение концентрации водорода
в остатке несжиженных газов может приводить к образованию взрыво-
опасных газовых смесей.
В хлоре, получаемом в электролизерах с твердым катодом и
диафрагмой, содержание водорода обычно невелико и не превышает
0,3—0,5%. При производстве хлора электролизом с ртутным като-
дом содержание водорода обычно бывает выше и при нарушениях
нормального хода технологического процесса может превышать
1—1,5%.
Опасности, возникающие в связи с образованием взрывоопасных
смесей, заставляют учитывать возрастание концентрации водорода
в процессе сжижения при определении коэффициента сжижения,
выборе параметров и технологической схемы процесса.
Смеси хлора с водородом и воздухом могут взрываться при со-
держании водорода более 4,2—5,5%. При нижнем пределе содержа-
ния водорода взрыв не носит характера детонации, которая наблю-
дается при содержании водорода 19—20% [17].
Содержание двуокиси углерода в газовой смеси мало влияет на
величину нижнего предела содержания водорода и характер про-
текания взрыва.
Обычно верхним пределом концентрации водорода в остатке
несжиженного газа считают 4%, это ограничивает допустимое зна-
чение коэффициента сжижения. При верхнем пределе содержания
водорода, равном 4%, допустимое значение коэффициента сжижения
будет тем выше, чем ниже содержание водорода в подаваемой на сжи-
жение газовой смеси.
На допустимую степень сжижения влияет также
таких газовых примесей, как двуокись углерода и воздух,
ющих как инертные разбавители.
Для расчета режима сжижения хлора, содержащего
водорода, в условиях, исключающих образование взрывоопасных
хлороводородных смесей, можно воспользоваться номограммой,
приведенной в работе [18].
Для наиболее полного сжижения хлора применяют двухступен-
чатый процесс. По достижении концентрации водорода 4% остаток
несжиженных газов разбавляют азотом и подвергают дальнейшему
сжижению при более низкой температуре или более высоком давле-
нии. Предложено проводить вторую^ стадию сжижения без разбавле-
ния смеси инертным газом, осуществляя процесс таким образом,
imi
наличие
действу-
примеси
21 Заказ 843
321
чтобы локализировать возможные участки взрывов /и обеспечить
сохранность аппаратуры даже при возникновении взрыва. Напри-
. мер, для этой цели предложено [19] проводить вторую стадию кон-
денсации, распределяя конденсируемую газовую смесь в виде мелких
пузырьков в системе, заполненной охлажденным жидким хлором.
Фирма «Уде» предложила вторую ступень сжижения по достижении
содержания водорода 4% проводить в толстостенной аппаратуре,
разделенной на небольшие камеры и рассчитанной на возможные
взрывы.
При этом между первой и второй ступенями сжижения уста-
навливаются дополнительные устройства для локализации возмож-
ного воспламенения газовой смеси во второй ступени [20].
Осложнения при сжижении, вызываемые загрязнением хлора
водородом, привели к неоднократным попыткам разработки рацио-
нального метода очистки хлора от водорода. Все предложенные ме-
тоды были основаны на взаимодействии водорода с хлором с образо-
ванием хлористого водорода, который легко отмывается от хлора
в орошаемых водой башнях.
Длительное время на заводах в ЧССР использовался способ тер-
мического выжигания водорода в хлоре [21]. Хлор подогревали
с помощью электрообогрева в кварцевых трубках до 280—320 °C.
При этом содержание водорода в хлоре снижалось до 0,1—0,3%
от начального 1,0—2,0%. После выжигания водорода хлор пода-
вали но свинцовым трубопроводам в игуритовые холодильники.
Расход электроэнергии на подогрев составлял 75 кВт-ч/т хлора.
Расход можно снизить, применяя рекуператор для использования
тепла газа, выходящего из реактора, для подогрева хлора, поступа-
ющего на очистку.
Выбор материалов, пригодных для изготовления реактора, реку-
ператора, подогревателя и трубопроводов, ограничен. Использова-
лись кварц, свинец и футерованная сталь. Однако необходимость
нагревания хлора до температуры около 300 °C и его последующее
охлаждение делает этот метод мало пригодным для широкого про-
мыш ленного использования.
Предложено проводить этот процесс на активированном угле
при 150—200 °C [22], а также при низких температурах с иницииро-
ванием процесса выжигания водорода в хлоре ультрафиолетовыми
лучами [23].
Однако этот способ очистки хлора от водорода не доведён до
промышленного использования. - .
Предложено также применение радиоактивного излучения для
инициирования процесса выжигания примесей водорода в хлоре [24]
(предпочтительно p-из лучение), однако данных о промышленном
использовании этих процессов не опубликовано.
В отличие от водорода и воздуха двуокись углерода значительно
более растворима в жидком хлоре (рис. 6-8). В обычных схемах сжи-
жения двуокись углерода рассматривается как инертная примесь,
удаляемая с несжиженным остатком газов. Специальная очистка
322
t-
жидкого хлора от СО2 обычно не предпринимается. Имеются предло-
жения по отдувкё СО2 от жидкого хлора газообразным хлором, по-
ступающим на сжижение [25]. При этом увеличивается содер-
жание СО2 в абгазах сжижения, что представляет интерес с точки
зрения снижения взрывоопасности
абгазов, содержащих водород.
С целью отдувки газообразный
хлор после компримирования и пред-
варительного охлаждения пропускают
противотоком жидкому хлору, вытека-
ющему из конденсаторов, в аппаратах
типа скрубберов, орошаемых жидким
хлором, или в барботажных аппаратах.
При этом газообразный хлор, посту-
пающий на сжижение, обогащается
двуокисью углерода. На первую сту-
пень сжижения поступает хлор, содер-
жащий 1% СО2 против начального со-
держайия 0,6%. Содержание СО2
в жидком хлоре после отдувки не пре-
вышает 0,05 мол,- %. Без отдувки со-
держание двуокиси углерода в жидком
хлоре, ПЪлучаемом на второй ступени
сжижения, может достигать
ной величины [26].
В случае применения
тельной очистки жидкого
СО2 [27] изменяется состав абгазов
и жидкого хлора. При двухступенчатой
коэффициентом сжижения 99,8 % и
/ •
Рио. 6-8. Растворимость дву-
окиси углерода в жидком
хлоре при различной темпе-
ратуре:
1 — при
3 — при —30 °C
5 — при 20 °C.
значитель-
дополни-
хлора от
60 °C; 2 — при —45 °C;
4 — при —15 °C;
схеме сжижения с общим
коэффициентом сжижения
При м е чание. При работе без очистки от двуокиси углерода жидкий хлор после
второй ступени содержит 3% Со2.
ь <ч
"1
на первой ступени 91,5% получены абгазы, состав которых при-
веден в табл. 6-4.
21* 323
За счет отдувки двуокиси углерода из жидкого хлора содержание
ее в абгазах первой и второй ступеней сжижения существенно возра-
стает. Снижается концентрация водорода и воздуха в абгазах за
счет разбавления их двуокисью углерода.
Треххлористый азот, образующийся в небольшом количестве
в электролизерах или на стадии охлаждения хлора в холодильниках
смешения при условии содержания в воде аммонийных солей или
аминов, обычно конденсируется и практически полностью попадает
в жидкий хлор. Как было указано ранее (см. главу 4), при испарении
жидкого хлора, загрязненного NG13, содержание последнего
в остатке неиспаренного хлора возрастает. В зависимости от усло-
вий испарения коэффициент разделения может составлять от 6
до 10.
Растворы треххлористого азота в жидком хлоре концентрацией
более 5% взрывоопасны. Если начальное содержание NCI3 в жидком
хлоре составляет 0,2%, то содержание 5% NC13 достигается при
объемном испарении тогда, когда остаток неиспаренного хлора будет
составлять 1,5—2,0% исходного количества. Поэтому при объемном
испарении жидкого хлора, содержащего даже малое количество
треххлористого азота, создается опасность взрыва в конце процесса
испарения. Обычно стремятся исключить возможность образова-
ния NC13 в хлоре. При наличии в жидком хлоре примеси NC13 необ-
ходимо проводить его полное испарение в проточном испарителе,
чтобы исключить возможность накопления треххлористого азота
в остатке неиспаренного хлора.
Примеси влаги в хлоре могут усиливать коррозионное разруше-
ние аппаратуры и хранилищ жидкого хлора. Исследование показало,
что в равновесной системе Н2О—С12 содержание влаги-в газовой
фазе вышег чем в жидкой, что приводит к повышению содержания
влаги в несжиженном остатке газов в процессе сжижения хлора.
В табл. 6-5 приведены данные по растворимости воды в жидком
хлоре при различной температуре и значения коэффициента распре-
деления а влаги между жидким хлором и газовой фазой.
При температурах выше 28,7 °C третьей фазой, находящейся
в равновесии с жидким хлором и паровой фазой, является жидкая
вода, при более низкой температуре — твердый гидрат хлора.
При давлении 6 ат и в интервале температур до 28,7 ®С твердый
гидрат хлора может образоваться в конденсаторах хлора. При полной
конденсации хлора максимально допустимая влажность не должна
превышать растворимости воды в жидком хлоре при температуре
конденсации. В практических условиях, когда 100%-ная конден-
сация не достигается, некоторое количество влаги уносится с нескон-
денсировавшимися газами.
Как видно из табл. 6-5, в широком интервале температур содер-
жание влаги в газовой фазе (абгазах) в 4,1—4,2 раза выше, чем
в жидком хлоре [28, 29]. Если содержание влаги в исходном хлоре
выше растворимости воды в жидком хлоре при температуре сжиже-
ния, то в процессе конденсации хлора, вследствие указанного явле-
324
:.Л-
Таблица 6-5. Растворимость воды в жидком хлоре [28]
Темпе- ратура, •с растворимость воды в жидком хлоре Равно- весное содержа- ние воды в газах Давление водяных паров,4 атм * 10~® а Равновесие
мол. % вес. ч./млн мол. %
5 50 0,21 530 0,87 123 4,1 G жидкой водой
40 0,16 400 0,65 72,7 4,1 То же
30 0,12 295 0,48 41,8 4,1 »
28,7 0,11 275 0,45 39 4,2 G жидкой водой и гидра-
0,094 0,40 том хлора
28,7 240 31 4,2 G гидратом хлора
25 0,098 250 0,41 21 4,2 G жидкой водой
20 0,076 190 0,32 9,85 4,2 G гидратом хлора
10 0,048 120 0,20 4,35 4,2 То же
0 0,029 78 0,12 1,8 4,2 »
—10 0,017 42 0,070 0,70 4,2 »
—20 0,0093 23 0,039 »
—30 0,0050 13 0,021 0,25 4,2 »
—40 0,0025 6 0,011 0,08 4,3 »
*
ния, в абгазах возрастает содержание влаги тем в большей мере,
чем выше степень сжижения.
Увеличение влажности газовой фазы в процессе сжижения
серьезно усложняет технологию сжижения хлора.
При сжижении хлора, высушенного по общепринятому серне-
Ill
кислотному методу, с повышением степени сжижения до 90% и вь
точка росы в абгазах сжижения возрастает до —30 °C и выше. По-
этому в конденсаторах наблюдается вымораживание влаги. Помимо
нарушения теплопередачи за счет намораживания «ледяной шубы»
создаются условия для образования в жидком хлоре отдельной вод-
ной фазы, что усиливает коррозионное разрушение аппаратуры [29].
Особое Значение осушка хлора приобрела в последнее время
в связи с внедрением схем с практически полным (100%-ным) сжи-
жением хлора.
Существенное снижение влажности поступающего на сжижение
хлора связано с серьезными трудностями. Поэтому предложены
схемы, по которым газообразный хлор после обычной осушки его
в башнях, орошаемых серной кислотой, поступает на первую ступень
сжижения. Абгазы первой ступени сжижения подвергаются допол-
нительной сушке под давлением сорбентами типа цеолита или моле-
кулярных сит [30—33] и подаются далее на вторую ступень для
сжижения при более низкой температуре. Применение таких схем
позволяет избежать образования отдельной водной фазы при глубо-
ком сжижении хлора в условиях низких температур и соответственно
исключить возможность интенсивной коррозии аппаратуры, трубо-
проводов и хранилищ жидкого хлора.
Небольшие количества хлорированных углеводородов, присут-
ствующие в исходном хлоре, практически полностью переходят
в жидкий хлор. Обычно содержание углеводородов невелико и не
вызывает неприятностей у производителей и потребителей жидкого
хлора. Однако в некоторых случаях содержание хлорорганических
примесей в жидком хлоре может значительно возрасти.
. При объемном испарении жидкого хлора происходит накопление
в остатке неиспаренного хлора хлорорганических примесей, имеющих
более высокие температуры кипения. Хроматографический анализ
этих примесей в кубовом остатке, составляющем 0,06—1,6% исход-
ного количества жидкого хлора, показал содержание следующих
хлорорганических соединений (в %):
г
GHC13 , . , . . 67,5
СС14 .........\ 23,0
C2Clfi......... 8,02
СН2С12 .......0,34
С2С14 ...»•• 0,22 '
Количество и состав хлорорганических примесей в жидком хлоре
зависят от условий производства, в частности от пропитки анодов,
применения в качестве конструкционных или защитных материалов
органических и полимерных продуктов.
Примеси тумана рартвора поваренной соли неполностью худа-
ляются на стадии охлаждения хлора и, попадая в аппараты осушки,
часто ухудшают работу системы осушки [34]. В результате взаимо-
действия этих примесей с серной кислотой образуется смесь части-
чек сульфата и хлорида натрия.
Туман серной кислоты и твердые частички сульфата и хлорида
натрия, хлорного железа и высокомолекулярных хлорорганических
соединений, проходящие в небольшом количестве через систему
очистки и фильтрации газообразного хлора перед компримирова-
нием, попадают в жидкий хлор и загрязняют его. При испарении
жидкого хлора эти загрязнения остаются в испарителе, что приводит
к необходимости периодической его чистки.
Ранее уже упоминалось о применении двухступенчатых схем
сжижения хлора, позволяющих исключить возможность образования
1 взрывоопасной концентрации водорода в абгазах и проводить про-
межуточную осушку абгазов с помощью сорбентов. Преимущество
Двухступенчатой схемы сжижения перед одноступенчатой заклю-
чается в снижении энергетических затрат. Это преимущество осо-
бенно важно при необходимости получить высокую степень сжи-
жения.
Как ранее рассматривалось, при сжижении чистого хлора про-
цесс сжижения может быть осуществлен вне зависимости от необ-
ходимой степени сжижения при постоянной температуре и давле-
нии, поэтому применение двухступенчатой схемы не рационально.
При сжижении технического хлора, содержащего примеси несжи-
жаемых газов, с увеличением необходимой степени сжижения необ-
г
,326
*Г.
51.
к
ходимо снижать температуру конденсации или повышать применя-
емое давление. И то и другое связано с дополнительными энергети-
ческими затратами. Поэтому для сжижения технического хлора
С высокой степенью сжижения целесообразно применять двухсту-
пенчатую схему, на первой ступени которой сжижается 80—90%
общего количества хлора при более высокой температуре сжижения,
а остающееся количество сжижается на второй ступени при более
низкой температуре конденсации. При этом основное количество
тепла процесса сжижения отводится на первой ступени, где
|&пользуется более дешевый холод на высоком температурном
уровне.
Такие двухступенчатые схемы сжижения с одинаковым давлением
газов на обеих ступенях и при различной температуре конденсации
в настоящее время широко используются в промышленности [35].
Возможны также двухступенчатые схемы сжижения с дополнитель-
ным компримированием газа между ступенями,, однако такие схемы
менее удобны [35].
Вторую ступень сжижения можно рассматривать как установку
для полезного использования абгазов сжижения. В этом случае
извлечение хлора из абгазов первой ступени сжижения производится
за счет более глубокого охлаждения. Эта же цель может быть до-
стигнута также сорбцией хлора из его смесей с инертными приме-
сями различными растворителями или' твердыми сорбентами с по-
следующей десорбцией концентрированного хлора и его сжижением.
Эти способы будут рассмотрены несколько позже.
ТЕХНОЛОГИЧЕСКИЕ СХЕМЫ ПРОИЗВОДСТВА
*1
f
В зависимости от условий производства применяются различные
технологические схемы сжижения хлора, отличающиеся применя-
емым давлением;, температурой конденсации, коэффициентов сжи-
жения хлора и соответственно используемой аппаратурой [36].
Основным критерием для выбора производственной схемы сжи-
жения, естественно, является стремление иметь минимальную стои-
мость сжижения. Помимо этого, выбор схемы во многом опреде-
ляется составом исходного хлоргаза и наличием или отсутствием
на заводе потребителя абгазов сжижения. Наличие в хлоргазе при-
месей водорода вынуждает принимать меры, исключающие возмож-
ность образования в процессе сжижения в абгазах взрывоопасных
смесей с водородом.
Применяемые на практике методы сжижения хлора можно разде-
лить на три типа: , -
1. Метод высокого давления, в котором сжижение производится
при сравнительно высоком давлении (обычно до 12 ат) и температуре
до 25—30 °C. При этом может быть достигнута степень сжижения
80—85% без применения специальных холодильных установок.
2. Комбинированный метод сжижения при давлении 3,0—5,0 ат
и температуре Минус 15 — минус 20 °C.
327
3. Метод глубокого охлаждения, в котором сжижение произво-
дится при давлении до 1,7—2,0 ат и температуре минус 35 — ми-
Приведенная классификация методов сжижения условна, так
как метод глубокого охлаждения является практически однйм из
вариантов комбинированного метода и отличается от последнего
меньшим давлением сжатия, что вызывает необходимость примене-
ния более низких температур.
Рис. 6-9. Принципиальные схемы сжижения хлора различными методами:
а — высокого давления; б — комбинированным; в — глубокого охлаждения; 1 — турбо-
компрессор, Р = 3,5 ат; 2 — турбокомпрессор, Р = 12 ат; з — конденсатор хлора с водя-
ным охлаждением; 4 — хранилище жидкого хлора; 5 — конденсатор хлора с охлаждением
от холодильной установки; 6 — холодильная установка (1от—15 до—20 °C); 7 .— компрес-
сор Р — 1 — 2,0 ат; 8 — холодильная установка (I — —40 4-60 °C).
Метод глубокого охлаждения в настоящее время мало приемлем
для многих производств, так как помимо требований экономики
производства давление хлора 1,7—2,0 ат недостаточно для многих
потребителей. Поэтому для сжатия и транспортирования хлора при-
меняются преимущественно компрессоры на давление 3,0—4,0 ат,
что соответствует комбинированному методу сжижения.
Температура сжижения, указанная для различных методов в за-
висимости от требуемой степени сжижения, состава хлоргаза и ре-
сурсов холода, может существенно изменяться. Помимо того, ни
один из указанных методов не пригоден для работы со степенью
сжижения 98—99%, для этой цели необходимо применять двухсту-
пенчатую схему сжижения с использованием на второй ступени
более низких температур.
Ниже будут рассмотрены наиболее распространенные технологи-
ческие схемы сжижения хлора.
При Сжижении сравнительно небольших количеств хлора и на-
личии на заводе потребителей абгазов сжижения применяются одно-
ступенчатые схемы сжижения, нашедшие широкое применение в про-
лые годы. На рис. 6-9 показаны принципиальные
мышленности в про
и
схемы одноступенчатого сжижения хлора методами высокого давле-
ния, комбинированным и глубокого охлаждения. Как было указано
328
ЗШЕ
Ir.
ранее, зти схемы мало пригодны для получения высоких коэ
циентов сжижения.
Для достижения коэффициента сжижения 98—99% методом высо-
кого давления с охлаждением конденсаторов водопроводной водой
потребовалось бы компримирование хлора до давления 25—30 ат,
а при использовании метода глубокого охлаждения при избыточном
давлении около 1 ат потребовалось бы применение холодильных
машин, работающих при температурах —60-4-— 70 °C.
1 г г
Сухой компримированный А б газ на санитарнуш ,
J --- ------- 1 очистку
азот
A
f
Хлоргаз
Вода
9
Вода
10
Рис. 6-10. Принципиальная двухступенчатая схема сжижения хлора комбини-
рованным методом при давлении 3,0—3,5 ат:
1 — турбокомпрессор для фреона; 2 — конденсатор фреона; 3 — поплавковые регуляторы
уровня; 4 — конденсатор хлора первой ступени; 5 — смеситель абгаза с азотом; 6 — тепло-
обменник; 7 — конденсатор хлора второй ступени; 8 — смеситель жидкого хлора; 9 — сбор-
ник жидкого хлора; 10 — погружной насос для перекачивания хлора; 11 — конденсатор
фреона; 12 — конденсатор-испаритель фреона; 13 — компрессора для фреона.
Давление компримирования .выше 12—15 ат не применяется
в хлорной промышленности. С повышением температуры растут за-
траты на получение холода. Поэтому в современных схемах сжиже-
ния хлора с высокой степенью сжижения, получивших распростра-
нение в промышленности, используется комбинирование повышен-
ного давления (3—12 ат) и охлаждения от —20 до —60 °C с помощью
холодильных установок.
В отличие от схем, изображенных на рис. 6-9, применяется двух-
ступенчатое сжижение хлора, что позволяет конденсировать 80—
90% хлора с использованием дешевого холода на уровне сравни-
тельно высоких температур и расходовать более дорогостоящий
низкотемпературный холод только на второй ступени конденсации,
где сжижается лишь 10—15% всего хлора.
На рис. 6-10 приведена двухступенчатая схема сжижения хлора
комбинированным методом. Хлоргаз после осушки компримируется
329
до 3,5 ат и подается на сжижение. Первая ступень сжижёния осу-
ществляется в конденсаторе, охлаждаемом испаряющимся фреоном,
поступающим с одноступенчатой холодильной установки при тем-
пературе —15 —25 °C. Абгазы после первой ступени разбавляются
азотом для предотвращения образования взрывоопасной смеси
с водородом и поступают в конденсатор второй ступени, охлажда-
емый фреоном из двухступенчатой холодильной установки при тем-
пературе —60 ч- —65 °C. По такой схеме достигается степень сжи-
жения 98—99% при сравнительно небольших затратах глубокого
холода.
схема сжижения хлора комби-
ПУ
Рис. 6-11. Принципиальная двухступенчатая
нированным методом при давлении 12,0—13,0 ат:
1 — хлор под давлением 3,5 ат после первой ступени компримирования; 2 — компрессор
рдя хлора на 12 ат; з — промежуточные водяные холодильники; 4 — конденсатор хлора
первой ступени с водяным охлаждением; 5 — смеситель для разбавлении абгазов; 6 — те-
плообменник для охлаждения азота; 7 — конденсатор хлора второй ступени; 8 — поплавко-
вый регулятор уровня; 9 — фреоновый компрессор; 10 — конденсатор фреона; 11 — сме-
ситель жидкого хлора; 12 — сборник жидкого хлора; 13 — погружной насос для жидкого
хлора.
J
Второй вариант двухступенчатой схемы изображен на рис. 6-11.
После первой ступени компримирования до 3,0—3,5 ат хлор подается
во вторую ступень компримирования^ где турбокомпрессорами или
винтовыми компрессорами сжимается до 12—13 ат. При таком давле-
нии основное количество хлора сжижается в конденсаторе С/водя-
ным охлаждением.
При температуре воды в летнее время 25—28 °C и перепаде тем-
пературы в конденсаторе 5 °C конденсация хлора на первой ступени
будет проходить при 30—33 °C. При этой температуре в зависимости
от концентрации хлора в исходном газе достигается коэффициент
сжижения 0,85—0,90. Для второй ступени сжижения используется
холод одноступенчатой фреоновой холодильной установки при тем-
пературе —15 4- —25 °C.
330
де-
<: л
£
(
*>
$
Высокая степень сжижения достигается при применении сравни-
тельно неглубокого охлаждения с малой затратой холода, так как
основное количество тепла конденсации отводцтся с охлаждающей
водой на первой ступени конденсации.
В отличие от приведенных ранее, в схеме, изображенной на
рис. 6-12, исключена специальная холодильная установка. Это упро-
щает схему производства и снижает необходимые затраты на его
организацию. Процесс сжижения проводится при давлении 12—
.13 ат, сжижение основного количества хлора, так же как ив
схеме
дутой: ; Яд газ
Хлор
Жидкий,
хлор
склада
Жидкий
*• хлор
ла склад
Рис. 6-12. Принципиальная технологическая схема двухступенчатого
нияz хлора без использования специальной холодильной установки:
7 — сухой хлоргаз под давлением 3,5 ат; 2 — компрессоры для хлора на 12
промежуточные водяные холодильники; 4 — конденсатор хлора первой ступени с водяным
охлаждением; 5 — смеситель длн разбавления абгазов; в — теплообменник для охлаждении
азота; 7 — конденсатор хлора второй ступени, охлаждаемый испаряемым хлором; 8 — сбор-
ник жидкого хлора; 9 — погружной насос длн жидкого хлора.
р=3,5зх
сжиже-
ат; з —
V
приведенной на рис. 6-11, происходит в конденсаторе, охлаждаемом
водопроводной водой.
Вторая ступень сжижения производится в конденсаторе, охла-
ждаемом испаряющимся жидким хлором. Часть сжиженного хлора
расходуется на испарение в конденсаторах второй ступени сжижения.
Испаренный хлор возвращается на всасывающую линию первой
ступени компримирования хлора. Испарение производится при давле-
нии около 1 ат, что соответствует температуре испарения —30
а/
и/-
В приведенных схемах не показана промежуточная осушка
хлора между первой и второй ступенями сжижения. Организация
промежуточной осушки абгазов перед подачей их на вторую ступень
сжижения позволяет исключить возможность вымораживания влаги
в конденсаторах второй ступени сжижения и образования отдельной
водной фазы в жидком хлоре, создающей условия для разрушения
аппаратуры и трубопроводов жидкого хлора; Такая промежуточная
осушка важна при применении глубокого холода (около —60 °C),
как это имеет место в схеме на рис. 6-10.
Предусмотренное в схемах двухступенчатого сжижения (см.
рис. 6-10—6-12) разбавление абгазов первой ступени сжижения азотом
излишне при сжижении хлора, содержащего небольшие примеси
водорода. В частности, при сжижении хлора, получаемого из цехов,
работающих по методу электролиза с диафрагмой, обычно нет необ-
, ходимости в разбавлении абгазов азотом и схема несколько упро-
щается.
Во всех схемах сжижения хлора должно быть обеспечено надеж-
ное разделение жидкого хлора и несжиженных абгазов. Это необхо-
димо, чтобы исключить попадание абгазов в емкости и хранилища
жидкого хлора и предотвратить возможность аварий при увеличении
содержания водорода в абгаза^ Для этой цели ранее между конден-
сатором и приемной емкостью для жидкого хлора устанавливали
гидравлические затворы, заполненные жидким хлором. В последнее
время стали применять поплавковые регуляторы уровня жидкого
хлора, закрывающие выход из конденсатора или промежуточного
сосуда при уменьшении в них жидкого хлора до определенного
уровня. Такие устройства более надежны, так как они исключают
попадание абгазов в хранилища даже при большом перепаде давле-
ния между конденсатором и хранилищем жидкого хлора.
Для обеспечения свободного стока жидкого хлора в приемные
хранилища в конденсаторе необходимо постоянно поддерживать
более высокое давление, чем в хранилище. Эту разницу давления
(0,5—1,0 ат) обычно поддерживают вручную или автоматически
с помощью дифференциального регулятора давления путем соедине-
ния газового объема хранилища с абгазной линией, в которой под-
держивается более низкое давление. .
Выбор наиболее рациональной и экономичной схемы сжижения
хлора зависит от многих факторов и определяется структурой по-
требления хлора на предприятии, наличием или отсутствием потре-
бителей низкопроцентного хлоргаза в виде абгазов, а также произ-
водств, для работы которых необходим чистый испаренный хлор.
Поэтому единой, общей для всех условий, оптимальной схемы сжи-
жения хлора не может быть предложено. В каждом отдельном случае
необходимо принимать наиболее рациональное решение.
Далее будут изложены некоторые общие соображения, которые
необходимо учитывать при конкретном выборе рациональной схемы.
Ниже приведены результаты технико-экономического сопоставления
методов высокого давлений, комбинированного и глубокого охла-
ждения при использовании одноступенчатой схемы сжижения [37].
Основной составляющей себестоимости жидкого хлора является
стоимость поступающего на сжижение хлоргаза, поэтому сравнение
всех методов проведено при одинаковом коэффициенте сжижения
и при условии, что абгазы сжижения находят полезное применение.
В табл. 6-6 приведены затраты на сжижение хлора различными
методами [37].
Наименьшие затраты на сжижение хлора получаются при исполь-
зовании метода высокого давления и максимальные — при методе
Таблица 6-6. Затраты на сжижение хлора
Затраты Метод высокого давления Комбин ир ов ан- ный метод Метод глубокого охлаждения
руб./т % руб./т % руб./т %
* Энергетические средства 3,57 46,3 5,14 54,3 8,91 60,7
Зарплата 0,79 10,2 0,79 8,3 1,30 8,8
Амортизация 0,87 11,3 1,01 10,6 0,97 6,6
Цеховые расхода . . . 2,52 32,4 2,52 26,8 3,49 23,9
Всего... 7 7,75 100 9,46 100 14,67 100
глубокого охлаждения. Это объясняется тем, что при методе высо-
кого давления снижаются энергетические затраты вследствие исполь-
зования воды для отвода тепла конденсации.
При отсутствии потребителя абгазного хлора на предприятии
возникает необходимость применения схем сжижения с коэффициен-
том сжижения, близким к единице, или оборудования установки для
извлечения хлора из абгазов сорбционными методами.
На новых предприятиях с большими мощностями по производству
жидкого хлора используются преимущественно двухступенчатые
схемы сжижения с общим коэффициентом сжижения 0,98—0,99.
Можно полагать, что наиболее экономичными должны быть двух-
ступенчатые схемы, работающие под давлением 10—12 ат с использо-
ванием на первой ступени сжижения в качестве холодильного агента
воды. / '
На второй ступени конденсация должна осуществляться при
температуре около —30 °C в конденсаторах, охлаждаемых испаря-
ющимся при атмосферном давлении жидким хлором или испаря-
ющимся фреоном от специальных холодильных установок.
ИЗВЛЕЧЕНИЕ ХЛОРА ИЗ ЕГО СМЕСЕЙ С ВОЗДУХОМ
ИЛИ ИНЕРТНЫМИ ГАЗАМИ
Ранее были рассмотрены двухступенчатые схемы сжижения хлора,
обеспечивающие практически полное сжижение хлора. Абгазы после
второй ступени сжижения содержат 0,5—2,0% от поданного на
сжижение хлора и обычно поступают на установки санитарной
очистки газов от хлора растворами щелочи или известковым молоком.
Получающиеся в установках растворы гипохлоритов натрия или
кальция стараются в дальнейшем использовать. В случае невоз-
можности их полезного использования проводят разру
ение актив-
и
ного хлора с помощью восстановителей, катализаторов разложения
гипохлоритов или путем солянокислотного разложения.
Однако нередко возникает необходимость использования хлоро-
воздушных смесей с низким содержанием хлора. При сжижении
333
J
хлора всегда получаются абгазы, содержащие некоторые количества
разбавленного хлора. Объем абгазов конденсации зависит от состава
исходного газообразного хлора и принятой схемы сжижения. На
одноступенчатых установках сжижения в абгазах остается обычно
до 10—15% поступившего на сжижение хлора. При этом состав
абгазов колеблется в широких пределах.
Если производится одноступенчатое сжижение хлора, получен-
ного электролизом с диафрагмой при 3,5 ат и температуре —154-
4~ —17 °C, тона 1 т сжиженного хлора выделяется примерно 120 кг
абгазного хлрра в виде газа, содержащего 62—67% С1а, 9—10% СОа
и 0,68—0,9% На. После второй стадии конденсации при давлении
3,5 ат и охлаждении до —60 °C абгазы имеют примерный состав:
13,4—22,4% С1а, 15,8—17,2% СОа, 1,2—1,8% Н2 и содержат 20—
25 кг хлора на 1 т сжиженного хлора.
Хлоровоздушные смеси получаются также при передавливании
жидкого хлора сжатым воздухом и при эвакуации остатков хлора
из цистерн, контейнеров и баллонов при их проверке перед заполне-
нием.
Хлор абгазов стараются использовать непосредственно для произ-
водства. Если это невозможно, хлор из хлоровоздушных смесей
коццентрйруют с целью получения жидкого хлора или использова-
ния 100%-ного газообразного хлора. Если не представляется воз-
можным использование или концентрирование разбавленного хлора
из абгазов, его обезвреживают, улавливая в санитарных колоннах
растворами щелочей.
Круг потребителей абгазного хлора ограничен. Наилучшими
потребителями разбавленного и загрязненного двуокисью углерода
. хлора являются производства бертолетовой соли," хлората натрия
химическим способом, хлораткальциевого дефолианта и некоторые
другие. Однако масштаб этих производств невелик. Производство
хлоратов натрия и калия развивается преимущественна по электро-
химическому способу.
Абгазы сжижения иногда используют для производства хлорной
извести, однако большое содержание двуокиси углерода в абгазах
и неравномерность подачи абгазов передавливания плохо отражаются
на качестве получаемой хлорной извести. .
При содержании хлора в смеси 60% и выше абгазы можно с успе-
хом использовать в производстве синтетической соляной кислоты.
При более низком содержании хлора смешивают электролитический
хлор с абгазами перед подачей их в печи синтеза HG1. При этом
образуется разбавленный хлористоводородный газ. Если хлорйстый
водород используется для гидрохлорирования органических про-
дуктов, его необходимо концентрировать на установке стриппинга.
Однако потребность в синтетической соляной кислоте весьма
ограничена, так как при производстве различного рода хлороргани-
ческих продуктов образуются большие количества попутного хло-
ристого водорода. Поэтому на многих крупных хлорных комбинатах,
имеющих большое количество попутного хлористого водорода, произ-
334.
515
водство хлористого водорода или соляной кислоты не может рассма-
триваться как реальный потребитель больших количеств абгазного
хлора.
Если прямое использование абгазного хлора для производства
невозможно, его концентрируют из разбавленных смесей с воздухом
и другими газами. Как ранее было рассмотрено, хлор из разбавлен-
ных смесей с инертными газами может быть сжижен глубоким охла-
ждением под давлением. Для этого смесь при давлении 3,0—12 ат
необходимо охлаждать до температуры —60 °C и ниже [38].
Если применение глубокого холода затруднительно, исполь-
зуются другие способы извлечения хлора из его разбавленных сме-
сей. Например, из смесей с воздухом хлор может быть выделен с по-
мощью различных жидких или твердых сорбентов (четыреххлори-
стогб углерода, воды под давлением, четыреххлористого титана,
хлоридов серы и др.) [39—41].
Фирма «Даймонд алкали» предложила способ селективной абсорб-
ции хлора четыреххлористым углеродом с последующим выделением
хлора потоком горячего пара [42, 43]. Получаемый таким способом
хлор содержит влагу, что затрудняет подбор материала для аппара-
туры и коммуникаций.
Если хлор удалять из растворителя в ректификационных колон-
нах при нагревании глухим паром, облегчается подбор доступных
и более дешевых конструктивных материалов [44]. Схема установки
по концентрированию хлора из разбавленных смесей сорбцией четы-
реххлористым углеродом приведена на рис. 6-13.
Растворимость хлора в четыреххлористом углероде возрастает
со снижением температуры (табл. 6-7), поэтому сорбцию обычно
ведут при температуре —15 —20 9С. Более глубокое снижение
температуры не рекомендуется, так как СС14 замерзает при —24 °C.
Таблица 6-7. Растворимость хлора в СС14
Давле-
ние,
мм
рт. ст.
Раство-
римость,
мол.
доли
Давле-
нве,
мм
рт. ст.
Раство-
римость,
мол.
доли
Давле-
ние,
мм
рт. ст.
Раство-
римость,
мол.
доли '
Темпе-
ратура,
®С
Раство-
римость *,
мол.
доли
При
124,2
271,3
409,2
515,0
И °C
0,062
0,140
0,211
0,267
При
84
181,5
271,0
364,0
512,5
760
0 °C
0,032
0,071
0,103
0,140
0,201
0,231
При
139,7
255,4
369,3
465,9
568,4
20 °C
0,026
0,054
0,077
0,101
0,124
25
40
50
60
70
80
90
0,119
0,079
0,060
0,052
0,043
0,034
0,030
* При 760 мм рт. ст.
При абсорбции хлора четыреххлористым углеродом выделяется
65 ккал/кг G12, поэтому применяют охлаждаемый абсорбер.
335
Абгазы, разбавленные воздухом для предотвращения образова-
ния взрывоопасной смеси при поглощении хлора, подаются в абсор-
бер, который выполнен в виде трубчатого теплообменника, ох ла-
Рис. 6-13. Принципиальная схема концентрирования хлора методом абсорбции
его 5 из абгазов четыреххлористым углеродом:
1 — абсорбер хлора; 2 — центробежный насос; 3 — холодильник СС14; 4 — теплообменник;
3 — ректификационная колонна для отгонки хлора; 6 — дефлегматор; 7 — кипятильник
колонны; 8 — конденсатор хлора; 9 — абсорбер паров СС14; 10 — холодильник гексахлор-
бутадиена; 11 — теплообменник; 12 — ректификационная колонна для отгонки СС14; 13—
дефлегматор; 14 — кипятильник колонны; 15 — конденсатор СС14; 16 — поступление рас-
сола; 17— отвод рассола.
5/7,04
о, оз
орг
^0,07
ОУ1 , Ор
Скорость газа, м/с
'Рис. 6-14. Зависимость коэффи-
циента скорости абсорбции хлора
четыреххлористым углеродом от
скорости газа.
чждаемого рассолом (температура —15 —20 °C), а затем на абсорб-
цию в насадочную часть аппарата, орошаемую четыреххлористым
углеродом при —15 —20 °C.
Содержание хлора в СС14, выхо-
дящем из абсорбера, зависит от усло-
вий проведения абсорбции: темпера-
туры, давления и содержания хлора
в исходном газе. Зависимость коэф-
фициента абсорбции хлора четырех-
хлористым углеродом от скорости
газовой смеси приведена на рис. 6-14.
При проведении абсорбции под
давлением до 2 ат и содержании
в исходном газе около 50% хлора
в выходящем из абсорбера растворе
можно получать до 12% хлора.
Отгонка хлора из раствора в СС14
проводится в ректификационной
колонне под давлением около 3,5 ат. При этом куб колонны обогре-
вается паром давлением 5 ат. Для образования флегмы жидкого хлора
336
3
дефлегматор колонны должен охлаждаться рассолом при темпера-
туре —15 + —20 °C.
Чистый четыреххлористый углерод из куба колонны возвра-
щается через теплообменник и холодильник на абсорбцию хлора.
Полученный из ректификационной колонны хлор содержит до
0,3% СС14 и может быть отобран в виде газообразного хлора или скон-
денсирован в холодильнике, как зто показано на рис. 6-13.
Непоглощенные инертные газы содержат небольшое количество
хлора и паров СС14. Для снижения потерь СС14 можно использовать
аналогичную абсорбционную систему, работающую с высокотемпе-
ратурным абсорбентом, например гексахлорбутадиеном. Гексахлор-
бутадиен кипит при 215 °C, его температура замерзания —21 °C.
Абгазы после первой установки абсорбции поступают на вторую,
где с помощью охлажденного гексахлорбутадиена улавливаются
остатки хлора и пары СС14. Поглощенные хлор и СС14 выделяются
из раствора в гексахлорбутадиене на ректификационной установке
и вновь возвращаются на первую ступень абсорбции.
При двухступенчатой схеме абсорбции потери хлора, СС14 и
гексахлорбутадиена очень невелики.
На 1 т жидкого хлора, извлекаемого из абгазов, затрачивается:
Холод (—15 ч- —20 °C), ккал . 300—500
Пар (6 кгс/см2), т .....0,7—0,8
Электроэнергия, кВт ч ... 10—15
Вода, м3 .......... 8—10
Схема может быть упрощена за счет исключения второй ступени
абсорбции гексахлорбутадиеном. Однако при этом увеличиваются
потери хлора и четыреххлористого углерода. Снижение потерь на
первой ступени абсорбции может быть достигнуто путем повышения
давления процесса и применения возможно более низкой темпера-
туры, но с учетом температуры замерзания СС14.
Если на абсорбцию поступают абгазы сжижения хлора с установки
глубокого охлаждения под давлением 1,3—1,7 ат, содержащие
примерно 55—70% С12, 18% СО2 и 1—4% Н2, то после абсорбции
при температуре —17 ч- —20 °C их состав изменится следующим
образом (в %):
С12 . . 1-5 Н2 . . 4—10
СО2 . . 40 СС14 . . 0,8—1,2
г ’ .
Потери на 1 т сжиженного хлора составляют около 2 кг хлора
и 2 кг СС14. Стоимость установки мощностью 10 тыс. т хлора в год
составит около 215 тыс. руб., а себестоимость концентрирования
абгазного хлоргаза в пересчете на 1 т жидкого хлора — 5—6 руб.
Основным недостатком этого метода является получение хлора,
содержащего около 0,3% СС14, что ограничивает число его потреби-
телей. Однако следует иметь в виду, что в хлоре диафрагменных
электролизеров также содержится от 0,05 до 0,20% хлорорганических
примесей.
22 Заказ 843
337
t /
Аналогичная одноступенчатая абсорбционная схема концентри-
рования хлора из его разбавленных смесей может быть осуществлена
с использованием в качестве абсорбента четыреххлористого ти-
тана [45].
Четыреххлористый титан при атмосферном давлении кипит при
136 °C, плавится при —23 °C, давление его паров при различных
температурах составляет [46]:
р, мм рт. ст
. . • « —13,9
. . . . 1,0
г,1 °C .....................80
р, мм рт. ст. 134
10 20 30 40 50 60
5,4 10,0 16,7 26,5 41,1 62,1
100 110 4 120 135
264,5 367,7 493,8 740,7
Растворимость хлора в четыреххлористом титане приведена
в табл. 6-8.
Хорошая растворимость хлора в TiCl4 и высокая температура
его кипения делают TiCl4 удобным для применения в одноступен-
чатых схемах абсорбции хлора из его разбавленных смесей.
Для абсорбции четыреххлористым титаном необходимо приме-
нять только тщательно высушенные хлоровоздушные смеси, так
как примеси влаги приведут к гидролизу TiCl4 с образованием
двуокиси титана, которая может забивать трубопроводы и аппара-
туру и загрязнять хлор примесями HG1.
Предложен способ концентрирования хлора из его разбавленных
смесей поглощением водой под давлением и последующей десорб-
цией [47]. Растворимость хлора в воде сильно изменяется в зави-
симости от давления и температуры (табл. 6-9).
Хлор поглощается водой под давлением в насадочном скруббере
и затем десорбируется из нагретого примерно до 100 °C раствора [48].
Эффективно комбинировать сорбцию хлора из разбавленных
хлорсодержащих газов холодной водой под давлением с последую-
щей подачей получаемых водных растворов хлора на башни для
охлаждения электролитического хлора. Схема такой установки по-
и
Таблица 6-8. Растворимость с хлора (в мол. долях)
при парциальном давления хлора р (в мм рт. ст.)
р
р
р
р
Пр и
69,6
140
200,3
260,3
359,1
457,0
509,0
545,3
625,8
10 °C
0,027
0,071
0,100
0,131
0,182
0,230
0,252
0,269
0,318
693,4
756,1
При
96,4
184,3
266,7
344,7
421,0
496,0
0,350
0,381
0 РС
0,038
0,070
0,102
0,141
0,162
0,184
При
141,4
275,0
405,0
530,9
650,4
765,0
1140,0
1672,0
2059,6
20 °C
0,031
0,062
0,091
0,108
0,140
0,162
0,251
0,359
0,436
2378,8
2909,2
3222,4
3617,6
При
2576,4
2986,8
3518,8
4111,6
4666,4
0,496
0,567
0,650
0,728
30 °C
0,423
0,484
0,557
0,641
0,711
338
к;
Таблица 6-9, Растворимость хлора в воде при различном давлении
ж температуре’(в г/л)
1 и — .... - ' " ' — .....
SJr 1
Fj- Л R
S «
св св
ifl-'- - J
w*
0.
10
20
Температура, °C
30
40
60
80
100
L 10
50
100
250
500
1000
2000
3000
4000
5000
0,488
0,679
1,717
2,79
*
* Выпадает
0,454
0,603
2,08
6,85
*
0,412
0,383
1,210
1,773
3,19
1,106
0,912
0,351
0,447
0,815
1,085
0,415
0,747
17,07
*
гидрат хлора.
2,69
4,30
13,02
18,73
30,8
3,61
10,22
14,47
18,84
1,914
2,80
7,14
2,08
3,07
6,38
12,54
15,26
9,52
11,42
Е*
[lit у *
I казана на рис. 6-15. Хлор
| холодной водой. Инертные газы поступают
и выбрасываются в атмосферу. При наличии в абгазах водорода по-
р вышенной концентрации их раз-
бавляют воздухом для предотвра-
щения образования взрывоопас-
ной смеси. Раствор хлора в воде
редуцируют и подают на орошение
колонн для охлаждения хлора,
выходящего из электролизеров.
При понижении давления и повы-
ении температуры воды в холо-
дильниках смешения большая
часть растворенного в воде хлора
десорбируется и поступает на
дальнейшую переработку вместе
с основным потоком хлоргаза.
Хлорная вода из холодильни-
ков смешения обесхлоривается
теми же способами, какие при-
няты при обычном использовании
охлаждения хлора (стр. 233).
Можно применять также схему с рециркуляцией воды. Горячая
вода после холодильников смешения не дехлорируется, а поступает
сначала на охлаждение в титановые холодильники и затем насосами
подается в скруббер для поглощения хлора из абгазов под давле-
нием. Путем подачи водного раствора хлора на орошение холодиль-
ников смешения цикл циркуляции воды замыкается.
абгазов под
I *
давлением поглощается
на санитарную очистку
Ь-’-.
?•
II
II
Лбгазы ни
санитарную
очистки _
/7ЯД
в нано-
Рис. 6-15. Принципиальная схема
концентрирования хлора абсорбцией
его из абгазов водой под давлением:
I — абсорбер для поглощения хлора
водой под давлением; 2 — редукционный ч
вентиль; з -- холодильник смешения; 4 ~~
колонна для удаления хлора из хлорной
воды. ’
Хлор из
электролиза
Хлор м
ч CyULKlf
холодильников смешения для
22*
339
При этом, как обычно, из цикла должен выводиться после пол-
ного дехлорирования избыток воды, образующийся вследствие кон-
денсации паров воды из хлоргаза в холодильниках смешения.
АППАРАТУРА УСТАНОВОК ДЛЯ СЖИЖЕНИЯ ХЛОРА
Компримирование хлора
Процесс компримирования хлора во многом аналогичен компри-
мированию других газов. Основные отличия обусловлены химиче-
скими свойствами и особенностями хлора. Значительная токсичность
хлора ставит повышенные требования к герметичности всей аппара-
туры, в том числе и компрессоров. Повышаются требования к плот-
ности сальниковых уплотнений особенно при применении сравни-
тельно высоких давлений (10—13 ат) [37]. Компримирование хлора,
как и любого другого газа, связано с повышением температуры.
При низкой температуре в атмосфере сухого хлора стойки многие
металлы. С ростом температуры стойкость металлов в сухом хлоре
снижается. При достижении так называемой температуры воспламе-
нения начинается сильно экзотермическая реакция металлов с хло-
ром. Температура воспламенения (в °C) некоторых металлов 149]
в атмосфере хлора приведена ниже:
Железо и сталь
Никель . . .
Свинец . . .
Медь ....
Титан . . .
—150
> 500
90—100
200
<20
Уже при повышении температуры хлора до 80—100 °C увели-
чивается скорость коррозии стали в атмосфере сухого хлора, по-
этому степень сжатия, допускаемая на одной ступени компримиро-
вания, ограничивается следующим условием: температура газовой
смеси не должна превышать 80—100 °C.
При начальной температуре хлора на всасывающей линии ком-
прессора 25 °C и верхнем допустимом пределе температуры 80 °C
допустимая степень сжатия на одной ступени составит 2,6.ХГакая
величина находится на нижнем пределе степени сжатия при компри-
мировании других газов [50]. Это означает, что в одноступенчатом
компрессоре практически можно получить хлор под давлением не
более 2,0—2,5 ат. В случае необходимости получить хлор более вы-
сокого давления следует проводить компримирование в две или бо-
лее ступени с промежуточным охлаждением хлора в межступенчатых
холодильниках.
Можно увеличить допустимую степень компримирования сни-
жением начальной температуры газа, подвергаемого компримиро-
ванию, или применением для изготовления деталей компрессорной
установки материалов, выдерживающих контакт с хлором при более
высокой температуре без заметной коррозии. ’
340
Для снижения температуры компримируемого хлора его можно
охлаждать в специальных холодильниках или путем вспрыскивания
F во всасывающую линию компрессора заданного количества жидкого
хлора [51, 52]. При этом за счет испарения жидкого хлора снижается
температура компримируемого хло-
ра, что позволяет достичь более вы-
сокой степени сжатия газа при той
же его конечной температуре.
Количество жидкого хлора,
вспрыскиваемого в газовую смесь,
ограничено необходимостью пол-
ного испарения жидкого хлора
с тем, чтобы парциальное давление
хлора в процессе компримирования
не достигало давления насыщенных
паров хлора при данном давлении и
температуре.
На рис. 6-16 показано изменение
давления и температуры в процессе
трехступенчатого компримирования
хлора при максимально допустимой
температуре 100 °C и температуре
хлора после межступенчатык холо-
дильников, охлаждаемых водой,
35 °C [53—55].
При обычном компримировании
предельно достигаемое давление со-
ставляет около 11 ат и при мак-
симально допустимом вспрыске жид-
кого хлора во всасывающую линию
ступеней компрессора около 22 ат.
Для увеличения коэффициента
сжатия необходимо затрачивать зна-
Рис. 6-16. Изменение темпера-
туры И давления в процессе
трехступенчатого компримирова-
ния хлора:
ABCDEFG — компримирование с ох-
лаждением в межступенчатых холо-
дильниках, < охлаждаемых водой;
A'B'C'D'E'F'G'— то же, но с мак-
симально допустимым вспрыском жид-
кого хлора; К — давление насыщен-
ных паров хлора; LM и НК — изо-
термы при 35 и 100 °C.
чительные количества сжиженного
хлора. При этом усложняется техно-
логическая схема и обслуживание
установки.
При применении поршневых ком-
прессоров попадание жидкого хлора
в цилиндры вследствие неточной его
дозировки во всасывающую линию
может привести к аварийному раз-
рушению оборудования.
Эти обстоятельства объясняют, почему такой способ повышения
степени сжатия хлора не получил применения в промышленности.
Увеличение степени сжатия на одной ступени при компримировании
хлора возможно лишь при использовании для изготовления деталей
компрессоров, теплообменников и трубопроводов, соприкасающихся
341
с горячим хлором, материалов, более коррозионно-стойких в этой
атмосфере, чем обычно применяемые стали. Применение никеля или
качественных никелевых покрытий позволяет повысить предельно
допустимую температуру нагревания хлора на ступени выше 80—
100 °C и соответственно увеличить степень сжатия более 2,6.
Дополнительные осложнения в процессе компримирования хлора
обусловлены тем, что применяемые для смазки компрессоров масла
хлорируются, осмоляются и теряют свои свойства. Пдэтому для ком-
примирования хлора используются компрессоры без обычных сма-
зочных масел. В качестве смазки для хлорных компрессоров нашла
применение концентрированная серная кислота. Низкая раствори-
мость хлора в концентрированной серной кислоте, удовлетворитель-j
пая стойкость обычных сталей в среде сухого хлора и серной кислоты
и удовлетворительные смазочные свойства создали условиц для ши-
рокого использования серной кислоты как смазки или запорной
жидкости. При снижении ее концентрации наступает интенсивная
коррозия сталей, поэтому тщательно следят, чтобы концентрация <
серной кислоты не снижалась менее 96%.
Впервые для компримирования хлора были использованы ком-
прессоры с жидким поршнем, известные как баденские. Жидкостью,
контактирующей с хлором, была концентрированная серная кислота,
передача давления осуществлялась через керосин, служивший ра-
бочей жидкостью компрессора. Затем были разработаны и получили
широкое распространение несколько типов поршневых двух- и трех-
стуненчатых компрессоров с сернокислотной смазкой цилиндров [7,
37]. Применялись двухступенчатые компрессоры фирмы «Сюрт»
производительностью 8 т/сут хлора с конечным давлением 6 ат, трех-
ступенчатые компрессоры фирмы «Амаг-Гильперт» производитель-
ностью 18 т/сут и давлением 6 ат и двухступенчатые компрессоры
фирмы «Эслингер>> производительностью 40 т/сут и давлением 8,6 ат
и ряд др. При параллельной работе обеих ступеней (компрессора
Болингер под давлением 2,8 ат производительность его достигала
60 т/сут. Это наиболее мощный поршневой компрессор для
хлора. '
Поршневые компрессоры с сернокислотной смазкой эксплуати-
руются и в настоящее время на некоторых старых установках, од-
нако вследствие невысокой мощности, громоздкости и неудобств,
связанных с применением сернокислотной смазки, они исполь-
зуются4 в небольшом объеме.
Известны также поршневые компрессоры без применения серной
кислоты [56, 57]. Это компрессоры Вюрцен производительностью
350 и 700 м3/ч при давлении 3,5 ат и производительностью 250 и
1500 м3/ч при давлении 12 ат. Отличительной особенностью компрес-
соров этого типа является применение графитовых поршневых ко-
лец, исключающих необходимость смазки поршневой группы. При-
ч менялись также поршневые кольца из тефлона [58, 59].
Были предложены лабиринтные поршневые компрессоры Зуль-
цер^-Эшер—Висс, также работащие без смазки и обеспечивающие
342
w-.
•VI
уплотнение поршня в цилиндре и штока за счет лабиринта с запор-
ным инертным газом [55].
В последнее время поршневые компрессоры для хлора уступают
место ротационным как более компактным, мощным и удобным
в эксплуатации. Применяются ротационные компрессоры с жидко-
стным поршнем, турбокомпрессоры и винтовые компрессоры. Во
всех типах ротационных компрессоров рабочее пространство, где
zzzzzzzz
SZZZE^ZZESZZZZE
Рис. 6-17. Схема турбокомпрессора с жидкостным поршнем типа РЖК-600/1,5:
1 — корпус; 2 — ротор; з — сальники.
/
происходит компримирование хлора, не имеет трущихся между со-
бой частей, поэтому отпадает необходимость в применении смазки.
В турбокомпрессорах и винтовых компрессорах исключена возмож-
ность загрязнения хлора посторонними примесями.
Смазочные масла применяются только для смазки деталей,
не соприкасающихся с хлором.
Ротационные компрессоры типа РЖК с сернокислотным запол-
нением выпускаются производительностью 650 и 1800 м3/ч при избы-
точном давлении до 1,5 ат. Устройство такого компрессора показано
на рис. 6-17. Характеристика ротационных компрессоров типа РЖК,
а также НЭШ, выпускаемых финской фирмой «Харкула» и итальян-
ской фирмой «Габионетти», приведена в табл. 6-10.
Таблица 6-10. Характеристика ротационных компрессоров
с жидкостным поршнем
Al
Компрессор * Частота вращения, об/мин ! Давление, нагнетания, ат 1 Производи- тельность, м* * 8/ч Потребляе- мая мощность, л. с.
1750
1750
1750
1750
720
720
Габионетти ...... .
Харкула Н-6 ........
Харкула Н-7 .. . . . .
РЖК-650 ........
РЖК-1800 ........
3,0 1350
2,8 980 130
3,15 966 138
2,8 1644 190
3,15 1614 200
2,5 650 55
2,5 1800 200
343
Ротационные компрессоры с жидким поршнем получили широ-
кое применение в хлорной промышленности. Схема установки та-
ких компрессоров показана на рис. 6-18. Охлаждение хлора произ-
водится циркулирующей серной кислотой, которая, в свою очередь,
охлаждается в водяном холодильнике.
Серная
кислотна
Рис. 6-18. Схема установки турбокомпрессора типа
РЖК:
1 — компрессор; 2 — смотровое стекло на линии подачи
серной кислоты в компрессор; 3 — сепаратор; 4 — холо-
дильник серной кислоты.
Значительно более экономичны по расходу электроэнергии тур-
бокомпрессоры. В СССР для сжижения хлора выпускаются турбо-
компрессоры типа ХТК на давление 3,5 и 12 ат.
Ниже приведена характеристика турбокомпрессора ХТК-2,5 —
3,5, выпускаемого в нашей стране [60]:
Производительность при условиях всасывания
: Р — 0,95 ат, t = 30 °C, м3/ч ............
Конечное давление, ат . . .................
Частота вращения ротора, об/мин............
Потребляемая мощность, кВт ................
2500
3,9
10 500
150
Компрессор четырехступенчатый с промежуточными холодиль-
никами между ступенями. Уплотнение между ступенями и на выводе
вала компрессора из корпуса осуществляется с помощью лабиринт-
ного устройства и*поддувки сухого инертного газа, предотвращаю-
щего попадание хлора в атмосферу помещения компрессорной. При
нормальной эксплуатации разбавление хлора за счет поддувки
инертного газа должно составлять около 0,2%.
Если хотят получить хлор давлением 12 ат, его под давлением
3,5 ат подают на второй компрессор.
При использовании турбокомпрессоров типа ХТК или анало-
гичных зарубежных типов во избежание возможной разбалансировки
и помпажа необходима тщательная очистка хлоргаза от аэрозолей
серной кислоты и других примесей. Обычно для этой цели приме-
няются фильтры с насадкой из стекловолокна, с помощью которых
остаточное содержание аэрозолей в хлоре снижается до 5 мг/м3.
344
За рубежом применяются турбокомпрессоры производитель-
ностью 6000 м3/ч с конечным давлением 3,5 и 12 ат [58, 59, 61]. Ана-
логичные компрессоры разрабатываются и в нашей стране.
Рис. 6-19. Схема работы вин-
тового компрессора ВК-9:
а —^устройство ступени винтового
компрессора ВК-9; б — схема ра-
боты: 1 — вторая ступень компрес-
сора; 2 — глушитель шума второй
ступени; з — холодильник первой
ступени; 4 — электродвигатель;
5 — глушитель шума первой сту-
пени; 6 — холодильник второй
ступени; 7 — первая ступень ком-
прессора; 8— глушитель шума на
всасывающей линии.
Вода
на сжижение
Вода
Адгаз на очистку
Вода
Сравнительно недавно стали широко использоваться для ком-
примирования хлора винтовые компрессоры, в которых как бы объе-
динены преимущества и объемных, и турбокомпрессоров [62]. Для
нормальной работы винтовых компрессоров не требуется столь
тщательная очистка хлора от аэрозолей, как для турбокомпрессоров.
Выпускаемый в СССР винтовой компрессор ВК-9 для хлора
имеет две ступени. Ниже приведена его характеристика [63]:
Производительность при условиях всасывания
Р = 0,95 ат и t = 30 °C, м3/ч............ .
Давление нагнетания, ат
3090
1,8
3,2
200
первой ступени ....
второй ступени ....
Потребляемая мощность, кВт
1
Компрессор снабжен водяными межступенчатыми холодильни-
ками для хлора и имеет лабиринтовое уплотнение вала с поддувкой
инертного газа. На рис. 6-19 показано устройство и схема работы
винтового компрессора ВКг9. Благодаря устойчивости в работе,
компактности и высокой производительности винтовые компрессоры
с успехом применяются в хлорной промышленности и в ближайшие
годы можно ожидать широкого их распространения.
II
Конденсация хлора
Как было ранее рассмотрено, в процессе конденсации хлора про-
исходит непрерывное изменение состава несконденсировавшейся
в данный момент газовой фазы. Следствием этого являются изменения
не только температуры ^конденсации
Длина, м
Рис. 6-20. Изменение температуры конден-
сируемого газа и хладо^гента по длине кон-
денсатора:
а — конденсация чистого хлора; б —конденса-
ция хлора с примесью инертного газа; 1 —
хлор; 2 — хладоагент.
но и ряда других факторов,
определяющих течение про-
цесса конденсации, при
прохождении газа через кон-
денсатор.
Изменяется объем остатка
несконденсировавшегося га-
за, его плотность, скорость
движения относительно по-
верхности теплопередачи.
У поверхности теплопере-
дачи образуется пленка не-
сжижаемых инертных газов,
затрудняющая поступление
хлора к поверхности тепло-
передачи. Процесс конденса-
ции значительно отклоняется
от режима, характерного для капельной или пленочной конденба-
ции. При этом за счет сопротивления диффузии возникает большая
разница между парциальным давлением хлорд в смеси и на поверх-
ности конденсации, что создает значительный перепад между тем-
пературой в объеме газа и поверхностной температурой конденсата,
покрывающего теплопередающую поверхность: На рис. 6-20 пока-
зано изменение температуры хладоагента и конденсирующегося
хлора вдоль конденсатора при конденсации чистого хлора и хлора,
содержащего инертные газы.
( На рис. 6-21 показаны температурные градиенты в пограничном
слое при конденсации чистого хлора и хлора, содержащего инерт-
ные примеси. В последнем случае, вследствие образования у поверх-
ности конденсации диффузионного слоя с высоким содержанием инерт-
ного газа, температурный градиент сильно возрастает. При сжиже-
нии хлора, содержащего инертные примеси, полезная разность тем-
ператур, при которой работает конденсатор, снижается.
Образование у поверхности теплопередачи пленки инертного
газа приводит к значительному уменьшению коэффициента тепло-
346
К передачи по мере увеличения степени сжижения хлора. Практически
среднее значение коэффициента теплопередачи в конденсаторах про-
мышленных типов близко к 40—50 ккал/(м2-ч- °С) [7, 53], в то время
как при конденсации чистого хлора, аналогично процессу конденса-
ции аммиака, коэффициент теплопередачи можно принять равным
600—800 ккал/(м2’Ч* °C). Изменение по мере конденсации хлора и
значения коэффициента теплопередачи, и разницы температур за-
трудняет расчеты конденсаторов для технического хлора, содержа-
щего примеси инертного газа [64, 65].
п
Расчет конденсаторов с учетом фактических теплофизических
свойстй паров хлора и смешанных с ним инертных примесей необ-
ходимо проводить постадийно с учетом непрерывного изменения
а
Рис. 6-21. Температурный градйент в пограничном
слое при конденсации чистого пара, (а) и смеси пара
с инертным газом (d):
1 — стенка трубки; 2 — пленка конденсата; 3 — пар; 4 —
диффузионная поверхность раздела; 5 — смесь пара с инерт-
ным газом.
<.
б
состояния конденсируемой смеси газов и охлаждающей среды.
Для получения окончательных результатов и сойпадения материаль-
ных и тепловых балансов расчеты приходится многократно повторять.
Такие расчеты трудоемки и облегчаются лишь при использовании
современной вычислительной техники.
Для расчета конденсаторов могут быть также использованы
данные по промышленным теплообменникам сходной конструкции,
используемым для сжижения хлораГ
Основное количество тепла обычно отводится в начальной зоне
конденсации, где концентрация инертных газов мала и влияние их
на коэффициент теплоперадачи невелико [66]. Для увеличения коэф-
фициента теплопередачи принимаются меры к созданию турбули-
зации потока газа, что позволяет снизить диффузионное сопроти-
вление в пленке инертного газа, а также применяют многоходовые
теплообменники, ступенчатую конденсацию, охлаждение конденса-
торов непосредственно испаряющимся хладоагентом и др.
Для конденсации хлора могут быть использованы любые про-
мышленные теплообменники. Конденсаторы должны быть рассчи-
таны на работу под давлением в зависимости от принятой схемы.
зшт
Толщина стенок трубок и аппаратов увеличивается с учетом воз-
можных коррозионных процессов. Ранее для конденсации хлора
широкд применялись аппараты, совмещавшие испаритель хладо-
агента (обычно аммиака) и конденсатор хлора в одной емкости, за-
полненной раствором хлористого кальция или хлористого натрия,
которые служили передатчиком холода от испарителя к поверхности
Ад газ
хлор
Рис. 6-22. Схема устройства совмещенного конденсатора хлора:
1 — корпус конденсатора; 2 — испарители аммиака; 3 — змеевики хлорного
конденсатора; 4 — коллектор жидкого хлора; 5 — сепаратор; 6 — мешалка
для рассола.
конденсатора хлора. Схема устройства такого совмещенного конден-
сатора показана на рис. 6-22. Для улучшения теплопередачи созда-
вали интенсивную циркуляцию рассола внутри аппарата. Как испа-
ритель аммиака, так и конденсатор хлора могут состоять из секций.
Такой многосекционный конденсатор с поверхностью конденсации
78 м2 имеет производительность 15 т/сут жидкого хлора.
Открытое зеркало рассола способствует усилению коррозии
поверхности конденсатора, соприкасающейся с рассолом. Для сни-
жения потерь холода аппарат покрывают слоем изоляции. Подоб-
ные конденсаторы применяются на ряде заводов и в настоящее время,
однако в новых мощных цехах сжижения начали преимущественно
использоваться кожухотрубчатые конденсаторы. Схема установки
такого конденсатора, охлаждаемого рассолом, показана на рис. 6-23.
Применяются как вертикальная, так и горизонтальная установка
кожухотрубчатых конденсаторов. При вертикальной установке обес-
печивается лучшая сепарация жидкого хлора от абгазов и наблю-
дается меньшее загрязнение поверхности теплопередачи, так как
возможные загрязнения . смываются жидким хлором,
по трубкам.
н
стекаю
щм
348
A*
Поступающий на сжижение хлор проходит внутри трубок, в меж-
трубноё пространство подается охлаждающий рассол. Для сниже-
ния коррозионной активности рассола к нему Добавляют пассивиру-
ющие компоненты, поддерживают слегка щелочную реакцию рассола
(pH —7,5—8,0) и предотвращают возможность насыщения рассола
кислородом воздуха. При. нарушении плотности конденсатора
хлор может попадать в систему циркулирующего рассола, что при-
водят к интенсивному коррозионному
трубопроводов. Поэтому необходимо
плотность конденсатора по отсутствию
активного хлора в циркулирующем
охлаждающем рассоле.
Наиболее удобно охлаждать конден-
сатор хлора путем непосредственного
испарения хладоагента. При этом упро-
щается схема установки, так как отпа-
дает необходимость в рассольном (или
какой-либо другой жидкости) цикле для
передачи тепла от испарителя хладо-
агента к конденсатору' хлора. Соот-
ветственно снижаются потери тепла и
создаются условия для получения более
высокого коэффициента теплопередачи,
что позволяет сделать конденсатор
более компактным.
Пока для сжижения хлора применя-
разрушению аппаратуры и
постоянно контролировать
II
ЗШЕ
А б газ
Жадкий
хлор в танк
Рис. 6-23. Схема установки
вертикального кожухотрубча-
Хладоагент
лись преимущественно аммиачные хо-
лодильные машины, было невозможно
использование конденсаторов хлора,
того конденсатора:
1 — конденсатор; 2 — сепаратор;
з — брызгоуловитель для жидкого
хлора.
>
охлаждаемых за счет испарения жидкого
хладоагента, так как случайные неплотности конденсатора приводили
бы к крупным авариям вследствие образования взрывоопасных смесей
хлора и аммиака. После того как в качестве хладоагентов стали
употреблять фреоны, оказалось возможным совмещение процес-
сов конденсации хлора и испарения фреона в одном теплообменном
аппарате [55, 66, 67]. В таких конденсаторах хлор поступает в труб-
ное пространство снизу, а несжиженные газы отводятся сверху.
Сжиженный хлор собирается в нижней части конденсатора и само-
теком стекает через регулятор уровня в промежуточный сборник.
Жидкий фреон подается в межтрубное пространство. Работа ведется
как с постоянным уровнем фреона в межтрубном пространстве, так
и с орошением поверхности теплопередачи жидким фреоном.
Холодильные установки
В процессе сжижения хлора газовую смесь необходимо охла-
дить до температуры конденсации, а также отвести выделяющуюся
при сжижении теплоту конденсации.
Если не стремиться к возможно более полному сжижению и при-
349
менять сравнительно высокое давление 10—12 ат, то сжижение можно
вести при температуре 30—35 °C. В этом случае для охлаждения
и отвода тепла конденсации используется вода. Температура воды
в средней полосе СССР в летнее врем^т обычно составляет около
25 °C, что позволяет осуществлять процесс сжижения с водяным
охлаждением при 3Q—35 qG. При наличии более холодной, например
артезианской, воды температура сжижения может быть понижена.
На практике водяное охлаждение используется на первых сту-
пенях двухступенчатых схем сжижения, работающих под давлением
10—12 ат (см. рис. 6-11). Для второй ступени сжижения водяного
охлаждения недостаточно, поэтому для получения желаемого коэф-
фициента сжижения необходимо применять холодильные установки.
При сжижении хлора при более низком давлении и для первых сту-
пеней требуется низкотемпературный хладоагент.
Для сжижения хлора могут быть использованы любые типы вы-
пускаемых промышленностью холодильных установок. В соответ-
ствии с принятой схемой сжижения и мощность^ установки выби-
рают тип холодильной машины и ее хладопроизводительность. Не-
обходимо учитывать, что в обычных схемах работы с передачей тепла
от конденсатора хлора к испарителю холодильной машины через
рассол температура испарения хладоагента должна быть на
5—8 °C ниже температуры конденсации.
Ранее цеха сжижения хлора имели свои холодильные установки.
. При применении совмещенных конденсаторов — испарителей ока-
залось удобнее соединять холодильную машину с конденсатором
хлора в одну производственную нитку. В последнее время цеха
сжижения хлора часто снабжают холодом из центральных завод-
ских холодильных станций. В этом случае в цеха подается холод
в виде охлажденного рассола, отработанный рассол , возвращают
на холодильную станцию.
Применяется также комбинированный способ снабжения холо-
дом цехов сжижения хлора. Холод для первой ступени сжижения
(на сравнительно высоком температурном уровне около —20°С)
обеспечивается за счет рассола, поступающего из центральной холо-
дильной станции; для второй ступени1 сжижения, где необходим
холод на уровне —50-^60 °C, создается локальная фреоновая холо-
дильная установка с непосредственным испарением фреона в хлор-
ном конденсаторе. -
Для получения холода в производстве жидкого хлора приме-
няются как ' компрессорные, так и абсорбционные холодильные
установки.
Абсорбционные холодильные установки для производства хо-
лода используют тепловую энергию л отличаются малым расходом
электроэнергии, поэтому их применение целесообразно при наличии
дешевых источников тепла [68—70]. Абсорбционные холодильные
установки потребляют максимальное количество пара в летние ме-
сяцы и минимальное — в зимние, что позволяет сглаживать сезон-
ные колебания в потреблении лара на химическом предприятии.
350
л
1£
i На абсорбционных аммиачных установках получают холод низких
Чемператур (—40 °C).
В По сравнении экономических показателей абсорбционных и ком-
Цпрессионных холодильных установок можно сделать вывод, что аб-
сорбционные установки малой мощности, работающие при высоких
^температурах испарения аммиака, не имеют преимуществ перед
В компрессионными. При низких температурах испарения аммиака
—25 °C и ниже) и высокой хладопроизводительности удельные
К капиталовложения на абсорбционные холодильные установки зна-
чительно ниже, чем на компрессионные [37, 68].
fe Компрессионные установки в СССР выпускаются с широким ди-
апазоном производительности по холоду и температур испарения [7] -
fe Кроме аммиачных применяются фреоновые компрессионные уста-
Вновки. Для получения температур до —30 °C используется фреон-21;
| для температур до —60 °C — фреон-12 и для температур до
fe—80 °C — фреон-22. Для получения температур ниже —304—40 °C
^применяются двух- и трехступенчатые холодильные установки.
I Для холодильных установок высокой хладопроизводительности
|и для работы при низких температурах в последнее время находят
Вприменение холодильные установки с турбокомпрессорами. При
|юборудовании мощных холодильных станций турбокомпрессорами
^снижаются удельные капиталовложения, а при низких температурах
fc испарения уменьшается на 10—20 % расход электроэнергии по
^сравнению с компрессорными станциями.
Фреоновые холодильные установки безопасны в работе, тогда
fe как при использовании аммиачных установок возможно образова-
| ние взрывоопасных смесей аммиака с воздухом при нарушении
I герметичности системы, заполненной хладоагентом.
LL* г
| Испарение жидкого хлора
| Перед употреблением жидкий хлор испаряют. Испарение жидкого
I хлора из цистерн, танков и контейнеров запрещено, так как при
|этом может происходить концентрирование треххлористого азота
| в остатке жидкого хлора. Ранее упоминалось, что присутствие не-
| больших количеств треххлористого азота в жидком хлоре при объем-
|ном его испарении может быть причиной крупных аварий.
В Испарение жидкого хлора можно проводить в теплообменни-
I кдх любой конструкции. Имеются две принципиально различные
[ схемы испарения жидкого хлора: в объемных и в проточных испа-
рителях.
| В проточных испарителях осуществляется полное испарение
| поступающего хлора. В качестве проточного испарителя могут быть
^использованы кожухотрубчатые теплообменники или конструкции
£ с змеевиком, в трубках которого происходит испарение жидкого __
L хлора. Проточные испарители Ьбеспечивают полную безопасность
f испарения хлора, даже в присутствии некоторого количества трех-
хлористого азота, так как при полном испарении исключена кон-
< центрация вредных примесей в количестве, приводящем к взрыву.
15
Кроме того, в случае применения проточных испарителей запас
жидкого хлора в испарителе невелик и автоматическое регулиро-
вание количества испаряемого в единицу времени хлора не ослож-
няется инерционностью системы. Поэтому для безопасного веде-
ния процесса испарения и облегчения его автоматического регули-
рования рекомендуется применять проточное испарение жидкого
хлора.
Объемные испарители обычно представляют собой емкость, снаб-
женную рубашкой для обогрева, но могут использоваться и тепло-
обменники других типов.
На входе в ijexa потребителей хлора обычно устанавливаются
рессивера для сепарации жидкого хлора, образующегося в зимнее
время в трубопроводах при передаче газообразного хлора на боль- .
шое расстояние. Нижняя часть рессивера оборудуется водяной ру-
башкой для обогрева и может служить испарителем жидкого хлора.
В объемных испарителях испарение происходит над зеркалом
жидкого хлора. Испарители могут работать периодически, если за-
литый первоначально в испаритель хлор полностью испаряется,
и с непрерывным пополнением испаряемого жидкого хлора. И в том,
и в другом случае в жидком хлоре, заполняющем испаритель, могут
накапливаться вещества, имеющие более высокую температуру ки-
пения, чем хлор, например треххлористый азот, что создает опас-
ность взрывов. Позто1Яу при применении объемных испарителей
непрерывного действия периодически проводят их полное опорожне-
ние, чтобы ограничить накопление примесей.
В объемных испарителях всегда присутствуют значительные
объемы жидкого хлора, их тепловая инерция велика и автоматиче-
ское регулирования процесса испарения жидкого хлора при значи-
тельных колебаниях потребности в хлоре затруднено.
Для обогрева испарителей обычно применяют горячую воду
(50—60 °C), получаемую в пароводосмесителях. При этом можно
получать испаренный ^лор давлением до 10—12 ат, что достаточно
для удовлетворения любых потребителей. Температура воды, пода-
ваемой на обогрев испарителей, обычно регулируется автоматически.
ХРАНЕНИЕ И ТРАНСПОРТИРОВАНИЕ ЖИДКОГО ХЛОРА
Некоторая часть жидкого хлора расходуется в цехах сжижения
на испарение с целью получения чистого газообразного хлора.
Основное количество сжиженного хлора передается потребителям.
Для транспортирования хлора на небольшие расстояния в пределах
одного завода обычно применяется трубопровод жидкого хлора.
Передача жидкого хлора по трубопроводу и испарение его потребите-
лем в ряде случаев выгоднее подачи испаренного хлора из централь-
ной станции.
Расчеты показывают, что стоимость трубопроводов и эстакад
для них при транспортировании жидкого хлора в 4—5 раз ниже,
чем трубопроводов испаренного хлора. Это объясняется применением
для трубопровода жидкого хлора труб малого диаметра.
352
Помимо того, при транспортировании испаренного хлора в зим-
нее время возникает опасность конденсации его в трубопроводе.
Для предотвращения попадания жидкого хлора непосредственно
4 потребителям последним приходится устанавливать дополнительные
сепараторы жидкого хлора с устройствами для его испарения, при-
с менять тепловую изоляцию трубопроводов и снабжать их паровыми
; спутниками для обогрева. Эти трудности отсутствуют при транспор-
тировании хлора в жидком виде [72]. Преимущество испарения хлора
L в цехе сжижения заключается лишь в возможности использования
- (хотя бы частично) тёпла испарения хлора с целью снижения расхода
холода на сжижение хлора.
а
Рис. 6-24. Схема танка для хранения жидкого хлора емкостью 125 мэ:
а — танк: 1 — штуцер для уровнемера ДУЕ-2; 2 — люк; 3— крышки люка с арматурой;
4 — скоростные клапаны; 5 — сифоны; 6 — штуцер для предохранительного клапана; б —
схема расположения штуцеров крышки люка: 1,5 — для выхода жидкого хлора; 2,4 —
для ввода жидкого хлора; 3 — для манометра; 6 — для подвода сжатого воздуха; 7 — для
сброса абгазов.
При транспортировании жидкого хлора по трубопроводу необ-
ходимо предпринимать специальные меры по обеспечению безопас-
ных условий эксплуатации трубопровода. При разрыве трубопро-
вода или нарушении его плотности возможно выделение в атмосферу
больших количеств хлора, что может привести к загазованности тер-
ритории и интоксикации персонала в районе аварии.
Правила устройства трубопроводов для жидкого хлора будут
рассмотрены ниже. В зависимости от объема потребляемого хлора
на далекие расстояния его перевозят в железнодорожных цистернах,
контейнерах и в баллонах. Есть сведения о перевозке жидкого хлора
водным транспортом в баржах специальной конструкции [73].
И производители, и потребители хлора создают склады для хранения
жидкого хлора. В связи с высокой токсичностью хлора при устрой-
стве складов и их эксплуатации должны быть соблюдены требо-
вания их безопасной эксплуатации [74—77].
Для хранения значительных количеств жидкого хлора исполь-
зуются горизонтальные танки емкостью от 15—20 до Г25 м3. На
рис. 6-24 показано устройство хранилища жидкого хлора емкостью
125 м3.
Для хранения малых количеств хлора применяются контейнеры
емкостью 500 и 1000 л и стальные баллоны емкостью от 20 до 55 л.
23 Заказ 843 353
Для потребителей хлора в лабораторных условиях применяют
баллоны емкостью до 1 л.
Устройство контейнера для жидкого хлора емкостью около
1000 л показано на рис. 6-25.
Танки, контейнеры и баллоны рассчитаны на рабочее давление
15 ат, что соответствует давлению насыщенных паров хлора при 50 °C.
Хранилища для жидкого хлора не реже чем раз в 2 года подвер-
гаются гидравлическому испытанию на давление, в полтора раза
превышающее рабочее. Хранилища должны отвечать всем требова-
ниям, предъявляемым к сосудам,
работающим под давлением [78].
Цистерны и контейнеры для
жидкого хлора
электросваркой, баллоны — путем
прокатки, т. е. являются цель-
нотянутыми. Сварные швы под-
вергаются термообработке для
снятия внутренних напряжений.
Баллоны периодически тариру-
ются водой, при увеличении объема
более чем на 3% от начального
баллоны отбраковываются.
Все виды тары снабжаются си-
фонными трубками, позволяющими
полностью опорожнять тару от
жидкого хлора. Баллоны снаб-,
жены одной сифонной трубкой,
контейнеры и цистерны двумя и
более. В отличие от емкостей для
других сжатых или сжцженных
газов на танках и цистернах для
жидкого хлора предохранительные
2030-
Рис. 6-25. Хлорный контейнер:
1 — защитная обечайка; 2 — защитный
колпак; з — кольцо стропальное; 4 — си-
фон; 5 —- бандажи; 6 — опора; 7 — кор-
пус.
изготавливаются
клапаны устанавливаются после за-
порного вентиля, так как для обеспечения надежной работы требуется
периодически проверять и ревизовать предохранительные клапаны.
Для гарантии безопасности устанавливаются два предохрани-
» тельных клапана и обеспечиваются такие условия, чтобы цри закры-
тии запорного вентиля на одном из клапанов второй обязательно
был в рабочем состоянии. Танки емкостью до 40 м3 имеют два сифона,
танки большей емкости — 4 сифона со скоростными клапанами, за-
крывающими выход жидкого хлора при случайных разрывах трубо-
проводов.
Железнодорожные цистерны для перевозки жидкого хлора по
устройству аналогичны танкам и снабжены защитным зонтиком
от нагревания солнечными лучами. Наиболее распространены цис-
терны грузоподъемностью 50 т; применявшиеся ранее цистерны на
15—20 т в настоящее время вытеснены более крупными. Циотерна
для перевозки жидкого хлора показана на рис. 6-26.
354
5\.
На танках и цистернах щтуцера устанавливают на крышке люка,
служащего для осмотра и чистки емкости. Наружная поверхность
Рис. 6-26. Железнодорожная цистерна для перевозки жидкого
хлора:
1 — котел; 2 — теневой зонт; 3 — колпай; 4 — предохранительный клапан.
Рис. 6-27.
рического
жидкого
2000 т.
Внешний вид
хранилища
t хлора
Для
емкостью'
цистерн окрашивается в светло-серый цвет с отличительной поло-
сой защитного цвета и снабжается надписями «Хлор», «Ядовито»
и «Сжиженный газ», наносимыми зе-
леной краской.
Перед заполнением жидким хлором
предусмотрена обязательная проверка
состояния контейнеров и баллонов и
замена вентилей отремонтированными
и прошедшими ревизию. Цистерны
проходят обязательную проверку каж-
дый раз, когда при возвращении в них
нет избыточного давления хлора.
В последнее время предложено хра-
нить сжиженный охлажденный хлор
в изолированных хранилищах под атмо-
сферным или небольшим избыточным
давлением [79—81]. Сообщается [82]
о применении для хранения жидкого
хлора сферических хранилищ емкостью
до 2000 т (рис. 6-27). Хранилища с двой-
ными стенками могут размещаться
в бетонной шахте, служащей аварийной
емкостью для жидкого хлора в случае
разрыва хранилища.
Оно рассчитано для хранения хлора под давлением, близким
к атмосферному, и должно работать при температуре около —34 °C.
Внутренний шар хранилища диаметром 13,7 м имеет толщину сте-
нок 23,1 мм и рассчитан на давление 2,32 кгс/см2 при темпера-
туре —45,5 °C. Зазоры между внутренним и наружным шарами
23*
355
заполнены изоляцией, через которую циркулирует тщательно высу-
шенный воздух.
Жидкий хлор из хранилища нельзя передавливать воздухом.
Для этой цели служат два центробежных насоса, вставленные в па-
трубки и опущенные почти до дна емкости. При демонтаже насоса
для ремонта обнажается только небольшая поверхность жидкого
хлора в патрубке. Насосы снабжены двойными сальниками с пода-
чей между сальниками сухого воздуха. Этим предотвращается воз-
можность проникновения хлора в атмосферу через сальники.
Минимальный запас жидкого хлора в хранилище 200 т. При даль-
нейшем снижении уровня хлора обнажаются вращающиеся части
насоса и начинается его вибрация. Кроме того, затрудняется под-
держание требуемой температуры в емкости. Максимальное запол-
нение хранилища не должно превышать (по проекту) 85,9% общего
объема. Контроль за уровнем жидкого хлора осуществляется с по-
мощью радиоактивного уровнемера с источником излучения137 Gs.
Низкая температура в хранилище поддерживается за счет испа-
рения хлора, пары которого отсасываются и затем после сжижения
вновь возвращаются в виде жидкости в хранилище. При хорошей
тепловой изоляции затраты холода невелики. При заполнении ем-
кости на 1700 т хлора расход его на испарение для поддержания по-
стоянной температуры составлял 2,5 т/сут. Если не отбирать газо-
образный хлор, температура хранилища в летнее время должна по-
вышаться на 0,55 °C в сутки.
При первоначальном заполнении хранилища необходимо соблю-
дать режим постепенного охлаждения емкости, чтобы исключить
возникновение напряжений в металле из-за большой разницы тем-
ператур в различных точках хранилища. Перед подачей в храни-
лище жидкий хлор охлаждают в теплообменнике до температуры
хранилища (до —344-----35 °C).
Для хранения больших количеств жидкого хлора предложено
использовать подземные полости, образующиеся при подземном
растворении поваренной соли. При этом жидкий хлор должен хра-
ниться под слоем насыщенного рассола высотой около 120 м, чтобы
создать достаточное давление [83].
При хранении жидкого хлора в танках их устанавливают в от-
дельные отсеки, снабженные приточной и вытяжной вентиляцией.
Каждый отсек отделяется от общего коридора склада газонепрони-
цаемыми дверями и имеет приемник для сбора всего количества
жидкого хлора из танка в случае аварии. Управление запорными
вентилями на хлорных танках должно быть дистанционным. Отсеки
снабжаются изолирующими противогазами. В отдельных случаях
допускается на заводах — производителях жидкого хлора установка
хранилищ для жидкого хлора вне помещения.
Допустимая емкость расходных складов у потребителей зависит
от размера потребления жидкого хлора [75]. При расходе до 0,5 т/сут
емкость склада не должна превышать квартальной потребности;
от 0,5 до 1 т/сут— 45-дневной; от 1 до 5 т/сут — 10-дневной;
356
г*
от 5 до 10 т/сут — 5-дневной; свыше 10 т/сут — 3-дневной. По-
рядок наполнения, перевозки и опорожнения емкостей с жидким
хлором подробно регламентирован [84].
Заполнение емкостей жидким хлором должно производиться из
расчета не более 1,25 кг жидкого хлора на 1 л объема. Такое ограни-
чение обусловлено высоким коэффициентом объемного расширения
жидкого хлора. При повышении температуры вследствие температур-
ного расширения газовый объем над жидким хлором уменьшается.
По достижении 100% заполнения емкости жидким хлором давление
в емкости повышается примерно на 10 ат на каждый градус даль-
нейшего повышения температуры.
Степень заполнения жидким хлором емкости и давление в ней
при нормальной загрузке (1,25 кг жидкого хлора на 1 л объема)
и при перегрузке на 5 и 10% против нормы приведены в табл. 6-11.
