/




























































































































































































































































































































































































Text
А.А.АБРАМОВ
ФЛОТАЦИОННЫЕ
МЕТОДЫ
ОБОГАЩЕНИЯ
Left blank
А.А.АБРАМОВ
ФЛОТАЦИОННЫЕ
МЕТОДЫ
ОБОГАЩЕНИЯ
Допущено Министерством высшего и среднего
специального образования СССР
в качестве учебника для студентов вузов,
обучающихся по специальности
«Обогащение полезных ископаемых»
)=Щ~=к МОСКВА "НЕДРА" 1984
УДК [622.765:622.34]@758)
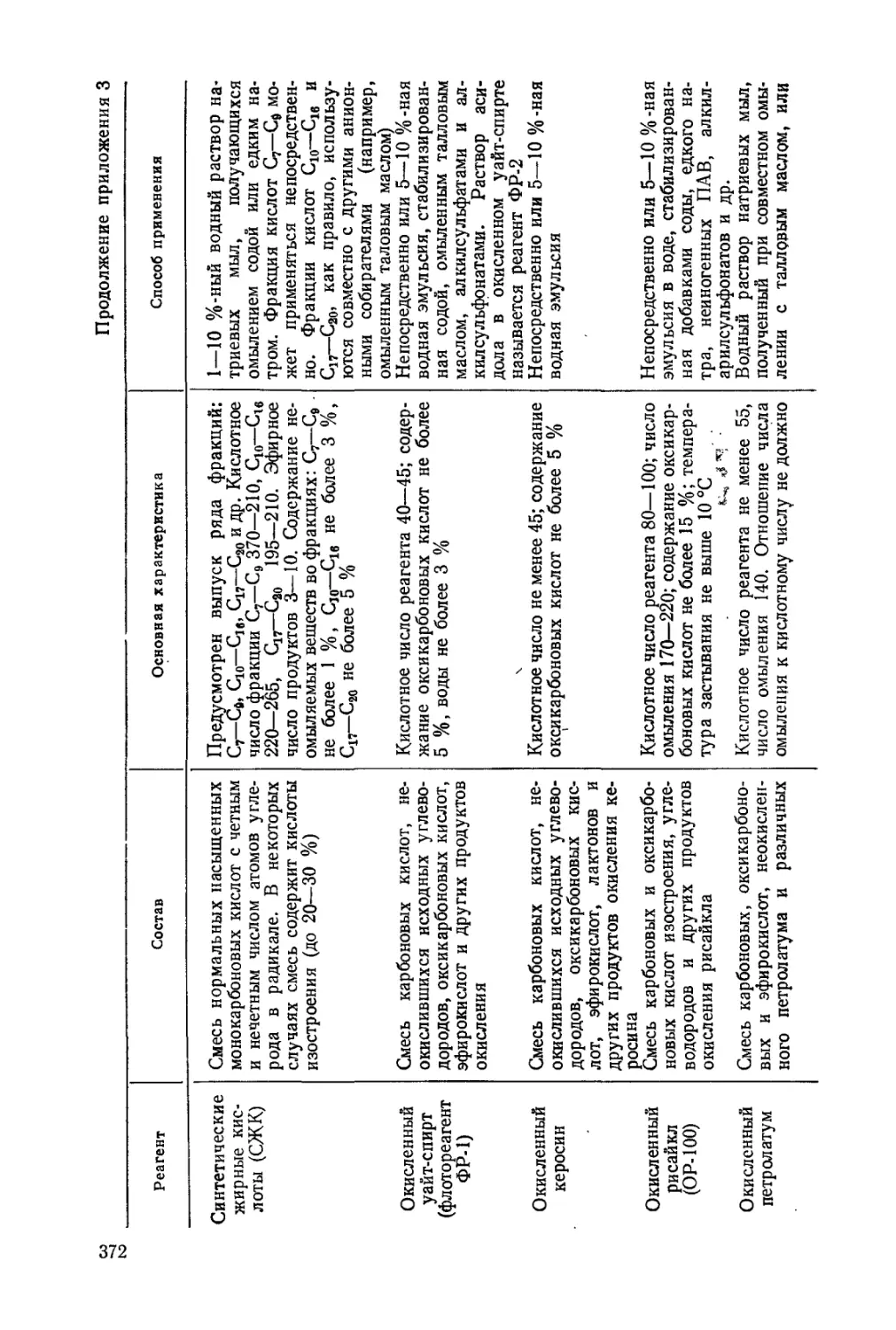
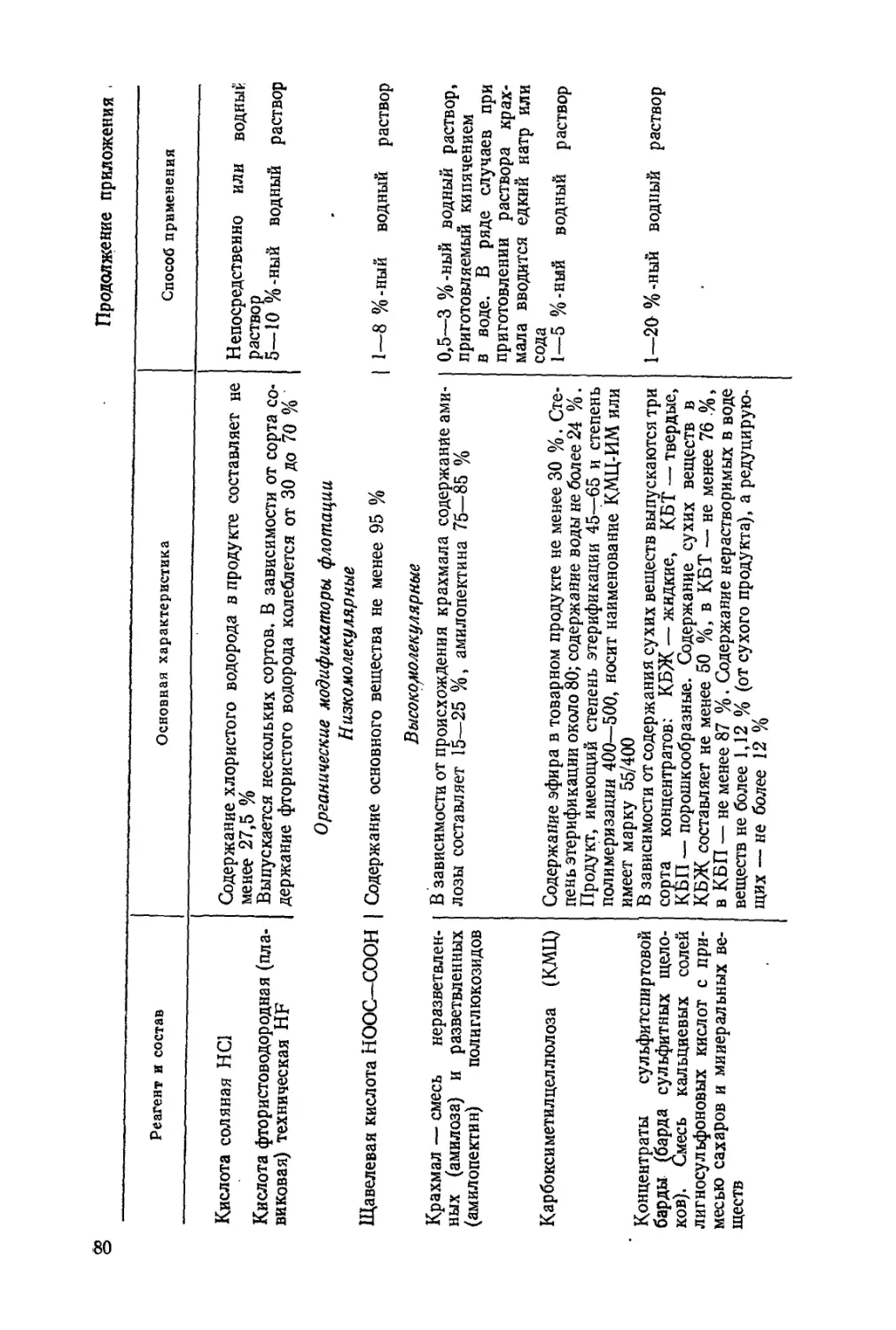
Абрамов А. А. Флотационные методы обогащения. Учебник для вузов. М.:
Недра, 1984,383 с.
Изложены основы теории флотационного процесса с позиций
современного состояния физической химии, физики твердого тела и других научных
дисциплин. Освещены вопросы технологии и практики флотации с
теоретическим обоснованием реагентных режимов, широко используемых в практике
селективной флотации различных руд. Показаны методы и пути оптимизации
и интенсификации флотационного процесса. Рассмотрены конструкции и
эффективность работы флотационных аппаратов.
Для студентов вузов, специализирующихся в области обогащения
полезных ископаемых.
Табл. 4, ил. 123, список лит.— 50 назв.
Рецензенты:
кафедра ОПИ ЛГИ,
лроф. д-р хим. наук В. И. Мелик-Гайказян (Курский политехн. ин-т).
2505000000-055
А 043@1)—84 ш'-т-**
I© Издательство «Недра», 108
ПРЕДИСЛОВИЕ
Важнейшей задачей, поставленной «Основными
направлениями экономического и социального развития СССР на 1981 —
1985 годы и на период до 1990 года», является удовлетворение
всевозрастающих потребностей народного хозяйства в цвет-
ных, редких, черных, благородных металлах, угле, удобрениях^
строительных материалах. Для решения этой задачи требуется
резкое увеличение объемов добычи, переработки и обогащения
полезных ископаемых. Основными в переработке многих типов,
полезных ископаемых являются флотационные методы
обогащения. Они используются, например, при обогащении более
95 % РУД цветных металлов, апатит-нефелиновых руд и других
типов минерального сырья. Возрастающее значение
флотационных методов обогащения в настоящее время обусловлено
вовлечением в переработку бедных, тонковкрапленных и трудно-
обогатимых руд и углей, необходимостью комплексного и более
полного их использования.
Учитывая важное значение флотационного процесса,
изучению его в целях совершенствования и интенсификации
уделяется большое внимание. На высоком научном уровне
проводятся исследовательские, конструкторские и проектные работы,
в институтах Механобр, ИПКОН АН СССР, Гинцветмет,
ИОТТ, ГИГХС, БИМС, ВНИИЦветмет, ЦНИГРИ, Унипро-
медь, ЦНИИОлово, Гипромашобогащение, Госгорхимпроект,
УкрНИИУглеобогащение и др. Значительный объем
теоретических и экспериментальных исследований выполнен учебными:,
вузами.
В результате проведенных исследований формируются новые
представления о физической и физико-химической сущности
явлений при флотации, разработаны принципе их
физико-химического моделирования, математические модели процессов,
созданы совершенные машины'и аппараты, технологические схемы
и режимы флотационного обогащения, предложены новые
процессы флотации и методы их интенсификации. Все это
позволяет на научной основе осуществить автоматический контроль,
и регулирование процессов коллективной и селективной
флотации, добиться высокого качества продукции, снизить потери:
ценных компонентов с отходами производства и повысить
комплексность использования сырья в условиях полного водообо-
рота и охраны окружающей среды.
Высокий технический уровень обогатительных
флотационных фабрик, оснащенных системами АСУ ТП и АСУП,
определяет необходимость подготовки специалистов, способных
в сложных производственных условиях творчески решать
вопросы совершенствования технологии флотационного обогаще-
3
ния, управления технологическим процессом и производством,
технически грамотно анализировать и добиваться высоких
технико-экономических показателей работы предприятий.
Назначение настоящего учебника — дать студентам
необходимые знания теоретических основ флотационного, обогащения
полезных ископаемых, детально ознакомить с технологией
флотации и флотационным оборудованием, методами оптимизации
и управления процессами, технико:экономическими
показателями флотационного обогащения различных типов
минерального сырья.
Учебник составлен в соответствии с учебной программой
дисциплины «Флотационные методы обогащения» для
студентов высших учебных заведений, обучающихся по специальности
0204 «Обогащение полезных ископаемых».
Автор выражает глубокую благодарность проф., д-ру хим.
наук В. И. Мелик-Гайказяну, доц., канд. техн. наук С. И. Гор-
ловскому и коллективу, кафедры обогащения полезных
ископаемых Ленинградского горного института за ценные советы,
критические замечания и указания, которые помогли улучшить
настоящее издание.
1. СУЩНОСТЬ И РАЗНОВИДНОСТИ
ФЛОТАЦИОННЫХ ПРОЦЕССОВ
Флотация является основным технологическим процессом
обогащения многих полезных ископаемых. В настоящее время
только в СССР работают сотни обогатительных фабрик, на
которых флотируют руды цветных, редких и черных металлов,
каменные угли, фосфатные руды, серу, полевой шпат, борные
руды, плавиковый шпат, калийные соли и другие полезные
ископаемые. Для многих, руд, особенно руд цветных и редких
металлов, нет другого технологического процесса обогащения,
который был бы в состоянии конкурировать с флотацией.
1.1. Сущность флотационного процесса
Как и'всякий процесс обогащения полезных ископаемых,
флотация основана на различиях в свойствах разделяемых
минералов. При этом используется различие в
физико-химических свойствах поверхностей минералов, а именно, различие
в их удельных свободных поверхностных энергиях.
Свободная энергия частицы' в "любой системе складывается
из ее потенциальной и поверхностной энергии. Первая из них
пропорциональна массе или объему частицы, т. е. диаметру
частицы в кубе, а вторая — ее поверхности, т. е. диаметру
в квадрате. Очевидно, что с уменьшением размера (диаметра)
частицы ее потенциальная энергия уменьшается в гораздо
большей степени, чем поверхностная энергия. Поэтому, как бы ни
была мала поверхностная энергия частицы по сравнению с
потенциальной, всегда можно получить частицы малых размеров,
у которых поверхностная энергия будет намного больше
потенциальной. Именно такие частицы участвуют во
флотационном процессе разделения. Крупность флотируемых частиц
обычно не превышает 0,6 мм, а при особых режимах
флотации— нескольких миллиметров [21].
Флотационная система является гетерогенной, включающей
твердую, жидкую и газообразную фазы, которые образуют
поверхности раздела: жидкость — газ (ж—г), жидкость —
твердое (ж—т) и твердое — газ (т—г). Каждая из поверхностей
раздела фаз характеризуется своим значением свободной
поверхностной энергии, появление которой обусловлено
неодинаковым притяжением молекул поверхностного слоя со стороны
соприкасающихся фаз (рис. 1.1). Так как молекулярные силы
имеют небольшой радиус действия, то предполагают [15], что
поверхностная энергия локализована в тонком поверхностном
слое, толщина которого ненамного превышает размеры двух-
трех молекул. В объемах соприкасающихся фаз 1 и 2 плотно-
5
Рис. 1.2. Влияние различия в
.диэлектрических постоянных D\—D2
соприкасающихся фаз на значение
удельной свободной поверхностной
энергии а на границе их раздела
(по данным К. А. Разумова [36])
ети энергий f\ и /2 постоянны; в поверхностном слое толщиной
h = h\+h2 плотность энергии возрастает, достигая максимума
на границе раздела фаз (см. рис. 1.1).
Значение удельной свободной поверхностной энергии о
зависит от разницы в значениях полярности соприкасающихся
фаз, т. е. разницы в значениях интенсивности действующих
в них молекулярных сил. .Мерой полярности фазы могут
служить такие ее свойства, как диэлектрическая постоянная, ди-
польный момент молекул и другие, так называемые
молекулярные, свойства фазы. Чем больше разница в значениях
полярности соприкасающихся фаз, тем меньше компенсируется
взаимодействие между молекулами и .ионами граничащих фаз
и тем больше удельная поверхностная энергия на границе их
раздела. По этой причине значение поверхностной энергии на
границе раздела двух полярных фаз, а также на границе
раздела двух неполярных фаз будет малое, а на границе раздела
полярной и неполярной фаз —большое.
Например, эоздух имеет низкую диэлектрическую
постоянную, поэтому на границе его раздела с разными жидкостями
удельная поверхностная энергия <тж-г будет более высокой для
жидкостей с большими диэлектрическими постоянными
(рис. 1.2). Значение 0ж-г для воды при 20 °С составляет
72,5 мДж'М-2 G2,5 мН-м), для жидких органических
веществ оно изменяется от 20 до 40 мДж-м-2 [13] (значения а
в единицах СИ совпадают со значениями а в единицах СГС —
Рис. 1.1. Схема сил молекулярного
притяжения на поверхности
раздела фаз и распределение
свободной энергии в поверхностном слое
Рис. 1.3. Кристаллические решетки графита (а) и галита (б)
эрг «см'2 или дин-см, приведенными в литературе прошлых
лет).
Энергия взаимодействия минеральных поверхностей с водой
и воздухом и значения поверхностной энергии на границе их
раздела определяются характером связей, разрушаемых при
дроблении и измельчении. Структурные элементы (атомы, ионы
или молекулы) кристаллической решетки минералов
располагаются с определенной закономерностью относительно друг
друга и существующие между ними связи отличаются по своей
прочности. Для прочных или сильных связей, таких, как
ионная, ковалентная, металлическая и других форм атомных
связей, энергия равна 80—800 кДж/моль. Молекулярная связь
значительно менее прочная. Энергия, необходимая для ее
разрушения, составляет всего* лишь 0,08—8 кДж/моль [6].
Водородная связь слабее химических, но прочнее молекулярных.
Если при разрушении кристалла рвутся сильные полярные
связи, то энергия взаимодействия поверхности с полярными
молекулами воды будет велика (значение от-ж — мало), а с
неполярными молекулами, например, воздуха — мала (значение
<тт_г — велико). В этом случае молекулы воды притягиваются
к поверхности твердого тела, хорошо смачивают ее и
поверхность становится гидрофильной. Если же при разрушении
кристалла происходит разрыв слабых связей, то образующаяся
неполярная поверхность слабее притягивает дипольные молекулы
воды и сильнее взаимодействует с воздухом, т. е. поверхность
становится гидрофобной.
-Характер обнажающихся на поверхности связей при
дроблении и измельчении минералов определяется строением их
кристаллических решеток.
Например, в графите (рис. 1.3, а) атомы углерода
расположены слоями, построенными в виде гексагональной сетки.
В плоскости этих слоев между атомами углерода существуют
ковалентны*1 связи, а связь между слоями (на плоскостях спай-
7
ности) осуществляется слабыми межмолекулярными силами.
Этим объясняются гидрофобность графита по плоскостям
спайности, где связь между находящимися в соседних слоях
атомами углерода молекулярная, и гидрофильность его на торцах
чешуек, где разрываются сильные атомные связи.
Сила взаимодействия поверхностей минералов с
молекулами или ионами окружающей среды зависит не только от
характера, но и от числа разрушаемых при измельчении
минералов связей, а также от положения атомов или ионов на
поверхности минерала. Так, например, в случае кубической решетки
кристалла NaCl (рис. 1.3, б) ион в положении А (внутри
кристалла) взаимодействует с ближайшими шестью противоио-
нами, окружающими его со всех сторон. Ион £, находящийся
на грани кристалла, взаимодействует только с пятью противо-
ионами и имеет одну ненасыщенную связь, которая может быть
им использована для взаимодействия с молекулами воды. Ион
В, находящийся на ребре кристалла, имеет две ненасыщенные
связи, а ион Г, находящийся в вершине, имеет три
ненасыщенные связи. Поэтому вершины и ребра кристалла являются
более активными при взаимодействии с водой, чем его грани.
Поверхность твердого тела, состоящего из атомов двух или
более видов, всегда энергетически неоднородна, и свободная
поверхностная энергия меняется от атома к атому. Поэтому под
свободной поверхностной энергией твердых тел понимают
среднюю величину для макроповерхности каждой из
кристаллических граней.
Разные минералы обязательно отличаются друг от друга
или составом, или строением кристаллической решетки и,
следовательно, имеют разные значения удельной поверхностной
энергии на границах раздела минерал — газ и минерал —
жидкость. Это приводит к тому, что минимум свободной энергии
флотационной системы в соответствии со вторым законом
термодинамики достигается для разных минералов при контакте
их с разными фазами. Так, для минералов с полярной
поверхностью энергетически выгодна поверхность раздела твердое —
полярная жидкость (вода), а для минералов с аполярной
поверхностью твердое — аполярное вещество (масло, газ), т. е.
разные минералы обладают различной способностью
закрепляться на рассматриваемых междуфазовых поверхностях. Это
создает условия для их разделения, что используется на
практике. По определению К. А. Разумова [36] флотационными
процессами называются процессы разделения
минералов, основанные на различной
способности этих минералов закрепляться на
междуфазовой поверхности. Частицу минерала,
закрепившуюся на междуфазовой поверхности, называют
флотирующейся, незакрепившуюся — нефлотирующей ся.
Флотация в отличие от других процессов обогащения
является процессом универсальным, так как не существует принци-
пиальных ограничений в отношении возможности ее
применения для разделения любых минералов, поскольку все они имеют
разные значения удельной свободной поверхностной энергии.
Универсальность флотационного процесса обеспечивается и тем,
что если «природная» разница в значениях удельной
поверхностной энергии у разделяемых минералов невелика и
недостаточна для эффективного флотационного разделения, то она
может быть увеличена с помощью специальных реагентов,
называемых флотационными, избирательное закрепление которых
па поверхности определенных минералов изменяет их
поверхностную энергию в заданном направлении.
Практика подтверждает принципиальную возможность при-.
менения флотации для разделения любых минералов и область
применения ее в различных отраслях народного хозяйства все
время расширяется.
1.2. Разновидности флотационных процессов
Флотационное разделение минералов может быть
осуществлено на следующих поверхностях раздела: жидкость — газ,
жидкость — жидкость, жидкость — твердое и газ — твердое.
Флотационное разделение минералов на поверхности
раздела жидкость — газ. Флотационное разделение минералов,
происходящее на плоской поверхности раздела вода — воздух,
получило название пленочной флотации. Исходная
смесь флотирующихся и нефлотирующихся частиц при этом
подается на водную поверхности сверху. Флотирующиеся частицы
удерживаются на поверхности и переносятся потоком к месту
разгрузки концентрата, а нефлотирующиеся тонут и удаляются
в виде хвостов. При кажущейся простоте этот процесс
достаточно чувствителен, малопроизводителен и в чистом виде не
получил промышленного применения.
Принцип пленочной флотации использован в настоящее
время при ф л отогравитац ионном способе
обогащения, широко применяемом в схемах доводки редкоме-
талльных концентратов. По данному способу смесь
относительно крупных частиц разделяемых минералов C мм и более)
сначала обрабатывается реагентами для повышения флотируе-
мости загрязняющих концентрат сульфидных минералов,
а затем поступает на концентрационный стол. Флотирующиеся
частицы сульфидных минералов плывут по поверхности воды
(пленочная флотация), а нефлотирующиеся минералы
разделяются на столе обычным способом: на тяжелую (например,
касситерит или вольфрамит) и легкую (силикаты, кварц)
фракции.
Производительность процесса резко возрастает, если при
разделении используют не плоскую поверхность раздела
жидкость — газ, а криволинейную поверхность пузырьков,
образуемых в пульпе, как в процессе пенной флотации и
различен
. ных ее модификациях. В этом случае флотирующиеся частицы
закрепляются на пузырьках и выносятся ими на поверхность
пульпы, образуя слой минерализованной пены. В зависимости
от способа насыщения пульпы пузырьками газа пенная
флотация подразделяется на4 несколько разновидностей.
При обычной пенной флотации, используемой в
настоящее время практически на всех флотационных фабриках,
газом является засасываемый или подаваемый под давлением
воздух, который диспергируется в пульпе на мелкие пузырьки
с помощью различных устройств.
При вакуумной флотации аэрация
пульпы-обеспечивается выделением воздуха из раствора. Процесс используется
для обогащения коксующихся углей и является перспективным
для флотации тонких шламов других полезных ископаемых.
Аналогичный процесс флотации получается, если сначала вода
насыщается воздухом под повышенным давлением, а затем при
атмосферном давлении выделяются пузырьки. Такая, флотация
с повышенным давлением, или компрессионная
флотация, используется для очистки воды от тонких
капелек нефти, которые закрепляются на поверхности
выделяющихся пузырьков и всплывают вместе с ними на поверхности
очистного сооружения.
Принцип ,компрессионной флотации получил развитие и
используется в настоящее время в разработанном в СССР
В. А. Малиновским процессе адгезионной сепарации,
которая включает в себя по существу три по-разному аппара-
турно оформленных метода: адгезионную каскадную
сепарацию (АКС), глубинную адгезионную сепарацию (ГАС) и
поверхностную адгезионную сепарацию (ПАС). Все- они
предназначены для извлечения из шахтных, сточных или оборотных
вод гидрофобных частиц, осадков и веществ в результате
адгезии их на поверхности выделяющихся из раствора пузырьков
газа и отделения образующихся флотационных комплексов от
объема жидкости или пульпы.
Образование пузырьков пара и выделение растворенного
в воде воздуха происходят и при кипячении пульпы. Флотация
кипячением применялась некоторое время для обогащения
графитовых руд.
При химической, или газовой, флотации пузырьки
газа образуются вследствие химического взаимодействия,
например, между загружаемой в пульпу кислотой и карбонатами
пустой породы. В этом случае флотирующиеся минералы
закрепляются на выделяющихся пузырьках углекислоты. Процесс
в течение ряда лет применялся в Австралии для переработки
отвалов хвостов отсадки, содержащих сфалерит.
При электрофлотации используется междуфазовая
поверхность образующихся при электролизе пузырьков водорода
или кислорода» крупность которых легко регулируется
изменением силы тока. Процесс может быть использован для флота-
Ю
ции мелких или весьма мелких частиц, а также при
осуществлении так называемой ионной флотации и ее
разновидностей (пенного фракционирования, флотации гидрофобных и
гидрофобизированных осадков, флотоэкстракции), если
поверхность раздела жидкость — газ используется для извлечения из
растворов иона и молекул органических соединений или
продуктов их взаимодействия с ионами или молекулами
неорганических соединений [19].
При пенной сепарации исходная пульпа,
предварительно обработанная реагентами, подается на пену или
аэрированную жидкость. Флотирующиеся частицы удаляются с
пеной, а нефлотирующиеся проходят сквозь пену под действием -
силы тяжести и разгружаются в виде камерного, продукта.
Процесс пенной сепарации предложен в СССР (в 1961 г.)
В.А.Малиновским и используется в настоящее время для
флотационного обогащения фосфоритовых, калийных и других типов
минерального сырья. Его применение оказывается особенно
эффективным при обогащении материалов, крупность которых
слишком велика для обычной пенной флотации, но мала для
эффективного гравитационного обогащения.
Процесс гидрообеспыливания также основан на
флотационных явлениях, происходящих на границе раздела
жидкость—'газ. В этом случае через запыленный воздух движутся
капельки воды. При столкновении частичек пыли с каплями
воды флотирующиеся частицы закрепляются на поверхности
капель (т. е. на междуфазовой поверхности раздела
жидкость — газ), а нефлотирующиеся частицы переходят внутрь
капель (т. е. в жидкую фазу).
Флотационное разделение минералов на поверхности
раздела жидкость — жидкость. На различной способности
минералов закрепляться на поверхности раздела вода — масло
основан процесс масляной флотации. Столкнувшись с каплями
диспергированного в пульпе масла и закрепившись на них,
флотирующиеся частицы удерживаются на поверхности раздела
масло — вода, а нефлотирующиеся частицы остаются в пульпе.
Если плотность масла меньше единицы, то его капельки вместе
с закрепившимися частицами всплывают на поверхность
пульпы, образуя слой минерализованного масла, который затем
удаляется. Если используется небольшое количество масла
с высокой плотностью, то образующиеся минерализованные
гранулы опускаются на дно, а нефлотирующиеся зерна выносятся
наверх восходящим потоком воды. Флотационный процесс в
таком исполнении называется грануляционным.
Процесс масляной флотации вследствие большого расхода
масла промышленного применения не получил.
Грануляционный процесс применяется до настоящего времени на некоторых
зарубежных фабриках для обогащения коксующихся углей.
Используемое в процессе флотации масло регенерируется при
коксовании угля.
И
Поверхность раздела жир — вода используется в
промышленных условиях для улавливания алмазов. При
обогащении на жировых поверхностях на дно желоба, по
которому течет содержащая алмазы пульпа, или на барабан
наносится слой вязкого жира. Алмазы закрепляются на
поверхности раздела жир — вода, а пустая порода сносится потоком
пульпы [36].
В процессе флотации при автоклавной плавке
серных концентратов используется способность частиц
пустой породы закрепляться на поверхности капель воды,
находящихся внутри расплава серы, т. е. на поверхности раздела
вода — расплав серы. Так как плотность нагруженных капелек
воды, меньше плотности расплава серы, то они поднимаются на
поверхность расплавленной серы, вынося с собой пустую
породу, в результате чего достигается очистка серы от
загрязняющих ее минеральных примесей.
Флотационные процессы на поверхностях раздела твердое —
жидкость и твердое — газ. Лабораторными исследованиями
установлено, что на поверхности раздела вода — стекло или
вода — металл могут закрепляться некоторые минералы. Такой
способ выделения минералов получил название флотации
твердой стенкой.
В настоящее время принцип флотации твердой стенкой
реализуется в так называемой флотации с носителем, когда
для повышения извлечения тонких гидрофобных частиц
в пульпу добавляют хорошо извлекаемые крупные частицы
(носители). Тонкие частицы закрепляются на поверхности час|иц
носителя и ^флотируются вместе с ними в пену. /
В качестве флотационного процесса на поверхности раздела
твердое — вода можно рассматривать коагуляцию
(слипание) минеральных частиц в пульпе, широко используемую в
технике для осветления шламовых вод. Роль твердой фазы в
данном случае играют слипшиеся минеральные частицы.
Аналогичным образом явление слипания твердых частиц
в аэрозолях и дымах можно рассматривать как процесс
их закрепления на поверхности раздела газ — твердое. Роль
твердой фазы здесь играют взвешенные частички пыли, а
газообразной — воздух или дымовой газ.
1.3. Значение флотационного процесса
Флотация способствует решению ряда важных
народнохозяйственных проблем, к числу которых относятся:
проблема расширения минеральных
ресурсов. До открытия флотационного процесса в эксплуатацию
вовлекались месторождения только богатых мономинеральных и
полимш/еральных крупновкрапленных руд, поддающихся
обогащению другими нефлотационными методами. Флотация
позволила вовлечь и эксплуатацию месторождения бедных, тон-
ковкрапленных комплексных руд;
12
проблема комплексного использования руд*
До применения флотации, например, из полиметаллических руд
удавалось получать только коллективные концентраты,
поскольку полезные минералы, обладающие близкими
физическими свойствами, нельзя было разделить гравитационными,,
магнитными и другими нефлотационными процессами
обогащения полезных ископаемых. Получаемые коллективные
концентраты обычно были непригодны для эффективного
металлургического передела, поэтому при их переработке некоторые
металлы и элементы терялись и полного использования всех
ценных компонентов руды не достигалось. При обогащении
ряда руд и особенно руд редких металлов, в которых полезные
минералы или часть их по плотности и магнитным свойствам
часто близки к минеральным породам, флотация является
практически единственным способом разделения минералов и
извлечения каждого из них в соответствующий концентрат; -
проблема обогащения шламов. При обогащении
многих полезных ископаемых гравитационными процессами
иногда значительная часть извлекаемых минералов
переизмельчается и Переходит в шламы/ До появления флотации эти
шламы не могли перерабатываться и поэтому направлялись
в хвосты или использовались в необогащенном виде.
Применение флотации для переработки шламов гравитационного
обогащения позволило значительно улучшить комплексное
использование оловянных руд, ценных и дефицитных коксующихся
углей и другого сырья.
Применение флотационного процесса непрерывно
расширяется. По количеству и разнообразию перерабатываемого сырья
флотация занимает первое место среди других технологических
процессов обогащения. Кроме того, флотацию широко
применяют в металлургии (для флотационного разделения файн-
штейна, отделения криолита от частиц угля и алюминия),
химическом производстве (для разделения хлористого аммония
и бикарбоната натрия), биологии (для разделения различных
видов бактерий), экологии (для удаления вредных веществ из
сточных вод или нефти и мазута с поверхности прибрежных
вод), сельском хозяйстве (для разделения различных семян),
геологии, медицине и других отраслях народного хозяйства.
Несомненно, что вследствие универсальности флотационного
процесса его значение будет все время возрастать.
Своими успехами за последние 50 лет флотация обязана
советским ученым: К. Ф. Белоглазову, О. С. Богданову,
Б. В. Дерягину, В. И. Классену, С. И. Митрофанову,
И. Н. Плаксину, К- А. Разумову, П. А. Ребиндеру, Д. Л.
Талмуду, А. Н. Фрумкину, М. А. Эйгелесу. Из зарубежных
исследователей в первую очередь необходимо отметить работы
А. М. Годена, М. Карта, М. Рея, А. Ф. Таггарта, И. В, Уорка,
Д. В. Фюрстенау. К настоящему времени СССР занимает
ведущее положение в области теории и технологии флотации.
15
ЧАСТЬ I
Основы теории минерализации пузырьков
при флотации
2. ТЕРМОДИНАМИКА ЭЛЕМЕНТАРНОГО АКТА ФЛОТАЦИИ
§ 2.1. Краткая характеристика раздела фаз,
участвующих в элементарном акте флотации
Основной и определяющей стадией флотационного процесса
является элементарный акт флотации, под которым
подразумевают закрепление единичного зерна на поверхности
раздела фаз.
К наиболее важным в практическом отношении фазам,
используемым при флотации, относятся вода* (жидкость),
минерал (твердое) и воздух (газ). Возможность элементарного
акта при этом зависит от свойств поверхностей раздела вода—
воздух (жидкость — газ) и минерал — вода (твердое —
жидкость) .
Свойства поверхности раздела вода — воздух определяются
главным образом значением ее удельной свободной
поверхностной энергии схг_ж и структурой поверхностного слоя
молекул воды на границе раздела газ — жидкость, которые, в свою
очередь, зависят от наличия и концентрации растворенных
солей, аполярных или гетерополярных органических соединений
в воде.
Наличие свободной поверхностной энергии на границе
раздела вода — воздух и стремление системы в соответствии со
вторым законом термодинамики к минимальным значениям ее
приводят к сокращению поверхности пузырька, находящегося
в.воде, до тех пор, пока капиллярное давление во'здуха или
газа в нем не приведет к равновесию системы. Давление рк
внутри пузырька, если его размеры превышают радиус
действия межмолекулярных сил, т. е. больше или равны 0,2 мкм
14], определяется в соответствии с первым законом
капиллярности по формуле
рк = аг_жA/Д + 1/р), B.1)
где /? и р — главные радиусы кривизны поверхности пузырька.
Если она сферическая, то /? = р = #ш и тогда
Рк=-2сгг_ж/Яш.
Значение рк существенно влияет на прочность закрепления
частицы на пузырьке, а также на жесткость поверхности
пузырька при изменении его размера.
14
Стремлением к уменьшению свободной энергии объясняется
коалесценция (слияние) пузырьков воздуха или капелек апо-
лярных масел в воде, поскольку это приводит к сокращению
поверхности раздела вода — воздух или аполярное масло — вода.
При коалесценции, например, двух одинаковых по размеру
пузырьков (или капелек) поверхность образующегося пузырька
(или капли) примерно на 25 % меньше исходной суммарной
поверхности пузырьков (капелек). Уменьшится при этом в
результате увеличения размера пузырьков или капли и значение
капиллярного давления в них.
Если в объеме воды присутствуют капельки аполярных
масел (углеводородных жидкостей) или гетерополярных
органических соединений (длинноцепочечных спиртов, жирных кислот,
аминов), применяемых при флотации в качестве реагентов, то
при столкновении с пузырьком они могут растекаться по его
поверхности.
Условием растекания (по Дюпре и Гарди) является
превышение работы адгезии Wa, т. е. взаимодействия молекул
органической жидкости и воды, над работой когезии WK>
т. е. взаимодействия молекул органической жидкости между
собой. Разность между ними получила (по В. Гаркинсу)
название коэ ф ф иди ент а растекания:
Учитывая, что в нашем случае Wa характеризует работу,
необходимую для разделения единицы поверхности раздела
масло — вода (м—в) на две поверхности вода — газ (в — г) и:
масло — газ (м—г), т. е.
Wa ■= <тв_г + (тм_г — ам_в,
a WK соответствует обратимой работе разрыва столбика масла
на две поверхности масло — газ, т. е.
получаем:
= <* в-г + <7м-г— СГМ_ в— 2<ТМ_Г = QB_r — @м-в + <Ум-г).
Для большинства используемых при флотации органических:
жидкостей коэффициент растекания s имеет положительное
значение, поскольку значение <тв-г, как правило, больше
значения Ом-в + сгм-г. Так, по данным В. Гаркинса [4], коэффициент
растекания для исследованных углеводородных жидкостей
изменяется от 0,2 до 6,8 мДж • м~2, для длинноцепочечных
спиртов — от 36,7 до 49 мДж • м~2 и для жирных кислот — от 24,6
(для олеиновой кислоты) до 45,7 мДж-м~2. Это означает, что-
их растекание по поверхности раздела вода — газ протекает
самопроизвольно и сопровождается уменьшением свободной
энергии.
1£
Рис. 2.1. Схема
обозначений к уравнению
Лапласа
^х- -Скорость растекания органической
жидкости возрастает с увеличением
значений коэффициента растекания и
падает с возрастанием кривизны
поверхности пузырька [4]. На скорость
растекания влияют вязкость
соприкасающихся фаз и наличие в них примесей.
Например, по данным Н. К. Адама,
даже такую нерастекающуюся
жидкость, как тяжелое парафиновое масло,
можно заставить растекаться по воде,
растворив в ней какую-либо жирную
кислоту или другое вещество,
повышающее адгезию между маслом и водой.
Процесс растекания сопровождается
обычно образованием довольно толстых пленок, которые после
взаимного насыщения соприкасающихся фаз на границе их
раздела могут распадаться, образуя на поверхности пузырька
в условиях равновесия монослой органической жидкости и
линзу избытка жидкости [4]. При закреплении пузырька на
частице избыток органической жидкости концентрируется по
линии контакта твердой, жидкой и газообразной фаз [7, 15].
Образование монослоя (или пленки) аполярного масла или
гетерополярного соединения на поверхности пузырька приводит
к существенному понижению равновесного значения ее
свободной энергии вплоть до значений, характерных для
поверхностей раздела органическая жидкость — газ. Динамические
условия пенной флотации вызывают нарушение адсорбционного
равновесия, например в результате вытягивания пузырька, и
возрастание свободной энергии его поверхности вплоть до
значений, соответствующих чистой поверхности раздела вода —
газ. Разница в значениях удельной поверхностной энергии в
динамических ад и равновесных стр условиях
Да = (Тд—ар
является количественной характеристикой неравновесности
процесса формирования адсорбционных слоев органического
реагента на поверхности пузырька и может достигать 10 мДж*м-2
и более [46].
Неравновесность адсорбционных слоев органических
реагентов на поверхности пузырька оказывает значительное влияние
на флотацию. Наблюдается, например, довольно тесная
корреляция между величиной Д<х и флотационной активностью
реагентов, их пенообразующей способностью, крупностью
флотируемых частиц и качеством продуктов флотации [22, 23].
Оценить степень равновесности адсорбционных слоев на
различных уровнях поверхности пузырька (в целях изучения
причин наблюдаемой корреляции и других явлений) можно,
используя ураншчшо Лапласа, являющееся основой математиче-
16
ской теории капиллярности. Данное уравнение описывает
форму легко подвижной границы раздела жидкость — газ (или
жидкость — жидкость), находящейся под действием только
капиллярных и гравитационных сил, и в преобразованном
(Д. К- Адамсом по инициативе Ф. Башфорта) виде для случая
симметричных капель или пузырьков (рис. 2.1) записывается
следующим образом [46]:
Ыр + b sincp/* - 2 + рг/62; B.2)
fb = (Dx-DJgWo, B.3)
где b — радиус кривизны поверхности в куполе пузырька; р —
главный радиус кривизны поверхности в плоскости
меридионального сечения в произвольной точке с координатами х и г;
<р —угол, образованный нормалью к поверхности пузырька
в этой.точке и осью симметрии пузырька; р — коэффициент,
характеризующий форму пузырька; D{ и D2 — плотность
соответственно нижней и верхней фаз; g — ускорение свободного
падения.
Определение а основано на оценке формы (коэффициента
Р) пузырька или капли по результатам совокупности различных
бесконтактных измерений, проводимых в определенной
последовательности на контуре их меридионального сечения, и
вычислений с использованием специальных таблиц (получивших
название таблиц Башфорта и Адамса), составленных по
результатам численного решения уравнения Лапласа для различных
значений ±р [46]. Безразмерный характер таблиц позволяет
применять их при исследовании границ раздела практически
с любыми размерами и значениями а и плотности граничащих
фаз. Использование их В. И. Мелик-Гайказяном позволило
установить в последние годы ряд важных зависимостей,
определяющих условия и прочность закрепления частицы на пузырьке
в процессах пенной флотации и сепарации [22, 23, 46].
Молекулы поверхностного слоя вследствие
неуравновешенности сил, действующих со стороны соприкасающихся фаз (см.
рис. 1.1), находятся в особом энергетическом состоянии.
Поэтому, как показано Б. В. Дерягиным, свойства жидкости,
находящейся в пограничном слое, отличаются от свойств
жидкости в объеме [4, 15].
Пограничные слои воды называют гидратными ело-
я м и. По сравнению с водой в объеме они имеют повышенную
вязкость и пониженную растворяющую способность, скорость
диффузии растворенных в них веществ меньше [7].
Особые свойства гидратных слоев обусловлены структурой
расположения в них молекул воды; при этом структура и
толщина гидратных слоев зависят от степени полярности
соприкасающихся фаз.
Структуру гидратного слоя, обеспечивающую минимальное,
значение свободной энергии системы полярная жидкость
17
Рис. 2.2. Структура гидратного слоя на поверхности пузырьков воздуха
в воде:
о. — чистого; б — покрытого маслом; в ~ покрытого гетерополярными молекулами: / —
масло; 2 -дипольные молекулы воды; 3 — гетерополярные молекулы; 4 ~ знак диполя
(вода) — аполярное вещество (воздух), можно представить
в виде слоя молекул воды на пузырьке, соприкасающихся
противоположными по знаку концами диполей (рис. 2.2, а), что
приводит к взаимной компенсации их дипольных моментов и
резкому ослаблению взаимодействия с молекулами воды в
объеме. В результате этого толщина гидратного слоя минимальна.
Замена поверхности раздела воздух — вода поверхностью
раздела аполярное масло—вода не изменит существенно
структуру и толщину гидратного слоя вокруг капельки масла или
пузырька воздуха, покрытого маслом (рис. 2.2, б).
Значительные изменения полярности поверхности и
структуры гидратных слоев наблюдаются при закреплении
(адсорбции) на пузырьке гетерополярных молекул, имеющих аполяр-
ный (углеводородный) радикал и полярную группу. Адсорбция
таких молекул сопровождается уравниванием полярностей
соприкасающихся фаз, уменьшением их разности и вследствие
этого понижением свободной поверхностной энергии системы
в результате особой ориентации гетерополярных молекул на
границе раздела газ — жидкость. Полярной частью они будут
обращены к полярной жидкости — воде, аполярным
радикалом — к воздуху.
Подобное расположение гетерополярных молекул на
поверхности раздела фаз обусловлено тем, что диполи воды, ак-
18
тивно взаимодействующие с полярными группами молекул,
практически не взаимодействуют с аполярными
(углеводородными) группами радикала и стремятся как бы вытолкнуть их
в воздушную фазу. При малом количестве гетерополярных
молекул на поверхности они будут располагаться почти в
плоскости поверхностного слоя, а при большом' количестве —
перпендикулярно поверхности раздела вода — воздух, т. е. будут
занимать положение, соответствующее минимальному значению
свободной поверхностной энергии [4].
Однако такая ориентация гетерополярных молекул
приводит к поляризации поверхности пузырька и вызывает
одинаковую ориентацию молекул воды, которая будет
распространяться, постепенно ослабевая, в глубь жидкости, пока не
оборвется тепловым движением. В результате этого вокруг
пузырька может образоваться значительный по толщине A0~6—
10~5 см) гидратный слой (рис. 2.2, в).
Аналогичное явление в присутствии гетерополярных
молекул наблюдается и на поверхности раздела капелька аполяр-
ного масла (или покрытого маслом пузырька)—вода,
поскольку в данном случае между пузырьком воздуха и капелькой
аполярного масла принципиальной разницы нет. Адсорбция
гетерополярных молекул уменьшает степень гидрофобности
поверхности раздела агюлярное масло — вода.
Образование толстых гидратных слоев вокруг пузырьков и
капель приводит, во-первых, к возрастанию жесткости их
поверхности и приближению ее формы к сферической. Поскольку
сфера является одной из наименее выгодных в
гидродинамическом отношении форм, скорость подъема пузырьков и капель
при этом уменьшится, а время пребывания их в пульпе
увеличится. Во-вторых, наличие гидратных слоев на поверхности
пузырьков и капель затрудняет их коалесценцию.
Термодинамическая вероятность ее может быть реализована только при
определенных затратах энергии активации, необходимой
для разрушения гидратных слоев, например, за счет
кинетической энергии столкновения пузырьков и капель. Все это
приводит к увеличению концентрации и устойчивости пузырьков и
капелек аполярных масел в пульпе.
Свойства поверхности раздела минерал —- вода зависят от
кристаллохимического строения минерала, определяющего
характер, число и расположение образующихся при измельчении
связей. Чем больше связей разрушено и чем они сильнее, тем
больше полярность и заряд поверхности.
В результате притяжения к заряженной поверхности ионов
щхшцюг^ (Ттротивоионов) на гранйце^азд^а^-
шШерал^г- вода ,]Жраз"уется д1ГсГи ной элект р и ч е с к и й
^J!JLjjI^C), схематически^изоВраженный на рис. 2.3-. Й~при- ^
веденном примере,jflaповерхности минерала анионов больше,
чем^лштионов^-И-они составляют внутреннюю обкладку
ДЭС (слой А). К заряженной внутренней обкладке ДЭС при-
19
Рис. 2.3. Структура двойного
электрического слоя и распределение
в нем термодинамического (р и
электрокинетического £
потенциалов (h — расстояние от
поверхности минерала)
Рис. 2.4. Зависимость свободной
поверхностной энергии 0 (а) и
емкости С двойного электрического
слоя (б) от потенциала (р
поверхности минерала в чистой полярной
жидкости G), в присутствии
поверхностно-активных молекул B),
катионов C), анионов D) и в апо-
лярной жидкости E)
-1ягива_юхся^ -лрютившоньт^ образуя внешню ю _itUJUUL ДЛУ.
ДЭС_ (сл_ои, К ilJQ. Противоионьь непосредственно прилегаю-
1Щ1е_к_внутр^еннеи[^обкладке ДЭС, прочно
связаны_с^наряженной поверхностью минера~ла и при движений^йнёрала в
жидкости перемещаются^ вместе с ним." Эти' противои~01ш~7Тас^ол^-
женные в..слое_/С) составляют^плотную часть внешиёРГоШлаЖкн'
ДЭС, или слой^Штёрна. Удаленные З^внутренне^Г^Тбтстгадки
противоионы связаны с ней слабее и. при перемещении мине-
рала в~~жидкасти orerafot от минерала. Зтот слой противоионов
(слой К') называют диффузным или слоем Гюи [4].
Поскольку поверхность частицы имеет заряд, в
пространстве, окружающем частицу, появляется электрическое поле.
Характер изменения потенциала этого поля по мере удаления от
поверхности минерала (при отсутствии специфических ионов)
изображен на рис. 2.3.
ДЭС может образоваться также на поверхности пузырька
(поверхности раздела вода—воздух) и капельки аполярного
масла в воде (поверхности раздела вода — масло) при
адсорбции на межфазной поверхности, например, гетерополярных
ионов или молекул.
20
Потенциал ср поля на межфазной поверхности называется
полным или термодинамическим, а на границе слоев Штерна
и Гюи — электрокинетическим или дзета-потенциалом £ (см.
рис. 2.3).
Значение ср-потенциала минерала зависит от концентрации
гютенциалопределяющих ионов (т. е. ионов, из которых состоит
минерал, и ионов, изоморфных им), тогда как значение
^-потенциала изменяется при изменении концентрации любых
электролитов в растворе.
Зависимость между свободной поверхностной энергией о,
потенциалом поверхности ср и ее зарядом г описывается первым
уравнением Липпмана
da/ftp = — е B.4>
и характеризуется электрокапиллярной кривой (рис. 2.4, а),
получаемой экспериментальным путем [13].
Из уравнения B.4) и электрокапиллярной -кривой (см.
рис., 2.4, а, кривая 1) следует, что максимальное значение а
соответствует нулевому значению е, т. е. минимальной
полярности поверхности, находящейся в контакте с высокополярной
жидкостью — водой, что вполне отвечает условию
максимальной разницы в полярности соприкасающихся фаз. Увеличение
заряда поверхности приводит к ее поляризации и уменьшению
разницы в полярности соприкасающихся фаз (минерала и
воды) и значения ст (см. рис. 2.4, а, кривая /). Восходящая
ветвь капиллярной кривой относится при этом к положительна
заряженной поверхности, а нисходящая — к отрицательно
заряженной. Если же минерал будет находиться не в воде^
а. в аполярной жидкости, то закономерность изменения
значений a при изменении ср будет прямо противоположной (см.
рис. 2.4, а, кривая 5).
Заряд е поверхности и определяемый им термодинамический
потенциал ср играют большую роль при взаимодействии
реагентов и молекул воды с минералом.
Так, например, отрицательный заряд поверхности
затрудняет адсорбцию анионов и облегчает адсорбцию катионов
поверхностно-активных веществ (ПАВ), резко уменьшающих
значение а, особенно при потенциалах, более отрицательных, чем
потенциал точки максимума (см. рис. 2.4, а, кривая 3).
Положительный заряд поверхности оказывает противоположное
действие (см. рис. 2.4, а, кривая 4). Адсорбция молекул ПАВ и
уменьшение о достигают максимального значения в области
потенциалов, близких к потенциалу нулевого заряда (см. рис. 2.4,
а, кривая 2). Адсорбция анионов предотвращается при
достаточно большом отрицательном, а катионов — положительном
заряде поверхности; адсорбция молекул предотвращается при
возрастании заряда любой полярности (см. рис. 2.4, а,
кривые 2—4).
21
Производная от заряда по потенциалу представляет собой
дифференциальную емкость С. С учетом
уравнения B.4)
С - де/ду = —д*о1ду2. B.5)
Полученное уравнение, известное как второе уравнение
Липпмана, широко используется для расчета значений а на
твердых поверхностях по результатам электрических
измерений (рис. 2.4, б).
В отсутствие ПАВ зависимость емкости С от потенциала ср
поверхности обычно имеет вид кривой / (см. рис. 2.4, б).
Сопоставление ее с электрокапиллярной кривой / на рис. 2.4, а
показывает, что в области потенциалов восходящей ветви
электрокапиллярной кривой емкость после первоначального резкого
спада продолжает уменьшаться, проходит через минимум,
отвечающий электрокапиллярному максимуму, а затем после
некоторого подъема сохраняется почти постоянной в широкой
области потенциалов нисходящей ветви электрокапиллярной
кривой.
Введение ПАВ влияет на ход не только электрокапиллярной
кривой (см. рис. 2.4, а, кривые 2—4), но и кривых изменения
заряда и емкости (см. рис. 2.4, б, кривые 2—4) в зависимости
от потенциала. В присутствии молекул, катионов и анионов
ПАВ кривые С—ф характеризуются минимумом, лежащим при
гораздо более низких значениях емкости, чем это наблюдается
для растворов поверхностно-инактивных электролитов. При
потенциалах, когда электрокапиллярные кривые/ полученные без
органического вещества и в его присутствии, сливаются
в одну общую кривую (см. рис. 2.4, а), на кривых С—ср (см.
рис. 2.4, б) наблюдается явно выраженный острый максимум,
отвечающий значению потенциала адсорбции-десорбции ионов
или молекул ПАВ на поверхности твердого.
Уравнения Липпмана B.4) и B.5), являющиеся основными
уравнениями теории электрокапиллярности,
электрокапиллярные кривые и кривые дифференциальной емкости могут быть
использованы и используются при изучении закономерностей
изменения состояния поверхности раздела твердой и жидкой
фаз под действием различных реагентов, в том числе
органических веществ. Например, по значению величины а и ее
зависимости от концентрации ионов и~ молекул ПАВ в растворе
можно рассчитать (по уравнению Гиббса) их адсорбцию на
исследуемой поверхности, по зависимости о от потенциала —
найти поверхностную плотность заряда, а по зависимости
адсорбции от потенциала — определить ориентировку молекул
в адсорбционном слое и характер их взаимодействия между
собой [4, 13].
Адсорбция ионов и молекул ПАВ на активных точках
поверхности и в ДЭС происходит в условиях конкуренции их за
место с молекулами воды.
22
Рлс. 2.5. Схема параллельной (а) и антипараллельной (б) ориентации
молекул воды на поверхности минералов (по Г. С. Стрельцыну [40])
Дипольные молекулы воды могут закрепляться
(адсорбироваться) на поверхности минерала в результате
электростатического притяжения их заряженными ионами поверхности,
межмолекулярного взаимодействия с поверхностью, образования
ъодородных и координационных связей с ионами или
молекулами поверхностного слоя. Структура гидратных слоев на
поверхности минерала при этом будет определяться
количественным соотношением разноименных зарядов, на которых -
оказывается возможным закрепление молекул воды, и их
расположением относительно друг друга.
При наличии на поверхности тела сильного
некомпенсированного электрического поля, отвечающего высокому значению
-ее потенциала, молекулы воды благодаря ион-дипольной связи
с поверхностными ионами поляризуются и ориентируются
одноименными полюсами диполей к поверхности, частично нарушая
водородную связь между молекулами воды. Такая ориентация
может распространяться, постепенно ослабевая, в глубь жид*
кости на большие расстояния, пока не оборвется тепловым
движением. В результате этого (в отсутствие органических
реагентов на поверхности), как и на поверхности пузырьков в
присутствии гетерополярных молекул (см. рис. 2.2, в), образуется
значительный по толщине гидратный слой односторонне
ориентированных молекул воды, которые благодаря поляризации
сильнее связаны с поверхностью тела и с молекулами воды в
объеме, чем молекулы воды в объеме друг с другом (рис. 2.5,
а). Такое состояние поверхности и строение гидратного слоя
отвечают ее гидрофильным свойствам [40].
При равномерном распределении разноименно заряженных
ионов и низких значениях потенциала строение гидратного слоя
23
на поверхности частиц должно быть иным. Так, например, на
поверхности ионных кристаллов NaCl или КС1 каждый ион
обладает некомпенсированным зарядом, который создает вокруг
иона сильное электрическое поле. Равенство числа
положительных и отрицательных зарядов на поверхности минерала и их
чередование приводят к тому, что адсорбированные на них
соседние молекулы воды ориентированы, противоположными
концами диполей. Антипараллельное расположение дипольных
молекул воды на поверхности ионного кристалла должно
приводить, как и на поверхности пузырьков в отсутствие гетеропо-
лярных ионов и молекул (см. рис. 2.2, а, б), к значительной
компенсации их дипольных моментов или непосредственно
соседними молекулами, или дополнительными молекулами воды
(рис. 2.5, б). Образование замкнутой связи между соседними
антипараллельно расположенными молекулами воды приводит
к ослаблению их взаимодействия с молекулами воды в объеме.
В результате чего поверхность частиц должна обладать
слабыми гидрофобными свойствами [40].
Кроме рассмотренных двух крайних типов гидратных слоев
должны встречаться гидратные слои, имеющие промежуточные
свойства. Степень гидрофобное™ или гидрофильности
поверхности при этом зависит в основном от степени
упорядоченности расположенных на ней разноименно заряженных ионов.
Причиной изменения соотношения разноименно заряженных
ионов на поверхности и возникновения заряда является или
преимущественная адсорбция из раствора входящих в состав
минерала (потенциалопределяющих) ионов одного знака
(например, при высокой концентрации их в растворе), или
преимущественный переход ионов одного знака с поверхности в
раствор. Последнее может быть следствием разницы в энергии
гидратации катионов и анионов минерала, а также в степени
их гидролиза при изменении рН среды и взаимодействия
с ионами и молекулами других веществ, которое можно
регулировать с помощью флотационных реагентов.
Во всех случаях увеличение заряда характеризует
возрастание числа одноименно заряженных частиц на поверхности и
образование гидратного слоя с параллельным расположением
молекул воды (см. рис. 2.5, а), вызывающим гидрофилизацию
поверхности. Вытеснение диполей воды с такой поверхности
ионами реагента наблюдается только в том случае, если они
более прочно связываются с минералом, чем дипольные
молекулы воды. Наибольшее ослабление связи между молекулами
воды гидратного слоя и молекулами воды в объеме достигается
при потенциале нулевого заряда, так как в этом случае на
поверхности возможно упорядоченное расположение ионов
противоположного знака и гидрофобизация поверхности обусловлена
в основном замыканием связей соседних антипараллельно
расположенных молекул воды на поверхности частицы (см.
рис. 2,5, б).
24
I
Рис. 2.6. Примерное-
изменение плотности е свободной
поверхностной энергии на
участках, покрытых собирателем
B) и свободных от него G)
Повышение степени гидрофоб-
ности минеральных частиц при
адсорбции на их поверхности гетеро-
полярных реагентов-собирателей,
полярная группа которых способна
взаимодействовать с
некомпенсированными связями, противоречит на
первый взгляд второму закону
термодинамики, так как ухудшение
смачиваемости поверхности
свидетельствует не о понижении, а об
увеличении разницы в полярности
соприкасающихся фаз и свободной
энергии поверхности раздела твер- <§[
дое — жидкость. Это кажущееся
противоречие можно объяснить [3],
если принять, что на поверхности
раздела твердое — жидкость при
адсорбции собирателя возникают
две границы (рис. 2.6): твердое —
полярная группа собирателя
(граница ^4) и аполярная группа
собирателя— вода (граница Б).
Поверхностная энергия границы £, обладающей гидрофобными
свойствами, будет больше поверхностной энергии исходной
границы раздела твердое — жидкость, а поверхностная энергия
границы А — значительно меньше, особенно если она отвечает
энергии химической связи полярной группы собирателя с
поверхностью. Благодаря этому общая свободная энергия всего
поверхностного слоя уменьшится, т. е. процесс закрепления
собирателя и гидрофобизация поверхности не противоречат
второму закону термодинамики. Высказанные соображения
иллюстрируются рис. 2.6, на котором изображено примерное
изменение плотности поверхностной свободной энергии на участках»
покрытых собирателем (кривая 2) и свободных от него
(кривая /). Благодаря взаимодействию полярной группы
собирателя с минералом общая поверхностная энергия всей системы
уменьшилась (площадь под кривой 2 меньше площади под
кривой 1). В то же время слабая связь аполярных групп
собирателя с водой приведет к созданию гидрофобной поверхности и
уменьшению смачиваемости твердого тела. Интенсивность
взаимодействия твердой и жидкой фаз можно оценить
калориметрически по теплоте иммерсионного смачивания [4, 46].
В присутствии капелек аполярного масла их закрепления на
поверхности гидрофильных минералов не происходит, если
такой процесс сопровождается увеличением свободной энергии
системы в результате замены поверхности контакта полярный
минерал — вода с малым значением стж-т (из-за малой
разности в полярности соприкасающихся фаз) поверхностью раздела
25
полярный минерал — аполярное масло с большим значением
Ож-т (из-за большой разницы в полярности этих фаз). По этой
же причине не должно происходить выделения на поверхности
гидрофильных минералов микропузырьков воздуха.
Закрепление капелек аполярных масел и выделение
микропузырьков воздуха может наблюдаться только на поверхности
достаточно гидрофобных минералов. В этом случае уменьшение
свободной энергии поверхности раздела масло — вода (или
воздух— вода) на площади контакта капельки или пузырька с
минералом превышает возрастание свободной энергии при замене
поверхности контакта минерал — вода поверхностью минерал—
масло (или минерал — воздух), т. е. сопровождается
уменьшением свободной энергии всей системы. Полное растекание
капелек аполярного масла или пузырьков воздуха на
поверхности известных минералов в воде невозможно, так как
полярность их поверхности всегда больше полярности масла или
воздуха [4]. * .
Уменьшением свободной энергии системы сопровождается
также слипание минеральных частиц в воде. По предложению
П. А. Ребиндера, все процессы слипания частиц чисто условно
можно разделить по механизму их протекания на флокуляцию
и коагуляцию [7, 15].
Флокуляция происходит в результате сцепления
(взаимодействия) аполярных групп гетерополярного
реагента-собирателя или углеводородных цепей аполярного масла,
закрепившихся на поверхности слипающихся природногидрофобных
или гидрофобизированных собирателем частиц. Она
протекает тем интенсивнее, чем гидрофобнее поверхность частиц и
больше выигрыш в свободной энергии системы при их
слипании.
Коагуляция может происходить в результате
взаимодействия поверхности даже гидрофильных минералов с
неорганическими электролитами. Она усиливается с возрастанием
ионной силы раствора (при подаче электролитов), вызывающей
сжатие диффузного слоя наружной обкладки ДЭС (см.
рис. 2.3, а) настолько, что частицы могут сблизиться до
расстояния, при котором становится возможным проявление сил
межмолекулярного взаимодействия между их поверхностями.
Естественно, что коагуляционные явления могут сопутствовать
флокуляциЪнным.
2.2. Изменение энергии прослоя воды между пузырьком
и частицей при элементарном акте флотации
Результаты исследований* А. Н. Фрумкина и Б. В. Дерягина
показали, что характер изменения удельной поверхностной
энергии е прослоя воды, находящегося между твердой и
газообразной фазами, при изменении его толщины h зависит от
степени гидрофобности твердой поверхности (рис. 2.7).
26
Рис. 2.7. Изменение плотности е е
свободной поверхностной энергии
прослоя воды между твердой
поверхностью частицы и пузырьком
воздуха при их сближении
Наиболее общий случай характеризуется кривой 2 (см.
рис. 2.7). В начальный период сближения пузырька и частицы
вода из прослоя удаляется легко и, пока гидратные слои
частицы и пузырька не пришли в соприкосновение, поверхностная
энергия прослоя будет оставаться постоянной и равной сумме
значений поверхностной энергии на границах раздела вода —
газ и вода — твердое: е = 0г-ж-гОж-т* С момента
соприкосновения гидратных слоев при толщине прослоя h\ дальнейшее
сближение пузырька и Частицы невозможно без дополнительной
работы на преодоление расклинивающей силы, равной величине
энергетического барьера е$.
В соответствии с теорией устойчивости лиофобных
(гидрофобных) коллоидов (известной как теория ДЛФО D]) и с
учетом природы межмолекулярных взаимодействий Б. В. Дерягин
выделяет три слагающие расклинивающего давления p(h) —
электростатическую pe(h), молекулярную pyi(h) и структурную
Pc(h):
P(h) = pe(h)-Pu(h) + Pc{h).
Составляющая pe{h) возникает в результате перекрытия или
деформации двойных электрических слоев, расположенных на
границах раздела жидкость — газ и жидкость — твердое, и
появления электрических зарядов. Составляющая pM{h)
характеризует притяжение в результате молекулярного взаимодействия
(дисперсионными силами) молекул водной прослойки как с
поверхностью твердого, так и между собой. Составляющая pc(h)
зависит от степени гидратированности (гидрофобности или
гидрофильное™) поверхности и проявляется, если взаимодействие
молекул воды между собой меньше, чем их взаимодействие
с поверхностью твердого тела [4, 7, 49].
Энергетический барьер уменьшается, если потенциалы
частиц и пузырьков сильно отличаются или их поверхности
противоположно заряжены, уменьшается радиус действия
электростатических сил (за счет сжатия диффузной части ДЭС при
увеличении ионной силы раствора в присутствии электролитов),
27
увеличивается степень гидрофобное™ частиц и уменьшается
толщина гидратных слоев на их поверхности. Наоборот, он
будет возрастать с уменьшением степени гидрофобности частиц и
с увеличением толщины гидратных слоев на их поверхности,
с ростом значений потенциалов при одноименном заряде
поверхности частиц и пузырьков [7, 49].
Преодоление энергетического барьера при толщине прослоя
h2 (см. рис. 2.7) вызывает перестройку гидратных слоев,
которая может сопровождаться их разрывом. Возникновение притя-'
жения между пузырьком и частицей с одноименными, но
различными по значению потенциалами является следствием
взаимодействия двойных электрических слоев на их поверхности.
В результате деформации диффузных частей ДЭС после
преодоления энергетического барьера происходит слияние их в один
диффузный слой одного знака, который притягивается с обоих
сторон к одноименно заряженным поверхностям другого
знака [7].
Сближение фаз до толщины /i3 происходит самопроизвольно
с уменьшением удельной свободной поверхностной энергии до
значения ек. Причем при толщине прослоя, равной Лз,
поверхностная энергия системы наименьшая и система находится в
состоянии устойчивого равновесия.
Прослой воды между газообразной и твердой фазами,
соответствующий минимальному значению поверхностной энергии
при закреплении частицы на поверхности раздела вода — газ,
называется остаточным гидратным слоем. Толщина его
изменяется от 1 до 150 нм. Он устойчиво связан с твердой
поверхностью и удаление его при дальнейшем сближении
пузырька и твердой поверхности приводит к значительному
возрастанию свободной энергии и сопряжено с большими
затратами внешней энергии (см. рис. 2.7).
Зависимость e=f(h) для гидрофильной поверхности твердой
фазы изображена кривой / (см. рис. 2.7). В этом случае при
сближении твердой и газообразнойkфаз при толщине прослоя
меньше суммарной толщины гидратных слоев пузырька и
частицы все время действует расклинивающая сила и
закрепление частицы на поверхности вода — газ невозможно.
Кривая 3 на рис. 2.7 соответствует полностью гидрофобной
поверхности твердой фазы. В этом случае энергетического
барьера нет и при толщине прослоев воды меньше суммарной
толщины гидратных слоев пузырька и частицы h\ \ но больше
hz между сближающимися фазами действуют силы
притяжения. На всем участке от h\ до h/ наблюдается
самопроизвольное сближение фаз. Толщина остаточного гидратного слоя после
закрепления частицы равна Л3'.
На практике такой случай реализуется редко даже для
минералов, обладающих естественной гидрофобностью, так как
чаще всего частица и пузырек воздуха заряжены одноименно.
В природе больше всего гидрофильных минералов, для кото-
28
рых зависимость e=f(h) характеризуется кривой /, и флотация
возможна только после предварительной гидрофобизации их
поверхности флотационными реагентами.
Рассмотренные закономерности изменения энергии прослоя
воды между пузырьком и частицей при элементарном акте
флотации справедливы и для случаев коалесценции пузырьков и
капелек аполярных реагентов, коагуляции и флокуляции
частиц, закрепления капелек гетерополярных и аполярных
реагентов на поверхности твердого и для других аналогичных
случаев.
2.3. Краевой угол смачивания и зависимость его
от удельной поверхностной энергии на границе
соприкасающихся фаз
Способность минералов закрепляться на поверхности
раздела воздух — вода (или в общем случае газ — жидкость)
зависит от степени полярности минеральной поверхности, энергии
взаимодействия ее с молекулами воды (жидкости) и
смачиваемости водой (жидкостью).
Гидратация поверхности минерала происходит, если энергия
взаимодействия ее с молекулами воды — энергия адгезии Wa —
больше энергии взаимодействия молекул воды друг с другом,
т. е. энергии когезии WK. Чем больше отношение WJWKj тем
лучше минерал смачивается водой и соответственно тем хуже
флотируется.
Смачиваемость поверхности твердого при соприкосновении
трех фаз (т, ж, г) характеризуется величиной равновесного
краевого угла, который представляет собой угол,
образованный поверхностью раздела двух фаз с поверхностью
третьей фазы. Его принято измерять через жидкую фазу и
обозначать символом 6р. Краевые углы смачивания тел различной
степени гидрофобности водой при различных положениях
соприкасающихся фаз приведены на рис. 2.8. Образующаяся
линия трехфазного контакта называется периметром
смачивания. Особенностью его является наличие трех сил
(обусловленных свободной поверхностной энергией на границе
раздела фаз), векторы которых направлены перпендикулярно
линии контакта трех фаз по касательной к поверхности раздела
каждых двух фаз.
Равновесный краевой угол 0Р является физико-химической
константой для соприкасающихся фаз и не зависит от их
размера и взаимного расположения, действия сил гравитации и
прочих факторов, не оказывающих влияния на значения
свободной поверхностной энергии на границе раздела фаз.
Равновесный краевой угол определяется условием равновесия сил,
действующих на границе раздела трех фаз:
бр = /(<7г-т, <ТЖ_Т, <ТГ_ЖХ.
29
Рис. 2.8. Равновесные краевые углы смачивания Gp тел (т) различной
степени гидрофобности водой (ж) и воздухом (г)
Например, для острого краевого угла (см. рис. 2.8, а) в
условиях равновесия аг-т = <Ьк-т + <Хг-ж cos0p, откуда
cos 0р = (ог_т—аж_т)/аг
B.6)
В свою очередь, для тупого краевого угла (см. рис. 2.8,6)
в условиях равновесия аж -т = <Хг-т + (Tr-Hccos A80—0Р) —
= 0r-T — (jr_mcos0p, откуда
cos 0Р = (<тг_т—аж_т)/аг_ж.
Для обоих рассмотренных случаев зависимость 0Р =
= /(аг-т, сгж_т, ащ_г), характеризующая смачиваемость твердой
поверхности жидкостью или газом, описывается одним и тем
же выражением, которое в литературе называется -законом
Юнга, правилом Неймана или вторым законом капиллярности
[4, 46].
Чем меньше смачиваемость минеральной поверхности водой,
тем больше степень ее гидрофобности и значение краевого угла
0Р. Абсолютно гидрофобные тела неизвестны. Наибольшей гид-
рофобностью из всех известных тел обладает парафин, краевой
угол которого равен 112°. В подавляющем большинстве случаев
практика флотации имеет дело с минералами, значения краевых
углов на поверхности которых меньше 90°. Такие же значения
наблюдаются и у так называемых краевых углов
избирательного смачивания, если вместо пузырьков воздуха или газа ис*
пользуются капельки аполярных масел [4].
2.4. Гистерезис смачивания ,
Явление гистерезиса, затрудняющего достижение
равновесного состояния, присуще многим физическим и
физико-химическим процессам, в том числе процессу образования
равновесного краевого угла, соответствующего минимальному значению
свободной энергии системы.
При перемещении периметра смачивания по поверхности
твердого тела обычно возникает противодействующая ему гисте-
30
а &г-ж
Рис. 2.9. Гистерезис смачивания твердого тела {г) водой (ж) и
воздухом (г)
резисная сила. Например (рис. 2.9,а), если жидкая фаза
наступает на газообразную, то возникает гистерезисная сила г|)ь
направленная внутрь жидкой фазы. Если же наступает
газообразная фаза на* жидкую, то возникает гистерезисная сила ^2,
направленная внутрь газообразной фазы. Гистерезисные силы ifi
и *|?2, возникающие при наступлении жидкой фазы на
газообразную и газообразной на жидкую,' неодинаковы. Различны и
образующиеся при этом гистерезисные краевые углы натекания
6Н и оттекания 0О (см. рис.2.9, а).
При наступлении жидкой фазы на газообразную при
некотором значении гистерезисного краевого угла натекания 9Н
наступит равновесие сил
<?ж-т + <?г-ж CQS 9н + ^1 = <*г-т, *
откуда
cos 6Н = (аг_т—0ж_т)/аг_ж—^/Ог-ж ^ cos 6р—Var->K.
Из последнего уравнения следует, что cos6H<cos0p и
8н>6р, т. е. при наступлении жидкой фазы на газообразную
гистерезисный краевой угол больше равновесного.
Аналогично выражение для гистерезисного краевого угла
оттекания 80 найдем из условия равновесия:
<*г-т + *Ф2 -=" <Ьк-т + <Уг-ж COS 60;
cos 0О = (а, _т—аж_т)/аг^ж + ф2/<Тг-ж = cos 9р +<ф2/сГг~ж.
Следовательно, cos0o>cos0p и 0о<0р, т. е. при наступлении
газообразной фазы на жидкую гистерезисный краевой угол
меньше равновесного.
Разность косинусов гистерезисных углов называют (по
П. А. Ребиндеру) гистерезисом смачивания
Ы ---- cos 80—cos 0н = cos 8Р +г|У<7г_ж—(cos 8Р—г|Уаг_ж) =
= (Ф2 + ЪУ<Тг-«. B-7)
Физико-химические причины, вызывающие гистерезис
смачивания, связаны в основном с формированием прграничных (гид-
31
ратных) слоев жидкости на поверхности раздела фаз при
наступлении водной фазы на газообразную и разрушением их при
наступлении газообразной фазы на водную. При этом
наблюдаются следующие закономерности: на гладких поверхностях
гистерезис меньше, чем на шероховатых; на более гидрофобных
поверхностях при наступлении водной фазы он больше, чем на
более гидрофильных; при наступлении жидкой фазы на
газообразную гистерезис больше, чем при наступлении газообразной
фазы на жидкую.
Гистерезисный краевой угол при закреплении пузырька на
горизонтальной поверхности твердого называют иногда углом
контакта 0К. В этом случае (рис. 2.9, б)
cos 0К = (аг_т—0ж_т)/ог_ж + 1|>/аг_ж = cos ер + ^аг_ж ■= cos 0Р + Л,
B.8)
где h — гистерезис смачивания.
Значения Ь! и h могут несколько различаться, так как в
уравнении B.7) используются крайние значения гистерезисных
углов 0Н и 0о, а в уравнении B.8) —постоянное вдоль всего
периметра контакта значение угла 0К, что более предпочтительно
[46].
Поскольку гистерезис смачивания (hr или h) определяется-
значениями гистерезисной силы (t|?, ty\ или я|J) и аг-ж> он
является чувствительным индикатором изменений, происходящих
на поверхности твердого тела и пузырька под влиянием
флотационных реагентов и внешних воздействий.
В результате образования по периметру контакта пузырька
с частицей легко подвижной каймы, например, аполярного
реагента и адсорбционной пленки его на поверхности пузырька
гистерезис смачивания обычно полностью исчезает [22, 23, 46].
Однако в этом случае вместо одной появляются три линейные
границы раздела, поверхность пузырька до подложки не
доходит и угол 0 характеризует наклон поверхности пузырька к
горизонту на уровне верхнего края каймы (рис. 2.9,б). Поскольку
в присутствии аполярного реагента значения аж-т уменьшаются,
это приводит, в соответствии с уравнением B.3), к увеличению
р (уплощению пузырька) и росту 0 (если к этому нет каких-
либо препятствий) без увеличения степени гидрофобности
поверхности.
Для вычисления значений краевого угла, независимо от того,
каким он является — гистерезисным или равновесным,
рационально пользоваться при проведении исследований уравнениями
B.2) и B.3), точно описывающими контуры любых капель и
пузырьков; при этом очевидно, что 0 = 180° — ср (см. табл. 2.1).
32
2.5. Термодинамический анализ возможности
элементарного акта флотации.
Показатель флотируемости
В соответствии со вторым законом термодинамики
элементарный акт флотации возможен, если свободная энергия Е2
системы после закрепления частицы на пузырьке меньше
свободной энергии Ei системы до закрепления частицы. В этом случае
система из I состояния (рис. 2.10) самопроизвольно переходит
во II состояние при условии, что на пути перехода нет
энергетического барьера или если системе временно сообщена энергия
(энергия активации), достаточная для его преодоления. Чем
больше разница в значениях свободной энергии в сравниваемых
состояниях, тем более вероятен переход в состояние с- меньшей,
энергией.
При принятых на рис. 2.10 обозначениях (S — площадь
контакта)
h\ = стг_ж ог_ ж ~\~ 0Ж_ т *Ьж—т»
Ео — 0р—ж "S г—ж
изменение поверхностной энергии системы при элементарном
акте флотации
Ь\ — Ь*2 = О г_ж(ог_ж""""*-> г-ж) т ^ж-т (<Ьж-т ^ ж—т) <Тг_*г Ог—Т.
Из рис. 2.10 видно, что 5Ж_Т — S/jK_T = Sr-T, тогда как
разность Sr-ж — S'r-ж нельзя принять равной Sr-r вследствие
деформации пузырьков, особенно маленьких, при закреплении на
них минеральных частиц.
Поэтому
Ех £2 — ^г—ж (^г—ж «Ь г—ж)~Г (<*ж—т~~~ <Тг—т) *Ьг—т.
Учитывая, что в равновесных условиях, по правилу Неймана
[выражение B.6)] ат-т — аг-т=—Яг-жсоэвр, получаем:
Ъ\ Ь2= <*г—ж («Ьг—ж <Ь г-ж) <*г—ж COS Op Or_T.
Разделив это выражение на Sr_T и обозначив
(Ei—£2)/5г-т = /7, найдем
F = (E!-E2)/Sr-T - ог_ж{ [(Sr-ж—S';_»)/Sr_T] —cos 0P}. B.9)
Величина F, характеризующая изменение поверхностной
энергии системы при элементарном акте флотации, отнесенное
к единице площади контакта газ — твердое, называется
показателем флотируемости. Система перейдет из I
состояния во II состояние (см. рис. 2.10) только при условии, что
F>0 (т. е. Ei>E2). Чем больше значение F, тем вероятнее
закрепление частицы на поверхности раздела жидкость — газ и ее
флотация.
2 Заказ № 1957 33
ж
Й"
$ж-т
I состояние Е состояние Шсостоянаё
Рис. 2.10. Схема состояния системы до- и после закрепления частицы на
пузырьке
При закреплении на пузырьках минеральных частиц,
размеры которых малы по сравнению с размерами пузырьков (что
наблюдается при обычной пенной флотации), т. е. если
деформация пузырьков мала и можно принять, что EГ_Ж —
— S'r-ж)/5г-т~ 1, выражение B.9) принимает вид
F = Gr^(l— COSBp)-,
B.10)
откуда следует, что, чем больше краевой угол, тем больше
показатель флотируемости. При 0Р = О показатель флотируемости
также равен нулю.
Разница в значениях краевого угла и показателя
флотируемости может привести к различному положению частицы на
поверхности раздела фаз (см. рис. 2.10): II состояние системы
соответствует так называемой «мокрой» флотации, III —
«сухой» флотации. Различаются они между собой только
значениями поверхностной энергии боковых граней (площадью Sq)
Частицы, т. е.
fc\ — С2 = СГЖ_Т Об—СГг—т «Ьб — Об (СГж-т — ®г—т).
Учитывая, что по правилу Неймана [выражение B.6)] ат-Т —
— аг_т= —аг-ж cos 0p, получаем
Е%—Е2 = — Se сТг-ж cos 0p
B.11)
Если краевой угол острый (О°<0Р<9О°), то на основании
уравнения B.11) Е\ — Е2<0 и Е\<Е%, т. е. переход из II
состояния в III (см. рис. 2.10) невозможен и возможна только
«мокрая» флотация.
При тупом краевом угле A8О°>0Р>9О°) на основании
уравнения B.11) Ех — Е2>0 и £i>£2. Поэтому система
самопроизвольно перейдет из II состояния в III и частица займет
положение «сухой» флотации (см. рис. 2.10).
34
2.6. Влияние пузырьков газа, выделяющихся
из раствора, на показатель флотируемости
При работе флотационной машины в пульпе создаются
значительные перепады давления. Поэтому, в соответствии с
законом Генри, в зонах повышенного давления происходит
растворение воздуха, а в зонах пониженного давления — его
выделение в первую очередь, как показано А. Ф. Таггартом, на
гидрофобной поверхности флотируемого минерала. Закрепление
частицы на пузырьке в этом случае будет сопровождаться коа-
лесценцией (слиянием) выделившегося на ее верхней грани
малого пузырька с большим (рис. 2.11). С учетом принятых на
рис. 2.11 обозначений можно записать:
Ei =■ аг_ж ог„ж + стг_ж оп + аж-т S ж-т + ®г-т *^м ~Ь А;
Jz 2 := аг_ж о г_ж + ог_т о г_т ~\~ Л,
где А — поверхностная энергия нижней и боковых граней,
которая не изменяется при закреплении частицы.
Отсюда
Ei—Е2 = Ог-ж (Sr—ж 4" Sn — о г-ж) + ^ж-т,Ь ж-.т + аг_т(ом ог_т).
B.12)
Учитывая, что SV-T=Sr-T— 5М, а также, что деформация
большого пузырька и изменение его объема при закреплении на нем
малой частицы незначительны, можно принять SV-ж—Sr-ж—
— Sr-T. После преобразований получаем
Ei — £2 = ^г-ж (Sn + 5r_T) + (°ж-т—аг_т)(ог_т- SM).
Так как на основании уравнения B.6) аж-т — ai—т —
= —ог-ж cos ер,
Ег—Е2 -- сгг-ж A —cos 0P) S^ + аг__ж (Sn + SM cos0P),
а показатель флотируемости для частицы, покрытой
пузырьками воздуха,
FB = (Ек — E2)/Sr„T = ог_ж A — cos 0р)+аг_ж (S„ + SM cos ep)/SM.
Первый член правой части уравнения для FB является
показателем флотируемости частицы, не покрытой выделяющимися
пузырьками газа [выражение B.10)], поэтому можно написать
Fh = F + *г-я (Sn + SM cos 6p)/Sr^T. B.13)
Из последнего уравнения следует, что выделение малых
пузырьков растворенного воздуха на поверхности частичек
увеличивает показатель их флотируемости. Это увеличение тем
больше, чем больше поверхность Sn пузырьков, выделившихся
на гранях флотируемых частиц. Сначала И. Свен-Нильсон
A937 г.), а затем В, И. Классен и др. [7, 15] экспериментально
2* 35
^—^^ *%-#<: Рис. 2-П. Схема состояния системы
л. до и после закрепления активиро-
г \ ванной микропузырьками частицы
] на пузырьке
Sb-гп J
о У
q m b
I состояние Л состояние
показали, что наличие микропузырьков на поверхности
минеральных частиц «катализирует» их прилипание к поверхности
более крупных пузырьков газа.
2.7. Показатель флотируемости при коалесцентном механизме
элементарного акта флотации
Коалесцентный механизм элементарного акта наблюдается
при флотации аполярных минералов с углеводородными
маслами. По данному механизму частичка, на гранях которой
имеется одна или несколько капелек масла, закрепляется при
столкновении с пузырьком, покрытым слоем масла, в результате
коалесценции капелек масла (рис. 2.12).
Формулу для показателя флотируемости FK коалесцентного
механизма элементарного акта, характеризующего уменьшение
поверхностной энергии системы на единицу площади контакта
газ — масло — твердое, можно получить, по К. А. Разумову,
следующим образом [36*].
На основании принятых на рис. 2.12 обозначений:
Ег = (TM_rSn + <*м- в«$п + а*-А + ^m-tSt -f ав_т (q — ST) + А;
£2 = ам-Л + стм_в5п + ам^+амН^ + Л.
Для малых по сравнению с пузырьком частиц деформация
пузырька при их закреплении незначительна и ею можно
пренебречь. Тогда Sn=Sn — q и после простых преобразований
получим
Ег-Е2 = о^в(д ^SM) + (aB^~aM.r)(q-ST). B.14)
Поскольку система вода — твердое — масло является
аналогом системы вода — твердое — воздух, в которой газовая фаза
заменена маслом, на основании правила Неймана [выражение
B.6)] можем записать:
где у — краевой угол на границе масло — вода — твердое.
$ж-т
>г-ж
1У°"
т
зб
Рис. 2.12. Схема состояния системы
до и после коалесцентного
закрепления частицы на1 пузырьке
/ состояние Л состояние
Подставляя найденное значение ав-т — сгм-т в выражение
B.14), получаем:
Ei—E2 = eM-.B(q + Su)—cr„_BcosY(?—ST);
Ег — Ег = аы_ъA — cosy) q +<j^b(Sm + ST cosy).
Первый член этого уравнения положителен, если y>0;
второй член положителен при всех значениях у, так как SM>ST.
Поэтому, если y>0, то Ех—Е2>0 и Е\>Е2. Следовательно,
система из I состояния перейдет во II, если поверхность минерала
хотя бы частично смачивается маслом (у>0).
Показатель флотируемости для коалесцентного механизма
закрепления будет равен
FK^(E1 — £2)/<7^0w_B(l— cosy) +oM_B(SM + ST cos y)lq. B.15)
При одном и том же значении первый член уравнения
B.15) остается для данного масла постоянным, второй же
возрастает при увеличении числа капель масла в единице объема
пульпы вследствие увеличения вероятности встречи капелек
с частицей и их числа на частице. Повышение дозировки масла
и степени его дисперсности приводит к увеличению показателя
флотируемости до тех пор, пока закрепившиеся на минеральной
поверхности капельки масла не начнут сливаться друг с
другом.
3. ФИЗИЧЕСКИЕ ОСНОВЫ ФЛОТАЦИИ
3.1. Условие равновесия частицы на плоской
поверхности раздела газ — жидкость
при пленочной флотации
При пленочной флотации или флотогравитации (рис. 3.1, а)
частица удерживается на плоской поверхности раздела газ —
жидкость флотационной силой Вф, под которой будем понимать
37
\Fm*Fb
tfi^B -
Рис. 3.1. Флотация, частицы на плоской поверхности раздела жидкость—газ
вертикальную составляющую сил поверхностного натяжения на
границе раздела жидкость — газ, приложенных к частице по
всему периметру Р смачивания:
^ф^Раж_г sin в, C.1)
где 8 — угол наклона поверхности жидкости к горизонту у
периметра трехфазного контакта.
Второй силой, направленной вверх, является сила Fr,
обусловленная гидростатическим давлением на нижнюю грань
частицы и равная, если пренебречь величиной h (см. рис. 3.1, а)у
весу вытесненной жидкости плотностью бж:
/^Vfirf, C.2)
где V — объем частицы; g— ускорение свободного падения.
Отрывают частицу плотностью бт от поверхности
направленные вниз внешняя сила FB и сила тяжести
Fv = V6rg. C.3)
В условиях равновесия
^ф + Fx — Ft + Ли
или с учетом выражений C.1) — C.3)
Раж_г sin 6 = V FТ— бж) g + FBt
т. е. сумма веса частицы, погруженной в жидкость, и внешней
отрывающей силы равна флотационной силе.
Критическое значение внешней силы FKJ?, вызывающей отрыв
частицы, характеризует прочность ее
закреплен и я:
FKp = Р<*ж-т sin О—V FТ—6Ж) g.
Если отрыв частицы происходит под действием только силы
тяжести, то FKV = 0 и
>Ma«-rSine—^(&г—6«)g-0, C.4)
где Рм — периметр, a VM — объем максимальной частицы, или
ф/^Q, C.5)
где Q — вес частицы в жидкости.
38
По уравнению C.4), впервые полученному С, Валентинером
[8], можно определить максимальный размер частицы, способной
удержаться на плоской поверхности раздела жидкость — газ.
Например, для частицы кубической формы PM = 4dMy Vm = ^m и
на основании выражения C.4)
dM - }Лаж_г sin 8/1F,-6Ж)£]. C.6)
Максимальная крупность зерен сульфидных минералов,
рассчитанная по формуле C.6), практически совпадает с
наблюдаемой при флотогравитационном обогащении черновых
оловянных и вольфрамовых концентратов [36]. Например, для
галенита при 6Т = 7,5- 103 кг-м, ож-г = 72,8 мН-м-1, 0 = 60° она
составляет
dM - ]А.72,8sin 60/G,5— 1,0IQ-3-9,81 = 0,0021 м = 2,1 мм.
Максимальная крупность зерен пирита в этих условиях
достигает 2,6 мм, а угольных частиц 8,6 мм.
Условие флотационного равновесия, записанное П. А. Ребин-
дером на основании уравнения C.4) для частиц произвольной
формы [46]
/У С | = I Яаж-гС05(ек + ф')/Fгф —6«)g |, C.7)
поскольку sin 9 = —cos @K+q/)> где q/ —угол формы (рис. 3.1,6)
позволяет оценить влияние различных факторов на процесс
пленочной флотации. Например, оно показывает, что [46]:
при уменьшении размера частиц соотношение F<f>/Q резко
увеличивается для всех частиц и поэтому переизмельчение
должно приводить к ухудшению селективности их разделения;
флотируемость частиц чешуйчатой и игольчатой форм
должна быть лучше флотируемое™ зерен изометрической
формы, имеющих меньшее значение периметра смачивания при
том же весе;
способствовать пленочной флотации и флотогравитации
частиц будут флотационные реагенты, увеличивающие гидрофоб-
ность их поверхности и гистерезис смачивания, но не
снижающие существенно значения сгж_г;
флотируемость частиц при прочих равных условиях
пропорциональна гидрофобности их поверхности и гистерезису
смачивания на ней, который может быть обусловлен, в частности,
острыми ребрами частиц.
Устойчивость закрепления частиц при пленочной флотации
или флотогравитации в практических условиях обеспечивается
трудной подвижностью трехфазного периметра контакта и
гистерезисом краевого угла 8, благодаря которому 'различные
волнения на поверхности жидкости, достигая периметра
контакта с частицей, не приводят к его перемещению, уменьшению
значений Р, F$ и отрыву частицы от поверхности,
а.компенсируются ростом угла 9 от 8' до 0" без изменения периметра кон-
39
такта (рис. 3.1,б). Флотационная сила Гф при этом временно
возрастает, удерживая частицу на поверхности раздела газ —
жидкость.
3.2. Факторы, влияющие на минерализацию пузырьков
и пенную флотацию частиц
Пенная флотация осуществляется в условиях турбулентного
(вихревого) движения пульпы, так как при ламинарном ее
движении частицы минералов стали бы осаждаться на дно
флотационной машины. При этом пульпа вместе с увлекаемыми
частицами и пузырьками перемещается по криволинейным
траекториям, что вызывает появление центробежных сил, под
действием которых пузырьки воздуха, имеющие плотность меньше
плотности пульпы, начинают двигаться в пульпе от периферии
вихря к его центру и одновременно всплывать. Частицы
минералов, плотность которых больше плотности пульпы, наоборот,
двигаются в пульпе от центра вихря к периферии и
одновременно тонут под действием силы тяжести. Противоположное
движение частицы и пузырька приводит к их встрече и
закреплению частицы на пузырьке.
Общую схему векторов ускорений и сил, действующих на
частицу при ее закреплении на пузырьке и криволинейном
движении минерализованного пузырька в пульпе, по К. А. Ра-
зумову [36], можно представить следующим образом.
Скорости пузырька и частицы до ее закрепления на
пузырьке складываются из скорости переносного движения
пульпы и скорости относительно движения их в пульпе. Так
как центробежные силы, возникающие при вихревом движении
пульпы, намного больше сил тяжести, то вертикальные
составляющие относительных скоростей движения пузырьков и частиц
малы по сравнению с радиальными составляющими этих
скоростей. Поэтому скоростями всплывания пузырьков и падения
частиц можно пренебречь и учитывать только скорости
радиального движения пузырьков к центру вихря, а частиц — к
периферии.
Вследствие относительного движения пузырька пульпа
обтекает его поверхность по линиям а—а (рис. 3.2), и частица
после столкновения с пузырьком (положение Л)' начинает
скользить по его поверхности к кормовой части. Вектор абсолютной
скорости скользящей частицы равен геометрической сумме трех
векторов: скорости vn переносного движения пульпы, скорости
vR относительного (радиального) движения пузырька в пульпе
и скорости v0 относительного движения частицы по пузырьку.
Вектор абсолютного ускорения частицы будет также равен
геометрической сумме трех векторов: ускорения переносного
движения пульпы; ускорения относительного движения
(скольжения) частицы по поверхности пузырька и ускорения Кориолиса,
направленного перпендикулярно к относительной скорости ча-
40
Рис. 3.2.
пузырьке:
R — радиус
зырька
вращения пульпы и
пу-
стицы в пульпе и
возникающего благодаря силе Кориоли-
са, обусловленной движением
тела относительно
вращающейся системы.
Предполагается, что скорость радиального
перемещения пузырька
постоянна, поэтому ускорение
относительного движения
пузырька в пульпе равно нулю.
При работе механической
флотационной машины
ускорение относительного
движения частицы по пузырьку во
много раз больше ускорения
переносного движения и кори-
олисова ускорения [36].
Поэтому в первом приближении
можно считать, что
абсолютное ускорение частицы равно ускорению скольжения частицы
по пузырьку и направлено от центра тяжести к центру
пузырька.
Отрывают частицу от пузырька центробежная сила Fi и
сила, обусловленная давлением воздуха F^ на контактирующую
с пузырьком грань частицы. Последняя равна сумме сил,
обусловленных гидростатическим давлением пульны на уровне
точки Л, давлением лобового сопротивления пульпы в точке Д,
возникающим вследствие движения пузырька в пульпе, и
капиллярным давлением вследствие поверхностного натяжения и
кривизны пузырька в точке А (см. рис. 3.2).
Препятствует отрыву частицы от пузырька флотационная
сила Рф и сила FM, обусловленная давлением пульпы на
нижнюю, обращенную в сторону пульпы грань частицы. Последняя
определяется разницей между гидростатическим давлением
пульпы на уровне нижней грани частицы и понижением
давления, обусловленным движением пузырька и частицы в пульпе.
В условиях равновесия сумма сил, отрывающих частицу от
пузырька и препятствующих ее отрыву, будет равна, т. е.
Fcp + F{i — Ft + FB.
Чем больше сдвинуто равновесие в сторону преобладания
сил прикрепления, тем прочнее контакт минеральных частичек
с пузырьком.
Механизм минерализации пузырьков при флотации
отличается большой сложностью. В процессе флотации участвуют
миллионы пузырьков и частиц и вероятность флотации каждой из
них зависит от вероятности отдельных этапов этого процесса.
К ним относятся: взаимодействие частиц с реагентами,
столкновение их с пузырьками", закрепелние на пузырьках, действие
41
Количество | I Время нахождения.
пузырьков ' ' ао
Вероятность
стомнобения
частиц с
пузырьками
Размеры
пузырьков
Интенсивность переме-
\шиванш■? пульпы
платность
и размерь/
частиц
Плотность
пульпы
^Вероятность
прилипания
i при
Столкновении
z
Вероятноещ
\оораз$ан1/я
минерализованного
пузырька
Условия
\столкновения
{Процессы в
\пенном слое
Фярма
частиц
| Гиёратцрован-
\ность паверхнос-
7и частиц
\Вывеление пузырь -
ков из раствора
Ферма а?регатов
минерал-пузырен
Природа поверх-
ности частиц
Сорбционный
слой реагентов
Рис. 3.3. Главные
В. И. Классену [15])
факторы, определяющие вероятность флотации (по
отрывающих сил, условия образования и разрушения сложных
аэрофлокул, процессы, происходящие в пенном слое, и "т. д.
В реальных условиях каждый этап осложняется тем, что в нем
участвуют частицы разных минералов, различающиеся
свойствами поверхности, формой, плотностью, размерами и др.
Поэтому взаимосвязь явлений единого флотационного процесса
весьма сложна и многообразна. Об этом наглядно
свидетельствует схема главных факторов, определяющих вероятность
флотации (по В. И. Классену), приведенная на рис. 3.3.
Вероятность Шф флотации рассматривается О. С.
Богдановым [44] и рядом других исследователей как произведение
вероятностей основных слагающих.событий: столкновения частицы
с пузырьком шс, закрепления на пузырьке шзакр, сохранения
частицы на пузырьке до выхода в пенный слой wCOxp и удержания
ее в пене до съема в концентрат wf.
Шф = ШсШзакр^сохр^.
Под столкновением понимается сближение пузырька и
частицы до расстояний, когда начинают преобладать не
гидродинамические, а поверхностные силы. Вероятность wc
характеризуется отношением массы частиц, столкнувшихся с
пузырьками в единицу времени, к общей массе частиц в пульпе.
42
Под закреплением понимается проявление
поверхностных свойств частицы и пузырька, заканчивающееся
образованием трехфазного контакта между ними. Вероятность ш>3акР
характеризуется отношением массы закрепившихся на пузырьках
частиц к массе частиц, столкнувшихся с пузырьками.
Под сохранением закрепившихся на пузырьках частиц
понимается сохранение трехфазного контакта между ними до
выхода в пенный слой. Вероятность шСохР характеризуется
отношением массы частиц, вынесенных в пену, к массе частиц,
закрепившихся на пузырьках.
Вероятностью ш/ удержания частиц в
пене.называется отношение массы частиц, извлеченных в концентрат,
к массе частиц, вынесенных в пену.
Чем больше вероятность каждого из перечисленных
событий, тем больше вероятность х&ф пенной флотации.
3.3. Вероятность столкновения частицы с пузырьком
Вероятность wc столкновения частиц с пузырьком зависит
от их скорости движения, количества, формы и размера; на нее
влияют также плотность частиц, вязкость и плотность среды.
Столкновение частицы с пузырьком определяется в
основном гидродинамическим режимом, для характеристики которого
используются: число (параметр) Рейнольдса Re,
характеризующее режим движения частицы и пузырька; число (критерий)
Стокса St, характеризующее инерционность частицы, и иногда
критерий Фруда Fr, характеризующий соотношение между
инерционными факторами, с одной стороны, и влиянием эффекта
закрепления и седиментации — с другой.
Вследствие увлечения частиц током жидкости (см. рис. 3.2)
с пузырьком' столкнется лишь часть частиц, двигающихся в
цилиндрической трубке, радиус поперечного сечения которой,
(площадью а0) равен сумме радиусов пузырька R и частицы г.
Наибольшее сечение (площадью ос) трубки радиусом /, из
которой все частицы осаждаются на пузырек, называется
сечением столкновения.
Отношение числа частиц, столкнувшихся с пузырьком,
к числу частиц, которые столкнулись бы с ним, если бы
двигались (как и в отсутствие пузырька) прямолинейно, а не по
- линиям тока, называют коэффициентом захвата Е.
Значение его, характеризующее вероятность wc столкновения
определяется соотношением площадей поперечного сечения стс и <т0:
Е - ас/а0 = nlVn (R + гJ - P/(R + r)\
Описание поля скоростей потока возле пузырьков всех
размеров, используемых при флотации, для определения
коэффициента захвата Е является сложной и не решенной до
настоящего времени задачей. Сложность ее обусловлена необходимо-
43
0,2 0,3 0,4 cL,mm О 0,25 0,5 0,75 d/д
Рис. 3.4. Влияние крупности d угольных частиц, крупности D пузырьков (а)
и соотношения d/D (б) на значение коэффициента Е захвата (по данным
Ю. Б. Рубинштейна)
стью учета всей совокупности существенных факторов, таких
как эффекты зацепления, гравитационного сноса,
инерционные, включая эффект присоединенной массы, а также эффекты,
связанные с нестационарностью и неоднородностью
набегающего на частицу потока, с вращением частицы и, наконец, с
неидеальностью обтекания пузырька жидкостью.
