/








































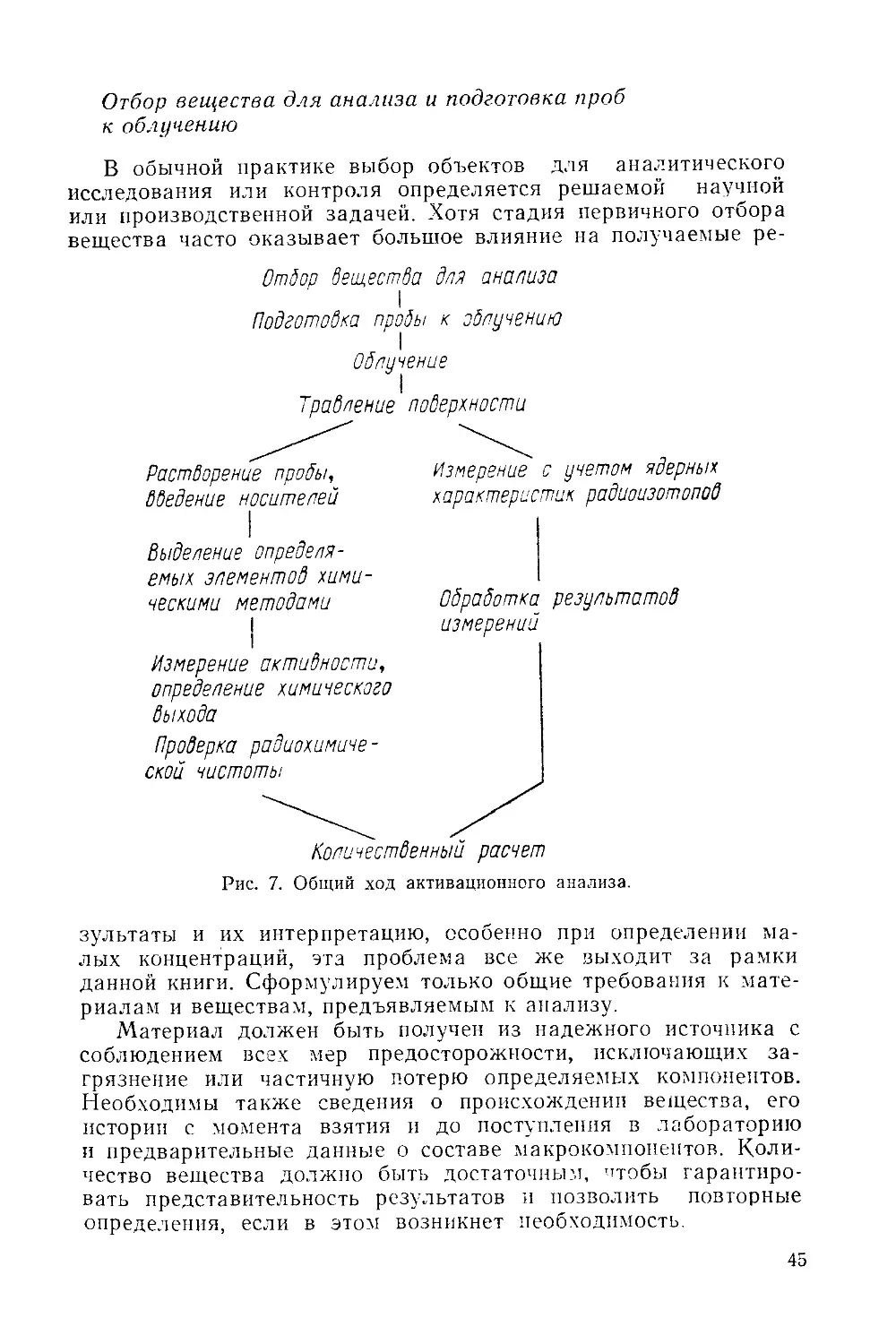














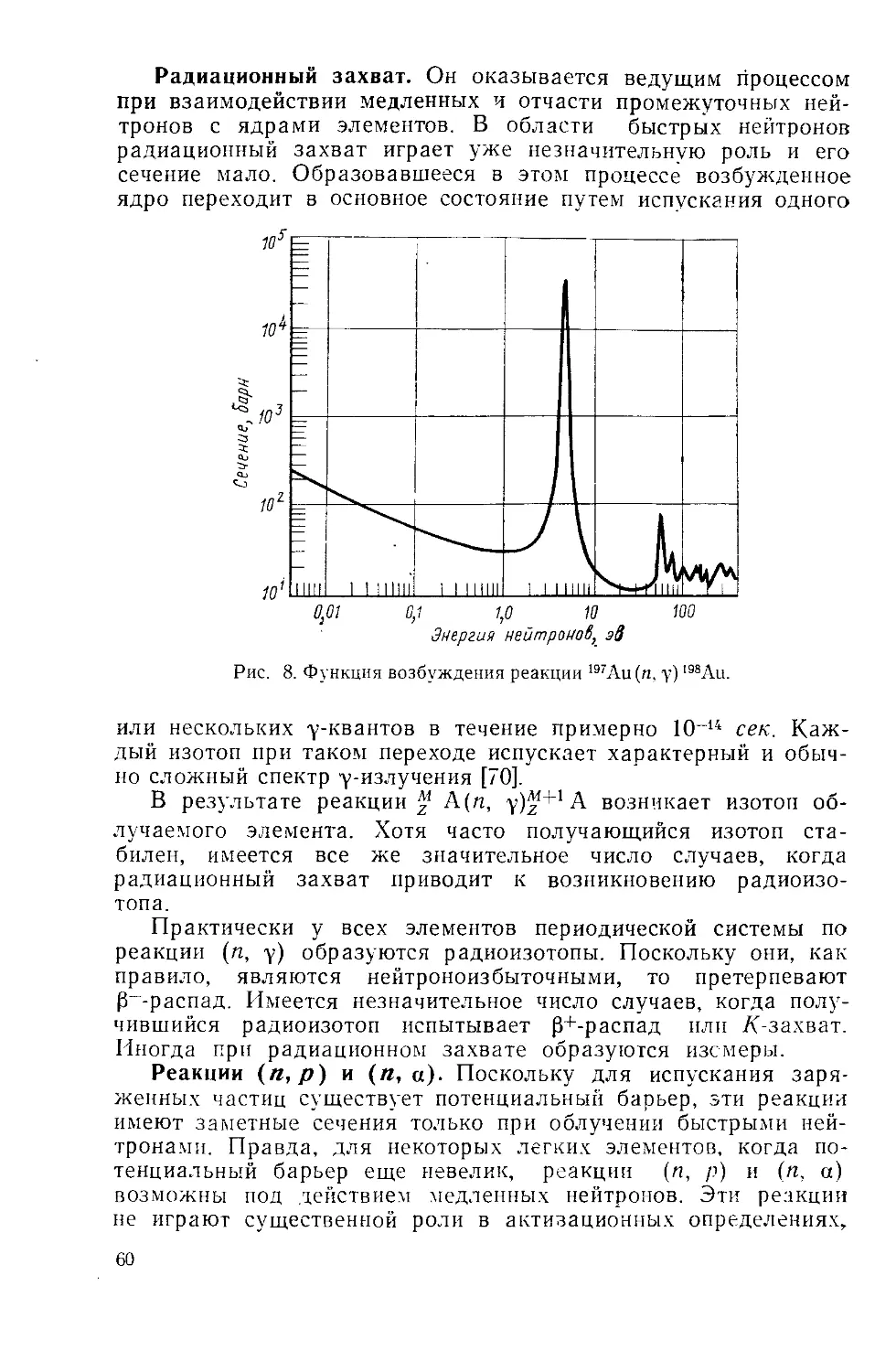








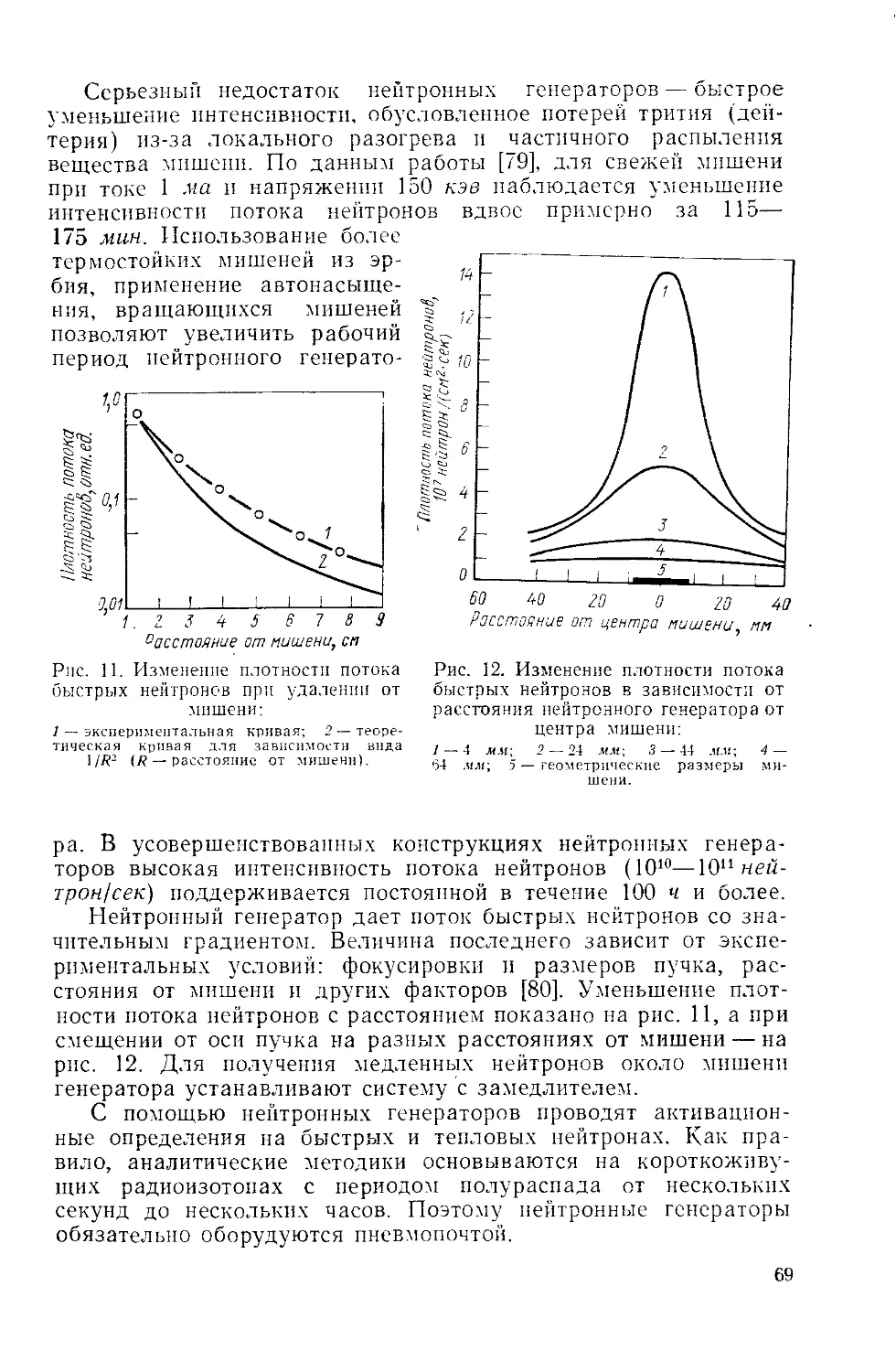




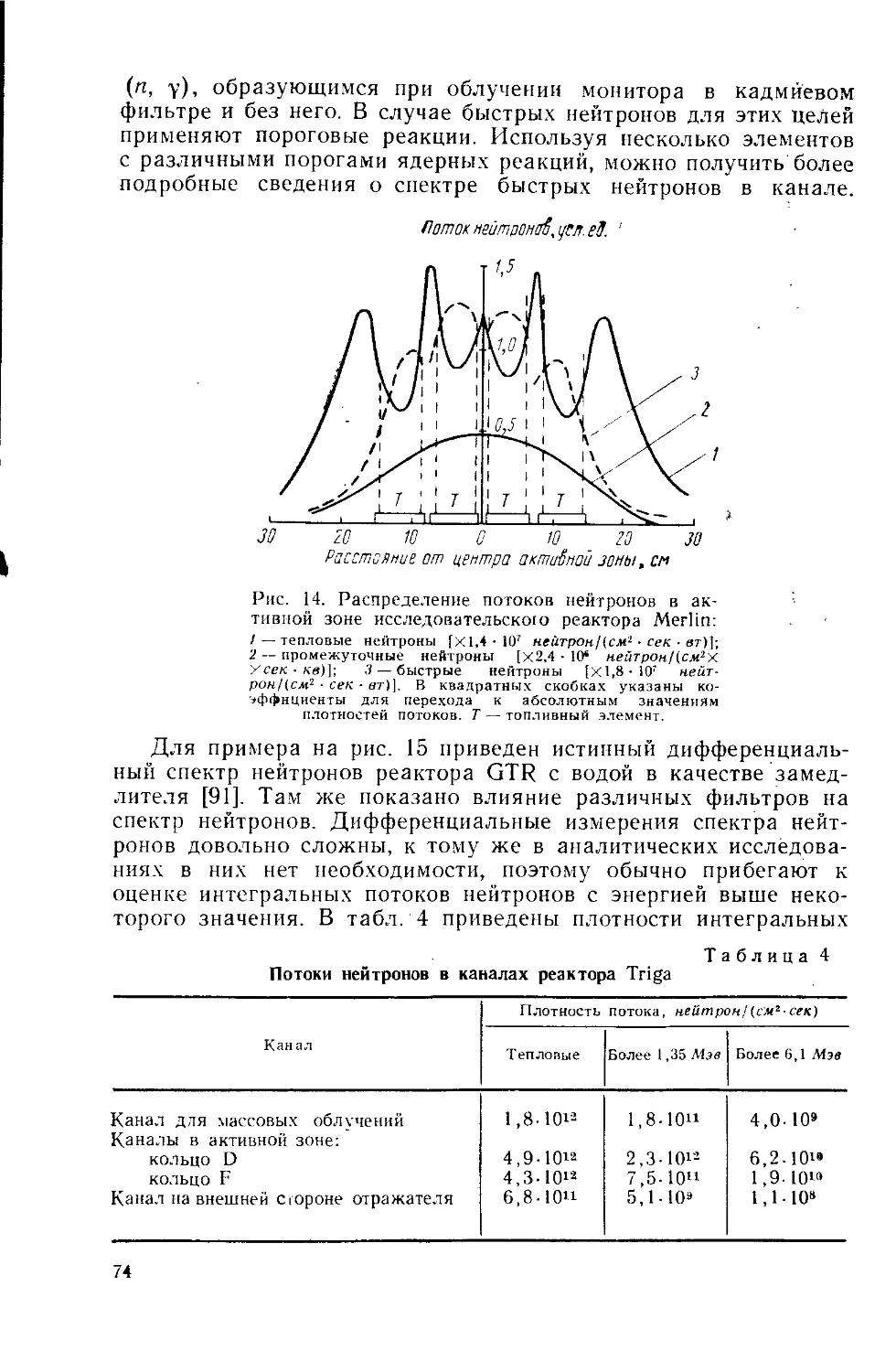













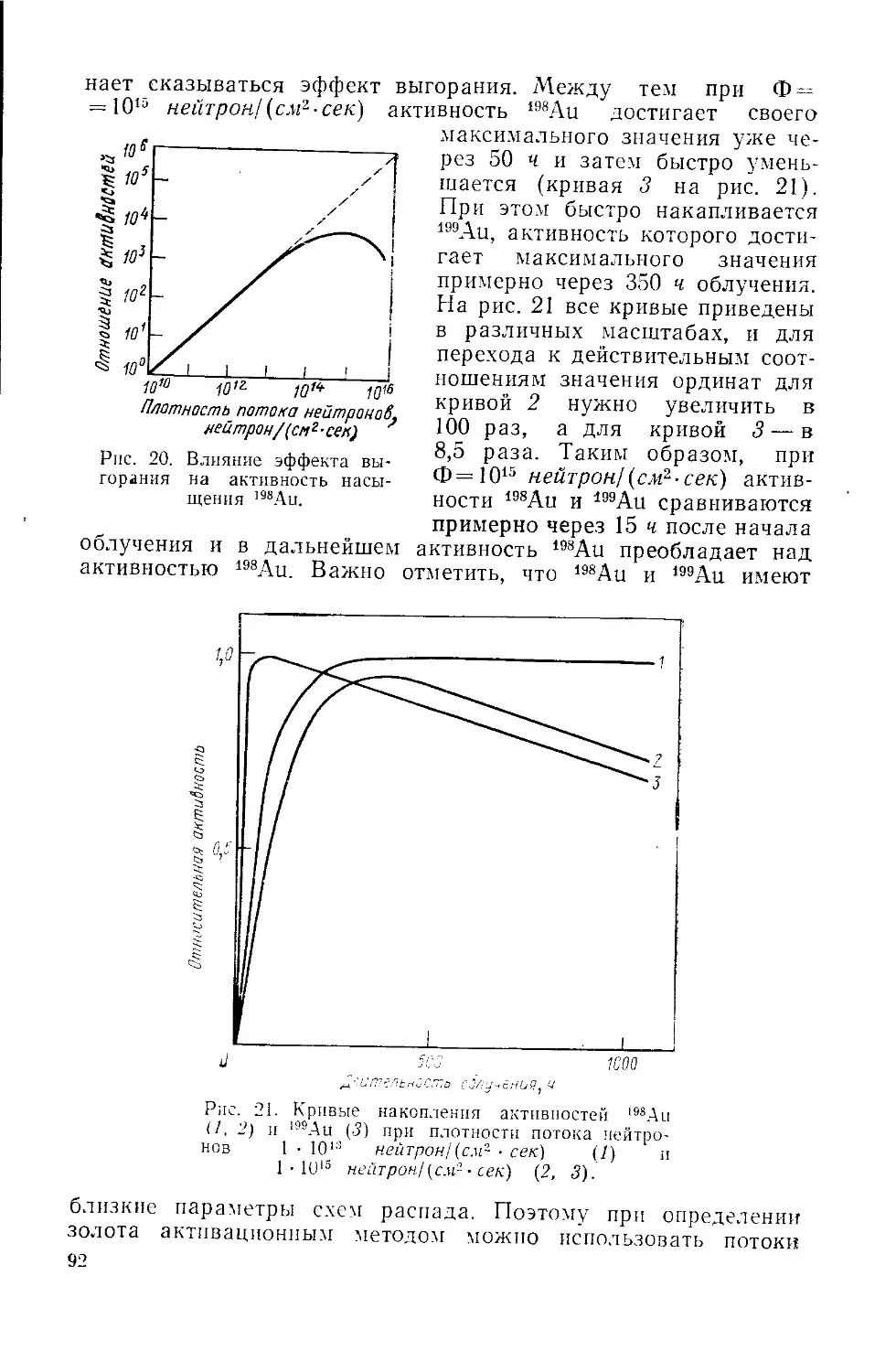


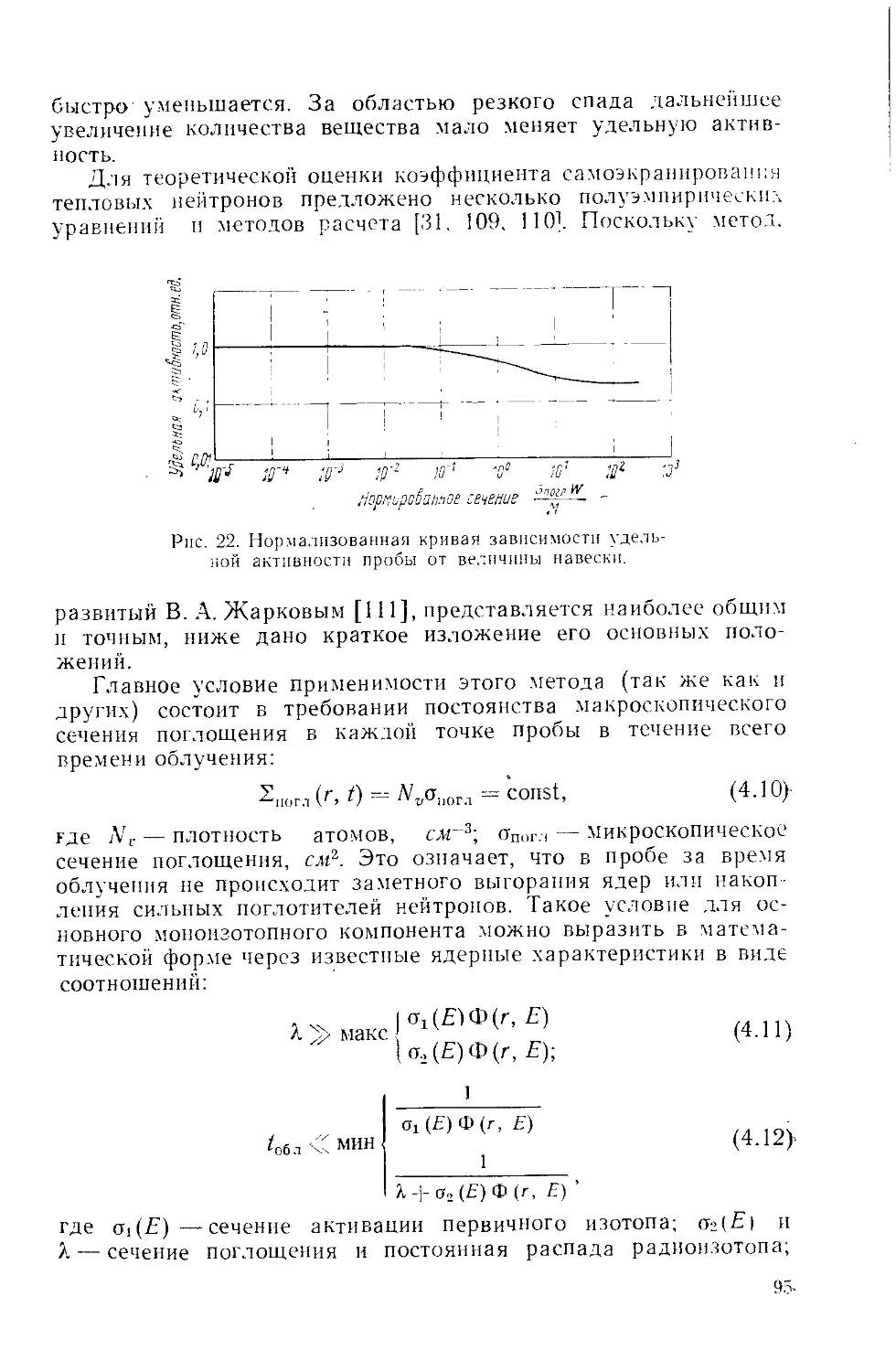















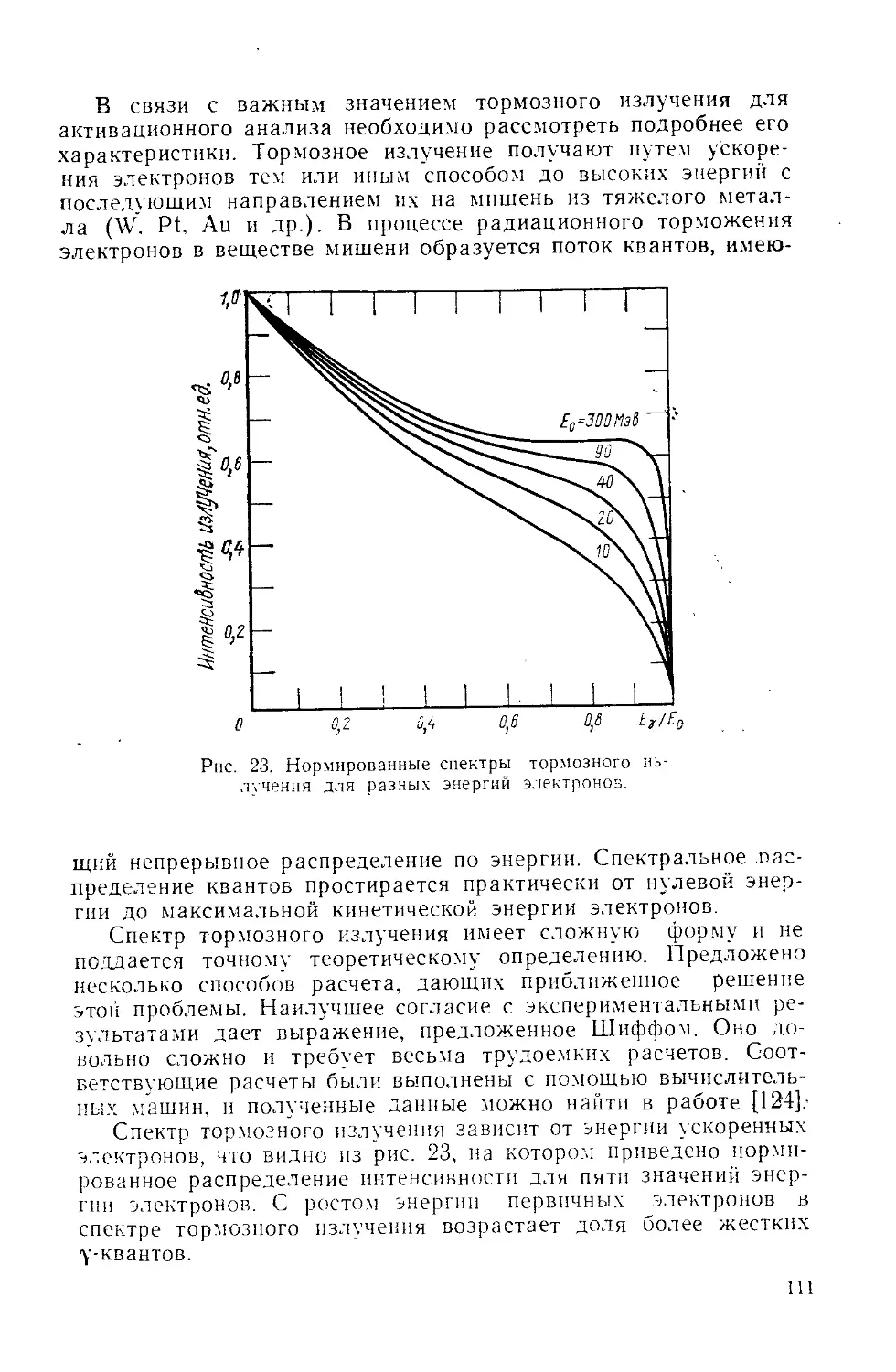
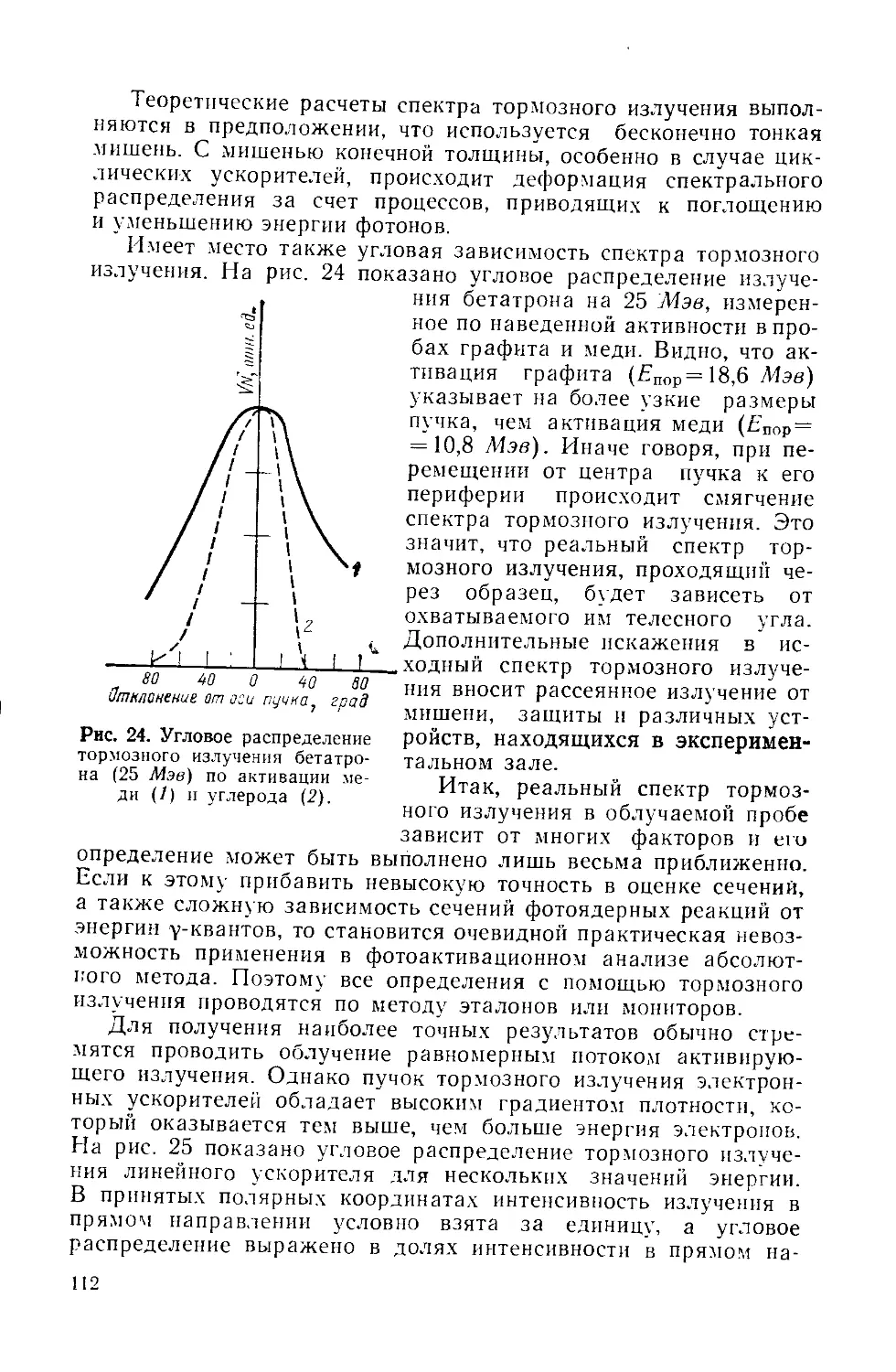
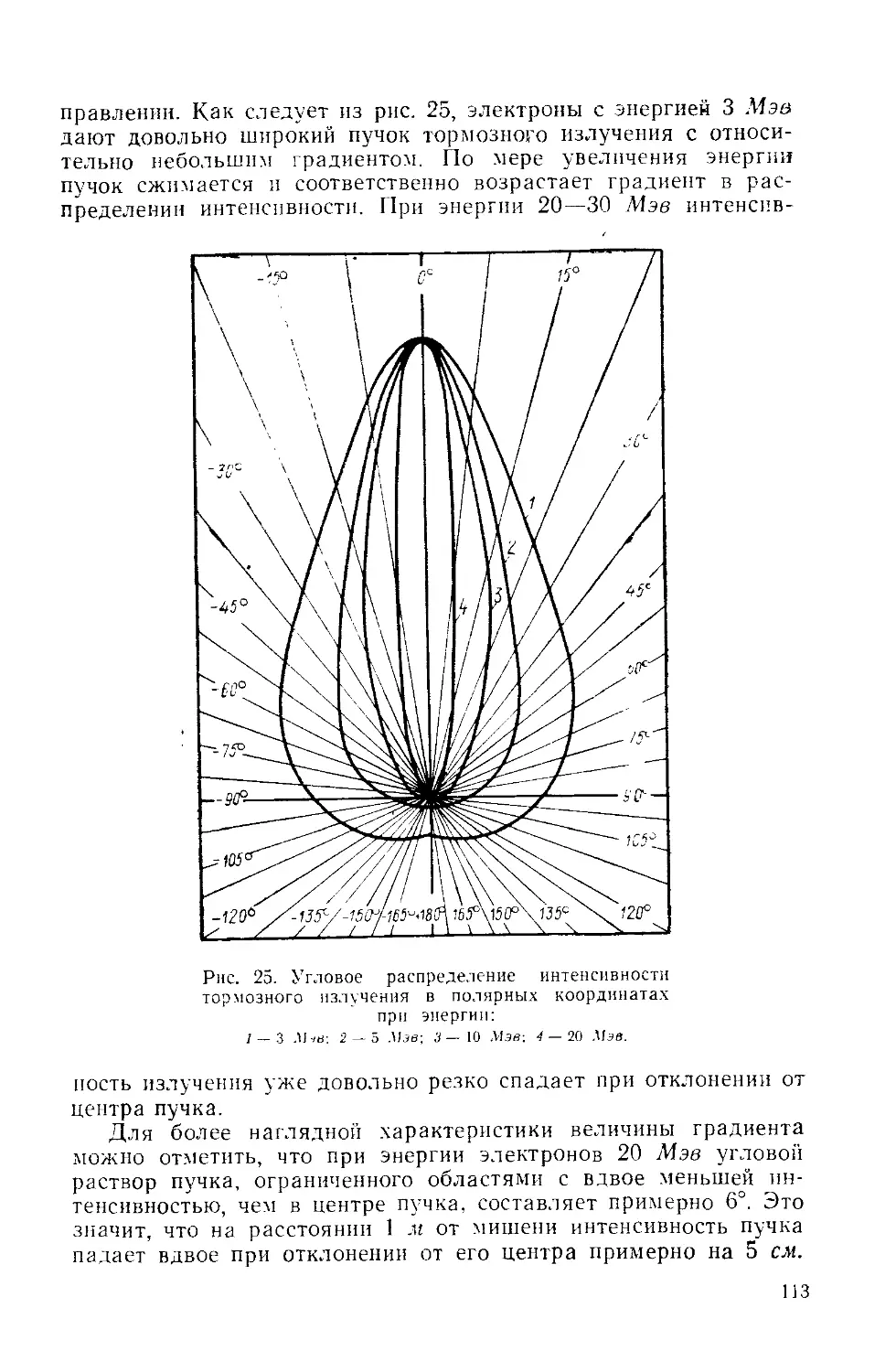
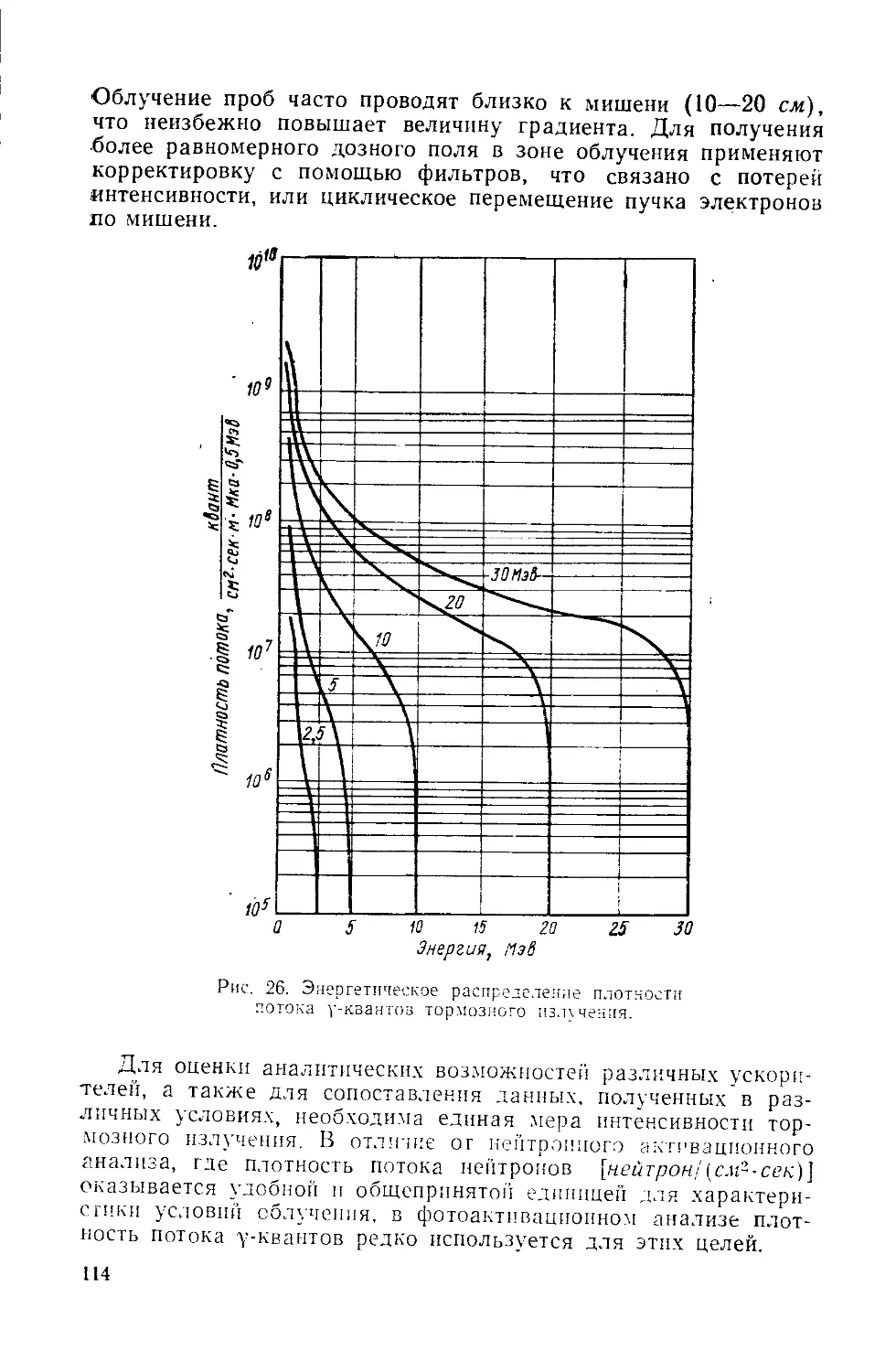
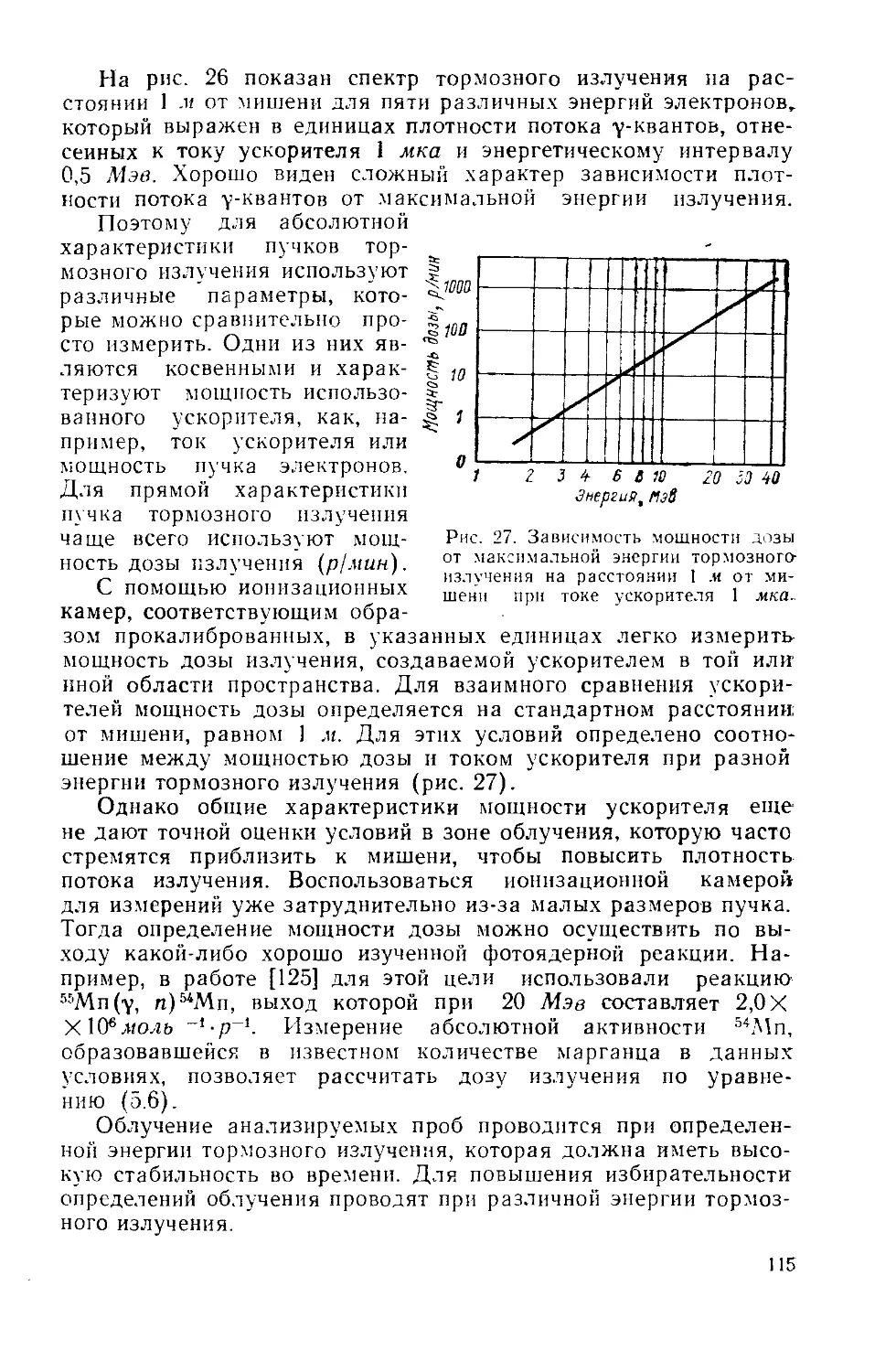