Таблица 6-11. Давление и степень заполнения жидким хлором
емкости при различных температурах
Температура, °C Избыточное давление, ат Степень заполнения, %
при нормальной загрузке при перегрузке /0 при перегрузке 10%
—33,6 0 80 84,6 88,4
—30 0,2 80,8 84,9 88,9
—25 0,5 81,5 85,5 89,6
—20 0,84 82,2 86,2 90,3
—15 1,23 82,8 86,9 91,1
—10 1,63 83,6 87,7 91,9
—5 2,14 84,3 88,5 92,7
0 2,66 85,1 89,4 93,6
5 3,25 86,0 90,2 94,0
10 3,95 86,8 91,1 95,5
15 4,75 87,7 92,0 96,5
20 5,62 88,6 93,0 97,5
25 6,63 89,6 94,0 98,5
30 7,75 90,6 95,1 99,6
35 8,95 91,6 96,4
40 10,50 92,8 97,4
50 13,70 95,1 99,5
60 17,60 97,8 -
69 22,0 100,0
Это же соотношение относится и к трубопроводам для транспорти-
рования жидкого хлора. Для предотвращения их аварийного раз-
рушения изолированные участки трубопроводов, закрытые с обеих
сторон задвижками, должны снабжаться устройствами для компен-
сации увеличения объема жидкого хлора при повышении темпера-
туры. Такие компенсаторы представляют собой емкости с газовой
подушкой.
Для передачи жидкого хлора из одной емкости в другую или пере-
дачи по трубопроводам на склад, для загрузки в железнодорожные
357
цистерны или потребителю применяется преимущественно пере-
давливание сжатым сухим воздухом. При этом давление сжатого
воздуха должно быть выше возможного давления в емкостях жидкого
хлора.
Помимо этого, на линии сжатого воздуха устанавливаются обрат-
ные клапаны, предотвращающие попадание хлора в воздушные ком-
муникации при случайном понижении давления в воздушной линии.
Для передавливания жидкого хлора применяются самостоятель-
ные компрессорные станции с давлением не менее 15 ат и осушкой
воздуха на силикагелевых, алюмбгелиевых или цеолитовых установ-
ках. Воздух для передавливания жидкого хлора должен содержать
не более 0,08 г/м3 влаги.
В среднем при передавливании воздухом 1 т жидкого хлора расхо-
дуется около 15 м3 воздуха и теряется 5 кг хлора. При передавли-
вании образуются большие объемы абгазов, содержащие около
20% хлора. Полезное использование этих абгазов затруднительно.
Сброс их в абгазную систему необходимо производить постепенно,
чтобы не создавать толчков давления в хлорной сети завода и рез-
ких изменений состава хлора в абгазном трубопроводе. В ряде слу-
чаев абгазы передавливания приходится уничтожать в санитарных
колоннах. Поэтому стараются применять другие способы транспорти-
рования жидкого хлора.
Вместо сжатого воздуха для передавливания жидкого хлора
используют газообразный хлор, подаваемый в хранилище под да-
влением. Для этой цели может применяться хлорный компрессор
либо напор создается путем испарения жидкого хлора под соответ-
ствующим давлением. Жидкий хлор может испаряться в отдельном
испарителе или в емкостях для жидкого хлора могут быть установ-
лены подогреватели. При этом в хранилище испаряется часть жидкого
хлора и повышается; давление паров до требуемой величины [85].
При использовании для передавливания жидкого хлора паров хлора
под давлением исключается образование разбавленного хлора, воз-
никающее при употреблении сжатого воздуха. В последнее время
разработаны и успешно применяются для перекачивания жидкого
хлора безсальниковые насосы с разделительной трубой [86, 87].
Трубопроводы для транспортирования жидкого хлора выпол-
няются сварными из стальных бесшовных труб и прокладываются
с уклоном для опорожнения трубопровода после перекачивания
жидкого хлора. На трубопроводах устанавливают, как было ука-
зано ранее, объемные расширители, а также скоростные клапаны
и арматуру для отключения трубопровода в случае необходимости.
На рис. 6-28 приведена схема защиты трубопровода жидкого
хлора от повышения в нем давления в случае нагрева при закрытых
вентилях на обоих концах трубопровода [72]. При повышении да-
вления на 25% сверх установленного рабочего предела срабатывает
один из двух предохранительных клапанов и жидкий хлор из трубо-
провода сбрасывается в буферную емкость, составляющую не ме-
нее 20% объема жидкого хлора, находящегося в трубопроводе.
358
Емкость обогревается горячей водой (не выше 50 °C). Испаренный
хлор из емкости подается через сбросный трубопровод хлора на ко-
лонны санитарной очистки.
Чтобы обеспечить свободный сброс хлора, после предохранитель-
_ ных клапанов нельзя устанавливать арматуру.
Для защиты трубопровода сжиженного хлора можно устанавли-
b вать буферную емкость, соединенную с трубопроводом сифоном,
’ опущенным до дна емкости. Объем парогазовой подушки над уровнем
жидкого хлора в этой емкости дол-
жен составлять не менее 20% объема
жидкого хлора в трубопроводе.
Схема установки такой буферной
емкости показана на рис. 6-29. Между
Рис. 6-28. Схема установки пред-
охранительных клапанов на тру-
бопроводе жидкого хлора:
Рис. 6-29. Схема установки на трубопро-
воде для жидкого хлора самокомпенсиру-
ющей емкости:
1 — хлорный танк; 2 — самокомпенсирующий
сосуд; з — потребитель жидкого хлора;
4 — запорная арматура танка.
1 — хлорный танк; 2 — запорная ар-
матура танка; з — запорная арматура
трубопровода; 4— защитная пластина;
5 — промежуточный сосуд; 6 — за-
порные вентили со спаренным штур-
валом, исключающим одновременное
отключение обоих предохранительных
НС
клапанов; 7 — предохранительные
клапаны; 8 — обогреваемая буферная
емкость. z
емкостью и трубопроводом не должно быть арматуры, уровень
жидкого хлора в емкости необходимо контролировать.
Может применяться также схема с компенсацией изменения
объема путем установки предохранительных обогреваемых баллонов.
Такие баллоны емкостью не менее 20% емкости трубопровода уста-
навливаются в высшей точке трубопровода и обогреваются горячей
водой при температуре не выше 60 °C. Баллон заполнен смесью воз-
духа с парами хлора. На рис. 6-30 показана схема установки такого
баллона на конце трубопровода у потребителя.
По правилам эксплуатации трубопроводов для жидкого хлора
запрещается закрывать арматуру на обоих его концах.
Для заполнения хлором баллонов и контейнеров организуются
специальные цеха розлива жидкого хлора. Такие цеха могут суще-
ствовать как при установках сжижения хлора, так и вне хлорных
заводов с использованием жидкого хлора, привозимого в цистернах.
Организация таких разливочных установок вне хлорных заводов
целесообразна в районах, отдаленных от производства жидкого
хлора, что увеличивает оборачиваемость баллонов и контейнеров
для жидкого хлора.
359
Рис. 6-30. Схема установки предохранитель-
ного баллона на трубопроводе хлора:
1 — испаритель; 2 — змеевик; з — предохранитель-
ный баллон; 4 — запорная арматура; I — линия
жидкого хлора; II — линия сжатого воздуха; III —
линия горячей воды; IV — линия пара; у — линия
испаренного хлора; VI — линия сброса.
Баллоны и контейнеры с промежуточного склада подаются на
опорожнение от остатков жидкого хлора и затем на эвакуацию хлора
с помощью хлорного компрессора. Остатки жидкого хлора соби-
раются и после анализа используются, а эвакуируемый из тары га-
зообразный хлор поступает обычно на санитарную колонну для по-
глощения. У освобожден-
ной от хлора тары вывора-
чиваются вентиля для ре-
монта и испытания, а бал-
лоны и контейнеры про-
мываются, сушатся и под-
вергаются тщательному
осмотру.
Баллоны и контейнеры,
нуждающиеся в окраске,
поступают на очистку по-
верхности, а затем в ка-
меру окраски и сушки.
После установки вен-
тилей происходит напол-
нение баллонов и контей-
неров жидким хлором.
Контроль наполнения осу-
ществляется по весу.
Контейнеры и баллоны
в установленные сроки
проходят гидравлические
испытания. < Баллоны и
контейнеры хранят в за-
крытых складах, состоящих из отсеков, которые снабжаются
устройствами для механизации приема и погрузки в вагоны бал-
лонов и контейнеров.
Перевозка жидкого хлора в цистернах, контейнерах и баллонах
по железной дороге строго регламентирована [88]. Хлор относится
к III группе — ядовитые газы. Перевозка баллонов с газами этой
группы должна производиться в вагонах или универсальных контей-
нерах без совмещения с другими грузами.
АВТОМАТИЗАЦИЯ КОНТРОЛЯ И УПРАВЛЕНИЯ ПРОЦЕССОМ
СЖИЖЕНИЯ ХЛОРА
г
Токсические свойства хлора и возможность возникновения ава-
рийного состояния в процессе сжижения, хранения и транспорти-
рования хлора предъявляют повышенные требования к автоматиза-
ции контроля и управления процессом сжижения хлора.
Непрерывность контроля и автоматическое устранение возни-
кающих отклонений от принятого режима являются основой обеспе-
чения безопасной эксплуатации производства, хранения и транспор-
тирования жидкого хлора.
3f0
Применяемые схемы автоматизации меняются в зависимости от
способа сжижения и местных условий. Ниже будут рассмотрены
основные решения по автоматизации контроля ц управления отдель-
ными стадиями производственного процесса.
В настоящее время все стадии процесса сжижения хлора автома-
тизированы в достаточной степени [89]. Наиболее распространен-
ные типы компрессоров для хлора: турбокомпрессор типа ХТК 2,5/3,5
и аналогичные типы зарубежных турбокомпрессоров, а также вин-
товые компрессоры снабжаются заводами-поставшиками приборами
и регуляторами, обеспечивающими автоматическое поддержание
стабильного заданного режима работы компрессора. Автоматически
регулируется давление хлора на всасывающем коллекторе и про-
изводительность компрессора в зависимости от выработки хлора
цехом электролиза. С помощью регулятора давления после конден-
сатора жидкого хлора и фазоразделителя поддерживается стабиль-
ное давление на стадии сжижения хлора, т. е. на напорной линии
компрессора.
Автоматическое поддержание температуры конденсации хлора
обычно осуществляется за счет автоматической стабилизации режима
работы холодильной установки и дополнительно путем установки
терморегулятора, увеличивающего или уменьшающего подачу хла-
доагента на хлорный конденсатор в зависимости от температуры
жидкого хлора в фазоразделителе.
Предложены различные схемы регулирования производитель-
ности установки конденсации хлора. При изменении концентрации
хлора предложено поддерживать заданную производительность со-
ответствующим снижением температуры сжижения или подачей
большего количества газообразного хлора для компенсации сниже-
ния коэффициента сжижения [14]. Во многих случаях целью автома-
тизации не является подержание постоянной производительности
цеха сжижения хлора.
Там, где цеха сжижения хлора выполняют роль буфера для хлора,
задачей автоматизации стадии сжижения будет автоматическое ре-
гулирование работы холодильных установок при увеличении или
снижении нагрузки на цех сжижения хлора.
Важной задачей является автоматический контроль состава ис-
ходного хлора и абгазов после первой и второй стадий сжижения,
а также автоматическое поддержание содержания водорода в абга-
зах на уровне, обеспечивающем безопасные условия производства.
Это особенно важно при. двухступенчатом сжижении с высоким
коэффициентом сжижения, но такой контроль следует применять
и при одноступенчатом сжижении, когда хлор поступает от цехов
электролиза с ртутным катодом. -
Для предотвращения образования взрывоопасной концентрации
водорода в абгазах применяется автоматическое разбавление послед-
него электролитическим хлором, сухим азотом или воздухом (в за-
висимости от применяемой схемы) по импульсу от газоанализатора,
определяющего содержание Н2 в абгазе.
361
и
Для контроля содержания хлора в осушенном хлоргазе, посту-
пающем на сжижение, в СССР разработаны автоматические фото-
метрические газоанализаторы типа УФ 6208 и для абгазов типа
УФ 6207. Для определения содержания водорода в абгазах может
быть использован дифференциальный термокондуктометрический га-
зоанализатор типа ТК-Г-18 [90]. Для автоматической сигнализации
предельного содержания водорода в хлоргазе может быть исполь-
зован менее точный прибор ТКГ-17. Для автоматизации процесса
испарения в проточных испарителях применяют комбинированное
автоматическое регулирование температуры горячей воды в испари-
теле и скорости подачи хлора в испаритель в зависимости от давле-
ния в линии испаренного хлора. Если жидкий хлор поступает в ис-
паритель под давлением сухого воздуха, подаваемого в хранилище
хлора, скорость подачи последнего в испаритель регулируется из-
менением давления воздуха. При использовании объемных испари-
телей хлора вследствие большой массы хлора в испарителе такой
прием не дает желаемых результатов.
Контроль за заполнением и опорожнением танков и емкостей
и за давлением в них осуществляется автоматически с передачей
показаний на центральный щит управления.
Для измерения количества жидкого хлора в танках применяются
манометрические весомеры и электротензометрические приборы.
Для измерения уровня жидкого хлора в хранилищах используются
также емкостные уровнемеры ДУЭ-2 и радиактивные уровнемеры
[91, 92].
Применение радиоактивных уровнемеров УР-4—УР-6 с кобаль-
товым источником требует специальных мер по защите обслуживаю-
щего персонала. Эти меры не нужны при использовании уровнемеров
УР-8 с источником излучения 137Cs. Подобные уровнемеры могут
быть вмонтированы внутрь хранилища для хлора. Для этой цели
в хранилище необходимо предусмотреть дополнительно два шту-
цера для установки уровнемера.
Основные показатели технологического процесса — давление,
температура, состав газов, расход хлора и энергетических ресур-
сов, уровни или количество жидкого хлора в хранилищах — пере-
даются на щит управления. Со щита управления производится
дистанционное управление арматурой танков для жидкого хлора
при приемке и расходовании жидкого хлора для отгрузки его в же-
лезнодорожных цистернах или передачи потребителям по трубо-
проводу.
II
II
ТЕХНИКА БЕЗОПАСНОСТИ
Вопросы техники безопасности приобретают в производстве
жидкого хлора особое значение в связи с тем, что при различных
аварийных ситуациях возможно выделение в атмосферу больших
количеств хлора из аппаратуры, трубопроводов и хранилищ жидкого
хлора. ~
362
Ниже приведены примерные данные о действии примесей хлора
в атмосфере на организм человека:
Концентрация хлора
в воздухе,
объемн ч./млн.
*
*
10—15
30
40—60
1000
1800
Действие на организм человека '
Легкие симптомы отравления после нескольких часов пребыва-
ния в атмосфере
Максимально допустимая экспозиция человека в атмосфере
хлора от 0,1 до 1 ч
Раздражение горла
Кашель
Опасно через 0,5 ч пребывания в атмосфере
Смертельно через 0,5 ч и менее пребывания'в атмосфере
Смертельно через 10 мин
Предельно допустимое содержание хлора в воздухе производ-
ственных помещений составляет 1 мг/м3, а в атмосферном воздухе
в населенных пунктах максимальное разовое содержание — 0,1 мг/м3 и
среднесуточное — 0,03 мг/м3.
Жидкий хлор очень широко используется в самых разнообразных
отраслях народного хозяйства; и безопасное его применение обес-
печивается лишь при точном соблюдении строго регламентированных
способов изготовления, проверки, испытания и обслуживания ап-
паратуры для сжижения и испарения хлора, емкостей для его хра-
нения и трубопроводов для его транспортирования.
Для безопасной эксплуатации должна быть исключена возмож-
ность образования взрывоопасной концентрации водорода в абгазах,
сжижения и попадания абгазов вместе с жидким хлором в емкости
для его хранения.
Опасность взрывов хранилищ или испарителей жидкого хлора
возникает при наличии в нем примесей треххлористого азота.
Высокий коэффициент объемного термического расширения
жидкого хлора может приводить к авариям при нарушении правил
заполнения хранилищ и эксплуатации трубопроводов. Меры, обес-
печивающие безопасную эксплуатацию, были рассмотрены ранее
при изложении технологических схем.
ЛИТЕРАТУРА
1. Пасманик М.И., С а с-с-Т нс ов ск.и й Б. А., Я ки и ен к о Л. М.
Производство хлора и каустической соды. Справочник. М., «Химия»,
1966.
2. Справочник химика. Т. I. М., Госхимиздат, 1962.
3. Справочник химика. Т. П. М., Госхимиздат, 1963.
4. Справочник химика. Т. III. М., «Химия», 1964.
5. Kirk R. Е., О thmer D. F., Encyclopedia of Chemical Technology,
v» 1 N. Y. 1952
6. Хлор жидкий. ГОСТ 6718—68.
7. С и и р и о в Л. А. Производство жидкого хлора. ОНТИ, 1935.
8. Chem. Week, 102, № 7, 28 (1968); 104, № 40, 35; № 8, 51; № 16, 55 (1969);
106, № 6 , 31 (1970). _
9. Chem. Eng. News, 47, № 5, 14; № 36, 85A; № 40, 35 (1969).
10. Chem. Techn., 21, Ml, 55 (1969).
363
11. Chem. Ing. Techn., 41, № 2, 87 (1969).
12. Chem. Ing., № 2, 34 (1969).
13. Пат. ФРГ 828239 (1952).
14. H ott e bom H., Chem. Ind. Techn., 37, № 6, 581 (1965).
15. Сасс-Тисовский Б.А. Производство хлора. M., Госхимтехиздат,
1933.
16. Якименко Л. М., Файнштейн С. Я. 50 лет химической науки
и промышленности. Сборник. М., «Химия», 1967. См. с. 163—175.
17. Сода то энсо, 15, № 2, 9 (1964).
18. Г е н и н Л. С., Смирнов Н. И., Хим. пром., № 6, 443 (1971).
19. Пат. ФРГ 1244737 (1967).
20. Chem. Ing. Techn., 33, № 4, 297 (1961).
21. Куба И. Доклад на конференции стран СЭВ по электролизу растворов
поваренной соли. Берлин, 1957.
22. Англ. пат. 1237568 (1971); канад. пат. 890819 (1972).
23. Sommers Н. A., J. Electrochem. Soc., ИЗ, № 3, 111; № 8, 185 (1966);
Р erugi n i G., Chim. e ind., 40, № 1, 16; № 2, 112 (1958); L i ska G.,
Weber H. D., Chem. Techn., 22, № 10, 602 (1970).
24. Якименко Л. M., Джагацпанян P. В., Ляскин Ю. Г.,
Филипов M. Т., 3 е т к и н В. И., авт. свид. СССР 347305 (1967);
Бюлл. изобр., № 24, 69 (1972); англ. пат. 1141134 (1967); франц, пат. 1515735
(1968). *
25. Пат. ФРГ 1266738 (1968).
26. Schmidt Н., Н о 1 z i n g e r F., Chem. Ing. Techn., 35, № 1, 37 (1963).
27. Hagemonn H., Chem. Ing. Techn., 39, № 12, 744 (1967).
28. К a t e 1 e a r J. A. A., Electrochem. Techn., 5, № 3—4, 143 (1967).
29. J. Electrochem. Soc., 113, № 3, Illa (1966); 113, № 8, 185 (1966).
30. Франц, пат. 1521128 (1968).
31. Англ. пат. 1125519 (1968).
32. Пат. ФРГ 1289520 (1969).
33. Япон. пат» 7006684 (1970).
34. N i с h о 1 J. Н., В г i n k J. A., Electrochem. Techn., 2, 233 (1964).
35. М i s с к е J., Р а у е г S., Chem. Ing. Techn.’, 39, № 12, 734 (1967); пат.
ФРГ 973113 (1959).
36. Winnacker К., К uchler L., Chemische Technologie, Bd. I, Anor-
ganische Technologie, Munchen, 1970.
37. Файнштейн С. Я. Жидкий хлор. M., «Химия», 1971.
38. Пат. США 3374637 (1968).
39. Пат. США 2750002 (1956).
40. Голланд, пат. 125296 (1968).
41. Франц, пат. 866780 (1944).
42. Пат. США 2765873 (1956).
43. Chem. Trade J., 140, № 3648, 1067 (1957).
44. Пат. США 3399537 (1968).
45. Байбеков М. К., Гель пери н Е. С. и др., авт. свид. СССР 216651
(1964); Бюлл. изобр., № 15, 23 (1968).
46. Фурман А. А., Р а б о в с к и й Б. Г. Основы химии и технологии
безводных хлоридов. М., «Химия», 1970.
47. J u d i J. W-, 8th Pacif. Northwest Industr. Waste Conf., Pullman Wasch,
1957, p. 81—84.
48. Bryson W. H. 11th Pacif. Northwest. Industr. Waste Conf., Cornwollis,
Oregon, 1963, p. 50, 147.
49. S с о n s e J., Chlorine, its Manufacture, Properties and Uses, N. Y., Rein-
hold, 1962.
50. Касаткин А. Г. Основные процессы и.аппараты химической техноло-
гии. М., «Химия», 1973.
51. Швейцар, пат. 327457 (1958).
52. Ensllik Е., Chem.-Ztg., 83, № 4, 122 (1959).
53. Ostertag A., Schweizerische Bauzeitung, 80, № 40, 677; 80, № 41.
704 (1962).
364
54. Z i g г 1 e r L., Chem. Ing. Techn., 22, № 11, 229 (1950).
55. AbelH.,Ozvegyl F., Technische Rundschau Sulzer, 50, № 3, 123 (1968),
56. Vogel K., Chem. Ing. Techn., 28, 260 (1956).
57. Kunzer W., Vogel K., Chem. Ing. Techn., 26, 555 (1954).
58. S о m m e r s H. A., Chem. Eng. Progr., 61, № 3, 94 (1965).
59. Hass K., Chem. Ing. Techn., 34, № 6, 337 (1962).
60. Компрессорное и холодильное машиностроение, ЦИНТИхимнефтемаш, № 2,
61.
3 (1967).
Chem.-Ztg., 88, № 12, 508 (1964).
С а кун И. А. Винтовые компрессоры. Основы теории, расчет, конструк-
ция. Изд. 2-е. Л., «Машиностроение», 1970.
Компрессорное и холодильное машиностроение, ЦИНТИхимнефтемаш,
№ 3, 2 (1966).
64. Н i е f 1 е A., Angewan. Chem., В8, 206 (1948).
Ozvegyl F., Schweizerische Bauzeitung, 83, № 9, 143 (1965).
63.
65.
Illi
В8, 206 (1948).
66. Смирнов H. И., Радун Д. В., Генин Л. С., Л омакинИ. Л.,
Хим. пром., № 9, 40 (1970).
67. К u b 1 i Н., N u renberger К., Chem. Age India, 12, № 6, 593 (1961).
68. Мартыновский В. С. Холодильные машины. М., Пищепромиздат,
1950.
69. Б а д ы л ь к е с Н. С., Д а н и л о в Р. Л. Абсорбционные холодильные
машины. М., «Пищевая пром.», 1966.
70. Бадылькес Н. С., Холодильная техника, № 3, 55 (1950).
71. Прейскурант № 23—02. Оптовые цены на оборудование холодильное и ком-
прессорное, вакуум-насосы, аппараты для производства кислорода и про-
дуктов разделения воздуха, оборудование для хранения газов. М., Прейску-
рантиздат, 1967.
72. Бережкове к и й М. И., СвистулевВ. М., Тимофеев А. Ф.,
Хайн П. Г., Хим. пром., № 10, 776 (1970).
73. Chem. Week, 89, № 22, 4 (1961); Chem. Eng., 65, 53 (1958).
74. Правила и нормы техники безопасности и промсанитарии для проектирова-
ния, строительства и эксплуатации производства жидкого хлора. М., «Хи-
мия», 1964.
75. Временные санитарные правила для проектирования, оборудования и содер-
жания складов для хранения сильнодействующих ядовитых веществ(СДЯВ)
в справочнике техники безопасности и производственной санитарии. Т. II.
Л., «Судостроение», 1965.
76. S о 1 о m a n Р., N a g п е Н., Le Chlore. Paris. Institut national de Secu-
rite pour la prevention des accidents du travail et des maladies, Profession-
nelles, I960:
77, The Chlorine Institute: Chlorine Manuel, New York, 1959.
78. Правила устройства и безопасной эксплуатации сосудов, работающих под
давлением. М., Металлургиздат, 1964.
79. Prahl К., Chem. Ing. Techn., 28, 56 (1956).
80/ Пат. США 2963874 (1960).
81. Chem. Proc., 45, № 3, 63 (1962).
82. Grona n C. S., Chem. Eng., 66, № 7, 76 (1959).
83. Пат. США 3151462 (1964).
84. Инструкция по наливу, перевозке, приемке, опорожнению и эксплуатации
цистерн для жидкого хлора. М., Госхимиздат, 1961.
85. Chem. Ind., № 48, 1584 (1958).
86. Chem. Proc., 50, № 10, 98 (1967).
87. Cojocaru L., Timm K., Chem. Ing. Techn., 39, № 2, 85 (1967);
KSB Techn. Berichte, № 14, 46 (1969).
88. Правила перевозки грузов. M., «Транспорт», 1967.
89. ЛомакинИ. Л., Радун Д. В., ЛевачевА. Г., БалашовЛ. Н.
Автоматизация хлорных производств- М., «Химия», 1967.
90. Газоанализаторы водорода в хлоре. Проспект ВДНХ, 1963.
91. Ромм Р. Ф., Т е п е р М. Е., Приборостроение, № 6, 29 (1958).
92. Радиоактивный уровнемер. Проспект ВДНХ, 1963.
365
ГЛ А В A 7
ХЛОРАТЫ ЩЕЛОЧНЫХ И ЩЕЛОЧНОЗЕМЕЛЬНЫХ МЕТАЛЛОВ
*
А
г
Многочисленные кислородные соединения хлора находят широ-
кое применение в технике и народном хозяйстве благодаря отбели-
вающим и окисляющим способностям.
Наибольшее применение находят соли кислородсодержащих ки-
слот, образуемых хлором и имеющих общую формулу HG10rt, где п
изменяется от 1 до 4.
Гипохлориты и хлориты — соли хлорноватистой (HG1O) и хло-
ристой (НС1О2) кислот широко используются в качестве дезинфи-
цирующих и отбеливающих средств. Промышленное производство
гипохлоритов и хлоритов осуществляется в основном химическим
способом. Растворы гипохлорита натрия частично и в насто-
ящее время получают электролитическим способом. Однако этот
способ получения, как будет указано ниже, связан со значительно
большими удельными расходами электроэнергии и поваренной соли
по сравнению с химическим способом, поэтому электрохимический
способ производства гипохлорита натрия находит практическое
применение только при малых масштабах производства, когда эко-
номические факторы не имеют основного значения.
К высшим кислородным соединениям хлора обычно отйосят
хлораты, перхлораты и хлорную кислоту. Производство этих соеди-
нений в последние годы получило большое развитие.
ПРОИЗВОДСТВО ХЛОРАТОВ ЩЕЛОЧНЫХ И ЩЕЛОЧНОЗЕМЕЛЬНЫХ МЕТАЛЛОВ
Хлорат калия впервые был получен Бертоле в 1786 г. хлориро-
ванием горячего раствора едкого кали и широко известен под назва-
нием бертолетовой соли.
Первоначально хлораты получали химическим способом по ме-
тоду/Либиха: хлорированием известкового молока с образованием
хлората кальция и последующим переводом его в хлорат натрия
(или калия) или хлорированием раствора NaOH (или КОН) с полу-
^чением непосредственно растворов соответствующих хлоратов.
Промышленная технология получения хлоратов электролити-
. ческим способом была разработана в конце прошлого столетия [1],
и впервые электрохимический способ производства хлоратов был
осуществлен в 1892 г. в Швейцарии и в 1896 г. во Франции.
366
II
Электролитический способ производства имеет ряд преимуществ
по сравнению с химическим способом, поэтому в настоящее время
производство хлоратов осуществляется преимущественно этим спо-
собом. В 1900 г. в мире было произведено около 17,5 тыс. т хлоратов,
из них около 65% электрохимическим способом. В 1940 г. мировое
производство хлоратов оценивалось [2] приблизительно в 150 тыс.т,
причем две трети всего производства приходилось на хлорат ка-
лия и только около одной трети на хлорат натрия. В1967 г. мировое
производство хлоратов щелочных металлов превысило 300 тыс. т [3],
причем доля производства хлората калия сильно сократилась, а хло-
рата натрия — выросла. \
Помимо хлоратов натрия и калия в промышленном масштабе
выпускаются также хлораты магния и кальция, используемые
главным образом как дефолианты.
Хлорат натрия получают практически только электрохимическим
окислением растворов поваренной соли. Хлорат калия получают
в результате обменной реакции между хлористым калием и хлоратом
натрия (получаемым электрохимическим способом) или хлоратом
кальция (получаемым хлорированием известкового молока). Хими-
ческий способ применяется в настоящее время только для производ-
ства хлоратов калия и кальция в тех местах, где имеется избыток
хлора или большие количества абгазного хлора, не используемого
для других целей.
Вначале в промышленности было оганизовано производство
хлората калия, применявшегося в основном при получении спичек
и пиротехнических изделий. Производство хлората калия в настоя-
щее время невелико.
Ниже приведены данные о масштабах производства хлората, ка-
лия в некоторых странах [4, 5]:
II
Страна
Индия
Япония
Франция
Произвол-
Год ство, тыс. т Страна
1959 15,0 Бразилия
1966 4,7 Италия
1965 4,4 Испания
1965 2,9
Производ-
Год ство, тыс. т
1959 3,4
1965 1,1
1963 1,2
Значительно большее развитие получило производство хлората
натрия, что объясняется его широким применением в народном
хозяйстве для получения дефолиантов, двуокиси хлора, используе-
мой при отбелке целлюлозы и тканей, обезвреживании воды и в дру-
гих областях, а также в качестве гербицида сплошного действия.
Ниже приведены данные о производстве хлората натрия в неко- *
торых странах [6—8]
Производ-
Страна Год ство, тыс. т
США . s . . . . 1971 178,2
Канада .......... 1967 87,0 *
Франция .... 1972 43,7
Швеция........ 1967 32,0 *
Произвол-
Страна Год . ство, тыс. т
Япония ..... 1965 29,8
Италия ..... 1965 4,2
Испания .... 1965 5,2 **
* Ориентировочные данные.
** Хлораты натрия и калия.
367
Увеличение производства хлората натрия в США и Франции за
последние годы видно из данных, приведенных ниже (в тыс. т):
США
Франция
1946 г. 1955 г. 1960 г. 1965 г. 1968 г. 1971г. 1972 г.
18,5 42,4 82,6 120,4 151,4 178,2 —
13,8 27,9 33,6 46,7 ~ — 43,7
Распределение хлората натрия между отдельными потребителями
и изменение удельного веса различных потребителей в балансе
производства и потребления за последние годы на примере США
показано в табл. 7-1.
Таблица 7-7. Распределение хлората натрия
между основными потребителями в США [9—12]
Области потребления
Доля в %
1955 1958 1961 1966
Производство двуокиси хлора для отбели-
вания ..............................
Производство других хлоратов и перхло-
ратов ..............................
Производство химических продуктов
Дефолианты и гербициды...............
Металлургия....................
Прочее .............................
41,1
18,8
5,1
26,1
2,6
6,3
68,0
16,0
11,0
5,0
Значительные количества хлората натрия используются в метал-
лургии при переработке урановой руды [13, 14]. Кроме того, хлорат
натрия может найти применение в качестве электролита при электро-
химическом методе обработки металлических изделий [15].
Хлораты магния и кальция используются главным образом в каче-
стве дефолиантов для предуборочного удаления листьев хлопчат-
ника при машинной уборке хлопка.
Высокая гигроскопичность хлоратов кальция и магния позво-
ляет применять их для этих целей без опасения возможных возго-
раний хлопка из-за некоторого наличия в нем хлоратов.
Обычно в качестве товарного хлорат-магниевого дефолианта при-
меняется смесь, получаемая сплавлением хлората натрия с хлори-
дами магния. Хлорат-кальциевый дефолиант получают хлорирова-
нием известкового молока, он состоит из раствора хлората кальция
и хлоридов натрия и кальция. Твердый хлорат магния используется
также как средство для осушки газов.
ПРОИЗВОДСТВО ХЛОРАТА НАТРИЯ
i
*
Натриевая соль хлорноватой кислоты NaClO3, молекулярный
вес 106,45, образует кристаллы кубической формы, т. пл. 248 °C
и плотность 2,490 Т/см3 при 15 °C. Теплота образования соли
368
83,60 ккал/моль, теплота плавления 5,29 ккал/ моль и теплота рас-
творения в 6400 частях воды 5,39 ккал/моль.
Хлорат натрия хорошо растворим в воде, растворимость его
сильно возрастает с повышением температуры (табл. 7-2).
Таблица 7-2* Растворимость хлората натрия в воде
1 Темпера- тура, °C Растворимость Темпера- тура, °C Растворимость
г/л г/100 г Н2О вес. о/ /о * г/л г/100 г нао вес. о/ /0
—15 1,380 580 72 41,9 60 1,514 920 155 60,8
0 1,389 612 79 44,1 70 1,536 969 171 63,1
10 1,409 667 90 47,4 80 1,559 1019 189 65,4
20 1,430 720 101 50,2 90 1,581 1069 209 67,6
25 1,440 745 107 51,7 100 1,604 1119 230 69,7
30 1,451 770 ИЗ 53,1 110 1,625 1170 257 72,0
40 1,472 820 126 55,8 120 1,649 1217 277 73,8
50 1,493 870 139 58,2 122 1,654 1225 280 74,1
в растворах хлористого натрия
натрия
1Ш1
Растворимость хлората
сильно уменьшается с ростом концентрации NaCl. При этом сни-
жается также температурный коэффициент растворимости хлората
натрия.
В табл. 7-3 приведена растворимость хлората натрия (в г/100 мл
раствора) в растворах поваренной соли.
Таблица 7-3. Растворимость NaC103 в растворах
поваренной соли [2]
Температура,
- °C
Содержание NaCl, %
20
20
40
60
80
100
66
75
102
57,4
65
70
77
41,8
42
44,0
системах
NaClO. - NaCl и КС1О3 - КС1
Растворимость в
приведена в работе [16].
К техническому хлорату натрия предъявляются требования, при-
веденные в табл. 7-4 [17].
Твердый хлорат натрия упаковывают в мешки из полиэтилено-
вой пленки толщиной 150—200 мкм или из поливинилхлоридной
пленки, вкладываемые в стальные барабаны емкостью 100 л. Раз-
решается упаковка в мешки из хлориновой ткани с вкладышем из
полиэтиленовой пленки. При этом масса хлората в одном мешке до
50 кг. Хлорат натрия перевозят также в виде пульпы в насыщенном
24 Заказ 843
369
Таблица 7-4. Технические требования к хлорату натрия
Марии
Содержание, % (не более)
твердый
жидкий
Хлораты в пересчете na^NaClOg
для твердого продукта (в пересчете на сухое
вещество), не менее
для жидкого продукта .
Хлориды в пересчете на NaCl
Сульфаты в. пересчете на SO4
Хроматы в пересчете на СгО4
Нерастворимый в воде остаток
Железо в пересчете на Fe
Влага ...............
99,5
0,3
0,1
0,04
0,01
0,015
0,06
99,0
0,7
0,3
0,10
0,05
0,015
3,0
45—65
0,7
0,3
0,20
0,05
0,015
Н. Н.
Цистерны
!1
растворе в стальных цистернах [18] с верхним сливом,
загружаются на 80 ±5% номинального объема.
При перевозке и хранении хлората натрия в виде пульпы резко
снижаются требования пожарной охраны и техники безопасности
по сравнению с твердым хлоратом натрия. Поэтому все потребители,
которые используют хлорат натрия в виде растворов, переходят на
его хранение и транспортирование в виде пульпы. При заполнении
и опорожнений цистерны используют зависимость растворимости
хлората натрия в воде от температуры: повышая ее, растворяют
твердую фазу и перекачивают раствор.
Хлорат натрия в промышленности получают преимущественно
электролизом водных растворов поваренной соли в электролизерах
без диафрагмы. Хотя электрохимическое производство хлората нат-
рия существует уже около 80 лет, исследования процессов образо-
вания хлоратов при электролизе водных растворов хлоридов щелоч-
ных металлов все время продолжаются. Ранее были установлены
основные зависимости электрохимического окисления водных рас-
творов хлоридов щелочных металлов до хлоратов в основном на
платиновых анодах [19—23].
Затем в связи с применением графита в качестве анодного мате-
риала продолжались исследования механизма процесса и техноло-
гии получения хлоратов [24—31]. В- последние годы продолжаются
работы [32—44] по исследованию механизма и кинетики процесса
получения хлората натрия окислением растворов поваренной соли
в процессе электролиза.
Первоначально в производстве хлоратов применялись платино-
вые, а затем магнетитовые аноды. В дальнейшем в производстве
хлоратов с успехом стали использовать графитовые аноды. В настоя-
щее время большое внимание уделяется процессу получения хлората
на анодах из перекиси свинца, а также на титановых анодах, покры-
тых активным слоем металлов платиновой группы или их окислов.
370
II
II
Замена материала анода всегда была связана с разработкой новой
технологии и определением оптимальных условий ведения процесса.
Процесс образования хлоратов при электролизе определяется
влиянием многих факторов: материала электродов (в основном ано-
дов), концентрации хлорида и хлората, значения pH электролита,
электродной и объемной плотности тока, температуры, добавок
к электролиту и др.
При электролизе концентрированного раствора хлористого нат-
рия без разделения электродных продуктов хлорат натрия обра-
зуется как в результате химического взаимодействия в растворе
первичных продуктов электролиза, так и их электрохимического
окисления.
Первичные процессы на электродах приводят к образованию на
аноде хлора и на катоде щелочи с выделением водорода
2С1-—2(?=С12 (7.1)
2Н2О4-2е = 2ОН’ + Н2 (7.2)
В результате смешения анодных и катодных продуктов на первой
стадии в зависимости от pH образуются гипохлориты или хлорно-
ватистая кислота
С12 + 2ОН- = С1О’ + С1- + Н2О (7.3)
С12 + ОН- = НС1О + С1- ' (7.4)
Последняя мало диссоциирована, поэтому концентрация ионов
СЮ’ в такой системе зависит от pH раствора [45].
При получении хлората натрия по электрохимическому меха-
низму происходит разрядка ионов СЮ’ на аноде
С1О-—е----> СЮ (7.5)
&
•I
Превращение СЮ в ионы хлората можно схематически изобра-
зить суммарным уравнением:
6С1О + ЗН2О 2C1Oj + 6H++4C1’ + 1,5O2 (7.6)
На образование одного грамм-иона С1О“ по реакциям (7.1) —
(7.3) необходимо затратить 2 Ф электричества, а на его разряд на
аноде по реакции (7.5) — еще один Ф. Всего для образования
двух грамм-ионов ClOj по реакциям (7.1) и (7.6) необходимо затра-
тить 18 Ф электричества, из которых 6 Ф расходуется на^выделение
кислорода, и выход хлората по току по такой схеме не превысит
66,6%.
Процесс электрохимического окисления ионов СЮ" в СЮ3 может
быть представлен в виде
6С1О- + 6ОН- + 6^2СЮу+4С1- + ЗН2О + 1,5О2 (7.7)
Доля, приходящаяся на электрохимический процесс, в общем
процессе образования хлората зависит от концентрации ионов гипо-
хлорита вблизи анода. Ее можно уменьшить, изменяя pH электро-
лита, его температуру, удельный объем электролита на единицу
24*
371
нагрузки, а также ограничивая или затрудняя диффузию ионов
гипохлорита к аноду.
Образовавшиеся в растворе гипохлорит и хлорноватистая кис-
лота взаимодействуют между собой с получением хлората по хими-
ческому механизму
2НС1О + С1О- = СЮз+2С1" + 2Н+ (7.8)
или
нею + 2С1О- = ClOj + 2С1- + Н+ (7.9)
Протеканию реакций (7.8) и (7.9) благоприятствуют слабокислая
реакция электролита и повышение температуры.
Максимальная скорость реакции (7.8) имеет место при pH—7,
а реакция (7.9) — при рН -^ 7,6. Константы скорости реакций (7.8)
и (7.9) близки между собой [43].
При получении хлората по химическому механизму отсутствуют
потери выхода по току на выделение кислорода и достигается высо-
кий выход хлората по току. Поэтому при реализации процесса
получения хлоратов на практике стремятся создавать условия для
протекания процесса преимущественно по химическому механизму.
Протеканию реакции по химическому механизму способствуют
повышение концентрации NaCl в прианодном пространстве, поддер-
жание pH на оптимальном уровне, увеличение температуры, затруд-
нение процессов диффузии ионов гипохлорита к аноду и возможно
быстрый вывод образовавшегося раствора гипохлорита и хлорно-
ватистой кислоты из межзлектродного пространства электролити-
ческой ячейки.
Если раствор поваренной соли подвергать электролизу без раз-^
деления образующихся на электродах продуктов, можно наблюдать
непрерывное изменение состава электролита. Вначале наблюдается
рост концентрации гипохлорита до достижения некоторой постоян-
ной величины за счет равенства скоростей образования гипохлорита
и окисления его до хлората по химическому и электрохимическому
механизму.
В процессе электролиза концентрация хлорида снижается, а хло-
рата возрастает.
Одновременно с основными процессами получения хлората на
катоде, аноде и в объеме электролита протекают побочные реакции,
снижающие полезное использование тока [32—34].
На катоде может происходить восстановление гипохлорита и
хлората до хлорида
СЮ" + Н2О + 2е > С1- + 2ОН’ (7.10)
СЮз +ЗН2О + 6е > СГ + 6ОН- (7.11)
Восстановление гипохлорита начинается при г0 = 0,86 В и хло-
рата при eQ = 0,63 В. Ион гипохлорита восстанавливается легче
иона хлората. Потери тока на восстановление гипохлорита и хло-
рата [35] по реакциям (7.10) и (7.11) определяются скоростью диф-
фузии ионов СЮ” и СЮз к катодной поверхности [36].
372
Потери выхода по току на аноде обусловлены протеканием ряда
процессов. При электрохимическом окислении ионов СЮ” до хло-
рата по реакции (7.7) 33,3% тока расходуется на выделение кисло-
рода. Кроме того, на аноде возможен разряд ионов ОНи или моле-
кул воды и выделение кислорода
2Н2О-4е----> О2 + 4Н+ (7.12)
Ч л
Потери выхода по току на разряд кислорода возрастают с уве- \
личением pH.
На платиновых, магнетитовых и перекисно-свинцовых анодах
возможно дальнейшее окисление полученного хлората до перхлората
CIC3 -НН2О-2е > СЮ4+2Ш (7.13)
На графитовых анодах не достигается потенциал, необходимый
для протекания реакции (7.13), поэтому окисления хлората до пер-
< хлората практически не наблюдается.
Снижение выхода по току происходит также за счет потери хлора
с газами электролиза при выделении растворенного в электролите
хлора [36]. Такие потери" тока и содержание хлора в газах электро-
лиза возрастают при снижении pH.
Потери выхода по току в результате катодного восстановления
и электрохимического окисления гипохлорита на аноде прямо про-
порциональны его концентрации, обратно пропорциональны плот-
ности тока, возрастают примерно на 2% с повышением температуры
на 1 °C и снижаются с ростом концентрации NaC103. При одинаковой
концентрации гипохлорита (СЮ” и НСЮ) эти потери не зависят
от pH в интервале его значений от 6,5 до 10 [32].
Такая зависимостыпозволяет считать, что скорость восстановле-
ния гипохлорита на катоде и окисления на аноде ограничивается кон-
векционной диффузией гипохлорита из объема к электродным по-
верхностям. Тогда для снижения потерь выхода по току по этой
причине следует уменьшать диффузию гипохлорита к электродам,
повышать плотность тока или увеличивать скорость циркуляции
раствора между электродами.
В объеме электролита возможно разложение гипохлорита с выде-
лением кислорода
2НС1О --> 2НС1 + О2 (7.14)
Этот процесс каталитически ускоряется при наличии в электро-
лите солей тяжелых металлов. Разложение может ускоряться также
при наличии в растворе железа, продуктов разрушения магнетито-
вых анодов или, например, кобальта, соединения которого в виде
сиккативов применяют при пропитке графитовых электродов льня-
ным маслом.
В щелочной среде возрастает концентрация ионов гипохлорита
и увеличиваются потери тока на катодное восстановление гипохло-
рита, а также на выделение кислорода по реакциям (7.7) и (7.12).
В кислой среде повышаются потери тока, обусловленные выделением
на аноде хлора.
Процесс электрохимического окисления хлорида натрия в хло-
рат сильно зависит от pH электролита [29, 30, 42].
На рис. 7-1 приведена зависимость от pH выхода по току по об-
;ему окислению на аноде (образованию хлората и гипохлорита, без
и
учета выделения элементарного хлора) и по получению хлората
натрия, учитывающая потери на катодное восстановление хлоратов
Рис. 7-1. Зависимость выхода по току от pH электролита при
40 ?С на платиновых анодах при га == 0,16 А/см2 (а) и на гра-
фитовых анодах при ia — 0,087 А/см2 (б):
1 — общее окисление на аноде; 2 — выход хлората натрия.
I
и гипохлоритов. Зависимость установлена для платиновых анодов
при следующих условиях процесса: анодная плотность тока ia =
= 0,16 А/см2, объемная плотность тока iv — 9 А/л, температура
40 °C.
На рис. 7-2 показано изменение потерь выхода хлората натрия
по току в зависимости от pH электролита, связанное с выделением
свободных хлора и кислорода на аноде и протеканием процессов
восстановления хлората и гипохлорита на катоде при тех же усло-
виях. Оптимальное значение выхода хлората по току достигается
при pH электролита около 6,5. При этом pH сумма потерь на выде-
ление элементарного хлора и кислорода на аноде имеет минимальное
значение. Потери на катодное восстановление сравнительно мало
изменяются с ростом pH. При снижении pH элактролита возрастают
потери на выделение свободного хлора, при увеличении pH возра-
стают потери на выделение кислорода.
При проведении процесса на графитовых анодах получаются
аналогичные зависимости выхода хлората по току и потерь выхода
по току от pH. Эти зависимости даны для плотности тока на аноде
0,087 А/см2, объемной плотности тока 8,3 А/л и температуры 40 °C
(см. рис. 7-2).
Оптимальное значение выхода хлората по току на графитовом
аноде наблюдается при pH 6, причем потери выхода по току
на выделение элементарного хлора и кислорода выше, чем на плати-
новом аноде в аналогичных условиях. Этим объясняются более вы-
374
сокие значения выхода хлората по току, получаемые на платиновых
анодах.
Оптимальное значение pH несколько меняется в зависимости
от объемной плотности тока и температуры электролиза. При пере-
ходе к высоким объемным плотностям тока и низким температурам
©нтимальное значение pH несколько сдвигается в кислую область.
Рис. 7-2. Зависимость потерь выхода по току от pH элек-
тролита при 40 °C на платиновых анодах при га = 0,16 А/см2
(а) и на графитовых анодах при га = 0,087 А/см2 (6):
1 — на выделение кислорода; 2 — на катодное восстановление; 8 —
на выделение элементарного хлора; 4 — на образование двуокиси
углерода.
Процесс электролиза обычно ведут в слабокислой среде при pH =
= 6,5 «е- 7,0.
С повышением температуры увеличивается скорость химического
образования хлората. Однако при этом снижается [44] стойкость
анодных материалов (графит, РЬО2 и др.). При применении графито-
вых анодов температуру электролиза не увеличивают более 40 °C.
При работе с анодами из РЬО2 температура может быть повышена
до 70-80 °C.
Увеличение удельного объема электролита, т. е. снижение объ-
емной плотности тока, благоприятно сказывается на выходе по току,
так как при этом снижается концентрация гипохлорита, и соответ-
ственно потери выхода по току.
Выход хлората по току и другие показатели процесса электро-
лиза сильно зависят от концентрации хлористого натрия в электро-
лите. На рис. 7-3 приведена зависимость выхода хлората но току
на графитовых анодах от концентрации хлорида натрия. В области
высоких концентраций NaCl изменение концентрации поваренной
соли мало сказывается на выходе хлората по току. Однако сни-
жение концентрации NaCl менее некоторой пороговой концентрации
приводит к значительному уменьшению выхода хлората по
току.
375
С повышением температуры электролиза значение пороговой
концентрации NaCl возрастает. Так, при 25 °C пороговая кон-
центрация составляет 100—110 г/л, а при 50 °C — около 150 г/л.
Со снижением концентрации NaCl менее пороговой при электро-
лизе с графитовыми анодами сильно возрастают потери тока на
окисление графита и соответственно увеличивается износ анодов.
300 250 200 750 700 50
Концентрация NaCl, г/л
Рис. 7-3. Зависимость выхода потоку хло-
рата натрия от концентрации хлорида
натрия на графитовых анодах:
1 — при 26,7 °C; 2 — при 48,7 °C; 3 — при
52 °C; 4 - при 65,8 °C.
Концентрация NaCl,
г/л
Рис. 7-4. Зависимость потерь вы-
хода по току от концентрации
NaCl на графитовых анодах:
1 — на выделение кислорода; ‘2 — на
образование СО2; 3 — на катодное
восстановление.
На рис. 7-4 приведена зависимость потерь выхода тока на выделе-
ние кислорода, образование двуокиси углерода и катодное вос-
становление от концентрации хлорида натрия при электролизе на
графитовых анодах в условиях плотности тока j.= 0,05 А/см2,
объемной плотности lv = 5 А/л и температуре 40 °C.
Сообщается [46], что при концентрации хлорида 120 г/л графито-
вые аноды изнашиваются вдвое быстрее, чем при концентрации
200 г/л. Износ сильно возрастает при снижении концентрации NaCl
менее 100 г/л. Износ графита увеличивается с ростом плотности, тока.
При электролизе на окисносвинцовых анодах также наблюдается
снижение выхода хлората по току по мере падения концентра-
ции NaCl в электролите, хотя и в Значительно меньшей степени,
что позволяет проводить процесс электролиза наг анодах из перекиси
свинца с большей степенью превращением NaCl в NaClO3, чем при
применении графитовых анодов.
Для снижения потерь тока на восстановление к электролиту
добавляют соли хрома [47, 48]. При восстановлении хромовой до-
бавки на катоде образуется слой из смеси трехвалентной окиси хрома
с хромовым ангидридом, выполняющей роль диафрагмы, которая
затрудняет диффузию ионов гипохлорита и хлората к катоду. За счет
этого снижаются потери тока на восстановление хлората и гипо-
хлорита.
376
b Действие добавок хромата в сильнокислых и сильнощелочных
растворах проявляется значительно слабее, что объясняется повы-
шенной растворимостью окиси хрома в этих растворах.
Аналогично действуют вводимые добавки солей кальция, магния, t
а также соединений ванадия и церия [48], однако практического
; применения эти добавки не нашли.
При добавлении солей кальция или магния на катоде образуются
плотные пленки осадка, которые приводят к повышению напряже-
ния на электролизере.
Действие добавок хромата связано также с ростом. плотности
тока в порах отлагающегося на катоде слоя хромовой пленки и уве-
личением скорости движения ионов СЮ- и СЮз, которая становится
г выше скорости конвекционной диффузии этих ионов к катоду [50].
Бихромат оказывает также буферное действие, поддерживая
оптимальную кислотность электролита за счет реакции
2СгС|“+2Н+ > Н2О + Сг2О?- (7.15)
Однако при применении графитовых анодов необходимо учиты-
вать, что добавление к электролиту хроматов приводит к усиленному
разрушению анодов. В присутствии хроматов увеличивается как
внутренний, так и наружный износ графитового анода [50—52].
Увеличение износа графитового анода может быть обусловлено лег-
костью выделения кислорода на поверхности графита в присут-
ствии адсорбированного хромата, а также прямым окислением гра-
фита хроматом.
На рис. 7-5 приведена зависимость износа графитовых анодов
и некоторых других показателей работы электролизера от концен-
трации хромата в электролите. Данные получены при сравнительно
кратковременных испытаниях, поэтому абсолютные значения износа
| графитовых анодов ниже стабильных, полученных при длительном
I проведении процесса. Особенно сильно проявляется влияние хро-
I мата на износ пропитанных графитовых анодов.
I Выход хлората по току и содержание кислорода в газах мало
| меняются при изменении концентрации Na2CrO4 в пределах от 5
р до 30 г/л. Поэтому, несмотря на увеличение выхода по току за счет
t снижения процессов катодного восстановления, с ростом концен-
J трации добавляемого к электролиту хромата ухудшаются общие
I показатели процесса электролиза в связи с увеличением износа
I графитовых анодов. Работы по изучению влияния добавок хроматов
L на процесс продолжаются и сейчас [52, 53].
I Первоначально в качестве анодного материала для получения
f хлоратов рассматривалась платина. Опубликовано большое число
। исследований процесса окисления хлорида до хлората на платино-
| вом аноде. Электрохимическое получение хлората натрия на плати-
I новом аноде может бытб осуществлено с выходом по току, близким
I к 100% . Однако высокая стоимость и дефицитность платины, а также
I значительные потери ее в производстве вынудили заменить платину
I другими анодными материалами.
ни
377
i . ' ’ -
j
। ,
До освоения производства искусственных графитовых электро-
дов применялись аноды из магнетита. Магнетитовые аноды исполь-
зовались для получения хлоратов калия до последнего времени [54].
Магнетитовые - аноды обладают малой электропроводностью, пло-
хими механическими свойствами, высоким напряжением на ячейке
и недостаточной стойкостью. В производстве хлората натрия магне-
титовые аноды часто трескаются и механически разрушаются при
5 10 15 20 25 30
Na2CrO4, г/л
а
Рис. 7-5. Влияние концентрации хромата на показа-
тели процесса электролиза:
а — непропитанные аноды, продолжительность опыта 7 сут;
б — аноды, пропитанные 15%-ным раствором льняного масла,
продолжительность опытов 20 сут.
W
86
82
i J—L j
III-
1 1
4
I 1 J—
I
1 1 r ; 1 -
10 15 20 25
Na2CrO4, г/л
повышении концентрации NaC103 более 200—250 г/л [2]. Износ
магнетитовых анодов составляет 2—3 кг/т хлората. Продукты раз-
рушения этих анодов загрязняют электролит примесями железа,
что способствует усиленному разложению гипохлорита в растворе
и снижению выхода по току. Вследствие высокого анодного потен-
циала на магнетитовых анодах может происходить окисление хло-
рат-иона до перхлората уже по достижении концентрации хлората
в электролите более 30 г/л.
С возникновением производства искусственных графитовых элек-
тродов для производства хлората натрия стали широко применять
графитовые аноды. На графитовых анодах процесс протекает при
более низких потенциалах, чем на магнетитовых или платиновых
анодах, и окисление хлората до перхлората не наблюдается.
Недостатком графитовых анодов является их недостаточная стой-
кость. В процессе электролиза графит окисляется до СО2. При этом
помимо химического износа наблюдается механический износ анода
и осыпание крупинок графита. Износ графитовых анодов' зависит
378
от условий протекания электролиза [50, 52, 55] и может колебаться
в пределах от 8—10 до 20—25 кг/т хлората натрия. Износ графитовых
анодов зависит также от потенциала анодного процесса. При достиже-
нии критического значения потенциала анода около 1,6 В (про-
тив Н. В. Э.) скорость коррозии графита резко возрастает
На рис. 7-6 приведена зависимость потенциала графитового анода
от плотности тока при условиях, аналогичных условиям работы
последнего электролизера в каскаде [состав электролита (г/л): NaCl —
110-115, NaClO3 — 520, Na2CrO4
достижения значения потен-
циала 1,6 В резко изменяется
наклон кривой, что соответ-
ствует изменению в ходе элек-
трохимического процесса на
аноде. .
Все факторы, способству-
ющие росту потенциала анода
до критического значения, вы-
зывают ускоренное разрушение
графитового анода. В частности,
повышение плотности тока до
1 кА/м2 привело к существен-
ному увеличению износа гра-
фитовых электродов, позтому
с целью снижения расхода гра-
фита целесообразно понижать
плотность тока до 0,7—
0,8 кА/м2. Следовательно, зна-
чение потенциала 1,6 В можно
рассматривать как верхний
предел интенсификации про-
цесса электролиза за счет повышения плотности тока в электро-
лизере с графитовыми анодами.
При работе с пропитанными анодами фактическая плотность тока
на аноде выше, чем в случае применения непропитанных анодов.
Поэтому (см. рис. 7-6) увеличение износа пропитанных анодов
наблюдалось при более низких плотностях тока, чем непропитанных
анодов [56].
При проведении электролиза при 40 ®С, конечной концентра-
ции NaCl в электролите 100—110 г/л* концентрации Na2C104.6—
8 г/л и плотности тока 1000 А/м2 расход графитовых анодов, пропи-
танных 15%-ным раствором льняного масла, составил: для графита
с первоначальной пористостью 25—30% и механической прочностью
200 кгс/см2 — 19 кг/т, для графита с первоначальной пористостью
20—25% и механической прочностью 300—350 кгс/см2 — 14 кг/т.
Качество графита оказывает существенное влияние на его удельный
расход.
— 25; pH = 5,8 4- 6,2]. После
25
i-д
—|------— । ।
100 500 2500 10000
Стационарные гальваноста-
Рис.
тические кривые для графитовых ано-
дов в условиях получения хлората
натрия при 40 °C:
1 — непропитанный графит; 2 — графит» про-
питанный 15%-ным раствором льняного
масла в четыреххлористом углероде.
379
При плотностях тока не выше верхнего предела интенсификации
основной причиной роста среднего напряжения на электролизерах
во время работы и соответственно расхода электроэнергии является
увеличение потерь напряжения в анодах за счет износа графитовых
анодов и значительного внутреннего разрушения графита. Эти
потери растут с увеличением плотности тока. Большое значение
приобретают конструктивные решения, обеспечивающие снижение
омических потерь в анодах при высоких плотностях тока. Это осо-
бенно важно, так как такая эффективная мера защиты от внутрен-
не'го износа, как возможно более полное закрытие пор графита про-
питывающим веществом, может при прочих равных условиях при-
вести к возрастанию анодного потенциала выше предельного.
Износ графитовых анодов увеличивается при повышении темпе-
ратуры, поэтому при применении графитовых анодов температуру
электролиза поддерживают не выше 40—45 °C. Ниже приведена
зависимость износа графитовых анодов и расхода тока на образо-
вание СО 2 от температуры при получении хлората натрия электро-
лизом при плотности тока 400 А/м2 [59]:
Темпера- тура, °C Концентрация NaCl, г/л Темпера- тура, °C Концентрация NaCl, г/л
120 240 120 240
Износ графитового 26 2 1 Расход тока на об- 26 2 1
анода, г/1000 А ч 43 8 3 разование СО2, 43 6 2,5
66 20 11 % 66 13 7,5
Рассматривался вопрос о применении охлаждаемых изнутри
графитовых анодов [60] с целью снижения расхода графита и воз-
можного увеличения плотности тока на анодах до 1,5 кА/м2, однако
практического решения он не нашел из-за громоздкости таких анодов.
В процессе электролиза при уменьшении концентрации хлорида
натрия стойкость графитовых анодов снижается, особенно при со-
держании NaCl менее 100 г/л [59].
Как показали исследования [51, 52, 55—61], потенциал анода
возрастает для непропитанных анодов особенно сильно при кон-
центрации NaCl в электролите менее 100—110 г/л. Для пропитанных
анодов сильный рост потенциала наблюдается уже при концентра-
ции NaCl ниже 140—150 г/л. Уменьшение концентрации NaCl при-
водит к росту потенциала выше критического и усиливает разруше-
ние графита. *
Стойкость графитовых анодов снижается с повышением плот-
ности тока [56]. Это объясняется увеличением анодного потенциала
и соответственно ростом скорости разряда кислорода и сгорания
анода.
380
Для увеличения стойкости графитовые аноды пропитывают льня-
ным маслом, хлорированным льняным маслом, хлорнафталином,
талловой олифой и другими веществами [62—65]. Чаще всего при-
меняют пропитку графитовых анодов раствором льняного масла
в четыреххлористом углероде. Необходимо учитывать, что пропитка
анодов приводит к уменьшению работающей поверхности графито-
вого анода, росту фактической плотности тока и увеличению потен-
циала анода при анодной поляризации. Поэтому при высоких плот-
ностях тока эффект пропитки анодов снижается, а иногда и вовсе
пропадает.
Графит может применяться также и в качестве катодного мате-
риала. Это целесообразно при биполярном включении электродов.
Для снижения напряжения выделения водорода на графите предло-
жено покрывать его слоем хрома или молибдена. Перенапряжение
выделения водорода на таких электродах близко к его значению для
соответствующих металлических катодов [66].
В последние годы разработаны [67, 68] и находят применение
в промышленности электроды из двуокиси свинца, которые могут
работать при более высокой температуре, чем графитовые. Аноды
из РЬО2 достаточно стойки в процессе электролиза, могут работать
при плотности тока 1000—3000 А/м2. При 60 °C, плотности тока
около 1500 А/м2 процесс электролиза можно проводить при напря-
жении 3,4—3^6 В с выходом по току 85—86%. Применение этих ано-
дов имеет большие преимущества по сравнению с графитовыми ано-
дами. Электролизеры с анодами из РЬО2 более компактны, вследствие
малого износа работают при практически постоянном во времени
тепловом и электрическом режиме, позволяют осуществлять более
глубокую переработку поваренной соли в хлорат, чем графито-
вые [69—70], и получать растворы, содержащие до 750 г/л хлората
при остаточном содержании поваренной соли 50 г/л и менее.
Двуокись свинца наносят на основу из титана, служащего для
подвода тока к активно работающему слою перекиси свинца. Пред-
ложено также наносить слой перекиси свинца на графитовые аноды
электролитическим способом из ванн с азотнокислым свинцом [71,
72]. Однако для практического применения таких анодов надо ре- <
шить задачу по предотвращению включения в электрохимический
процесс графитовой основы электрода. В противном случае не может
быть обеспечена долговечность работы такого анода.
Для получения растворов хлората натрия с малым содержанием
хлоридов, пригодных для непосредственного использования в произ-
водстве двуокиси хлора, удобно пользоваться анодами из двуокиси
свинца. При этом несколько снижается вы±од хлората по току, однако
стойкость анодов из двуокиси свинца существенно не снижается.
Срок службы анодов из двуокиси свинца примерно в 4 раза больше,
чем анодов из графита. Расход РЬО2 составляет обычно 0,8—1,0 кг/т
хлората натрия.
По сравнению с двухступенчатым процессом преимущество элек-
тролиза на анодах из двуокиси свинца заключается в том, что
381
растворы не загрязняются продуктами разрушения графитовых ано-
дов на первой ступени электролиза и отпадает необходимость в рас-
ходе платины на второй ступени. Отмечается, что, несмотря на высокое
содержание хлората, окисление его в перхлорат не наблюдается.
j Добавки солей хрома к электролиту не только снижают процесс
катодного восстановления, но и затрудняют анодное окисление хло-
рата до перхлората.
В последнее время возобновился интерес к применению анодов
на основе металлов платиновой группы, а также двуокиси руте-
ния, нанесенных на титановую основу электрода [73—75]. Для более
полного превращения хлорида натрия в хлорат (90—97%) можно
завершать процесс электролиза на анодах из металлов платиновой
группы [76].
Большой интерес представляет использование анодов,- образо-
ванных нанесением на титановую основу активного слоя, содер-
жащего смешанные окислы рутения и титана. Такие аноды имеют *
низкое значение потенциала при высоких плотностях тока и позво-
ляют проводить электролиз с высоким выходом хлората по току.
Расход тока иа выделение кислорода невелик и содержание кисло-
рода в электролитических газах ниже, чем при использовании ано-
дов из двуокиси свинца. Сообщается [77] о промышленном приме-
нении анодов такого типа. При плотности тока 3 кА/м2 и температуре
около 67 °C процесс электролиза протекает при анодном йотенциале
1,4 В по сравнению с потенциалом 1,63 В на графитовых и 1,85 В
на двуокисносвинцовых анодах [77].
Были предложены также аноды из окислов кобальта, нанесен-
ных на основу из титана или другого стойкого в этих условиях
металла [78].
В качестве катодов при монополярном включении электродов
\ I применяется обычная сталь. Рассматривалась такЩе возможность
ч использования в качестве катодов никель-хромовых сталей или
обычной стали с хромовым покрытием [33], чтобы исключить необ-
ходимость добавления к электролиту солей хрома. Однако такие
предложения в промышленности не были использованы. При би-
полярном включении электродов катодная поверхность опреде-
ляется конструкцией биполярного электрода и может быть графи-
товой или титановой.
Предпринимаются попытки снизить напряжение на хлоратных
электролизерах за счет применения катодов, работающих с кисло-
родной деполяризацией [79]. На пористых катодах, содержащих
катализаторы из соединений металлов платиновой группы, хро-
мовой кислоты и др., за счет сгорания подаваемого на ^электрод
кислорода с выделяющимся на катоде водородом можно снизить
напряжение до 1,2 В.
Процесс электролиза водных растворов хлорида натрия в элек-
тролизерах без диафрагмы используется также Для получения рас-
творов гипохлорита натрия, применяемых для отбеливания и дезин-
фекции. Как отбеливатель раствор гипохлорита натрия предпочти-
382
У
J
тельнее хлорной извести или растворов гипохлорита кальция, так
как при применении последних в аппаратах образуется шлам, а на
отбеливаемом материале могут образовываться осадки нераствори-
мых солей кальция, ухудшающие равномерность отбелки и качество
получаемого материала. Наилучшие результаты получены при при-
менение в качестве анодов платины или титана, покрытого активным
слоем из металлов или окислов металлов платиновой группы (Pt,
It, Ru и др.). Графитовые аноды недостаточно стойки й этих условиях.
Процесс электролиза необходимо проводить в условиях, пре-
пятствующих превращению
хлорноватистой кислоты
в хлорат как по электро-
химическому, так и по хи-
мическому 1 механизму, а
также по возможности
снижающих потери на
катодное восстановление
гипохлорита. Для сниже-
ния скорости превращения
гипохлорита в хлорат по
химическому механизму
электролизу необходимо
подвергать нейтральные
растворы NaCl при низкой
температуре. Необходимо
полученного гипохлорита натрия и
Концентрация NaCIO, е/х?
Рис. 7-7. Зависимость выхода по току и
удельных расходов электроэнергии и NaCl
от концентрации гипохлорита натрия:
1 — выход по току; 2 — удельный расход NaCl;
— удельный расход электроэнергии.
ограничить перемешивание
электролита, чтобы не на-
рушать кислый прианод-
ный слой электролита,
в котором образуется малодиссоциированная хлорноватистая кислота.
При повышении плотности тока возрастает доля тока, расходу-
емая на образование гипохлорита.
Повышение концентрации NaCl в электролите способствует сни-1
жению потенциала выделения хлора и таким образом увеличивает;
выход гипохлорита по току, однако приводит к возрастанию расходаj
поваренной соли. <
Если проводить электролиз периодическим способом, для каждог©
из выбранных режимов электролиза будет сначала наблюдаться
рост концентрации гипохлорита до определенной величины, по
достижении которой становятся равными скорости образования
и потерь активного хлора на катодное восстановление, преобразо-
вание в хлорат и химическое разложение гипохлорита.
Уровень предельной концентрации увеличивается с понижением
температуры, ростом концентрации NaCl и плотности тока, зависит
от pH, конструкции электролизера и анодного материала.
Для снижения катодного восстановления обычно применяют
добавки солей кальция, поскольку примеси солей хрома нежела-
тельны в процессе отбелки.
388
Выход гипохлорита по току, удельный расход электроэнергии
и хлорида натрия на 1 кг активного хлора в растворе изменяются
при всех прочих равных условиях в зависимости от концентрации
образующегося гипохлорита натрия и NaCl в применяемом рассоле.
На рис. 7-7 показано изменение этих параметров в зависимости
от концентрации NaClO при исходной концентрации NaCl в рассоле
около 15%.
Даже при малых концентрациях гипохлорита натрия (10—15 г/л)
расход электроэнергии примерно в 2 раз^, a NaCl в 6—10 раз выше,
чем при химическом методе получения гипохлорита натрия из каусти-
ческой соды и элементарного хлора. Поэтому электрохимический
способ получения гипохлорита натрия не нашел широкого приме-
нения в промышленности, он^ имеет важное техническое значение
лишь как одна из стадий производства хлоратов электрохимическим
способом.
ТЕХНОЛОГИЧЕСКИЕ СХЕМЫ ПРОИЗВОДСТВА
Промышленное производство хлората натрия электрохимическим
способом осуществляется по разнообразным технологическим схе-
мам, многие из которых зависят от местных условий производства
или патентных соображений и не цредставляют особого интереса.
Ниже будут рассмотрены лишь некоторые основные технологические
схемы производства.
Периодические схемы производотва
Если раствор хлорида натрия длительное время подвергать элек-
тролизу в электролизере без диафрагмы в условиях, благоприят-
ствующих образованию'хлоратов, то в результате концентрация NaCl
будет непрерывно снижаться за счет конверсии его в хлорат, а кон-
центрация хлората — возрастать.
В начале электролиза в электролите, состоящем из чистого рас-
твора поваренной соли, начнет увеличиваться концентрация гипо-
хлорита, пока не достигнет стационарного состояния, а концентра-
ция NaCl будет снижаться за счет окисления до гипохлорита, а затем
до хлората. При изменении состава электролита меняются и
электрохимические показатели процесса электролиза. Так, с пони-
жением концентрации NaCl в растворе уменьшается выход хлората
по току, причем особенно сильно при концентрации > NaCl менее<
100—120 г/л. При изменении концентрации С1О“ и СЮз изменяются
потери тока на катодное восстановление. Ход изменения состава
электролита и расхода тока на целевой, побочные и вредные про-
цессы приведен на рис. 7-8 и 7-9.
При периодической схеме в процессе электролиза происходит
снижение выхода хлората натрия по току. При соблюдении опти-
мального режима по pH раствора и температуре имеется возмож-
ность вести процесс в течение всего цикла с выходами по току,
близкими к оптимальным.
Конец процесса определяется в зависимости от материала анода.
Для графитовых анодов обычно процесс прекращают при снижении
концентрации поваренной соли до 100—120 г/л, так как при дальней-
шем проведении процесса наблюдается сильное снижение выхода
хлората по току и резкое увеличение износа графитовых анодов.
На анодах из перекиси свинца, цлатины или других металлов
этой группы может достигаться более глубокая конверсия NaCl
в NaG103 без сильного снижения выхода по току.
После прекращения электролиза растворы из электролизеров
сливаются, затем их вновь заполняют раствором NaCl и снова ведут
процесс электролиза. -
Рис. 7-8. Изменение состава
электролита при периодическом
электролизе:
1 — поваренная соль; 2 — хлорат на-
трия; 3 — гипохлорит.
Рис. 7-9. Изменение выхода по току хло-
рата натрия при периодическом электро
лизе:
1 — общее окисление на аноде; 2 — выход хло
рата натрия по току.
Смесь растворов хлората и хлорида натрия направляют на пере-
работку для выделения чистого хлората'натрия. Переработка за-
ключается в разрушении активного хлора в растворах, очистке рас-
творов от взвесей графитовой пыли (при применении графитовых
электродов) и выделении хлората натрия.
Растворы хлорид-хлоратных щелоков поступают на выпарива-
ние. При концентрировании растворов частично выпадает и отде-
ляется в виде кристаллов NaCl, а раствор далее подается в кристал-
лизаторы, где при охлаждении выделяются кристаллы NaC103.
г
Непрерывная схема с выпаркой
Неудобства периодической схемы общеизвестны, поэтому пред-
ложены разнообразные варианты непрерывных схем производства
хлората натрия. В настоящее время в производстве хлората натрия
используются почти исключительно различные варианты непрерыв-
ной схемы электролитического окисления NaCl в NaG103.
Поскольку с ростом концентрации хлората и соответственно
при снижении концентраций хлорида ухудшается выход по току,
работу электролизеров организуют по каскадной схеме. Для этого
25 Заказ 843 385
несколько электролизеров включаются последовательно по току
электролита, и для каждого электролизера каскада устанавливаются
стационарные значения концентрации NaCl и NaC103 и соответ-
ствующие этим концентрациям значения выхода по току и других
электрохимических параметров процесса.
С увеличением числа ступеней каскада суммарный выход по току
возрастает, приближаясь к максимальному, который получают при
периодическом процессе электролиза. Практически значительный
рост суммарного выхода по току наблюдается для числа электроли-
зеров в каскаде 4—6, так как при дальнейшем увеличении числа
электролизеров в каскаде повышение суммарного выхода по току
невелико. 1
Для поддержания оптимального значения pH в электролизеры
каскада подают необходимое количество соляной кислоты.
Имеются предложения по созданию конструкции многосекцион-
ных электролизеров с внутренним каскадом на пути движения
электролита [80].
В зависимости от условий выделения кристаллов хлората натрия
различают две технологические схемы производства. В первой схеме
электролизу подвергается исходный раствор поваренной соли и из
последнего электролизера каскада получают смешанный раствор
хлората натрия концентрацией около 350—370 г/л и хлорида натрия
концентрацией около 100—120 г/л. Для выделения хлората натрия
из такого раствора его после разрушения активного хлора и отде-
ления от графитового шлама концентрируют в выпарных аппаратах
до содержания 850—950 r/л хлората натрия. При этом основная
часть NaCl выпадает в виде кристаллов, которые отделяются на цен-
трифугах от маточника и возвращаются вновь в производство для
приготовления исходного раствора, поступающего на электролиз.
Концентрированные растворы хлората натрия после отделения кри-
сталлов NaCl поступают на кристаллизацию, которая проводится
в вакуумных кристаллизаторах с охлаждением раствора за счет
испарения влаги под вакуумом или в кристаллизаторах, охлажда-
емых водой или холодильными растворами.
Кристаллы хлората натрия отделяются на центрифуге и маточ-
ник возвращается вновь в цикл.
По такой схеме на кристаллизацию поступает концентрирован-
ный раствор хлората натрия и кристаллизация может быть прове-
дена без использования холодильной установки.
По второй схеме производство хлората натрия осуществляется
без выпарки растворов. Концентрация хлората в выходящих из
электролизеров щелочах составляет примерно 500—550 г/л, поэтому
для кристаллизации хлората требуются низкие температуры, полу-
чаемые с помощью холодильной установки. Маточные растворы после
отделения кристаллов хлората натрия содержат примерно 200—
250 г/л хлората и 100—120 г/л NaCl. Их донасыщают поваренной
солью и после корректировки значения pH и концентрации хромата
падают на питание каскада хлоратных электролизеров.
Приведенные выше две схемы выделения кристаллов хлората
натрия имеют много различных вариантов, используемых в про-
мышленности.
Пар
Рис. 7-10. Технологическая схема производства длората натрия с применение^
выпарки:
1 — бак для приготовления и очистки исходного электролита; 2 — фильтр для электролита;
3 — насос; 4 — напорный бак для электролита; 5 — электролизеры; в — сборный бак элек-
тролита; 7 — аппарат для окончательного дехлорирования электролита; 8 — второй корпус
выпарки; 9 — первый корпус выпарки; 10 — фильтры для отделения NaCl; 11 — фильтр
для упаренного раствора; 12 — приемник упаренных щелоков; 13 — кристаллизатор; 14 —
паро-вакуумная установка; 15 — фильтр для хлората натрия; 16 — сборник маточника;
17 — бункер для соли; 18 — сушилки хлората натрия; 19 — колонка для отмывки водорода
от хлора; 2о — колонка для промывки водорода водой; 21 — вентилятор для водорода;
22 — контактный аппарат; 23 — холодильник; 24 — компрессор.
На рис. 7-10 показана принципиальная схема производства хло-
рата натрия [81] с применением выпарки. В качестве сырья ис-
пользуется природная соль. Сюда же на стадию приготовления
25*
387
электролита подается обратная соль, выделяемая в процессе выпарки,
и маточники от кристаллизации хлората натрия. Полученныц рас-
твор очищают от ионов кальция добавлением раствора соды и от
ионов магния — добавлением щелочи. В случае необходимости
электролит очищают от сульфатов осаждением хлористым или угле-
кислым барием. Для осветления и фильтрации рассола может быть
использована такая же аппаратура, как и при очистке рассола
в производстве хлора и каустической соды. К чистому рассолу до-
бавляют хроматы натрия или калия и соляную кислоту.
При применении чистой соли, полученной выпаркой очищенного
рассола или другим каким-либо способом, стадия очистки рассола
упрощается.
Готовый электролит следующего примерного состава: 280 г/л
NaCl, 40—80 г/л NaC10g, 3—6 г/л Na2Gr2O7 при pH 6—7 пропу-
скают через каскад из 4—6 электролизеров, включенных последо-
вательно по электрическому току и по току жидкости. При прохожде-
нии электролита концентрация хлорида натрия в нем снижается,
ч,
а концентрация хлората возрастает.
Состав электролита, вытекающего из последнего электролизера
каскада, зависит от степени превращения хлорида в хлорат. При
применении графитовых анодов остаточное содержание хлорида
натрия обычно составляет не менее 100—120 г/л. В этих условиях
содержание хлората натрия 350—375 г/л. Часто для снижения рас-
хода графитовых анодов и повышения выхода по току доводят оста-
точное содержание NaCl до 130—150 г/л.
В процессе электролиза, вследствие частичного выделения хлора,
увеличивается значение pH, поэтому для поддержания оптимальных
условий в электролизеры подают 10%-ный раствор соляной кислоты.
Содержание активного хлора в электролите, вытекающем из кас-
када электролизеров, обычно составляет около 3—4 г/л.
Основное количество гипохлоритов превращается в хлораты
за счет самоокисления хлорноватистой кислоты в сборнике электро-
лита. Для ускорения процесса по реакциям (7-8) и (7-9) электролит
подогревают. Затем его подщелачивают и проводят химическое
обесхлоривание добавлением восстановителей (формиата натрия,
карбамида или других реагентов).
Для выделения хлората натрия в твердом виде растворы упа-
ривают в двухступенчатой выпарной системе. Раствор поступает
вначале во второй Корпус выпарной системы, находящийся под ва-
куумом и обогреваемый соковым паром после первого корпуса.
Здесь концентрация NaGlO3 увеличивается примерно до 500 г/л.
Затем раствор перекачивается в первый корпус выпарной системы,
обогреваемый свежим паром. Выпадающий при выпаривании хло-
рид натрия отделяют от раствора на центрифугах и возвращают
в цикл для приготовления исходного электролита.
В результате упарки получают растворы, содержащие около
900—950 г/л хлората натрия и 80—90 г/л NaCl. При 90—100 °C
эти растворы насыщены по хлориду натрия и близки к насыщению
388
по хлорату натрия. После фильтрования и отделения взвешенных
частиц графита и других примесей раствор поступает на кристал-
лизацию.
На рис. 7-10 показан вакуум-кристаллизатор, где охлаждение
кристаллизуемого раствора осуществляется за счет испарения
влаги в вакууме. Могут применяться и другие типы кристалли-
заторов, в частности с охлаждением рассолом.
Кристаллы хлората натрия отделяют от раствора на центрифу-
гах или фильтрах и, в случае необходимости, подвергают осушке.
Маточные растворы возвращаются в цикл для приготовления исход-
ного электролита.
Некоторые потребители с успехом применяют влажный хлорат
натрия. При использовании хлората натрия для получения двуокиси
хлора предпочитают его перевозить и хранить в виде пульпы в на-
сыщенном растворе соли.
Получаемый в процессе электролиза водород промывают от при-
меси хлора раствором щелочи и тиосульфата натрия в насадочных
колоннах, от брызг щелочи — водой и подают на каталитическую
очистку от кислорода. После охлаждения и конденсации избыточной
влаги чистый водород может быть направлен потребителям.
Применяют также технологические схемы производства хлората
натрия без выпарки электролитических щелоков (рис. 7-11). При
этом в процессе электролиза необходимо получать значительно более
концентрированные растворы, содержащие 500—550 г/л и более
хлората натрия. Для кристаллизации твердого хлората натрия
из таких растворов необходимо применять рассольное охлаждение.
Исходный электролит, поступающий на питание каскада электро-
лизеров, получают донасыщением чистой поваренной солью маточ-
ных растворов после кристаллизации хлората натрия. Электролит
«одержит —200 г/л NaCl, —340 г/л NaC10g и 3—8 г/л Na2CrO4.
Поддержание pH =• 5,64-5,8 в электролизерах каскада осуще-
ствляется добавлением 10%-ной соляной кислоты.
Из каскада электролизеров получают растворы при pH = 5,8 ч-
4-6,2, содержащие 520—540 г/л NaClO3, 100—110 г/л NaCl, 3—
$ г/л Na2CrO4 и —3 г/л NaClO.
Ниже приведен примерный состав электролита по электролизе-
рам в четырех- и шестиступенчатом каскаде (в г/л):
Ступени каскада I II III IV V VI
При четырехступенчатом каскаде
NaCl .......... 172 149 126 105 — —
NaClO3 ........ 380 430 480 530 — —
При шестистуиеичатом каскаде
NaCl .......... 180 165 150 135 120 105
NaClO3 ........ 362 395 429 462 499 530
Если электролизеры работают с графитовыми анодами, то при
плотности тока 700^—1000 А/м2 и температуре 40 °C начальное напря-
жение на электролизерах равно 3,3—3,6 В, а среднее за тур работы
анодов 3,8—4,2 В. Выход хлората по току составляет 80—85%
389
и расход электроэнергии постоянного тока в расчете на товарный
хлорат натрия — 7200—7900 кВт*ч/т. Электролизные газы содержат:
4,5—5,5% О2, 0,3-1% С12 и 1—2% СО2.
Срок службы графитовых анодов зависит от "положения электро-
лизера в каскаде.
Показатели работы электролизеров и всей схемы в целом могут
изменяться в зависимости от конкретных условий работы. Ниже
приведены опубликованные. в литературе основные показатели
производства хлората натрия по схейе без применения выпарки [82]
и с кристаллизацией хло-
рата при минусовых тем-
пературах.
Хлоратные электроли-
зеры с графитовыми ано-
дами работали при плот-
ности тока 700—1000 А/м2.
Питающий рассол содер-
жал 330—340 г/л NaGlO3;
190-200 г/л NaCl и 8-
10 г/л Na2CrO4.
Электролитические ще-
лока, вытекающие из кас-
када электролизеров,
держали 500—520
NaGlO3 и 100—110 г/л
NаС1. Электролиз прово-
дили при температуре
электролита 38—42 °C и
составил 83 % . Начальное напряжение на электролизере 3,5—3,8 В,
конечное напряжение перед выключением 5,2—6;3 В.
В ходе электролиза напряжение на электролизере возрастает
вследствие износа анодов вначале медленно, а в конце тура — бы-
стро.
На рис. 7-12 приведена зависимость напряжения на последнем
Рис. 7-12. Изменение напряжения за тур
работы электродов на последнем электро-
лизере каскада из четырех аппаратов при
i = 920 А/ма.
со-
г/л
pH = 5,8—6,4. Средний выход по току
электролизере каскада из четырех аппаратов от продолжительности
работы при плотности тока 920 А/м2.
В зависимости от конечного напряжения, при котором электро-
лизер отключается для смены анодов, изменяется среднее напряже-
ние на электролизере за тур работы, а также удельный расход элек-
троэнергии и графитовых анодов. При более полном срабатывании’
графитовых анодов соответственно снижается удельный расход
графита, однако возрастает среднее напряжение на электролизере
и удельный расход электроэнергии. Экономически выгодная степень
срабатывания анодов рассчитывается при условии, чтобы затраты
на производство были минимальными.
При плотности тока 750 А/м2 в зависимости от степени срабаты-
вания графитовых анодов и места электролизера в каскаде среднее
напряжение на электролизере может колебаться от 3,7 до 4,0 В,
391
расход графита от 12,8 до 30 кг/т и удельный расход электроэнергии
от 6700 до 7300 кВт-ч/т.
Очистка водорода от примесей хлора и кислорода может быть
осуществлена аналогично тому, как описано на стр. 389.
Для обеспечения большей безопасности работы на стадии очистки
водорода от кислорода электролитические газы можно разбавлять
очищенным водородом, возвращая часть водорода после очистки
от кислорода и охлаждения обратно в цикл для снижения содер-
жания кислорода в сме^и, поступающей в контактные печи. В тех
случаях, когда водород не может быть рационально использован
на предприятии, его выбрасывают в атмосферу. При этом электроли-
тические газы разбавляют инертными газами — азотом или дву-
окисью углерода в зависимости от местных условий. Можно приме-
нять для этой цели воздух, однако требуется подача минимум 25—
30-кратного количества воздуха по отношению к продуцируемому
в электролизерах водороду. При разбавлении газов воздухом воз-
можен повышенный унос брызг электролита из электролизеров и
усложняется санитарная очистка от хлора большого объема газов,
выбрасываемых в атмосферу.
Электролитические щелока, выходящие из каскада электролизе-
ров, содержат 3—4 г/л активного хлора. Они поступают в баки-
приемники, где за счет протекания реакции (7.8) содержание С12
снижается до 1,5—2,0 г/л. Для дальнейшего разрушения активного
хлора растворы подогревают [83, 84] в титановом теплообменнике
до 65—75 °C и пропускают через две или три колонны, продуваемые
для перемешивания жидкости сжатым воздухом. При этом за 4—6 ч
содержание активного хлора снижается примерно до 100 мг/л.
Окончательное обесхлоривание проводят химическим способом
путем добавления формиата натрия или других восстановителей.
После полного обесхлоривания и подщелачивания раствора до со-
держания щелочи 0,5—1,5 г/л щелока поступают в механические
сепараторы, гравитационные отстойники или на фильтры для отде-
ления графитового шлама.
Исходное содержание графитового шлама в щелоках, вытека-
ющих из последнего электролизера каскада, составляет около 200—
300 мг/л. После отстойников содержание шлама в осветленном рас-
творе снижается до 20—50 мг/л в зависимости от продолжительности
отстоя.
При фильтровании раствора через пористые фильтры (пористый
фторопласт или металлокерамические фильтры) остаточное содер-
жание шлама обычно снижается до 8—12 мг/л. В ^сепараторах можно
достичь хорошего осветления раствора, однако требуется большое
число сепараторов и возрастают затраты на ремонт и обслужива-
ние оборудования.
Осветленные щелока подогревают, донасыщают чистой поварен-
ной солью и подают на кристаллизацию.
Совместная растворимость хлората и хлорида натрия в при-
сутствии хромата натрия приведена на рис. 7-13.
392 '
Раствор хлорид-хлоратных щелоков после отделения графито-
вого' шлама проходит подогреватель и при 40—50 °C поступает
в донасытитель, где концентрация NaCl возрастает до 140—156 г/л,
затем проходит осветлитель, промежуточный бачок и поступает
в верхнюю часть центральной трубы классифицирующего кристал-
лизатора. В нижнюю часть трубы с помощью насоса подается охла-
жденный в противоточном холодильнике до 8—10 °C маточный рас-
твор, содержащий 340—376 г/л NaC103 и 150—175 г/л NaCl. За счет
350
О 100 200 300 000 500 ООО 700 ООО 900
Растворимость NaCIOg , г/л
Рис. 7-13. Изотермы растворимости в системе NaClO3—NaCl—Н2О в присут-
ствии 10 г/л Na2CrO4 и 1 г/л NaOH:
I — область ненасыщенных растворов; II — область растворов, насыщенных по NaCl;
III — область растворов, насыщенных по NaGlO3.