Результаты экспериментальных исследований и расчета по
приближенным формулам показывают, что коэффициент
захвата возрастает с увеличением размера d и плотности частиц
и убывает с ростом размера D пузырька (рис. 3.4,а). Это
объясняется тем, что, чем больше диаметр D пузырька, тем
сильнее искажается поле скоростей жидкости и увеличивается
вероятность огибания частицей (вместе с потоком жидкости)
пузырька без столкновения с ним [37].
Характер гидродинамического взаимодействия полей,
создаваемых движущимися частицами и пузырьками, зависит от
соотношения размеров частицы и пузырька. При увеличении
отношения d/D эффективность захвата Е сначала растет за счет
увеличения сил инерции и эффекта зацепления, а затем падает.
Последнее связано с тем, что поле скоростей жидкости,
определяющее обтекание, изменяется таким образом, что для
пузырька, соизмеримого с частицей, как бы увеличивается
вероятность огибания ее вместе с потоком. В результате этого
зависимость Е от d/D имеет экстремальный характер (рис. 3.4,6).
При уменьшении диаметра пузырька область экстремума
смещается в сторону меньших размеров частиц.
Инерционные силы уменьшаются с уменьшением размеров
частиц и при определенном критическом значении числа Стокса
StKp частицы уже не могут преодолеть силы вязкости и столк-
44
нуться с пузырьком только за счет сил инерции. Минимальный
размер dm\n частиц можно рассчитать при этом по формуле
rfmin - V/"l8vSJKReStKp/t;FT—бж) , C-8)
где v — кинематическая вязкость среды; v — скорость
движущейся в потоке частицы перед встречей ее с пузырьком. Так,
частицы рудных минералов [6Т=E—7) 103 кг*м~3] крупностью
менее 15 мкм и угля FТ = 2-103 кг-м~3) крупностью менее
75 мкм не могут столкнуться с пузырьком, имеющим диаметр
около 1 мм, если действуют только силы инерции [37]. Действие
других сил, вызывающих закрепление таких частиц на
поверхности пузырьков, рассмотрено ниже.
Для частиц обычной флотационной крупности значение ws
тем больше, чем выше концентрация частиц и пузырьков в
единице объема пульпы.
3.4. Вероятность закрепления частицы на пузырьке
Вероятность а/заКр закрепления частицы на пузырьке
зависит как от гидродинамических параметров процесса (скорости,
размера и формы пузырька и частицы, времени их контакта при
соударении, массы частицы), так и от свойств поверхности
пузырька и частицы, для регулирования которых используются
флотационные реагенты.
Основной потенциальный барьер при закреплении частицы
на пузырьке обусловлен необходимостью понизить толщину
гидратной прослойки между ними в каком-либо месте до
критического значения (см. рис. 2.7).
Для крупных частиц это достигается за счет инерционных
сил. Вероятность закрепления частицы на пузырьке возрастает
с увеличением плотности частиц и силы удара ее о пузырек.
Оптимальная скорость столкновения частиц флотационной
крупности, по данным Ф. Шпетла и Ф. Дедека, находится в
пределах 2—10 см/с. При большей скорости частицы отскакивают
от упругой поверхности пузырька [37, 44].
Если инерции частицы недостаточно для разрушения водной
прослойки в месте удара, то закрепление ее может произойти
при последующем скольжении по пузырьку [44]. Условия
скольжения зависят от силы удара и угла падения частицы. Чем
больше угол, иод которым частица ударяется о поверхность
пузырька, и чем дальше точка удара от вертикальной оси, тем
меньше нормальная составляющая силы удара, способствующая
разрушению гидратной прослойки, и больше тангенциальная
его составляющая, под действием которой частица отскакивает
от поверхности пузырька или скользит по ней. С увеличением
тангенциальной составляющей вероятность закрепления частиц
на пузырьке уменьшается.
45
Для мелких частиц, обладающих малой инерцией,
скольжение по поверхности пузырька является основным механизмом
их закрепления [37].
«Безынерционные» частицы могут соприкоснуться с
поверхностью пузырька, например, вследствие возникновения
прижимной силы, обусловленной тем, что выше экваториальной
плоскости линии тока жидкости все же стремятся приблизиться к
пузырьку и увлекают за собой частицы. По достижении
критической толщины водной прослойки между частицей и пузырьком
начинают действовать дальнодействующие молекулярные и
электростатические силы, которые могут привести к притяжению и
преодолению энергетического барьера с образованием
трехфазного периметра смачивания. Вероятность закрепления но такому
механизму, однако, не может быть большой, так как частице
трудно подойти близко к поверхности пузырька.
Второй причиной закрепления «безынерционных» частиц
может быть образование завихрений в кормовой области
пузырька, также способствующих сближению с ним мелких
частиц. Подсос частиц на корму пузырька в результате
возвратно-вихревого движения жидкости может наблюдаться только
в присутствии гетерополярных молекул, обеспечивающих
жесткость пузырька, и значении числа Re более 20 [44]. При
возникновении стационарных вихрей создаются особые чисто
гидродинамические условия соударения на корме сферического
препятствия. По такому механизму могут закрепляться только те
частицы, диаметр которых меньше толщины гидратного слоя
пузырька, т. е. крупностью несколько микрометров.
Коллоидные частицы, ионы и молекулы гетерополярных
соединений могут достичь поверхности пузырька благодаря
диффузии [37].
Минимальное время контакта, необходимое для
закрепления частицы на пузырьке, принято называть временем
индукции [15, 49]. Для крупных частиц контакт при ударе
значительно эффективнее, чем при скольжении. Для мелких частиц
время контакта определяется временем скольжения их по
пузырьку. Для средних по размеру частиц общее время контакта
состоит из времени удара (более эффективного) и времени
скольжения (менее эффективного). Чём гидрофобнее
поверхность частицы, тем меньше требуются сила удара ее о пузырек
и время контакта с пузырьком, обеспечивающие преодоление
энергетического барьера.
Исследованиями с применением скоростной киносъемки
установлено, что определяющей для .образования комплекса
пузырек.— частица при соударении и скольжении является
кинетическая энергия обоих компонентов.
Экспериментальная кривая, характеризующая вероятность
ЗДзакр закрепления частиц разной крупности на пузырьках
воздуха, носит экстремальный характер (рис. 3.5). Максимальное
значение ш3акР соответствует частицам крупностью 50—90 мкм.
46
Рис. 3.5. Влияние крупности d
частиц на вероятность о>закр их
закрепления на пузырьках воздуха:
1 — мартит (по данным М. Ф.
Емельянова, И. И. Максимова); 2 — пирит (по
данным В. Д. Самыгина, В. С. Черти-
лина, В. П. Неберы); 3 — кварц (по
данным Д. Анфаноа, Д. Китченера)
Снижение шзакр при уменьшении крупности частиц обусловлено
в основном снижением их кинетической энергии, что затрудняет
преодоление энергетического барьера и прорыв водной
прослойки между пузырьком и частицей. Причиной снижения шзаКр
при увеличении крупности частиц сверх оптимальной является
недостаточное для разрушения водной прослойки время
контакта частицы с пузырьком в результате отскакивания и отрыва
частиц от пузырька уже после прорыва водной прослойки из-за
больших значений кинетической энергии крупных частиц.
Агрегаты частица — пузырек могут образоваться также в
результате выделения пузырьков растворенного воздуха на
частицах [15]. При 8>0° зарождающемуся пузырьку легче
отодвинуть молекулы воды от твердой поверхности, чем оторвать их
друг от друга с образованием полости пузырька. Интенсивность
выделения пузырьков растворенного воздуха на поверхности
частиц возрастает с увеличением его содержания в воде, степени
гидрофобности частиц и турбулентности перемешиваемой
пульпы. Устанавливая определенную скорость перемешивания
пульпы, можно осуществить селективное выделение
растворенных газов на поверхности частиц флотируемых минералов.
Пузырьки возникают из раствора на поверхности частиц
всех размеров. Однако наиболее важное значение такой
механизм образования агрегатов пузырек — частица имеет при
флотации мелких («безынерционных») частиц, обладающих
развитой поверхностью и большей вероятностью выделения на ней
пузырьков растворенного воздуха, соприкосновение которых
с пузырьками в пульпе весьма затруднительно.
За счет кинетической энергии преодолевается энергетический
барьер, равный e6dl (где d\ — площадь контакта газ —
твердое). Поэтому прорыв гидратной прослойки облегчается при
наличии на поверхности частицы выступов и острых граней
с малым значением dK. Причем, чем гидрофобнее будет выступ,,
тем меньше усилий нужно приложить для уменьшения
гидратной прослойки до критического значения, легче осуществить
прорыв ее в одном месте и обеспечить закрепление частицы на
О 80 fSff d}MnM
47
пузырьке с образованием трехфазного периметра смачивания.
Роль гидрофобных выступов могут играть закрепившиеся на
поверхности минеральных частиц капельки углеводородных
масел или выделившиеся из раствора микропузырьки
растворенного воздуха [7, 15], а также имеющиеся на поверхности
пузырьков тонкие частицы гидрофобных продуктов
взаимодействия флотационных реагентов с растворенными солями в пульпе
[49, 50].
Гидрофобизация поверхности частицы при адсорбции гетеро-
полярных ионов или молекул собирателя, уменьшая значение
энергетического барьера, способствует преодолению его при
соударении частицы с пузырьком. Учитывая это и используя
представления теории бинарных соударений, Ю. Б. Рубинштейн
предложил следующую формулу [37]:
^акР -^exp l~(U0-vQc)/UK]. C.9)
Формула C.9) учитывает зависимость величины шзаКр от сте-
рического фактора Я, характеризующего влияние взаимного
расположения частиц и пузырьков при столкновении; эффекта
деминерализации пузырьков % и соотношения энергии,
необходимой для прорыва гидратной прослойки между пузырьком и
частицей (характеризуемой разностью барьерной энергии £/0 и
энергии, определяемой характеристикой v и расходом Qc
собирателя), и энергии Uк, характеризуемой кинетической энергией
и свойствами частицы.
Изменение энергетических параметров процесса закрепления
частицы на пузырьке связано с изменением величины /?,
которая характеризует активность собирателей с точки зрения их
энергетических свойств, способствующих преодолению частицей
энергетического барьера.
. Вид зависимости C.9) подтвержден результатами
исследований флотации угольного шлама различными собирателями [37].
Условия, обеспечивающие наибольшую вероятность
закрепления на пузырьках частиц одних минералов и наименьшую
вероятность закрепления других, создаются изменением физико-
химических и физических факторов флотации с помощью
флотационных реагентов и изменением физических параметров
реализации процесса во флотационных аппаратах.
3.5. Вероятность сохранения закрепившихся
на пузырьке частиц
Вероятность шСОхР сохранения закрепившихся на пузырьке
частиц зависит от прочности прилипания, определяемой
состоянием поверхности, весом и формой части и условиями всплыва-
йия минерализованных пузырьков в пульпе. При высокой
турбулентности потоков пульпы большую роль играют
инерционные силы, отрывающие частицы от пузырьков.
В статических условиях на частицу диаметром и высотой dy
закрепившуюся на пузырьке (рис. 3.6,а), помимо сил Рф, Fr и
48
Fl 0 5 10 ISV-lirfcH3
Рис. 3.6. Схема сил, действующих на частицу, закрепившуюся на пузырьке
объемом V (а), и их значение при ^ = 0,1 мм (б):
1 — О=70 мН; 2 — сг=50 мН; 3 — а*=30 мН (по данным В. И. Мелик-Гайказяна [22])
.FT, как в предыдущем случае( см. рис. 3.1,а), действует
дополнительная отрывающая капиллярная сила FK. Появление ее
обусловлено избыточным давлением рк газа внутри пузырька,
рассчитываемого по уравнению B.1), на площади контакта
пузырька с частицей (см. рис. 3.6,а):
FK^0,25nd*pK = 0,25nd*a^T(VR + 1/р). C.10)
Поскольку давление газа в пузырьке постоянно и
определяется кривизной поверхности пузырька в купольной его части,
т. е, оно равно 2аж..г/&, а гидростатическое давление в
жидкости по высоте h (от купола пузырька до его основания)
растет до значения hbmg, очевидно, что на такую же величину
должно падать капиллярное давление газа в пузырьке на
площади его контакта с частицей, т. е.
рк = 2ож^т1Ь—кЬжц. C.11)
Учитывая это, А. Н. Фрумкин заменил выражение C.10) для
FK тождественным ему выражением
FK =—nrf«pK-=^-ndaBa«-r/6-A6«g), C.12)
4 4
так как экспериментально определять значения Ь и h гораздо
проще, чем R и р.
В условиях равновесия F$ + FT=Fi + FK и с учетом
выражений C.1) —C.3) и C.12) можно записать
Рож^г sin 9 + У6ж£= Vbrg + -|- я^2Bаж~г/Ь—/гбж^),
4
49
или с учетом принятых н_а рис. 3.6, а обозначений и после
некоторых преобразований:
ш*аж_г sin в - — nd? (в*—6Ж) g + — nd2 {2вж_г1Ь — Ag6«). C.13)
4 i < ■. 14
Так как второй член уравнения C.13) представляет собой
вес Q частицы в жидкости, то можно записать, что в условиях
статического равновесия
^-Q+Fk. C.14)
Анализ уравнения C.13), полученного на основании
представлений А. Н. Фрумкина и Б. Н. Кабанова [13], и уравнения
C.14) позволяет отметить следующее [46]:
в отличие от пленочной флотации [уравнение {3.5)],
отрывать частицу от пузырька будет не только вес Q, но и
капиллярная сила FK [уравнение C.14)], обусловленная избыточным
давлением газа в пузырьке. Результаты проведенных расчетов
[22, 23] показывают (рис. 3.6, б), что при закреплении на
пузырьках частиц флотационной крупности капиллярные силы
отрыва FK в зависимости от условий могут в десятки раз
превосходить силы отрыва, обусловленные весом {Q = FT—Fr)
закрепившейся на пузырьке частицы;
удельный вклад капиллярной силы отрыва FK резко
возрастает с уменьшением размера пузырька в связи с увеличением
кривизны его поверхности и значения капиллярного давления
в нем, как это следует из уравнения C.11). По результатам
расчетов, проведенных В. И. Мелик-Гайказяном для частиц
флотационной крупности, диаметром и высотой 0,1 мм и
значений 0Ж_Г=72,8 мН-вг-1, 6т = 5-103 кг-м-3, бж = 996,94 кг-м-*,
g = 9,8 м-с-2, соотношение Fv/Q при уменьшении размера
пузырьков с 5 до 0,5 мм возрастает с 7 до 147;
незначительный вклад веса частицы в силы отрыва ее от
пузырька отвечает гораздо меньшей крупности частиц при
пенной флотации по сравнению с пленочной. При этом уменьшение
размера пузырька, сопровождающееся увеличением вклада Fu
и соответственно уменьшением вклада Q в силу отрыва,
приводит к уменьшению, а увеличение размера пузырька — к
увеличению крупности частиц, способных удержаться в условиях
статического равновесия на поверхности одиночных пузырьков
газа. Если максимальная крупность частиц галенита в
рассмотренных ранее условиях пленочной флотации составляет 2,1 мм,
то на пузырьках размером 5 мм она составляет менее 1,1 мм,
а на пузырьках размером 0,5 мм — всего 0,12 мм;
снижение стж-г под действием поверхностно-активных
реагентов практически не снижает прочности закрепления частицы
на пузырьке в статических условиях. Если в уравнении C.4),
характеризующем равновесие частицы на плоской поверхности
раздела газ—жидкость, уменьшение ош~г приводит к
уменьшению только левой части уравнения, то в уравнении C.13), ха-
50
рактеризующем равновесие, частицы на пузырьке, при
уменьшении (тж-г наблюдается одновременное уменьшение как левой,
так и правой частей уравнения. Поэтому изменение значений
<7Ж- г не оказывает влияния (рис. 3,6, б) на расстояние между
кривыми /\j) и Fk, т. е. прочность закрепления частицы на
пузырьке в статических условиях не снижается. Более того, при
уменьшении значения сгж-г и, следовательно, /^ [уравнение
C.1)], но увеличении размера пузырька, вызывающем
уменьшение кривизны его поверхности и, следовательно, значений
рк и FK [уравнения C.11) и C.12)], может происходить в
соответствии с уравнением C.14) увеличение значений Q и размера
частицы, т. е. может иметь место обратная по сравнению с
пленочной флотацией зависимость между /^ и Q при изменении
значения 0Ж_Г.
Отмеченные особенности свидетельствуют о существенном
различии условий равновесия частицы на пузырьке (см.
рис. 3.6, а) и плоской поверхности раздела жидкость — газ (см.
рис. 3.1, а).
Условия равновесия частицы на неподвижном пузырьке,
характеризуемые уравнением C.13) и другими аналогичными
ему по своей сущности уравнениями [8, 15, 44], не могут быть
непосредственно применимы к реальным флотационным
условиям, так как в этом случае на частицу и пузырек действуют
значительные дополнительные, например инерционные, силы
(см. рис. 3.2).
По мнению Н. В. Матвеенко [21], для расчета
равнодействующей Fq капиллярных и инерционных Ft сил в динамических
условиях флотации достаточно учесть F$, F{ и FK, т. е.
или с учетом принятых на рис. 3.7, а обозначений
F0 = яа<хж_г sin 0 — Kd3 (ST—бж) С па2 Bаж_г/# — /г£бж),
4
где С — ускорение отрыва частицы от пузырька, а К —
безразмерный коэффициент пропорциональности между кубом
диаметра d частицы и ее объемом V, т. е. K—V/d*. Для
кубических частиц /С=1, а для шара /(=0,52. Для других форм
частицы при проведении расчетов следует, принимать вероятное
среднее значение /( = 0,75 [21].
Разделив все члены уравнения на na = nxd (где х —
безразмерный коэффициент, характеризующий отношение диаметра
площади контакта к диаметру частицы и имеющий для частиц
неправильной формы среднее значение 0,25 [21], получим
уравнение в форме удельных сил на единицу длины трехфазного*
периметра контакта пузырька и частицы:
F = a^rsinQ — (K/n.K)d2(8T — бж)С -х4Bаж_г/Я —Ag»*).
4
51
Рис. 37. Закрепление частицы на всплывающем пузырьке (а) и значение
относительной максимальной крупности dKp флотируемых частиц
(рассчитанной Ы. В. Матвеенко [21]) (б) при постоянных условиях в = 90°; аж-г =
=7 • 10—4 Н/см, Я~&=2 мм, К—0,75, и=0,25и ускорении их отрыва от
пузырька, м/с2:
/ — 300; 2 — 200, 3 — 150; 4—100; 5 — 50; 6 — 10. Заштрихована область действия
механических флотационных машин стандартного ряда
Если принять, что в каждом флотационном аппарате или
машине максимальное ускорение С отрыва частицы от
пузырька постоянно [21] и, следовательно, инерция отрыва
возрастает пропорционально d2y то при некотором критическом
значении dlv силы отрыва станут равны силам прикрепления и
флотация прекратится. В этом случае F = 0 и уравнение
принимает вид
аж-rSinG—(/С/яхLр(бт—бж)С |-х4рBаж-г/Я —*£бж) = 0.
C.15)
Полученное уравнение но своему содержанию близко к
уравнению C.13), полученному на основании представлений
А. Н. Фрумкина и Б. Н. Кабанова, и отличается от него только
тем, что силу тяжести Q = FT—Fr заменила сила инерции Fi
твердой частицы. Рассчитанные по уравнению C.15) значения
максимального размера минеральных частиц, флотируемых на
одиночных пузырьках при различных ускорениях отрыва их от
пузырьков, приведены на рис. 3.7, б.
В современных механических и пневмомеханических
флотационных машинах наибольшее ускорение, равное 100—340 м/с2
и определяющее критическую массу флотируемых зерен,
частицы получают на периферии импеллера [21]. Рассчитанные
по уравнению C.15) для этих условий значения dKp при
флотации углей, алмазов, золота, медных минералов, галенита,
сильвина в машинах механического типа (при /С=0,75 и х —0,25)
оказались близки к фактическим, получаемым на различных
обогатительных фабриках [21]. Снижение ускорений частиц
52
Рис. 3.8. Максимальная крупность частицы галенита при флотации ее на
одиночном пузыре (а), группой пузырьков (б), в условиях флокулярной
флотации (в)
в зоне минерализации пузырьков до 1 g в новых конструкциях
машин позволит повысить крупность флотируемых минералов
в 5—6 раз по сравнению с достигаемой в стандартных
механических флотационных машинах (см. рис. 3.7, б и 3.8, а).
Дальнейшее увеличение крупности частиц возможно
благодаря увеличению периметра смачивания (и значения ^ф на
единицу длины его), например, закреплением нескольких
пузырьков на различных участках поверхности крупной частицы
(рис. 3.8, б) или в результате образования аэрофлокул группы
слипшихся частиц и пузырьков разной крупности (рис. 3.8, в)у
как это наблюдается при так называемой флокулярной
флотации. Для образования аэрофлокул необходимы высокая ги-
дрофобность поверхности минеральных частиц, хорошая аэри-
рованность пульпы при тонком диспергировании пузырьков
воздуха и относительно слабое перемешивание пульпы в зонах
всплывания аэрофлокул и отстаивания пены, учитывая
непрочность образующихся аэрофлокул. Флокулярной флотации
способствуют большое содержание флотируемых частиц в единице
объема пульпы и оптимальное выделение газов из раствора.
В результате уменьшения ускорений частицы и увеличения
периметра смачивания максимальная крупность частиц галенита
может быть увеличена с 0,2 до 1,4 мм (см. рис. 3.8), а
угольных частиц —до 13 мм [21].
Ускорение частицы и периметр смачивания влияют на dKp
значительно сильнее, чем значение 6, поскольку dKp
пропорционален корню квадратному из sin6 [см. уравнение C.15)].
Поэтому снижению 0 с 90 до 1° соответствует весьма медленное
уменьшение dKp. Во всех случаях максимальная крупность
частицы обратно пропорциональна плотности флотируемого
минерала (см. рис. 3.7, б).
В динамических условиях пенной флотации система
частица— пузырек, двигаясь в турбулентном потоке пульпы,
испытывает всплески отрывающих усилий, которые в десятки
раз могут превышать вес частицы. Механизм компенсации их
53
и предотвращения деминерализации пузырьков при этом трудно
объяснить на основании уравнения C.15), поскольку для
вычисления истинных значений ускорений С и внешних
инерционных сил отрыва Fi необходимо знать параметры
гидродинамических потоков пульпы, воздействующих на систему пузырек —
частица, что является пока нерешенной задачей.
Неоднозначность в интерпретацию получаемых результатов будут вносить
также трудноопределяемые в каждом конкретном случае
коэффициенты К и х и полное отсутствие учета неравновесности
физико-химического состояния поверхности пузырьков в
динамических условиях флотации в присутствии органических
реагентов.
Механизм предотвращения деминерализации пузырьков при
всплесках отрывающих усилий не может быть объяснен и
явлением гистерезиса смачивания, т. е. сопротивлением пузырька
сокращению периметра его контакта с частицей в результате
возникновения противодействующих тангенциально
направленных гистерезисных сил. Для доказательства этого, приравняв
правые части уравнений C.10.) и C.12) и решив полученное
уравнение относительно высоты h пузырька перед его отрывом,
когда 2|/?|^|—р|, получим соотношение
h - B/b—sin в/а) аж-г/Fж£),
хорошо согласующееся с экспериментальными данными [46],
хотя при его выводе гистерезис не учитывается. Кроме того,
исследования В. И. Мелик-Гайказяна показали, что уравнение
C.13) справедливо не только на уровне периметра контакта
пузырька, но и на любом его уровне по высоте, когда о
гистерезисе говорить нельзя. Если, однако, допустить, что один или
несколько молекулярных слоев поверхности пузырька у
периметра контакта все же жестко связаны с подложкой, то разрыв
поверхности пузырька (и его отрыв) произойдет над этими
слоями, а на поверхности подложки может остаться лишь
остаточный микропузырек [46]. О несущественной роли гистерезиса
при пенной флотации (в отличие от пленочной)
свидетельствует также упрочнение контакта между пузырьком и частицей
(приводящее к повышению крупности флотируемых частиц)
в присутствии аполярных реагентов, обычно полностью
устраняющих гистерезис смачивания [22, 23].
Флотационные реагенты являются основным средством
воздействия на физико-химические свойства поверхности
пузырька и частицы. Очевидно, упрочнение контакта между ними
невозможно при постоянстве аж-г на всей поверхности
пузырька. Например, на основании уравнения C.14) для двух
пузырьков одинакового объема
т. е. в состоянии равновесия всякому приращению />
соответствует такое же приращение силы FK и упрочнение контакта
54
IQ+f
Рис. 3.9. Схематическое изображение агрегата пузырек — частица до
отрыва (а), во время отрыва (б) и изменение значения аг-ж по высоте
пузырька (в) при отрыве
(измеряемого максимальным весом частицы) не наблюдается
[46]. Упрочнение становится возможным, когда рост /^ не
сопровождается таким же ростом FK, т. е. когда на поверхности
пузырька . возможны неравновесные состояния, приводящие
к локальному росту аж г. Явление локального роста ;аж_г с
градиентом до 50 Н-м-2 на вытягиваемых участках поверхности
пузырька в присутствии аполярных реагентов установлено
В'. И. Мелик-Гайказяном [46].
Многократное упрочнение контакта пузырька с частицей при
воздействии на нее отрывающих усилий в присутствии
аполярных или гетерополярных реагентов, понижающих сгж-г, по
В. И. Мелик-Гайказяну [23, 22], будет происходить вследствие
быстрого роста флотационной силы />. Этот рост обусловлен
резким увеличением стж-г на растягиваемой вокруг частицы
кольцевой поверхности пузырька вследствие замены
поверхности раздела масло — вода на поверхность раздела воздух —
вода, обладающей большим значением а^-г, а также
вследствие одновременного увеличения угла 6 (рис. 3.9, а, б).
Значение FK при этом остается практически неизменным.
Локальное растяжение поверхности пузырька у частицы,
сопровождающееся разрывом адсорбционных пленок аполярного
или гетерополярного реагента, приводит к изменению значения
(Тж-г только на небольшом участке кольцевой поверхности
пузырька (рис. 3.9,б). Поэтому увеличение угла 6' во время
отрыва вызвано не только действием отрывающей силы /, но и
ростом <х^_г, приводящим к прогибу поверхности пузырька для
компенсации недостатка капиллярного давления в нем,
обусловленного пониженным значением сгж-г на остальной
(нерастянутой) его поверхности. В соответствии с уравнением B.1)
для сохранения постоянства значения рк в пузырьке увеличение
ськ-г должно сопровождаться уменьшением на такую же вели-
55
чипу суммы Aу7?4-1/р). Поскольку даже небольшое
вытягивание поверхности пузырька у периметра его контакта с
отрываемой частицей приведет к уменьшению R и росту 1//?, чтобы
обеспечить необходимое уменьшение алгебраической суммы
A/7? +1/р), величина 1/р должна убывать вплоть до
отрицательных значений. Поверхность пузырьки при этом выгнется и
6 возрастет до 6' (рис. 3.9, б).
Одновременный рост аж_г и 6 обеспечивает заметное
упрочнение контакта, противодействующее отрывающей силе f (см.
рис. 3.9, б). Оценить значение его можно через соотношение
флотационных сил во время отрыва F^ и до отрыва />.'
^^Ф = ^-г«!пе'/(аж_г5те).
Так как во время отрыва 6'->90°, а отношение сгж_гДуж-г
может достигать значения 2, максимальное упрочнение при
этом
Если 6 = 3°, то значение F'^ перед отрывом будет почти
в 40 раз больше значения F$. Этим в значительной мере
определяется положительное влияние добавок углеводородных
масел при флотации крупных частиц.
Упрочнение контакта является временным, так как
нарушенная адсорбционная пленка восстанавливается с течением
времени и система приходит в исходное состояние. Скорость
восстановления адсорбционного равновесия на растянутой
поверхности пузырька зависит от поверхностной активности, вяз;
кости, общей концентрации реагента и некоторых других
параметров, определяющих его кинетические характеристики.
Величина упрочнения контакта пузырька с частицей
пропорциональна разности значений поверхностного натяжения
Л(Уж~г в динамических а£_г и статических ож_г условиях
(рис. 3.9, в):
Да =<уд —ас
ж—г ж—г ж—г*
являющейся количественной мерой неравновесности, которая
возникает в адсорбционных слоях на различных участках
поверхности пузырька под влиянием внешних воздействий. Чем
более поверхностно-активным будет реагент, тем больше
локальный рост вж-г на вытягиваемых участках поверхности
пузырька и, следовательно, больше значение сгж-г и упрочнение
контакта между пузырьком и частицей. По данным А. Ф. Таг-
гарта и А. М. Годена,
где d — диаметр частицы; k и т — эмпирические константы,
т. е. прочность контакта и крупность флотируемых частиц
возрастают с увеличением контакта и крупность флотируемых ча-
56
стиц возрастает с увеличением Авт-Т [22, 23]. Следует, однако*
учитывать, что чрезмерно большая величина Ааж-Г сглаживает
разницу в степени гидрофобное™ частиц разделяемых
минералов и ухудшает селективность их флотационного разделения.
Таким образом, для повышения вероятности сохранения
закрепившихся на пузырьке частиц необходимо следующее:
применение соответствующих флотационных реагентов,
обеспечивающих гидрофобизацию поверхности; образование аэрофло-
кул и многократное упрочнение контакта флотируемых зерен
с пузырьками; возможно более полное устранение
турбулентности потоков пульпы в зоне всплывания пузырьков или
обеспечение всплывания минерализованных пузырьков в
восходящих потоках, близких к ламинарным.
3.6. Вероятность удержания частиц в слое пены
Значения вероятности <о/ удержания частиц в слое иены
в промышленных условиях колеблются в пределах 20—55 %
[44]. Значительная часть ефлотированных частиц выпадает из
пены и возвращается в пульпу при ударе пузырьков о пенный
слой, коалесценции пузырьков в пене, движении пены к
сливному порогу и ее съеме. Основной причиной низкой вероятности
удержания частиц в слое иены являются разрушающее
воздействие турбулетных потоков пульпы на слой пены и коалесцен-
ция пузырьков.
Повысить значение Wf можно снижением степени
турбулентности потоков пульпы в подпенном слое, увеличением
скорости и полноты удаления пены с поверхности пульпы,
количества подаваемого во флотационную машину воздуха, степени
его дисперсности, степени минерализации пузырьков, а также
увеличением расходов реагента-собирателя и пенообразователя
при оптимальном соотношении их концентрации в пульпе.
Регулированием процессов, происходящих в пенном слое,
можно существенно влиять на скорость и селективность
(избирательность) флотации.
3.7. Необходимый размер пузырьков
при пенной флотации
Необходимый размер пузырьков при пенной флотации
должен удовлетворять условиям:
подъемная сила F& минерализованного пузырька объемом
Vn должна быть больше сил инерции Fu противодействующих
подъему;
плотность бп минерализованного пузырька должна быть
меньше плотности бж пульпы, так как иначе он не будет
всплывать;
скорость v подъема минерализованных пузырьков должна
быть оптимальной и равной по данным практики E-И5)Х
57
ХЮ 2 м/с. При меньшей скорости пузырьки не успевают
всплывать на поверхность пульпы и большая их часть переходит
в хвосты. Более высокие скорости подъема минерализованных
пузырьков требуют получения крупных пузырьков, что связано
с уменьшением поверхности раздела жидкость — газ и
вероятности флотации.
По первому условию минимальный необходимый размер
минерализованного пузырька определяется из условия равенства
между подъемной силой Fa пузырька, равной весу вытесненной
жидкости, и силой инерции F* частицы:
Уп6ж£>Ы3Fт-6ж)С. C.16)
Выражая объем пузырька через его диаметр D
Vn^-nDm C.17)
и подставляя полученное выражение для Vn в уравнение C.16),
находим
D^y^C Fт~6ж)/(я£бж).
Результаты расчетов в диапазоне ускорений С от g до 30g
для частиц минералов с критической крупностью и плотностью
6Т= C,5-И7,5) -103 кг-м-3 дают диаметр пузырьков D = 2,2-f-
-7-1 мм. Для всплывания с несколькими частицами нужны
более крупные пузырьки.
По второму условию необходимо, чтобы
вп = 0п/У„<бж, C-18)
где Qn — вес минерализованного пузырька, практически равный
весу частиц, закрепившихся на пузырьке. Объем закрепившихся
частиц можно найти как произведение значений площади
пузырька nD2, занятой частицами, и средней толщины слоя
частиц, равной среднему диаметру частиц d. Поэтому вес
минерализованного пузырька, если пренебречь весом воздуха, будет
равен
Qn-nD2d6Ta, C.19)
где a — коэффициент минерализации пузырьков,
характеризующий отношение площади поверхности пузырька, покрытой
минеральными частицами, к площади его поверхности и
равный 0,03—0,3.
Подставляя в уравнение C.18) Qn из уравнения C.19) и Vn
из уравнения C.17), получаем
бп = 6а^6т/£><6ж,
откуда
Г>>6а46т/8Ж. C.20)
Результаты расчетов по формуле C.20) при бт = 7,5-103 кг-
-м^3, d=0,05 мм, бж—1,2-103 кг-м-3 и <х=0,15 показывают, что
диаметр пузырьков не должен быть меньше 0,28 мм.
58
Определим теперь размер, воздушного пузырька, исходя из
третьего условия, предусматривающего оптимальную скорость
подъема минерализованного пузырька [v= E-ь- 15)< 10~2 м/с].
Скорость подъема пузырька в жидкости можно вычислить
по формуле Аллена
о=2бо^(«ж-б„)/|х, . ■ (з. ,
где \i — вязкость пульпы. Допуская, что вязкость пульпы равна
вязкости воды @,001 Па-с) и подставляя в уравнение C.21)
значение 6n = 6adST/D, получаем уравнение для определения
диаметра пузырька по заданной скорости его подъема
1,77- 10в6жО3—10,6- 106od6TD2 —v*= 0. C.22)
Результаты расчетов по уравнению C.22) для тех же
значений a, d, дж, бт, что и в предыдущем случае, показывают, что
при у = 5-10~2 м/с D = 0,6 мм, а при v= 15» 10~2 м/с D = l,27 мм.
Таким образом, минимальный размер транспортирующих
пузырьков при флотации зерен обычной крупности должен
быть не менее 0,6 мм, а при флотации зерен критической
крупности— не менее 1—2 мм. Однако следует учитывать, что по
результатам исследований И. Свен-Нильсона и В. И. Классена
[7, 15] очень мелкие пузырьки, закрепляясь на поверхности
флотируемых частиц, активируют их прилипание к более
крупным пузырькам, имеющим достаточную подъемную силу и тем
самым повышают их скорость флотации. Причиной этого
являются как увеличение показателя флотируемости [см. уравнение
B.13)], так и повышение эффективности прорыва гидратной
прослойки между пузырьком и частицей при наличии на ее
поверхности тонких пузырьков газа, играющих роль гидрофобных
выступов. Поэтому желательно одновременное присутствие
в пульпе как очень мелких, например выделяющихся из
раствора (для активации), так и крупных транспортирующих
пузырьков.
Большое влияние на скорость флотации оказывает
соотношение размеров пузырьков и частиц. Поверхность, изобража-
ощая экспериментально полученную зависимость кинетической
константы К флотации от крупности угольных частиц и
пузырьков имеет область экстремумов (рис. 3.10).
Все кривые K—f(d) в плоскостях, соответствующих
определенному размеру пузырька (£>г = const), асимметричны, что
связано с постепенным уменьшением значения К при увеличении
крупности частиц и резким спадом его при уменьшении размера
частиц ниже оптимального. Максимальный размер dKp
угольных частиц при /(->0 можно оценить по уравнению C.15), а
минимальный их размер dmin — по уравнению C.8).
При увеличении пузырьков на кривых K=f(D) в
плоскостях, соответствующих определенному размеру частицы (di =
59
Ь^мют*
Рис. 3.10. Зависимость кинетической константы К флотации от крупности d
угольных частиц и D пузырьков воздуха (по Ю. Б. Рубинштейну)
= const), наблюдается монотонное уменьшение значений К.
Причем влияние размера пузырьков на флотируемость
угольных частиц разной крупности различно и проявляется наиболее
сильно для средних классов крупности (см. рис. 3.10). Можно
полагать, что самая высокая эффективность мелких пузырьков
при флотации частиц различной крупности связана с большей
вероятностью столкновения их с частицами, а также с
развитой поверхностью раздела жидкость —газ и с увеличением
времени их пребывания в объеме пульпы из-за меньшей скорости
подъема. Только при уменьшении крупности пузырьков ниже
допустимого предела, когда их подъемная сила становится
недостаточной для транспортировки частиц, наблюдается резкое
падение их активности. Минимальный размер пузырьков при
этом можно оценить с учетом оптимальной скорости их
подъема по уравнению C.22).
Сопоставление значений скорости подъема пузырьков с
эффективностью их минерализации показывает коррелирован-
60
ность этих параметров. При увеличении, скорости подъема
пузырька (при увеличении его размера) сверх оптимальной
{E—15)* Ю-2 м/с] эффективность извлечения угольных частиц
всех классов крупности уменьшается.
С увеличением интенсивности перемешивания до
определенного предела при флотации тонких частиц мелкими
пузырьками скорость флотации может увеличиваться, тогда как при
флотации крупных частиц во всех случаях скорость флотации
уменьшается при любом увеличении интенсивности
перемешивания.
• Уменьшение эффективности флотации при увеличении
относительной скорости движения пузырька (например, вследствие
увеличения его размера) и частицы (например, при
увеличении интенсивности перемешивания пульпы) сверх критического
значения согласуется с уменьшением вероятности закрепления
из-за избытка кинетической энергии частицы, приводящего
к отрыву ее или упругому соударению с последующим
отскакиванием от пузырька. Поэтому крупные частицы (d>0,4 мм для
угля и d>0,l мм для минералов цветных металлов) гораздо
лучше флотируются при прямоточном режиме движения
частиц и пузырьков в пульпе (характеризующимся резким
уменьшением относительной скорости их движения), чем при проти-
воточном. Этому способствует также то, что при движении
суспензии снизу вверх крупные частицы движутся медленнее
потока и время пребывания их в камере увеличивается. Однако
уменьшение времени пребывания пузырьков в камере и
соответственно времени минерализации и числа столкновений
частиц с пузырьком при прямоточном движении твердой и
газообразной фаз вызывает в результате понижение флотируемо-
сти более мелких классов. Все это приводит к необходимости
обеспечения различных аэродинамических режимов и разного
времени нахождения крупных и мелких частиц при флотации
t37]. Решение данных задач может быть связано как с
созданием в одном аппарате потоков двух направлений, так и с
разделением исходного материала на песковую и шламовую
фракции с последующей раздельной флотацией их в машинах или
аппаратах специальной конструкции.
3-8. Характеристика процесса пенной сепарации и сил,
действующих на частицу в пенном слое
При пенной сепарации пульпа или раствор, содержащие
разнообразные гидрофобные, гидрофобизированные и
гидрофильные частицы, а также поверхностно-активные вещества,
поступают на подготовленную любым способом пену сверху.
Гидрофобные и гидрофобизированные частицы и поверхностно-
активные вещества концентрируются в верхних слоях пены,
.а гидрофильные частицы удаляются из пены потоками
жидкости, поступающей на пену с пульпой [20].
61
Такой принцип осуществления флотации отличается от
рбычной пенной флотации исключительно благоприятными ус-*
ловиями для быстрого образования комплексов гидрофобная
частица — воздушный пузырек.
Это обусловлено, во-первых, высокой вероятностью wc
столкновения пузырьков с частицами при подаче пульпы на
поверхность сплошного слоя пены.
Во-вторых, в условиях встречного движения воздушных
пузырьков вверх, а частиц вниз, когда пульпа как бы
фильтруется через пену, в ней создаются исключительно
благоприятные условия для необходимого времени контакта между
гидрофобными частицами и воздушными пузырьками и тем самым
обеспечивается высокая вероятность w3aKX) закрепления частиц
на пузырьках.
Высоким значениям вероятностей столкновения и
закрепления частиц на пузырьках способствует также более
развитая, чем при обычной пенной флотации, поверхность, на
которой могут . закрепляться гидрофобные частицы. Они могут
закрепляться не только на внешней поверхности пены,
внутренней и внешней поверхностях пузырьков, но и на
высокоразвитой поверхности гидрофобных частиц и веществ, уже ранее
закрепившихся на воздушных пузырьках. Следовательно, при
пенной сепарации наблюдаются явления, на которых основана
не только пенная, но и пленочная, масляная флотация, а также
флотация твердой стенкой. При этом создаются условия для
аэрофлокулярной и групповой флотации, возрастает
вероятность флотации крупных и труднофлотируемых зерен.
Почти полное отсутствие центробежных сил, стремящихся
оторвать гидрофобную частицу от пузырька в пене, в свою
очередь, обеспечивает высокое значение вероятности wCoxp
сохранения закрепившихся частиц на поверхности пены, пузырьков,
гидрофобных частиц и веществ.
Время разделения минеральных зерен в пенном слое
определяется скоростью выпадения из пены гидрофильных частиц,
причем крупные частицы выпадают за 1—2 с, а мелкие — за
7—10 с [20, 46]. Быстрое извлечение (в течение нескольких
секунд) из поступающей на пену пульпы гидрофобных частиц и
отсутствие заметного разрушения флотационных комплексов
в пене под действием динамических сил обеспечивают высокую
вероятность Wf удержания частиц в слое пены.
Схема, иллюстрирующая особенности пенной сепарации по
сравнению с флотацией из объема пульпы, приведена на
рис. 3.11.
Пенная сепарация обладает рядом преимуществ по
отношению к обычной пенной флотации из объема пульпы. К ним
относятся [20, 21, 46]:
возможность извлечения из пульп крупнозернистых
гидрофобных частиц. Максимальная крупность успешно
флотируемых зерен при пенной сепарации возрастает, например, для
62
Подача
пульпы на
пенный
слой
Минимальная,
динамичность
Отсутствие
Влияния предельно
минерализованных
пузырьков
Длительный
контакт частщ с
пузырьками
Отсутствие Г
Отрывающих салу1
Почти полное
отсутствие
трения между •
частицами
Отсутствие
разрушения флото-
номпленсов за
счет
динамических сил
Почти полное
отсутствие
изменения свойств
поверхности
частиц в печном
слое
Высокий кпд
машин
Способность
пены
удерживать крупные
гидрофобные
зерна
Высокая
скорость
разделения
Рис. 3.11. Особенности условий пенной сепарации (по О. М. Кнаусу, Р. И.
Гуревичу, Ю. П. Уварову и Е. Н. Никитину)
сильвина до 4 мм, для фосфорита до 1,5 мм, для угля до 10—
13 мм, для марганцевых и сульфидных минералов до 2 мм, т. е.
она в 5—7 раз больше, чем при пенной флотации из объема
пульпы;
резкое сокращение времени флотационного разделения
гидрофобных и гидрофильных частиц, что определяет большую
производительность машин пенной сепарации и позволяет
предотвратить протекание некоторых нежелательных реакций
в пульпе во время флотации;
отсутствие необходимости во взвешивании частиц в пульпе,
что значительно сокращает энергозатраты по сравнению с
флотацией из объема пульпы и уменьшает истирание зерен
хрупких минералов при флотации;
возможность извлечения гидрофобных частиц при любом
разбавлении пульпы (от Т:Ж=1:1 до Т:Ж= 1:100 и более).
Пенная сепарация начинает находить применение при
обогащении различных полезных ископаемых. Учитывая
отмеченные особенности, в первую очередь целесообразно ее
применение при обогащении крупнозернистых материалов, в
межцикловой флотации, при обогащении очень разбавленных или очень-
плотных пульп, для получения грубых концентратов.
Широкому распространению пенной сепарации в настоящее время
препятствует конструктивная недоработанность ее
аппаратурного оформления и отсутствие учета в практических условиях
основных закономерностей разделения минералов в пенном
слое.
Процесс пенной сепарации основан на разделении частиц
при их прохождении сверху вниз через слой движущейся пены,
образуемой на поверхности аэрируемой жидкости.
Флотационная сила, с которой пена воздействует при этом на частицу
с массой т и тормозит ее движение вниз, совершаемое под
действием силы тяжести mg навстречу восходящему потоку
пузырьков, характеризует несущую способность F
пенного слоя. Для гидрофобных и гидрофильных частиц она
63
Рис. 3.12. Характер кривых несущей способности F пены в зависимости
от глубины // погружения в нее гидрофильной А н" гидрофобной В частиц
значительно различается. Для гидрофобной частицы F^>mg и
поэтому она будет находиться на поверхности или в объеме
пены; для гидрофильной частицы F<mg и она, опускаясь,
будет проходить сквозь пену. Неодинаковое соотношение сил F и
mg для частиц с различными поверхностными свойствами и
в результате этого неодинаковое торможение, которое
оказывает пена частицам различных минералов, является основой
их разделения при пенной сепарации.
Значения сил, действующих на частицу на различных
уровнях пены, неодинаковы. Однако характер их распределения,
в зависимости от глубины Н погружения частицы в пену и под-
пенный слой, имеет во всех случаях принципиально одинаковый
вид (рис. 3.12), независимо от формы, размера и поверхностных
свойств частиц [46].
В положении 1 частица находится еще в воздухе над пеной
и F = 0. При соприкосновении с пеной частица с краевым
углом менее 90° стремится из положения 2 перейти в состояние
«мокрой флотации», т. е. она смачивается, втягиваясь в пену, и
сила F приобретает отрицательное значение. В положении 3
продвижению частицы вниз противодействует максимально
прогнувшаяся под ней поверхность пены и F достигает
наибольшего значения. При погружении частицы в пену
(положение 4) значение F сначала резко, а затем постепенно
уменьшается до тех пор, пока частица не достигнет нижнего уровня
пены. На границе пены с подпенным слоем (положение 5) F
снова проходит через максимум и при дальнейшем движении
64
частицы в аэрированной жидкости (на участке 6) F сохраняет
практически постоянное значение. Для сравнения на рис. 3.12
приведены также значения силы Архимеда FAi действующей на
частицу в неаэрированной жидкости (положение 7), и
флотационной силы Fu для пленочной флотации (положение 8) этой
же частицы.
Значение силы F на всех уровнях пены зависит от
состояния поверхности частиц. Сила F значительно возрастает с
увеличением степени гидрофобности (см. рис. 3.12, кривая В) и,
наоборот, падает с возрастанием степени гидрофильное™ (см.
рис. 3.12, кривая А) их поверхности, хотя все кривые F = f(H)y
независимо от состояния поверхности частиц, имеют
практически одинаковый вид.
Результаты проведенных исследований [46] с частицами
разной формы и размера подтвердили пленочный характер их
флотации в положении 3 (см. рис. 3.12) и возможность
разделения на поверхности раздела воздух — пена зерен
максимальной крупности. В соответствии с особенностями пленочной
флотации максимальная крупность частиц в большой степени
зависит от гистерезиса их смачивания и значения сгж-r.
Обработка частиц реагентом (например, мазутом), вызывающим
трудноподвижность периметра трехфазного контакта и
образование больших гистерезисных краевых углов, приводит к
увеличению силы F и крупности флотируемых частиц. Наоборот
значения F и крупность частиц уменьшаются в присутствии
поверхностно-активных веществ (например, ОПСБ), вызывающих
уменьшение значений аж_г и флотационной силы, в
соответствии с выражением C.1). Максимальные значения силы F на
границе раздела воздух — пена (см. рис. 3.12) и
соответствующее им максимальное значение соотношения F/Q, по условию
П. А. Ребиндера, для пленочной флотации [уравнение C.7)]
позволяют достичь на этой границе наибольшей селективности
разделения крупных частиц, обладающих неодинаковыми
поверхностными свойствами.
Сила F для гидрофобных частиц на поверхности раздела
воздуха — пена (см. рис. 3.12 положение 3) часто оказывается
больше флотационной силы Fn на границе раздела воздух —
вода (положение 5), хотя плотность верхних слоев пены
гораздо меньше плотности воды. Это позволяет, по мнению
В. И. Мелик-Гайказяна [46], считать, что частица
удерживается не только поверхностью пены, но и внутренней
поверхностью пузырьков, контактирующих с пеной и частицей
(рис. ЗЛЗ, а).
Под действием силы тяжести частица смещается вниз и
устанавливаются такие гистерезисные углы натекания и отте-
кания, при которых векторы аж_г наружной и внутренней
поверхностей пузырьков становятся параллельными и направлен*
ными вверх (см. рис. 3.13, а), и пена через периметры контакта
с частицей оказывает соответствующее противодействие ее
3 Заказ № 1957
65,
Рис. 3.13. Схема сил, действующих на частицу, находящуюся на
поверхности (а) и в объеме (б) пены
движению вниз. При этом наибольшее противодействие
окажут пары векторов 1 и 2 у верхнего периметра контакта
частицы перед отрывом. Действие пар векторов 3 и 4 в
вертикальном направлении меньше, чем у пар / и 2, а в
горизонтальном направлении компенсируется капиллярным давлением
рк газа в пузырьках на площадь контакта с частицей. Еще
меньше влияние на торможение частицы, оказываемое парами
векторов 5 и б. Результирующая для них будет равна
гидростатической силе выталкивания частицы вверх, которая
незначительна из-за малой плотности пены [46].
Гораздо меньшие значения силы F и несущей способности
пены для частиц с гидрофильной поверхностью (см. рис. 3.12,
кривая А) обусловлены тем, что в этом случае векторы о,ж-г
у каждой прослойки направлены в противоположные стороны
или пересекаются друг с другом под большим углом.
Равнодействующая их мала, а вертикальная составляющая для
боковой поверхности частицы при отсутствии гистерезиса
смачивания близка к нулю. Частицы сильнее втягиваются в пену по
сравнению с гидрофобными (см. рис. ЗЛ2, положение 2) и,
поскольку несущая способность пены для них меньше, проходят
они ее значительно быстрее.
Отрываясь от поверхности раздела воздух — пена, частица
переходит из состояния пленочной флотации (см. рис. ЗЛ2,
положение 2) в состояние обычной пенной флотации
(положение 4). Действие пар 1, 2, 3 векторов аж-г (рис. 3.13, б) при
этом заметно ослабляется капиллярным давлением рк газа
в пузырьках на площадь контакта с частицей, и сила F
воздействия пены на частицу уменьшается более чем в 2 раза,
приобретая минимальные значения (см. рис. 3.12, участок 3—4). По
результатам исследований В. И. Мелик-Гайказяна,
эффективной с точки зрения торможения движения частицы через слой
пены является не весь периметр контакта с ее поверхностью,
а удвоенная длина проекции его периметра на нормаль к на-
66
правлению движения. Если пузырьки пены покрывают
поверхность частицы в несколько ярусов, то суммарная эффективная
длина периметров пузырьков в каждом ярусе будет равна
удвоенной длине периметра частицы, а число ярусов может быть
оценено по отношению высоты частицы к среднему диаметру
пузырьков пены.
Значительное уменьшение силы F, удерживающей частицу
в пене, вызывает такое же уменьшение максимальной крупности
флотируемых зерен. Поэтому процесс пенной сепарации в
конструктивном отношении необходимо осуществлять таким
образом, чтобы крупные и тяжелые частицы при своем движении
в пене сверху вниз не достигали «слабого» участка пены и тем
более не должны преодолевать его, двигаясь снизу вверх [46].
По данным Л. А. Климовича, В. И. Зеленова и других
исследователей, тяжелые частицы, прошедшие пену сверху вниз,
вновь в пену не извлекаются.
Значительное возрастание силы F в положении 5 (см.
рис. 3.12) частицы на границе раздела пена — подпенный слой
обусловлено, во-первых, возникновением направленных сил,
которые создаются межфазным натяжением как только на
границу раздела попадают частицы и образуют периметр
трехфазного контакта, и, во-вторых, возрастанием гидростатических
сил в связи с большей плотностью аэрированной жидкости по
сравнению с плотностью пены в ее нижних слоях. Поэтому
повышенное содержание крупных частиц на границе пены с под-
пенным слоем вполне закономерно как для частиц, выпавших
из выше расположенного «слабого» участка пены, так и для
частиц, попавших на эту границу из аэрированной жидкости.
Несущую способность пены на всех уровнях пенного слоя
можно значительно понизить, если провести ее орошение
водой или подать на нее сильно обводненное питание.
Наблюдаемое при этом возрастание скорости нисходящих потоков,
турбулентности, их в связи со встречным движением пузырьков, и
в конечном счете, отрывающих усилий заметно снижает выход
частиц в пенный продукт, причем это снижение особенно
заметно для менее гидрофобных частиц (рис. 3.14, кривая 1), чем
для более гидрофобных (рис. 3.14, кривая 2).
Уменьшение значений F при возрастании обводненности
питания оказывает особенно вредное влияние на пенную
сепарацию крупных частиц. Поэтому, чтобы обеспечить эффективное
извлечение крупных и тяжелых частиц в пенный продукт,
питание, подаваемое на пену, не должно быть обводненным.
Пенная сепарация имеет общие черты как с пленочной, так
и с пенной флотацией. Это создает определенные трудности
в выборе флотационных реагентов.
Например, возможность флотационного извлечения крупных
частиц при пенной сепарации определяется целиком
механизмом пленочной флотации на границе раздела воздух — пена.
В связи с этим реагенты, используемые при пенной сепарации
3*
67
Рис. 3.14. Влияние обводненности
исходного питания (Ж: Т) на
извлечение 8 слабогидрофобных A)
и сильногидрофобных B) частиц
при пенной сепарации (по данным
[46])
^ О 5 10 15 Ж-Г
крупных частиц, должны, как и при пленочной флотации, ги-
дрофобизировать поверхность частиц, увеличивая гистерезис
смачивания, вызывающий трудноподвижность периметра
трехфазного контакта, и не быть, по возможности, поверхностно-
активными, чтобы не снижать значение <тж-т и флотационной
силы ^ф, как это следует из выражения C,1).
Извлечение мелких частиц в отличие от крупных при пен-,
ной сепарации определяется уже не столько гистерезисным,
сколько капиллярным механизмом в объеме пены, как и при
обычной пенной флотации. Применяемые реагенты в этом
случае должны гидрофобизировать поверхность частиц, уменьшая
гистерезис смачивания, и быть в достаточной мере
поверхностно-активными, чтобы обеспечить необходимое упрочнение
контакта пузырек—частица при динамических воздействиях на
флотируемую частицу.
Поскольку для извлечения крупных и мелких частиц
требуются различные реагенты (в связи с различным
преобладающим и определяющим механизмом их флотации при пенной
сепарации) предлагается [46] исходное питание
классифицировать и после раздельного перемешивания с реагентами
крупнозернистый и несколько обезвоженный материал подавать на
пену, а необезвоженный мелкозернистый материал — в объем
пены, подпенный слой или в объем аэрируемой жидкости.
Заглубление аэрирующего устройства способствует обычно
выходу в пенный продукт не только мелких, но и крупных частиц
благодаря созданию более спокойных условий в пене.
3.9. Условия повышения крупности
флотируемых частиц
Повышение крупности флотируемых частиц может иметь
Важное значение по следующим причинам:
не надо измельчать руду тоньше только потому, что
крупные частицы свободных минералов не могут быть подняты
пузырьками на поверхность пульпы. Повышение крупности dKp
в основной флотации в 3—4 раза приведет к сокращению
потерь ценных минералов в виде переизмельченных зерен к сок-
68
ращению энергетических затрат на измельчение примерно
в 2 раза. Применение флотационных машин с малыми
ускорениями при перемешивании пульпы приведет также к
сокращению расхода электроэнергии;
повышается производительность не только измельченного
отделения, но и отделения сгущения, фильтрования и сушки и
тем самым снижается стоимость обогащения;
крупнозернистые концентраты лучше для дальнейшей
переработки (например, автоклавная плавка крупнозернистых
концентратов самородной серы протекает более эффективно) или
они дороже ценятся (например, крупные частицы графита
больше ценятся, чем мелкие);
в некоторых случаях возможно повышение селективности и
улучшение технологических показателей флотации.
Для повышения крупности флотируемых зерен необходимо
увеличить вероятности шзаКр их закрепления, wCoxp сохранения
частиц на пузырьках и Wf удержания их в пене. Практически
это осуществляется следующим образом:
применением специальных конструкций флотационных
машин, характеризующихся малой турбулентностью движения
пульпы и небольшим значением инерционных сил отрыва
частицы от пузырька, что приводит к резкому увеличению
значения аУсохр. Данные рис. 3.7 (кривая 6) показывают, что
наибольшая крупность минеральных частиц достигается при
минимальном ускорении их отрыва от пузырька, т. е. при отсутствии
перемешивания пульпы, когда на частицу действует только
сила тяжести. Такой случай практически реализован,
например, в машинах пенной сепарации и частично в аэролифтно-
механических флотационных машинах;
созданием в некоторых конструкциях флотационных машин
(например, в машинах КС) спокойных восходящих и близких
к ламинарным потоков пульпы, облегчающих подъем крупных
частиц при резком уменьшении инерционных сил отрыва и
увеличении значения шС0Хр;
использованием принципа подачи пульпы на пенный слой
(как это реализовано в машинах пенной сепарации), что
приводит к резкому увеличению значений wc, ш3акр и а>Сохр",
повышением прочности пены с помощью специальных
веществ и создания особых устройств для ее быстрого, но
осторожного съема, т. е. создания условий, увеличивающих
значение Wf\
уменьшением удельных (приходящихся на единицу длины
трехфазного периметра прилипания) сил отрыва в условиях
особого режима перемешивания и аэрации пульпы.,
обеспечивающего закрепление нескольких пузырьков на различных
участках поверхности частицы. Это достигается, например,
в условиях так называемой аэрофлокулярной флотации при
осторожном перемешивании и интенсивной аэрации пульпы
мелкими пузырьками. Образование аэрофлокул, образованных
69
из частиц и пузырьков разных размеров, сопровождается
резким увеличением длины периметра трехфазного контакта,,
уменьшением удельных сил отрыва, но требует повышения ги-
дрофобизации поверхности. Повышение степени гидрофобностк
крупных частиц достигается при использовании смеси гетеро-
полярных и аполярных реагентов, например керосина,
трансформаторного, соляровогб и других масел.
Осуществление перечисленных мероприятий позволяет
увеличить . крупность флотируемых зерен сульфидных минералов
до 2—3 мм, а угля до 6—13 мм. При этом крупность
флотируемых частиц зависит не только от плотности, но и от степени
гидрофобности и формы частиц. Минералы, обладающие
высокой естественной гидрофобностью и малой плотностью, можно
флотировать при большей крупности зерен. Зерна округлой
формы флотируются хуже, чем зерна чешуйчатой формы.
3.10. Условия флотации тонких частиц
Худшая флотируемость тонких частиц может быть
объяснена следующими физическими и физико-химическими
причинами:
малой вероятностью встречи с пузырьками воздуха, так
как при обтекании пузырьков воздуха потоком пульпы очень
мелкие частицы в отличие от крупных относятся этими
потоками и не встречаются с пузырьком;
недостаточностью кинетической энергии тонких частиц для
преодоления энергетического барьера при разрушении гидрат-
ной прослойки между пузырьком и частицей при их встрече;
недостаточностью площади поверхности пузырьков для
закрепления на них тонких зерен при малой аэрации и большом
содержании шламистых частиц;
более высокой степенью гидрофильности отдельных тонких
частиц вследствие, например, окисления или их агрегирования
в результате, например, коагуляции с такими же частичками
минералов породы.
Повышение эффективности извлечения тонких частиц
достигается:
селективной флокуляцией их до оптимального размера с
помощью соответствующих реагентов. Скорость флотации таких
флокул часто больше скорости флотации частиц обычной
флотационной крупности;
использованием «носителя», на поверхности которого по
механизму селективной флокуляции закрепляются тонкие
частицы флотируемого минерала. В качестве «носителя»
используются зерна оптимальной крупности либо природно-гидрофоб-
ных веществ, либо искусственно гидрофобизированных, в
частности более крупных фракций извлекаемого минерала.
Вероятность столкновения таких зерен с пузырьком неизмеримо
больше, чем одних тонких частиц. В зависимости от значения
70
Рис. 3.15. Возможные механизмы а
флотации частиц A) на зернах но- а я .
сителя B) ч Воздух
*т
сил сцепления тонких частиц с поверхностью зерен «носителя»
возможны два механизма флотации (рис. 3.15). По первому
механизму (рис. 3.15, а) флотируется зерно «носителя» вместе
с закрепившимися на нем топкими частицами, что
наблюдается при значительной прочности закрепления тонких частиц
на поверхности зерна. По второму механизму флотируются
только тонкие частицы (рис. 3.15, б), а зерно «носителя» не
флотируется из-за малых сил сцепления между его
поверхностью и тонкими частицами;
применением специальных конструкций флотационных
машин, аэрация пульпы в которых сопровождается выделением
воздуха из раствора. В этом случае возникновение пузырьков
на мельчайших гидрофобных частицах вследствие большой их
удельной поверхности наиболее вероятно [15].
4. КИНЕТИКА И МОДЕЛИРОВАНИЕ
ФЛОТАЦИОННОГО ПРОЦЕССА
4.1. Кинетика флотации
Кинетика флотационного процесса характеризуется
зависимостью извлечения г флотирующегося минерала от
времени t, т. е. е=/@- Производная d&/dt представляет собой
скорость флотации в данный момент времени и
определяется тангенсом угла наклона кривой s—f{t).
Скорость флотации является важной технологической
характеристикой процесса. Она определяет производительность
флотационных аппаратов, позволяет судить об изменениях
условий флотации и анализировать их влияние на процесс
коррективной или селективной флотации.
Анализ закономерностей изменения скорости флотации во
времени является весьма перспективным направлением
исследования и оптимизации флотации. По результатам анализа
можно, например, обосновать оптимальное распределение
съема пены по фронту флотации, сопоставить и оценить фло-
тируемость разных минералов в разных условиях, выявить
действие тех или иных факторов флотации.
В последнем случае можно воспользоваться анализом
изменения удельной скорости флотации во времени. Расчет
значений удельной скорости флотации проводят по уравнению
К- Ф. Белоглазова, для вывода которого используется следую-
71
щее общее уравнение процесса минерализации пузырьков при
флотации [36]: ^#^г*
Uxl&t = 2фааКр# (*0 —х),
DЛ)
где х — число частиц, закрепившихся на поверхности
пузырьков к моменту времени t; z — коэффициент
пропорциональности; фзакр — коэффициент эффективности закрепления; N —
число способных минерализоваться пузырьков в единице
объема пульпы: х0 — число частиц, находящихся в пульпе и
подлежащих флотации.
Скорость dxjdt закрепления частиц на пузырьках
пропорциональна числу частиц, способных прилипнуть к пузырькам
(хо—х), числу пузырьков N и эффективности закрепления
частиц на пузырьках фзакр.
Перенеся величину (х0—х) влево, a dt— вправо и
проинтегрировав, получим:
х t
J dxl(X0 — X) = 1П [ДСо/(*о — *)] = z I # фэакрД*.
О О
Разделив числитель и знаменатель первого интеграла на х&
и заменив х/хо на s (в долях единицы), получим:
ln[l/(i-£j\
In [1/A— e)]-zftf<p3aKp<tf.
о
Величина 1п[1/A—е)] может быть названа
коэффициентом удельной скорости флотации. Кривые
изменения коэффициента удельной скорости флотации в координатах
1п[1/A—е)]—t могут иметь разнообразную форму (рис. 4.1),
Прямолинейная форма кривой (кривая 1) свидетельствует
о постоянной скорости флотации, выпуклая (кривая 2)—об
уменьшении скорости к концу
флотации, вогнутая (кривая 3) — об
увеличении. Для случаев резкого изменения
концентрации реагентов в пульпе
(например, сульфидных ионов)
характерна экстремальная форма кривой
скорости флотации (кривая 4).
Сопоставление кривых изменения
скорости флотации с вещественным
составом флотируемого материала и
значениями концентраций реагентов
в пульпе позволяет сделать
обоснованные рекомендации по
оптимизации флотационного процесса и повы-
г>™ л 1 Yofto,„fll, ,„we сить его селективность.
Рис. 4.1. Характер изме- „
нения коэффициента удель- Для количественной оценки селек-
ной скорости флотации тивной флотации предложен целый
72
ряд критериев или коэффициентов. На практике наиболее
широко используются только два из них [25, 36]:
оценка селективности процесса флотации по суммарному
извлечению ценных компонентов руды в одноименные
концентраты. Так, при получении из руды трех концентратов индекс
селективности
т]:-е1 + е2 + ез;
оценка селективности процесса флотации, по К. Ф. Бслогла-
зову,
t| = lg [1/A -ex)]/lg [1/A -82)],
где ei и £2 — извлечение в один концентрат разделяемых
компонентов.
4.2. Модели флотации
Кинетическая модель процесса флотации необходима для
разработки научных принципов расчета и конструирования
новых флотационных аппаратов, проектирования флотационных
фабрик с учетом требований контроля и автоматизации,
построения систем управления процессом в режиме
автоматического регулирования.
К настоящему времени предложено несколько типов
моделей флотации [37, 44].
Модель четырех состояний основана на том, что частицы,
поступившие в некоторый момент времени во флотационную
машину, могут находиться в свободном состоянии в пульпе
{рис. 4.2, состояние 1) или пене (состояние 4), закрепиться на
пузырьках в пульпе (состояние 2) или пене (состояние 3),
перейти в концентрат (пенный продукт—С) или хвосты (г).
Вероятности переходов между состояниями описываются
алгебраической системой обыкновенных дифференциальных
уравнений (для состояний /—4):
йтгШ = — (/C2iх + КалЛ-KZfi)mx + /Ci,2^2 + К1Лтг + т0; )
4тгШ = К% iffij—(Klt 2 + К*, 2) т2; I ,4 2\
dmzldt ^ Kz,2m2—(K4tS + Кс,*)тъ + Кз,*™*, [
<W<W = KMml +-^4,3^3 — (K3,4 + Kl, 4+^0,4)^4, I
где rtii — масса частиц в t-м состоянии в момент времени t\
Kt — константы скоростей переходов, представляющих собой
вероятности совершения переходов из одного состояния в другое
в единицу времени.
При установившемся непрерывном процессе, когда dm;/d£=
=0 (где 1=1, 2, 3, 4), можно найти trti и извлечение е частиц
в концентрат. Однако практическое применение модели четырех
состояний затруднительно из-за необходимого определения
большого числа параметров для описания кинетики флотации
73
J Рис. 4.2. Диаграмма переходов
^ и ?Г^: частиц из одного состояния в дру-
хбостыA)<^Ц f|J у>канцентрат(С) гое в машине непрерывного дей-
/^ ствия
-7 — J
^питание @)
частиц, обладающих даже одинаковой вероятностью переходов
(одного класса флотируемости) во флотационной машине.
Недостатком рассматриваемой модели является также отсутствие
описания процессов, происходящих в пенном слое [44].
В двухфазной модели флотации пульпа (фаза р) и пена
(фаза f) рассматриваются как идеально перемешиваемые фазы,
в каждой из которых частицы находятся только в одном
состоянии [44]. Двухфазная модель может быть получена из модели
четырех состояний, если в уравнениях системы D.2)
состояния 1 и 2 заменить одним состоянием р, а состояния 3 и 4 —
одним состоянием пены f. Тогда система уравнений,
описывающих кинетику флотации в двухфазной модели будет выглядеть
следующим образом:
drrip/dt = — Kfpftip + Kpftrif—КгргПр + m0;
dntf/dt = KfPnip — Kpftrif—Kcfffif
Данной модели в основном присущи те же недостатки, что и
предыдущей.
Однофазная модель флотации является наиболее
распространенной упрощенной моделью, изучению которой посвящено
большое число экспериментальных исследований. В этом
случае, как и в уравнении D.1), учитываются частицы только в
одном состоянии — свободных частиц в пульпе. Разделив обе
части уравнения D.1) -на xQ и учитывая, что х/хо — г, получаем:
d6/d*=2<p3aKptf(l—6). D.3)
Если свойства флотируемого минерала и условия флотации
ПОСТОЯННЫ (фзакр=: COnst, iV = COnst), TO
de/d/ = *(l—в), D.4)
т. е. скорость флотации должна быть прямо пропорциональна
массе флотируемого материала A—е) и характеризоваться
вероятностью k флотации в единицу времени, отражающей все
основные этапы флотационного процесса; столкновение частиц
с пузырьком wc; закрепление на пузырьке ку3акр; сохранение
частиц на пузырьке до выхода в пенный слой адСохр и удержание
их в пене до съема в концентрат Wf.
Интегрируя уравнение D.4), получаем уравнение, известное
как уравнение Белоглазова:
е=1— ехр(—Ы). D.5)
Если часть полезного компонента тесно связана с пустой
породой (например, в виде изоморфной примеси, тонкодисперс-
74
ных включений) или находится в неизвлекаемых минералах, то
предельное извлечение будет меньше 100% (еп<1). В этом
случае уравнение D.4) примет вид
<te/d£ = &(en—в).
После интегрирования получим
* = еп[1— ехр(—Ы). D.6)
В реальных условиях флотируемость отдельных зерен
полезного минерала непостоянна, так как она зависит от многих
условий: крупности и формы зерен, степени окисленности,
наличия тонкодисперсных вкраплений пустой породы или других
минералов. Кроме того, часть полезного минерала всегда
находится в сростках, состав которых может быть различным.
В первую очередь извлекаются в пену наиболее легко
флотируемые зерна, поэтому флотируемость остающихся в пульпе
частиц непрерывно понижается. Флотируемость зерен
изменяется также по ходу процесса вследствие изменения
концентрации реагентов в пульпе. Только по этим причинам простейшие
уравнения D.5) и D.6) для реальных условий обычно
неприменимы. Кроме того, линейность данных уравнений скорости
флотации может быть нарушена флокуляцией частиц в объеме
пульпы и процессами, протекающими в зоне пены.
Накопленный экспериментальный материал показывает, что
скорость флотации гораздо чаще описывается уравнением
<le/ctf = К A —е)« = A —е) К A — е)«-«, DГ7)
в котором показатель степени п изменяется от 1 до 6 [36].
Сопоставление уравнения D.7) с уравнениями D.3) и D.4)
позволяет считать, что в реальных условиях фзакр и k являются
функцией содержания и флотируемости оставшихся в пульпе,
зерен:
Поскольку обычно п—1^1, очевидно, что значение k (и
<Рзакр) с уменьшением содержания в пульпе флотируемых
зерен также уменьшается.
Заменяя в уравнении D.7) kl(l— e)n~l на функцию f(k)
распределения частиц по флотируемости и интегрируя его,
получаем:
оо
«=f [1— ехр(—#)]f(fe)dft.
о
Если учесть также разную продолжительность пребывания
различных частиц в камере, то
ОО ОО
€ = J f [I —exp (_«)] f (k) E @ dkdt,
о о
где E(t)—функция распределения материала по времени
пребывания частиц в камере.
75
Предложены модели с учетом и других видов неоднородно-
стей. Так, например, в модели Кинга одновременно
учитываются распределения по крупности, минеральному составу
зерен определенной крупности, поверхностной активности частиц
определенной крупности и минерального состава. Трудности
использования таких моделей обусловлены сложностью расчета
принятых функций распределения параметров процесса [37].
Развитие моделей кинетики флотации связано с попытками
учета влияния размеров камер, скоростей потоков, условий
перемешивания пульпы и других факторов.
Ввести в модель процесса перемешивание в пространстве
позволяет применение методов теории массопереноса.
Уравнение массопереноса для одномерного случая записывается в виде
dCldt= —vdCldx + Dxd*C/dx29 D.8)
где С — концентрация частиц; v — скорость движения потока;
х — пространственная координата; Dx — коэффициент
диффузии. Первый член правой части уравнения описывает перенос
частиц жидкостью, второй — диффузионный поток, вызванный
градиентом концентрации частиц. В случае флотации
уравнение D.8), описывающее изменение концентрации минеральных
частиц, должно быть дополнено членом, характеризующим
данный процесс.
Например, если все частицы однородны по свойствам,
гидродинамика аппарата неизменна, соблюдаются условия
свободной флотации частиц, выход концентрата незначителен и
объем пульпы можно считать постоянным, уравнение будет
выглядеть следующим образом [37]:
dC/dt = —vdC/dx + Dxd2C/dx2—KC,
где К — константа скорости флотации.
В более сложных случаях уравнение также усложняется
или заменяется системой аналогичных уравнений.
Степень описания процесса флотации предложенными к
настоящему времени моделями недостаточна; они не позволяют
с необходимой точностью предсказывать результаты флотации.
Параметры моделей пока можно определить только
экспериментально. Причиной этого являются многогранность,
многофакторность флотационного процесса и отсутствие достоверного
аналитического описания гидродинамики флотационных
аппаратов, физико-химических условий флотации, скорости
процесса минерализации пузырьков и их движения, процессов
в пенном слое, материального баланса твердого в камере
флотационной машины с учетом скорости минерализации
пузырьков, переноса и перемешивания в камере [37, 44].
ЧАСТЬ II
Флотационные реагенты и механизм
их действия на поверхности
раздела фаз при флотации
5. НАЗНАЧЕНИЕ И ОБЩАЯ ХАРАКТЕРИСТИКА
ФЛОТАЦИОННЫХ РЕАГЕНТОВ
5.1. Назначение и классификация
флотационных реагентов
■ Возможность флотационных явлений обусловлена
разницей в удельных свободных поверхностных энергиях на границе
раздела фаз./В обычной наиболее важной в практическом
отношении пенной флотации участвуют три фазы: вода,
минерал, воздух. [Назначением флотационных реагентов является
направленное изменение поверхностной энергии на границе
раздела этих фаз в целях изменения показателя флотируемости
разделяемых минералов, числа и размера пузырьков воздуха,
прочности пены. Прогресс в области флотационного
обогащения в значительной мере определяется совершенствованием
реагентного режима, улучшением способов использования
флотационных реагентов, разработкой и внедрением новых
эффективных реагентов и их сочетаний. 1
Флотационные реагенты могут быть органическими или
неорганическими соединениями, а также их растворами или
смесями. Одни из них образуют в водной фазе пульпы истинные
растворы ионно-молекулярной дисперсности, другие
растворяются весьма слабо, образуя коллоидно-дисперсные растворы,
эмульсии и тонкие взвеси. Номенклатура реагентов
насчитывает сотни наименований.
1 Современная классификация предусматривает разделение
: флотационных реагентов в зависимости от их роли при флота-
: ции на следующие группы:
>- пенообразователи, представляющие собой различные
гетерополярные органические соединения, которые в
результате их адсорбции на поверхности раздела жидкость — газ
облегчают диспергирование воздуха на мелкие пузырьки,
препятствуют слиянию пузырьков и повышают прочность пены;
собиратели, представляющие собой органические
вещества, способные закрепиться на поверхности извлекаемых
минералов и резко увеличить их флотируемость;
депрессоры, или подавители, к которым относятся
реагенты, понижающие флотируемость тех минералов,
извлечение которых в пенный продукт нежелательно в данной
операции;
77
активаторы, к которым относятся реагенты,
способствующие закреплению собирателя на поверхности, гидрофобизации
поверхности и флотации извлекаемого минерала;
регуляторы среды, к которым относятся реагенты,
влияющие на процессы взаимодействия собирателей,
депрессоров и активаторов с минералами. Основное назначение их
. состоит в регулировании ионного состава пульпы, процессов
диспергирования и коагуляции тонких шламов.
Депрессоры, активаторы и регуляторы среды часто относят
к одной группе и называют модификаторами, поскольку
s один и тот же реагент может выполнять различную роль при
\ флотации.
/~ Ко всем флотационным реагентам предъявляются
следующие требования: селективность действия, стандартность
качества, дешевизна и недефицитность, нетоксичность, удобство
в применении (устойчивость при хранении, хорошая
растворимость в воде, отсутствие неприятного запаха при хранении и
применении и т. д.).
Направленное изменение поверхностной энергии раздела фаз
под действием флотационных реагентов достигается в
результате их химических взаимодействий в объеме жидкой фазы и
адсорбции на поверхности, возможность протекания которых
зависит от природы и состояния межфазной поверхности и
реагентов в пульпе.
5.2. Состояние флотационных реагентов
в жидкой фазе пульпы
Загружаемые в пульпу реагенты могут находиться в ней
в виде ионов, молекул или выпадать в осадок. Для расчета
концентрации ионных и молекулярных форм реагентов в растворе,
как и при термодинамическом анализе возможных реакций на
поверхности минералов, используют следующие уравнения и
зависимости:
уравнение константы равновесия
„ [Р]*[С]° ,
1А]а[В]ь'
зависимость между стандартной свободной энергией
образования и константой равновесия
^реакции — 2j (^конечная— А/7начальная) = —RT In К =—1,364 lg K\
зависимость между стандартной свободной энергией
образования и стандартным электродным потенциалом
А^реакцни - E°nF = 23,06я£°;
78
зависимость между измеренным потенциалом реакции,
стандартным потенциалом и константой реакции
Г F° I Rr In lD]d[C]C -Eo 1 °'059 1r lD?lCf
t^t + nF Ш [Л]»»]* + n g [A]«[B]* '-
где /С — константа равновесия; [А]у [В], [С], [D] — концентрация
продуктов А, В, С, D реакции; а, 6, с, d — стехиометрические
коэффициенты; А/70 — стандартная свободная энергия
образования; R — газовая постоянная; Т — абсолютная температура;
Е° — стандартный электродный потенциал; п — число
электронов; F — число Фарадея; Е — потенциал реакции.
Состояние в растворе одно-одновалентных реагентов. К
одно-одновалентным реагентам относятся многие
реагенты-модификаторы и собиратели (например, цианид калия, олеат
натрия, алкилсульфаты, алкилсульфонаты, ксантогенаты щелочных
металлов, соли первичных аминов и др.). Они являются обычно
солями сильных оснований и слабых кислот или, наоборот,
слабых оснований и сильных кислот и в зависимости от
значения рН подвергаются гидролизу или диссоциации.
Так, при подаче в пульпу цианида калия, олеата натрия
и других аналогичных солей сильных оснований и слабых
одноосновных кислот справедливо равновесие
НАп^Н+ + Ап-, E.1)
где НАп — слабодиссоциирующие синильная или олеиновая
кислоты; Н+—ион водорода; Агг — ион цианида или олеата.
Константа К\ равновесия уравнения E.1) диссоциации
равна ,
„ [Н+ЦАП-]
Kl " [НАп] '
откуда
[НАп] = JH±lIAnZL. E.2)
Общая концентрация Со ионных и молекулярных
компонентов реагента в растворе равна сумме их концентраций
Со = [НАп] + [Ап-1 = [H+\iAn ] + [An-] = [An-] (-
тогда
КЫ-Ш-КП-ЯШ-. E.4)
Изменение значений [Ап~] и [НАп] неорганических реагентов-
модификаторов при изменении рН раствора можно представить
79
Рис. 5.1. Состояние одно-одновалентных неорганических (а) и
органических (б, в) реагентов в растворе
в виде диаграмм (рис. 5.1, а). Например, при рН 6 молекулами
HCN представлено 99,5 % цианида и только 0,5 % в виде
свободных ионов CN-. При рН 12, наоборот, 99,8 % цианида
представлено ионами CN- и только 0,2 % молекулами HCN.
При расчете состояния органических реагентов, например
олеата натрия в растворе, уравнениями E.3) и E.4) можно
пользоваться, если найденная по уравнению E.4)
концентрация молекул не превышает растворимости олеиновой кислоты
в воде B-10-5 моль/л).
В противном случае концентрацию молекул олеиновой
кислоты в растворе следует принять постоянной и равной по
значению растворимости олеиновой кислоты, поскольку избыток
ее выделяется из раствора в виде отдельной фазы с
образованием эмульсии.
Концентрацию олеатных ионов в растворе в этом случае
можно рассчитать по уравнению
А «ЛНАп]_
[Н+]
(где /Сг — константа равновесия реакции диссоциации
олеиновой кислоты), полученному из выражения E.2) после
подстановки вместо НАп значения растворимости олеиновой кислоты.
Концентрация олеатных ионов при этом не зависит от расхода
собирателя и определяется только значением рН пульпы.
Количество олеиновой кислоты, находящейся в виде отдельной
фазы в жидкой фазе пульпы, определяется по разности между
общей концентрацией загружаемого собирателя (олеата натрия
или олеиновой кислоты) и общей концентрацией в растворе
ионов и молекул олеиновой кислоты. Характер изменения
концентрации ионных и молекулярных компонентов анионного
собирателя в зависимости от рН раствора изображен на рис. 5.1,6.
При подаче в пульпу солей слабых оснований и сильных
кислот, например солей первичных аминов, в растворе
протекает реакция
RNH3+^RNH2 + H+,
80
константа равновесия которой
^ _ [RNHsHH+l
[RNH+]
Отсюда
„NHJ-JSJ^L „ли ВНН+,- 'H+1'RN"'' . F.5)
LH+] Аз
Учитывая, что общая концентрация Со ионных и
молекулярных компонентов в растворе
С» = [RNH3+] + [RNH2] - [RNH3+] + *3 [*"+1 =
_[НМ*](ЛИ±^.).
получаем:
[rnh>+J = t£jt*T <«>
IRNH.1- *''■""*) —5- С°1Н+1 - °* . E.7)
IH+] [Н+] [H+I + Кэ [H+J + /Г, V
Уравнения E.6) и E.7) для расчета концентрации ионов
и молекул амина в растворе используются в случаях, когда
предельная растворимость RNH2 (равная, например, для лау-
риламина 2,0-10~5 моль/л) не превышена.
Иначе для определения концентрации ЯЫНз+ применяется
уравнение E.5), в котором вместо [RNH2] используется
значение предельной растворимости RNH2 в воде. Концентрация
RNH2, находящегося в виде коллоидной (отдельной) фазы,
определяется как разность между стехиометрической
концентрацией и суммарной концентрацией RNH3+ и растворенного RNH2.
Характер изменения концентрации ионных и молекулярных
компонентов катионного собирателя в зависимости от рН
раствора изображен на рис. 5.1, в.
Сравнение диаграмм, изображенных на рис. 5.1,6 и в,
показывает, что ионная форма анионного собирателя (например,
олеата) преобладает в щелочной, а катионного (например,
амина) —в кислой и слабощелочной средах.
Состояние в растворе одно-двухвалентных реагентов, К
одно-двухвалентным реагентам относятся jjikhsl
реагенты-модификаторы, как сода Na2,C03, сернистый натрий Na2S и другие
аналогичные соли сильных оснований и слабых двухосновных
81
кислот. Для определения их состояния в пульпе необходима
рассмотреть следующие равновесия и равенства:
НАп-=ё*Апа- + Н+;
/с4 =
[Ап»-][Н+]
[НАп~]
Н2Ап=^=НАп- + Н+;
[НАп-]
#4
[An*-];
К,
[НАп-] [Н+]
[Н2Ап]
[Н+][НАп~1 _ [Н+]а[Ап2-]
E.8>
E.9)
E.10)
E.11)
[Н2Ап] ' '-<•-—* къ KtK&
Общая концентрация ионных и молекулярных компонентов
реагента в растворе:
С0 = [Апг-] + [НАп-] + [Н2Ап]. E.12)
Подставляя соответствующие выражения для [НАп-] и
[Н2Ап], получаем:
С0 = [Ап2-]
[Ап2-](-
Откуда
[An2-]
[Н+][Ап«-] [Н+]'[Ап«-] _
*«*, + *8 [Н+] + [Н+]« ч
К*Къ . У
КьКьС0
[НАп-]
[Н2Ап]
К«*5+*в1Н+] + [Н+]» '
К*КЪ + Кь№+]+[П+]* '
[н+рс,
К«*,+ *,[Н+Ц-[Н+]« '
E.13)
E.14)
Графическое изображение изменения значений [An2-], [НАп-]
и [Н2Ап] при изменении рН пульпы приведено на рис. 5.2, а.
рн рН
Рис. 5.2. Состояние одно-двухвалентных (а) и одно-трехвалентных (б)
реагентов в растворе
82
Состояние в растворе одно-трехвалентных реагентов. К
таким реагентам относятся, например, фосфаты, арсенаты щщу-
гие соли сильных оснований и слабых трёхосновных кислот.
Для -получения расчетных формул концентрации различных
ионов и молекул реагента в растворе необходимо рассмотреть
следующие равновесия и равенства:
HAna-*±Ans- + H+;
к [Ап'-]ГН+] = [Н+НАпЗ-]
[НАп*-] ' /Се
Н2Ап-^=НАпа- + Н+;
к _ [HAn'-][H+] . [H+][HAn'~] . [Н+ЩАп»-] .
7 [HaAn-] 2 J K7 К«К,
Н8Ап=?±Н2Ап- + Н+;
к [Н.Ап-][Н+] [H3Anl^ tH+HH2An-] = [Н+]з
8 [Н3Ап] tf8 *,К7*.
Общая концентрация ионных и молекулярных компонентов
реагента в растворе
Со = [An8-] + [HAn8-] + [Н2Ап-] + [Н3Ап].
Подставляя соответствующие выражения для [НАп2~],
[НгАп-] и [Н3Ап], получаем:
Си-[.\п*-] 1 [ннЧ[Ап3~1 | [Н+НАп'-] [Н+]ЧАпз-]
= [Дпз-j ( KoW» + *т*. 1Н+] + К» [Н+]а + [Н+]» \
V КвКтКв )
Откуда:
[An3-] = К,К,К,С0 E 15)
AVC7/C8 + К7/С8 [Н+] + К8 [Н+]2 + [Н+]3
/НАп2-] = /С7К8[Н+] С0 я
гн Ап-1 = *,[н+]ч:, .
2 ^в/С7/С8 + ^7/С8[Н+] + /С8[Н+р+[Н+]3 '
1Н3Ап] = [*^> .
К&К* + К7К9 [Н+] + К8 [Н+]2 + [Н+]3
Графическое изображение изменения значений [An3***], [HAn2*"],
[Н3Ап], [Н2Ап~] при изменении рН. пульпы приведено на
рис. 5.2,6.
Состояние в растворе многоосновных солей сильных кислот.
К таким солям относятся в первую очередь используемые в
качестве реагентов-модификаторов при флотяпии со-тш тяжядых
83
металлов (железа, алюминия, свинца, меди, цинка и др,)« При
подаче их или переходе"в пульпу в результате растворения мп
неральных компонентов протекают реакции гидролиза или
диссоциации в зависимости от значения рН раствора. Примени
тельно к солям, например трехвалентных железа или алюми
ния, необходимо учитывать протекание следующих реакций:
Me (ОНK *± Ме3+ + ЗОН-; К9 = [Ме3+] [ОН-]3; E.16)
МеО№+^Ме3-ь + ОН-; Кг*= ^о^1 ; <бЛ7>
Me (OH)f ^ МеОН2+ + ОН"; К* = [Ме°Н2+] [°Н^] ;
[Ме(ОН)+]
Me (ОНK ^ Н2МеОГ + Н+; К12 - [Н2МеОГ] [Н+]; E.18)
Н2МеОГ^МеОГ + Н20; К1г- г LMe0a J ; E.19)
[H2Me(V]
Н2О^Н+ + ОН-; /Cw = [H+][OH-]. E.20)
Рассматривая совокупность выражений для констант
равновесий /Сэ — Kw как систему уравнений и решая ее
относительно [Н+], найдем:
[Мез+]^ *.[Н+Р . [Ме0№+] = -^Ш1]1;
[Me(OH)fl- Kf+J ;
^10АцЛог
[Н2МеОГ] = -^-; [МеОГ] = Al2*13 ; [ОКГ]
L J [н+] L J [н+i ' L J [H+]
По разности между количеством загруженного в пульпу
реагента и суммарной концентрации С его растворенных
компонентов
С = [Ме3+] + [МеОН2+] + [Me (OH)f| + [Н2МеОГ] + [МеОГ]
можно определить количество реагента, выпавшего и виде
осадка Ме(ОНK.
Состояние в растворе комплексообразующих реагентов.
К комплексообразующим реагентам относятся_циаш1Ды, Р"А&г
.jOLOgj, £оли аммония и некоторые другие соединении,
применяемые приГ^флотацийГМетодику расчета их состояния и ригпюре
84
рассмотрим на примере совместного присутствия в пульпе
цианида и меди. В этом случае протекают реакции:
Cu(CNF^Cu+ + 2CN-; Kn= 'Cu+HCN-12
[Си (CNO]
Си (СЫ)Г ** Си (CN)F + CN-; К1в =
[Cu(CN^] [CN~]
Си (CNM_=^ Си (CN)!~ + CN~; Kv =
HCN^CN- + H+; ^-=-Ш1ШО-
[Cu(CNJ-]
[Си (CN)g~] [CN-]
[Си (CN)f-]
V18
[HCN]
E.21)
E.22)
E.23)
E.24)
Используя выражения для констант равновесия К\ъ — К\ь
и уравнения баланса по меди и цианиду:
Сси = [Си+] + [Си (CNJ~] + [Си (CN)|-] + [Си <CN)M; E.25)
Ccn = [HCN] + [CN~] + 2 [Си (CNO] + 3 [Си (CNK2~] + 4 [Си (CNKr],
E.26)
получаем систему уравнений, решая которую находим
зависимость между концентрациями цианидных и водородных ионов
[CN-] = /([H+]). E.27)
Решая уравнение E.27) одним из методов математики,
найдем значения [С№~] при различных значениях рН.
Используя их в системе уравнений [E.21) — E.26)] для Kis — Kis, Ccu
и C(CN), находим значения концентрации остальных ионных и
молекулярных компонентов [HCN], [Cu(CNJ~], [Cu(CNK2~],
[Cu(CNL~]~~nPH любых
интересующих нас значениях рН или
концентрациях медь- или цианидсодер-
жащих соединений в растворе.
Состояние в растворе реагентов,
обладающих
окислительно-восстановительными свойствами. К таким
реагентам относятся суль^г1щми1яьл +^;
J™jL„..собиратели^ соли ^железа,
хрома, марганца и некоторые
другие модификаторы "флотации. При
их подаче в пульпу реакции
гидролиза, диссоциации и осаждения
сопровождаются реакциями
окисления и восстановления соединений.
При расчете состояния таких
реагентов в растворе необходимо
сначала определить границы устойчи-
+0,5
+ 0,3
-0,1
-0,3
-0,5
кч
"
Fe(
* *
:°зЧ
■^
Л20 + Н2
F
\.
X
sN
• Ji20'+02 f
3
jFe^js
N
*4^
0 2 4 6 8 10 pti
Рис. 5.З. Границы
устойчивости солей железа (при общей
концентрации растворенных
карбонатных компонентов
в растворе 10~5 моль/л)
85
вости возможных соединений в координатах: окислительно мое»
становительный потенциал Е—рН, а затем рассчитать концеит^
рации растворенных компонентов для каждой облип и их еу-
ществования.
Применительно, например, к солям железа, возможными
продуктами осаждения которых при загрузке в пульпу миля-
ются гидроксид трехвалентного железа Ме(ОНK, гидроксид
двухвалентного железа Me (ОН) 2 и карбонат двухвалентного
железа МеСОз, такой расчет можно сделать следующим
образом. Условия межфазного перехода Ме(ОНJ^Ме(ОН)з
описываются на основании реакции
Me (ОНJ + Н20 *t Me (OHK + Н+ + е
уравнением ее потенциала
£19 = Я?9—0,059рН,
а условия межфазного перехода МеСОз^Ме(ОНK
описываются на основании реакции
MeCOs + 3H30 ** Me (ОН)8 + СОз~ + ЗН+ + е
уравнением потенциала
.^2о -= ^20 + 0,059 lg [COi"] -3■ 0,059рН.
Значение рН межфазного перехода Ме(ОНJ^МеСОз
можно определить исходя из условия Ех9 = Е2о по уравнению
H==_L ^""fa+J-ig[ooS-],
* 2 0,059 ^ 2 L J
Все реакции во флотационных системах протекают в
пределах электрохимической устойчивости воды, верхний предел
которой характеризуется разложением воды с выделением
кислорода по реакции
2Н20^02 + 4Н+ + 4е;
Еп = £21 + — • 0,059 lg [02] [Н+]4 - 1,23- 0,059рН,
4
а нижний предел — разложением воды с выделением водорода:
Н2=^2Н+ + 2е;
£22==£°22 +^.0,0591gJ^i^ = 0-0,059pH.
2 [Н2]
Границы устойчивости солей железа, полученные в
результате расчетов по приведенным реакциям и уравнениям при об-
:щей концентрации растворенных карбонатных компонентов
10~5 моль/л, изображены на рис. 5.3.
>86
Ионный состав раствора в области существования Me (ОН) %
найдем, используя следующие реакции и уравнения:
Ме(ОНJ^Ме2+ + 20Н-; /C2i = [Me2+] [ОН-]2; E.28)
Me (ОН)+ ч*Ме2+ + ОН-; /С22 = [Ме'+] [0Н~~] ; E.29>
[Me (OH)+]
Me (ОН)а ч* НМеОГ + Н+; /С23 - [НМеОГ] [Н+]; E.30>
НМеОГ^ МеОГ + Н+; Ки = [Ме0М [н+] . {531>
[НМеО^]
Рассматривая совокупность выражений E.28) — E.31) для
констант равновесий К21 — Кяа как систему уравнений и решая
ее относительно [Н+], найдем:
[Ме2+] = IlllpL. [Me (OH)+] = -^3_;
[НМе02_] =
^23
[Н + 1 '
[МеОГ] = ^^£L. [ОН-] = -£=-.
[н+]2 [н+]
Для расчета ионно-молекулярного состава раствора в
области существования МеСОз, помимо уравнений E.20), E.29) —
E.31) для Kw, K22 — ^24, используется также уравнение
/С2в = [Ме+2][СОГ],
полученное на основании реакции
МеС03 ^Me2+ + COf~,
и уравнения E.9), E.11), E.12), в которых под НгАп, НАп~
и An2- понимаются соответственно НгС03, НС03~ и С032~,
а под Со — суммарная концентрация карбонатных компонентов-
в растворе. Решив полученную систему уравнений относительна
[Н+], находим:
[Ме2+] =-^_ (ад8 + /СБ [Н+]+ [Н+]2);
[Ме (°Н)+] ° КМ*сан+] (К*К*+К* И + tH+]2);
[НМеОГ] = -^ " [МеОМ = J&*}-; ,[01Г] =-^-.
Значения [С032~], [НС03~] и [Н2С03] в растворе
рассчитываются по уравнениям E.13) — E.14).
Для расчета ионного состава раствора в области
существования Ме(ОНK используются реакции и уравнения E.16) —
E.20).
По разности между количеством загруженного в пульпу
реагента и суммарной концентрацией его растворенных компо-
87
нентов можно определить количество реагента, выпадмкшито
в виде Ме(ОНK, Ме(ОНJ или МеС08.
Соотношения между концентрациями двух- и трехвалентных
ионов металла в любой области значений потенциала Е и pll
пульпы определяются на основании реакции
Ме2+^:Ме3+ + е
уравнением
^ = ^+0,059 lgi^.
5.3. Адсорбция реагентов на поверхности раздела фаз
Адсорбцией называется ловьннвн&е!4У&няшшжвние
концентрации -того или иного вещества на поверхности раздела
фаз. Процесс адсорбции всегда протекает в сторону
уменьшения свободной энергии системы в соответствии со вторым
законом термодинамики.
Адсорбция на поверхности раздела фаз веществ,
обладающих различными молекулярными свойствами, приводит к
изменению разницы в полярности соприкасающихся фаз и,
следовательно, к изменению поверхностной энергии на. границе
их раздела. В соответствии с правилом, сформулированным
П. А. Ребиндером, вещество С может адсорбироваться на
поверхности раздела фаз Л и В, если оно в результате своего
присутствия в поверхностном слое будет уравнивать разность
полярностей этих фаз. По этой причине при адсорбции гетеро-
полярных молекул происходит их ориентация на границе
раздела фаз: полярной частью молекулы обращены к более
полярной фазе, а неполярной — к менее полярной.
При адсорбции на поверхности раздела жидкость — газ
небольшого количества гетерополярных молекул они
располагаются почти в плоскости поверхностного слоя, а при большом
количестве — перпендикулярно поверхности раздела, например,
вода—воздух (см. рис. 2.2в), т. е. занимают положение,
соответствующее минимальному значению свободной
поверхностной энергии.
Адсорбция на поверхности раздела жидкость — газ
описывается взаимосвязанными уравнениями Гиббса, Лангмюра и
Шишковского.
Уравнение Гиббса характеризует соотношение между вели-
■ чиной адсорбции Г и изменением поверхностной энергии
раствора о:
RT \дС 1st •
где С — концентрация растворенного вещества, молп/л; R —
газовая постоянная, равная 8,314 Дж/(К*моль); Oa/tH:
-частив
ная производная поверхностной энергии по концентрации (при
постоянной площади поверхности S и температуре 7),
которую называют поверхностной активностью.
Вещества, значительно понижающие поверхностную
энергию, называются поверхностно-активными; для них
да/дС<0. Поверхностно-активными являются спирты,
альдегиды, кетоны, карбоновые кислоты и другие соединения.
Вещества, мало влияющие на поверхностное натяжение,
называются поверхностно-инактивными; для них
да/дС>0. К ним относятся электролиты, сахар и др.
Для количественной характеристики адсорбции используется
уравнение Лангмюра
где Гоо — количество адсорбированного вещества при
насыщенном поверхностном слое, моль/см2; 7 — коэффициент,
характеризующий среднюю продолжительность пребывания молекул
или ионов реагента на поверхности, численно равный
разбавлению равновесного раствора, при котором Г = 0,5Гоо, л/моль;
С — концентрация поверхностно-активного вещества в растворе
в состоянии равновесия, моль/л.
Зависимость между поверхностной энергией раствора и
концентрацией растворенного вещества может быть установлена
по уравнению Шишковского
о—а0^= — ЫпA + аС),
где о и во — поверхностная энергия раствора соответственна
концентрацией С и чистого; а, Ъ — постоянные.
Адсорбция реагентов на поверхности раздела минерал —
раствор, приводящая к изменению ее свободной энергии,
является основным средством регулирования флотируемости
минералов.
Адсорбция реагентов на минеральной поверхности протекает
всегда в условиях конкуренции с молекулами воды и
сопровождается понижением свободной энергии системы. Это понижение
может быть обусловлено, во-первых, химическим
взаимодействием с образованием ионных, ковалентных, координационных
связей между ионами или молекулами реагента и химически
ненасыщенными атомами или радикалами на поверхности
раскола минерала; во-вторых, межмолекулярным
взаимодействием силами Ван-дер-Ваальса, электростатической
поляризации, электрического изображения. Первый случай получил
название химической адсорбции, второй — физической адсорбции..
Они являются крайними случаями процесса адсорбции,
описываемого единым квантомеханическим уравнением.