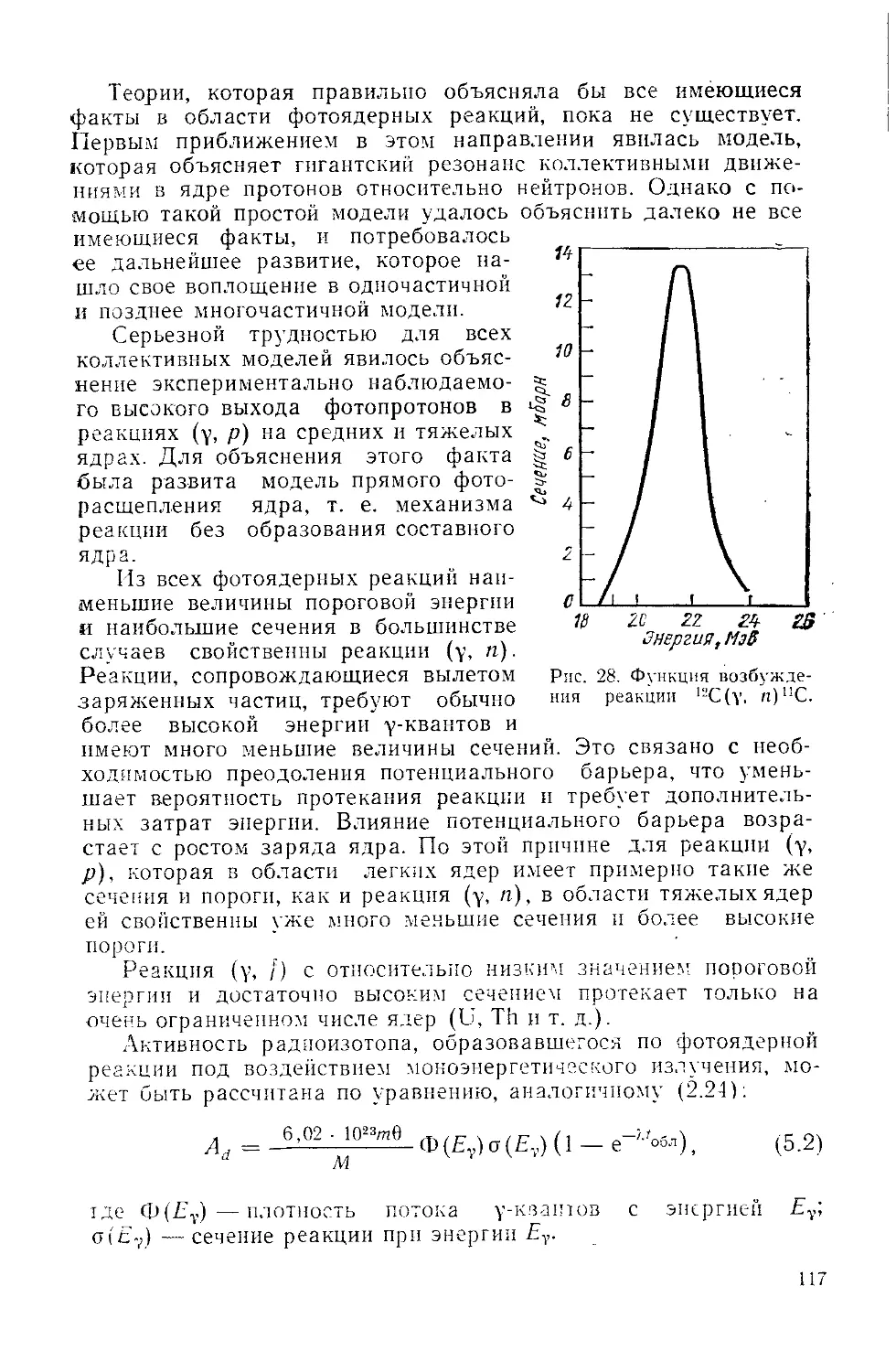


















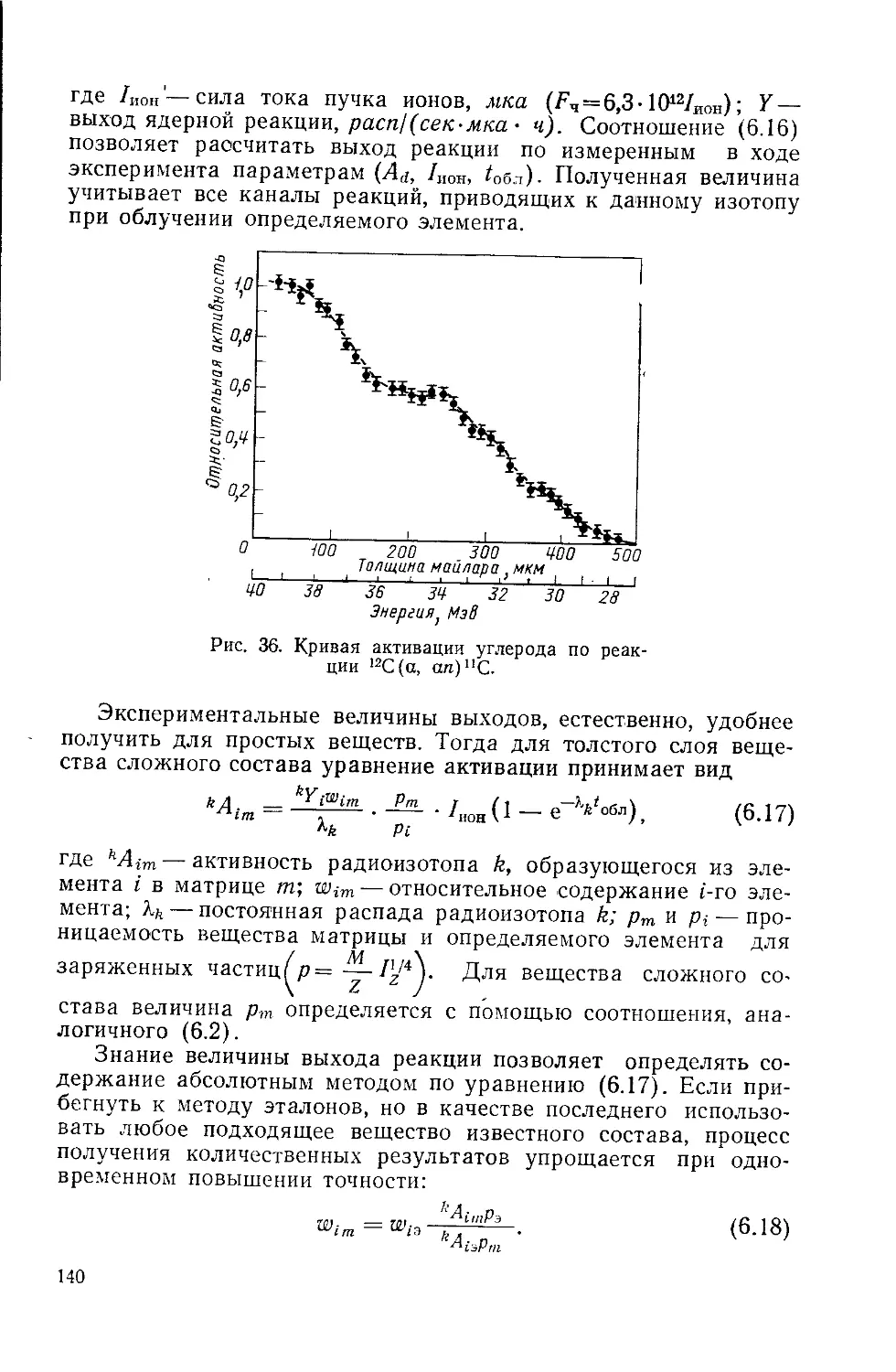







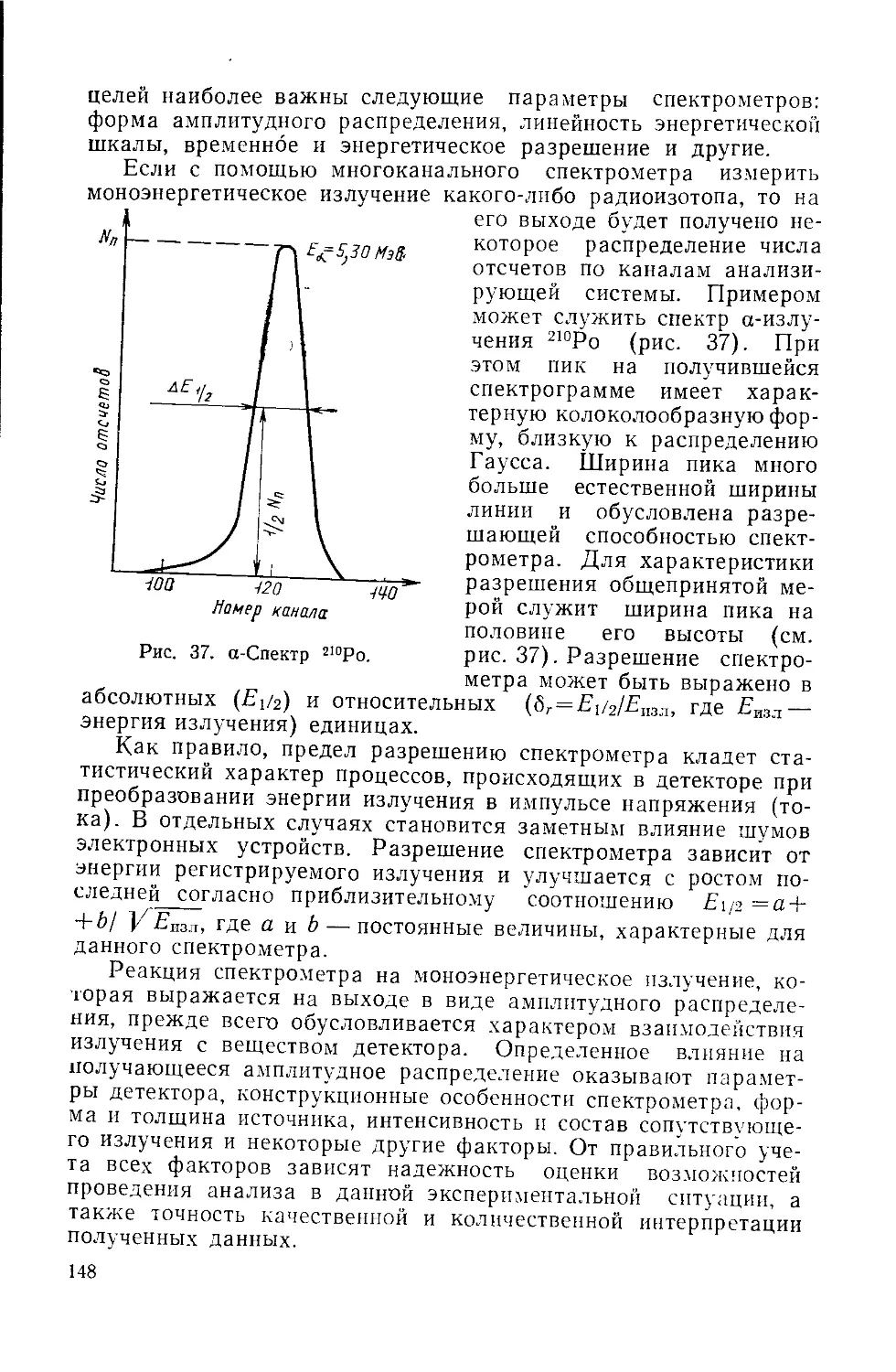


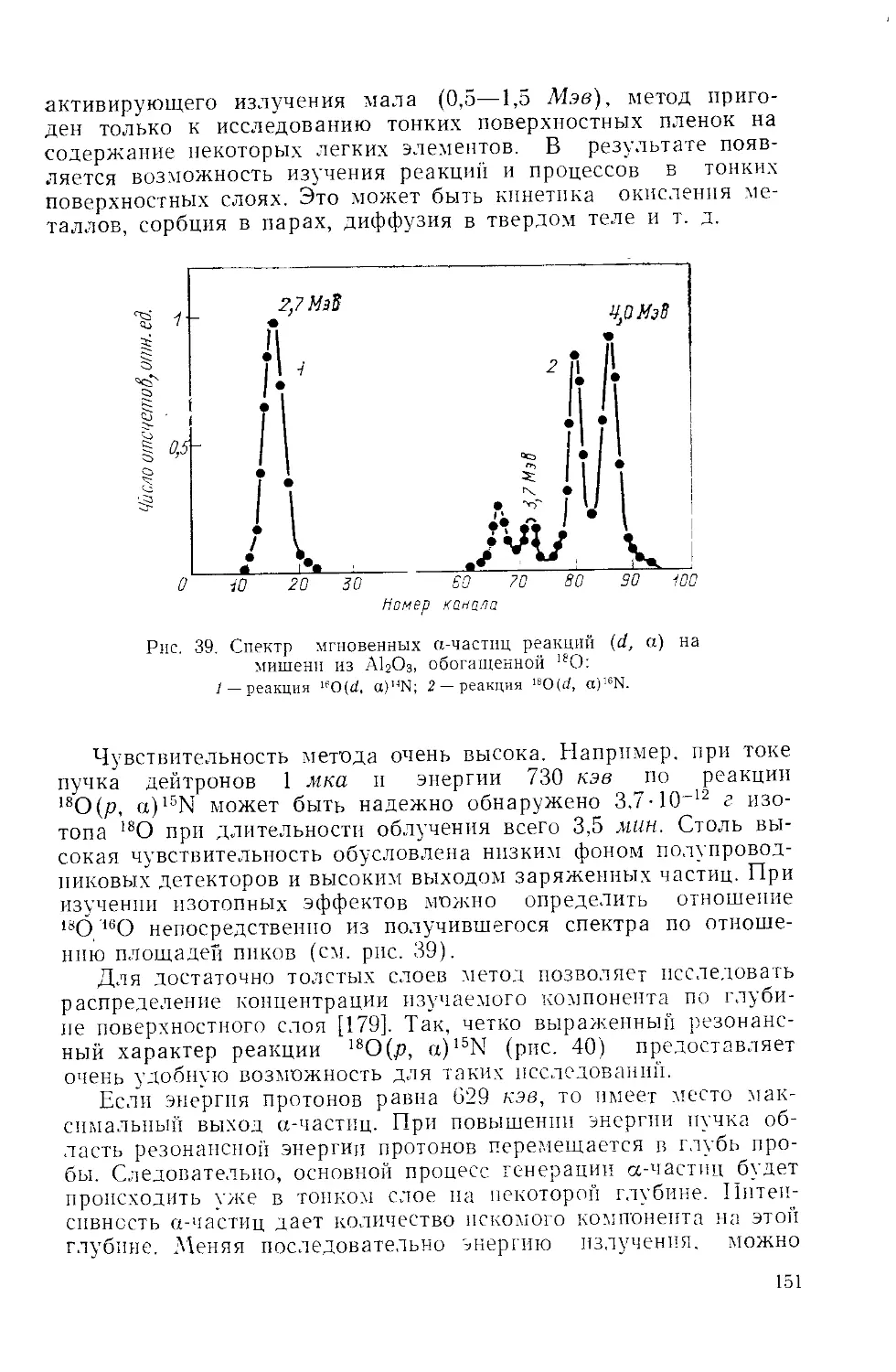
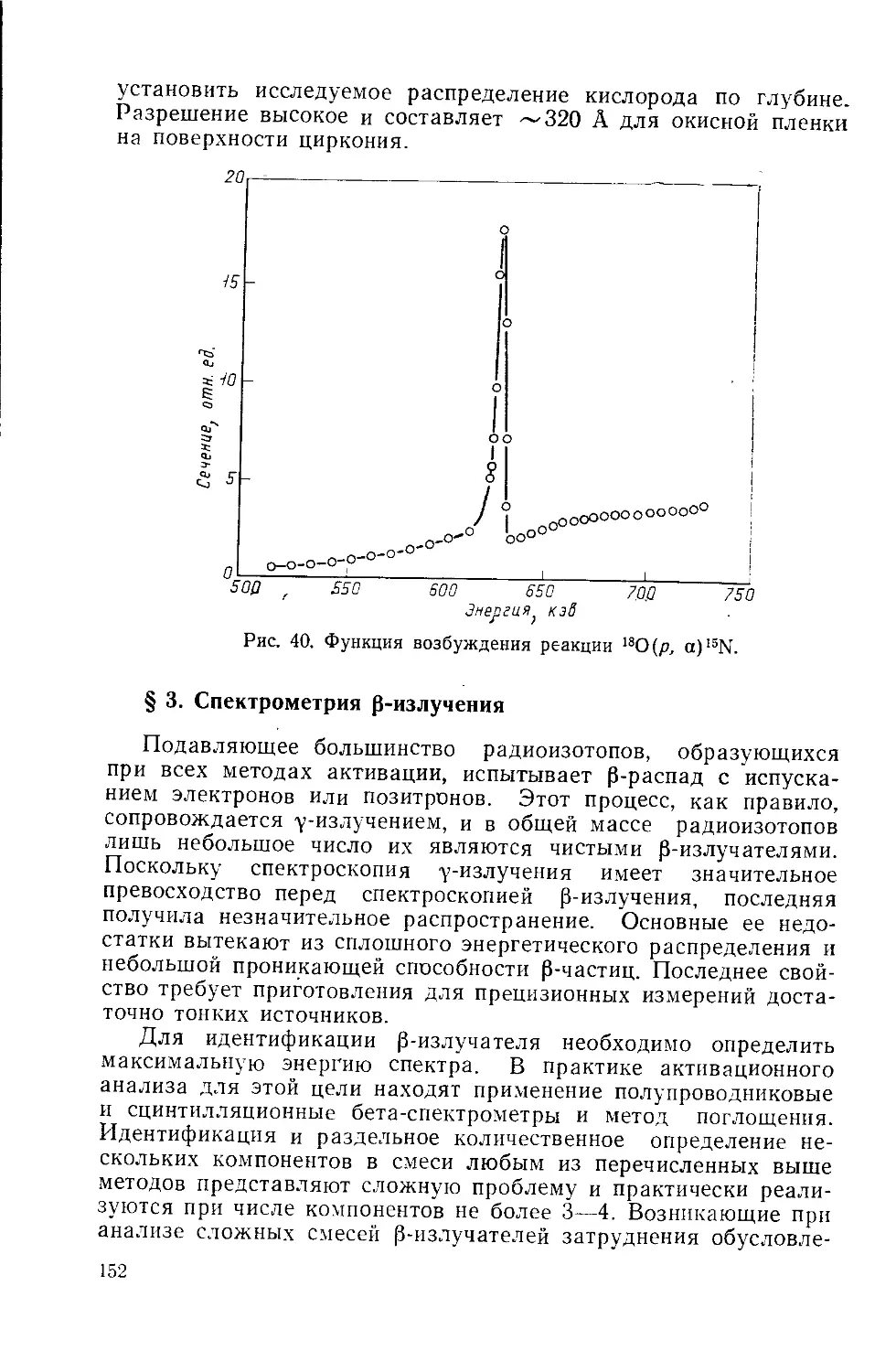








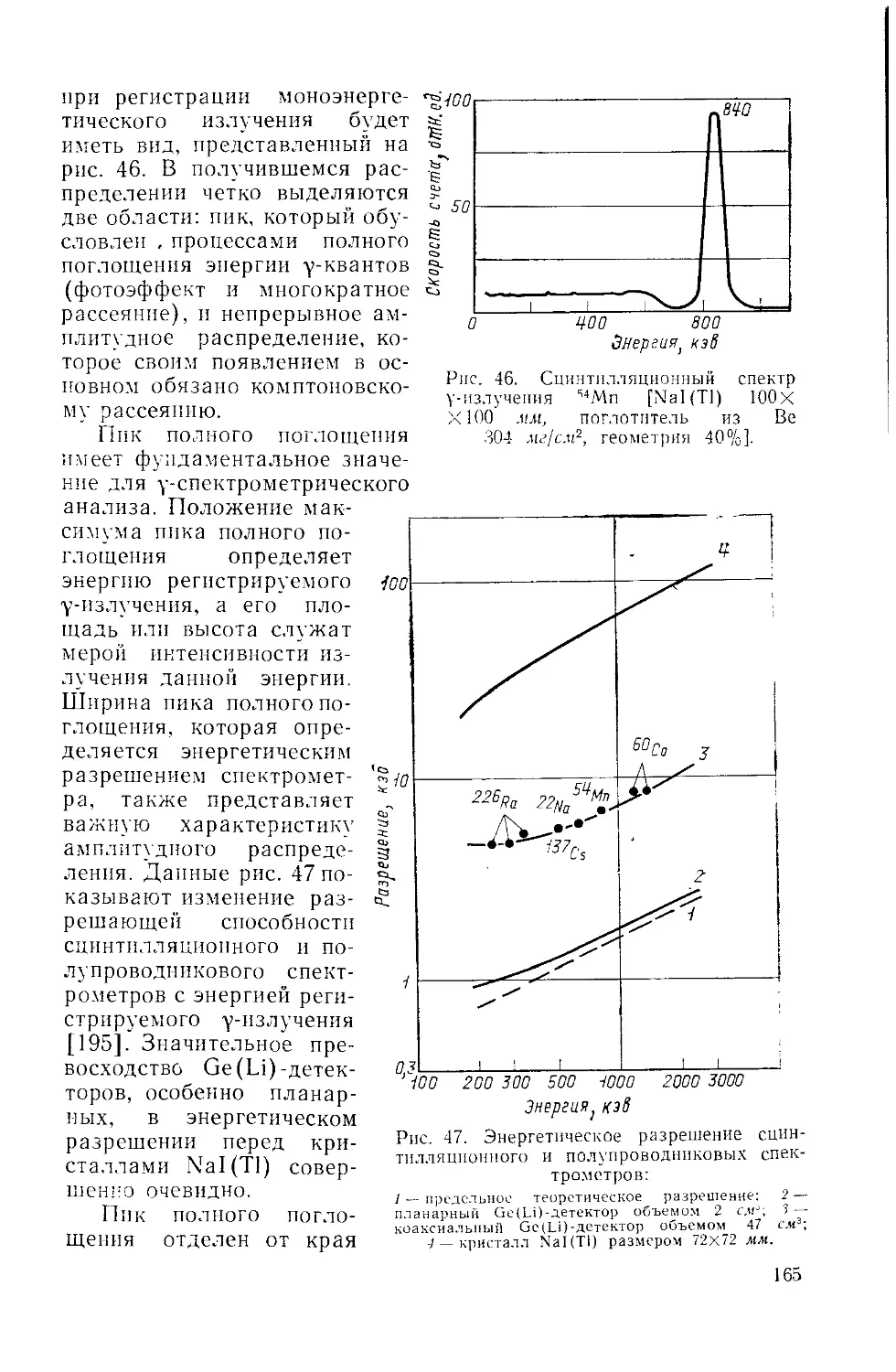









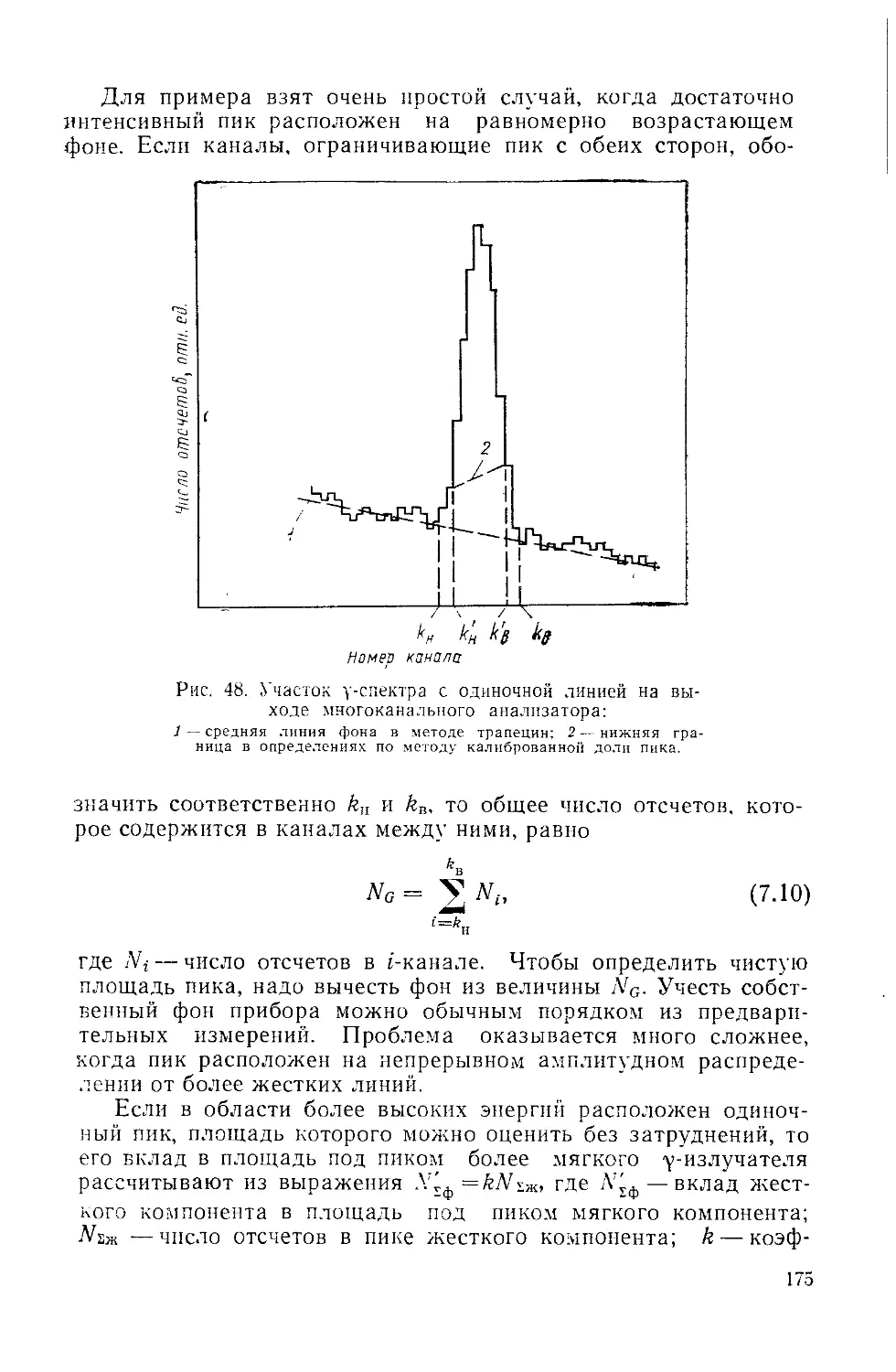


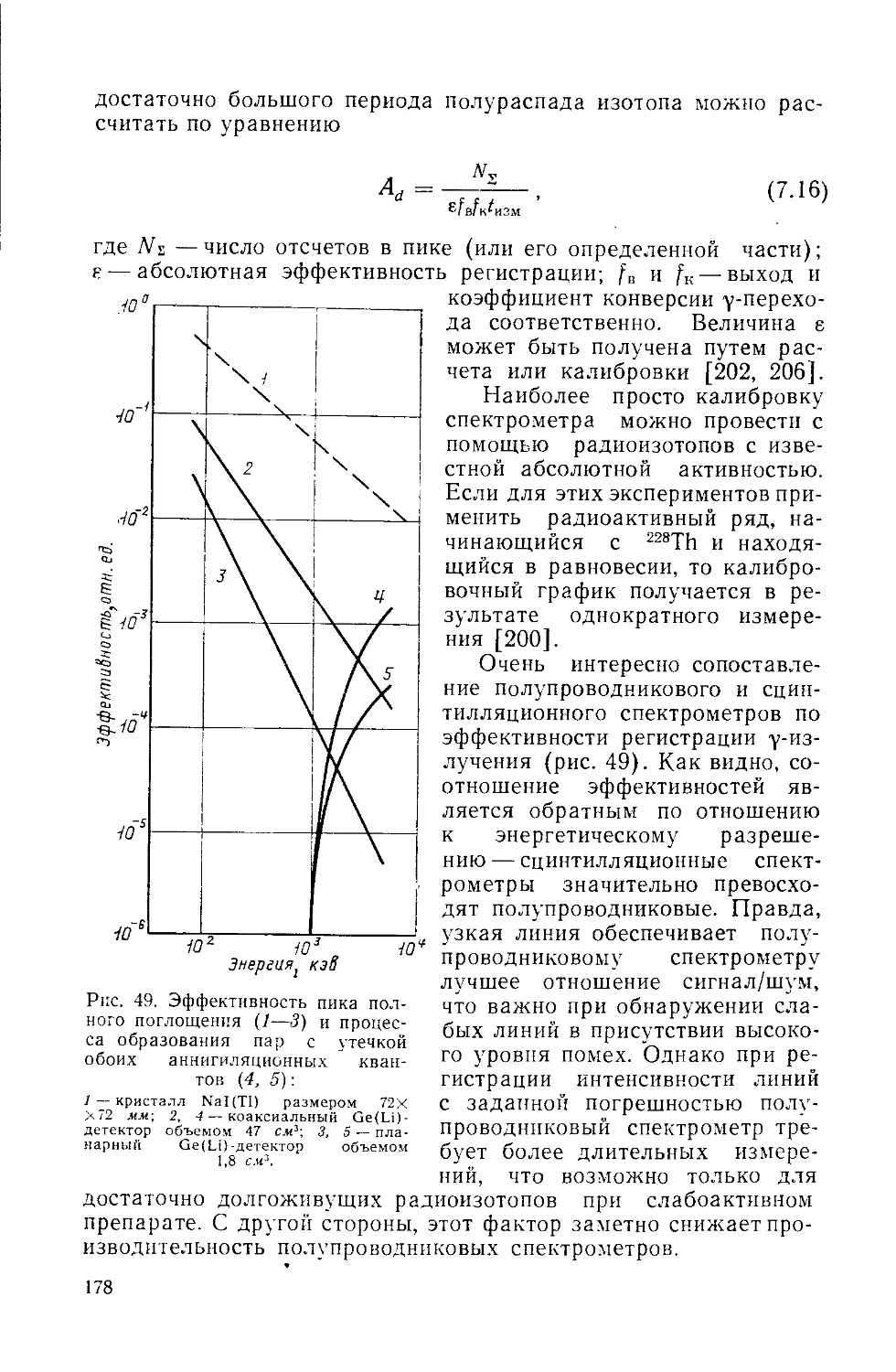

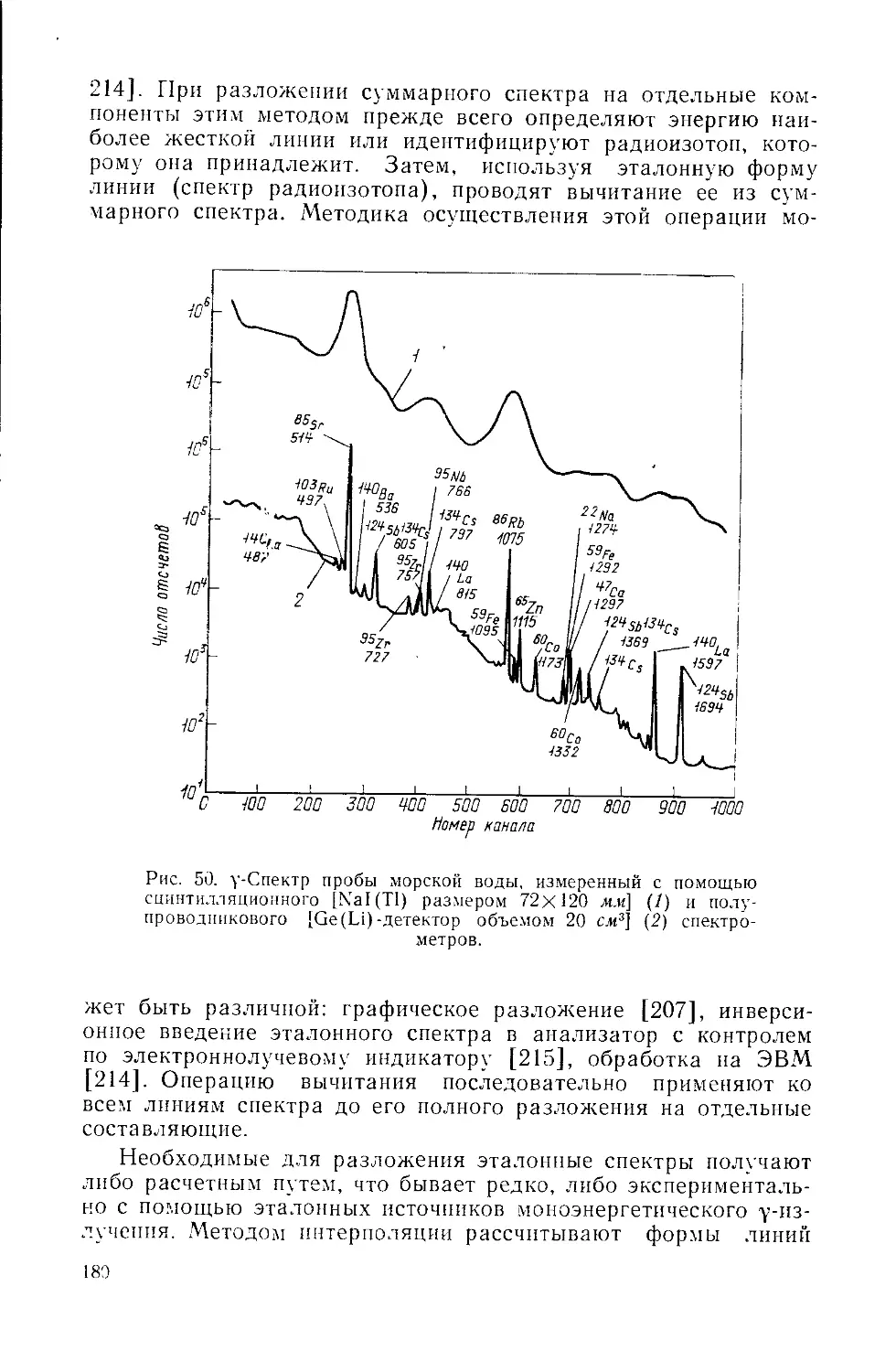


















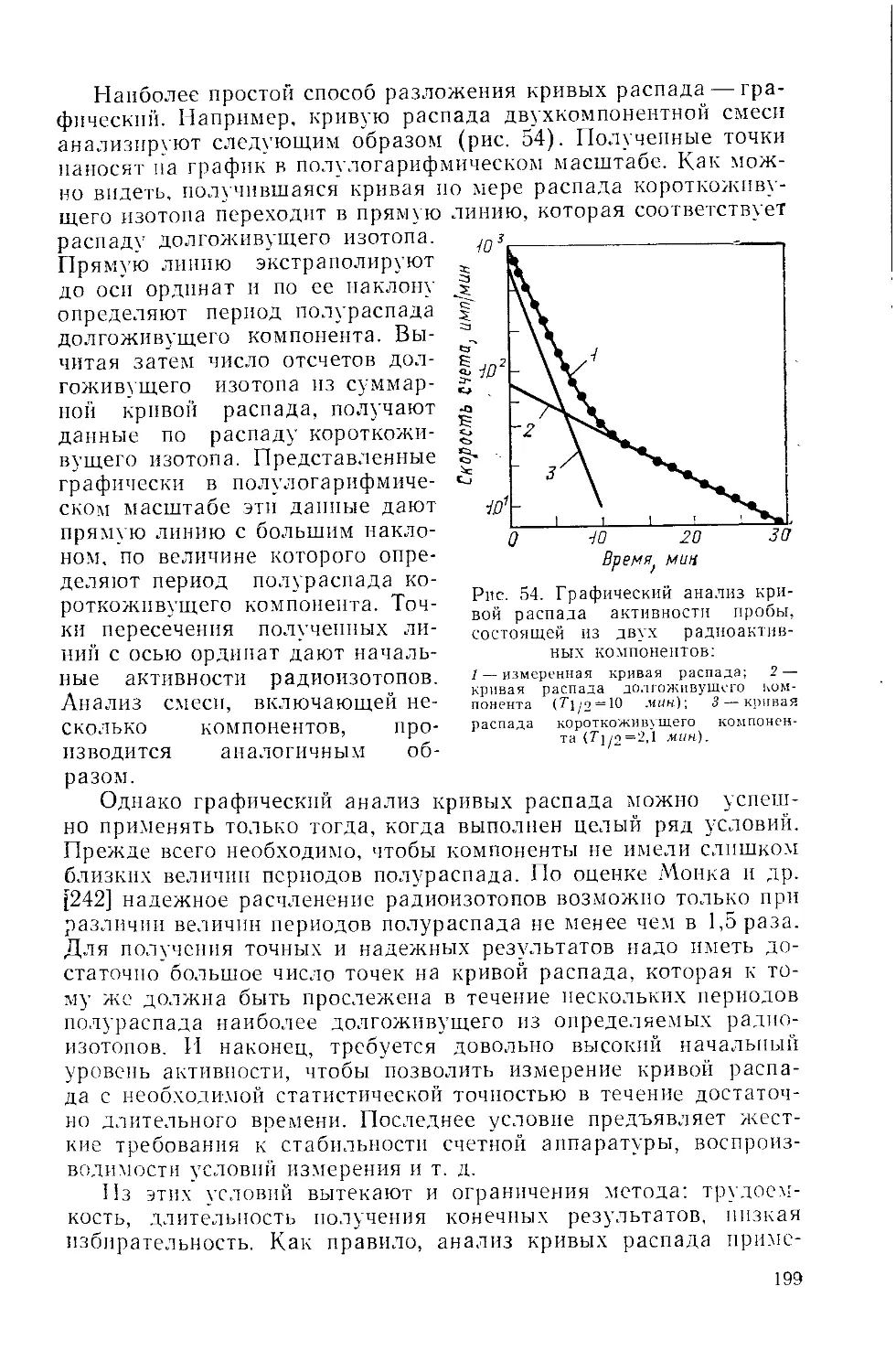































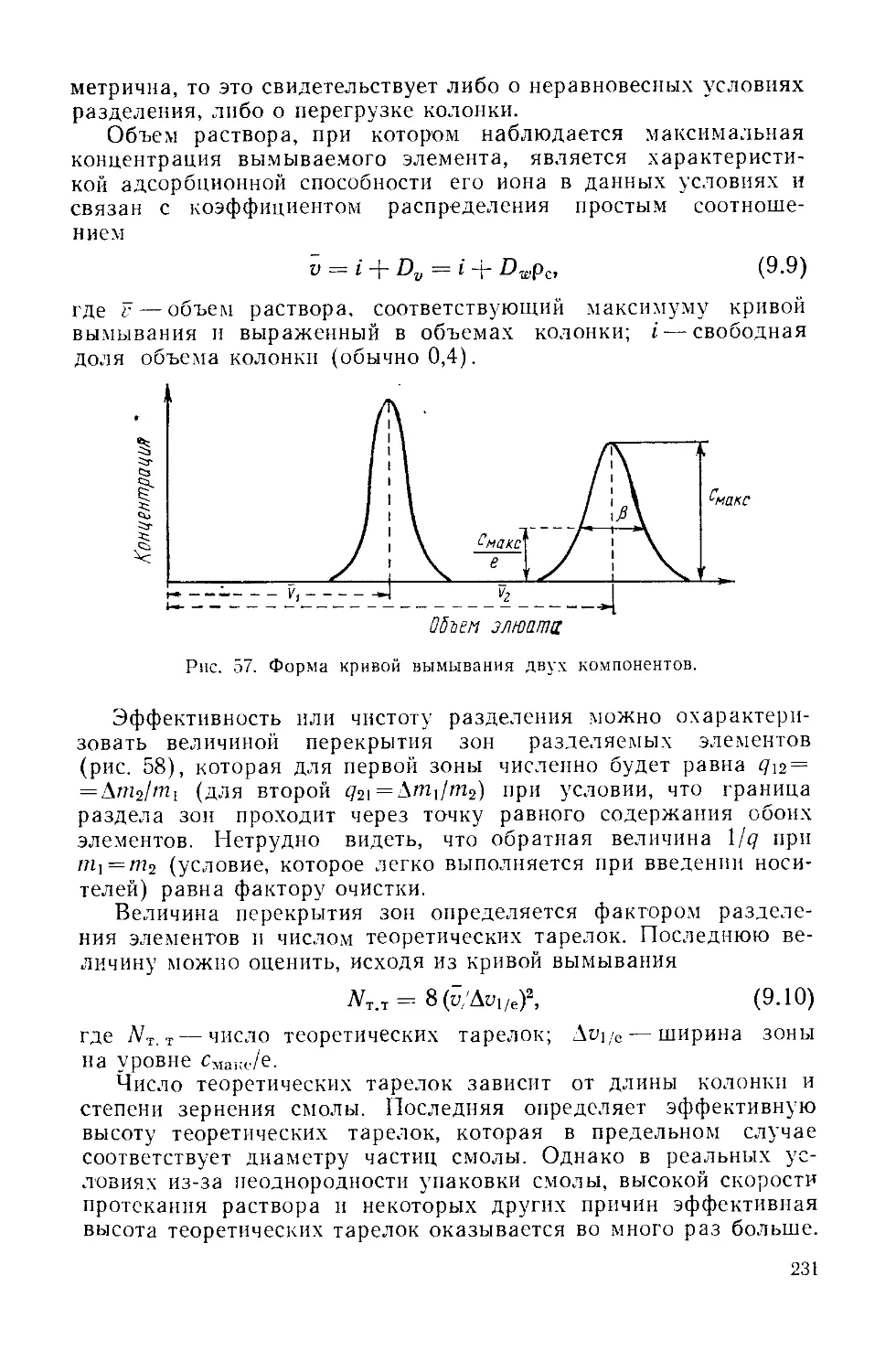
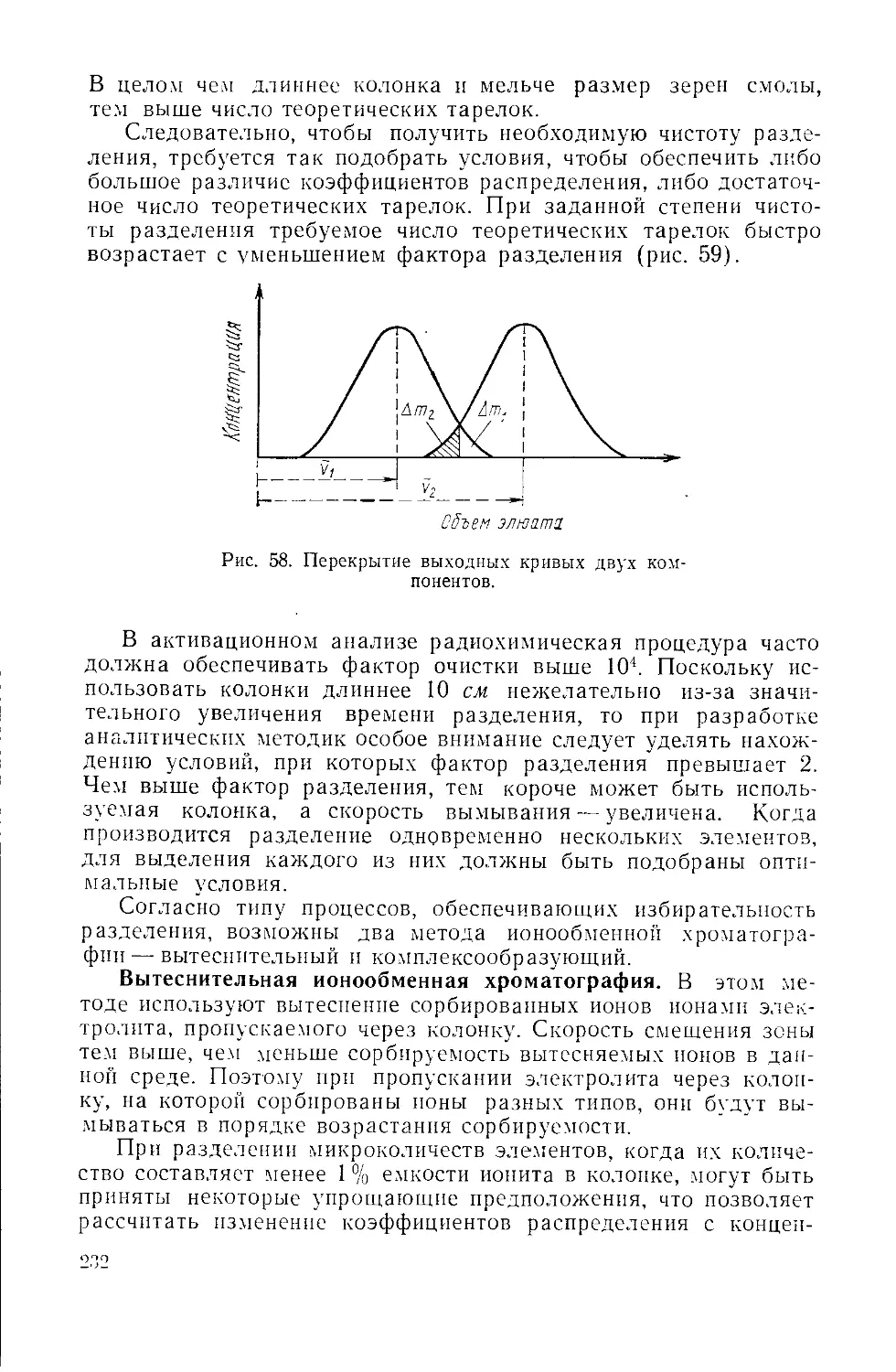
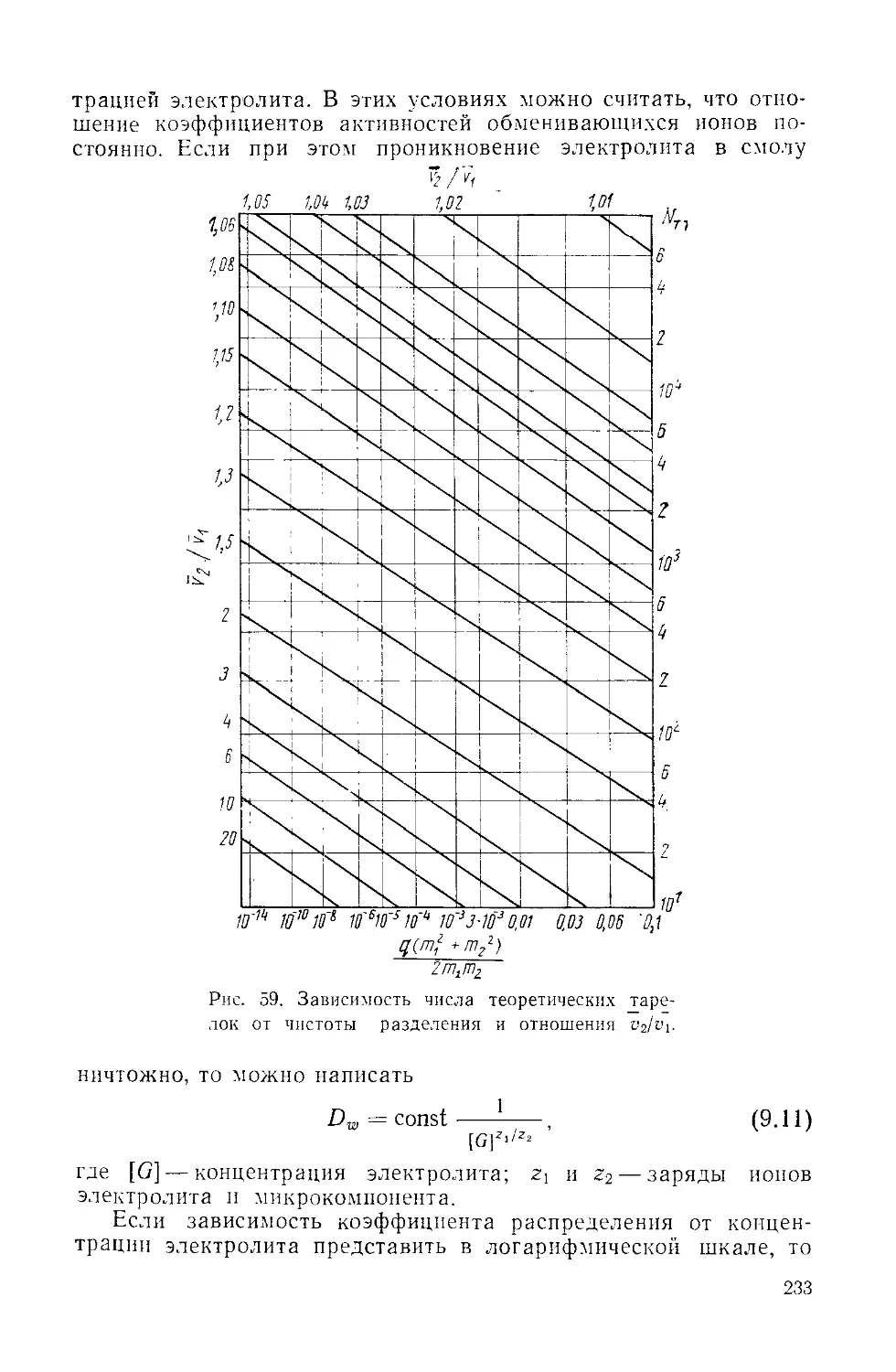
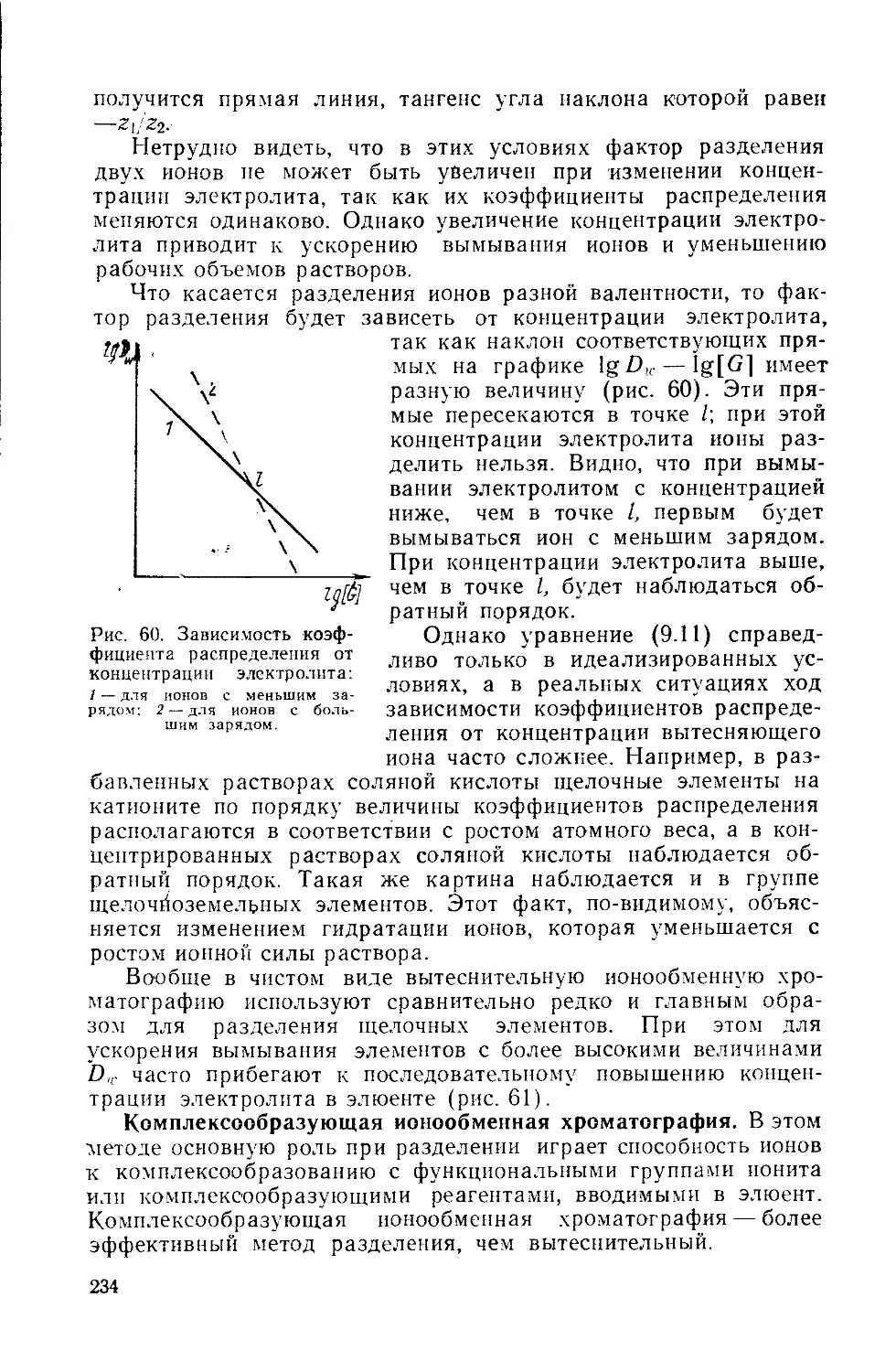
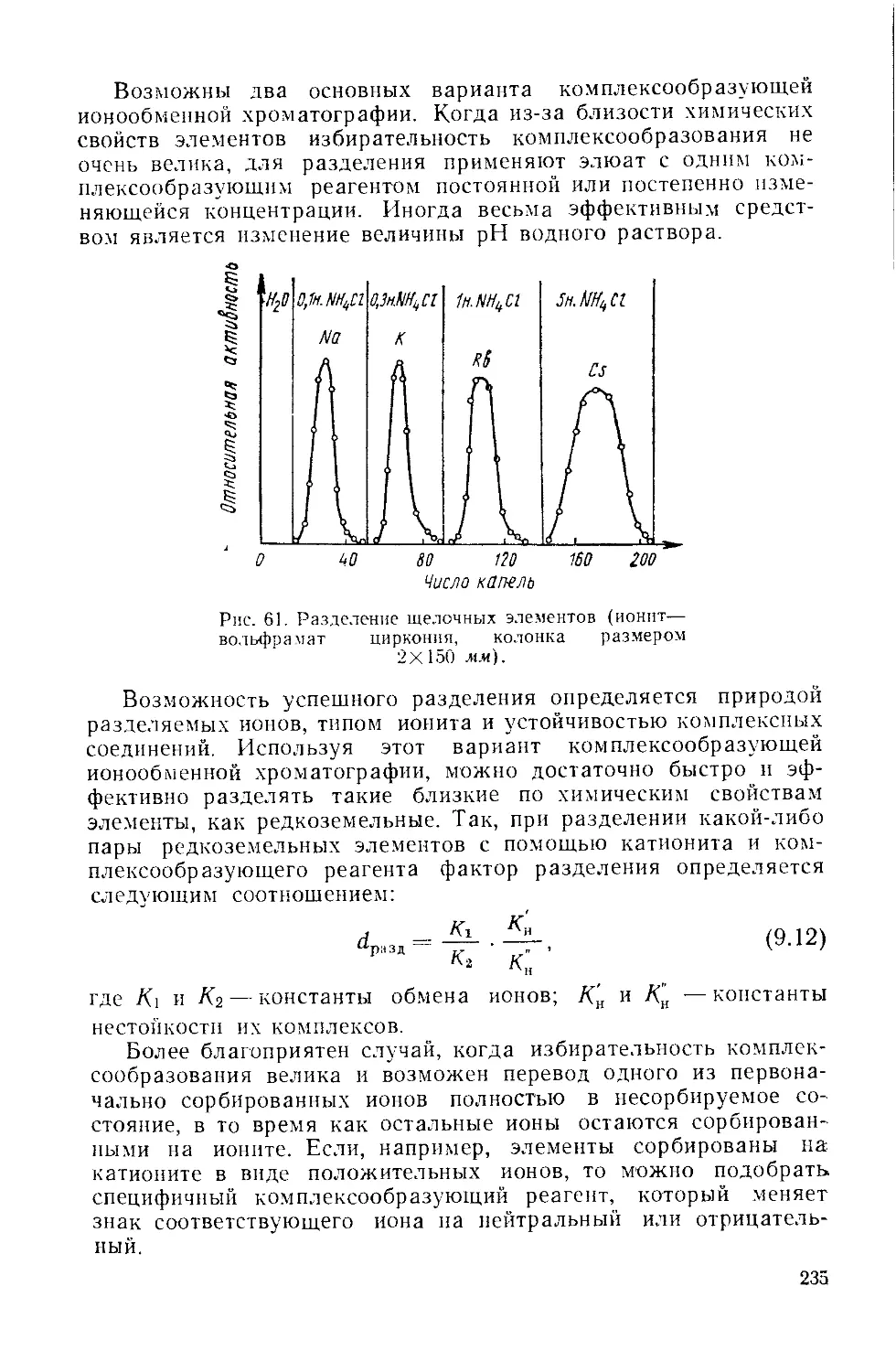




