циркуляции маточного раствора поступающий в кристаллизатор
раствор охлаждается.до —3 4 5 °C и отводится тепло кристалли-
зации хлората натрия. Образование кристаллов хлората натрия
и их рост до необходимых размеров происходят в классифицирующем
конусе кристаллизатора. Схема и конструкция кристаллизатора
позволяют регулировать соотношение питающего и циркулирующего
раствора, т. е. тепловой баланс процесса и линейную скорость вос-
ходящего потока в зоне взвешенного слоя, и таким образом влиять
на количество образующихся центров кристаллизации, рост кристал-
лов и гранулометрический состав конечного продукта.
Отвод выделяющихся кристаллов хлората натрия на центрифугу
в виде пульпы, содержащей 15—20% твердой фазы, производится
из нижней конусной части кристаллизатора через автоматический
клапан, обеспечивающий загрузку центрифуги и поддержание необ-
ходимого уровня жидкости в кристаллизаторе при непрерывной
циркуляции маточника в системе. Отфильтрованный на центрифуге
хлорат натрия срезается ножом и транспортером передается в прием-
ный бункер и далее на расфасовку в барабаны.
393
II
li
Получаемый продукт довольно однороден по гранулометриче-
скому составу. Большое количество крупных кристаллов (более
100 мкм) обеспечивав# хороший отжим маточника и высокое каче-
ство продукта по содержанию основного вещества и хлорида при
минимальной промывке (50 л/т). Изучение кристаллов под микро-
скопом показало, что они имеют правильную кубическую форму,
При поддержании в трубках холодильника скорости движения
маточного раствора 1,8—2 м/с инкрустация труб холодильника про-
исходит медленно и холодильник требуется промывать только через
14—20 ч непрерывной работы.
Предварительное донасыщение щелоков поваренной солью перед
подачей их в кристаллизатор необходимо для снижения раствори-
мости хлората натрия и, следовательно, для более полного его выде-
ления в процессе кристаллизации.
Маточник после подогрева поступает на второе донасыщение
поваренной солью и затем после корректировки pH и содержания
хромата натрия возвращается вновь на питание электролизеров
каскада.
Приведенная схема/в зависимости от местных условий может
иметь варианты. Например, если в качестве сырья используется
не твердая соль, а рассол, возможно сочетание описанной схемы
с частичной выпаркой щелоков, подаваемых на кристаллизацию.
Тогда отпадают стадии донасыщения циркулирующих щелоков до
и после кристаллизации, но появляется стадия частичной выпарки
растворов с целью выведения из системы избытка воды, введенной
со свежим рассолом.
В производстве хлората натрия приходится иметь дело с очень
агрессивными слабокислыми средами, содержащими активный хлор.
Для облегчения выбора конструкционных материалов все операции
по фильтрованию, упариванию растворов й кристализации проводят
после разрушения примеси активного хлора в растворе. Для раство-
ров, содержащих активный хлор, применяют металлическую аппа-
ратуру, защищенную гуммированием, футеровкой керамическими
плитками или пластическими массами. Хорошую стойкость в этих
условиях показывают титаСн и его сплавы [85, 86].
Для достижения максимального выхода по току необходимо стре-
миться, чтобы процесс получения хлоратов протекал по химическому
механизму, а электрохимическое окисление гипохлорита и хлорно-
ватистой кислоты было ограничено. С этой целью предложено f
вести электролиз таким образом, чтобы процессы, протекающие
в самом электролизере, свести по возможности только к продуциро-
ванию гипохлорита, а дальнейшее окисление гипохлорита и хлорно-
ватистой кислоты до хлората осуществлять в специальном реакторе,
вынесенном из электролизера [381. Тогда электролиз можно прово-
дить, например, на графитовых анодах при температуре около 40 °C,
а конверсию гипохлорита и хлорноватистой кислоты в хлорат —
при повышенной температуре, получая суммарно высокий выход
хлората по току [87}. Для такого процесса необходима интенсивная
1
4s
циркуляция электролита по циклу электролизер — реактор с тем,
чтобы образующийся активный хлор быстро выводился из электро-
лизера в реактор [88]. Подобная схема предложена для электроли-
зеров не только с графитовыми анодами, но и с платинотитано-
выми [89] и с окисносвинцовыми.
При работе с окисносвинцовыми анодами или с анодами с актив-
ным слоем из Металлов платиновой группы температура электролиза
может быть увеличена до 70 °C и выше, что упростит технологиче-
скую схему и аппаратуру.
Для снижения потерь тока на катодное восстановление и электро-
химический процесс образования хлоратов на аноде предложено
также проводить электролиз в электролизерах с диафрагмой и внеш-
ней циркуляцией электролита через реактор, в котором происходит
образование хлората по химическому механизму [90].
Если хлорат натрия используется для получения двуокиси
хлора, отпадает необходимость выделения чистого твердого хлората
натрия. В этом случае производственная схема упрощается за счет
исключения стадии кристаллизации. При производстве хлората
натрия в виде раствора особое преимущество имеют электролизеры
с анодами из двуокиси свинца или с активным слоем из металлов
платиновой группы, так как на них достигается более глубокое пре-
вращение хлорида в хлорат, и остаточное содержание NaCl в рас-
творе может быть снижено без чрезмерного ухудшения электрохими-
ческих показателей до 30—50 г/л. г
Предложено комбинировать электролизеры с графитовыми ано-
дами для работы на первых ступенях электролитического каскада
при высокой концентрации NaCl в электролите и электролизеры
с анодами, имеющими активный слой из металлов платиновой группы,
для последних ступеней каскада, где содержание NaCl в электролите
может составлять 10—3% конечной концентрации
трия [76].
хлората на-
в производ-
соды, анало-
КОНСТРУКЦИИ ЭЛЕКТРОЛИЗЕРОВ
Первичные процессы, протекающие на электродах
стве хлоратов и при получении хлора и каустической
гичны. Однако в отличие от производства хлора и каустической соды,
где одним из основных требований к конструкции электролизеров
является возможно более полное разделение выделяющихся на элек-
тродах продуктов, в производстве хлоратов необходимо добиваться
возможно более полного взаимодействия выделяющегося на аноде
хлора со щелочью, образующейся у катода. Наблюдаемое некоторое
выделение элементарного хлора, уносимого в виде примеси с газами
электролиза, приводит к потерям выхода по току и к необходимости
соответственно подкислять электролит.
Поэтому при конструировании электролизеров для производства
хлоратов стремятся обеспечить возможно лучший контакт между
анодными и катодными продуктами электролиза. Подавляющее
п
395
большинство конструкций электролизеров для получения хлоратов
не имеет диафрагмы для разделения образующихся продуктов.
Предлагались также конструкции электролизеров с диафрагмой [91,
92J для производства хлоратов. При этом исходили из того, что не-
избежное при электролизе без диафрагмы образование в электролите
ионов гипохлорита и молекул хлорноватистой кислоты приводит
к снижению выхода хлората по току как за счет анодного окисления
ионов СЮ" до хлората по электрохимическому механизму, так и за
счет катодного восстановления гипохлорита и хлорноватистой кис-
лоты.
Для снижения потерь выходов по току предлагали проводить
процесс электролиза с разделением электродных продуктов в элек-
тролизере и смешивать их вне электролизера в реакторе, где могут
быть созданы оптимальные условия по температуре и pH для проте-
кания реакции образования хлората. Электролит должен циркули-
ровать в системе электролизер — реактор. Насколько нам известно,
практического применения такой способ работы электролизера не
получил.
Проводились исследования по использованию переменного тока
для электролиза горячих водных растворов хлоридов с целью полу-
чения хлоратов [93]. Неоднократно предлагались схемы, в которых
при наружной циркуляции электролита проводили его хлорирова-
ние перед подачей вновь в электролизер [94—96] для регулирования
кислотности электролита.
Однако снижение концентрации активного хлора в электролите
может быть достигнуто также и в электролизерах без диафрагмы
за счет создания интенсивной циркуляции электролита через вынос-
ной реактор. Преимущества, полученные при использовании электро-
лизера с диафрагмой и заключающиеся в снижении концентрации
гипохлоритов в электролите, не оправдывают усложнения кон-
струкции электролизера, увеличения напряжения и расхода элек-
троэнергии за счет введения в конструкцию диафрагмы.
В электролизерах без диафрагмы предусматриваются меры по
улучшению перемешивания катодных и анодных продуктов, главным
образом путем внутренней циркуляции электролита, возника-
ющей вследствие выделения водорода на катоде.
Для поглощения хлора, выделяющегося на аноде, предлагали
в электролизере Ауссигер ферейн [97, 98] размещать электроды
в нижней его части, тогда над ними находился бы большой слой
электролита. При прохождении пузырьков хлора через этот слой
и при хорошем перемешивании восходящим потоком водорода про-
исходит более полное поглощение хлора. Однако в современных кон-
струкциях это предложение не используется.
Предлагали также располагать электроды горизонтально [46].
В большинстве конструкций электролизеров стараются исполь-
зовать подъемную силу выделяющегося на катоде водорода для со-
здания внутренней циркуляции электролита. Газожидкостная смесь
поднимается в пространстве между анодами и катодами, после
396
отделения пузырьков газа электролит опускается вниз в той части
сечения электролизера, которая свободна [от газовых пу-
зырьков.
Применяются также конструкции с наружной циркуляцией
электролита через выносной реактор, в котором должно протекать
окисление ионов С1О“ до хлората. При этом возможна как естествен-
ная циркуляция электролита за счет подъемной силы пузырьков
водорода, выделяющегося на катоде [99], так и принудительная —
с помощью насоса [100].
Для быстрого и полного вывода образовавшегося в ячейке гипо-
хлорита и хлорноватистой кислоты стараются поддерживать интен-
сивную циркуляцию и скорость движения электролита в ячейке
около 0,3—0,6 м/с [99].
В электролизерах с графитовыми анодами температура должна
быть не выше 30—40 °C. Для охлаждения внутри электролизеров
устанавливаются холодильники. В большинстве конструкций ис-
пользуются водяные змеевики, которые с целью защиты от коррозии
соединяются электрически с катодом и работают как катоды со
сравнительно небольшой плотностью тока, необходимой для катод-
ной защиты металла змеевиков. Применяются также охлаждаемые
катоды, хотя в целом это значительно усложняет конструкцию катода
и электролизера. При наружной циркуляции электролита через
выносной реактор регулирование температуры осуществляется обычно
в наружных теплообменниках, устанавливаемых на пути циркуля-
ции электролита перед поступлением его в электролизер.
Для изготовления катодов, холодильников и корпуса электро-
лизера применяется сталь. Стальные детали, погруженные в элек-
тролит, часто защищают от коррозионного разрушения с помощью
катодной поляризации при условии, что деталь работает в качестве
катода при плотности тока, достаточной для катодной защиты.
Детали и части деталей электролизеров, находящиеся в электроли-
зере выше уровня электролита — в газовом Ьбъеме, а также днище
электролизера, где может осаждаться шлам, препятствующий нор-
мальной катодной защите, необходимо защищать гуммированием,
футеровкой или слоем полимерных материалов, стойких в этих усло-
виях.
Изучено [100а] поведение титана, платины и стальных катодов
в хлорид-хлоратных растворах. Плотность тока катодной защиты
стальных поверхностей должна быть выше предельного диффузион-
ного тока процесса восстановления гипохлорита. Титан и платина
в хлорид-хлоратных растворах в присутствии активного хлора
пассивны и без наложения тока поляризации.
Конструкции электролизеров для получения хлората могут
быть различными в зависимости от применяемого материала анода
(магнетит, графит, двуокись свинца, металлы платиновой группы
или окись рутения, нанесенные на титановую основу электрода),
способов включения электродов (моно- или биполярное) и способа от-
вода избытка тепла, внутренней или наружной системы циркуляции
397
электролита, применяемой плотности тока, материалов, исполь-
зуемых для изготовления деталей электролизеров, и других фак-
торов.
В начале развития электрохимического Метода производства
хлоратов были предложены конструкции электролизеров как с моно-
так и с биполярным включением электродов. В биполярных конструк-
циях предусматривались преимущественно платиновые электроды.
Однако в промышленности долгое время находили применение в ос-
новном только конструкции с монополярным включением электро-
дов как более простые в изготовлении, обслуживании и ремонте.
Только в последние годы в связи с большими успехами в области
разработки новых электродных материалов и созданием конструк-
ционных материалов, стойких в условиях электролиза растворов
поваренной соли с получением хлоратов, интерес к электролизерам
с биполярным включением электродов вновь возрос.
Созданы и используются в промышленности конструкции бипо-
лярных электролизеров для получения хлоратов. Такие электроли-
зеры позволяют создавать агрегаты с большим числом последо-
вательно включенных ячеек и большой суммарной эквивалентной
нагрузкой. При этом легко осуществляется регулирование pH
и температуры в ячейках при интенсивной циркуляции электролита
через наружные холодильник и реактор, что резко сокращает число
точек регулирования температуры и подачи соляной кислоты в цикл
и открывает возможность практически полной автоматизации под-
держания оптимального технологического режима в процессе элек-
тролиза.
Применялись разнообразные конструкции электролизеров для
получения хлоратов. Многие из них аналогичны и отличаются друг
от друга лишь деталями устройства. При применении в качестве
анодов графитовых пластин их располагают рядами между сталь-
ными катодами, которые обычно снабжают перфорацией. Для охла-
ждения используются холодильники из стальных труб, соединенных
с катодом для защиты от коррозии. В случае применения стержневых
графитовых или магнетитовых анодов корпус электролизера делят
на квадратные отделения разделительными стенками, служащими
катодами. Внутри отделения помещают аноды. Разделительные стенки
обычно не доходят до дна и кончаются ниже уровня электролита,
что обеспечивает возможность циркуляции электролита во всех
отделениях в электролизере.
Старые конструкции электролизеров, схемы их устройства и тех-
нические показатели приведены в литературе [2,101—103], поэтому
коротко будут освещены только те конструкции, которые можно
рассматривать как представителей определенных направлений в кон-
струировании аппаратуры.
Одной из первых промышленных конструкций был электролизер
Корбена [104] с платиновыми биполярно включенными электродами,
укрепленными на эбонитовых рамках, который работал при плот-
ности тока 2400 А/м2, температуре около 75 °C и расстоянии между
' 398
электродами 12—15 мм. Напряжение на ячейке составляло 5,0—
5,5 В.
На начальном этапе развития производства хлоратов электро-
химическим способом использовались также и другие аналогичные
типы конструкций [105].
Хлоратный электролизер Ангела, _ схема которого приведена
на рис. 7-14, представляет собой простую и рациональную кон-
струкцию с монополярными графитовыми анодами. В стальном,
преимущественно защищенном, корпусе электролизера размещены
аноды из графитовых плит, закрепленные в крышке электролизера
и имеющие верхний подвод тока. Катоды из стальной сетки крепятся
к катодной раме и могут быть при демонтаже удалены в собранном
Рис. 7-14. Хлоратный электролизер Ангела:
1 — графитовые аноды; 2 — катоды; 3 — катодная рама; .
4 — корпус электролизера; <5 — крышка.
виде. Катоды расширяются кверху для увеличения межэлектродного
расстояния в верхней части ячейки и более равномерного распреде-
ления тока по поверхности анода. Это позволяет предотвратить об-
разование в верхней части графитового анода шеек вследствие повы-
шенного износа этой части анода. При плотности тока 300^—400 А/м2
температура электролизера устанавливалась без специального охла-
ждения не выше 50 °C.
Примером конструкции с магнетитовыми анодами может служить
монополярный электролизер со стальным защищенным корпусом
типа Биттерфельд [106]. Магнетитовые аноды и катоды из стальных
листов с отверстиями прикреплены к крышке, которая разделена
на четыре секции [46, 81J, в каждой секции 36 анодов. Холодильные
змеевики располагались в средней части корпуса электролизера
и были защищены от коррозионного разрушения катодной поляри-
зацией. Электролизеры с магнетитовыми анодами работали при тем-
пературе около 70 °C й расстоянии между электродами около 10 мм
со следующими показателями [81]:
Нагрузка, А ..........
Напряжение, В ....
Плотность тока
анодная, А/м2 . . .
объемная, А/л ...
Выход по току, % . .
Расход анодов, кг/т . .
12 000
От 3,6 до 4,2
200
85—95
399
Рис. 7-15. Электролизер для получения
хлората натрия с охлаждаемыми катодами:
1 — битумная заливка; 2 — анодный короб;
3 — прокладка; 4 — токоподводящая шина;
5 — крышка; 6 — корпус; 7 — анод; 8 — катод-
ный комплект; 9 — гуммировка.
Имеется аналогичная конструкция электролизера Биттерфельд
с графитовыми анодами [81, 101].
Электролизеры на нагрузку 20 000 А с анодной плотностью
тока около 350 А/м2 и объемной плотностью тока около 3 А/л при
температуре до 40 °C работают с выходом по току 82% при напряже-
нии 3,4 В [81].
Электролизер фирмы «Вестерн электрокемикал К°» [46] также
состоит из стального защищенного корпуса, в котором размещены
графитовые аноды, закрепленные в бетонной крышке электролизера.
Катоды выполнены из стальных листов. Между электродами распо-
ложены охлаждающие змее-
вики, соединенные электри-
чески с катодами. Электро-
лизеры работают при 30—
40 °C, анодной плотности
тока от 100 до 1000 А/м2 с вы-
ходом по току 75% и напря-
жением 2,8—3,5 В [46, 81].
Фирма «Персон» предло-
жила электролизер с гра-
фитовыми анодами цилинд-
рической формы, покрытыми
слоем перекиси свинца, оса-
жденной электрохимическим
способом из азотнокислого
электролита. Электроды рас-
полагаются вертикально в
цилиндрическом или прямо-
угольном корпусе, служащем
одновременно катодом. Элек-
тролизер может быть ис-
пользован также для полу-
чения гипохлоритов и пер-
хлоратов [69]. Преимуществом такого электролизера является
более полная конверсия поваренной соли (остаточное содержание
около 50 г/л NaGl) и соответственно возможность получения более
концентрированных растворов хлората натрия — до 750 г/л. Тур
работы электродов составляет до 2 лет [81]. Аналогичные результаты
получены при использовании анодов из двуокиси свинца, нанесен-
ной на титановую основу электрода [39, 69].
В Советском Союзе для промышленного получения хлората на-
трия нашли применение несколько типов электролизеров без диа-
фрагмы с графитовыми анодами, рассчитанные и работающие в интен-
сивном режиме при электродной плотности тока от 700 до 1000 А/м2
и объемной плотности тока от 7 до 15 А/л. Все электролизеры имеют
верхний подвод тока к анодам. В табл. 7-5 приведены основные
показатели применяемых в СССР конструкций электролизеров с гра-
фитовыми анодами для получения хлората натрия [107].
400
Таблица 7-5. Основные показатели хлоратных электролизеров
с графитовыми анодами
Показатели
Нагрузка, кА . . ...................
Среднее напряжение, В . ............
Анодная плотность тока, А/м2 . ...
Объемная плотность тока, А/л . . .
Выход по току, % ................
Температура электролита, °C ....
Расход воды на охлаждение, м3/ч (темпе
ратура воды 25—27 °C) ..............
Межэлектродное расстояние, мм . . .
Масса (без электролита), т .....
Габариты в плане (без штуцеров), мм
Высота, мм .........................
Электролизеры
I II Ш IV
20 25 25 37
4,2 4,2 3,9 4,2
1000 1000 690 1020
10 10 10 15
83—85 83—85 83—85 82—84
35-45 35-45 35—45 35—45
7,0 8,5 8,5 12,5
10 10 10 10
5,5 6,4 7,6 7,6
2030 X 2040 X 2060 X 2060 X
X 1435 X 1740 X 1900 X 1900
1700 1800 1900 1900
Электролизеры типов I и II имеют охлаждаемый катод и отли-
чаются друг от друга нагрузкой и рабочей поверхностью электро-
дов. Электролизеры III и IV типов имеют неохлаждаемые катоды.
Охлаждение осуществляется с помощью холодильных змеевиков,
вмонтированных внутри электролизера. Эти электролизеры разли-
чаются между собой нагрузкой за счет изменения плотности тока.
На рис. 7-15 приведена схема устройства электролизера с охла-
ждаемым катодом. Электролизер состоит из гуммированного корпуса,
в котором размещены два катодных комплекта и графитовые аноды
(48 штук на 20 кА, 60 штук на 25 кА). Каждый катодный комплект
на резиновых прокладках крепится к одной из двух гуммированных
крышек электролизера, каждая крышка имеет два короба, в которых
размещаются анодные шины; головки анодов присоединяются к анод-
ным шинам с помощью небольших шинок. Герметизация всех зазо-
ров в коробах крышек производится с помощью бетона и битума,
заполняющего всю полость короба. Шины катодного комплекта
выведены через отверстия крышки и уплотнены резиновой про-
кладкой.
Катодный комплект с крышкой, анодами, шинами и герметизи-
рованными коробами представляет собой один сменный узел, кото-
рый на резиновой прокладке крепится к корпусу электролизера.
Катодный комплект выполнен в виде двух рядов полых плоских
коробок — катодов, объединенных общим коллектором. Во внутрен-
ние полости катодов поступает вода для охлаждения. Коллектор
используется одновременно для распределения воды и тока по отдель-
ным катодам. Верхняя часть коллектора, расположенная в газовом
пространстве, гуммирована.
Катодные шины обоих катодных комплектов объединены над
крышкой общей токОподводящей шиной, к которой присоединяют
26 Заказ 843 401
анодные шины смежного электролизера и шунтирующий выключа-
тель электролизера.
Электролит вводится в электролизер через штуцер в нижней
части электролизера и распределительную винипластовую трубу.
Электролит выводится через штуцер в верхней части корпуса элек-
тролизера. Штуцер для вывода газов расположен в крышке электро-
лизера.
а
шг/
Рис. 7-16. Электролизер для получения хлората натрия на нагрузку 15 кА:
а — продольный разрез; б — поперечный разрез; 1 — охлаждаемый катодный комплект;
2 коллектор для подачи и отвода охлаждающей воды; 3 — уравнительная катоднан шина;
4 — корпус электролизера; 5 — анодные плиты; 6 — распределительная шина; 7 — анод-
ные шины; 8 — сборная аноднан шина.
Корпус электролизера катодно поляризован, что обеспечивает
защиту корпуса от коррозионного разрушения при местных нару-
шениях гуммировки [108). Такой способ защиты корпуса электро-
лизера делает его практически йеизнашиваемым. Срок службы
гуммировки корпуса не менее четырех лет. Кроме того, гуммирован-г
ный корпус, имея достаточное шламовое пространство, позволяет
не производить промывку электролизера от шлама в течение всего
тура работы до замены анодов.
На рис. 7-16 показан продольный и поперечный разрез такого
электролизера на нагрузку 15 000 А. Электролизер монтируется
из отдельных секций электродных элементов и предназначен для
работы при объемных плотностях тока около 7 А/л и электродных
плотностях 700—800 А/м2.
Электродные элементы располагаются в полости корпуса доста-
точно плотно, не оставляя места для размещения холодильников,
необходимых для отвода избыточного тепла процесса. Отвод избы-
точного тепла в данной конструкции производится с помощью
охлаждаемого стального катода. Кроме того, охлаждение катода
улучшает показатели процесса, снижая катодное восстановление.
В качестве катодного материала используется углеродистая
сталь, из которой свариваются в виде полых прямоугольных коро-
бок катодные элементы. Коробчатые катодные элементы объединяются
402
в катодные секции гребенчатого типа. При этом вертикально стоящие
катодные элементы приваривейотся к двум горизонтальным коллек-
торам. Верхний^ коллектор разделен поперечной перегородкой; одна
его часть служит для подачи воды к катодным элементам, а дру-
гая — для отвода воды. Одна группа катодных элементов секции
параллельно питается водой из пбдающей части верхнего коллектора.
Вода стекает в нижний коллектор, откуда по другой части катодных
элементов поступает в отводную часть верхнего коллектора.
Крайние катодные элементы в секции изготовляются в виде
перфорированных стальных листов.
К верхнему коллектору подведены два водяных патрубка и токо-
подводящий ниппель, проходящий через крышку. Водяные трубы
служат также для подачи и распределения тока по коллектору,
откуда он переходит на катодные элементы. Над крышкой электро-
лизера патрубки и ниппель объединены распределительной медной
шиной, подсоединенной к уравнительной шине электролизера. <
Катодные секции подвешиваются в электролизере к раструбу кор-
пуса на специальных лапах.
Электролизер комплектуется из нескольких секций. Для одной
секции должна быть принята типовая нагрузка, тогда более крупные
электролизеры могут быть получены путем объединения в одном
корпусе нужного числа секций. Так можно создавать нормальный
ряд электролизеров все возрастающих мощностей. Секционное
построение электролизера облегчает монтажные и ремонтные опе-
рации.
В качестве анодов используются плоские графитовые плиты,
широко применяемые в хлорной промышленности.. Аноды имеют
верхний токоподвод. Анодные ниппели выходят через крышку
и анодными шинками подсоединяются к медной сборной шине,
в свою очередь подсоединяемой к уравнительной шине следующего
по ходу тока электролизера.
Анодные плиты подвешиваются при монтаже с помощью вини-
пластовых стержней на катодных элементах так, чтобы каждая анод-
ная плита входила в зазор между двумя соседними катодными эле-
ментами, связанными в гребенку. Между анодами и катодными
элементами вставлены винипластовые распорные стержни для
предотвращения замыкания.
Электродная секция, состоящая из катодной группы с установлен-
ными на ней анодами, подвешена на лапах к борту корпуса и пере-
крывается бетонной крышкой. Проходы катодных и анодных нип-
пелей в крышке герметизируются бетоном и замазкой. Крышка всего
электролизера составляется, таким образом, из ряда крышек над
секциями. Стыки крышек между собой, а также периметр собранной
общей крышки уплотняются замазками. Такой способ сборки элек-
тролизера позволяет заменить любую секцию электролизера без
нарушения соседних.
Корпус электролизера — прямоугольной формы, сварен из листо-
вой стали. В верхней части корпуса имеется раструб, к одной из
26* , 403
длинных сторон которого приварена уравнительная стальная шина.
Корпус и раструб с внутренней стороны гуммированы.
К уравнительной шине с одной стороны присоединяются сборные
анодные шины предыдущего электролизера, а с другой — сборные
катодные шины данного электролизера. Кроме того, эта шина слу-
жит для присоединения шунтирующих устройств.
Электролизеры объединяются в каскады из 4—6 электролизеров.
Срок службы анодов зависит от положения электролизера в каскаде.
Ниже приведена продолжительность пробега графитовых анодов
ухудшенного качества (в сут) при различной плотности тока в за-
висимости от положения электролизера в каскаде:
Ванна
каскада
При 700 А/м1 2 При 800 А/м2
I 520 440
II 370 330
Янна
каскада При 700 А/м* При 800 А/м2
III 290 260
IV 210 186
В III и IV типах электролизеров применены неохлаждаемые ка-
тоды. Для охлаждения электролита служат специальные холо-
Рис. 7-17. Электролизер для получе-
ния хлората натрия с неохлаждаемыми
катодами:
1 — битумная заливка; 2 — катодная шина;
з — крышка; 4 — корпус-катод; 5 — гумми-
ровка; в — поддон; 7 — холодильник; 8 —
анодная шина.
дильники, размещенные в про-
межутках между электродными
элементами. Холодильники вы-
полнены из стальных труб и для
защиты от коррозионного раз-
рушения соединены с катодами.
Схема такого электролизера
изображена на рис. 7-17.
Электролизер может рабо-
тать при нагрузке 25 кА и
плотности тока 700 А/м2 или
при 37 кА и плотности тока
1000 А/м2.
Электролизер состоит из
корпуса — катода, 88 графито-
вых анодов, трех катодно-по-
ляризованных трубчатых хо-
лодильников, гумированного
днища, крышки и ошиновки.
В одной из модификаций кон-
струкций катодный комплект
выполнен вместе с крышкой
в виде сварного узла.
АноДы с помощью винипластовых фиксаторов навешиваются на
катодные листы и после установки крышки присоединяются к че-
тырем анодным шинам. Герметизация анодных выводов произво-
дится так же, как и в электролизерах I и II типов.
Напряжение на электролизере зависит, при прочих равных
условиях, от применяемой плотности тока и качества графитовых
анодов. На рис. 7-18 приведена зависимость среднего напряжения
404
на электролизере за тур работы анодов от применяемой плотности
тока.
Все перечисленные конструкции электролизеров имеют верхний
токоподвод к анодам. Хлоратные электролизеры с нижним токо-
подводом не нашли широкого применения вследствие меньшей на-
дежности и более высокого среднего напряжения. Недостаточная
надежность этих электролизеров объясняется тем, что случайные
дефекты в гуммировке анодно-поляризованного днища или в изоля-
ции анодных контактов (битумом или бетоном) могут привести
к очень интенсивной коррозии.
Определенный интерес предста-
вляет конструкция электролизера
с боковым токоподводом, так как она
позволяет увеличивать мощность
электролизеров за счет их высоты
без изменения габаритов в плане.
Кроме того, вследствие более корот-
ких путей тока по графиту снижается
среднее напряжение на электроли-
зере. Такой электролизер был испы-
тан на нагрузку 20 кА, однако он, так
же как и электролизер с нижним
токоподводом, недостаточно надежен
и поэтому не нашел применения
в промышленности.
Монополярные конструкции элек-
тролизеров с графитовыми анодами
и анодами из РЬО2 для получения
хлората натрия продолжают совер-
шенствоваться. Нагрузка на элек-
тролизеры повышена до 60 кА, увеличивается применяемая плот-
ность тока [109].
Публикуются сообщения: об установке [110] и разработке новых
конструкций электролизеров с графитовыми анодами [111], электро-
лизеров с интенсивной циркуляцией, создаваемой путем подачи газа
для создания эрлифта или с помощью механических побудителей
циркуляции в виде пропеллеров [111, 112].
В последние годы в связи с появлением новых' анодных и кон-
струкционных материалов получили широкое развитие работы по
созданию электролизеров с биполярным включением электродов.
Предложены разнообразные варианты конструкций биполярных
электролизеров [113, 114] и отдельных узлов, в частности электрод-
ных комплектов к ним [115, 116]. Разрабатываются конструкции
электролизеров как с графитовыми, так и с малоизнашивающимися
металлическими анодами. Получили распространение биполярные
электролизеры большой мощности с графитовыми анодами [117,
118]. В таких электролизерах с потребляемой мощностью в одном
аппарате до 1500 кВт обеспечена интенсивная принудительная
Рис. 7-18. Зависимость среднего
напряжения от плотности тока
на хлоратном электролизере с гра-
фитовыми анодами, пропитан-
ными 15%-ным раствором льня-
ного масла:
1 — графит с пористостью 20—25%
и механической прочностью 300—
350 кгс/см*; 2 •— то же, с пористостью
25—30% и механической прочностью
около 200 кгс/см2.
405
Таблица 7-6. Показатели работы хлоратных электролизеров
* Биполярное, включение электродов.
** Напряжение на электролизере.
циркуляция электролита [100], что способствует быстрому выводу
гипохлорита из электролизных ячеек [119] и снижению расхода гра-
фитовых анодов. Разработаны конструкции электродов [120] и кор-
пуса электролизера [121], защита деталей-электролизера от корро-
зии [122], изучены другие особенности ведения процесса [123].
Обслуживание подобного электролизера может быть полностью
автоматизировано, так как интенсивная циркуляция электролита
позволяет поддерживать температуру и pH электролита в ячейках
на оптимальном уровне.
Сообщается, что применение интенсивной циркуляции электро-*
лита в биполярном электролизере через выносной реактор позволяет
повысить выход хлората по току до 95% и снизить расход графито-
вых анодов до 6,7 кг/т хлората натрия [124—126].
Канадская фирма «Дриден хемикаль» разработала биполярный
электролизер на нагрузку 30 кА и напряжение до 100 В [126]г
выполненный из поливинилхлорида в качестве конструкционного
материала. В электролизере фирмы «Кребс» с интенсивной внутрен-
ней циркуляцией электролита [112] создается быстрый проток элек-
тролита между электродами, что позволяет при использовании гра-
фитовых анодов увеличить выход по току до 87—90% при высокой
анодной плотности тока [81].
В последнее время вновь предлагают использовать платину или
металлы платиновой группы в качестве анодного материала в би-
полярных электролизерах для получения хлоратов. Предложены?
электролизеры с платинотитановыми [73, 89, 127—129] и окисно-
рутениевыми анодами [130] на титановой основе и стальными като-
дами.
Фирма «Кребс», разработала конструкцию электролизера NC-12
с титановыми анодами, покрытыми активным слоем на основе бла-
городных металлов платиновой группы (рутений) [131, 132]. Элек-
тролизер работает с естественной циркуляцией электролита. Пре-
имущества такого электролизера заключаются в высоком выходе
хлората по току, низком удельном расходе электроэнергии и невы-
соком содержании кислорода в водороде и в безопасности обслужива-
ния электролизера.
В табл. 7-6 приведены показатели работы некоторых описанных
в литературе конструкций электролизеров.
ПРОИЗВОДСТВО ХЛОРАТА КАЛИЯ
*
Хлорат калия КСЮ3 (калиевая соль хлорноватой кислоты НС1О3)
был открыт в 1786 г. французским химиком Бертоле и получил на-
звание бертолетовой соли. Молекулярный вес хлората калия 122,55.
Бертолетова соль образует бесцветные моноклинические пластин-
чатые кристаллы, плотность 2,344 г/см3 и температура плавления
370 °C. Бертолетова соль пожаро- и взрывоопасна: при нагревании
до 400 °C разлагается с выделением кислорода. В присутствии не-
которых окислов (MnO2, Fe2O3 и др.) хлорат калия разлагается
407
с выделением кислорода уже при 150—200 °C, с органическими и легко
окисляющимися веществами образует чувствительные к механи-
ческим воздействиям (трению, удару) и нагреванию взрывчатые
смеси. Чувствительность к взрыву возрастает в присутствии бромата
калия. Смеси бертолетовой соли с аммонийными солями, аминами,
гидразинами могут самовозгораться при хранении.
Бертолетова соль — токсическое вещество, при введении в орга-
низм действует на кровь, переводя гемоглобин крови в метгемо-
глобин, и приводит к образованию тромбов из продуктов распада
красных кровяных телец.
Бертолетова соль применяется в производстве спичек и в пиро-
технике. Ранее она использовалась как компонент в производстве
взрывчатых веществ.
Технические требования к бертолетовой соли [133] приведены
ниже:
Содержание, % - 1-й сорт 2-й сорт
КС1О3 в пересчете на сухое вещество, не менее 99,85 99,6
Примеси, не более
влага.................................... 0,05 0,05
нерастворимые в воде вещества . . . 0,01 0,04
хлориды в пересчете на СаС12 .... 0,03 0,05
сульфаты в пересчете на CaSO4 . . . 0,025 0,04
KBrOg ................................... 0,008 0,025
щелочи в пересчете на СаО........ 0,01 0,02
органические вещества........... 0,005 0,01
тяжелые металлы ......................... 0,0005 0,005
Fe ...................................... 0,002 0,004
Бертолетова соль упаковывается аналогично хлорату натрия.
Влажная бертолетова соль, содержащая до 7% влаги, может упако-
вываться в сухотарные деревянные бочки, выложенные оберточной
бумагой.
В производстве спичек применяется так называемая пудрирован-
ная соль, получаемая размолом кристаллов бертолетовой соли.
Таблица 7-7\ Растворимость КС Юз в^ воде '
Темпера- тура, °C ь dl Растворимость Темпера- тура, °C 4 4 Растворимость &
г/100 г Н2О ©^ г/100 г н2о *4© ©^
0
10
20
25
30
40
50
1,021
1,033
1,045
1,051
1,058
1,073
1,092
3,3
5,2
7,3
8,6
10,1
13,9
18,5
5,01
6,96
8,0
9,3
12,1
15,6
60 1,115 23,8
70 1,139 30,2
80 1,169 37,6
90 1,192 46,0
100 1,219 56,75
104 1,230 60,0
19,4
23,4
27,4
31,6
35,9
37,6
408
4
I При размоле остаток на сите 01 должен составлять не более 10%
| для 1-го сорта и не более 55% для 2-го.
f Остаток на сите 0056 должен быть соответственно не более 23%
[ для продукта 1-го сорта и 95% ~ для 2-го сорта.
Растворимость бертолетовой соли в воде приведена в табл. 7-7.
В присутствии КС1 растворимость бертолетовой соли снижается.
J Ниже приведена растворимость КС1О3 в водных растворах КС1
f при 20 °C:
КС1, г/л. КСЮ», г/л „•20 ai КС1, г/л КСЮз, г/л .20
0 77,1 1,050 130 23,5 1,103
10 58,0 1,050 140 22,5 1,108
20 49,0 1,050 150 21,5 1,113
30 43,0 1,050 160 21,0 1,119
40 39,5 1,054 170 20,5 1,124
50 36,5 1,058 180 20 1,130
60 34,0 1,064 190 20 1,135
70 32,0 1,070 200 20 1,140
80 30,0 1,075 210 20 1,145
90 28,0 1,081 220 20 1,156
100 27,0 1,086 240 20 1,161
110 25,5 1,091 250 20 1,168
120 24,5 1,098
Растворимость в системе КС1—КС1О3—Н20 изучена подробно
для широкого интервала температур [134].
Хлорат калия может быть получен при электролизе водных рас-
творов хлористого калия. Процессы, протекающие при этом, ана-
логичны описанным выше йри получении хлората натрия.
Изучалось влияние температуры, добавок СаС12 [135] и значе-
ния pH [136] на выход КС1О3 при электролизе растворов хлористого
калия.
Особенности электрохимического способа получения хлората
калия обусловлены значительно более низкой растворимостью КС1О3
в воде и растворах КС1 по сравнению с хлоратом натрия.
Для получения более концентрированных растворов хлората
калия проводят электролиз при повышенных температурах, однако
при использовании графитовых анодов наблюдается их усиленное
разрушение. Поэтому в электролизерах для получения КС1О3 ранее
применяли преимущественно магнетитовые аноды или аноды из
платиновых металлов. Даже в этих условиях концентрацию КС1О3
в электролитических щелоках не увеличивают более 150—200 г/л,
чтобы избежать кристаллизации твердой соли в охлаждаемых частях
электролизеров, коммуникаций и арматуры.
Изучали электролиз растворов хлористого калия на графитовых
анодах, покрытых слоем двуокиси свинца [137].
Так же, как и при производстве хлората натрия, электролиз
может осуществляться периодически или непрерывно. В последнем
случае применяют проток электролита через каскад из 4—6 электро-
лизеров. В электролитических щелоках, вытекающих из электролизе-
ров, разрушают гипохлорит и хлорноватистую кислоту нагреванием,
409
/
затем добавляют восстановители, фильтруют и выделяют хло-
рат1 калия кристаллизацией при охлаждении. Маточный раствор
после донасыщения хлористым калием возвращают вновь на элек-
тролиз. Раствор, подаваемый на электролиз, имеет следующий при-
мерный состав: 250 г/л KG1, 50 г/л КС1О3 и 2—6 г/л К2Сг2О4.
Применяют также частичное упаривание электролитических
щелоков перед подачей их на кристаллизацию для повышения кон-
центрации KGlOg. *
Предварительное упаривание позволяет работать в электролизе-
рах с более низкой концентрацией КС1О3, что особенно важно при
использовании графитовых анодов, когда повышение температуры
электролиза выше 40—50 °C нежелательно.
Описано производство хлората калия электролизом растворов
KQI в электролизерах с графитовыми анодами и стальными ка-
тодами [138]. В электролизеры поступает раствор, содержащий при-
мерно 250 г/л КС], 50 г/л КС1О3 и 2 г/л К2Сг2О4 при pH окоДо 5,5.
При прохождении через электролизеры 5—10 г/л хлористого калия
превращается в хлорат и гипохлорит. Раствор из электролизеров
при pH — 6,9 поступает в реакторы, где в течение примерно 7 ч основ-
ная масса гипохлорита превращается в хлорат. Затем раствор охла-
ждается до 15—20 °C распылением в колонне, продуваемой возду-
хом, при этом кристаллизуется хлорат калия. Кристаллы отделяют
на центрифуге. Маточник донасыщают хлористым калием для воспол-
нения его расхода и возвращают вновь на питание электролизеров.
При такой схеме съем хлората калия с 1 м3 циркулирующего
б системе раствора не превышает 8—16 кг. Это приводит к необхо-
димости устанавливать громоздкую аппаратуру для обеспечения
циркуляции электролита, превращения гипохлорита в хлорат и
охлаждения раствора для кристаллизации хлората калия.
Для получения товарного продукта часто проводят перекристал-
лизацию хлората калия.
Хотя прямое получение хлората калия путем электролиза нашло
применение в промышленности [138, 139Ьи имеется значительное
число патентов и предложений по разработке этого метода [140’—
142], однако наибольшее распространение приобрело получение
хлората калия обменным разложением хлората натрия и хлористого
калия [143]
NaC103 + KCl = KC103 + NaCl (7.16)
При этом хлорат натрия получают по одной из описанных ранее
схем, а электролитические щелока после разрушения активного
хлора и удаления взвеси продуктов разрушения анодов поступают
на обменное разложение с хлористым калием [143].
Вследствие малой растворимости при низких температурах хлорат
калия после охлаждения растворов выпадает в виде кристаллов
и отделяется от маточника на центрифугах.
Маточники после корректировки pH и концентрации NaCl и
хроматов могут быть вновь направлены на электролиз для получения
410
хлората натрия. Количество хлорида калия, подаваемого на стадию
обменного разложения, должно регулироваться таким образом,
чтобы избыток KCI был небольшим и исключалась возможность
кристаллизации КС1О3 в электролизерах, аппаратуре для разруше-
ния активного хлора и в коммуникациях.
Если на реакцию обменного разложения подается природная
соль хлористого калия, циркулирующий раствор необходимо очи-
н
щать от накапливающихся в цикле солеи кальция, магния и суль-
фатов.
Рис. 7-19. Примерная технологическая схема получения хлората калия обмен-
ным разложением NaC103 с КС1:
1 — аппарат для донасыщения рассола; 2 — центробежные насосы; 3 — аппарат для очистки
рассола от солей Са, Mg и S04; 4 — фильтр; 5 — напорный бак рассола для корректиро-
вания pH и содержания хроматов; в — напорный бак соляной кислоты; 7 — каскад элек-
тролизеров; 8 <— аппарат для разложения активного хлора в щелоках; 9 — фильтр для
шлама; 10 — реактор для обменного разложения; 11 — кристаллизатор; 12 — сгуститель
центрифуги; 13 — центрифуга; 14 — сборный бак маточника.
, S
S
*
Примеси бромистокислого калия к бертолетовой соли понижают
ее стабильность. Поэтому необходимо исключать возможность за-
грязнения ими бертолетовой соли. Если в исходном сырье содер-
жится бром, необходима специальная очистка раствора от этой
примеси. С этой целью броматы разрушают соляной кислотой и от-
дувают бром воздухом.
На рис. 7-19 показана примерная технологическая схема произ-
водства хлората калия обменным разложением электролитических
щелоков производства хлората натрия хлористым калием.
Маточные щелока после отделения хлората калия донасыщают
поваренной солью для восполнения механических потерь соли в про-
изводстве. Теоретически хлорид натрия не должен расходоваться,
411
так как его количество, потребляемое при получении хлората натрия
в электролизерах, компенсируется NaCl, образующимся при обмен-
ном разложении.
После донасыщения растворы очищают от солей кальция и магния
добавлением соды и щелочи, а также в случае необходимости от
сульфатов добавлением хлорида или карбоната бария. После остаи-
вания рассол фильтруют и подают в напорные баки электролиза,
где производится корректировка pH и содержания хроматов.
Электролитические щелока после выхода из каскада электроли-
зеров обесхлориваются, нейтрализуются добавлением щелочи, от-
фильтровываются от шлама и поступают на обменное разложение
с хлористым калием.
Кристаллы хлората калия выделяются в кристаллизаторе и отде-
ляются на центрифуге от маточного раствора, который возвращается
вновь в цикл. После перекристаллизации хлорат калия отгружается
как товарный продукт или поступает на сушку и последующий раз-
мол для получения пудрированной бертолетовой соли.
Если в качестве сырья применяются очищенные концентрирован-
ные растворы хлористого калия и натрия, стадия очистки от солей
кальция, магния и сульфатов может быть ограничена только неболь-
шой частью (10—15%) циркулирующего раствора для вывода посте-
пенно накапливающихся в цикле загрязнений. При этом будет воз-
растать объем находящейся в цикле воды, которую необходимо
удалять на выпарной установке.
Не потерял значения и химический способ получения хлората
калйя, заключающийся в хлорировании известкового молока и об-
менном разложении получаемых щелоков хлористым калием. Рас-
творимость солей в системе Са(С1О3)2 — КС1—Н2О изучена [ 139]
и благоприятна для выделения кристаллов КС1О3. При химическом
способе производства в качестве отхода получают растворы хло-
ристого кальция, которые не всегда находят потребителя.
Химический способ получения хлората калия уступает место
электрохимическому, и в последнее время нет сообщений о строитель-
стве новых крупных производств по химическому методу.
Однако необходимо учитывать, что производство хлората калия
химическим способом, является идеальным потребителем низкокон-
центрированного хлора. Этот способ может использоваться на круп-
ных хлорных заводах для поглощения абгазов сжижения и пере-
давливания жидкого хлора. Поэтому, особенно при наличии по-
требителей отходного хлористого кальция, химический йетод
производства хлората калия в зависимости от местных условий
может быть удобным и рентабельным и в настоящее время.)
ПРОИЗВОДСТВО ХЛОРАТОВ КАЛЬЦИЯ И МАГНИЯ
Хлораты кальция и магния нашли применение в сельском хо-
зяйстве в качестве дефолиантов и десикантов. В нашей стране
хлораты кальция и магния используются для предуборочного удале-
ния листьев хлопчатника и высушивания хлопчатника на корню.
412
1
X *
Хлорат кальция
/
Хлорат кальция выцускается под товарным названием «хлорат-
хлорид кальциевый дефолиант» в виде раствора относительной
плотностью с?|° не менее 1,5, насыщенного по хлористому натрию
и при комнатной температуре не насыщенного по хлорату и хлориду
кальция.
Товарный хлорат-хлорид кальциевый дефолиант, представля-
ющий собой жидкость без запаха (допускается муть и окраска),
должен содержать (в г/л ) [144]:
Са(С1О3)2 428 ± 8
СаС12, не более 380
NaCl, не более » .......... 45
Допускается выпадение в осадок не более 40 г/л NaCl в пересчете
на сухую соль.
Хлорат-хлорид кальциевый дефолиант перевозят в железнодо-
рожных цистернах с нижним сливом.
Получение хлората кальция хлорированием известкового молока
известно очень давно и широко применялось при производстве
хлоратов щелочных металлов, главным образом хлората калия
по методу Либиха. Процесс хлорирования протекает по суммарной
реакции v
6Са(ОН)2+бС12 = Са(СЮз)2 + 5СаС12 + 6Н2О (7.17)
При этом образуется смесь хлората и хлорида кальция, содержащая
на 1 моль хлората кальция 5 молей хлорида.
Было предложено много способов концентрирования или очистки
растворов хлората кальция, получаемых хлорированием известко-
вого молока [145, 146], или выделения основных хлоратов и хлори-
дов кальция [147—148].
Изучение совместной растворимости хлората и хлорида кальция
в водных растворах [149, 150] показало, что разделение этих солей
кристаллизацией из растворов мало пригодно для промышленного
использования. Предложено проведение обменной реакции между
СаС12 и NaG103 в среде ацетона, однако способ не нашел приме-
нения [151].
Для повышения концентрации хлората кальция и увеличения
соотношения хлорат : хлорид в пользу хлората пользуются хими-
ческими методами очистки, в частности осаждением части кальция
в виде карбонатов с помощью соды
ч
CaC12+Na2GO3 = GaCO3 + 2NaCl (7.18)
При этом карбонат кальция и значительная часть хлорида натрия
выпадают в осадок. После отделения осадка получают растворы
хлорат-хлорид кальциевого дефолианта.
Электролизом растворов хлористого кальция [152—154] можно
пр аналогии с производством хлората натрия получать хлорат-
413
хлорид кальциевые растворы с высоким соотношением хлората и
хлорида.
При электрохимическом окислении растворов хлористого кальцйя
' до хлората на катоде обычно образуются осадки гидроокиси или
оксихлоридов кальция, затрудняющие протекание процесса. Для
предотвращения образования осадков необходимо принимать спе-
циальные меры: снижать pH раствора, вводить интенсивное Пере-
мешивание, периодически изменять направление протекания тока
в электролизере и др. [152—154].
* Поэтому этот способ получения хлората кальция не получил
применения.
Хлорат магния
< Хлорат магния используется в народном хозяйстве как дефолиант
для предуборочного удаления листьев хлопчатника при машинной
уборке хлопка. В отличие от хлорат-хлорид кальциевого дефолианта
хлорат магния выпускается в твердом виде и упаковывается в мел-
кую тару, что значительно более удобно для потребления в сель-
ском хозяйстве.
Хлорат магния упаковывают в барабаны из черной стальной ч
жести емкостью 20 л или в бумажные битумированные дублирован-
ные пятислойные мешки емкостью 20 кг с вкладышами из поли-
этиленовой или поливинилхлоридной пленки толщиной не менее
150 мкм.
Хлорат магния, применяемый в качестве дефолианта для пред-
уборочного удаления листьев хлопчатника, поставляется в виде
пластин или чешуек от желтого до светло-коричневого цвета и должен
отвечать следующим требованиям [155]:
Содержание \
естиводного хлората магния Mg(C10g)2 * 6Н2О, % . *
не растворимого в воде остатка, %, не более....
Температура начала плавления, сС, не менее .......
Хлорат магния может быть получен электролизом водных рас-
творов хлористого магния [156], однако трудности, связанные
с осаждением гидроокиси магния на аноде, делают этот метод мало
пригодным для практического использования.
Предложено получать хлорат магния хлорированием гидроокиси
или карбоната Магния [157] по реакции
6Mg(OH)2 + 6CI2 = 5MgCl2 + Mg(C103)2 + 6Н2О
6Mg(CO3)2 + 6С12 = 5MgCl2 + Mg(CO3)2+ 6CO24-3O2
Uli
0f6
44
(7.19)
(7.20) ,
растворы
По этому методу можно получать хлорид-хлоратные
магниевых солей, содержащие 70—100 г/л хлората магния и 300—
350 г/л хлорида магния. Дальнейшая обработка щелоков с целью
получения более богатых хлоратом, магния растворов затруднена,
414
1
так как обе соли хорошо растворяются в воде и трудно поддаются
разделению кристаллизацией.
Предложено экстрагировать хлорат магния из таких растворов
ацетоном [158], однако процесс оказался мало пригодным для про-
мышленного использования.
Применяется более простой метод получения хлорат-магниевого
дефолианта, состоящий в смешении хлората
магния в расплавленном состоянии [159].
Хлорат магния образуется при обменном
разложении
2NaC103 + MgCl2 • 6Н2О -
= Mg(C103)2 • 6H2O + 2NaCl
натрия с хлоридом
Рис. 7-20. Зависимость
температуры плавления
хлорат-магниевого дефо-
лианта от соотношения
хлорида магния и хло-
рата натрия (в г-экв).
(7-21)
Технологическая схема производства
хлората
водный хлорид
сталлизационной воде и к нему в
с мешалкой дозируется твердый
натрия. Обменную реакцию проводят при
НО—120 °C. Выпадающий хлорид натрия
остается в виде суспензии в расплаве шести-
водного хлората магния и избытка хлорида
магния.
Расплав подается на барабанные кри-
сталлизаторы, охлаждаемые водой. Готовый
продукт в виде чешуек упаковывается в ба-
рабаны.
Температура плавления получаемого дефолианта зависит от
соотношения хлорида магния и хлората натрия, взятых для прове-
дения обменной реакции. На рис. 7-20 приведена зависимость тем-
пературы плавления получаемого дефолианта от
MgCl2-6H2O и NaClO3. Для получения продукта, "температура
плавления которого должна быть не ниже 44 °C, соотношение
MgCl2-6H2O : NaClO3 (в г-зкв) должно быть не менее 1,2. Чтобы
избежать получения малогигроскопичного хлората калия, ограни-
чивают содержание КС1 в хлориде магния, применяемом в качестве
сырья.
магния
проста [160].
магния плавится
Шести-
в кри-
реаКтор
хлорат
соотношения
Франц, пат. 179413 (1886).
Промышленный электролиз водных растворов. М.
8, 1404 (1968).
№ 16, 10 (1967). v
И, № 8, 413 (1966).
. Rep., 190, № 3, 9 (1966); 193, № 10, 3, 26 (1968).
Госхимиздат, 1959.
3. Hemijska Ind., №
4. Chem. Weekly, 11,
5. Chem. Ind. News,
6. Oil, Paint and Drug
7. Rev. prod, chim., 7, № 1377' 52 (1968).
8. Oil, Paint and Drug. Rep., 193, № 4, 25 (1968).
9. Chem. a. Rubber, 6, № 8, 7 (1959).
415
10. Шумахер И. Перхлораты: свойства, производство и применение. Пер.
с англ. М., Госхимиздат, 1963.
11. Walde Н., Chem. Ing. Techn., 41, 185 (1969).
12. Chem. Eng. News, 39, № 33, 44, 47 (1961); 40, № 9, 21 (1962).
13. Chem. Trade J., 151, № 3927, 477 (1962); 155, № 4041, 711,713 (1964).
14. Can. Chem. Proc., 46, № 6, 54, 65, 67 (1962).
15. Chem. Week, 99, № 17, 1, 81 (1966).
16. Nallet A., P a si s R. A., Bull. Soc. chim. France, № 3, 488, 494 (1956).
17. Хлорат натрия. ГОСТ 12257—66.
18. Chem. Week, 81, № 13, 83 (1957).
19. Foerster F., M ii 11 e r E., Z. Electrochem., 8, 8, 515, 633, 665 (1902);
9, 171, 195 (1903).
20. F о e r s t e r F., Elecktrochemie .wassriger Losungen, Leipzig, 1905.
21: Muller E., Корре P., Z. Electrochem., 17, 421 (1911).
22. Foerst er F., Elektrochemie wassriger Losungen, Leipzig, 1923.
23. F о er st er F., Trans. Am. Electrochem. Soc., 46, 23 (1924).
24. L i n a r i A., La Pietra L., Chem. e ind., 19, 693 (1937); 20, 590 (1938).
25. Lowy A., Chim. et ind., 40, 31, 1066 (1938).
26. P i t m a n A. L. et al., Chem. Met. Eng., 44, 302 (1937); 45, 692 (1938).
27. Imagava H., J. Electrochem. Soc. Japan, 18, 382 (1950); 20, 28, 489,
-571 (1952); 21, 520 (1953).
28. V a 1 e г a V., Trans. Faraday Soc., 49, № 1, 1338 (1953); 60, № 8, 1450
(1964); J. Electrochem. Soc., 104, № 3, 162 (1957).
29. Яковлева E. И.,РозентальК. И., Филипов T. С., ЖФХ,
30, 937 (1956).
30. Филипов Т. С., Яковлева Е. И. Труды 4-го совещания по элек-
трохимии. М., изд-во АН СССР, 1956. См. с. 257.
31. D'Ans J., Frend Н. Е., Z. Electrochem., 61, № 1, 10 (1957).
32. Hammer L., Wranglen G., Electrochim. acta, 9, № 1, 1—16 (1964).
33. N a d a i T., T a k e i T., J. Electrochem. Soc. Japan, 24, № 12, 557 (1956);
25, № 2, 55; № 3, 110; № 7, 373; № 10, 517 (1957); 28, № 4—6, 66, 97 (1960).
34. N a d a i T.Jakei T., Denki kagaku, 24, 557 (1956); 25, 373, 517 (1957).
35. В e с e л о в с к а я И. E. и др., ЖПХ, 37, № 1, 76 (1964).
36. Imagava Н., Denki kagaku, 25, 607 (1957); 29/632, 787 (1961); J. Elec-
trochem. Soc. Japan, 29, № 10, 697 (1961).
37. Wrangler G., Tekn. Tidskrift., 92, 197 (1962).
38. Beck T. R., J. Electrochem. Soc., 108, № 3, 68c (1961); 113, № 3, 111c
(1966); 116, № 7, 1038 (1969); Beck T. R., Brannland K.,
J. Electrochem. Soc- 119, № 3, 320 (1972).
39. Nam Ch. W., SekineT., Denki kagaku, 37, № 3, 207 (1969); Nam Ch.
W., Dachan Hwahak Hwoejee, 13, № 2, 165 (1969).
40. I b 1 N., Z a n d о 1 d D., J. Electrochem. Soc., 115, № 7, 713—720 (1968).
41. Schumacher J., J. Electrochem. Soc., 116, №2, 68c (1969).
42. Ф и л и п о в T. С. и др., Электрохимия, 2, № И, 1273 (1966); 4, № 1,
20 (1968); 5, № 7, 866 (1969); Кокоулина Д. В., Кришта лик
Л. И., Электрохимия, 7, № 3, 346 (1971); Якшич М. М., Дестич А. Р.,
Николич Б. Ж., Электрохимия, 8, № И, 1573 (1972).
43. Флис И.Е., Быняев М. К., ЖПХ, 30, 339 (1957).
44. I Ъ 1 N., L a n d о 1 d D., Chem. Ing. Techn., 39, № 12, 706 (1967); J. Elec-
trochem. Soc., 113, № 3, Illa (1966); Jaksic M., Nikolic B., J.
Appl. Electrochem., 2, № 4, 337 (1972); Landold D.,Ibl N., Electro-
chem. Acta, 15, № 7, 1165 (1970).
45. F a i r G. M., G о r d о n M., J. Am. Water Works Assoc., 40, 1051 (1948).
46. Bauer R., Chem. Ing. Techn., 34, № 5, 376 (1962).
47. Герм. пат. 110505 (1898); Muller E., Z. Electrochem., 5, 469 (1899);
6, 398 (1900); 8, 909 (1902).
48. Герм. пат. 235706 (1920).
49. Wagner C., J. Electrochem. Soc., 101, 181 (1954).
50. Филипов T. С., Эбериль В. И., Агапова Р. А., Разы-
граева Г. Н., ЖПХ, 40, № 11, 2488 (1967).
416
51. Э бери ль В. И., К ок оу л ина Д. В., Кришталик Л. И.»
Елина Л. М., Электрохимия, 5, № 3, 336 (1969).
52. А г а п о в а Р. А., Е л и н а Л. М., ЖПХ, 44, № 6, 1302 (1971).
53. Such omel J., Chem. Prum., № 2, 47 (1970); Marzec S., Przem.
Chem., 50, № 8, 449 (1971).
54. Matsumura T. et al., Electrochem. Techn., 6, № 11—12, 402 (1968).
55. W e iuer R., Klein G., Chem. Ing. Techn., 29, № 5, 339^1957).
56. Э б e p и л ь В. И., Филипов Т. С., ЖПХ, 40, № 11, 2482 (1967).
57. Э б е р и л ь В. И., К у п о в и ч Ф. В., Электрохимия, 6, № 3, 332 (1970).
58. Э б е р и л ь В. И.,' Елина Л. М., Электрохимия, 6, № 6, 782; № 7»
1010 (1970).
59. Janes М., Trans. Electrochem. Soc., 43, 92, 23 (1947); Groggins P. H.
et al., Chem. Met. Eng., 44, 302 (1937); 45, 692 (1938); № 7, 468 (1940).
60. Англ. пат. 679339 (1952).
61. Агапова Р. А., Е л и н а Л. М., ЖПХ, 44, № 7, 1514 (1971).
62. Р i s z е z е k L., Chem. Stosowana, Ser. А14, ' № 1, 39 (1970).
63. В la s i а к E., Przem. Chem., 46, № 11, 675 (1967); 47, № 3, 154 (1968).
64. Jaksic M. M., Csonka J. M., Electrochem. Techn., 5, №9—10,
473 (1967); 6, № 11-12, 397 (1968).
65. S j о d i n B., Wranglen G., Electrochem. Acta, 10, 203 (1965).
66. J a к s i с M. et al., Electrochem. Soc., 116, № 3, 394; № 5, 684; № 9,
1316 (1969); J aksic M., Hemijska Ind., № 3, 24, 502 (1969).
67. Скрипченк о В. И., Д р оз децка я Е. П., Ильин К. Т., авт.
свид. СССР 281435 (1967); Бюлл. изобр., № 29, 33 (1970); Хим. пром., № 12,
910 (1971).
68. И з и д и н о в С. О., Веселовский В. И., Электрохимия, 6, № 11,
1621 (1970).
69. Англ. пат. 1104078 (1968); Chem. Eng., 72, №15, 82 (1965).
70. Т h i a g a raj a n N., Narasimhan К. C., Udupa N. V. К.,
Chem. Ing. Techn., 43, № 4, 216 (1971).
71. Udupa N. V. K. et al., Indian J. Techn., 4, № 10, 305 (1966); 9, № 7,
257 (1971); Chem. Proc. Eng., 52, № 6, 11 (1971).
72. Пат. ФРГ 1182211 (1964).
73. Newberry J. R. et al., J. Electrochem. Soc., 116, № 1, 114 (1969).
74. Feeck J,, Chem. Ing. Techn., 43, № 4, 173 (1971).
75. Канад, пат. 640131 (1962).
76. Франц, пат. 1350880 (1964); пат. ФРГ 1260447 (1968).
77. Brit. Chem. Eng., 15, № 5, 601 (1970); 16, № 2/3, 149 (1971); De N ora О.,
Chem. Ing. Techn., 43, № 4, 182 (1971).
78. Япон. пат. 27741 (196В).
79. Бельг, пат. 630224 (1962); япон. пат. 2888 (1965); англ. пат. 991079 (1965);
пат. США 3203882 (1965); 3329594 (1967); канад. пат. 755602 (1967); пат.
ФРГ 1261837 (1968).
80. Пат. США 3350286 (1967).
81. W i h n а с к е г К., Kuchler Z., Chemische Technologie, Bd. 1, Anor-
ganische Technologie, Munchen, 1970.
82. Агапова P.A., Эбериль В. И., Елина Л. М., Степнова
3- И., Хим. пром., № 12, 912 (1970).
83. Макаров С. 3., Ш р а й б м а н С. С. Вести, техн.^и эконом, информ.,
ВНИИТЭхим, № 2, 47 (1959).
84. Ш р а й б м а н С. С. и др., ЖПХ, 41, № 4, 734 (1968).
85. Шварц Г. Л., Макарова Л. С., Хим. маш., № 6, 18 (1962).
86. Мамылихина М. В., Романушкина А. Е., Защита металлов,
4, № 5, 603 (1968).
87. Jaksic М. et al., Hemijska Ind., № 8, 109, 1397, 1404 (1968).
88. Англ. пат. 757761 (1956); пат. СССР 364156 (1967); Бюлл. изобр., № 4,
148 (1973).
89. Англ. пат. 1104728 (1968).
90.
Франц, пат. 1502793 (1967); пат. США 3516918 (1970).
27 Заказ 843
417
91.
92.
93.
94.
95.
96.
97.
98.
99.
Франц, пат. 1502455, 1502519 (1967).
Англ. пат. 1161677, 1161678 (1969).
Joshi К. М., J. Indian Chem. Soct, 33, № 9, 653 (1956).
Англ.
Пат.
Швед
Герм. пат.
Chem.-Ztg.,
Англ. пат. 1185507 (1970); канад. пат. 851695 (1970); пат. США 3475313
(1969).
Канад, пат. 696941 (1964).
Т е т е р е в а Л. Г., Электрохимия, 8, № 1, 14 (1972).
пат.
США
пат.
644318, 647749 (1950).
2628935 (1953).
143029 (1953).
418945 (1930).
51, 81 (1927).
100. Канад, пат. 696941 (1964).
100а. Г и н с б у р г В. И., Тетерева Л. Г., Электрохимия, 8, № 1, 14 (1972).
101. Matthes F., Wehner G., An organ isch-technische Verfaren, Leip-
102.
zig, 1964.
103.
Хомяков В. Г., Маш о в е ц В. И., Кузьмин Л. Л. Техноло-
гия электрохимических производств. М., Госхимиздат, 1949.
Wasilews’ki L., Kepinski J. et al., Technologia cloru i zwiazkow
chloru, Warszawa, 1963.
104. Франц, пат. 226257 (1892); 238612 (1894).
105. Англ. пат. 393 (1901).
106. Ullmanns Enzyklopedie der technischen Chemie, Bd. 5, 1954, S. 525.
107. Якименко Л.М., Файнштейн С. Я., Хорошилин В. Г.,
М е л ь ников М. А., Е л и н а Л. М., Э б е р и л ь В. И. Технология
। производства хлората натрия. Доклад на научно-техническом совещании
специалистов стран СЭВ по вопросам электролиза растворов поваренной
соли. Краков, 17—23 января 1967 г.
108. Добров Ю. В., Е л и н а Л. М., Защита металлов, 3, № 5, 618 (1967).
109. Jaksic М., J. Electrochem. Soc., 117, № 3, 74с (1970).
110. Chem. Trade J., 139, № 3607, 138 (1956).
111. Jaksic M. M. et al., Technika (Белград), 23, № 8, 1397 (1968).
112. Англ. пат. 948287 (1964); пат. ФРГ 1197852 (1966).
113. Пат. США 3298946 (1967); 3451914, 3463722 (1969).
114. Канад, пат. 800596 (1968).
115. Англ. пат. 1009459 (1965).
116. Канад, пат. 740864 (1966); 828147 (1969).
117. Can. Chem. Proc., 50, № 7, 85, 92 (1966); Chem. Eng., 74, № И, 136 (1967).
118. Chem. Week, 98, № 22, 132 (1966).
119. Канад, пат. 590810 (1963).
120. Канад, пат. 599769 (1963).
121. Канад, пат. 715550 (1965).
122. Канад, пат. 696942 (1964).
123. Канад, пат. 696943, 696944 (1964); 736459 (1966).
124. Chem. Week, 100, 132 (1967).
125. Rev. prod, chim., 70, № 1353, 12 (1967).
126. Hass K., Tromm W., Chem. Ing. Techn., 40, № 12, 557 (1968).
127. Chem. Eng., 72, № 15, 82 (1965).
128. Англ. пат. 959498 (1964).
129. Нидерланд. па^. 6510232 (1966). •
130. Can. Chem. Proc., 50, №12, 52 (1966); Brit. Chem. Eng., 15, №5, 601 (1970).
131. Europ. Chem. News, 417, № 16, 13 (1970); Inform, chim., № 68, 66 (1968).
132. Grigore.scu T., Celul. si hirt, 18, № 12, 587 (1969).
133. Соль бертолетЪва техническая. ГОСТ 2713—70.
134. Donald, J. Chem. Soc., 1967, 1325.
135. Pei ChuanChow, Hua Hsuch Kung Geh, № 4, 27 (1957).
136. Ching M i n g S u n, Hua Hsuch Kung Geh, № 2, 31 (1958).
137. Kama ch a n d r a n N., Indian chem. Eng., 8, № 1, 6 (1966).
138. W h i t e N. C., Trans. Electrochem. Soc., 92, 15 (1947).
139. О s a k, a N i s h i o, Bull. Chem. Soc. Japan, 5, 181 (1930).
140. Франц, пат. 1156356 (1956).
418
ч
141. Rayagopal К., S r irangan P. В., Altech (Мадрас), 7, 21 (1957).
142. M u г a к a mji T.
143. ~
Rec. Ocean. Works in Japan, № 3, 155 (1959).
[вещ. пат. 323310 (1957); Schumacher, J. Chem. Eng. Progr., 43,
№ 4, 177 (1947).
144. Хлорат-хлорид кальциевый дефолиант. МРТУ-6-01—1969.
145. Пат. США 1854405 (1932); 1887809 (1933); 1893740 (1933); 1949204 (1934).
146. Герм. пат. 583106 (1933).
147. Англ. Цат. 617778 (1949).
148. Швейц, пат. 267089 (1951).
149. Макаров С. 3., В ольнов П. П., Изв. АН СССР, Отд. хим. наук,
№ 2, 201 (1951).
150. Вольнов П. П., Изв. секции физ.-хим. анализа, ИОНХ им. Курни-
кова, 24, 277 (1954).
151. Мартынов Ю.М., авт. свид. СССР 106874 (1957).
152. J ok ogam а Т., J. Chem. Soc. Japan, 58, 647 (1955).
153. Япон. пат. 10519 (1956); 17812 (1961); 16252 (1962); герм. пат. 688368 (1940);
пат. ФРГ 957937 (1957).
154. Воронин Н. Н., Шахова М. А., Изв. Киев, политехи, ин-та, 20,
20—36 (1957).
155. Хлорат магния (дефолиант). ГОСТ 10483—66.
156. Вржосек Г. Т\, Б о р м а ш е н к о И. Б., Л и с о г о р А. И. Труды
Киев, политехи, ин-та, т. 38, 1962. См. с. 97. . ,
157. Герм. пат. 449583 (1921).
158. Пат. США 2075179 (1934).'
159. Якименко Л. М., Мартынов Ю. М., Фурман А. А., а^т.
свид. 109667 (1957).
160. Мартынов Ю. М., Якименко Л. М., М а т в е е в М. А., Фур-
ман А. А., Хим. пром., № 7, 420 (1958).
Г Л А В A 8
ХЛОРНАЯ КИСЛОТА И ПЕРХЛОРАТЫ
S
Впервые хлорная кислота и ее калиевая соль — перхлорат
калия — были получены в 1816 г. Стадионом [1]. Первоначально
перхлорат калия был выделен при термическом разложении хлората
калия, затем, несколько позже, получен электролизом насыщенного
раствора хлората калия на платиновых электродах.
Хлорная кислота была получена в 1835 г. Берцелиусом при
электролизе соляной кислоты, а позже — при электролизе водного
раствора двуокиси хлора и взаимодействием перхлората калия
о серной кислотой [2].
В первой половине XIX в. были выделены и изучены перхлораты
многих металлов [3]. Электрохимическое производство этих солей
было запатентовано Карльсоном в 1890 г. [4].
Первое промышленное производство перхлоратов электрохими-
ческим способом было создано в Швеции (Мансбо) в 1893 г. В начале
XX в. было организовано промышленное производство перхлоратов
во Франции, Швейцарии, США и Германии, однако масштаб произ-
водства этих солей был невелик и их мировая выработка до первой
мировой войны не превышала 2000—3000 т/год [51.
Перхлораты и хлорная кислота применялись в аналитической
химии, в пиротехнике; было предложено их использование для
производства взрывчатых веществ [6]. Последнее положило начало
развитию производства перхлоратов во время первой мировой
войны. Так, в Германии за годы войны было выработано около
20 тыс. т перхлоратов [5], а мировое производство возросло до
50 тыс. т/год. После окончания войны производство перхлоратов
резко сократилось и получило новое развитие только в годы второй
мировой войны.
В связи с перспективами использования перхлоратов в ракетной
•технике производство их стало развиваться в США, Канаде, а затем
и в некоторых других странах. В 1945 г. производство перхлоратов
в США достигло 20 тыс. т/год. В это время определились достоин-
ства перхлората аммония при использовании его для нужд ра-
кетной техники [5], поэтому производство перхлората аммония
получает преимущественное развитие по сравнению с перхлоратом
калия и других металлов.
420
Поскольку хлорная кислота, перхлорат аммония и некоторые
другие перхлораты являются стратегическими материалами, точных
данных о масштабах их производства в литературе нет. Уровень
производства перхлората аммония в начале 60-х годов оценивается
в пределах 50—100 тыс. т/год [7].
ХЛОРНАЯ КИСЛОТА
Свойства хлорной кислоты
Хлорная кислота, НСЮ4, одна из наиболее сильных неоргани-
ческих кислот (подробный обзор свойств хлорной кислоты и ее соеди-
нений приведен в работах [8, 9]).
Безводная хлорная кислота представляет собой бесцветную
подвижную жидкость, сильно дымящую во влажной атмосфере.
Вязкость хлорной кислоты при 20 °C равна 0,795 сПз, т. е. меньше
вязкости воды [10]. Безводная хлорная кислота очень реакционно-
способна, при соприкосновении со многими органическими веще-
ствами взрывается. При хранении в условиях комнатной темпера-
туры малостабильна и медленно разлагается. При частичном раз-
ложении хлорная кислота окрашивается продуктами разложения
в желтовато-бурый цвет. Такая кислота опасна при хранении, так
как может самопроизвольно взрываться.
Молекулярный вес хлорной кислоты 100,465, в парах она в
основном мономерна [И, 12], в жидком состоянии возможна ассоциа-
ция молекул хлорной кислоты вследствие образования межмолеку-
лярных водородных связей [13, 14].
Наиболее вероятно образование димерных молекул с замыканием
цикла типа [8]:
В литературе приводятся различные данные о стандартной энер-
гии образования хлорной кислоты и ее солей, что связано с трудно-
стями ее экспериментального определения. Ниже указаны наиболее
достоверные сведения об энтальпии хлорной кислоты и некоторых
се солей (в ккал/моль):
Соединение . Состояние
НС1О4 ............. Жидкое
НС1О4-Н2О .... Кристаллическое
НС1О4*2Н2О .... Жидкое
HC104-aq ..... Раствор
LiC104 ....... Кристаллическое
NaClO4 ............ »
Н? 298 Литература
—8,62 [15, 16]
—90,28 [8]
—160,97 [8]
—29,77 [16]
—30,87 [17]
—89,98 [16]
—90,89 [18]
—90,68 [16]
-91,464 [19]
421
КС104 . . .
RbC104 . .
CsG104 . . .
Mg(C104)2 .
Ca(C104)2
Ba(C104)2
NH4C104‘ . .
G(NH2)3C104
N2HsG1O4 .
N02C104 . .
Cl207 . . .
Кристаллическое.
s
»
»
»
»
' »
»
»
»
»
»
—101,93
—103,22
—104,38
—105,07'
—134,07
—173,94
—205,3
—69,54
—74,10
-41,24
—8,89
59,2
[16]
[18]
[20]
[20]
[16]
[16]
[16J
[16]
[8]
[22]
[23]
[24]
Безводная хлорная кислота плавится при температуре около
—102 °C [25—27], теплота плавления равна 1,657 ккал/моль [251,
энтропия плавления — 9,67 кал/(моль • град) [8]. Температура
Н2о 10 30 50 70 90 НСЮ4
Состав, мол % HCIO4
Рис. 8-1. Диаграмма плавкости системы вода — хлорная кислота.
кипения безводной хлорной кислоты при атмосферном давлении опре-
делена путем экстраполяции v и равна примерно 110е G, теплота
испарения — 10,4 ккал/моль [8]. G водой хлорная кислота образует
ряд гидратов:
Темпера-
тура плав-
Гидрат ления, °C
' НС104 . .............—102
НС104-0,25Н20 . . —73,1
НС1О4-Н2О . ... 49,905
ПС1О4-2Н2О .... —20,65
422
Темпера-
тура плав-
Гидрат ления, °C
НС1О4-2,5Н2О . . . —32,1
НС1О4-ЗН2О .........—40,2
НС1О4-3,5Н2О ... —45,6
НС1О4-4Н2О .... —57,8
Диаграмма плавкости системы вода — хлорная кислота приве-
дена на рис/8-1 [8, 28—31].
По своим свойствам моногидрат хлорной кислоты существенно
отличается от других гидратов. Предлагалось рассматривать моно-
гидрат хлорной кислоты как соль ониевого катиона Н3О+, т. е.
как перхлорат оксрния, являющийся по строению аналогом пер-
хлората аммония [8, 32—36].
I
Концент-
рация
НС1О4,
вес. %
В табл. 8-1 приведена плотность водных растворов хлорной
кислоты [12, 37—40].
* \ / -
Таблица 8-1. Плотность водных растворов хлорной кислоты (в г/см3)
при различной температуре
- S Температура, °C
-25 0 15 20 30 50 75
•• -’Г, .
I
10
20
30
40
50
60
70
80
90
95
100
1,0637 1,0597 1,0579 1,0539 1,0437 1,023
—— 1,1356 1,1279 1,1252 1,1200 1,1075 1,096
1,2312 1,2168 1,2067 1,2033 1,1965 1,1821 1,160
1,3308 1,31*11 1,2991 1,2947 . 1,2866 . 1,2703 1,251
1,4528 1,4255 1,4103 1,4049 1,3944 1,3752 1,350
1,5908 1,5580 1,5386 1,5327 1,5218 1,4994 1,470
1,7306 1,6987 1,6736 — -- ! 1,6344 1,617
1 - I— 1 । 1,7540 1,727
— - ч г > 1 , 1 1,7720 1,738
1,8043 1 . 1,7515 1,704
1 — 1,8077 1,7676 , 1 II 1,7098
Водные растворы хлорной кислоты обладают хорошей электро-
проводностью и используются как электролиты для проведения
электрохимических процессов, в частности для получения хлорной
кислоты [28, 41—44]. .
В табл. 8-2 приведены данные по удельному электрическому
сопротивлению водных , растворов хлорной кислоты различной
концентр ации [ 28 ].
Теплота растворения хлорной кислоты в воде приведена в
в табл. 8-3.
Давление паров безводной хлорной кислоты (в мм рт. ст.) при
различных температурах может быть найдено по формуле [8]:
S
lgp=----^-+9,1366 (8.1)
Температура кипения растворов хлорной кислоты различной
концентрации при давлении 18 мм рт. ст. составляет [45]:
Концентрация, НСЮ4, вес» % '...... 100 94,8 ‘ 92,0 84,8 79,8 70,5
Температура кипения, °C . . . ... . . 16,0 24,8 35 70 92 107
Состав пара над растворами хлорной кислоты низких кон-
центраций (ниже 72%) обогащен парами воды, при высоких
423
Таблица 8-2- Удельное электрическое сопротивление
водных растворов хлорной кислоты, Ом * см
Темпера-
тура, °C
Концентрация НСЮ4, вес. %
50
40
30
20
10
о
—10
—20
—30
—40
—50
2,207 1,272
2,428 1,397
2,715 1,562
3,100 1,776
3,628 2,072
4,420 2,488
3,102
- - —
1,028
1,132
1,262
1,436
1,665
1,992
2,464
3,176
1,001
1,106
1,240
1,414
1,647
1,968
2,436
3,133
4,250
6,21
10,41
1,154
1,286
1,452
1,670
1,964
2,376
2,982
3,919
5,505
8,44 .
1,540
1,725
1,961
2,275
2,705
3,320
4,242
5,742
8,402
13,82
27,10
2,401
2,704
3,084
3,575
4,227
5,129
6,418
11,59
вления
хлорной кислоты
при 25 [15, 19]
Концент-
рация
исходного
раствора
НС1ОЪ
мол. %
Мольное отношение Н2О/НС1О4 -АН, ккал/моль Концент- рация исходного раствора нею*, мол. % Мольное отношение Н2О/НС1О4
в исход- ном растворе после разбав- ления в исход- ном растворе после разбав- ления
-дя,
ккал/моль
100
99,36
97,70
97,00
96,90
90,48
84,35
80,32
74,04
71,19
61,43
60,60
58,99
0,0000
0,0064
0,0235
0,0309
0,0320
0,1052
0,1855
0,2450
0,3506
0,4046
0,6278
0,6502
0,6952
811
763
794
794
804
727
800
802
776
819
803
735
801
21,15
21,07
20,94
20,92
20,89
19,98
19,23
18,86
17,77
17,19
13,37
12,80
12,48
58,34
55,42
50,59
46,08
40,98
36,59
32,98
29,78
24,60
20,21
15,02
10,29
6,332
0,7141
0,8044
0,9766
1,1701
1,4402
1,7329
2,0321
2,358
3,065
3,948
5,656
8,152
14,794
826
796
906
771
645
817
760
653
711
812
216,6
270,8
459,9
12,09
10,60
7,98
7,80
7,78
7,24
5,27
4,29
2,68
1,46
0,393
—0,067
—0,129
концентрациях — ^лорным ангидридом. При концентрации 72,4%
хлорная кислота образует с водой азеотропную смесь. На рис. 8-2
приведен состав пара над растворами хлорной кислоты различной
концентрации [8]. Отклонение состава пара от состава жидкости
в области концентраций, близких к 100%, объясняется наличием
равновесия между хлорной кислотой, ее^ангидридом и моногидратом:
ЗНС1О4 С12О7+НС1О4« Н2О (8.2)
Давление паров моногидрата очень невелико.
424
Рис. 8-2. Зависимость состава пара над
растворами хлорной кислоты от состава
жидкой фазы.
Безводная хлорная кислота — сильный окислитель. Элементар-
ный фосфор и сера окисляются ею до фосфорной и серной кислот [46].
Иод окисляется хлорной кислотой, бром, хлор, а также бромистый
и хлористый водород не взаимодействуют с хлорной кислотой даже
при нагревании [47, 48].
При контакте безводной хлорной кислоты с органическими веще-
ствами, способными легко окисляться, происходит бурная реакция,
сопровождающаяся взрывом. С хлороформом и четыреххлористым
углеродом хлорная кислота смешивается без взаимодействия, однйко
затем наблюдается протекание медленной реакции с разложением
кислоты [8]. Путем прямого взаимодействия органических веществ
с хлорной кислотой получены
метил- и этилперхлораты [49],
а также эфиры хлорной ки-
" слоты [50].
НС1О4, как сильная ки-
слота, помимо солей с метал-
лами, может образовывать
с другими неорга-
ническими и орга-
ническими кисло-
та ми ацилперхло-
? р а т ы, например перхлорат
| нитрония NO2-C1O4.
| Попадание хлорной ки-
| слоты на кожу вызывает по-
1 явление болезненных, долго
не заживающих ожогов.
Поскольку при хранении
f кислота медленно разлагается, ее стараются готовить непосред-
ственно перед использованием.
: Нестабильность концентрированной хлорной кислоты объяс-
няется распадом присутствующего в ней хлорного ангидрида и на-
| коплением образующихся в растворе активных молекул низших
окис лов хлора, обладающих свойствами свободных радикалов.
г. Стабильность хлорной кислоты может быть повышена добавками
I ингибиторов — веществ, дезактивирующих активные частицы про-
дуктов распада. К ингибиторам относятся в частности органические
соединения с трихлорметильной группой [51], например трихлор-
уксусная кислота и четыреххлористый углерод.
Добавление ингибиторов в количестве до 2—3% от массы кислоты
Й не меняет механизма разложения хлорной кислоты, но увеличивает
на несколько порядков длительность индукционного периода раз-
ложения [8].
Разработка способов стабилизации хлорной кислоты продол-
жается и в настоящее время [52].
* При действии на безводную хлорную кислоту сильными водо-
отнимающими средствами можно получить ангидрид хлорной
425
при комнатной температуре хлорная
кислоты Cl 2О7, образующийся также в небольшом количестве при фо-
тохимической или термической реакции между хлором и озоном.
Чистый хлорный ангидрид — бесцветная жидкость с темпера-
турой плавления —90 ± 2 °C; иногда при получении, а также при
хранении он окрашивается низшими окислами хлора.
Если к фосфорному ангидриду, охлажденному до —78 °C, при-
ливать медленно безводную хлорную кислоту, не допуская повыше-
ния температуры смеси более —30 °C, получают чистый хлорный
ангидрид [53].
В отличие от других окислов хлора и хлорной кислоты ее ангид-
рид ' отличается сравнительно низкой химической активностью.
При комнатной температуре он не реагирует с серой, фосфором и
бромом, растворяется без взаимодействия в четыреххлористом угле-
роде, при попадании капли ангидрида на дерево или бумагу он
испаряется без взрыва. Однако при работе с ангидридом хлорной
кислоты (особенно загрязненным) так же, как и с безводной хлор-
ной кислотой, необходима большая осторожность вследствие воз-
можности взрывов.
Хлорная кислота может служить удобным сырьем для получения
различных неорганических и органических перхлоратов. Путем
нейтрализации хлорной рислоты можно получать перхлораты любых
металлов, гидразина и других органических оснований. Для боль-
шого числа перхлоратов, выпускаемых в ограниченном масштабе
и используемых в качестве реактивов, производство их через хлор-
ную кислоту наиболее удобно и экономично. Однако, например,
перхлорат висмута не может быть получен взаимодействием метал-
лического висмута с концентрированной НСЮ4, так как реакция
проходит со взрывом [54]. В определенных условиях реакция ней-
трализации хлорной кислоты соответствующими основаниями может
оказаться целесообразной не только для получения перхлората
магния, алюминия, бериллия и других металлов, но также и для
получения перхлоратов щелочных металлов и аммония.
Хлорная кислота и перхлораты находят широкое применение
в аналитической практике. Хлорная кислота используется при коли-
чественном определении калия осаждением в виде малорастворимой
соли — перхлората калия. Как сильный окислитель хлорная кис-
лота используется для окисления и разрушения органических ве-
ществ (влажное сожжение), для окисления руд. Кроме того, хлор-
ная кислота применяется в качестве растворителя, как среда для
неводного титрования, для разрушения протеинов при биологиче-
ских анализах, а также как добавка к электролиту в гальванотех-
нике и при электролитической обработке металлов.
Хлорную кислоту разрешается перевозить только в виде водных
растворов концентрацией примерно до 70% (до азеотропа). В случае
применения безводной хлорной кислоты, ее следует получать на
месте потребления.
i
426
Способы получения хлорной киолоты
Впервые соли хлорной кислоты и затем сама кислота были полу-
чены химическим путем [1] из хлората калия. При термическом
разложении хлоратов одновременно с выделением кислорода обра-
зуются соответствующие перхлораты. Однако этот способ получения
перхлоратов, так же как и прямой синтез хлорной кислоты из Н2О,
О3 и С12 [45] облучением смеси (длина волны 2537 А) не нашел про-
мышленного применения.
Наиболее удобно получение перхлоратов и хлорной кислоты
путем анодного окисления ионов СЮ3 или С1" в водных раство-
рах. При прямом получении хлорной кислоты электролизу подвер-
гаются растворы соляной кислоты или хлора, а при получении ее
через перхлораты проводят электрохимическое окисление водных
* растворов хлоратов щелочных металлов с последующей обработкой
образующегося перхлората сильной минеральной кислотой.
