/



































































































































































































































































































































































































































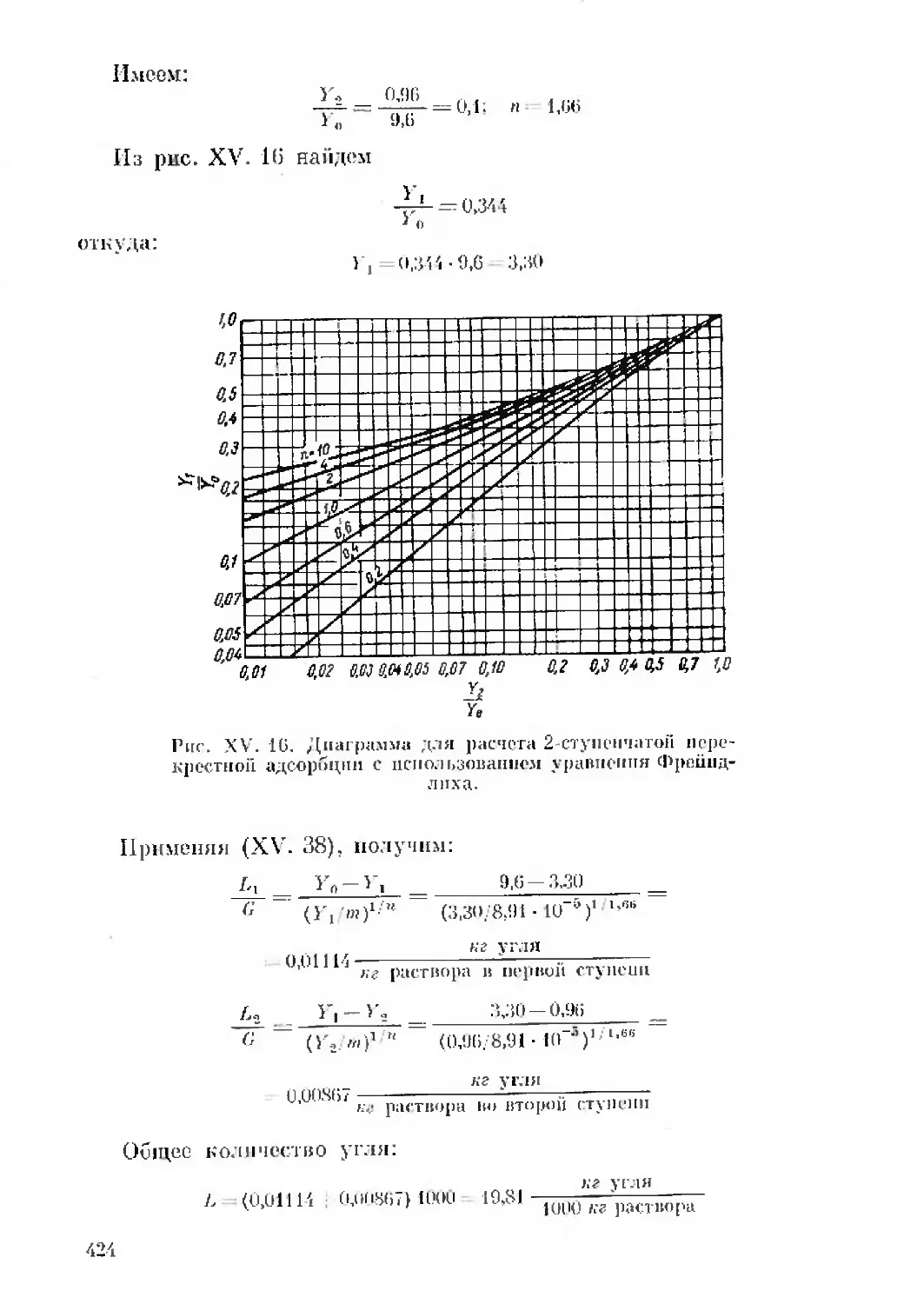


































































































Text
А.М. БАТУНЕ Р
ПРОЦЕССЫ
И АППАРАТЫ
ОРГАНИЧЕСКОГО
СИНТЕЗА
И БИОХИМИЧЕСКОЙ
ТЕХНОЛОГИИ
«ХИ МИЯ»
1366
ДК 66.01 : 66.021 : 577.1
В книге описываются методы расчета процессов
и аппаратов органического синтеза и биохимической
технологии на основе химической кинетики и термо-
динамики. Излагаются принципы составления мате-
риального и теплового балансов описываемых процес-
сов и аппаратов. Приведенные здесь многочисленные
примеры взяты из лабораторной и заводской практики
и являются типичными. Аналогично этим примерам
решаются задачи, с которыми постоянно встречаются
химики в своей практической деятельности.
Книга предназначается для инженеров-химиков,
производственников, исследователей и проектиров-
щиков. Она может также служить пособием для сту-
дентов химико-технологических вузов и аспирантов.
14-2
Предисловие
Любой процесс должен рассматриваться в связи с его
кинетикой и термодинамикой. С помощью учения о кинетике
исследуются скорость и механизм процессов и реакций, а термо-
динамика дает возможность найти условия получения максималь-
ного выхода продуктов, для которого впоследствии может быть
установлен оптимум.
Излагаемый в книге теоретический и расчетный материал по
процессам и аппаратам сопровождается выводами конкретных
закономерностей и числовыми примерами, которые составляют его
неотъемлемую часть.
Говоря об аппаратуре, мы всегда имеем в виду протекающие
в пей процессы, которые обусловливают конструкцию, размеры
и режим работы аппарата. Таким образом, расчет аппарата в дан-
ном случае есть не что иное, как расчет процесса.
В отношении энзиматических процессов в биохимической тех-
нологии следует отметить, что успешное использование их в про-
мышленности, а также их дальнейшее развитие не могут быть
осуществлены в отрыве от физической химии и химической тех-
нологии.
Работая в области химической технологии, например биоло-
гически активных веществ, трудно в настоящее время обойтись
без методов и многих процессов, используемых в органическом
синтезе. Биологически активные вещества можно изменить и улуч-
шить. т. е. повысить их эффективность, синтетическим путем.
Кроме того, во многих случаях экономически более выгодным
оказывается получение этих веществ чисто химическим способом
или же но комбинированной схеме. Поэтому научно-технические
сведения о процессах и аппаратах целесообразно дать в едином
плане как для органического синтеза, так и для биохимической
технологии.
Книга в таком виде издается впервые. Все замечания i поже-
лания читателей будут приняты с благодарностью.
Автор
1* з
Перевод единиц измерения некоторых величин
в единицы Международной системы (СИ)
Величины Единицы изме- рения, исполь- зуемые в книге Единицы измерения в СИ Соотношение между единицами измерения
Вес (сила тяжести) ?ггс н 1 кгс = 9,81 н
Вязкость и коэф- н сек/м- 1 спз 1 Ю-3 н • сек/м2
фициент дина- мический кй/.ч • сек 1 ге/,и - сек — = 0,102 кге • сек/м2 = = 1н> сек/м2
Давление кгс/с.н’2 пли ат кге/м2 или л.л and. ст. .ил рт. ст. атл н/м~ 1 кгс.'с.н2 - 9.81 104 н/м2 1 кге/.ч2^ 9,81 н/.и2 1 .н.ч. рт, ст. = 133,3 м/м2 1 ат.и=. 10,1 • 10'1 н/м2
Длина МК А 1 .1IK 1 • Ю-6 .4 1А = 1 -10“10 .и
Натяжение но- дик/см н /.и 1 дин/см = 1 10-3 н/м
верхностпоо
Объем л, дм2 И3 1 а 1,000028 Aw3 ^1 10’3 .и3
Скорость угловая об/мин ра д/ сек 1 об/мин = рад сек 30 '
Тепловой поток ккал.ч вт 1 к ка л / ч = 1,1 (>3 вт
Теплоемкость удельная массо- вая ккал/кгград дж/кг град 1 ккал/кг•град = = 4187 дж/кг- град «4,19 кдж/кг - град
Теплоотдачи коэф- фициент, тепло- ккал ;м- .ч.г рад вт/м- - град 1 ккал/м? - ч град = — 1,163 вт/.и’2 • град
передачи коэф- фициент
Т еп лоиро водности коэффициент ккал/м. ч-град вт. /м град 1 ккал/.ч • ч • град - 1.163 вт/м- град
'1 оплота удельная “(фазового пре- К К(1-Л-j fie- дж/ кг 1 ккал/кг 4187 дж/кг^ «4,19 кдж/кг
образования)
Энтальпия удель- ная ккал/кг дж/кг 1 ккал/кг = 4187 дж/кг «=4.19 кдж/кг
Энтропия удел!*- к кал/кгград дж/кг • град 1 ккал/кг•град —
пая =«4187 дж/кг • грид ^;4,19 кдж/кг град
4
Глава I
Общие сведения
о технологических
и тепловых расчетах
реакционных аппаратов
Реакционная аппаратура может быть подразделена
на следующие группы:
1) емкостные аппараты, производительность которых опре-
деляется величиной их реакционного объема и временем прове-
дения процесса;
2) тепловые аппараты, производительность которых опреде-
ляется поверхностью теплообмена и скоростью подвода и отвода
тепла;
3) массообмспные аппараты, производительность которых опре-
деляется величиной поверхности фазового контакта.
По организационному принципу проведения процессов аппа-
раты подразделяются па непрерывно действующие и периодиче-
ского действия.
Непрерывно действующие аппараты характеризуются един-
ством времени завершения всех стадий процесса, протекающего
в аппарате.
Периодически действующие аппараты характеризуются един-
ством места завершения всех стадий процесса.
Технологические расчеты
Емкостные аппараты
Для расчета количества и емкости аппаратов рассма-
триваемого тина необходимо знать объем веществ, перерабаты-
ваемых в сутки па данной стадии процесса, время проведения
процесса и его организационный характер.
Процесс проводится периодически. Задан объем ве-
ществ. перерабатываемых в сутки, и время проведения процесса.
Примем следующие обозначения:
Рсут
Ка
- 2%
I а
— объем веществ, перерабатываемых в сутки;
— полный объем аппарата;
— рабочий объем аппарата;
— степень заполнения аппарата;
т — время проведения процесса;
а — суточная мощность производства, выраженная числом
операций, проводимых на данной стадии в течение
суток;
р — мощность одного аппарата, выраженная числом опера-
ций, проводимых в нем в течение суток;
— необходимое число рабочих аппаратов;
— число устанавливаемых аппаратов;
nip
т
6 — резерв мощности аппаратуры.
Суточная мощность производства рассчитывается по формуле:
V CVT
а- '
М
Суточная мощность одного аппарата:
?4
К
Зная а и ₽, легко определить необходимое число рабочих аппа-
ратов по уравнению:
(X _ I сутт
Количество устанавливаемых аппаратов с учетом резерва
мощности б:
т—- /ир (1 4-0,01 6)
В расчетах исходят из емкости одного аппарата Га и вычис-
ляют общее их количество пли же задаются количеством аппаратов
и вычисляют емкость одного аппарата по формуле:
... . 1суТт (1-0,01 6)
а
Величина <р равна 0,7—0,85 для аппаратов, в которых про-
цессы не сопровождаются вспениванием, и 0,4 -0,6 для аппара-
тов, в которых процессы сопровождаются вспениванием и кипе-
нием.
Резерв мощности аппаратуры для обычных условий прини-
мается равным 10—15%, в особых случаях и при серьезных обос-
нованиях он может быть значительно увеличен.
Процесс проводится непрерывно. 1. Заданы объем пе-
рерабатываемых в секунду веществ ГСС|. и продолжительность
пребывания материала в аппарате тсск.
6
Рабочий объем всей аппаратуры V находится по формуле:
V = Г cvj;TCei;
Очевидно, что необходимое число аппаратов составляет:
fit IS — ---
I' аЧ;
Таким образом
т = /Нр (1 -г 0,01 б) = ГсекТсск 7 6)
г а <р
откуда:
I- I секТсск (1-р0,01 б)
• а - --------------
nttf
2. Заданы те же величины, что и в предыдущем случае, и ско-
рость движения веществ в аппарате и? (в м/сек).
По объему перерабатываемых в секунду веществ определяется
площадь поперечного сечения аппарата / (в .н2)
у 1' сСк
IV
а по продолжительности пребывания материала в аппарате —
высота или длина аппарата L (в м):
Л = ик
Если найденная площадь поперечного сечения аппарата ока-
жется слишком большой, то устанавливается т аппаратов, соеди-
ненных параллельно. При этом каждый аппарат имеет длину Л
и площадь поперечного сечения /.’?«.
Если же слишком большой окажется длина, то устанавли-
вается т аппаратов, соединенных последовательно. При этом
каждый аппарат имеет площадь поперечного сечения f
и длину Ыт.
3. Заданы те же величины, что и во втором случае, и режим
движения в реакционной зоне аппарата.
Режим движения жидкости определяется величиной критерия
Рейнольдса Re. Запишем критерий Рейнольдса в виде:
Re *^10’ (1.1)
где rr / 5 — гидравлический радиус аппарата, л;
S — периметр поперечного сечения аппарата, л;
о — плотность, кг/л3;
и — вязкость реакционной массы, спз.
Подставим в формулу (I. 1) значение и? из уравнения расхода
W = Гсек
7
получим:
Отсюда:
Вс = ±1!>У<?. ю»
/В
_ -^УсекО 103
rc Re и
Для аппаратов с внутренним диаметром d, поперечное сечепие
которых имеет форму окружности
d ’ , л d-
следовательно:
Re rtii
Зная d и" Усек, легко найти ш:
Л d~
Длина реакционной зоны аппарата определяется по формуле:
I
L =ц?т
Тепловые аппараты
При расчете реакционных аппаратов, производитель-
ность которых определяется поверхностью теплообмена, всегда
имеют заданными и поверхность теплообмена F.
Процесс проводится непрерывно. 1. Заданы Ксек,
F и w.
Площадь поперечного сечения аппарата:
* f — ^сС|
W
Кроме того, имеем
SL
следовательно:
(1-2)
5 ~ wF
Умножив левую и правую части уравнения (I. 2) на 4, получим:
—"э —
(1-3)
S "и ” 1₽/'
С помощью формулы (1.3), задаваясь величиной L. можно
вычислить эквивалентный диаметр с/0 и найти площадь попереч-
ного сечения аппарата.
8
2. Заданы VCCK, F и т.
Рабочий объем аппарата:
Рр — I' cci;T
Очевидно, что
следовательно:
Усек? (1.6)
Разделив обе части уравнения (I. 4) на 5, получим:
/ __ ______ I'ccifT
.S’ “ SL ~ F
или
V , _ ZlF(;CKT
Д' 3 F
Масео обменные аппараты
При технологическом расчете реакционной аппара-
туры, производительность которой определяется величиной поверх-
ности фазового контакта, должны быть известны VCCK, ик поверх-
ность фазового контакта Л’в.
Площадь поперечного сечения аппарата, реакционный объем
которого заполнен насадочными телами, рассчитывается но фор-
муле:
4 —
' wx,
где х — коэффициент свободного сечепия аппарата, т. е. отно-
шение площади поперечного сечения аппарата, заполнен-
ного насадкой, ко всей площади поперечного сечения.
Объем аппарата находится как объем насадочных тел:
I* - Fk .
la- —
где о — удельная поверхность насадочных тел, Л12/.н3.
Высота (длина) аппарата:
_ Кч __ 1'иц}'л
i I' сек°
Тепловые расчеты
Тепловой расчет реакционных аппаратов в основном
сводится к составлению теплового баланса процесса, определению
количества подводимого или отнимаемого тепла, определению
9
расхода теплоносителя или охлаждающего агента, вычислению
поверхности теплообмена.
Нами будут рассмотрены тепловой баланс процесса и про-
граммированный подвод или отвод тепла.
Тепловой баланс
Тепловому балансу всегда предшествует материальный
баланс. В наиболее общей форме уравнение, выражающее тепловой
баланс (в ккал/'ч или ккал/опсрация) химического процесса, можно
представить так:
(Л4-<>24-<?з - Ci 4- th 4- <?« (1 ">)
где Qt — тепло, вносимое в аппарат с перерабатываемыми мате-
риалами,
Q., — тепло, подводимое теплоносителем и ш отнимаемое от
аппарата охлаждающим агентом;
Q;i — тепловой эффект процесса;
Qi — тепло, уносимое из аппарата с продуктами реакции;
Q:> — тепло, расходуемое на нагревание отдельных частой
аппарата (при непрерывном процессе Qb можно не учи-
тывать):
Q6 — тепли, теряемое аппаратом в окружающую срс IV.
Равенство (I. 5) справедливо как для процессов, протекающих
при нагревании, так и для процессов, идущих при охлаждении
Однако в последнем случае Q-, имеет отрицательное значение.
Основная величина Q„ находится из разности:
Количество тепла, вносимое в аппарат с перерабатываемыми
веществами и уносимое с продуктами реакции, может быть опре-
делено но формуле:
Qt , — f‘cl СC.,c.,f2- . . .
•
где G — количество вещества, кг:
с — теплоемкость, ккал, кг • грав\
t — температура. С
Величины G берутся по данным материального баланса. Тем-
пературы обычно заданы регламентом. В том случае, когда зна-
чения нужных теплоемкостей отсутствуют в справочной литера-
туре, их приходится рассчитывать.
Расчеты входящих в тепловой баланс величин Qb и 0<; подробно
рассматриваются в курсе «Основные процессы и аппараты хими-
ческой технологии» и поэтому здесь не приводятся.
Тепловой эффект процесса Q3 представляет собой суммарное
количество теп ia, которое выделяется пли поглощается при нро-
10
текапии химических реакций и сопровождающих их физико-
химических процессов (испарение, плавление, растворение и т. д.).
В соответствии с законом Гесса и его следствиями мы имеем
следующие методы расчета тепловых эффектов химических реак-
ций
1. Чтобы вычислить тепловой! эффект реакции, пользуясь
данными теплит образования соединений, необходимо п,з суммы
теплот образования продуктов реакции вычесть сумму теплот
образования исходных веществ.
2. Чтобы вычислить тепловой эффект органической реакции,
пользуясь теплотой сгорания соединений, необходимо из суммы
теплот сгорания исходных веществ вычесть сумму теплит сгора-
ния образующихся продуктов реакции.
3. Тепловой эффект химической реакции может быть также
определен посредством алгебраического комбинирования уравне-
ний! отдельных реакций! с учетом их тепловых эффектов (см. гл. X I).
4. Тепловой эффект реакции может быть определен ио кон-
стантам равновесия реакции.
В ряде случаев для расчета тепловых эффектов приходится
пользоваться данными об энергии связи в молекулах реагиру-
ющих веществ. Под энергией связи подразумевается количество
энергии, которое необходимо для разрыва этой связи.
Первый из указанных выше методов расчета тепловых эффек-
тов реакций является основным.
В тех случаях, когда величины теплот образования взаимо-
действующих соединений в справочной литературе отсутствуют,
их приходится вычислять по тсплотам сгорания, которые для
многих органических продуктов определены экспериментально.
Если для какого-либо органического соединения известна те-
плота сгорания //с, то его теплота образования q() (в ккал/моль)
может быть вычислена также по закону Гесса, который в данном
случае можно представить уравнением: г
g0-94.38C-HJU9H-M(Br + Cl-LI |-N-r O)-|-41,4 F -j 69,38-?с (1.6)
где С, Н, Bi и г. д. — числа атомов углерода, водорода, брома и
т. д. в молекуле данного органического
соединения.
Равенство (I. (») справедливо в тех случаях, когда продуктами
сгорания органических соединений являются газообразные СО2,
Cl2, Nl, и SO2, жидкие 1LO и бром, тверды и иод. а IIF находится
в растворе
Если же продуктами сгорания будут водные растворы НВг.
IICI, П\О3 и ILSOj, то определяется ио формуле:
7о = 91,38 с -J- 31.19 11-L 3,63 Вг 3,27 С1 , 0(1-0) +
Н 13,61 N -г 41,4 F — 139,1 S 7с
11
Для большого числа органических соединении. с которыми
имеют дело в производстве, данные относи только тенлот сгорания
отсутствуют в справочной литературе, поэтому эти теплоты опре-
деляются расчетным путем.
Основное положение расчета тенлот сгорания органических
соединений состоит в том, что связь между атомами образуется
всегда за счет не одного, а двух электронов, которые после этого
являются общими для двух атомов
При сгорании углеводорода происходит смещение электронов
от атомов углерода и водорода к атомам кислорода, в результате
чего атомы углерода и водорода становятся электроположитель-
ными и соединяются с электроотрицательными атомами кисло-
рода. Количество тепла, выделяющегося при смещении одного
электрона от атома углерода или водорода к атому кислорода,
составляет 26,05 ккал (теория Караша).
Таким образом, теплота сгорания простейших органических
соединений qc (в ккал моль) может быть вычислена ио уравнению:
*?с = 26,05я (*-7)
где п — число перемещающихся электронов.
Пример. Вычислить теплоту сгорания метана, исходя из уравне-
ния реакции горения*
С11*4-2О2=СО2-F2ILO
При условном изображении электронов точками реакцию горения мотана
можно записать в следующем виде:
11
1ГС-П4-^: :^=л- -с- .(»л_П:О:Н
1 О: :О °- Л" ,(,~Н;():11
II
Здесь все электроны, участвующие в образовании связей между атомом
углерода и атомами водорода, смещаются при образовании новых связей
атомов водорода и углерода с атомами кислорода, т. е. п — 8. Следователь ио
ус= 2(3,U5 • 8 208,4 ккал/моль
что мало отличается от найденной экспериментальным путем величины
210,8 ккал/моль.
Теплота сгорания более сложных углеродов (с двойными
и тропными связями), а также многочисленных замещенных пе
может быть вычислена ио формуле (I. 7), так как но гипотезе
Караша электроны атомов углерода и водорода данных соеди-
нений несколько смещены по отношению к тому положению,
какое они занимают в молекулах простых углеводородов. Поэтому
и теплота сгорания несколько изменяется в большую или меньшую
сторону. Эти изменения учитываются введением так называемых
поправок для всевозможного вида связей атома углерода в моле-
куле, причем каждой такой связи соответствует определенная
поправка, независимая от поправки на другие связи в молекуле
12
(правило аддитивности). Формулу для определения теплоты
сгорания в общем виде можно представить так:
9с = 26,05n-|-^V t \ (1.8)
где £ — число одноименных заместителей;
Д — тепловая поправка на связь с заместителем.
Пользуясь формулой (I. 8). можно вычислить теплоту сгора-
ния большинства жидких органических веществ, если будут
известны числовые значения тепловых поправок Л (табл. I. 1).
В тех случаях, когда требуется вычислить теплоту сгорапия орга-
нических соединений, находящихся в другом агрегатном состо-
янии, следует ввести соответствующие дополните пятые поправки
на теплоты плавления и испарения (парообразования).
С учетом поправки па агрегатное состояние даг теплота обра-
зования веществ будет равна
^о=
Общие формулы тепловых поправок на агрегатное состояние
приведены в табл. I. 2.
Для определения теплот испарения дПС11 (в ккал кг) могут
быть использованы следующие формулы:
Клаузиуса — Клапейрона
. ---1 Р-1
1g----
Кистяковского
(4,575 1g Т -г 8.75)
УИС» —----------у---------
Грутопа
__ JJ- Т к
Viren — Л ПСП "
где plt р., — давление паров жидкости при 7\ и 7’2 ° К соответ-
ственно;
М — молекулярный вес (молекулярная масса) испаряемом
жидкости;
Тк — температура кипения жидкости, °К;
Лисп — постоянная величина, равная 20 22.
Кроме того, для соединений, разлагающихся ниже темпера-
туры кипения (температура кипения неизвестна), рассчитывают
<7нсп (в ккал/кг) по эмпирической формуле:
240о
tfucii —
где о' — сумма валентностей атомов, входящих в состав соеди-
нения.
13
Таблица 1.1
Числовые значения тепловых поправок
Группы, заместители и связи д. Примечание
Связь между алифатическим и ароматическим радикалами Ar—R —3.5 При сгорании атомов углерода, один из которых принадле- жит к ароматическому ради- калу, а другой —к алифати- ческому, будут перемещаться все электроны, имеющиеся в этих атомах
Связь между ароматическими радикалами Аг—Аг Этиленовая связь С=С —6,5 При ci орании атомов углерода, входящих в разные арома- тические ядра, будут пере- мещаться все электроны, имеющиеся в этих атомах. Число тепловых поправок в случае сгорания соедине- ний, представляющих собой конденсированные ядра аро- матических углеводородов, равно числу «спаек» ядер
в г/пс-соединеииях — 16,5 —
в трямс-соединсинях +13 —
Связь между ароматическим радикалом и вшшлыгыми или ацетиленовыми группами Аг—СН=СН2 или Аг—Сг. СН —6,5 —
Двойная связь в цикло СИ а Н2С СП 11 Н2С СП +6,5
Ацетиленовая связь —с=сн + 4 Л —
Группа —С = С—, нс связан- ная с атомами водорода +33,1 —
Связь между первичным али- фатическим радикалом и гидроксильной группой (пер- вичные спирты) R-OII + 13 При сгорании углерода, свя- занного с гидроксильной группой, перемещаются толь- ко три электрона. Не пере- мещается электрон, участ- вующий в образовании сняли между углеродом и кислоро- дом, а также электрон водо- рода, содержащегося в гидро- ксильной группе
14
Продулжение табл. 1. 1
Группы, заместители и связи д, ккал/молъ Примечание
Связь между вторичным али- фатическим радикалом и гидроксильной группой (вто- ричные спирты) R Чсп-оп Связь между третичным али- фатическим радикалом и гидроксильной группой (тре- тичные спирты) R\ R-C-O1I RZ . Связь между ароматическим радикалом и гидроксиль- ной группой Аг—ОИ Алифатические и ароматиче- ские эфиры (Аг) R—О—R (Аг) Альдегидная группа в алифа- тических и ароматических соединениях (Ar) R-CHO Кето-груипа в алифатических и ароматических соединениях (Аг) R-CO-R (Аг) а-Кетокпслоты О R—С^СООН + й’5 4-3,0 +3,5 +19,5 +13 +6,5 + 13 При сгорании углерода, свя- занного с гидроксильной группой, перемещаются только три электрона. Це перемещается электрон, уча- ствующим в образовании связи между углеродом и кислородом, а также элек- трон водорода, содержаще- гося в гидроксильной группе Как и в спиртах, при сгора- нии углерода, связанного с кислородом, перемещаются три электрона В углероде альдегидной груп- пы и кето-группы при сго- рании перемещаются два электрона Тепловую поправку вводят, 0 если группа R —С— связана с группой—СООЦ, в осталь- ных случаях не требуется тепловой поправки на группу —СООН. При сгорании угле- рода карбоксильной группы перемещается только один этектрои, так как остальные уже «затрачены» на образо- вание связи между угле ро- дом и кислородом
15
Продолженис табл. J. 1
Группы заместители и связи л, ккал/.чо-гг> Примечание
Оксикислоты к 4-6,5 Тепловую поправку вводят, R
ХС(ОН)-СООН if если группа ^С(ОП)—свя- R зана с группой —СООН, все сказанное о кетокмелотах справедливо и для окепки- слот
Г руина О II R-C— + 6,5 Тепловую поправку вводят, О (1 если группа R—С— связана с такой же группой, кроме того, вводят две поправки па кето-группу, т. е, +6,5 • 2
Циклопропановые кольца в карбоновых кислотах соон + 13
Циклобутановые кольца в кар- боновых кислотах -С-С- '1 1 . -С—СП—соон + 13 —
Лактоны типа НгС—СП.,-СНо-С=0 1 -о—1 +13 —.
Ангидриды карбоновых кислот О 1 R—С\ О R-C/ II О -10
Сложные алифатические эфиры R—COOR + 16.5
16
П родолжсиис табл. J. 1
Группы, замссгптсли и связи А. КОДЛ .* At 0-<Ь Примечание
Первичные ароматические амины ArNlL Первичные алифатические амины И NI L Вторичные ароматические амины Ar-NH—Аг Вт орпчныс алифатические амины R-NII—R Т ретичиые ароматические амины Аг /N-Ar Аг Третичные алифатические амины К \n-b в Связь между ароматическим радикалом и азотом в аминах Замощенные амиды тина В В—IIN—С=О Связь между углеродом и ннтрилыюи трупной —C = N в алифатических и аромати- ческих соединениях +6.5 + 13 +13 -1-19.5 +19.5 26 —3,5 +6.5 +16.5 В атоме углерода, связанном с аминогруппой, при сгора- нии перемещаются все элек- троны; при подсчете числа перемещающихся электроне в учитываются также элек- троны водородных атомов, непосредственно связанных с азотом. Это же справед- ливо и для вторичных и для третичных аминов Жирноароматические вторич- ные амины рассматриваются как вторичные ароматиче- ские Жпрноа рематические третич- ные амины рассматриваются как третичные ароматпче окне Кроме поправки на соответст вуюгцую аминогруппу (см. выше), вводят тепловые ио правки па каждую связь между ароматическим ради- калом и азотом В атоме углерода, связанного с азотом в нптрильной груп- пе, при сгорании перемеща- ются все электроны
- Л. М Батунер.
17
17 родолжение табл. 1. 1
Группы, заместители и связи Л. ккал/люло Примечание
/Хроматические нитрилы Аг—С = Х —6.5 Вводятся две поправки: па связь между углеродом и группой —C=N и на собст- венно ширильную группу
Алифатические изонптрплы R-N=C 4-33,1 —-
Нитрогруппа — NO2 в алифа- тических и ароматических соединениях 4 13 При сгорании углерода, свя данного с питрогруппой, пе- ремещаются не все электро- ды, а па один электрон меньше. Этот электрон оста- ется при азоте нитрогруппы, который восстанавливается в процессе горения
Сульфо группа —S03II в аро- матических соединениях —23,4 Сказанное о нитрогруппе спра- ведливо и для сульфогруппы
Хлор в алифатических соеди- нениях —7,7 При сгорании в атоме угле- рода, связанном с галогеном, перемещаются не все элек- троны, а на одни меньше
Хлор в ароматических соеди- нениях —6.5 —
Бром и алифатических соеди- нениях 4-16,5 —
Бром в ароматических соеди- нениях —3,5 —
Иод в алифатических и неко- торых ароматических соеди- нениях 4-42 -—
Таб ища 1. 2
Тепловые поправки qac на агрегатное состояние веществ
Исходное вещество Величина gai, для получаемых продуктов
газ (пар) жидкость твердое вещество
Газ (пар) -Сн + Сн исх 4 псп +<?пл4 71(СГ|
Жидкость ~<i. 0
Твердое вещество .... н <п-Cs -оисх ‘ пл
Примечание 'V|tcn — теплота испарении (парообразования). кпол.'люль;
7|К1 — теплота плавления, ккал! моль; верхний индекс «п» относится к получаемому про-
дукту, верхний индекс «исхо —к исходному веществу.
18
Теплота плавления дпл (в ккал кг) находится по формуле:
И" П.Г
У ПЛ А ПЛ м
где /<[1Л — постоянная величина, равная 9 ч-11 для органических
соединении и 5 ч-7 для неорганических соединении;
'/’„.j — температура плав геМпя, ’К
Имеется следующая зависимость:
^-=К
I к
где А' — постоянная, равная 0,58 для органических соединений
и 0,72 для неорганических соединении.
Таким образом:
Vl>r„ 22ГК = 22ГК
10/ пл 10 и,-»Ь Гк
НЛП
Уп.з - 0,20 упсп
Теплота растворения ^раегв (,} ккал кг) находится по уравнению:
4,575 Г, Т., 1g
Ураств •-= (Г* —Л/ "
где С’,. С., — растворимость вещества при температуре 7\ и Т2
ZK соответственно.
Вычислим теплоты сгорания и образования некоторых органи-
ческих соединении.
Пример. Вычислить теплоту образования нитробснзолсульфо-
кяслоты C61J4NO.,SO3H.
Имеем:
« = 4- 44-2--34-4 • 1 = 26
AX0i = 13; ьхо,= 1
Aso,n = —-;Vi; &so»n = 1
Получим:
9с - 26,05-26 4- 1 -13 — 1 -23,4 — 666,9 ккал/моль
Таким образом, теплота образования нитробензол сульфокислоты соста-
вляет:
9О = 95,38-6 4- 34,19-5 4* 69,3-1 — 666,9 = 139 ккал/моль
Пример. Вычислить теплоту образования фенола CeII5OH.
Имеем:
п — 5 • 4 -р 1-3 4- б 1 — а
Аои “ 3,5; =он = 1
о*
19
Следовательно:
fjc = 26,05-28 “ 1-3,5 = 731,5 ккал/лоль
(jo = 94,38-6 + 34.19-6 — 731,5 = 41,5 ккал/моль
Пример. Вычислить теплоту образования бензойной кислоты
С6ПдСООН.
Имеем:
„ = 6-4 -|- 1.1 + 5.1 = до
*ЛГ- А] = —L\r- Al ~1
Таким образом:
<?с = 26,05-30 — 1-3,5 = 778 ккал/моль
5о = 94,38-7 34,19-6 — 778 = 87,8 ккал/.ноль
Пример. Вычислить теплоту образовапия анилина CeII5NiI2.
Имеем:
«=6-44-7-1 = 31
Лхн,=2 +6>5; £XIIj = 1
Таким образом;
ffc = 26,05-31 1-6,5 = 814,05 ккал/моль
9о = 94,38-6 4- 34,19-7 — 814,05 = —8,44 киал/молъ
Пример- Вычислить теплоту образовапия хлорбензола С6115С1.
Имеем:
п =5-441-3+5-1 = 28
Aci“ —6,5; ?с) = 1
Следовательно:
$с — 26,05-28 — 1-6,5 = 722,9 ккал/моль
<7о = 94,38-6 + 34,19-5 — 722,9 = 14,33 ккал/'моль
Пример. Вычислить теплоту образовапия нафталина СщН8.
Имеем:
п = 10-4 + 8-1 = 48
^Аг - Аг — ^6,5, £Аг _ Лг =2
Таким образом:
qc = 26,05-48 + 2 (—6,5) = 1237,4 ккал/моль
qn = 94,38-10 + 34,19-8 — 1237,4 =— 19,0 ккал/моль
Пример. Вычислить теплоту образовапия антрацена С^Н^СН)/^!^.
Имеем:
п = 14-4 + 10-1 = 66
^Аг-А1~ —
Получим;
(jo — 26,05-66 — 3,5-4 = 1705,3 ккал/моль
д„ = 94,38-14 + 34,19-10 — 1705,3 = —42,0 ккал/молъ
20
Пример. Вычислить теплоту сгорания антрахинона
СОЧ
хсо
Имеем:
п =<12-4 < 2-2 8-1 = 60
\\г- AI ~ “8,5: *Лг_ д| = 4
Дсо=’^! есо=2
Следовательно:
дс — 26,05-60 -J- 4 (—3,5) -у- 2-6,5 = 1562 ккал.моль
Определение теплот образования солей с помощью
теплоты нейтрализации. 1. Сульфокислоты ароматических угле-
водородов по своим кислотным свойствам приближаются к таким
сильным кислотам, как IIC1, HNO3 и H2SO4, которые при взаимо-
действии с сильными основаниями выделяют на 1 г-экв водорода
одинаковое количество тепла, равное 13,7 -4-13,9 ккал. Зная
теплоту нейтрализации сульфокислот, можно легко вычислить
теплоту образования их солей.
Примем следующие обозначения теплот (в ккал.'моль):
д.. — теплота образования кислоты;
— теплота образования соли;
?ос» — теплота образования основания;
7н — теплота нейтрализации;
дв — теплота образования воды.
По закону Гесса для реакции образования соли
CBH5SO31I 4- NaOII = CeII5S03Xa + II2O -Ни
имеем
или
«М - ''S = 9|. -t- tfoc,, - <7R = Av
следовательно:
Vm = ^s-I-av
Пример. Вычислить теплоту образования натриевой соли нцтро-
б ензол су л ьфок 11 слоты (!e 114NОsS() s Ха.
Теплота образования нитробепзолсульфокислоты д^ — 139 ккал/моль.
Теплота образования натриевой соли иитробензолсульфокнслоты:
<7-ц 139 57,9 = 196,9 ккал/моль
где ;\д = 57,9 ккал'моль — тепловая поправка (по справочным данным)
2. Теплей а нейтрализации карбоновых кислот близка к теплоте
нейтрализации сульфокислот и равна 12,9 ккал моли.
21
Пример. Вычислить теплоту образования натриевой соли бензой-
ной кислоты (’.eII6COONa.
Теплота образования бензойной кислоты равна 8/,<8 кклл'.моль.
Рассчитываем тепловую поправку, подставляя справочные данные:
\q = qu q0CI| — qu = 12,9 -J- 112,7 — 68,38 = 57,2 ккал-моль
Теплота образования натриевой соли бонюипой кислоты:
87,8 -j- 57,2 145 ккал/моль
3. Гидроксильная группа, находящаяся в ядре ароматического
углеводорода, по своему кислотному характеру соответствует
слабой кислоте, теплота нейтрализации которой равна
—1,3 ккал .моль.
Таким образом можно определять теплоты образования фено-
лятов и нафтолятов.
Пример. Вычислить теплоте образования фенолята натрия
СвН5ОХа.
Теплота образования фенола равна 41,5 ккал/моль.
Тепловая поправка:
\<7 '/и 'Г 7осн — <7н — ' ГЗ -|- 112,7 68,38 - 43 ккал/моль
Теплота образования фенолята натрия:
41,5 43 = 84.5 ккал Смоль
4. Амины ароматического ряда и жнрпоароматические амины —
слабые основания. Нейтрализация их минеральными кислотами
сопровождается выделением тепла. Теплота нейтрализации равна
9,11 ккал!моль.
Пример. Вычислить теплоту образования солянокислого
апцлппа CeHsNII2-НС1.
Теплота образования анилина равна —8,44 ккал/моль.
Тепловая поправка:
Дд == I- 7цс1 — ''в = VI + “ 0 = 31 Л 1 ккал/моль
1де 7нс1 ~ ккал/моль.
Теплота образования солянокислого анилина:
—8,44 + 31,11 — 22,G7 ккал/моль
Программированный подвод или отвод тепла
(термокинетика химических реакций)
Пусть в аппарате периодического действия протекает
процесс при постоянной температуре по схеме:
А > X -НР
Общее количество тепла изменяется во времени в соответствии
с изменением количеств взаимодействующих веществ, которые
могут быть определены па основании законов химической кине-
тики.
Примем следующие обозначения:
а — начальная концентрация пли количество веществ А;
х — концентрация или количество получаемого продукта X
в любой момент времени;
т — время;
т1; — продолжительность процесса;
к — константа скорости реакции.
Для реакции первого порядка, как известно, имеем следующее
кинетическое уравнение:
~=-^к(а-х) (1.9)
Уравнение (I. 9) надо проинтегрировать при начальных усло-
виях:
г = 0 при т=0
Для решения уравнения (I. 9) находим
х — а (1 —
Если т(; — конечный момент времени,
а — значение концентрации для этого момента, то
лкт=«(1—е
Очевидно, что количество выделенного или поглощенного тепла
прямо пропорционально степени превращения Л в вещество X,
и следовательно:
Q — (рк = ср (1 — е ПХ* ) (1. 10)
где гр — коэффициент пропорциональности.
Для любого момента времени т равенство (I. 10) может быть
переписано так:
<?t = <p(l-₽-'tT) (I II)
Разделив (I. 10) на (I. И) и перенеся Q вправо, получим фор-
мулу, выражающую закон тепловыделения и теплопоглощения
в реакционном аппарате:
= Q kx U-12)
1— е к
Поскольку количество теплоносителя или охлаждающего
агента прямо пропорционально величине Q, то расход их должен
меняться во времени в соответствии с равенством:
। - kT
и т II'-- \т (1.13)
1 — е
23
где И7 — расход теплоносителя пли охлаждающего агента на весь
процесс;
IVX — количество теплоносителя или охлаждающего агента,
расходуемого за время т.
Для аппаратов непрерывного действия будем иметь:
где 1К — конечная длина пли высота аппаратов, равная произве-
дению скорости па время.
Пример. Некоторый химический процесс, проводимый в аппарате
периодического действия, протекает согласно уравнению реакции первого
порядка. Начальная концентрация исходного продукта равна единице,
степень превращения 0,96, константа скорости реакции А- — 0,000895 сек-1,
а общее количество выделяющегося тепла (при 96% превращении) составляет
10 000 ккал. Вычислить данные для построения графиков тепловыделения
и расхода охлаждающей воды, если известно, что процесс проводится при
постоянной температуре.
Продолжительность процесса, протекающего по уравнению реакции
первого порядка, может быть найдена по известной формуле:
тв b 2,3031g а_х~' о,()ооз95
1 _
1—0,96
3600 сек
По у равней ню находим значение (в ккал) для отрезков времени, соот-
ветствующих 10, 20. 30, 40 и 50 мин с момента начала реакции:
Qio — 10 000 1- 1- _ <) у । q- — OjOOOfiSS - Й00 9 7 fg-l),0(11)855 3000 = 10 000- 1—0,582 1 — 0,04 — 4350
<>20- 10 000 1 — 1 — 9 у jg-0,000855-1200 9 718 ~ <,,и00й9’'3609 = 10 000 1—0.342 1—0,04 = 6850
(?ЗИ ‘ 10 000 1- 1- 9 У jg-OjOdOSSd 1800 9 718 ” 0,000895'3600 10 000 1-0,2 1—0,04 = 8330
Q ю = 10 000 1 — 1- 9 -|g-0,000s»3-2400 9 71g-0»000895’3600 = 10000- 1-0,116 1—0,04 = 9200
<>30 = 10 000 - 1 1 — 2 7^ ~ 0,000695 ’3000 9 7jg-0,00085u- 3800 = 10 ooo - 1-0,068 1 —0,04 = 9700
Общий расход охлаждающе!! воды:
где г1( = 20° С — конечная температура воды;
/л 12е С — начальная температура воды.
24
Расход охлаждающей воды в каждый данный момент времени опреде-
ляется по формуле ([. 13), соответственно чему можно найти значение ПЛТ
(в кг) для 10, 20, 30, 40 и 50 .пип с момента
начала реакции:
,25ОТЗ^>=857
1 — 0.2
1Узо 1250v =1040
1 —0.04
и- i-i-n 4—0.116 .....
"1-0,04 ^lbU
На основании полученных данных
можно построить графики тепловыделе-
ния и расхода охлаждающей воды. На
рис. 1. 1 представлен расход охлаждающей
воды в соответствии с кинетикой реакции.
Для реакции второго порядка
количество получающегося в любой
момент времени продукта в соответ-
Рис. I. 1. Расход воды, соот-
ветствующим кинетике реак-
ции первого порядка.
ствии с уравнением кинетики бхдет
равно *:
ab(eahx-ebhx)
Х aeukx — bebhr
где а и b — количества или концентрации исходных веществ.
Это значение х подставляется в уравнение (I. 10) и (1. 11):
. / сЛт.. ЬкХ,.\
ab\e l' — e h)
akx,. bkx,.
ае •' — be
ab(e<lhx-ebhx)
Q acahx-bebhx
Таким образом:
( ala l>hx\ f akx,. , Ькх.Д
О fAe ~c /v*c -be ‘J
( akx , bkx\ / al<x,c bkx,.\
—be )\e '—e )
Для аппарата непрерывного действия получим:
( akl bkl\{ akl,. , bhlu\
О — <) yg Afle —be K)
/ ahi г bit! X/ akl,. bhL.X
{ae —be )\c K—e ")
(1.14)
* См- Л. M. 1, а т у ire p и M. E. II о » и п. Математические методы
в химической технике, ГосхиМиздат, 1063, стр. 84.
25
Пример. Рассмотрим кинетические и тепловые соотношения ре-
акции омыления уксусиоэтилового эфира щелочью:
СН3СООСЛ16 -I- NaOH = CII3C00Na -L C2II5O1I
Опытные данные приведены в табл. I. 3
Таблица 1.3
Опытные данные
Время т от начала опыта, лаги Темпера- тура, Количество IICJ (0,01 и.), пошедшее па торможение реакции, .п.г Количество NaOH (0,01 и ), пошедшее на обратное титрование, .ил
10 30,0 10 1,0
20 30.2 10 28
зо 30.1 10 4.0
(>5 29.8 10 4,6 '
80 30,0 10 4,8
3 суток 20,0 10 5,0
Поскольку рассматривается реакция второго порядка, используем для
определения константы скорости реакции формулу:
2.3< 3 (а-х)Ь
т (а — Ь) ® (Ь — х) а
где а = l.j .w.i - начальное количество щелочи;
к = 10 мл — начальное количество эфира;
а — х — количество свободной щелочи;
b — х — количество свободного эфира;
Таким образом:
2.303
т (1— o.oi
lg(),67
а~-х
Ь — х
^-lg0,67
а — .г
Вычисляем к с помощью расчетной табл. 1.4.
Таблица 1.4 \
Расчет константы скорости реакции
т» лил а — х Ь — х , . а — а 0J57 *7 b — х л а — х 0,е' Ь-х Ь, моль-* -Muir'
10 9,0 4,0 1.49 0,17 12*,0
20 7,2 2,2 2,20 0.34 11,9
30 6,0 1,0 4,00 0,60 14.0
65 5,4 0,4 13,50 1.13 12,2
80 5,2 0.2 26,00 1/12 12,5
Среднее значение копегаиты скорости реакции А-Ср — 12,5 моль 1-.иип~1.
Тепловой эфяект реакции:
~ ^ClbCOONa “* 9с,П0ОП ~ ^СТЬСООСДГ» — Я.Х'аОП
где
^CHaCOONa “ ^СНэСООН l?ll + #Х.ЮП — '4
Следовательно:
f/p= ^CsHjOII + ?СН,СООП + 12,9 — 68,38 — ?СН,СООСгТ14
Теплоты образования:
^С1ТаСООСгн4 = 94,27 • с+34,16 И — дс = 94,27 4 + 34.16 -8 — 540 =
= 110,4 ккал/моль
<7стьсоон = 94,27 2+34,16 • 4 —209,4 - 115,88 ккал/моль
?CtH40H = 94,27 -2+34,16-6 — 328 ~ 65,52 ккал/моль
Тепловой эффект реакции:
др =•- 65,52 + 115,88 + 12.9 — 68,38 — 110,4 = 15,52 ккал/моль
Количество тепла, выделяющегося в любой момент времени:
/ ahx bkx\ ( айт,, , дйт.Л
= 1е —е Дае к-5е
^ / айт Ьйт \ / akx , l>hx\
\е и— е к) \ае —be )
Подставляя а = 1,5 Ь, получим:
( 1,5МТ Мт\ . _ 1,5ЬЛт ЫЩ\
О гЛ* ~е Ж1.5е к-<? к) _
' т ' / 1,5Мт,. Ыет.Д . (. _ 1,5 bhx W<t\
(е ‘'—с —с )
Ы'.х / 0,5 bkx Л Ькх.. {. „ 0,5 Мт,. \
е (е —1) 6 1411,5* *-1)
— v МТ / 0.5^/tT.. Д МТ/ 0,5МТ
_е ’Де ь —1/е Ц.бе —V
0,5 Мт__J
Считая на 1 кг уксусноэтилового эфира, найдем:
1000-15,5
^т ya "* 1,5ео,5Ьйт_ । “'J'J i,5e0’’Мт_
Так как
л_.. 0,5-0,01-12.5 АЛ/1.
0,о Ькх =-------------т =•- 0,041л т
1,5
окончательно получим:
0,0-115 Т .
Q . 255 -----------—
Т LSc'M-iu-T-!
Для определения (?т воспользуемся расчетном табл. I. 5.
Па основе полученных данных строим график тепловыделения, соответ-
ствующего кинетике реакции омыления уксусноэтилового эфира щелочью
(рис. 1. 2).
Таблица 1. 5
Расчет
S D,0-'» 1 5 т х «J SO1»3 31 1 'J1 'ЛГ(Г' р 1 О с 7 <а> 7 2
0 0 0 1 1.50 0 0,50 0
10 0.415 0.178 1.50 2.20 0,50 1,25 102
20 0.830 0,360 2.29 3.44 1.29 2,44 135
30 1.245 0,535 3,50 5,25 2,50 4,25 150
40 1.600 0,690 4,<80 7,50 3,80 6,20 157
50 2.080 0,860 7,25 10,90 6,25 9,90 163
60 2.490 1,070 11,70 17,60 10,70 16.60 168
Рис. I. 2. Тепловыделение,
соответствующее кинетике ре-
акции омыления уксусноэти-
лового эфира щелочью (реак-
ция второго порядка).
Термокинетику химических реакций сложного характера рас-
смотрим на примере каталитического разложения серпокислой
соли о-метоксифепилдиазоння.
Синтетическое производство гваякола из о-анн.зидина осно-
вано па следующей схеме:
1) получение серпокислой соли о-анизидипа
28
2) диазотирование сернокислой соли о-аиизидипа
II..SO, 4- 1ЬЬ04-г 2Ха.\О-. —>
SO*"4-Xa.,SO -г-4Н„О
4 -4
3) каталитическое разложение сернокислой соли о-метокси-
фенилдпазонпя
j X „-j-l 1 jSO.j
В качестве катализатора разложения сернокислой соли о-мсто-
кс ифеиплдиазоппя применяется медный купорос.
Пример. Скорость разложения сернокислого о-метокенфеннл-
дпазоипя определялась по объему азота, выделяющегося при разложении
соли диазол ня. Опытные данные при температуре 96° С приведены в табл. I. 6.
Таблица 1. 6
Опытные данные
Время т от начала опыта, мин Количество вызолившегося азота, с.к* Количество азота, приведенного к нор- мальным условиям, см* Степень разло- жения соли Диазовня, % 1<1. .чнн“*
2 2,6 2,5 5,7 0.029
4 5,3 5,0 11,4 0,031
6 8.3 7,9 18.0 0.033
8 11,7 11,1 25.2 0.036
10 15.1 14.3 32.5 0039
42 18,5 17,5 39,8 0,042
14 21.7 20,6 46,8 0,045
16 25.2 23.8 54.1 0.048
18 28.3 26.8 61.0 0,052
20 31,4 29,7 67,6 0,056
24 36,5 34.6 78.7 0,06-4
30 41,8 39,6 90,1 0.076
оо 46,4 44,0 100,0 —
Величина Ai представляет собой константу скорости реакции, вычи<л< и-
ную с помощью уранеипя реакции первого порядка Как следует из табл. 1 б,
зта величина изменяется и не может характеризовать процесс.
29
Используем б данном случае следующее видоизмененное уравнение для
константы скорости реакции, усложненной процессами неизвестного харак-
тера:
2.303 , А В + х'
т (А + В)1% А-х' ‘ В
где т — время от начала опыта;
А — начальная концентрация исходного вещества, т. е. соли диазония
(выраженная в количестве миллилитров азота, выделившеюся при
полном разложении раствора);
В — величина, характеризующая особенности процесса;
х1 — количество вещества, вступившего в реакцию (количество милли-
литров азота, приведенного к нормальным условиям прн т).
Среднее значение константы скорости реакции А-Ср = 0,0022 .чин'1.
Тепловой эффект реакции разложения сернокислого о-метоксифепил-
диазонпя.
?Р -= *HtSO4 + 2?о- % - 2?в
где <?', д0 — теплота образования гваякола и дпазосоедипения соответ-
ственно, ккал/моль.
Таблица I. 7
Расчет константы скорости реакции
MUM В Л 2.3 Л В-Ьх' 1в Л . в±^- Л, JHtW1
Т(А-Ц) А — X' В G А —X' В
2 И 44 0.0210 2,5 1,30 0,114 0,0024
4 11 44 0,0100 5,0 1,64 0,215 0.0022
6 11 44 0.0070 7,9 2,09 0,318 0,0022
8 И 44 0.0050 11.1 2.73 0,436 0.0022
10 11 44 0,0040 14.3 3,40 0,532 0,0021
12 и 44 0.0035 17,5 4.30 0.633 0,0022
14 11 44 0,0030 20.6 5,40 0.732 0,0022
16 и 44 0.0026 23.8 6,90 0.840 0,0022
18 11 44 0,0023 26,8 8.78 0,950 0,0022
20 и 44 0,0021 29,7 11,37 1,056 0.0022
24 11 44 0,0017 34,6 19,40 1,288 0,0022
30 11 44 0.0014 39.6 46.00 1.662 0.0023
Для определения теплоты образования диазососдписпия напишем урав-
нение реакции диазотирования о-аппзпдппа:
ОСИ,
4-2H.,SO.,-|-2XaN().»
ОСП*
ОСН,
Теплоты образования по справочным данным (в ккал/моль) ^uNO. “
= 83,2; = 326; ?в = 68,38; ffHjS0< = 192,2.
Теплота образования о-анпзпднпа:
п- 4-7 + 9 — 2 — 35
ЛОСН,
7с 26.05 35 г 26 — 937,75 ккал/моль
сд>-= 94,38-С + 34,19 II —?(5 94.38-74 34,19 - 9 — 937,75 - 30.62 кка i/моль
Для дпазосоеднпснпя имеем:
п -- 4-14 + 14-1 — 4 = 66
В справочной литературе нет тепловой поправки па азогруппу, поэтому
определяем ее с помощью экспериментальных данных следующим образом.
Рассмотрим суммарную реакцию диазотирования анилина с последующим
сочетанием:
2C6ILN1L- Na\O.,+ НС1 -> СеН5К = .XCeII4ML + NaCI4 2HsO+gp
Тепловой эффект реакции получения п-аминоазобепзола эксперимен-
тально определен В. В. Свептославскпм: сщ = 43,2 ккал/моль.
Теплота сгорания п-амнноазобепзола равна 1574 ккал/моль.
Теплота образования п-ампноазобензола:
до = 94,38-12 4- 34,19-11 — 1574 — —65,35 ккал/моль
Теплота образования анилина равна —8,44 ккал/моль Таким образом,
тепловой эффект реакции образования п-аминоазобензола:
= [-65,35 4- 97,7 + 2-68,38] - [2 (-8,-44) + 83,2 {- 39,3] =
— 63,5 ккал/моль
Следовательно, тепловая поправка на азогруппу:
63,5 — 43,2 = 20,3 ккал/моль
Сумма тепловых поправок для дпазососдинения:
20,3 + 19,5-2 = 59,3
Теплота сгорания
дс — 26,05-66 + 59,3 = 1778,6 ккал/моль
Теплота образования дназососдппения:
- 94,38 14 34,19 14 + 69,3—1778,6 90,6 ккал/моль
Теплота образования гваякола:
п — 4 7 + 7 — 3 = 32
ЛОСН, ж 10,5’ дон = 3,5
qc = 26,05-32 4* 23 = 856,6 ккал/моль
и
д'о — 94,38 • 7 4- 34,19 8 -856,6 77,6 ккал/моль
I силовом эффект реакции:
Чр = (192,2 ! 2-77,6) - (90,6 + 2-68 38) = 120 ккйл.'.ноль
31
При использовании формулы (1. 14) примем:
а = 5,оЪ
где а — начальное количество воды;
Ь — начальное количество диазососдннения.
Тогда будем иметь:
р.5 bkx_ebkr) b (5>5gS,5bhXb-^kT^
( о.б bhx,. Ькх.Л { h.ibkx Ъкх\
(с ,ч — с ^)Ъ\Ь,эе —е )
f мин ння о-метокепфенилдиазония (реакция
сложного характера).
Таблица 1.8
Расчет Qx
т, .KUM 0.(11 4 X ]g е0.014 т с0,014т 0,5еО><’14т е0,П14 г_ । 5.5 е<6014 T_j # v кка-i /кг
0 0 0 1 5,50 0 4.50 0
5 0.070 0.0304 1.075 5,61 0.075 4 61 83.4
10 0,140 0,0606 1.165 6,40 0,165 5,40 166,0
15 0.210 0.0910 1.230 6.75 0.230 5,75 217,0
20 0.280 0.1230 1.330 7,30 0.330 6,30 284,0
25 0.350 0.1520 1,420 7,80 0,420 7,80 335,0
30 0,420 0.1850 1,580 8.45 0,530 7,45 386,0
Па основе данных табл. I. 8 строим график тепловыделения, соответ-
ствующего кинетике реакции разложения о-метоксифепилдиазония (рис. 1. 3).
Глава II
Кинетика гомогенных
химических реакций
и процессов
в проточных аппаратах
Материальный баланс проточных
систем
В условиях проточных систем закон сохранения мате-
рии, как известно, может быть выражен простым соотношением:
Приход = Убыль 4* Накопление
Для химических систем, находящихся в движении, этот закон
выражается с помощью бесконечно малых величин в виде диф-
ференциальных уравнений, где в качестве переменных служат
концентрация, время и положение массы в пространстве.
При установившемся состоянии в любой точке аппарата кон-
центрация не зависит от времени; в этом случае рассматриваются
только две переменные.
Пример. В аппарате содержится Fo л раствора с концентрацией
реагента Со. Другой раствор с концентрацией С\ поступает в реактор со
скоростью u'i; жидкость выходит из аппарата при скорости и>2. Масса в аппа-
рате хорошо перемешивается, так что концентрация распределена равномерно
и ее значение в потоке у выхода такое же, как в баке.
Кинетическое уравнение реакции:
где г — скорость химической реакции;
k — константа скорости реакции;
п — порядок реакции.
Для материального баланса за промежуток времени с?т получим:
Приход —
Убыль = Cw2dr 4- rVrfr = (Cu?2 AVCn) dr
Накопление d (CV) = (Fo 4- (tz’i ^2) T1 dC -|> C (u>t — u>2) dr
Таким образом
Cju?! dr = (c’u?3 + kVCn) «т + [Vo + (и?! — u’2) r] dC -f- С (и?х — u>2) dr
3 Л. M. Еатунер.
33
Преобразуя, получим:
dC __________________
dx Vo + (m>j — tr.2) т Го 4- (u^ — w2) x
При Ci = 0 равенство (П. 1) приводится к уравнению Бернулли; после
подстановки z = С1~п найдем
(П.1)
A _L_ _ = (1 _,А
dx ' + U 4
Эго уравнение линейное с постоянными коэффициентами и легко решается.
При реакции первого порядка и — w2 будем иметь:
C^ — jw 4- feV0) C’q = w -Р A-Fo
ClW - (w Д- ЛУ0) С Ио
Пример. Реакционная смесь протекает в трубчатом аппарате
сечения 5 при постоянной скорости и>. Вследствие концентрационного гра-
диента здесь имеет место также диффузия в направлении оси аппарата в соот-
ветствии с законом Фика:
Скорость — — DS -Я-
аЬ
где D — коэффициент диффузии;
L — длина аппарата;
S — площадь.
При установившемся состоянии:
1 [риход = wSC dx — DS dx
Убыль = wS (С 4- dC) dx—DS
dx
Накопление — —kCn dV dx -—kCn S dL dx
Следовательно:
wdC — Dd (~}-kCndL = Q
\dL J
или
D w № 1 k^n — О
D -d&~W 'dL^^ "°
Для случая, когда n = 1, получим линейное дифференциальное уравне-
ние второго' порядка, решение которого
/1с" т?!,Л4-2?с_’",£
где mi и т2 — корни характеристического уравнения
Dm-— wm -р к — О
Величины А и В являются постоянными, которые могут быть определены
из начальных условий.
34
Батарея реакционных аппаратов
с мешалками
Графический метод расчета
Эффективность работы реакционной батареи зависит
от числа ступеней, размеров отдельных ступеней и от интенсив-
ности перемешивания. Если переметивание достаточно, то кон-
центрация во всем объеме реакционной массы для данной ступени
равномерна и на выходе она такая же, как внутри аппарата.
Рис. II. 1. Схема для расчета батареи реакционных аппаратов.
Обращаясь к схеме, изображенном на рис. II. 1, составим
материальный баланс применительно к m-му реактору:
= С mCm “F ~|---—•
ИЛИ
Скорость прихода = Скорость убыли + Скорость накопления
где F — объемная скорость потока;
V — объем аппарата применительно к одной ступени;
С — концентрация;
г — скорость реакции.
При стационарном режиме объем реакционной массы посто-
янен и скорость накопления равна нулю. Таким образом, имеем
m __
dr
Для равномерного потока F:
r7>i~ р Ст-1 (И- “)
v т г т
Скорость реакции может быть выражена, как функция кон-
центрации одного из реагентов, т. е.
r=kf(C) (II. 3)
или для отдельных ступеней:
rm—kf(Cm) (II. 4)
3* 35
Учитывая данную концентрацию при поступлении массы в тот
или другой аппарат батареи, можно получить путем совместного
решения (II. 2) н (II. 4) значение концентрации для соответству-
ющей ступени.
Прежде чем перейти к рассмотрению этого решения, поста-
раемся получить уравнение (II. 2) другим путем. Так как скорость
реакции в аппарате т постоянна, то ее можно представить как
отношение конечного приращения концентрации и времени.
Средняя продолжительность пребывания массы в аппарате т
равна:
В течение этого
изменяется от Cm_i
равна
Г,п= ” A? Vm/Fm_x ~
что идентично уравнению (II. 2).
Для графического расчета запишем уравнение (II. 5) в таком
виде:
промежутка времени концентрация массы
до Ст. Следовательно, скорость реакции
ЛС1 Ст —Стп-i Ст—1 — Ст /т»
Применительно к концентрации Gn-i у входа в аппарат урав-
нение (II. 6) представляет линейную зависимость между концен-
трацией па выходе и скоростью реакции. Прямая линия пересе-
кает абсциссу в точке Ст_г и имеет наклон —l/0w. Однако не все
значения гт и Ст находятся между собой в таком соответствии,
а только те, которые также отвечают кинетическому уравнению
(II. 4). Таким образом, точка пересечения этой линии с кривой,
изображающей зависимость скорости реакции от концентрации,
определяет Ст. На рис. II. 2 показан ход решения. После того
как определена, индексы в уравнении (11.6) повышают на 1
и повторяют графическое решение с целью определения кон-
центрации Ст+1 на выходе в следующей ступени.
Если размеры аппаратов одинаковы, то продолжительность
пребывания материала в пих одна и та же и прямые линии, пред-
ставляющие уравнение (II. 6), параллельны.
Этот метод применим только в тех случаях, где скорость реак-
ции выражается как функция одной переменной.
Для сложных реакций используется аналитический метод.
Пример. Реакция
2А С + D
36
должна быть проведена в батарее реакторов непрерывного действия, снаб-
женных мешалками. Объемная скорость потока 100 мя/ч. Начальная кон-
центрация вещества Л составляет 1,5 к.ноль/.иЛ причем концентрации веществ
С и D в исходном растворе равны нулю.
Коэффициент скорости прямой реакции 10 л3/к.ноль -ч, константа термо-
динамического равновесия Яр = 16,0.
Для случая, когда желательно достигнуть 80% равновесного превра-
щения, определить:
1) объем реактора Гг (если применяется реактор);
2) число аппаратов, расположенных в батарее последовательно, если
объем каждого из них равен 1/10 объема реактора, рассматриваемого при-
менительно к варианту 1.
Рис. II. 2. Ход расчета батареи реакционных аппаратов
с мешалками при графическом методе.
Обозначим через з-р равновесное количество продукта реакции С или D.
Тогда будем иметь
откуда
Далее:
ХР
Ар (1,5 —2гр)3
= 0,667.
= 16,0
х» — 0,8-0,667 = 0,533
Cai = 1,5 - 2-0,533 = 0,433
Кинетическое уравнение имеет следующий вид:
_«к=/£(с«-_ «Л) = 1О Г(1,5_2
Значения, соответствующие Са = 1,5 — 2х и — dCJdx, изображены
на рис. II. 3.
Для единичной ступени
.2
16 J
г —10 (1,5-2-0.533)2 - -^-7
АС1 = АС _ 100(1,5 — 0.433)
АТ р т1/ Zj I' у»
откуда
Гг=62,7 .и3.
Если Vr Для каждого аппарата батареи составляет 62,7*0,1 =а 6,27 ,н3,
то наклон линии материального баланса на рис. II. 3,6 равен:
Рис. II. 3. Ход расчета:
а — реактор (единичная cry ноль); б — батарея аппаратов.
Интерполируя применительно к Са — 0,433, найдем число ступеней
(аппаратов):
„ , 0.52 - 0433 _оп
3 1 0,52 — 042
Для единичной ступени в дополнение к предыдущему расчету объема
аппарата на рис. II. 3, а изображено построение, из которого находим зна-
чение наклона
т — —1,60
и
т, 100 .... ,
V,- = — = —-= 62,;> .'I3
in l,b
.что хорошо совпадает с первым результатом.
Аналитический метод расчета
Напишем уравнение (II. 6) в виде
Cm-!-— Cm—i (П. 7)
Это уравнение может быть решено методом итерации*. Возьмем,
например, кинетическое уравнение первого порядка: г = кС.
Тогда
Ст -р ЛОщСтп = Cm (1 — ' С m-i (II. 3)
* См. Л. М. Б ат у пер, М. Е. П о з и и, Математические методы
в химической технике, Госхимиздат, 1963, гл. XX.
38
или
г __ CWi-i
Ст~ Ц-АОт
и в частности
Со
Со
14-ло.» (H-A-ej (ц-л-е2)
и т. д.
Если продолжительность пребывания массы в аппаратах оди-
накова, то
С - С°
Ст- (1 + Л-9)“
Для кинетических уравнений не первого порядка, например
для г = кС'-, будем иметь:
или _______________
„ —1 — j/* 1 ~т~'^A'OntC‘m 1
Таким образом
„ -1 i М т 4*0,6',,
61 _ 2А-ех
И г. д.
Более сложные процессы могут быть также обработаны ана-
литически. Пусть, например, имеем ряд реакций
Л1 Л1
Л —► В+С; В4-С-> D
причем СЬо — Ссо. Кинетические уравнения:
га = к}Са
ГЬ = ^1^(1
Соотношения между концентрациями в последовательных сту-
пенях (аппаратах) получаются путем подстановки в (П. 7). Таким
образом:
//- \ (Cft)m-1
, > n—
1 + AxUm
(Q)?n 4- [A-2 (Ct,);n- A-! (Ca)m] 0wt = (Cb)^
39
В соответствии с (II. 9) все значения (Со)?71 могут быть выра-
жены через начальную величину:
(См)т —
(Ср)о
(1+^0)т
(П. 10)
Значения, вычисленные с помощью (II. 10), подставляются
в ряд уравнений для Ств, из которых первым является следу-
ющее:
(п.и)
Бензол
14 мл/мин
1007. С6Нб
Кислота 48ми/лин
8.01 мол. % HNO3
7,21 мол. 7. HNO,
90.05 мол. %С6Н6
0,2503 моль
c6h5no2
Рис. II. 4. Схема материальных потоков
процесса нитрования бензола.
Уравнение (11.11) легко
решается относительно
(G>)jl-
Пример. Нитро-
вание бензола нитрующей
смесью из водных растворов
азотной и серной кислот было
проведено в пятиступенчатом
реакторе с мешалкой. Условия,
при которых протекала реак-
ция, даны на рис. П. 4. Темпе-
ратура 30° С поддерживалась
с помощью водяной рубаш-
ки. Объем этой системы во
время реакции оставался по-
стоянным. В соответствии
с указанными па схеме пото-
ками водная фаза составляла
31,414 моль! л смеси, а органи-
ческая фаза — 2,5151 моль/л
смеси.
Объем массы для каждой ступени равен 103 лм. При данной концентра-
ции серной кислоты скорость реакции определяется концентрацией азотной
кислоты в водной фазе и концентрацией бензола в органической фазе. На
рис. II. 5 представлены результаты опытов, выполненных в системе аппара-
тов периодического действия с концентрацией серной кислоты 20,5 мол. %.
Определить к. и. д. ступени и сравнить работу этого реактора с про-
ведением процесса в единичной ступени.
Пусть А представляет азотную кислоту, а В — бензол. Продолжитель-
ность реакции в каждой ступени:
п W3 . .....
U —ГГ-=1>Ь61 .WHW
62
Конечные концентрации даны на рис. 11-4, а их значения для отдель-
ных ступеней найдем методом подбора.
В ступени 1:
С(_, 1 = 90,05 мол. %
Сдо = 8,01 мол. % — 0,0801-31,414 = 2,5162 моль/л смеси
Принимаем:
Са 1 = 7,80 мол. % — 0,078-31,414 — 2,4503 моль/л смеси
По рис. II. 5;
п — 0,0422 моль/л-мин,
П0 = 0,0422 -1,661 = 0,0701 моль/л
40
Таким образом, имеем
Саг = Сао - гх0 = 2,5162 - 0,0701 = 2,4461
Эта величина нс согласуется с принятой выше концентрацией Са j =
= 2,4503. Для других проб расчеты представлены следующим образом:
примем Cai- 7,79
или 2,4472
fl--- 0,0419
^0 = 0,0696
Са j = 2,4466
примем 7,78
или 2,4440
0.0416
0,0691
2,4471
мол. %
моль/л смеси
моль-л - ашн
моль/л смеси
моль/л смеси
Интерполируя, получим Сп1 = 7,788; примем 7,79 мол. %. Аналогично
найдем:
... 2,2648 + 0,0(596 1П_ Q ft, „
Съ 2 = '--9 51-5|----*00 92,8 мол. % оензола
Результаты вычислений для остальных теоретических ступеней при-
ведены в табл. И. 1.
41
Таблица II. 1
Результаты вычислений
Сту- пень Азотная кислота Бензол Конвер- сия, .моль/ л смеси Нитро- бензол, .мОЛЬ; .г, смеси
водная фаза, мол. % моль/л смеси скорость реакции г, .моль/л-лшн органиче- ская фаза, мол. % .4 ОЛЬ 'Л смеси
0 8,01 2,5162 — —— —. —
1 7,79 2,4466 0,0419 90,05 2,2648 0,0696 0,2503
2 7,59 2,3846 0,0373 92.82 2,3344 0,0620 0,1807
J 7,41 2,3180 0,0341 95,56 2,3934 0.0566 0,1187
4 7,24 2,2656 0,0315 97,80 2,4530 0.0524 0,0621
5 7,09 2.2165 0,0294 99,88 3,5054 0,0491 0.0097
6 — — — 100.0 2,5151 — 0
Для пятой теоретической ступени из расчета имеем Саь = 7,09 мол. %,
что меньше измеренного значения 7,21.
Следовательно, число теоретических ступеней равно:
,v/. । Са 4 — Сп г,_Л t 7,24 — 7,21
7,09
а ,1 'а 5
Таким образом, общий к. и. д. реакционной ступени составляет:
4,2
100 = 84%
O»v
Для того чтобы сравнить единичною ступень с реактором, следует рас-
считать объем аппарата, который необходим для получения той же кон-
версии ц:
11 = (0,0801 — 0,0721) 31,414 = 0,2513 моль/л
Са ~ 7,21 мол. %
Q, = 90,05 мол. %
г = 0,029 моль/л• мин
0 = 5-1,661 = 8,305 мин
Следовательно, полезный
-2L
гб
ооьем одноступенчатого аппарата равен
0,2513 .
8,305-0,029 " '°4
в то время как при пяти ступенях имеем:
5-0,103 = 0,515 л
При проведении сложных реакций распределение продукта
в аппаратах зависит от метода работы и может повлиять па его
выбор. Ниже исследуется в этой связи сложная (деградирующая)
химическая реакция.
42
Сравнительная оценка эффективности
периодической и непрерывной работы
аппаратов для сложных
(деградирующих) химических реакций
При проектировании производственных процессов
химической технологии на основе лабораторных работ возникает
вопрос о том, как должен быть организован процесс — периоди-
чески или непрерывно. В последнем случае необходим еще выбор
реактора, который может быть трубчатым или в виде батареи
аппаратов с мешалками.
Мы будем решать так>ю задачу: какой процесс дает наиболь-
ший выход ценного продукта?
В производстве органических продуктов выход часто является
наиболее важной оценкой при сопоставлении нескольких ме-
тодов. (
В любой части реакционной массы, проходящей через труб-
чатый аппарат, имеет место непрерывное изменение состава —
такое же, как при периодическом процессе. Однако элементарные
слои жидкости на различных расстояниях от оси трубчатого аппа-
рата имеют различную скорость движения массы, и здесь может
происходить молекулярная диффузия от одной части потока
жидкости к другой. Поэтому не все молекулы имеют одинаковое
время пребывания в аппарате. В реакционной среде около оси
аппарата реакция может протекать недостаточно полно,
а жидкость, движущаяся у стенкп, реагирует долго, особенно при
деградирующих процессах в ламинарных потоках.
В условиях турбулентного движения продолжительность пре-
бывания реакционной массы в трубчатом аппарате невелика,
поэтому справедливо предположение о том, что в данном случае
жидкость имеет «поршневое» движение. В турбулентном потоке
процессы, идущие в трубчатых аппаратах, по своей химиче-
ской кинетике подобны соответствующим периодическим про-
цессам.
Совершенно иная картина наблюдается в реакторах непре-
рывного действия с мешалками. Если перемешивание достаточно
эффективно, то состав реакционной массы у выхода будет таким
же как внутри аппарата Поэтому стационарные концентрации
реагирующих веществ здесь значительно ниже, чем соответству-
ющие средние концентрации в аппаратах периодического действия
или трубчатой системе.
Выравнивание концентрации в результате перемешивания
непрерывно поступающих компонентов с реакционной массой
в аппарате вызывает уменьшение градиентов концентрации н,
следовательно, снижение скоростей массообмепа и химической
реакции. Так, например, если смешиваются равные объемы реаген-
тов А и В, имеющих концентрацию 10 единиц, то в начальный
43
Рис. II. 6. Зависимость
концентрации реагентов
при их перемешивании
в аппарате периодиче-
ского действия от вре-
мени.
момент их взаимодействия концентрация каждого из них равна
5 единицам и после этого она постепенно падает (рис. II. 6). Пусть
продолжительность процесса такова, что конечная концентрация
составляет 0,5 единицы, т. о. выход 90%.
Если аппарат непрерывного действия с мешалкой работает
с такими же начальными растворами и достаточно велик для того,
чтобы дать такую же степень превращения, то концентрации
на выходе должны быть 0,5 единицы, и эти значения будут в любом
месте аппарата при условии совершенного перемешивания в нем
реакционной массы.
Таким образом, в то время как реак-
ция в аппарате периодического действия
и трубчатом аппарате протекает с посте-
пенным падением концентрации от 5
до 0,5 единицы, реактор непрерывного
действия с перемешиванием работает все
время при концентрации 0,5 единицы
(точка Q на рис. II. б). Начальные рас-
творы А и В смешиваются с реакционной
массой, состав которой соответствует
точке Q, сразу же при поступлении в аппа-
рат и никогда не достигают более высо-
кой концентрации.
Средняя концентрация реагентов и
средняя скорость реакции в аппарате не-
прерывного действия с перемешиванием
намного меньше, чем в реакторе пери-
одического действия и трубчатом аппа-
рате при одинаковых условиях питания
ц степени завершенности реакции. Поэтому объем аппарата не-
прерывного действия с перемешиванием должен быть увеличен
по сравнению с реакторами периодического действия и трубчатым
для одного и того же выхода продукта. Для уменьшения этой
разницы удобно пользоваться двумя или большим числом аппа-
ратов, расположенных последовательно; в этом случае происхо-
дит ступенчатое падение концентраций.
Деградирующими реакциями называются такие химические
реакции, при которых ценный продукт перерождается тем или
иным путем в балластный материал. Так, например, при хлори-
ровании бензола по реакции
r k k
CBII6 -!-> С6ЩС1 — > c6h4ci2
мы можем, наряду с цепным продуктом монохлорбензолом, полу-
чить также ди- и трихлорпроизводные.
При синтезе важною продукта формальдегида может про-
исходить перерождение его в углекислый газ.
В общем виде такие реакции могут быть представлены уравне-
ниями:
А + В —> X
Х + В —> Y
А + В X
Пример. Для последовательно протекающих реакций
А Л-.> в ±L_> с
имеются следующие численные значения:
А'х = 0,35 «Г1; А-2 = 0,13 ч-1
Са о = 4 кмоль/.4s; СЪ 0 = Сс о = 0
Найти максимальную концентрацию вещества В, которая получается:
1) при периодической работе реактора;
2) для одноступенчатого реактора непрерывного действия с мешалкой;
3) для двухступенчатого аппарата непрерывного действия с мешалкой.
Кинетические уравнения:
—k С
dx -hiCa
rb=--=ВД-А-1Са
Решение (II. 13) найдем, учитывая, что СЬо— 0. Таким образом*:
кСао (с~Л|Т -e~ktX)
к* — ку
Дифференцируя (И. 14) для максимума, получим
ЛСь_= КСдр (_А -й,т+/ -fe,T)
dx кп — к.
СЬ=
го =
(И. 12)
(П-14)
(11.13)
откуда:
In (ki/k2) ___ In (0,35/0,13) 55
Т" Ач-А’г
0,35-0,13
(ео,13 • fl,65 _ е0,ЗБ 4,65) = 2,23
1. Следовательно:
/z> v _ 0,35-4
(Самане—0>35_ 0Лз
Для исследования работы одноступенчатого реактора непрерывного
действия подставим уравнение (II. 12) в (II. 9), а уравнение (II. 13) в (II. 8).
Получим:
f. _ сао
1+М
Сь 1 + (A'jCb J — kiCa i) 9 = Сь о “ 0
* См. Л. М. Б а т у не р и М. Е. П о з и п, Математические методы
в химической технике, Госхимиздат, 1963, гл. III и V.
45
илп
Cbl =
_____*1бд qQ_____
(14-*i0)(14-*26)
(П.15)
ри дифференцировании для максимума будем иметь
(1+М) (1+*20) -е [*! (1 fc20)+к2 (14- лхб)] = о
откуда:
°=/^=/оет=«
2 (Сь } - 0>35-4-4,7
й' '‘С'С i/макс — 77 "i1 'л or—/л , г. .—7~~~ = 1,54
(14-0,35 • 4,7) (1 4-0,13 4,7)
Рис. II. 7. Интерполяция расчетных значений
для любого .числа ступеней реактора непрерыв-
ного действия с мешалкой.
.. Для рассмотрения работы двухступенчатого реактора' непрерывного
действия снова применим уравнения (II. 8), (II. 9) и (II. 13):
Съ 2 (1 4- *20) - =____ куСа °0
<1 + *1б)2 (1-г*10)(14-*2О)
илп
с. __ *1^аоО[24-(А 4-*а)0]
2 [(1Н-*10)(14-М)]2
Максимум найдем методом подбора следующим образом*
О 0 0,5 1,0 1,5 2,0 2,5 3,0 5 10
Cbt. 0 1,003 1,500 1,725 1,815 1,820 1,782 1,505 0,892
откуда следует, что
(б1>з)макс — 1,82
При использовании только бесконечно большого числа ступеней в реак-
торе непрерывного действия можно достигнуть такого же максимального
46
выхода промежуточного продукта В, как в аппарате периодического дей-
ствия.
Полученные в результате расчета значения могут быть интерполированы
для любого числа ступеней, как показано па рис. II. 7. Здесь максимальная
концентрация изображена относительно обратной величины числа ступе-
ней 1/т-
В том случае, когда желательно получить максимальную концентрацию
вещества В. реактор периодического действия является наиболее эффектив-
ным для использования его при осуществлении сложного процесса.
Неу становивш ееся состояние
в батарее реакционных аппаратов
Если батарея реакционных аппаратов включается
в работу или выключается, а также если в пей совершается переход
к другому режиму, то концентрации в отдельных реакторах изме-
няются с течением времени.
Часто представляет интерес
значение продолжительности
этого состояния для пере-
хода к установившимся усло-
виям работы. В другом слу-
чае бывает необходимо опре-
делить, как долго изменяются
концентрации в отдельных
реакторах, чтобы достигнуть
же л ател ьпых к онцент р аций.
Такого рода переменные
или переходные условия опи-
сываются уравнением, пред-
ставляющим материал ьпы й
баланс m-го реактора (см.
рис. II. 1). При использова-
нии и постоянном потоке
между ступенями оно может
быть представлено так:
AC т I Ст ,
Ст~1
(И. 16)
Уравнение (II. 16) было
6
Вне. II. 8. Степени приближения пере-
ходного состояния к условиям устано-
вившегося процесса (батарея рекциоп-
ных аппаратов).
решено в общем виде для многих
случаев реакций первого порядка.
На рис. II. 8 показаны степени приближения переходного
состояния к условиям установившегося процесса для необрати-
мой реакции первого порядка с нулевыми начальными концен-
трациями во всех реакторах, а также с одинаковыми значениями
продолжительности реакций в аппаратах.
47
Пример. При 0 = 40 мин, к — 0,4 и с числом реакторов,
равным 4, требуется найти время для выхода реакционной массы из послед-
него аппарата с получением 90%-ной степени приближения к концентрации
при установившемся режиме.
По рис. II. 8 имеем
(1-F А-0) V (1 -4-0,4 -10) т
откуда
т= 12,8 мин.
Прн более общих условиях может быть составлена и решеиа система
дифференциальных уравнений, подобных (II. 16), для каждого реактора
в батарее.
Рассмотрим случай для трехступенчатой реакционной батареи, где каж-
дый реактор наполнен раствором, имеющим свою концентрацию. Пусть Со —
концентрация раствора при его поступлении в батарею, а х, у и z — концен-
трации в отдельных реакторах. Для первого реактора уравнение (II. 16)
примет следующий вид:
dx . х Сп
~dr + им?)
или
где а = A-j 1/0г.
Аналогично для других реакторов:
^+^-5- (И-1ЭД
dz и
= f (U.20)
Мы получили линейные дифференциальные уравнения с постоянными
коэфф ициентами.
Решение (II. 17);
Подставляя в (II. 19), получим:
dy
dx
gp
0i02tz
Л1
e,
(H. 21)
Решение (II. 21):
Co ,
' 0o (i> — a)
0 =
в-вт+ЛГИ
. __gp Ay
48
Для последнего реактора получим:
। ______________I_____р - Ъг-Л2 р-Ъх
dx 1 О^ОзаЬс (М)3(Ь-п) 1 03
- —___£о__ '__________dl________с~ах-I- —Л'г е~bv -L-
GjOjOgabc О.»О8 (& — а) (с — а) 03 (с— Ь)
л _.________Со______________________________^2
3 “° О^О^Ьс 0.2U3(t —n) (с-«) 03(c —t)
При равенстве между значениями 0 и А для всех ракторов имеем а =
— Ъ = с и в этом случае решения будут:
.. С$ । / В\Х
J \ О
-г #2) е"ат
Со . ( Ihx2 , В3т ,
= 0»а3 \ 20* "г 6
В —V -С°
-~Уо 02«2
р —z Сп
° езйз
Для реакций выше первого порядка формального решения дифферен-
циальных уравнений нс существует. Пригодные для этого случая способы
решения иллюстрируются в следующем примере.
Пример. Реакция второго порядка проводится в двухступен-
чатой батарее аппаратов. Пусть х и у представляют концентрации в реакто-
рах. К началу процесса они содержат растворы с концентрациями Со =
= = уо = 1; такая же концентрация и в поступающей жидкости. Реакторы
одинаковых размеров, причем 0 = 1 н к = 0,5.
Кинетические уравнения:
dx
dr
4-x4-O,5zs =
1
dy , , Л„ о
^•+у+0лу-=«
Уравнение (II. 22) может быть записано так:
f dx
J 1 — а: — 0,5 а:2
(11.22)
(И.23)
(Н. 24)
Интегрирование производится непосредственно. Результаты изображены
на рис. (II. У).
4 Л. М. Батунср.
49
Уравнение (II. 23) должно решаться приближенными методами *. Чтобы
получить данные для подстановки в ряд Тейлора, найдем:
^о = 1
хо I *0 0,5 х' = 0,5
Рис. II. 9. Результаты вычислений:
1 — первая ступень; 2 — вторая ступень.
*оа) = 4" -а'Х" - (го)Е - Ч'Ч = 1 >5
Уо = 1
Уо=хо-Уо--°’5?о=-0.5
^ = Ч-Уо-^о = О>5
Ч" = Ч “ У0У0 “ ( У'о)2 0,25
У4>=Ч ” ~ Ч'' - мГ - (Ч)2 - 2ЧЧ = ~ 1
Принимая Л = Дт = 0,1, будем иметь
01=1 + 0,1 (-0,5) + -^ 0,5+ 0,25 + ^1 (-1)+ . . . =0,953
= 0,962 — 0,953 - 0,5 • 0.9532 ==—0,445
У.> ~ у о + 2ky'i =1 + 2 • 0,1 (—0,445) = 0,911
у', = 0,928-0,911 -0,5 0,911s = -0,398
г/з = 0,953 + 2 0,1 (— 0,398) = 0,873
и т. д.
* См. ,1. М. Б а т у и е р и М. Е. II о з и н, Математические методы
в химической технике, Госхимиздат, 1963.
59
Результаты вычислении представлены в табл. II. 2 и изображены на
рис. II. 9.
Таблица II. 2
Результаты, вычислений
т зс V -У'
0 1,000 1.000 0,500
0,1 0,962 0,953 0,445
0,2 0,928 0,911 0,398
0,3 0,896 0.873 0,358
0,4 0,869 0,839 0,321
0,5 0,846 0,809 0,290
0,6 0,829 0,781 0,257
0,7 0,815 0,758 0,230
0,8 0,802 0,735 0,203
0,9 0,791 0,717 0,183
1.0 0,782 0,699 0,161
1,2 0,767 0.671 0,129
1,4 0,758 0,647 0,098
1,6 0,752 0,632 0,080
1,8 0,746 0,615 0,042
2,0 0,743 0.616
Установившееся состояние достигается после двухчасовой работы.
Реактор периодического действия
с переменным объемом
реакционной массы
Скорость изменения содержания компонента i во время
реакции как с постоянным, так и с переменным объемом массы:
1 dNx = 1 __ 1 VdCH-CidF
ri~ V dr ~ V с/т ~ V * dr
Для системы с постоянным объемом имеем:
dCi
с/т
Если пользоваться мольной конверсией вместо концентрации,
то для реактора с переменным объемом массы удастся установить
одночленное выражение для скорости реакции. Однако такое
упрощение может быть эффективным, если принять ограничение,
что объем реакционной массы изменяется линейно с конвер-
сией, т. е.
(11.26)
4* 51
где с, — изменение объема системы до и после полной конверсии,
доли единицы.
Пример. Рассмотрим изотермическую реакцию в газовой фазе:
А —> 4В
При проведении реакции с чистым А получим:
Однако при наличии 50% инертных веществ два объема реагирующей
смеси дают в случае полном конверсии пять объемов конечном смеси. Таким
образом, писем:
5 — 2
—2—-1.0
Величина Еа учитывает как стехиометрические соотношения,
так и присутствие инертных веществ. Отмечая, что
JV0=^Ya(((l-Xa)
в сочетании с (II. 26) найдем
„ _ .Va _ Л'ло(1-*'й) Z, 1 —Л'а
а~ V ~ -Са0 14-£яХя
или
С а о 1 4- ?а Ха
что представляет соотношение между конверсией и концентрацией
для систем с переменным объемом (или переменной плотностью),
удовлетворяющей принятой линейности (II. 26). С учетом этих
соотношений уравнение (II. 27), написанное для реагента Л, при-
мет следующий вид:
1 dNa 1 Л’по<7(1—Л’я) Са о dXа
Ги~ ~ dr - V0(l-]-^aXa) dx ~ l + taXa' dx
(II. 28)
Это равенство обрабатывается значительно легче, чем (II. 25).
После интегрирования получим
• f dXa -х
e0J (l + bXa)(-ra)
(П.29)
Рассмотрим специальные примеры для этого выражения, где
могут быть применены интегрирование и графическое решение.
1. Реакции нулевого порядка. Для реакции
пулевого порядка скорость изменения любого реагента Л не зави-
сит от его концентрации:
___ 1 dNa Сд о &Ха ,
а~ V ‘ dx l + £aXa * dx
(П.ЗО)
Учитывая (II. 29), получим:
с «о f In (1 -Г SaXa) = In = Ат
J 1т$аЛв G« £а I о
е
2. Р с а к ц и и первого порядка. Для моно-
молекулярной реакции первого порядка скорость изменения реа-
гента Л составляет
1 <Wa
-Га---1Т'~&Г~кСа
или в единицах конверсии при сочетании с (II. 27) и (II. 28):
__г СЯ О tfXg A*€?g (1 — Xg)
1Т СоХд dx 1-рсаХа
Интегрируя, получим
Г -Т^Г- -1п(1-Ха) = Ат
О
или в значениях общего объема системы с учетом (II. 26):
-1п(1-^)=Ат (II 31)
\ Sa^o/
3. Р е а к ц и и второго порядка. Для бимо-
лекулярной реакции второго порядка
2А —> Продукты
или
Л 4 Б —> Продукты; Сао —СЬо
скорость изменения реагента следует равенству
-,о=лс>л(А)г
В сочетании с (II. 26)—(II. 28) оно примет в значениях конверсии
такой вид:
, 4а о </Ха _ / 1—Ап \‘“
а 1 ; М'а ~ 00 k 1-rGaXg J
Отделяя переменные, разлагая по частям и интегрируя, мы
получим:
* t
J dx“ = ^7^° + ~ х»> - кс«
О
Реакции n-го порядка. Для реакций /г-го
порядка типа
_г —___L _ kc f 1 Ха \П / J т О9\
а V ~ dr ~кСа АМ1тУ'а/ (II.3-)
уравнение (II. 29) приводится к выражению
*а
( (Ц-^аХд)”’1 г п~Ч-т
~(1-Ха~ а а°
которое не может быть непосредственно решено.
Для реакций n-го порядка и других реакций мы должны инте-
грировать (II. 30) графически, пользуясь соответствующим кине-
тическим уравнением. Применяемый здесь способ подобен общему
приему, который предназначен для интегрального метода анализа
работы реакторов периодического действия с постоянным объемом.
Трубчатые аппараты
Скорость реакции
Скорость реакции, конечно, не зависит от того, осуще-
ствляется ли она в аппаратах периодического или непрерывного
действия. Однако реакции, идущие в потоке, обычно не протекают
при постоянном объеме и, таким образом, здесь появляются неко-
торые осложнения, связанные с определением времени реакции.
При отсутствии какого-либо изменения плотности реакционной
массы время реакции составляет:
Vr
% 1F/Q
где Vr — объем реактора;
W — массовая скорость потока;
о — плотность.
Величина, обратная т, носит название пространственной ско-
рости 0, т. с.
(н.зз)
В частности, если реакционная масса представляет газовую
среду, то здесь имеет место значительное изменение плотности,
и, стало быть, время реакции зависит от степени завершенности
процесса.
Рассмотрим реакцию, протекающую в газовой фазе по схеме:
«Л4-М4- . . . —> AP4-'Q4-
54
Пусть б обозначает приращение числа молей системы на моль
иступившего в реакцию вещества А. Будем выражать все величины,
считая их па единицу питающей массы. Таким образом, величина
па есть число молей вещества А на единицу поступившей массы,
которая может быть выражена в молях, кубических метрах,
килограммах и т. д.
В точке реактора, где площадь поперечного сечения 5 и длина
(или высота) L, линейная скорость равна:
IV («г о 4-М RT
S ' Р
где п<0 — общее число молей в начальной смеси.
Vr
п n-dn
х х +dz
Рис. II. 10. Схема для вывода уравнения потока в трубчатом
аппарате непрерывного действия.
Время для прохождения бесконечно малой длины dL равно
dL SdL — dVr Л Г чм
и ~ W[nt(i-\-br)KTiP И' (пго + дх) ДГ/Р ' •' J
В таком виде это выра/кение не может быть проинтегрировано
для получения времени реакции, если некоторые из переменных
не будут исключены. Исключим время. Из определения скорости
реакции имеем:
? dX % / I 1 «>Г X
</т = = —7-----1 ГЬу (11 • Зэ)
гр г (nt от-дг) Hi lP
Подставляя в (II. 34), получим после преобразования:
V х
о
Мы получили основное уравнение проточного реактора. Ввиду
его ва/кности приведем и другой вывод уравнения (II 36).
Обращаясь к схеме изображенной на рис II 10, рассмотрим
элемент объема реактора dVr, в котором скорость реакции равна г.
Конверсия в этом пространстве составляет rdVr, так как г есть
скорость изменения на единицу объема. Применяя закон сохра-
нения материи, получим
Приращение — Приход — Убыль
г dVr — И>»а — И7 («а — dna} = —И7 dna = П7 dx
Это равенство может быть приведено к уравнению (II. 36).
Если IF выражается в таких же объемных единицах, как
и Fr, то левая часть (II. 36) есть обратная величина простран-
ственной скорости.
Теперь рассмотрим несколько примеров использования урав-
нении потока.
Для реакции первого порядка, А —> (б 1) В, кинетиче-
ское уравнение:
к(пал — х) _ к(пал — х)
V ПТ , х ч
— (П* о-М
При постоянных давлении и температуре уравнение потока
принимает следующий вид:
_ f ДГ(И(0-дх)
IV J кР(пао — х)
S- [_Jl4.(„,o+6„ov)ln_2!i^
Л/ L па о—х
Истинное время контакта получается из (II. 35). Таким обра-
зом:
т = f * ‘ f_^= ‘ 1п_^>_
J rV kJ па0~х к па0 — х
о о
В качестве другого примера возьмем реакцию второго порядка,
для которой
кпапь _ к (па ft — х) (пь п — г)
V- Г ПТ . , . ,]2
[ — («/о —6х)
Подставляя в (II. 36), получим:
Уг __ 1 / П Т Y2 Гфод. (nf о па а)~ Пп о__________>_
И’ к \ Р ) L ,1а о — ,1Ь о лво~пЬо *
, (»<о-?-б»Ьо)г 1П "Ъ г.
«оо-нЬо tlbn — х.
Истинное время контакта:
Т dx ПТ Р («г o'i-бх) dx
J rV кР J («а о х)(ль<) х)
Л-О а-о
ПТ Г 1 г1Ъп —х б , , . Л . .Iх
=- ТТЛ----------Г п1 01“ ----------1п ("« О — *) — Т— 0 — х)
кР (Нд q — иь q) L о—х па о п1> о J хо
56
Пример. Обратимая реакция А 4 > 2В проводится при
322° С и 0,2 атм в трубчатом реакторе. Питающая смесь газов содержит
30 мол. % вещества А с остатком инертных материалов и поступает в аппарат
со скоростью 5 клсмь/ч. Кинетическое уравнение:
где к = 1.GG сек 1 — коэффициент скорости реакции;
Ар = 0,0055 — константа равновесия.
Требуется определить:
1) объем реактора;
2) пространственную скорость при условии 75%-иого выхода от рав-
новесного состояния реакции.
Отнесем все величины к 1 к.чоль питающей массы. Таким образом:
1
и« о- 0,3
Имеем
0.082.за5«+»)=мз(1 ! х)
Для равновесного состояния получим
ПЬ j ____________________(2л?равн)3___________
1'иа / равн — хравп) (0,3 — Хравн)
откуда Хравн = 0,2.
Далее скорость реакции
0,3 — х 1
243(l-f-z) ”” 0,0055
= 0.0066
Это выражение интегрируется с помощью правила Симпсона; подынтеграль-
ные величины приведены в табл. II. 3.
Следовательно:
— 167 (15,92 j-4 17,38 — 2 10,69) = 148 м3/кхолъ сек
Vr~ 148И’ = ^0,21 л3
5 • 24,2
3600 0,21
— 0.016 сек 1
Таблица II. 3
Значения подынтегральных величин
П од ынте rpa.i ы i а я вел и чина
0 3.33 __
0,025 — 3,79 —
0.05 — — 4,33
0,075 5.15 —
0,10 — — 6,36
0,125 — 8.44 —
0,15 12,59 — —
Всего . . . 15.92 17,38 10,69
Пример. Две группы исследователей изучали кинетику химиче-
ской реакции разложения А, для которой действительно стехиометрическое
соотношение:
Л—>R 4 S 4- Т
Используя,чистое вещество Л при 650s С и 1,2 атм, группа I установила,
что для получения 95% конверсии в трубчатом реакторе необходимо про-
странственное время 6 = 2,7 мин применительно к условиям поступа-
ющего питания.
Группой И при тех же экспериментальных условиях и конверсии най-
дено время пребывания т = 1 мин.
Определить размер реактора, который необходим для обработки
33,3 м3/мин вещества А при 560° С и 1,2 атм с 95% конверсией, применяя
поочередно опытные результаты, полученные группами I и II.
1. Использование данных группы I. Из (II. 33)
имеем:
V = 6г0 ~ 2,7-33,3 = 90 л?
где v0 = H7q .
2. И с н о л ь з о в а и и е данных группы II. Необ-
ходимый размер реактора может быть определен из величины времени пре-
бывания в нем реакционной массы с помощью (II. 26) и (II. 31). Итак, при
= 2 получим:
Л'а/ г 0.95
. /, С dX a r f а
т-1-CaoJ (1 + |вХа)(-г„) J (1-г2Ха)(-/й)
о
Xaf O.es
- V п Г dX° С <*Ха
J J (i-j-гх»)(—r„)
о о
Поскольку —га не определена, мы не можем продолжить решение на
основе данных группы II для отыскания размера реактора. Примем допол-
нительно, что изучаемая реакция разложения второго порядка относительно А
а^кСа
1 — Ха \£
1 -I )
58
Отметим, что эта информация не требуется для случая, когда_используется
пространственное время вместо времени пребывания. Для т и В будем иметь:
Xaf
т=1 f ‘
с
г ьаЛ а 7 у-
__)2 аЛа
*af
1 f (i + £«w ,Y
*CaoJ (1-A'a)2 ' '
0
Интегрируя и подставляя Ara = 0,95 и ga = 2, найдем:
НСао===Еа 1п(1-Ха)-Нёа-Н)
1-Хо 51
Из (II. 75):
АОСа 0 = 2са (£а -И) In (1 - Ха) -г (§й +1 Г- + ЦХа = 138,8
После разделения и подстановки т = 1 мин получим
следовательно
к 138,8
е=^г
У=ЗЯ,Я-1^2-»<90.»3
01
Этот пример показывает, что при £ 4- 0 использование 6 с целью определе-
ния объема реактора упрощает расчеты и приводит непосредственно к резуль-
тату при наименьшей информации по сравнению с применением т. Кроме того,
видно, что эти величины но могут быть взаимно заменяемыми.
Перепад давления
Из рассмотрения составляющих энергетического ба-
ланса следует, что напор за счет подъема жидкости и механиче-
ская работа редко могут иметь какое-либо значение при проекти-
ровании реакторов; скоростной напор также пренебрежимо мал.
Таким образом, перепад давления равен потере напора за счет
трения.
Применительно к элементу объема реактора или его
длины dL будем иметь:
(11.37)
где / — коэффициент трения;
D — диаметр аппарата.
После некоторых подстановок уравнение (II. 37) можно при-
вести к более удобному для пользования виду. Для критерия
Re в пределах 5000 < Re < 20 000, которые обычно имеют место
59
в реакторах, коэффициент трения определяется следующим соот-
ношением:
/ТР = 0,046 Не - °’2 = 0,046 (
где ц — вязкость;
W — массовая скорость.
Кроме того, линейная скорость и может быть исключена при
использовании равенства
IV — 0,785 Z)aQU
Уравнение (11. 37) приводится к виду:
dP.> 0,036 IV1 V’2 dL^i}
Плотность реакционной массы является переменной величиной
и зависит от давления и степени завершенности реакции. Для
идеальных газов:
М_ РМ_ = РМ^_ = о
1 V НТ НТnt ' НТ (fiio + M
где Мо — молекулярный вес (молекулярная масса) питающей
смеси.
Таким образом
dp - 0.036 ГУ1 (nf[,+6x) dL=0
Pg№Mont „
Подставляя в это равенство значение dL из уравнения (И. 36)
для проточного реактора
Liz dx
Ц—- = Г dVT = 0,785 D*r dL
-“о
(II-38)
получим:
dp . 0,0461Г2У’8ЛГ (nf 0 -fir)
PgD^nt „
^ = 0
(П. 39)
где х — количество вещества, вступившего в реакцию, моль-моль
загрузки.
Для удобства выражение (II. 39) может быть записано так:
,п + бг dr ,. т ,
АР 4- п 101------и (11.40)
7jrtto г
где
0,046 1Уг’ер0,2Л7’ ,тг
« “------------ IL 41)
^6-8^2
60
Способ решения уравнения (II. 40) может быть показан при-
менительно к кинетическому уравнению первого порядка:
Подставляя (И. 42) в (II. 40), получим:
/ W = 0
0 («а о —х)
ИЛИ
рз rJ= ря f + (I j 43)
knt о J гга о х
о
Выполняя интегрирование для изотермических условий, най-
дем:
Р3 = Р? - Г’Н'56’ (“ 1-’)’ +
+2*4 .("о.”*) “ 2Ч о*Е. <11 «»
При яо = О будем иметь:
рз = р, _ ЗоЛ£ Г („ + 6„ )S ln _ 0,5 (,»(я„ „ _ +
Л'г*?о L ;{ао ж
-? 2Ь2па о (пй 0 — х) — 2б«/ ох — 1,5 о (11.45)
Выражения (II. 44) и (II. 45) дают искомое соотношение между
давлением и величиной конверсии я, которое может быть подста-
влено опять в (И. 38) с целью онредеделения Ут./Ил.
Для реакций более высокого порядка дифференциальное урав-
нение (II. 40) решается численным методом или графически.
Пример. Газовая реакция А —+• 2В проводится при 334° К
в трубчатоА! реакторе, диаметр которого равен 61 .им. Питающая масса содер-
жит 50 мол. % вещества Л с остатком инертных материалов. Молекулярные
веса реагента А и инертных веществ, соответственно, равны 40 и 20. Скорость
загрузки 4000 кг/ч. Давление при поступлении в аппарат составляет 5,2 атм;
константа скорости реакции 2000 ч'1, вязкость 0.075 кг/м -ч.
Определить зависимость между конверсией, перепадом давления и объ-
емом реактора.
Подставляя имеющиеся данные в (II. 41), получим:
0,046 • 4000s’8 • 0,075°’2 0,082 • 334 ,
а —---------------------—----------------= j ;jOO
1,28-10е'0,061е’6 30“
Используя (II. 43). найдем:
3-1500-0082-334 f <L^Ldx= )42_01.G fM
2000 J 0,5—x J 0,o —x
« о
61
Интегрирование выполняется с помощью .численного метода решения
дифференциальных уравнений. Находим искомую зависимость между давле-
нием и конверсией, которая приведена в табл. II. 4.
Наконец, применяя (II. 38), получим для объема реактора
147? Г f 1+х , 4000-0,082-334 f Ц-х
r J/o/£ J />(0,5 — x) ’ 30-2000 J P (0,5 —x)
о 0
1-4-x
P(0,5 —x)
dx
Это уравнение также решается численным методом; результаты даны
в табл. II. 4.
Таблица II. 4
Результаты вычислений
X (1+хР U ,5 - х у 0 р, «пме 1 +х X у 0 Уг, ж»
Р (0,5 - х)
0 2,00 0 5,2 0,384 0 0
0.05 2,45 0.11 5,1 0,457 0,0216 0,040
0,10 3,02 0,25 5,0 0,550 0,0475 0,088
0,15 3.87 0,42 4.8 0,684 0,0784 0,145
0,20 4.80 0,64 4,6 0,870 0,1170 0,216
0,23 6,22 0,89 4,4 1,135 0,1650 0,306
0,30 8,45 1,26 3,9 1,665 0,2340 0.432
0,35 12,1 1,77 зл 2,900 0,3380 0,626
Определение искомых параметров для модели
при исследовании производственных процессов
Для процесса охлаждения воды спроектирован новый
тип трубчатого теплообменного аппарата. Данные о гидравличе-
ском сопротивлении потоку воды в таком теплообменнике отсут-
ствуют. Предполагается испытать модель теплообменника, длина
которого составляет 1/10 длины производственного аппарата.
Скорость воды в производственном аппарате 756 л!мин при
средней температуре 49° С.
1. Какая скорость воздуха при 15,7° С и давлении 1 атм
в модели может создать условия, подобные таковым в производ-
ственном аппарате.
2. Если перепад давления в модели для скорости потока,
установленной в 1, равеп 760 лш вод. ст., то какой перепад
давления будет в производственном аппарате.
62
Для воды при 49° С:
v = 0.0567 > 10“5 № 'сек
2» 987 кг/м3
Для воздуха при 15,7° С:
v = 0.147 • 10“4 л* 2/«гк
о= 1,22 кг/м3
1. Для динамического подобия производственного аппарата
и моде [и необходимо, чтобы числа Рейнольдса были равны между
собой.