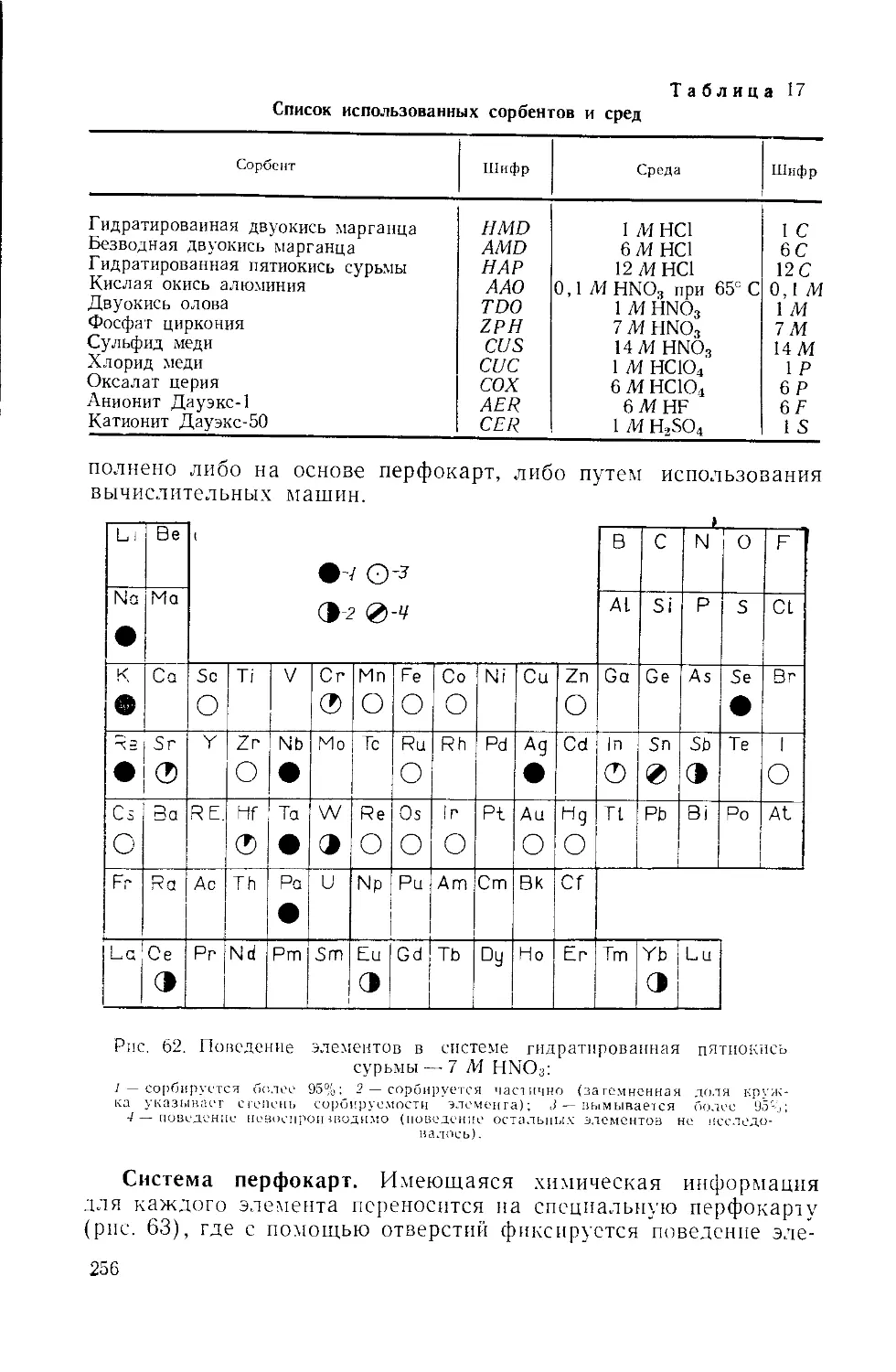

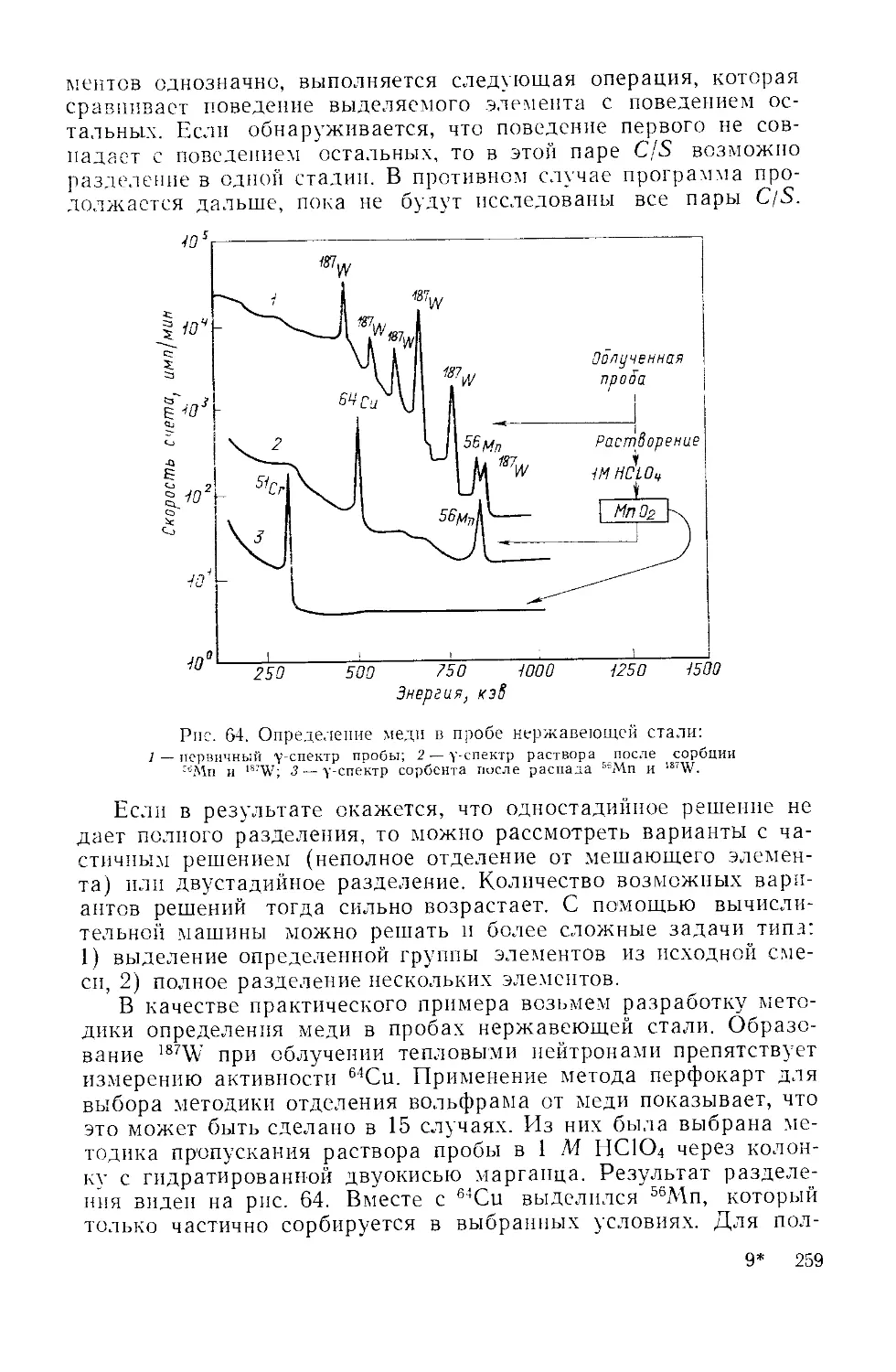








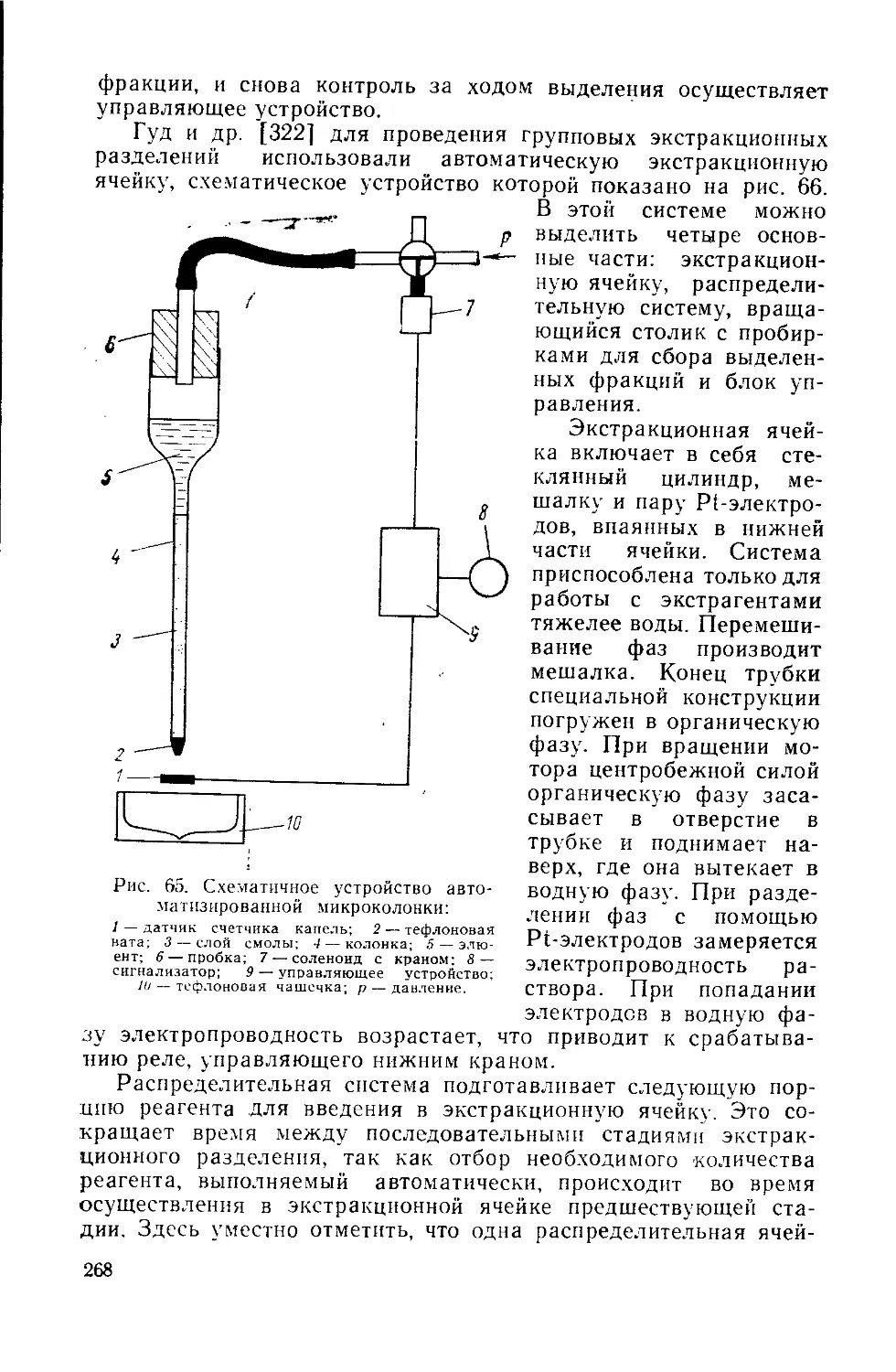
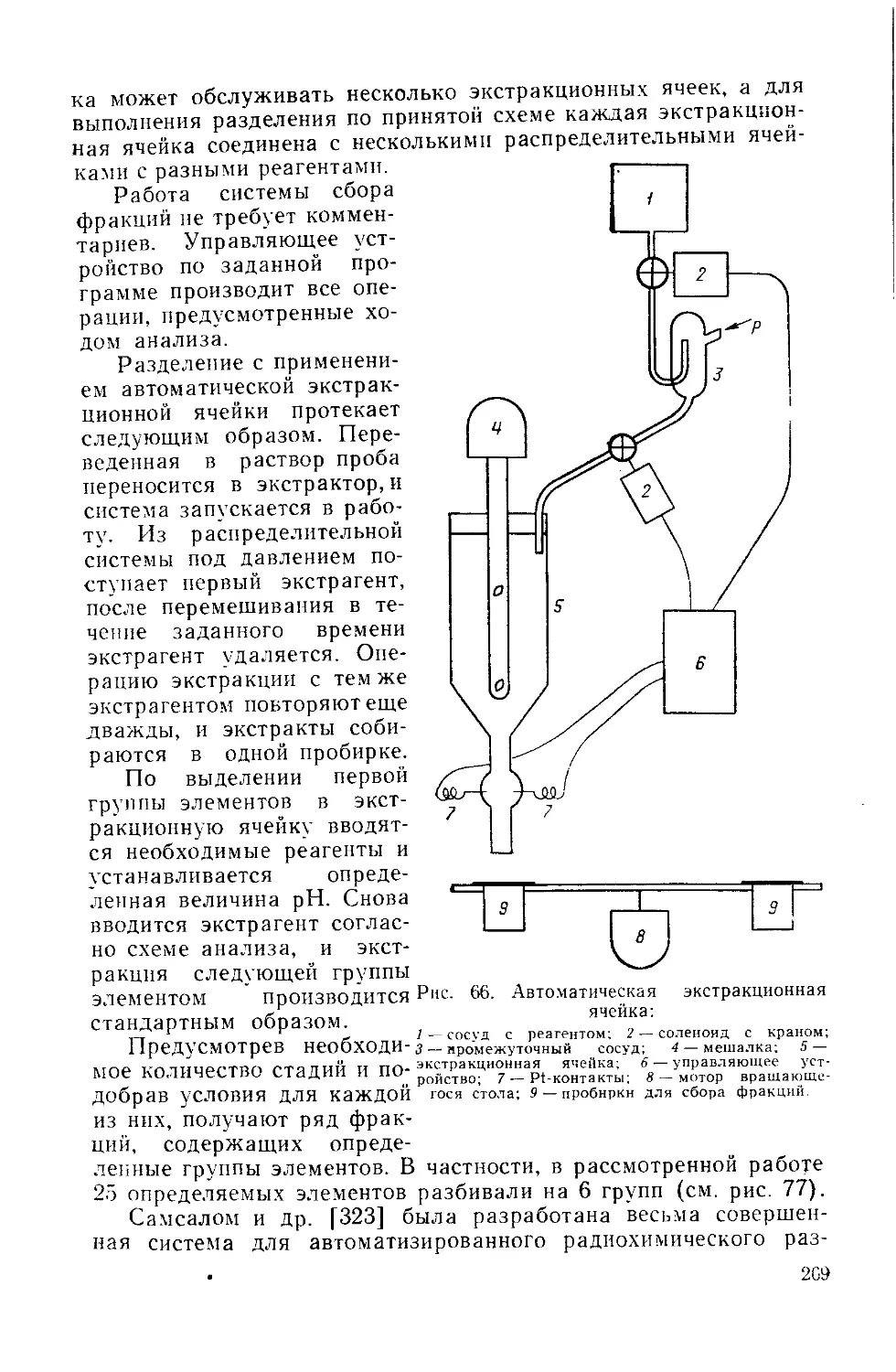