Электролиз растворов соляной кислоты исследован в рабо-
тах [56—58]. Первоначально при прямом получении хлорной кис-
лоты электролизу подвергали только разбавленные воДные растворы
соляной кислоты при низких температурах [59], так как уже при
применении 1 н. раствора практически вся соляная кислота расхо-
дуется на получение хлора и хлорная кислота практически не обра-
зуется. При использовании 0,1 н. раствора солйной кислоты поло-
вина ее окисляется до хлорной кислоты и половина — до хлора [60].
При применении в качестве электролита 0,5 н. раствора HG1 полу-
чали хлорную кислоту концентрации 20 г/л. При 18 °C и плотности
тока 90 А/м2 напряжение на ячейке составило 8 В и расход электро-
энергии 28 кВт*ч ца 1 кг 60%-ной хлорной кислоты. Недостатками
этого метода были получение очень разбавленных растворов хлор-
ной кислоты и большие затраты на ее концентрирование. Было пред-
ложено трехступенчатое окисление с использованием 0,5 н. кислоты,
сообщалось об использовании этого способа в промышленности [5].
Окисление ионов хлора до хлорной кислоты протекает прй высо-
ком потенциале 2,8—3,0 В на платиновом или титановом, плакиро-
ванном платиной, аноде. Процесс окисления соляной кислоты по
суммарному выражению
НС14-4НаО—8е = НС1О4+8Н+
протекает через несколько стадий образования промежуточных
кислородных соединений хлора, сорбируемых на аноде. На катоде
выделяется водород.
По аналогии с получением озона электролизом концентрирован-
ных кислородсодержащих кислот было предложено проводить
окисление соляной кислоты до хлорной, подвергая электролизу
разбавленные растворы соляной кислоты в концентрированной 4—
6 н. хлорной кислоте [61, 62].
Ход процесса электролиза зависит от концентрации в электро-
лите хлорной и соляной кислот, температуры электролиза и-плотности
*
тока [62—66]. На рис. 8-3, а приведена зависимость выхода
продуктов электролиза по току от концентрации соляной кислоты
в электролите. Данные получены при условиях 20 °C, 4 н. НС1О4
при анодном потенциале 2,8 В [63].
Наиболее высокие выхода, хлорной кислоты по току получены
при концентрации соляной кислоты 0,8—2 н. При снижении кон-
центрации соляной кислоты выход НС1О4 по току уменьшается за
счет повышения выхода кислорода по току. При повышении кон-
центрации- НС1 более 2 н. возрастает расход тока на выделение
хлора и выход хлорной кислоты по току резко снижается. Значение
оптимальной концентрации соляной кислоты в электролите повы-
шается с уменьшением температуры электролиза и зависит также
от концентрации хлорной кислоты в электролите.
% А, %
Рис. 8-3. Зависимость выходов по току продуктов электролиза от концентрации
НС1 в 4 н. НС1О4 при 20 °C и Е — 2,8 В (а) и от температуры раствора, содер-
жащего 4 н. НС1О4 + 1 н. НС1 при Е = 2,8—3,0 В (б):
1 — НС1О4; 2 — С12; 3 — О2.
Процесс анодного окисления соляной кислоты до хлорной сильно
зависит от температуры. На рис. 8-3, б показана зависимость выхода
продуктов электролиза по току от температуры раствора [63] при
содержании в электролите 4 н. НС1О4 и 1 н. НС1 и потенциале анода
2,8—3,0 В. С понижением температуры выход хлорной кислоты по
току возрастает, а хлора и кислорода соответственно снижается.
Расход платины зависит от температуры электролиза и умень-
шается с понижением температуры. Содержание примесей в хлорной
кислоте зависит от чистоты исходной соляной кислоты и применения
достаточно коррозионно-стойких конструкционных материалов для
изготовления электролизеров, трубопроводов и аппаратуры.
Для получения хлорной кислоты высокой чистоты ее следует
очищать от ионов хлора. Очистка может быть осуществлена электро-
химическим способом, т. е. возможно более полным окислением
примесей С1" до хлорной кислоты, однако при этом по мере снижения
концентрации ионов хлора выход хлорной кислоты по току сни-
жается и приближается к нулю при достаточно полной очистке рас-
твора от примеси соляной кислоты.
428
Очистку хлорной кислоты можно производить также отгонкой.
Хлорная кислота может быть получена анодным окислением
растворенного электролите хлора [57]. В качестве электролита
может применяться 4—6 н. раствор хлорной кислоты [65, 67].
Сообщается, что на платиновых анодах и серебряных катодах элек-
тролизеры на нагрузку 3,5 кА при плотности тока 2 кА/м2 и темпе-
ратуре О °C работали при напряжении 4 В [68]. Процесс протекает
по суммарному выражению:
С12
8Н2О —> 2НСЮ4+7Н2
(8.3)
Этим способом можно получать очень чистую кислоту, поскольку
со стороны не вводится каких бы то ни было загрязняющих приме-
сей. Часть получаемой кислоты возвращается вновь в пройзводствен-
ный цикл, часть отбирается и после перегонки выпускается в виде
60—70%-ной товарной кислоты.
Предложено получать хлорную кислоту анодным окислением
водных растворов хлоратов в трехкамерном электролизере с двумя
диафрагмами [69]. В анодном пространстве электролизера с плати-
новым анодом и стальным катодом можно получить 2 н. хлорную
кислоту, а в катодном пространстве соответствующую щелочь.
Сообщается [7] о применении для окисления хлористого водо-
рода или хлора в электролите — хлорной кислоте — электролизе-
ров фильтр-прессного типа с диафрагмой из пластмассовой сетки.
Рамы электролизера выполнены из поливинилхлорида, аноды из
платиновой фольги и катоды из серебра. Электролизеры на нагрузку
5 кА работают при плотности тока 2,5 кА/м2, напряжении на ячейке
4,4 В и выходе по току около 60%. Получаемая при этом кислота
отличается высокой чистотой.
Хотя есть указания, что хлорная кислота, полученная прямым
электрохимическим методом, используется для производства раз-
личных перхлоратов [69], с успехом применяется также обратный
J <3 . . ... . •
II
и щелочноземельных металлов. При этом перхлораты получают окис-
лением водных растворов хлоратов. Один из первых промышленных
методов получения хлорной кислоты был основан на реакции между
перхлоратом калия и серной кислотой [2]
КСЮ4+H2SO4=НСЮ4-Ь KHSO4
(8.4)
Хлорную кислоту отгоняли под вакуумом. При этом может быть
получена хлорная кислота высокой концентрации, близкой к без-
водной, если применять достаточно концентрированную серную
кислоту. Реализация этого процесса в промышленности связана
с трудностями, обусловленными сложностью аппаратурного офор-
мления, ограниченностью конструкционных материалов, для работы
в смеси хлорной и серной кислот и необходимостью проведения
отгонки хлорной кислоты под вакуумом. Поэтому предлагаемый
процесс целесообразен только при получении безводной хлорной
кислоты.
429
Водные растворы хлорной кислоты получают при взаимодей-
ствии перхлората калия с кремнефтористоводородной кислотой
в водном растворе
KCi04+HSiF6=HC104 + KSiFe (8.5)
f
После отделения осадка плохо растворимого кремнефтористо-
водородного калия [3] фильтрованием разбавленные растворы хлор-
ной кислоты могут быть концентрированы и затем подвергнуты
перегонке в виде азеотропной 72%-ной кислоты. Однако осадок
кремнефтористоводородного калия плохо фильтруется, что затруд-
няет практическое использование этого метода.
Аналогичные результаты могут бьг£ь получены при обработке
раствора перхлората бария серной кислотой. При этом выпадает
осадок труднорастворимого сульфата бария
Ba(C104)8 + HaS0j = 2HC104+BaS04 (8.6)
В последние годы для организации производства хлорной кислоты
в промышленном масштабе использован процесс, основанный на
взаимодействии перхлората натрия с соляной кислотой [5}
NaC104+HCl=HC104 + NaCl (8.7)
/ . -
Способ был предложен [70] и изучен давно [71]. Однако техни-
ческие трудности, связанные с его промышленной реализацией,
были преодолены только в последнее время [72].ч
Концентрированные растворы перхлората натрия обрабатывают
концентрированной соляной кислотой или газообразным НС1. Выпа-
дающие в процессе реакции кристаллы хлорида натрия отделяют
фильтрованием. При выпаривании полученного раствора, содер-
жащего около 32% НС1О4, отгоняют сначала избыток соляной кис-
лоты. Полученную при этом 35%-ную соляную кислоту возвращают
на первую стадию процесса. Концентрация хлорной кислоты
в остатке повышается до 39%. При дальнейшем упаривании до
57% НС1О4 полностью отделяется НС1, улавливаемая в виде слабой
соляной кислоты. Загрязненная хлорная кислота после дополни-
тельной отгонки воды отгоняется в виде азеотропной смеси, содер-
жащей до 70—72% хлорной кислоты.
Растворимость, давление паров воды и вязкость в системе
NaCIO4—НС1О4—Н2О изучены достаточно подробно [73]. Выделя-
ющиеся в кубе отгонного аппарата кристаллы NaC104 возвращаются
на первую стадию производственного процесса.
Практически на всех стадиях аппаратура работает в условиях
очень агрессивных сред, содержащих в различных соотношениях
хлорную и соляную кислоты.
Я. Для обеспечения промышленного производства необходим тща-
тйльный подбор коррозионно-стойких материалов. Сообщается об
использовании стеклянной, эмалированной или остеклованной аппа-
ратур^, в частности для вакуумной дистилляции хлорной кислоты.
*?:£•
va-.
На некоторых стадиях процесса с успехом можно применять аппараты
из титана. л
При комбинировании этой схемы получения хлорной кислоты
с производством перхлората натрия выделяемые отходы хлорида
натрия, загрязненного хлорной кислотуй или перхлоратом натрия,
могут быть возвращены на производство перхлората натрия. После
вакуумной дистилляции
можно получить 70—72% -
ную хлорную кислоту высо-
кой степени чистоты.
Для получения безводной
хлорной кислоты применяют
перегонку в вакууме смеси
технической примерно 70% -
ной хлорной кислоты с трех-
четырехкратным по объему
количеством дымящей серной
кислоты [74]. Выход безвод*
ной хлорной кислоты зависит
от условий ведения процесса,
в частности от исходной
Рис. 8-4. Схема установки для получения
безводной хлорной кислоты:
1 — труба из кварцевого стекла или пирекса;
2 — электрообогрев; з — капельная воронка;
4 — приемник отработанной смеси кислот; 5 —
трубка для отвода па^ов хлорной кислоты; 6 —
приемник-конденсатор безводной хлорной кислоты.
концентрации хлорной ки-
слоты, соотношения реаги-
рующих олеума и азеотроп-
ной хлорной кислоты, темпе-
ратуры и вакуума, при кото-
рых протекает вакуумная отгонка. Ниже приведены выходы (в%}
безводной хлорной кислоты при вакуумной отгонйе (остаточное дав-
ление 2—3 мм рт. ст.) в зависимости от объемного соотношения
реагентов и температуры процесса, *Ъри содержании в азеотропной
жидкости 71% хлорной кислоты и применении 60%-ного олеума [8]:
нею* : SO3
1': 0,8 ’
1 : 1,2
1 : 1,4
1 : 1,7
При 7 О °C
50
67
При 100 °C
60
79
81,5
82
С увеличением объема олеума, подаваемого на смешение, проис-
ходит более полное обезвоживание хлорной кислоты и увеличивается
выход безводной кислоты, который возрастает также с повышением
температуры. Однако при температуре выше 100 °C получается
кислота, окрашенная низшими окислами хлора, образующимися
за счет термического разложения НС1О4. При этом хлорная кислота
становится нестабильной и дальнейшее обезвоживание становится
опасным из-за возможности взрывного разложения кислоты. По-
этому 100 °C является верхним температурным пределом для прове-
дения процесса обезвоживания хлорной кислоты методом вакуум-
ной перегонки [8].
, 431
При чрезмерном увеличении объема олеума также возникает
опасность взрыва, так как возможно образование очень нестабиль-
ного ангидрида хлорной кислоты.
Предложен непрерывный процесс получения безводной хлорной
кислоты обезвоживанием азеотропа с помощью олеума при вакуум-
ной отгонке [75]. Схема лабораторной установки показана на рис. 8-4,
однако такой принцип может быть использован и для создания более
крупных установок. В самом аппарате всегда присутствует неболь-
шое количество смеси кислот, что уменьшает опасности, связанные
с возможными взрывами. При смешении кислот требуется охлажде-
ние смесителя во избежание перегрева смеси и возможного терми-
ческого разложения хлорной кислоты.
ПЕРХЛОРАТЫ МЕТАЛЛОВ
Свойства перхлоратов
Соли хлорной кислоты так же, как и хлорная кислота, — соеди-
нения, богатые кислородом. Многие перхлораты в отличие от хлор-
ной кислоты обладают достаточной стабильностью. Такие соли,
как перхлораты щелочных металлов и, главным образом, перхло-
рат аммония широко используются в качестве окислителей для
ракетных топлив и в пиротехнике. Перхлораты щелочноземельных
металлов обладают высокой гигроскопичностью, поэтому они обычно
не применяются ни в ракетной технике, ни для пиротехнических
целей. Перхлорат магния широко используется как очень эффектив-
ный осушитель.
В табл. 8-4 приведены [5, 76] основные свойства перхлоратов
щелочных и щелочноземельных металлов, а также перхлората
аммония.
В табл. 8-5 указана растворимость перхлоратов щелочных метал-
лов и аммония в некоторых органических растворителях при 25 °C.
Чистые перхлораты металлов в обычных условиях — достаточно
стабильные соединения. В контакте с органическими соединениями
или веществами, способными окисляться, перхлораты становятся
огне- и взрывоопасными. Поэтому в процессе производства, хра-
нения, транспортирования и применения перхлоратов необходимо
исключить контакт этих солей со смазочными материалами или веще-
ствами, способными окисляться. Перхлораты чувствительны к уда-
рам, трению и другим инициирующим воздействиям. Наблюдались
случаи взрывов (перхлората аммония) при растирании соли в ступке.
При обращении с перхлоратами необходимо соблюдать осторожность.
При попадании перхлоратов или их растворов на одежду, ее следует
немедленно тщательно вымыть. Перхлораты разрушают кожу и дей-
ствуют на слизистые оболочки.
Перхлораты щелочных и щелочноземельных металлов (за исклю-
чением перхлората лития) не имеют определенной температуры
плавления, так как при их нагревании происходит частичное разло-
ГН
, 432
ч
Таблица 8-4. Свойства перхлоратов Щелочных и Щелочноземельных металлов
28 Заказ 843
' Таблица 8-5. Растворимость перхлората аммония и щелочных
металлов в органических растворителях [5]
(г/100 г растворителя)
Растворители NH*C1O4 - LiClO* NaClO* КСЮ* RbClO* CsClO*
Метиловый спирт . .
Этиловый спирт . .
w-Пропиловьтй спирт
«-Бутиловый спирт
Изобутиловый спирт
Ацетон ......
Уксусноэтиловый эфир
Этиловый эфир . . .
6,862
1,907
0,3865
0,017
0,1272
2,26
; 0,032
Нераств.
182,25
151,76
105,00
79,31
58,05
136,52
95,12
113,72
51,355 0,1051 0,06 0,093
14,705 0,012 0,009 0,011
4,888 0,01 0,006 0,006
1,864 0,0045 0,002 0,006
0,786 0,005 0,004 0,007
51,745 0,1552 0,095 0,15
9,649 He 0,0015 ‘раствори: 0,016 м Нераств.
жение и образование смеси неразложившегося перхлората с продук-
тами его разложения. Поэтому приведенные в табл. 8-5 данные
о температуре плавления необходимо рассматривать как темпера-
туры плавления эвтектических смесей соответствующих перхлоратов
с продуктами их разложения.
Перхлорат лития плавится при 247 °C. При этой температуре
не замечено его разложения, оно начинается с заметной скоростью
лишь при 400 °C и выше.
Все перхлораты щелочны± и щелочноземельных металлов, кроме
перхлоратов калия и аммония, образуют гидраты. Из перхлоратов
щелочных металлов наиболее гигроскопичен перхлорат лития,
гигроскопичны также соли щелочноземельных металлов. С аммиаком
перхлораты образуют аммиакаты различного состава.
Перхлораты представляют собой белые кристаллические соли.
При температуре фазового превращения ромбическая форма'кристал-
лов, свойственная этим солям при обычных температурах, переходит
в кубическую. Не изменяет своей кристаллической формы только
перхлорат лития.
и
Способы получения перхлоратов металлов
Промышленное производство перхлоратов осуществляется в на-
стоящее время исключительно электрохимическим способом •— окис-
лением водных растворов хлоратов [5] — или через хлорную кис-
лоту, получаемую также электрохимическим окислением соляной
кислоты. Хотя многие перхлораты образуются при непосредствен-
ном окислении водных растворов их хлоратов или хлоридов, практи-
чески таким способом получают только перхлорат натрия. Перхло-
раты калия, аммонии и некоторых других металлов удобнее полу-
чать обменным разложением перхлората натрия с соответствующей
солью калия, аммония или других катионов. Прямое получение пер-
хлората калия затрудняется вследствие его малой растворимости.
Непосредственное электрохимическое окисление хлорида или хло-
434
рата аммония до перхлората не применяется по условиям техники
безопасности. Получение перхлоратов щелочноземельных металлов
электролизом сопряжено с теми же трудностями, которые были рас-
смотрены для случая получения хлоратов этих металлов. Термическое
разложение хлоратов можно проводить так, чтобы при этом образо
вался соответствующий перхлорат. Так, разложение хлората на-
трия может протекать по уравнению
2NaC103—> NaClO4+NaCl + O2| (8.8)
4NaC103—> 3NaC104 + NaCl (8.9)
Процесс образования перхлората при термическом разложении
хлората натрия может* протекать при 400—600 °C. Из плава, содер-
жащего перхлорат, хлорид и некоторое количество неразложивше-
тося хлората натрия, необходимо выделять перхлорат дробной кри-
сталлизацией. Нераэложившийся хлорат натрия возвращается вновь
на разложение.
Предложено также экстрагировать перхлорат натрия из твердой
смеси его с NaCl и NaC103 жидким алкилфосфатом 180].
Хотя предложения о термическом методе производства делались
и в самое последнее время [81, 82], однако в промышленности ой
не нашел применения из-за опасности взрыва и обесценивания части
хлората, особенно при протекании реакции (8.8).
Перхлораты могут быть получены также химическим окисле-
нием хлоратов, например свежеосажденной двуокисью свинца [83].
Окисление ведут в кислой среде. *Для улучшения протекания про-
цесса и последующего фильтрования предложено добавлять НС104
[84]. Однако способ химического окисления хлоратов, насколько
известно автору, не применяется в промышленности.
До последнего времени получение перхлората натрия осуще-
ствлялось исключительно окислением водного раствора хлората
натрия. Прямое окисление хлорида натрия до перхлората не было
рекомендовано и практически до последнего времени не использо-
валось в промышленности [85]. Это объясняется тем, что нецеле-
сообразно с экономической точки зрения вести процесс окисления
хлорида до перхлората на платиновых анодах. Поэтому предлагали
начальные стадии окисления осуществлять на более дешевых и до-
ступных анодных материалах. Так, для этой цели использовались
графитовые аноды, а полученный при этом хлорат отделяли кристал-
лизацией и окисляли до перхлората на платиновых анодах.
В последние годы разработан процесс подучения хлората и пер-
хлората окислением на анодах из перекиси свинца, что создало
условия для одностадийного ведения процесса без промежуточной
стадии выделения хлората натрия. Этот способ, по-видимому, скоро
получит широкое применение в промышленности. Высказываемое
неоднократно мнение [7, 68] о нецелесообразности проведения пря-
мого окисления хлорида натрия до перхлората вследствие полу-
чения более низких выходов по току в настоящее время уже
нельзя считать обоснованным.
435
Перхлорат натрия
Перхлорат натрия хорошо растворим в воде. Зависимость его
растворимости от температуры приведена в табл. 8-6, а совместная
растворимость с NaCl — в табл. 8-7.
Таблица 8-6. Растворимость NaClO^ в воде [86]
Температура,
°C
Плотность,
г/см3
Содержание NaC10<
Твердая фаза
г/100 г Н2О
О
15
25
28
50
50,8
15
25
38
55
75
100
143
1,663
1,683
1,713
1,749
1,758
1,757
1,757
1,756
1,757
1,758
62,87
65,63
67,82
70,38
73,26
73,30
71,68
73,21
72,83
73,94
75,01
76,75
79,04
169,32
190,95
210,75
237,61
273,97
253,11
273,27
268,05
283,73
300,16
330,11
№С1О4-Н2О
№C104-H20-NaC104
NaClO4 (метастабильное
состояние)
NaClO4
Анодное окисление хлората натрия до перхлората протекает
по уравнению:
С10д + Н20 >С1О4+2Н+4-2е (8-10)
По механизму реакции окисления хлоратов нет единого мнения,
исследования его продолжаются и в настоящее время. Наиболее
обоснованным представляется механизм реакции, основанный на
предположении о разряде на аноде хлорат-иона с образованием
радикала С1О3, который, взаимодействуя с водой, образует перхлорат:
2С10д
2С10з + Н20
2C1Oj + H2O -
Такое представление было высказано е
2С1О3 + 2е
—> HCIO4+HCIO3
> НС1О4+ НС1О3 + 2е
(8.11)
(8.12)
(8.13)
в начале столетия [87—
89] и подтверждается рядом последних работ [90—93]. По-видимому,
процесс идет через образование промежуточных комплексов из раз-
ряжающихся радикалов С1О3 и молекул воды [89], при распаде
которых кислород воды войдет в состав иона СЮ3, тогда как в состав
иона C10J будет входить только кислород, содержащийся в хлорате.
Приведенный механизм подтверждается результатами исследо-
вания процесса окисления хлоратов до перхлоратов в водных рас-
творах, меченных тяжелым изотопом кислорода 18О. В работе [94]
отмечается, что 18О входит сначала в состав хлората и только затем
436
Таблица 8-7. Совместная растворимость NaC10< и NaCl [86]
Тем пер а-
Tyga,
Плотность,
г/см3
Концентрация, г/100 г Н2О
Твердая фаза
NaClCh
NaCl
15
25
38
50
75
100
1,663
1,683
1,683
1,713
1,713
1,756
1,755
1,757
1,757
1,758
1,757
1,664
1,532
1,567
39,31
80,15
123,38
157,99
169,32
190,95
210,75
207,74
237,61
234,97
2^5,73
278,41
300,16
296,36
330,11
324,86
237,07
107,54
83,3
25,44 )
17,02
10,21 J
6,7
0
° I
0 I
4,27
0
3,55
0
2,97
0
3,32
О
3,77
4,92 )
6,55 !•
17,71 J
NaCl
NaCl + NaClО4 Н2О
NaClOi • Н2О
NaC104 • Н2О
NaC104 H2O-f-NaCl
NaC104 • H2O
NaC104 H2O + NaCl
NaC104
NaC104+NaCl
NaC104
NaC104 + NaCl
NaC104
NaC104+NaCl
NaCl
по мере окисления хлората переходит в состав перхлор-аниона.
Это не должно наблюдаться при окислении ионов C10J атомарным
кислородом или при реакции между радикалами С1О3 и ОН.
Необходимо, однако, учитывать, что при переходе от одного
анодного материала к другому (например, от платины к перекиси
свинца) может меняться и механизм протекания процесса [92, 95].
Второй возможный механизм протекания процесса заключается
в окислении ионов хлората кислородом, образующимся при разряде
ионов ОН' и выделяющимся или сорбированным на аноде [96—99]:
2ОН- > М — О4-Н2О4-2е (8.14)
ClOg+M-O —> ClOj+M (8.15)
C10f-f-20H- —> СЮ4+Н2О + 2е (8.16)
Вторая конкурирующая реакция выделения молекулярного кис-
лорода может быть записана в виде
2М — О —> 2М + О2 (8.17)
Эта реакция должна ускоряться с понижением концентрации
хлората в электролите. При протекании реакции по уравнению
- > 437
(8.16) выход по току должен зависеть от условий подвода достаточ-
ного количества ионов СЮ; к поверхности анода. Выход по току
будет снижаться с уменьшением концентрации С1О3 и плотности
тока [7].
На основе полярографических исследований предложено также
объяснение процесса окисления хлората до перхлората, исходя
из одновременного разряда ионов СЮ; и ОН" [100]. Образующиеся
при разряде ионов СЮ; радикалы обладают высокой активностью
и окисляются кислородом, выделяю
щмся при разряде ОН до пер-
и
хлоратного радикала, который принимает электрон
в ион СЮ;.
Для протекания процесса предложен также
риант [97]:
гон- —> 2ОН + 2е
и превращается
следующий ва-
(8.18)
OH + C1OJ—> ОН’ + СЮз (8.19)
ОН + СЮз —> НСЮ4 —> С1О7+Н+
OH-+C1OJ > С1О7 + Н+ + 2е
< (8.20)
(8.21)
Л Для электрохимического окисления хлората до перхлората
необходимы электродные материалы с высоким анодным потенциа-
лом. На графитовом аноде образование перхлоратов практически
не наблюдается; на магнетитовом аноде образование перхлоратов
незначительно. Наилучшим материалом для анодов является глад-
кая платина, на которой благодаря ее высокому потенциалу образо-
вание перхлоратов происходит с высоким выходом по току. Основ-
ным недостатком, ограничивающим применение платины, является
ее дороговизна и дефицитность.
Для снижения расхода платины применяют платинотитановые
или платинотанталовые аноды [68, 101], образуемые плакированием,
например, титановой основы тонкой платиновой фольгой. Платино-
вая фольга толщиной 20—40 мкм приваривается к каркасу анода,
причем она может покрывать только часть поверхности анода.
Расход платины зависит от условий проведения электролиза
и может достигать 7—10 г/т перхлората [7]. Расход платины можно
уменьшить снижая температуру электролиза й глубину превраще-
ния хлората в перхлорат.
По мере уменьшения концентрации хлората в электролите за счет
его окисления выход по току медленно снижается, а расход платины
несколько возрастает до достижения предельной концентрации хло->
рата (около 50 г/л). При снижении концентрации хлората ниже
предельной выход по току резко снижается, а расход платины сильно
возрастает.
Для предотвращения потерь выхода по току на катодное восста-
новление к электролиту добавляют соли хрома аналогично произ-
водству хлоратов. Механизм действия этих солей такой же, как и при
438
II
получении хлоратов. Предложено также в качестве добавки, сни-
жающей потери тока при получении перхлоратов, использовать пер-
сульфат щелочного металла например, Na2S2O8 от 1 до 10 г/л
[102J, однако о промышленном применении персульфатов сообще-
ний нет.
Вместо платины в качестве анодного материала применяется
также двуокись свинца [102—105], осаждаемая электролитически
из растворов азотнокислого свинца с различными добавками. Дву-
окись свинца может быть нанесена на графит [106—108] или ти-
тан [104, 109]. В последнем случае необходимо принимать меры
по предотвращению образования переходного сопротивления между
титановой основой и активным слоем из двуокиси свинца вследствие
окисления титана в процессе электролиза.
На электродах из двуокиси свинца выход по току меньше, чем
на платиновых, однако вследствие более низкого напряжения удель-
ные расходы электроэнергии не выше, чем на платиновых ано-
дах [110]. При использовании анодов из РЬО2 добавки хроматов
, приводят к снижению выхода по току [85, 111], поэтому применяются
добавки фтористых солей, которые предполржительно повышают
перенапряжение выделения кислорода на анодах из РЬО2 [112—
114]. На электродах из РЬО2 процесс проводят при плотности тока
до 2000—2500 А/м2, температуре 50—70 °C и напряжении на элек-
тролизере 4,5—5,0 В [114].
Предложена прямая схема получения перхлората натрия из
хлорида электролизом с перекисносвинцовыми анодами без проме-
жуточного выделения хлората. При степени превращения хлорида
в хлорат 99% достигается выход по току 65—76% [110].
Рассматривалась также возможность применения в качестве
анодного материала двуокиси марганца [97], однако сообщений
о промышленном использовании анодов из МпО2 не было.
В процессе окисления хлората до перхлората с изменением кон-
центрации хлората изменяются электрохимические показатели элек-
тролиза, в частности, снижается выход по току и возрастает удель-
ный износ анодов как платиновых [115, 116], так и из перекиси
свинца. Для уменьшения потерь выхода по току и материала анодов
процесс обычно проводят в каскаде электролизеров, последовательно
включенных по току жидкости. Так же, как и в производстве хлора-
тов, каскад обычно состоит из четырех-пяти электролизеров. По-
скольку электрохимические показатели процесса ухудшаются при
унижении концентрации хлората в электролите ниже 50 г/л на пла-
тиновых анодах и ниже 100 г/л на анодах из перекиси свинца, весь
процесс окисления разделяют на две стадии: продукционную и за-
вершающую очистную. На первой стадии концентрация хлората
натрия выше критической и электрохимические характеристики1
мало меняются. На завершающей стадии с понижением концентра-
ции хлората натрия снижается выход по току, возрастает доля
тока, затрачиваемого на выделение кислорода, и увеличивается
удельный расход анодов.
439
Ниже приведена примерная зависимость выхода перхлората
натрия по току (в %) на различных анодах от глубины срабатывания
хлората в электролите [68]:
Конечная концентрация
NaC!O3, г/л
5
80
Платиновые аноды
90
95
Аноды из РЬОг
85
90
о
При переработке электролитических щелоков на товарный про-
дукт — перхлорат натрия или перхлораты других металлов, полу-
чаемые
ющими
точного содержания хлората натрия. Примеси хлората натрия к пер-
обменным разложением перхлората натрия с соответству-
солями, необходима тщательная очистка растворов от оста-
Зависимость на-
ла электроли-
Рис. 8-5.
пряжения
зере от температуры.
хлорату снижают стабильность получа-
емых продуктов, что особенно важно для
перхлората аммония [117]. Очистка от
хлоратов обычно производится путем его
солянокислого разложения
NaC103 + 2НС1 = NaCl + i/2Cl2 +
+ С1О2 + Н2О (8.22)
Глубину срабатывания хлоратов и их
остаточную концентрацию в электролити-
ческих щелоках выбирают исходя из эко-
номических соображений, с учетом наи-
меньших затрат. Обычно остаточное со-
держание хлората составляет 5—10 г/л.
Электролиз проводят при слабокислом
электролите (pH ~6,5).
При электрохимическом получении перхлоратов в промышлен-
ных условиях показатели производства зависят от таких факторов,
как температура, pH, состав электролита и концентрации хлоратов
и перхлоратов, анодная плотность тока, материал анода и добавок
к электролиту. Теоретические исследования не позволяют в на-
стоящее время количественно рассчитать влияние этих факторов
на основные показатели процесса, поэтому для практического опре-
деления оптимальных условий работы пользуются эксперименталь-
ными данными.
Первоначально при окислении хлората до перхлората процесс
электролиза рекомендовали проводить при пониженных темпера-
турах (10—30 °C) с целью получения более высоких выходов по
току 1118]. При повышении температуры электролиза наблюдается
некоторое снижение выхода по току и увеличение расхода анодного
материала (платины, РЬО2) [5]. При этом повышается электро-
проводность электролита и снижается напряжение на электролизере.
На рис. 8-5 показана зависимость напряжения на ячейке электроли-
зера от температуры для электролизеров двух типов [68], работа-
ющих при различных температурах.
440
При повышении температуры сильно снижается напряжение
на электролизере и, несмотря на некоторое уменьшение выхода
по току, достигается снижение удельного расхода электроэнергии.
Поэтому рабочую температуру электролиза принимают с учетом
этих противоположно действующих факторов из экономических
соображений. Современные установки обычно работают при темпе-
ратуре 40—60 °C [68].
Рекомендуется работать со слабокислыми электролитами, чтобы
уменьшить потери выхода по току за счет разрядки ионов ОН“
с выделением кислорода [89]. При pH <7 выход по току практи-
чески не зависит от pH [97]. На анодах из РЬО2 выход по току полу-
чается более высоким в щелочных электролитах [119]. Имеются
сообщения [68] о том, что на платиновых анодах при pH 10
получали выход по току 95%.
Необходимо учитывать, что в прианодном слое электролита зна-
чение pH всегда значительно ниже, чем в объеме за счет разряда
ионов ОН- или образования ионов Н+ при разряде молекул воды,
или при взаимодействии с радикалами C10J в процессе получения
перхлората. Поэтому прианодный слой электролита в производствен-
ных условиях практически имеет кислую реакцию.
Для снижения расхода тока на выделение хлора на аноде и умень-
шения затрат кислоты предложено поддерживать pH электролита
путем его подкисления в процессе электролиза хлорной кисло-
той [120]. При этом расход НС1О4 на 1 т перхлората составит 4,8—
6,5 кг.
Как указывалось ранее, при любом из перечисленных выше меха-
низмов окисления хлората до перхлората получению высоких выхо-
дов по току должна способствовать высокая концентрация хлората
в электролите. По мере умень;
и
ения концентрации хлората в элек-
тролите выход по току должен снижаться, особенно после достиже-
ния предельной критической концентрации хлората. Зависимость
выхода по току от концентрации хлората при проведении электро-
лиза на платиновых анодах и на анодах из перекиси свинца без
добавок фторидов приведена на рис. 8-6 [68]. При введении добавок
фторидов выхода по току на анодах из РЬО2 значительно возрастают.
В табл. 8-8 приведены данные о зависимости выхода по току от кон-
центрации хлората на различных анодах.
Таблица 8-8. Выход по току при различной концентрации NaC103 [121]
Концентрация NaClО8, г/л Выход по токуг % Концентрация NaCIOa, г/л Выход по току, %
начальная конечная .начальная конечная
Платиновые аноды
602
293
197,6
100
39,8
3,9
87,4
82,4
65,4
Аноды из РЬО2
606
186
198
100
75
27,1
33,9
49,1
441
Плотность тока почти не влияет на выход по току, но напряже-
ние на электролизере и расход энергий сильно зависят от этой вели-
чины. На рис. 8-7 приведена зависимость напряжения на промышлен-
ных электролизерах [68] от плотности тока для платиновых анодов.
"Естественно, что напряжение на
его конструкции.
электролизере зависит также от
Конечная концентрация
NaClO3,a//z
и
Рис. 8-6. Зависимость выхода по
току от конечной концентрации
хлората натрия:
1 — платиновые аноды; 2 — аноды из
РЬОг без добавок фторидов.
Рис. 8-7. Напряжение на
электролизере при различ-
ной плотности тока.
Учитывая высокую стоимость платины, обычно работают на пла-
тиновыхг анодах при более высокой плотности тока 4—5 кА/м2,
несмотря на повышенный расход электроэнергии. При использова-
нии анодов из перекиси свинца плотность тока составляет 2—
2,5 кА/м2.
Общая схема производства перхлората натрия зависит также
от того, в каком виде должен выпускаться конечный продукт. Если
товарным продуктом является твердый перхлорат натрия, электро-
4
литические щелока после электролизеров упариваются и из них за-
тем кристаллизацией выделяют твердый кристаллический перхлорат
натрия. Условия кристаллизации перхлората натрия определяются
совместной растворимостью хлората и перхлората (рис. 8-8) [68L
Прямая линия, пересекающая поле рисунка, характеризует изме-
нение концентрации хлората и перхлората в ходе процесса электро-
лиза по мере окисления хлората.
Маточные растворы после кристаллизации могут вновь возвра-
щаться на стадию упаривания или же передаются обратно на элек-
тролиз. При такой схеме значительно снижаются требования к чи-
стоте электролитических щелоков и к возможным загрязнениям
их хлоратами, хроматами и другими примесями. При этом отпадает
необходимость очистки электролитических щелоков от остатков
неокисленного хлората, а также от хроматов (при платиновых ано-
дах) или фторидов (при анодах из РЬО2), вводимых в электролит для
снижения потерь выхода по току в процессе электролиза. Пока
442
содержание этих примесей ниже их совместной растворимости с пер-
хлоратом натрия при его температуре кристаллизации они остаются
в маточном растворе и достаточно полно удаляются при фильтрова-
нии и промывке кристаллов NaC104. Некоторое количество приме-
сей может оставаться в перхлорате натрия в виде отдельных включе-
ний капель маточника внутри кристаллов.
Растворимость, г ЫасЮд / ЮОг Н2О
Рис. 8-8. Растворимость в системе NaC103—NaC104—Н2О (R — ме-
тастабильная четверная точка для NaC103, NaC104, Н20 и льда).
В случае необходимости получения соли высокой чистоты при-
меняют перекристаллизацию технического продукта. При этом, как
указывалось ранее, маточные растворы возвращаются на пригото-
вление исходного электролита. При такой схеме в исходном электро-
лите возможно значительное содержание перхлората. В Процессе
электролиза не обязательно добиваться высокой степени конверсии
хлората в перхлорат, так как остаточный хлорат вновь возвращается
на электролиз с маточником после кристаллизации. Аналогично нет
необходимости очищать растворы от хроматов или фторидов, по-
скольку они также возвращаются с маточными растворами на ста-
дию приготовления электролита.
При увеличении остаточной концентрации хлоратов в электро-
лите, отбираемом из электролизеров, процесс можно вести с более
высоким выходом по току (стр. 441);
Выделяющийся на катоде водород всегда загрязнен кислородом.
При использовании платиновых анодов выход перхлората до току
выше и содержание кислорода в электролитическом газе ниже, чем
на электродах из двуокиси свинца.
443
На начальных продукционных стадиях окисления, пока концен-
трация хлората вн
се критической, содержание кислорода в водо-
и
роде в зависимости от условий работы составляет около 4—7%
на платиновых и около 5—12% на перекисносвинцовых анодах.
При дальнейшем снижении концентраций хлората в электролите
на очистной стадии окисления выход перхлората по току резко
снижается, а содержание кислорода в водороде соответственно
возрастает.
Водород из электролизеров очистной стадии очень сильно за-
грязнен кислородом и после разбавления выбрасывается в атмо-
сферу. Водород из электролизеров продукционной стадии может
быть использован после очистки от примесей хлора и кислорода,
аналогично тому, как это делается в производстве хлората натрия.
В процессе электролиза (особенно на очистных стадиях каскада)
на аноде параллельно с выделением кислорода образуются также
небольшие количества озона, который уходит с газами из электро-
лизера. К таким электролизным газам предъявляются повышенные
требования техники безопасности, они не должны попадать в атмо-
сферу производственного помещения.
Если перхлорат натрия используется для последующей перера-
ботки в перхлорат аммония, необходима тщательная очистка элек-
тролизных растворов от содержащихся в них загрязнений, лак как
маточные растворы после кристаллизации перхлората аммония
не могут быть, возвращены на электролиз из-за содержания в них
ионов аммония.
Обычно потребители предъявляют высокие требования к чистоте
перхлората аммония. Весьма нежелательна примесь хлората аммо-
ния, поэтому растворы перхлората натрия, поступающие на обмен-
ное разложение для получения NH4C1O4, должны быть тщательно
очищены от остатков неокисленного хлората.
При использовании перхлората натрия в производстве NH4C1O4
стремятся достичь более полного окисления хлората до перхлората,
чтобы при химической очистке растворов разрушалось меньшее
количество хлората. Остаточная концентрация хлоратов в электро-
химических щелоках после очистной стадии электролиза опреде-
ляется из экономических соображений и обычно не превышает 5—
10 г/л [7]. . ~
Процесс электролиза может проводиться как периодическим,
так и непрерывным способом. Периодический способ в настоящее
время не применяется. При непрерывном способе электролиза (так же,
как и в производстве хлоратов) применяют каскадное соединение
электролизеров с целью получения более высокого выхода перхло-
рата по току. Принципиальная технологическая схема производства
перхлората натрия электролитическим окислением растворов хло-
рата с выпуском твердого NaGIO4 приведена на рис. 8-9.
Имеется большое число патентов и предложений фирм по кон-
струкциям электролизеров для получения перхлоратов, однако
приведенные в литературе промышленные данные относятся только
444
к некоторым старым, маломощным конструкциям электролизеров.
Обычно применяют электролизеры без диафрагмы. Практически
все конструкции электролизеров, применяемых для получения пер-
хлоратов до последнего времени были монополярного типа. Однако
в последние годы появились предложения по биполярным кон-
струкциям.
Маточник
Рис. 8-9. Принципиальная технологическая схема производства
перхлората натрия с выпуском твердого NaC104:
1 — приемник растворов NaG10s; 2 — каскад перхлоратных электролизеров;
з — сборник раствора NaG104; 4 — выпарной аппарат; 5 — фильтрование рас-
твора; 6 — кристаллизатор; 7 — центрифуга; 8 — сушка NaG104.
Во всех старых конструкциях электролизеров предусмотрены
аноды из платины. В настоящее время применяются также платино-
in
титановые аноды, однако в последние годы платина все больше усту-
пает место анодам из двуокиси свинца, нанесенной на титан. Отме-
чается ограниченное применение анодов из двуокиси свинца на гра-
фитовой основе [68].
Выбор катодного материала зависит от материала применяемых
анодов. При использовании платиновых анодов наиболее удобны
стальные катоды; для предотвращения катодного восстановления
хлоратов применяют добавки солей хрома. .Для анодов из РЬО2
добавление хроматов недопустимо и рекомендуются катоды из нер-
жавеющей стали. Материалом для изготовления корпуса может
служить тот материал, что и для катодов; корпус электролизера
должен быть катодно поляризован. Части корпуса электролизера,
не защищенные катодной поляризацией, должны быть гуммированы
или футерованы. Для отвода тепла и поддержания низкой темпера-
туры электролизеры обычно снабжаются холодильниками. В по-
следних конструкциях электролизеров с биполярным включением
электродов предложено регулировать температуру путем интенсив-
ной циркуляции электролита через наружный теплообменник.
' На рис. 8-10 приведена одна из первых конструкций электроли-
зеров для получения перхлоратов. Электролизер состоит из бетон-
ного корпуса, в котором размещены цилиндрические электроли-
тические элементы с бронзовыми катодами; платиновые аноды
расположены внутри катода.
Такой же принцип устройства электролизера из отдельных ци-
линдрических электролитических элементов использован в электро-
лизере фирмы «Кардокс», изображенном на рис. 8-11. Катод электро-
лизера состоит из стальных труб, сваренных со стальной крышкой.
Внутри каждой трубы размещен платиновый анод. Для обеспечения
естественной циркуляции электролита трубы катода в верхней и
445
нижней части снабжены отверстиями. Электролизер имеет холо-
дильник.
В электролизере Шумахера (рис. 8-12) используются аноды
из платиновой жести, закрепленные между стеклянными стержнями.
Стальной корпус- электролизера снабжен устройствами для водя-
ного охлаждения [122—123].
Рис. 8-10. Схема устройства электролизера
(«Шедд», Франция):
I — аноды; 2 — катоды; 3 — подвод тока; 4 — изо-
лятор; 5 — бетонный корпус.
Рис. 8-11. Электролизер фирмы
«Кард оке»:
1 — стальная крышка; 2 — стенка
корпуса; 3 — нитевидный анод;
4 — трубка катода; 5 — охлади-
тель; 6 — отверстия для циркуля-
ции; 7 — изолятор.
Ле ретон электролита
/ Вход
fохлаждающей
/ воды____
Вход электролита
с
и, л u'v н ггажз
\ 22 И » 22ЧЙСТ » /МУГЯ/МГ»' 22 22
». 22 22 22" 22>j\ 22 22 22 JJ J} ' Ц-П*
с
d
С
1
т
Рис. 8-12. Электролизер Шумахера:
1 крышка; 2 — анодная сборка; 3 — стальной кожух; 4 — коробка для охлаждающей
воды; 5— трубки; 6 — анод из платиновой фольги; 7 — стеклянные стержни; 8 — каналы
длн охлаждающей воды; 9 — медные шины; 10 — охлаждающая рубашка.
В табл. 8-9 приведены некоторые показатели промышленных
электролизеров.
В промышленности в настоящее время применяются более мощ-
ные и совершенные электролизеры с нагрузкой до 25—50 кА, однако
достаточной информации о технических деталях в литературе нет.
>
446
Таблица 8-9. Показатели электролизеров для получения
перхлората натрия [7]
7
•'г
Показатели
4 Шедд Паси- фик Энжи- ниринг Американ. Поташ, корпорейшен 1 . г КарДокс
продук- ционная очистная
Пла-
тина
Биттер-
фельд
Пла-
тина
на меди
РЬ02
ft*’.
Материал
анода
Нагрузка, А
Плотность тока, А/м2
Напряжение, В* Л .
Бронза
1500—
3500
4500
Нерж,
сталь
1500
4,75
500
500
500
3100
3100
5200
Пла-
тина
12 000
3000
Выход по току, %
Температура, °C .
pH ..............
Расход платины, г
Режим работы
95
60
9—10
85
45
90
70
40—45
6,8
97
50
10
85-90
35
Непре-
рывный
Перио-
диче-
ский
Непре-
рывный
Концентрация, г/л
NaGlO3, начальная
NaGlOз, конечная
NaG104, начальная
NaG104, конечная
300
80
700
1100
700
0
1000
600
100
о
50,0
100
500
600
650
20
0
800
400—500
40—50
50—100
1000
Л
7
$•7
1 *
гЛ?
К
катода . .
,•5'
й
IW',
•i
и .
7
Предложены многосекционные электролизеры для
£
7^
производства
перхлоратов и хлоратов [124]. В таких электролизерах создается
внутренний каскад, позволяющий увеличить выход по току без
необходимости устройства каскада электролизеров. Такое устрой-
ство, по-видймому, может быть целесообразным только при очень
малом масштабе производства.
Предложены монополярные электролизеры с анодами из РЬО2
для,получения хлоратов и перхлоратов [125]. Электролизеры должны
работать при сравнительно невысокой плотности тока 1240 А/м2
с добавками NaF в количестве 0,2—2 г/л для увеличения выхода
по току.
Для электролизеров с платиновыми или платинотитановыми
анодами предложена работа со скоростью движения электролита
не менее 1 м/с при применении высокой плотности тока (более
2000 А/м2) [126], предложены электролизеры с защитой от корро-
зии футеровкой из пластифицированных смол [127] и для работы
с повышенными плотностями тока [128].
В последнее время разрабатываются конструкции биполярных
электролизеров с охлаждаемыми электродами [129], а также злектро-
лизерор с платинотитановыми анодами и полностью заполненными
447
электролитом ячейками [130]. В последней конструкции пред-
усмотрено отделение газов от электролита в наружном сепараторе,
установленном на пути вне
ней циркуляции электролита.
и
Применение биполярных электролизеров с интенсивной наружной
циркуляцией электролита улучшает условия безопасности работы
электролизеров, так как внутри электролизера отсутствуют газовые
пространства, в которых могут образоваться взрывоопасные газовые
смеси. Облегчается также регулирование температуры и PH элек-
тролита, поскольку можно легко автоматизировать подачу воды
на холодильник и поддерживать необходимое значение pH. При
интенсивной циркуляции все эти операции централизуются в одной
точке на линии внешней циркуляции.
Перхлорат аммония
Из всех солей хлорной кислоты наибольшее распространение
получил перхлорат аммония, используемый в качестве окислителя
и составляющий около 75 вес. % смесевых твердых ракетных то-
плив [5, 7].
Перхлорат аммония кристаллизуется из водных растворов в виде
бесцветных, безводных кристаллов орто- и ромбической структуры.
Он стабилен, малогигроскопичен и в чистом виде безопасен.
Растворимость NH4C1O4 в воде и совместная растворимость
NH4C1O4 с NaC104 и NaCl приведена в табл. 8-10—8-12.
Таблица 8-10. Растворимость NH4CIO4 в воде [131, 132]
Темпера-
тура, °C
Содержание NH<C1O*
вес. %
г/100 г НгО
Темпера-
тура, °C
Содержание £Ш*С10<
вес. %
г/100 г Н2О
—2,72
О
15,2
25
34
9,84' (9,8)
10,73 (10,74)
15,95
19,89 (20,02)
23,32
10,91 (10,86)
12,02 (12,03)
18,98
24,83 (25,03)
30,41
45,1
55,2
65,1
75
84,7
27,64 (28,02)
31,55
35,37
39,05 (39,45)
42;54
38,20 (38,93)
46,09
54,73
64,07 (65,15)
74,03
Примечание. В скобках приведены данные [132].
Перхлорат аммония в промышленности получают практически
двумя способами, из которых наиболее простым является нейтрали-
зация хлорной кислоты аммиаком? [134]
НС1О4 +NH3 = NH4C1O4 (8.23)
Такой метод предусматривает получение хлорной кислоты элек-
трохимическим окислением НС1 или хлора в электролите из хлорной
кислоты. Получаемая кислота может быть загрязнена ионами хлора
и при использовании для производства очень чистого перхлората
аммония должна быть очищена электролитически или отгонкой при-
месей в виде НС1. При очистке хлорной кислоты электролитически
448
*
г мл;
Л’/ •
ч • т'
1
Таблица 8-11- Совместная растворимость NaC104 и NH4CIO4 в воде [432]
Концентрация,
г/100 г H2O
Концентрация,
г/100 г HaO
••Лгу;-
O'
NaC104
NH4CIO4
Твердая фаза
Твердая фаза
4
NaC104
NHaC104
60 °C
0
12,28
40,01
121,13
217
208,6
25
19,92
14,77
9,46
8,02
4,86
NH4C1O4
' nh4cio4 +
+ NaC104-H20
NaC104 • H2O
0
19,96
43,86
74,5
154,97
288,33
289,11
50,6
41,67
31,58
17,14
NH4C1O
NH4C104 + NaCl О
NaG104
Таблица. 8-12. Совместная растворимость NH4CIO4 и NaCl в воде [133]
Концентрация,, г/100 г Н2О Твердая фаза Концентрация, г/100 г Н2О
Nad * NH4CIO* NaCl NH4CIO4
Твердая фаза
0,09
7,16
13,9
21,5
30
35,15
35,4
35,9
13,3
19,2
30,2
NH4C1O4
35,4
36,2
36,2
36,1
26,8
10,8
0
50 °C
NaCl + NH4C104
NaCl
20
20,3
14,2
5,8
0
41,8
38,3
36,6t
35,8
80 °C’
NaCl + NH4C1O
50 °C
NaCl
0
21,1
NH4C1O
35,45
36,7
37,6
66,7 ]
60,4 J
58,5 J
57,45
10,6 1
0 f
NH4C1O4
NaCl + NH4C10
NaCl
г
2
О
О
и
на последних стадиях, при очень малом содержании ионов G1",
практически происходит разложение воды на Н2 и О2 и процесс
окисления хлор-иона до хлорной кислоты идет с малым полезным
использованием тока, при повышенном расходе платиновых анодов.
Получаемые после нейтрализации концентрированные растворы
перхлората аммония охлаждают для выделения кристаллов, которые
отделяют на центрифуге или фильтре от маточного раствора^
Маточные растворы после упаривания также возвращаются
на кристаллизацию.
Целесообразность такого метода производства NH4C1O4 зависит
от производственных затрат на электрохимическое получение хлор-
ной кислоты. Существенным недостатком этого способа является
29 Заказ 843
449
необходимость применения платины в качестве анодного материала.
Чтобы избежать затрат платины, предложено получать перхлорат
аммония через хлорную кислоту, которую в свою очередь полу-
чают [135] обработкой водного раствора перхлората натрия серной
кислотой и последующей отгонкой хлорной кислоты в вакууме
15—400 мм рт. ст. После нейтрализации 50—72%-ной кислоты аммиа-
ком образующиеся растворы перхлората аммония передают на ста-
дию кристаллизации.
Второй способ получения перхлората аммония заключается
в обменном разложении растворов перхлората натрия аммонийными
солями различных кислот. Этот способ нашел преимущественное
развитие в настоящее время в ряде стран, поскольку разработаны
экономичные способы получения NaC104, в том числе и методы без
использования платины в качестве анодного материала. Предло-
жено [136] много вариантов обменного способа получения перхло-
рата аммония. NH4C104 может быть получен двойным разложением
растворов перхлората натрия хлористым аммонием [5, 137] по
реакции
NaC104+NH4Cl = NH4C104+NaCl (8.24)
f
f * •
После охлаждения раствора выделяются кристаллы NH4C1O4,
а хлорид натрия остается в растворе. При упаривании маточных
растворов NaCl отделяется в виде кристаллов и после промывки
от NH4C1O4 может быть возвращен в производственный цикл.
Этот метод был видоизменен [138] путем замены хлористого аммо-
ния соляной кислотой и аммиаком. При этом растворы перхлората
натрия обрабатывают соляной кислотой или хлористым водородом,
а также аммиаком по реакции:
NaC104 + НС1 + NH3 = NH4C1O4 + NaCl (8.25)
Получаемый в качестве отхода NaCl не может быть использован
в качестве товарного продукта, так как даже после промывки за-
грязнен перхлоратом аммония. При совмещении производств пер-
хлората и хлората хлорид натрия может быть использован в произ-
водстве хлората, что позволяет иметь производство без неисполь-
зуемых отходов.
Предложено проводить обменное разложение перхлората натрия
с бикарбонатом аммония [139].
NaC104+NH4HCO3 = NH4C1O4+NaHCO3 ' (8.26)
Выделяемый при этом бикарбонат натрия может быть переведен
в кальцинированную соду.
Процесс может проводиться также с карбонатом аммония в атмо-
сфере СО 2 под давлением до 20 ат [140].
Вместо бикарбоната аммония двойное разложение растворов
NaC104 может проводиться газообразной двуокисью углерода и
аммиаком [141].
2NaC104 + 2NH3 + СО2 + 2Н2О ~ 2NH4C1O4+Na2CO3 (8.27)
450
Л'
’iF>
sT’
ik! .<•> •
' -»
:V
<•
fr;
w
•Джу
Ж
э:
В
После отделения кристаллов перхлората аммония маточные
растворы обрабатывают СО2 и образующийся бикарбонат выпадает
в осадок. Преимуществом всех методов обмена с карбонатами аммо-
ния является получение в качестве отхода кальцинированной соды,
однако масштабы производства перхлората аммония несоизмеримы
с масштабами производства кальцинированной соды. Поэтому при
сравнительно малом масштабе производства дополнительные за-
траты, связанные с использованием карбонатной схемы, не окупаются
стоимостью получаемой кальцинированной соды.
Предложено обрабатывать перхлорат натрия фосфорной кислотой
и аммиаком [142] с получением перхлората аммония и натрийаммо-
нийфосфата
NaC104 + Н3РО4+2NH3 = NH4C1O4+NaNH4HPO4 (8.28)
Взаимодействие между перхлоратом натрия и солью аммония,
например сульфатом аммония, можно проводить в безводном рас-
творителе, например в жидком аммиаке [143], с получением без-
водного NH4C1O4. В жидком аммиаке можно получать перхлорат
аммония взаимодействием перхлората калия с хлористым аммо-
нием [144]. При этом хлористый калий выпадает в осадок, а перхло-
рат аммония выделяют из раствора.
Из многочисленных вариантов проведения обменного разложения
перхлоратов' щелочных металлов с солями аммония промышленное
применение получил только обмен с хлористым аммонием, а также
обработка перхлората натрия соляной кислотой и аммиаком. Раз-
делению продуктов реакции — NH4C1O4 и NaCl — способствует
более сильная температурная зависимость растворимости NH4C1O4
в воде по сравнению с зависимостью растворимости поваренной
соли [133].
Принципиальная схема производства перхлората аммония об-
менной реакцией между перхлоратом натрия, аммиаком и соляной
кислотой приведена на рис. 8-13 [5]. При проведении процесса осо-
бые требования предъявляются к чистоте исходных реагентов и рас-
творов во избежание накопления загрязнений в производственном
цикле. Растворы перхлората натрия, поступающие на обменное
разложение, подвергаются тщательной очистке от примеси хроматов,
тяжелых металлов и хлоратов.
К чистоте соляной кислоты и аммиака также предъявляются
повышенные требования.
При обменной реакции получают раствор, содержащий примерно
30% перхлората аммония и 15% хлористого натрия. Температура
в реакторе поднимается за счет тепла нейтрализации соляной кис-
лоты аммиаком. Для регулирования температуры и предотвращения
ее чрезмерного повышения применяют рециркуляцию маточных '
растворов после кристаллизации через реактор. Температуру в реак-
торе поддерживают на таком уровне, чтобы не достигалось насыще-
ние раствора по перхлорату аммония и он не выпадал в осадок.
Значение pH растворов после реактора поддерживается на уровне 5,5.
29* 451
*
f
Применяется двухступенчатое регулирование pH, на первой сту-
пени — изменением количества подаваемой в реактор соляной кис-
лоты* на второй ступени — корректировкой pH в промежуточном
сборнике, подачей нужного количества аммиака.
Большое значение придается кристаллизации перхлората аммо-
ния, которая может проводиться как периодическим, так и непре-
рывным способом. Регулирование размеров кристаллов и их ка-
чества достигается изменением степени перенасыщения растворов
Рис. 8-13. Принципиальная схема производства перхлората аммония по обмен-
ной реакции между перхлоратом натрия, аммиаком и соляной кислотой: t
1 — бак для приема NaG10< из цеха электролиза; 2 — бак для очистки раствора от хроматов
и тяжелых металлов; 3 — бак для очистки раствора от хлоратов; 4 — реактор; 5, 10 — кри-
сталлизаторы; 6 — центрифуги для кристаллов NH4G1O4; 7 — аппарат для выпарки маточ-
ных растворов; 8 — центрифуга кристаллов NaCl; 9 — бак для растворения.
в кристаллизаторе. При применении непрерывной схемы кристал-
лизации для этой цели регулируют температуру кристаллизации,
скорость рециркуляции жидкости и высоту слоя кристаллов; при
периодическом способе кристаллизации — изменяют скорость охла-
ждения. Применяются разные типы кристаллизаторов. В литературе
имеются указания на применение вакуум-кристаллизаторов с ре-
циркуляцией жидкости [5].
Предложены разнообразные методы и конструкции аппаратов для
получения перхлоратов аммония и калия в виде округлых кристал-
лов [145]. Округлые кристаллы имеют преимущества перед обычными
кристаллами в производстве смесевых ракетных топлив. Они улуч-
шают технологические свойства топливных смесей.
Перед подачей на центрифугу пульпа сгущается и содержит при-
мерно 40% кристаллов перхлората аммония и около 10% перхло-
рата аммония и 13% хлорида натрия в растворе. После центрифуги-
рования получают перхлорат аммония влажностью около 5%.
Если требования к чистоте перхлората аммония невысоки, кристаллы
после центрифуги промывают и передают на сушку. Для улучшения
отмывки кристаллов от маточных растворов может применяться
репульпация осадка в чистом растворе перхлората аммония. При
повышенных требованиях к, чистоте необходима перекристаллизация
продукта, как это показано на рис. 8-13. Маточные растворы после
452 '
Л','.'.
/перекристаллизации постепенно загрязняются примесями (в основ-
ном хлоридами) и периодически сбрасываются в цикл первой кри-
сталлизации.
Маточный раствор после первой кристаллизации поступает на
выпарку, где по мере удаления из раствора воды выпадает хлористый
натрий. Процесс выпарки регулируется так, чтобы перхлорат аммо-
ния оставался в растворе, вновь возвращаемом на первую кристал-
лизацию. При центрифугировании кристаллов хлористого натрия
нельзя охлаждать растворы до температуры начала кристаллиза-
ции NH4G1O4. Иначе выделяемый хлорид натрия содержит большое
количество NH4G1O4,
Поваренную соль после промывки от перхлората аммония и
очистки от ионов аммония возвращают на производство хло^та
натрия.
Кристаллы перхлората аммония после центрифугирования и
промывки поступают на сушку и рассев. Просеянный NH4G1O4
загружают в контейнеры с силикагелевыми патронами и отправ-
ляют потребителям. !
Для облегчения и удешевления хранения и транспортирования
перхлората аммония его сушку можно производить на месте потре-
бления. В этом случае транспортируют влажный перхлорат аммония,
содержащий около 5% влаги. При этом снижаются требования к таре
и складским помещениям.
Важно исключить возможность загрязнения перхлората аммония
продуктами коррозии материалов аппаратуры, трубопроводов и
арматуры. Поэтому для изготовления аппаратуры и трубопроводов
необходимо применять материалы, стойкие к растворам перхлоратов
при температурах, значении pH и содержании примесей, характер-
ных для этого производства.
Преимущественное применение в качестве конструкционных
материалов нашел титан и его сплавы, специальные стали типа ЭИ-448,
ЭИ-924. Пригодны также эмалированные аппараты и трубопроводы,
однако при этом необходимо следить, чтобы в перхлорат аммония
не попадали частички эмали при возможных сколах или местном
разрушении защитного слоя эмали.
Перхлораты других металлов
Из разнообразных перхлоратов металлов в значительных коли-
чествах выпускаются только перхлораты лития и калия. Прочие
перхлораты используются как реактивы и производятся в крайне
ограниченных количествах в основном из хлорной кислоты и соот-
ветствующих окислов или гидроокисей металлов.
Перхлорат магния используется как эффективный осушитель
и производится в ограниченных масштабах также в основном через
хлорную кислоту.
Некоторые перхлораты (Mg, Zn) можно получать растворением
соответствующих металлов в хлорной кислоте, например, при
453
нагревании висмута в концентрированной хлорной кислоте про-
цесс протекает со взрывом [54, 146].
Перхлорат лития содержит наибольшее количество кислорода
по сравнению с другими перхлоратами. Поэтому ЫС104 представляет
интерес как источник чистого кислорода [147] и возможный окисли-
тель ракетных топлив.
Основные свойства ЫС1О4 были приведены ранее (стр. 433).
Изменение растворимости перхлората лития с температурой и области
существования различных гидратов ЫС104 приведены в табл. 8-13;
совместная растворимость LiG104 и NaCl дана в табД. 8-14.
Таблица 8-13* Растворимость LiC104 в воде [148J
ЫС1О4
ысю4
Твердая фаза
Твердая фаза
О
10
20
25
30
40
64,6
77,9
89,2
92,3
94,3
95,1
94,8
93,2
29,9
32,88
35,95
37,48
38,87
41,97
50
55
60
62,5
65
66,32
66,67
70
70,3
42,65
48,99
56,13
59,95
63,59
72,32
100
122,22 •
150
166,67
185,7
196,9
200
233,3
236,7
LiG104'3H20
92,53 *
93,2
97,3
108,9
120,7
148,5
149,0
149,3
145,75 *
167,5
172
236
(Т. пл.)
70,33
70,5 '
71
72,8
75
85
85,45
87,5
91,04
91,11
100
237
239 .
244,8
267,6
300
LiC104-3H20 4
+ LiC104H20
LiC104H20
LiC104H20 4
+ LiC104
LiC104
LiC104
I
• Тройная точка.
Перхлорат лития гигроскопичен. При 25 °C и относительной
влажности 40% он погло
аёт за 1 ч0,1% воды [76], при дальнейшем
и
хранении на влажном воздухе расплывается.
Перхлорат лития может быть получен электрохимическим окис-
лением хлората лития [149]. Электролиз проводят при плотности
тока 2 кА/м2 и температуре более 20 °C. Возможно также прямое
окисление хлорида лития до перхлората без стадии промежуточного
выделения хлората лития [150].
Предложено много вариантов получения перхлората лития через
хлорную кислоту или ее соли.
Перхлорат лития можно получать действием хлорной кислоты
на гидроокись, карбонат или хлорид лития с отгонкой образую
;ейся
п
при этом HG1 при нагревании реакционной смеси.
!L
Ш* Таблица 8-14* Совместная растворимость LiClO* и NaCl [5]
.у
Концентрация,
г/100 г H2O
Концентрация,
г/100 г Н2О
Твердая фааа
Твердая фаза
•а*!' V
F?-.
LiGlO*
NaGl
LiGlO
NaGl
20 °C
0
30,39
49,89
51,42
56,13
♦ 0
47,53 '
68,86
68,83
69*14
72,32
35,87
22,02
14,94
36,37
23,38
19,18
14,66
6,76
0
NaCl
0
36,83
63,15
92,56
37,12
26,49
21,22
16,56
60 °C
NaCl
LiC104-3H,0
40 PG
90,78
90,4
93,8
11,36
6,42
0
NaCl +
+ LiC104;3H20
ЫС1О4 -ЗН2О
NaCl
80 °C
+ ЫСЮ4 ЗН2О
LiC104-3H»0
0
30,52
66,74
107,7
133,5
129,2
126,4
38,3
21,9
16,8
15,3
10,75
5,5
0
NaCl
LiC104-3H20
И
У
0
При воздействии гидроокиси или карбоната лития на перхлорат
аммония можно получить чистый перхлорат лития [151]. Для уда-
ления аммиака реакцию между перхлоратом аммония и LiOH ведут
при 150—250 °C, при проведении реакции с карбонатом лития,
температура поддерживается на уровне 250—300 9С.
Предложена также обменная реакция между перхлоратом натрия
и|хлоридом лития, смешиваемыми в стехиометрическом соотношении
Таблица 8-15• Растворимость КС1О4 в воде |5]
Темпера-
тура, °C
Плот-
ность,
г/см’
Растворимость
КС104
вес. %
г/100 г
Н8О
Темпера-
тура, °C
Плот-
ность,
г/см®
Растворимость
KG1O*
вес. %
г/100 г
Н2О
0
10
15
20
25
30
40
50
60
70
1,005
1,0076
1,0085
1,0096
1,017
0,75
1,05
1,33
1,65
2,03
2,5
3,6
4,9
6,8
9,2
0,76
1,06
1,35
1,67
2,07
2,57
3,74
5,15
7,3
10,1
75
80
90
100
120
140
180
200
225
250
265
1,036
1^0681
10,36
11,8
15
18,2
25
32,5
46
52,5
60
67
70
11,56
13,4
17,7
22,25
33,3
48,1
85,2
110,5
150
203
233
455
f + %
Таблица 8-16- Совместная растворимость KCIO4 и NaCl [153]
* -- <
Плот-
ность,
г/см3
Концентрация
г/100 г HtO
Твердая
фаза
' КСЮ* NaCl
Плот-
ность,
г/см3
Концентрация
г/100 г Н,О
КСЮ* NaCl
Твердая
фаза
1,005
1,214
1,209
1,013
1,207
J
1,198
1,017
1,205
1,185
При 0 °C
0,76 0
1,01 35,86
0 35,9'
П р и 25 °C
2,07 О
2,22 35,73
0 36,3
При 50 °C
4,95 0
4,5 36,52
КС1О4
ксю4 +
+ NaCl
NaCl
КС104
ксю4 +
+ NaCl
NaCl
КСЮ4
КС1О4 +
+ NaCl
NaCl
1,036
1,207
1,176
1,068
1,116
1,153
1,19
1,216
1,201
1,183
1,164
При 75 °C
11,55 О
9,32 41,47
0. 37,9
При 100 °C
22,2
19,75
17,6
15,92
14,51
11,99
5,1
О
КС1О4
КС104 +
+ NaCl
NaCl
О
10,44)
20,751
30,81 J
38,37
62,99)
38,87)
40 J
КС1О4
ксю4 +
+ NaCl
NaCl .
NaCl
О
[152]. Продукты реакции *- перхлорат лития и хлорид натрия —
разделяются дробной кристаллизацией.
Перхлорат калия плохо растворим в воде. В табл. 8-15 приведена
растворимость КС1О4 в воде и в табл. 8-16 совместная растворимость
перхлората калия и хлорида натрия.
Рис. 8-14. Принципиальная схема
производства перхлората калия
обменным методом:
1 — приемник растворов NaClO3; 2 —
каскад перхлоратных электролизеров;
3 — сборник растворов > NaClO*;
4, 11 — кристаллизаторы КС1О4;
5, 12 — центрифуги для кристаллов
КСЮ*; 6 — аппарат для выпарки
маточников первой кристаллизации;
7 — центрифуга для NaCl; 3 — рас-
творитель КС1; 9 — растворитель
КС1О4; 10 — фильтр раствора КС1О4;
13 — сушка. -
Перхлорат калия может быть получен электрохимическим окис-
лением [154]. Вследствие очень малой растворимости производство
его электролизом хлорида или хлората калия связано с образова-
нием твердой фазы в электролизерах.
Обычно перхлорат калия получают обменным разложением пер-
хлората натрия с хлоридом калия.
NaC104+KCl==KC104+NaCl (8.29)
456/
!
*
де
fc
’V
V:
ii.^
Г,
.> ।
V.:
Процесс проводят при повышенной температуре таким образом,
Втобы перхлорат калия оставался в растворе, а значительная часть
(Образовавшегося хлорида натрия выпадала в осадок. После отде-
ления NaCl растворы охлаждают и отфильтровывают кристаллы
^перхлората калия, которые далее подвергаются сушке. Хлорид
^натрия после промывки может быть возвращен на производство
Клората^натрия, айалогйчно тому, как это делается в производстве
Г " —
ж
м
WH4C1O4.
J Принципиальная схема производства перхлората калия пока-
зана на рис. 8-14.
ПЕРХЛОРАТЫ НЕМЕТАЛЛОВ
t г ь
Хлорная кислота может образовывать перхлораты с различными
соединениями. Интерес к таким перхлоратам связан с высоким
содержанием в них кислорода и большим тепловым эффектом, полу-
чаемым при использовании этих соединений в качестве окислителей
в ракетном топливе. К перхлоратам неметаллов можно отнести также
Дм перхлорат аммония, описанный ранее.
| Свойства некоторых наиболее интересных неметаллических пер-
хлоратов приведены в табл. 8-17 [76].
Таблица 8-17- Свойства неметаллических перхлоратов
В
bf.- K ?
Свойства .
NO2C104 NOC1O4 n2h<* •2НС1О* N2H4. • НСЮ4 NHsOH* •C104
CN3He-
• сю4
О
Ж? Ж
fe.
Ж-;’?
Ж'
Ж
fe
Ли?1
Содержание кислоро-
да, вес. % . . .
Молекулярный вес
Температура, °C
плавления . .
фазового превра-
щения . . .
разложения . .
Плотность при 25 °C,
г/см3 . . . . . .
Теплота образования
при 25 °C
ккал/моль . . .
ккал/г-атом О2
66,0
120
•j'8,88
61,8
100
2,169
-41,8
54,9
232,978
170
—70,1
132,513
137
145
1,939
59,9
133,497
' 40
159,54
240
180
180
350
74,1
—66,5
-13,3
™10^6
некоторыми нео ргани-
носкими и органическими кислотами образуются солеобразные
соединения, получившие название ацилперхлоратов.
Из сильных неорганических кислот наиболее стойкие соединения
получаются из азотной и азотистой кислот. Соответствующие соеди-
нения фосфорной кислоты менее стабильны, а соединение хлорной
кислоты с серной Н 2SO4-2HC1O распадается на исходные кислоты
при температурах выше —92 °C.
Перхлорат нитрония — белое кристаллическое вещество, образует
моноклинные кристаллы. При комнатной температуре давление
ларов перхлората нитрония ниже 0,05 мм рт. ст.
J
При взаимодействии хлорной
кислоты
457
л
Перхлорат нитрония очень гигроскопичен: в течение 2 ч пребы-
вания на влажном воздухе поглощает до 25% влаги [155]. При этом
перхлорат нитрония гидролизуется, распадаясь на хлорную и азот-
ную кислоты:
NO2C1O4+H2O3=HNO3 + HC1O3 (8.30)
Получающаяся при гидролизе хлорная кислота образует гидраты.
Перхлорат нитрония хорошо растворяется в безводной азотной
кислоте; в нитрометане растворимость при комнатной температуре
составляет Q,08 М, а в четыреххлористом углероде и в хлороформе
0,01 М. В отсутствие влаги NO2C1O4 достаточно устойчив и не чув-
ствителен к удару и трению; заметное разложение его начинается
при температурах выше 70 °C. При нагревании более 135 °C быстро
разлагается без взрыва с выделением NO2.
Перхлорат нитрония бурно взаимодействует с большинством
органических соединений уже при низкой температуре, в ряде
случаев реакция сопровождается самовоспламенением и взрывами.
Поэтому при работе с перхлоратом нитрония необходимо тщательно
избегать контакта его с органическими соединениями. Перхлорат
нитрония совмещается с ограниченным ассортиментом продуктов,
в том числе с нержавеющей и черной сталью, стеклом, тефлоном,
полиэтиленом, непластифицированным поливинилхлоридом, фтор-
углеродными маслами и смазками [76].
Токсическая активность его определяется действием продуктов
распада, т. е. HNO3, НС1О4, С1О2, окиси азота. Имеются указания
о возможности использования перхлората нитрония в качестве
окислителя в ракетных топливах [76, 155].
Перхлорат нитрония может быть получен взаимодействием без-
водных азотной и хлорной кислот [155, 156].
HNO3 + 2HC1O4=NO2C1O4+HC1O4.H2O (8.31)
При этом выделяется смесь кристаллов перхлората нитрония
и моногидрата хлорной кислоты. Перхлорат нитрония отделяют
дробной кристаллизацией из его растворов в безводной азотной кис-
лоте или в нитрометане. Остатки маточного раствора после филь-
трования могут быть удалены отгонкой нитрометана или хлориой
и азотной кислот при нагревании в вакууме.
При взаимодействии азотной кислоты с хлорной молекула азот-
ной кислоты после присоединения протона теряет молекулу воды,
образуя ион NO2
HNO3 + H+ = NO++H2O (8.32)
Поэтому формула продукта, образуемого взаимодействием хлор-
ной и азотной кислот — перхлората нитрония — записывается
в виде NO|-C1O4.
Чистый перхлорат нитрония может быть получен взаимодействием
безводной хлорной кислоты с избытком пятиокиси азота [8, 1551
N205+3HC104=2N02C104+HC104-H20 (8.33)
2N2O5 + нею* • Н2О = N02C104-f- 3HNO3 (8.34)
458
Разработан способ получения перхлората нитрония при реакции
двуокиси хлора и озона с окислами азота [157].
2С102+Кл04+4О3=2N02C104+40 2
(3.35)
Имеются сообщения [158] о производстве перхлората нитрония
этим способом на опытно-промышленной установке для испытания
его в качестве окислителя в ракетных топливах. Взяты патенты на
производство перхлората нитрония [159].
Предложено получать сложные соли перхлората нитрония и
пиридина, которые совмещаются со многими органическими, соеди-
нениями [160].
Перхлорат нитрозила— NO-G1O4 можно рассматривать как
ацилперхлорат азотистой кислоты. Перхлорат нитрозила образует
бесцветные кристаллы орторомбической формы, аналогичные кри-
сталлам перхлората оксония. Он гигроскопичен, под действием воды
разлагается с образованием хлорной кислоты и выделением окислов
азота, со многими органическими веществами энергично реагирует
с самовоспламенением и взрывом. Перхлорат нитрозила термически
менее стоек, чем перхлорат нитрония.
Перхлорат нитрозила может быть получен взаимодействием без-
водной хлорной кислотй с трехокисью азота
N203+3IIC104=2NOC104+IIC104. Н20
(8.36)
Можно также получить NO -С104, пропуская NO2 или эквимоле-
кулярную смесь NO и NO 2 через водный раствор хлорной кислоты
концентрацией выше 53% [161] или через смесь водного раствора
NaG104 с концентрированной серной кислотой [162].
С гидразином хлорная кислота образует два перхлората. Моно-
перхлорат гидразина N2H4*HG1O4 получают нейтрализацией
гидразина или -гидразин-гидрата хлорной кислотой до pH = 3,2.
N2H4-HC1O4— белое кристаллическое вещество, легко детониру-
ющее от удара или трения. Чувствительность безводного перхлората
к удару аналогична чувствительности инициирующих веществ.
Диперхлорат гидразина образуется аналогично моноперхлорату
полной нейтрализацией гидразина или гидразин-гидрата [163]
хлорной кислотой. При этом можно получить продукт чистотой
более 99%. В качестве обычных примесей в диперхлорате гидразина
присутствуют 0,15—0,40% моноперхлората и 0,04—0,22% свобод-
ной хлорной кислоты [164].
Предложен способ получения перхлората гидроксиламмония
. для применения в качестве окислителя [165], сообщается о произ-
водстве опытных партий перхлората гуанидина для тех же целей [166].
С органическими соединениями хлорная кислота образует пер-
хлораты аминов, сложные эфиры и др. О практическом применении
этих соединений в литературе нет сообщений.
Литература
1. S/tadion F., Gilberts Ann. Phys., 52, 197 (1816).
2. S t a d i о n F., Ann. chim. phys., 8, № 2,-406 (1818).
3. Serullas G., Ann. chim. phys., 45, № 2, 270 (1830); 46, № 2, 297, 323
(1831).
4. Швед. пат. 3614 (1890).
5. Шумахер И., Перхлораты. Свойства, производство и применение.
Пер. с англ. М., Госхимиздат, 1963.
6. Швед. пат. 8487 (1897).
7. W ihn a cker К., К itchier L., Chemische Technologic. Bd. I, Anor-
ganische Technologic, Munchen, 1970.
8. P о c.o ловскийВ. Я., Химия безводной хлорной кислоты, М., «Наука»,
1969; Исследование высших кислородных соединений хлора. Докт. дисс.,
ИОНХ АНСССР, 1969.
9. Зиновьев А. А., Бабаева В. П., ЖНХ, 3, № 6, 1428 (1958).
10. Зиновьев А. А., Росоловский В. Я., ЖНХ, 5, 2564 (I960).
11. Акишин П. А., В и лк ов Л. В., Р о с о л о в с к и й В. Я., Кристал-
лография, 4, 353 (1959). -
12. Щ у к а р е в С. А., Андреев С. Н., Б ал и я е в а Т. Г., ДАН СССР,
144, 606 (1962); Вести. ЛГУ, Сер. физ.-хим., № 4, 128 (1962).
13. Hahtzsch А., Вег., 60, 1933 (1927).
14. Taketa Т., J. Sci. Hiroshima Univ., А19, 133 (1955).
15. Кривцов Н. В., Росоловский В. Я., Зиновьев А. А.,
ЖНХ, 5, 772 (1960).
16. В о р о б ь е в А. Ф., П р и в а л о в а Н. М., М о н а е нк о в а А. С., Ску-
ратов С. М., ДАН СССР, 135, 1388 (1960); Воробьев А. Ф., П р и -
в ало в а Н. М., ЖФХ, 34, 1142 (1960); Скуратов С. М., Во-
робьев А. Ф., Привалова Н. М., ЖНХ, 7, 677 (1962).
17. J о h n s о n W- Н., Gilliland A. A., J. Res. Nat. Bur. Stand.,
65A, 63 (1961).
18. G i 1 l i 1 a n d A. A., Johnson W. H., J. Res. Nat. Bur. Stand., 65A,
67 (1961).
19. V a nd erf.ee С. E., Swanson J. A., J. Phys. Chem., 67, 285 (1963).
20. Pitzer ’K.S., J. Am. Chem. Soc., 60, 1828 (1938).
21. Витку M. M., H e p 1 e r L. G., J: Phys. Chem., 64, 686 (1960).
22. Ш и д л б в с к и й А. А., Семишин В. И., Шмагин Л. Ф.,
ЖПХ, 35, 756 (1962).
23. Gilliland A. A., J. Res. Nat. Bur. Stand., 66A, 447 (1962J.
24. P о с о л о в с к и й В. Я., Кривцов Н. В., Зиновьев А. А.,
ЖНХ, 5, 778 (1960).
25. 3 и н о в ь е в А. А., Р о с о л о в с к и й В. Я., ЖНХ, 3, 2382 (1958).
26. Т г о w Ь г i d g е J. С., Westrum Е. F., J. Phys. Chem., 68, 42 (1964).
27. Dahl A. J., Trowbridge J.C., Taylor R. C., Inorg. Chem.,
2, 654 (1963).
28. Brickwedde L. H., J. Res. Nat. Bur. Stand., 42, 309 (1949).
29. Wolf G., C h r i s t of z i k P-, J. prakt. Chem., 1, № 4,.237 (1955).
30. Mascherpa G., Pavia A., Potier A., C. r., 254, 3212 (1962).
31. Ma scher pa G., C. r., 257, 3414 (1963).
32. V о 1 m e r M., Liebigs Ann., 440, 200 • (1924).
33. Richards R. E., S m i t h J. A. S., Trans. Faraday Soc., 47, 1261 (1951).
34. Kakiuchi Y., Shono H. et al., J. Chem. Phys., 19, 1069 (1951).
35. Komatsu H., Kigochi K., J. Phys. Soc. Japan, 7, 102 (1952).
36. Kakiuchi Y., Komatsu H., J. Phys. Soc. Japan, 7, 380 (1952).
37. M a r k h a m A. E., J. Am. Chem. Soc., 63, 874 (1941).
38. 3 и н о в ь e в А. А., Б а б a e в а В. П., ЖНХ, 2, 2188 (1957).
39. Росоловский В. Я., Зиновьев А. А., Прохоров'!!* А.,
ЖНХ, 5, 692 (1960).
40. К oh пег Н., Gressmann М. L., Z. phys. Chem., 144, 137 (1929).
460
ЯИЬ . Усанович М. И., Сумарокова Т.Н., ЖОХ, 17, 1415, 1422
ДИКс (1947); Acta Phisicochim, 21, 836 (1946).
:1ДВр2. Linde E., Z. Electrochem., 30 (55), 255 (1924).
К л очко M. А., Курбанов H. Ш., Изв, сектора физ.-хим. анализа,
О 24, 237 (1954).
ЛЕШ. С е м ч е н к ох Д. П., А ппенин Н. П., Ушакова К. Л., Труды
/fEtF Новочеркас. политехи, ин-та, 34, № 48, 47 (1956).
JEEt5/ Van Wyk Н. J., Z. anorg. Chem., 48, 1 (1906).
ЛКй. к о S с о е Н. Е., J. Chem. Soc., 16, 82 (1863).
V orland er D., К aascht E., Ber., 56, 1162 (1923).
®^8. Миссан A. E., С у x о тин A. M., ЖНХ, 4, 606 (1959).
Meyer J., Spormann W.,Z. anorg. Chem., 228, 341 (1936). .
иВЙЮ. Зиновьев А. А., Захарова И. А., Кондрацкая Г. П.,
Ж. ЖНХ, 3, 2390 (1958).
Mfel. Мисс а н А. Е., Сухо т и н А. М., ЖНХ, 4, 606 (1959); 3 и и о в ь е в
!Ий А. А., Б а б а е в а В. П.,,ЖНХ, 6, 271 (1961); Бабаева В. П., Терми-
. ческое разложение хлорной кислоты. Канд, дисс., ИОНХ АНСССР,
fc 1963.
Ж|2. Пат. США 3258309 (1966).
ЯвЗ. Зиновьев А. А., Росоловский В. Я., ЖНХ, 1, 2596 (1956);
«К Росоловский В. Я., ЖНХ, 5, ,2148 (1960).
мЕВ4: F i с h t е г F., J еппу Е., Helv. chim. acta, 6, 225 (1923); N i c h о 1 -
Яр son' D., Reedy J., J. Am. Soc., 57, 817 (1935).
ЯШ. Пат. США 2925368 (1960).
liSr56. Семченко Д. П.,Ильин К. Г., Труды Новочеркас. политехи, ин-та,
111 19, №33, 95 (1948); 34, № 48; 25, 29 (1956); 47, № 61, 139 (1958).
®7. Muller W., Jonck Р., Chem. Ing. Techn., 35, № 2, 78 (1963).
ЛЕбб; Ильин К. Г., С к р и п ч е н к о В. И., Изв. вузов. Хим. и хим. технол.,
7, 572 (1964).
«р., Пат. США 1271633 (1918).
jjffefeO. Goodwin Н. М., Walker Е. С., Trans. Electrochem. Soc., 40, 157,
к 377 (1922); Chem. Metal. Eng., 25, 1093 (1921).
'^№1. Mathers F. C., Proc. Indian Acad., 63, 138 (1953).
:^№'^2. О закономерностях анодных процессов в HCIO4 и смеси HCIO4 + НС1 на
flip платиновом электроде. НИФХИ им. Л. Я. Карпова, Инф. карта ЦСИФА
ДВЕ;>№ 5653—67; Касаткина Э. В. и др., Электрохимия, 3, 1034 (1967);
5, 139 (1969).
ЯВбЗ. Шим он и с И. В., Рак о в А. А., Веселовский В. И., Электро-
Ж; химия, 6, № 2, 163, 169 (1970); 6, № 7, 1073 (1970); 7, № 5, 670 (1971).
К64. Шимонис И. В., Канд, дисс., НИФХИ им. Л. Я. Карпова, 1964.
1|Й55. К р а с и лова Т. Я., Касаткин Э. В., Веселовский В. И.,
Электрохимия, 6, № 3, 356 (1970).
Красилова Т. Я., Касаткин Э. В., Розенталь К. И.,
Яр- В ес'е л овский В. И., Электрохимия, 5, 44 (1969).
ЯК 67. Пат. ФРГ 1031288; пат. США 2846383 (1958); англ. пат. 818335 (1959);
Mfc-; Z. Electrochem., 11, № 1, 31 (1971).
Мр 68. Legendre A., Chem. Ing. Techn., 34, № 5, 379, 383 (1962); япон. пат^
IB 1360 (1972).
MP 69. Newnam К., Mathers F., Trans. Electrochem. Soc., 75, 271 (1939).
Hp 70. Kreider D., Am. J. Sci., 49, № 3, 443 (1895).
в 71. Mathers F., J. Am. Chem. Soc., 32, 66 (1910).
72. Пат. США 2392861 (1946); 3056656 (1962); франц, пат. 1261824 (1961).
Bp 73. Черных Л. В., Иванов В. В., Алексеева Э. А., ЖНХ,
₽ 15, № 7, 1922 (1970).
74. S m i t h G. F., J. Am. Chem. Soc., 75, 184 (1953); Mascherpa G.,
Ж C. r., 252, № 12, 1800 (1961). —
75. Зиновьев A. A., Ж. неорг. хим., 3, 1205 (1958).
76. С a p н e p С., Химия ракетных топлив. Пер. с англ. М., «Мир», 1969.
77. Gordons., С amp bell С., Anal. Chem., 27, 1102 (1955).
В 78. V or 1 a n d е г D., К a a s с h t Е., Вег., 56В, 1157 (1923).
' 461
79.
80.
81.
82.
83.
84.
85.
86.
МарковичМ., Ж- физ. хим., 62, 827 (1958).
Пат. США 3151935 (1964).
В о s с h A. v., At en А. Н. W., J. Am. Chem. Soc., 75, 3835 (1953).
А 2733982 (1956).
Пат. С
О t t Е., Chem. Вег., 86, № 9, 1065 (1953).
Англ. пат. 878904 (1961); пат. США 3038782 (1962); канад. пат. 642202 (1962).
Sugin о К., . Yamchita М., J. Electrochem. Soc. Japan, 15, 61
(1947); Sugino К., Bull. Chem. Soc. Japan, 23, 115 (1950).
Cornec E., Dickely J., С. R., 184, 1555 (1927); C. A., 21, 3296
(1923).
87. О c h s 1 i W., Z. Electrochem., 9, 807 (1903).