Величины, входящие в критерий Рейнольдса, представлены
в табл. II. 5.
Таблица II. 5
Величины, входящие в критерий Рейнольдса
Величины
Модель
Длина..............
Объемная скорость
потока, м3/мии . .
Скорость ..........
Кинематическая вяз-
кость, м2/сек . . .
Число Рейнольдса . .
L
Q
Q
/,*
0,147 • 10"<
L Q ।
L2 0,147-10"4
Производственный аппарат
10L
0,756
0,756
МО/,2
0,0567 • 10“5
10L A75(L---------1-----
1007J2 0,0567 IO’5
Из равенства чисел Рейнольдса получим:
10-0,756 0,147 • 10~4
100-0,0567- 10“5
== 2,0 м3/мим
Таким образом, скорость воздуха в модели должна быть
равна 2j0 м3/мии.
2. Если между моделью и производственным аппаратом суще-
ствует динамическое подобие, то должны быть равны между собой
числа Эйлера (II. 6).
Так как числа Эйлера равны между собой, то имеем:
А _ 760 987 • 0,756* о „ .
1,22-2,0* =8'8~ Мд' ст-
Мы нашли величину перепада давления в производственном
аппарате, которая соответствует перепаду давления 760 мм вод. ст.
Для модели.
63
Таблица II, 6
Величины, входящие в критерий Эйлера
Величины Модель Пр оизводствевный аппарат
Длина ....... L 10L
Перепад давления, .«.и вод. ст. . . . 760 ДР
Скорость 2,0 0,756
Z,2 100L2
Плотность, кг/.и3 . . 1,22 987
Число Эйлера . . . 760j? аря
1,22 (2,0/Г2)2 987 (0,756/1 OOP2)2
Трубчатые аппараты с ламинарным потоком
реакционной массы
В проточных реакторах, где турбулентность отсут-
ствует, в любом поперечном сечении аппарата имеет место разность
продолжительности пребывания массы. В ламинарном потоке,
как известно, градиент его скорости является параболическим
с максимумом в центре.
Несмотря на то, что средняя скорость реакции в ламинарном
потоке такая же, как в других потоках, конверсия все же раз-
лична. Положение осложняется диффузией. Вследствие того, что
у стенок аппарата продолжительность пребывания массы увели-
чивается, продукты реакции образуются с более высокой концен-
трацией и диффундируют по направлению к центру в то время,
как реагенты диффундируют по направлению к стенкам. Резуль-
татом такого процесса является частичная компенсация недоста-
точной турбулентности в потоке. Наличие свободной конвекции
также благоприятствует достижению вихревого потока.
Некоторые понятия о степени влияния этого эффекта могут
быть получены при рассмотрении упрощенного случая реакции
первого порядка (радиальной и осевой диффузией пренебрегаем).
Скорость распределения потока в соответствии с уравнением
Пуазейля для ламинарного потока равна:
*=2ит (11.46)
где ит — средняя скорость;
R — радиус трубчатого аппарата.
Рассмотрим бесконечно малый элемент аппарата на расстоя-
нии R' от центра с площадью поперечного сечения dA и длиной dL.
64
При установившемся состоянии приход равен убыли и, таким
образом, для реакции имеем (рис. II. 11):
/ dC \
иС (1А = и ( С -Ь ~ dL ) J.4 -h AC dЛ dL
\ <‘L /
После упрощения и подстановки (II. 4G) получим
4^+а-с-о
aL
2и?я
Для вховнога
сеченця:
Um
Рис. II. 11. Схема для расчета трубчатого аппарата с лами-
нарным потоком массы.
Интегрируя, найдем
1» —
2н,н
Это равенство выражает концентрацию в точке на расстоянии /?'
от центра трубчатого аппарата. Среднее значение концентрации
может быть определено посредством численного интегрирования
для всего поперечного сечения аппарата. Такого рода расчеты
были выполнены для турбулентного и ламинарного потоков;
в табл. II. 7 представлены сравнительные данные для реакции
первого порядка при отсутствии диффузии.
Очевидно, что величина, определяющая разность концентра-
ций для процессов в этих двух потоках, зависит от относительных
значений коэффициента скорости реакции, скорости потока
и длины реакционном зоны аппарата.
В промышленных трубчатых реакторах, работающих в усло-
виях ламинарного потока, обычно имеется свободная конвекция.
В этом случае она создает подобие турбулентного движения.
а л. я. Батунср.
65
Таблица II. 7
Сравнительные данные для турбулентного и
ламинарного потоков в трубчатом аппарате
f реакции иерчого иоривка при отсушетчич <)«(/>-
фузии)
l<L С родня я к он центра цп я в выходящем потоке
ламинарный поток турбулентный ноток
0,00 0,9810 0,9802
0.10 0,8328 0,8187
0.50 0/1432 0.3679
1,00 0,2195 0,1353
2,00 0,0(503 0,0183
Однако в лабораторных установках с малыми диаметрами трубча-
тых аппаратов разница между концом грациями в потоках может
быть весьма существенной. Поэтому при получении опытных
данных в лаборатории для больших масштабов производства
рекомендуется поддерживать турбулентный режим работы.
Следует отметить, что неравномерность течения реакции,
вследствие распределения потока, может дать совершенно нежела-
тельный эффект. Например, при полимеризации вязких смесей
более широкое распределение молекулярных весов будет полу-
чаться в реакторе с ламинарным потоком по сравнению с интен-
сивно перемешиваемой реакционной системой С другой стороны,
при наличии побочных и in последовательных реакции недостаток
в контроле их продолжительности может привести к тому, что
будет необходимо использовать реактор с мешалкой.
Для расчета профиля скоростей ламинарного потока может
быть использовано следующее выражение:
» _ /о[Ф(-г)]-/п(Ф(х)/Г/Яв„|
U Л|Ф(х)] 1 ‘
где и — скорость потока на любом расстоянии от оси трубопро-
вода;
U — осевая скорость потока;
и /2 — модифицированные функции Бесселя первого рода*,
зависящие от х — расстояния сечения потока от входа
в трубопровод.
Значения Ф (х) даны в табл. II. 8 в зависимости от (х Dnn) Re
Эти величины изображены также на рис. II. 12.
* См Л. М. 1> а т у и с р, М. Е. П о з н и, Математические методы
в химической технике, Госхимиздат, 1963, стр. 277—295.
66
Таблица II. 8
Значения Ф(х)
Ф (-V) Л'/Оцг1 Rc Ф (.V) х> Ппн Не
20,0 0,000205 2,5 0,01788
11,0 0,00083 2.0 0,02368
8,0 0,001805 1,4 0.0341
6,0 0,003576 1,0 0,04488
5,0 0,00535 0,6 0,06198
4,0 0,00838 0,4 0,0760
3,0 0,01373
Уе
Рис. II. 12. Зависимость Ф (я) от (я/Лцц); Нс
Пример. Жидкость протекает в трубе с внутренним диаметром
53,4 ,н.и. Число Рейнольдса 1000.
Используя (II. 47), рассчитаем профиль скоростей на расстоянии 190,5 .«.и
от входного сечения трубы.
Построим также график для профиля скоростей, показывающий зависи-
мость Н //?вн от и/U, и сравним с профилем установившихся ламинарных
скоростей в соответствии с формулой (II. 46):
и — 2мт
Имеем:
х— 190,5 л.н; 4/ви = 53»4 ил
Re = 1000
5*
67
Таким образом:
•t j DjSU
Re
190,5
53,4 • 1000
3,58 IO"3
По рис. II. 12 получим: Ф (о?) — 6,0.
Из таблицы для функций Бесселя:
Zo (6,0) = 67,234
(fi50) = 46,787
u/U
Рис, II. 13. Результаты вычислений:
1 - по формуле (П. 47): 2 — по формуле (И. 46); 3 — ось трубы.
Таблица //. Q
Результаты вычислений
И’ ЛВ|( <i-<i Е' KlHF u ~T~
по формуле' (II- 47) Устанопшппийся поток по фор- муле (II. 46)
1,0 67,23 0 0
0,95 51,17 0,344 0,193
0,90 39,01 0,604 0,38
0,83 29,79 0,779 0,555
0,80 22.79 0,949 0,72
0,75 17,48 1,065 0.875
0,70 13,44 1,130 1,02
0,65 10,70 1,210 1,155
ОЛИ) 8,03 1,268 1,28
0.53 6,24 1,304 1,343
0,50 4.88 1,337 1.50
0,40 3,05 1,372 1.68
0,30 1.99 1,397 1,82
0,20 1.39 1,408 1,92
0.10 1,09 1,413 1,98
0 1,0 1,415 2.0
68
Определим значения Io (0,0 Н'/Нвн) ДЛ» различных отношении Н'/Лвн
и вычислим uiU из (II. 47).
Например, при R'lRvn — 0,95 имеем:
6,0-0,95 = 5,70
4 (5,70) = 51,17
Из (II. 47) получаем:
и 67,23-f-251,17
~и =-----4W87-----= 0'3'“
Результаты для остальных значений Я7/?вн приведены в табл. II. 9
и изображены на рис. II. 13.
Реакция при непрерывном транспортировании
взаимодействующих веществ
под постоянным давлением
Рассмотрим необратимую реакцию в общем виде:
а А -4- b В 4- сС —>d D 4- еЕ 4- /Е (11-48)
где а, Ь, с — стехиометрические количества молей взаимодей-
ствующих веществ Л. В и С.
Пусть Л'а представляет число молей, имеющихся в момент т;
тогда _Va dr выражает число молей Л, вступающих в реакцию
в единицу времени. С увеличением т величина А’я уменьшается,
следовательно, производная dNa dx отрицательна. Эта производ-
ная очевидно пропорциональна присутствующему числу молей
вещества А, т. е. пропорциональна Лгй.
Уравнение (II. 48) может быть написано в таком виде:
Л 7- (л — |) А 4- ЬВ -Г сС -| ₽Е 4- /F
Следовательно, дифференциальное уравнение, выражающее
скорость превращения вещества Л при постоянной температуре,
примет вид:
—AW (11.49)
где С — концентрация в молях па единицу объема.
Уравнение (U. 49) представляет собой общее уравнение ско-
рости реакции в гомогенных системах.
При применении уравнения (II. 49) к реакции, протекающей
изотермически в условиях непрерывного процесса с постоянным
давлением и изменяющимся объемом, возникает дополнительное
осложнение вследствие того, что время контакта представляет
собой функцию скорости движения газа п длины реакционно i
трубки. Для этих условий типичен следующий пример.
69
Нри.чер. Для необратимой реакции второго порядка разложс
ния ацетальдегида
2СН3СП0 —> 2CII.J -j- 2СО (II. 30)
найдено значение константы скорости при 518°С:
А' = 0,331 л/моль • еск
Реакция осуществляется при давлении 1 атм путем испарения альде-
гида в сосуде, с последующей подачей паров в один конец реакционной трубки,
изготовленной из кварца, и удалением продуктов реакции из другого конца.
Содержимое реакционной трубки поддерживается при 318е С. Требуется
установить зависимость степени завершенности реакции от длины реакцион-
ной зоны трубки.
Обозначим через Аг число молей альдегида, поступающего непрерывно
в реакционную трубку за 1 ч, а через т — время прохождения паров альде-
гида через реакционную трубку. Пусть пз общего числа молей Л’йо исход-
ного альдегида некоторая часть, равная А'й моль, остается неразложеииой
по истечении какого-либо промежутка времени с момента начала разложения.
Рис. II. 14. Схема для расчетов.
. Применение уравнений (11.49) и (11.50) дает
э.у
±^=-KNaCa (И.51)
Для решения этого уравнения следует иск почить одну из переменных.
Целесообразнее, однако, выразить переменные Са итчерез переменный объем
реакционной зоны, который обозначим Еп.
Пусть V есть общий объем неразложнвшегося альдегида и продуктов
его разложения, соответствующих Аа в любой момент после начала разло-
жения.
Пользуясь уравнением состояния идеального газа и стехиометрическим
уравнением, получим:
пт
Г” 7р-[А'.,.+(*<,.-*«)!
Если х — расстояние от входного конца трубки до некоторого ее сечения
(рис. 11. 14), то объем трубки на протяжении длины х будет Л’х,
где S — поперечное сечение трубки.
Следовательно:
f/E R S dx
Примем, что скорость паров в любом сечении трубки является постони
ной; линейная скорость U массы газа, проходящего путь dx за промежуток
времени dx, будет равна
dx н _ V
dx S dx S
79
откуда
«Т= -р- (1х =
dVlt
И
(П. 52)
Величина F представляет собой объем вещества, образующегося после
ввода в реакционную трубку .¥п молей альдегида в единицу времени, и мо-
жет рассматриваться как объем газов, проходящих в единицу времени через
сечение .X.
Уравнение (II. 52) устанавливает зависимость между продолжитель-
ностью т контакта во время реакции в трубке и объемом трубки Гд.
Остается выразить Са через .¥«.
Как известно
Z*. П _____ Z Т |
° V ~ RT{2Na-Na) (IL j3)
Подстановка уравнений (II. 52) и (11. 53) в уравнение (II. 51) дает:
Мд { р у/ х
dVR И2 Л \ RT )\ 2Na-Na )
Разделение переменных приводит к уравнению:
В задаче требуется определить мольну ю долю разложившегося альдегида
при заданном объеме реакционной трубки I R, т. е. А'а/Х .
Обозначим
ад ~ /
тогда уравнение (II. 5'1) примет следующий вид
Интегрируя левую часы, этого уравнения по / в пределах от 1 до /, а правую
часть но Г;? в пределах от 0 до получим, пренебрегая незначительным
изменением Р вследствие трения в трубке
4r„ (II. Ху,
Эта формула дает возможность вычислить / для данной скорости поступления
альдегида в молях в единицу времени при использовании реакционной трубки
с объемом Vjj.
Пусть внутренний диаметр трубки составляет 3,3 с.ч, длина ее 80 с.и
и скорость поступления альдегида 50 г/ч. Подставив сюда численные данные,
указанные в условии задачи, будем иметь:
А" 0.331 л .ноль • сек
50
Л'»." заю-м = х,в’"г* ми">!№К
Р — 1 атя
71
Т = 518 н 273 701 к
В = 0,08203 л атм/.чоль град
Лао \ Л71 ) 1R 3,16 1(Г4’ \0,08206 - 761
Уравнение (II. 55) при этих числовых данных принимает вид
З-у-9,2 1g/ + / = -0,17
Решая это уравнение, получим / = 0,87, т. е. при прохождении через реак-
ционную трубку 13% альдегида окажется разложенным.
Определение длины
трубчатого реакционного аппарата
Рассмотрим элементарный объем реакционного аппа-
рате! dV длиной dz. При наличии химической реакции в этом слу-
чае для материального баланса будем иметь:
Приток — Убыль — Источник — Сток — Приращение (II. 56)
Обозначим одни из компонентов реакционной массы через Л
и примем время реакции в рассматриваемом объеме с?т.
Приток и убыль представляют массовые количества вещества
при поступлении и выходе из рассматриваемой зоны реактора
за время dr. Они составляют изменение количества А в пределах
между элементарным объемом и окружающей средой.
Источник выражает массу компонента А, который появляется
внутри элементарного объема за время dr, а сток — количество
компонента А, исчезающего за этот же промежуток времени
вследствие химической реакции. Для установившегося состояния
приращение равно нулю.
Примем следующие обозначения:
F — скорость потока, кг'сек;
ха — число молей А, вступающего в реакцию в любой точке
аппарата па кг жидкости, моль: кг;
га — скорость убывания А в любой точке аппарата на еди-
ницу объема, моль м3-сек;
па — число молей Л в любой точке аппарата на кг жидкости,
моль, кг;
пао — число молей А при поступлении в аппарат на кг жидко-
сти, моль/кг;
— объем реактора, .и3.
Для промежутка времени dx материальный баланс относи-
тельно А и объема dVR в соответствии с (II. 56) и при учете того,
что приращение и источник равны нулю, составит:
г., Гг„ <HFna) .1
« * <12
(11.57)
-ToilVR=^
a
ПЛИ
—<l(Flta)^ra<iVR
(П.58)
При установившемся состоянии величина F — постоянная,
и мы имеем:
па~ ,1а9~~~ ха
Следовательно:
(II. 59)
(11.60)
dFn„ = Fdna — Fdx(l
Подставляя (11.59) в (11.58), получим:
Fdxa = radVR
Пример. Смесь озона с кислородом проходит через трубчатый
реактор при 1 атм и 100° С со скоростью 0,0155 м/сек.
Принимая, что в начальной смеси 10% озона, определить длину реак-
тора. которая необходима для 50%-го разложения озона.
Реакция может быть написана в таком виде:
2О3 —> 30;
и константа скорости реакции в уравнении
г = к (Оя)2
составляет
к = 8,6-10 2 м'\ кмоль- сек
Используя (И. 60), найдем объем реактора:
х
rR=-F
С
0
(II. G1)
Скорость потока равна
0,0155 • 1 - 0,1 1 ,о . 0,0155 1 • 0.9 • 1
f------------- 48+—0|082.373
0,082 • 373
32 9,001557 кг/сск
0.0155-1 -0,1 -1
0,082 • 373
0,001557 кг питающей смеси
Из каждого моля О3, вступающего в реактор, образуется ;!/е моля О2.
Общее число молей в питающей реактор смеси составляет 0,02976 молъ/кг
смеси. В точке, где х молей О2 па 1 кг питающей смеси оказывается разложив-
шимся. общее число молей будет:
= 0,02976
кмоль
Концентрация 03 раина:
СОз
«о-F -у
&
и
X
"<>. О,—*
«/(ЯТ//>)
73
Скорость реакции:
0.086 (»0- Оа-Д)г
(«о + ^)2(^7’/Р)2
кмоль/м3 • сек
(11.62)
Подставляя (II. 62) в (II. 61), получим:
Примем следующие обозначения;
по О»~й =0,002976 кмоль О3/кг питающей смеси,
«ф— Ь = 0,02976 моль/кг питающей смеси:
с = 0,5;
я = 0,002976/2 = 0,001488.
После этого подынтегральное выражение представим в таком виде:
(Ь-|-ся)2 Ь2 . 2Ьса: , с2х2
(я —х)2 (я — я)2 (« — я)2 1 (я —х)2
Получим;
(я —я)2 [ я —я Jo
25сх . ... Г| / \ , а 1*
-----— ах = 2ос In (я — я) Ч-----
(а— я)2 I ' а — х Jo
с2я2 , •> Г/ , „ , , , я2 "Iх
-----— ах = —с- (я — я) — 2» In (а — я)-----
(я—я)2 ' ' а — х Jo
причем
Ь- = (2,976 • 10~2)2 = 8,837-10"4
а — х = 0,002976 — 0.001488 = 0,001488
(я - я)2 « 2,214-10-е
Ь2 = (2,976-НГ2)2 - 8,857-IO"4
а — х = 0,002976 — 0,001488 = 0,001488
(а - х)2 = 2,214- 10“в
2box = 2-0,02976-0,5-0,001488 = 4,428-Ю“5
с-я2 = 0,23 (1,488-10-3)2 = 0,5535 10-е
25с = 2-0,02976-0,5 = 0,02976
In (« — я) = In 0,001488 = -6,511
In Я = In 0.UO2976 = —5,818
с2 = 0,25
я2 = 8,857-Ю-0
74
Первый интеграл:
Г ь2
I 7-----ГТ йх = 8.8;>7 • 10“4
J («-«)-
1 1
1,48-10“3 2,976-10-»
= 2,98G -10"1
Второй интеграл:
I <?*= 2,976 • 10-2 [-6,511 + 5,818 +
J («—xf L
, 2,976-10-» 2.976-10"3 1 ЛО..1Г .n_.
''W8 7iF - 2,976-10-»-] = °’913G ‘ W '
Третий интеграл:
7—dx= —0,25
(я —xy
0,001488 - 0,002976 - 5,952- JO-3 (-6,511 + 5,88)-
/ 8,857 • 10“6 8,857 IO’8 \
\ 1,488-10-» 2,976 10“» /
= 8,48 • IO"5
Таким образом, уравнение (11. 63) может быть написано так:
V„ = °’00bj;j' ' 3'3)~ (0,2976 + 0,009136 + 0,000085) = 5,18 №
UJJoo * 1
Длина трубчатого реакционного аппарата с единичной площадью попе-
речного сечения равна £ 5.2 .и.
Установившееся состояние
Реактор с обратным смешением
Проектное уравнение для реактора с обратным смеше-
нием получается из следующего равенства
Приток = Отток + Расход по реакции (II. 64)
Если Fao = ц+Со есть мольная скорость компонента А при
поступлении в реактор (рис. II. 15), то по отношению ко всему
его объему будем иметь:
Приход А — 1‘ао (1 — ^ао) = 7'а0
Отток Л = Fa = Fao (1 — Ха)
моль реаги-
г> * , . рующего А
Расход Л по реакции—(—га) I =—г+------------
‘ 4 / Время X Объем
реакционной
\ жидкости
Объем реактора,
занятого реакци-
онно!! жидкостью
Подставляя в (11.64), получим:
/' а (Да = ( — га) I'
После преобразования найдем
о а о —га
75
или
<‘о — га
где Ха п га определяются мри условиях выходящего потока, кото-
рые соответствуют таковым внутри реактора.
В более общем случае, когда питание, для которого принят
индекс «О», поступает в реактор частично конвертированным
(индекс г) и уходит при условиях, для которых индекс обозначим
через /, будем иметь
I7 __ __ -У af <п
Fa о vt)Ctl о (—ru)f
ПЛ II
бц о (Xaf )
(II. 66)
Caf-^a
Vf
)f - (' )
Fo
Рис. ii. 15. Схема для расчета реактора с обрат-
ным смешением при установившемся состоянии.
Реактор с обратным смешением не имеет ничего общего с перио-
дически действующей установкой. Легкость обработки опытных
данных, полученных при использовании реактора с обратным сме-
шением, благоприятствует его применению при изучении кинетики
сложных реакций, для которых требуется дифференциальный
анализ.
Пример. Реакция в жидкой фазе
протекает в реакторе с обратным смешением, объем которого составляет
120.1. Константы скорости реакции (в с'.чоль-.иин):
Ал = 7; /го — 3.
В реактор поступают два питающих потока с равными объемами, причем
один из них содержит 2,8 .цель А/л, а другой имеет 1,6 .чоль В/.г. Желательно
получить 75%-иую конверсию применительно к лимитирующему компоненту.
Определить скорость каждого потока. Плотность реакционной массы в реак-
торе можно принять постоянной.
76
Концентрации компонентов питающей смеси (в моль'л):
<ао = М
С.д0 = 0,8
Go = о — I)
Для 75%-нон конверсии компонента В состав реакционной массы в реак-
торе и на выходе будет в (моль/л):
Са = 1,4 —0,6 = 0.8
£ь = 08—0,6- 0.2
Сг = о,б; С4 = о,б
Таким образом, скорость реакции в аппарате составляет. — rt, = — =
— k\CaCi) — kzCrCs = 7-0,8-0,2 — 3-0,6-0,6 — 0,04 моль л-мин.
При постоянной плотности реакционной .массы, когда £ =• 0, выражение
(II. 65) примет такой вид:
g ___ с д О — (д
~ V ~ —Га
Следовательно, объемная скорость реакционном массы при входе и вы-
ходе из реактора
илп 4 л/мин для каждого потока питающей смеси.
Пример. На основе приведенных данных найти соответствующее
уравнение скорости реакции разложения в газовой фазе
Л —> В S
протекающей изотермически в реакторе с обратным смешением.
Дано (результаты пяти опытов):
0 по начальным хслониям,
мин...............'........ 0,423 5.10 13,5 44,0 102
Ха (для Саь (',002 кмоль/м3) 0,22 0,63 U.75 0.88 0.96
Скорость реакции найдем с помощью (II. G5)
„ ( ft о А а
и представим в табл. II. 10. Теперь испытаем уравнение типа
-г - kCn
которое при постоянном давлении и = I принимает вид:
-Г Z-Г / V
° '"° и М'а ) ~ ‘ “° (1
После логарифмирования получим:
77
Таблица II. IQ
Расчет скорости реакции
Время, ли я 1-У„ С’ Y 1 ! ~га
0.423 0,22 0.78 0,639 °’0°2, Q 22= 104 10-5 0,423
5,10 0,63 0.37 0,227 Д^О,63 24,7-10-5 5,1
13,5 0,75 0.25 1,143 -^^-0,75=11,1 - IO"’ 13,5
192 0,96 0,04 0,021 °^2 0,96^1- IO"5
Используя данные табл. II 10,
построим графическую зависимость,
чтобы выяснить, не является ли
она прямой. Рис. II. 16 показывает,
что принятое нами уравнение ско-
рости реакции представляет опыт-
ные данные при п= 1,4. По нему
же получим:
кСа 0= к (0.002 клольЛи3) =
= 2 10-;* к.«е.«./,и3 • .чин
Следовательно, уравнение ско-
рости реакции получится в таком
виде:
Рис. II. 16. Зависимость — га от
1-Хд
н-х«
Трубчатый реактор
В трубчатом реакторе состав жидкости изменяется
по длине пути ее движения; следовательно, материальный баланс
для компонента реакции должен быть составлен для бесконечно
малого элемента объема dV. Таким образом, для Л получим
(в людь/время):
Поток — Отток -f- Расход но реакции (II. 67)
78
Обращаясь к рис. II. 17, видим, что для объема dV
Приток А = Fa
Отток А = Fa -г а
Расход Л
Подставляя эти величины в (11.67), получим
^=(^ + ^c) + (-r0)dP
Расстояние по длине реактора
Рис. II. 17. Схема для расчета трубчатого реактора
при установившемся состоянии.
Отмечая, что
а ~ 1 п о (1 X а) ] = I' а о а
при подстановке найдем
/’а о <1Ха = (—/«) (И. tiS)
Это и есть уравнение для расчета Л в бесконечно малой части
реактора с объемом жидкости dVa, Для реактора в целом данное
выражение должно быть проинтегрировано. Скорость питания Fa(j
постоянна, но га зависит от концентрации материалов или от
конверсии. Группируя соответственно величины, получим
V xaf
= г (1Х<1
J г а О J ~ га
о I)
Ха!
И Г <ixa
Go J га
о
79
или
(II. 69)
Равенство (fl. G9) позволяет определить размер реактора для
данных скорости питания и конверсии. При сопоставлении (II. 65)
с (II. 69) находим, что га в трубчатом реакторе изменяется, а в ре-
акторе с обратным смешением га постоянна.
Для более общего случая, когда питающая смесь частично
конвертирована (индекс i) и выходит с конверсией соответственно
/, будем иметь:
Рао С^а с/’о J —га
ИЛИ
^af
o = -=rco f (П.7О)
,;о r J —га
Трубчатый реактор может быть использован для исследования
кинетики химических реакций. Графический анализ работы труб-
чатого реактора подобен таковому для периодических систем
с двумя модификациями; 0 используется вместо т, и в соответ-
ствующем выражении для скорости реакции должно быть учтено
изменение плотности жидкости. Поэтому анализ проточных систем
здесь усложняется. Однако в том случае, когда £ — 0, этот прием
становится идентичным тому, который применяется при исследо-
вании систем с постоянным объемом, /(ля необратимых реакций
н-го порядка
А‘а
°"7"” 17^ (П'71)
J АСП
0
и с учетом расширения, пропорционального конверсии, получим
,, Nа А д 0 (1 A'fl) р 1 — Хд
а~ V “ HofH-BnA'J -Gc<’ 1-|-^Ха
поэтому
А'а
О г------- [
Для реакций нулевого порядка равенство (II. 72) приводится
к виду:
7» 0 — Сп |;А а
(11.73)
80
Для необратимых реакций первого порядка:
А’О = (1 -j- tn) й* -7 у £«А a (II. 74)
1 — л д
Для необратимых реакции второго порядка с одним реагентом
Л пли двумя реагентами с одинаковыми концентрациями:
Для обратимых реакций с инертными веществами или без них,
которые протекают по схеме
Л ^=>. ?R
в соответствии с кинетикой реакции первого порядка, имеем:
А-10 = i„ (i _л-хд)
где
Для всех остальных кинетических уравнений рекомендуется
графическое интегрирование. Оно выполняется путем построения
зависимости —1,'гЛ от Х(1 и вычисления площади под этой кривой
между соответствующими пределами.
Пример. Разложение фосфина в гомогенной газовой среде идет
при 650* С
4РН, (г) —> Р4(г) 6IL
со скоростью реакции первого порядка:
—ГРН3 = (ЮЛг) Срц,
Определить размер трубчатого реактора, работающего при 650° С
si 4,6 flWLu, для 80%-ной конверсии питающего газа, с которым подается
1.82 «.ноль чистого фосфина в 1 ч.
Обозначив Л — РП3, Н = и S = Н2, будем иметь
4А —> R -1- <>S
с кинетическим \равнением
— га = (Ю/ч) Са
Объем трубчатого реактора в соответствии с (11. 69):
Л fl
,, ,, f fOK(l р
* — /' а о ~ — Г« о ~Т7-
J —'a -J '• '-а
О о
При постоянном давлении
6 л. м. Батунср.
81
C.i едовател ыю:
о Г 1 ~|~ 5а Xл
кСа о J 1
о
dXa
Для принятых обозначений имеем:
Fa о = 1,82 кмоль!ч; к — 10/ч
7 — 4
= 0,75; Х«=0.9
4
Таким образом, объем реактора составляет.
1 49 г 1 1
V= (.0N0.0W8 [(14-0.75)In —-0.75-0,8] -М .«»
Пример. Для элементарной газовой реакции, протекающей
изотермически л трубчатом аппарате, имеем:
fci
А + В -^±. R
h 2
1. Составить проектное уравнение для питающей смеси из Л, В, 11
п инертных веществ.
2. Найти 0 для питания, состоящего из эквимолярных количеств А
и В.
В первом случае имеем:
„ I. Z’ f. г. f .________________ I. ° & I- г
—' а — Л at- ь Л оС г — 1 “у ’ у Л 2 у
При постоянном давлении с учетом расширения и конверсии А в долях
единицы получим:
_ т_ А,щ> " ' и и**-,*1- ~ ' и у - - « у— < - _ г. ~ » у * - ц и - - I
1 lzo (1 “г^яА'о) 1' о (i'i 5aXa) * l'o (1 г =aXa)
Принимая Л/ — СЬо:Сао, Л/' = Сга’Са0, найдем:
, , (1-Ха)(Л/-Х») _ V -Аг;
й° (1+М'«) - "° l-|-S«Xa
проектное уравнение для трубчатых реакторов приме)
а
Следовательно,
в ид
„ f (/Х„
/
____________________ Г ____ (1 + ^Хц)2 rfXn_____________
J —ra J кгСа с (1 ~ A'a) (-V7 А «) — />'. (.17 + А'а) (1 + СаХа)
о_______________________________________________________о
В этом выражении относится к инертным газам в питающей смеси.
При отсутствии инертных газов и эквимолярных с о от ношен них в пита-
ющей смеси будем иметь Са 0 = Qo; C'rn = 0; .1/ — 1; Л7' = 0; — -0,5-
82
Следовательно, выражение для трубчатого реактора приводится к виду
_________(1-0.5Лл)2</Л'п_________
АдСа о (1 — %а)’ к^Х-а (1 —О.э -^а)
Графическое вычисление этого интеграла дает необходимое пространствен-
ное время.
Не изотермические реакции в потоке
Химические реакции в потоке, идущие при колебании
температуры, исследуются методами, аналогичными тем, которые
были описаны выше для периодических процессов.
Тепловой баланс в основном составляется с целью определения
зависимости между степенью конверсии и температурой, а допол-
нительно, и коэффициентом скорости реакции. Имея эти сведения,
можно решить у равнение проточного реактора Рассмотрим хими-
ческую реакцию
А-гВ—>C-J-D
Для теплообмена на 1 кмоль питающей массы в трубчатом
реакторе будем иметь:
X
о
где Тт — температура теплоносителя;
D — диаметр реактора;
d\ г — элемент поверхности теплообмена.
Тогда тепловой баланс может быть представлен в таком виде:
п т
I (яп 't ,!l> qCi, Нц Sc — - hj oCrf) dl о I (IT -}-
7 6 1ь
T T T
-rp'bo—j) I Ct, (IT r(«cu-bT)J Cc <CT — (//./ 04-x) j C\i(lT + '
1ъ Тъ 1ь
-| Х(\//)Г---|А(Гт —7)-у- (11.76)
* Ъ jj v Г
6*
83
Если теплота реакции вычислена при начальной температуре
7’0, то
т т
J* («а <+« + «ь оСъ + «с о<+1- nt[QCa) dT -| -ж | AC dr + х (ДЯг)7-о —
те т 0
где
iJ V t
О
АС - Сс “hCd — Су — С[у
Пример. Смесь идеальных газов поступает в трубчатый реактор
со скоростью 2,52-Ю-3 кмоль/сек при 834° К. Давление в аппарате остается
постоянным при 49- 10ч и. Внутренний диаметр труб реактора 0,102 .«.
Схема реакции:
Л -|- В —> D
Имеем:
Т, °К.................. 778 805 834 801 890
к, м3!кмоль сек .... 1,355 2,64 5,20 10,15 19,40
Состав питающей смеси (вмол. %): Л 40; В 40; инертные вещества 20.
Теплоемкость (в дж/кмолъ-град): реагенты 25,1 • 103; продукты реак-
ции 41,87* 10я; инертные вещества 20,9 -10:). Величина ДЯГ =
= 53,5- 10е <?ж/к.«смь Л при 278° К. Найти:
1) объем реактора в зависимости от конверсии для вещества А при адиа-
батном процессе;
2) объем реактора в зависимости от скорости теплопередачи, необходи-
мой для поддержания постоянной температуры 834° К;
3) степень конверсии в зависимости от объема реактора для случая,
когда начальная температура 834° К, коэффициент теплопередачи 5 ккал/.ч2 X
Хч-град и Тт = 890Q С постоянна.
Для расчета принят 1 л.иоль питающей массы.
Кинетическое уравнение реакции:
пд 0 — X
ВТ f 1 S X
0-I’M
0,1 -х
8,315- 103 Т (1—ж)
49 101
к / 0,4 — -г- \ -
(0,017 Г)2 \ 1 -х )
Применяя (11.76), получим:
(0,4-50,2* 103-1-0,2-20,9- 10я) (834-278) = (0,4-х) 30,2 -103 +
+ -41,87 • 103х + 0,2 • 20,9 • 103 (Г-278) + 53,5 106х-
4-28-4 f 890 - Т
0,102 J г
о
dx
II.'1 и
«<)0— Т
13,5 1041 — 53,5 • 10»х +1120 —--- dx
J г
__________________________о____________
24,3- 10 я- 8.3- 103х
84
1. В этом случае теплообмен отсутствует; слсдопатслыю:
Г = 278
13,5- 10е — 53,э- 10*ж
24,3-10:,~ 8.3- 10:‘.г
(П. 77)
Применяя уравнение проточного реактора, найдем
Vr= ,v f = 2,52.10, f (0'01I7r)2 (*=*-} to
Jr J It \ 0,4 — x /
о
Величина Fr определяется с помощью численного интегрирования этого
выражения при использовании (II. 77). На основании полученных данных
составим табл. II. 11.
Таблица П, Ц
Расчетные данные
Т h-10"® 1 г X f 0 Vr, .м»
0 834 18,73 0,0625 0 0
0,01 813 11.55 0,0990 0,00081 0,00735
0,02 792 6,86 0,1635 0.00209 0,01895
0,03 772,5 4,55 0,2420 0,00412 0,03740
0,04 752 2,62 0,4140 0,00724 0,06570
0,05 730 1,56 0,6760 0.01245 0,11300
2. При изотермическом процессе подставим Т — 834е К в уравнение
теплового -баланса, которое преобразуем к следующему виду:
х
<? = 1120 [ ±L90TS34> (!х = (24.3.103 - 8,3 10:,х) (834 - 278) -
J г
о
— 13,5 10"-|- 33,5 10°.1- = 49 • W"x
где Q — общее количество тепла па 1 кмоль питающей смеси, которое пере-
дается системе ко временя, когда конверсия равна х.
Так как внутренняя поверхность трубы, считая па 1 .«3 объема реактора,
равна 4/£>, то скорость передачи тепла будет равна:
W dQ ШГ dQ rD dQ .,.e rD
4 4 н , dx 4 dx 4
49-10".0,102 5,2 /0,4 — х V / 0,4— х \
4 ’ (0,017 - 834)2 \ 1 — х ) ° 1-х )
(II. 78)
85
Таким образом:
т. Г dx 2,52 • 10"« (0,017 • 834)2 f/ 1—ж V,
о
X
<".74)
О
В табл. II. 12 приведены расчетные данные, полученные с помощью урав-
нении (II. 78) и (II. 79).
Таблица II. 12
Расчетные данные
X (о*,ГД)* 0 Vr, м* Q, .м!-ч
0 6,25 0 0 1750
0,05 7,35 0,34 0,031 1490
0,10 9,00 1,49 0,135 1200
0,15 11,55 2,17 0,197 945
0,20 16,00 3.86 0.350 683
0,25 25,00 5,52 0,500 437
0,30 49,00 9,46 0,860 223
3. Примем приращение Дж = 0,02, так как при больших значениях
этой величины в данном случае численное интегрирование становится оши-
бочным.
Выполним расчеты для двух точен. Имеем:
ж0 = 0; То = 834; /сп = 5,2
1 (0,017-834)“ / 1-0 V ...
---—- 4 ------I = Z41J
То 5,20 \ 0,4 — 0 7
890-Го _ (890-834)240 _
То ' 1,0
Для ^ = 0,02:
примем Г1 = 811°К
Ад = 3,211
— = 388
'г
11 - 388 (890 - 811) = 30,7 - I О3
1120 f I dx = 1120 • 0,02 • 0,5 (30,7 • 103 -|-
+ 13,44- 103) =495-103
Л -278 +
. 13,5 106 -107 10'* + 595 10s
' 24,14 • 103
= 825° К 7-811° К
примем 7\ 814° К
Ад =2,98
4-=426
Г1
lt = 426 (890 - 814) = 32,4 • Ю3
1120 f I dxr. 1120 • 0,02 0,5 (32,4 • 103 +
•I 13,44-103) = 514-Ю3
?!- 278 +
, 13,5 10е —107 104 + 514 - 103 _
1 24,14-103
= 813,5= К
86
Интегрируя, найдем 7’1=812°К и Zt = 30,1 - 10н-
Для а:2 = 0,04:
, примем Га = 803° К
/г2 — 2,46
— = 550
ri
73=46,4-103
1120 J1 dx = 1120 (13,44 • 10’ -J-
-г 4 • 30,1 • 10’4-46,4 10э) 1348• 10я
72=274-
, 13,5 10е — 214 • 10* 4 1348 103 „
' 24 103 ~
= 807,5° К
примем Т2 = 805° К
/с, = 2,62
/2 =42,6 103
/0,02
I dx. 1120 (13.44 103 4-
+ 4 - 30.1 103 + 42,(5 Ю3) = 1320 • 103
Л =27 4-
13,5 108—214 10*4-1320-10я
24-Ю3
= 806.5° К
Интерполирование дает Т2 = 803° К.
Объем реактора определяется на равенства:
Г,-Ж f f
J >' J к \ 0,4 — х /
о
Продолжая расчеты для остальных точек, получим данные, приведенные
в табл. II. 13.
Таблица II. 13
Расчетные данные
X Т к г у 0 1'г.
0 834 5,20 225 „
0,02 811 3,09 373 —
0.04 803 2,52 486 14,6 0,0368
0.06 804 2,55 522 — —
0.08 804 2,55 565 36 0,0906
0,10 808 2,79 566 — ——
0,12 811 3,00 583 58,3 0,1470
0,14 814 3,21 607 —
0.16 816 3,38 650 83 0.2095
0,18 810 3,66 670 — —.
0,20 822.5 4,02 725 110 0,278
0,22 827 4,50 766 — —
0,24 830 4.85 861 140,5 и,35 4
0,26 836 5,55 948 — —
0.28 842 6,41 1072 180 0,453
0,30 848 7,45 1269 — —
0,32 857 9,02 ' 1579 198,5 0,580
0,34 865 11,10 9195 — —
0,36 875 14,20 3710 300 0,755
87
Определение кинетической модели
Две проблемы делают отыскание действительного меха-
низма реакции сложным. Во-первых, реакция может протекать при
сочетании нескольких механизмов. Во-вторых, данному кинети-
ческому соотношению могут соответствовать несколько механиз-
мов реакции. Решение этих вопросов сложно и требует основатель-
ного изучения химия рассматриваемых веществ. Оставляя это
в стороне, посмотрим, как испытать соответствие между принятым
механизмом и экспериментальной кинетикой реакции. Для того
чтобы испытать гипотетический механизм, включающий в себя
набор последовательных элементарных реакций, мы должны сопо-
ставить принимаемые кинетические уравнения с эксперименталь-
ными кинетическими уравнениями. При этом допускаем следующее.
1. Если компонент i участвует в нескольких реакциях, то
общая скорость его изменения есть сумма скоростей его изменения
в каждой из элементарных реакций, т. е. rit Сбщ =
2. Так как промежуточные соединения присутствуют в малых
количествах, то их скорости изменения в системе за коротким про-
межуток времени не могут быть значительными. Поэтому прини-
маем их равными нулю. Это — метод приближения к установив-
шемуся состоянию. Такого рода аппроксимация необходима
при решении связанных с процессом математических вопросов,
и она оправдана том, что предполагаемые на основе этого метода
результаты очень часто согласуются с экспериментом. Подбор
данных с целью отыскания механизма реакции иллюстрируется
в следующем примере.
Пример. Реакция
2А |- В = Л2В
была изучена кинетически. (’.корпеть образования продукта в соответствии
с экспериментом хороню определяется следующим равенством:
0,72 0,72 (А]3 (В]
- ' 1 1-2 (А]
(П.80)
Какой механизм реакции устанавливается уравнением (И. 80), если
(благодаря химическому исследованию реакции известно, что промежуточное
соединение состоит главным образом из совокупности молекул реагиру-
ющих веществ и ценной реакции нс происходит?
Предположив, что здесь элементарная реакция, для СС скорости получим:
=*МА]2(В) (11.81)
Однако сопоставление (11.80) и (11.81) убеждает нас в том, что это
псэлемснтарная реакция.
Перейдем к испытанию различных механизмов п моделей и установим,
какая из них даст выражение для скорости реакции, подобное по виду эксие-
рнмепталыю найденной зависимости.
88
Начнем с простой двухступенчатой модели и при неудаче рассмотрим
более сложные трех-, четырех- и пятиступенчатые модели.
Модель I. Примем двухступенчатую обратную схему,
которая включает в себя образование промежуточного вещества Л'. Это
вещество невозможно обнаружить, поэтому его присутствие предполагается
только в малых количествах. Таким образом, имеем
А, г , 113
2А 7=^ Л„; Л.,-Н В Л.В
ht - - k*
(11.82)
где предусмотрены четыре элементарные реакции:
2А — > А' (11.83)
А;— > 2Л (11.84)
л; + в—->Л2В (11.85)
Л2В—->Л'4-В (11.86)
Отнесем значения А- в каждом случае к любому из расходуемых компо-
нентов; например, Zj относится к А, величина йа — к Л* и т. д. Теперь со-
ставим равенство Для т. о. для скорости образования А,В. Так как этот
компонент входит в (11.85) и (11.86), то общая скорость его изменения
ость сумма отдельных скоростей. Имеем:
К] (В1-Л-4|Л,В( (11.87)
Все величины в равенство (II. 87) за исключением А' могут быть най-
дены. h сожалению, не определяется экспериментально; следовательно,
если оно присутствует, то в чрезвычайно малом, не поддающемся изменению
количество. Поэтому концентрацию этого вещества нужно заменить концен-
трациями А, В пли Л>В, которые могут быть измерены. Из четырех элемен-
тарных реакций, включающих в себя А., найдем:
=-1 АЧ [А]'2 —А\, [л;]-А3 [д'] [BJ-? А., [А„В] (II. 88)
Вследствие малой концентрации, можно принять, что [А.,] достигает
установившегося или равновесного состояния в пределах очень короткого
промежутка времени Следовательно, скорость изменения вещества Л2 равна
нулю или
(П. 89)
Это и есть аппроксимация установившегося состояния. Из (II. 88) и (11. 89)
найдем:
р1[ЛР-ЬМА2В)
(II. 90)
89
Подстановка в (II. 87) дает скорость образования AfiB в измеряемых
количествах. Итак:
4-Мз[А]ЧВ1-М1(Л2В]
Г^’ =' A-s + MB] (IL 91)
В поисках модели, совпадающей с наблюденной кинетикой, можно
ограничить более общую модель путем произвольного подбора величины
различных констант скорости реакции. Так как (И. 91) но своему виду пе
соответствует (II 80), то посмотрим, не будет ли сходства при некоторых
упрощениях. Так, если Л2 очень мала, то это выражение приводится к виду
^ = у*11Л12 (И. 92)
для реакции второго порядка относительно А и независимой от В. Если А-4
очень мала, то rQtb приводится к виду
_ (М3/2А-г)(Л]ЧВ]
1-j-(A's.'Av) [HJ
(11.93)
Ни одно из этих специальных выражений не совпадает с экспериментально
найденным кинетическим уравнением (II. 80). Таким образом, гипотетиче-
ский механизм рассматриваемой реакции (II. 82) является некорректным.
Модель II. Так как пата первая модель дала выражение
для скорости реакции (II. 93), в какой-то мере подобное равенству (11.80),
то испытаем для модели II такой механизм реакции, который был бы близок
к модели I.
Испытаем механизм реакции
i<,
А 4- В Л13'
ha
ЛВ'Ч-Л Л.В
k 4
(II. 94)
По аналогии с тем, что имеем выше, найдем:
^ = А‘3[ЛВ'][А1-А-4[А,В]
(II. 95)
Исключим [АВ'] в выражении (II. 95). С помощью аппроксимации уста-
новившегося состояния получим
rab’ 0= \ fA 1 [В] — А’., [ A В'] - А., [Л В'] [Л] + к4 [ Л.В ]
откуда:
[дв^ АЩЬда (п.<ж
А 2‘Г л а 1'4
Подставляя (11.9ft) в (II. 95) с целью исключения концентрации про
межуточного вещества, получим:
_ M\,[A]4B] + mja2b|
Г(^~ А^+МА]
Применим ограничение к этой общей модели.
С очень малым значением для Л, ио <учнм:
Сравнивая (II. 80) с (II. 98), видим, что они идентичны. Таким образом,
реакция может быть представлена следующим механизмом
\ Ч-В “t АВ'
Ъг (II. 99)
АВ' + Л — -> Л2В
Константы скоростей реакций могут быть определены только при нали-
чии дополнительной информации. Константа равновесия К$ = ki/k« и есть
необходимая информация.
Нам удалось представить данные в виде уравнения, которое
точно совпало с выражением для теоретического механизма реак-
ции. Часто большое число уравнений может удовлетворить набору
экспериментальных данных одинаково хороню. Во избежание
потери корректного механизма рекомендуется при испытании
различных теоретически полученных уравнений с помощью экспе-
риментальных данных использовать статистические методы ана-
лиза.
Глава III
Кинетика гетерогенных
химических реакций
и процессов, протекающих
в реакционных аппаратах
Реакции и процессы
в системе жидкость — жидкость
Для удобства рассмотрим газовую и жидкую фазы:
пусть А представляет собой реагент в газовой фазе, а В — рсагепт
в жидкой фазе. Если мы в действительности имеем дело с двумя
жидкостями вместо газа и жидкости, то просто принимаем здесь
газовую фазу как вторую жидкую фазу и соответственно изменяем
терминологию.
Представим процесс в виде
А Ъ В ---> 11 родукты
с противоточным движением фаз.
Материальный баланс в дифференциальной форме для проти-
воточного движения реагентов:
6' Л’ = L(!Xk = Gd(^-\
b \Ри /
— d ( = d ( G 'Pa = — — d (
b \CuJ \ P ) b \ cu J
(III.l)
где G = (”pu!P— мольная скорость потока п'нертных веществ
в газовой фазе, поднимающейся вверх, па 1 .и~
поперечного сечения аппарата;
Л = Ь'Сц/Соощ —мольная скорость потока инертных веществ
в жидкой фазе, направляющейся вниз, на 1 .и2
поперечного сечеппя аппарата;
G', IJ — мольная скорость всего газа и всей жидкости
на 1 .v2 сечения аппарата;
Ya — Ра/Рц— количество молей А на 1 .ноль инертного ве-
щества в газе;
Ха = Са/Си — количество молей А па 1 .моль инертного ве-
щества в жидкости.
92
Индекс и в уравнении (III. 1) означает носитель пли инертный
компонент в фазе.
С учетом обозначений найдем:
- • +Ри ) (1П
С’общ=Сд + Сь4' • • 4-Си J
r?y = d df,u (IH 3)
V Pu J ft
dXa = d(~\ = Ca dCu — Cu dC„ (111. 4)
\ct4/ cl
Для параллельных потоков (движение газа сверху вниз)
величина G заменяется значением — С по всей колонне.
Составы реакционной массы в любой точке трубчатого аппарата
могут быть найдены в значениях для конечных условий процесса
путем интегрирования уравнения (III. 1).
Таким образом, имеем:
11 “ й,) Ь Л₽и Риb U’u Q
= G'pa O'pai 1 / L'Cb L[CLt \
P 1> b k С’оСщ Собп. i > * ’ ,J'
Уравнения (III. 5) дают состав реагентов Л п Н в фазах по
всему трубчатому аппарату.
13 специальных случаях (III. 1) и (III. 5) могут быть упро-
щены. Например, когда все составляющие в реакционной массе
разбавлены, то ри Р, Си СОбщ и для материального баланса
в дифференциальной форме получим
dpa = - \ (!СЬ (HI 6)
г общ
и для конечных условий:
~р {Ра Ра 1) =--(И Г. 7)
Другой случай имеет место, когда полностью отсутствует инерт-
ный компонент или носитель. Ниже рассматривается одна из
многих возможных форм, которые материальный баланс может
принимать в специальных случаях.
Отмечая, что кинетические уравнения
гь 1 dX а 1 (INb _
b S Pb ж r/t Cb j * Ра bS dx
Ра ж b 1 Pai (111.8)
1 1- 1
г а~ — /•а ж Гь _ b 1 И 1 " 5 ’ а 1^(1 г (IN д dr f'a гРп (И1. 9)
93
1
I! Hal
к а г к а я;
Ра
(ГН. 10)
относятся к единичной межфазной поверхности, найдем для
расхода А:
GdYa=-^-
Mo.ni проре-
агировавше-
го Л
Единичная
межфазная
поверхность
Межфазная
поверхность
Единичный
объем
Объем
элемента
аппарата
= (—га) adh
(III. 11)
где гал гь — скорости реакции;
А'аг, Ло ж—коэффициенты массоперсдачи в газовой и жидкой
фазах;
Hai = pa[/Cai — коэффициент распределения па поверхности раз-
дела фаз;
D — коэффициент диффузии;
__^’р >в)бсз реакции ________________ 1Й Xq Т k/Dam _ th тх0
я;)с реакцией__________________тХй
.г0 = Da .|{ 1 ка -,к
а — поверхность фазового контакта, приходящаяся на единицу
объема аппарата;
h — высота аппарата.
После преобразования и интегрирования (III. 11) получим
в значениях Л и 13:
Я 2 A 1
I dY« = £ f ‘1XJ> (Ш.12)
J (—О<) « b J (—га) а
Уа 1 Ь2
где dya и (кеь определяются с помощью формул (III. 3) и (III. 4).
Для разбавленных систем имеем:
2 1
£ f tlP“ _ L С (}съ
i* J ( ra)a ЬСобЩ J (—Гд) и
i 2
(Hl. 13)
Высота аппарата может быть найдена, если величину скорости
реакции в (III. 11) и (III. 12) заменить соответствующим выраже-
нием из (III. 8)—(III. 10). 13 большинстве случаев интеграл
должен быть определен либо графически, либо численным методом.
94
Для того, чтобы определить, какое из двух выражении — (1П. 8)
или (T1I. 9) —должно быть применено для какого-либо данного
случая, нужно знать концентрации реагентов и физические свой-
ства используемых жидкостей. 'Гем не менее, можно сделать при-
ближенную оценку следующим образом.
Для массопередачп компонента через жидкость:
/>;к Ю~5 Г.и- ci'/,'
Л'о & 10-2 С-»|
Для газовой фазы:
1)г 0,1 см'1 со:
х$ 10-'- e.v
Следовательно, для движущих сил в единицах концентрации
будем иметь
А' = кН Г ъ 10 с.и со:
‘ 1 з-о
Подстановка этих значений в формулу
показывает, что в том случае, когда
Рч ___ г Ю b ;к
ПТ ь
для решения вопроса может быть использовано упрощенное кине-
тическое уравнение (ГН. 9). Следует, однако, иметь в виду, что
(ГП. 12) есть только порядок оцениваемой величины. Если реа-
гент В нерастворим в инертной жидкости и пре вставляет собо i
тоже жидкость, то в этом случае жидкой пленки не будет и ско-
рость процесса полностью определяется сопротивлением газовой
пленки. Таким образом, уравнение (III. 9) остается в силе.
Пример. Концентрация нежелательной примеси А в воздухе
должна быть уменьшена с 0,1 до 0.02% нутом абсорбции газа чистой водой
или кислым раствором различной крепости.
Найти высоту колонны для противоточной абсорбции:
1) в чистой воде;
2) в крепком растворе кислоты с концентрацией Су 0,81 клолъ/.и3;
3) в разбавленном кислом растворе с концентрацией Су = 0,034 кмолъ/м3;
4) в кислом растворе с концентрацией Су - 0.135 «.иоль/.м3.
Реакция
А (г) В (ж) —> Продукты (ж)
имеет место в жидкой фазе и протекает быстро; продукты реакции остаются
в жидкости.
Для используемых насадочных тел:
ка i-ч = 33,6 кмолъ/ч м3'апг. ка ;кл 0,1 ч-1
95
Растворимость вещества А в чистой воде: Нп — 0,12 ат-лл/кмоль~
Скорости потоков жидкости и газа (в кмоль/м2-ч) : L' — 63,5; Сг = 9,1
(при Р = I ат).
Мольная плотность жидкости: Собт “ 59 кмиль/м3.
Примем, что коэффициенты диффузии веществ Л и В в воде равны.
Таким образом:
Ь'а я; — kl> ;к =
1. На рис. III. 1, а показаны известные нам количественные соотноше-
ния. Так как мы имеем дело с разбавленными растворами, то можем восполь-
зоваться упрощенной формой материального баланса:
Ра~ Рт
1)
с общ
Ра - 0,0002 = —^4 (Са - Са J
v»l * OiJ
ра —0,0002 = 0,1185 Са ~ 0,12 Са
Рис. III. 1. Данные для расчета.
Концентрация вещества Л
в жидкости па выходе:
_ 0,001 0,0002
аъ 0,12 0.12
0,0067 клюль/.и3
Выберем некоторое число парциальных давлений вещества Л в колонне,
определим соответствующие им концентрации в жидкости, вычислим равновес-
ные парциальные давления ра и затем найдем значение общей движущей
силы физической абсорбции. Эти расчеты представлены в табл. HI. 1.
Общий коэффициент массолередачи в расчете па объем колонны соста-
вляет
_!_=_1_ 4. ,|.W2 = ,.03
А’а1л ^Г« ’ W1 33,6 ' 0,1
откуда А'а г — 0,812 кмоль/ч-.и3-ат.
96
Таблица HI. 1
Результаты расчетов
Ра Са Ра^11а('а Р = Ра ~ Ра
0,0002 0 0 0,0002
0,0006 0,0034 0,0004 0.0012
0,0010 0.0067 0,0008 0,0002
Для высоты колонны из (III. 9) и (III. 13) имеем:
2
л = — f dPa ° С (}р” —
Р J (—га) а J ^ага‘-^Ра
ра1
0)001
= 9,1 J 0,812 • 0,0002 4,> "
0,0002
Здесь жидкая пленка составляет 95% сопротивления массе передаче.
Следовательно, можно допустить, что жидкая пленка контролирует этот
процесс.
2. Эта часть примера решается следующим обрамим (см. рис. III. 1, б):
составляем материальный баланс и находим концентрацию кисяоты в выхо-
дящем из аппарата потоке; устанавливаем, какая из двух модификаций кине-
тического уравнения должна быть использовала, затем определяем высоту
колонны.
Для разбавленных растворов с быстрой химической реакцией из (III. 7)
имеем:
LP
Ра — Ра 1 = , — (О> 1
W УОоищ
или
Ра-0,0002 = 9д3,1.'!9 (0,84 -Сь)
Ра = 0,12-0,84 — 0,12 Сь 4- 0,0002 = 0,101 — 0.12 Сь
Концентрация нанрореагпровавшего D па выходе из колонны:
_ 0,101-0,0010 П£)О , 3
Сь 2 ------012-----= 0,83 КМОЛЬ х
Для верха колонны имеем (в кмыъ-ч-.ч3)
А-агара = 33,6-0,0002 = 0,0067
А-,К«СЬ = 0,1 -0,84 = 0,084
для низа колонны
к а Ра = 33,6-0,001 = 0,0336
А';1;йС’ь = 0,1 -0,83 « 0,083
Па обоих концах колонны кагра <* к-,кСъ', таким образом, сопротивление
газовой фазы контролирует процесс и для использования должно быть при-
нято уравнение (III. 9).
7 Л М. Батунер.
97
Из (III. 9) и (III. 13) имеем:
J’O2
, Л f dPa j' (lPa
P J a P J A« ,«pa
VQ 1
0,001
0,0002
В физической абсорбции жидкая фаза контролирует процесс. Однако
при наличии химической реакции контролирующее сопротивление может
быть другим. Действительно, мы в этом случае видим, что только газовая фаза
оказывает влияние на скорость процесса, химическая же реакция служит
только Для устранения сопротивления жидкой пленки.
3. По аналогии с предыдущей частью примера из (III. 7) найдем
(рис. III. 1, в)
L Р
Ра Ра 1 = z.. (G, 1 Q.)
v ОС общ
или
Ра - 0.0002 (0,034 - Сь)
VJ* 1 VU
ИЛИ
Ра —0,12 (0,034 — Сд) "г 0,0002 — 0,0043-0,12 Сь
Таким образом, концентрация непрореагпровавшего В па выходе из
колонны составляет:
„ 0,0043 —0,0010 0,0033 лЛО„ , ,
СЪз=-------(Щ------= -Qj2 = 0,021у к.чолъ. ,113
Для верха колонны имеем (в к.иоль/ч-лг3)
Л-е гар« = 33.0 • 0,0002= 0,00672
к^рСь - 0,1- 0,034 = 0,0034
для низа колонны
ka v»Pa = 33,6 • 0,001 = 0,0336
А-^яб’ь =0,1- 0,0275 = 0,00275
На обоих концах колонны имеем А‘а гРа.^> А’;|{С(>; следовательно, внутри
жидкой пленки имеет место химическая реакция и здесь должна быть исполь-
зована формула (III. 8), причем
_ " Н «О ь Ч~ Ра
а~ 1/А‘аг I Яа/А’-.к
Определим несколько значений ИаСъ -Ь Ра (табл. III. 2)
Таблица 111.2
Расчет НаСь-ГРа
Cjj (из материального баланса) нась=о,12С6 WJ’e
0,0002 0,0334 0,0040 0,0042
0,0006 0,0302 0,0036 0,0042
0,0010 0.0275 0,0032 0.0042
98
Таким образом, высота колонны в соответствии с (III. 8) и (III. 13)
будет равна
Ра2
. _ G Г dpa ____ G i‘ 1 кат-~- И aj k-tg
P J —raa L* J p«
0,0010
f 1/33,6 + 0,12/0,1
J 0,0042
0,0002
0,001
4pe = 9,1 J
0,0002
0,029+1,2 ,
0,0042 (P(l
2,2 -w
•4. Составляем материальный баланс. В любой точке внутри колонны
имеем (рис. III. 1, г)
ра - 0,0002 = 913>1'.*9 (0,135 - Сь) = 0,12 (0,135 - Сь) = 0,0159 - 0,12 Сь
11.1 н
ра = 0,0002 — 0,0159 — 0,12 Сь = 0,0161 — 0,12 Сь
Из этою выражения найдем для низа колонны:
Cb2 =
0,0161—0,0010
0,121
=0,125 кмоль, '.и3
Для верха колонны имеем (в кмоль/ч -.и3):
АаГера = 33,6-0,0002 = 0,00672
ктаСЬ = 0,1 -0,135 -= 0,0135
для низа колонны
кага ра = 33,6-0,0010 = 0,0336
. kiKaCb = 0,1 -0,125 = 0,0125
Для верхней части колонны каГра к»;Съ и поэтому здесь должна быть
использована формула (III. 9). Для низа колонны, поскольку мы имеем
/‘ягра > к}цСь, применим формулу (III. 8).
Теперь найдем условие, при котором зона реакции достигает поверхности
раздела фаз с изменением вида кинетического уравнения. Эго произойдет
в том месте, где
Откуда
A‘f4 гРа — к-,цСь ИЛИ 33,6 ра г -— 0,1 Ср
С b = 33(5 ра г
Решая совместно с материальным балансом
найдем
откуда
р„ г = 0,0161 - 0,12 Сь
Ра г ~ 0,0161 — U, 12 -336ра [’
ра г = 0,00039
Скорость реакции получим из табл. III. 3. Как и следовало ожидать,
при Ра = 0,00039 вычисленные с помощью (111. 8) и (111. 9) скорости реакции
имеют одинаковые значения.
99
Таблица III. 3
Расчет скорости реакции
Ра сь "аСЬ “aCb+l'a ka г« или 1 Н)° 1
1 + “а
кйТа Аа;к°
0,00020 33.6 0,00672 149 (Ш. 9}
0,00039 — — — 33 6 0,01327 74,4
0,00039 0.133 0.0160 0,0164 0,0287 0,01327 74,41
0,00070 0,131 0.0157 0,0164 0,0287 0,01327 74,4 (Ш. 8)
0,00200 0,1285 0,0154 0,0164 0,0287 0,01327 74,4
Высота колонны может быть найдена графическим интегрированием
пли аналитически следующим образом:
0.00039
Г dPa
0,001
Г 1/АЯ + /чкСв (i
J Ра~{-11аС[)
0,00039
Л- Лиерх-г/<ш1з =
J каг“Ра
0,0002
0.001
91 Г 0,308-1-1.9
= • 2,303 [1£? 0,00034- 1g 0,0002] + 9,1 —7-7777^—dpa
33,0 I 0.0164
0,00039
= 0,189 +0,41 ~ 0,0 •«
Рассмотрим случаи когда химическая реакция контролирует
процесс в колонном аппарате с противоточным или прямоточным
движением жидкостей. Напишем уравнение реакции в таком виде
А (г) |-ЬВ(ж) —> Продукты
при следующем ограничении относительно материального баланса:
количество непрореагировавшего Л в жидкости мало по сравнению
с А в газовой фазе. Непрореагпровавший компонент В движется
вниз с жидкостью, гт непрореагпровавший А поднимается вверх
с газом и материальный баланс в соответствии с (111. 1) остается
в силе.
Отмечая, что каждый моль вещества Л, реагирующего в жидко-
сти, заменяется молекулой свежего Л из газового потока и сочетая
материальный баланс с кинетическим уравнением (Ш. II), полу-
чпм: • - - " X / J
„ LdXb /Мо-ni нсвро-Х G di а = —-—= реагировав- 1 \ шею Л / /Объем жидкойХ / фазы \ / Объем \. .. 1 Объем жид- | \жидкости/ 1 у кости J (III. 14)
Так как скорость реакции в этом случае отнесена к единице
объема жидкости, то объемная доля реагирующей фазы / в (Ill. 14)
100
появляется вместо межфазной поверхности на единицу объема
в уравнении (III. 11).
проектное уравнение:
После интегрирования получим искомое
2 Л tj j
G Г dY L Г <1X1.
^'<1 1 3
где dYa п d.Xa определяются из (111. 3) и (111. 1), а — гп предста-
вляет соответствующее выражение для скорости реакции.
Уравнение (III. 15) может быть различным по своему виду
в зависимости от физических условий процесса. Рассмотрим,
например, распылительную колонну с разбавленным раствором
вещества В в виде капель, причем реагент Л присутствует в газе.
Если условия таковы, что концентрация А в газе приблизительно
постоянна во всей колонне, то (111 15) приводится к виду:
Cbi
hА Г А9^ц) = __L— Г А. _ А£_?_!„ Al (Ш.2в)
V J кСаСь b/A.C>iCfi6rn J <-b ^-'bi •
c b -i
Приводимый ниже пример иллюстрирует другой вид проект-
ного jравнения, который обусловлен спецификой применяемого
материал ыюго баланса.
Пример. Бензол подвергается хлорированию путем прог тот оч-
ного контактирования с потоком чистого газообразного хлора. Реакция
СеНс(и<) + С1г(е) —>СсПЛС1(ж)4 НС1(г)
медленная, алиментарная, необратимая п протекает и жидкой фазе между
растворенным хлором к бензолом. С учетом дополнительных упрощений:
мольная плотность жидкости постоянна (С'Общ = const);
давление и газовой фазе постоянно (Р = const);
движение в обоих потоках «поршневое»;
количество растворенного п пспрореагироиавтпего хлора в жидкости
незначительно;
растворимость IICI в жидкости мала;
величина 7/tt постоянна;
скорость реакции С12 с СЙНЙС1 пренебрежимо мала,
составить уравнение для высоты колонны в зависимости от переменных
системы.
Пусть А — хлор, В — бензол, С — мопохлорбепзил, D — хлористый
водород.
Материальный баланс не может быть составлен с учетом L и G, так как
здесь отсутствуют инертные вещества. По заметим, что ла каждый моль
используемого хлора образуется 1 .ноль MCI, поступающего в газовую фазу.
Таким же образом общая скорость мольного потока жидкости не изменяется,
так как па каждый моль реагирующего бензола получается 1 .моль хлор-
бензола. Следовательно, общие скорости мольных потоков газа и жидкости
остаются без изменений, т. е. L’ и G' постоянны, и материальный баланс
в соответствии с (Ill. 1)
/ С' ара \ _____£ / L'Cb \
X I’ ) \ б-обгц /
10!
примет вид:
-^-dPa~—J^-dCb (111.27)
Р LoGlll
Сочетая с кинетическим уравнением, отнесенным к единице объема реа-
гирующей илп сплошной фазы
—Гд — —г & = кСаСь
мы получим:
-7Г dPa = —7^~— = kC(i Cbf dh
i О'общ
где / — объемная (Оля жидкости для реагирующей фазы.
После соответствующего преобразования и интегрирования найдем
высоту колонны
P<t 2
У _ dla Г dp(f ____ L Нп Р dCf, . .
Ч? J РаС'ъ */Соищ J РаС'ъ * ’
Pai
причем и Сь связаны между собой посредством материального баланса.
Интегрируя уравнение материального баланса в дифференциальной форме
(III. 2/), найдем для любой точки в колонне:
Сь-<%бщ- G'Cpm (pa-pai) (HI - 29)
Подстановка (III. 29) в (Ill. 28) и последующее интегрирование дают
к _ -С7/„ |п Рах\Р-(С'/Ь')(Р-рп J]
/С’общд [PH-(G'/L') ра г] Р-
или в мольных долях:
h= [1 Ya J 1,1 Уй 1 [] - T" (1-Уа)] (J11- 30)
где
Диффузия, сопровождающаяся химический реакцией.
При поглощении газа раствором процесс диффузия газа в рас-
твор сопровождается химической реакцией первого порядка,
скорость которой пропорциональна концентрации растворенного
в жидкости газа. Скорость диффузии в жидкости принимается про-
порциональной градиенту концентрации.
Требуется найти функциональную зависимость, выражающую
изменение концентрации растворенного газа но толщине диффу-
зионного слоя.
В любой плоскости, перпендикулярной к направлению диффу-
зии, условия процесса одинаковы. Выделим в пограничном слое
элемент толщины dx, ограниченный плоскостями, параллельными
плоскости раздела фаз и проведенными на расстоянии х и х — dx
от этой плоскости; составим материальны и баланс для этого
элемента. Площадь элемента примем равной единице.
102
Скорость диффузии в точках плоскости, отстоящей от плоскости
раздела фаз па расстоянии х, будет равна
ах
где D — коэффициент диффузии;
с — концентрации газа в жидкости на глубине х.
Так как концентрация уменьшается в направлении диффузион-
ного потока, то коэффициент диффузии взят со знаком минус.
Количество газа, иродиффунднровавшего за время dr будет
равно:
Приход ——D — (1т
(IX
Аналогично, количество газа, продиффунднровавшего через
противоположную границу элементарного слоя, отстоящую от
плоскости раздела фаз на х f- dx, будет
Убыль *- —I)
так как градиент концентрации в этой плоскости будет равен
de (х -I- dx) de
dx dx
Во время диффузии через элементарный объем газ взаимодей-
ствует с жидкостью со скоростью, пропорциональной его коли-
честву, находящемуся в этом слое.
Так как объем рассматриваемого элемента равен dx, то коли-
чество диффундирующего вещества через элементарный объем
получится уможением этого объема на концентрацию с. Однако
скорость химической реакции пропорциональна концентрации
и, следовательно, равна кс, т. е.
de
dx
= kc
где к — константа скорости реакции.
Таким образом, количество диффундирующего газа, вступа-
ющего в химическую реакцию, в элементарном объеме dx за время
dx составит kcdxdx.
Если процесс диффузии считать установившимся, то при соста-
влении материального баланса ио обычной схеме
Приход— Убыль = Приращение
следует принят!, приращение равным нулю. Поэтому уравнение
материального баланса будет иметь следующий вид:
—(4—'j+ <^ (1т—Ice dx ch—О
dx L\ dx J \ dx )
103
После упрощения получим:
-й-=4с (1U.31)
dr2 L)
Для решения уравнения (III. 31) обозначим к/) через я'2;
уравнение примет вид: c"(z) — а2с (z) = О.
Обозначим у (р) изображение функции с (z), которую будем
считать оригиналом. Используя операционное исчисление, най-
дем. что изображение у (р) удовлетворяет следующему алгебраи-
ческому уравнению:
Р~У — Р'^0 — РЪ. — a'2lf = й
Решая ого относительно у (р), получим:
, х Р2 , ра
J u ' и р2 —а2 1 а р2 — а-
Пользуясь соотношениями таблицы изображений и оригиналов
функции, найдем оригинал, т. с.
с (.г) = ад ch ах -J- sh «х-
Здесь zc - const есть значение функции с (z) при z — 0. а z2 —
значение ее производной, т. е.
яд = с' (о) — = const
ах
Если z0 и Zjl считать произвольными числами, то найденная нами
функция является общим интегралом уравнения. Эту функцию
можно представить в виде:
c(z)= (Ш.32)
Пусть нам известна концентрация газа в пограничном слое,
а также в’слое, расположенном на расстоянии I от пограничного.
Допустим, что с — сд при х == 0 и с — с2 при х — I. Тогда:
<д = Ад + Ад; <д = к^14- - <а (III. 33)
Решая уравнения (III. 32) и (III. 33) относительно Ад н Ад.,
получим!
_ cs — I с1-<^—с-г
14 2 sh al ’ 2 2 sh al
Подстановка и k2 в общий интеграл дает:
i । 1 п ' c->5hT/ 4- x-Нт Т/
c2 sh ax -|- sh a (I—.r) fl) f D
sh al /
Sb |/ T I
104
Обратимые процессы. Если два одновременно идущих
процесса взаимозависимы и, противодействуя один другому,
дают противоположные результаты, то они являются обратимыми.
Пусть вещество С превращается в другое вещество X, причем
эта реакция — первого порядка. В то же время X переходит
обратно в С также в результате реакции первого порядка.
Через сих обозначим эквивалентные количества С и X в ми-
мент т.
Требуется найти константы скоростей реакций и к.,.}
Уравнение материального баланса для вещества X имеет вид:
<1х = /rjC dx — к2х dr (III. 34)
Уравнение (111.34) содержит три переменные, по так как
общее число эквивалентов обоих веществ не изменяется ни в той,
пи в другой реакции, то
•£ л (' = ;^oTQi
где ж0, с0 — значения а: и с при т = 0.
Исключив с из (III. 34) получим:
-7-=А1(си-|-^)-(/ь1 + Л.) х (111.35)
Дифференциальное уравнение (III. 35) имеет два важных
свойства. Во-первых, из самого уравнения видно, что с увеличе-
нием х скорость реакции dx:dr будет уменьшаться, оставаясь
при этом положительной. С другой стороны, из физико-химиче-
ских соображений следует, что система будет стремиться к состоя-
нию равновесия.
При равновесии, т. о. когда скорости прямой и обратной реак-
ции равны, dx dr = 0. Обозначив х при равновесии черех хсо,
получим;
*«»*•>,+*£~<с»+-г'») (111.36)
Обозначив далее
у — х... —ж
преобразуем дифференциальное уравнение (III. 35) в уравнение
(111.37)
показывающее, что скорость реакции-пропорциональна степени
отклонения системы от состояния равновесия и что это — процесс
первого порядка относительно переменной у, которая определяет
эту степень отклонения.
1 оз
Чтобы найти константы скоростей реакций, необходимо сна-
чала, пользуясь экспериментальными данными, определить их
отношение. Затем находят их сумму, пользуясь уравнением
(III. 37). Зная сумму и отношение констант, -легко вычислить
значения самих констант.
Пример. Уксусная кислота и этиловый спирт вступают в реак-
цию с образованием уксусноэтилового эфира и воды:
СЩСООН + СЛ15()Н CH;iCOO(Ul, -I- П.,0
Процесс обратим, причем протекающие в обоих направлениях реакции
второго порядка.
Наблюдалось, что при условиях, когда
т = 0 64 ос сутки
Количество кислоты = 1 0,750 0,333 Л«М ь
» спирта — 1 0.750 0,333 »
» эфира = 0 0,250 0,667 »
0 воды — 0 0,250 0,667 )>
Требуется составить уравнение кинетики этого процесса.
Как обычно, сначала составим общее уравнение для такого рода процес-
сов.
Обозначим через х, г/, и и г количества в ,щць, соответственно, спирта,
кислоты, эфира п воды в момент т: при т — 0 эти величины будут, соответ-
ственно, х0, у0, м(( и г0.
Тогда дифференциальное уравнение скорости превращения спирта
<7х
д-- = — l‘\xy ' - Ь” (III. 38)
u- L
Чтобы исключить у, и ii v, воспользуемся стехиометрическими соотно-
шениями
,Г0 — X = у0 — у = ц — г((1 = f — г,,
из которых следует:
У = * у() — х0; « = х0 w0 — х; г --= х0 [- r0 — х (III. 39)
Пользуясь (III. 39), приводим дифференциальное уравнение (111.38/
к виду:
fix
— = ex- 4- Ьх -р с (III. 40)
где
а — к-2 — k'i
b = (7а - 2Ы х0 - Л-1Уа - А-2»о - Лаг-о (Ш- 41)
с =-- к д (х0 4- м0) (х0 [- ц()
Обозначим через значение х при равновесии. В этом случае
4'^=0 (111.42)
Вычитая (111.40) из (111.42), получим
106
Разложив правую часть этого уравнения на множители, имеем:
—-^7 = cGct»-a;)(;c+xoO+v) (HI 43)
Для решения уравнения (III. 43) используем уравнение (III. 38), напи-
санное для равновесного состояния. Из (111. 38) при dr dr 0 находим
(II 1.44)
*1 «оеА»
Из этого уравнения следует, что если известны предельные значения перемен-
ных, то можно найти отношение констант скоростей реакций. Кроме того,
если мы не в состоянии получить даже из опыта какое-нибудь одно из этих
предельных значений, то тем не мопсе можем сделать паши вычисления зна-
чительно более простыми, введя это отношение как одно из неизвестных.
Обозначим такое отношение через т:
т = (111.45)
Теперь обратимся к дроби Ь.'«, находящейся во втором множителе правой
части уравнения (III. 43). Пользуясь значениями а и b (III 41) н вводя т
из (Ill - -45) находим
— = ——г [ (1 — 2м) х0 — — ти0 — ти0 ] (III 4(>)
Из формулы (III. 4(5) следует, что если известны равновесные значения
переменных и их начальные значения, то известно также и Ь;а. Уравнение
(III. 43) в этом случае можно написать так:
где
Пользуясь вышеприведенными данными для спирта и кислоты, находим
из (III. 44)
, ,> 1
А1 = 4Л-3 или т — —
Подставляя в уравнение (III- 47) найденные значения постоянных, при
ведем его к виду:
В примере были использованы такие данные, что предел,
к которому стремилась переменная, мог быть найден непосред-
ственно из опыта. Если такие данные отсутствуют, то задача
усложняется, во-первых. но причине возникновения чисто мате-
матических затруднений, во-вторых, потому, что данные сами
по себе недостаточны. Следующий пример дает один из возможных
методов решения такого рода задач.
Пример. Дпфепплхлормстан вступает в реакцию с этиловым
спиртом с образованием хлористоводородной кислоты и эфира. Предпола-
гается, что реакция идет но схеме:
RC1 4- R'Oll IIC1 -г RON' (III. 48)
107
Желательно проверить это предположение и определить константы ско-
ростей прямой и обратной реакций. Имеются следующие данные:
Т, шт . . 13 119 142 162 182 212
Количество НС!, ло.щ . . 0.00346 0,02680 0,0309 0.0343 0.0370 0,0-418
Очевидно, что здесь равновесие нс было достигнуто даже в каком-либо прибли-
жении.
Количество спирта было настолько велико (около 21 лоль), что в процессе
реакции оно оставалось практически постоянным. Начальное количество
дифенилхлормстапа т0 = 0,09966 .моль.
Примем следующие обозначения:
х — число молей дифенилхлормстана ко времени т;
у — число молей НС1, а также молей эфира, ко времени т.
При т —> тот величина ?/ —> у^. *
Если реакция соответствует уравнению (III. 48), то дифференциальное
уравнение задачи следующее:
= (111.49)
где /ci и /та — константы.
Из стехиометрического соотношения имеем: я 4- у — х„, вследствие чего
х может быть исключен и уравнение (III. 49) приобретает вид:
^- = fri(^-!/) = W (III. 56)
В состоянии равновесия у = г/то п tly/dx = 0
°=Мхо~-'/о=)-М«
Вычитая (III. 51) из (III. 50) получим
(Ш.51)
(111.52)
Непосредственно из (III. 51):
Полагая
7
kL=
XQ-!f<n
приведем дифференциальное уравнение (III. 52) к следующему виду:
77^^ (Усс--'/) С'/ + Р)
Интегрируя это уравнение, придем к зависимости:
1П'/Со“-'/ 1П I/ СО
Сущность метода дальнейшего решения задачи состоит теперь в том,
чтобы, задаваясь значениями для неизвестного у^., построить графики для
108
v 4- р ..
---относительно т. При этом, если одно из выоранных значении ут
окажется действительно правильным и если гипотеза о природе реакции
верна, то в результате мы должны получить ряд точек, расположенных на-
столько близко к прямой липни, насколько позволяет точность эксперимен-
тальной работы. На рис. III. 2 показаны результаты для пробных значений
y_c, равных 0,070, 0,075, 0,080 и 0,083 и обозначенных соответственно
X , I, < и >•
Верхняя и нижняя кривые дают весьма незначительную вогнутость,
направленную, соответственно, вверх и вниз, хотя отклонение от прямой
линии здесь в каждом случае находится в пределах экспериментальных
ошибок.
Определенные для четырех проб значения Д-2 составляют 0,0170, 0,0118,
0,0082 и 0,0064. Таким образом, первое значение превышает последнее
более, чем в два раза.
Вис. III. 2. Результаты для пробных значе-
ний уто.
На основе такого подбора данных можно прийти к единственному заклю-
чению, что если реакция действительно протекает по уравнению (III. 48),
то константа вероятно, находится либо в интервале между полученными
наибольшими и наименьшими значениями, либо вблизи от ппх.
Чем ближе к равновесным данные, полученные опытным путем, тем более
точно может быть определена к$ этим методом, так как дифференциальное
уравнение, (III. 50) показывает, что влияние Аг2 возрастает с увеличением
значения у.
Константа ki вычисляется легче; приведенные выше данные вполне
достаточны для определения ее значения, так как Ад является константой,
которая оказывает большее влияние в начале реакции. Нетрудно убедиться,
что все четыре пробы дают почти одно и то же значение Ад.
Последовательные процессы. Рассмотрим также про-
цессы, являющиеся результатом двух более иростых процессов,
следующих один за другим. Примером таких процессов могут
служить превращения радиоактивных веществ.
Предположим, что какое-либо вещество, начальное количество
которого равно g0, превращается в другое вещество, а последнее,
в свою очередь, также изменяется, причем скорости этих двух
реакций различны. Примем, что это — реакции первого порядка.
Пусть
g — количество исходного вещества ко времени т;
я — количество первого продукта, образовавшегося ко вре-
мени т по первой реакции;
У — количество второго продукта, образовавшегося ко времени т.
100
Тогда х — у есть количество первого продукта, получившегося
ко времени т.
По условию задачи в процессе протекания реакций скорость
изменения х пропорциональна £, т. е. пропорциональна g0 — х.
а скорость изменения у пропорциональна х — у. Поэтому диффе-
ренциальные уравнения для х и у будут:
dx dy
= — я — = Л-а(.т-?/)
Первое из этих двух дифференциальных уравнений может быть
сразу же проинтегрировано:
* = go(l~e"ft,T) (Ш.53)
Значение х подставим во второе дифференциальное уравнение.
При этом получим:
-^ + *2y = ^0(i-^ftlT) (III. 54)
Уравнение (III. 54) принадлежит к типу линейных уравнений.
Решением его будет
„ , &Л'2 “*1Т fin*! Л-Лг-С
~~к^Ге
(III. 55)
Уравнение (III. 55) является уравнением кинетики образова-
ния конечного продукта в результате двух последовательных
процессов первого порядка.
Иногда возможно предпринять раздельное или независимое
исследование двух реакций и, следовательно, найти константы
их скоростей /сх и А\2.
Пример. Предположим, что исходное вещество — совершенно
чистое и количество его равно единице, т. е. g0 = 1. Предположим также,
что полный анализ образца был сделан ко времени т и оказалось, что к этому
времени содержание наличного количества первого продукта равно 0,4,
второго продукта 0,3, а, следовательно, содержание исходного пепзмепив-
шегося вещества также 0,3. Примем, что т — 3, т. е. пусть мы нашли с по-
мощью анализа, что х — у = 0,4 и у — 0,3 при г — 3. Покажем, как может
быть решено трансцендентное уравнение (111.55) относительно постоянной
к?, если Ап рассматривать как известную величину.
Решим уравнение (III- 55) относительно е~йгТ, а множитель ₽“'<1Т
исключим путем подстановки его значения из (III. 53)
В этом уравнении вес величины, кроме к2, известны. Для его решения по-
строим на миллиметровой бумаге график функции е“/12Т, рассматривая
Лг как переменную и принимая т равной 3. Далее таким же образом построим
график правой части уравнения (III. 56). Мы получим прямую линию, кото-
рая пересечется с графическим изображением функции , вообще
говоря, в двух точках (рис. III. 3). Одна из этих точек будет иметь своей
110
абсциссой значение, рапное уже найденному значению ki. Другая точка будет
иметь своей абсциссой значение искомой к».
После того, как первое приближение для к-2 найдено, можно уточнить
это значение, применяя метод иск гедовательных приближений. Для этого
представим уравнение (III. 56) в виде
о
А'и / У — *
п подставив в его правую часть вместо к? найденное приближенное значение,
получим второе приближение для А-г, которое в ряде случаев оказывается
более точным, чем первое. Подставляя это _кч
второе приближенное значение для к-2 е
снопа в правую часть, получим третье при-
ближение. В ряде случаев этот процесс
оказывается сходящимся и позволяет опре-
делить к-2 с любой степенью точности.
Применив этот метод к приведенным выше
числовым данным, получим ki —
- In 0,3 = 0,4. Уравнение (III. 56)
оправдается в
C"3fts= — A-2-J-0,7 (II 1.57)
Построением графика определяем пер-
вое приближение к корню: к% — 0,35.
Запишем теперь уравнение (III. 57)
в виде
/,•„ = 0,7 —с-3"2
и найдем второе приближение, иод ста
вляя в правую часть этого уравнения
А’9* = 0.35. Имеем = 0,34995. Подставляя второе приближенное значе-
ние снова в правую часть уравнения, получим третье приближение A-f3^ =
= 0,34999. При пользовании этим методом, конечно, нужно учитывать точ-
ность, с которой заданы постоянные, входящие в уравнение.
Покажем теперь, что в двух последовательных процессах первого по-
рядка количество первого продукта возрастает до максимума и затем надает
до нуля. Найдем также максимальное количество первого продукта и зна-
чение т, при котором этот максимум достигается.
Вычитая (III. 55) из (III. 53) получим:
А’о — /С,
(III. 58)
Для нахождения максимума х — у нужно приравнять производную
от х — у пулю; решая относительно т полученное при этом уравнение, най-
дем:
При таком значении времени т достигается максимальное количество
первого продукта.
Ill
Чтобы найти значение максимального выхода первого продукта, подста-
вим значение т в формулу (III. 58). Получки
kt
[А'« 1
*‘2 J
По этой формуле молпю вычислить х — у, т. е. максимальный выход первого
ироду кта
Процессы смешанного типа. Параллельные, обратимые
и последовательные процессы очень часто протекают совместно.
Число возможных сочетаний весьма велико, но методы их анализа,
в общем, подобные приведенным выше, могут быть использованы
и в этом случае для составления дифференциальных уравнений,
которые, однако, ire обязательно окажутся с разделяющимися
переменными илп линейными.
Следующим пример дает представление о ходе решения, связан-
ного с указанными процессами
Пример. При пропускании водяного пара через раскаленный
уголь получается водяном газ, который представляет собой смесь водяного
пара, двуокиси углерода и водорода.
Имеются следующие опытные данные но составу' газовой смеси
Объемная .юля пара .... 1,000 0,906 0,709 0,556 0,376 0,056
Объемная доля СО., .... 0.000 0,041 0,100 0,123 0,099 0,0242
Требуется показать, что эти результаты достаточно хорошо согласуются
с нреднотожением, что реакции протекают в соответствии с уравнениями
(А)С + 2П2О=СОг-|-2Н.;
(Б)С-4-Н.,О^.СО + И2
(B)C-j-CO.,« 2СО
а также с предположениями, что все три реакции — первого порядка н что
ни одна из них при условии опыта не. является в заметной степени обра-
тимой.
Определить также отношение констант скоростей реакций.
Пусть Ал, 1;2 и к3 — константы скоростей, соответственно, реакции
(А), (Ь) п (В).
Двуокись углерода образуется в (А) и используется в (В)- Обозначив
через х и у объемы пара в СОг ко времени т, мы, па основе сделанных пред-
положений, получим дифференциалыюе уравнение для переменной у
~ -Адг -А’зУ (111.59)
Однако пар используется как в реакции (А), так и в (В), причем обе они
и jvt одновременно и должны рассматриваться па основе принципов, уста-
новленных для побочных процессов, между том как (В) — последовательная
реакция.
При составлении уравнения (III 59) Ад была выбрана таким образом,
чтобы она соответствовала скорости образования одного объема С()2, образу-
ющегося из двух объемов пара. Следовательно, дифференциальное уравне-
ние для переменной х будет:
= — 2А-Гс — fr„z (III. GO)
С целью исключения с7т разделим (III. 59) на (III. 60)
dy _ kjX—ksU
dx —2krx— k2x
Разделив числитель и знаменатель па Ач и приняв обозначения
Г> 7* Q _ , fro
0=7? " "=2 | тг
.получим
(hj рг/ — х
dx их
Это уравнение является линейным относительно у.
Значения а и |> могут быть найдены методом подбора н, следовательно,
отношения fr3/fri и А'г/A'i становятся известными.
Можно было бы предположить, что реакция (Л) — второго порядка,
но это привело бы к такому дифференциальному уравнению, которое нс со-
гласуется с экспериментальными данными. Следует отметить, что эта реакция
протекает на поверхности угля и отличается от гомогенных газовых- реакции.
Некаталитические реакции и процессы
в системе жидкость — твердое вещество
Химическая стадия процесса в значительно большей
степени зависит от температуры, чем физические стадии. Таким
образом, проведение опытов при нескольких температурах даст
возможность легко установить различие между пленочной диффу-
зией и химической реакцией в качестве контролирующих процесс
стадий.
Кинетический анализ показывает, что время, необходимое для
достижения одинаковой степени конверсии частиц с различными,
по неизменяющимнея размерами определяется таким образом:
для контролирующей пленочной диффузии
т.2 03 / 7?2 \ 0,3 ч- 2,0
для контролирующей диффузии в твердой массе
т2 _ 0.2 / К., у
тг Oj \ 77 j )
для контролирующей химической реакции
(111.61)
(III. 62)
т.2 0.,
Т1 “О?
/7.,
*1
(Ш.СЗ)
где 0 — время, в течение которого завершается реакция с твердой
частицей;
Н — начальный радиус частиц.
Следовательно, опыты ио кинетике с частицами неодинаковых
размеров дают возможность установить различие между
8 Л. М. Натулей.
ИЗ
реакциями, в которых контролирующими являются химическая
или физическая стадии.
Рассмотрим питание, состоящее из твердых частиц неодина-
ковых размеров. Обозначим через т количество твердого вещества,
обрабатываемого в единицу времени. Кроме того, пусть F (Л\)
представляет количество материала (с размером частиц
питающего реактор.
Из анализа процесса диффузии через газовую пленку для
любого момента т вытекает, что
о
(III. (И)
где — конечное значение радиуса твердых частиц.
Соотношение (111. 64) можно представить в значениях степени
конверсии (в долях единицы), отметив, что
4 з
__У _ Ооъем пепрорсагпроиалпгсго ядра _ 3 ’ с
ь Общий объем частицы 4
—
>
Таким образом:
Для случая, когда процесс контролируется диффузней через
твердую массу, применяя закон Фика, получим:
В значениях степени конверсии, выраженной в долях единицы,
найдем:
“=! ^3(1-Л'ь)*/’-Ь2(1-Ль)
В том случае, когда химическая реакция контролирует про-
цесс, для кинетики по схеме
А (ж)Ц-ЬЛ (гн) —> Жидкие продукты
—> Твердые продукты
—> Жидкость и твердые продукты
будем иметь:
1 <lNl, Л (lNtl ,, z.
i.-vf Л "
Написав Л'ь в значениях уменьшающегося радиуса, получим
равенство
1 / ° , г
—---- О, т ЛГГ, ;-fl, — = Ьк „
лл,.2 -ъ с (lx Le> </т сн'йг
‘ с
114
которое при интегрировании приводится к виду:
ус у
—Qb I (h'c 13 Ьк^цСа г | dx
ii о
или
Время 0, необходимое для полного завершения реакции, может
быть найдено, когда гс ~ 0:
^А"тпС« г
Уменьшение радиуса или увеличение степени конверсии час-
тицы в значениях 0 определяется с учетом кинетического уравне-
ния в таком виде:
y-'-V 1-(1-Л'ь}‘/з (III.(15)
Принимаем, что реакция идет сначала на поверхности твердо!'!
частицы. Затем зона реакции передвигается вглубь твердого
тела, оставляя позади полностью конвертированный материал
и инертную твердую массу. Последняя носит название «золы».
Следовательно, в любой момент с течением реакции здесь суще-
ствует переагирующее ядро материала, которое постепенно сокра-
щается в своем объеме.
Если Ит наибольший размер частиц в питании, то для частиц
с пепзменяющимся размером имеем:
F=^ V?’(/?t)
а;=0
При «поршневом» потоке время пребывания тр в реакторе
является одинаковым для всех твердых частиц. С. учетом этого
явления, а также кинетики контролирующего сопротивления,
можно найти степень конверсии Хь (R-^ для любого размера ча-
стиц Rt. Тогда средняя степень конверсии Хь твердых частиц,
оставляющих реактор, может быть получена путем соответству-
ющего суммирования отдельных степенен конверсии. Итак:
Среднее значение
для доли
непрореагиро-
вавшего В
Доля пенрореагироняв-
шего реагента в твер-
дых частицах размером
(До in лита- \
ющен смеси 1
с размером I
частиц Ki '
или
1-АЬ = ^[1- (нрн 0^А-^1)
8*
115
илп
1-хь= 2 (III.66)
Я(ТР-6)
где /?{Т( — радиус наибольшей частицы, завершившей пол-
ностью реакцию в аппарате.
Пример. Питание, состоящее ил
30% твердых частиц с радиусом 50 мк
40% » » о KJO »
30% » » » 200 »
поступает непрерывно в тонком слое на вращающуюся решетку, через кото-
рую перекрестным током проходит реакционный газ. Соответственно планиру-
емым условиям, необходимое для полной конверсии время составляет 5, 10
и 20 мин применительно к трем размерам частиц в твердом питании.
Найти степень конверсии твердых частиц при продолжительности их
пребывания и реакторе, равной 8 мин.
Принимаем, что твердая масса находится в сплошном потоке, а газ
поступает с постоянным составом. Таким образом, для смешанного питания
можно применить (111.66):
I -Л'ъ= (-1 -Ль (100)! [1 -Л’ь (200)1 ——
г Г
где
--- 0,30 и 0 (50) = 5 мин
1 (*°0) = 0,40 и 0 (100) = 10 мин
—(у,-- = 0,30 и 0 (200) = 2U мин
Так как для трех размеров частиц имеем
Ri : Н2 : R3 = 0i : 02 : 03
то в соответствии с (III. 63) устанавливаем, что здесь химическая реакция
контролирует процесс и зависимость между конверсией и временем опреде-
ляется выражением (III. 65)
Подставляя в (III, 66), получим:
(ДЛЯ R Ц>«) (ДЛЯ К—200)
0,0032 4 0,0048-= 0,068
Следовательно, конверсия твердого вещества равна 93,2%. Отметим,
что частицы с наименьшим радиусом полностью конвертируются и поэтому
не учитываются при суммировании в (III. 66).
116
Кинетику диффузии к системе жидкость — твердое вещество
л кинетику растворения твердых тел в жидкости рассмотрим па
конкретных примерах.
Пример. Дно реакционного аппарата покрыто слоем слежав-
шейся твердой массы. Для промывки дна аппарат залит водой. Найти зави-
симость между концентрацией растворяющегося в аппарате вещества и вре-
менем т для точки, находящейся на расстоянии х от дна аппарата.
Напишем уравнение диффузии:
дс , , д-с
-х- А"
дт. дх-
(III.67)
Обозначим с1; концентрацию вещества в растворе па дне аппарата. Най-
дем решение уравнения (III. 67), удовлетворяющее условиям:
с ск: х — 0: т>0]
с ----- 0; х > 0; т = 0 J
Обозначим через и (.г, р) изображение, функции с (х, т)
Lc (т, т) — и (х, р)
(III. 68)
'1огда, переходя от оригинала с к изображению м в уравнении (III. 67)
и учитывая (III. 68), получим:
d-u р
dr'- ~~ h- М
(111.69)
Мы получили линейное уравнение с постоянными коэффициентами отно-
сительно функции и.
Общее решение (III. 69) есть
_|/ZFX l/jE7x
н = Лс ’ fcl -f-b’Z **
Так как концентрация ие увеличивается неограниченно при бесконечном
возрастании х. то
В-- 0
Далее, так как с = ск при х -- 0, мы должны иметь
Ск А
Следовательно,
U=3 С|;С
Для определения функции с по се изображению^», примем а — х/Л;
тогда ““
«=Свв’я1>; с= L~1cKe~n V p
Обращаясь к таблице оригиналов функций и их изображений, получим;
<1
2Г‘т
Z,-1e“"»p = l------4=- I e-"sd>i
1 1 J
117
Отсюда для искомой концентрации с в аппарате получим следующее,
выражение:
I
2<1 I
’ du
V
тело, имеющее форму шара, рас-
творяется в химически активной жидкости, причем па растворение 1 лоль
твердого вещества расходуется I .ноль растворителя. Требуется составить
уравнение для определения коэффициента скорости растворения твердого
венц етва.
Пусть г0 обозначает радиус шара в начале процесса при г — 0, а г —
радиус этого >ке, тела в момент времени т. Обозначим через I’ объем I моль
растворяемого вещества, через х — число молей вещества, растворившегося
ко времени т,'и через « — начальное число молей растворителя.
Скорость растворения в момент т пропорциональна поверхности фазового
контакта S и количеству оставшегося к моменту т растворителя а — х.
Так как объем шара равен лг3, то объем х молей растворившегося вещества
О
равен Г'х
Пример. Гомогенное твердое
откуда
г
1
з 31 \ 3
и
4л
Поверхность Л’ к моменту т равна
тяг- или
а ЗГх
° 4л /
Скорость процесса растворения будет равна
dx
dx
d.X / г - ( Я
~7~ 1лК ( го
dx \ °
з
(U-Z)
откуда
dx
.з З1'х
и
4л
где А' —
Для
ловки:
искомый коэффициент скорости растворения,
интегрирования уравнения (III. 70) сделаем
следующие по ц’.та-
я(| dl.i =
Тогда получим:
3174л = 6; а0— Ьх ;3
"о--3 . >г
~1 . j (I X
и
b
dx
'i.tA-’t
(а0 — Ьх) 3 (я—.г)
118
Подставив принятие выше значения в подынтегральную функции»,
получим;
f dz f________________dz________ 1 Г dz 1 f (z + 2n)dz
3 «а-г:‘ (м-;)(л2фя:-|-:-) n-J n-z n- J н-4-пз~гй
j
Первый из :пих интегралов равен — -—г In (w— г). Второй интеграл
преобразуем следующим образом;
Исходный интеграл оказывается, таким образом, равным
Возвращаясь к переменному х, получим:
(«у— ab) 3
("п — Ъх) 3 + («<» — М 3
1
(«о -й&) 3 “ (й<> — М
(III.71)
Для определения постоянной интегрирования С следует переменные
величины взять при т — 0, т. е. положить х О п г -- г,,.
Уравнение относительно искомой величины К не. является трансцен-
дентным. Значение А легко вычисляется, если входящие в ото уравнение
буквы заменить их численными значениями. По если К задано и требуется
определить а.-, то относительно х уравнение (111. 71) трансцендентно, так как
содержит х под знаками трапецепрентных функций —логарифма и нрк-
ташепса.
Реакции и процессы
в присутствии твердых катализаторов
Для исследования кинетики каталитических реакции
может быть использован любой тин реактора. Наиболее удобным
является проточный реактор, работающий при установившемся
состоянии.
Дифференциальное уравнение, выражающее скорость исчезно-
вения реагента Л при установившемся состоянии, может быть
представлено так
моль Л в пнтаппи
с (/ о> -----~-------
ч».н> прореагпровавшо! о Л
моля А в питании
( ’a)dl
(111.72)
113
Проточный реактор для катализа рассматривается как диф-
ференциальный, если скорость протекающей в нем реакции
одинакова во всех точках аппарата. Этот же аппарат определяется
как интегральный, если скорость реакции изменяется внутри
реактора. Дифференциальный метод анализа химических процес-
сов используется на основе данных, полученных с помощью
дифференциального реактора. Скорости реакции в дифферен-
циальных реакторах находятся прямым путем, и в атом случае
мы имеем:
V
а и
ИЛИ
(Н1.7.3)
•ализамвр
Ряс. III. 4. Проточная система прп
установившемся состоянии.
oQ.*ao
При интеграл (.пом методе анализа процессов механизм реак-
ции с его соответствующим кинетическим уравнением подвер-
гается испытанию путем интегрирования кинетического уравне-
ния для проточных условии
реактора. Какой пз реакто-
ров следует полагать пред-
почтительным, можно уста-
новить только после сопо-
ставления их преимуществ
и недостатков примени-
те.! ьно к особенностям иссле-
дуемой реакции. Вообще,
дифференциал ьпый реактор
более пригоден для иссле-
дования сопротивлений диффузии в потоках и химической реак-
ции в то время как интегральный реактор используется с наи-
большим эффектом при изучении кинетики пленочной диф-
фузии.
Проточная система при установившемся состоянии, изображен-
ная па рис. 111. 4. представляет собой модификацию дифферен
ииалытого я интегрального реакторов. Эта установка заключает
в себе большую часть достоинств как дифференциального, так
и интегрального реакторов и исключает их недостатки; она со-
стоит из рециркуляционной цели, содержащей определенное
количество катализатора. Пусть ноток реагента при постоянном
составе л постоянной скорости подается в цель реактора. Примем,
что скорость реакции одинакова во всех точках в слое катализа-
тора Для любого опыта. ]аким образом, это устройство подобно
дифференциальному реактору.
120
При отсутствии рециркуляции ХЙ1 = Ха0, и мы имеем:
(~r<t)cp~ Ха/,ТЛа0 (Ш.79
1 / г а О
где (—?п)ср изменяется для некоторой сродней конверсии потоков
при их поступлении и выходе из слоя катализатора. Следовательно
%af << ХПср < А й j
(Ш.75)
Так как разность Ха/ — А'„ , может иметь большое значение,
то в этом случае использование средней скорости реакции при-
водит к серьезной ошибке.
Теперь нрпмГм, что часть продукта рециркулирует. С увеличе-
нием скорости рециркулирующей жидкости питание при поступ-
лении в аппарат смешивается с большим объемом потока про-
дукта реакции и поэтому величина t будет приближаться
к значению Хд/. Из (III. 75) видно, что при этом ошибка, связан-
ная с принятием постоянной скорости реакции в катализаторе,
соответственно уменьшается. Итак, при высокой степени рецирку-
ляции мы будем иметь:
Ха'-йга° (Ш.76)
v 1*а о
где скорость реакции измеряется в значениях, соответствующих
условиям выходного потока для выбранного объема катализатора.
Этот тип реактора носит название рециркуляционного реактора,
но он может рассматриваться как реактор с обратным смешением.
Пример. Каталитическая реакция
А —>4В
протекает при 3,2 атм и 117° С в трубчатом аппарате, который содержит
катализатор в количестве 10 c.v3. Питание, состоящее из частично конверти-
рованного продукта, подается со скоростью г — 20 л/ч в расчете па пепро-
реагировавшее вещество А.
Результаты пспыгани и:
(7авход, моль/л . 0,100 0.080 0.060 0,010
с«выход» моль/л..................... 0,081 0,070 0,055 0,038
1. Найти кинетическое уравнение реакции
2. Используя кинетическое уравнение, определить объем трубчатого
реактора (в л) с целью получения 35%-ной конверсии Л в В для скорости
питания 2000 моль Л/ч при 3,2 am.it и 117е С (390s К).
3. Повторить решение 2 без использования кинетическою уравнения,
но путем графического интегрирования проектного уравнения.
Решаем первую из поставленных задач.
Так как максимальное изменение относительно среднего значения
концентрации составляет 8%, то можно рассматривать аппарат как диф-
ференциальный реактор с применением (III. 73) для отыскания скорости
реакции.
121
Расчетные
"вход г* « 0 г «ВЫХОД с «ер* .моль /.г А' = «ВХОД - С„ j «ВХОД о ( 1 — “выход Y во
са о
'выход с, . , «выход • 1 а о
. , . ^«ВХоД ‘--«е а о
1 0,84 0,092 1 — 1 14-3 ° 1 - 0.84 л 14-3-0,84
0,8 0,70 0,075 0.0588 0.0968
0.0 0,55 0.0575 0,1429 <>.1698
0.4 0.38 0,039 0,2727 0.2897
,конверсию Для всех опытов на чистое вещество \ ппц 3 *> ото
и 111 С, иудеи „меть: 1 '
' « о Ра о 3»2______мо. ti> Л
Пк А* Г 0.082-390 ’ л жидкости
и
Fa о Са о1’ — 0,1 • 20 —
— 2 моль А/ч
С учетом изменения плот-
ности во время реакции за-
висимость между концентра-
цией и конверсией выразится
так:
Рис. III. 5. Определение кинетического
уравнения реакции А —> 4В.
Y 1 Сд/Сдо
Ifse (Сд. о)
•'ДО 1а == 3 для принятого
основания (чистое Л).
В таил. III. 4 представлен
ход расчетов. При построении
графика для —га относи-
тельно Са мы получим прямую
линию, проходящую через на-
чало координат, что свиде-
жепия первого порядка. Скорость этой
из рис. Ill, 5 в таком виде:
тельствует о наличии разло-
реакцин может быть определена
1 d.\'a {
-Г-Н96
л жидкости
ч • л катализатора
„ воль А
а л жидкости
Решаем вторую задачу.
122
данные
Таблица Hi. 4
^^0 вы ход Х°вход дха _ ЛХД 1 ч-л —га ’ моль А°ср
а Wo0 0,01 л 2 .чо.гъ/ч
0,0455 0Ю455 0,01/2 0,110 0,02275
0.0380 7,6 0,1316 0,0778
0,0269 5,4 0,186 0,1563
0,0170 3,4 0,294 0,2812
Необходимый объем катализатора определяется с помощью проектного
уравнения для трубчатых реакторов. Таким образом, имеем
Л'д—о,з&
(i моль пен popcar. Л
.ноль Л
.«ель яепрореаг. Л
- гп »-------------------
t,-' ч-л катализатора
1’нс. III. 6. Графическое интегрирование
для определения объема катализатора.
Применительно к кинетическому уравнению реакции первого порядка
мы получим:
Г
123
Используя уравнение кинетики для вычисления интеграла, найдем
1' а о
“С а о
Подставляя в эту формулу известные величины, получим:
2000
.ноль А
л жидкости Wo 1 МОЛ1‘ A 'j
ч • л катализатора J \ л жидкости /
= 140 л катализатора
Решаем третью задачу.