Принципиальное отличие химической адсорбции от
физической состоит в том [6], что при химической адсорбции
происходят электронные переходы между адсорбентом и адсорбатом*
89
которые образуют единую квантомеханическую систему с
общим электронным хозяйством. При физической адсорбции
адсорбируемая молекула или ион и решетка адсорбента могут
рассматриваться как две независимые системы, взаимодействие
которых представляет собой слабое возмущение;
адсорбированное соединение при этом химически не изменяется. Отличие
химической и физической форм адсорбции определяет следующие
их характерные особенности:
при химической адсорбции ион или молекула реагента
связываются с- поверхностью более прочно, чем в случае физической
адсорбции. Если при химической адсорбции энергия связи
адсорбированной частицы с поверхностью, твердого соответствует
теплоте образования типичных химических соединений и
достигает 1 эВ, то при физической адсорбции она соизмерима с
теплотой . сжижения или кристаллизации адсорбируемого вещества
и составляет лишь 0,01—ОД эВ;
химическая адсорбция более избирательна и специфична, чем
физическая. Так как закрепление реагентов при химической
адсорбции происходит в первую очередь на наиболее активных
участках поверхности, где больше ненасыщенных сильных связей
(вершины, ребра, незавершенные грани роста кристалла), то
она характеризуется и большей неравномерностью
распределения адсорбированного реагента на поверхности по сравнению
■с физической адсорбцией;
химическая адсорбция в отличие от физической требует
обычно большой энергии активации и поэтому с повышением
температуры скорость ее возрастает, тогда как скорость
физической адсорбции уменьшается, небольшая прочность связи
адсорбированного вещества с адсорбентом при физической
адсорбции приводит к ее обратимости, тогда как десорбция хемо-
сорбированного реагента может быть осуществлена в
основном только при вытеснении его другими ионами, способными
•образовать менее растворимое поверхностное соединение. Это
не исключает возможности перемещения хемосорбированных
частиц по поверхности. Однако преодолеваемые при этом энер-
тетические барьеры меньше энергии связи частицы с решеткой
минерала.
Характер взиамодействия реагентов с минералами и форма
их адсорбции зависят от состояния минеральной поверхности и
реагента в пульпе. Сорбционные явления на поверхности гете-
рополярных кристаллов, к которым относится большинство
минералов, могут быть разделены на следующие виды [44]:
хемосорбция неизоморфных ионов или молекул и
гетерогенная химическая реакция;
первичная адсорбция собственных и изоморфных ионов,
которая разделяется на первичную обменную адсорбцию и по-
тенциалопределяющую адсорбцию;
вторичная адсорбция, которую можно подразделить на
ионную и молекулярную.
$0
а
R"
И+ R~M+
Ж
R"
М+1
шМ
бГ
R-
RJM+
M+R-
г
М+
m+|r~
R~M+
е
M^JR~|Nn
R"M+ RH
Рис. 5.4. Формы хемосорбции:
а, б — слабая связь; е> г — прочная
акцепторная связь; д, е — прочная донор-
ная связь
Следует учитывать, однако, что
в реальных процессах трудно
четко определить границу
между ними, как и между
физической и химической
адсорбцией, протекающей в водной
среде.
Хемосорбция ионов и
молекул, неизоморфных
кристаллической решетке
адсорбента, ограничивается
поверхностным слоем. Энергии
возникающей химической связи,
характер которой■зависит от
электронной структуры
взаимодействующих веществ, недостаточно для отрыва
поверхностных атомов минерала от кристаллической решетки.
Различают [6] две формы хемосорбции — слабую и прочную.
При слабой хемосорбции связь между частицей и решеткой
осуществляется без участия свободного электрона или дырки, на
при существенном затягивании их в решетку. Значение диполь-
ного момента при этом может на несколько порядков
превышать значение дипольного момента, индуцируемого при
физической адсорбции. При прочной хемосорбции хемосорбированная
частица удерживает около себя свободный электрон
(акцепторная, или прочная гс-связь) или свободную дырку (донорная„
или прочная р-связь) кристаллической решетки. В обоих
случаях электрон или дырка принимают непосредственное участие
в образовании связи.
В качестве примера на рис. 5.4 схематически показаны
различные формы хемосорбции для частицы С.и кристалла типа
MR, состоящего из однозарядных ионов М+ и R~. Свободный
электрон в таком кристалле означает нейтральное состояние
М, блуждающее по ионам М+ решетки, а свободная дырка —
нейтральное состояние R, которое переходит от одного к
другому иону R~ решетки.
На рис. 5.4, а и б показана слабая хемосорбционная связь*
образованная в результате втягивания валентного электрона
адсорбируемой частицы в поверхностную зону кристалла
(рис. 5.4, а) или электрона из аниона решетки на
адсорбированную частицу (рис. 5.4,6).
На рис. 5.4, виг показана прочная акцепторная связь,
обусловленная захватом свободного электрона решетки
адсорбированной частицей и образованием совместно с ее валентным
электроном прочной двухэлектронной связи. Эта связь на
рис. 5.4,в соответствует чисто ковалентной, а на рис. 5.4, г —
чисто ионной связи. На рис. 5.4, д я е показаны аналогичные
случаи при образовании прочной донорной связи в результате
захвата свободного электрона частицы решеткой.
91
Различные формы хемосорбции могут переходить друг
ъ друга в результате локализации и делокализации свободного
электрона или дырки на адсорбированной частице или вблизи
нее. Хемосорбционная' способность поверхности и
относительное содержание на поверхности различных форм хемосорбции
зависят от положения уровня Ферми [6].
Хемосорбция является необходимой, предварительной
стадией для образования новой объемной фазы в результате
гетерогенной химической реакции.
Условия образования новой фазы определяются отношением
произведений растворимости соединений до и после
взаимодействия минерала с реагентом, т. е.
Cia/C^1-L1/L2= const,
где С\ и С2—концентрация конкурирующих ионов; пх и п2 —
их валентность; L\ и L2 — произведения растворимости
соответствующих соединений.
Толщина пленок новой фазы зависит от количества
реагентов и времени их контакта с минералом. Примером
химического взаимодействия реагента с минералом может послужить,
например, образование пленки сульфида меди на малахите
ъ растворе сернистого натрия, приводящее к изменению
первоначальной окраски минерала.
Чем больше соответствие параметров кристаллических
решеток образующейся пленки и минерала, тем выше прочность
закрепления пленки на минерале. При хорошем их
соответствии в месте контакта образуется как бы твердый раствор
вещества пленки с минералом. Слабее удерживаются пленки,
структурно не связанные с минералом, и значительно слабее —
пленки, образованные налипанием шламов из раствора.
Большое значение при флотации имеет обменная адсорбция
ионов. Адсорбцию их во внутренней обкладке двойного
электрического слоя называют первичной, а во внешней —
вторичной.
Первичная адсорбция может осуществляться или
в результате достройки кристаллической решетки минерала
ионами раствора, или в результате замены ионов
кристаллической решетки ионами раствора. В обоих случаях адсорбция
подчиняется правилу Панета — Фаянса, в соответствии с
которым она имеет место в том случае, когда адсорбируемый ион
с ионом минерала образует менее растворимое соединение, чем
сам минерал. Поэтому адсорбированные ионы прочно связаны
с кристаллической решеткой минерала. Поскольку достраивать
кристаллическую решетку и обмениваться с ионами решетки
могут только собственнные ионы минерала или близкие им по
^свойствам, то первичная адсорбция является специфической
(избирательной).
Если при адсорбции наблюдается эквивалентный обмен
ионов, когда количество и заряд поглощаемых поверхностью и
ч92
переходящих в раствор ионов равны и электростатическое
состояние поверхности не изменяется, то она называется
первичной обменной адсорбцией. Такая адсорбция
обычно представляет собой адсорбцию ионов, способных
образовать с одним из ионов кристаллической решетки минерала
изоморфное соединение. Она может не ограничиваться
поверхностным слоем, но идти и на глубину. Примером первичной
обменной адсорбции может служить активация сфалерита ионами
меди. Ионы меди вытесняют ионы цинка с поверхности
сфалерита, в результате чего образуется поверхностное соединение
сульфида меди, более активно взаимодействующее с ксантоге-
натом, чем сфалерит.
Неэквивалентный обмен ионов, обусловленный обычно
различной способностью ионов к гидратации, поляризуемости или
избытком одних ионов по сравнению с другими и т. д., получил
название потенциалопределяющей адсорбции, так
как она приводит к изменению заряда и термодинамического
потенциала поверхности. Потенциалопределяющая. адсорбция
ограничивается долями мономолекулярного слоя из-за сильного
отталкивания ионов, заряды которых не компенсированы
ионами противоположного знака на поверхности.
В случае обменной адсорбции ионов в общем виде и вне
зависимости от природы взаимодействия она может быть
выражена уравнением Фервея — Никольского
Г^/ГУ"*^ KC\fniIC¥\
Предложен ряд упрощенных вариантов. Например, для
ионов одинаковой валентности
iyra=kcjc2.
Для количественной характеристики адсорбции потенциал-
определяющих ионов используется уравнение
Г = а ± 6 In С,
где а и Ъ — константы.
Закрепление ионов и молекул во внешней обкладке
двойного электрического слоя носит название вторичной
адсорбции. Величина ее зависит от природы адсорбирующихся
ионов. Из ионов одинаковой валентности максимальную
адсорбционную способность проявляют ионы наибольшего
радиуса, так как такие ионы в большей степени поляризуются,
сильнее притягиваются поверхностью и меньше гидратируются
лри одном и том же значении заряда. При адсорбции ионов
различной валентности сильнее притягиваются ионы, которые
несут больший заряд.
Вторичная адсорбция может иметь место как в
упорядоченном слое противоионов, так и в диффузном. Ионы
упорядоченного слоя более прочно связаны с минералом, чем ионы
диффузного слоя, но менее прочно, чем ионы, адсорбирующиеся во
93
внутренней обкладке. Предполагается, что адсорбция катион-
ных собирателей на кварце и других силикатах происходит
в упорядоченном слое внешней обкладки. Ионы,
адсорбирующиеся в диффузном слое, очень слабо связаны с поверхностью
минерала и поэтому практически не оказывают влияния на его
флотируемость.
Для описания закономерностей вторичной адсорбции
используется чаще других уравнение Фрейндлиха
Т^КС1/т,
где т — постоянная величина больше единицы.
Часто уравнение Фрейндлиха используют в
логарифмической форме
lgr-lgK+l/mlgC-a + Z>lgC.
Следует иметь в виду, что при современном состоянии
теории адсорбции, нереально проводить количественные расчеты
ее величины на неоднородных поверхностях, характерных для
минералов. Приведенные уравнения служат для приближенной
оценки адсорбции.
При использовании нерастворимых в воде жидких
реагентов взаимодействие происходит в результате смачивания:
капельки реагента, сталкиваясь с минералом, частично
растекаются по его поверхности, что так же, как и адсорбция
растворимых, реагентов, изменяет флотационные свойства минералов.
6. ВЛИЯНИЕ ОСНОВНЫХ ФАКТОРОВ НА СОСТОЯНИЕ
ПОВЕРХНОСТИ МИНЕРАЛОВ
В ОТСУТСТВИЕ СОБИРАТЕЛЕЙ
Состояние поверхности минералов зависит от их
химического состава, особенностей кристаллической структуры,
электрофизических свойств и ионного состава жидкой фазы пульпы.
Знать состояние поверхности основных минералов необходимо
для достоверного анализа механизма действия реагентов с
поверхностью минералов, выявления причин и закономерностей
взаимного влияния минералов на их окисление, активацию и
сорбцию собирателя в процессах измельчения и флотации.
6.1. Влияние рН и окислительно-восстановительного
потенциала пульпы на состояние поверхности
минералов
Состояние поверхности силикатных минералов рассмотрим
на примере кварца Si02.
В кварце каждый атом кремния связан с четырьмя атомами
кислорода, а каждый атом кислорода — с двумя атомами крем-
94
О о 0он О но onmw-wM
Рис. 6.1. Кристаллическое строение кварца (а) и поверхность его раскола
в воздухе (б) и воде (в)
ния (рис. 6.1, а), что схематически можно изобразить
следующим образом:
Si02]m = Si—О—Si = [SiO*]n,
где три связи при Si означают, что находящийся на
поверхности атом кремния связан с тремя атомами кислорода решетки
кварца, обозначенной символами [Si02]m и [Si02Jn.
При разрушении кристаллов кварца на их поверхности
обнажаются атомы кремния и кислорода с ненарушенной связью,
образующие так называемые силоксановые группы Si—О—Si,
и разрываются связи между кремнием и кислородом с
образованием ненасыщенных связей при каждом атоме, а именно
ISi02]m=Si—О— и —Si=[Si02]« (рис. 6.1, б).
Если разрушение происходит в воде, то по каждому месту
разрыва связей поглощается по одному иону Н+ и по одному
иону ОН~ (рис. 6.1, в) и поверхность кварца покрывается си-
ланольными группами^ Si—ОН:
^[st02]m-Si-0H + OH-St=[StO2]n.
Приближенно можно считать, что 50 % поверхности раскола
кварца занято силоксановыми группами и 50%—силаноль-
ными, причем суммарное взаимодействие диссоциированных и
недиссоциированных силанольных групп с водой в 1,5 раза
•больше, чем молекул воды между собой [39].
Заряд поверхности кварца зависит от значения рН пульпы.
Например, в кислой среде при большом количестве водородных
ионов протекает реакция
!Si02]m KSi-OH + H+ -* [Si02]m = Si+ + НА
в результате чего поверхность кварца оказывается
положительно заряженной. В щелочной среде при большом количестве
95
Рис. 6.2. Влияние рН на значение ^-потенциала кварца (У), ^-потенциала
поверхности кальцита B), значение его g-потенциала (З), а также на
концентрацию С ионов Са2+ D), UCOJ E), СО|~ (в), СаОН+ (8) и молекул
Н2С03 G) в растворе в присутствии кальцита
гидроксильных ионов поверхность кварца заряжается
отрицательно в результате протекания реакции
^[5i02]m^SL-0-+H20.
О характере изменения потенциала поверхности кварца при
различных значениях рН можно судить по изменению значений
его ^-потенциала (рис. 6.2, кривая 1), являющегося частью
термодинамического потенциала ф (см. рис. 2.3, б), поскольку
характер изменения обоих потенциалов в рассматриваемых
условиях при рН от 2 до 11 совпадает. Энергия взаимодействия
с молекулами воды и гидрофилизация поверхности кварца
возрастают с увеличением степени диссоциации силанольных групп
и значения потенциала поверхности.
Состояние поверхности карбонатных минералов
щелочноземельных металлов можно рассмотреть на примере кальцита
СаС03.
Свежеобнаженная поверхность данного минерала
представлена катионами кальция и карбонатными анионами. От
соотношения этих потенциал определяющих ионов на поверхности
зависят ее заряд, потенциал и значение свободной поверхностной
энергии минерала. Изменить количественное соотношение по-
тенциалопределяющих ионов на поверхности можно
изменением концентрации их в растворе. Это можно сделать, изменяя
количество загружаемого кальций- или карбонатсодержащего
реагента или изменяя состояние самого реагента в растворе,
поскольку соотношение концентраций кальцийсодержащих
ионов Са2+ и СаОН+, а также молекулярной Н2С03 и ионных
96
НСОГ и СОз" форм углекислоты в растворе определяется
значением рН в соответствии с реакциями E.8), E.10), E.29).
Для расчета ионного состава раствора при различных
значениях рН в присутствии кальцита необходимо учесть
уравнения E.9), E.11), E.20), E.29) и уравнения баланса:
Сса-[Са2+] + [СаОН+];
[Са2+] + [СаОН+] - [СО§-] + j НСОГ] + [Н2С03]-С0,
где Сса—суммарная концентрация кальцийсодержащих
компонентов: С0 — суммарная концентрация углеродсодержащих
компонентов в растворе вследствие растворения углекислоты
воздуха или подачи реагентов (например, соды).
Решая полученную систему уравнений, находим:
К22 [Н+] AН+]« + /С5[Н+] + KtK6) [COf-f- К<К5КЮС0 х
х [Н+] [СО*-] -К4К5К2& (К22 [Н+] + Ка) - 0. F.1)
Использовав в системе перечисленных выше уравнений
рассчитанные по уравнению F.1) значения [С02~3], найдем
концентрацию остальных компонентов раствора:
[Са2 +1 - Ki* ; [Са (ОН)+1 ^^ ;
1 J [со32-] l j к22 [н-] [со|-]
[н+1 IcoM r „ n [н+|2 Гсо§-1 •
[он-!
к,
W
[н+]
В свою очередь, потенциал поверхности кальцита можно
рассчитать на основании реакции
Са+С02-^гСаС03 + 2в
(А/70реакции =—601,51 кДж) ПО ураВНСНИЮ
£-- 3,11— 0,5- 0,059 lg [COf-].
Результаты проведенных расчетов и экспериментальных
определений значений ^-потенциала поверхности кальцита при
регулировании рН раствора едким натром и концентрации
растворенной углекислоты воздуха С0=Ю~5 моль/л приведены на
рис. .6.2.
Уменьшение значений рН приводит к уменьшению
концентрации ионов СОз~ в растворе (см. рис. 6.2, кривая 6) и на
поверхности и отрицательных значений потенциала кальцита
(кривая 2). Увеличение рН, наоборот, сопровождается увеличением
концентрации ионов COf~~ в растворе и на поверхности и
отрицательных значений потенциала минерала.
При повышении рН, вызывающем подавление гидролиза и
увеличение концентрации СОз~ наблюдается также рост
4 Заказ № 1957
97
Щпл
Bf5
0,3
0
-ofi
-аз
:j;-b \ \Ог+^о|
: А \
Fe(OH)j \ j
ч \
^ \
Ч^ \
\v \ \
*ДФ\ \ >е(°НЬ
YVU^^\ \
АХлчл
\s\ \ W К
"\ f\Fe(°HJ
1 L _lV_L _\\ i
If Z Ч 6 8 10 ЯрН
8- Ю рН
си*+\ \
ке(он)Д Ре(онK \
1^к. \
lcu20,Fe2>ss^ \
Cii2@HJC03
Fe@HK
4Fe2++Fe@NK
Cu(OH)z "j
Fe(OHj3 1
K^ ^\o^^^e^^2°+Fe^3 i
L >V\ ^^^<:4^CU2s+FeC?34Xs^
^Ctt^eS^ х*чКл;и25+РеИ2
Г h+W ^\ ^<Щ^)г
/2 pH 0
«7 /2 pW
Рис. 6.3. Влияние рН и окислительно-восстановительного £7*-потенциала
раствора на состояние поверхности галенита (а), пирита (б), халькозина (в),
борнита (г), сфалерита (д) и пентландита (е)
отрицательных значений ^-потенциала (см. рнс. 6.2, кривая 3),
Наружная обкладка двойного электрического слоя при этом
формируется из ионов Са2+, Na+ и Н+. Наоборот, при снижении
рН и уменьшении концентрации потенциалопределятощих ионов
С02~з в растворе (в результате смещения равновесия в сторону
гидролиза) в случае кальцита достигается не только изоэлек-
Трическая точка*, но и перезарядка его поверхности, и в роли
противоионов уже выступают отрицательно заряженные ионы
СОз™, НСО~з и ОН~. Действие содовой среды и среды
сернистого натрия на g-потенциал поверхности кальцита
принципиально не отличается от действия среды едкого натра (см.
рис. 6.2, кривая 3} и приводит лишь к незначительному сдвигу
максимальных отрицательных значений ^-потенциала в сторону
менее щелочной области. При этом максимальные
отрицательные значения ^-потенциала поверхности кальцита ' по
сравнению со средой едкого натра несколько возрастают.
Существенным отличием сульфидных минералов от
силикатов и карбонатов является зависимость состояния их
поверхности не только от значения рН, но и от значения окислительно-
восстановительного потенциала Eh раствора, значение которого
зависит от концентрации растворенного кислорода и
соотношения окисленных и восстановленных форм реагентов в пульпе
[1, 3]. Результаты экспериментальных исследований
показывают, что значения ^Л-потенциала жидкой фазы пульпы как
в лабораторных, так и в промышленных чусловиях таковы, что
свежеобнаженная поверхность сульфидных минералов в
неизменном виде существовать не может. Она термодинамически
неустойчива и под действием растворенного кислорода воздуха
окисляется с образованием различных продуктов окисления
[3, 25, 29].
д е
4* 99
с Так, поверхность галенита в обычных условиях
представлена (рис. 6.3, а) при рН меньше 1,6 сульфидом свинца и эле-1
ментарной серой; при рН от 1,6 до 6,74 сульфатом свинца; при
рН от 6,74 до 9,06 карбонатом свинца; при рН от 9,06 до 12
гидратокарбонатом свинца; при рН больше 12 начинается
растворение поверхности с образованием НРЬО~2 [1].
Полученные результаты по изменению состава окисленных соединений
свинца в нейтральной и щелочной среде. можно использовать
для оценки состояния не только галенита, но и таких
свинцовых минералов, как англезит и церуссит.
При высоких положительных значениях £Л-потенциала
раствора преимущественным продуктом окисления сульфидов
железа (пирита, марказита, пирротинов) является гидроокись
железа. При менее положительных (или более отрицательных)
значениях £7г-потенциала могут образовываться или карбонат,
или гидрозакись железа [1]. О граничных условиях
существования различных продуктов окисления можно судить по
диаграмме Eh—рН для пирита (рис. 6.3, б). Подобные диаграммы
для других сульфидных минералов железа отличаются от
приведенной только значениями потенциалов начала окисления
сульфидов [1].
Продуктами окисления не содержащих железа сульфидов
меди (ковеллина, халькозина) в области положительных
значений JS/i-потенциала раствора могут быть гидратокарбонат,
гидроокись двухвалентной или окись одновалентной меди, а в
области более отрицательных значений f/i-потенциала
раствора— металлическая медь или сульфид одновалентной меди-
(рис. 6.3, в). На поверхности железосодержащих сульфидов
меди (халькопирита, борнита), помимо названных окисленных
соединений меди, присутствуют также окисленные соединения
железа (рис. 6.3, г).
Диаграммы для разных сульфидов меди отличаются друг
от друга только значениями потенциала межфазного перехода
сульфид меди — окисленное соединение металла (или
металлов). Полученные диаграммы Eh—рН для сульфидных
минералов меди [1], в том числе изображенные на рис. 6.3, могут быть
использованы также для оценки состояния самородной меди,
куприта, малахита и азурита при различных значениях рН и
£/г-потенциала раствора [1, 3].
Основным продуктом окисления сульфида молибдена —
молибденита является гидратированная трехокись молибдена
МоОз • Н20. Кристаллохимические особенности молибденита
позволяют предполагать, что его окисление протекает главным
образом по краям незавершенных плоскостей роста и торцевым
участкам кристаллов.
Преимущественным продуктом окисления сульфидов цинка
примерно до рН 5 является карбонат цинка, а при более
высоких значениях рН — гидроокись цинка (рис. 6.3> д).
Слабокислая, нейтральная и слабощелочная среды характеризуются од-
100
новременным присутствием на поверхности обоих названных
продуктов окисления. На поверхности железистых образцов
сфалерита (марматита) в состав продуктов окисления, помимо
гидрата или карбоната цинка, входят в зависимости от
значений рН и £/ьпотенциала гидроокись, гидрозакись или карбонат
железа (рис. 6.3, е).
Диаграмма состояния основного сульфида никеля — пент-
ландита в координатах Eh—рН приведена на рис. 6.3, е. Она
показывает, что основным окисленным соединением никеля на
поверхности минерала является его гидроксид. Аналогичные
результаты получены [1] на другом сульфиде никеля — хизлеву-
дите Ni3S2.
Диаграммам Eh—рН для различных сульфидных минералов
присущи следующие особенности:
образование элементарной серы на сульфидной поверхности
минералов возможно лишь в кислой восстановительной среде
и в значительной мере зависит от концентрации
серосодержащих компонентов в растворе;
возможность окисления сульфидной поверхности
определяется f/i-потенциалом раствора, значение которого в условиях
флотации практически всегда более положительно, чем
необходимо для окисления сульфидов, причем вероятность
окисления возрастает с увеличением значения рН пульпы.
Характер конечных продуктов окисления не изменится, если
сера минерала будет окисляться не до S02~4 или S202~3, а до
других сульфоксидных ионов.
Значения £/г-потенциала (при рН 7—12) суспензий
различных сульфидных минералов (см. кривые Eh на рис. 6.3) и
промышленных пульп находятся в области существования РЬС03
или РЬ3(ОНJ(СОзJ — для сульфида свинца; Fe(OHK —для
сульфидов железа; Zn(OHJ— для сульфидов цинка;
Си2(ОНJС03 или Си (ОН) 2 — для несодержащих железа
сульфидов меди и этих же окисленных соединений меди в смеси
с Fe(OHK— для железосодержащих сульфидов меди;
Мо03Н20 — для молибденита; Ni(OHJ — для хизлевудита;
смеси Ni(OHJ с Fe(OHK— для пентландита.
В обычных условиях перечисленные соединения являются
конечными продуктами окисления сульфидов. В этом случае
потенциалы начала окисления свежеобнажеипой поверхности
минералов являются потенциалами межфазного перехода
сульфидов в конечные продукты окисления и их можно рассчитать
на основании реакций
MeS +1H20 + fXp~ -> Me (OH)a X, + pSnO£- + yH+ + те F.2)
по уравнениям вида
Е - Е° - -^- 0,059рН + -£- 0,059 lg [SflCtfr] —
т т
-J- 0,059 lg[Xp-j. F.3)
т ■ - - . .•
1G1
Рис. 6.4. Принципиальная схема
состава поверхности окисляющегося
сульфидного минерала:
/ — неизмененный сульфид; II, III —
соответственно промежуточный и конечный
продукт окисления; X — анион SO ~~
SO^~", СО!*-, ОН~~ и т. д.
Значения потенциалов поверхности сульфидных минералов,
рассчитанные по уравнению F.3) и определенные
экспериментально (например, по кривым спада потенциала после
предварительной катодной поляризации), практически совпадают (см.
рис. 6.3, а, б), подтверждая достоверность образования на
поверхности перечисленных окисленных соединений [1, 3].
Вполне очевидно, что процесс окисления свежеобнаженной,
например при измельчении, поверхности сульфидных
минералов до конечных продуктов включает в себя образование
промежуточных продуктов (типа сульфидо-карбонатов, сульфидо-
гидратов и др.)- Можно полагать, что образующаяся окисная
пленка в этом случае состоит из тонкого (порядка
мономолекулярного) плотно прилегающего слоя промежуточного
продукта окисления и наружного пористого слоя конечного
продукта окисления (рис. 6.4).
Растворимость поверхностного (промежуточного)
соединения является промежуточной между растворимостью самого
минерала й растворимостью объемной фазы конечного продукта
окисления. Необходимую при анализе механизма действия
реагентов равновесную концентрацию ионов по отношению к
промежуточному продукту окисления можно рассчитать без
всяких допущений из условия равновесия сульфид — окисленное
соединение [3].
Например, закономерность изменения концентрации ионов
Fe2+, равновесных промежуточному продукту при окислении
пирита FeS2 до гидроокиси трехвалентного железа Fe(OHK>
может быть определена из выражений для потенциалов Е
одинаково справедливых в этих условиях реакций:
FeS2 + 8Н20 ч* Fe2+ + 2SOJ- + 16Н+ + Не;
£и= +0,362— —0,059рН + —0,059 lg'fSOM +
7 7 l j
+ +0,059 lg[Fe2+];
14
Fe2+ + 3H20 ^r Fe (OHK + 3H+ + e;
£14= 1,056—3-0,059 pH — 0,059 lg[Fe2+].
Поскольку в рассматриваемых условиях £12 = £i4,
приравнивая выражения для них, получаем уравнение, характеризующее
< х
;ме
-Г
;Х
:Х
ме;
Ж
тпкт
102
значение [Fe2+] в условиях перехода Ре5г-ИРе(ОН)з при
различных значениях рН (при [SO2~~4]==10_3 моль/л):
lg[Fe2+] = llL--^-pH. F.4)
10
Аналогично на основании выражений для потенциалов Е
реакций:
FeS2 + 4Н20 ^ FeOH+ + S2032~ + 7Н+ + бе;
ЕХ1 - 0,526 + — 0,059 lg[FeOH+] — — 0,059 рН +
б б
+ ^ 0,059 lg|S202-];
FeOH+ + 2Н20 ^ Fe (ОН)8 + 2Н+ + е\
Е19 -- 0,546- 0,059 lg [FeOH+] — 2.0,059 рН
при Е\7 = Е{9 получим уравнение
lg [FeOH-] = 0,29- ± рН _ ± lg [sfit] , F.5)
характеризующее значение [FeOH+] в условиях перехода
FeS2-^Fe(OHK при различных значениях рН пульпы.
Возможность расчета концентрации любых интересующих
нас ионов, равновесных поверхностным соединениям
окисляющихся в условиях измельчения и флотации сульфидов,
позволяет провести достоверный анализ механизма активации и
действия реагентов на минеральной поверхности.
Окислению в процессе добычи и переработки подвергаются
и каменные угли. Одно из возможных поверхностных
соединений угля с кислородом имеет следующую структуру [7]:
О О О
/\ /\ /\
-с с —с с—с с—
II II II II II II
В зависимости от степени метаморфизма углей и
интенсивности процесса окисления на поверхности могут образовываться
не только карбонатные —С = 0, но и другие, например,
карбоксильные —СООМ группы. Активное взаимодействие этих
групп с молекулами воды приводит к гидрофилизации
окисленных участков поверхности. Степень гидрофилизации
поверхности увеличивается при диссоциации поверхностных групп с
изменением рН пульпы.
Особенностью ионных кристаллов растворимых солей
(NaCl, KC1 и др.) является равномерно чередующееся
расположение противоположно заряженных ионов как в самом кри-
103
сталле (см. рис. 1.3, б), так и на обнаженных плоскостях при
потенциалах нулевого заряда. Последнее обусловливает
антипараллельное расположение молекул воды на поверхности
ионного кристалла (см. рис. 2.5, б), что приводит к частичному
замыканию противоположно расположенных концов соседних
молекул друг с другом. Такое замыкание на себя водородных и
электростатических (дипольных) связей молекул воды,
сорбированных на ионных кристаллах, ослабляет связи этих молекул
с объемной водой, что и приводит к частичной гидрофобизации
поверхности ионных кристаллов в воде. Проведенные
исследования по флотации частиц хлористого .натрия в насыщенном его
растворе подтвердили значительную гидрофобизацию
поверхности флотируемых частиц, которую нельзя отнести за счет
высаливающего эффекта [40].
6.2. Влияние изоморфизма и электрофизических
свойств минералов на состояние их поверхности
Изоморфизм характеризуется замещением определенных
атомов или ионов кристаллической решетки данного минерала
другими атомами или ионами. Замещающие атомы или ионы
имеют близкий размер (атомный или ионный радиус). Разница
в ионных радиусах у замещающих ионов не превышает 15 %.
Чаще всего наблюдается изовалентный изоморфизм,
когда замещаются ионы, атомы или группы атомов
одинаковой валентности. В этом случае замещение происходит по изова-
лентным рядам, соответствующим вертикальным направлениям
Периодической системы элементов Д. И. Менделеева
(например, Rb+-^Cs+; Nb5-VTa5+; Sr2+-^Ba2+ и т. д.). Однако
известно много случаев гетеровалентиого
изоморфизма, когда происходит замещение ионов разной
валентности. Здесь могут замещать друг друга соседние элементы
Периодической системы, расположенные по диагонали (слева —
вниз направо). Эти элементы имеют близкий размер
(например Ca2+-^Y3+; Mg2+-+Sc3+; Na+-^Ca2+ и т. д.).
При гетеровалентном замещении электростатическое
равновесие внутри кристалла нарушается, поэтому для соблюдения
принципа электронной нейтральности гетеровалентный
изоморфизм всегда сопровождается замещением или вхождением
других ионов. Сумма валентностей у замещающих ионов
обязательно должна быть равна сумме валентностей замещенных.
Изоморфные примеси наблюдаются у многих минералов.
Например, в сфалерите ионы цинка часто замещаются ионами
железа и кадмия, в вольфрамите происходит замещение железа
марганцем; в апатите наблюдается замещение кальция
редкоземельными элементами; во флюорите кальций замещается
иттрием и т. д. Количество изоморфных примесей бывает
различно, в ряде случаев оно оказывается настолько большим, что
учитывается в написании химической формулы. Например,
104
вольфрамит.имеет формулу (Fe, Mn)W04, а пентландит— (Ni,
Fe)9S8. Изоморфная примесь оказывает заметное влияние на
состав и структуру продуктов окисления, поверхностные и
флотационные свойства минералов. Известно, например, что флоти-
руемость сфалерита зависит от содержания в нем железа, а
присутствие даже небольших количеств ионов меди в сфалерите
вызывает повышение его флотоактивности.
Большое значение для характеристики поверхностной
активности минералов имеют их электронные свойства, связанные
с наличием в минералах подвижных электронов и дырок. Эти
свойства наиболее детально изучаются на полупроводниках и
их часто называют полупроводниковыми свойствами.
Полупроводниковыми свойствами обладает большое число
минералов: многие сульфиды, селениды, теллуриды, арсениды,
оксиды ртути, куприт, флюорит, сера и др.
Анализ структур электронных оболочек атомов и
кристаллических структур различных соединений приводит к выводу,
что полупроводниковыми свойствами обладают соединения,
в состав Которых входит по крайней мере один элемент из IV—
VII групп Периодической системы элементов Д. И.
Менделеева.
Носители тока в полупроводниках — электроны и дырки —
освобождаются в результате теплового движения, а также под
воздействием света.
Электроны поставляют основные атомы решетки и примеси,
имеющие большую валентность, чем соответствующий
компонент решетки. Обычно основное количество электронов
поставляют примеси, хотя число их атомов гораздо меньше, чем число
атомов решетки.
Если примеси имеют электроотрицательный характер, то
они присоединяют электроны атомов кристаллической решетки,
образуя узлы с недостатком электронов — дырки, которые
могут переходить в результате теплового движения от одного
атома к другому.
В зависимости от преобладающей роли свободных
электронов или свободных дырок в переносе тока различают
полупроводники n-типа с электронным типом проводимости и р-типа
с дырочным типом проводимости.
Полупроводниковые свойства минералов оказывают
большое влияние на характер адсорбции (см. рис. 5.4),
распределение ее форм и общую величину [6, 48, 49].
Кроме того, проводимость минералов и значение потенциала
их поверхности определяют характер изменения состояния
поверхности минералов под действием кислорода воздуха и воды
уже в процессах добычи, транспортировки, дробления и
измельчения полезного ископаемого. Установлено, например, что
скорость окисления сульфидов, находящихся в контакте, зависит
от разности их потенциалов. Сульфид с более отрицательным
потенциалом окисляется быстрее, чем в отсутствие контакти-
105
Рнс. 6.5. Влияние рЙна потенциал <р
начала окисления меди A),
халькозина B), борнита C), ковеллина
D), халькопирита E), галенита F),
сфалерита G), марказита и
пирита (8), миллерита (9), пентландита
и хизлевудита G0), пирротина (П),
никеля A2) и железа A3) при
окислении серы минералов до S2O3
[3]
Wfr7 8 9 ~W 11 рН
рующего сульфида, и, наоборот, сульфид с более
положительным потенциалом окисляется медленнее.
На основании экспериментально подтвержденных
результатов расчетов потенциалов свежеобнаженной поверхности
сульфидов (рис. 6.5) по уравнениям вида F.3) на основании
реакций типа F.2) сульфидные минералы по начальной скорости их
окисления можно расположить в следующий ряд: пирро-
тин>пентландит и хизлевудит>миллерит>марказит и пи-
рит>сфалерит>галенит > халькопирит > ковеллин >> борнит>
>халькозин. Глубина окисления поверхности и конечная
скорость процесса определяются уже структурой образующихся
пленок.
Значение потенциала минералов оказывает большое
влияние не только на возможность и скорость их окисления (см.
рис. 6.5), строение гидратного слоя на поверхности (см. рис. 2.5)
и степень ее гидрофильности, но и на сорбцию ионов и молекул
реагентов. Большие отрицательные значения потенциала
затрудняют физическую адсорбцию минералом анионов и
облегчают адсорбцию катионов. Положительный заряд оказывает
противоположное действие. Адсорбция молекул достигает
максимального значения в области потенциалов, близких к
потенциалу нулевого заряда, и уменьшается до нуля при больших
зарядах поверхности [13].
Таким образом, изоморфизм и электрофизические свойства
минералов могут значительно изменять состояние их
поверхности и тем самым влиять на взаимодействие с реагентами,
адсорбцию реагентов и флотируемость минеральных частиц.
6.3. Влияние кристаллохимического строения минералов
на состояние их поверхности
Влияние кристаллохимического строения минералов на
состояние их поверхности рассмотрим на примере' талька,
имеющего слоистое строение (рис. 6.6). Поверхность спайности
структурных пакетов талька (см. рис. 6.6, а), состоит только из
атомов кислорода силоксановых групп Si—О—Si (см. рис. 6.6,
б), погасивших свои валентные связи с атомами кремния. По-
106
Рис. 6.6. Поперечный разрез структурных пакетов fa) и плоскости
спайности (б) талька
этому структурные пакеты талька слабо связаны друг с другом.
Взаимодействие атомов кислорода силоксановых групп с водой
также определяется в основном дисперсионными силами и
водородной связью. При этом энергия взаимодействия атомов
кислорода силоксановых групп с водой в 2 раза меньше энергии
взаимодействия молекул воды друг с другом [39], лто является
причиной гидрофобности плоскостей спайности талька.
Поверхность измельченных зерен талька должна состоять
как из плоскостей спайности, почти полностью насыщенных
в валентном отношении и вследствие этого частично
гидрофобных, так и из торцевых участков, образующихся в результате
разрыва алюмокремнекислородных слоев и представляющих
поверхность ненасыщенных связей алюминия, кремния и
кислорода. Ненасыщенная поверхность торцевых участков талька
интенсивно реагирует с водой с образованием на ней гидроксид-
ных групп, причем водород присоединяется к атому кислорода
на поверхности, а гидроксид — к атомам кремния и алюминия.
В результате вся поверхность торцевых участков оказывается
покрытой гидроксидными группами, которые являются
основными центрами обратимой физической адсорбции воды,
протекающей путем образования водородных связей между ОН-груп^
пами поверхности и кислородом адсорбированных молекул
воды.
Знак и значение заряда, а также наличие катионов на
торцевой поверхности талька в значительной степени
определяются значением рН раствора. Это обусловлено как амфотер-
ными свойствами самого алюминия, так и зависимостью
степени диссоциации Si—ОН и А1—ОН групп, расположенных на
107
торцевых участках минеральных !зерен, от концентрации
водородных ионов.
Таким образом, поверхность измельченных зерен талька и
других подобных ему минералов должна состоять как из
частично гидрофобных участков, являющихся причиной флотируе-
мости без собирателя, так и сильно гидратированных участков.
Учитывая, что сильно гидратированная поверхность торцевых
участков является и наиболее активной по,отношению к
реагентам, вводимым в целях изменения состояния поверхности
рассматриваемого минерала, флотируемость талька
определяется главным образом, состоянием поверхности торцевых
участков и возможностью их^ гидрофобизации под действием тех или
иных флотационных реагентов [3].
Анализ кристаллической структуры и характера
обнажающихся связей при разрушении кристалла позволяет провести
предварительную оценку поверхностных свойств минерала..
В частности, предложенная Г. С. Стрельцыным, а затем
А. М. Годеном [8] систематизация структур кристаллов по
типам связи атомов и групп атомов между собой, учитывающая,
что кристаллы разрушаются в основном по
кристаллографическим плоскостям с ослабленными силами, позволяет [1, 8, 40,
44]:
объяснить природную гидрофобность таких минералов, как
графит, самородная сера, тальк;
оценить характер взаимодействия собирателя с минералами
и выявить причины гидрофобизации их поверхности;
объяснить явления избирательного взаимодействия
флотационных реагентов на различных плоскостях одного и того же
кристалла;
объяснить различную гидрофобизацию разных минералов
в присутствии одного и того же реагента;
предсказать в ряде случаев наиболее вероятный тип
собирателя, способного погасить некомпенсированные связи на
минеральной поверхности^ и обеспечить тем самым ее
гидрофобизацию;
объяснить особенности гидратации и строение гидратных
слоев на поверхности различных минералов.
Учет особенностей кристаллической структуры минералов
и характера связей на поверхности необходим при
исследовании механизма действия реагентов при флотации несульфидных
минералов, структура и состав поверхности которых не
меняется существенно под действием ионных и молекулярных
компонентов жидкой фазы пульпы. При этом следует учитывать, что
при измельчении руды разрушение минералов происходит не
строго по плоскостям спайности. Как показывают наблюдения,
плоскости разрушения минералов являются бугристыми и
содержат много макро- и микротрещин и пор. В результате этого
отдельные структурные элементы кристалла, расположенные
в разных частях образованной поверхности, имеют разную проч-
108
-ность связи с решеткой минерала и разную насыщенность, т,_е,
-поверхность энергетически неоднородна. В пределах
флотационной крупности энергетическая неоднородность поверхности
крупных, и мелких зерен примерно одинакова [44].
7. СОБИРАТЕЛИ И МЕХАНИЗМ ИХ ДЕЙСТВИЯ ПРИ ФЛОТАЦИИ
7.1. Строение молекул и классификация собирателей
В качестве собирателей предложено большое число
органических соединений. Классификация основных групп
собирателей, наиболее широко используемых при флотации различных
типов минерального сырья, приведена на рис. 7.1.
Поскольку собиратели применяются для гидрофобизации
поверхности минералов, то в состав их молекул в
обязательном порядке входят аполярные группы атомов. Если молекулы
собирателя состоят только из углеводородов, то такие
собиратели называются аполярными, неполярными,
углеводородными маслами,
Гораздо чаще при флотации используют гетерополяр-
ные собиратели, молекулы которых кроме углеводородного
радикала алифатического или реже циклического ряда, т. е.
аполярной (или неполярной) части, имеют и полярную группу.
Полярная группа собирателя определяет его химические
свойства и - способность закрепляться на полярных минералах,
поэтому она называется еще солидофильной или
функциональной группой. Поперечное сечение ее и вследствие этого
Собира т ели
Аполярные
Углеводородные масла,
керосин и др.
Неионогенные
Дисульфиды, /пиано карда -
маты, альдегиды и эфиру
ксантогеновых и амиды
карбоновых кислот
С неопределенным химичее-
ким составам
Смоли, сульфированный и
окисленный керосин и др.
Ионогенные катионные-.
Первичные, вторичные и
трети vHi/е амины, амма -
ниевые,' фосфониевые и ар-
сониевые соединения
Оксигидрильные
(органические производные
кислородсодержащих кислот)
4TS
111
i*&
II
[ Сульфгидрильные ~]
(органические производные
1 тиокислогп и сероводорода)
\Т
*
LL
f$l
St Л
a^
4&
1-1
S^t
n 1
Рис. 7.1. Классификация основных групп собирателей
109
площадь, занимаемая одной молекулой на поверхности
минерала, обычно больше поперечного сечения углеводородной цепи,
равного 18,5 «Ю-20 м2. Так, например, в условиях монослойного
покрытия на одну молекулу (или один ион) приходится для
ксантогената—29-10-20 м2, для лауриламина — 20,3.10~20 м2.
Прочность закрепления гетерополярного собирателя на
поверхности определяется энергией связи функциональной группы
с минералом, зависящей от ее характера и природы минерала,
а также энергией дисперсионного взаимодействия
углеводородных радикалов в адсорбционном слое, возрастающей с
увеличением длины углеводородного радикала [44]. Эффективность
действия собирателя поэтому может быть изменена или
изменением характера функциональной группы, или изменением
углеводородного радикала.
Собиратели с неопределенным химическим
составом (смолы, сульфированный и окисленный керосин и др.)
представляют собой смесь аполярных и гетерополярных
органических соединений.
В зависимости от характера полярной группы гетерополяр-
ные собиратели могут быть ионоген ным и, т. с. обладать
способностью к диссоциации, или н е и о н о г е н н ы м и, т. е.
не обладать такой способностью.
Из неионогенных гетерополярных собирателей наибольшее
распространение получили:
диксантогениды
S S
^ ч
R — О — С — S— S—С —0 —R ?
тионокарбонаты
S
Rj —0-C —NH—R2,
тиоангидриды ксантогеновых кислот
S S
Ч •
^—о—с—s —с—о—к2 9
эфиры ксантогеновых кислот
f?1 — О — С — S— R2.
Если при диссоциации ионогенного гетерополярного
собирателя углеводородная часть молекулы входит в состав аниона,
то он называется анионным, если в состав катиона — катил
онным собирателем. Строение молекул гетерополярных
анионных (на примере бутилового ксантогената калия) и катион-
110
а
б
в
Рис. 7.2. Структура гетерополярных молекул катионного (а), анионного (б)
собирателей и стереохимическая модель иона бутилового ксантогената (в)
ных (на примере хлористого лауриламина) собирателей,
а также состав анионов, катионов и стереохимическая модель
иона бутилового ксантогената. изображены на рис. 7.2.
Наиболее важное практическое значение из катионных
собирателей имеют органические производные аммиака — амины.
В зависимости от числа атомов водорода, замещенных в
аммиаке на углеводородный радикал R, различают первичные
RNH2, вторичные R2NH, третичные R3N амины и четвертичные
аммониевые основания R4NC1.
Из анионных собирателей наиболее важное практическое
значение имеют органические производные угольной,
фосфорной, серной, мышьяковой кислот и соответствующих им тиокис-
лот, а также алкилгидраксамовые кислоты и их соли.
Органические производные кислородсодержащих кислот,
характеризующиеся наличием в их полярной части или оксигид-
рильной группы —О—Н (у кислот) или группы —О—Me (у
солей), получили название оксигидрильных
собирателей. Важнейшими представителями таких собирателей,
применяемых в основном для флотации несульфидных минералов,
являются:
производные угольной кислоты — карбоновые кислоты и их
мыла
' ^ (Л
R — С— ОН(Ие),
производные серной кислоты — алкил- (арил-) сульфаты '
О
R—о—s—он(ме)
Ч
о
111
и сульфонаты
R — S — ОН(Ме),
\
О
производные фосфорной и мышьяковой кислот —алкил-
(арил-) фосфорные
R —О О
\/
Р
R—о он(мё),
фосфоновые
фосфиновые
R О
\ /
Р
/\
но он,
R /О
\/
Р
/■\
R ОН,
\ /
и арсоновыс ^s кислоты, а также алкилгидроксамо-
/ \
но он
вые кислоты и их соли
R О
С
R NH —ОН(ме).
.Органические производные тиокислот и тиаспиртов,
характеризующиеся наличием у них сульфгидрильной группы —S—Н
(у кислот) или группы —S—Me (у солей), объединяются под
общим названием сульфгидрильные собиратели. Эти
собиратели применяются в основном для флотационного
извлечения сульфидных минералов, а также благородных металлов
112
и самородной меди. Важнейшими представителями сульфгид-
рильных собирателей являются:
производные дитиоуголыюй кислоты — ксантогенаты
S
//
R —О —С —S —Me,
дитиофосфорной кислоты — дитиофосфаты (аэрофлоты)
R—О S
Р
R —О S—Н(Ме),
тиоспиртов — меркаптаны и тиофенолы R—S—Н(Ме),
дитиокарбаминовой кислоты — дитиокарбаматы
/
N —С
S — Me.
Оптимальная длина углеводородного радикала R в общем
случае зависит от характера полярной группы собиратели и
увеличивается с возрастанием ее полярности. Например, если
радикал сульфгидрильных собирателей содержит обычно 2—5
атомов углерода, то у алкилгидроксаматов длина углеродного
радикала насчитывает уже 7—8 атомов углерода, а у
органических производных карбоновых кислот — не менее 12 атомов
углерода [44].
7.2. Влияние длины аполярной цепи собирателя
на прочность его закрепления
Прочность закрепления собирателя, характеризуемая
работой, которую необходимо затратить на десорбцию его с
поверхности минерала, имеет большое значение при флотации.
Влияние длины аполярной цепи собирателя на прочность.его
закрепления рассмотрим на примере десорбции
неорганическими ионами анионных собирателей, имеющих одинаковую по-'
лярную группу, но разную длину углеводородной цепи.
Схематическое изображение связей и поверхностных энергий, которые
исчезают или вновь образуются при этом, приведено на рис. 7.3.
Процесс десорбции сводится к замене ионов собирателя на
поверхности неорганическими ионами и обратному процессу
в объеме пульпы [36].
113
а
^1
taSSf
5$л
*^
IQ
I №
о
Do
0 0
0
I»
I IF
^
°E6
Ъ
Os
s£
4
0/
\
\^>
<if££
ъ
0
.X.
Si
£J
M
с
О
Ъ О
If*
0 О
7
<= ^
<ь о
0 Ъ
I состояние системы
Л состояние система/
Рис. 7.3. Схема десорбции анионного собирателя с длинной (а) и короткой
(б) углеводородной цепью (по К. А. Разумову [36]):
/Г, — энергия взаимодействия солидофильной группы аниона собирателя с катионами
минерала (не зависит от длины цепи, так как солидофильная группа одна и та же);
/Г/, и Е2" — энергия дисперсионной связи между углеводородными цепями собирателя,
закрепившегося на минерале (возрастает с увеличением цепи); Е% и Е3" — свободная
поверхностная энергия на границе раздела вода — торцевые концы углеводородных
цепей; 1и — энергия гидратации той части неорганических ионов, которая при десорб--
ции собирателя перейдет на минерал; Е$ — энергия взаимодействия неорганических
ионов, закрепившихся на минерале, с катионами минерала; £fi --энергия взаимодействия
диполей воды с закрепившимися на минерале неорганическими ионами (энергия
гидратации); Я? — энергия взаимодействия диполей воды с полярными группами анионов
собирателя, находящихся в растворе (энергия гидратации); Еь' и £8" — «свободная»
поверхностная энергия на границе раздела вода—боковые поверхности углеводородных
цепей, зависящая от числа десорбированных ионов собирателя и длины их цепей (в
адсорбированном слое вследствие большой плотности сорбции ионов собирателя условно
принимается равной нулю)
Разница в суммарных анергиях десорбции собирателей
с длинной и короткой цепями
Е— Е -- (е'2~-El) ~\: 1£8— Ее).
Так как дисперсионная связь между длинными цепями
сильнее, чем связь между короткими цепями, то £/2>£//2- Разность
(£'г—Е) можно считать прямо пропорциональной увеличению
длины цепи. Возникновение «поверхностных энергий» Е'ъ и £"8,
как и энергий Е'$ й £"з, объясняется тем, что диполи воды
сильнее взаимодействуют между собой, чем с углеводородной цепью,
в результате чего молекулы воды, контактирующие с углеводо-
1 родной цепью, обладают некоторой избыточной потенциальной
энергией по сравнению с молекулами воды, находящимися
в объеме водной фазы.
Таким образом, более прочное закрепление собирателей
с большей длиной цепи зависит не только от разницы в
энергии дисперсионной связи между углеводородными цепями
закрепившихся на минерале молекул, но и от разницы в значе-
П4
ниях свободной поверхностной энергии, появляющейся при
переходе углеводородных цепей с минерала в воду. Увеличивая
длину углеводородного радикала, можно усилить
собирательное действие собирателя. Однако избыточное увеличение
прочности закрепления собирателя обычно приводит к ухудшению
селективности (избирательности) его действия при флотации.
7.3. Аполярные собиратели
Аполярные реагенты не имеют в составе своих молекул со-
лидофильной группы и поэтому лишены возможности химически
фиксироваться на поверхности минералов. Они представлены
углеводородными жидкостями главным образом нефтяного
происхождения. Закрепление их на минеральной поверхности может
происходить только по механизму избирательного смачивания
с образованием дисперсионных межмолекулярных сил между
углеводородными цепями реагента и поверхностью минерала.
Термодинамическая возможность закрепления аполярных
реагентов на поверхностях различной степени полярности
определяется в общем случае конечным выигрышем энергии
вследствие разницы энергий поверхностей раздела масло — вода,
масло — твердое и твердое — жидкость до и после закрепления
капелек реагента на минеральной поверхности. Очевидно
выигрыш энергии будет тем больше, чем больше аполярность и^гид-
рофобность минеральной поверхности.
Исследованиями А. Н. Фрумкина и др. [13] показано, что
адсорбция нейтральных и не образующих химической связи с
поверхностью молекул аполярных собирателей тем больше, чем
меньше заряд, т. е. полярность поверхности. Максимум
адсорбции таких молекул совпадает с областью нулевого заряда
ловсрхности (см. рис. 2.4). Поэтому в настоящее время
аполярные реагенты используются в качестве самостоятельных
собирателей только при флотации минералов, обладающих
природной гидрофобностью, таких, как сера, графит, уголь,
молибденит, тальк и др.
Являясь химически инертными веществами, углеводородные
масла, особенно парафинового ряда, практически нерастворимы'
в воде и их подают в пульпу обычно в виде эмульсии.
Капельки эмульгированного масла при столкновении с
пузырьками воздуха обычно полностью растекаются по их
поверхности, поскольку это сопровождается уменьшением
свободной поверхности энергии системы примерно на 5—10 мДж-м~2.
На поверхности известных минералов полное растекание масла
невозможно. Неполное растекание капелек масла происходит
лишь на поверхности минералов, обладающих природной
гидрофобностью или гидрофобизированных гетерополярными
собирателями. На поверхности гидрофильных минералов
закрепления капелек масла не происходит, поскольку такой процесс
сопровождался бы повышением свободной энергии системы.
115
Если частичка, на гранях которой имеется одна или не-
•■ сколько капелек масла, столкнется с .пузырьком, покрытым
слоем масла, то она закрепится на пузырьке по коалесцснтному
механизму (см. рис. 2.12). При этом увеличение дозировки
, масла и степени его дисперсности приводят к увеличению
показателя флотируемое™ FK [см. уравнение B.15)] до тех пор,
пока закрепившиеся на минеральной поверхности капельки
масла не начнут сливаться друг с другом.
Закрепление углеводородных масел на минеральной
поверхности приводит не только к увеличению показателя флотируе-
шости FK, но и к многократному упрочнению контакта пузырька
-воздуха с частицей (при воздействии на нее отрывающих
усилий) вследствие быстрого роста флотационной силы />.
Последнее обусловлено резким увеличением поверхностного
натяжения а на растягиваемой вокруг частицы кольцевой
поверхности пузырька вследствие замены поверхности раздела
масло — вода па поверхность раздела воздух — вода,
обладающей большим значением поверхностной энергии, а также
вследствие одновременного увеличения угла 0 (см. рис. 3.9,6, е.).
Упрочнение контакта является временным. Скорость
восстановления адсорбционного равновесия на растянутой
поверхности пузырька зависит от поверхностной активности, общей
концентрации плотности, коэффициента рефракции,
температуры кипения, вязкости собирателя и других параметров,
определяющих кинетические характеристики собирателя, что
необходимо учитывать при разработке реагентных режимов
флотации минералов. Для флотации тонких частиц обычно
рекомендуется применять легкие масла плотностью 0,82—
0,87 г/см3 при 20 °С и вязкостью C—10) • 10 м2/с при 50 °С;
для флотации крупных частиц и сростков — тяжелые масла
плотностью 0,88—0,93 г/см3 и вязкостью A0—50) • 10 м2/с [29].
Наличие капелек аполярного собирателя на поверхности
минеральных частиц резко сокращает время индукции и
значительно увеличивает скорость их прилипания к пузырьку. Это
связано с тем, что в* этом случае, во-первых, разрыв гидратной
прослойки происходит между двумя гидрофобными
поверхностями (аполярной капли и покрытого слоем масла пузырька),
когда процесс практически не имеет энергетического барьера
(см. рис. 2.7, кривая 2), и, во-вторых, капля имеет малую
площадь сечения и обладает хорошей «прокалывающей»
способностью по отношению к гидратной прослойке между
пузырьком и частицей.
По указанным причинам капельки масла становятся также
центрами селективной флокуляции природно гидрофобных или
гидрофобизированных тонких частиц между собой и с более
крупными частицами того же минерала при достаточной
загрузке аполярного собирателя в пульпу. Образующиеся
агрегаты имеют большую скорость флотации, чем разрозненные
тонкие частицы, так как вероятность их столкновения и закреп-
116
ления на пузырьках -выше, чем вероятность столкновения
несфлокулированных тонких частиц.
На явлении селективной флокуляции под действием аполяр-
пых масел основаны «агломерационная», «флокулярная» и
другие аналогичные виды флотации.
Помимо применения в качестве собирателей при флотации
природно гидрофобных „минералов, аполярные собиратели
используются в качестве добавок к гетерополярным собирателям
для получения более хрупкой пены, обеспечивающей повышение
качества концентратов.
Причиной повышения эффективности действия гетерополяр-
ных собирателей при добавке углеводородных масел является
не только многократное упрочнение контакта частицы и
пузырька, но и некоторая дополнительная гидрофобизация
минеральной поверхности в результате частичного растекания
закрепляющихся капелек масла по углеводородным концам ге-
терополярного собирателя. Аналогичный эффект достигается
в конечном счете при использовании углеводородов, в
частности керосина, для растворения высокомолекулярных карбоно-
вых кислот, например олеиновой. При этом в несколько раз
увеличивается вероятность взаимодействия труднорастворимого
гетерополярного собирателя с минеральной поверхностью.
Молекулы собирателя располагаются на поверхности капель масла
таким образом, что полярные группы их обращены в сторону
водной фазы. Такое расположение молекул гетерополярного
собирателя способствует интенсивному эмульгированию и
образованию большого количества капелек масла в пульпе. При
столкновении их с минеральными частицами происходит
взаимодействие полярных групп с поверхностью, а масло
растекается по углеводородным цепям закрепившегося
гетерополярного собирателя, повышая общую гидрофобность поверхности.
В качестве аполярные собирателей применяются керосин,
различные смазочные масла и другие углеводородные
продукты перегонки нефти (веретенное, трансформаторное,
соляровое масла и др.). Расход их при флотации велик и в
зависимости от сортности реагента, содержания флотируемого
минерала в руде, степени измельчения руды, плотности пульпы и
других параметров составляет от 300 до 2000 г/т руды.
Повышенный расход углеводородных масел по сравнению
с гетерополярными растворимыми собирателями объясняется
образованием на поверхности пузырьков относительно толстых
пленок масла, а на поверхности минералов — капель. Кроме
того, углеводородные масла находятся в пульпе в виде
эмульсий, а не растворов, поэтому для увеличения вероятности
встречи собирателя с частицами минералов необходим больший
расход собирателя. Интенсивное эмульгирование масла в пульпе
является одним из путей снижения его расхода и,
следовательно, концентрации в сточных водах до предельно
допустимой, равной 0,03 мг/л.
117
В ряде случаев для облегчения эмульгирования масел
целесообразно подавать во флотацию небольшое количество
поверхностно-активных веществ — эмульгаторов, а для очистки
поверхности капель масла от шламов породы —реагенты-пеп-
тизаторы [29].
7.4. Гетерополярные собиратели с неопределенным
химическим составом
К собирателям этой группы относятся разные смолы
(каменноугольная, древесная, торфяная, сланцевая), дегти,
сульфированный и окисленный керосин, контакт Петрова,
нейтрализованный черный контакт (НЧК) и некоторые другие
вещества, имеющие сложный состав. Они могут содержать высшие
спирты R—ОН, карбоновые кислоты R—GOOH, сложные эфиры,
углеводородные масла и другие органические вещества.
Механизм действия рассматриваемых собирателей аналогичен
механизму действия аполярных масел в присутствии гетеропо-
лярных собирателей.
Собиратели с неопределенным химическим составом
используются при флотации все реже и реже, так как они
нестандартны по качеству; дефицитны, поскольку часто являются
ценным сырьем для химической промышленности; обладают
обычно не только собирательными, но и пенообразующими
свойствами, что затрудняет регулирование процесса;
характеризуются высоким расходом вследствие большого содержания
в них неактивных или малоактивных составных частей, напри-
мер углеводородов; часто содержат фенолы и другие спирты
ароматического ряда, вредные для рыб и животных, что
приводит к необходимости строительства дорогостоящих очистных
сооружений.
7.5. Катионные собиратели
К катионным собирателям относятся амины и соли аминов,
органические аналоги аммониевых соединений, органические
сульфониевые, арсониевые и фосфониевые соединения, в
которых сера, мышьяк или фосфор замещает атом азота в
аммониевых соединениях, а также другие собиратели, при ионизации
которых углеводородный радикал и полярная группа остаются
в катионе.
Из катионных собирателей наибольшее практическое
значение в настоящее время имеют только органические
производные аммониевых соединений. Наибольшее" распространение из
них получили гидраты, солянокислые и уксуснокислые соли
первичных аминов и четвертичные соли аммония.
Соли первичных аминов хорошо растворяются в воде и
диссоциируют на ионы в кислой и слабощелочной средах. При
высоких значениях рН эти собиратели находятся в молекулярной
118
форме, в которой они, как правило, плохо растворимы.
Состояние первичных аминов в зависимости от рН изображено на
рис. 5.1,0. Примерно до рН 9 амин представлен ионами, а при
более высоких значениях рН — молекулярной формой амина
RNH2, часть которого находится в растворенном виде, а часть —
в виде коллоидной фазы. Для лауриламина, например,
концентрация молекул в растворе не превышает 2- 10~5 моль/л [3].
Катионные собиратели легко флотируют кварц, полевые
шпаты, слюды, фосфаты, сульфиды. Ряд флотируемости
минералов катионными собирателями обычно бывает обратным по
отношению к ряду флотируемости анионными собирателями
типа карбоновых кислот.
Собирательные свойства катионных собирателей зависят от
длины углеводородной цепи. Оптимальное число атомов
углерода в цепи катионного собирателя обычно равно 12—14 [3,
44].
До настоящего времени существуют разные мнения о
механизме действия катионных собирателей (аминов) и причинах,
вызывающих адсорбцию их на минеральной поверхности. Одни
исследователи считают, что гидрофобизация минералов
обусловлена закреплением ионов, другие — молекул амина. В обоих
случаях выдвинуты гипотезы как химического взаимодействия
ионов или молекул катионного собирателя, так и физической
адсорбции их на минеральной поверхности. Каждая из гипотез,
основанная на результатах опытов с небольшой группой
минералов, как правило, отражает лишь одну из характерных
сторон механизма коллектирующего действия аминов и не может
объяснить всех экспериментальных фактов. При этом
большинство гипотез не указывают конкретные причины, вызывающие
адсорбцию собирателя на поверхности минералов.
Причинами адсорбции поверхностно-активного иона
катионного собирателя могут быть: кулоновское притяжение иона
электростатическим полем минеральной поверхности,
поляризация адсорбента ионом, электростатическая поляризация иона
полем поверхности и неполярные силы Ван-дер-Ваальса. В свою
очередь, адсорбция молекул амина, полярная группа которых
обладает постоянным диполем, зависит от возможности
проявления сил дипольного и дисперсионного взаимодействия,
образования водородной связи или металламинных комплексов
(типа аммиачных) Men[RNH2]m.
Таким образом, принципиально возможна адсорбция на
минеральной поверхности как ионов, так и молекул амина, что
согласуется с экспериментальными данными и выводами ряда
работ [3]. -
При этом адсорбция молекул амина на поверхности
приводит к образованию краевых углов, значения которых не
меньше, чем при адсорбции ионов амина [1]. Максимально
плотная упаковка ионов и молекул собирателя в адсорбционном
слое, способствующая более прочному закреплению их на по-
П»
верхности в результате дисперсионного взаимодействия апо-
лярных цепей сорбирующихся частиц, достигается при
соответствии параметров кристаллических решеток минерала и амина.
Соответствие может иметь решающее значение в тех случаях,
когда прочность связи полярной группы амина с поверхностью
минерала недостаточна, как, например, в случае растворимых
солей.
Преимущественная адсорбция иолов или , молекул амина
в общем случае определяется значением, знаком и градиентом
электростатического поля, природой и характером связей на
минеральной поверхности и состоянием собирателя в растворе.
Так как различные минералы обладают различной электронной
структурой и состав их поверхностного слоя может меняться
(например, при изменении рН раствора), вызывая изменение и
характеристик электростатического поля поверхности, то все
это должно оказывать большое влияние на величину и
прочность адсорбции как ионов, так и молекул амина.
По этой причине собирательные свойства аминов и их
солей в значительной мере зависят от щелочности пульпы.
Например, кварц и окислы железа лучше флотируются при рН
6—8, берилл, сподумен и полевой шпат (альбит) при рН .9—
9,5, сульфидные минералы при рН 9,5—10,5, окисленные
цинковые минералы (смитсонит, каламин^ при рН 10,5—11,5,
кальцит при рН> 11,5.
Удовлетворительное объяснение закономерностей гидрофоби-
зации и флотации различных минералов катионными
собирателями можно получить только при одновременном учете в
каждом конкретном случае состояния минеральной поверхности,
знака и значения ее заряда и состояния самого амина в
растворе [1, 3]. Факторами, определяющими возможность гидро-
фобизации поверхности под действием положительно
заряженных ионов амина, являются отрицательный заряд поверхности
и преимущественно ионная форма состояния амина в растворе;
гидрофобизация же под действием молекул амина возможна
при относительно малом заряде поверхности (близком к
нулевому) и молекулярной форме амина.
Неблагоприятное влияние возрастания заряда поверхности
минерала на адсорбцию амина легко объяснить, если
воспользоваться выражением для энергии конденсатора, которому
можно уподобить двойной электрический слой (ДС):
<о<2л/Е2/е, G.1)
где со — энергия конденсатора; / — эффективная толщина ДС;
Е — плотность заряда; е—диэлектрическая проницаемость.
Поляризация, т. е. сдвиг потенциала в любую сторону от
точки нулевого заряда поверхности, приводит к образованию
двойного электрического слоя. Чем больше поляризация, тем
•больше плотность заряда. Из выражения G.1) следует, что
при данном значении Е энергия конденсатора уменьшается
120
с ростом е. Стремление системы к уменьшению энергии
вызывает втягивание в ДС молекул воды, обладающих более
высокой диэлектрической проницаемостью, чем адсорбированные
ионы или молекулы амина. Вместе с тем вытеснение
(десорбция) адсорбированных крупных ионов или молекул амина
должно уменьшать эффективную толщину ДС, что также вызывает
уменьшение энергии конденсатора. Пока поляризация мала, на
поверхности превосходят силы электростатического притяжения
молекул воды. При более высокой поляризации наступает
десорбция органических ионов и молекул. Поверхность любого
твердого тела энергетически неоднородна. Ее можно
рассматривать как серию элементарных площадок с различной
работой адсорбции на каждой из них. Поэтому наблюдается не
очень резкая граница адсорбции — десорбции амина.
Значение заряда поверхности, например, сульфидных
минералов свинца, меди, железа и сульфидизированной поверхности
самородного висмута определяется концентрацией потенциал-
определяющих сульфидных ионов [S2-] в пульпе.
Зависимость между потенциалом минеральной поверхности
и [S2] в пульпе (с учетом процесса окисления сульфидных
ионов в приэлектродном слое до элементарной серы) найдем на
основании реакций [3]:
Pb T2S2~ ^ PbS-^So Me; £- — 0,72 -0,0295 lg IS2"]; G.2)
FeS + 2S°^±FeS2 + S°-f Ae\ E= — 0,635—0,0295 lg[S2-]; G.3)
Cu2S + 2FeS + 2S2" ^ 2CuFeS2 + S° + 4г;
E= —0,663 — 0,0295 lg[S2"]; G.4)
2Bi + 4S2~ ^ Bi2S3 + S°^8tf; £- —0,683—0,0295 lg[S2~]. G.5)
Уравнения G.2) — G.5) позволяют провести оценку влияния
потенциала минеральной поверхности на адсорбцию
собирателя. Так, используя их для расчета потенциала поверхности
минералов при различных [S2-], экспериментальные данные
(рис. 7.4, а) можно представить в виде, изображенном на
рис. 7.4, б. Учитывая, что между плотностью адсорбции
собирателя и флотируемостью минералов в данном случае существует
тесная зависимость, можно считать, что потенциал
поверхности является основным фактором, определяющим адсорбцию
катионного собирателя (лауриламина) и флотируемость
минерала. Различные его значения для разных минералов при
одной и той же [S2-] в пульпе [уравнения G.2) —G.5)] являются
основной причиной различий их сорбционных (см. рис. 7.4, б) и
флотационных (рис. 7. 4, в) свойств.
Максимальная адсорбция собирателя и флотируемость всех
минералов наблюдается при одном и том же потенциале,
равном 0,4—0,45 В, а полная десорбция собирателя и подавление
флотации —при 0,55—0,6 В (см. рис. 7.4,6, в). Различные
значения плотности адсорбции лауриламина на разных минера-
121
-14 40 -£ lg[S2"] -0,2 '0,3 -W -0,5Е(н;вшфЪ 0 10 20 SO Г,с/*
Рис. 7.4. Влияние значений концентрации сульфидных ионов (а) и
потенциала Е поверхности (б) на плотность адсорбции Г (в долях условного
монослоя) лауриламина (а, б) и плотности его адсорбции на флотнруе-
мость у минералов (в):
1 — галенит; 2 — пирит; 3 — халькопирит; 4 — самородный висмут
лах при одном и том же значении потенциала поверхности
обусловлены, можно полагать, различным градиентом
электростатического поля поверхности вследствие различия параметров
кристаллической решетки минералов и степени энергетической
неоднородности их поверхности.
Поглощение минеральной поверхностью ионов амина из
кислых и нейтральных растворов протекает обычно гораздо
менее интенсивно, чем поглощение молекул амина из щелочных
растворов. Можно полагать, что данное явление обусловлено
возникновением активационного барьера при адсорбции ионов
амина.
При приближении к поверхности адсорбирующийся ион
амина оказывается под действием двух противоположно
направленных сил: притяжения к поверхности и отталкивания от
поверхности, обусловленных уже адсорбированными
одноименно заряженными ионами амина (рис. 7.5). Соотношение
между этими противоположно действующими силами меняется
с расстоянием от поверхности, что при определенной
плотности сорбционного слоя ионов амина может привести к
образованию около поверхности энергетического барьера. При
закреплении на поверхности незаряженных молекул амина силы
отталкивания между полярными (дипольными) группами
значительно меньше, поэтому адсорбция молекул катиоиных
собирателей должна протекать гораздо интенсивнее, чем сорбция их
ионов. Совместная адсорбция ионов и молекул амина также
предпочтительнее, чем только ионов, так как внедрение
молекул между адсорбированными на поверхности ионами амина
позволяет снизить электростатическое отталкивание между
полярными группами последних (см. рис. 7.5).
Плотность слоя катионного собирателя, обеспечивающая вы-
122
Минерал
} Вода
Рис. 7.5. Схема сил при
адсорбции молекул (/) и ионов B)
амина на минеральной
поверхности
сокое извлечение, для разных
минералов колеблется в
широких пределах, % условного
монослоя: кварц — 5; полевой шпат,
биотит, сподумен, берилл — 8—
10; мартит-гематит—15;
магнетит—50 [44],
Анализ влияния плотности
адсорбции лауриламина на фло-
тируемость сульфидных
минералов показывает, что сорбцион-
ный слой физически
адсорбированного собирателя на всех
минералах обеспечивает более
эффективную их флотацию, чем
сорбционный слой металламин-
ных соединений, являющихся продуктом химического
взаимодействия молекул амина с поверхностью пирита и
халькопирита в сильнощелочной среде. Необходимая для полной
флотации сульфидов плотность сорбции амина составляет в первом
случае 25—35 % (см. рис. 7.4, в), во втором 85—95 % условного
монослоя. При физической адсорбции амина флотируемость
минералов определяется плотностью его адсорбции и не зависит
от того, закрепляется он в виде ионов или молекул.
Из катионных собирателей практическое значение имеют
собиратели, содержащие 11—18 атомов углерода в цепи. В
настоящее время в СССР производится катионный собиратель
марки АНП-2, представляющий собой смесь солянокислых
первичных алифатических аминов изостроения с числом атомов
углерода в радикале. Q2—Ci8 и первичные амины на основе
высших жирных кислот с Си—С20. Первичные амины с
радикалами нормального строения в США производятся под
названием Аэромины C035 и 3037), в ФРГ — Флотигамы.
Выпускаемый в СССР Каталин А является четвертичным аммониевым
основанием с арильным радикалом С!2—Ci8.
Катионные собиратели начали применяться сравнительно
недавно, но их значение для практики флотации непрерывно
возрастает. В настоящее время они используются для
разделения сильвина КС1 и галита NaCl при избирательной
флотации сильвина в насыщенном растворе солей; для флотации
кремнезема из грубых фосфатных концентратов и железных
руд; при отделении полевого шпата от кварца в присутствии
фтористоводородной кислоты; для флотации окисленных
цинковых минералов после их сульфидизации; для селективной .
флотации минералов руд редких металлов и в других случаях
флотационного обогащения. Расход собирателя составляет
50—200 г/т. Предельно допустимая концентрация (ПДК)
в сточных водах аминов жирного ряда с С7—Сэ составляет
0,1 мг/л; с Сю—Ci6 —0,004 мг/л; с С16—С20—0,03 мг/л.
123
7.6. Оксигидрильные собиратели
Оксигидрильные собиратели широко применяются при
флотационном обогащении редкометалльного и химического сырья.
Из них наиболее изучены и получили широкое распространение
органические производные угольной, серной, фосфорной и гид-
роксамовой кислот.
Органические производные угольной' кислоты — карбоновые
кислоты и их мыла имеют общую структурную формулу
О
R—с—он(ме).
Радикал R у жирных кислот представляет собой
зигзагообразную цепь, в вершинах которой расположены атомы
углерода. Каждое углеводородное зерно СН2 удлиняет общую цепь
на 1,26- 10~ш м. Углеводородная цепь непредельных жирных
кислот, например, олеиновой или линолевой, изогнута у
двойной связи.
Растворимость жирных кислот в воде уменьшается с
удлинением цепи на одну группу СН2 в 4,25 раза, но увеличивается
с повышением температуры. Зависимость состава водных
растворов мыл и критической их концентрации, при которой
начинают возникать сложные образования — мицеллы, от
температуры изображена на рис. 7.6.
Жирные кислоты в растворах частично образуют димерные
молекулы
О ... НО
^ \
R—С С —R
\ •
ОН... О
вследствие взаимодействия полярных групп дипольных молекул.
- Состояние карбоновых кислот и их мыл в растворе зависит
от рН пульпы (см. рис. 5.1,6). В отличие от катионных
собирателей (см. рис. 5.1, в) в кислой и нейтральной среде они
представлены не ионной, а в основном молекулярной формой.
Так как растворимость жирных кислот в молекулярной форме
очень мала, то в кислых пульпах основное колцчество реагента
находится в нерастворенном виде и образует эмульсию.
Высокая концентрация анионов может быть получена только в
щелочных пульпах. Однако она не может быть больше ККМ —
критической концентрации мицеллообразования (равной для
олеата Ю-3 моль/л), превышение которой приводит к
выпадению избыточного собирателя в виде ионных, а затем
молекулярных мицелл, представляющих собой коллоидные образова-
124
Рис. 7.6. Влияние
температуры Т и концентрации
С водных растворов
на их состав
мыл
ния (из 50—300 конов или молекул,
например, олеата натрия).
Состояние реагента в условиях
равновесия определяется только
значением рН пульпы и не зависит от
того, добавляется ли реагент в виде
кислоты или в виде мыла. Однако
степень дисперсности эмульсии, если
она '-образуется, больше в том случае,
когда реагент добавляется в виде
мыльного раствора.
Собирательное действие карбоно-
вых кислот и их мыл находится в
прямой зависимости от степени
дисперсности этих реагентов в водной среде.
Чем выше степень их дисперсности
(вплоть до ионов и молекул), тем
лучше результаты флотации.
Увеличение степени дисперсности собирателя в пульпе
достигается в основном следующим образом:
уменьшением степени гидролиза и увеличением ККМ за
счет увеличения числа двойных связей в молекуле собирателя;
продувкой эмульсии жирной кислоты кислородом или
воздухом для образования в ней гидроперекисной группы;
увеличением растворимости собирателя за счет
разветвления углеводородной цепи при достаточной ее длине;
повышением рН пульпы и флотацией большинства
минералов в слабощелочной или щелочной средах;
предварительным омылением жирных кислот, особенно в тех
случаях, когда пульпа имеет невысокую щелочность;
применением органических растворителей, например
керосина или спирта;
эмульгированием в присутствии стабилизирующих добавок,
например алкилсульфатов, сульфоиола, эфиров полигликолей
и других соединений;
предварительной обработкой мыла жирной кислоты жидким
стеклом перед подачей ее в процесс;
подогревом пульпы.
Расход жирных кислот колеблется от 0,2 до 0,7 кг/т. Он
уменьшается по мере понижения растворимости образуемых на
минералах поверхностных соединений собирателя. Это
объясняется, в основном, тем, что с уменьшением растворимости
образующихся на поверхности минералов соединений уменьшается
бесполезный (но неизбежный) расход собирателя на создание
равновесной концентрации его в растворе.
Большим недостатком жирных кислот и мыл является
зависимость их собирательных свойств от жесткости воды, которая
определяется концентрацией в ней ионов Mg2+ и Са2+.
Образование с этими ионами труднорастворимых мыл приводит
125
к потерям собирателя и нарушению селективности процесса. Па*
этому в ряде случаев, особенно при флотации руд редких
металлов, применяют умягченную воду [49, 50].
Из карбоновых кислот наиболее изучена как собиратель
олеиновая кислота С17Н33СООН. Она является ненасыщенной
кислотой и имеет одну двойную связь посередине углеводородной
цепи. Соответствующая ей насыщенная кислота называется
стеариновой и имеет формулу С17Н35СООН. Промышленное
значение имеют также аналоги олеиновой кислоты с больщим числом
двойных связей:C17H3iCOOH—-линолевая (две двойных связи),
С17Н29СООН — линоленовая (три двойных связи).
Олеиновая кислота дорогой и дефицитный реагент, поэтому
в качестве заменителей ее используют более дешевые талловое
масло или сульфатное мыло, нафтеновые и синтетические
жирные кислоты, окисленные углеводороды, отходы масло-жировых,
и химических производств [29, 44].
Жирные кислоты и их заменители являются хорошими
собирателями большого числа минералов. Ими особенно хорошо
флотируются не содержащие кремнезема соли
щелочноземельных металлов [кальцит СаС03, флюорит CaF2, шеелит CaW04>
апатит Са5(Р04)з (F, C1, ОН), барит BaS04, витерит ВаСОз]
и карбонатов черных металлов (сидерит FeC03, родохрозит
МпСОз), несколько хуже окислу железа (гематит Fe203,
магнетит Fe304, бурые железняки Ре2ОзмН20 и др.) и марганца
(пиролюзит МпОг и др.). Силикатные минералы флотируются
жирными кислотами в том случае, если на их поверхности
имеются катионы, способные образовать с собирателем
труднорастворимые поверхностные соединения.. Чистый кварц жирными
кислотами не флотируется и для его флотации необходима
предварительная активация катионами щелочноземельных или
тяжелых металлов.
Олеиновая кислота и заменители — малоселективные
собиратели, но селективность их действия можно повысить путем
подбора оптимальных дозировок реагентов-модификаторов.
Органические производные серной кислоты — алкилсульфаты
и алкилсульфонаты представляют собой соединения,
содержащие углеводородный радикал и сернокислотный остаток. Если
остаток соединяется серой непосредственно с углеродом
радикала R, то соединение называется сульфоновой кислотой, а соль
ее — алкилсульфонатом
О
R —S —0—Me;
\
Если углерод радикала связан с серой через кислородный
«мостик», то такое соединение называется сульфоэфиром или:
алкилсерной кислотой, а соль — алкилсульфатом
126
о
R—0 — S —0 —Me.
Ч
О
Алкилсульфаты и сульфонаты являются анионными
собирателями-пенообразователями, способными флотировать
несульфидные минералы. Они обладают также моющими свойствами
и входят в состав различных стиральных порошков, например,
«Новость», «Прогресс» и др.
Так как алкилсерная и сульфоновая кислоты относятся
к сильным, то их соли мало гидролизуются. Это позволяет
применять алкилсульфаты и сульфонаты в кислых пульпах.
Высокая растворимость солей по сравнению с солями высших кар-
боновых кислот позволяет, в свою очередь, использовать
алкилсульфаты и сульфонаты при флотации в жестких водах.
Алкилсульфаты («Новость», алкилсульфаты вторичных
высших спиртов) и сульфонаты («Сульфонол НП»,
сульфированные нефтепродукты) хорошо флотируют барит и применяются
в качестве собирателя для флотационного его извлечения из ше-
елито-баритовых, свинцово-баритовых и других баритсодержа-
щих, например, полиметаллических, руд ряда месторождений.
Алкилсульфаты применяются также для флотации
железосодержащих минералов при переработке бедных руд, для
извлечения железосодержащих и других минеральных примесей из
стекольного песка и полевого шпата в кислой среде, при
селективной флотации руд редких металлов.
Удельный расход алкилсульфатов и сульфонатов примерно
такой же, как и карбоновых кислот, но промышленное значение
при флотации полезных ископаемых намного меньше.
Органические производные фосфорной и мышьяковой
кислот. При замене одного или двух атомов водорода в гидро-
ксильных группах ортофосфорной кислоты
получают
моно-
R
НО П
Р
/\
НО ОН
или диалкилфосфоряые
—0 0 R—
Р или
/ \
НО ОН R-
кислоты
-0 0
Р
/\
-0 ОН
127
которые [например, изооктилфосфат натрия (C8Hi70JPOONa]
оказались довольно эффективными собирателями при флотации
касситерита, апатита, фосфорита, пирохлора и циркона.
Положительной особенностью алкилфосфорных кислот является их
малая чувствительность к солям жесткости.
При замене одной гидроксильной группы в молекулах ор-
тофосфорной или мышьяковой кислоты на радикал получают
моноалкил-(арил-)фосфоновые
R ,0
\ /
Р
НО DH
или алкил-(арил-)арсоновые
R О
\'
As
/ \
н0 ОН кислоты.
При замене двух гидроксильных групп в молекуле орто-
фосфорной кислоты на радикалы получают диалкил-(арил-)-
фосфиновые кислоты
Р
R ОН
Алкилфосфоповые, алкиларсоновые (например, толуолар-
соиовая) и диалкилфосфиновые кислоты с радикалами Сб—С8
используются для селективной флотации касситерита Sn02.
Алкилгидраксамовые кислоты и их соли с общей
структурной формулой
R—С — NH—0Н(Ме)
и длиной радикала С7—С9 являются основой разработанного
в СССР весьма перспективного реагента ИМ-50. Гидраксамо-
вые кислоты, входящие в реагент ИМ-50, имеют константу
диссоциации около 2- 10~10 и являются довольно устойчивыми
соединениями при различных концентрациях в диапазоне рН
3—10. Они обладают сравнительно слабой поверхностной
активностью, что является несомненным их достоинством, спо-
128
собны образовывать труднорастворимые соединения (с
произведением растворимости 10~10— Ю-20), а также ~устойчивые
комплексные соединения с рядом катионов металлов на
поверхности минералов.
Комплексообразующие свойства гидроксамовых кислот
обусловлены наличием в их функциональной группе двух донор-
ных атомов — азота и кислорода, что приводит к образованию
между ними и катионами решетки координационных связей
R — C = 0 R— С = 0
\
Me wm
\
Н—14 —О Н—N —D
Прочность образующихся гидроксаматных комплексов за^
висит от катиона минерала. Она не велика при взаимодействии
с катионами щелочноземельных металлов (Са2+, Ва2+ и др.)» но
значительна при взаимодействии с переходными металлами
(Ti, V, Mn, Zn, Nb, Hf, Та и др.) и катионами высокой
валентности (Al3+, Fe3+). При этом прочность гидроксаматных
комплексов с Nb5+, Та5+, Sn4+ не меньше, чем с Fe3+.
Возможность образования устойчивых комплексных
соединений *с титаном, ниобием, танталом и оловом позволяет
использовать реагент ИМ-50 в качестве селективного собирателя
при флотации пирохлоровых, лопаритовых, перовскйтовых, кас-
ситеритовых и других руд, полезные минералы которых
содержат в своем составе перечисленные металлы.
Механизм действия оксигидрильных собирателей.
Современные представления о силах, вызывающих адсорбцию
поверхностно-активных веществ, позволяют полагать, что и молекулы,
и ионы оксигидрильных собирателей могут закрепляться на
минеральной поверхности путем как физической, так и химической
их адсорбции [1,3, 44].
Например, физическая адсорбция ионов собирателя может
быть обусловлена проявлением сил кулоновского притяжения и
поляризации, а физическая адсорбция молекул — сил диполь-
ного взаимодействия и возможности образования водородной
связи. В обоих случаях прочности закрепления будет
способствовать дисперсионное взаимодействие аполярных цепей
собирателя как между собой, так и с поверхностью адсорбента.
Физическая адсорбция ионов и молекул жирнокислотных
собирателей должна характеризоваться не очень прочной связью
сорбированных частиц с минеральной поверхностью и
допускать перемещение их как* по поверхности минерала, так и
в объеме раствора.
Химическое взаимодействие между ионом собирателя и кат
тионом, расположенным на поверхности минерала, или ионами,
тяжелых и щелочноземельных металлов в объеме раствора
5 Заказ № 1957
129}
обусловлено возможностью образования ковалентной или
координационной связи между полярной группой собирателя и
этими ионами.
Образующиеся в результате химического взаимодействия
соединения являются труднорастворимыми. Например,
значения произведения растворимости составляют для образующегося
в объеме раствора олеата магния 1,6-Ю-11, олеата бария
1,2-10~12, олеата кальция 4-10~13, олеата свинца 1,6-10~17,
олеата железа 2*10~30. Растворимость поверхностных
соединений еще ниже.
На основании работ Волькенштейна можно считать, что
закрепление молекул собирателя на минеральной поверхности
также возможно, в результате образования слабой одноэлект-
ронной связи или удержания свободного электрона или
свободной дырки кристалла и образования совместно с валентным
электроном хемосорбированной частицы весьма прочной двух-
электронной связи. Кроме того, свободный электрон решетки
может привести к разрыву связи внутри молекулы, т. е. к ее
диссоциации, и прочному закреплению образовавшегося иона
собирателя на поверхности.
Преобладание того или иного вида и формы закрепления
зависит от природы и состояния собирателя в растворе.
Например, закрепление ионов собирателя на поверхности за счет
физической и химической их адсорбции можно ожидать, когда
поверхность минерала обладает большой долей ионных
связей и имеет положительный заряд, а собиратель представлен
ионной формой. И наоборот, преобладание металлических и
гомеополярных связей и наличие электронов и дырок в решетке
минерала, близкий к нулевому заряд и присутствие собирателя
в молекулярной форме приводят к возрастанию «вклада»
молекул собирателя в общую плотность его адсорбции на
минеральной поверхности.
Такие представления о механизме закрепления оксигидри-
льных собирателей вполне согласуются с образованием в ряде
случаев многослойных покрытий и с возможностью частичной
отмывки сорбированного собирателя дистиллированной водой.
Совместное нахождение ионов и молекул жирных и гидро-
ксамовых кислот на поверхности минералов подтверждаются
результатами исследований методом инфракрасной
спектроскопии, результатами расчетов на основании определений
емкости двойного электрического слоя, результатами исследований
с применением избирательных растворителей. Например,
исследованиями адсорбции жирнокислотных собирателей на
металлической меди (при различной степени окисления),
флюорите, барите, гематите и других минералах, проведенными
с применением инфракрасной спектроскопии, установлено, что
в общем случае сорбционный слой на поверхности представлен
хемосорбированным поверхностным, например, олеатом
металла, поверх которого физически соосорбируются нормальные
130
О 10 20 30 *tf 50 60 70 80 90J3,%
Рис. 7.7. Влияние содержания р физически адсорбированной олеиновой
кислоты в сорбционном слое на флотируемость у галенита A), халькопирита
B) и пирита C) при постоянной общей плотности адсорбции олеата
E0 % условного монослоя)
или замещенные олеаты металла, олеат натрия и молекулы
олеиновой кислоты.
До последнего времени различные исследователи считали
причиной флотации, как правило, только одну из форм сорбции.
Результаты исследований [1, 3] показали, что собиратель,
закрепившийся только химически или. сорбированный только
физически, обеспечивает низкую флотационную активность
минеральных частиц. Плотность собирателя, необходимая для
полной флотации минералов, в обоих случаях зависит от гидро-
фильности поверхности и обычно составляет несколько услов^
ных монослоев.
Наиболее эффективная флотация минералов, не
обладающих естественной гидрофобностью, наблюдается, когда в
сорбционном слое на минеральной поверхности присутствуют
одновременно и химически закрепившийся и физически
сорбированный собиратель. Причем доля как той, так и другой формы
его закрепления в сорбционном покрытии должна составлять
(в зависимости от используемого собирателя и флотируемого
минерала) не менее 10—30 % общей плотности сорбции
собирателя. При соблюдении этого условия необходимая для
полной флотации сульфидных минералов плотность общей
адсорбции, например олеата, не превышает 0,4—0,5 условного моно--
слоя (рис. 7. 7.).
5*
131
/
V
f
т
1 £
й
6
J
fv\
?a
Рис. 7.8. Влияние концентрации С
эмульгированного керосина на флотируемость
пирита (а) и халькопирита (б) без
предварительной обработки их олеатом (уо)
и после предварительной обработки
олеатом и удаления физически
сорбированных молекул олеиновой кислоты (у)
О 0,1 0,2' 0,3 0 0,5 С,мг/Л
Высокая флотационная активность сорбционного покрытия,
состоящего как из химически, так и физически закрепившегося
собирателя, подтверждается многими экспериментальными
результатами. Например, на неактивированном кварце олеат
закрепляется только в виде физически адсорбированных молекул
олеиновой кислоты и при плотности адсорбции их 0,6 условного
монослоя минерал не флотируется. Предварительная
обработка кварца солями железа, приводящая к закреплению 65—
70 % сорбированного олеата в виде «олеата железа» и 30—
35 % в виде молекул олеиновой кислоты, позволяет весьма
быстро ефлотировать кварц практически при той же плотности
общей адсорбции собирателя @,55 условного монослоя).
Роль физически адсорбированных молекул олеиновой
кислоты могут играть молекулы аполярных собирателей.
Например, предварительно обработанные олеатом пирит и
халькопирит после удаления из сорбционного слоя собирателя физически
адсорбированных молекул олеиновой кислоты флотируются
плохо (рис. 7.8). Однако даже незначительные добавки
эмульгированного керосина приводят к такой же бурной
флотации пирита и халькопирита, как и естественно гидрофобных
минералов. Те же минералы без предварительной обработки их
олеатом флотируются при добавке эмульсии керосина во много
раз хуже.
7.7. Сульфгидрильные собиратели
К основным сульфгидрильным собирателям относятся:
органические производные дитиоугольной кислоты (ксанто-
генаты)
132
5
R—о—С ;
s — Me
органические производные дитиофосфорной кислоты (дитио-
фосфаты)
R—О S
V/
р ;
R—0 S—Me
органические производные дитиокарбаминовой кислоты (ди-
тиокарбаматы)
R S
\ /
N —С
/ \
R S — Me,
органические производные сероводорода (меркаптаны и тио-
фенолы) R—S—Н.
Ксантогенаты щелочных металлов имеют наиболее важное
для практики значение. Они являются устойчивыми, хорошо
растворимыми в воде соединениями и обладают собирательной
способностью по отношению к самородным металлам и
сульфидам тяжелых металлов, на поверхности которых образуются
труднорастворимые ксантогенатные соединения.
Растворимость ксантогенатов тяжелых металлов убывает
в следующем порядке: ZnKx2>PbKx2>CuKx>AgKx, где Кх —
ксантогенатный ион
S
R—О —С—S— •
Уменьшение растворимости ксантогената улучшает флоти-
руемость минерала, поэтому ряд флотируемости сульфидов
ксантогенатами является обратным по отношению к ряду
растворимости ксантогенатов: ZnS<PbS<Cu2S<Ag2S.
Уменьшение или увеличение числа атомов серы в полярной
группе ксантогената изменяет энергию химической связи ее
с катионом минерала. Например, тритиокарбонат R—S—CSSMe
обладает еще более сильными собирательными свойствами, чем
ксантогенат, тогда как монотиокарбонат R—О—COSMe гораздо
слабее ксантогената, поскольку введение сильно полярной
133
группы =С = 0 вызывает довольно резкое увеличение
растворимости солей монотиокарбоната с ионами тяжелых металлов.
С увеличением длины углеводородной цепи растворимость
ксантогенатов уменьшается, а их флотационная активность и
гидрофобизирующее действие при одинаковой плотности
адсорбционного слоя возрастают. Свертывание углеводородной
цепи в кольцо ослабляет гидрофобизирующее действие.
Разветвление цепи при большом числе атомов углерода в ней
усиливает собирательную способность реагента. Селективность
действия ксантогенатов уменьшается при увеличении длины цепи.
Ксантогенаты являются солями ксантогеновых кислот,
константа диссоциации которых составляет около 3 • 10~2. В
нейтральной и щелочной средах они полностью диссоциируют на
составляющие их ионы
R —О—CSSK^R —О—CSS- + K+.
В слабокислой среде (рН 4—7) ксантогенаты гидролизуются
R—О—CSS- + H+^tR—О—CSSH,
а при рН<4 начинают разлагаться с образованием исходных
веществ при получении ксантогенатов: спирта, сероуглерода и
щелочи
R_0—CSSK + Н20 ч* ROH + CS2 + КОН.
Продукты разложения ксантогената не обладают
собирательными свойствами, поэтому флотацию желательно вести
в щелочной среде. При флотации в слабокислой среде ксанто-
генат лучше подавать отдельными порциями. В очень кислых
пульпах ксантогенаты не эффективны.
Ксантогенаты могут окисляться до диксантогвнида
S - S S
// # Ч
2R—О—С—S —^^R —О—С —S —S —С—О—R + 2е.
Скорость процесса окисления Кх~ до Кх2 кислородом
воздуха в объеме раствора незначительна. Она несколько увели-
чиваетсй в присутствии растворенной углекислоты воздуха,
в растворах соды и извести и резко возрастает в присутствии
металлической, сульфидной поверхности, ионов некоторых
металлов. Окисление может протекать, например, по следующим
реакциям:
2Кх- + Н20 + 0,5О2 - Кх2 + 20Н-;
2Кх- + Н20 + 02 = Кх2 + Н20- + ОН-.
На основании работ И. А. Каковского реакции окисления
ксантогената можно считать обратимыми. Предполагается, что
окисление ксантогената до диксантогенида на поверхности судь-
134
фидных минералов осуществляется хемосорбированным
кислородом без участия катионов минералов. Роль минерала
сводится к тому, чтобы сделать более вероятным электронный
обмен между ксантогенатом и кислородом, т. е. снизить энергию
активации процесса окисления ксантогената. Каталитические
свойства сульфидов по отношению к реакциям
окисления-восстановления обусловлены их полупроводниковыми свойствами.
Реакции окисления-восстановления ксантогената на
минеральной поверхности играют большую роль в процессе гидрофобиза-
ции и флотации минеральных частиц.
Учитывая обратимость реакции Кх~—Кх2, не имеет
принципиального значения загружается собиратель в виде
ксантогената или диксантогенида. Диксантогениды, входящие
составной частью в американский реагент Минерек, являются, как
и ксантогенаты, собирателями сульфидов и самородных
металлов. Эффективность их действия зависит от степени
дисперсности реагента в пульпе. Она возрастает при подаче
диксантогенида в виде тончайшей эмульсии, которая может быть получена
эмульгированием самого диксантогенида или путемv окисления
ксантогената в пульпе, например, гипохлоритом натрия.
При взаимодействии ксантогенатов с фосгеном в водной
среде образуются тиоан гидриды ксантогеновых кислот
с общей структурной формулой
S S
R—О —С —S—С—О —R ,
представляющие собой, как и диксантогениды, неионогенные
маслянистые жидкости. Тиоангидриды оказались весьма
эффективными собирателями цементной меди, хорошими
собирателями молибденита, самородной меди и благородных металлов,
сульфидов меди, содержащих мышьяк и сурьму, а также тон-
коизмельченных сульфидов меди и цинка. В СССР
синтезирован реагент СЦМ-2, имеющий формулу
S О
^ /
с4н9—о—c-~s—ь — о—сн3.
При взаимодействии ксантогенатов с галлоидным алкилом^
образуются э ф и р ы ксантогеновых кислот
R— О—CSSK + C1 — R'->R_О—CSS—R' + KC1.
Они представляют собой маслянистую жидкость, почти не
растворимую в воде, и являются более слабыми собирателями
сульфидов меди, свинца и цинка, чем ксантогенаты. Однако эти
эфиры сами по себе и в сочетании с углеводородными маслами
являются более сильными собирателями молибденита, чем
135
углеводородные масла. Об этом свидетельствует практика
применения аллилового эфира бутилксантогеновой кислоты
(реагента АБ-1) в СССР и аллилового эфира амилксантогеновой
кислоты (реагент S-3302) за рубежом. Кроме того, под
действием эфиров ксантогеновых кислот иногда наблюдается
некоторое улучшение извлечения медных минералов, обработанных
ксантогенатом.
При взаимодействии ксантогенатов и алифатических аминов
получаются диалкилтионокарбаматы с общей
структурной формулой R—О—CS—NH—R'. В СССР нашел широкое
применение изопропилметилтионокарбамат, известный под
названием НТК, а в США — изопропилэтилтионокарбамат
СН5 S
\ /
сн— о — с
/ \
СН3 NH —С2Н5 ,
известный под названием «реагент Z-200». Оба реагента
обладают довольно сильным собирательным действием на медные
минералы и активированный сфалерит и слабым — на пирит.
Исследования Гинцветмета подтверждают перспективность
применения тионокарбаматов при проведении коллективной медно-
цинковой и селективной медной и цинковой флотации из пирит-
ных руд. Как и эфиры ксантогеновых кислот,
диалкилтионокарбаматы в сочетании с углеводородными маслами оказывают
более сильное собирательное действие на молибденит, чем сами
углеводородные масла.
Если с амином взаимодействуют соли не дитиоугольной
(ксантогенаты), а тритиоугольной кислоты, то образуются так
называемые S, N-диалкилдитиокарбаматы с общей
структурной формулой R—S—CS—NH—R'. Они могут
использоваться при флотации медных, медно-молибденовых и медно-
никелевых руд. За рубежом находит применение реагент R-10,.
представляющий собой циклогексилдитиокарбамат.
Из органических производных дитиофосфорной кислоты —
дитиофосфатов наибольшее практическое значение имеют
жидкие крезиловый (СНзСбН^гРБгН и ксиленоловый, а также
твердый содовый бутиловый (C4H90JPS2Na дитиофосфаты.
В дитиофосфатах центральный атом фосфора сильнее
смещает электронное облака серы по направлению к центру, чем
углерод в молекуле ксантогената. Связь фосфора с серой
прочнее, а связь серы с металлом слабее, что является
причиной повышения растворимости дитиофосфорных солей по
сравнению с ксантогеновыми. Поэтому как собиратели
дитиофосфаты слабее ксантогенатов, но более устойчивы в кислых
средах и не окисляются (по данным Б. А. Степанова) кислородом
136
воздуха, содержащимся в пульпе. Поскольку сульфиды железа
флотируются дитиофосфатами несколько слабее, чем сульфиды
цветных металлов, то дитиофосфаты более селективны при
флотации сульфидов меди в присутствии цинковых и железных
сульфидов.
Жидкие дитиофосфаты содержат некоторое количество
крезола и обладают не только собирательными, но и пенообра-
зующими свойствами. Твердый содовый дитиофосфат пенооб-
разующими свойствами не обладает. В жидких и сухих дитио-
фосфатах содержатся остатки сероводорода или сернистого
натрия, поэтому они обладают небольшим сульфидизирующим
действием по отношению к частично окисленным сульфидам.
Диалкил- и диарилмонотиофосфаты с общей
структурной формулой
*-\/ »-ч/
Р или Р
/ \ /\
R—0 0 — Me R — О S—Me
в отличие от дитиофосфатов при флотационных концентрациях
не образуют нерастворимых осадков с ионами тяжелых
металлов. Вследствие этого с помощью монотиофосфатов можно
достичь высокого извлечения цементной меди из кислых, пульп
при малом расходе собирателя, а также высокой селективности
флотации сульфидов меди или цинка от пирита при
значительно меньших рН, чем с дитиофосфатами или ксантогена-
тами.