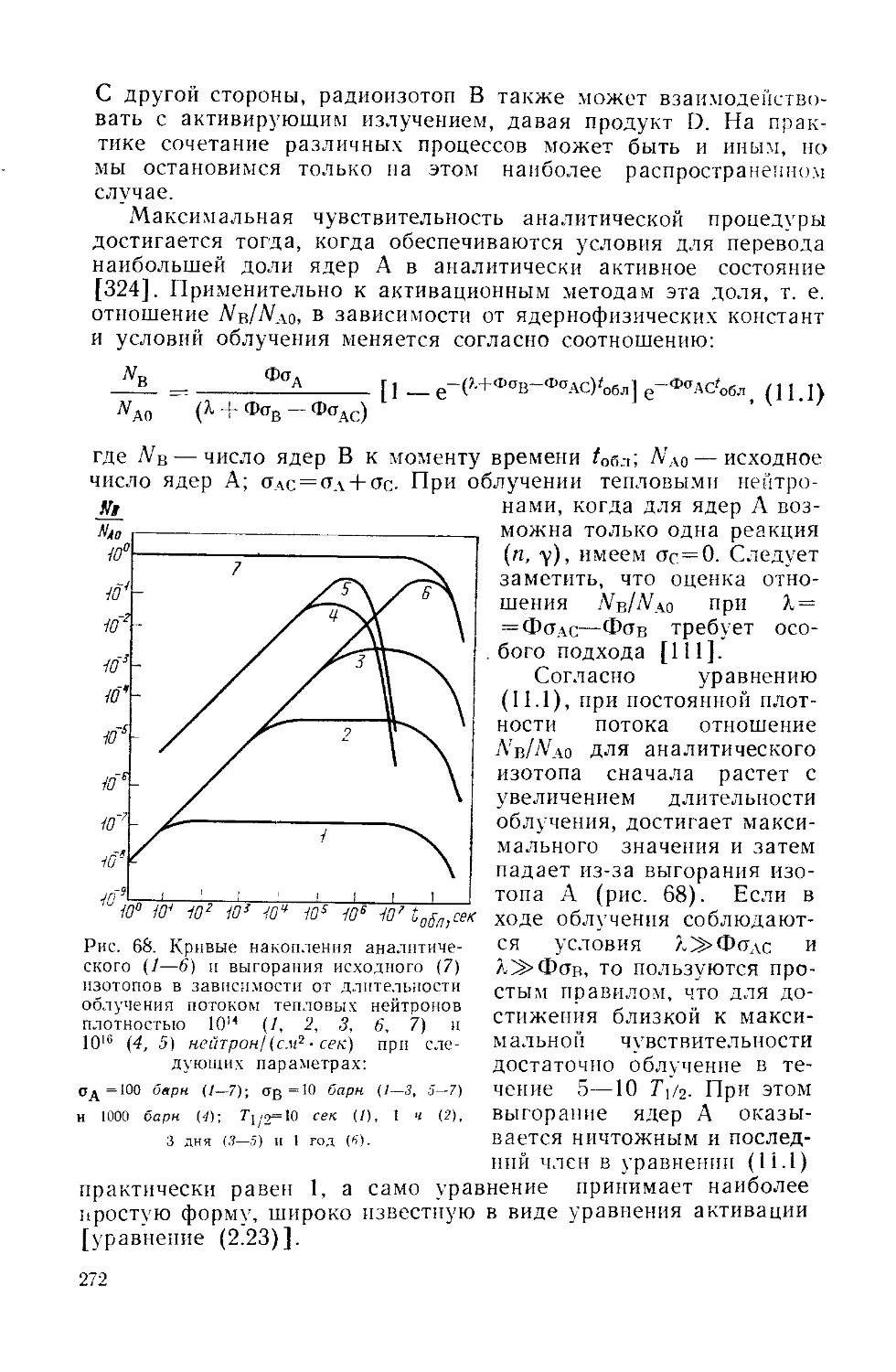

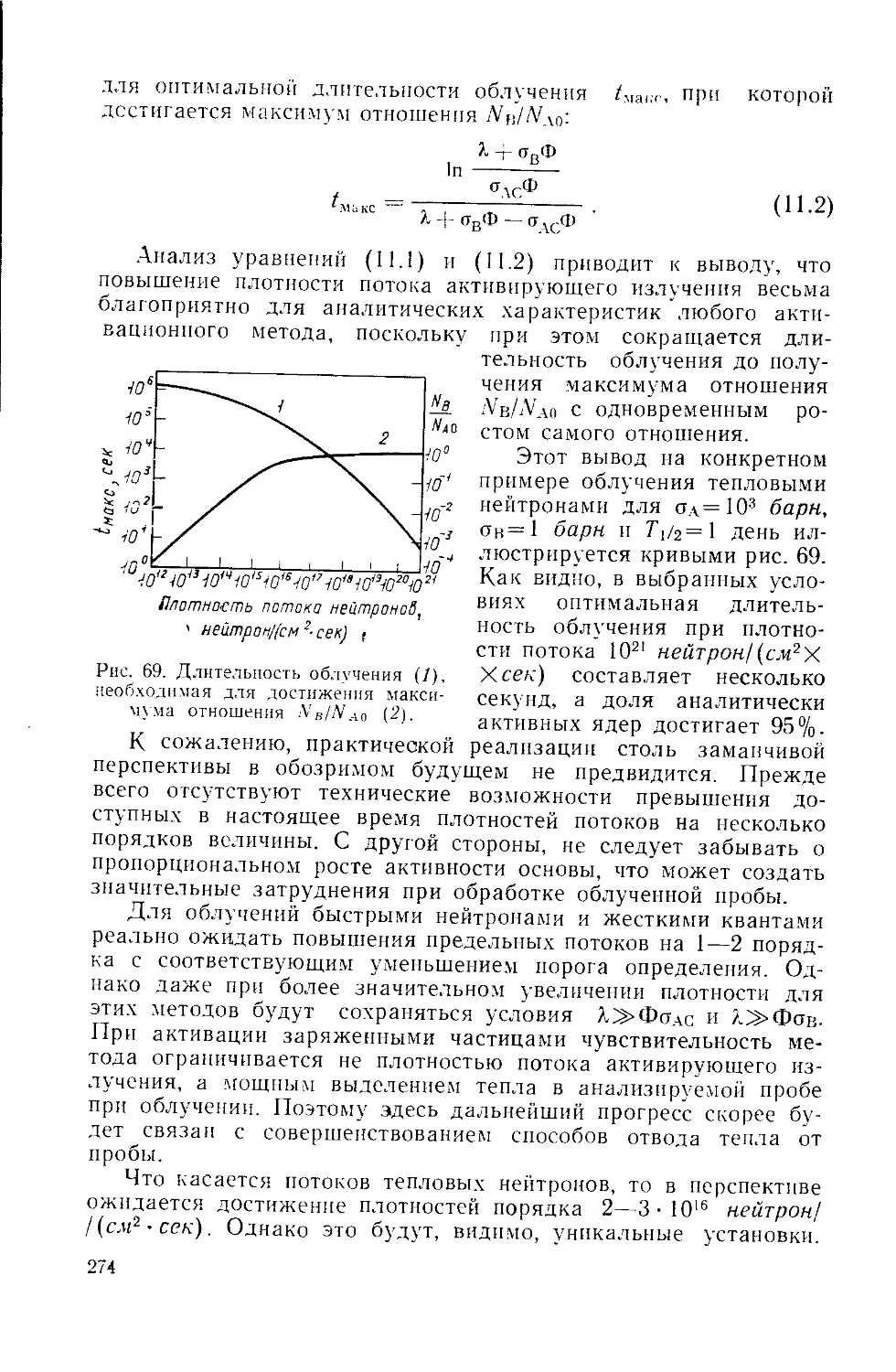

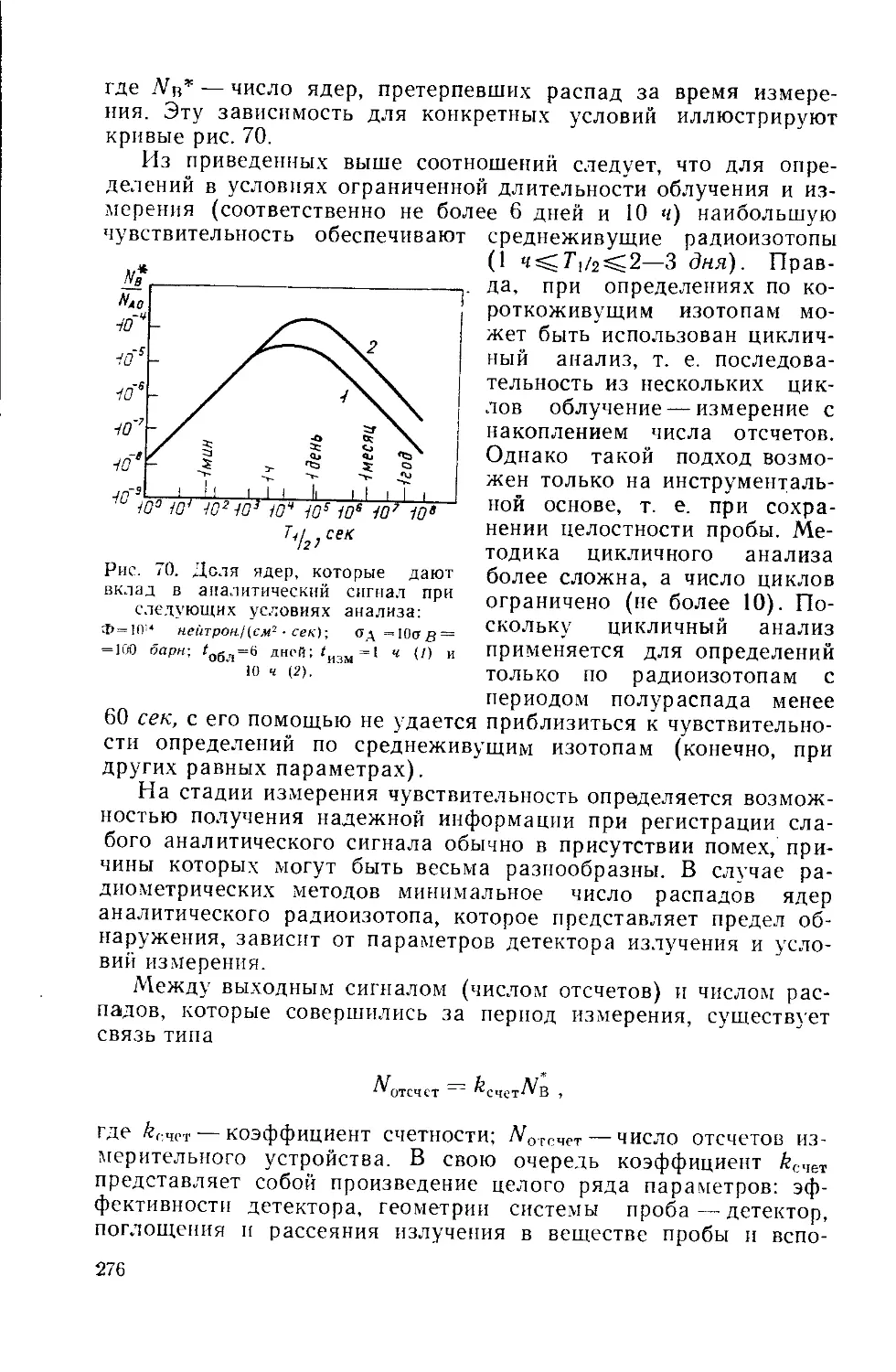



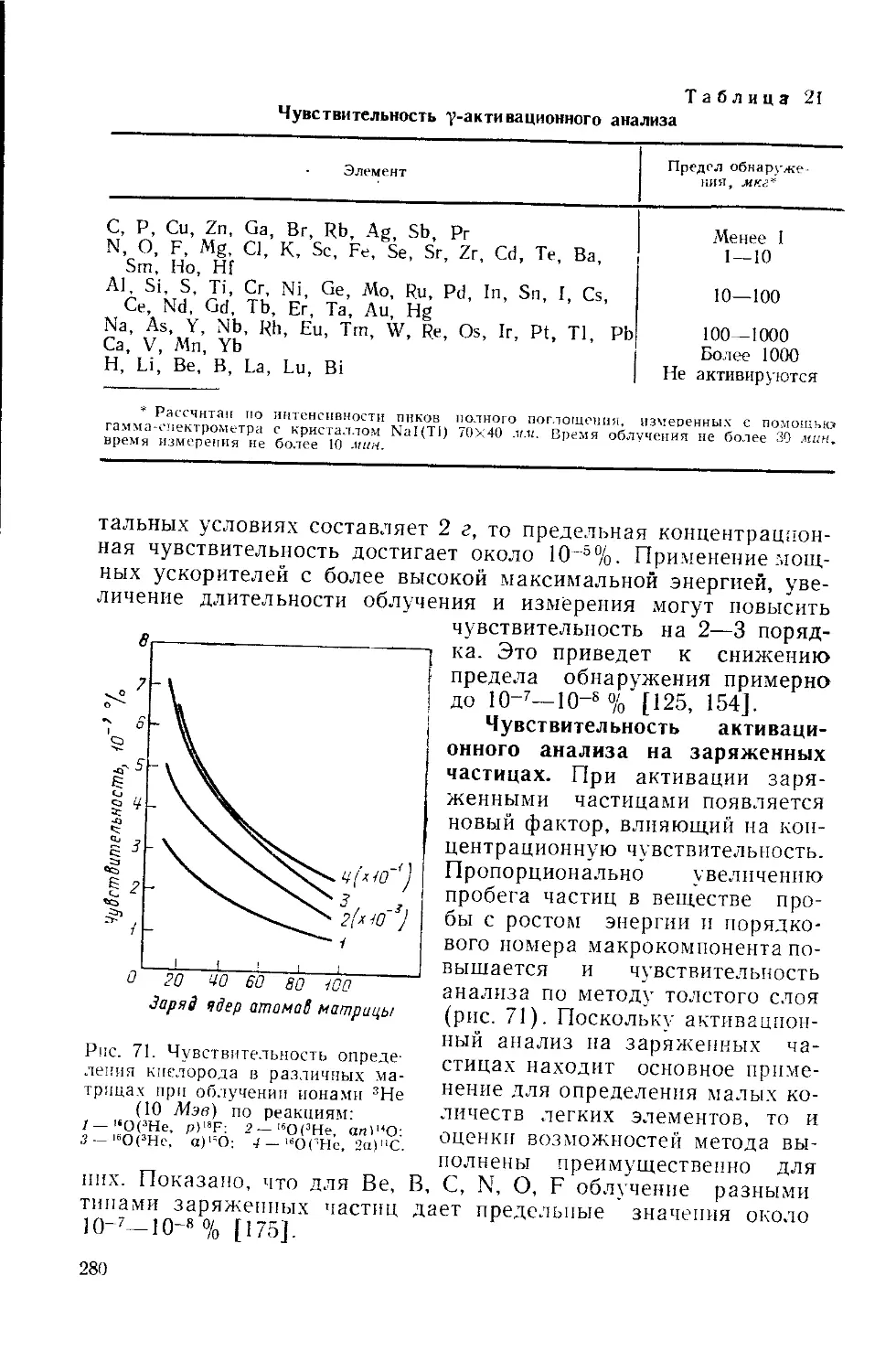










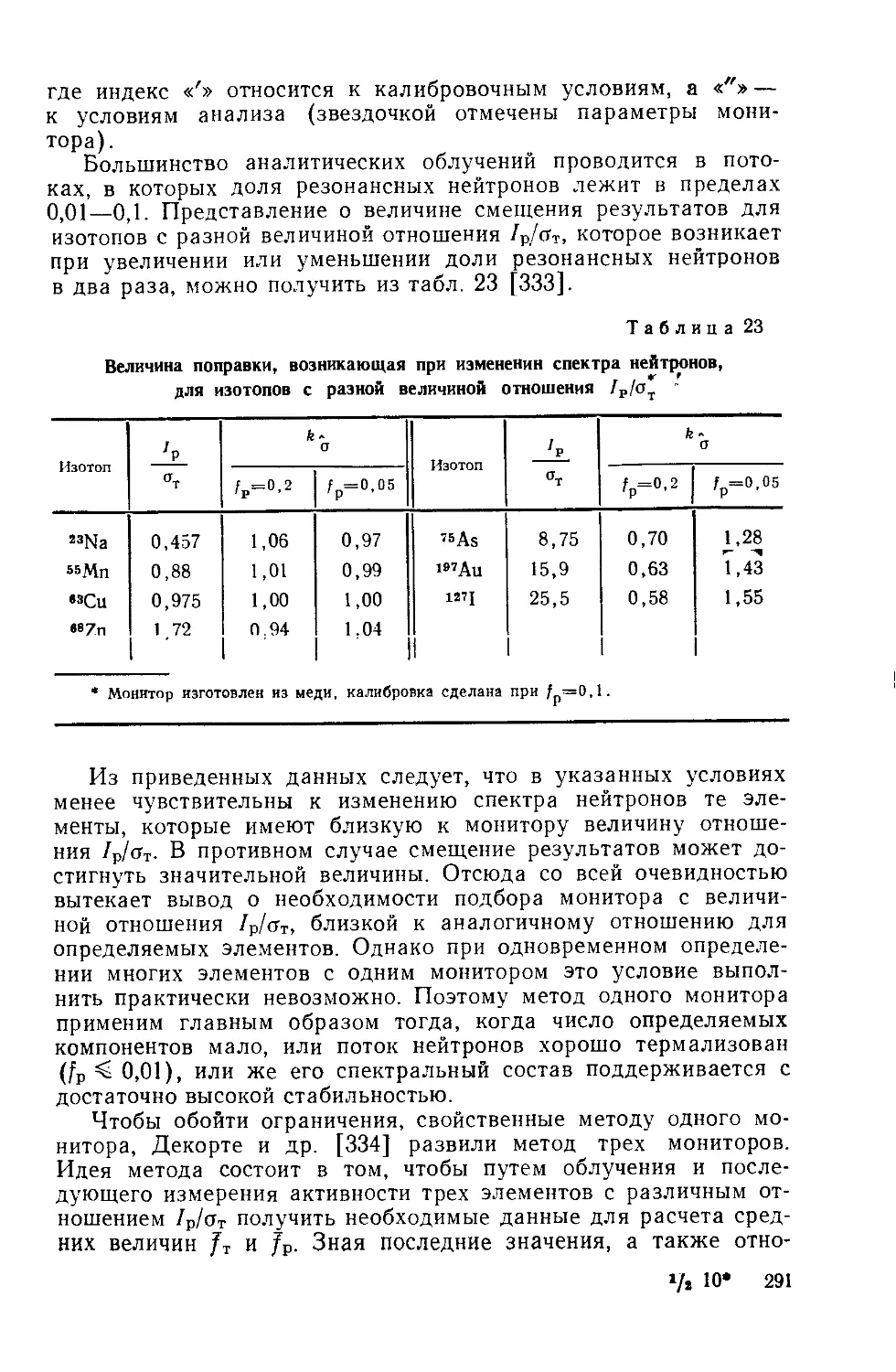














































Similar




Text
Р. А. КУЗНЕЦОВ
АКТИВАЦИОННЫЙ
АНАЛИЗ
Издание второе, переработанное
и дополненное
МОСКВА АТОМИЗДАТ 1974
УДК 543.53
Р. А. К у з н е ц о в. Активационный анализ.
Изд. 2-е. М., Атомиздат, 1974, с. 344.
Книга посвящена изложению теории и методики
активационного анализа — одного из ведущих методов
современной аналитической химии. В ней в системати-
зированной и обобщенной форме рассмотрены основные
принципы и классификация активационных методов, а
также дано описание общего хода активационного ана-
лиза, его инструментального и радиохимического ва-
риантов и различных методических вопросов. Приведе-
ны наиболее важные характеристики источников акти-
вирующего излучения, измерительной аппаратуры и
некоторых вспомогательных устройств. В заключитель-
ном разделе книги разобраны основные аналитические
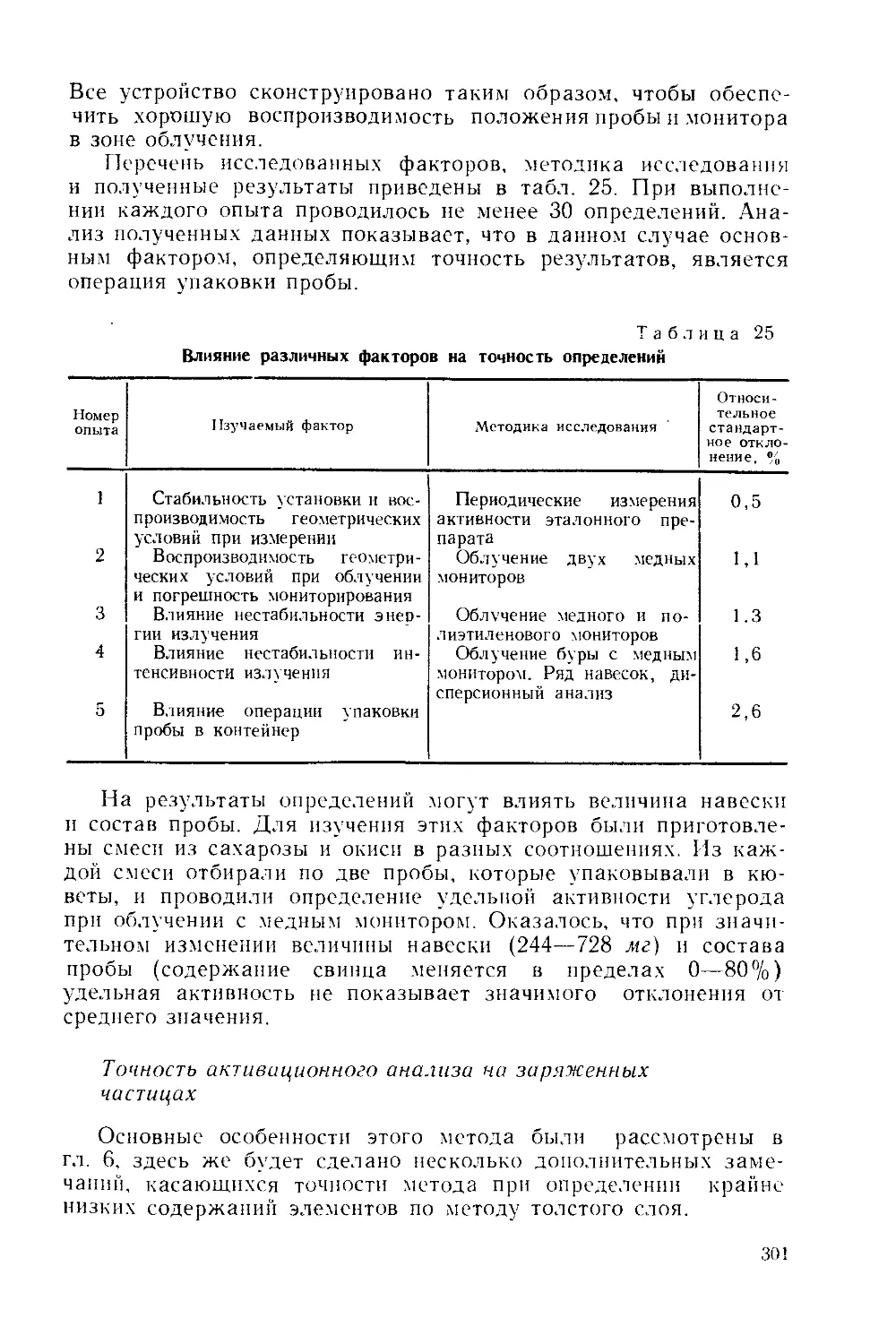
характеристики активационных методов: чувствитель-
ность, избирательность и точность.
Рисунков 80, таблиц 26, библиография 384 назв.
К
0257—050
034(01)—74
50—74
© Атомиздат, 1974.
ПРЕДИСЛОВИЕ
Даже беглое сравнение этого издания книги с предшествую-
щим показывает их значительное различие. И причина столь
серьезной переработки книги заключается не только в естест-
венном желании автора усовершенствовать свое произведение,
но и в необходимости отразить те глубокие изменения, которые
претерпел метод за период между двумя изданиями.
Основное содержание книги составляет систематическое из-
ложение теории и методики активационного анализа. Поскольку
этот метод вырос на стыке ряда наук — ядерной физики, радио-
химии и аналитической химии, то при изложении его основ не-
избежно приходится затрагивать вопросы, относящиеся к раз-
личным аспектам перечисленных научных дисциплин. Естест-
венно, что многие вопросы при этом рассматриваются весьма
кратко и в самой общей форме. Если при чтении книги возник-
нет необходимость в более детальном изучении какого-либо
вопроса, то следует обратиться к монографической или обзорной
литературе, на которую даны ссылки.
Опыт практического применения активационного анализа
в данной книге затрагивается лишь в той степени, которая не-
обходима для того, чтобы продемонстрировать значение метода
и его широкие аналитические возможности. Ряд конкретных
применений метода использован в качестве примеров для иллю-
страции тех или иных положений теории и методики активацион-
ного анализа.
На основе имеющихся рекомендаций внесены изменения в ис-
пользуемую терминологию, относящуюся к измерению ионизи-
рующих излучений («Сборник рекомендуемых терминов».
Вып. 76. М„ «Наука», 1968), представлению результатов ана-
лиза (ГОСТ 16263—60) и экстракции («Ж. аналит. химии»,
1971 г„ т. 26, № 5, с. 1021).
Во втором издании заметно увеличился общий объем книги
и изменилось распределение материала по главам. Как и прежде,
первая глава является вводной. Первая часть ее дает краткий
обзор исторического развития активационного анализа и ста-
новления его основных направлений и методов, а вторая при-
звана иллюстрировать роль и значение метода при решении
3
различных аналитических проблем. Принципы метода рассмот-
рены в главе второй. В третьей главе затронуты важные мето-
дические вопросы, такие, как методы измерения ионизирующих
излучений, оценка пределов обнаружения и точности радиомет-
рических методов, общий ход активационного анализа и т. д.
Основные активационные методы, их особенности и характе-
ристики, наиболее перспективные области применения, пара-
метры используемых источников активирующего излучения
составляют предмет четвертой — шестой глав. Следующие две
главы касаются различных аспектов и методов инструменталь-
ного активационного анализа, а в главах девятой и десятой речь
идет об альтернативном радиохимическом варианте.
Заключительная, одиннадцатая глава дает обобщенную
характеристику аналитических возможностей активационного
анализа. Здесь обсуждаются чувствительность, избирательность,
и точность его различных методов. Материал этой главы важен
для практических приложений активационного анализа и может
служить основой для его сопоставления с другими аналити-
ческими методами.
Книга рассчитана на химиков-аналитиков, но может быть
полезна аспирантам и студентам радиохимических специаль-
ностей, а также тем специалистам, которые в своей работе стал-
киваются с проблемами анализа. Автор надеется, что эта книга
даст возможность не только познакомиться с современным со-
стоянием этого перспективного метода, но и окажет помощь
при его практическом освоении.
Глава
1
ВВЕДЕНИЕ
§ 1. Исторический обзор
Возникновение и развитие активационного анализа бази-
руется на выдающихся достижениях целого комплекса наук,
связанных с исследованием ядра, ядерных реакции и радио-
активности. Особое влияние на этот процесс оказали ядерная
физика и радиохимия. Важную роль сыграло также стремитель-
ное развитие ядерной техники.
История открытия радиоактивности и последующих событий,
приведших к гигантским последствиям в науке и технике, до-
вольно хорошо известна [1], и нет необходимости на ней оста-
навливаться. Отметим только два открытия, непосредственно
подготовивших почву для возникновения активационного
анализа.
В 1919 г. Резерфорд на примере взаимодействия а-частиц
с ядрами азота доказал возможность осуществления ядерных
реакций. А в 1934 г. Ирен и Фредерик Жолио-Кюри при облу-
чении алюминия а-частицами полония получили искусственный
радиоизотоп фосфора. В течение последующего года было ис-
следовано уже более пятидесяти новых радиоизотопов. Быстрый
прогресс в изучении ядерных реакций и искусственной радио-
активности довольно скоро привел к их использованию в анали-
тических целях.
Это произошло в ходе обстоятельных экспериментов венгер-
ских химиков Хевеши и Леви по изучению радиоизотопов редко-
земельных элементов, образующихся при облучении нейтронами
Ra—Be-источника, когда ими в 1936 г. была показана возмож-
ность определения примеси Dy в препаратах Y [2]. Позднее
Сиборг и Ливенгуд [3] применили дейтроны, ускоренные в цикло-
троне, для определения примеси Ga в железе.
Новый этап в развитии активационного анализа начался
после появления ядерных реакторов, первый из которых был по-
строен под руководством Ферми и запущен в 1942 г. Высокие
потоки тепловых нейтронов в реакторах позволили достигнуть
исключительно высокой чувствительности определения многих
элементов. Это достоинство метода сразу же нашло применение
на практике для определения малых концентраций элементов
в материалах, представляющих большой интерес для новых
5
отраслей техники. Причем в первую очередь развитие самой
ядерной техники потребовало анализа материалов, используе-
мых для строительства реакторов, на содержание примесей
элементов, сильно поглощающих нейтроны. С этого момента
облучение тепловыми нейтронами в реакторах становится веду-
щим методом активационного анализа.
Значительному развитию нейтронного активационного ана-
лиза способствовала разработка малогабаритных, относительно
недорогих нейтронных генераторов. Первые примеры использо-
вания этих приборов для аналитических целей относятся
к 1953 г.
Несмотря на много меньшие значения нейтронных потоков
и соответственно более низкую чувствительность, применение
нейтронных генераторов оказалось очень перспективным для
определения некоторых легких элементов и Для разработки
инструментальных методов, обладающих высокой экспресс-
ностью и надежностью.
Что касается внедрения заряженных частиц и жестких фото-
нов в аналитическую практику, то первоначально оно происхо-
дило весьма медленно. Этот факт прежде всего был связан
с незначительным распространением и высокой стоимостью со-
ответствующих ускорительных установок. При этом по чувст-
вительности анализа они значительно уступали реакторам. Лишь
успехи в конструировании ускорителей стимулировали более
интенсивное развитие этих методов, начиная примерно с сере-
дины 60-х годов.
По способу выполнения аналитического определения в акти-
вационном анализе развились два основных направления. В од-
ном из них избирательность анализа основывается на ядерно-
физических свойствах элементов и образующихся радиоизотопов.
Поскольку при этом сохраняется целостность анализируемой
пробы, то это направление получило название инструменталь-
ного активационного анализа.
Значительное развитие инструментальный подход получил
в связи с появлением сцинтилляционного метода спектрометрии
ионизирующих излучений. Длительное время он даже составлял
основу инструментального активационного анализа. Однако
с середины 60-х годов в практику активационного анализа вхо-
дит спектрометр с Ge(Li)-детектором. Его замечательная осо-
бенность состоит в высоком энергетическом разрешении, которое
на порядок превышает разрешение сцинтилляционных детек-
торов.
Главные достоинства инструментальных методов — быстрота,
малая трудоемкость и высокая экономичность анализов. Значи-
тельно расширило возможности инструментального подхода
создание быстрых транспортирующих систем для перемещения
анализируемых проб в зону облучения, а затем к измеритель-
ному устройству. Поскольку длительность транспортировки в
с.
быстрых системах составляет доли секунды, это позволило
широко использовать в аналитических целях короткоживущие
радиоизотопы.
Большие возможности для инструментального направления
открыли вычислительные машины. Значительно ускорив прове-
дение расчетов, они позволили математическими средствами
решить две очень важные для активационного анализа проб-
лемы, касающиеся выбора оптимальных условий и методики
инструментального анализа, а также повышения точности и
надежности обработки многокомпонентных спектров. Важным
следствием применения вычислительных устройств явилось так-
же создание полностью автоматизированных систем для вы-
полнения активационного анализа практически без участия
оператора.
Однако инструментальные методы испытывают и определен-
ные затруднения, связанные с ограниченной временной и энер-
гетической разрешающей способностью имеющихся приборов,
сложностью обработки результатов измерений, помехами от
сильноактивирующихся компонентов анализируемой пробы.
Альтернативный подход связан с разложением пробы и по-
следующим применением химических процедур, избирательность
которых определяется уже физико-химическими свойствами эле-
ментов. Это направление получило название радиохимического.
Поскольку радиохимическое выделение может сочетаться
с интегральными методами регистрации ионизирующего излу-
чения радиоизотопов, то это часто обеспечивает более высокие
чувствительность и точность, чем в случае инструментального
подхода. В то же время выполнение радиохимических процедур
делает анализ более трудоемким и длительным.
До середины 50-х годов радиохимический подход явно пре-
обладал. Лишь появление сцинтилляционных гамма-спектро-
метров привело к вытеснению радиохимического метода из тех
областей, где инструментальный подход имел неоспоримые пре-
имущества.
Первоначально радиохимическое разделение обычно основы-
валось на методе осаждения, существенный недостаток кото-
рого— соосаждение вместе с выделяемым элементом посторон-
них радиоэлементов. Поэтому для получения правильных и
надежных результатов требуются длительные операции радио-
химической очистки, что сильно увеличивает трудоемкость и дли-
тельность анализа и делает невозможным использование корот-
коживущих радиоизотопов. Так, в ранних работах по актива-
ционному анализу, включающих радиохимическое разделение,
применялись только радиоизотопы с периодом полураспада
более 1 ч.
Большие возможности перед радиохимическим направлением
открыли успехи в развитии таких быстрых и прогрессивных
методов разделения, как экстракция, ионообменная хроматогра-
7
фин и др. В этих методах практически отсутствуют или не
играют столь решающей роли явления, аналогичные соосажде-
нию. Они часто просты по выполнению, высокоизбирательны
при правильном выборе условий и позволяют быстро выделять
определяемый элемент в радиохимически чистом виде. Часто
имеются хорошие возможности для автоматизации процесса
разделения.
Новые методы позволили заметно ускорить разделение эле-
ментов. Мейнке на большом числе практических примеров до-
казал даже возможность экспрессных радиохимических разделе-
ний при активационном анализе различных объектов [4]. На вы-
деление элемента при этом требуется всего несколько минут.
В результате граница распространения радиохимического вари-
анта сместилась к радиоизотопам с периодом полураспада,
начиная примерно с 2 мин.
Важное значение для радиохимического подхода приобрел
метод субстехиометрического выделения, который применитель-
но к активационному анализу был предложен и развит Ружич-
кой и Стары [5]. Субстехиометрический метод заметно упрощает
процедуру радиохимического разделения при одновременном
повышении избирательности.
Некоторое время, когда уже вполне сформировались оба
направления активационного анализа, они развивались парал-
лельно и даже несколько противопоставлялись друг другу.
Однако вскоре было обнаружено, что сочетание обоих методов
часто дает исключительно хорошие результаты. В частности,
комбинированные методы позволили создать быстрые и надеж-
ные методики определения большого числа элементов в пробах
любого химического состава.
В заключение несколько слов о развитии активационного
анализа в СССР. В предвоенные годы исследования в области
ядерной физики, радиохимии, естественной и искусственной
радиоактивности в нашей стране находились на высоком
уровне [6]. Впервые на интересные возможности, которые откры-
вает использование искусственной радиоактивности в аналити-
ческих целях, обратил внимание А. А. Гринберг [7]. Однако
Великая Отечественная война не позволила развернуть намечен-
ные исследования.
Непосредственно перед началом войны в Радиевом институте
Б. С. Айдаркиным и др. [8] была разработана методика фото-
нейтронного определения бериллия в рудах. Источником жест-
кого у-излучения служил радий. Фактически это была первая
в мировой практике попытка применить фотоядерные реакции
в аналитических целях, но результаты этой работы были опуб-
ликованы только в 1957 г.
В послевоенный период исследования в области активацион-
ного анализа были возобновлены в 1953 г. под руководством
II. П. Алимарина [9, 10]. Однако их развертывание на этом
8
этапе происходило медленными темпами. Перелом наметился
в начале 60-х годов, когда заметно возросла интенсивность тео-
ретических исследований и технических разработок, а также
расширилась сфера практических приложений метода.
§ 2. Основные особенности и области применения
активационного анализа
Активационный анализ — один из ведущих методов современ-
ной аналитической химии, который находит все расширяющееся
применение в различных областях науки и техники [9, 11]. Основ-
ные особенности и аналитические возможности этого метода
главным образом определяются тем обстоятельством, что акти-
вационный анализ основывается на ядерных взаимодействиях
и свойствах возбужденных (радиоактивных) атомных ядер.
Из ядернофизической сущности метода сразу же вытекает одна
из важнейших аналитических особенностей — нечувствительность
к химическому состоянию атомов определяемого элемента в
исследуемом объекте, т. е. активационный анализ способен
давать только общее (валовое) содержание элемента в пробе
и без привлечения дополнительных химических средств не по-
зволяет раздельно определять элемент в разных химических
формах.
С другой стороны, современная техника предоставляет в рас-
поряжение аналитика целый набор источников активирующего
излучения, с помощью которых можно осуществлять актива-
ционное определение практически всех элементов периодической
системы. Нередко эта способность метода реализуется на прак-
тике в схемах анализа, предусматривающих определение значи-
тельного числа элементов из одной небольшой навески, что сразу
же дает обширную аналитическую информацию.
Некоторые виды излучений, как применяемые для воздейст-
вия на ядра элементов, так и испускаемые последними в ядер-
ных процессах, обладают достаточно высокой проникающей
способностью, что позволяет проводить инструментальный ана-
лиз представительных по массе проб. Причем успех инструмен-
тального подхода главным образом связан с тем фактом, что
один из основных видов ионизирующего излучения радиоактив-
ных ядер (у-излучение) является характеристическим, что дает
возможность по параметрам излучения (энергия, период полу-
распада и интенсивность) проводить идентификацию элементов
и их избирательное количественное определение.
Активационный анализ относится к числу наиболее чувстви-
тельных аналитических методов [11, 12]. Предел обнаружения
для большинства элементов находится в интервале 10-7—10-11 %.
При этом анализ по крайней мере сплошных твердых проб сво-
боден от необходимости введения поправки на холостой опыт,
поскольку количество выполняемых операций до облучения про-
9
бы может быть малым, к тому же все возможные поверхностные
загрязнения могут быть удалены травлением облученной пробы
перед проведением конечных измерений. Химическая обработка
облученной пробы, когда в этом появляется необходимость,
может проводиться с помощью обычных аналитических реаген-
тов, так как небольшие количества нерадиоактивных примесей
в них уже не могут повлиять на результаты анализа. Конечно,
при этом следует избегать радиоактивных загрязнений, но это не
представляет особой проблемы при тщательно спланированной
методике работы и аккуратном выполнении аналитических опе-
раций. Постоянный радиометрический контроль может быть ис-
пользован для своевременного выявления радиоактивного за-
грязнения реактивов, посуды, рабочего места и т. д.
Отмеченная особенность активационного анализа позволяет
отнести его к наиболее надежным методам определения малых
содержаний элементов в самых разнообразных объектах. По
этой причине он часто рассматривается как арбитражный метод
в области крайне низких концентраций элементов. В то же время
у активационного анализа нет каких-либо существенных кон-
центрационных ограничений и он с одинаковым успехом может
работать в широкой области — от предела обнаружения до
макроконцентраций. При этом точность получаемых результатов
достаточно высока и погрешность в большинстве случаев нахо-
дится в интервале 1 —10 отн.%.
Конечно, активационный анализ, как и любой другой анали-
тический метод, не свободен от затруднений и помех, которые
неблагоприятным образом сказываются на его характеристиках
и могут серьезным образом осложнить выполнение анализа.
Поэтому для достижения максимального эффекта требуется
хорошее понимание сущности метода и его аналитических воз-
можностей.
Важно также обратить внимание на то обстоятельство, что
отмеченные высокие аналитические параметры часто могут быть
достигнуты только С ПОМОЩЬЮ ДОВОЛЬНО Л'ШОГОГО и сложного
в эксплуатации оборудования. Успехи современного активацион-
ного анализа во многом обусловлены прогрессом в разработке
и совершенствовании таких видов ядернофизического оборудо-
вания, как ядерные реакторы, различные типы ускорителей за-
ряженных частиц, многоканальные анализаторы, средства
регистрации ионизирующих излучений и т. д. [13]. Все более
широкое распространение получают вычислительные машины,
которые позволяют оптимально планировать режим анализа,
облегчают и ускоряют обработку полученной информации, берут
на себя функцию оперативного управления ходом аналитиче-
ского определения.
В процессе практического применения методов активацион-
ного анализа всегда приходится учитывать радиационную опас-
ность от воздействия ионизирующего излучения на организм
10
человека. А это предъявляет специфичные требования к мето-
дике анализа и оборудованию лабораторий [14]. Кроме того,
необходимо особо подготавливать специалистов для работы в
области активационного анализа. Совокупность указанных выше
факторов создает определенный барьер на пути широкого ис-
пользования активационных методов в рядовой аналитической
работе. Практика показывает, что наиболее благоприятные воз-
можности для их применения создаются в специальных исследо-
вательских центрах, оснащенных необходимым оборудованием
и укомплектованных квалифицированными кадрами. Описание
подобных лабораторий и основных направлений научных иссле-
дований в них систематически публикуется в J. Radioanalyt.
Chemistry.
Хотя потенциально активационные методы могут быть по-
лезны при решении широкого круга аналитических проблем
и эти возможности постоянно реализуются на практике, примеры
чего можно найти в периодической литературе по химии, однако
четко выделяются два направления, где применение активацион-
ного анализа наиболее эффективно. Имеются в виду определе-
ние содержания малых компонентов в различных природных и
промышленных объектах и экспрессный инструментальный ана-
лиз в области средних и высоких концентраций (10-4—100%).
Детальное рассмотрение практических применений активацион-
ных методов не входит в задачу данной книги, и поэтому ниже
для иллюстрации их аналитических возможностей приведены
только некоторые наиболее интересные примеры.
Развитие современной техники вовлеко в сферу производства
значительное число новых материалов и обусловило система-
тическое и непрерывное повышение требований к их качеству
[9, 15]. Как уже упоминалось, реакторостроение было одной из
первых отраслей техники, которая предъявила высокие требо-
вания к чистоте материалов, используемых в качестве горю-
чего (U), замедлителя (тяжелая и обычная вода, графит) и для
изготовления различных конструкций и устройств внутри актив-
ной зоны (Zr, Al, Be и Др.). Эти материалы не должны содер-
жать элементов с высоким сечением поглощения тепловых ней-
тронов. В ряде случаев требуется также отсутствие элементов,
дающих при облучении долгоживущие радиоизотопы с жестким
у-излучением. Для всех этих случаев были разработаны соответ-
ствующие активационные методики, позволившие получить тре-
буемую чувствительность определения (10-4—10~6%) для боль-
шей части нежелательных примесей.
Более высокие требования к чистоте используемых материа-
лов выдвинула быстро развивающаяся электронная промышлен-
ность. Содержание различных примесей в полупроводниковых
материалах лимитируется уровнем 10—6—10-8% [9, 16]. Основные
материалы для полупроводниковой техники — германий и крем-
ний; в последнее время важную роль начинают играть некото-
рые другие элементы (Sb, In, Ga, As и др.) и их соединения.
Серьезные требования при производстве полупроводников были
предъявлены и к чистоте ряда вспомогательных материалов.
Задача контроля чистоты материалов при производстве полу-
проводников не является единственной. Например, важное зна-
чение имеет точное определение концентрации легирующих
добавок, специально вводимых в используемый материал в коли-
честве 10~4—10~5%, или изучение равномерности их распреде-
ления. Активационные методы внесли существенный вклад в
решение многих аналитических проблем, возникших в ходе ста-
новления и дальнейшего развития производства полупроводни-
ковых приборов [9, 17].
Высокая чувствительность и надежность активационных
методов привели к их широкому использованию в научных ис-
следованиях, которые связаны с изучением крайне низких кон-
центраций элементов. При этом активационный! анализ часто
позволяет быстро накапливать обширную аналитическую инфор-
мацию, представляющую исключительный интерес для целых
областей науки.
Прежде всего здесь следует отметить большой вклад в гео-
химию и космохимию [17, 18]. Изучение содержания малых кон-
центраций элементов в земных породах дает сведения о геохими-
ческих процессах и может оказать помощь при поисках место-
рождений редких и рассеянных элементов. Активационный ана-
лиз применяется для исследования вариаций изотопного состава
некоторых элементов и для ядерной геохронологии. В последнее
время активационный анализ оказывается одним из ведущих
методов при изучении химического состава вещества, доставлен-
ного с поверхности Луны [19].
Велико значение этого метода для биологии, медицины и
смежных дисциплин [20. 21]. Эти науки заинтересованы в уста-
новлении содержания малых компонентов, роль которых в био-
логических процессах часто велика. Кроме того, такие исследо-
вания позволяют оценить воздействие некоторых элементов на
живой организм при попадании через продукты питания или
при хроническом отравлении на производстве. Можно также
изучать процессы миграции элементов в организме и накопле-
ния их в отдельных органах. Аномальное содержание какого-
либо элемента может указывать на нарушение процессов обмена
и, таким образом, давать дополнительную информацию при
диагностике заболевания.
Инструментальный вариант активационного анализа, часто
обладая достаточно высокой чувствительностью при одновремен-
ном определении широкого круга элементов, ускоряет процесс
получения аналитических данных и в то же время сохраняет
исследуемый объект. Эти качества важны, например, для архео-
логии. Результаты химических анализов дают сведения о составе
археологических находок, из которых могут вытекать весьма
12
важные для археологии и истории выводы о технологии изго-
товления различных изделий, об экономике, торговых связях,
источниках сырья и т. д.
Широко применяется активационный анализ и в криминали-
стике [22]. Исследование незначительных материальных следов,
обнаруженных на месте преступления, может представить убе-
дительные доказательства в пользу наличия или отсутствия при-
частности подозреваемых к преступлению. Активационным мето-
дом можно проверить предположение об отравлении некоторыми
веществами, разоблачить подделку и провести ряд других ис-
следований.
Намечаются широкие перспективы применения активацион-
ных методов в сфере материального производства. Здесь круг
аналитических проблем, в решение которых они могут внести
существенный вклад, весьма обширен и простирается от поиска
полезных ископаемых до оперативного контроля за качеством
готового продукта. При этом наибольшее значение имеют такие
параметры метода, как возможность проведения инструменталь-
ных и экспрессных определений. Естественно, что аналитическое
оборудование, предназначенное для работы в производственных
условиях, должно быть достаточно простым и надежным [23, 24].
По этой причине основными источниками активирующего излу-
чения являются относительно недорогие радиоизотопные источ-
ники и нейтронные генераторы. Из-за более слабой интенсив-
ности этих источников область аналитических концентраций не
опускается ниже 10-3—10-4%.
Для многих случаев индустриального применения были раз-
работаны весьма быстрые и экономичные методики, роль кото-
рых особенно велика при массовом анализе проб близкого со-
става. Так, в геологии активационные методы анализа горных
пород и руд позволяют ускорить поиск полезных ископаемых.
Их значение велико и для горнообогатительной, металлургиче-
ской и некоторых других отраслей производства.
Причем аналитические определения могут производиться как
обычным способом отбора проб с последующей обработкой в
аналитической лаборатории, так и непосредственно на ленте
конвейера, в трубопроводе, на месте залегания и т. д. Возможен
также дистанционный автоматический активационный анализ
внутри геологической скважины, на морском дне и даже на по-
верхности других планет [25].
Если говорить об анализе методом отбора проб, то наиболее
ярким примером может служить активационное определение
кислорода в продуктах металлургического производства. Анализ
здесь требует всего нескольких минут, в то же время он обеспе-
чивает достаточно высокую чувствительность и надежность
определения [26]. Анализ на конвейере или в потоке может быть
успешно использован для оперативного управления технологи-
ческим процессом [24].
13
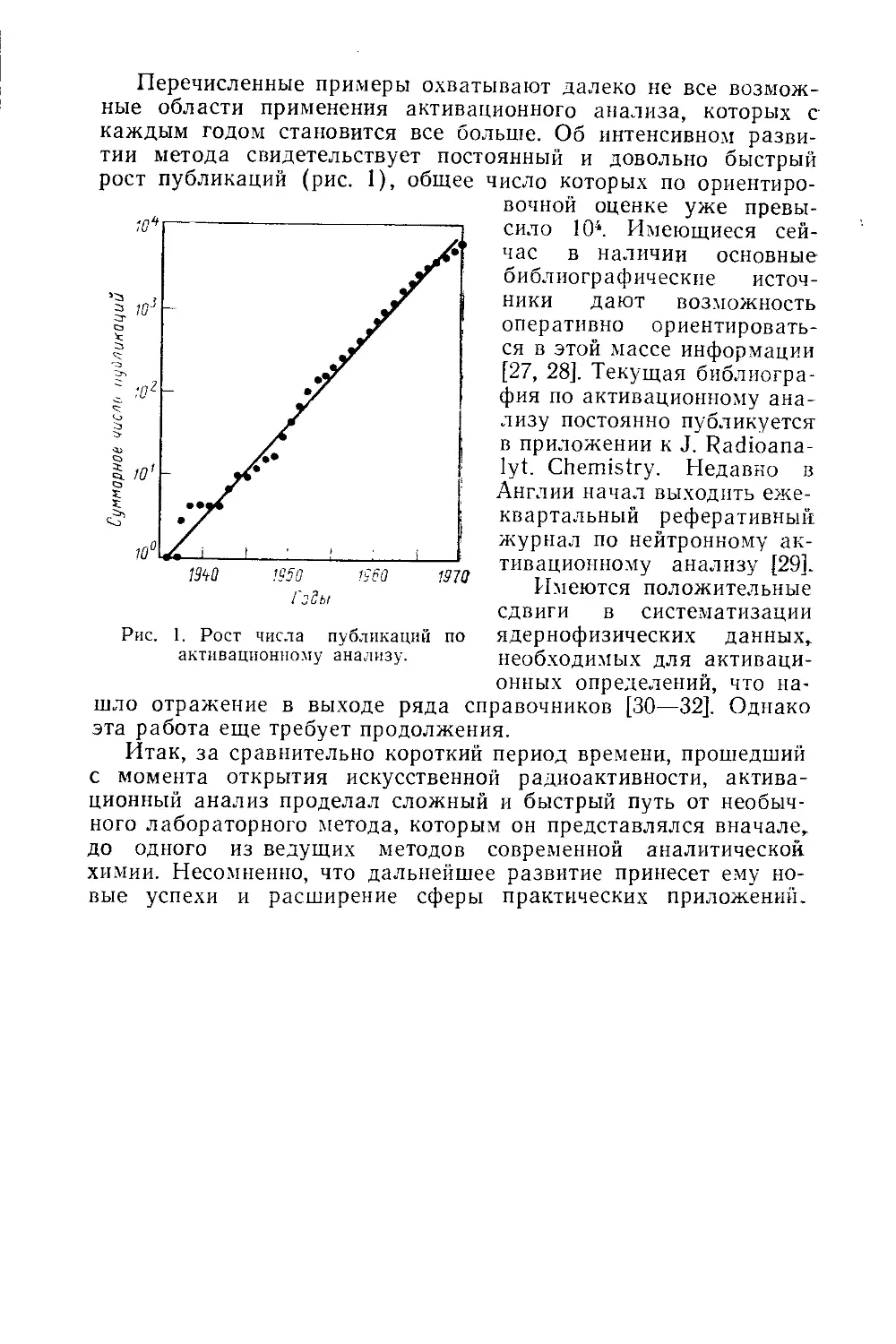
Перечисленные примеры охватывают далеко не все возмож-
ные области применения активационного анализа, которых с
каждым годом становится все больше. Об интенсивном разви-
тии метода свидетельствует постоянный и довольно быстрый
рост публикаций (рис. 1), общее
Рис. 1. Рост числа публикаций по
активационному анализу.
число которых по ориентиро-
вочной оценке уже превы-
сило 104. Имеющиеся сей-
час в наличии основные
библиографические источ-
ники дают возможность
оперативно ориентировать-
ся в этой массе информации
[27, 28]. Текущая библиогра-
фия по активационному ана-
лизу постоянно публикуется
в приложении к J. Radioana-
lyt. Chemistry. Недавно в
Англии начал выходить еже-
квартальный реферативный
журнал по нейтронному ак-
тивационному анализу [29].
Имеются положительные
сдвиги в систематизации
ядернофизических данных,
необходимых для активаци-
онных определений, что на-
шло отражение в выходе ряда справочников [30—32]. Однако
эта работа еще требует продолжения.
Итак, за сравнительно короткий период времени, прошедший
с момента открытия искусственной радиоактивности, актива-
ционный анализ проделал сложный и быстрый путь от необыч-
ного лабораторного метода, которым он представлялся вначале,
до одного из ведущих методов современной аналитической,
химии. Несомненно, что дальнейшее развитие принесет ему но-
вые успехи и расширение сферы практических приложений.
Глава
2
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ
АКТИВАЦИОННОГО АНАЛИЗА
§ 1. Ядерные реакции
Активационный анализ основан на ядерных взаимодействиях,
поэтому при выполнении анализа исследуемую пробу облучают
потоком ядерных частиц или квантов с достаточной энергией.
В результате взаимодействия активирующего излучения с яд-
рами элементов возможно протекание различных процессов,
которые приводят к изменению состояния облучаемых ядер.
Изменение может произойти в составе нуклонов и (или) в энер-
гетическом состоянии ядра.
Ядерные взаимодействия имеют квантовомеханическую при-
роду. Это создает значительные трудности в развитии строгой
теории ядерных взаимодействий, и пока для их описания ис-
пользуют различные упрощенные модели, которые более или
менее удовлетворительно отражают течение ядерных процессов
и допускают приближенный расчет различных параметров ядер-
ных взаимодействий [33, 34]. Однако для аналитических приме-
нений точное знание механизма ядерных процессов не обяза-
тельно, а все необходимые параметры определяются экспери-
ментально.
Для общего представления отметим модель составного ядра,
механизм прямого взаимодействия и кулоновское возбуждение.
-Модель составного ядра исходит из предположения, что процесс
ядерного взаимодействия состоит из двух независимых стадий:
1) поглощения бомбардирующей частицы и распределения ее
энергии между нуклонами ядра; 2) испускания частицы (иногда
нескольких), которая уносит значительную долю избыточной
энергии. Время жизни составного ядра ДО-18—10~14 сек) значи-
тельно превышает интервал времени, необходимый бомбарди-
рующей частице для прохождения ядра (10~20—1023 сек).
Образовавшееся конечное ядро часто оказывается в возбуж-
денном состоянии и переходит в основное состояние путем ис-
пускания одного или нескольких у-квантов, Длительность этой
стадии составляет 1012—10-11 сек. Последовательность про-
цессов в модели составного ядра можно представить так:
А + (2.1)
I
В + у
15
где А и В — исходный и конечный продукты ядерного взаимо-
действия; а и b— бомбардирующая и испускаемая частицы;
С* — составное ядро и В* — возбужденное состояние конечного,
ядра. По механизму составного ядра протекает большинство
ядерных взаимодействий, используемых в аналитических
целях.
В прямом взаимодействии бомбардирующая частица пере-
дает одному или нескольким нуклонам энергию, достаточную
для оставления ядра. Сама бомбардирующая частица тоже
может покинуть ядро, но уже с меньшей энергией. После пря-
мого взаимодействия конечное ядро часто остается в возбуж-
денном состоянии.
Кулоновское возбуждение ядра происходит при прохождении
заряженной частицы в непосредственной близости от ядра.
Вероятность такого процесса очень мала, и поэтому он не пред-
ставляет какого-либо существенного значения для аналитиче-
ских применений.
Протекающие ядерные реакции обычно записывают в сокра-
щенной форме. Например, для реакции (2.1) это будет
А (а, Ь)В. Как видно, в такой записи фиксируются только исход-
ное и конечное ядра, бомбардирующая и испускаемая частицы.
Испускание у-излучения обычно не отмечается, правда, реак-
ция радиационного захвата нейтрона (п, у) составляет исклю-
чение.
Исходный продукт в уравнении (2.1) представляет собой
стабильное ядро определенного изотопа, которое характери-
зуется соответствующими значениями Z (заряд) и М (массовое
число). В качестве бомбардирующих частиц используются ней-
троны, протоны, а-частицы, дейтроны, ядра 3Н и 3Не, а также
у-кванты.
Далеко не всегда взаимодействие ядер типа А с потоком
бомбардирующих частиц а протекает только одним способом.
Возможны взаимодействия, которые могут идти несколькими
конкурирующими способами (каналами реакций):
(В + b (I)
| С + xb (II)
A+tzjD (III) (2.2)
| А* + a' (IV)
Ia + я (V)
На протекание ядерных процессов накладывают определен-
ные ограничения законы сохранения, которые позволяют также
установить четкую взаимосвязь между исходным и конечным
состояниями. Для рассматриваемой проблемы наиболее сущест-
венным представляется закон сохранения электрического за-
ряда, числа нуклонов и энергии [33].
16
Количество возможных каналов ядерного взаимодействия
при энергии бомбардирующих частиц не выше 40—50 Мэв
обычно невелико (не более 4—5). Очень часто преобладающую
роль при этом для данной энергии бомбардирующих частиц
играет один или два канала. Уменьшение энергии облучения
приводит к сокращению числа возможных каналов ядерных
реакций.
В реакциях, представленных уравнением (2.2), испускаемыми
частицами являются обычно нейтроны, протоны и а-частицы,
значительно реже дейтроны, ядра 3Н и 3Не [процесс (I)]. Имеют
место и процессы, которые протекают с испусканием двух или
трех частиц [процесс (II)] или не дают вторичной частицы
[процесс (III)]. В последних двух реакциях уравнения (2.2) бом-
бардирующая и испускаемая частицы идентичны. Однако про-
цесс (IV) идет с переводом исходного ядра в более высоко-
энергетическое состояние А* (неупругое рассеяние) с последую-
щим излучением энергии возбуждения в виде у-кванта. Про-
цесс (V) не приводит к какому-либо изменению внутреннего
состояния ядра (упругое рассеяние) и поэтому не может быть
использован для аналитических целей. Правда, легкие ядра
(!Н и 2Н) в упругих солкновениях с быстрыми частицами могут
~ приобрести энергию отдачи, достаточную для осуществления
X^s. некоторых ядерных реакций.
\i Возникший в результате ядерного взаимодействия продукт
^большинстве случаев представляет собой изотоп исходного или
V ' соседних элементов и характеризуется новым сочетанием Z
и А1. Наиболее глубокими изменениями исходного ядра отли-
чается реакция деления. Очень часто конечное ядро в своем
основном состоянии оказывается неустойчивым и претерпевает
Y-x радиоактивный распад с переходом в новое, чаще всего стабиль-
) ное ядро.
Как и химические реакции, ядерные реакции протекают с вы-
делением (экзоэнергетические) или поглощением (эндоэнергети-
ческие) энергии. Последние могут происходить только в том слу-
чае, если энергия активирующего излучения превышает некото-
рый уровень (порог реакции). Для реакций с участием частиц
величина порога £пор связана с энергетическим эффектом реак-
ции соотношением
т„ -1- М.
£nOp = [QJ--^-A (2.3)
где та—масса бомбардирующей частицы; Л1А— массовое число
ядер A; [Q] — абсолютное значение энергии реакции. Для реак-
ций под действием у-квантов £nnp = [Q].
К пороговым относят также реакции, протекающие с уча-
стием заряженных частиц (бомбардирующих или испускаемых),
так как им приходится преодолевать кулоновский барьер ядра,
величина которого зависит как от заряда Ze облучаемого, (конеч-
17
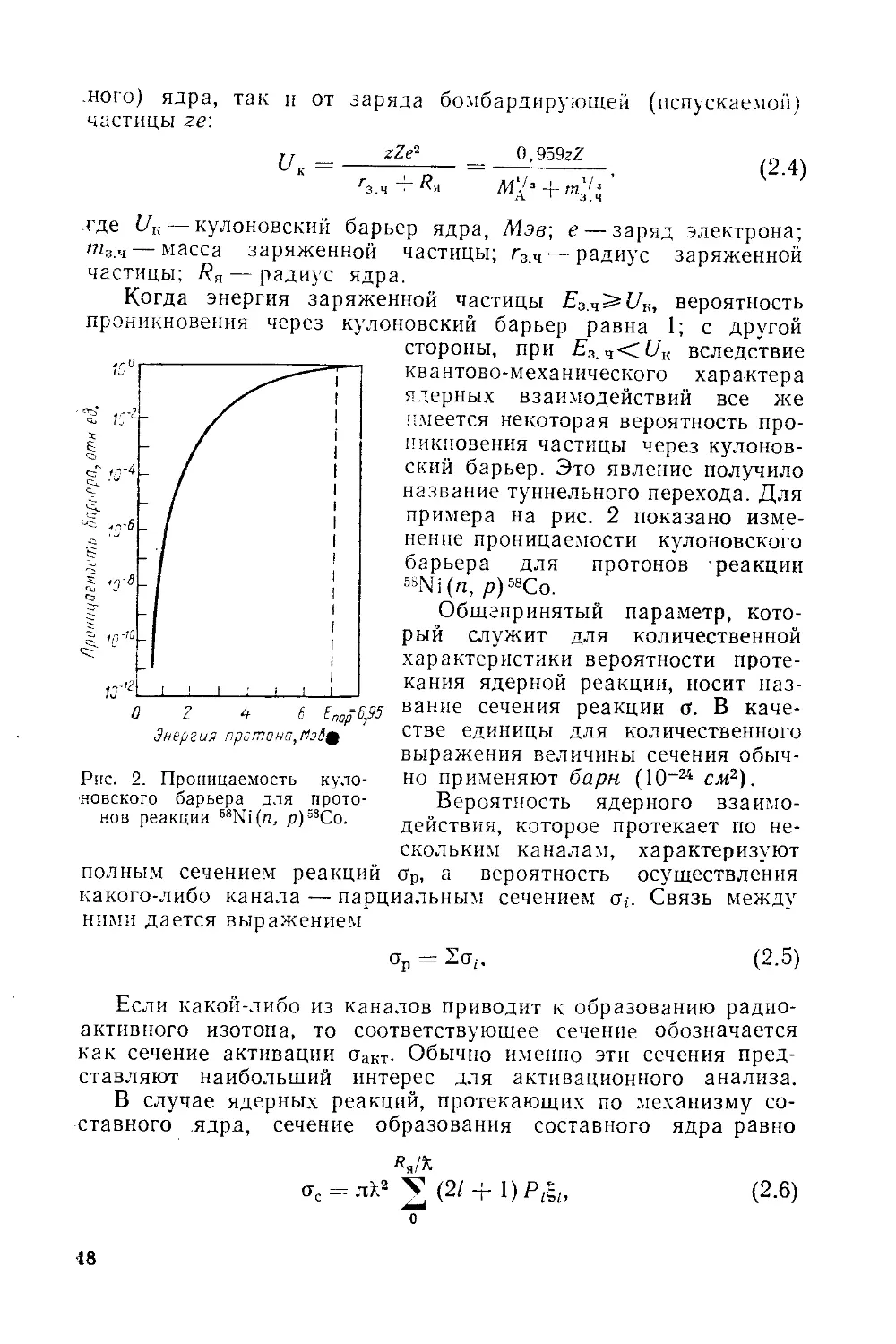
Рис. 2. Проницаемость куло-
новского барьера для прото-
нов реакции 68Ni(n, р)58Со.
.кого) ядра, так и от заряда бомбардирующей (испускаемой)
частицы ze\
гт __ zZe1- O,959zZ
\J к — - ————— , I Z.. “т J
гз.ч M'J3 4- m'J *
A 1 3.4
где UK — кулоновский барьер ядра, Мэв\ е — заряд электрона;
^з.ч—масса заряженной частицы; гзч-—радиус заряженной
частицы; Рн— радиус ядра.
Когда энергия заряженной частицы E3.4^UK, вероятность
проникновения через кулоновский барьер равна 1; с другой
стороны, при Е3.Ч<Д1; вследствие
квантово-механического характера
ядерных взаимодействий все же
.имеется некоторая вероятность про-
никновения частицы через кулонов-
ский барьер. Это явление получило
название туннельного перехода. Для
примера на рис. 2 показано изме-
нение проницаемости кулоновского
барьера для протонов реакции
5SNi(n, р)58Со.
Общепринятый параметр, кото-
рый служит для количественной
характеристики вероятности проте-
кания ядерной реакции, носит наз-
вание сечения реакции о. В каче-
стве единицы для количественного
выражения величины сечения обыч-
но применяют барн (10-24 см2).
Вероятность ядерного взаимо-
действия, которое протекает по не-
скольким каналам, характеризуют
сгр, а вероятность осуществления
какого-либо канала — парциальным сечением сгг-. Связь между
ними дается выражением
полным сечением реакций
о-р = 2о,-,
(2-5)
Если какой-либо из каналов приводит к образованию радио-
активного изотопа, то соответствующее сечение обозначается
как сечение активации стакт- Обычно именно эти сечения пред-
ставляют наибольший интерес для активационного анализа.
В случае ядерных реакций, протекающих по механизму со-
ставного ядра, сечение образования составного ядра равно
ос = лк* У (2/ + l)PZ5Z, (2.6)
О
<8
где Л—длина волны де Бройля бомбардирующей частицы, по-
деленная на 2 л; / — квантовое число; Pi — вероятность проник-
новения через кулоновский и центробежный барьеры; — веро-
ятность прилипания частицы с моментом^/ к ядру. Максималь-
ная величина сечения будет достигнута при Л=1 и £г=1, тогда
ас,макс = лХ2 V (2/4-1). (2.7)
о
Получившееся выражение представляет собой арифметиче-
скую прогрессию, сумма членов которой равна л(7?я+М2, следо-
вательно,
Ос,макс = Л (7?я + т)2. (2.8)
Из уравнения (2.8) вытекает, что для быстрых частиц (£я^
Ос, мане = л7?я, т. е. совпадает с геометрическими размерами
ядра. Однако это предельная оценка, поскольку действительная
величина параметра s/<71 и не поддается точному теорети-
ческому определению.
Для характеристики ядерных реакций часто применяют еще
один параметр — выход ядерной реакции, представляющий собой
отношение числа происшедших актов ядерного взаимодействия
к числу прошедших через мишень бомбардирующих частиц.
Скорость ядерной реакции в заданных условиях облучения
определяется соотношением
dNn
= Фа^А, (2.9)
ре А'в— число атомов продукта реакции; А?д — число исходных
атомов; Ф — плотность потока активирующего излучения,
частиц!(см2-сек). Тогда выход ядерной реакции:
dN-o /dt
У = —5— = oNa.
Ф
Это определение выхода справедливо для облучения
моноэнергетического излучения. Если поток излучения
ризуется сплошным энергетическим распределением или имеет
место изменение энергетического спектра при прохождении
излучения в веществе мишени, то в уравнении (2.10) следует
использовать интегральные величины:
£
макс
Y = Na [ Фо (£) о (E)dE,
о
где Фо(£)—спектральное распределение активирующего излу-
чения при единичном значении параметра, измеряемого в ходе
эксперимента; а(Е)—функция возбуждения ядерной реакции.
Последняя показывает изменение сечения реакции с энергией
(2.Ю)
потоком
характе-
(2.Н)
19
.активирующего излучения, она часто меняется довольно слож-
ным образом.
Процедура экспериментального определения выхода реакции
и его изменения с энергией излучения обычно гораздо проще,
чем функции возбуждения. Для возможности сравнения выходов
реакций в различных экспериментальных условиях их значения
относят к определенному количеству вещества мишени (грамм,
моль и т. д.). Выход реакции более наглядно выявляет имею-
щиеся аналитические возможности метода в случае сложного
хода кривой возбуждения, применения активирующего излуче-
ния со сплошным энергетическим спектром или при облучении
проб, толщина которых превышает пробег бомбардирующих
частиц. В частности, знание функций Y(£) весьма полезно в
.практической аналитической работе с пучками тормозного излу-
чения или заряженных частиц при решении таких вопросов, как
оценка величины взаимных помех и чувствительности опреде-
ления элементов, выбор оптимальной энергии облучения и т. д.
В заключение необходимо дать определение активационного
анализа, в котором кратко сформулирован общий принцип ме-
тода. На основе такого определения строится общая классифи-
кация активационного анализа и проводится четкая граница
между ним и другими ядернофизическими методами. В данной
монографии к активационному анализу отнесены все методы
исследования качественного и количественного состава вещества,
которые основаны на измерении интенсивности и энергетиче-
ского распределения ионизирующего излучения, возникающего
вследствие индуцированного изменения состояния ядер эле-
ментов.
В аналитических целях находит применение регистрация
ядерных частиц и квантов, испускаемых как непосредственно
в процессе ядерного взаимодействия (мгновенное излучение),
так и при распаде образовавшихся радиоактивных ядер (задер-
жанное излучение). При этом сфера приложения второго метода
из-за определенных преимуществ значительно шире.
§ 2. Радиоактивный распад
Образовавшиеся в процессе ядерных взаимодействий не-
устойчивые ядра испытывают радиоактивный распад, превра-
щаясь в конечном счете в стабильные ядра. Скорость распада
совокупности большого числа ядер определенного типа (радио-
изотопа) определяется соотношением
dN^dt = —^NB, (2.12)
где Л’в — число радиоактивных ядер; 7.— постоянная распада.
Интегрирование этого уравнения дает экспоненциальный
закон распада радиоизотопа:
Net = NBoe-uPacn, (2-13)
2Э
где jVjjo—исходное число радиоактивных ядер; — число
радиоактивных ядер, оставшихся после распада в течение вре-
мени fpacn-
Постоянная 7. является характерной величиной для данного
радиоизотопа, так же как и период полураспада Л/г, связан-
ный с к соотношением
74=-^^. (2.14)
Практический интервал значений периодов полураспада
радиоизотопов простирается в широких пределах, примерно
от 108 сек до 1015 лет. По периоду полураспада радиоизотопы
удобно разбить на шесть групп [35]: 1) очень короткоживущие,
Л/2<0,01 мин-, 2) короткоживущие, 0,01 мин^Т^г^ 10 мин;
3) среднеживущие, 10 лшн^Л/г^! день; 4) долгоживущие,
1 день^Л/2^1 год; 5) очень долгоживущие, T’i/2>1 год;
6) крайне долгоживущие, T’i/2>106 лет.
Наибольшее применение в активационном анализе находят
радиоизотопы 2—4-й групп, очень редко—1-й группы и совсем
не используются радиоизотопы 5—6-й групп.
Имеется шесть основных видов радиоактивных процессов:
а-распад, [3-распад, у-излучение, деление ядер, испускание
запаздывающих нейтронов и протонный распад. Последний вид.
распада представляет только специальный интерес и в дальней-
шем упоминаться не будет. Наибольшее значение для актива-
ционного анализа имеют у-излучение и |3-распад. Роль осталь-
ных видов распада более ограниченна.
Радиоактивный распад радиоизотопа обычно представляет