Для определения объема катализатора выполним графическое интегри-
рование проектного уравнения из задачи 2, используя табличные данные
для Г( -гп) относительно Хк. Из рис. 111.0 имеем:'
о.зг
ЙЛ""/'
--------=• 0,0/ол
—‘а
о
Следовательно:
TZ (win •U0’ib А ч"л катализатора \
Р = 2CKJ0 ------- ( ОЛРл------------;--5— = 14/ л катализатора
\ ч / \ моль А /
Пример. Каталитическая реакция А —> Ш исследуется
в трубчатом аппарате при использовании различных количеств катализатора.
Скорость питающей смеси при 3,2 «ям/ и 117° С составляет 20 л/ч вещества А.
Концентрации вещества Л в выходящем потоке определены для каждого
опыта:
Объем используемого
катализатора, смг . . 20 40 80 120 160
Невыход» •«о-И’/л . . . 0,074 0,060 0.044 O.ilj.j О.П29
1. Найти объем трубчатого аппарата (в .;) с целью получения 35%-иой
конверсии А в В для питающей скорости, равной 2000 нольЛ/ч при 3,2 атм
и 117“С.
2, Найти кинетическое уравнение для реакции, используя интегральный
.метод анализа.
Из примера .1 (см. стр. 121) имеем для всех опытов;
Спо = 0,1 .ноль А/л жидкости (:аза)
Л* о=2 молъ А/ч; £а = 3
Так как во время опытов концентрация существенно изменяется, то
экспериментальный реактор должен рассматриваться как интегральный
реактор.
Необходимый объем реактора может быть найден непосредственно
из графика для величины Ха относительно V/Fu 0. Из табл. 111. 5 и*рис. III. 7
найдем при 35%-ной конверсии
V
-р— = 0.07
‘‘а о
124
Таблица HI. 5
Расчетные данные
V. .1 катали- за гора у * а и л катализатора ч С п вых од са о _г - dx“ « , / V \ ’ / 1 \ * а ° / л'.о./и Л
1 * ?а г « 0
.ноль А
ч-л
0 0 1 0 0.4
0.02 0,01 0,74 0,0808 0.043 '
0.04 0,02 0,60 0,1429 5,62
0,08 0.04 0,44 0,2415 4,13
0,12 0.01 i 0.35 0,3170 3.34
0,1(5 0.08 0,19 0,3790 2.715
и для скорости питания 2000 .моль Л ч
,, I л катализатора-'/ X/,,,,,, моль Л \ .
I = [ 0,0/----------г-5----И 2000 --------) = 140 л катализатора
\ моль ‘\ /\ ч }
Проводим интегральный анализ.
Применим скатала уравнение скоро-
сти реакции первого порядка. Если оно
окажется неудовлетворительным, то испы-
таем другое кинетическое уравнение.
Имеем:
Обращаясь к кинетическому уравне-
нию Для вычисления интеграла, получим:
* = -7^== (I -=ft)ln—L^- i„.\„
* а о 1 — Л <1
После подстановки значений ga, С,, в
й /ц 0, найдем:
V л катализатора
ыоль К-ч
Рис. III. 7. Определение Г;7-\0
для Ха = 0,35.
( 4 In
Построив зависимость между величинами, взятыми в скобки, мы получим
постоянную пропорциональности к. Вычислим эти величины в табл. III.fi
Для соответствующих точек и изобразим их на рис. III. 8, из которого видно,
что искомая зависимость является линейной.
Таблица III. 6
Расчетные данные
i 111 1 1-Л'й ЗА'а \ 111 ! — з А' 1-Ап Vr, .г катали- затора к 20
0.0808 0.3372 0,2424 0,0948 0,02 0,001
0,1429 0,6160 0.4287 0,1873 0,04 0,00'2
0,2415 1,1080 0,7245 0,3835 0.08 0,004
0,3170 1,5268 0,9510 0,5758 0.12 0,000
0,3700 1,9080 1,1370 0,7710 0,16 0,008
Таким образом, мы можем сделать вывод о том, что квиетическое уравне-
ние для реакции первого порядка удовлетворяет опытным данным. С уче-
том Л-, вычисленной но рис. IlI. 8, получим:
.1 Ж1 д кости
' катализатора
.ч».ть /1
л жидкости
.ноль А
ч • .< катализатора
Рис. III. 8. Определение постоян-
ной к.
Механизм гетерогенного ка-
тализа весьма сложен. Проточ-
ные реакции такого тина со-
стоят, по крайней мере, из
следующих пяти стадий:
1) диффузия взаимодейству-
ющих молекул к поверхности
катализатора;
2) адсорбция реагентов на
поверхности;
3) реакция па поверхности;
4) десорбция продуктов ре-
акции;
5) диффузия продуктов ре-
акции в проточную среду.
Обычно предполагается, что
только один из этих процессов
оказывает существенное сопро-
тивление реакции. Рассматри-
вая адсорбцию, примем, что
скорость адсорбции какого-либо вещества в любой момент про-
порциональна его парциальному давлению р и долевой части
(1 — 0) поверхности, которая осгаегся свободной к этому вре-
мени.
Таким образом:
G=*iP(l-0)
где 0 — часть поверхности, покрытой ацсорбтивом.
126
С другой стороны, скорость десорбции пропорциональна
только части поверхности О, использованной для контакта фа::,
г. о.
При равновесном состоянии скорости адсорбции и десорбции
равны между собой Поэтому
V (1-6) = 4-^6
или
где К — ку. к_х— константа равновесия для адсорбции.
Обозначим через Оа и (-)ь части поверхности катализатора,
покрытой двумя разными молекулами газов А и В. Неиспользо-
ванная в этом случае поверхность будет (1 — (-)и — (%) и мы
получим:
,-ia ~ к-уаРа (1 — ©а — &b)
Г-1а к _ 1а6д,
?1Ь = A ibPb (1
Г-1Ь~
Скорости адсорбции и десорбции при равновесном состоянии
равны между собой, и решениями для этих условии будут:
________А' аРа____
1 + Я ара т К i)Pi,
0. ==___- г КьРъ
1 “Г А аРа “Г А ЪРЬ
Скорость реакции, протекающей на поверхности катализатора,
пропорциональна количествам реагентов на поверхности и, следо-
вательно, поверхностям, занимаемым каждым из них. Например,
для реакции А -.- В # М 4- N получим:
£ @ Q. ____________к\К дРдКъРЬ__________' __
(1 + А ира -р К 1}рь-J- Ктрт т Л пРп)“
________________kpaph ____________
(1 -г АаРа 4- КьРЪ 4- Ктрт + А прп)’-
Пример. Каталитическая реакция на поверхности 2Л —> В
имеет место при давлении 6 ат. Вначале присутствует 20 мол. % вещества Л,
остальное — инертная масса. Экспериментальные значения скорости реакции
и данные о парциальном давлении представлены в табл. III. 7.
Предполагается, что только диффузия вещества Л к поверхности ката-
лизатора и скорость поверхностной реакции являются темп факторами,
которые существенно влияют на общую скорость процесса. Вещество В
не адсорбируется.
127
Таблица III. 7
Экспериментальные и расчетные данные
ра г Г Pai Pai/г Pa/* r
1,2 58,5 0,775 0,1013 0,1570
1,0 47,5 0,655 0,0961 0,1465
0,8 35,5 0,540 0,0909 0,1340
0,6 24,35 0,423 0.0857 0.1213
0,4 13,9 0,299 0,0802 0.1О71
0,2 4,96 0,164 0,0735 0,0897
Найти константы кинетического уравнения. Известно, что коэффициент
диффузии кга ~ 137,5 для условий опыта.
Уравнения поверхностной реакции и массоиередачи имеют вид:
Рис. III. 9. Сравпепие зависимостей
ра г; I г и Pai/'Уг соответственно от г
II Pai-
r = kta (Par —Pat) =
= 137.5 (par-Pm) (Ш.78)
I’ табл. III. 7 приведены
также значения p[ti. вычислен-
ные из уравнения (III. 78).
Запишем уравнение
(III. 77) в таком виде:
Pai 1 , Kg „
77-/-Л7Г-
Зависимость рар |7 г от ра!-
(рис. III. 9) является прямой
линией, что подтверждает пра-
вильность принятого меха-
низма реакции, кинетическое
уравнение которой:
22GP-;
Г ’ (1 + 0,69 pfti)2
Для сравнения на
рис. HI. 9 дополнительно по-
казана также зависимость
Par/Fr 01' Par. которая нс
представляет прямой линии.
Пример- Двуокись
азота с целью ее утилизации
подвергается разложению при
каталитическом влиянии пла-
тиновой поверхности. Некото-
рые измерения при 741° С по-
казаны в табл. III. 8.
128
Таблица HI. 8
Эксперимен та лъиые да и и ыс
г РО2 РХОг 1/г (по расчету)
315 10 85 26,7
750 20 75 55,9
1400 30 65 73,1
2250 40 5.5 97,7
3450 50 45 142,8
5150 60 35 196,0
Кислород (В) и, возможно, двуокись азота (А) адсорбируются. Поэтому
необходимо проверить два кинетических уравнения:
дра __ ^Ра
(Ш.79)
dt 1 -г аРа 4 & ЬРЬ
(1Ра _ кРа
с!т 1~г^ЬР1>
Представим эти уравнения в таком виде:
___Ра I 4- л я.°а + КЬРЪ--.< । r>„ ,
У — — =---------------------Е-— Л -j- Вра -г С Рь
(III. 80)
(111.81)
к
РЪ 1-гКъръ . . _
У =- — =-------£----= Л -|- ВрЬ
Производные 1/r = —dx,!dp, приведенные в таблице, определены числен-
ным методом. Константы кинетических уравнений рассчитаны с помощью
способа наименьших квадратов*.
Для уравнения (III. 81):
2 (Я 4 Вра 4 Сръ — у)2 = mi и
Состав inn три производных относитеilho Л, В и С и приравнивая их
нулю, получим после преобразования:
пЛ42?Ур„4С ^{/ = 0
лУрй4Ь'2р‘4С’2^Рь-2^=о (HI.82)
А 2 2 ^“2 ръу=°
В табл. III. 9 дапы вычисленные для Л, В и С различные коэффициенты.
11 одетанляя, найдем:
6 Л 360/? - 21 ОС — 29700 О
30ОЛ -|- 23 3007? -|- 10 850С— 1 641 000 =-- О
2 ЮЛ —10 8502? 4 9 Ю0С — 1197 000 -= 0
* См. Л. М. 1J а т у н е р и М. Е. Поз и и, Математические методы в хи-
мической технике, Госхимяздат, 1963, гл. XVII.
9 Л. М. Батунер.
129
Таблица III. 9
Вычисленные значения коэффициентов
Ръ Ра'г Ра РиРь Р'ъ Р,Т<
10 85 26,7 2 270 7 200 850 100 19 300 22 700
20 75 55.9 4 191 5 600 1 500 400 314 000 83800
30 65 73.1 4 752 4 230 1 950 900 309 000 143 000
4о 55 97,7 5 374 3 озо 2 200 1600 296 000 215 ООП
50 45 142.8 6 426 2 020 2 250 2500 289 000 321 000
GO 35 190.6 6 660 1220 1 100 3600 240 000 412 000
210 360 — 29 G73 23’300 10850 9100 1 641 000 1 197 000
Решение уранпопий даст:
Л = |0.',77, В 88,8 и С = —4,08
Для уравнения (III. 82)
н.4 - В У, рь — У, у = О
Л V Pb-r В V pl — У рьу ~= О
2 ЮЛ 4-9100/7-1 197 500=0
После решения получим:
Л = 1790 и В = 90,4
Так как отрицательные
постоя иные не имеют физи
ческою смысла, то только
уравнение (111 80) удовлет-
воряет экспериментальным
данным и кинетическое урав-
нение составляет:
0,0005bра
1 + 0,0506рь
На рис. III. 10 дана зави-
симость, построенная по спо-
собу наименьших квадратов
для опытных данных.
Регенерация катализа-
Рис. III. 10. Зависимость pNOi/r от p0j,no- тора в псевдоожижепном
строенная по способу наименьших квадра- слое. В исевдоожижеппом
тов для опытных данных. потоке можно различить
несколько стадии
1. При малых скоростях потока частицы твердого остаются
неподвижными и перепад давления увеличивается, как обычно.
с возрастанием скорости
2 С увеличением скорости достигается критическое состояние,
когда перепад давления соответствует весу насадочного слоя.
130
частицы поднимаются, отделяясь одна от другой; насадочный
слой начинает расширяться.
3. После дальнейшего увеличения скорости потока перепад
давления остается, в основном, постоянным, но расширение слоя
продолжается.
Если поток газовый, то образуются пузырьки, которые про-
ходят через массу, и система в аппарате напоминает собой кипя-
щую жидкость.
4. При высоких скоростях свободная поверхность исчезает
и частицы непрерывно удаляются из аппарата, причем перепад
давления возрастает со ско-
ростью. Это состояние теку-
чести соответствует газовому
состоянию потока. Оно часто
называется псевдоожижением
с разбавленной фазой в отли-
чие от плотной фазы, кото-
рая существует при наличии
наблюдаемой свободной по-
верхности.
Принципиальная схема
аппарата с псевдоожижен-
ным слоем показана па
рис. III. 11, а.
В плоском дппще аппа-
рата имеются отверстия, че-
рез которые подают газ.
Газ барботирует через слой
а
Рис. III. 11. Схемы аппаратов с псе-
вдоожиженным слоем:
« — одноступенчатый аппарат: б — многосту-
пенчаты н.
материала и вызывает его
«кипение». Постоянный уровень материала поддерживается с по-
мощью порша, через который перетекает сыпучий материал.
Минимальная толщина слоя, обеспечивающая устойчив] ю работу
аппарата, 150—200 мм. В тех случаях, когда хотят обеспечить
противоток между газом и материалом, аппарат делают состоящим
из нескольких полок, по которым перетекает материал (рис.
III. 11, <5). В режиме кипящего слоя перерабатываются частицы
до 6—8 мм.
Минимальная скорость потока СМш1 (в кг/ч-лг) может быть
получена с помощью эмпирической формулы:
6’ =.-45 2OOZ)1-80
АШИ ±,Л
1<?(»ТП —(?)
J10-88
(I1I.S.)
где Dтп — диаметр частиц твердой массы, мк;
р — плотность жидкой (газовой) фазы, кг Л(3;
ртв — плотность твердых частиц, кг.м3;
р. — вязкость жидкости (или газа), кг!м-ч.
9*
131
Пример. Катализатор, на котором получен осадок угля, под-
вергается регенерации путем сжигания угля в потоке воздуха.
Общая скорость процесса регенерации г (в кмоль/кг катализатора-ат-ч)
определяется скоростью поверхностной реакции и скоростью диффузии
кислорода к поверхности частиц катализатора:
r= krCTpt = kd (p — Pi)
где Cr — количество угля на регенерируемом катализаторе;
р — парциальное давление кислорода в газе;
Pi — парциальное давление кислорода па поверхности раздела фаз.
Коэффициенты скорости реакции и диффузии определены в таком виде:
A-d =---пгт 2,87- IO-4
IgA-,.— 10 300—
где G— скорость потока, кг/.и2-ч;
D4 — размер частиц, лк;
I — абсолютная температура, °К.
Отработанный катализатор, содержащий 1,6% угля, должен быть
регенерирован до 0,2%. Катализатор поступает при температуре 483е С
и не может быть нагрет выше 565° С-
Регенерирующий газ поступает прн 37,7° С и давлении 3,16 кгс/см1.
Теплоемкость твердой массы и газа равна 0,25 ккал/кг-град. Теплота сгорания
угля составляет 7750 ккал/кг. Свойства газа могут быть приняты такими,
как для воздуха.
Определить необходимые соотношения между содержанием кислорода
в поступающем и выходящем газах, скоростями потоков и толщиной взвешен-
ного слоя. Для того чтобы обеспечить надлежащую регенерацию примем,
что воздух поступает с 20% избытком.
Расчет ведем на 100 кг регенерированного в 1 ч катализатора.
Данные:
рвозд = 1.65 кг/.и3 при 4,22 ат и 565° С;
Ввози= 0.158 кг/м ч,
Пц — 44,5 .«к =0,0000445 м
Количество сгорающего углерода составляет:
99,8 — 0,2 = 1,42 кг/ч или 0,118 кмоль/ч
Количество подводимого кислорода:
1,2-0,118 = 0,142 кмоль/ч
Теплота реакции:
1,42-7750 = 11000 ккал/ч
Обозначим через а мольную долю кислорода в газе при сто поступлении
в аппарат. Общее количество газа равно:
0.142 - 29 4.11
--------=-------кг/ч
а а
132
Изобразим материальные потоки па схеме:
Газ
4,11/а кг/ч; 37,7° С
Отработанный катали-
затор 101,42 кг ч; 483° С
Выходящий газ
4,11 /а -4-1,42 кг/ч
Регенерированный ката-
лизатор 100 кг/ч; 565° С
Тепловой баланс:
откуда
101,42 ( 483 — 37,7) 0,25 +11 000 =
Риг. III. 12. Зависимость степени флюидизации от
коэффициента расширения насадочного слоя Nf
при различных диаметрах частиц.
а — 0,0008
Количеству поступающего газа:
0,0608 ‘' 68,0 Кг/<4
Из формулы (III. 83) найдем;
Г 4.53- II)4(44,5 • Ю-*1)1’82 [1,65 (2560—1.65)]0,₽4
°МИП~-----------—— --------------------------------
О,1580,88
кг
= idi ——
.и- ч
Пусть G = 10 СМИП = Ю-7,17 = 71,7 кг/.н2-ч.
По рис. III. 12 для коэффициента расширения насадочного слоя имеем
7 ~= 1,17, а по рис. III. 13 порозпость равна с.) — 0.58 при минимальном
потоке. Следовательно:
1-^ , 0,42
ь — 1 ту— 1 - — - — 0,64
А/ 1,1/
{.’слой — (1 —0,64) 2560 = 920 ке/.ч9
133
Ad = 2,87 • 10-* = 0.00495
•44, S1’”
lgAr = W300—A*r»= 7.0-4
Обозначим:
F — 68,5 кг/ч ностуиающого газа;
М — количество катализатора в реакторе, кг;
Рис. 111. 13. Зависимость порозпо-
сти с/ от диаметра частиц Ъч.
клоль кислорода nt t>p
И----------------------------ГГ—
кг поступающего газа /’
Исключая />i с помощью двух
кинетических уравнении (стр. 132).
получим:
Р
г~ 1/A-d 4-1 1. ,-Сг ~
Рп
"" «ln(i/Ad4’l/ArCr)
Таким образом:
М == 4,22F ; 4,22 • 68,5 ~ 289,0 кг
Поперечное сечение реактора составляет:
F 68,5
G 71,7
=0 95
Толщина взвешенного слоя:
Л/
289
ОслойЗ 920-0,95
Автокаталитические реакции. Если во время реакции
одно из взаимодействующих веществ действует как катализатор,
то такой процесс носит название автокатализа. Рассмотрим
случай, когда превращаемое вещество действует как катализатор.
Тогда константа скорости реакции зависит от концентрации
вещества, действующего как катализатор. Полагая эту зависи-
мость линейной, примем се равной / - (а — .г). Тогда для
скорости реакции найдем:
- (А- -с к' (« — х)] (а — х)
Разделение переменных, интегрирование и подстановка
= а — х дают:
р dx' '*”* ’ч* р d:
Т |А- А' («- х)1 (а-х) “И =
Разложим подынтегральное выражение па элементарные дроои:
1 _ 1 к'
[А 4 А'з]з kz к (к k'z)
Отсюда получим
Г dz _ f dz , к' С dz
Te —J (к 4- k'z) z Г J T^^FJ A-4- A-T —
= — 2-In 3+4- In (A- ±k'z) -LC-4-ln ~t/~ ч C =
fl n H fj
In
A- t-к' (a -z)
a — x
Постоянную интегрирования С найдем из условия: х ~ 0 при
т = 0. Ито нам дает:
Подставляя в формулу, определяющую т, вместо С ее значение,
получим:
1 Г к-гк'(а-х) к-]-к'а 1 1 а [А'4- к’ (а — х)]
T= -j- In — — — 1 и — ------ = — In ——---- / '
к j. а—х a J к (а — х) (А- 4- А «)
В том случае, когда катализатором является получающееся
в результате мономолекулярпой реакции вещество, дифферен-
циальное уравнение принимает’ вид:
— = (А- ]-А'х)(а — х}
Как и выше, имеем:
f dz 1 Г dz к' dz
~ I (& + *' (а—2)] г — ~ к-^-к'а J ~z к^к'а J (A-J-A' (а - г)] “
1 f d [A-f- к' (а-з)] fdzl ’1 (| „ ,
7~ ,, , ,----т1 — — = . , г, (in А -А- (а —г) —1пг} =
А т-kz J к-^-к (а—z) J г к-{-к а 1 1
1 k-i-k'(a-z) 1 A-I-A'a: L/1
к 4 A- fl z A- + A « a—x
Используем начальное условие: x = 0 при т = 0:
Таким образом, окончательно имеем:
1 Г А-т-А-'х к I 1 . ti(k-i-k'x)
Т “ < I /, ’’I--------1П — - III ——----—
А 4- А-'а |_ «— “J к т к а к (а — X)
Адиабатические реакции. Если а — начальная моль-
ная -“концентрация реагирующего вещества, а х — его убыль
135
к моменту времени г, то о случае мономолекулярпой необратимой
адиабатной реакции х оказывается связанным с т следующим
уравнением
причем К является
температуры 7’аос-
уже нс константой, а следующей функцией
К =е
Температура, которую принимает реакционная масса в момент,
когда прореагировало х моль/л, находится с последней величиной
в такой зависимости:
(а — ж) qb = Q (Tl;— Т) (II Г. 84)
где q — тепловой эффект реакции;
b — объем реакционной массы;
<2 — теплоемкость, отнесенная ко всей реакционной массе;
7’1; — конечная температура.
Найдем время, которое потребуется для того, чтобы темпера-
тура реагирующей массы повысилась от 7\ до Т2.
Поскольку переменными являются х и Т, то дифференцируя
равенство (III. 84), найдем:
d Т qb dx
dr Q dx
Исключая отсюда и из исходного дифференциального уравне-
ния dx/dr, получим:
Используя (III. 84), исключим отсюда (« — х):
dT
dx
= f~'r {Тк-Т)
Разделяя переменные, получим:
JL dT
dx c'E-el Тк—Т
После интегрирования находим
Этот ннтс грал невозможно выразить через элементарные функции.
Применяя формулу Симпсона, найдем его приближенное значение,
136
приняв за пределы интегрирования 7\ и Т2. Разобьем промежуток
интегрирования Т., — 7\ на два частичных промежутка. Тогда
/г = Z2~~ Г|- , а значения подынтегральной функции в точках 7\,
Г.-О Гл т
Д1 ' Т„ оудут:
9 " -
Подставляя эти величины в формулу Симпсона, найдем
Ота формула используется при исследовании скоростей каталити-
ческого разложения химических соединений.
Полунепрерывные процессы. Некоторые процессы осу-
ществляются таким образом, что реагенты поступают в аппарат
с различной скоростью. В одном случае вся масса реагентов за-
гружается в аппарат быстро, а в другом — питание производится
постепенно. Пли же один реагент может загружаться у входа про-
точного аппарата, а остальные инжектируются в нескольких ме-
стах по длине реактора.
Примерами такого рода процессов могут служить регенерация
подвижного катализатора, где воздух инжектируется в нескольких
точках реактора во избежание повышения температуры, а также
полимеризация олефинов при каталитическом воздействии пленки
фосфорной кислоты, распределенной в зернах твердой массы.
Целью таких операций является ограничение тепловых эффектов
или улучшенное распределение продукта. Они также требуются
для ограничения растворимости или же рециркуляции частично
отработанных материалов пли разбавителей.
Из всего разнообразия этих процессов рассмотрим только
некоторые из них.
Пример. Реакция
А — В —> Продукты
протекает изотермически в реакторе с мешалкой. Реагент А, находящийся
в растворе при концентрации Сяо, загружается в аппарат с обкомом Fu;
после этого реагент В подается в аппарат насосом со скоростью IP мя/ч в виде
раствора, имеющего концентрацию С’ь0.
Во время реакции раствор не выходит из реактора и плотность массы
не изменяется.
Требуется найти зависимость между временем и количеством пепрореагп-
ровашпего вещества А, присутствующего в баке.
137
Имеем:
L(l у
1' x( 1,0 (y,an ,?a)
6^ -----------j--------
I I't. + irr
Таким образом,
<7wa _IZ ,,-z- z, ^7,’« (li tCj0 — I o^no —
——Г-----—'►la' u- -----------г;--—--------—
ЙТ V0-f-llT
Мы получили уравнение следующего вида:
<Ь/ __ У (я ~г Ъх 4* c.v)
dx d -Т ex
которое является уравнением Абеля п решается численными методами (ем.
выше).
Пример. Реактор с мешалкой, объем которого ГС1, наполняется
реакционной массой, состоящей из веществ А и 13 в растворе.
Реакция идет по схеме:
А -г В —> 11 родукты
Раствор вещества В с концентрацией Cbw нагнетается в аппарат со ско-
ростью IK ,н3/ч. Конечный раствор выходит из пего с той же скоростью.
Определить ход реакции.
Составляя материальный баланс для вещества Л за промежуток вре-
мени dx. получим:
Приход = Убыль -|- Накопление
О = Н Т,’a dr + rV'o с/т -г Vo dCb
Для вещества 13
1 гСщи<ix = wcb (1х + г dx -г (1Съ
Следовательно,
(’vcbw - вс; - wocacb) dx v0 <icb
-(WCa - WQCaCb) С/r- 1-; c/crt (III. 85)
Разделив, найдем:
dCn ll Cg - kVtiC(lCh
dC b 11- С{ду И С b — AV aC b
Мы опять получили уравнение \бс 1Я, которое также решается численными
методами для отыскания зависимости между Си и Сь- После этого уравнение
(П1. 85) может быть проинтегрировано для определения связи между вре-
менем и концентрациями.
Пример. Газ В поступает в реактор с мешалкой, в котором на-
ходится раствор вещества Л при объеме Р'(|. Растворимость газа 13 является
ограниченной, по не зависит от присутствия растворенных веществ. Найти
зависимость между временем и переменной скоростью питания, необходимой
для но.щержания раствора в состоянии насыщения нерсагирукицим веще-
ством В.
Обозначим через Cbl) постоянную концентрацию вещества В.
Материальный баланс для вещества В:
Приход = Убыль 4 Накопление
И’ dr = к VvCaCao dx |- 0 (111.86)
138
Для вещества А имеем:
О — 1' oCqC о '!т 4- V (> d(’a
(П1.87)
Интегрируя (III. 87), получим:
Са=Саое ОТ
Подставляя в (111.86), найдем
П- = кСаСь о = kV0Cn ,}СЬ „е ~ кСЬ от
что представляет искомую зависимость.
Фильтрация суспензий во многих случаях может быть отнесена
к полунепрерывным процессам. Наиболее важные факторы, влияющие па
этот процесс: 1) перепад давления; 2) площадь фильтрации; 3) вязкость филь-
трата; 4) сопротивление фильтрующею слоя (осадка); 5) сопротивление филь-
трующей ткани и начального слоя осадка.
Д в и ж е л и е ж и д к о с т и через ф и л ь т р у ю щ и й с л о и.
В данных условиях мы имеем, в отличие от других случаев, движение жид-
кости через фильтрующий слой при непрерывном увеличении его высоты.
Таким образом, если давление фильтрации постоянное, то скорость потока
будет постепенно уменьшаться и, наоборот, если скорость потока должна
быть постоянной, то давление должно постепенно увеличиваться.
Вследствие того, что частицы, образующие при фильтрации осадок,
обычно достаточно малы, а скорость движения фильтрата незначительна,
мы имеем здесь почти всегда условия ламинарного течения. Поэтому для всех
случаев процессов фильтрации действительна формула:
1 rfV ________ е3 АР
A dr ~ 5 (1 - <?)а
(Hi, 88)
где Л — общая площадь поперечного сечения слоя твердых частиц;
V — объем жидкости, протекающей за время т;
ДР — перепад давления;
I — толщина слоя;
е — породность;
ц — динамический коэффициент вязкости жидкости;
S — удельная поверхность насадочного материала.
Фильтрующие осадки могут быть несжимаемыми и сжимаемыми. 13 случае
несжимаемого осадка обусловливаемое им сопротивление движению филь-
трата почти не зависит от перепада давления в слое, а также скорости его
образования. С другой стороны, при сжимаемом осадке увеличение перепада
давления или скорости потока способствует образованию более плотного
осадка с большим сопротивлением. Рассмотрим несжимаемые осадки, для
которых е в уравнении (111. 88) может быть принята постоянной.
Тогда величина —----у-^-
а (1 —еу S-
будет выражать свойства частиц, из которых
состоит осадок, и, следовательно, должна быть для данного материала посто-
янной. Таким образом, мы можем написать:
1 (IV _ ДР
А dx HilI
(111. 89)
(111. 90)
139
Формула (111.89) является основным уравнением фильтрации. Постоянная
/? дает удельное сопротивление и численно равна перепаду давления, необ.хо
димом у для получения единичной скорости потока фильтрата, имеющего еди-
ничную вязкость, через слои осадка, объем которого составляет единичный
куб. Размерность R будет L-s.
Зависимость м е жд у то л щи п о по с а дк а и о б ъе м о м
ф и л ь т р а т а. Количество твердого вещества в фильтрующем осадке:
т = (1 —с) И/р8
где os — плотность твердых частиц.
Количество жидкости, остающейся в осадке:
т- с Al о
где у — плотность фи п»трата.
Если J есть (массовая) доля твердых частиц в начальной суспензии, то
J _ (I—е) Л/Qs ___ Масса твердых частиц в фильтрующем осадке
1 — J (I' + e/lOy Масса фильтрата-)-масса жидкости в осадке
ил н
(1-./) (1 - е) .4/о5/Гу + ЛсУ/с
Следовательно:
, JP'y
Л [(l-J)(l-e)o5_yeQ]
(IIL 91)
Обозначив объем осадка, образующегося при прохождении единичного
объема фильтрата, через
Г = (Т-J) (1-6>)у8-Л'О (111'92)
получим:
Подставляя в (III. 89), найдем:
с/Г _ Л- ДР
dx PuVr
(HI. 93)
(HI. 94)
Если сопротивление потоку к началу процесса равно пулю и объем V
фильтрата проходит в период времени т, то (III. 94) может быть проинтегри-
ровано при условии, что зависимость между Р, I' п т известна. Рассмотрим
следующие два важных случая.
I. Фильтрация проходит при постоянной скорости.
Имеем:
ь'Г Г
—:— = — COIlst
ат т
Следовательно
I* _ Л~ :\р
х BiiVv
или
т Лиг т.
“Г “ Л* АР (III. 95)
откуда находим, что разность ДР прямо пропорциональна объему V.
140
2. Фильтрация проходит при постоянном давлении.
Интегрируя (Ill. 94), получим:
V- _ А-ХРт
2 ~ Пии
или
т /?р.п
Т~2.42ЛР V
(Ш.96)
(111. 97)
Отсюда следует, что для фильтрации при постоянном давлении сущест-
вует линейная зависимость между V2 и т или между т/V и V.
В формулах (III. 96) и (III. 97) было принято, что фильтрация прово-
дится при постоянном давлении за весь период процесса, т. с. V = 0 при т =
= 0. Однако в большинстве случаев перепад давления ДР в начале процесса
постоянно увеличивается и давление приводится к его конечному значению
только через некоторое время тх, в течение которого проходит фильтрат
в количестве Ух- Тогда после интегрирования (111. 94) получим
Таким образом:
2'Р \Р
(Т- l-д) (V- 4- 21\) - (т- Tt)
илп
Т~~Т1 flp / р- 1,’ \ 1 /II J QQX
Г-1\ 2.4 s ДР [ l) ‘ Л2 ДР (111.98)
Нетрудно видеть, что между I - и т, а также между -р—р— и V — |‘д,
устанавливается линейная зависимость. Здесь т — тх представляет время,
в течение которого фильтрация протекает при постоянном давлении;
V — Vi — соответствующий объем полученного фильтрата.
Фильтрация суспензии с учетом совмест-
ного влияния ткани и осадка. Гидравлическое сопротивление
движению жидкости, оказываемое тканью, не может быть определено, так’как
носледняя всегда содержит в себе частицы осадка. Совместное сопротивле-
ние ткани с включенными в нее твердыми частицами осадка значительно
превышает сумму пх отдельных сопротивлений.
Предположим, что фильтрующая ткань и начальные слон осадка по своему
гидравлическому сопротивлению эквивалентны толщине L осадка, получа-
ющегося при дальнейшей стадии процесса. Тогда, обозначив через ДР раз-
ность давления но общей высоте фильтрующего слоя, по lyqiiM в соответствии
с уравнениями (III. 89) и (III. 93):
1 (IV
А ' dx /?ц(И-Л)
.I2 \Р
&Р
<!У
ах
~t-L
(Ш. 99)
оПи
Уравнение (111.99) можно проинтегрировать в пределах т = 0, V <= 0
н т ~ п, V = У1 при постоянной скорости фильтрации и в пределах т =* тх,
I == Их при постоянном давлении фильтрации.
Для постоянной скорости фильтрации имеем:
Г1 /Г2 \Р
141
или
или
Т1 _ Z?LLP у I РрР
Т7~ Л2 1 ’ -I ^Р
(/2_иЛ£г т
1 1 V 1 Яцу 1
(111. 100)
(III. 101)
Для постоянного давления фильтрации найдем:
1 (г. у;), М из А Р о' '>)= Ч) (III. 102)
или v Ztpt’ (111. 103)
или Г —Т1 Яцр . / у у \ । 1 j_ ZZjlZ, U 11 л2 др Л ЬР (III. 104)
И — Vj 2Л-&Р
Таким образом, получена липойпая зависимость
между t — у
и V -
— Уп причем тангенс угла наклона пропорционален удельному гидравли
ческому сопротивлению, как и. в случае движения жидкости через слой
осадка — формула (III. 98), с тем отличием, что здесь линия на рисунке нс
будет проходить через начало координат.
Рис. III. 14. Схема
Точка пересечения прямой с осью ординат даст
возможность определить эквивалентную толщину
осадка за счет сопротивления фильтрующей ткани,
хотя такого рода результаты не могут быть воспро-
изведены из-за того, что это сопротивление суще-
ственно зависит от начальных условий опыта,
С ж и м а е м ы е ф и л ь т р у ю щ и е
осадки. Вследствие гидравлических потерь на
трение, возникающих при движении фильтрата
через слой осадка, мы здесь будем иметь градиент
давления. При взаимодействии твердых частиц
с жидкостью осадок уплотняется. Поскольку уплот-
няющая сила, проявляемая жидкостью, распростра-
няется к частицам ио глубине осадка, изменение ее
к расчету сжимаемых
фильтрующих осад-
ков .
будет происходить от нулевого значения па сво-
бодной поверхности осадка до' максимального па
фильтрующей ткани.
В действительности уплотняющее давление за-
висит от структуры осадка и природы контакта
между частицами, но оно может быть выражено как функция разности давле-
ний на поверхности осадка Р2 и на глубине z, т. е. Р2 (рис. III 14). Для
породности в любой точке будем иметь:
cz-/(Z’.,-P2)
Нетрудно видеть, что порозность уменьшается ио мерс перехода от
свободной поверхности к фильтрующей ткани при одновременном увеличении
гидравлического сопротивления, В этом случае уравнение (III. *89) примет
вид:
1 с/У 1 dPz
Л с/r Z?zp dz
(III. 105)
где Hz — удельное сопротивление в данной точке.
142
Равенство (III. 105) можно написать так:
1 dV = сг______________________1_ dPz
Л t/т 5 (t — ег)'~ S2 p dz
(HI. 106)
Бесконечно малая толщина слоя осадка dz должна быть связана с малым
объемом фильтрата <7V, проходящего во время образования слоя осадка.
Так как осадок сжимаем, то объем его слоя, получающегося при прохождении
единичного объема фильтрата, пе будет постоянным, однако весовое его коли-
честно (масса) с почти не зависит от условий, при которых образуется осадок.
Имеем:
</= (IV —---Ц---—
(1 -~ez) L’s л
Из уравнения (II. 106) найдем:
1 dV ez (1—ег) оЛЛ 1 dPz
А dX 5(1—№ с р </Р
^L = —___________(ПТ. 107)
dx 5(1—с5) Л’“ pc 41' рс/Л dV
где
jf 5 (1 ~«z) 5-
ezQs
Удельное гидравлическое сопротивление R отнесено к потоку фильтрата
па единичное количество массы осадка, образующегося на единичной площади,
в то время как R имело своей основой поток фильтрата, проходящего через
единичный объем осадка. Сопоставление формул (III. 89) u (III. 107) показы-
вает, что для сжимаемого осадка
cR = vR, т. е. И - R —
с
и размерность R будет L-2 IAM 1 = M“XL.
Из уравнения (III. 107) имеем для любой фильтрации суспензии:
V Р.
J dx pc J fiz
о Fi
Так как по толщине осадка величина dVjdx является постоянной величи-
ной, то получим:
dV
dx
Fi
Л2 Г dl>2
1>с J Rz
F»
(III 108)
Выше было показано, что Rz есть функция разности давлений Р2 — Pz
и что она нс зависит от абсолютного значения давления.
Напишем:
Г/2--= Т{ (Л.- вгГ'
где //' но зависит от Р>.
143
Т огда:
Pi Pi
f dPz — 1 Г (jpz — 1 (/JJ —_ 1
J bz ~Ti' J (fi2~^z)n' —Tv i—«' ~Tv i -ri'
i*2 1 *2
Таким образом:
«V _ Л'2___________AP _ Л- АР _ Л2 AP
f'T ~1’рс7? (l-и') AP’1' — ГрГ/f Pn' ~ FucZ?
где
Ii = (1 — n) /f; H -- П APn'
Выражение (III. 109) может быть проинтегрировано таким же образом,
как и для несжимаемого осадка, причем формула для несжимаемого осадка
здесь также остается в силе, если vli заменить величиной сВ.
Оптимальны й режим фильтрации сусле н-
з л й. Оптимальная толщина лепешки на фильтрпрессе зависит от сопроти-
вления, оказываемого фильтрующим осадком, и от времени, расходуемого
на разборку и сборку аппарата. Получение более тонких фильтрующих осад-
ков приводит к ускорению процесса фильтрации, по вместо с тем здесь имеет
место более частая разборка фильтра и, следовательно, увеличенный расход
времени на проведение процесса фильтрации. Для фильтрации при постоянном
давлении из (III. 102) имеем:
т _ Лиг? Т7 , НцЬ
V 2.42 ДР ' ~ А АР ~
Р,У-Р.2
(ТП. НО)
где Z?j и В2 — постоянные.
Решая уравнение (III. 109) относительно т, получим:
т = В Д'2 — B2V
Обозначим через т' время для разборки и сборки фильтрпресса, причем
оно не зависит существенно от толщины лепешки. Общая продолжительность
процесса фильтрации, при котором получается фильтрат объемом V, будет
т 4- т', и общая скорость фильтрации составит:
V
Величина П’ф будет максимальной, если «ПГф/г/И = 0. Дифференцируя
П’ф по V, и приравняв производную нулю, найдем
откуда:
РД’2 + B.2V г' — 7 (2 7?!V •/?,) = и
т' = BLV*
11 ром ы в к а о с а д к а и а ф и льтрпресса х. Для
промывания осадков на фильтрпрессах могут быть применены «простой» и
«полный» способы. При первом способе промывная жидкость проходит но
тому же пути в каналах осадка, по которому проходила суспензия; в этом
случае имеет место эрозия осадка. По второму способу промывки жидкость
поступает в отдельные каналы, расположенные в так называемых промывпых
плитах, и протекает через осадок по всей его толщине сперва в обратном на-
правлении, а затоми том же направлении, что и фильтрат. Площадь промывки
вдвое меньше площади фильтрации и, кроме того, промывная жидкость здесь
должна пройти дважды через слой осадка. Таким образом скорость промывки
составляет, примерно, четвертую часть конечной скорости фильтрации.
Ра с чет ф и i ь т р а ц и и на основе опыт и ы х
дан п ы х. Фильтрация суспензии производится на фильтрпрессе с 12 ра-
мами; площадь рамы составляет 0,1 лг, толщина ос равна 25 .ил. В течение
первых трех минут давление фильтрации постепенно увеличивается до ко-
нечного значения 4,0 атя, причем скорость фильтрации поддерживается
постоянной. После начального периода фильтрация проводится при постояв
ном давлении и по истечении следующих 15 мин завершается образованием
осадка. Затем следует в течение 10 .ним- промывка осадка водой при давлении
3 атя по способу «полной» промывки. Требуется определить объем фильтрата
и необходимое количество воды для промывки.
Предварительно была проведена фильтрация образца суспензии на ла-
бораторном листовом вакуум-фильтре площадью в 0,05 .ад2 при вакууме
500 .н.ад рт. ст. Объем фильтрата, собранного за первые 5 мин, составляет
250 лл, а в конце следующих 5 .чин. — 150 .мл. Осадок несжимаем; сопроти-
вление фильтрующей ткани ла вакуум-фильтре и фильтрпрессе принимается
одинаковым.
На вакуум-фильтре процесс фильтрации проводился при постоянном
давлении. Поэтому в соответствии с (111. 102) имеем:
1Л .. ДУМ2
— I - - т
V /11И,-
11а фильтрпрессе сперва получается фильтрат в количестве при постоян-
ном скорости фильтрации за время Тр после чего процесс протекает при посто-
яппом давлении. Пользуясь формулой (III. 101), найдем:
1 у
F
------— т
-Kjxy
Далее, из (III. 102) имеем:
(f2-F;) + 2 —
' 1/ у
Опытные данные фильтрации:
т = 5 .чин',
F = о,23 л;
.4 = 0,05 л2;
Подставляя эти величины, пайдсм
Нии
т 10 .ипи;
v= 0,40 •<;
Ар 0,7 атя
и. in
0>252 + 2 • 0,05 — 0,25= 2 ’0,/ ’ШЬ‘
и
0// + 2- 0,05—0,4 =
у
Я.Ш>-
2 0,7-0,05°- 10
Нии
0,0625 + 0,025 — U1.1,
v H\w
0,0350
0,160+0,04
У /?ИУ
Ю Л М. Патупер.
145
откуда:
L/u — 3.5; R = 0,116
Объем фп и,трата Vlt собранного в течение периода работы фильтрпресса
при постоянной скорости фильтрации, может быть найден из равенства (при
Л = 24-11.1 = 2,4; ХР = 4: т= 3)
Г?+2Л.ЗЖ„*_™3
И III
H-H-VU7!- 600 о
откуда;
V\ ~ 21.
Для периода фильтрации при постоянном давлении имеем т — Ti —
— 15 ,иин.
Можно написать
(V2 -441) -1-16,8 (Г —21) = 400 • 15
или
V2+ 16,81'-5204 = 0
откуда V — 71л.
Конечная скорость фильтрации в соответствии с уравнениями (III- 89)
и (III. 93):
и Ф ____________„
Й1Ц,(Г + ДД\ ».IW(71+M-3.5)
Так как скорость промывки равна конечной скорости фильтрации,
то скорость промывки при 4,0 атм равна 0,62, а при 3,0 атм — 0,46 л/мин.
Следовательно, расход промывной воды в течение 1U мин составляет:
6'в = 0,46-10^4.6 л
К о м б и и п р о в а п п а я ф и л ь т р а ц и я. Суспензия,
содержащая 0,9 кг твердого (плотность 3,0 кг/дм:!) на 1 кг воды, подается на
барабанный фильтр, длина и диаметр которого 0,6 ,w. Барабан делает один
оборот за шесть минут, и 20% фильтрующей поверхности его находится в не-
посредственном соприкосновении с суспензией в любой момент времени.
Нужно определить толщину осадка на барабане при вакууме 500 .ад рт. ст..
если скорость получения фц 1ьтвата составляет 273 кг/ч, а порозность осадка
0,5.
Некоторое время спустя работа барабанного вакуум-фильтра приоста-
навливается и процесс фильтрации осуществляют на фильтрпрессе с поверх-
ностью рамы в 0,1 м~. На разборку фильтра затрачивается 2 мин, па сборку
также 2 мин, причем 2 .ими требуются дополнительно для удаления осадка
с каждой рамы. Если фильтрация проводится при давлении 17,5 атм с той
же скоростью, которая была для вакуум-фильтра, то нужно найти, какое
минимальное число рам потребуется в этом случае и какова их толщина.
Осадок принят несжимаемым и сопротивлением фильтрующей ткани можно
пренебречь.
1. Барабанный вакуум фильтр.
Фильтрующая поверхность барабана:
Л л 0.6-0,6 = 0,36 л №
146
Скорость фильтрации:
П'ф = 273/60= U,90455 .и3 'лип
Объемная скорость образования оса ц«а:
0,00455 - 0,9 -U- • = -2^?= 0,00274 л3/мин
0>о 3,0 1,66
Ироде икитслыюсть соприкосновения фильтрующей поверхности с су-
спензией равна 6-0,2 =1,2 мин.
Объем осадка, образующегося за один оборот барабана, составляет
0,00274-6 = 0,0164 л3.
Толщина осадка:
0,0164 • 1000
0,36 • 3,14
= 14,5
.1131
Далее из уравнений (III. 89) и (III. 94) имеем;
с?И _ ДР Л ХРЛ2
с/т — Ар/ — ЯрУу
При постоянном давлении из (III. 96) получим
2
V2 = Л/МЧ= А' \РЛ2т
Лрг;
ил и
(0,00455 - 6)2 = А' 0.66 (0,36 л)2 1,2
откуда
К = 0,00073
2. Фильтрпресс.
Пусть фильтрпресс имеет и рам толщиной 6 .иль Продолжительность
полного цикла фильтрации составляет;
=- 0,00455 .«3/.и.нл
2 = Тф-Ь 2ft-f-4
где тф — время фильтрации, мин.
Общая скорость фильтрации:
У»
ТФ + “Zl л- 4
гДе Уф — полный объем фильтрата в течение одного цикла. Д ыее:
Объем рам фильтрпресса
I'd) =-: --------------г;---------т-----;--------- 1,66 ьс
4 Ооъсм лепешки X Единица ооъема фильтрата
где 6 — толщина лепешки осадка.
1^= 0,00073 • 17.5 (2 - 0,1 • п)2 тф = 1,66 гг&2
откуда:
!.«№ .
Тф= O.W073-t7,7,-4-O,0l-«^-0'll:bl "
Таким образом, получим:
Л Л-,/-- 1,66 nt)
0,00100 О.0О5 б2 + 2»-Н 4
10*
147
Упрощая, найдем
0,0054 62 + 2» т 4 = 0,00365 пб
откуда:
4 J 0,0054 6’-
0,03656 + 3
Величина п является минимальной при d«/</6 = О, т. о. когда
(0,0365 6 + 2) 0,11 6 — (4 + 0.0054 62) 0,0365 = 0
пли
0,000207 б2 + 0,022 6 — 0,1460 = 0
Решая это квадратное уравнение, получим 6 ~ 45 .u.w. Следовательно,
п — 4,15, и минимальное число рам составляет 5.
Глаза IV
Химическая кинетика
энзиматических процессов
Сведения о кинетике и механизмах энзиматических
процессов и реакции получены, главным образом, благодаря много-
численным работам по кинетике химических реакции.
В общем случае для скорости реакции, как известно, имеем:
dx
где Л и В — реактанты;
к и Р — постоянные.
Общий порядок такой реакции составляет:
п — а — р — ...
Следует отметить, что не все реакции протекают таким образом.
Для энзиматических реакций, в частности, найдено, что их ско-
рость изменяется с концентрацией реагента S (субстрата или пита-
тельной среды) по закону:
где а. и I) — постоянные при данной температуре.
Зависимость (IV. 1) не соответствует простому порядку реакции,
однако для некоторых условий он может быть выявлен. Так,
например, если Ъ [SI 1, то скорость
г - a [S]
и реакция первого порядка относительно субстрата. Если b [S1
>> 1, то скорость
а
v'~ Т
149
и, следовательно, реакция нулевого порядка относительно суб-
страта.
С целью получения необходимого кинетического уравнения
энзиматических процессов часто используется приближенный
метод решения дифференциальных уравнений, известный под на-
званием способа установившегося состояния. Последний основан
па предположении о том, что некоторые промежуточные про-
дукты, как. например, свободные радикалы и энзимосубстратиые
комплексы во время реакции подвергаются медленному изменению.
Если [XJ есть концентрация такого промежуточного вещества,
то будем иметь:
-^Д = 0 (IV. 2)
ат
Такие соотношения приводят к значительному упрощению
дифференциальных уравнений и позволяют легко получить ре-
шение. Пусть мы имеем, например, следующую схему реакции
А + В X
А-,
Дифференциальные уравнения кинетики, соответствующие этим
реакциям, будут:
= Ь1Л11 В]—Лг (XJ (IV. 3)
£2 X ti-Ti
[А] [В] == k_t [Х]-/гг [X] (IV.4)
с, X
^ = Л-2[Х] (IV. 5)
Для точной обработки задачи необходимо исключить [X J и
решить конечное дифференциальное уравнение для получения
IE] в виде функции от т. Однако несмотря па простоту, это не
может быть выполнено известными математическими приемами.
Способ установившегося состояния предусматривает использова-
ние (IV. 2) и, следовательно, из (IV. 4) найдем:
ЛИЛ] [В]-/с_1[Х]-Л2[Х] = 0 (IV. 6)
Для концентрации [X] получим
[Х] = А»1АП[А] В) (1V. 7)
^—1 Г" ^2
Подстановка этого значения в (IV. 5) дает:
</[Р] _ ЛУГАНО]
-ItT- кг (lV<8)
150
Обозначив начальные количества А и В через а и Ь и количества
продукта Р ко времени т черех ж, получим для 1\ . (5 следующее
уравнение
б'х _ /д/л, (и — х) (Ь — х)
ТТ ' . А'.
которое легко решается простыми методами.
Реакции с одним субстратом
Энзиматические реакции состоят из двух стадии: первая
стадия представляет образование комплекса между энзимом Е
и субстратом S:
(I) E + S ES
it-,
а вторая — разложение комплекса с образованием продуктов
реакции Р, причем здесь происходит регенерация энзима
(II) ES — > P-f-E
При высоких субстратных концентрациях энзим насыщается
субстратом и практически полностью находится в виде комплекса.
Поэтому увеличение концентрации субстрата не вызывает даль-
нейшего увеличения его содержания в комплексе. Следовательно,
скорость реакции, пропорциональная концентрации комплекса,
не зависит от концентрации субстрата.
При малых субстратных концентрациях, с другой стороны,
наибольшая часть энзимных молекул находится в свободном
состоянии и только незначительная часть их связана с субстратом.
В этом случае количество образующегося комплекса пропорци-
онально количеству субстрата и. таким образом, скорость образо-
вания продуктов реакции, т. с. скорость реакции (II) пропорци-
ональна концентрации субстрата.
Скорость образования комплекса складывается из трех вели-
чии. Комплексы образуются посредством реакции (I), идущей
слева направо, скорость которой Ад [Е] IS]; они удаляются в ре-
зультате обратной реакции (I), протекающей со скоростью k_t {ES],
и реакции (II) со скоростью Ад [ESI-
Суммарная скорость образования комплексов, равная пулю,
представляется в соответствии с методом установившегося состо-
яния так:
MEHSJ-fc-JESl-A-JES] 0 (iV. !>)
Концентрация свободного энзима [EJ равна [Е|„— {XI, где
[Е]() — начальное количество энзима, которое составляет:
[И I» IE]-HESJ
151
Следовательно, из (IV. 9) имеем
([Е]о- [ES]) [SJ-(A-_rr А-.2) [ES] О
откуда
fES1- МЕНЯ]
11 I fcjsj
аким образом, скорость реакции:
v = kt [ES] =
М-2 [EMS]
Л-1 +/‘‘-2 + [S]
л2 /t. 1а,, [EJo [Si
MIEIofS]
1 + A' [SJ
(IV. 10)
где К = A-i (/r_i k2).
Если через а обозначим начальное количество субстрата, а че-
рез х его количество ко временит, то (IV. 10) приводится к такому
виду:
<1* = 1'*' (д ,г)
с/т 1 Г-А (а—х)
где V = к.2 [Е]о.
Решение (IV. 11) дадим для предельных условий и для общего
случая.
Если начальная концентрация субстрата достаточно мала
(/<а 4С 1), то (IV. 11) приводится к такому виду:
и мы имеем
l'A’=—In—(IV. 12)
т а —X
Если начальная концентрация субстрата достаточно велика
{Ка 1), то (IV. 11) приобретает следующий вид:
dx ,,
di
(IV. 13)
Интегрирование (IV. И) для общего случая дает:
Гт= ( Г-гт-7-!—--I-1
J L А (а — х)
dx -j- const
где постоянная интеграции определяется с помощью
условий: х — 0 при т = 0.
Решением будет:
граничных
или
(IV. 14)
152
При малом значении К выражение (IV. 11) приводится
к (IV. 12), в то время как при большом значении К оно обращается
Обратимые реакции с одним субстратом
Схема рассматриваемой здесь реакции имеет следующий
вид:
Л, k..,
Е [ S ES ЕТ Е-J-Т
I
где S и Т — субстраты.
Это уравнение соответствует, например, реакции гидролиза
Р-метплглюкозида, причем S представляет глюкозид, а Т — глю-
козу; концентрация метилового спирта, вследствие его большого
избытка, может быть принята постоянной во время реакции.
В соответствии с гипотезой, принятой для метода установив-
шеюся состояния, будем иметь
[S]-f-А_3 (ЕТ] — (АдА’.2) [ES]О (IV. 15)
= [EJ П’1+А4 IES] -(//,+/. _,) [ЕТ] --0 (IV. 16)
Так как общая энзимная концентрация
{EJo = [EH-[ES]-HET1
то (IV. 15) и (IV. 16) представляются в таком виде:
Ад ([Е]о —[ES| —[ЕТ]) [S] +А_3 [ЕТ] —(А’.^г А2) [ES] = О (IV. 17)
А-; ((Е]о-[ ES J-[ ЕТ ]) [ Т] Н- А-2 (ES ] - (А-; 4- 7г _[ЕТ] = 0 (IV. 18)
Решая (IV. 17) и (IV. 18) относительно [ES] и [ЕТ], получим
[ES]
[Е]о
__________________^G-2-I-^)1S]4-a-_.X|T]______________
“ <A2-a-_XiА(^+*-+<,)[S]-r<[Т]
[ЕТ|
ГЕЬ
_ ___________ A-A [S)-j-A;(fc_,+A2)(T]______
+ + А-1'(А-_1-|-Л,Ч-А-_2)[Т]
153
Суммарная скорость реакции слепа направо:
r=A-2[ES]-A2[ET] =
= ___^________________________
’ l-M'-i - А\(М V+aQ ISJ+AX^-p^ + ^JlT]
(IV. 19)
Равенство (IV. 19) может быть написано так:
17<|S] ГЛ''[Т]
" Ц-Л-(5]-1 Л-'ITJ
(IV. 20)
M*g—fc-«—
i-q
Из (IV. 20) видно, что при работе с чистым субстратом S (т. е.
при Т = 0) начальная скорость реакции будет:
VA'IS]
' l + A’ISJ
Аналогично для начальной скорости реакции с чистым Т най-
дем:
!-!-/< [Т]
Таким образом, мы получили уравнения, подобные (IV. 11)
для необратимых реакций; копстапты здесь имеют другие значе-
ния.
По-прежнему обозначим через а начальную концентрацию S,
равную сумме концентраций S и Т в любой момент; ати концентра-
ции могут быть обозначены, соответственно, через а — х и х.
Тогда для скорости реакции найдем:
<Лг _ УК (а — х)— V'K'x
dx 1 -|- К (« — z) -j- Л' 'я
Если л-р — концентрация Т при равновесном состоянии, то
VK («—Л’р)== ГЯ>р (IV. 21)
Уравнение скорости реакции может быть записано так:
dx VKa — (VК -г V"К) х
dx 1 — А'я.(Л''— А’) х
Используя (IV. 21), получим:
dx __ VKa—VKaxKcp
dx 14-K«-|-(A'— K)x
_________________VKa (xp—x)_____________
(1 -|- Л'о) xp -г (A'' — A') xp - j - (A' — К') xp (.rp —x)
Обозначив величины отклонения от равновесного состояния
— х через z, получим
~ “ (1 + Aft) хр -|- (А — А) х* + (А - A'') xpS
Это уравнение подобно (IV. 11) и его решение соответствует ра-
венству (IV. 13). В том случае, когда К — К', изменение z со
временем т следует закону реакции первого порядка.
Скорость реакций при торможении
за счет субстрата
Для некоторых реакций с одним субстратом найдено,
что они протекают со скоростью, которая не отвечает уравнению
(IV. И), причем с увеличением концентрации субстрата скорость
реакции проходит через максимум, а затем уменьшается.
Установлено, что энзиматические реакции связаны с участием
двух групп на поверхности энзима. Для протекания реакции не-
обходимо, чтобы молекула данного субстрата имела доступ к этим
двум участкам; реакция задерживается в том случае, когда моле-
кула субстрата привязывается к каждому участку. Это положение
может быть изображено в таком виде:
Здесь ES обозначает комплекс, в ’котором S присоединен
к участку, благоприятствующему течению реакции, в -то время
155
как SE представляет комплекс, в котором присоединение тормозит
реакцию. Тройной комплекс SES содержит на каждой стороне
молекулу субстрата. Примем, что SE и SES инактивны. Для
уравнений равновесия имеем:
[ESJ - A;s[E][Sl (IV. 22)
(SEl-AJEHS] (IV. 23)
[SES]=ysA\[E’[S]i (IV. 24)
Общее содержание энзима составляет:
I ЕJo- [Е] + [ ESJ + [SEJ + [SES] = [Е] (14 A's JS] + [SJ 4-KSK'X [S]2)
(IV. 25)
Из (IV. 22) и (TV. 25) получим
IESJ ---------AslEI,l|S1-,----
l-r(Xs + /f,)[Sl+^,ISl5
Следовательно, для скорости реакции найдем:
V - А., [ ES] = —-- д ---- (I v. 26)
l + (As-r^T)[SJ4^[Sl
Из (IV. 26) видно, что скорость реакции равна нулю при очень
высоких концентрациях субстрата, а максимальная будет достиг-
нута при [S]- — 1/KSK .
Представим (IV. 26) в таком виде:
Л [SJ
1 [SJ-j-C [SJ®
(IV. 27)
где Л.ВиС — постоянные.
Если а — начальное количество субстрата и а — х предста-
вляет его количество ко времени т, то для (IV. 27) будем иметь:
d.v ________Л (а — х)_____
dr 1 J-Z/ (a—z)4 С (а — .г)2
После интегрирования получим:
В
Л
V'0-1’
dr const
где постоянная интеграции определяется из условия: т — 0 при
х = 0.
В результате найдем:
, , G. В с
Лт = 111-------г —Г ж 4- “Г ах
а—х 1 Л 1 Л
Сх2
тг
Обозначим у = а — х, тогда, последнее равенство примет сле-
дующий вид:
/1т - In 4 г -г - У) + -у « (« —'/) — ~
у -4 /1 2/1
Процесс торможения энзиматических
реакций
Посторонние примеси оказывают влияние на скорость
энзиматических реакций и в общем случав замедляют их протека-
ние. Такие вещества называются ингибиторами. В этом случае сле-
дует предположить, что эпзим образует комплекс ES с субстратом и
комплекс EI с ингибитором, по пе может образовать комплекс,
в котором к энзиму были бы присоединены I и S. С другой стороны,
при неконкурентной ингибиторной реакции субстрат и ингибитор
связываются на различных участках энзима с образованием ком-
плекса IES.
Для рассматриваемой реакции имеем:
E-J-S ES
Е-1-1 EI
h-i
ES —> E-j-P
При установившемся режиме [см. (IV. б) J
откуда: A-1[E|[S]-(fr_1+A2)fESJ = 0 [ESJ = A'[E][SJ (IV. 28)
где К ~ кх (/с_! т /г2)-
Аналогично для EI найдем
МЕНЕ = *-i [EI]
откуда: [EI] =Л\[Е][1]
где Ki = ki/k_i.
Общее содержание энзима составляет:
(Е]о = [E]4-[ES]-f-[EI] = [EJ (1[S]-i-A\[I]) (VI. 2U)
Используя (IV. 28) и (IV. 29), получим для уравнения скорости
реакции: v-k [ES1 — M'[E]ft [S] 1 + A|S| + A-.(I|
157
или
УК [S|
1 + A4SJ + /4[I]
(IV. 30)
где К = k2 [Е]о.
Равенство (IV. 30) запишем в таком виде
dx (я — х)
dr 1 -|-Л' (« — х) -j- Л' ;.г
Интегрирование (IV. 31) дает:
1 т = ( Г ,, . —-4-1 -i- Д3---— I dx + const
J L К (а — .г) Л а — х J
Учитывая граничные условия х — 0 нри т — 0. найдем:
Для неконкурентного ингибиторного режима схема реакции
принимает такой вид:
Константа скорости к2 относится к реакции разложения ком-
плекса ES па Е + продукты, причем величины А" представляют
константы равновесия; А,, например, относится к реакции
ES-- 1 ESI и равна IESI1/IES] (И. Нетрудно видеть, что
Для уравнений равновесных состояний имеем:
[ES] А [Е] [S]
[KI] =Ki [EJ [I]
[esi]-a'-[es][I]-/<a:;[E][S][ij
(IV. 32)
Общее содержание энзима:
[E]0=r[EJ4-[ES]-Mi:iJ-r[ESI] = [E](l-pA[S]-I- [II + KK[ [S] [ I])
(IV. 33)
158
(IV. 34)
Из (I\ . 32) и (IV. 33) получим:
[ES] -------- Is]----------
1+Л'[Ь]+А-.[1] + ЛЛ\[Э][1]
Таким образом, уравнение скорости реакции приводится к сле-
дующему виду:
v « Л, [ESJ ==---—----------------
l-bA4S] + /QlI] + A'<(SHlJ
При —Ki из (IV. 34) будем иметь
КА JEMS]
(l-f-K (SJ) (i-fA* [1J)
или
dx k.2K (Е]0 (« — .г)
~dX~ Н-гА («-x)J [Ц Л>]
Интегрирование (IV. 35) дает:
КА [Е]от=(! +А\«) In—J-Ax-J-Zt'/fj JL
Реакции с двумя конкурирующими
субстратами
(iV. 35)
(IV. 3(5)
Этот случай отличается от реакции с двумя субстратами
тем, что последние вступают во взаимодействие между собой.
Примером такой реакции может служить действие эстеразы на
смесь £)- и Z-форм эфира.
Имеем следующую схему реакций:
где S и Г — субстраты,
а А и к — константы скоростей реакций первого порядка <ля
процессов разложения ES и ЕТ.
Остальные константы связаны с процессами адсорбции и де-
сорбции; так, например, символ JS] указывает на то, что Е
взаимодействует с S при скорости ks [El [S]
В данном случае воспользуемся методом установившегося
режима. Будем иметь для ES
/•s[E][SJ-(/._s^/.)[ES|=O
Это равенство может быть записано так:
[ESJ- A'JEHSJ (IV. 37)
где Л'3 = А-3 (/г_в А-).
Аналогично найдем для ЕТ:
[ET] = AHFim (IV. 38)
i,ie А/ т А:).
Общее содержание энзима:
(E]0=[EJ-MESbr(ET] (IV. 39)
Подставляя (IV. 37) и (IV. 38) в (IV. 39), получим:
[Е]о= [EJ (1 + /G [Sbi-AO ГЛ) (IV. 40)
Из (IV. 37) и (IV. 40) следует. что
1Е$] =
______А $ [Ejo (Sj
1 ~ A s [S j -г А\ ['1 j
(IV- 41)
п скорости реакций для субстратов S и Т соответственно:
Отметим, что
_ j. <t?ci _ AAS [Е]о [S]
' “A|bS,-T7x7tsJ+K7T=iT
,=. гг. УК<|ЕЫТ]
H-^[SJ-rAt[T]
о _ 1'\А\ [S]
ЛА',[Т]
(IV. 42)
(IV. 43)
(IV. 44)
Это выражение можно записать так:
I dif V'sA's 1 dz
у dx ~~ VfKf г dx
где у и z — количества S и 'I ко времени т.
Приведем (IV. 45) к такому виду:
1 (Itf 1 dz
г — - —— = — - --
!7 dx z dx
где г = lW<r%A's.
После интегрирования (IV. 46) получим:
(IV. 4G)
, а . Ь
rig—= 1g —
(IV. 47)
где а и b — количества реагентов при т = 0.
160
Освобождаясь от логарифмов, найдем:
(IV. 48)
(IV. 49)
Таким образом, (IV. 43) примет следующий вид:
dx I A'-f- Л' fb (у; л)
Проинтегрируем (IV. 49):
1* —— 1 1 -1 Ktb г_{
< if
К
(IV 50)
Учитывая граничные условия: у == а при т = 0, получим:
т
а —У ,
ь
1
гл.
1 ® । АД) /г г\
(IV. 51)
Примем для количества продукта х — и — у; тогда
Т - -р---- . ;т- 1П —--------}- —[лГ - (л - x/J
I s * 1.,А.« a—.t K„ftrr
(IV. 52)
Реакции между двумя субстратами
Многие энзимы действуют каталитически в реакциях
между двумя субстратами. В таких случаях исходные вещества
связываются с энзимом в таком виде:
1
Напишем уравнения равновесных состояний:
[ES] = K, (EIIS] (IV. 53)
[ЕТ] =/Q [Е] П1 (IV. 54)
fEST] = ^A';|E][SH'l] . (IV. 55)
Общее содержание энзима:
II J(j- (E|-4ES] + |ET|-MEST] = (r )(1 -PA^fSl-F*/ (Т (SJITI)
( V. 56)
И л. М. Батувср.
161
Из (IV. 55) и (IV. 56) для концентрации EST имеем:
[EJ fS) [TJ
[ЕЬ1] ' 1-PAJS] !-A-z[T]+A'X[S][T]
Таким образом, скорость реакции составляет:
k"KsK' [Е]о [S] [Т]
и” к" [ EST] == -----------------------—------------
1 Ч'A' [S]А [Т]-j-АГ A'f [S][T]
(IV. 57)
При Kt = Kt, т. e. в том случае, когда комплекс ES связывается
с Т так же легко, как Е с 'Г, уравнение скорости реакции (IV. о8)
приводится к такому виду:
„ A-"AsA-HE]r,[S][T] |V
Если один из субстратов находится в большом избытке, то (IV. о9)
приводится к (IV. 25), решение которого дано выше; для других
условий задача становится более сложной.
Решим (IV. 59) для относительно простого случая, когда
начальные концентрации двух субстратов равны между собой.
Тогда концентрации во время процесса остаются равными и могут
быть приняты равными у, а (IV. 59) можно представить так:
Jf/ Р'А.еА'(Г/2
где V - к" IE]0.
Проинтегрируем (IV. 60):
Гг = —
-| const.
+ , *
KsKtij * KsKty-
Kt , 1
А«А( - KsK{y ,
При граничных условиях: у — а для т — 0, получим:
,г , 1 а — ц
1-т = —, ' . ' In " ’ '
A SA t
A’sA'j
ИЛИ
Е = Г.- Lbl
AsAj а — х
(IV. 60)
(IV. 61)
(IV. 6.2)
(IV. 63)
а
где х = а — у — количество продукта ко времени т.
Первые два члена соответствуют выражению (IV. 14), в то
время, как последний член равенства (IV. 63) имеет такой же вид,
как для случая бимолекулярной реакции.
Если К9а и Ktti достаточно малы по сравнению с единице]"!,
то последний члец будет преобладать в уравнении (IV. 63), которое
162
тогда характеризует реакцию второго порядка. Если же эти
величины имеют большие значения, то рассматриваемый член
равенства (IV. 63) исчезает для данных стадий реакции; кинетика
реакции отвечает закону пулевого порядка.
Инактивация энзима с течением
реакции
Скорость инактивации (потери активности) энзима
может быть настолько значительной, что ее необходимо учесть
при изучении продолжительности каталитических реакций. Ки-
нетика процессов инактивации соответствует, главным образом,
скорости реакций первого порядка.
Для данного процесса наблюдаются следующие случаи: 1) суб-
страт не оказывает никакого влияния; 2) субстрат защищает энзим
против инактивации, т. с. энзимо-субстратный комплекс теряет
свою активность медленнее, чем свободный энзим и 3) энзим теряет
свою стойкость в присутствии его субстрата.
Общий вид кинетического уравнения [см. (IV. 25)]:
dx _ k2K [Е]' (я —Ж)
dx l-j-A" (я — .г)
(IV. 64)
где [E]q — общее содержание энзима в любой момент (эта величина
постепенно уменьшается).
В соответствии с законом распада будем иметь для инактива-
ции:
1Е|С —|Е|,/_,'Т
где к — константа скорости реакции первого порядка.
Поэтому для (IV. 25) но. учим:
dx VK {a — x}e~hT ....
dr = х) <’'*>>
где V — к2 [Е]о — начальная скорость.
Интегрируя (IV. 65) с учетом граничных условий : для х = О
при т = 0, найдем:
VK j const
VA>-,1T , , , , к„. , . , ft r t, VK
------ »= — In (« — ж)- Kx4-const = In H Aj:-7—
fc----------------------------------------------a — x 1-к
= --p/fa. (IV. 66)
К Й — X
11*
163,
Уравнение (IV. 66) отличается от (IV. 14) только дополнительным
множителем в левой части. Если начальное количество субстрата
достаточно мало, то учитывается только первый член правой части
равенства; при больших количествах субстрата имеет значение
лишь второй член, ио крайней мере для начальных стадий реакции.
При т = сю равенство (IV. 66) принимает такой вид:
где — концентрация субстрата в конце реакции.
Для случая, когда субстрат полностью защищает энзим о г
инактивации, схема реакции может быть представлена в таком
виде:
ht
Е S ES
где D — ннактивный энзим, образующийся из Е по закону реак
ции первого порядка с постоянной /л
Для упрощения примем, что ES не подвергается инактивации
в значительной степени.
Применяя метод установившегося режима для ES, ио ivmhm
[ES]--=K[EHS]
где [Е] — количество свободного энзима в любой момент;
Л' ’ /q +
Если [Е]о представляет общее количество активного шзима
к мом опту т, то
[Е8] л (lE]0— IES]) [S]
Общее количество свободного энзима в любой момент равно
[Е]о — [ES] и, таким образом, в соответствии с законом распада
имеем:
-^ = 4lEI„-|i:si) (IV. es)
Скорость убывания субстрата:
—~ A-2[ES] (IV. 69)
164
Таким образом, протекание реакции характеризуется решением
уравнений (IV. 68) и (IV. 69).
Примем следующие обозначения:
р — Л («1 — р) у (IV. 70)
—in r-Ki^ni—р) (IV. 71)
—У = к*Р (IV. 72)
где p = [ES], m = [Ej; и у [SJ.
Граничные условия составляют: т 0, у — a, hl — ?н0,
где а — начальная концентрация субстрата и тп — концентрация
всего энзима.
Из (IV. 70) и (IV. 71)
—т' =
к }/п
1 - Ку
а из (IV. 70) и (IV. 72):
(IV. 73)
Следовательно:
(IV. 74)
Интегрируя (IV. /4) при гранн’шых условиях: у а, т т0,
наИдем:
т --------I;- 111 — (IV. 75)
Л'2Л у
Энзим полностью исчезает и реакция останавливается при
a k..Kin„ _
In----— ——----- <l\. «.)
или при
(,v- 77)
Продолжительность реакции определяем при комбинировании
(IV. 73) и (IV. .75):
у
к..К у
УТ К у
(IV. 78)
Интегрируя (IV. 78), получим.
?дЛгюу/ч _ ,,//
u а
(IV. 79)
(,!1о — -г-.-
165
Тогда (IV. 79) примет следующих! вид:
, С 14-А аяг 5 . . .
йп-г= I ,---------+ const
J z In z
Интегрирование с учетом граничных условий: z = e
дает
А-.,т=—— + (flln — )1 —In ( 1 — In -
Keq L \ a / J x 'I
при т = О,
ГДО
Й
co
табулированный интеграл.
Глава V
Моделирование химической
и биохимической кинетики
В инженерной практике встречаются две группы техни-
ческих задач, отличающихся своей постановкой. К первой из них
относятся задачи, в которых требуется получить численный ре-
зультат решения математической задачи для использования его
в каком-либо исполнительном органе. Во второй группе задач
требуется исследовать характер какого-либо процесса, оценить
влияние изменения того или иного параметра па ход процесса,
сопоставить» различные варианты однотипных конструкций. Для
решения первой группы задач используются вычислительные —
«счетно-решающие» приборы и устройства, а второй — моделиру-
ющие установки.
Наиболее важным классом машин непрерывного действия
являются электронные машины, предназначенные для интегри-
рования систем обыкновенных дифференциальных уравнений.
Эти машины называются электронными интеграторами или элек-
тронными моделирующими установками. Последнее название
объясняется тем, что с помощью этих установок воспроизводятся
математические зависимости, описывающие (моделирующие) изме-
нение различных систем. В электроинтеграторах математические
действия 'осуществляются с помощью электрических решающих
схем, а участвующие в решении задачи величины изображаются
в виде напряжений.
Электронные интеграторы относятся к числу современных
быстродействующих математических машип.
Принципиальной особенностью электроинтеграторов является
то, что они имеют в качестве независимой переменной текущее
время, в частности, и при решении дифференциальных уравнений,
описывающих быстро протекающие процессы.
167
Основным для электронных моделирующих установок являете я
коэффициент усиления, изображаемый обычно так, как показано
на рис. V. 1.
Усиление, или отношение выходного напряжения к входному
напряжению, обычно колеблется в пределах между 20 0U0 и не
сколькими миллионами. Если выходное напряжение не превышай*!
100 в. то на входе оно будет меньше 5 мв. Вследствие большого коэф-
фициента усиления напряжение па входе усилителя, таким обра-
зом. б 1ИЗКО к нулю.
,ход -_ГХ_____________выход В структурной .модели эле-
и менты независимы и соединя-
ются между собон при нодго-
Рис. V. I. Коэффициент усиления. ювке модели к решению каж-
дой конкретной задачи. Семей-
ство решений представляется в виде диаграмм на экране элек-
тронного осциллографа.
Начальные условия, то есть начальные значения моделиру-
емых переменных величин, должны быть заданы на выходах
интеграторов модели в виде напряжений. с которых начинаете <
отработка решения задачи. Сопротивления и емкости присоеди
няются к усилителю для образования устройств, е помощью ко-
торых выполняются сложение, умножение и интегрирование.
Для сложения и умножения составляется цепь в таком виде,
как показано на рис. \ . 2. Сила тока, проходящего через сопро-
Рис. V. 2. Схема для сложения и умножения.
тпвлепие, равна величине падения напряжения, разделенной на
сопротивление. Таким образом, имеем:
для
Сила тока -1 —
ДЛЯ /С
Сила тока = — г—-
Jit о
ДЛЯ Па
Сила тока = —° —-
Ид
Поскольку вход усилителя присоединен к сетке лампы, то
ток ие может пройти в усилитель. Поэтому сила тока, проходящего
К'.н
через сопротивление /?«, равна сумме токов, и (\щих через и
*'.> <1.- 'i—!'п • f,u
На 1 ~~Н7~
Кроме того, так как величина е0 принята равной нулю, то
можно написать:
еа _ ei I f2
На ~ R1 "Г R.
“НЪ+тМ <v”
Рис. V. 3. Схема для интегрирования.
Для интегрирования используется схема, изображенная на
рис. V. 3.
Сила тока, проходящего через емкость, представляется здесь
выражением С {dE <Zr), где С-— величина емкости, а Е — напря-
жен не в емкости.
Составляя, по-прежнему, равенство для токов, получим:
Так как е0
<Цеа)
(1Х
(V.2)
1роцеес умножения осуществляется с помощью потенциометра
(рис. \ . 4).
Отметим, что при составлении структурных схем из звеньев,
основанных па приицине отрицателbiioii обратной связи, следует
учитывать, что каждое звено, производя математическую операцию
169
дает результат с обратным знаком. Интегрирующий усили-
тель, как показано на рис. V. 5, есть усилитель с включенной па-
раллельно емкостью и сопротивлением, ведущим к входному току.
Должны быть также предусмотрены провода от зажимов для «на-
чальных условий» с целью фиксирования значения х при т 0.
Рис. V. 4. Структурная схема
потенциометра.
Пример. Рассмотрим дне
последовательно идущие химические
реакции первого порядка
А —> В —> С
для которых имеем
(V’3)
^L^z-k# (V.4)
(V.5)
где у и г — концентрации веществ,
соответственно, Л, В и С-
Способ решения задачи вытекает из основных понятий о моделирующей
установке.
рименяя указанные выше принципы к решению задачи химической
кинетики, начнем с уравнения (V. 3)
Выходное напряжение интегрирующего усилителя пусть выражает зна-
чение концентрации х. Тогда напряжение у входного сопротивления и соот-
ветствии с, (V. 2) представляет - с/х/с/т.
Из (V. 3) имеем:
Рис. V. 5. Интегрирующий усилитель (схема для .?).
С помощью потенциометра получим kLz из .г. Условие, при котором
—dx/dx должно быть равно Apr, получается путем соединения точек, предста-
вляющих эти величины (см. рис. V. 5).
Использование емкости 1 мкф и сопротивления 1 Ми.ч требует, чтобы
время было выражено в секундах.
Структурная схема для у изображается подобным же образом. Операции
сложения и интегрирования выполняются посредством таких же усилителей.
Пусть выходная величина равна —у, и, следовательно, сумма входных зна-
чении будет dyldx.
170
Рис. V, <S. Результиру-
ющие кривые.
Время
Согласно (V. 4):
dlf
dT
Лдх -Аду
Напряжение, представляющее Ада;, уже опроде.н-на. н —А-,.у получается
пл —>/ с помощью другого потенциометра, как изображено па рис. V. б,
при использовании указанных сопротивлении и емкостей.
Наконец, моделирование величины з выполняется в результате npnpai: in-
па имя выходной величины третьего усилителя значению z. а входной — про-
изводной г/з/г/т: отметит, что
dz
dr
—It.,it
Напряженно, соответствующее —Аду, уже определено. Эта часть струг;
турпоп схемы показана на рис. V. 7.
Решение, обычно, начинается с того, что три переключателя, которые шун-
тируют усилители, открывают последовательно. Решение подучается в виде
трех кривых, изображающих зависимость напряжения от времени, которая
является аналогией для связи между концентрацией и временем.
На рис. V. 8 представ лены кривые, полученные для начальных условно:
ж —100. у =s—0 при Ад-- 0.4 и Ад=0..’5
Кинетические, уравнения были решены также аналитически и на p:ic. V. 8
для сравнения показаны расчетные точки.
Моделирование, как метод воспроизведения какой либо системы
или процесса, происходящего в живом организме, представляет
самостоятельное направление в развитии биологической электро-
ники. С помощью моделирования, как было показано выше,
удастся выявить механизм некоторых яв leiinii, проверить пра-
вильность теоретических представлений об их происхождении и
даже предсказать существование зависимостей. которые ранее
не наблюдались.
Электронные модели созданы также для объяснения электро-
кардиограммы, механизма цветного зрения, проведения реакций
центральной нервной системы.
Техника может позаимствовать у организма многое из того,
что он приобрел на длинном нуги биологической эволюции Ито
составляет содержание особого, весьма перспективного напра-
вления современной техники, которое называют бионикой.
Глава VI
Термодинамические
потенциалы в инженерно-
химических расчетах
Функции потенциала и равновесие
Равновесие может быть определено как состояние, при
котором все силы сбалансированы и результативная движущая
сила для любого изменения равна нулю. Обратимый процесс с ма-
тематической точки зрения является предельным случаем, когда
движущая сила стремится к пулю. Движущая сила всегда может
быть выражена некоторой функцией потенциала. Таким образом,
истинный обратимый процесс характеризуется равновесными усло-
виями. при которых не существует движущей силы.
Известно, что работа, которая получается в результате любого
изменения состояния системы, будет максимальной в том случае,
если это изменение протекает обратимо. При спонтанных (само-
произвольных) процессах мы. имеем конечную движущую силу,
поэтому опи необратимы. В таких процессах работа, совершенная
системой, меньше той теоретической величины, которая была бы
при наличии обратимого изменения. Если система переходит от
состояния 1 к состоянию 2 с помощью необратимого или самопроиз-
вольного процесса, то мы будем иметь в соответствии с первым за-
коном термодинамики
Дб’о —(VI. 1)
где индекс «5» обозначает необратимый или спонтанный процесс.
Для обратимого процесса:
Ч=^-"'л (VI. 2)
Изменение внутренней энергии XU по зависит от пути, по ко-
торому протекает процесс, и поэтому
(VI. 3)
173
Таким образом
@R ~11 Л = — И S
ИЛИ
^K — <?S=^R — (VI. i)
Так как обратимая работа больше, чем спонтанная, то
1Ил-Н’5>0 (VI. 5)
п поэтому
Это означает, что тепловой эффект обратимого процесса должен
быть больше, чем тепловой эффект, связанный со спонтанным
процессом. Величина \V{( — JV представляет энергию, теряемую
вследствие спонтанного характера любого необратимого процесса.
Тепло, теряемое системой, должно быть воспринято окру кающей
средой (пли наоборот). Так как окружающая среда по сравнению
с системой является бесконечной, то температура среды может
рассматриваться неизменной При этих обстоятельствах тепло
обмен с окружающей средой представляет собой обратимый процесс.
Рассмотрим систему, подвергающуюся необратимому расшире-
нию в изотермических условиях с переходом от состояния 1 к со-
стоянию 2. С целью ограничения необратимости в пределах про-
цесса внутри системы примем, что окружающая среда находится
при температуре системы, т. е. что разность температур, необходи-
мая для передачи тепла, является бесконечно малой величиной.
Обозначим количество тепла, переходящего от окружающей среды
к системе, через а производимую работу через ИР .
При вычислении изменения * энтропии системы укажем, что
мы можем связывать передаваемое тепло с изменением энтропии
только для обратимых процессов. Если QR тепло, которое погло-
тилось системой при переходе от t к 2 в результате обратимого
процесса, то изменение энтропии системы составляет Q ‘Т. Выше
было указано, что теплообмен между окружающей средой л систе-
мой происходит обратимо, даже в том случае, когда процессы
внутри системы протекают необратимо. Поэтому передача ген га
Q от окружающей среды обуславливает изменение энтропии
I I
----— в последней (аосолютпый знак используется здесь во
избежание смешения знаков, которое может возникнуть при сов-
местном рассмотрении системы и окружающей среды, когда знаки
величин теплоотдачи различны в зависимости от се направления).
Таким образом, полное изменение энтропии составляет:
Д5 — ДД’сист Т ----уг"" (V1.7)
17>
Разделив (VI. 4) на Т, получим:
<?л Qs _ И'д--П я (VI. 8)
т т т
Следовательно, для спонтанного (самопроизвольного) процесса
при постоянной температуре полное изменение энтропии (система
плюс среда) равпо количеству теряемой энергии, разделенному на
температуру. Так как для самопроизвольного процесса
Т
то будет справедливо неравенство
Д5>0 (VI. 9)
Если процесс обратим, то l'VR равна и
Д5 = 0 (VI. 10)
Сопоставляя последние два выражения, получим
ДЛ О (VI. 11)
причем знак неравенства или равенства зависит от необратимости
или обратимости процесса.
Так как движущая сила обратимого процесса равна пулю,
то говорят, что (VI. 11) является критерием равновесия процесса.
Если для данной системы можно найти любое изменение со-
стояния, которое вызывает изменение энтропии Д5, величина
которой больше нуля, то такое изменение будет иметь конечную
движущую силу и это изменение будет происходить самопроиз-
вольно. Однако, если для всех изменений состояния AS равна
нулю,, то система находится в состоянии полного равновесия.
Необходимо подчеркнуть, что AS есть сумма изменений эн-
тропии системы и окружающей среды.
Значение ДS только для системы не является полным крите-
рием равновесия; оно может быть отрицательным, равным пулю
или положительным, но не дает никакого указания относительно
движущей силы и равновесия системы с окружающей средой.
Имеются и другие, более удобные критерии для исследования
равновесных процессов. Из определения энтропии мы имеем для
обратимого изменения системы при постоянной температуре
Т следующее соотношение:
T&S = QB (VI. 12)
где ~~ тепло, поглощаемое системой, обратимо; все величины
в (VI. 12) относятся только к системе.
Применяя (VI. 6), найдем:
Г v$>«s (VI !3)
В общем случае (VI. 12) и (VI. 13) дают
Qi-Т \ST^o (VI. 14)
причем индекс «7’» указывает на изотермический процесс, а знаки
равенства и неравенства используются, соответственно, для обра-
тимого и необратимого случаев применительно к рассматриваемой
системе. Учитывая дополнительно еще одно orраниченпе для равно-
весного состояния, а именно, постоянство давления, будем иметь
QP~MI
Подставляя в (VI. 14), получим
ДЯТ р — Г Л.УТ 0 (V1. 15)
но при постоянной температуре
\И — Т \S \F
и (V!. 15) может быть записано так:
(VI. Ri)
что служит критерием равновесия только системы.
Из (VI. 16) находим, что если при каком-либо изменении со-
стояния происходит уменьшение анергии системы, то существует
конечная движущая сила цля этого изменения, которое будет иметь
место самопроизвольно. Если SF для данного изменения равно
пулю, го система находится в состоянии равновесия.
Свободная энергия и химические реакции
Если некоторые химические вещества вступают во
взаимодействие с образованием продуктов и, стало быть, величина
изменения свободной энергии является отрицательной, т. е сво-
бодная энергия системы уменьшается, то мы найдем в соответствии
с (VI. 16), что изменение может быть самопроизвольным, так как
здесь имеется копечная движущая сила процесса. Далее будет
показано, что (VI. 16) может быть использовано при равновесном
состоянии системы для расчета равновесных концентраций ве-
ществ в реакционной массе, полагая в этих условиях А/ равным
пулю.
Но аналогии с энтропией для племен гарных веществ в их наи-
более стабильной форме при 25^ С и единичной фугативиости сво-
бодная энергия произвольно приравнивается нулю. Свободная
энергия образования соединения может быть найдена из этих
17(5
сравнительных значений. Свободные энергии образования отдель-
ных соединений могут быть также применены для отыскания изме-
нения свободной энергии химической реакции. Так как значения
свободных энергий образования соединений табулированы при
единичной фугитивности, то вычисленное с их помощью изменение
свободной энергии носи г название стандартного изменения сво-
бодной энергии и обозначается XF с указанием температуры, на-
пример. X/'os-
Изменение свободной энергии представляется в таком виде:
А/-’ -У *i(VJ)i
(VI. 17)
где v —етехпомегрпческнй коэффициент.
Пример. Определить измени пае свободной энергии для следу-
ющей мо.1'он•(.кой реакции:
со (г) - 1 /20., (г) СО., (г)
при 2л С II I а/им.
Из таблицы инеем <гандаргные свободные энергии образования соедине-
ний (в ККЦ.1 l.U‘>.U>)'
\F для СО (г)
\/- > О. (г)
\1'с -> СО., (г
> О.,(Г)
•> СО., (г)
—32,8079
О
—9 4.2598
Значения г, равны —1, —1/2, -~1, соответственно, для СО, О, и СО...
Из (VI- 17) найдем:
\/'= - — | (—32.8079) — 1 /2(0) •[ 1 ( -94,2598) —(И.4519 ккал/моль
Мы получили большое значение для AF°. Эго показывает, что в данной
реакции существует большая движущая сила, которая должна привести к ее
самоп ропзво юному течению. Однако термодинамика нс дает никаких све-
дений относительно сопротивлении процессам или относительно их скоростей:
она предоставляет только информацию о движущей силе, которая существует.
Окись углерода и кислород могут смешиваться при комнатной температуре
бс.з всякого взаимодщ ствия в течение многих лет, если не будет преодолено
сопротивление реакции. Тепло может возбу (ить реакцию так же, как подходя-
щий катализатор, но при отсутствии средств, с помощью которых начинаете;!
реакция, последняя побудет иметь места с поддающейся измерению скоростью
при 29.8° К, хотя известно, что существует мощная движущая сила.
Применение третьего закона
термодинамики
При отсутствии данных о свободной энергии образова-
ния соединений расчет ХЕ для рассматриваемой реакции может
быть осуществлен другим способом, основанным на так называ
емом третьем законе термодинамики (Периста). Этот закон утвер-
ждает, что для .любой химической реакции при абсолютном нуле
изменение энтропии равно нулю и разность между теплоемкостями
продуктов и реагентов при абсолютном нуле исчезает. Таким
12 Л. М. 1 атунер.
177
образом, при Т = 0 имеем AS = 0 и АС'Р —0 для твердых тел и
жидкостей. Для газов ДС’Рт и 0 стремится к конечной величине.
Закон Нерпста иногда выражается так: AF и А// стремятся друг
к другу асимптотически при приближении температуры к абсо-
лютному нулю.
Третий закон термодинамики дает возможность определить
абсолютные значения энтропии вместо их изменений относительно
произвольно выбранной величины. Абсолютные значения энтропии
могут быть определены путем калориметрических измерений илп
с помощью спектроскопических данных при использовании кван-
товой механики и кинетической теории. В настоящее время име-
ются обширные таблицы абсолютных значений энтропии; часто
данные этих таблиц применяются па практике при отсутствии
сведений о свободной энергии образования соединений. Ji этом
случае используется соотношение
,\F — \П — Т АЛ’
при постоянной температуре.
Для химического равновесия будем иметь
,\F° = Ай° — Г А5°
Значение А5° для реакции может быть найдено из таблиц абсо-
лютной энтропии, так как
Пример. Определить с помощью третьего закона термодинамики
стандартное изменение энергии для следующей реакции при 25° С:
СО (г) + 1/2О2(г) СО-2(г)
Данные из справочных таблиц:
ДЯ° А Л'°
СО (г)............—26/1157 47,301
0-2 (г) ............ О 49,003
СОг (г)...........—94,0518 51.061
Для этой реакции v, равно —1, —1/2, + 1 соответственно для СО, О2
и СОл. Следовательно:
ДЯ° = -1 (-26,4157) -1/2 (0) + 1 (-94,0518)
и
А5° = —1 • 47,301 —0,5 49,003+1 • 51,061 = —20,7415 кал/милъ град
Окончательно имеем
i\F° = -67,6361 -298,17 —= — 61/1516л-кал/.Иоль
1000
Это значение совпадает с результатом, полученным в предыдущем при-
мере.
178
Свободная энергия, фугативпость и активность. Имеем
tlFT = d RT In f
При интегрировании этого выражения получим:
\F=IIT In А
/1
Последнее равенство можно записать в таком виде:
+ In-А (VI. 18)
где величины с индексом « »относятся к стандартному состоянию,
а значения без индекса к частному случаю.
Отношение / /° называется активностью и обозначается
(VI. 19)
Таким образом (VI. 18) приобретает следующий вид:
F = F°-\-RT\aai (VI. 20)
Для любого химического процесса будем иметь:
1 "Л
г i г
или
Л/’-- ЛГ°-!-v in (li (VI. 21)
i
Однако при химическом равновесии А/ = 0, и в этом случае
(VI. 21) составляет:
= — У ViHT hi'/,. (VI. 22)
г
Равенство (VI.22) имеет большое значение для проблем, свя-
занных с химическим равновесием; оно может быть использовано
Для определения свободной эпергиц образования химических
соединений по данным об их растворимости.
Пример. Требуется определить свободную энергию образовапия
твердого хлористого натрия. В этом случае будем иметь:
1) КаС1(г) —> .\иС1(н-н, л. р); А/’’° = 0.
Символ «п-и, н. |м> обозначает «исионизированный в насыщенном растворе*';
-1верд1.1й хлористый натрин находится в равпов гном состоянии с этой же
солью в насыщенном растворе; А/-’ = 0.
Таким образом:
\7° {.ХаС1 (н-и, n-p)j = AF [ХаС1 (т)]
2) ХаС1(п-п, и. р) —> Ха'Ч-СГ; ДЕ=0.
11*
179
Из (VI. 22) следует:
ЛЁ* (Na+)-j- \7 (Cl-J— X/^f.XaCl и и. и. |>)] . - VV;Kr 1п«,
.'») Na+ -> элемент; —Д/’с = —(—62.588 ккал/'е-атом)- Эта реакция
обратна прямой реакции образования иона из элемента в стандартном состо-
ЯНИН.
4) Ci- -----> I 2С.1г; —ДС’° —(—31,245 нкал/г-атим).
Складывая с обратными знаками указанные величины, найдем для свободной
энергии образования твердого хлористого натрия из элементов:
.Ха 1/2С1. ---->.XaCI(T): \1' \Е ’ - 93.933 г У v./ГГ hi "i
Заменим член У, чдЯТЧп/ц выражением НТ 1п JZy, где у — коэффициент
активности, а Л/ — молярноетъ, определяемая числом молей NaCl на 100, _>
воды.
Молярность насыщенного раствора NaCl при 25° С составляет 6.12.
При этой концентрации ко>ффпцпепг активное!и у равен 4,4. Следовательно:
НТ In Му 298 In (6,12 • 4.4) = 1,95 ккал :г-яол
Имеем:
Х/'’°= —93.933-[ 1,95 = --91,98 ккал/г-.vti.i
Следует отметить, что значение ВТ in Му обычно мало по сравнению
с другими величинами, входящими в расчет. Поэтому даже большая погреш-
ность в определении у по может дать существенную ошибку для значения
свободной энергии образования твердого материала.
' Изменение свободной энергии
с температурой
Выше были описаны два метода расчета изменения
свободной анергии для химической реакции; в одном из них ис-
пользу отся таблицы стандартной свободной энергии образования
соединений, а в другом — таблицы абсолютной энтропии и тон.ют
образования соединений. Эти способы расчета дают изменение стан-
дартной свободной энергии реакции при 25и С и 1 атм (или.
точнее, при единичной активности). В действительности многие
реакции при этой температуре имеют незначительную или нулевую
скорость и на производстве они осуществляются при более вы-
соких температурах. Поэтому важно иметь возможность опреде-
лять изменение свободной энергии реакции при этих температурах.
Такой расчет связан со следующим равенством
dF = —S dT + v dp
и для реакции при постоянном давлении получим:
—5
(VI. 23)
180
Для свооодной энергии имеем
Г = V — Т S -г рг
пли
/' и - TS
откуда:
Подставляя (\ I. 24) в (VI. 23), пандем:
Так как нас интересует расчет изменения свободной энергии
с температурой, т,; вместо F и П подставим \Е и \Н\
(dXF\ _ ЬГ — М/
I дТ )р~~ Г
Разделим (VI. 26) па Т:
7<?.W’\ I А/' А//
V дТ ),> т т-~~ ’ т~
или
Интегрируя (\ I. 28), будем иметь
АЛ’ Г хп
Т ~~ I 7-
*7
и
(VI. 2G
(VI. 27)
(VI. 28)
(VI. 23}
где / — постоянная интегрирования; Д 7/ сеть функция Т
Для Д/7 имеем:
т
Ml = у XCpdT 4- II0
о
Из (VI. 29) получаем:
т
Т ^о~г \ с!7'
-г- = - -------' (VI. 30)
Исследование реакции
для производства формиата натрия
Определим имеет ли промышленное значение произ-
водство муравьинокислого натрия (формиата натрия) по реакции:
NaOH (т)4-СО(г) НСОО.Ха (т)
181
Найдем из справочных таблиц значения свободной энергии
образования для следующих соединений:
А/'э, ккал/люль
СО (г) —32,808
НСОО.Ха........................ Отсутствует
Х'аОН (т)......................... —90,600
Свободная энергия для ПСООХа может быть определена
по методу, описанному па стр. 177. Данные о растворимости этого
соединения имеются в справочниках. Мы найдем, что
^НСООХа -“147,00 кка.1,/лень
Для реакции при 25° С:
А/'= т= —147,00 [(—90,600) 4- ( -32,808)] - 2:1,59 .иа.гь
большое отрицательное значение \FC указывает па то, что
реакция при этой температуре имеет бо гыную движущую силу и,
следовательно, может быть признана перспективной для произ-
водства рассматриваемого здесь продукта. Однако мы нс распола-
гаем данными о скорости реакции. С увеличением температуры ско-
рость этой реакции увеличивается; каково же влияние темпера-
туры па движущую силу? Нам необходимо определить SF^ при
более высоких температурах.
[айдем значение А// (теплота образования) и Ср для трех
соединений:
\//° с}>
НСООХа (т) .... —155,030 18.3
ХаОП (т) .... —101,9110 12.5
<’-O(i)............ -26,416 6,3424 1,8363-10"3Т —
— 0,2801 - Ю-СГ-
Пз приведенных данных имеем:
A7/.>3s 26,654 кл’йл/.ноль
ЛС’р -0,54 — 1,84-10’3 Т
для \6'р отброшен член с i - ввиду отсутствия точных данных
о теплоемкости твердых тел.
Таким образом:
т
\1/т — |' ДС’Р <1Т //, — 0,34 Г — 0.92 11Г3 72
и
При Д//ог,.5 —26 654 кад/люль для постоянной интегри-
рования найдем
//о 26-411 КЯЛ/МОЛЪ
и будем иметь:
Л//т = -26 411 —0,54 Т -0,92 - ПГ’Г
182
Применяя (VI. 30). получим:
т т
.\FT ,,, г С _26Z.1I— 0,54 Т — 0,92-КГ3?’* , т
— — } -тТ~(П }--] -----------------------—-----------— dl |/ =
о о
9(; 411
=---------У----J 0,54 In Т г0,92 • 10-37’ |-/
И
AfT = - 26 411+0,54 7 In Г + 0,92 Ю^Г + УГ
Так как А/ж^ = —23 592 кал/моль, то, подставляя это зна-
чение в последнее равенство, найдем для постоянной интегриро-
вания /
—23 592- —26 411 +0,54 • 298 In 298 + 0,92 • 1(Г3 • 298= + 298/
откуда:
/ =6.1
Следовател ыю:
Д/'г= - 26 411 । 0,5-4 Т In Т -i 0,92 • Ю^ГЧ-б,! Т
При Т - 573° К или I = 300° С найдем:
Д/^,^26 411 +0.54-573 In 573 + 0,92- Ю"3 5732 + 6,1 • 573 — —20 647 кал/молъ
Движущая сила при более высокой температуре несколько
уменьшилась, но остается еще весьма значительной и поэтому
применение реакции для производства формиата натрия предста-
вляет большой интерес.
Пример. Рассмотрим другой случай изменения движущей силы с тем-
пературой для реакции
СЛ!, (г) + Н2О (О С„Н/)Н (г)
Воспользуемся третьим законом термодинамики. Найдем из таблиц
с л ед у ниц! I с д а и и ы с:
ДЛ2Э8 ЧД<Э8
С.Н, (Г)........................... 12,496 52,45
Н.,0 (г) —57,798 45.106
CJI5OI1 (г)........................—56,240 67,4
Из приведенных данных для рассматриваемой реакции имеем
ДЯ20в =— 10.938 ккял/.иняь
ЛЛ’2()8 -—30,156 кал/моль • грае/
Тогда
298
Д/.‘= =л//° — Т \SJ = —10,938 — (—30.16) -1,94 ккал/моль
Реакция при 25° С им’ет малую движущую силу. Принимая, что ЛН°
и AS0 остаются постоянными, определим значение Л/-’0, для последующих
1 83
температур (табл. VI. [). Это равносильно тому, что примем ЛСр = 0 (в дан-
ном случае \С}) мала, поэтому допускаемое упрощение в носит .малую oiiiiiokj }.
Таблица VI. 1
Определение А/" для различных
температур
I, 'C T AS innl) ЛЬ”, ккал. .чо.гл
0 - 8,24 2,70
23 —9,00 — 1.94
50 —9,75 -1,19
100 -11,20 -1-0,30
2Ck.l -14,24 —3,30
Движущая сила исследованной реакции уменьшается с увеличением
температуры и нетрудно видеть, что А/-’0 проходит через пулевое значение
ири температуре ~10(Р С. Действительно:
\Г°=.\Н° — Г \Л’О = 0
и
Подставляя соответствующие значения, получим:
—10 938
. — 303° К или 90' (’
--30,16
Соотношение между А/*”
и константой равновесия К
Рапос было показано, что критерием равновесия для
химической реакции является пулевое значение изменения сво-
бодной энергии. Рассмотрим теперь более подробно изменение
свободной энергии применительно к уравнению химической реак-
ции в общем виде:
«Л-г^П^П,4-мМ
Изменение свобо (ной энергии получим, если из свободной
энергии продуктов вычтем свободную энергию реагентов. Примем,
что система находится в состоянии равновесия и бесконечно малые
количества реагентов превращаются в продукты реакции; вели-
чины будут —da, —db, -\-dl, ~-dnt. Изменение свободной
энергии с ост а в л я ет
V ViF{
или
Здесь используются парциальные свободные анергии, поскольку
мы имеем смесь.
При равновесии dF = О
J/i'l + dmFm — daFa — dbl'i> = О
Разделив это равенство на da, полупим
^гь=о (VI.31)
Но относительные количества взаимодействующих веществ опре-
деляются стехиометрическим уравнением, поэтому
(Н I dm т db Ь
du <i ’ da a ’ da a
Таким образом, нз (VI. 31) путем умножения па а получим:
IF т F,ti-aF а —bFi^Q (\ 1.32)
Парциальная свободная энергия для каждого вещества соста-
вляет:
= (VI. 33)
Подставляя (VI. 33) в (VI. 32), найдем
I (F°t~BT + 1п«,„)-« (F'a^HT In %)-
in «6)-- о
ИЛИ
(/1) -j- mf‘h — a F*a — bFf^ [- RT (Ии at |- m In am — л In «Q — b In аь) 0
Первое слагаемое этого выражения представляет изменение сво-
бодной. энергии для случая, когда взаимодействующие вещества
и продукты реакции находятся в стандартных состояниях и по-
этому обозначает стандартную свободную энергию реакции.
Для второй группы слагаемых имеем:
v; In rt. = In (d'>
Таким образом:
или
.\F°=—R1 In HflV
где символ II обозначает произведение.
Величина Пар носит название константы равновесия А; сле-
довательно
&F° = —RT In К
(VI. 34)
185
Зто равенство справедливо для всех случаев равповес него состоя-
ния, независимо от того, взаимодействуют ли твердые, жидкие
или газообразные вещества.
Равновесие в газовых реакциях
Так как в соответствии с (VI. 19) активность предста-
вляет собой отношение фугитивности к фугитивности в стандарт-
ном состоянии, а стандартное состояние для газа есть единичная
фугитивность, то для газа фугитивность равна активности. Сле-
довательно, формула (VI. 34) действительна для газовых реакций
при написании постоянной равновесия как через активность,
так и фугативпость, т. е. имеем:
А'а — К f
Как только константа равновесия определена с помощью сво-
бодной анергии, мы сразу можем решить вопрос о равновесных
концентрациях. Для пой цели должна быть учтена фугативпость
каждой составляющей равновесной смеси. Фугативпость соста-
вляющей /г в смеси будет:
где индекс «г» относится к идеальному газу.
Подставляя в формулу для константы равновесия в общем виде»
получим
А\-= П/;= П'/рПРЧ exp I 27;
з о
Так как общее давление постоянно, то
i. vj
\lPvi=P3
Для того чтобы вычислить /</, требуются данные о парциаль-
ных мольных объемах. Практическое решение задачи упрощается
таким образом. Имеем:
где Д — фугативпость чистого компонента к при температуре
и общем дав н пип системы.
Используя коэффициент активности
186
получим:
(VI 35)
Применим (VI. 35) для А'/:
V V. 2 V .
^ = A*,/'V^> } (VI. 36)
где
^ = Ну/ и Лу IIYp
Д гя идеальных газов А* равно единице и может быть записано
так
К' = I Ir/J; 11Рv J I (Уj^)vj (V1.37)
По ijjP есть парциальное давление компонента /. Поэтому
(VI. 37) приводится к виду:
А'' — 11 P'-i - Ktjpl (VI. 38)
Формула (\l.36) дает возможность определить равновесный
выход продукта для газовых химических реакции при любых
температуре и давлении.
Рассмотрим следующее уравнение реакции:
и Л -|-Ь В = /« М
Напишем (\ I. 36) в таком виде:
ч1п
(VL39)
УаУь
где Ди — V vj
j
Используя (VI. 31) в экспоненциальном виде, получим
А'/ ,-л^/лг
и так как
АЛ}= -Т У.$7
то
КГ-е^-/Це-^‘^1{Т (VI.40)
Сопоставляя (VI. 39) с (\ I 40), получим:
Величина у™ есть мера выхода продукта для данной химиче-
ской реакции. Выражение (VI. 41) соответствует принципу Ле-
Шателье, согласно которому для эндотермической реакции с уве-
личением температуры возрастает выход продукта, а для реакции
187
< уменьшающимся числом молей повышение давления увеличивает
указанный выход. Действительно для эндотермической реакции
АН численно положительна и мы можем в (VI. 41) написать
I
eMtuiHT
Очевидно, что с увеличением 1 уменьшается значение с
п поэтому ут, т. е. выход продукта, оудет возрастать. Далее,
нетрудно видеть, что при уменьшении числа молен \п будет
численно отрицательна и для такой реакции пусть, например,
п = — I. Тогда в (VI. 41) будем иметь:
Гак как теперь давление содержится в числителе, то с увели-
чением общего давления возрастает выход продукта AJ.