Гидролизованные аэрофлоты (реагенты ТФ)
представляют собой сочетания моно- и дитиофосфатов. Они
являются более избирательными собирателями тонкоизмельченных
сульфидов, чем дитиофосфаты. В частности, они оказывают
сильное собирательное действие на медные минералы,
цементную медь и активированный медью, сфалерит, более слабое
собирательное действие на галенит и еще более слабое на пирит.
Органические производные дитиокарбаминовой кислоты —
ёитиокарбаматы, например диэтилдитиокарбамат натрия
СоНс О
\ X
N—С ,
/ \
С2Н5 S— Na
являются более сильными собирателями для сульфидов, чем
соответствующие ксантогенаты, но из-за дороговизны не нашли
промышленного применения. Дитиокарбаматы легко окисля-
137
ются растворенным в пульпе кислородом до дитиокарбамидов
по реакции, аналогичной реакции окисления ксантогенатов
4 (QHeJa NCSSNa + Оа + 2Н20 ^ 2 [ (С2Н5J NCSS]2 + 4NaOH.
Органические производные сероводорода —меркаптаны и
тиофенолы обладают сильными собирательными свойствами по
отношению к сульфидам и некоторым окисленным минералам
цветных металлов, например церусситу, малахиту и др.
Способность флотировать окисленные минералы объясняется суль-
фидизирующим действием этих реагентов. Неприятный запах —
одна из главных причин, по которой меркаптаны и тиофенолы
не нашли промышленного применения в СССР. В США для
флотации сульфидов меди и цинка используется меркаптобен-
зотиозол (имеющий два изомера)
А
V
С — N — Н
/\с-
Р С C=S или
С —N
V
О С —SH
s s
Из всех реагентов при флотации руд цветных металлов
главными реагентами-собирателями являются ксантогенаты и
дитиофосфаты. Остальные собиратели используются весьма
в ограниченном масштабе, главным образом в качестве
добавок к основным собирателям.
Расход сульфгндрильных реагентов при флотации несколько
возрастает с увеличением содержания в руде флотируемого
минерала и уменьшением крупности его частиц, но в основном
он зависит от степени окисления сульфидов, наличия в пульпе
ионов тяжелых металлов, связывающих собиратель в
нерастворимые соединения, и содержания минералов пустой породы,,
интенсивно сорбирующих собиратель (например, углистых
сланцев). Обычно суммарный расход сульфгндрильных
собирателей при основной и контрольной флотации составляет 30—
100 г/т руды, но при неблагоприятных условиях он может
возрасти до 200—300 г/т. ,
Механизм действия сульфгидрильных собирателей.
Результаты исследований показали, что при адсорбции из растворов,
находящихся в контакте с атмосферой, сорбционный слой ксан-
тогената на поверхности различных сульфидов представлен по
крайней мере двумя, а чаще тремя формами его закрепления
(рис. 7.9).
Первый монослой ксантогената, закрепившегося на поверх-,
ности сульфида, является хемосорбционным и представлен не
чем иным, как давно известным по гипотезе Д. А. Шведова
сульфидоксантогенатом. Вышележащие слои адсорбированного
ксантогената представлены нормальным, или полным, ксанто-
138
КХ
if ж
Ка;2
И
Рис. 7.9. Схема состава сорбционного слоя ксантогената на галените:
J — неизмененный сульфид; II — сульфидоксантогенат; /// — ксантогенат свинца; IV —
.диксантогенид
генатом свинца. Третья форма закрепления ксантогената
представлена продуктом его окисления — молекулами диксантоге-
нида. Такой состав сорбционного слоя на поверхности
сульфидных минералов наблюдается и в тех случаях, когда в качестве
собирателя используется не ксантогенат, а продукт его
окисления — диксантогенид [1, 3].
Последнее подтверждает обратимость реакции 2Кх-^Кх2 и
•свидетельствует о том, что на сульфидной поверхности
существует определенное равновесие между концентрациями ксан-
тогенатных ионов и молекул диксантогенида. Закрепление
собирателя в этих условиях обусловлено не только способностью
полярной группы ксантогената образовывать прочную
химическую связь с ионами тяжелых металлов и катионами
кристаллической решетки сульфидов, но и способностью аполярной
цепи собирателя к дисперсионному взаимодействию с молекул
лами диксантогенида (и другими углеводородными
радикалами).
Предложенные в разное время гипотезы механизма
собирательного действия сульфгидрильных собирателей отдавали
предпочтение, как правило, лишь одной из форм их адсорбции,
считая влияние остальных на флотацию несущественным.
Например, по химической теории, предложенной А. Ф. Таг-
гартом, предполагается, что успешная флотация минералов
обусловлена протеканием гетерогенной химической реакции
обменного типа с образованием труднорастворимых ксантогена-
тов соответствующих металлов. При этом возможность и
-направление реакции можно рассчитать по произведениям
растворимости соответствующих реакций в объеме раствора.
Однако с этой точки зрения трудно объяснить, например, случаи
адсорбции собирателя и флотации минералов, когда
соответствующее произведение растворимости еще не достигнуто, плохую
флотируемость окисленных медных и свинцовых минералов
даже при значительном превышении произведения
растворимости нормальных ксантогенатов свинца и меди. Гипотеза не
учитывает образования диксантогенида, не может объяснить при-
130
чин флотации минералов одним аполярным маслом или
пенообразователем.
В гипотезе, выдвинутой Д. А. Шведовым, предполагается,
что гидрофобизация поверхности сульфидных минералов и их
флотация при взаимодействии с ксантогенатом обусловлена
образованием сульфидоксантогенатов, прочно связанных с
кристаллической решеткой минерала. Если образуются полные
ксантогенаты металлов, то они теряют связь с
кристаллической решеткой, легко отслаиваясь при механическом
воздействии, и не оказывают собирательного действия на минеральную
поверхность.
Гипотеза Д. А. Шведова согласуется с худшей (по
сравнению с сульфидами) флотируемостью ксантогенатами
окисленных минералов цветных металлов. Так как растворимость суль-
фидоксантогената может быть меньше растворимости полного
ксантогената, то она объясняет и случаи адсорбции ксантоге-
ната и флотации сульфидов, когда произведение растворимости
полного ксантогената металла еще не достигнуто. В остальном
данной гипотезе присущи те же недостатки, что и химической
теории.
По гипотезе, предлагаемой А. А. Голиковым, основное
действие ксантогената сводится к очистке поверхности
сульфидных минералов от окисленных пленок, а гидрофобизация
поверхности и флотация сульфидов являются следствием
смачивающего действия диксантогенида — продукта каталитического
окисления ксантогената кислородом на их поверхности.
Необоснованное исключение А. А. Голиковым из рассмотрения влияния
химически закрепившегося собирателя на результаты флотации
вызвало справедливые замечания и сомнения в достоверности
и справедливости выдвинутой их гипотезы.
Помимо рассмотренных гипотез механизма собирательного
действия ксантогената, предложены также гипотезы
ионообменного механизма, гидролитического механизма и другие
менее известные.
Результаты исследований влияния различных форм
адсорбции ксантогенатов на флотируемость минералов показали [1],
что, как и в случае с оксигидрильными собирателями, ксанто-
генат, закрепившийся только химически или адсорбированный
в виде диксантогенида только физически, обладает низкой
флотационной активностью. Необходимая для полной флотации
плотность адсорбции в обоих случаях составляла обычно
несколько условных монослоев.
Эффективное закрепление частиц минералов на пузырьках
воздуха и их флотация при плотности адсорбции собирателей,
составляющей доли условного монослоя, наблюдается, когда
на поверхности одновременно присутствует как химически, так
и физически адсорбированный собиратель. Причем доля
каждого из них должна быть не меньше 10—30 % общей плотности
адсорбции собирателя на минеральной поверхности (рис. 7.10).
140
80 Д%
Рис. 7.10. Влияние содержания р
физически адсорбированного диксантогенида
в сорбционном слое на флотируемость у
галенита A), халькопирита B) и
пирита C) при постоянной общей
плотности сорбции бутилового ксантогената
E0 % условного монослоя)
1
г
10
8
6
4
2
уйм
нео5х
О
4 Гсм^
v ' пеоох
Рис. 7.11. Зависимость между
необходимой для полной
флотации различных образцов
галенита плотностью
адсорбции Г^еобх (в Д°лях
условного монослоя) одного
химически закрепившегося
собирателя и плотностью
адсорбции Гц"^х ксантогената при
одновременном присутствии
в сорбционном слое
химической и физической форм его
закрепления
Как и в случае с оксигидрильным собирателем, данное
положение подтверждается многими экспериментальными
результатами. Например, на сульфидизированной поверхности окис*
ленных цинковых минералов (смитсонита и каламина) и на
поверхности неактивированного сфалерита ксантогенат
представлен только химической формой его закрепления (ксанто-
генатом цинка) и названные минералы практически не
флотируются. Предварительная обработка их медным купоросом,
вызывающая образование диксантогенида (до 20—40 % общей
сорбции ксантогената), обеспечивает практически полную их
флотацию при той же плотности общей сорбции собирателя на
поверхности, что и до активации @,8—0,9 условного монослоя).
Флотационная активность как смешанной «пленки»
собирателя, так и сорбционного слоя, представленного только продук-
141
тами его химического взаимодействия, зависит от исходной
гидрофобное™ минеральной подкладки.
Зависимость между необходимой для полной флотации
плотностью адсорбции одного химически закрепившегося ксан-
тогената и плотностью адсорбции собирателя при
одновременном присутствии в сорбционном слое химической и физической
форм его закрепления для галенитов с различной степенью
окисления поверхности приведена на рис. 7.11.
Чем меньше степень окисления галенита, степень «ионно-
сти» и гидратации его поверхности, тем меньше необходимая
плотность адсорбции собирателя. Однако при любой степени
окисления поверхности галенита флотационная активность
сорбционного слоя собирателя, представленного обеими
формами его закрепления, выше флотационной активности одного
химически закрепившегося ксантогената (см. рис. 7.11).
Причем это различие тем больше, чем меньше степень окисления
поверхности минерала.
Небольшие значения необходимой адсорбции собиратели,
часто наблюдаемые на практике, в значительной мере
объясняются наличием обоих форм его адсорбции на поверхности
хорошо флотирующихся минералов. Кроме того, при пенной
флотации в ряде случаев роль физически сорбированного
собирателя могут играть молекулы пенообразователя.
7.8. Роль форм адсорбции собирателей при флотации
Высокая флотационная активность «смешанного» покрытия
собирателя, содержащего как химические, так и физические
формы его адсорбции, наряду с низкой флотационной
активностью только химически закрепившегося или только
физически адсорбированного собирателя позволяет предполагать, что
каждая из форм адсорбции выполняет разные функции,
обусловленные механизмом и кинетикой закрепления минеральной
частицы на пузырьке воздуха.
Самопроизвольный разрыв гидратной прослойки при
закреплении частицы на пузырьке на основании кривых Фрумкина —
Дерягина (см. рис. 2.7) должен происходить в том случае,
когда толщина прослойки достигнет критического значения,
которое весьма сильно зависит от степени гидрофобности
поверхности частицы и свойств поверхности пузырька. Чем больше
гидрофобность поверхности частицы, тем при большей толщине
гидратной прослойки может произойти самопроизвольный ее
разрыв, сопровождающийся закреплением частицы на
пузырьке.
Основной потенциальный барьер при закреплении частицы
на пузырьке обусловлен необходимостью уменьшить толщину
гидратной прослойки между ними в каком-либо месте до
критического значения. На основании кривой Фрумкина—
Дерягина в наиболее общем случае (см. рис. 2.7, кривая 2) для за-
1,42
крепления на поверхности вода — газ минеральная частица
должна преодолеть за счет своей кинетической энергии
энергетический барьер, равный e^d2, где е§ — удельная поверхностная
энергия прослоя воды; d2— площадь контакта газ — твердое.
Очевидно, это легче сделать над выступами с малым значением
d2> чем над плоской поверхностью. Последнее вытекает также из
результатов исследований П. А. Ребиндера, по которым гидрат-
ная оболочка легче прорывается на участках наибольшей
кривизны. Чем гидрофобнее выступ, тем меньше усилий нужно
прилагать для уменьшения гидратной прослойки до
критического значения и тем легче осуществить прорыв ее в одном
месте.
В настоящее время можно считать установленным,
что.физически адсорбированный собиратель не растекается по при-
родно гидрофобной или гидрофобизированной собирателем
поверхности минеральных частиц, а образует капельки.
Последнее подтверждено не только теоретическими, но и
многочисленными экспериментальными исследованиями [46]. Причем при
образовании смешанного сорбционного покрытия собирателя
точки химического его закрепления служат как бы центрами
конденсации физически сорбирующихся молекул и
образования микрокапелек молекул поверхностно-активного или апо-
лярного реагента (например, олеиновой кислоты, диксантоге-
нида, «гемимицелл» катионных собирателей, керосина и др.).
Образующиеся на минеральной поверхности микрокапельки
органических реагентов должны способствовать локальному
уменьшению прослойки жидкости между частицей и
пузырьком. .Жидкая микрокапелька должна быть более эффективна,
чем твердый выступ, так как в этом случае уменьшение водной
прослойки между минеральной частицей и пузырьком воздуха
требует меньших затрат энергии. Последнее обусловлено тем,
что граница жидкая капелька — вода может увлекаться водой,
вытекающей из прослойки, в то время как вода «прилипает»
к неподвижной поверхности твердого выступа и уменьшение
водной прослойки требует свершения большей работы против
сил вязкости.
При достижении критической толщины гидратной прослойки
между гидрофобной микрокапелькой и пузырьком воздуха
происходит ее разрыв. Локальный разрыв прослойки может
сопровождаться переходом части поверхностно-активного вещества
из жидкой капли во вновь образующуюся поверхность раздела
вода — воздух и быстрым распространением периметра
контакта пузырька по гидрофобизированной поверхности твердого.
Этому способствует капиллярное давление пузырька, которое
«раздувает» кольцевой участок пузырька у трехфазного
периметра, обладающий (вследствие концентрации на нем
органического вещества капли) меньшим значением дт-т> чем
остальная поверхность пузырька. Чем больше степень гидрофобности
поверхности, тем больше вероятность и скорость закрепления
143
частицы на пузырьке. Площадь контакта при этом, очевидно,
определяется максимумом убыли поверхностной энергии и
геометрией частицы.
Такой механизм закрепления частицы на пузырьке делает
понятным сосредоточение физически адсорбированного
собирателя по периметру трехфазного контакта, что способствует не
только образованию, но многократному упрочнению контакта
между пузырьком и частицей по механизму, предлагаемому
В. И. Мелик-Гайказяном [22, 23], По данным С. И. Крохина,
повышенная концентрация реагента у трехфазного контакта
возникает одновременно с его образованием и существенно
повышает прочность прилипания пузырьков к минералам.
Рассматриваемый механизм закрепления частицы на
пузырьке позволяет полагать, что основная роль физически
сорбированного собирателя заключается в том, чтобы обеспечить
эффективный разрыв гидратной прослойки между минеральной
частицей и пузырьком воздуха. Однако это приведет к
закреплению частицы на пузырьке только в том случае, если
поверхность ее в достаточной степени гидрофобна или гидрофобизи-
рована собирателем и обеспечивает возможность
распространения на ней трехфазного периметра контакта пузырька
воздуха. Например, закрепление на поверхности капелек апо-
лярного реагента (керосина) приводит к резкому повышению
флотируемости только таких минералов, как самородная сера,
чешуйчатый графит и молибденит, поверхность которых
обладает естественной гидрофобностью. Гораздо хуже флотируются
сульфиды цветных металлов и практически не флотируются
еще более гидрофильные карбонатные минералы.
Гидрофобизация минеральной поверхности и создание тем
самым условий для распространения по ней трехфазного
периметра контакта пузырька является, можно полагать, основным
назначением химической формы закрепления собирателя.
Использование в качестве собирателей полярно-аполяриых реа-
гейтов, полярная группа которых способна к химической
фиксации на поверхности флотируемых минералов, преследует
именно эту цель: нейтрализовать на минеральной поверхности все
те некомпенсированные связи, которые ответственны за
взаимодействие ее с дипольными молекулами воды, вызывают их
притяжение и приводят к образованию гидратной оболочки, и в то
же время за счет аполярной части реагента добиться того,
чтобы энергия взаимодействия молекул воды с поверхностью
минерала, покрытой углеводородными радикалами собирателя,
была бы гораздо меньше энергии взаимодействия молекул воды
друг с другом, т. е. гидрофобизировать поверхность минерала.
Вполне естественно, что необходимая для гидрофобизации
плотность сорбции химически закрепившегося собирателя
зависит от характера обнажающихся связей на плоскостях
раскола минералов, равномерности их компенсации собирателем,
прочности его закрепления и т. д.
144
Ограничение роли химически закрепившегося собирателя
вызывается тем, что без физически сорбированного собирателя
в сорбционном покрытии потенциальная (термодинамическая)
возможность закрепления пузырька воздуха на естественно
гидрофобной и на гидрофобизированной химически
закрепившимся собирателем поверхности реализуется с большим
трудом. Поэтому эффективная флотация минералов в общем
случае обеспечивается только при наличии и оптимальном
соотношении обеих форм закрепления собирателя в сорбционном
покрытии. Необходимая для полной флотации минералов
плотность общей адсорбции собирателя при этом значительно
понижается.
Последнее в значительной мере объясняется отмечавшейся
выше возможностью перехода части гидрофобизирующего
реагента из капли на трехфазный периметр контакта пузырька
в процессе закрепления его на поверхности минеральной
частицы и повышением прочности закрепления частицы на
пузырьке при этом, а также некоторой «цементацией» физически
сорбированным реагентом слоя химически закрепившегося
собирателя и повышением общей степени гидрофобной
поверхности.
Возможность хорошей флотации минералов при высоких
значениях адсорбции одного химически закрепившегося
собирателя следует, очевидно, связать с энергетической
неоднородностью минеральной поверхности и термодинамической
выгодностью закрепления собирателя в первую очередь на
выступах: углах и ребрах частицы. В результате этого на
гидрофобизированной поверхности имеются еще более
гидрофобные выступы, обеспечивающие эффективные прорыв гидрат-
ной прослойки между пузырьком и частицей. О большом
влиянии углов и прочих выступов при закреплении частиц на пузырьке
свидетельствует и тот факт, что окатанные зерна, например
самородной серы и молибденита, гораздо хуже флотируются,
чем зерна этих минералов, имеющих неправильную форму (по
данным Н. С. Петрова и Л. И. Гросмана).
8. АКТИВАТОРЫ И МЕХАНИЗМ ИХ ДЕЙСТВИЯ ПРИ ФЛОТАЦИИ
8.1. Назначение и основные механизмы
активирующего действия реагентов-модификаторов
_ Селективная флотация минералещ, как правило, не может
быть осуществлена с применением только собирателя и
пенообразователя. Она обесдечивается использованием реагентов-
модификаторов^ средаГкоторых важное значение имеют
реагенты-активаторы. В качестве реагентов-активаторов
применяют, как . правило, неорганические соединения: кислоты,
щелочи, соли щелочноземельных и тяжелых металлов, комплек-
145
сообразующие соединения и т. д. Основным назначением их
применения является обеспечение закрепления собирателя на
поверхности и ее гидрофобизация в целях эффективной
флотации извлекаемого минерала.
Результаты изучения роли химической и физической форм
сорбции собирателей позволяют рассматривать активацию
флотации минералов как создание условий, обеспечивающих
образование смешанного сорбционного слоя собирателя наряду
с повышением степени исходной гидрофобности минеральной
поверхности. Это достигается применением
реагентов-активаторов, вызывающих:
химическую очистку поверхности минералов от депресси-
рующих пленок и обнажение элементов кристаллической
решетки, способных к взаимодействию с собирателем и
образованию на них сорбционного слоя необходимого состава;
хемосорбцию ионов на поверхности, которые становятся
центрами закрепления собирателя;
гетерогенную химическую реакцию, приводящую к
образованию объемных пленок, поверхность которых является
благоприятной для образования необходимого сорбционного
покрытия собирателя.
8.2. Активирующее действие реагентов
путем химической очистки поверхности минералов
Примером такого механизма флотации минералов является
активирующее действие кислот.
Например, серная кислота активирует флотацию окисленных
пиритных руд. При сильном окислении сульфидов железа их
поверхность экранируется объемной пленкой конечных
гидрофильных продуктов окисления, не обладающих ни ионной, ни
электронной проводимостью. По этой причине взаимодействие
ксантогената с сульфидной поверхностью протекает весьма
слабо и закрепившийся собиратель не способен перекрыть гид-
рофилизирующее действие продуктов окисления. После добавки
кислоты гидрофильные окисленные соединения железа
растворяются, обнажая сульфидную поверхность, взаимодействие
ксантогената с которой приводит к образованию сорбционного
слоя собирателя необходимого состава, обеспечивающего
эффективную флотацию минерала.
Для активации никеленосных пирротинов по такому же
механизму лучшие результаты могут быть получены при
использовании многоосновных кислот, например щавелевой, или
свежеприготовленных растворов сернистой кислоты.
Растворение поверхностных пленок кислотами повышает
флотируемость касситерита, ильменита, вольфрамита,
флюорита и других минералов оксигидрильными собирателями.
Применение, например, плавиковой кислоты для активации берилла
объясняется растворением гидрофильных кремнекислородных
146
остатков на поверхности и обнажением катионов бериллия, что
приводит к увеличению доли хемосорбционного закрепления
олеата и образованию оптимального сорбционного слоя
собирателя на минерале [44].
Активирующим действием по такому же механизму при
флотации с оксигидрильными собирателями могут обладать
также щелочи и комплексообразующие соединения (цианиды,
фосфаты, фториды и др.)- Например, повышенная
концентрация едкого натра приводит к частичному растворению
силикатов, что используется для избирательной активации сподумена.
В свою очередь цианиды могут являться активаторами сильно
окисленных сульфидов меди, так как окисленные соединения
меди легко растворяются цианидами. Однако процесс
активации следует вести осторожно. Избыток цианида, сверх
необходимого для растворения окисленной пленки, приведет к
депрессии сульфидов меди при флотации их с сульфгидрильными
собирателями.
8.3. Активирующее действие реагентов
путем хемосорбции ионов на поверхности
Наиболее известными примерами данного механизма
активирующего действия реагентов являются активация
силикатных минералов солями щелочноземельных (кальция, бария и
др.) и тяжелых (свинца, меди, железа и др.) металлов,
активация сфалерита и сульфидов железа солями меди, активация
алюмосиликатов плавиковой кислотой в присутствии катион-
ных собирателей.
Активирующее действие солей щелочноземельных и
тяжелых металлов на силикатные минералы имеет особенно
большое значение при селективной флотации несульфидных
минералов оксигидрильными собирателями. Минералы, не имеющие
на своей поверхности катионов, способных образовать
труднорастворимые мыла, например кварц, не могут флотироваться
такими собирателями. Исследования показали, что на чистом
кварце из растворов олеата адсорбируются только молекулы
олеиновой кислоты и эта адсорбция, не содержащая хцмически
закрепившегося собирателя, не приводит к флотации. Однако
кварц легко активируется катионами двухвалентных и тяжелых
металлов, после чего начинает флотироваться олеатом или
другим оксигидрильным собирателем. По активирующей
способности М. А. Эйгелес [49, 50] располагает их в следующий ряд:
Fe3+>Al+3>Fe2^>Ca2+> Mg2+.
Механизм активации кварца ионами металлов и
последующей гидрофобизации его собирателем состоит в следующем [3].
Поверхность кварца [Si02]m в воде, как было показано
ранее, покрыта силанольными группами =Si—ОН и
схематически ее можно изобразить как [Si02]m=Si—ОН. В присутствии
катионов металлов в пульпе водород силанольной группы
4 147
ее
5,k 5,8 6,1 6,6 1,0
, /СаОН+] x
80
60
70
10
О
M
io zo so ko so 6or;%
[CaOH+]
при акти-
Рис. 8.1. Зависимости между значениями Ig—— и lg
ЬГ0-Г * [Н+1
вации кварца солями кальция (а) и между плотностью адсорбции Г (в
долях условного монослоя) кальция и флотируемостью у кварца (б) олеатом
натрия (8,68- Ю-5 моль/л)
замещается продуктом частичного гидролиза катиона металла
МеОН+ по реакции
[Si02]m^Si-0-H-
—Me—OH + H+,
МеОН+ -^ [Si02]m = SiO—
константа равновесия которой
/С-
[ [SiQ2]m = SiO — Me — ОН] [Н+]
|[Si02]m = SiO- H] [MeOH+]
(8.1)
(8.2)
Если учесть, что в процессе активации концентрация
активированных центров ([SiCblm^SiO—Me—ОН) на поверхности
кварца численно равна плотности адсорбции катионов металла
Г, а концентрация нсактивированных центров ([SiC^Jm^Si—
—О—Ы) при этом равна разности между предельной
плотностью сорбции активирующих катионов Го, равной обычно
условному монослою, и измеренной Г, то выражение (8.2)
константы равновесия реакции (8.1) примет вид
Г [Н+]
/С-
к
Г0—Г [МеОН+]
В логарифмическом виде
Г
-^IgK + lgJ^HlL,
г & ^ & [н+]
между значениями lg
[МеОН+1
Го
зависимость . ^, „ в „ „
Го-Г [11+]
представляет собой уравнение прямой линии, что для случая
активации кварца катионами кальция подтверждается
148
(рис. 8.1, а) результатами обработки экспериментальных дан-'
ных, приведенных, например, в работе [44].
Хемосорбированный на кварце катион удерживается одной
валентной связью с поверхностью, другая валентная связь
удерживает гидроксильный ион ОН-, который легко
замещается на анион Агг собирателя:
[Si02]m = SiO—Me—ОН + Ап" -^ [Si02]m =
= SiO—Me—An + OH-.
В результате такого замещения поверхность кварца
оказывается покрытой хемосорбированным собирателем, поверх
которого могут закрепляться путем физической адсорбции
молекулы собирателя. Образование такого сорбционного слоя
собирателя обеспечивает эффективную флотацию кварца.
Необходимая для полной флотации кварца олеатом натрия
(8,68 • 10~5 моль/л) плотность адсорбции кальция на его
поверхности составляет 50 % условного монослоя (рис. 8.1, б)г
что отвечает значению lg — J-^6,76 (см. рис. 8.1, а).
[Н+]
Следовательно, значение необходимой для активации и полной
флотации кварца концентрации СаОН+ ионов при всех
значениях рН раствора можно рассчитать по уравнению
g [СаОН+]необх = 6,76—рН.
Аналогичен механизм действия оксигидрильных
собирателей при флотации не только силикатных минералов (кварца,
полевого шпата и др.), активированных другими катионами,
например трехвалентного железа или, алюминия, но и при
флотации касситерита. При взаимодействии олеиновой кислоты
с чистой поверхностью касситерита она адсорбируется только
в молекулярной форме и минерал не флотируется. Если же
поверхность касситерита активирована ионами поливалентных
металлов, то образуются хемосорбированные соединения
собирателя, поверх которых, по данным С. И. Полькина [34], могут
закрепляться десятки и сотни монослоев физически
адсорбированных соединений и молекул собирателя, т. е. образуется
сорбционный слой, необходимый для эффективной флотация
минерала.
Активирующее действие солей тяжелых металлов на
флотацию сульфидных минералов играет важную роль при
селективной флотации с сульфгидрильными собирателями.
Практически на всех обогатительных фабриках,
перерабатывающих полиметаллические руды, для активации сульфидов
цинка используется медный купорос (CuS04'5H20). Плохая
флотируемость неактивированного сфалерита, несмотря на
наличие хемосорбированного собирателя, объясняется
невозможностью окисления ксантогенатных ионов до диксантогенида
149>
вследствие высокого отрицательного заряда поверхности
данного минерала [1].
Образование активирующего соединения на поверхности
сфалерита в присутствии медного купороса обусловлено
замещением цинка на изоморфный ион меди
ZnS + Cu2+ «£ CuS + Zn2+. (8.3)
Реакция (8,3) прекращается после образования на
поверхности сфалерита монослоя CuS [44].
Условия активации сфалерита ионами меди описывается на
основании реакции (8.3) уравнением
lg [Си2+]не0бх - -11 + lg [Zn2+]. (8.4)
Так как в нейтральной и щелочной средах [Zn2+J
контролируется произведением растворимости гидроокиси цинка (см.
рис! 6.3, д)
Zn (ОНJ + 2Н+ & Zn2+ + 2Н20 (8.5)
и -описывается на основании реакции (8.5) уравнением
lg[Zn2+]- + 11,7-2 рН, (8.6)
то после подстановки выражения для lg[Zn2+J из уравнения
(8.6) в уравнение (8.4) получим
g[Cu2+]„eo6x - +0,7-2рН. (8.7)
Имеющаяся концентрация ионов меди [Си2+]Имеющ при
подаче медного купороса в пульпу, в свою очередь,
контролируется произведением растворимости Си2(ОНJС03 при рН<9 и
Си(ОНJ при рН>9 (см. рис. 6.3, в, г), а именно
Си2 (ОНK С03 + 2Н+ ^r 2Cuf+ + 2Н20 + СО2-,
lg [Си2+]имеющ = -2,78-рН-0,5lg [СО2-]
или с учетом выражения E.13) для [С032-]:
lg[Cu.+1,mOT _ -2,78-рН-0,5 lg (кл+£*$ + т^ i (8.8)
Си (ОНJ + 2Н+ & Си2+ + 2Н20;
№и2+]Ймеющ- +9,2-2pH. (8.9)
Возможность активации сфалерита определяется
соотношением [Си2+]имеющ/[Си2+]Необх. Результаты расчетов показывают
(рис. 8.2, а, кривая /), что при всех рН значение [Си2+]имеющ
больше [Си2+]Необх и это неминуемо должно приводить к
активации сфалерита ионами меди. Необходимо учитывать при
этом, что превышение имеющейся концентрации ионов меди
над необходимой еще не определяет закономерностей
адсорбции меди на сфалерите. По результатам исследований [44J,
адсорбция ионов меди на этом минерале подчиняется уравнению
J 50
Лангмюра и поэтому скорость 100
адсорбции (или за
определенный промежуток времени плот- ^ 80
ность адсорбции) меди (см. ц^
рис. 8.2, а, кривая 3) вполне ^
закономерно уменьшается в М
связи с резким понижением
[Си2+] в растворе (см. рис.
8.2, а, кривая 2) при
увеличении рН в соответствии с
уравнениями (8.8) и (8.9).
Наличие изоморфной примеси
железа в сфалерите не
оказывает влияния на закономерности
адсорбции ионов меди на его
поверхности [30, 44]. Несмотря
на снижение [Си2+] в
растворе, при всех значениях рН
наблюдается активация и
успешная флотация сфалерита (см.
рис. 8.2, а, кривая 4). Это
подтверждается и практикой
флотации сульфидов цинка.
Однако вследствие весьма
малых значений [Си2+] в
пульпе необходимое время
активации в промышленных
условиях может достигать 20—40
мин [2, 25].
Загрузка медного купороса
в пульпу может вызвать акти
SO
30
20\
V ю
4Ю\
* 81
42
46
-20
0
2\~
42
0
2,5
2,0
1,5
1,0
а
\ *Sl
1 \
1 '
I L
Чг
/
4
]N
уд
1/гК\! I
_ 46L 0,5l
7 8 9 W 11 pH
Рис. 8.2. Влияние рН на
соотношение [Си2+]имеющ/[Си2+]Нвобх A),
[Си2+]име ю щ B), плотность адсорб-
ции Г ионов меди на поверхности
C) ■ и изменение флотируемостиг
(прирост извлечения Де) D) сфа-
вацию и других сульфидных лерита (а) и пирита (б)
минералов. Условие
активации, например, пирита — одного из наиболее распространень
сульфидов железа — описывается на основании реакции
FeS2 + Cu2+ ^ CuS + So + Fe2+
уравнением
Jg [Си2+]необх = -6,77 + lg [Fe2+]
или с учетом выражения F.4) для Ig[Fe2+]
lg[Си2+]йеобх = -6,77 +Н.4 g-pH=+ 4,63 - -?jj- рН. (8.10>
15 15
Оценка возможности активации пирита с использованием
уравнений (8.8) и (8.9) для расчета значений [Си2+]Имеющ и
уравнения (8.10) для расчета значений [Cu2+Jfie06x (рис. 8.2, б,
кривая /) позволяет обосновать наблюдаемые на практике
адсорбцию ионов меди (см. рис. 8.2, б, кривая 3) и изменение
151
флотируемое™ пирита (см. рис. 8.2, б, кривая 4) при подаче
медного купороса, поскольку их изменение при различных
значениях рН подчиняется общей закономерности (см. рис. 8.2, б,
кривые I, 3, 4).
Вытеснение ионами меди ионов цинка и железа объясняется
тем, что сульфид меди CuS менее растворим, чем сульфиды
цинка и железа.
Установлено, что если сульфид металла менее растворим,
то и ксантогенат этого металла также будет менее растворим.
Поэтому для сульфидных минералов всякая растворимая соль
металла, дающего при взаимодействии с ионами серы менее
растворимый сульфид, является активатором. Например, для
сфалерита активаторами могут быть не только коны меди, но
и ионы свинца, серебра, и ртути, так как PbS, AgS и HgS
менее растворимы, чем ZnS. Закрепление ксантогената на
активированном вышеперечисленными металлами сфалерите
происходит более прочно и при меньших концентрациях собирателя
в пульпе. Закрепление ксантогената на активированном
сфалерите сопровождается одновременным образованием диксанто-
генида в сорбционном слое собирателя [1], что приводит к
эффективной флотации минерала.
Таким образом, если основной причиной активации
силикатных минералов является обеспечение возможности хемо-
сорбции собирателя, то при активации сфалерита
обеспечивается возможность образования в сорбционном слое собирателя
физически адсорбированных молекул диксантогенида.
Расход активирующих солей составляет 50—400 г/т.
Скорость их взаимодействия с минеральной поверхностью зависит
от значения рН, изменяющего состояние реагента в жидкой
фазе пульпы и состояние минеральной поверхности.
Например, при активации силикатных минералов скорость
и значение адсорбции активирующих ионов обычно возрастают
с увеличением рН, так как при этом увеличивается
концентрация ,однозамещенных гидратов щелочноземельных металлов
(например, ВаОН+, СаОН+ и др.) и число силанольных групп
на поверхности, в результате чего увеличивается вероятность
протекания реакции (8.1).
При активации сульфидных минералов медным купоросом,
наоборот, максимальные скорость и значение сорбции меди
наблюдаются в слабокислой и нейтральной средах, отвечающих
наибольшей концентрации ионов Си2+. В щелочных пульпах
образуется малорастворимая гидроокись меди Си(ОН)г,
концентрация ионов Си2+ резко уменьшается (см. рис. 8.2,
кривая 2) и скорость активации падает.
Активирующее действие комплексных соединений имеет
важное значение при флотации алюмосиликатов (берилла,
полевых шпатов) с катионным собирателем в присутствии
плавиковой (фтористоводородной) кислоты HF [44].
По гипотезе М. X. Букенгема активирующее действие пла-
152
виковой кислоты обусловлено взаимодействием ее с группами
= А1—ОН на поверхности алюмосиликатов по реакциям:
]-Al-OH + 2H++2F-^t] = Al—F-)H+ + H20;
]=А1-ОН + 2Н№]-А1 — F-)H+ + H20,
в результате протекания которых гидроксильные группы
замещаются ионами фтора с образованием координационной связи
между ионами фтора и алюминия. Образующееся на
поверхности комплексное соединение алюминия с фтором способно
замещать свой ион водорода на ион катионного собирателя —
первичного алифатического амина RNH3+
[-Al-F-)H+ + RNH3+^]-AI-F-)RNH3+ + H+,
что приводит к гидрофобизации поверхности алюмосиликатов
и эффективной их флотации.
Если в присутствии плавиковой кислоты в пульпе
последовательно протекают реакции:
Si02 + 4Н+ + 6F" ^ SiF|~ + 2Н20;
SIFJ-+ RNH3+^t RNH2SiF^,
то образующийся комплексный ион RNH3SiF6~ способен также
активировать флотацию алюмосиликатов [44] в результате
взаимодействия с катионом алюминия на их поверхности
] = А1—ОН + RNH3SiF^ + Н+ ^ ] = А1—SiFeRNH2 + Н20.
Таким образом, конечным результатом взаимодействия
катионного реагента (амина) с алюмосиликатами в присутствии
плавиковой кислоты является образование комплексного
соединения, в состав которого входят алюминий, фтор и амин.
8.4. Активирующее действие реагентов
путем гетерогенной химической реакции
Примером данного механизма активации является
образование объемных сульфидных пленок на поверхности
окисленных минералов свинца, меди и цинка под действием сульфи-
дизаторов. В качестве сульфидизаторов могут применяться
любые растворимые сернистые и гидросернистые соединения
щелочных и щелочноземельных металлов и аммония: Na2S, NaHS,
K2S, KHS, (NH4JS, NH4HS, CaS, BaS. Из них наиболее
широко на фабриках используется Na2S, как наиболее дешевый и
доступный. *
Без предварительной сульфидизации окисленные минералы
свинца, меди и цинка сульфгидрильными собирателями
практически не флотируются, несмотря на значительную плотность
адсорбции химически закрепившегося собирателя на их
поверхности [1].
155
e/f%rCf%
80
so
¥o
zo
V w
О Z ¥ 6 8 W 12 1b t,MUH
l§[Sz'l
-1026
Y-1166
-17.06*
Y-20^6
L~23;86
Рис. 8.З. Изменение концентрации сульфидных ионов [S2~], общей
плотности адсорбции Г ксантогената (в долях условного монослоя), содержания С
диксантогенида в сорбционном слое (в долях общей сорбции собирателя),
извлечения е и скорости К флотации церуссита в присутствии сульфидиза-
тора в зависимости от времени t флотации (концентрация бутилового
ксантогената калия 20 мг/л, рП 9,5)
Результаты исследований роли сульфидизатора [I]
позволяют считать, что резкое возрастание флотируемости
окисленных свинцовых и медных минералов после их предварительной
сульфидизации обусловлено, во-первых, повышением степени
гндрофобности минеральной поверхности до обработки ее
собирателем, во-вторых, появлением возможности образования
покрытий собирателя, состоящих как из химически
закрепившегося ксантогената, так и физически адсорбированных на нем
молекул диксантогенида.
Ярко выраженная зависимость флотируемости, например,
сульфидизированного церуссита от наличия и содержания
диксантогенида в сорбционном слое собирателя на поверхности
иллюстрируется рис. 8.3. Излишне высокая [S2~] в растворе
в течение первых минут предотвращает образование
диксантогенида и флотацию сульфидизированного церуссита, несмотря
на наличие на его поверхности химически закрепившегося
ксантогената. Появление диксантогенида в сорбционном слое
собирателя после соответствующего понижения [S2~] вызывает
бурную флотацию церуссита (см. рис. 8.3, кривые С, е, К).
При этом общая плотность адсорбции Г собирателя
изменяется весьма незначительно. Дальнейшее уменьшение концен-
154
Рис. 8.4. Сопоставление
теоретических значений
[S2-] G—3),
обеспечивающих переход
окисленных свинцовых
соединений в сульфид
свинца, и
экспериментальных значений' [S2-]
D—8), обеспечивающих
максимальную
скорость флотации
свинцовых минералов:
/ — карбонат, гидратокар-
бонат и молнбдат свинца;
2 — фосфат свинца; 3 —
хромат свинца; 4 — церус-
сит и галенит; 5 —
вульфенит; 6 — англезит; 7 —
свинцовые минералы
(галенит, церуссит и англезит)
при флотации окисленных
и сульфидно-окисленных
руд пяти месторождений
СССР; 8 — пироморфит
Ц [§г~],моль/л
трации сульфидных ионов приводит к уменьшению скорости
флотации К, несмотря на резкое возрастание общей
плотности адсорбции Г собирателя на поверхности минерала.
Аналогичные результаты получены на других свинцовых
минералах (окисленном с поверхности галените, англезите,
вульфените, пироморфите) и минералах меди (азурите,
куприте, малахите и окисленном с поверхности халькопирите).
Во всех случаях максимальная скорость флотации сульфи-
дизированных минералов соответствует оптимальной [S2~]
в пульпе, значение которой существенно зависит от рН пульпы
и не зависит от степени окисления руд и от того, с какого
месторождения взяты минералы или руды, если свинец или медь
в них представлены такими же минералами.
При сопоставлении экспериментальных значений
оптимальной [S2~] при флотации свинцовых минералов с результатами
теоретических исследований установлено [1], что оптимальная
[S2~] при всех значениях рН и для всех исследованных
свинцовых минералов соответствует теоретически необходимой
[S2-], обеспечивающей возможность равновесия между
окисленными и сульфидными соединениями свинца (рис. 8.4).
Для церуссита, вульфенита, галенита и англезита,
поверхность которых до сульфидизации представлена карбонатом или
гидратокарбонатом свинца, зависимость оптимальной [S2-] от
155
80\
10
ВО
50\
щ
ш
°1/
дгко/
Я®
V*
/з
' 1
•
®^
ч°
о\о>ч
Ар
L
ч\
• *
\°
-з -z
[S*J,
О +1
имеют.
i-Z +3
ц :?-цмеюц
Рис. 8.5. Влияние соотношения
[52-]ИМеющ/[52']Необу в пульпе на
извлечение е общей A) и
окисленной B) меди в коллективный мед-
но-молибденовый концентрат
межцикловой флотации Алмалыкской
обогатительной фабрики и общей
меди C) из смешанных руд Снеги-
ревского месторождения
рН пульпы при флотации одинакова; для пироморфита —
фосфата свинца эта зависимость другая (см. рис. 8.4). В обоих
случаях значения оптимальной [S2~] при различных рН
соответствуют началу интенсивного образования диксантогенида
в сорбционном слое собирателя на поверхности сульфидизиро-
ванных свинцовых минералов. При агитации или в точке
подачи сульфидизатора [S2-] должна быть примерно в 100 раз
больше, чем при флотации. Во всех случаях [S2~], такова,
что ее вполне можно измерить, используя в качестве датчика,
например, сульфидсеребряный или аргентитовый электрод.
Оптимальная [S2~] при сульфидизации и флотации
окисленных минералов меди (малахита, азурита, куприта и
окисленного халькопирита) значительно превышает необходимую для
образования на их поверхности CuS или СшЭ. При
использовании в качестве собирателя ксантогената наблюдается
определенная зависимость между [S2-]0«thm, [Кх~] и рН. Например,
для бутилового ксантогената (ButKx) она имеет для всех суль-
фидизированных минералов меди следующий вид:
lg коптим = -16 + lg [ButKx-] + рН.
(8Л1)
Пленка сульфида на окисленном минерале не прочна в
механическом и химическом отношениях. Она сравнительно легко
отслаивается при перемешивании пульпы и окисляется
растворенным в воде кислородом. Поэтому скорость флотации с
течением времени уменьшается. Для повышения извлечения
применяется повторная сульфидизация. Расход сульфидизатора
колеблется в пределах 200—1000 г/т, но если в пульпе
присутствуют такие поглотители ионов S2-, как сульфат цинка или
железа, тонкие шламы лимонита и некоторых других
минералов, то расход может значительно возрасти.
156
Очевидно, что при постоянном расходе сульфидизатора
IS2~] в пульпе при флотации вследствие неоднородности
вещественного состава перерабатываемых руд величина
переменная. Слишком низкая [S2-] в пульпе не обеспечивает
необходимой сульфидизации окисленной поверхности минералов,
тогда как избыточная [S2-] приводит к подавлению флотации
этих минералов (см. рис. 8.3, 8.5). Поэтому необходим
автоматический контроль и регулирование [S2~] в пульпе при
флотации свинцовых минералов в соответствии с зависимостью
lS2~]=f (pH) (см. рис. 8.4), а при флотации медных
минералов— в соответствии с уравнением (8.11).
*. ДЕПРЕССОРЫ И МЕХАНИЗМ ИХ ДЕЙСТВИЯ ПРИ ФЛОТАЦИИ
9.1. Назначение и основные механизмы
действия реагентов-депрессоров
Применение реагентов-депрессоров является основным
средством получения максимальной селективности прц
флотационном разделении минералов с близкими свойствами.
Если для эффективной флотации минералов необходимо
соблюдение двух условий: гидрофобизации поверхности,
например, за счет погашения некомпенсированных валентностей
минеральной поверхности собирателем и возможности
закрепления на гидрофобизированной подкладке микрокапель
физически адсорбированного собирателя, то для подавления их
флотации необходимо несоблюдение хотя бы одного из этих
условий. Это обеспечивается обычно применением
реагентов-депрессоров, в качестве которых на практике наиболее широко
используются щелочи, цианиды, сернокислый цинк, сернистый
натрий, сернистая кислота и ее соли, смесь сернокислого
железа и сульфата натрия, двухромовокислые соли, жидкое
стекло, некоторые органические
высокомолекулярные и другие а .. . , .....
соединения. Основной труд- №/}//?}Щ ^ w//////////A
ностыо при выборе депрессирую- б ' ' *
щих реагентов является их не- tyW""/s%L.__^.№wwmh<
достаточная избирательность по в *' *" у'
отношению к разделяемым ми- ' k^/M^/M \$)Ш$}к
нералам. * я **k ^
Основные механизмы депрес- ? пузырек
сирующего действия реагентов т^^тгп
можно свести к следующим: w?///^?/}k _
1. Растворение поверх- ' ■"/
ностных соединений со- 1 -собиратель; Ж-депрессор
бирателя и создание ус- 0 П1 -
тт ~ J, т, л „«~ ™, Рис- 9.L Схема механизмов де-
лови и, препятствующих Прессирующего действия реаген-
закр.еплению собирателя тов
157
на поверхности минерала. Примером такого
механизма является депрессирующее действие цианистых солей на
флотацию сульфидов меди или активированного медным
купоросом сфалерита. В отсутствие депрессора собиратель
закрепляется на их поверхности. Добавка в пульпу цианистых солей
приводит к разрушению ксантогенатных соединений меди
вследствие образования прочных медноцианистых комплексных
ионов Cu(CN)-2, Cu(CNJ-3, Cu(CNK-4 по реакции
МеА] ~<ЧК\ + aCN- -> MertSm] + pCu (CN)r + ?Kx-.
В результате этого обнажается гидрофильная поверхность
минерала и его способность флотироваться утрачивается (рис.
9.1, а).
2. Вытеснение ионов собирателя ионами
депрессора, образующими с ионами, минерала
труднорастворимое гидрофильное соединение.
Примером такого механизма является депрессирующее
действие гидроксильных ОН- и сульфидных S2~ ионов,
конкурирующих с ионами собирателя Кх~ и замещающих их на
поверхности (рис. 9.1, б):
Me„Sm] —MepKxv + «ОН- -* Me„Sm] —МерОНа + уКх~;
Ме^]—ЩКху + aS2- -* Me„Sm] —Ме^а + ?Кх".
3. Повышение степени гидрофильности
минеральной поверхности без вытеснения
собирателя. Энергетическая неоднородность минеральной
поверхности вызывает неравномерное распределение собирателя по
ней. Средняя гидрофобность, например, сульфидной
поверхности определяется гидрофобностью участков,.покрытых
собирателем (МеаКхр), и гидрофобностью чистой поверхности
(Меа SaOj/), не занятой собирателем. При дооавке депрессора
Апр~ он может закрепляться на свободных участках
поверхности, резко увеличивая степень их гидрофильности (рис. 9.1, в):
Me„S„
—АВДхр
—MeaSxOy + 7Апр" -* Me„S1
J —- MeaKxp
п^т
—MeaKxp
—MeaAnv + SxQj'
—MeaKxp
Это приводит к увеличению средней гидрофильности
поверхности без вытеснения-собирателя и флотируемость
минерала ухудшается. Примерами такого механизма является
депрессирующее действие хроматов и фосфатов на галенит.
4. Закрепление на поверхностидепрессируе-
мого минерала гидрофильных неорганических
или. органических частиц. Тонкодисперсные и
коллоидные частицы всегда значительно больше ионов или молекул
158.
собирателя. Закрепляясь на свободных от собирателя участках
ловерхности, они перекрывают гидрофобизирующее действие
собирателя (рис 9.1, г). Контакт между пузырьком и частичкой
и ее флотация поэтому становится невозможными. По такому
механизму могут депрессировать флотацию -минералов жидкое
стекло, осадки продуктов взаимодействия реагентов,
высокомолекулярные органические вещества.
9.2. Депрессирующее действие щелочных реагентов
В качестве щелочных реагентов чаще всего используют
соду, известь и едкий натр. В присутствии анионных
собирателей с их помощью можно подавить флотацию практически
всех минералов. Поскольку сульфиды меди и активированный
медным купоросом сфалерит обладают наибольшей, а
сульфиды железа наименьшей устойчивостью к депрессирующему
действию щелочных реагентов, при флотационном разделении
данных минералов чаще всего и
используются щелочные
реагенты.
Депрессируют щелочные
реагенты в основном по второму
механизму. Повышение
концентрации гидроксильных ионов
в пульпе приводит к вытеснению
ионов ксантогената из сорбцион-
ного слоя на поверхности
сульфидов (рис. 9.2).
Необходимое соотношение
концентраций ионов собирателя
X" и гидроксильных ионов ОН-,
обеспечивающее прилипание
пузырька воздуха к поверхности
минералов, отвечает общему
выражению [8]
ЦГ,МОЛЬ/СМг
[Х']=т[ОНГ]у9
в котором численные значения
коэффициента
пропорциональности т и другой
характеристической константы у. зависят от
флотируемого минерала и
используемого собирателя.
Расчетные уравнения
необходимой концентрации собирателя
в пульпе при флотации каждого
конкретного минерала могут
-39
-7 -6 -S-t* ~J ~2 -/ 0 +1 i-Z t3+H-
1л[Кх-3
Рис. 9.2. Влияние соотношения
концентраций ксантогенатных
[Кх~] и гидроксильных [ОН-]
ионов в растворе на плотность Г
адсорбции ксантогената на
поверхности пирита (по данным
A.M. Годена [8]):
/ — для бутилового ксантогената на
зыряновском пирите ио результатам
г -Г „ г=/ (рН) при различных [Кх~1; 2~
бЫТЬ ПОЛучеНЫ Путем ТерМОДИНа- Для этилового ксантогената на шь
J J r рите по результатам Г=/ (рН)
VII1V «V y\*>*j"~
мического анализа возможных различных [Кх-1
при
159
[YiX] [Kx~72 fKx'Ty (кх7
pHi pH£ pH3 pH
Рис. 9.З. Влияние концентрации ксантогената [Кх~] при постоянных
значениях рН (а) и значений рН при постоянных [Кх~] (б) на флотацию
(извлечение е) минералов
реакций собирателя в приэлектродной зоне минерала и
сопоставления полученных результатов с экспериментальными
данными [1, 3].
Количественные зависимости между необходимой [Х~] и рН
пульпы требуются, во-первых, для объективного анализа и
совершенствования процессов селективной флотации руд и, во-
вторых, для использования их в качестве задания
функциональным блокам систем автоматического контроля и
регулирования флотационных процессов на обогатительных фабриках.
Рассмотрим наиболее важные в практическом отношении
зависимости между концентрацией ксантогенатных ионов [Кх~]
и значением рН пульпы при флотации основных сульфидных
минералов.
Зависимость'[Кх~] = 1 (рН) при флотации сульфидов свинца.
Экспериментально установлено, что каждому значению рН
отвечает своя [Кх~]необх, при которой обеспечивается полная или
максимальная флотация сульфидов свинца (рис. 9.3, а), и,
наоборот, каждой [Кх~] в пульпе отвечает вполне определенное
критическое значение рН, превышение которого вызывает
резкое падение флотируемости минерала (рис. 9.3, б).
Совокупность таких данных, полученных при флотации образцов
галенитов многих месторождений, позволила установить
количественную зависимость [Кх^необх—/(рН) (рис. 9.4, а).
160
Рис. 9.4. Влияние рН на необходимую для полной флотации 1аленитов
разных месторождений концентрацию [Кх~]Необх этилового (Л) и бутилового
(Б) ксантогенатов (а), а также избытка и недостатка ксантогената в пульпе
по сравнению с [Кх~]пеобх на извлечение ерв свинца (б) на Белоусов-
ской (/—4), Зыряновской E, 6), Алмалыкской G) обогатительных фабриках
и канадской фабрике «Сулливан» (8)
Сопоставление результатов теоретических исследований
с экспериментальными (см. рис. 9.4, а) приводит к выводу, что
[Кх~], необходимая для полной флотации галенита, равна
минимальной, обеспечивающей образование ксантогената свинца
на поверхности галенита при данном значении рН. Она
практически одинакова для галенитов различных месторождений и
может быть рассчитана для значений рН от 7 до 9,5 по
уравнению, полученному из условия равновесия РЬКх2^РЬС03>
а для значений рН от 9,5 до 12 —по уравнению, полученному
из условия равновесия РЬКх2^РЬ3(ОНJ(С03J, т. е. из
условия равновесия продукт окисления^РЬКх2 [1, 3].
Данный вывод подтверждается также хорошим совпадением
полученных результатов с приводимыми А. М. Годеном
экспериментальными значениями [Кх~], равновесной ксантогенату
свинца в водном растворе [8], с данными Уорка и Кокса по
зависимости между критической [Кх~] и рН раствора [38] и,
наконец, с результатами промышленных исследований (рис
9.4,6).
Зависимость [Kx~] = f(pH) при флотации сульфидов
железа. Результатами теоретических исследований по
обоснованию экспериментально полученной зависимости (рис. 9.5, а)
6 Заказ .No 1957
161
Рис. 9.5. Влияние рН на необходимую для полной флотации пиритов G),
марказита C) и пирротинов B) разных месторождений концентрацию [Кх~*]
этилового (А) и бутилового (Б) ксантогенатов (а) и результаты
экспериментальное проверки уравнения (9.4) на Алмалыкской и Зыряновской
обогатительных фабриках (б):
•Ф — IgJC полной флотации; Д— IgK полной депрессии
между минимальной необходимой для полной флотации
сульфидов железа (пирита, марказита и пирротина) [Кх~]необх и
рН пульпы установлено [1], что она не может быть объяснена
образованием на поверхности нормальных ксантогенатов двух-
или трехвалентного железа. Ее нельзя также объяснить, если
предположить, что на поверхности сульфидных минералов
железа образуются более прочные и труднее растворимые (чем
нормальный ксантогенат) основные ксантогенаты
трехвалентного железа типа Fe(OHJKx и Fe(OH)Kx2 [1].
Экспериментальные зависимости [Kx-]neo6x=f(pH) для
сульфидов железа (пирита, марказита и пирротина) можно
объяснить, если предположить, что [Кх~]необх определяется [FeOH+]
в приэлектродном слое в условиях перехода FeS2 и FeS
:в Fe(OHK (основной продукт окисления сульфидов железа)
и S2032~ [1, 3].
Например, для пирита в этих условиях FeOH+ .описывается
уравнением F.5). Прибавив к его левой и правой частям
lg[Kx~], найдем расчетное уравнение [Кх~]необх при различных
рН:
3g [Кх-]веобх = {-0,29+ lg [FeOH+] [Кх~] } + ± рН +
+ -i-lg[S20§-]. (9.1)
162
Аналогичным образом могут быть получены зависимости
[Кх^необх^ДрН) и для других сульфидов железа. Они имеют
общий вид
lg [Кх-]необх = [\gK + lg [FeOH+] [Kx-]) +
+ apH + pl£[S2Or], (9.2)
где значение IgK зависит от термодинамических
характеристик минералов, а значения а и р — от химического состава
минералов.
Произведение [FeOH+][Kx~] = Ко = const для каждого
минерала при использовании в качестве собирателя этилового
ксантогената составляет: для пирита 3,5-10~п; для марказита
2,2-1 О*-11; для пирротина 1Д*10-10. Значения Ко при
использовании бутилового ксантогената примерно в 10 раз меньше, что
хорошо согласуется с теоретическими представлениями о
влиянии длины аполярной цепи собирателя на его активность.
Разные значения /Со для разных сульфидов железа
свидетельствуют о том, что постоянство произведения [Fe(OH)+][Kx~] не
является следствием образования на минеральной поверхности
или в приэлектродном слое основного ксантогената железа
Fe(OH)Kx.
Как и в случае галенита, необходимая для максимального
извлечения того или иного сульфида железа [Кх~] практически
не зависит от того, с какого месторождения отобран минерал.
Однако полученные по уравнениям (9-1) и (9.2) расчетные
значения [Кх~]необх являются справедливыми, если концентрация
ионов кальция в пульпе еще недостаточна для образования
кальцийсодержащих соединений на поверхности сульфидных
минералов железа. В противном случае, как, например, при
регулировании рН известью, необходимо учитывать наличие
кальцийсодержащих ионов в пульпе. К настоящему времени
установлено, что депрессирующее действие извести
обусловлено не только повышением рН пульпы при ее подаче, но и
адсорбцией ионов кальция на поверхности сульфидов железа,
что приводит к ее цементации и предотвращению адсорбции
на ней собирателя [1, 3].
Цементацию поверхности сульфидов железа кальцийсодер-
жащими соединениями можно рассматривать как следствие
химического взаимодействия продуктов окисления серы
минерала на поверхности с ионами Са(ОН)+ для точек, занятых
сульфоксидными анионами, по реакции
-Щ ■ (S А) + Са (ОН)+ г - Fte] • S„Om ■ Са (ОН)
и для точек, занятых катионами железа, по реакции
-Fe] + + SrtOm +Са (ОН)+ ^ - Fe] • SnOw • Са (ОН).
6*
16Э
Возможность цементации сульфидной поверхности кальций-
содержащими соединениями в присутствии Кх~ ионов в пульпе
определяется на основании реакции
-Fe]-Kx + SBOgr + Ca(OH)+ z£ -Fe].SnOm.CaOH + Kx-, (9.3)
т. е. соотношением концентрации ионов Кх~, сульфоксидных и
СаОН+.
Используя те же предпосылки, что и при выводе уравнения
(9.1), на основании реакции (9.3) находим соотношение между
концентрациями ионов Кх_, Н+ и СаОН+ в пульпе, которое
определяет граничные условия флотации сульфидов железа.
В общем виде соотношение имеет вид
lg [Кх~] - а рН + р lg [СаОН+] - lg /С.
Результаты экспериментальной проверки расчетного
уравнения для пирита и марказита
lg [Кх-] - 4- РН + 4- lg'[CaOH+] = lg/C (9.4)
bo
в лабораторных и промышленных условиях на обогатительных
•фабриках (рис. 9.5, б) подтвердили его справедливость [1, 3].
В условиях, соответствующих полной депрессии сульфидов
железа, представленных преимущественно пиритом, lg /C= 15,2,
а в условиях, соответствующих полной их флотации, lg/C=ll,8
(см. рис. 9.5, б).
Зависимость [Кх~]=/(рН) при флотации сульфидов меди.
В результате исследований [1] установлено, что условие
образования ксантогената одновалентной меди в приэлектродном
слое сульфидов меди нельзя использовать для теоретического
обоснования экспериментально полученных зависимостей
1Кх~]=/(рН) при флотации халькозина, борнита, ковеллина и
халькопирита (рис. 9.6, а). Вывод не изменится, если исходить
из условия образования СиКх не в приэлектродном слое
минералов, а в объеме раствора [1, 3].
Если бы необходимая для полной флотации сульфидов меди
1Кх~] определялась возможностью образования на их
поверхности СиКхг, то все они требовали бы для своей флотации одной
и той же [Кх~] в пульпе. Однако результаты флотационных
исследований показывают (см. рис. 9.6, а), что необходимая
для полной флотации халькопирита, борнита, ковеллина и
халькозина [Кх~] разная и закономерности ее изменения при
изменении рН пульпы существенно различаются для разных
минералов. Следовательно, условие образования СиКх2 на
минеральной поверхности не может быть принято в качестве
критерия для разработки количественной зависимости между
необходимой [Кх~] и значением рН .пульпы при флотации
сульфидных минералов меди.
Экспериментальные зависимости [Кх~]=/(рН) можно
теоретически обосновать, если использовать те же предпосылки, что
164
!- '.
1
1 X
sg4. >3
<§Й*
^^\® t
&>
v^y
<
| i
л.
1 <Я \
5
sO\j
X
■?*,
>K<3> I
Ж^]
^
N%^
*V ^^w ч I
1 \. 1
« 5*
i
^
4>
I
S
<ъ
и при выводе расчетных уравнений [Кх~]=/(рН) для
сульфидов железа [1].
Наиболее полно экспериментальные данные (см. рис. 9.6, а)
описываются уравнениями, которые имеют следующий общий
вид:
для не содержащих в своем составе железа сульфидов меди
(халькозина, ковеллина)
lg [Кх-]ЙСОбх ^ f lg К + а --п— — р lg [CuOH+] [Kx-] ] + YPH, (9.5)
[ 0,059 J
где Е° — стандартный электродный потенциал образования
ксантогената меди;
для железосодержащих сульфидов меди (борнита,
халькопирита)
lg [Кх-]необх - (lg К + a lg [Cu+] [Kx-] + р lg [FeOH+] X
X[Kx~]}+VpH. (9.6)
Как следует из уравнений (9.5) и (9.6), флотируемость
халькозина и ковеллина определяется значением [СиОН+][Кх~],
а флотируемость железосодержащих сульфидов меди (борнита
и халькопирита) значением [FeOH+][Kx~].
Различные значения Ко=[СиОН+][Кх~] для халькозина и
ковеллина, а также Ко—[FeOH+][Kx~] для борнита и
халькопирита не позволяют считать постоянство произведений
[MeOH+JKx-] для каждого отдельного минерала при
необходимой [Кх~] в жидкой фазе пульпы следствием образования на
минеральной поверхности или в приэлектродном слое
соединений типа МеОНКх. Можно полагать, что определенное
значение этого произведения, как и в случае сульфидов железа,
является условием поддержания определенного значения
потенциала поверхности и, тем самым,, определенной и
достаточной концентрации диксантогенида Кхг в приэлектродном слое
сульфидных минералов, чтобы пассировать их поверхность
(экранировать восстановительное действие поверхности) ксан-
тогенатными соединениями и за счет этого обеспечить
интенсивную адсорбцию Кх2 на химически закрепившемся ксанто-
генате, образование «смешанного» сорбционного слоя
собирателя и эффективную флотацию минерала [1, 3].
Сопоставление результатов определения флотируемое™,
значения и форм адсорбции собирателя показывает, что
контролирующим условием полного (максимального) извлечения
минерала при флотации является не значение адсорбции,
а возможность интенсивного образования диксантогенида на
гидрофобизированной химически закрепившимся кёантогенатом
поверхности [1, 3].
Условием интенсивного образования диксантогенида
является равенство имеющейся в пульпе и рассчитанной [Кх~] (см.
рис. 9.6, б, кривая 5). [Кх~] в пульпе при этом может быть не-
166
достаточной для образования на минеральной поверхности
нормального ксантогената металла (как, например, при
флотации сульфидов железа), может соответствовать его
образованию (так, например, при флотации сульфидов свинца) или
быть на несколько порядков выше (как, например, при
флотации сульфидов меди).
Следует отметить, что изменение адсорбции собирателя и
флотируемость минералов подчиняются разным
закономерностям. В частности, плотность адсорбции химически
закрепившегося собирателя не определяет флотируемость минерала,
закономерность ее изменения при различных концентрациях
собирателя и рН раствора более сложна, чем известные изотермы
адсорбции Лангмюра, Фрейндлиха или Темкина (см. рис. 9.6,6,
кривые 1—4).
Радиометрическими измерениями также установлено, что
одно и то же значение необходимой [Кх~] для полной флотации
одинаковых минералов различных месторождений не
исключает различия в плотностях адсорбции собирателя на их
поверхности. Причем адсорбция ксантогената определяется в
основном степенью окисления образцов минерала. С уменьшенном
степени окисления уменьшается и плотность адсорбции
ксантогената, но возрастает скорость флотации сульфидного
минерала.
9.3. Депрессирующее действие сульфидов
щелочных металлов
■ Сульфиды щелочных металлов, например сернистый
натрий Na2S, являются сильными депрессорами для всех
сульфидов цветных, черных и редких металлов, кроме молибденита
MoS2. Депрессия сульфидов протекает в основном по второму
механизму: сульфидные ионы вытесняют ксантогенатные ионы,
так как растворимость сульфидов тяжелых металлов
значительно меньше растворимости их ксантогенатов.
Сульфиды щелочных металлов широко используются в
практике флотации полиметаллических руд для предотвращения
взаимоактивации сульфидов в процессе измельчения, для
десорбции собирателя с поверхности сульфидных минералов
перед их разделением и в процессе самого разделения, например,
медно-молибденовых концентратов. Эффективность каждой из
этих операций зависит от того, насколько концентрация
сульфидов ионов в пульпе близка к оптимальной.
Количественные зависимости между концентрациями
сульфидных, водородных и ксантогенатных ионов для условий
полной флотации сульфидов, полной депрессии их флотации,
а также для условий полного предотвращения сорбции
собирателя на поверхности сульфидных минералов могут быть
получены на оснований тех же теоретических положений, что и
при разработке зависимостей [Kx~]=f(pH).
167
Например, минимальная необходимая для полной флотации
галенита [Кх^] может быть рассчитана из условия образования
нормального ксантогената свинца
Kx-] = V[S2-]/d//C2, (9.7)
где К\ и Къ— значения произведений растворимости
соответственно ксантогената и сульфида свинца.
Полная депрессия флотации галенита достигается, если
[Кх~] в пульпе на 3,5 порядка меньше концентрации,
необходимой для образования ксантогената свинца [1], а
предотвращение адсорбции собирателя на его поверхности — если [Кх~]
на 6 порядков меньше.
При наличии ионов S2" в пульпе возможно взаимодействие
их с ионами FeOH+ на поверхности сульфидов железа с
образованием FeS:
FeOH+ + S° + Н+ ^ FeS + Н20;
IgK^lg- \ -27,5. (9.8)
[FeOH+] [s2~] [Н+]
После добавления к обеим частям уравнения (9.8) lg[Kx~]
найдем общую для всех сульфидов железа зависимость [Кх~] =
= /([SnpH):
g [Kx-] -- (- 27,5 -f lg [FeOH+] [Кх~]} + lg [S2~j - pH.
Аналогичным образом можно получить такие зависимости
для сульфидов меди [1, 3].
Общая для всех сульфидов железа и меди зависимость
минимальной необходимой [Кх~] от (S2~) игрН имеет следующий
общий вид:
lg[Kx-]^[lg/C4lg{MeOH+][Kx-]]4-lg[S2~]-pH. (9.9)
При этом флотируемость всех сульфидов железа в
присутствии ионов S2- в пульпе зависит от значения /C0=[FeOH+]
[Кх~], а сульфидов меди — от значения /Со = [СиОН+][Кх~].
Подставляя в уравнение (9.9) значения lg /С и вместо
lg[MeOH+][Kx~] его значения, установленные paiTee при
определении зависимостей [Кху] = /:(рН), можно получить
необходимые расчетные уравнения для каждого из сульфидов железа
и меди. Из этих уравнений следует, что наибольшей
устойчивостью к подавляющему действию, ионов S2" обладают
халькозин и марказит, наименьшей — халькопирит и пирротин [1,2].
Полная депрессия флотации сульфидов меди и железа
в присутствии сульфидных ионов достигается, если [Кх~]
в пульпе на 3 порядка меньше концентрации, необходимой для
полной флотации рассматриваемых минералов, а
предотвращение сорбции собирателя на их поверхности — если [Кх~]
примерно на 6 порядков меньше. Типичные результаты, полу-
168
Рис. 9.7. Влияние соотношении имеющейся и рнетиоре. и крншчш-кон кпн
центраций ионов S*~ в присутствии сернистого ширин (а) и .шнчечшн 1цК
в присутствии цианида F) на плотность адсорбции \\ химически закрепим-
шегося собирателя и Гф физически адсорбированного диксантогенида,
а также на флотируемость пиритов уп (а) и халькозинов у* (б) разных
месторождений при различных концентрациях бутилового ксантогената
в растворе (от 5,33 • 10 6 до 5,33 * Ю-4 моль/л)
ченные на образцах пиритов двух месторождений, изображены
на рис. 9.7, а.
Радиометрическими измерениями плотности сорбции и
количественными определениями форм закрепления собирателя
на поверхности сульфидов свинца, меди и железа установлено,
что при наличии в пульпе S2~-hohob (как и в их отсутствие)
полной флотации минералов соответствует интенсивное
возрастание содержания диксантогенида в сорбционном слое, а
полная депрессия их флотации характеризуется полным
отсутствием диксантогенида на поверхности (см. рис. 9.7, а).
Плотность адсорбции химически закрепившегося собирателя не
определяет закономерностей флотации минералов. Например,
при практически одной и той же плотности его адсорбции
флотируемость халькозина может изменяться от 0 до 100 % [1].
Поскольку количественные зависимости между
концентрациями ионов Кх \ Н+, S2~ являются справедливыми для
минералов, отобранных с разных месторождений, они могут быть
использованы для построения систем автоматического контроля
и регулирования, например, процессов флотации сульфидов
169
меди из руд, разделения медно-молибденовых концентратов,
десорбции собирателя с поверхности сульфидных минералов
малыми расходами сернистого натрия.
9.4. Депрессирующее действие цианидов
В качестве цианидов на практике используются NaCN, KCN,
Ca(CNJ или технический продукт цианплав, содержащий
42—47 % цианидов в пересчете на NaCN.
Цианиды довольно легко депрессируют сульфиды железа и
активированный ионами меди сфалерит^ несколько труднее —
халькопирит. и только при очень больших расходах —
вторичные сульфиды меди (ковеллин и халькозин).
Депрессия флотации минералов в присутствии цианидов
осуществляется по первому механизму. Цианиды растворяют
ксантогенатные соединения на поверхности депрессируемого
минерала вследствие образования прочных комплексных ионов
с катионами железа FefCNJe4- или меди Cu[CN2~]. Образование
таких ионов приводит к понижению концентрации свободных
ионов железа и меди в пульпе до такой степени, что
произведение из их концентраций [Меп+] и ионов ксантогената [Кх~],
например для меди [Си+][Кх~], становится меньше
произведения растворимости «поверхностного» ксантогената металла,
который начинает поэтому растворяться. Поскольку при
избытке ионов CN~ катионы железа и меди все время
связываются в комплексные ионы, растворение протекает до полного
разрушения ксантогенатных соединений на поверхности
сульфидов меди, железа и активированного сфалерита. Во всех
таких случаях действует правило: «Если цианид с металлом
сульфида способен дать прочный комплексный ион, то
ксантогенаты- этих металлов будут растворяться в цианиде, а
сульфиды этих металлов будут ими депрессироваться» [36].
По данным И. А. Каковского, основные металлы могут быть
разделены в зависимости от растворимости их ксантогенатов
в водных растворах цианидов на три группы:
I группа — свинец, висмут, олово, сурьма, мышьяк, рубидий.
Эти металлы не образуют комплексных ионов с цианидами, их
ксантогенаты не растворяются в цианидах и сульфиды этих
металлов не депрессируются цианидами;
II группа — ртуть, кадмий, медь. Эти металлы образуют
с цианидами комплексные ионы средней прочности.
Ксантогенаты этих металлов обладают средней растворимостью, а
сульфиды их депрессируются цианидами лишь при высоких
загрузках;
III группа —цинк, палладий, никель, золото, железо. Эти
металлы образуют прочные комплексные ионы с цианидами.
Ксантогенаты этих металлов легко растворяются в цианидах
и сульфиды их легко депрессируются цианидами при малых
загрузках.
170
Внутри каждой группы металлы расположены и порядке
унеличения растворимости их ксантогенптон н цианидах. При
чтом, чем дальше отстоят один от другого металлы, тем легче
разделяются сульфиды этих металлом. Например, легко и при
широком диапазоне расхода цианида может быть достигнуто
отделение сульфидов свинца от сульфидом цинка, а тем более
от сульфидов железа. Разделение сульфидом сиинца и меди
происходит труднее и требует высоких расходом цианида.
Особенно большие трудности предеiаилнет разделение сульфидов
соседних металлов: меди и цинка. При малых расходах
депрессора не достигается достаточного подавления сфалерита, <Jto
приводит к значительным потерям цинка в медном
концентрате, а при увеличенных нагрузках начинает депрессироваться
халькопирит и увеличиваются потери меди в цинковом
концентрате [2].
Результаты селективной флотации еще более ухудшаются
при наличии в руде растворимых в цианиде медных минералов
(карбонатов и вторичных сульфидов меди), так как в этом
случае резко снижается концентрация в пульпе свободных
ионов CN~ и возрастает концентрация медных ионов,
активирующих сфалерит.
К общим недостаткам цианидов как реагентов относятся
следующие:
они частично растворяют и дснрессируют золото, что
вызывает потери;
они являются сильным ядом и при их использовании
следует особо строго соблюдать правила техники безопасности,
г удаляемые из фабрики хвосты и сточные воды необходимо
обезвреживать;
их можно использовать только в щелочных пульпах, так
как в кислой среде выделяется синильная кислота HCN,
являющаяся сильнейшим ядом.
Несмотря на эти недостатки, цианиды до сих пор
достаточно широко используются при селективной флотации
полиметаллических руд и разделении коллективных, например
свинцово-медных, концентратов, содержащих вторичные суль-.
фиды меди. Возникающие при этом затруднения обусловлены
не столько недостатками цианидов как флотационных
реагентов, сколько отсутствием контроля необходимой концентрации
цианистых соединений в пульпе.
Необходимые количественные зависимости между
концентрациями определяющих селективность процесса ионных
компонентов в пульпе можно получить, используя те же
теоретические предпосылки и численные значения [МеОН+][Кх_] = /Со,
что и при разработке количественных зависимостей [Кх~] =
=/(рН) H[Ss-]=f([Kx-],pH).
Количественные зависимости между концентрациями циа-
нидных [CN], ксантогенатных, водородных ионов и металлциа-
нистого комплекса [Me(CN)mn_] для условий полной флотации
171
и депрессии сульфидов меди и железа имеют следующий
общий вид [1, 3]:
lg ^^~~ + -18 [Кх-1 ± Р РН ± у lg [SrtO-] =
= [lg /С + т lg [MeOH+] [Kx-] ] = const. .
Флотируемость всех минералов меди в присутствии
в пульпе цианидных ионов, как и в присутствии сульфидных
ионов, контролируется значением произведения [СиОН+][Кх~],
а флотируемость сульфидов железа — значением произведения
[FeOH+][Kx~]. При этом основным продуктом окисления серы
сульфидных минералов меди и железа, за исключением
халькопирита, в присутствии цианидных ионов являются ионы
S2O32"". Сера халькопирита должна окисляться в приэлектрод-
ном слое до S032~ ионов. Основным металлцианистым ионом
для сульфидов железа является Fe(CNN4~, для сульфидов
меди — Си (CN) 2- [1].
Значение произведения [МеОН+][Кх~] в граничных условиях
полной депрессии флотации сульфидов меди и железа в
присутствии цианидных ионов на 3 порядка меньше, чем его
значение в граничных условиях полной флотации этих минералов.
Как и в ранее рассмотренных случаях, условиям полной
флотации сульфидов меди и железа соответствует интенсивное
образование диксантогенида на их поверхности, а полной
депрессии флотации — отсутствие диксантогенида в сорбционном
слое, хотя- плотность Сорбции химически закрепившегося
собирателя может быть в этих условиях еще весьма значительной
(рис. 9.7, б).
Результаты промышленных исследований, проведенных на
одной из обогатительных фабрик, показали, что
регулирование процесса цианидного разделения свинцово-медного
концентрата на фабрике сводится практически к поддержанию
условий депрессии халькозина. Последнее является вполне
закономерным, так как в исходном свинцово-медном концентрате
содержится 8—19 % халькозина, обладающего но сравнению
с другими сульфидами меди наибольшей устойчивостью к
подавляющему действию цианидных ионов.
С учетом численных значений постоянных членов расчетное
уравнение соотношения концентраций ионных компонентов,
обеспечивающего полную депрессию флотации халькозина
в присутствии цианидных ионов и бутилового ксантогената
в пульпе, можно представить в более удобном для
практического пользования виде [1].
|g_[CMCN|]_= Alg|ButKx-]—LpH.
[CIST]2 3 6
В этом виде оно может быть использовано в качестве
задания функциональному блоку системы автоматизации цикла
172
p;iчленения свинцово-медного концентрата на фабрике. Пред-
станин это уравнение в виде
1д [CN-] = —0,43 + — lg [ButKx '] |
-i--i-lg[Cu(CNFJ+ JLpH,
можно предложить несколько путей совершенствования
процесса и снижения расхода цианплава. К основным из них
относятся: удаление излишней [Кх~] из жидкой фазы пульпы;
понижение концентрации медноцианистого комплекса в пульпе;
уменьшение значений рН пульпы.
9.5. Депрессирующее действие кислот
Депрессирующее действие кислот, как и цианидов,
обусловлено главным образом их дезактивирующим действием на
поверхность, особенно силикатных минералов.
Например, серная кислота используется для депрессии
кварца при флотации оксигидрильными собирателями, плави-
ковая депрессирует слюду при флотации кмтиоииым <иГ>ирате-
лем, а молочная-—слюду при флотации сульфидин жедо.ча
сульфгидрильными собирателями. В сиою очередь, димоннйн
кислота депрессирует кварц, слюду и полевой шпат при фло*
тации барита жирными кислотами. Расход кислоты при
флотации составляет 250—2000 г/т.
9.6. Депрессирующее действие солей щелочных,
щелочноземельных металлов и алюминия
Соли натрия, калия и кальция используются в качестве
депрессоров при разделении полевых шпатов. Натриевые или
кальциевые соли при флотации аминами подавляют натриевые
полевые шпаты благодаря избирательному закреплению на их
поверхности изоморфных ионов натрия или кальция. По этой
же причине соли калия избирательно депрессируют флотацию
калиевых полевых шпатов. Депрессия минералов связана как
с вытеснением собирателя, так и с гидрофилизацией
поверхности сорбирующимися катионами.
Соли калия и алюминия могут использоваться при
флотации катионным собирателем для отделения бериллийсодержа-
щей слюды от калиевых слюд, а соли магния — для выделения
литийсодержащей слюды из смеси слюд [44]. Депрессирующее
действие катионов солей основано на том же принципе, что и
при разделении полевых шпатов.
Избирательное вытеснение амина с поверхности окислов
железа в результате закрепления катионов кальция
используется для разделения окислов железа и кварца.
173
9.7. Депрессирующее действие хроматов и бихроматов
Бихроматы К2СГ2О7, ЫагСг207 и хроматы КгСг04 являются
депрессорами галенита при флотации сульфгидрильными
собирателями, барита и кальцита — при флотации оксигидрильными
собирателями.
При подаче депрессоров в жидкой фазе пульпы
устанавливается равновесие между бихроматными Сг2072"* и хроматными
Сг042~ ионами
Cr202- + H20 ;? 2CrOJ- + 2H+.
При малой концентрации депрессора в пульпе закрепление
ионов Сг042~ на поверхности протекает на местах, не занятых
собирателем, и не сопровождается его вытеснением, т. е.
осуществляется по третьему механизму депрессии (см. рис. 9.1, в).
Замещение хроматными ионами на поверхности галенита суль-
фоксидных ионов SnOm2~, на поверхности барита сульфатных
ионов и на поверхности кальцита карбонатных ионов вызывает
сильную гидратацию минералов. Она оказывается настолько
значительной, что минерал перестает флотироваться, несмотря
на то, что около 30 % его поверхности еще покрыто
собирателем. При высокой концентрации депрессора в пульпе и
длительном воздействии депрессия может сопровождаться
вытеснением собирателя и протекать уже в соответствии со вторым
механизмом депрессии (см. рис. 9.1, б).
Наибольший практический интерес представляет
применение бихромата для разделения свинцово-медных концентратов,
когда используется не только различная способность
минералов к образованию на их поверхности хроматных соединений,
но и различная способность к окислению бихроматом. Д. В.
Сологубом и С. И. Митрофановым показано [29], что окисление
галенита бихроматом происходит при рН< 10,5ч-11, а
халькопирита и пирита — при рН<6-ь8. Окисление галенита
сопровождается образованием на его поверхности хроматных
соединений, тогда как на поверхности халькопирита и пирита хроматы
не образуются. Эти различия и являются основными
причинами селективной депрессии галенита бихроматом при
расходе его 0,5—2 кг на 1 т концентрата. Лучшая депрессия
галенита достигается в среде, близкой к нейтральной, когда на
его поверхности образуется наиболее устойчивая пленка
хромата свинца.
Способ разделения свинцово-медных концентратов с
использованием двухромовокислых солей имеет следующие
недостатки: не депрессируется галенит, активированный ионами
меди; отчасти подавляется борнит, что увеличивает потери
меди в свинцовом концентрате; подавляется сфалерит, что
затрудняет получение в камерном продукте высококачественного
свинцового концентрата в случае небольшого содержания
свинца в исходном продукте.
174
9.8. Депрессирующее действие фосфатов
Такие фосфаты, как тринатрийфосфат Na3P04, триполифос-
фат Ыа5РзОю, гексаметафосфат (ЫаРОз)б, пирофосфат натрия
Na4P207, применяют обычно при расходе 250—2000 f/t для
депрессии флотации минералов, содержащих катионы
щелочноземельных металлов на своей поверхности. Депрессия
осуществляется в основном по третьему механизму (см. рис. 9.1, в)
в результате гидрофилизации поверхности труднорастворимыми
продуктами взаимодействия фосфорных соединений с
катионами поверхности.
' По данным ряда исследований [44], фосфаты могут депрес-
сировать также флотацию галенита. При действии небольших
количеств фосфата на галенит имеет место хемосорбционный
процесс, не сопровождающийся вытеснением собирателя. При
высокой концентрации фосфата и значительном окислении
галенита может наблюдаться гетерогенная химическая реакция
с образованием труднорастворимого фосфата свинца и третий
механизм депрессии перейдет во второй. Поскольку сульфиды
меди (халькопирит, борнит и халькозин), в отличие от
галенита, адсорбируют незначительное количество фосфата и не
депрессируются, принципиально возможна селективная
флотация сульфидов свинца и меди, например, при разделении
коллективного свинцово-медного концентрата.
9.9. Депрессирующее действие ферри- и ферроцианидов
Ферри- и ферроцианиды K3Fe(CNN и K4Fe(CNN могут
успешно применяться для депрессии вторичных сульфидов меди
(халькозина, борнита).
Анион Fe(CNNn~ депрессора в значительных количествах
адсорбируется этими сульфидами, меньше халькопиритом, еще
меньше галенитом и в незначительных количествах сфалеритом
и пиритом. Причиной его адсорбции является образование
ферри- и ферроцианидов тяжелых металлов при
взаимодействии с катионами окисленных соединений на поверхности
сульфидов. При этом на всех минералах наблюдается аналогичная
зависимость между адсорбцией депрессора и депрессирующим
действием реагента (рис. 9.8). На поверхности обычно более
окисленных вторичных сульфидных минералов (по сравнению
с халькопиритом) адсорбируется большее количество Fe(CNNn~
и соответственно наблюдается более эффективная депрессия их
флотации.
Депрессия минералов в присутствии ферри- и
ферроцианидов не сопровождается вытеснением собирателя и может быть
обусловлена только гндрофилизирующим действием
депрессора, адсорбированного свободными участками минеральной
поверхности, т. е. допросе и я осуществляется в основном па
третьему механизму (ом. рис. 9.1, в).
175
гЬ
*р о
4Q* ^
'К
i
Л
i
ДА /
пт 2
о т 3
&J
Значительную роль в депрес-
сирующем действии ферри- и
ферроцианидов играет
налипание на поверхность депрессируе-
мых минералов тонких частиц
нерастворимых «смешанных»
феррицианидов. Такие ферри-
цианиды образуются при
взаимодействии реагента с
катионами тяжелых металлов
пульпы и содержат прочно
связанные феррицианиды щелочных
и тяжелых металлов типа
nMe3[Fe(CNN]2'mK3Fe(CNN.
При контакте тончайшей
частицы «смешанного» феррициа-
нида с поверхностью минерала
катионы калия, находящиеся на
поверхности «смешанного» фер-
рицианида, могут заменяться
катионами тяжелого металла,
находящегося на поверхности
минеральной частицы. Такая
реакция образования феррицианида
тяжелого металла приводит
к возникновению химической
связи между поверхностью
минерала и частицей «смешанного»
феррицианида.
Обязательным условием
эффективности образования таких
связей является соответствие структур поверхности минерала
и осадка. Депрессор селективно закрепляется и подавляет
флотацию только минералов с определенной структурой их
поверхностного слоя. Например, активированный сфалерит, несмотря
на наличие катионов меди на его поверхности, не адсорбирует
осадки фер|рицианидов тяжелых металлов и не депрессируется
в их присутствии, тогда как вторичные сульфиды меди
(халькозин, ковеллин) такими осадками депрессируются достаточно
эффективно.
Свойство ферри- и ферроцианидов селективно депрессиро-
вать вторичные сульфиды меди используется при разделении
свинцово-медных, медно-цинковых и медно-молибденовых
концентратов и продуктов, в которых медь представлена главным
образом халькозином или борнитом. Наиболее эффективная
депрессия всех сульфидов меди при этом наблюдается в
слабокислой и нейтральной средах. Повышение рН более 7,5—7,8
приводит в связи с разрушением феррицианидов тяжелых
металлов к резкому ухудшению депрессии флотации минералов
&А
80
60
40
20
19105моль/мг
Рис. 9.8. Влияние плотности
адсорбции Г феррицианида Fe(CN)Q~
на флотируемость y
халькопирита A), борнита B) и
халькозина C) с различной степенью
окисления их поверхности при
расходе бутилового ксантоге-
ната 100 г/т (по данным работы
{44])
176
меди. Иногда ферри- и ферроцианиды используются также
в качестве селективного депрессора сульфидов железа при
отделении их от сульфидов кобальта и никеля. Расход
депрессора 50—100 г/т руды.
9.10. Депрессирующее действие цинкового купороса
и его сочетаний с другими реагентами
Цинковый купорос, обычно в сочетании с содой, едким
натром, сульфитом или цианидом, применяется для депрессии
сфалерита или вторичных сульфидов меди (но методу
Ю. И. Еропкина) при флотации свинцово-цинковых, свинцово-
медных, медно-цинковых и свинцово-медно-цинковых руд.
Депрессирующее действие цинкового купороса на сфалерит
связывают с налипанием коллоидных цинксодержащих осадков
на его поверхности. Такие осадки образуются в пульпе в
результате взаимодействия загруженного цинкового купороса
с растворенной углекислотой воздуха и содержат в своем
составе гидроксильные и карбонатные ионы. В нейтральной и
щелочной средах цинксодержащие осадки налипают на
поверхность всех сульфидных минералов (галенита, пирита,
халькопирита, сфалерита), но сильное депрессирующее действие они
оказывают только на сфалерит. Причиной селективности де-
прессирующего действия цинкового купороса может быть
одноименный катион в осадке и на поверхности сфалерита, что
улучшает закрепление осадка на минерале и способствует
образованию более плотного и более прочного шламового
покрытия на сульфидах цинка по сравнению с другими сульфидами.
Наибольшая депрессирующая способность осадков
наблюдается в момент их образования, когда кристаллическая
решетка еще не сформировалась и ненасыщенные валентные
связи осадка компенсируются ненасыщенными связями
поверхности сфалерита. Кристаллические осадки, например
Zn(OHJ, получаемые при дополнительной подаче едкого
натра в пульпу (до рН<11), не депрессируют флотации
сфалерита. В кислой среде цинковый купорос также не является
депрессором для сфалерита, так как в этих условиях осадков не
образуется. Минимальной растворимостью и максимальной
устойчивостью осадки гидроокиси цинка обладают в объеме
пульпы и на поверхности сфалерита при значениях рН 9—9,5
(рис. 9.9, кривая /). Именно в этой области значений рН, по
данным П. Г. Хаджиева и Л. И. Гросмана, достигаются лучшие
результаты депрессии сфалерита цинковым купоросом (см. рис.
9.9, кривые 3 и 4) и максимальная селективность флотации.
Прочность связи осадка с поверхностью, как и
селективность его налипания, очевидно, определяется числом взаимно-
компенсированных связей при закреплении и соответствием
параметров кристаллических решеток осадка и минералов. Во
всех случаях, однако, она гораздо меньше прочности связи хе-
мосорбированного ксантогената, поэтому закрепление осадка
177
Рис. 9.9, Влияние рН
на значение суммарной
концентрации С иопон
2n2^, ZnOIP; IIZnO"~
и ZnC;;-в растворе (I)
в присутстнии осадка
гидроокиси цинка, его
электрокинетический
потенциал I B), флота-
руемость у ириродио
активированного
сфалерита п среде едкого
натра Г>о:* депрессора
C) и с цппкоиым
купоросом П,.'М i/л)
в среде едкого натра
D) и в содовой среде
E) при исходной
концентрации оЛ'тплоиого
ксантогената 12 мг/л
(по данным II. Г
Хаджиева и Л. \\ I
роема на)
" 8 10 рН
протекает без вытеснения собирателя с поверхности.
Депрессия минерала осуществляется в основном по четвертому
механизму депрессии, т. е. закрепление гидрофильных частиц осадка
перекрывает гидрофобизирующее действие аполярных цепей
собирателя, размеры которых значительно меньше размеров
частиц осадка (см. рис. 9.1, г).
В зависимости от конкретных особенностей вещественного
состава перерабатываемого сырья могут использоваться смеси
цинкового купороса с различными реагентами.
Смесь цинкового купороса и соды используется главным
образом для флотационного отделения сульфидов меди и
железа от сульфидов цинка в процессе обезмеживания и обезже-
лезнения черновых цинковых концентратов, получаемых при
флотации полиметаллических руд. Л. И. Гросман и П. Г.
Хаджиев показали, что в условиях депрессии сфалерита цинковым
купоросом в содовой среде (см. рис. 9.9, кривая 5) образуются
аморфные осадки основного карбоната цинка с размером
частиц от десятых долей микрометра до нескольких
микрометров. Эффективность депрессии сфалерита такими осадками
уменьшается при повышении температуры пульпы, повышении
рН пульпы более 10,5, добавке в пульпу жидкого стекла,
введении в пульпу ионов меди, увеличении продолжительности и
интенсивности перемешивания, увеличении расхода собирателя,
т. с. при всех мероприятиях, вызывающих ослабление прочно*
сти связи или удаление осадка с минеральной поверхности.
*»/о
ГЬ>\
SO]
60\
Ш
10\
-5
-15
-Z5
'35
гщь,ммь/л
О L-45L-77l
\--7
b-sh
178
В оптимальных условиях расход цинкового купороса
составляет 2—4 кг/с, а соды—около 1 кг/т чернового концентрата.
Смесь цинкового купороса с едким натром представляет
собой щелочной раствор цинкатов (рН>11).^По данным
В. А. Конева, цинкаты хорошо депрессируюг сфа*5ертт, не
оказывая депрессирующего действия на галенит, халькопирит и
пирит. При отделении сульфидов меди от сфалерита цинкаты
дают более высокие результаты по сравнению с цианидами,
так как в отличие от цианидов они не снижают флотируемости
сульфидов меди. Механизм депрессирующего действия
цинкатов недостаточно исследован. Предполагается [17], что в
момент загрузки цинкатные ионы [Zn(OHL]2~~ избирательно
закрепляются на сфалерите через атом кислорода гидроксильной
группы вследствие одинаковой электронной конфигурации и
геометрической структуры цинкатных ионов и элементов
кристаллической решетки сфалерита, близости размеров и
одинаковых центральных атомов тетраэдров цинкатных ионов и
сфалерита. Расход цинкового купороса при цинкатном методе
депрессии сфалерита составляет 2,5 кг/т коллективного свин-
цово-медно-цинкового концентрата (в пересчете на безводную
соль), расход едкого натра — 3 кг/т.
Смесь цинкового купороса с цианидом применяется весьма
широко при флотации сульфидных руд. В результате
протекания реакций
a Zn2+ + р ОН + у С02- ?. Zna (ОН), (С03)у ;
Zn2-4-2Ci\T~^ Zn(CNJ;
Zn(CNJ + 2CN- г Zn(CN)J-
форма нахождения цианида и цинка зависит от молярного
соотношения загружаемых реагентов смеси. Если молярное
отношение KCN к ZnS04 в загружаемой смеси меньше 2, то
реагенты в пульпе находятся в основном в виде осадков гидрато-
карбоната цинка Zna(OH)p(C03)v и цианида цинка Zn(CNJ-
При молярном отношении реагентов больше 4 они
присутствуют в пульпе только в виде растворимых цинкцианистых
ионов Zn(CNL2~ и свободных цианидных ионов CN~. При
промежуточных значениях молярного соотношения цианида и
цинкового купороса в пульпе находятся все виды осадков и
растворимых соединений реагентов и их свободных ионов. В
зависимости от соотношения молекулярных и ионных форм цианида
и цинка в пульпе депрессирующее действие смеси цианида с
цинковым купоросом может быть обусловлено:
V дезактивацией сфалерита в результате снижения
концентрации ионов меди в растворе в соответствии с реакциями
Си+ + Zn (CNJ- ? Си (CN)" + Zn (CNJ,
Си+ + Zn (CNJ <* Си (CN)- + Zn2 *-,
Cu++2CN- <*Cu(CNO;
179
растворением образовавшихся на поверхности
активированного сфалерита и пирита соединений собирателя (первый
механизм депрессии);
предотвращением образования поверхностных соединений
собирателя на вторичных сульфидах меди в результате
образования неактивного по отношению к собирателю
«поверхностного сфалерита»
Cu„Sm] Cu2S 4- Zn (CNJ- -> CunSm j ZnS + 2 Cu (CN)r, (9.10)
налипанием гидрофильных тонкодисперсных осадков
цианида и гидратокарбоната цинка (четвертый механизм
депрессии).
Соотношение (по массе) цианида к цинковому купоросу
при флотации полиметаллических руд колеблется от 1:10 до
1 :2 (режим Шеридана—Гризвольда). Эта смесь при обычных
значениях рН пульпы 7,5—9, создаваемых содой, сильно де-
прессирует сфалерит и сульфиды железа; чистый галенит этой
смесью не депрессируется, а халькопирит депрессируется
только при больших расходах. Поэтому смесь цинкового
купороса и цианида широко применяется в практике селективной
флотации полиметаллических сульфидных руд для отделения
галенита от сфалерита и сульфидов железа; халькопирита от
сфалерита и сульфидов железа; галенита от халькопирита и
сульфидов железа; галенита от вторичных сульфидов меди
(ковеллина, халькозина, борнита). В последнем случае
разделение может быть достигнуто, если сернокислый цинк и
цианид смешиваются в стехиометрическом отношении,
необходимом для образования цинкцианистых ионов Zn(CHL2~ (метод
Ю. И. Еропкина), обеспечивающих протекание реакции (9.10).
При флотации полиметаллических руд расход цианида
составляет от 25 до 150 г/т руды. При разделении коллективных
медно-свинцовых концентратов расход цианида, добавляемого
для депрессии вторичных сульфидов меди, возрастает до 1 —
2,5 кг/т коллективного концентрата. Расход цинкового
купороса изменяется от 50 до 1500 г/т руды.
Смесь цинкового купороса с цианидом и аммиаком может
быть использована в качестве селективного депрессора
сфалерита и пирита при отделении их от сульфидов свинца.
Поданным В. А. Конева, применение данной смеси реагентов на
свинцово-цинковой обогатительной фабрике Алмалыкского
ГМК позволяет получать более высокие технологические
показатели разделения свинцово-цинкового концентрата, чем со
смесью цинкового купороса с цианидом в режиме Шеридана—
Гризвольда. Механизм депрессирующего действия смеси не
должен принципиально отличаться от механизма действия
смеси цинкового купороса с цианидом.
9.11. Делрессирующее действие сульфоксидных
соединений и их сочетаний с другими реагентами
В качестве сульфоксидных соединений используются
сернистая кислота H2S03, ее соль Na2S03, тиосульфат Na2S0203,
гидросульфит натрия Na2S204. Образующиеся в пульпе при
подаче этих реагентов сульфоксидные ионы S032~, S2032~, S2O42-
и продукты их гидролиза термодинамически неустойчивы и
постепенно окисляются до конечного продукта — иона SCU2-.
В процессе окисления, сопровождающегося поглощением
кислорода из пульпы, они переходят друг в друга, поэтому при
подаче любого сульфоксидного соединения в пульпе
присутствует смесь сульфоксидных ионов и изменяется только
соотношение их концентраций.
Чистые растворы сульфоксидных соединений обладают
слабой депрессирующей способностью. Например, по данным
Л. А. Глазунова и С. И. Митрофанова [29], полной депрессии
флотации сульфидов не наблюдается вплоть до концентрации
сульфита или тиосульфата натрия в растворе, равной 1000—
2000 мг/л. Наиболее сильно депрессируется сфалерит,
несколько слабее халькопирит; галенит в этих условиях
практически не депрессируется.
Механизм действия сульфоксидных соединений,
обладающих восстановительными свойствами, изучен недостаточно. Де-
прессирующее действие их, по мнению А. Ф. Таггарта и
И. А. Каковского, протекает по первому механизму депрессии,
по мнению С. М. Ясюкевича — по третьему механизму
депрессии. В. М. Арашкевич и Ф. И. Нагирняк связывают депресси-
рующее действие этих реагентов с их воздействием на
устойчивость ксантогената, а С. И. Митрофанов и Л. А. Глазунов —
с поглощением кислорода и изменением
окислительно-восстановительного потенциала пульпы [29].
Наиболее высокие технологические показатели селективной
флотации достигаются при использовании в качестве
депрессора смеси сульфоксидных соединений с солями тяжелых
металлов.
Смесь сульфита натрия и сульфата железа депрессирует
сфалерит и пирит, но не депрессирует халькопирит и ковел-
лин, поэтому она не может быть использована для отделения
сульфидов меди от сульфидов цинка и железа. В узком
диапазоне рН (от 5,5 до 6,2) эта смесь оказывает депрессирующее
действие на галенит и поэтому может быть применена для
разделения сульфидов меди от галенита. Смесь сульфита натрия
и сульфата железа несколько активирует халькопирит,
вследствие чего результаты разделения халькопирита от сфалерита
и пирита при ее использовании получаются более высокими по
сравнению с цианидной технологией.
Расход реагентов в смеси при разделении коллективных
медно-свинцовых концентратов составляет 1—2 кг/т, а при
181
разделении медно-цинково-пиритных концентратов — около
3 кг/т концентрата.
Исследованиями К. Г. Бакинова установлено, что более ус-
тойчнвая депрессия галенита при разделении свинцово-медных
концентратов достигается заменой сульфита тиосульфатом
натрия.
Механизм депрессирующего действия смеси реагентов
изучен недостаточно. К. Г. Бакинов связывает его с налипанием
на поверхность минералов тонкодисперсных осадков,
состоящих из гидратов закисного и окисного железа; В. А. Конев —
с образованием комплексных дисульфитоферритных или суль-
фитосульфатоферритных ионов; С. И. Митрофанов — с
поглощением растворенного кислорода из пульпы вследствие
окисления сульфоксидных ионов до сульфатных [36].
Смесь сульфита натрия с цинковым купоросом широко
используется при селективной флотации медно-цинково-пиритных
руд и концентратов, а также сильно активированных свинцово-
цинковых и полиметаллических руд. Эта смесь слабее депрес-
сирует сфалерит, чем цианиды, но если в руде содержится
значительное количество растворимых в цианиде вторичных
сульфидов меди или присутствуют, хотя бы в малых количествах,
карбонаты меди, то хорошая депрессия сфалерита цианидами
не достигается. В таких случаях лучшие результаты
получаются при использовании в качестве депрессора сфалерита и
нирита смеси цинкового купороса с сульфитом натрия. Такая
смесь оказывается наиболее эффективной и в тех случаях,
когда сульфиды цинка сильно активированы соединениями свинца
и подача цианидов практически не оказывает влияния на их
флотируемость. Расход цинкового купороса 0,5—2 кг/т,
сульфита натрия 0,3—1 кг/т.