сложное явление и включает в себя несколько процессов и ста-
дий. Каждый радиоизотоп имеет определенное сочетание видов
распада и различных характеристик, которые объединяются
в понятие схемы распада. По важнейшим параметрам схемы
распада (вид излучения, период полураспада, энергия излучения
и т. д.) осуществляется идентификация радиоизотопа, поэтому
эта проблема представляет исключительный интерес для актива-
ционного анализа.
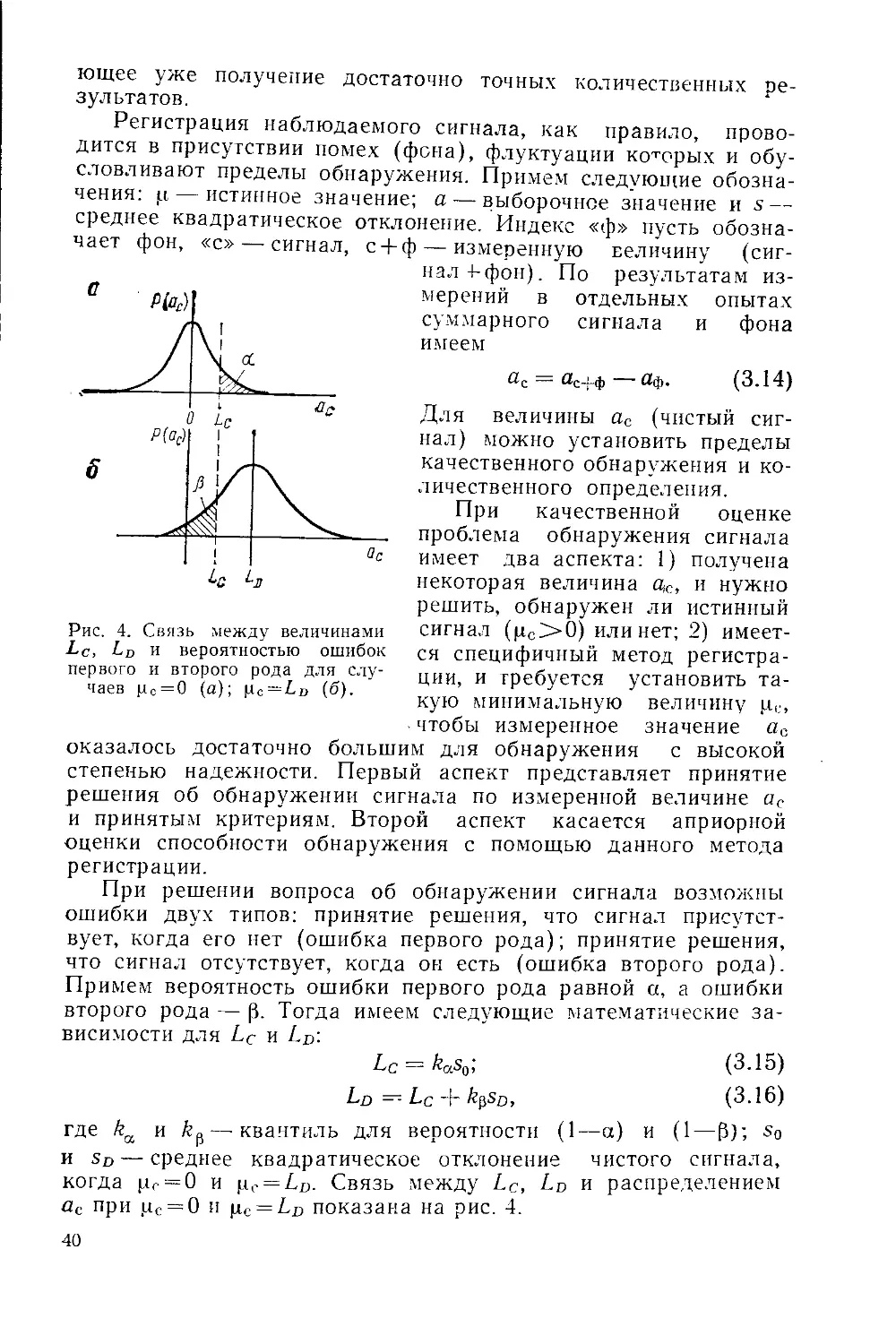
и-Распад. Отдельные радиоизотопы, принадлежащие к огра-
ниченному числу наиболее тяжелых элементов, испускают
а-частицы. При этом заряд ядра уменьшается на две единицы,
а масса на четыре (рис. 3,а). Энергия а-излучения радиоизото-
пов имеет дискретный характер и лежит обычно в пределах
4—7 Мэв. Вообще количественное определение по а-активным
радиоизотопам редко применяется в активационном анализе.
Значительно большее применение а-излучатели нашли в каче-
стве изотопных источников нейтронов и а-частиц.
р-Распад. К p-распаду относятся процессы, приводящие
к изменению заряда исходного ядра на ±1 при сохранении
массового числа М. При p-распаде может происходить испуска-
21
ние электрона (см. рис. 3,а, б, г), позитрона (см. рис. 3,д) или
захват электрона с одной из орбит атома (см. рис. 3, е).
Важная особенность [3-распада состоит в том, что р~- и
Р+-частицы, испускаемые радиоизотопом, имеют непрерывный
спектр энергий. Сплошной характер p-спектра обязан процессу
случайного распределения энергии перехода между одновремен-
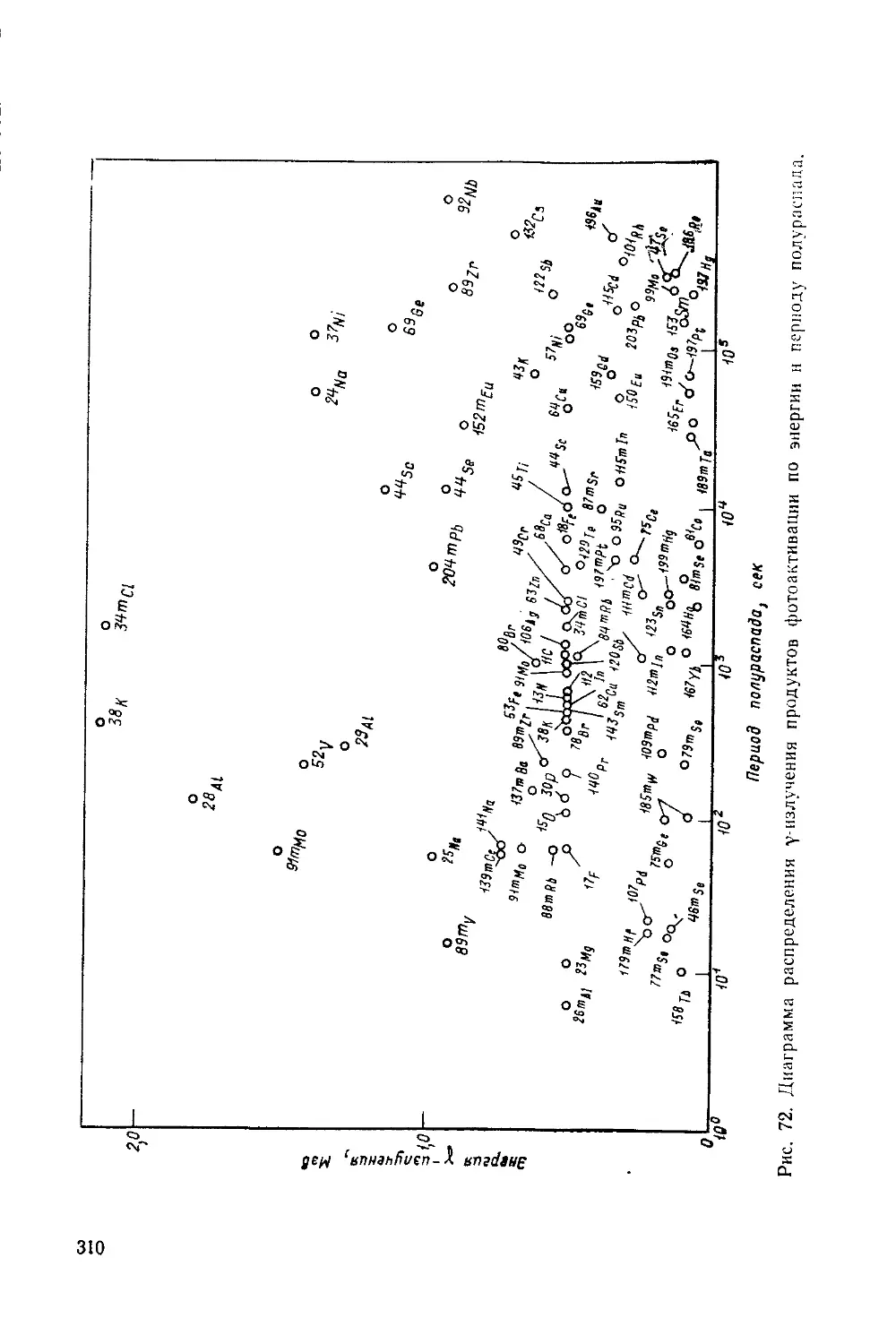
Рис. 3. Схема распада радиоактивных ядер:
« — радиоактивная цепочка из и а-излучателей; б— р—-распад, сопровож-
дающимся у-излучением; в — изомерный переход; г — р--распад с последующим
каскадным у-излучением; д — позитронный распад; е — электронный захват.
Справа или слева от уровня приведен период полураспада, около линии пере-
ходов указаны вид распада, энергия излучения (кэв) и выход.
но испускаемыми р-частицей и нейтрино. Однако для каждого
3-перехода характерна определенная максимальная энергия
Р-частиц.
Формы спектров электронных и позитронных излучателей не-
сколько различаются тем, что у последних выше доля частиц
с высокими энергиями. Другой особенностью позитронного рас-
пада является аннигиляционное излучение. При аннигиляцион-
ном взаимодействии позитрона и электрона образуются два
у-кванта каждый с энергией 0,511 Мэв, разлетающиеся в про-
тивоположных направлениях.
22
При электронном захвате энергию перехода почти целиком
уносит нейтрино. Поскольку отсутствуют эффективные методы
регистрации нейтрино, электронный захват обычно труднее
обнаружить, чем другие виды распада. Это можно сделать
только по вторичным излучениям, сопровождающим захват
электрона. Одним из них является характеристическое рентге-
новское излучение, возникающее при заполнении вакантной
электронной оболочки. Электронный захват часто выступает
как конкурирующий процесс для позитронного распада.
у-Излучение. Ядро, образовавшееся в результате процессов
а- или (3-распада, часто оказывается в возбужденном состоянии.
Разрядка возбужденного состояния обычно осуществляется
через у-излучение (см. рис. 3,6), причем период полураспада
у-переходов очень мал и составляет около 1013 сек. Однако
имеется значительное число случаев, когда в силу действия
правил отбора мгновенный переход оказывается затрудненным
и возникает состояние с измеримым периодом полураспада.
Такие состояния носят название метастабильных пли изомерных
(см. рис. 3, в). Изомерные состояния могут возбуждаться также
непосредственно в ядерных взаимодействиях.
У некоторых радиоизотопов разрядка возбужденного состоя-
ния происходит путем испускания нескольких у-квантов, часто
последовательно друг за другом (см. рис. 3, г). Последний слу-
чай носит название каскадного перехода, который составляет
важную особенность схемы распада.
у-Излучение ядер имеет дискретный характер с очень узкой
шириной линии и в принципе может служить основой для одно-
значной идентификации радиоизотопа. Обычно возможности
у-спектрометрического метода ограничивают разрешающая спо-
собность детекторов и некоторые другие факторы.
Конкурирующим по отношению к у-излучению является про-
цесс внутренней конверсии, при котором энергия возбуждения
ядра передается электронной оболочке, в результате чего по-
является моноэнергетический электрон конверсии. Как и в слу-
чае электронного захвата, заполнение образовавшейся вакансии
приводит к характеристическому рентгеновскому излучению.
Выше основные виды радиоактивных процессов были рас-
смотрены на примере радиоизотопов с простыми схемами рас-
пада. В действительности же схемы распада часто являются
сложными и включают несколько конкурирующих ядерных пере-
ходов. Тогда в схеме появляется еще один параметр — выход
данного вида излучения, который показывает, сколько событий
определенного типа приходится на 100 актов распада.
Процесс распада исходного (материнского) радиоизотопа
может привести к образованию нового (дочернего) радиоизотопа
со своей схемой распада (см. рис. 3, а). Цепочка последователь-
ных радиоактивных распадов может включать и большее число
радиоизотопов.
23
К настоящему времени в различных процессах получено и
исследовано громадное количество радиоизотопов. Анализ на-
копленных данных показывает, что в этой массе отсутствуют
радиоизотопы с полностью идентичными схемами распада
[36, 37]. Это значит, что схема распада является индивидуальной
характеристикой радиоизотопа и может служить основой для
его однозначной идентификации. Этот факт во многом опреде-
ляет высокую специфичность активационного анализа.
Надо отметить, что определение всех параметров схемы рас-
пада, которые необходимы для однозначной идентификации,
представляет не всегда простую задачу и может быть выполнено
в ходе аналитического определения. К тому же параметры изме-
рительных приборов часто далеки от совершенства и ограничи-
вают возможность определения характеристик распада с необ-
ходимой точностью. Поэтому в трудных случаях, когда физи-
ческие средства не позволяют зарегистрировать излучение нуж-
ного радиоизотопа и надежно идентифицировать его из-за помех
со стороны других радиоизотопов, приходится прибегать к хими-
ческой обработке пробы. Сочетание избирательности химиче-
ского выделения с использованием физических параметров схем
распада представляет наиболее надежный путь для идентифика-
ции радиоизотопа.
§ 3. Уравнение активации
Ранее было показано, что количественное определение воз-
можно по мгновенному и задержанному излучениям. В первом
методе исследуемая проба помещается в поток активирующего
излучения и проводится измерение интенсивности вторичного
излучения. Количественное соотношение между числом вторич-
ных частиц в единицу времени Аг и количеством изотопа выте-
кает из уравнения
Аг =-- грФстЛ'д, (2.15)
где ц,— выход вторичного излучения.
Однако в практике активационного анализа основной метод
получения количественных результатов состоит в измерении
интенсивности ионизирующего излучения радиоизотопов, образо-
вавшихся в процессе облучения пробы потоком активирующего
излучения. При этом измерение проводится в отсутствие акти-
вирующего излучения и, как правило, вне зоны облучения.
В этом случае также имеет место строго пропорциональная за-
висимость между экспериментальными параметрами, которые
регистрируются по ходу анализа, и количеством определяемого
элемента.
Установим эту зависимость для наиболее простого случая —
облучения моноизотопного элемента потоком моноэнергетиче-
ских частиц. Пусть в этих условиях протекает ядерная реакция
24
A (a, b) В, которая характеризуется определенной величиной
сечения активации. Конечный продукт этой реакции радиоакти-
вен и имеет постоянную распада Тогда скорость накопления
ядер радиоизотопа будет определяться двумя процессами:
скоростью образования в результате ядерных взаимодействий
н скоростью исчезновения при радиоактивном распаде:
dN
—^ = Фо.,ЛА-Мв. (2.16)
dt
Количество атомов радиоизотопа В, накопившихся за опре-
деленное время облучения /Обл, можно получить интегрирова-
нием уравнения (2.16). Проведем интегрирование в предполо-
жении, что величины Ф, оакт и Ад сохраняют постоянное значе-
ние в процессе облучения. Это означает, что плотность потока
активирующего излучения постоянна во времени и в любой
точке объема пробы, что в ходе облучения не происходит изме-
нения энергии активирующего излучения и не наблюдается
уменьшения числа ядер облучаемого изотопа. При таких усло-
виях интегрирование уравнения (2.16) приводит к выражению
А'в^ а-кт -А-(1 —е-л/обЛ). (2.17)
Л
После облучения производится измерение числа распадов
за определенный промежуток времени бгам, что дает зависимость
= ФаааТЛд (J __ е-^обл) (1 — е“?‘1пм) (2.18)
/v
Если отношение бим/Е 1/2 мало, т. е. число атомов радиоизо-
топа за время измерения уменьшается в незначительной степени,
то имеем (1—е"~?',г,13м) ~бб1зм, и уравнение (2.18) приобретает вид
(2.19)
Ч13М
где Ad — скорость распада или активность радиоизотопа.
Нетрудно видеть, что при длительности облучения, значи-
тельно превышающей период полураспада радиоизотопа
(б>бл Ti/2), из соотношений (2.18) и (2.19) получаем
Ах = Фаа1пЛУА, (2-20)
где Ах — активность насыщения.
Выражение (2.20) совпадает с ранее рассмотренным уравне-
нием (2.15) для интенсивности мгновенного излучения. Отсюда
следует, что только при достижении насыщения активность
радиоизотопа и интенсивность мгновенного излучения при про-
чих равных условиях совпадают. Это значит, что необходимые
результаты быстрее можно получить путем измерения мгновен-
ного излучения, чем в случае предварительного облучения до
насыщения и последующего измерения задержанного излучения.
К сожалению, при практическом осуществлении этого метода
встречаются серьезные трудности, главным образом связанные
с необходимостью регистрировать вторичное излучение в при-
сутствии много более интенсивного потока активирующего излу-
чения. Имеются и другие серьезные ограничения.
Уравнение (2.19) было выведено для моноизотопного эле-
мента; если элемент многоизотопный, уравнение принимает вид
Лd - Фоак10Уэл (1 - е-^обл), (2.21)
где Уэл — исходное число атомов элемента; 0 — доля активи-
рующего изотопа в естественной смеси изотопов.
Переходя к выражению количества облучаемого элемента
в граммах, получаем
Ad = А— Ф^ПхФСТактб (1 _ е-мобл), (2.22)
где тх — весовое количество элемента; М — атомный вес эле-
мента.
Уравнение (2.22) дает активность радиоизотопа в момент
окончания облучения. Если измерение активности производится
через некоторый промежуток времени /раСп, то необходимо
учесть распад радиоизотопа:
Adt = - 6’°2 ' 102^тхФОаКТ9 (1 _ е-МобЛ)е-Мрасп. (2.23>
Уравнение (2.23) представляет собой наиболее простую
форму уравнения активации, которое справедливо при соблю-
дении принятых выше условий. Однако на практике эти условия
редко выполняются в полной мере. В соответствии с ограниче-
ниями, вводимыми условиями эксперимента, видоизменяется
форма уравнения активации и приобретают несколько иной
физический смысл некоторые входящие :в него параметры. Ряд
таких частных форм уравнений активации, действительных для
специфичных условий облучения, будет рассмотрен по ходу даль-
нейшего изложения.
Из приведенных выше соотношений следует, что по существу
активационный анализ — метод изотопного анализа, так как в
результате ядерной реакции на определенном изотопе элемента
возникает радиоизотоп с характеристической схемой распада.
Активность этого радиоизотопа пропорциональна содержанию
изотопа, из которого он образуется. Поэтому рассчитать коли-
чество элемента по уравнению активации можно только в том:
случае, если известен изотопный состав элемента.
Как показали тщательные исследования, изотопный состав
почти всех элементов периодической системы постоянен в по-
давляющем числе естественных объектов. Однако в некоторых
случаях процессы радиоактивного распада, изотопные и другие
26
эффекты в природных условиях приводят к определенным ва-
риациям изотопного состава.
В настоящее время производство обогащенных изотопов по-
лучило широкое развитие. Наблюдались случаи, когда отходы
от производства обогащенных изотопов поступали в продажу
без отметки об измененном изотопном составе [38]. Все пере-
численные факты заставляют тщательно анализировать проб-
лему постоянства изотопного состава исследуемого вещества
и реактивов, используемых для приготовления эталонов, при
практических применениях активационного анализа. С другой
стороны, эта особенность активационного анализа находит при-
менение для определения изотопного состава элементов.
Поскольку многие элементы состоят из нескольких стабиль-
ных изотопов, при их облучении возникают различные продукты
ядерных реакций. Если учесть, что взаимодействие с каждым
изотопом, в свою очередь, может протекать по нескольким кана-
лам, то при облучении одного многоизотопного элемента часто
возникает несколько различных радиоактивных продуктов. При
этом в зависимости от типа и энергии активирующего излуче-
ния возможно образование радиоизотопов как исходного эле-
мента, так и соседних. Сложность протекающих процессов,
с одной стороны, затрудняет проведение анализов, а с другой —
предоставляет больше возможностей для подбора наиболее под-
ходящих условий анализа.
Как правило, идентификация и количественное определение
ведутся путем измерения активности радиоизотопа, который
в данных условиях анализа обеспечивает наибольшие избира-
тельность, чувствительность и точность анализа. В дальнейшем
для краткости этот радиоизотоп будет называться аналитиче-
ским. При этом следует помнить об относительности такого
обозначения, так как в других условиях анализа может ока-
заться, что в качестве аналитического лучше использовать дру-
гой радиоизотоп, получающийся из исследуемого элемента.
Практический интерес может представить вид уравнения
активации для переменной во времени плотности потока акти-
вирующего излучения, что соответствует случаю облучения про-
бы с помощью источника, излучение которого нестабильно во
времени. С учетом того факта, что плотность потока является
функцией времени Ф(/), решение дифференциального уравне-
ния (2.16) в общем виде дает следующее выражение для наве-
денной активности на конец облучения:
Чбл
Ad = Л,А<тЛе“/7°бл Ф(1)еи<И. (2.24)
6
Как следует из уравнения (2.24), величина Ал может быть
рассчитана, если известен закон изменения плотности потока
излучения во времени. Частный случай, когда излучение источ-
97
ника испытывает гармонические колебания около среднего зна-
чения, разобран в работе [39].
Возможна и другая ситуация, когда по условиям работы
источника активирующего излучения облучение пробы имеет
прерывистый характер, т. е. режим активации состоит из после-
довательной цепочки периодов облучения и распада. Если при
этом соблюдается постоянство длительности отдельных облуче-
ний и интервалов между ними, то уравнение активации имеет
вид [40]
(1 _ e“?’(o6.i) (] _ е—,’п0обл~(расп)')
Adn = /УАФо 3------------------------------->- , (2.25)
1 __е /‘гобл"г'расп)
где — активность радиоизотопа в конце п циклов облуче-
ния; Na—число атомов активирующегося изотопа. Приведен-
ные в работе [40] таблицы позволяют ускорить расчеты актив-
ности. В определенных условиях уравнение (2.25) переходит
в более простые выражения, что облегчает расчет наведенной
активности.
Глава
3
МЕТОДИКА АКТИВАЦИОННОГО
АНАЛИЗА
§ 1. Измерение ионизирующих излучений
Регистрация излучений, испускаемых в ядерных процессах,
или при радиоактивном распаде, дает необходимые сведения
для количественных расчетов, а часто и для идентификации
определяемого элемента. Обстоятельное изложение вопроса
можно найти в книгах [41—43], поэтому отметим только неко-
торые моменты, представляющие особый интерес для актива-
ционных определений.
Наиболее важные и широко используемые методы регист-
рации основаны на преобразовании энергии регистрируемой
частицы (кванта) в электрический импульс, который затем
обрабатывается и фиксируется' специальными электронными
устройствами. В качестве преобразователей выступают различ-
ного типа детекторы излучений, действие которых основано на
процессах ионизации или возбуждения атомов (молекул) веще-
ства при прохождении через него заряженных частиц При этом
у-квапты и нейтроны регистрируются через ионизирующее дейст-
вие вторичных частиц, возникающих при их взаимодействии
с веществом.
Газонаполненные детекторы. Это обширный класс детекторов,
характеристики которых зависят от конструкции, состава и дав-
ления наполняющего газа и напряжения на рабочих электродах.
При движении заряженной частицы в рабочем объеме детектора
происходит ионизация атомов (молекул) газа. Под действием
приложенного напряжения образовавшиеся электроны и поло-
жительные ионы собираются на соответствующих электродах,
давая импульс напряжения (при работе в импульсном режиме).
Амплитуда импульса может быть пропорциональна энергии
первичной частицы (пропорциональный счетчик) или практи-
чески не зависеть от первичной ионизации (счетчик Гейгера—
Мюллера).
Газонаполненные детекторы преимущественно применяются
для регистрации а- и [3-частиц. Если в состав рабочего газа
детектора ввести газообразный BF3, то он становится чувстви-
тельным к тепловым нейтронам.
Сцинтилляционные детекторы. В них происходит возбужде-
ние и ионизация атомов (молекул) сцинтиллятора под воздейст-
29
вием ионизирующего излучения. Энергия возбуждения испус-
кается затем в виде квантов света, которые регистрируются
фотоэлектронным умножителем (ФЭУ). Последний преобразует
световую вспышку в поток электронов и усиливает его, давая
на сопротивлении нагрузки импульс напряжения.
Сцинтилляторами служат некоторые неорганические и орга-
нические вещества в газообразной, жидкой и твердой формах.
Среди них наиболее широкое применение находят моно-
кристаллы йодистого натрия, активированные таллием Nal(Tl).
Реже используются органические монокристаллы (стильбен,
антрацен и др.) и пластмассовые сцинтилляторы (терфенил в
полистироле), а также жидкие сцинтилляторы (раствор тер-
фенила в ксилоле).
Сцинтилляционные детекторы в той или иной степени чувст-
вительны ко всем видам излучений. Путем подбора сцинтилля-
тора и условий измерения можно получить систему, чувстви-
тельную преимущественно к одному виду излучения. Так, моно-
кристаллы Nal(Tl) реагируют на а-, 0- и у-излучение. Однако
два первые из них можно отфильтровать, поместив между де-
тектором и источником подходящий поглотитель.
Полупроводниковые детекторы. Они работают на принципе,
аналогичном газовым детекторам. В веществе детектора со
сравнительно низкой проводимостью создается электрическое
поле. Когда заряженная частица проникает в рабочий объем
детектора, она затрачивает энергию на образование электронно-
дырочных пар. Под действием электрического поля образую-
щиеся заряды собираются на электродах, давая на нагрузке
импульс, амплитуда которого пропорциональна энергии, поте-
рянной в рабочем объеме детектора. Если пробег частицы пол-
ностью укладывается в пределах рабочего объема, то амплитуда
импульса пропорциональна полной энергии частицы.
Имеется несколько типов полупроводниковых детекторов,
среди которых наибольший интерес представляют барьерные и
диффузные. Глубина рабочего слоя барьерных детекторов не
превышает 1 мм, поэтому их можно применять для регистрации
тяжелых заряженных частиц и мягкого 0-излучения. Рабочий
объем диффузных детекторов много больше, поэтому они при-
годны для измерения у- и жесткого 0-излучения.
Диффузные детекторы готовят из кремния или германия
с дырочной проводимостью (p-типа), которую компенсируют
путем диффузии лития (донор). Область с компенсированной
плотностью примесей донорного и акцепторного типов представ-
ляет рабочий объем детектора. Наиболее эффективны к у-излу-
чению Ge (Li)-детекторы. Однако их использование и хранение
требуют охлаждения до температуры жидкого азота.
Основные характеристики детекторов: общая эффективность,
мертвое время и энергетическое разрешение. Каждый детектор
имеет определенное сочетание этих параметров, которые показы-
30
вают его возможности при регистрации ионизирующего излуче-
ния. Эффективность — это отношение числа импульсов регистра-
ции к числу частиц, попавших в рабочий объем детектора.
Мертвое время отражает длительность процессов, происходящих
в детекторе при одном акте регистрации. В пределах мертвого
времени детектор не способен к нормальной реакции на появле-
ние в рабочем объеме новой частицы. Энергетическое разреше-
ние определяет способность детектора к раздельной регистрации
частиц близких энергий (см. § 1 гл. 7). Некоторые параметры
детекторов приведены в табл. 1.
203 Таблица 1
Сводка основных параметров детекторов *
Детектор Мертвое вре- мя, сек. Энергетическое разрешение, %
а—5 Мэв ₽=1 Л1эв 7=0,65 Л136
Счетчик Гейгера—Мюлле- ра (самогасящийся) 2-10-4 * *
Пропорциональный счет- чик Около IQ—4 1,5 Около 6
Органический сцинтилля- тор IO-9 ~6
Кристалл Nal (Т1) 2,5-10-’ — — 7,5
Полупроводниковый по- верхностно-барьерный ю-9 0,2—0,5 0,5—1
Полупроводниковый Ge (Li) ю—7 В — — 0,5
* Указаны предельные величины.
Измерительные установки. На выходе детекторов получается
распределение импульсов, которое содержит всю полезную ин-
формацию о регистрируемом излучении. Для перевода этой ин-
формации в форму, пригодную для дальнейшей математической
обработки, необходимы специальные устройства [44—46]. Наи-
более простое из них — счетная установка, которая служит для
подсчета числа импульсов поступающих от детектора за.
время измерения.
В счетном режиме могут использоваться все детекторы, отме-
ченные выше. Однако у многих из них амплитуда импульса
Нимп пропорциональна энергии Еиз.-т, потерянной зарегистриро-
ванной частицей (квантом) в рабочем объеме детектора,
т. е. имеет место зависимость Упмп = &£из.т, где k — коэффициент
пропорциональности. Поэтому амплитудное распределение им-
пульсов от таких детекторов содержит в себе информацию об
энергетическом спектре регистрируемого излучения. Анализ та-
ких распределений проводят с помощью дискриминаторов и ана-
лизаторов.
3V
Интегральный дискриминатор представляет собой электрон-
ную систему, пропускающую на счетное устройство только те
импульсы, амплитуда которых превышает порог дискриминации
Ед. Это означает, что такой прибор фиксирует частицы с энер-
гией выше определенного значения. Изменяя порог дискрими-
нации, можно устанавливать нужную граничную энергию.
Дифференциальный анализатор позволяет регистрировать
только те импульсы, амплитуда которых лежит в заданном
интервале ДЕд, что в энергетических единицах соответствует
разности энергий ДЕПЗл. Если величину ДЕд сделать достаточно
малой и последовательно увеличивать нижний порог дискрими-
нации, то получим дифференциальный амплитудный спектр
Ni = f(Vn). Из этого спектра при соответствующей обработке
можно извлечь весьма ценную информацию о качественном и
количественном составе зарегистрированного излучения.
Наиболее совершенные приборы — многоканальные ампли-
тудные анализаторы. Эти приборы обычно работают на прин-
ципе амплитудно-временного преобразования. Амплитуда посту-
пившего на вход анализатора импульса преобразуется в про-
порциональное ей число стандартных импульсов. Это число
соответствует номеру канала анализатора, в котором происхо-
дит регистрация отсчета.
Эффективность регистрации. Из уравнений (2.15) и (2.23)
вытекает возможность количественных определений по интенсив-
ности мгновенного излучения или активности радиоизотопа.
'Однако из-за действия ряда факторов только некоторая доля
излучения может дать полезный сигнал на выходе измеритель-
ного устройства. Тогда при отсутствии помех будет справед-
ливо соотношение
nz = M;, (3.1)
где П{ — скорость счета прибора; е — эффективность регистра-
ции; Ad — активность радиоизотопа.
Эффективность характеризует конкретную измерительную
ситуацию и обусловливается несколькими факторами. Прежде
всего на эффективность регистрации сильное влияние оказывают
геометрические параметры системы детектор—источник: раз-
меры и форма детектора и источника, расстояние между ними,
особенности их взаимного расположения. Большую роль могут
играть и эффекты, связанные с поглощением и рассеянием иони-
зирующего излучения в веществе источника, упаковке детектора
и других вспомогательных конструкционных материалах (покры-
тие детектора, подложка источника, поглотители и т. д.). Кроме
того, надо знать параметры схемы распада радиоизотопа или
возбужденного уровня, т. е. выход излучения, коэффициент кон-
версии и т. п. В заключение важно подчеркнуть большое значе-
ние эффективности детектора, которая обычно зависит от энер-
32
гии излучения. В таких случаях требуется измерение спектраль-
ной чувствительности детектора.
Учет всех перечисленных факторов пли уменьшение их влия-
ния до пренебрежимого уровня часто представляет сложную
проблему, которая не всегда может быть разрешена быстро и
с необходимой точностью. Поэтому, как правило, прибегают
к проведению измерений в фиксированных условиях, поддержи-
ваемых постоянными во всей серии проводимых определений.
Тогда количественные оценки можно выполнять абсолютным
или относительным методом. В первом из них калибруют прибор
по эффективности с помощью препаратов, абсолютная актив-
ность которых известна. Во втором последовательно измеряют
активности пробы и эталона.
Статистическая погрешность при измерениях радиоактив-
ности. В радиометрических методах сигнал, несущий полезную
информацию, обладает следующими свойствами:
1) радиоактивный распад ядер, а следовательно, и регист-
рируемое излучение (сигнал) относятся к случайным явлениям,
подчиняющимся статистическим закономерностям;
2) сигнал имеет дискретный характер и выражается в числе
отсчетов (импульсов) регистрирующего устройства;
3) сигнал изменяется во времени в соответствии с законом
радиоактивного распада.
Перечисленные свойства рассматриваемого сигнала приводят
к особой форме распределения результатов измерения, которое
только в определенных условиях переходит в нормальное (47].
Последнее, как известно, наиболее распространено в других
(нерадиометрических) методах анализа [48, 49].
Пусть облучение дало Nw радиоактивных ядер с постоян-
ной распада Л. Тогда вероятность распада определенного числа
ядер Nd за время измерения задается законом биномиаль-
ного распределения
Р (v ) =----^22----(1 _ е~?7изм)ЛД (е“л/.пм)л'во-/3 2)
d 0Тво-М/)!У/!
Наиболее вероятное среднее число распадов .Vj равно
^ = №о(1-е-х/"зМ). (3.3)
Среднее квадратическое отклонение числа распадов от среднего
рассчитывается из выражения
44 • (з-4)
Г \ ЛВ0 /
Анализ показывает, что биномиальное распределение, если его
применять для статистической обработки числа отсчетов регист-
рирующего устройства, переходит в распределение Пуассона
2 Р А. Кузнецов
33
при соблюдении хотя бы одного из следующих условий: а) ис-
ходное число радиоактивных ядер достаточно велико (Лгво> 1);
б) длительность измерения мала по сравнению с периодом полу-
распада радиоизотопа (/изм^Л/г); в) эффективность регистра-
ции низка (е<<1). Тогда закон распределения вероятности числа
отсчетов приобретает форму
(3.5)
Это распределение при малом числе отсчетов несимметрично,
но с их ростом становится все более симметричным относительно
среднего значения.
Для распределения Пуассона лучшим приближением к сред-
нему числу отсчетов является величина (Л\ + 1), а для среднего
квадратического отклонения имеет место равенство хЛ-= У Ni+ 1.
Однако если Nt велико (более 100), то соответствующие пре-
образования приводят к хорошо известному' закону Гаусса:
Р W) = - —- е~(Л'^Л’‘')2/2^. (3.6)
\' 2nNt
Здесь наблюдается полная симметрия относительно Ni=Ni.
Наилучшим приближением к истинному среднему числу отсче-
тов Ni является сама измеренная величина Ni, а среднее квад-
ратическое отклонение равно s= V ЛД Тогда относительная
погрешность равна Лд. Поскольку для долгоживущих
радиоизотопов справедливо соотношение Л\- = П(/Изм, где П; —
скорость счета, то получаем s,.=l/KшДзм- Следовательно,
уменьшение относительной погрешности требует набора доста-
точно большого числа отсчетов, что приводит к соответствую-
щему увеличению длительности измерения. Для короткоживу-
щих изотопов такой способ повышения точности неприменим,
и поэтому, чтобы достигнуть максимального эффекта, необхо-
димо должным образом планировать условия облучения и изме-
рения. Возможная альтернатива состоит также в цикличном
способе облучения и измерения с суммированием числа отсчетов.
Поправка на фон. При радиометрических измерениях в по-
лучаемые результаты необходимо вводить поправку на число
фоновых отсчетов, которые возникают в результате действия
космического излучения и радиоактивных загрязнений окружаю-
щей среды. Фон прибора определяется в отдельном опыте в от-
сутствие исследуемого препарата. В результате скорость счета
радиоизотопа П; получается как разность между скоростью счета
суммарного эффекта и скоростью счета фона Пф:
п( — п1+ф — Пф. (3.7)
34
Среднее квадратическое отклонение скорости счета равно
8 = V П('4-ф + Пф , (3-8)
а относительная погрешность
/ ”‘+ф '
«/д ф — «Ф
Поправка на мертвое время. Используемые измерительные
установки обладают ограниченной временной разрешающей
способностью, т. е. они тоже имеют мертвое время. Поэтому
временное разрешение регистрирующей системы в целом опре-
деляется параметрами основных элементов—детектора и элек-
тронных систем. О мертвом времени детекторов речь уже шла
(см. табл. 1). Что касается электроники, то для простых счет-
ных устройств разработаны весьма быстродействующие элек-
тронные системы, мертвое время которых не превышает 10 4—
10-9 сек [46]. Правда, наибольшее распространение полечили
устройства с временным разрешением около 10 6 сек. В боль-
шинстве практических случаев этого вполне достаточно.
Хуже обстоит дело со сложными анализирующими устрой-
ствами типа многоканальных анализаторов, которые представ-
ляют важнейший элемент наиболее совершенных спектромет-
рических установок. Их мертвое время лежит в пределах
1Q-5—10 :i сек, что накладывает опретеленные ограничения на
возможности практического применения.
При высокой интенсивности излучения истинную скорость
счета, т. е. число частиц, попавших за единичный интервал вре-
мени в чувствительный объем детектора, следует рассчитывать
по соотношению
= , (З.Ю)
где rii — зарегистрированная скорость счета; тм — мертвое вре-
мя установки.
Уравнение (3.10) справедливо только для источников с по-
стоянной интенсивностью. Если его интенсивность меняется в
ходе измерения, т. е. в источнике содержатся короткоживущие
изотопы, то поправка на просчеты может быть рассчитана толь-
ко при известном законе изменения интенсивности во времени.
Поскольку во многих случаях- активационных определений этот
закон не известен и к тому же может быть достаточно сложным
из-за наличия нескольких радиоизотопов в смеси, поправки на
просчеты не могут быть корректно рассчитаны. Тогда остается
единственный путь получения правильных результатов — подбор
таких условий анализа, чтобы регистрируемая активность не
превышала определенного уровня.
2
35
§ 2. Методы получения количественных результатов
Абсолютный метод. Конечным результатом всякого количе-
ственного метода анализа должны быть данные о содержании
определяемого компонента в анализируемой пробе. При актива-
ционном анализе количественные определения возможны на ос-
новании соотношений (2.23) и (2.15). Ниже речь пойдет в ос-
новном только о первом из них.
Исходя из уравнения (2.23), по измеренной абсолютной
активности Adi, известным условиям облучения и измерения
(Ф, Л>бл, ^расп ) и табличным значениям ядерных параметров
((Такт, Л1, 0, Z) можно рассчитать количество определяемого эле-
мента. Такой подход получил название абсолютного метода.
Несмотря на кажущуюся простоту, на практике абсолютный
метод в строгом виде используется очень редко из-за трудности
достижения точных результатов. Прежде всего далеко не всегда
можно с требуемой точностью определить плотность потока ак-
тивирующих частиц в зоне облучения каким-либо прямым фи-
зическим методом.
Так, в случае заряженных частиц для этих целей можно
использовать измерение тока пучка. Для нейтронных же пото-
ков приходится прибегать преимущественно к облучению мони-
тора, т. е. известного количества какого-либо элемента, имею-
щего благоприятные активационные характеристики, с после-
дующим расчетом Ф по уравнению (2.23).
В абсолютном методе весьма важным обстоятельством яв-
ляется строгое постоянство потока активирующего излучения во
времени и по объему облучаемой пробы. Желательно постоян-
ство потока излучения в течение целой серии аналитических
определений, тогда абсолютный метод имеет значительное пре-
имущество и может сэкономить массу времени. К сожалению,
такая ситуация встречается крайне редко, и поэтому для кон-
троля за потоком активирующего излучения требуется облучение
монитора при каждом определении. Но тогда имеется много
оснований рассматривать этот вариант как частный случай
метода мониторов. К тому же не всегда точно известны величи-
ны сечений ядерных реакций, которые сильно зависят от энер-
гии активирующего излучения, и поэтому даже небольшое из-
менение энергетического спектра может привести к значитель-
ной погрешности.
Достаточно сложную проблему представляет определение
абсолютной активности, особенно если измерения проводятся по
Р-излучепию. Для этого прибегают либо к помощи 4л-счетчиков,
либо к введению большого числа поправок при измерениях с
помощью счетчиков других типов. Значительно более просто и
точно абсолютную активность можно измерить с помощью гам-
ма-спектрометра, предварительно прокалиброванного по эф-
фективности.
36
Метод эталонов. Многие трудности, свойственные абсолют-
ному методу, отпадают, если одновременно с анализируемой
пробой облучить точно известное количество определяемого
элемента—эталон. После облучения может быть использован
инструментальный или радиохимический вариант. В обоих слу-
чаях активность эталона и исследуемого препарата (пробы)
измеряют в одинаковых условиях. При необходимости вводят
поправки на химический выход, радиоактивный распад и т. д.
Содержание определяемого элемента при этом рассчитывают
из простого соотношения
тх _ Ах
тэ Аэ
(З.Н)
где тх и тэ — количество элемента соответственно в пробе
и эталоне, а Ах и Аэ — соответствующие активности (скорости
счета).
Нетрудно видеть, что в случае метода эталонов нет необхо-
димости точно знать плотность потока активирующего излуче-
ния, лишь бы она была постоянной по всему объему, занимае-
мому пробой и эталонами. Отпадает также требование к по-
стоянству интенсивности активирующего излучения во времени.
Погрешность величины сечения активации и изменения в энер-
гетическом спектре активирующего излучения уже не оказы-
вают влияния на конечные результаты. Абсолютные измерения
заменяются относительными, которые в целом много проще и
часто точнее. Все это вместе взятое заметно облегчает прове-
дение активационного анализа и повышает точность опреде-
ления.
Метод мониторов. В последнее время значительное распро-
странение получил метод мониторов, который в конечном счете
приобрел право на самостоятельное существование. Его сущ-
ность состоит в контроле за условиями облучения, главным
образом за интенсивностью активирующего излучения, каким-
либо подходящим способом. При этом важно, чтобы между
показаниями монитора Лм и средней плотностью потока акти-
вирующего излучения Ф в пробе имела место определенная
функциональная связь типа
(3.12)
где /гм — коэффициент пропорциональности.
Если можно с высокой точностью рассчитать или опре-
делить экспериментально в калибровочных опытах, то из вы-
ражения (3.12) рассчитывается Ф и открывается возможность
применения абсолютного метода. Однако если kv оценить не-
возможно или сложно, и в то же время без затруднений можно
обеспечить его постоянство в проводимой серии определений,
то применяют следующую методику.
37
Облучают вместе монитор и эталон и получают величины
и Аа в заданных условиях анализа. Затем с тем же мони-
тором (или аналогичным) облучают пробу и получают вели-
чины ДМЛ. и ДЛ.. Важно подчеркнуть, что Ах и Аа определяют
с теми же предосторожностями, что и в методе эталонов. Из
уравнений (3.1 i) и (3.12) не представляет труда получить
выражение для количественных расчетов:
А А
тх=-- ~~тэ. (3.13)
Рассмотренный метод можно распоостранить на несколько
определяемых элементов, и тогда при анализе проб отпадает
необходимость каждый раз одновременно облучать и затем
измерять соответствующее количество эталонов.
В качестве монитора обычно используют определенное ко-
личество элемента, обладающего благоприятными активацион-
ными характеристиками. Например, для активационного ана-
лиза на тепловых нейтронах часто используют мониторы из
золота, марганца, кобальта, меди и других элементов, а также
некоторые сплавы (например, сплав кобальта с алюминием).
Исходный материал должен быть в такой форме, чтобы из
него без труда можно было бы сделать необходимое количество
стандартных мониторов (определенной массы и формы). Чаще
всего для этого используется проволока или фольга. Не сле-
дует упускать из вида и возможность повторного использо-
вания монитора после выдержки.
Частный случай метода мониторирования — использование
в качестве монитора одного из компонентов пробы (или добав-
ляемого к пробе). Последний поэтому носит название внутрен-
него монитора.
Для мониторирования могут быть использованы и другие
методы. Например, в случае заряженных частиц можно изме-
рять ток пучка в ходе облучения. Иногда с помощью различ-
ных счетных устройств регистрируют интенсивность вторичного
или рассеянного излучения, величина которой пропорциональ-
на основному потоку. Правда, можно отметить, что подобные
методы мониторирования находят сравнительно ограниченное
применение.
Метод мониторов имеет определенные преимущества в це-
лом ряде случаев и прежде всего тогда, когда интенсивность
активирующего излучения (поток активирующих частиц или
квантов) в данной серии анализов меняется от одного облуче-
ния к другому. Метод мониторирования заслуживает особого
внимания при серийном определении большого числа элемен-
тов в одной пробе, когда эталоны в каждом анализе требуют
больших затрат труда и времени на различные подготовитель-
ные операции и измерение активностей. Ограничивающим фак-
тором может быть и недостаток места в контейнере для облу-
38
чения. Иногда одновременное облучение и измерение пробы и
эталона оказываются невозможными или трудными, как, на-
пример, при проведении анализа по короткоживущим изото-
пам или при использовании источников с сильным градиентом
потока активирующих частиц.
Недостатки метода мониторов связаны с необходимостью
строго выдерживать интервалы времени в ходе анализа и с
зависимостью конечных результатов от нестабильности интен-
сивности и спектра активирующего излучения при облучении.
Эти факторы оказывают влияние на величину kM. Однако тща-
тельное планирование и выполнение анализа позволяют в зна-
чительной степени уменьшить действие перечисленных фак-
торов..
§ 3. Оценка пределов обнаружения
Под пределом обнаружения понимается минимальное коли-
чество элемента, которое еще может быть надежно обнаружено
в данных условиях анализа. Таким образом, предел обнару-
жения ограничивает область применимости данного метода со
стороны низких концентраций, т. е. задает чувствительность
метода *. Поскольку в активационном анализе сигналом, несу-
щим количественную информацию, является ионизирующее из-
лучение радиоактивных ядер, нижняя граница определяемых
концентраций зависит от минимального числа распадов, кото-
рое может быть зарегистрировано с достаточной надежностью.
В связи с важностью проблемы многие исследователи за-
нимались анализом факторов, влияющих на чувствительность
активационного анализа, проводили теоретическую и экспери-
ментальную оценки пределов обнаружения элементов в различ-
ных условиях определения. Однако результаты этих исследова-
ний не всегда строго сопоставимы из-за отсутствия общеприня-
той терминологии и различий в выборе критериев для оценки
пределов обнаружения. Критический разбор этой проблемы
выполнили X. Э. Гунне, Л. Л. Пелекис [50] и Курри [51].
Для решения различных задач, связанных с оценкой воз-
можностей аналитического метода для определения минималь-
ных количеств вещества, вводятся три величины: 1) критиче-
ский предел обнаружения Lc, с помощью которого можно толь-
ко принять решение о том, указывает ли полученный результат
на присутствие определяемого компонента; 2) предел обнару-
жения Ld — служит границей уверенной регистрации элемента;
3) порог определения LQ — представляет значение, обеспечива-
* Вообще термин «чувствительность» в значении предела обнаружения
не рекомендуется к употреблению, и сделанное отступление представляет
дань давней традиции.
39
ющее уже получение достаточно точных количественных ре-
зультатов.
Регистрация наблюдаемого сигнала, как правило, прово-
дится в присутствии помех (фона), флуктуации которых и обу-
словливают пределы обнаружения. Примем следующие обозна-
чения: ц истинное значение; а — выборочное значение и s —
среднее квадратическое отклонение. Индекс «ф» пусть обозна-
чает фон, «с» — сигнал, с + ф — измеренную величину (сиг-
нал 4-фон). По результатам из-
мерений в отдельных опытах
суммарного сигнала и фона
имеем
Рис. 4. Связь между величинами
Lc, Ld и вероятностью ошибок
первого и второго рода для слу-
чаев Цс=0 (a); pc=i.D (б).
ас = ас+ф — Оф. (3.14)
Для величины ас (чистый сиг-
нал) можно установить пределы
качественного обнаружения и ко-
личественного определения.
При качественной оценке
проблема обнаружения сигнала
имеет два аспекта: 1) получена
некоторая величина цс, и нужно
решить, обнаружен ли истинный
сигнал (цс>0) или нет; 2) имеет-
ся специфичный метод регистра-
установить та-
величину Це,
значение ас
с высокой
ции, и требуется
кую минимальную
чтобы измеренное
оказалось достаточно большим для обнаружения
степенью надежности. Первый аспект представляет принятие
решения об обнаружении сигнала по измеренной величине ас
и принятым критериям. Второй аспект касается априорной
оценки способности обнаружения с помощью данного метода
регистрации.
При решении вопроса об обнаружении сигнала возможны
ошибки двух типов: принятие решения, что сигнал присутст-
вует, когда его нет (ошибка первого рода); принятие решения,
что сигнал отсутствует, когда он есть (ошибка второго рода).
Примем вероятность ошибки первого рода равной а, а ошибки
второго рода — |3. Тогда имеем следующие математические за-
висимости для Lc и Ld:
Lc = kaS0', (3.15)
Ld — Lc -T kfiSo, (3.16)
где ka и — квантиль для вероятности (1—а) и (1—ft); «о
и Sp — среднее квадратическое отклонение чистого сигнала,
когда рс = 0 и Цс = Тр. Связь между Lc, LD и распределением
ас при Цс = 0 и цс = Тв показана на рис. 4.
40
Оба предела (Lc и LD) непригодны с точки зрения количе-
ственных определений, когда нужен результат, близкий к ис-
тинному значению. Поскольку в этом случае погрешность дол-
жна быть малой частью определяемой величины, то для порога
определения имеем
LQ = kQsQ, (3.17)
где kQ=l/srQ (sr Q — требуемое относительное среднее квад-
ратическое отклонение).
При измерениях радиоактивности, если сигнал достаточно