88. Foerster F., Electrochemie Wasseriger Losungen, Leipzig, 1905.
89. К nibbs К. V., PalfrCeman H., Trans. Faraday Soc., 16, 402
(1920).
90. Sugino K., J. Electrochem. Soc., 26, 185 (1953).
91. S u g i n o К., А о у a g i S., J. Electrochem. Soc., 103, № 3, 166 (1956);
Bull. Chem. Soc. Japan, 26, 185 (1953).
92. ObaraS., Sekine T., Denki kagaku, 37, № 4, 252 (1969).
93. D e N о г a et al., J, Electrochem. Soc., 116, № 1, 146 (1969).
94. Франчу к И. Ф., Изв. АН СССР, Отд. хим. наук, № 1, 63 (1963).
95. О b а г a S., Denki kagaku, 36, № 4, 291 (1968).
96. Bennet С. W., М а с k F. L., Trans. Electrochem. Soc., 29, 323 (1916).
97. Hackett J. W. et al., Developpement d'une methode de format! on
electrochimique de perchlorare a partir de chlorate sans emploi de platine,
Washington, U. S. Depart, of Commerce, 1953.
98. С о n w а у В. E., Theory and Principle of Electrode Processes, New York,
R. ou a Id pross 1965
99. Grotheer M. P., С о о k E. H., Electrochem. Techn., 6, № 5—6, 321
(1968).
Яковлева E. И., Розенталь К. И., Филипов Т. С., ЖФХ,
30, 937 (1956).
Канад, пат. 640131.
100.
101.
102.
103.
104.
105.
106.
107.
108.
109.
НО.
111.
112.
ИЗ.
114.
115.
119.
120.
121.
Пат. ФРГ 1076094 (1960).
Schumacher J. С. etal., J. Electrochem. Soc., 105, № 3, 151 (1958);
116, № 2, 68 (1969).
(1971).
Пат. США 2512973 (1956); 2840519 (1958); 2872405 (1959).
Udupa Н. V. К., Naras imhan К. С., Indian Chem. Eng., 2,
№ 2, 66 (1960); U d и г a N. V. К. Chem. Age India, 22, № 1, 21
Chem. Weekly, 5, № 4, 8 (1960); Chem. Eng., 67, № 9, 107 (1960).
Chem. Trade J., 157, № 4080, 211 (1965); нидерланд. пат. 6509556
Пат. США 2945791.
(1967).
al., Chem. Ing. Techn., 41, № 24, 1301
в T., Denki kagaku, 37, № 4, 258;
(1969).
6, 407
J. Electro-
№
N a g a 1 i n g a m N. et
Nam Ch. W., S e k i n
(1969).
Schumacher J. C.,
chem. Soc., 105, № 3, 151 (1958).
Пат. США 3493478 (1970).
S u g i n о К., J. Electrochem. Soc. Japan, 26, 185 (1958).
OsumaT., FujiC., J. Electrochem. Soc., 116, № 2, 203 (1969).
Ильин К. Г., Скрипченко В. И., Дроздецкая Е. П.
Труды Новочеркас. политехи, ин-та, 197, 56 (1969).
Пат. США 3475301 (1969).
С., J. Phys. Chem., 64, 1309 (1960).
Chem. -Ztg., 17, 90 (1898); C oul.eru M., Chem.-Ztg.,
116.
117.
118. W in t el er F. Z.
15, 213 (1906).
Narasimham К. C., Sundararajan S.,
J. Electrochem. Soc., 108, 798 (1961).
Пат. США 2772229 (1956); англ. пат. 769102 (1957).
GriggerJ.C., MillerH.C., L о о m i s E.
Soc., 105, 100 (1958).
Udupa H. V. K.,
D., J. Electrochem.
462
'^4
, Chem. Met. Eng., 51, 108 (1944); Trans. Electro-
(1947).
(1949).
(1967).
(1968).
(1968).
(1968).
Schumacher J.
chem. Soc., 92, 45
Пат. США 2475157
3350286
1104078
1104728
3403091
123.
Ж124.
g 126.
Ш127.
>128.
129.
Ci
у*
•t.
.4
Atti. Accad. Lincei, 30, № 5, 11
Пат. США
Англ. пат.
Англ. пат.
Пат. США
Канад, пат. 846805 (1970).
Нидерланд. пат. 6510232 (1966); канад. пат. 800596 (1968); белы. пат.
667203 (1966).
130. Пат. США 3463722 (1969).
131. Ma zzucchelli А., I
270 (1921).
132. Freeth F., Rec. trav. chim., 43, 475 (1924). •’
433. S c hu macher J., Stern D., Chem. Eng. Progr., 53, № 9, 428 (1957).
134. Франц, пат. 1380293 (1964).
135. Пат.. США 3307903 (1967).
436.
137.
138.
139.
140.
141.
442.
143.
Пат. США 3254947 (1966); 3288560 (1966); 3518173 (1970).
Япон. пат. 1609 (1963).
Chem. Eng., 62, № 12, 334 (1955); пат. США 2739873 (1956).
JamadaS. et al., J. Chem. Soc. Japan, Ind. Sect., 66, № 8 (1963).
Пат. США 3128121 (1965).
Пат. США 1274377 (1918); 3105735 (1963); англ. пат. 912953 (1962).
Япон. пат. 11803 (1962); пат. США 3154380 (1964); пат. ФРГ 1207926 (1965).
Франц, пат. 1492058 (1967); англ. пат. 1148125 (1969); пат. США 3502430
(1970).
Пат. США 2972514 (1961).
Франц, пат. 1289722 (1962); 1362622 (1964); англ. пат. 934536 (1963); пат.
США 3498759 (1970); пат. ФРГ 1132847 (1962).
Пат. США 3615179 (1971).
MarkowitzM. М. et al.,
№ 4, 321 (1964).
.4
144.
145.
146.
147.
148.
149.
150.
151.
152.
153.
154
155-
156.
157.
158.
159.
160.
161.
462.
163.
164.
165.
166.
Мочалов E., Изв. Казан.
1, 24 (1934).
Пат. США 3020124 (1962).
Пат. ФРГ 1080000 (1958); пат.
Ind. Eng. Chem., Prod. Res. Developm. 3
J. Am. Chem. Soc., 50, 1650 (1958).
хим. -технол. ин-та им. А. М. Бутлерова
Пат. ФРГ 1080000 (1958); пат. США 2998299 (1961).
Пат. США 3102784 (1964); 3175979 (1965).
С о г n е s Е., N е u m е i s t е г A., Caliche, 10, 492
5091 (1929).
Warasimban К. С., Indian. J. Techn., 5, № 11,
Age India, 19, № 7, 535 (1968).
God d a rd D. R., Hughes E. D., Ingold C.
480 (1946); J. Chem. Soc., 1950, 2559.
941 (1941).
J. W. T., Can. J. Res., 18B, 358 (1940).
(1960).
$
(1929); С. А., 23,
368- (1967); Chem.
К., Nature, 158,
Hantzsch А., Вег., 58B,
Gordon W. E., S p i n k c
Chem. et Ind., 83, № 3, 368
Пат. США 3186790 (1965).
Пат. США 3442726 (1969).
Markowitz М. М. et al.
J. Am. Chem. Soc., 79, 3659 (1957).
al., Z. anorg. allg. Chem., 259, 154 (1949).
Пат. США 3436172 (1969); -3450492 (1969).°
Caruso R., Loprest F. J., Lum A., J. Phys. Chem., 69, 1716
(1965).
Пат. США 2768874 (1956); 3420621 (1969).
Chem. Eng., 67, № 14, 68 (1960).
ГЛАВА*
ХЛОРИСТЫЙ ВОДОРОД И СОЛЯНАЯ КИСЛОТА
СВОЙСТВА ХЛОРИСТОГО ВОДОРОДА
Хлористый водород НС1 (мол. вес 36, 46) в обычных условиях
представляет собой бесцветный газ с резким запахом, на воздухе
дымит, образуя с влагой воздуха туман.
При —85,1 °C хлористый водород сжижается при атмосферном
давлении и переходит в твердое состояние при температуре —114,2 °C.
Твердый хлористый водород существует в двух модификациях:
ромбической, стойкой при температуре ниже —174,75 °C, и куби-
ческой.
Водный раствор НС1 (соляная кислота) в чистом виде бесцветен.
Примеси железа, хлора или органических веществ, присутствующие
в технической соляной кислоте, окрашивают ее в желтый цвет.
Сухой хлористый водород не оказывает коррозионно-активного
действия на металлы, соляная кислота разрушает металлы. В боль-
ших концентрациях хлористый водород токсичен, соляная кислота
разъедает кожу и мышечную ткань.
Основные свойства хлористого водорода приведены ниже:
Объем 1 моль газа ................................... 22,25
Плотность газа при нормальных условиях, кг/м3 . . . 1,6391
Плотность газа по отношению к воздуху................. 1,2679
Плотность жидкого хлористого водорода, г/см3
при т. кип......................................... 1,187
» т. кр.......................................... 1,273
Температура, °C
плавления ......................................... —114,2
кипения ......................................... —85,1
Критические константы
температура, °C..................................... 51,4
давление, атм....................................... 81,6
плотность, г/см3.................................... 0,42
Поверхностное натяжение на границе НС1 жидк. — НС1цар>
дин/см
при —110,0 °C .................................. 27,87
при —101,4 °C .................................. 26,25
при —92,9 °C .................................24,718
Теплота, кал/моль
плавления при —114,2 °C ............................. 476
парообразования при —85 °C ......................... 3860
разбавления (при бесконечном разбавлении) ... 17 400
Теплота образования газа при ,25 °C, ккал/моль .... —22,063
Свободная энергия образования, ккал/моль.............. —22,8
464
г
В
в .
ЦЧ «г ’ •
I
дат
ОТ
1-
Показатель преломления
жидкого НС1 ..................................
газообразного НС1 ........................
Электропроводность жидкого НС1 при —96 °C,
Ом’1-см“1 .............1 ....... .
Дипольный момент (при температурах от —84 до
300 °C) ...................................
Диэлектрическая проницаемость газа
при 0 °C......................................
при 21 °C.................................
Диэлектрическая проницаемость жидкого НС1
при —113,2 °C ................................
при —90,42 °C ............................
при —15 °C................................
при 27,7 °C ......................... . .
Энергия ионизации, эВ.........................
1,00045
110-8
1,03
1,0046
1,0038
№
Tisii
11,80
9,12
Под давле-
нием
12,85
Давление насыщенных паров HG1 в широком интервале темпе-
ратур приведено в табл. 9-1.
Таблица 9-1 >. Давление насыщенных паров хлористого водорода
iff
'И;
Температу- ра, °C Давление, мм рт. ст. Температу- ра, °C Давление, мм рт. ст1. Температу- ра, °C Давление, мм рт. ст.
Твердый хлористый
водород
—200,7
—196,5
—190,5
—183,5
—175,1
—164,9
—151,8
—114,2
—108,8
—108,53
—108,0
—105,2
—80
—75,3
—71,4
—70
—60
—55
—50,5
—40
ю-«
10"5
10-4
10" 3
10" 2
10“1
1
—140,67
—140,33
—137,95
—136,1
—133,95
—130,15
—127,35
5,58
5,89
7,87
10
13,08
20,44
27,82
- —124,75
—123,8
—118,85
—118,15
—117,6
—114,5
—114,2
(тройная
точка)
37,05
40,0
66,92
72,21
75,6
100
103,71
Жидкий хлористый водород
103,71
159,3
162,9
168,5
200,0
—103,87
—100,5
—99,2
—96
-91,31
229,8
289,9
317,1
503,4
526,4
—90,69
—88
—87,12
—85
(Т. кип.)
Жидкий хлдристый водород
ат
1,32
1,80
2,00
2,Q3
3,45
4,26
5,00
7,55
—36,1
-35
—31,7
—30
—20
—8,8
0
5,9
А
ат
8,53
8,73
10
10,62
14,53
20
25,46
30
17,8
20
25,0
27,9
36,2
40
50
51,4
(Кр. т.)
545,5
640,3
674,1
760
ат
40
41,58
46,42
'50
60
64,52
78,48
81,6
30 ЗМказ 843
465
Таблица 9-2. Плотность твердого и жидкого хлористого водорода
Температу-
ра, °C
Плотность,
г/см*
Температу-
ра, °C
Плотность,
г/см*
Температу-
ра, °C
Плотность,
г/см*
Ромбическая
форма НС1 тв.
—273,15 1,54
—195,15 1,507
—192,15 1,503
Кубическая
форма НС1 тв.
—178,15 1,55
—174,15 1,49
—166,15 ' 1,469
Жидкий НС1
—104,5
—101,2
—98,2
—92,2
—89,8
—85,8
1,2438
1,2347
1,2242
1,2127
1,2038
1,1937
Давление паров хлористого водорода в интервале температур»
от —158 до —НО °C можно определять по формуле:
\gP = A~B/T
(9.1)
где р — давление насыщенного пара, мм рт. ст.; Т — абсолютная температура
°К; А — 8,4430; В — 1023,1.
Плотность твердого и жидкого хлористого водорода приведена
в табл. 9-2.
В табл. 9-3 приведена плотность жидкого хлористого водорода
при температурах выше его температуры
и его насыщенного пара
кипения и атмосферном давлении.
Зависимость плотности р жидкого хлористого водорода от тем-
пературы выражается уравнением
р= 1,187+0,00318 (ТКип“П
, (9.2)
где Ткип — температура кипения, ?К; Т — температура измерения плотност и
?К.
Плотность газообразного хлористого водорода при различных
температурах и давлении может быть определена по номограмме,,
приведенной на рис. 9-1. Для определения плотности Необходимо
на номограмме
соединить точки,
характеризующие
температуру
и давление газа. Пересечение прямой с кривой плотности дает иско-
мую величину.
Таблица 9-3» Плотность хлористого водорода
Температу-
ра, °C
Плотность, г/см* Температу- Плотность, г/см*
жидкости пара ра, °C жидкости пара
—85,03
—80
-60
-40
-30
—20
1,191
1,178
1,122
1,063
1,031
0,997
0,0025
0,0032
0,0083
0,017
0,023
0,032
0
20
40
51,4
(Кр. т.)
0,924
0,831
0,697
0,424
0,054
0,097
0,180
0,424
466
Ниже приведены относительная вязкость жидкого хлористого
водорода (вязкость воды при температуре 22 °C принята за еди-
ницу):
Температура, °C ....... —112,35 —101,45 “90,95
Относительная вязкость . . . 0,59 0,53 0,447
и вязкость газообразного НС1 при давлении 1 атм:
Температура, °C
Вязкость газо-
образного HG1, сП
0
20
50
54
0,0133
0,0143
0,0158
0,0160
Температура, °C
100
150
200
250
Вязкость газо-
образного HG1, сП
0,0183
0,0208
0,0230
0,0253
30*
467
Для определения , вязкости
при различных температурах
газообразного хлористого водорода
можно пользоваться формулой
1+С/То
(9-3>
Лу —Лто
где rjy — вязкость при температуре Т (°К); т)То — вязкость при температуре
TQ — 273 °К; С — константа Сюзерленда.
Для газообразного НС1 при 0—250 °C константа С — 360.
Удельная теплоемкость хлористого водорода при 15 °C и постоян-
ном давлении составляет 0,1939 кал/(г-°С), отношение Ср : Cv —.
1,41. В интервале от 0 до 1700 °C удельную теплоемкость хлори-
стого водорода можно определить по формуле:
Ср = а + &Т (9.4)
где а = 0,1805; Ь = 2,687 -10~5; Т — абсолютная температура, °К.
Погрешность
определения составляет 1,5%.
В табл. 9-4 приведена мольная теплоемкость твердого и жидкого
хлористого водорода.
Таблица 9-4. Мольная теплоемкость С р хлористого водорода
при постоянном давлении [ в ккал/(моль • °C)]
Температу- ра, °с ; СР а Температу- ра, °C СР Температу- ра, °C СР Температу- ра, °C СР
Ж и д к и и
—250,65
—248,20
—246,00
—243,70х
—240,15
—237,25
< —235,65
—233,05
—228,40
—223,05
1,74
2,13
2,65
2,77
3,16
3,51
3,71
4,05
4,54
4,93
в е р д ы й
—217,05
—211,05
—205,35
—203,05
—191,05
—186,88
—182,91
—178,60
-176,25
—172,10
НС1
5,31
- 5,57
6,06
6,33
7,07
7,34
7,53
8,00
8,15
9,53
—168,60
-165,35
-162,15
—140,70
—136,65
-130,65
-126,45
—119,75
—116,65
9,74
9,80
9,94
10,98
11,30
11,50
11,82
11,90
12,12
—108,85
—104,25
—101,75
—99,05
-94,35
—92,05
—91,95
—85,35
-85,00
(Т. кип.)
НС1
14,18
14,97
14,88
15,03J
15,07
15,10
15,25
15,25
15,25
Ниже, в табл. 9-5, приведена мольная теплоемкость газообраз-
ного НС1 при различной температуре.
Таблица 9-5. Мольная теплоемкость газообразного НС1
[в ккал/(моль • еС)]
Температу-
ра, °C
Температу-
ра, °C
Температу-
ра, °C
0
100
200
500
1200 \
2000
5,00
5,09
5,27
5,46
6,13
6,9
26,85
326,85
526,85
726,85
926,85
6,95
7,07
7,28
7,54
7,78
1126,85
1326,85
1526,85 /
1726,85
7,98
8,14
8,28
8,38
468
jgkvr-.
ife?
|№r.'.
Br
^Растворимость хлористого водорода в воде рассмотрена ниже,
ртворимость НС1 (в мол. долях) в органических растворителях
& 20 °C приведена ниже:
|Клогексан ............
^трихлорэтилен . . ,
йрахлор
ксан .................
лорэтилен
ран . . .
^омоформ
щхлорэтан
0,0154
0,0163
0,0181
0,0197
0,0206
0,0296
0,0306
0,0310
Хлорбензол . .. .
Бромистый этил .
Бензол . 1......
Хлороформ . .
Толуол ....
Диэтиловый эфир
Метиловый спирт
Этиловый с
II!
[рт
0,0315
0,0348
0,0425
0,0444
0,0507
0,402
0,437
0,467
| Теплота растворения хлористого водорода в органических рас-
Ворителях составляет 3—4 ккал/моль.
рас-
воде
| СВОЙСТВА СОЛЯНОЙ КИСЛОТЫ (1-5)
I-
I Хлористый водород хорошо растворяется в воде, образуя
Ьоры соляной кислоты. Растворимость хлористого водорода в
Ьвисит от температуры и парциального давления хлористого водо-
рода в газовой смеси [6].
| В табл. 9-6 приведена растворимость хлористого водорода в воде-
гри различных условиях.
Таблица 9-6. Растворимость НС1 в воде при различных условиях
Температу-
ра, °C
---------------
lit-
Раствори-
мость, вес. %
Температу-
ра, °C
Раствори-
мость, вес. %
Давление
HG1, мм рт. ст.
Раствори-
мость, вес. %
Sr
i *
П* ъ
е;'
fl',
[<-
Ji
I* ь
—24
—21
—18,3
—18
—15
—10
—5
0
4
табл.
50,3
49,6
49,0
48,9
48,3
47,3
46,4
45,15
44,36
атм
8
12
14
18
23
30
40
50
60
43,88
43,28
42,83 х
42,34
41,54
40,23
38,68
37,34
35,94
9-7 приведены максимальные
При 0
60
100
150
200
300
400
500
600
760
1000
1300
38,00
39,65
40,69
41,42
42,46
43,28
43,88
44,44
45,15
46,12
47,22
концентрации
соляной
кислоты, которые можно получить сорбцией хлористого водорода
водой из газовых смесей в зависимости от температуры сорбции
|.и концентрации НС1 в газовой смеси.
£ Значения парциального давления хлористого водорода и воды
t над растворами соляной кислоты различной концентрации в интер-
вале температур от 0 до 100 °G приведена в табл. 9-8 [7—9].
..ч
В табл. 9-9 приведены коэффициенты абсорбции хлористого водо-
рода водой, т. е. объем НС1, приведенный к нормальным условиям
469
Таблица 9-7* Зависимость максимальной концентрации соляной кислоты (в %)
от температуры абсорбции и концентрации НС1 в газе
* Температу- ра абсорб- ции, °C Концентрация НС1 в газовой смеси, %
5 10 20 . 30 50 70 90
5 10 35,4 33,2 36,1 35,5 38,6 38,0 40,0 39,4 41,9 41,3 43,2 42,5 44,1 43,4
' 45 32,6 34,9 37,3 38,7 40,6 41,8 42,7
20 32,0 34,2 36,6 38,0 39,9 41,1 42,0
25 31,3 33,6 35,9 37,4 39,2 40,4 41,3
30 30,4 32,9 35,2 36,5 38,4 39,6 40,6
40 29,2 31,5 33,8 35,1 37,0 38,1 . 39,0
50 .28,0 30,0 31,8 33,6 35,4 36,5 37,4
(О °C и 760 мм рт. ст.), который может быть растворен в одном объеме
воды при данной температуре и давлении НС1 1 ат.
Растворимость HG1 в серной кислоте (в мг/100 г) различной
концентрации при 25 °C и парциальном давлении НС1, равном
760 мм рт. ст., приведена ниже:
CHtSO4, %
76,43
81,87
86,76
89,31
90,69
Растворимость
358,8
142,0
97,4
92,0
92,2
ch2SO4, %
92,20
94,14
97,36
98,65
100,00
Растворимость
99,6
108,2
143,2
197,1
401,5
состава газа
СССР выпу-
кислоте зависит от
абсорбции НС1. В
Содержание примесей в соляной
и чистоты воды, используемой для
скается несколько сортов соляной кислоты, качество которых регла-
ментируется соответствующими ГОСТами и техническими условиями.
Реактивная соляная кислота (ГОСТ 3118—67) — бесцветная,
прозрачная, дымящая на воздухе жидкость. Для реактивной кис-
лоты сорта «чистая», технической синтетической соляной кислоты
(ГОСТ 857—69) и технической соляной кислоты (ГОСТ 1382—69)
допускается желтоватая окраска.
Выпускается также ингибированная соляная кислота
(ТУ 6-01-714—72), содержащая 19-25% НС1 и 0,8-1,0% ингиби-
тора, допустимые примеси хлорида мышьяка (в пересчете на As)
0,01—0,015%. Цвет ингибированной кислоты светло-коричневый.
Скорость растворения стали марки Ст. 20 в ингибированной кислоте
при 20 °C составляет 0,15 г/(м2*ч), что соответствует утоныпению
»стенок стальной емкости, в которой находится кислота, на 0,16 мм/год.
Кроме того, производится особо чистая соляная кислота, к кото-
рой предъявляются весьма жесткие требования в отношении мини-
мально допустимого содержания примесей. Концентрация НС1
в пищевой соляной кислоте должна быть не менее 31 %.
В табл. 9-10 приведены основные технические требования, кото-
рым должны отвечать выпускаемые сорта соляной кислоты.
470
Таблица 9-8* Парциальное давление р хлористого водорода
и воды над растворами соляной кислоты
j(b мм рт. ст.)
GS
8
10
12
14
16
18
20
22
24
30
36
38
40
42
44
6
8
10
12
14
16
18
20
22
30
Температура, °G
оz
О
0 15 20 25
ЛНС1 ^н2О ^НС1 ^н2о РНС1 Рн2о *>НС1 ^Н2О
0,000023 0,000044 0,000084
0,000018 0,000131 0,00024 0,00044 —*
0,000066 4,18 0,000425 11,7 0,00076 15,9 0,00131 21,8;
0,000118 ' 0,00104 0,00178 0,0031 ->
0,00042 3,84 0,00232 10,7 0,00395 14,6 0,0067 20,0'
0,0009 0,0052 0,0088 ь 0,0145
0 0024 3,39 0,0118 9,65 0,0196 13,1 0,0316 18,0
0,0056 0,0265 0,0428 « 0,0685
0,0135 2,87 0,060 8,26 0,095 11,3 0,148 15,4
0,0316 2,62 0,132 7,50 0,205 10,3 0,32 14, Г
0,0734 2,33 0,294 6,75 0,45 9,3 0,68 12,6>
0,175 2,05 0,66 6,03 1,00 8,3 1,49 11,4
0,41 1,76 1,47 5,21 2,17 7,21 3,20 9,95-
1,0 1,50 3,36 4,54 4,90 6,32 7,05 8,75
2,4 1,26 7,60 3,88 10,6 5,41 15,1 7,52
5,7 1,04 16,8 3,25 23,5 4,55 32,5 6,37
13,1 0,85 36,8 2,70 50,5 3,81 68,5 5,35>
29,0 0,68 78 2,19 105 3,10 142 4,41
63,0 0,53 158 1,75 210 2,51 277 3,60
130 0,41 307 , 1,37 399 2,00 515 2,88
253 0,31 500 1,06 709 1,56 900 2,30
510 > • ‘
Температура, °C
30 35 40 50
£НС1 2>Н2О ^НС1 Рн8о ЗДС1 3>Н2О едс1 Рн2о
0,000151 _ а. 0,000275 0,00047 0,00140
0,00077 0,00134 0,0023 I» 0,0064
0,00225 29,1 0,0038 39,4 0,0062 50,6 0,0163 86,0
0,00515 « 0,0085 0,0138 0,0344 -
0,0111 26,8 0,0178 35,5 0,0282 47,0 0,069 80,0
0,0234 , 0,037 — 0,058 р « 0,136 1
0,050 24,1 0,078 31,9 0,121 42,1 0,275 72,0
0,106 » 0,163 0,247 * 0,55 1
0,228 20,6 0,345 27,5 0,515 36,4 1,11 62,5
0,48 19,0 0,72 25,1 1,06 33,3 2,21 57,0
1,02 17,1 1,50 22,8 2,18 30,2 4,42 52,0
2,17 15,4 3,14 20,4 4,5 27,1 8,9 46,7
4,56 13,5 6,50 18,0 9,2 24,0 17,5 41,5
9,90 11,8 13,8 15,8 19,1 21,1 35,7 36,5
21,0 10,2 28,6 13,7 39,4 18,4 71 32,0
4715
Продолжение табл. 9-8
Температура, °C
30 35 40 50
S THG1 РН2О РНС1 Рнао РНС1 ?H2O PHG1 РН,0
32
34
36
38
40
42
44,5
92
188
360
627
8,70 60,0 11,7 81 15,7
7,32 122 9,95 161 13,5
6,08 246 8,33 322 11,4
5,03 465 6,92 598 9,52
4,09 830 . 5,68 • —W 7,85
3,28 4,60 V 6,45
141
273
335
955
27,7
24,0
20,4
17,4
14,5
12,1
1 Температура, °C
‘во 70 80 90 100
РНС1 Рн2О PHCI * Рн2о РНС1 Рн2о РНС1 РН2О РНС1 РН,О I1
2
4
6
&
10
12
14
16
18
20
22
24
26
28
30
32
34
36
38
40
42
0,00380
0,0165
0,040
0,081
0.157
0,305
0,60
1 0,171
2,3
4,4
8,6
16,9
32,5
64
124
238
450
860
139
130
116
102
93,5
85,6
77,0
69,0
60,7
53,5
46,5
40,5
34,8
29,6
25,0
21,2
0,0100
0,0405
0,094
0,183
0,35
0,66
1,25
2,40
4,55
8,5
16,3
31,0
58,5
112
208
390
720
220
204
185
162
150
138
124
112
99,0
87,5
• 76,5
66,5
57,0
' 49,1
42,1
35,8
0,0245
0,095
0,206
0,39
0,73
1,34
2,50
4,66
8,6
15,6
29,3
54,4
100
188
340
623
333
310
273
248
230
211
194
173
154
136
120
104
90 .
77,5
67,3
57,2
J
.S
Таблица 9-9. Коэффициент абсдрбции НС1 водой
Температура,
°C
Коэффициент
абсорбции
К онцентрация
HG1,
вес. %
Температура
0
4
8
12
525,202
494,722
480,288
471,336
45,148
44,361
43,82а
43,277
14
18
18,25
0,058 0,132
0,21 - 0,46
0,44 492 0,92
0,82 - 1,64
1,48 463 2,9
2.65 » 5,1
4,8 425 9,0
8,8 16,1
15,7 374 28
28,1 345 49
52 317 90
94 290 157
169 261 276
309 234 493
542 207 845
970 184 —
161 п—
140 —I.
Л 120 " '
105
89,2 —
Коэффициент
абсорбции
462,375
451,222
450,660
435,034
715
677
625
550
510
467
426
388
349
310
275
243
212
182
158
135
Концентрация
HG1,
вес. %
42,829
42,344
42,281
41,536
472
а б лиц a 9-10. Основные технические требования к соляной кислоте
i*
ft;
£
&
в
Показатели
Плотность, г/см3 . . .
концентрация НС1, вес.
^Примеси, %, не более
нелетучий остаток .
сульфаты ...............
серная кислота (в пе-
ресчете на SO3)
свободный хлор . .
тяжелые металлы
группы H2S
железо ....
Техническая
I
4
Реактивная
w
мышьяк
и
о
Не менее
31
0,005
0,02
0,0002
* I
0,4
0,6
0,03
0,01
Не нор^
миру-
ется
То же
0,001
0,0002
0,0006
0,0002
0,0002
0,00005
0,000005
1,174—1,1885
35,0—38,0
0,002
0,0005
0,001
0,0002
0,0005
0,0001
0,00001
и хранят в таре
0,01
0,002
0,002
0,001
0,001
0,0005
0,00002
из прозрачного
Реактивную кислоту перевозят
бесцветного стекла с притертыми пробками (0,25—10 кг). Пробку
покрывают снаружи пергаментной бумагой и заливают гипсом
(при перевозке до 2 кг кислоты гипс можно заменять менделеевской
замазкой). Каждую склянку снабжают этикеткой с необходимыми
обозначениями.
Соляную кислоту для пищевой промышленности перевозят и
хранят в стеклянных бутылях емкостью до 30 л с чистыми стеклян-
ными или деревянными пробками. Снаружи пробки обвязывают
плотной бумагой и заливают гипсом или специальной мастикой, не
содержащей железа. Горло закупоренной бутыли обвязывают хлоп-
чатобумажной тканью или плотной бумагой. Бутыли помещают
в корзины или деревянные обрешетки со стружкой или соломой.
Каждую бутыль снабжают биркой.
Техническая соляная кислота транспортируется в специальных
стальных гуммированных цистернах и контейнерах, а также в сте-
клянных бутылях емкостью до 40 л, помещаемых в деревянные
обрешетки или корзины, которые выложены соломой или стружками.
Верхние края обрешетрк и корзин должны доходить до горла буты-
лей. Бутыли закрывают притертыми стеклянными или тщательно
обожженными глиняными пробками, перекрывающими горло бу-
тыли. Сверху пробка заливается специальной мастикой. Для об-
вязки горла бутыли применяют пеньковую ткань. По договоренности
с потребителем пеньковую ткань можно заменить крафт-бумагой.
Кроме ящиков-обрешеток с металлическими угольниками, обеспе-
чивающими нужную жесткость, для перевозки бутылей с синтети-
. 473
ческой кислотой можно использовать ивовые корзины (за исключе-
нием перевозок водным транспортом). Бутыли в корзинах обяза-
тельно обкладывают стружкой или соломой.
Тара для синтетической технической соляной кислоты должна
удовлетворять таким же требованиям, как и для технической кис-
лоты.
Ингибированную синтетическую кислоту перевозят в стальных
железнодорожных цистернах или автоцистернах и хранят в сталь-
ных резервуарах.
Плотность водного раствора соляной кислоты (в г/мл) при 25 °C
хюжно определить из выражения:
р=1 + С/2ОО (9.5)
f
где С — концентрация НС1 в соляной кислоте, вес. %.
Плотность соляной кислоты различной концентрации при 15 еС
приведена в табл. 9-11.
Таблица 9-11. Плотность соляной кислоты (при 15 SC)
Концентрация HCI
Концентрация НС1
Плотность,
г/см*
Плотность,
г/см’
вес.
вес. %
1,000
1,010
1,020
1,030
1,040
1,050
1,060
1,070
1,080
1,090
1,100
0,16
2,14
4,10
6,15
8,16
10,17
12,19
14,17
16,15
18,11
20,01
1,6
21,6
42,1
63,3
84,9
106,8
129,2
151,6
174,7
197,4
220,1
0,044
0,593
1,155
1,737
2,328
2,929
3,544
4,158
4,784
5,414
6,037
1,110
1,120
1,130
1,140
1,150
1,160
1,170
1,180
1,190
1,200
21,92
23,82
25,75
27,66
29,57
31,52
33,46
35,38
37,23
39,11
243,3
266,8
291,0
315,4
340,1
365,6
391,5
417,5
443,1
469,3
6,673
7,317
7,981
8,648
9,327
10,03
10,74
11,45
12,15
12,87
При измерении плотности соляной кислоты при других темпера-
турах в значение плотности вносят следующие поправки*:
Плотность, г/см’ Поправка на 1 °C
1,000—1,040 0,0002
1,041—1,085 0,0003
1,086—1,120 0,0004
1,121—1,155 0,0005
1,156—1,200 0,0006
* Если плотность
туре выше 15 °C,
HI
НС1 измеряется при темпера-
поправку прибавляют к ре-
зультату измерения; если измерение проводят
при температуре ниже 15 °C — поправку вычи-
тают.
474
к
* I
£
Г
if!
??
fe
,si
Л-
Для выражения плотности, измеренной при требуемой темпера*
£ре через плотность при 15 °C можно воспользоваться номограммой^
риве денной на рис. 9-2.
Пример пользования номограммой. Плотность соляной кислоты, измеренная
ри 40 °C, равна 1,146 г/смэ. Соединив точку, соответствующую 40 РС на темпе-
атурной шкале, с точкой 1,146 на шкале р, продолжаем соединительную пря-
ую до пересечения со шкалой р15. Значение плотности кислоты при 15 °C
аходим в точке пересечения. Проводя от нее горизонтальную линию до пере-
гчения со шкалами концентрации, находим значения концентрации НС1 (соот-
ветственно в % и в г/л).
Плотность Концентрация
a a HCI
<tO
15
20
25
30
50
co
1,10
1,09
1,08
1,07
1,06
1,05
1,20-
1,19'
I,18
7,77
1,16
1,15
из
К?
7,20-,
1,19-
1,13 -
1,16'-
7,74-
1J3-
1,10-
1,09—
1,08-
1,07-
1,06-
1,05-
35-
30-
20 —
15 —
Рис. 9-2. Номограмма для определения плотности
кислоты при различных температурах.
k50
bOO -
350-
300
250
200
150
соляной
Вязкость соляной кислоты при 20 °C приведена в табл. 9-12
при различных температурах в табл. 9-13.
Таблица 9-12. Вязкость соляной кислоты при 20 РС (в сП)
Концент-
рация
HO,
Вязкость Концент- рация НС1, вес. % Вязкость f Концент- рация НО, вес. % Вязкость Концент- рация НО, вес. %
Вязкость
0,00 1,005 14,29 . 1,251 22,37 i 1,452
3,76 1,065 17,59 1,323 23,50 1,482
5,00 1,080 17,95 1,333 23,91 1,500
7,39 1,125 20,00 1,36 25,43 1,555
8,79 1,168 20,80 1,408 26,94 1,611
10,89 1,187 21,43 1,430 29,90 1,731
32,80
36,53
39,14
40,61
1,779
1,870
2,004
475
Таблица 9-13. Вязкость соляной кислоты при различной температуре (в сП)
f
..... .™—I ' II4 I *!!
Концентрация ilGl, вес. %
Температура, °C
15 25 35 45
8,14
16,13
23,05
1,260
1,420
1,0284
1,181
1,364
0,8575
1,0006
1,170
0,7118
0,8536
1,006
Таблица 9-14. У^еяь-вля теплоемкость соляной кислоты [в кал/(г*°С)]
Температура, °C
0,0 3,3 20. 20,5 40,4 60
60,5
Концент-
рация
НС1.
вес. %
4 0,9282 0,9286 0,8425
10,2 0,8264 IW 0,8348 0,7765 —- 0,8541
15,5 0,7520 0,7627 0,7107 II "
16,8 0,72 0,74 - 'W— 0,78
21,5 0,6851 * 0,6966 0,6662 0,7230
25,8 1 0,6437 0,6570 0,6220 0,6858
,38,9 0,61 1 1 “ 0,631 1 0,67
31,7 0,5973 — 0,6097 0,6044 0,6455
33,6 0,58 — 0,591 1 0,638
.33,7 0,5665 * 0,5825 II — 0,6357
41,4 0,55 II —“ ч —“ 0,61
Рис. 9-3. Номограмма
Для определения те-
плопроводности со-
ляной кислоты:
7 —для 5—15%-ной НС1;
2 — для 15—30 % -ной
HGL
Теплоемкость соляной кислоты приведена в табл. 9-14.
К Теплопроводность соляной кислоты по отношению к теплопровод-
Е ности воды, принятой за 100%, приве-
В дана ниже:
К Концентрация НС1, вес. % 12,5 25 38
К Относительная теплопровод-
11 ность . . . .......... 87,0 79,4 72,6
К Теплопроводность водных растворов
В юоляной кислоты можно определить по
В номограмме, приведенной на рис. 9-3.
В Пример пользования номограммой. Для
В определения теплопроводности водных раство-
i ров соляной кислоты, содержащих от 5 до 15%
Ж НС1, проводят прямую из точки, соответству-
В ющей заданной концентрации, через точку 1 jw
Б пересечения со шкалой теплопроводности. Для
I определения теплопроводности растворов, содер-
В жащих от 15 до 30% НС1, прямую проводят
В’- через точку 2.
I Хлористый водород образует с водой
г .азеотропную смесь, при атмосферном да-
I влении она кипит при температуре 108,5 °C.
| В зависимости от давления состав азеотропной смеси HG1 и Н2О
несколько изменяется (табл. 9-15).
! Температура кипения соляной кислоты различной концентрации
f при атмосферном давлении приведена на рис; 9-4, а температура
к замерзания в табл. 9-16.
Количество тепла, выделяющегося при абсорбции ЙС1 водой
f с получением соляной кислоты различной концентрации, приведено
I в табл. 9-17 [9].
* J
’• и
Ji Таблица 9-15 > Состав азеотропных смесей HCl + вода при различном
Концентрация HCI,
г/100 г кислоты
Рис. 9-4. Температура ки-
пения соляной кислоты
различной концентрации.
4
Таблица 9-16. Температура замерзания растворов соляной кислоты
Концентрация
HG1
Концентрация
НС1
Т.
замерз.,
°C
Твердая фаза
Т.
замерз.,
°C
Твердая фаза
48,81
47,53
46,39
45,21
43,93
42,49
41,41
40,38
39,17
37,97
36,68
35,33
33,74
31,99
31,87
31,24
28,19
23,89
22,81
47,02
44,67
42,67
40,68
38,64
36,26
34,86
33,40
31,75
30,20
28,56
26,94
25,11
23,20
22,90
22,40
19,35
15,48
14,58
—17,50
—18,00
—19,75
—22,45
—26,25
—25,65
—24,85
—24,85
—25,4
—26,95
—29,3
—32,45
—37,05
—42
—44,25
—46,2
—63,5
—79,5
• —66,5
НС1-2Н,0 21,46 13,48 —58,5 Лед
То же 20,48 12,70 —52,6 »
» 19,49 11,94 —46,1 »
» 18,56 11,23 —41,75 »
» 17,36 10,36 -36,65 »
НС1-ЗЩО 16,18 9,52 —32,2 »
То же 14,97 8,68 —27,6 »
» 13,75 7,86 —23,55 »
12,46 7,02 —19,1
» 11,24 6,26 -16,75 »
» 10,04 5,51 —13,75 »
» 8,95 4,85 —11,55 »
» 7,90 4,23 -9,45
» 6,89 3,65 —8,0 »
» 5,91 3,10 —6,3 »
» 4,82 2,50 -4,7 »
» 3,32 1,69 —3,0 »
» 2,07 1,04 -1,6 »
Лед 1,20 0,60 -1,0 »
Таблица 9-17 • Теплота растворения НС1 в воде
ь
Концентрация НС1, вес. % Теплота растворения, кал/г раствора Концентрация НС1, вес. % Теплота растворения, кал/г раствора
при 42 °C при 62 °C при 42 °C при 62 °C
2 —— 10,2 20 89,7 91,7
4 19,9 22 97,4 99,5
6 29,3 29,7 24 105,1 106,9
8 38,9 39,5 26 112,3 113,9
10 48,0 48,6 28 118,7 120,5
12 56,9 57,5 30 124,9 126,9
14 65,4 66,5 32 130,7 132,1
16 73,7 75,1 34 135,9 137,8
18 81,7 83,4 36 140,6
При растворении хлористого водорода в бесконечно большом
количестве воды при 15 °C теплота растворения составляет
478
Таблица 9-18* Теплота бесконечного разбавления растворов
соляной кислоты при 25 ?С
Концентрация
разбавляемого
раствора, моль
НгО/моль НС1
Теплота
разбавления,
ккал/моль
Концентрация
разбавляемого
раствора, моль
HjO/моль НС1
Теплота
разбавления,
ккал/моль
3
5
10
12
15
20
4,47
2,76
1,46
1,25
1,05
0,85
25
50
100
200
400
1600
0,73
0,433
0,343
0,249
0,181
0,090
17,4 ккал/моль НС1. При разбавлении соляной кислоты водой выде-
ляется тепло. В табл. 9-18 приведены значения теплот разбавления
соляной кислоты водой.
ПРОИЗВОДСТВО ХЛОРИСТОГО ВОДОРОДА
и соляной кислоты
Развитие производства хлористого водорода и соляной кислоты
и изменение соотношения различных методов производства были
рассмотрены ранее в 5-й главе. Показано, что во всех промышленных
странах с развитием производства органических хлорпродуктов,
получаемых заместительным хлорированием углеводородов основ-
ное количество хлористого водорода и соляной кислоты стали полу-
^егося хлористого водорода. Старые методы
чать из побочно образую
п
получения хлористого водорода из хлористого натрия и серной
кислоты, а также прямым синтезом из хлора и водорода потеряли
ведущую роль. После разработки способов очистки попутного хло-
ристого водорода и соляной кислоты, получаемой из него, от орга-
нических примесей открылись широкие возможности для исполь-
зования побочного хлористого водорода.
Сульфатные хлористый водород и соляная кислота производятся,
как правило, там, где имеется потребность в сульфате натрия. Произ-
* водство синтетического хлористого водорода сохраняется главным
образом там, где к качеству хлористого водорода и соляной кислоты
предъявляются повышенные требования. Часто заводы организуют
производство синтетической соляной кислоты для использования
абгазов сжижения хлора, если нет других потребителей абгазов,
а схема сжижения не предусматривает полного или почти полного
сжижения хлора.
Все способы получения соляной кислоты осуществляются в две
стадии: а) получение хлористого водорода и б) абсорбция хлористого
водорода водой с образованием соляной кислоты.
Если хлористый водород используется непосредственно, вторая
стадия — абсорбция хлористого водорода отпадает.
479
СПОСОБЫ ПОЛУЧЕНИЯ ХЛОРИСТОГО ВОДОРОДА ;
Из многочисленных способов получения хлористого водорода
ниже будут рассмотрены только следующие три метода, которые
представляют наибольший интерес и практически используются
в промышленности: а) сульфатный метод; б) синтез НС1 из хлора
и водорода; в) получение хлористого водорода как побочного продук-
та при производстве органических и неорганических продуктов.
Сульфатный метод получения хлористого водорода
Сульфатный метод получения хлористого водорода является
одним из старейших процессов в химической промышленности. Он
служил основой прЬизводства соды по методу Леблана. При этом
длительное время хлористый водород не находил применения и
являлся отбросом производства. Впервые он начал использоваться
для получения соляной кислоты в 1827 г.
В дальнейшем производство соды развивалось по аммиачному
способу; сульфатный способ получения хлористого водорода при-
менялся и применяется только тогда, когда возникала потребность
в сульфате натрия. Поэтому в странах неэкономических районах,
имеющих запасы природного сульфата, удобные для разработки,
сульфатный метод получения хлористого водорода не нашел при-
менения.
Сульфатный метод основан на взаимодействии хлористого натрия
с концентрированной серной кислотой по реакциям:
NaCl+H2SO4=NaHSO4 + НС1 (9.6)
NaCl+NaHSO4=Na2SO4+HCl (9-7)
Процесс проходит с поглощением тепла, поэтому реагирующую ч
массу необходимо нагревать. Реакция протекает при 500—550 °C
с образованием твердого сульфата натрия и газовой смеси, содержа-
щей хлористый водород в различной концентрации в зависимости
от способа производства. Промышленные способы осуществления
процесса взаимодействия H2SO4 и NaCl можно разделить на 2 типа.
По первому типу реакция проводится во вращающихся печах
при прямом соприкосновении реагирующих веществ с продуктами
горения для достижения необходимой температуры. При этом полу-
чают сульфат натрия высокого качества, однако выделяющийся
хлористый водород сильно разбавлен продуктами сгорания топлива.
Преимуществом вращающихся печей является их сравнительно
высокая производительность. Печь диаметром 1,5 и длиной 12 м
вырабатывает 25 т/сут сульфата и обеспечивает хлористым водородом
производство 40—42 т 27,5 % -ной соляной кислоты. Недостаток таких
печей заключается в низком содержании хлористого водорода в га-
зовой смеси (около б%), что ограничивает возможности получения
концентрированной соляной кислоты.
480
L
Процессы второго типа проводятся в муфельных реакторах,
снабженных гребками для перемешивания реагирующей массы
(рис. 9-5).
Муфель печи, в которой осуществляется реакция, выполняется
из чугуна или из огнестойкого и кислотоупорного шамотного кир-
пича и обогревается топочными газами. Для перемешивания реаги-
рующей массы и передвижения ее по направлению к выгрузочному
отверстию муфель снабжен мешалкой с гребками, выполняемыми
NaCl-
Рис. 9-5. Механическая сульфатная печь:
1 — бункер; 2 — шнековый питатель; з — ввод серной кислоты; 4 — муфель; 5 — вал;
в — гребки; 7 — зубья; 8 — приемный бункер; 9 — трубчатая мельница.
большей частью из термосилида [10]. Сульфат через выгрузочное
отверстие поступает в бункер и далее в охлаждаемую водой шаровую
мельницу.
Печи муфельного типа более сложны в устройстве, и эксплуата-
ция их обходится дороже вращающихся печей, но в печах муфель-
ного типа получают хлористый водород высокой концентрации.
При разложении поваренной соли 93%-ной серной кислотой
теоретически можно получить газ, содержащий 83% хлористого
водорода и 17% паров воды или в пересчете на сухой газ чистый
100%-ный НС1. Практически всегда происходит разбавление реак-
ционного газа за счет подсоса воздуха и топочных газов, так как
31 Заказ 843 481
/
в муфеле поддерживается небольшое разрежение ' (несколько
мм вод. ст.). Поэтому содержание хлористого водорода в пересчете
на сухой газ составляет обычно 30—50%.
Внутри муфеля температура поддерживается в пределах 500—
550 °C, температура топочных газов, обогревающих муфель, обычно
составляет 950—1000 °C. Из-за трудности равномерного обогрева
реакционной массы до необходимой температуры возможно ухудше-
ние качества сульфата натрия. Хлористый водород и соляная кис-
лота, получаемые сульфатным способом, обычно загрязнены приме-
сями, вносимыми с серной кислотой (мышьяком, железом).
Для получения 1 т НС1 (в пересчете на 100%) расходуется (в т):
Поваренная соль (97%) ...... 1,7—1,9
Серная кислота (93% H2SO4) ... 1,45—1,52,
Условное топливо (7000 ккал/кг) в му-
фельных печах ..................... 0,36—0,44
Предложено также проводить реакцию между поваренной солью
и серной кислотой в реакторах с псевдоожиженным слоем [11].
В реактор подается нагретый хлорид
натрия и поток горячих газов, содер-
жащих пары или мелкие капли серной
кислоты. Сульфат натрия, образу-
ющийся на поверхности кристаллов
поваренной соли, удаляется вместе
с газовым потоком, отделяясь от кри-
сталлов NaCl за счет истирания в псе-
вдокипящем слое. Принципиальная
схема производства по такому способу
показана на рис. 9-6., Концентрация
НС1 в реакционном газе здесь невелика
(не превышает 5 %).
Для получения сульфата и хлори-
стого водорода вместо серной кислоты
можно использовать смесь SO2 и О2,
пропуская ее через слой кристаллов
поваренной соли, подогретой до необ-
трмпературы. Аналогично
описанному ранее процессу взаимодей-
ствия NaCl и H2SO4, предложено [121
получать хлористый водород и сульфат
натрия, калия или кальция в псевдо-
хлоридов перечисленных выше металлов
Соль
на
Кабсор-
берам
\ Воздух
5s
в
Na2SO4
Топлибо
H2SOa
Рис. 9-6. Схема получения
сульфата и НС1 в j аппарате
с кипящим слоем:
1 — реакционный аппарат; 2 — ХОДИМОЙ
бункер-питатель; <? — топка; < —
воздуходувка; 5 — испаритель
серной кислоты; в —- бункер;
7 — сепараторы; S —- горелкк.
III
ожиженном слое реакцией
с двуокисью серы и кислородом:
NaCl+SO24-0,5O2 + H2O = NaHSO4+HCl (9.8)
NaCl + NaHSO4—> Na2SO4+HCl (9.9)
Однако этот процесс, известный под названием процесса Хар-
гривса, не получил широкого применения в промышленности.
482
|< v Продолжаются работы по усовершенствованию аппаратуры для
| получения сульфата натрия и хлористого водорода из NaCl и серной
I кислоты [13,14]. Предложена следующая схема: процесс ведут в двух
последовательно включенных реакторах, причем во втором реакторе
массу для более полного превращения нагревают в токе горячего
газа [15].
Синтез хлорнстсго водорода из элементов
Синтетический хлористый водород получают сжиганием водо-
рода с хлором по уравнению:
Н2 + С12 2НС14-44120 кал (9Л0)
Константу равновесия этой реакции при различной температуре
можно определить из выражения:
lg^p = -^—-—0,5331gT-2,42 (9.11)
где Т — абсолютная температура, °К. ‘ )
г
Доля хлористого водорода X, диссоциированного при данной
температуре, может быть определена из выражения:
-(1-Х)2
Р”
(9.12)
Значение степени диссоциации хлористого водорода при раз-
личных температурах приведено ниже:
Температура* °C
20
400 *
700
Доля диссоцииро
ванного HG1
2,95 -10“17
4,3540-8
. 9,75-IO"8
Температура, °C
1200
1700
2200
Доля диссоцииро-
ванного НС1
5,32-10’4
3,77-10-3
1,22-10-2
Диссоциация хлористого водорода наблюдается в заметной сте-
пени только при температурах выше 2000 °C.
При обычной температуре, в отсутствие катализаторов или све-
товых лучей, реакция между С12 и Н2 не идет. Реакция может быть
инициирована нагреванием, путем воздействия катализаторов или
светового облучении'и протекает затем по цепному механизму со /
взрывом.
Реакция хлора с водородом является типичным примером фото-
химической реакции. При облучении световыми лучами с длиной
волны 4800 А квантовый выход высок и составляет 104—10е. Фото-
химическое инициирование реакции предлагалось использовать для
выжигания примесей водорода в хлоре [16].
Тщательно высушенные С12 и Н2 не вступают в реакцию между
собой. Примеси кислорода снижают скорость реакции между С12
и Н2, по-видимому, вследствие соединения кислорода с атомарным
31*
483
я
водородом и возрастания вероятности обрыва ценной реакции С12
и Н2. В технике используют термическое инициирование реакции,
сжигая водород с хлором в факеле горелки. Спокойное течение реак-
ции без взрывов обеспечивается равномерным поступлением хлора
и водорода и смешением их только в факеле пламени горелки.
Процесс синтеза хлористого водорода из элементарных веществ
протекает с выделением большого количества тепла. При сжигании
водорода в хлоре в соотношении, близком к стехиометрическому,
температура в факеле горелки достигает 2200 °C.
При всех конструкциях печей для синтеза хлористого водорода
акел пламени не должен непосредственно касаться стенок аппарата
во избежание очень быстрого их перегорания. Даже при соблюдении
этого условия стенки аппаратов необходимо охлаждать для пред-
отвращения чрезмерно быстрого выхода аппаратов из строя. Однако
при охлаждении стальных или других металлических аппаратов
нельзя допускать понижения температуры внутренних стенок ниже
точки росы во избежание конденсации влаги и образования на поверх-
ности аппарата пленки соляной кислоты, быстро разрушающей
металлические части печи. Необходимо учитывать, что точка росы
концентрированной кислоты при том же парциальном давлении
водяных паров на несколько десятков градусов выше, чем чистой
воды. Это обусловлено сильным снижением парциального давления
паров воды над концентрированной кислотой. Хлориды металлов,
образующиеся при коррозии и растворяющиеся в кислоте, могут
дополнительно снижать парциальное давление паров воды над кис-
лотой и повышать температуру образования жидкой фазы на стенках.
Парциальное давление паров воды в синтетическом хлористом
водороде зависит от влажности исходных хлора и водорода, а также
от содержания в них кислорода. Свободный кислород соединяется
с водородом, образуя пары воды. Двуокись углерода также может
частично восстанавливаться до окиси углерода с образованием
некоторого количества паров воды.
Состав синтетического хлористого водорода зависит от загрязне-
ний, содержащихся в исходных хлоре и водороде. Некоторые за-
грязнения вносятся в продукты реакции в результате коррозионного
разрушения стенок аппаратов для синтеза и охлаждения газов.
Наиболее вероятно загрязнение хлористого водорода хлоридами
металлов, попадающими в виде возгона. Поэтому, если потребители
хлористого водорода предъявляют высокие требования к его ка-
честву, применяется графитовая или кварцевая аппаратура.
В хлоре обычно содержатся пары воды, азот, кислород, водород,
двуокись и окись углерода. С водородом обычно помимо паров воды
вносятся примеси кислорода и азота.
Как было указано, кислород полностью, а двуокись углерода
частично восстанавливаются с образованием паров воды. Остальные
газовые примеси полностью переходят в состав полученного хлори-
стого водорода. Так как синтез хлористого водорода ведут в избытке
водорода (до 5%), он разбавляет образующийся хлористый водород.
Помимо упомянутых примесей с хлором и водородом могут по-
ступать небольшие количества хлорированных углеводородов, брызг
и тумана серной кислоты, которыми газы загрязняются при серно-
кислотной осушке или компримировании ротационными компрес-
сорами с сернокислотным заполнением.
Примеси серной кислоты могут в печах синтеза восстанавливаться
вплоть до сероводорода, что следует учитывать при применении
хлористого водорода для процессов каталитического гидрохлори-
рования, поскольку серосодержащие соединения отравляют ката-
лизатор.
В этом случае хлор необходимо тщательно очищать от брызг
и тумана серной кислоты в электрофильтрах или специальных филь-
трах с насадкой из стеклянной тонковолокнистой композиции.
Если для синтеза'хлористого водорода использовать испаренный
хлор и электролитический водород с минимальным избытком, то
можно получить газ, содержащий до 99% HGL Примерное содер-
жание примесей в таком продукте будет следующим (в %):
Водород .......0,6—0,7 Двуокись углерода . —0,05
Азот ........ —0,2 Окись углерода . . —0,01
Пары воды......—0,1
При применении электролитического хлора содержание примесей
будет более высоким.
Получение в печах синтеза полностью сухого хлористого водо-
рода практически невозможно, так как в исходных газах содержится
некоторое количество кислорода и даже в случае применения для
синтеза сухих газов хлористый водород увлажняется за счет воды,
образующейся в печи во время синтеза.
Вместо водорода для получения синтетического хлористого водо-
рода может быть использован светильный газ или смесь окиси угле-
рода с парами воды [17].
В последнем случае в пламени горелки происходит конверсия
окиси углерода в двуокись с образованием водорода и последующим
синтезом НС1:
СО + Н2О + С12 = 2НС1 + СО2 (9.13)
При этом надо учитывать возможность внесения дополнительных
загрязнений вместе со светильным газом или окисью углерода и
ухудшения качества хлористого водорода и соляной кислоты.
Предложено получать синтетический НС1 из хлорокислородной
смеси и горючего, например природного, газа. При этом часть горю-
чего газа расходуется на сгорание с кислородом, а остальная часть
на соединение с хлором с образованием НС1 [18].
Тепло, выделяющееся при синтезе хлористого водорода, частично
отводится через стенки аппарата; остальная его часть уходит с горя-
чими газами из печи. Перед поступлением на абсорбцию газы должны
быть дополнительно охлаждены. Стремятся не допускать снижения
температуры стенок печи менее 140—200 и повышения более 400—
485
500 °C. Для охлаждения стенок аппарата в печах малой мощности
(производительность до 10—15 т/сут) можно применять естественное
воздушное охлаждение [19]. При использовании печей большой
мощности необходимо водяное охлаждение.
В качестве материала для изготовления печей преимущественно
используется сталь, применяются также графитовые и кварцевые
печи. Однако размеры и соответственно производительность квар-
цевых аппаратов ^ограничены, кварцевая аппаратура очень дорога
и трудна в ремонте и обслуживании. Эти недостатки, но в меныпей
степени, присущи и графитовой аппаратуре, хотя графитовый печи
еще находят применение в промышленности. Печи из неметалли-
ческих материалов используются преимущественно для получения
хлористого водорода повышенной чистоты, когда недопустимо за-
грязнение НС1 хлоридами металлов.
Как указывалось ранее; синтез НС1 проводят в небольшом из-
бытке водорода, за счет чего достигается отсутствие хлора в хло-
ристом водороде и соляной кислоте. Помимо этого, избыток водорода
постоянно обеспечивает в печи восстановительную атмосферу, что
способствует снижению коррозионного разрушения^стальных и гра-
фитовых печей. Пленка малолетучего хлористого железа, отлага-
ющаяся на металлических поверхностях в печи в среде Н2, предохра-
няет металл от коррозии. Если в газовой смеси создается избыток-
хлора, образуется легко возгоняющееся хлорное железо, и процесс
коррозии стальных стенок ускоряется. В графитовых печах при
избытке хлора происходит хлорирование стенок с образованием
летучих продуктов.
Предложены и используются в промышленности разнообразные
конструкции печей. Простейшая из них представляет собой цилин-
дрический аппарат с горелкой, расположенной по его оси. Иногда
аппарат выполняют в виде двух конусов, соединенных широкой
частью. Такая форма позволяет отдалить стенки печи от факела пла-
мени в наиболее горячей зоне печи. В большинстве конструкций
печей факел пламени направлен снизу вверх, однако есть много
конструкций с обратным направлением пламени. Печи большой
мощности снабжаются водяной рубашкой для охлаждения.
В некоторых конструкциях в верхних зонах сжигания устанавли-
ваются теплообменники различного типа для дополнительного охла-
ждения газов до 150—160 °C [20].
На рис. 9-7 показаны устройства цилиндрической печи с водяной
рубашкой и печи с дополнительными холодильниками для охлажде-
ния хлористого водорода.
При нарушении нормальной работы печи внутри ее может обра-
зоваться взрывоопасная хлороводородная смесь. Эта смесь может
образоваться при случайном погасании пламени в горелце, при боль-
ших колебаниях в подаче хлора и водорода. Для предотвращения
разрушения при взрыве печи снабжаются взрывными панелями, при
разрушении которых снижается давление. Смеси взрывоопасной
концентрации могут возникать также в аппаратуре для абсорбции,
где вследствие поглощения НС1 возрастает концентрация не всту-
пивших в реакцию компонентов.
Предложены и используются в промышленности конструкции,
в которых в одном аппарате объединены печь для синтеза хлористого
водорода и абсорбер для получения соляной кислоты. Абсорбер
соляной кислоты. Абсорбер
Рис. 9-7. Схемы устройства печи для синтеза хлористого водо-
рода с водяной рубашкой (а) и с холодильником (б):
1 — корпус печи; 2 — водяная рубашка; 3 — горелка; 4 — смотровой люк;
5 — холодильник.
может быть расположен под печью для синтеза с направлением пла-
мени сверху вниз [21] или над печью [22]. В последнем случае над
факелом горелки устанавливается колокол, предотвращающий по-
падание'кислоты на факел горелки.
Попутный или абгазный хлориотый водород
Основное количество хлористого водорода во всех промышленных
странах получают в виде побочного продукта в различных хими-
ческих производствах. В зависимости от производства, в котором
образуется хлористый водород, изменяется его концентрация и со-
став. В большинстве случаев хлористый водород загрязнен продук-
тами, получаемыми в рассматриваемом процессе или используемыми
в качестве сырья. В некоторых процесса^ получаемые газы разба-
влены инертными газами и иногда парами воды- Состав примесей
и загрязнений индивидуален для каждого процесса. Ниже рассма-
триваются некоторые наиболее характерные процессы, протекающие
с образованием больших количеств хлористого водорода.
1. Процессы заместительного хлорирования углеводородов или
их производных
RH+C12 —>• RC1+HC1
(9.14)
487
К этой группе относится хлорирование метана с целью получения
хлористого метила, метиленхлорида, хлороформа и четыреххлори-
стого углерода, бензола в производстве хлорбензола, этилового
спирта для получения хлораля, керосина в производстве сульфо-
нола и др.
В этих процессах хлористый водород помимо хлорорганических
примесей может содержать также элементарный хлор. Концентра-
ция НС1 в абгазном хлористом водороде от производства хлормета-
нов невысока и составляет обычно не более 20%. В остальных упо-
мянутых процессах выделяется концентрированный хлористый
водород (88—99% HG1).
2. Процессы пиролиза хлорпроизводных углеводородов
RH-RiCl —> R=Ri+HCl (9.15)
К наиболее важным и крупнотоннажным производствам этого
типа относится получение винилхлорида пиролизом дихлорэтана,
трихлорэтилена из тетрахлорэтана и разложение нетоксичных
изомеров гексахлорана с получением трихлорбензола. В процессах
пиролиза хлорированных углеводородов обычно получается высоко-
концентрированный хлористый водород. Иногда он загрязнен хло-
ром, если хлор применяется в качестве инициатора при пиролизе.
3. Хлористый водород образуется при конденсации Фриделя-
Крафтса
RH4-R1CI -А1-\ R — Rj+HCI (9.16)
Примером такого процесса является конденсация хлоркеросина
с бензолом в производстве сульфонола, при которой получают высо-
коконцентрированный хлористый водород.
4. Процессы гидрофторирования хлорпроизводных углеводоро-
дов:
RC1 + HF > RF + HC1 (9.17)
В производстве фреонов и фторопластов получают хлористый
водород, загрязненный фтористым водородом и требующий специаль-
ной ОЧИСТКИ. ’
5. Значительным источником НС1 являются установки по сжи-
ганию хлорорганических отходов:
СпНтС1р+ (п + -24“£’) °2 —* вСОа+РНС1+ -2 Р-Н2О (9.18)
Образующийся хлористый водород очень разбавлен инертными
газами, продуктами сгоцания топлива и парами воды. Обычно его
удается перерабатывать только на разбавленную соляную кислоту.
6. Процессы высокотемпературного, гидролиза хлоридов ма-
гния [23, 24]
MgCl2 + H2O—> MgO-f-2HCl (9.19)
или железа [25]
2FeCl3-{-3H2O ---> Fe2O3 + 6HCl
(9.20)
488
Гидролиз хлорида магния приобретает большое значение при
получении MgO для нужд производства огнеупоров, особенно в связи
с развитием процессов переработки карналитовых руд, которые
сопровождаются получением KG1 и маточных растворов, бога-
тых MgCl2. Большое значение имеет также гидролиз FeCl3 в связи
с широким распространением солянокислотного травления металлов.
Аналогично может быть проведен гидролиз хлористого кальция
с получением хлористого водорода [26].
Хлористый водород при пиролитическом гидролизе хлоридов
магния, железа или кальция получается сильно разбавленным
инертными газами и парами воды, что затрудняет его рациональное
использование. При регенерации соляной кислоты из хлорного же-
леза травильных растворов удачно решается проблема использования
разбавленной кислоты, получаемой абсорбцией НС1, так как
для травления металлов применяются обычно разбавленные рас-
творы соляной кислоты.
Для получения обычной стандартной соляной кислоты или
100%-ного хлористого водорода из газов пиролиза хлоридов необ-
ходимо применять специальные приемы, которые будут рассмотрены
несколько ниже.
Хлористый водород может быть получен также и во многих дру-
гих производствах: при производстве фосфатов калия действием
фосфорной кислоты на 'КС1, активной двуокиси кремния — аэро-
сила — из SiCl4l монохлоруксусной кислоты из трихлорэтилена и др.
Для использования абгазной соляной кислоты в качестве товар- -
ной и абгазного хлористого водорода для гидрохлорирования в боль-
инстве случаев необходима очистка от загрязнений, а в ряде слу-
чаев и концентрирование НС1. Универсального метода очистки абгаз-
ного хлористого водорода от примесей не может быть предложено.
Способы очистки изменяются в зависимости от производства, в ко-
тором получается абгазный НС1, и загрязняющих его примесей.
Примеси и загрязнения в абгазном хлористом водороде могут
быть разбиты на несколько основных групп:
1) инертные газы: азот, водород, метан и др.
2) органические и хлэрорганические примеси (обычно высоко-
молекулярные соединения практически не растворяются в соляной
кислоте, но низкомолекулярные растворяются в значительной сте-
пени);
3) примеси кислого характера, например Cl2, HF, SO2 и др.;
4) механические примеси типа SiO2 в производстве аэросила;
5) пары воды.
Для каждой группы примесей имеются свои приемы и методы
очистки.
Инертные примеси нерастворимы в соляной кислоте, обычно их
можно отделить в процессе абсорбции НС1 водой при получении
соляной кислоты. Если требуется получить 100%-ный хлористый
водород, необходима последующая десорбция НС1 из соляной кис-
лоты.
in
489
t
Предложены также способы получения 100%-ного хлористого
водорода адсорбцией HG1 различными сорбентами с последующей
десорбцией НС1 при нагревании. В качестве адсорбентов предло-
жены сульфаты или фосфаты меди, свинца и кадмия [27—30], хлор-
окись железа [31], цеолиты [32], хлорид или бромид тетраэтилам-
мония [33]. Сорбцию проводят при обычной температуре, а десорб-
цию при нагревании до 100—120 °C. Сорбция хлористого водорода
сульфатами металлов (например, свйнца или меди) сопровождается
образованием соединений типа PbSO4-2HCl или CuSO4*2HC1 и выде-
лением значительного количества теплзц близкого к теплоте раство-
рения НС1 в воде.
В практике используют обычно абсорбцию НС1 водой, что удо.бг
нее и проще в аппаратурном оформлении. Экономические расчеты
процесса извлечения НС1 из разбавленных газовоздушных смесей
(около 7,5% HCI) с помощью хлористого тетраметил аммония, цео-
лита и сульфатов свйнца и меди показали, что этот способ значи-
тельно дороже водной абсорбции [34].
Для отделения хлорорганических примесей от абгазного хлори-
стого водорода широко используется охлаждение абгазов перед
подачей их на абсорбцию. Хлорорганические примеси, как правило/
имеют намного более высокую температуру кипения по сравне-
нию с НС1. Поэтому, охлаждая абгазы до минусовых температур т
можно добиться высокой степени конденсации органических и хлор-
органических примесей и снизить их содержание в хлористом водо-
роде до 0,01—0,001%. Так, охлаждая до —30 °C абгазы от произ-
водства хлораля перед подачей на абсорбцию, достигают снижения
содержания хлораля в соляной кислоте до 0,004% [35]. Однако глу-
бокое охлаждение абгазов не всегда экономически целесообразног
оно иногда может сопровождаться кристаллизацией и инкрустацией
теплообменных поверхностей. Поэтому чаще ограничиваются охла-
ждением абгазов в водяных холодильниках.
Для удалений хлорорганических примесей, имеющих невысокую
температуру кипения, с успехом применяют адиабатическую абсорб-
цию [36, 37]. При этом, вследствие проведения абсорбции при высо-
кой температуре, растворимость хлорорганических соединений в со-
ляной кислоте заметно снижается, ц эти примеси уносятся с инерт-
ными газами из абсорбционной колонны [38, 39]. Этому способствует
также то, что некоторые примеси образуют с водой, азеотропные
смеси, отгоняющиеся вместе с инертными газами.
С помощью адиабатической абсорбции можно получать соляную
кислоту с меньшим содержанием хлорорганических примесей по
сравнению с абсорбцией при обычной температуре, однако такая
соляная кислота все же не удовлетворяет требованиям многих потре-
бителей/
Для дополнительной очистки применяют отдувку примесей хло-
ристым водородом, инертными газами или перегретым паром [37].
При этом можно снизить содержание примесей в абгазной * кислоте
от производства хлорбензола в 10—20 раз [40]. Дополнительная
490
j 4
очистка может быть достигнута кипячением соляной кислоты в ниж-
ней части абсорбционной колонны [41—43]. Вместе с примесями
частично отгоняется HG1, и его концентрация в кислоте снижается
it
л
Для очисткиГот хлорорганических продуктов, легкосорбируемых
водными растворами соляной кислоты, применяют промывку абгаз-
ного хлористого водорода соляной кислотой [44]. При этом полу-
чают 10—25% загрязненной кислоты, для которой необходимо найти
потребителя или дополнительно подвергать ее очистке.
f Для очистки абгазного хлористого водорода от плохо растворимых
в воде органических примесей (например, четыреххлористого угле-
рода) вместо соляной кислоты можно применять высококипящие
абсорбенты, не растворяющие или плохо растворяющие НС1. К ним
относятся гексахлорбутадиен, трихлорбензол или высококипящие
парафиновые масла [45].
Для очистки абгазносо хлористого водорода от органических при-
месей предложено также сжигать их , в окислительной среде. При
этом к абгазиому хлористому водороду добавляют газы, содержащие
водород и избыток кислорода [46, 47], а также некоторое количество
хлора х[48]. При сжигании смеси происходит окисление органиче-
ских примесей. Метод отдувки прймесей из соляной кислоты инерт-
ными газами или кипячением мало эффективен для хорошо раство-
римых в кислоте примесей* Примеси неорганических солей [49],
метанола, фенола, крезолов и уксусной кислоты [50—52] предло-
жено удалять из соляной кислоты с помощью ионообменных смол,
однако этот способ очистки вряд ли может быть экономически целе-
сообразным для крупного производства.
Для очистки хлористого водорода от примесей, имеющих кислот-
ные свойства, нет единого способа, поэтому нужны специальные
приемы ддя удаления каждого вида примеЬей.
В абгазный хлористый водород попадает хлор вследствие непол-
ноты реакции хлорирования или использования хлора, как ини-
циатора реакции дегидрохлорирования. Всегда стремятся достичь
возможно полного использования хлора в процессах хлорирования
за счет применения избытка хлорируемого соединения или путем
пропускания абгазов хлорирования дополнительно через реактор
с хлорируемым соединением.
При адиабатической абсорбции, вследствие малой растворимости
хлора в горячей соляной кислоте, может быть получена кислота
со сравнительно небольшим содержанием хлора 0,002% даже при
наличии в абгазе около 8% хлора [39]. Хлор может быть отогнан
из соляной кислоты теми же приемами, что и легколетучие органи-
ческие соединения, т. е. продувкой инертными газами или кипя-
чением [36]. Для полной очистки от свободного хлора к соляной
кислоте добавляют гидразин и его производные, а также гидроксил-
амин в виде, неорганической или органической соли [53, 54].
Предложено много других способов очистки хлористого водорода
от свободного хлора, в частности абсорбцией хлора четыреххлористым
491
углеродом [45] или путем адсорбции на угле при пониженной
температуре [55], а также взаимодействием хлора с окисью углерода
и парами воды — каталитической реакцией, протекающей на акти-
вированном угле с образованием НС1 и СО2 [56].
Для очистки хлористого водорода от HF можно промывать газо-
вую смесь соляной кислотой, содержащей хлористый кальций.
При этом образуется малорастворимый Са F2, который выпадает в оса-
док [57]. Фтористый ,водород предложено также сорбировать на
окислах СаО [57а], А1 (ОН)3 [58], силикагеле, а-А1203 или сили-
катах [59]. Для очистки хлористого водорода от SO2 предложены
отмыв SO2 концентрированной серной кислотой, окисление SO2
хлором до H2SO4 [60], адсорбция активированным углем при пони-
женной температуре [55], очистка от серной кислоты и SO2 с по-
мощью А1С13 [60].
Иногда абгазный хлористый водород загрязнен механическими
примесями, что имеет место в производстве сульфата натрия (осо-
бенно при проведении процесса в аппаратах с псевдоожиженным
слоем), а также активной двуокиси кремния гидролизом SiCl4,
где в газах содержится мелкая пыль SiO2. Такие загрязнения отде-
ляют от газа сепарацией или фильтрованием.
В ряде случаев абгазный хлористый водород разбавлен большим
количеством паров воды, например при пирогидролитическом разло-
жении хлоридов магния и железа, в производстве пирофосфата калия
и других случаях. Из такого абгазного хлористого водорода нельзя
обычными приемами абсорбции получить концентрированную соля-
ную кислоту. Возможно образование разбавленной кислоты, для
переработки которой в концентрированный НС1 или стандартную
соляную кислоту необходимы специальные методы обезвоживания
с применением водоотнимающих средств.
Некоторые способы очистки абгазного хлористого водорода
и соляной кислоты, получаемой из абгазов различных хлороргани-
ческих производств, рассмотрены в работе [61].
СПОСОБЫ ПОЛУЧЕНИЯ соляной кислоты
Соляную кислоту получают абсорбцией хлористого водорода
водой. Процесс растворения НС1 в воде протекает интенсивно с вы-
делением большого количества тепла. По способу отвода выделя-
ющегося тепла различают два основных способа абсорбции хло-
ристого водорода [62, 63]: изотермический — с охлаждением абсор-
бера и абсорбента — и адиабатический, при котором поглощение HCI
протекает при высокой температуре и тепло реакции отводится за
счет испарения воды.
Иногда применяют смешанную адиабатическо-изотермическую
абсорбцию г когда на отдельных стадиях применяется адиабатический
режим и отвод тепла осуществляется путем испарения йоды, исполь-
зуемой для абсорбции, а на других — процесс протекает при пони-
женной температуре и постоянном принудительном охлаждении.
492
Каждый из этих способов имеет свои преимущества и недостатки.
Вопрос о целесообразности применения того или иного способа
обычно зависит от условий производства и требований к проведению
процесса.
Вода на Охлаждающая
Инертные вева
газы 1
mi ммнннмш Iwiiifl
Вода
7/УЛ
Изотермическая абсорбция
При изотермической абсорбции тепло отводится в результате .
непосредственного охлаждения абсорбера или с помощью холодиль-
ника, через который циркулирует кислота.
Способ изотермической абсорбции использовался с начала заро-
ждения производства соляной кислоты. Поскольку с понижением
температуры уменьшается и парци-
альное давление НС1 над водными
растворами соляной кислоты, то
путем изотермической абсорбции
можно полнее извлекать НС1 из га-
зов и получать соляную кислоту
более высокой концентрации. При
этом вместе с хлористым водородом
сорбируются также некоторые ле-
тучие примеси. Изотермическую
абсорбцию целесообразно применять
для получения концентрированной
кислоты и для переработки хлори-
стого водорода низкой концентрации.
# У становки для изотермической
абсорбции должны иметь сравни-
тельно большие поверхности тепло-
передачи для отвода тепла, поэтому
они более громоздки и требуют
больших капитальных затратно сра-
внению с установками для адиаба-
тической абсорбции.
Первоначально для установок изотермической абсорбции приме-
нялось разнообразное керамическое оборудование типа турилл,
целяриусов, башен, охлаждаемых снаружи водой. Низкая тепло-
проводность керамики обусловила применение главным образом
аппаратов с абсорбцией поверхностью воды или кислоты, запол-
няющей туриллы, целяриусы или другие аналогичные аппараты.
Развитие поверхности абсорбции с целью интенсификации процесса
было ограничено величиной теплопередающей поверхности и воз-
можностями отвода тепла. Абсорбция может проводиться в аппаратах
колонного типа, причем необходимо иметь несколько абсорбционных
колонн с промежуточными охлаждением кислоты между ними.
С появлением оборудования из игурита и импрегнированного
графита керамические аппараты для получения соляной кислоты
были практически полностью вытеснены. Высокая теплопроводность
адсорбцию I
Соляной
кислота
Рис. 9-8. Схема устройства блоч-
ного изотермического абсорбера.
493
углеграфитовых материалов позволяет интенсифицировать [про-
цесс абсорбции НС1.
Наиболее распространены блочные абсорберы из импрегнирован-
ного графита, представляющие собой теплообменник блочного типа
отверстиями и каналами для
о
с расположенными крест-накрест
прохождения реагирующей смеси хлористого водорода и соляной
кислоты и для охлаждающей воды. На рис. 9-8 показано устройство
такого абсорбера. В качестве абсорберов применяются также труб-
494
чатые теплообменники из игуритовых труб (рис. 9-9). Абсорберы
из импрегнированного графита и игурита позволяют работать с га-
зами при сравнительно высокой температуре (до 300 °C) [64].
Очень хорошим материалом для аппаратуры производства соля-
ной кислоты является тантал. Он стоек в соляной кислоте различных
концентраций во всем интервале температур, применяемых в про-
мышленности. Однако из-за высокой стоимости
и малой доступности тантала известны только
единичные случаи его использования.
Абсорберы, работающие по принципуУпада-
ющей пленки, применялись давно, однако
изготовление этих аппаратов из стекла или
эмалированной стали ограничивало область их
применения лишь в установках малой произ-
водительности. Разработка углеграфитовых
материалов позволила изготавливать много-
трубчатые абсорберы с падающей пленкой
[65, 66]. В таких абсорберах внутри трубок
по стенкам прямотоком с газом стекает пленка
абсорбента — соляной кислоты, а в межтрубном
пространстве протекает охлаждающая вода.
Пленочные абсорберы целесообразно приме-
нять для поглощения JIC1 из высококонцент-
рированных газов. В таких абсорберах проис-
ходит недостаточно полное поглощение НС1,
поэтому на выходе из аппарата необходимо
устанавливать хвостовую насадочную колонну
для улавливания непоглощенного хлористого
водорода. Примерная’схема[установки с пленоч-
ными абсорберами показана [66] на рис. 9-10.
Если способом изотермической абсорбции
кислоту из абгазов хлорорганических производств, содержащих
пары этих продуктов или других растворимых в соляной кислоте
примесей, то кислота содержит большое количество загрязнений.
Это объясняется тем, что при контакте абгазов с холодной кислотой
может наступать конденсация примесей, содержащихся в абгазах.
Растворимость этих примесей в холодной кислоте выше, чем в горя-
чей. Кроме того, при конденсации хлорорганических примесей могут
образоваться трудно расслаивающиеся смеси, что затрудняет отде-
ление органических загрязнений.
Инертные
газы
НС1
Вода.
.Вода на
< 1
абсорбцию
[ЙонцентрироВа нна я
*
кислота
Рис. 9-10. Схема уста-
новки для абсорбции
хлористого водорода
с пленочным абсор-
бером:
1 — хвостовая колонна;
2 — абсорбер второй
ступени; з — абсорбер
первой ступени.
получают соляную
Адиабатическая абсорбция
При адиабатической абсорбции образующаяся соляная кислота
нагревается за счет тепла реакции до температуры кипения, поэтому
абсорбция производится практически кипящей кислотой. Отвод
избыточного тепла реакции осуществляется путем испарения части
воды из соляной кислоты. Если на абсорбцию подавать 100%-ный
495
НС1 при О °C и вести процесс абсорбции в адиабатических усло-
виях водой при исходной температуре 20 °C, температура жидкости
будет возрастать по мере поглощения хлористого водорода
и, при концентрации образующейся соляной кислоты около
16,5% НС1, достигнет температуры кипения этой кислоты 107 °C.
При продолжении процесса абсорбции НС1 и повышении концен-
трации соляной кислоты температура абсорбции будет определяться
точкой кипения кислоты соответствующей концентрации. Сначала,
Вода на
адсорбцию
Инертные
-з газы
2| __Ю0% Н2О 100°С
----О % HCJ 100°С
---5% HCI 10Т>С
— 10% HCI
---15% HCI
20% HCI
--25% HCI
--30% HCI
103°С
Ю6°С
109*0
105°С
90°С
--35% НС1
-Д^—100% HCI о°с
Гнс1
вО°С
Кислота
Рис. 9-11. Схема распределения темпера-
тур и концентрации соляной кислоты по
высоте адиабатического абсорбера:
1 — температура; 2 — концентрация соляной
кислоты.
по мере повышения концент-
рации кислоты до азеотроп-
ной, температура возрастет
до точки кипения азеотропа
(до 108,5 °C), а затем пони-
зится в соответствии с умень-
шением температуры кипения
более концентрированной ки-
слоты.
При проведении процесса
абсорбции хлористого водо-
рода в аппарате колонного
типа с противотоком газа
и кислоты происходит опре-
деленное распределение тем-
ператур и концентрации со-
ляной кислоты по высоте
колонны. На рис. 9-11 при-
ведено такое распределение
для случая абсорбции 100%-
ного хлористого водорода,
поступающего в колонну при
0 °C, водой, подогретой до
100 °C. При других темпера-
турах газа и воды, поступаю
;их на абсорбцию, распределение тем-
ператур несколько изменяется, но общая закономерность остается
и
такой же.
В каждой точке колонны парциальное давление хлористого водо-
рода приближается к равновесному для системы НС1—Н2О при
данной температуре, однако оно остается несколько выше рав-
новесного. Без осуществления противотока по методу адиабати-
ческой абсорбции нельзя обеспечить достаточно полное извлече-
ние НС1 из газов, поступающих на абсорбцию, так как при темпера-
туре кипения соляной кислоты парциальное давление хлористого
водорода сильно возрастает с увеличением концентрации кислоты.
Поступающие на абсорбцию газы кроме хлористого водорода
и инертных примесей часто содержат пары воды. Для определения
условий абсорбции и предельно возможной концентрации получаемой
соляной кислоты основное значение имеет соотношение парциаль-
ных давлений паров воды и хлористого водорода. Наличие инертных
496
газов в смеси имеет меньшее значение. Сумму парциальных давлений
паров воды и хлористого водорода, равную разности между общим
давлением газов и парциальным давлением инертных газов в смеси,
иногда называют «приведенным давлением».
На рис. 9-12 показано соотношение парциального давления НС1
и суммы парциальных давлений НС1 и Н2О при различных темпера-
турах для соляной кислоты концентрацией 5—20%, т. е. ниже азео-
тропной. На рис. 9-13 приведены аналогичные данные о соотноше-
нии давления паров воды и суммы парциальных давлений НС1 +
+ Н2О для соляной кислоты концентрацией выше азеотропной [67].
Рис. 9-12. Отношение парциального давления НС1 к сумме давления
НС1 + Н2О для концентрации 5—20% НС1.
При противотоке воды и газа, содержащего хлористый водород,
в условиях адиабатической абсорбции и достаточной высоты абсорб-
ционной колонны можно достичь практически полного извлечения
хлористого водорода из газа, так как выходящая из колонны смесь
водяного пара и инертных газов близка к равновесию с практически
чистой водой при 100 °C. Концентрация кислоты внизу колонны
зависит от парциального давления хлористого водорода в поступа-
ющем на абсорбцию газе, состава и температуры газа и соответ-
ственно температуры получаемой кислоты [68].
На рис. 9-14 приведены расчетные соотношения концентраций HG1
в жидкой и газовой фазе по высоте колонны при адиабатической
абсорбции чистого 100%-ного хлористого водорода при атмосферном
давлении. Из рисунка видно, что максимально достигаемая кон-
центрация соляной кислоты составляет около 35% HG1. Кривая
32 Заказ 843 497
37,5 % -ной кислоты пересекает равновесную кривую, что свидетель-
ствует о невозможности в этих условиях получения такой ки-
слоты [67].
При изменении концентрации НС1 в газах, поступающих на аб-
сорбцию, путем разбавления их инертными примесями условия аб-
сорбции несколько меняются. На рис. 9-15 показано соотношение
концентраций хлористого водорода в жидкой и газовой фазах при
различном парциальном давлении хлористого водорода в исходном
} ммрт. СЛЪ
Рис. 9-13. Отношение парциального давления паров Н20 к сумме
давлений НС1 + Н2О для концентраций 25—38% НС1.
J
I
газе. При достаточной высоте колонны обеспечивается практически
полная абсорбция НС1 и концентрация кислоты внизу колонны будет
определяться в основном соотношением хлористого водорода и воды,
поступающих в колонну.
Концентрация НС1 в жидкости возрастает от верха колонны
к низу. Максимальная температура в колонне наблюдается там, где
концентрация хлористого водорода в кислоте соответствует азео-
тропной смеси. Этой точке соответствует температура кипения азео-
тропной кислоты (около 108,5 °C). Выше и ниже этой точки темпе-
ратура в колонне снижается и соответствует температуре кипения
кислоты данной концентрации. Температура соляной кислоты,
выводимой из колонны, зависит от содержания инертных газов
в смеси: чем выше содержание инертных газов, тем ниже температура
кислоты. Поскольку потери тепла через стенки в общем тепловом
балансе невелики, температура и концентрация кислоты мало зави-
сят от изменения нагрузки в широких пределах.
1'1
f1
i
а*
V
Л
498
о 60
100 г-
.о
^80
в го
о ю го jo 4о
Содержание HCf 6 жидкости%
Рис. 9-14. Расчетное распре-
деление концентрации НС1
в жидкой и газовой фазах по
высоте адиабатической ко-
лонны при абсорбции чистого
НС1 в условиях атмосферного
давлении:
1 — 25% НС1; 2 — 30% HCI;
5 — 35% НС1; 4 — 37,5% НС1;
5 — равноверная кривая.
Колоннц адиабатической абсорбции соляной кислоты могут
работать со значительно меньшей плотностью орошения жидкостью
по сравнению с насадочными колон-
нами в других абсорбционных процес-
сах. Из-за отсутствия теплопереда-
ющих поверхностей установка для
адиабатической абсорбции более ком-
пактна, снижаются затраты на обору-
дование установки. Как было указано
ранее, при адиабатической абсорбции
легколетучие органические примеси и
хлор только в небольшой степени
сорбируются горячей соляной кисло-
той, их основное количество уносится
с газами цз абсорбера и может быть
выделено из абгазов. При этом обра-
зуется более чистая кислота, чем при
изотермической абсорбции.
Концентрация получаемой соляной
кислоты зависит от содержания HG1
в исходном газе и температуры кислоты.
При благоприятных условиях теорети-
чески может быть получена кислота,
содержащая до 35% HG1. Практически
в промышленных условиях равновес-
ное состояние между жидкой и газовой фазами не достигается, по-
этому получают соляную кислоту, содержащую 30—32% НС1.