Смесь сульфита натрия с цинковым купоросом и цианидом
используется для усиления депрессии сфалерита. Она
применяется обычно вместо режима Шеридана—Гризвольда при
флотации сульфидных руд. В связи с уменьшением удельного
расхода цианида при использовании смеси наблюдается, как
правило, более эффективная флотация халькопирита.
Смесь сульфита натрия с медным купоросом депрессирует
флотацию практически всех сульфидов, сопутствующих
молибдену. В настоящее время ведутся промышленные испытания
использования этой смеси для разделения медно-молибдено-
вых концентратов.
Помимо рассмотренных смесей реагентов предложено
использовать при отделении халькопирита от галенита,
сфалерита и пирита смеси сульфоксидных реагентов с
трехвалентным хромом, с цинковым купоросом и аммиаком и^ некоторые
другие сочетания. Можно полагать, что при использовании
таких смесей третий и четвертый механизмы депрессии играют
значительную роль в общем механизме депрессии флотации
сульфидных минералов.
182
~ 9.12. Депрессирующее действие жидкого стекла
В общем случае жидкое стекло можно рассматривать как
соль поликремневых кислот с общей формулой mSi02- лЫа20.
Отношение т/п называется модулем жидкого стекла. Состав
полиионов может быть разнообразным, вплоть до простых
ионов SiCV".
При обработке жидкого стекла растворами кислот
получается так называемое активированное жидкое стекло,
содержащее значительное число взвешенных в воде частиц
кремнекислоты. Например, для простейшего случая
Siq- + 2H+->Si02(H20).
Состав растворов и степень дисперсности частиц кремнекис-
лоты зависит от рН, разбавления раствора и времени стояния
его после приготовления. Чем выше щелочность, тем больше
степень дисперсности. При очень высоких рН могут быть
получены истинные растворы, содержащие простые ионы, при
низких рН получаются растворы, содержащие полиионы и
частицы кремнекислоты.
Многокомпонентность растворов жидкого стекла, изменение
их состава во времени (вследствие процессов гидролиза и
коагуляции) обусловливают непостоянство депрессирующих
свойств этого реагента, если условия приготовления его
растворов и сроки их хранения неодинаковы.
Механизм депрессирующего действия жидкого стекла
сложный и изучен недостаточно полно. Предполагают, что при
добавке жидкого стекла могут иметь место одновременно три
механизма:
вытеснение анионов собирателя ионами HSi03", Si032~ и
ОН~ (второй механизм);
закрепление силикатных ионов без вытеснения ионов
собирателя (третий механизм);
налипание гидрофильных коллоидных частиц кремнекислоты
на поверхности минералов (четвертый механизм).
Преобладание того или иного механизма зависит от состава
растворов жидкого стекла. Например, при грубодисперсных
растворах, которые получаются при высоком модуле жидкого
стекла и низкой щелочности пульпы, более вероятен четвертый
механизм, так как в этом случае в пульпе содержится большое
количество коллоидных частиц кремнекислоты. При понижении
модуля и повышении щелочности пульпы в растворе
уменьшается концентрация, осадков кремнекислоты и возрастает
концентрация полиионов и простых силикатных ионов. Поэтому
может иметь место закрепление их на поверхности минерала
путем обменной адсорбции сначала без вытеснения собирателя,
а затем с вытеснением его ионов с поверхности минерала. На
депрессирующее действие жидкого стекла влияет также и тем-
18$
пература пульпы. С увеличением температуры степень
дисперсности жидкого стекла повышается, соответственно изменяется
и депрессирующее его действие.
Жидкое стекло является малоселективным депрессором и
при больших расходах может депрессировать все минералы. Но
все же депрессирующее действие жидкого стекла на отдельные
классы минералов различное и при анионном собирателе оно
уменьшается в следующей последовательности: кварц;
силикаты; окислы; карбонаты и не содержащие кремнезема соли
щелочноземельных металлов; сульфиды; минералы,
обладающие высокой естественной гидрофобностью.
Разделение минералов, принадлежащих к одному классу,
с использованием жидкого стекла представляет значительные
затруднения. Повышение селективности действия этого
реагента достигается следующими способами: точным подбором
расходов соды, жидкого стекла и собирателя; обработкой
пульпы жидким стеклом при повышенной температуре;
добавкой в пульпу солей поливалентных металлов или
предварительно приготовленной смеси растворов жидкого стекла и солей
поливалентных металлов, например железа или алюминия;
использованием активированного жидкого стекла;
предварительной кислотной обработкой пульпы.
Жидкое стекло применяется при селективной флотации
несульфидных минералов, для депрессии силикатных шламов при
сульфидной флотации особенно сильно разрушенных руд, при
флотации серных, графитовых и других руд. Обычный расход
жидкого стекла составляет 100—500 г/т.
9.13. Депрессирующее действие органических реагентов
В качестве органических депрессоров используются
соединения, в которых практически отсутствуют гидрофобные
углеводородные радикалы, но имеется большое количество
полярных групп, способных прочно удерживать по несколько
молекул воды.
В зависимости от характера полярных групп различают:
I неионогенные органические депрессоры, имеющие в своем
составе гидроксильные —ОН, котонные =С = 0 или альдегид-
0
Бые 0 —Н группы. По данным С. И. Горловского, И. И.
Ванеева и Н. В. Зашихина, гидроксильная группа связывает три
молекулы, а альдегидная группа — две молекулы воды;
ионогенные анионные органические депрессоры, имеющие
0
/
в своем составе карбоксильные —G —ОН,
184
сульфоксильные —$ —ОН
\
о
о
или сульфатные —g — g—дц
\
группы. По данным тех же авторов, карбоксильная группа
способна удерживать четыре молекулы воды. Ионизация этих
депрессоров лучше происходит в щелочных средах;
иоиогенные катионные органические депрессоры, имеющие
в своем составе аминогруппу — NH2. Ионизация таких
депрессоров происходит лучше в кислой среде;
ионогенные амфотерные органические депрессоры,
содержащие в своем составе полярные группы, характерные как для
анионных, так и катионных депрессоров. В щелочной среде
амфотерные депрессоры ведут себя как анионные, а в кислой —
как катионные депрессоры.
Из всех органических депрессоров при флотационном
обогащении полезных ископаемых наиболее широко применяются
реагенты, относящиеся к группам неионогенных и ионогенпых
анионных реагентов.
Например, растворимый крахмал и декстрин, относящиеся
к группе неионогенных депрессоров, применяют для депрессии
флотоактивных силикатов при флотации сульфидных руд;
окислов железа при обратной флотации как катиопными, так и
анионными собирателями железных руд; молибденита при
селективной флотации медно-молибденовых концентратов; галенита
при флотации сильвинитов. Имеются также сведения о
возможности использования таннина и таннидов для депрессии барита
и кальцита. В некоторых случаях крахмал способствует
депрессии халькопирита и сфалерита в нейтральной среде,
одновременно улучшая флотацию галенита.
В свою очередь, карбоксиметилцеллюлоза (КМЦ), сульфат
и сульфонат целлюлозы, относящиеся к группе анионных
депрессоров, являются селективно действующими подавителями
флотоактивных силикатов,, таких, как тальк, серицит, хлорит
и др., и широко используются при флотации медно-никелевых
руд. С помощью сульфитцеллюлозных экстрактов (СЦЭ)
можно избирательно депрессировать кальций и барит при
отделении их от флюорита. Лигнинсульфоновые кислоты в
сочетании с фтористым натрием успешно применяются при
селективной флотации сподумен-берилловых руд и разделении
коллективных концентратов [44].
185
Депрессирующее действие органических реагентов
недостаточно селективно. Для повышения селективности необходимо
опытным путем подбирать ионный состав пульпы. Расход
органических депрессоров обычно колеблется от 50 до 400 г/т.
Механизм депрессирующего действия органических
соединений на флотацию минералов изучен недостаточно. Установлено,
что подавление флотации сопровождается адсорбцией
органического депрессора на минералах, однако вытеснения
собирателя с поверхности не наблюдается. Гидрофилизация
поверхности обусловлены тем, что размеры гидрофильных молекул
депрессора значительно превышают размеры углеводородных
радикалов и экранируют их гидрофобизирующее действие.
-о-'
<^o>Lo-j
0 и '
/ \
н н
V
0 С
\ /\
н н н н н
V \/
о о
\/
Лсдерхнсетъ к8арца
н Ш0 н
-'<> н>'-о-
~-0<K_H0><h
о н
Ch2
С=0
6
-•'Л
н н
Y
н н
•••• /
о
1/Н
H0\L0.
г/
сн2
о
сн2
C--Q
о
I
^7Fe;
осиной
_Vh ■ hoV0_
-(Г
*Пс.ве з х чос^ь г ем am и та
т Fe >;////>
Рис. 9.10. Принципиальные схемы закрепления элементарных звеньев
крахмала на кварце (а~в) и карбоксиметилцеллюлозы на гематите (г—е) по
С. И. Горловскому [10] (водородные связи, показанные пунктиром, даны
только для некоторых групп минералов)
186
Так, при нейтрализации солей кальция содой или фосфатом
необходимо предотвратить протекание реакции
Са2+ + 2 An- T- СаАп2, A0.1)
что возможно при условии
К'= — < ! или Са2+ < *пр , A0.2)
*пр [Са2+] [An"]2 [An"]2
где Kf и /(пр — соответственно константа равновесия реакции
A0.1) и произведение растворимости СаАп2.
Если для осаждения солей кальция используют соду, то
[Са2+] определяется условиями равновесия их с осадком СаСОз.
На основании ранее проведенного расчета ионно-молекулярного
состава раствора в области существования МеСОз (см.
разд. 5.2) получаем
Са2+] = _К»_(КаКь + Къ +] + [н+]2), A0.3)
где /С25 — произведение растворимости СаСОз; Ка и Къ —
вторая и первая константы диссоциации Н2СО3, Ссо— суммарная
концентрации С032~, НССЬ и Н2СОз. Используя выражения
A0.2) и A0.3), получаем
С1 > К^1]2 (К4К5 + /С. [Н+] + [H+1V (Ю.4)
В присутствии фосфатов [Са2+] определяется условиями
равновесия их с осадком фосфата кальция Саз(Р04Ь:
Са3(Р04J ^ ЗСа2+-ь 2POJ-;
XM=[Ca»+]a-[PQi-]s или |Са2+]з = ^_.
Используя выражения A0.2) и E.15) для [Р03~4], получаем
е% > к»\Ап~^-(к^кв+к,кш [н+] + к* \и+\2+ [н+]3J,
K6K7KsKup
A0.5)
где Ср0 — суммарная концентрация Р03~4, НР02~4, Н2РО~4 и
НзР04; /С2б — произведение растворимости Са3(Р04J; Кб, Ki,
К% — третья, вторая и первая константы диссоциации Н3Р04.
Уравнения A0.4) и A0.5) могут быть использованы в
качестве задания функциональным блокам систем регулирования
расхода соды или фосфата при кондиционировании
промышленных, оборотных вод или жидкой фазы флотационной пульпы
при использовании оксигидрильных собирателей.
189
Ионы, депрессирующие флотируемые минералы. К таким
ионам относятся, например, сульфидные ионы, избыточная
концентрация которых наблюдается в пульпе после сульфидизации
окисленных цинковых минералов перед их флотацией и после
осуществления десорбции собирателя с поверхности минералов
коллективного концентрата перед его разделением.
Нейтрализация депрессирующего 'действия избытка ионов серы
достигается добавками солей тяжелых металлов, используемых в
данном случае в качестве регуляторов среды.
Оптимальное значение остаточной [S2-] после
нейтрализации определяется природой флотируемого в последующей
операции минерала, концентрацией собирателя и значением рН
пульпы. Если, например, флотируются сульфиды свинца, то
значение [S2~] может быть рассчитано на основании уравнения
(9.7) по формуле
[S2-] ^4ЧКх-]2; A0.6)
если же флотируются сульфиды железа или меди, то [S2-}
можно рассчитать по формуле
lg [s2-] = {-lg/C-lg [МеОН+] } [Кх-] + lg [Kxi + рН, A0.7)
полученной на основании уравнения (9.9).
Уравнения A0.6) и A0.7) после подстановки
соответствующих значений lg К и lg [MeOH+] [Кх~] также могут быть
использованы в качестве задания системам автоматического
регулирования расхода реагентов-регуляторов, нейтрализующих
избыточную концентрацию S2- ионов в пульпе при десорбции
собирателя с поверхности коллективного концентрата.
Ионы, активирующие флотацию депрессируемых минералов.
Например, ионы меди нежелательны при свинцовой флотации
свинцово-цинковых руд, так как они активируют сфалерит. Для
связывания их применяют цианиды или в небольшом
количестве сернистый натрий. Для нейтрализации активирующего
действия солей щелочноземельных металлов на минералы пустой
породы при флотации с оксигидрильными собирателями
применяют соду, фосфатные соединения, жидкое стекло.
На основании данных, приведенных на рис. 8.1, £,
предотвращение флотации одного из главных силикатных
минералов— кварца обеспечивается, если адсорбция солей кальция
на его поверхности не превышает 20 % условного монослоя, что
Г 0 2
при значении lg = lg- ! = —0,6 соответствует
значению lg[CaQH+] =5,56 (см. рис. 8.1, а)
или
lg [СаОН+] = 5,56—рН.1* ' A0.8>
Если для осаждения солей кальция применяют, например,
соду, то концентрация кальцийсодержащих ионов определяется
190
условиями равновесия их с осадком карбоната кальция.
Воспользовавшись полученной ранее (см. разд. 5.2) при расчете
ионно-молекулярного состава раствора в области
существования МеСОз формулой для концентрации ионов МеОН4 в
рассматриваемых условиях, найдем, путем подстановки данного
выражения для [СаОН4] в уравнение A0.8), зависимость между
суммарной концентрацией С0С карбонатных компонентов СОз2~,
НСО~з, Н2СО3 и концентрацией водородных ионов в пульпе:
l8[V-(^5 + iC5[H4-] + [H^'I 5>б6_
ИЛИ
18СЦ^-5.5б1+18 «Л + М^ч-К]' +2рН.
L ^22 J A4K5
Аналогичным образом можно получить зависимость между
суммарной концентрацией С0р ионов Р043~, НРС>42~, НгР04~,
Н3РО4 и концентрацией водородных ионов для случая
осаждения солей кальция фосфатами [3].
Предотвращение активации сульфидов цинка ионами меди
при подаче сернистого натрия основано на образовании
труднорастворимого сульфида меди CuS. При этом соотношение
между концентрациями ионов меди [Си24-] и сульфидных ионов
[S2-] контролируется выражением для константы равновесия
реакции:
CuS^Cu2+-|-S2~;
lg[S2-]^lgK27-»lg[Cu2+], " A0.9)
Активация сфалерита не наблюдается, если [Си2+] в пульпе
меньше необходимой, описываемой уравнением (8.7), т. е.
lg[Cu2+] < + 0,7—2pH. A0.10)
Подставляя выражение для [Си2+] из уравнения A0.10)
в уравнение A0.9), получаем расчетное уравнение
минимальной необходимой [S2 ], предотвращающей активацию сульфидов
цинка ионами меди:
lg[S2~] >lg/C,7-0f7 + 2pH.
Достоверность этого уравнения подтверждена в
промышленных условиях [1, 2].
10.3. Регулирование окислительно-восстановительного
потенциала пульпы
Окислительно-восстановительный f/i-потенциал пульпы
может оказать значительное влияние на состояние поверхности
минералов, на скорость протекания реакций окисления-восста-
Sfe А Рис. 'lO.f. Влияние окислительно-
восстановительного потенциала Eh
или концентрации кислорода
в пульпе [02] на краевой угол
смачивания 0 или извлечение е
сульфидов при флотации
£Л,[02]
новления (например, в системе ксантогеыат — диксантогенид)
поверхности сульфидных минералов, на соотношение
окисленных и восстановленных форм реагента в объеме пульпы. Eh-no-
тенциал пульпы можно регулировать подачей окислителей
(например, перекиси водорода, перманганата и др.) или
восстановителей (сульфита, тиосульфата и др.), электрохимической
обработкой пульпы или ее аэрацией.
Количественная зависимость между гидрофобностью или
извлечением сульфидов при флотации и значением £/г-потенциала
пульпы, регулируемого продолжительностью аэрации и
изменением концентрации кислорода в пульпе (рис. 10.1), показывает,
что при флотации существует оптимальная концентрация
кислорода в пульпе и оптимальные значения £/ьпотенциала.
Различные сульфиды по-разному взаимодействуют с
кислородом. Например, пирит и, особенно, Пирротин
взаимодействуют быстро, а халькопирит и другие сульфиды медленнее.
Поэтому при большом содержании пирротина в руде, он
поглощает кислород в первую очередь, а другие минералы из-за
недостатка кислорода окисляются медленно и флотация их может
идти вяло. Дополнительная аэрация в таких случаях
оказывается весьма полезной. Так, осуществление операции накислоро-
живания пульпы перед цинковой флотацией на Зыряновской
обогатительной фабрике позволило повысить скорость
флотации и извлечение цинка в концентрат. На других фабриках
оказывается полезным, наоборот, подогрев пульпы, приводящий
к уменьшению концентрации кислорода в пульпе и
возрастанию отрицательных значений £/1-потенциала.
10.4. Регулирование процессов диспергации
и коагуляции шламов
Во флотационных пульпах часто наблюдается коагуляция
тонких шламов и их налипание на более крупные частицы.
Налипание как гидрофильных, так и гидрофобных шламов
приводит к депрессии флотации крупных частиц. Гидрофильные
частицы предотвращают разрыв гидратной прослойки между
частицей и пузырьком, а гидрофобные, закрепляясь на пузырьке,
192
отрываются при подъеме пузырька от крупных частиц,
оставляя их в пульпе (см. рис. 3.15, б).
Коагуляция тонких частиц в большинстве случаев является
неселективной. При этом слипаются шламистые частицы
различных минералов, приводя к образованию искусственных
сростков и нарушению селективной флотации тонких частиц.
Для предупреждения неселективной коагуляции и
налипания тонких частиц на крупные применяют реагенты,
получившие название диспергаторов. В качестве диспергаторов обычно
применяются жидкое стекло, фосфаты, крахмал, сернистый
натрий и некоторые другие реагенты.
К настоящему времени имеются примеры селективной
коагуляции ошламованных минералов, когда слипаются только
частицы одного и того же минерала. Такая коагуляция,
достигаемая с помощью соответствующих, в большинстве случаев
органических реагентов позволяет повысить извлечение тонких
частиц в одноименные концентраты.
--11. ПЕНООБРАЗОВАТЕЛИ И МЕХАНИЗМ ИХ ДЕЙСТВИЯ
Большая поверхность раздела газ — жидкость воздушных
пузырьков, на которой происходит закрепление минеральных
частиц, и необходимая прочность минерализованной пены на
поверхности пульпы обеспечиваются при флотации с
применением пенообразователей (вспенивателей).
11.1. Строение и физико-химические свойства
пенообразователей
В качестве реагентов-пенообразователей наиболее широко
применяются гетерополярные поверхностно-активные вещества,
содержащие полярную (водоактивную) и неполярную (воздуш-
ноактивную) части. Вещества такого типа способны
адсорбироваться на границе раздела вода — воздух, ориентируясь
полярной группой к воде, а неполярной к воздушной фазе.
Полярные группы пенообразователей могут быть не
ионизирующиеся или слабоионизирующиеся, такие, как —ОН,
= С = 0, —СН = 0; ионизирующиеся анионные, например
О D О
— с —он, — о—s—он, — s —он
\ \
о о
и ионизирующиеся катион ные — NH2, =N. Аполярная
часть молекулы может быть представлена алкильным или
арильным радикалом. Молекулы пенообразователей содержат
7 Заказ № 1957
193
обычно один углеводородный радикал и одну или небольшое
число полярных групп.
Адсорбция пенообразователя сопровождается изменением
поверхностной энергии границы раздела вода — воздух.
Поверхностная активность пенообразователей а = да/дС зависит от
характера полярной группы и строения углеводородного
радикала.
Например, при одинаковом радикале карбоновые кислоты
обладают большей поверхностной активностью, чем спирты.
Однако для практических нужд лучше пользоваться спиртами
с неионизирующимися полярными группами — ОН, чем карбо-
новыми кислотами, полярная группа которых —СООН может
ионизироваться с изменением рН, резко изменяя пенообразую-
щую способность, а также взаимодействовать с поверхностью
многих минералов, изменяя их флотируемость. Эти недостатки
карбоновых кислот присущи и другим пенообразователям
с ионизирующимися полярными анионными и катионными
группами.
При увеличении длины углеводородной цепи на одну группу
= СНя поверхностная активность пенообразователей
увеличивается по правилу Траубе примерно в 3 раза. Причем лучшими
пенообразующими свойствами обладают соединения с нераз-
ветвленной углеводородной цепью. Используемые на практике
пенообразователи содержат, как правило, от 5 до 12 атомов
углерода в цепи, а их растворимость составляет обычно 5—
0,2 г/л. Гетерополярные вещества с меньшим числом атомов
углерода в радикале не применяются из-за малой их
поверхностной активности, а с большим числом атомов углерода в
радикале — из-за чрезмерно малой их растворимости.
11.2. Роль и механизм действия пенообразователей
Действие реагентов-пенообразователей при флотации
обусловлено главным образом адсорбцией их на поверхности
раздела жидкость — газ и в гораздо меньшей степени на границе
раздела жидкость — твердое.
Адсорбция пенообразователей на границе раздела
жидкость — газ позволяет изменять коалесцентную способность
воздушных пузырьков и степень их дисперности в пульпе,
скорость подъема пузырьков, структурно-механические свойства
оболочек воздушных пузырьков и прочность пены. Адсорбция
на границе раздела жидкость — твердое оказывает иногда
нежелательное влияние на прочность закрепления частиц на
пузырьках, а также на свойства и характер трехфазных
флотационных пен.
Уменьшение коалесцентной способности и средней
крупности пузырьков. Интенсивность процесса флотации определяется
площадью поверхности раздела жидкость — газ, которая при
одном и том же количестве воздуха увеличивается с увеличе-
194
нием дисперсности воздушных пузырьков. Однако этот процесс
сопровождается резким увеличением свободной поверхностной
энергии и поэтому аэрированная пульпа является неустойчивой
в термодинамическом отношении. При столкновении пузырьки
коалесцируют, т. с. сливаются в более крупные, общая площадь
поверхности их и свободная энергия системы уменьшаются. По
этой причине чистые жидкости не могут образовывать
устойчивую пену.
В присутствии пенообразователя процесс коалесценции
резко замедляется, так как в результате адсорбции на
поверхности раздела жидкость — газ пенообразователь образует
ориентированный слой молекул, полярные концы которых гидра-
тируются диполями воды (см. рис. 2.2, в). Этот гидратиро-
ванный слой приводит к повышению механической стойкости
оболочек пузырьков и препятствует их слиянию при
столкновении друг с дугом, что позволяет сохранить в пульпе более
мелкие пузырьки.
Характерная зависимость средней крупности пузырьков
воздуха от концентрации пенообразователя в пульпе изображена
на рис. 11.1.
Уменьшение скорости подъема пузырьков. В отсутствие
пенообразователя в пульпе пузырьки не имеют на своей
поверхности структурно устойчивой гидратной оболочки (см. рис. 2.2,
а), поэтому при всплывании в пульпе они легко деформируются
и принимают форму, выгодную в гидродинамическом
отношении. В результате этого скорость их подъема возрастает. Иначе
обстоит дело с пузырьками воздуха, заключенными в довольно
«жесткую» гидратную оболочку и имеющими вследствие этого
форму, близкую к сферической. При подъеме такие пузырьки
меньше деформируются, у них меньше возможностей принять
выгодную в гидродинамическом отношении форму, поэтому
скорость их подъема гораздо меньше скорости подъема пузырьков
такого же размера в чистой воде. Скорость подъема пузырьков
в присутствии пенообразователя близка к теоретической
скорости подъема сферического пузырька.
Снижение скорости подъема пузырьков под действием
пенообразователей приводит к увеличению содержания воздуха
в пульпе и тем самым увеличивает вероятность их
столкновения с минеральными частицами.
Повышение прочности пены. Пеной называется
концентрированная эмульсия газа в жидкости. Если она не содержит
твердых частичек, то ее называют двухфазной, если содержит—
трехфазной. В обоих случаях пенообразователь увеличивает
прочность пены.
Причина этого заключается в следующем:
довольно прочная ассоциация молекул воды около
полярных групп пенообразователя препятствует стеканию воды из
прослоя h (рис. 11.2) при выходе пузырьков в пенный слой. Чем
больше гидратированы полярные группы пенообразователя, тем
7*
195
Jk
Рис. 11.1. Влияние концентрации С Рис. 11.2. Стабилизация пузырька
пенообразователя на средний раз- молекулами пенообразователя
мер D пузырьков в пульпе
медленнее стекает вода с поверхности пузырька между
адсорбционными слоями пенообразователя и тем прочнее и устойчивее
пена;
ассоциация молекул воды полярными группами
пенообразователя затрудняет испарение жидкости из тонкой прослойки
между пузырьком и атмосферой, тем самым затрудняя
дальнейшее ее утоньшение и разрушение пузырька;
адсорбционный слой пенообразователя увеличивает
эластичность пузырька и его сопротивляемость разрушению при
случайных механических воздействиях.
Последнее обусловлено тем, что при внезапном растяжении
пузырька концентрация молекул пенообразователя в зоне
растяжения (см. рис. 11.2) уменьшается и вызывает увеличение
поверхностного натяжения и тем самым появление сил,
препятствующих дальнейшему растяжению поверхности в данном
месте. «Прочность» пузырька автоматически возрастает как раз
в том месте, где возникает опасность разрыва пленки и
разрушения пузырька.
Степень эластичности пузырька зависит от характера и
концентрации пенообразователя в пульпе.
Исследованиями установлено, что для пенообразователей,
имеющих одну и ту же полярную группу, радикал является
определяющим в формировании пузырьков максимальной
устойчивости. Например, по данным -С. В. Дуденкова, у реагентов
с полярной группой — ОН, у которых углеводородный радикал
состоит из алифатической цепи нормального строения,
оптимальной является цепь из восьми атомов углерода (рис. 11.3),
для реагентов типа фенола — циклический радикал с одной
группой СНз и для циклогексанолов — радикал с
алифатической цепью, содержащей более трех атомов углерода. С
увеличением или уменьшением радикала эффективность
пенообразователей уменьшается и увеличивается значение их
минимальной концентрации, при которой предотвращается разрушение
(коалесценция) пузырьков (см. рис. 11.3).
196
Рис. 11.3. Зависимость объема V
пены в растворе спирта при
постоянной его концентрации 5 • 10~4 моль/л
и минимальной концентрации С
спирта, вызывающей прекращение
разрушения (коалесценции)
пузырьков воздуха от числа п атомов
углерода в углеводородной цепи
молекулы спирта (по данным [29])
При уменьшении радикала это обусловлено чрезмерно
быстрым выравниванием плотности адсорбционного слоя на
поверхности пузырьков вследствие увеличения подвижности молекул
пенообразователей, а также неспособностью их значительно
изменять поверхностное натяжение при адсорбции и десорбции
в связи с уменьшением поверхностной активности
пенообразователей с меньшим размером радикала. Уменьшение
стабилизирующего действия на пену пенообразователей, радикал
которых больше оптимального, обусловлено уменьшением их
растворимости, снижением концентрации и подвижности молекул
пенообразователя в пульпе.
Для каждого пенообразователя существует своя
оптимальная концентрация, при которой обеспечивается наиболее
эффективная стабилизация пены. Значение ее соответствует
оптимальной скорости выравнивания плотности адсорбционного
слоя в зоне растяжения после деформации пузырька. При
высоких концентрациях пенообразователя скорость выравнивания
плотности будет чрезмерно высокой, а «эластичность»
пузырьков — очень низкой, поэтому насыщенные, растворы вообще не
пенятся. При очень низких концентрациях пенообразователя
недостаточные «эластичность» пузырьков и прочность пены
обусловлены тем, что поверхностное натяжение раствора мало
отличается от поверхностного натяжения чистой воды и
изменение его при растяжении пузырька не может быть значительным.
Кроме того, при малой плотности адсорбционного слоя
полярные группы молекул пенообразователя удерживают меньшее
количество молекул воды.
На эффективность действия пенообразователя при всех
прочих равных условиях могут оказать влияние изменения
температуры и рН пульпы. Изменение температуры вызывает
изменение растворимости пенообразователя, концентрации и
подвижности его молекул в пульпе, что приводит к изменению
скорости выравнивания плотности адсорбционного слоя на
300
250
200
150
100
50
О
I
тн. к
~
<
?л
L.
с,
V;
\
у
\
/\
1
{
1
\
г
/-
\
\
\
L
;
'
МОЛ
N»
3
197
пузырьках и тем самым к изменениям их «эластичности» и
прочности пены. Изменения рН оказывают влияние на эти же
процессы вследствие изменения степени диссоциации или
дисперсности пенообразователей в пульпе. Наиболее эффективным
путем стабилизации пенообразования в практических условиях,
характеризующихся довольно широким изменением целого ряда
параметров, является применение смеси пенообразователей.
Повышение прочности закрепления пузырька на частице.
Пенообразователи, являясь поверхностно-активными веществами,
закрепляются не только на поверхности раздела жидкость—газ,
но и на поверхности раздела жидкость — твердое, активно
влияя на гидрофобизацию поверхности и флотируемость многих
минералов. Например, основная масса сульфидных минералов
может быть сфлотирована одним пенообразователем без
добавки собирателя.
Причинами изменения флотируемости минералов под
действием пенообразователя могут быть:
повышение степени гидрофобности минеральной
поверхности за счет углеводородного радикала адсорбированного
пенообразователя, полярная группа которого образует с катионами
или атомами поверхности химические, водородные связи или
удерживается силами дипольного взаимодействия;
образование с собирателем, закрепившимся на поверхности
комплексов, способствующих разрыву гидратной прослойки
между пузырьком и частицей и закреплению минеральных
частиц на пузырьках воздуха^
повышение устойчивости закрепления пузырьков на
частицах не только за счет уменьшения поверхностного натяжения
на поверхности раздела жидкость — газ и капиллярного
давления в пузырьках, отрывающего пузырек от частицы, но и
многократного упрочнения контакта пузырек — частица в
присутствии молекул пенообразователя по механизму, предлагаемому
В. И. Мелик-Гайказяном;
повышение степени дисперности собирателя в пульпе,
улучшающее его собирательное действие.
Присутствие пенообразователя в растворе практически
всегда способствует увеличению сил прикрепления воздушного
пузырька к минеральной поверхности, причем относительное
увеличение этих сил зависит от класса применяемого
пенообразователя, а их абсолютное значение — от типа собирателя.
11.3. Стабилизация и гашение пены
флотирующимися частицами
Разрушение слоя флотационной пены, представляющей
собой трехфазную систему, состоящую из пузырьков воздуха,
водных прослоек и твердых частиц, происходит в результате
коалесценции пузырьков в пенном слое и разрушения их на
поверхности. Флотирующиеся частицы в зависимости от сте-
198
пени их гидрофобное™ могут
как ускорять, так и
затруднять эти процессы.
Например, если краевой
угол 0 на поверхности
флотирующейся частицы меньше \^ ^Щ[ ^^^чашйщ
90°, то должна иметь место
«мокрая» флотация (см. ~_^SZS~.~ ^Ь>>у__^~в~од~а
рис. 2.10, II состояние), при
которой флотационные силы
выталкивают частицу из га- _ п, г . Л , а
- „ . J Рис. 11.4. Стабилизирующее дси-
зообразнои фазы в жидкую. ствие частиц на пену
Если такая частица
оказывается между двух пузырьков
воздуха, то она выталкивается из обоих пузырьков и
затрудняет их сближение и коалесценцию (рис. 11.4). Если такие
частицы оказываются на поверхности пены, то вода как бы
«всасывается» между зернами, чтобы обеспечить им «мокрую»
флотацию, тем самым предотвращая утоныпение водной
оболочки пузырьков и их разрушение.
Если краевой угол 0 на поверхности флотирующейся
частицы больше 90°, то должна происходить «сухая» флотация
(см. рис. 2.10, III состояние). В этом случае флотационные
силы, действующие на частицу, находящуюся между двух
пузырьков воздуха, «втягивают» частицу в каждый из пузырьков,
приводя к их коалесценции. В свою очередь, наличие таких
частичек па поверхности пены способствует утоньшению водной
оболочки пузырька, чтобы обеспечить «сухую» флотацию
частиц на его поверхности, и вызывает разрушение (гашение)
пены.
Практика флотации обычно имеет дело с минералами,
краевой угол на поверхности которых меньше 90°, поэтому
флотирующиеся частицы стабилизируют пену. Особенно стабильны
пены, содержащие большое количество сравнительно
гидрофильных, но еще флотирующихся тонких частиц (шламов), что
приводит иногда к так называемой «плывучке». Однако при
флотации минералов, обладающих естественной флотируе-
мостью, краевой угол которых больше 90°, наблюдается
гашение пены. Вызвать гашение пены могут также тонкие частицы
сильногидрофобных продуктов взаимодействия, например
анионных собирателей с катионами растворимых солей пульпы,
а также капельки аполярных масел (например, керосина),
подаваемых в качестве реагентов или попадающих в руду в
процессе горных работ. Преодолеть гасящее действие гидрофобных
осадков и капель аполярных масел удается дополнительной
подачей пенообразователя, молекулы которого при этом
ассоциируют аполярными радикалами с аполярным веществом осадка
или капли, а полярные группы, направленные наружу,
вызывают гидратацию их поверхности.
199
Следует отметить, что флотирующиеся частицы механически
затрудняют сток воды между пузырьками и тем самым
способствуют стабилизации пены при всех значениях краевого угла.
11.4. Вторичная концентрация минералов в пене
Турбулентные потоки пульпы и всплывающие пузырьки
выносят в нижнюю часть пены не только частицы флотируемого
минерала, но и частицы пустой породы или депрессируемого
минерала. В результате разрушения и коалесценции пузырьков
наблюдается вторичная концентрация минералов в пене.
Обогащение верхних и обеднение нижних слоев пены происходит
вследствие вымывания частиц породы или депрессируемого
минерала стекающей между пузырьками водой. Стекание воды
приводит к сужению каналов и толщины водных прослоек, что
вызывает, особенно при наличии прочно закрепившихся на
пузырьках зерен флотируемого минерала, торможение частиц
породы и образование из них пробок. Для очистки пены от
частиц породы в таких случаях применяют орошение водой. Это
позволяет увеличить толщину прослоек между пузырьками
в пене и за счет уменьшения несущей способности пены на всех
ее уровнях облегчить вымывание особенно крупных частиц
породы или депрессируемого минерала обратно в пульпу. В ряде
случаев применение орошения позволяет значитс^льно повысить
качество концентрата или заменить одну (иногда две) пере-
чистную операцию флотации.
11.5. Пенообразователи, применяемые на практике
Из большого числа соединений, предложенных в качестве
пенообразователей, наибольшее распространение получили ге-
терополярные соединения с неионизирующейся полярной
группой, сосновое масло и синтетические реагенты, ОПСБ, Т-66,
МИБК, циклогексанол, Э-1, ТЭБ, Д-3 и др.
Сосновое масло состоит в основном из терпеновых
спиртов и углеводородов (терпенов). Наиболее активной частью
его является терпинеол СюНпОН, содержание которого для
сортов А, Б и В составляет соответственно 60, 50 и 40 %,
в масле ИМ— не менее 60 %, в масле Вахтан — не менее 44 %.
Сосновое масло, обладающее слабыми избирательными
способностями, ранее широко применялось при флотации всех типов
минерального сырья. Расход его составляет 10—100 г/т. В
настоящее время область применения соснового масла
значительно сужена синтетическими реагентами.
Из синтетических реагентов хорошо зарекомендовал себя
особенно при селективной флотации сульфидов свинца
циклогексанол, имеющий структурную формулу
200
-он
Нашел широкое применение при селективной флотации
полиметаллических и других руд метилизобутилкарбинол
(МИБК)—вторичный гексиловый спирт, имеющий
структурную формулу
Н Н Н
I I I
н.с — с — с — с —он
I I I
uii? Н СН3
Расход МИБК составляет 10—50 г/т.
По исследованиям Гинцветмета перспективным
пенообразователем при флотации сульфидных руд является третичный
гексиловый спирт
СН5
I
сн3—сн2—сн2—&—он
Щ
Реагент ВВ-2 (вспениватель ВНИИЦветмета) состоит из
алифатических двух- и трехатомных спиртов и используется
при флотации руд цветных металлов (при расходе 50—200 г/т)
вместо токсичного крезола и дефицитного циклогексанола.
Близок по пенообразующей способности к спиртовым
пенообразователям реагент Э-1, представляющий собой смесь
монобутиловых эфиров полиэтиленгликолей с общей структурной
формулой С4Н9—[—О—СН2—СН2—]п—ОН, где п = 2, 3, 4.
Реагент используется па Лениногорской фабрике при расходе его
в 5—6 раз меньше ранее применявшегося крезола.
Не сильно отличается по пенообразующей способности от
реагента Э-1 пенообразователь ОПСМ, являющийся
смесью монометиловых эфиров полипропиленгликолей
СН3
сн3—[— о— сн2— сн—]п— он,
где л= l-f-4. По составу он близок к известному американскому
Дауфросу-250. Реагент хорошо растворим в воде и малотокси-
201
чен. При меньших расходах (примерно, в 2 раза) обеспечивает
получение практически тех же показателей флотации, что и
циклогексанол.
Более сильным пенообразователем, чем Э-1 и ОПСМ,
является ОПСБ, представляющий собой смесь монобутиловых эфи-
ров полипропиленгликолей
СН3
ЦН9-[-р-сн2—сн-]п-он,
где /г=1-г-4. Этот реагент в отличие от других
пенообразователей может обеспечивать при умеренном или малом расходе
хорошее пенообразование в присутствии углеводородных масел.
В связи с этим он обладает особыми преимуществами перед
другими пенообразователями при флотации молибденовых,
медно-молибденовых, медных и других руд, которые флотируют
обычно с добавками углеводородных масел. Применение ОПСБ
при селективной флотации обычно нецелесообразно. Реагент
хорошо растворим в воде и малотоксичен, расход его меньше
расхода соснового масла.
К сильным пенообразователям относится и триэтокси-
б.ута н (ТЭБ)
СН3— СН —СН2 — СН — 0 — С2Н5
о —с2н5 о — с2н5
Расход его при флотации в 2—3 раза меньше расхода
соснового масла. Преимуществом ТЭБ является то, что в водных
растворах он в течение нескольких дней разлагается с
образованием безвредных продуктов, вследствие чего хвостовые воды,
содержащие ТЭБ, не требуют специальной очистки.
Реагент Д-3, представляющий собой диметилфталат со
структурной формулой
н3о—о оо о —сн.
\/ \/
с с
по силе действия уступает реагентам ТЭБ и ОПСМ и не
превосходит их по селективности. Может использоваться при
селективной флотации простых полиметаллических руд. За
рубежом получили распространение дипропил- и дигексилфталаты
при расходе их 25—75 г/т.
202
Пенообразователь Т-66, представляющий собой смесь
различных веществ — преимущественно одноатомных спиртов
пиранового и диоксанового ряда, а также некоторых гликолей,
является в СССР одним из основных пенообразователей.
Причины этого — довольно постоянный состав, несмотря на
сложность и многообразие компонентов Т-66, малая токсичность,
хороший технологический эффект на различных типах руд при
сравнительно небольших расходах реагента. В ближайшее
время предстоит замена его реагентом-аналогом Т-80 и
пенообразователем ЛВ, получаемым из того же исходного сырья.
Масло X представляет собой смесь циклических спиртов
(циклогексанола и дициклогексанола), эфиров карбоновых и
дикарбоновых кислот, углеводородов. Этот нерастворимый
в воде продукт широко используется при флотации углей при
расходе 100—300 г/т. Достоинством его является устойчивость
к пеногасящему действию аполярного собирателя
(углеводородных масел).
Реагент ДС-РАС представляет собой смесь алкиларил-
сульфонатов с 5—11 атомами углерода в радикале, т. е.
является апионпоактивным пенообразователем. Он хорошо
растворим в воде, обладает сильным пенообразующим и заметным
собирательным свойствами. В связи с этим он нашел применение
при флотации барит- и свинецсодержащих руд, а также при
жирнокислотной флотации доломита из фосфатных руд в
кислой среде.
Реагенты ОП-4, ОП-7, ОП-10 являются оксиэтилирован-
ными алкилфенолами и по строению аналогичны реагенту Э-1.
Значение п в реагентах ОП-4, ОП-7, ОП-10 равно соответственно
4, 7 и 10. По своему назначению реагенты ОП являются
эмульгаторами пенообразователей и собирателей. Их добавка B0—■
40 г/т) уменьшает расход собирателя, понижает устойчивость
пены до необходимого предела, улучшает диспергацию
шламовых флокул, что повышает качество концентрата и
производительность фильтров.
В последнее время многие фабрики (особенно зарубежные)
для более устойчивого ведения технологического процесса
применяют или смесь пенообразователей (сочетая их в одном
цикле либо используя разные пенообразователи в разных
циклах), или смесь пенообразователя с так называемыми
реагентами-регуляторами пены (различными маслами и отходами
химических производств).
ЧАСТЬ III
Технология флотации
12. ОСНОВНЫЕ ХАРАКТЕРИСТИКИ ВЕЩЕСТВЕННОГО СОСТАВА
ПОЛЕЗНЫХ ИСКОПАЕМЫХ И ИХ ВЛИЯНИЕ НА ФЛОТАЦИЮ
К основным характеристикам вещественного состава полез-,
ных ископаемых, определяющим технико-чкопомическис
показатели обогащения, относятся: содержание и флотирусмость
денных компонентов; минеральный состав; характер
вкрапленности и срастания минералов; наличие в них изоморфных
примесей; вторичные изменения минералов вследствие окисления,
выветривания и взаимоактивации [2].
12.1. Содержание ценных компонентов
Руды, угли и другие полезные ископаемые представляют
собой смесь ценных компонентов, каждый из которых может
найти применение в народном хозяйстве. Степень извлечения
каждого компонента в отдельный концентрат зависит от его со-.
держания в руде. При прочих равных условиях извлечение
возрастает с увеличением содержания данного компонента в руде.
Это обычно обусловлено тем, что содержание неизвлекаемой
части компонента в руде более или менее одинаково и с
увеличением общего содержания флотируемого компонента
увеличивается доля его извлекаемой части. Однако при переработке
на фабрике различных сортов руд такой связи может и не быть,
если окажется, что в рудах с более высоким содержанием
извлекаемого компонента он представлен труднофлотируемымн
или неизвлекаемыми минералами, а в рудах с небольшим
содержанием данного компонента —легкофлотируемыми
минеральными разностями. ?
12.2. Минеральный состав
Технологические показатели извлечения каждого
компонента из руды и качество получаемых концентратов зависят от
минерального состава руды. Это, во-первых, связано с тем, чтй
каждый металл или элемент может быть представлен разными
минералами, обладающими разной флотируемостью. Например,
медь в рудах может быть представлена легкофлотируемыми
сульфидными минералами (халькопиритом, борнитом, халько-1
зином и ковеллином), гораздо хуже флотируемыми окислен-;
ными минералами (малахитом, азуритом, купритом) и
практически неизвлекаемыми при флотации хризоколлой и
алюмосиликатами меди. Разные группы минеральных форм требуют,
204
разных реагентных режимов и при их совместном присутствии
Трудно обеспечить оптимальные условия флотации всех
минералов. Поэтому в технологической схеме обычно
предусматривают раздельное флотационное извлечение; например, сначала
Сульфидных, а затем окисленных минералов. Изменение
соотношения минеральных форм в сторону увеличения труднофло-
тируемых разностей извлекаемого компонента приводит
к уменьшению его извлечения в концентрат.
Во-вторых, возможность селективной (избирательной)
флотации зависит от степени близости физико-химических свойств
разделяемых минеральных компонентов; трудности
осуществления селективной флотации возрастают при разделении
минералов с одинаковым анионом или катионом. Например, если
флотационное отделение сульфидных минералов от несульфидных
Является обычно простой операцией, то разделение сульфидов
протекает гораздо труднее. Трудности селективной флотации
минералов с одинаковым катионом или анионом обусловлены
еще и тем, что разные минеральные формы одного и того же
металла или элемента обладают различными свойствами. Если,
например, отделение галенита от халькопирита протекает
довольно легко, то отделение его от халькозина требует особых
условий.
В-третьих, селективная флотация осложняется при наличии
в рудах легкофлотируемых алюмосиликатов и при
значительном содержании шламистых минералов и пород, обладающих
большой поглотительной способностью по отношению к
флотационным реагентам. Например, флотационное извлечение
окисленных минералов свинца из сильно разрушенных руд
становится практически невозможным, если в них присутствуют
г окислы и гидроокислы марганца. Трудной задачей является
также эффективная депрессия флотоактивных силикатов, резко
понижающих содержание извлекаемого компонента в
концентрате.
12.3. Влияние генезиса руд
Условия образования полезных ископаемых (генезис) опре-
*^ дел я ют их строение, характер кристаллизации, изоморфизм,
Скорость и степень окисления и электронные свойства
минералов.
Например, сульфидные руды, образующиеся в результате
раскристаллизации расплавленных магм или осаждения
сульфидных минералов из горячих водных растворов отличаются
у плотностью, крупнокристаллическим строением и не имеют пор.
''. Окисленные же руды, образовавшиеся в процессе окисления и
выщелачивания сульфидных руд, характеризуются обычно
мелкокристаллическим строением и большим количеством пор,
заполненных охристо-глинистым материалом. При измельчении
таких руд охристо-глинистый материал образует большое коли-
205
чество так называемых первичных шламов, оказывающих
вредное влияние на флотацию. t :
Генезисом определяется содержание изоморфной примеси
в минералах. Широкое изменение содержания изоморфного
железа в сфалерите и пентландите, молибдена в шеелите,
марганца в вольфрамите оказывает большое влияние на
необходимые условия активации и депрессии изоморфных
разновидностей минерала. Изоморфизм является основной причиной
наличия в рудах легко- и труднофлотируемых разностей одного
и того же минерала.
От генезиса месторождения зависят соотношение
концентраций электронов и дырок и характер изменения уровня Ферми
у полупроводниковых минералов. Исследование влияния их на
флотируемость сульфидных минералов показало [1], что
изменение концентрации электронов в поверхностном слое
минералов не требует изменения установленных ранее соотношений
концентраций реагентов в граничных условиях флотации (см.
зависимости [Kx~] = f (pH), Kx~ = f ([S 2], рН) и др.), но может
значительно повлиять на максимально возможное извлечение
минералов в концентрат. Причиной этого является нарушение
условий образования диксантогепида на поверхности при
высокой концентрации электронов или, наоборот, дырок в
поверхностном слое минералов [1]. Повысить извлечение минерала при
высокой концентрации электронов можно, например, используя
реагенты-окислители, а при высокой концентрации дырок —
реагенты-восстановители, понижающие их концентрацию в
поверхностном слое до оптимальных значений [1].
12.4. Вторичные изменения минералов
Вторичным изменениям могут быть подвергнуты как рудные
минералы, так и минералы вмещающих пород.
Наиболее важные изменения минералов пустой породы
связаны с окремнением, каолинизацией, хлоритизацией и серици-
тизацией их поверхности. Каолинизация и серицйтизация
являются, например, основными процессами изменения полевых
шпатов, тогда как для железомагнезиальных минералов
наиболее характерна хлоритизация. В процессе вторичных
изменений происходят унификация поверхностных свойств
различных породных минералов при возрастании общей степени их
гидрофобности и образование громадного количества легкофло-
тируемых серицито-хлоритовых шламов даже при сравнительно
крупном измельчении. В результате этого возрастают
трудности депрессии пустой, породы, предотвращения вредного
влияния шламов и получения богатых концентратов.
Вторичные изменения рудных, например сульфидных,
минералов связаны в основном с их окислением и
взаимоактивацией.
Окисление сульфидов в зоне окисления месторождения или
206
в процессе добычи, транспортировки, дробления и измельчения
руды приводит к образованию на их поверхности более
полярных соединений, чем сами сульфиды. При взаимодействии с
собирателем па таких поверхностях образуются рыхлые
гидрофобные шламы, затрудняющие флотацию сульфидных зерен.
Наилучшие результаты в этом случае дает предварительная
сульфидизация окисленной поверхности сульфидных минералов.
Наибольшие трудности при селективной флотации может
вызвать взаимоактивация минералов как в самом
месторождении, так и в процессе подготовки руды к флотации. Особенно
трудно поддаются селекции сульфидные руды из зон
вторичного обогащения, когда поверхность практически всех
сульфидов железа, цинка, свинца покрыта пленками вторичных
сульфидов меди. В этом случае флотируемость всех минералов
становится одинаковой. Разделение их удается осуществить только
после очень топкого измельчения при ограниченном развитии
активирующих пленок на поверхности или только после
удаления их с помощью соответствующих растворителей.
12.5. Характер вкрапленности
и необходимая крупность измельчения
Необходимая конечная крупность измельчения полезных
ископаемых или продуктов их обогащения должна
удовлетворять следующим требованиям:
размеры зерен флотируемого минерала не должны выходить
за определенные пределы крупности, за которыми невозможно
эффективное их закрепление на пузырьках воздуха и флотация;
основная масса флотируемых минералов должна находиться
в свободном виде, т. е. быть освобожденной из сростков с
пустой породой (перед коллективной флотацией) и из сростков
минералов друг с другом (перед селективной флотацией).
Крупность получаемых концентратов должна отвечать
предъявляемым к ним требованиям (кондициям). Например,
апатитовые концентраты должны содержать не более 14 % класса
+ 0,15 мм.
В .большинстве случаев не удается достигнуть полного
раскрытия всех сростков, представленных обычно срастанием зерен
соизмеримых размеров, пленками одного минерала на
поверхности зерен другого, эмульсионными включениями или
прожилками одного минерала в другом и другими, более сложными
формами срастания минералов. При флотации приходится
отделять частицы, более насыщенные включениями извлекаемого
минерала, от менее насыщенных ими зерен. Для полного
раскрытия всех сростков потребовалось бы слишком тонкое
измельчение всей руды с сильным переизмельчением минералов, что
экономически и технологически нецелесообразно. Поэтому
каждая руда имеет свою экономически выгодную степень
измельчения. Чем выше содержание полезных минералов в руде,
207
больше производительность фабрики и крупнее вкрапленность
извлекаемых минералов, тем желательнее более полное
раскрытие сростков.
12.6. Управление качеством полезных ископаемых,
поступающих на флотационное обогащение
Полезные ископаемые, поступающие на флотационное
обогащение из различных участков месторождения, обычно резко
отличаются по вещественному составу, физико-механическим
свойствам и обогатимости, тогда как высокие и устойчивые
показатели работы фабрики во многом определяются
стабильностью состава поступающих в процесс обогащения руд.
Зависимость недоизвлечения, например, меди и цинка от среднего
квадратического отклонения их содержания в руде на одной
из медно-цинковых фабрик изображена на рис. 12.1.
Соответствие качества планируемой к переработке
(«проектной») руды, оптимальной для выбранных схем рудоподго-
товки и обогащения, и фактически поступающей на обогащение
обеспечивается: предварительным технологическим изучением
руд месторождения, в результате которого должно быть
осуществлено технологическое картирование месторождения;
перспективным планированием и оперативным управлением качеством
руды в процессе горно-транспортных работ; рудничным и
фабричным усреднением качества руды.
Наличие данных технологического картирования позволяет
составить графики отработки месторождения с учетом всех
особенностей вещественного состава и обогатимости руд различных
участков и горизонтов, разработать и создать применительно
к этим участкам гибкие технологические схемы и режимы,
обеспечивающие получение высоких показателей обогащения и
комплексное использование сырья.
Если руды, угли или другие полезные ископаемые,
подлежащие флотационному обогащению, характеризуются резкими
колебаниями их вещественного состава, то они усредняются и
перерабатываются обычно по технологическим сортам. При
невозможности такой переработки полезных ископаемых
обеспечивается тщательное усреднение их перед обогащением.
Шихтовка руды с автоматизированным поддержанием
заданного соотношения сортов руд в смеси используется, например,
на Зыряновской и некоторых японских фабриках. Усреднение
руды осуществляется благодаря организации разгрузки бункера
главного корпуса системой одновременно работающих
питателей при атоматическом регулировании расхода руды,
поступающей с отдельных питателей. Дальнейшее усреднение питания
флотации достигается при подаче измельченной руды в общий
пульподелитель между секциями [2].
Мероприятия по усреднению качества руды и продуктов
обогащения, обеспечивая стабилизацию технологического процесса
208
Рис. 12.1. Зависимость недоизвле- .
чения Де меди G), цинка B) от
среднего квадратического отклоне- 2
ния а их содержания в руде
О 0,2 О/г 0,6 6
на оптимальном уровне и эффективную работу систем
автоматизации, позволяют повысить производительность фабрики (на
5—8%), извлечение металлов (на 0,5—5%) и снизить расход
реагентов (на 10—15%).
13. СХЕМЫ ФЛОТАЦИИ
Под схемой флотации понимают определенную
последовательность операций флотации и их сочетание с операциями
измельчения и классификации. При разработке и выборе схем
флотации учитывают характер и размер вкрапленности
полезных минералов, их содержание в руде и флотируемость,
наличие и характер шламов, требования к качеству концентратов,
необходимость комплексного использования сырья при
минимальных затратах на обогащение.
13.1. Классификация операций флотации
Первая операция флотационного извлечения минералов
одного или нескольких металлов называется основной
флотацией. В результате ее проведения обычно не удается получить
кондиционный концентрат и отвальные хвосты вследствие
близости флотационных свойств разделяемых минералов,
недостаточного раскрытия их сростков, несовершенства флотационных
аппаратов. Получаемые некондиционные (бедные, грубые)
концентраты и богатые хвосты подвергаются, иногда после их до-
измельчения, повторной флотации.
Операция повторной флотации концентрата основной
флотации называется перечисти ой, а операция повторной
флотации хвостов — контрольной. Цель перечистной флотации
концентрата — повышение его1 качества до необходимого по
содержанию основных компонентов и загрязняющих примесей.
Цель проведения контрольной флотации — получение бедных по
отношению к извлекаемым минеральным компонентам хвостов
флотации.
Число перечистных и контрольных операций зависит от
содержания флотируемых компонентов в исходном материале, их
флотируемости и требований, предъявляемых к концентрату
и хвостам. Число перечистных операций обычно тем больше, чем
выше требования к концентрату, ниже содержание извлекаемых
209
Руда
Кмассиф икац ия
+
Оснодная флотация
Iпере ч и cmная I контрольная
♦ ♦
И пере чистная
Д контрольная
Концентрат
/6 осты
Рис. 13.1. Развернутая схема цикла флотации с предварительным
измельчением исходного материала
минералов в исходном материале и лучше их флотируемость.
В противоположных условиях увеличивается число
контрольных операций флотации и уменьшается число перечистных.
В большинстве случаев число контрольных операций не
превышает двух-трех, а перечистных — двух-четырех.
Совокупность основной, контрольных и перечистных
операций называется циклом флотации. В схеме может быть
несколько циклов флотации. Они именуются по получаемому
в них концентрату: свинцовый, медный, цинковый, медно-свин-
цовый, медно-цинковый и т. д.
Конечными продуктами каждого цикла флотации являются
получаемые концентраты и хвосты. Все остальные продукты,
циркулирующие внутри схемы, называются
промежуточными продуктами или промпродуктами. Каждый
цикл может включать в себя одну или несколько операций доиз-
мельчения промпродуктов. Промиродукты обычно возвращаются
в предыдущую операцию, но они могут направляться и
в другие операции. Во всех случаях промпродукты стремятся
подать в ту операцию, в которую поступает материал, примерно
с таким же содержанием извлекаемых минералов. В ряде
210
случаев промпродукты перерабатываются в отдельном цикле.
Это может быть связано, например, с большим содержанием
в них шламов, необходимостью создания особых условий доиз-
мельчения и флотации и с рядом других причин.
Развернутая схема цикла флотадии, состоящего из
основной, двух контрольных и двух перечистных операций,
представлена на рис. 13.1. В схеме флотации все пенные продукты всегда
изображаются слева, а камерные — справа от наименования
операции флотации. В любой операции схемы минеральные
частицы должны иметь выход в оба конечных продукта цикла
флотации, чтобы случайно попавшие в операцию частицы могли
перейти в результате перечистных и контрольных операций
в соответствующий конечный продукт.
13.2. Стадиальность схем флотационного обогащения
Под стадией обогащения понимается совокупность операций
измельчения, классификации и флотации, в результате которых
получается один или несколько конечных продуктов обогащения
(концентраты, хвосты).
Число стадий обогащения зависит от размера и характера
вкрапленности минералов и склонности руды к ошламованию.
По одностадиальным схемам обогащаются лишь руды с
относительно равномерной вкрапленностью и мало шламующиеся.
К таким рудам относятся некоторые богатые медные руды,
шеелитовые, баритовые, флюоритовые, берилловые и сподуме-
новые руды ряда месторождений. При обогащении этих руд по
одностадиальной схеме они измельчаются до необходимой
крупности и обогащаются без дополнительного доизмельчения
каких-либо продуктов флотации, например по схеме рис. 13.К
При неравномерной вкрапленности полезных минералов и
склонности их к переизмельчению применяются
многостадиальные схемы измельчения и флотации. Схемы усложняются с
увеличением сложности характера вкрапленности и склонности
к переизмельчению полезных минералов.
Двух- и трехстадиальные схемы могут быть представлены
следующими тремя принципиальными вариантами.
1. Выделение после относительно грубого измельчения в I
стадии хвостов и бедного концентрата, подвергаемого доизмель-
чению и флотации во II стадии (рис. 13.2, а) или во II и III
стадиях (рис. 13.2,6). По таким схемам обогащаются некоторые
медно-пиритные, полиметаллические, медно-молибденовые и
графитовые руды, в которых полезные минералы тесно связаны
между собой или с небольшим количеством пустой породы, но
их агрегаты могут быть легко отделены от остальной части
вмещающих пород уже при грубом измельчении. Это позволяет
резко снизить стоимость измельчения, поскольку измельчению
до конечной крупности и перефлотации подвергается только
концентрат.
211
Руда
г\Измельчение и
\ Vклассификация I
Медная
флот ац и я
Пиритный
концентрат
Руда
^Измельчение и
^классификация I
Коллект ивная
медно-ци пк о 8а я
флотация
■^Измельчение и , И/-
) классификация U т ^оосты
\ Измельчение и
Q классификация д
Медный
Пере чист на я концентрат
Me Зная
флот а ция
\Измельчение и
) классификациям
Цинковая флотация
Медный
концентрат
ЦинкоЗый
концентрат
Пиритный
концентрат
Рис. 13.2. Принципиальные двухстадиальная (а) и трехстадиальная
схемы флотации с доизмельчением бедного концентрата
7 ■ Руда
^Измельчение и
\J классификация I
свинцоЗая
флотаиия
1 Измельчение и
/^классификация Л
0 Руда
!
г\ Измельчение и
Vy] классификация I
I сЗинцово- медная
флота и ия
Свин цоЗая
флотация
г*\Измельчение и
КУклассифи-
Т нация Л
ЛсЗинцово-медна я
я
\ флот а и и *
w
Свинцовая
флота и и я
i \ | Свинцовый Медный Измельчение и
' I концентрат концен- классифика-
Цинковая тРаю W*m
флот а кия
]
Хвосты
Цинковый
концентрат
цинково - пиритная
флотаиия
Перечистные
операции
цинковая
флот а и и я
Хвосты
Свинцовый
концентрат
Ц инхо вы й Пир ит ны и
концентрат концентрат
Рис. 13.3. Принципиальные двухстадиальная (а) и трехстадиальная
схемы с доизмельчением богатых хвостов флотации
I Измельчение и
О классификация I
6
Основная
флотация
Перечистная
Сгущение
1 h
фЦ классификация Е
\ ^Измельчение
Флотация Я
промпродунта
У
V
Перечцшная
Сгущение
Слад 8
\ оборот
Руда
i
О Измельчение I
Классификация
\ 100%-0,15мм (
Основная
флотация
Хвосты
Меёный
\ концентрат
Рис. 13.4. Дпухстадиальная (а)
чением промпродуктов флотации
Медный
f концентрат
трехстадиальная
Х9сстыу
с доизмель-
2. Выделение после относительно грубого измельчения в I
стадии готового или достаточно богатого концентрата и богатых
хвостов с доизмельчением и флотацией их во II стадии (рис.
13.3, а) или во II и III стадиях (рис. 13.3,6). По таким схемам
могут обогащаться некоторые, например полиметаллические,,
медно-никелевые, медные и другие, руды, в которых наряду
с крупной вкрапленностью имеются тонкие вкрапления
полезного минерала во всей массе вмещающей породы. Применение
стадиальных схем в этом случае является реализацией принципа
«не дробить ничего лишнего» и позволяет резко снизить
переизмельчение полезных минералов, улучшить селективность их
разделения и тем самым повысить технико-экономические
показатели обогащения.
3. Выделение после предварительного измельчения в I
стадии готового концентрата, бедных хвостов и промпродукта
. с большим содержанием сростков, который доизмельчается и
подвергается флотации во II стадии (рис. 13.4,а). В СССР
такие схемы применяются обычно в более сложном исполнении,
когда доизмельчаются и другие продукты флотации (рис.
13.4,6). Такие схемы позволяют избежать переизмельчения,
снизить стоимость обогащения и повысить качество
концентрата.
Таким образом, применение стадиальных схем позволяетл
учесть особенности вещественного состава флотируемых руд и
получить за счет снижения ошламования полезных минералов
технологические, а за счет снижения стоимости измельчения и
повышения извлечения металлов экономические преимущества
по сравнению с одностадиальной схемой обогащения.
2ia
13.3. Схемы с раздельной обработкой
и флотацией песков и шламов
Основной целью применения таких схем при переработке
руд с большим содержанием первичных шламов или быстро
шламующихся в процессе измельчения является
предотвращение вредного влияния шламов при флотации. Причинами их
вредного влияния является следующее:
1. Резкое изменение поглотительной способности пульпы по
отношению к флотационным реагентам. Так как шламы
обладают развитой поверхностью и большой адсорбционной
способностью, то даже незначительное изменение их содержания
приводит к резким изменениям концентрации основных реагентов
в пульпе. Поддержание необходимой концентрации реагентов
в этих условиях становится затруднительным, а при отсутствии
систем автоматического контроля и регулирования практически
невозможным. Недостаточная концентрация собирателя в пульпе
приводит к возрастанию потерь особенно крупных частиц
флотируемого минерала в хвостах, а недостаток депрессора — к
нарушению селекции и ухудшению качества концентрата.
Попытка обеспечить заведомо достаточную концентрацию
собирателя в пульпе за счет больших его расходов также приводит,
как правило, к ухудшению селективности флотации.
2. Неселективная коагуляция шламистых частиц полезного
минерала и минералов пустой породы, что приводит: при
значительном содержании в агрегате частиц полезного минерала —
к переходу его в пенный продукт и загрязнению концентрата
тонкими фракциями пустой породы; при значительном
содержании в агрегате частиц пустой породы — к переходу его в
камерный продукт и возрастанию потерь тонких частиц полезного
минерала в хвостах флотации. В свою очередь, налипание
тонких частиц породы на крупную частицу флотируемого минерала
вызывает гидрофилизацию* поверхности частицы и подавление
ее флотации. Если прилипшие к крупной частице тонкие шламы
полезного минерала достаточно гидрофобны, то пузырек может
к ним прилипнуть. Однако устойчивость слипания пузырька
с частицей минерала будет весьма невысокой — пузырек легко
оторвется, увлекая с собой часть шламового покрытия (см.
рис. 3.15,6). В обоих случаях наблюдается снижение извлечения
крупных частиц флотируемого минерала.
3. «Бронирование» пузырьков тонкими частицами и
изменение прочности пены. При большом содержании тонких частиц
флотируемого минерала в обычных условиях флотации
поверхность пузырьков «бронируется» тонкими частицами, однако
поверхности раздела вода — воздух недостаточно для
эффективной флотации всех гидрофобных тонких частиц. Кроме того,
к «бронированным» пузырькам плохо прилипают флотируемые
частицы нормальной крупности. По этим причинам скорость
флотации как тонких, так и крупных частиц резко уменьшается,
214
Руда
\Измельчение
\б5-70°/о-0рЬми
Т
н л а с с и фи нация
(Шла мы
85-90%-0,ОПмм
Основная флотация
Песни
25-30%'0Шмм
к о н траль пая
ч!ж
Основная
фл о щация
Ко н т р о л ьная
J перечистная
\ Доизмель venue '
\30%-О07**М!И
Ш пе ре ч истная
i
Хвосты
И леречистная Т \<
I — J \90%-О,О7Ьмм
Медный
концентрат
Рис. 13.5. Схема обогащения сульфидных медных руд на Джезказганской
обогатительной фабрике
а потери извлекаемых минералов в хвостах увеличиваются.
Большое содержание слабогидрофобных шламов приводит к
образованию трудноразрушаемой пены, что также вызывает
нарушение селективности флотации.
Вредное влияние шламов при небольшом их содержании
удается предотвратить с помощью реагентов-пеитизаторов,
предотвращающих образование шламовых покрытий на минералах
и пузырьках, дробной подачи реагентов, позволяющей
поддерживать более постоянную их концентрацию в пульпе, реагентов,
снижающих поглотительную способность шламов (сернистого
натрии, гексаметафосфата и др.).
При значительном содержании в руде тонких шламов схема
обогащения может предусматривать предварительное
разделение руды на песковую и шламовую фракции, раздельную их
обработку реагентами в разных режимах и последующую
совместную флотацию обеих фракций. Таким способом,
предложенным В. А. Глембоцким [7], удается учесть различные
физические и физико-химические свойства минералов песковой и
шламовой фракций и оптимизировать их реагентную обработку
перед флотацией.
215
Еще более широкие возможности представляются при
использовании схем не только с раздельной обработкой песковой
и шламовой фракций, но и с раздельной их флотацией. По
таким схемам работают, например, медная Джезказганская (рис.
13.5), медно-молибденовая Сорская и некоторые другие
обогатительные фабрики.
Схема с раздельной обработкой и флотацией песковой и
шламовой фракций позволяет:
практически полностью устранить вредное влияние тонких
частиц сульфидных минералов и концентрацию в шламовой
фракции минералов, склонных к переизмельчению;
осуществить селективную флокуляцию тонких частиц
полезных минералов с помощью реагеитов-флокулянтов или
эмульсии аполярных реагентов;
создать наиболее благоприятные условия флотационного
извлечения полезных минералов как из шламовой, так и
песковой фракций. Например, для флотационного извлечения тонких
частиц могут быть использованы флотация с
минералами-носителями, флотация газами, выделяющимися из раствора,
Руда
ГЛ измельчение I
Классификация 1
Слив
t+2-tf%-0,071+мм
)
Измельчение U
Т
классификация II
\\60%-0f01hMM
Основная медно-
молибденовая флотация
1 переч истная Классификация
к ♦ \Шламы \ }/
Сгущение
класс иф ика ци я
\ 80-85%
\у \-0,074мм
ЛМпере чистные
шзмель-
иение
Основная
флотация пес но в-
К о н т р ильная
Классифи к ация
Г
т LH пере ч истнь/е
}
\
Моизмель-
чение
Коллективный модно-
молибЗеновьш концентрат
Отвальные
хвосты
Рис. 13.6. Схема обогащения медно-молибденовых руд на Балхашской
обогатительной фабрике
216
электрофлотация. В свою очередь, флотация песковой фракции
может быть проведена в специальных аппаратах (машинах
пенной сепарации, кипящего слоя и др.). Кроме того, при
флотации каждой фракции могут быть установлены оптимальные
плотность пульпы, режимы перемешивания, условия аэрации и
пенообразования, время флотации.
Все это позволяет получать максимальное извлечение
металлов, что и является причиной широкого применения на фабриках
схем с раздельной флотацией песков и шламов.
В некоторых случаях, особенно при переработке медных
шламистых руд, хорошие результаты флотационного
обогащения могут быть получены по схеме с дофлотацией песковой
фракции хвостов флотации после их обесшламливания. При
дофлотации доизвлекаются полезные минералы, находящиеся
в сростках с пустой породой и обычно теряемые с отвальными
хвостами обогатительных фабрик. Полученный концентрат
направляется на доизмельчение совместно с иромпродуктами
пли концентратами рудного цикла. Осуществление такой схемы,,
например, на Балхашской (рис. 13.6) и Чорух-Дайронской
обогатительных фабриках позволило значительно сократить потери
меди в хвостах и повысить общее извлечение ее в концентрат
и тем самым комплексность использования руд.
При флотационном обогащении фосфоритов, марганцевых,,
сильно ожелезнепных окисленных полиметаллических руд и
некоторых руд редких металлов получаемые, например в
результате промывки, шламы направляют сразу в отвал, .
несколько извлечение ценных компонентов из них обычно является:
проблемой, решение которой существующими методами
экономически нецелесообразно.
13.4. Схемы коллективной и селективной флотации
При флотационном обогащении многокомпонентных руд,,
например полиметаллических, могут применяться схемы
прямой селективной флотации (рис. 13.7,а),
коллективно-селективные (рис. 13.7, б, в) и коллективной флотации с последующей
селекцией (разделением) коллективного концентрата (рис.
13.7,г). Выбор схемы зависит от содержания полезных
компонентов в руде, их вкрапленности, флотируемости, характера
/вмещающих пород. В каждом конкретном случае он
производится с учетом результатов анализа технологических и
экономических преимуществ различных вариантов схем. .. (
По схеме прямой селективной флотации (см. рис. 13.7, а)
производится последовательное выделение полезных минералов
в отдельные концентраты, что возможно, если подавляемые
в начале флотации минералы поддаются последовательной
активации. Обычно сначала выделяют легкофлотирующиеся
минералы, затем труднофлотирующиеся.
Недостатками таких схем являются:
217
■— необходимость измельчения всей руды до достаточно полного-
раскрытия сростков полезных минералов между собой и с
минералами пустой породы;
__ необходимость установки большого числа флотационных
машин, поскольку основной поток пульпы
последовательно'проходит все циклы флотации;
- большой расход реагентов, поскольку в каждом
последующем цикле необходимо радикально менять не только
соотношение концентраций реагентов в пульпе, но и их номенклатуру,
чтобы избирательно нейтрализовать депрессирующее действие
реагентов и обеспечить активную флотацию только
определенной группы минералов в каждом цикле;
трудность селективной флотации близких по своим
флотационным свойствам минералов, например сульфидов меди и
гнпнца;
трудности осуществления полного в^дооборота на обогати-
iivihimft фабрике.
Отмеченные недостатки схем прямой селективной флотации
являются причиной более широкого распространения на
практике коллективно-селективных схем флотации (см. рис. 13.7, б,_б).
и схем с предварительной коллективной флотацией всех
полезных минералов (см. рис. 13.7, г). Последние особенно
перспективы дли обогащения бедных руд с агрегатной
вкрапленности) полезных минералов. В этом случае основная масса
отвальных хвостов (а иногда и все хвосты) выделяется в
коллективном цикле после грубого измельчения, обеспечивающего
раскрытие сростков агрегатов полезных минералов с пустой
породой. Тонкому измельчению, необходимому для разрушения
агрегатов полезных минералов, подвергается лишь небольшое
количество коллективного концентрата, после десорбции с его
поверхности собирателя.
Применение, коллективно-селективных схем_ и схем с
предварительной -коллект1?внЪй"^Хбтац^ по сравнению
со схемой прямой селективной флотацией руды:
- -снизить затраты на измельчение благодаря возможности
выделения пустой породы в коллективных циклах флотации при
грубом измельчении руды;
.-• сократить фронт флотации в результате сокращения числа
'циклов флотации, через которые проходит основной поток
пульпы. /Так, по схеме на рис. 13.7,6 он проходит три цикла;
по схеме на рис. 13.7, в — два цикла и по схеме на рис. 13.7, г -—
только один цикл флотации;
-снизить эксплуатационные затраты на 30 %;
- осуществить полный водооборот в коллективных циклах
флотации и в результате этого сократить расход реагентов.
Важным достоинством этих схем является также
возможность выделения минералов, близких по флотационным
свойствам, в отдельные коллективные концентраты и применения для
их разделения специальных режимов/Так, например, сульфиды
219.
Руда
Цикл I коллективов и
флота ци и
* 1
Д е с о р if и и я
с о б~и р am ел я
Никл IT коллект ивн о й
флотации
Доиомельчение
Цикл свинцово-медной
флота ции
Дикл разделен и я
свинцово- мед мог о
концентрата
Хвосты I
оизмель чение
Цикл цинпово-пцритнай
флотации
Хвосты Л
\ \
Свинцовый медный I
квнцентоат концентрат "
иикл разделения
цинково -пиритнога
концентрата
f » .
Цинковый Лиритньш
концентрат концентрат
Рис. 13.8. Принципиальная схема обогащения полиметаллических руд
свинца и меди практически всегда выделяются сначала в
коллективный свинцово-медиый концентрат.
В последние годы все чаще используется естественное
различие во флотируемости извлекаемых минералов одного и того
же металла. Например, для полиметаллических руд,
содержащих как легко-, так и труднофлотирующиеся разности
сульфидов цинка и железа, может быть использована схема,
изображенная на рис. 13.8. По этой схеме в первом приеме
коллективной флотации при малых расходах собирателя извлекаются
сульфиды свинца, меди и легкофлотируемые разности
сфалерита и пирита. Поскольку сфалерит и пирит не активированы
солями меди, они довольно легко могут быть отделены от
сульфидов свинца и меди (после десорбции и измельчения) в цикле
свинцово-медной флотации и объединены с цинково-пиритным
концентратом, полученным во втором приеме коллективной
флотации, проводимом при значительных расходах собирателя и
с добавкой медного купороса для извлечения труднофлотиру-
емых разностей сульфидов цинка и железа. Для флотации всех
сульфидов в коллективном цикле пришлось бы увеличить
расходы собирателя и активатора труднофлотируемых минералов,
что значительно затруднило бы последующую селективную
флотацию полученного коллективного концентрата.
220
Трудности применения коллективных и
коллективно-селективных схем флотации связаны в основном с подготовкой
коллективного концентрата к разделению, включающей операции
десорбции собирателя с поверхности минералов коллективного
концентрата и доизмельчение его до необходимой крупности.
13.5. Комбинированные схемы
В связи с усложнением вещественного состава руд,
возрастанием трудности их обогащения и необходимостью повышения
комплексности использования сырья все большее применение
находят комбинированные схемы. В таких схемах используются
или различные методы обогащения, или комбинация методов
обогащения и металлургии. Применение комбинированных схем
позволяет в ряде случаев обеспечить радикальное решение
проблемы извлечения ценных компонентов из некоторых видов
минерального сырья.
Так, например, использование операций выщелачивания и
флотации обеспечивает эффективную переработку труднообо-
гатимых сульфидно-окисленных полиметаллических руд (рис.
13.9). Другим примером является переработка окисленных и
сульфидно-окисленных медных руд методом Мостовича, по
которому окисленная медь выщелачивается, цементируется
железной стружкой н флотируется вместе с имеющимися
сульфидами меди.
\Измельчение и
)классификация
Коллективная флотация
сульфидных и ширенных минералов
свинца, мед и и цинка
Ч
Выщелачивание
окисленных минералов меди и
ц инк а
Очистка злегтролита
от меди и железа
\вементная
1 медь
Гидроокись
\ железа
Флотация
сульфидов
-^Измельчение и
классификация
Электролиз
цинка
Оборотный
Хзлектролит
УСатодныи
у цинк
Разделение коллектив-
ног о сульфидного кон-
ц е н трата
Свинцовый
Медный
Цинковый
Флотация
окисленных свинцовых
минералов
к о н ц е н лп р а т ь/
. Окисленный
^свинцовый
Хвосты
Рис. 13.9. Комбинированная схема переработки труднообогатимых
сульфидно-окисленных полиметаллических руд
221
Примерами комбинированных схем, предусматривающих
пирометаллургические и флотационные операции, являются:
процесс сегрегации, включающий предварительный обжиг руды
с последующей флотацией восстановленной до металла меди;
плавка на файнштейн с последующим его флотационным
разделением на никелевой и медной концентраты; флотационное
извлечение меди из шлаков медной плавки [2, 26].
Комбинированные схемы, предусматривающие
электромагнитное обогащение и флотацию, рациональны при обогащении
некоторых типов железных руд; схемы, предусматривающие
операции магнитного, гравитационного и флотационного
обогащения, применяются для доводки некоторых вольфрамовых,
оловянных и других концентратов при наличии в них
сульфидов, апатита и железных минералов.
14. РЕЖИМЫ ФЛОТАЦИИ
14.1. Классификация минералов по флотируемости
Предложенная М. А. Эйгелесом [49] классификация
минералов по флотируемости позволяет сгруппировать их следующим
образом:
I. Неполярные минералы, обладающие высокой естественной
гидрофобностью (самородная сера, графит, каменный уголь,
молибденит, тальк, озокерит), — флотируются аполярными
собирателями, например керосином.
II. Самородные металлы (золото, серебро, платина, медь),
сульфиды цветных, черных и редких металлов и близкие к ним
по флотационным свойствам теллуристые (теллуриды) и
селенистые (селениды) металлы — легко флотируются сульфгид-
рильными собирателями.
III. Окисленные минералы цветных металлов (карбонаты
свинца, цинка и меди, сульфат свинца, окислы меди и др.) —
флотируются сульфгидрильными собирателями после сульфи-
дизации.
IV. Соли щелочноземельных металлов, не содержащие крем-
некислоты и карбонаты черных металлов (апатит, фосфориты,
кальцит, шеелит, флюорит, барит, сидерит, родохрозит и др.)» —
легко флотируются оксигидрильными собирателями без
предварительной активации.
V. Окислы железа, марганца, олова (магнетит, гематит, пси-
ломелан, пиролюзит, касситерит и др.)—флотируются без
активации оксигидрильными собирателями, но значительно хуже,
чем минералы предыдущей группы.
VI. Силикаты и кварц. Многие из минералов этой группы
флотируются анионными оксигидрильными . собирателями.
Иногда требуется предварительная активация, если на гранях
или поверхностях разлома минерала ие имеется достаточного
222
количества катионов, способных образовывать с анионным
собирателем труднорастворимые или прочные комплексные
соединения. Такие минералы часто лучше флотируются катионными
собирателями. Более детальное исследование флотационных
свойств минералов данной группы приведет в дальнейшем
к разделению ее на отдельные подгруппы.
VII. Растворимые в воде минералы: галит NaCl, сильвин
КС1, карналлит KCbMgCl2-6 Н20, бишофит MgCl2-6 H20 и
др. Минералы этой группы флотируются из насыщенных
растворов, называемых «маточником» или «рапой», обычно
катионными и гораздо реже анионными собирателями.
14.2. Флотация руд, содержащих минералы
с высокой естественной гидрофобностью
Общая характеристика особенностей флотационного
обогащения руд с аполярными минералами. Высокая естественная
гидрофобность таких минералов, как графит, самородная сера,
каменный уголь, обусловлена особенностями их
кристаллических структур (см. рис. 1.3, а, рис. 6.6). Краевой угол
смачивания составляет: для каменного угля 60—90°, для самородной
серы 75—85°, для графита 55—75° и для талька 52—69°.
Довольно большие колебания значений краевого угла смачивания
являются следствием изменения соотношения на макроповерх-
иости минерала высокогидрофобных плоскостей спайности и
гидрофильных торцовых участков кристалла.
Несмотря на высокую естественную гидрофобность угля,
самородной серы, графита и талька, термодинамическая
вероятность их флотации реализуется плохо. Эффективная флотация
минералов достигается только после подачи аполярных
собирателей, которые закрепляются на поверхности в виде линз или
капелек, обеспечивая прорыв гидратной прослойки между
частицей и пузырьком при их встрече и способствуя
многократному упрочнению образовавшегося контакта. Флотационная
активность аполярного собирателя обычно тем выше, чем больше
содержание в нем непредельных соединений и ароматических
углеводородов, чем лучше он эмульгирован и чем выше исходная
гидрофобность флотируемых частиц.
Исходная гидрофобность, определяемая соотношением
гидрофобных и гидрофильных участков на поверхности, зависит от
крупности кристаллизации, совершенства кристаллических
форм, степени окисления и характера включений пустой
породы.
При неблагоприятных условиях (например, если графит и
тальк представлены скрытокристаллическими разностями,
а уголь довольно сильно окислен с образованием
карбонильных = С =Ои карбоксильных =СООН групп, активно
взаимодействующих с водой) доля гидрофильных участков на
поверхности становится значительной и средняя гидрофобность частиц
223
невысокой. Для гидрофобизации полярных участков
поверхности необходима добавка гетерополярных реагентов. Поэтому
в общем случае наиболее эффективной при флотации
рассматриваемой группы минералов является смесь или сочетание двух
типов реагентов: поверхностно-активных веществ с гетерополяр-
ным строением молекул и малорастворимых в воде аполярных
веществ. Оптимальное соотношение их определяется
характером перерабатываемого сырья и устанавливается
экспериментально. Расход смеси составляет 0,5—1,5 кг/т.
Помимо гидрофобизации полярных участков поверхности
флотируемых частиц, добавка поверхностно-активных реагентов
улучшает эмульгирование аполярных реагентов, аэрацию
пульпы и образование устойчивой иены. Лучшие результаты
получены с реагентами, имеющими в качестве полярной группы
гидроксил (пенореагент, синтетические спирты, кубовые остатки
производства бутилового спирта, метилизобутилкарбинол,
сосновое масло и др.). Можно полагать, что их закрепление на
полярных участках минеральной поверхности обусловлено
образованием водородных связей.
Минералы первой группы обычно совместно в рудах не
встречаются, поэтому задача обогащения сводится к отделению
их от пустой породы при минимальных расходах на измельчение
и реагенты и минимальном времени флотации.
К технологическим особенностям флотационного обогащения
руд, содержащих минералы первой группы, следует отнести
следующее:
1. Необходимость преодоления вредного влияния
тонкодисперсных глинистых шламов, гипса, растворимых органических
веществ или битуминизированной пустой породы. Для этого
применяются жидкое стекло (до 2 кг/т), пирофосфат, тринат-
рийфосфат или сульфитцеллюлозный щелок. Они пептизируют
тонкие шламы и подавляют флотацию битуминизированной
пустой породы.
2. Необходимо учитывать, во-первых, что более мягкие
полезные минералы при измельчении режутся острыми краями
твердых зерен пустой породы и это может привести к
ухудшению качества конечного, например графитового, концентрата.
Во-вторых, при взаимотрении естественно гидрофобных
минералов и пустой породы в мельницах минералы пустой породы
приобретают флотационную активность, что снижает
эффективность перечистных операций флотации и ухудшает качество
получаемых концентратов.
3. Сера, графит, уголь и тальк имеют относительно малую
плотность, поэтому максимальная крупность флотирующихся
зерен для них несколько выше, чем для других минералов и
составляет при флотации в машинах механического типа около
0,5 мм. Учитывая это, а также «монометаллический» характер
руд, содержащих названные минералы, весьма перспективным
следует считать применение для их флотации машин пенной се-
224
парации. В этом случае крупность сфлотированных зерен может
достигать нескольких миллиметров, что позволяет получать
максимальный выход, например, более ценных сортов
крупночешуйчатого графита.
Учитывая особенности вещественного состава, при флотации
угля и серных руд применяют простые схемы, а при флотации
графита и талька — многостадиальные схемы с многократным
доизмельчением и дофлотацией получаемого концентрата.
Серные концентраты содержат 60—90 % серы,
графитовые — 93—95 % углерода, угольные — не более 7,5—9,5 % золы,
тальковые — 88—89% нерастворимого остатка. Извлечение
полезных минералов составляет: из серных руд 80—95 %, из
графитовых руд и каменных углей более 90%, из тальковых руд
82—83%.
Наибольшие масштабы в настоящее время приобрела
флотация угольной мелочи. Затраты на переработку 1 т составляют
в среднем по Донбассу от 70 коп. до 1 руб. 50 коп., по
Кузбассу— 51 коп. и по Карагандинскому бассейну — 48 коп. [7].
Флотация графитовых руд. Флотируемость- графита зависит
от крупности его кристаллов, характера примесей и степени
окисления поверхности. Флотацию его проводят обычно с
керосином и пенообразователем (сосновым маслом, Т-66 и др.)
в щелочной (рН 8—10) или кислой (рН 4—5) среде,
создаваемой содой, известью или серной кислотой [44]. Для депрессии
минералов породы, особенно карбонатных, применяют жидкое
стекло; для депрессии флотации слюды (при наличии ее
в руде)—фтористый натрий. Чешуйчатая форма графита,
относительно небольшая его плотность и естественная гидрофоб-
ность позволяют флотировать крупные частицы.
В зависимости от крупности кристаллизации графита
графитовые руды делятся на чешуйчатые (с размером чешуек
больше 0,2 мм), плотнокристаллические (меньше ОД мм) и
скрытокристаллические (меньше 0,001 мм). Легче всего
обогащаются чешуйчатые руды, гораздо труднее — плотно — и
скрытокристаллические. Более медленная флотация зерен скрыто-
кристаллического графита, помимо большей гидрофильности
их поверхности (по сравнению с крупными чешуйками), часто
бывает обусловлена также присутствием в скрытокристалличе-
ских рудах органических веществ, оказывающих депрессирую-
щее действие на флотацию графита.
К схемам флотационного обогащения графитовых руд
предъявляются три основных требования: минимальные расходы на.
измельчение, максимальный выход крупночешуйчатого графи-,
тового концентрата, низкая зольность концентратов. С целью
сокращения расходов на измельчение основная масса отвальных!
хвостов выделяется уже в операциях основной (рудной)
флотации после грубого измельчения (до 45—55 % класса —0,074 мм)»;
поскольку для графитовых руд характерна агрегатная вкрап-,
ленность. Измельчению до конечной крупности подвергается
8 Заказ № 1957
225
только концентрат. Чтобы увеличить выход более дорогого
и дефицитного крупнозернистого графитового концентрата
(класса ±0,2 мм), используются многостадиальные схемы
с удалением породы по мере ее освобождения из сростков с
графитом. Для снижения содержания золы в концентратах
используется большое число перечистных операций, основанных на
хорошей флотируемости графитовых частиц. Обычно черновой
концентрат подвергается шести-восьми перечистным операциям
и двум-четырем операциям доизмельчения.
Готовый концентрат, содержащий 93—95 % углерода, с
извлечением более 90 % сушат и классифицируют на отдельные
сорта по крупности. Отходы флотации используют обычно как
низкосортный литейный графит.
Флотация серных руд. Серные руды характеризуются обычно
довольно крупной вкрапленностью (более 0,1 мм) самородной
серы при содержании ее более 4 %. В качестве собирателя при
флотации самородной серы применяют керосин и другие
нефтепродукты @,5—1,5 кг/т), в качестве пенообразователя —
сосновое масло, Т-66 @,1—0,4 кг/т), в качестве депрессоров
породы и вредных примесей (гипса, глинистых шламов и
битуминозных веществ)—жидкое стекло (до 2 кг/т) и фосфаты
(пирофосфат Ыа4РгОт или тринатрийфосфат Na3P04).
После измельчения до 0,25—0,8 мм руда подвергается
основной и контрольной флотации при содержании твердого
в пульпе около 25%, а полученный черновой концентрат —
одной— четырем перечистным (при содержании твердого в пульпе
17—20%). Крупная фракция хвостов (±0,074 мм)
используется в сельском хозяйстве для подкисления почв [14]. В
зависимости от содержания в руде вредных примесей серные
концентраты содержат 60—90% серы при извлечении ее 80—95%.
Серные концентраты направляются в автоклавную плавку для
получения «комовой» серы. Чтобы повысить эффективность
плавки, стремятся к получению крупнозернистых концентратов.
Хвосты плавки возвращаются на фабрику.
Флотация угля. Флотации подвергаются шламы (крупностью
менее 0,5 мм) гравитационного обогащения всех коксующихся
углей и значительной части газовых (например, в Донбассе)
углей и антрацитов [43]. При этом обеспечивается не только
их обогащение, но и своевременный вывод тонких илистых
частиц из водно-шламовых схем переработки углей.
Различные ингредиенты угля обладают разной флотиру-
емостью. Блестящие разновидности их — витрен и кларен —
флотируются лучше матовых — дюрена и фюзена. Поэтому при
высоком содержании блестящих ингредиентов флотируемость
органической массы угля увеличивается. Флотируемость углей
ухудшается с возрастанием степени их окисленности,
увеличением содержания минеральных примесей (и особенно
глинистого материала), а также степени их дисперсности и
равномерности распределения в органическом веществе.
226
Эффективность флотационного обогащения углей в
значительной мере определяется подготовкой пульпы к флотации,
в процессе которой решаются следующие задачи:
получение однородного материала путем смешивания всех
потоков шламов, поступающих на флотацию, в специальной
емкости (демпфере) или радиальном сгустителе;
предотвращение поступления на флотацию крупнозернистого
материала (более 0,8—0,5 мм), обычно теряемого в хвостах
флотации, путем классификации шлама в низконапорных
классификаторах— гидроциклонах, на дуговых или вибрационных
грохотах;
обеспечение необходимого контакта пульпы с реагентами,
отличающимися высокой вязкостью и малой растворимостью,
в специальных аппаратах или устройствах (например, агрегате
«Каскад»).
Почти на всех фабриках СССР в качестве собирателей
применяют аполярные ароматические реагенты ААР-1, ААР-2, реже
керосин или его ароматизированную фракцию АФ-2, а в
качестве пенообразователей — высшие спирты, масло X и пеноре-
агент. За рубежом в качестве аполярных реагентов используют
газовые, дизельные, парафиновые и нефтяные масла, креозоты
и дистиллированные каменноугольные масла.
Поверхностно-активными реагентами служат крезолы, сосновое масло, фенолы,
ксиленолы, мстилизобутилкарбинол [43].
Расход аполярных реагентов колеблется от 500 до 1800 г/т,
гетерополярных — от 20—70 г/т (спирты) до 270 г/т (масло X).
Изменение соотношения расходов аполярного и гетерополярного
реагентов является одним из средств регулирования процесса
и качества продуктов обогащения. Например, снижение
зольности концентратов достигается увеличением доли собирателя.
Повышение степени окисленности исходного шлама и его
разбавления требует, наоборот, увеличения доли гетерополярного
пенообразователя.
Для шламов с высоким содержанием глинистого материала
целесообразна дробная подача реагентов, которая на фабриках
ограничивается обычно двумя или тремя точками: 70—75 %
общего расхода реагентов подается в узел подготовки пульпы
к флотации, остальное количество — в камеры флотационных
машин. Повышение эффективности действия и снижение
расхода реагентов достигаются при подаче их в виде гидрозолей
или аэрозолей, для устойчивости которых используют
добавки— эмульгаторы [31]. Снизить расход высокомолекулярных
и вязких реагентов и повысить эффективность их действия
можно, повысив температуру пульпы до 27—28 °С [43].
Полезными оказываются предварительная обработка аполярного
собирателя олефинами, воздействие на него ионизирующего
излучения, добавки сернистого натрия (при флотации окисленных
углей и антрацитов), предварительная аэрация пульпы с целью-
образования в ней «активирующих» микропузырьков [45].
8*
22Г,
Оптимальная плотность пульпы при флотации углей зависит
fc основном от их зольности, крупности, дисперсности
минеральных примесей, наличия глинистого материала. При обогащении
шламов легкой и средней флотируемости содержание твердой
фазы в пульпе составляет 120—200 г/л. При большом
содержании в шламах глинистого материала плотность пульпы
понижается до 100 г/л и менее. Флотация таких пульп сопровождается
снижением производительности флотационных машин и
увеличением расхода реагентов, однако при этом значительно
улучшаются селективность процесса и качество концентрата
вследствие уменьшения механического выноса илистого материала
в пенный продукт [43].
Степень влияния рН пульпы на результаты флотации углей
разной стадии метаморфизма зависит от наличия на их
поверхности функциональных групп. Показатели обогащения углей
марки К обычно незначительно улучшаются, углей марки Г
ухудшаются, а углей марки А практически не изменяются с
повышением щелочности пульпы [31]. Несмотря на весьма
разнообразный состав природных и оборотных вод, значение рН на
обогатительных фабриках не выходит за пределы 7,5—8,2 [43].
На большинстве фабрик применяют «прямые» схемы
флотации, по которым пенный продукт всех камер объединяют
в общий концентрат, а хвосты удаляют из последней камеры.
При флотации угольных шламов повышенной зольности (более
18—20%) наиболее распространенной (например, в ФРГ)
является схема с перечистной флотацией концентрата последних
камер. Гораздо реже используется схема с перечистной
флотацией всего концентрата основной флотации.
Технологические показатели флотации колеблются в
широких пределах в зависимости от общей зольности исходного
питания, шламуемости угля и содержания в нем наносной
золы. Зольность концентрата составляет 2,5— 10 %, а хвостов —
60—80 % [43].
Флотация талька. Исходным сырьем для получения
флотационных тальковых концентратов являются тальковые породы
(например, талькомагнезитовый камень), содержащие
серпентин, хлорит, магнезит, доломит и 45—75% талька Mg5(OHJ
[Si^io]. Качество получаемых концентратов определяется
чистотой, оцениваемой содержанием нерастворимого остатка,
белизной, крупностью и содержанием железа, придающего тальку
желтоватый цвет. В зависимости от качества тальковые
концентраты используются как наполнители при производстве
бумаги, резины или электроизоляционных материалов.
Необходимая крупность измельчения при флотации составляет около
0,15 мм. Технологические показатели несколько улучшаются
при стадиальном обогащении. В качестве собирателя
используются обычно керосин (до 200 г/т), в качестве
пенообразователя— сосновое масло (до 100 г/т). Наиболее гидрофобные
образцы чешуйчатого талька могут флотироваться одним пено-
228
образователен в кислой среде, создаваемой серной кислотой.
Волокнистые модификации талька и плотные разновидности
требуют обязательного применения собирателя. Трудно
флотируется так называемый окисленный тальк, поверхность
которого загрязнена окислами железа. Применение небольших
количеств серной кислоты для снижения рН пульпы до 6—6,5
улучшает селективность флотации талька.
Схемы флотации состоят из основной, контрольной и двух-
трех перечистных операций флотации и обеспечивают
содержание в концентрате 88—89 % нерастворимого остатка при
извлечении 82—83 % талька. Хвосты тальковой флотации могут
служить сырьем для получения из них путем жирнокислотной
флотации кондиционных магнезитовых концентратов [7].
14.3. Флотация сульфидных руд
и самородных металлов
Сульфидные руды являются основным источником получения
цветных металлов. Их флотация наиболее изучена, а
разработанные технологические режимы позволяют в большинстве
случаев добиться наиболее целесообразного распределения
основных металлов и сопутствующих им элементов (кадмия, селена,
теллура, рения, серебра, золота и др.) в концентраты или
продукты, из которых они наиболее эффективно извлекаются в
металлургическом производстве. Это позволяет повысить
извлечение всех металлов и добиться комплексного использования
рудного сырья.
Флотация медных и медно-молибденовых руд. К
промышленным относятся руды, содержащие более 0,35—0,4 % меди.
Медь в рудах в основном представлена халькопиритом CuFeS2,
борнитом CuBFeS4, халькозином C112S и небольшим количеством
E—15%) окисленных медных минералов. Неизменным
спутником являются сульфиды железа (пирит FeS2 и пирротин FeS),
содержание которых в рудах изменяется от нескольких
процентов (в медных порфировых рудах) до 65—85 % (в медистых пи-
ритах). С пиритом часто ассоциируют золото, серебро и
металлы платиновой группы [2]. Практически единственным
извлекаемым молибденовым минералом в медных рудах является
молибденит.
Все обогатительные фабрики, перерабатывающие медные и
медно-молибденовые руды, используют сульфгидрильные
собиратели: ксантогенаты, дитиофосфаты, тионокарбаматы и др.
Карбоновые кислоты, мыла, ал кил сульфаты, амины, также
хорошо флотирующие все сульфиды, гораздо менее селективны
по отношению к минералам пустой породы, чем
сульфгидрильные собиратели, и поэтому пока не нашли практического
применения.
Сульфидные минералы меди обладают высокой
флотационной активностью в широком диапазоне рН: от 6 до 14. Причем
229
вторичные сульфиды меди флотируются лучше первичных и де-
прессируются при более высоких значениях рН пульпы. Это
объясняется тем, что минимальная необходимая для полной
флотации вторичных сульфидов концентрация собирателя
значительно меньше, чем для халькопирита.(Когда концентрация,
-например, ксантогената в пульпе равна Только минимальной
необходимой для халькопирита, она уже в несколько раз
превышает ее для вторичных сульфидов меди, что приводит к
большей вероятности закрепления на них собирателя, гидрофобиза-
ции поверхности и флотации.репрессирующее действие на
флотацию сульфидов меди оказывают цианидные, сульфидные и
гидроксильные ионы.
Отличительной особенностью флотационного поведения
сульфидов железа по сравнению с сульфидами меди является
гораздо более высокая их чувствительность к депрессирующему
действию цианидных и гидроксильных ионов, что используется
на практике. "
Особенности гидрофобизации и флотации молибденита
близки к таковым для минералов, обладающих естественной
гидрофобностью, к которым он относится в силу особенностей
своего кристаллического строения. Поскольку доля поверхности
сильно гидрофобных плоскостей спайности, обнажающихся при
измельчении, обычно во много раз больше поверхности
гидрофильных торцовых участков чешуек молибденита, для его
эффективной флотации достаточно небольших расходов аполяр-
ного собирателя. Обычные подавители сульфидов на
молибденит (при обычных расходах)практически не действуют. В то же
время молибденит депрессируется органическими коллоидами —
крахмалом и декстрином, тонкими шламистыми частицами
слоистых алюмосиликатов (сланцев, талька и др.), обладающих
гидрофобностью по плоскостям спайности, и гидрофобизирован-
ными шламами сульфидных минералов (например,
халькопирита). С увеличением степени измельчения возрастает доля
торцовых участков и необходимость их гидрофобизации ионоген-
ными собирателями. Одновременно возрастает зависимость
флотируемое™ гидрофобизированпого таким образом молибденита
от наличия и концентрации в пульпе обычных депрессоров.
Например, если в качестве ионогешюго собирателя применяется
дитиофосфат, то депрессирующее действие реагентов на
молибденит возрастает в следующем порядке: едкий натр, феррици-
анид натрия, сернистый натрий, цианид натрия [29].
Отмеченные особенности флотационного поведения
минералов меди, железа и молибдена используются при флотации
РУД [2].
Флотационное обогащение медных и медно-молибденовых
руд производится по разветвленным технологическим схемам.
Отвальные хвосты удается выделить после сравнительно
грубого измельчения руды (до 50—65% класса — 0,074 мм)
вследствие агрегатной вкрапленности полезных минералов. Чер-
230
•ювде концентраты или промпродукты (или и те и другие) для
р&сцрытш сростков перед следующей стадией обогащения доиз-
цельчаются до 85—95 % класса — 0,074 мм. В настоящее время
Я*ХОдят все более широкое применение схемы с раздельной
флотацией песковой и шламовой фракций и с дофлотацней
песцовой фракции хвостов [2, 25].
Основной задачей при обогащении медных и медно-пирит-
яых руд является отделение сульфидов меди от сульфидов
железа, представленных обычно пиритом. Для депрессии сульфи- f
дов железа (пирита) применяют известь, иногда с небольшими '
добавками цианида, не вызывающими депрессии флотации
медных минералов. Режим селективной флотации зависит от
содержания в руде пирита. Прямая селективная флотация (рис.