велик, величина ас представляет хорошее приближение для
щ (нормальное распределение), а дисперсия сигнала равна
Sc = Sc+ф Зф = (яс + &ф) Н------. (3.18)
п
В этом выражении предполагается, что среднее значение фона
получено из п определений. Тогда соответствующие выражения
для величин Lc, LD и Lq принимают вид
Lc = k-,s0 = ka (аф + s|)1/2; (3.19)
Удобные для практических оценок выражения можно получить,
если принять а=р = 0,05 и fee=10 (табл. 2). Особенно простые
выражения получаются, когда число отсчетов фона a$ велико
(нижняя строка табл. 2).
В приведенных выражениях для различных пределов связь
с длительностью измерения выражена в неявной форме. Чтобы
увидеть зависимость чувствительности от времени, необходимо
перейти от интегральных величин Lc, LD, LQ и Оф к скоростям
счета. Тогда, например, для предела = в случае высо-
кого фона получим выражение
/п = 3’29/Яф/изм = 3,29 1/ (3.22)
Гизм у Фзм
Таким образом, предельная скорость счета долгоживущего изо-
топа уменьшается с увеличением длительности измерения про-
порционально V /изм.
Однако для короткоживущих изотопов имеется оптимальное
время измерения, когда соответствующие пределы оказыва-
ются минимальными. Анализ этой проблемы сделан Стерлин-
ским [52]. Оптимальное время измерения зависит от уровня
41
Таблица 2
Формулы для оценки Lc , LD и LqB различных экспериментальных условиях
Предельные величины, отсчеты
Условия измерения Lc lD LQ
Парные наблюдения* («ф = °Ф) Точно определенный фон** (5ф = 0) Нулевой фон (аф = 0) Высокий фон*** 2,33 \/у 1,64 |+ф 0 1,64/аф (аф> 0) 2,71+4,65/5ф 2,71+3,29/^ 2,71 3,29/5; (аф> 67) 50 (1+Г 1+_5L]V2| 1 L 12,51 J 5о|1+р+ДФ_у/2| 100 ю /Оф (аф> 2500)
’Длительности измерения сигнала и фона равны.
*’Величина получена как среднее из многократных определений (длительные
измерения фона).
**’Большое число отсчетов фона за время измерения сигнала при точно определенном
фоне.
фона прибора пф. Когда это значение достаточно велико, опти-
мальное время составляет примерно два периода полураспада
измеряемого радиоизотопа (рис. 5). Если фон прибора мал, то
оптимальная длительность измерения увеличивается.
Для наглядности на рис. 6 приведены значения различных
пределов, рассчитанные для конкретного случая, в котором
производится измерение долгоживущего у-излучателя в течение
10 мин с эффективностью 10% при фоне детектора 20 от-
счет/мин. Для сравнения там же показаны значения эмпириче-
ских пределов, полученные с помощью наиболее часто исполь-
зуемых критериев для оценок чувствительности различных ме-
тодов. Как видно, результаты перекрывают почти три порядка.
Это убедительное свидетельство в пользу того положения, что
оценку чувствительности метода необходимо делать па основе
единых критериев.
В активационном анализе очень часты ситуации, когда по-
меха состоит не только из фона прибора, но и вклада некото-
рых радиоактивных компонентов облученной пробы, излучение
которых регистрируется (частично или полностью) вместе с
излучением исследуемого компонента. Если вклад интерфери-
рующего излучения может быть оценен, то при расчете соответ-
ствующих пределов необходимо использовать величину
аф+и = цФ + аи, (3.23)
42
где а„ — число отсчетов интерферирующего излучения за вре-
мя регистрации. Отсюда следует вывод, что появление интер-
ферирующего излучения равносильно увеличению фона изме-
рительной установки и, следовательно, связано с потерей чув-
ствительности.
Рассмотренные выше выражения дают соответствующие
пределы в виде числа отсчетов
аналитических определений
эти величины представляют
интерес только тогда, когда
они выражены в единицах
массы или числе атомов.
Связь устанавливается с по-
мощью уравнений (2.23) и
(3.1). Тогда, например, для
предела обнаружения, выра-
женного в весовых категориях,
или скорости счета. Однако для
индекс предела
Рис. 6. Значения различных преде-
лов обнаружения:
Lq — критический предел обнаружения;
L& — предел обнаружения; Lq — порог
определения; 1 — 8 индексы эмпирических
пределов, где / — 1$ф; 2—0,1^; 3 —
2%- 4-38ф; 5 - 35ф+3зс; 6 - 2Яф; 7-
1000 расп/мин-, 8—100 pacnjcen (йф— чис-
ло отсчетов фона; $ф и sc — среднее
квадратическое отклонение соответственно
фона и сигнала).
Рис. 5. Зависимость предела об-
наружения от длительности изме-
рения для короткоживущих изо-
топов.
получим для обобщенной формы, учитывающей интерферирую-
щее излучение (^а=^р), следующее уравнение:
+ 2ka [аФ + S|+ + ®иГ/2
=---------------------------- ---------- —. (3.24)
6,02 1023 Л1-1 Фаакт0е)1-1(1—е обл)(1 —е ' изм) е " Расп
Если помеху оказывают несколько радиоизотопов, то вели-
чины а„ и $2 нужно соответственно заменить на Sa„ и Ss2 .
Когда время измерения фона (хф = а(»,) и мешающего излучения
равно времени измерения сигнала, то корень квадратный в чи-
слителе равен [2аф + 2аи]1/2. С другой стороны, когда аф и аи
получены как среднее из многократных измерений (s|<^a$ и
s- Сйц), этот корень равен [аф + аи]1/2.
43
Практически интересный предельный случай имеет место,
когда интенсивность интерферирующего излучения велика и
справедливы условия !га < V аи и йф<ап [53]. Тогда для оди-
накового времени измерения после подстановки развернутого
выражения для ап получаем уравнение
2йа[6,02- 10232Фтиаакт X
то ------------------------------------
6,02- Ю'«Фожт0Л1-18 (1—е~“обл)х~1Х
Q ___ е Vo6ji) _____ е Vh3m) е ^и^распр/2
(1 __ е~^изм) е~^расп
(3.25)
Таким образом, в присутствии интерферирующего излуче-
ния чувствительность метода с увеличением плотности потока
растет только пропорционально ]/ф. Увеличение в пробе со-
держания интерферирующего элемента снижает чувствитель-
ность метода к определяемому элементу. Такая ситуация часто
встречается в случае инструментального варианта, когда не
удается обеспечить необходимую избирательность определения.
§ 4. Общий ход активационного анализа
Схема анализа
Каждое аналитическое определение, выполняемое активаци-
онным методом, складывается из нескольких последовательных
стадий. Среди них есть обязательные для всех случаев актива-
ционных определений, другие же привлекаются при решении
ограниченного круга задач. Количество стадий и их характер,
совокупность которых представляет схему анализа, зависят от
исследуемого объекта, требуемой чувствительности определе-
ния, числа определяемых элементов, вида активирующего из-
лучения и других факторов.
Как уже отмечалось, в активационном анализе выделяются
два основных варианта схем анализа — инструментальный и
радиохимический. Основные стадии анализа твердых проб в
обоих случаях показаны на рис. 7.
Следует подчеркнуть, что каждая стадия, отмеченная на
рис. 7, дает только общее представление о выполняемой опе-
рации, которая в конкретной схеме анализа может иметь весь-
ма специфический характер. Обсуждение особенностей кон-
кретных методик трудно совместить, поэтому общий ход ана-
лиза здесь будет показан на примере определения малых кон-
центраций элементов (менее 10—4%) при облучении тепловыми
нейтронами реактора. Методические особенности других важ-
ных случаев будут отмечены в соответствующих разделах.
44
Отбор вещества для анализа и подготовка проб
к облучению
В обычной практике выбор объектов для аналитического
исследования или контроля определяется решаемой научной
или производственной задачей. Хотя стадия первичного отбора
вещества часто оказывает большое влияние на получаемые ре-
0m дор вещества для анализа
Подготовка правы к облучению
Облучение
Травление поверхности
Растворение пробы,
введение носителей
Выделение определя-
емых элементов хими-
учетом ядерных
характеристик радиоизотопов
ческими методами
Измерение активности,
определение химического
выхода
Обработка результатов
измерений
Проверка радиохимиче-
ской чистоты
Количественный расчет
Рис. 7. Общий ход активационного анализа.
зультаты и их интерпретацию, особенно при определении ма-
лых концентраций, эта проблема все же выходит за рамки
данной книги. Сформулируем только общие требования к мате-
риалам и веществам, предъявляемым к анализу.
Материал должен быть получен из надежного источника с
соблюдением всех мер предосторожности, исключающих за-
грязнение или частичную потерю определяемых компонентов.
Необходимы также сведения о происхождении вещества, его
истории с момента взятия и до поступления в лабораторию
и предварительные данные о составе макрокомпонентов. Коли-
чество вещества должно быть достаточным, чтобы гарантиро-
вать представительность результатов и позволить повторные
определения, если в этом возникнет необходимость.
45
В принципе активационный анализ на тепловых нейтронах
не предъявляет каких-либо особых требований к агрегатному
состоянию вещества. Пробы можно облучать з твердом, жид-
ком и газообразном состоянии непосредственно после взятия
вещества на анализ. Более того, когда требуется определение
крайне малых содержаний, любые манипуляции с веществом
перед облучением весьма нежелательны, так как это потенци
альный источник загрязнений.
Масса пробы, отбираемая для облучения, определяется
рядом причин: количеством имеющегося материала, макси-
мальной концентрационной чувствительностью, уровнем радиа-
ционной опасности от облученной пробы, степенью самоослаб-
ления потока нейтронов и т. д. В большинстве случаев при
облучениях в реакторе масса пробы составляет примерно 1 г.
Иногда для повышения концентрационной чувствительности
применяют навески большей массы, однако навески более 10 г
в реакторах облучают очень редко. Лишь при облучении с по-
мощью источников со средней и низкой плотностью потока ней-
тронов применяемые навески достигают 100 г. При облучении
в реакторе весьма успешно используют навески в несколько
миллиграммов и даже микрограммов.
При отборе пробы следует избегать попадания посторонних
загрязнений, источниками которых могут быть пыль из поме-
щения или различные вещества с инструментов, используемых
для отбора пробы. Пыль — серьезный источник загрязнений, к
тому же трудно контролируемый. Состав пыли сильно ме-
няется в зависимости от местных условий. Особенно разнооб-
разным может быть состав пыли в химических лабораториях,
где ассортимент используемых для работы веществ очень ши-
рок.
Для уменьшения опасности загрязнений пробу желательно
отбирать в отдельном помещении, в котором не работают с
радиоактивными веществами и макроколичествами определяе-
мых элементов. Пробы отбирают в специальном герметичном
боксе из органического стекла. Для отбора твердых проб ис-
пользуют шпатели или пинцеты из полимерных материалов.
Жидкие пробы отбирают полиэтиленовой пипеткой.
Навеску пробы переносят в ампулу, которая герметизи-
руется. Герметичная упаковка гарантирует сохранность пробы
при транспортировке и в ходе облучения, а также предохраняет
пробу от попадания посторонних веществ из окружающей
среды.
В качестве материала для ампул чаще всего используют
кварц, полиэтилен и алюминий. Любой из этих материалов
устойчив к облучению и слабо поглощает тепловые нейтроны;
их можно получать достаточно чистыми. В то же время у каж-
дого материала есть свои особенности. Алюминий, например,
обычно содержит больше примесей, чем другие материалы, при
46
наличии в потоке быстрых нейтронов в нем по реакции (п, а)
образуется 24Na с жестким у-излучением. Полиэтилен стано-
вится хрупким после непродолжительного облучения и не вы-
держивает нагревания.
Перед отбором пробы ампулы промывают для удаления
поверхностных загрязнений. Для этого используют различные
растворители, неорганические кислоты или их смеси и воду.
Все эти реагенты подвергают специальной очистке [54]. Очень
чистую воду получают при пропускании ее через колонку с ка-
тионитом и анионитом с последующей перегонкой в кварцевом
аппарате. Кислоты обычно очищают многократной дистилля-
цией. Наиболее чистые кислоты получают, растворяя соответ-
ствующие газообразные продукты в воде, очищенной описан-
ным выше способом. Алюминиевые ампулы, например, очи-
щают последовательным промыванием бензолом, азотной кис-
лотой и водой. Полиэтиленовые и кварцевые ампулы промы-
вают бензолом или ацетоном, горячей смесью кислот HNO3 +
+ H2SO4 или царской водкой и водой. Нельзя промывать ампу-
лы хромовой смесью, так как ионы хрома сильно сорбируются
стенками ампулы и плохо удаляются при последующем про-
мывании. Промытые ампулы высушивают в герметичной си-
стеме при нагревании.
Следует иметь в виду, что пробы твердых веществ после
облучения часто можно подвергнуть травлению, в результате
чего поверхностные загрязнения удаляются. Однако для жид-
костей такая процедура исключена, и поэтому требуется осо-
бая осторожность при их отборе. При анализе жидкостей на
содержание малых количеств элементов встречается еще одна
трудность, связанная с адсорбцией определяемых элементов на
стенках ампулы. Адсорбция может привести к частичной или
даже полной потере определяемого элемента.
Оригинальный способ облучения жидких или гелеобразных
биологических проб в замороженном состоянии предложен Бру-
не и Вепзелом [55]. Этот прием позволил избежать основных
ограничений, связанных с облучением жидких проб или их
предварительной обработкой. Предложенная методика сводится
к быстрому замораживанию пробы в кварцевой или полиэтиле-
новой ампуле с помощью жидкого азота. Ампулы затем за-
кладывают в специальное термостатирующее устройство, за-
полняемое сухим льдом (/ = —78° С). Устройство вводится в
канал реактора, где происходит активация пробы. Показано,
что при плотности потока тепловых нейтронов 2•1012 ней-
трон; (см-- сек) длительность облучения с таким устройством
может достигать 15 ч. При этом температура внутри пробы
не поднимается выше —40° С. После облучения поверхностный
слой пробы стаивается и удаляется.
Развитием этого способа облучения является заморажива-
ние небольшого объема анализируемого раствора на неболь-
47
шой полоске из чистого алюминия, уложенной на кусочек су-
хого льда. Раствор на полоску наносится с помощью микропи-
петки. После замораживания полоска сворачивается и поме-
щается в контейнер, охлаждаемый сухим льдом или погружае-
мый в гелиевый криостат.
В некоторых случаях возникает необходимость проведения
перед облучением дополнительных операций. Для твердых тел
наиболее часто встречающаяся операция — измельчение пробы.
Порошкообразная проба быстрее разлагается, если приме-
няется радиохимический вариант, и дает лучшую воспроизво-
димость геометрических условий при инструментальном ана-
лизе. Иногда измельчение большого исходного образца необ-
ходимо для отбора средней пробы. Порошкообразная проба
требуется также при анализе сильных поглотителей нейтронов,
чтобы можно было использовать разбавление наполнителем,
слабо поглощающим нейтроны.
Для жидкостей чаще всего применяют упаривание, которое
позволяет уменьшить объем анализируемого раствора или да-
же полностью удалить жидкость. При анализе биологических
объектов используют высушивание и озоление. В ряде случаев
для концентрирования определяемых элементов прибегают к.
химическим методам — ионному обмену, экстракции, соосаж-
дению и др.
Иногда необходимость в предварительной обработке выте-
кает из специфики активационного анализа. Например, опре-
деление малых количеств редкоземельных элементов в уране
невозможно из-за образования радиоизотопов этих элементов
при делении урана. Поэтому И. П. Алимарин и др. [56] приме-
нили методику предварительного удаления урана (около 1 г)
методом ионообменной хроматографии.
Е. М. Лобанов и др. [57] удаляли возгонкой основную массу
мышьяка, который мешает инструментальному определению
золота. Для этого навеску пробы (около 20 мг) помещали в
отверстие угольного стержня и прокаливали 20 сек в пламени
вольтовой дуги. При такой обработке удалялось 99 процен-
тов As.
Предварительное выделение магния из биологических проб
в виде бромсодержащего внутрикомплексного соединения было
использовано Смазером и др. [58] для повышения чувствитель-
ности определения. После разложения пробы из полученного
раствора экстрагировали магний в виде комплекса с 5,7-ди-
бром-8-оксихинолином. Отделение комплекса от избытка ре-
агента осуществляли с помощью бумажной хроматографии.
Пятно с комплексом магния вырезали и облучали тепловыми
нейтронами. После облучения определяли количество брома
по-методу эталонов, а зная стехиометрический состав комплекса,
рассчитывали количество магния. Поскольку бром имеет более
благоприятные активационные характеристики (сечение акти-
48
вации и период полураспада), повышается чувствительность
определения магния и таким образом облегчается выполнение
анализа.
Однако любые дополнительные операции, выполняемые с
пробой перед облучением, увеличивают опасность загрязнений
и сводят на нет одно из преимуществ активационного анализа.
При проведении таких операций, как упаривание, высушивание
и сожжение, имеется опасность потерь летучих компонентов.
Вообще говоря, активационный анализ не предъявляет ка-
ких-либо специфических требований к операциям по предвари-
тельной подготовке и концентрированию по сравнению с дру-
гими методами анализа малых концентраций элементов. По-
этому здесь можно опустить подробное описание приемов ра-
боты по подготовке пробы, концентрированию, предотвраще-
нию загрязнений и потерь летучих компонентов и т. д., сослав-
шись на соответствующие источники [17, 54, 59, 60].
Эталоны
Очень важная стадия для большинства нейтроноактиваци-
онных определений — приготовление эталонов определяемых
элементов. Обычная процедура состоит в растворении извест-
ного количества металла или чистой соли с последующим раз-
бавлением до необходимой концентрации.
Исходные вещества должны быть марки ч. д. а. или х. ч.
Независимо от этого желательно проверить небольшое количе-
ство каждого используемого вещества предварительной акти-
вацией на присутствие посторонних элементов [61]. Для раство-
рения и последующего разбавления применяют неорганические
кислоты и воду, подвергнутые специальной очистке [54].
Обычно готовят растворы первичных эталонов, которые
имеют относительно высокую концентрацию элемента (более
100 мкг/мл). Эталонные растворы легко гидролизующихся
элементов подкисляют в необходимой степени или вводят ком-
плексообразователи. Концентрацию основного компонента в
растворе рассчитывают на основе исходных данных и получив-
шихся объемов, но лучше провести независимое определение
каким-либо подходящим, достаточно точным методом.
Первичные эталонные растворы используют в течение дли-
тельных сроков и поэтому хранят в герметичных условиях в
бутылях из стекла, полиэтилена или тефлона, периодически
проверяя концентрацию. Например, показано [62], что при кон-
центрации элемента 10—25 мкг/мл длительное хранение в те-
чение двух лет в темноте в стеклянной бутыли с притертой
пробкой не привело к какому-либо заметному изменению кон-
центрации 14 исследованных элементов.
Для облучения эталоны могут быть приготовлены индиви-
дуально для каждого элемента или в виде группового эталона.
49
В последнем случае смешивают первичные эталонные раство-
ры с таким расчетом, чтобы получить заданные концентрации
элементов [61, 63].
Дальнейшая процедура состоит либо в разбавлении эталон-
ного раствора до меньшей концентрации и отборе аликвоты,
либо во взятии небольшого объема раствора с помощью микро-
пипетки. Количество элемента в эталоне удобнее брать на
два-три порядка выше расчетного предела обнаружения в дан-
ных условиях, что обычно соответствует 10~3—10-9 г. Большие
количества элементов не следует брать из-за опасности само-
экранирования активирующего излучения и слишком высокого
уровня наведенной активности. Растворы эталонов отбирают в
отдельные ампулы из кварца или полиэтилена, которые пред-
варительно тщательно промывают. Эталоны можно облучать
либо непосредственно в растворе, что бывает редко, либо после
высушивания. Возможен другой способ приготовления этало-
нов, когда небольшое количество эталонного раствора перено-
сят на полоску из алюминия или фильтровальной бумаги. По-
сле высушивания полоску с эталоном запаивают в ампулу или
сверху и снизу обкладывают полосками из алюминиевой фоль-
ги и сворачивают в трубочку. Очевидно, методика с высушива-
нием пригодна только для нелетучих компонентов. Например,
при такой операции отмечена [62] потеря ртути.
Возможен ряд других способов получения эталонных пре-
паратов. В качестве эталона можно использовать вещество
близкого состава к исследуемой пробе, в котором содержание
определяемого элемента известно. Однако такие случаи крайне
редки. Иногда эталон в подходящей форме добавляют к от-
дельной части исследуемой пробы, об этом методе речь пойдет
ниже. Эталон можно готовить и путем нанесения определенно-
го количества эталонного раствора на инертный носитель
(А120з, MgO и т. д.). Тогда он может иметь тот же объем, что
и проба.
Облучение
В большинстве случаев облучение потоком тепловых ней-
тронов проводят в цилиндрических устройствах (каналах).
Несколько таких каналов (чаще всего вертикальных) распола-
гаются в различных участках активной зоны источника ней-
тронов. В соответствии с расположением каналов меняется и
плотность потока нейтронов в канале. Диаметр канала нахо-
дится в пределах 30—70 мм, хотя в специальных случаях он
может выходить за эти пределы.
Для облучения пробы и эталоны укладываются в контейнер,
который загружается в канал и извлекается из него с помощью
специального механического устройства. Контейнеры для облу-
чения в реакторах изготовляют из чистого алюминия. Размеры
50
активной зоны реакторов позволяют в одном канале одновре-
менно облучать несколько контейнеров, причем в один контейнер
в зависимости от размеров может вместиться до нескольких де-
сятков проб. Каналы с механической загрузкой и выгрузкой
используются преимущественно для длительных облучений (от
часов до нескольких недель).
Для коротких облучений служат каналы, которые оборуду-
ются пневмотранспортным устройством (пневмопочтой) [64].
В пневмопочте проба и эталон (монитор) укладываются в чел-
нок, который перемещается в зону облучения и обратно с по-
мощью давления или разрежения. Скорость перемещения со-
ставляет 10—40 м/сек, поэтому после облучения проба за очень
короткое время может быть доставлена к месту дальнейшей
обработки или к измерительному устройству. Обычно в ка-
нале с пневмопочтой одновременно облучается только одна
проба.
Пробы и эталоны, подготовленные для облучений, уклады-
вают в алюминиевый контейнер с таким расчетом, чтобы по
возможности исключить погрешность за счет градиента потока
нейтронов. Контейнер вводят в канал источника нейтронов и
облучают в условиях, наиболее оптимальных для решения по-
ставленной аналитической задачи. Наиболее простой случай —
определение с предельной чувствительностью одного элемента
в матрице, обладающей низким сечением поглощения тепловых
нейтронов. Поскольку плотность потока тепловых нейтронов за-
висит от параметров имеющегося в наличии источника, остается
только определить длительность облучения.
Как уже отмечалось, для достижения максимальной актив-
ности при заданной плотности потока нейтронов требуется об-
лучение в течение 5—10 периодов полураспада аналитического
радиоизотопа. Однако иногда приходится ограничиваться более
кратковременным облучением, что может обусловливаться ре-
жимом работы источника нейтронов, слишком большим перио-
дом полураспада радиоизотопа, сильной активацией пробы и
другими причинами.
Многие элементы при облучении дают два или более радио-
изотопа, имеющих различные ядерные характеристики, в част-
ности период полураспада. Выбор того или иного радиоизотопа
в качестве основы для определения зависит от нескольких фак-
торов. Здесь же будут только сопоставлены особенности анализа
по короткоживущим и достаточно долгоживущим радиоизо-
топам.
Применение короткоживущих изотопов открывает для акти-
вационного анализа интересные возможности [65, 66]. В некото-
рых случаях из-за благоприятного сочетания ядерных характе-
ристик определение по короткоживущему радиоизотопу дает
более высокую чувствительность, а для отдельных элементов
при облучении тепловыми нейтронами это единственный способ
51
определения, ибо активация таких элементов приводит к обра-
зованию только короткоживущих изотопов.
С помощью короткоживущих изотопов удается создавать ис-
ключительно экспрессные методы анализа, так как длительность
облучения до насыщения является короткой, а воемя, затрачи-
ваемое на промежуточные операции и измерение, по необходи-
мости должно быть мало. Непродолжительное облучение часто
позволяет избежать заметной активации основы, что упрощает
анализ.
Малый период полураспада дает возможность сравнительно
просто и быстро идентифицировать радиоизотопы по кривой рас-
пада. Иногда короткоживущий изотоп обладает излучением, бо-
лее благоприятным для измерения. При инструментальном ана-
лизе имеется возможность повторной активации пробы после
распада короткоживущих изотопов, что позволяет увеличить
точность определений.
Однако применение короткоживущих изотопов имеет и свои
ограничения, которые в основном связаны с быстрым уменьше-
нием их активности. Очевидно, что работа с короткоживущими
изотопами должна проводиться непосредственно у источника
излучения, который оборудован системой для быстрой транспор-
тировки облученных проб. Все вспомогательные операции, в том
числе и химическое разделение, должны быть быстрыми. Осо-
бенность использования короткоживущих изотопов заключается
также в ограничении числа элементов, одновременно определяе-
мых из одной навески.
Средне- и долгоживущие изотопы позволяют проводить ана-
лиз в более спокойной обстановке, часто в лабораториях, уда-
ленных от реактора, и одновременно определять значительное
число элементов. Длительность облучения достаточно велика и
определяется стремлением достигнуть желаемой чувствитель-
ности по наиболее долгоживущим изотопам, но в то же время
облучение более одной недели используют очень редко.
Выдерживание пробы перед обработкой или измерением
приводит к распаду мешающих короткоживущих изотопов, что
особенно важно, когда активация матрицы и мешающих ком-
понентов дает короткоживущие продукты. Иногда для получе-
ния данных о возможно большем числе элементов проводят
анализ параллельных проб анализируемого материала по ко-
ротко-, средне- и долгоживущим изотопам. Соответственно
выполняют несколько облучений разной длительности в соче-
тании с определенным временем выдерживания пробы перед
измерением.
Иногда к подготовке пробы и условиям облучения предъяв-
ляются и некоторые специфические требования, которые станут
ясными из дальнейшего текста. Здесь же уместно отметить
два дополнительных момента, связанных с облучением в
реакторе.
52
При работе реактора выделяется тепло, и поэтому темпера-
тура в активной зоне повышена. В исследовательских реакто-
рах температура в активной зоне обычно невелика и часто не
превышает 50° С. Облучаемые пробы должны выдерживать
длительное пребывание в условиях повышенной температуры.
Особенно это относится к жидкостям, которые, как правило,
облучают запаянными в кварцевые ампулы. Чтобы в ампуле
под действием температуры не могло развиться высокое дав-
ление, способное разорвать ее, ампулу заполняют жидкостью
менее чем на 1/3 объема. Запаянную ампулу испытывают на
устойчивость к давлению, нагревая ее в течение 24 ч при тем-
пературе 100° С.
При облучении в реакторе пробы подвергаются также зна-
чительному радиационному воздействию со стороны интенсив-
ных потоков у-квантов и нейтронов. Хотя в целом радиацион-
ное изменение пробы не влияет на результаты активационного
анализа, имеются два процесса, которые могут вызвать не-
которые затруднения. Под воздействием облучения многие
вещества разлагаются (иногда с выделением газообразных про-
дуктов). Если при этом возникнет большое давление, то это
может вызвать взрыв ампулы во время облучения или при ее
вскрытии. В таких случаях, если возможно, пробы лучше облу-
чать в открытой ампуле.
Другая трудность связана с тем, что при облучении неко-
торые вещества, главным образом органические, полимеризу-
ются, превращаясь в соединения, которые плохо разлагаются,
если необходимо применить радиохимическую обработку. Что-
бы обойти это затруднение, можно применить облучение мень-
шей интегральной дозой или провести предварительную подго-
товку пробы.
Анализ облученной пробы
В результате облучения в анализируемой пробе возникает
целый ряд радиоизотопов, которые получаются из изотопов
элементов, входящих в состав пробы либо в качестве примесей,
либо макрокомпонентов. Значит, дальнейшая задача состоит в
том, чтобы измерить интенсивность излучения нужного радио-
изотопа на фоне излучения других радиоизотопов, присутству-
ющих в пробе. Эта задача может быть решена либо с помощью
средств и методов ядерной физики, либо путем применения
химического выделения соответствующих элементов.
Первый способ — основа инструментального варианта акти-
вационного анализа. Для получения необходимой избиратель-
ности определения в инструментальном варианте широко при-
меняют вариацию условий облучения — тип активирующего
излучения, его энергию и т. д. и особенности схем распада
определяемых радиоизотопов — вид и энергию излучения, пе-
риод полураспада и т. д.
53
Поскольку с помощью физических средств при благоприят-
ных условиях радиоизотопы можно идентифицировать и коли-
чественно измерить непосредственно в облученной пробе, то
нет необходимости в какой-либо обработке пробы между об-
лучением и измерением. При быстрой доставке пробы на изме-
рение получаются исключительно экспрессные методы анализа,
часто использующие радиоизотопы с периодом полураспада
всего в несколько секунд. Другие достоинства инструменталь-
ного варианта — малая трудоемкость и высокая экономичность
анализа. В сочетании с современной вычислительной техникой
инструментальные методы образуют полностью автоматизиро-
ванные системы для активационного анализа [45].
Однако в практическом применении инструментального ме-
тода имеются и определенные трудности, связанные с ограни-
ченной разрешающей способностью используемы?: приборов,
сложностью обработки результатов измерений и невозможно-
стью применить этот метод к определению малых концентра-
ций элементов в объектах, которые сильно активируются в
процессе облучения. Особенности этого метода и его примене-
ние подробно рассматриваются в гл. 7 и 8.
Что касается радиохимического варианта, то его можно
применять для определения большей части элементов периоди-
ческой системы независимо от основы пробы. Метод в целом
более чувствителен и точен, чем инструментальный, и позво-
ляет определять большое число элементов из одной навески.
Недостатки метода — трудоемкость и длительность. По этой
причине в радиохимическом варианте находят применение в
основном средне- и долгоживущие радиоизотопы
д>30 мин). Лишь экспрессные методики химического разделе-
ния позволяют использовать короткоживущие изотопы (7'i/27>
>2 мин).
В результате облучения анализируемая проба становится
радиоактивной, а следовательно, и источником ионизирующего
излучения, которое представляет определенную радиационную
опасность. Следует заметить, что при активационном анализе
интенсивность полного излучения пробы не всегда одинакова
и зависит от условий облучения. Так, мощность экспозиционной
дозы у-излучения пробы сравнительно мала после облучения
большинства материалов потоками тепловых нейтронов плот-
ностью менее 1011 нейтрон!(см2-сек). В этом случае часто бы-
вает достаточно применить простую защиту в виде экранов
из свинца и некоторые несложные методы работы.
При плотности потоков нейтронов выше 1013 нейтрон!(см2Х
Хсек) мощность экспозиционной дозы облученной пробы за-
висит от ее состава. При анализе материалов, дающих при об-
лучении короткоживущие радиоизотопы, пробу можно выдер-
жать до распада высокоактивной основы, прежде чем начинать
какую-либо обработку. Не представляет особой опасности ра-
54
бота с образцами, основа которых активируется слабо (Si,
SiO2, Be, С и др.). При работе с сильно активирующимися ма-
териалами, дающими при облучении достаточно долгоживу-
щие радиоактивные изотопы с жестким у-излучением (Ge,
GaAs, Sbin и др.), радиационная опасность велика и требуется
провести ряд мероприятий по обеспечению безопасных условий
работы, а это усложняет анализ. Поскольку в некоторых слу-
чаях образующаяся активность проб может составлять десятки
кюри, то ручная обработка их исключается. Поэтому на пер-
вых стадиях анализа приходится работать в специальных за-
щитных боксах, снабженных манипуляторами [67].
Вполне естественно, что эти первые стадии должны преду-
сматривать отделение определяемых элементов от высокоактив-
ных компонентов, после удаления которых дальнейшая работа
может проводиться уже с применением простых защитных
средств. Для удаления высокоактивных компонентов наиболее
подходят химические методы, которые требуют минимума руч-
ных операций и являются в то же время наиболее избиратель-
ными.
Последовательность операций, выполняемых после облуче-
ния, примерно следующая. Ампулы извлекают из алюминиевого
контейнера и вскрывают. Первыми, как правило, поступают в
обработку ампулы с пробами. При анализе твердых проб, об-
лученных в виде сплошного куска или крупнокристаллического
порошка, первой операцией обычно является поверхностное
протравливание. Тонкоизмельченные пробы травлению не под-
вергают. Обработка пробы реагентами, растворяющими тонкий
поверхностный слой, позволяет удалять загрязнения, сорбиро-
ванные на нем во время отбора и подготовки к облучению.
Однако такая простая обработка не всегда эффективна. Так,
травление соляной кислотой поверхности германия для удале-
ния меди не дало желаемого эффекта даже при растворении
до 20% исходной массы пробы [68]. Некоторые элементы силь-
но цементируются на свежей поверхности германия, поэтому
лишь применение специальной методики травления до и после
облучения позволило избавиться от поверхностных загрязнений
медью, золотом и серебром и получить воспроизводимые ре-
зультаты анализов.
Полезными методами удаления поверхностных загрязнений
после облучения могут оказаться механическое удаление тон-
кого слоя или сочетание травления с полировкой. Хороший
эффект может дать введение в раствор, используемый для
травления, удерживающих носителей определяемых элементов.
После травления проба направляется либо на инструмен-
тальный анализ, либо на радиохимическую обработку. В по-
следнем случае ход анализа состоит из ряда стадий. Предва-
рительная стадия включает в себя перевод пробы в раствор,
введение носителей, контроль валентного состояния. Основная
55
стадия предусматривает выделение определяемых элементов в
отдельные фракции с использованием различных химических
методов. Вспомогательная стадия призвана обеспечить высо-
кую радиохимическую чистоту выделенного препарата путем
проведения дополнительных химических операций. На конт-
рольной стадии с помощью физических средств и методов про-
водится идентификация радиоизотопов и делается заключение
о радиохимической чистоте препарата. Заключительная стадия
состоит в определении химического выхода и измерении актив-
ности аналитического радиоизотопа. Более подробно радиохи-
мический метод рассмотрен далее, в гл. 9 и 10.
Затем эталоны вымывают из ампул, в которых они облуча-
лись, с помощью реагентов с введенными в них носителями
соответствующих элементов. Если эталоны были нанесены на
бумажную или алюминиевую полоску, то последние также
переводят в раствор, обрабатывая кислотами. Если активность
эталонного раствора велика, то его разбавляют до определен-
ного объема и отбирают аликвотную часть.
В дальнейшем раствор эталона можно пропустить через ту
же химическую обработку, какая применяется для фракций,
выделяемых из проб, но чаще эталон подвергают только не-
большой радиохимической очистке. Иногда после облучения
эталоны измеряют без какой-либо предварительной подготовки.
Однако в этом случае следует учесть различия в геометриче-
ских условиях при измерении и убедиться в отсутствии помех
от примесей в подложке или материале ампулы.
Получение конечных результатов
Активности препаратов, выделенных радиохимическим ме-
тодом из проб и эталонов, измеряют на установках для регист-
рации ионизирующего излучения. Хотя для этой цели можно
применять различные методы, наибольшее распространение
получило измерение у-излучения с помощью сцинтилляцион-
ных счетчиков и p-излучения с помощью простых торцовых
счетчиков.
Вследствие большой проникающей способности у-излучения
можно измерять активность как твердых, так и жидких проб.
Следовательно, измерения можно проводить сразу же после
выделения препарата в виде осадка или в растворе. Важно
только, чтобы строго соблюдались геометрические условия при
измерении эталона и препарата. Это означает, что в случае
жидких препаратов и.х необходимо перенести в стандартные
кюветы, которые заполняют раствором до одинакового уровня;
твердые препараты должны иметь определенные объемы и
площадь. Состав раствора или твердого осадка обычно не
влияет на результаты измерений, если у-излучение достаточно
56
жесткое. Это может быть существенным лишь для мягких у-пз-
лучателей.
Объемы жидких препаратов не следует делать слишком
большими, так как это ухудшает геометрические условия изме-
рения и уменьшает эффективность регистрации. Для получения
максимальной чувствительности лучше пользоваться сцинтил-
ляционными детекторами с «колодцем». При таком методе из-
мерения препарат в стандартной упаковке вводят в отверстие
(колодец), проделанное в сцинтилляционном кристалле. В ре-
зультате обеспечивается геометрия, близкая к 4л. Однако даже
в этом случае для получения точных результатов необходима
стандартизация геометрических условий.
Более сложная подготовка требуется, если необходимо по-
лучить точные результаты при измерении 0-излучения, так как
при этом могут возникнуть погрешности за счет поглощения
и рассеяния 0-частиц в веществе источника, подложке и окру-
жающих конструкционных материалах. Поэтому точные изме-
рения по 0-излучению требуют перевода определяемого элемен-
та и эталона в одинаковую химическую форму при равенстве
массы вещества на мишенях. Геометрические условия измере-
ния должны быть строго одинаковы или предварительно про-
калиброваны.
Измеряемые препараты обычно наносят ровным слоем па
стандартные мишеньки. В частности, для этого удобны алю-
миниевые тарелочки, в углубление которых помешают препа-
рат. Для перенесения препарата на измерительную мишеньку
можно использовать взвесь осадка в какой-либо жидкости с
последующим высушиванием. Жидкость подбирают так, чтобы
при высушивании осадок располагался ровным слоем, который
бы при этом не растрескивался и не отслаивался.
Перед измерением препараты необходимо высушить. Соеди-
нение, выработанное для конечного определения, не должно
быть гигроскопичным. Для ускорения подготовки препарата
лучше всего переносить его на мишеньку с помощью легколе-
тучих жидкостей. Подготовка препарата значительно упро-
щается, если количество носителя мало (менее 1 мг). Умень-
шение количества носителя позволяет также уменьшить объе-
мы растворов, получающиеся в процессе разделения, и увели-
чить скорость разделения. Более подробное описание методики
измерения 0-активных препаратов дано в работе [43].
Анализ заканчивается расчетом количества определяемых
элементов в анализируемой пробе (см. § 2 гл. 3).
Глава
4
НЕЙТРОННЫЙ АКТИВАЦИОННЫЙ
АНАЛИЗ
§ 1. Основные виды взаимодействий нейтронов
с атомными ядрами
Как известно, нейтрон — элементарная частица, не имею-
щая электрического заряда. Нейтроны и протоны — составные
части ядра, в котором они прочно связаны ядерными силами.
Отсутствие электрического заряда исключает взаимодействие
нейтрона с электронными оболочками атомов и кулоновским
полем ядра. Поэтому для проникновения нейтрона в ядро нет
потенциального барьера и взаимодействие может произойти
даже при небольшой энергии нейтрона.
Взаимодействия нейтронов с ядрами элементов весьма об-
ширны и разнообразны, их характер зависит от энергии ней-
тронов и структуры ядра [69]. Согласно энергии нейтроны
разделяют на несколько групп, границы между которыми в
некоторой степени условны:
Холодные
Медленные:
тепловые
резонансные (надкад-
миевые)
Промежуточные
Быстрые
Сверхбыстрые
£„<0,005 эв
0,005 эв< Ег1< 0,4 эв
0,4 эв< Еп< 1000эв
1кэв< Еп< 500кэв
0,5 Л1эв< Еп"'. 50Мэв
Еп> 50Л1,?в
Холодные и сверхбыстрые нейтроны представляют только
специальный интерес и не находят применения в аналитических
целях.
Основные виды взаимодействий нейтронов проявляются при
столкновениях с ядрами. В результате столкновения нейтрон
может быть просто отклонен в поле ядерных сил или может
быть захвачен ядром с образованием составного ядра. В пер-
вом процессе, который называется упругим рассеянием, сум-
марная кинетическая энергия нейтрона и ядра в результате
взаимодействия не меняется и ядро остается в основном со-
стоянии. Упругое рассеяние играет очень важную, роль при
замедлении нейтронов.
Второй процесс, связанный с захватом нейтрона ядром,
более сложен и может привести к целому ряду ядерных пре-
58
вращений. Если ядром был поглощен нейтрон с кинетической
энергией то образующееся составное ядро оказывается в
возбужденном состоянии, причем энергия возбуждения Е'=
= £„ + ?,.зз, где егв — энергия связи нейтрона в составном ядре.
Энергия отдачи ядра мала, и ее обычно не принимают' во
внимание.
В зависимости от энергии возбуждения и свойств составного
ядра переход в более низкое энергетическое состояние может
совершаться различными путями. В соответствии с характером
распада составного ядра выделяют следующие процессы*:
а) радиационный захват (я, у);
б) расщепление с вылетом заряженных частиц (п, р) и
(я, а);
в) эмиссия нейтронов (я, 2л);
г) деление ядра (я, [);
д) неупругое рассеяние (я, я').
Вероятность образования составного ядра характеризуется
полным сечением, а вероятность осуществления какого-либо
определенного процесса — парциальным сечением. Очень часто
случается, что основной вклад в полное сечение при данной
энергии нейтронов дает только один из перечисленных процес-
сов. При изменении энергии нейтронов меняется и вклад от-
дельных процессов в полное сечение.
Полное сечение зависит от энергии нейтрона, причем ход
зависимости для большинства элементов имеет общие черты.
Наибольшие значения полного сечения наблюдаются для теп-
ловых нейтронов; с ростом энергии нейтронов оно уменьшается.
Для медленных нейтронов у большинства изотопов полное се-
чение меняется обратно пропорционально скорости нейтрона с.
Такой ход зависимости сечения получил название закона 1/о.
При дальнейшем увеличении энергии нейтронов полное сече-
ние продолжает уменьшаться, приближаясь по величине к гео-
метрическому сечению ядра.
Монотонный ход зависимости полного сечения от энергии
нейтронов во многих случаях нарушается при резонансном по-
глощении нейтронов ядрами, которое наблюдается при совпа-
дении энергии возбуждения с одним из энергетических уровней
составного ядра. В области резонанса сечения иногда дости-
гают очень высоких значений. Для примера на рис. 8 приве-
дено изменение сечения радиационного захвата медленных
нейтронов золотом.
Четко выраженные пики имеют место только при низкой
энергии нейтронов. По мере роста их энергии число резонанс-
ных пиков увеличивается, расстояние между ними умень-
шается, а ширина резонансных уровней возрастает. В резуль-
тате происходит слияние пиков в одну плавную кривую.