При \н О, т. е. для реакции с постоянным числом молен
из (VI. 41) вытекает, что увеличение давления не оказывает ника-
кого в 1ИЯИНЯ па выход продукта. Для (VI. 41) это положение оправ-
дано тем, что Р (Ап — 0) — 1, но А\. есть также функция давления
и если этот коэффициент уменьшается при увеличении общего
давления, то выход продукта с увеличением давления увеличится
даже в том случае, когда Ап 0. Известны многие реакции, для
которых эта зависимость справедлива.
Равновесный выход этанола
Определим равновесный выход этанола при 1 атм
и 100е С, получающегося но реакции
CJI4 (г)тН,0 (г) (АН;,ОН (г)
если взаимодействующие вещества представляют эквимолекуляр-
ную смесь. Эта реакция была рассмотрена в примере па стр. 183
и вычисленные там данные мы используем для этого процесса.
Изменение свободной энергии составляет
«5
302 /.кч.-.гъ
Примем, что в реакцию вступает 1 моль С^Н.р ио условию
задачи мы должны для начала иметь также 1 моль Н2О. Пусть .г
молен С.,Н[ превратились в этанол. Составим таблицу для расчета
(табл. VI. 2).
Из (VI. 36) имеем:
Л'у = А уА
188
Таблица !1.2
Данные для расчета
Начальное числи молей V MCKIbtlUC изменение Число .молей при р;>П!Ц|- ИССНОМ СОСТОЯ- НИИ СИСТРАН»!
С.Н, . 1 —1 — X 1—X
il.,0 . 1 —1 •—X 1-х
CJI5OU — i -1 T-r X
Всего . . , . . 2 —1 —.г 2-х
При 1 атм. для данной реакции коэффициенты фуга гн внести
компонентов очень близки к единице (поэтому Л\ 1; Xv>
з
= -!)•
Следовательно
Л f Кр- Kypi
и так как Р — 1 атм, то имеем
A z- _ А у
Далее, из (VI. 31) найдем
А- _ Усдиоп __ х/(2—х) __ ж (2 — х) _ -302 i.w-sjs
Ус,Н.УЦ4) 1 х I х (I—ХУ
> — X ' 2-х
откуда
X - 0.223
т. е. степень конверсии этилена в данных условиях составляет
22,3%.
Из данного примера видно, что даже при положительном зна-
чении AFC для реакции в написанном виде имеет место некоторая
конверсия в состоянии равновесия. Положение о том, что отрица-
тельное значение AFJ указывает на наличие движущей силы реак-
ции, остается совершенно правильным; чем (больше отрицатель-
ное значение Л/ , тем больше движущая сила н степень завершен-
ности реакции.
При положительном значении AF° движущая сила связана,
главным образом, с реакцией, идущей справа налево, но даже
и в этом случае равновесный выход желательного продукта может
быть существенным.
IS'J
Реакция получения синильной кислоты
из ацетилена и азота
Определим имеет ли промышленное значение процесс
получения синильной кислоты по реакции
С2П, (Г)-Х2 (г) 2HCN (г)
Решение задачи состоит в том, чтобы определить равновесный
выход продукта с изменением как температуры, так и давления
и сделать соответствующее заключение. Найдем сперва для
данной реакции. Необходимые сведения приведены в табл. VI. 3.
Таблица VI. 3
Данные для расчета
Соедине- ние ЛГ°, кл;ал/л[о,гь дя\ ккал/люлъ S< / .wo.16 СР
а Ъ 10* С - 1(1“ d-10-*
еде 50,000 54,194 47,997 11,94 4.387 -2,322
N.i 0 0 45,767 6,4492 1,4125 —0.0807 —
HCN 28.7 31.2 48,29 5,974 10,208 —4,317 —
При 25° С (298° К) имеем:
Д/’М8-2-28,7-101’ — 50,000 103 —0= 7,4 10я
Положительное значение А/’, хотя и малое, показывает, что
для температуры 25° С движущая сила не способствует течению
реакции в желательном направлении. Определим температуру,
при которой имеет место равновесное состояние системы, т. е.
когда движущая сила равна нулю:
Ы™ = ду/о — Т \S° = 0
и
Мы получили очень высокую температуру, причем IICN дис-
социирует при значительно более низких температурах. Следует
иметь в виду, что ДЯ° и Д6’° изменяются с температурой и, таким
образом, значение температуры, при которой Д/7" = 0 может быть
совершенно отличным от полученного выше при грубом прибли-
жении.
Вычислим AF при других различных температурах.
Имеем:
Ср для HCN . . . 2(5,974 + 10,208- 1и"3Т— 4,317- 10"сГ2)
Ср для СЛ......... 11,94 + 4,387 • 10“37’ - 2,322 1CR7 ~2
С'р для N2 . . . . 6,4492 т 1,4125 - 10~3Г —0,0807 - 1()~°Г2__________
У ViCPi= АС? = —6,441 -I-14,616 • 10-3Г-8-553 • Ю'6?'2-2,322 • Ю’?’"3
190
Применим (VI. 30) для отыскания зависимости AF0 от темпе-
ратуры:
т т
I1 //„-LfACprfr
Л 7? О I 1
* ♦л
О
Подстановка для АСР с последующим интегрированием дает
1 1G1 - -1ГР
Л/-’° = Яо 6,441 111 Т - 7,308 • 10-3 Г2 + 1,438 Ю"6 7’3 1,101 1и 4-IТ
Имеем:
Л//.ж = /70 - j- J А6'р dT == II ь - 6,441 298-1-7,308 10“3 298“ —
6
-2,851 1О-« 2983 — 2,3217 ~11Ь = 2 • 31 200 — 54 194 = 8 206 кал!моль
Решая относительно //0, получим:
II0= 10 330
При использовании ранее вычисленной AF298 и Яо пайдем
постоянную интегрирования:
/=—43,1
Следоватсл т.п о:
AFT 103304-6.441 In Г-7,308- КГ3/2 4-1,438 - 10-|!Р—1lG1 4,10". _ 43, j Т
Константа равновесия определится из (VI. 34):
Кт=е~^т/пт
В табл. VI. 4 приведены значения А?1’, и Кт для нескольких
температур
Таблица VI. 4
Значения М<'°г и К т для различных
температур
t, сс Л Е°, к цал/ .но ль к Г
0 7.49 1,00 иг®
100 7.21 5,9(1-10"5
200 6,98 5,95 • ИГ1
300 6,75 2.66 • IO-3
400 6,49 7,80 • IO"3
500 6,27 1,69 • Ю-2
191
Вследствие диссоциации HCN температуры > 300 С учиты-
вать ио будем для данной реакции.
Согласно принципу Ле-Шатслье, давление не оказывает
влияния на равновесное состояние этой реакции, так как здесь
число молей остается без изменения. Однако определим выход
продукта при 300° С и различных давлениях, используя равенство
(VI. 39):
'J KyJ,Stl
Так как Х?л 0, то P'v I. Таким образом:
,. 3.-е. • i Vs
Л ц — -тт----
Л у
Примем, что для начала реакции взята эквимолекулярная
смесь С2П.» н в количестве J моль для каждого компонента.
Пусть х обозначает число молей С2Н2, израсходованного во время
реакции.
Сведем в таблицу данные для расчета (таб i. VI. 5).
Таблица 17,.j
Данные для расчета
Соединение Начальное число молей V Мольное изменение Число МЧЛСЙ При раннипссном* состоянии системы
ел No IH.N 1 1 < 1 1 1 I- —-Г —т 2г 1— .г 1—Д’ j.r
Всего ’) (1 и 2
По-прежш му напишем:
Унсх(Зх/2)2
2 2
V*
I 7?
Далее
_Тнсх
Ус.нЛ'х.
Значения коэффициентов активности ? при Т = 373 К ц
различных давлениях могут быть найдены и таблицах.
В табл. VI. С даны результаты расчетов.
192
Таблица VI. 6
Результаты расчета
<1?П VC2H2 vncx KY К), .V
Г) too 1,00 0,9!) 0,98 O.U.I271 0.025
10 1,00 1.00 0.97 0,9 i O.0028', li.i 121;
25 1,01 0.9!) 0.92 0.85 0.00313 0.027
50 1,02 0,97 0,85 0,73 О.||:)3(И 0,<»2:1
100 1,04 0,95 0,72 0.52 0.003 И (1,035
200 1,10 0,90 0,55 0,31 0,00858 U.O'ii
Влияние давления на равновесный выход продукта в этом
случае обусловлено тем, что система не является идеальной.
Исследование показывает, что с увеличением давления выход
продукта улучшается, но все же он недостаточен для промышлен-
ного использования рассматриваемого здесь процесса.
Следует отметить, что при использовании избыточного количе-
ства одного из исходных компонентов представляется возмож-
ность в соответствии с законом действующих масс сдвинуть равно-
весие вправо и получить 100%-ную конверсию второго компо-
нента. Однако мы бы имели продукт, загрязненный примесями.
13 Л л. Кцтунер
Глава VII
Процессы хлорирования
Условия проведения процессов
Хлорирующими агентами являются: 1) газообразный
хлор; 2) жидкий хлор; 3) хлористый сульфурил; 4) соли хлорно-
ватистой и хлорноватой кислот; 5) хлористый водород в смеси
с воздухом или кислородом.
Температура влияет на ход процессов хлорирования: она
повышает константу скорости процесса и в ряде случаев сказы-
вается на его направлении.
Ароматические соединения, как правило, хлорируются при
низких температурах (20—70° С), так как повышение температуры
способствует образованию больших количеств полихлорзамс-
щенных, получение которых в большинстве случаев нежела-
тельно.
[еоретически давление нс оказывает влияния па ход процес-
сов хлорирования даже при условии проведения процесса в паро-
вой фазе, так как из двух молекул взаимодействующих веществ
получаются также две молекулы продуктов реакции.
Материальный баланс хлоратора
и технологические расчеты
Независимо от того, какие атомы водорода будут заме-
щаться атомами хлора (в ядре или в боковой цени), процесс хло-
рирования может быть представлен уравнением:
1Шп + лС12 = пс1?1-|-«н(:1 (VII. 1)
,М 71-71 У! п 71 ЗС.-»
где М — молекулярный вес (молекулярная масса) хлориру-
емого продукта;
1 94
п — число атомов водорода, заменяемых атомами хлора;
Мп(1,2,з) — молекулярный вес хлорзамещенных.
Введем следующие обозначения:
G — количество 100%-го хлорируемого продукта, кг;
1У — содержание влаги в хлорируемом продукте, кг;
G± — количество образующихся монохлорзамещенных, кг;
тц — выход монохлорзамещенных, масс, доли;
G., — количество образующихся дихлорзамещенных, кг;
р, — выход дихлорзамещениых, масс, доли;
G3 — количество образующихся трихлорзамещеппых, кг;
1]3 — выход трнхлорзамещснных, масс, доли;
Gy — количество технического хлора, расходуемого при хлориро-
вании. кг;
с — концентрация хлора в техническом хлор-газе, %;
р — давление паров хлорируемою сырья при температуре
хлорирования, леи рт. ст.;
Р — общее давление в хлораторе, мм рт. ст.
Соответственно уравнению (VI1. 1) количество образующихся
монохлорзамещенных:
Количество получающихся дихлорзамещенных:
Количество получающихся трихлорзамещеппых:
В процессе хлорирования, соответственно количествам обра-
зовавшихся хлор замещенных, расходуется хлора (в кг):
Образуется хлористый водород в количестве (в кг):
, ЗПз)
jW
Полученный хлористый водород будет частично гидратирован
влагой, находящейся в исходном продукте, с образованием соля-
ной кислоты.
Если получаемая соляная кислота будет иметь концентрацию
Си, то количество ее (в кг) составит
Gl< " 1-0>01*к
13*
195
и, следовательно, количество хлористого водорода, связанного
с водой, будет
В результате хлорирования останется ненрореагнровавшего
хлора:
<7т0,01 с—С'
Кроме того, к массе инертных газов, содержащихся в техни-
ческом хлоре, прибавится хлористого водорода в количестве
— Ск 0>01 Ск
Общее количество газов, уходящих из хлоратора, составит:
0,01 ск)
В процессе хлорирования имеет место отгонка хлорируемого
сырья. Количество хлорируемого сырья (7ИСП (в кг), которое будет
испаряться и уходить из хлоратора вместе с газовой смесью,
может быть нап щно по формуле:
_ РфЛ/
G"“- (P-W).»/, 61
где 6ТГ — общее количество газов, уходящих из хлоратора, кг;
Мг — средний молекулярный вес смеси газов;
ср — степень насыщения газов парами хлорируемого сырья;
М — молекулярный вес сырья.
Кроме того, в результате хлорирования останется непро-
реагировавшего сырья (в кг)
6’ (1 —1Д-112-П,)
Из зтого количества кг удаляется из хлоратора вместе с ухо-
дящими газами.
В итоге материальный баланс (в кг) процесса хлорирования
можно представить следующим образом:
Загружено
Хлорируемое сырье ......................... (j
Кода (и сырье).............................It
Хлор-газ (технический) ....................(7Т
Итого: б'оощ
II олучено
В жидкой фазе:
мо11ох.1орзаме;Ц|‘!И1Ыс ..............' (i}
дп.х лорзамещеииыс ........................ (!<,
три хдорзамсщсипые.......................... <73
iii'iipopeaiпропавшее сырье .... G (I — —1]2 — Г].)— С'псп
соляная кислота . ,.......................... GK
Итого ... <7а
Н-б
В газовой фазе:
пепрореагировавший хлор ................. G,r 0.01 с — G'
хлористый водород........................С"—б?к0.01 ск
ипсртпые газы .......................... GT(1 —0.01с)
хлорируемое сырье............................ Сцеп
Итого......... б’б
Всего.........Gа |-Go—('общ
При хлорировании получаются моно-, дн- и трихлорзамещеп-
ные продукты. Соотношение между их количествами может быть
установлено на основании законов химической кинетики
Примем следующие обозначения:
а — начальная концентрация хлорируемого продукта, мол. доли;
х — концентрация прохлорированного продукта, мол. доли;
у — убыль мопохлорзамещенпого (концентрация ди- и три-
хлор замещенных), мол. доли;
с — концентрация хлора, мол. доли;
п — порядок реакции по хлору;
т — время;
— константа скорости реакции монохлорирования;
/л> — константа скорости реакции дпхлорировапня.
Исходя из того, что реакции образования хлорзамещеипых
имеют одинаковый порядок и протекают последовательно, можно
написать следующие кинетические уравнения:
— = к.Сп (а — х) (VII. 2)
а Г
(VII. 3)
Разделив (VII. 3) па (VII. 2), получим:
«I _ Ы*~*>... к ’-2. (VII. 4)
г/.г (« - .г) а — х
Представим это уравнение в следующем виде:
(« — х) cfy — (А'х — /.'у) dx = U (VI1. 5)
Уравнение (\ II 5) принадлежит к типу уравнении первого
порядка и первой степени с линейными коэффициентами, а именно:
(Ле !- By 4- С) (U. -|- {(!х + Ну Л) dy = О
где коэффициенты при dx и dy линейны относительно х и у.
Для приведения (VII. 5) к уравнению с разделяющимися
переменными, применим подстановку
х — ц> -f- т; dx = dw
у = v~-tt‘, dy = dv
197
где w и v — повыс переменные, а т и п — постоянные числа,
которые выбираются так, чтобы опи удовлетворяли уравнениям:
Л т Bn 4- С = О
Ст -г Ни -|- L = О
В результате такой подстановки в уравнение (VII. 5) получим
(« — w — т) do — к (w-}-m — у — zz) die = О
причем постоянные величины должны равняться нулю
—т ~ а — 0; — кт 4* кп = О
откуда:
т= д; т=п, г. е. т — п = а
Таким образом, подстановка х = и? -|- а и у — v + а приводит
уравнение (VII. 5) к виду:
(а — w—a) do—(А (w~{-a)— к (г-;-0)] </«' = 0
ИЛИ
(—kw4- Ar) div — w dv = 0 (VII. 6)
Сделаем еще одну подстановку
v = uw; du - и dw 4- и> du
Тогда уравнение (VII. б) примет следующий вид:
[—кю к (ton) ] dw — uw div —wz du 0
После упрощения (разделив на w), найдем:
(—к 4- ки — и) dw = w du
Мы пришли к уравнению с разделяющимися переменными:
du: _ du
tv (к — 1) и — к
Интегрируя это уравнение (предварительно умножив обе части
на к—1), получим:
(к — J) In ty г= In [(А — 1) и — к ] 4- С
Общий интеграл исходного уравнения получим, если вернемся
после некоторого преобразования к прежним переменным х и у:
(А— 1) In (д— х) « In [(1 -A) j—у4-1] 4-С (VII. 7)
При т — 0 имеем х = 0, у = 0. Отсюда находим
С — — (1 — к) In я
Подставляя это значение С в (VII. 7), получим:
1«J8
Освобождаясь от логарифмов, найдем:
а — х
Обозначим концентрацию ненрореагировавшсго продукта а—х
через xnt т. е.
а — х = Яц
а концентрацию мопохлорзамещеппого х—у через жм, т. е.
X — У — хя
п решим последнее уравнение относительно xv. Мы получим
формулу для выхода монохлорзамещсппого по заданной концен-
трации непрохлорированного сырья:
1-fc “ i — lc
Определим, при каком значении функция а;м имеет макси-
мум. Для этого найдем производную и приравняем ее пулю
_ & „l-к h-i I Л
“ 1-Л-и
(VII. 8)
жм
1—к
Отсюда определим а'ц
1
.гп = ак
Максимальное значение получим, внося это значение ян
в (VII. 8):
k
(VII. 9)
т — пк l~k
'’макс —
Пример. Вычислить максимальное содержание хлорбензола
в хлорированной жидкости, которое может быть достигнуто при хлорирова-
нии бензола, если известно, что соотношение констант скоростей реакций
образования полихлоридов и хлорбензола составляет:
-^-=fr 0.118
Для решения используем формулу (VII. 9). Начальная концентрация
(в мол. долях) хлорируемого бензола а 1. Следовательно:
n.iis
жмма1;с := 1 0.118 1-0,118 =-0,75 МОЛ. доли
Пример. Жидкий бензол подвергается хлорированию в аппарате
периодического действия. При условии, что реактор спабжеи мешалкой,
обеспечивающей полный расход поступающего хлора, и выделяющийся из
аппарата газ представляет только хлористый водород, определить количество
хлора, необходимого для получения максимального выхода мопохлорбепзола.
Реакция протекает изотермически при 55° С с отношением для констант
скоростей реакций
Л*1/Л« = 8.0 и А. А-3 30,0
199
кричем Ад откосится к реакции 1, /г2 — к реакции II и Аэ — к реакции III.
уравнения для которых приведены ниже:
(I) CeHe4-Ch —* CeHjCl + HCl
(II) СЙНЙСН-С12 —-> C6H.5Cl2-i-HCl
(III) С,Н«С18-гС12 —> CcII3Cl34-HCl
Примем для расчета 1,0 .ноль бензола и введем следующие переменные
для определения состояния системы ко времени т:
р — мольная доля присутствующего хлора:
q — мольная доля присутствующего бензола;
г — мольная доля присутствующего моиохлорбепзола;
s — мольная доля присутствующего дихлорбензола:
t — мольная доля присутствующего трихлорбензола.
Тогда будем иметь
7 !-г-Н-Н= 1 (VII. 10)
и Общий расход хлора составит:
(VII. 11)
Теперь примем, что объем реакционной массы 1‘ является постоянным.
Таким образом, скорость реакции для бензола представляется так:
V-^=-klPq (VII. 12)
Скорость образовапия каждого продукта будет:
= (VII. 13)
(Is
V ^=ktpr-k3ps (Vll. li)
F -k3ps (VII. 15)
Время т может быть делим уравнения (VII. 13 исключено как переменная величина, если мы раз- ) и (VII. 15) па уравнение (VII. 12). Следовательно: — = = (VII. 16) dq k\q 8q
Мы получили однородное дифференциальное уравнение первого порядка,
которое может быть решено путем подстановки.
Пусть r = i?q (VII. 17)
поэтому £ <vn is>
Подстановка (VII. 17) и (VII. 18) в (VII. 16) с последующим интегрнро-
ванном даст:
1п$ = 1п с-у In (7с 4-8) (VII. 19)
где С — постоянная интегрирования.
200
Освобождаясь от логарифмов, найдем:
(VII. 20)
Д'ля т = 0 имеем г/ = 1. г 0 и С = 8S,/7
После преобразования (VII. 20) получим:
г= 8 (д‘ 'в — <?)/7
(VII. 21)
Аналогично из уравнении (VII. 12) п (VII. 11) можно показать, что
ds х г
dq ~ 240<? 8q
При исключении г с использованием
(VII. 21) выражение (VII. 22) приводится
к дифференциальному уравнению, которое
может быть решено посредством интегриру-
ющего множителя. Мы получим:
S = (29,? - 239''1,8 -Ь210'/'
(VII. 23)
fc/’u расходуемого мооа
Для любого значения q можно определить
соответствующие значения г и х из (VII. 21) и
(VII- 23), а затем значение I из (VII. 10). Ана-
логично с помощью (A1I.I1) определяется
Рис. VII. 1. Результаты вы-
числений;
I —бенгпл; •? —монпхларбен-
зол; .1 —днхлорбензол; -4 —
трцх.ъ рбенаол.
общее количество расходуемого хлора- Эти
стадии расчета являются арифметическими и поэтому но приводятся де-
тально; результаты вычислений подытожены па рис. А 11. I, где мольная
доля каждого галоиднзпрованпого углеводорода изображена относительно
общего количества расходуемого хлора. Количество хлора, необходимого чля
максимального выхода моиохлорбеплола, в гоответст1.нц с рис. А II. 1 соста-
вляет 1 .ноль хлора на 1 ,шь загружаемого ни шла.
Пример. Для химического процесса l.'.'n ч чистого вещества Л
в жидком состоянии должны постукать непрерывно в первый аппара т ба га-
рей из двух одинаковых но размерам реакторов с vi шал .амп, которые
работают последовательно. При условии, что в этих аппаратах темперагура
поддерживается постоянной таким образом, что hmcci место реакция
определить общий объем установки, котт.ц 1Я может дать маю пма.и.!1ый вы-
ход продукта В. Константы скоростей реакций составляю! /,| (i,1 и k2 —
= 0,05 мип~1 при температуре реакционной массы, плотноеiь которой по-
стоянна н равна 9HU к?!м\
Пусть концентрации веществ А, В н (1 при выходе и;: любого реактора п
равны, Соответственно, Са ?l, Ct tl и (\ }|. Тогда дли материального баланса
Иримсч1итс.тыю к стадии г. в соответствии с общим понятием об уравнениях
в конечных разностях будем иметь:
для компонента А
^с, п-i
(VII. 24)
где т — продолжительность пребывания жидкости в реакторе;
201
для компонента В
Q, и -1 - сь,»=* А »-*А.. (VII. 2S)
Решение уравнения (VII. 25) выполняется обычным путем. Примем
А-Хг = а и Л2т = р. Тогда имеем:
(Я-а)Сс п-Св1П_1==0 (VII. 26)
(Л1+а)-1](7й>п = 0 (VII. 27)
или
Са,п=К& (VII. 28)
где Ki — произвольная постоянная, а рх = 1/(1 4* а).
Подстановка для Са п из уравнения (VII. 28) в (VII. 25) после преобразо-
вания дает:
[(14- ₽) Е -1) Cb „ _ х = пК(VII. 29)
Общим решением уравнения (VII. 29) будет:
Cb,n--=K2^ (VII. 30)
где К2 — произвольная постоянная, а = 1/(1 -f- р).
Пользуясь теорией уравнений в конечных разностях для частного реше-
ния (VII. 29) мы можем написать:
b> w'1= (1-4-Р) /с — 1 I= (l + 0)(h-l = P-а (VIL 31)
Таким образом, для общего решения получим:
(V11.32)
Так нак в реактор поступает чистое вещество А при п — 0, Са = Са 0
Сь = °, то
^ = ^«.0 (VII. 33)
(VH. 34)
После подстановки этих значении в уравнение (VII 32) найдем:
(vic зад
Искомое условие должно дать максимальную величину концентрации
Сь п для п ~ 2 при некотором значении т. Згот максимум легче найти путем
варьирования п при фиксированной величине т, чем, наоборот, при изме-
нении т для постоянного значения д.
Продифференцируем (VII. 35) п результат приравняем иутю:
~ («Г'« с,- С<’=) G <™- 3“)
Получим:
У* In pi
о” I“Q-2
202
Так как
11 1 =________________________
^1— 1 + « — 1 + 0,1 ’ 1 + p U,05 т
то
/1 + 0,05 TV In (1 +0,05 т)
\ 1 + 0,1 т J 1п(1+0,1т) ' ‘ 1 '
Применяя для решения (VII. 37) методы, описанные в курсах для при-
ближенного решения алгебраических уравнений, найдем:
т = 0,456 мин
Таким образом, будем иметь:
Объем реактора________ V _
Объемная скорость питания 4540 ’Ь
960 60
Оптимальный объем каждого из реакционных аппаратов составляет:
«03ЛЗ
Пример- Используя приведенные ниже данные, получить
приближенно значение для диаметра труб, устанавливаемых в реакторе с не-
подвижным слоем катализатора, который используется для синтеза хлори-
стого винила из ацетилена и хлористого водорода. В трубах находится хлори-
стая ртуть в качестве катализатора, осажденного на углерод с размером
его частиц 0,25 .w.w. Тепло реакции должно быть применено для образования
пара при 121° С с целью поддержания процесса на его последних стадиях.
Этот процесс осуществляется мри условии, что температура внутренней по-
верхности труб поддерживается при постоянной температуре 130Q С.
Теплопроводность массы катализатора X = 6,0 ккал/ч-м^град.
Теплота реакции при температуре слоя катализатора Д д = 2570 ккал/моль.
Насыпная плотность катализатора о = 2880 кг/л3.
Скорость регищии является функцией температуры, концентрации и
различных коэффициентов адсорбции, ио для предварительной оценки
примем, что скорость реакции выражается так:
•ПОЛЬ
г =г0(1 + ЛТ)
ч- иг катализатора
где г0 — 0,12; А — 0,0133, а Т — температура (в сС) над уровнем 93°' С.
Максимально допустимая температура катализатора, при которой еще
обеспечивается его удовлетворительное действие, составляет 265° С (т. е
Т = 172° С).
Примем следующие обозначения:
И'п — удельная скорость потока, кг/ч-.и‘г поперечного сечения реактора;
R — радиус трубы, .и;
х — радиальная координата, .и;
г — осевая координата от начала поступления реакционной массы,
ср — теплоемкость газа, ккал/кг-град.
Составляя тепловой баланс применительно к элементарному объему,
получим.
.и
дТ
Приход — —2лд+ 6з |-2лхбжИ'пСр2’ -; 2\qr
Убыль — —2лх?„ 6z + Г —2лхЛ 6s + 2лхбхИ''цСр ( Т
dx их I dx \
дх.
203
Приращение равно нулю, так как процесс протекает в установившемся
состоянии. 11о
Приход — Убыль = Приращение
Разделив все члены этого равенства па 2л?1.тйгбг, найдем:
д2Т . 1
<Хгг ' х
<>Т IFnCp дТ Q Лдг
дх ~ Л ’ dz '
(VII. 38)
Мы получили дифференциальное уравнение в частных производных,
устанавливающее зависимость между температурой и размерами реактора.
Для предварительной оценки примем, что температура насадки достигает
максимума для всех значений радиусов при одной и той же величине г, счи-
тая от места поступления реакционной массы в аппарат. Тогда для данного
радиального сечения насадочного слоя OTidz будет равна нулю, несмотря
па то, что действительные температуры изменяются с изменением .т. Темпе-
ратура будет максимальной по осп трубы и она по должна превышать 265" С.
Следовательно, по осп трубы дТЮх будет также равна нулю, но эта производ-
ная имеет конечное значение для всех радиусов, величина которых больше
нуля. Таким образом, дифференциальное уравнение (VII. 38) в частных
производных становится обыкновенным дифференциальным уравнением
для данного частного значения г, т. е.
4^Ч-'Т--Т" + ^£2(1 + -47’) = 0 (VII.39)
dx- 1 2 dx 1 а
Выражение (VII 39) легко может быть приведено к Бессолевому уравне-
нию пулевого порядка с последующим его решением. В этом случав сначала
должно быть использовано преобразование Лапласа.
Представим (VII. 39) в таком впде:
х-^- + 4^4-1^(1 + ЛГ)х = О (VII.40)
ах- ' ах ' f„
Применяя теорию дифференциальных уравнений и операционное исчи-
сление, получим:
l G =~ [{>2т ^-рт <°> -г =~р2г' +2рт -т
(VII. 41)
L(4r) = pZ (р)-7’(0) (VII-42)
Также имеем:
= (VII. 43)
К А / 1
Далее:
£ хТ\ = г (,,) (VII. 44)
\ Л / Л
Подставляя изображения (VII. 41) — (VII. 44) для соответствующих
величин в уравнении (VII. 40), получим после упрощения:
rir(^L_i_ / f (VI1 45)
fZp Р“-Г<? /'2(Р'-г<2)
где Q = ц&дгцА/л. и Р = рЛдг0/А.
204
Уравнение (VII. 45) может быть решено при использовании интегриру-
ющего множителя с получением следующего результата:
Ир2-l-е <?/>
Для перехода к оригиналам используем таблицы; решением будет:
Т (VII. 4В)
где С — произвольная постоянная интегрирования, которая может быть
вычислена.
При х = 0, Jh(xV Q) — 1 и Т = 172е С получим 172 — С —1/.4. По
А = 0,0133; 1/Л = 96,5 и С = 172 + 96,5 = 268,о. Далее
= 2880 • 2570 • 0,0133 0,12
6,0
и __ _______
/<)' = /1500 =38,8
Уравнение (VII. 46) приводится к виду:
Г = 268,5 Jft (38,8 х)-96,5
Для границы х = Л имеем Т — 56е С. Таким образом
56 - 268,5 Jo (38,8 х) — 96.5
или
У0(38,8Л) = -±±£=.-0,565
268,0
Из таблицы для Бесселевых функций имеем
38,8 Л = 1,4
откуда
'114
Л 0,036 м или 36 л.и
38>8
Тепловой баланс хлоратора
Тепловой баланс хлоратора (в ккал'ч пли ккал'опера-
ция) можно представить следующим равенством:
Cl -Г <>2+(?3 = Qa Т <?5 + <?б + (?-
где — тепло, поступающее в аппарат с хлорируемым сырьем;
(Т — тепло, поступающее в аппарат с хлором;
Q.t — тепловой аффект процесса хлорирования;
(jj — тепло, содержащееся в продуктах реакции (жидкая
фаза);
Qs — тепло, удаляющееся из аппарата вместе с отходящими
газами;
Q6 — тепло, отнимаемое охлаждающим агентом;
Q7 — тепло, теряемое в окружающую среду.
205
Мы будем рассматривать величину
Тепловой аффект процесса хлорирования Q,> (в ккал/моль) скла-
дывается из теплоты реакции ()р, теплоты растворения хлори-
стого водорода в воде <?раСтв и теплоты испарения (парообразова-
ния) сырья (последняя величина отрицательная):
(?з — <?р т красть — (?пси
Теплота реакции хлорирования <?р (в ккал!моль) вычисляется
обычным способом по закону Гесса:
Vp==9o-<-rn'?nci-rt'?C!I
где — теплота образования продукта хлорирования;
(/0 — теплота образовапия хлорируемого сырья;
711с1 — теплота образования хлористого водорода (22 ккал/моль);
<7СЬ — теплота образования молекулярного хлора (0 ккал/моль);
п — число атомов водорода, замещенных при хлорирова-
нии атомами хлора.
Таким образом:
(?р-<-?о + 22«
Определив но этой формуле теплоту реакции для монохлорирова-
ния gPl, для днхлорировапня др. и для трихлорирования дРг,
найдем суммарное количество тепла, выделяющегося в процессе
протекания реакции (в ккал):
(2Р- 10011 </yi + -у- 7ps — -щ-
Количество тепла, выделяющегося при растворении хлори-
стого водорода в воде (в ккал/кг), может быть вычислено по эмпи-
р ичес к ой ф о р м ул с:
*7раств = 477.5 m —157 in-
где т — растворимость хлористого водорода, кг IlCl/кз воды.
Растворимость хлористого водорода в воде зависит от темпе-
ратуры растворения и парциального давления хлористого водо-
рода. Эта зависимость в достаточной степени хорошо следует
уравнению:
м = (0.304-0,0016/) /А1*
где t — температура растворения, °C;
р — парциальное давление хлористого водорода в газовой
смеси, .н.н pin. ст.
206
Общее количество тепла, выделяющегося при растворении
хлористого водорода в воде (в ккал), выразится так:
Qрасти = б'к 0,01 f к (/расти
где <7,{ — количество получающейся соляной кислоты, кг;
ск — концентрация соляной кислоты, %.
Теплота парообразования (в ккал) хлорируемого сырья может
быть вычислена обычным методом:
О пенсией
где Сцеп — количество испаряющегося сырья, кг;
дпС11 — теплота парообразования, ккал 'кг.
О хлораторах
Хлораторы можно подразделить па следующие группы:
1) хлораторы для хлорирования ядра жидких ароматических
углеводородов;
2) хлораторы для хлорирования боковой цепи ароматических
углеводородов;
3) хлораторы для хлорирования твердых (высокомолекуляр-
ных) ароматических углеводородов в среде растворителей.
Хлораторы первой группы могут быть поверхностно-
пленочными, барботирующими или распиливающими аппаратами
(температура хлорирования 20—100° С); материал — сталь и
чугун, причем эти материалы ускоряют процесс и подвергаются
незначительной коррозии.
Хлораторы второй группы. При выборе хлоратора
второй группы нужно учесть, что условия хлорирования боковой
цели ароматических соединений отличаются от условий хлориро-
вания ядра тем, что здесь процесс проводится при температуре
кипения хлорируемой жидкости и чаще всего при отсутствии ката-
лизатора (катализатор хлорирования ядра — железо — не дол-
жен присутствовать в реакционной массе даже в самых незначи-
тельных количествах). Материалом для изготовления хлораторов
в этом случае служит гомогенно освинцованная сталь, эмалиро-
ванный чугун и ряд кислотостойких неметаллических материалов.
Аппаратами для хлорирования служат, главным образом, реак-
торы с мешалками, а также колонны.
Хлораторы третьей группы. В этом случае условия
проведения процесса хлорирования соответствуют условиям хло-
рирования боковой цени жидких ароматических углеводородов.
Отличительной особенностью здесь являются: 1) более вязкая
консистенция реакционной массы: 2) температура реакции, пе
превышающая точки кипения применяемых растворителей, и
207
3) возможность применения чугуна и стали в качестве материала
для аппаратов, представляющих собой реакторы с мешалками.
Для хлорирования бензола применяют хлораторы непрерыв-
ного действия. Такой аппарат представляет собой вертикальный
Рис. VII. .3. Хлоратор периодиче-
ского действия;
S — :;<.'рпус; 2 — руи.тшка; .7
1 — о чр Нигер.
мешалки;
Парогазовая смесь
Рис. VII. 2. Хлоратор непрерывного
действия в производстве хлорбен-
зола;
1 — иерхняя !>.icniH|iei:!l.'oi 4;srn. хлора-
тора. часть хлиртгора; Л —
4 rn£ij£a термпметра; 5
Чугучч.Н! И 'ЧН-Т!:, ’1 [ЯГ J НСр ДЛЯ Cliy-
ci«i i.eaaiai: 7
цилиндрический сосуд, футерованный внутри диабазовой плиткой
(рис. VII. 2). В нижней части колонны установлена чугунная
решетка, на которой уложена насадка из смеси керамических
и стальных колец. Стальные кольца одновременно служат катали-
затором. Верхняя часть хлоратора имеет расширение, которое
способствует разделению прохлорироваппой жидкости и газа.
208
Жидкий бензол и газообразный хлор подают иод решетку,
и смесь движется снизу вверх прямотоком. Хлорирование произ-
водится в избытке бензола при температуре кипения, поэтому от-
вод тепла осуществляется испаряющимся бензолом. Хлорирован-
ная жидкость выходит через боковой штуцер в верхней части
аппарата.
Газ, состоящий из хлористого водорода и ларов бензола,
поступает из верхней части хлоратора в специальные холодиль-
ники, где конденсируется основная масса бензола.
На рис. VII. 3 показана конструкция хлоратора периодиче-
ского действия. Для защиты хлоратора от коррозии рекомендуется
все его детали покрыть асбовинилом, а стальной вал н барботаж-
ную трубу обмазать диабазовой замазкой с одновременной обмот-
кой стеклотканью.
14 Л. М. Батуиср.
Глава VIII
Процессы сульфирования
Сульфированием называют процесс замещения водо-
родного атома в органическом соединении на группировку атомов
SO3H. Процесс сульфирования осуществляется путем обработки
сульфируемых продуктов различными сульфирующими агентами.
Исходными продуктами при сульфировании служат, главным
образом, ароматические углеводороды, амино- и окси и рои вводные
ароматического ряда и др.
В качество сульфирующих агентов применяются:
1) серная кислота различных концентраций. главным образом,
купоросное масло и моногидрат;
2) олеум различных концентраций с содержанием свободного
S0;j от 0 до 60%;
3) хлорсульфоновая кислота ($(.%! IC1);
4) бисульфаты некоторых металлов.
В ирон родственных условиях из всех перечисленных сульфи-
рующих агентов наиболее часто находят применение серная
кислота и олеум, значительно реже хлорсульфоновая кислота
и только в особых случаях пользуются бисульфатами металлов.
Условия проведения процессов.
Ход и направление процесса сульфирования зависят от тем-
пературы , продолжитсяьностн процесса, воздействия катализа-
торов, а также от концентрации сульфирующего агента.
Температура в процессе сульфирования играет значительную
роль. Если для многих химических процессов температура опреде-
ляет лишь скорость реакции, то при проведении процесса сульфи-
рования изменение температуры сказывается не только на ско-
рости реакции, но н па самом характере получаемых продуктов.
Серная кислота может быть сульфирующим агентом лишь в том
случае, когда концентрация ее превышает некоторую определен-
210
цую величину, обозначаемую через л. Если понижение концентра-
ции перейдет предел я, то сульфирование приостанавливается
Н. следовательно, при сульфировании нужно брать такое количе-
ство серной кислоты, чтобы в конце процесса концентрация ее
была не меньше л.
Величина л не может быть определена теоретически, она на-
ходится для каждого частного случая сульфирования экспери-
ментально применительно к данному сульфируемому продукту.
Расход сульфирующего агента складывается из двух величин:
первая из них отвечает стехиометрическому соотношению, а вто-
рая — необходимому избытку, обеспечивающему оптимальные
условия процесса сульфирования.
Обозначим:
S — количество SO3 в сульфирующем агенте в начале про-
цесса, масс, доли;
р — количество SO3 в сульфирующем агенте в конце процесса,
масс, доли;
G — количество сульфирующего агента, «г;
<7С — количество сульфируемого продукта, ка;
М — молекулярный вес (молекулярная масса) сульфируемого
продукта.
Напишем в общем виде уравнение реакции сульфирования:
HHn + nS03 —> R(SO3II)n
Общее количество серного ангидрида составляет G5.
Расход серного ангидрида па образование сульфокислоты по
стехиометрическому соотношению выразится величиной:
r п 80
с-лГ
где 80 — молекулярный вес серного ангидрида;
п — количество сульфогрупп, вступающих в сульфируемый
продукт.
После сульфирования в реакционной массе остается серной
кислоты с заданной концентрацией SO3, равной
Составим уравнение баланса серного ангидрида:
откуда
(7(5-o) = Gc^-(1-q)
jkl
Таким образом, расход сульфирующего агента составляет:
г (, «80(1 —о)
211
На практике в большинстве случаев в сульфируемых продук-
тах содержится то пли иное количество воды, наличие которой при
сульфировании вызывает разбавление сульфирующего агента.
Поэтому при определении расхода сульфирующего агента необ-
ходимо учитывать некоторую добавку его с целью устранения
разбавляющего действия воды.
Обозначим:
W — количество влаги, вносимой с сырьем, кг;
GmCg — добавочное количество сульфирующего агента, кг.
Количество добавляемого сульфирующего агента должно быть
таким, чтобы вносимая с сырьем влага образовала сульфирующий
агент с концентрацией SO... равной у, т. е. должно иметь место
равенство
б'дОб5=(14'-Ьб’лои)п
откуда
С учетом этом добавки общий расход сульфирующего агента
оп р ед е л итс я р а ве i ic г в ом:
G =
п 80 (1 — р)
с V(S- 0 "
И’
с
Л'-о
Пример. Определить расход купоросного масла (92% 11,80.,)
для сульфирования 800 кг нафталина, если конечная концентрация II2SO4
в масс, долях SO:, равна 0,6. Влага в исходном нафталине отсутствует.
Содержание S()3 в сульфирующем агенте в начале сульфирования:
Обозначим:
Сс — количество нафталина (800 кг);
у — конечная концентрация сульфирующего агента (0,6 масс, доли);
А/ — молекулярный нес. нафталина (128);
п — количество сульфогрупп, вступающих в нафталин (1).
Расход купоросного масла на сульфирование:
_ 80n^c (1 -Q) _ 80 • 1 800 (I —П.6)
Л/ (5 — о) 128(0.75-0,6) “1 J ™
Расход купоросного масла но реакции:
CioIK-piUSOi —> С1ОП78О3П 4-Н.,0
128 98 208 18
составляет
98 1
800 728'0Ж= 605 ,!г
Таким образом, фактический расход куноросного масла в два раза больше
требуемого но стехиометрии.
212
Расчет кислотных смесей
4
В практике сульфирования в качестве сульфирующих агентов
могут быть использованы лишь товарные сорта серной кислоты:
олеум, моногидрат и купоросное масло.
Олеум выпускается сернокислотными заводами только в двух
концентрациях с 20 и 65% содержанием свободного SO3. Купорос-
ное масло имеет концентрацию 92—94% H2SO4.
Однако требуемая производственными регламентами концен-
трация сульфирующего агента часто не совпадает с концентрацией
товарной серной кислоты. Отсюда п возникает необходимость
приготовления серной кислоты заданной концентрации из ее
товарных сортов путем смешения последних.
При расчете кислотных смесей в процессе сульфирования
приходится иметь дело со смешением: 1) двух кислот различной
концентрации или олеума с олеумом; 2) кислоты с водой; 3) кис-
лоты с олеумом;
1. Смешение двух кислот
Обозначим:
q — концентрация крепкой серной кислоты, %;
с, — концентрация слабой серной кислоты, %;
с — заданная концентрация серной кислоты, %;
— количество крепкой серной кислоты, кг;
С., — количество слабой серной кислоты, кг;
G — количество серной кислоты заданной концентрации, кг.
Балансовое равенство моногидрата:
6 -|- (72с3= Ч- 62) С
Откуда:
По условию имеем:
Ct~G2=G
Решая совместно два последних уравнения, получим:
Ci=G C-Cz (VIII. 1)
q—
G^G — GX (VIII. 2)
2. Смешение кислоты с водой.
Если заменить слабую кислоту водой (с2 = 0), то формулы
(VIII. 1) и (VIII. 2) примут вид:
Ga=G-Ct
3. Смешение олеума с серной кислотой (ослабление олеума).
Обозначим:
с0 — концентрация ослабляемого олеума, % избыточного SO3;
213
о/.
/о.
количество ослабляемого олеума,
с — концентрация получаемого олеума, % избыточного SO3;
с,- — концентрация серной кислоты
G — количество получаемого олеума, кг;
GI; — количество серной кислоты, кг.
Количество
воды, находящейся в серной кислоте, равно
„ 100—Ск
к 100
□то количество
воды свяжет серный ангидрид ио реакции
ILO4-SO3 —-> H2S04
IS 80 98
в количестве:
r 100-Ск 80 „ 100—ск ...
С“^00--18 = С“—Йб-Ш Кг
Балансовое равенство процесса смешения по SOs
Г со Г 100—си , Gc
'° 100 к 100 1,14 100
ИЛИ
G gCg — 4,44 (?! (100 — Сд) = С с
По условию имеем;
Gq+Gk=G; 0?о= G — GK
(VIII. 3)
Подставляя значение Go в равенство (VIII. 3), получим
(С - Gi:) е0-4,44 GK (100 —ск) = Gc
откуда необходимое для разбавления олеума количество сер пой
кислоты составляет
г — G <со—с)
15 О,+4,44 (100- ск)
а количество ослабляемого олеума будет
G0=G-GK
4. Смешение серной кислоты с олеумом (укрепление серной
кислоты).
Обозначим-
ск— концентрация укрепляемой серной кислоты, %;
с — концентрация получаемой при смешении серной кислоты, %;
с0 — концентрация олеума, % избыточного SO3;
GK — количество укрепляемой серной кислоты, кг;
G — количество серной кислоты, получаемой после смешения,
кг;
Go — количество олеума, кг.
214
Количество свободного сорного ангидрида в олеуме:
- со
го
rft 100
Количество моногидрата серной кислоты, образующегося при
взаимодействии избыточного серного ангидрида с водой:
__ вв ____ . р С0
100 80 ’ 0 100
Балансовое равенство по серпом кислоте:
хт 100 Ср 99 S С Ср I z1 <-И л С
° 100 1 11 - о 100 * 100 “G 100
<4)
ИЛИ
Gp (100 — сс) -|-1,225 б?фС0-|- бкск — Gc
Кроме того
G0-}-Gk= g
Из двух последних равенств получим:
г G(c — cv)
° 100 •}-0,225 ср — ск
и
<?к — G— 6'0
5. Графический расчет простых случаев смешения кислот.
Расчетные формулы для простых случаев смешения (смешение
двух кислот) иногда для скорости подсчета па практике заменяют
графиком.
Если построить параллелограмм или прямоугольник, провести
его диагонали и в углах, прилежащих к левой стороне, поставить
концентрации смешиваемых кислот, а концентрацию получаемой
кислоты поставить в точке пересечения диагоналей, то отноше-
ние разностей концентраций, получаемых вычитанием величин
по диагоналям, равно отношению количеств смешиваемых кислот.
Пример. Из 60 и 20%-ной кислот требуется получить кислоту
30% в количество 200 иг. Определить количества
с концентрацией ILSO
составил Ю1ЦИХ к пел от.
Имеем по графику
Таким образом:
G60 = 200 = 50 кг; С20 = 200 = 150 кг
215
Материальные балансы
Сульфирование серной кислотой
или олеумом
В общем виде реакция сульфирования может быть пред-
ставлена так.
UHrt-h»S03--=R(S03II)rt
М1 Н 80 М г
где Мд — молекулярный вес сульфируемого продукта;
п — число атомов водорода, замещаемого при сульфировании
сульфогруппами;
— молекулярный вес образующейся сульфокислоты.
Кроме того, обозначим:
Са — количество сульфируемого продукта, кг;
W — количество воды, вносимое с сульфируемым продуктом,
кг;
G — количество сульфирующего агента, кг;
I] — выход по сульфированию, масс, доли;
S — содержание 8О3 в сульфирующем агенте, масс, доли;
р — конечная концентрация сульфирующего агента, масс,
доли SC)3;
Дл — количество примесей, загружаемых с сырьем, кг;
— количество побочных продуктов, образующихся в ре-
зультате сульфирования, кг.
Количество сульфокислоты, образующейся в результате про-
цесса:
С»7Л Ч
Количество сульфирующего агента уменьшается на величину
израсходованного ангидрида, т. е. на
После сульфирования реакционная масса будет содержать суль-
фирующий агент в количестве:
Материальный баланс (в кг) представляется следующим обра-
зом:
Загружено
Сульфируемый продукт............................ с,-
Вода с сульфируемым продуктом.................. И'
Примеси с сульфируемым продуктом..............
Сульфирующий агент с концентрацией S............. С
Итого................СиС,1Ц
216
Получено
Сульфокислота
Сульфирующий агент с концентрацией q G <7С —
-‘К
Примеси с сульфируемым продуктом . . gj
Примеси, образующиеся в результате суль-
фирования .............................. А'г
Итого........... б’опц
Количество образовавшихся примесей получается по разности
между количествами загружаемых и полученных продуктов:
Г и 80
/Л» = Собщ — ^Gc ~j' Л + G — Gc И - -(- <д
Сульфирование хлорсульфоновой кислотой
Имеем для суммарного уравнения реакции:
RH„-| 2nS03HCl = R(SO2Cl)n-!-»H.1S04 MIC1
Ml ‘2?! 116,5 ,’J 1 ?! ?1 S(!. a
где Aft — молекулярный вес сульфируемого продукта;
Л/2 — молекулярный вес образующегося арилсульфохлорида.
Обозначим:
Gc — количество сульфируемого продукта, кг\
И7 — количество воды в сульфируемом продукте, кг\
— количество примесей в сульфируемом продукте, кг',
G — количество загружаемой хлорсульфоновой кислоты, кг;
g2 — количество серной кислоты в хлорсульфоповоп кислоте,
кг;
ga — количество примесей при сульфировании, кг'.
T] — выход продукта.
Количество сутьфохлорида:
Количество серной кислоты:
w!)H
6с-л7Г
Количество хлористого водорода:
.V
Кроме того, серная кислота и хлористый водород получаются
в результате взаимодействия хлорсульфоповоп кислоты с влагой,
имеющейся в сульфируемом продукте:
SOJiCl-HLO — > H2SO.t НИИ
in;,5 is 98
217
При наличии в сульфируемом продукте W воды образуется
•серной к^£Л0ТЫ
и хлориц101Ч) водорода
... 36,5
Расхс\дуется хлорсульфоновой кислоты:
18
Расхо хлорсульфоповой кислоты по основной реакции:
г 2п 116,5
с~ Му
Бала1|С хлорсульфирования может быть представлен следую-
щим обр413ОМ:
Загружено
^Удьфируемый продукт........................... Сс
*4’нмеси с сульфируемым продуктом.............
“°да с сульфируемым продуктом .................И'
Снятая кислота (и хлорсульфоновой кислоте) .... ,с-2
Получено
^Рдлсульфохлорнд ..............
Сс‘Ьиая кислота ...............
•^Аэрпстый водород ............
^лорсульфоновая кислота (нспро-
Ьсагировавшая).................
ПРцмссл с сульфируемым про-
Ауктом ........................
НРдмссм при сульфировании (но
йазпости) ..... ...............
Итого................бг’общ
Итого.......................... (?оГ,щ
Тепловой баланс сулъфуратора
Сульфирование серной кислотой или олеумом
Тепловой баланс (в ккал!ч пли ккал/операция) сульфу-
ратора м1)жно в общем виде представить так:
218
где Qy — тепло, поступающее в аппарат с обрабатываемыми
материалами;
<2й — тепло, поступающее в аппарат с теплоносителем;
— тепловой эффект процесса;
— тепло, содержащееся в продуктах реакции;
— тепло, идущее на нагревание аппарата;
— тепло, теряемое аппаратом в окружающую среду.
Величина (Л, является характерной для процесса сульфиро-
вания и поэтому должна быть специально рассмотрена.
Тепло процесса сульфирования складывается из теплот реак-
ции, выделения SO3 и растворения сырья (в ккол):
Qs — (?р "Ь (?выд Ч- Срасте
где Qp — теплота реакции;
(?выд — теплота выделения SO3 из сульфирующего агента;
(?раств — теплота растворения сульфируемых продуктов;
Определение (?р. По закону Гесса имеем (в ккал.1 моль):
7р= 2 .- Ако и— 2. Апач
Представим реакцию сульфирования в таком виде:
RH + SO3 —> RSO3H - 7р
Тогда
Ар ~ As-Ао~ Aso3
или
Ар = <7S —<70 —103,2
гДс — теплота образования сульфокислоты;
— теплота образования сульфируемого продукта;
— теплота образования серного ангидрида (103,2 ккал'моль).
Теплота выделения SO3 из сульфирующего агента. Для вычис-
ления этой теплоты могут быть применены формулы, определя-
ющие теплоту смешения сорного ангидрида с водой, так как эта
теплота равна по величине (и противоположна но знаку) теплоте
выделения SO3 из серной кислоты и олеума.
При смешении серном кислоты с водой выделяется тепло в ко-
личестве (в ккал'моль серной кислоты):
17,86 а:
а: + 1,798
где х — число молей воды, приходящихся на 1 .толь серной кис-
лоты после разбавления.
Согласно другой формуле при смешении у кг воды с 1 кг сер-
ного ангидрида выделяется тепло в количестве (в ккал:к? ЯО3):
504,2 if , 0,714 (t — 15) у
" У -г 0,2013 у + 0,002
219
где t — температура, при которой осуществляется процесс сме-
шения.
С целью упрощения расчетов по теплотам смешения кислот
используется следующая формула:
5/ =9ы>-г(* — зо) а
где qt — теплота смешения при температуре t° С, ккал-кг воды;
— теплота смешения при 50° С (из таблиц);
а — температурная поправка, значения которой помещены
в таблице.
Таблица
Количество тепла, выделяющегося при смешении
J кг воды с ВО3 при 50е С
Концентрации SOs в сульфирующем агенте, масс, доли Количество тепла при смешении при 5(1° С qSo, ккал/кк воды Тепловая поправка, а
0,94 1683 5,1
0,92 1576 4,6
0,90 1502 4,2
0,8В 1440 3,7
0,86 1390 3,4
0,84 1-343 3,0
0,8163 1270 2,5
0,80 1195 2,3
0,78 1120 2,0
Ио закону Гесса имеем:
f
?выд ^см ^см
где г/см — теплота смешения серного ангидрида с водой при обра-
зовании сульфирующего агента с концентрацией о;
qCM — теплота смешения серного ангидрида с водой при обра-
зовании серной кислоты с концентрацией S.
Таким образом, тепловой эффект процесса сульфирования
составляет:
„ _ 10006’с .. ... ,
(?з — —у--7р (1 —•$) 7выд I (/паств
Обычно (/рас™ (теплота растворения сырья) принимается равной
нулю.
Пример. Вычислить тепловой аффект процесса сульфирования
бензола в ко; ичесгве 1001) кг при I — 50° С, если в качестве сульфирующего
агента применяется моногидрат серной кислоты, а отработанная кислота имеет
концентрацию серного ангидрида р = 0,6 масс. доли.
220
Расход моногидрата па сульфирование W00 яг бензола:
„ «8()(1-С) 1000-1 (1-0,0)
-V(A-o) “ 78(0,81вЗ-(ЙГ
где М — 78 — молекулярный лес бензола;
S = 0,8163 — концентрация S03 в моногидрате.
'Гоплота реакции сульфирования:
— г;о —103,2 134,4 —(—4,1) —103,2 = 35,3 ккал;молъ
где <fs 134,4 ккал-моль — теплота образовапия бе изол сульфокислоты
q0 — —4,1 к/гял/.иель — теплота образования бензола.
Теплота выделения SO3 из сульфирующего агента:
<?пэд=%- ?о = С15—1270= — 655 кл-дл/nv воды
а’Де -- 615 ккал/кг воды | теплоты смешения
<lf} — 1270 ккал/кг воды j ПРП 50 С (из гаолнцы)
Тепловой эффект процесса сульфирования:
, 1000(7с . „ п
(?3--------Qp + & (1 — S) 1/выд — (?р.эств —
= w90J000 -5,3 — 1 goo (1 _ 0,8163) G55 г 0 = 224 000 ккал
io
Величина врасти принята равной пулю.
Сульфирование хлорсульфоновой кислотой
Для сульфирования хлорсульфоновой кислотой теп-
ловой баланс может быть представлен следующим образом:
<?i Ч- Q*.+<?з = Q1 - Q* i ('« + Q-
где при сохранении прежних обозначений Q-t — тепло, уносимое
из аппарата хлористым водородом.
Величина Q7 (в может быть найдена обычным путем как
произведение массы, теплоемкости и температуры хлористого
водорода, уходящего из сульфуратора:
<?7
где с — теплоемкость хлористого водорода, ккал к.?;
t — температура хлористою водорода, С,
—’"Is'/ — количество хлористого водорода, выделяюще-
гося при сульфировании (см. материал виши
баланс).
221
Тепловой эффект процесса сульфирования хлорсульфоновой
кислотой складывается из теплоты реакции сульфирования и
теплоты разложения хлорсульфоновой кислоты водою, находя-
щейся в сульфируемом сырье:
(Л) RII + SO31 (CI = RSO.,C1 -I- 1ISO
(Z>) SO3IIC1 -I- II2O I LSO.J - 11C1
Количество тепла, выделяющегося при протекании процесса
ио уравнению (Л), найдем обычным способом, сделан небольшое
допущение, которое вызвано отсутствием опытных данных отно-
сительно теилот образования арнлеульфохлоридов. В техниче-
ских расчетах без особой погрешности можно допустить, что теп-
лота образования арилсульфохлорида настолько меньше теплоты
образования арилсульфокислоты. насколько теплота образования
хлорсульфоновой кислоты меньше теплоты образования серной
кислоты.
Если обозначить разность между теплотами образования
серной кислоты и хлорсульфоновой кислоты через Л, то теплота
реакции (А) может быть выражена (в ккал/моль) так:
где дв — теплота образования воды (68,38 ккал /моль);
г/ч — теплота образования соответствующей данному сульфо-
хлориду арилсульфокислоты;
— теплота образования сульфируемого сырья;
д* — теплота образования хлорсульфоновой кислоты
(140,2 ккал/лнмь).
Кроме того, но условию имеем:
Л — ?НгКО4 — '7х' ®HtSO* Л
где qu — теплота образования сорном кислоты (192,2 ккал/моль)
Подставляя значение qx, получим:
?( .А) = vB -! tfs - f’n - ?н,5Ол = ?s - % 4- 68,38 -192,2
Далее:
|7(7;)=//цгйо<л-'/нС1-'7х — 7В = 192,2-1-22 — 68=38 — (40,2 ~ 5,6 ккал/моль
Складывая (Л) и (Г>), получим (в ккал:моль):
222
Таким образом, количество тепла (в кнад/операция), выделя-
ющегося при сульфировании хлорсульфоновой кислоты, со-
ставляет:
ft=1000 = (£ ?(л,+ол IV )
Аппаратурное оформление смешения
кислот
Для смешения кислот применяются типовые котлы
и чаны, выполненные из кислотостойкого материала.
Следует различать три случая смешения при сульфировании:
1) приводящее к образованию разбавленной кислоты (<75%
HoSO.J; 2) приводящее к образованию концентрированной кис-
лоты (75—100% H2SO4) и 3) приводящее к образованию олеума.
В первом случае смешение можно проводить в открытом ап-
парате, который выполнен из материала, стойкого к воздействию
серной кислоты. Этот аппарат может быть снабжен либо мешалкой,
либо воздушным барботером.
В случае смешения, приводящего к образованию концентри-
рованной кислоты, проведение процесса в открытом аппарате
и перемешивание с помощью барботажа воздухом нежелательны,
так как и то и другое способствует поглощению кислотой влаги
из воздуха и, следовательно, се разбавлению.
При смешении олеума открытые аппараты и перемешивание
сжатым воздухом неприменимы вследствие легкости десорбции
серного ангндрц щ.
Для получения разбавленной кислоты применяются баки из
кислотостойкого материала. Эти баки снабжаются змеевиками
для охлаждения и воздушными барботерами для перемешивания.
Для смешения олеума или кислот, приводящего к образованию
олеума или кислот других концентраций, применяются закрытые
чугунные или стальные аппараты.
О сульфураторах
Сульфураторы могут быть разделены на две группы.
В первую группу входят тс аппараты, в которых процесс проте-
кает при температурах ниже 1б(Р С. Ко второй группе относятся
сульфураторы, в которых процесс протекает при высоких темпе-
ратурах (> 160° С).
Сульфураторы нерпой группы имеют греющее приспособление
в виде паровой рубашки, рассчитанной в большинстве случаев
на рабочее давление в 4 ат, сульфураторы же второй группы
имеют теплообмоиивающую поверхность в виде змеевиков. Как
тс, так п другие сульфураторы снабжены якорными пли
99”.
Рис. VIII. 1. Сульфуратор с рубашкой:
1 — крышка; l' — корпус: -? — мешалка; t — рубашка.
Рис.VIII. 2.Сульфуратор
с рубашкой и змееви-
ками:
I — крышка; 2 — рубашка;
3 — .змеевик; 4 — мс.пальа.
пропеллерными мешалками в зависимости от консистенции сульфо-
массы. Как правило, пропеллерные мешалки применяются при
сульфировании жидких и расплавленных веществ, якорные — при
сульфировании твердых веществ.
При сульфировании обычно используется концентрированная
серная кислота, которая не вызывает коррозии черных металлов,
поэтому сульфураторы изготовляют из чугуна или углеродистой
стали.
На рис. A Ill. 1, представлен чугунный сульфуратор с рубаш-
кой. Аппарат имеет чугунную якорную мешалку» вращающуюся
со скоростью 40—50 об мин.
При высоких температурах процесса применяется сульфура-
тор в виде котла со стальной рубашкой, в которой расположены
змеевики, залитые свинцово-сурьмянььм расплавом (рис. VI11. 2).
Обогрев аппарата производится паром 10—12 ат, проходящим по
змеевику. Для удаления реакционной массы по трубе переда-
вливания применяется сжатый воздух, поэтому аппарат рассчиты-
вают на дав 1ение 3—4 ат.
45 Л. М. Багунср.
Глава IX
Процессы нитрования
Нитрованием называют процесс замещения водород-
ного атома, главным образом, в ядре ароматических соединении
на группировку NO>.
В качестве исходных материалов для нитрования служат
углеводороды ароматического ряда, а также их замещенные.
В качестве нитрующих агентов применяются: 1) азотная кис-
лота; 2) меланж, представляющий собой смесь азотной и серной
кислот; 3) селитра в смеси с серной кислотой; 4) окислы азота и др.
Действующим началом в процессах нитрования, независимо
от того, какой из нитрующих агентов применяется, служит азот-
ная кислота. В общем виде уравнение, выражающее процесс
нитрования, может быть представлено следующим образом.
RII + IIN03= КХО.. + 1LO
где НН — соединение, подвергаемое нитрованию;
RNO2 — пнтросоединспие, полученное в результате нитрования.
Условия проведения процессов
Как правило, нитрование ведут при невысоких темпе-
ратурах (0—80° С). Особенность влияния серной кислоты па про-
цесс нитрования заключается в ее каталитическом и водоотпима-
ющем действии. Нитрующий агент, т. е. пнтросмесь, состоящая
из азотной и серной кислот, а также воды, должна содержать
некоторый минимум серной кислоты, без которого процесс нитро-
вания протекает несовершенно даже в том случае, когда серная
кислота заменяется азотной, и общая кислотность нитрующей
смеси остается без изменения. Азотная кислота в нитрующей
226
смеси берется в количестве, превышающем теоретическое на 1 —
10%.
Соотношение количеств серной кислоты и поды в нитрующих
смесях должно быть строго определенным, так как от него зависит
ход нитрования. Даже очень малые изменения в соотношении
количеств серной кислоты и воды приводят к значительному
уменьшению выходов.
Обозначим:
Сц — количество нитруемого продукта, кг;
d — число нитрогрупп, вступающих в ароматическое ядро
при нитровании;
Л — количество технической азотной кислоты, загружаемой
па нитрование, кг;
а — содержание HN03 в технической азотной кислоте,
масс, доли;
(?5 — расход серной кислоты на нитроваппс, кг;
S — содержание H,,SO4 в серной кислоте, масс, доли;
р — содержание H3SO4 в отработанной серной кислоте,
масс. доли.
В состав полученной после нитрования отработанной серной
кислоты войдут:
1) вся серная кислота, которая загружается в аппарат;
2) вода, находящаяся в технической азотной кислоте;
3) вода, выделяющаяся в процессе нитрования.
Количество воды (в кг) в азотной кислоте, идущей на нитрова-
ние:
Лг(1-«)
Количество воды, выделяющейся при нитровании, определится
из стехиометрического уравнения:
RI Id 4- d IINOS -'г R(N'O.,)d -J- dH8O
АГ «Тез Mi dis
Согласно этому уравнению и принятым выше обозначениям коли-
чество воды, выделяющейся при нитровании, будет:
18d
Таким образом, количество отработанной серной кислоты:
Напишем теперь равенство, выражающее баланс II2SO4 при
нитровании (без учета потерь)
+ (1 —«)-|--тт-18сЯ С?
L ЛХ J
15*
227
откуда
ЛГ (1 —
18г/ ]
Л/ J
Численные значения р для наиболее распространенных случаев
нитрования приведены в табл. 1Х-1.
Таблица IX. I
Значения Q и $
Нитруемое вещество (?, масс, доли .-С1
Бензол (мононитрат) . . . 0,775 1-3
Толуол » 0.711 1
Хлорбензол » ... 0.816 1
Нафталин » ... 0,671 1
Бензол (динитрат) .... 0,871 10
В практике нитрования приходится определять не только
расход водоотнимающего агента, но и состав нитрующей смеси
по заданному составу отработанной кислоты.
Обозначим в дополнение к предыдущему:
G — количество нитрующей смеси, кг\
I — содержание азотной кислоты в нитрующей смеси, %;
т— содержание серной кислоты в нитрующей смеси, %;
$ — избыток азотной кислоты по сравнению с теоретическим
расходом, %.
Количество моногидрата азотной кислоты (в кг), находящейся
в нитрующей смеси, составляет
Согласно уравнению реакции нитрования расход азотной кислоты
ла проведение процесса нитрования может быть представлен
равенством:
(IX. 2)
П.з (IX. 1) п (IX. 2) имеем:
6'”-^(И-0.01£) (IX.3)
Содержание серной кислоты (в кг) в нитрующей смеси:
G —
100
Количество отработанной кислоты (в кг), состоящей
и серной кислоты:
, 100—I , „ 18(I
и м
100
Палане по моногидрату серной кислоты
т / 100 -I . 18^ \
6Тю = (с-ПйГ+с"Лг)°
из которого следует, что
т — (100 — 1)q 18(7
6-----100----==6u1F9
Из (IX. 3) и (IX. 5) получим:
7
из поды
(IX 4)
получим
______________ _______________ 3.5(1-7-0.01 g)
W —(100 —Об ~~ 1&> Q
Решая последнее уравнение относительно величины т,
формулу:
_ [1 3,5(1--0,01 =)]Р
3,5(1-|-0,01 £) 1 10°9
или
т = kl Ъ
где
к
и Ь — ЮОо
-3,5 (1 — 0,01 ^)| q
3.5(1 ,-О.оП)
Формула (IX. 6) называется «уравнением состава нитрующей
смеси». Рассматривая это уравнение, мы можем сделать следующие
выводы:
1) для успешного проведения процесса нитрования необхо-
димо применять нитрующие смеси, в которых содержание НХ'О3
и HoSO.j связано прямолинейной зависимостью;
2) состав кислотных компонентов для нитрующих' смесей,
применяемых для нитрования одного и того же продукта (т. е.
для заданных £ и р), может быть самым различным в зависимости
от наличия составляющих частей, но, однако, таким, чтобы со-
держание серной и азотной кислот строго соответствовали
нению состава».
«урав-
Расчет нитрующих смесей
При расчетах нитрующих смесей обычно бывают задан-
ными следующие величины: общее количество смеси, ее состав
и состав всех исходных ее частей. Искомыми величинами явля-
ются количества исходных веществ, входящих в состав смеси.
В наиболее общем случае нитрующая смесь составляется
из трех веществ, в состав которых входят азотная и серная кислоты
и вода.
229
Примем следующие обозначения;
G — количество нитрующей смеси, кг;
I — содержание азотной кислоты в приготовляемой смеси, %;
т — содержание серной кислоты в приготовляемой смеси, %;
п — содержание воды в приготовляемой смеси, %;
G, — количество составляющей А, идущей на приготовление
пптросмссп, кг;
1и — содержание азотной кислоты в составляющей А, %;
ма — содержание серной кислоты в составляющей Л, %;
пя — содержание воды в составляющей А, %;
Сь — количество составляющей 13, идущей па приготовление
нитросмеси, кг;
1Ь — содержание азотной кислоты в составляющей 13, %;
гпь — содержание серной кислоты в составляющей 13, %;
пь — содержание воды в составляющей 13, %;
б’с — количество составляющей С, идущей на приготовление
нитросмеси, кг;
1С — содержание азотной кислоты в составляющей С, %;
тс — содержание серной кислоты в составляющей С, %;
пе — содержание воды в составляющей С, %.
Сумма количеств всех составляющих должна равняться ко-
личеству приготовленной смеси, т. е.
+ G . (IX. 7)
Количество азотной кислоты, которое будет находиться в нит-
росмеси:
Gl= Gа1аGЪ1Ъ-v-Gcic (IX. 8)
Количество серной кис юты в нитросмсси:
Gт = Gum„ -L- Gb^b + Gcmc (1X. 0)
Количество воды в нитросмсси:
Gп = Gaita -l-G^b-r Gcnc (IX. 10)
Решим совместно уравнения (IX. 7) — (IX/9). Иснользуя
теорию определителей, получим:
1 1 1
| А | =* 1а
lb lc = Uа — 1ъ) (та-ть) — (i«—1С) («я-mb)
та тс
Таким образом, имеем:
1 1 1
Z 1Ь 1С
т nib rilc
(la — lb) ("'a— "?c) ~ (la — lc)^na—nib)
(Ic — /) (mc — nib) — (G — b>) Q»c — "0
(la lb) (^a. ^c) (1ц lc) (,fJa l^b)
230
1 I 1
la I lc
->_________________ m >nc____________
(la — h>l ("Щ — Wc) — (la ~ M (wa ~ >!lb)
G (lc — l) (f»a —»?<)—— (Wr —w)
’ (la —lb) ('»« — mc)- (Za — lc) —
1 1 1
la lb I '
G __________________»>a Wt> m_____________________
{l-t — lb) (,na tllc) (la lc) (m" l,lb)
,,___(l<i—l) (»>b — >»(i) — (lb — la) ('»« —"0
(la — lb) ("’« — »>c)~(la — lc) — (ma ~ >»b)
Если вместо серной кислоты применяется олеум, то формула
для расчета не меняется при условии, что концентрация олеума
будет выражена в % серной кислоты. Для пересчета концентрации
олеума, выражением в % свободного серного ангидрида, на кон-
цептратцпо серной кислоты применяется следующая формула:
/л'= 100-|-0,225 а
где и — содержание свободного серного ангидрида в олеуме %;
т' — концентрация олеума, выраженная в % моногидрата
серной кислоты.
Пример. Рассчитать состав нитрующей смеси с содержанием
16% ПХО3 п расход кислот, идущих на ее приготовление, для нитрования
1200 кг хлорбензола (Л/ — 112,5) на мононитрохлорбензол при р = 0,70
н избытке Н.ХО3 против теории ? = 1%. Нитрующая смесь составляется
мз меланжа, 20%-го олеума (104,5% Н2Й().,) и отработанной кислоты.
Состав меланжа: 85% HN03; 10% H2SO.,; 5% Н2О.
Состав отработанной кислоты: 70% IIaSO4; 30% II 2().
1. Расход моногидрата азотной кислоты на нитрование:
<7^(1 -I-0,91 £) = 1200 1 (^.12 S'01 ' П ~ 678 №
где 6П ~ 1200 кг — количество нитруемого хлорбензола;
d — 1 — число нитрогрупп, вступающих в соединение;
ь = 1% — избыток азотной кислоты ио сравнению с теоретическим.
2. Уравнение состава нитрующей смеси:
т —
(1—3,5(1-?-0,01 ?)] о
3,5(1 + 0,01?)
1-L 100/) =
= [1-3.5 (1-0.01-1)] 0,7 „
3,5(1 — 0,01-1) ^ + 100-0,/— О.оОЗ/ , /0
Подставляя заданную величину I — 16, получим значение т:
/// = -0,503- 16 | 70 61,95%
231
3. Расход нитрующей смеси.
По заданной концентрации М.\О3 в нитрующей смеси I = 16% опреде-
ляем общее количество нитрующей смеси:
Таким образом, для состава нитрующей смеси имеем:
Н.\О3 I ~ 16%; HoSOj т = 62%; Н2О п 22%
меланж
UNO, Z(l =--• 85%; ILSO.1 та = 10%; Н2О па = 5%
олеум (20%-пый)
HN03 % U, H2SOt гл£> = 104ii3?ti 1 П2О «ь —0
отработанная кислота
ЦД'О3 Zc = 0, IL>SO4 ™с = 70%, Н20 пс = 30%
4. Подставляя соответствующие значения в расчетные формулы, полу-
пим:
расход меланжа
Г /9-0 (° 1 <70 ~ 104>5) ~ (° ~ 0) <70 ~ G2) - "OS г,
Сга. (I0—70)_(85_()) (10_J04.5) ' ‘
расход олеума
,, (0 -1G) (W - 70) - (85 - 0) (70-G2) _
' b - (85 _ о) (10 - 70) — (85 — 0) (10-104,5) 1
расход отработанной кислоты
(85-16) (104,5 -10) - (0-85) (10- 62) _
с ' (85—0)(1О—70) —(85 —О) (10—104,5) J ‘
Проверяя общее количество смеси, имеем
G - G а -|- 6% -)-• Gс = 795 -|- 400 -р 3055 = 4250 *>‘й
что соответствует заданному количеству нитрующей смеси.
Тепловой баланс процессов смешения
кислот для приготовления
нитрующей смеси
Определение количества выделяющегося тепла и скорости
охлаждения является главной частью расчета смешения кислот.
Количество поступающего в аппарат тепла (2 (в ккал) можно
представить равенством:
<2'—eacata—с ъсъ^ъ~\~ ессЛс "г Q р
(IX. II)
где Са, Съ, Gc — количества составляющих, кг;
съ> С‘с — теплоемкости составляющих, ккал!'кг • град;
ta, С — начальные температуры составляющих, СС;
<2р — количество тепла, выделившегося при смешении,
ккал.
232
Количество тепла Q" (в ккал) в продуктах смешения:
<)"=Gci-|-Qx (IX. 12)
где G — количество смеси, кг;
с — теплоемкость смеси, ккал'кг -град;
I — начальная температура смеси, С;
()Л. — количество тепла, отнимаемого охлаждающим агентом,
ккал.
Приравнивая правые части равенств (IX. II) и (IX. 12) и ре-
шая полученное уравнение, относительно Qx, получим:
(Лк = G aeala “Ь G 1,СЪ1Ь 'Г бссЛ “г Get
Если известно Qx, то продолжительность охлаждения и расход
охлаждающего агента определяют обычным путем.
Тепло, выделяющееся при образовании нитрующей смеси,
может быть вычислено как теплота смешения 100 %-поп серной
кислоты, 100%-ной азотной кислоты и воды. Для этого введем
понятие об исчерпывающем разбавлении кислот.
Исчерпывающим называют такое разбавление кислот водой,
при котором дальнейшее разбавление уже но сопровождается
тепловым эффектом (300 масс. ч. воды па 1 масс. ч. кислоты).
Пусть при исчерпывающем разбавлении 1 кг серной кислоты
выделяется S() ккал, при исчерпывающем разбавлении 1 кг
азотной кислоты — 7nxos ккал и ПРИ исчерпывающем разбавле-
нии 1 кг смеси данного состава — qa ккал (например, для состав-
ляющей Л).
Исчерпывающее разбавление рассматриваемых кислот и кис-
лотных смесей может быть проведено двумя способами.
п - с тп
Во-первых, могут иыть исчерпывающе разоавлепы Ga кг
серной кислоты и Gn кг азотной кислоты. При этом общее
количество выделяющегося тепла (в ккпл.) составляет:
/1 _ !t,(t , < г 1г> »
'A юр • « щи ^пхо-,
Во-вторых, разбавление может быть произведено следующим об-
разом. 1 {начале смешиваются Са ~~ кг серной кислоты, кг
Ivu 1VU
• z-1 (I
азотнои кислоты и Ga кг воды; при этом выделяется количество
тепла Qa ккал. Затем полученная смесь исчерпывающе разбавляет-
ся водой с выделением Gfitpt ккал тепла и, таким образом, общее
количество выделяющегося тепла (в ккал) составляет:
Qi -- Qu г б
Поскольку в первом и во втором случаях процесс закапчивается
исчерпывающим разбавлением исходных кислот, то количества
выделяющегося тепла Qr и Q., равны между собой, т. о.
f ___ у-» Л I у-, 1(1
Va" ^aVa—?П;501 , va<?пх°з
Последнее равенство дает возможность найти количество тепла,
которое получается при образовании составляющей смеси из
неразбавленных кислот, т. с.
а '= С> а TJT ?HsSO, + Ga Г/ДХО,G а^а <1X. 13>
Аналогично могут быть вычислены и количества тепла, выде-
ляющегося при образовании составляющих В и С из 100 %-пой
серной кислоты, 100%-ной азотной кислоты и воды. Пусть эти
количества тепла будут Qb и Q,-.
Применяя формулу (IX. 13) к нитрующей смеси, получим:
e=GW’».so<+6'no’HKo.-C’ СХ.14)
где q — теплота исчерпывающего разбавления нитрующей смеси,
ккал/кг.
Следовательно, при приготовлении нитрующей смеси из 100%-
ной серной кислоты, 100%-ной азотной кислоты и воды выдели-
лось бы количество тепла, равное Q ккал. Однако нитрующая
смесь готовится не из 100%-ных кислот, а из кислотных смесей,
при получении которых из 100 %-пых кислот уже выделились ко-
личеств а тепла (?«, (% и Qc.
Понятно, что количество тепла Qp (в ккал), выделяющегося
при приготовлении нитрующей смеси из кислотных смесей будет
меньше на сумму Qa — Qb “г Qc, т. е.
(?р = Q—Qa—Qb— Qc
Сопоставляя последнее равенство с формулами (IX. 13) и
(IX. 14), т. е. заменяя Qa, Qb и Qc их значениями, получим:
п _г ( т г 1 \
^/n.sox "щи v ?ихо=<“йю— q) ~
с ( та х „ \
— ° а (^НэЬО, [( K J "Г ^ПХОз Хю ~ 4(1) ~
( тЪ , 1ь \
~ tfihso* । ^пх'озТуо — % у —
Z’ (,Г‘с I ... \
I ?Нг8О4 Ю0 % у
После умножения и соответствующего сокращения, получим:
<2р = Ga<la 4~ Gb'/b + Gcifc — G'l (1X. 15)
2М
Для расчета теплоты разбавления водой кислотной смеси
рекомендуется формула А. П. Плаповского:
Здесь
... 312» л m
а n-j-98,3 ’ У 32-4» ’ Х т-Н
где I — содержание азотной кислоты в смеси, %;
т. — содержание сорной кислоты в смеси, ‘.’б;
п — содержание воды в смеси, %.
Если одной из составляющих смеси является олеум, то необ-
ходимо к вычисленному тепловому эффекту прибавить теплоту
гидратации серного ангидрида водой, которая составляет
255 ккал кг серного ангидрида.
Пример. Вычислить количество тепла, выделяющегося при
смешении 100 кг 100%-иой серной кислоты п (00 кг 80%-пой азотной кислоты.
В результате смешения получается смесь следующего состава (в ь-г):
Серная кислота .................. 100
Азотная кислота................ . 80
Вода ............................ 20
т. о. т — 50; / = 40 и /г =10.
Теплота исчерпывающего разбавления серной кислоты:
f/d = 183 ккал;кг (из таблиц)
Теплота исчерпывающего разбавления 80%-ной азотной кислоты:
tjj> = 111 — 47,2 = 03,8 ккал/кг
где 111 ккал/кг — теплота исчерпывающего разбавления 100%-ной азотной
кислоты;
47.2 ккал/кг — теплота разбавления 100%-пой азотной кислоты до кон-
центрации 80% (из таблиц).
Теплота исчерпывающего разбавления полученной смеси:
а _ 82,2 1 — Ьх 1 — 0,555 • 0, где ... 312» а — 111 - п -j- 98.5 , . ккал — = 102 Z : Зь кг разоавленном смеси = 111 — Д12'J0 — 82,2 10-1-98,э
‘ 1 /о п — 49 82,2 100-Г 49
т Х т.л-1 50 . 50-г 40
Тепловой эффект процесса смешения:
<>р = Caf!a -Г Gt/lb — Gq^= 100-183 - 100 -63,8-200-102 = 4280 ккал
235
Пример. Определить тепловой эффект смешения 4000 кг 20%-го
олеума и бш.)0 кг 50%-ной азотной кислоты.
Находим состав (в кг) смешиваемых кислот:
Серная кислота 4000 - 0,8 = 3200 1 ....
Серный ангидрид 4000-0,2 ~ 800 j
Азотная кислота . . . ООО -0,5 — 3000 | .тт
Вода ...............6000-0,5-3000 | 111 >
Состав полученной /моей (в кг)
Серная кислота . . .
Азотная кислота . , .
Вода ................
98
3200-1- 800-3-— 4180
о U
3000
18
3000 - 800-377 = 2820
oU
т. с. ?,i = 41,8%
I 30%
п= 28,2%
Теплота гидратации серного ангидрида:
<?г= = 255 • 800 =204 000 ккал
где 71 = 255 ккал/кг — теплота гидратации сериого ангидрида;
Ci = 800 кг — количество серного ангидрида.
Теплота исчерпывающего разбавления 100%-ной серной кислоты:
да — 183 кклл/кг
Теплота исчерпывающего разбавления 50%-пой азотной кислоты:
7й =111 — 94 = 17 ккал/кг
где 111 ккал/кг — теплота исчерпывающего разбавления 100%-ной азотной
кислоты;
94 ккпл/кг — теплота разбавления 100%-ион азотной кислоты до концен-
трации 50%,.
Теплота исчерпывающего разбавления полученной смеси ио формуле
А. II. 11лайовского:
--— =-----------—------= 53,2 икал:кг разбавленной смеси
1—Cr 1—0,30-0,а83------1
где
« = 111
312 .312-28,2 _
7/4-98,5 1 28,2 + 98,5 1 ,J
Тепловой эффект процесса смешения:
= О ц7и т Сl>Qb ~Q G 7 — 4180 • 183 -~
+ 6000 17 -г 204 000-10 000 53.2 = 530 000 ккал
236
Материальный баланс процесса
нитрования
Обозначим:
Glt — количество нитруемого продукта, кг;
G — количество нитрующей смеси, кг;
I— содержание азотной кислоты в нитрующей смеси, о;
т — содержание серной кислоты в нитрующей смеси, %;
1] — выход по нитрованию в масс, долях от теоретического;
d — число нитрогрупп, вступающих в молекулу углеводорода
после нитрования;
gL — количество воды в нитруемом продукте, кг.
Для процесса нитрования имеем:
R11</ + d IINO.,-> R (NO-»)d - d П.,0
М i d 63 M i cl its
где Л/j — молекулярный вес нитруемого продукта;
Л/2 — молекулярный вес нитропродукта;
Количество пптропродукта составляет (в кг):
zr ЛД,
с'11л7Г’1
Расход азотной кислоты (в кг):
d ()3
611 лл
Количество выделяющейся воды (в кг):
г <*18
611 ла
Количество нспрореагировавшен азотной кислоты (в кг):
е 1________________________Г d&>
6 100 6,1 ДА
Количество серной кислоты останется равным первоначальному.
Общее количество воды в нитромассе после нитрования (в кг):
„ 100 — I — т , d 18
°—Йо-------+Й’г6“-7Г
Таким образом, для материального баланса процесса нитрова-
ния имеем (в кг):
Загружено
Нитруемое сырье ................6'ц
Влага с сырьем . .................gt
Нитрующая смесь ................G
Итого . . . б’(_|0[ц
237
II случено
Иитропродукт .................
Азотная кислота ..............
Горная кислота ...............
Вода .........................
Примеси (по разности).........
6 "37? Ч
(' . I_Q 71 вЗ
кю Л/д
»г
6 "ПнГ
100-Z-»i , ,HS
о г^-гбн-^
Итого . . . Gooih
Определение констант скоростей одновременного образования
о-, ле- н п-изомеров в процессах нитрования.
Реакции образования о-, м- и //-изомеров в процессах нитро-
вания — второго порядка, причем скорости реакций пропорцио-
нальны как концентрации нитробензола, так и концентрации
азотной кислоты.
Найдем константы скорости всех трех одновременно протека-
ющих реакций:
NO., NO3
П+JWO, \ ЧГХ0'+Н,0
V
NO., NO.,
I I
П+1И0э^-П + П,0
NO., NO.,
I I
fY-lINOs^^HI+lLO
V
I
NO.,
Пусть:
с — число эквивалентов нитробензола, оставшегося ко вре-
мени т;
с — число эквивалентов азотной кислоты, оставшейся ко
времени т;
.т, У, ’ — числа эквивалентов о-, м- и «-продуктов ко времени т.
Рассматриваемые три одновременно идущих процесса харак-
теризуются следующей системой кинетических уравнений:
dx , , dy , , dz . , ,,
—— — h\cc ; ———/.’..се; —— — к.,сс (IX. 1 и)
с/т 1 с/т “ Jt
.238
Далее, один эквивалент азотной кислоты расходуется па каж-
дый эквивалент любого из трех образующихся продуктов; то же
имеет место и в отношении нитробензола. Отсюда имеем:
е=с0 — (.т + у-гг) И с'== с' — (я + у-|-z)
где с0 и с\) — начальные количества исходных реагентов (при
т = 0).
Разделив последовательно одно дифференциальное уравнение
на другое и проинтегрировав, получим:
х : у : г~ Ад : А-.,: Ад
Учитывая, что молекулярные веса о-, м-, и п-соедипепии одина-
ковы, можно заменить отношение чисел эквивалентов отношением
массовых количеств.
Опытным путем найдено, что если пользоваться тремя экви-
валентами азотной кислоты на один эквивалент нитробензола
в начале процесса, то но истечении 20 мин, расходуется половина
нитробензола и что к тому же времени получающиеся о-, м- и
н-соединеппя дпнитробснзола присутствуют в массовых соотноше-
ниях, соответственно равных 6,4; 93,5 и 0,1. Поэтому
х : у : z = Ад : kz : А3 = Ь,4 : 03,5 : 0,1
Складывая три дифференциальных уравнения (IX- 16) и прирав-
нивая х - у -J- z = и, получим:
= (А*1 + к2 + *3) (С0 ~ М) (% “11)
Решение задачи сводится к решению уравнения
которое дает
^ = А'(со-“)(сг“)
или
откуда
Ь основу расчета А положим 1 кг-эквива icht нитробензола
(сй) и 3 кг-эквивалепта азотной кислоты (с0). Тогда получим:
А=., ..... fig (1 — 0,5) —1g (3 — 0,5)—1g-^-1 = 21,10-5 с<?№*
U —J) zU UU L J J
239
Следовательно.
•JI . |П-3
1,33.10-ь
к.= 21° (13,5= 19.04 - «Г3
91 . 1()“&
/»з=- кю— 0.1 0,021-10->
Ten^ioeoii баланс процесса нитрования
Применим следующие обозначения (в ккал, ч пли
- — rs-e*M.MZ, * **vnx
ккал- операция):
Qi тепло, вносимое с сырьем;
Qi — тепло, вносимое с нитрующей смесью;
Qa тенлот,,j эффект процесса нитрования;
Qi тепло, уносимое нитромассой;
(?г> количе(.тво тепла, отнимаемого охлаждающим агентом;
Qa — количество тепла, расходуемого па нагревание отдельных
частей аппарата и теряемого в окружающую среду.
Для балансового уравнения можем написать
<2i t-e>4-(^QH-<?6 + <2«
Характерных! является тепловой эффект процесса нитрования
(в ккал);
(?з = Q [j “J" (?а — (?выд
где Qp — теплц реакции нитрования;
Qa — тепло разбавления нитрующей смеси реакционной во-
дой;
(?выд тепло выделения азотпой кислоты из нитрующей смеси.
Гепло реакция нитрования может быть определено по закону
lecca. В соотвс%с.ТВцН с уравнением
RHd + <? HN03 —> К (NO..) d -J- d 1ЬО
имеем (в ккал/м1)ЛЬу
7р = <7и-г<17в - <?о - J'/IIXO3 = <?н - <70 Ь
ч- d (68/1—41.6) = <?„ — Vo 4- 26.8 d
(IX. 17)
где gQ — теплота образования нитруемого сырья;
7пхо3 ““ теп;*ота образования 100%-ной азотной кислоты
01,6 ккал!моль);
(?„ — геп.юта образования питропродукта;
gn — теп.’юта образования воды (158,38 ккал!молъ);
d — чис.[О нптрогрупп.
240
Тепло нитрования всего количества сырья определится форму-
лой
В данном случае при определении теплоты реакции нитрова-
ния взята теплота образования 100%-ной азотной кислоты, что не
вполне соответствует действительности, так как азотная кислота
нитрующей смеси частично гидратирована.
Для того чтобы уравнение (IX 17) оставалось юйствптель-
ным для любого случая нитрования, т. е. для любой степени
гидратации азотной кислоты, следует представить ход процесса
нитрования следующим образом.
Вначале из нитрующей смеси выделяется 11)0% азотная кислота
и при этом затрачивается количество тепла Выделяющаяся
реакционная вода разбавляет оставшуюся смесь (серную кислоту)
до концентрации р(в масс, долях) с выделением количества тепла Qa.
Для определения ^Оыд воспользуемся изложенным выше
методом и применим формулу (IX 15), согласно которой при
смешении Gv кг азотной кислоты с (G — (7,) кг водной серной
кислоты, содержащейся в нитрующей смеси С кг, выделяется тепло
в количестве:
<?нмд= V + (<? - G.x) 4S - СV
где 7y — теплота исчерпывающего разбавления азотной кислоты,
ккял; кг*,
7 — теплота исчерпывающего разбавления водной серной
кислоты, ккал кг\
(j — теплота исчерпывающего разбавления применяемой ни-
трующей смеси, ккал. кг.
Теп юта разбавления сорной кислоты Qa в соответствии
с формулой (13 13) может быть найдена так:
Qa (G~G^<ls"G^
где 6’0 — количество отработанной кислоты, кг;
//,, — теплота исчерпывающего разбавления отработанной
кислоты, ккал кг.
I аким образом
Qa ~ <%ыд = G Ч ~ G .V '/.V ~ G и %
и, следовательно, в общем виде тепловой эффект процесса нитро-
вания представится равенством:
10006н , „
Q3 -= l/t <Zp - б- 6 V/ v -
Пример. Вычислить тепловой эффект реакции нитрования
100D бензола (Д/= 78) нитрующей смесью, взятой в количестве 5611 кг.
16 л м. Батунср.
211
если и.чврзтпо, чю концентрация серной кислоты
о - 0,78, а нитрующая смесь содержит
в отработанной кислоте
Азотной кислоты
Серпом кислоты
Воды...........
24,6% (/=24,6)
59,4% (/и = 59,4)
16,0% (а =16,0)
Количество отработанной кислоты — 5020 кг (отработанная кислота
нс* содержит H.NO3). Теплота образования бензола <?0 = —4,1, нитробензола
дп = 28,8 ккал/моль.
Теплота реакции нитрования:
- <7п — <7« -г 26,8 28,8 — (—4,1) + 26,8 -= 59,7 ккпл/.ноль
Теплота исчерпывающего разбавления нитрующей смеси по формуле
Л. 11. Плановского:
1 — 1>.т 1 — Q,31 • 0,705
где
а= 111
312» _ 312-16
п — 98,5 164-98,5
п о9,4 п
х = -----Г = •'п~/"~й77 ~
ш “г / а9,4 — 24,6
Теплота исчерпывающего разбавления азотной кислоты (из таблиц)
<7д7=ГИ ккал.кг
Теплота исчерпывающего разбавления отработанной кислоты, вычислен-
ная по формуле А. II. Плановского для х = 1, р = О,78 и /г =22, составляет
г/„ = 89 к кал/кг.
Тепловой эффект реакции нитрования:
п 1006% 1000 • 1060 rn „ ,
Q3 = - <7P + бч - GA.r/jV - =----------59, < +
4-5614-91—5611-0,246- 111—5020-89 2“ 746 000 ккал
О нитраторах
Нитраторы представляют собой баки с мешалками; размеши-
вающими приспособлениями служат мощные турбинные и пропел-
лерные мешалки. Температура проведения процессов нитрования
О—1.00" С. причем процесс протекает с выделением больших
количеств тепла. Вследствие этого при нитровании приходится
иметь дело с охлажденном. Следует отметить, что перегревы
реакционной .массы связаны с опасными последствиями (вплоть до
взрыва) и поэтому интенсивное охлаждение должно рассматри-
ваться как важнейшее условие для нормального протекания
процесса.
Мощные нитраторы имеют охлаждающую поверхность, выпол-
ненную в виде змеевиков и трубчаток. Нитраторы чаще всего вы-
полняются из чугуна, реже для их изготовления применяют ооыч-
нуго сталь и специальные стали.
При попадании воды в реакционный объем пптратора может
произойти взрыв, поэтому элементы охлаждающей поверхности
необходимо оформить так, чтобы была исключена возможность
попадания охлаждающих агентов в нитромассу. Последнее обстоя-
тельство заставляет отказаться от подачи охлаждающей воды иод
давлением. Если охлаждающая вода будет не нагнетаться в охла-
ждающие элементы аппарата, а
просасываться через них, то
в случае разъедания перегородок
Рис. IX. 2. Нитратор;
j - корпус аппарата; 2 —рубашка; 3 —
крышка; 4 — защитный чулок из нержа-
веющей стали; 5 — вал с мешалками; о’ -
стакан; 7 —змеевик.
Рис. IX. 1. Нитратор:
7 — корпус с ноли исты .ч it Стенкамп; 2
руиашка: з — двойная теплообменная
трубка; 4 — пропеллерная мешалка.
она нс попадет в реакционный объем и не вызовет тяжелых аварий.
На рис. IX. 1 показан нитратор периодического действия
со стальной рубашкой для охлаждения и внутренним устройством
для перемешивания среды. Для увеличения поверхности тепло-
обмена стенки аппарата сделаны волнистыми. С целью дополни-
тельного охлаждения в аппарате установлены двойные теплообмен-
ные трубки. Реакционная масса но окончании процесса переда-
вливается сжатым воздухом, поэтому аппарат рассчитывают па
давление 3—4 ат.
Другой чугунный аппарат со стальной рубашкой (рис. IX. 2)
оборудован внутри змеевиком для дополнительного охлаждения
и вставным цилиндром, внутри которого две пропеллерные ме-
шалки перемешивают среду по всему объему аппарата, создавая
циркуляцию вокруг змеевика.
16* _____________
Глава X
Процессы восстановления
нитросоединений
и щелочного плавления
Восстановление нитросоединений
Аминированием посредством восстановления на «ывагог
процессы восстановления питросоедипений, нитрозосоединений
и других азотсодержащих групп, находящихся в ядре аромати-
ческих соединении.
В качестве восстанавливающих агентов служат, главным
образом, чугунная стружка, цинковая пыль и сернистые ще-
лочи.
При проведении процесса восстановления чугунной стружкой
реакционная масса представляет собой многофазную систему,
имеющую две жидких фазы (иптропродукт и водный раствор
электролита) и одну твердую фазу (чугунная стружка). Поэтому
здесь необходимо интенсивное перемешивание реакционной массы.
Часто восстановление ведут либо при кипении или при темпера-
туре, близкой к кипению.
Материальный баланс процессов
восстановления чугунной стружкой
Примем следующие обозначения:
Gn — количество шпр осоед и нения. кг\
сп — концентрация нитросоединсння, %;
— количество воды, кг;
б’н — количество соляной кислоты, кг;
с[; — концентрация соляной кислоты, %;
Qtp — содержание Fe в чугунной стружке, %;
G13B — количество извести, кг\
б'сгр — количество чугунной стружки, кг.
241
I стадия. Протравление чугунной стружки соляной
кислотой по реакции:
2IICl-j'-Fe I-eCL + H.,
2-36,5 55,8 126,8 2
Количество израсходованного железа (в кг):
100
55 8
2 36,5
= 0,76
ск
100
Образуется хлористого железа (в кг):
ск 126,8 , ,,
100 '2-36,5 ь 100
Образуется водорода (в кг):
_£к_
100
0,0274 6К
Ск
100
II стадия. Восстановление нитросоедппепия:
4RNO;-;-9Fe-Hn<.O -> 4RN1L + 3Fc8O4
4MJ( 9-55,8 4-18 4Ма 3-231,5
где Л/и — молекулярный вес нптросоединения;
Л/а — молекулярный вес амина.
Израсходовано железа (в кг):
с„ 9-55,8 с Ч
6,1 100 4Мц 1 Ь " 100 Л/п
где т) — выход но стадии восстановления.
Израсходовано воды (в кг):
С11 4-18 _с„ Ч
Cjl 100 4Л/Н 1 18 11 100 М„
Образуется аминопродукта (в кг):
Образуется закись-окнсп железа (в кг):
г C[I ' 231.-ч . „ „ с» В
Gn 100 4 Мн 1=1 /,J,G 6и 100 ‘ Мн
По окончании восстановления растворенное хлористое железо
осаждают в виде гидрата действием извести по реакции:
FeCl.,-;-Ca(OII)-, -> Fe(OH);,+CaCL
I2<J,S 7i 89,8 ill
245
При наличии в реакционной массе 1,74 кг хлористого
железа израсходуется Са(0Н)3 (в кг):
1,74 GK
ск
100 ’
74
126,8
- 1.01 G
100
железа (в кг):
89.8 _____ с
Образуется гидрата закиси
1,/4 6,{ W0' 126,8 — 1,23 Ь,; loo
Образуется хлористого кальция (в кг):
а 7/ fi Cl, 1 11 < го f' СК
’ 1 'к 100” 126,8 ЮО
Считая, что для нагревания реакционной массы будет израс-
ходовано Gn кг пара (острого), представим материальный баланс
(в кг) восстановления следующим образом:
Загружено
Нитрон роду кт ............ б'н
Вода...........................6’в
Соляная кислота............ GK
Чугунная стружка...............GCTI)
Известь .......................(} изВ
Конденсат ................. 6'п
Итого .... (?общ
И о./учено
Аминопродукт .... G,,<n КИШц 4
Закпсь-окпсь железа 173.6 (7„ ———п 100Л/„ 1
Гидрат закиси железа 1 94 С Ск "° 6,5 100
X юристьп! кальций 1,52 G к 100
Водород . Непрореагировавшее железо и примеси с 0,0274 6’и -g- НМ
............
1!и;,а ..............с«+с"+с«1Ли<Г-'86"тач
Примеси (по разности) д
Итого.......С’общ
:246
Тепловой баланс процессов
восстановления чугунной стружкой
Учитываем две стадии: протравление и восстановление.
Стадия протравления. Имеем (в ккал'ч или ккал опе-
рации):
где <21 — тепло, поступающее с греющим паром;
(Л — тепло, поступающее с обрабатываемыми материалами;
<2.ч — тепловой эффект реакции;
(2t — тепло в продуктах после протравления;
(25 — теп.ю, расходуемое на нагрев;
(Л — тепло, теряемое в окружающую среду.
Для определения теплового эффекта реакции напишем:
где GiZ — количество соляной кислоты, кг;
ск — концентрация соляной кислоты, %;
qp — теплота реакции протравления, ккал'молъ.
Величина tjr) может быть найдена но закону Гесса:
= ?FeCJ s + ?Н a— Fe 2?I<G[
где 7ГеС1 — теплота образования хлористого железа (100,1
ккал! моль);
б/1Г — теплота образования водорода;
^Fe — теплота образования железа (0 ккал/молъ);
<7ПС[ — теплота образования хлористого водорода (39,4
ккал/молъ)
Подставляя числовые значения теплот образования в последнее
уравнение, получим
^=100,1—2-39,4=21,3 ккал/2 моль ИС1
или
10,65 ккал/моль НС1
Таким образом:
<?з= б'1; 10,65 = 2,85 GKci;
3>6а
Стадия восстановления. Для теплового эффекта (в ккал)
реакции восстановления наппшем:
п 1000<7р
или
г. слЮ#р
—ОКТ 1
где qp — теплота реакции, ккал/молъ нптропродукта.
247
Теплота реакции г/р в соответствии с уравнением реакции
восстановления может быть найдена так:
«р = «пр - «п + °’75 «то. --’25 «Ге - «в
где 9пр — теплота образования амипопродукта;
<7„ — теплота образования питропродукта;
^тс о. — теплота образования закись-окисп железа (27.08
ккил:моль);
<]Vi, — теплота образовапия железа (0 ккал!моль);
г/в — теплота образования воды (68,38 ккал'моль).
Получим (в ккал /моль):
«р=?пр—«п —48
Технологическая схема
восстановления нитросоединений'
Схема непрерывного восстановления питрососднпснип (нитро-
бензола) чугунной стружкой в воде дана на рис. X. 1.
Рис. X. 1. Схема непрерывного восстановления нитробензола чугунной
стружкой:
1 — ,з напорные баки; 1 >) регулирующие ротаметры; 7 гместг.ть; .у насос;
,v — реактор для 1к?сстан11и.|ен1гя ннтрибепаола; it) — питатель Для чугунной струил;::;
11 — регулятор числа обиротов двигателя; 12, 11 двигатели; 7-7 — вычислительное
устройство; 7.5 — датчик прибора для определения магнитной проницаемости р«-акцион-
itnii массы; hi — per j.’iHTnp расхода анилиновой коды: 77 —регулятор расхода пара; is —
прибор для определения нптрпбедеола в парах анилина; 79 —кон ичшат -р; 2Н, 21 —
iicHo.tHiiTe.itiiibie механизмы, переключающие враны.
248
Автоматические приборы регулируют процесс по двум факто-
рам: магнитные свойства реакционной массы и содержание иитро-
сосдннепия в парах, отгоняющихся из редуктора (реактора для
восстановления). С ценно ускорения реакции восстановления
в редуктор вводят самый активный электролит — хлористый
аммоний, чугунную стружку и анилиновую воду.
Магнитная проницаемость реакционной массы зависит от
содержания в ней недоокисленноп стружки и концентрации
шлама, которые регулируются ротаметром, изменяющим количе-
ство анилиновой воды, загружаемой со стружкой.
Фотоколориметрический прибор сигнализирует о повышении
содержания невосстановленного нитросоединения в жидкости и
автоматически переключает направление потока готового про-
дукта из одного насоса в другой.
Щелочное плавление
Щелочным плавленном называют процесс взаимодейст-
вия металлических солей ароматических сульфокислот со щелоча-
ми к числу наиболее распространенных промежуточных продук-
тов, получаемых при щелочном плавлении, относятся фенол и
Р-нафтол.
Исходным сырьем являются металлические соли органических
сульфокислот, применяемых в виде растворов, наст и сухих
продуктов. В качестве минерального сырья в процессах щелоч-
ного плавления используют NaOII (расплавтонный или раство-
ренный), а также KO1I.
Одним из наиболее существенных факторов, влияющих на
ход процесса, является качество солей сульфокислот и качество
ще ючп. Щелочной агент должен быть по возможности свободным
от примесей минеральных солеи, так как последние при плавке
не растворяются в расмлавленпой щелочи и находятся в впде
комков в расплавленной массе и препятствуют перемешиванию
массы, что приводит к местным перегревам п авариям.
> спешное проведение плавки во многом зависит от консистен-
ции массы, находящейся в аппарате. Наиболее подвижная кон-
систенция достигается в тех случаях, когда в качестве щелочных
агентов применяются но расплавленные щелочи, а растворы щело-
чо! . Однако при этом реакционный аппарат до гжеп работать по i,
давлением. Величина его соответствует давлению паров воды
при данной температуре. Поддержание подвижной консистенции
плава при применении для плавки щелочных растворов может
быть достигнуто также при наличии обратного холодильника,
который возвращает в аппарат испарившуюся воду и этим поз-
воляет поддерживать требуемую консистенцию раствора без
применения автоклава.
249
Материальный баланс процесса щелочного
плавления
Примем следующие обозначения:
бн.е — количество натриевой соли органической сульфокислоты,
кг;
1УВ — количество воды, вносимой с сырьем, кг;
g — количество примесей, вносимых с сырьем, кг;
G — количество едкого натра, кг;
— количество воды, вносимой с NaOlI, кг;
S' — количество примесей, вносимых с едким натром, кг;
Л/„ с — молекулярный вес натриевой соли органической сульфо-
кислоты, кг;
Л/1:Р — молекулярный вес продукта реакции, кг;
1) — выход по плавке, масс, доли (от теории).
Суммарное уравнение щелочного плавления:
R(SO3Na)n + 2п.\аОП = R (ОNa),t -J- пNa*SО3 -J- н112О
-Чн с 271-40 _М.|р n12G «18
Образуется (в кг):
фенолята (нафтолята) натрия
сульфита натрия
воды
Расходуется едкого натра (в кг):
Остается непрореагировавшего едкого натра (в кг):
При плавке испаряется много воды, количество которой должно
быть установлено опытным или расчетным путем.
Для определения количества воды (в кг), испаряющейся
с поверхности, может быть использована формула Дальтона:
R псп — К Ft (/?,[ — рь)
где /< — коэффициент пропорциональности, кг.!м2 • ч • сд.ч рт.сгп.-,
F — поверхность испарения жидкости, at2;
250
т — время испарения, ч;
ри — давление паров жидкости при температуре испарения,
jj.it рт. ст.-,
ра — парциальное давление паров жидкости в окружающем
воздухе, Л1.н рт. ст.
Для неподвижного воздуха значение К определяется по
формуле:
где а — коэффициент (для воды а = 0,026);
t — температура испарения, °C.
Если воздух или жидкость находятся в движении, то для К
имеем:
К = л (1 г -4-Л <c0,J
\ 2/3 7
где т — скорость жидкости или воздуха, м/сек.