14.1, я) с получением в хвостах медной флотации готового пи-
ритного концентрата возможна, если содержание пустой породы
В руде не превышает 10—15%. В иных случаях кондиционный
пнритный концентрат может быть получен или путем
последовательной селективной флотации сульфидов меди и железа
(рис. 14Л,б), или в результате разделения коллективного
медно-пиритного концентрата (рис. 14.1,<з). Медно-молибдено-
вые руды обычно содержат небольшое количество пирита, и
их флотация производится по общепринятой схеме (рис. 14.1,г),
предусматривающей коллективную флотацию медных
минералов и молибденита и последующую селективную флотацию
медно-молибденового концентрата.
коллективная медно-пиритная или медно-молибденовая
флотация" осуществляется в нейтральной или слабощелочной среде *
при содержании твердого в пульпе 25—35%. Слабощелочная |
^£Деда создается или * содой, или сернистым натрием @,1— ,
0,3 кг/т), подаваемым Для активации флотации окисленных мед-
~"йых минералов. Повышению качества коллективного
концентрата и извлечению в него металлов способствует применение
депрессоров пустой породы —''жидкого стекла, гексаметафос-
фата, КМЦ и других реагентов. Для усиления действия сульф-*
гидрильного собирателя и более полного извлечения сростков и ^
молибденита используются добавки аполярных масел, подава-,..
емых в виде эмульсии. В качестве пенообразователей в этом
случае применяют спиртовые реагенты (сосновое масло, ОПСБ,
метилизобутилкарбинол), пенообразующее действие которых
изменяется незначительно в присутствии аполярных
собирателей.
За последние годы возросло применение при коллективной
медно-молибденовой флотации более селективных собирателей:
содового дитиофоефата или этилового ксантогената обычно
ji сочетании с диксантогенидом, минереком, тиоангидридом
ксантогеновых кислот (например, СЦМ-2), тиоэфиром ксанто-
геновых кислот (например, реагентом S-3302) или диалкилтио-
яокарбаматом (например, реагентом Z-200). Такие сочетания,
относительно слабо флотирующие пирит, позволяют увеличить
231
«раАечение молибдена в коллективный концентрат и
обеспечивают более эффективное его последующее разделение на мед-
ный и молибденовый концентраты.
Разделение сульфидов меди и сульфидов железа при
селективной флотации руды (см. рис. 14.1, а, б) или коллективного
медйо-пиритного концентрата (см. рис. 14.1, в) производится
в сильнощелочной среде (рН 11—12), создаваемой известью.
Небольшие добавки цианида оказываются необходимыми
только в следующих случаях {2]:
е!сли в руде или коллективном концентрате содержится
молибденит и во избежание его депрессии нельзя повышать рН
пульпы более 11,5;
если часть сульфидов железа представлена арсенопиритом^
недостаточно хорошо депрессирующимся в известковой среде;^'
ерли сульфиды железа сильно активированы, что обычно
наблюдается при наличии в руде сажистых образований
вторичных сульфидов меди.
Готовый пиритный концентрат, содержащий 49—50 % серы
и не! более 1 % суммы цветных металлов, получается по схеме
на рис. 14.1, в в виде хвостов селективной флотации. Данная
схема, обеспечивающая извлечение пирита до 80%,
экономичнее рхемы, предусматривающей флотацию пирита из хвостов
медной флотации (см. рис. 14.1,6). Трудности получения пирит-
яого' концентрата по последней схеме обусловлены
необходимостью понижения рН всей рудной пульпы для активации
депонированного известью пирита. Для этого можно
использовать ; серную кислоту, углекислоту отходящих дымовых газов
Металлургических заводов, кремнефтористый натрий,
сернокислое (Железо или другие реагенты, снижающие значение рН
лулЦш до 8—9. Иногда лучшие результаты дает
предварительное Сгущение с последующим разбавлением свежей водой или
кислыми рудничными водами.-. При флотации медно-молибде-
иоаык руд на ряде фабрик самостоятельный пиритный
концентрат Получают из хвостов перечистной флотации коллективного
мед*ф-молибденового концентрата.
Иногда пиритные концентраты содержат заметные
количества кобальта, минералы которого (линнеит, кобальтин, смаль-
тин й др.) обычно тесно связаны с сульфидами железа. Если
они фободны, то их можно сфлотировать из пиритного или
коллективного медно-пиритного концентрата, депрессируя сульфиды
меди и железа сернистым натрием C—5 кг/т).
Дйя разделения коллективных медно-молибденовых
концентратов используются методы, основанные на депрессии как
молибденита, так и сульфидов меди и железа [2, 25].
Так, на ряде зарубежных фабрик («Артур», «Магна», «Силь-
вер Белл») при селективной флотации медно-молибденового
ковдейтрата депрессируют молибденит декстрином. Для
повышения качества конечного концентрата камерный молибденсо-
Д«ряшщий продукт обезвоживается и подвергается селектив-
! 233--
ному обжигу при температуре около 300 °С, после чего из него
флотируется молибденит.
На других обогатительных фабриках для разделения медно-
молибденового концентрата применяются методы, "основанные
на депрессии сульфидных минералов меди и железа и активной
флотации молибденита. Депрессия флотации сульфидов медиг
и железа при этом достигается с помощью следующих
реагентов: " ~
сульфида Na2S, гидросульфида NaHS натрия или их смеси
в соотношении 1:1. Разделение происходит в сильнощелочнойг
среде (рН 11—12) при высоких расходах реагента B—20 кг/г
коллективного концентрата),- обеспечивающих высокую
концентрацию сульфидных ионов в пульпе. Благодаря этому
достигается десорбция собирателя с поверхности сульфидов меди
и железа и депрессия их флотации;
Ноукс, представляющего собой продукт взаимодействия
P2S5 с NaOH. Механизм депрессирующего действия
сульфидных ионов в данном случае дополняется гидрофилизирующим
действием фосфатных ионов при закреплении их на
поверхности депрессируемых минералов. На возможность сокращения
расхода сернистого натрия при добавлении фосфатов
(например, тринатрий фосфата) указывал С. В.. Дуденков. Средний-
расход реагента Ноукс составляет около 5 кг/т коллективного
концентрата. Значение рН при разделении 8—10,5;
Анимол Д, представляющего собой продукт взаимодействия
AS2O3 с Na2S, в результате которого образуются сульфидные
и арсенатные ионы. Механизм депрессирующего действия
данного реагента аналогичен реагенту Ноукс. Средний расход
реагента Анимол Д 9—10 кг/т коллективного концентрата;
цианида @,5 кг/т) совместно с цинковым купоросом или
ферроцианидом A —1,5 кг/т) в слабощелочной среде (рН 7—8);
смеси сульфита натрия с медным купоросом при расходе
каждого реагента около 3 кг/т коллективного концентрата.
Выбор режима депрессии зависит от вещественного состава
коллективного медно-молибденового концентрата, типа
собирателя в коллективном цикле и флотоактивности минералов^ Чем
больше меди в концентрате представлено вторичными
сульфидами, выше степень окисленности минералов, больше сильного»
собирателя на поверхности минералов исходного концентрата,,
тем труднее получить хорошие технологические показатели. За
редким исключением высокая эффективность разделения медно-
молибденовых концентратов по одному из перечисленных
способов достигается только после предварительного разрушения
большей части собирателя [2] с помощью:
окислительной пропарки сгущенного коллективного
концентрата при температуре 85—93 °С в течение 1—4 ч в известковой
среде при расходе извести 0,8—1,2 кг/т концентрата. При этом
часто возникает необходимость дополнительной подачи в
пропарку кислорода или воздуха. Окисление и разрушение ксан-
234
тогената сопровождается одновременным окислением поиррхио-
сти депрессируемых сульфидов меди и железа;
окислителей, таких, как перекись водорода @,5—1 кг/т)
<; гипохлоритом (около 2 кг/т) или перманганат натрия (до
10 кг/т концентрата);
низкотемпературного обжига медно-молибденового
концентрата. ^
На большинстве фабрик в настоящее время используется
окислительная пропарка, поскольку применение окислителей
сильно усложняет процесс, а обжиг является неудобной и
дорогостоящей операцией.
При незначительной степени окисления минералов хорошие
результаты могут быть получены при совмещении подогрева
и селективной флотации с использованием сернистого натрия.
В этом случае пульпа подогревается до 70—80 °С острым
паром в каждой камере флотационной машины. Флотация зерен
молибденита осуществляется на пузырьках пара. Внедрение
такой технологии на Балхашской фабрике позволило снизить
расход сернистого натрия с 21 до 3 кг/т коллективного
концентрата при некотором улучшении технологических показателей
селективной флотации.
Дри селективной флотации медно-молибденового
концентрата основная и контрольная операции флотации проводятся
в плотных пульпах, содержащих иногда до 40 % твердого, а пе-
речистные операции флотации молибденового концентрата
(число которых может достигать 7—12)—в разбавленных,
содержащих до 12—15 % твердого.
В молибденсодержащих рудах в качестве сопутствующих
компонентов часто присутствуют висмут и рений. Если рений
обычно находится в виде изоморфной примеси в молибдените,
то висмут представлен сульфидными формами (например,
висмутином Bi2S3) и самородным висмутом, легкофлотируемыми
■сульфгидрильными собирателями и переходящими в
коллективный медно-молибденовый концентрат. Поскольку сернистый
натрий или сульфит натрия в сочетании с медным купоросом,
применяемые при разделении медно-молибденовых
концентратов, депрессируют флотацию висмутсодержащих минералов,
они переходят в камерный продукт селекции,
представленный обычно сульфидами меди и железа. Селективная флотация
с получением отдельного висмутового продукта может быть
осуществлена путем:
депрессии висмутовых минералов сернистым натрием (при
рН около 11) и флотации сульфидов меди и железа катионным
•собирателем A00—150 г/т);
флотации висмутовых минералов сульфгидрильным
собирателем при депрессии сульфидов меди и железа цианидом.
При тонком прорастании и присутствии меди в висмутине
в виде изоморфной примеси целесообразно.применение
комбинированного метода, включающего операции выщелачивания и
•235
V7
w
JO
20
10
wcj/dffl
I
•
ol
•P
>
p /^
/•O
•
• /
®J
w
20
Рис. 14.2. Зависимость между
производительностью фф фабрики и
производительностью Qp труда
одного рабочего на медных A), мед-
но-молибденовых B) и медно-цин-
ковых C) фабриках
цементации висмута на
железе.
При 'переработке медно-
молибденовых руд извлечение
меди в медный концентрат,
содержащий 21,5—49 % меди,,
составляет 80—95%.
Молибденовые концентраты
содержат 51—58 % молибдена при
извлечении его 50—86 %. При
переработке медных руд
содержание меди в концентрате
составляет 21,5—34,3 % при
извлечении в концентрат 88—
91%.
Медные и медно-молибде-
новые обогатительные фаб-
q^mbicj/cym Ри™ характеризуются наибо-
ф лее высокой
производительностью труда на одного
рабочего в год. Зависимость ее от
пр оизводител ьности фабрики
приведена на рис. 14.2.
Флотация медно-цинковых
пиритных руд, Медно-цинко-
во-пиритные руды, с точки зрения режима флотации, относятся
к одному из наиболее сложных типов. При этом сплошные
(массивные) руды, содержащие более 70 % сульфидов,
являются более труднообогатительными по сравнению с
вкрапленными рудами, содержание сульфидов в которых менее 50%.
Трудности селективной флотации с получением медного,,
цинкового и пиритного концентратов из таких руд обусловлены:
сложным и довольно тесным взаимопрорастанием части
сульфидов, для раскрытия которых требуется очень тонкое
измельчение. Например, для вкрапленных сульфидных руд Урала
необходимая крупность измельчения составляет 90—96 %
класса — 0,074 мм, а для сплошных колчеданных руд 90—94 %
класса —0,043 мм. При существующей технике измельчения
половина потерь меди и цинка в хвостах и разноименных
концентратах приходится на сростки, тогда как другая половина
потерь сульфидов этих металлов обусловлена их
переизмельчением;
близостью флотационных свойств сульфидов меди и
активированных ионами меди сульфидов цинка. В обоих случаях на
поверхности образуются медьсодержащие соединения
собирателя. Избирательное разрушение и предотвращение
образования таких соединений на сульфидах цинка в условиях
селективной флотации требуют тонкой регулировки соотношения
концентраций реагентов в пульпе;
236
высокой флотационной активностью сульфидов железа в
рудах;
неодинаковой флотируомостыо разновидностей сфалерита,
причинами которой могут быть различное содержание в &их
изоморфной примеси железа (от 0 до 20 %), кадмия (до 2,5 %),
индия, галия (например, увеличение содержания изоморфного
железа в сфалерите повышает его чувствительность к депрес-
сирующему действию извести), а также различная степень
природной активации сульфидов цинка в различных участках
одного и того же месторождения.
Кроме того, следует учесть, что сфалерит активируется не
только при подаче медного купороса, но и под действием
катионов тяжелых металлов, находящихся в равновесии с продуктами
окисления или растворения других сульфидов. Особенно
сильная активация его наблюдается в присутствии вторичных
сульфидов и окисленных минералов меди, что является основной
причиной особых трудностей флотационного разделения сульфидов
меди и цинка при переработке руд зоны вторичного обогащения.
Используемые на практике варианты схем селективной
флотации медно-цинково-пиритных руд (рис. 14.3) учитывают
особенности их вещественного состава, относительное содержание
в них вторичных сульфидов меди, степень активации сфалерита
и окисленности сульфидов железа [2].
Так, схема прямой селективной флотации (рис. 14.3, а)
применяется для переработки первичных вкрапленных и сплошных
колчеданных руд, в которых сульфиды цинка мало
активированы.
При резко выраженной их природной активации, большом и
переменном содержании в руде растворимых солей меди и
шламистого материала, сложном взаимопрорастании
разделяемых сульфидов более рациональна схема с предварительной
коллективной флотацией сульфидных минералов (рис. 14.3,6).
При неравномерной вкрапленности сульфидов меди
эффективной оказывается схема с предварительным выделением
части их в готовый концентрат (рис. 14.3,в), а при наличии
в руде легко- и труднофлотируемых разностей сфалерита —
схема с двумя приемами коллективной флотации (рис. 14.3,г).
В первом приеме медные минералы вместе с хорошо
флотирующейся частью сфалерита и пирита отделяются от
остальных сульфидов, что облегчает дальнейшую селективную
флотацию этих минералов.
Характерной особенностью схем при всех вариантах
селективной флотации медно-цинково-пиритных руд является много-
стадиальность измельчения и флотации, использование
межцикловой флотации. Обязательным условием при флотации
рассматриваемых руд является применение слабых собирателей,
малоактивных по отношению к пириту, и «голодных» режимов
дозировки смеси слабого и сильного собирателей (иногда
разных собирателей в разных циклах флотации). Большой расход
237
собирателей в коллективном и медном циклах приводит к
резкому снижению эффективности действия подавителей, несмотря
на увеличение их расхода, и значительному возрастанию потерь
металлов в разноименных концентратах.
Промышленные способы селекции минералов меди и цинка
основаны главным образом на депрессии сульфидов цинка. Для
этого используются различные сочетания следующих реагентов:
цианида, растворяющего ксантогенатные соединения меди на
поверхности сфалерита; сернистого натрия, связывающего ионы
меди и предотвращающего активацию сфалерита; сульфоксид-
ных соединений (сернистой кислоты, сульфита натрия,
бисульфита аммония), изменяющих окислительно-восстановительный
потенциал пульпы и скорость окисления сульфидных ионов и
минералов в пульпе; цинкового или железного купороса,
образующего с гидроксильными, карбонатными, цианидными и суль-
фоксидными ионами в определенных условиях соединения, гид-
рофилизующие поверхность сфалерита; щелочи (соды или
извести). Применяемое сочетание реагентов обеспечивает
депрессию и сульфидов железа. Следует отметить, что содержание
цианида в реагентных смесях постепенно уменьшается и к
настоящему времени на некоторых фабриках полностью
исключено.
Повышению селективности процесса при флотации
сульфидов меди способствуют [2, 25]:
поддержание оптимальных значений рН, равных при
измельчении 7—8 и при флотации 9—10. При таких значениях рН
достигается лучшая флотируемость сульфидов меди и более
полная депрессия сульфидов цинка и железа.
аэрация пульпы перед селективной флотацией медных
минералов, вызывающая окисление и понижение флотируемости
пирита и пирротина. На некоторых фабриках, перерабатывающих
руды, с большим содержанием сульфидов железа, II стадия
измельчения осуществляется в замкнутом цикле с аэраторами.
Для усиления подавления флотации сульфидов железа аэрация
пульпы проводится в известковой среде при рН 9—10.
Гораздо реже при селекции минералов меди и цинка
используются режимы, основанные на депрессии флотации сульфидов
меди.
Первый из них основан на применении ферроцианида, деп-
рессирующего флотацию вторичных сульфидов меди, и
практически непригоден для селективной флотации медно-цинковых
руд, в которых медь представлена первичными сульфидами.
Второй режим основан на различной скорости окисления
сульфидов меди и активированного сфалерита в присутствии
большого количества пирита. Он нашел применение на ряде
японских фабрик, на которых коллективный медно-цинковый
концентрат, содержащий 7—8 % влаги, складируется на
несколько дней на открытом воздухе для окисления медных
минералов, после чего репульпируется горячей водой до 35—40%.
239
Затем из него при температуре 50—60 °С флотируются
сульфиды цинка. Медный концентрат получается в виде камерного
продукта.
В циклах цинково-пиритной и пиритной флотации сульфиды
отделяются от пустой породы. Флотация проводится в среде,
близкой к нейтральной. Для активации сфалерита и депресси-
рованного цианидом пирита подают медный купорос @,1—
0,3 кг/т). Активация сульфидов цинка может быть
осуществлена также с помощью кремне-фтористого натрия.
Флотация сульфидов цинка в циклах разделения цинково-
пиритного концентрата и цинковой флотации предшествует
аэрация пульпы в известковой среде при рН не ниже 11 в
целях депрессии сульфидов железа. Повышение извлечения цинка
и качества цинкового концентрата способствует подогрев
пульпы до 40—65 °С. При наличии в руде или цинково-пирит-
ном концентрате арсенопирита и сильноактивированного пирита
целесообразно для их депрессии добавлять после активации
сульфидов цинка медным купоросом небольшое количество
цианистой соли. В этих условиях флотируемость активированного
сфалерита не только не снижается, но может даже несколько
возрасти [30].
Извлечению сульфидов железа из хвостов цинковой
флотации предшествует нейтрализация действия извести с
использованием методов и реагентов, применяемых при флотации
медных руд.
В ряде случаев не удается избежать загрязнения цинкового
концентрата сульфидами меди и железа. Поэтому для ях обез-
меживания и обезжелезнения используется «обратная»
флотация по методу, предложенному Л, Б. Дебривной. В этом
случае цинковый концентрат подвергают операциям десорбции и
доизмельчения, после чего из него флотируют сульфиды меди
и железа в содовой среде (при рН 9,5—10) с депрессией
сфалерита цинковым купоросом B—4 кг/т концентрата). В
результате этого достигается значительное улучшение^ качества
цинкового концентрата, получаемого в виде камерного продукта.
Медные концентраты, получаемые на фабриках, содержат
17—31% меди при извлечении 73—97%; цинковые
концентраты— 50—62% цинка при извлечении 67—92%. Зависимость
между производительностью фабрики и производительностью
труда одного рабочего приведена на рис. 14.2.
Флотация полиметаллических руд. К основным сульфидным
минералам полиметаллических, обычно вкрапленных, руд
относятся галенит, халькопирит и другие сульфиды меди, сфалерит,
пирит и пирротин. Часто присутствующие в рудах золото и
серебро связаны в виде тонкодисперсной вкрапленности с
пиритом, хилькопиритом, галенитом и, в меньшей степени,
сфалеритом. Нередко в рудах содержится значительное количество
барита.
Для полиметаллических руд характерна агрегатная вкрап-
240
ленность сульфидов. Для освобождения агрегатов сросшихся
сульфидов из сростков с пустой породой обычно достаточно
измельчения руды до 45—55 % класса —0,074 мм, тогда как для
раскрытия сульфидов из^ агрегатов необходимо тонкое
измельчение до 90—100 % класса —0,074 мм. Данное обстоятельство
является одной из основных причин широкого распространения
на практике коллективно-селективных схем (см. рис. 13.7,6, в)
и схем коллективной флотации с последующим разделением
коллективного концентрата (см. рис. 13.7, г, 13.8), которые, как
было показано ранее, обладают рядом технологических и
технико-экономических преимуществ по сравнению со схемой
селективной флотации.
В результате флотации стремятся получить кондиционные
медный, свинцовый, цинковый и пиритный концентраты с
максимальным извлечением в них одноименных компонентов.
Вредными примесями в свинцовых концентратах являются
цинк и медь. Содержания металлов в свинцовых концентратах
первого и последнего сорта следующие, %: свинца не менее
70 и 30, цинка не более 2,5 и 12, меди не более 1,5 и 4. Вредная
примесь в цинковых концентратах — железо. Кондиции для
первого и последнего сорта цинкового концентрата, %: цинка не
менее 53 и 40, железа не более 7 и 16. Для медных
концентратов свинцово-цинковой промышленности в зависимости от сорта
установлены следующие содержания цветных металлов, %:
меди не менее 20 и 11, свинца не более 7 и 19, цинка не более
6 и 19.
Основными собирателями при флотации полиметаллических
руд являются ксантогенаты и производные ксантогенатов: мине-
реки (диксантогениды) и тионокарбоматы. В большинстве
случаев используется смесь ксантогенатов с различной длиной
углеводородной цепи и комбинация двух или более
пенообразователей.
Коллективная флотация всех сульфидов проводится в
содовой среде при значениях рН 8—9 и интенсивном пенообразо-
вании. Снижению потерь металлов в хвостах способствует
подача большей части собирателя (до 70%) в циклы
измельчения.
Легкофлотируемые слоистые алюмосиликаты (типа сери-
цито-хлоритовых или оталькованных сланцев), содержащиеся
в руде, загрязняют коллективный концентрат. Повышение
качества коллективного концентрата в этом случае достигается:
, дофлотацией концентрата в слабокислой среде (рН 4—5)
в присутствии КМЦ, жидкого стекла, гексаметафосфата или
кремнефтористого натрия, обеспечивающих в этих условиях
глубокую депрессию минералов пустой породы;
дофлотацией концентрата при высоких значениях рН и
концентрации сульфидных ионов в жидкой фазе пульпы,
обеспечивающих глубокую депрессию сульфидов вследствие десорбции
собирателя с их поверхности и интенсивную флотацию слоистых
241
алюмосиликатов пустой породы, причиной которой является
переход полярных силанольных групп =Si—ОН на их
поверхности в этих условиях в силоксановые =Si—О—Si = ,
обладающие весьма слабой сорбционной способностью по отношению-
к молекулам воды. Данную операцию можно осуществить
в процессе десорбции собирателя с поверхности коллективного^
концентрата сернистым натрием по методу А. С. Конева перед,
отмывкой концентрата от избытка реагентов [2].
Процесс подготовки коллективного концентрата к
разделению предусматривает десорбцию собирателя с его поверхности:
и доизмельчение концентрата до необходимой крупности. Для"
десорбции собирателя используется один из следующих
способов:
интенсивное перемешивание концентрата без аэрации с
сернистым натрием C—6 кг/т) с последующей отмывкой десорби-
рованного собирателя и избытка сернистого натрия (метод.
А. С. Конева);
перемешивание с сернистым натрием (до 2 кг/т) и
активированным углем (до 1 кг/т) без последующей отмывки;
перемешивание только с активированным углем (до 2 кг/т}
без отмывки.
Первый способ применяется при большом, а последний —
при малом избытке собирателя в коллективном концентрате.
Другие методы десорбции собирателя (сгущение и
фильтрование, пропарка коллективного концентрата в щелочной среде;
обжиг) имеют ограниченное применение. При доизмельченин
коллективного концентрата переход с шаров на рудную галк>
позволяет в ряде случаев значительно сократить (в 1,5—2 раза)
расход сернистого натрия при десорбции и стабилизировать
процесс последующей селекции.
Отделение сульфидов свинца и меди от сульфидов цинка и
железа и пустой породы в цикле свинцово-медной флотации
осуществляется довольно легко. В большинстве случаев
флотация ведется в слабощелочной среде (рН 7—9), создаваемой
обычно с помощью соды и реже извести, при расходах цианида
15—100 г/т и сернокислого цинка в количестве, в 5—10 раз
превышающем расход цианида натрия (режим Шеридана —
Гриссвольда). Частичная или полная замена цианида сульфок-
сидным реагентом или применение различных продуктов
взаимодействия солей.цинка с сульфоксидными и щелочными
реагентами [ZnS203, ZnS204, Na2Zn(S204J, (NH4JZn(S204J, цин-
каты и некоторые другие соединения] в ряде случаев по? ^ляют
значительно повысить извлечение меди, сократить ее потери
в цинковом концентрате и тем самым исключить необходимость
проведения операции обезмеживания цинкового концентрата.
При депрессии активированных солями свинца сульфидов
цинка хорошие результаты достигаются с помощью
комбинации сернистого натрия, сульфоксидного реагента и'цинкового
купороса.
242
Способы разделения свинцово-медных концентратов можно
свести к следующим основным реагентным режимам,
основанным:
на флотации сульфидов меди и депрессии галенита
реагентом-окислителем (бихроматом, перекисью водорода, перманга-
натом, .хлорной известью) в слабокислой или слабощелочной
среде (рН 6—8) при расходе 0,5—2,5 кг/т концентрата;
сернистой кислотой с бихроматом или крахмалом или с бихроматом
и крахмалом;, сульфитом или тиосульфатом натрия с солью
тяжелого металла (цинковым или железным купоросом, трех-
хлористым железом или алюминием) при значении рН 5,5—
6,2. В присутствии вторичных сульфидов меди используется
сульфит натрия с бихроматом;
на флотации галенита и депрессии сульфидов меди
цианидом в щелочной среде (рН около 10) иногда с небольшими
добавками сернистого натрия @,3—0,5 кг/т концентрата); смесью
цианида и цинкового купороса в соотношении, необходимом для
образования комплексной цинкцианистой соли Na2Zn(CNL
(метод Ю. И. Еропкина); красной кровяной солью при расходе
3—7 кг/т концентрата; органическим соединением «Конго
красный».
Выбор метода разделения свинцово-медного концентрата
зависит от его вещественного состава. Если сульфиды меди
представлены главным образом халькопиритом, то эффективное
разделение концентрата может быть достигнуто при
использовании практически всех реагентных режимов, основанных на
депрессии галенита или меди. Присутствие вторичных
сульфидов меди (барнита и халькозина) приводит к нарушению
селекции. Более эффективными методами разделения при
значительном содержании вторичных сульфидов меди в концентрате
являются цинкцианистый метод (метод Ю. И. Еропкина) и
методы, основанные на применении ферроцианида и реагента
«Конго красный».
Цинковые минералы, содержащиеся в коллективном
концентрате, при цианидных методах разделения переходят в
медный концентрат, а при применении сульфоксидных реагентов —
в свинцовый. В последнем случае их можно извлечь из
камерного продукта флотацией после дополнительной подачи извести
до рН 11—12, медного купороса, собирателя и
пенообразователя, а также после аэрации камерного продукта с известью
в течение 30 мин и последовательной подачи смеси сульфита
с бихроматом C:2), медного купороса, собирателя и вспени-
вателя.
Реагентные режимы пиритной, цинково-пиритной и цинковой
флотации не отличаются принципиально от режимов при
флотации медно-цинково-пиритных руд.
Повышение качества цинковых концентратов
осуществляется (после десорбции.собирателя):
обессвинцеванием их путем дофлотации галенита после пе-
243
ремешивания цинковых концентратов в щелочной среде с
цианидом и цинковым купоросом;
обессвинцеванием, обезмеживанием и обезжелезнением по
методу Л. Б. Дебривной: в содовой среде с цинковым
купоросом, часть которого можно заменить более дешевым железным
купоросом. Вместо соды и цинкового купороса можно
использовать цинкаты, предложенные В. А. Коневым [17].
Вследствие сложности руд и применения довольно сложных
схем и режимов, необходимых при флотации
полиметаллических руд, технологические показатели их обогащения на ряде
фабрик, особенно зарубежных, относительно невысоки.
Извлечение металлов в одноименные концентраты составляет, %: для
меди 67—92, для свинца 66—89, для цинка 73—94.
Флотация медно-никелевых руд. Сплошные и вкрапленные
сульфидные медно-никелевые руды являются основным
источником получения никеля. Помимо основных ценных
компонентов (никеля, меди, серы) они часто содержат такие
сопутствующие элементы, как золото, серебро, кобальт, палладий,
платину, селен, теллур и другие редкие и рассеянные элементы,
тесно связанные с сульфидами основных металлов. Сульфиды
никеля представлены пентландитом (Fe№)9S8, миллеритом NiS
и никеленосным пирротином, имеющим магнитную
(моноклинный пирротин) и немагнитную (гексагональный пирротин)
разновидности; сульфиды меди — халькопиритом CuFeS2,
кубической разновидностью его — кубанитом и иногда талнахиток.
Трудности селективной флотации сульфидов меди и никеля
и удаления железа из никелевого концентрата обусловлены:
широким изменением степени изоморфного замещения
никеля железом (от 10 до 42 %) и кобальтом в пентландите и
железа никелем (до 3 %) в пирротине, что приводит к изменению
поверхностных и флотационных свойств основных никельсодер-
жащих минералов;
изменением соотношения кристаллических разновидностей
пирротина (моноклинного и гексагонального) и сульфидов меди
(халькопирита и кубанита), обладающих различными
флотационными свойствами;
трудностью активации никельсодержащих минералов после
их депрессии в цикле медной флотации;
тонкой неравномерной вкрапленностью и тесным
взаимопрорастанием ценных компонентов;
наличием в рудах легкофлотируемых слоистых
алюмосиликатов (например, талька, хлорита, оталькованного
серпентинита и других подобных им минералов породы).
В связи с перечисленными трудностями к настоящему
времени наибольшее распространение получили схемы,
предусматривающие предварительное получение коллективного медно-
никелевого или медно-никелево-пирротинового концентратов.
Помимо общеизвестных преимуществ, применение таких схем
позволяет:
244
устранить загрязнение концентратов тугоплавкой пустой
породой и несколько усреднить вещественный состав подлежащего
дальнейшему разделению сульфидного продукта;
легче осуществить стадиальное обогащение с межцикловой
флотацией в рудном цикле и раздельную флотацию песков и
шламов при переработке шламистых медно-никелевых руд;
повысить комплексность использования сырья в результате
попутного извлечения металлов платиновой группы, золота,
серебра и кобальта в цикле коллективной флотации благодаря
использованию сильных реагентов-собирателей без применения
какого-либо специального оборудования или с использованием
его (например, шлюзов для улавливания крупных зерен
металлов платиновой группы).
Флотационное извлечение сульфидов меди и никеля в
коллективный концентрат осуществляется с применением сульф-
гидрильных собирателей. В СССР применяют бутиловые ксан-
тогенат A00—200 г/т) и дитиофосфат A00—200 г/т), на
фабриках Канады и Финляндии — амиловый или смесь амилового
и изопропилового ксантогенатов. Для повышения извлечения
сульфидов никеля из руд к настоящему времени предложены
алкилтритиокарбонаты, ксантогенаты, изготовленные на основе
2-диалкиламиноэтиловых спиртов и гидроокисей 2-оксиэтилтри-
алкиламмониевых соединений, нефтяные сульфиды. Однако эти
собиратели пока не нашли промышленного применения из-за
их дефицитности или токсичности.
Для депрессии флотоактивных силикатных минералов
пустой породы используются следующие органические депрессоры
A50—400 г/т): карбоксиметилцеллюлоза (КМЦ), сульфоэфиры
целлюлозы (СЭЦ), этансульфонатцеллюлозы (ЭСЦ), медноам-
миачные растворы целлюлозы. На зарубежных фабриках
применяют также гуартек и кукурузный декстрин. При флотации
в щелочной среде (рН 7,5—9,5), создаваемой содой @,5—
3 кг/т), лучшие результаты получены с применением КМЦ.
Наиболее эффективная депрессия флотоактивных силикатов
достигается в слабокислой среде (рН 3—5), создаваемой
серной, щавелевой или сернистой кислотами. Предпочтительными
депрессорами в этом случае являются СЭЦ и ЭСЦ.
Слабокислая среда является также предпочтительней, чем
щелочная, для активации флотации пирротина, которая
осуществляется медным купоросом (до 50 г/т) или медноаммиач-
ным комплексом Си (NH3L -БСЫ-НгО, образующимся при
смешении аммиака и медного купороса в соотношении 2:1.
Коллективные концентраты содержат не менее 3,5 % никеля
и не более 15—20 % окиси магния, входящей в состав
силикатов породы. В коллективный концентрат достаточно полно
извлекаются платиносодержащие минералы. Худшей флотируемо-
стью обладают ферроплатина и купроплатина, на поверхности
зерен которых образуются гидрофильные соединения железа
или меди, а также бреггит, стибиопалладинит, сперрилит и не-
245
которые другие минералы, содержащие платину. Для их более
полного извлечения или создают в контрольной медно-никеле-
вой флотации особые условия (подача дополнительных
реагентов, подогрев пульпы и др.), или улавливают их из хвостов
флотации гравитационными методами обогащения (с применением
шлюзов, отсадочных машин и других аппаратов) {2].
Если отношение содержания меди к содержанию никеля
в коллективном концентрате меньше 2, то его целесообразно
подвергнуть плавке на файнштейн, который затем направляется
на флотационное разделение по методу И. Н. Масляницкого.
Файнштейн состоит из халькозина СигЭ, хизлевудита NisS2 и
небольшого количества сплава меди и никеля. После
измельчения его примерно до 50 мкм, подачи ксантогената A,0—1,3 кг/т
-файнштейна) и пенообразователя флотируется сульфид меди
в сильнощелочной среде (рН около 12), создаваемой обычно
едким натром; в камерный продукт переходят сульфид никеля
и сплав никеля с небольшим количеством меди. Процесс
характеризуется высокой эффективностью и является ярким
примером решения трудной проблемы применением комбинированной
схемы, предусматривающей операции металлургии и
обогащения.
Коллективный медно-никелевый концентрат подвергается
непосредственному разделению, если соотношение содержаний
меди и никеля больше 2. Селекция концентратов после их доиз-
мельчения основана на довольно эффективной депрессии пент-
ландита и пирротина в щелочной известковой среде (рН 9—
12), не влияющей на флотируемость халькопирита.
Предварительная пропарка концентрата при температуре 80 °С позволяет
понизить расход извести и улучшить условия селективной
флотации. Показатели селективной флотации улучшаются также
при добавлении совместно с известью сернистого натрия,
сульфита натрия или цианида. На некоторых зарубежных фабриках
для усиления депрессирующего действия извести применяют
небольшие добавки органических коллоидов и цианида.
Если часть меди в концентрате представлена кубанитом и
талнахитом, депрессирующимся в сильнощелочной среде, то
селективная флотация минералов меди осуществляется в
нейтральной или слабокислой среде (рН 5,5—7,5) в присутствии
сульфита натрия (до 700 г/т), после предварительной аэрации
в течение 20 мин для окисления и депрессии никельсодержащих
сульфидов.
Повышению общего извлечения никеля и комплексности
использования сырья на фабриках способствует распространение
технологии выделения никеленосного пирротина в
самостоятельный концентрат, который либо присоединяют к основному
никелевому концентрату, либо подвергают металлургической
переработке с извлечением из него меди, никеля, железа и
других сопутствующих элементов. Извлечение пирротина
осуществляется из хвостов медно-никелевой флотации после ак-
246
тивации медным купоросом A00 г/т) или по комбинированной
магнитно-флотационной схеме с применением магнитных
сепараторов с высокой напряженностью магнитного поля в начале
процесса [2].
Содержание меди в селективных медных концентратах до*
стигает 30 %, а никеля в никелевых концентратах составляет
4—10,5%. Коллективные медно-никелевые концентраты
содержат 5—10 % меди и 7—12 % никеля при среднем извлечении
в них 82,5 % меди и 82Д % никеля. Пониженное извлечение
никеля по сравнению с извлечением меди объясняется тем, что
часть его находится в нефлотируемых силикатах и сульфидах,,
эмульсионно вкрапленных в пустую породу.
Достигнутая на медно-никелевых фабриках
производительность труда составляет 12—51 тыс. т руды на одного
работающего.
Флотация сульфидных руд мышьяка, сурьмы и ртути.
Основными промышленными минералами в таких рудах являются
киноварь HgS, антимонит Sb2S3 и арсенопирит FeAsS. Другие
сульфиды сурьмы (джемсонит, буланжерит, тетраэдрит и др.)
и мышьяка (реальгар, аурипигмент и др.) обычно
практического значения не имеют. Различные сочетания сульфидов
мышьяка, сурьмы и ртути в рудах порождают большое число
типов руд: от монометаллических (например, ртутных) до-
комплексных, часто 'содержащих золото, серебро или
флюорит [32].
Киноварь легко флотируется с помощью ксантогенатов или
дитиофосфатов обычно без предварительной активации,
которую можно осуществить солями свинца или меди A00—
250 г/т). Известь и цианид, подавляя пирит, не снижают
извлечения киновари. Трудно окисляясь кислородом воздуха,
киноварь может быть сфлотирована в присутствии пенообразователя
и углеводородного масла. Депрессируется она сернистым
натрием. Поскольку жидкое стекло также оказывает депрессирую-
щее действие на флотируемость киновари, для пептизации
охристо-глинистых шламов при флотации ртутных руд применяюг
органические вещества типа лигнинсульфонатов E0 г/т).
Антимонит флотируется ксантогенатами только после акти-.
вации его солями меди или свинца в слабокислой или
нейтральной среде (рН 4—7,4). В щелочной среде медный купорос
оказывает на антимонит депрессирующее действие. Сурьмяные
минералы, содержащие в кристиллической решетке катионы
свинца (буланжерит) или меди (тетраэдрит), не требуют
предварительной активации. Крахмал и щелочь депрессируют
антимонит. Эффективно депрессируется антимонит цианидом лишь
при рН около .8. Обладая более высокой по сравнению с
киноварью окисляемостью, антимонит может быть депрессироваи
с помощью окислителей. Неокислеяные зерна антимонита
флотируются пенообразователем, углеводородными маслами или
диксантогенидом без предварительной активации.
247
Арсенопирит по флотационным свойствам близок к пириту:
легко окисляется, теряя способность к флотации. При
флотации с ксантогенатами он депрессируется щелочью и цианидами
132].
Селективная флотация сульфидных руд мышьяка, сурьмы и
ртути осуществляется обычно с применением сульфгидрильных
собирателей по коллективно-селективным схемам.
Селекцию коллективного обычно золотосодержащего пи-
ритно-арсенопиритного концентрата и кеков цианистых заводов
примерно того же состава можно осуществить после десорбции
собирателя с поверхности концентрата и перемешивания его
с окислителем (пиролюзитом, перманганатом, кислородом
воздуха) для депрессии арсенопирита [32].
Селекцию коллективного концентрата, получаемого при
флотационном обогащении сурьмяно-мышьяковых руд, можно
осуществить:
депрессируя арсенопирит и пирит щелочью, цианидом и
цинковым купоросом и флотируя антимонит;
депрессируя антимонит сернистым или едким натром и
флотируя пирит и арсенопирит ксантогенатом с небольшими
добавками медного купороса.
Гораздо реже применяется прямая селективная флотация
пирита и арсенопирита из руды ксантогенатом с добавлением
медного купороса и едкого натра с последующей
активацией антимонита солями свинца и флотацией его
ксантогенатом.
Селекция коллективных мышьяково-ртутных концентратов,
содержащих арсенопирит и киноварь, легко достигается
депрессией арсенопирита известью. Флотационное разделение сурь-
мяно-ртутных концентратов основано на значительно более
высокой окисляемости антимонита по сравнению с киноварью.
Активным реагентом-окислителем, обеспечивающим
эффективную депрессию антимонита в этом случае, является, по данным
П. М. Соложенкина, перекись водорода в сочетании с
хромпиком, не влияющими существенно на флотацию киновари.
При совместном присутствии в руде или в коллективном
концентрате сульфидов мышьяка, сурьмы и ртути может быть
принята следующая последовательность их флотации. От
антимонита и арсенопирита киноварь отделяется в щелочной среде;
после извлечения киновари флотируется антимонит в
присутствии медного купороса. Для депрессии арсенопирита
добавляются жидкое стекло и цианид. В последнюю очередь
флотируется арсенопирит после активации его сернистым натрием. По
такой схеме получаются три кондиционных концентрата:
ртутный, сурьмяный и мышьяковый.
Ртутные концентраты содержат 10—30 % ртути при
извлечении ее 75—95 %. Сурьмяные концентраты должны содержать
не менее 30 % сурьмы и не более 0,25 % мышьяка. Сурьмяные
и мышьяковые концентраты часто содержат благородные ме-
248
X -/ о_
таллы, стоимость которых может намного превышать стоимость
основных металлов или элементов в концентратах.
Флотация самородных металлов. Из самородных металлов
в рудах встречаются золото, серебро, платиноиды, медь, реже
висмут и ртуть. Наибольшее практическое значение имеет
флотация золота и меди. Серебро обычно тесно связано с
сульфидами и при флотации извлекается вместе с ними, а платиноиды
извлекаются в основном гравитационным способом [12].
Самородное золото никогда не бывает химически чистым и
содержит до 50% примесей серебра, ртути, меди, висмута,
свинца и некоторых других элементов. Оно часто встречается
в сульфидных рудах тяжелых цветных металлов. Часть его
находится в более крупных выделениях, другая часть тесно
ассоциирует с сульфидами и реже находится в виде очень тонкой
вкрапленности в кварце. Наличие золота в сульфидах повышает
их флотируемость [2, 25].
Свободное мелкое золото легко флотируется ксантогенатами
при рН 7—9. Примеси меди и железа, образуя на поверхности
зерен окисленные соединения, ухудшают флотационные
свойства золота. Частичное удаление этих образований оттиркой
в плотной пульпе ведет к возрастанию адсорбции ксантогената
на поверхности частиц золота и улучшению их флотации.
Известь, сернистый натрий и цианиды депрессируют золото по
такому же механизму, как и при флотации сульфидов [12].
На золотоизвлекательных фабриках флотация обычно
применяется в сочетании с процессами металлургии, основным из
которых является цианирование [12].
В комбинированных схемах флотация применяется в первую
очередь для первичной концентрации золота из убогих руд
в концентрат с извлечением в него 90—93 % золота, который
поступает далее на цианирование или плавку. Флотация
измельченной руды до 65—85 % класса —0,074 мм
осуществляется с применением смеси ксантогената и дитиофосфата или
смеси ксантогенатов с различной длиной углеводородной цепи
при общем расходе 100—200 г/т. Депрессия легкофлотируемой
пустой породы достигается с помощью органических реагентов
(типа КМЦ), поскольку жидкое стекло и крахмал при больших
расходах подавляют флотацию золота. Обязательным является
применение гравитационных аппаратов для улавливания
свободного золота в циклах измельчения и перечистной флотации.
Для этого между мельницей и классификатором устанавливают
отсадочную машину или флотационную камеру с ловушкой для
золота. В цикле перечистной флотации черновой концентрат
пропускают через короткоконусный гидроциклон или щелевой
шлюз (концентратор), слив которых подвергают перечистной
флотации. Готовым является продукт, состоящий из песков
гидроциклона или концентрата шлюза, в которых
концентрируются труднофлотируемые частицы золота (крупные зерна с
покровными образованиями, *пластинки со вкованными в их по-
249
верхность минералами породы, сростки и др.), с трудом
перешедшие в черновой концентрат и легко теряемые в перечист-
ной флотации, и концентрата перечистной флотации слива
гравитационных аппаратов.
Флотация может применяться также для удаления из руды
вредных для последующего цианирования примесей, таких, как
сульфиды мышьяка, сурьмы, селена и теллура. Для
исключения вредного блияния этих сульфидов применяется обжиг,
однако, так как обжиг всей руды обходится дорого, руда
подвергается коллективной флотации с извлечением в концентрат
сульфидов и золота. В обжиг и на последующее цианирование
поступает только небольшое количество коллективного
концентрата. При наличии в золотых рудах минералов меди, также
оказывающих вредное влияние на процесс цианирования,
получают коллективный медно-золотой концентрат,
направляемый в плавку.
Флотация позволяет повысить комплексность использования
руд с извлечением из них, кроме золота, других ценных
компонентов (меди, серебра, свинца, барита, селена, теллура
и др.). Например, применение флотации после цианирования
позволяет доизвлекать теллуриды золота и золотосодержащий
пирит. Для активации их пульпу обрабатывают в специальных
условиях сернистым газом при рН v6,3. После
самопроизвольного увеличения рН до 7 и дополнительной активации медным
купоросом в результате флотации получают золото-пиритно-
теллуровый концентрат, который после обжига может быть
снова направлен на цианирование.
В некоторых золотых рудах теллур и висмут присутствуют
главным образом в виде самостоятельных минералов тетради-
Коллешибный
концентрат
Сгущение
Доиз-
Сода(до рН8-8,5) X*мелъчение
ЦианидY SO г/т)
w
Теллуро- 8и смуглявая флотация
Сода, цианиЗ *
СлиВ
- Воборот
г- у— Залото-пиритныи
1-Шлеречистные концентрат
Теллуро-Висму- 1 промпроВуш
\тоВыи » (BoSapom)
концентрат r y
Рис. 14.4. Схема селективной флотации минералов теллура и висмута
коллективного золото-пиритного концентрата
250
мина и висмутина. В условиях коллективной флотации теллур
и висмут достаточно полно переходят в концентраты, однако
при цианировании или обжиге последних они практически
теряются. Извлечение теллура и висмута из коллективного
концентрата в отдельный продукт можно осуществлять
селективной флотацией по схеме на рис. 14.4. Отделение теллура и
висмута от золотоносного пирита и свободного золота основано на
депрессии последних цианидом в содовой среде [12].
При переработке медных и полиметаллических руд режим
устанавливается таким образом, чтобы максимальное
количество золота флотировалось в медные или свинцовые
концентраты, из которых оно легко извлекается при металлургическом
переделе. Этому способствует, например, применение бесциа-
нидных методов флотации. Однако технологические
особенности руд и режимы обогащения, оптимальные для извлечения
основных металлов, не всегда позволяют решить проблему
повышения извлечения золота без организации его извлечения из
пиритных концентратов (после их обжига). В отдельных
случаях из общей массы пирита удается выделить богатый
золотосодержащий продукт, проводя флотацию при рН 9,5 в
содовой среде с добавками сернистого натрия.
Флотационное извлечение мелких зерен самородной меди
из руд может быть осуществлено в слабокислой, нейтральной
или слабощелочной среде с помощью сульфгидрильных
собирателей и дисульфидов.
Флотация металлической меди производится на фабриках,
перерабатывающих труднообогатимые окисленные медные
руды по методам Мостовича или сегрегации.
Метод Мостовича, получивший за рубежом название
«процесс LPF», предусматривает выщелачивание окисленной
меди серной кислотой C—40 кг/т), осаждение (цементацию)
меди, перешедшей в раствор, измельченным до 0,2 — 0,5 мм
губчатым железом или чугунной стружкой B—30 кг/т) по
реакции
CuS04 + Fe = \ Си + FeS04
и флотацию металлической меди совместно с сульфидами меди
и других металлов, которые не растворяются при
используемых в методе Мостовича расходах серной кислоты.
Выщелачивание измельченной до 60—70% класса — 0,074 мм руды
производят при обычной температуре или при подогреве острым
паром до 60—70 °С в течение 20—80 мин, цементацию меди —
в течение 15—20 мин, остаток непрореагировавшего
растворителя выделяют из хвостов флотации магнитной сепарацией.
Флотацию проводят в слабокислой или кислой среде (рН
2—5). Так как ксантогенат в кислой среде разлагается, то
лучшие результаты флотации получают при использовании
реагента СЦМ-2 D0—50 г/т), гидролизованного дитиофосфата
(около 100 г/т) или минереков. Во всех случаях флотация
251
цементной меди замедляется и становится неустойчивой, если
концентрация ионов хлора в пульпе превышает 100—200 мг/л.
Показатели обогащения зависят от полноты растворения меди.
Концентрат обычно содержит около 50 % меди при извлечении
€0—85%.
Процесс сегрегации включает в себя
восстановительный обжиг труднообогатимой окисленной медной руды в
присутствии кокса A—3 %) и поваренной соли A—3 %) при
температуре 700—800 °С в течение примерно 1 ч. В процессе
обжига соединения меди восстанавливаются до металлической,
образуя частички флотационной крупности, которые
флотируются в слабокислой среде сульфгидрильными собирателями
после охлаждения без доступа воздуха и доизмельчения.
А. А. Зубковым и Б. А. Степановым показана возможность
эффективной флотации металлической ртути из ступпы
—продукта металлургической переработки ртутных руд и
концентратов. По флотационным свойствам металлическая ртуть
аналогична самородному золоту и хорошо извлекается
сульфгидрильными собирателями и продуктами их окисления (например,
диксантогенидом) при оптимальных значениях рН, равных 5—7.
Сернистый натрий, известь, цианид, крахмал, КМЦ,
снижение концентрации кислорода в пульпе депрессируют флотацию
ртути. Флотация ступпы обеспечивает практически полный
перевод в товарный металл металлической ртути (в виде пенного
продукта и остатка на дне машины).
14.4. Флотация окисленных и смешанных руд
цветных металлов
Краткая характеристика руд. Окисленные и смешанные
(сульфидно-окисленные) руды являются трудным объектом
обогащения. Это обусловлено в основном следующими
причинами [2]:
полезные минералы в таких рудах представлены
сульфидами, карбонатами, сульфатами, силикатами, фосфатами, арсе-
натами и другими минеральными соединениями, обладающими
различной флотируемостью. Поэтому очень трудно подобрать
универсальный реагентный режим, обеспечивающий
эффективное извлечение всех минеральных форм каждого металла в
одноименные концентраты, тем более что для таких труднофлоти-
руемых минералов, как хризоколла, плюмбоярозит и некоторые
другие, способы флотационного извлечения еще не
разработаны;
ассоциация полезных минералов с пустой породой
(особенно с гидроокислами железа) бывает иногда настолько
тесной, что исключает раскрытие части минералов при
экономически и технологически приемлемой степени измельчения;
окисленные и смешанные руды, как правило, сильно
разрушены, выветрены и ожелезнены, что является причиной боль-
252
шого содержания в них растворимых солей и
охристо-глинистых шламов, оказывающих на процесс флотации резко
отрицательное влияние.
Режимы флотационного извлечения окисленных минералов
-свинца, меди и цинка из руд. Несмотря на большое количество
предложенных режимов для флотационного извлечения
окисленных минералов цветных металлов, на практике
используется и позволяет получать устойчивые технологические
показатели только режим сульфидизации с последующей флотацией
сульфгидрильными или катионными собирателями. Только
лредварительная сульфидизация позволяет в какой-то мере
преодолеть трудности, связанные с особенностями
вещественного состава этих руд вследствие:
унификации поверхностных свойств окисленных минералов
в результате их сульфидизации;
нейтрализации вредного действия растворимых солей
тяжелых металлов в результате их осаждения в виде
труднорастворимых сульфидов;
дезактивации минералов пустой породы в результате
удаления с их поверхности отслаивающихся свежеобразованных
•сульфидов активирующих катионов, поскольку параметры
кристаллических решеток сульфидов и минералов породы не
совпадают;
пептизации охристо-глинистого материала.
При сульфидизации окисленных минералов на них
образуется сульфидная пленка. Скорость ее роста зависит от природы
минерала, концентрации сульфидизатора, времени контакта
с ним, ионного состава жидкой фазы пульпы и ее температуры
[25].
Наиболее легко сульфидизируются карбонаты меди
[малахит CuC03-Cu(OHJ, азурит 2CuC03-Cu(OHJ] и свинца (це-
руссит РЬСОз). При их сульфидизации не требуется установки
контактных чанов, и сульфидизатор загружается
непосредственно во флотационную машину в несколько приемов при
общем расходе 0,5—1,5 кг/т [46]. Сульфидизация карбонатов меди
лучше протекает при низкой щелочности пульпы, а церуссита
при рН 9,5. Англезит PbS04 также легко сульфидизируется,
особенно при оптимальном значении рН 7,5—8,2, но требует
более длительного перемешивания (до 10 мин) с сульфидиза-
тором. Ионы хлора замедляют сульфидизацию карбонатов и
«сульфатов свинца и меди, а ионы кальция и магния могут
оказать депрессирующее действие на их флотацию в результате
образования на сульфидизированной поверхности зерен
осадков СаС03 и CaS04 по реакциям:
МеС03] + Са2+ + S2- -> MeS] - CaCOs;
MeS04] + Са2+ + S2- -> MeS] - CaS04.
Добавка солей аммония NH4C1 или (NH4JS04 повышает
растворимость солей кальция и магния и уменьшает их вред-
253
ное влияние на сульфидизацию и флотацию. При большом
содержании в руде растворимых солей и шламов сульфидизация
успешно протекает только после предварительного их удаления
путем промывки крупнодробленой руды.
Другие окисленные минералы свинца [вульфенит РЬМо04,
крокоит РЬСг04, ванадинит Pb5(V04KC], пироморфит
Pbs(P04KCl, миметезит Pb5(As04KCl] и меди (куприт Cu20)
сульфидизируются хуже даже при оптимальных значениях рН
5,5—8. Плюмбоярозит Pb-Fe6[(OHN(S04K] и силикаты меди
не сульфидизируются и при флотации теряются в хвостах.
При флотации окисленных минералов свинца и меди
применяются простые схемы, предусматривающие основную,
контрольную и две-три перечистные операции флотации. Расход
сульфидизатора зависит от свойств извлекаемых минералов и
наличия в руде гидроокислов железа, марганца и других
соединений, вызывающих каталитическое окисление или поглощение
сульфидизатора. Обычно он изменяется от 0,5 до 3 кг/т. Чтобы
понизить значение рН при флотации и приблизить его к
оптимальному, не превышающему 9,5, вместо Na2S частично или
полностью загружают NaHS. В качестве собирателя
используют обычно ксантогенаты. Применение высших или смесей
низших и высших ксантогенатов позволяет повысить извлечение
минералов свинца или меди и сопутствующих им благородных
металлов. Иногда лучше результаты обеспечивает применение
меркаптанов. Наиболее успешное подавление флотоактивных
силикатов породы достигается с помощью КМЦ, жидкого
стекла или гексаметафосфата [2].
Основной причиной потерь свободных окисленных
флотирующихся минералов свинца или меди в хвостах флотации
являются большие отклонения концентрации сульфидных и ксанто-
генатных ионов в пульпе от оптимальной из-за резких
изменений вещественного состава перерабатываемых руд.
Автоматический контроль и регулирование расхода
реагентов в соответствии с уравнением (8.11) при флотации сульфи-
дизированных минералов меди и зависимостью, изображенной
на рис. 8.4, при флотации сульфидизированных минералов
свинца позволяет повысить извлечение окисленных минералов
в концентрат, осуществить эффективную совместную флотацию
сульфидов и окислов каждого металла и сократить расход
реагентов.
Медь, представленная карбонатами, на 80—90 %
извлекается в концентрат, содержащий 18—25 % меди. Свинец,
представленный церусситом и англезитом, на 85—90 % извлекается
в концентрат, содержащий 50—60 % свинца. Потери в хвостах
обусловлены труднофлотируемыми минералами меди (хризо-
коллой CuSi03-nH20) и свинца (плюмбоярозитом и
значительной частью пироморфита, ванадинита и миметезита).
Из всех методов, предложенных для извлечения окисленных
цинковых минералов [смитсонита ZnC03 и каламина
254
Zn4Si207(OfIJ'H20] из руд наиболее перспективными
являются методы Дэвиса — Андреевой и Рея.
Метод Дэвиса — Андреевой заключается в предварительной
-сульфидизации обесшламленной плотной пульпы (Т:Ж=1:1)
при температуре 50—70 °С, активации медным купоросом и
последующей флотации ксантогенатом и дитиофосфатом после
разбавления пульпы холодной водой. Метод Рея
предусматривает флотацию окисленных цинковых минералов катионньш
собирателем ^(первичными алифатическими аминами) при рН
10,5—11,5 после перемешивания пульпы с сернистым натрием
при обычной температуре. Расход сульфидизатора в обоих
случаях 3—6 кг/т, расход собирателя 150—300 г/т. Пустая порода
депрессируется жидким стеклом или КМЦ C00—400 г/т).
Недостатками метода Дэвиса — Андреевой,
ограничивающими его применение в промышленном масштабе, являются
неприменимость этого метода для флотации руд с высоким
содержанием железа, недостаточная эффективность процесса по
отношению к силикатам цинка, а также необходимость
подогрева всей пульпы до 50—70 °С. Метод Рея лишен этих^
недостатков и обеспечивает получение более высоких и стабильных
технологических показателей. В общем случае извлечение
окисленных цинковых минералов в концентрат, содержащий 38—
43 % Цинка, определяется возможностью механического
разделения полезных минералов от пустой породы и возможностью
предотвращения вредного влияния растворимых солей и шла-
мов. На зарубежных фабриках оно составляет 76—88 % цинка
от исходного питания цикла окисленной цинковой флотации.
При одновременном присутствии в рудах сульфидных и
окисленных минералов обычно применяется схема,
предусматривающая циклы сульфидной флотации и разделения
сульфидов, окисленной свинцовой или свинцово-медной флотации и
окисленной цинковой флотации. Такой схеме присущи
следующие недостатки:
большой расход реагентов, высокая стоимость которых
заставляет в ряде случаев отказываться от извлечения из руд,
например, минералов окисленного цинка;
необходимость тщательного контроля реагентного режима,
что не всегда оказывается возможным вследствие резких
изменений вещественного состава окисленных и смешанных
полиметаллических руд;
большие неизбежные потери полезных минералов со шла-
мами, удаление которых из процесса перед флотацией,
например окисленных цинковых минералов, является обязательной
операцией;
недостаточно высокие технологические показатели.
Комбинированные схемы обогащения. Результаты многих
исследований показывают, что применение только
обогатительных методов при переработке окисленных и смешанных
полиметаллических руд не может обеспечить комплексного извлече-
255
Руда
\
П ром ывка
крупно дробленой р уды
Т
Кол л о к т ивная
флотация с у л ьаэ и б о б.
Коллективный с^шОшй %%%%№%,%%%&?&*
концентрат/на злектротер- ца ме$и или ииика
мическун? обработку или \
разделение)
Удаление минералов пустой породы из
коллективного окисленного концентрата
\ Хвосты
в отдал
Минералы пустой породы
(карбонаты и сульфаты
кальция, магния и бария, выщелачивакие окисл енных
легкофлотируемые с или- минералов меди и цинка
^кать/J
Очистка электролита
от меди и ж ел е за
Г Г
Цементная
медь
Гидроокись
железа
Флотация минералоб
окисленного свинца
N
Окисленный ■
|Р6 концентрат
Хвосты
Электр о л из цинка
Катодный
цинк
Оборотный
электролит
Рис. 14.5. Принципиальная комбинированная схема переработки труднообо-
гатимых сульфидно-окисленных полиметаллических руд
ния из них ценных компонентов. Для этого необходимо
предусматривать в схеме металлургические операции.
Один из возможных вариантов комбинированной схемы
изображен на рис. 14.5. Он предусматривает получение двух
коллективных концентратов: сульфидного, получаемого с сульф-
гидрильным собирателем, и окисленного, получаемого с окси-
гидрильным собирателем (олеатом натрия или ректификатом
таллового масла). Сульфидный концентрат после десорбции и
доизмельчения поступает на разделение. Окисленный
концентрат загрязнен карбрнатами и сульфатами щелочноземельных
металлов и поэтому подвергается сначала облагораживанию.
Для этого он перемешивается с сернистым натрием и содой.
После чего без удаления жидкой Фазы пульпы и отмывки
реагентов и при поддержании концентрации сульфидных ионов не
256
Черновой коллективный окисленный
или сульфидно-окисленный сдинцодо-
медно-цинковый концентрат
Na2S (k-8'г/л)
)t^
Перемешивание
Eмин,Т:Ж~1:2)
Na2C03 A-2 г/л)
Флотация A0мин)
Пере меши в ание
E'мин, T'.?K~1:Z)
НагС05Щ /г/л)
У /
Флотация (8мин)
Пенный продукт Камерный продукт
(кардонаты и сульсраты ( ffoz а тый коллективный
щелочноземельные ме- концентрат)
таллсв)
Рис. 14.6. Схема флотации карбонатов и сульфатов щелочноземельных
металлов из коллективного окисленного или сульфидно-окисленного свинцово-
медно-цинкового концентрата, полученного флотацией с оксигидрильным
собирателем
менее 0,5—1 мг/л проводится флотация минералов породы при
депрессии сульфидных и сульфидизированных окисленных
минералов (рис. 14.6). После удаления загрязняющих примесей
коллективный окисленный концентрат подвергается
выщелачиванию. Медь и цинк из сернокислотных растворов выделяется
электролизом, а остаток выщелачивания, представленный
окисленными свинцовыми минералами, направляется на плавку
(см. рис. 14.5).
Представляет интерес также вариант комбинированной
схемы (см. рис. 13.9) с получением одного коллективного
сульфидно-окисленного концентрата, который после
предварительного облагораживания (см. рис. 14.6) поступает на
сернокислотное выщелачивание. Флотационное отделение сульфидов от
окисленных свинцовых минералов по данной схеме
производится или непосредственно в процессе выщелачивания
окисленных меди и цинка, или из остатков выщелачивания в кислой
среде при рН 1,5 без добавки собирателя. Окисленный
свинцовый концентрат получается в виде хвостов сульфидной
флотации. При необходимости окисленные свинцовые минералы
можно извлечь в высококачественный свинцовый концентрат
путем флотации с алкилсульфатом в кислой среде.
[$г1не
менее @,5-1'мг/л)
9 Заказ № 1957
257
В обоих вариантах комбинированных схем (см. рис. 13.9 и
14.5) исключается необходимость обесшламливания рудного
материала, реагентный режим и управление процессом
предельно просты, а извлечение основных и сопутствующих
металлов и элементов в конечную продукцию металлургических
заводов является максимальным.
Повышение комплексности использования труднообогати-
мых монометаллических окисленных и смешанных медных руд
достигается комбинированием в схеме операций флотации и
выщелачивания (метод Мостовича) илц пирометаллургии
(метод сегрегации). Метод сегрегации может быть использован
также для извлечения никеля из окисленных труднообогати-
мых никелевых руд [2].
U.S. Флотация несульфидных руд
солеобразных минералов
Краткая характеристика основных минералов. Из
несульфидных солеобразных минералов наиболее важное
промышленное значение имеют барит BaS04, флюорит CaF2, кальцит
СаС03, магнезит MgC03> доломит CaC03-MgC03, апатит
Са5(Р04)з (F, C1, ОН), шеелит CaW04, повеллит СаМо04. Все
они являются солями щелочноземельных металлов и поскольку
их кристаллическая решетка носит ионный характер, степень
исходной гидрофильности поверхности данных минералов
больше, чем у сульфидов.
Гидрофобизация и флотация минералов довольно легко
обеспечивают в присутствии оксигидрильных собирателей
(мыл, органических кислот, алкилсульфатов, алкилсульфонатов
и др.), образующих с катионами щелочноземельных металлов
на поверхности трудиорастворимые соединения. Ксантогенаты
не образуют таких соединений и не обладают собирательными
свойствами по отношению к несульфидным минералам. Катион-
ными собирателями наиболее легко флотируется барит.
Минералы обладают близкими флотационными свойствами.
Особенно трудно разделить минералы с одинаковым катионом
в кристаллической решетке (например, кальцит, флюорит и
шеелит). Их разделение и отделение от минералов породы
(кварца, силикатов, других карбонатов, гидроокислов железа)
производится с применением жидкого стекла, карбоната и
сульфида натрия, солей алюминия, меди или железа, щелочных
пирофосфатов, метафосфатов и полифосфатов, органических
реагентов (таннина, декстрина, лигносульфоновых кислот
и др.) в тщательно отработанных условиях по щелочности или
кислотности пульпы, температуре, плотности пульпы,
длительности предварительной обработки реагентами и т. д.
Наиболее крупным объектом флотационного обогащения
являются фосфатные, шеелитовые, флюоритовые и баритовые
руды.
25&
Флотация барита из руд. Барит относится к легкофлотируе-
мым минералам. Его флотируемость олеиновой кислотой и
смесью карбоновых кислот улучшается в щелочной среде (при
рН>7). Соли двухвалентных (меди, свинца, цинка) и
трехвалентных (железа, алюминия) металлов оказывают депрессиру-
ющее действие на флотацию барита. Смесь жидкого стекла и
хромата калия депрессирует барит при небольших расходах.
Органические реагенты (таннин, крахмал, декстрин, глюкоза,
сульфитцеллюлоза) депрессируют барит только в
сильнощелочной и кислой среде, являясь активаторами его флотации при
рН 7—9.
Барит флотируется обычно в щелочной среде олеиновой
кислотой, талловым маслом в смеси с керосином, сульфатным
маслом, нафтеновыми кислотами или алкилсульфатами при
расходе 0,5—1,5 кг/т. Наибольшей селективностью обладают ал-
килсульфаты с аполярной цепью, содержащей 15—17 атомов
углерода.
Легче всего барит извлекается из руд, пустая порода
которых представлена кварцем и силикатами, легко депрессирую-
щимися уже при небольших расходах жидкого стекла,
несколько активирующих флотацию барита.
Расход депрессора резко возрастает (до 1,5 — 4 кг/т) с
возрастанием в руде содержания карбонатов кальция и магния.
Расход всех реагентов снижается, если в качестве" собирателя
применяется дитиофосфат A00—150 г/т), обеспечивающий,
кроме того, возможность флотации в жесткой воде без
предварительного обесшламливания. Поскольку повышенная
концентрация жидкого стекла оказывает депрессирующее действие и
на флотацию барита, то при значительном содержании
карбонатов кальция и магния в руде оказывается целесообразной
обработка или промпродуктов в отдельном цикле, или чернового
баритового концентрата по методу Н. С. Петрова. Метод
Н. С. Петрова заключается в пропарке предварительно сгу:
щенного (до 50—60 %) твердого концентрата в течение 30—
60 мин в растворе жидкого стекла @,3—2 %) при температуре
80—85 °С, разбавлении холодной водой до 25—40 °С и
последующей флотации барита. В процессе пропарки собиратель де-
сорбируется с поверхности загрязняющих концентрат частиц
кальцевых минералов и их флотация практически полностью
депрессируется.
Если в руде содержится значительное количество
окисленных минералов железа, то флотацию барита проводят в
содовой среде (при рН 11) карбоксильным собирателем; в
качестве депрессора применяются окислы железа метасиликата
натрия @,5—1 кг/т).
В большинстве случаев барит извлекается из
полиметаллических руд. Получаемые баритовые концентраты используются
или в химической промышленности, или в качестве
утяжелителя при бурении нефтяных скважин. Высокосортные концент-
9»
259
раты для химической промышленности содержат до 95 %
барита. Плотность концентрата для нефтяной промышленности
должна быть 4,1—4,3 г/см3, а содержание класса —10 мкм не
более 5—7 % [41].
Схемы флотационного извлечения барита сравнительно
просты. Они предусматривают обычно основную, контрольную
и две-три перечистные операции. Иногда концентрат последней
перечистной флотации подвергается классификации в
гидроциклонах, пески которого являются концентратом для
нефтяной промышленности. Из слива гидроциклона после двух-трех
перечистных операций получают концентрат, пригодный для
химической промышленности.
Флотация флюорита из руд. Флюорит извлекается из
силикатных и карбонатных флюоритовых, барито-флюоритовых,
а также сульфидных руд и руд редких металлов. Он довольно
легко флотируется оксигидрильными собирателями:
олеиновой кислотой, олеатом натрия, аэрозолями ОТ и МА (диалкил-
сульфосукцинатом натрия), алкилсульфатом. Максимальная
адсорбция собирателя и флотируемость минерала
наблюдаются при рН 6. Жидкое стекло снижает адсорбцию анионного
собирателя. Лимонная кислота депрессирует флотацию
флюорита. Соли алюминия особенно в смеси с жидким стеклом и
органические реагенты (декстрин, лигнинсульфонаты и Др.),
депрессирующие барит и кальцит, даже несколько активируют
флотацию флюорита. По результатам лабораторных
исследований разделение флюорита и кварца может быть осуществлено
с применением катионного собирателя; причем при рН 1—3
флотируется флюорит, а при рН 11 —14 — кварц.
На флотацию поступают или исходная руда, или хвосты
гравитационного обогащения (в целях выделения крупновкрап-
ленных флюорита и барита), или хвосты сульфидной флотации.
Если порода представлена силикатными минералами,
высокое извлечение флюорита достигается при небольших расходах
@,2—0,3 кг/т) оксигидрильного собирателя и жидкого стекла.
Для повышения селективности флотации флюорита из
карбонатных руд депрессия кальцита осуществляется обычно
в сильнощелочной среде, создаваемой едким натром @,4—
0,6 кг/т), с последовательной подачей жидкого стекла с
модулем 2,6—2,8 @,45—0,6 кг/т) и соли алюминия @,6 — 0,8 кг/т).
Дополнительная подача декстрина @,6 кг/т), лигнинсульфо-
ната или других подобных им органических реагентов
усиливает депрессию кальцита и активирует флотацию флюорита.
Селективность флотации может быть повышена подогревом
пульпы- до 40—50 °С и некоторым усложнением схемы в
результате введения дополнительных перечистных операций или
выделения промпродуктов для переработки в отдельном цикле.
Наибольшие трудности возникают при флотации барито-
флюоритовых руд. В этом случае возможна как схема прямой
селективной флотации барита и флюорита, так и схема с пред-
•260
Руда
л.
\
Измельчение
^50~70%f07^MM
Сода (WQ-Щг/т), жидкое стекло
(ЗО0-6ОО?/т)
Перемеш.иванйе
Алкилсульфат н-атрияA00^00г/т)
лк
Цикл баритовой
флотации
Баритовый
концентрат
\Цикл флнуоритовой
флотаци и
Жидкое стекло G0О-70ООг/т),
олеиновая кислота G50 -200г/т)
Флюоритовый
концентрат
Руда
Отвальные
хвосты
^Измельчение
'6О-7О%-0,07¥мм
Декстрин E00-7000 г/т)
Перемешивани е
щикл флюоритовой флотации
(у\\ 6,9-7)
MZ
Крахмал,
хромпик,
олеиновая кислота,
сосновое масло
Флюоритовый
концентрат
щикл баритовой
флотации
Лимонная кислота,
олеиновая кислота,
сосновое масло
MZ
баритовь/и
концентрат
Отвальные
хвость)
Рис. 14.7. Принципиальные схемы селективной флотации барито-флюорито-
вых руд -
варительной коллективной флотацией обоих минералов и
дальнейшим выделением в пенный продукт флюорита.
Варианты схем селективной флотации изображены на
рис. 14.7. ч
По первому из них (см. рис. 14.7, а) вначале флотируют
барит при небольших расходах (менее 0,4 кг/т) алкилсульфата,
а затем — флюорит олеиновой кислотой @,2 кг/т) в
присутствии жидкого стекла @,8— 1 кг/т).
По второму варианту схемы (см. рис. 14.7, б) сначала
флотируют флюорит при депрессии барита хромпиком и
органическими реагентами (крахмалом и декстрином), а затем — барит
при депрессии минералов пустой породы лимонной кислотой.
Коллективный барито-флюоритовый концентрат
разделяется при депрессии барита таннином и солью закисного железа,
декстрином и бихроматом калия, КМЦ и сернокислым
алюминием, лигнинсульфанолом, декстрином или крахмалом.
Селективность разделения иногда может быть улучшена
использованием слабокислой среды (рН 4,6—4,8) и подогревом пульпы
до 40—45 °С. Флотационные флюоритовые концентраты
содержат 92—98 % CaF2 и используются для получения плавиковой
кислоты и фтористых солей. Вредными примесями в них
являются кремнезем (не более 1,5—3 %) и кальцит (не более 2—
3 %). Извлечение флюорита в концентрат в зависимости от
состава руд колеблется от 78 до 91 %. Вместе с флюоритом
переходят в концентрат связанные с ним редкоземельные
элементы.
Флотация фосфатных минералов из руд. К основным
промышленным фосфатным минералам относятся апатит,
фосфорит и монацит. Если апатитовые и фосфоритовые руды являются
основным источником получения фосфатных удобрений, то
редкоземельный фосфат (монацит) является основным
промышленным источником получения тория из ториевых руд.
Апатит представляет собой фосфат кальция переменного
состава с общей формулой Са5(С1, F, ОН) (Р04)з, содержащий
от 41 до 42,2 % Р2О5, фосфориты представляют собой
скопления тонко дисперсных фосфатов типа <хСа3 (Р04) 2 ■ pCaF2 -
• уСаСОз, содержащих 35—36 % Р205 в осадочных породах. Это
вещество является цементом в желваковых фосфоритных
рудах или, наоборот, цементируется нефосфорным цементом
в зернистых или ракушечных фосфоритах.
Апатит и фосфорит обладают, близкими флотационными
свойствами. Они легко флотируются оксигидрильными
собирателями. Однако невысокая стоимость минералов требует
применения дешевых реагентов. Поэтому наиболее широко
используются заменители жирных кислот или мыл: сульфатное мыло,
талловое масло, смесь жирных кислот, торфяная смола и
другие реагенты, часто в смеси с аполярными собирателями.
Апатит может быть сфлотирован также аминами.
Апатитовый концентрат для производства суперфосфата
262
должен содержать не менее 39,4 % Р2О5, а концентрат для
производства термофосфата 20—22 % Р2О5. Вредными
примесями в фосфатных концентратах являются окислы железа,
алюминия, карбонаты магния и кремнезем [14].
Апатит из апатито-нефелиновых руд Хибинских
месторождений, содержащих 15—18 % Р2О5, извлекается флотацией
в естественной щелочной среде (рН 9,3—9,7), обусловленной
присутствием нефелина (Na, KhAUSisOs-ftSiC^, смесью
технических продуктов (сульфатного мыла и отходов мыловаренного
производства) при расходе 0,2—0,4 кг/т с добавками едкого
натра @,1—0,2 кг/т) для омыления собирателя и жидкого
стекла @,015—0,1 кг/т) для пептизации шламов и. депрессии
минералов породы. Расход жидкого стекла может возрасти
в несколько раз, если в процессе горных работ для
предотвращения смерзания руды в нее добавляют соль (хлористый
натрий). При флотации руд (особенно окисленных) с повышенным
содержанием шламов для регулирования свойств пены и
повышения смерзания руды в нее добавляют соль (хлористый нат-
оксипропиленовых соединений (например, реагентов ОП-4 или
альфапол-4). Получение кондиционного по крупности (не
более 14% класса —0,15 мм) и по содержанию C9,4—39,5 %
Р2О5) концентрата с извлечением в него 95—96 % фосфора
достигается по простой одностадиальной схеме после двух-трех
перечистных операций [9].
Для повышения комплексности использования
апатито-нефелиновых руд хвосты флотации обесшламливаются, а затем
при рН 10,5 и расходе около 1,5 кг/т смеси собирателей
(сырого и дистиллированного таллового масла, окисленного петро-
латума) флотируют в коллективный концентрат ванадийсодер-
жащий эгирин, сфен и титаномагнетит. Камерный продукт
представляет собой нефелиновый концентрат, содержащий не
менее 29 % А1203.
Флотационное извлечение апатита из гематитовых руд
осуществляется при рН 8,7 с применением в качестве собирателя
эмульгированной смеси таллового и машинного масел A:2)
при небольшой добавке алкиларилсульфонатов с общим
расходом 0,9 кг/т. Депрессия окислов железа обеспечивается
подачей жидкого стекла @,2 кг/т).
С целью повышения эффективности флотации апатита из
апатито-карбонатных железных руд для депрессии карбонатов
(кальцита, доломита) рекомендуется применение соды (до
2 кг/т) с жидким стеклом @,75 кг/т) или каллогено-таннидным
реагентом @,2 кг/т) или применение смеси оксиэтилированных
кислот С7 — С2о> соды, крахмала и жидкого стекла (в
соотношении 1:2:2:5) при общем расходе смеси 2,5 кг/т.
Трудности флотационного извлечения фосфорита из руд
обусловлены его тонкой вкрапленностью, наличием карбонатов
кальция и магния, растворимых солей и шламистого
материала.
263
При отсутствии в руде карбонатов щелочноземельных
металлов разделение фосфорита и силикатной породы из обесш-
ламленного материала может быть осуществлено или прямой
анионной флотацией фосфорита, или обратной флотацией
силикатов аминами @,2—0,5 кг/т) с небольшой добавкой
крахмала. Шламы присоединяются к фосфоритному концентрату.
Наличие в пульпе глауконита нарушает избирательность
флотации фосфорита, поскольку глауконитовые шламы депрес-
сируют фосфорит, а фосфоритовые шламы активируют
глауконит. Эффективное обогащение кварцево-глауконитовых фосфа-
тизированных песков достигается применением
предварительной магнитной сепарации руды, обеспечивающей выделение
слабомагнитного глауконита в отдельный продукт. В свою
очередь, селективная флотация пеобесшламленных глинисто-глау-
конитовых фосфоритных руд (например, Егорьевского
месторождения) становится возможной только после
предварительного обжига крупнокусковой руды при температуре 700—
1000 °С, в процессе которого фосфорит переходит в
минеральные формы с кристаллической структурой апатита и более
высокой флотационной активностью при сохранении низкой фло-
тируемости минералов породы: кварца и глауконита [14].
При небольшом содержании карбонатов в руде может
использоваться технологическая схема, разработанная в
лабораторных условиях для кингисепских фосфоритов (см- 14.8, а) и
состоящая из двух циклов: анионного и катионного. При кати-
онной доводке полученного из кингисепских руд F—7 % Р2О5)
концентрата в результате удаления силикатов содержание
Р2Оп возрастает до 31—33 % при извлечении фосфора 77—
80 %• Если основную флотацию проводят с катионным
собирателем и фосфориты остаются в хвостах, то их сгущают,
добавляют жидкое стекло для депрессии фосфоритов и флотируют
карбонаты талловым маслом или другими техническими
мылами и продуктами.
При большом содержании карбонатов в руде сначала
флотируют карбонаты мылами жирных кислот или синтетическими
жирными кислотами (С10 — Q6) при депрессии фосфоритов
фосфорной кислотой в слабокислой среде (рН 4,5 — 5), а затем
в щелочной среде, создаваемой содой, флотируют фосфориты
смесью жирной кислоты и аполярного собирателя (рис. 14.8, б).
Флотации фосфоритов предшествует операция обесшламлива-
ния. По такой схеме из руд Каратау B2—23 % Р205)
получают фосфоритовый концентрат, содержащий 29 % Р2О5 при
извлечении в него фосфора около 90 %.
Если карбонаты представлены в основном кальцитом, то его
можно сфлотировать алкилсульфатом без подачи реагентов для
депрессии фосфорита, и после нескольких перечистных
операций получить кондиционный карбонатный продукт D0 % СОг).
Фосфорит флотируется из хвостов в присутствии жидкого
стекла с использованием обычно жирных кислот C00—500 г/т).
264
Руда
6
X
Измельчение
Сода A-1,5кг/т),
жидкое стекло @,8 к г/т),
таллсвоемасло A'кг/т),
соляровое масло {?,2кг/лт)
/
L-f
U и к л
фосфоритной флота и и а
Обе с тмламли3a nи е
Хвосты
Слив
I Серная кислотаB~^5кг/т)
Перемешивание
1
С г у щ е н и е
J | АНП-2@,2-0^кг/т)
Силикатный
продукт
(8 отвал)
л
л
Цикл флотации сили-\
катов породы
Руда
6-
Фосфоритовый
концентрат
) Из мель чение
^ -0,15 мм
Обе с шл а м л и В а пае
Шламы
\\/
Фосфорная кислота Fкг/т),
СЖКСю-См @,3кг/т)
Цикл флота ции каро'оиа/под\
ш ГрН Ь,8-5)
Карбонатный
продукт
X
Сода A,5кг/т),
жидкое стекло @,5к г/т),
/лалло воемасла {f/7/rг/лгу,
керосин @,5кг/т)
Z
шикл флотации фосфо-
ритов (у\\ 7,6-8)
Фосфоритовый
ксни?нп77)ат
Хвосты
Рис. 14.8. Принципиальные схемы селективной флотации фосфоритовых руд
Кингисепа (а) и Каратау (б)
Флотация монацита с сульфированными растительными
маслами в кислой среде и алифатическими аминами с крахмалом
в щелочной среде применяется для доводки гравитационных
монацитовых концентратов, получаемых при обогащении тори-
евых руд или из хвостов флотации медно-молибденовых руд.
Для флотационного отделения монацита от сопутствующих
ему циркона, ильменита и граната при переработке россыпей
можно использовать прямую или обратную флотацию
монацита. Прямая флотация монацита проводится с эмульсией
олеиновой кислоты при депрессии сопутствующих минералов
серной и фосфорной кислотами, а также едким натром. При
обратной флотации гранат, ильменит и циркон флотируют
аминами в солянокислой среде при депрессии монацита фтористым
натрием [34].
Монацит из хвостов сульфидной флотации коренных
монацитовых руд извлекают флотацией олеиновой кислотой
A,8 кг/т) при депрессии пустой породы жидким стеклом^
A,4 кг/т) и кальцинированной содой (рН 8,5). После трех пе-
речистных операций с применением жидкого стекла монацито-
вый концентрат содержит 50 % Th02 и Th203 [34].
Флотация шеелитовых и вольфрамитовых руд. Шеелитовые
и вольфрамитовые руды являются источником получения
вольфрама, используемого в основном для производства
легированных сталей. Поэтому к вредным примесям в получаемых
концентратах относятся фосфор, мышьяк и другие элементы,
ухудшающие качество сталей.
Шеелитовые концентраты должны содержать 50—60 %
W03 и получают их только флотацией. Флотационные
вольфрамитовые концентраты, содержащие 30—50 % W03, Получают
гораздо реже и в основном из шламов гравитационного
обогащения.
Основные минералы вольфрама — шеелит CaW04,
вольфрамит (Mn, Fe)W04 (иногда гюбнерит MnW04, ферберит FeW04)
флотируются собирателями типа жирных кислот @,1—0,4 кг/т)
в щелочной среде (рН 9—10), создаваемой содой A—5 кг/т).
Для подавления пустой породы, представленной в основном
силикатами (кварцем, полевыми шпатами, слюдами и другими
минералами), используется жидкое стекло A—7 кг/т), кремне-
фтористый натрий, бихромат или декстрин. Удаление
сульфидов, содержащихся иногда в исходной руде или продуктах
гравитационного обогащения, производится в начале процесса
с применением сульфгидрильных собирателей.
Получаемые черновые концентраты загрязнены
кальциевыми минералами (кальцитом, апатитом, флюоритом и др.),
некоторыми силикатами (топазом, слюдами и др.), остатками
сульфидных минералов и поэтому направляются после
предварительного сгущения до 50—60 % твердого в цикл доводки
концентрата.
Доводка черновых шеелитовых концентратов, содержащих
266
3—5 % W03 и обычно некоторое количество окисленного
молибдена, осуществляется в мировой практике по
разработанному в СССР методу Петрова. Сущность метода заключается
в избирательной десорбции собирателя с поверхности
карбонатов кальция, магния и силикатных минералов в процессе
пропарки черновых концентратов в 3—4 %-ном растворе жидкого
стекла при 85—90 °С в течение 30—60 мин. В результате этого
минералы породы депрессируются, а шеелит и обычно
сопутствующий ему повеллит СаМ604 сохраняют свою флотируемость.
После разбавления холодной водой до 25—30 % твердого и
двух-трех перечистных операций флотации получают
кондиционный концентрат. Если черновой концентрат содержит
значительное количество барита, флюорита, апатита или легкофло-
тируемых алюмосиликатов, полной депрессии которых в
условиях метода Петрова обычно не достигается, то он поступает
на дальнейшую обработку в целях удаления вредных примесей
фосфора (апатита), фтора (флюорита), серы (барита).
До настоящего времени некондиционный по фосфору шеели-
товый концентрат выщелачивают соляной кислотой. При этом
вместе с апатитом растворяется повеллит и, частично, шеелит и
для выделения из раствора вольфрама и молибдена необходимо
применять гидрометаллургические методы. В то же время
известно, что минералы с анионами SO|~ (например, целестин,
барит), РО|- (например, апатит)-, F~ (например, флюорит)
можно отделить от минералов с анионами WO|~ (например,
шеелита, вольфрама, гюбнерита), МоО^- (например, повеллита),
SiO|~ (силикатов) флотационным путем по методу Л, И. Грос-
мана.
По этому методу концентрат сначала перемешивается в
кислой среде (при рН 1,5—2), создаваемой соляной, серной,
фосфорной или щавелевой (но не плавиковой) кислотами. При
этом образованные на поверхности минералов с анионами
WO|~, МоО^- и с адсорбированными на поверхности силикатов
катионами мыла разлагаются, катионы переходят в раствор,
на поверхности образуется как бы пленка гидрофильной
вольфрамовой, молибденовой или кремневой кислоты, и минералы
теряют флотируемость. В этих же условиях минералы с
анионами SO^, РО^~ и F" эффективно флотируются алкилсульфа-
том или алкилсульфонатом @,2—0,3 кг/т), обеспечивая
получение в камерном продукте кондиционного по вредным примесям
шеелитового концентрата.
Использование метода Л. И. Гросмана позволило решить,
например, проблему разделения барито-шеелитовых
концентратов и повысить комплексность использования руд Чорух-Дай-
рона. Получаемые шеелитовые концентраты могут содержать
некоторое количество силикатов. Их отделение от минералов
вольфрама и молибдена может быть осуществлено после
267
отмывки от кислоты флотацией шеелита и повеллита,
восстанавливающих флотируемость при доведении рН до 6—7.
На отечественных обогатительных фабриках из руд,
содержащих ОД— 0,4 % W03 и 0,03—0,1 % окисленного молибдена,,
получают концентраты, содержащие 60—70 % W03 при
извлечении 80—90 % и 8—10 % окисленного молибдена при
извлечении около 65 %.
При доводке флотационных вольфрамитовых концентратов
вместо метода Петрова может быть использована дофлотация
чернового концентрата или в слабокислой среде (при рН 5—6),
или после пропарки с кремнефтористым натрием при 80—85 °С.
Значительное улучшение качества концентрата достигается
благодаря депрессии силикатных минералов и значительной
части флюорита. Для дальнейшего удаления вредных примесей
можно воспользоваться методом Л. И. Гросмана.
В Механобре разработана технология обогащения
вольфрамовых руд с применением реагента ИМ-50, позволяющего уже
на стадии основной флотации достаточно полно удалить в
хвосты не только кварц и полевой шпат, но и слюду, флюорит, эии-
дот, ожелезненпые силикаты и окислы железа [44]. При
значительном содержании в руде флюорита основная флотация
проводится в слабокислой среде (рН 5,5 — 6,5), а перечистные —
при рН 5. Оптимальное значение рН в обычных условиях
равно 8—9. Применение небольших расходов G0—80 г/т) крем-
нефтористого натрия и реагента ОП-4 заметно улучшает
селективность процесса. Доводка полученного концентрата
осуществляется дофлотацией его в сильнокислой среде (рН 1,5) с
небольшими добавками реагента ИМ-50. При флотации пгламов
гравитационного обогащения вольфрамитовых и гюбнеритовых
руд, содержащих 0,11—0,43% W03, получены концентраты
с содержанием 30 — 50 % W03 и извлечением в них 65 — 74 %
вольфрама.
14.6. Флотация окислов металлов
Краткая характеристика минералов-окислов. Группа
окислов весьма многочисленна и многообразна. В настоящее время
флотация применяется для извлечения из руд окислов железа,
марганца, хрома, титана, тантала, ниобия, олова, алюминия,
урана, тория и некоторых других металлов.
По сравнению с предыдущими группами минералов окислы
обладают более высокой степенью гидрофильности, требуют
более тщательного регулирования условий флотации, а некоторые
из них — применения сложных схем и новых, более
эффективных реагентов для осуществления процессов активации,
дезактивации минералов при их селективной флотации.
Флотация железных руд. Для обогащения железных руд
используются в основном методы магнитной сепарации и
гравитации. Флотация применяется главным образом для обогаще-
268
ния тонковкрапленных гематитовых, мартитовых и бурожелез-
няковых руд, извлечения тонковкрапленных слабомагнитных
окислов железа из хвостов магнитной сепарации, доводки
бедных концентратов гравитационного обогащения и получения
«суперконцентратов» для нужд аккумуляторной
промышленности и порошковой металлургии.
К основным промышленным минералам в железных рудах
относятся магнетит Fe304, гематит и мартит Fe203, гетит и
водные окислы железа, объединяемые под общим названием
«бурые железняки» и с общей формулой Fe203 -A120H,5-3, и сиде- .
рит FeC03. Все они могут флотироваться оксигидрильными
собирателями. Так как железные руды являются относительно
дешевым сырьем, то при их флотации используются наиболее
дешевые собиратели из группы жирных, нафтеновых кислот и
оксикислот или побочные продукты различных отраслей
промышленности, из которых наибольшее распространение
получили талловое масло и сульфатное мыло. Чтобы несколько
понизить чувствительность жирных кислот к солям жесткости,
применяются добавки эмульгаторов (типа реагентов ОП) и ал-
килсульфатов. При высокой концентрации солей жесткости
в пульпе более целесообразно применение нечувствительных
к ним оксикарбоновых кислот. Повышение селективности окси-
гидрильиого собирателя при флотации железных руд
наблюдается при увеличении числа двойных связей в его аполярной
цепи или введении атома азота в его молекулу. .
Флотируемость , минералов железа понижается с
увеличением содержания связанной воды и уменьшением плотности
структуры. В обычных условиях наблюдается следующий ряд
их флотируемости оксигидрильными собирателями: сидерит>
>гематит, мартит>магнетит>гетит. Предварительное
окисление магнетита, приводящее к увеличению содержания
трехвалентных ионов железа в поверхностном слое, улучшает его
флотируемость.
Гидроксильные ионы, катионы щелочноземельных металлов
и жидкое стекло депрессируют, а сода активирует флотацию
всех железных минералов. Органические депрессоры —
крахмал, КМЦ и другие аналогичные реагенты подавляют
флотацию окислов железа как при анионном, так и катионном
собирателях. Отмеченные особенности флотационного поведения
минералов железа используются при их отделении от породы,
представленной обычно кварцем, полевым шпатом,
амфиболами, пироксенами, хлоритами, гранатом, эпидотами,
оливинами, кальцитом в различном соотношении в зависимости от
генезиса месторождения.
При флотации железных руд применяются три основных
метода: прямая анионная, обратная анионная или обратная ка-
тионная флотация [41, 42].
Прямая анионная флотация окислов железа
обычно проводится при значениях рН, равных 6—7 в опера-
269
циях основной и контрольной флотации и 5,5 — 6 в перечист-
ных операциях. Для регулирования рН применяется серная
кислота (до 0,8 кг/т). Иногда флотация проводится в содовой
среде (рН до 9,5—10). Для депрессии минералов породы
загружается жидкое стекло или коллоидная кремневая кислота
(до 1 кг/т). Селективность действия жидкого стекла
увеличивается при добавке солей алюминия, меди и др. Если в руде
содержится апатит, предпочтительна флотация в слабокислой
среде при добавках фторидов или фторсиликатов.
Для нейтрализации действия солей жесткости,
связывающих собиратель, активирующих пустую породу и* депрессирую-
щих окислы железа, дополнительно подаются небольшие
количества фосфатных соединений, сернистого натрия и других
реагентов. Расход собирателя зависит от пористости полезных
минералов и достигает 1 кг/т при флотации наиболее
пористых из них — гидроокислов железа, флотирующихся менее
эффективно, чем гематит и мартит. При наличии в руде силикатов
железа, значительных количеств апатита, кальцита и ангидрита",
переходящих при прямой анионной флотации в концентрат и
загрязняющих его, более целесообразно применение обратной
флотации.
Обратная анионная флотация минералов породы
производится в сильнощелочной известковой среде (при рН
около 11). Железный концентрат получается в виде камерного
продукта. Депрессия флотации минералов железа
обеспечивается: депрессирующим действием гидроксильных ионов,
подачей органического депрессора — крахмала, КМЦ, лигнинсуль-
фоната или другого аналогичного им реагента при расходе
0,6—1 кг/т и, наконец, депрессирующим действием ионов
кальция. В то же время ионы кальция играют роль активатора
силикатов породы. К достоинствам обратной анионной флотации
относится возможность использования жесткой воды при
расходе собирателя 0,2—0,6 кг/т.
Обратная катионная флотация силикатных
минералов породы проводится в содовой среде при рН 8—9 аминами
или их солями @,2—0,4 кг/т). Для депрессии окислов железа
применяются крахмал, декстрин, таннин @,5—1 кг/т). Вместо
них могут использоваться менее дефицитные сульфитцеллюлоз-
ный щёлок, отходы мукомольного производства, КМЦ и другие
органические депрессоры. Вредное влияние на процесс катион-
ной флотации оказывают ионы железа и алюминия,
адсорбирующиеся в первую очередь на силикатных минералах,
вызывая тем самым их депрессию.
Схемы флотации железных руд достаточно просты. Они
предусматривают обычно основную, иногда контрольную
флотацию и две-три перечистных операции. Результаты обогащения
всеми тремя методами флотации железных руд примерно
одинаковы. Выбор метода в каждом конкретном случае
определяется стоимостью передела.
270
К характерным особенностям флотационного обогащения
железных руд относятся:
большая чувствительность процесса флотации к
интенсивности перемешивания пульпы, с уменьшением которой
селективность процесса возрастает;
необходимость применения возможно более плотных пульп
при флотации;
необходимость предварительного удаления шламов при
значительном содержании их в руде, чтобы избежать излишнего
поглощения реагентов и нарушения селективности
флотационного процесса. Предотвратить потери железных минералов со
шламами в ряде случаев удается с помощью реагентов
(например, крахмала, тапиока), вызывающих селективную
коагуляцию окислов железа.
Качество получаемых флотационных железных
концентратов зависит от состава руд. Так, концентраты, получаемые при
флотации магнетито-гематитовых руд, содержат не менее 62—
65 % железа; при флотации бурожелезняковых руд 43—50 %
железа, при флотации сидеритовых руд 30—35 % железа.
Советскими исследователями показана возможность получения
в результате обратной флотации сверхбогатых
«суперконцентратов», содержащих более 70 % железа. Во всех случаях
содержание вредных примесей в концентратах должно быть не
более, %: серы 0,1—0,3; фосфора 0,15—0,2; мышьяка 0,07 — 0,1;
свинца 0,01—0,015. Извлечение железа в концентраты зависит
от содержания его в рудах A3—35 %) и изменяется от 65 до
85%.
Флотация хромитовых руд. Флотационные свойства
единственного промышленного минерала хрома—хромита FeO-Cr203
близки к флотационным свойствам окислов железа.
При прямой анионной флотации хромита для депрессии
основных минералов пустой породы (серпентина и оливина)
в кислой среде используются фтористый и кремнефтористый
натрий, а в щелочной среде — КМЦ, сульфитцеллюлозные
щёлоки и жидкое стекло. При наличии в руде кальцита
дополнительно подают небольшие количества гексаметафосфата
натрия. Обратную катионную флотацию минералов породы
проводят после предварительного обесшламливания в
сильнощелочной среде (рН больше 12) с применением в качестве
собирателя первичных алифатических аминов.
Получаемые хромитовые концентраты используются в
производстве феррохрома.
Флотация марганцевых руд. Флотации подвергаются только
шламы (—0,1 мм) гравитационного и магнитного обогащения
марганцевых руд. Вследствие высокой пористости полезных
минералов, наличия в них включений глины и сильной ошламо-
ванности как минералов марганца, так и пустой породы,
марганцевые шламы являются трудным объектом для
флотационного обогащения.
271
Основные промышленные минералы марганца пиролюзит
Мп02, браунит Мп203, манганит Мп203-Н20, псиломелан-вад
(М.п02)т*(№пО)п (Н20)р, родохрозит МпС03 характеризуются
высокой гидратированностью поверхности, легко шламуются и
обладают худшей флотируемостью по сравнению с окислами
железа. Предварительная кислотная обработка резко снижает
степень гидратированности минералов марганца, приводя
к улучшению флотируемости и повышению скорости их
флотации при снижении расхода собирателя. В качестве собирателя
используются те же реагенты и продукты, что и при флотации
железных руд (талловое масло, окисленные углеводороды,
нафтеновые кислоты, побочные продукты нефтеперерабатывающих
заводов, содержащие карбоновые кислоты и их мыла). Их
собирательная способность возрастает при добавке углеводород-,
ных масел и эмульгаторов и предварительном эмульгировании
собирателя. В общем случае в присутствии оксигидрильного
собирателя флотируемость окислов марганца уменьшается в
следующем порядке: браунит>манганит>пиролюзит>псиломе-
лан-вад [41, 42].
Флотационное отделение минералов марганца от минералов
породы, представленных в основном кварцем, полевыми
шпатами, глиной, кальцитом, доломитом, песчаниками,
глауконитом, производится по схемам и режимам, принципиально не
отличающимся от схем и режимов прямой и обратной анионной
флотации железных руд.
Оптимальные значения рН, равные при прямой анионной
флотации 7—9 и при обратной 9—10, создаются обычно содой
и лишь иногда едким натром. Собиратель подают порционно
при общем расходе его 2—3 кг/т. При наличии карбонатов
марганца их флотируют перед флотацией окислов марганца по
схеме прямой анионной флотации. Для депрессии породы
используется жидкое стекло @,4—0,7 кг/т). Флотации
предшествует обесшламливание по классу —10—15 мкм,
сопровождающееся большими потерями марганца в шламовой фракции.
По этой причине разработка эффективного режима
флотационного обогащения марганцевых материалов должна включать
в качестве обязательного этапа селективную коагуляцию или
флокуляцию марганцевых минералов неорганическими и
органическими реагентами.
Селективная коагуляция или флокуляция позволяет резко
снизить потери марганца при обесшламливании или вообще
исключить эту операцию из схемы, как, например, при так
называемой эмульсионной или агломерационной флотации, когда
центрами коагуляции являются капельки эмульгированного
собирателя. Эффективная флотация таких флокул требует
особого гидродинамического режима работы и особой конструкции
флотационной машины. Наиболее подходящими для этих целей
в настоящее время являются машины для пенной сепарации,
флокулярной флотации и, возможно, колонные флотационные.
272
В результате флотации получают марганцевые
концентраты, содержащие 22—46 % марганца, при извлечении его
в концентраты 80—90 % от обесшламленного питания
флотации, но только 40—70 % от исходного материала. Содержание
фосфора в концентратах не должно превышать 0,2 %.
Флотация минералов титана и циркония из руд и россыпей.
В коренных и россыпных месторождениях основными
титановыми минералами являются ильменит FeTi03, рутил ТЮг, иль-
менорутил (Ti, Nb, Fe) 02, перовскит СаТЮ3 и сфен CaTiSi20b.
Из минералов циркония промышленное значение имеют циркон
ZrSi04 и бадделеит Zr02.
Флотация титановых и циркониевых минералов применяется
при переработке коренных руд и россыпей, продуктов их
гравитационного и магнитного обогащения, для доводки и
разделения коллективных титано-циркониевых концентратов.
Титановые и циркониевые минералы флотируются жирно-
кислотными собирателями предельного и непредельного рядов,
их смесями, техническими продуктами (мылонафт, окисленный
петролатум) часто с добавками аполярных масел.