Процесс упругого рассеяния через составное ядро не рассматривается.
59
Радиационный захват. Он оказывается ведущим процессом
при взаимодействии медленных и отчасти промежуточных ней-
тронов с ядрами элементов. В области быстрых нейтронов
радиационный захват играет уже незначительную роль и его
сечение мало. Образовавшееся в этом процессе возбужденное
ядро переходит в основное состояние путем испускания одного
Рис. 8. Функция возбуждения реакции 197Au (п, у)198Au.
или нескольких у-квантов в течение примерно 10”14 сек. Каж-
дый изотоп при таком переходе испускает характерный и обыч-
но сложный спектр у-излучения [70].
В результате реакции^1 А(и, у)^+1 А возникает изотоп об-
лучаемого элемента. Хотя часто получающийся изотоп ста-
билен, имеется все же значительное число случаев, когда
радиационный захват приводит к возникновению радиоизо-
топа.
Практически у всех элементов периодической системы по
реакции (и, у) образуются радиоизотопы. Поскольку они, как
правило, являются нейтроноизбыточными, то претерпевают
[7 -распад. Имеется незначительное число случаев, когда полу-
чившийся радиоизотоп испытывает 0+-распад или А-захват.
Иногда при радиационном захвате образуются изсмеры.
Реакции (п, р) и (п, а). Поскольку для испускания заря-
женных частиц существует потенциальный барьер, эти реакции
имеют заметные сечения только при облучении быстрыми ней-
тронами. Правда, для некоторых легких элементов, когда по-
тенциальный барьер еще невелик, реакции (п, р) и (п, а)
возможны под действием медленных нейтронов. Эти реакции
не играют существенной роли в активационных определениях,
60
но в отдельных случаях могут служить источниками интенсив-
ных потоков заряженных частиц.
При облучении быстрыми нейтронами сечения реакций (п,
р) и (и, ct) имеют для легких и средних ядер более высокие
значения, чем для тяжелых, что связано с влиянием потенци-
ального барьера ядра. Так, при энергии нейтронов 14 Мэв
сечения этих реакций составляют десятые доли барна и посте-
пенно уменьшаются до тысячных долей барна для тяжелых
ядер.
По реакции А(п, р)^-\ & образуется изобар исходного
ядра, который почти всегда радиоактивен и, будучи нейтроно-
избыточным, претерпевает р~-распад, превращаясь снова в
исходное ядро. Продукты реакции ** А (и, а) ^~23 А также почти
всегда радиоактивны и испытывают р_-распад.
Реакция (п, 2п). Для освобождения из ядра дополнитель-
ного нейтрона требуются затраты энергии, поэтому реакции
этого типа всегда пороговые. Для большинства ядер порог
реакции (п, 2п) находится в области 6—12 Мэв. По реакции
z А(п, 2п) z ’ А образуется нейтронодефицитный изотоп исход-
ного элемента, который часто бывает радиоактивным и
обычно распадается путем позитронного распада. Для нейтро-
нов с энергией 14 Мэв сечение реакции (/?, 2п) возрастает с
увеличением атомного номера ядра примерно от 0,01 барн для
легких ядер до 1—2 барн для элементов с Z>50.
Деление ядер. Некоторые наиболее тяжелые ядра вследст-
вие возрастания кулоновского отталкивания протонов в ядре
оказываются энергетически неустойчивыми и способными к са-
мопроизвольному или происходящему под воздействием ядер-
ного облучения делению на два осколка. При облучении мед-
ленными нейтронами реакция (п, /) наблюдается только на
ядрах 235Н и 233U. Быстрые нейтроны вызывают также деление
23sU И 232П.
Образующиеся продукты деления являются изотопами мно-
гих элементов, находящихся в середине периодической систе-
мы примерно от Zn до Cd. Поскольку изотопы обычно содер-
жат избыток нейтронов, они радиоактивны и испускают Р~-ча-
стицы.
Неупругое рассеяние. В процессе (п, п') нейтрон отдает
часть своей энергии для возбуждения ядра. Положение пер-
вого возбужденного состояния зависит от атомного номера
ядра. У тяжелых ядер неупругое рассеяние наблюдается при
энергии нейтронов более 0,6 Мэв, а у легких — выше 1 Мэв.
Сечение неупругого рассеяния тоже зависит от атомного
номера ядра и энергии нейтронов. Оно возрастает при перехо-
де от легких ядер к тяжелым и с ростом энергии нейтронов.
При этом сечение неупругого рассеяния меняется не очень
сильно — в пределах 0,6—3 барн.
61
При переходе из возбужденного состояния в основное ис-
пускается у-квант с энергией, характерной для каждого ядра.
В очень небольшом числе случаев неупругое рассеяние при-
водит к образованию изомеров [71].
Следует отметить, что в реакциях (п, у), (п, р), (п, а) и
(п, [) в области медленных нейтронов происходит поглощение
нейтрона и испускание вместо него другой частицы, кванта или
быстрых нейтронов. Эти реакции выводят медленные нейтроны
из потока и поэтому их суммарное сечение объединяют под
названием сечение поглощения. При облучении некоторых проб
поглощение медленных нейтронов оказывается настолько зна-
чительным, что приводит к заметному изменению энергетиче-
ского спектра и плотности потока. Это создает некоторые за-
труднения при выполнении анализов (см. § 4 этой главы).
§ 2. Источники нейтронов
Основные типы и характеристики источников нейтронов.
Получение нейтронов в свободном состоянии в виде достаточно
интенсивных потоков возможно только в результате двух ос-
новных процессов: ядерных реакций и деления (спонтанного
или вынужденного). Имеется значительное число ядерных ре-
акций, в ходе которых освобождаются нейтроны, но все они
требуют источника первичного излучения с достаточно высокой
энергией. Таковыми могут быть некоторые радиоизотопы или
ускорители заряженных частиц.
Радиоизотопные источники преимущественно основываются
на ядерных реакциях типа (а, п) и (у, «). Из ускорителей
для получения нейтронов чаще других используются ускорители
дейтронов [реакция (d, «)]; протонов [реакция (р, п)1 и элек-
тронов, тормозное излучение которых позволяет осуществлять
реакцию (у, и). Последние две реакции требуют ускорения за-
ряженных частиц до энергий выше 1,5 Мэв, в то время как ре-
акция (d, п) имеет высокое сечение уже при энергии дейтронов
около 100 кэв.
Наиболее интенсивные потоки нейтронов возникают в ядер-
ных реакторах, в которых нейтроны освобождаются в ходе цеп-
ной реакции, основанной на процессе вынужденного деления
ядер некоторых тяжелых элементов. Как известно, в этом про-
цессе на каждый поглощенный первичный нейтрон возникает
2—3 вторичных нейтрона. При определенных условиях цепная
реакция деления становится самоподдержнваюшимся процессом,
который сопровождается выделением большого количества ней-
тронов и энергии.
Для аналитического применения основными параметрами ис-
точников нейтронов представляются плотность потока нейтронов
в зоне облучения и энергетический спектр нейтронов. В боль-
шинстве элементарных процессов возникают быстрые нейтроны,
62
поэтому для снижения их скорости до тепловой поток прихо-
дится пропускать через слой вещества, содержащий легкие
элементы (замедлитель). С точки зрения практической работы
важное значение имеют и такие характеристики источника, как
стабильность плотности потока и его энергетического спектра во
времени, величина градиента, возможность транспортировки,
безопасность эксплуатации л т. д. Немалую роль шрают и та-
кие факторы, как сложность обслуживания и доступность для
отдельных аналитических лабораторий.
Радиоизотопные источники. Это малоинтенсивные и сравни-
тельно доступные источники, в которых плотность потока ней-
тронов обычно не превышает 105 нейтрон/(см2-сек). Подробные
сведения об основных технических характеристиках радиоизо-
топных источников, а также наиболее важные данные по их
изготовлению и применению можно найти в работах [72—74].
В источниках, основанных на реакции (а, п), используются
радиоизотопы естественных радиоактивных семейств и некото-
рые искусственные элементы, преимущественно трансурановые
(табл. 3). Другим компонентом источника является бериллий,
Т а блица 3
Наиболее важные радиоизотопные источники и их основные параметры
Источник Реакция Л/2 Выход, нейтрон Максимальная энергия ней- тронов, Л1эв Средняя энергия ней- тронов, Мэв
сек-кюри
зюРо—Be a,n 138 дней 2,5-106 11,3 5,0
22-Ас—Be a,n 21,7 года 2,6-10’ 13 4,7
226Ra—Be a,n 1600 лет 1,7-10' 13 4,7
239Pu__ Be a,n 24 360 лет 2,310» 11 5,0
23spu—Be a,n 86,4 года 2,5-10» 11,3 5,0
244 Am—Be a,n 458 лет 2,7-106 11,5 5,0
244Cm—Be a,n 18,1 года 2,4-106 — —
i24Sb—Be Y Л 60,1 дня 1,6-10в — 0,024
252Cf Спонтанное 2,65 года 4,4-109 — 2,3
деление
на ядрах которого протекает реакция 9Ве (а, п)12С. В связи с
малым пробегом а-частиц в веществе такие источники обычно
приготавливают путем тщательного перемешивания порошкооб-
разных препаратов а-излучателя и бериллия. Приготовленную
таким образом смесь помещают в герметичную стеклянную или
металлическую ампулу, объем которой рассчитан на выделение
гелия. Выход нейтронов, а также в какой-то степени и энергети-
ческий спектр (а, п)-источников зависят от способа его приго-
товления. Недостаток некоторых радиоизотопных источников
(Ra—Be, Ac—Be) состоит в высоком уровне жесткого у-излуче-
ния, что несколько осложняет проблему защиты от излучения.
63
Энергетические спектры (а, п)-источников мало отличаются
друг от друга и обычно имеют сплошное распределение с мак-
симумом в области 4—6 Мэв. Основная масса нейтронов заклю-
чена в интервале 1—8 Мэв.
Источники нейтронов, основанные па реакции (у, я), исполь-
зуют значительно реже, чем (а, и)-источники. С одной стороны,
это связано с тем, что выходы фотонейтронных источников
малы из-за низкого сечения реакции (у, п). С. другой стороны,
реакция (у, п) является эндоэнергетической и может протекать
только в случае, если энергия у-квантов превышает энергию
связи нейтрона в ядре. Поэтому для фотонейтронных источников
необходимо использовать жесткие у-излучатели. Среди стабиль-
ных изотопов наименьшими значениями энергии связи нейтрона
отличаются 9Ве (1,67 Мэв) и 2Н (2,23 Мэв). Эти элементы обычно
используют для приготовления фотонейтронных источников.
Фотонейтронные источники получают, помещая у-активный
препарат внутрь цилиндра или шара из бериллия, или в рас-
твор тяжелой воды. Интенсивность фотонейтронного источника
определяется мощностью источника у-излучения и толщиной
слоя вещества мишени. Особенность фотонейтронных источников
состоит в том, что они в основном монохроматические и имеют
энергию нейтронов в области десятков и сотен килоэлектрон-
вольт.
В табл. 3 отмечен только 124Sb—Be-источник, так как его
применяют чаще, чем другие фотонейтронные источники. Одна-
ко короткий период полураспада и необходимость мощной
защиты от у-излучения — серьезные недостатки этого источ-
ника.
Как источник нейтронов 252Cf отличается весьма благоприят-
ными параметрами. Средний период полураспада, высокий вы-
ход, низкая интенсивность сопровождающего у-излучения и ма-
лые размеры активного вещества выделяют этот источник среди
других изотопных источников. Однако 252Cf пока относительно
дорог, и его получают в ограниченных количествах. Как следует
из изложенного выше, большинство радиоизотопных источников
непосредственно испускают быстрые или промежуточные нейт-
роны. Для получения тепловых нейтронов источник необходимо
поместить внутрь какого-либо замедлителя. При конструирова-
нии систем для активационного анализа в качестве замедлителя
обычно используют парафин и очень редко воду.
Быстрые нейтроны при прохождении через замедлитель те-
ряют энергию в результате упругих и неупругих столкновений
с ядрами замедлителя. Поэтому по мере продвижения потока
нейтронов в замедлителе быстро возрастает доля медленных
нейтронов и уменьшается доля быстрых. Следовательно, наи-
больший поток быстрых нейтронов находится в непосредствен-
ной близости от источника, а максимальный поток тепловых
нейтронов — на некотором оптимальном расстоянии от источни-
64
ка, которое зависит от типа замедлителя и начального энергети-
ческого спектра нейтронов. Например, для Ra—Ве-источника,
помещенного в воду, поток тепловых нейтронов имеет макси-
мальную плотность на расстоянии 4—5 см от источника.
Схема устройства, предназначенного для активационного
анализа с источником нейтронов небольшой интенсивности, при-
ведена на рис. 9. Ампулу с нейтронным источником помещают
в центре парафинового блока. Облучение смешанным потоком
Рис. 9. Схема устройства для облучений с
радиоизотопным источником нейтронов:
/ — защита; 2 — кадмиевый канал; 3 — кассета с про-
бой для облучения смешанным потоком нейтронов;
4 — канал для облучения тепловыми нейтронами:
5 — свинцовый диск; 6 — источник нейтронов; 7 — па-
рафиновый блок.
быстрых и медленных нейтронов проводят в центральном ка-
нале. На некотором расстоянии от центрального канала по ок-
ружности располагают каналы для облучения тепловыми нейт-
ронами. Иногда в системе предусматривают специальный канал,
стенки которого выложены кадмием (Cd-канал). Этот канал
располагают как можно ближе к источнику. Поскольку кадмий
поглощает все тепловые нейтроны, активация элементов проис-
ходит под действием нейтронов с энергией выше 0,4 эв.
Недостатком рассмотренной системы для облучения яв-
ляется большой градиент потока нейтронов в каналах. Поэтому
для точных определений необходимы эталоны, имеющие те же
состав и объем, что и анализируемые пробы. С помощью не-
скольких радиоизотопных источников, располагаемых опреде-
ленным образом вокруг центрального канала, можно получить
систему с более однородным потоком нейтронов. Плотность по-
тока тепловых нейтронов в целом довольно низка и в некоторой
степени зависит от устройства системы для облучений. Ориен-
тировочно можно указать, что получающаяся плотность обычно
на 2—3 порядка меньше интенсивности источника. При облуче-
3 Р. А Кузнецов
65
ниях быстрыми нейтронами можно получить более высокие
значения плотности потока, так как пробу можно поместить
ближе к источнику, но градиент при этом возрастает.
Изотопные источники находят некоторое аналитическое при-
менение, однако из-за низкой плотности потоков нейтронов ме-
тоды определения с их помощью имеют низкую чувствитель-
ность (более 0,1%) и ограничиваются элементами с наиболее
высокими сечениями активации. По этой причине радиоизотоп-
ные источники обычно применяют для экспрессной инструмен-
тальной оценки содержания макроколичеств благоприятных
элементов.
Особый интерес вызывает применение изотопных источников
для решения некоторых геологических задач [75]. Подобные ис-
следования всегда требуют проведения массовых анализов проб
с достаточной точностью и по возможности наиболее быстрыми
методами. Следует также отметить использование изотопных
источников для каротажных исследований скважин.
Повышение интенсивности изотопных источников представ-
ляет большой интерес, и в этом направлении достигнуты неко-
торые успехи. Так, облучение в реакторах большой мощности
позволяет получать препараты 124Sb с высокой удельной актив-
ностью и большой суммарной активностью. Собранное на основе
такого препарата устройство может обеспечить поток тепловых
нейтронов средней интенсивности [76].
Основу конструкции составляет диск из бериллия. В отвер-
стиях этого диска, расположенных по окружности относительно
центрального канала, помещают ампулы с радиоизотопом
124Sb. При общей активности 1000 кюри в центральном канале
объемом 35 сл3 получается поток тепловых нейтронов плот-
ностью до 1-Ю8 нейтрон) (см2 сек). В центральном канале плот-
ность потока меняется на ±1% в вертикальном направлении и
±4% по горизонтали. Это наиболее дешевый источник с такой
плотностью потока тепловых нейтронов. Конечно, такой источ-
ник требует громоздкой защиты из-за высокой интенсивности
у-излучения, но все же главный его недостаток состоит в отно-
сительно коротком периоде полураспада 124Sb, что требует по-
стоянных усилий на проведение повторной активации сурьмы.
Спад интенсивности составляет 1,2% за 24 ч. Развитие ядерной
энергетики и технологии привело к получению в значительных
количествах ряда трансурановых элементов, отдельные радио-
изотопы которых обладают весьма благоприятными параметра-
ми для приготовления радиоизотопных источников средней ин-
тенсивности. Так, приготовлен 244Ст— Be-источник, который
содержит 0,63 кюри [77]. Этот источник имеет интенсивность
нейтронного излучения 1,25-108 нейтрон/сек. Видимо, будут до-
ступны и более интенсивные источники, поскольку ожидается,
что производство 244Ст в США к 1980 г. достигнет около
10 кг/год.
СП
Намечаются хорошие перспективы для получения 252Cf в зна-
чительных количествах. Уже сейчас приготовляемые источники
содержат до 0,37 мг этого радиоизотопа [74], что позволяет по-
лучить источник с плотностью потока тепловых нейтронов
9,7-10б нейтрон (см2-сек).
Нейтронные генераторы. Низковольтные нейтронные генера-
торы портативны, относительно недороги и просты в обращении.
Поскольку нейтронный генератор во время работы не испускает
у-излучения, то создание защиты, обеспечивающей безопасные
условия для персонала, относительно несложно. К тому же в
выключенном состоянии генератор нейтронов полностью безо-
пасен, а это может оказаться большим достоинством, если по
условиям эксплуатации его часто приходится транспортировать
с места на место. Иногда важным достоинством нейтронных
генераторов оказывается их способность работать в импульсном
режиме. Поэтому нейтронные генераторы стали доступными от-
дельным аналитическим лабораториям и в значительной степени
позволили расширить области применения нейтронного актива-
ционного анализа.
Для получения нейтронов в этих устройствах используются
две экзоэнергетические реакции взаимодействия дейтерия с дей-
терием или тритием: 2H(d, «)3Не и 3H(d, /г)4Не. В этих реак-
циях образуются быстрые монохроматические нейтроны, энер-
гия которых зависит от энергии дейтронов и угла вылета нейт-
ронов по отношению к падающему пучку. При низкой энергии
дейтронов (£<;~0,1 Мэе) в прямом направлении по первой
реакции выделяются нейтроны с энергией 2,8 Мэв, а по второй
14,5 Мэв.
Сечение реакции 2H(d, п)3Не плавно увеличивается с ростом
энергии дейтронов и в области 1—3 Мэв имеет широкий макси-
мум, где величина сечения составляет 0,1 барн. Функция воз-
буждения реакции 3Н(<7, /?)4Не имеет резко выраженный резо-
нансный ход. Сечение достигает максимума (5 барн) при энер-
гии дейтронов 109 кэв и при этом более чем на два порядка
превышает сечение реакции 2H(d, л)3Не. Поэтому она находит
преимущественное использование в низковольтных генераторах
нейтронов.
Обычно в этих установках пользуются методом прямого ус-
корения ионов дейтерия в постоянном электрическом поле
(рис. 10). Высокое напряжение получают с помощью электро-
статических генераторов, каскадных генераторов и высоковольт-
ных трансформаторов. Поскольку напряжение, подаваемое на
ускорительную трубку, не превышает 200 кэв, то конструиро-
вание источников высокого напряжения несложно и их можно
изготовить весьма компактных размеров.
Ионы дейтерия получают в ионном источнике, в котором
молекулярный дейтерий, вводимый с помощью специальной
системы, подвергают ионизации. Образующиеся положительные
67
ионы дейтерия втягиваются электрическим полем в ускоритель-
ную трубку, где им сообщается необходимая энергия. Для по-
лучения равномерного электрического поля и для обеспечения
хорошей ^фокусировки ионного пучка ускорительные трубки со-
бирают из нескольких секций, каждая из которых состоит из
электрода специальной формы и керамического изолятора. На-
пряжение на электроды подается от высокоомного делителя.
Рис. 10. Схема устройства нейтронного генератора с заземленной мишенью:
1— источник высокого напряжения; 2 — пульт; 3— переходной трансформатор;
4 — источник ионов; 5 — конденсор; 6 — фокусирующие электроды; / — ускоритель-
ная трубка; <3— вакуумный насос; 9 — вентиль; 10 — квадрупольная линза; 11 — ди-
афрагма; 12 — танталовый клапан; 13— водяное охлаждение; 14 — мишень.
Ускоренные ионы дейтерия падают на мишень, содержащую
дейтерий или тритий. Чаще всего мишени изготовляют из ти-
тана или циркония, либо путем напаивания фольги, либо напы-
лением металлов на подложку из меди, никеля или другого
металла. Процесс изготовления мишени завершается обезгажи-
ванием при нагревании до 600—700° С в вакууме с последую-
щим охлаждением в атмосфере дейтерия или трития.
По конструктивному оформлению ускорительной трубки
нейтронные генераторы подразделяются на непрерывно откачи-
ваемые и отпаянные. Первые из них применяются в лаборатор-
ных условиях и могут работать в непрерывном и импульсном
режимах. Отпаянные нейтронные трубки дают потоки нейтронов,
меньшей интенсивности и чаще всего эксплуатируются в им-
пульсном режиме. Они особенно полезны для проведения акти-
вационного анализа в полевых условиях [78].
Нейтронные генераторы — источники нейтронов средней ин-
тенсивности. Выход нейтронов на 1 мкк ионов дейтерия с энер-
гией 100—200 кэв достигает 108 нейтрон/сек. Промышленные
образцы нейтронных генераторов устойчиво работают при ион-
ном токе в несколько сот микроампер, что обеспечивает интен-
сивность потока нейтронов 109—1010 нсйтрон]сек. Замедление
быстрых нейтронов дает потоки тепловых нейтронов с плот-
ностью 107—108 нейтрон](см1 сек). Предложены конструкции
нейтронных генераторов, работающих при полном рабочем токе
2—5 ма (интенсивность до 1012 нейтрон]сек).
68
Серьезный недостаток нейтронных генераторов — быстрое
уменьшение интенсивности, обусловленное потерей трития (дей-
терия) из-за локального разогрева и частичного распыления
вещества мишени. По данным работы [79], для свежей мишени
при токе 1 ма и напряжении 150 кэв наблюдается уменьшение
интенсивности потока нейтронов вдвое примерно за 115—
175 мин. Использование более
термостойких мишеней из эр-
бия, применение автонасыще-
ния, вращающихся мишеней
позволяют увеличить рабочий
период нейтронного генерато-
Рис. 11. Изменение плотности потока
быстрых нейтронов при удалении от
мишени:
1 — экспериментальная кривая; 2 — теоре-
тическая кривая для зависимости вида
I//?2 (R — расстояние от мишени).
Рис. 12. Изменение плотности потока
быстрых нейтронов в зависимости от
расстояния нейтронного генератора от
центра мишени:
1 — 4 мм; 2— 24 мм; 3 — 44 мм; 4—
64 мм; 5 — геометрические размеры ми-
шени.
ра. В усовершенствованных конструкциях нейтронных генера-
торов высокая интенсивность потока нейтронов (1010—1011 ней-
трон/сек) поддерживается постоянной в течение 100 ч и более.
Нейтронный генератор дает поток быстрых нейтронов со зна-
чительным градиентом. Величина последнего зависит от экспе-
риментальных условий: фокусировки и размеров пучка, рас-
стояния от мишени и других факторов [80]. Уменьшение плот-
ности потока нейтронов с расстоянием показано на рис. 11, а при
смещении от оси пучка на разных расстояниях от мишени — на
рис. 12. Для получения медленных нейтронов около мишени
генератора устанавливают систему с замедлителем.
С помощью нейтронных генераторов проводят активацион-
ные определения на быстрых и тепловых нейтронах. Как пра-
вило, аналитические методики основываются на короткоживу-
щих радиоизотопах с периодом полураспада от нескольких
секунд до нескольких часов. Поэтому нейтронные генераторы
обязательно оборудуются пневмопочтой.
69
Ускорители электронов в качестве источников нейтронов.
Пока что в роли источников нейтронов ускорители электронов
выступают эпизодически. По интенсивности нейтронных потоков
они не уступают нейтронным генераторам. Электронные уско-
рители могут работать длительное время без падения интенсив-
ности, однако энергетические затраты на производство нейтро-
нов много больше, чем у нейтронных генераторов. Энергетиче-
ское распределение получающихся нейтронов оказывается
сплошным, причем максимальная энергия нейтронов соответст-
вует разности между максимальной энергией тормозного излу-
чения и величиной порога используемой фотоядерной реакции.
Из этого обстоятельства вытекает принципиальная возможность
регулирования спектра нейтронов путем изменения энергии
ускорителя.
Рассмотрим имеющиеся в литературе данные по использо-
ванию ускорителей электронов в качестве источников нейтро-
нов. Так, электростатический ускоритель на энергию 3 Мэв при
токе электронов 3 ма способен создать плотность потока тепло-
вых нейтронов около 5-107 нейтрон!(см2 • сек) [81]. Мишенью
служит бериллий в форме куба с ребром 15 см.
Линейный ускоритель (£ТОрм=30 Мэв, /=145 мка) с ми-
шенью из тантала (9,76 г/см2) или вольфрама (56 г/см2) спо-
собен генерировать поток быстрых нейтронов (£„>2 Мэв)
плотностью около 1-1010 нейтрон/(см2 • сек) [82]. Распределение
нейтронов вокруг мишени изотропно, поэтому пробы можно
облучать только нейтронами, исключая активацию тормозным
излучением, которое узким пучком проходит через мишень в
прямом направлении.
Микротрон типа МР-30 (£ТОрм = 30 Мэв, 1=15 мка) способен
быть источником быстрых нейтронов интенсивностью 1012 нейт-
рон/сек и создавать поток тепловых нейтронов с плотностью
1010 нейтрон/(см2-сек) [83].
Ускорители ионов в качестве источников нейтронов. Сейчас
ведутся еще предварительные исследования возможностей раз-
личных ускорителей ионов как источников нейтронов для акти-
вационного анализа [84]. Для получения нейтронов в ядерных
реакциях с протонами, дейтронами и 3Не лучшие результаты
дает бериллиевая мишень. С помощью толстой бериллиевой
мишени выход нейтронов достигает следующих величин:
3Не(20 Мэв) — 1,3-109 нейтрон/(сек• мка); р (14 Мэв)—6Х
ХЮ10 нейтрон/(сек-мка); d (7,5 Мэе) — 1,4-1010 нейтрон/(секу.
Умка). Средний ток современных ускорителей составляет
100 мка, следовательно, можно превратить ускоритель ионов в
достаточно интенсивный источник нейтронов.
Угловое распределение потока нейтронов анизотропно, ос-
новная масса нейтронов движется в направлении пучка. Энер-
гетическое распределение сплошное, однако максимальная энер-
гия ограничена и зависит от вида и энергии ионов. В указанных
70
выше условиях она составляет для 3Не 26 Мэв, для р и d —
8 Мэв.
Широкое использование ускорителей ионов для нейтронного
активационного анализа, несомненно, будет зависеть от про-
гресса в конструировании простых в управлении, небольших по
габаритам и недорогих ускорителей.
Нейтронные размножители. Это подкритическая сборка, ко-
торая может увеличивать первичный поток нейтронов от изо-
топного источника. Примером может служить промышленная
конструкция нейтронного размножителя СО-1 [23]. В активную
зону СО-1, выполненную в виде цилиндра диаметром 240 мм и
высотой 280 мм, загружается двуокись урана (обогащенная по
235U), диспергированная в полиэтилене. Отражателем служат
полиэтилен и графит. Начальный поток нейтронов задается
Ро — Be-источником активностью 65 кюри.
Тепловая мощность размножителя составляет 0,5 вт, макси-
мальное значение плотности потока тепловых и быстрых нейт-
ронов в центре активной зоны равняется соответственно 2,5-107
и 7-Ю7 нейтрон! (см2-сек). Нейтронный размножитель оснащен
тремя вертикальными каналами диаметром 52 мм и одним го-
ризонтальным каналом диаметром 51 мм, оборудованным пнев-
мопочтой. Все экспериментальные каналы располагаются в зоне
графитового отражателя. Биологическая зашита размножителя
состоит из слоя свинца толщиной 118 мм, слоя парафина с
5%-ным содержанием карбида бора и слоя воды. Последние
два слоя имеют толщину по 288 мм. Вертикальные каналы
оборудованы специальным защитным устройством, позволяю-
щим перегружать пробы при работающем нейтронном размно-
жителе.
Ядерный реактор. В отличие от нейтронного размножителя
ядерный реактор — критическая система, в которой осуществ-
ляется самоподдерживающаяся, управляемая цепная реакция
деления ядер урана. Спектр нейтронов, выделяющихся в про-
цессе деления, заключен в широком энергетическом интервале
от небольших энергий вплоть до 25 Мэв. Средняя энергия нейт-
ронов деления равна примерно 2 Мэв, а наиболее вероятная
энергия — 0,72 Мэв. Доля нейтронов с энергией более 0,1 Мэв
составляет около 99% общего потока нейтронов деления; 66%
потока лежит между 0,5—3 Мэв. Выше 3 Мэв поток нейтронов
уменьшается почти экспоненциально с ростом энергии.
Реакторы в зависимости от энергии нейтронов, служащих
для поддержания цепной реакции, разделяются на три типа: на
быстрых, промежуточных и тепловых нейтронах. В первом типе
реактора энергетический спектр нейтронов в активной зоне бли-
зок к спектру нейтронов деления. В реакторах остальных двух
типов обязательно содержится определенное количество замед-
лителя, поэтому средняя энергия нейтронов в них смещена в
область более низких энергий по сравнению со спектром деле-
71
ния и они имеют максимальную интенсивность потока нейтро-
нов соответственно в промежуточной или тепловой области.
В пределах перечисленных основных типов разработано мно-
го различных конструкций реакторов, среди которых наиболь-
шее применение для аналити-
ческих целей получили иссле-
довательские реакторы на теп-
ловых нейтронах [85]. Изложе-
ние основ ядерной техники и
описание различных конструк-
ций реакторов можно найти в
специальной литературе [86,
87]. Здесь же рассмотрены
только те системы и парамет-
ры реакторов, которые наибо-
лее важны для их аналитиче-
ских применений. Параметры
реактора находятся в тесной
связи с его конструкцией, по-
этому далее дается описание
гетерогенных реакторов с
обычной водой в качестве за-
медлителя, преимущественно
используемых для аналитичес-
ких целей.
Активная зона реактора, в
которой протекает самопод-
держивающийся процесс деле-
ния, представляет собой си-
стему трубок, расположенных
определенным образом (чаще
всего по окружности) в кожу-
хе реактора. В каждую трубку
вводится несколько стержней,
содержащих уран (топливные
элементы). Общее количество
урана (обычно обогащенного
по 235U) должно быть несколь-
ко выше, чем необходимо для
достижения критичности. Уп-
равление работой реактора осуществляется с помощью стерж-
ней из материала, сильно поглощающего нейтроны. Схема одной
из простых конструкций реактора приведена на рис. 13.
В налаженном реакторе запуск в работу осуществляется за
несколько минут путем извлечения регулирующих стержней до
определенного уровня. С помощью стержней можно менять мощ-
ность реактора, предельное значение которой обусловлено его
конструкцией и возможностью отвода выделяющегося тепла.
Рис. 13. Схематичное устройство ре-
актора Triga:
1 — система -подъема проб; 2 — привод ре-
гулирующего стержня; 3 — система конт-
роля; 4— система центрального канала;
5 — центральный экспериментальный ка-
нал; 6 — регулирующий стержень; 7 — от-
ражатель; 8 — алюминиевый бак; 9—
ионная камера; 10 — система вращения
проб; И — трубка для транспортировки
радиоизотопов; 12 — пневмопочта.
72
В свою очередь, мощность реактора определяет предельную
плотность потока нейтронов в экспериментальных каналах, рас-
полагаемых в различных точках активной зоны реактора. Так,
в небольших маломощных ядерных реакторах плотность потока
нейтронов составляет 1010—1011 нейтрон) (см2-сек), а в наибо-
лее мощных современных исследовательских реакторах получают
потоки быстрых и тепловых нейтронов до 1015 — 5-1015 нейт-
рон/ (см2• сек) [88].
При работе в стационарном режиме ядерные процессы, про-
исходящие в реакторе, можно схематично представить такой
последовательностью. Ядра 235U в топливных элементах, погло-
щая тепловые нейтроны, испытывают деление с испусканием в
среднем 2,44 нейтрона. Поскольку эти нейтроны быстрые, для
поддержания цепной реакции они должны быть замедлены. По-
этому, покидая топливный элемент, они попадают в замедли-
тель (легкая или тяжелая вода, графит, бериллий), где посте-
пенно теряют энергию и приходят в тепловое равновесие со
средой. Диффундируя в активной зоне, часть из них снова по-
падает в один из топливных элементов, вызывая новое деление.
Согласно этой схеме, полный поток нейтронов в активной
зоне реактора состоит из трех основных групп: 1) быстрых
нейтронов, выделяющихся при делении; 2) резонансных и про-
межуточных нейтронов с энергией между 0,4 эв — 1 Мэв, полу-
чающихся в процессе замедления; 3) тепловых нейтронов. Коли-
чественное соотношение между потоками нейтронов этих трех
групп зависит от конструкции активной зоны реактора, положе-
ния канала для облучения, типа замедлителя и ряда других
факторов. Обычно реальный энергетический спектр нейтронов
меняется от реактора к реактору и даже от канала к каналу в
пределах одного реактора. Изменение потоков различных групп
нейтронов по активной зоне легководного реактора Merlin при-
ведено на рис. 14 [89].
Наибольший поток быстрых нейтронов наблюдается в топ-
ливных элементах в центре активной зоны, он быстро падает по
мере удаления от топливных элементов. С другой стороны, поток
тепловых нейтронов максимален в промежутках между топлив-
ными элементами. Пространственное изменение потока резо-
нансных нейтронов значительно меньше, чем для быстрых и
тепловых нейтронов.
Значения плотностей потоков основных энергетических групп
нейтронов в экспериментальных каналах, как это будет пока-
зано ниже, представляют большой интерес для практических
применений активационного анализа. Обычный способ оценки
спектрального состава потока нейтронов состоит в облучении в
исследуемом канале фольг из элементов с хорошо изученными
ядерными характеристиками и расчете плотности потока из из-
меренной наведенной активности [90]. Плотности потоков тепло-
вых и резонансных нейтронов определяют по продуктам реакции
73
(п, у), образующимся при облучении монитора в кадмиевом
фильтре и без него. В случае быстрых нейтронов для этих целей
применяют пороговые реакции. Используя несколько элементов
с различными порогами ядерных реакций, можно получить более
подробные сведения о спектре быстрых нейтронов в канале.
Поток нейтрона^, рол. еЛ. '
Расстояние, от центра активной зоны, си
Рис. 14. Распределение потоков нейтронов в ак- '
тивной зоне исследовательского реактора Merlin:
/—тепловые нейтроны (Х1.4’107 нейтронЦсм'1 • сек • вт)];
2 — промежуточные нейтроны [х2,4 • 10е нейтрон!(см2Х
Усек-кв)]; 3 — быстрые нейтроны [х 1,8 • 10г нейт-
ронЦсм2 сек • вт)]. В квадратных скобках указаны ко-
эффициенты для перехода к абсолютным значениям
плотностей потоков. Т — топливный элемент.
Для примера на рис. 15 приведен истинный дифференциаль-
ный спектр нейтронов реактора GTR с водой в качестве замед-
лителя [91]. Там же показано влияние различных фильтров на
спектр нейтронов. Дифференциальные измерения спектра нейт-
ронов довольно сложны, к тому же в аналитических исследова-
ниях в них нет необходимости, поэтому обычно прибегают к
оценке интегральных потоков нейтронов с энергией выше неко-
торого значения. В табл. 4 приведены плотности интегральных
Таблица 4
Потоки нейтронов в каналах реактора Triga
Кан ал Плотность потока, нейтрон !(смг-сек)
Тепловые Более 1,35 Мэв Более 6,1 Мэв
Канал для массовых облучений 1,8-1012 l,8-10ii 4,0-109
Каналы в активной зоне: кольцо D 4,9-1012 2,3-1012 6,2-101»
кольцо F 4,3-1012 7,5- Юч 1,9-101»
Канал на внешней стороне отражателя 6,8-104 5,1-10» 1,1-10“
74
потоков нейтронов в различных каналах реактора Triga [92].
Этот реактор имеет мощность 250 кет и дает максимальный по-
ток тепловых нейтронов 4,9-1012 нейтрон/(см2 сек).
Итак, в экспериментальных каналах внутри активной зоны
реактора полный поток нейтронов через облучаемую пробу со-
стоит из тепловых, резонансных (промежуточных) и быстрых
Рис. 15. Дифференциальный спектр нейтронов ре-
актора GTR:
/ — через фильтр из ,0В толщиной 1,1 г/см-: 2 — через
кадмиевый фильтр толщиной 0,5 мм; 3 — через фильтр
из естественного бора толщиной 243 мг/см:: '/ — нор-
мальный спектр нейтронов в реакторе.
нейтронов. В аналитической практике нередки ситуации, когда
по тем или иным причинам возникает необходимость в направ-
ленной трансформации нейтронного потока. Например, облуче-
ние в фильтре из сильного поглотителя тепловых нейтронов
(кадмия или бора) обеспечивает активацию пробы только резо-
нансными и быстрыми нейтронами. Значительно усилить поток
быстрых нейтронов можно с помощью уранового конвертера.
Анализируемую пробу помещают в камеру из 235U и затем ка-
меру вводят в канал реактора. Внутри камеры получается
поток быстрых нейтронов такой же величины, что и поток теп-
ловых нейтронов в месте облучения. Если камера достаточно
толстая, то уран поглощает все тепловые нейтроны и поток
внутри камеры состоит в основном из нейтронов спектра деле-
ния и промежуточных нейтронов.
75
С другой стороны, иногда требуется уменьшить влияние ин-
терферирующих реакций на быстрых нейтронах. Тогда канал
для облучения размещают в других зонах реактора — отража-
теле или специальной тепловой колонне. Последняя является
призмой из замедлителя (обычно графита), примыкающей к
активной зоне или отражателю. При диффузии по тепловой
колонне происходит постепенное замедление быстрых и проме-
жуточных нейтронов и смещение спектра в область тепловых
нейтронов. Однако получение более чистого потока тепловых
нейтронов связано со значительным падением плотности потока
вследствие утечки и поглощения нейтронов. Так, в отражателе
плотность потока составляет примерно 1/10 плотности потока в
центре активной зоны, а в тепловой колонне падение составляет
уже три порядка.
Как важную деталь некоторых реакторов можно отметить
ловушку нейтронов, которая представляет собой полость в цент-
ре активной зоны, заполненную слабо поглощающим нейтроны
замедлителем [93]. Ловушка нейтронов чаще всего имеет форму
шара диаметром 10—15 см, который заполняется водой. Плот-
ность потока тепловых нейтронов в ловушке оказывается в 3—4
раза выше, чем в активной зоне. В ловушке можно облучать
пробы ограниченных размеров, имеющих небольшое макроско-
пическое сечение поглощения (Мтпогл ~ 1 см2).
Как правило, реакторы работают в стационарном режиме,
их рабочий период продолжается 5 дней (с начала до конца
рабочей недели). Конечно, возможны и другие рабочие циклы в
зависимости от проводимых исследований. Стабильность плот-
ности потока в стационарном режиме высокая.
Отдельные конструкции реакторов допускают работу в им-
пульсном режиме. Такой реактор способен в одном импульсе
создать мощный интегральный поток нейтронов. Например, ре-
актор Triga в импульсе, длительность которого равна 15 мсек,
развивает интегральный поток 4,5-Ю14 нейтронам2 [85]. У оте-
чественного растворного реактора ИИН-З в центральном канале
интегральный поток нейтронов за импульс составляет 8Х
Х1014 нейтрон/см2 [23]. Облучение в импульсном потоке нейтро-
нов сокращает длительность облучения и создает преимущества
в активации очень короткоживущих радиоизотопов.
В заключение несколько слов о реакторе РГ-1 (ИВВ-3), спе-
циально спроектированном для аналитических применений [23].
Это гетерогенный реактор с горючим из двуокиси урана с
10%-ным обогащением по 235U. Активная зона набирается из
кассет и погружается в алюминиевый бассейн диаметром
1500 мм и глубиной 3500 мм. Отражателем служит графит, а
теплоносителем — вода с естественной циркуляцией. При работе
на полную мощность (5 кет) температура воды не превышает
45° С. Для обеспечения радиационной защиты реактор углублен
в землю и сверху накрыт чугунной плитой толщиной 460 мм.
76
Всего реактор имеет 11 каналов, из них два оснащены пнев-
мопочтой. Максимальная плотность потока тепловых нейтронов
достигает 2-Ю11 нейтрон/ (см2-сек). При введении системы при-
нудительного охлаждения мощность реактора может быть зна-
чительно увеличена [94].
§ 3. Методы нейтронного активационного анализа
Нейтронный активационный анализ распадается на три ме-
тода: активационный анализ на тепловых, резонансных и быст-
рых нейтронах. Такая классификация прежде всего обусловлена
большими различиями в характере взаимодействия основных
энергетических групп нейтронов с веществом и частично мето-
дическими особенностями аналитических определений, выпол-
няемых с их помощью. Аналитические возможности этих мето-
дов существенно различны, поэтому они преимущественно на-
ходят применение для решения разнородных аналитических
задач и, следовательно, в определенном отношении дополняют
Друг друга.
Следует обратить внимание также на специфические особен-
ности нейтронных потоков, получаемых с помощью широко
используемых источников: а) сплошное энергетическое распре-
деление, которое часто охватывает широкий энергетический ин-
тервал; исключение в этом отношении составляют потоки быст-
рых нейтронов от нейтронных генераторов; б) первичный спектр
нейтронов (обычно быстрых) фиксирован, т. е. не поддается
регулированию, и задается имеющимся источником; в) поток
нейтронов одной энергетической группы, как правило, сопровож-
дается потоками нейтронов других энергетических групп;
г) спектральное распределение активирующего нейтронного из-
лучения часто претерпевает заметное возмущение при взаимо-
действии нейтронов с веществом пробы и окружающими вспо-
могательными и конструкционными материалами.
Отмеченные особенности нейтронных потоков имеющихся
источников указывают на некоторые затруднения в проведении
избирательной активации и на потенциальные источники погреш-
ностей и поэтому должны тщательным образом учитываться
при выполнении анализов нейтронным активационным методом.
В отдельных случаях с помощью сравнительно простых средств
удается улучшить характеристики нейтронного потока (сформи-
ровать поток нейтронов с требуемым энергетическим спектром,
подавить нежелательную компоненту и т.д.). Это определенным
образом расширяет возможности метода, но не дает радикаль-
ного решения, которое состоит в необходимости иметь интен-
сивный источник моноэнергетических нейтронов с переменной
энергией.
Ядерные реакции под воздействием нейтронов вследствие по-
рогового характера и резонансного хода кривых возбуждения
предоставляют значительные возможности для проведения изби-
рательной активации элементов. Однако отсутствие подходя-
щего источника нейтронов не позволяет реализовать эти возмож-
ности в полной мере. Правда, экспериментальная ядерная фи-
зика располагает несколькими методами получения потоков
моноэнергетических нейтронов с переменной энергией [69]. Од-
нако необходимые для этого устройства весьма сложны, а полу-
чающиеся потоки нейтронов имеют низкую интенсивность. По-
этому применение их для аналитических целей весьма ограни-
ченно.
Активационный анализ на тепловых нейтронах
Облучение тепловыми нейтронами, несомненно, является
ведущим методом активационного анализа. Такое значение этот
метод приобрел из-за нескольких благоприятных факторов. Не-
сомненное достоинство активационного анализа на тепловых
нейтронах состоит в том, что при облучении большинства эле-
ментов протекает только одна ядерная реакция (га, у), в ре-
зультате которой образуется радиоизотоп исходного элемента.
Это ограничивает число радиоизотопов, образующихся при
облучении сложных объектов, а отсутствие реакций, связанных
с изменением заряда ядер, исключает взаимные помехи элемен-
тов. Большинство радиоизотопов испытывает 0-распад, сопро-
вождающийся у-излучением. При этом различие параметров
схем распада выше, чем у радиоизотопов, получающихся при
других методах активации.
По реакции (га, у) происходит образование радиоизотопов
у подавляющего числа элементов периодической системы, что
придает методу определенную универсальность. Кроме того, ана-
литические определения возможны не только по излучению ра-
диоизотопов, но и по мгновенному у-излучению радиационного
захвата, а это определенным образом расширяет возможности
метода.
Сечения реакции (га, у) часто имеют высокие значения, что,
в свою очередь, приводит к высокой чувствительности определе-
ния. В этом отношении активационный анализ на тепловых