1*1
Жидкая фаза (% HCI)
Рис. 9-15. Кривые равновесия в системе НС1—Н2О при
давлении:
1 — 760 мм рт» ст.; 2— 608 мм рт. ст.; 3 — 456 мм рт. ст.; 4 —
304 мм рт. СТ.; 5 — 152 мм рт. ст.
32*
Впервые адиабатическая абсорбция была предложена для полу-
чения соляной кислоты Гаспаряном [69] и быстро нашла широкое
применение в промышленности [36, 70, 71]. Хотя путем адиабатиче-
ской абсорбции нельзя получить соляную кислоту высокой концен-
трации, этот способ широко применяется для переработки абгазного
хлористого водорода после хлорорганических производств. В по-
следнем случае часто получают соляную кислоту, пригодную для
применения некоторыми потребителями без дополнительной очистки.
Инертные газы
^Вода на
Рис. 9-16. Совмещенный
абсорбер:
1 — адиабатический абсор-
бер; 2 — конденсатор.
Рис. 9-17. Схема установки для адиабатической
абсорбции НС1 из отходящих газов хлорорганиче-
ских производств:
1 — адиабатический абсорбер; 2 — конденсатор; з — раз-
делитель; 4— хвостовая колонна; 5 — разделитель водной
и органической фаз; 6 —сборник органической фазы; 7 —
сборник водной фазы; 8 — насос; 9 — отдувочная колонна;
10 — теплообменник; 11 — сборник кислоты; 12 — насос.
Для абсорбции используются обычно колонны с насадкой из
колец Рашига. Материалом для изготовления колонн служит импре-
* гнированный графит. Могут применяться также металлические
защищенные колонны или колонны из фаолита.
Для определения размеров колонн могут быть использованы
известные методы расчета ректификационных колонн непрерывного
действия [71, 72]. Высота абсорбционной колонны или число тре-
буемых теоретических тарелок зависит от концентрации НС1 в газе,
концентрации получающейся кислоты и допустимого проскока
хлористого водорода на выходе из колонны.
Вытекающую из низа колонны горячую кислоту охлаждают для
удобства хранения, перевозки и работы с ней. Известна конструкция
адиабатического абсорбера, совмещенного с конденсаторам для
отходящих из абсорбера паров воды (рис. 9-16). Над адиабатическим
абсорбером с насадкой расположен конденсатор. При этом в про-
цессе конденсации паров воды происходит дополнительное улавли-
500
вание небольших количеств хлористого водорода, уносимого абга-
зами вместе с парами воды, конденсат затем поступает на абсорб-
цию HCL Однако такая схема работы может быть применена только
для абсорбции чистого хлористого водорода.
Установка для адиабатической абсорбции хлористого водо-
рода из абгазов хлорорганических производств показана на
рис. 9-17.
Рис. 9-18. Схема получения концентрированной соля-
ной кислоты из абгазов.
1 — колонна адиабатической абсорбции; 2 — конденсатор;
з — отделитель газов; 4 — разделитель органической и водяной
фаз; 5 — холодильник; 6 — сборник кислоты; 7 — насос; 8 —
изотермический абсорбер; 9 — сборник концентрированной ки-
слоты.
Пары хлорорганических веществ уносятся из колонны и вместе-
с парами воды поступают в конденсатор. После конденсатора проис-
ходит разделение органической и водной фаз. Органическая фаза
может возвращаться в производственный цикл, а слабокислый вод-
ный конденсат после нейтрализации поступает на очистные сооруже-
ния или передается на абсорбционную колонну, как показано на
рис. 9-17.
Горячая соляная кислота, вытекающая из колонны адиабатиче-
ской абсорбции, содержит небольшие количества органических за-
грязнений. Содержание примесей в соляной кислоте, полученной
из абгазов хлорорганических производств, зависит от характера
технологического процесса, при котором образуется абгазный НС1.
При использовании процесса адиабатической абсорбции хлористого
водорода из абгазов производства хлорбензола получаемая соляная
кислота обычно содержит не более 0,01—0,02 вес. % органически
связанного хлора, а после производства метиленхлорида хлори-
рованием метана получается соляная кислота, содержащая 0,2—
0,4 вес. % органических примесей,
501
В большинстве случаев абгазная кислота от производств хлор-
органических продуктов по качеству отвечает требованиям к кислоте
2-го сорта (МЁТУ 6-01-193—68):
Состав
1Г
J L
Содержание, %
. Не менее 27,5
. Не более 0,02
. Не более 0,02
. Не более 0,8
торорганических продуктов
HCI . . . ...............
Свободный хлор...........
Железо ..................
Органически связанный хлор
Абгазная кислота от производства
без специальной очистки, кроме того, содержит 1—2% HF.
Для удобства хранения и транспортирования кислоту охлаждают
водой в теплообменнике.
Поскольку температура кислоты Внизу колонны зависит от ее
концентрации (т. е. от соотношения НС1 и воды, подаваемой на
абсорбцию), подачу воды можно легко регулировать автоматически
по температуре внизу колонны.
На рис. 9-18 показана установка для получения высококонцен-
трированной соляной кислоты из абгазов хлорорганических произ-
водств. Установка состоит из двух ступеней абсорбции. Первая
ступень адиабатической абсорбции используется для получения
кислоты, содержащей 30—32% НС1 и очищенйой от хлорорганиче-
ских загрязнений. После охлаждения такую кислоту направляют
в колонну изотермической абсорбции, где она донасыщается чистым
хлорцстым водородом при пониженной температуре [73].
ПОЛУЧЕНИЕ 100%-Н0Г0 ХЛОРИСТОГО ВОДОРОДА
ПУТЕМ СТРИППИНГ-ПРОЦЕССА
Хотя хлористый водород в ряде методов его производства сразу
получается достаточно чистым и концентрированным, он может
быть использован без дополнительной очистки лишь отдельными
потребителями. Хлористый вбдород 95—99%-ный может быть полу-
чен синтезом из элементарных веществ, либо как абгазный при пиро-
лизе хлорпродуктов, в производстве сульфанола и др. В таких слу-
чаях помимо его очистки от органических примесей требуется иногда
только дополнительная сушка и компримирование газа для транс-
портирования и преодоления противодавления, создаваемого на
участках потребления.
Для большинства потребителей газообразного хлористого водо-
рода обычно необходим тщательно осушенный газ, и только отдель-
ные производства, использующие, например, НС1 для целей выса-
ливания из водных растворов, могут использовать влажный хло-
ристый водород. Осушка хлористого водорода необходима также
для снижения коррозионной активности газа. Тщательно высушен-
ный хлористый водород может транспортироваться по стальным тру-
бопроводам.
Для осушки хлористого водорода можно применять сернокислот-
ные установки, аналогичные установкам для осушки хлора. Если
502
при сернокислотной осушке (98,3—72,3% H2SO4) дополнительно
охлаждать газ до —10 °C, то содержание влаги в хлористом водороде
можно снизить до 0,005% [61, 74].
Предложены и другие способы осушки влажного хлористого*
водорода, в частности охлаждением под давлением [75], с помощью
алюмогеля, силикагеля, безводного хлористого алюминия и других
водоотнимающих средств [63]. Широкое распространение получила
осушка хлористого водорода охлажденной концентрированной соля-
ной кислотой. Этот метод будет подробнее рассмотрен ниже.
Компримирование хлористого водорода может проводиться рота-
ционным компрессором с сернокислотным заполнением типа РЖК
или НЭШ.
Однако в большинстве случаев возникает задача подучения чи-
стого высокопроцентного (около 100%) хлористого водорода из его
смесей с различными газами и парами воды. Выделение НС1 из его
газовых смесей с помощью твердых сорбентов [27—32] мало исполь-
зуется в промышленности. Наиболее распространена сорбция НС1
водой с последующей его десорбцией. Этот способ, получивший• на-
звание стриппинг-процесса, позволяет получать чистый сухой хло-
ристый водород под давлением [76].
Стриппинг-процесс заключается в разделении бинарной смеси
НС1—Н2О ректификацией. Полное разделение этой смеси простой
ректификацией невозможно, так как образуется азеотропная
смесь НС1 и воды, которая при атмосферном давлении кипит при
108,5 °C и содержит около 20—22% НС1 [77].
При ректификации кислоты, содержащей более 20% НС1, из
верхней части колонны будет отводиться чистый хлористый водо-
род, а из куба — азеотропная смесь. Выходящий из колонны хло-
ристый водород охлаждается в обратном холодильнике сначала
водой, а затем рассолом до —13 4- —15 °C. При этом содержащаяся
в хлористом водороде влага конденсируется и образует с HG1 кон-
центрированную соляную кислоту, которая в виде флегмы возвра-
щается в колонну. Парциальное давление паров воды над охлажден-
ной концентрированной соляной кислотой невелико, поэтому осушку
отходящего из колонны хлористого водорода проводится до оста-
точного содержания влаги менее 0,01%.
Содержание влаги в хлористом водороде можно снизить за счет
применения более глубокого холода, однако надо учитывать, что
при температурах ниже —17 °C происходит образование твердого
гидрата хлористого водорода, забивающего теплообменники.
Стриппинг-процесс можно проводить под давлением, получая
хлористый водород сразу под необходимым давлением без установки
специальных компрессоров [78]. При этом для обогрева куба ко-
лонны необходимо применять пар более высокого давления, а вся
аппаратура стриппинга-процесса должна быть рассчитана на соответ-
ствующее давление.
Отбираемая из куба колонны азеотропная соляная кислота по-
дается на установку абсорбции НС1 из его смесей с инертными газами
к
и после донасыщения вновь возвращается на ректификацию в стрип-
пинг-установке. Чем больше концентрация HG1 в соляной кислоте,
поступающей на стриппинг-процесс, тем выше извлечение хлористого
водорода из соляной кислоты за один проход ее через ректификацион-
ную колонну, а меньше количество кислоты, которую надо пропу-
стить через стриппинг-установку для получения 1 т чистого НС1
и соответственно ниже удельный расход тепла и электроэнергии.
Поэтому для достижения максимальной производительности
стриппинг-установки пр чистому хлористому водороду и минималь-
ных удельных затрат энергетических ресурсов на производство целе-
сообразно для получения возможно более концентрированной соля-
ной кислоты донасыщать азеотропную кислоту путем изотермиче-
ской абсорбции. Однако, как уже было сказано, при этом получается
соляная кислота с большим количеством загрязнений. Необходимо
учитывать, что в стриппинг-процессе вместе с хлористым водородом
будут практически полностью отгоняться все легколетучие примеси,
. содержащиеся в соляной кислоте, которая поступает на ректифика-
цию. Этому способствует образование низкокипящих азеотропных
смесей кислоты со слабо растворимыми в воде органическими при-
месями.
Для получения 1 т чистого хлористого водорода в зависимости
от концентрации НС1 в исходной кислоте (30—35% НС1) необходимо
пропустить через ректификационную колонну от 8 до 4,5 т кислоты.
Практически все легколетучие примеси из этой кислоты перейдут
в состав получаемого хлористого водорода. Таким образом, в стрип-
пинг-процессе не происходит очистки хлористого водорода от лету-
чих примесей, а, наоборот, возможно загрязнение ими, до 5—10 раз
большее по сравнению с исходной соляной кислотой.
Для получения достаточно чистого хлористого водорода необ-
ходимо на стриппинг-процесс подавать соляную кислоту, содержа-
щую в 5—10 раз меньше примесей против допускаемых для чистого
хлористого водорода. Например, для получения хлористого водорода,
содержащего не более 0,01% СС14, на установке стриппинга, пере-
рабатывающей 30%-ную соляную кислоту, содержание СС14 в послед-
ней должно быть не выше 0,001%. Поэтому к качеству соляной кис-
лоты, поступающей на ректификацию в стриппинг-установках,
всегда предъявляются высокие требования.
На установках стриппинга, перерабатывающих абгазы хлор-
органических производств, применяют обычно адиабатическую аб-
сорбцию для донасыщения азеотропной кислоты и дополнительную
очистку полученной концентрированной кислоты путем отдувки
примесей инертными газами или кипячения. Это приводит к сниже-
нию концентрации НС1 в кислоте, поступающей на ректификацию,
ухудшает технико-экономические показатели производства, но по-
зволяет получить хлористый водород требуемого качества.
Для улучшения энергетических показателей установки стрип-
пинга предложен ряд усовершенствований в схеме ректификацион-
ной установки. Предложено направлять конденсат концентрирован-
504
11
ной соляной кислоты из холодильников для хлористого водорода
в специальную укрепляющую часть ректификационной колонны [781,
а также орошать укрепляющую часть колонны искусственной флег-
мой — 40—42%-ной соляной кислотой, специально получаемой путем
донасыщения части исходной кислоты хлористым водородом, выхо-
дящим из колонны [62].
Установки стриппинга работают в среде горячей соляной кислоты,
поэтому к конструкционным материалам предъявляются очень
жесткие требования. Для изготовления теплообменной аппаратуры
Рис. 9-19. Схема установки
для получения 100%-ного
хлористого водорода из
абгазо?:
1 — адиабатический абсорбер;
2 — конденсатор; 3 — отдели-
тель тазов; 4 — разделитель
фаз; 5 — отдувочная колонна;
6 — сборник кислоты; 7 — на-
сос; 8 — теплообменник; 9 —
кипятильник; 10 — ректифика-
ционная колонна; 11 — водя-
ной холодильник; 12 — рас-
сольный холодильник.
пригодны только углеграфитовые материалы и тантал. Поскольку
тантал мало доступен для применения в промышленности, для изго-
товления аппаратуры установок стриппинга применяется практи-
чески только импрегнированный графит. Трубопроводы изгото-
вляются также из фторопласта. Керамика и стекло мало подходят
для изготовления установок, работающих под давлением.
На рис. 9-19 приведена принципиальная схема получения
100%-ного хлористого водорода на стриппинг-установке. Для пере-
работки абгазов хлорорганических производств в схеме дополни-
тельно предусмотрена стадия очистки соляной кислоты, путем от-
дувки загрязняющих хлорорганических примесей.
ПЕРЕРАБОТКА АБГАЗНОГО ХЛОРИСТОГО ВОДОРОДА,
СИЛЬНО РАЗБАВЛЕННОГО ПАРАМИ ВОДЫ
Значительные количества абгазного хлористого водорода, полу-
чаемого в виде побочного продукта, разбавлены парами воды. К про-
изводствам, в которых получаются разбавленные парами воды газо-
вые смеси, содержащие НС1, относятся термический гидролиз хло-
ридов магния, получение пирофосфата калия и др. Основную трудность
505
б переработке таких абгазов представляет повышенное содер-
жание в них паров воды. В зависимости от соотношения содержания
паров воды и НС1 в этих газах меняются способы их возможной пе-
реработки. Если соотношение Н2О : НС1 < 2,9, способом адиабати-
ческой или изотермической абсорбции можно получить кислоту
концентрации выше азеотропной. Из такой кислоты может быть
получен 100%-ный хлористый водород ректификацией на установке
.стриппинга.
При более высоком содержаний паров воды простыми способами
абсорбции нельзя получить кислоту концентрации выше азеотропной.
Разбавленная соляная кислота (10—20% НС1), как правило, не
находит сбыта в народном хозяйстве и нуждается в переработке ее
на концентрированный хлористый водород или стандартную соляную
кислоту. Если на том же предприятии имеются ресурсы концентри-
рованного хлористого водорода, разбавленная абгазная соляная
кислота может быть использована вместо воды для абсорбции хло-
ристого водорода с выпуском стандартной товарной кислоты. Однако
в ряде случаев такая возможность отсутствует.
Рассмотренный в предыдущем разделе способ получения 100%-
ного НС1 ректификацией концентрированной соляной кислоты пред-
усматривает донасыщение азеотропной соляной кислоты хлористым*
водородом. Если потребитель 100%-ного хлористого водорода рас-
положен далеко от источников абгазного НС1, производство ослож-
няется необходимостью транспортирования большого количества
соляной кислоты и перевозки в обратном направлении азеотропной
кислоты для ее донасыщения.
Вопрос может быть fрешен организацией переработки соляной
кислоты с полным превращением содержащегося в ней НС1 в газо-
образный 100%-ный хлористый водород.
Парциальное давление НС1 над разбавленными растворами соля-
ной кислоты невелико и возрастает при приближении к концентра-
ции азеотропа. Поэтому разбавленная соляная кислота, содержа-
щая НС1 меньше, чем в азеотропе, может быть сконцентрирована
ректификацией. При этом в верхней части колонны будет отбираться
практически чистая вода, а из куба колонны отводиться, так же как
и в стриппинг-процессе, азеотропная кислота.
Предложено выделять 100%-ный хлористый водород из азео-
тропной соляной кислоты ее ректификацией под давлением 60—
127 ат и соответственно при температуре 280—325 °C [79].
С повышением давления состав азеотропной кислоты изменяется.
Содержание НС1 в азеотропной смеси при указанных давлениях
составляет 2,8—0,1%. Применение давления около 100 ат при тем-
пературе примерно 300 °C встречает большие трудности в связи
с отсутствием материалов, пригодных для конструирования необ-
ходимой аппаратуры. Поэтому для получения хлористого водорода
из разбавленных растворов соляной кислоты и из азеотропной
кислоты сочетают ректификацию с использованием водоотнимающих
средств [67, 80]. '
.506
Ill
Предложено много вариантов осуществления такого процесса
с концентрированной серной кислотой в качестве водоотнимающего
средства [81—86].
Серная кислота связывает воду, а выделяющийся хлористый
водород отводится из верхней части колонны. Для осушки хлори-
стого водорода кроме серной кислоты применяют концентрирован-
t ную охлажденную соляную кислоту (стр. 503). Разбавленная сер-
ная кислота должна быть сконцентрирована и вновь возвращена
в цикл или ее следует постоянно донасыщать за счёт введения сер-
ного ангидрида. Сочетание серной кислоты с соляной создает труд-
ности в подборе достаточно коррозионно-стойких материалов. По-
этому для практического использования более приемлемы способы.
f основанные на применении в качестве водоотнимающих средств кон-
J центрированных растворов СаС12 или MgCl2 [87—89].
L Описана методика расчета колонны для экстрактивной ректифи-
f кации соляной кислоты с использованием в качестве водоотнима-
ющего средства концентрированных растворов хлористого кальция.
Процесс может быть осуществлен также в две стадии. На первой
стадии хлористый водород выделяется из концентрированной соля-
ной кислоты (30—35% НС1) в обычных ректификационных установ-
ках, а азеотропная кислота иэ куба колонны первой стадии посту-
пает на вторую стадию экстрактивной ректификации с раство-
рами СаС12. Газообразный хлористый водород из второй колонны
поступает в первую, откуда отбирается после осушки. На получе-
ние 1т хлористого водорода из 21 %-ной соляной кислоты расхо-
дуется 4,85 т соляной кислоты, 25 т 55%-ного раствора СаС12 и 2 т
пара [90].
Разбавленный раствор хлористого кальция необходимо концен-
трировать для возвращения в цикл. Раствор СаС12 содержит некото-
рое количество соляной кислоты, поэтому его концентрирование
возможно только в углеграфитовых аппаратах. При концентрирова-
нии раствора СаС12 хлористый водород летит вместе с парами воды,
загрязняя конденсат. При этом потери хлористого водорода соста-
вляет несколько процентов от вырабатываемого количества НС1.
Кроме потерь продукции образуются очень слабые растворы соляной
кислоты, которые усложняют проблему очистки сточных вод.
Разбавленные растворы СаС12 перед передачей их на концентри-
рование могут быть нейтрализованы известковым молоком с образо-
ванием хлористого кальция, при этом облегчается решение вопроса
сточных вод производства, и выпарку разбавленных растворов СаС12
можно проводить в стальной аппаратуре. Объем раствора хлори-
стого кальция, циркулирующего в системе, будет все время возра-
стать, поэтому его избыток следует выводить для реализации на
стороне. При наличии на предприятии производства СаС12 избыточ-
ные растворы хлористого кальция могут быть переданы на это произ-
I водство. z
g Для проведения экстрактивной ректификации желательно при-
менять возможно более концентрированные растворы хлористого
507
кальция, однако надо учитывать, что 55—56%-ные растворы СаС12
кристаллизуются при температуре, близкой к комнатной. Чтобы
исключить кристаллизацию СаС12 в трубопроводах, арматуре и свя-
занные с этим нарушения в работе аппаратов, нужны специальные
меры. Поэтому верхний предел концентрации растворов СаС12
обычно лежит около 52—55%, а разбавленных растворов около 42%.
На рис. 9-20 приведена примерная схема установки для экстрак-
тивной ректификации соляной кислоты с концентрированными рас-
творами хлористого кальция. Установки такого типа могут исполь-
Рис. 9-20. Схема экстрактивной ректи-
фикации соляной кислоты с растворами
хлористого кальция:
1 — холодильник; 2 — ректификационная
колонна; з — нейтрализатор; 4 — выйар-
ной аппарат; 5 — холодильник раствора
СаС12.
зоваться для производства
100%-ного хлористого водорода
из привозной соляной кислоты.
Технико-экономические рас-
четы, проведенные Бабаян Е. Л.
с сотр., показали, что себе-
стоимость 100%-ного хлори-
стого водорода, полученного
из привозной абгазной соляной
кислоты, близка к себестоимо-
сти хлористого водорода, полу-
ченного синтезом из электро-
литического хлора и водорода.
Экстрактивная ректифика-
ция соляной кислоты может
проводиться также с растворами
MgCl2 [91]. Такая схема осо-
бенно удобна для получения
сухого хлористого водорода из
разбавленных отходящих газов от процесса термического разложения
хлористого магния. При концентрировании разбавленных растворов
MgCl2 их можно нейтрализовать с помощью Mg (ОН)2, а избыточ-
ное количество MgGl 2 возвратить на термическое разложение.
Парциальное давление паров воды над растворами MgCl2 сильно
снижается с увеличением концентрации хлорида магния (рис. 9-21).
Одновременно снижается также растворимость хлористого водо-
рода в растворах MgCl2 (рис. 9-22). Поэтому целесообразно работать
€ высококонцентрированными растворами MgCl2. Однако необхо-
димо учитывать, что в насыщенных хлористым водородом концен-
трированных растворах MgCl2 выпадает твердая фаза гексагидрата
хлористого магния. Температура начала кристаллизации зависит
•от концентрации MgCl2 в растворе и составляет для растворов MgCl2:
°с
30
32
сс
60
63
При использовании концентрированных растворов MgCl2 не
следует снижать температуру в производственном цикле до темпе-
ратуры начала кристаллизаций.
508
Давление, паров воды , мм ртп.
60 70 80 30 700
Температура, °C
Рис. 9-21. Парциальное давле-
ние паров воды над раство-
рами MgCl2:
Рис. 9-22. Растворимость
НС1 в растворах MgCl2:
1 — 3,35% MgCl2; 2 — 10,05%;
з — 14,88%; 4 — 20,29%; 5 -
25,24%; 6 — 30,18%; 7 — 34,96%
MgCI2.
7—30% ШС12; 2 — 32%; 3 —
34%; 4 — 36%; 5 — 38%: 6 — 40%;
7 — 42 % MgCl2. .
Пир
Adz азы
1ОО °/о
HCI
Инертные
Осушенные газы
газы
А зеотроп (.
кислоты ---\ Нейтральный
раствор MgC12 7 оастоор MgCl2
Рис. 9-23. Схема получения 100%-ного НС1 из абгазов,
содержащих большое количество паров воды:
1 — сушильная колонна; 2 — колонна адиабатической абсорбции;
3 —ректификационная колонна; 4 — холодильник; 5 — выпарной аппа-
рат; 6 — кипятильник; 7 — нейтрализатор.
Предложен вариант схемы переработки разбавленных, богатых
парами воды, абгазов, содержащих хлористый водород, в котором
предусмотрено поглощение основного количества паров воды из
газов в колонне, орошаемой раствором MgCl2. Из схемы на рис. 9-23
видно, что из осушенного, абгаза хлористый водород извлекается
путем адиабатической абсорбции [67]. Полученная концентрирован-
ная соляная кислота перерабатывается на 100%-ный хлористый
водород в стриппинг-процессе. Разбавленные растворы MgGl2 ней-
трализуются и упариваются.
При переработке абгазов, сильно разбавленных парами воды,
потери хлористого водорода от растворения его в растворах MgCl2
сильно возрастают. При соотношении Н2О : HG1 в абгазах, равном 13,
практически весь HG1 теряется.
ПОЛУЧЕНИЕ ЖИДКОГО ХЛОРИСТОГО ВОДОРОДА
Аналогично хлору хлористый водород может быть сжижен.
В жидком виде его удобно использовать в ряде производств. Хло-
ристый водород кипит при атмосферном давлении при температуре
—85,11 °С^ При 50 °C давление паров HG1 над жидким хлористым
водородом составляет 78,48 ат. Поэтому в отличие от жидкого хлора
для конденсации хлористого водорода требуются значительно боль-
шие давления или более низкие температуры. Аналогично сжиже-
нию хлора сжижение хлористого водорода возможно как способом
глубокого охлаждения, так и способом высокого давления или ком-
бинированием компримирования с охлаждением [92, 93].
Способ глубокого охлаждения может применяться для получе-
ния небольших количеств чистого хлористого водорода, когда кон-
денсация НС1 осуществляется под небольшим избыточным давлением.
Впервые жидкий хлористый водород был получен в Германии
примерно в 1930 г. [63, 94]. Концентрированный хлористый водород,
содержащий 95—96% HG1, после осушки в башнях, орошаемых сер-
ной кислотой, сжимался четырехступенчатым поршневым компрес-
сором до 60 ат и конденсировался в теплообменнике, охлаждаемом
водой. ,
Долгое время хлористый водород сжижался лишь в ограничен-
ном количестве для удовлетворения нужд мелких потребителей в су-
хом чистом 100%-ном хлористом водороде. Поэтому для транспорти-
рования его использовались стальные баллоны емкостью до 30 кг.
Сообщается о выпуске технического жидкого хлористого водорода,
содержащего более 99,7% HG1, не более 0,2% воды и незначительное
количество инертных газов, а также особо чистого хлористого водо-
рода, содержащего не менее 99,95%, в стальных емкостях на 5
и 500 кг, испытываемых при давлении 155 и 210 ат [95].
Разработаны специальные цистерны емкостью 45 м3 для хранения
и транспортирования жидкого хлористого водорода [96]. Цистерны,
толщина стенок которых 16 мм, были рассчитаны на рабочее давле-
ние 42,2 кгс/см3, и снабжены тепловой изоляцией толщиной 250 мм.
' 510
Цистерны рассчитаны на сохранение температуры жидкого хлори-
стого водорода в пределах от —45,5 до —17,7 °C в течение 30 сут.
Для этого помимо тепловой изоляции цистерны были снабжены запа-
сом твердой двуокиси углерода или локальными холодильными
установками.
В связи со значительными затратами на сжижение й транспортй-
рование жидкого хлористого водорода трудно предполагать, чтобы
в ближайшее время он нашел применение в масштабах, близких
к масштабам использования жидкого хлора.
1 4
МАТЕРИАЛЫ, СТОЙКИЕ В СРЕДЕ ХЛОРИСТОГО ВОДОРОДА
И СОЛЯНОЙ КИСЛОТЫ
Подбор конструкционных и защитных материалов для аппаратуры
и трубопроводов в производствах хлористого водорода и соляной
кислоты встречает большие трудности. Количество материалов,
достаточно стойких в этих средах, ограничено.
В сухом хлористом водороде при комнатной или близких к ней
температурах удовлетворительно стойки ряд металлов и их сплавов.
С повышением температуры стойкость металлических материалов
постепенно снижается до определенной для каждого металла темпе-
ратуры. При температуре выше предельной скорость коррозионного
разрушения быстро возрастает и материал уже не может считаться
стойким в этих средах. Максимальные температуры, допустимые
при длительной работе в среде сухого хлористого водорода, для
различных металлов и их сплавов [971 приведены ниже:
Темпера-
тура, °C
Платина .............................. 1200
Золото ...................f........... 870
Вольфрам . ;..............' .......... 600
Никель................................. 510
Инконель (80% Ni; 14% Сг; 6% Fe) . . .
Хастеллой ............................
Нержавеющая сталь
Углеродистая сталь
Монель-металл (65—67%
0-1,4% Fe; 0,15% С)
Серебро...............
Медь .................
Ni; 29—30% Си;
, 480
450
400-430
260
230
230
95
При температурах до 200—250 °C для изготовления аппаратуры
с успехом может применяться углеродистая сталь, при более высо-
ких температурах — нержавеющая сталь или никель. Для хранения
и перевозки жидкого хлористого водорода можно применять сталь-
ные баллоны, бочки или цистерны.
В среде влажного хлористого водорода и соляной кислоты любой
концентрации при температуре вплоть до температуры кипения
из металлических материалов хорошо стоек тантал. При невысокой
концентрации соляной кислоты и небольшой температуре удовлет-
ворительно стоек титан и некоторые его сплавы. Однако в высоко-
концентрированных растворах соляной кислоты и при температурах
выше 40—50 °C титан и его сплавы уже не стойки. Умеренно стойки
в солянокислых средах сплавы никеля с молибденом типа хастеллой,
которые применяются для изготовления некоторых деталей аппара-
тов, насосов и арматуры.
Ранее для изготовления аппаратуры и трубопроводов применя-
лись практически только керамические материалы, стекло и кварц.
Керамические плиткй и кислотоупорный кирпич в настоящее время
широко используется для защиты стальных емкостей при хранении
соляной кислоты.
Керамика неудобна как материал для изготовления аппаратуры
и трубопроводов из-за малой механической прочности, плохой
теплопроводности и низкой стойкости при колебаниях температуры.
Поэтому керамика уступила место новым материалам.
Стеклянные трубопроводы в настоящее время широко приме-
няются в промышленности для растворов соляной кислоты. Кварц
и изделия из него вследствие высокой стоимости используются
лишь в исключительных случаях, когда необходимо получать очень
чистую соляную кислоту.
Среди неметаллических конструктивных и защитных материалов
наибольшее распространение нашли различные углеграфитовые
материалы, защитная гуммировка и новые полимерные материалы.
Импрегнированный графит и прессованные углеграфитовые мате-
риалы типа игурит используются для изготовления разнообразных
блочных и трубчатых теплообменников, абсорбционных колонн,
трубопроводов, насосов и другой аппаратуры для работы с соляной
кислотой любой концентрации, а также для работы под давлением
несколько атмосфер. Гуммировка широко применяется для защиты
от коррозионного разрушения железнодорожных и автомобильных
цистерн для перевозки соляной кислоты, а также как подслой под
футеровку крупных стационарных емкостей и аппаратов.
Из синтетических полимерных материалов широко используется
фторопласт для изготовления трубопроводов и деталей аппаратуры.
В ближайшее время можно ожидать широкого распространения
стеклопластиков на основе полиэфиров, полипропилена и других
полимерных материалов [98].
ЛИТЕРАТУРА
1. ПасманикМ. И., Сасс-ТисовскийБ. А., Якименко JL М.
Производство хлора и каустической соды. Справочник. М., «Химия», 1966.
См. с. 176.
2. Справочник химика. Т. I—III. М., «Химия»* 1962—1964.
3. Gmelins Handbuch der Anorganische Chemie^ Sist. № 34, 8 Aufl., Berlin, 1962.
4. L a n d о 1t - В о г n s t e i n, Zahlenwerte und Funktionen aus Physik —
Chemie — Astronomie — Geophysik und Technik, 6 Aufl., Berlin, 1960.
5. Meddor J. W.,A. Comprehensive Treatise on Inorganik and Theoretical
Chemistry, Supplement II, Longmans, Green and Co,, 1956.
6. SchmidtA., Chem. Ing. Techn., 25, № 8—9, 455 (1953).
512
9.
10.
ZeisbergF. С., Intern. Critical Tables, Bd. Ill, 1928. См. с. 301.
Othmer D. F., Ind. Eng. Chem., 34, 952 (1942).
Van Nuys С. C., Trans. Am. Inst. Chem. Eng., 39, № 5
11.
t 13.
s 14.
' 15.
'I 16.
17.
1 18.
химическая технология. T. 1. M., Госхимиздат, 1952.
Пат. США 2706145 (1955).
Пат. США 2706144 (1955).
Р г a n s о n G., Chem.-Ztg.,
Пат. США 3363977 (1968).
Англ. пат. 1145390 (1969).
Sommers Н. A., Chem.
chem. Soc., 113, № 3, 111; №
40, № 1, 16; № 2, 112 (1958).
Пат. США 3085860 (1961); пат. ФРГ 1119832 (1961).
91, № 20, 751 (1967).
Eng. Progr., 61, № 3, 94 (1965);
8, 185 (1966); Perugini G
СССР 175045 (1965).
Chem.-Ztg., 84, № 7, A195 (1960).
663 (1943).
А. Общая
J. Electro-
Chim. е ind..
Мокрова H. Г., авт. свид.
L 19. Chem.-Ztg., 84, № 7, А195 (1960).
| 20. Пат. ФРГ 1098493 (1961); англ. пат. 713199 (1954); пат. ГДР 65562 (1969).
21. Chem. Proc., 8, № 9, 28 (1962).
t 22. Chem. Trade J., 148, № 3849, 553 (1961).
[ 23. Англ. пат. 734080 (1955).
Пат. США 3447901 (1969).
' A., Blech., 13, № 5, 264 (1966).
й л о в Ф. К. и др., Хим, пром.,
е А. Н., Chem. Trade J., 112, 97
пат. 450648 (1943).
пат. 1012419 (1952).
[А 2356345 (1943); 2397760, 2397769
25.
27.
28.
30
31
Г 33.
Г 34.
Миха
Maud
Бельг.
Франц.
Пат. CI
Пат. США 3131027 (1962).
Пат. США 3001607 (1961).
Пат. США 2717199 (1954).
№ 8, 595 (1964).
(1943).
(1945).
35.
политехи, ин-та, № 52, 125 (1969).
Пат. США 2490454 (1949).
OldershawC. F., S i m ens о
№ 7, 371 (1947).
Сб. науч, трудов
et al., Chem. Eng.
£ 37. View eg R., Chem. Techn., 14, 107, 728 (1962)? 4
t 38. H e 1 1 e m a n n s G., Sei bring H., Chem. Ing. Techn.,
39.
40.
? 41.
Пермского
Progr., 43,
1046 (1966).
Пат. ФРГ 1006400 (1957); 1124022 (1962); англ. пат. 849055 (1960).
Пат. США
Пат. ФРГ
Япон. пат.
38, № 10,
3140244 (1962).
1171878 (1964).
22773 (1968).
3387430 (1968).
2730194 (1956).
Ь 43.
! 44.
| 45. Пат. США 2558011 (1951); 2841243 (1958).
Англ. пат. 695064 (1953).
Япон. пат. 11651 (1956).
Англ. пат. 695063 (1953).
Пат. ФРГ 1161855 (1964).
Англ. пат. 797163 (1958).
Пат. США 2684331 (1954); 2911363 (1959).
Пат. США
Г 47.
Г 49.
I 50.
| ,51.
I 52. Ind. Eng. Chem., 45, 228 (1953).
t 53. Англ. пат. 739144, 742037 (1955); пат. США 2787525 (1957).
| 54- Chem. Age, 69, ИЗО (1953).
К 55. Пат. ФРГ 1024064 (1958).
Ь.' 56. Пат. США 2426914 (1947).
Г 57. Пат. США 2555278 (1951); франц, пат. 1129260 (1957).
Р 58. Пат. США 3140916 (1961); франц, пат. 14374^3 (1965).
33 Заказ 843
513
59. Франц, пат. 1129026 (1957); пат. США 3074773 (1963); англ. пат. 960261
(1964); япон. пат. 33213 (1971).
60. Пат. США 2416011 (1947); канад. пат. 437940 (1946); 880788 (1971).
61. О с у м и Я., Сода то энсо, 15, № 8, 281; № 9, 327 (1964).
62. Р амм В. М. Абсорбционные процессы в химической промышленности.
М., Госхимиздат, 1951.
63. Synowiec J., Przem. Chem., 39, № 8, 478 (1960); Chernik, 13, № 7—8,
285 (1960).
64. Heyder W., Chem. Techn., 3, 195 (1951); 7, 249 (1955).
65. DobratzC. J. et al., Chem. Eng. Progr., 49, № 11, 611 (1953).
66. G а у b о г d W. H., Miranda M. A., Chem. Eng. Progr., 53,139 (1957).
67. Vieweg R., Chem. Techn., 12, № 4, 188 (1960).
68. Hachnik B., KochH., Chem. Techn., 20, № 12, 738 (1968).
69. Г а с п a p я н A. M., Труды Ереванского института им. К. Маркса,
№ 1, 59 (1941), № 2 (1942).
70. HunterF. L., Trans. Am. Inst. Chem. Eng., 37, 350, 741 (1941).
71. Kan tyka T. A., ’ H incli ef f H. R., Trans. Am. Inst. Chem. Eng.,
50, 236 (1954); Ind. Chem., 31, 32 (1955).
72. Haase B.,_ Koch H., Chem. Techn., 20, № 12, 738 (1968); V ie w eg R.,
Chem. Techn., 12, № 9, 525 (1960).
73. Kolbe E., В г a d m a i г F., Chem. Ind. Techn., 35, 262 (1963).
74. Новосельцева H. А., Захарова Л. E., Наги-Заде H. M.,
Генин Л. С., Хим. пром., № 12, 932 (1969).
75. Пат. США 3260059 (1966).
76. Brumbaugh С. С. et al., Ing. Eng. Chem., 41, 2165 (1949).
77. Hawliez еЪ J., Chem. Stosowana, 6, 369 (1962).
78. Англ. пат. 778421 (1958).
79. Пат. США 3394056 (1968).
80. WinnackerK., KiichlerL., Chem. Techn., Bd., I, Anorganische
Technologie, Munchen, 1970.
81. Канад, пат. 437940 (1942).
82. Пат. США 2316633 (1940); 2351461 (1941); 2367301 (1944); 2492706 (1946);
2511072 (1950).
83. Mari yama Т., Sakayori T., J. Chem. Soc. Japan, 64, № 10,
1877 (1961).
84. Пат. ФРГ 1257757 (1968).
85. Бельг, пат. 747709 (1970).
86. Grew er T., Chem. Ing. Techn., 43, № 11, 655 (1971).
87. Англ. пат. 668706 (1947); 669671 (1949).
88. Пат. США 2357095 (1944); 2764532 (1956).
89. Франц, пат. 2036388 (1971).
90. Тышецкая О. В., Гринштейн И. М. Сб. трудов Всесоюзного
науч.-исслед. ин-та гидролизной и сульфитно-спиртовой пром-сти, 14,
225, 280 (1965); Сыркина И. Г., Крашенинникова А. А.,
За ли он о В. М., ЖПХ, 45, № 6, 1230 (1972).
91. Пат. ГДР 61793 (1968).
92. Сода то энсо, 15, № 11, 403 (1964).
93. Пат. США 2463188.
95. W a s i 1 е w s k i L., К e г i n s k i J. et al., Technologia cloru i zwiazkow
Chloru, Warszawa, 1963.
95. Кэиекйти Камням a, Kagaku to koge (Chem. and Chem. Ind.),
23, № 5, 582 (1970).
96. Chem. Eng., 67, № 17, 58 (1960).
97. Brown H. M. et al., Ind. Eng. Chem., 39, 839 (1947).
98. Chem. Proc., 33, № 2, 20 (1970).
L>s.
J J •
f ГЛ A В A 10
ПРОИЗВОДСТВО ХЛОРИДОВ. Кандидат техн, наук А. А.
L"
урман
Заметное количество хлора и соляной кислоты расходуется на
-получение хлоридов. В производствах хлористого алюминия, хлор-
ного железа и хлоридов фосфора может быть непосредственно исполь-
зован осушенный электролитический хлор, для получения четырех-
хлористого кремния применяют только испаренный жидкий хлор.
Четыреххлористый титан обычно получают на титано-магниевых
комбинатах, используя анодный хлор, выделяющийся при электро-
лизе расплава хлористого магния. Для получения хлоридов цинка
и марганца применяют соляную кислоту.
Безводные хлориды, а также растворы некоторых хлоридов на-
ходят широкое применение в различных отраслях промышленности
и народного хозяйства, в частности в качестве катализатора орга-
нического синтеза (А1С13, FeCl3, TiCl3), как исходное сырье для произ-
водства кремнийорганических соединений и полупроводниковых
материалов (SiCl4, Si2Cl6), а также нового антидетонатора (МпС12)
для получения металлического титана и пигментной двуокиси ти-
тана (TiCl4), для получения фосфорорганических соединений (РС13,
РОС13), при обработке древесины (ZnCl2) и др.
Ассортимент выпускаемых промышленностью хлоридов непре-
рывно расширяется, увеличивается также объем их производства.
Особое внимание в последнее время уделяется получению хлоридов
повышенной чистоты.
ХЛОРИСТЫЙ АЛЮМИНИЙ
Физико-химические свойства
Безводный хлористый алюминий представляет собой бесцветные
шестигранные пластины.
Хлористый алюминий может образовывать димерные молекулы,
устойчивые в различных агрегатных состояниях. До 440 °C пары
хлористого алюминия соответствуют соединению А12С16, в интер-
вале’440—800 °C димер сосуществует с мономером, при 800—1000 °C
стабильной формой является мономер. Плотность пара хлористого
алюминия при различных температурах равна:
t, °C ....... 218 350 758 943 1244
Плотность, г/см3 . . 9,19 9,34 4,80 4,56 4,25
33* 515
Температура сублимации хлористого алюминия равна 180,2 °C,
тройная точка соответствует температуре 192,6 °C и абсолютному
давлению 2,26 ат. Плотность кристаллического хлористого алюми-
ния равна 2,465 г/см3, жидкого А12С16 при 190 °C — 1,33 г/см3,
а при 230 °C — 1,23 г/см3.
Основные термодинамические константы по данным [1—3]:
•и
Теплота образования, кал/моль
А1С13 (т)............................—166 800
А1С13 (г)........................... —137 875
Изобарно-изотермический потенциал, кал/моль "
А1С13 (т) ....... ...................—149 140
А1С13 (г).......................... —135 634
Теплоемкость, кал/(моль град)
А1С13 (т) . ....................... 21,6
А1С13 (г).......................... 16,8
Энтропия, кал/(моль-град)
А1С13 (т)........ . . ........... 27,46
А1С13 (г) ........................ 75,56
Теплота плавления А12С16 при 465,80 °К,
кал/моль . ......................... . 17 000
Теплота испарения А12С16, кал/моль
при 298 °К ............................ 14 319
при 433 °К ........................ 9 700
Теплота сублимации А12С16, кал/моль
при 298 °К ...................... 28 800
при 453 °К ........................ 26 700
Безводный хлористый алюминий на воздухе дымит, выделяя
небольшие количества хлористого водорода, в очень разбавленных
водных растворах он в значительной степени гидролизуется. Рас-
творимость хлористого алюминия в воде при 15 °C составляет 41,13%.
Плотность растворов различной концентрации:
Концентрация А1С13,
вес. % ............ 5 10 20 30 40 41,13
Плотность, г/см3 .... 1,0361 1,0734 1,1537 1,2422 1,3415 1,354
Хлористый алюминий растворим также во многих неводных рас-
творителях: фосгене, ацетоне, хлороформе и др.
л
С аммиаком^хлористый алюминий образует аммиакаты с различ-
ным числом молекул аммиака, наиболее изучено соединение со-
става A1C13*6NH3. Дляк хлористого алюминия характерно образо-
вание комплексных соединений со многими неорганическими хлори-
дами и оксихлоридами, в частности установлено существование
устойчивых соединений состава А1С13 • РОС13, А1С13 • FeCl3,
A1C13-NH4C1, AlCl3-SnCl2 и др. В большей степени проявляется
взаимодействие хлористого алюминия с органическими соединениями,
чем обусловлена роль хлористого алюминия как катализатора в про-
цессах органического синтеза [4].
Из низших хлоридов алюминия известно соединение состава А1С1.
Монохлорид алюминия может быть выделен при температуре 800—
1200 °C при действии паров хлористого алюминия на алюминий [5].
516
углерода
р"' / '
| Монохлорид алюминия — очень неустойчивое соединение, энер-
| гично разлагается под действием кислорода, азота, окиси
Г и паров воды.
качестве
синтеза
изомери-
и
и
I Применение хлориотого алюминия
Г Хлористый алюминий нашел широкое применение в
| катализатора разнообразных процессов органического
| (реакция алкилирования, ацилирования, гидрогенизации,
К зации и др.). Каталитические свойства А1С13 используют при произ-
I водстве смазочных масел и моторных топлив, синтетического кау-
| чука; А1С13 применяют для очистки нефти и масел от серы [4].
К В СССР основное количество вырабатываемого хлористого алю-
р миния расходуется в производстве этилбензола, изопропилбензола,
| сульфоно ла, синтетических красителей, синтетического каучука,
при крекинге и очистке нефтепродуктов.
Г Растворы хлористого алюминия, частично нейтрализованные
| до образования основных хлоридов типа А1я (ОН)3/г_х С1х, приме-
। няют в качестве коагулянта при очистке питьевой воды [6], как
закрепитель керамических форм для литья по выплавляемым
| делям [7, 8], в текстильной и мыловаренной промышленности и
мо-
ДР.
ме-
к
г-
Tt
Теоретические основы получения хлориотого алюминия
Хлористый алюминий может быть получен хлорированием
талла, окиси алюминия или природных алюмосиликатов (каолина,
боксита).
Наиболее энергично протекает взаимодействие хлора с алюми-
нием. По данным [9], алюминий с предварительно обработанной
поверхностью начинает реагировать с хлором при температуре
около —20 °C, тогда как при наличии окисной пленки взаимодействие
начинается только при 200—300 °C [10]. Скорость хлорирования
алюминия практически не зависит от давления и мало изменяется
при повышении температуры. Энергия активации этой реакции со-
ставляет всего 200 кал/моль [11].
Большой тепловой эффект и высокая скорость хлорирования
алюминия осложняют отвод тепла и препятствуют созданию высоко-
производительного аппарата. С целью эффективного отвода избы-
точного тепла подробно изучены различные способы хлорирования
| алюминия в расплаве солей. Алюминий весьма медленно реагирует
| с хлором в расплаве NaCl—А1С13. Однако добавление 5% хлорного
железа к этому расплаву вдвое увеличивает скорость хлорирования
алюминия при 200 °C [12]. Предложен [13, 14] способ хлорирования
алюминия в расплаве в присутствии переносчика хлора (например,
хлорного железа) под давлением 3—4 ат с одновременной ректифи-
кацией тпо лученного продукта. Хлористый алюминий при 250 °C
и соответствующем давлении находится в жидком состоянии, что
позволяет за счет интенсивного испарения А1С13 и последующего
' ' 517
.к.
•' А |
возврата части его в реактор в виде флегмы отводить все избыточное
тепло реакции.
Более благоприятные условия создаются в случае гидрохлори-
рования алюминия в расплаве солей. Использование хлористого
водорода вместо хлора заметно снижает тепловой эффект реакции.
Установлено [15, 16], что алюминий взаимодействует с хлористым
водородом с достаточно высокой скоростью при 160—180 °C, причем
с повышением температуры скорость реакции понижается. Так,
при возрастании температуры от 180 до 450 °C количество прореаги-
ровавшего алюминия при прочих равных условиях уменьшается
более чем в 2 раза. Это явление автор [16] связывает с аналогичной
зависимостью растворимости хлористого водорода в расплаве NaAlCl4
от температуры.
Предложен ряд способов хлорирования алюминия в кипящем
слое. По одному из патентов [17], кипящий слой частиц алюминия
создают инертным газом, а хлор вводят в среднюю часть образующе-
гося слоя. Температура процесса должна быть не выше 600 °C.
Согласно патенту [18], порошкообразный алюминий подают в слой
инертного материала (например, песка) и тщательно регулируют
заданный температурный режим в различных точках реакционной
зоны.
Основные преимущества применения алюминия в качестве исход-
ного сырья для производства хлористого алюминия заключаются
в значительном сокращении числа стадий технологического процесса
и в небольших габаритах печи хлорирования. Однако возникают
серьезные трудности в создании условий для интенсивного отвода
тепла, в выборе конструкционных материалов для хлоратора, осо-
бенно для хлороподводящего устройства. При хлорировании в ки-
пящем слое всегда имеется реальная угроза расплавления отдельных
частиц алюминия за счет местных перегревов и нарушения тем самым
режима псевдоожижения.
Для использования в промышленности представляется более
приемлемым способ гидрохлорирования алюминия в расплаве
NaCl—А1С13.
Хлорирование окиси алюминия может быть представлено урав-
нением
А12О3 + ЗС12 2А1С13+з/2О2
Вероятность протекания этой реакции определяется разностью
значений изобарно-изотермических потенциалов хлорида и окисла.
В расчете на 1 моль А1С13 эта разность составляет
Д& — Д&! — Д<?2 =—149,2 — (—0,5 378) — 4-39,8 ккал/моль
Положительное значение AG указывает на то, что хлорирование
окисла может идти только при условии одновременного удаления
продуктов реакции или связывания одного из них, в частности кис-
лорода.
Исходя из этих термодинамических предпосылок хлорирование
окисных соединений алюминия осуществляют обычно в присутствии
518
восстановителей (угля, кокса, окиси углерода, фосгена). Температура
начала реакции хлора с окисью алюминия, по данным [19], соста-
вляет 1250 °C, а в присутствии угля 700 °C [20]. Однако Спицын [21]
считает, что добавление восстановителя не снижает температуру
начала хлорирования, а только увеличивает скорость и полноту
реакции. Первой стадией процесса, как предполагает автор [21],
является вытеснение кислорода из окисла хлором, а второй стадией —
связывание , кислорода восстановителем, что смещает равновесие
реакции в сторону образования хлорида.
Температура г °C
Рис. 10-1. Скорость хлорирования раз-
личных форм А12О3:
1 — аморфный глинозем (С1а СО); 2 —
аморфный глинозем (СОС1г); 3 — боксит
(С1г + СО); 4 — корунд (С1, + СО).
Рцс. 10-2. Д иффе ренциал ьно-те р-
мическая кривая нагревания као-
лина.
Изучение роли и степени активности восстановителя при хлори-
ровании окиси алюминия стало предметом ряда исследований [22—
24]. Наиболее эффективным восстановителем является углерод
птп
затем следуют фосген и окись углерода. Однако уголь связывает
кислород преимущественно в виде СО, что дает незначительный
тепловой эффект, тогда как при взаимодействии окиси углерода
с кислородом выделяется почти в три раза больше тепла. Это позво-
ляет вести процесс хлорирования без подвода тепла извне.
Применение фосгена эффективно только при 600—650 °C, т. е.
когда степень диссоциации его не превышает 60%.
Важную роль в процессе хлорирования играет структурная моди-
фикация окиси алюминия или минералогический состав алюмо-
силикатного сырья. Наименее реакционноспособен а-глинозем, обра-
зующийся при высокотемпературном обезвоживании гидроокиси
алюминия, более активными являются гидраргилит и у-глинозем [24,
25]. На рис. 10-1, по данным [24], приведена скорость хлорирования
различных видов сырья. Исходя из повышенной активности аморф-
ной окиси алюминия предложен [26] способ хлорирования у-глино-
зема в кипящем слое смесью СО + С12 в присутствии катализатора
хлоралюмината натрия NaAlCl4.
Заметное влияние на хлорируемость алюмосиликатов оказывает
температурный режим прокаливания исходного сырья. Например,
519
изучение хлорируемости каолина, обезвоженного при различных
температурах, сравнение этих данных с дифференциально-термиче-
ской кривой нагревания каолина (рис. 10-2) показывает, что деги-
дратацию каолина следует проводить в интервале 660—970 °C.
При более высоких температурах в прокаленном каолине появляются
малоактивные модификации окиси алюминия — силиманит, муллит
и др. [27—30].
Для промышленного получения хлористого алюминия практиче-
ский интерес представляют условия преимущественного хлорирова-
ния отдельных окислов, содержащихся в каолине. В зависимости
от конкретных условий и потребности народного хозяйства может
оказаться экономически выгодным увеличение или уменьшение
выхода побочного продукта — четыреххлористого кремния. Ско-
рость хлорирования SiO2 и А12О3 в смеси всегда выше скорости хло-
рирования индивидуальных окислов, т. е. наблюдается явление
взаимного катализа [31]. По данным [22, 27], в интервале 550—
800 °C глинозем и кремнезем хлорируются в том соотношении, в ка-
ком они находятся в исходном сырье. О распределении хлора при
более высоких температурах имеются противоречивые данные. Так,
в работе [28] указывается, что выше 900 °C выход А1С13 снижается,
а выход SiCl4 заметно возрастает. Между тем, как следует из много-
летней промышленной практики, с повышением температуры вы-
ход SiCl4 уменьшается и достигает минимального значения при 1000—
1200 °C. Это расхождение объясняется тем, что при хлорировании
в шахтной печи при 1000—1200 °C снижение выхода SiCl4 происхо-
дит не за счет снижения скорости хлорирования кремнезема, а вслед-
ствие протекания вторичной реакции
2Al2O3+3SiC14 3SiOa+4AlCl3
Выход четыреххлористого кремния может быть заметно увеличен,
если в каолиновые брикеты добавить 10—15 вес. % каменноуголь-
ного кокса и снизить температуру прокаливания брикетов до 800 °C.
При этом степень хлорирования SiO2 возрастает с 12 до 28% [22].
С целью извлечения SiCl4 из отходящих газов изучена адсорбция
четыреххлористого кремния твердыми сорбентами [32], а также пред-
ложен и внедрен на одном из заводов абсорбционный/метод извлече-
ния с использованием в качестве растворителя керосина, предвари-
тельно очищенного от непредельных соединений [33, 34].
Производство хлористого алюминия
Промышленное получение хлористого алюминия было начато
в США в 1915 г. по способу Мак Афи [35]'. Процесс заключается
в хлорировании при температуре около 900 °C брикетов, пригото-
вленных из боксита, восстановителя (уголь или нефтяной кокс)
и связующего (расплавленный битум). *
В Германии производство хлористого алюминия было организо-
вано по способу Вурстера [36] с использованием в качестве восста-
520
f
новителя окиси углерода. В некоторых странах, особенно при не-
больших масштабах производства, получают хлористый алюминий
хлорированием алюминия или алюминиевых отходов.
Выпуск безводного хлористого алюминия в США составляет 38—
40 тыс. т/год, кристаллогидрата А1С13-6Н2О и растворов хлористого
алюминия — около 26 тыс. т/год.
В СССР разработка промышленного способа получения хлори-
стого алюминия из отечественного сырья была начата в конце 20-х
годов и завершилась в 1935 г. пуском первого в стране цеха хлори-
стого алюминия по способу Казарновского [37]. Способ основан
на хлорировании каолиновых брикетов в присутствии окиси угле-
рода в шахтной печи.
В соответствии с ГОСТ 4452—66
двух сортов:
Содержание, %
А1С13, не менее ........*
TiCl4, не более ..........
FeCl3, не более *.....
Размер частиц, мм, не более . .
II
хлористый алюминий выпускают
I сорт
99,0
0,5
0,05
II сорт
98,5
0,8
0,15
Растворы хлористого алюминия, согласно ТУ БУ-Х-101—68,
содержат не менее 350 г/л А1С13. Способ получения растворов хло-
ристого алюминия основан на растворении гидрата окиси алюминия
в соляной кислоте с последующей нейтрализацией избыточной кис-
лоты. Если растворы А1С13 применяются в качестве коагулянтов,
нейтрализацию производят известью. Для получения закрепителя
керамических форм в литейном производстве нейтрализацию произ-
водят алюминиевым порошком [38].
Сырье [39, 40]. В состав брикетов, приготовленных для произ-
водства хлористого алюминия, входит каолин и технический гли-
нозем.
Каолин — тонкодисперсная пластичная порода, основной соста-
вляющей которого является минерал каолинит AlgOg^SiOg^HaO.
Первичные каолины, образующиеся в результате выветривания
обычно «подвергают обогащению (отмучиванию). Основные место-
рождения каолина в СССР — Кыштымское (Урал), По ложе кое и
Просяновское (УССР), Трошковское (Сибирь). В производстве хло-
ристого алюминия преимущественно используют обогащенный каолин
кыштымского месторождения, выпускаемый в соответствии с ГОСТ
4193—63 следующих сортов:
Содержание, %
I сорт
II сорт ill сорт
А12О3, не менее .... • 37
Fe2O3, не более 1
Fe2O3 + TiO2, не более с 1,6
Влага, не более 20
36 35
1,2 2,5
1,9 3,0
20 20
Для определения пригодности каолина его обычно испытывают
на водопоглощение, пористость и прочность брикетов в сочетании
с пробами на хлорируемость в стандартных условиях. Например,
521
г
II
35—36
47-48
115—120
32—33
тымский каолин, прокаленный при 800 *С, характеризуется
следующими данными:
Водопоглощение, % ...........................
Кажущаяся пористость, % ....................
Предел прочности при сжатии, кгс/см2 ........
Степень извлечения А12О3 (хлорирование в течение 5 ч
при 1000 °C) . .'...................... k
Глинозем a-формы выпускают нескольких марок по ГОСТ 6912—64.
Содержание примесей колеблется от 0,1 до 0,25% SiO2, от 0,02 до
0,04% Fe2O3 и от 0,02 до 0,04% СаО.
Технологическая схема и отдельные стадии производства [41,42].
Способ получения хлористого алюминия включает несколько стадий.
Получение окиси углерода в специальных генераторах газифика-
цией каменноугольного кокса кислородом. Суммарно основные реак-
ции горения и газификации выражаются уравнениями
С + О2 — СО2 + 97 650 кал /моль
C + V2O2 = 00 + 29 430 кал/моль
СО + 1/2О2 = СО2 + б8220 кал/моль
СО2 + С —2СО—38 790 кал/моль
Равновесное содержание СО и СО2 в газовой смеси зависит ^рт
температуры, при 925 °C оно составляет соответственно 96 и 4%.
Поскольку двуокись углерода проходит в генераторе через слой
кокса, раскаленного до 1300—1600 °C, реакция восстановления про-
текает с большой скоростью и высоким выходом.
В генераторе возможны также побочные реакции компонентов
ихты с влагой, летучими углеводородами, серой и другими при-
месями, содержащимися в коксе
с+н2о==со+н2
С+2Н2О = СО2 + 2На
ЗС + 2Н2О = СН4+2СО
Летучие углеводороды и влага выделяются главным образом
в верхней части генератора во время сушки и разогрева кокса,
практически не достигая зоны газификации.
При установившемся температурном режиме получается газ сле-
дующего состава (в %):
СО, не менее ............. 92
СО2, не более.............. 2
СН4+На, не более......... 4
N2 ....
О2, не более
Температура газа на выходе из генератора в период розжига
Должна быть примерно 100 °C, а при установившемся режиме —
около 250 °C. Понижение температуры на выходе из генератора
указывает на уменьшение ее в зоне газификации и сопровождается
заметным увеличением содержания двуокиси углерода.
522
II
СН4+2Оа = СОа + 2На О
S + Оа = SOa
S + Ha = H2S
0,4
к"
3
•IJ
$
Я
н
3
II
II
Приготовление, сушка и прокаливание каолиновых брикетов.
Сырой каолин и глинозем замешивают с водой и паром до получения
пластичного теста. Количество добавляемого глинозема составляет
10—20% массы каолина, содержание влаги 30—40%.
Брикеты сушат топочными газами при 300 °C в сушилке конвейер-
ного типа с непрерывно движущимися перфорированными ков-
[ами [43]. После сушки содержание влаги в брикетах снижается
до 17-18%.
Высушенные брикеты прокаливают в шахтных печах при 800—
850 °C для удаления гигроскопической влаги (3—4%) и связанной
воды (около 14%). При прокаливании каолинит (Al2O3‘2SiO2*2H2O)
необратимо переходит в метакаолинит (Al2O3’2SiO2), а также ча-
стично разлагается на А12О3 и SiO2.
В некоторых цехах хлористого алюминия внедрен способ полусу-
хого приготовления брикетов. По этому способу брикетирование
каолина осуществляют без добавления воды под давлением. Брикеты
полусухого прессования имеют форму сплюснутого шара с размером
по большой оси около 50 мм и по малой — 30 мм. Содержание сво-
бодной влаги в брикетах 19—22%, вместо 4% в случае предвари-
тельной сушки в камерах сушилки. В связи с этим возникает необ-
ходимость соответствующего изменения режима в прока л очных печах.
Однако чрезмерная интенсификация процесса прокаливания
(особенно повышение температуры более 970 °C) крайне нежела-
тельна, так как при этом происходят фазовые превращения, заметно
снижающие хлорируемость каолина. Содержание влаги в прока-
ленных брикетах должно быть не более 0,7%.
Хлорирование каолиновых брикетов проводят в непрерывно дей-
ствующей шахтной печи газообразным хлором в присутствии окиси
углерода.
Основная реакция хлорирования
А12О3+ЗС1а+ЗСО =* 2 AICI3+ЗСО2
Кроме окиси алюминия с хлором заметно взаимодействует и
окись кремния
SiOt -Ь 2CI2 + 2CO =• SiCl4+3CO2
В шахтной печи в различных температурных зонах протекают
также побочные реакции
Т iOa+2С13+2СО == TiCli+2СОа
Fe2O3+3Cl2+3CO = 2F0C13+3CO2
Н2О +С12 + СО =» 2НС1+СО2
СО2+С=2СО
СО + С12 = СОС12
Температура в реакционной зоне поддерживается в’|преде-
лах 1000—1250 °C за счет тепла реакции. Для регулирования
523
11
температуры к каолиновым брикетам, загружаемым в печь, добавляют
10—15% подсушенного кускового кокса. Эндотермическая реакция
восстановления двуокиси углерода до окиси углерода в, присутствии
углерода позволяет отвести часть тепла, а также заметно снизить
расход генераторного газа.
Реакционные газы, выходящие из печи хлорирования при 450—
500 °C, кроме хлористого алюминия содержат хлориды кремния,
титана, железа, щелочных и щелочноземельных металлов, а также
двуокись и окись углерода, хлористый водород, хлор, азот, кисло-
род и фосген. Хлориды щелочных и щелочноземельных металлов
и частично хлориды железа образуют с хлористым алюминием рас-
плав, затвердевающий при 160—170 °C. Поскольку температура
реакционных газов выше 170 °C, плав находится в жидком состоянии
и его выводят через специальный шламоотделитель. Примерный
состав шлама: 70—80% А1С13, 5—8% FeCl3, 5—10% А12О3, 5—8%
(КС1 + NaCl).
Печь хлорирования работает под небольшим избыточным давле-
нием (5—10 мм вод. ст.). • .
Конденсация паров хлористого алюминия. Перед входом в кон-
денсатор* реакционные газы должны иметь температуру 220—300 °C.
Конденсаторы охлаждаются водой, температура ее на выходе из
рубашки должна быть не выше 45 °C.
В аппарате происходит конденсация паров хлористого алюминия
и хлоридов железа. В зависимости от качества каолина технический
продукт содержит 94—96% А1С13 и 2,5—4,0% FeCl3.
Очистка технического хлористого алюминия от примеси хлор-
ного железа основана на сублимации хлористого алюминия из рас-
плава NaGl—А1С13 в присутствии алюминиевой стружки. При этом
хлорное железо восстанавливается до хлористого, которое, как менее
летучее соединение, остается в расплаве. Хлорное железо может
восстанавливаться алюминием и до металлического железа, послед-
нее в свою очередь также будет действовать как восстановитель
3FeClg+Al == AlClg + 3FeCl2
FeCl3-f-Al == A1C13 + Fe
2FeCl3 + Fe = 3FeCl2
Очистку хлористого алюминия проводят в специальной реторте,
обогреваемой топочными газами. В нее загружают хлористый алю-
миний и около 8 вес. % поваренной соли. Увеличивая температуру
до 150—170 °C, расплавляют смесь солей. В полученный плав равно-
мерно подают технический хлористый алюминий и просушенную
алюминиевую стружку в количестве 0,5—1,5% массы загруженного
хлористого алюминия. Хлористый алюминий отгоняют при 180—
200 °C.
По мере накопления в реторте отработанного плава прекращают
загрузку хлористого алщминия, повышают температуру до 200—
230 °C и отгоняют из плава остатки хлористого алюминия. Для более
524
полного извлечения А1С13 рекомендуют загружать в аппарат допол-
нительное количество алюминиевой стружки с целью восстановле-
ния хлористого железа до металлического железа. При этом осво-
бождается часть хлористого алюминия, связанного в виде соедине-
ния 2А1С13-FeCl2.
Примерный состав отработанного плава: 66% А1С13, 25% FeCl2,
1% FeCl3, 5% нерастворимого остатка и З% металлического алюми-
ния. Шлам сливают и разлагают водой в токе азота.
В процессе приготовления плава при загрузке и отгонке хлори-
стого алюминия в реторту также непрерывно подают азот для пред-
отвращения образования взрывоопасной смеси кислорода И водо-
рода. Водород выделяется при взаимодействии НС1 и алюминия.
Хлористый водород в свою очередь образуется при действии на хло-
ристый алюминий влаги, вносимой в реторту с загружаемым сырьем
или в результате подсоса влажного воздуха. Продувка азотом улуч-
шает транспортирование паров очищенного хлористого алюминия.
Очистка отходящих газов. На выходе из конденсаторов техни-
ческого хлористого алюминия
состав (в
газы имеют примерно следующий
объемн. %):
СО2 • » • »
НС] . . . .
со . . . . .
SiCl4 .. . .
н2 • • • • •
40
13
10
»
TiCI4 ...........0,3
О2 ..............3,2
CL, ..... ... 1,7
А1С13, г/м3 .... 40
FeCl3, г/м3 . . . 0,08
извлечения четыреххлористого
имеется установка для
очистка отходящих газов проводится после этой стадии.
Если
кремния,
В камере отходящих газов с помощью быстро вращающихся
мешалок создается туман мельчайших капелек воды, что обеспечи-
вает хороший контакт газа с водой'и очистку его от SiCl4, TiCl4, НС1
и мелких частиц А1С13. Для удаления из отходящих газов хлора
в камеру подают также сернистый ангидрид, который восстанавли-
вает хлор до хлористого водорода
SO2+H2O = H2SO3
H2SOs+С1а+Н2О « H2SO4 + 2НС1
Кислый раствор, содержащий продукты гидролиза хлоридов,
после нейтрализации сливают в канализацию.
Технологическая схема производства хлористого алюминия пока-
зана на рис. 10-3. Каолин и глинозем хранятся в специальном углу-
бленном складе, отапливаемом во избежание смерзания каолина
в зимнее время. Каолин подается грейферным краном в приемный
бункер питателя, а затем ковшовым транспортером в бегуны мокрого
помола, куда поступают также вода и глинозем. Все ингредиенты
тщательно перемешиваются до образования тестообразной массы,
влажность которой должна быть не менее 30 и не более 40%.
После бегунов каолиновое тесто подается в горизонтальную
брикетную машину, где дополнительно перемешивается и затем
525
выдавливается через круглые мундштуки диаметром 30 мм. Дви-
I жущиеся перед мундштуками проволочные ножи режут тесто на
цилиндрические брикеты длиной 40—60 мм, поступающие на ленты
брикетоукладчиков. Укладчики доставляют брикеты к ковшам
конвейерных сушилок. Бесконечно движущаяся цепь с заполнен-
ными ковшами трижды проходит вертикальную шахту сушилки
и поступает в ее горизонтальную часть, из .которой брикеты выгру-
жаются в бункеры, расположенные над прокалочными печами.
В цехах полусухого брикетирования каолиновое тесто подают
по ленточному транспортеру в ручейковый пресс для формирования
брикетов. Туда же подают глинозем. Полученные брикеты отсеи-
вают от мелочи на вибросите, подают в шахтный подъемник, а затем —
в прокалочные печи, представляющие собой шахты, выложенные
шамотным кирпичом и заключенные в стальные кожухи. Внутрен-
ний диаметр шахты 900 мм, высота 4500 мм. Необходимая темпера-
тура в печах поддерживается горячими топочными газами, проходя-
щими через слой брикетов. Прокаленные брикеты ссыпаются по на-
клонным течкам в ковши конвейера, транспортирующего брикеты
сначала в смесительный бункер для приготовления шихты, а затем
в печь, хлорирования.
Продвигаясь вниз в печи хлорирования, шихта нагревается за
счет тепла реакции и выходящих из печи газов. Температура в реак-
ционной зоне составляет 1150—1250 °C. Смесь хлора и окиси угле-
рода через коллектор и фурмы поступает в печь и движется вверх
навстречу шихте.
Прохлорированные брикеты (отходы) выгружаются в специаль-
ную камеру. Отходящие газы проходят очистную колонну и выбрасы-
ваются в атмосферу. Перед выгрузкой из камеры прохлорированные
брикеты продуваются азотом для удаления адсорбированного
хлора.
Реакционные газы из печи хлорирования поступают по газоходу,
состоящему из двух скрещенных труб, в холодильник, футерованный
изнутри двумя слоями диабазовой плитки, а оттуда через шламоот-
делитель — в конденсаторы. Шлам из шламоотделителя периоди-
чески спускается в разлагатель, где обрабатывается водой.
Из холодильника реакционные газы поступают в конденсатор,
охлаждаемый водой. На стенках труб конденсатора осаждаются
твердые частицы хлористого алюминия. Снятые с помощью мешалки
кристаллы А1С13 ссыпаются в бункер конденсатора и оттуда выгру-
жаются в контейнеры.
Из конденсатора отходящие газы, содержащие неосевшую пыль
хлористого алюминия, поступают в наклонную трубу, где за счет
снижения скорости газов дополнительно улавливается пылевидный
продукт.
Весь технический хлористый алюминий собирают в контейнерах
и направляют в отделение очистки, где его загружают в специаль-
ные реторты. Одновременно туда же добавляют алюминиевую
стружку. Отгоняемые из реторты пары хлористого алюминия
527
поступают в конденсаторы очищенного продукта. Готовый A1G13 по-
дается на мельницу, а из нее — в барабаны.
Газы после конденсаторов технического и очищенного продуктов
подвергаются санитарной очистке в камере отходящих газов.
Технологическая схема производства хлористого алюминия вклю-
чает также стадию улавливания SiGl4 из отходящих газов. На
рис. 10-4 изображена установка для абсбрбции — десорбции SiCl4
с помощью керосина.
Рис. 10-4. Схема установки для извлечения SiCl4 керосином из отходящих
газов производства хлористого алюминия:
1 — рукавный фильтр из стекловолокна; 2 — механический абсорбер; з — напорный бак;
4 — ротаметр; 5, в, 11, 15* 16 — холодильники; 6 — подогреватель; 7 — куб дистиллятора;
9 — сборник керосина; 10, 13 — насосы; 12 — сборник сырца; 14 — ректификационная
колонна; 17 — сборник готового продукта.
Расходные коэ
ициенты, а также количество побочных продук-
тов и отходов производства на 1 т очищенного хлористого алюминия
составляют:
Сырье, кг
каолин (40% А12О3) .
глинозем (98% А12О3)
хлоргаз (100%) . .
кокс................
Электроэнергия, кВт-ч . . .
Пар, Мкал............... .
Вода, м3 ..................
Побочный продукт (SiCl4), кг
Отходы, кг
прохлорированные брикеты
шлам из шламоотделителя
шлам из реторты ....
ИЗО
215
1350
370
12
700
0,6
200
100
650
70
200
У У
528
t
II
Очистка хлористого алюминия ,
К чистоте хлористого алюминия, применяемого в полупровод-
никовой технике, в производстве особо чистого алюминия, алюмо-
гидридов и других продуктов, предъявляются повышенные требо-
вания. Так, при электроосаждении алюминия из расплава А1СГ3 —
NaCl наличие 10"3—10"4 вес. % железа ухудшает качество катод-
ного осадка, снижает выход по току. Для изготовления монокристал-
лов рубина, используемых в квантовой радиоэлектронике, приме-
няется окись алюминия, полученная из особо чистого хлористого
алюминия.
Из препаративных способов известна очистка А1С13 с помощью
четыреххлористого титана [44]. Растворимость хлористого алю-
миния в TiGl4 при 137 °C равна 283 г/л, а при 25 °C — только 17 г/л,
между тем как хлорное железо почти нерастворимо в TiCl4. Очи-
щаемый А1С13 растворяют в четыреххлористом титане при нагрева-
нии в аппарате с обратным холодильником, а из охлажденного
фильтрата выделяют кристаллы хлористого алюминия центрифуги-
рованием. Для удаления следов TiCi4 полученный продукт возго-
няют. Аналогично можно очищать хлористый алюминий кристалли-
зацией из спирта, сероуглерода или четыреххлористого угле-
рода [45]. V
Наиболее эффективна очистка хлористого алюминия путем ректи-
фикации. Поскольку тройная точка хлористого алюминия соответ-
ствует температуре 192,6 °C и давлению 2,26 ат, ректификация про-
водится под давлением й при повышенной температуре. Впервые
этот способ описан в патенте [46] и подробно изучен советскими
исследователями [47—49]. Авторы разгоняли смесь А1С13 (90%)
и FeCl3 (10%) в стеклянной ректификационной колонне высотой
70 см, заполненной стеклянными кольцами Фенске. В колонне
ективностьЬ 7 теоретических тарелок удавалось отогнать около
99% загруженного хлористого алюминия. Дистиллят содержал
менее 0,01% FeGl3.
Еслиг разгонке подвергается предварительно очищенный хлори-
стый алюминий, содержащий 0,05—0,001% FeGl3, то путем ректи-
икации можно получить продукт повышенной чистоты (5 • 10"4 %
FeGI3 [47,48]. По сообщению [48], этот способ был проверен также
на полупромышленной ректификационной колонне, изготовленной
из углеродистой стали. Ректификационная часть колонны диаме-
тром 76 мм и высотой 3 м была заполнена керамическими кольцами
Рашига. Эффективность колонны составляла примерно 10 теорети-
ческих тарелок. Разгонке подвергался технический хлористый
алюминий, содержащий 2—6% хлоридов железа.
Качество полученного продукта отвечало требованиям ГОСТ
Л
Потери хлористого алюминия с кубовыми остатками в зависи-
мости от режима разгонки составляли 3—10%, в то время как в про-
мышленных условиях потери при очистке равны 20—25%.
34 Заказ 843
529
Предложен способ [50] очистки хлористого алюминия возгонкой
в токе газа-носителя, например хлористого водорода, при 240 °C.
Содержание примеси железа в полученном продукте составляет
5*10“4%.
Чистый безводный А1С13 также получен [51] прямым синтезом
из предварительно очищенных простых веществ: электролизного
хлора (электролиз соляной кислоты в ванне с иридиевым анодом)
и алюминия марки А99.
По другому способу [52] алюминий взаимодействует с парами
четыреххлористого олова, полученного обработкой расплава олова
хлором.
Чистые кристаллы хлористого алюминия размером от милли-
метра до нескольких сантиметров получены [53] по двухстадийному
процессу: сначала А1С13 очищают сублимацией в потоке аргона,
затем пары хлористого алюминия направляют в стеклянный сосуд,
где создаются условия для роста
образующихся кристаллов.
ЧЕТЫРЕХХЛОРИСТЫЙ КРЕМНИЙ
Физико-химичеокие свойства
Четыреххлористый кремний
бесцветную жидкость, дымящую
Молекула SiCl4 имеет тетраэдрическую структуру с атомом крем-
ния в центре, в жидком состоянии четыреххлористый кремний не
представляет собой прозрачную
во влажном воздухе.
ассоциирован.
В табл. 10-1 приведены физико-химические свойства SiCl4 при
различных температурах [54].
Таблица 10-1. Плотность р, вязкость т) и поверхностное натяжение (a) S1CI4
при различных температурах
°C
р, г/см8
г, °C
•И. сП
°C
а, дин/см
-5,4
—2,1
21,7
33,7
39,1
40,5
41,6
49,3
66,3
66,7
80,9
82,6
1,5313
1,5248
1,4764
1,4522
1,4401
1,4365
1,4341
1,4185
1,3815
1,3809
1,3485
1,3442
—6,0
0,5
5,0
10,0
15,5
20,0
31,0
44,3
60,2
70,0
80,9
0,6246
0,5706
0,5548
0,5293
0,4925
0,4823
0,4279
0,3825
0,3369
0,3103
0,2881
23,0
25,0
30,0
35,0
40,0
45,0
50,0
55,0
19,28
19,13
18,50
17,85
17,31
16,78
16,24
15,73
Температура кипения SiCl4 при атмосферном давлении составляет
57 °C, температура плавления —68 °C, показатель преломления
530
— 1,41079. Давление насыщенного пара четыреххлористого
кремния при различных температурах приведено в табл. 10-2.
Таблица 10-2'. Давление насыщенных паров SiCl4
при различных температурах
р>-
мм рт. ст.
i, °C
Р,
мм рт. ст.
р,
мм рт. ст.
—68,0
—64,0
-60,0
—56,5
—51,0
-47,0
-42,5
—36,6
—32,7
—27,0
1,0 —25,0 19,0
1,5 -21,0 24,5
2,0 —15,3 34,5
2,5 —10,0 45,5
3,5 —5,3 58,0
5,0 0 77
6,5 5 98
9,5 10 124
11,5 15 153
17,0 20 191
Основные термодинамические константы
Теплота образования, кал/моль
SlG14 (ж) ....
SiCl4 (г) ....
Изобарно-изотермическ
кал/моль:
SiGl4 (ж) . .............
SiCl4 (г) .....................
Теплоемкость, кал/(моль-град)
SiCl4 (ж) .................
SiCl4 (г) ................... .
Энтропия, кал/(моль-град)
SiCl4 (ж) .....................
SiCl4 (г) . ...................
Теплота испарения SiCl4 (ж), кал/моль
Теплота плавления SiCl4 (т), кал/моль
25
30
35
40
45
50
55
57
60
по данным
—154 000
—146 810
—137 970
—137 290
32,4
21,7
57,3
79,2
6900
1840
' 235
287
346
419
501
599
709
760
839
[1, 551:
t, °C
м
В четыреххлористом кремнии заметно растворяется газообраз-
ный хлор: при 0 °C в 1 г SiCl4 растворяется 0,131 г хлора, при пар-
циальном давлении хлора 1 атм растворимость хлора в SiCl4 при 0 °C
составляет 14,04 вес. %, при 20 °C — 7,63 вес. %. С жидким хлором
четыреххлористый кремний смешивается в любых отношениях.
Эвтектическая точка смеси SiCl4—С12 на кривой температуры за-
мерзания соответствует —117 °C и содержанию хлора 86%. Четы-
реххлористый кремний смешивается с TiCl4J РОС13, ВС13, СС14,
а также со сложными и простыми эфирами органических кислот.
При взаимодействии с водой четыреххлористый кремний практи-
чески полностью гидролизуется. При этом в зависимости от соот-
ношения SiCl4 : Н2О, кислотности среды, температуры, скорости
перемешивания и других факторов образуются промежуточные
соединения, содержащие различное число гидроксильных групп
или атомов хлора и отличающиеся структурой. Конечный продукт
34*
531
1
гидролиза (силикагель) представляет собой высокомолекулярный
гидроксиполисилоксан. .
- При нагревании смеси паров четыреххлористого кремния
и сухого кислорода образуется гексахлордисилоксан Si2OCl6
(т. кип. 137 °C и т. пл. —28,1 °C). При температуре красного кале-
ния SiCl4 частично образует с водородом силикохлороформ, а при
пропускании смеси SiCl4 и водорода над раскаленным углем полу-
чается металлический кремний.
Четыреххлористый кремний реагирует с аммиаком при комнат-
ной температуре с образованием аммиаката SiCl4*6NH3. Восстано-
вление SiCl4 металлами (цинком, алюминием, бериллием, магнием)
протекает при 200 —300 СС. Взаимодействие SiCl4 с железом начи-
нается только при температуре красного каления.
Наряду с четыреххлористым кремнием известны соединения
низшей валентности (SiCl, SiCl2) и высшие хлориды кремния (поли-
хлорсиланы) гомологического ряда SinCl2rt+2. Полихлорсиланы
с п 3—5 при комнатной температуре —бесцветные маслянистые
жидкости, SieCl14 —твердое вещество, Si10Cl22—вязкая жидкость,
Si25Cl52 —пластичная масса. ч
Все полихлорсиланы энергично гидролизуются, легко окисляются
на воздухе. Атомы хлора в полихлорсиланах вступают во все реак-
ции, свойственные хлору в четыреххлористом кремнии,
р • ’г
'f
Применение четыреххлориотого кремния
Четыреххлористый кремний применяют главным образом для
получения различных кремдийорганических соединений. Наиболее
освоен в промышленности процесс получения эфиров ортокремневой
кислоты, используемых в качестве высокотемпературных теплоно-
сителей и пеногасителей, для приготовления специальных смол,
пластификаторов и смазок. Эфир ортокремневой кислоты (этилси-
ликат) широко применяется при изготовлении форм для литья по
выплавляемым моделям.
Четыреххлористый Кремний используют при получении кремний-
органических полимеров взаимодействием SiCl4 с металлооргани-
ческими соединениями [56, 57].
Значительные количества четыреххлористого кремния расхо-
дуются для получения аэросила — высокодисперсной двуокиси крем-
ния, которая служит наполнителем резины. Для этой цели SiCl4
сжигают в водородном пламени [58] или гидролизуют водой при
повышенной температуре [59].
Четыреххлористый кремний может быть использован как дымо-
образующее вещество. Дымообразующий эффект сильно возрастает
при добавлении аммиака [60].
Для получения полупроводникового кремния применяют SiCl4
особой чистоты. В последние годы в полупроводниковой технике
нашел применение также Si2Cl6.
532
Теоретические основы получения четыреххлориотогорсремния
Четыреххлористый кремний получают преимущественно хлори-
рованием элементарного кремния или его сплавов (например, ферро-
силиция).
Механизм взаимодействия кремния с хлором подробно изучен
Мартиным 161] и схематически может быть представлен следующим
образом:
-Si—
— Si—
— Si—
I
— Si^
-Si-
цепь
Si-ато-
мов
Cl— Si—Cl
Cl—Si—Cl
I
Cl-Si-Cl
Cl-Si-Cl
Cl—Si—Cl
I
Cl-Si-Cl
I
I стадия
хлорирова-
ния
SiCl3 SiCl3 SiCl4
Cl-Si-Cl SiCis SiCl4
I
SiCl3 SiCl4 SiCl4
SiCl3 SiCl4 SiCl4
Cl-Si-Cl —i SiCl3 +C1. SiCl4
I I —*
SiCl3 SiCl3 SiCl4
II стадия
хлорирова-
ния
1
III ста-
дия хло-
рирова-
ния
конеч-
ная ста-
дия хло-
рирова-
ния
Температура начала хлорирования кремния, рассчитанная по
предложеннрй [9] эмпирической формуле, равна —40 °C [41].
Между тем, по экспериментальным данным [62, 63], кремний начи-
нает хлорироваться при 200 —240 °C; Добавление в качестве катали-
заторов хлоридов калия, кальция, алюминия или их смесей позво-
ляет снизить температуру процесса до 140 °C [63]. В условиях,
предотвращающих образование на поверхности металла окисной
пленки, удалось осуществить хлорирование кремния в шаровой
мельнице при комнатной температуре [64, 65].
Чем ниже температура хлорирования, тем больше вероятность
образования высших хлорсиланов. По данным [61], при температуре
хлорирования 300—310 °C образуется 4% Sf2Cle, при 250—260 °C
4,6%, при 180—200 °C —8,6%. В присутствии катализатора
(2% КС1) и при 160 °C в продукте хлорирования содержится 79%
полихлорсиланов (из них 64,3% Si2Cle и 14,7% Si3Cl8) [63]. При
температуре выше 450 °C образовавшиеся на первой стадии Si 2С1в
или Si3Cl8 взаимодействуют с хлором и практически полностью пре-
вращаются в SiCl4.
Кинетика реакции кремния с хлором изучена в недостаточной
степени. В одной из работ [66] авторы проводили исследования мето-
дом раздельного калориметрирования. Установлено, что скорость
хлорирования зависит от чистоты кремния: чем чище кремний, тем
ниже порядок реакции и выше энергия активации (соответственно
15 и 24 ккал/моль). Зависимость выхода SiCl4 от температуры при
хлорировании ферросилиция, по данным [62], представлена на
рис. 10-5.
533
При хлорировании ферросилиция наряду с SiCl4 образуются
побочные продукты. По данным работ [56, 62], следует ожидать пре-
имущественного образования хлорного железа. Практика промыш-
ленного производства четыреххлористого кремния не подтверждает
этого предположения. Исследованиями [67] установлено, что со-
держание соединений железа различной валентности (в % от общего
количества) в продуктах хлорирования ферросилиция находится
в прямой зависимости от температуры реакции:
t, °C Fe8+ Fe2+ FeMpT
400 61,2 38,8 —
600 62,3 37,7 —
800 55,6 44,4 —
1000 26,5 36,5 37,5
1200 13,5 33,6 52,9
Увеличение количества металлического железа при температуре
выше 1000 °C может быть объяснено в первую очередь сильным вос-
становительным действием субхлорида кремния (SiCl2), образующе-
гося только при 1000—1200 °C.
Температура хлорирования, °C
Рис. 10-5. Зависимость выхода SiCl4
от температуры хлорирования. ’
700 000 7700 7300
Температура, °C
Рис. 10-6. Хлорирование различных
кремнийсодержащих материалов:
1 — аморфный кремнезем; 2 — кристоба-
лит; 3 — кварц.
Известно, что при хлорировании как кремния, так и силицидов,
выделяется большое количество тепла, что затрудняет создание
высокопроизводительных процессов. В связи с этим усилия иссле-
дователей направлены на поиски условий, благоприятствующих
отводу избыточного тепла. Предложен [68] способ хлорирования
ферросилиция в расплаве хлоридов железа и щелочных металлов.
Подробно изучен [69, 70] механизм этого процесса и показана роль
попутно образующихся хлоридов железа как переносчиков* хлора.
Хлорирование ферросилиция в расплаве NaCl— FeCl3 протекает
в две стадии
1. Si+4FeCi3 4FeCl2+SiCl4
Fe + 2FeGl3=3FeGl2
2. 2FeCl2 + Cl2 = 2FeCl3
Оптимальная температура процесса 600—650 °C, нижний предел
обусловлен возможностью кристаллизации расплава, верхний пре-
дел— увеличением уноса хлоридов натрия и железа реакционными
газами.
Фирма «Монсанто» [71] запатентовала способ получения SiCl4
хлорированием в кипящем слое тонкоизмельченных кремния и инерт-
ных добавок (уголь, окись алюминия, кварц). Роль таких добавок
сводится к предотвращению спекания шихты вследствие возможного
образования небольшого количества плава хлоридов щелочных
и щелочноземельных металлов. Некоторые уточнения режима хло-
рирования в кипящем слое изложены в патентах [72, 73].