Оптимальные значения рН флотации всех минералов находятся в
области, близкой к нейтральной. Они могут быть сфлотированы и
алкилсульфатами, но только в кислой среде. Для повышения
селективности процесса при флотации рутила используют
сочетание жирных кислот с аминами. Наиболее флотоактивным
является циркон, затем рутил, несколько хуже флотируется
ильменит.
Сильными депрессорами титановых минералов являются
жидкое стекло и крахмал в содовой среде. Депрессированный
этими реагентами рутил можно активировать, подавая серную
кислоту до нейтральных значений рН, а перовскит — подавая
хромат или бихромат натрия. Депрессирующее действие на
флотацию рутила и ильменита оказывает продувка пульпы
азотом. В свою очередь, депрессия циркона достигается с помощью
кремнефтористого натрия, жидкого стекла, сернистого натрия
и медного купороса при1 расходе более 0,25 — 0,7 кг/т.
Селективность разделения титановых и циркониевых минералов
улучшается после предварительной кислотной обработки, в
результате которой с поверхности минералов не только
удаляются поверхностные образования (например, гидроокислы
железа, соли и др.), но и образуются новые соединения,
активирующие или депрессирующие флотацию минералов. Большое
значение имеет вид кислоты. Так, если серная кислота
активирует флотацию всех титановых минералов, то соляная кислота
избирательно депрессирует флотацию рутила.
Для депрессии минералов породы применяют фтористый
натрий, небольшие расходы жидкого стекла или значительные
расходы соды и жидкого стекла, избыток которых удаляют
в процессе предварительного обесшламливания пульпы. Обес-
шламливание, как и предварительное удаление сульфидных
273
минералов, является обязательной операцией, от тщательности
проведения которой зависит качество концентратов.
Флотацию ильменита из хвостов магнитной сепарации ти-
таномагнетитовых руд проводят при значениях рН, равных 6—
6,5 в основной флотации и 5—5,5 в перечистпых операциях.
Значения рН регулируются серной кислотой. Для депрессии
породы (граната и апатита) применяется фтористый натрий
или хромпик (до 0,2 кг/т).
Флотации перовскита обычно предшествует удаление
кальцита, поскольку перовскит флотируется жирными кислотами
после обработки его серной кислотой, последующей отмывки
(для удаления солей кальция и магния) и доведения значений
рН до оптимальных, равных 5,2—6. Кислотную обработку и
последующую отмывку в ряде случаев можно исключить, если
в качестве собирателя использовать реагент ИМ-50 (до
0,5 кг/т). Флотация в этом случае осуществляется в
слабокислой среде (рН 5—6,5). Минералы породы депрессируются
жидким стеклом (до 0,2 кг/т).
Рутил встречается в рудах обычно вместе с цирконом и
другими минералами. Для разделения, например, наиболее
часто встречающейся ассоциации рутил — циркон — ильмент
разработаны следующие методы:
депрессия рутила и ильменита в содовой среде жидким
стеклом (до 0,5 кг/т) или крахмалом (до 0,1 кг/т) и флотация
циркона жирнокислотным собирателем (до 5 кг/т). После
нейтрализации щелочности серной кислотой (до рН 7) флотируются
рутил и ильменит;
флотация циркона жирнокислотным собирателем при рН
8—9 после обработки коллективного концентрата газообразным
азотом, глубоко депрессирующим ильменит и рутил;
флотация циркона мылом при рН 11,4—11,5 после
предварительной промывки коллективного концентрата кислотой;
флотация циркона при рН 1,5—2 после предварительной
обработки коллективного концентрата раствором мыла @,2—
1 кг/т) и последующей промывки кислотой A0—15 кг/т);
флотация рутила и ильменита при рН 5,5—6 оксигидриль-
ным собирателем @,5—1 кг/т) или при рН 2 катионным
собирателем при депрессии циркона кремнефтористым натрием
B—3 кг/т). Последующее разделение рутила и ильменита
может быть достигнуто путем депрессии ильменита щавелевой
кислотой @,2 кг/т) и флотации рутила при рН 3,5—4.
Получаемые ильменитовые концентраты содержат, %; 38—
45 Ti02, не более 53,6 Fe203 и 2,5 SiC>2. Рутиловые концентраты
должны содержать не менее 94% ТЮг. Вредными примесями
в концентратах являются сера и фосфор. В цирконовых
концентратах содержание минералов циркония должно быть не
менее 75—80%.
Флотация тантало-ниобиевых руд. Тантало-ниобиевые руды
обогащаются в основном гравитационными методами. Флотация
274
Руда
На-катионированная вода
I Измельчение
[50%-О,О7Ьмм
Сода @,7кг/т), . *
жидкое стекло @,25кг/т), Обесшламливание
олеиновая кислота | ■" 1
@,15 к г/т)
Jl!
Цикл срлотаци и
кальцита ^рн Ц2)
Т
Аальцитовь/и продукт
Шлама/
t*z
АНП~7ч-(Оч2кг/т)
Цикл фл от а ц и и
слюды(№ 9,2)
\
ha-катиониро-
ванная вода
Слюдяной
продукт
Отмывка от реагентов
:м
Сернистый натрий@,05кг/т)
олеиновая кислота (во/кг/т)
\Шламы
Цикл пироаслор-цирк о новой
флот а и и и
Хлористый
натрий GJ кг/т),
серная кислота ( 7 кг/т),
алкилсульфот A,1 кг/т)
Кислотная обработка концентрата Полевошпатовый
E-8%ный р-р ПгЪОьвО'60мин) кониентрат
От мы Вка от кислоты
кремнефрюристыи
натрий @,3кг/т),
серная кислота/ф-кг/т)
илкижулырат/Щкг/т)
ч
_^д
Цикл доводки лирахлор-
цирконовога концентрата
(ph2,t-70°C)
г*
Слив
Эгирин -авгитовый
продукт
\Ц и к л флотации
ц и рко на •
*.
Серная кислотаA,5кг/т),
алкилсульфат (ЦО£кг/т),
АНП-14 (Ц03кг/т)
шикл флотации
п ироэслора
Ма г ни т на я
с е /7 а р-а ц и я
\ i .
Титаномагнети- Цирка новый /7ирохлоровый
товый продукт концентрат концентрат
Рис. 14.9. Принципиальная технологическая схема флотации пирохлор-цир-
коновых руд
используется для извлечения тантало-ниобиевых минералов из
хвостов и шламов гравитационного обогащения и тонковкрап-
ленных руд [34].
К основным минералам тантала и ниобия относятся
танталит (Fe, Mn).Ta206, колумбит (Fe, Mn) • Nb206, пиро-
хлор (Na, CaJ-(Nb, TiJ06-[F, ОН], микролит (Na, CaJX
Х(Та, TiJ06.[F;~OH] и.лопарит (Na, Се, Са) . (Nb, TiH3. Они
находятся в различном сочетании с полевыми шпатами, нефе-
275
лином, кварцем, биотитом, пироксеном, апатитом, кальцитом,
флюоритом, ильменитом, магнетитом, эгирином, сфеном,
цирконом, рутилом, сульфидными минералами.
Необходимость комплексного использования сырья и
близость флотационных свойств многих разделяемых минералов
требуют применения сложных схем флотации. В первых циклах
обычно производится удаление аполярных, сульфидных
минералов, слюд, минералов щелочноземельных металлов.
Дальнейшие циклы включают флотацию основных ценных окислов и
силикатов, доводку полученных концентратов и их разделение,
в процессе которых используются операции кислотной и
щелочной обработки различных продуктов, отмывки и обесшламлива-
ния. В качестве примера на рис. 14.9 приведена
принципиальная схема флотации пирохлор-цирконовых руд, разработанная .
в ВИМСе 149].
По данной схеме из измельченной и обесшламленной руды
предварительно флотируется кальцит (жирнокислотным
собирателем), а затем слюды (катионным собирателем). После
отмывки хвостов от жидкого стекла и солей жесткости N-катиони-
рованной водой флотируют с помощью олеиновой кислоты пи-
рохлор, циркон, сфен, ильменит, магнетит, гранат, эгирин при
депрессии полевых шпатов сернистым натрием. После
кислотной обработки получаемого пенного продукта и отмывки
выщелоченных с поверхности катионов F3+ и А13+ флотацией алкил-
сульфатом в присутствии серной кислоты и хлористого натрия
удается освободиться от основной массы породы, главным
минералом которой является эгирин. Отделение циркона,
ильменита и магнетита от пирохлора и остатка породных минералов
производится также флотацией алкилсульфатом, но в
присутствии кремнефтористого натрия. Отделяя ильменит и магнетит от
циркона магнитной сепарацией, получают богатый цирконовый
концентрат и титаномагнетитовый продукт. Пирохлоровый
концентрат получается в результате дофлотации хвостов цирконо-
вой флотации с добавками катионного собирателя и алкил-
сульфата.
По другому варианту схемы предусматриваются [34]:
коллективная флотация всех минералов олеиновой кислотой
в щелочной среде, создаваемой едким натром и содой с
получением в хвостах флотации полевошпатового концентрата;
перечистная флотация коллективного концентрата с содой и
сернистым натрием с получением в камерном продукте эгирин-
авгитового продукта;
дофлотация полученного концентрата с содой и жидким .
стеклом, обеспечивающим депрессию всех минералов, за
исключением кальцита, и получение кальцитового продукта;
флотация слюды из камерного продукта катионным
собирателем;
кислотная обработка камерного продукта и доводка его.
Разделение полученного пирохлор-цирконового концентрата
276
может быть осуществлено так же, как и в предыдущей схеме
(см. рис. 14.9).
В последние годы широкое применение при флотации руд
редких металлов получил комплексообразующий собиратель —
реагент ИМ-50. Принципиальная технологическая схема
флотации ниобийсодержащих минералов с ИМ-50 из пирохлоровых
руд и продуктов гравитационного обогащения, разработанная
в Механобре, изображена на рис. 14.10, а. Схема
предусматривает после удаления карбонатов и сульфидов получение нио-
биевого продукта (концентрата) для химико-металлургической
переработки и кондиционного полевошпатового концентрата.
Ниобиевый продукт содержит, помимо пирохлора, минералы
титана (ильменорутил, ильменит, сфен) и циркон и в принципе
может быть разделен на пирохлоровый, титановый и цирконо-
вый концентраты. В свою очередь, разделением сульфидно-
кальцитового продукта можно получить кальцитовый и
сульфидный концентраты. ,
Принципиальная схема флотации лопарита с реагентом
ИМ-50 из шламов гравитационного процесса приведена на
рис. 14.10,6. Она позволяет отделить флотацией лопарит и эги-
рин от основной массы пустой породы после предварительного
удаления апатита. Флотационное отделение лопарита от эги-
рина производится после кислотной обработки и отмывки при
депрессии эгирина щавелевой кислотой и гексаметафосфатом..
Лопаритовые концентраты должны содержать 8 % Nb2Os,
пирохлоровые — 30% Nb205. Вредными примесями в них
являются фосфор (не более 0,05%) и двуокись кремния (не
более 1,5%).
Флотация оловянных руд. Использование флотации для
дополнительного извлечения олова и повышения комплексности
использования оловосодержащих руд является одной из
основных тенденций развития технологии их обогащения [35]. В
первую очередь флотации подвергаются шламы гравитационного
обогащения кварцевокасситеритовых и сульфидно-касситерито-
вых руд, потери олова с которыми могут достигать 25—30%.
Основным промышленным минералом олова является
касситерит Sn02, иногда присутствуют небольшие количества стан-
нина Cii2FeSnS4. Сопутствующие им минералы обычно
представлены кварцем, турмалином, окислами железа, карбонатами,,
железистыми хлоритами, алюмосиликатами, сульфидами,
соотношение которых меняется при изменении типа руды [35].
Сложность проблемы селективной флотации касситерита от
минералов породы и получения богатых концентратов
обусловлена^ основном следующим:
главные разделяемые минералы — касситерит и кварц, не
флотирующиеся без предварительной активации поверхности
катионами металлов, с которыми мог бы взаимодействовать
анионный собиратель, активируются практически одними и
теми же катионами, поставляемыми в пульпу поверхностью
277
других минералов породы. Это приводит к нивелированию
флотационного поведения разделяемых минералов;
степень активации и характер активирующих касситерит
катионов изменяется в зависимости от состава сопутствующих
минералов и генезиса месторождения.
Решение проблемы селективной флотации касситерита
ведется как в направлении изыскания новых эффективных
собирателей для касситерита, так и по пути подбора условий
избирательной дезактивации и депрессии минералов пустой породы.
В нейтральной и щелочной средах избирательной
дезактивации минералов достигнуть обычно не удается. Этим
объясняется невозможность эффективного отделения касситерита в
данных условиях от ожелезненных или активированных кварца,
полевого шпата, железосодержащих силикатов, окислов железа
и других минералов, имеющих на поверхности катионы,
взаимодействующие с обычно применяемыми при флотации
оловянных руд и шламов оксигидрильными собирателями.
Избирательная дезактивация " может быть осуществлена
в кислой среде. Возможность ее обусловлена разной энергией
связи катионов на поверхности разных минералов и разной
скоростью их выщелачивания из поверхностного слоя. В
результате этого наблюдается разная скорость дезактивации для
разных минералов. Она зависит от характера катиона, природы
минерала, значения рН, свойств кислоты и ряда других условий.
Для избирательной дезактивации поверхности минералов
породы используются серная, соляная, щавелевая, лимонная и
другие кислоты. Их действие усиливается применением
реагентов-депрессоров: жидкого стекла, <гексаметафосфата, кремне-
фтористого натрия и их сочетаний.
Необходимым условием селективной флотации касситерита
является тщательное обесшламливание исходного питания
основной оловянной флотации и иногда вторичное обесшламлива-
,ние грубого концентрата перед перечистной флотацией или его
доводкой. Вместе со шламами удаляются растворимые соли,
затрудняющие избирательную дезактивацию и депрессию
минералов породы. Сульфиды, если они присутствуют в исходном
питании, удаляются предварительной флотацией сульфгидриль-
ными собирателями обычно в слабокислой среде (рН 6—6,5).
В качестве собирателя при флотации касситерита
предложено использовать большое количество органических
соединений, однако практическое применение получили только карбо-
новые, арсоновые, фосфоновые, алкилгидроксамовые, сульфоян-
тарные кислоты и их производные.
Удовлетворительные результаты флотации касситерита
карбоксильным собирателем получают только на относительно
простых силикатных рудах с низким содержанием турмалина и
гидроокислов железа. Значения рН основной и контрольной
флотации касситерита находятся в диапазоне 5—5,7, а в пере-
чистных операциях 4,2—4,8. В этих условиях минералы породы
279
уже в достаточной степени дезактивированы, а касситерит еще
сохраняет флотируемость. При более высоких значениях рН
нарушается селективность, а при более низких — депрессируется
флотация касситерита. В качестве собирателя на одной из
фабрик СССР используется эмульсия таллового масла с керосином
A:1) при расходе до 2,8 кг/т. Получаемый концентрат после
пяти перечистных операций содержит 10—12 % олова при
извлечении его 60—70 % от питания.
Эмульсия карбоксильного собирателя @,7—1 кг/т) с
большим количеством аполярного масла C—4 кг/т) используется
в специальном процессе обогащения сильно шламистых и
бедных оловосодержащих материалов — «агломерационной»
флотации. В процессе длительного перемешивания D0—60 мин)
плотной пульпы F0—70 % твердого) со смесью собирателей
гидрофобные частицы агрегируются с капельками масла,
которые затем всплывают на воздушных пузырьках и удаляются
в концентрат. Процесс исследуется и может оказаться
полезным особенно для извлечения шламистых частиц касситерита.
В целях повышения качества получаемых с жирными
кислотами флотационных оловянных концентратов разработан метод
обратной флотации их. После термической обработки при
температуре 300—350 °С, вызывающей термическое разложение
собирателя, концентрат репулышруется и из него флотируют
кварц и другие силикатные минералы катионным реагентом
в щелочной среде (рНП), создаваемой известью и едким
натром. Для депрессии касситерита используются отходы
мукомольной промышленности. Камерный продукт содержит 40—
60% олова при извлечении 60—75%. Метод пока не нашел
применения из-за громоздкости его аппаратурного оформления.
Более селективным собирателем, по сравнению с карбоно-
выми кислотами, являются алкил- и ариларсоновые кислоты,
с общей формулой
О
R—As — ОН
■\
ОН
Применение, например, паратолиларсоновой кислоты (на
фабриках ЮАР) и реагента SM-119 (на австралийской
фабрике) позволило получать флотационные концентраты с
содержанием олова 17,7—31% при извлечении 69,2—59%.
Флотация осуществляется в слабокислой среде (рН 5—6) с
добавками (около 1 кг/т) жидкого стекла и фтористого натрия.
Применение арсоновых кислот ограничивается необходимостью
тонкого измельчения (поскольку крупные зерна флотируются
плохо), значительным их расходом @,4—1 кг/т), токсичностью
и высокой стоимостью (в 15 раз дороже олеиновой кислоты).
280
Фосфоновые кислоты с общей формулой
О
R —Р —ОН
\
ОН
по имеющимся данным являются более эффективными
собирателями по сравнению с олеиновой и арсоновой кислотами, ал-
килсульфатами и алкилфосфатами. При использовании алкил-
фосфоновых кислот @,1—0,3 кг/т) или смеси фосфоновой (ОД —
0,25 кг/т) и жирной кислот @,35—0,8 кг/т) для флотации
касситерита оптимальное значение рН пульпы составляет 2,5—3,5,
а температуры 35—45 °С. Для повышения селективности
используется смесь @,7—1,5 кг/т) жидкого стекла с кремнефтори-
стым натрием. Получаемые концентраты содержат 6—49 %
олова при извлечении 85—55 % от исходного материала.
В последние годы на ряде зарубежных фабрик («Уануни»,
Боливия, «Уил Джейн», Великобритания) в качестве
собирателей при флотации касситерита стали применять производные
сульфоянтарной кислоты (соединения сульфосукцинатного и
сульфосукципаматного типа), известные под названием «Аэро-
промотер-845» («Аэрозоль-22»), «Алкопол-540». Флотация
касситерита этими реагентами может осуществляться обычно при
рН от 2 до 10. В присутствии ионов железа и кальция
предпочтительнее.использование кислой среды. Основная касситерито-
вая флотация осуществляется обычно при рН 2—2,3, перечист-
ные операции — при рН 2,2—4. Лучшие результаты по
отделению касситерита от турмалина и железных минералов
достигаются при использовании в качестве их депрессора
лимонной кислоты @,04—0,15 кг/т) или смеси жидкого стекла и
кремнефтористого натрия. В этом случае удается снизить
расход собирателя до 0,5—0,6 кг/т и несколько повысить рН
пульпы при флотации (с 2—2,3 до 2,5—3). Из руд и шламов,
содержащих около 1 % олова, получают после двух-трех пере-
чистных операций концентрат с содержанием 9—20 % олова
при извлечении 60—89 %.
В СССР разработана технология флотации касситерита из
труднообогатимых шламов гравитационного обогащения с
применением реагента ИМ-50. Принципиальная схема флотации
касситерита (рис. 14.11) предусматривает первичную
концентрацию минералов олова за счет практически полного удаления
в хвосты кварцево-полевошпатовой породы и частично
турмалина при флотации эмульсией дистиллированного таллового
масла (до 0,75 кг/т), стабилизированной 2 %-ным ОП-4 в
слабокислой среде (рН 5,5—5,7), создаваемой кремнефтористым
натрием @,9—1,2 кг/т). Доводка грубого концентрата, при
которой происходит отделение касситерита от железосодержащих
281
. Шламы
гравитационного обогащения
Сгущение и
обесшламливани е
Эмульсия JJTM и УЛ-ч-7 крем-
несртористьш натрий
Коллективная флотация
касситерита и
железо с о дер жащ а ос
минералу 8
шмамы
Активированный уголь,
серная кислота
"ЧУ
Месорбция жирнокис-
м от ног о
собирателя A0°С),
кислотная ^оораб'отка
Селективная
флотация касси-
терита (W 2fi'2j8)
Серная кислота,
щавелевая кислота, ИМ'бО
Оловянный
концентрат
^6-8%Sr\0z)
П е р е ч и с т н ы е
ГрН 2)
/Г^
Десорбция ИМ-50
мевным
купоросом
Серная кислота,
щавелевая кислота, ИМ So
\ у
Л обо дна аловян-
нога
концентрата
__^ .
Оло бянныи концентратG2-73% Sn O^J
От бальные хвосты
Рис. 14.11. Принципиальная схема флотации касситерита из шламов
гравитационного обогащения
минералов (турмалина, гидроокислов железа, ожелезненной
породы), проводится с собирателем ИМ-50 @,2—0,3 кг/т)
после десорбции части жирной кислоты с поверхности минералов
активированным углем при подогреве пульпы до 70 °С.
Железосодержащие минералы в цикле доводки депрессиру-
ются щавелевой кислотой (до рН=^2,8—3). Расход
дорогостоящей щавелевой кислоты может, быть значительно снижен в
результате замены части ее серной или соляной кислотой. После
двух-трех перечистных операций концентрат содержит 6—8 %
олова при извлечении 75—85 % от операции флотации шламов,
содержащих ОД % олова. Дальнейшее повышение качества
концентратов достигается повторной доводкой их после
предварительной десорбции ИМ-50 с минералов породы медным
купоросом.
Получить флотационным путем кондиционный концентрат,
содержащий 60—40 % олова, ни на одной фабрике не удается.
При флотации касситерита из шламов. отечественные фабрики
работают с получением концентратов, содержащих 8—10 %
олова. На зарубежных фабриках флотируется более богатое
сырье и концентраты содержат 17—30 % олова при
извлечении 35—70%. Такие концентраты поступают-обычно на фью-
мингование.
Для повышения качества бедных флотационных концентра-
. тов предложен ряд комбинированных методов. Так, И. Н. Шор-
шером разработан метод, по которому поверхность зерен
касситерита восстанавливается водородом и окисью углерода при
500—900 °С до SnO или Sn203, а затем сульфидизируется
сероводородом. Сульфидизированный касситерит после
активации ионами свинца или меди флотируется сульфгидрильными
собирателями в высококачественный концентрат. Предложен
также процесс, предусматривающий предварительный босстано-
-вительный обжиг бедного концентрата и последующую
флотацию металлических частиц олова. Эти способы обеспечивают
получение высоких технологических показателей, но вследствие
сложности их не получили пока промышленного
применения.
Флотация бокситов. Бокситы представляют собой смесь гид-
раргиллита АЬОз'ЗШО, диаспора и бемита (разновидностей
А120з'Н20) с каолинитом, кварцем, минералами железа
(лимонитом, гематитом, сидеритом, пиритом) и титана
(ильменитом, рутилом). Обогащение их позволяет улучшить показатели
химического и металлургического переделов и за счет
вовлечения в эксплуатацию некондиционных бокситов расширить
сырьевую базу алюминиевой промышленности [18]. Бокситовые
концентраты, поступающие на производство глинозема и
глиноземного цемента, должны содержать не менее 28—30 % АЬОз
при отношении А120з: Si02=4 :5,6, а предназначенные для
производства электрокорунда—не менее 49—52% А1203 при
отношении АЬОз: Si02 = F—15) : 1.
283
Наиболее эффективным собирателем гидратированных
окисей алюминия является олеиновая кислота @,4—0,6 кг/т). На
практике часто пользуются ее смесью с талловым и машинным
маслом (или керосином). Обязательным условием при
флотации является снижение содержания растворимых солей
(кальция, магния, железа, алюминия) и диспергирование пульпы.
Для этой цели используются сода, едкий натр, сернистый
натрий, фосфатные соединения (метафосфат, гексаметафосфат,
иирофосфат натрия), жидкое стекло и крахмал, оказывающие
одновременно и депрессирующее действие на минералы породы.
Предварительное введение в пульпу р.еагента ОП-7
способствует резкому уменьшению расхода собирателя и усиливает
депрессию глинистых шламов. Оптимальное значение рН 7,5—9,5.
Более высокие значения рН требуются при высоком
содержании в руде каолинита [18].
При кондиционировании бокситов производится удаление из
них пиритной серы сульфгидрильными собирателями,
органических примесей аполярным собирателем, карбонатов железа
жирными кислотами.
При обогащении бокситов возможно попутное получение
ряда продуктов: каолинитового, железорудного, титанового, пи-
ритного с промышленным содержанием в них металлов или
элементов, т. е. комплексное использование сырья.
Флотация урановых руд [34]. Промышленное значение в
урановых рудах имеют окислы (уранинит, смоляная обманка),
карнотит, браннерит и коффинит. Все они флотируются жирными
кислотами или их смесью с аминами в слабокислой (рН5—5,7)
или слабощелочной (рН 7—7,5) среде. В качестве депрессоров
пустой породы применяются кремнефтористый натрий и
жидкое стекло.
Флотацию часто используют в сочетании с другими
процессами обогащения и металлургии. Например, ее применяют для
удаления из урановых руд сульфидов, карбонатов и других
минералов, часто представляющих самостоятельную ценность или
препятствующих эффективному протеканию последующих
гидрометаллургических процессов извлечения урана из руд.
При флотации карнотитовых руд ее применяют для получения
карбонатной и силикатной фракций, которые затем раздельно
выщелачивают соответственно содой и кислотой.
14.7. Флотация силикатов
Краткая характеристика силикатов. Силикатные минералы
весьма разнообразны. Они всегда содержатся в добываемых
рудах-и часто в значительных количествах. С развитием
технологии флотационного обогащения все большее число силикатных
минералов переходит из разряда породных в разряд полезных
минералов.
Трудности селективной флотации силикатов обусловлены
m
близостью их флотационных свойств. Селективность флотации
достигается путем избирательной активации или дезактивации
разделяемых минералов в результате кислотной или щелочной
обработки, обработки плавиковой и серной кислотами,
фтористым и кремнефтористым натрием, жидким стеклом и другими
реагентами. Широко практикуются использование умягченной
воды, предварительная промывка руды и обесшламливание
пульпы перед флотацией, оттирка (очистка) поверхности
перемешиванием в плотной пульпе и другие приемы, повышающие
селективность флотации силикатов анионными и катионными
собирателями.
К настоящему времени наибольшее распространение и
значение приобрела флотация полевых шпатов, берилловых и спо-
думеновых руд, алюминийсодержащего сырья и силикатов при
обратной флотации железных, фосфатных и других типов руд.
Флотация полевых шпатов. Полевые шпаты представляют
собой главным образом изоморфные смеси KAlSi308,
NaAlSi308 и CaAl2Si208, имеющие одинаковую
кристаллическую структуру и весьма близкие химико-физические свойства.
При получении полевошпатовых концентратов из пегматитов,
полевощпатовых песков и гранитов, основными минералами
которых являются полевые шпаты и кварц, решаются следующие
основные задачи.
1. Удаление минеральных примесей, из которых наиболее
часто встречаются биотит, мусковит, серищгг, ильменит, окислы
железа и некоторые другие минералы. Обычно их удаляют при
коллективной флотации в слабощелочной (рН 8,5—9,2) или
нейтральной среде при использовании в качестве собирателя
смеси реагентов разных классов. Основой реагентной смеси
являются жирные кислоты с добавкой алкилсульфоната; при
повышенном содержании в руде слюды добавляется амин, при
наличии сульфидов — сульфгидрильный собиратель. С целью
повышения комплексности использования сырья удаление
примесей может быть произведено последовательной флотацией
железосодержащих (темноцветных) минералов и ожелезненных
зерен кварца и полевого шпата анионным собирателем чили
смесью анионного и катионного собирателей (при отношении
8:1) в нейтральной, слабощелочной или слабокислой среде и
минералов слюды катионным собирателем @,1—0,2 кг/т) после
понижения рН пульпы до 5 серной кислотой. Иногда цикл
флотации слюды предшествует циклу флотации железосодержащих
минералов.
2. Отделение полевых шпатов от кварца. Наиболее
селективной является флотация полевых шпатов катионным
собирателем @,2—0,3 кг/т) в сильнокислой среде (рН 2—3) после
обработки пульпы фтористоводородной кислотой A—2 кг/т) в
целях депрессии кварца и активации полевых шпатов. Дорогую
фтористоводородную кислоту можно заменить смесью NaF
с серной или соляной кислотой. Кварцевый концентрат, полу-
285
чаемый в камерном продукте, должен содержать по кондициям
стекольной промышленности 98—99 % кварца и не более 0,05—
0,08 % железа.
3. Разделение полевых шпатов. Работами Уралмеханобра и
Механобра установлено, что при использовании катионных
собирателей полевые шпаты с повышенным содержанием КгО
(ортоклазовые, микроклиновые) избирательно депрессируются
КС1, с повышенным содержанием Na20 (альбитовые) —NaCl,
а с повышенным содержанием СаО (анортитовые)—СаСЬ-
Обычно депрессируют флотацию той разновидно.сти полевых
шпатов, содержание которой в коллективном концентрате
является наиболее высоким. Концентрация депрессирующей соли
в цикле селективной флотации должна быть высокой F—*
10 г/л) и расход ее при использовании оборотных вод
составляет 5—10 кг/т. Расход катионного собирателя не превышает
0,1—0,2 кг/т.
Полевошпатовые концентраты используются в керамической
и абразивной промышленности. Суммарное содержание КгО и
ЫагО должно быть около 12 %, а содержание СаО не более 2 %.
При переработке пегматитов и кварцево-полевошпатовых
песков извлечение полевых шпатов в концентраты достигает 95—
98%, а при флотации гранита — колеблется в пределах 62—
92 %. Наибольшее применение имеют калиевые полевошпатовые
концентраты с отношением K20:Na20>2.
Флотация бериллиевых руд [34]. Флотации подвергаются
мелко- и тонковкрапленные бериллиевые руды. Основной
промышленный минерал — берилл BesAblSieOis] близок по своим
флотационным свойствам к встречающимся в рудах актино-
литу, турмалину, топазу, кварцу и полевому шпату. В рудах
присутствуют также слюды, флюорит, сульфиды и некоторые
другие минералы. Для получения кондиционных берилловых
концентратов, содержащих 8—10 % ВеО, применяют щелочную
или кислотную схемы флотации.
Флотации берилла по кислотной схеме предшествуют
следующие циклы:
коллективной флотации сульфидных минералов сульфгид-
рильным собирателем в слабощелочной, нейтральной или
слабокислой средах;
флотации флюорита (при значительном содержании его
в руде) небольшими добавками оксигидрильного собирателя
с одновременной подачей жидкого стекла для депрессии
силикатов;
флотации слюды катионным собирателем @,2—0,3 кг/т)
в кислой среде (рН 3—4), создаваемой серной кислотой B—
4 кг/т), или в сильйощелочной среде (рН 10), создаваемой
содой или едким натром. Переходящая в концентрат кальциевая
слюда — Маргарит с промышленным содержанием бериллия —
может быть выделена в селективный концентрат при дофлота-
ции коллективного слюдяного концентрата в кислой или ще-
286
лочной среде в присутствии хлористого алюминия @,5—0,7 г/л),
обеспечивающего депрессию всех остальных слюд;
обработки хвостов слюдяной флотации фтористоводородной
(плавиковой) кислотой A,5—2 кг/т) в смеси с серной кислотой
@,5 кг/т) для активации берилла и полевого шпата и
депрессии кварца.
Последующее флотационное извлечение берилла из
подготовленного таким образом матерала осуществляется по
коллективной или селективной схемам флотации.
Коллективная берилл-полевошпатовая флотация
проводится с катионным собирателем @,15 кг/т) с получением в
камерном продукте кварцевого концентрата. Отделение берилла
от полевого шпата производится двумя способами:
флотацией берилла анионным собирателем @,1—0,2 кг/т)
после трехкратной отмывки катионного собирателя с
поверхности коллективного концентрата слабым раствором соды
@,04 кг/т руды) и обесшламливания (по классу—15 мкм);
флотацией берилла в кислой среде (до 2 кг/т серной
кислоты) нефтяным сульфонатом после обработки коллективного
концентрата в плотной пульпе E0 % твердого) гипохлоритом
@,2—0,9 кг/т) и отмывки.
Получаемый берилловый концентрат может быть объединен
с маргаритовым, а полевошпатовый концентрат, получаемый
в камерном продукте, может быть подвергнут флотационному
разделению на калиевую и натриевую разновидности в
растворе соли натрия или калия по рассмотренной выше
технологии.
Селективная флотация берилла из хвостов слюдяной
флотации производится жирными кислотами в щелочной среде
с выделением в хвосты кварца и полевого шпата. Черновой
берилловый концентрат обычно загрязнен актинолитом,
амфиболами и другими минералами и поэтому требует доводки,
которая осуществляется после пропарки с содой (примерно
0,5 кг/т), жидким стеклом или сернистым натрием @,1 —
0,2 кг/т) при 85 °С.
Поскольку коллективная схема флотации берилла позволяет
более комплексно использовать минеральное сырье, селективная
схема применяется только при высоком содержании в руде
сильно разрушенного полевого шпата.
По щелочной схеме флотационное извлечение берилла из
руды осуществляется после ее щелочной обработки. Если в руде
присутствуют турмалин и гранат (более флотоактивные, чем
берилл), то их удаляют до щелочной обработки флотацией
жирными кислотами (при малых расходах) в содовой среде.
Обработка руды щелочными реагентами (едким натром или
сернистым натрием) производится для активации берилла, пеп-
тизации шламов и депрессии минералов породы в результате
предотвращения активации их солями железа и других
металлов. После щелочной обработки в плохой пульпе и удаления
287
шламов и избытка щелочи путем промывки Na-катионирован-
ной водой до рН 8—8,5 берилл флотируется жирными
кислотами @,2—0,4 кг/т) в содовой среде. Для повышения
эффективности действия собирателя его вводят в пульпу в виде
подогретой до 80—85 °С эмульсии.
Черновой концентрат, загрязненный слюдами, кварцем,
полевыми шпатами, остатками турмалина, граната и др., после
перечистной флотации в содовой среде (рН 9,5) подвергают
двукратной пропарке с содой или с содой и сернистым натрием
для депрессии загрязняющих берилловый концентрат
минеральных примесей.
Технологические показатели флотации берилла по
кислотной и щелочной схемам близки: извлечение бериллия в
концентрат составляет 70—80%- Однако кислотная схема
обеспечивает более высокую степень комплексности использования
минерального сырья. Недостатком кислотной схемы является
применение плавиковой и серной кислот, усложняющих
обслуживание флотационного процесса, требующих применения
кислотоупорного оборудования и более строгого соблюдения правил
техники безопасности. Недостатками щелочной схемы являются
необходимость подогрева пульпы и применение умягченной
воды.
Флотация литиевых руд [34], Флотация является основным
методом обогащения литиевых и комплексных руд,
содержащих тонковкрапленные литиевые минералы: сподумен
LiAI[Si2C>6], лепидолит, амблигопит, циннвальдит и петалит.
Наиболее распространена и изучена флотационная технология
обогащения сподуменовых руд, обычно содержащих кварц,
слюды, полевые шпаты, иногда окислы железа, берилл и
другие минералы.
Сподумен легко флотируется с помощью собирателей окси-
гидрильного и катионного типа после щелочной обработки руды
в плотной пульпе E0—70 % твердого) и последующей отмывки
щелочного раствора и шламов (класса —15 мкм).
Предполагается, что активация флотации сподумена обусловлена
выщелачиванием кремнезема из поверхностного слоя минерала и
удалением с него шламистых покрытий. Крахмал, декстрин,
сернистый натрий и жидкое стекло являются сильными
депрессорами сподумена, тогда как хлорная известь, депрессирующая
берилл и полевые шпаты, не ухудшает его флотацию.
Флотационное обогащение сподуменовых руд осуществля-
гтся по одной из следующих технологических схем:
прямая анионная флотация сподумена жирными
кислотами и их мылами @,2—0,5 кг/т) при рН 6,5-8,5 после
предварительной щелочной обработки пульпы едким натром
A—3 кг/т) в течение 0,5—1 ч, обесшламливания и промывки.
Депрессия сопутствующих минералов достигается связыванием
активирующих ионов (например, железа) в щелочной среде
в труднорастворимые соединения. Селекция ухудшается, если
288
в процессе щелочной обработки используются кремнефтористый
натрий, жидкое стекло или гексаметафосфат. Перечистную
флотацию чернового сподуменового концентрата проводят с
добавлением жидкого стекла и квебрахо или молочной кислоты.
Резкое повышение качества концентрата может быть получено
при его дофлотации кремнефтористым натрием в кислой среде,
однако оно сопровождается понижением извлечения сподумена;
обратная флотация, в которой сначала флотируют
кварц, полевой шпат и слюды катионным собирателем в
щелочной среде (рН 10—11) при депрессии сподумена и минералов
железа известью и декстрином. Сподуменовый концентрат
получают в камерном продукте из хвостов катионной флотации
после их промывки, обесшламливания, перемешивания плотной
пульпы G0% твердого) с плавиковой кислотой @,1—0,2 кг/т)
и флотации минералов железа натриевыми солями смоляных
кислот @,5—1 кг/т). Пенный продукт катионной флотации
также поступает на разделение: в слабокислой среде,
создаваемой серной кислотой, из него удаляют слюды и получают
слюдяной концентрат, затем после промывки обесшламливания й
обработки плавиковой кислотой и флотации катионным
собирателем в пенном продукте получают полевошпатовый концен-
центрат, а в камерном — кварцевый;
коллективная флотация сподумена и слюд смесью
жирных кислот и катионного собирателя в слабощелочной среде
с последующим разделением коллективного концентрата
флотацией слюды в сернокислой среде. Из хвостов коллективной
флотации можно выделить полевошпатовый концентрат флотацией
катионным собирателем в присутствии плавиковой кислоты.
В камерном продукте остается кварц.
Конечные сподуменовые концентраты I сорта содержат не
менее 4%, а II сорта — не менее 3 % окиси лития. Извлечение
лития в концентраты составляет 55—70%. Важнейшим и
необходимым условием переработки сподуменовых руд является
комплексное их использование. По этой причине наиболее
предпочтительными являются последние две схемы.
Флотация алюминийсодержащих силикатов. К алюминийсо-
держащим силикатам относятся в первую очередь минералы
андалузитовой группы: андалузит, кианит, силлиманит,
имеющие общую химическую формулу Al203'Si02, и нефелин
Na[AlSi04]. Концентраты этих минералов используются для
производства силумина, огнеупорных и кислотоупорных изделий и
в качестве исходного сырья алюминиевой промышленности.
Андалузитовые руды следует измельчать в галечных
мельницах, чтобы предотвратить активацию минералов породы
железом. Флотации предшествует обесшламливание, если в руде
присутствуют легкоизмельчаемые минералы пустой породы и
шламы легкофлотируемых силикатов. Андалузит флотируют
жирными кислотами @,3—1,3 кг/т) в содовой среде E—
8 кг/т). Для депрессии кварца, слюды, железных, титановых и
Ю Заказ № 1957
289
некоторых других минералов подают крахмал @,1—0,2 кг/т)
и сульфитный щёлок @,1—0,2 кг/т), активной частью которого
ябляется лигнинсульфоновая кислота.
При флотационном обогащении кианитовых руд песко-
вая и шламовая части флотируются раздельно. Флотация
кианита осуществляется жирными кислотами и их солями @,5—
1,0 кг/т) в слабощелочной среде, создаваемой содой @,5—
1 кг/т) или едким натром @,2—0,3 кг/т) с небольшими
добавками жидкого стекла @,2—0,3 кг/т) для депрессии основной
массы породы. Очистка кианитового концентрата от биотита и
граната производится в перечистных операциях в слабокислой'
среде (рН 6,6—5,2). При наличии в руде сульфидов они
удаляются в начале процесса.
При флотации силлиманитов ых руд жирнокислотньши
собирателями @,2—0,6 кг/т) в качестве регуляторов флотации
и депрессоров породы используются сода, едкий натр, пирофос-
фат натрия и жидкое стекло. Флотация силлиманита
производится обычно после предварительного обесшламливания и уда^
ления железных и титансодержащих минералов.
. Получаемые концентраты при флотационном извлечении из
руд минералов андалузитовой группы содержат 55—57 % А1203
при извлечении 70—90 % •
Основным источником получения нефелиновых
концентратов являются обесшламленные и сгущенные хвосты
апатитовой флотации, содержащие (помимо 70 % нефелина) остатки
апатита C%), эгирин B0%), сфен, титаномагнетит и другие
минералы. Кондиционный нефелиновый концентрат,
содержащий не менее 29 % А120з и не более 60 % класса +0,088 мм,
получается в камерном продукте коллективной флотации всех
сопутствующих минералов заменителями олеиновой кислоты (тал-
ловым маслом, окисленным петролатумом и др.) в щелочной
среде (рН 10—10,5), создаваемой едким натром. Коллективный
концентрат (в целях комплексного использования сырья)
подвергается разделению. На основании лабораторных
исследований предложено несколько технологических схем. Одна из них
предусматривает следующие операции с использованием в
качестве собирателя алкилсульфата: флотацию апатита при
рН6,8—7 с добавкой соды или едкого натра и собирателя
@,25—0,3 кг/т); флотацию сфена при рН 5,4—5,5 с добавкой
серной или соляной кислоты и собирателя @,5 кг/т); титано-
магнетитовую флотацию при рНЗ и нефелиновую флотацию*
при рН 4 без добавки собирателя.
14.8. Флотация растворимых солей
К основным минералам растворимых солей относятся
сильвин KCI, галит NaCl, карналлит KCl-MgCl2 -6H20, лангбейнит
K2S04-2MgS04, бораты и-некоторые другие. Наибольшее
распространение получила флотация сильвинитовых руд, в кото-
290
рых, кроме сильвина и галита, содержатся в небольших
количествах хлориды магния (бишофит, карналлит) и
нерастворимые минералы, представленные обычно глиной и гипсом [11].
Флотация растворимых солей производится анионными и
катионными собирателями в насыщенных растворах,
называемых маточником. При флотации сильвинитовых руд маточник
содержит около 10 % КС1, 20 % NaCl и имеет плотность
1,27 г/см3. На фабриках соблюдается полный водооборот:
маточник регенерируется при сгущении и фильтровании как
концентрата, так и хвостов флотации.
Высокая ионная сила жидкой фазы пульпы накладывает ряд
особенностей при флотации растворимых солей.
Первая особенность связана с характерным влиянием
ионного состава маточника на флотируемость минералов..По
правилу Стремовского флотируемость минерала ухудшается, если
его растворимость в маточнике данного состава уменьшается, и,
наоборот, флотируемость возрастает при увеличении
растворимости минерала. Например, гипс не флотируется алкилсульфа-
том в водном растворе, но флотируется при добавках NaCI,
увеличивающих его растворимость. Поэтому изменение состава
маточника может послужить средством повышения
селективности и эффективности флотации.
Вторая особенность выражается в необходимости избегать
значительных перепадов температуры пульпы при флотации.
Изменение температуры маточника может вызвать выпадение
из него новой твердой фазы (при понижении температуры) или
растворение поверхности разделяемых минералов (при
повышении температуры), изменение степени дисперсности собирателя
и характера действия реагентов. Эти явления приводят к
увеличению расхода реагентов, неустойчивости процесса и
ухудшению технологических показателей обогащения.
Третья особенность заключается в ограничениях при
выборе собирателя. Собиратели не должны образовывать
труднорастворимых солей с катионами маточника и высаливаться под
действием его высокой ионной силы. К таким собирателям
относятся короткоцепочечные жирные кислоты с числом атомов
углерода 7—9, реагенты типа алкилсульфатов,
нечувствительных к солям жесткости, и катионные собиратели (амины)
с числом атомов углерода в алифатической цепи 13—16. Эти
собиратели обеспечивают достаточно эффективную флотацию
минералов растворимых солей, но только при нахождении их
в маточнике в ионной форме. Учитывая зависимость состояния
собирателей в растворе от значения рН, флотация с
применением жирнокислотных собирателей должна производиться* при
рН больше 6, а аминами — при рН меньше 8. Алкилсульфаты
при используемых на практике значениях рН находятся в
ионной форме и слабо зависят от рН среды. Значения рН
регулируются обычно малыми добавками соды, едкого натра или
соляной кислоты.
10*
291
Помимо перечисленных общих особенностей, флотационное
обогащение каждого типа руд имеет свои особенности.
Наибольшие трудности широко распространенной флотации
сильвинитов ых руд определяются наличием в них
глинисто-карбонатных шламов. Предотвращение вредного действия
шламов на флотацию и загрязнение ими получаемых
концентратов достигается:
флотацией шламов, скоагулированных полиакрила мидом
A0—15 г/т), с применением в качестве собирателя, например,
окисленного уайт-спирита (до 150 г/т) с небольшой добавкой
керосина. Флотация шламов значительно интенсифицируется
при подаче собирателя в виде аэрозоля благодаря более
равномерному его распределению;
применением реагентов (крахмала, декстрина, производных
лигнина, целлюлозы и других реагентов), гидрофилизирующих
поверхность шламов и депрессирующих их флотацию;
обесшламливанием по классу —10, —40 мкм.
Содержащиеся в шламах сильвин и галит извлекают выщелачиванием и
противоточной декантацией (промывкой).
Сильвинитовые руды обычно имеют относительно крупную
вкрапленность и их измельчают до 0,8 мм. Однако
употребление калийного (сильвинового) концентрата значительно
облегчается по мере увеличения его крупности. В связи с этим
применение флотационных машин типа пенной сепарации,
пригодных для флотации крупнозернистых материалов, является
совершенно необходимым, позволяющим не только повысить
качество продукции, но и снизить себестоимость переработки
руды.
Разделение сильвина и галита при флотации может быть
осуществлено с помощью анионного или катионного собирателя.
При использовании жирных кислот флотируется галит после
предварительной активации его растворимой солью свинца.
Вместе с галитом флотируется- основная масса загрязняющих
сильвин примесей — глины, гипса, ангидрита. Это позволяет
получать в камерном продукте сильвинитовый концентрат
высокой чистоты. При использовании в качестве собирателя ал-
килсульфатов или алкиламинов флотируется, наоборот,
сильвин, а галит и минералы породы депрессируются крахмалом,
КМЦ и другими органическими реагентами-полимерами.
Важнейшим свойством полимеров при этом является их
активирующее действие на флотацию сильвина, обусловленное, по мнению
В. А. Арсентьева и С. И. Горловского, уменьшением
адсорбции собирателя глинистой породой за счет создания
препятствий доступу, например, амина в межплоскостные
пространства глинистых минералов. Причем катионноактивные
полимеры являются более эффективными активаторами флотации
сильвина, чем анионноактивные. В качестве активаторов могут
быть использованы и катионноактивные реагенты с низкой
молекулярной массой (например, из серии реагентов ЛГИ).
292
Наибольшее распространение на обогатительных фабриках
нашлет первичные алифатические амины. Их селективность
обусловлена, по мнению Д. В. Фюрстенау, соответствием ионных
радиусов полярной группы амина и катиона калия A,33-
* 1(Н° м) на поверхности сильвина. В то же время ионный
радиус катиона натрия @,98-10~10 м) слишком мал, чтобы его
место на поверхности галита могла занять положительно
заряженная полярная группа иона амина RNH+з. Флотацию
сильвина проводят в плотной пульпе C5—40 %. твердого) по
простым схемам. Расход амина составляет 40—100 г/т, КМЦ —
около 500 г/т. Продолжительность флотации 8—10 мин в
слабокислой или нейтральной среде. Сильвиновый концентрат
содержит 92—95 % КО при извлечении 84—90 %.
Флотация калийно-магниевых минералов (поли-
галита, лангбейнита, каинита) осуществляется обычно
низкомолекулярными жирными кислотами С7—Сэ в нейтральной
среде (рН около 7) при депрессии минералов пустой породы
полиакриламидом и жидким стеклом A,5 кг/т). Получаемый
концентрат содержит 17—19% КгО при извлечении 75—80%.
14.9. Ионная флотация
Термином «ионная флотация» обозначают группу
флотационных процессов, основанных на использовании поверхности
раздела жидкость — газ для извлечения из растворов ионов и
молекул органических соединений или продуктов их
взаимодействия с ионами или молекулами неорганических соединений.
Они пригодны для извлечения веществ, концентрация которых
в растворе может составлять 10~2—Ю-8 моль/л [19].
Терминология этих процессов еще не установилась. К
основным разновидностям их относятся: пенное фракционирование,
ионная флотация, флотация гидрофобных осадков, флотация
гидрофобизированных осадков, флотоэкстракция [19].
Пенное фракционирование основано на способности
поверхностно-активных ионов и молекул адсорбироваться на
поверхности пропускаемых через раствор пузырьков газа и
концентрироваться в получаемом пенном слое. Данный процесс
используется для решения актуальной проблемы очистки сточных и
природных вод от органических веществ, и в первую очередь от
моющих веществ например, алкилбензосульфонатов), не
поддающихся биологическому разрушению.
При извлечении из сточных вод 90—95 %
поверхностно-активных веществ объем пенного продукта в несколько сотен раз
меньше объема исходного продукта. Текущие расходы на
очистку 1 м3 сточных вод не превышают 1 коп.
Ионная флотация основана на притяжении извлекаемого
иона противоположно заряженными ионами собирателя,
закрепившимися на поверхности барботируемых через объем
раствора пузырьков воздуха или газа. Для извлечения катионов
293
применяются анионные, для анионов — катионные собиратели.
Между полярной группой собирателя^ и ионом действуют
электростатические силы притяжения или связи, характерные для
координационных соединений.
Ионная флотация применяется для извлечения из растворов
анионов ортофосфорной кислоты четвертичными аммониевыми
основаниями, анионов Сг2072~ бромистым цетилэтилдиметилам-
монием, катионов радиоактивных металлов алкилбензосульфо-
натами, алкилсульфатами, аминополикарбоксильными
кислотами и другими анионными собирателями.
Четыре-пять соединенных последовательно противоточных
колонн для ионной флотации обеспечивают степень очистки
порядка 108—1010. Ориентировочная стоимость обработки 1 м3
стоков, содержащих радиоактивные вещества, составляет 0,8—
1 долл. Если уран присутствует в растворе в виде уранила
U022+, то он не флотируется катионным собирателем типа
[R4N]+Br-. Однако если ион уранила путем добавки
карбонатных ионов перевести в уранилтрикарбонатный анион
[и02(СОз)з]4~, то его можно извлечь тем же собирателем,
благодаря образованию на поверхности пузырька комплекса
[R4N]4+-[U02(C03K]4~. Анионы С032~ в данном случае играют
роль активаторов, как и катионы поливалентных металлов при
флотации кварца анионными собирателями.
Флотация гидрофобных осадков основана на способности
многих цветных и редких металлов образовывать с алкилксан-
тогенатами, алкилмеркаптанами, дитизоном и другими
веществами труднорастворимые в воде соединения, осадки которых
имеют ярко выраженный гидрофобный характер и могут быть
сфлотированы после добавки пенообразователя.
Процесс применяется для извлечения ксантогенатом ртути
из промывных вод при рН 4,5 в концентрат, содержащий
21,4% ртути при извлечении 89,7%. Он рекомендован для
извлечения никеля и кобальта из стоков гидрометаллургических
заводов и из растворов выщелачивания пиритных огарков,
меди из шахтных вод и других металлов из различных
растворов. Расход ксантогената во всех случаях близок к стехиомет-
рическому, соответствующему образованию нормальных ксан-
тогенатов металлов. Регенерация ксантогената осуществляется
обработкой полученного концентрата щелочью: металлы
выпадают в виде гидроокиси, а щелочной раствор
регенерированного ксантогената поступает в оборот.
Показана принципиальная возможность извлечения железа,
меди, цинка, свинца, никеля, кобальта, палладия, ванадия,
марганца, церия и селена из разбавленных (Ю-5—10~4 моль/л)
водных растворов при использовании в качестве селективных
реагентов диаминобензидина, гидроксихинолина, а-нитрозо-р-
нафтола, купферона, оксимов.
Ленинградским горным институтом установлено, что,
используя в качестве собирателя натриевые мыла синтетических жир-
294
ных кислот Сю—Ci6, можно извлекать медь, цинк, никель,
кобальт и железо из разбавленных растворов
гидрометаллургической переработки полиметаллических руд и продуктов их
обогащения. Разработанная селективно-коллективная схема
предусматривает предварительную флотацию железа при рН 2,2—
2,6 и последующую флотацию цветных металлов в
коллективный концентрат при рН 8,5—9. После обезвоживания,
оплавления концентратов при температуре 60—70 °С и обработки их
серной кислотой собиратель регенерируется и отделяется от
концентрированного сульфатного раствора железа или цветных
металлов. Суммарное извлечение металлов в растворы
составляет 97—99 %.
Флотация гидрофобизированных осадков основана на
предварительном осаждении извлекаемых металлов и последующей
гидрофобизации полученного осадка собирателем. К
настоящему времени показана возможность: извлечения ионов
тяжелых металлов из сточных вод промышленных предприятий
путем осаждения их ферроцианидом калия и последующей
флотации осадка с желатином; извлечения цинка из стоков
вискозного производства после осаждения его в виде сульфида и
последующей флотации с лауриламином; селективного
извлечения тяжелых металлов из растворов путем ступенчатого
повышения рН, приводящего к последовательному выпадению
металлов в виде гидроокиси, и стадиальной флотации
выпадающих осадков. Например, при низких значениях рН можно
извлечь гидроокись железа, а при последующем ступенчатом
повышении рН выделить гидроокиси меди, цинка, никеля и
кобальта. Флотация гидрофобизированных осадков обладает
рядом технологических преимуществ: меньшим расходом
собирателя, высокой скоростью процесса, низкой чувствительностью
к присутствующим электролитам.
Флотоэкстракция основана на аккумулировании в слое
органического растворителя на поверхности аэрируемого раствора,
гидрофобных соединений, транспортируемых из объемаь
раствора воздушными или газовыми пузырьками. Она является
комбинацией флотации с жидкостной экстракцией ц при
одинаковой эффективности с жидкостной экстракцией требует
значительно меньших расходов экстрагента (собирателя) и
растворителя. Область применения флотоэкстракции непрерывно
расширяется.
Перечисленные разновидности ионной флотации позволяют
обеспечить высокую эффективность очистки сточных вод и
предотвратить загрязнение окружающей среды, повысить
извлечение ценных компонентов и комплексность использования
перерабатываемого сырья.
295
14.10. Направления совершенствования режимов флотации
Совершенствование режимов флотации достигается в
первую очередь осуществлением автоматического контроля и
регулирования ионного состава жидкой фазы пульпы,
регулированием состава продуктов и скорости взаимодействия реагентов
на минеральной поверхности и в объеме пульпы, применением
электрохимической, магнитной, ультразвуковой, термической
обработки пульпы и растворов реагентов, использованием
режимов скоростной, флокулярной флотации, пенной
сепарации и др.
Автоматический контроль и регулирование концентрации
реагентов в пульпе. Основной причиной, не позволяющей получать
на обогатительных фабриках максимально возможные
технологические показатели при флотации, является изменение
вещественного состава перерабатываемых руд по общему содержанию
в них металла, поглотительной способности минеральной
поверхности всех компонентов руды по отношению к реагентам,
содержания растворимых солей, примесей в промышленной
воде и т. д. Изменения вещественного состава компенсируются
изменением расхода реагентов.
Осуществляемое в настоящее время на многих
предприятиях дозирование реагентов по производительности или объему
пульпы является всего лишь средством упорядочения расхода
реагентов на фабрике. Основанная па этом система
автоматического регулирования расхода реагентов не учитывает
изменений вещественного состава руды и поэтому не может быть
достоверной. Система регулирования расхода реагентов по
содержанию металлов в руде должна быть более эффективной.
Однако надежность такой системы регулирования довольно
низка, так как в ней не учитывается основная причина
изменения расхода реагентов — изменение поглотительной или сорб-
ционной- емкости минеральной поверхности всех компонентов
измельченной руды по отношению к загружаемым реагентам.
К настоящему времени установлено, что все изменения
вещественного состава перерабатываемых руд отражает
концентрация загружаемых реагентов в пульпе, и доказано, что
соотношение концентраций реагентов в пульте определяет
результаты флотации. Поэтому контроль и регулирование
концентраций реагентов в пульпе является практически единственным
путем создания надежных и эффективных систем автоматического
поддержания оптимального реагентного режима и получения
максимально возможных показателей флотации при
минимальном расходе реагентов.
Основой систем автоматизации флотационного процесса по
ионному составу пульпы являются рассмотренные ранее
теоретически и экспериментально обоснованные количественные
зависимости между концентрациями реагентов в пульпе в
оптимальных условиях регулируемого процесса.
296
Дозатор
\ксантогената\
Объект регулирования
(процесс флотации)
Датчан
б пульпе
Функциональный
блок,
вырабатывающий зад иси ~
масть [кк-]=фн)
X
Регулятор
Вторичный]
Рис. 14.12. Принципиальная схема
системы автоматического
регулирования расхода ксантогената при
флотации
\Коллектц§ный |
медно-молиб- [■
\дено8ыи
концентрам
и
—*"
\
Цикл разделения медно-
молибденового концентрата 1
\
'
Датчик
\ции тонов
1 S2*
V
и
V
г
\
Датчик
рН
<
t 1
. Датчик
концентра -1
ции ионов
* 1
Ъукциональный блок\
]ырабаюыдающий за -
Ыимость
т
Регулятор
Рис. 14.13. Возможная
принципиальная схема системы
автоматического контроля и регулирования
расхода сернистого натрия при
разделении коллективного медно-мо-
либденового концентрата
Например, количественные зависимости необходимой
концентрации ксантогената [Кх~]=/(рН) при флотации различных
сульфидных минералов могут, быть реализованы в системе,
принципиальная схема которой изображена на рис. 14.12.
При работе системы сигнал датчика рН пульпы
направляется в функциональный блок, где вырабатывается необходимое
для данного рН значение концентрации ксантогената.
Полученное значение сопоставляется в регуляторе с текущим
значением [Кх~], измеряемым датчиком концентрации ксантогената.
В зависимости от знака рассогласования дозатор ксантогената
уменьшает или увеличивает расход собирателя в процессе
флотации.
Внедрение такой системы на фабрике позволит не только
значительно понизить расход собирателя, но и улучшить
технологические показатели обогащения в результате резкого
уменьшения коэффициента вариации {Кх~] в пульпе, повышения
однородности коллективных концентратов по плотности адсорбции
собирателя на поверхности минералов, улучшения вследствие
этого условий подготовки концентратов к разделению и
стабилизации процесса самого разделения.
В свою очередь, зависимости [Kx~]=f (pH, (S2~]) могут быть
использованы для построения систем автоматического контроля
и регулирования процессов флотации окисленных и смешанных
полиметаллических руд, селективной флотации сульфидов
297
1 »■
г
Дозатор
цианида
■*
Флотация
\
Датчик
соотношения
Л„№№Ы
1
'
Регулятор
—Г"
Датчик
концентрации
ионов
1
'
~Т
Датчик
рн
|
Функциональный блок, ]
вырабатывающий
зависимость
rn.ufr.uixi . \
ч
£CN
Г'
ЫЩп
Рис. 14.14. Возможная принципа-
альная схема системы
автоматического контроля и регулирования
процесса цианидного разделения
свинцово-медного концентрата
свинца и меди от сульфидов цинка и железа, десорбции
собирателя с поверхности коллективных концентратов и разделения
медно-молибденовых концентратов с сернистым натрием.
В качестве примера на рис. 14.13 приведена возможная
принципиальная схема системы автоматического контроля и
регулирования расхода сернистого натрия при разделении
коллективного медно-молибденового концентрата. Принципы
работы данной системы аналогичны предыдущей (см. рис. 14.12).
Поскольку из всех сульфидов меди наибольшей [S2~] для своей
депрессии требует халькозин, то использование в качестве
задания системе расчетного уравнения [S2~] для данного минерала
обеспечит эффективную депрессию всех сульфидов меди. В то
же время задаваемая [S2~] еще не вызывает ухудшения
флотации молибденита и это обеспечивает возможность
эффективного разделения медно-молибденового концентрата при
сравнительно небольшом расходе сернистого натрия.
Зависимости [Cu(CNJi/[CN-]2=/([H+], [Kx~] могут быть
использованы в качестве задания функциональному блоку
системы, например, при оптимизации процесса разделения свин-
цово-медных концентратов по возможной принципиальной
схеме, изображенной на рис. 14.14.
По данной схеме сигналы с датчиков концентраций ионов
Кх~" и рН пульпы, установленных во флотации, поступают
в функциональный блок, где рассчитывается необходимое
значение lg[Cu(CNJ~/CN-]2. Сигнал от датчика, измеряющего
текущее значение этого же параметра в пульпе, поступает в
регулятор, который сравнивает его с входным сигналом
функционального блока и, в зависимости от знака рассогласования,
воздействует на дозатор цианида таким образом, чтобы это
рассогласование стало равным нулю.
Важными достоинствами систем автоматизации, основанных
на контроле и регулировании концентраций реагентов в пульпе,
являются:
возможность использования их на любой обогатительной
фабрике в соответствующем цикле флотации. Например, разра-
298
ботанные количественные зависимости [Кх~]=/(рН) при
флотации сульфидов свинца являются справедливыми не только для
Зыряновской фабрики, но и фабрики «Сулливан» (Канада) и
других фабрик (см. рис. 9.4, б)\
для реализации зависимостей между оптимальными
концентрациями реагентов в пульпе на фабриках необязательно
применение дорогостоящих вычислительных машин — достаточно
простейшего функционального блока, т. е. оптимизация реагент-
ного режима может быть осуществлена на любой фабрике без
существенных затрат, что особенно важно для фабрик малой
производительности.
Оптимизация состава сорбционного слоя собирателя на
минеральной поверхности. Максимальная скорость флотации
минералов наблюдается, когда на их поверхности имеется
оптимальное соотношение химически и физически адсорбированного
собирателя. Это соотношение, в зависимости от типа
перерабатываемой руды, может регулироваться различными методами.
К основным из них относятся следующие [1, 2].
1. Применение доцолнительной подачи эмульсии аполярных
органических соединений при флотации различных руд сульф-
гидрильными, оксигидрильными и катионными собирателями.
Это необходимо в первую очередь при флотации минералов,
поверхность которых излишне химически активна по отношению
к собирателю (например, сульфидов меди по отношению к ксан-
тогенату) или, наоборот, характеризуется недостаточно полной
компенсированностыо валентных сил собирателем.
Как избыточная, так и недостаточная плотность продуктов
химического взаимодействия на поверхности затрудняет
образование необходимого количества физически адсорбированного,
например, диксантогенида. Подача эмульсии аполярных
соединений, в качестве которых наиболее перспективны различные
аполярные масла, аллиловые эфиры ксантогеновых кислот, ор-
ганополисилоксаны, восполняя недостаток физически
адсорбированного собирателя, увеличивает число возможных мест и
вероятность прорыва гидратной прослойки. между частичками
флотируемого минерала и пузырьками воздуха и повышает
вследствие этого вероятность извлечения их в концентрат.
О значительном увеличении скорости прилипания
минеральных частиц, покрытых капельками аполярных реагентов, к
пузырьку воздуха свидетельствуют результаты ряда работ.
Флотационная эффективность аполярных масел возрастает с
увеличением их расхода и степени дисперсности эмульсии до тех пор,
пока не появится опасность образования на минеральной
поверхности сплошной пленки загружаемого реагента, когда
поверхность утрачивает «прокалывающую» способность по
отношению к гидратной прослойке между частицей и
пузырьком воздуха. «Прокалывающая» способность эмульгированного
масла оказывается особенно полезной при флотации шламистых
минералов, поскольку позволяет избирательно сфлокулировать
299
гидрофобизированные зерна флотируемого минерала. При
флотации крупнозернистого минерала большое значение
приобретает повышение прочности контакта пузырек — частица в
присутствии аполярных масел.
2. Использование сочетания собирателей при флотации.
Комбинация собирателей является универсальным средством для
получения необходимого соотношения в каждом конкретном
случае химически и физически адсорбированного собирателя на
поверхности флотируемого минерала.
В частности, при использовании в качестве собирателей
ксантогенатов, это оказывается возможным благодаря
различной способности их низших и высших гомологов к окислению на
минеральной поверхности до диксантогенида. В настоящее
время на практике используются различные сочетания
этилового, изопропилового и бутилового ксантогенатов. Доля
каждого ксантогената обычно в бинарной смеси определяется
экспериментально. Разное соотношение ксантогенатов в смеси на
разных фабриках обусловлено разным соотношением дырок и
электронов в' поверхностном слое флотируемых минералов [1].
Соотношение химически и физически закрепившегося
реагента в сорбционном слое можно регулировать также, применяя
различные типы собирателей, например сульфгидрильные- и ка-
тионные жирнокислотные, и сульфгидрильные или катионные и
жирнокислотные, в зависимости от характера
перерабатываемого сырья. Примеры успешного- использования таких смесей
собирателей все чаще появляются в печати.
3. Применение электрохимической обработки растворов
собирателей. При катодной или анодной электрической обработке
раствора изменяются состояние и состав находящегося в нем
собирателя. Например, при электрохимическом окислении
растворов ксантогената на аноде происходит окисление ионов
собирателя до молекул диксантогенида. Десорбированный
с анодного электрода вследствие высокого положительного
заряда на нем диксантогенид находится в состоянии молекулярной
дисперсии. Выход диксантогенида увеличивается с повышением
концентрации собирателя в растворе и напряжением,
подаваемым на электроды. Частичным электрохимическим -окислением
растворов ксантогената удается компенсировать замедленную
стадию реакции образования диксантогенида на минеральной
поверхности в обычных условиях и обеспечить оптимальное
соотношение форм сорбции собирателя на поверхности
флотируемых минералов.
Замедление реакции Кх~-^Кх2 вызывается
неблагоприятным соотношением дырок и электронов на их поверхности. Чем
выше концентрация электронов, тем больше должна быть
степень предварительного электрохимического окисления раствора
ксантогената перед его подачей в процессе флотации.
Положительный эффект электрохимической обработки катионного
собирателя может быть обусловлен образованием малоустойчивых
300
свободных радикалов, возможно, с некоторым химическим
изменением реагента, что приводит к повышению его активности
наряду с увеличением степени дисперсности собирателя в
растворе.
4. Применение рентгеновского, радиоактивного облучения
или электрического заряжания минеральных частиц. В
результате такой обработки на поверхности и в объеме минералов
изменяется количество дефектов кристаллической решетки, что
приводит к изменению концентрации и соотношения носителей
тока: электронов и дырок. В результате этого меняется
кинетика окислительно-восстановительных реакций на поверхности
и состав сорбционного слоя собирателя на ней [48].
Оптимизация крупности и состава поверхности пузырьков.
Интенсификация флотационного процесса за счет «активизации»
воздушных пузырьков может быть достигнута одним из
следующих приемов.
1. Создание условий, обеспечивающих интенсивное
образование в пульпе микропузырьков воздуха. Этим достигается
принципиально та же цель, что и при добавке к основному
собирателю эмульсии аполярного масла. Микропузырьки воздуха,
как и микрокапельки аполярных реагентов, выделяются в
первую очередь на гидрофобных и гидрофобизированных
собирателем поверхностях, повышая «прокалывающую» способность их
по отношению к гидратпой прослойке между частицей и яу-
зырьком воздуха, способным поднять ее в пену. Показатель
флотируемости Fb и вероятность извлечения частиц при этом
увеличиваются. Это следует из полученного ранее уравнения
B.12).
Чем интенсивнее идет образование микропузырьков воздуха,
тем выше скорость флотации и, в ряде случаев, извлечение
флотируемого минерала. При высокой концентрации в пульпе
очень мелких пузырьков и одновременном присутствии крупных
частицы больших размеров могут выноситься на поверхность
пульпы аэрофлокулами, образующимися из частиц и пузырьков
разной крупности. На крупную частицу налипают мелкие
пузырьки воздуха, к которым с помощью тонких частиц
флотируемого минерала прилипают другие пузырьки. Средняя плотность
аэрофлокулы меньше плотности пульпы и она всплывает на ее
поверхность.
Возможность флотации крупных зерен, наряду с
эффективной флотацией тонких частиц мелкими пузырьками, является
достоинством предложенной Г. С. Бергером аэрофлокулярной
флотации. Аэрофлокулярный режим флотации является одним
из решающих факторов извлечения, например, крупных зерен
фосфорита и апатита. Для создания условий аэрофлокулярной
флотации необходима высокая степень гидрофобизации
минеральной поверхности, которая достигается сочетанием
собирателей с гетерополярным строением молекул и аполярных
углеводородов оптимальной вязкости.
301
2. «Активация» воздушных пузырьков. М. А. Эйгелесом и
М. Л. Воловой установлено [49, 50], что продукты
взаимодействия реагентов в пульпе при определенной их крупности и
плотности сорбции на пузырьках могут активировать элементарный
акт и скорость флотации. Этот эффект наблюдается при
замедлении процесса роста частиц труднорастворимых продуктов
взаимодействия реагентов и неполном заполнении ими
поверхности пузырьков. Можно полагать, что он обусловлен
«прокалывающим» действием закрепившейся частички на гидратную
прослойку между пузырьком и флотируемой частицей.
«Активация» пузырьков наблюдается обычно при дополнительной
подаче в пульпу органических реагентов-стабилизаторов
коллоидной системы, в качестве которых в зависимости от особенностей
флотационной системы могут использоваться алкиларилсуль-
фонаты, оксиэтилированные алкилфенолы и другие
соединения.
По данным В. А. Глембоцкого [7], «активация» пузырьков
может быть достигнута также в результате ультразвукового
воздействия на них. Причиной «активации» пузырьков при этом
может служить процесс деагрегирования воды, не позволяющий
образовываться на поверхностях раздела фаз устойчивому ги-
дратному слою, что неминуемо приводит к уменьшению
энергетического барьера при элементарном акте флотации.
Регулирование окислительно-восстановительного потенциала
и состояния жидкой фазы пульпы.
Окислительно-восстановительный потенциал Eh жидкой фазы пульпы определяет
вероятность и скорость протекания реакции взаимодействия
поверхности минералов с ионными и молекулярными компонентами
раствора. Приближение его к оптимальному позволяет
устранить или существенно замедлить скорость протекания
нежелательных реакций, стабилизировать ход флотационного процесса
и повысить технологические показатели при сокращении
расхода реагентов. Регулирование £71-потенциала пульпы может
быть осуществлено несколькими способами [1, 2, 46]:
подачей реагентов-окислителей или
реагентов-восстановителей, что широко применяется при селективной флотации медно-
цинковых руд и разделении медно-молибденовых
концентратов;
электрохимической обработкой пульпы без добавления
специальных реагентов-восстановителей (или
реагентов-окислителей). К настоящему времени получены положительные
результаты испытаний данного метода регулирования £7г-потенциала
на полиметаллических и сурьмяных рудах [47];
-термической обработкой пульпы. Примером данного способа
является паровая флотация при разделении
медно-молибденовых концентратов на Балхашской обогатительной фабрике.
Аэрация пульпы пузырьками пара при повышенной
температуре позволяет сократить концентрацию в ней кислорода, резко
замедлить скорость окисления сульфидных ионов, депрессирую-
.302
щих флотацию сульфидов меди, и обеспечить стабильный ход
процесса при малых расходах депрессора.
Регулирование состояния жидкой фазы пульпы в ряде
случаев удается осуществить ее магнитной обработкой.
Имеющиеся данные показывают, что предварительная магнитная
обработка пульпы может изменить смачивающую способность
воды, растворимость в ней газов и их активность при окислении
сульфидных минералов, активность и избирательность действия
на флотацию реагентов (жидкого стекла, цинкового купороса
и др.) и технологические показатели флотации. Однако
наблюдаемые явления пока в достаточной степени не объяснены, что
затрудняет использование магнитной обработки пульпы для
повышения показателей флотации.
Оптимизация подготовки коллективных концентратов к их
разделению, В последние годы все чаще при подготовке
коллективных концентратов к разделению используют термическую
и нпогца ультразвуковую их обработку.
Причинами более высокой^ эффективности разделения
коллективных концентратов после предварительной термической
их обработки (пропарки, варки, подогрева) могут быть:
возможность термического разложения избытка собирателя
(например,- при пропарке медно-молибденовых концентратов);
более интенсивное протекание реакции, приводящей к
подавлению флотации депрессируемых минералов (при доводке
шеелитовых концентратов по методу Петрова);
возможность разнонаправленного изменения
термодинамических функций разделяемых минералов (при пропарке медно-
никелевых концентратов перед их разделением), когда флоти-
руемость одних минералов улучшается, а других — ухудшается
вследствие различной закономерности изменения, например,
энтропии минералов от температуры.
Эффективность ультразвукового разрушения
адсорбционных слоев реагентов на минералах при подготовке
коллективных концентратов к разделению зависит от плотности
адсорбционного слоя, его состава, природы минерала, его крупности,
продолжительности и других параметров ультразвуковой
обработки. К настоящему времени показана принципиальная
возможность разделения после ультразвуковой деструкции
коллективного свинцово-пиритного, медно-пиритного, свинцово-медного
и других концентратов. Использование ультразвука в
промышленных условиях пока затрудняется отсутствием
промышленных образцов необходимых аппаратов.
15. ФЛОТАЦИОННЫЕ МАШИНЫ И АППАРАТЫ
Для флотационного обогащения созданы сотни машин и
аппаратов, из которых лишь несколько десятков нашли широкое
промышленное применение. Общим для всех современных кон-
303
струкций флотационных машин является использование в
качестве рабочего агента воздуха в виде мелких пузырьков,
образуемых в пульпе тем или иным способом. Минерализация
воздушных пузырьков происходит при непосредственном
столкновении их с частицами, скольжении частиц по поверхности
пузырьков, выделении пузырьков на поверхности частиц и
сочетании этих явлений. Относительная роль способов
минерализации зависит от применяемых способов аэрации и конструкций
флотационных машин.
15.1. Классификация флотационных машин
Флотационные машины различаются по конструктивным
признакам, способу аэрации и технологическому назначению.
В большинстве случаев их классификации за определяющий
признак принимают способ аэрации [24, 44].
По этому признаку флотационные машины могут быть
разделены на следующие группы [24]:
механические, в которых аэрация пульпы осуществляется
засасыванием воздуха из атмосферы мешалками различных
конструкций;
пневмомеханические, обеспечивающие аэрацию пульпы
сжатым воздухом, подаваемым в машину от вентиляторов,
воздуходувок или компрессоров, диспергирование которого
осуществляется мешалками или виброустройстзами различной
конструкции;
пневмогидравлические с самоаэрацией или принудительной
подачей сжатого воздуха, в которых для диспергирования
применяются различные гидравлические устройства;
пневматические с аэрацией пульпы сжатым воздухом,
подаваемым через патрубки или пористые перегородки;
электрофлотационные с аэрацией жидкости пузырьками,
выделяющимися при электролизе;
флотационные машины с изменяемым давлением, аэрация
в которых обеспечивается выделением растворенных газов из
пульпы при снижении давления над ней;
комбинированные, в которых пульпа аэрируется
несколькими способами.
Аэрирующие устройства устанавливаются в емкостях ко-
рытного, колонного и камерного типов.
Флотационные машины корытного типа представляют собой
ванну, вытянутую в длину. Исходная пульпа поступает с одного
конца ее и выходит с другого в виде хвостов. Пену удаляют
в желоба по всей длине ванны через боковые борта (обычно
самотечным способом). Уровень пульпы регулируют скоростью
разгрузки хвостов.
Флотационные машины колонного типа представляют собой
вертикальные устройства круглого, прямоугольного или
эллипсовидного сечения. Концентрат удаляется с верхней, а хвосты —
304
с нижней частей колонны; исходное питание поступает обычно
в среднюю часть.
Флотационные машины камерного типа состоят из
отдельных камер, в каждой из которых устанавливается один или
несколько аэраторов. В зависимости от способа продвижения
пульпы из предыдущей камеры в последующую машины
подразделяются на камерные, прямоточные камерные или камерно-
прямоточные.
В камерных машинах уровень пульпы регулируется в
каждой камере. Пульпа из одной камеры в другую попадает через
специальный разгрузочный карман. Образующийся в полости
работающего импеллера небольшой вакуум обеспечивает
возможность подсоса в аэратор промпродуктов флотации.
Благодаря этому в одной машине можно осуществить несколько
технологических операций. Недостатками камерных машин
являются: более сложный надзор из-за необходимости
регулирования уровня пульпы в каждой камере; ограничение
производительности машины по потоку производительностью импеллера;
непостоянство аэрации при колебаниях потока пульпы.
В прямоточных камерных машинах, в которых пульпа течет
по длине машины самотеком, уровень пульпы регулируется
только в последней камере, а одинаковый дебит проходящей
через аэратор пульпы обеспечивает постоянство ее аэрации. Это
исключает „недостатки, присущие камерным машинам. Для
прохода пульпы в межкамерных перегородках по ширине всей
камеры имеются большие отверстия, нижний уровень которых
находится на уровне надымпеллерного диска, а верхний — на
300—400 мм ниже уровня пульпы. Недостатком прямоточных
машин является понижение уровня пульпы вдоль машины, из-за
чего в каждой камере устанавливается своя высота пенного
порога и своя высота лопастей пеносъемника.
Камерно-прямоточные машины собираются из секций,
состоящих из нескольких камер. Первая камера называется
всасывающей. Пульпа в нее подается непосредственно на
импеллер, а остальные камеры работают как прямоточные.
Уровень пульпы регулируется в последней камере каждой
секции.
Кроме того, существуют так называемые монокамерные
флотационные машины, состоящие из одной камеры. Эти машины
обычно устанавливают на сливе мельницы, между ней и
классифицирующим устройством, или перед основным фронтом
флотации.
Камерными обычно бывают машины механического и
пневмомеханического типа, корытными — машины всех других
типов, колонными — машины пневматического типа.
15.2. Требования, предъявляемые к современным
флотационным машинам
Практика промышленного применения флотационных машин
для обогащения различных полезных ископаемых и в других
областях техники, исследование процессов, происходящих во
флотационных машинах при пенной флотации и изучение
гидроаэродинамики машин позволяют сформулировать
следующие основные требования к современным конструкциям
флотационных машин.
1. Равномерная по всему объему аэрация пульпы при
высокой степени диспергирования воздуха и оптимальном
соотношении тонкодисперсных и более крупных (несущих) пузырьков.
2. Все твердые частицы в пульпе должны находиться во
взвешенном состоянии и в условиях тесного контакта с
пузырьками воздуха. Максимальная частота столкновения частиц
с пузырьками должна протекать при минимальных
относительных скоростях их движения, но при достаточном для полной
минерализации пузырьков пути их движения в пульпе.
3. Всплывание минерализованных пузырьков должно
проходить в относительно спокойной (безвихревой) среде или в
восходящем потоке пульпы (что улучшает флотацию крупных
частиц и агрегатов).
4. Должно обеспечиваться оптимальное соотношение между
объемом флотационной пены и скоростью ее удаления. Если эта
скорость чрезмерно велика, то не обеспечивается возможность
возврата частиц пустой породы, механически Захваченных
пузырьками, из пены в пульпу и качество концентрата
ухудшается. Если же скорость удаления пены недостаточна, то из-за
деминерализации пены снижается извлечение.
5. Непрерывность флотации, т. е. непрерывная подача
питания и непрерывная разгрузка сфлотированных и несфлотиро-
ванных частиц.
6. Возможность регулирования высоты уровня пульпы и
пены, внутрикамерной циркуляции и аэрации пульпы.
Кроме этих требований, к флотационной машине, как и ко
всякой другой, предъявляются общетехнические требования:
надежность в работе, высокая износоустойчивость деталей,
малая энергоемкость, дешевизна, простота конструкции и т. д.
Работу флотационных машин характеризуют
технологические и технико-экономические показатели: извлечение и
содержание полезных компонентов в концентратах и хвостах,
продолжительность и стоимость флотации, производительность,
удобство ремонта, занимаемая площадь на единицу
производительности и т. д.
306
15.3. Процессы диспергирования воздуха
и аэрации пульпы во флотационных машинах
Диспергирование воздуха и аэрации пульпы во
флотационных машинах достигаются с помощью механического
воздействия на струю засасываемого или подаваемого под давлением
воздуха, пропускания воздуха сквозь мелкие отверстия,
выделения из объема пульпы растворенных в ней газов или
разложения воды электролизом.
Диспергирование воздуха механическим воздействием —<
основной способ аэрации пульпы в машинах механического,
пневмомеханического и пневмогидравлического типов.
Перемещение струй воздуха и жидкости в аэрирующих устройствах
этих машин характеризуется крайней турбулентностью. Это
приводит к появлению сил, различных по значению и
направлению, обеспечивающих не только разрыв струи воздуха и
образование мелких пузырьков, но и разрыв жидкости с
образованием кавитационных полостей, заполняемых парами воды
или растворенными газами. Эффективность диспергирования
возрастает, а крупность образующихся пузырьков d
уменьшается при увеличении скорости v перемещения струи воздуха
относительно жидкости, турбулентности движения и
уменьшении поверхностного натяжения аг-ж на границе раздела
жидкость—газ. Размер пузырьков при этом качественно
характеризуется уравнением
где D и К' — начальный размер и коэффициент сопротивления
пузырьков; р и р'— плотность соответственно жидкости и газа.
Среднее значение d пузырьков в механических флотационных
машинах около 1 мм [24]. По данным X. Шуберта, высокая
дисперсность пузырьков достигается при использовании аэраторов,
создающих большое количество мелкомасштабных
турбулентных импульсов в объеме жидкости [24].
Струя воздуха вводится в пульпу обычно с помощью
импеллера. Чтобы выбросить пульповоздушную смесь в камеру,
импеллер должен создать статистический напор несколько
больший, чем напор Нп пульпы в камере на уровне лопаток
импеллера.
Развиваемый импеллером статический напор Яс по теории
действия центробежных машин определяется уравнением
Hc^vV2g,
где *ф — коэффициент, зависящий от конструкции импеллера и
условий работы (для импеллера без статора г|э=0,5-ь0,7, со
статором -ф = 0,75-г-0,85); V — окружная скорость импеллера.
307
Импеллер начинает засасывать воздух только при некоторой
критической скорости vo, отвечающей условию
Напор Я, вызывающий аэрацию пульпы, равен избыточному
над критическим #п:
При постоянном дебите пульпы и различных значениях
окружной скорости вращения импеллера объем
выбрасываемого в камеру воздуха определяется отношением
<г;/<г;=м^-^ж-уо).
где QB' и QB" — объем выбрасываемого воздуха при скорости
соответственно V\ и v2\ K\ — коэффициент пропорциональности.
Аэрация пульпы в камере пропорциональна QB, поэтому
ai/a>B=K(of-o2)/(t|-^)f
где а,\ и а2 — аэра
ности импеллера
0 2 4 ап,м3/мин
Рис. 15.1. Влияние
объема Qn потока
в полость
импеллера на объем QB
засасываемого
воздуха в камерах
флотационных машин
ФМ-5 A), ФМ-6 B)
и ФМ-7 C) (по
данным В. А. Рундквис-
та)
ция пульпы. При постоянной производитель-
по пульпе аэрация прямо пропорциональна
разности квадратов фактической и
критической окружной скорости вращения
импеллера.
Импеллер не может засасывать воздух,
если его полость целиком заполнена
пульпой. В то же время при встречающихся на
практике значениях скорости вращения
импеллер не может работать как
вентилятор, так как давление воздуха на выходе
из импеллера недостаточно для
преодоления противодействующего давления
пульпы. Поэтому для каждой заданной
окружной скорости импеллера, превышающей
v0, существует оптимальный поток пульпы
через импеллер Qoiit, при котором аэрация
будет максимальной (рис. 15.1).
Увеличение окружной скорости
усиливает аэрацию, но при этом возрастают
расход мощности и значения сил инерции,
отрывающих частицы от пузырьков;
поэтому окружная скорость вращения
импеллера у современных флотационных машин
не превышает 10 м/с [24].
Аэрация возрастает при уменьшении
критической скорости и0, определяемой
308
давлением столба пульпы в камере. Поэтому уменьшение
высоты камеры приводит к увеличению аэрации. При увеличении
высоты камеры аэрирующий узел необходимо переводить в
режим поддува, гарантирующий голодный режим его по жидкому
и неизменность потребляемой мощности. Это особенно целе-
собразно при использовании камер большого объема.
Образование пузырьков при прохождении воздуха через
пористые перегородки (ткани, перфорированную резину, пористую
керамику и др.) является основным способом диспергирования
воздуха и аэрации пульпы в машинах пневматического типа.
Если при выходе газа из отверстия du пористой перегородки
образуется пузырек диаметром df объемом л:с?3/6, то он будет
отрываться под действием архимедовой силы
^а^^ж—Sr)gnd3/6
и удерживаться силой Fu поверхностного натяжения,
действующей по периметру дй„ отверстия,
F =nd с
II П Ж—Г*
Предельный диаметр пузырька при отрыве найдем из
условия FA — F„ по формуле
из которой следует, что повышение дисперсности воздушных
пузырьков может быть достигнуто снижением поверхностного
натяжения и диаметра отверстий в пористой перегородке.
Избыточное давление входящего газа по сравнению с
выходящим из аппарата обусловлено необходимостью преодолеть
противодействие избыточного давления внутри пузырька (лап-
ласова давления), статического давления столба жидкости
в аппарате, сопротивлений при прохождении отверстий в
пористой перегородке. При небольших размерах отверстий в
пористой перегородке избыточное давление подаваемого воздуха
может быть значительно выше статического давления жидкости
в аппарате. На практике стремятся поддерживать такие
избыточное давление и расход воздуха и применять пористые перс-
городки с таким размером отверстий, чтобы можно было
получить тонкодиспергированные пузырьки воздуха.
При средней крупности пузырьков 3—4 мм степень аэрации
в пневматических флотационных машинах колеблется от 15
до 35 % [24]. При истечении сжатого воздуха через движущиеся
пористые перегородки или в движущиеся потоки пульпы
размеры пузырьков уменьшаются вследствие срезающего
воздействия потоков пульпы [15].
Образование пузырьков при выделении газов из раствора
является основным процессом аэрации пульпы во
флотационных машинах с изменяемым давлением (вакуумных и
компрессионных).
309
В соответствии с законом Генри, растворимость газов
растет при повышении давления на жидкость и, наоборот,
уменьшается при понижении давления, приводя к выделению
избыточно растворенных газов в виде пузырьков. При этом размер
«dmin пузырьков, способных существовать в воде, находится
в прямой зависимости от значения сгж-г и в обратной от
величины АС пересыщения раствора газами или снижения давления
Ар при условии начальной насыщенности раствора газами:
<U - 2<тж_г/(*ЛС) в 2аж_г/Лр, A5.1)
где k — постоянная в уравнении Генри. Число п зародышей
пузырьков; образующихся в единице объема за единицу времени,
определяется по уравнению Я. Б. Зельдовича [15]:
где в — коэффициент, D — аналог коэффициента диффузии,
К — постоянная Больцмана, Т — абсолютная температура.
С учетом уравнения A5.1)
л=(МС/2(Тж-гL bD*j8nD/(KT).
Отсюда следует, что число зародышей пузырьков газов,
выделяющихся из раствора, тем больше, чем выше' пересыщение
раствора газами и чем меньше поверхностное натяжение на
границе раздела жидкость—газ.
По данным Н. Ф. Мещерякова [24], при вакууме 933 гПа и
флотационных концентрациях пенообразователя (соснового
масла) из 1 л воды выделяется 90—96 % растворенных газов
общим объемом 300 мл. При крупности пузырьков 0,1—0,3 мм
их суммарная поверхность в 1 л воды велика и составляет 6—
18 м2.
Выделение растворенных газов улучшается в присутствии
тонких гидрофобных частиц, поскольку, чем гидрофобнее
поверхность, тем слабее связь ее с молекулами воды, и,
следовательно, работа на образование зародышей микропузырьков и
последующего роста их, связанная с разрывом связей между
молекулами на поверхности раздела минерал—вода, меньше
работы по разрыву связей между молекулами воды.
Явления растворения и выделения растворенных газов
наблюдаются также в пульпе механических, пневмомеханических
и пневмогидравлических машин, характеризующихся большими
лерепадами давления особенно в зоне аэрационного устройства.
Образование пузырьков газов при электролизе является
основным процессом аэрации пульпы в электрофлотационных
машинах.
Минимальное напряжение, необходимое для начала
электролиза воды, составляет обычно 1,6—1,7 В. На поверхности
жатода выделяются пузырьки водорода, на поверхности анода —
310
пузырьки кислорода. Крупность образующихся пузырьков
зависит от условий электролиза, степени шероховатости и
кривизны поверхности электродов, поверхностного натяжения на
границе раздела фаз и температуры пульпы [7, 13].
15.4. Механические флотационные машины
Во всех аэрационных узлах флотационных машин
засасывание воздуха из атмосферы и образование пульповоздушной
смеси, выбрасываемой под действием центробежных сил в
камеру, обусловлено образованием небольшого вакуума в полости
вращающегося импеллера. В качестве импеллеров
используются мешалки различных конструкций (дисковые с радиально
расположенными лопатками, стержневые типа беличьего
колеса с осевыми насосами внутри них и др.)- При этом аэрация
пульпы определяется окружной скоростью импеллера и
конструктивными особенностями аэрирующих узлов и камер
механических флотационных машин.
В СССР наибольшее распространение получили
механические флотационные машины конструкции Механобра ФМ
(СССР) с вместимостью камер от 0,14 до 6,3 м3. Стандартная
машина ФМ собирается из двухкамерных секций:. первая
камера является всасывающей, вторая прямоточной (рис. 15.2).
В случае необходимости машина может собираться из одних
всасывающих камер или из звеньев, состоящих из одной
всасывающей и нескольких прямоточных камер.
В каждой камере устанавливается блок аэраторов, который
полностью монтируется на заводе и является самостоятельным
конструктивным узлом. Блок аэратора (см. рис. 15.2) состоит
из вертикального вала 10 с насаженным на нем импеллером.
Импеллер представляет собой диск 19 с шестью радиальными
лопатками 17. Вал вращается внутри трубы 2, верхний конец
которой закрыт наглухо. В нижней части труба расширяется и
к ней крепится надымпеллерный диск 9 с лопатками статора 16,
расположенными под углом 60° к радиусу. Направляющие
лопатки статора способствуют превращению тангенциальной
составляющей динамического напора пульпы в статический,
увеличивая тем самым аэрацию. Радиальный зазор между
лопатками импеллера и статора не должен превышать 5—8 мм.
Исходная пульпа из приемного кармана / поступает в
аэратор по трубе 20, а воздух — по трубе 3. Для внутрикамерной
циркуляции надымпеллерный диск имеет круглые отверстия,,
расположенные по окружности над лопатками 17 импеллера.
Кроме того, для регулирования внутрикамерной циркуляции
в нижней части трубы 2 имеется отверстие 18, которое
прикрывается заслонкой 14. Тягой 5 она устанавливается в таком
положении, чтобы был обеспечен оптимальный поток пульпы на
импеллер, необходимый для достижения максимальной
аэрации.
ЗН
гО 19 18 17 16 15 7fy 13 12 11 fO
Рис. I5.2. Флотационная машина конструкции Механобра с всасывающими
(а) и прямоточной F) камерами
Для всасывания промпродуктов в каждой камере может
быть установлен патрубок, идущий от центральной трубы к
передней стенке камеры. В тех камерах, куда промпродукт не
поступает, патрубок не устанавливается, а отверстие в
расширенной части вертикальной трубы закрывается пробкой 15.
Пенный продукт удаляется в сборный желоб.
Всасывающая и прямоточная камеры разделены
перегородкой 4, В каждой второй камере секции или в последней камере
прямоточной машины имеется устройство для регулирования
уровня пульпы и удаления камерного продукта (хвостов).
Основная часть пульпы переливается через отверстие 13 в
боковой стенке 12 камеры и поступает в приемный карман
следующей камеры. Чтобы вместе с камерным продуктом не уходила
пена, разгрузочное отверстие экранировано перегородкой 6.
Для регулирования высоты слоя пены в камере (секции)
или, что то же, уровня пульпы разгрузочное отверстие со
стороны межкамерного кармана прикрыто заслонкой 11,
положение которой регулируется устройством 8. Для разгрузки
крупных частиц (песков), находящихся в нижнем слое пульпы,
внизу межкамерной перегородки 12 имеется небольшое отверстие,
которое может перекрываться шибером при опускании его
тягой 7.
Для создания спокойной зоны пенообразования
предусмотрен успокоитель, состоящий из радикальных Г-образных
пластин, расположенных вокруг статора и прикрепленных ко дну
камеры. Для устранения застаивания пены в задней части
камеры и ускорения пеносъема задняя стенка выполнена
изогнутой в сторону пенного порога, лопасти пеносъемщика имеют
шарнирную подвеску.
312
Рис. 15.3. Флотационная машина-
с кипящим слоем ФМ6, ЗКС
К недостаткам машин ФМ .
относятся большой износ
лопаток статора и сильные
восходящие потоки пульпы,
вызывающие бурление и
нарушение процесса пенообразо-
вания, что имеет особенно
большое значение при
флотации руд с низким содержанием
полезного компонента. Однако
машины отличаются большой
производительностью по
потоку пульпы и засасываемому
воздуху; по конструктивным
параметрам они находятся на
уровне лучших зарубежных
образцов.
Флотационная машина
с кипящим слоем ФМ6,ЗКС (СССР) отличается по конструкции
(рис. 15.3) от флотационных машин ФМ, во-первых, тем, что
внутри камеры на высоте 500—550 мм от дна камеры 2
устанавливается решетка / из уголков, живое сечение щелей
которой составляет 18—20 % всей ее площади. Во-вторых, на
передней стенке камеры с внешней или внутренней стороны
устанавливается сходящийся желоб 5 постоянного сечения,
соединяемый трубой 6 с колпаком надымпеллерной трубы 3. Желоб
служит для отбора через щель 4 циркуляционного потока на
импеллер из верхней зоны камеры.
Решетка обеспечивает гашение турбулентности потоков них
равномерное распределение но всему горизонтальному сечению
камеры. В результате этого над решеткой создается кипящий,
или взвешенный, слой минеральных частиц и воздушные
пузырьки вместе с потоками жидкости .движутся по
криволинейным каналам, образуемым витающими частицами. Это
обеспечивает многократное столкновение пузырьков с частицами
минералов и более длительное время их контакта, чем в машинах
ФМ. Наряду с этим резкое уменьшение турбулентности потоков
в зоне минерализации и флотации позволяет свести к минимуму
деминерализацию воздушных пузырьков, а наличие восходящих
потоков, направленных к пенному порогу, ускоряет вывод
минерализованных пузырьков из камеры. Все это позволяет
повысить скорость флотации и иногда крупность флотируемых
частиц. Кроме того, пульпа, возвращаемая на импеллер 8 и
статор 7 через щель 4У имеет низкую плотность и не содержит
крупных абразивных частиц, что способствует увеличению
срока службы аэратора.
Машина ФМ6,ЗКС предназначена в основном для
обогащения горно-химического сырья флотационной крупности, но
может быть использована и для руд цветных металлов.
313:
.^J
\C\
Vf
V
Рис. 15.4. Аэраторы флотационных
машин «Денвер-М» (а), «Бут» (б),
«Гумбольдт» (в)
Отличительной особенностью аэратора флотационной маши-
ны «Денвер Суб-А» (США) с вместимостью камеры до 11,3 м3
по сравнению с аэратором машины ФМ является радиальное
расположение статора. Это уменьшает бурление пульпы, но
приводит к тому, что глубокие машины (с глубиной камеры
1235 мм) .могут работать только с подачей воздуха от
воздуходувки.
Флотационная машина «Денвер-М» (США) отличается
(рис. 15.4, а) от предыдущей наличием на одном валу двух
импеллеров: центробежного импеллера 1 (типа «Денвер
Суб-А») и осевого четырехлопастного пропеллера 5.
Центробежный импеллер, имеющий больший диаметр, расположен на
глубине 800 мм, осевой — на глубине 1200 мм от верха камеры.
Аэрация пульпы обеспечивается в результате засасывания
воздуха из атмосферы через центральную трубу 3 в зону верхнего
импеллера.
Пульповоздушная смесь, выбрасываемая центробежным
импеллером 1 через лопатки 2 статора 4, частично увлекается
осевым импеллером 5 вниз, обеспечивая тем самым в нижней
части камеры перемешивание и аэрацию пульпы. В нижней
зоне камеры подсасываемый в трубу 6 воздух не только
дополнительно диспергируется, но и частично растворяется.
Растворенный воздух при выходе из трубы выделяется на
поверхности гидрофобных частиц в виде микропузырьков,
которые облегчают последующее прилипание к частицам крупных
пузырьков при их прохождении в зоне интенсивной аэрации,
создаваемой центробежным импеллером /.
Флотационные машины этого типа применяют для флотации
тяжелых или крупнозернистых материалов, например
золотосодержащих песков, свинцово-цинковых руд, а также для
флотации углей.
314
Флотационная машина «Бут» (США) с вместимостью
камеры до 12,5 м3 также имеет на одном валу два импеллера,,
установленных на разных уровнях от дна машины (рис. 15.4 6).
Надымпеллерный диск 4 с радиальными лопатками 5
монтируется над верхним импеллером 2, который выполнен в виде
крестовины трапециевидного сечения и предназначен для
засасывания (через трубу 3) и диспергирования воздуха. Нижний
импеллер 1 имеет форму пропеллера и обеспечивает
взвешивание частиц минералов в нижней части камеры и подачу их
в зону аэрирующего действия верхнего импеллера 2. Аэрацион-
ный узел обеспечивает хорошую циркуляцию пульпы и
достаточно интенсивную ее аэрацию в средней части камеры 4
машины.
Недостатками флотационных машин «Денвер-М» и «Бут>
являются сложность конструкции аэратора, недостаточная
аэрация в нижней части камеры, трудность создания с подобным
типом аэратора^машин с большой вместимостью камер. В
настоящее время машины «Денвер-М» серийно не выпускаются.
Флотационная машина «Гумбольдт» (ФРГ) с вместимостью-
камеры до 10 м3, имеет (рис. 15.4, в) импеллер 5,
представляющий собой наклонный диск, на котором сверху и снизу
расположены лопатки. Над импеллером помещена статорная плита
2, на наружной кромке которой имеются короткие радиально
расположенные ребра 4 (статор), между которыми находятся
отверстия 3. При вращении импеллера 5 создается резкая смена
давления и разрежения, в результате чего достигается
интенсивное перемешивание и аэрация пульпы. Флотационные
машины применяются для флотации калийных солей, железных
руд-и углей.
Прямоточная флотационная машина «Вормен» (Австралия)
имеет аэрационный узел (рис. 15.5, а), состоящий из
пальцевого ротора 3, крепящегося на полом валу 4У статора 2 и
успокоителя 1. Ротор представляет собой диск с прикрепленными
к нему пальцами круглого или фигурного сечения,
расширяющимися к нижней части. Оси пальцев расходятся книзу и имеют
наклон 45° в направлении, противоположном направлению
вращения ротора. Статор представляет собой диск с лопастями^
расположенными по окружности в виде отдельных дуг. Высота
лопастей по направлению к периферии уменьшается.
Успокоитель состоит из четырех секторов с эвольвентными лопастями
различных радиусов.
Преимуществом машины «Вормен» является возможность
переработки грубозернистой пульпы, так как интенсивный
турбулентный поток обеспечивает взвешивание крупных частиц,
что облегчает их контакты с пузырьками воздуха.
Удельная производительность и технологические показатели
. машины выше, чем у флотационной машины «Денвер Суб-А».
Машины «Вормен» применяются на угольных фабриках
Японии, США, а также на рудных фабриках Австралии.
315
Рис. 15.5. Аэрационные узлы флотационных машин «Вормен» (а), «Фагер-
грен» (б), МФУ (в)
Прямоточная флотационная машина «Фагергрен» (США)
(рис. 15.5, б) имеет аэратор (типа 1 + 1), выполненный из
цельнолитых (из износостойкой резины или синтетических
материалов) ротора 3 и статора 4.