При проведении процесса щелочного плавления в автоклавах
И исп = 0.
Материальный баланс (в кг):
Загружено
Натриевая соль сульфокислоты .............GfI.e
Вода с натриевой солью сульфокислоты . . , Н''п
Примеси с натриевой солью сульфокислоты g
Едкий натр . ....................... G
Вода с едким натром .....................И'
Примеси с едким натром....................g'
Итого . . . GoOm
Получено
Фенолят натрия. . .
Сульфит натрия. . .
Едкий натр (сотая
шнйся).............
Вода ... ...........
При меси............
Примеси, получаю-
щиеся в результате
реакции (но раз-
ности) .............
Л/~ нр
И н. с.
126/»
Л/н. с
>1
с 80га
'“е
18га 1т.
н.с“Т7-----Л—Н псп
н. с
gx
Итого .... Собщ
251
Тепловой баланс процесса щелочного
плавления
Работа плавильных котлов может быть разделена на
две самостоятельные стадии: 1) плавление твердого NaOlI пли
упаривание его раствора; 2) собственно плавка.
I стадия. Имеем (в ккал/ч или ккал/операция):
Q|-| Сз-+
где <21 — тепло, получаемое аппаратом от теплоносителя;
(Л, — тепло, поступающее с обрабатываемыми продуктами;
< 2з — тепло, расходуемое на плавление твердого ХаОН илп
упаривание его раствора;
< 2, — тепло, содержащееся в продуктах реакции;
Q-o — тепло, расходуемое на нагрев аппарата;
< 2С — тепло, теряемое в окружающую среду.
Рассмотрим методы вычисления Q3. Если применяется твер-
дый NaOlI, то
(?3=6’Г/1|Л
где G — количество расплавляемого NaOU., кг;
£пл — теплота плавления твердого ХаОН (40 ккал кг).
В случае применения для плавки в открытых котлах раствора
ХаОН, который подвергается предварительному упариванию,
определение величины Q3 сводится к вычислению теплоты испаре-
ния воды из щелочного раствора. В практических расчетах удобно
пользоваться для этих целей специальными диаграммами.
Если требуется обезводить раствор ХаОН. с концентрацией
q до с.2, то, определяя но диаграмме величины теплот обезвожива-
ния и находят расход тепла (в ккал) па испарение ио фор-
муле:
<2з = щ)
где G' — количество обезвоживаемого каустика, кг 100%-ного
NaOH.
II стадия. Здесь остается в силе прпве leiinoe выше
равенство, в котором величина ()3 (тепловой аффект реакции)
имеет положительное значение:
~ (>з=(?4 -Ь&4-(?в
В данном случае Q3 — тепловой эффект плавки, который
складывается из теп юты реакции, теплоты изменения концентра-
ции щелочного агента и теплоты испарения воды (при плавке
в открытых котлах), т. е.
Qa —Сиси
252
где (?р — тепло реакции;
(?и;пг — тепло изменения концентрации щелочного агента;
Сиси — тепло испарения воды.
Мольная теплота реакции (в ккаллолъ) может быть вычис-
лена из закона Гесса по стехиометрическому уравнению:
Яр = 7ф - V с + «'/Xa£so, Т «?„ - ffxaoii
где — теплота образования фенолята натрия;
г/1ЬС — теплота образования натриевой соли сульфокислоты;
^xa-so — теплота образования сульфита натрия (262 ккал .ноль);
qfs — теплота образования воды (68,38 ккал'моль)]
<7Ха0П — теплота образования расплавленного NaOH (102,7
ккал 'моль);
п — число сульфогрупп.
Подставляя числовые значения теплот образования, получим:
<?р=--'7ф-'7м.с ! 125.G»
Теплоты образования фенолятов и пафталятов вычисляют,
пользуясь теплотами централизации.
Теплота изменения концентрации щелочного агента может
быть вычислена при помощи диаграммы, на оси ординат которой
нанесены теплоты растворения в воде твердого NaOH (в ккал'кг
NaOH), а па осп абсцисс — содержание щелочи (в %).
При плавке NaOH целиком выделяется из водного раствора.
Ото выделение требует затраты тепла (в ккал)
где G" — количество загружаемого 100%-го ХаОН, кг;
q" — теплота разбавления 100 %-го NaOH до концентрации
исходного раствора, ккал’кг NaOH (из диаграммы).
По окончании плавки непрорсагнровавший NaOH, количество
которого составляет G'", будет растворяться в воде с образованием
раствора концентрацией с. Процесс растворения ХаОН сопро-
вождается выделением тепла (в ккал)'.
где q" — теплота образования NaOH в растворе, концентрация
которого равна с, ккал’кг NaOH (из диаграммы).
Следовательно, в итоге при изменении концентрации щелоч-
ного агента тепловой аффект составит:
Qw* G',!q!‘~G"<f
Тепло испарения воды определяется по известной формуле:
(?псн=М 7исп
253
Рие. X. 2. Котел для плавления резорцина:
1 — крышка; 2 — корпус; з — перемешивающие устрой-
ства.
Таким образом, тепловой эффект процесса щелочной плавки,
отнесенный ко всему количеству загружаемого материала, может
быть выражен так:
st _f 1000 7р /т/у„т п
V3 — Он. с —77--*1 । <7 — од — И дпсп
Аг и. с
Тепловой баланс плавок, проводимых под давлением, отли-
чается от тепловых балансов плавильных котлов лишь отсут-
ствием расхода тепла на испарение воды.
Аппарат для щелочного плавления
В процессах щелочного плавления образуются вязкие и
застывающие массы, для обработки которых применяют аппараты
с мощными перемешивающими устройствами (рис. X. 2). Котел
сделан из жаростойкого чугуна. Внутри котла имеется вал с
шестью лопастями. Между лопастями помещают треугольные пере-
городки, служащие для перетирания вязкой реакционной массы
при вращении мешалки. Мешалка делает 50 об/мин.
Для обогрева аппарата применяют редукционные катушки,
одетые снаружи корпуса. Аппарат снабжен мощным приводом.
Глава XI
Процессы диазотирования,
азосочетания
и нитрозиро&ания
Рассматриваемые процессы протекают с выделением
больших количеств тепла и по допускают повышения температуры
сверх требуемых пределов от 0 до 10 °C. Вследствие этого необхо-
димо энергичное охлаждение реакционной массы, которое может
быть достигнуто с помощью льда или охлаждающих растворов.
Диазотирование. Диазотированием называют про-
цесс взаимодействия солей ароматических аминов с азотистой!
кислотой, который! приводит к образованию малостойких про-
дуктов — дпазосоедииепий.
Исходными продуктами во всех процессах диазотирования
служат первичные ароматические амины. Диазотирующим агентом
во всех практически важных случаях является нитрит натрия.
Процесс диазотирования можно представить в таком виде:
RN’I.,- НХ-! ХаХОл ИХ -> RX X [-NaX '-2ILO
I
Лзосочетапис. Лзосочетапием называют процессы взаи-
модействия дпазосоединепий с аминами пли оксисосдппепиями
ароматического ряда, приводящие к образованию азосоединений:
RXoX-J-HlHY -> RN^XR^’-HX
Азосочетание проводится в кислой или щелочной среде, при-
чем в щелочной среде сочетанию подвергаются оксисоодннепня,
а в кислой — амины.
Интрозпроваипс. Нптрозироваппем называют процесс
замещения водородного атома в ядре ароматического углеводо-
рода группировкой атомов ХО (пнтрозогруплой), который про-
текает при непосредственной обработке сырья нитрозирующим
агентом.
Сырьем, подвергаемым пптрозироваппю, служат преимущест-
венно ароматические амины и гпдрокси.тнроизводные. Питрози-
р гющим агентом во всех практически важных случаях служат
металлические соли азотистом кислоты (главным образом, нитрит
натрия), которые в кислой среде легко выделяют азотистую кис-
лоту, реагирующую с органическими соединениями в момент
выделения:
RH-J-IINOS=RNO !-П„О
Тепловой баланс процессов диазотирова-
ния, азосочетания и нитрозирования
Общее уравнение теплового баланса (в ккал'ч или
ккал/опсрация):
С1-НО2-1'Сз=<Л-ЬС5
где(?1 — тепло, поступающее в аппарат с материалами;
Q., — тепло, поступающее в аппарат от окружающей среды
(потерн холода);
(Л, — тепловой эффект процесса;
— тепло, содержащееся в продуктах реакции;
ф- — тепло, отнимаемое от реакционной массы охлаждающим
агентом.
Характерными для указанных процессов являются методы
определения величин Q2, Q3 и «25-
Потери холода в окружающую среду Q., определяются количе-
ством водяных паров, которые, находясь в воздухе, при сопри-
косновении с поверхностью холодной жидкости диффундируют
в эту жидкость, конденсируются в ней и, таким образом, вносят
некоторое количество тепла. Потерн холода наружной поверх-
ностью аппарата практически приближаются ’ к пулю и потому
в технических расчетах могут пс учитываться.
Количество водяных паров (в кг), диффундирующих из воз-
духа в охлаждающую жидкость, определяется уравнением:
G=KFx (рв—р1К)
где К — коэффициент пропорциональности, кг '.и- ♦ ч • льчрт. ст.-,
F — поверхность (зеркало) охлажденной жидкости,
т — время, ч;
pR — парциальное давление паров воды в воздухе, леи рт. ст.;
Р-.г. — давление паров воды над охлажденной жидкостью,
рт. ст.
Коэффициент пропорциональности А' может быть найден при
помощи следующей формулы:
К «= 0.026 (1
где t-n — температура охлажденной жидкости, °C.
17 Л. М. Батуне]). 25.
Зная количество конденсирующихся водяных паров, можно
подсчитать потери холода (в ккал):
(г 1 • tiK)
где i — энтальпия водяных паров прп температуре воздуха,
ккал!кг.
Величина @5 может быть вычислена по разности, когда все
остальные составляющие теплового баланса определены, т. е.
+ +
Зная находят расход охлаждающего агента, например,
льда:
jy = —2»—
л 804-1
где /ч — конечная (минимальная) температура реакционной
массы.
Величины Q3 являются характерными для каждого процесса
и поэтому должны быть рассмотрены отдельно.
Тепловой эффект процессов диазотирования
Основой для тепловых расчетов в данном случае явля-
ются опытные данные тсплот диазотирования, которые для боль-
шинства ароматических аминов найдены экспериментально и
опубликованы В. В. Свентославским (1917 г.). Он эксперимен-
тально нашел теплоты отдельных реакций, суммируя которые
можно представить любой процесс диазотирования. К этим реак-
циям относятся:
разложение соли
RNHa-HCl -> RNH24-HC1—<2п.а
нейтрализация диазогпдратов
RN N 4- IIC1 -> RN = N Н.,0 4- qa. д
ОН С1
диазотирование
RNHs4-HN0.2 -> RN = N-J-lbO-rb
он
разложение нитрита натрия соляной кислотой
NaNO., |-НС1 —> ГЧ аС1-| HNO.»4-3»45 кка,л!.чолъ
Сложив левые и правые части этих уравнений, получим после
сокращения:
RN4I.,-ИС14-.\аХ0.4 -IIC1 -> RN = N'+NaC14-2ir..O4-
I
Cl
-|- Яд-Нк. д—?п. а4-3/15 ккал!.коль
£58
Таким образом, теплота реакции диазотирования определяется
так (в ккал/молъ)
<?1> '7;сЬ9н.д—7н.а 3,45
или (в ккал'ч, ккпл/операцпя)
где Gt — количество загружаемого ароматического амина,
кг/операция (или кг/ч);
Ci — содержание чистого вещества в исходном диазотируемом
сырье, масс, доли;
— молекулярный вес исходного амина;
ц — выход по реакции диазотирования, масс. доли.
Если при диазотировании берется избыток нитрита натрия
(6, % от теории), то в результате разложения его выделится коли-
чество тепла (в ккал ч или ккйл/операция):
« .. 69 6 1000-3,45 3.45 6
Vp-GiS -v/j * WO * 69 ~ lCl Ml
Таким образом, общее количество тепла, выделяющегося при
диазотировании, может быть представлено так:
<?„- <?;+?;=(юооч?,>+з.««)
В табл. Xf. l помещены тепловые эффекты отдельных реакций
для наиболее широко распространенных первичных ароматиче-
ских аминов:
Таблица XI. 1
Тепловые эффекты (в ккал/моль) отдельных реакций в процессах
ди азот ирона иим
Походные амины #п.а #н.д #д
Аминоазобензол 1,77 10,28 12.56
Анилин 7.4 11,68 18.52
о-Аиизидин 7,5 13,01 18.38
о-Мптроаиилии 1,81 8,0 12.47
р-Нитроанилин 1,81 8,0 12,53
Сульфаниловая кислота 3,15 8.5 15.01
о-Толуидин 7.49 12,0(> . 19.97
р-Толуидин 7,6 13 0 18.29
При отсутствии экспериментальных значений теплот диазоти-
рования в расчетах приходится принимать таковые по аналогии
с известными теплотами диазотирования.
17*
259
Тепловой эффект процессов азосочетания
Так же как и ио теплотам процессов диазотирования
В. В. Свентославский приводит опытные данные по теплотам
отдельных реакций, сумма которых дает общее выражение для
процесса азосочетания и, следовательно, его тепловой эффект.
К этим отдельным реакциям относятся:
азосочетапие
RN=N—ОН-J-IIKI -> RN=NRI4-H.2O-Hm
изомеризация дпазосоединений
RN = N -> RN = NOJI +?„
ОН
нейтрализация диазогидратов
RX = N + 1IC1 -> H.ZH-RX = Х + <7]г д
I I
ОН С1
Реакцию нейтрализации диазогидратов напишем в таком виде:
RN N-I ПоО -> НN = N + ИС1 - $п. л
I ‘ 1
Cl Oil
Складывая левые и правые части приведенных равенств, получим
суммарное уравнение, которое выражает процесс азосочетания
RN = i\J-HRI -> RN=XIII '-НС!
I
Cl
тепловой эффект которого составляет:
•<7a_l_7ii—йл. д
Тепловые эффекты отдельных реакций в процессах азосочета-
пия с Р-пафтолом приведены в табл. XI. 2.
Таблица. XI. 2
Тепловые. эффекты (в ккал/моль) отдельных
реакций в процессах азосочетания с ф-иафтолоМ
Диазотированные амины Ч-л + 'Ок Д
о-Аип.чпдин ...... 33,06 13,01
А ПИЛ!! К 30,93 11.68
Антрани юная кислота 38,3 1 10 00
п-Иафти.ымпн 30,89 12.86
о-Толуиднн 30,43 12,06
р Толуидин 30,65 13.0
260
Следует отмстить, что теплоты азосочетания различных диазо-
соединений с одной п той же азосоставляющей мало изменяются
при замене одного диазосоединения другим и, следовательно,
в технических расчетах (при отсутствии данных) можно брать
теплоту сочетания диазосоедпнений с оксипроизводными как
величину постоянную в пределах цифр, указываемых н табл. XI. 2.
Теп левой эсрфект процессов нитрозирования
При определении теплового эффекта нитрозирования
следует пользоваться опытными данными, которые для распрост-
раненных иигрозируемых продуктов опубликованы 13. В. Свеито-
славским.
Отметим, что теплоты нитрозирования близки к теплотам азо-
сочетания.
Имеются экспериментальные данные относительно теплот сго-
рания наиболее распространенных нитрозопродуктов (табл. XI. 3).
Пользуясь этими данными, легко вычислить теплоту образования
питрозосоединений и, следовательно, определить теплоту реакции
нитрозирования обычным путем.
Таблица XI, 3
Теплоты сгорания (е ккал/моль) нитрозопродуктов
Теплоты
Продукты сгорания
Ди нитрозо резорцин (т нерпы н) 528,5
} (ифе пил кето н си м 1(526,8
р-Питрозодифени замни 1526,9
а-1111 гроз о - а -и афто.т 1163,5
а-Нитрозо-р-пафто.т . 1166,7
Р-Н итрозо-а-нафтол 1167,4
р-Нптрозофспол 713,5
Аппараты для процессов
диазотирования, азосочетания
и нитрозирования
Па рис. XI. 1 показан стальной чан для диазотирова-
ния, футерованный диабазовой плиткой. Крышка чана и стальная
мешалка гуммир> готся.
В чане для охлаждения реакционной массы рассолом может
быть размещен змеевик из свинцовых труб или же из стальных
труб, покрытых эмалью, а также титановые змеевики.
Слив реакционной массы осуществляется посредством днище-
вого затвора (рис XI. 2), который состоит из гуммированного
корпуса и конической гуммированной пробки. Па рис. XI. 3
представлен чугунпый эмалированный аппарат с рубашкой для
охлаждения рассолом.
Рис. XI. 1. Чаи для диазотирования:
1 — корпус; 2—футеровка; з — крышка; 4 —гуммированная мешалка;
3 — дннщепой спуск.
Аппарат для непрерывного диазотирования (рис. X 1.4).
Исходные вещества (например, анилин, соляная кислота и раствор
нитрита натрия) непрерывно подаются в два кольцевых капала
262
1 и 2, расположенные один над другим; к ним присоединены
патрубки 6 и 7. Из этих каналов потоки распределяются по ради-
Рис. XI. 2. Днищевой гуммированный спуск (затвор).
альным канавкам (лоткам) 3. Аппарат снабжен рубашкой 4 с па-
трубками 5 для охлаждения реакционной массы водой или рас-
солом.
Процесс диазотирования
протекает по мере движения ре-
акционной массы по канавке
(лотку) в тонком слое.
В даппом случае имеем про-
цесс теплопередачи в сочета-
нии с химической реакцией
или же химическую реакцию,
протекающую в тонком слое
Рис. XI. 3. Чугунный эмалирован-
ный аппарат для диазотирования.
1 — чугунный эмалированный корпус;
2 — рубашка; 3 — мешалка; 4 — гильза
для термометра; 5 — штуцер для слива
реакционной массы.
263
при одновременной теплопередаче. Количество тепла, отводимого
от реакционной массы, примем пропорциональным градиенту
।
1‘пс. XI. 4. Аппарат для непрерывного диазотирован; я.
J, - кнльцекыс капилы; .? — радиальные канавки (лчтки); ; — ру-
башка; 5 —7 — патрубки.
концентрации вновь образующегося продукта и тепловому эф-
фекту реакции (тепловыделению).
Рассмотрим в реакционной массе две единичных’ площадки,
параллельные дну лотка и расположенные на расстояниях ft- и
h -j- dh от уровня реакционного слоя. Количество тепла, выде-
ляющегося через эти площадки за время di будет:
Л de , _, Г de . / de \' ,
<? Q Л
где 7 — тепловой эффект реакции, ккал • .и- кг ч;
с — концентрация вновь образующегося вещества, кг л3.
Если рассмотреть элементарный столбик реакционной массы,
основания которого совпаду ют с упомянутыми выше единичными
площадками, то количество тепла, выделяющегося из этого объема
за время d т, будет равно
То же количество тепла, очевидно, пропорционально количе-
ству образующегося в этом объеме вещества, т. е. пропорцио-
нально dedh.
Из дифференциального уравнения скорости химической реак-
ции
(где к — константа скорости реакции, л3,кз • ч;
Я п />5 — концентрации исходных продуктов, кг л’’) следует:
de dli = к (Л — с) (В — с) dh dr
Отсюда находим:
«о'/ -jp- ~ к <Л — г) ""
где — тсплодиффузионный коэффициент пропорциональности,
кг • муккая. • .н3.
Последнее уравнение можно представить в таком виде:
В результате получим дифференциальное уравнение второго
порядка, устанавливающее функциональную зависимость между
h и с.
Кроме того, данный процесс характеризуется дифференциаль-
ным уравнением теплопроводности в движущейся среде:
d?i dt
где а А/ср — коэффициент темиератч ро про водности, .ч2. ч.
2l>5
На границе жидкой пленки и стопки аппарата имеет место
теплоотдача, поэтому
а = — Z ——
i —t0
где а — коэффициент теплоотдачи от реакционной массы к стенке,
ккал'м'1 • ч • град;
t0 — температура стенки, °C;
t — температура жидкости, °C.
При диазотировании в топком слое режим движения жидкости
в радиальных канавках (лотках) может быть ламинарным или
волновым. В том и другом случае уравнение одномерного движе-
ния имеет вид:
du: dp . d и?
"^е-ет—
В итоге получим следующее дифференциальное уравнение,
описывающее термохимический процесс диазотирования в тонком
слое
dp
d'-i dt
й -гт = w —
dx- dx
die dp . (Pw
и условие па границе
dx * dx2
я I dll )
а = — л-------
Совместное решение этих уравнений методом подобия дает кри-
териальное уравнение тепло- (или термо-) кинетики непрерывного
процесса диазотирования в тонком слое, и, вообще, физико-
химических процессов, протекающих в подвижных слоях -или
пленках жидкости.
Применяя масштабные множители, получим следующие кри-
териальные зависимости
«.’.ту
---— = Re
М-
_ 1
р Fr • Ей
причем критерий Фр уда характеризует силу тяжести, а критерий
Эйлера — перепад давления.
Далее имеем:
1гЛ т ,
----= Ре
«о
260
где к — постоянная, характеризующая скорость реакции, ч-1,
k — толщина слоя реакционной массы, .и;
а0 — теплодиффузионный коэффициент пропорциональности;
q — тепловой эффект реакции, ккал мг;кг • ч.
Критерий диазотирования Di характеризует рсакпионпопро-
водность взаимодействующей среды, отнесенную к количеству
тепла, отводимого с 1 at2 поверхности теплообмена в 1 ч, при про-
текании процесса в движущемся слое жидкости. Наконец:
При геометрически подобной границе потоков:
Nu=/0[Rc, (Fr- Eu), Ре', Di]
где /о — функция, устанавливаемая экспериментально.
Критерий Ре' можно заменить более удобным критерием
Прандтля:
Получим:
Nn = /]Rc, (Fr-Eu), Гг', Di]
Приводя критериальную зависимость к степенной функции,
находим
N и = 4 (Fr • Ен)” Гг'г Di2
где ф, т, п, ?•, z — постоянные и отвлеченные числа, устанавли-
ваемые экспериментально.
Искомой величиной является коэффициент теплоотдачи па
поверхности раздела жидкости и внутренней стопки рубашки
Этот процесс с целью определения расхода к должеп быть рас-
смотрен дополнительно с гидродинамической точки зрения. Мы
имеем здесь установившееся прямолинейно-параллельное течение
вязкой несжимаемой жидкости при наличии одной твердой пло-
ской стенки и одной свободной границы, причем вторая жидкость —
газ на свободной поверхности имеет сравнительно малую плот-
ность.
В теории динамики вязкой несжимаемой жидкости доказы-
вается, что в этом случае на свободной поверхности раздела
нормальная составляющая напряжения должна быть равна
267
постоянному давлению, а касательная составляющая должна оора-
щаться в нуль. Таким образом, вдоль свободной границы давление
не будет зависеть от х. т. е.
^ = 0 (XI. 1)
их
Следовательно, здесь перепада давления вдоль течения не
может быть и само течение может происходить при наклоне твер-
дой стенки к горизонту, т. е. под действием силы тяжести.
Так как траектории всех частиц строго прямолинейны и парал-
лельны между собой, то
и.-у = 0; w2 = 0 (XI. 2)
При этом предположении из уравнения несжимаемости найдем:
-^ = 0 (XI. 3)
дх
Таким образом, единственная проекция вектора скорости
вдоль всей траектории будет оставаться постоянной и может
изменяться только в поперечном к траекториям направлении.
При использовании (XI. 2) и (XI. 3) дифференциальные урав-
нения движения жидкости упростятся:
(XI. 4)
Обозначим угол наклона твердой стенки (дна) к горизонту
через О н выберем ось х параллельно направлению стенки. Гак
как проекция силы тяжести жидкости на ось х будет равна
gx=gsinO
то первое уравнение (XI. 4) при учете (XX. 1) и предположении, что
скорость не зависит от координаты z, представится в виде:
сРшх ё n /ч-i -х
. , —----—sin 0 (XI. э)
ci>/~ v
Вследствие условия прилипания имеем
при у = 0 (XI. (j)
н так как на свободной границе величина вязкости на сдиннцу
площади должна обращаться в нуль, то соответственное граничное
условие для скорости будет:
=0 (при ч /') (XI. 7)
Общее решение дифференциального уравнения (XI. 5) имеет
вид:
=* — siu ° У' Т" C1V + С2
На основании граничных условий (XI. 6) и (XI. 7) получим:
Q =
v
sin О
г-2 ^2
»
Таким образом, решением рассматриваемой задачи будет
— -у-; sin О (21/Л — I/2)
Максимальная скорость имеет место на свободной границе
Расход v равен
''Л2
И'маьс — “й—S|n О
h
f , 1 t
г — u\ at/ — —— • ---------sin 6.
J v
о
Глава XII
Процессы, протекающие
под давлением
Высокие давления позволяют увеличивать скорость
реакций, сдвигать химическое равновесие в желательном напра-
влении, уменьшать объем перерабатываемых материалов (в слу-
чае взаимодействия газов), проводить процессы взаимодействия
жидких веществ при температуре, значительно превышающей
их температуру кипения.
Промышленность органических полупродуктов использует ме-
тоды высоких давлений для проведения целого ряда основных
процессов, в первую очередь для проведения процессов гидролиза,
аминирования и алкилирования.
Характеристика процессов гидролиза,
аминирования и алкилирования
Гидролиз — ото процесс обмена заместителей, нахо-
дящихся в ядре или в боковой цепи ароматических углеводородов,
на гидроксильную группу. Продуктами, подвергаемыми гидролизу,
служат главным образом хлорпроизводпые и амины ароматиче-
ских соединений. Веществами, способствующими гидролизу и
применяемыми в технике, являются водные растворы щелочей,
кислот и некоторых минеральных солей. Полагая, что действу-
ющим началом в процессе гидролиза является вода, суммарное
уравнение для него можпо представить следующим образом:
RX-HI20 ROH-I ИХ
где X — заместитель, обмениваемый па ОН.
Аминированием называется процесс обмена различных
заместителей, находящихся в ядре или боковой цепи ароматиче-
ского соединения, на аминогруппу.
270
Из органических соединений, подвергаемых аминированию,
следует отметить, в первую очередь, хлорпроизводные и гидро-
ксплпронзводпые. Аминирующими агентами являются водный
раствор аммиака, сульфит аммония, хлористый аммоний, угле-
кислый аммоний и ряд других аммонийных солей.
Уравнение процесса аминирования можно представить в
таком виде:
RX4-2NH3 —> RK1I2 + MI4X
где х - ото ОН, Cl, SO3H, NO2 и т. д.
Алкилированием называется процесс замещения во-
дородных атомов аминогрупп и гидроксильных групп ароматиче-
ских углеводородов алкильными радикалами.
Алкилирующими агентами являются спирты, галондалкилы
и эфиры.
Алкилирование почти всегда проводится в присутствии кислот
или щелочей, причем кислоты применяются при алкилировании
аминов, а щелочи — при алкилировании гидроксильных произ-
водных.
Суммарные уравнения процесса алкилирования:
(1) RNH,4-XAlk = R.XHAIk + HX
(2) ROH4-XAlk=ROAlk + IIX
где Aik — алкильный радикал;
X — кислотный радикал.
Для проведения перечисленных процессов в технике приме-
няются автоклавы, аппараты змеевикового типа (трубчатые) и
специальные аппараты, рассчитанные па работу под давлением.
Автоклавы. Аппараты змеевикового
типа
Автоклавами называются различные реакционные ап-
параты, работающие под давлением >-6 ат. Они представляют
собой стальные литые котлы, чаще всего вертикальные, со сфери-
ческими днищем и крышками.
На практике применяют следующие соотношения основных
размеров автоклава:
Нд = Нц: Иц<Йк<2Рц
где 7?д — радиус кривизны днища;
Л и — радиус цилиндрической части автоклава;
/?в — радиус кривизны крышки.
Высота цилиндрической части автоклава, как правило, при-
равнивается диаметру цилиндрической части, что и позволяет
271
установить все основные размеры аппарата по его полной емкости,
так как радиус цилиндрической части (в лкч) может быть вычис-
лен по формуле:
Иц .575 р V
где V — полный объем автоклава, л3.
Рис XII. 1 Автоклав литой:
1 — корпус автоклава; 2 — крышка; 3 — вкладыш на клелото-
стойкоп стали; 4 — мешалка; 5 — гильза для терм-ок-тра; <: —
сальник; 7 — труба передавливания.
В качестве материала для изготовления автоклавов исполь-
зуется преимущественно стальное литье, причем для работы при
низких давлениях, когда условия механической прочности не
272
имеют решающего значения, могут быть
использованы автоклавы сварные, изго-
товленные из листовой стали.
В тех случаях, когда материалы,
перерабатываемые в автоклавах, ока-
зывают коррозионное действие па
стальное литье, применяются все же
стальные автоклавы, но при этом они
снабжаются вкладышами предохраня-
ющими их стенки от воздействия реак-
ционной массы.
Вкладыши для автоклавов предста-
вляют собой металлические сосуды,
форма которых соответствует форме;
того автоклава, в который они вста-
вляются. Чаще всего их изготавливают
из стали и чу гуща, покрывая вмут
рентою поверхность защитным слоем
другого металла или эмали. Обычно
приходится иметь дело с вкладышами,
внутренняя поверхность которых освин-
цована или эмалирована кислотостой-
кой пли стойкой к щелочам эмалью.
На рис. XII. 1 изображен автоклав
из качественной углеродистой стали.
С целью создания условий для тепло-
передачи пространство между вклады-
шем и корпусом аппарата заполняется
легкоплавким металлом (сплав сурьмы
и свинца). Автоклав снабжен пропел-
лерной мешалкой.
Трудности, связанные с вводом
вала в аппарат высокого давления
через сальник, привели к созданию
конструкций с герметизированным при-
водом. На рис. XII. 2 представлен
бессальниковый автоклав. Ротор элек-
тродвигателя из немагнитного мате
риала здесь укреплен непосредственно
на вал мешалки. Иго отделяют от ста-
тора защитной гильзой и приводят
в Движение вращающимся магнитным
полем статора.
Аппараты .змеевикового типа.
Непрерывное проведение процессов
гидролиза, аминирования, алкилирова-
ния и других делает целесообразным
18 Л. М Еатунер.
Рис. XII. 2. Автоклав с
приводом без сальника (ро-
тор электродвигателя рас-
положен внутри автоклава,
статор снаружи).
применение трубчатых аппаратов, которые легко выдерживают
высокие давления и, кроме того, позволяют проводить процесс
без специальных размешивающих приспособлений, поскольку
Рис. XII. 3. Схема проведения процессов под
давлением в трубчатых аппаратах;
I — напорный бачок; 2 — измеритель расхода;
3 — насос; -1 — змеевиковый подогреватель; 5 —
трубчатый реакционный аппарат; с — дроссельные
вентили.
небольшое сечение труб может обеспечить турбулентный режим
движения жидкостей, а вместе с этим и энергичное пх перемеши-
вание.
Па рис. XII. 3 представлена схема установки со змеевиковым
аппаратом, работающим под высоким давлением.
Давление жидкости в реакционном
аппарате при ее термическом
расширении (термодинамика
жидкости)
Если зависимость между независимым переменным х
и его функцией у задана аналитическим выражением, не разрешен-
ным относительно у, т. е. если задано уравнение / (я, у) = О,
то говорят, что это уравнение определяет у как неявную функцию
от х.
Дифференцирование функции / (х, у) = 0 дает:
df — ^-dx-\-^—dy О
Ох Оу
Отсюда получаем формулу для производной неявной функции!
<У
dy _ dx
dx of
dy
276
Аналогичные соотношения могут быть получены для функции
с любым числом переменных. Если мы имеем функцию / (а^, х2,
xs...,x,i, z) = 0 несли примем Яр .т2, хл за независимые пере-
менные, az — за их функцию, то частная производная от z по х^
выразится следующим образом:
й/
OZ
Пусть, в частности, имеем функцию
/(^ у, z) = 0
На основании формулы (XII. 1) получим
Перемножая эти формулы, придем к следующей зависимости:
ду \ / dz \ / дх \
) z \ ду ) х \ <9з /у
(XII. 2)
Экспериментально установлено, что физическое состояние
однородной жидкости определяется заданием двух из трех пере-
менных величин, фигурирующих в уравнении состояния: темпера-
туры t, объема v и давления р. Между этими переменными суще-
ствует зависимость:
/(р, г, 0 = 0 (XII. 3)
Уравнение (XII. 3) называется уравнением состояния жидко-
сти. Так как три переменных р, v, I связаны одной зависимостью
(XII. 3), то одну из этих переменных можно рассматривать как
функцию двух других.
Применим формулу (XII. 2) к уравнению состояния (XII. 3).
Мы получим;
/ др \ / ди \ / dt \ _ j
\ di> Jt \ dt )р \ др Д.-
или
/ ди \
/_£р\ 1 - (XII 41
\ ди Jt \ dt Jp / dt \ \ dt Jv / du \
\ Jv \ dp Jt
Воспользуемся формулой (XII. 4) для определения величины
давления, развивающегося при нагревании жидкости, которая
занимает весь объем закрытого реакционного аппарата. Пусть
жидкостью является реакционная масса, нагреваемая от 40 до
50 С. Пренебрегая расширением сосуда, этот процесс можно
считать протекающим при постоянном объеме.
После интегрирования но I равенства (XII. 4) получим:
/ ди \
Pi—Pl=— I ~/"Х / dl (XII. 5)
J \ др )t
Используя определения коэффициента объе того расширения
и коэффициента сжатия
формулу (XII. о) можно написать следующим образом:
—
(XII. 6)
Подынтегральная функция в формуле (XII. 6) зависит от
давления и температуры, и уравнение (XII. 6), строго говоря,
есть интегральное уравнение, так как искомое давление содержит-
ся под знаком интеграла. Однако в рассматриваемом интервале
изменения температуры коэффициенты сс и fl, являющиеся функ-
циями t и р. с достаточной степенью точности могут быть приняты
постоянными.
Коли, например, о: —- 0,3 • 10“3 грсиГ1 и 0 = 38 10_<3 сшГ1,
/х = 40 'С, I., — 50 С, то, решая уравнение (ХИ. 6), получим:
0,30-Ю"3
---------— 1и
33-10-*
3000
38
79 ат
Тепловые балансы процессов гидролиза,
аминирования и алкилирования
Независимо от того, как проводятся указанные про-
цессы (периодически пли непрерывно), тепловые соотношения
целесообразно рассматривать отдельно для стадии химического
взаимодействия и стадии отгонки иопрореагировавших продуктов
при спуске давления.
276
Тепловой баланс стадии химического
взаимодействия
Тепловой баланс (в ккал;ч или ккал.'операция) стадии
химического взаимодействия может быть написан так:
<?> - + С>г.Ш
где <21 — тепло, вносимое в аппарат с перерабатываемыми мате-
рии ымк;
< 2-> — тепло, поступающее в аппарат с теплоносителем;
< 2з — тепловой эффект процесса;
< 2, — тепло, содержащееся в продуктах реакции в конце
процесса;
< 2& — тепло, расходуемое па нагревание отдельных частей
аппарата;
< 26 — тепло, теряемое аппаратом в окружающую среду.
Рассмотрим способ вычисления теплового эффекта процесса (?3.
Тепловые эффекты процессов вычисляются различно в зависи-
мости от их химической природы и характера реагирующих
веществ, поэтому рассмотрим отдельно тепловые эффекты про-
цессов гидролиза, аминирования и алкилирования.
Тепловой эффект процессов гидролиза можно считать
равным сумме теплоты реакции и теплоты изменения концентра-
ции едкого натра.
Теплота реакции (в ккал!моль) определяется по закону Гесса
в соответствии с уравнением реакции
RC1 -;-2Л’аОП = КОХа 4-NaCl + 11,0
с л ед у ю I ц! I м образом:
= ^чех Л' V.xaCl " “'ГхаОН
где <7[[р — теплота образования продукта гидролиза, ккал/моль;
Ччсх — теплота образовапия исходного органического со-
единения , ккал!моль j
< 7x-ici — теплота образования растворенного хлористого нат-
рия (96,6 ккал моль);
(]а — теплота образования воды (68,38 ккал;моль};
</ХаО11 — теплота образования 100%-го едкого натра
(102,7 ккал, моль).
Подставляя в предыдущее равенство численные значения
теплот образования, получим:
— 7i!ex — 40,4
Теплота разбавления щелочи водой, выделяющейся в резуль-
тате реакции, в данном случае может быть найдена как теплота
277
изменения концентрации (избытка) щелочного агента в процессах
щелочного плавления (см. гл. X).
Тепловой эффект процессов аминирования склады-
вается из теплоты реакции и теплоты изменения концентрации
аминирующего агента, т. е. раствора аммиака.
Теплота реакции аминирования хлорпроизводных в соответ-
ствии с уравнением реакции
RC1 -h 2NII3 = К N IL -| • N11 i('l
может быть найдена по закону Гесса:
где qQ — теплота образования амппопродукта;
д0 — теплота образования аминируемого продукта;
^nh*ci теплота образования хлористого аммония (71 ккал моль)-
7ХН— теплота образования газообразного аммиака (II
ккал/моль).
Таким образом, имеем (в ккал/моль):
?р=?о“%+49
Теплота изменения концентрации аминирующего агента (избы-
точного) может быть вычислена аналогично теплоте изменения
концентрации щелочен в процессах щелочного плавления, а также
щелочного агента при гидролизе.
Следовательно, изменение концентрации аминирующего агента
сопровождается тепловым эффектом </113Ч (в ккал! кг воды), вели-
чина которого определяется разностью ио уравнению:
9113м— — 91
где — теплота растворения аммиака с образованием копцел
трацни исходного раствора;
q2 — теплота растворения непрореагировавшего аммиака.
Теплота изменения концентрации аминирующего агента,
отнесенная ко всему количеству загруженного в аппарат
аммиачного раствора (и выраженная, таким образом, в ккал),
может быть вычислена по формуле:
<?изм = Сами (1 —0,01 с) <?П:>М
где Сами — количество аммиачного раствора, «з;
с — начальная концентрация раствора, %.
Последнее равенство справедливо лишь для тех процессов
аминирования, которые протекают без выделения воды. С учетом
выделяющейся при реакции воды теплота изменения концентра-
278
цпп аммиачного раствора (в ккал/кг воды (в загрузке)] может
быть найдена так:
?иам=(1 — 4l
где G' — количество реакционной воды, приходящейся на 1 кг
воды, загружаемой в аппарат.
Величины и могут быть взяты из таблиц в справочниках.
Тепловой эффект процессов алкилирования является
теплотой только реакции, так как теплота изменения концентра-
ции алкилирующих агентов незначительна и может не учиты-
ваться во всех случаях за исключением алкилирования фенолов
в щелочной среде.
Теплота реакции (в ккал моль) алкилирования аминов метило-
вым спиртом в кислой среде в соответствии с уравнением реакции
HNH„ - НС1 + СНЭОИ= RNH • ИС1 |-Н20
сн3
может быть найдена таким образом:
Яр — ?м. а“ ?с. а ?в“ VcibOU
где </.ч.а — теплота образования моноалкилпроизводиого амина;
qc а — теплота образования солянокислой соли амина;
qB — теплота образования воды;
7СН он — теплота образования метилового спирта.
Аналогично вычисляют теплоты реакции алкилирования фе-
нолов.
Для вычисления теплот реакции по этому уравнению можно
пользоваться не теплотами образования солей аминов, а теплотами
образования самих аминов, так как теплоты нейтрализации алки-
лированных и неалкилированных аминов почти одинаковы и,
следовательно, сокращаются.
Тепловой баланс стадии отгонки
непрореагировавших веществ
при спуске давления
Тепловой баланс процессов гидролиза, аминирования
и алкилирования в стадии отгонки летучих непрореагировавших
продуктов при спуске давления может быть представлен равен-
ством:
— (Gj.— Go) с2£2"I* G-Д + Qu
где GL — количество реакционной массы перед спуском давле-
ния, кг;
279
— теплоемкость реакционной массы нерол спуском давле-
ния, ккал кг • град;
— температура реакционной массы перед спуском давле-
ния, С;
<?2 — теплоемкость продуктов, оставшихся после отгонки
легколету’шх веществ, ккал кг град;
!.> — температура оставшихся продуктов, °C;
6'.2 — количество испаряющихся продуктов, кг;
I — энтальпия получающихся паров, ккал кг;
Qn — тепло, теряемое в окружающую среду, ккал.
Из последнего равенства находят либо количество отгоняю-
щихся продуктов С2, задаваясь температурой Л, либо температуру
t2, задаваясь величиной С'.,.
Определение i в случае отгонки смеси летучих компонентов
(например, аммиак и вода) осложняется необходимостью предва-
рительного вычисления состава образующихся паров.
Состав паров, образующихся в результате испарения жидко-
сти, может быть вычислен по уравнению простой днетиллянни:
где — начальное количество смеси летучих компонентов
в реакционной массе, кг;
X — состав жидкой смеси летучих компонентов;
У — состав паров, соответствующий составу жидкости.
Решение приведенного выше уравнения в общем случае дости-
гается лишь путем графического интегрирования.
Простая дистилляция
Если при многократной одноступенчатой дистилляции жидкой
смеси испаряется каждый раз только бесконечно малая часть жидкости, то
в результате мм получим так называемый процесс простои дистилляции.
Гипюрные системы. Пар, выходящий из аппарата, находится
в .побои момент в равновесном состоянии с жидкостью, ио непрерывно изме-
няется в своем составе. Таким образом, здесь мы имеем бесконечно малые
величины.
Пусть в какой-либо момент при дистилляции в аппарате находится L
.ноль жидкости состава .¥ мол. долей (ио компоненту А) и испаряется г/Л.моль
с получением равновесного пара состава У. Остаточная жидкость L - йЛ
будет иметь состав X — dX. Материальный баланс по компоненту А:
LX = У* dL -J- (Л - dL) (X — <7Х)
Пренебрегая дифференциалом высшего порядка, получим
dL _ dX
L ~Y'—X
230
которое интегрируем следующим ооразом:
}' -VF
Г (IL , F С dX
у--х (хи./)
И- Л'и..-
где /•' — пиело молей в загружаемой смеси состава Х^-:
1Г — число молен в остаточной жидкости состава _V^-.
Выражение (XII. 7} известно под названием уравнения Гелен. Решение
(XII. 7) обычно составляется графически.
Средний состав дистиллята может быть определен из материального
баланса:
xxF=/)yfJ-;.irxu. (XI!. 8)
Используя значение относительной летучести
С1 =
У (1-Х)
X (1-У)
найдем ыры подстановке в (XII. 7):
Последнее равенство приводится к такому виду:
C£1^f(i-x1v)
(ХИ. 9)
Л/рп.иср. Жидкость, содержащая 50 мол. % гептана (Л) и 50
мол. % октана (В), подвергается простой версионно при 1 ат‘, количество
испаряющейся жидкости составляет 60 мол. % от начальной смеси. Опреде-
лить составь! дистиллята (В) и кубового остатка (И')- Для расчета примем
F — 100 .ноль начальной смеси.
Имеем:
Хр 0,5; J9 = 60 .ноль; Н'=40 .коль
Из (XII- 7) получим:
In
100
40
Данные о равновесном состоянии системы:
X 0,50 0.46 0.42 0.38 0.34 0,32
У 0,689 0,864 0,608 0,567 0,523 0.497
1 ... 5,29 5,32 5,32 5,35 5,50 5,65
При построении графика принимаем значения X в качестве абсппссг
а значения !/()• — X) в качестве ординат. Начиная от XF = 0,5, опреде-
ляем площадь иод кривой и как только ос величина достигает значения
281
0,91(1, интегрирование прекращаем; при этом ^»33 мол. долей гептана
и купоном остатке.
Средний состав дистиллята получим с помощью (XII. 8)
100 0,50 = 60УП +40 0,33
откуда У'р = 0,614 мол. долой гептана.
Так как для данной системы при 1 ат среднее значение а = 2,16, то из
(XII. 9) имеем
109 •()..>_„ . 100(1 -0,5)
4ОХЩ ^le40(l-¥w)
откуда методом подбора найдем Х^ = 0,33.
Многокомпонентные системы. Идеальные растворы. Примени-
тельно к многокомпонентным системам, образующим идеальные жидкие
растворы, для любых двух компонентов может быть написана формула
(ХИ. 9). Обычно выбирается один компонент, относительно которого опре-
деляются летучести. Так, например, для вещества i с относительном лету-
честью ctlB получим
FXbf
iiz у aiB £ П,-y
’ Anv и
и
x\\r= 1,0
где XiF n X^v — мол. доли вещества i, соответственно, в начальной смеси
и кубовом остатке.
Лрн.иер. Жидкость, содержащая 50 мол. % бензола (А), 23 мол. %
толуола (В) и 25 мол. % о-ксилола (С), подвергается простой перегонке
при 1 ат, причем количество испаряемой жидкости составляет 32,2 мол. %
от начальной смеси. Определить составы дистиллата и кубового остатка.
Средняя температура перегонки будет несколько выше температуры кипения
начальной смеси.
Примем температуру перегонки равной 100 °C. Исправления могут
быть сделаны впоследствии путем повторных вычислений температуры кипе-
ния кубового остатка, причем значения а с изменением температуры остаются
почти постоянными Давление паров рассматриваемых веществ при 100 сС
приведены в табл. XII. 1; относительные летучести определены по толуолу.
Таблица X/f. 1
Данные для расчета
Вещество Даплепнс пара при 100° С. Л.И рга. Ой. а Хр, мол. доли
Л 1'170 1370/550 = 2,49 0,50
В 530 1,0 0,25
С 290 О.ЗС! 0,25
Принимая за основу 100 .ноль смеси, будем иметь:
I) — 32,5 .моль и IK = 67,5 .ноль
282
Из (XII. 9) получим:
для Л
. 100-0,50 „,п1 100-0,25
1g рт г V — = 2,49 1g -----------
0/,о Аауу Ь/,о A^iv
для С
, 100 • 0,25
1g -—-—-----
07 ,,j
= 0,3641g
100 0,25
67,.» А{ду
Кроме того:
c w' r "i- % c iv —1'0
Решая совместно эти уравнения методом подбора, получим
'^avv -0,385, ^biv — 0,285; ^cw—0,335
Сумма этих значений составляет 1,005 вместо 1,00, что является вполне
у довлев верительным.
Состав дистиллята определится из материального баланса для каждого
компонента. Баланс относительно компонента А
100 - 0,50 = 32,5 УаП 4- 67,5 0,385
откуда:
KoD-0,7-42
Аналогично получим:
УЬр = 0,178 и Ус£) = 0,073
Кинетика реакции получения
изопропилбензола алкилированием бензола
пропиленом
При алкилировании бензола пропиленом в присутствии
Л1С13 образовавшийся изопропилбензол подвергается дальней-
шему алкилированию с образованием диалкилбепзола, который,
взаимодействуя с пропиленом, дает триалкилбензол и т. д.,
т. е. реакция алкилирования является последовательной. Она
протекает через образование ряда продуктов по схеме: бензол —>
—> моноалкилбепзол —> диалкилбензол —> триалкилбензол —> тетр-
алин лбензол и т. д.
Реакция осуществляется в сложной многофазной системе
газ — жидкость — катализатор (один из реагентов вводят в жид-
ком виде, а другой — в виде газа). Так как непосредственное
превращение возможно только на поверхности диспергирован-
ного в- жидкости катализатора, то газ, прежде чем прореагиро-
вать, должен раствориться в жидкости. Поэтому активной кон-
центрацией газа является его концентрация в растворе, пропор-
циональная давлению. При достаточно интенсивном перемешива-
нии и барботаже газа через реакционный объем можно считать,
что концентрация газа в ходе всего опыта не меняется. Следова-
тельно, изменение скорости реакции в ходе опыта определяется
изменением концентрации жидких веществ.
283
Для упрощения кинетического анализа реакции алкилирова-
ния допустим, что эта реакция двухстадийпая и обе стадии реак-
ции необратимы.
Тогда рассматриваемую реакцию можно представить так:
Л —> В —> с
где Л — бензол;
В — моиоалкилбеизол (изопропилбензол);
С — полпалкилбеизолы (диизопропилбензол, трнизоиро-
пплбепзол и т. д.);
/'1 и г2 — скорости реакций.
Как показано выше, скорость каждой реакции алкилирования
меняется только в зависимости от изменения концентраций жид-
ких продуктов, так как концентрация пропилена во время реак-
ции не меняется. Поэтому можно принять, что скорости первой
и второй реакций соответственно пропорциональны концентра-
циям бензола и моноалкилбензола и могут быть записаны в таком
виде.
?*£ — И Г с —
т. е. обе реакции являются мопомолекулярными.
Для последовательных мономолекулярных реакций было уста-
новлено, что изменение концентраций веществ Л и С во времени
Соответственно уменьшается и увеличивается, а концентрация
промежуточного продукта В проходит через максимум.
Константы скоростей первой и второй реакций можно найти
следующим образом.
Напишем
dm
’1 I' dr
где т — количество прореагировавшего вещества;
V — объем реакционной массы;
т — время.
Если в исходной реакционной смеси содержалось а моль А и не
было В и С, а к моменту времени т прореагировало X моль; то
оста юсь (а — X) молъ вещества Л.
Тогда в произвольный момент времени концентрация Л будет
Количество образовавшегося вещества не равно X люлъ,
так как часть его превратилась в С. Обозначим число молей В,
превратившегося в С к моменту т, через У. Тогда в произвольный
момент времени концентрация вещества В будет
284
Таким обра ом, имеем
dx к а — Х _ с/У , А' —У
?1 И с/т 4 И ’ 7 '~ К с/т ‘2 И
НЛП
dX
(1т
А-! (« --V)
с/У
с/т
= Ь(Х-У)
Решение этой системы дифференциальных уравнений дает:
Х--а(1-е"А'т) (XII. 10)
У = а ( 1 - —k- e~A’T-j- . l‘l . с“*гТ>) (XI1. 11)
\ Л-j /ц — h\ /
Если X = ft '2, т. е. половина вещества А превратилась в В и С,
то из уравнения (XII. 10) найдем:
~ Л1Г, .
Os 1 In 2
е =-Х ИЛИ Ay=^L (XII. 12)
где т1/г — время полупревращения вещества Л.
Зная т,, из эксперимента, можно рассчитать Ау. Значение k.t
можно определить следующим образом. Из уравнений (XII. 10)
п (XII. 11) получим:
х-у *1Я .(е-^_с-**т) (XII. 13)
Формула (XII. 13) дает зависимость количества образующе-
гося вещества В от времени. Когда количество В проходит через
максимум, то:
ДИ-=0
с/т
Поэтому, продифференцировав по т правую часть уравнения
(XII. 13) и приравняв производную нулю при т = тД!а1;С,
найдем:
(-Аус “ Л1Тм!>кс йАмцкг:) 0
Так как постоянный множитель
то
—Аус /;1Т^ьс _ц А._|р ЛЛмакс = у
А.-2е-ЛзТмцкС^ Аус“Л1Т-':;,1:с
285
II
11-1 А • In А*о / v Т Т л / т
<макс = г—— (XIL 14>
A-j-А-о
Уравнения (XII. 12) и (XII. 14) можно использовать для опреде-
ления Ад и к2.
Таким образом, если экспериментально найдено время полу-
превращения вещества Л и время максимального выхода проме-
жуточного продукта В, то, используя приведенные соотношения-,
можно определить константы ско-
рости реакции.
Парис. XII. 4 изображена за-
висимость мольного состава реак-
ционной массы от времени, по-
лученная на основе эксперимен-
тальных данных. Из рис. XII. 4
следует, что образование кумола
достигает максимума при продол-
жительности
При этом в
содержится 0,6
0,35 моль кумола и 0,05 моль по-
лпалкилбензолов. Для получения
такой смеси должно прореагиро-
вать 0,4 моль бензола.
0.35
мола при этом составляет =
= 0,875 моль па 1 моль прореагировавшего бензола.
Пропилен при этих условиях почти полностью затрачивается
на образование моноалкилбензола.
Константа ку связана с временем полупревращения вещества Л
соотношением (XII. 12). По рис. XII. 4 находим время полупре-
вращения бензола т1Х = 32 мин, время
кумола TMaltC = 25 мин. Следовательно:
ку = = 0,022
т, 32
Рис.
ного
MO.IL-
массы
Зависимость
реакционной
времени.
XII. 4.
состава
от
Следовательно, выход ку
In ку~ —3,8
Используя (XII. 14), найдем время
промежуточного продукта:
In Ад —In к,
Тмакс~
3,8 — In А-
реакции 25 мин.
реакционной массе
моль бензола,
максимального
выхода
.и пн 1
максимального
выхода
0,022 —Ла Л, —0,022
Для определения /с2 удобно, задавшись несколькими
пнями к2, близкими к ку, рассчитать тмакс по уравнению (XII. 14).
По полученным таким образом результатам можно построить гра-
фик зависимости к2 от тчакс и из пего, приняв тмакс — 25,
найти к2.
к
зиачс-
286
В табл. XII. 2 приведены даппые для построения графической
зависимости.
Таблица XII. 2
Данные для построения зависимости к2 от Тмакс
kx, .м«н“* In ftt In ftt-J- 3,8 fts —0,022 тмакс1
0,025 —3.68 0,12 0 003 40
0.04 —3,22 0,58 0,018 32,2
0,05 -3,0 0,8 0,028 28,6
0.07 —2,65 1,15 0,048 24
Таким образом, имеем:
Ад =0,022 1 и Ад 0,062 мшг1.
Глава XHI
Биохимический процесс
ферментации
Получение ценных продуктов с помощью аэробных
микроорганизмов при их размножении в питательной среде до-
стигло в настоящее время производственных масштабов. В связи
с этим в промышленности возникает проблема аэрации культуры.
При организации производственного процесса в любом случае
одна из главных задач заключается в том, чтобы выяснить, какие
переменные факторы влияют па выход продукта. В аэробных фер-
ментациях доставка воздуха к микроорганизму и служит той
переменной, которая может повлиять на ход процесса. Для общих
случаев найдено, что с увеличением скорости подвода кислорода
к активно размножающейся культуре повышается выход продукта,
по крайней мере, до тех нор, пока какой-либо другой процесс,
отличный от абсорбции кислорода, не начинает контролировать
скорость роста микроорганизмов и образование продукта.
Динамика роста микроорганизмов
Мы здесь встречаемся с такой закономерностью, кото-
рой подчиняются многие явления природы Когда в органическом
или неорганическом мире протекает процесс, при котором какой-
либо фактор постоянно сам, вследствие своей деятельности, возра-
стает, в таком случае то. что возникает в каждый момент, сейчас
же проявляет функцию действующего фактора. Определение
последовательности такого рода возрастания основывается па
Y It
показательное функции е .
Если активная масса в количестве с в течение определенного
промежутка времени возрастает на р%, то приращение будет
р
с -р— , а возросшая к концу этого периода времени масса:
288
Масса Cj к концу 2-го периода времени делается равной:
Мм приходим, таким образом, к общей формуле, что по исте-
чении п периодов времени масса будет
(XI И. 1)
Пусть р. 100 -/>, тогда
( / к vO
С —-с lim ( 1 -|-) ।
I \ в / J
где п бесконечно увеличивается. Далее, пусть будет
Тогда
к 1 о
— =- -Т- ИЛИ п О/ь
я о
II, когда 6 стремится к бесконечности (так как п -> со), в пределе
получается
С=сек (ХШ.2)
Итак, равенство (XIII. 2) показывает, что, если возрастание
величины активной массы в каждый момент пропорционально
самой величине, и за единицу времени эта величина с возрастает
па А: = р. 100, то для определения количества активной массы
к концу единицы времени величину С необходимо умножить па
показательную функцию еЛ. Очевидно, что к концу второй единицы
времени активная масса вырастет до
Сек = сскек = се'к и т. д.
Следовательно, величина активной массы к концу промежутка
времени т единиц будет равна
С—секх
Напишем
A- = lny (XII!. 3)
П
и=ек (XI II. 4)
Графическое изображение показательной функции (т. е. ее
кривая) дано на рис. XIII. 1.
Переход от (XIII. 3) к (XIII. 4), т. е. от логарифма к показа-
тельной функции, состоит в том, что в первом случае к рассматри-
вается как функция от ?/, а в другом, наоборот, у рассматривается
1 Л. М. Батуне)).
289
как функция от к. Когда к принимает значения от 0 до со, вели-
чина eh принимает значения от 1 до со и увеличивается очень
быстро.
Такой же результат можно получить следующим образом.
Обозначим количество микроорганизмов, имеющихся в данный
момент, через С. Тогда скорость их размножения будет, очевидно,
clC th. Согласно условию, дифференциальное уравнение задачи:
-^- = А-С (XIII. 5)
где к — коэффициент пропорциональности между скоростью раз-
множения микроорганизмов и их числом.
Рис. XIII. 1. Графическое изображе-
ние показательной функции.
Рис. ХШ. 2. Динамика роста мице-
лия и образования стрептомицина
в процессе ферментации:
1 — стрептомицин; 2 — мицелии; з — ки-
слород.
Пусть, например, имеем:
при Т = О
при т = 3
С = 100
С = 200
(XIII. 6)
(XIII. 7)
Равенство (XIII. 6) — это начальное условие задачи. Равенство
(XIII. 7) — дополнительное условие для определения постоянной к.
После разделения переменных получим:
Проводя интегрированно и обозначая для удобства при по це-
лующем потенцировании постоянную интеграции через In с, по-
лучим: '
290
ПЛИ
In С = /rr-L- In с (X11 Г. 8)
Разрешая уравнение (ХП [. 8) относительно искомой функции С,
т. с. потенцируя, получим:
С=г^-1пе=Д)пе = С(,Ат
так как по определению логарифма с,|1С = с.
Следовател ыю:
С’=--сеАт (XIII. 9)
Па рис. XIII. 2 представлена динамика роста продуцента
н образования стрептомицина в процессе ферментации. Результаты
исследований показывают, что максимальное количество кисло-
рода, поглощаемого средой, соответствует 12 ч, а максимальный
прирост биомассы относится к 36 ч.
Однако образование продукта, т. е. стрептомицина, происхо-
дит в промежутке между 12 и 44 ч; стрептомицин не образуется
ко времени максимальной дыхательной активности. Действительно,
максимальное накопление стрептомицина в культуральной жидко-
сти происходит, как видно из рис. XII 1.2, в период низкой
дыхательной активности.
Все кривые роста на рис. XIII. 2 соответствуют рассмотренной
выше показательной функции е .
Сопротивление массопередаче
в процессах аэрации
Так как аэрация питательной среды происходит, глав-
ным образом, путем использования растворенного кислорода
живой клетки, то процесс доставки его к последней представляет
собой явление массоиередачи молекул кислорода из газовой фазы
через культуральную жидкость к среде внутри клетки. Сопроти-
вление такой массопередаче может возникнуть вследствие факто-
ров, указанных па рис. XIII. 3. Опи подразделяются па две груп-
пы, одна из которых состав шет сопротивление растворению ки-
слородных молекул, а другая — сопротивление при вдыхании
кислорода клеткой.
Таким образом, имеем:
IZ/tj — сопротивление газовой пленки, образующейся в про-
странстве между массой газа и поверхностью раздела газа и жид-
кости;
1//с, — сопротивление за счет поверхности раздела газа и
жидкости (в жидкость проникают только те молекулы кислорода,
которые обладают достаточной энергией, остальные отражаются
обратно к газовой фазе);
19*
291
1. А’з — сопротивление жидкостной пленки, возникающей со
стороны массы жидкости;
1//ь4 — сопротивление но пути прохождения через жидкость;
l.'A’j — сопротивление жидкостной пленки, окружающей клет-
ку;
Жидкостная пленка. ।
7//rj
В&Зырек г
ьоздухп к
Газовая пленка, 1/к, [
/ I
/ Поверхность разёела,1/кг^
Сопротивление жидкостной
Внутриклеточное
сопротивление, //ке
I пленки
прохождения
жидкости, 1/к4
Сопротивление за счет
реакции, 1/к7
Рис. XIII. 3. Факторы, обусловливающие сопротивление мас-
соперсдаче в процессах аэрации.
1 А'е — внутриклеточное сопротивление, которое зависит от
характера роста микроорганизмов;
1//с- — сопротивление за счет реакции (сопротивление взаимо-
действию кислородных молекул с клеточными энзимами при их
дыхании).
Подвод кислорода к культурам
при перемешивании
При установившемся состоянии движение молекул
кислорода через газовую пленку происходит с такой же скоростью,
с какой молекулы кислорода проходят через жидкостную пленку.
Следовательно, для общей массонередачи будем иметь:
ЛГА-А-г(^г-И)«*ж(^-С>к)
(XI11. 10)
где рг — парциальное давление кислорода в газе;
С}К — средняя концентрация кислорода в жидкости;
/т,. — коэффициент массонередачи в газовой пленке;
/си; — коэффициент массонередачи в жидкостной пленке;
Pi и С’;. — значения парциального давления и концентрации со-
ответственно у поверхности раздела фаз, где имеет место
равновесие.
При решении (XIII. 10) необходимо знать парциальное давле-
ние и концентрацию растворенного кислорода у поверхности
раздела фаз, значения которых могут быть получены только гра-
292
фическим интерполированием. Поэтому обычно для уравнения
массопередачи используют общие коэффициенты:
А-л = л\. (Рг- /V) = * ж (Се - C;ii) (X111. 11)
где А\— скорость массопередачи;
— парциальное давление кислорода в газе, находящемся
в состоянии равновесия с жидкостью концентрации С’ж;
С’с — концентрация кислорода в жидкости, находящейся в со-
стоянии равновесия с газом давления рс.
Таким образом, разность рг — р<: есть общая движущая сила,
выраженная в единицах парциального давления, а С\. — Ст;1: —
общая движущая сила в единицах концентрации для жидкости.
Так как скорость массопередачи зависит от величины межфаз-
ной поверхности а, то ее следует учесть в рассматриваемом здесь
уравнении. В общем виде выражение для скорости растворения
кислорода в ферментационной жидкости примет следующий вид:
Лд ;=Л>(рг-р^К,^(Сс-Сгк) (ХП~)
Установлено, что при абсорбции газа контролирующее сопро-
тивление оказывает жидкостная пленка (!. А3 1. А^) и поэтому
можем представить (XIII. 12) в таком виде:
de
(XIII. 13)
илп
^ = Л-н:/1(С--6’и;) (XIII. 14)
где С’ — концентрация в насыщенном состоянии;
Cifi — фактическая концентрация растворенного кислорода.
При аэрации культуры в установившемся состоянии скорость
доставки кислорода путем растворения его в жидкости должна
быть равна скорости потребления его микроорганизмом. Эта
скорость потребления выражается так:
Q?OS = К(С" - Сyr) (X 11 1. 13)
или
СД
= г- Г (ХИГ. 16)
где Сс — концентрация кислорода в клетке;
— расход кислорода.
Величина может быть использована для определения ско-
рости абсорбции кислорода в жидкости, она полезна при оценке
работы аппарата для аэрации культур. В частности, если приход
кислорода больше его убыли, т. е.
А'ил(С'--С}1{)>Сс^Ог (X11I. 17)
то говорят, что аэрация протекает соответственно процессу фер-
ментации.
293
Минимальная аэрация, необходимая для данной культуры,
определяется формулами (XIII. 15) и (XIII. 16).
Величина А';к« носит название коэффициента эффективности
аэрации и используется для определения характеристик фермен-
татора.
Измерение коэффициента эффективности аэрации Кта осу-
ществляют следующим образом. Для ферментатора, наполненного
бульоном, определяется скорость растворения кислорода. Вклю-
чают аэрацию и перемешивание и периодически отбирают пробы
для определения количества растворенною кислорода с помощью
полярографа. После этого коэффициент эффективности аэрации
может быть получен из уравнения (XIII 11), которое при инте-
грировании дает:
In (С‘ — C;i;) = — A’}K«t+const (XIII. 18)
Построив график в координатах 1g (С* — Сж) и т (времени),
мы получим прямую линию, откуда:
/<>кл — —2,303 (наклон липин)
На абсорбцию кислорода в культуральной жидкости воздей-
ствуют следующие факторы:
1) перемешивание;
2) высота слоя жидкости в аппарате;
3) распределение воздуха;
4) скорость воздушного потока;
5) растворимость кислорода;
6) физические свойства ферментных жидкостей.
Перемешивание способствует эффективности аэрации тремя
способами:
1) увеличивает поверхность массопередачи, так как разби-
вает поток воздуха на мелкие пузырьки;
2) благодаря циркуляции жидкости в быстрых вихревых
потоках задерживает исчезновение воздушных пузырьков из
жидкости, увеличивая тем самым время контакта;
3) увеличивает турбулентный сдвиг, уменьшая тем самым тол-
щину плепкп жидкости; перемешивание может также преодолеть
сопротивление диффузии в реакционной среде и способствовать
теплопередаче.
Поскольку указанные воздействия невозможно отделить друг
от друга, соотношение между эффективностью аэрации и переме-
шиванием является полностью эмпирическим.
Установлено, что коэффициент эффективности аэрации про-
порционален расходуемой мощности па единицу объема
Кпа=сР°>м (ХШ. 19)
где с — постоянная;
Р — мощность на единицу объема реакционной среды.
294
Соотношение (XIII. 19) справедливо при достаточной турбу-
лентности, которая определяется числом Рейнольдса в следующем
виде:
где 1)м — диаметр мешалки;
Дг — скорость вращения мешалки;
q — плотность жидкости;
II — вязкость жидкости.
Для достаточной турбулентности число Рейнольдса должно
быть >105.
Попытки улучшить растворение кислорода путем прибавления
к валу второй мешалки показали, что возросшая мощность не
приводит к увеличению эффективности аэрации.
При постоянном расходе мощности эффективность аэрации
быстро понижается с увеличением объема жидкости. Ото явление
особенно заметно при низких скоростях воздушного потока
(400 мл:мин) и становилось меньше при наибольшей скорости
воздушного потока (6000 мл.'мин).
Влияние высоты слоя жидкости па эффективность аэрации
определяют путем сравнения с эффективностью при Z D = 1
(где Z — высота слоя жидкости; D — диаметр аппарата) для
данных условий работы.
При постоянном потоке газа и постоянной мощности на еди-
ницу объема жидкости эффективность аэрации возрастает с уве-
личением отношения Z/Z) (для Z D = 2 — в 1,4 раза).
Для разбивания воздушного потока па ряд мелких пузырьков
применяются обычно два способа:
1) использование аэраторов из нержавеющей стали или по-
ристой керамики;
2) переметывание с большой скоростью через одиночное сопло
или ряд сопел.
Наиболее широко применяемыми воздушными распредели-
телями для фермептаторов являются перфорированные трубки,
используемые либо самостоятельно, либо в сочетании с перемеши-
ванием. Такие распределители имеют форму колец или крестов,
диаметр которых составляет приблизительно 3/,1 диаметра ме-
шалки.
Тип избираемой системы «перемешивание — аэрация» зависит
от данных рассматриваемой ферментации. Применение аэрации
с мелкими пузырьками без механического перемешивания имеет
то преимущество, что опо уменьшает расход энергии и сокращает
капитальные затраты. Однако успешная ферментация требует
не только соответствующей аэрации, по также тщательного пере-
мешивания микроорганизма со средой. Следовательно, без пере-
мешивания можно обойтись лишь там, где одна аэрация в
295
состоянии ооеспечить достаточное перемешивание как. например,
в жидкости с низкой вязкостью и малым количеством твердых
веществ.
Для ферментатора с механическим перемешиванием имеется
такая зависимость:
-ЛиА*37
где Л — постоянная;
гс — линейная скорость воздуха.
Найдено, что при высоких скоростях воздушного потока
(120.Ч Ч) эффективность аэрации больше не увеличивается из-за
«захлебывания» мешалки. Действие «захлебывания» изменяется
в зависимости от тина и скорости вращения мешалки, причем
для мешалок с. малым числом оборотов (лопастные) «захлебывание»
наступает при скорости воздуха 20 .ч/ч, что значительно меньше
той, которая была получена для турбинных мешалок.
Растворимость кислорода почти не зависит от общего давления
газа, но прямо пропорциональна парцпалыюму давлению кисло-
рода в газовой фазе (закон Генри):
6"
Для культуральной жидкости, насыщенной воздухом при
температуре 25° С, величина С' составляет примерно 0,20 .имолъ л
п, поскольку составляет 0,21 .ii.w рт. ст., то константа Генри???
будет приближенно равна единице. Низкая растворимость ки-
слорода является важным фактором при рассмотрении вопроса
подачи кислорода к дышащим культурам.
Из закона Генри следует, что увеличение парциального дав-
ления кислорода в системе приводит к более высокой раствори-
мости кислорода. Jto достигается либо подачей воздуха под да-
влением, либо барботированием воздушио-кислородиых смесей
или даже чистого кислорода. Обычно ферментаторы работают иод
давлением, что способствует уменьшению загрязнений посторон-
ними микроорганизмами.
Вопрос о влиянии физических свойств ферментных жидкостей
на скорость подачи кислорода мало освещен в литературе. Харак-
теристики пульны сильно влияют па скорость растворения кисло-
рода в ферментной жидкости. Кроме того, эти характеристики из-
меняются в процессе ферментации. Изменения, которые могут
происходить, касаются вязкости и поверхностного натяжения.
Обе эти величины воздействуют на эффективность аэрации, по-
скольку они влияют на размер пузырьков (поверхность массооб-
мена), турбулентность в жидкости и на условия сопротивления
граничной плепкп.
Установлено, что ферментные жидкости'но могут быть охарак-
теризованы ньютоновым законом iрения. Исследования показы-
вают, что напряжение внутреннего трения в жидкости увеличи-
вается с возрастом культуры и, следовательно, с количеством
микроорганизма.
О ферментаторах
Общин вид цеха ферментации представлен па
рис. XIII. 4, схема контроля и автоматического регулирования
дана на рис. XIII. 5.
Схема ферментатора с материальными потоками изображена
на рис. XI1 [. (j.
Рас. Х111. 1. Общин вид цеха ферментации.
Объем ферментатора может быть определен но уже приведен-
ным в гл. 1 зависимостям. Основным фактором, определяющим
объем, является время, которое может быть найдено из кинетики
процесса.
Расход энергии для стандартных ферментаторов колеблется
в продолах 0,18—1.8 кет на JOOO.z массы, а скорость воздушного
потока 15—60 .и ч.
Для определения расхода энергии в ферментаторе может быть
использована следующая формула:
Р-
где Р — расход энергии;
Q — плотность массы;
Л — число оборотов мешалки;
— диаметр мешалки;
с — постоянная.
297
Общий расход энергии для подачи воздуха и перемешивания
па 1 л массы в основном по зависит от размера ферментатора, по
отношение мощности, потребляемой для перемешивания, к мощ-
ности, необходимой для подачи воздуха, значительно умень-
шается с увеличением объема ферментатора.
Дубление i ферментаторе
Рис. XIЛ. 5. Схема контроля и автоматического регулирования процесса
ферментации:
1 —регулирующий клапан на впздуишиш: с мембранным пневмоприводом; 1' —отпор
импульса давления иа воздушнике; .5 -соленоидный вентиль; 4 — оак с неиогаептс.и-м:
5 — цапни! регулятора уровня иены (термометр сопротивлении); >> — ферментатор; 7 —
термометр Сопротивления; S — регулирующий клапан на линии подачи холодной иоды:
'.t — угольный фильтр; ю — регулирующий клапан на липни подачи воздуха в фермен-
таторе; и — сужающее устройство (диафрагма); 12 — мемиранпый, оесшкальный диф-
манометр е индукционным датчиком; 1:1 электронный расходомер тина ОЛНД-'К:
it — приборы па щите уираиления; 15 — панель диета пцкоп кого управления подачей
сжатого воздуха; 16 —арматура сигнальной лампы с желтым стеклом; /- —арматура
сигнальной лампы с красным стеклом; 7Л' — реле времени; ю — реле промежуточное:
20 — кнопка управления мешалкой ил цеха; 21 —магнитный пускатель; 22 — кнопка
управления мешалкой со щита; 23 — регулятор уровня пены и давления (электронный
мост); 24 панель дистанционного уираиления выходящего воздуха; /.•; — регулятор
температуры (электронный мост); 26 панель дистанционною управления подичай
воды; 27 — приборы по месту.
298
Для сравнения эффективности аэрации различных систем
аэрации и перемешивания применяется следующее соотношение:
где С — расход кислорода, л.;
L — объем жидкости, л;
Р — мощность, кет;
т — время, ч.
Рис. XI11. 6. Схема ферментатора
Расход энергии только для механического перемешивания
(с поправкой на мощность, потребляемую приводом и трением
в сальнике) значительно уменьшился при использовании воздуха
(вследствие более низкой плотности реакционной массы)
Стерилизация воздуха
Очистка воздуха от любой примеси состоит из двух стадий. Первой
стадией является выделение частиц из потока воздуха под действием тех или
иных сил. Второй стадией является закрепление частиц (выделенных из
потока) па поверхности тел, смоченных жидкостями.
299
В слое набивки (канитель, плетеная сетка, трубка, вязанная из метал-
ической проволоки и свернутая по спирали, металлические кольца и т. д.)
процесс очистки газа следует представлять себе протекающим так: поток газа
дробится на отдельные струйки, которые делают большое количество поворо-
тов и меняют направление и скорость своего течения; при этом частицы под
действием определенных сил выделяются из потока.
Частицы, выделившиеся из потока, достигают поверхности жидкости
(набивка, смоченная какой-либо жидкостью) и, имея достаточную кинети-
ческую энергию, пробивают поверхность жидкости и остаются в пей. Другие
частицы пс могут преодолеть сопротивление поверхностного натяжения жид-
кости и отскакивают от поверх-
ности жидкости или остаются на
ней. Такие плавающие частицы, если
они смачиваются этой жидкостью,
насыщаются сю, а затем погружа-
ются в слой жидкости. Если частицы
не смачиваются пли плохо смачива-
ются, то флотационная сила вытал-
кивает их из жидкости и они могут
быть вторично подхвачены потоком
воздуха.
Рассмотрим случай движения
частицы в элементарном приводи
ней ном канале.
Вудом считать, что частица пред-
ставляет собой шар. На частицу,
движущуюся в потоке по криволи-
нейной проекции, действует центро-
бежная сила инерции. Нод действием
этой силы частица начинает дви-
гаться относительно газа в напра-
влении действия центробежной
силы. Такое движение, в свою оче-
Рис. XIII. 7 Схема действия па
частицу в криволинейном потоке
(плоская задача).
редь, вызывает появление силы со-
противления газовой среды. Силой тяжести и подъемной стой пренебре-
гаем, так как они направлены в противоположные стороны, а частица имеет
небольшую массу. Силу противодавления не учитываем, так как она незна-
чительна ио величине.
Таким образом, рассматриваемый случай движения частицы в криволи-
нейной струйке снедей к задаче, когда внешние силы, действующие на частицу,
лежат в одной плоскости (рис. XIII. 7).
В данном случае, применяя принцип Даламбера, уравнение движения
частицы в криволинейном канале можно представить следующим образом;
rt-
fi- —
(XIII. 20)
гн«?2 „
где ----- — цеитрооежпая си ia инерции;
и-
ы 1!о ----сила сопротивления;
т масса частицы:
г — радиус, по которому движется частица в данное мгновение;
/ — коэффициент сопротивления частицы;
— поперечное сечение частицы;
n<j — плотность воздуха:
и — нормальная составляющая скорости движения частицы;
и? — тангенциальная составляющая скорости движения частицы.
300
Представим
уравнение (XIII. 20)
в полярных координатах
(IG
dr
и = ——
ат
и выразим массу m и площадь F через плотность и размеры частиц:
л.63
m ----------р; г
(i
лб2
где
q — массовая плотность частиц:
6 — диаметр частицы;
О — угол поворота;
т — время.
Так как скорость движения частицы небольшая, то можно принять,
что
24 24\
Не «6
где
: v кинематическая вязкость воздуха;
Во число Рейнольдса.
Подставляя приведенные формулы в уравнение (XIII- 20). получаем:
лй’пг2 ( d() 24vntfi2g0 ( dr V
dr )
(Jr \ с/г
dr
(XIII. 21)
w
с/т ’
<1Т
После преобразования уравнения (XIII. 21) и замены с/О/с/т через со (угло-
вая скорость), получим дифференциальное уравнение:
dr й2ды
(XIII. 22)
_ <70
г I8vq<,
Для нашего случая можно полагать, что угловая скорость в элементар-
ном канале будет постоянна и тогда, интегрируя уравнение (XIII. 22) в пре-
делах от (12 до 01 и от Г2 до ri, получим:
In r„ — In ri = (0й — 0х)
18vq0
Л’-ofti
Ооозначая —(0.,—(J.) через In Л. можем написать что
ihvpu 4 - *
In г.> — In rt = In Л
или
_JnA. г _ге-\пА
-с , Tj — г2е
Вычитая левую и правую части последнего равенства из г-2, окончательно
получим
=r.(i
(XIII. 23)
или. подставляя в уравнение (XIII. 23) значение Л, получим пробег частицы s
но направлению действия силы инерции:
/ _ CigQ<0 (Оа-Чj)
S = г., \ 1 — с iev°°
(XIII. 24)
301
Применим уравнение (XIII. 24) для ок редел сипя зависимости содержания
частиц в очищенном воздухе от высоты слоя набивки, через которую прохо-
дит воздух.
Из опыта известно, что по мере прохождения воздуха через слой набивки
изменение содержания частиц в воздухе :авпсит от высоты слоя набивки,
пройденной им, или, что то же, от числа поворотов струек воздуха. Эта за-
висимость показана в общем виде па рис. XIII. 8,
Надо иметь в виду, чго для рассматриваемого случая действительные
перемещения частиц и размеры каналов малы, поэтому величину можно
считать постоянной. Как показывают проведенные опыты, содержание частиц
при прохождении воздуха через первые слои набивки резко уменьшается.
Ио мере прохождения воздуха через слой набивки
интенсивность падает.
Из рис. XIII. 8 видно, что изменение содср
жання частиц в воздухе в зависимости от нуги,
пройденного струйкой газа в набивке, можно пред-
ставить следующим дифференциальным уравнением:
dy
-7- = —1 У
ах
где i] — коэффициент, характеризующий интен-
сивность процесса очистки воздуха от
частиц;
у — содержание частиц в воздухе;
х — путь, пройденный воздухом в набивке.
Д 1я 1] в случае, если капал квадратного сече
пня и центробежная сила направлена перпендику-
лярно одной из сторон, имеем:
г» (1—е"111
ч=₽^—Г
где Ь — сторона квадрата;
Р — коэффициент эффективности удара частицы
о поверхность жидкости.
Для криволинейного канала круглого сечения будем иметь:
_Й-1П -Ц
-в-----(XI11.27)
(XIII. 25)
Рис. XIII. 8. Изме-
нение содержания ча-
стиц в газе в зависи-
мости от высоты слоя
набивки.
(XI11. 20)
'1 = P
Подставив значение 1] из (XIII. 2G) в (XIII. 25), получим для капала квад-
ратного сечения
dy
dx
Произведем
разделение переменных:
b
b
У
-1П А\
------- dx
Интегрируя,
получим
In
'/
с-1пЛ
ь
при X = 0, у —
тогда
С — In у,, л уравнение примет вид:
1пу —!пу0 — р
х
b
302
II. lit
In -У- -
г/о
Окончатся ыю:
-Р
Для случая криволинейного канала круглого сечения, подставив значе-
ние коэффициента I] но уравнению (XIII. 27) в уравнение (XIII. 25) и произ-
ведя интегрирование, получим:
Рис. XIII. 9. Схема для составления теплового баланса.
Изменение содержания частиц в во.щу хе в зависимости от высоты набивки
получено экспериментально и представляется в таком виде:
'/ = '/ос“ЙЛ’
где /. — коэффициент, определяемый из эксперимента;
х — число поворотов струек газа, равное, например, при применении
металлических колец числу рядов колец.
Теплообмен в процессах стерилизации воздуха рассмотрим па конкретном
примере.
ilpn.vpp. Источник горячего воздуха должен быть получен путем пропу-
скания холодного воздуха через нагретую цилиндрическую трубу с диамет-
ром 102 .ил;, и длиной 1,22 .и. Температура трубы поддерживается при 315° С
но всей длине. Для воздуха ср — 0,22 ккал/ка-г/як>, А - 0,03 ккял/.и-ч-гро</,
Q = 0,/2.) «Ли".Скорость воз ty ха w = 27 м3/ч. Температура поступающего
воздуха 2 Г" С.
Коэффициент теплоотдачи составляет
а = 24,6 г-0”1 ккал /.к2 ч • град
где х — расстояние по длине трубы, считая or се входного конца.
Принимая, что передача тепла имеет место вследствие теплопроводности
газа по оси, за счет движения массы потока в осевом направлении, а также
теплоотдачи от стопки трубы найти т мпературу выходящего газа.
Используя приведенные выше обозначения и рис. XIII. 9, мы можем
составить следующий тепловой баланс:
Приход
Для теплопровод- ту-
пости ........... —). Л
dx
С потоком массы
газа.............. ц-оср?'
Для теплоотдачи
от с гонки . . .
Убыль
- л dT <J
— t.A—---г -г-
ах ах
H-’QCp
лОа (7’cr— Т) bx
где Л — площадь поперечного сечения трубы;
D — се диаметр;
Т — температура воздуха, СС.
Так как при установившемся состоянии накопление равно пулю, то
- 4- л/Ja (7\.т-Т) б-г = -ЛЛ (-а.1 W-|-
(/-Е J </.t \ dx J
(т , 'IT . \
+^(^4-^-^
Таким образом,
Wcr“ Л Л -g- I rt-oCp g
Преобразуя, получим:
d-T »’pcp di . л/9а ,,,4 л
dx* ТТ~ТТГ( J-u
Подставляя t = 7\r — T, будем иметь:
d't irtv-г, dt 4a
--------=-£. --------1 = 1)
dx'- 7.Л (Ar 7. D
Несло подстановки чистовых значении найдем:
d*l 27-0.24-0,725 dt 1,22- 24.6 x"0,s .
(Z-r- 0.03 -0,785-0.1U22 * dx 0,03-0,102 ‘ ° ' ’
Примем г = с.2 для привезеппя коэффициента при t к рациональному
виду:
s — (I -г 39800^) — 32800^/ = о
dz
Применение рядов для решения этой задачи по является эффектпвш.ьм,
потому что в уравнения (X11I. 28) дна коэффициента очень велики по сравне-
нию с коэффициентом при d'Hidx2.
Вторая производная обусловлена наличием теплопроводности газа.
Если ею пренебречь (см. пите обоснование), то (ХП1. 28) приводится к урав-
нению:
-^--г 0,825 л:~(,'~л1 =0
которое может быть решено нутом разделения переменных. Таким образом,
Из граничного условия имеем
t 315-21 294J С (яри х 0)
следовательно,
t 294с-1 ,|55 v°’5 (XI! 1.29)
Для температуры воздуха будем иметь
I 315 —
304
и на выходе из трусы опа составляет:
Твих = 315-294с-1-268.5= С
После того, как в уравнении (XIII. 28) при его решении была исключена
вторая производная, мы можем проверить правильность такого действия пу-
тем сопоставления этого слагаемого с о цюй из остальных величин.
Двухкратное дифференцирование (XIII. 29) дает:
• ,65 • 294
dt 1.65 294 _0 - . v(i,5
dx 2
Таким образом:
(Pt
dx-
19900 19Ч00л:~и’5 3980CU- r 24200a:0’5
dx
Это отношение мало, исключая такие случаи, когда х является очень малым,
и поэтому принятое выше допущение оправдано.
Абсорбция этилового спирта
из смеси его паров с углекислым газом,
выделяющимся из ферментатора
В производстве этилового спирта путем ферментации мелассы
выделяется углекислым газ, содержащий лары спирта, который в настоящее
время теряется. 1 аз из фермента-
торов выходит при 1 гпи.м, со сред
вен температурой 26,6е С и ври
состоянии равновесия с водным
раствором спирта концептрацией
6,5%. Расход газа составляет
426 м?1ч.
Определить размеры абсор-
бера.
Парциальные давления эта-
нола и воды над разбавленными
растворами спирта показаны на
рис. XIII. 10. Давление паров
этанода для чисто! воды, есте-
ственно, равно нулю.
Интерполируя для 6,5%-го
этанола при 26,6° (I (давление
паров чистой воды равно
26,3 .ч.ц рт. ст.), получим р;)Т =
— V,5 _ii.it рш. ст. и Р] 1г<) ;
= 25,5.4.11 рт. ст. Следовательно,
для поступающего в абсорбер газа
имеем:
Да Зление парод бодь.\ мм рт ст
Рис. XIII. 10. Равновесные парциаль-
ные давления для растворов спирта
в воде:
Ь'риная Масс. % С2Н(ОН Мол долл С.ЩОН
= 0,00986 мол. (олеп этанола
-0 Л. м. Батуиср,
305
Расход газа, поступающего в абсорбер:
273 1 ,_о
42ь • —г-г = 11,3 КМ0.1 ь: ч
2/3-j-26,о 22,1
Количество этанола в газе:
17,3 • 0,00986 = 0,171 к-иоль/ч или 7,85 кг/ч
Количество водяных паров в газе:
1713 —0.58 л-.поль/ч
/ ЬО
или 10,5 кг/ч
Количество С02 в газе
17.3 — 0,171 — 0,58 =16.55 клюлъ/ч или 725 лт ч
Общее (массовое) количество газа
7254-1015-1 7,85= 743.4 кг,!ч
Для газа, выходящего из абсорбера, временно прямом, что этанол пол-
ностью удаляется, его давление 1 атм и газ насыщен нарами воды. Абсорбция
углекислого газа пренебрежимо мала.
Количество СО« в выходящем газе составляет 16.55 к.иолъ'ч или 725 кг/ч.
Количество водяных паров:
,._г 26,3
16>о5 —————- = 0,->Ь7 к.иоль; ч
i 6U — 26,3
Общее количество выходящего из абсорбера газа:
16,55 4 0,567 = 17,12 к.иоль’ч
Средняя скорость газа:
17,34-17,12
-------—-----— 0,21 Я.ИОЛЬ : Ч
Баланс по энтальпии или тепловой баланс. Ниже приведены значения
теплот растворения жидкого этанола в воде при 26,6° С:
.тэт, мол. доли......... 0,05 0,10 0,15 0,20 0,2:5
Т оплота растворения,
ккал, кмоль этанола —1860 —1525 — 1120 —812 —595
Для сравнения примем температуру 26,6°
Напишем балансовое уравнение для адиабатного процесса (Q — 0)
в таком виде:
"Д— « G« (I'i-H'c,)
где II { — энтальпия жидкости в нобоп точке колонны, ккал; кмоль:
II — энтальпия жидкости при поступлении в колонну, ккал/кмоль
L — скорость жидкости, к.моль/ч-зг.
L-г — скорость жидкости ври поступлении в колонну, кмоль/ч- м'2;
Н(} — энтальпия газа, ккал/ кмоль газа;
G — скорость газа, кмоль/ч-.н2;
H(ii — энтальпия газа у выхода из колонны;
306
Gi — скорость газа у выхода из колонны, кмоль/ч - ,ч2;
Gs— скорость газа, свободного от извлекаемого вещества, к.нсыь/ч ..и2;
7/г; — энтальпия свободного от извлекаемого вещества газа в любой
точке колонны, ккал/клоль газа;
Ис — энтальпия свободного от извлекаемого вещества газа у выхода
из колонны, ккал/к.молъ газа.
Содержание водяных паров в газе будет почти постоянным по высоте
абсорбера. Пренебрегая каким-либо изменением физического тепла самого
газа (газ находится при постоянной температуре 26,б С), мы найдем, что его
энтальпия приходится только за. счет теплоты парообразования содержа
щегося в газе этанола при 26,6° С, которая составляет 9950 кктм. к.ио.гь эта-
нола. Обозначив через Y содержание этанола в п.чгмь. клдль СОо, получим
Из материального баланса но этанолу имеем:
fis(r-5’2) = bs(X-X2) = isX
где — скорость жидкого растворителя, клоль/ч •2;
X — концентрация этанола в жидкости, кладь. кг растворителя.
Так как поступающая в абсорбер жидкость является чисток водой при
26,6° С, то П^ = 0. Поэтому
II т - 995ОХ
L Ls
Пусть А' — 0,01 к.ио.п, этанола.'к.чоль воды, тогда
, , л, 0,01 ... ,
L.-L^— 1,01 и х - 0,09989
° л 1,01
Экстраполируя табличные данные, найдем, что в этом случае теплота
растворения этанола в воде составляет —2140 яилл/клодь этанола. Для расчета
количества тепла па 1 кл<мь растворе» умножим эго значение на Х/(1 4- А')
AIIд = -2140 ~4 = -21,2 ------—----------
° 1.01 клсолъ раствора
Средний молекулярный вес (молекулярная масса) раствора:
„ 0.01-46,05-М 18.02
л/ср —
1,01
Теплоемкость раствора с,= 1,0 ккал'кг-г рад
Для мольной энтальпии раствора состава У при температуре имеем
где — температура сравнения.
Таким образом, получим
II L=* 1,0 (<L — 2G.6) 18,31—21,2
и
[ 1,0 26,6) 18,31 — 21.2] 1,01 9950 - 0.01
откуда:
tL 33-с
20* 307
Из рис. Nili. 11 найдем, что при этих температурах и концентрации
парциальное давление парой этанола равно 5,0 ,ч.и p/п. ст. Следовательно:
5.0
У-)Т "^ТТГ ~ 0,00658
i 60
Аналогично имеем:
0 0,00497 0,00989 0,01475
26,6 30,0 33,2 36,3
0 0,00302 0,00658 0,01316
'’О
ЧС>
З-ЭТ • г.................
Г, С ....................
Утг......................
Рис. XIII. И. Адиабатическая равновес-
ная кривая.
Этим данным соответствует
равновесная кривая на
рис. XIII. 11.
Диаметр абсорбе-
ра. Для насадки используем
керамиковые кольца Рашига
диаметром 25,4 .и.ч. Среднее
значение тангенса угла на-
клона равновесной кривой
т = 0,73.
Для экономичной скоро-
сти жидкости в абсорбере при
инмаем:
Таким образом, имеем:
L'G = 1,6-0,73 =
=-1,17 л’-воль иоды/клголь газа
или
1,17 • 17,21 = 20,1 к.но.гь. ч.
20,1 • 18,02 = 364 кг, ч
Внизу абсорбера при полном поглощении паров этанола скорость жид-
кости равна
364 + 7,85 = 372 кг.'ч
Далее
0g ~ 743,4/426 = 1,78 кг/.ir1
1000 кг/.н3
Для графика, изображенного иа рис. XIII. 12, имеем
L / 0g\°’5_ 372 / 1,78 \о,5
g ( or / — \ юоо)
,)тому значению иа оси абсцисс при «захлебывании» соответствует ордината
0,2, а при нормальной нагрузке 0,072. Для колец Рашига диаметром 25,4 .u.u
из справочной таблицы имеем:
«. с3 -- 538
еде и — удельная поверхность сухой насадки, .иг/.ч3;
= — морозность насадки (безразмерппая ве шчпна)-
Вязкость жидкости 0.05 cn.j.
308
При условии «захлебывания»? абсорбера получим:
0.2^oGQf \о-б /о,2
(«л3) вГ-J =\
1,27-10я» 1,78-lOOOV.s П|_. ,
--------------------I = 911 ;? ка '.н-
538 • O.950’2 /
ч.
Диалогично при нормальной нагрузке получим G —5450 кг/м2-ч
Предусматривая возможность дальнейшего увеличения производитель-
ности абсорбционной установки, принимаем
С = 3900 кг.'-и2 ч; L = 3900 • 1955 кг W ч
i 43/1
Рис. XIII. 12. Зависимость состояния «захлебывания» от
характеристики насадки:
1 — захлебывание для неупорядоченной насадки; с — пияшнп предел
нагрузки.
II инцадь поперечного сечения абсорбера
откуда диаметр
абсорбера:
1)
743,4
3900 -
0,2 -и-
1/-^ =
I 0,785
0,5 л
Высота насадим абсорбера. Примем г/2 0,0001 мол. долей паров
этанола. При таких разбавленных смесях уравнение рабочей линии с обо-
значением концентраций в мольных долях может быть написано так
G («,— у2) = L (.г, — х.)
Зоо
пли
откуда
17,21 (0,00986 — 0,СЮ01) =- 20.1 Gq-0)
17,21 • 0,00976
= 0,00835 мол. долей
Наибольшее число ступеней массопередачи, обычно, находится в топ
части абсорбера, где имеются разбавленные растворы; тайгоне угла наклона
равновесной кривой соответственно этому концу аппарата равен 0,607 (в точке
ж = 0,007). Поэтому определяем постоянную абсорбции Я применительно
к последнему:
L., 20,1
/1 = —— =------- —------= 1,94
0,607- 17,21
Кроме того, имеем:
= „.опт _
yL — znXj f/j 0,00086
По рис. XIII. 13 находим число ступеней массопередачи .V — 3,0.
Высота I1L, эквивалентная ступени массопередачи для жидкой
фазы. Средняя температура жидкости:
33,2 4- 26,6
1 — 29,9 °C
Коэффициент диффузии этилового спирта в воде при ж = 0,001 и 10 С
составляет 0,8- 10-i с.иУсек.
Вязкость воды при 10° С и 29,9" С соответственно равна 1,308 н 0,80 cnj.
В соответствии с теорией абсолютных скоростей получена- стсдующая эмпи-
рическая зависимость:
Т
Г^аЬИЬ
const
где Т — абсолютная температура, СК;
D— коэффициент диффузии для вещества Л и среде В, с.п-Усск;
iij> — вязкость растворителя В, слз.
Таким образом, коэффициент диффузии этанола в воде при 29,9е С:
D _= 29,9-1-273
ь 10 1-273
1225-0,8.10"5 = 1,4-
Для критерия Шмидта имеем
Рь _ 0,008
С£— еь/>ь — 0,991- i,i- иг-’ "='и,>
где — вязкость жидкости, г/с.ц-сек;
ql — плотность жидкости, г/см3;
f)L — коэффициент диффузии, см'-/сек.
Зависимость высоты Hj от характеристики процесса абсорбции опреде-
ляется следующей эмпирической формулой
- Ф
Sc,”;5
где /->’ — массовая скорость жидкости, кг/ч-м'~.
310
.Значения эмпирических констант Ф и ц можно найти в работе Гирш
фе.тьдера. Пирда и Сиотпа *:
Ф =0,0022: ц =0.22
Рис. XIII. 13. Диаграмма для определения числа ступеней по
абсорбции и десорбции.
Применяя эмпирическую зависимость, найдем:
/Л = 0,0022 (~ Г"' Sc0-5 = 0,0022 | )"’’"5750'3 = 0,22 .и
/ V 2’83 /
где itf — вязкость жидкости, ке;.ч-ч.
* J. О. Il i г s с h f е 1 d е г, R. К. В i г d and L. L. Spot z Trans.
A. S. M. E., 71, <J2I, (1949); Cliein. Bev. 44, 205 (1949).
311
Высота Hf;, эквивалентная ступени масеоисредачи для газонов
фазы. Для определения коэффициента диффузии паров н газовой фазе вос-
пользуемся следующей формулой, которая полечена на основе киnc-rnuecKOii
теории газов:
Dab =
0,0009292 Т*'г
Р (гчъ)~ I/ (А-Г/е^))
Рис. XIII. 14. .Зависимость / (А-7’/е) от /г7'/€.
где /)„ь — коэффициент диффузии, с.и^/сек;
Т — температура, °К;
1/гг, Мь—• молекулярные веса, соответственно, А и В;
р — давление, ши;
r„b — Постоянная, А;
— энергия молекулярного взаимодействия, лрг;
А — постоянная Больцмана (1,38-Hr6 эрг/гргЩ);
/ (ЛУ’/^ль) — функция, определяемая по рис. XIII. 14.
Величины и смогут оыть определены для каждого компонента эм-
пирически:
£/*= 1,39 7’ (XIII. 30)
Г - 0,835 г’j; (XII 1.31)
ГД°. 7’— температура кипения при нормальных условиях. ‘К:
5 кр — критический объем, c.ir'/.vo.ia.
312
Кроме того, для некоторых веществ эти величины могут быть найдены
КЗ следующей таблицы:
? . л, °к г. Л £.'А, ° К а г, Л
Воздух . . 97,0 .”.,67 СП, .... 136.5 3,882
Н-. .... 23,3 2.968 О, .... 113,2 3,433
X.> 91.46 3,681 СО .... 110,3 3,590
со., . ’ . 190 3,996 Аг .... 124,0 3,410
ГС, О .... 220 3.879 Хе .... 35,7 2,80
NO .... 119 3,470 Нс .... 6,03 2,70
Температура
кипения
его критический объем 1G7
этанола при
,5 с.чЛ.иоль. Из
нормальных условиях
(XIII. 30) имеем
351,4е К,
= 1,39-351,4 488= К
а из (XIII. 31)
г = 0,835 • 167,5П,33 = 4.59 А
Для углекислою гача найдем:
£'А-= 190 и г = 3,996 А
Среднее геометрическое значение \!.к равно
и
£/* = /488 190 = 304° К
*7’/s =
273-1 29,9
304
= 0.995
С помощью рис, XIII. 14 найдем:
Далее:
/(А-77$)=0,72
Для коэффициента диффузии паров этанола
только получим:
в углекислом газе оконча-
ил ц
^ЭТ., СО: -
0,0009292 Гэ/! (+ -уу-—
\ *«эг jWCOt /
I> (Cm, Со2)“ * / COS
0,0009292(273 —29,9)1,а ( )
________________________\ 4Ь,Оа 44 /
1 • 4,292 0,72
— 0,0785 см2,'сек
ою785 -S=M28;t м’-‘‘ч
Вязкость углекислого газа при 29,9° С равна = 0,015 са.?. Для кри-
терия Шмидта имеем
0,015-3,6
0.78 • 0,028;
= 1,075
313
Для определения высоты ступени абсорбции в газовой фазе воспользуемся
ел ед угощен эмпирия еской ф о рмул ой:
"в
Постоянные величины р и о (<х = 3):
Кельна Рашига (в лмг): ₽ С
9,5 0,45 0,47
25,4 0,39 0,58
0,32 0,51
38,1 0,38 0.66
0,38 0,40
50,8 0,41 0.45
Седловидные тела (в м.м):
12,7 ................................... 0,30 0,74
0,30 0,24
25,4.................................... 0,36 0,40
38,1 ................................... 0,32 0,45
Таким образом, для высоты, эквивалентной ступени абсорбции в газовой
фазе получим
= gGP gco,5 = 3 3900o,3S
CG’ 19550’58
l,U750,5 = 0,965 -u
Высота насадки в абсорбере. Высота насадки, эквивалентная
'Ступени абсорбции, применительно к обеим фазам составляет
Я, О 99
Я = Яс + -^-=0.965 + ^ = 1,1.м
и, окончательно, для общей высоты насадки получим:
Z = ЛГЯ= 8,0 • 1,1 =8,8 л
Для полной высоты абсорбера примем
Z = 10,0 л
.причем 0,6 л используется для нижней части аппарата под насадкой и 0,6 м
для верхнего конца абсорбера над насадкой.
Глава XIV
Экстракция
Жидкостная экстракция
В процессах экстракции участвуют но крайней мере три ве-
щества, а именно: два компонента, подлежащие разделению, и рас-
творитель. Поэтому экстракционная система в своей простейшей
форме является тройной.
Вместе с тем мы должны рассмотреть и бинарные смеси в связи
с понятием о фазовом равновесии в процессах экстракции жид-
костей.
J—о—I-
75 % А
25% Б
50 % В 63% В
Рнс. XIV. 1. Графическое изображение концентраций
двух веществ для гомогенной системы.
При комбинировании компонентов возможно получить систему,
состоящую из одной жидкой фазы или двух несметипвающихся
фаз, при условии, что компоненты являются жидкостями.
Если два вещества Л и В при смешении образуют только одну
фазу, то они называются гомогенными, смешивающимися.
Состав гомогенной бинарной смеси можно представить графи-
чески точкой на линии, концы которой изображают чистые компо-
ненты Л и В (рис. XIV. 1). Например, точка С изображает гомо-
генную бинарную смесь состава 60% В и 40% Л.
Вещества А и В могут смешиваться и не в любых отношениях.
Тогда они носят название ограниченно смешивающихся. В этом
случае при определенных соотношениях Л и В смесь образует две
фазы, которые не смешиваются между собой.
315
Каждая фаза состоит из насыщенного раствора одного компо-
нента в другом. Если компонент Л прибавляется постепенно к дан-
ному количеству компонента 13, то точка, дающая общий состав
смесей в любой момент, будет передвигаться от точки В но напра-
влению к Л (рис. XIV. 2).
Возможно, что после прибавления первых капель вещества А
никакого разделения не наступит, так как для достижения насы-
щенного состояния необходимо прибавить достаточное количество
вещества А.
Если состояние насыщения представляется точкой С\ то это
означает, что все смеси, состав которых изображается некоторой
точкой на участке между Е и С, являются однородными. пре щта-
вляющими ненасыщенные растворы компонента А в компоненте В.
Гомогенная сомогенная
X *
4 8° '<? 6
гетерогенная
Рис. XIV. 2. Графическое изображение концентраций двух
веществ для гетерогенной системы.
Наоборот, вещество В может добавляться каплями к веществу
Л. Тогда точка, изображающая состав смеси, будет передвигаться
от Л по направлению к В. В определенный момент и здесь дости-
гается состояние насыщения.
Если состав насыщенного раствора изображается точкой D
(рис. XIV. 2), то любая точка между Л и D будет представлять
однородную смесь, которая должна рассматриваться как нена-
сыщенным раствор вещества В в А.
Таким образом находим, что на рис. XIV. 2 отрезки ВС и AD
изображают предел однородной бинарной системы веществ А
и В.
Участок DC дает только гетерогенные смеси двух веществ.
Любая смесь общего состава, которая изображается точкой
на участке линии DC, будет распадаться на две фазы; но какой бы
в этом случае пи был состав смеси, составы фаз всегда опреде-
ляются точками С и D. Причем массовое (или объемное) соотно-
шение их будет различным, в зависимости от общего состава смеси.
Правило пропорциональности
в бинарных системах
Смесь общего состава, которая отвечает точке Е
(рис. XIV. 2), расслаивается на две фазы С и D. Причем количества
этих фаз пропорциональны длинам отрезков DE и ЕС.
31S
Следовательно, для точки Е можно написать следующее со-
отношение:
фаза С : фаза D — DE : ЕС
Это доказывается следующим образом: допустим, что массовая
доля компонента В в фазе D равна лд, а в фазе С — х>.
Допустим также, что суммарный вес бинарной смеси Е равен
g и что эта смесь расслаивается на gi масс. ч. фазы D и g., масс. ч.
фазы С.
Очевидно
Количество В в фазе С g2.r.,
Количество В в фазе />=g1.rI
Общее количество В
В соответствии с требованиями материального баланса по-
следняя величина должна равняться количеству В в исходной
смеси (равной g.r). Поэтому
пли
"1-Г1 ~ g‘2^2 = U1 -Г й) -Т
откуда следует, что
gi х2 — х ЕС
g2 х — х1 DE
Таким образом
gr __ ЕС _ фаза D
g2 “ />/? — фаза С
Тройные системы
Общий состав тройной системы можно всегда изобра-
зить точкой па равностороннем треугольнике.
Если точку внутри равностороннего треугольника соединить
с точками, лежащими на каждой из его сторон таким образом,
чтобы соединяющие линии были параллельны соответствующим
сторонам треугольника, то сумма длин соединяющих линий будет
постоянна и равна стороне треугольника
На рис. XIV. 3 из точки Е проведены параллельно сторонам
треугольника соединяющие линии ЕР, LQ и ЕЕ.
Очевидно, что ЕР = CS; EQ — QS; ЕЁ — QA, а в сумме мы
получим сторону треугольника Л С.
Предположим, что состав тройной смеси выражается отноше-
нием отрезков ЕР, EQ и ЕЕ.
Если длину стороны треугольника принять за 100%, или за
единицу, го (липы этих отрезков выражают содержание компо-
нентов в процентах или в долях единицы
317
'Гак, например, все смеси, состав которых задай точкой па
прямой ЛЕ, нс содержат компонента С. Все смеси, состав кото-
рых представлен точкой па линии (ЛУ, содержат 30% компонента
С, так как точка Q представляет бинарную смесь Л и С, содержащую
30% С, а точка U — бинарную смесь В и С, содержащую также
30% С.
Все смеси, состав которых изображается точками на линии
RS, содержат 50%i компонента А, а состав всех смесей, содержа-
Z7
Рис. XIV. 3. Диаграмма для расчета тройней
смеси.
щих 20% компонента В, может быть представлен точками на
линии РТ.
Линии QU и РТ пересекаются в точке Е. Это значит, что точка Е
представляет тройную смесь, имеющую состав: 50% Л, 20% Ji
и 30% С.
Аналогично можно показать, что тропная смесь, соответству-
ющая точке D, содержит 25'% А, 35% В и 40% С.
Каждая сторона треугольника представляет бинарную смесь,
а каждая точка внутри него — тройные смеси.
Правило пропорциональности
для треугольной диаграммы
При расслаивании смеси Q (рис. XIV.-4) на фазы Р
и R действительны следующие положения:
1) точки на треугольной диаграмме, представляющие состав
исходной смеси Q, а также составы фаз Р и II, находятся па одной
прямой;
318
2) количества фаз Р и 7? относятся, как длины отрезков QP
и QP. что можно доказать следующим образом.
Предположим, что масса исходной смеси (7 равна g.