Представляет интерес способ хлорирования гранулированной
смеси кремния, двуокиси кремния и углерода [741. Соотношение
компонентов в шихте подбирают таким образом, чтобы количество
тепла, выделяющееся при хлорировании кремния, соответствовало бы
количеству тепла,/необходимому для инициирования и поддержания
эндотермической реакции хлора с двуокисью кремния и углеродом.
Это позволяет вести автотермический процесс при постоянной тем-
пературе без отвода тепла.
Природное окисное сырье не нашло широкого применения для
промышленного производства четыреххлористого кремния. Хлори-
рование двуокиси кремния может осуществляться только в присут-
ствии восстановителя. На скорость хлорирования и степень извле-
чения SiO2 влияет как температура, так и химическая активность
различных модификаций кремнезема [75—77]. Так, температура
начала хлорирования аморфного кремнезема в присутствии угля
равна 730—740 °C, кварца — 1220 °C [76]. Степень хлорирования
при 1100—1150 °C в присутствии угля (1 : 1) в течение 1 ч составляет
для песка 1,62%, плавленого кварца 15,2%, аморфного кремнезема
40,56% [77]. Зависимость выхода SiCl4 от температуры для некото-
рых разновидностей SiO2 показана на рис. 10-6. Реакция аморфной
SiO2 с хлором в присутствии угля замедляется при 850—1000 °C,
а затем резко ускоряется, что объясняется превращением аморфной
кремневой кислоты в кристобалит.
Из всех видов природного сырья наибольший интерес для произ-
водства четыреххлористого кремния представляют диатомит и тре-
пел, так как они являются аморфной разновидностью кремнезема
и обладают сильно развитой поверхностью. В лабораторных усло-
виях проводилось хлорирование брикетов, приготовленных из диа-
томита, древесного угля и сульфит-целлюлозного щелока. При 1100—
1150 °C выход продукта достигал 46—50% [78].
Предложен [79] способ хлорирования диауомита в среде расплав-
ленных солей. Вследствие каталитического влияния хлоридов ще-
лочных металлов температура реакции снижена до 750—800 °C
и создается хороший контакт между измельченной шихтой (диатомит,
уголь) и хлором в среде расплава. Способ был проверен на опытной
установке производительностью 1 т/сут.
535
Производство четыреххлориетого кремния
Промышленное получение четыреххлористого кремния начато
в 30-х годах, когда нашли широкое применение кремнийорганиче-
ские соединения. Мощность первых установок — около 1 т/сут SiCl4,
II
мощность современных цехов достигает 15 тыс, т/год.
Основным промышленным способом получения SiCl4 как в Со-
ветском Союзе, так и за рубежом является хлорирование кремния,
ферросилиция или карбида кремния в шахтных печах. Некоторое
количество четыреххлористого кремния получают в качестве побоч-
ного продукта в производствах хлоридов титана, алюминия и цир-
кония.
В соответствии с ГОСТ 8767—58 к четыреххлористому кремнию
предъявляются следующие требования:
Плотность, г/см3 ....................................1,48—1,50
Температура, °C
начала перегонки, не менее ..................55
конца перегонки, не более . . ................... 59
Остаток после перегонки, %, не более ................ 2,5
Содержание железа (Fe), %, не более ............ . . 0,001
Сырье. Кремний. В промышленности кристаллический кремний
получают из кварца в шахтных дуговых электрических печах мо
и
ностью 2000—5000 кВА; в качестве восстановителя используют
древесный уголь или нефтяной кокс. На получение 1 т кремния рас-
ходуется около 1400 кВт-ч электроэнергии.
В соответствии с ГОСТ 2169—69 выпускается кремний четырех
марок (КрО, Кр1, Кр2 и КрЗ), содержащий от 95,5 до 99% Si. Коли-
чество железа, алюминия и кальция колеблется от 0,5 до 1,5%.
Ферросилиций — сплав железа с кремнием. Среднекремнистый
(43—50% Si) и высококремнистый (72—95% Si) ферросилиций вы-
плавляют в шахтных дуговых электрических печах. Малокрем-
нистый (9—15% Si) ферросилиций выплавляют в доменных печах.
В соответствии с ГОСТ 1415—61 выпускают три марки ферроси-
лиция следующего состава (%):
Si Мп Сг р s
Си-90
Си-75
Си-45
........... 87—95 0,5 0,2 0,04 0,04
........... 74—80 0,7 0,5 0,05 0,04
........... 40—47 0,8 0,5 0,05 0,04
В производстве четыреххлористого кремния используют только
ферросилиций марок Си-75 и Си-90.
Технология получения четыреххлористого кремния. Технологи-
ческая схема процесса (рис. 10-7) включает следующие стадии:
1) измельчение сырья и составление шихты; 2) хлорирование; 3) по-
следовательная конденсация твердых возгонов и четыреххлористого
кремния; 4) ректификация четыреххлористого кремния; 5) очистка
отходящих газов. Некоторые отличия схем, принятых различными
заводами, обусловлены типом основного оборудования, в частности
хлоратора.
536
В качестве сырья применяется шихта, содержащая 70% метал-
лического кремния и 30% ферросилиция марки Си-75 или только
ферросилиций марки Си-90; размер кусков ферросилиция составляет
примерно 50—60 мм. Желательно, особенно при хлорировании в го-
ризонтальной печи, чтобы сырье имело следующий гранулометри-
ческий состав:
Л
Размер зерен, мм....... 25—30 50—80 80—120
Содержание, % ..... 5—10 25—30 65—70
Сырье такого состава способствует равномерному распределению
хлора по сечению печи и предотвращает сильные перегревы при
взаимодействии хлора с ферросилицием, обладающим развитой
поверхностью.
ю
Рис. 10-7. Технологическая схема производства четыреххлористого кремния:
1 — щековая дробилка; 2 — ковшовый элеватор; 3 — грохот; 4 — шахтный подъемник;
5 — загрузочный бункер; 6 — печь, хлорирования; 7 — конденсатор хлорного железа;
& — скруббер; 9 — кипятильник; 10, 14 — холодильники; 11 — сборник технического
продукта; 12 — ректификационная колонна; 13— дефлегматор; 15 — сборник готового
продукта; 16 — разлагатель твердых отходов; 17 — камер.». гидролиза; 18,— колонна для
поглощения отходящих газов.
и
Ферросилиций дробят на щековой дробилке, затем ковшовым
элеватором подают на грохот, где установлены сита разных разме-
ров. Крупные куски из верхнего сита возвращаются на повторное
дробление. Мелочь, прошедшую через последнее сито, отбрасывают
только в том случае, если хлорирование проводят в горизонтальной
печи.
Дробленый ферросилиций с помощью шахтного подъемника про-
дают в загрузочные бункера, из которых он самотеком поступает
в подогреватель сырья (в период пуска печи). Сырье при 300—400 °C
загружают через определенные промежутки времени в печь хло-
рирования. _
Печь хлорирования (рис. 10-8) представляет собой двухконусный
стальной аппарат шахтного типа с водяной рубашкой. Поверхность
охлаждения около 7 м2. Диаметр широкой части печи 900 мм, узкой
480 мм, общая высота печи 3300 мм. Нижний конус печи имеет мень-
шую высоту (650 мм) и футерован диабазовой плиткой в один слой.
537
Печь заполняют на высоту 600—700 мм кусками битого и предвари-
тельно высушенного графита, который служит подушкой для рас-
пределения хлора. Непосредственно на графит загружают ферро-
силиций, поддерживая высоту слоя шихты в пределах 250—350 мм.
Производительность печи — около 2 т/сут SiCl4.
Для хлорирования ферросилиция используют испаренный жидкий
хлор. Газообразный электролитический хлор, содержащий примеси
Рис. 10-8. Двухконусный аппарат для
хлорирования ферросилиция.
4
основная масса твердых возгонов.
двуокиси углерода и кисло-
рода, окисляющие кремний,
для этой цели не приме-
няется. Хлор подают в печь
через распределительный
коллектор. Реакция хлори-
рования протекает с выде-
лением большого количества
тепла, которое отводится во-
дой через стенки аппарата.
Температура в зоне реакции
колеблется в пределах 800—
1200 °C.
Реакционные газы из
печи хлорирования посту-
пают в конденсатор, состо-
ящий из двух вертикальных
труб с общим конусным
бункером. Внутренний диа-
метр труб 400 мм, высота
5200 мм, объем бункера 2 м3.
Внутри труб расположены
скребковые мешалки, снима-
ющие со стенок сконденси-
ровавшиеся хлориды. Трубы
и бункер снабжены рубаш-
ками. В рубашку первой
трубы подают воду для охла-
ждения, в результате чего
конденсируется и осаждается
Вторую трубу конденсатора
и конусный бункер во избежание конденсации в них паров четырех-
хлористого кремния обогревают паром, поддерживая температуру
S0—120 °C. Всего в конденсаторе улавливается примерно 90—95%
твердых хлоридов и других возгонов.
Окончательную очистку паров четыреххлористого кремния от
твердых хлоридов и других примесей проводят в дополнительном
«ухом конденсаторе, обогреваемом паром, либо в скруббере, ороша-
емом четыреххлористым кремнием. При мокрой очистке достигается
практически полное удаление всех твердых частиц, однако такая
установка громоздка и требует дополнительных энергетических затрат.
538
В скруббере, представляющем собой вертикальную колонну
с распределительными тарелками, реакционные газы движутся про-
тивотоком жидкому четыреххлористому кремнию, который промы-
вает газы, увлекая твердые частицы. Поступающая в кипятильник
суспензия подвергается отпариванию. Пары SiCl4 . после конден-
сации вновь возвращаются на орошение скруббера, а скапливающиеся
в кипятильнике твердые хлориды и другие частицы периодически
выгружаются.
Пары четыреххлористого кремния конденсируются в трубчатых
холодильниках, охлаждаемых последовательно водой и рассолом.
Полученный сырец подвергают ректификации в системе, состоящей
из перегонного куба, ректификационной колонны, дефлегматора,
змеевиковых холодильников и сборников кубовых остатков и гото-
вого продукта. Ректификационная колонна представляет собой
стальную трубу, заполненную керамическими кольцами размером
50x50x5 мм. Вначале, для удаления растворенного в сырце газо-
образного хлора, змеевиковый холодильник включают как обратный
и нагревают смесь до тех пор, пока температура паров после дефлег-
матора не достигнет 55 °C. После этого переключают холодильник
и отбирают основную фракцию SiCl4 в сборники готовой продукции.
Отбор готового продукта прекращают, когда температура паров
достигнет 75 °C.
В качестве отходов получают твердые хлориды и частицы, оса-
ждаемые при сухой и мокрой очистке реакционных газов. Эти отходы
в настоящее время не используются, а разлагаются водой в спе-
циальных емкостях.
Отходящие газы различных стадий процесса (конденсации и рек-
тификации, разложения твердых и кубовых остатков) направляют
сначала в камеру гидролиза четыреххлористого кремния, выпол-
няемую из кирпича и футерованную изнутри диабазовой плиткой.
Для лучшего соприкосновения газов с водой камера разделена на
отсеки, в которых вращаются распылители, создающие плотную
завесу из брызг воды. Непоглощенные отходящие д?азы (в основном
хлор) удаляют из камеры воздушным эжектором и передают в ко-
лонну, орошаемую известковым молоком. Кислые шламовые воды
из разлагателя направляют в отстойники-нейтрализаторы.
На некоторых зарубежных предприятиях [57, 80], а также на
одном из отечественных заводов в производстве четыреххлористого
кремния применяют горизонтальные печи хлорирования (рис. 10-9).
При этом изменяется и конденсационная система, практически
составляющая одно целое с хлоратором.
Диаметр горизонтальной части печи 600 мм; с одной стороны зта
часть печи закрывается дверцей, охлаждаемой водой, а с другой —
переходит в первую конденсационную трубу. Печь имеет также не-
большую вертикальную трубу, заканчивающуюся заслонкой, через
которую загружают ферросилиций. Горизонтальная и вертикальная
трубы, а также штуцер для ввода хлора снабжены рубашками для
охлаждения. Температура охлаждающей воды на выходе из рубашек
53&
вертикальной и горизонтальной труб должна быть не выше 60—
69 °C, а на выходе из рубашек шнека конденсационной трубы, из
Ферросилиций
Вода
Вода
Пар
SiCI4 t
4 Твердые
хлориды
ЛЛ1\ЛЛ1\Й
дверцы и хлорного штуцера — не
выше 50—55 °C. При соблюдении
этих условий можно предотвра-
тить коррозионное разрушение
аппаратуры.
Конденсационная часть уста-
новки состоит из трех располо-
женных друг над другом горизон-
тальных труб, также снабженных
рубашками для охлаждения. В пер-
вой и второй трубах размещены
шнеки со скребками, непрерывно
снимающими со стенок твердые
Возгоны. К концу первой трубы
подсоединен длинный абшайдер
диаметром 300 мм, соединенный
патрубком со второй конденсаци-
онной трубой. Абшайдер служит
для сбора твердых возгонов, ниж-
ний его конец закрыт шибером и
оборудован пневматическим вибра-
Рис. 10-9. Горизонтальный аппарат
для хлорирования ферросилиция:
1 — реактор; * 2 — загрузочное устрой-
ство; 3 — горизонтальные конденсаторы;
4 — спиральные транспортеры твердых
хлоридов: 5 — абшайдер; 6 — труба аб-
шайдера для ^вывода твердых хлоридов;
7 — сборник.
ном не отличаются от этой же
приведенной выше схеме (см. рис. 10-7).
Расходные коэффициенты на 1 т SiCl4:
тором для разрыхления осадка на
стенках. Между второй и третьей
трубами имеется гидрозатвор для
поддержания постоянного давле-
ния в системе. Хвостовая часть
конденсационной системы и рек-
тификационная колонна в основ-
части установки, показанной на
о? Й
§
Хлор, т \....................... ..
Ферросилиций (Си-90), т......... .
Шихта, т1
кремний Кр1 .......................
ферросилиций Си-75 .............
Известковое молоко, т ..........
Вода промышленная, м3........... .
Азот, м3 ...........................
Электроэнергия, кВт ч ..............
Пар, Мкал s ............. .
1,41
0,215
0,15
0,064
0,14
200
30к-40
300
1-1,5
Очистка четыреххлористого кремния
При получении монокристаллов кремния, кремниевых эпитакси-
альных пленок, специальных оптических стекол и особо чистой
двуокиси кремния к исходному сырью — четыреххлористому крем-
нию — предъявляются более высокие требования. Суммарное содер-
жание примесей в таком сырье не должно превышать 10’7 —
10"8 вес. %.
В техническом продукте содержание примесей (считая на эле-
мент) примерно следующее (в %): по 1—3*10“4 алюминия, железа,
кальция; по 1—5-10'5 магния, титана, свинца, олова, сурьмы,
фосфора й по 1—5-Ю"6 меди, марганца и бора.
Известные в настоящее время способы глубокой очистки нельзя
считать универсальными, в каждом случае Удаляется лишь группа
примесей. В зависимости от требований к качеству конечного про-
дукта могут сочетаться различные методы глубокой очистки.
Хорошо изучена ректификационная очистка четыреххлористого
кремния [81—90]. Некоторые авторы [82, 83] определяли предель-
ную степень очистки SiCl4 от таких примесей, как соединения каль-
ция, магния, меди, фосфора и железа с помощью ректификации.
Подробно исследовано [84—90] равновесие жидкость — пар для
систем четыреххлористый кремний — примесь и на основании полу-
ченных данных определены соответствующие коэффициенты разде-
ления, а также разделяющая способность насадочных и тарельчатых
колонн.
Ректификация довольно эффективна для очистки преимущественно
от неполярных и малополярных примесей, неограниченно раство-
римых в SiCl4. К ним относятся хлориды и оксихлориды фосфора,
титана, ванадия, бора, олова и сурьмы. Растворы таких компонен-
тов в SiCl4 по свойствам приближаются к регулярным растворам;,
относительная летучесть этих растворов может быть с достаточной
точностью рассчитана по уравнению Гильденбранда — Вуда. При-
меси соединений кальция, магния, железа, алюминия, марганца,
свинца и других дают квазиазеотропные смеси с SiCl4 и ректифика-
цией практически не отделяются.
Описаны в лйтературе экстракционные методы очистки четырех-
хлористого кремния [91—94]. Предложено [91] экстрагировать при-
меси концентрированными серной и фосфорной кислотами при 20 °C.
После очистки содержание соединений железа, меди, бора и титана
снижается примерно в 5 раз. В качестве высокополярного неоргани-
ческого экстрагента может применяться треххлористая сурьма [92].
Большая область расслаивания и высокая относительная летучесть
в системе SiCl4—SbCl3, а также значительная растворимость неко-
торых хлоридов в SbCl3 позволяют очищать тетрахлорид кремния
методом экстрактивной ректификации или путем последовательной
экстракции и ректификации. При этом достигается удовлетвори-
тельная очистка от железа, алюминия, титана, кальция и меди.
К органическим экстрагентам относятся уксусная кислота и ее ан-
гидрид [93]. Для удаления примеси бора предложено [94] использо-
вать фенол.
Более глубокая очистка четыреххлористого кремния достигается
адсорбционными методами. Изучена [95] адсорбция ВС13 и РС13
из четыреххлористого кремния силикагелем, активированной, окисью
541
алюминия, кокосовым углем и цеолитами различных марок. Наилуч-
шие результаты получены при использовании силикагеля или акти-
вированной окиси алюминия с зернами размером 0,075—0,175 мм,
а также на силикагеле, полученном при гидролизе SiCl4. Эффектив-
ность очистки повышается, если до адсорбции четыреххлористый
кремний обрабатывают хлором или хлором в смеси с хлори-
стым алюминием. Хлор окисляет РС13 до РС15, а при добавле-
нии хлористого алюминия образуется комплексное соединение
РС15-А1С13.
Для удаления серы SiCl4 рекомендуется обрабатывать медными
стружками. После предварительной обработки четыреххлористого
кремния медными стружками, хлором и А1С13 его подвергают адсорб-
ционной очистке. Автор [95] утверждает, что при этом достигается
глубокая очистка от бора, фосфора, а также от хлоридов тяжелых
металлов.
Физико-химические факторы, определяющие адсорбцию SiCl4,
подробно изучены в работах [96—99]. Исследована динамика адсорб-
ции хлоридов алюминия, меди, железа, бора,, титана и бинарных
растворов в четыреххлористом кремнии при различных скоростях
потока раствора, высоте слоя адсорбента и температуре (от —25
до +40 °C). Наиболее вероятной стадией, лимитирующей процесс
адсорбции хлоридов из четыреххлористого кремния, является вну-
тренняя диффузия. Определены адсорбционные коэффициенты и вы-
явлено наличие двух областей адсорбции, причем за счет особо актив-
ных центров поглощается не более 2—5% общего количества адсор-
бированного вещества.
Полученные данные позволяют рассчитывать промь
in
ленные
адсорбционные установки. Исследована [100] динамика и статиче-
ская активность некоторых промышленных адсорбентов: силикагеля
марки АСМ, активированной окиси алюминия, березового угля БАУ,
активированного угля СКТ и цеолита 4А при удалении РС13. К наи-
более эффективным адсорбентам относятся
активированная окись
алюминия и силикагель. Изучена также адсорбционная очистка SiCl4
в паровой фазе [100].
К адсорбционным методам очистки SiCl4 примыкают методы ча-
стичного гидролиза, использующие адсорбционную активность поли-
кремневых кислот в момент их образования. Частичный гидролиз
осуществляют добавлением к четыреххлористому кремнию различ-
ных влагоносителей (кристаллогидраты некоторых солей, гидрати-
рованная целлюлоза или вода в любом агрегатном состоянии)
[Ю1-Ю4].
Имеются сообщения о методах, основанных на восстановлении
отдельных примесей в условиях, при которых четыреххлористый
кремний остается без изменения. Например, при обработке SiCl4
порошкообразным цинком и соляной кислотой образуется атомарный
водород, восстанавливающий хлориды некоторых металлов [105].
Для удаления примесей углерода, фосфора, бора, железа, мышьяка
и сурьмы предложено восстанавливать их в присутствии металлов
542
>
| с сильно развитой поверхностью (губчатые алюминий, натрий, цинк),
к нагретых ниже температуры пиролиза SiCl4 [106].
Подробный обзор методов глубокой очистки четыреххлористого
i- кремния приведен в работе [107].
К ЧЕТЫРЕХХЛОРИСТЫЙ ТИТАН
Физико-химические свойства
Четыреххлористый титан — бесцветная прозрачная дымящая на
воздухе жидкость. Молекула TiCl4 представляет собой правильный
тетраэдр, в центре которого находится атом титана, а в вершинах —
атомы хлора.
Плотность р и вязкость ц жидкого четыреххлористого титана
составляют:
t, °C, р, г/см* ть П
—10 1,7784 0,0117
0 1,7609 0,01012
+Ю 1,7446 0,00912
t, °C, р, г/см® Т|, П
20 1,7277 0,00826
40 1,6937 0,00702
100 1,5891 —
личных температурах:
°C р, мм рт. ст. t, °C] *р,
—13,9 1,0 30
10 5,4 40
20 10.0 50
-Плотность паров TiCl4 по воздуху равна 6,836, плотность кри-
сталлов 2,03 г/см3 (при —60 °C).
Температура плавления TiCl4 составляет —23 °C, температура
кипения при атмосферном давлении 136 °C. Давление паров при раз-
мм рт. ст. t, °Cj р, мм рт>3ст. t, °C р, мм рт. ст.
16,7 60 62,1 110 367,7
26,5 80 134,0 120 493,8
£41,1 100 264,5 135 740,7
Основные термодинамические константы по данным [1,551:
TiGl* (г)
—176 270
TiGl* (ж)
186 000
Теплота образования, кал/моль
Теплота испарения (409 РК),
кал/моль ....................
Теплота плавления (245 ° К),
кал/моль ....................
Изобарно-изотермический потен-
циал, кал/моль ..............* —169 950
Энтропия, кал/(моль-град) . , 60,4
Теплоемкость, кал/(моль • град) 35,7
—167 490
84,4
22,9
При взаимодействии е водой четыреххлористый титан полностью
гидролизуется, реакция протекает бурно с образованием объемистого
; осадка. С кислородом TiCl4 начинает реагировать при 500—600 °C.
: Водород и некоторые металлы восстанавливают TiCl4 до TiGl3 цлй
TiGl2, а в определенных условиях до элементарного титана. Железо
I* при температуре ниже 700 °C не взаимодействует с TiCl4.
543
Растворимость хлора в четыреххлористом титане при атмосфер-
ном давлении составляет [108, 109]:
г, °C . . .................... 0 20 40 60 80 100 136
Растворимость хлора, вес. % » . . . 11,5 7,60 4,10 2,40 1,80 1,10 0,03
При парциальном давлении хлора 1,5 атм и 20 °C растворимость
хлора равна 11,16 вес. % [110].
Подробный обзор физических и термодинамических свойств TiCl4
приведен в работе [111], обзор реакций TiCl4 с органическими и не-
органическими веществами дан в работе [112].
Низшие хлориды титана. Существуют три кристаллических моди-
фикации (а, у, 6) фиолетового треххлористого титана и (J-модифика-
ция коричневого TiGl3 [113—117]. Фиолетовые кристаллы имеют
форму пластин, коричневые кристаллы могут быть выделены в виде
игл только при низких температурах.
Треххлористый титан впервые был получен в 1846 г. Все способы
получения треххлористого титана сводятся к восстановлению TiCl4
различными восстановителями, а именно: водородом [118—121],
титаном [41, 115, 122—124], кремнием [125, 1261, натрием [127—
’ 129}, алюминием [130—134] и другими элементами [130, 135, 136].
Треххлористый титан возгоняется без разложения при 425 °C
и давлении менее 1 мм рт. ст.,
при
постепенном повьг
it
ении темпера-
туры от 425 до 440 °C и давлении 0,001 мм рт. ст. можно получить
сублимированный TiCl3 в виде прозрачных фиолетовых пластин.
Двухлористый титан образует черные гексагональные кристаллы.
Температура плавления 1035 °C. Двухлористый титан можно полу-
чить термическим разложением трёххлористого титана или восста-
новлением TiCl4 титаном [114, 137—139].
Применение четыреххлориотого титана
Четыреххлористый титан применяется главным образом для
получения металлического титана. Впервые титан достаточной
чистоты был получен восстановлением четыреххлористого титана
натрием. Более широкое распространение получил процесс Кролля,
основанный на восстановлении TiCl4 магнием [140].
Значительные количества четыреххлористого титана расходуются
для получения пигментной двуокиси титана [141].
Четыреххлористый титан применяется также в качестве ката-
лизатора при полимеризации этилена и при алкилировании арома-
тических углеводородов [142, 143]. Полученный восстановле-
нием TiCl4 треххлористый титан применяется в качестве катализатора
при полимеризации олефинов-, в частности в производстве полипро-
пилена [143].
Теоретические основы получения четыреххлористого титана
Для получения TiCl4 применяют главным образом окисное сырье
{рутиловые концентраты, титановые шлаки). В отдельных случаях
^используют также карбиды и нитриды титана.
544
тальных исследований [144,
CJ
Рис.
на скорость хлорирования TiO
ООО 500 000 700 800
Температура, °C
10-10. Влияние температуры
2*
f В отсутствие восстановителя хлорирование двуокиси титана
[; начинается при температуре выше 800 °C с выделением кислорода,
причем степень хлорирования не превышает 1% [19]. При очень
большом избытке хлора и температуре выше 1200 °C максимальный
выход TiCl4 составляет около 40%.
Заметнее хлорируются в отсутствие восстановителя низшие
окислы титана. В результате проведенных расчетов и эксперимен-
145] установлено, что хлорирование
[. окиси титана начинается при температуре около 300 °C, а в интер-
/ вале температур 400—700 °C степень хлорирования достигает 50%.
j Практически полное хлорирование окислов титана происходит
только в присутствии восстановителя, который связывает выде-
;• ляющийся кислород. Ряд исследова-
' телей [146—150] изучали термодина-
J мические основы реакций хлориро-
< вания двуокиси титана в присутствии
I твердого и газообразного восстано-
L вителей с целью определения теоре-
t тических равновесных парциальных
давлений реагирующих веществ и
j продуктов реакции. Была устано-
влено, что до 500—600 °C реакция
L хлорирования с восстановителем
идет преимущественно с образова-
j нием двуокиси углерода, выше 700 °C
j преобладает окись углерода, а в ин-
! . тервале 900—1000 °C кислород дву-
v окиси титана связывается с образованием почти исключительно
f окиси углерода.
t Влияние температуры на скорость хлорирования смеси двуокиси
f титана и кокса показано на рис. 10-10. Реакция начинается при
[ 445 °C, до 550 °C при линейной скорости хлора 16 см/мин процесс
идет в кинетической области, при более высоких температурах
L переходит в диффузионную область. Энергия активации в кинемати-
г ческой области равна 37 500 кал/моль, в диффузионной области —
I около 2000 кал/моль [144, 151, 152].
Исследования механизма хлорирования двуокиси титана в при-
| сутствии восстановителя представлены в работах [153—155]. Авторы
I считают, что скорость хлорирования зависит от поверхности контакта
| зерен двуокиси титана с частицами угля, а лимитирующей стадией
| процесса является переход молекул промежуточного продукта,
I образовавшегося на угле, на поверхность зерен двуокиси ти-
I тана.
I Хлорирование титановых шлаков связано с рядом особенностей,
| обусловленных сложным химическим и минералогическим составом
I шлаков. На скорость хлорирования шлаков влияет содержание
I в них низших окислов титана (TiO, Ti2O3) и изменение фазового
I состава шлака в результате окисления [156; 157]. Наличие в шлаках
В*
! 35 Заказ 843 , 545
lllll
около 5—8% окиси кальция и окиси магния ускоряет процесс
хлорирования. Такое влияние объясняют внедрением катионов Саа+
и Mg2+ в межбазисные плоскости углерода (восстановителя), что
ослабляет связи углерода в решетке и тем самым увеличивает его
реакционную способность.
Степень хлорирования содержащихся в шлаках FeO, А12О3,
MgO, СаО и МпО зависит от температуры, при 800 °C эти окислы
извлекаются полностью. Извлечение кремнезема увеличивается при
повышенном содержании Ti2O3 в шлаках и уменьшается при нали-
чии более 11% MgO. Степень хлорирования основного компонента —
двуокиси титана — В интервале 500—800 °C колеблется от 98 до
99,5% [1561.
В производстве четыреххлористого титана обычно применяют
65%-ный хлоргаз, образующийся в электролизерах при получении
магния. Установлено, что разбавление хлора воздухом не влияет
на скорость хлорирования двуокиси титана, а также на качество
получаемого TiCl4 [1581. Однако наличие кислорода в хлоре вызы-
вает сгорание части кокса (восстановителя), увеличивает количество
выделяющегося тепла, что ограничивает производительность хло-
ратора. Предложен способ [159], заключающийся в том, что разба-
вленный хлор абсорбируют четыреххлористым титаном, а затем
при нагревании выделяют концентрированный хлор и направляют
его на хлорирование.
Для промышленного получения TiCl4, в частности для правиль-
ного выбора конструкции аппарата, футеровочных материалов и для
определения максимальной производительности реактора, предста-
вляют существенный интерес термодинамические расчеты макси-
мальной температуры хлорирования титановых шлаков в шахтной
электропечи. Показано [160], что при адиабатическом хлорировании
шлаков хлором, подогретым до 800 °C, и отношении в реакционных
га^ах СО : СО2 = 9:1 теоретическая максимальная температура
процесса составляет 1187 °C. В тех же'условиях при использовании
65%-ного хлора максимальная температура хлорирования возра-
стает до 1310 °C. Следовательно, нет опасений, что при интенсифи-
кации процесса в шахтной печи будет превышена допустимая с точки
зрения термической стойкости огнеупоров температура.
Существенным недостатком применяемых для хлорирования ти-
тановых шлаков шахтных электропечей является необходимость
подвода электроэнергии через боковые графитированные электроды,
что вызывает частые газовыделения при нарушении уплотнений
и остановки печей на ремонт. В последнее время разработана кон-
струкция шахтного хлоратора непрерывного действия без внешнего
подогрева. Производительность такого аппарата примерно вдвое
выше по сравнению с шахтной электропечью [161].
Хлорирование титансодержащего сырья в среде расплавленных
солей нашло широкое распространение [162—165] и все более вытес-
няет процессы, основанные на хлорировании брикетов в шахтных
печах.
У
546
f На скорость и степень хлорирования в расплаве влияют следу-
> ющие факторы: температура, скорость подачи хлора, высота слоя
расплава, плотность, вязкость и Поверхностное натяжение рас-
плава, свойства и степень помола восстановителя, способ загрузки
шихты в реактор и характер распределения твердых частиц в объеме
расплава.
При хлорировании в эквимолекулярной смеси NaCl и КС1 опти-
мальная температура процесса составляет около 900 °C. Большое
значение имеет дисперсность восстановителя, лучшие результаты
получены при размере частиц менее 0,1 мм [162]. При разбавлении
хлоргаза до концентрации 40% не наблюдается заметного снижения
скорости реакции, однако дальнейшее разбавдение до 20% умень-
шает скорость хлорирования в 2 раза. Установлено, что в присут-
ствии хлоридов железа и алюминия в расплаве скорость хлориро-
вания увеличивается в несколько раз, что объясняется каталити-
ческим действием этих хлоридов [163]. Предполагают [165], что
двуокись титана хлорируется в расплаве нерастворенным хлором,
а главным образом — хлорным железом, находящимся в расплаве
в виде иона FeClJ.
Более резко на скорость хлорирования влияет поверхностное
натяжение расплава. При уменьшении поверхностного натяжения
от 172 до 65 дин/см скорость реакции возрастает в 25 раз [166].
Исследования по хлорированию в кипящем слое описаны в ра-
ботах [167—170]. Наиболее пригодным сырьём для таких процессов
является карбид или нитрид титана, представляющие собой туго-
плавкие .неспекающиеся материалы. Карбид и нитрид титана хлори-
руются при значительно более низкой температуре (250—400 °C),
чем кислородные соединения титана, не требуется добавление вос-
становителя. Отсутствие кислорода исключает образование окси-
хлоридов, что позволяет получать более чистый TiCl4 [171].
Производство четыреххлориотого титана
ж
Промышленное производство четыреххлористого титана полу-
чило развитие в 40-х годах, когда Кролль разработал магнийтерми-
ческий метод получения титана из TiCl4.
Основной способ промышленного производства четыреххлори-
стого титана как в СССР, так и за рубежом состоит в хлорировании
брикетов из титансодержащего сырья и угля в шахтных электро-
печах. В нашей стране широко используется также процесс хлори-
рования в среде расплавленных солей. Ограниченное применение
нашли процессы хлорирования титанового сырья в кипящем слое,
а также способы, основанные на предварительной обработке иль-
> менита, титанового шлака или рутила с целью получения карбида
титана и его последующего хлорирования.
Сырье [111, 172, 173]. В природе титан встречается преимуще-
ственно в виде кислородных соединений. Он входит в состав более
130 титансодержащих минералов. Промышленное значение имеют
и
35* 547
рутил, ильменит, аризонит и титаномагнетит, а при комплексной
переработке также сфен, перовскит и лопарит. В СССР имеются
крупнейшие месторождения ильменита, перовскита и титаномагне-
тита.
Наиболее удобным сырьем для получения TiCl4 является рутил,
в котором содержится от 91 до 99% TiO2? однако объем добычи ру-
тила ограничен и стоимость его относительно велика.
Хлорирование перовскитовых и сфеновых концентратов, содер-
жащих 25—35% СаО, связано с повышенным расходом хлора и
приводит к попутному образованию легкоплавкого хлорида кальция.
Ильменитовый концентрат обычно также не используется для
непосредственного хлорирования. Его подвергают обезжелезива-
нию. Наиболее широко для этой цели применяется восстановитель-
ная плавка титановых концентратов в электродуговой печи. Конеч-
ными продуктами плавки являются чугун и титановый шлак. В за-
висимости от условий плавки состав шлака колеблется в следующих
пределах (в %):
TiO2 . . . 85—90 MgO . . . 1,7—3,0
FeO ... 2,5—6,5 А12О3 . . . 1,0-3,5
SiO2 . . . 2,5—5,5 MnO . . . 1,0—1,5
СаО . . . 0,75-4,0
В СССР титановые шлаки являются основным сырьем для полу-
чения четыреххлористого титана.
Технология получения четыреххлористого титана [173—176].
Процесс получения четыреххлористого титана включает четыре
стадии: подготовку сырья, хлорирование, конденсацию и очистку
технического продукта. В зависимости от выбранного способа произ-
водства (хлорирование в шахтных электрических печах, в расплаве
солей, в аппаратах КС) отличаются лишь отдельные стадии. Так,
подготовка сырья заключается либо в приготовлении брикетов из
титансодержащего сырья и кокса, либо в измельчении этих компо-
нентов и составлении шихты. Меняется состав реакционных газов
(в частности, соотношение СО : СО2), вследствие чего различны
условия и режим конденсации. Очистка технического продукта
одинакова независимо от способа производства. , -
Хлорирование в шахтпнъгх печах. Шихта, поступающая на хлори-
рование в шахтные печи, брикетируется. Для обеспечения высокой
степени хлорирования компоненты шихты предварительно измель-
чают. Титановый шлак дробят последовательно в шнековой и в ко-
нусной дробилках, затем размалывают в шаровой мельнице. Нефтя-
ной кокс также дробят сначала в шнековой, затем в молотковой дро-
билке. Более тонкий размол нефтяного кокса получают в молотко-
вой мельнице с воздушной сепарацией. Около 80% измельченного
шлака должно проходить через сито с отверстиями 0,1 мм, такое же
количество восстановителя должно проходить через сцто с отвер-
стиями 0,015 мм. В качестве связующего при брикетировании при-
меняют сульфит-целлюлозный щелок, каменноугольный’ пек или
И'"“
548
патоку. Количество связующего зависит от давления при брикети-
ровании, помола исходных компонентов и температуры нагрева
шихты. Так, при использований ' вальцовых прессов (давление
300 кгс/см2) в шихту добавляют 7—8% каменноугольного пека,
смесь нагревают перед прессованием до 90 °C. Если шихту нагре-
вать до 100—110 °C, в нее можно добавлять только 5—6% пека.
Количество сульфит-целлюлозного щелока составляет 10—14%.
Сырые брикеты после прессования содержат 50—60% TiO2, 18—
22% С, 5,3—5,8% летучих приме-
сей и 1,5—1,6% влаги.
Для удаления остаточной влаги
и летучих органических примесей
сырые брикеты подвергают коксо-
ванию (прокаливанию без доступа
кислорода). Для полного удаления
летучих брикеты, приготовленные на
сульфит-целлюлозном щелоке, нагре-
вают до 500—600 °C, а брикеты на
каменноугольном пеке до 800—
io
т
10-11. Шахтная электриче-
печь для хлорирования ти-
Вконден -
'опционную
систему
о&р о О Qpfo Чр с? о?оо
Для прокаливания брикетов при-
меняются промышленные печи не-
скольких видов. При небольшом
масштабе производства используют
камерные печи периодического дей-
ствия. Брикеты вводят через съем-
ный свбд в закрытых контейнерах
из жаропрочной стали. Поскольку
во время прокаливания брикеты
неподвижны, они не истираются и
мелочь не образуется.
К печам непрерывного действия
относятся туннельные и ретортные.
В последних обычно имеются четыре
керамические реторты, в верхней
части которых происходит сушка и
нагрев брикетов, а в нижней — прокаливание и
В прокаленных брикетах содержится 20—22%
лее 0,2% летучих, прочность брикетов колеблется в пределах
100—120 кгс/см2.
Хлорирование прокаленных брикетов проводят в шахтной элек-
тропечи (рис. 10-11), состоящей из стального кожуха, изнутри по-
крытого диабазом на жидком стекле и футерованного специальным
низкопористым шамотным кирпичом. В нижней части печи разме-
щены два ряда угольных электродов (по три электрода в каждом
ряду). Пространство от подины до уровня, на 400—700 мм выше верх-
них электродов, заполнено насадкой из угольных цилиндров, слу-
жащих электрическим Сопротивлением.
Рис.
ская
танового сырья:
1 — электроды; 2 — фурма для по-
дачи хлора; 3 — очистной лаз; 4 —
распределительный конус; 5 — золот
никовый питатель; <
7 — футеровка; 8 — брикеты; 9~~ ко
жух из листовой стали;
пая насадка;
расплава.
11 — летка
6 — патрубок*
10 — уголь-
для слива
удаление
углерода
летучих,
и не бо-
По разогретой насадке стекают расплавленные хлориды метал-
лов, которые собираются в нижней, зоне печи. Расплав периодически
выгружают через сливную летку в изложницы. Над нижним рядом
электродов расположено несколько фурм, через них в печь посту-
пает хлор. Расход подводимой к электродам электроэнергии опреде-
ляется количеством тепла, необходимого для подогрева хлора до
750—800 °C, поддержания хлоридов в расплавленном состоянии
и для компенсации потерь тепла в окружающую среду. Обычно ниж-
ние электроды все время находятся под напряжением, а верхние
отключаются после выхода печи на режим. Для измерения темпера-
туры по высоте печи устанавливают несколько^ термопар.
В крышке печи расположены футерованный патрубок для реак-
ционных газов, отверстия для шуровки, и люк со взрывной мембра-
ной. В центре крышки укрепляется загрузочное приспособление,
состоящее из герметичного приемного бункера, питателя с золотни-
ковым затвором и распределительного конуса. Загрузочное устрой-
ство позволяет проводить непрерывную загрузку брикетов без пре-
кращения подачи хлора.
Важное значение для процесса хлорирования в шахтной печи
имеет правильное распределение и движение шихты и газов, поддер-
жание оптимального температурного режима по всей высоте печи.
В нижней зоне от подины до фурм, где собираются расплавленные
хлориды, температура должна быть порядка 600—700 °C; между
фурмами и верхним уровнем угольной насадки поддерживают тем-
пературу 700—800 °C. В эту зону поступают расплавленные хло-
риды из реакционного пространства, здесь же происходит предва-
рительный разогрев хлора. Температура в реакционной зоне коле-
блется от 800 до 1150 °C. По мере накопления над поверхностью на-
садки непрохлорированного остатка зона хлорирования переме-
щается по направлению к своду печи. Границы реакционной зоны
зависят также от температуры поступающего хлора: при подаче
холодного хлора зона реакции растягивается, при подаче нагретого
хлора — сокращается.
При хлорировании в шахтных печах достигается довольно полное
(не менее 97—98%) извлечение титана. Степень извлечения других,
окислов из шихты зависит от температуры хлорирования и свойств
извлекаемого компонента. Например, если окись кремния находится
в виде кварца, степень хлорирования SiO^ составляет 10—20%,
а окись кремния, входящая в состав силиката, хлорируется на 80%
и более. Окись алюминия в виде корунда хлорируется незначительно,
алюмосиликаты — почти полностью.
Производительность шахтной электропечи определяется ее вну-
тренним диаметром и высотой слоя шихты. Верхний предел произ-
водительности с учетом устойчивости футеровки в зоне реакции
ограничен максимально допустимой температурой, которая само-
произвольно возрастает (до определенного уровня) за счет экзотер-
мичности процесса. Продолжительность пребывания брикетов в печи
колеблется от 8 до 12 ч, средняя скорость опускания шихты от 0,1
550
до 0,5 см/мин, удельный съем TiClrc 1 м2'сечения печи составляет
2—2,5 т/сут. * .
Режим работы печи чувствителен к высоте слоя шихты. При за-
вышенном слое шихты между образующимися хлоридами и окис-
лами, входящими в состав шлаков, протекают вторичные реакции.
Кроме того, возгоны высокоплавких хлоридов осаждаются на бри-
кетах верхних слоев шихты и увеличивают сопротивление печи.
При заниженном уровне шихты ^прореагировавший хлор взаимо-
действует с окисью углерода, образуя фосген, который конденси-
руется вместе с TiCl4. Низкий уровень шихты обусловливает увели-
чение пылеуноса возгона хлоридов и возрастание температуры
реакционных газов на выходе из печи. ?
От температурного режима печи и высоты слоя шихты зависит
также отношение СО : СО2 в отходящих газах. Чем выше эти пара-
метры, тем больше относительное содержание СО, Если для хлори-
рования применяют анодный хлоргаз, в отходящих газах содержится
азот. Примерный состав газовой смеси после конденсационной
системы (в %):
СО .......... 59 О2 ..... . 1,5
N2 ........ 28 СОС12 . . . .' 0,5
СО а ........ 7 С12 Следы
НС1 ..... 3
Окись углерода, содержащаяся в отходящих газах, может образо-
вывать взрывоопасную смесь при смешении с воздухом, проникаю-
щим в печь. Вследствие этого хлоратор и аппараты конденсационной
системы должны работать под небольшим избыточным давлением.
Хлорирование в расплаве солей. Накопление непрореагировавшего
остатка в шахтных печах создает весьма серьезные затруднения.
Так, в случае применения титанового концентрата с повышенным
содержанием кальция или других видов титанового сырья (перов-
скиты и др.) хлорирование в печах шахтного типа становится невоз-
можным из-за спекания шихты хлоридом кальция.
Распространенный в Советском Союзе способ хлорирования
в среде расплавленных солей особенно важен при использовании
титанового сырья, содержащего значительное количество примесей,
образующих легкоплавкие хлориды. В качестве среды при хлори-
ровании применяют расплав хлоридов калия и натрия. Эвтектиче-
ская смесь этих солей плавится при 660 °C. На титаномагниевых
комбинатах в производстве TiCl4 используют отработанный расплав
магниевых ванн примерного состава (в %):
KG1 ..... 75—80 MgCl2 .... 5—7
GaGlo......8-10 FeGL + MnGL 1—3
NaCl .... 5—10
Г
Хлоратор представляет собой прямоугольную шахту, выложен-
ную шамотным кирпичом. Шахты бывают однокамерными и много-
камерными. Производительность одного аппарата может достигать
551
f
50 т/сут TiCl4. В нижней части хлоратора имеются фурмы для подачи
хлора в газораспределительные устройства. В боковые стенки вмон-
тированы графитовые токоподводящие электроды для поддержания
электролита в расплавленном состоянии в пусковой период или при
временной остановке хлоратора. Для слива плава имеются летки.
В верхней крышке хлоратора размещены штуцера для загрузки
шихты и для добавления расплава, а также патрубок для отвода
реакционных газов.
Особенностью конструкции хлоратора являются теплоотводящце
элементы, позволяющие интенсифицировать процесс за счет отвода
избыточного тепла. Элементы представляют собой графитовые блоки,
внутри которых проходят стальные охлаждаемые водой штанги.
Шихту (титановый шлак и нефтяной кокс) загружают на поверх-
ность расплава или вдувают ее под уровень расплава через канал
в боковой стенке хлоратора. Для проведения хлорирования в рас-
плаве важное значение имеет степень помола восстановителя. Опти-
мальный размер зерен нефтяного кокса равен 0,1 мм. При более
крупном помоле не обеспечивается достаточное извлечение титана,
при более тонком помоле наблюдается заметный унос восстановителя
реакционными газами.
Кислород окислов, содержащихся в титановом шлаке, в условиях
хлорирования-в расплаве взаимодействует с восстановителем пре-
имущественно до СО2, а не СО, как это происходит в шахтных печах.
В результате повышается концентрация TiCl4 в реакционных газах
и соответственно улучшаются условия конденсации. Резкое умень-
шение содержания СО увеличивает безопасность процесса и снижает
вероятность образования фосгена.
В ходе хлорирования расплав постепенно обогащается хлори-
дами кальция, магния и других металлов, в результате чего повы-
шается его вязкость и ухудшаются условия реакции. В связи с этим
необходимо периодически обновлять плав. При сливе части плава
попутно выводится непрохлорированный остаток, что является важ-
ным преимуществом этого процесса по сравнению с хлорированием
в шахтных печах. Отработанный расплав имеет примерно следующий
состав (в %):
КС1 ..........30-40
MgCL ....... . 25—35
FeCi;+ FeCl3 + MnCl2 10-20
С ......... 7—9
SiO2..........3—&
NaCl ....... 2—5
СаС12.........2—4
TiO2........ .0,5—1
Таким образом, хлорирование титансодержащего сырья в ра-
сплаве солей позволяет создавать высокопроизводительные аппараты,
отказаться от громоздких операций приготовления и прокаливания
брикетов, частично улучшить условия конденсации, резко снизить
концентрацию СО в отходящих газах и непрерывно выводить непро-
хлорированный остаток.
К недостаткам способа относится увеличение потерь титана с от-
работанным расплавом за счет вторичных реакций TiCl4 с кис лоро-
552
дом или окислами шихты, а также в результате уноса мелких ^пыле-
видных) частиц шихты с реакционными газами и др. Возрастает
также количество твердых возгонов, что повышает нагрузку на пы-
левые камеры. В дальнейшем усовершенствовании нуждаются кон-
струкции хлораторов, в частности их теплоотводящие устройства.
Хлорирование в кипящем слое. Использование аппаратов кипя-
щего слоя для хлорирования титансодержащего сырья — одно из
возможных направлений интенсификации производства четырех-
хлористого титана.
В аппаратах с кипящим слоем достигается эффективный массо-
и теплообмен, быстрое выравнивание температуры по всему слою и
высокая скорость процесса даже при сравнительно низких темпера-
турах. Так же, как при хлорировании в расплаве, при ведении про-
цесса в кипящем слое отпадает необходимость брикетирования шихты
и создаются условия для осуществления непрерывного процесса.
Аппарат КС для хлорирования титансодержащего сырья пред-
ставляет собой цилиндрическую шахту, футерованную плотным
динасовым кирпичом. В ее нижней части расположена газораспре-
делительная решетка. Кроме того, для лучшего использования хлора
и более полного извлечения титана в аппарате имеется несколько
полок, размещенных друг над другом. Исходная шихта поступает
на верхнюю полку, где частично хлорируется поступающим снизу
непрорёагировавшим хлором, и через сливной канал пересыпается
на следующую полку. Наиболее интенсивно протекает хлорирование
на нижней полке.,
Размеры частиц титанового сырья и восстановителя должны
быть подобраны с учетом плотностей этих материалов таким образом,
чтобы под воздействием газового потока не происходило преимуще-
ственного выноса из слоя одного из компонентов шихты.
При повышенном содержании в сырье кальция, магния, марганца
и других примесей образование хлоридов этих металлов может при-
вести к увеличению слипания частиц и нарушению режима кипя-
щего слоя. В результате резко изменяются условия массо- и тепло-
обмена и возникает необходимость остановки процесса. Поэтому
хлорирование шлаков, включающих значительные количества окис-
лов кальция и магния, проводят при 600 °C, т. е. при температуре,
не выше температуры образования наиболее легкоплавкой эвтекти-
ческой смеси получающихся хлоридов. Скорость процесса в таких
условиях заметно уменьшается.
При хлорировании автоклавного или флотационного рутиловых
концентратов, которые почти не содержат примесей, образующих
плавкие хлориды, температуру процесса можно повысить до 900—
1000 °C и тем самым увеличить его скорость.
Эффективным сырьем для хлорирования в аппаратах кипящего
слоя являются карбид титана и оксикарбонитрид (последний обра-
зуется попутно при карбидизации в промышленных условиях).
Порошкообразный карбид титана представляет собой тугоплавкий
неспекающийся материал, т. пл. 3140 °C. Он может хлорироваться
553
J
без восстановителя. При этом отпадает необходимость в составле-
нии шихты и главным образом в подборе такого^ гранулометриче-
ского состава хлорируемой массы, при котором можно избежать
избирательного уноса ее отдельных . компонентов. Хлорирование
протекает с достаточной скоростью при более низких температурах
(300—400 °C).
Как и для всех процессов в кипящем слое, серьезная проблема
заключается в устранении пылеуноса. Для улавливания пыли при-
меняют циклоны, размещаемые в верхней широкой части аппарата.
При использовании выносного циклона его обогревают для пред-
отвращения конденсации низкокипящих хлоридов. Уменьшение
пылеуноса может быть также достигнуто с помощью специальной
. инертной насадки.
Производительность аппаратов КС в зависимости от температуры
хлорирования составляет 5—10 т TiCl4 в сутки на Г м2 сечения
печи. , /
Конденсация и разделение хлоридов. В продуктах хлорирования
титансодержащего сырья наряду с TiCl4 присутствуют другие хло-
риды и газы: SiCl4, CCI4, VOC13, А1С13, FeCl3, FeCl2, СаС12, fMgCl2,
МпС12, СО, СО2, НС1, N2, СОС12 и др. Одна из самых трудных ста-
дий процесса — выделение четыреххлористого титана из такой мно-
гокомпонентной системы. Задача осложняется тем, что соединения,
входящие в состав реакционных газов, могут взаимодействовать
между собой с образованием твердых и жидких растворов. Кроме
того, все эти хлориды гигроскопичны и при попадании влаги гидро-
лизуются. Поэтому непременным условием нормальной работы си-
стемы конденсации является надежная герметичность аппаратов'
и коммуникаций.
В производстве четыреххлористого титана используют различные
способы конденсации хлоридов. Схема раздельной конденсации
основана на применении системы конденсаторов, рукавных фильтров
и других аппаратов; в каждом из них поддерживается определен-
ная температура. Парогазовая смесь из реактора поступает сначала
в два последовательно соединенных конденсатора. На входе в пер-
вый конденсатор температура смеси 500—600 °C, на входе во второй
300—350 °C и на выходе из второго конденсатора 120—180 °C.
Для уменьшения высоты конденсаторов их делают двухходовыми,
с внутренними перегородками. Слой возгонов, отлагающийся на
стенках конденсаторов, предохраняет аппарат от действия хлора,
но в то же время снижает коэффициент теплопередачи от парогазо-
вой смеси наружному воздуху. Стенки конденсатора и нижнего ко-
нуса очищаются от возгонов с помощью скребков или цепи, закре-
пленных на вращающемся валу. К горловине нижних конусов кон-
денсатора присоединены шнеки для непрерывной выгрузки. Если
выгрузка возгонов производится периодически, под основанием
конуса устанавливают шиберный затвор и герметично присоединяют
кюбель. Для предотвращения конденсации паров TiGl4 кюбель обо-
гревают.
:554
ft
7
t
Состав твердых возгонов зависит от способа производства и ка-
чества сырья. При хлорировании титановых шлаков в шахтных
электрических печах возгоны содержат главным образом хлориды
магния, железа, марганца, алюминия и др. При хлорировании
в расплаве солей увеличивается содержание хлоридов натрия и
калия.
Для окончательной очистки парогазовой смеси от твердых частиц
. после конденсаторов устанавливают рукавные фильтры из стекло-
волокна. Очень важно поддерживать в фильтрах постоянную тем-
пературу, несколько превышающую точку росы TiGl4. Более высокая
температура вызывает разрушение фильтрующей ткани и способ-
ствует проскоку паров А1С13, при понижении температуры происхо-
дит конденсация TiCl4, стеклянная ткань покрывается вязкой массой,
что увеличивает давление в системе. Обычно в конденсаторах ула-
вливается 50—60% всех возгонов, в фильтрах 40—50%. После филь-
тров количество твердых частиц в газах составляет 1,5—2 г/м3.
В последние годы начали применять так называемые солевые
фильтры, представляющие собой колонны с насадкой из кусков
поваренной соли. Температура в колонне 350—450 °C. Хлориды
железа и алюминия, содержащиеся в парогазовой смеси, проходя
колонну, образуют с NaCl легкоплавкие эвтектические соединения
типа NaAlCl4 и NaFeCl4. Расплав стекает вниз и периодически
удаляется из колонны.
После полного отделения твердых хлоридов парогазовая смесь
поступает в конденсаторы жидких хлоридов.
Схема совместной конденсации состоит в том, что выходящую
из реактора парогазовую смесь резко охлаждают и из нее одновре-
менно конденсируются твердые и жидкие хлориды. Охлаждение
проводят в оросительных конденсаторах, где в качестве орошающей
жидкости используется охлажденный четыреххлористый титан. Оро-
сительный конденсатор состоит из двух труб, соединенных внизу
общим конусом. В верхней части каждой трубы установлены фор-
сунки для разбрызгивания TiCl4. Полнота конденсации и улавли-
вания твердых хлоридов определяется плотностью орошения и тем-
пературой газов на выходе из конденсатора (последняя обычно не
превышает 70 °C). Освобожденный от твердых частиц газовый поток
. направляют в холодильники для конденсации оставшегося TiCl4
(10—20%). Первые по ходу холодильники охлаждают водой, послед-
ний — рассолом.
Образовавшаяся в оросительном конденсаторе пульпа стекает
в промежуточный бак, из которого часть TiCl4 с помощью погружного
насоса подаётся на орошение; остальную пульпу собирают в сгусти-
теле для отделения четыреххлористого титана от твердых хлоридов.
Осветленный хлорид направляют в сборник технического продукта.
Сгущенная пульпа поступает в испаритель для отгонки оставше-
гося четыреххлористого титана (примерно 6—7% общего количе-
ства TiCl4). Для этого в испаритель добавляют поваренную соль
и нагревают массу до 500 °C. Четыреххлористый титан отгоняется,
555
а твердые возгоны образуют с хлористым натрием при 500 °C расплавг
который периодически удаляют через летку.
В промышленной практике обычно применяют комбинированную
схему конденсации, включающую сухие конденсаторы для отделе-
ния основной массы твердых хлоридов и оросительные конденса-
торы, где улавливаются только уносимые после первых аппаратов,
твердые хлориды. На рис. 10-12 показана технологическая схема
конденсации и разделения хлоридов в производстве четыреххло-
ристого титана.
Вода Рассол
Рис. 10-42. Технологическая схема конденсации и разделения
хлоридов:
1 — пылевые камеры; 2 — рукавный фильтр; з — контейнеры для твердых
хлоридов; 4 — оросительные конденсаторы; 5, 6 — холодильники; 7 — по-
гружные насосы; 8 — сгустители;' 9 — шнеки для подачи шлама; J о —
фильтры.
После конденсационной системы отходящие газы содержат в ос-
новном СО и СО2 и немного примесей Cl2, TiCl4, SiCl4, HCI, SOC12,
COC12 и др. Перед выбросом в атмосферу газы подвергаются сани-
тарной очистке в скрубберах, орошаемых водой или известковым
молоком. При новь
in
ении содержания SiCl4
в отходящих
газах
становится выгодной его утилизация. Для этой цели перед санитар-
ными скрубберами устанавливают абсорберы, орошаемые холодным
четыреххлористым титаном. Полученную смесь TiCl4 и SiCl4 напра-
вляют на ректификацию.
Очистка четыреххлориотого титана
Технический четыреххлористый титан содержит растворенные
и взвешенные примеси. К ним относятся газы (N2, С12, СОС12),
хлориды некоторых металлов (А1С13, FeCl3, SiCl4, SnCl4, СаС12,
556
JVIgCl2), оксихлориды (VOCL, TiOCl2, SOC12) и органические соеди-
нения (гексахлорбензол, хлорацетилхлориды, четыреххлористый
углерод). Органические примеси попадают в четыреххлористый титан
из восстановителей и связующих, применяемых при брикетировании
шихты (нефтяной кокс, каменноугольный пек, Сульфитный щелок).
Фосген образуется при взаимодействии непрореагировавшего хлора
с окисью углерода. Оксихлорид титана выделяется в результате
гидролиза TiCl4 при его контакте с влажным воздухом.
Состав и количество примесей зависят не только от качества ис-
ходного сырья, но также от условий ведения процесса, например
температуры хлорирования и конденсации. Чем ниже температура
в аппаратах конденсационной системы, тем выше содержание газо-
образных и легколетучих примесей, так как растворимость СОС12,
Cl2, НС1 и СО2 в четыреххлористом титане увеличивается с пониже-
нием температуры. Твердые хлориды, наоборот, мало растворимы
при низких температурах. Некоторые примеси (SiCl4, СС14, VOC13
и CS2) смешиваются с TiCl4 в любых соотношениях.
Фактическое содержание примесей в техническом TiCl4 колеблется
в следующих пределах (в вес. %):
Si .... 0,01—0,3 TiOCl2 . . 0,04—0,5
Al .... 0,01—0,1 СОС12 . . 0,005—0,15
Fe .... 0,01—0,02 Cl2 .... 0,03—0,08
V ..... 0,01-0,1 S ..... 0—0,03
Четыреххлористый титан очищают от взвешенных частиц фильтро-
ванием или центрифугированием, а от растворенных примесей —
фракционированной перегонкой.
Присутствие А1С13 в четыреххлористом титане способствует кор-
розионному разрушению аппаратуры, особенно при нагревании.
Предложено [177—179] переводить А1С13 в нерастворимый оксихло-
рид добавлением определенного количества воды, а затем отделять
его вместе с другими взвешенными частицами при фильтровании.
Такие примеси, как оксихлорид ванадия, довольно трудно от-
делить ректификацией из-за близости температур кипения VOC13
и TiCl4 (соответственно 127 и 136 °C). Предложено восстанавли-
вать VOC13 до менее летучего соединения VOC12. Наибольшее рас-
пространение получил способ восстановления VOC13 медью (добавле-
ние медного порошка или пропускание паров TiCl4 через медную
сетку). Наряду с оксихлоридом ванадия восстанавливаются соеди-
нения серы, хлорное олово и оксихлорид хрома. С медью взаимодей-
ствуют также органические соединения типа хлорацетилхлоридов,
образуя на поверхности металла нерастворимые в TiCl4 соединения.
За рубежом в качестве восстановителя широко применяются
сероводород [180—182] и другие активные сульфиды (меркаптаны,
трехсернистая сурьма). От образующихся осадков четыреххлори-
стый титан отделяют декантацией, фильтрованием или перегонкой.
Рекомендуют обработанный сульфидом четыреххлористый титан
пропускать через колонну, заполненную известью [183].
557
Предложен способ непрерывной очистки четыреххлористого ти-
тана от VOC13 водородом [184]. Технический TiGl4, t содержащий^
0,3—0,4% VOC13, испаряют, а перегретые до 700 °C пары взаимо-
действуют в колонне с большим избытком водорода. Выходящую
из аппарата парогазовую смесь орошают охлажденным жидким TiCl4.
При этом конденсируется часть газообразного TiCl4 вместе с неле-
тучими примесями (в том числе VOC13); основная масса газа, выходя-
щего из колонны, подвергается более глубокому охлаждению, в ре-
зультате чего получают четыреххлористый титан, содержащий ме-
нее 0,008% VOC13. Часть TiCl4, сконденсировавшегося при ороше-
нии, перегоняют отдельно.
По данным [185], глубокая очистка от VOC13 достигается при
пропускании паров TiCl4 через солевой расплав, который содержит
хлористый калий, обезвоженный карналлит или отработанный элек-
тролит производства магния. В присутствии восстановителя (уГоль,
металлический титан) VOC13 восстанавливается до VC13, без восста-
новителя образуется малолетучее соединение K2VOC14. В обоих
случаях очищенный TiGl4 содержит 0,003—0,001% ванадия.
На некоторых отечественных заводах технический TiCl4 очищают
следующим образом. Четыреххлористый титан заливают в реактор
и включают мешалку. Через загрузочное устройство добавляют
увлажненную поваренную соль (или влажный активированный
уголь), количество влаги в которой достаточно для превраще-
ния A1G13 в нерастворимый оксихлорид алюминия. Затем в аппарат
загружают медный порошок в'количестве 0,5% от массы TiCl4 (при
содержании 0,1% ванадия) и перемешивают содержимое реактора
в течение 0,5 ч. Далее суспензию перекачивают на предварительное
отстаивание или фильтрование. В качестве фильтрующего материала
применяют стеклоткань, асбестовую набивку, керамику и др. Остав-.
' шийся на фильтре меднованадиевый кек направляют на отпарива-
ние TiCl4 или гидролиз, а отфильтрованный четыреххлористый ти-
тан — на ректификацию.
Ректификацию четыреххлористого титана осуществляют в колон-
нах из нержавеющей стали с дырчатыми тарелками или тарелками
со щелевидными прорезями. В первой колонне отделяется SiCl4.
В нижней части колонны обычно поддерживают температуру 139—
140 °C при давлении примерно 800—900 мм рт. ст., в верхней —
соответственно 60—70 °C и остаточное давление 20—25 мм рт. ст.
Неконденсирующиеся газы (СО2, С12, N2, СОС12) направляются
из дефлегматора через гидрозатвор в систему газоочистки. Четырех-
хлористый кремний подвергают повторной дистилляции и исполь-
зуют как товарный продукт.
Очищенный от SiCl4 и летучих компонентов четыреххлористый
титан поступает во вторую ректификационную колонну, где, .про-
ходя через насадку, освобождается от оксихлоридов титана и дру-
гих высококипящих примесей. Температура в верхней части колонны
составляет 136 °C. Очищенный четыреххлористый титан содержит
около 0,004% V, 0,006% Si, 0,004% Fe, 0,004% Al, 0,001-0,002% О2.
1
Физико-химические основы процессов разделения продуктов,
образующихся при хлорировании титансодержащего сырья, и очистки
TiCl4 от примесей подробно изложены в работах [111, 176, 1861.
В последние годы предложены методы более глубокой очистки
четыреххлористого титана. Представляет интерес сорбционная
очистка от примесей [187—190]. Четыреххлористый титан обрабаты-
вают различными веществами (животными или растительными жи-
рами, органическими кислотами, спиртами и др.) и нагревают смесь
до обугливания. Примеси адсорбируются продуктами обугливания
и удаляются при фильтровании. Получают TiCl4 высокой чистоты.
Рекомендуется [191 ] очищать четыреххлористый титан полукоксом,
предварительно обработанным водяным паром и двуокисью углерода.
Для освобождения от высококипящих соединений кремния (на-
пример, гексахлордисилоксана) TiCl4 может быть обработан [192]
фторирующими агентами (КНF2, BF3, TiF4). При этом гексахлор-
дисилоксан, температура кипения которого близка к температуре
кипения TiCl4, переходит во фторсодержащее соединение с более
низкой температурой кипения и легко отделяется при перегонке.
Описан [193] способ получения четыреххлористого титана высо- .
кой чистоты, основанный на сочетании ректификационной и адсорб-
ционной очистки. Ректификацию проводят в стальной насадочной
колонне, футерованной фтороцластом-4. Насадку изготовляют из
проволочной никелевой спирали. Разделительная способность ко-
лонны равна 60—80 теоретическим тарелкам. Средняя фракция (80%
загруженного продукта) передается в адсорбционную колонну,
выполненную из нержавеющей стали с внутренним покрытием из >
фторопласта-3. В качестве адсорбента используют силикагель с удель-
ной поверхностью 700—750 м2/г и содержанием примесей не более
1 10“ 4%.. Очищенный четыреххлористый титан содержит менее
4-10“4% каждой примеси/
Подробно изучена статика и динамика адсорбционной очистки *
TiCl4 на промышленных сорбентах. Показано [194] i что при исполь-
зовании трехслойного адсорбционного фильтра, состоящего из акти-
вированного угля БАУ, активной окиси алюминия А-1 и особо чи-
стого силикагеля АСМ, достигается высокая степень очистки. Так,
при 9-10-4 вес. % начального суммарного содержания контроли-
руемых примесей (РС13, РОС13. AsCl3, SnCl4, FeCl3, S2C12, CS2)
содержание примесей в очищенном продукте составляет не более
4-10“6 вес. %.
4 /
ХЛОРИДЫ ФОСФОРА
Физико-химичеокие свойства [195—197]
К числу хлоридов фосфора, имеющих промышленную ценность,
относятся треххлористый, фосфор РС13, пятихлористый фосфор РС15,
хлорокись фосфора РОС13. Известен также двухлористый фос-
фор Р2С14.
559
Треххлористый фосфор — прозрачная, бесцветная, дымящая на
воздухе жидкость, ее плотность при О °C равна 1,612,,при 21 °C —-
1,575 г/см3; плотность паров (по отношению к воздуху) равна 4,75.
Температура кипения РС13 равна 76 °C, давление паров при разных
температурах составляет:
Л °C ..................... . —20 —10 0 10 20 30 40 50
р, мм рт. ст................. 38 63 101 156 234 341 486 674
Теплота образования A/7°98 (ж) = —75,8 ккал/моль, теплота
испарения 7,28 ккал/моль, теплоемкость Ср '== 28,7 кал/(моль*град)
в интервале 298—348 °К.
Треххлористый фосфор растворим в сероуглероде, бензоле,
четыреххлористом углероде, эфире. При действии холодной воды
•он гидролизуется с образованием фосфористой и соляной кислот.
Под воздействием горячей воды продуктами гидролиза могут быть
Н3РО4, РН3 и белый фосфор. РС13 относительно легко взаимодей-
ствует с кислородом и серой, образуя соответственно хлорокись
или сульфохлорид фосфора.
Пятихлористый фосфор — белые кристаллы, имеющие иногда
зеленый оттенок из-за выделения хлора, возгоняются в присутствии
воздуха при 160 °C. При незначительном повышении давления он
плавится при 167 °C. Плотность твердого вещества при 160 °C равна
1,601 г/см3. Плотность паров пятихлористого фосфора значительно
ниже расчетного значения, равного 7,22, вследствие диссоциации
на РС13 и С12. При 300 °C степень диссоциации РС15 достигает 96% ,
а плотность пара (по отношению к воздуху) составляет 3,65. Давле-
ние паров пятихлористого фосфора при различной температуре:
Л °C'....................... 90 100 110 120 130 140 150 160
р, ммрт. ст................. 18 35 67 117 191 294 445 670
Теплота образования ДЯ°«8 (тв) “ —106,5 ккал/моль, теплота
испарения 15,5 ккал/моль, теплоемкость Ср = 33,0 кал/(моль-град)
в интервале 298—432 °К.
При взаимодействии с водой РС15 гидролизуется с образова-
нием фосфорной и соляной кислот. В случае недостатка воды или
при действии водяных паров образуется хлорокись фосфора и соля-
ная кислота. Пятихлорцстый фосфор растворим в четырёххлористом
углероде, со спиртами образует хлоралкилы, с кислотами — хлор-
ангидриды соответствующих кислот.
Хлорокись фосфора — бесцветная жидкость с острым запДхом,
во влажном воздухе сильно дымит. Плотность РОС13 при 25 °C равна
1,648 г/см3, температура кипения 107 °C, относительная плотность
паров при 150 °C равна 5,33.
Теплота образования ДЯ298 (ж) = —144 ккал/моль, теплота
испарения 8,4 ккал/моль, теплоемкость Ср ~ 22,12 + 3,60-10’3 Т
[в кал/(моль>град) при 298—1000 °К].
Хлорокись фосфора является полярным растворителем, раство-
ряет как органические (например, СС14), так и неорганические соеди-
.560
нения (NaCl, NH4C1). Конечными продуктами гидролиза РОС13
являются фосфорная и соляная кислоты.
Все хлориды фосфора являются токсичными веществами, спо-
собными вызвать острые и хронические отравления.
Применение хлоридов фоофора
Треххлористый фосфор является прежде всего исходным сырьем
для получения пятихлористого фосфора, хлорокиси фосфора, фос-
фористой кислоты и сульфохлорида фосфора.
Все хлориды фосфора применяют для производства широкого
ассортимента фосфорорганических производных. К ним относятся
инсектициды для сельского хозяйства, наполнители смазочных
масел и топлив, красители, пластификаторы, растворители, стабили-
заторы, лекарственные вещества и др.
Пятихлористый фосфор применяют также как катализатор в ре-
акциях циклизации и перегруппировок. Вступая в реакцию с хло-
ристым аммонием, он образует фосфорнитрилхлорид, являющийся
исходным сырьем для получения огнестойких полимеров.
Во многих органических синтезах вместо РС15 предпочитают
использовать хлорокись фосфора. Кроме того, РОС13 применяют
для производства негорючих гидравлических жидкостей, ионооб-
менных смол, а дакже в качестве катализатора для получения неко-
торых красителей.
Теоретические основы получения хлоридов фоофора
Наиболее энергично протекают реакции образования хлоридов
фосфора из элементарных веществ. При избытке фосфора прямой
синтез из белого фосфора и хлора приводит к образованию треххло-
ристого фосфора, в избытке хлора образуется пятихлористый фосфор
Р4+6С12—>4РС13
РС13+С12 —► РС15
Возможна также побочная трудноконтролируемая реакция
6РС15 + Р4—> 10РС13
При синтезе хлорокиси фосфора из элементарных веществ
P4+2O2 + 6CI2 —> 4РОС13
полагают, что часть фосфора окисляется до Р2О5, оставшийся фосфор
хлорируется сначала до РС13, затем до РС1б. Процесс завершается
реакцией
V
.л
Р2О5+ЗРС15 —> 5РОС13
Синтез хлоридов фосфора из элементарных веществ сопрово-
ждается выделением большого количества тепла, наиболее экзотер-
мична реакция образования хлорокиси фосфора (939 ккал/кг РОС13).
36 Заказ 843 561
Для обеспечения спокойного протекания процесса и создания усло-
вий интенсивного теплосъема предложено осуществлять синтез РОС13
в среде жидкой хлорокиси фосфора [198], а синтез РС13 — вереде
треххлористого фосфора [199].
Прямое хлорирование природных фосфатов значительно удеше-
вило бы процесс получения хлоридов фосфора. Костяной уголь (смесь
фосфата кальция и угля), окись углерода и хлор начинают реаги-
ровать при 180 °C, а при 330—340 °C реакция идет быстро и нацело.
Процесс проходит через промежуточные фазы образования мета-
фосфата кальция
Са3(РО4)2 + 2СО + 2С12 > Ca(PO3)2-f-2CO2+2СаС12
Са(РОз)2+4СО + 4С12 —> 2РОС13 + 4СО2 + СаС12
Са3(РО4)2 + 6СО + 6С12----> 2РОС13 + 6СО2 + ЗСаС12
В работе [200] указанные температурные условия не подтвер-
ждаются. При хлорировании гранулированных апатита или костя-
його угля смесью хлора и окиси углерода при 360—380 °C удавалось
достичь незначительного выхода хлорокиси фосфора (порядка 5%
по израсходованному хлору). С достаточной скоростью хлорирова-
ние фосфата идет лишь при 750—900 °C. Такие же результаты при-
ведены в работе [201].
В результате хлорирования фосфата кальция возможно образо-
вание РОС13, PG13 и РС15. Химизм процесса, включающего стадию
образования элементарного фосфора с последую
и
[им его хлорирова-
нием, может быть, по данным [202], представлен следующими урав-
нениями
2Са3(РО4)2 + 6С12+8С —> Р4+6СаС12 + 8СО2
Р4 + ЮС12 —> 4РС15
РС15—> pci3-f-CI^
Са3(РО4)2 + 6С12 + ЗС —-> 2РОС13+ЗСаС12 + ЗСО2
Са3(РО4)2 + 6С12+4С —> 2РС13+ЗСаС12 + 4СО2
Са3(РО4)2+8С12 + 4С ---> 2РС15 + ЗСаС12+4СО2
Преимущественно хлорирование фосфата кальция идет до Р0С13.
Хлорокись фосфора легко может быть превращена в другие хлор-
производные фосфата. Если пары РОС13 пропускать через колонну
с раскаленным докрасна углем, образуется треххлористый фосфор.
При действии хлора РС13 легко превращается в РС1Ь.
Хлорирование фосфата кальция всегда сопровождается образо-
ванием хлористого кальция, который закрывает поры угля, обвола-
кивает частицы фосфата и препятствует дальнейшему проникнове-
нию хлора. Это обстоятельство является главной причиной противо-
речивых данных отдельных авторов об оптимальных условиях хло-
рирования фосфата кальция.
562
Получившие в последние годы развитие способы хлорирования
в расплаве солей позволяют в значительной степени устранить эти
трудности, если процесс получения хлоридов фосфора из природ-
ного сырья осуществлять в среде хлористого кальция при 850—
900 °C, т. е. выше температуры его плавления. Подробно изучены
[203—205] механизм и кинетика хлорирования фосфатов в расплаве
хлоридов натрия, калия и кальция, влияние концентрации и ско-
рости подачи хлора, влияние количества и степени измельчения
восстановителя и др.
Производство хлоридов фосфора
В промышленности треххлористый фосфор получают из элемен-
тарных веществ — хлорированием белого фосфора. При малых мас-
штабах производства используют также красный фосфор. Хлориро-
вание может быть осуществлено как периодически, так и непрерывно.
Производительность современной установки достигает 500 кг/ч
и более.
Хлорокись фосфора получают главным образом окислением трех-
хлористого фосфора кислородом или воздухом. Ограниченное приме-
нение находит способ, основанный на взаимодействии РС15 и Р2О5
или РС13, С12 и Р2ОБ.
Мировое производство хлорпроизводных фосфора составляет
160—180 тыс. т/год, причем следует учитывать, что выпускаемый
треххлористый фосфор расходуется на получение хлорокиси фосфора
и пятихлористого фосфора. Выпуск РОС13 в США в 1970 г. составлял
33 тыс. т [206].
По данным [207], к хлоридам фосфора предъявляются следу-
ющие технические требования:
Содержание, %
РС13 . ...............................
РС15 .. ..............................
РОС13 ................................
Свободный хлор .............
Fe . . . .............................
Ni....................................
Тяжелые металлы (в пересчете на РЪ) . .
Пределы кипения, °C
начало........................ .
конец ............................
Остаток, % ............
РС1з РС15 POCh
99,5 Следы 0,2
— 99,5 —
— - 99,0
— — Следы
— 0,0005 0,0005
— 0,001 0,001
— 0,0005 —
74,5
79,0
0,5
106,0
108,5
0,5
фосфора р е г л аментир у е тс я
В СССР качество треххлористого
ТУ 6-02-574—70, хлорокиси фосфора — СТУ 53-244—62.
Сырье [195, 208]. Фосфор существует в трех основных модифика-
циях: белый, красный и черный. Белый фосфор имеет специфический
запах, светится в темноте, хорошо растворим в сероуглероде, жидком
аммиаке, бензоле; практически нерастворим в воде. На воздухе легко
воспламеняется, поэтому его хранят под водой. В присутствии при-
месей технический белый фосфор окрашен в желто-бурый цвет. При
563
*
36*
нагревании без доступу воздуха до 270—300 °C белый фосфор пере-
ходит в красный.
В жидком и парообразном состоянии (до 800 °C) фосфор четырех-
атомен, при более высоких Температурах распадается на двух-
атомный.
Производство элементарного фосфора осуществляется электро-
термическим восстановлением фосфорита или апатита коксом в при-
сутствии кварцевого песка. Для получения 1 т фосфора расходуется
вода
Рис. 10-13. Технологическая схема производства треххлористого фосфора;
1 — приемный бак для фосфора; 2 — мерник фосфора; 3 — сборник фосфора; 4 — гид^о-
затвор; 5 — растворитель фосфора; 6 — погружной насос; 7 — хлоратор; 8 — полочная
колонна; о — обратный} конденсатор; 10 — холодильник; 11 — приемник сырца; 12 —
дохлоратор; 13 — конде^атор; 14 — приемник готового продукта.
7—10 т фосфата, 1,4—1,6 т кокса и до 3 т песка. Расход электроэнер-
гии составляет 14—17 кВт-ч.
В производстве фосфора и его соединений особо, важно соблюде-
ние специальных мер предосторожности. Воспламеняясь, белый
фосфор вызывает болезненные, долго не заживающие ожоги. Обож-
женную кожу следует промыть раствором перманганата кадия или
сернокислой меди. Горящий фосфор тушат двуокисью углерода,
раствором CuSO4 или песком.
Технология получения РС13, РС15 и РОС13 [199, 208—210].
Технологическая схема производства треххлористого фосфора
представлена на рис. 10-13. Процесс состоит из следующих стадий:
1) приготовление раствора фосфора в треххлористом фосфоре; 2) хло-
рирование; 3) дистилляция и до хлорирование РС13-сырца; 4) очистка
отходящих газов и нейтрализация сточных вод.
564
возможно хлорирование
Рис. 10-14. Аппарат для по-
лучения PClg.
Разделение стадий приготовления раствора фосфора и хлориро-
вания этого раствора позволяет осуществлять процесс по полуне-
прерывной цли непрерывной схеме. В аппарате-растворителе готовят
насыщенный 12%-ный раствор фосфора в треххлористом фосфоре
и с помощью погружного насоса его непрерывно перекачивают в хло-
ратор. Избыток раствора по переливной линии возвращается в рас-
творитель.
Концентрацию фосфора в хлораторе поддерживают на уровне 6—
9%. При содержании фосфора менее ।
до РС1б.
В этом случае при последующей
загрузке фосфора произойдет бурная
реакция с пятихлористым фосфором,
сопровождающаяся резким повышением
давления, что может привести- к раз-
рыву аппаратуры. Если содержание
фосфора будет превышать его раство-
римость в треххлористом фосфоре (12,6%
при 65 °C), возможно образование двух
жидких фаз; поступающий в реактор
хлор начнет взаимодействовать непо-
средственно с нижним слоем фосфора,
что также приведет к бурному течению
реакции или взрыву. Показателем под-
держания требуемой концентрации
фосфора может служить температура
кипения раствора- (78—80 °C), так как
насыщенный раствор кипит при 82 °C,
а чистый РС13 — при 76 °C.
Процесс хлорирования ведут при
температуре кипения раствора и избы-
точном давлении 0,1—0,15 ат.
Образующиеся в хлораторе пары
треххлористого фосфора под давле-
нием поступают в колонну, верхняя
садку из керамических колец, а затем направляются в конден-
саторы. Для очистки пары РС13 в колонне орошают флегмой. В слу-
чае необходимости может быть осуществлена вторичная дистилляция
или дохлорирование сырца в следующем ‘ аппарате с целью удаления
следов фосфора, уносимых отгоном в процессе хлорирования.
Тепло реакции расходуется главным образом на испарение воз-
вращаемого в виде флегмы треххлористого фосфора. Стенки колонны
охлаждаются водой, подаваемой в рубашку. В качестве обратного
холодильника рекомендуется использовать оросительный кожухо-
трубчатый теплообменник, что предотвращает возможность попада-
ния воды в реактор в случае неисправностей в теплообменнике.
Пятихлористый фосфор получают в оросительных ' башнях,
внутренняя поверхность которых покрыта свинцом (рис. 10-14).
царга которой имеет на-
565
1
Реакция идет между абсолютно сухими жидким треххлористым
фосфором и подаваемым противотоком хлором. В реакционной
массе поддерживается избыток хлора. Кристаллический пятихло-
ристый фосфор выводится через низ башни с помощью шнека.
Используется в промышленности и периодический способ. Смесь,
содержащую 1 часть РС13 и 1 часть СС14, вводят в реактор с во-
дяной рубашкой, оборудованный быстро вращающейся мешалкой
и плотной крышкой с обратным холодильником. Над уровнем
жидкости вводят хлор, по завершении реакции из хлоратора выво-
дят суспензию кристаллов пятихлористого фосфора в четыреххло-
ристом углероде. Суспензию фильтруют, кристаллы сушат горячей
водой, циркулирующей в рубашке фильтра. Четыреххлористый угле-
род возвращают в процесс.
Промышленный способ получения хлорокиси фосфора заклю-
чается в окислении треххлористого фосфора воздухом или кислоро-
дом в стальных , вертикальных аппаратах, освинцованных внутри.
Кислород подается в аппарат по свинцовому барботеру спирального
типа, имеющего по всей поверхности спирали отверстия диаметром
6—8 мм. Окисление ведут при 40—65 °C. Температурный режим
поддерживается и регулируется изменением скорости подачи кисло-
рода, а также количества и температуры воды, подаваемой на охла-
ждение внешних стенок аппарата.
Каталитическое действие на процесс окисления оказывают Н3РО3
или Н3РО4, которые частично образуются при взаимодействии РС13
или Р0С13 с влагой, содержащейся в кислороде или азоте. В случае
затухания процесса в реактор специально добавляют некоторое
количество фосфорной кислоты. Процесс замедляется в присутствии
железа, меди и некоторых других металлов.
Очистка хлоридов фоофора
Для получения фосфидов металлов и особенно для легирования
кремниевых, германиевых й других полупроводников необходимы
соединения фосфора высокой степени чистоты.
Предложен [211, 212] способ получения треххлористого фосфора
высокой чистоты из элементарных веществ, причем фосфор дважды
перегоняли в вакууме в присутствии поверхностно-активных ве-
ществ, хлор испаряли из баллонов. Получен треххлористый фосфор,
содержащий (в %): 4,2-10“б Fe, 5,2-Ю”4 Al, 2,7-10"5Si и 5,2-10’7Си.
Последующая двукратная перегонка РС13 не оказывает существен-
ного влияния на качество продукта.
Японские авторы [213, 2141 предложили очищать РС13 и РОС13
дистилляцией в насадочной колонке высотой 35 см. В качестве на-
садки применяли куски кварца или стекла. Для повышения эффек-
тивности очистки, особенно от серы, в колонну добавляли хлористый
алюминий, активированный уголь, порошок железа или меди (по
0,5—2 г на 40 мл образца). Получены препараты с суммарным содер-
жанием всех примесей 1-10“4%.
566
ХЛОРНОЕ ЖЕЛЕЗО
Физико-химические свойства
Безводное хлорное железо образует гексагональные кристаллы
фиолетово-черного цвета с зеленоватым отблеском. Пары хлорного
железа окрашены в желто-коричневый цвет.
Плотность паров хлорного железа при 440 °C составляет 11,14 г/л,
что соответствует соединению Fe2Cle. В интервале 440—450 ~°С
плотность пара постепенно снижается до значения, соответствующего
соединению FeCl3. Относительная плотность кристаллов безводного
хлорного железа d^0*8 == 2,804.
Температура плавления FeGl3 равна 306 °C, температура кипе-
ния 317 °C.
Давление паров хлорного железа составляет:
°C .... 200 240 253 271 285 310 370
р, атм .... 0,012 0,019 0,045 0,131 0,283 0,835 2,04
Основные термодинамические константы по данным [1, 55, 215]
Теплота образования, кал/моль
FeCl3 (т) ..........................-96 360
FeCl3 (г) .......... . . . .........-63 087
Изобарно-изотермический потенциал,
кал/моль
FeCl3 (т) ..........................—80 240
FeGl3 (г) .................... —63 329
Теплоемкость, кал/(моль-град)
FeCl3 (т) ........................ 24,33
FeCl3 (г) 16,63
Теплота плавления Fe2Cl6 при 576 °К,
кал/моль . ................................. 21 392
Теплота испарения Fe2Gl6 при 597 °К,
кал/моль ................................... 12 590
Хлорное железо растворимо в воде, метиловом и этиловом спир-
тах, ацетоне, в этиловом и изопропиловом эфирах, менее растворимо
в сероуглероде, бензоле, нерастворимо в глицерине, этилацетате.
Данные о растворимости FeCl3 в воде приведены на рис. 10-15.
При 20 °C насыщенный водный раствор содержит 47,9 вес. % FeCl3.
Плотность растворов хлорного железа разной концентраций
при 25 °C составляет:
Концентрация FeCl3,
вес. % ............. 1,7 8,7 16,2 25,8
Плотность, г/см3 . . 1,0106 1,0718 1,1428 1,2381
43,6
1,4611
Нась
in
ценный
раствор
1,7934
Вязкость растворов хлорного железа заметно выше вязкости
растворов других хлоридов. Так, вязкость насыщенного раствора
при 25 °C составляет 0,3164 сП.
Водные растворы хлорного железа при нагревании гидроли-
зуются, причем степень гидролиза и состав образующихся продуктов
567
зависят от температуры и концентрации растворов. Конечным
продуктом гидролиза является соединение FeCl3-nFe(OH)3.
При исследовании термохимического разложения безводного
хлорного железа установлено, что в токе азота до 400 °C в получен-
ном продукте содержится 89,6% FeCl3 и 10,2% FeCl2, а в продукте,
Растворимость моль/100моль Н2О
Рис. 10-15. Растворимость FeCl3 в воде
при различной температуре.
полученном после нагревания
FeCl3 в токе воздуха до 400 °C,
найдено 93,5% FeCl3 и 6,3%
Fe2O3 [216].
При взаимодействии FeC]3
с Fe2O3 образуется оксихлорид
железа FeOCl. Условия обра-
зования, структура и другие
свойства этого соединения по-
дробно описаны в работах
[217—219].
Применение хлорного
железа
Хлорное железо применяют
главным образом для очистки
промышленных и сточных вод,
а также для обезвоживания
осадков на станциях аэрации.
Хлорное железо применяют в качестве катализатора в процессах
органического синтеза, для протравки металлических деталей перед
нанесением гальванопокрытий, его можно добавлять к портланд-
цементу для ускорения процесса схватывания, употребляют в фар-
мацевтической промышленности, в медицине, в качестве пигмента
для окраски асбоцементных плиток или других облицовочных мате-
риалов и др.