Статор представляет собой цилиндр с отверстиями овальной
формы, на внутренней поверхности которого между отверстиями
проходят вертикальные ребра, имеющие форму полуцилиндра.
Статор крепится к воздушной трубе 7, внутри которой
проходит вертикальный вал с ротором 3 на нижнем конце. Ротор
316
представляет собой в сечении форму звезды с 8 или 10
радиальными лопастями («лучами»), которые на периферии имеют
трапециевидное или цилиндрическое утолщение. Статор короче
ротора, благодаря чему аэратор обеспечивает хорошее
взвешивание минеральных частиц в камере.
При вращении ротора пульпа, засасываемая снизу, и
воздух, засасываемый сверху через трубу 7, смешиваются и
выбрасываются в зону интенсивных ударов между статором и
ротором. В этой зоне воздух тонко диспергируется и выводится
в горизонтальном направлении потоками пульпы в окружающее
статор пространство. Пузырьки воздуха поднимаются на
поверхность в пенный слой, а деаэрированная пульпа
возвращается по боковым стенкам обратно, в зону ротора.
Для создания на поверхности пульпы спокойной зоны пено-
образования на трубе 7 установлен конический
перфорированный колпак 5. Для усиления циркуляции пульпы в камере 6
установлено перфорированное ложное днище / с
циркуляционной трубой 2, через которые пульпа засасывается ротором
вверх.
Горизонтальная направленность потоков пульпы,
выбрасываемых ротором, и высокая их аэрированность позволяют вести
процесс флотации в машинах при низком уровне пульпы в
камере, имеющей большую ширину, что обусловливает низкий
удельный расход электроэнергии (не более 2 кВт*ч/т руды).
Пена из камер удаляется с двух сторон самотеком.
Флотационные машины «Фагергрен» (или «Вемко-Фагер-
грен») выпускаются с вместимостью камер до 28,3 м3 и
являются одной из наиболее распространенных конструкций машин,
применяемых для флотации руд и углей [24, 44].
Прямоточная флотационная машина МФУ (СССР) с
вместимостью камеры до 10 м3 и двусторонним съемом пены
(рис. 15.5, в) имеет в каждой камере 4 по два блок-аэратора 2,
состоящих из центробежного 3 и осевого 1 импеллеров,
расположенных на общем валу 9, центральной трубы 8 с отверстием
10 для воздуха. Импеллеры помещены в аэрационную камеру
14, верхняя часть которой представляет собой крышку 13 с
лопатками, выполняющую роль статора.
Пульпа подводится к нижней части центробежного
импеллера 3 с помощью пульповода 7 и к верхней — через кольцевые
отверстия 12 между диском импеллера и статора и через
отверстия 11 между диском импеллера и центральной трубой. Здесь
она аэрируется и затем поступает на осевой импеллер /, где
пульповоздушная смесь смешивается с частью неаэрированной
пульпы и выбрасывается во флотационную камеру 4 через
успокоитель 6 с перфорированной поверхностью. Для аэрации
верхнего слоя пульпы в камере и стабилизации работы осевого
импеллера часть высоко насыщенной воздухом пульпы
выбрасывается через щели 5 аэрационной камеры непосредственно во
флотационную камеру 4. В результате этого в машине осуще-
317
ствляется послойная аэрация пульпы на уровне центробежного
и осевого импеллеров.
Двухстадиальный принцип аэрации пульпы и полное
разделение зон аэрации и флотации, позволяющее создать
необходимый гидродинамический режим в каждой из этих зон,
стабильная и достаточно высокая степень аэрации пульпы, наличие
восходящих потоков в камере являются положительными
особенностями машины МФУ2-63, широко используемой при
флотации углей [43].
Преимуществами механических флотационных машин по
сравнению с другими типами являются их хорошие
гидродинамические параметры, универсальность применения и
пригодность для использования в любых технологических схемах,
отсутствие потребности в дополнительных „ источниках воздуха.
К недостаткам механических машин относятся:
непостоянная аэрационная характеристика, зависящая от степени износа
импеллера и статора; отсутствие регулирования количества
воздуха в зависимости от потребностей технологического
процесса; сложность конструкции; относительно высокая
энергоемкость и металлоемкость; довольно быстрый износ статора и
импеллера.
15.5. Пневмомеханические флотационные машины
Особое значение, которое приобрели пневмомеханические
машины в последние годы, обусловлено возможностью
создания камер большой вместимости вследствие разъединения
в них операций подачи воздуха и диспергирования его с
одновременным перемешиванием пульпы мешалками различных
конструкций.
Из пневмомеханических машин с пальцевым аэратором
наибольшее распространение получили в СССР флотационная
машина ФПМ с вместимостью камеры до 6,3 м3, а в
зарубежной практике машины «Аджитейр» с вместимостью камеры
до 22,6 м3. Машины являются
прямоточными и имеют
принципиально одинаковый
аэрирующий узел (рис. 15.6). На
полый вал / насажен
конический (у машин ФПМ) или
плоский (у машин
«Аджитейр») импеллер 2, по
окружности которого -на
расстоянии 20—30 мм друг от друга
вертикально расположены
стержни (пальцы) небольшой
длины. Импеллер окружен ста-
Рис. 15.6. Флотационная машина Т0РН°Й решеткой
(успокоитесь пальцевым аэратором лем) с радиальными лопастя-
318
Рис. 15.7. Аэраторы флотационных машин «Денвер ДР» (а), «БФР» (б) и
ФПМУ6.3 (в)
ми 5. Сжатый воздух по воздухопроводу через полый вал
подается под крышку импеллера от воздуходувки низкого
давления (ЫО5—1,5-105 Па).
Эффективная диспергация воздуха и аэрация пульпы■
осуществляются при прохождении их между стержнями
вращающегося импеллера и при ударе о радиальные лопасти статор-
ной решетки, обеспечивающей также гашение турбулентных
потоков, выбрасываемых импеллером, вращающимся с
окружной скоростью 6—8,5 м/с.
Достоинствами машины по сравнению с механическими
являются возможность регулирования аэрации в каждой камере,
меньшие энергоемкость, стоимость ремонта и время флотации
(на 20—30%) в основных операциях; недостатками —
невозможность флотации крупнозернистого материала,
необходимость полной выработки камер при замене блок-аэратора.
Прямоточные машины ФПМ12,5 (СССР) с вместимостью
камеры 12,5 м3, ФПМ25 (СССР) с вместимостью камеры 25 м3
и «Денвер ДР» (США) с вместимостью камеры до 36,1 м3
имеют принципиально одинаковый центробежный аэратор
(рис. 15.7, а). Нижняя часть воздушной трубы 1, в которой
вращается вал импеллера 3, помещена внутрь открытого
конуса 2, к нижней части которого присоединяется статор 4.
Труба и конус соединены между собой вертикальными
ребрами. Такая конструкция обеспечивает создание кольцевого
пространства между трубой и цилиндром.
При работе машины пульпа засасывается через кольцевое
пространство между трубой и цилиндром, а воздух нагнетается
по трубе. Пульповоздушная смесь, насыщенная хорошо
диспергированными пузырьками воздуха, выбрасывается через
статор по всей поверхности днища камеры, преобразуясь
затем в равномерные потоки, направленные вверх и
способствующие подъему пузырьков к поверхности.
Применение машин ФПМ12,5, ФПМ25 и «Денвер ДР»
позволяет несколько повысить крупность флотируемых частиц, так
как при больших объемах пульпы, циркулирующей через им-
319
пеллер, увеличивается скорость восходящих потоков,
обеспечивающих эффективное взвешивание минеральных частиц.
Машины ФПМ12,5 и «Денвер ДР» широко применяются на
обогатительных фабриках, перерабатывающих калийные соли,
фосфатные, медные, цинковые, молибденовые и другие руды,
а также при флотации углей и других полезных ископаемых
[41,43].
Основной конструктивной особенностью прямоточной
флотационной пневмомеханической машины БФР (Швеция) с
вместимостью камеры до 8 м3 является подача воздуха в зону
перемешивания в виде струй топкодисперсных пузырьков.
Аэрационный узел этой машины (рис. 15.7, б) состоит из
импеллера 4, резинового кольца 2, сопел и диспергатора 3.
Импеллер представляет собой плоский диск с радиальными
лопатками с обеих сторон. Кольцо из специальной мягкой резины
имеет форму тора и устанавливается по периферии надымпел-
лерного диска. В нижней части кольца имеются или несколько
рядов прорезей длиной 2—5 мм, или большое число мелких
отверстий. Диспергатор, представляющий собой кольцо с
радиальными лопатками овальной формы, служит для
дополнительной диспергации воздуха и равномерного распределения
его и пульпы по камере. Воздух подается от компрессора или
воздуходувки и расход его регулируется вентилем /.
Получение тонкодиспергированного воздуха положительно
сказывается па эффективности флотации. Широкому
распространению машины препятствуют сложность конструкции
аэратора, относительно малый объем поступающего в камеры
воздуха, довольно высокие энерго- и металлоемкость.
Прямоточная флотационная машина ФПМУ6,3 (СССР)
с вместимостью камеры 6,3 м3 имеет комбинированный блок-
аэратор (рис. 15.7, в). Воздух поступает через полый вал /
в ступицу осевого импеллера 5; заключенного в трубу 6.
Радиальные отверстия 7 полого вала служат для равномерного
распределения воздуха в полости конического пальцевого
импеллера 3. Кроме того, воздух может быть подан через
патрубок фурмы 4 непосредственно на лопатки осевого импеллера.
Вокруг пальцевого импеллера установлен статор 2 с
укороченными лопатками, улучшающими диспергирование воздуха и
способствующими снижению турбулентности пульпы в камере.
Сочетание в блоке-аэраторе центробежного пальцевого и
осевого импеллеров позволило добиться эффективного
диспергирования больших объемов воздуха (до 300 м3/ч) и
удовлетворительного распределения его по всему сечению камеры.
В прямоточной флотационной машине ФПМ (СССР) с
коническим аэратором с вместимостью камеры до 6,3 м3 аэратор
(рис. 15.8, а) представляет собой полый усеченный конус 4У
закрытый сверху диском 2 и установленный на полом валу 3
вершиной вниз. На внешней поверхности конуса имеются
стержневые рифли 1.
320
Рис. \5.8. Аэраторы флотационных машин ФПМ с коническим аэратором (а),
«Минимет» (б), OKI6 и ОК38 (в, г).
Воздух подается внутрь полого конуса, выходит через
отверстие в его нижней части и за счет собственной подъемной
силы устремляется вверх по внешней расширяющейся
поверхности, попадая в пристенный слой, в котором он движется
вместе с потоками перекачиваемой жидкости. При этом благодаря
расширению конуса слой воздуха утоньшается и на него с
возрастающей интенсивностью воздействуют неровности
поверхности, за которыми образуются каверны, при замыкании которых
происходит эффективное диспергирование воздуха, Пульповоз-
душные потоки в верхней части тела вращения по периметру
основания выбрасываются в окружающую среду.
Аэратор отличается простотой конструкции, низкой
энергоемкостью и высокой удельной производительностью. Машина
с коническими аэраторами применяются в основном при
флотации горнохимического сырья [24, 28].
В прямоточной флотационной машине «Минемет»
(Франция) с вместимостью камеры до 12,2 м3, широко применяемой
на обогатительных фабриках Франции, импеллер (рис. 15.8, б)
И Заказ JVe I957
321
выполнен в виде корзины, стержни 5 которой расположены на
поверхности двух противоположно направленных конусов,
находящихся на одной оси. Угол конусности составляет 15—25°.
Верхний диск 4 импеллера сплошной, а в нижнем / имеются
циркуляционные отверстия 6. Импеллер устанавливается на
полом валу 3, в нижней части которого имеются отверстия 2
для подачи воздуха внутрь импеллера.
Импеллер флотационной машины, как правило, работает
без статора, достаточно равномерно распределяет воздух и
перемешивает его с пульпой. Съем пены односторонний и
осуществляется самотеком.
Аэратор прямоточных флотационных машин ОК16 и ОК38
(Финляндия) с вместимостью камеры 16 и 38 м3 состоит
(рис. 15.8, в, г) из лопастного ротора 1 и радиального статора
2. Ротор 1 представляет собой диск, к которому снизу по кругу
крепятся 10 элементов. Каждый элемент состоит из двух
радиальных лопастей сложного профиля и имеет V-образную
форму. Лопасти соседних элементов параллельны и между
ними имеются щели, из которых воздух, подаваемый через
полый вал 3, выходит в камеру.
При вращении ротора пульпа со дна камеры засасывается
вверх в полость между радиальными лопастями и выходит
в верхней части ротора. Точки выхода пульпы и воздуха из
полости ротора чередуются попеременно по кругу, но на
выходе из него смешиваются. Образованная пульповоздушная
смесь выбрасывается между лопатками статора в камеру.
Аэратор обладает хорошими аэрационными характеристиками
и машина ОК16 нашла широкое применение в Финляндии.
В вибрационной флотационной машине
пневмомеханического типа (рис. 15.9, а), разработанной в СССР, аэратор
Рис. 15.9. Флотационные машины вибрационная (й) и «Максвелл» (б)
322
представляет собой диск 1 с конусными отверстиями, который
крепится на полой штанге 2. Аэратор приводится в движение
от электромагнитного вибровозбудителя 4, При вибрации
дискового аэратора через конусные отверстия происходит
движение пульпы от аэратора к днищу камеры, затем к боковым
стенкам и снова к аэратору. Воздух поступает в аэратор через
штуцер 3 и полую штангу. Применение вибрационного
движения рабочего органа во флотационных машинах вместо
вращательного может обеспечивать значительное снижение расхода
электроэнергии и уменьшение износа рабочих органов.
Флотационная машина «Максвелл» (Канада) представляет
собой (рис. 15,9,6) чан вместимостью от 4,25 до 56,6 м3, в
котором пульпа перемешивается лопастным импеллером 2, а
воздух подводится снизу через трубу / с обратным резиновым
клапаном и специальной 'распределительной головкой. Пенный
желоб 5 расположен внутри чана и закреплен на четырех
успокоительных пластинах 4. Исходное питание поступает по трубе
6\ камерный продукт выходит через трубу 3.
В других флотационных машинах, созданных на основе
агитационных чанов, воздух подается через кольцевой воздуховод,
гуммированный резиной с отверстиями для выхода воздуха и
помещаемый внутрь чана.
Преимуществом машин типа «Максвелл» является
возможность переработки в них сравнительно крупнозернистых пульп
без оседания частиц при наличии в верхней части камеры
спокойной зоны. Отличительной особенностью их являются
небольшой расход подаваемого воздуха @,12—0,33 м3/мин на
1 м3 объема пульпы) и слабая зависимость времени флотации
от времени пребывания пульпы в камере (чане) [44]. Машины
устанавливают обычно дополнительно к основному фронту
флотации и в них выполняется либо операция
концентрирования пульпы с одновременным снятием в пену богатой
«головки» концентрата, либо дофлотация хвостов обычных машин.
Достоинствами пневмомеханических машин являются:
достаточная простота их конструкции; постоянство аэрационной
характеристики, не зависящей от износа рабочих органов;
возможность регулирования расхода воздуха в широком
диапазоне; небольшая металлоемкость; меньший расход
электроэнергии; большой срок службы аэратора; простота
эксплуатации.
К недостаткам пневмомеханических машин относятся:
невозможность организации покамерной регулировки уровня
пульпы; необходимость использования всасывающего
механического блока для перекачки промпродуктов; необходимость
применения воздуходувок для нагнетания воздуха.
11*
323
15.6. Пневмогидравлические флотационные машины
К этой группе машин относятся флотационная машина
с циклонными аэраторами, эжекторная флотационная машина,
а также флотационная машина «Апатит».
Во флотационной машине с циклонными аэраторами
(рис. 15.10) аэраторы представляют собой центробежные
форсунки, в которых распыление подаваемого под давлением
в 1,5 - 104 Па воздуха происходит под воздействием вихревых
потоков жидкости, вытекающей из циклона в виде веера. В
качестве жидкости используется пульпа, подаваемая в аэратор.
Объем циркулирующей пульпы составляет 25—30 %
вместимости машин. Циклонные аэраторы расположены вдоль машин
корытного типа по осевой линии на расстоянии 1200 мм друг
от друга. Пена удаляется с двух сторон самотеком через
пенные пороги или пеносъемники. Хвосты отводятся через
разгрузочный карман.
Во флотационной машине «Апатит» (СССР) в качестве
аэраторов используется газо-жидкостной аппарат, в который
под давлением 2,5* 105—3,5-105 Па подаются воздух и вода
в соотношении 15:1. Аэраторы монтируются в три ряда вдоль
продольной оси днища ванны флотационной машины. Машина
предназначена для переработки апатито-нефелиновых руд
■с большим выходом пенного продукта [9,24].
В эжекторных флотационных машинах часть пульпы
подается насосами в аэратор эжекторного типа, засасывающий
воздух из атмосферы. Аэрированная пульпа выбрасывается внутрь
флотационной камеры и соответствующими устройствами
равномерно распределяется по всему ее объему [24].
Пневмогидравлические флотационные машины оказываются
в ряде случаев весьма эффективными, но не получили широ-
Цсходная лумьпа
Ваздух- из воздуходувки
Рециркуляция пумьпы
Рис. 15.10. Флотационная машина с циклонным аэратором:
1 — ванна; 2 — насос; 3 — циклонные аэраторы
324
кого применения из-за необходимости установки большого
числа насосов, значительного износа рабочих частей и
высокого расхода энергии, достигающего 5—6 кВт» ч/т
перерабатываемой руды.
15.7. Пневматические флотационные машины
В пневматических флотационных машинах пульпа
аэрируется и перемешивается сжатым воздухом. К основным типам
таких машин относятся аэролифтные, колонные, циклонные и
пенной сепарации.
В мелких аэролифтных машинах (рис. 15.11, а) подаваемый
из ресивера 1 под давлением 1,2- 104—3- 104 Па воздух, выходя
из трубок 6, установленных в ванне 4У поднимается между
продольными стенками аэролифта 5 и смешивается с пульпой,
понижая ее плотность в этой зоне. Вследствие возникающей
разности гидростатического давления, пульпа выбрасывается из
аэролифта и падает между стенками аэролифта и
перегородками 2. В аэролифте и в зоне падения происходит интенсивное
перемешивание воздуха с пульпой и его диспергирование.
Аэрированная пульпа вытекает из зоны падения через отверстия
в перегородках. Толщина слоя пены, образующегося между
перегородками и стенками машины, регулируется хвостовыми
порогами 3 или накладками на них.
Пульпа циркулирует в ванне машины под действием
аэролифта и течет вдоль машины под напором поступающего в
машину потока. При высоте ванны машины около 900 мм,
расстоянии между пенными порогами 1300 мм и между стенками
аэролифта 160—200 мм расход воздуха составляет от 4 до
13 м3/мин на 1 м длины машины.
Рис. 15.11. Мелкая (а) и глубокая (б) аэролифтные машины
325
По сравнению с механическими и пневмомеханическими,
эти машины отличаются простотой конструкции и малой
металлоемкостью. Их недостатком являются низкие удельная праиз-
водительность @,75—3 т-м2/ч) и коэффициент использования
воздуха, а также недостаточная эффективность при работе на
зернистых тяжелых пульпах.
Принцип действия глубоких аэролифтных машин
аналогичен предыдущим. Глубокая аэролифтная флотационная машина
(АФМ2,5) конструкции Механобра (рис. 15.11, б) с глубиной
ванны 2,5 м отличается от других типов аэролифтных машин
конструкцией аэролифта 2 и воздухопровода 1, имеющего
в нижней части аэраторы 3 с резиновыми щелевыми затворами,
гарантирующими воздухопроводную систему от попадания
в нее пульпы.
Эти особенности глубокой аэролифтной машины сделали ее
удобной в эксплуатации. Производительность по потоку пульпы
достигает 10 м3/мин, расход воздуха 5—7 м3/мин на 1 м длины
машины при средней крупности флотируемого материала и до
10 м3/мин при повышенной его крупности; давление воздуха
2,5-104—3-104 Па.
Машина имеет ряд преимуществ по сравнению с мелкими
аэролифтными машинами: более высокую аэрацию пульпы и
лучшую диспергацию воздуха; более высокий кпд аэролифта,
увеличивающий интенсивность циркуляции и перемешивания
пульпы и позволяющий флотировать крупнозернистый
материал; более высокую производительность на единицу объема
машины; занимает значительно меньшую площадь. Расход
энергии для АФМ2,5 приблизительно на 40 % меньше, чем для
мелких аэролифтных или механических флотационных машин.
Машину рекомендуется применять при простых схемах
обогащения и одновременно большом выходе пенного продукта.
В последние годы в СССР и за рубежом испытываются.
в качестве флотационных аппаратов пневматические
флотационные колонны (рис. 15.12, а). Высота их меняется от 2 до
10 м, а сечение может быть круглым, эллиптическим или
прямоугольным. Исходная пульпа по пульпопроводу 5 подается
в среднюю часть колонны 4, X воздух из ресивера / под
необходимым давлением A,4 - 105 Па) вводится в аэратор 6 из
пористого материала с отверстиями размером от 5 мкм до
2,5 мм.
Флотация в колонной машине осуществляется при противо-
точном движении воздушных пузырьков и потоков пульпы.
Пульпа движется вниз к разгрузочному патрубку 7 навстречу
всплывающим пузырькам. Воздушные пузырьки образуют на
поверхности колонны пену, которая орошается для удаления
частиц пустой породы водой из трубы 3. Пена отводится по
трубе 2. Скорость нисходящих потоков пульпы должна быть
меньше скорости всплывания воздушных пузырьков.
Превышение этой скорости приводит к локальному скоплению пузырь-
326 •
Рис. 15.12. Колонная флотационная машина (а) и машина для пенной
сепарации (б). --
ков, их коалесценции и периодическому выбросу воздушных
пробок.
Флотационная колонна имеет ряд преимуществ, по
сравнению с обычными флотационными машинами: она занимает 5—
10 % площади, необходимой для обычных машин; не имеет
движущихся частей, что снижает расход электроэнергии и
затраты на обслуживание; может быть легко автоматизирована
и управляться с центрального пульта. Основные трудности,
возникающие при работе колонных машин, связаны с забивкой
аэраторов.
В циклонных флотационных машинах, имеющих форму
циклона, создается вращательное движение пульпы,
подаваемой насосом; воздух поступает через пористые перегородки и
патрубки.
Во флотационной машине пенной сепарации (рис. 15.12, б)
пульпа, обработанная реагентами, подается сверху через
загрузочное устройство 1 и приемные желоба 3, обеспечивающие
равномерное распределение пульпы по всей длине
флотационной машины на ее правую и левую стороны. Пульпа в желобах
подвергается разжижению и аэрации воздухом, эжектируемым
при работе брызгал 2, и воздухом, подаваемым через резино-
327
вые пористые трубки, установленные в этих желобах. Затем
пульпа поступает па пенный слой, образуемый в результате
подачи сжатого воздуха (под давлением около 1,5 * 105 Па)
через трубчатые резиновые аэраторы 4 с пористыми стенками,
установленные на 150—200 мм ниже пенных порогов.
Гидрофобные минеральные частицы закрепляются на поверхности
воздушных пузырьков, а гидрофильные частицы под действием
силы тяжести попадают на дно камеры и разгружаются через
разгрузочное устройство 6. Сфлотированпые частицы
загружаются через пенные пороги в концентрационные желоба 5.
Принципиально новый способ подачи пульпы в машину,
обеспечивающий максимальную вероятность флотации при
минимальных значениях инерционных сил, позволяет значительно
увеличить скорость флотации и повысить крупность
флотируемых частиц в 3—4 раза по сравнению с обычными
флотационными машинами. Недостатком машин пенной сепарации
является необходимость классификации (в ряде случаев),
тщательного перемешивания с реагентами и аккуратной подачи
материала на пенный слой.
Выпускаемые в СССР флотационные машины ФПС16 и
ФП16 предназначены главным образом для флотации крупных
классов горно-химического сырья и легкофлотируемых
материалов, не требующих длительного времени флотации. В отличие
от ФПС16, в машине ФП16 возможна организация дофлотации
материала путем неоднократной подачи его на пенный слой
встроенным аэролифтом. Необходимое время флютации может
быть получено увеличением числа камер в машине.
К достоинствам машин пневматического типа относятся:
предельная простота конструкции; отсутствие вращающихся
частей, быстроизнашивающихся деталей и узлов; малая
металлоемкость; простота эксплуатации. Недостатками их являются:
необходимость применения воздухонагнетательных установок
для подачи воздуха и насосов для перекачки промиродуктов;
ограниченность применения (только для простых схем
флотационного обогащения).
15.8. Флотационные машины с изменяемым давлением
К этой группе машин относятся вакуумные и
компрессионные флотационные машины.
В вакуумных флотационных машинах пульпа,
предварительно обработанная реагентами и насыщенная воздухом,
поступает в вакуумную камеру, в которой (вследствие снижения
давления над пульпой) на поверхности гидрофобных частиц
выделяются пузырьки растворенного воздуха, приводя к их
флотации. Гидрофильные частицы осаждаются на дно камеры и
разгружаются в сборник хвостов.
Применение вакуумной флотации позволило бы повысить
эффективность разделения тонких шламов, однако конструк-
328
тивные трудности ограничивают их применение в настоящее
время несколькими угольными фабриками в Великобритании.
В компрессионных машинах флотация осуществляется в
результате выделения очень мелких пузырьков при снижении
давления над пульпой. Пульпа перед подачей в машину
подвергается насыщению сжатым воздухом при избыточном
давлении 3- 105—4- 105 Па, а затем подается в ванну машин, в
которой при падении давления над пульпой до атмосферного
происходит выделение растворенных газов в виде
микропузырьков непосредственно на поверхности частиц.
Компрессионные машины широко применяются для флотационной очистки
сточных вод промышленных предприятий.
15.9. Электрофлотационные машины
Для ионной флотации и очистки сточных вод в последние
годы начинают широко применять электрофлотационные
машины, аэрация пульпы в которых осуществляется в результате
электролиза водной части пульпы. При электролизе
выделяются очень мелкие пузырьки @,05—0,2 мм) водорода и
кислорода, обеспечивающие эффективную флотацию тонких взвесей
и хлопьев. Принципиальная схема одной из разновидностей
конструкции электрофлотационной машины представлена на
рис. 15.13. Применение электрофлотационных машин для
очистки сточных вод оказалось не менее эффективным, чем
компрессионных. Затраты электроэнергии в обоих случаях
составляют 0,3—0,4 кВт-ч/м3 очищаемой жидкости.
15.10. Основные факторы, влияющие на эффективность
работы флотационных машин при пенной флотации
Па эффективность работы флотационных машин при
пенной флотации оказывает влияние большое число обычно
взаимосвязанных параметров.
Аэрированность пульпы является одним из главных
факторов, определяющих эффективность работы флотационных
машин. Она зависит от расхода вводимого воздуха, степени его
диспергирования, распределения в объеме пульпы,
интенсивности перемешивания и плотности пульпы, типа и расхода
реагента-пенообразователя.
Эффективность работы флотационной камеры
непосредственно определяется ее аэрируемым объемом, который в
различных типах флотационных машин различен (рис. 15.14).
Содержание воздуха в аэрируемом объеме может изменяться от
0 до 30%.
В промышленных условиях аэрация характеризуется
объемом воздуха, проходящего в единицу времени через единицу
площади горизонтального сечения флотационной камеры или
через единицу объема пульпы. В различных типах флотацион-
329
,->-
\гЪ
° - ° °« °_ 0_~Z? .0 О О О п"
о—о-
о _„
Й2
О О—О — °— 0-~ о — О-О- О-
v:
jL
Рис. 15.13. Электрофлотационная машина
i — выпрямитель; 2 — труба для подачв исходного раствора; 3 — пеноеъемник-, 4 ■
желоб; 5— электроды; 6'— патрубок для отвода очищенного раствора
ных машин она изменяется обычно от 600 до 1300 л/(м2-мин).
С увеличением аэрации скорость флотации, как правило,
возрастает. Например, время флотации вкрапленных сульфидных
руд, необходимое для достижения одинакового извлечения,
обратно пропорционально корню квадратному из степени
аэрации. Расход вводимого или засасываемого в пульпу воздуха
определяется типом машины и режимом ее работы. Расход
воздуха в пневматических и пневмомеханических
флотационных машинах регулируется автоматически. Однако для
получения максимального технологического эффекта важен не общий
объем воздуха, а его дисперсность, определяющая суммарную
площадь поверхности воздушных пузырьков и скорость их
всплывания в пульпе.
Дисперсность воздушных пузырьков зависит от типа
аэраторов концентрации и типа пенообразователя. В механических
& Воздух 6 Воздух
Воздух
Рис. 15.14. Аэрируемый объем
- Воздух в различных флотационных
^ камерах:
а — пневматическая с. пористым
днищем; б —глубокая аэролифт-
ная; в — пневмомеханическая;
г — пневмомеханическая с
вращающимся ротором; д —
механическая с импеллером стержневого
типа; е — механическая с
импеллером радиального типа [24]
330
Рис. 15.15. Влияние плотности
пульпы . (содержание твердого Т) на
различные параметры и показатели
П флотации:
е — извлечение полезных компонентов
в концентрат; 0 — содержание полезных
компонентов в концентрате; <7П~
удельная производительность машины по
пульпе; 4 р— удельная производительность
машины по руде; еп- удельный расход
электроэнергии по пульпе; ер— удельный
расход электроэнергии по руде; а — аэ-
рированность пульпы; 7ф ~~ флотируе-
мость крупных зерен; *рф — Флотируе-
мость мелких зерен; t — время
пребывания пульпы во флотационной машине;
Ср— объемная концентрация реагента
при постоянном его расходе; / —
истираемость поверхности зерен
1 ^
Т
и пневмомеханических флотационных машинах она возрастает
с повышением интенсивности перемешивания пульпы
импеллером. Однако очень сильное перемешивание приводит не только
к увеличению дисперсности пузырьков и труднорастворимых
в воде реагентов, аэрированное™ пульпы и вероятности
столкновения частиц с пузырьками, но и к увеличению сил отрыва
от пузырьков, уменьшению вероятности флотации крупных
зерен, увеличению расхода электроэнергии. Па практике
известны -случаи, когда уменьшение скорости вращения
импеллеров флотационных машин (например, rta Кировской апатито-
нефелиновой обогатительной фабрике) улучшает
технологические и экономические показатели работы фабрик.
Равномерность распределения пузырьков в объеме пульпы,
определяющая коэффициент полезного использования
флотационной камеры, зависит от интенсивности перемешивания
пульпы и типа машины.
Плотность пульпы, характеризуемая на практике
содержанием твердого в единице объема пульпы или соотношением
массы твердой и жидкой фаз в пульпе (Т:Ж), оказывает
большое влияние на целый ряд параметров и показателей
флотации (рис. 15.15).
С увеличением плотности пульпы (при постоянной
производительности фабрики) увеличивается время пребывания пульпы
во флотационной машине и концентрация реагентов в пульпе
(при условии постоянства их расхода на единицу твердого
в пульпе).
Содержание полезных компонентов в пенном продукте
с увеличением плотности пульпы, наоборот, непрерывно
падает. Основными причинами этого являются: механический
вынос минералов пустой породы; уменьшение флотируемости
331
крупных зерен из-за возрастающего воздействия со стороны
окружающих частиц и увеличения вероятности их отрыва от
пузырьков; увеличение флотируемости мелких частиц не только
флотируемых минералов, но и пустой породы вследствие
увеличения вероятности их столкновения с пузырьками.
Содержание шламов при повышении плотности пульпы может
возрастать в результате увеличения истираемости, особенно хрупких,
минералов.
Максимальные значения удельной производительности и
минимальные значения удельного расхода электроэнергии по
твердому наблюдаются при некоторых оптимальных значениях
плотности пульпы.
Извлечение полезных компонентов в концентрат и аэриро-
ванность также имеют экстремальную зависимость от
содержания твердого в пульпе. Наиболее высокое извлечение и
максимальная аэрация, обусловленная лучшей диспергируемостью
и наиболее равномерными распределением воздушных
пузырьков, наблюдаются обычно при содержании твердого в пульпе
20—30%.
Выбор значений плотности пульпы на практике
определяется главным образом, крупностью и плотностью флотируемого
материала, назначением операции флотации и требуемым
качеством продуктов флотации. Содержание твердого в пульпе
возрастает до 35—40 % с увеличением крупности и плотности
флотируемого материала и уменьшается до 10—15% при
большом содержании шламов и малой плотности обрабатываемого
материала. Для снижения потерь в хвостах в операциях
основной и контрольной флотации используются более плотные
пульпы, содержащие 25—40 % твердого. В перечистных
операциях целью которых является повышение качества
концентрата, применяют более разбавленные пульпы, содержащие
обычно 10—25 % твердого.
При флотационном обогащении руд по сложным схемам
для поддержания оптимальной плотности пульпы в каждой
операции приходится иногда применять специальные меры по
разбавлению (например, концентратов перед перечистной
флотацией) или сгущению продуктов (например, промпродуктов
или коллективных концентратов) перед дальнейшей их
флотационной переработкой.
Крупность флотируемого материала определяет при прочих
равных условиях эффективность работы флотационных машин.
Ранее было показано, что условия эффективной флотации
тонких и крупных зерен различны и требуют для своей
реализации специальных конструкций машин. По этой причине
использование одного типа машин при переработке материала
широкого диапазона крупности приводит к тому, что основные
потери полезных минералов в хвостах обогатительных фабрик
происходят вследствие плохой флотации крупных или тонких
зерен или и тех и других. Поэтому представляется целесооб-
332
разным при обогащении
руд по сложным схемам,
предусматривающим циклы
флотации крупно- и тонко-
измельченных продуктов,
применять различные типы
флотационных машин. Это
позволит получить в
каждом цикле максимально
возможные
технологические показатели и
обеспечить высокую скорость и
низкую стоимость
флотации.
Стремление создать
наиболее благоприятные
условия флотации крупных и
тонких частиц выражается
во все большем
распространении на
обогатительных фабриках схем с
раздельной флотацией песко-
вой и шламовой части
флотируемого материала.
Скорость потока пульпы
значительно влияет на
эффективность работы
флотационных машин. Это
влияние обусловлено продольным перемешиванием в них пульпы.
В процессе такого перемешивания часть зерен флотируемого
материала выносится в хвосты и, следовательно, потери не
могут быть компенсированы более длительным пребыванием
в пульпе других частиц. Вследствие этого изменение
содержаний флотируемого минерала по фронту флотации зависит от
соотношения скоростей флотационного извлечения частиц и
продольного перемешивания пульпы.
В пневматической или аэролифтной машинах, в ванне
которых пульпа перемешивается относительно слабо, эти
скорости сопоставимы и содержание полезного компонента плавно
убывает от точки подачи питания к разгрузочному концу
машины (рис. 15.16, кривая 2). Однако время t2 флотации,
необходимое для получения хвостов с одинаковым содержанием рп'
в них извлекаемого компонента, в этих машинах больше
времени t\ в машинах периодического действия, кинетика
флотации в которых (см. рис. 15.16, кривая /) не осложняется
перемешиванием частиц с потоком пульпы.
В импеллерной машине перемешивание настолько сильное,
что содержание полезного компонента в пульпе каждой
камеры практически постоянно и меняется скачкообразно при
Рис. 15.16. Зависимость содержания ра
полезного компонента в пульпе от
времени / флотации в машине
периодического действия {1), пневматической или
аэролифтной B), механической
камерной с малой вместимостью C) и
механической камерной с большой
вместимостью D)
333
15.11. Технологические особенности
ленной сепарации
В отличие от обычной пенной флотации увеличение
продолжительности процесса пенной * сепарации сопровождается
непрерывным повышением качества пенного продукта и
уменьшением как выхода его, так и извлечения в него флотируемых
минералов (рис. 15.18). Причем характер наблюдаемых
зависимостей е, р, y=f(t) не зависит от крупности разделяемых
частиц. В связи с этим под временем пенной сепарации
подразумевается время, необходимое для достижения заданной
полноты разделения при допустимых в данной операции потерях
флотируемого материала.
Время ленной сепарации определяется, в основном,
скоростью прохождения гидрофильных частиц через слой пены и
составляет обычно несколько секунд. Высокая скорость
разделения обуславливает большую удельную производительность
машин пенной сепарации, которая в расчете на аэрированный
объем достигает (в машинах ФПС) 100—120 т/(м3-ч) и более,
что в 20 с лишним раз превышает удельную
производительность механических флотационных машин. Продолжительность
флотации в них зависит от разности в скорости аэрации пульпы
у места ее загрузки и сливного порога. Чем она больше, тем
выше скорость горизонтального перемещения и удаления пены,
больше извлечение, но хуже качество концентрата.
Для повышения качества концентрата можно применять
адгезионный съем пены, орошение ее водой или растворами
флотационных реагентов, регулирующих свойства пены. При
образовании устойчивых аэрофлокул, дальнейшее повышение
качества концентрата обеспечивается повторной сепарацией
его только после предварительного разрушения
образовавшихся флокул.
Экспериментальная кривая е = /(/) на рис. 15.18 хорошо
описывается экспоненциальным уравнением
г = ехр(—wt)y
где w — вероятность выпадения флотируемых частиц из пены,
с-1.
Значение w увеличивается с повышением крупности и
плотности частиц, но уменьшается с повышением гидрофобное™ их
поверхности. Однако данное уравнение не включает в себя
характеристики пены и частиц и не позволяет судить о влиянии
того или иного фактора па условия разделения минералов при
пенной сепарации.
Особенно важное значение при пенной сепарации имеет
характер поступления на пену жидкости или пульпы, так как
это связано с проникновением частиц на определенную
глубину пены [20].
336
Рис. 15.18. Влияние
продолжительности / пенной сепарации на выход
Y пенного продукта, содержание Р
в нем и извлечение е флотируемого
компонента
При малой толщине слоя пены или слишком больших
потоках пульпы, пробивающих пенный слой и проникающих
в толщу аэрированной жидкости, в пене задерживаются
только наиболее легкофлотируемые гидрофобные частицы. Чем
глубже под пену проникает поток исходной пульпы, тем меньше
вероятность флотации увлекаемых им частиц и тем ниже
конечное извлечение.
Чтобы погасить кинетическую энергию поступающей на
пену исходной пульпы и уменьшить проникновение ее в подпен-
ные слои, в зону поступления пульпы подается сильный
восходящий поток пузырьков и жидкости. В результате этого
исходная пульпа входит в пенный слой под небольшим углом и
несет на своей поверхности уже готовые флотационные
комплексы.
Эффективность процесса пенной сепарации возрастает
с увеличением плотности исходной пульпы как в результате
уменьшения динамичности потоков, так и в результате
увеличения несущей способности пенного слоя.
В связи с незначительным временем пенной сепарации,
исчисляемой несколькими секундами, исходный материал не
может быть обработан реагентами во время ее проведения.
Кондиционирование пульпы должно быть полностью закончено до
ее подачи во флотацию.
При обогащении руд, измельченных до обычной
флотационной крупности, перемешивание с флотационными реагентами
осуществляется обычно в контактных чанах при содержании
твердого в пульпе 40—50 %.
При пенной сепарации крупнозернистых материалов их
целесообразно предварительно обесшламливать и
кондиционировать в очень плотных пульпах, содержащих до 80 %
твердого, при относительно высоких концентрациях реагентов
(в том числе аполярных) в оставшейся жидкости. Для обес-
шламливания и сгущения применяются механические
классификаторы, грохоты, гидроциклоны; для перемешивания с
реагентами— двухвальные и барабанные смесители.
337
При пенной сепарации материалов широкого диапазона
крупности более высокие показатели обогащения достигаются
при раздельном предварительном перемешивании песковой и
шламовой фракций с реагентами [46].
По мнению В. А.. Малиноаского [20], перемешивание пульпы
с реагентами перед пенной сепарацией при любой крупности
материала целесообразно осуществлять методом
вибрационного контактирования, при котором в отличие от агитационного
не поток пульпы, а каждая быстро двигающаяся частица
находится в контакте с реагентами. При этом ускоряется
протекание диффузионных процессов, что сокращает необходимое
время контакта частиц с реагентами с нескольких минут до
нескольких секунд.
Применение пенной сепарации и вибрационного
контактирования открывает новые возможности и по управлению
реакциями взаимодействия минералов с флотационными
реагентами различного типа. Например, при строго определенном
соотношении концентраций реагентов в пульпе можно обеспечить
перед пенной сепарацией их контакт в течение любого времени,
начиная с секунды и кончая минутами, а затем сфлотировать
гидрофобные минералы за 3—10 с, исключив протекание
в пульпе нежелательных реакций [20].
15.12. Выбор флотационных машин
При выборе машин для оснащения обогатительных фабрик
исходят главным образом из свойств руды, возможностей
получения максимальных технологических показателей,
минимальных энергетических затрат, простоты регулирования и
эксплуатации.
За рубежом при обогащении руд по простым схемам,
применяют механические машины «Фагергрен»,
пневмомеханические «Аджитейр» или аэролифтные, удельные затраты
электроэнергии в которых обычно ниже, чем в механических с
центробежными импеллерами. Они проще в регулировке и надежнее
в эксплуатации [24, 41, 44]. При обогащении
полиметаллических руд по сложным схемам переработки обычно
используются механические машины, которые обеспечивают
транспортирование промпродуктов без установки дополнительных
насосов. В некоторых случаях на фабриках при обогащении таких
руд устанавливают несколько типов машин. Например, на ряде
полиметаллических фабрик США применяют машины
«Денвер», «Фагергрен» и «Аджитейр» [24, 41, 44].
В СССР в настоящее время могут быть рекомендованы
к широкому промышленному использованию следующие
машины:
механические ФМ — в сложных схемах флотации,
требующих установки большого числа всасывающих камер и
тщательного покамерного регулирования выхода пенного продукта.
338
Они обычно используются также при флотации
крупнозернистые материалов;
пневмомеханические ФПМ — в простых схемах флотации
при крупности перерабатываемого материала не менее 40 %
класса — 0,074 мм с максимальной крупностью зерен до 1 мм;
аэролифтные АФМ и механические МфУ — в простых
схемах флотации, не требующих высокой селективности, с
большим выходом пенного продукта.
Современной тенденцией является разработка новых
конструкций флотационных машин с камерами большой
вместимости. Такие машины получили широкое распространение на
медных, медно-молибденовых, медно-иикелевых и
молибденовых фабриках США, Канады, Чили, Австралии,
оборудованных крупными мельницами (вместимостью до 150 м3 и более).
Технологические показатели, получаемые во флотационных
машинах с камерами большой вместимости, не уступают
показателям стандартных машин при более низких удельных
капитальных затратах и эксплуатационных расходах и более
стабильном технологическом процессе. Высокая
производительность новых флотационных машин с камерами большой
вместимости позволяет укрупнить секции, сократить
коммуникации и вспомогательное оборудование, уменьшить число точек и
приборов автоматического контроля и управления
технологическим процессом, повысить производительность труда. Следует
отметить, что в большинстве новых машин регулирование
уровня пульпы и пены автоматизировано.
Большой интерес представляет применение колонных
флотационных машин и машин «Максвелл» вместимостью до 65 м3.
Например, замена обычных механических флотационных
машин на канадской фабрике «Опемиска» колонными с
вместимостью колонны около 60 м3 снизила расход электроэнергии
и затраты на ремонт и обслуживание более чем в 2 раза.
Установка флотационных машин «Максвелл», как правило, в
начале процесса позволяет: выделить богату-ю «головку»
концентрата и за счет этого предотвратить ошламование раскрытых
минералов при стадиальном измельчении; интенсифицировать
процесс в последующих операциях флотации за счет
предварительной аэрации, длительного контактирования пульпы с
реагентами и стабилизации питания по вещественному составу;
наиболее эффективно увеличить фронт*флотации без
расширения производственных, площадей.
Для повышения эффективности обогащения за счет
флотации крупнозернистых частиц целесообразно применять
флотационные машины с кипящим слоем и машины пенной
сепарации. Пенную сепарацию в первую очередь следует применять
для извлечения из руды и хвостов частиц крупнее 0,074 мм,
например в межцикловой флотации на крупнозернистых пульпах
и для обогащения песковой фракции хвостов.
339
Питание
t
Основная ср/ютация
л
1
I перечистная Контрольная
i » *
Е перечистная
1
Хвосты
Концентрат
д
Еоере- с
чист-
к на я
Питание
1
ое-
речист-]
к мая
ГТ
Основная (
срлотация
Контрольна я
Хвосты
Концентрат
в Питание
L
\Inepe-
част-
\ ная
I ое- о-
речист-
\ мая
Основная
тлотация
Контрольная
Хвосты
л
Концентрат
Рис. 16.1. Распределение операций флотационной схемы (а) при
использовании механических, пневмомеханических (б) и пневматических (в)
флотационных машин
деление легко осуществить, если используются механические
флотационные машины, аэраторы которых легко справляются
с подсосом промежуточных продуктов флотации. Сложную
схему труднее осуществить, если используются
пневмомеханические машины. В этом случае, чтобы избежать установки
насосов, для подсоса промпродуктов в первых камерах машины
можно устанавливать аэраторы механических флотационных
машин. Подача промпродуктов .также может производиться
специальными пульпоподъемниками, установленными в
отдельных камерах или в конце желоба. При использовании
флотационных машин пневматического типа осуществление, такой
схемы оказывается возможным только с помощью насосов,
342
как это изображено на рис. 16.1, в. Данное обстоятельство
учитывается при выборе флотационных машин для оснащения
процесса флотации.
16.2. Устройства для подготовки пульпы к флотации
При необходимости длительного перемешивания пульпы
с реагентами, ее аэрации или кондиционирования перед
флотацией устанавливаются контактные чаны, аэрирующие
устройства, аппараты или агрегаты для подготовки пульпы.
Контактные чаны (рис. 16.2) предназначены для
перемешивания пульпы с реагентами. Они представляют собой емкость
. 4 цилиндрической формы, в центре которой вращается вал 2
с мешалкой 7 осевого типа. Вал находится в трубе 3 с
отверстиями 6 для циркуляции пульпы, что обеспечивает
равномерное распределение реагентов в объеме пульпы. Исходная пульпа
по трубе / поступает в верхнюю часть центральной трубы и
затем на мешалку, а подготовленная пульпа удаляется через
сливную трубу 5.
Степень однородности и подготовленности пульпы к
флотации определяются временем и интенсивностью перемешивания
пульпы в контактном чане. Необходимое время перемешивания
пульпы в контактном чане определяет его вместимость V, (м3),
рассчитываемая по формуле
V^Q(l/bT+R)t№.
где Q — производительность, т/ч; 6Т — плотность твердого, т/м3;
■R — плотность пульпы (Ж: Т); t — необходимое время контакта,
мин, определяемое необходимой длительностью процесса
взаимодействия реагентов с поверхностью минералов. Для уголь-
ных пульп оно составляет 1—2 мин при содержании твердого
в пульпе 35—40 % и 3—4 мин при 20 % твердого [43]. Для
рудных пульп оно может изменяться от 1 до 60 мин [2,42].
Вместимость чанов при этом
изменяется от 0,3 до 47 м3 [43].
Интенсивность перемешивания
возрастает при использовании
плохораствбримых в воде
реагентов, при подготовке
грубозернистых пульп,- при
необходимости обдирки (оттирки)
поверхностей минералов от
покрывающих их пленок
загрязняющих примесей или
реагентов.
Аэрация пульпы в
процессе подготовки к флотации,
например сульфидных руд
цветных металлов, осуществляется
Пищание
Рис. 16.2. Контактный чан
343
Рис. 16.3. Аппарат для подготовки пульпы «Каскад».
обычно или в первых камерах механических флотационных
машин, или в специальных аэрирующих устройствах,
представляющих собой монокамеры механических, пневмомеханических
или пневматических машин разнообразной конструкции.
Аппарат для подготовки пульпы «Каскад» (рис. 16.3)
широко, применяется для кондиционирования флотационных
угольных пульп, в процессе которого смешиваются все потоки
шлама, поступающего на флотацию, производится обработка
пульпы реагентами, подаваемыми в виде аэрозоля, интенсивно
перемешиваются взаимодействующие фазы и пульпа
равномерно распределяется по флотационным машинам.
Все поступающие потоки пульпы направляются в смеситель
■5, в котором благодаря тангенциальному подсоединению
патрубков 9 образуется вихрь, обеспечивающий необходимое
смешение потоков. Из смесителя пульпа проходит через отверстия
конуса 10 и веерообразным потоком поступает на верхнюю
344
штампованную решетку //, растекается по ее поверхности и
через щели направляется, в нижнюю часть колонны 12.
Реагенты из дозатора 4 поступают в устройство для
приготовления аэрозоля, представляющее собой быстроходный
вентилятор-ротор 5, приводимый во вращение электродвигателем
7 через клиноременную передачу 6. Образующаяся под
механическим воздействием ротора и потоков воздуха аэрозоль
реагентов, пройдя фильтр 3, по трубе / подается в колонну 12
под решетку 11 навстречу потоку струй пульпы, что позволяет
достаточно равномерно распределять реагенты по всему объему
пульпы. Для чистки решетки предусмотрено отверстие 13.
Протекая в нижнюю часть колонны, пульпа с реагентами
проходит через жалюзийную решетку 14, где происходит
дополнительное ее перемешивание, и поступает в смесительную
воронку 17.
Нижняя часть колонны представляет собой пульподелитель,
на боковых стенках которого имеются отверстия 19. К стенкам
прикреплены сливные коробки 15 с поворотными заслонами 16
и ручками 2. К сливным коробкам крепятся запорные
клапаны 18 для регулирования скорости истечения подготовленной
пульпы из колонны через сливные коробки, максимальное
число которых может достигать восьми при максимальной
производительности аппарата «Каскад» 8000 м3/ч.
В основе нового аппарата для кондиционирования пульпы
АКП-1600 производительностью 1600 м3/ч использован в
несколько измененном конструктивном оформлении тот же
принцип интенсификации подготовки пульпы к флотации, что и
в аппарате «Каскад».
16.3. Оборудование и эксплуатация
реагентных площадок
Реагентные площадки располагаются выше уровня
флотационных машин, чтобы обеспечить самотек растворов
флотационных реагентов от реагентных питателей до точек подачи
их в процесс. Растворы реагентов в питатели поступают из
расходных бачков, обычно устанавливаемых за фронтом
реагентных питателей и соединенных реагентопроводами с pea-
гентным отделением.
Сеть трубопроводов для реагентов должна обеспечивать
максимальную гибкость питания реагентами во всех возможных
точках потребления. Реагенты подаются в центральную трубу
контактных чанов или в межкамерные перегородки
флотационных машин.
Реагентные питатели должны обладать высокой точностью
дозирования- реагента в процесс, легкостью и достаточно
широким диапазоном регулирования расхода реагента, простотой
конструкции и обслуживания, износостойкостью и
коррозионной стойкостью, быть дешевым в изготовлении.
345
К настоящему времени разработано и используется на
фабриках для дозирования растворов реагентов большое число
автоматизированных питателей, схема действия которых
приведена на рис. 16.4..
В автоматизировавшем скиповом питателе АКПР-1 угол
наклона стаканчика скипа ограничивается штоком,
упирающимся в лекало. Положение лекала, встроенного в
исполнительный механизм, определяет производительность питателя.
В питателе АКПР-2 подача регулируется переменой угла
наклона стаканчика путем измерения длины цепи Галля,
выполняющей функцию тяги кривошипно-шатунного механизма. Один
конец гибкой тяги закреплен в рамке скипа, другой перекинут
через звездочку, установленную на кривошипе, и закреплен
на лекале исполнительного механизма. Подача питателя
ППЭМ-1176 зависит от числа ходов скипового подъемника
в единицу времени.
В автоматизированных ковшовых питателях 5-АДР и 6-АДР
подача регулируется изменением угла наклона ковша. На один
привод 6-АДР могут быть установлены от 1 до 12
регулируемых блоков, каждый из которых предназначен для
обслуживания одной точки.
'Мембранный питатель МПР-642 имеет две мембранные
камеры, которые попеременно заполняются и опорожняются
при помощи распределительного устройства. Перемещение гиб-
- ких стенок камер определяет степень их заполнения в
соответствии с требуемой подачей.
В клапанном и игольчатом питателе АПИ-1 подача
реагента регулируется изменением площади живого сечения
дросселя при перемещении иглы по лекалу исполнительного
механизма. В питателе АДР-1 вся система регулирования вместе
с исполнительным механизмом установлена на поплавке в
ванне с реагентом, чтобы устранить влияние колебаний уровня
раствора в питающем баке на подачу питателя. В импульсном
питателе ПРИ-1, представляющем собой односедельный клапан,
объем дозируемого реагента является функцией времени
открытия клапана, управляемого электромагнитной системой.
В порционных питателях ПР-2 и АДБ-1 двухседельный
клапан сочленен с мерной емкостью, которая заполняется при
подаче импульса и опорожняется после его снятия. Заполнение
мерной емкости в питателе ПР-2 ограничивается воздушной
трубкой; в питателе АДБ-1 фиксация уровня раствора реагента
в мерной емкости осуществляется электродом. Частота
импульсов определяет подачу питателей.
Диафрагмовый питатель ДПР-1 является вертикально
расположенным диафрагмовым насосом с регулируемым ходом,
тогда как сильфонный дозатор малых расходов представляет
собой небольшой сильфонный насос с регулируемым ходом.
Наибольшее распространение из них получили питатель
ПР-2 для дозирования растворов реагентов, не содержащих
346
твердых частиц, питатель 5-АДР для дозирования чистых и
загрязненных осадками растворов, блочный питатель 6-АДР.
для подачи небольших объемов невязких реагентов
(детергент, терпинеол, ксиленол и др.).
Для дозирования жидких вязких реагентов применяются
шкивные питатели. Расход реагента, снимаемого скребком с
поверхности шкива диаметром 0;4 — 0,5 м, вращающегося в ванне
с вязким реагентом, регулируется изменением ширины или
числа работающих скребков. Шкивные питатели выпускаются
с одним, тремя и шестью шкивами при вместимости одной
ванны 10 л.
Для сокращения расхода труднорастворимых аполярных
реагентов и повышения эффективности их действия при
флотации используют эмульгирование таких реагентов в воде в
эмульгаторах различной конструкции. Наиболее широкое
распространение получили ультразвуковые и струйные эмульгаторы.
В применяемом на угольных фабриках струйном
двухступенчатом эмульгаторе СЭД:2 эмульгирование реагента
осуществляется струей воды диаметром 1,5 мм, вытекающей с большой
скоростью. Затем эмульсия разбавляется и дозируется в
процесс входящим в комплект поплавковым клапаном [43].
При необходимости дробной подачи реагентов-собирателей
и реагентов-пенообразователей и распределения их по
технологическим точкам подачи используются распределители
реагентов.
Широко применяемый в угольной промышленности
распределитель реагентов РР-3 (рис. 16.5). состоит из верхнего и
нижнего делителей и
закрыт сверху кожухом 4.
В состав верхнего делителя
входят делитель 13,
подвижный лоток 14 и
делительная воронка 6\ в состав
нижнего делителя —
делительная воронка 7 и
корпус 8. Обе делительные
воронки имеют по 6 сливных
щелей, расположенных на
одинаковом расстоянии
друг от друга. Воронки
подвешены на валу 5,
вращение которого
осуществляется через коническую
передачу 2
электродвигателем 3.
Реагент по трубе /
поступает в делительную во-
Рис. 16.5. Распределитель реагентов ронку ft которая, вращаясь
рр„3 равномерно распределяет
Pea
гент
348
его через сливные щели по периферии. Затем он перетекает в
делитель 13 цилиндрической формы, разделенный перегородками
пополам. Одна половина делителя соединена патрубком 12 с
воронкой //, откуда реагент поступает в аппарат подготовки
пульпы АПП, а из второй половины реагент по патрубку 10
сливается в делительную воронку 7 нижнего делителя. Объем
реагента, поступающего в ту или иную половину делителя,
регулируется изменением положения подвижного лотка 14.
Поступающая в делительную воронку 7 нижнего делителя часть
реагента выливается через ее щели в корпус, разделенный
перегородками на сектора, число которых определяется числом
флотационных машин, питаемых через распределитель. При
вращении воронки происходит равномерная подача реагентов во
все сектора, а из них — в машины, число которых может
изменяться от 2 до 8. При остановке одной из машин подача
реагента в нее перекрывается краном 9.
Для дозирования реагентов в твердом виде используются
питатели по конструкции аналогичные питателям руды:
ленточные, тарельчатые, вибрационные и другие.
16.4. Кондиционирование ионного состава
промышленных и оборотных вод
Состав поступающей на фабрику промышленной или
оборотной воды должен удовлетворять требованиям
технологического процесса. В связи с уменьшением запасов чистой воды
и требованиями по охране окружающей среды доля оборотных
и промышленных вод, характеризующихся переменным
составом в них вредных примесей, возрастает. Сохранить высокие
показатели флотационного обогащения в этом случае можно
только путем предварительного кондиционирования состава
используемых вод.
Кондиционирование является избирательной очисткой
оборотных и промышленных вод, в процессе которой удаляются
с последующей их утилизацией только те компоненты, которые
оказывают вредное влияние на процесс селективной флотации
данной фабрики, и сохраняются необходимые для протекания
флотационного процесса.
Простейшим случаем кондиционирования промышленных
вод является регулирование их щелочности и поддержание
значения рН, близкого к необходимому при флотации. Такая
задача может возникнуть при использовании на фабрике смеси
кислых рудничных и сильнощелочных фабричных вод.
При использовании большого количества рудничных вод
для флотации сульфидных руд может оказаться необходимой
очистка их от ионов меди или других металлов методами
цементации или ионной флотации. При флотации руд редких
металлов часто требуется очистка oil солей щелочноземельных
металлов, которая осуществляется ионообменной очисткой на
природных или синтетических ионообменных материалах.
349
Наиболее сложные задачи возникают при
кондиционировании оборотных вод, поступающих на селективную флотацию
полиметаллических руд. В- этом случае оно должно
осуществляться в соответствии с количественными зависимостями
между необходимыми концентрациями реагентов в оптимальных
условиях процесса, куда подается оборотная вода.
Кондиционирование необходимо вести с применением систем
автоматизации, аналогичных изображенным на рис. 14.12.—14.14. При
таком кондиционировании вод мо^но значительно снизить
расход реагентов без ухудшения технологических показателей
обогащения при полном водообороте на фабрике.
16.5. Разрушение флотационной пены
Необходимость осуществления специальных мероприятий
по разрушению флотационной пены может быть обусловлена
необходимостью, например, повысить эффективность
подготовки коллективного концентрата.к разделению, улучшить
условия транспортирования пенного продукта, снизить потери
ценных компонентов в операциях обезвоживания. Для
разрушения пены используются:
механические методы, основанные на применении
импеллеров или мешалок, изготовленных в виде специальных блоков
(центробежных пеноразрушителей) и расположенных над
уровнем пульпы в зумпфе или чане. Пена должна поступать
прямо па импеллер. Центробежные пеноразрушители хорошо
зарекомендовали себя на апатито-нефелиновых фабриках:
физико-химические способы, основанные на применении
растворов различных реагентов
(кислот, солей, неорганических
коллоидов, аполярных масел),
вызывающих разрушение пены;
вакуумные и эжекторные
устройства;
звуковые, ультразвуковые,
электрические и термические
способы, для осуществления
которых применяются звуковые
генераторы, сирены, искровой
разряд или перемешивание
пенного продукта в чане с
добавкой острого пара.
Наиболее простым и
достаточно надежным в большинстве
случаев является механический
способ разрушения пены.
Однако практика применения
механических ' пеногасителей при
флотации углей показала их
непригодность для разрушения
На бакуум-дзимьтры
Выпуск
Рис. 16.6. Схема вакуумно-меха-
нического способа пеногашения
350
вязких пен [43]. Поэтому значительное распространение на
углеобогатительных фабриках получил вакуумно-механический
способ пеногашения при поддержании вакуума в пределах
150—300 Па [43].
По данному способу (рис. 16.6) флотационный концентрат
поступает в закрытый резервуар /, в который глубоко в слой
пены опущена всасывающая труба, соединяющая резервуар
с ресивером 2. Под действием вакуума, создаваемого вакуум-
насосами 3, пена из резервуара поступает в ресивер, где,
ударяясь об отбойную стенку, гасится как вследствие удара, так и
под действием разрежения. Погашенный продукт направляется
в сборник 6 концентрата, оборудованный гидрозатвором 7,
и далее с помощью центробежного насоса 5 на
вакуум-фильтры 4.
Недостатком вакуумно-механического способа пеногашения
является сложность и громоздкость системы, высокая
энергоемкость, значительные капитальные и эксплуатационные
затраты.
16.6. Контроль и регулирование флотационного процесса
Контроль и управление технологическим процессом
флотационного обогащения являются основными задачами
автоматизированной системы управления технологическими
процессами АСУ ТП обогатительной фабрики. Ее подсистемами
являются [16,41—43]:
централизованный непрерывный контроль значений
технологических параметров и работы основного оборудования.
Системы автоматического контроля процессов флотации
применяются для измерения расходов пульпы и реагентов, уровня
пульны во флотационных машинах или чанах, уровня и цвета
пены в машинах, температуры и плотности пульпы, расхода
воздуха, концентрации ионов в жидкой фазе пульпы и
реагентов в их растворах, содержания металлов в твердой фазе
пульпы и некоторых других параметров. Получаемая
информация обеспечивает представление о ходе процесса и
используется для ручного или автоматического управления им;
низовые локальные системы автоматического регулирования,
осуществляющие стабилизацию или программное
регулирование технологических параметров процесса;
оперативный учет и математическая обработка измеряемых
величин с учетом имеющихся функциональных зависимостей;
необходимая результирующая информация, например по
балансу металлов, может выдаваться автоматически;
управление технологическими агрегатами и механизмами,
в том числе управление поточно-транспортными системами и
централизованным пуском и остановкой технологической цепи
.аппаратов и маогин. Их пуск осуществляется, начиная с конца,
а остановка — с начала схемы цепи аппаратов;
351
оптимальное управление, осуществляелюе путем расчета и
реализации регулирующих воздействий на задатчики
локальных систем по заданным алгоритмам управления в целях
оптимизации флотационного процесса.
Контроль и управление технологическим процессом
осуществляются с операторского или диспетчерского пунктов цеха
или фабрики.
16.7. Охрана труда и техника безопасности
на флотационных фабриках
Кроме общих правил техники безопасности, на
флотационных фабриках должны соблюдаться специальные правила,
связанные с применением флотационных реагентов.
Хранение реагентов производится в специализированном
помещении — хранилище. Жидкие, сухие реагенты и известь
хранятся раздельно; причем горючие реагенты — в помещении
для огнеопасных материалов, а. ядовитые — в изолированном
помещении, удовлетворяющем требованиям "санитарной
инспекции.
Дробление -реагентов перед их растворением допускается
только по особому разрешению санитарной инспекции и охраны
труда и производится в специальном изолированном
помещении. Система вентиляции должна обеспечивать санитарные
нормы по концентрации пыли в помещении.
Подача флотационных реагентов в сухом виде (особенно
цианида и сернистого натрия) в процессе флотации
запрещается, так как это может привести к запылению воздуха
флотационным реагентом и появлению опасности отравления
рабочих.
Растворять реагенты можно только в специальном
помещении реагентного отделения. Подача реагентов в растворные
чаны производится с рабочих площадок, расположенных на
уровне верхней части чана, через спускные трубы.
Растворять ядовитые реагенты и хранить готовые растворы
необходимо в изолированном помещении, удовлетворяющем
требованиям специальных инструкций. Растворяются они в
герметично закрытой аппаратуре с местной вытяжной
вентиляцией и с исключением ручных операций. Баки с ядовитыми
реагентами должны быть закрыты крышкой на замке. Питатели
ядовитых реагентов должны быть автоматическими, закрыты
кожухом с замком, а разводящие их трубопроводы — глухими
по всей длине и окрашенными в яркий цвет. Место ввода
ядовитого реагента в пульпу должно обеспечивать быстрое и
равномерное его перемешивание со всем объемом пульпы. Место
ввода необходимо ограждать таким образом, чтобы
исключалась возможность контакта рабочих с концентрированными
растворами реагентов до их полного перемешивания со всем
объемом пульпы.
352
При опорожнении и очистке растворных чанов и расходных
баков из-под дитиофосфатов, растворов цианидов, сернистого
натрия, ксантогенатов рабочие должны надевать противогазы
и после работы с перчаток, обуви и спецодежды удалять
остатки реагентов. Для хранений, обезвреживания и стирки
спецодежды рабочих, имеющих дело с ядовитыми реагентами,
должны быть изолированные помещения. Тара из-под ядовитых
реагентов хранится отдельно от прочей. Перед отправкой она
обезвреживается в соответствии с инструкцией.
Оборудование и организация работ по хранению,
приготовлению и использованию реагентов должны исключать
возможность соприкосновения раствора таких реагентов, при
смешении которых выделяются в рабочую зону ядовитые газы
(например, смешение цианистых растворов с кислыми). На боках
и таре с ядовитыми реагентами делаются установленные
надписи и рисунки.
На- рабочих местах должны быть инструкции по технике
безопасности и аптечки с противоядиями.
17. ОСНОВНЫЕ ТЕХНОЛОГИЧЕСКИЕ
И ТЕХНИКО-ЭКОНОМИЧЕСКИЕ ПОКАЗАТЕЛИ ^
РАБОТЫ ФЛОТАЦИОННЫХ ФАБРИК
Технологические и технико-экономические показатели
флотационного передела определяются большим числом
технологических параметров. Они возрастают с увеличением степени
учета особенностей вещественного состава руд и углей,
повышением эффективности работы флотационных машин,
улучшением организации контроля и регулирования технологического
процесса на обогатительной фабрике повышением
комплексности использования сырья.
Уровень техники флотационного обогащения руд
оценивается качеством концентратов и степенью извлечения металлов
в одноименные концентраты. Данные по извлечению металлов
на некоторых фабриках медной и никелевой подотраслей СССР
приведены на рис. 17.1. Технологические показатели
обогащения, получаемые на более труднообогатимых рудах в СССР, не
уступают показателям обогащения, получаемым на
зарубежных фабриках. Комплексность использования сырья на примере
свинцово-цинковых руд, иллюстрирует рис. 17.2.
Экспуатациошше расходы по операциям флотации
включают стоимость флотационных реагентов и расходы, связанные
с работой флотационных машин, насосов, приготовлением и
подачей реагентов.
Расход флотационных реагентов на 1 т перерабатываемой
руды, определяющий затраты на их применение, зависит от
состава руд, сложности флотационной схемы и изменяется в
довольно широких пределах (табл. 17.1).
12 Заказ № 1957
35а
Фабрит-^ zj^z0^z^s^
ганская --Щ^гР^
Балхаш- к skQS .1, „^^ - #.#^=
Алмалык- \ $$%
екая * «'
Рис. 17.1. Рост извлечения металлов на передовых обогатительных фабриках
медной и никелевой подотраслей
Лиритный
концентрат
\Cepa
Свинцово-
ццнковые
РуЗы
Медный I
концентрат
медь
телл^
селен
золото
ceaeoW
Химический |
\состав руды \
РЬ
Zn
Си
Fe
Cd
Bl
AS
s
MO
Ba
5b 1
Sn
se
CO
те
Tn
П 1
Au
AQ
Баритовый
концентрат
Сера
Оловянный
концентрат]
Олово
\Висмут
Теллур
Вольфрам
Золото- и
cepefrpo-
содержащие
продукты
Рис. 17.2. Диаграмма комплексности использования свинцово-цинковых руд
Остальные эксплуатационные расходы определяются
главным образом значениями удельной вместимости флотационных
машин, необходимой для переработки 1 т руды, и
производительности Q фабрики. При удельной вместимости флотационных
машин V=0,05 м3/(т-сут) и производительности фабрики по
руде более 4 млн.т/год расходы на операции флотации (без
354
Таблица 17.1
Расход реагентов на 1 т перерабатываемой руды на фабриках США
Руда
Медная
Медно-порфировая
Медно-свинцово-цинковая
Медно-цинково-пиритная
Свинцово-цинковая
Свинцово-цинково-серебря-
ная
Цинковая
Пл авиковошп атовая
Золото-серебряная
Модификаторы
4,031
1,590
0,499
4,549
0,442
1,040
1,684
7,417
1,246
Активаторы
0,039
0,055
0,357
0,207
0,469
0,247
0,473
Депрессоры
0,003
0,027
0,313
0,095
0,134
0,252
0,035
1,347
Собиратели
0,043
0,032
0,058
0,155
0,075
0,137
0,033
0,501
0,132
Вспенива-
тели
0,024
0,041
0,0.19
0,048
0,044
0,089
0,029
0,161
0,019
Флокулянты
0,003
0,014
0,006
0,002
0,003
0,007
0,001
0,015
0,014
Средний
4,084
1,693
4,622
1,232
0,843
1,935
0,420
9,749
1,112
стоимости реагентов) составляют 0,051 руб/т исходной руды.
При постоянной производительности Q = const с увеличением
V расходы по цеху флотации возрастают пропорционально
отношению V: 0,05. При изменении Q должен вводиться
поправочный коэффициент Кх в соответствии со следующей
зависимостью между Q и К\:
0, млн. т/год ^4 3 2 1 0,5 0,2
Кх 1 1,03 1.13 1,33 1,5 1,8
Если обозначить через а (руб/т) стоимость флотационных
реагентов, то полные эксплуатационные расходы С (руб/т) по
операциям флотации можно подсчитать по формуле
С = а+ 0,051 (W0,05)/Ci.
Расход электроэнергии по цеху флотации при Q^4 млн.
т/год и У=0,05 м3/(т-сут) составляет 2,6 кВт-ч/т исходной
руды. Для других условий расход электроэнерии
Расход технологической воды зависит от разбавления в
отдельных операциях флотации и числа операций и изменяется
от 2 до 6 м3/т. При полном использовании оборотной воды
расход свежей воды для технологического процесса. может быть
уменьшен до 1—1,5 м3/т. Полный расход воды складывается
из расхода ее в самом технологическом процессе и расхода на
смыв полов, промывку оборудования при внезапной остановке
или перед ремонтом, расхода в бытовых и административно-
хозяйственных помещениях и т. д. Полный расход воды на
флотационных фабриках составляет обычно 4 — 8 м3/т.
12*
355
Таблица 17.2'
Расход электроэнергии, воды, измельчающих материалов на 1 т руды
Руда
Медная
Медно-порфир овая
Медно-свинцово-цинковая
Медно-цинково-пиритная
Свинцово-цинковая
Свинцово-цинково-серебряная
Цинковая
Пл авиковошпатовая
Золото-серебряная
Электроэнергия,
кВт-ч
17,63
16,09
18,63
23,92
21,49
29,39
15,76
70,66
17,10
Вода,
л
3129
2983
2128
3359
3024
3379
2086
6569
2796
Стержни,
кг
0,17
0,307
0,099
0,139
0,139
0,432
0,119
—
0,1706
Шары,
кг
0,496
0,652
0,143
0,310
0,124
0,772
0,1
0,405
0,157
Футеровка, кг
0,0559
0,075
0,048
—
0,025
0,114
0,021
0,143
0,011
Расход электроэнергии, воды и стали для различных типов
руд, перерабатываемых, например, на флотационных фабри*
ках США, приведен в табл. 17.2.
Капитальные затраты по цеху флотации при Q>4 млн.
т/год и значение У=0,05 м3/(т«сут) составляют 0,165 руб. на
1 т годовой производительности по руде. Для других условий
капитальные затраты
Р = 1,1 • 0,165 (V/0,05) KiK2 = 3,63V/CX/C2,
где 1,1—поправка на неучтенные расходы, a /G — поправка,
зависящая от места строительства и равная 1,15 —1,25 для Мур-
Таблица 17.3
Эксплуатационные расходы, капитальные затраты и расход
энергии для флотационных фабрик
Операция
Крупное, среднее, мелкое дробление,
грохочение, транспортирование,
складирование крупной руды
Бункерование руды-в цехе измельчения
я измельчение до 70—75 % — 0,074 мм
Флотация (без стоимости реагентов)
Сгущение*
Фильтрование*
Сушка без стоимости топлива*
Складирование и отгрузка
концентратов*
Удаление и укладка хвостов*
[луатацион-
р а сходы,
г руды
О 4>Ю
а 2 >»
Ода
0,09—0,11
0,51—0,77
0,051
0,09—0,22
0,13
0,13
0,05—0,1
0,1—0,2
н « a
е - л
- Я д Ь ^i
с &&5Л
t^nSfflC
0,4-0,6
1,0-1,17
0,165
0,6—1,10
0,15—0,30
0,54
0,2—0,4
0,5-1,2
:од электро-
гии,
ч/т руды
3£н
f3 я«
Они м
1,0—1,2
12—20
2,6
1—2
2—3
3,0
—
* На 1 т продукта, поступающего в операцию обработки.
356
Таблица 17.4
Основные технико-экономические показатели флотационных фабрик
большой производительности
Показатель
Годовая производительность по руде,
млн. т
Удельный расход на 1 т руды:
электроэнергия, кВт-ч
вода производственная, м3
стальфутеровочная для дробилок и
мельниц, кг
стержни и шары, кг
условного топлива на 1 т
концентрата, кг
Удельные показатели на 1000 т
годовой производительности фабрики по
Руде: 5
объем производственных зданий
(без транспортных галл ерей), м3
стоимость 1 м3 здания (средняя),
руб.
масса технологического
оборудования, т
установочная мощность
электродвигателей, кВт
Коэффициент использования
оборудования по главному корпусу —
рабочее время 0/
календарное время
Производительность на 1 трудящегося
по руде, тыс. т/год
Капитальные затраты на 1 т годовой
производительности по руде, руб.
Стоимость обогащения 1 т руды, руб.
В том числе стоимость реагентов на
1 т руды, руб.
медная
5—20
20—30
2,5—5,0
0,2—0,3
1,2—1,6
20—28
100—200
12—21
1,7—2
5—8
90—93
6—14
3,2—5
1,2-1,6
0,2—0,5
Руда
полиметаллическая
2—4
23—40
4—6
0,2—0,4
0,7—1,6
17—22
120—360
15—23
2,3—2,6
7—11
90—93
4—6
5,2-8,2
1,7—3,1
0,5—0,9
апатитовая
22,7
21,0
4,3
0,13
0,9
17,5
117
! 19
1 U
' 5,6
93,5
12,5
5,6
1,5
0,07
манской области и Приморского края; 1,02—1,08 для
Кавказа, Средней Азии и Казахстана; 1—для остальных районов
СССР.
Распределение эксплуатационных расходов, капитальных
затрат и расхода электроэнергии, по данным института Меха-
нобр, для отдельных переделов флотационных фабрик
приведены в табл. 17.3.
Основные технико-экономические показатели для
флотационных фабрик даны в табл. 17.4.
18. ПЕРСПЕКТИВЫ И НАПРАВЛЕНИЯ ДАЛЬНЕЙШЕГО РАЗВИТИЯ
ФЛОТАЦИОННОГО ПРОЦЕССА
Дальнейшее развитие флотационного процесса обусловлено
возрастающими потребностями народного хозяйства в
минеральном сырье. Для повышения степени комплексности его
использования необходимы дальнейшее совершенствование
технологии, интенсификации флотационного процесса, применение
нового и модернизированного оборудования, механизация и
автоматизация обогатительных фабрик.
Решение перечисленных задач невозможно без дальнейшего
развития теоретических представлений во флотации.
В области теории флотации наблюдается стремление перейти
от качественных представлений к количественным
закономерностям. При проведении исследований ставятся цели: обнаружить
закономерности протекания реакций в процессе гидрофобиза-
ции и флотации минералов ксантогенатами, дитиофосфатами,
жирными и алкилгидраксамовыми кислотами, алкилсульфа-
тами; дать количественное описание процесса разрыва гидрат-
ной прослойки между пузырьками и поверхностью; дать
количественное описание кинетики процесса флотации.
Необходимость перехода от качественных представлений к
количественным закономерностям обусловлено стремлением разработать
теоретически обоснованные принципы синтеза новых
эффективных флотационных реагентов, оптимизации и автоматического
регулирования флотационного процесса.
Очень важным является применение более строгих и
объективных методов исследования флотационных систем и
особенно механизма действия реагентов при флотации.
Из теоретических методов исследования следует особо
подчеркнуть более широкое использование термодинамического
метода. Он применяется, например, для исследования реакций
между различными собирателями и ионами металлов при
различных значениях рН и в присутствии реагентов-депрессоров;
для исследования состояния твердой поверхности при
различных значениях рН и окислительно-восстановительного
потенциала раствора; для изучения механизма действия собирателей
при флотации различных минералов. В результате
термодинамических исследований получен целый ряд необходимых
констант, сделан вывод о перспективности применения при флота*
ции сочетаний собирателей, дана объективная оценка
возможного состояния поверхности многих минералов, которая
существенно отличается от общепринятых представителей.
Термодинамический метод исследования совершенно необходим при
разработке количественной физико-химической модели
процессов селективной флотации, которая позволит коренным
образом решить проблему оптимизации флотации, повышения
технико-экономических показателей работы обогатительных
фабрик и комплексности использования сырья.
358
Из экспериментальных методов исследования особого
внимания заслуживают методы, основанные на измерении
электрохимических характеристик поверхности твердого в жидкой
среде. В отличие от других методов, они позволяют
исследовать непосредственно поверхность раздела твердое — жидкость,
а не поверхность твердое — газ (как при обычной методике
использования радиоактивных изотопов), твердое — твердое
или твердое — органическая жидкость (как при использовании
инфракрасной спектроскопии ИКС) или в растворах
растворителя (как при использовании электронного парамагнитного
резонанса ЭПР или ИКС) и т. д. Из электрохимических
методов наиболее перспективны методы измерения емкости
двойного электрического слоя, вольтамперометрия, метод спада
потенциала.
Например, применение метода измерения емкости двойного
электрического слоя при исследовании кинетики уточнения и
разрыва гидратной прослойки между пузырьком и ртутью в
присутствии собирателя позволяет учесть форму закрепления
реагента и, в какой-то мере, его ориентировку на поверхности
раздела фаз, проследить кинетику изменения напряжения
отрыва пузырька при различных значениях потенциала
поверхности. Этот же метод может быть использован для
определения закономерностей адсорбции ионных и молекулярных форм
собирателя.
Применение вольтамперометрии при линейном измерении
напряжения в совокупности с обычными методами измерения
краевого угла и флотации позволило получить интересные
данные по физико-химическому состоянию флотационной системы
сульфидный минерал — сульфгидрильный собиратель. В свою
очередь, метод спада потенциала обеспечивает получение
надежных данных по состоянию поверхности сульфида, которые
необходимы для достоверного теоретического анализа
механизма взаимодействия ее с флотационными реагентами.
Наибольший эффект при исследовании достигается при
одновременном использовании методом термодинамики, элекрохимии и
флотации.
Значительный интерес представляют электрохимические
исследования полупроводниковых материалов, поскольку они
позволяют выявить роль области пространственного заряда
сульфидов, концентрации электронов и дырок и их влияние на
кинетику и механизм взаимодействия реагентов с поверхностью,
плотность адсорбции и соотношение форм сорбции собирателя.
Результаты исследований подтверждают наличие определенных
соотношений между элекронными свойствами минералов,
электрохимическими свойствами поверхностей раздела твердой и
жидкой фаз, адсорбции собирателя и степенью гидрофобности
поверхности. Использование даже несовершенных моделей
позволяет говорить о возможности целенаправленного изменения,
например, степени дефектности кристаллической решетки, кон-
359
центрации электронов, адсорбции реагентов и в конечном счете
флотируемости минералов. Для этого можно воспользоваться
радиационными методами, ультразвуковой, электрохимической
обработкой и некоторыми другими методами физического
воздействия на пульпу. Заслуживает внимания использование
температурной обработки при разделении коллективных
концентратов. Повышение эффективности селективной флотации
в этом случае может быть обусловлено различными
закономерностями изменения термодинамических характеристик
разделяемых минералов.
Работы по флотационным реагентам направлены на
изыскание нетоксичных новых, более избирательных собирателей,
пенообразователей, депрессоров и активаторов для различных
типов минерального сырья, чтобы расширить ассортимент и
повысить качество применяемых реагентов. Перспективно
применение ионообменных смол для регулирования ионного состава
пульпы.
Конечной целью совершенствования флотационных
процессов является разработка безотходной технологии,
обеспечивающей полное и комплексное использование перерабатываемого
минерального сырья в условиях полного водооборота.
Количество и ассортимент сырья, подвергаемого
флотационному разделению на отдельные слагающие его компоненты»
будут увеличиваться и расширяться. Флотация найдет еще
более широкое применение для разделения промпродуктов
металлургического и химического производства, извлечения ценных
компонентов из разбавленных растворов, очистки сточных вод>
извлечения органических веществ из растений и горючих
сланцев, очистки и сортировки семян, очистки растворов сахара,
бумажных пульп, виноградных вин, текстильных волокон и
решения других народнохозяйственных задач.
СПИСОК ЛИТЕРАТУРЫ
1. Абрамов Л. А. Теоретические основы оптимизации селективной
флотации сульфидных руд. М., Недра, 1978.
2. Абрамов А. А. Технология обогащения руд цветных металлов. М„
Недра, 1983.
-3. Абрамов А. А., Леонов С. В., Сорокин М, М. Химия флотационных
систем. М., Недра, 1982.
4. Адамсон А. Физическая химия поверхностей. М., Мир, 1979.
5. Барский Л. Л., Плаксин Я. Н., Тюрникова В. Я. Комплексное
обогащение молибденовых руд. М., Недра. 1965.
6. Волькенигтейн Ф. Ф. Физико-химия поверхности полупроводников. М.,
Наука, 1973.
7. Глембоцкий В. А., Классен В. Я. Флотационные методы обогащения.
М., Недра, 1981.
8. Годен А. М. Флотация. М., Госгортехиздат, 1959.
9. Голованов Г, А. Флотация Кольских апатитсодержащих руд. М.,
Химия, 1976.
10. Г орловский С. Я., Хайнман В. #. Об особенностях действия
высокомолекулярных флокулянтов.— Обогащение руд, 1961, № 4, с. 24—29.
11. Желнин Л. Л. Теоретические основы и практика флотации калийных
солей. М„ Химия, 1973.
12. Зеленое В. Я. Методика исследования золотосодержащих руд. М.,
Недра, 1978.
13. Кинетика электродных процессов/ А. Н. Фрумкин, В. С. Багоцкий,
3. А. Иофа и др. М., МГУ, 1952.
14. Классен В. Я. Обогащение руд химического сырья. М., Недра,
1979.
15. Классен В. Я., Мокроусов В. А. Введение в теорию флотации. М.,
Госгортехиздат, 1959.
16. Клебанов О. Б, Реагентное хозяйство обогатительных фабрик. М.,
Недра, 1976.
17. Конев В. А., Конев В. Л„ Разумов К. Л. Депрессирующее действие
цинката натрия на сфалерит.— Обогащение руд, 1966, № 1, с. 4—9.
18. Кузнецов В. П., Волова М. Л. Лафиренко В. Е. Обогащение
бокситов. М., Недра, 1978.
19. Кузькин С. Ф., Гольман Л. М. Флотация ионов и молекул. М., Недра,
1971.
20. Малиновский В, А. Элементы основ пенной сепарации. В кн.: Пенная
сепарация. М., ВЗПИ, 1971, с. 4—15.
21. Матвеенко Я. В. Пенная сепарация полезных ископаемых. М., Недра,
1976.
22. Мелик-Гайказян В. Я. О механизме действия аполярных реагентов
при пенной флотации.— Обогащение руд, 1970, № 3, с. 38—43.
23. Мелик-Гайказян В. Я., Емельянов Н. Я., Глазунова 3. Я. О
капиллярном механизме упрочнения контакта частица — пузырек при пенной
флотации,—Обогащение руд, 1976, № 1, с. 25—31.
24. Мещеряков Н. Ф. Флотационные машины и аппараты. М., Недра,
1982.
25. Митрофанов С. Я. Селективная флотация. М., Недра, 1967.
26. Митрофанов С. Я., Барский Л. А. Самыгин Я. Д. Исследование
полезных ископаемых на обогатим ость. М., Недра, 1974.
27. Найфонов Г. Б. Флотация титановых минералов при обогащении
комплексных титаносодержащих руд. Л., Наука, 1979.
28. Новые конструкции флотационных машин и опыт их применения.
Серия «Горнохимическая промышленность», М., НИИТЭХИМ, 1977.
361
29. Основы теория и практика применения флотационных реагентов/
С, В. Дуденков, Л. Я. Шубов, Л. А. Глазунов и др. М., Недра, 1969.
- 30. Околович А. М., Фигуркова Л. И. Особенности флотации сфалерита
из полиметаллических сульфидных руд. М,, Наука, 1977.
31. Пикат-Ордынский Г. Л., Острый В, Л. Технология флотационного
обогащения углей. М., Недра, 1972.
32. Плаксин И. И., Мясникова Г. Л. Околович Л. М. Флотационное
обогащение мышьяково-пиритных руд. М., АН СССР, 1955.
33. Плаксин Я. Я., Шафеев Я Ш., Чантурия В/Л. Взаимосвязь между
энергетическим строением кристаллов минералов и их флотационными
свойствами. VIII Международный конгресс по обогащению полезных
ископаемых. Л., 1969, т. II.
34. Полькин С. И. Обогащение руд и россыпей редких металлов. М.,
Недра, 1967.
35. Полькин С. И., Лаптев С. Ф. Обогащение оловянных руд и
россыпей. М, Недра, 1974.
36. Разумов К. Л. Флотационный метод обогащения. Л., ЛГИ, 1975.
37. Рубинштейн Ю. Б,, Филиппов /О. Л. Кинетика флотации. М., Недра,
1980.
38. Сазерленд К. Л., Уорк И. В. Принципы флотации. М., Метал лург-
издат, 1958.
39. Стрельцын Г. С. Влияние кристаллической структуры на
взаимодействие поверхности твердых тел с водой.— В кн.: Тр. н.-техн. конф. ин-та
Механобр., Л., 1968, т. 1, с. 183—204.
40. Стрельцын Г. С. Гидрофобизирующее действие неорганических
электролитов на природно-гидрофилъные минералы.— В кн.: Тр. н.-техн. конф.
ин-та Механобр, Л., 1968, т, 1, с. 171—182.
41. Справочник по обогащению руд. М., Недра, 1982—1983.
42. Справочник по обогащению руд черных металлов. М., Недра, 1980.
43. Справочник по обогащению углей. М., Недра, 1984.
44. Теория и технология флотации руд/ О. С. Богданов, И. С.
Максимов, А. К. Поднек и др. М., Недра, 1980.
45. Тюрникова В. Я. Повышение эффективности действия собирателей
при флотации руд. М., Недра, 1971.
46. Физико-химические основы теории флотации/ О. С. Богданов,
А. М. Гольман, И. А. Каковский и др. М., Наука, 1983.
47. Флотационное обогащение руд и некоторые вопросы геологии
полиметаллических месторождений восточного Забайкалья/С. Б. Леонов, Б. П.
Санин, Г. Д. Русецкая и др. Иркутск, 1971.
48. Чантурия В. А., Шафеев Р. Ш. Химия поверхностных явлений при
флотации. М., Недра, 1977.
49. Эйгелес М. А, Основы флотации несульфидных минералов. М., Недра,
1964.
50. Эйгелес М. Л. Реагенты-регуляторы во флотационном процессе. М.„
Недра, 1977.
ОГЛАВЛЕНИЕ
Предисловие . . . . г s :
1. Сущность и разновидности флотационных процессов . > !
1.1. Сущность флотационного процесса I
1.2. Разновидности флотационных процессов ■
1.3. Значение флотационного процесса ' 1!
ЧАСТЬ I, Основы теории минерализации пузырьков при флотации . . 1
% Термодипамика элементарного акта флотации 1
2.1. Краткая характеристика раздела фаз, участвующих в
элементарном акте флотации 1
2.2. Изменение энергии прослоя воды между пузырьком и
частицей при элементарном акте флотации 2
2.3. Краевой угол смачивания и зависимость его от удельной
поверхностной энергии на границе соприкасающихся фаз ... 2
2.4. Гистерезис смачивания .3
2.5. Термодинамический анализ возможности элементарного акта
флотации. Показатель флотируемости 3
2.6. Влияние пузырьков газа, выделяющихся из раствора, на
показатель флотируемости 3
2.7. Показатель флотируемости при коалесцентяом 'механизме
элементарного акта флотации 3
3. Физические основы флотации 3
3.1. Условие равновесия частицы на плоской поверхности раздела
газ — жидкость при пленочной флотации . . . ' 3
3.2. Факторы, влияющие на минерализацию пузырьков и пенную
флотацию частиц 4
3.3. Вероятность столкновения частицы с пузырьком 4
3.4. Вероятность закрепления частицы на пузырьке 4
3.5. Вероятность сохранения закрепившихся на пузырьке частиц . 4
3.6. Вероятность удержания частиц в слое пены 5
3.7. Необходимый размер пузырьков при пенной флотации ... 5
3.8. Характеристика процесса пенной сепарации и сил,
действующих на частицу в пенном слое 6
3.9. Условия повышения крупности флотируемых частиц .... 6
ЗЛО. Условия флотации тонких частиц 7
4.-Кинетика и моделирование флотационного процесса 7
4.1. Кинетика флотации -. 7
4.2. Модели флотации ..*..„ 7
ЧАСТЬ II. Флотационные реагенты и механизм их действия на
поверхности раздела фаз при флотации 7
5. Назначение и общая характеристика флотационных реагентов ... 7
5.1. Назначение и классификация флотационных реагентов ... 7
5.2. Состояние флотационных реагентов в жидкой фазе пульпы . 7
5.3. Адсорбция реагентов на поверхности раздела фаз ..... 8
6. Влияние основных факторов на состояние поверхности минералов в
отсутствие собирателей 9
6.1. Влияние рН и окислительно-восстановительного потенциала
пульпы на состояние поверхности минералов 9
6.2. Влияние изоморфизма и электрофизических свойств минералов
на состояние их поверхности 10
6.3. Влияние кристаллохимического строения минералов на
состояние их поверхности 10
7. Собиратели и механизм их действия при флотации .10
38
ОГЛАВЛЕНИЕ
Предисловие . . . . г г . . , , . 3
1. Сущность и разновидности флотационных процессов 5
1.1. Сущность флотационного процесса 5
1.2. Разновидности флотационных процессов 9
1.3. Значение флотационного процесса 12
ЧАСТЬ I. Основы теории минерализации пузырьков при флотации . . 14
2. Термодинамика элементарного акта флотации 14
2.1. Краткая характеристика раздела фаз, участвующих в
элементарном акте флотации 14
2.2. Изменение энергии прослоя воды между пузырьком и
частицей при элементарном акте флотации 26
2.3. Краевой угол смачивания и зависимость его от удельной
поверхностной энергии на границе соприкасающихся фаз ... 29
2.4. Гистерезис смачивания '. 30
2.5. Термодинамический анализ возможности элементарного акта
флотации. Показатель флотируемости 33
2.6. Влияние пузырьков газа, выделяющихся из раствора, на
показатель флотируемости 35
2.7. Показатель флотируемости при коалесцентном механизме
элементарного акта флотации 36
3. Физические основы флотации 37
3.1. Условие равновесия частицы на плоской поверхности раздела
газ — жидкость при пленочной флотации ..." 37
3.2. Факторы, влияющие на минерализацию пузырьков и пенную
флотацию частиц 40
3.3. Вероятность столкновения частицы с пузырьком 43
3.4. Вероятность закрепления частицы на пузырьке 45
3.5. Вероятность сохранения закрепившихся на пузырьке частиц . 48
3.6. 'Вероятность удержания частиц в слое пены 57
3.7. Необходимый размер пузырьков при пенной флотации ... 57
3.8. Характеристика процесса пенной сепарации и сил,
действующих на частицу в пенном слое 61
3.9. Условия повышения крупности флотируемых частиц .... 68
ЗЛО. Условия флотации тонких частиц 70
4.-Кинетика и моделирование флотационного процесса 71
4.1. Кинетика флотации : 71
4.2. Модели флотации . .' 73
ЧАСТЬ II. Флотационные реагенты и механизм их действия на
поверхности раздела фаз при флотации 77
5. Назначение и общая характеристика флотационных реагентов ... 77
5.1. Назначение и классификация флотационных реагентов ... 77
5.2. Состояние флотационных реагентов в жидкой фазе пульпы . 78
5.3. Адсорбция реагентов на поверхности раздела фаз ..... 88
6. Влияние основных факторов на состояние поверхности минералов в
отсутствие собирателей 94
6.1. Влияние рН и окислительно-восстановительного потенциала
пульпы на состояние поверхности минералов 94
6.2. Влияние изоморфизма и электрофизических свойств минералов
на состояние их поверхности 104
6.3. Влияние кристаллохимического строения минералов на
состояние их поверхности 106
7. Собиратели и механизм их действия при флотации ~. 109
381
* ■ 7.1. Строение молекул и классификация собирателей 109
7.2. Влияние длины аполярной цепи собирателя на прочность его
закрепления ; . 113
7.3. Аполярные собиратели 115-
7.4. Гетерополярные собиратели с неопределенным химическим
составом 118
7.5. Катионные собиратели . 118-
7.6. Оксигидрильные собиратели 124
7.7. Сульфгидрильные собиратели 132
7.8. Роль форм адсорбции собирателей при флотации 142
8. Активаторы и механизм их действия при флотации 145
8.1. Назначение и основные механизмы активирующего действия
реагентов-модификаторов 145-
8.2. Активирующее действие реагентов путем химической очистки
поверхности минералов 146-
8.3. Активирующее действие реагентов путем хемосорбции ионов
на поверхности 147
8.4. Активирующее действие реагентов путем гетерогенной
химической реакции .153-
9. Депрессоры и механизм их действия при флотации 157
9.1. Назначение и основные механизмы действия
реагентов-депрессоров ■ . ; . 157
9.2. Депрессирующее действие щелочных реагентов 159
9.3. Депрессирующее действие сульфидов щелочных металлов . .167
9.4. Депрессирующее действие цианидов 170
9.5. Депрессирующее действие кислот . 173.
9.6. Депрессирующее действие солей щелочных, щелочноземельных
металлов и алюминия 173
9.7. Депрессирующее действие хроматов и бихроматов 174
9.8. Депрессирующее действие фосфатов 175
9.9. Депрессирующее действие ферри- и ферроцианидов .... 175
9.10. Депрессирующее действие цинкового купороса и его
сочетаний с другими реагентами 177
9.11. Депрессирующее действие сульфоксидных соединений и их
сочетаний с другими реагентами 181
9.12. Депрессирующее действие жидкого стекла 183
9.13. Депрессирующее действие органических реагентов . . . 184
10. Регуляторы среды и механизм их действия - при флотации . . . .187
10.1. Регулирование рН пульпы 188
10.2. Удаление из жидкой фазы пульпы нежелательных ионов . .188
10.3. Регулирование окислительно-восстановительного потенциала
пульпы 191
10.4. Регулирование процессов диспергации и коагуляции шламов 192
11. Пенообразователи и механизм их действия 193
11.1. Строение и физико-химические свойства пенообразователей . 193
11.2. Роль и механизм действия пенообразователей 194
11.3. Стабилизация и гашение пены флотирующимися частицами . 198
11.4. Вторичная концентрация минералов в пене 200
11.5. Пенообразователи, применяемые на практике 20О
ЧАСТЬ III. Технология флотации 204
12. Основные характеристики вещественного состава полезных
ископаемых и их влияние на флотацию 204
12.1. Содержание ценных компонентов 204
12.2. Минеральный состав 204
12.3. Влияние генезиса руд 205
12.4. Вторичные изменения минералов 206
12.5. Характер вкрапленности и необходимая крупность измельчения 207
12.6. Управление качеством полезных ископаемых, поступающих
на флотационное обогащение 208
13. Схемы флотации 209
13.1. Классификация операций флотации 209
13.2. Стадиальность схем флотационного обогащения 211
13.3. Схемы с раздельной обработкой и флотацией песков и шламов 214
13.4. Схемы коллективной и селективной флотации 217
■ 13.5. Комбинированные схемы 221
14. Режимы флотации 222
14.1. Классификация минералов по флотируемости 222
14.2. Флотация руд, содержащих минералы с высокой
естественной гидрофобностью 223
14.3. Флотация сульфидных руд и самородных металлов .... 229
14.4. Флотация окисленных и смешанных руд цветных металлов . 252
14.5. Флотация несульфидных руд солеобразных минералов . . . 258
14.6. Флотация окислов металлов : 268
14.7. Флотация силикатов : 284
14.8. Флотация растворимых солей 290
14.9. Ионная флотация 293
14.10. Направления совершенствования режимов флотации . . . 296
15. Флотационные машины и аппараты 303
15.1. Классификация флотационных машин 304
15.2. Требования, предъявляемые к современным флотационным
машинам 306
15.3. Процессы диспергирования воздуха и аэрации пульпы во
флотационных машинах 307
15.4. Механические флотационные машины 311
15.5. Пневмомеханические флотационные машины 318
15.6. Пневмогидравлические флотационные машины 324
15.7. Пневматические флотационные машины 325
15.8. Флотационные машины с изменяемым давлением 328
15.9. Электрофлотационные машины 329
15.10. Основные факторы, влияющие на эффективность работы
флотационных машин при пенной флотации 329
15.11. Технологические особенности пешюй сепарации 336
15.12. Выбор флотационных машин 338
15.13. Расчет необходимой вместимости машин при флотации . . 340
15.14. Основные направления совершенствования флотационных
машин 340
16. Организация работы флотационного отделения фабрики 341
16.1. Распределение операций флотации по флотационным машинам 341
16.2. Устройства для подготовки пульпы к флотации 343
16.3. Оборудование и эксплуатация реагентных площадок . . . 345
16.4. Кондиционирование ионного состава промышленных и
оборотных вод 349
16.5. Разрушение флотационной пены 350
16.6. Контроль и регулирование флотационного процесса . . . .351
16.7. Охрана труда и техника безопасности на флотационных
фабриках . . 352
17. Основные технологические и технико-экономические показатели
работы флотационных фабрик 353
18i Перспективы и направления дальнейшего развития флотационного
процесса 358
Список литературы 361
Приложение 1. Технические^ характеристики флотационных машин СССР 363
Приложение 2. Технические"* характеристики зарубежных флотационных
машин 364
Приложение 3. Характеристика основных реагентов-собирателей .... 368
Приложение 4. Характеристика основных флотационных
реагентов-пенообразователей . : : 376
Приложение 5. Характеристика основных флотационных
реагентов-модификаторов . . : 378
Александр Алексеевич Абрамов
ФЛОТАЦИОННЫЕ МЕТОДЫ ОБОГАЩЕНИЯ
Редактор издательства О. И. Паркани
Переплет художника С. Л. Салтанова
Художественный редактор О. Н. Зайцева
Технический редактор Е. В, Воробьева
Корректор К. С. Торопцева
ИБ № 4922
Сдано в набор 05.10.83. Подписано в печать 04.01.84. Т-03404. Формат
60X90Vi6. Бумага книжно-журнальная. Гарнитура «Литературная». Печать
высокая. Усл. печ. л. 24,0. Усл. кр.-отт. 24,0. Уч.-изд. л. 25,0. Тираж 4500 экз.
Заказ № 1957/8819-11. Цена 1 р. 20 к.
Ордена «Знак Почета» издательство «Недра», 103633, Москва, К-12,
Третьяковский проезд, 1/19
Ленинградская типография № 4 ордена Трудового Красного Знамени
Ленинградского объединения «Техническая книга» им. Евгении Соколовой Союз-
полиграфпрома при Государственном комитете СССР по делам издательств,
полиграфии и книжной торговли. 191126, Ленинград, Социалистическая ул., 14