Обозначим массу фазы Р через gt, а фазы /7 — через g2. Из
рис. XIV. 4 следует, что
количество 13 в фазе P = gyPP
количество В в фазе 7? - g2RAr
количество В в смеси ц gQM
И
gi PL | g.J(N =5(Ш
Рис. XIV. 4. Иллюстрация к правилу про-
порциональности для треугольной диаграммы.
откуда после подстановки gi = g — g2 получается:
g KN—PL g _ PS
g2 ~ или g2 QO
Из рис. XIV. 4 следует также* что
количество А в фазе Р gtPU
количество Л в фазе И -g.,UW
количество Л в смеси Q gtyX
И
giPbT-|-g2KIV = gC)x
После подстановки gr = g — g2 получим:
g Pl! — HW g PS
gi ~ PU-Qx ИЛИ g2 “ PO
Здесь получаются два треугольника, они подобны между
собой и углы равны. Следовательно, точка Q находится па прямой
РР. Зто доказательство первого положения.
319
Теперь перейдем к доказательству положения второго:
количество С в фазе Р — gxPD
количество С в фазе R — g>RK
количество С в смеси Q =
и
g.PD^g.PK^gQC
Подстановка g = gx -г- g2 даст:
Рис’ XIV. 5. Обобщение правила Рис. XIV. (5. Прямоугольный тре-
пропорциональпости. угольник для правила пропорции'
лальпости.
Следовательно
g, ПК IIG BQ
g.2 ~ EG ~~ EI) ~ QP
причем gx — количество фазы Ру;
g.2 — количество фазы II.
Следовательно, фаза Р : фаза II — RQ : QP.
Изложенное выше доказательство правила пропорциональности
является общим и поэтому оно справедливо для случаев, когда
к смеси двух компонентов добавляется третий
Если к смеси А н С, состав которой выражается точкой Р на
рис. XIV. 5, добавлен компонент В. то состав полученной тройной
смеси выразится точкой Q па прямой линии RP, причем
Количество В QP
Количество Р QB
что вытекает из прави та пропорциональности.
320
Вместо равностороннего треугольника может быть применен
и прямоугольный треугольник (рис. XIV. б). На нем точка Я
изображает тройную систему, состоящую из 0,5 масс. ч. С и
0,2 масс. ч. А.
Содержание компонента В здесь определяется по разности
и в нашем случае составляет 0,3 масс. ч.
Диаграмма экстракции
Чтобы установить общую картину разделения тройных
смесей, рассмотрим эффект прибавления вещества С к смеси из
двух компонентов (рис. XIV. 7).
Принимаем, что вещества Л и С, а также В и С полностью сме-
шиваются, но вещества Л и В являются ограниченно смешива-
ющимися.
Все бинарные смеси
Ли В, составы которых
и зображаются точ к oil
между Р и (Л распада-
ются на две фазы, при-
чем составы последних
с оот ветств уют точкам
Г п Q.
Рассмотрим смесь,
общин состав которой
определяется точио й
D на рис. XIV. 7. Эта
смесь разделяется на
фазы Р и (2 в отноше-
нии двух отрезков
DQ : DP.
Если компонент С
ирибавл яется ка iи я ми
к смеси Z), то отно-
изменяться. Количество одной из фаз
шение фаз будет постепенно
уменьшается до тех пор, пока, наконец, достигается состояние,
при котором в результате прибавления достаточного количества С
к смеси D исчезает фаза с наибольшим содержанием компонента В
и смесь становится однородной. Это состояние изображается
на диаграмме точкой D\ представляющей точку смешения
Л, В, С.
При прибавлении компонента С к другой гетерогенной бинар-
ной смеси (Е, F, G . . .) имеется возможность для любой бинарной
смеси подобрать такое количество С, которое обеспечило бы по-
лучение однородной тройной смеси (Е', F', G' . . .).
Таким образом при «титровании» большого числа бинарных
смесей между Р и Q с помощью растворителя С мы получим ряд
21 Л. М. Батуиер.
321
точек смешения, образующих плавную кривую QD'E’F'G'P
(рис. XIV. 7). Эта линия называется Синодальной кривой.
Следовательно, внутри площади, ограниченной линией PQ
и кривой QD’E' . . . Р, система всегда гетерогенпа (зона рассла-
ивания). Остальная часть диаграммы представляет однородную
систему.
В зоне расслаивания любая система разделяется па две сосу-
ществующие фазы, составы которых изображаются точками па
Синодальной кривой. Линия, соединяющая точки состава двух
таких фаз, называется линией сопряжения. Тангенс угла наклона
такой линии сопряжения изменяется в соответствии с природой
компонентов смеси (рис. XIV. 8 — XIV. 10). Линия PQ будет
линией сопряжения.
Коэффициент распределения
Под коэффициентом распределения одного из компонен-
тов двухфазной системы мы подразумеваем отношение концентра-
ций этого компонента в двух фазах.
На рис. XIV. 8 — XIV. 10 коэффициент распределения компо-
нента С между фазами Р и Q выражается следующим отношением:
Концентрация С в фазе Р _ PW
Концентрация С в фазе Q QV
Для случая, представленного на рис. XIV. 8, на
рис. XIV. 9 к = 1; на рис. XIV. 10 к < 1.
Из рис. XIV. 8 следует, что
РТ
коэффициент распределения Л между Р и Q = —у
v Y . он
коэффициент распределения Л между Q и Р = -~г
PS
коэффициент распределения В между Р и Q*=
коэффициент распределения В между Q и
Коэффициент распределения в тройной системе в общем слу-
чае нс имеет постоянного значения и зависит от состава фаз.
Это может быть проверено, если прибавлять постепенно веще-
ство С к гетерогенной смеси Я, состоящей из веществ Л и В
(рис. XIV. 11) и будем определять состав равновесных фаз. Тогда
общий состав гетерогенной смеси будет изображаться точками
на линии RRjR.zRalijRs-
Гетерогенные системы, составы которых представлены этими
точками, распадаются на равновесные фазы Р и (?, Pt и Q1} Р,г
и (?2, Р3 и <2з и т- fi-
ll результате прибавления компонента С к смеси R составы
фазы с высоким содержанием В изменяются по кривой QQiQzQaQiQb,
а составы фаз с высоким содержанием Л — по кривой
Р1\Р.Р3Р4Р„
Таким образом, составы двух фаз сближаются друг с другом
по мере того, как состав гетерогенной системы изменяется по
Рис. XIV. 8 Диаграмма с липнем
сопряжения.
Рис. XIV. 9. 'Диаграмма с линией
сопряжения
Рпс. XIV. 10. Диаграмма с ли-
нией сопряжения.
Рис. XIV. 11. Определение состава
равновесных фаз с прибавлением тре-
тьего компонента.
липни RC. В точке пересечения линии RC с бимодальной кривой
остается только одна фаза, т. е. смесь становится однородной.
Мы получили критическую точку смешения К на синодальной
кривой, где составы двух фаз становятся одинаковыми.
21* 323
Концентрация вещества С для двух тождественных по составу фаз
в критической точке одинакова (равна Л'5). т. е. коэффициент
распределения С (также Л и В) равен 1.
На рис. XIV. 12 представлена диаграмма тронной системы:
анилин — гептан — толуол при 25° С.
Толуол
ГАгцпак 80 80 40 20 Анилин
Рис. XIV. 12. Диаграмма тройной системы:
анилин — гептан — толуол при 25° С.
В табл. XIV. 1 приведены экспериментальные данные отно-
сительно состава равновесных фаз, из которых видно, что значение
коэффициента распределения к возрастает с увеличением концен-
трации толуола в рассматриваемых фазах.
Таблица XIV. J
Состав (в объема. равновесны.г фаз
Анилиновая фаза Гентановая фаза k
анилин толуол ген тан анилин толуол гептан
93.2 — 6,8 5,4 - 94,6
84.8 4,2 11,0 8,1 5,1 86.8 0.82
67.2 13,5 19.3 15,0 15,3 69.7 0,88
54,5 18,5 27,0 24,8 19.6 55.6 0,94
Определение критической точки смешения
и интерполирование линий сопряжения
Па рис. XIV. 13 предполагается, что расположение
Синодальной кривой, а также линий сопряжения а, Ь, с известно.
Проведем через точки /9, Е, Е, G, II и 1 прямые, параллельные бо-
ковым сторонам треугольника.
Прямые, выходящие из точек равновесных фаз Г) п Е, пересе-
каются соответственно в точках Лг и Л". Линии, выходящие из
точек F и С, пересекаются в точках О и О’ и т, д.
Рис. XIV. 13. Диаграмма для интерполирования ли-
ний сопряжения.
Соединив между собой все указанные точки пересечения, по-
лучим вспомогательную линию P(EV . . . Х'О'Р', пересекающую
Синодальную кривую в критической точке смешения К.
Положение критической точки, как видно, зависит от поло-
жения линии сопряжения.
Пусть теперь требуется установить, какая фаза равновесна
с фазой /?. Проводим через точку В линию параллельно ЕС и на-
ходим на вспомогательной линии точку пересечения (Л
Из точки Q проводим прямую параллельно /16'. 'Гочка пересе-
чения последней с бипедальной кривой дает состав фазы S,
равновесной с фазой R, а линия RS, следовательно, представляет
линию сопряжения. Понятно, что положение точки S можно было
бы определить посредством точки Q'.
Построение диаграммы экстракции
При экстрагировании важно, чтобы к разделяемой
смеси прибавлялся растворитель, обладающий способностью к се-
лективному (избирательному) извлечению одной из составных ча-
стей.
Таким образом в процессе экстракции получаются тройные
системы, состав которых может быть представлен треугольной
диаграммой.
Рис. XIV. 14. Построение диаграммы экстрак-
ции.
При построении диаграммы экстракции символ S, обознача-
ющий растворитель, удобно располагать справа на одной линии
с точкой Л, изображающей один из компонентов, ограниченно
смешивающийся с растворителем.
Второй компонент В, полностью смешивающийся с раствори-
телем, следует поместить на диаграмме в точке R (рис. XIV. 14).
Кроме того, принимаем, что к означает коэффициент распре-
деления компонента В между фазой, богатой растворителем, и фа-
зой с высоким содержанием компонента Л.
Если растворитель постепенно прибавлять к смеси из А и В
(точка С па диаграмме, рис. XIV. 14), то общий состав тройной
смеси будет изменяться по линии CS.
Если смешиваются равные части растворителя и смеси С, то
общий состав тройной смеси представляется точкой F таким
326
образом, что FC — FS. В этом случае гетерогенная смесь F будет
состоять из двух фаз, составы которых даются точками Р и (Л
Фаза Р с высоким содержанием растворителя называется
экстрактивной фазой, а фаза Q с низким содержанием раствори-
теля — рафинатной фазой.
При удалении растворителя из экстрактивной фазы (промыв-
кой, дистилляцией) получается свободный от растворителя экс-
тракт, состав которого определяется точкой Е.
Таким же путем рафинат R получается из рафинатной фазы Q.
Следовательно, процесс экстракции жидкости сводится к тому,
что начальная смесь С распадается на две смеси Е и /?.
Сравнивая их с исходной смесью С, находим, что смесь Е
получается с более высоким содержанием В, а рафинат R содер-
жит меньше компонента В.
Обратное явление имеет место в отношении компонента Л.
Экстракция, как видно из диаграммы, имеет своим результатом
частичное разделение компонентов смеси.
Из диаграммы представляется возможным получить следующее
количественное соотношение:
Экстрактивная фаза Р QF
Рафинатная фаза Q ЕР
и
Экстракт Е С/?
Рафинат Н СЕ
Таким образом, с помощью диаграммы мы можем рассчитать
состав экстракта и рафината.
Пример. Рассчитать экстракцию 100 ,«.г смеси состава С.
Расчет может быть выполнен двумя способами:
1) на основе данных об экстракте и рафинате;
2) на основе данных об экстрактивной и рафипатпон фазах.
Из рис. XIV. 15 следует, что
Экстракт (£) СП
Рафинат (Я) СЕ
Измерив отрезки СР и СЕ, найдем
СП : СЕ = 0.509
отсюда имеем:
экстракт
0,509
1,509
100 = 33,7 мл
рафинат
Т~АиТ 100 = 6G.3 мл.
1,509
327
Из рис. XIV. 15 также, следует, что экстракт состоит из 68.0 ч. В и 32 ч. Л
и что рафинат содержит в себе 10,7 ч. В и 89.3 ч. А, и далее
/22.9 -и.< В
33,7 л'-< экстракта состоят из
\10.S мл Л
z 7,1 мл В
мл рафината coctohi из
х59,2 .«л Л
Начальная смесь С состоит из 30 ш В и 70 ли
Ч 7,1
в экстракт перешло 100 7621%. а и рафинат —-
цента В от его исходного количества.
А. Следовательно,
Iin) = 23,7"(l комио-
Рпс. XIV. 15. К примеру для расчета экстракции
100 лл смеси состава С.
Для компонента Л находим, что 100= 15,4«„ исходного количества
59 2
перешло в экстракт и 100- 81,6% в рафинат. Эти количества могут быть
вычислены также с помощью равновесных фаз следующим образом:
Экстрактивная фаза (Р)__ FQ
Рафинатная фаза (^) “ FP
Измерив отрезки линий, получим:
-gt=i«
Известно, что получено путем смешения ранных объемов С и 5. Прини-
мая, что исходное количество С составляет 100 .ил, найдем общий объем фаз
328
I 84
(paniii.ni C + 5) равным 200 мл. Поэтому экстрактивная фаза (Р) 200-=
- 129,6 мл, а рафинатная фаза (0 - у-^у- 200 70,4 дм.
Из построения на рис. XIV. 15 далее имеем, что экстрактивная фаза
(Р) содержит 8.3% Л н 17,7",, В, так что
,10,8 лгл .А
129-6 мл экстрактивной фазы содержит f
'22,9 мл В
Рафинатная фаза (0 содержит 83,9% А и IOJ",, В. отсюда
,59.2 мл А
70.4 .чл рафинатной фазы содержит f
4 7,1 мл В
что полностью совпадает с. предыдущим расчетом.
Изданных расчетов вытекает, что рафинат /{ содержит в себе значительно
большее количество А, чем в исходной смеси С, по можно также отметить, что
этот компонент, получающийся таким путем, является еще недостаточно чи-
стым. Более высокая степень чистоты может быть достигнута при дальнейшем
прибавлении растворителя к рафинату /f (пли к рафинатной фазе 0. Если
растворитель прибавляется в таком количестве, что общий состав смеси изо-
бражается, например, точкой !' (рис. XIV. 15), то составы образующихся
фаз будут соответствовать точкам Р' и О', причем после удаления раствори-
теля мы получим новый состав рафината /?'- Этот рафинат состоит из компо-
нента А, более, чем 96%-пой чистоты.
Чтобы определить количества компонентов в двух фазах, мы можем
применить расчеты, аналогичные приведенным выше. При этом должно быть
принято, что здесь другая начальная смесь Р ( = 66,3 .ил) распадается при
экстрагировании на фазы Е' п /{'.
Прибавляя растворитель к смеси /V' (или 0), получим после разделения
фаз и удаления растворителя рафинат, содержащий Л еще большей чистоты,
чем в /{'. Таким образом, компонент А может быть получен из начальной смеси
С слюбой степенью чистоты. Что касается компонента В,то ясно (рис. Х1А . 15),
что максимальная чистота, которая может быть получена для этого компо-
нента, дается составом в точке, находящейся на касательной, проведенной
из 5 к синодальной кривой. При данных условиях процесса и температуре
высшая степень чистоты этого компонента отвечает составу с содержанием
76% вещества 11.
Экстракция с перекрестным током
На производстве применяются следующие способы
экстракции;
1) экстракция с перекрестным током;
2) противоточная экстракция;
3) экстракция с двумя растворителями;
4) противоточная экстракция с орошением.
Наиболее распространенными являются первые дна метода
экстракции.
Экстракция с перекрестным током (рис. XIV. 16) представляет
собой процесс, при котором растворитель прибавляется к разделя-
емой смеси, а затем после установления равновесия и образования
329
двух фаз отделяется экстрактивная фаза, а рафинатная фаза
снова обрабатывается растворителем.
Процесс повторяется до тех пор, пока не получится рафинатная
фаза с определенным составом.
Каждая обработка растворителем называется экстракционной
стадиен или ступенью.
Рафинатная фаза содержит в себе продукт, получающийся
перекрестной экстракцией сырья F в конце третьей стадии.
Выше были приведены соображения о методе определения на
треугольной диаграмме точек для равновесных фаз. 11 а рис. XIV. 17
этот метод иллюстрируется применительно к трехступончатой
экстракции с перекрестным током.
Рис. XIV. 16. Схема экстракции с перекрестным
током.
Рафинат, получающийся в конце 3-й стадии (рафинат ///),
содержит в себе, как видно из диаграммы, незначительное коли-
чество компонента В, которое, естественно, может быть еще умень-
шено, если применить дополнительные стадии экстракции. Однако
эффективность перекрестной экстракции определяется не только
числом ступеней экстракции. Из диаграммы видно, что коэффи-
циент распределения (и отсюда положение линий сопряжения)
также является важным фактором, наряду с относительным рас-
ходом растворителя, т. е. отношением количества растворителя
к количеству загружаемой смеси.
Недостатком перекрестной экстракции является то, что здесь
в чистом виде может быть получен только один из компонентов
бинарной смеси. Еще одни недостаток этого метода экстракции
заключается в том, что с увеличением чистоты продукта (компо-
нент Л) уменьшается его выход, так как при каждой стадии вместе
с экстрактом выделяется некоторое количество компонента Л.
При перекрестной экстракции, наконец, имеет место значи-
тельный расход растворителя, так как для каждой ступени при-
ходится добавлять чистый растворитель, что вызывает также
дополнительные затраты на регенерацию ценного растворителя.
Поэтому перекрестная экстракция применима только тогда, когда
выполняются следующие условия:
330
1. I {«экстрагируемый ценный компонент необходимо получить
в возможно более чистом виде, но не с максимально возможным
выходом.
2. Коэффициент распределения экстрагируемого компонента
очень высок по сравнению с коэффициентом распределения очи-
щаемого цепного продукта.
3. Если не требуется регенерации растворителя пли процесс
регенерации является дешевым.
Экстракт Ш
80
Экстракт 1
SO
20
80 SO 00
Экстракт 2
00
20
Раракат I В
Рафинат П £
Раффнат Ш ° J
л-
2-Л ступень ~
3-я ступем
8
Рис. XIV. 17. К иллюстрации метода расчета трехступепчатой
экстракции перекрестным током.
Расчет1 перекрестной экстракции. Распределение ком-
понента между двумя фазами представляется величиной, извест-
ной под названием коэффициента экстракции этого компонента:
& __ Количество компонента и экстрактивной фазе
Количество компонента в рафинатной фазе
Имеется следующее соотношение между коэффициентом экс-
тракции Е, коэффициентом распределения k и объемами экстрак-
тивной фазы V и рафинатной фазы и:
331
Можно показать, что доля данного компонента исходной смеси,
остающегося в рафинатной фазе (неэкстрагировапная часть), вы-
ражается следующим образом:
Экстрагируемая часть того же компонента равна:
Таким образом. при первой экстракционной ступени
(рис. XIV. 17) получаются рафинатная фаза /ц и экстрактивная
фаза Л\. Примем следующие обозначения'
т, — концентрация В в /?,;
т. — концентрация В в Л’,;
г — объем рафинатной фазы Л\;
И — объем экстрактивной фазы
Имеем:
ш'! — общее количество В в рафинатной фазе:
1'а:2—общее количество В в экстрактивной фазе.
Таким образом, из общего количества В в начальной смеси
(равной кд?, г Кт.,) в рафинате остается:
(XIX. 1)
v -Д
Далее, по определению /г х2/л\\ следовательно:
(XIV. 2)
При n-стуненчатой экстракции перекрестным током
а доля экстрагированного В соответственно равна 1 — (f пли
(а- ip — 1
(/;-Н)п
(XIV. 4)
Но формулам (XIV. 3) и (XIV. 4) можно также рассчитать
состав и количество конечного рафината для компонента А. Од-
нако, величина А*, для этого компонента изменяется довольно
332
сильно. Так, согласно рис. XIV. 17, для различных экстракцион-
ных ступеней коэффициент распределения равен:
в первой ступени
= 0,100
78,0
A(i во второй ступени —0,071
84,8
А’л в третьей ступени ‘Д’ 0,003
<S 1,3
А’а (ср) - 0,078.
Соотношение фаз для этих трех экстракционных ступеней
можно определить при помощи рис. XIV 17.
Они соответственно равны: 2.4: 1,7; 1,5. Средняя величина
в этом случае равна:
у
-(Ср)-: 1.8
Если цредпо, ожить, что в этом процессе ере (няя величина
А\, — 1,25 (рис. XIV. 17), то
Еа = 0,078-1,8 0,14
£в = Аа—-125-1,8 2.25
и
Доля пеэкстрагиронанного Л:
*»= ^ДДГ^"^"=0'075 “7,5%
Доля неэкстрагированиого В:
ф* ------—— = . *ч 0,0291 it.ur 2,91%
(/ц, + 1)П
Пусть 100 г исходного раствора состоит из 30 ч. В и 70 ч. А.
Тогда по найденным фа и ср6 можно рассчитать:
количество неэкстрагировапного Л
70 _ ._...
Ж"0'"'
количество неэкстрагировапного В
30
100
•2,91
0.87 ч.
Всего..........48,12 ч
Сумма неэкстрагировапных частей компонентов Л и В соста-
вляет рафинат.
Таким образом, в результате 3-ступенчатой экстракции мы
получаем рафинат состава
100 = 98,2% А
^-10°-= 1.8% В
с выходом 48,12%.
Если в рассматриваемой области треугольной диаграммы
имеют место большие изменения величин к и отношения объемов
Рис. XIV. 18. Зависимость q> от п при
различных значениях Е.
V/v при переходе от одной
к другой ступени,то в этом
случае целесообразно произ-
водить расчет экстракцион-
ного процесса графическим
путем (см. ниже) или же сде-
лать определенное допуще-
ние в отношении этих вели-
чин.
С этой целью формулу
(XIV. 33)можно видоизменить
следующим образом:
1
9 U4-1) (£' + 1) (/:" + !).. .
где Е, Е', Е" . . . — коэффи-
циенты экстракции в после-
довательных ступенях.
Чтобы определить вели-
чины (ри и (pfc для данных
условий экстракции с пере-
крестным током, можно ис-
пользовать график, на кото-
ром приведены расчетные
линии зависимости между (р
и п при постоянной вели-
чине Е.
На рис. XIV. 18 представлены подобные зависимости для
величин в пределах от 0,01 до 10%. Каждая линия рассчитана
по формуле (X V. 3) при постоянном значении Е и переменной
величине п.
Пример. Смесь, состоящая пз 30% уксусной кислоты и 70%
хлороформа, подлежит разделению с таким расчетом, .чтобы получить хлоро-
форм почти полностью очищенным от уксусной кислоты. Экстрагирование
проводится посредством воды в качестве растворителя. Для экстракции
зл
применен перекрестный ток с относительным расходом растворителя 2:1,
т. с. па 1 масс. ч. исходного раствора используются 2 масс. ч. растворителя.
В полученной тройной смеси содержание уксусной кислоты не должно
превышать 10% по массе. Такая концентрация уксусной кислоты будет только
на I ступени. Па последующих ступенях опа уменьшается.
Для коэффициента распределения ка принимаем величину, равную 5.
Если коэффициент распределения известен, то имеем:
Е 2
Еа = к а = а "у = 10
Теперь нужно установить максимально допустимое количество уксусной
кислоты в конечном раф! нате. Если это количество равно, например, 0,01 а
(на 100 г исходной смеси), то доля пеэкстрагированной уксусной кислоты
должна быть равна
фа = -^100 = 0,033%
Согласно рис. XIV. 18, для получения такой величины фо требуется
4 ступени. Таким образом, рис. XIV. 18 даст возможность быстро определить
.число ступеней, если нам известно <ра.
При графическом расчете перекрестной экстракции исполь-
зуется правило пропорциональности, рассмотренное выше.
Из рис. XIV. 17 имеем:
Рафинат I KF
Экстракт I DF
Если количество загружаемой смеси составляет 100 ч. (рафинат
I экстракт I — 100), то
Рафинат I ____ К1<’
100— рафинат / DF
Изморив длины отрезков KF и DF, получим рафинат I —
= 66,1 ч. от исходной смеси.
Для второй ступени экстракции будем иметь:
Рафинат II DL
Экстракт II DG
Кроме того, так как рафинат II экстракт II = рафинат
I (= 66,1 ч.), то
Рафинат 77 _ DL
06,1 —рафинат /7” DG
Определив длины отрезков DL и DG, получим: рафинат II =
= 55,4 масс. ч. загрузки.
Третья ступень экстракции дает:
Рафинат 777 __ GM
Экстракт III GI1
333
После измерения указанных отрезков найдем: рафинат ///
48 1 масс. ч. загрузки.
На рис. XIV. 17 состав конечного рафината (рафинат ///)
определяется точкой II : 1,8% В и 98,2% А.
Определение необходимого числа ступеней
при использовании ограниченного количества
р астворителя
Расчеты, приведенные выше, были основаны на том,
чю относительный расход растворителя является произвольным
независимо от числа ступеней экстракции.
Если растворитель должен быть регенерирован и если экстракт
нужно извлечь из экстрактивной фазы, то количество растворителя
должно быть ограниченным. В связи с этим во шикают два вопроса:
1) какая часть общего количества растворителя должна быть
использована для каждой ступени экстракции?
2) зависит ли эффективность экстракции от числа ступеней?
К первому вопросу.
Если мы примем, что распределение растворителя, например,
между двумя ступенями экстракции неизвестно, то значение <р
будет (см. формулу (XIV. 3)|
1
{£’ |-!)(£'+ !)
или
1
Так как величина Е — Е' постоянна для данного общего колнче
сгва растворителя, то из этой формулы следует, что ср имеет мини-
мальное значение в том случае, если произведение ЕЕ' является
максимальным. Но это произведение будет максимальным, если
дифференциал d (ЕЕ') = 0; так как
и -ь !:’ - с
или
С —Е
то
</[£ (С —A’)J d (ЕС-Е-) О
откуда
f'clE 2EdE 0 паи Е --
но
Е \-Е'- с
поэтому
Е = Е‘
Таким образом, экстракция с ограниченным количеством рас-
творителя наиболее эффективна, когда па каждой из двух ступеней
используется половина общего количества растворителя.
33.1
Применяя вышеизложенное к каждой паре ступеней, можно
сделать вывод, что в случае экстракции с перекрестным током,
наиболее экономичным является использование равных коли-
честв растворителя на каждой ступени.
Л’о второму вопросу.
Пусть наиболее выгодное число ступеней равно и, тогда коли-
чество растворителя. приходящегося на каждую ступень, соста-
вляет И п и при равных значе-
ниях Л’ во всех ступенях мы будем
иметь:
Здесь Е — коэффициент экс-
тракции при одно-
ступенчатом процес-
се с нспользованием
всего количества
растворителя Г.
Предельное значение величины
(f при /? — > ос равно:
Если Е 1. то предел <р при
п —> со близок к 1.
Если ЕУ> I, то значение <р
становится более приемлемым.
На рис. XIV. 19 показана за-
висимость ср ц п для значений
равных 0,1; 0.5; J; 't.
Рис. XIV. 19 Зависимость ср от п
при различных значениях Е.
Очевидно, что при низкой величине Е распределение всего
количества растворителя между несколькими ступенями не даст
соответствующего эффекта.
Такое распределение становится полезным только при Е
> — 0.5.
Противоточная экстракция
На рис. XIV. 20 представлена схема противоточной
экстракции. Растворитель S поступает из приемника 2 на верхнюю
часть экстракционной колонны 3. а исходная смесь F из сбор-
ника 7 подается снизу. Под влиянием разности плотностей этих
двух жидкостей создаются противоположно направленные
потоки. Полученный раствор (экстрактивная фаза Е) поступает
-- .'1. М. Ват упер. ;>,, V
в дистилляционную колонну /, в которой отгоняется растворитель
и откуда выгружается экстракт. Раствор фазы Н выходит с
некоторым количеством растворителя из верхнем части аппарата и
поступает в дистилляционную колонну 5, где отгоняется раство-
ритель и получается рафинат 11.
Растворитель, полученный в результате дистилляции фаз Е
и IE возвращается в приемник 2 н оттуда идет снова на экстрак-
цию. Процесс протекает непрерывно. Для этой схемы учтены
следующие предпосылки:
1. Плотность растворителя больше плотности исходной смеси.
Если же плотность смеси больше плотности растворителя, то точки
раствор
Рис. XIV. 20. Схема противоточной экстракции:
1, 5 — дистилляционная колонии; 2 —приемник. з —:j:;c-
тракцпонная kd.iokiui; 4 — сборник.
в местах поступления F и S па рис. XIV. 20 должны быть пере-
ставлены в обратном порядке.
2. Температура кипения растворителя ниже температуры кипе-
ния компонентов смеси. Если имеет место обратное соотношение,
то экстракт и рафинат в колоннах 1 и 5 получаются но как кубо-
вые остатки, а как дистиллят.
Одним из преимуществ противоточной экстракции по сравне-
нию с перекрестной является то, что при противоточной экстрак-
ции получается только один экстракт, в то время как при пере-
крестной экстракции образуется столько экстрактов, сколько
имеется ступеней в процессе.
В результате экстракт от противоточной экстракции полу-
чается с более высоким содержанием компонентов, которые должны
б ыт ь э кстр а гл р ов а н ы.
Противоточная экстракция может быть осуществлена и в сме-
сителях с отстойниками по прилагаемой схеме (рис. XIV. 21).
Здесь жидкость подается насосом или же самотоком при каскад-
ном расположении аппаратов.
33S
Для противоточной экстракции, кроме температуры, могут
быть указаны следующие переменные величины:
1) число ступеней экстракции;
pffC&fWP
Гис. XIV. 21. Противоточная экстракция со смесителями
и отстойниками.
2) отношение массы растворителя к массе исходной смеси;
3) состав рафинатной фазы;
4) состав экстрактивной фазы Л.
Рис. XIV. 22, Расчет числа ступеней при противоточной экстракции:
а — диаграмма; б — схема;
1—3 - экстракторы.
Указанные переменные, как правило, не могут быть выбраны
произвольно, так как они взаимно связаны между собой. Любая
пара из этих двух переменных может быть выбрана произвольно,
но тогда остальные две переменные величины фиксируются самим
процессом.
22*
339
Обычно выбираются составы фаз Е и Z?, а число стуиеиеп экс
тракции и массовое отношение растворителя к смеси определяется
ио рис. XIV. 22.
Предположим. что при противоточной экстракции исходная
смесь F разделяется на рафинат, имеющий состав, изображенный
точкой /?, и на экстракт, отвечающий точке Е.
Отсюда следует, что в результате экстракции мы должны
получить рафинатную фазу Рх и экстрактивную фазу Q3.
Проведем линии PXQ3 и FS. Точка пересечения Л/t щет массо-
вое отношение растворителя и исходной смеси, необходимое для
разделения смеси па фазы Pf и т. с. экстракция должна про-
водиться при соотношении
О )>аетн'Ч>»тС1н Д/1!'
бсмесн *1^| 5
Из правила пропорциональности имеем:
Экстрактивная фаза Q-, _ .IfjP,
Рафинатная фаза Р,
Далее проводим линии FQ3 и HS, которые при продолжении
пересекаются в рабочей точке Ji
Из проводим линию сопряжения PtQi, точку соединяем
с И ц линию //(/j продолжаем до точки пересечения Р2 на бимо-
дальной кривой. Из этой точки Р., проводим линию сопряжения
Р-iQi и (?2 соединяем с // и т. д. Такого рода построение продол-
жаем до тех нор, пока не получим Р3 для рафинатной фазы, равно
весной заданной экстрактивной фазе Q3.
Число линий сопряжения, которые были таким образом про
водепы па треугольной диаграмме, дает число теоретических сту-
пеней, необходимых для экстракции в данных условиях.
Приведенное выше построение доказывается следующим
образом.
Гак как при установившемся состоянии объем каждой фазы
на данной ступени остается постоянным, то в единицу времени
из каждой ступени вытекает столько жидкости, сколько поступает
с соседней ступени (рис, XIV. 22. б).
Поэтому имеем:
^3-F=Q2-P3 rpj-Л, -л —/>,{ -//) (XIV..',)
Если Q;i — F II, то состав II может быть изображен таким
образом (рис. XIV. 22, а), что
//:/’= l’Q3:Q3H
Однако из формулы (XIV. 5) следует, что Q3 — F - Q2 — P.t.
т. е. состав смеси Q., — Р3 должен изображаться также точкой II,
следовательно:
11 : 1>3 - : (J..11
340
Аналогично мы можем найти подобные соотношения и дли
других членов формулы (XIV. 5), откуда следует, что положение
точки // определяется с помощью правила пропорциональности
и, таким образом, является точкой пересечения линий, соединя-
ющих (Л( с Г; (Л с Ра; <21 с Р3 и <’• ‘S’-
Из рис. X. 1\ . 22 следует, что невозможно получить экстрактив-
ную фазу с концентрацией компонента В более высокой, чем та,
которая соответствует точке (?т, если мы принимаем, что линия
PmQm есть линия сопряжения, проходящая через точку /'".Таким
образом, экстрактивная фаза, например, состава Q здесь нере-
альна при протнвосточной экстракции, так как ее состав изобра-
жен точкой, расположенной выше точки Qm.
Рис. XIV. 23. (Лома для иллюстрации аналитического
расчета противоточной экстракции:
I, 2. ... /и. н —акстракторы.
Аналитический расчет противоточной экстракции.
Противоточная экстракционная установка состоит из /г ступеней
(рис. XIV. 23).
При установившемся состоянии в экстрактор т поступает Л
молен экстрактивного растворителя и Я/ молей рафинатного
растворителя. После перемешивания и последующего отстаива-
ния рафинат направляется в экстрактор т 1, а экстракт
в аппарат т— I. Какого рода процесс имеет место в каждом
экстракторе. Исходный рафинатный раствор, идущий в экстрак-
тор /, состоит из Л/ молей растворителя, в котором на один моль
содержится У'о молей извлекаемого вещества В
Начальное количество экстракта, подаваемого в аппарат п.
состоит из L молей экстрактивного растворителя, в котором па
один моль содержится X)i+1 молей извлекаемого вещества В.
Рафинат и экстракт представляют собой несмсшива1О1циеся
жидкости.
В каждом экстракторе достигается равновесное состояние,
которое может быть выражено так:
Xm = kYm (XIV. G)
Определим зависимость между составом для данной степени
экстракции и содержанием извлекаемого вещества в начальном
растворе.
Из материального баланса для ступени т будем иметь:
11 рЦХОД В = Л/ К,п_! + L X т х!
\ иы.ть В Л/> ?,г-|-/>Л (л
Приращение равно 0. Следовательно:
•Wm-i Н~ £ A ni+i — -МУ?л-|- LAm (XIV, 7)
Сопоставляя (XIV. 6) и (XIV. 7), получим:
У,»+1 - (Л’о -Ь1) У„г-I- -0 (XIV. 8)
Равенство (XIV. 8) есть разностное уравнение *.
В данном случае решением однородного уравнения будет -
У=срт
и, таким образом, для (XIV. 8) найдем
p--(L'o-H)£+£0^=0
Корни этого уравнения:
Р1 = 1 и ₽.,~£0
Общее решение. (XIV. 8):
у f _t_ Г гт
Постоянные Сл и С2 могут быть определены из следующих
краевых условий:
У=У0 при w-0
У = J^7+1 — Уп+1 П pH т = n -f- 1
к
Для искомого выражения окончательно получим:
у^-го _ С-i
Хгг+1 — I О /J*1 + 1 — 1
Для т ~ п найдем:
л’Г'-i
где ф — количество В, ушедшее с экстрактом;
гр = 1—ip =---—
En + l— 1
и *
(<р — количество примесей вещества В в рафинате).
Па рис. XIV. 24 представлена зависимость гр от числа ступе-
ней п применительно к противоточной экстракции при нескольких
значениях Е.
" .1. М. Ь а т у к с р и М. Е. 11 о з и н. Математические методы
в химической технике, Госхнмиздат, If)(i3, стр. 174—179.
342
имеет ли массовое (объемное) отпо-
п
Рис. XIV. 24. Зависимость ф от п при
различных значениях Е (противоточная
экстракция).
Минимальный расход растворителя. Выход рафината данного
состава зависит от количества применяемого растворителя.
С уменьшением количества растворителя увеличивается выход
продукта, но при этом возрастает число ступеней экстракции.
Вопрос сводится к тому,
шение количеств раство-
рителя и экстрагируемой
смеси минимальное значе-
ние. В этой связи рассмот-
рим рис. XIV. 25.
Если исходная смесь F
экстрагируется при отно-
шении количеств раство-
ригеля и смеси , то
для получения рафината
состава И необходима
только одна ступень экс-
тракции. При уменьшении
количества растворителя
(точки Л/.,, и т. д.) мы
получим экстрактивные
фазы Е2 и Е3 . . . , в кото-
рых содержание экстракта
увеличивается с пониже-
нием отношения количеств
растворителя и смеси.
Из диаграммы видно,
что при постоянном со-
ставе рафинатной фазы
число ступеней экстрак7
цпи будет постоянно уве-
личиваться.
Проведем па диаграмме
линию сопряжения Ет11т,
которая при продолже-
нии проходит через точку
F. Тогда точка Ет будет
изображать конечную экстрактивную фазу с максимально воз-
можной концентрацией экстракта. Следовательно, при противо-
точной экстракции невозможно получить экстрактивные фазы,
состав которых находится в пределах между точками К в Л'т,
так как оперативная точка Н лежит в этом случае на продолжении
линии сопряжения, т. е. па липни ЕтНт.
Применяя графический метод расчета числа ступеней с полу-
чением фаз и Ет, мы най (ем, что вспомогательные линии,
выходящие из оперативной точки //, почти совпадают с линиями
сопряжения и, таким образом, для получения экстрактивной
фазы состава Е1Ц потребовалось бы бесконечное число таких линий.
Мы можем сделать следующий вывод: если на треугольной
диаграмме соединигь точки, изображающие составы исходной
смеси и растворителя, а также точки, дающие состав фазы ЕП1
с максима гьной концентрацией экстракта и состав фазы /?,, то
точка пересечения полученных линий отвечает минимальному
отношению количеств растворителя и смеси
Рис. XIX 25. Диаграмма для определения условии про-
тивоточной экстракции с минимальным расходом раство-
рителя.
Естественно, что отношение количеств растворителя и смеси
может быть н ниже, чем то, которое соответствует точке 1/4, но
тогда рафинат будет менее чистым (точки Л/' и /?' на рис. XIV. 25).
Пример. Исходная смесь в количестве ИМ ?1-,; подвергается
экстрагированию. Массовая доля экстрактивных веществ ,сн — 0.25, а массо-
вая доля рафината 0,75. Растворитель принят чистым .rs — О. Данные
о составе равновесных фаз приведены в табл. XIV. 2.
Экстракция осуществляется противоточным способом с двумя ступе-
нями. Состав по компоненту А конечной экстракгпниоп фазы АД равен 6,40.
Для нанесения табличных данных па диаграмму воспользуемся в этом слу-
чае прямоугольным треугольником, где точка /’ отвечает составу sa U.25,
-г{, — 0,75. а точка А’г составу уа — 0,40 (рис. XIV. 2(>)-
Проведем линию, соединяющую точку /•’ с /?2, продолжим ее влево. . 1 пипя
сопряжения проводится через точку АД в соответствии с табличными
данными.
Соединяем точки Е и .S’ линией E.S и устанавливаем (иона произвольно)
состав конечной рафинатной фазы /Д.
Таблица XIV. 2
Состав равновесных фал
•v« vb •V . J'a
0,000 0,950 0,050 0,000 0.100 0,900
0,095 0.925 0.050 0.079 0.101 0,820
0,050 0.899 0,051 0,150 0,108 0.742
0,075 0,873 0,052 0.210 0.115 0,675
0,100 0,846 0.054 0,262 0.127 0,611
0,125 0.819 0,056 0.300 0,142 0,558
0,150 0.791 0,059 0.338 0,159 0,503
0,175 0.763 0.062 0,365 0.178 0,457
0,200 0,734 0,066 0.39O 0.196 0,414
0.250 0,675 0,075 0,425 0.246 0,32.9
0,300 0.611 0.089 0,445 0,280 0,275
0,350 0.5-44 0.106 0,450 0,333. 0,217
0,400 0.466 0.134 0,430 0.405 0.165
0,416 0.434 0,150 0,416 0.434 0,150
'Каким образом, мы iio.’ijчи.111 еще одну линию сопряжения E\R\. Про-
должим липни Е\1<2 и SRi влево. Если нее три линии E1R2; Slii; Е%Е пере-
секаются и общин точке, то найденная выше точка /?1 норна. В противном
случае следует принято другое значение для /? । н повторить процесс постро-
ения диаграммы снова.
Рис. XIV. 26. Расчет минимального расхода рас-
творителя при двухступенчатой противоточной
экстракции.
Искомый результат является приемлемым, если /fi и /щ отвечают сле-
дующим составам;
^ = 0,121 уа -0,295
хь= 0,824 ^ = 0,140
xs = 0,055 ys = 0,565
Па диаграмме при пересечении липни SF и EPii получим точку jf.
Следовательно, отношение количеств растворителя и смеем будет
О с F х
TT^^HTs илп loo = 0,198
Минимальное количество растворителя составляет:
х — |9 8 кг Растворителя
1 100 кг исходной смеси
Па диаграмме находим следующие данные:
1) исходная смесь F : = 0,25;
2) конечная экстрактивная фаза Ей : уа — 0,40;
3) промежуточная экстрактивная фаза ' Уа — 0,295;
4) конечная рафинатная фаза Hi : ха — 0,121;
5) промежуточная рафинатная фаза Кг ; 'Oi = 0,211.
Количества рафииатпой и экстрактивной фаз могут быть получены из
следующих материальных балансов.
Первая ступень
Е1-л/?1==:1$ф-я2 (XIV. 8а)
Компонент, А
0,295 Ех-г 0,121 Ех = 0,0 6т-|-0,211 Яа (XIV. 86)
Вторая ступень
Е2 + Яй==Е1 + /’ (XIV. 9)
Компонент Л
0,40 Е3 + 0,211 = 0,295 Е\ + 0,25 F (XIV. 10)
Из (XIV. 7) и (XIV. 10) для F = 100 и S = 19,8 имеем (в кг)
Ег = 38.33
Ей-= 37,63
Ki= 82,15
100,68
при минимальном расходе растворителя.
Экстракция компонентов
из сильно разбавленных растворов
Перекрестный ток. Содержание вещества Р в водном
растворе составляет 1%. Компонент Р экстрагируется раствори-
телем В при 20° С.
1. Определить степень экстрагирования компонента Р, если
100 кг начального раствора обрабатываются растворителем в коли-
честве 150 кг.
2. Определить степень экстракции вещества Р в условиях
трехстадийного проведения процесса с использованием для каж-
дой ступени 50 кг растворителя.
346
Данные о равновесном состоянии системы:
К# р
х'^----------- ... .0 0,001011 0,00246 0,00502 0,00751 0,00998 0,0204
кг во. и.!
=. .0 0,000807 0,001961 0,00456 0,00686 0,00913 0,01870
кг растворит.
1. Имеем:
хр 0,01:
Хе
F
0,01
1 — 0,01
= 0,0101
кг Р
кг поды ’
/'=100 кг
Рис. XIV. 27. Расчет экстракции компонентов из
сильно разбавленных растворов (перекрестный ток).
Па рис. XIV. 27 из точки F проводим линию FD с тангенсом
угла наклона —0,66 до пересечения с равновесной линией
в точке D, где
х — 0,00425 и
?/' = 0,00380
кг Р
кг жидкости
Следовательно, количество экстрагированного вещества 1?
составляет
А'- 99 (0,0101—0,00425) =• 0,580 кг
т. е. 58% от его начального количества в растворе.
2. Для каждой стадии имеем:
Проводим па рис. IV. 27 из точки F рабочие линии с тангенсом
угла наклона - 1,98, конечная концентрация будет ().(К)34
и количество экстрагированного вещества Р равно
g 99 (0,0101 0,0034) о(663 h-г
т. е. 66,8% от начального количества в растворе.
Противоток. Содержание вещества Р в воде состав-
ляет 1%. Раствор в количестве 1000 кг ч подвергается противо-
точному экстрагированию растворителем В при 20° С с тем, чтобы
довести содержание Р до 0,1%.
Рис. XIV. 28. Расчет экстракции из сильно разба-
вленных растворов (противоток).
Определить:
1) минимальный расход растворителя в 1 ч;
2) число теоретических ступеней при условии расхода 1150 кг:ч
растворителя.
Данные; о равновесном состоянии системы (см. стр. 347) пзобра--
жены на рис. XIV. 28.
1. Отсюда имеем:
!' — Ю00 кг/ч; xt. — 0,ul
Л -= 1000 (1 - 0,01) - 990
0,01
F’ 1 — 0,01
= 0,0101
кг Р
кг воды ’
•Гу-^ 0.001
0,001
1 — 0,001
—- 0,001001
кг Р
кг воды
348
Содержание растворителя В во всех экстрактивных фазах
11 содержание компонента А (воды) во всех рафинатных фазах
здесь постоянны.
Составляя баланс для компонента Р, получим:
Л2^. I Ву\
Последнее равенство есть уравнение прямой линии с тангенсом
угла наклона Л Б, проходящей через точки ({/' и .г),) и ^г/' - О;
О-
Начало рабочей линии находится в точке L (уг — 0, х'у -
0,1)01001) и для бесконечно большого числа ступеней эта линия
проходит через точку К на равновесной кривой при х'^.
Из рис. XIV. 28 находим у'к 0,0093.
11 потому
0,0093-0
/Лнн! 0.0101 -0,001001
И
п /1 __ 990 (1(,п кг растворителя
йм1’11 1.021 1,021 <JbJ ч
2. Имеем
В 1150 кг/ч] Л/В = 990/1150 = 0,860
Таким образом:
/ 0,0101 —0,001001
откуда:
, кг Р
If. _ 0,1)0782------------
i кг растворителя
Проведя рабочую линию через точку д’^), найдем число
ступеней равным
•V = 8,9 9
Аппараты для жидкостной экстракции
Для жидкостной экстракции имеются два основных
типа установок:
1. Одиос т у н с п ч а т ы е у станов к и, в которых
протекает процесс экстракции с последующим отстаиванием
смеси.
349
Рис. XIV. 29. Смесительный
аппарат для экстракции:
I — перегородка, 2 — мешалка.
2. Многост у и е н ч а т ы е уста п о в к п, в которых
совмещаются многие ступени экстракции. Последние являются
наиболее важными.
Одноступенчатые установки. В периодических процес-
сах перемешивание, отстаивание и декантация могут быть осу-
ществлены в одном экстракторе. Для непрерывных процессов
требуется несколько аппаратов. При перемешивании экстраги-
руемых жидкостей важно создать такие условия; чтобы в даль-
нейшем разделение смеси не было
продолжительным. Для перемеши-
вания жидкостей применяются сме-
сители (агитаторы), а также струн-
ные аппараты (инжекторы).
Смесительные аппараты
(агитаторы). Такого рода аппараты
состоят из баков для жидкостей
с мешалками (рис- XI\ 29) В этом
аппарате имеются внутренние пере-
городки во избежание завихрения
жидкости. Высота наполнения жид-
кости в аппарате должна быть равна
диаметру экстрактора. Для непре-
рывной экстракции могут быть при-
менены каскадно расположенные
аппараты с внутренними циркуляци-
онными стаканами (рис. XIV. 30).
Смесительные инжекторы.
Объем этих устройств относительно
мал и поэтому продолжительность
перемешивания здесь незначитель-
ная. Они особенно пригодны для
жидкостей с малой вязкостью и низким поверхностным натя-
жением, при которых происходит быстрое диспергирование.
На рис. XIV. 31 показаны конструкции таких смесителей.
Следует отметить, что эксплуатационные расходы здесь велики,
так как при подаче жидкостей требуется большой перепад давле-
ния и, следовательно, увеличенный расход энергии.
Па рис. XIV. 32 изображена схема инжекторного насоса,
в котором жидкость под высоким напором используется для пере-
качивания другой жидкости с низким напором.
Здесь: hs — напор во всасывающей! трубе;
hj — напор жидкости, проходящей через сопло;
ws — скорость всасываемой жидкости;
Wj — скорость нагнетающей жидкости;
uz — скорость смеси;
Лиап — высота напора.
350
Рис. XIV. 30. Схема работы батареи экстракционных аппаратов:
j — станин; s — перелив
Приток жидкости в сечении /.
Для нагнетающего сопла:
(XIV. 11)
А;
4-3
Qg 2g
Рис. XIV. 31. Смесительные инжекторы:
J — форсунка; 2 — отверстие.
Д in всасывающей ли-
пин:
(XIV. 12)
В и р о с т р а п с т в о
м е ж д v с о ч е н и я м и
I и Н.
I leiipepiiiBiiocTi»:
W"; । I’s'V's ' I’cj/'-r'1
L— площадь поперечного
сечения в начале диф-
фузора;
а — площадь поперечного
сечения в разрезе I,
и;, — площадь попереч-
ного сечения сопла.
При Q-- по-
лучим:
w2=,r;^7-!-^-J-(xiv.i3)
К о л и ч е с 1’ в о д в и ж е и и я
(!\ - Р2) И 2 = "cpi'M2 - ; - Qsw|«5
Рис. XIV. 32. Схема действия инжектора.
У;- 2д — о;р
при
будем иметь:
aj
л.,
(XIV. 11)
В пространстве
Энергетическим баланс:
г>. и~
м с ж Д у сеч е и и я м и // и III.
о
о
где Л — потери напора
Таким образом, для
в диффузоре.
высоты нагнетания папдом:
/'паи
р., , »; /,
$g r 2g g
Обозначив через 1]ца[1 к. п. л. диффузора, получим:
(XIV. 15)
/'нац
и
0g ’ 11,13,1 2
Рл
(XIV. 16)
Пример, Вола от источника на высоте 30 .и используется в инжек-
торе для иодачи 0,3 .ч3/«?к жидкости из другого места, расположенного па
высоте всасывания —1,5 .ц.
Сечения нагнетающего сопла и всасывающей трубы, соответственно,
равны 0,0045 .и2 и 0,045 №.
Необходимо определить объем питающей жидкости и высоту подачи
эжектора.
К. п. д. диффузора составляет 70%.
Если объем потока во всасывающей трубе 0,3 м9/сск, то скорость его
движения:
0,03 .
_бЛ45‘ = Ь’Ь5 Л/СС,;
Из (XIV. 12) имеем:
Р
Qg ь 3g
Используя (XIV. 11), найдем
О
Ру
-- hj — —I = 30 + 3,74 = 33,74 .«
2g Qg
откуда: ____________
u?j— ]z33,74 • 2 • 9,81 =25,75 м/сек
Расход нагнетающей жидкости:
0,0045 • 25,75 0,116 №/л?н
Равенство (XIV. 13) дает:
Таким образом, для высоты нагнетания из (XIV. 16) получим:
/'пап — 3,0 .ч
23 л. М. Батупср,
35
Смешение и эмульгирование
Пусть смеситель загружен веществами А и В, имеющими
поверхность раздела (рис. XIV. 33). Назначение аппарата состоит
в том, чтобы увеличить первоначальную поверхность раздела
между слоями. Это действие можно рассматривать как перемещение
частпц в трехмерном пространстве.
Рпс. XIV. 33. Схема к исследова-
нию смешения и эмульгирования
методом теории вероятностей.
Рис. XIV. 34. Распределение эле-
ментарных объемов в загрузке.
Количественное соотношение между величиной поверхности
раздела S в данный момент и продолжительностью перемешива-
ния т характеризуют кинетику процесса перемешивания.
Допустим, что за время т была развита поверхность раздела,
отношение которой к максимально возможной поверхности раз-
дела обозначим Yx. При этом полагаем, что характеристика пере-
мешиваемых веществ: вязкость, удельный вес и г. п., а также
режим процесса, остаются постоянными.
Приращение величины поверхности раздела пропорционально
разности между максимальной и фактической поверхностью раз-
дела (процесс первого порядка), а следовательно:
Ъ+1 = \ть(1 VT) (XIV. 17)
где s — коэффициент пропорциональности.
354
Обозначив
—у-с (XIV. 18)
представим зависимость (XIV. 17) в следующем виде:
^t+i ~%х )
Отсюда следует, что
(XIV. 19)
Согласно (XIV. 17), имеем:
Z0 — 1 - I у
Для преобладающего большинства случаев перемешивания
отношение первоначальной величины поверхности раздела к макси-
мально возможной будет пренебрежимо мало, поэтому можно
принять Уо = 0.
Отсюда следует, что
Zo =: 1
вследствие чего зависимость (XIV. 19) можно переписать так:
Zt=(l-s)T
Возвращаясь к переменной Yt, получим:
1-Гт=(1-0г«^,п
Приняв
будем иметь:
Л-1-^тС
Величина S поверхности раздела в момент времени т будет
равна:
5 = 5р(1-с-гС) (XIV. 20)
где Sj) — максимально возможная поверхность (при т = со).
Формула (XIV. 20) недостаточна для определения S, потому что
значение Sp не может быть определено экспериментально.
Так как перемешивание рассматривается как процесс пере-
мещения частиц в трехмерном пространстве, то для оценки этого
процесса величинами, которые могут быть найдены эксперимен-
тально, необходимо основываться на степени распределения
исходных веществ во всем объеме загрузки.
Другими словами, мы должны оцепить полпоту перемешива-
ния путем наблюдения за распределением элементов поверхности
раздела во всей массе материала.
Если объем всей загрузки разделить па большое число эле-
ментарных равных объемов ДУ (рис. XIV. 34), а поверхность
23* 35Д
на п равных элементов Л 5 (рис. XIV. 35), то результат перемеши-
вания может быть выражен величиной, характеризующей равно-
мерность распределения элементов развитой поверхности раздела
среди этих элементарных объемов. Кроме того, для этой цели
достаточно, чтобы в любой части элементарного объема присут-
ствовал по крайней мере один элемент поверхности раздела.
Пусть п будет большое, по определенное число. По мере увели-
чения поверхности раздела величина АХ = S/n возрастает. Веро-
ятность Ui того, что элемент поверхности Д/?,- будет находиться
в некотором определенном элементарном объеме (кубике), про-
Рис. XIV. 35. Распределение поверхности па п рав-
ных элементов AS.
порциональиа величине Д5Ь следовательно, Ui — kASit где/с —
коэффициент пропорциональности.
Так как все элементы поверхности раздела равны между собой,
а элементарный объем является случайно выбранным, то это соот-
ношение остается справедливым для всех элементов поверхности
раздела, независимо от их номера, т. е.
Ui к AS
где i может изменяться от 1 до п.
Вероятность того, что по крайней мере один из элементов дан-
ной поверхности раздела попадает в кубик AF, оценивается в соот-
ветствии с известными теоремами теории вероятности *.
Применяя эти теоремы к рассматриваемой системе, найдем,
что вероятность тою, что по крайней мере один из элементов
* J. М Б а т у и о р и М. Е. II о.ч и п, Математические методы
в химической технике, 1963, гл. XV.
351>
данной поверхности попадет в кубик ДР', выразится следующим
образом:
1 л 1 /_1 -1
при п —> <х’ правая часть равенства (XIV. 21) стремится
к 1 — tJ "S- *
Отсюда следует, что Р = 1 — е~™.
Заменяя 5 его выражением из формулы (XIV. 20), найдем
/* (XIV. 22)
Формула (XIV. 22) выражает вероятность попадания в слу-
чайно выбранную часть объема загрузки по крайней мере одного
из элементов поверхности раздела, полученной при перемешива-
нии в течение времени т.
Формула (XIV. 22) остается справедливой, если в ней Р заме-
нить вероятностью Рх, определяющей ту часть общего количества
элементарных объемов, которая состоит из кубиков, содержащих
по крайней мере один из элементов поверхности раздела. Следо-
вательно
(XIV.23)
Обозначим через х число кубиков вещества Л в системе, име-
ющей объем V. Тогда:
У ; Л = и
где t? — число единиц объема, содержащих вещество Л.
Очевидно, что полнота перемешивания может быть оценена
числом v единиц объема, который содержит распределенный
материал Л.
Предположим, что для определения kSp и с в уравнении
(XIV. 23) были взяты образцы объемом Уо.
Согласно уравнению (XIV. 22), вероятность отсутствия одного
из компонентов смеси в любой части объема V’o определяется выра-
жением е ~ ,iSp 1 _° ” х<:).
Вероятность отсутствия одного из компонентов смеси в
объеме v будет
Следовательно, можно записать:
[ _/iS (1-г?- Г') |®Л'о
('Vh-’-b р I (XIV. 24)
где (Р^Е является значением той доли общего числа объемов и.
которая состоит из объемов v, содержащих компонент Л.
Таким образом, в том случае, когда (^т)£ принимается как
конечное значение для удовлетворительного результата псреме-
шивапия, можно получить искомое время, необходимое для дости-
жения этого результата, решая относительно т уравнение (XIV. 24),
в котором значения kSp и с ранее были определены путем взятия
пробы Уо.
Уравнения (XIV. 23) и (XIV. 24) применимы к любым смеси-
тельным системам, так как при работе всех смесительных устройств
и систем преследуется одна цель — увеличение поверхности
раздела фаз и предусматривается возможность перемещения ком-
понентов поверхности раздела к отдельным составляющим объема
загрузки.
Промывка масла водой. Пусть 1,89 .и3 масла должны
быть промыты 0,340 лг3 водного раствора, содержащего 10,0 кг
поваренной соли (соль служит для высаливания масла во избе-
жание его потерь). Методом для определения содержания воды
в отбираемых пробах смеси может быть анализ на ион хлора.
В конце первой минуты перемешивания отобрано некоторое
число проб смеси объемом = 25 см3 каждая. Найдено, что
0,2 этих проб содержат по менее 10 згг хлорида натрия. В конце
пятиминутного перемешивания доля проб, содержавших пе менее
10 мг хлорида натрия, составляет 0,75.
Условием удовлетворительности перемешивания является нали-
чие не менее 10 мг NaCl в пробах, составляющих по своему коли-
честву 0,9 р, т. е. 0,9 общего количества проб объемом и. Здесь v
является частным от деления общего объема смеси на количество
10 миллиграммовых порций хлорида натрия, содержащегося в за-
грузке.
Какой промежуток времени потребуется, чтобы выполнить
эти условия?
Для решения задачи пользуемся уравнением (XIV. 23):
0,2=1—
0,75= 1 - С-йзР(1-е~5С)
Из этой системы уравнений находим: 1 1 1 1 1П 1-0.2 . ... 1,1 1-0,75 1 -е~ 1—с~1С
Отсюда имеем
In 1,25
следовательно 1 —<?“6С^6,22 —0,22е-с
Пусть !/ = с~с
358
после преобразования полупим:
у5 —€,,22 у Ь 5.22= О
ИЛИ
У5 — у — 5,22 у -ь .5,22 = О
Разлагая левую часть этого уравнения на множители, будем
иметь:
(У -1) (у‘ + У3 + У-+У - 5,22) = О
Так как у 1, то решение задачи сводится к решению урав-
нения у'1 !• у3 + у2 + у — 5,22 = 0, один из корпей которого,
очевидно, лежит в пределах между 1 и 2. Решая это уравнение
методом подбора, найдем у = 1,109. Следовательно, е~с = 1,109 и
Далее, так как общин объем смеси равен 1,89 + 0,34 = 2,23 .я3,
то
v = 2,23 е.и3/10 мг
Подставляя найденные нами величины в формулу (XIV. 24),
придем к уравнению
0,9 = 1 — [ е3>°« О - 1 аоет )]2,23/26
Для решения этого уравнения запишем его так:
2,045 (1 — 1,109х) = 1 п 0,1
2а
или
1,109 х =13.62
Отсюда найдем;
т=25,2 мчи
Итак, для получения заданной степени смешения потребуется
приблизительно 25-минутное перемешивание компонентов.
Смешение твердых материалов. В смесительном бара-
бане, имеющем постоянную скорость вращения, смешиваются
0,062 № газовой сажи с 0,10 .w3 углекислого кальция.
Опробование содержимого, которое было произведено после
того, как барабан сделал 10 оборотов, показало, что 20% проб
объемом в 1 ел/3 каждая содержали по мепес 1 мг углекислого
кальция. Пробы такого же объема были взяты после 20 оборотов
и показали, что 86,5% проб содержат не менее 1 мг угле-
кислого кальция. Удовлетворительная степень перемешивания
359
характеризуется 95%-иым наличием объемных единиц у, содержа-
щих но меньшей мерс 1 мг углекислого кальция, где v опреде
ляегся как частное от деления общего объема загрузки на общее
число миллиграммов присутствующего углекислого кальция.
Насыпная плотность углекислого кальция 320 кг лг!.
Требуется определить необходимое число оборотов барабана
для достижения заданных условий перемешивания.
Пользуясь формулой (XIV. 23), мы можем написать
0.2
0,865= 1 —'СС)
Для преобразования tfrnx уравнений получим
111 1—0,2 1П 1-0,865
|_r-uie - 1 Р-
откуда находим с = —0,2075. Подстановка этого значения в фор-
мулу
даст kSp — —0,0319.
Значение v для данного случая будет
0.1-3211-10“ = °’№'П8 taC0>
Применяя формулу (XIV. 24), получим
fJ0,031p (1-е°’2П75 т)1п,misos Т
откуда т — число оборотов, необходимое для получения заданной
степени смешения, оказывается равным 47.
Осаждение
Скорость отстаивания эмульсии зависит от разности
плотностей жидкостей, от вязкости сплошной среды, а также от
размеров капель диспергированной массы. Стабильные эмульсии
образуются при диаметрах диспергированных капель от 1 до
1,5 .«к. Диспергированная масса с диаметром капель в 1 .war
и больше отстаивается быстро.
Па рис. XIV. 3G изображен отстойник непрерывного действия.
Иногда применяются центрифуги для ускорения процессов отста-
ивания.
360
Следует отметить, что центрифуги не влияют на скорость
укрупнения капель диспергированной фазы, такое укрупнение
может быть достигнуто путем пропускания смеси через пористые
вещества.
Рис. XIV. 3(5. Отстойник непрерывного действия.
Многоступенчатые установки
Оросительные колонны. Эти аппараты являются са-
мыми простыми для непрерывного контактирования жидкостей.
Легкая Легкая
Рис. XIV. 37. Оросительные колонны.
Они состоят из полых цилиндров с устройствами для подвода
и отвода жидкостей.
На рис. XIV. 37, а изображен аппарат, где диспергирована
легкая жидкость. Тяжелая жидкость выходит через патрубок Л.
361
Высота слоя этой жидкости зависит от общей высоты жидкости
в колоппс. В аппарате поддерживается поверхность раздела
в сечении В. Отвод D используется для периодического удаления
йены п загрязнений.
При уменьшении высоты патрубка /1 поверхность раздела
жидкостей понижается (рис. XIV. 37, б); теперь диспергированной
становится тяжелая жидкость, а легкая жидкость является сплош-
ной. Обычно в качестве диспергированной фазы применяется
Главная поверх-
ность раз-
дела раз
Стек
Легкая
живк&ЛЭо
нервюрил' мч -л f
(силцалля) лиг ле я
Легкая
жиЗквсвм
Тяжелая
жиднт^ь
Слои ~
легкой
жидкости
(Тяжелая
жид.чесмь
Рис. XIV. 38. Колонна с насад- Рис. XIV. 39. Колоппа с ситчатыми
кой. тарелками.
та жидкость, которая проходит через колонну с наибольшей ско-
ростью.
Колонны с насадкой. Па рис. XIV. 38 представлена
экстракционная колонна с насадкой. Размер насадочных тел
должен быть меньше 1/8 диаметра колонны.
Колоппы с ситчатыми тарелками эффективны в ка-
честве экстракторов как по своей производительности, так и по
степени экстрагирования. На рис. XIV. 39 изображена колонна
с ситчатыми тарелками.
Легкая жидкость проходит через отверстия и поднимается
через сплошную тяжелую жидкость с образованием слоя жидкости
нод каждой тарелкой. Диаметры отверстий от 3 до б .н.ч. Скорость
жидкости через отверстия от 0.1 до 0.3 м’сек.
Пульсирующие колонны. Для небольшой производи-
тельности эти экстракторы (рис. XIV. 40) весьма эффективны.
В колонне имеются ситчатые тарелки (отверстия 1 мм), через
которые жидкость почти не проходит. При пульсации одной из
Легкая
Легкая
жадкость
Разделительные
тарелка
Тяжелая
жидкость
Легкая
жаёкесть
Peptpspapo-
' ванная (а/т-
vami.я} тарел-
ка
Рис. XIV. 40/ Пульсиру-
ющая колонна.
Решетка
fpfa.wa#
мешалка
Яерегареока
Тяжелая |
жидкость
Рис. XIV. 41. Колоппа с. ме-
шалкой.
двух жидкостей последние приходят в движение п перемеши-
ваются. Аппарат предусмотрен для 1000 пульсаций в минуту.
Пульсация осуществляется диафрагмовым насосом или с помощью
ультразвука.
Колоппы с мешалками. В этих колоннах имеются та-
релки с продольными перегородками (рис. XIV. 41).
Центробежные экстракторы
Экстрактор Подбельпяка состоит из перфорированной
(ситчатой) пластины, свернутой в спираль вокруг горизонтального
вала, вращающегося со скоростью 2000—5000 об/мин. Жидкости
нагнетаются в аппарат через вал. Причем тяжелая жидкость напра-
вляется к центру спирали, а легкая к периферии. После этого
жидкости проходят через спиральную пластину в противополож-
ных направлениях при некотором диспергировании одной фазы
в Другую, вследствие их движения через отверстия. Жидкости
удаляются через вал (рис. XIV. 42).
363
Экстрактор Подбсльняка пригоден д легко эмульгиру-
ющихся фаз с небольшой разностью их плотностей. Эти аипа-
Рис. XIV. 42. Центробежный экстрактор Подбельпяка - Фл-
лшшосьятща.
Рис. XIV. 43. Общий вид центробежного экстрактора.
ратьг применяются для ускорения (порядка секунд) процесса
при большом числе ступеней экстракции. Производительность
100 ООО л’ч. Общий вид аппарата показан на рис. XIV. 43.
Аппарат Лувсста — трехступенчатый экстрактор с вер-
тикальным вращающимся валом (3800 об/мин). Жидкости пе-
ремешиваются при прохождении через отверстия на валу для
каждой ступени (см. рис. XIV. 44).
Тяжелая жидкость
Легкая жидкость
Легкая жидкость
Рис. XIV. 44. Аппарат Лувеста.
у? МОЛЬ/у "2
растворимого
Рис. XIV. 45. Схема к рас-
чету высоты (длины) кон-
тактных экстракторов.
Высота (длина) контактных экстракторов
Для высоты экстрактора имеем
Z ~ Nil'
где Z — высота;
N — число ступеней экстракции;
I! — высота, эквивалентная ступени экстракции.
(XIV. 25)
Рассмотрим схему экстр акции, изображенную на рис. Х! \ . 45.
Для бесконечно малой высоты dZ поверхность фазового контакта
па единицу площади поперечного сечения колонны обозначим
через dS м'2/м~ сечения колонны.
Скорость массопередачи при экстракции может быть пред-
ставлена следующим образом:
d (Их) = d (Еу) = КR (х - X-) dS = КЕ {у* — у) dS (XIV. 26)
где В — скорость потока рафинатной фазы, кмолъ/м*
Е — скорость потока экстрактивной фазы, кмолъ/м2 «ч;
365
х — мольные доли извлекаемого материала в рафинатной
фазе;
у — мольные доли извлекаемого материала в экстрактивной
фазе;
х — концентрация извлекаемого вещества в рафинатной фазе
при равновесии с экстрактивной фазой состава у;
у' — то же в экстрактивной фазе при равновесии с рафинатной
фазой состава х;
Л'д — коэффициент массопередачп в рафинатной фазе,
кмоль/м~ • ч мол. доля;
КЕ — коэффициент массопередачп в экстрактивной фазе,
кмолъ/м2 ч • мол. доля.
Обозначим поверхность фазового контакта через а.«2/,м3; сле-
довательно,
dS—adZ (XIV. 27)
Величина а объединяется с коэффициентами массопередачп
в виде KRa и KRa.
Как рафинатная фаза, так и экстрактивная фаза изменяются
по высоте колонны. Но если взаимная растворимость обеих жидко-
стей постоянна, то R (1 — х) также остается постоянной. Поэтому
d{Rx)^R{i-x)d-^— = -^~ (XIV. 28)
Если концентрация извлекаемого вещества изменяется значи-
тельно по высоте колонны, то величина должна быть
более постоянной, чем KR, причем (1 — x) v представляет среднее
значение концентрации вещества в рафинатной фазе.
Тогда уравнение (XIV. 2G) примет следующий вид:
или
R dx К ца (I £) jj (•£ х') dZ
1 —X (I
(I —х)м dx К Ra(l—x)yidZ
1 —X х — х* R
(XIV. 29)
(XIV. 30)
Интегрируя (XIV. 30), получим число ступеней массопередачп:
где
X /j* (1 — £)
(XIV. 31)
(XIV. 32)
выражает высоту, эквивалентную ступени экстракции или массо-
передачи.
366
Для (1 — х)Л1 имеем:
При умеренно разбавленных растворах эта величина соста-
вляет:
(I- х)м = (*-?') Hl-*) (XIV 33)
Подставляя (XIV. 33) в (XIV. 31), найдем:
К1
= I (XIV. 34)
J Х—Х 2 1—
Xs
Интеграл уравнения (XIV. 34) вычисляется графически.
Если х принимается в массовых долях, то в правой части
(XIV. 34) будет дополнительно:
±1П М*-—1)+»
2* ^(г—1) |-1
где г — отношение молекулярных весов нерастворимой п раство-
римой частей в рафинатной фазе.
Если концентрации извлекаемого вещества выражаются так
, _______ кг С___________
— кг рафинатного остатка
и
, кг С
у =-----------------------
кг экстрактивного остатка
то для ступеней экстракции будем иметь:
(XIV. 35)
Кроме того, если равновесный и рабочий графики прямо-
линейны в пределах рассматриваемых концентраций, то можно
показать, что в данном случае применимы средние логарифмиче-
ские разности конечных концентраций.
Ж,—?/( —У-2
тг Л — -- —
П (х — х‘)у1 ‘ Е (у’—н)м
(XIV. 36)
Здесь имеют место следующие эквивалентные соотношения:
/?(^ —х2) = /Г(у1 —y2) = /<flaZ (x — z')yi^KEaZ (у*— у)м (XIV.3/)
367
Если для данной экстракционной системы действителен закон
Генри: у = тх и равновесный график есть прямая линия, про-
ходящая через начало координат, то аналогично случаю в про-
цессах абсорбции мы получим:
(XIV. 38)
где т — константа Генри.
Пример. Определить число ступеней массопередачи (экстракции)
для процесса противоточной экстракции уксусной кислоты из водного рас-
твора изопропиловым эфиром. Концентрация уксусной кислоты в начальном
Таблица XIV. 3
Значение X и .т*
X X* 1 X - л-
0.30 0,230 14,30
0,25 0,192 17.25
0,20 0,154 20,75
0,15 0,114 27,80
0.10 0.075 40,00
0,05 0,030 50,00
0,02 о 50,00
растворе составляет 30%; экстраги-
рованная жидкость содержит 2%
уксусной кислоты.
Выразим ,т и у в массовых до-
лях уксусной кислоты. Имеем:
= zp = 0,30; г/2 = 0;
а\,-0,02; ^ = 0,10
Расчетная диаграмма изобра-
жена па рис. XIV. 46, откуда с по-
мощью рабочей и равновесной ли-
ний находим значения z и z- при раз-
личных значениях у.
Величина площади под кривой,
построенной на диаграмме: х (абс-
368
цпсса) и . 1 . (ордината) в пределах от ж 0,3 до х = 0,02, соста-
илист 8,40.
Поскольку взаимная растворимость изопропилового эфира и воды для
этих растворов совершенно незначительна, мы можем принять, что
Применяя формулу (XIV. 34) с дополнением, получим число ступеней
экстракции
jV = 8,40 + -J-1h
1—0,02 , 1 . 0.02(0,3
1—0,30 2 0,30(0,3
Пример. Проведены опыты по экстракции диэтиламина из воды
с помощью толуола в насадочной колонне диаметром 0,15 .и и высотой 1,2 м.
Насадочными телами являются кольца Рагпига 12,7 .и.м. Скорости пото-
ков водного раствора диэтиламина и толуола составляют, соответственно,
6,2 З13/.и2-ч и 0,92 ,и3/.и--ч. Температура экстракции 30,8э С. Концентрации
диэтиламина в водном растворе:
1) при входе в колонну 0,252 к.ио.<ь/.и3;
2) при выходе из колонны 0,231 кмоль-м\
Концентрации диэтиламина в толуоле:
1) при входе в колонну 0;
2) при выходе из колонны 0,137 к.иоль/.и3.
При данной температуре коэффициент распределения диэтиламина
(т. е. концентрация диэтиламина в воде, разде юиная на концентрацию диэтил-
амина в толуоле) составляет 1,156.
Диспергированной фазой является толуол.
1. Определить экстракционные характеристики насадки для данных
условии.
2. Вычислить концентрации потоков, выходящих из колонны с высотой
насадки 2,5 .и.
1) Пусть водный раствор — рафинатная фаза, а толуольный раствор —
экстрактивная фаза. Тогда будем иметь:
СЕ,- 0.137; 6'^ = 0
CRt- 0.252; С„=0,23!
Из (XIV. 37) получим:
V 0 Д’Э ~ А Ea/j (('Е ~ 6£) ц
где
, л;3 экстракта ,, , ,
0,92-------г-?-----, Z = 1,2 м
.и - • ч
Далее
CEi = /п Сд( 0,865 • 0,252 -= 0,217
-4 -I- М. Батуне;». 309
и
С' =-• mCfi = 0,865 • 0,231 = 0,200
Jt2 *'»
. (0.2-0)-(0,217-ОД37) д 0132 кмом,л
1П 0,217—0,137
Таким образом, имеем
0,92 (0,137 - 0) = К'Еа -1,2 « 0,132
откуда:
0,92 - 0,137 _ „г
= 1,20,132 =0,'Э5
КЛ4ОЛЬ
.И3 Ч • ЕСе
Применяя к рассматриваемому случаю формулу K'Ra = тКЕа
найдем: / кмолъ К па 0,86а • 0,795— 0,687 , .,, к зР • ч
Имеем: КЕа = СК'Еа
где С — мольная плотность экстрактивной фазы, кмоль/м?-
Для указанных разбавленных растворов плотность и средний молеку-
лярный вес экстрактивной фазы могут быть приняты такими же, как для
чистого толуола, соответственно, 856 кг/м3 и 92,1.
Следовательно, мольная плотность
856 _оо кмоль
~ 92,1 “ ’ .и3
Таким образом
„ ,Л «.ноль
А „а = 9,3 0,79э — 7,40 —г-------------
.«•* ч • мол. доля
Количество экстрактивной фазы:
к.ноль
Е = VFC = 0,92 9,3 = 8,55 —5--•
•с м~ - ч
Высота насадочных тел, эквивалентная ступени экстракции:
Е
КЕа
8,55
7Л0
= 1,16
31
Мы также получаем:
0,92
0,795
= 1,16
м
370
Для характеристики насадки по рафинатной фазе имеем:
V» 6.2
/7 р = —°'0 Л1
R Кпа 0.68/
2) Пусть CRj — концентрация днлтиламииа в воде, а — концен-
трация диэтиламина в толуоле при выходе пз колонны с высотой насадки
2,5 .и.
Используя (XIV. 38), получим:
_ 6,2 =
«к д 0,865 • 0,92
N 11R 9,0
9,5
’ 0,277
Ri
0,252; С,. =0
-*J2
Следовательно
1п
0,277— —
Г 0 252
(1-7,78) 1-7,78
откуда
С р — 0,224 к моль!.и®
*‘2
Материальный баланс для диэтиламппа
или
откуда
1Е (СЕ, ~~ СЕг) — Р В (CR,
0,92 (С’£1 —0) =- 6,2 (0,252 — 0,224)
CEi = 0,189.
Определение диаметра экстракционной
колонны
Безпасадочная колонна. Для экстракции уксусной кис-
лоты из воды в качестве растворителя используется изопропило-
вый эфир. Процесс осуществляется в безпасадочной колонне непре-
рывного действия.
Скорости потоков: 5,1 м'Л эфира/ч и 3,4 .«3 разбавленного рас-
твора уксусной кислоты/ч. Плотность разбавленного раствора
уксусной кислоты составляет 1008 кз/.м3, его вязкость 3,1 спз.
Плотность эфирной фазы 730 кг/м3. Величина поверхностного
натяжения эфирной фазы 13 дин! см. Определим диаметр колонны.
В колонне диспергированию подвергается эфирная фаза не
только потому, что опа подается в значительно большем объеме,
по также вследствие ее легкой воспламеняемости и в этом случае
работа аппарата будет более безопасной.
24*
371
Чтобы обеспечить получение капель одинакового размера,
принимаем скорость прохождения эфира через распределительное
отверстие, равное 0,09 м/сек.
Следовательно, площадь отверстии будет:
Жда=0Ю158 Л*
Диаметр отверстии во избежание забивания их принимаем
равным 4.75 льн. Тогда число отверстий в распределителе соста-
вляет:
Имеем:
о = 13 дин/см; d# = 0,00475 Ж рр~3,1
Ор =1НО8 кг/м3; дэф = 730 кг/м*
До— 1008 —730 278 кг.'.и3; г= 0,09 л/сек
Приведенные выше данные связываются следующей эмпири-
ческой формулой, дающей возможность найти диаметр колонны:
<4 , 396</V>517pf,'f-‘9
Ду '
'+ + 0,01255
где .1% — объем сферической капли диспергированной фазы.
Таким образом, будем иметь:
13.0 - 0.00475 , 396 0.004751’1'2 • 0,0<Л547 • 3,1Л’27Я
278 ‘ 2781’3
откуда найдем диаметр капли dK = 0,007 лг.
Кроме того:
^=41=1,5
Гр 3,4
Скорость движения непрерывно протекающего раствора в мо-
мент «захлебывания» определяем с помощью эмпирической фор-
мулы:
320 \ог'•-*
278
= —---------------------------------------— Ы',0 .4 ч
1(1,483 3,10,07Г’ 10О8°'5-г 0.007и’"5С 730"’5 • 1,5"’Ча
Принимая скорость сплошной массы раствора в колонне рав-
ной 40% от предельной скорости, получим:
Площадь поперечного сечения колонны
f ..... , = 0,1 об л-.
22,4
откуда диаметр колонны 79 — 0,45 ль
Насадочная колонна. Для насадочной колонны най-
дено, что сумма скоростей движения раствора и эфира равна неко-
торой постоянной величине н, таким образом, допустимая скорость
)'’й
i '
Рис. XIV. 47. Диаграмма для расчета диаметра на-
с ад очной колонны.
потока диспергированной фазы .увеличивается с уменьшением
скорости сплошной фазы. Зависимость, изображенная на
рис. XIV. 47, позволяет вычислить диаметр колонны для экстрак-
ции жидкостей.
Пусть, например, процесс экстракции, рассмотренный выше,
проводится в насадочной колонне, заполненной кольцами Рашига
размером 25,4 .иль I о-прсжпсму эфир диспергирован. Найдем
Диаметр колонны.
Для данных колец Рашига доля свободного объема g = 0,685
и а!%А — 540 (из таблиц). Следовательно:
= ГИО-0,685s =173
Имеем для оси абсцисс:
373
Па рис. XIV. 47 этой величине соответствует значение орди-
наты при режиме «захлебывания» 320. Таким образом
ЗЗОаНр 320-173-3,1 .
а? = ------и—Н ------=----------------= За.0 ч
4 _L / ±Р* у-512 (1 + 1 .50,5)2 1008
1 1 I 1-’ I I *-Р
L \ 1 Р / J
Принимая скорость движения жидкости 40% от предельной,
получим:
u-р г- о,4.35= 14 м /ч
Площадь поперечного сечения колонны:
Диаметр колонны:
/)- 0,55 .«
Многоярусная колонна с ситчатыми тарелками. Тяже-
лая жидкость протекает вдоль каждой тарелки через капельно-
жидкую массу, направляясь к сливному порогу, а оттуда па ниже-
лежащую тарелку.
Скорость прохождения жидкости через отверстие рекомен-
дуется принять па основе опытных данных в пределах от 0,12
до 0,3 м/сек. При более пнзких скоростях диаметр колонны ста-
новится без всякой необходимости чрезмерно большим и но все
отверстия тарелки могут работать. Вместе с тем при высоких
скоростях прохождения легкой жидкости через отверстия обра-
зующиеся в большом количестве мелкие капли уносятся через
сливные устройства вслед за тяжелой жидкостью.
Определим диаметр ситчатой колонны применительно к усло-
виям тон же экстракции, которая была рассмотрена выше.
Отверстие ситчатой тарелки принимаем равным
+ - 0,00475 л
Площадь, занимаемая отверстием:
г 0,785 • 0,00475“ = 0,0000176 у2
Скорость жидкости в отверстиях принимаем
Иу--0,212 м/сек или F() = 760 л.ч
Для эфира в количестве 5,1 м3/ч количество отверстий на одной
тарелке
760 0,0000176
Располагая отверстия тарелки по углам равносторонних тре-
угольников, стороны которых составляют 19,1 лг.м, получим
площадь отверстий равную 0,118 .и2.
374
Колонна будет в состоянии «захлебывания», если значитель-
ные количества диспергированных капель будут уноситься через
слив со сплошной фазой.
Таким образом, скорость движения жидкости в сливном устрой-
стве ь’й должна быть меньше скорости осаждепия всех частиц,
кроме самых мелких капель с диаметром ниже 0,785 мм. Для
определения скорости жидкости в сливпом устройство приравняем
последнюю скорости осаждения эфирных капель размером d =
— 0,785 зьн.
Данные:
Рр—11,2 кг.'.и• ч; g — 1,27- 10s .w/ч2; До=278 « .и3
Пользуясь формулой Стокса, получим:
gAorf2 1,27-10е-278-0,00078.=? .
1,‘‘=,Л^=----------18ЛЁ2--------=1“ ', '1
Для разбавленной жидкости в количестве 3,4. м3.'Ч площадь
поперечного сечения сливного устройства равна
3 А
= .,.2
111 ’
Следовательно, площадь поперечного сечения колонны, заня-
того сливным устройством и местом для отверстий, составляет
2 0,0314- 0,118 = 0,180 лг2
Отверстия находятся по крайней мере на расстоянии 25 льн
как от стопки колонны, так и от сливного устройства. Поэтому
общая площадь поперечного сечения колонии будет 0,215 at2,
откуда диаметр колонны D -- 0,52
Экстракция твердых тел
Процессы экстракции твердых тел могут быть под-
разделены на три группы;
1) экстрагируемая масса и растворитель имеют направленную
скорость (противоточное или прямоточное экстрагирование; леп-
тонные и шнековые экстракторы);
2) экстрагируемая масса и растворитель не имеют направлен-
ной скорости (аппараты с мешалками);
3) растворитель имеет направленную скорость, а экстраги-
руемая масса — не имеет (диффузионное экстрагирование; диф-
фузор).
Ленточный экстрактор Де-Смета
Экстрактор состоит из стального корпуса 1. Экстра-
гируемый материал перемещается внутри аппарата па ленточном
транспортере 4. Шибер, снабженный указателем 23, регулирует
375
высоту слоя экстрагируемого материала от 0,6 до 1.2 .и
(рис. /XIV. 48).
Звездочки 3 вращают лепту. Над слоем материала расположен
ряд распылителей 7. Под лептой расположен ряд приемных воро-
нок 6 для мисцеллы, соответствующих количеству ступеней экс-
тракции. Каждая воронка питает соответствующий центробежный
Материал
Л шнековому испарителю
Рис. XIV. Ленточный экстрактор Де-Смета;
] — корпус; ;* — загрузочный бункер; <J—зпездочкн; 4 — ленточный транспортер;
J — разгрузочный оункер; - приемные воронки; 7 — 1(1 — форсунки (распылители);
JJ. 17 —насосы; 12, 13 —рыхлители, 11 —щеточный барабан; 15 —вращатель; la
ciTBi.p: is — подогреватели; /у — расходомер; 20 — сифонный аппарат; 21 - электро-
магнит; 2с — качаю 11’пйсн желоб; :?.у — указатель: 21, 25 — клапанные устройства.
насос 77, который в свою очередь питает соответствующий рас-
пылитель.
Каждая ступень экстрактора имеет осушительную зону для
некоторого стока растворителя. Верхний слой материала па уча-
стке осушительной зоны прочесывается грабельными рыхли-
телями 72.
Экстрагируемый материал, пройдя электромагнит 2/, через
затвор 16 поступает в бункер 2, откуда медленно увлекается лен-
точным транспортером. -11ри движении ленты материал подвер-
гается орошению через форсунки 7 и распылители 8 сначала
крепкой мисцеллой, а затем мисисллой все болео слабой концеп-
376
грации. Перед разгрузочным бункером 5 материал через фор-
сунку 9 орошается чистым растворителем, поступающим пз резер-
вуара оборотного растворителя через подогреватель 78 и рас-
ходомер Z-9.
Обработанный материал, сваливающийся с лепты в бункер J,
с помощью вращателя /5 подается в шнековый! испаритель.
При последовательном переливании мисцеллы из воронки
в воронку, свежий растворитель, насыщаясь извлекаемым мате-
риалом, приобретает прогрессивно возрастающую концентрацию.
Конечная, наиболее насыщенная ценным веществом, мисцелла
перетекает в сборную воронку, откуда направляется в мисцслло-
сборник.
Из сифонного аппарата 20 через форсунку 10 периодически
подастся чистая фильтровальная мисцелла для очистки сетки.
Мутная мисцелла, собирающаяся в нижней воронке, насосом /7
через подогреватель 18 подается в качающийся желоб 22 и оттуда
па свежий слой материала. На конце разгрузочного участка лепты
имеется рыхлитель 13. Для очистки сотки устроен щеточный бара-
бан 14. В целях автоматического поддержания уровня материала
в бункере имеются специальные клапанные устройства: 24 для
верхнего и 2а для нижнего уровня.
Корпус экстрактора па всех важных участках имеет смотровые
окпа, позволяющие наблюдать за орошением материала и состо-
янием слоя его.
Расчет экстракции на ленточном аппарате
Де-Смета
Рассмотрим единичную ступень экстракции твердой
массы.
Твердая ыасса
В кг нерастворимого остатка
В кг (А + С)
Твердая масса после экстракции
$кг В/кг ТА*С)
УРкгС/кг(А+С)
Растворитель
rfp кг раствора ТА * С)
Раствор
В. кг раствора (а*- С)
В кг нерастворимого остатка
В, кг (А ^С)
Ц кг В/кг (а * С)
yf кг С/кг /А* С)
T(j№ С/к? (Д-t С)
zf кг С/кг (А+ С)
Рис. XIV. 49. Схема к расчету экстракции твердых тел.
Схема процесса, изображенная па рис. XIV. 49. включает
в себя смешение твердой массы с растворителем и отделение нерас-
творимой фазы от раствора.
377
Примем следующие обозначения:
А — чистый растворитель;
С — растворимое вещество;
— отношение количеств твердого остатка и раствора (zlJ-Q;
F — количество растворимого вещества и растворителя в экстра-
гируемой массе;
R — количество растворителя и растворимого вещества в жид-
Рис. XIV. 50. Схема материального
баланса экстракции твердых тел.
кой фазе;
Е — количество растворителя
и растворимого веществе!
в экстрагируемой твер-
дой массе;
х — концентрация раствори-
мого вещества в рас-
творе, масс, доли;
у — концентрация раствори-
мого вещества в смеси,
масс. доли.
Таким образом, имеем:
B^NfF^-ExN1 (XIV. 39)
баланс по компоненту С:
1'Ур + = Л1?/1 + /?Л(Х1У. 40)
Баланс по компоненту Л:
Баланс по раствору:
F . = £1-1-7?! = ^
Твердая races
В кг нерастВоринаго/ ч
F кг(А*С)/ч
кг В/кг М
yF кг С/кг (А О
РастВор
Р, кг [A
г кг С/кг (А-С)
*г
У,
Гис. XIV. 51. Схема батарейной экстракции твердых тел:
Г — 3, ... Np — экстракторы.
Перечисленные соотношения изображены па рис. XIV. 50.
Точка отвечающая составу всей смеси, должна находиться
па прямой липни, соединяющей 7?п и F.
Точки и /?!, изображающие потоки у выхода системы,
располагаются на концах липни сопряжения, проходящей
через Л/Р
378
При контактировании обрабатываемой твердой массы со све-
жим растворителем извлекается дополнительное количество цеп-
ного вещества.
Расчеты для дополнительных ступеней выполняются анало-
гично расчетам первой ступени лишь с том отличием, что обраба-
тываемая масса становится каждый раз исходным сырьем для
последующих стадий экстракции.
Для противоточной экстракции твердых тел представлена
схема, изображенная на рис. XIV. 51.
Г> а л а н с но р а с т в о р у для п р о т и в о т о ч п о й
э к с тр а к ц ни т в о р д ы х т о л
= W,-F/?V =Л7 (XIV. 42)
Величина Л/ представляет гипотетическую, — свободную от
твердой массы В, — смесь, которая получается при смешении
экстрагируемой твердой массы
с растворителем.
Равенство (XIV. 42) может
быть преобразовано так:
F-Л^Гд,
. ' 7> 1 П+1
Для первых трех ступеней
имеем:
F — Пу — II
Точка Н представляет посто-
янную разность в потоке (обычно
отрицательная величина между
ступенями). На рис. XIV. 52 эта
разность может быть представлена
в точке пересечения линий FHX
и Е 11 при их продолже-
*'р+1
Гис. XIV. 52. Равновесная диа-
грамма для процесса экстракции
твердых тел.
нпп в соответствии с характери-
стиками принятых координат. Так как выходящие из каждой
ступени потоки характеризуются практически линией сопряже-
ния, то 1‘\ будет находиться в конце линии сопряжения, прохо-
дящей через точку Линия, проведенная от Ех к II, дает
И Т. д.
Пример. Твердая масса при поступлении па экстракцию содер-
жит 20% продукта С и должна выходить пз аппарата при полупроцентном
содержании последнего, считая на сухой остаток. Расход растворителя соста-
вляет 1 кг А па 1 кг твердой массы. Раствор, движущийся через материал,
свободен от твердых частиц за исключением первой ступени, где обогащенный
раствор содержит 10% твердого во взвешенном состоянии. Определим число
ступеней экстракции
379
Опыты показывают, что количество раствора, остающегося в твердой
массе после 6 мин дренажа, зависит от содержания продукта С в растворе:
Содержание V. в растворе, %............ О 20 30
1 N, кг раствора.'кг нерастворимой
твердой массы .................. . 0,58 0,66 0,70
В этом случае линии сопряжения вертикальны, так как х = у .
Представим данные о дренаже в виде табл, XIV. 4.
Таблица XIV. 4
Данные для расчета
Содержание С в растворе, <v /О 1/Л’, иг раствора в осадке ?гг твердой массы .V у /Л , кг/С
иг твердой массы
О 0,58 1,725 0
20 0,66 1,515 0,132
30 0.70 1,429 0,210
Расчет ведем па 1 кг твердой массы, поступающей в аппарат.
В з а т р у ж а е м о и смеси:
В — 0,8 кг нерастворимого осадка;
/’ = 0,2 кг продукта С;
ЛГр — 0,8/0,2 — 4,0 кг нерастворимого осадка/кг С;
tjF = 1,0 масс, доля С.
В р а с т в о р и т е л с;
В =1,0 кг Л; .rv =0 масс, доля С
>+i np+i
3 к с т р а г и р о в а и пая т в с р да я мае с а.
Содержание С на 1 кг нерастворимого твердого остатка составляет:
При интерполировавши между равновесными данными получим:
-.с кг твердого
/V лг — 11/10 1
кг раствора
Потери нерастворимого твердого остатка с жидкостью равны:
0,8 - 0,1 = 0,08 кг
Далее:
г 0,72
/г = 1———= 0,420 кг раствора в осадке
‘'р Ll.rlo
Количество продукта С, удерживаемого осадком:
0,00503-0,72 = 0,00362 кг
Количество растворителя, удерживаемого осадком:
0,420 — 0.00362 ~ 0.416 кг
у — „ q)(j[)S(; масс, долей С в растворе с конечным осадком
0,420
380
Раствор.
Количество растворится я:
1 — 0,416 = 0,584 иг
Количество продукта С:
Рис. XIV. 53. Диаграмма для расчета числа сту-
пеней экстракции твердых тел.
Имеем:
/Д 0,584 + 0,196 = 0,780 кг осветленного раствора
= ~ 0,252 масс, долей С в жидкости
v j / oU
0,08 __ кг нерастворимого твердого осадка
0,780 ’ кг раствора
Для расчета числа ступеней экстракции пользуемся рис. XIV. 53.
Точка И1 представляет раствор в виде суспензии и поэтому она сме-
щена в точке N . Точка Н располагается, как обычно, и число ступеней
найдем равным 5.
Экстракция в шнековых (винтовых)
аппаратах
В шнековых аппаратах процесс экстракции может
осуществляться противоточным или прямоточным способом
(рис. XIV. 54).
Переход извлекаемого вещества из сырья в растворитель
можно рассматривать как с точки зрения уменьшения его
381
содержания в сырье, так и с точки зрения увеличения концен-
трации в растворителе. Найдем выражение для концентрации
извлекаемого вещества по длине аппарата. Обозначив время сопри-
косновения растворителя с обрабатываемым веществом через т,
получим:
de
— = к(е-с) (XIV. 43)
dx
— — = X (с' - с) (XIV. 44)
где с — концентрация извлекаемого вещества в растворе;
х — содержание его в обрабатываемой массе;
с — концентрация извлекаемого вещества, содержащегося на
поверхности обрабатываемой массы;
к и К — постоянные коэффициенты.
Тбеpda я
масса
Рис. XIV. 54. Шнековый аппарат.
Будем считать, что с и х связаны между собой прямой про-
порциональностью
ас'; с' = (XIV. 45)
Подставив в формулы (XIV. 43) и (XIV. 44) значение с , получим:
А =± (XIV. 46)
с/т \ а / '
= (XIV. 47)
с/т \ а / '
382
Интегрирование уравнений (XIV. 46) и (XIV. 47) дает решение
вопроса экстракции в шнековых аппаратах. Обозначим через U
и и, соответственно, массовые количества обрабатываемой массы
и растворителя, поступающие в единицу времени в аппарат. Обо-
значим концентрации извлекаемого вещества в обрабатываемой
массе и растворителе в точке поступления последних в аппарат, —
соответственно через х() и с0. Обычно с0 = 0. Количество вещества,
отдаваемого обрабатываемой массой и получаемого растворителем,
равпы между собой:
(х0 — х) U = (с—с0) и (XIV. 48)
При с0 — 0 получим:
х = х0—Jj~c (Xiv. 49)
Подставив значение ж в (XIV. 46), получим:
Умножим обе части уравнения (XIV. 50) на — 4-1),
имеем: {—уг + 1 \ de \ аб ) / ч , \ — '‘1 / г т I 1 хп и } 77“ С — С a V.U
После интегрирования получим:
In
а
----- 1
uU ‘
(XIV. 51)
=~ч^+<)+с
Постоянную интегрирования С найдем, исходя из того сообра-
жения, что в начальный момент т = 0 концентрация с также
равна нулю. Это нам дает
С—In
(X
Отсюда:
10 [v-(7^+‘)=-Ч^+,)+1пV <xlv-52>
Далее, обозначив время пребывания растворителя в аппарате
через тс, а время пребывания обрабатываемой массы через тА-,
получим:
Разделив первое уравнение па второе,, имеем:
de к dxc
dx К dxx
383
Из соотношения (XIA . 48) найдем:
de U
dx it.
Сравнивая между собой два последних равенства, получим:
U _ A- dTc
и К dxx
Коэффициенты к и К являются постоянными. Установим,
в каком случае они будут равны между собой. Этому условию
удовлетворяет равенство:
utc = (7тх
(XIV. 53)
Принимаем, что к и Л' £ , где F — поперечное сечение
аппарата, ар — постоянная, зависящая от природы обрабаты-
ваемого вещества, растворителя и температуры. Подставим эти
значения к и К в (XIV. 52), предварительно записав это равенство
следующим образом:
После подстановки получим:
Так как их = г, где v — объем экстракционной системы но
растворителю (воде), то
где l= ~ — длина аппарата.
Обозначив и!U через р, получим:
(o-(-a)j — —-|т"(0 1-“)
или
Обозначив
и решая последнее уравнение относительно с, получим оконча-
тельно следующее выражение для концентрации с извлекаемого
вещества по длине аппарата
(X1V. 54)
384
Экстракция в аппарате с мешалкой
Объем экстрактора У непрерывного действия опреде-
ляется так:
V =
где Ут — производительность аппарата, л1Л'лш«;
т — продолжительность пребывания смеси в аппарате, мин.
Значение т находим из уравнения кинетики процесса экс-
тракции.
Пример. Пусть в лаборатории проведена пробная экстракция
продукта из массы, содержащей 52% ценного вещества. Количество взятой
пробы 25 г. Количество растворителя (бензола) 100 г. При этом были полу-
чены следующие данные:
Бремя, мин ...........
г/100 г бензола . . .
0 10 20 30 40 50 60 70 80 90
0 2.15 3.4 4.32 5.10 5,73 0.32 6,78 7,04 7,42
Рис. XIV. 55. Определение порядка процесса экс-
тракции твердых тел.
Растворимость продукта (предельная) в бензоле составляет 11,7г/100г
бензола. Найдем зависимость, которая представляла бы полученные резуль-
таты. Сначала было принято, что скорость извлечения продукта пропорци-
ональна количеству остающегося нерасторопного продукта, что предпола-
гает процесс первого порядка. Для того чтобы это проворить, была построена
диаграмма (рис. XIV. 55), па которой точками представлена мависимость
логарифма числа, выражающего количество нерастворенного продукта от
времени. Если провести через эти точки линию, то опа окажется кривой,
а по прямой, что означает, что экстракция продукта бензолом не является
процессом первого порядка.
25 Л. М. Батунер.
385
Предположим, что скорость извлечения продукта пропорциональна коли-
честву перастворснпого продукта, а также разности между фактической
концентрацией раствора в данный момент и концентрацией его в состоянии
насыщения, т. е. производная
где IV — количество перастворснпого продукта ко времени т, г;
Л' — концентрация продукта в насыщенном растворе, г/lQO г;
С — концентрация продукта в растворе ко времени т, г/ЮО г.
Полученное дифференциальное уравнение содержит три переменных IV,
С и т. Но IV и С связаны между собой в силу того, что общее количество про-
дукта равно
С-ЬIV = 25-0,52 = 13 г
Отсюда
И = 13 — С и dW — —dC
Следовательно.
dC
-±- = К (13-С) (11,7-0
ЙТ»
В результате интегрирования имеем:
Г 13 1
2,303 |jg(13-0-lg(ll,7-Cj-lg =(13-11,7)ЛЧ
Если эта зависимость действительно выражает данный процесс,
, 13—С
то построив диаграмму для величины 1g —— относительно т, мы должны
получить при использовании опытных данных ряд точек, лежащих почти
на прямой линии
Такая прямая в действительности и получается на графике с достаточной
степенью точности.
Расчет экстракции в диффузорах
На рис. XIV. 56 изображен типовой диффузионный
аппарат, а на рис,zXIV. 57 — диффузор с гидравлическим
затвором.
На рис. XIV. 58 представлена диффузорная батарея.
Примем следующие обозначения'
т — количество ценного продукта в твердой массе к моменту т, кг\
Л — высота слоя твердой массы в аппарате, .и;
Л — фильтрующая поверхность, .и2;
V — объем твердой массы, .w3;
х — количество ценного продукта в растворе, кг;
и — объем раствора, л(3;
с — концентрация раствора к моменту г, кг '.ч3;
т — время от начала процесса экстракции, мин-,
IV— объемная скорость движения раствора, м3/мин • з<А
При постоянном режиме работы аппарата можно принять,
что концентрация цепного продукта в растворе прямо нронор-
380
циональпа его концентрации в твердой массе в любой момент
процесса, т. е.
dx кт
~dv ~ V
(X1Л . 55)
В следующий момент концентрация раствора изменится на
некоторую величину de, тогда будем иметь
Рис. XIV. 56. Диффузионный
аппарат:
i — крышка; 2 — днище ,з — сет-
ка; -1 — корпус.
Рис. XIV. 57. Диффузор с гидравлическим
затвором: ,
1 — контргруз; 2 — гидравлический цилиндр;
3 — шарнир; 4 — нижний фильтр; _ 5 — кольцо;
6 — сетка; 7 — тяга.
ИЛИ
de = — ду- dx
(XIV. 56)
При постоянной скорости раствора имеем
Ф- = ЛН' (XIV. 57)
ах
Из этого равенства, а также из отношения dx.’dv — с, следует, что
(It
-р- = Л1\с (XIV. 58)
аХ
25*
387
Подставляя (XIV. 58) в формулу (XIV. 56), получим:
, de А*Н- tit
dc=--------------- или -----~-----------—-
I с Л
Интегрируя, получим:
1нс
(XIV. 59)
где с' — концентрация раствора в начале процесса.
Рис. XIV. 58. Батарея диффузионных аппаратов:
1 — теплообменник; 2 — диффузионный аппарат; з — сетка.
Уравнение (XIV. 59) может быть представлено в следующем
виде:
_ Мут
с—с'с L (XIV. GO)
Определив для каждого отдельного случая экспериментально
значение можно установить при помощи формулы (XIV. 60)
концентрацию раствора в любой момент от начала процесса,
а также влияние изменения скорости экстракции и толщины стоя
массы на ход процесса.
Критериальное уравнение
проточной экстракции
В экстракционных процессах диффузии почти неизменно
сопутствует движение либо жидкости — растворителя, либо
экстрагируемой массы, или той и другой. Эти факторы влияют
существенным образом на процесс экстрагирования. Поэтому
383
уравнение диффузии оказывается недостаточным для характери-
стики экстрагирования в проточной системе. В нашем случае
имеет место движение жидкости — растворителя.
Дифференциальное уравнение для диффузии в движущейся
среде имеет вид:
д- с д-с д'-с \ де де , дс
Т^- I —ПТ Ь ~~— ) = «’.V Т" - -л- ” гг2 -----
дх- ду- 1 dz- J дх - ду dz
(XIV. 61)
где с — концентрация вещества в растворе;
D — коэффициент диффузии.
Уравнение (XIV. (И), описывающее распределение концен-
трации в движущейся среде, аналогично дифференциальному
уравнению конвективной теплопередачи.
Так как, кроме концентрации, переменной является еще ско-
рость, то уравнение (XIV. 61) должно рассматриваться в сово-
купности с дифференциальными уравнениями движения жидкости
и неразрывности (сплошности) потока при экстрагировании.
Уравнение движения в данном случае (жидкость несжимаемая)
имеет вид:
(XIV. 62)
При установившемся движении —= 0.
Для вывода уравнения сплошности применительно к проточной
экстракции примем следующие обозначения:
с — концентрация извлекаемого вещества в жидкости. кг/м'3;
z — концентрация извлекаемого вещества в обрабатываемой
массе, ка/.ч8;
V — количество проходящей через аппарат жидкости,
т — объем жидкости на единицу сечения аппарата (в №) и па
погонный метр длины (или высоты) аппарата, м3/м*-м;
п — объем обрабатываемой массы в том же выражении, лг3Лн2-at.
Обозначим через dx бесконечно малое приращение пути х,
проходимого потоком вдоль аппарата за время Лт. Рассмотрим
часть аппарата между сечениями в точках х и х -|- dx.
Через с обозначим среднее значение с в течение времени Ат.
Тогда сУАт представляет собой поступающее через сечение
в точке х за время Ат количество извлекаемого вещества и с de
равно среднему .значению с в точке х 4- dx в течение времени Лт.
Отсюда (с de) УДт равно количеству извлекаемого вещества,
выходящего через сечение в точке х -j- dx за время Лт.
389
В результате вычитания находим, что увеличение количества
извлекаемого вещества в пределах пространства между сечениями
в точках жиж -г dx за промежуток времени Дт составляет — Vdc Дт.
Допустим, что с' — среднее значение концентрации с в пре-
делах пространства, соответствующего отрезку пути dx.
к моменту т.
Тогда mc'dx выражает количество извлекаемого вещества,
содержащегося в жидкости в этом пространстве к моменту т.
Пусть z равно среднему значению концентрации z для обра-
батываемой массы в пространстве, соответствующем отрезку dx
к моменту т.
Тогда nz dx равно количеству извлекаемого вещества, содер-
жащегося в обрабатываемой массе для этого пространства к мо-
менту времени т.
Ко времени т 4- Ат величина с' становится равной с — Ас'
и z’ принимает значение z Дг'.
Поэтому выражение
[wi (с' -}- Ас') — п (z' -[- \s')] dx
представляет собой общее количество извлекаемого вещества ко
времени т Дт. Следовательно
[иг (с' { Ac')-[-n (z'-f-Дг')] rfx—(тс' ) ггз') «х = [иг Ac' 4~н \='] с?г
есть увеличение количества извлекаемого вещества за промежу-
ток времени Ат.