Хлорное железо является энергичным хлорирующим агентом,
вследствие чего оно может быть использовано для избирательного
извлечения отдельных компонентов руд, например из полиметал-
лических сульфидных руд. .
Теоретические основы получения хлорного железа
Синтез хлорного железа по уравнению
I
2Fe + 3Cl2 ► 2FeCl3
kt
сопровождается побочными реакциями восстановления или терми-
ческого разложения FeCl3
2FeCl3-f-Fe "► 3FeC12
2FeCl3----► 2FeCl2 + Cl2
568
1Ш8
Суммарно процесс можно выразить уравнением:
6rFe-j-£>Cl2 -> cFeCl3-|-dFeCl2еС12
Коэффициент е учитывает не только количество хлора, выделя-
ющегося при термическом разложении FeCl3, но и не израсходован-
ного при реакции.
Реакция восстановления FeCl3 протекает довольно энергично.
При нагревании хлорного железа с избытком железа в запаянной
трубке при 200 °C происходит полное превращение FeGl3 в FeCl2.
Термическое разложение хлорного железа может происходить
как в твердой, так и в газовой фазе. Механизм, кинетика и условия
разложения хлорного железа подробно рассмотрены в работах
[220—223].
Степень разложения паров хлорного железа с повышением темпе-
ратуры уменьшается. По мере перехода димера в мономер (т. е. при
температуре выше 600 °C) термическая устойчивость хлорного же-
леза возрастает в большей степени.
Побочные реакции, приводящие к образованию хлористого же-
леза, особенно вредны для промышленного процесса. Вследствие
малой летучести хлористое железо остается на поверхности метал-
лического железа и процесс практически прекращается. Наблю-
дается также загрязнение готового продукта примесью низшего
хлорида.
В отличие от алюминия железо в меньшей степени пассиви-
руется окисной пленкой, так как окись железа имеет небольшую
плотность и при действии хлора может превращаться в
наличия восстановителя. Взаимодействие Fe2O3 и С12
энергично уже при 400 °C.
Хлорирование железа сопровождается выделением
количества тепла, что затрудняет регулирование процесса. Пока-
зано [224], что если к железу добавить древесные опилки или уголь
и применить для хлорирования разбавленный хлор, то температура
реакции сильно понизится. Исследована [225] зависимость выхода
хлорного железа от общего количества подаваемого хлора и от
количества хлора, расходуемого на образование хлоридов (т. е. за
вычетом непрореагировавшего хлора), а также зависимость проскока
хлора от температуры реакции.
Сопоставление полученных данных позволило заключить, что
оптимальной температурой образования FeCl3 является 400—425 °C;
при более высоких температурах проскок хлора уменьшается,
но соответственно возрастает количество образующегося хлористого
железа. Разбавление хлора азотом (в пределах от 1 : 5 до 1 : 20)
не влияет заметно на выход FeCl3. Установлено, что при разбавлении
хлоргаза азотом уменьшается возможность восстановления FeCl3
железом или его термического разложения (вследствие сокращения
времени пребывания FeCl3 в зоне реакции).
Хлорирование окислов железа редко используется для промыш-
ленного получения хлорного железа. Однако при хлорировании
FeCL без
о
протекает
большого
569
комплексного природного сырья, содержащего Fe2O3 и FeO, хлорное
железо может образоваться в качестве побочного продукта. В отсут-
ствие восстановителя окись железа хлорируется более энергично,
чем закись. Так, степень хлорирования за 4 ч составляет 37,7%,
a FeO в аналогичных условиях 5,45% [226].
При добавлении восстановителя (например, древесного угля)
резко снижается энергия активации, причем по сродству к хлору
окислы проявляют противоположные свойства. Так, закись железа
хлорируется с достаточной скоростью уже при 200 °C, тогда как для
окиси железа требуется температура 300 °C.
' \
Производотво хлорного железа
Хлорное железо выпускают в виде безводной соли, шестивод-
ного кристаллогидрата и растворов.
Первые промышленные установки были пущены в Германии: для
получения шестиводного хлорного железа в 1923 г., безводного
продукта — в 1933 г. '
В США объем производства хлорного железа (безводного и шести-
водного) составляет около 40 тыс. т, в Японии 7,5 тыс. т [227].
В СССР производство безводного хлорного железа было организо-
вано после войны.
В соответствии с ГОСТ 11159—65, качество безводного хлорного
железа должно отвечать следующим требованиям (в вес. %):
I сорт II сорт
FeCl3, не менее ...............
FeCl2, не более ...............
Нерастворимый остаток, не более . . . .
97 95
1 2
2 Не определяют
Растворы хлорного железа получаются как отходы производства
бромсодержащих веществ и при улавливании абгазного хлора рас-
творами хлористого железа. По ТУ 6-01-334—69 и ТУ 6-602—70
выпускают растворы, содержащие 30 и 40% FeCl3.
Кристаллогидрат FeGl3?6H2O выпускают только реактивный
в соответствии с ГОСТ 4147—65 (марки «хч», «чда» и «ч»).
Технология получения безводного хлорного железа хлорирова-
нием стального скрапа включает следующие стадии: подготовку
сырья, составление шихты, хлорирование, конденсацию паров
хлорного железа и очистку отходящих газов от пылевидного про-
дукта и хлора.
Технологическая схема процесса представлена на рис. 10-16.
Шихта состоит из смеси мелкого и крупного стального лома в со-
отношении 1:1. При длине более 500 мм лом режется специальными
ножницами. Шихта периодически загружается в хлоратор через
верхнюю крышку.
Хлоратор представляет собой стальной вертикальный цилиндри-
ческий аппарат. Он состоит из 6 царг: первые четыре царги имеют
рубашки для охлаждения водой, пятая царга внутри футерована
570
диабазовой плиткой, шестая имеет крышку и служит для загрузки
сырья. В нижней части хлоратора вмонтирована колосниковая ре-
шетка, также охлаждаемая водой. Внутренний диаметр хлоратора
0,5 м, высота около 6 м.
Рекомендуется поддерживать шихту на уровне газохода и не
снижать этот уровень более чем на 0,5 м. Хлор подают в нижнюю
часть печи по стальному трубопроводу. Расход хлора в зависимости
от заданной производительности колеблется от 75 до 120 м3/ч.
Вода
6
Рис. 10-16. Технологическая схема производства
хлорного железа:
1 — хлоратор; 2 — конденсаторы хлорного железа; з — ка-
мера для отделения ныли; 4—скруббер; 5—циркуляционный
бак; б — барабаны для готового продукта.
Для предотвращения образования FeGl2 содержание хлора в от-
ходящих газах должно поддерживаться в пределах 3—12%. В отсут-
ствие крупного стального лома печь может работать на мелком ломе,
что увеличивает реакционную поверхность. Вследствие этого воз-
растает количество образующегося хлористого железа, поэтому необ-
ходимо повышать расход хлора, поддерживая определенный избыток
его в отходящих газах. Рекомендуется также разбавлять хлор, но
не ниже 88—90%, так как процесс может замедляться из-за окисле-
ния железа кислородом воздуха.
Температура в реакционной зоне достигает 650—750 °C. Периоди-
чески (1—2 раза в смену) печь очищается от шлака через люк первой
царги. Шлак содержит в основном окись железа, окислы других
металлов, присутствующих в виде примесей в ломе, а также некото-
рые трудно летучие хлориды.
Образующиеся в хлораторе пары хлорного железа вместе с избыт-
ком хлора поступают по газоходу в конденсаторы.
Конденсатор для хлорного железа состоит из двух стальных аппа-
ратов прямоугольного сечения, соединенных между собой в нижней
части общей камерой. В камере расположено разгрузочное устрой-
ство, представляющее собой полый вал, охлаждаемый водой, на
571
который насажены гребки. Конденсатор имеет рубашку для охла-
ждения водой, поверхность охлаждения каждого аппарата равна
16,8 м2.
В первом по ходу газа аппарате конденсируется примерно 2/3,
а во втором около х/3 всего количества полученного продукта.
Хлорное железо выгружается из конденсатора с помощью шнека
в железные барабаны или деревянные бочки емкостью 100—110 л.
Из конденсатора отходящие газы, содержащие мельчайшие ча-
стицы хлорного железа, поступают в пылевую камеру — стальной
прямоугольный аппарат с коническим дном, внутри которого имеется
перегородка для изменения направления потока газа. Объем ка-
меры 16 м3.
Благодаря изменению скорости и направления потока в камере
пыль отделяется от газовой фазы. Мелкие частицы хлорного железа
оседают в нижней части камеры и собираются в барабанах. При общей
производительности установки 10 т/сут из пылевой камеры выгру-
жается около 350 кг/сут.
Окончательная очистка газов от пыли производится в колоннах,
орошаемых водой. Раствор циркулирует в них до тех пор, пока
концентрация хлорного железа не достигнет примерно 500 г/л. По-
лученные растворы хлорного железа используются для внутризавод-
ских нужд (обезвреживание ртути, фосфорсодержащих стоков и др.).
Хлор, выходящий из башен мокрой пылеочистки, улавливается
в санитарных колоннах, орошаемых известковым молоком. В состав
отходящих газов входят следующие компоненты (в объемн. %):
До улавливания
С12 ................. 3,7—14,2
СО2 .............. 1,2—3,4
FeCl3 ............. 0,003-0,01
После улавливания
120—250 (мг/м3)
0,0005—0,0007
Расход сырья и материалов на 1 т хлорного железа составляет:
Стальной лом, кг ...................400
Хлор, кг ...........................920
Известь обожженная, кг ...... 135
Электроэнергия, кВт-ч .............45
Вода, м^ .........................20
ХЛОРИСТЫЙ цинк
Физико-химические свойства
Безводный хлористый цинк представляет собой белые, весьма
гигроскопичные кристаллы.
Температура плавления равна 318 °C. По данным [228], ZnCl2
существует в двух модификациях аир, плавящихся соответственно
при 315 и 326 °C.
Плотность плавленого ZnCl2 составляет 2,904 г/см3, твердого
2,79 г/см3.
572
Температура кипения равна 732 °C. Давление насыщенного пара
ZnCl2:
°C .............. 428 481 508 536 566 584 610 648 689 732
р, мм рт. ст. ... 1 5 10 20 40 60 100 200 400 760
Основные термодинамические константы ZnCl2 [1,229]:
Теплота образования, кал/моль
ZnCl2 (т) ..................
ZnCl2 (ж) ...................
Изобарно-изотермический потенциал,
кал /моль
ZnCl2 (т) ..................
ZnCl2
Энтропия,
ZnCl2
ZnCl2
Теплоемкость,
ZnCl2 (т)
ZnCl2 (ж)
Теплота плавления, кал/моль
ZnCl2 (т) ............
Теплота испарения, кал/моль
ZnCl2 (ж)..............
(ж)...........
кал/(моль град)
(т) ..........
(ж)...........
кал/(моль • град)
—99 550
—98 600
—88 220
-75 930
38,02
32,76
18,3
24,0
5 540
30 900
Хлористый цинк характеризуется высокой растворимостью в воде
(рис. 10-17). При 25 °C концентрация насыщенного раствора 81,2%,
при 100 °G она равна 86% ZnCl2. При низких температурах (от
Рис. 10-17. Взаимная
растворимость в системе
ZnCl2—Н2О.
Содержание^ бес %
Плотность растворов хлористого цинка при 19,5 С равна:
—62 до +28 °C) из растворов выделяются кристаллогидраты с 1,
1х/2, 2г/2, 3 и 4 молекулами воды, при температуре выше 28 °C в очень
узком интервале концентраций из раствора кристаллизуется без-
водный хлористый цинк.
30
1,291
60
1,740
573
10 15 20 25'
1,091 1,137 1,187 1,238
40 45 50 55
1,420 1,488 1,566 1,650
Концентрация ZnCl2, вес. % . 5
Плотность, г/см3 ............1,015
Концентрация ZnCl2, вес. % . 35
Плотность, г/см3 ...... 1,352
В разбавленный растворах хлористый цинк гидролизуется:
Zna+ + H2O ХГГ ZnOH+4-H+
При упаривании концентрированных растворов хлористого цинка
или при нагревании кристаллов до 250 °C в результате гидролиза
могут образоваться соединения типа ZnCl2*xZnO*yH2O. Описана
большая группа соединений основных хлоридов цинка х Zn(OH)2-
•у ZnCl2 с различным соотношением х : у [230, 231].
Применение хлориотого цинка
Хлористый цинк применяют самостоятельно или в смеси с фено-
лом или хроматом для цропитки шпал, для приготовления дезинфи-
цирующих и бальзамирующих жидкостей; при пайке, для получения
минеральных красок в производстве органических красителей;
хлористый цинк используют также в качестве водоотнимающего
средства, катализатора в некоторых химических процессах (получе-
ние ионообменных смол, гидрокрекинг каменного угля и др.).
Быстро твердеющая смесь концентрированного раствора хлори-
стого цинка с окисью цинка является основой зубных цементов.
Свойство раствора хлористого цинка растворять клетчатку исполь-
зуют в производстве пергаментной бумаги.
Теоретические основы получения хлористого цинка
Металлический цинк при комнатной температуре не реагирует
с хлором. По данным [9], взаимодействие начинается при 440 йС.
Реакция бурно развивается при более высоких температурах, когда
образующийся хлористый цинк возгоняется и удаляется с поверх-
ности металла. z
Окись цинка, в отличие от некоторых других окислов, может
взаимодействовать с хлором в отсутствие восстановителя. По рас-
четам [229], изобарный потенциал реакции
ZiiO(t)-|-С12 ——> ZnCl2(r)-|-l/2O2
при 600 °C равен —3434 кал/моль, с повышением температуры термо-
динамическая вероятность реакции увеличивается, в частности
при 900 °C ДО = —11 393 кал/моль. По экспериментальным дан-
ным [19], температура начала реакции хлора с окисью цинка
равна 300 °C.
Изучена кинетика хлорирования окиси цинка хлористым водо-
родом и хлором в интервале 150—750 °C [232]. Показано, что при
температуре выше 500 °C окись цинка полностью превращается
в хлорид как при действии хлористого водорода, так и хлора. Зна-
чительное влияние на скорость хлорирования оказывает удаление
образующегося хлористого цинка из зоны реакции.
Безводный хлористый цинк может быть также получен при
взаимодействии сульфида цинка с хлором [233], хлористым водо-
родом [234], фосгеном [235]
ZnS-j-Clg — 1 '> ZnCl2-|-S
ZnS+2HCl—> ZnCl2-|-H2S
ZnS+COCl2—> ZnCl2 + COS
574
я
В промышленности хлористый цинк получают в виде концентри-
рованных растворов. Реакции растворения цинка и окиси цинка
в соляной кислоте изотермичны и протекают энергично.
При сушке растворов полностью удалить влагу не удается из-за
сильной гигроскопичности хлористого цинка. Кроме того, в резуль--
тате гидролиза сушка сопровождается образованием основных
солей [236]. Ниже приведено содержание основных солей и влаги
в ZnCl2 в зависимости от температуры обезвоживания:
Темпера- Нераствори- Содержание
тура, °C мый остаток, % воды, %
200 0,65 12,23
220 0,70 9,57
250 1,59 4,03
Темпера- Нераствори- Содержание
тура, °C мый остаток %, воды, %
280 1,78 2,92
310 1,78 2,34
340 1,94 ' ’ 1,18
Производство хлористого цинка
Хлористый цинк производят в виде плава,
содержащего не ме-
нее 97% основного вещества, и в виде растворов концентрации от 40
до 49% ZnCl2. В последние годы отечественная промышленность
начала выпускать чешуйчатый и гранулированный хлористый цинк.
Объем производства хлористого цинка в США составляет 23—
24 тыс. т, в Японии около 14 тыс. т [237].
По ГОСТ 7345—68, к хлористому цинку предъявляются следу-
ющие требования:
Марка В
Марка А
Марка В
I сорт
II сорт
Внешний вид .........
Содержание, %
хлористого цинка, не менее .
железа, не более............
сульфатов, не более . . . .
остатка, нерастворимого в соля-
ной кислоте, не более . .
свинца, не более . . . . . .
меди, не более . . . . . . .
мышьяка, не более *........
Концентрация водородных ионов
(pH), не менее
Плав-или
пластинки
серого
цвета
97
0,3
0,05
Прозрачный бес-
цветный или
светло-желтый
раствор
Мутный
раствор бу-
рого цвета
0,1
49
0,008
Отсут-
ствует
0,01
0,002
0,002
0,001
3,7
1йит
цинка I
сорта используют
Сырье. Для получения хлористого
металлический цинк марок ЦО И Ц1 (ГОСТ 36-40—65), для низших
сортов — цинковую изгарь, золку, гартцинк (ГОСТ 1639—67).
Технологическая схема получения хлористого цинка. Существуют
два способа получения растворов хлористого цинка: действием
575
хлористого водорода на цинк в колоннах, орошаемых водой и обработ-
кой цинковых отходов технической или абгазной соляной кислотой.
По первому способу хлористый водород из печей синтеза посту-
пает через нижний боковой штуцер в реакционную колонну, запол-
ненную дробленым цинком. Догрузка цинка производится перио-
дически через верхний люк колонны/ Уровень слоя цинка поддер-
живается примерно постоянным (недогруз по высоте не должен пре-
вышать 0,5—0,6 м).
Сверху в колонну подается вода. Хлористый водород и вода дви-
жутся противотоком, образующаяся соляная кислота вступает
в реакцию с цинком. Раствор хлористого цинка самотеком через
гидрозатвор поступает в приемный бак, откуда насосом подается
в колонну нейтрализации, загруженную кусковым цинкомЛ По
достижении pH менее 3,7 раствор хлористого цинка поступает в сбор-
ник, а затем перекачивается в отстойник. Отстой длится 12—15 ч,'
после чего нейтрализованный и осветленный раствор перекачивается
в баки готового продукта.
Отходящие из реакционной колонны газы, содержащие водород,
пары воды, хлористый водород и инертные газы, отмываются водой
от НС1 и сбрасываются в атмосферу. Кислая вода с содержанием
не более 1 г/л НС1 нейтрализуется или сливается в канализацию.
По второму способу процесс проводят в реакторах с мешалкой.
Для предотвращения бурного течения процесса, что может привести
к интенсивному выделению водорода и тепла, металлический цинк
или золку загружают в смесь соляной кислоты и хлористого цинка
плотностью не менее 1,3 г/см3.
При достижении плотности раствора 1,5 г/см3 и pH среды не
менее 3,7 прекращают перемешивание, подачу цинка и азота, сли-
вают часть готового продукта в приемный бак, а в реактор добавляют
свежую порцию соляной кислоты.
Полученные растворы хлористого цинка направляют непосред-
ственно потребителям или упаривают. Упарку осуществляют в чу-
гунных котлах, обогреваемых топочными газами или водородным
пламенем. Температура раствора в процессе упарки постепенно
повышается до 360 °C. Пары воды из аппарата отводятся вакуум-
насосом в конденсаторы. Полученный расплав хлористого цинка
разливается в герметично закрывающиеся барабаны.
Разработан способ получения гранулированного хлористого цинка
из 45—50%-ных растворов в аппаратах кипящего слоя. В качестве
теплоносителя используют топочные газы или подогретый азот.
Для предотвращения сплавления хлористогд цинка температура
теплоносителя под решеткой не должна превышать 250—260 °C.
576
?
ЛИТЕРАТУРА
1. Fichte R., Die Therm odynamischen Eigenschaften der Metallchloride,
VEB Verlag Technik, Berlin, 1953.
2. Термические константы неорганических величин. М., изд-во АН СССР, 1949.
3. Нестеренко В. Б., Зиновьев А. В., Ба ж и н М. А., Изв.
АН БССР, Сер. физ.-техн, наук, № 2, 32 (1967).
4. Томас Ч. Безводный хлористый алюминий в органической химии.
М., Издатинлит, 1949.
5. К 1 е m m W., Voss Е., Geierberger К., Z. anorg. Chem.,
256, 15 (1948).
6. К у л ь с к и й Л. А. Теоретические основы и технология кондициони-
рования воды. Киев, «Наукова Думка», 1971.
7. Перевозкин Ю. Л., Канд, дисс., М., ВЗПИ, 1968.
8. Перевозкин Ю. Л., Фурман А. А., Коган В. М., Вакс-
ман П. А., авт. свид. СССР 240952 (1968); Бюлл. изобр., № 13 (1969).
9. LemarchandsM., LemarchandsE., J а с о b М., Bull. Soc.
chim. France, № 53—54, 1139 (1933).
10. Koch W., Metallwirtschaft, 10, 69 (1931).
11. Evans M., Mem. Proc. Manchester. Lit. Phil. Soc., 79, 13 (1935); C. A.,
30, 7981 (1935).
12. Грошев Г. JI., Канд, дисс., ГГУ им. Н. И. Лобачевского, 1967.
13. Рабовский Б. Г., Генин Л. Ш., Фурман А. А., Спек-
тор И. Э., Коган В. М., авт. свид. СССР 169093 (1963); Бюлл.
изобр., № 6 (1965).
14. Коган В. М., Р а б о в с к и й Б. Г., Ф у р м а н А. А., Хим. пром.,
№ 5, 365 (1967).
15. Грошев Г. Л., Р а б о в с к и й Б. Г., Щ е г о л ь Ш. С., авт. свид.
СССР 146301 (1960); Бюлл. изобр., № 8 (1962).
16. Грошев Г. Л., Хим. пром., № 3, 187 (1970).
17. Пат. США 3222127 (1965); Off Gaz., 821, № 1, 264 (1965).
18. Пат. США 3446579 (1969); Off. Gaz., 862, № 4, 1191 (1969).
19. К a n g г о W., Jahn R., Z. anorg. allg. Chem., 210, № 6, 325 (1933).
20. W a s m u h t R., Angew. Chem., 43, 98 (1930).
21. Спицын В. И., Гвоздева О. М. Хлорирование окислов и природ-
ных соединений. М., изд. Института прикладной минералогии, 1931.
22. Будников П. П., Z. anorg. allg. Chem., 37, 100 (1924).
23. Воронин Н. Н., Га линкер И. С., ЖХП, 7, № 2—3, 143 (1930).
24. Treadvel W.,Terebesi D., Helv. chim. acta, 15, 1053,1353 (1932).
25. Подойников К. В., ЧерныйА. Г., ЖПХ, 38, 2625 (1965).
26. Hille J., Durrwachter W., Angew. Chem., 72, № 22, 850 (I960).
27. А д а д у p о в И. E., ЖПХ, 5, № 21—22, 1989 (1928).
28. KaczorowskiA., Przem. Chem., 20, № 8—9, 221 (1936).
29. P a б о в с к и й Б. ЖПХ, 33, № 3, 540 (1960).
30. Келлер Э. К., Л е о н о в А. И., Усп. хим., 22, 334 (1953).
31. Антоневич Н. К. Труды ВНИИкерамики, вып. 45, 3 (1934).
32. Щеголь Ш. С., Кулакова О. Я., Рабовский Б. Г., Труды
по химии и химической технологии (Горький), вып. 3, 533 (4958).
33. Рабовский Б. Г., Щ е г о л ь Ш. С., Труды по химии и химиче-
. ской технологии (Горький), вып. 1, 81 (1958).
34. Рабовский Б. Г., Щеголь Ш. С. и др., авт. свид. СССР 118493
(1956); Бюлл. изобр., № 6 (1959).
35. М с A f е е A., Ind. Eng. Chem., 21, 670 (1929).
36. Wurst er С., Angew. Chem., 43, 877 (1930).
37. Казарновский И. А., авт. свид. СССР 23970 (1931); Вести, ком.
по изобр., № 11 (1931).
38. Перевоз к и н Ю. Л., Фурман А. А., Коган В. М., Вакс-
м а н П. А., авт. свид. СССР 260624 (1968); Бюлл. изобр., № 4 (1970),
39. Самойлов В. Ф., Мельников И. И., Каолин. Вып. И, М.,
Госгеологиздат, 1951.
37 Заказ 843 577
40. Одинцов М. М. Сырьевые ресурсы тонкокерамической промышлен-
ности СССР и пути их использования. Сборник. М., изд-во, АН СССР,
1948. См. с. 224.
41.
42.
43.
44.
45.
46.
Л7.
48.
49.
50.
51.
52.
*
53.
54.
55.
56.
Фурман А. А., Рабовский Б. Г., Основы химии и технологии
безводных хлоридов. М., «Химия», 1970.
Федотьев П. IL, Чижик А. А., Труды ГИПХ, вып. 20, 76
(1934).
Оренштейн Э. И., Хим. пром., № 8, 592 (1967).
Пат. США 2502327 (1950).
Пат. США 1647446 (1927).
Пат. США 2387228 (1945).
Данов С. М.,. Д е в я т ы х Г. Г., А н и с и м о в Г. А., Труды по
химии и хим. технологии (Горький), вып. 4, 727 (1961).
Данов С. М., А н и с и м о в Г. А., Труды по химии и химической
технологии (Горький), вып. 2 (8), 215 (1963).
Девятых Г. Г., Данов С.• М., Труды по химии и химической
технологии (Горький), вып. 1, 1 (1962).
Наумова Т.Н., Б ы к о в а И. Г. ЖПХ, 43, № 1, 164 (1970). .
Стороженко В. Н., ЖПХ, 42, № 4, 780 (1969).
Фомин В. К., ЗвездинА. Г., КетовА. Н., авт. свид. СССР
316654 (1970); Бюлл. изобр., № 30 (1971).
Seegm idler D., Inorg. Nucl. Chem. Letters, 6, № 12, 885 (1970).
Нисельсон Л. А., Пугачевич П. П., Соколова Т. Д.,
Бедердинов Р. А., ЖНХ, 10, вып. 6, 1297 (1965).
КубашевскийО., ЭвансЭ. Термохимия в металлургии. М.,
Издатинлит, 1954.
Андрианов К. А.
издат, 1955,.
Кремнийорганические соединения. М., Госхим-
5/. БажантВ., ХваловскиВ., РатоускиИ. Силиконы.
с чешек.
М., Госхш
здат,
1960.
58.
59.
60.
Wagner Е., В г iinner Н., Angew? Chem., 72, 744 (1960).
Николина В. Я., Хим. пром. Укр., № 4, 3 (1966).
Вейцер Ю. И., Лучинский Г. П. Маскирующие . дымы.
Госхимиздат, 1947.
Пер.
М.,
61. М а г t i n G., J. Chem. Soc., 105, 2836 (1914).
62. Андрианов К. А., ДАН СССР, 28, 66 (1940).
63. Англ. пат. 712279 (1954); пат. США 3376109 (1965); РЖХим, 1969,
20Л115п.
реф.
64.
65.
66.
67.
68.
69.
70.
Koster A., Angew. Chem., 69, 563 (1957).
Пат. ФРГ 1045985 (1959); РЖХим, № 13, 53029 (1960).
Фельдштейн Н. С., Горбунов А. И. и др., ЖФХ, 43, № 3,
71.
72.
73.
74.
75.
76.
77.
78.
747 (1969).
Рабовский Б. Г., Фурман А. А., Титова М. В., ЖПХ,
39, № 10, 2203 (1966).
Ф у р м а н А. А., Т и т о в а М. В., авт. свид. СССР 136329 (1960);
'Бюлл. изобр., № 5, (1961).
Фурман А. А., Рабовский Б. Г., Торопов М. С., Ти-
то в а М. В., Горелова В. А., Хим. пром., № 5, 359 (1968).
Фурман А. А., Рабовский Б. Г., Торопов М. С., Ти-
това М. В., Горелов а В. А., Карталов Б. В., авт. свид. СССР
181633 (1965); Бюлл. изобр., № 10 (1966).
Англ. пат. 815276 (1959).
Пат. ФРГ 1048892 (1959); С. А., № 11, 108241 (1961). (
Пат. США 3173758 (1961); РЖХим, 1966, реф. 9Л93п.
Пат. США 3197283 (1965); Off. Gaz., 816, № 4, 1473 (1965).
Schwarz R., Z. anorg. allg. Chem., 76, 422 (1912).
GriinerE., ElodJ., Z. anorg. allg. Chem., 195,269 (1931).
Будников П. П., КречЭ. И,, ЖПХ, 9, № 1, 1225 (1936).
Snacu Р.. Voichesen Р., б vahesion A., Studii si certe-
tari de Chimie, 3, № 3-4, 195 (1955).
578
80.
82.
83.
84.
Фурман Д. А., М и ч к о в а 3. И. авт. свид. СССР 211524 (1956);
Бюлл. изобр., № 8 (1968).
Биллитер Ж. Промышленный электролиз водных растворов. Пер.
с нем. М., Госхимиздат, 1959, См. с. 345.
Петров Д. А., Жукова Л. К. Вопросы металлургии и физики
полупроводников. Сборник. М., изд-во АН СССР, 1957, См. с. 18.
Litton F., Anderson Н., Electrochem. Soc., 101, 287 (1954).
Akerman К., Brafman M. etal., Rep. Inst, badan jadrow, PAN,
№ 294, 16 (1961).
мета л лур гия», № 6
Изв. вузов, Сер. «Цветная
86.
87.
89.
124 (1960).
Нисельсон Л. А., Изв. вузов, Сер. «Цветная
, Черняев В. Н., Мартынов Ю. М., Изв.
вузов, Сер. «Цветная металлургия», № 4, 8 (1961).
“ ~ , ЖНХ, 5, 1139 (1960).
., ЖНХ, 7, № 2, 360 (1962).
Стол я р о в Ю. И./ЖФХ,
металлургия», № 2, 135 (1960).
90.
36, № 7, 1521
91.
133 (1963).
Пат. США 2844441 (1958).
(1962).
Л. А., Изв. вузов, Сер. «Цветная металлургия», № 2
93.
94.
95.
96.
35, № 10, 2161 (1962).
Пат. ФРГ 942000 (1956).
Пат. США 3403003 (1968); Off. Gaz., 854, № 4 (1968).
TheuererH., J. Electrochem. Soc., 107, № 1,
Мартынов Ю. M.,
29 (1960).
ЖФХ, 3
№
97.
98.
1038 (1964).
Мартынов Ю. М.,
26 (1965).
ЖФХ
39,
№
99.
872 (1966).
100.
1354 (1966).
Мартынов Ю. М.
Мартынов Ю. М.
ЖФХ,
ЖФХ,
40
40
№
№
6
101.
102.
103.
104.
105.
106.
107.
Хим.
Пат.
Пат.
Пат.
Пат.
Пат.
Пат.
пром., № 5, 41 (1962).
США 2820698 (1958).
ФРГ 1028543 (1959).
ФРГ 1046582 (1959); РЖХим, № 23, 92918 (1960).
ФРГ 1072227 (1960).
ФРГ 922466 (1955).
США 2931709 (1960).
и д у с И. И., Нисельсон Л. А. Тетрахлор- и трихлорсилан.
М., «Химия», 1970.
ДелароваН. И. и др., Изв. АН СССР, Сер. «Металлургия и топливо»,
№ 4, 33 (1960).
Деларова Н. И. и др. Труды ВАМИ, 46, 116 (1960); 53, 111
(1964).
KrieveW., Mason D., J. Phys. Chem., 60, № 3, 374 (1956).
Войтович Б. А., Барабанова А. С. Физико-химические основы
разделения продуктов хлорирования титансодержащих материалов. Киев,
«Наукова думка», 1969.
112. Slawisch A., Chem. -Ztg., 92, № 9, 311 (1968).
113. N a 11 a G., Corradini P. et. al., Rend. Classe sci. fis., mat. e. nat.,
24, 121 (1958).
108.
109.
110.
111.
115. Klemm W.
116. N a t ta G.,
399 (1961).
Z. anorg. allg. Chem., 249, 198 (1942).
Z. anorg.. allg. Chem., 253, 218 (1947).
AllegraG., J. Polymer Sci., 51
579
117.
118.
119.
120.
121.
122.
123.
124.
125.
126.
127.
128.
129.
130.
131.
132.
133.
134.
135.
136.
137.
138.
139.
140.
141.
142.
143.
144.
145.
146.
147.
148.
149.
150.
151.
152.
153.
154.
155.
156.
157.
580
N a t't a G., PasquonJ. et al., J. Polymer Sci., 51, 387 (1961); Хим.
и технол. полимеров, № 4, 3 (1962).
Sher fey J., J. Res. Nat. Bur. Stand., 46, 299 (1951).
Англ. пат. 881955 (1959).
Итал. пат. 577808 (1958).
Пат. ФРГ 1142159 (1960); РЖХим, 1965, реф. 24Л115п.
Исино Тоси о , Танура X и д з о, Н а к а га в а О сам у,
J. Chem. Soc. Japan, Ind. Chem. Sect., 64, № 8, 1344 (1961).
Ф у на к и, У тим у pa, J. Chem. Soc. Japan, Ind. Chem. Sect., № 8,
538 (1954).
5
85 (1957).
Бюлл. изобр.,
Н e l 1 е г W.,
J Г» О. о. fU, О Ъ X • J.J. - , J. Ъ. Л AU. J V р ЧЛ* л-лл
Труды института металлургии им. Байкова, вып. 1,
авт. свид. СССР 142639 (1960);
№ 22 (1960).
V., Лаврова В. Б., Хим. пром., № 5, 42 (1966).
Р о 1 а п у М., Trans. Faraday Soc., 32, 633 (1936).
Резниченко В. А. Титан и его сплавы.
М., изд-во АН СССР, 1963, См. с. 172.
Яблоков Ю. С.,
Титан и его
Вып. 9.
сплавы.
Вып. 8. М., изд-во АН СССР, 1962. См. с. 135.
Ruff О., Neuman nF., Z. anorg. allg. Chem., 128, 81 (1923).
Пат. США 3001951 (1961).
Пат. США 3010787 (1961).
Пат. ФРГ 1271095 (1968); Ausz. Pat., 14, № 26, 2438 (1968).
Пат. США 3451768 (1968); Off. Gaz., 863, № 4, 1174 (1969).
В i 1 1 у М., В г a s s е u г Р., С. R., 200, 1765 (1935).
Этлис В. С., авт. свид. СССР 115998 (1958); Бюлл. изобр., № И,
26 (1958).
Ehrlich Р., Hein Н., KuhniH., Z. anorg. allg. Chem., 292,
139 (1957).
Tadenuma H., Ikeda H., Fujita K., J. Chem. Japan, Ind.
Chem. Sect., 59, № 3, 356 (1956).
Гопиенко В. Г., ЖПХ, 33, № 11, 2600 (1960).
К г о 1 1 W., J> Trans. Electrochem. Spc., 78, 35 (1940).
Хазин Л. X. Двуокись титана. M?, «Химия», 1970.
Cullinane N., L е у s h о n D., J. Chem. Soc., № 8, 2942 (1954).
Корнеев H. H. и др. Комплексные металлоорганические катализа-
торы. М., «Химия», 1969.
Резниченко В. А., Солом а х а В. П. Титан и его сплавы.
Вып. 4. М., изд-во АН СССР, 1960. См. с. 39.
Зеликман А. Н., Сегарчану Т., ЖОХ, 26, № 3, 625 (1956).
Памфилов А. В., ХудяковА. С., ШтандельЕ. Г., ЖОХ,
5, № 5, 605 (1935).
i
।
Штандель Е. Г., ЖПХ, 9, 1770
ЖОХ, 7, № 8,
ЖОХ, 7, № 22,
• >
II
>
Rowel" "о pd e^W.,“j?Metals77/№ И, 1189 (1955).
Морозов И. С., Стефанюк С. Л., ЖНХ, 3, № 10
А.. Соломаха В. П. Титан и
1781 (1936).
1264 (1937).
2760 (1937).
2366 (1958).
его сплавы.
• 1
Вып. 5. М., изд-во АН СССР, 1961, См. с. 115.
С ер як о в Г. В., Вакс С. А., Желтова В. В., Страш унЕ. П.
ЖНХ, 12, № 1,8 (1967).
СеряковГ. В., В а к с С. А., ЖелтоваВ.В., СтрашунЕ. П.
Хлорная металлургия (Гиредмет). Сборник XXIV, 1969. См. с. 30.
С ер як о в Г. В., Вакс С. А., Страшун Н. Е., Туляков Н. В.
ЖПХ, 43, № 8, 1653 (1970).
Резниченко В. А., Соломаха В. П. Титан и его сплавы.
Вып. 5. М., изд-во АН СССР, 1961. См. с. 102.
Васютинский Н. А., БережкоА. В., Шалло О. Я.,
Г
Сидоренко А. П. Металлургия и химия титана. Т. IV. М., «Метал-
лургия», 1970. См. с. 73.
158. Оксюзов В. А., Зотикова А. Н. Титан и его сплавы. Вып. 8.
М., изд-во АН СССР, 1962. См. с. 98.
159. Байбеков М. К., Г а л ь п е р и н Е. С. и др., авт. свид. СССР
216651 (1964); Бюлл./изобр., № 15 (1968).
160. Г а л и ц к и й Н. В. Титан и его сплавы. Вып. 8. М., изд-во АН СССР,
1962. См. с. 101.
161. Хлопков Л. П. и др. Металлургия титана. М., «Металлургия»,
1970. См. с. 30.
162. Ким Мен Рин, Мелентьев Б. Н. Титан и его сплавы. Вып. 5,
изд-во АН СССР, 1961. См. с. 120. !
163. Безукладников А. Б., Вильнянский Я. Е. Титан и его
сплавы. Вып. 5. М., изд-во АН СССР, 1961. См. с. 135.
164. Безукладников А. Б., ЖПХ, 40, № 1, 31 (1967).
165. Безукладников А. Б., ЖПХ, 40, № 2, 291 (1967).
166. Зезянов С. П., Ильичев В. А., ЖПХ, 39, № 10, 2174 (1966).
167. Сулейменов Э. Н., Мачкасов Е.И., Пономарев В. Д.
Труды института металлургии и обогащения АН Каз.ССР, 9, 32 (1964).
168. Мачкасов Е. И. и др., там же, 13, 43 (1965).
169. Мачкасов Е. И. и др., там же, 18, 58 (1966).
170. К е 11 е г i d g е L, Brit. Chem. Eng., 10, №7, 452 (1965).
171. M e e p с о н Г. А. и др. , Цветная металлургия, № 5, 108 (1961).
172. Крамник В. Ю. Металлургия титана. М., «Металлургия», 1968.
См. с. 59.
173. Сергеев В. В., Галицкий Н. В., Киселев В. П. Метал-
лургия титана. М., «Металлургия», 1964.
174. С е р я к о в Г. В*., X о м я к о в П. П. Металлургия титана. М., «Ме-
таллургия», 1968. См. с. 129.
175. Основы металлургии. Т. 3. М., Металлургиздат, 1963. См. с. 271.
176. Коршунов Б. Г., СтефанюкС. Л. Введение в хлорную ме-
таллургию редких элементов. М., «Металлургия», 1970.
177. Пат. США 2600881 (1952).
178. Англ. пат. 712295 (1954).
179. Англ. пат. 777539 (1957). ,
180. Пат. США 2758009 (1956).
181. Kiyoshi A., J. Mining Inst. Japan, 71, 687 (1956).
182. В* а о с h С., Kaczmarek, U.S. Bur. Min. Rep. Invest., №5141,
76 (1955).
183. Пат. США 2819147 (1958).
184. Пат. США 3108854 (1963).
185. МорозовА. И., ЖПХ, 42, № 4, 757 (1969).
186.
Морозов И. С. Применение хлора в металлургии редких и цветных
металлов. М., «Наука», 1966.
187. Англ. пат. 656098 (1951).
188. Пат. США 2754256 (1956).
189. Пат. США 2879131 (1959).
190. Пат. США 2920016 (1960).
191. Пат. ЧССР 97317 (1960).
192.
193.
194.
195.
196.
США 2927843 (1960).
Пат.
Мартынов Ю. М.,
пром., № 2, 132 (1969).
Хим.
Хим., пром., № 10, 759 (1969).
Ван Везер. Фосфор и его соединения. М.
ВерятинУ. Д. и др. Термодинамические свойства неорганических
Издатинлит, 1962.
веществ. М., Атомиздат, 1965.
197. Mellor’s Compr. Treat. Inorg. Theoret. Chem., 8, 998—1079 (1958).
198. Ma з ель В. А., Гольдберг M. Б., Труды ГИПХ, вып. 20, 41
(1934).
581
199.
авт. свид. СССР 146298
200.
201.
(1962); Бюлл. изобр., № 8 (1962).
Рождественский Б. A.t Труда ГИПХ, вып. 20, 47 (1934).
Liebig J., Wehner G., Chem. Techn., 12, № 5, 254 (1960).
Hartley F., J. Appl. Chem., 2, № 24, 1955 (1952).
П ечк os ский В. В., Сафронов А. Л., ЖПХ, 39, № 6, 1225
204.
(1966).
Тетерев ков А. И., ПечковскийВ. В., Борисова H. В.
Изв. АН БССР, Сер. хим. н., № 3, 54 (1969).
Т е т е р е в к о в А. И. и др., Изв. вузов, Сер. хим. и хим. технол., 13,
№ 5, 668 (1970); № 6, 827 (1970).
Chem. Eng. News, 47, № 36, 53А (1969).
~ Технология минеральных солей. Ч. II. М., «Химия»,
1970. См. с. 938—956'.
206.
207. П о зин М. Е.
208. К i г k Е., OthmerD., Encyclopedia of Chemical Technology, v. 10,
N. Y., 1953, p. 481.
209. Ullmann's Encyklopadie der technischen Chemie, Bd. 13, 1963, s. 560—565.
210. Perot G., Chim. et ind., 87, № 1, 120 (1962).
211. Черных В. Я., Таланов Н. Д., Смирнова И. Н., Труды
по химии и химической технологии (Горький), вып. 2 (8), 220 (1963).
212. Черных В. Я. и др. Промышленность, химических реактивов и особо-
чистых веществ. Вып. 3 (9) М., изд. НИИТЭхим, 1965. См. с. 24.
Chem. Soc. Japan, Ind. Chem. Sect.,
Soc. Japan, Ind. Chem.,
чистых веществ. Вып. 3 (9) M.,
213. Н aka нэ Масанори и др
73, № 4, 682, А39 (1970).
214. Наканэ Масанори и др.,
Sect., 73, № 5, 883, А51 (1970).
215. Де вятовская Л. И., Виль
Вып. 8. М., Госхимиздат, 1959. См. с. 23.
J. Chem.
Д. E., Труда УНИхим.
216. ПечковскийВ. В., Воробьев Н. И., ЖНХ, 9, № 1, 12 (1964).
217. Schafer Н., Z. anorg. allg. Chem., 259, 54, 75, 265 (1949); 260, 127,
279 (1949); 261, 142 (1950); 264, 249 (1951).
218. Gold staub M., Bull. Soc. Franc, min., 58 (1935).
219. Латина 3. И., Фурман А. А., ЖПХ, 43, № 4, 830 (1970).
220. Kangro W., Peterson E., Z. anorg. allg. Chem., 261, 157 (1950).
221. Kangro W., В erenst of f H., Z. anorg. allg. Chem., 263, 316 (1950).
222. Wilson I., Gregory N., J. Phys. Chem., 62, № 4, 433 (1958).
223. И,л ь и ч е в В. А., Вл адимир ова А. М. Титан и его сплавы.
Вып. 5. М., изд-во АН СССР, 1961. См. с. 233.
224. Ададуров И. Е., Хим. пром., 6, № 3—4, 203 (1929).
225. Gaby J., Diss. doct. Sci. techn., Ecole polytechn. feder; Zurich, 1965.
226. ИващенцевЯ. И., Ахмазова H. А.,Белобородо в а Л. Г»
Труда Томского гос. ун-та, 170, 146 (1964).
227. Chem. Market Abstr., 55, № 1, 128 (1963); 56, № 1, 76 (1964).
228. В es s on J., Grange P., C. r., 238, 2002 (1954).
229. Герасимов Я. И., Крестовников А. Н., Ш а х о в А. С.
Химическая термодинамика в цветной металлургии. Т. 1. М., Металлург-
издат, 1960.
230. Аксельруд Н. В., Спивакове кий В. Б., ЖНХ, №2, 269 (1958).
231. Hof f man J., Austral J. Chem., 21, № 6, 1439 (1968)..
232. ИващенцевЯ. И., Шматова Л. В., Труды Томского гос. ун-та,
, 157, 171 (1963).
233. Biswas A., Khundkar М., J. Indian Chem. Soc., Ind. and News
Ed.,’14, 29 (1951).
234. Б p ицк e E. В., КапустинскийА. T., Веселовский Г. K.
Z. anorg. Chem., 213, 65 (1933).
235. C h a u v e n e t, C. r., 152, 87 (1911).
236. Ефремовы. H., В о л ь ф Ф. Ф. Сборник работ по химической
технологии минеральных веществ. Свердловск, 1929.
237. Хим. пром, за рубежом., № 5 (1967); № 3 (1968).
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абгазы
при конденсации хлора 334
при сжижении хлора 323. 324,
337
состав 537
Абсорбер
адиабатический 496, 499
блочный изотермический 493
кожухотрубчатый 494
пленочный 495
совмещенный 500, 501
Абсорбционная очистка
хлора из абгазов 335, 337, 491,
492
от хлор органических соединений
490, 491
хлористого водорода 493 сл.,
499
четыреххлористого кремния 528,
541, 542
Автоматизация процесса
выпарки щелоков 261
сжижения хлора 360 сл.:
электролиза 247 сл.
Адиабатическая абсорбция НС1
495 сл.
Азобензол 118
а
Азот, растворимость в жидком хлоре
316
Алюминий, хлорирование в расплаве
517, 518
Амальгама(ы)
восстановительная способность
117, 118
очистка рассола от ядов 225, 227
перенапряжение водорода 37,
38
равновесный потенциал 37
разложение 14, 39 сл., 117, 118
— , интенсификация 39, 167 сл.
э. д. с. 40, 41
♦
Амальгама(ы)
яды см. Амальгамные яды
Амальгамная проба 206
Амальгамные яды
влияние на разряд Н2 107
выделение из рассолов 225, 227
состав 38
Аммиакаты 516
Аммиачные холодильные машины 349
Анодное окисление 429
Аноды
графитовые см. Графитовые ано-
ды
магнетитовые 57, 378
малоизнашивающиеся 73 сл.,
153 сл., 186, 187
металлические 16, 17, 22, 154 сл.,
166, 167
монолитные для электролиза
НС1 289
окиснокобальтовые 382
окиснорутениевые 34, 67, 79 сл.,
87, 382
окисносвинцовые 376, 381
отливки из магнетита 57
падение напряжения 55, 56
пассивирование 78
перенапряжение выделения хло-
ра 87
перфорация 66, 96
платиновые см. Платиновые ано-
ды
подвод тока 56
поляризационные кривые 79, 80
потенциалы 77, 78, 84, 85, 379
потери напряжения 97
пробой 74
в производстве хлората натрия
370, 371
проницаемые металлические 67
размер 65
*
583
Аноды
разрушение 35
на титановой основе 22
угольные блоки 57
устойчивость 76, 77, 79 сл.
Анолит
донасыщение 195, 196, 200
кислотность, регулирование 59сл.
концентрация соли, расчет
108 сл.
обесхлоривание 220, 221
температура, влияние напряже-
. ния 136
Аппарат(ы)
для активного хлора 394
— влажного хлора 238, 239
с кипящим слоем 553, 554, 576
для сжижения хлора 340 сл.
Фредер кинга 270
Асбест
волокно 53
диафрагменная бумага 51, 52
хризотиловый 51
Аэросил 532
ч
Баланс напряжения
электролизеров БГК-17 136, 137
электролитической ячейки в со-
ляной кислоте 285, 286
Бассейн-растворитель поваренной со-
ли 203, 204
Башни оросительные 565
Бертолетова соль см. Хлорат калия
Биполярные электролизеры 149, 292,
398 сл.
Вакуум-кристаллизатор 387, 389
Вода(ы)
очистка 209
растворимость в жидком хлоре
325
— хлора 339
сточные, очистка от ртути 274,
275
хлорная, обезвреживание 233
Водород
взрывоопасные концентрации с
хлором 321
выжигание примесей 322 ч
компримирование 241
очистка от примесей 392
— — ртути 240
“ — хлора 322
перенапряжение выделения 88,
89
побочный продукт 18
пределы воспламеняемости 32
Водород
растворимость в жидком хлоре
316
свойства 31, 32
сжигание с хлором 483
содержание в хлоре 230
Воздух, очистка от ртути 241
Вращающиеся печи 480
Выпарные аппараты
с естественной циркуляцией 255
с кожухотрубчатыми подогре-
вателями 254
для концентрирования каусти-
ческой соды 256
с подвесной греющей камерой
255
с принудительной циркуляцией
256, 257
Выпарные системы
аппаратура см. Выпарные ап-
параты
выбор разности температур 254
депрессия температур 253, 254
инкрустации, удаление 254
Выпрямительные агрегаты
полупроводниковые 243
ртутные 242
Вязкость
водорода 31
жидкого хлора 309
растворов хлорного железа 567
соляной кислоты 475, 476
хлора 25
хлористого водорода 467, 468
хлорной кислоты 421
четыреххлористого кремния 530
четыреххлористого титана 543
Газонаполнение анолита и католита
49, 50-
Гексахлорбутадиен, абсорбция хло-
ра 337
Гидразина перхлораты 459
Гидраты
перхлоратов 434
хлорной кислоты 421 сл.
Гипохлорит калия, применение 9»
366
Гипохлорит натрия 366
восстановление на катоде 372,
373
выход по току 383, 384
образование при. электролизе
106
окисление на аноде 371, 372
получение 382
разложение 373
584
Гипс (Сульфат кальция)
выделение при электролизе 225,
224
модификационный состав 219
растворимость в рассоле 217,
218, 223 сл.
Графит
в анодах см. Графитовые аноды
импрегнированный 494, 495
разрушение 59, 61
свойства 58, 59
удельный расход 60
шлам, содержание в щелоках
392
Графитовые аноды
биполярные 289
выход по току хлората натрия
374 сл.
двойной токоподвод, схема 71
изменение напряжения 391
износ 58 сл., 376, 377, 380
кислотность анолита, влияние
на износ 59 сл.
межэлектродное расстояние, ре-
гулирование 164, 165
основа, материал 74
охлаждение 380
перенапряжение выделения хлора
59, 86 сл.
перфорация 66, 96
плотность тока 379
подвод тока 67, 69 сл.
потенциал выделения хлора 88
пропитка 63, 64, 381
, разрушение хроматами 377
расход 379
расчет выхода по току, прибли-
женный метод 106
стойкость 62, 63
титановые токоподводы 67
устройства для опускания 72 сл.
форма 64, 65
химический износ, скорость 64
электролиз НС1 286, 287
Давление паров
безводной хлорной кислоты 423
воды над растворами NaOH 252
' — —-------и NaCl 111
-------MgCl2 509
— — соляной кислотой 498
начала конденсации хлора 317
перхлората нитрония 457
пятихлористого фосфора 560
над твердым хлором 309, 310
треххлористого фосфора 560
хлора 25, 26
хлористого водорода 465, 466,
471, 472, 497
хлорного железа 567
Давление пара
четыреххлористого кремния 531
— титана 543
Двуокись- титана, скорость хлори-
рования 545
Двуокись углерода
образование при электролизе 106
растворимость в хлоре 312, 323
Двуокись хлора, производство 368
Дезинфицирующие вещества 366 сл.
Деполяризация катода 382
Дефолианты 367, 368, 413 сл.
Диаграмма
Молье 26, 27
тепловая для диафрагменного
электролизера 115, 116
Диафрагменные электролизеры
баланс напряжения 98
БГК 125 сл., 129
Биллитер — Лейкам 43
биполярные 148 сл., 151, 155
с вертикальной диафрагмой 103
ВМ-50 151
выход по току 100 сл.
с газозащитными оболочками 43
с горизонтальной диафрагмой
102, 103
ДА 138 сл.
Даймонд 125, 145 сл.
ДАУ 148
ДС 155
. концентрация соли в анолите,
расчет 108 сл.
с малоизнашивающимися ано-
дами 153 сл.
материальный баланс 108 сл.
напряжение и температура, влия-
ние плотности тока 136
Ниссо 150, 151
объем католита 108, 109
общее напряжение 83, 84
падение напряжения 97
Песталлоци 43
показатели работы 128, 134,
139, 142, 147, 152, 155,
158 сл.
пропитка анодов 63, 64
с проточными диафрагмами 43 сл.
разделение продуктов 41 сл.
размещение 245, 246
секционное устройство 151
серии, организация работы 138,
242
тепловая диаграмма 115, 116
тепловой баланс 113 сл.
удельный расход электроэнер-
гии 133
585
Диафрагменные электролизеры
фильтр-прессного типа 149
Хантер-Отис-Блю 149
Хукер 125, 139 сл.
Элинкор 46 с металлическими I Защита от коррозии 228
анодами 155 | аппаратуры под давлением 242
Диафрагмы
асбестовая 43, 51, 52
из волокнистого материала 51
выход по току 103
горизонтальная 51
ионообменные 282
колокол 42, 43
материалы для изготовления 51,
ш
сопротивление 4
196, 200
металлизированная 50
насасывание, схема установки
52 сл.
насосная 150
— с добавками латекса 51
осажденная, характеристика 54
падение напряжения 97 сл.
пористость 43
приготовление 52, 125
протекаемость 44 сл.
—, влияние давления 47
проточная 43 сл.
распределение концентрац:
лочи по высоте 104
регенерация 46
электрическое
Донасыщение
анолита 195, ‘
рассолов 216,
щелоков 394
Едкий натр см. также Каустическая
сода
давление паров воды над раство-
рами 111, 252
качество 29 сл.
концентрация раствора, изме-
нение 48, 49
коэффициент диффузии 45
температура кипения растворов
251
теплота растворения 252
удельная электропроводность
растворов 91, 92
Едкое кали
качество 30
температура кипения растворов
теплота растворения 252
Железо, хлорирование .569
Жидкий хлор 316
испарение 351, 352
получение 314 сл.
свойства 309 сл.
Жидкий Хлор
хранение и транспортирование
352 сл.
греющие камеры 257
заземленные вставки 243
контактной части анодов 71
осушительной аппаратуры 234
плавильных котлов 269
платиновых покрытий 76, 77
электролизеров 146
Заместительное хлорирование угле-
водородов 487, 488
Износ анодов 378 сл.
Изотермическая абсорбция хлори-
стого водорода 493
Ионообменные смолы, очистка рас-
творов от ртути 275
Испарители хлора 351, 352
Кальцинированная сода
производство 18, 19, 281, 282
расход при очистке рассола 218
Кальция сульфат, растворимость в
NaCl 217, 218, 223
Карбид титана, хлорирование 553,
Карбонат кальция, растворимость
в рассоле 207
Катодная поляризация Электроли-
зера 402
Катодное восстановление хлоратов
374, 376
Катоды
V- и W-образные 52
кислородная деполяризация 382
конструкция блока 130
материалы 39, 382
охлаждаемые 400, 401
потенциал 89, 90
в производстве перхлората на-
трия 445
равновесные потенциалы 84, 85
разветвленный 129
ртутный, способы создания 156,
157
скелетный никелевый катали-
затор 41
в электролизерах 126, 140, 145,
146
Каустическая сода
глубокая очистка 249
обезвоживание (плавка) 267 сл.
очистка 14, 264 сл.
Каустическая сода
производство см. Каустическая
сода, производство
распределение между потреби-
телями 13, 14
состав 266
температура кипения 268
товарные формы 249
транспортирование 249
чешуированная 269
Каустическая сода, производство
вывод сульфатов 251, 261 сл.
выпарка щелоков, схемы 257 сл.
непрерывная плавка с даутермо-
вым обогревом 270
плавка в аппаратах Фредер-
кинга 270
схемы 194 сл.
удельный расход электроэнер-
гии 83
л '
Кислород
выход по току 375, 428
растворимость в жидком хлоре
316
Коалин
брикеты 523, 524
обогащенный 521, 522
Колонны экстрактивной ректифи-
кации 507
ч
Компрессоры
ВК-9 345, 346
с жидким поршнем (баденские)
342, 343
лабиринтные поршневые 342, 343
ротационные 343, 344
типа РЖК и НЭЖ 236
фирм <<Амаг-Гильперт», «Сюрт»
и «Эслингер» 342
хлорные 342, 344, 345 '
Компримирование хлора 235, 236
Конденсаторы
двухходовые с внутренними пе-
регородками 554
кожухотрубчатые 348, 349
оросительные 555
расчет 347
совмещенный для хлора 348
Конструкционные материалы
коррозионно-стойкие 239, 243
печи 486
в производстве активного хлора
394
— — хлористого водорода и со-
ляной кислоты 511, 512
— — хлорной кислоты 430, 431
для хлоратных электролизеров
397
Контейнеры для хлора 354
Коэффициент(ы)
абсорбции НС1 водой 472
диффузии хлоридов и гидрооки-
сей 45
извилистости пор диафрагмы 97
объемного расширения хлора 311
протекаемости пор 45, 54
сжимаемости хлора 311, 317,
318, 320
скорости абсорбции хлора СС14
336
теплопередачи в промышленных
конденсаторах 347
увеличения сопротивления диа-
фрагмы 44
— — электролита 96
Кремний, механизм взаимодействия
с хлором 533
/Тролля-процесс восстановления 544
Льняное масло 63
Мак А фи способ получения А1С13
520, 521
Малоизнашивающиеся аноды 73 сл.,
153 сл., 186, 187
IMS
Материальный баланс
разлагателя 113
электролизера 108 сл.
Металлические аноды 16, 17, 22,
154 сл., 166, 167
Механизм
взаимодействие кремния с хло-
ром 533
окисление гипохлорита 371, 372
— хлората натрия 436 сл.
хлорирования TiO 2 545
Механическая сульфатная печь 481,
482
Напряжение
баланс в БГК-17 136, 137
влияние износа анода 391
— плотности тока 144, 442
— на температуру 136
— температуры 440, 441
изменение за тур работы 137
пробоя защитного слоя 74
снижение деполяризацией 296
на электролизере БК Г 128, 134
----Де Нора 291"
----с МИА 153
— — Хукер 144
— ячейке, влияние плотности'
тока 290
Насасывание диафрагмы 52 сл.
587
Насадочные колонны для осушки
хлора 235
Натрий металлический,
118
получение
Натрия сульфат
растворимость в рассоле 223,
262, 263
— в щелочном растворе 251
содержание в рассоле 262
удаление из электролитических
щелоков 251, 261 сл.
Нитрозила перхлорат 459
Нитрония перхлорат 457 сл.
Нормальные потенциалы
выделения хлора и водорода
297
катодный на графитовых элект-
родах 298
Обесхлоривание рассола 220 сл., 392
Окись углерода, выделение 522
Оксихлорид ванадия 557 сл.
Осветлители рассола
Дорра 211 сл.
ОВР-ПШ 209, 212
ЦНИИ 209, 211
со шламовым фильтром 22
Осушка хлора 234 сл., 325
Отбеливатели 9, 366 сл.
Очистка
абсорбция см. Абсорбционная
очистка
воды 209
водорода
от примесей 392
. от ртути 240, 241
от хлора 322
воздуха от ртути 241
каустической соды
жидким аммиаком под да-
влением 265, 266
кристаллогидратный способ
267
электрохимический способ
267
рассола см. Очистка рассола
от ртути сточных вод 274, 275
хлора 236 сл., 323
хлористого водорода из отхо-
дящих газов 489 сл.
хлорной кислоты от хлора 428,
429
четыреххлористого кремния
540 сл.
Очистка рассола
от амальгамных ядов 225
аппаратура 209, 211 сл.
Очистка рассола
от гипса 223 сл.
глубокая от сульфатов 218
дехлорирование 220, 221
известково-содовая 207
ионообменными смолами 207, 219
карбонизация 210, 211
коагулянты и флокулянты 209
осветление 216, 217
подземная 200, 201
из природной соли, схема 222
содово-каустическая 206 сл.
от сульфатов 217 сл., 261 сл.
технологические схемы 200
фильтрование 213, 214
флотация 215
для электролиза с диафрагмой
206 сл.
— — —ртутным катодом 219 сл.
Пенные аппараты для осушки хло-
ра 235, 236
Перекись натрия 118
Перекись свинца 442
Перенапряжение на электродах
выделения водорода 88 сл.
— хлора 85 сл.
Л
Перхлорат аммония
качество 444
промышленное получение 448 сл.
растворимость 448, 449
свойства 433
Перхлорат калия
взаимодействие с H2SiF6 430
получение 452
производство обменным спосо-
бом 456, 457
растворимость в воде 455, 456
свойства 433
Перхлорат лития
получение 454
растворимость в воде 454, 455
свойства 433
Перхлорат натрия
выход по току 440 сл.
обменное разложение 450 сл.
переработка в перхлорат ам-
мония 444
получение 373, 435 сл.
растворимость в воде 436, 437
твердый, получение 445
Перхлораты
гидраты 434
неметаллов 457 сл.
получение 427, 434 сл.
применение 430, 432
свойства 433
стабильность 432
588
Печи
синтеза НС1 486, 487
хлорирования
двухконусная 438
горизонтальная 539, 540
кипящего слоя 547
в расплаве 551, 552
ретортные 549
туннельные 549
шахтные 546, 548 сл.
Пиролиз хлоруглеводородов 483
Плавильные горшки 268
Плавка (обезвоживание) кау стиче'
ской соды 267 сл.
Платина 377
расход на 1 т С12 78
— при электролизе хлората 433
— — — хлористого водорода
428
Платиновые аноды 34, 377
выход по току хлората натрия
375
износ 76 сл.
пассивирование 78
из платино-иридиевого спла-
ва'57, 58
потенциал 77, 78
на титановой основе 76 сл., 154
Плотность
водорода 31
графитовых пропитанных ано-
дов 64
жидкого хлора 309, 310
кристаллического А1С13 516
неметаллических перхлоратов
457 .
паров А1С13 515
плавленого хлористого цинка 572
поваренной соли 197
пятихлористого фосфора 560
растворов хлористого алюминия
516
— — цинка 573
— хлорной кислоты 423
соляной кислоты 474, 475
треххлористого фосфора 560
хлора 25
хлората натрия 368
хлористого водорода 464, 466,
467
хлорного железа 567
четыреххлористого кремния 530
— титана 543
Поваренная соль
каменная 199
качество 202'
коэффициент диффузии 45
Поваренная соль
обратная, очистка от амальгам-
ных ядов 18
перевозка 202
подземное выщелачивание 18, 22
раствор см. Рассол
растворение методом
ба 200
гидровру-
растворимость в воде 197, 198
202, 203
— в растворе NaOH 250
самосадочная 199
состав 199
удаление из щелоков 250, 251
физико-химические свойства 197-
Поверхностное' натяжение
на границе жидкий хлор — па-
ры 311
хлористого водорода 464
четырех хлористого кремния 530
Подвод тока в электролизерах 67 сл.
Показатели работы
компрессора В К-9 .345
— ХТК-2,5 344
электролизеров
БГК-13 128
БГК-17 и БГК-50 134
биполярного большой мощ-
ности 152
с графитовыми анодами 176
Даймонд 147
ДС 155
диафрагменных 156 сл.
Р-20 176, 187
Р-101 174, 187
с ртутным катодом 187
для соляной кислоты 290, 293
Хукер 142, 143
Полиакриламид 22, 209, 219
Политермы растворимости едких ще-
лочей 31
Предельно-допустимые концентра-
ции
водорода в воздухе 321
ртути 271, 272
хлора в воздухе 363
Пределы воспламеняемости хлора,
водорода и воздуха 32
Природное фосфатное сырье, пря-
мое хлорирование 562
Промышленные адсорбенты 542
Протекаемость диафрагмы 44 сл.
Пятихлористый фосфор
качество 563
образование 561
применение 561
производство 565, 566
свойства 560
589
Разделение электродных продуктов
41 сл., 56, 57
Разлагатели
вертикальные 39, 40, 168
горизонтальные 39, 40, 167 сл.
интенсификация разложения 39,
40
материальный баланс 113
насадка 39, 40, 168
с погружной насадкой 168, 169
скрубберный 168, 169
тепловой баланс 117
э. д. с. амальгамного элемента
40
Рассол
автоматизация приготовления
и очистки 228
выпарка 224 сл., 264
донасыщение 216, 220, 227, 228
качество 206 '
коррозионная активность 228
обедненный, состав 220
обесхлоривание 220 сл., 392
обратный, получение 250
осветление 208, 209
подземная очистка 200, 201
пороговая концентрация 376
приготовление 199 сл.
растворимость кальция 207
— сульфата натрия 263
— хлората натрия 369
состав подземных и искусствен-
ных 200
схема получения методом гидро-
вруба 200
— приготовления и очистки 222,
224, 225
сырье для электролиза 194 сл.
удаление гипса 223, 224
удельная электропроводность 90
электролиз см. Электролиз
Растворимость
взаимная С12 и СО2 312
водорода, азота и кислорода
в жидком хлоре 316
воды в жидком хлоре 325
гидрата хлора в воде 28
гидроокиси железа в щелочном
растворе 209
двуокиси углерода в жидком
хлоре 323
едкого кали в воде 31
едкого натра в воде 31
карбоната кальция в рассоле 207
карбоната магния в щелочном
растворе 207
перхлората аммония 448
перхлората калия 455, 456
перхлората лития 454, 455
Растворимость
перхлората натрия 436
перхлората нитрония 458
перхлоратов в воде 433
— в органических растворите-
лях 434
поваренной соли в воде 198
— — в щелочном растворе 250
пятихлористого фосфора 560
н системе NaCl — Н2О 197
-----NaCl — КС1 — Н2О 205
совместная см. Совместная рас-
творимость
сульфата кальция , в рассоле
217, 218, 223
сульфата натрия в рассоле 251,
262, 263
треххлористого фосфора 560
хлора
в воде 28, 339
в растворах NaOH 28
— четыреххлористом ти-
тане 338, 544
— четыреххлористом угле-
роде 335
хлората калия 408
хлората натрия 369
хлористого алюминия в TiCl4 529
хлористого водорода 469
— — в растворах MgCl2 509
хлористого калия 198
хлористого цинка 573
хлорного железа 567, 568
Растворители соли 203, 204
Расходные коэффициенты
на 1 т очищенного А1С13 528
— 1т хлора 300, 337
— 1т хлористого, водорода 482
— 1т хлорного железа 572
— 1т четыреххлористого крем-
ния 540
в электролизерах БГК 128, 129,
134
Реактор с кипящим слоем 489
Регенерация ртути
из водорода и газовых сбросов
271
— сточных вод 271, 272, 274,
275
шлама 272 сл.
Регулирование межэлектродного рас-
стояния 21, 72 сл., 164 сл.
Ректификация хлористого алюми-
ния 529
Ртути сульфид, выделение и пере-
работка 273, 274
Ртутяо-выпрямителЬные агрегаты-
242
590
Ртуть
окисление в каскаде колонн
402, 403
потери при электролизе 271
предельно-допустимые нормы 271
регенерация см. Регенерация
ртути
удаление из водорода 240, 241
Серии электролизеров 241 сл.
Серная кислота
осушка хлора 234 сл. h
— хлористого водорода 507
растворимость НС1 470
Сернистая ртуть, образование 221
Сжижение хлора
автоматизация 360 сл.
аппаратура 340 сл., 350, 351
влияние примесей 316, 317
— треххлористого азота 324
выбор рациональной схемы 332,
333
при высоком давлении 329
выход 319
двухступенчатое 326 сл.
затраты 333
комбинированный способ 329,
350
коэффициент 317, 318, 320
разделение хлора и абгазов 332
режим 321, 322
способы 327
температура 317
без холодильной установки 331
Сжижение хлористого водорода 510,
511
Системы
NaClO3—NaClO4—Н2О 443
ZnCl2—Н2О 573
t
Скорость
коррозии стали в хлоре 340
окисления титана в ПТА 74
отстаивания суспензий 208
разряда водорода 107
растворения металлического ни-
келя 300
фильтрации электролита 102
хлорирования алюминия 517, 519
Скруббер(ы)
для осушки хлора 234,^ 235
— очистки SiCl4 от твердых
хлоридов 538, 539
Совместная растворимость
хлора и двуокиси углерода 312
хлората и хлорида калия 409
хлората и хлорида натрия 393
NH4C1O4 и NaCl 449
КСЮ4 и NaCl 455
Совместная растворимость
LiC104 и NaCl 455
NaClOg и NaClO4 443
NaClO4 и NH4C1O4 449
NaC103 и NaCl 437
Соляная кислота
абгазная 12, ,489, 502
вязкость 475, 476
давление НС1 над растворами
497
качество 470,,473, 502
концентрация по высоте абсор-
бера 499
концентрированная из абгазов
501, 502
максимальная концентрация 470
окисление 305
переработка в хлор 12
плотность 474, 475
получение 492 сл.
— из абгазов 334, 335
—, установки 500
производство 283
равновесие жидкой и газовой
фаз 499
регенерация из травильных рас-
творов 489
состав азеотропной смеси 477
температура замерзания и кипе-
ния 477, 478
температура и концентрация по
высоте абсорбера 496
теплоемкость 476
теплопроводность 476, 477
теплота разбавления 479
транспортирование и хранение
473, 474
экстрактивная ректификация
507, 508
электролиз 19, 20, 282 сл.,
427 сл.
электропроводность 287
Стабильность
перхлоратов 432
хлорной кислоты 421, 425
Степень
диссоциации НС1 483
перфорации- анодов 81, 82, 96
Сточные воды, очистка от ртути
274, 275
Стриппинг-процесс получения
100%-ного НС1 502 сл.
Сульфатный способ получения хло-
ристого водорода 480 сл.
Танки для хранения хлора 353, 356
Температура
воспламенения металлов в хлоре
340
591
t
Температура
замерзания соляной кислоты 473
кипения
водорода 31
высококонцентрированной
!• каустической соды 268
растворов едкого кали 252
— — натра 251
— хлорной кислоты 423
треххлористого фосфора 560
хлора 25
хлористого водорода 464
хлористого цинка 573
хлорного железа 567
хлорокиси фосфора 566
четыреххлористого кремния
530
электролитических щелоков
251
конденсации хлора 317, 346
максимально допустимые для ра-
боты в среде НС1 511
. начала кристаллизации MgCl2
508
плавления
водорода 31
гидратов хлорной кислоты
422
неметаллических перхло-
ратов 457
перхлоратов 433
поваренной соли 197
хлора 25
хлорат-магниевого дефо-
лианта 415
хлористого водорода 464
хлористого цинка 572
хлорного железа 567
хлорной кислоты 422
четырех хлористого кремния
530
четыреххлористого тита-
на 543
разложения
неметаллических перхлора-
тов 457
перхлоратов 433
хлорирования кремния 533
Теплоемкость
жидкого хлора 311, 312
мольная поваренной соли 197
пятихлористого фосфора 560
соляной кислоты 476
треххлористого фосфора 560
удельная поваренной соли 197
хлористого водорода 468
хлорокиси фосфора 560
четыреххлористого титана 543
Теплопроводность
водорода 31
Теплопроводность
соляной кислоты 476, 477
хлора 25
Теплота
образования
неметаллических цер хло-
ратов 457
перхлоратов 433
пятихлористого фосфора 560
треххлористого фосфора 560
хлората натрия 369
хлористого цинка 573
хлорного железа 567
хлорокиси фосфора 560
четыреххлористого титана
543
парообразования хлора 310
плавления
хлората натрия 369
хлористого цинка 578
хлорной кислоты 423
хлорного железа 567
четыреххлористого титана
543
разбавления соляной кислоты
479
растворения
хлористого водорода 478
хлорной кислоты 423, 424
Техника безопасности при обслу-
живании
электролизеров 242, 243
Титан
получение 544
четыреххлористый см. Четырех-
хлористый титан
Титановые шлаки, хлорирование
545 сл., 551 сл.
Токоподводящие стержни 67 сл.
Транспор тир ов ание
жидкого хлора 352, 353
соляной кислоты 473, 474
хлора 234
Требования к качеству
абгазной соляной кислоты 502
бертолетовой соли 408
газообразного хлора 29
графитовых пропитанных анодов
64
едкого кали 30
едкого натра 29 сл.
жидкого хлора 312
коалина обогащенного 521
очищенного рассола 206
перхлората аммония 444 сл.
поваренной соли 202
пятихлористого фосфора 563
соляной кислоты 470, 473, 502
треххлористого фосфора 563
592
Требования к качеству
хлор ат-хлорид кальциевого де-
фолианта 413
хлората магния 414
хлората натрия 370
хлористого алюминия 521
хлористого калия 205
хлористого цинка 575
хлорного железа 570
хлорокиси фосфора 563
Треххлористый азот
в жидком хлоре 230, 324
образование 28, 229, 230
свойства 28, 29
Треххлористый титан, свойства 544
Треххлористый фосфор 560, 563 сл.
Трубопроводы, защита от повыше-
ния давления 358, 359
Тунговое масло 63
Турбокомпрессоры 236, 361
Удельная теплоемкость перхлоратов
433
Удельная электропроводность
растворов NaCl 90
--------и NaOH 91
соляной кпслоты 287, 288, 302
хлористого водорода 465
электролитических щелоков 91,
92
Удельное электрическое сопротивле-
ние
графитовых пропитанных ано-
дов 64
растворов хлорной кислоты 423,
424
электролитов 91, 92, 97
Ферросилиций, хлорирование 534
Фильтры
волокнистые 237
насыпные 214
пористые 392
рамные 214, 215
рукавные из стекловолокна 555
солевые 555
трубки из фторопласта 215
электростатические 237
Флотореагенты 205
Фриделя — Крафтса процесс хло-
рирования 488
Фтористый водород, удаление из
хлористого водорода 492
Химические способы получения хло-
ра 12, 19, 303 сл.
38 Заказ 843
Хлор
абсорбция из абгазного НС1
335, 491, 492
— четыреххлористым титаном
333
— — углеродом 335
активный, аппаратура 394
активный, восстановление 107,
392
взаимная растворимость с СО2
312
взаимодействие с кремнием, ме-
ханизм 533
выход по току 102, 103, 428
гидраты 26
гидролиз в щелочном растворе 35
давление начала конденсации 317
диаграмма Молье 26, 27
жидкий см. Жидкий хлор
компримирование 235, 340 сл.
конденсация 346 сл.
контроль содержания 362
концентрирование, схема 336,
339
окисление 427, 429
осушка 234 сл., 325
отдувка СО 2 323
очистка 229 сл., 236, 237, 322,
485
перенапряжение 86
получение химическими способа-
ми 12, 19, 303 cjf.
потенциал выделения 87, 88
предел ьно-допустимая кон цент-
рация 363
применение 10, 313, 314, 316
примеси 326
производство см. Хлор, про-
изводство
равновесная концентрация 304
распределение между потреби-
телями 13, 14
растворимость
в воде 339
воды 325
двуокиси углерода 323
в четыреххлористом титане
338, 544
в четыреххлористом угле-
роде 335
розлив 359, 360
свойства 25 сл.
сжигание с водородом 483
содержание водорода 107
состав после сушильных башен
236
— — электролизеров 229
степень заполнения емкостей 357
температура конденсации 317
токсичность 363
593
Хлор
удаление из рассолов
Хлор, производство
высококонцентрированный 238
комбинированный способ 227
окисление соляной кислоты МпО2
280
развитие 10 сл.
схемы 194 сл.
удельный расход электроэнер-
гии 82, 83 /
- электролиз рассолов 280 сл.
— расплавов хлоридов 281
— - соляной кислоты 282 сл.
Хлорат калия
масштабы производства 367, 368
получение 366, 367, 409 сл.
потребители 368
растворимость 408, 409
— совместная с КС1 409
свойства 407, 408
схемы производства 411, 412
Хлорат кальция (Хлорат-хлорид
кальциевый дефолиант) 368
413, 414
Хлорат магния (Хлорат-магниевый
дефолиант) 368, 414, 415
Хлорат натрия
выделение 388
выход по току 374 сл., 385
катедное восстановление 374,
376
качество 370
непрерывное производство
385 сл.
образование 106
объем производства 367, 368
периодическое производство 384,
385
применение 367
производство 370 сл.
разложение 435 сл.
растворимость 369
свойства 368, 369
совместная растворимость с NaCl
393
схемы производства 386 сл.
хранение 369, 370
энтальпия 421, 422
Хлоратные электролизеры
Ангела 399
аноды 395
Ауссигер ферейн 396
биполярные 398, 405
Биттерфельд 399
с графитовыми анодами 400 сл.
без диафрагмы 396
Карбена 398
каскады 404
Хлоратные электролизёры
конструкционные материалы 397
с наружной циркуляцией элект-
ролита 397
с неохлаждаемыми катодами 404
охлаждение 397, 400, 401
плотность тока 405
показатели работы 399, 401,
406, 407
с титановыми анодами 407
Хлораторы 551, 552
Хлориды, применение 515
Хлорирование
алюминия в расплаве 517, 518
железа 569
заместительное углеводородов
487, 488 ,
карбида титана 553, 554
окиси цинка 574
окислов железа 569, q 70
прямое природного фосфатного
сырья 562
сульфида цинка 574
титановых шлаков 545 с л.,
551 сл.
Хлористый алюминий
безводный 530
каталитическая диссоциация 305
качество 521
коагулянт 209
конденсация паров 524
окисление 305
очистка 524, 525, 529, 530
плав, состав 525
получение 517 сл.
применение 517 .
производство 520 сл., 526, 527
растворимость в TiCl4 529
свойства 515
Хлористый аммоний (нашатырь),
электролиз 19
Хлористый барий 217
Хлористый водород
абгазный 12, 283, 284, 485 сл.,
489, 505 сл.
абсорбция 490
адиабатическая абсорбция 495 сл.
вязкость 467, 468
давление паров 465, 466
жидкий, получение 510, 511
изотермическая абсорбция 493 сл.
коэффициент абсорбции 472
окисление кислородом 303 сл.
осушка 502, 503
очистка 489 сл.
парциальное давление над со-
ляной кислотой 497
переработка в хлор 12, 20* 284
плотность 466, 467
Хлористый водород
получение 12, 20
примеси 489
растворимость в воде 469
— — растворах MgCl2 509
— — серной кислоте 470
свойства 464, 465
синтетический 479, 483 сл.
степень диссоциации 483
100%-ный, получение 502 сл.,
509, 510
сульфатный метод получения
480 сл.
теплоемкость 468
теплота растворения 478
токсичность 464
Хлористый калий
качество 203 сл.
совместная растворимость с КС1О3
409
Хлористый кальций, расход при
очистке рассола 218
Хлористый натрий
давление водяного пара над рас-
творами 111
конверсия 385
пороговая концентрация рас-
твора 376
совместная растворимость с хло-
ратом 393
электрохимическое окисление
374
Хлористый цинк 572 сл.
Хлориты 118, 366
Хлорная известь 9, 313
Хлорная кислота
ангидрид 425, 426
безводная, получение 423, 431,
432
взаимодействие с N2O3 459
выход 428, 431
гидраты 421, 422, 458
диаграмма плавкости 423
очистка от хлора 428, 429
плавкость 422
плотность растворов 423
получение 427 сл.
применение 420, 421, 426
свойства 421 сл., 425
состав пара над растворами 424,
425
стабильность 421, 425
токсичность 425
Хлорноватистая кислота --
диссоциация 36
образование 35
превращение на аноде 372
Хлорное железо
безводное 570 сл.
Хлорное железо
гидролиз 567
коагулянт 209
применение 568
производство 570 сл.
свойства 567, 568
Хлорные электролизеры фильтр-
прессного типа 429
Хлорный ангидрид 425, 426
Хлорокись фосфора 560 сл.
Холодильники
для водорода 170
графитовые 234
керамические для хлора (цел-
ляриусы) 232
поверхностные титановые 22,
232, 234
смешения 22, 232 сл.
Холодильные установки 350, 351
Хранение хлора 353 сл.
Хранилище сферическое для хлора
355, 356
Хроматы,’”1 потери тока 376, 377♦
438, 439
г
Центрифуги 259
Цистерны железнодорожные для
хлора 355
Циркуляция электролита 396 сл.
Четыреххлористый кремний
гидролиз 531, 532 539
глубокая очистка 541, 542
извлечение из отходящих газов
528
очистка от твердых хлоридов
538, 539
применение 532
растворимость хлора 531
ректификация 541 .
свойства 530, 531
Четыреххлористый титан
абсорбция хлора 333
высокой чистоты 559
конденсация и выделение из
смеси 554 сл.
очистка 556 сл.
— А1С13 529
применение 544
примеси 557
производство 547 сл.
растворимость хлора 338, 544
Четыреххлористый углерод 335,
338
Экстрактивная ректификация со-
ляной кислоты 507 сл.
595
38е
Электрическое сопротивление диа-
фрагмы 45, 54
Электродные материалы 286, 287
Электроды см. также Аноды, Ка-
тоды
графитовые, потенциалы 298
монополярные, устройство 150
Электролиз
выход по энергии 108
растворов двухвалентной меди
299, 300
растворов поваренной соли см.
также Электролиз с диа-
фрагмой, Электролиз с
ртутным катодом
автоматизация 247 сл.
каскадная схема 385, 386,
389, 390
состав электролита, изме-
нение 385
растворов хлористого аммония
19
— — калия 409, 410
— — никеля 300
растворов хлоридов щелочных
металлов 33 сл.
расчет выхода по току 106
соляной кислоты 427 сл.
анодные процессы 285
косвенные методы 297 сл.
. прямые методы 19, 20,
282 сл.
в смеси с азотной 296
схема установки 294 сл.
электроды, выбор 286
Электролиз с диафрагмой
давление фильтрации 47, 48
концентрация щелочи 48, 49
протекаемость диафрагмы 44 сл.
способы интенсификации 20, 21
сырье 15
удельные капиталовложения 14,
15
Электролиз с ртутным катодом*
общее напряжение 83, 84
преимущества и недостатки ме-
тода 14 с л.
размещение 246, 247
расход электроэнергии 15
серии 242
способы интенсификации 20, 21
Электролизеры
биполярные 21, 447, 448
с диафрагмой см. Диафрагмен-
ные электролизеры
единичная мощность
защита от
коррозии
износ анодов 58 ел.,
кислотность анолита
21
146
391
59 сл.
Электролизеры
напряжение, влияние межэлект-
родного расстояния 39
для перхлората натрия 445, 446
подвод тока 67
показатели работы 447
в производстве хлората
с внутренним каскадом 386
напряжение 391, 392
срок службы анодов 391
разложение амальгамы 39
регулирование межэлектродного
расстояния 21, 72 сл.
с ртутным катодом см. Электро-
лизеры с ртутным катодом
сборка 138
секционные 21
для соляной кислоты 290 сл.,
302
для электролиза растворов двух-
валентной ртути 301 сл.
— — — хлорида меди 300
— — — никеля 300, 301
Электролизеры с ртутным катодом
аноды, размер 65
баланс напряжения 99
биполярные 185
с вертикальным катодом 170,
171
с горизонтальным катодом 157
с графитовыми анодами 176
Де Нора 69, 167, 179
дисковый типа Хонзберга 170,171
корпуса 162, 163
Кребе 176 сл., 187
Матисон 169
материальный баланс ИЗ
с МИА 186
показатели работы 174, 176,
183, 184
потеря напряжения 96
Р-20 174 сл.
Р-101 171 сл.
развитие конструкции 185, 188
расположение электродов 65
' регулирование межэлектродного
расстояния 164 сл.
Сольве 167, 178, 179, 187
СУ, СА и СДМ 184, 187
тепловой баланс 117
устройство 156, 157
фирмы «Асахи Гласс Ко.» 157,
184, 185, 187
— «Куреха» 181, 182, 187
— «О лин-Матисон» 180, 181, 187
—• «Уде» 166, 182 сл.
— «Хехст-Уде» 187
Электролит(ы)
влияние pH на выход по току
374
596
Электролит(ы)
газонаполнение 66, 92, 96
давление паров воды 112
движущая сила циркуляции 92,
93
коэффициент увеличения сопро-
тивления 96
напряжение 90 сл.
для получения хлората натрия
388
примерный состав по каскад-
ной схеме 389
pH, влияние потенциала ано-
да 77
расчет концентрации соли 108 сл.
скорость фильтрации 102, 103,
105
смесь соляной и серной кислот
287 4
состав 97
удельная электропроводность
90 сл.
Электролит(ы)
удельное электрическое сопро-
тивление 91, 92, 97
Электролитические щелока
двухстадийная выпарка 257, 258
обесхлоривание химическим спо-
собом 392
осветление 392
состав 250
температурная депрессия 253
удельная электропроводность 91,
92
упаривание 250, 253, 254
Электрохимическое производство
хлора и каустической соды
см. также Хлор, произ-
водство, Каустическая со-
да, производство
автоматизация контроля и упра-
вления 23
интенсификация 20
комбинированный метод 17, 18
ЯКИМЕНКО ЛЕОНИД-МАРКОВИЧ
ПРОИЗВОДСТВО ХЛОРА,
КАУСТИЧЕСКОЙ СОДЫ
И НЕОРГАНИЧЕСКИХ ХЛОРПРОДУКТОВ
Редактор И. В. Лебедева
Технический редактор В. М. Скитина
Художник Е. В. Бекетов
Корректоры Л. В. Гав рилина, Н. А. Ваничкова
Т-09049. Сдано в наб. 15/11 1974 г. Подп. в печ. 5/V 1974 г.
Формат бумаги 60х90‘/и- Бумага тип. № 2. Усл. печ. л. 37,5.
Уч.-изд. л. 40,68. Тираж 3500 экз. Зак. 843. Изд. № 412.
Цена 2 р. 24 к.
Издательство ,,Химия**. 107076, Москва, Стромынка, 23
Ленинградская типография № 6 „Союзполиграфпрома" при
Государственном комитете Совета Министров СССР по делам
издательств, полиграфии и книжной торговли. \
196006, Ленинград, Московский пр., 91.
готовится
Н ВЫПУСКУ
В 1975 Г.
ИЗДАТЕЛЬСТВО „ХИМИЯ"
ПРИКЛАДНАЯ ЭЛЕКТРОХИМИЯ
ИЗД. 2-е, ПЕРЕРАБОТАННОЕ И ДОПОЛНЕННОЕ
Под редакцией Н. Т. Кудрявцева
36 л., 10 000экз« ц. 1 р. 50 к» в пер.
Книга является вторым изданием учебника для
студентов химико-технологических специальностей
вузов (первое издание выпущено в 1949 г.).
В первой части книги рассматривается произ-
водство химических источников электроэнергии
(гальванических элементов, свинцовых и щелочных
аккумуляторов), во второй — технология получения
водорода, кислорода, хлора, щелочей, некоторых
кислот, солей и органических соединений. Третья
часть посвящена технологии электрометаллургиче-
- ских процессов, гальванотехнике и гальваностегии.
Книга может быть полезна также студентам,
специализирующимся в области металлургических
процессов, и инженерно-техническим работникам
электрохимических и электрометаллургических про-
изводств.
Предварителъные заказы на . книгу можно оформить
в магазинах, распространяющих научно-техническую лите-
ратуру. -