Приравнивая вновь полученное выражение первоначальному,
получим:
V tie Ат -I- т Ac' dx -|- п As' dx —О (XIV, 63)
Разделим уравнение (XIV. 63) на с?хДт и каждый из этих мно -
жителей устремим к нулю. После предельного перехода найдем
окончательно:
де де dz
р д. ,н -к — = 0
dx дх дх
(XI V. 64)
Уравнение (XIV. 64) и есть уравнение непрерывности (сплош-
ности) для рассматриваемого здесь процесса проточного экстра-
гирования.
Уравнения для одномерного движения:
de „ d-e
m — = D
dx dx-
dm
dp d’u
if -r- Q= og— — 4 ii—;
dx dx dx-
de de , dz
V -y- 4- tn п -4— ' (I
dx dx dx
(XIV. 65)
(XIV. 66)
(XIV. 67)
390
Совместное решение уравнений (XIV. 65)—(XIV. 67) дает иско-
мое математическое выражение для моделирования процесса
проточного экстрагирования. Примем масштабные множители
в соответствии с теорией подобия
х' :с АЬ id’ W = Л™; х’ X ~ Лт, V -^v
nt' Ап’ п' А . е' т= ло; s' = ил’ — = А< * ц в
т. п дп g
де' — /1 Ос' да = "Arad ст
дх1 их grad сх’ irt' дх
dz' д2 = л дю' дю — А grad го
От' дх grad z' дх' ' дх
Из этих уравнений имеем:
^gradCj. Aradc^A A^dc
-^grad Ст С)Х ^gradc./ х <•’—А
Далее:
grad г ^г — ^grad zA Az^z
Aradw dw — Arad цЛ — At\ du?
Таким образом, имеем:
дсг aic'
Ox' Лс дх'2 Ас
де /1^ ’ с?-с ^2
дх дх-
дсг dz'
дх' Л с дх' Лг
дс ~ ~А~ ’ dz ~ Л~
----- L ----------- т
дх дх
дю'
дх' ^гох
&Ю ~ At
дх
д-ю'
Л у-л
А1
Дифференциальные уравнения диффузии для модели напишем
в соответствии с уравнениями (XIV. 65)—(XIV. 67), причем
391
входящие сюда величины, в отличие от величин для производствен-
ного аппарата, обозначим со штрихами
В эти уравнения мы можем, согласно приведенным выше соотно-
шениям, подставить
.г' = Atx', гг' = Лт' = Л т; с' = Лсе
о' = Л о; ш' -- Атпг; п' = 1)’ = ADf)
где величины без штриха относятся к производственному
аппарату.
Таким образом, для модели получаем из уравнения (XIV. 0-5):
1 дс л л А
A.aw —----— — AnD
А { dx v
dic
Уравнение (XIV. 06) дает:
дю
71 о
Л/
d-w
дх-
Из уравнения (XIV. 67) имеем:
Лс дс Ас de Иг oz
ЛvV —------— - Л„,т —— ——- А ..п —--— = О
v Ai дх ' tn И йт п Л дх
По аналогии с теплопередачей можем написать выражение
для коэффициента экстракции аэк:
йг' t
дх.’
с
Toi да
л^лг
Ч. “"л]л;
с —со
392
Принимая во внимание только такие процессы, для которых
значения масштабных множителей перед производными можно
было бы сократить, получим:
= (XIV. 08)
'h
-'lu’r
л- л‘="‘Л’ = л7 - (XIV. to)
Чт7 = '|».# = '|,.-Т- (XIV. 70)
' ‘ Z1T -*т
-3 Г>
(XIV. 71)
Следовательно, если численные значения масштабных множи-
телей удовлетворяют уравнениям (XIV. 68)—(XIV. 71), то в диф-
ференциальных уравнениях эти множители могут быть сокращены
и для модели остается система дифференциальных уравнений,
которые полностью идентичны уравнениям производственного
аппарата.
Представим уравнения (XIV. 68)—(XIV 71) в более удобной
форме. Из уравнения (XIV. 68) имеем:
.'Itc —
At
Отсюда следует
wl w'l*
D ~ ~1У~
T. e. имеем аналог критерия подобия Пекле:
Из уравнения (XIV. 69):
А «:•! и.-Л- (
или
wZo iv'l'n'
—- ~-------7^- — Re
И н
а к
Из уравнения (XIV. 60) получаем:
. Лс . Ас
-*г —;— — Лгп -:—
Л1 Ах
393
или
<1’1
Vr _ У'т'
ml т'Г
Критерий Фй характеризует кратность обмена растворителя.
Далее:
или
Г'ст У'с'т'___
--Г- — -
гиз п I Z
Критерий подобия Ф2 характеризует степень экстрагирования.
Имеем также:
или
тс т'с’
nz n'z’
ИЛИ
^L = 5kL=Nu'
D W
т. e. получаем аналог критерий Нуссольта.
Его численные значения для всех потоков с одинаковыми Re,
Ре' и т. д. равны между собой и зависят только от значений этих
величин.
Следовательно, при геометрически подобной! границе потоков:
Xu' =/0 (Re, Ре/, ФР Ф2» Фэ)
где /0 — функция, устанавливаемая экспериментально.
Вместо критерия Ре' можно ввести и другой, который полу-
чается в результате иного комбинирования уравнений. Весьма
удобным является аналог критерия Рт', а именно:
wl
Re ml U
v
где v — коэффициент кинематической вязкости.
Критерий Рт' содержит в себе только физико-химические
постоянные вещества, находящегося в движении.
Тогда уравнение проточной экстракции можно написать так:
Nu' = / (Re- Рг', ФР Ф2, Ф3)
где f — другая функция.
394
Искомой величиной служит коэффициент экстракции аэк на
поверхности раздела фаз:
Nii'/}
«:»{ =--у-
L
где «эк — коэффициент экстракции, л.ч;
I — характерный линейный размер, at;
D ~ коэффициент диффузии, м~/ч.
Зависимость между критериями подобия обычно представляется
в виде степенных функций; в данном случае имеем:
Nu' =i|!
где ф, а, Ъ, с, d и е являются постоянными и отвлеченными чис-
лами, определяемыми экспериментально.
Глава XV
Адсорбция
Адсорбция — частный случай более общего явления,
называемого сорбцией. Сорбция — это поглощение веществ, вхо-
дящих в состав одной фазы, другой, более плотной фазой, назы-
ваемой сорбентом.
Нод адсорбцией обычно понимают поглощенно разреженного
вещества поверхностью чаще всего твердого поглотителя, назы-
ваемого адсорбентом.
Резкой границы между явлениями адсорбции и абсорбции
провести нельзя, так как известны случаи поглощения твердыми
фазами, при которых процесс адсорбции сопровождается процес-
сом абсорбции с образованием твердых растворов.
Адсорбционная волна
Рассмотрим случай адсорбции ценного вещества из
бинарного раствора, находящегося в жидком или газообразном
состоянии. Обозначим концентрацию извлекаемого вещества через
Со; жидкость пли газ проходят непрерывно сверху вниз через
слой адсорбента, свободный к моменту начала процесса от адсорб-
гива.
Верхний слои твердого адсорбента, находясь в соприкоснове-
нии с раствором, поглощает извлекаемое вещество сперва доста-
точно быстро, а остатки его удаляются при прохождении раствора
через нижние слои адсорбента. Поток жидкости или газа при
выходе из аппарата практически свободен от растворенного ве-
щества, концентрацию которого обозначим через Са (XV. 1).
По мере прохождения жидкости через слой твердой массы
зона адсорбции передвигается вниз подобно волне со скоростью,
которая очень мала по сравнению с линейной скоростью жидкости.
Через некоторый промежуток времени твердая масса насыщается
примерно наполовину извлекаемым веществом. 1(о концентра-
ция Сь в выходящем потоке все еще равна пулю.
В дальнейшем нижняя часть зоны адсорбции достигает осно-
вания слоя адсорбента и концентрация извлекаемого вещества
в потоке жидкости на выходе резко увеличивается до значения Сс.
В этом случае говорят, что система начинает работать с «проско-
ком» Причем рассматриваемая .здесь концентрация теперь воз-
Рлс. XV. I. Схема адсорбционной волны.
растает быстро и по мере того, как зона адсорбции проходит через
основание слоя адсорбента приближается к начальному значе-
нию Со.
Кривая выходной концентрации в промежутке между с и d
называется кривой «проскока».
Если раствор продолжает двигаться вниз, то поглощение будет
незначительным, так как адсорбент практически находится пол-
ностью в равновесии с раствором. Эти кривые концентрации назы-
ваются иногда адсорбционными волнами, а зона адсорбции носит
название фронта адсорбции. II, наконец, имеется так называемый
критический минимум высоты слоя адсорбента, уменьшение кото-
рого вызывает очень быстрое возрастание концентрации раствора
на выходе из аппарата.
По мере прохождения зоны адсорбции через слой адсорбента
концентрация адсорбтива в последнем непрерывно увеличивается
аналогично противоточному процессу с бесконечным числом сту-
пеней.
Кинетика адсорбции в аппарате
с неподвижным слоем адсорбента
Пусть кривая «проскока», изображенная на рис. XV. 2,
получена в результате прохождения раствора через слой адсор-
бента со скоростью G кг/ч'М2 и с начальной концентрацией Уо кг
извлекаемого вещества на 1 кг растворителя.
Через некоторый промежуток времени количество потока на
выходе из аппарата составляет ТУ кг/м1 поперечного сечения
аппарата. Кривая проскока представляет линию с крутым из-
менением хода и поэтому концентрация извлекаемого вещества
в потоке быстро возрастает (примерно от 0 до ее начального зна-
чения) .
Рис. XV. 2. Кривая проскока для адсорбиру-
емого вещества.
Некоторая величина Ув на диаграмме выбрана произвольно
для обозпачеяия концентрации проскока. Причем адсорбент рас-
сматривается насыщенным при условии, когда концентрация рас-
твора достигла некоторого значения У , близкого к У .
Рассмотрим величину 1УВ, обозначающую количество жидкости,
прошедшей к моменту появления проскока, а также вид кривой
в промежутке между WB и WE.
Количество жидкости, которое прошло через твердый слой
с момента появления «проскока», равно
”'а = иу-И'в
Зона адсорбции с постоянной высотой Za представляет ту
часть насадочного слоя,’в котором происходит изменение кон-
центрации от Ув до YЕ в любой момент времени.
398
Обозначим через та время, необходимое для смещения зоны
адсорбции, измеряемой в середине фронта. Тогда будем иметь:
та = —^- (XV. 1)
Обозначив через т время от начала процесса до того момента,
когда адсорбтив достигнет конца слоя адсорбента (срок службы
адсорбента), получим:
ть- — (XV. 2)
Если высота слоя адсорбента Z (в л), ат,,- время, необходи-
мое для образования зоны адсорбции, то
Za = Z \ (XV. 3)
ТЛ’ — I/,’
Количество извлекаемого вещества, остающегося в зоне адсорб-
ции от момента образования «проскока» до состояния насыщения,
равно U кг извлекаемого вещества на 1 № поперечного сечения
аппарата Эта величина изображена на рис. XV. 2 заштрихован-
ной частью площади
«д-
Г (Уо— (XV. 4)
С другой стороны, при полном насыщении количество извлека-
емого вещества будет У0Жа кг/м2.
Следовательно, в точке «проскока», когда зона адсорбции на-
ходится еще внутри аппарата, адсорбирующая способность ее
составляет:
I (У.-У)ЛГ
U _ ИД;____________
1 oil а ©И' (1
(XV. 5)
Величина / = 0 при полном насыщении рассматриваемой зоны.
Тогда время образования зоны в верхней части насадочного слоя
тр должно быть в основном равно та, т. е. времени распростране-
ния зоны па расстояние, равное ее высоте. Если же f = 1 и адсор-
бент почти не содержит извлекаемого вещества, то время образо-
вания зоны адсорбции должно быть очень малым с приближением
к нулю. Эти граничные условия представляются следующим ра-
венством:
Т;,.=--(1-/)То (XV. 6)
Формулы (XV. 3) и (X.V. 6) дают:
гу г/ rf 11 С7
Тд-(1-/)тЛ д Н£-(1-/)И„
(XV. 7)
399
Адсорбер высотою Z содержит в расчете па единицу площади
поперечного сечения Zq кг твердой массы, где о — насыпная
плотность адсорбента.
Если бы система была в равновесном состоянии при полном
насыщении адсорбента с концентрацией X,f кг извлекаемого ве-
щества/кг адсорбента, то количество извлекаемого вещества соста-
вляло бы ZpX"r кг. В точке «проскока» зона адсорбции Zft еще
находится внутри аппарата у его основания. По остаточная масса
насадочного слоя Z — Z„ уже насыщена.
Рис. XV. 3. Схема стационарной адсорбции
газа:
а — схема цетеков; б — рашювеспая кривая и рабочая
линия.
Поэтому количество извлекаемого вещества (в кг) к моменту
образования «проскока» будет:
(Z-Za)oXT^ZaQ}Xr
Степень насыщения выражается так:
(7'-7'а)^т + Ха^ХТ Z-(i-f}Za
1 _ _------z----- (Х v •8)
Зона адсорбции в неподвижном слое адсорбента в действитель-
ности передвигается через твердую массу. Вообразим, что твердая
масса поглотителя проходит вверх в противоположном направле-
нии к раствору с достаточной скоростью таким образом, что зона
адсорбции остается стационарной (рис. XV. 3).
439
Пз материального баланса будем иметь:
С(Го-О) -Ь(Л’Т-О)
или
Л _ г„
с хг
Так как рабочая линия проходит через начало координат, то
на любом уровне аппарата концентрация извлекаемого вещества
в растворе У и в адсорбенте X находятся в таком соотношении:
GY = LX
(XV. 11)
При бесконечно малой высоте dZ имеем:
G dY =KYa (У — У') dZ (XV. 12)
Число ступеней адсорбции:
». ( dY Za ________ Za
J У—У ~~ II ~~ G!Kva
(XV. 13)
Для любого значения Z < Za, принимая Я постоянной с из-
менением концентрации, получим д. я Z при любом значении У:
J у - У"
Z 1Г'-И’В ул
На (XV. 11)
f (1Y
J у —У
у в
С помощью (XV. 14) представляется возможность построить
кривую «проскока» нутом графического вычисления интегралов.
Продолжительность
неустановившейся адсорбции
Воздух при t — 2(5.6° С и 1 ат. с влажностью 0,00267 кг
воды/яз сухого воздуха подвергается обезвоживанию путем про-
пускания его через аппарат с неподвижным слоем силикагеля, на-
сыпная плотность которого 670 кз-'.w3. Диаметр частиц силикагеля
в среднем 1,73.«.и, а наружная поверхность частиц равна 2,16.н2,'кг.
Высота слоя адсорбента 0,6 .я. Массовая скорость 480 кг сухого
воздуха ,Н“ -ч.
Процесс адсорбции протекает изотермически. Точка «проскока»
соответствует степени обезвоживания, наступающего в тот
26 л. м. Патунер. 401
момент, когда выходящий воздух имеет влажность 0,0001 кг влаг и'кг
сухого воздуха.
Адсорбент считается отработавшим, если влажность воздуха
при выходе из аппарата составляет 0,0024 кг влаги'кг сухого
воздуха.
Коэффициент массопередачп выражается следующим образом:
Рис. XV. 4. Данные о равновесном
состоянии системы.
У_. а столбец 2 — соответственно
Куа ж= :jUW6’«><35 .
1 .«•’ ч • Д1
Г „, „ - воды
А а = 3460 ----5-гг
й .uJ-4-AX
Определим время, необходи-
мое для достижения точки
«проскока». Данные о равно-
весном состоянии системы
изображены на рис. XV. 4.
Влагосодержание в выхо-
дящем из аппарата воздухе
настолько мало, что рабочая
линия на диаграмме может
быть проведена через начало
координат при пересечении
равновесной кривой в точке
/о = 0,00267 кг влаги/кг воз-
духа*.
Кроме того, мы имеем
Y = 0,0001 и У£ - 0,0024.
В табл. XV. 1 столбец 1
даст значения Yдля рабочей
линии в пределах между Yn и
значения Y’, взятые на рав-
новеспой кривой для одних и тех же значений X. Пользуясь
этими величинами, составляем столбец 3.
Построив кривую в координатах Y (абсцисса) и у 1 - (орди-
ната), выполним графическое интегрирование между каждым
значением У в табл. XV. 1 и Ув для получения столбца 4, где
представлены числа ступеней адсорбции соответственно отдель-
ным значениям У. Общее число ступеней массоперсдачи, согласно
формуле (XV. 13), равно Лг — 9,304.
Разделяя каждую величину в столбце 4 на 9,304, найдем зна-
чения для столбца 5 в соответствии с формулой (XV. 14). Данные
столбца 6 получены путем деления каждой величины столбца I
на Уо.
Па рис. XV. 5 изображена так называемая кривая «про-
скока».
402
Таблица XV’, /
Данные для расчета
Y Y- 1 Г dY W — И'д VV а У у«
У - у- J У-у-
1 2 3 4 5 с
ув -0,0001 0,00003 14300 0 0 0,0374
0,0002 0,00007 7700 1,100 0.1183 0,0749
0,0004 0,00016 4160 2,219 0,2365 0,1198
0,0006 0,00025 3030 2.930 0,314 0,225
0,0008 0,00041 2560 3,487 0,375 0,300
0,0010 0,00057 2325 3,976 0,427 0,374
0,0012 0,000765 2300 4,438 0,477 0,450
0,0014 0,000995 2470 4,915 0,529 0,525
0,0016 0,00123 2700 5,432 0,584 0,599
0,0018 0.00148 3130 6,015 0,646 0,674
0,0020 0,00175 4000 6,728 0,723 0,750
0,0022 0,00203 5880 7,716 0,830 0,825
У = 0,0024 0,00230 10000 9,304 1,000 0,899
Формула (XV. 5) может быть представлена в таком виде:
откуда следует, что / равно пло-
щади под кривой на рис. XV. 5.
Графическое интегрирование
дает / — 0,530. При массовой
скорости воздуха, равной
480 ка/лГ’-ч, для коэффициента
адсорбции имеем:
Куп=3000-480°’133 = 44400
* .и3 • ч • ДУ
II
Рис. XV. 5. Диаграмма для графи
ческого интегрирования в расчетах
адсорбции газа.
Л .« = 3460
кг • воды
.и3 • ч ДХ
По рис. XV. 4
Хт = 0,0858
кг - воды
кг геля
26*
-•03
Применяя (XV. 10), найдем:
= У0С = 0,00267 480 _
Хт ~ 0,0858
Среднее значение тангенса угла наклона равновесной кривой
-= 0,0185
откуда
тС 0,0185-480 п _
ТГ = —ITS ~
Из теории диффузии имеем:
по газовой фазе
п G 48о ,, „ <,
Куа - 44400 - 0,01131
II, - -Л- 12,8 — = 0.0035 .u
но твердой фазе
К tJi 3460
Высота, эквивалентная ступени адсорбции, составляет
И = nG -| — IIL = 0,011 -ь 0,7 • 0,0035 = 0,01345 л
Из (XV. 13) имеем:
Za = NH = 9,304 • 0,0134 = 0,125 .и
Поскольку высота слоя адсорбента равна 0,6 м, то из (XV. 8)
найдем степень насыщения адсорбента
Z-(i-f)Za _ 0,6-(1-0,53)0,125 по
Z “ 0,6
илп 90%.
Для насадочного слоя силикагеля имеем 0,6 ,и3Лм2 поперечного
сечения.
Количество силикагеля;
0,6 • 670 — 402 кг/.и2
При 90%-пом насыщении содержание влаги в адсорбенте будет
402 • 0.90 • 0,0858 = 31 кг поды /.и.2
С воздухом поступает;
480 • 0,00267 - 1,28 кг виды /л2 • ч
404
Следовательно, время, необходимое для достижения указан-
ного выше содержания влаги в воздухе («точки проскока») равно:
31
1,28
24,2 ч
Кондиционирование воздуха.
Увлажнение воздуха в потоке
Экспериментальное изучение процессов диффузии для
газов и жидкостей в аппаратах с пасадкой и с движением парал-
лельно плоским стенкам приводит к критериальным формулам
для массопередачи, представленным в табл. XV. 2 н на соответ-
ствующем ей рис. XV. 6.
Таблица XV. 2
Крит»риальныс формулы массопередачи
Кри- вая Условия движения потока Л' У ИПСДСЖЫ sc
1 Внутри труб Re * • J — Sc ’3-- 0,6 -4- 3000
J D fl M
« Г г ' = 7Asc-
2 Движение газов че- рез пучок труб Re a,.P , J Г) - Q M 0,6 4- 2,6
Газовый поток, омывающий сфери- ческое тело Движение газов па- раллельно плоским стоикам Движение жидко- стей через насадку Rc-ScV’ Re Нс & u,.d 10-:‘ I 0.6 2,7 0.6 2,1 10690
3 4 5 JD - }) n(.P ,, z,rio- = „ Scn,6s io-1
6 Движение газов че- рез насадку Re Jn-1CH = = io-1 ''ЛГ 0,6
В табл. XV. 2 приняты следующие обозначения:
— безразмерный комплекс величин [/ (Не, Sc)];
— коэффициент массопередачи для жидкостей;
«с. — коэффициент массопередачи для газов;
405
Рис. XV. ti, Графическое изображение зависимостей для расчетов кондиционирования воздуха:
/ —б1 — номера кривых по табл. XV.
— коэффициент массопередачи для газов;
d — диаметр частиц;
Р — давление;
— расход газа;
Л, — расход жидкости;
j — порозность насадки.
Определим скорость испарения воды из сосуда, установлен-
ного в виде дна для газохода, через который проходит воздух
со скоростью 6 м.'сек. Площадь поперечного сечения квадрат-
ного сосуда 0,09 at'2; температура поверхности воды 37,8° С. Тем-
пература воздуха 60° С при 1 атм, парциальное давление водяных
паров в воздухе 0,0315 шпм.
Плотность воздуха:
оп = 1.21)3 = 1 ,06 кг/.н»
2/3 — 60
Вязкость воздуха р = 0,0195 спз, или [1”= 0,07 кг'м-ч. Длина
стенки сосуда I — 0.3 at.
Критерий Рейнольдса:
Р.С _ iSt = оз-в-иу-жю = 97300
р 0.0/
Коэффициент диффузии I) для паро-воздушной смеси при
25,9° С составляет;
1} — 0.258 см2/сек
В пересчете па 60° С получим:
D ₽- 0.258 (-£'.? Г *— 0.306 с.ч2/сек
\ 213 — 2о,9 /
или
/9 = 0.11 .«2,4
Критерий Шмидта:
Чг М _ °’07 _пг
оиЯ 1.06-0.11
По рис. XV. 6 и табл. XV. 2 пайдем (линия 4)
J D = 0.0037 = П(;^~С1> Sc2/ 3
° м
причем
,, 6-3600-1,06 клю.ть
6 » = ~М~ =------------- '
где М = 28.7 — средний молекулярный вес воздуха.
Парциальное давление водяных паров па поверхность воды
при 37,8° С:
Рдг~ 0’0475 «т.51
407
Парциальное давление водяных паров в газовом потоке:
рл q = 0 >0315 ат м
Парциальное давление воздуха па поверхность раздела фаз:
1 — 0,0-475 = 0,9525 атм
Парциальное давление воздуха в газовом потоке:
pIiG = 1 — 0,0315 0,9685 атм
Следовательно, среднее парциальное давление воздуха
будет:
0,9685 + 0,9525 Лпг|
Рп.ср =-------------= 0,961 атм
Коэффициент массопередачп составляет:
0,0037 -С'и 0,0037-796 , клоль
<+ = -------гт- =-------тт-^4,18—;--------
Т'в.срЬс'3 0,961-0,6'3 .w-4-fim.u
Количество испаряющейся воды в соответствии с уравнением
Л л = аСг^РА
будет:
na = aG 777 4118 (0,0-475 — 0,0315) -0,67
Скорость испарения воды:
L = 0,67 0,09 18,02 = 0,98 кг/ч
Определение количества адсорбента,
размеров адсорбера
и перепада давления
Регенерационная установка должна улавливать 350 кг ч
паров этилацетата из смеси с воздухом, в котором содержание
указанных паров составляет 28,8 ка/1000 № воздуха при <32.5° С
л 1 ат. Адсорбентом является активированный уголь. Средний
диаметр частиц 0,00273 .и. Плотность активированного угля
720 кг/м3. Поглотительная способность активированного угля
равна 0,45 кг иаров/кз угля до состояния «проскока».
Насыпная плотность активированного угля 480 кг Aw3.
Продолжительность процесса адсорбции, включая и время
регенерации адсорбента, равна 3 ч. Определим необходимое коли-
чество активированного угля, размеры насадочного слоя угля
и перепад давления в нем.
В течение 3 ч пары этилацетата адсорбируются в количестве:
3-350 = 1050 кг
408
Количество активированного угля, необходимого для насадки,,
равно
1050/0.45 = 2350 кг
Принимая для установки 2 аппарата, из которых один в работе,
а другой используется для регенерации адсорбента, получим
общее количество угля:
2-2350 — 4700 кг
Объем насадочного слоя в аппарате будет:
2350/480 = 5 -и3
Устанавливая высоту слоя активированного угля 0,45
получим площадь его сечения:
5/0,45=11,1 м-’; F = 2,1 X 5,2 .м2
Перепад давления в слое активированного угля может быть
вычислен с помощью следующей эмпирической формулы:
ДР _ 150(1-^) , „„
Z ‘(1-1)6'=- Re
где ДР — перепад давления, «/at2;
Z — высота насадочного слоя угля, л;
Е, — порозность насадочного слоя;
d — средний диаметр частиц, .и;
G — фиктивная массовая скорость газа, кг‘м'2-ч;
ог — плотность газа, ка/.м3.
Вязкость воздуха при 32,5° С и 1 ат : ц = 0,018 спя или
0,0620 кг/м-ч.
Плотность воздуха 1,158 кг/м3.
Объем твердых частиц в насадочном слое активированного
угля:
480/720 = 0,067 .«3
Для свободного пространства в насадочном слое имеем
§ = 1-0,667 = 0,333
Объемная скорость воздуха:
350 - 1000
28,8
= 12180 я’/ч
или
12180* 1,158
11,1
= 1265 кг/м2 • ч
Линейная скорость 0,304 м/сек.
Число Рейнольдса:
Re=
dC
V
0,00273-1265
“ 0.0620
409
Подставляя найденные значения получим:
АР • 36002 • 0.3333 0,00273 1,158 _ 150 (1 - 0,333) , л
0,45(1 —0,333) 12б5а ~ 56
откуда перепад давления в слое адсорбента составляет
ДР = 1180 н.лг или 115,8 лм водяного столба.
Переходный режим работы аппаратов
с неподвижным слоем адсорбента
Процесс адсорбции может быть рассмотрен при анализе
переходного режима в диффузионных аппаратах.
Примем следующие обозначения:
у — мольная доля адсорбируемого пара в газе;
х — мольная доля адсорбируемого пара в твердой массе;
у — мольная доля адсорбируемого пара, находящегося в равно-
весном состоянии с твердой массой, содержащей х мол.
долей пара;
п — число ступеней адсорбции;
Z — высота аппарата, считая от места поступления газового
потока;
Н — высота, эквивалентная одной ступени адсорбции (массо-
обмена);
О — время, необходимое для массообмена в объеме, соответ-
ствующем одной ступени адсорбции;
г = G4h\
G — количество твердой массы в аппарате;
h — количество пара в аппарате;
t — время;
G — массовая скорость газового потока;
а — поверхность фазового контакта;
к — коэффициент пропорциональности (коэффициент диф-
фузии) .
Дифференциальные уравнения материального баланса
дх . . .
О’ -^-= — ка (у- —у}
G^==ka
Эти уравнения могут быть представлены в более удобном виде,
если использовать следующие величины:
Z Zka а кН
п=-л-^—’ е= —
41и
Тогда получим:
'6 4т = — ("* — У)
UL
ди
В дальнейшем мы будем, кроме того, пользоваться следующей
величиной для времени (безразмерной)-
mt
т—ЙГ
Следовательно, рассматриваемые дифференциальные
пня окончательно приводятся к виду
уравне-
(XV. 15)
причем т определяется
Система уравнений
влена таким образом:
Принян
дУ
~х~ = У —у
дп
(XV. 16)
из равновесного соотношения:
у* — тх + b
(Х\ . 15) —(XV. 17) может быть
предста-
дх
т —
дХ
ду
дп
U — еп + х (у — тх — Ь)
(XV. 20)
найдем последовательным дифференцированием, что
ду дх '
и — тх — о 4--т
пп дп
9U
дц е
д-Ц
дхдп
дх , д дх
т т —----------—
дп 1 дх дп
дх
т т—
дп
дх . д дх
| дп ' дп дх
дх д
т -------—
дл; t • \
w ~ — (У — У)
— — е
т
~ — тс
= _ еп + т У = + т {тхл.Ь_у) и
дп
Примем, что при т = 0 концентрация пара в твердом адсор-
бенте распределена равномерно, т. е.
X (л, 0) = уи-const (XV.22)
411
н что содержание пара в газе при поступлении в аппарат посто-
янно. т. е.
у (0, т) — const (XV. 23)
Дополнительные граничные условия могут быть получены при
использовании (XV. 15) и (XV. 16). Так. при т = 0 мы имеем в соот-
ветствии с (XV7. 16)
[.7 ("> о)] --= 'П.то-Т h — У («» 0)
И ПОЭТОМУ
1/(«,(!) Л’ы0-|-р — (/пл,:| h у)е“л (XV. 24)
Аналогично из (XV. 15) имеем
[/ня (0, r)J у() —/«.?:((), т) — b
б* t-
Следовательно,
пгх (0, т) = уц -1 Ь — (уй — тхь — b)e~x (X V. 25)
Наконец, вследствие (XV. 24) и (XV. 25), с учетом (XV. 21),
.найдем:
и («. 0) = у0- рп.г0 Н- fe) (XV. 2G)
С(0, т)^0-(тж0 + Ь) (XV. 27)
Применяя теперь преобразование Лапласа, получим *:
тх (л, т) — тх0
у0 — (лг.г0 + Ь)
(XV. 28)
dr
у(». T) —(mx0-]-b)
Уо~ (тяод- b)
(XV. 29)
где /€ — функция Весселя мнимого аргумента.
Уравнения (XV. 28) и (XV. 29) воспроизведены для различных
значений п на диаграммах (рис. XV. 7 и рис. XV. 8). На абсциссах
этих диаграмм отложены значения т. Точки на осях ординат соот-
ветствуют левым частям (XV. 28) и (XV. 29), причем первая из
этих диаграмм даст возрастание концентрации адсорбируемого пара
в твердом адсорбенте, а вторая — уменьшение со сержания пара
в газовом потоке. Для пользования ими необходимо знать следу-
ющие величины:
Z Z ка /н I
П = 1Г='~С~ ' Т="ЙГ
* Л. М. ]] ат у в с р, АТ. Е. И о з и н. Матвматичсские методы
в химической технике, Госхммиздат, 19(>3, стр. 189—214.
412
(q <-°xtlJ)-0A
Рис. XV. 7. Диаграмма для решения уравнения (XV. 28).
-(mx0*b)
Рис. XV. 8. Диаграмма для решения уравнения (XV. 29).
Обозначим:
При адсорбции влаги из воздуха силикагелем найдено для
высоты, эквивалентной ступени массопередачи (адсорбции)
где d — размер частиц силикагеля;
и — вязкость воздуха.
Равновесное влагосодержаппе для силикагеля равно:
х'~ 0,55 Р'/Р или Р' = 1,82 хР (XV. 31)
где х — содержание влаги в адсорбенте;
Р' — парциальное давление водяного пара в газе, находя-
щемся в равновесном состоянии с влагой адсорбента, атм\
Р — давление водяного пара при данной температуре, апгм
Величина Р':Р есть относительная влажность газа при равно-
весном состоянии системы.
Для сушки воздуха при атмосферном давлении в соответствии
с законом Дальтона имеем:
М,р
Р'=У -ту-= 1,62^ (XV. 32)
где — молекулярный вес воздуха;
Л/2 — молекулярный вес адсорбтива;
Р — общее давление (1 атм).
Сопоставляя (XV. 31) и (XV. 32), получим:
1,82
у’ = , .... Рх = тх (X V. 33)
1 >Ь2
где
»г=1,122Р
Для специального случая изотермической адсорбции водяного
пара силикагелем из (XV. 30) имеем:
а = -^ = -£- = 0,703а (-у-) (XV. 34)
Из (XV. 30) и (XV. 34) имеем:
о т т Gm 1,122 GP
К — —— — 'I •— ————- — —.... -—
' Саде ЬР h G / tlG \ 1,22 G Р 0,703 а [ 1 \ И / Саде Оадс^ С’адс^ 0,780 GPa f (IG \ -°-51 ,v„ = 1 J (XV. 3.)) Саде \ Ц / 415
Пример. Воздух пропускается через слои силикагеля толщи-
ной 0,3 .« при 26.6" С с относительной влажностью <30% (У„ ~ 0,0179 кг/кг
сухого воздуха).
Размер частиц силикагеля в среднем составляет d = 0,00273 .ч; насып-
ная плотность силикагеля gaac — 025 кг/м3. Определить:
1) влажность выходящего из аппарата воздуха по истечении 2 ч;
2) среднюю влажность выходящего воздуха за период времени, рав-
ный 2 ч\
3) распределение влаги в слое адсорбента к концу периода времени,
равного 2 ч;
4) среднее содержание влаги в адсорбенте к концу двухчасового периода
работы аппарата.
Имеем:
Саде 625 кг/м3
Qb --1,18 лг/.ч3
G 30,5 -1,18 = 36 кг/м3 мин
Ив = 111 • I0-5 кг/м • мин
Из справочной таблицы найдем величину удельной поверхности сили-
кагеля:
а = 930 л2/.и9
Следовательно:
,dG 0,00273 36 • К»5
----- =------77Д------= оЬ>1
И 111
и
Давление водяного пара при 2G,6C С:
Р = 0,0345 атм
1. Из (XV. 34) имеем:
а = ~ ~ = 0,703 • 930 - 0,1025 = 67 л"»
С* //
Используя (XV. 35), получим:
0 =~ = 0,789 • 930 0,1025 -0,145 ли/Г»
* гО 62а
Таким образом:
т=0/ = -^=0,115-120 = 17,4
/0
п = а/ = 0,30 i • 67 = 20,5
Ио рис. (XV. 8) найдем
У/Уо=0,34
откуда: У = 0,34 • 0,0179 = 0.0061 Кг- В°-НЛ кг сухого воздуха
2. Для определения средней влажности воздуха! при выходе из аппарата
необходимо проинтегрировать значения конечных влажпостей за период,
416
равный 2 часам. Величины У вычисляются для различных отрезков времени
таким же образом, как на стр. 416;
/, мин т - У
0 0 0 0
20 2.90 0 0
40 5,80 0.003 0.00005
GO 8,74 0,016 0,00029
80 11.60 0.065 0.00116
100 14,50 0,185 0.00331
120 17,40 0-340 0.0061и
Рис. XV. 9- Графическое интегриро-
вание в расчетах адсорбции влаги
силикагелем.
Рис. XV. 10. Распределение влаги
в слое адсорбента.
Графическое интегрирование значении У'относительно / дает (рис. XV. 9);
Y = 0,0010 кг/кг сухого воздуха
Эта величина соответствует ординате для площади прямоугольника, связан-
ного с количеством клеток под кривой.
3. Распределение влаги в слое адсорбента в конце двухчасового периода
работы аппарата найдем с помощью рис. XV. 7.
Имеем:
Z 120 .имя; т= ₽/ = 17,4; У„ = 0,0179
Из (Х\ . 31) получим
0,55 • 0.80 0,44
Значения счхиветствующие величинам и = aZ, определяются
но рис. XV. 7 для постоянной абсциссы т= 17,4, а именно:
Z 0 п — <tZ О .т/х0 •1.00 X 0,440
о.об 4.1 1,00 0,440
0.12 8,2 0,92 0,405
0,18 12,3 0,78 0,344
0.24 16,4 0,57 0,250
0,30 20.5 0.30 0,132
2/ Л. М. Батупер.
417
Распределение влаги в слое адсорбента показано графически на
рис. XV. 10.
4. Среднее значение влаги в слое адсорбента к концу двухчасового пери-
ода работы можно найти путем графического интегрирования значений .с
относительно Z; оно составляет
хср = 0,348 кг влаги/кг сухого адсорбента.
Рекуперационная установка
для растворителей
Пары бензола должны быть адсорбированы из воздуха
на рекуперационной установке при пропускании воздуха сверху
вниз через слой силикагеля при 21е С и атмосферном давлении.
Массовая скорость воздуха 36,6 кг сухого воздуха/.ад2• .инн.
Период работы адсорбента желательно установить в течение
60 мин с минимальной степенью извлечения бензола из воздуха,
равной 90%.
Воздух при поступлении в аппарат содержит 0.90 объемн. %
бензола. Размер (диаметр) частиц силикагеля в среднем соста-
вляет d = 0,0039 .ад; морозность адсорбента равна 0,5; насыпная
плотность оадс ~ 625 кг,'.ад3.
Для адсорбции до 20% адсорбгпва от веса силикагеля количе-
ство поглощаемого бензола прямо пропорционально парциаль-
ному давлению р бензола в воздухе согласно уравнению х =
- 1,67 р'!р. Давление бензола р равно 0,125 ат» при 21° С.
1. Определить толщину слоя адсорбента.
2. Определить содержание бепзола в силикагеле наверху,
посредине и внизу слоя адсорбента по истечении 60 мин.
1) Имеем:
d Т= 0.0039 .и; а — 665 №/.нэ
р. = 111 • 10~5 кг/м • мин', G — 36,6 кг/м2 мин
Таким образом:
dG = 0,0039 • 36,6 • 10д 12д
и 111
И
Из (XV. 30) получим:
(dG \ - в,61
— ) 0,703 • 665 • 0,0825 = 38,6
Р /
Для отыскания значения § определим величину т следующим
образом. Так как по закопу Дальтона:
Л/-2
418
и
то
Аналогично (XV. За) имеем
= цю Ср = «ДО-ЗМИМУ. = ол_ J!uir
<5Ь,Ь
Следовате льно:
т = р/= 0/i5 • 60 = 27,0
Имеем:
У,/Уд 0,1 (при 90% извлечения бензила)
По рис. XV. 8 при т — 27,0 получим:
п = uZ = 36
Таким образом:
. Z = = 0 935~ 1,0 Л-
38,6
2) Для определения
местах слоя адсорбента
содержания бензола в указанных выше
воспользуемся формулой:
Из предыдущего имеем:
а =38,С- т = 27,О
Верх Середина Низ
Z . . . 0 0,500 1
aZ . . 0 19,300 38,6000
z/.ru (из рис. X V. 7) . . 1,00° 0,830 0,0790
X , . . ('.120 0,099 0,0095
Жидкостная адсорбция
Из раствора надлежит удалить с помощью адсорбции
находящуюся в нем примесь (пигмент), причем адсорбентом слу-
жит уголь. Была проведена серия лабораторных опытов, которые
заключались в том. что отдельные образцы начального раствора
перемешивались с адсорбентом, взятым в различных количествах,
до установления равновесного состояния при постоянной темпе-
ратуре.
27*
419
Начальный pacmlop^-^ >> Конечный раствор
Г₽ _ UfwT_ 'ёежш адсорбент
*——%Х^7—
Отработанный х? Ls
адсорбент Х-0
Рис. XV. 11. Схема и диаграмма
жидкостной адсорбции с одной сту-
пенью.
Рис. XV. 12. Схема и диаграмма
2-ступенчатой перекрестной адсорб-
ции при минимальном расходе угля.
Рис. XV. 13. Схема и диаграмма 2 сту
нснчатой противоточной адсорбции.
Ниже приведены опытные данные:
Отношение
кг угля 0 0,0(11 0,004 0.008 0,02 0,04
к.: раствора Концентрация раствора при рав- но ноевом состоя- нии, уел. ед. . . 9,6 8,(5 6.3 4.3 1.7 (|,7
Конечная концентрация раствора
должна составить 10% от началь-
ного ее значения, г. е. 0,96.
Определить количество свежего
угля на I000 кг раствора:
I) для адсорбции с одной сту-
пенью (рис. XV. II);
2) для 2-ступепчатой перекрест-
ной адсорбции при минимальном
расходе угля (рис. XV. 12);
3) для 2-ступенчатой противоточ-
ной адсорбции (рис. XV. 13).
Экспериментальные данные дол-
жны быть приведены к такому виду,
который позволил бы построить изо-
терму равновесия процесса адсорб-
ции. С этой целью обозначим через У
число единиц удаляемого продукта
на 1 кг раствора, а через X — число
1 кг угля. Растворы в отношении
Рис. XV. 14. Графическое
определение констант в урав-
нении Фрейндлиха.
единиц этого вещества на
примеси являются разба-
вленными и поэтому раоочие. липни на диаграмме в координатах
X — У являются прямыми.
Результаты опытов представлены в табл. XV. 3.
Таблица XI . 3
Результат ы on ытов
Отношение, кг угла . уел, ед. кг pacujctpa в состоянии равпоисспн уел, ед- Кг угли
кг раствора
0 9,(5
0,001 8,6 (9,6-8,6)/0,001 -1000
0,00-4 6,3 (9.(5—6,3)/0,004 825
• 0,008 4,3 663
0,020 1,7 395
0,040 0,7 223
Построив зависимость на логарифмической сетке, получим
прямую линию (рис. XV. 14). Тангенс угла наклона этой линии
421
п — 1,66; при х - 663 имеем У = 4,3. Таким образом, дли урав-
нения изотермы Фрейндлиха имеем:
т. ——— = 8,91 • 1Гг5
С63ЪОС
Следовательно, уравнение Фрейндлиха принимает такой вил:
У = 8,91 • 10“3Л'1>6в
Адсорбция с одной ступенью
уел. ед.
Имеем У. -9.6. Далее У. = О ДО • 9.6 = 0.96 -пе1ЦесТ1!а .
и 1 кг раствора
Пусть G = 1000 кй раствора и L — количество угля. Так как
здесь используется свежий уголь, то Х'л — 0.
На рис. XV. 15 точка Л изображает состав начального рас-
твора во время контакте» со свежим углем, а точка Н находится
на равновесной кривой и даст концентрацию примеси в конечном
растворе. Утой точке соответствует АД = 270.
U
4
BfWpgfi
fisnsvrioe
содержант
пигнентя.
ПЗрС-МЖ?
него мех?
JO
начвямпе,
северкам-
гьгиенч^
гео
800 fOOD
400 600
л
Рис. XV. 15. Диаграмма для расчета жидко-
стной адсорбции.
Из материального баланса имеем
I, У Г,— > ! O.f) — О,9() . . Кй тгля
— — —---------гД- — —--------:~ и,0.’2----------:------
6 Aj —Хо 2/0 — 0 ь-2 растдора
и общее количество составляет
Л 0,032-1000=32,0
кг угля
1000 к-2 jiacTi-.vpa
422
Мы можем также воспользоваться уравнением Фрейндлиха
}• „«А®
(XV. 37)
откуда для конечного равновесного состояния имеем:
Ь _ Уп-У,
(r
Таким образом
(XV. 38)
Д_ _ hi - >'i ________________________9,6 — 0,96_____________ Q f).^9 кг угля
{' (У1/т},/'1 (0.96/8.91 • 10"’ь)1,'1,С6 ‘ " кг раствора
откуда:
L — 0,032- 1000--32,0 кг угля/1000 кг раствора.
Двухступенчатая перекрестная адсорбция
Минимальное общее количество угля может быть най-
дено ио рис. XV. 15 методом подбора. Отмстив точку С на равно-
весной кривой, проведем рабочие линии ЛС и DB; величины Ll
и L., вычислим посредством уравнении материального баланса
для первой и второй ступеней:
^(Уо-Л)=^(Х1-Хо) (XV. 39)
G ОТ - l’s) = L2 (А\ - Ао) (XV. 40)
где и L2 — расходы угля, соответствующие первой и второй
ступеням.
Положение точки С на графике изменяется до тех нор, пока
сумма Lx и L2 по станет минимальной. Точка С на рис. XV. 15
отвечает ее конечному значению с координатами Х\ = 565, =
= 3,30 при Х.2 — 270 (в точке В).
Из (XV. 39) и (XV. 40) имеем
G
1000 (9,6 — 3.30)
565 — 0
У г-У 2
А4 —А'о
= 11,14 кг
L2= G
1000(3,30-0.96)
= 8,Ь7 кг
270 — 0
откуда:
11,14 + 8,67 = 19,81
кг угля
1000 кг раствора
Кроме того, используя уравнение Фрейндлиха, применим
рис. XV. 16.
423
Имеем:
О,яв
9,6
= 0,1.
1,6b
Из рис. XV 16 найдем
откуда:
4Л = 0,344
1 о
Fj 0,344-9,6 3,30
Y,
n
Рис. XV. 16. Диаграмма для расчета З-стуиенчатой пере-
крестной адсорбции с использованием уравнения Фрейнд-
лиха.
Применяя (XV. 38). получим:
Lx = Ур-К, = 9,6-3,30 =
G ~ (Kl'w)1/W ~~ (3,30,8.91 10"5)’
иг угля
0,011 14--------------------:--------
/.г раствора в первой ступени
L. ~ У; - X., _ .3,30-0.96
6 ~~ (Уг. " ~~ (0,96.8,91 Ю-5)1'1’66
кг угля
0,00867---------------------—--------
кг раствора во второй ступени
Общее количество угля.
кг угля
1. (0.011И (1.00807) 1(ХЮ 10.81
4’4
Двухступенчатая противоточная адсорбция
Имеем:
Г0«=9,б: У» = 0,96: А'п=0
Рабочая линия па рис. XV. 15 изображается путем подбора
до тех пор, пока между этой .нишей и равновесной кривой нс будет
двух ступеней. Находим, что для точки Е
Ряс. Х\. 17. Диаграмма для расчета 2-сгупенчатоЙ
противоточной адсорбции с использованном уравне-
ния Фрейндлиха.
Из уравнения материального баланса следует
G о~ - НИЮ (ОД» — 0,96) рэд г;г угля
-V— Л',, 675 — 0 10(H) кг раствора
В отом случае также имеем:
Пользуясь уравнением Фрейндлиха, найдем:
Л = Г„-Т,_ =-----------0.В—О^В = _!±уг^ _
G (>1 •'"')1;" (4,42/8,91 КГ’)1'1-’0 кг раствора
L - 0,01280 10(10 12,80 -- 5 ------
Ji ДЮ кг раствора
Непрерывный процесс адсорбции
Частные коэффициенты массопсрелапл в жидкости (или
газе) и твердой массе при адсорбции водяных паров из воздуха
посредством силикагеля в неподвижном слое составляют:
KYd- 3000(/<>,а5 (газовая фаза)
А „а =3460—----------— (твердая фаза)
й .и- • ч • ДА
где С — массовая скорость газа, кг/.ч‘2>ч.
Кажущаяся плотность силикагеля равна (570 кг. мл. Средний
диаметр частиц силикагеля 1,7 л.и. Наружная поверхность частиц
силикагеля 2,35 м-,кг.
Определить высоту противоточного адсорбера непрерывного
действия для сушки воздуха при t = 26,6° С и атмосферном
давлении.
Начальное содержание влаги в воздухе 0,005-------——--------.
1 * кг сухого воздуха
Конечная влажность равна 0,0001----------—-------.
кг сухого воздуха
Силикагель при поступлении в аппарат — сухой. Скорость
движения массы силикагеля 2270 кг/лг-ч. Скорость воздуха
4540 cvXOr'° воздуха
.И- ч
Для данных концентраций влаги в воздухе равновесная изо-
терма адсорбции может быть принята прямолинейной, уравнение
которой имеет следующий вид:
Имеем:
У =0.(118Г> у
0,005
кг поды
кг сухого воздуха
кг воды
кг сухого воздуха
Г2 0,001
0 = 4540 —
Л “ ч
_______кг воды_______
кг сухого адсорбента
426
Лз уравнения материального баланса получим:
-.G
-j- Л о — 4540
0,005 — 0,0001
2270 —
сухого адсорбента
Далее, У2 = 0 — влажность воздуха, находящегося в равно-
весном состоянии с силикагелем при его поступлении в аппарат:
у; = 0,0185 А\ =- 0,0185 • 0,0098= 0,0001815 --~g П°ДЫ-
кг сухого воздуха
Для определения средней логарифмической разности имеем:
У L — У,*= 0,005 — 0,0001815 = 0,00482
У2 — }<= 0,0001 — 0 = 0,0001
и
0,00482-0,0001
' ср- 0,00482
П 0,0001
0,001217
Число ступеней адсорбции:
v_ yt-y2 _ 0,005 -0,0001 _
ЛУ(:р 0,001217 '
'Гак как данные о частных коэффициентах адсорбции получены
при неподвижном слое адсорбента, то для аппарата с движущимся
адсорбентом нам необходимо определить относительные скорости
воздуха и твердого адсорбента.
Лилейная скорость потока твердой массы, движущейся вниз,
будет:
2270/670 -3,38 л/ч
Плотность воздуха при 26,6° С и 1 атл составляет 1,18 кг/м3
н, таким образом, линейная скорость воздуха, поднимающегося
вверх, будет:
4540/1,18 = 3875 -ч/ч
Относительная линейная скорость воздуха и твердого мате-
риала равна
3875-j-3,38—3878 м/ч
Относительная массовая скорость воздуха:
- 3878 • 1,175 = 4580 кг/.ч2 • ч
Высота, эквивалентная ступени адсорбции для газовой фазы:
_ С _ G _ 4540 п
//глз — —77 —----------— —-----------0,030 м
кум 3UOOG'0’6’ 3000 • 4580°’55
Высота, эквивалентная ступени адсорбции для твердой фазы:
Кроме того:
mG O.OI85-4540 Л _
---=------------= 0 (I,' i
L 2270
Для общего коэффициента массопередачи имеем высоту, экви-
валентную ступени адсорбции
И = //-г» и,03() 0,037 0,65 0.0G0 .4
Следовательно. высота слоя адсорбента равна
Z \Н 4,03 -0,000 0.242 л
Промышленные установки
и аппараты для адсорбции
Процесс периодической адсорбции активированным
углем и силикагелем может производиться различными способами.
Широкую известность получил так называемый четырехфазный
способ. При работе по этому способу весь цикл состоит из 4 ч
фаз:
I фаза — насыщение активированного у гля пог ющаемым ком-
понентом ;
II фаза — десорбция поглощаемого компонента из актнвпро
ванного угля;
111 фаза — сушка активированного угля;
1\ фаза — охлаждение активированного yr ui.
Паровоздушная смесь пропускается через слой активирован-
ного угля до состояния насыщения (1 фаза). По окончании насы-
щения паровоздушная смесь направляется во второй аппарат,
а из насыщенного слоя активированного угля при обработке его
водяным паром выделяется поглощенное вещество, т. е. произ-
водится десорбция (II фаза).
Смесь паров воды и вы щлеп'юго из угля вещества наира
ваяется на конденсацию и затем регенерированное вещество отде-
ляется от воды. Отделение от воды производится пли отстаиванием
(если вещество не смешивается с водой) пли ректификацией (если
вещество смешивается с водой). Влажный, после десорбции, уголь
сушится горячим воздухом (111 фаза).
После сушки горячий уголь охлаждается холодным воздухом
(IV фаза). ’
Но окончании IV фазы цикл начинается снова.
Разработан также двухфазный способ адсорбции, по которому'
нагретую паровоздушную смесь пропускают через горячий вл аж
428
iibiii активированный уголь. При этом одновременно с поглоще-
нием происходит и подсушивание угля. Затем через активирован-
ный уголь пропускают холодную
паровоздушную смесь. Здесь одно-
временно с поглощением происходит
и охлаждение поглотителя.
Итак, при двухфазном методе ра-
боты параллельно с поглощенном:
происходит и высушивание и охла-
ждение поглотителя (I фаза); по
окончании адсорбции производят де-
сорбцию водяным паром (II фаза),
после чего в горячий и влажный
уголь вновь пропускают нагретую
паровоздушную смесь и цикл начи-
нается снова.
Работы опытных и производ-
ственных установок показывают,
что наиболее экономичным с точки
зрения энергетических расходов
является двухфазный метод работы.
Кроме того при двухфазном методе
значительно повышается производи-
те л ьпость уставов ок.
Периодический процесс адсорб-
ции производится в вертикальных
Рис. XV. 18. Вертикальный
аппарат для проведения про-
цесса адсорбции:
Л —адсорбер; г' —Слои активиро-
ванного угля: - центральная
труба для подачи иарп-иоздушнои
смеси при адсорбции и воздуха
при сушке н охлаждении; J -- бар-
ботер для иодачи острого пара при
десорбции: .5 — штуцер дли вы-
хода инертных ИО OTIIOIHCHtllQ
к поглотителю газон при адсорбции;
/> — штуцер для выходи паров при
десорбции.
и горизонтальных
аппаратах. Одним
являет я аппарат,
цилиндрических
из первых аппаратов вертикального типа
изображенный па рис. XV. 18.
Рис. XV. 19. Горизонтальный аппарат для проведения
ироцесса адсорбции:
1 адсорбер; 2 слои аитпиприпаиного угля; .1 — штуцер для
подачи iiapo-Bn.xiyiiiHiiti смеси при адсорбции; I штуцер ДЛИ
отвода инертных газов при аде .'рицин; 5 барб< тер для ко-
дами острого пара при десорбции; 6' — штуцер для отвода паров
при десорбции.
В настоящее время существует стремление уменьшать высоту
угольного слоя в адсорберах периодического действия. Это дает
возможность уменьшить расход энергии па преодоление сопроти
вления при прохождении паро-воздушной смеси через аппарат.
429
На рнс. XV. 19 изображен адсорбер горизонтального типа с неболь-
шой высотой слоя угля.
Сокращение количества фаз при периодической адсорбции
дало большой экономический эффект. Очевидно, еще больший
с движущимся
активированного
Рис. XV. 20. Вертикальная
колонна с движущимся
сверху вниз слоем активи-
рованного угля:
1 — адсорбционная колонна;
з — десорбционная колонна;
3—Г 2, 16— штуцеры; 13 —
трубка; 14—сепаратор; 13—
теплообменник.
эффект должен дать переход на не-
прерывный способ работы. В настоя-
щее время непрерывный процесс ад
сорбции осуществляется обычно в вер-
тикальной колонне
сверху вниз слоем
угля (рис. XV. 20).
Снизу через штуцер 4 в адсорбци-
онную колонну / подается паро-воз-
душная смесь. В колонне осуще-
ствляется тесный контакт между акти-
вированным углем и паровоздушной
смесью. В этой колонне происходит
адсорбция поглощаемых веществ. Не-
использоваппая часть смеси выводится
из верхней части колонны через шту-
цер 5. Насыщенный поглощаемым веще-
ством уголь из нижней части адсорб-
ционной колонны поступает в верхнюю
часть десорбционной колонны 2. В де-
сорбционной колонне происходит не-
прерывная десорбция поглощаемых ве-
ществ нз угля острым водяным паром,
поступающим в колонну через шту-
цер 8. Десорбционные пары удаляются
нз колонны через штуцер ,9. Выходящий
из нижней части десорбционной ко-
подхватывается струей
полны горячий и
воздуха. поднимается
влажный уголь
по трубке 13 и
подается вновь в верхнюю часть адсорбционной колонны.
В верхней части установки над адсорбционной колонной
имеется обычно циклон для очистки воздуха от мелких частиц
угля. Недостатком указанных адсорберов непрерывного действия
является то обстоятельство, что в них происходит механическое
истирание частиц угля и образование угольной ныли.
Типовая схема адсорбционной установки
периодического действия
На рис. XV. 21 изображена типовая схема промыш-
ленной адсорбционной установки для улавливания паров органи-
ческих веществ из их смеси с воздухом.
Гис. XV. 21. Типовая схема адсорбционной установки периодического действия:
1 — фильтр; 2 — гравийный огнепреградитсль; .?—предохранитель; 4 — вентилятор; .5 — холодильник; в — калорифер; 7 — кон-
денсационный горшок; S — адсорбер; а — конденсатор; ;о — жидкостные манометры; п — пружинные манометры; /^—ртутные
термометры.
Смесь в большинстве случаев содержит значительное коли-
чество ныли, могущей засорить адсорбционную установку. По-
этому ио пути из цеха на адсорбционную установку паровоздуш-
ную смесь обычно пропускают через фильтр 7.
Гак как смесь паров органических веществ с воздухом является
взрывоопасной, то по длине трубы устанавливаются огпепрегра-
дителп 2 н предохранительные компенсаторы 3 с разрывными
мембранами. В большинстве установок вентилятор 4, транспорти-
рующий паровоздушную смесь, устанавливается за огнепрегра-
дителем и компенсатором.
Выходящая из вентилятора паровоздушная смесь делится
на две части: одна часть охлаждается в водяном холодильнике 5,
другая нагревается в калорифере 6.
Охлажденная часть паровоздушной смеси подается в адсор
бор при насыщении, а нагретая — при сушке. На некоторых
установках охлаждения и нагревания паровоздушной смеси не
производится и последняя после вентилятора поступает в адсорбер.
В адсорбер через специальный патрубок при необходимости
(возникновение пожара) можно ввести воду. Смесь паров воды
if органического вещества из адсорбера 8 направляется в конден-
сатор 9, откуда выходит жидкий рекуперат и вода.
Фильтр для очистки паровоздушной смеси. Из всех
методов пылеочистки наибольшее применение имеет метод очистки
паровоздушно!! смеси фильтрованием через слои насадки (кирпич-
ный щебень керамиковые пли металлические кольца и т. и.).
Одним из преимуществ подобных фильтров является незначи-
тельное сопротивление, оказываемое прохождению газа и, следо-
вательно, незначительный расход электроэнергии на фильтрова-
ние.
Насадку целесообразно смачивать для того, чтобы к его поверх-
ности прилипали частицы пыли (висциповое масло).
Огпепреградители и предохранители-компенсаторы. Эф-
фективно действующим огиепреградителем является слой тепло
емкого материала. Этот теплоемкий материал, отнимая тепло
у горящего потока, тем самым препятствует распространению
пламени.
В качестве такого теплоемкого материала широко применяется
гравий. В гравийных огпепреградитслях обычно применяется
гравий со средним размером частиц от 5 мм и выше. Обычная
толщина слоя гравия от 100 до 150 льи. Следует предусмотреть,
однако, не только средства предупреждения пожара и взрыва,
но и меры, предотвращающие разрушения, если взрыв уже про-
изошел. Для этой це hi па трубопроводе, по которому проходит
паровоздушная смесь, или же на корпусе самого огнспрсградп-
теля имеются патрубки с мембранами, разрывающимися при
взрыве. Па рис. XV. 22 изображен предохранитель-компенсатор,
устанавливаемый на трубопроводе. 1 ароноздушпая смесь ирохо-
«2
днт последовательно ио трупам /, 2, 5; на концах труб ./ п 3
имеются мембраны /. Таким образом, возникшая в трубке i
взрывная полна выбьет мембрану, но нс попадет в трубку 3 п не
вызовет разрушении установки. Материалом для изготовления
мембран служат жесть, латунь, свинец и т. п.
Вентилятор. Транспортирование паровоздушной смеси
осуществляется с помощью центробежного вентилятора высокого
давления или же газодувки. В большинстве случаев вентилятор
устанавливается до адсорбера (см. рис. XV. 21) и тогда давление
в последнем оудет несколько
установках, однако, вентиля
вливается за адсорбером.
При таком расположении
давление в адсорбере будет
ниже атмосферного и пары
огне- или взрывоопасной
паровоздушной смеси не смо-
гут просасываться через не-
плотности аппарата в поме-
щение цеха.
Вентилятор рекомеп-
выше атмосферного. На некоторых
гор (из предосторожности) устана-
Рис. XV. 22. Иррдохраиитслг.-комиен-
сатор. устанавливаемый на трубопро-
воде:
1 :у трубы: t — мембраны.
дуется устанавливать за ад-
сорбером также и тогда, когда поглощаемое вещество кор-
родирует металл. В этом случае при нормальном технологи-
ческом режиме установленный за адсорбером вентилятор будет
отсасывать воздух, уже не содержащий корродирующего веще-
ства.
Д 1Я обеспечения пожарной безопасности моторы отделяют
от вентилятора газонепроницаемой перегородкой, а отверстие,
через которое проходит вал, уплотняют специальным сальником.
Моторное отделение должно иметь обособленный ход.
Температура паровоздушной смеси при прохождении через
вентилятор повышается па 7—10° С.
Холодильник. Охлаждение паровоздушной смеси после
вентилятора производят обычно в трубчатых холодильниках.
Коэффициент теплоотдачи от паровоздушной смеси к стенке
невелик. Поэтому с этой стороны часто увеличивают число ходов
холодильника. Иногда число ходов увеличивают и со стороны
охлаждающей воды. Даже при наличии многоходовых холодиль-
ников величина поверхности теплообмена в большинстве случаев
достаточно велика (около 200 м~ и более). Обычно охлаждающую
воду подают в трубы, а паровоздушную смесь в межтрубпое
пространство.
Адсорберы. Адсорберы изготовляются из листовой
стали толщиной от 8 до 10 мм. Па рис. XV. 23 изображен цилин-
дрический сварной адсорбер вертикального тина с приварным
сферическим днищем и сферической крышкой па болтах.
28 Л. М. ВатунерЛ
433
Лдсороер имеет центральную трубу 2 для ввода паровоздунг
ной смеси. На уровне шва между цилиндрической частью адсор
Рис. XV. 23, Вертикальный ци-
линдрический сварной адсорбер:
1 — корпус; 2 — центральная труба;
з — колосники; 4 — колоснокивал
решетка; 5 — загрузочные люки; с —
разгрузочные люки; 7—приварное
кольцо; я—]з, 16—1s, 10 — патруб-
ки; 14 — гильза термометра; 15 —
ото йник брызг; 17 — цилиндрический
горшок.
В патрубке для выхода
бора и днищем приварены колос-
ники 3, на которые положена
разборная чугунная колоснико-
вая рспнтка 4. Па колосниковую
решетку кладут обычно еще ме-
таллическую сетку, иногда в не-
сколько рядов. На сотку помещают
слой гравия с размером кусков
5—50 льч Общая высота слоя гра-
вия около 100 .м.и. На гравий
кладут слой угля, который сверху
покрывают металлической сеткой.
Иногда между слоями гравия и
угля также помещают металличе-
скую сетку. Загрузку гравия и
угля производят через загрузоч-
ные люки 5 на крышке адсор-
бера. На уровне колосниковой
решетки расположены разгрузоч-
ные люки 6 для выгрузки гравия
и угля.
Центральная труба привар-
ным кольцом 7 из угловой стали
па своем нижнем конце опирается
па колосниковую решетку. Верх-
ний конец центральной трубы
болтами соединен с патрубком <S’,
приваренным в центре сфериче-
ской крышки адсорбера. Острый
нар подастся в адсорбер снизу
через патрубок-барботер 9, рае-
положенный кольцом в сфериче-
ском днище.
В крышке адсорбера имеются
патрубки 1.0, 11, 12 и 13 для
предохранительного клапана, ма-
нометра. выхода паров при де-
сорбции в конденсатор и выхода
в атмосферу воздуха, освобожден-
ного от поглощаем ы х веществ.
паров при десорбции установлена
гильза термометра 14. Кроме того, гильзы термометров устано-
влены также в цилиндрической части адсорбера.
В патрубке для выхода паров имеется отбойник брызг 13.
Предохранительный клапан устанавливается обычно па давление
(абсолютное) 1.5 ат. Кроме перечисленного, на крышке адсорбера
имеется патрубок 16, заглушенный свинцовок мембраной. В центре
сферического днища приварен цилиндрический горшок /7, в кото-
рый стекает конденсат, образовавшийся во время нагревания
адсорбера острым паром. Конденсат отводится из нижней части
горшка через патрубок 18. Кроме того, в горшке имеется патру-
бок /.9, через который в адсорбер подается вода в случае загора-
ния угля.
Рис. XV. 24. Горизонтальный цилиндрический
сварной адсорбер;
1 —2, •/. в, il — lti — патрубки; .? — барботер; .? —
гильза термометра; 7 — люки; а — колосники; 9 — ко-
лосцикоиая решетка; 10 — разделитель.
На рис. XV. 21 изображен сварной цилиндрический адсорбер
горизонтального тина с двумя приварными сферическими дни-
щами.
Разборная чугунная колосниковая решетка ,9 опирается на
колосники 8. В верхней части адсорбера имеются три люка 6'
для загрузки гравия и угля. Подача паровоздушной смеси произ-
водится через патрубок I в верхней части левого днища. В правом
днище ниже колосниковой решетки имеется патрубок 2 для отвода
в атмосферу воздуха, освобожденного от поглощаемых веществ.
Острый пар поступает через патрубок в левом днище в барботер 3.
При десорбции пары выходят через патрубок 4, расположенный
в правом днище выше угольного слоя.
Та новая схема адсорбционной
установки непрерывного действия
с движущимся слоем адсорбента
На рис. XV. 25 изображена схема адсорбционной установки
с движущимся слоем активированного угля для разделения газо-
вой смеси па три фракции: легкую, промежуточную и тяжелую.
Разделительная колонпа / состоит из трех зон: адсорбционной
зоны I, где происходит взаимодействие между газовой смесью
28* 4.35
и свежим активированным углом; десорбционной зоны 7/7, где
поглощенные углом вещества выделяются вследствие нагревания
угля; ректификационной зоны //, где выходящие из десорбцион-
Исходная
смесь
додо
Легкая
фра-^о^я
.. Вода
4
нагоеОав
щей аген<п
г
27-
-15
Поссу* ты
~релл^-
г .15 Saaui
И. ап г.
оар
Гяжсмз
фракция
Нвгзеёа
орлий
а ген-'
ИГ
П-
**UUL vu- jf.
Язи
6
.10
:.ар
р "
Ч4
нагребаюсь-5.
зге.ню
HocDs5aH)u4iiL
М-ЧГ;
3
\в II
Д- f3 fг'
Y» J
Рис. XV. 25 Гиповая схема ад
сорбционной установки непрерыв-
ного действия с движущимся слоем
адсорбента.
/ — разделительная колонна; j — те-
плообменники. / — распредели гсльны'*
тарелки j — питаюшин механизм. ».
гидрозатп'р: 7, s, is, ja патрубки;
!> — питатель; mi подъемник: ii
сборник: /.? иептплктпр; п бункер;
/.» — рсактиплтир.
ной зоны вещества вытесняют
из угля менее сорбируемые
компоненты. По высоте ко-
лонны в местах вво ia и вывода
газа имеются четыре распреде-
лительные тарелки 4.
Исходная газовая смесь по-
ступает иод вторую сверху рас-
пределите 1ьпую тарелку и
поднимается в адсорбционную
зону, находящуюся между пер-
вой и второй (сверху) тарел-
ками. 13 адсорбционной зоне
поглощаемая часть газовой
смеси адсорбируется углем, а
нопоглощениая часть, называ-
емая легкой фракцией, выво-
дится в верхней части колонны
из-под первой сверху тарелки.
.Легкая фракция иногда на-
зывается также верхней пли
головной.
Из адсорбционной зоны
угол!» опускается вниз в ректи-
фикационную зону, находя-
щуюся между второй! и четвер-
той тарелками сверху.
В рассматриваемой схеме
промежуточная фракция отби-
рается из-под третьей тарелки.
Из ректификационной зоны
уголь опускается в десорбцион-
ную пли отпарную зону, нахо-
дящуюся иод четвертой сверху
тарелкой.
В десорбционной зоне уголь
нагревают и одновременно про-
дувают острым перегретым водяным паром Встречая горячий
уголь, пар практически не конденсируется, а лини» выдувает
десорбируемые вещества. Смесь десорбированных веществ < водя-
ным паром (тяжелая фракция) выходят из-под четвертой сверху
тарелки.
Горячий уголь из зоны десорбции выводится вниз через спе-
циальный питающий механизм 5 в так называемый гидрозатвор б
предотвращающий попадание пара в подъемную систему. Выпу-
скаемы ii через гидрозатвор уголь газовым подъемником 10
подается в бункер И, расположенный над колонной.
Циркуляция транспортирующего газа в газовом подъемнике
осуществляется с помощью вентилятора 13. Поступающий в (бун-
кер уголь на некоторых установках подсушивают, пропуская
через него часть легкой фракции, вводимой в бункер через трубки
холодильника 2. Из бункера уголь ссыпают в холодильник 2.
из холодильника в адсорбционную зону, и цикл начинается снова.
Для поддержания в бункере постоянного уровня в него доба-
вляют уголь.
Разделяемый газ может содержать трудно десорбирующиеся
вещества. В этих случаях часть угля из бункера направляется
в реактиватор /J — аппарат, нагреваемый до температуры, более
высокой, чем десорбер колонны. В реактиваторе также тщательно
обрабатывают уголь острым водяным паром, как и в десорбцион-
ной зоне. Продукты реактивации и пар отводят из верхней части
реактиватора.
Холодильник. Холодильник представляет собой вер-
тикальный 1 рубчатый теплообменник. По трубкам проходит уголь
и часть легкой фракции. В межтрубиос пространство подают снизу
вверх охлаждающую воду.
Десорбер и реактиватор. Десорбер, так же как п хо-
лодильник, представляет пучок вертикальных труб, ввалi.повин-
ных в две трубные решетки. По трубам проходит уголь и водяной
нар. а в мс.жтрубпое пространство полается нагревающий агент,
характер которого зависит от необходимой температуры десорб-
ции. В качестве нагревающего агента применяется водяной нар
или нары органического теплоносителя — смеси днфепплокенда
с дефпиплом.
Конструкция реактиватора аналогична конструкции десор-
бера.
Распределительные тарелки. Распределительные та-
релки устанавливают для обеспечения равномерного распреде-
ления газа по сечению Колоппы, наибольшего соприкосновения
между газом и поглотителем, а также для предотвращения уноса
частиц угля выходящими газами.
Распределительные тарелки представляют трубную решетку,
в отверстия которой ввальцовывают трубы длиной 0.25—0,6 .я
и диаметром 10—50 ,u.w (рис. XV. 26).
Тарелка монтируется в колонну патрубками вниз.
Питающий механизм. Пи тающий механизм (меха
нпзм выгрузки) имеет очень важное значение, так как обеспе-
чивает постоянную скорость угля и его равномерное распределение
по всему сечению колонны.
Регулирование скорости подачи твердых частиц можно осуще-
ствить различными степенями открытия задвижки (рис. XV. 27).
437
На рис. XV. 28 изображен питатель с вращающейся звездой кон.
Подача твердых материалов регулируется числом оборотов зве-
здочки.
Рас. XV. 26. Распределительная тарелка.
Па рис. XV. 29 представлена аппаратурная схема адсорбцион-
ной установки периодического действия для жидкостей.
Рпс. XV. 27. Питающий ме-
ханизм с задвижкой.
Рпс. XV.,28. Пита-
ющий механизм с вра
ща toincii ся зв ездоч -
коп.
Пз смесителя, где происходит адсорбция примеси из жидкости,
суспензия подается насосом на фильтр-пресс, откуда очищенная
жидкость поступает в приемник.
438
Рис. XV. 29. Схема адсорбционной установки периодиче-
ского действия для жидкостей:
1 — адсорбер, 2 — фильтрпресс; з — сборник фильтра.
Рис. XV. 3U. Аппарат для
непрерывной 2-ступенчатой
противоточной адсорбции:
з — корпус; з — рубашка:
з —мешалка; > —стакан: J
рубашка стакана; с — стойка;
2 — труба для псрсданлиьа-
шш; 8 — вад мешалки.
Аппарат для непрерывной 2-ступенчатой противоточной адсорб-
ции изображен па рис. XV. 30, адсорбционная установка непре-
рывного действия для жидкостей с противоточным движением
взаимодействующих масс — на рис. XV. 31.
*—• Уголь
-►—о- Жидкость
-и—о- Уголь и жидкость
---- Воздух
----дары жидкости
Рис. XV. 31. Схема непрерывней 2-ступоп-
чатоп противоточной адсорбции:
1 — .шпарит .тля перемешивания; s — вакуум-
фильтр: 3 ciinpiiiii: очищенного раствора: 7 —сбор-
ник загрязненного раствора; > печь Герссгоффа;
'» — регенератор; ? — печь; 8 — конденсатор; о —
циклоп.
Здесь смесители непрерывного действия с внутренними цирку-
ляционными стаканами связаны с (барабанными фильтрами. Обра-
ботанный адсорбент поступает в печь типа Герресгоффа для реге-
нерации. а отгула через бункер снова в аппарат для адсорбции.
Глава XVI
Ионный обмен
Ионный обмен
в процессах водоподготовка
(Периодический процесс)
Аппарат для ионного обмена
периодического действия
На рис. XVI. 1 показан Н-катионитовый фильтр диа-
метром 1600 зьн и общей высотой 5030 Этот аппарат снабжен
двумя тарельчатыми плитами, за-
жатыми между фланцами. На пли-
тах имеются отверстия диаметром
20 лмц па которых закрепляются
дренажные колпачки. Вся внутрен-
няя поверхность фильтра, поверх-
ность плит и других внутренних
деталей фильтра гуммируются слоем
резины толщиной 5 льм. На случай
заклинивания щелей дренажных
колпачков зернами катионита, пре-
дусмотрен подвод сжатого воз (уха
для их продувки.
Расчет процесса
водоподготовки.
Кинетика
ионного обмена
в процессах
водоподготовки
Рассмотрим процесс водо-
подготовки, который состоит в уда-
лении из воды металлических попов
путем обмена их на ион натрия, ко-
торым заряжена ионообменная
смола.
Рис, XVI. 1. Аппарат для ионного обмена
периодического действия.
fcefSod £sc'fr'.
регенерационного раствора
и oinSoSSoBc; аосле Eopt/x-
/тени}! кат ирнцта^
/!pnufi .ч&пи рнитп
Гарел еиотоя алита
ngamS с>?0 „0 0 р
Обратная реакция осуществляется при регенерации ионита
посредством обработки его раствором поваренной соли.
Скорость ионного обмена при постоянной температуре и по-
стоянной скорости потока, согласно экспериментальным данным,
пропорциональна произведению концентрации, например, ионов
кальция в воде и квадрату концентрации натрия в смоле.
Таким образом, имеем:
для скорости прямой реакции
z-j Адиг- (XVI. 1)
для скорости обратной реакции
г2= — — ^'ЛХаЧ)3 Р (XVI. 2)
Суммарная скорость реакции
гп = Адмг2 — А» (Ya'?)2 Р (XVI. 3)
где р — содержание кальция в смоле, эквивалент на ед. массы;
и — содержание натрия в смоле, эквивалент на ед. массы;
q — содержание патрия в воде, эквивалент иа миллион
ед. массы воды;
и — содержание кальция в воде, эквивалент на миллион
ед. массы воды;
— эквивалент извлекаемого из воды кальция/ед. массы X
Хед. времени;
гп — эквивалент кальция, переходящего из смолы в рас-
твор/ед. массы-ед. времени;
/•„ — суммарная скорость понижения жесткости воды, экви-
валент ед. массы-ед. времени.
уа — коэффициент активности для иона патрия в растворе;
при низких концентрациях уа = 1.
Найдено, что константы скорости реакции не зависят от ско-
рости потока, но возрастают с уменьшением размеров частиц
ионообменной смолы, а именно:
Mein........ 2-14 14—20 20—28 28—35
Ад.......... 1,62 1,91 2,29 2,86
Константа равновесия для данной реакции
Л‘ж-Ь- = 241000
Таким образом, в период понижения жесткости воды скорость
обратной реакции пренебрежимо мала.
Переменные u, v, р и q являются функциями расстояния х
и времени т. Будем находить выражения для и и и в зависимости
от х и т. Для элементарного слоя ионообменной смолы толщи-
442
пой dx па расстоянии х с единичной площадью сечения, располо-
женного нормально к направлению потопа, имеем
С, / du \ , ( др j \ । <?в'т ди ,
юД—a?‘'9rfr-=c“"'4W',Tr!-TiF лЛ’”т (XVI
где рпас — насыпная плотность ионообменной смолы;
р„ — плотность воды;
G — скорость воды, кг'м--мтг,
г/ — объем, занимаемый водой в смоле.
Последний член равенства (XVI 4) представляет собой изме-
нение содержания кальция в воде для свободного пространства
ионообменной смолы, которое достаточно мало но сравнению
с количеством протекающей воды. Пренебрегая этой величиной,
получим:
Л = _2!rSs±.±!. (XVI 5)
()х (j их
Так как каждый эквивалент нона кальция при переходе к смоле
заменяет собой эквивалент натрия, то бу щм иметь:
или
Р — /’в=г11—и
де др
дх дх
(XVI •;)
(X VI 7)
При сопоставлении (\\1. 1), (XVI. 5) и (XVI. 7) найдем:
ди. ,
— — auv-
(Wf.S)
дх
1 Щ
Й=1(М“^- ц ь=-к
Задача свелась к интегрированию системы двух дифферен-
циальных уравнепи i XV I. 8) частных производных с двумя неиз-
вестными функциями и и V.
Нам пу;кпо найти то решение этой системы, для которого
к (II, т) - иу, г (z, 0) --- г0 (X V1. 9)
Для получения этого решения заметим, что из уравнения (XVI. 8)
с 10 1ует. что
д (bti) (! (а у)
дх дх
а это означает, что avdx -f- bit dr есть полный дифференциал неко-
торой функции / (х, т). Если эту функцию мы найдем, то и и и
определяется формулами:
1 df 1 д/
— —— ‘ и — — —
а д.т. * Ь dx
(XVI. 10)
Легко составить дифференциальное уравнение, которому удо-
влетворяет функция / (х, т). Если в (XVI. 8) заменить их выраже-
ниями через / при помощи формул (XVI. 10), то мы придем к урав-
нению:
д-f df / df
(t ----~
<Jx t/т Лг \ dx
определяющему функцию /. Это уравнение запишем в виде:
у/ Й / 1 \
- “- / -— I
дх----------------дх I д] I
\ ~дх /
(XVI. 11)
Найдем, при каких дополнительных условиях нужно инте-
грировать это уравнение. Положив в первом равенстве (XVI. 10)
т = 0, мы, на основании второго условия (XVI. 9), найдем:
df
дх
r—ч
аг„
(XVI. 12)
Интегрируя это равенство по х, имеем:
/ (*, о) = «еъх
(XVI. 13)
где ci — произвольное постоянное.
Точно так же из второго уравнения (XVI. 10) находим:
— / (0, т) = 1ш0-|-с.
(XVI 14)
Полагая в условии (XVI. 13) х -= 0, а в условии (XVI. 14)
т = 0, найдем <д = с2. Значит, уравнение (XVI. II) нужно инте-
грировать при условиях:
f(x, О) = агиж4-с; / (0, т) = Ьн0 -|-с (XVI. 13)
Интегрирование уравнения (XVI. 11) по т дает:
' = -^+ФМ; -Й-=ф(^=7 <XVI-,B)
Здесь Ф (я) означает произвольную функцию, зависящую
только от х. Для нахождения этой функции положим в последнем
уравнении т — 0. Тогда, используя (XVI. 12) и (XVI. 15),
получим:
Л
ЯРЙ= -у—---------
Ф (х) — ДГ0Х — с
Отсюда определяем функцию Ф (х):
Ф(зН
г0
Уравнение (XVI. 16) обращается, таким образом, в следующее
обыкновенное дифференциальное уравнение:
df ___________а__________
dx , I
ог0.г-|-с4-------/
г-0
НЛП — Г0Х ;----------;--------— (Х\ 1. Il)
df ” а ‘ аг0 а '
Интегрируя это линейное относительно х уравнение, найдем
его общее решение:
7*0 — 1Ч (/ — 6О-0.Т — <•) ф (т)
где ф (т) — произвольная функция одного т.
Для нахождения функции ф (т) положим .г = 0 и восполь-
зуемся вторым условием (XVI. 15). Найдем:
ip (т) -- Ьиоеот 4- се0 — In &иот
Следовательно, функция / определяется следующим неявным
уравнением:
/г0—In (/—агох —e) = twveoT-rct’o — In 6w0t (XVI. 18)
Для нахождения зависимости, определяющей функцию v,
воспользуемся уравнением (XVI. 17). Принимая во внимание
первую зависимость (XVI. 10), найдем
• , + + 1 I
/=<ц;ох4-с-}------
Со v
Внесем это выражение для / в формулу (XVI. 18). Мы получим
следующее уравнение, определяющее функцию и:
—-----1- — Ьчп»йе
(XVI. 19)
Функцию и можно выразить через функцию v следующим обра-
зом. Продифференцируем равенство (XVI. 19) по т и на основании
1
уравнения (XVI. 8) заменим па Ьи.
После ряда преобразований получим:
1
i’Wo1сД
(XVI. 20)
Уравнения (XVI. 19) и (XVI. 20) дают окончательные решения
системы (XVI. 8).
Для упрощения примем:
1?на<Л11;оа'
- - 6.
X — —^П(|1уТ —- к
И = ^2--1
I-
{XVI 21)
l’i:c. XVI. 2. Диаграмма дня расчета пошгого обмена в прессах водоподготовки.
Тогда из (XVI. 19) и (XVI. 20) получим:
In 7, -} z =iln 54-5 — г
v _ 1
~ 1+Z
и _ 1 + 1/5
I'o 1 + 1/2
(XVI. 22)
С целью ускорения расчетов для практических задач построена
диаграмма (рис. XVI. 2) в соответствии с уравнениями (XVI. 22),
где переменные представлены в виде безразмерных групп г, S,
S/Г И и.их>.
Определение остаточной жесткости воды
Желательно определить остаточную жесткость воды
после того, как ионообменный фи 1ьтр проработал в течение 16 ч,
вода проходила через слон попита толщиной 0,912 .и со скоростью
16,35 л!м-‘Мин. Начальная жесткость воды соответствует 300 час-
тям углекислого кальция на миллион частей воды, что составляет
6,0 кг-экв попа кальция на 1000000кг воды. Начальное содержа-
ние заменяемого натрия в ионообменной смоле эквивалентно
57 250 г СаСО3/.мЛ
Насыпная плотность пойнта 450 кг .и3; размер зерен ионита
14—20 м<чп. (пли 0,000974 .м).
Имеем:
Оцас= 459 кг/л3; х= 0,912 л
Ад I 91 мин 1 (см. стр. 442); G = 16,35 кг .и2-мин
(57 ‘>50 \ / '> \
—-— ) [ —— 1 — 0,00255 кл-лкв \а+/кг смолы
100 J \ 45U J
ии = 6,0 кг-зкв Са** па 1000 000 кг поды
т = 16-60=960
Из (XX I- 21) получим:
10° • 450 • 1,91 • 1ЦЮ2552 • 0.912
г—--------77------- — -------------... .,Ч — 31 ,и
6 16,33
5 = = 1,91- 6,0 • 0,00255 900 = 28,1
Используя (XVI. 22), будем иметь:
и 1Ц
Далее:
In Z + Z = In 5 + 5 — г = 1п 28,1 + 28,1 - 31,3 = 0,14
2 = 0,62
и
«о
1,0356
2,61
0,397
Следовательно, искомая величина и составит и = 0,39? *6,0 -=
2,33 кг-экв Са2* на 1 000 000 кг воды, оставляющей ионообмен-
ный фильтр в конце 1(5 ч.
Так как г — 31,3 и S — 28,1, то Sr — 0,898. Значение н'и0,
соответствующее этим данным, равно 0,36 (см. рис. XVI. 2).
Определение константы скорости реакции
для ионного обмена
в процессах водоподготовки
Определить константу скорости реакции /;, для ионо-
обменного фильтра. Толщина слоя ионообменной массы 0,912 л
с Quae 450 ка/л13. Максимальная ионообменная емкость соста-
вляет 37 250 г СаСО3 па 1 .м3 смолы. Вода, содержащая начальную
жест кость в количестве 300 частей углекислого кальция па мил-
лион частей воды, проходит через фильтр со скоростью
16,35 лм--мин в течение 14,1 ч, причем конечная жесткость воды
составляет 7 частей СаСО3 на миллион частей воды.
Имеем:
% 6.00; 0,00255; G 16.35 кг'м-~мип
- —0,0233; т= 84(5 -ww«
"о
S 6-16,35-846
Г 1U*-450-(>.00255-0.912
На рис. XVI. 2 точка пересечения S,r — 0,79 с и'и0 0,0233
дает значение г = 32
Таким образом, искомая величина составляет:
}. __ г(‘____________32- 16.35______
4 106о игх 10е-450-0,00255--0,912 - ’
|и Олас о
Моделирование ионообменных с^ильтров
В ионообменной колонне протекают в основном три
процесса, а именно: проточная экстракция, реакция обменного
разложения, сопровождающегося диффузией, и фильтрация, т. с.
движение жидкости через слой ионита.
Для количества вещества, вступающего в реакцию ионного
обмена, в элементарном объеме за время dx будем иметь:
g —d (г dx) = kf (с) dx dr
где к — постоянная величина, характеризующая природу реакции.
Составляя материальный баланс таким же образом, как для
диффузии, сопровождающейся химической реакцией (см. стр. 103),
получим уравнение:
de Г / de \ ( de \ 1
_/9 __ и ( ) • d ( t ) t/T__ /.-/ е}х dx - 0
dx L \ dx } \ dx J J '
Упрощая, придем к дифференциальному уравнению второго
порядка:
.т
Т)
Как л для проточной экстракции (см. стр. 390), имеем допол-
нительные дифференциальные уравнения, описывающие процесс
ионного обмена в колонне:
de
Применим метод масштабных множителей для приведения диф-
ференциального уравнения ионного обмена (XVI. 23) к крите-
риальному виду:
с' — Лсс; х* = Л jx; к'— -Af(k; D' -
Таким образом, для модели из дифференциального уравнения
получим:
Л Д' . (Pc _ AkkAcf (с)
Л] '
или
1 _ Ле
Отсюда находим
kl- k’lr-
D ~ D'
т. с. критерий, характеризующий реакцию обменного разложе-
ния . сопровождающуюся диффузией.
Обозначим критерий ионного обмена через UQ, тогда
Следовательно, критериальное уравнение рассматриваемого здесь
процесса, вытекающее из представленных выше дифференциаль-
ных уравнений, принимает следующий вид:
Nu ~ £ Нса'Гг,Л<1ф'(1^'Ф®'Uj' (X VI. 24)
где ф, а , Ъ’, с', d', с', h‘ — постоянные и отвлеченные числа, уста-
навливаемые экспериментально.
29 л М. Батупср. 'ViO
Соображения, приведенные выше, могут быть использованы
для моделирования ионообменных фильтров. С этой целью рас-
смотрим ионообменную реакцию
NaCl-fr Кат[Н] Кат [NaJ-|-IlCl
Так как степень завершенности процесса ионного обмена пред-
ставляет собой заданную максимально возможную величину,
то критерий подобия Ф3 может быть опущен в экспериментальных
исследованиях.
Примем также следующие обозначения:
w— фиктивная скорость жидкости, лг'сск;
I — конечная высота попита, .и;
v — кинематический коэффициент вязкости раствора, л2 •сек;
р—динамический коэффициент вязкости раствора, кгл-сск;
j — объем раствора, профильтрованного до «проскока»
л3/л2 • сек-,
с — концентрация извлекаемого вещества в жидкое ги, кг .и3;
z — содержание извлекаемого вещества в твердой массе, кг л:;;
т — объем жидкости па единицу сечения фильтра и на 1 .и
высоты аппарата, л3/л‘2-л;
п — объем попита на единицу сечения фильтра и па 1 л высоты
аппарата, л3 м~ • л;
а — коэффициент массопередачп, л3.и’2-сек, или мсек-,
D — коэффициент диффузии, лг/сек;
т — время, сек;
q — плотность раствора, кг/л3.
Коэффициент массообмсна а, входящий в критерий Нуссельта,
определяется по формуле:
г;
U ~ F • Лет
где G — количество поглощенного вещества, кг;
— поверхность зерен катионита, л2;
Дс — средняя движущая сила ионообменного процесса;
т — время фильтрования, сек.
Значения Ас вычисляются по формуле:
2,3031g.—!Ц-
Ьо — г
где г — динамическая емкость до «проскока» для единицы объема
катионита, кг/л3;
«и — статическая емкость, отвечающая состоянию равновесия,
отнесенная к единице объема катионита, кг л3.
450
Значения вычисленных критериев подобия представлены
в табл. XVI. 1.
Зависимость (XVI. 24) может быть найдена путем обработки
экспериментальных данных.
Определение коэффициента а . Используя эксперимен-
тальные значения критерия Хи при переменных величинах Нс
получим:
lg Nil = lgC!-b«' lg Re
Рис. XVI. 3. Зависимость
Nu от Re.
Рис. XVI. 4. Зависи-
мость Хи от J’г.
Изооразия зависимость между Ren Xu графически на логариф-
мической сетке (рис. XXI. 3); значение показателя степени равно:
а'= 0.92
Определение коэффициента Ь’. Определяем экспери-
ментально Хи при переменных значениях Рг. Имеем:
lg Nu — lgC24-d Ig Рг
Из рис. XVL 4 найдем:
Ъ'~ —2,90
29*
431
Вычисление крите
W С,-10* D- 10l« л-10« V-10* c z a Ac
0,0013 4,28 2,3 1.8 967 0,575 18,5 1000 0,025 h. 15,8
0,0033 3,78 2,3 1,8 870 0,575 16,4 1000 17,3
0,00GG 3.38 2,3 1,8 780 0,575 14,6 1000 18,5
0,0099 3,18 2.3 1,8 750 0,575 13,8 1000 19.1
0,0013 4.20 2,7 1,19 440 1.15 18,3 1000 0,05 h. 15,8
0,0033 3,43 2.7 1.19 390 1.15 14.8 1000 17,5
0,0066 3.34 2.7 1,19 360 1,15 14,5 1000 18.4
0,0099 3.18 2,7 1,19 356 1,15 13,8 1000 19,1
0,0013 4.1 3.2 1.2 230 2.3 17,8 1002 0,1 в. 16
0.0033 3,4 3.2 1,2 189 2,3 14.8 1002 18,4
0,006G 3,1 3,2 1,2 186 2,3 13,5 1002 19,2
0,0099 2,94 3,2 1.2 160 2,3 12,8 1002 19,7
0,0013 3,5 3,9 1,21 110 4,6 15,2 1006 0,2 и 17-5
0,0033 2,97 3,9 1.21 9u 4.6 12.9 1006 19,7
0,0066 2,97 3,9 1,21 83 4,6 12,9 1006 19,7
0,0099 2.72 3.9 1.21 82 4.6 1,18 1006 20.4
452
Таблица XVI.1
рисе подобия
X rf a-10® ft 10® Re Pr Nu Ф< Ф; L’d
NaCl 14 500 18,8 78 4.2 10 5100 3,04 18,6 0.220 2 440
5 150 16,7 180 11,1 25 5100 7.05 16,8 0,248 6 270
2 250 14,8 346 17.2 50 5100 13,6 15,1 0,280 10 500
1 500 14 472 22,3 75 5100 18,5 14,5 0,295 12 500
NaCl 6 750 18,5 166 7,38 10 4350 5,52 8,7 0,256 3 520
2 320 15,1 360 17.2 25 4350 12,0 7,5 0,318 8220
1100 14,7 715 34.4 50 4350 23,6 6,9 0,322 16 800
720 14,0 975 47,5 75 4350 32,7 6,8 0.340 22 600
NaCl 3 450 18,1 321 12,4 10 3680 ’ 9,04 4,4 0.530 5 000
980 15 870 31,8 25 3680 22,0 3,6 0,640 12 800
520 13,7 1350 58,3 50 3680 38,0 3>5 0,700 23 400
320 13,0 1980 123,0 75 3680 55,5 3,0 0,740 49 500
NaCl 1460 15,4 586 34,0 10 3060 13,5 2,4 1,27 11 300
510 13,0 1268 72,0 25 3060 29,6 1,7 1.50 24 (ii iO
250 13.1 2560 134.0 50 3060 59,4 1,6 1,50 44 500
170 11.9 3320 17,2 75 3060 77,0 1,5 1,64 5 700
453
Рис. XVI. 5. Зависимость Nu
от Ф£.
Определение коэффициента с'. Определяем экспери-
ментально Nu при переменных значениях Фг Производя анало-
гичные построения, найдем
из рис. XVI. 5:
с’ — —6,20
Определение ко-
эффициента d'. Из рис.
XVI. В находим:
d' = 5,40
Определение ко-
эффициента Л'. Определяем
Nu при переменных значе-
ниях критерия ионного
обмена н.о. 1 (ользуяс ь
рис. XVI. 7, получим:
h’~ 1
Таким образом, Кри-
те риалыюе уравнение про-
цесса ионного обмена меж-
ду раствором хлористого натрия и смолой имеет следующий вид:
tri ХМ-* / V ‘2,fi0 / Ут \-6,20 / Усг \5,10 / hl'2 \
v / \ D ) \ ml ) \ nlz / \ U /
454
Полученное уравнение позволяет определить характер и сте-
пень влияния переменных факторов па данный процесс ионного
обмена и, следовательно, установить рациональный режим работы
фильтра.
Ионный обмен
в производстве антибиотиков
и в химических процессах
(Непрерывный процесс)
Характеристика процесса ионного обмена
в производстве стрептомицина
В производстве стрептомицина применяются периоди-
ческие процессы сорбции и десорбции на карбоксильной смоле
КК-4П-2 в солевой форме. Они имеют следующие недостатки.
1. При сорбции и десорбции стрептомицина с течением вре-
мени в аппарате (колонне) образуется в прогрессирующем коли-
честве отработанная смола, которая становится балластом
(рис. XV. 1). На рис. XV. 1 зона сорбции представляет постепенно
убывающую высоту эффективного слоя ионита в аппарате.
2. Для периодических процессов сорбции и десорбции тре-
буются относительно большие количества смолы в обороте.
3. Возможность получения смолы с достаточно высокой актив-
ностью при сорбции, а также концентрированных растворов стреп-
томицина при десорбции в этом случае практически исключается.
4. При большой толщине слоя смолы (2—3 .w) последняя
уплотняется с образованием местных ходов для жидкости, кото-
рые обусловливают низкий коэффициент использования смолы.
Продолжительность проведения основных процессов ионного
обмена стрептомицина очень велика, что способствует разложе-
нию нестойкого продукта.
Аппарат для непрерывного ионного обмена
в производстве стрептомицина
Перечисленные выше недостатки устраняются при
непрерывном процессе ионного обмена стрептомицина, который
предлагается осуществить в аппарате, изображенном па
рис. XVI. 8.
Ионообменная смола находится в ковшах L1 ленточного аппа-
рата, который представляет собой фильтр с медленно движущейся
непрерывной цепью 8. Фильтр состоит из отдельных секций-сту-
пеней. Ковши снабжены снизу и сверху перфорированными пла-
стинами, которые служат днищем и крышкой и, таким образом,
455
Рис. XVI. 8. ,’Гепточпыц аппарат для иппрсрывного ионного обмена:
1 — вил; 2 — зпеадьчки; .ч —крепление; 4 — кожух; .5 — крышка; в -разбрызгиватель; 7 — камера; $ — цепь; а — соедини-
тельная трубка; /а —приемная воронка; и цнвш-фнльтр; 12 — оуикср; J-'f — заслонка; 14}— '.питатель; /а — шкив; //> — сцеп-
ление; 17 — груз; м — спуск; i'j -стопка; 20 — насос; 21 — вентиль.
смола находится в непрерывном движении оез выгрузки в течение
1.5—2 лет до полного истощения.
Каждая фильтрующая ступень включает в себя разбрызгива-
тель 6’, филыр //, приемную воронку 10 и насос 20.
Во время своего продвижения смола орошается раствором
стрептомицина, и при атом происходит сорбция. Каждая воронка
питает соответствующий насос, который, в свою очередь, питает
соответствующий распылитель. Каждая ступень аппарата имеет
осушительную или дренажную зону для некоторого стока
жидкости.
Исходный раствор стрептомицина направляется в аппарат
через его пятую ступень (но расчету) и проходит в противоточном
по отношению к смоле направлении при перекрестном орошении
ее потоком жидкости в фильтрующих ступенях. Освобожденная
от стрептомицина жидкость выходит из аппарата через его первую
ступень. В шестой ступени аппарата происходит промывка
смолы водой.
Следующие две ступени, согласно расчету, служат для десорб-
ции стрептомицина из смолы раствором серной кислоты.
На этом же аппарате с помощью фильтрующих ступеней при
транзитном движении смолы происходит ее промывка водой
и перевод водородной формы смолы в натриевую, после которого
ионит снова непрерывно поступает вместе с перфорированными
ковшами в сорбционную зону аппарата. Фильтрующие ковши
со смолой непрерывно омываются внизу водой, которая исполь-
зуется в качестве промывной жидкости для соответствующих
ступеней процесса.
Бункер 12 и отверстие внизу кожуха предусмотрены, соответ-
ственно, для загрузки свежей смолы ири пуске системы и выгрузке
отработавшей смолы в конце периода непрерывной работы уста-
новки (1.5—2 года).
Привод ведущих звездочек 2 осуществляется через редуктор.
Тяговые звездочки 2 вращают перфорированные ковши, причем
скорость вращения звездочки можег меняться в широком диапа-
зоне, благодаря наличию вариатора скоростей.
Материалом для установки .может служить сталь марки
ЭН-530.
Сорбция стрептомицина
Определение скорости движения ионообменной смолы
иа лейте. Имеем:
1. Скорость движения жидкости 0,9лс.и'-ч (но данным
завода).
2. Количество раствора стрептомицина, поступающего в аппа-
рат К = 5250 л.ч (по данным завода).
157
мг-экв Sir
3. Коэффициент массопередачи К 0,154
мг-Dt: в SU‘
г - ч-------—
л.
4. Начальная, концентрация стрептомицина л растворе С\ =
3000 сд{.и.1= 12,2 мг-зкв ЯЛт/л (по данным завода).
5. Выход па стадии сорбции 99% (по данным завода).
6. Конечная концентрация стрептомицина в растворе при
выходе нз аппарата С2 — 0.01 • 12,2 0,12 мг-зкв.л.
Рис. XVI. 9. Построение равновесной и рабо-
чей линий для расчета сорбции стрептомицина.
Количество стрептомицина, сорбированного смолой.
СОСТЛ-
вляе г
5250 (12,2 — 0,12) = 63 500 мг-экв/ч
Содержание стрептомицина в смоле после его регенерации
принимаем V2 — 0,2 мг-экв Str/a ионита.
Строим рабочую линию. На рис. XVI. 9 отмечаем точку
(С.,, Х2). При минимальном отношении количеств ионита и рас-
твора рабочая линия проходит через точку при значении X —
.> мг зкв Str
3,0.) -------, соответствующем равновесной концентрации
г ношпа 1 1
раствора С\ 12,2. Тогда минимальная скорость движения твер-
дой массы ионита будет:
63 5<>о
-2^-—=18 200 ?.ч = 18,2 ч
3,6-) — 0,2
Принимая избыток смолы равным 20%, получим действитель-
ную скорость движения ионообменной смолы на лепте:
18,2 - 1,2 = 21,8 кг/ч
Из баланса по стрептомицину
63 500 = 21 800 (X,—0,2)
н.идем:
А'ц = 3,1 .чг-экв Str/г ионита
где — действительное содержание стрептомицина в ионооб-
менной смоле.
Изобразим па рис. XV 1.9 точку (С’,>, А\). Соединяем точки
(Cj, XJ и (6\, А\) прямой линией, которая при данных низких
концентрациях является рабочей линией.
Число ступеней ионного обмена ленточного аппарата
в зоне сорбции стрептомицина и количество ионита на лепте.
Количество ионита на ленте может быть определено с помощью
кинетического уравнения:
»S2oHac=-^-J с —С" (XVI. 25)
ۥ1
S’Zo.^c — количество ионита (смолы) на ленте, кг;
S — площадь, занимаемая лептой;
Z — высота слоя смолы на ленте;
Риас — насыпная плотность смолы;
А’ -0,15 \
мг-л>;в Str
.чг-икс Str
г ч---------—
л
Для любой концентрации С между имеем соответств у гощес р авновесиое емое с помощью равновесной кривой (см. стрелки на рис, XVI. 9) С .... 0,2 0,5 0,8 1 2 С” .... 0 0,1 0,1 0.12 0,2 С\ и С., на рабочей линии значение С~, оиределя- прп том же значении X 4 6 8 10 12 0.6 1 1,6 2.8 4,5
I .... 5,0 2,5 1,43 1,2.’ > 0,556 0,294 А 9 0,156 0,139 l!,I39
С—С'
Строим кривую в координатах С и 1 и вычисляем графи-
чески интеграл в пределах от С, до С2. Площадь под кривой
(рис. XVI. 10) составляет 5.
Следовате ы.ио, число ступеней ионного обмена для .ленточ-
ного аппарата в зоне сорбции стрептомицина .¥ 5. Количество
459
ионообменной смолы, которое должно постоянно находиться
на ленточном аппарате в зоне сорбции, равно
SZq1wc --- 170000 г-- 170 кг
О, К)!
Определение длины ленточного аппарата в зоне сорб-
ции стрептомицина. Объем смолы:
112. = 0,680 л3
2 0
где 250 кг'м3 — насыпная плотность смолы в набухшем состоянии.
Рис. XVI. 10. Диаграмма для графического инте-
грирования в расчетах сорбции стрептомицина.
Высота слоя смолы на лепте Z = 0,25 .w; тогда площадь ленты
в зоне сорбции стрептомицина
е 0,680 _...
.8 = —-—=2,/2 -U-
0,2.)
Ширина ленты 0,8 .и. Таким образом, длина ленты:
2.72
Z - ‘
0.8
Объемная скорость движения смолы:
^= 0.OS7 •«’?.
Линейная скорость ионообменной смолы (ленты):
1Г
0,087
0.25 • 0,8
= 0.435 .и/ч
460
Длина ленты для дренажа между ступенями ленточного аппа-
рата, в зоне сорбции:
Г = «1 гт _= /(. 0/135 - 0,25 = 0,14 м
где к = 4 — число промежутков для ступеней аппарата;
т = 0,25 ч — продолжительность дренирования раствора для
каждой ступени.
Общая длина лепты в зоне сорбции стрептомицина составляет
£соРб= г-Ы= 3,4 1-0,44=3,81 -«
Десорбция стрептомицина
Кинетическое уравнение применительно к непрерыв-
ному процессу ионного обмена для ленточного аппарата в зоне
десорбции стрептомицина. Интегрирование кинетического урав-
нения (XVI. 25) в случае, когда рабочая и равновесная липни
являются прямыми, даст
V (С3 - G) = K.szGfiac (С- - С)ср (XV1. 26)
причем (С‘ — C)clt — средняя логарифмическая разность движу-
щих сил па концах аппарата в зоне десорбции стрептомицина, т. с.
(С* — С)ср
(с;-с,)-(с;-с,)
Число ступеней ионного обмена и смолосодержапие
для ленточного аппарата в зоне десорбции стрептомицина. Для
обмена 63 500 мг-экв Str/ч требуется такое же количество мг-экв
Н+/ч. При 27%-ном использовании серной кислоты последняя
должна содержать:
63 ,Ю0 _ мг.экв н+/ч
0,2/
Или
235 000 ... , . о
—=181 л/ч 1,3 н. серной кислоты
JL
Имеем:
г-о п г 635(Ю_->гП str
С ]_ — U II Go— — u<Jv —
lol Л
Кроме того:
X,- 0,2 и Х2 = 3,1 ST Slr
^Чере.з точки (Cj Xj и (С; Х2) на рис. XVI. 11 проводим
рабочую линию.
Для определения смолосодсржания па ленточном аппарате
в зоне десорбции используем формулу (XVI. 26):
(6s Ci)» А52()цас (6” — Оср
Имеем (рис. XVI. 11):
Cj-t’j- 40-0 = 40
-И<?-.9Л’в Str
„ мг-зкб Str
С„ = 620 — 350 = 270 ---------
Таким образом:
Рис. XVI. 11. Равновесная и рабо-
чая линии для расчета десорбции
стрептомицина.
Подставляя это значение
в уравнение (XVI. 26). найдем:
63 500 - 0,0056 (5Zqii;1(.) 158.5
Отсюда получаем величину
смолосодержания. на ленточ-
ном аппарате в зоне десорб-
ции стрептомицина:
f..z 63 500
'1,ас^Жб6-158,5
~ 72 000 г = 72 л?
Число ступеней для ленточ-
ного аппарата в зоне ионооб-
менной десорбции стрептоми-
цина:
230
7V — —~- = 1,45 ^2 ступени
lao.a
Длина ленточного
аппарата в зоне дес орбцип
стрептомицина. Обтд'ы смолы:
~ 0,288 .ч3
где 250 кг.'м5 — насыпная плотность смолы.
Высота слоя смолы па ленточном аппарате Z — 0,25 м. Сле-
доватол ьно:
Ширина лепты 0,8 -it. Таким образом, длина ленточного аппа-
рата в зоне десорбция:
Длина ленты для дренажа между ступенями аппарата:
Г =: riirt = 1 0,435 • 0,25 = 0,11 .и
где п — 1 — число промежутков между ступенями;
и.' = 0,43.'» .и ч — скорость движения ленты;
т — время, ч.
Общая длина ленты в зоне десорбции:
•£<дсс ~ —1,45*1-0,11 = 1,56 .’I
Длина ленточного аппарата для зон сорбции и десорбции
стрептомнцппа составляет:
^cupu“i- ^дсс ~ 3,84 -f- 1,5(> - 5.40 >,5
Ионный обмен медного купороса в башне
при его регенерации из раствора
Рассмотрим следующий пример.
Из разбавленного отбросного раствора надлежит извлечь медь
с помощью ионного обмена. Концентрация CuSO, в растворе соста-
вляет 21) .на-акд Си2*/л; скорость потока раствора 37 850 л'ч.
Для осуществления данного процесса ионного обмена предусма-
тривается установка непрерывного действия, где раствор и регене-
рированный попит проходят через вертикальную башню в про-
тивоположных направлениях. Степень обмена Си2* равна 0,99.
Этот процесс может быть осуществлен и на ленточном аппарате
с перекрестно-противоточным движением фаз.
Регенерация ионита производится во второй башне при проти-
воточном контактировании с 2н. I-LSO.,.
В примере для расиста приняты:
II з в л е ч е н и e Си2*. Скорость движения жидкости
2.2 л/е,и--ч; скорость массопередачи 2,0 мг-экв Си2*/ч*а ио-
нита -мг-экв Си2+. л.
Регенерированный ионит содержит 0,30 мг-экв Си2+/а ионита
(X.,). причем его количество составляет 1,2 Л/, где М — мини-
мальное отношение количеств ионита и раствора.
Р е гене р а ц и я и опита. Скорость движения жидкости
0,17 л. см~-ч, скорость массопередачи 0,018 мг-экв Си2*;ч-<? ио-
нита -мг-экв Си2*.'.г.
Степень использования кислоты 70%.
Равновесное состояние при ионном обмене Си2*—П+ для кон-
центраций 20 и 2000 мг-экв катиона/л изображено на рис. XVI 12
и XVI. 13.
Требуется определить скорость прохождения ионообменной
массы и се количество в каждой башне.
1. II з в л е ч сине Си2*. Известно, что скорость потока
раствора 37 850 л ч; концентрация раствора 20 мг-экв Си2* л,
С.2 = 0,01 -20 = U,20 мг-экв Си2*/л.
463
Количество обмениваемой Си2* составляет:
37 850 (20—0,20) 750 000 мг-лкз/ч
Величина Х’2 = 0. 0 мг-экв Сн2*/г ионита. На рис. Х\ 1. 12
отмечаем точку (С2, Х2). При минимальном отношении количеств
ионита и раствора и бесконечно большой высоте башни рабочая
линия проходит также через точку Р при А — 4,9, соответству-
ющую равновесной концентрации С\. Тогда минимальная скорость
движения твердой массы будет:
750 000
1Ю11П"
Гис. XVI. 12. Равновесное состоя-
ние при полном обмене Си2* — Ц+
для концентрации 20 .иг кати-
она/л.
Умножив па 1,2, получим:
Рис. XVI. 13. Равновесное состоя-
ние при ионном обмене Си2* — II*
для концентрации 2000 мв-экв ка-
тиона л.
1(53 000- 1,2 = 196 000 г/ч
Баланс по меди дает
750 000 = 19G (Xi — 0,30)
откуда:
А'1-4,12 мг-мм Си2*/г ионита
Изобразим на том же трафике точку (С\. Х^), после чего про-
ведем прямую линию, которая при данных низких концентрациях
является рабочей линией.
Количество ионита в башне может быть определено с помощью
кинетического уравнения, которое получается при составлении
материального баланса:
V de = — (С — С') d (SZо) (XVI. 27)
где V — количество жидкости, л/ч;
С — концентрация Си2* в растворе, мг-экв.'л раствора;
4G4
С' — концентрация Сн‘2+ в растворе при равновесии с твер-
дой массой, мг же. л раствора;
Л Л y у /
—----коэффициент массопередачп, мг-энв ч -г ионита-мг-экв л\
К — коэффициент массонерсдачп в жидкости, мг-окс.ч -.if- X
X мг-лкв/л;
а — поверхность частиц пойнта, .и2 .и3;
р — плотность повита, г.'елг3;
SZo — количество пойнта в башне, г;
Л — площадь поперечного сечения башни, c.v ;
Z — высота башни. с.н.
Преобразуя это уравнение и интегрируя его. получим:
с,
' <!С
Ка 1 С —С'
О •/
Се
Для С на рабочей линии между С1 и С., имеем соответствующие
значения С\ определяемые с помощью равновесной кривой при
том же значении X (таб г. XVI. 2).
2'аб.гица XI 1.2
Дачные оля построения равновесной кривой
С 2и 1С 12 S 1 1 0.»
С* 2/. 1.9 0,5 0.25 0,10 0,05 0.02 0
1 С —С' 0.0568 0.0710 0,6870 0.129 0,25(5 0,513 1.02 •'•°
Строим кривую в координатах С (абсцисса) и ——и вычисляем
графически интеграл в пределах от С\ до С... Площадь под кривой
(рис. XVI. 14) составляет 5,72. Подставляя это значение в урав-
нение (XVI. 27), найдем:
37 850 5,72
108 300 г
2. Рогене р а ц и я и о и и т а. Для обмена 750 000 .иг-эк<?
Си-+/ч требуется такое, же количество мг-лкв Н+/ч При 70%-ном
использовании серной кислоты, последняя должна содержать:
750 ОЭи
0.7
I 071 Обо мс-^кв Н+;ч
или
1 071 пои
2000
53G л 2н. JOSOt ч
30 л, м. Ватунср.
5 65
Имеем:
и
Кроме того:
CL О
750 000 ,/лп „ о.
С., = ———— — 1400 мг-экв Си24- л
5.j(>
А\ 0,30
,, , . ле Си-4-
п А <, = 4,12------------
г повита
Отмечаем на графике (рис. XVI. 13) точки (С^ Х'1)и(б’2, Х2)
и проводим рабочую линию.
Интегрирование кинетического уравнения (XVI. 27) в данном
случае (где как рабочая, так и равновесная липин являются пря-
мыми) дает
Рис. XVI. 14. Графическое опре-
деление интеграла.
V(C3-G) ^~SZQ(C--C)m
(XVI. 28)
где (С* — С)-п — средняя логариф-
мическая разность движущих сил
на концах башни:
^ — ^ = 120-0 120
с; - С. = 1700-1100 = .300
Л
И
(0 *--С)гп
300-120 г л?-.™<?Си2+
, зоо =1J0”’-----------л-----
Подставляя это значение в уравнение (XVI. 28), найдем
750000= 0,013 (SZo) 190,5
откуда получаем количество ионита в регенерационной башне:
5Хо= 212 000 г, или 212 кг
Содержание Си2+ в выходящем из аппарата растворе увели-
чилось в
1400 20 70 раз
что равнозначно испарению из начального раствора 37 500 л
воды ч.
Глава XVII
Сушка
Сушка с распылением материала
Сушка во взвешенном состоянии
Многие влажные вещества, которые транспортируются
пневматически, могут быть высушены во взвешенном состоянии.
Ирк этом транспортирующий газ предварительно нагревается
настолько, чтобы он мог служить осушителем. Такого рода су-
шильные аппараты состоят из вертикальной трубы, в которой
зернистый пли измельченный материал, находясь в токе нагре-
того воздуха или газа, высушивается во взвешенном состоянии
(рис. XVII. I).
Воздух, засасываемый вентилятором Z, подается после подо-
грева в теплообменнике 2 в трубу 4, откуда он поступает в гидра-
влическую ловушку 5, циклон 6 и через рукавный фильтр .9 уда-
ляется в атмосферу. Материал поступает в стояк (трубу) непре-
рывно через патрубок 3 и выгружается с помощью затворов 7 и 8.
В трубе рассматриваемого здесь сушильного аппарата проте-
кают следующие процессы: движение массы, тепло- и влагообмеп.
На частицу в сушильной трубе действует сила тяжести и сила
сопротивления, которую оказывает поток среды. Рассмотрим
шарообразную частицу (рис. XVII. 2) в восходящем потоке газа,
скорость которого составляет С-г.
Примем следующие обозначения:
— вес частицы, н;
IV — сила сопротивления, н;
dru — диаметр частицы, .и;
pin — плотность частицы, кале3;
С’и, — коэффициент сопротивления;
«г — скорость газа, м сек;
//•гв — скорость частицы, м‘сек;
рг— плотность газа, кг. .и3.
30* iG7
Коэффициент сопротивления С является функцией числа
Рейнольдса. Шар сохраняет свое положение в том случае, когда
скорость газа равна скорости падения частицы, которую она имела
бы в неподвижной среде.
Скорость восходящего потока пвпт, при которой шар находится
в покос, носит название подъемной скорости витания; эта вели-
чина получается при условии, что G4 — IV. .
I-’пс. XVII. 1. Схема сушки во взвешенном со-
стоянии:
1 — пентил «тор; 3 — теплообменник; з —загрузочный
патрубок; 4 -труба; .5 — гидравлическая ловушка;
G — циклоп; 7, 8 — затворы; а — рукавный фильтр.
Для птв = 0 имеем:
»внт = 1/ -1 l^TH (XVII. I)
V о С \ Пр /
При увеличении скорости газа частица уносится в потоке
со скоростью Птв = нг — нвпт.
На рис. XVII. 3 представлена зависимость скорости витания
от температуры газа для различных величии частицы.
Тепло- н массоперсдача. Па рис. XVII. 4 представ-
лены данные для конвективной теплопередачи в потоке воздуха.
На оси ординат указаны значения критерия Пуссельта
л
468
Рис. ХЛ II. 2. Силы, действу-
ющие па шарообразную ча-
стицу в восходящем потоке
газа.
Рис. XVJ.I. 3. Зависимость скорости вита
ния от температуры газа для различных
величин частицы.
Рис. XVII. 4. Зависимость Хи от Не при конвективной
теплопередаче в потоке воздуха.
а на оси абсцисс — величины критерия Рейнольдса
rc^2^I!L
v
причем и. равна разности между скоростями газа и частицы.
С помощью эмпирических исследований для критерия Нус-
сельта найдено следующее выражение
Nu ~ 2 + 0,552 Pr’/sRc’ '' = (X VII. 2}
Количество тепла, отнимаемого от частиц при их движении
с потоком газа, составляет
(XVII. 3)
где а — средний коэффициент теплоотдачи; при этом принимаем,
что с/тп — const.
Количество тепла, необходимого для испарения влаги с поверх-
ности частицы
Я
Q ~ е-гп (А'1-XX) L (XVII. 4)
и
где L — теплота парообразования, ккал;кг\
Хг и X, — содержание влаги в твердых частицах, масс. доли.
Продолжительность сушки равна:
A'\~-Ys (XVII.5)
v AfCp
При малых значениях критерия Рейнольдса (Re 0) будем
иметь в соответствии с (XVII. 4):
Хи=—(XVII. В}
Используя (XVII. (5) для (XVII. 5) получим:
<п0тп(А, — AJ
высушивания твердых частиц в вертикальной
1 = йгпт (XVII. 8)
скорость, при которой сначала неподвижная
ходит в вертикальной трубе за время т.
Длина пути для
трубе г
где нтв — средняя
частица
Для этой величины имеем:
нтп =. k (nTu)j]-»]«4 /• I
470
где
(Чтв)макс -
иг Ь'внт
применительно к наибольшей частице высушиваемого материала.
При длительной сушке к^1.
Количество газа для сушки определяется из следующего
соотношения:
('цС{, (/ft — /к)
где 6’ц — количество сухого
воздуха;
Ср — теплоемкость сухого
воздуха при постоян-
ном давлении.
На рис. XVII. 5 дана за-
висимость времени сушки и
длины вертикальной трубы от
диаметра частиц в соответствии
с формулой (Х\ II. 5).
Средняя удельная
паропроизводителытость. Для
сушилок с распылением мате-
риала следует дополнительно
определить поверхность частиц,
которые в любой момент на-
ходятся в трубе на 1 .я3 ее объ-
ема, а затем количество тепла,
передаваемого в 1 ч этим части-
цам. Поверхность шарообраз-
ных частиц:
С^тв
где g — порозность.
Рис. XVII. 5. Зависимость времени
сушки т (кривая 1) и длины верти-
кальной трубы I (кривая i?) от диа-
метра частиц </тв.
Количество тепла, передаваемого частицам в 1 .ч3 сушильной
трубы, составляет
(iv— ив “—п------- Аб’р (Х^ IГ. 10)
«ТВ
причем
। тв Qj- С
('ll Qtb А (С— 1)
где
С и г
~ йтВ
Таким образом, имеем:
г><7 <7v = . (' тв Or . С’ V(-p- (XVII. 11)
в {?ти (-z 1)
471
Средняя пар о производительность сушильного аппарата с рас-
пылением материала:
т
(XVII. 12)
Пример. Определить время сушки, иаронропзволительпость
и длину вертикальной трубы сушильного аппарата, работающего с распыле-
нием влажного материала. Данные:
диаметр частиц I .и.«;
температура воздуха при входе в аппарат GOO3 С;
температура воздуха при выходе из аппарата 1.”0° С;
... кг
начальная влажность материала А, = 2,ь ------------------;
1 кг сухого остатка
конечная влажность материала Х* = 0.6 -------------------;
кг сухого остатка
Плотность материала, считая на сухой остаток рт1! — 500 кг/.v';
температура на поверхности высушиваемого материала 70° С,
Плотность маюриала в начальном и конечном состояниях:
Qi = Qtb (1-1- ХО = 500 ( I ! - 2,6) =1800 «г/.u3
Qa — ртв (1 "I- Xj) — 500 (1 + 0,6) = 800 кв;.u3
Сре. [ияя плотность влажного материала:
1800 + 800 1n,v.
Qcp=------------- = B0U к.
Средняя разность температур:
^cp —
600— 150
In
600 — 70
150 — 70
-237е С
В сушильном аппарате средняя температура воздуха равна
’ I' "Г ^ср 70 + 237 = 307° С
По рис. XVII. 3 скорость витания частиц диаметром 1 .и.и в токе воздуха
при 300° С составляет
Квит —о»5 в^'- сек
Для числа Рейнольдса имеем:
5,5 • 0.001
33,5 10"в
Re
164
где
v принята для средней температуры (307 + 70),Я? as+ДГ С.
По рис. XVII. 4 находим для критерия Пуссельта
Nu=8
Используя (XVII. 5), получим:
557 0,001 • 500 (2,6 0,6) __
г ——----------- • 3600 л-л сек
6 - 250 237
где
и-
л „ 0.032
372
Длила сушильной трубы может быть найдена из (XIV. 8):
i = 5,5 • 5.5= 30 л
Определим с помощью (XVII. 11) и (XVII. 12) среднюю величину удель-
ною парообразования:
— би ^ТК {?!' С Af
<lv~ ldTV ’ г;8 *’ л(с-1) Л ср
Отношение б'тв/Ср найдем па основании теплового баланса:
б тп (X’ 1 X >) L = б цС р (Zл — £к)
Таким образом:
б'тв 0,244 ((>00 —150) _
557 (2,6-и,б) = Л
где Ср — 0,244 кклл.5;г-гр«й — теплоемкость воздуха при £ср 300э С.
Плотность воздуха @г = 0,62 кг/мя.
Принимая, согласно опытным данным, k = 1 и С — 2, получим:
^-2-237-162
Сушка с распылением растворов а суспензий
Для определения среднего диаметра капли, получа-
ющейся при распылении жидкости и суспензии, может быть ис-
пользована следующая эмпирическая форму ia:
585/о / р. \о-15 ( 100^ У’6
/Лер — - / ——го9/ —7==-] —_ (XIV. 13)
(l I (?г \ У J \ 'Т /
где /Эср — средний размер капли, лгк;
сг — поверхностное натяжение, дин!см;
и — относительная скорость между газом и жидкостью.
м/сек;
рг — плотность жидкости, г/с.и3;
[I — динамическая вязкость жидкости, г/см-сек;
— объем
иР — объем
Для удельной
жидкости в единицу времени, л!ч\
газа, отнесенный к единице времени, л.ч.
поверхности частиц F (в .н2/л) имеем:
6000
d
(XVII. 14)
частиц, мк.
где d — диаметр
Па рис. XVII. 6 представлена зависимость F от d.
Коэффициент теплоотдачи определяется из формулы!
Nu = -^- = 2+APr1/:,Re,/1
473
В сушильной камере распылительного аппарата (рис. XV4I. 7)
значение чисел Рейнольдса очень малы. Поэтому мошем написать
откуда найдем
Хи г 2
и
Принимая к - 0,02л ккал 'ле-ч ’град, получку следующие вели-
чины для я в зависимости “от d:
d, мн ........... 1 10 ЮО
а, нка.1, .V- - ч - град .... 3Dii!.)O 5090 500
11 родолшитсльность сушки:
аппарата.
Решение (XVII. 15) дает:
т = С.гВ
где
ciFAZcp 'Л -:1
.(XVII. 1б)
^ср
^11 —*К
In '-U—L
tK-i‘
(XVII. 17)
причем t темпераiура па поверхности высушиваемого мате-
риала.
Л (I3
'Гак как и
’•'тн (. Ртв и F — л d'-y то для продол-
жительности процесса сушки получим
(XVII. 18)
Более удобно пользоваться
материала р; вместо
соотношение
Т11> между
СР
। величиной плотности жидкого
которыми имеется следующее
Qi
(?тг: л ।
(XVII. 19)
Следовател ыю:
Часто бывает целесообраз-
нее принимать вместо перепада
температуры среднюю логариф-
мическую разность величины
.А А ср перона а
« А/ср
влажности.
а \Л'срЛ
ст
<х ___ Zt.
Ср du Ср
Из
получим
\fcy
Рис. XVII. 8. Зависимость продол-
жительности сушки т от диаметра
капли </.
где Ср — теплоемкость газа
при постоянном давлении.
Но опытным данным k = 0,9.
Тогда (XVII. 20) примет следующий вид:
т
и
L
р
р
т=М75т^^в,н
Для практических случаев при сушке воздухом путем распы-
ления материалов в камере могут быть приняты следующие
данные: /. 0.027 ккал м-ч -град:. Ср = 0,241 ккал кг-град\ <ч ,.
= 1200 кг'.u3; Xj — АТ = 0,95; 1 Н~ Х\ = 2. Тогда для т (в ч)
получим:
где диаметр капли d принят в метрах. Уто соотношение изобра-
жено на рис. XVII. 8 для различных значений ДА\:р или Д/<р.
Л том случае, когда диаметр капли сильно уменьшается,
полученная выше формула должна быть изменена с учетом пере-
менной поверхности частиц.
Принимая, что уменьшение объема тела равно объему испарив-
шейся жидкости, получим следующее дифференциальное урав-
нение:
, / d3 • л \ п d3 л ,
-сЦ —(Д = —Vcp.dr (XVII.23)
Продолжительность сушки:
1
С LrtQi За'3
(».«« Я-Д|ср (XVII.24)
Подставляя а = 2/Jd, найдем:
т = 75—— f d d (d) = Lqi
д/н> J 8a Д«ср
При использовании перепада влажности ДХср:
_ «.-4)
8?.. ЛХ'ср
(XVII. 25)
где рг — плотность испаряющейся жидкости.
Величина плотности жидкого материала р может быть при-
равнена р;.
Размеры сушильной камеры распылительного
аппарата. Скорость капли в вертикальном потоке газа скла-
дывается из двух частей, а именно, из скорости падения Us
в неподвижной среде и скорости сушильного агента:
<11. _ — (?г).С , j
dr 18р-9,81 1 '
(XVII. 26)
где р — плотность капли, кг.-м3;
. рг — плотность газа, кг’м3',
п — динамическая вязкость, кг/м-сек.
Первое выражение правой части (XVII. 26) представляет Us,
.определенную на основании закона Стокса, a Ur есть средняя
скорость газа.
Имеем:
Для случая с уменьшением диаметра капли из (XVII. 25) найдем
476
Исключая рг в (XVII. 26), получим:
di~____f4
4/,ЛГср 18.u -9.8l
и
8/. Л/ср
Диаметр сушильном камеры аппарата
где объем сушильной камеры V (в .w3) равен
V
в
Qu
причем GK — массовая скорость воздуха, кг сек.
Так как
Г (jTil 1---- А о)
('В ~ ------~----
Д' о Л- J
где А\ и Х2 — начальная и конечная влажность высушиваемого
материала;
хх и .г., — начальное и конечное вла» осодержанпе воздуха,
то
Р — ^'ТП 1 —
Qn (т.2 — jct)
Расчет распылителя для жидкостей. Очень часто
для распыления жидкостей применяется устройство, изо-
браженное на рис. XVII. 9.
Ряс. XVII. 9. Устройство для распыления жидко-
стей.
Угол 0 между просверленным отверстием и вертикалью,
от которого зависит место падения раствора в камере, опреде-
ляется следующим образом. Пусть Ц есть расстояние сопла от
верхнего края рабочей части камеры, а длина г — расстояние
места падения струи от осп сопла. Тогда в соответствии с законом
свободного падения тел при продолжительности полета т получим:
II = irr cos 0 -|-
прнчем
г =_ ю т sin О
Исключая т и учитывая, что
получим поело подстановки Л = 2ф3/г:
Cig о
где h — бысота напора н сопле;
ср — коэффициент сжатия струп (ср = 0,85);
Л — 2*0,853й = 1,457г.
Сушка твердых тел газом,
проходящим через слой материала
Кинетика сушки
На рис. XVII. 10, а изображен аппарат для сушки
газом, проходящим через слой материала параллельным током,
па рис. XVII. 10, б — аппарат, в котором газ проходит через
полочные решетки последовательно.
Рассмотрим слой твердой массы, через который проходит газ
с влажностью Уд при скорости 6’с кг сухого газа 'м~ч. Максималь-
ная скорость сушки Hzn.i4ah.c (в кг влагп/.ч3 -ч) будет в том слу-
чае, если газ яри выходе из аппарата насыщен парами жидкости
при температуре адиабатного насыщения и имеет, следовательно,
влажность У(1Д:
11 «Чмакс " (YaII ~ У1)
(XVII. 27)
Обычно газ уходит с влажностью У2 и скорость сушки в любой
момент времени будет
В'в.-1= Сс(Г2-У) (XVII. 28)
Для бесконечно малого сечения слоя материала, где влажность
газа изменяется па dY и конечная его влажность составляет У,
скорость сушки равна
вл “ = Оу dS (Уа11 1 )
(XVII. 29)
478
где cir — коэффициент массопередачи в газовой фазе, кг испаря-
ющейся влаги/.м2-ч ДУ;
6’ — поверхность контакта фаз, .и’2’.и2 поперечного сечения
слоя материала.
Обозначим через а поверхность фазового контакта па единицу
объема слоя материала, высота которого з; тогда получим:
Рис. XVII. 10. Аппараты дтя сушки твердых тел газом, проходящим через
слой материала:
а — параллельное движение потоков газа; б — последовательное движение газа
через полочные решетки;
2 вентилятор; 2 нагреватель; з —няправляницип :<о;кух; 4 полочная решетка;
заслонка для рециркуляции воздуха, И воздухоитвод.
Уравнение (XVII. 29) примет следующий вид Ys г f с/Г р ау adz у _у = (XVII. 30) J 1 all 1 J *-'с 11 о
откуда имеем: , У«Я-,1 «уя* In ?У_—— (XVI 1.31) гп№12 1,С
где У — число ступеней массопередачи в газовой фазе рассматри-
ваемого слоя материала.
Средней движущей силой для процесса испарения влаги будет
средняя логарифмическая разность величин Уд// — ц YqH —
— У2. Сопоставляя уравнения (XVII. 27), (XVII. 28) и (XVII. 31),
получим
и'вл X-2-V1
в-’макг У ail
1- уД" *.д = 1
2 all 1 1
(XVII. 32)
479
Последнее равенство выражает скорость сушки РРВЛ при усло-
вии, если а а и Лг известны. Величина А" установлена для спе-
циальных случаев, рассматриваемых ниже.
Сушка твердого материала без пор
Размер частиц от 2,00 до 0,075 ji.it применительно
к высоте слоя материала 5 > 11.5 зьм. Для этого случая постоян-
ная скорость сушки определяется с помощью уравнения (XVII. 27).
Выражение (XVI Г. 32) может быть использовано как для постоян-
ной, так и для переменной скорости сушки. Величина а изменяется
с содержанием влаги, и поэтому более удобно определять число
ступеней сушки посредством эмпирической формулы:
Л'=^ (XVII. г,)
где d — диаметр частиц, лг.;
Риас — насыпная плотность материала, кг сухого мате-
риал a.
Gc — массовая скорость газа, кг газа/лг-ч;
р, — вязкость газа, ка/лг-ч;
X — содержание влаги в твердой массе;
— критерий Рейнольдса.
Пример. Летчика кристаллического осадка подвергается сушке
воздухом, проходящим через слои этого материала.
Частнцы лепешки нс обладают пористостью, их средний диаметр 0,203 .и.ч.
Вследствие нерастворимости осадка в воде равновесная влажность его ни-
чтожно мала. Толщина слоя осадка составляет 17,8.и.в; насыпная плотность
материала 1360 к;: сухой массы/.»3. Начальная влажность осадка 2,5"(|,
а конечная 0.1%. Скорость прохождения воздуха через с.’юй материала
853 кг сухого воздуха/.»2-ч при 32,2° (’ и 50%-иой влажности.
Определить время сушки.
Имеем:
0,025
Л j = —--- 0,0256 иг воды/?;.’ cvxon массы
• 1 — O.lJJo
f^u oci] 0.001QU1 us воды/кг сухой массы
С помощью диаграммы I--d найдем:
1'5 = 0.01.58 кг воды/ка сухого воздуха;
taH = 23,9’ С;
= 0,0190 кг воды/кг сухого воздуха.
Для среднего значения потока воз ivxa имеем:
- c-r - Q-./К01.580.0190
.'С = 8оЬ j-h.bj-----“------
867,85 кг/.и2-ч
Применяя (XVII. 2/), получим:
И71зл11акс — 853 (0,0190 — 0,0138) 2,74 кг испаряемой воды/.ц?. »
480
23.85 t- 32,2
Вязкость воздуха при средней температуре —----------—-----= 28° С соста-
вляет у, — 0,018 спз, или 0,0425 кг/.и-ч.
Таким образом, для критерия Рейнольдса имеем:
o (1Сс 0.000203-867,85 .
Re —------=-------...г-------= 4,14
у 0,0425
Используя (XVII. 33), получим число ступеней сушки:
jY = 0,332 • 4,14°*215 (X -1360 • 0,0178)°’” _д ] *0,«4
о,ооо2оз°’35 ° ’
Скорость сушки в соответствии с (XVII. 32) будет:
ИЪл = 2,74(1-е-^’1А'°^1)
Следующие данные дают значения скорости сушки И’вл в зависимости
от X в соответствии с последним уравнением
X, кг воды/яг сухой массы 0.0256 0,02 0,015 0,010 0,008 0,006 0,004 0,002 0,001
Ч’вл. кг воды /л2 ч . . . 2,722 2,720 2,680 2,605 2,560 2,438 2,240 1,830 1,360
По приведенным данным на рис. XVII. 11 изображена кривая скорости
сушки.
Продолжительность сушки определится при нахождении площади под
кривой, построенной в координатах 1/1Г относительно X:
Величина площади под этой кривой составляет 0,0097. Так как
Лр
—— -= 1360 0,0178 = 24,2 кг сухой массы/.ч2
то время сушки составляет т = 24,2-0,0097 0,235 ч, пли —14 зеин.
31 л. м. Батунср. 481
Сушка пористых тел
Размер частиц от 3,0 до 20,0 льн применительно к вы-
соте слоя материала от 10,0 до 65 ЛМ1. Во время сушки при постоян-
ной скорости газ уходит ненасыщенным и эта скорость процесса
определяется посредством (XVII. 32). Величина а,, может быть
найдена из формулы:
ay = Jf/;c/ScI/a (XVII. 34)
причем JD в свою очередь вычисляется с помощью кривых па
рис. XV. 6. Поверхность контакта фаз может быть приравнена
величине поверхности частиц. При сушке твердых тел воздухом
критерий Шмидта Sc = 0,6.
Пример. Гранулы влажного пористого катализатора, имеющие
форму цилиндров диаметром 13,45 ,ч.ч и высотой 12,85 .«.и, подвергаются
сушке в аппарате при пропускании воздуха через слой материала. Катализа-
тор в слое высотой 50,8 .ч.и, считая на сухой остаток, расположен на ситча-
тых противнях. Скорость прохождения воздуха равна 3900 кг сухого воз-
духа/.и3-ч при 82° С, влажность воздуха 0,01кг воды/кз сухого воздуха.
Насыпная плотность катализатора 605 кг сухого материала/.»3, а поверхность
его частиц составляет 282 .и3/.»3 слоя катализатора.
Определить скорость сушки, а также влажность и температуру воздуха
при выходе из аппарата в течение периода постоянной скорости сушки.
Имеем:
1'1 = 0,01 кг воды/кз сухого воздуха
Из диаграммы / — cl находим влажность воздуха в состоянии насыщения
0,031 № воды/кз сухого воздуха
при соответствующей температуре адиабатного насыщения /пЯ = 32,2° С.
Средняя скорость воздуха:
6С = 3900-р 3900 0,01 = 3978 кз/.»3 ч.
Средняя вязкость воздуха: р = 0,019 спз, пли 0,0685 кг/.»-ч.
Поверхность частицы катализатора:
/ = 0,785 • 0,01345s 2 -|-3,14 0,01345 • 0,01285 = 0,00083 м2
Диаметр сферической частицы с такой же поверхностью составляет:
г,=|/йфГ=ад163 „
Найдем число критерия Рейнольдса;
~ dGc 0,0163-3978 Л,_
Кривая 6 на рис. XV. 6 дает JD — 0,06.
Критерий Шмидта Sc для системы воздух — водяной пар Sc = 0,6.
Используя (XVII. 34), получим:
0,06-3900 ооп , , ...
nv =-----—----=330 кг воды/.»2 • ч ДУ
Y 0,6 13
482
Число ступеней сушки равно:
„ al-GZ 330 282 • 0,0508 . о,
G'o 3000
Максимальная скорость сушки в соответствии с (XVII. 27):
И влманс = Сс (Ган - Л) = 3900 (0.031 - 0,01) = 82 кг воды/.и2 . «
Применяя (XVII. 32), получим
И ВЛ 1 о ~~ (Ш1 в —1.2]
82 — 0,031--0,01 ~ —
откуда 11 пл = 57,5 кг испаряемой воды/.u2-ч.
Влажность воздуха при выходе из сушилки:
У.Л=0,0162 кг воды/кг сухого воздуха
С помощью диаграммы / — d определяем температуру выходящего
из аппарата воздуха: t — 66,5е С
Так как
£е/Л = бО5 0,508 = 30,8 кг сухой твердой массы/.н2
т<> из уравнения материального баланса имеем для скорости сушки
... А 57,5 ,
-----И пл — -- —тг - 1.87 кг воды; кг cvxoro катализатора • ч,
«Г ‘ Ас 30.8
Камерная сушилка
Одним из неудобств при пользовании камерной сушилкой
является неравномерность распределения остаточной влаги в конечном про-
дукте. Это обстоятельство обусловливается тем, что в камерных сушилках
воздух распределяется неравномерно при сто движении внутри аппарата.
Такого рода недостаток может быть устранен применением большого объема
доздуха со скоростью 3—6 м/сек. Для уменьшения расхода тепла на практике
свежий воздух используется в незначительном количестве при рециркуля-
ции так называемого отработанного воздуха, составляющего во многих слу-
чаях 80—95% от общего потока. Следует, однако, отметить, что при этом
влажность воздуха внутри сушилки увеличивается по сравнению с влаж-
ностью свежего воздуха. Поэтому необходимо внутри аппарата соответственно
повысить температуру и надлежащим образом изолировать его стенки,
причем не только для уменьшения потери тепла в окружающую среду, но
и для предотвращения возможности конденсации пара на внутренней поверх-
ности стенок камерной сушилки.
Рассмотрим анализ работы камерной сушилки, изображенной на
рис. XVII. 12. Аппарат содержит в каждом отделении по 1(1 противней высо-
той 38 .о; расстояние между противнями составляет 102 .им. Ширина каж-
дого противня 0,93 ,и с общей площадью для высушиваемого материала 14,0.и2.
Желательно, чтобы для воздуха, поступающего в зону I, температура сухого
термометра была 93° С и влажность 0,05 кг воды,'кг сухого воздуха. Темпе-
ратура атмосферного воздуха 26,6° С, влажность его составляет 0,01 кг
воды/кг сухого воздуха. Скорость воздуха при его поступлении в пространство
.между противнями равна 3.0 м/сек. Количество удаляемой влаги 27,2 кг
воды/ч. Определим количество циркулирующего воздуха и состояние его
в различных частях камерной сушилки.
Имеем:
У'= 0,05 кг воды/кг сухого воздуха; =93°С
31* 483
Объем влажного воздуха:
( « . У' \ Н-273 1 / 1 , 0,05 \ .... 934-273 1
\ Л/„ Л/ь ) 273 ’ Р \ 28,97 “г 18,02 ) 273 ’ 1
= 1,125 .ч3/кг сухого воздуха
Величина свободной площади для прохождения воздуха между про-
тивнями:
/ = 0,93 (0,102 - 0,038) 11 = 0,654 л2
Объемная скорость потока воздуха между противнями
3 • СО - 0,654 = 118 л3/.ним.
Рис. XVII. 12. Камерпая сушилка:
I—IV — зоны; 1 — подогреватель; г — решетка; 2 — пентилятор.
пли
118/1,125=105 кг сухого воздуха /мин
в зоне /.
Скорость испарения влаги равна
27,2/60 = 0,454 кг/мин
и влажность в зоне II будет
0,054-0,454/105 0,0543 кг воды/кг сухого воздуха
Считая, что в аппарате протекает адиабатная сушка, температура воз-
духа в зоне II может быть найдена с помощью адиабатной липни, проходя-
щей через точку, отвечающую состоянию воздуха в зоне /. Таким образом,
из диаграммы I — //для полученного выше У2 — 0,0543 имеем — 84,5 ’ С.
Состояние воздуха в зоне IV, а также выходящего из сушилки воздуха,
должно быть таким, как в зоне I. Следовател1.по, мы можем составить для
сушилки полный баланс но воде в следующем виде:
О’с (0,05— 0,01) - 0,454 кг испаряемой воды/лык
484
откуда
б’с 11,35 кг поступающего и выходящего сухого воздуха/лни
Общее количество воздуха в зонах III а IV составляет
10511,35 = 116,35 кг сухого воздуха/.иия
и в зоне IV объем влажного воздуха должен быть равным 1,125 ,и3/кг сухого
воздуха.
Объемная скорость воздуха, проходящего через вентилятор:
116,35 • 1,125=131 лз/.нин
Количество циркулирующего воздуха:
100 = 90,0%
Г16,а
Энтальпия воздуха в зоне II (и в зоне /) в соответствии с диаграммой
I — d равна 52,25 ккал/кг сухого воздуха.
Значение энтальпии свежего воздуха также находим из диаграммы I — d
равным 12,42 ккал/кг сухого воздуха.
Энтальпия смеси в зоне III определится из равенства:
55,25-105 4-12,42-11,35 с. , „ _г„ „
---------—— ----------= 51 ккал/кг cvxoro воздуха
116,35
Так как влажность воздушной смеси 0,05, то по диаграмме I — d темпе-
ратура сухого термометра будет 78,5° С.
Расход тепла в подогревателе, пренебрегая потерями:
116,35 (55,25 —51,0) = 49,5 ккал/мин
Температура точки росы воздуха в зонах /, III и /V, по диаграмме I — d
равна 40,3° С, а в зоне II она составляет 41,7° С. Таким образом, стенки ап-
парата должны быть достаточно хорошо изолированы для того, чтобы внутри
камерной сушилки не было температуры <^41,7° С.
Глава XVIII
Дисперсионный анализ
в органическом синтезе
и биохимической технологии
Дисперсионный анализ применяется при изучении
влияния совокупности факторов на результаты наблюдения пли
опыта. Ценным свойством дисперсии является то, что если в про-
цессе имеется несколько факторов, каждый из которых вносит
какую-то долю в величину дисперсии конечного продукта, то
общая дисперсия равна сумме составляющих дисперсий. При
этом предполагается, что факторы действуют независимо
Указанное свойство аддитивности дисперсии позволяет раз-
ложить дисперсию по составляющим факторам, относительная
важность которых может быть оценена.
Обоснование метода
сравнения дисперсий
Пусть из очень большой нормальной совокупности,
имеющей дисперсию н2, взяты две независимые случайные выборки
объемов nt и п.у. Обозначим дисперсии выборок через p'j и id:
Отношсние дисперсий выборок называется показателем досто-
верности
/7 = Р[/р- (XVI 11.1)
и подчиняется определенному закону распределения, завися-
щему только от объемов выборок и п2. Этот закон распрсделе-
пия позволяет при заданных nL и вычислить вероятность того,
что отношение дисперсий в силу случайных причин превзойдет
заданное число tt. Если эта вероятность окажется достаточно
малой, то соответствующее число F можно считать пограничным,
показателем достоверности в том смысле, что в случайных выбор-
ках отношение дисперсий практически не должно превзойти его
(по принципу практической невозможности маловероятных со-
бытий).
Исходя из общих соображений и точности постановки задачи,
можно заранее условиться о том, какую вероятность считать до-
статочно малой:
Р = 0,0.7. Р 0,01; Р 0,001
Для каждого из указанных трех чисел можно при заданных
и вычислить соответствующий пограничный показатель досто-
верности, который мы будем обозначать так:
(.тля Р 0.05); (для Р = 0,01); Р01 (для Р = 0,001)
В справочных таблицах приведены значения этих показателей
для различных объемов выборок, причем там приняты обозначе-
ния
A-j —г. — —1
Например для пх = 6, = 15, т. е. для kt = 5 и к.> = 14 при-
ведены три числа: 2,96; 4,69 и 7,92. Это означает, что если объемы
выборок равны г?! = 6, п2 — 15, то вероятность того, что отно-
шение их дисперсий превзойдет, например, число 4,69, равна 0,01
(очень мала). Вероятность же того, что это отношение будет
больше 7,92, равна 0,001 (чрезвычайно мала).
Установленный закон распределения отношения дисперсий
случайных выборок из нормальной совокупности позволяет оце-
нить степень расхождения двух выборок.
Дисперсионный анализ по партиям
продукта
Допустим, что некоторый процесс состоит из двух ста-
дий. В первой стадии над металлом, поступающим партиями по
300 кг, производится операция Л. Эта операция не является слу-
чайной, и получающийся продукт можно охарактеризовать,
в частности, дисперсией и2 . Во второй стадии процесса каждая
партия в 300 кг делится на три партии по 100 кг, каждая из кото-
рых подвергается затем операции В. Эта операция также нс
является точно воспроизводимой, и конечный продукт характери-
зуется дисперсией [ip. Задача состоит в рассмотрении общей вели-
чины дисперсии конечного продукта и разложении се на компо-
ненты рД и [ip. Выполнив это, мы будем в состоянии решить, какая
487
часть процесса потребует более тщательного контроля для того,
чтобы уменьшить величину общей дисперсии. Для определения
р2г и р.|{ мы приступаем прежде всего к расчету величия /гр,2* ~
и pj} (в общем случае, когда каждая основная партия подразде-
ляется па п более мелких).
Величина /гр2^ -г имеет т — 1 степеней свободы, где
т— число основных партий; р'^ имеет т (п—1) степеней сво-
боды *. Поскольку р\ существует, величина пр2* -р р2{ должна
быть больше рв; мы можем легко проверить это по методу отно-
шения дисперсий.
В табл. XVIII. 1 приведены числовые значения рассматривае-
мого свойства продукта для каждой партии (всего их тп, в пашем
случае 3-10 — 30); значения даны столбцами, по три в каждом.
Любой столбец содержит данные по трем партиям, полученным
пз одной основной. Для упрощения арифметических вычислений
из всех данных была вычтена одна и та же постоянная; это смеще-
ние нулевой точки пе влияет на относительную изменчивость.
Вычисления ведутся следующим образом.
(1) Складываются квадраты данных наблюдения:
(-3)Ч-(-4)24-(-ЗГ + .. .-И2 I-22 |-12 =204
(2) Складываются квадраты итогов по столбцам; полученный
общий итог делится па число данных наблюдения в каждом
столбце:
(~8)g I (-12)2 !-(-8)2 . . .-|-02-;-12
* Числа, стоящие в знаменателях исправленных дисперсий, носят
название чисел степеней свободы и играют большую роль л дисперсионном
анализе. Эти числа указывают на число независимых отклонении, по кото-
рых! вычислены соответствующие дисперсии; они имеют тот же смысл, кото
рый им придают в физической химии. Число степеней свободы равно числу
отклонения, по которому вычисляется дисперсия, уменьшенному па число
соотношений, наложенных на эти отклонения.
4Ь8
(3) Квадрат общего итога по всем данным наблюдения делится
па общее число наблюдений:
(—3—4—3—. . . ' N-2-H)-
3d
Для удобства дисперсионного анализа полученные данные
сводятся в табл. Х\ III. 2.
Таблица XVIII. 2
Сводные данные
ИСТОЧНИК)! дисперсии С у аг ма квад рат о в Число степе- ней слободы Среяние lillJl.ipiiTOn Компоненты дисперсии
Между столбнами По столбцам (2)-(3) 160.54 (1)-(2) = 43.33 т — 1 - 9 тп— гн = 20 17,84 2.17 "В л Вц вЬ
Итого .... (1)-(3) = 203.87 нш — 1 = 29 — —
Здесь т — число столбцов;
п — число данных наблюдения в столбце;
ид — дисперсия вследствие различии между столицами;
u‘jf — дисперсия но столбцам.
Гри суммы квадратов являются разностями между соответ-
ствующими величинами (I), (2) п (3). рассчитываемыми ранее.
Числа степеней свободы выводятся следующим образом.
М е ж д у с г о л б ц а м и. Число столбцов равно т, поэтому
число степеней свободы составляет т — 1.
II о с т о л б ц а м. В каждом столбце п данных наблюдения;
каждый столбец поэтому дает п — I степеней свободы; всего
имеется т столбцов, поэтому общее число степеней свободы равно
т (« — I) — т/г — т.
О б щ е е число степеней свободы равно общему числу
наблюдений без единицы.
Среднее квадратов равно соответствующей сумме квадратов,
деленной на число степеней свободы.
В последнем столбце табл. XVIII. 2 показано, что среднее
квадратов между столбцами оценивает величину пид -|- ц^,
а среднее квадратов по столбцам оценивает pj_. Таким образом,
если u5v действительно существует, то первое значение средних
квадратов должно быть заметно больше второго.
Мы желаем проверить, существенно ли превышает среднее
квадратов между столбцами среднее квадратов по столбцам.
Это можно проверить ио отношению дисперсий. Определяем
в (Х\ II . I) отношение большего среднего квадратов к меньшему
484
/17,84 о о‘Л " г
(-yiy = 8,22 i и обращаемся к таолице значении / для nt степенен
свободы большей дисперсии (в данном случае пл = 9) и п.> степе-
нен свободы мепыпей дисперсии (в данном случае л.» - 20).
Если наше значение F больше указанного в таблице, то результат
более велик, чем уровень, приведенный в таблице. В данном слу-
чае, например при w, — 8 (в таблице пет значений для nt =~ 9)
и /г., — 20, величина /•' имеет значения 2,45; 3,56 и 5,44 для уров-
ней значимости соответственно в 5; I и 0,1 % . Наш результат (8, 22)
свидетельствует о значимости выше 0,1 %. Установив, таким обра-
зом, что среднее квадратов между столбцами значительно больше
среднего квадратов по столбцам, т. е. что дисперсия между столб-
цами существует, приступим к ее вычислению.
Имеем
«Рд + Рв = 17,84
pj{ = 2,17
где п — число данных наблюдения в столбце (в данном случае 3).
Поэтому
Полная величина дисперсии в наших исходных данных, таким
образом, составилась из двух компонент: р^ — 5,22 — вслед-
ствие различий между столбцами (в данном случае столбцы пред-
ставляют основные партии сырья) и р® = 2,17 — вследствие
изменчивости ио столбцам (в данном случае в пределах основных
партий. 1. е. между партиями продукта, составляющими
основную).
Исследование многоступенчатых
процессов
При попытке уменьшения случайных флуктуации
в процессе главное внимание должно быть уделено стадии А.
Общая дисперсия конечного продукта процесса (в момент
исследования) равна = р^ -г р){ 5,22 -Н 2,17 = 7,39. Та-
ким образом, его стандартное отклонение равно j/7,39. Паша
оценка разброса от среднего значения, в пределах которого
должны находиться 95% всех данных наблюдения, выражается
числом ±1,96 )/7,39 — ±5,3. Ото подтверждается и в резуль-
тате рассмотрения исходных данных: из тридцати данных наблю-
дения наименьшее составляет —5. а наибольшее —6; общий раз-
брос равен И.
Допустим, что все усилия были направлены па уточнение
процесса Z?; работа прошла вполне успешно, так что р^ умень-
490
шилось до нуля. Полная величина дисперсии теперь стала бы
— 5,22. стандартное отклонение 2,29 и разброс 95'% данных
наблюдения ±1.96-2,29 ±4,5. Поскольку первоначальный
разброс был ±5,3, улучшение оказалось незначительным.
Если бы усилия были направлены на уменьшение случайных
флуктуаций в стадии /1 и нам удалось бы свести п- к нулю, общая
дисперсия была бы 2,17, а стандартное отклонение 1,47;
разброс 95% данных наблюдения стал бы ±1,96-1.47 = ±2,9;
полный разброс 5.8.
Таким образом, до проведения детального физико-химического
исследования многоступенчатого процесса, очевидно, желательно
определить путем показанного дисперсионного анализа, какие
ступени больше всего влияют на случайные флуктуации, п со-
средоточить внимание па них. Выгода такого метода состоит в том,
что стадии процесса, рассмотрение которых признано нецеле-
сообразным, могут содержать большое число переменных, каждая
из которых при ином подходе потребовала бы исследования. Ди-
сперсионный анализ заставляет вначале сосре щточиться па струк-
туре процесса, прежде чем переходить к исследованию отдельных
переменных.
Дисперсионный анализ
при неравных столбцах
Число данных наблюдения в каждом столбце иногда
бывает неодинаково: тогда прием, показанный в разделе стр. 487,
непосредственно неприменим. В табл. XVIII. 3 приведены данные
о продукции, полученной из отдельных производственных уста-
новок завода до выхода их из строя из-за коррозии. Данные рас-
пределены но реакционным аппаратам.
Та б лица X1-711.,1
Данные о продукции
Аппарат Полученная продукции Итого Сумма
/1 84; ВО: 50; 47; 24 265 .53.0
11 (>7: 92; 95: 40; 98; 60; 104; 8G 705 78,3
С 4G; 93; 1(Ю — *'* 79.7
С первого взгляда можно было бы прийти к выводу, что аппа-
рат Л в среднем работает с более низкой производительностью,
чем аппараты Н и С.
Анализ начинаем с расчета следующих величин.
(1) Сумма квадратов наблюдений
842 - - GO- - 40- - . . . — 93- ± 100 95 7( >9
491
(2) Сумма квадратов итогов каждой строки, деленных на число
наблюдений в строках:
2G52
&
•»о
л •zy- = 88310
(3) квадрат общего итога, деленный на общее число:
(265-1-705-
ГТ"
2:«П*
=85981
Составим таблицу дисперсионного анализа (табл. XVIII. 4).
Таблица Л V111. 4
Сводные данные
Источники дисперсии Сумма квадратов Число степеней сиойоди Средине квадратов
Между а и па ратами Но аппаратам . . (2) — (3) = 2329 (1)-(2)= 7399 и 1164 528.5
Итого (1)-(3)-9728 16 —
Число степеней свободы определяется следующим образом.
М е ж д у а п и а р а т а м и — на единицу меньше числа
аппаратов.
11 о а п и а р а т а м — ио каждому аппарату на единицу
меньше числа наблюдений, относящихся к нему, т. с. 4 8 —
-г 2 = 14.
Итоговое — на единицу меньше общего количества на-
блюдений.
Сопоставляя среднее квадратов между аппаратами и но аппа-
ратам, получаем отношение дисперсий 1164/5285 = 2,2 при числах
степеней свободы п2 = 2 и п2 = 4. Это число намного ниже зна-
чения, соответствующего уровню значимости в 5%; таким образом,
нет достаточных основании считать, что аппарат А имеет более
низкую производительность, чем остальные аппараты.
В данном случае мы более всего заинтересованы получить
средние но трем аппаратам. Применим дисперсионный анализ
для проверки существенности кажущегося различия между
средними. В более общем случае при проведении анализа ио
партиям и меж IV партиями, как это делалось в двух предыдущих
разделах, может понадобиться вычислить обе компоненты диспер-
сии Среднее квадратов по аппаратам, как и прежде, определяется
492
величиной р.^ = 528,5. Расчет дисперсии между аппаратами
осложняется, если размеры партий (столбцов) неодинаковы.
Исли Лг — общее число наблюдений, п — число наблюдений
по каждой партии для i — 1, 2, ..., к (где к — число партий),
то среднее квадратов между аппаратами (между партиями или
столбцами) определяется как
Л(&-1) “А
Заметим, что в случае, когда все партии (столбцы) одинако-
вого размера, т. е. все = /г, уравнение упрощается:
pjA'p — кл" о ,j о п
nk{k-t) №-Ц«л—П»+Рл
В нашем примере величины щ составляют 5; 9; 3; -V — 5 4-
-j- 9 4- 3 = 17; к = 3. Поэтому:
Ш.. — I / (a 8 | j ) •> .» _ .,,, п с
>4,а --17(3—1)---- -и«' '' На 118 Нв + ИЛ
Вычитая р’4 = 528,5, получим р = — 124,3.
Разложение дисперсии на составляющие
по строкам и столбцам
и остаточную дисперсию
Часто показатели могут быть распределены по катего-
риям не только по партиям (столбцам), но одновременно и по
строкам. Формально мы будем иметь дело с двойной классифи-
кацией; каждый индивидуум принадлежит к заданной строке
и заданному столбцу. Например, в экспериментах по нитрации
целлюлозы могли применяться nt различных концентраций кис-
лоты (соответствуют строкам) и п., различных значений темпера-
туры кислоты (соответствуют столбцам).
Например, для изучения влияния некоторых факторов на
качество продукции мы испытываем эти факторы в различных
вариантах и после каждого испытания измеряем некоторый ко-
личественный параметр изготовленной продукции. В результате
произведенных испытаний мы получим ряд значений измеряемого
параметра, который будем называть в дальнейшем результатив-
ным (в данной связи) признаком. Значения (показатели) резуль-
тативного признака, полученные путем проведенных испытаний,
образуют «статистический комплекс». Внешне это выражается
оформлением статистического комплекса в виде специальной
комбинационной таблицы. Например табл. XVIII. 5 составлена
для случая четырехфакторного комплекса.
493
Таблица XVIII. 5
Оформление ста спастического четырс.гфакторного комплекса
л, Л 2 Л, л* А&
В, вг Ва в, Вг В3 И, 7К И з В. вг Jh Bl в» вз
С1
/Л
IB *
Do и
б’з 1В
/>2
сй IB
D.,
Здесь:
Л15 Л,, Л3, Л4, Л6 — варианты первого фактора Л;
BL, В.,, В3 — варианты второго фактора В;
С\, С2, С3, Сл — варианты третьего фактора С;
/?i, Do — варианты четвертого фактора D.
13 каждой клетке проставляются показатели (значения) резуль-
тативного признака, отвечающие соответствующей комбинации
испитываемых факторов.
Например, результаты испытания комбинации вариантов:
Л3 — фактора Л; Вг — фактора /?; С\ — фактора Ст; D± — фак-
тора D надо поместить в клетку табл. XVIII. 5, отмеченную
звездочкой.
Кроме организованно взятых на учет (испытываемых) факторов
Л, В, С, D, на результативный признак оказывают влияние мно-
гие неучитываемые в рассматриваемом статистическом комплексе
посторонние факторы. Это влияние проявляется в виде колебаний
значений показателей внутри каждой клетки комбинационной
таблицы (если каждая комбинация вариантов факторов испыты-
вается несколько раз). При этом условии оказывается возможным
определить дисперсию случайной вариации результативного при-
знака для того, чтобы с ней можно было сравнить дисперсию груп-
повых средних по отдельным факторам.
Дисперсионный анализ как раз и состоит в оценке отношения
дисперсии, характеризующей систематические колебания груп-
повых средних ио отдельным факторам, к дисперсии, характери-
зуй
зующей случайное колебание показателей результативного при-
знака. Для этой цели производится разложение общей вариации
совокупности результатов наблюдения (показателе!! комплекса)
на частные вариации, обусловленные воздействием отдельных
факторов и их комбинаций, и на остаточную вариацию.
Сравнивая с дисперсией этой остаточной вариации дисперсии
групповых средних по факторам и их комбинациям и оценивая
их расхождения указанным выше способом (см. стр. 486) мы
можем подтвердить влияние исследуемых факторов на результа-
тивный признак.
Обработка полученной при учете результатов испытания ком-
бинационной таблицы методом дисперсионного анализа значи-
тельно упрощается, если мы имеем дело с равномерным ком-
плексом. Статистический комплекс называют равномерным, если
каждая комбинация вариантов факторов Л, /?, С, D... испыты-
вается строго одинаковое число раз. При этом в каждой клетке
комбинационной таблицы будет одинаковое число показателей
результативного признака. Это число мы будем называть повтор-
ностью опыта и обозначать буквой т. Общее число показателей
в равномерном комплексе равно произведению чисел вариантов
учтенных факторов на число т:
N = rnabed
где «, /?, с, d— соответственно числа вариантов факторов Л, В,
С, D (например, в приведенной комбинационной
таблице а — 5; b ~ 3; с — 4; d — 2; число по-
казателей N = 12077?).
Для простоты изложения и записи мы рассмотрим двухфактор-
ный равномерный комплекс (табл. XVII1. 6). Обозначим через А
и В взятые на учет факторы, а и b — числа их вариантов,
тп — повторность опыта. Значения результативного признака —
показатели — будем обозначать буквой х с индексами.
Таблица XVIII. 6
Оформление статистического двухфакторного комплекса
A, 4 A a
1>1 (1) (2) И) *1L> *11’ - ’ *11 (1) (2) («0 *1-2. *12 *12 (1) (2) On) *10’ *1«> • •• *1«
Hi (I) (2) (•«) *21- *21 *21 (1) (2) ("') (I) (2) 0«) *2« *2«» - • ’ *20
I>1> (I) (2) (/«) *b i> *fc *b i (1) (2) («) 2» Л'Ь 2» • • •» 2 . . . (1) (2) (/«) *7и.ч -i *7>ft
495
Число всех показателей:
Л’ =« mab
Для того чтобы найти общую вариацию и разложить ее на
нужные нам части, мы должны вычислить средние, найти откло-
нения и составить суммы квадратов этих отклонений.
Средние вычисляем отдельно по вариантам факторов, но их
комбинациям и общую по всему комплексу.
Средняя величина показателен по каждой отдельной комби-
нации вариантов учитываемых факторов (средняя величина
показателей в клетке /го столбца г-ц строки комбинационной
таблицы) равна:
Средняя величина показателей по каждому варианту фактора
/1 и Н (средняя величина показателей в у-м столбце или в г-й
строке комбинационной таблицы) равна
Н 1 — (-И! j -J- jV,2J — М(, j)
Li — — (Д/ j j, 2 -j- ... — Alja)
Общая средняя но всему комплексу (но всей комбинационной
таблице) равна
v 1 V - 1 V п 1 V z _ ' V V „
} — 1 г = 1 i j
Указанные средние представлены в табл. XVIII. 7.
Таблица XVIII. 7
Средние но комплексу *
.11 Я 2 Л а
/Л *12 ЛЛа
Л'.21 #е2 - . -
Вь Мь\ ‘-ИгД /Л Mba На Lb М
* Здесь Д9, Д9, . . . , Д9 означают показатели результативного приз-
нака при r/t испытаниях комбинации х-го варианта фактора В и у-го ва-
рианта фактора Л.
496
Полученные три ряда средних позволяют вычислить четыре
ряда отклонений.
1. Общие отклонения — отклонения всех показателей от об-
щей сроднен М:
и — х — М
сумма всех этих отклонений равна пулю:
2» = ^(ж--М)== ЛГЛ/=О
2. Средние отклонения по фракциям /1 и В — отклонения
средних по отдельным вариантам этих факторов от общей средней:
Очевидно, и .здесь суммы отклонений равны нулю:
2 а;= 2 = 0; 2 = 2 Li -I,M = 0
J = 1 5—1 i =1 i = 1
3. Средние отклонения по комбинациям факторов — отклоне-
ния средних от общей средней М:
V г J = Л/ij — М
Здесь также
У У, .Vр--аЪМ = 0
о
Для того чтобы эти средние отклонения характеризовали влия-
ние на результирующий признак только совместного действия
комбинаций вариантов факторов (Л/, В^, в них надо внести
поправку па изолированное действие тех вариантов (Л у и Я;),
которые действуют в соответствующей клетке (гД.'Зто дает испра-
вленные средние отклонения по комбинациям факторов
И'Чу= Fv-(«; + Pi) =-АЛ>-(/О + ^) + ^
4. Остаточные отклонения — отклонения показателей х от
соответствующей частной средней внутри клетки:
Составим теперь суммы квадратов этих отклонений, распро-
страненные па все показатели (XVIII. 7) (мы будем называть
такие суммы квадратов варьированием).
Общее варьирование (суммирование но всем отклонениям):
S У ««
32 л. м. Ватунср.
Варьирование под влиянием а вариантов фактора Л:
Варьирование под влиянием b вариантов фактора В:
ъ
5в=-г2₽1='»»(Й+й+ • • • +Й)
г-=1
Варьирование иод влиянием ab комбинаций вариантов фак-
торов zl и В:
Суммирование ио отклонениям, вызванным иод влиянием
неучтенных факторов:
~ 2L zij
Вследствие равномерности рассматриваемого комплекса, суммы
квадратов отклонении (варьирования) связаны соотношениями,
аналогичными приведенным выше соотношениям между самими
отклонениями
или короче
5 = SA -J- SK 4- SAIi }- sz
Это соотношение устанавливает разложение общего суммирования
£ на частные суммы 5’ Sи остаточную сумму 5 , т. е.
позволяет из общей суммы 5 выделить подсуммы, учитывающие
иод влиянием факторов zl и В, и их сочетания.
Схема дисперсионного анализа
Значение числа степенен свободы выясняется из следующей
теоремы, которую мы примем без доказательства. Если статисти
ческий комплекс представляет собой случайную выборку из
неограниченной нормальной совокупности, то отношения
дисперсий
498
подчиняются каждое распределению, указанному я разделе 2,
с параметрами, равными соответственно числам степеней свободы
сравниваемых дисперсий:
Л1=/сЛ=а- 1; ki = kU ~b~ 1; — ]) — О
Следовательно, показатели достоверности F , F можно
оценить при помощи справочных таблиц при соответствующих
значениях кл и к>.
Для удобства анализа полученные данные сводятся обычно
в таблицу, типа табл. XVIII. 8.
Таблица XVIII. 8
Данные для дисперсионного анализа
Источник дисперсии Варьиро- вание (сумма квадратов откло- нений) S Число степе- ней свободы И Среднее квад- ратов (дис- персия) В2 Отношение дисперсий I- Предельное отношение (но таблицам)
Fi Foi
Фактор А . . Фактор В . . Комбинация факторов АВ Случайная з (остаточная) Общая . . . . Л’д ^АИ Sz S к д = а — 1 кв--=б-1 КАП=КЛ ‘ КВ к2 — Л' — ab Л’-1 РА Рв Рав Рг II 1 £ 1 ’L ”1 — —
КАВ Kz * АВ — р; — —
Выводы при помощи дисперсионного анализа о влиянии фак-
торов /1 и В или их комбинации на результативный признак
можно делать на основании следующего соображения.
При условии обеспечения случайности влияния всех остаточ-
ных факторов на результативный признак (рандомизация опыта)
мы можем считать дисперсией случайного признака. Если
о
|Г\
окажется, что, например. F .=——> F то с надежностью, пре-
р:
вышающоп 0,99, мы должны считать расхождение между и
р- не случайным, а закономерным, и существенным влияние
32*
фактора Л па результативный признак. Важно отметить, что для
применимости дисперсионного анализа чрезвычайное значение
имеет случайность отбора дапных.
Анализ данных о влиянии состава
и времени вулканизации на качество
продукта
Рассмотрим анализ некоторых экспериментальных дан-
ных о влиянии состава и времени вулканизации па качество про-
дукта.
Результативным Признаком выбрало сопротивление разрыву
в кас/сл2.
Изучаемые факторы испытывались в вариантах:
время I — 20', 25', 30', 40'. 60' (в лш«);
состав С в шести вариантах (А’1, Лг2, Л’З, -V4, А'5, Дг6).
Здесь пас интересуют только отклонения, поэтому в табл. XVIII. 9
внесены показатели результативного признака, уменьшенные
на одно и то же число (130 кгс,'смг).
Таблица XVIII. 9
Сопротивление разрыву, умшыпенное па 130 кгс!смг
№ состава С 20' 25' 30' 40' СО'
Г 22» 22. 17» *22 28, 25, 16, 39 19, 29, -15, 22 13, —9, —14, 16 —4, 35, 25, 27
2 16. и, 14» 13 33, 23, 33, 35 20. —13, —7, —6 21, 6, 23. 14 Ct 1 о о с*
3 -а» в, 10» 18 14, 2, -9, 2 8, -1, 12, 4 0, —3, -7, 2 -9, 2. -4, —1»
4 -19, —14, -1 SO t- 1 1 — 14, -13, -10, -И 2, 12, 10, —5 —ПК —2, -1, —4
5 —14, -26, —7, —И) — 12, —It!. <1, —5 СО Ю 1 1 OJ — 1 1 (7, —2П, 14, 12 —2, —15, - 19. -4
с -3, —IS. - 13, —3 1 а * 1 -!• оо • -I, -29, 1, 2 (|, —5. 2, —15 11 1 ’ Гч? —
Число всех показателен:
*N -4-5-6 120
Сумма квадратов всех показателей:
V х2 = 26 422
500
Вычислим групповые средние. Для удобства вычислений мы
составим табл. XVI11. 10 сумм показателей, из которых средние
получаются непосредственным делением па соответствующие мно-
жители, указанные в таблице (так как в каждой клетке таблицы
имеется 4 показателя, то их сумма равна учетверенной их средней;
аналогично сумма всех показателей в строке таблицы равна сред-
ней, умноженной на 4-5 — 20 и т. д.). Общая ере шяя равна:
Составим табл. XVIII. 10 сумм квадратов чисел но данным
табл. XVIII. 9. При помощи табл. Х\ III 10 подсчитаем общее
отклонение
Л’ _= V — 12СШ2 26 422 480 25 942
Таблица А17//. 10
Сумма квадратов чисел
4.V 20' 25' 30' 4 0' 60' 20 Г,
Ke 1 83 108 55 16 81 343
№ 2 54 129 —6 64 16 257
X» 3 15 9 23 —8 —21 18
.V 4 —36 — 16 -48 . 19 — 23 -104
№ 5 - 57 -27 —49 23 -40 -150
Х 6 - 37 — 17 — 27 —18 -25 —124
24// 22 186 —52 96 -12 240
(4.U)’ 20' 25' 30' 40' G-J' (20Ь)г
Ле 1 6889 11 664 3025 256 6501 117 649
.№ 2 2916 16 641 36 4096 256 66 049
Л« 3 225 81 529 64 441 324
№ 4 1296 256 2304 361 529 10816
№ 5 3249 729 2401 529 1600 22 500
№ 6 1369 289 729 324 (525 15 376
(24//)* 484 34 596 2704 9216 111 —
Суммы отклонений по вариантам времени вулканизации (между
столбцами):
5\-21 V. //j-12v.V/3
} 1
484 34 5962704 — 9216 — 111
----------------------------jS<5 ИМ.
1
501
Отклонение по вариантам состава (между строками):
5с = 20 У Л;-120ЛГ2
i=l
117 649'66 049 324 + 10816 22500 - 15 376 ,
= --------------------—----------------------480 11 1-эЬ
Сумма квадратов отклонении средних но всем комбинациям
вариантов обоих факторов подсчитывается так:
Л’ис11 4^ Л/Ь~ ,2оЛ/* 173(17,5 — 480 17087,5
Отклонение по комбинациям вариантов обоих факторов
Л’Т(.=5)1Г1|-(УТ 17 088-(11156 1484) 4448
Остаточное отклонение:
Л\»гт S - Sbch = -5 9 42-17 088 8854.
Составим сводную таблицу дисперсионного анализа
(табл. XVII Г. II).
Таблица \ 1 /// Ц
CcoJuuc данные для дисперсионного анализа
Нс ГОЧШ1К11 дисперсии Варьирование (сумма квад- ратов) 8 Число сте- пеней свободы К Средние квадраты» (дисперсия) И“ ] юказа- геаь дисперс- ности F Табличные значении
F, f 0 1
Между столицами (врем:: вулкани- зации т) . . 1 484 4 371 3.77 3.32 4,62
Между стрекали (состав (') . . 11 156 й 2231 22.67 3.02 4.10
Взаимодействие Т II С .... 4 448 20 222 4 2.26 1.92 2,33
Ост.тго шал . 8 854 90 98.4 —— Т —
Итога . . . 25 942 119 — — — —
В последнем столбце приведены данные из справочной таблицы
при /с, 1; 5; 20 (соответственно); к2 =~ 00. Сравнение с ними
данными позволяет с (слать следующие выводы: испытанные
шесть вариантов состава существенно различно влияют на сопро-
тивление резины разрыву, что подтверждает влияние процент-
ного содержания испытываемого вулканизирующего вещества
(с надежностью 0.999). Гем самым мы получаем возможность
502
выделить лучшие варианты (.V 1 и № 2) и в дальнейшем сравнивать
только эти варианты между собой путем сопоставления их сред-
них показателей и притом по разному для разных вариантов со-
става.
Проведем более детальное исследование влияния состава и
времени вулканизации па сопротивление разрыву по выделен-
ным составам № 1 и № 2 как наиболее выгодным. Для этого
сравним между собой средние значения сопротивления разрыву
по составу № 1 и Л® 2 при различных значениях времени вулка-
низации т.
Общие средние для состава Ле 1 и для состава № 2 равны
соответственно
г 343 ....
1,‘1а
Таким образом, средние различаются па
Ij 1 — — 4,3
Оценим достоверность различия этих средних. Мы знаем, что
дисперсия средней равна рЛ’гс, где р'2 — дисперсия распределе-
ния каждого значения, ап — число элементов, из которых берется
средняя. Поэтому
Дисперсия разности равна:
Таким образом, нормированное отклонение:
z . z-i~^ 1,4
.иь,-ьа Г'9,84
Надежность вывода полученного различия средних оцениваем
но таблице при /с - коСт = 90. Это дает Р •< 0,9.
Таким образом, у пас нет уверенности в существенности рас-
хождения средних значений сопротивлений разрыву по составам
Ле 1. и Лё 2. Сравним еще средине но составам № 1 и Ле 2 для
разных отрезков времени вулканизации т.
108-;-129
Для т 23' среднее АГ =------—— 29,625
О
Для т 20' среднее Л/" —А ‘ ,м- = 17,125
О
Эти средние различаются па
М'—М” 12,5
503
Для оценки достоверности этого различия найдем дисперсию:
Нм
ti2 ___ Ноет _ 98,4
- В м 8- — у “
12.3
м"-"Bji'"гВм" ^z,"6
и составим нормированное отклонение
12j_
B.M'-.w" /24,6
По таблице при /с — 90 этому значению t соответствует вероятность
большая, чем 0,98. Следовательно, с такой вероятностью вывода
мы можем утверждать, что время вулканизации т = 25' для со-
ставов № 1 и № 2 дает существенно лучшие результаты, чем время
т = 20' (а значит и другие значения т, так как для них средние
еще меньше, чем для т — 20').
Опытная сушка образцов
синтетического материала
Определение остаточной влажности образцов синте-
тического материала после сушки заключалось в нахождении
потери веса 10-граммовых проб после нагревания их при высокой
температуре в течение короткого промежутка времени.
С целью выявить эффективность этого метода определения
влажности было решено произвести отбор проб и анализы на
основе следующих четырех факторов.
1. Аналитики — работы для сопоставления выполняются
шестью аналитиками.
2. Время — каждый аналитик испытывает пробы одного и
того же образца двумя периодами.
3. Отбор проб — пять различных проб отбираются из раз-
личных слоев продукта в сушильном аппарате.
4. Повторность — каждый аналитик проводит все испытания
дважды.
Общее число анализов, осуществляемых каждым аналитиком,
составляет
6Х2X5X2 120
Экспериментальные данные приведены в табл XVIII. 12 и
XVIII. 13.
Рассматривая табличные результаты, можно сделать следу-
ющие заключения.
1. Между повторными анализами наблюдается лучшая схо-
димость сравнительно с результатами различных аналитиков.
Поэтому воспроизводимость повторных анализов не может слу-
жить критерием эффективности метода анализа продукта на
влажность.
504
Таблица XVII/. 22
Влажность синтетического материала (% н'1Лл;постиХ 100)
Пробы д и алит и ки Сред- нее
А Л с D Е F
1 1) 15,5 16,5 (1-й день) 32,2 31,8 (1-й день) 29,2 28,7 (2-й день) 36.1 36.0 (2-й день) 29,0 29,0 (3 й День) 27.0 26.9 (3-й день) I 30.0
2) 22,0 22.0 (4 йдень) 33,1 33.0 (4-й день) 35.0 35.0 (6-й день) 41.5 40.5 (6-й день) 33,6 32.4 (5 йдснь) 27,0 27,1 (5-и депь)
И 1) 26,1 25.9 (1-й день) 32 >4 31,7 (1-й день) 26,0 26.2 (2-и день) 35,3 34,7 (2-й день) 30,0 30,0 (3 йдень) 23.1 29.0 (3-й депь) II 30,3
2) 20,0 20,0 (4-й Юпь) 33,1 32,7 (4-йдень) 33,1 33,0 (6-й день) 41.2 40,8 (6 йдснь) 35.1 35.0 (5 йдень) 29,6 28,5 (5-йдепь)
Ill 1) 39,2 32,7 (1-й депь) 33,0 33,1 (1-й де ль) 25.8 24.3 (2-й день) 32.4 31,6 (2 йдень) 31.0 30.9 (3-й день) 24.3 25.6 (3 йдснь) III 32,2
2) 33.0 33.0 (4-йдень) 33.1 34,6 (4-йдень) 32,2 32,1 (П-йдепь) 42,0 42,0 (G-и день) 35.3 34,6 (5 йдснь) 31.7 30.5 (5-й день)
IV 1) 21.2 21.0 (1-й день) 30,0 30,0 (1-й день) 19.7 18.3 (2-й День) 34,3 33.8 (2-й день) 32.6 31.6 (3-й день) 26,5 25,5 (3-й день) IV 30.3
2) 21.7 20.4 (4 йдснь) 38.0 38,0 (4-й день) 35,6 34.5 (6-й день) 38,0 40.0 (6 йдснь) 37,0 36.9 (5-й день) 31,6 30,5 (5-й день)
V 1) 21,0 21,1 (1 йдснь) 30,2 29.7 (1 йдснь) 24.1 24.0 (2-й день) 34,7 33,3 (2 й день) 31.3 30,8 (3 й депь) 25.2 24.8 (3-й депь) V -30,9
2) 20.0 20,0 (4-й день) 35.1» 34,5 (4 н день) 36,2 36,0 (6 йдень) 39.5 39,5 (6 й день) 36,0 37,8 (5-й день) 32,6 31,5 (5 й день)
( рРДВСС Общее средине 23,9 33-0 29.5 37,3 33,1 27.6 30.7
505
Таблица XVI/I. 13
1)ксперчментальные данные
Образцы Общее среднее А н а л и г и к и
А С i) /•; F Общее Среднее
I 7G.9 10,0 130,1 32,52 127,9 31,97 154.1 38.52 124,0 61,0 108.0 27,0 720,1 39,0
11 Общее Среднее 92,0 23,0 129.9 32,47 118,3 29.57 152.0 38.0 130.1 32,52 104.2 26,05 726,5 30.27
III Общее Среднее 131,9 32,97 133,8 33.45 114.4 28,6 148,0 37,9 131.8 32,95 112.1 28.02 772.0 32.17
IV Общее Среднее 84,3 21,07 136,0 34.0 108,1 27.02 146.1 36,52 138,1 34,52 114.1 21,52 726,7 39,28
V Общее Среднее 94,1 23.52 130,0 32,5 120,3 30,07 146,2 36,55 137,9 34.47 114,1 28.52 742.G 30,94
Общие Средние • - - • 478,3 23,915 659,8 32.99 589,0 29,45 746,4 32,32 661,9 33,095 552.5 27,625 3687,9 30,732.;
2. Имеется постоянное увеличение влажности при переходе
от одного периода анализа к другому.
3. Партия продукта, подвергаемого сушке, однородна в своей
массе, так как разности между средними значениями влажности
незначительны для пяти образцов.
Гипотеза, что 6 аналитиков по отличаются в своей работе
по определению влажности искусстве иного волокна, испытывается
дисперсионным анализом.
Общее число степеней свободы составляет 119; последние
распределяются следующим образом:
Между шестью аналитиками................................ 5
Между 5 пробами ................................................ 4
Между периодами анализов ....................................... 1
Между повторными анализами (69 и шторных анализов) ............6.)
Взаимодействие можду пробами и аналитиками (5- 4) .............20
Взаимодействие между пробами и периодами анализа................ 4
Взаимодействие между аналитиками и периодами анализа .......... .5
Взаимодействие между аналитиками, пробами и периодами
анализа (5x4x1 = 20) ...................................29
Итого..............119
50(5
Для дальнейших вычислений составим на основе предыдущей
таблицы данные для двухфакторной таблицы, обобщая и усредняя
результаты всех четырех анализов, принадлежащие одному ана-
литику по каждому образцу:
Отклонение но вариантам аналитиков:
478.32 f-659,82 1-589,0'2-J-74(>,42-r 661,92-;-552,52 3687.22 1О„ л
= 223о06
Отклонение ио вариантам проб
720,12 -f- 726,5- -|-772,0’2-- 72G.73 -|- 742,(г 3687,92 „
Лп — -------------------—---:--------------------:--- /.-S.zfl
Отклонение но ко м б и и а ц и я м в а р н а и т о в
двух фактор о в
Сумма квадратов • по всем комбинациям вариантов обоих
факторов:
Тогда отклонение по комбинациям вариантов между аналити-
ками и пробами будет
Ла, и = 2825,89 - 2237.06 - 73,20 515.(53.
Далее составим табл. XVIII. 14 на основе экспериментальных
данных.
Таблица XVIII. 14
Оора (отк* лкспериментальныл динныт
Период (время) Л я а л и т к к it Всего
/1 н с D Е F
1 234,2 314,1 240.3 342,1 306,2 251,9 1695,1
2 214,1 345.7 342,7 41)4,0 355,7 300,(5 1992.8
Итого . . . 3687,9
Отклонение но вариантам периодов времени:
1695,124- 1992,82 3687,9'2
Сумма квадратов но всем комбинациям вариантов обоих
факторов:
Лисп
ЗИ,!2-р. . . 3GK7.9-
Ti-------------10-12'
Так как сумма квадратов но вариантам аналитиков опреде-
лена выше, то сумма отклонений по комбинациям вариантов обоих
факторов между аналитиками и периодами равна:
5а,« 3187,35 — 738,55 — 2237,06 211,74
Т а ил ии а XVIII. 7-7
Обработка .} кслери.« е нт а .г ън ы .г дан н ы х
Период (прпмя) 11 [1 О 1) ы Всего
J П ш IV V
1 337,9 344,4 357.9 324,5 330,4 16954
2 382,2 382,1 4144 402,2 412.2 1992,8
Итого . . . 3687,9
Составим табл. XVIII. 15, из которой находим сумму квадратов
по всем комбинациям вариантов обоих факторов
Сумма отклонений по комбинациям вариантов обоих факторов
между пробами и временем:
Л'п. в — 876,13— 73,20 — 738,55 64,30.
Теперь остается вычислить сумму квадратов для 60 разностей
между анализами в количество 60 пар, а также сумму квадратов
по всем комбинациям взаимодействия между тремя факторами.
Пользуясь таблице i экспериментальных данных, получим
Sana.i-(16,5—15,5)2 |-(32.2— ЗЕВ)2-!-. . . - 21,70
Сумма квадратов по всем комбинациям вариантов трех фак-
торов:
5юсп = 15>52 -|-16,52-Р32,22-| 31.8s+- . . .-- ^4055.71
120
Сумма отклонений по комбинациям вариантов трех факторов
составляет:
, П, в = 4055,71 — 21,70 - 2237,06 - 73,20 - 738.55 -
— 64,30 - 211,74 - 515.63 193,53
Составим сводную таблицу дисперсионного анализа
(табл. XVIII. 16).
При выборе среднего квадрата ошибки (дисперсии ц-) для
определения показателя дисперсности F исследователю надлежит
решить, руководствуясь условиями опыта и особенностями про-
цесса, какой источник дисперсии должен быть принят во внима-
ние. Взаимодействия, указанные в табл. XVI 1’1. 16, нс имею)
физического смысла в данном эксперименте и поэтому продета
508
Таблица XVIII. fG
Сводные данные для дисперсионного анализа
Источник дисперсии Варьиро- вание (су мма квадра- тов) Число степеней свободы Средние квадратов (диспер- сия Щ) ЛУказа- тель лис- пу ре- пости F Значи- мость
Между аналитиками . . . 2237,06 5 447,41 22.25 Всроят-
Между пробами 73,20 4 18.30 18,30 пость <0.01 Нот
Период (время анализа) 738,55 1 738,55 36,73 Вероят-
Между повторными анали- зами 21,70 60 0.362 пость < 0.01
Взаимодействие: проба—аналитик . . . 515,63 20 25,78 — —
проба —время 64,30 4 16,07 — —
аналитик—время . . . 211,74 5 42,35 —- —
Проба - аналитик - время 193,53 20 9,67 —- -—
Всего . . . 4055,71 119 — — —
За счет взаимодействия (ошибки) 985,20 49 20,106 — —
валют собой ошибки, которые обусловливают дополнительные
дисперсии.
Начальные выводы в основном подтверждаются и дисперсион-
ным анализом. Однако здесь мы имеем возможность установить
степень достоверности того или другого заключения относительно
влияния рассматриваемых факторов.
Желая иметь оценку применяемого метода определения влаж-
ности продукта, исключим влияние таких факторов, как период
времени анализа и различия между аналитиками, тогда остаточ-
ная дисперсия, равная 20 0 дает (с достоверностью в 95%) ошибку
± 0,09%.
Исследование влияния качества
промежуточного соединения
Н-кислоты на выход продукта
Для опыта взяты шесть образцов промежуточного ве-
щества. представляющих различные серин в производстве про-
дукта. Из каждого образца в лаборатории были изготовлены пять
проб продукта.
509
Выход продукта для пробы определялся в граммах продукта
стандартной окраски.
В табл. XVII1. 17 приведены опытные данные:
Таблица XVIII. 17
Экспериментальные данные
Лй образца Н-кис- лоты Выход п роду к та Среднее
1 1545, 1440, 1440, 1520, 1580 1505
2 1540, 1555, 1490, 1560, 1495 1528
3 1595, 1550, 1605, 1510, 1560 1564
4 1445, 1440, 1595, 1465, 1545 1498
5 1595, 1630, 1515, 1635, 1625 1600
6 1520, 1455, 1450, 1480, 1445 1470
Общее среднее ..... 1527,5
По-прежнему для дальнейших расчетов используем показа-
тели, уменьшенные па одно и то же число 1400 г. Составим
табл. XVIII. 18.
Таблица XVIII. 1S
Обработка опытных данных
.V- образца Н-кис- лоты Выход — 1 400 (1) Сумма (2) Сумма квадра- тов (3>s »/5 (',) Сумма квадратов между образ- цами (2) — -(3) (5) Число степеней свободы
1 145; 4о; 40; 120; 120 525 71 025 55 125 15900 4
2 14о; 155; 90; 160; 95 640 86 350 81 920 4 430 4
3 195; 150; 205; 110; 160 820 140 250 134 480 5 770 4
4 45; 40; 195; 65; 145 490 66 900 48 020 14 880 4
5 195; 230; 115: 235; 225 1000 210 000 200 000 10000 4
6 120; 55; 50; 80; 45 350 28 350 24 500 3 850 4
Общее — 3825 602 875 544 04') 58 830 24
510
Из табл. XVIII. 17 и XVIII. 18 имеем:
Сумма квадратов отклонений от средних
(сумма квадратов между образцами)
Число степенен свободы .........
Дисперсия внутри образцов .........
S2/30 . . . .".....................
Сумма квадратов между образцам» ....
Сумма квадратов но 30 наблюдениям . . .
Составим таблицу дисперсионного
58 830
24
58 830/24 = 2 451
3 825-/30 = 487 687.5
5-4-4 0-45 — 487 687.5 = 56 357.5
602 875 — 487 687,5 = 115 187,5
анализа (табл. XVIII. 19).
Таблица XVIII.19
Сводные данные для дисперсионного анализа
Источник дисперсии Сумма квадратов Число степеней свободы Диепе рейн Показа- тель дис- персности Табличные значения
ГО5 J 01
Между образцами 56357,5 5 11272 4,6 2,62 3,90
Внутри образцов 58830.0 24 2 -451 —- —
Мы приходим, таким образом, к следующему выводу: испы-
танные пять образцов Н кислоты различно влияют с достовер-
ностью 0,995 па выход продукта.
Следует отметить, что имеют место также ошибки при отборе
проб, но они малы.
Дисперсионный анализ
в производстве антибиотиков
При испытании образцов пенициллина были получены
данные о содержании продукта в растворе, сведенные в таблицу.
Определение производилось с помощью метода диффузии в агар.
Для этого агар заливался в чашку Петри с диаметром 9 е.н, в кото-
рую были вставлены G цилиндров (диаметром ~0,i см) на равном
расстоянии один от другого. В эти цилиндры были налиты рас-
творы пенициллина для сравнения, после чего чашка с образцами
выдерживалась в термостате в течение определенного промежутка
времени.
Пенициллин диффундировал в массу агара, образуя круговую
зону задержки роста микроорганизмов, которая легко поддава-
лась измерению. Диаметр этой зоны находится в известном соот-
ношении с концентрацией пенициллина в растворах.
В проведенных опытах шесть образцов пенициллина сравни-
вались в двадцати четырех чашках. В таблице даны значения диа-
метров (в juj) зон замедленного роста микроорганизмов. Для
удобства расчетов из каждого результата наблюдения вычиталась
постоянная величина 20 мм.
511
Каждый образец пенициллина испытывался 24 раза. При
исключении суммы отклонений между чашками мы составили
табл. XVП1. 20 дисперсионного анализа между образцами и
внутри них обычным способом.
Таблица XVIII. 20
Данные для дисперсионного анализа между образцами
и внутри них
Чашка Л- Образцы пенициллина Общее Среднее
I 2 3 5 6
1 7 О 6 3 3 1 24 3.83
2 7 3 6 3 3 1 23 3.83
3 5 1 5 4 4 0 19 3,17
4 6 3 5 3 3 0 20 3.33
5 5 2 6 •> 3 0 18 3,00
(> 4 2 5 3 2 —1 15 2,50
7 4 0 3 1 2 -1 9 1.50
8 б 2 6 4 4 1 23 3.83
9 4 1 4 2 2 0 13 2,17
10 4 1 4 3 2 —1 13 2,17
11 6 3 6 4 4 1 24 4.00
12 5 2 6 4 4 0 21 3,50
13 6 4 6 4 5 2 27 4,50
14 6 3 6 3 3 0 21 3.50
15 6 3 э 4 4 2 24 4.00
16 5 2 5 3 3 0 18 3.00
17 5 1 4 3 3 0 16 2,67
18 5 2 4 3 3 -1 16 2.67
19 4 1 3 1 1 — 1 9 1.50
20 6 3 (5 4 4 -1 9 1.50
21 5 1 4 2 2 —2 12 2,00
22 5 2 5 2 2 0 16 2.67
23 5 1 4 2 4 — 1 14 2.33
24 4 1 4 2 1 —2 10 1.67
Общее . . . 124 47 118 69 71 —1 428 —
Общее
Среднее . . . 547 1.96 4,92 2.88 2.96 —0,04 среднее . . . 2.97
Обозначив общее среднее через z, среднее образцов (в столб-
це) через Ci и число сосудов (т. е. число в каждом столбце) через г,
получим сумму квадратов между образцами
С
Г у (Ci — х')~
i Л
для с — 1 степеней свободы, где с — число образцов (столбцов).
Применительно к данному случаю эта величина составляет 449
при 5 степенях свободы.
Подобным же образом мы можем представить, что таблица
состоит из 24 номеров чашек, каждая из которых подвергается
шестикратному испытанию; вычислим сумму квадратов между
чашками (строками). Если среднее строки обозначим через /?},
то сумма квадратов будет
е V
при г — 1 степенях свободы. Эта сумма составляет 106 при 23 сте-
пенях свободы.
Сумма квадратов для всех данных в таблице равна 590. Имеем
№/144 = '4282/ 144 = 1272
Общая сумма квадратов — [72 З2 — ... -г Г2 + (—2)’2] —
— 1272 -- 590; степени свободы 143.
Сумма квадратов между чашками = (232 232 — ... К2
-}- 102) : 6 — 1272 — 106; степени свободы 23.
Сумма квадратов между образцами = 11242 472 -г ... -
— 712 + (—I)2] : 24 — 1272 = 449; степени свободы 5.
Используя эти величины, составим дисперсионную табл.
XVIII. 21.
Таблица XVIII. 21
Сводные данные для дисперсионного una.tit.ui
Источник дисперсии Сумма квадратов Число степеней скободы Дисперсия И» Показа гель дисперс- ное ги Табличные 3!К1ЧС!ПС1
/’os Гл|
Между образцами 449 5 89,8 299,3 2,21 3.112
Между сосудами За счет взаимодей- ствия (ошибки) 106 35 21 115 4.6 0.31) 15,3 1,52 1,79
Всего . . . 590 143 — — — — -
Остаточная сумма квадратов и соответствующее число степе-
нен свободы получаются путем вычитания нз общего итога.
В результате дисперсионного анализа опытных данных мы
можем сделать вывод о том, что представленные образцы пени
цнллппа существенно отличаются один от другого.
Литература
Альдере Л.. Жидкостная экстракция, ИЛ. 1962.
А р и с Р., Оптимальное проектирование химических реакторов, ИЛ,
1963.
Батунер Л. М., Позип М. Е., Математические методы в хими-
ческой технике., «Химия», 1963.
Берк и а п Б. Е., Промышленный синтез хлорбензола, Госхпм-
издат. 1957.
Б е р к м а п Б. Е., Сульфирование и щелочное плавление в промыш-
ленности органического синтеза. Госхимнздат, 1960.
Беркман Б. Е., Промышленный синтез ароматических нитросо-
единеннй и аминов, «Химия». 1964.
Б р а й н е с Я. М., Подобие и моделирование в химической и нефте-
химической технологии, Гостоитехиздат, 1961.
Брей Дж., Уайт К.. Кинетика и термодинамика биохимических
процессов. ПЛ, 1959.
Введенск и й Л. Термодинамические расчеты нефтехимических
процессов, Гостоитехиздат, i960.
Бей лас С., Химическая кинетика и расчеты промышленных реак-
торов, «Химия», 1964.
В и in и е в с к и й Н. Е., Г л у х а и о в II. П., К о в а л е в И. С.,
Аппаратура высокого давления с экранированным электродвигателем,
Машгиз. 1961.
Ворожцов II. 1!.. Основы синтеза промежуточных продуктов и
красителей, Госхимнздат. 1955.
Додж Б. Ф., Химическая термодинамика в применении к химиче-
ским процессам и химической технологии. ПЛ, 1950.
3 а х а р i> с в с к и и М. С., Кинетика химических реакций, Изд,
ЛГУ. 1959.
К Иоффе И. И.. Письмен -1. М.. Инженерная химия гетерогсп-
пого катализа. «Химия». 1965.
К а р а петь я и ц М. X., Примеры и задачи по химической термо-
динамике. Росвузиздат, 1963.
К а р а п е т ь я и ц М. X.. Химическая термодинамика. Госхпмиз-
дат, 1953.
К а с а т к и п А. Г., Основные процессы и аппараты химической тех-
нологии, Госхимнздат, 1960.
514
Каф а ров В. В., Процессы перемешивания в жидких средах,
Г ос х 11 м из да т, 1947.
Кафа р о в В. В., Основы массопередачи, «Высшая школа», 1962.
Л а щ и и с к и и А. А , Т о л ч и и с к и й А. Р , Основы конструи-
рования и расчета химической аппаратуры, Машгиз, 1963.
Лейдлер, Кинетика органических реакций, «Мир», 1966.
Л ы к и в М. В., Сушка распылением, Н|пценромпздат, 1955.
Май оф пс Л С., Химия и технология химико-фармацевтических
препаратов, Медгнз, 1964.
II а х од Ф , Шуберт Д , Ионообменная технология, ИЛ. 1939.
Обнов ленск и й II. А., К о р о т к о в II. А., Г у р е-
в и ч A. Л.. И л ь и и В. В., Основы автоматики и автоматизации хими-
ческих производств, «Химия». 1965.
Павлов К. Ф., Роман ков И. Г.. II о с к о в А. А., При-
меры и задачи по курсу процессов и аппаратов химической техполо! пн
«Химия», 1964
II а с ы и с к и й А. Г.. Биофизическая химия, «Высшая школа», 1963.
II л а и о в с к и й А. II., Г у р е в и ч Д. А., Аппаратура промыш-
ленности органических полупродуктов и красителей, Госхимнздат, 1961.
Р а м м В. М., Абсорбционные процессы в химической промышлен-
ности, Госхимнздат, 1951.
Р ейхсфельд В. О., Е р к о в а Л. II. Оборудование производ-
ства основного органического синтеза и синтетических каучуков. «Химия».
1965.
Робертс С., Динамическое программирование в процессах хими-
ческой технологии и методы управления, «Мир», 1965.
Гоманков 11. Г., Р а ш конская 11. Б., Сушка в кипящем
слое, «Химия», 1964.
Самсонов Г. В., Сорбция п хроматография антибиотиков. Изд.
ЛИ СССР, 1960.
Сер пион ова Е. И , Промышленная адсорбция газов и паров.
Госхимнздат, 1956.
Смирнов II. В., Д у н н п - В а р к о в с к и й 11. В., Курс
теории вероятностей и математической статистики. «Наука». 1965.
Тимофеев Д. II.. Кинетика адсорбции, Изд. АН СССР, 1962.
Черноусов Н. И-, Кутин Л II., Федоров В Ф..
Герметические химико-технологические машины и аппараты Машинострое-
ние, 1965.
Ш к о р о п а д Д. Е., Л ы с к о и с к и й И. В.. Центробежные
экстракторы. «Машиностроение», 1962.
Эмануэль Н. М., К пор ре Д. Г., Курс химической кинетики,
«Высшая школа», 1962.
Я р о в е п к о В JI.. Пах м а и о в и ч Б. М.. 1Ц с б л ы
кип 11. II., Сенкевич В В., Непрерывное брожение в ацегоно-
бутиловом производстве, 1963.
А г i s В., Introduction to the Analysis of Chemical Reactors, N. Y.. 1965.
I) a n c k w e r Is P. V., Trans. Faraday Soc., 46, 300 (1950); AIChE J..
1, 456 (1955).
Denbigh K. G.. The Principles of Chemical Eguilibrium. 1955.
Frost A A, Pearson R. G., Kinetics and Mechanism, X. Y..
1961.
Grogins P. II., United Processes in Organic Synthesis. X Y., 195s.
К ha r a sell M. S., J. Res. Xat. Виг. Stand., 2 (1929).
Levcnspiel O.. Chemical Reaction Engineering, X. Y. — London,
1962.
Steel R. (ed.). Biochemical Engineering, London, 1958.
33*
515
Содержание
Глава I. Общие сведения о технологических и тепловых
расчетах реакционных аппаратов....................... 5
'Технологические расчеты ....................................... 5
Емкостные аппараты .................................. 5
Тепловые аппараты ........................... ..... 8
Массообмешгые аппараты .............................. 9
Тепловые расчеты ............................................... 9
Тепловой баланс ............................................... 10
Программированный подвод или отвод тепла (термокине-
тика химических реакции) ........................... 22
Глава II. Кинетика гомогенных химических реакций и
процессов в проточных аппаратах ......... 33
Материальный баланс проточных систем........................... 33
Батарея реакционных аппаратов с мешалками...................... 35
Графический метод расчета........................... 35
Аналитический метод расчета..........................38
Сравнительная оценка эффективности периодической и
непрерывной работы аппаратов для сложных (дег-
радирующих) химических реакций..................... 43
Неустановившееся состояние в батарее реакционных аппаратов ... 47
Реактор периодического действия с переменным объемом реакцион-
ной массы ................................................. 51
Трубчатые аппараты ................................. 54
Скорость реакции ................................... 54
Перепад давления ................................... 59
Определение искомых параметров для модели при иссле-
довании производственных процессов ....... 62
Трубчатые аппараты с ламинарным потоком реакционной
массы .............................................. 64
Реакция при непрерывном транспортировании взаимодей-
ствующих веществ под постоянным давлением .... 69
Определение длины трубчатого реакционного аппарата 72
Установившееся состояние . , .................................. 75
Реактор с обратным смешением.................. 75
Трубчатый реактор .................................. 78
516
Пеизотермичесжие реакции в потоке............................... S3
Определение кинетической модели ................................ 88
Гласа Ill. Кинетика гетерогенных химических реакции
и процессов, протекающих в реакционных аппаратах 92
Реакции и процессы в системе жидкость — жн (кость............... 92
11скаталитические реакции и процессы в системе жидкость — твердое
вещество ..................................................... ИЗ
Реакции и процессы в присутствии твердых катализаторов......... 119
Гласа IV. Химическая кинетика энзиматических процессов 149
Реакции с одним субстратом.................................... I >1
Обратимые реакции с одним субстратом........................... 153
Скорость реакций при торможении за счет субстрата.............. 155
Процесс торможения энзиматических реакций...................... 157
Реакции с двумя конкурирующими субстратами..................... 159
Реакции между двумя субстратами................................ 161
Инактивация энзима с течением реакции.......................... 163
Г ла. а К. Моделирование химической и биохимической ки-
нетики ........................................ 167
Глава VI. Термодинамические потенциалы в инженерно-
химических расчетах ............................. 173
Функции потенциала и равновесие............................. 173
Свободная энергия и химические реакции ....................... 176
Применение третьего закона термодинамики ...................... 177
Изменение свободной энергии с температурой .... ............... 180
Исследование реакции для производства формиата натрия.......... 181
Соотношение между AF° и константой равновесия К ............... 184
Равновесно в газовых реакциях.................................. 186
Равновесный выход этанола ......................... 188
Реакция получения синильной кис п>ты нз ацетилена и
азота ......................................... 190
Глава VII. Процессы хлорирования .................. 194
Условия проведения процессов .................................. 194
Материальный баланс хлоратора if технологические расчеты .... 194
Тепловой баланс хлоратора ..................................... 205
О хлораторах................................................... 207
Гласа VIII. Процессы сульфирования ......... 210
Расчет кислотных смесей ....................................... 213
Материальный балансы ..................................... . . 216
Сульфирование с< рной кислотой или олеумом......... 216
Сульфирование хлорсульфоновой кислотой ............ 217
Тепловой баланс сульфуратора .................................. 218
Сульфирование серной кислотой или олеумом......... 218
Сульфирование хлорсульфоновой кислотой............. 221
Аппаратурное оформление смешения кислот........................ 223
О сульфураторах ............................................... 223
Гласа IX. Процессы нитрования ................. 226
Условия проведения процессов................................... 226
Расчет нитрующих смесей........................................ 229
Тепловой баланс процессов смешения кислот для приготовления нитру-
ющей смеси .................................................... 232
Материальный баланс процесса нитрования........................ 237
517
Тепловой баланс процесса нитрования ............................. 240
О иитраторах .................................................... 242
Глава X. Процессы восстановления пнтросоедпнснпй и
щелочною плавления............................. 244
Восстановление нитрососдинепий .................................. 244
Материальный баланс процессов восстановления чугун
пой стружкой .................................. 244
Тепловой баланс процессов восстановления чучуяиой
стружкой ...................................... 247
Технологическая схема восстановления нитросоедипепий .248
Щелочное плавление .............................................. 249
Материальный баланс процесса щелочного плавления 250
Тепловой баланс процесса щелочного плавления .... 252
Аппарат для щелочного плавления............... 255
Глава XI. Процессы диазотирования, азосочетания и
нитрозпрования ................................ 25G
Тепловой баланс процессов диазотирования, азосочетания и ннтрозп
ронанля ....................................................... 257
Тепловой аффект процессов диазотирования............. 258
Тепловой эффект процессов азосочетания............... 260
Тепловой эффект процессов цнтрозпровапня ............ 261
Аппараты для процессов диазотирования, азосочетания и пнтрозирова-
нпя ........................................................... 261
Глава. XII. Процессы, протекающие под давлением . . 270
Характеристика процессов гидролиза, аминировапия и алкилирова-
ния .......................................................... 270
Автоклавы. Аппараты змеевикового типа........................... 271
Давление жидкости в реакционном аппарате при ее термическом рас-
ширении (термодинамика жидкости) .............................. 274
Тепловые балансы процессов гидролиза, аминирования и алкилиро-
вания ......................................................... 276
Тепловой баланс стадии химического взаимодействия . . 277
Тепловой баланс стадии отгонки пепрореагировавшпх
веществ при спуске давления ................... 279
Простая дистилляция ............................................. 280
Кинетика реакции получения изопропилбензола алкилированием бен-
зола пропиленом ............................................. 283
Глава XIII. Биохимическим процесс ферментации . . . 288
Динамика роста микроорганизмов................................... 288
Сопротивление массопередаче в процессах аэрации.................. 291
Подвод кислорода к культурам при перемешивании................... 292
О ферментаторах ................................................. 297
Стерилизация воздуха ............................................ 299
Абсорбция этилового спирта из смеси его паров с углекислым газом,
выделяющимся из ферментатора .................................. 305
Глава XIV'. Экстракция .............................. 315
Жидкостная экстракция ........................................... 315
Правило пропорциональности в бинарных системах . . 316
Тройные системы...................................... 317
Правило пропорциональности для треугольной диа-
граммы ........................................ 318
Диаграмма экстракции ................................>21
Коэффициент распределения ........................... 322
518
Определение критической точки смешения и шпернолпро-
ванис линий сопряжения ............................... 324
Построение диаграммы экстракции .................... 32G
Экстракция с перекрестным током..................... 329
Определение необходимого числа ступеней при пс.ноль-
зоваппи ограниченного количесл на растворителя . . , 336
Противоточная экстракция ........................... 337
Экстракция компонентов из сильно разбавленных раство-
ров ........................................... 346
Аппараты для жидкштпой экстракции................ 349
Смешение и эмульгирование........................ 334
Осаждение. ......................................... 36и
Многоступенчатые установки ......................... 361
Центробежные экстракторы ......................... 363
Высота (длина) контактных экстракторов ....... 365
Определение диаметра экстракционной колонны .... 371
Экстракция твердых тел....................................... 375
..Ч ситочный экстрактор Де-Смета................. 375
Расчет экстракции на ленточном аппарате Де-Смета . . 377
Экстракция в шнековых (впнтоиых) аппаратах....... 381
Экстракция в аппарате с мешалкой................. 385
Расчет экстракции в диффузорах................... 386
Критериальное уравнение проточной экстракции . . . 388
Глава XV. Адсорбция ................................ 396
Адсорбционная волна............................................ 396
Кинетика адсорбции в аппарате с неподвижным слоем адсорбента . . 398
Продолжительность пеустаиовпвгаснс.я адсорбции ................ 401
Кондиционирование воздуха. Увлажнение воздуха в потоке .... 405
Определенно количества адсорбента, размеров адсорбера и перепада
давления .................................................... 408
Переходный режим работы аппаратов с неподвижным слоем адсор-
бента ......................................................... 410
Ре.кунерацпонная установка для растворителей................... 418
Жидкостная адсорбция .......................................... 419
Адсорбция с одной ступенью......................... 422-
Двухступенчатая перекрестная адсорбция ............. 423
Двухступенчатая противоточная адсорбция ........... 425
Непрерывный процесс адсорбции.................................. 426
Промышленные установки и аппараты для адсорбции................ 428
Типовая схема адсорбционной установки периодического действия 430
Типовая схема адсорбционной установки непрерывного действия
с движущимся слоем адсорбента.................................. 435
Глава XVI. Ионный обмен ........................... 441
Иопный обмен в процессах водоподготовки........................ 44!
Аппарат для ионного обмена периодического действия 441
Расчет процесса водоподготовки. Кинетика ионного об-
мена в процессах водоподготовки.................... 441
Определение остаточной жесткости воды.............. 447
Определение константы скорости реакции для ионного
обмена в процессах нодоно (готовки................. 448
Моделирование ионообменных фильтров ...... 448
Ионный обмен в производстве антибиотиков и в химических про-
цессах ....................................................... 455
.Характеристика процесса ионного обмена в производстве
стрептомицина ................................ 455
Аппарат для непрерывного ионного обмена в производ-
стве стрептомицина ........................... 4э5
519
Сорбция стрептомицина............................. 457
Десорбция стрептомицина........................... 461
Ионный обмен медного купороса в башне rr-i его регене-
рации на раствора ................................. 463
Гласа А \ II. Сушка............................... 467
Сушка с распылением материала................................. 467
Сушка но взвешенном состоянии................................. 467
Сушка с распылением растворов и суспензии ........ 473
Сушка твердых тел газом, проходящим через слой материала . . 478
Кинетика сушки ................................... 478
(.ушка твердого материала без пор................. 480
('.ушка пористых тел ............................. 482
Камерная сушилка ................................. 483
Глава XV///. Дисперсионный анализ в органическом син-
тезе и биохимической технологии.................... 486
Обоснование метода сравнений дисперсий........................ 486
Дисперсионный анализ но партиям продукта...................... 487
Исследование многоступенчатых процессов........................490
Дисперсионный анализ при неравных столбцах.................... 491
Разложение дисперсии на составляющие по строкам п столбцам и
остаточную дисперсию.......................................... 493
Схема дисперсионного анализа ................................. 498
Анализ данных о влиянии состава и времени вулканизации на каче-
ство продукта ................................................ 504
Опытная сушка образцов синтетического материала.............. 504
Исследование влияния качества промежуточного соединения 11-кис-
лоты па выход продукта........................................ 509
Дисперсионный анализ в производстве антибиотиков.............. 511
Лев Менделееич Батунер
Процессы и аппараты органического
синтеза и биохимической технологии
с 520 УДК (56.01 : 66,0,21 : 577 1
Темплам 1966 г. п. 84
П-0ательс»1во «Д’клеил», Ленинградское Omdcacxu'*,
Невский пр., 2s
Редактор Ю. К. Кузнецов
Техн, редактор Ф. Т. Черкасская
Корректоры: I/. />. Генгут, Л. А. Любочич
М-1 5792. Формат G(ix(HJl,'is. Бумага Д7 2. Псч. л. 32.5. Уч-изд. а. 29.5
Тираж 7 5011 »КЗ. Цепа 1 р. GO к. Заказ 712.
Лсипнградскаа типография .\» 14 «Красный Печатник» Главполнграфнрома
Комитета но печати при Совете Министров СССР
Московским проспект, 91
ОПЕЧАТКИ
Стра- ница Ст рока Я <i печатано Должно быть
4 Таблица Вязкость и Вязкости
Н2С ll,(f
г. Таблица 1 1 I
П2С II tC
,1 • ill, I
II .11 ,11 IJCN
1X 1 ’[.ШЛИЦ. 1 ‘ ^Н.Т ‘ ’иги 'll.’t ' 'licit
1 •-! 2 гни iy — 3 = 28
-X Таблица I. .j е—«мчи х _ j /!,« 414 I _ [
» и Рис- 11 4 (1,25(13 .МОЛЬ 1.ц11*.ХО3 i ,2593 .«o.iii,'.! Colli.XD,
4. -1>х ’1 f-uT
',9 2 снгрху ' •‘В (б <) ' (6 .!)
3 сверху 1 dAa t ,,vo
• V ’ г(Т \ ill
’1S э спилу ।
.'9 2, 4, 8, 1 —
IИ. I 3 | (1
г.игруу /
*;п 3 снизу / ”'
н ' 9 сверху 49 - 1 и* ;г i'.'-IM* Л/Л!1
24, 25 спер- а /.кил .v.f -ч-грпи 28,4 «гп/.и*-грид
х7 9 снерху Тг = 2~ T, = 278 -г
|1>К • pnliMV 1 1 = «,У* —Jbw’
(III. :il>)
1. :! ,3 сверху (И k.iio,ii>, >;. ил I'a.TtCi.rrri- (к клсолt>>.• i:a тал n.iaropa • ч)
р;1 -ат • ч)
1'. 1 фпрму 1.1
1111 • .* ) V ‘ v )
1 . < 2*> сверху (III. 109) (III. 1 !<)
1 4 j 1 3 <и илу = Т1 T|
(.•! Формхла . _L j; _ A— i 4-Aj
(IV м
1 • J 7 гигрчу '< It' 1
4 —
ФормX.I. -fAvr('
(IX. ГО
2 »u I и Г111ГА > Il(Xdt)d > K(X'>±)d
2 1 12 снизу (13.I3) (l \. 1 1.
:js2 формула । <lx <>x
IXIV. 47/ :
:: <ч и з 'чипу । <!x dt
| 1 3 си нау (W 34 1 (W. {.n
Л М Батукер.