нейтронах превосходит другие методы активационного анализа.
Дополнительным благоприятным фактором является нали-
чие целого набора источников нейтронов, которые перекрывают
широкий диапазон плотности потока тепловых нейтронов. Не-
которые источники доступны отдельным аналитическим лабора-
ториям и даже для эксплуатации в полевых условиях. К тому
же объем активной зоны источников нейтронов достаточно ве-
лик, что позволяет одновременно облучать большое число проб.
Это весьма важное обстоятельство при массовых анализах.
Между тем в практических ситуациях активационный анализ
на тепловых нейтронах часто сталкивается с трудностями и ог-
78
раничениями, значительная доля которых обусловлена несовер-
шенством источников нейтронов или измерительных устройств,
а определенная часть имеет принципиальный характер. Обсуж-
дению некоторых из этих проблем посвящен следующий па-
раграф.
Заслуживает внимания и вопрос о форме уравнения актива-
ции, поскольку приведенное ранее уравнение (2.23) справедливо
для моноэиергетического излучения, а реальный спектр тепло-
вых нейтронов охватывает широкую область энергий, следуя
распределению Максвелла. Так как сечение реакции (п, у) так-
же зависит от энергии нейтронов, то в уравнении активации
произведение Фоакт должно быть заменено интегралом
со
fO(E„)aaKT(En)dE, (4.1)
о
где Ф(£„)—энергетическая зависимость плотности потока
нейтронов; <гакт(^п)—функция возбуждения реакции. Приво-
димые в справочниках сечения активации 02200 соответствуют
наиболее вероятной энергии нейтронов (при нормальной темпе-
ратуре 0,025 эв, что соответствует скорости нейтронов
2200 м!сек).
Можно показать, что интеграл (4.1) для реакции, сечение
которой следует закону 1/ц, равен произведению Фт022оо, в кото-
ром Фт = нт022оо, где пт— общая плотность тепловых нейтронов.
Это означает, что по числу происходящих взаимодействий поток
нейтронов с распределением Максвелла равноценен моноэнерге-
тическому потоку нейтронов с энергией 0,025 эв, но с той же
общей плотностью нейтронов. Для тех немногих ядер, у которых
сечение реакции не подчиняется закону 1/ц, интеграл (4.1) за-
меняется произведением ФтОггоой', где g— коэффициент, учиты-
вающий степень этого отклонения [95].
Как было отмечено ранее, поток тепловых нейтронов, полу-
ченный замедлением быстрых нейтронов, обычно сопровож-
дается потоком резонансных нейтронов. Последние тоже дают
некоторый вклад в реакцию радиационного захвата, и поэтому
для расчета наведенной активности следует применять эффек-
тивное сечение активации:
°эФФ = ст22оо + • Ль (4.2)
. „ , гп(£)dE
где /р—резонансный интеграл, определяемый как /р = |.
о’ 1 ч :
Для определения отношения Фр/Фт достаточно измерить
кадмиевое отношение для какого-либо элемента (детектора) с
известными О2200 и /р. Кадмиевое отношение рассчитывают по
79
активации небольшого количества этого элемента при облуче-
нии в фильтре из кадмия (толщиной 0,5—1 мм) и без него. Оче-
видно, что в отсутствие кадмия активация элемента происходит
при участии тепловых и резонансных нейтронов, а с фильтром —
только при участии последних, так как фильтр из кадмия пол-
ностью поглощает все нейтроны с энергией менее 0,4 эв.
На основании облучения и последующего измерения актив-
ности имеем
Ар Фт<*2200 Ч~ Фр^р (4
4р Фр^р
где Lea—кадмиевое отношение; Лт и Лр — активность радио-
изотопа, обусловленная соответственно действием тепловых и
резонансных нейтронов.
Из уравнения (4.3) видно, что кадмиевое отношение зависит
от чувствительности детектора к тепловым 02200 и резонансным
/р нейтронам и поэтому оказывается неодинаковым у разных
детекторов.
Если кадмиевое отношение определено экспериментально, то
при известных 02200 и /р отношение плотностей потоков тепло-
вых и резонансных нейтронов можно рассчитать по уравнению
-^=-^(Ecd-l). (4.4)
Для подобных определений очень удобны детекторы, у кото-
рых сечение радиационного захвата подчиняется закону 1/и.
Соответствующий расчет показывает, что для таких детекторов
отношение 022ооДр=2, поэтому
—j-Oca-l). (4-5>
Кадмиевое отношение внутри активной зоны графитового или
тяжеловодного реактора для детекторов типа 1/у равно при-
мерно 33, что соответствует отношению Фт/Фр=16.
Очень часто кадмиевое отношение для используемых условий
облучения определяется по активации золота. Можно подсчи-
тать, что для отмеченных выше условий кадмиевое отношение
золота будет равно 2. Кадмиевое отношение для других детек-
торов можно выразить через кадмиевое отношение золота:
= 1 + (Еса - I) - — —, (4.6)
^рст22Э0
где один штрих в индексе относится к золоту, а два штриха — к
другому элементу.
80
Активационный анализ на резонансных нейтронах
Активационный анализ на резонансных нейтронах имеет
много общего с активационным анализом на тепловых нейтро-
нах, так как в обоих случаях ведущую роль играет реакция
радиационного захвата. Однако этому методу свойственны оп-
ределенные особенности, связанные главным образом со специ-
фикой взаимодействия резонансных нейтронов с ядрами эле-
ментов и способами осуществления облучений.
Как было отмечено ранее, у некоторых изотопов сечение ре-
акции (и, у) равномерно падает с ростом энергии нейтронов
согласно закону 1/и. Однако у многих изотопов функция воз-
буждения при определенной энергии нейтронов имеет резонанс-
ные пики, в области которых сечение радиационного захвата
достигает исключительно высоких значений и может более чем’
на два порядка превосходить сечение активации для тепловых
нейтронов. В принципе такой характер функции возбуждения
предоставляет интересную возможность для проведения избира-
тельной активации элементов. Но она не может быть реализо-
вана из-за отсутствия подходящего источника моноэнергетиче-
ских нейтронов с переменной энергией.
Для аналитических определений доступны только резонанс-
ные нейтроны, получающиеся главным образом в процессе за-
медления быстрых нейтронов. Наибольшей интенсивности пото-
ки резонансных нейтронов достигают в ядерных реакторах, но
при этом они всегда сопровождаются интенсивным потоком теп-
ловых нейтронов. Поток резонансных нейтронов имеет сплошное
распределение, а плотность потока изменяется по закону 1/£„.
Уже сам ход зависимости плотности потока резонансных
нейтронов от энергии способствует большей активации элемен-
тов, резонансы у которых расположены в области низких энер-
гий, так как здесь плотность потока резонансных нейтронов
выше. Воздействовать на избирательность активации резонанс-
ными нейтронами можно путем применения фильтров, которые
видоизменяют энергетический спектр нейтронов в желаемую
сторону.
Облучение резонансными нейтронами преимущественно при-
меняют для повышения избирательности определения элементов,
проявляющих наиболее интенсивное взаимодействие с резонанс-
ными нейтронами. Это, естественно, сокращает круг определяе-
мых элементов и сопровождается понижением чувствительности
по сравнению с активационным анализом на тепловых нейтро-
нах того же источника. Более сложна методика проведения не-
которых стадий аналитических определений (главным образом
облучений).
Активационный анализ на резонансных нейтронах представ-
лен тремя вариантами, различие между которыми вытекает из
методики анализа: 1) облучение с нерезонансным фильтром;
8Е
2) облучение с резонансным фильтром; 3) анализ по времени
замедления нейтронов. Эти методы способствуют определению
элементов с четко выраженными нейтронными резонансами, сре-
ди которых можно отметить: As, Mo, Ru, Rh, Pd, In, Sb, I, Nd,
Sm, Tb, Lu, Ta, Re, Pt, Au, Hg, U.
Облучение с нерезонансным фильтром. Этот метод приме-
няется для подавления тепловой компоненты нейтронного пото-
ка [96]. Наиболее удачный фильтр для такой цели — кадмиевый.
Слой кадмия толщиной 0,5—1,0 мм пропускает только около
4-10~4 первичного потока тепловых нейтронов.
Применение кадмиевого фильтра приводит к уменьшению
удельной активности элемента в такое число раз, которое равно
его кадмиевому отношению в принятых условиях облучения.
В результате повышается избирательность определения элемен-
тов с высоким резонансным интегралом в присутствии элемен-
тов с низким резонансным интегралом. Экспериментальное оп-
ределение кадмиевых отношений для 42 элементов, проведенное
Н. А. Дамбургом и Л. Л. Пелекис в канале реактора ИРТ-2000
с кадмиевым фильтром толщиной 0,5 мм, дало величины, кото-
рые лежат в интервале от 2 до 240 [96]. Следовательно, приме-
няя этот метод, в предельном случае можно достигнуть повы-
шения избирательности активации почти на два порядка.
Иногда в качестве материала для фильтра используют бор,
который имеет высокое сечение поглощения медленных нейтро-
нов, подчиняющееся закону \/v. Ослабление потока моноэнер-
гетпческих нейтронов следует соотношению
—= e~Nvonot\nx (4.7)
Фо
где Фо и Фл — соответственно исходная плотность потока нейтро-
нов и плотность после прохождения слоя вещества толщиной х;
Л’г — число атомов на единицу объема поглотителя; Опогл — се-
чение поглощения.
Экспоненциальная зависимость ослабления потока нейтронов
от величины сечения поглощения при изменении последней по
закону \/v приводит к тому, что борный фильтр обеспечивает
достаточно четкую границу поглощения, ниже которой поток
нейтронов ослабляется в значительной степени. Увеличение тол-
щины фильтра перемещает границу поглощения дальше в об-
ласть более высоких энергий, что приводит к возрастанию сред-
ней энергии нейтронов, проходящих через фильтр (см. рис. 15).
Борный фильтр приходится применять в сочетании с кадмиевым
в связи с тем, что необходимо уменьшить количество тепла, вы-
деляющегося в результате экзоэнергетической реакции
10В(п, a)7Li. Активация элементов нейтронами реактора с
фильтром 0,5 мм Cd+ 2 мм ВС4 (бор природного изотопного
состава) исследована в работе [96]. С таким фильтром удельная
-82
активность элементов меняется от 2 до 900 раз, т. е. заметно
больше, чем в случае одного кадмиевого фильтра.
Облучение с нерезонансным фильтром — один из эффектив-
ных методов повышения избирательности инструментального
метода. Примером может служить работа Борга и др. [97] по
определению марганца в биологических объектах, которые со-
держат значительные количества натрия и хлора. Анализируе-
0,5 1,0 1,5 0,5 1,Q 1,5
Энергия, пзб
Рис. 16. Сцинтилляционные спектры у-излучения биологи-
ческой пробы, облученной неотфильтрованным (а) и от-
фильтрованным (б) потоками нейтронов.
мые пробы облучали в алюминиевом контейнере, стенки которо-
го были выложены бором (обогащенным 10В до 93%). Тепловые
нейтроны отфильтровали кадмиевым фильтром, покрывающим
снаружи алюминиевый контейнер.
Толщина борного фильтра была подобрана таким образом,
чтобы обеспечить поглощение нейтронов с энергией менее 6 эв.
Облучение отфильтрованным потоком увеличило отношение
удельных активностей марганца и натрия в 7 раз, а марганца
и хлора в 14 раз по сравнению с облучением тепловыми нейт-
ронами. Влияние фильтра хорошо видно из рис. 16.
При облучении резонансными нейтронами анализируемая
проба не должна содержать значительных количеств легких
элементов (в основном водорода), которые могут привести к
замедлению некоторой части нейтронов. Например, удельная
активность индиевой фольги, облученной отфильтрованным с
83
помощью кадмия (0,5 мм) потоком нейтронов, возрастает в
1,44 раза в присутствии 120 мл воды [96].
Облучение с резонансным фильтром. Избирательность акти-
вации резонансными нейтронами можно несколько повысить,
если в дополнение к кадмию в фильтр ввести резонансный по-
глотитель нейтронов. Естественно, что при облучении пробы в
комбинированном фильтре заметно уменьшается активация того
компонента, который совпадает с резонансным поглотителем.
Это явление можно использовать либо для подавления актива-
ции мешающего компонента, либо для получения количествен-
ных оценок для определяемого компонента по изменению его
активности при облучении с кадмиевым и комбинированным
фильтрами.
Именно последний вариант был практически разработан
Р. Г. Гамбаряном и А. С. Штанем [98]. Причем для достижения
максимальной избирательности метод облучения с резонансным
фильтром сочетается с у-спектрометрией. Тогда методика ана-
лиза включает следующие операции. Анализируемую пробу
делят на две равные части. Одну из них облучают с кадмиевым
фильтром, другую — с комбинированным. После измерения
у-спектров обеих частей пробы получают разностный спектр.
Интенсивность аналитической у-линии в разностном спектре
пропорциональна содержанию определяемого элемента.
Ограничения такой методики связаны с применимостью к
простым системам с известным качественным составом и увели-
чением статистической погрешности, поскольку конечный резуль-
тат получается как разность соизмеримых величин. При облу-
чении неизбежно происходит сильная активация резонансного
фильтра, что затрудняет процесс извлечения пробы и повторное
использование фильтра.
Анализ по времени замедления нейтронов. Идея этого мето-
да применительно к аналитическим целям была предложена
Р. Г. Гамбаряном и др. [99]. Если через замедлитель из тяже-
лого элемента пропустить импульсный поток быстрых нейтронов,
то потеря энергии ими будет происходить практически синхрон-
но. Поэтому энергия нейтронов в потоке оказывается пропор-
циональной длительности процесса замедления. Когда энергия
потока становится равной энергии резонансного уровня вещест-
ва, введенного в замедлитель, происходит интенсивное поглоще-
ние нейтронов, которое сопровождается испусканием мгновен-
ного у-излучения (рис. 17). Интенсивность последнего служит
мерой количества резонансного поглотителя, а длительность
интервала замедления может быть использована для дополни-
тельной идентификации элемента (рис. 18).
Соответствующие оценки показывают, что чувствительность
определения отдельных элементов этим методом при использо-
вании нейтронного генератора с интенсивностью 10'° нейтрон/сек.
достигает 2 • 1 (У 3 %.
84
Рис. 17. Зависимость интенсивности у-излучения радиационного за-
хвата от длительности замедления для Ag, Pt и Pd.
Рис. 18. Длительность замедления нейтронов в
.свинце до энергии основных резонансов некото-
рых изотопов.
Активационный анализ на быстрых нейтронах
Для быстрых нейтронов (£„>0,5 Мэв) сечение реакции
(лг, у) уже мало, и основную роль в их взаимодействиях с ядра-
ми элементов играют пороговые реакции типа (п, р), (и, а),.
(п, 2п) и (и, п'). Величина порога для большинства реакции
составляет несколько мегаэлектронвольт, а функции возбужде-
ния имеют максимум при энергии нейтронов 8—20 Мэв [31].
Типичным примером может слу-
жить функция возбуждения ре-
акции 27Al(n, a)24Na, порог кото-
рой равен 3,27 Мэв (рис. 19).
Сечения ядерных реакций на
быстрых нейтронах не превыша-
ют нескольких барн и в боль-
шинстве случаев лежат в преде-
лах 0,01—1 барн. Более слабое
взаимодействие быстрых нейтро-
нов с ядрами по сравнению с
тепловыми приводит к двум
важным аналитическим последст-
Рис. 19. Функция возбуждения ре- виям. Во-первых, значительно па-
акции -'А1(л. a)24Na (Q= дает чувствительность определе-
=—3,1 Мэв). ния, а во.ВТОрЬ1Х> уменьшается
влияние вещества пробы на по-
ток нейтронов (эффект экранирования).
При облучении быстрыми нейтронами активируется подав-
ляющее большинство элементов периодической системы, и по-
этому в отношении круга определяемых элементов активацион-
ный анализ на быстрых нейтронах не уступает активационному
анализу на тепловых нейтронах. Однако более низкая чувстви-
тельность предопределяет основное применение этого метода к
определению элементов, для которых активационный анализ на
тепловых нейтронах неблагоприятен (О, N, S, F и др.), и для
массового инструментального анализа различных объектов со
сравнительно высоким содержанием компонентов.
Взаимодействие быстрых нейтронов с ядрами элементов
может протекать по нескольким каналам, которые часто связа-
ны с изменением заряда ядра. Эта особенность ядерных взаимо-
действий быстрых нейтронов, с одной стороны, облегчает под-
бор подходящей ядерной реакции, а с другой, представляет
собой источник взаимных помех при определении соседних
элементов. Облучение быстрыми нейтронами можно проводить
с помощью различных источников, основные характеристики
которых суммированы в табл. 5.
Наибольшее значение для активационного анализа на быст-
рых нейтронах приобрели нейтронные генераторы на основе
реакции ;!Н (d. п)4Не. Поток нейтронов от этих источников моно-
86
Таблиц, 5
•Основные характеристики различных источников быстрых нейтронов
Тил источника Ядерная реакция Максимальная энергия ней- тронов, Мэв Спектр нейтроноп Плотность потока ней - троной’ . yeiiinpiiH СМ - ’С* <
Изотопные источники sBe (а, п)12С 13 Сплошной 10‘
Нейтронный генератор 2Н (d, п) 3Не ~3 Моноэнер- гетический 107
:tH (d, п) 4Не ~14 То же 1019
Реактор Деление ~20 Сплошной 1012—1013
Ускорители электронов (Т. п) Зависит от энергии из- лучения » 101’
Ускорители** ионов (тол- стая мишень) Разные Зависит от типа иона и его энергии » 1011
’Приведены предельные величины для имеющихся в настоящее время источников.
*’Ускорители ионов с тонкой мишенью позволяют получать потоки моноэпергетических
•нейтронов с переменной энергией, но выход нейтронов при этом много ниже.
хроматичен и имеет достаточно высокую интенсивность.. Наи-
более мощный .‘Kiuinnn быстрых нейтрон'?» — рряктор Однако
поток нейтронов в реакторе смешанный, т. е. содержит медлен-
ную компоненту. Присутствие в потоке медленных нейтронов
приводит к определенным затруднениям, так как продукты ре-
акции (п, у) вследствие более высоких сечений оказываются
значительно активнее радиоизотопов, образующихся в резуль-
тате реакций на быстрых нейтронах. Поэтому для подавления
тепловой компоненты потока приходится использовать фильтры
из кадмия или других материалов. Однако это лишь частично
решает проблему, так как интенсивность потока резонансных
нейтронов при этом уменьшается в незначительной степени.
Ускорители, как источники быстрых нейтронов, имеют хоро-
шие параметры. Хотя энергетическое распределение быстрых
нейтронов сплошное, доля медленных нейтронов в потоке
ничтожна и не мешает анализам. Источники нейтронов этого
типа пока не получили широкого распространения.
Аналитические возможности активационного анализа на
быстрых нейтронах многократно исследовались для самых раз-
ных экспериментальных условий, причем основное внимание
было уделено нейтронным генераторам. Прежде всего был изу-
чен круг определяемых элементов и образующихся изотопов, а
также чувствительность метода, которая с распространенными
нейтронными генераторами в настоящее время не превышает
1 мкг [100, 101].
87
Анализ помех, встречающихся при активационном анализе-
с 14-А4эв нейтронами, был выполнен Мазуром и Ольдхамом.
[102]. Было отмечено, что на стадии облучения основным источ-
ником помех являются конкурирующие ядерные реакции, кото-
рые вместе с некоторыми другими полезными сведениями сум-
мированы в форме, удобной для практического использования.
Исследование возможностей активационного анализа на
нейтронах с энергией 2,8—3,0 Мэв показало, что в этом случае-
круг определяемых элементов уже, а достигаемая чувствитель-
ность много ниже (более 10 мг) [71]. Низкую чувствительность
дает облучение быстрыми нейтронами изотопного источника.
Быстрые нейтроны реактора обеспечивают наиболее высокую-
чувствительность, которая на один-два порядка может превос-
ходить чувствительность, достигаемую с нейтронным генерато-
ром (14-Мэв нейтроны) [65]. Однако сложность работы со сме-
шанным потоком нейтронов затрудняет широкое применение
реакторов для активационного анализа на быстрых нейтронах.
Множественность каналов и наличие конкурирующих ядер-
ных реакций, а также некоторые особенности схем распада про-
дуктов активации оказывают неблагоприятное влияние на из-
бирательность активационного анализа на быстрых нейтронах,
особенно в его инструментальном варианте [102, 103]. Учитывая
пороговый характер многих реакций, можно иногда исключить
помехи от конкурирующей ядерной реакции путем применения
нейтронов соответствующей энергии. Примером такого случая
может служить определение фтора по реакции 19F(n, a)16N
(Епор~3 Мэв) в присутствии кислорода, который дает тот же
радиоизотоп по реакции !6O(n, p)16N (ЕПор= 10,5 Мэв). При
имеющемся различии в порогах эта задача может быть решена
при облучениях нейтронами, энергия которых лежит чуть ниже
порога интерферирующей реакции. При этом можно использо-
вать три способа: 1) подбор источника с соответствующей
энергией нейтронов; 2) применение источника, который допу-
скает регулировку максимальной энергии нейтронов; 3) измене-
ние энергетического спектра быстрых нейтронов при пропуска-
нии через замедляющие фильтры. Как следует из данных
табл. 5, возможности первого способа весьма ограничены, к
тому же для проведения разнообразных определений требуется
наличие в лаборатории нескольких источников нейтронов.
Весьма перспективен второй способ [82], который при нали-
чии одного источника позволяет плавно регулировать энергию
нейтронного потока. Однако сплошной характер нейтронного
потока затрудняет избирательную активацию. Поэтому раз-
дельное определение этим способом возможно только при зна-
чительном различии в порогах основной и интерферирующей
ядерных реакций (несколько мегаэлектронвольт).
Последний метод состоит в пропускании потока моноэнерге-
тических нейтронов (обычно 14 .Мэв) через водородсодержа-
88
щий материал, что приводит к замедлению нейтронов с соот-
ветствующим изменением энергетического спектра. Поскольку
-функции возбуждения ядерных реакций различны, облучение
пробы в исходном и измененном потоках может позволить раз-
дельное определение двух компонентов [104]. Однако из-за силь-
ного ослабления потока нейтронов фильтром и небольшой ве-
личины эффекта подобный метод, видимо, не может иметь осо-
бой практической ценности.
§ 4. Некоторые особенности и источники погрешности
при облучении тепловыми нейтронами
Градиент потока
Уравнение активации (2.23) справедливо для облучения в
условиях равномерного потока активирующего излучения. В то
же время источники нейтронов дают потоки с некоторым гра-
диентом. Поэтому в уравнение активации следует подставлять
среднее значение плотности потока Ф, которое является функ-
цией размеров и формы пробы и распределения плотности
потока в объеме, занимаемом пробой. Однако такая замена
может быть сделана только для гомогенных проб. В противном
случае полученный результат будет отличаться от среднего зна-
чения, причем отклонение может быть тем сильнее, чем выше
градиент потока и негомогенность пробы. Так, атомные реакто-
ры обладают сравнительно небольшим градиентом. Например,
в реакторе Merlin градиент равен 2% на 10 см в вертикальном
направлении и 1,8 см в горизонтальном [105], а в реакторе
Triga в обоих направлениях составляет 5% на 2,4 см [92]. Зна-
чительный градиент плотности потока нейтронов наблюдается у
нейтронных генераторов.
Для исключения погрешности за счет градиента потока при-
ходится принимать специальные меры предосторожности или
использовать особую методику анализа, которые определяются
величиной градиента и конструктивными особенностями источ-
ника нейтронов. В потоках с небольшим градиентом стремятся к
размещению пробы и эталона (монитора) по возможности бли-
же друг к другу. Иногда даже рекомендуют оборачивать ампу-
лу с пробой несколькими фольгами с нанесенным на них эта-
лоном. Фольги располагают определенным образом, что в
результате позволяет оценить среднее значение активности эта-
лона. Более радикальным средством исключения погрешности
является применение особого вращающего устройства, которое
позволяет облучать большое число проб и эталоны в усреднен-
ном потоке [92].
Если градиент потока источника постоянен во времени, то
можно применить метод мониторов. При этом необходимо обес-
печить четкую фиксацию как взаимного расположения пробы и
89
монитора, так и их положения в определенном месте нейтрон-
ного поля источника. Кроме того, анализируемые пробы всегда
должны занимать определенный объем, а для калибровочных
облучений нужны эталоны, имеющие те же геометрические раз-
меры, что и анализируемые пробы.
Облучение изотопов с высоким сечением активации
в интенсивных потоках тепловых нейтронов
Основное уравнение активации (2.23) было выведено в пред-
положении, что в ходе облучения число ядер активирующегося
изотопа практически не меняется и нет никакого другого про-
цесса, кроме радиоактивного распада, который приводил бы к
исчезновению образовавшихся ядер. Такое предположение яв-
ляется приближенным, так как очевидно, что число облучаемых
ядер должно неизбежно уменьшаться за счет вступления части
из них в ядерную реакцию; как говорят в таком случае, изотоп
«выгорает».
Однако в большинстве случаев при имеющихся сейчас в на-
личии потоках активирующих излучений и реально доступных
длительностях облучений вследствие умеренных величин сече-
ний реакций доЛя выгоревших ядер оказывается ничтожной, и
этим процессом можно пренебречь. Однако при облучении теп-
ловыми нейтронами, когда имеются мощные источники с очень
высокой плотностью потока, а некоторые изотопы обладают
большими сечениями, эффект выгорания может оказаться суще-
ственным.
Уменьшение числа ядер при облучении подчиняется закону
NAt = NAoe^<t’n<yt°6si, (4.8)
где Л'л/ — число оставшихся ядер; Л\о—начальное число ядер.
Подсчет на примере золота (197Аи, о = 96 барн') для облучения
в течение трех суток в максимально доступном потоке [Ф„ =
= 1015 нейтрон! (см2 сек)] показывает, что в этих условиях вы-
горит всего 2,5% ядер 197Аи. Однако при увеличении длитель-
ности облучения доля выгоревших ядер быстро возрастает.
Сильнее эффект выгорания сказывается и для изотопов с более
высокими сечениями. Так, для 168Yb, сечение активации которого
максимально (оакт= 1,1 • 104 барн), расчет показывает, что в
указанных условиях останется только около 5,5% исходного
количества ядер.
Однако последний пример экстремальный в отношении как
сечения активации, так и плотности потока. По оценке
И. А. Маслова [106], при Ф„ = 1014 нейтрон/ (см2 - сек) эффект
выгорания наблюдается только при сечениях порядка 1000 барн
и выше. Для обычно используемых потоков с плотностью
1012—1013 нейтрон)(см2 сек) и времени облучения несколько
суток выгорание ядер является довольно редкой проблемой.
90
В табл. 6 указаны изотопы, для которых эффект выгорания
при облучении в соответствующем потоке нейтронов превышает
3% [Ю7]. Длительность облучения соответствует достижению
насыщения (для короткоживущих изотопов), но не превышает
30 дней.
Таблица 6
Изотопы с заметили степенью выгорания
Плотность потока нейтронов, нейтрон ’(см2-сек) Л’д/ Иютоиы. для которых <"0,97 •v А 0
1042 154Еи, Н»Та
101- 1-iiEu, ’s2Ta
10й is-iEu, ы-Eu, ie»Yb, 477Lu, 192Ta, 192Ir, i«Au, isimHg, 197Hg
101» 60Co. 73Se, 114mIn. 134Cs, l34Ba, 1S3Sm, 15*Eu, '»2Eu, 153G4 Kioyb, i"Dv, i65Dv, i66Ho, i«»Yb, 175Yb’, i7»Lu, 477bu, i^Hf, i8*Hf, 182Ta, i8i\y is«Pe n^Os i92Ir 194Ir i9:,Pt 198Au l97”'Hg, 1&7Hg
В табл. 6 наряду с исходными стабильными изотопами от-
мечены и некоторые вторичные радиоизотопы, так как они име-
ют высокие сечения захвата нейтронов и для них существен
эффект выгорания. Это значит, что эти ядра имеют заметную
вероятность перехода в другие ядра не только при радиоактив-
ном распаде, но и путем вступления в ядерную реакцию с нейт-
ронами. Примером может служить активация золота:
197Аи---[Д2]---_ 198Ап ---[Д2]-----> 199Ди
96 барн о2=26000 барн ।
Ti/2—2,7 дня 15 дня
198Hg 199Hg
Изотоп ,98Au имеет очень высокое сечение захвата тепловых
нейтронов, поэтому эффект выгорания оказывает сильное влия-
ние на ход накопления этих ядер. Ниже будет рассмотрено
уравнение активации, действительное в таких случаях (см. § 1
гл. 11). С помощью уравнения (11.1) было рассчитано относи-
тельное изменение активности 198Ап с ростом плотности потока
нейтронов при /Обл = 27 дней (рис. 20). При ФйИО13 ней-
трон-(см2-сек) линейное увеличение активности насыщения с
ростом Ф нарушается, что свидетельствует о большом влиянии
эффекта выгорания.
На рис. 21 приведены кривые накопления активности ,98Аи
и 199Аи при двух значениях плотности потока. При Ф = 1013 ней-
трон, (см2-сек) кривая накопления для 198Ап имеет типичную
форму с выходом на плато, т. е. достигается активность насы-
щения. Правда, при дальнейшем увеличении Фбл также начи-
91
нает сказываться эффект выгорания. Между тем при Ф==
= 1015 нейтрон//см2-сек) активность 198Аи достигает своего
Плотность потока нейтронов.
нейтрон/(смг-сек) 7
Рис. 20. Влияние эффекта вы-
горания на активность насы-
щения 198Аи.
максимального значения уже че-
рез 50 ч и затем быстро умень-
шается (кривая 3 на рис. 21).
При этом быстро накапливается
199Аи, активность которого дости-
гает максимального значения
примерно через 350 ч облучения.
На рис. 21 все кривые приведены
в различных масштабах, и для
перехода к действительным соот-
ношениям значения ординат для
кривой 2 нужно увеличить в
100 раз, а для кривой 3 — в
8,5 раза. Таким образом, при
Ф=1015 нейтрон//см2-сек) актив-
ности 198Ап и 199Аи сравниваются
примерно через 15 ч после начала
облучения и в дальнейшем активность 198Аи преобладает над
активностью 198Аи. Важно отметить, что 198Аи и 199Аи имеют
Рис. 21. Кривые накопления активностей 198Аи
(/, 2) и |99Аи (3) при плотности потока нейтро-
нов 1 • 1013 нейтрон!(см- сек) (/) и
1 • 1015 нейтрон/(см2 сек) (2, 3).
близкие параметры схем распада. Поэтому при определении
золота активационным методом можно использовать потоки
92
нейтронов вплоть до 1015 нейтрон' (см2- сек) без заметных огра-
ничений чувствтельностп за счет эффекта выгорания, нужно
только проводить измерение излучения того радиоизотопа, ко-
торый дает наибольшую активность в условиях анализа. Мож-
но применить и интегральную регистрацию излучения обоих
радиоизотопов.
Кривые на рис. 20 и 21 построены для чисто теплового по-
тока нейтронов. Если в потоке содержится заметная доля резо-
нансных нейтронов, то следует учитывать большой резонансный
интеграл 197Аи, что должно привести к более заметному про-
явлению эффекта выгорания.
Возмущение потока нейтронов веществом пробы
Введение анализируемой пробы в равномерный поток нейтро-
нов неизбежно приводит к определенным изменениям (возму-
щению) энергетического спектра и плотности потока нейтронов
как в окружающей среде, так и внутри пробы. Величина эф-
фекта возмущения и его характер зависят от параметров окру-
жающей среды в зоне облучения, состава пробы, ее массы и
исходного спектра потока нейтронов.
В конечном счете возмущение потока нейтронов веществом-
пробы приводит к изменению удельной активности компонентов
по сравнению с облучением в невозмущенном потоке. При этом
в методе эталонов (мониторов) возникает систематическая
погрешность, зависящая от различия эффектов возмущения в-
пробе и эталоне (мониторе). В некоторых случаях влияние
возмущения потока нейтронов на конечные результаты оказы-
вается очень большим, и тогда требуется принятие определен-
ных мер по ограничению эффекта или введение соответствующей
поправки.
При облучении в потоке тепловых нейтронов общий эффект
возмущения обусловлен действием трех процессов:
1) депрессией потока нейтронов в окрестностях пробы, свя-
занной с утечкой нейтронов при их поглощении веществом про-
бы в условиях диффузионной среды;
2) экранированием внутренних слоев пробы в результате
поглощения нейтронов ядрами атомов внешних слоев при их
диффузии внутрь пробы;
3) рассеянием нейтронов, которое сопровождается измене-
нием траектории движения нейтронов и их энергетического со-
стояния.
Тогда коэффициент возмущения потока Ев, определяемый
как отношение удельной активности компонента в пробе к
удельной активности этого компонента при облучении в невоз-
м\щепном потоке, будет равен произведению трех компонентов,
каждый из которых учитывает влияние одного из отмеченных
93:
факторов:
FB =- hihip’ (4.9)
где Ад — коэффициент депрессии; /:)—коэффициент экраниро-
вания; ф — коэффициент рассеяния.
Эффект возмущения потока в активационном анализе часто
является ограничивающим фактором и источником трудно учи-
тываемых погрешностей из-за сильной зависимости эффекта как
от характеристик окружающей среды и параметров облучаемой
пробы, так и от энергетического спектра и углового распреде-
ления нейтронного потока. Как было отмечено, потоки нейтро-
нов реальных источников состоят из трех основных компонентов:
тепловых, резонансных и быстрых нейтронов. Поскольку роль
каждой из этих групп в рассматриваемых процессах различна,
дальнейшее изложение будет вестись с учетом этого факта.
Следует подчеркнуть, что эффект возмущения обусловлен в
первую очередь макрокомпонентами пробы и их характеристи-
ками по отношению к поглощению и рассеянию нейтронов. В то
же время рассматриваемый процесс активации может быть
связан с любым макро- или микрокомпонентом, определение
которого в данной пробе представляет интерес. Различия в ходе
зависимости сечения поглощения, рассеяния и активации рас-
сматриваемых компонентов от энергии нейтронов приводят к
тому, что коэффициент экранирования, а следовательно, и ко-
эффициент возмущения при облучении в смешанных потоках
нейтронов часто оказываются индивидуальными величинами,
т. е. действительными только для определенного изотопа.
Вследствие значительного влияния эффекта возмущения на
точность получаемых результатов рассмотрению этой проблемы
посвящено большое число исследований. Предложен ряд мето-
дов для экспериментального учета эффекта возмущения или
ограничения его величины определенным пределом. Развива-
ются и расчетные методы оценки необходимых поправочных
коэффициентов. Однако последний путь связан с принятием
определенных предположений, идеализирующих условия облу-
чения, что приводит к выражениям, не всегда дающим доста-
точно надежные оценки.
Тепловые нейтроны. Анализ эффекта возмущения обычно
проводят на примере активации элемента, который составляет
основу пробы. Тогда коэффициент /э превращается в коэффи-
циент самоэкранирования. Экспериментально самоэкранирова-
ние легко обнаружить и количественно оценить, если облучить
пробы из какого-либо элемента с постепенно возрастающей мас-
сой и рассчитывать удельную активность (рис. 22) [108]. Пока
количество вещества мало и отсутствует самоэкранирование,
удельная активность остается постоянной величиной. Однако
при достижении некоторого предела влияние поглощения ней-
тронов начинает сказываться и величина удельной активности
44
быстро уменьшается. За областью резкого спада дальнейшее
увеличение количества вещества мало меняет удельную актив-
ность.
Для теоретической оценки коэффициента самоэкранированпя
тепловых нейтронов предложено несколько полуэмпирически.х
уравнений и методов расчета [31. 109, 1101. Поскольку метол.
Рис. 22. Нормализованная кривая зависимости удель-
ной активности пробы от величины навески.
развитый В. А. Жарковым [111], представляется наиболее общим
и точным, ниже дано краткое изложение его основных поло-
жений.
Главное условие применимости этого метода (так же как и
других) состоит в требовании постоянства макроскопического
сечения поглощения в каждой точке пробы в течение всего
времени облучения:
2,югл (г, 0 = Vim.1 = const (4.10)
где Nr — плотность атомов, or'1; стп<>г.ч — микроскопическое
сечение поглощения, см2. Это означает, что в пробе за время
облучения не происходит заметного выгорания ядер или накоп-
ления сильных поглотителей нейтронов. Такое условие для ос-
новного моноизотопного компонента можно выразить в матема-
тической форме через
соотношений:
Л
известные ядерные характеристики в виде
[Маодг, £)
] о'2(£')Ф(г, £);
(4.И)
Абл
Oi (£) Ф (г, £)
1
(4.12).
X + со (£) Ф (г, Е) ’
активации первичного изотопа; о2(Е| и
где Oi(E)—сечение
X — сечение поглощения и постоянная распада радиоизотопа;
95-
•А>бл — время облучения. В случае многоизотопного основного
элемента или пробы сложного состава справедливость соотно-
шений (4.11) и (4.12) должна быть проверена для всех эле-
ментов и изотопов.
При расчете активности основного компонента при нали-
чии самоэкранирования вследствие неравномерного распределе-
ния плотности потока нейтронов по объему пробы и ужестчения
спектра нейтронов уравнение (2.24) запишется в виде
А (/) = J (1 — е^бл), (4 13)
где
/=П2акт(^)Ф(г, E)drdE;
2ai.T(£) = A^i(£).
Двойной интеграл /, имеющий смысл скорости радиацион-
ного захвата нейтронов, что приводит к образованию радиоизо-
топа, можно выразить через коэффициенты экранирования и
депрессии:
= 2а^Ф0Мд. (4.14)
где 5акт — усредненное по максвелловскому спектру макроско-
пическое сечение активации; V — объем пробы; Фо —плотность
невозмущенного потока; /э— коэффициент экранирования; 1гя —
коэффициент депрессии.
Для проб произвольной геометрии коэффициенты экраниро-
вания и депрессии можно рассчитать, используя следующие
соотношения:
/э = -^—; (4.15)
^-погл-^
4 =-----(4.16)
1 + 47v
'Здесь ф — средняя по максвелловскому спектру вероятность
поглощения нейтрона в пробе при изотопном падении; 2ПОгл —
усредненное по максвелловскому спектру макроскопическое се-
чение поглощения вещества пробы; X=4V/S, где S — площадь
поверхности пробы; fv—параметр, зависящий от свойств окру-
жающей пробу среды и практически не зависящий от поглощаю-
щих свойств пробы.
Соотношения (4.15) и (4.16) для коэффициента /э справед-
ливы только для случая, когда сечения поглощения и активации
зависят от энергии нейтронов одинаковым образом, например
оба подчиняются закону 1/и. Это условие удовлетворяется для
большинства элементов [95]. В противном случае для расчета
:96
коэффициента j., необходимо применять более общее соотно-
шение:
/Э = ^|П2^-, (4.17)
^-актХ
где черта означает усреднение по максвелловскому спектру.
Дл я ^погл справедливо соотношение
2;огл =- уГ^~ё(Г) 2ПОГЛ, (4.18)
где Т — температура пробы, К°; 2Догл — макроскопическое се-
чение поглощения при энергии kg(T) (k — постоянная Больц-
мана); g(T)—коэффициент, учитывающий отклонение энерге-
тической зависимости сечения от закона 1/г>. Аналогичное_вы-
ражение может быть написано и для сечения активации Пакт-
Для оценки коэффициентов /э и йд в данных условиях
анализа значения функции ф, рассчитанные с помощью вычи-
слительной машины для проб разной формы, приведены в ра-
боте [111]. Необходимый для оценки коэффициента депрессии
параметр Д может быть рассчитан по одному из следующих
приближенных соотношений:
шар:
5; (4.19)
4 ?.тр R + La
круглая фольга:
„ , ( --0.85 -5- \
= ± .А и - е ;д /• (4.20)
4 Др
бесконечный цилиндр
f = JL W ДО (4 21)
/V 4 Др' ' K^RiLJ ’
где /_;[ и лТр — длины диффузии и переноса тепловых нейтронов
в окружающей пробу среде; R — радиус шара (фольги, цилин-
дра): Л)о(л') и A’i(.v) — модифицированные функции Бесселя.
При облучении в канале, линейный характерный размер ко-
торого существенно превышает размер пробы, а также для
проб, малых по сравнению с длиной переноса нейтронов в
окружающей среде, йд=1. Эти условия часто реализуются на
практике.
Хотя приведенные выше соотношения для коэффициентов Д
и йд выведены для активации макрокомпонента, эти коэффи-
циенты сохраняют свое значение и для любых микрокомпонен-
тов, конечно, с учетом отмеченных особенностей в ходе зависи-
4 Р А Кузнецов 97
мости сечения активации микрокомпонента и сечения поглоще-
ния вещества пробы от энергии нейтронов.
Необходимо отметить, что рассмотренный выше метод оцен-
ки эффекта возмущения предполагает, что эффектом рассеяния
нейтронов можно пренебречь (2Рас<^2погл). Это справедливо
в подавляющем числе случаев. Однако если основа пробы со-
держит значительные количества легких элементов, это ведет к
некоторому изменению поглощающих свойств пробы. Для тепло-
вых нейтронов этот эффект учитывается коэффициентом zp:
zp =-------—1-------------. (4.22)
•^погл Т ‘-рас
Резонансные нейтроны. Радиационный захват резонансных
нейтронов также дает вклад в активацию элементов, и поэтому
возмущение их потока веществом пробы должно быть принято
во внимание. Для резонансных нейтронов эффект депрессии
несуществен (йд=1), и основное влияние на ход активации
оказывает их поглощение макрокомпонентами пробы.
Если в число макрокомпонентов входят сильные поглотители
резонансных нейтронов, то коэффициент экранирования должен
учитывать ослабление потока резонансных нейтронов. Тогда
полный коэффициент экранирования /э для данного изотопа
можно выразить через коэффициент экранирования тепловых
нейтронов /т, коэффициент экранирования резонансных нейтро-
нов /р и кадмиевое отношение для него в данных условиях
облучения таким образом:
h = (1 - тЦ А + -л—/р- (4.23)
Методы оценки /т были рассмотрены в предшествующем
разделе. Что касается методов оценки /р, то это довольно слож-
ная проблема, поскольку при этом необходимо учитывать резо-
нансные пики и их взаимное расположение у макрокомпонентов
и определяемого элемента, рассеяние нейтронов в матрице и
ряд других факторов. Какой вклад в полный коэффициент
экранирования при определении золота и .меди в шариках из
серебра дают тепловые и резонансные нейтроны, можно видеть
из данных табл. 7.
Серебро — сильный поглотитель резонансных нейтронов
(^ пог л = 1900 барн); золото имеет высокий резонансный интеграл
активации (/р==1560 барн), а медь слабо взаимодействует с
резонансными нейтронами (/р = 4,4 барн). Сравнение коэффици-
ентов экранирования показывает, что коэффициент К для меди
практически определяется поглощением серебром тепловых ней-
тронов, в то время как для золота существенную роль играет
поглощение серебром резонансных нейтронов. По оценке Хог-
дала [110], влияние резонансных нейтронов на процесс актива-
98
Таблица 7
Коэффициенты экранирования при определении золота и меди в шариках
из серебра (кадмиевое отношение для золота равно 2,6)
Масса, мг Радиус, мм /'т р % Содержание, о/ /0
Си Аи Си Ла Си Аи
8,00 0,56 0,90 0,97 0,17 0,90 0,62 1,20 0,91
48,11 1,03 0,82 0,95 0,14 0,83 0,56 0,92 0,97
149,2 1,50 0,75 0,93 0,13 0,76 0,51 0,86 0,97
703,6 2,53 0,63 0,89 0,10 0,64 0,43 1,04 1,02
1794 3,46 0,55 0,87 0,09 0,56 0,37 1,14 1,18
ции становится незначительным, если кадмиевое отношение для
определяемого элемента в данных условиях облучения боль-
ше 50.
Быстрые нейтроны. Из-за малости сечения реакции радиаци-
онного захвата их прямой вклад в активацию элементов мал.
Однако их замедление веществом пробы может увеличить поток
тепловых нейтронов, что приведет к усилению активации опре-
деляемых элементов. Этот эффект существен только при облу-
чении проб, основу которых составляют легкие элементы, в
каналах, где доля быстрых нейтронов в общем потоке значи-
тельна. Так, по данным работы [112], усиление активации ко-
бальта в водных растворах составило 5% Для объема 1,5 мл
и 12% для 30 мл.
Для экспериментального учета или ограничения эффекта
возмущения потока нейтронов на результаты анализа предложен
ряд методов. Самый простой из них состоит в том, чтобы в
качестве эталонной использовать пробу того же состава, массы
и формы, что и анализируемая, но с известным содержанием
определяемых элементов. Поскольку эффект возмущения в
этом случае в обеих пробах одинаков, он не оказывает влияния
на конечный результат. Получить пробу с известным содержа-
нием можно либо методом добавок, введя в анализируемую
пробу известные количества определяемых элементов, либо
используя пробы, для которых есть аналитические данные, по-
лученные независимым методом.
Так, для определения истинного содержания иридия в родии
была использована следующая методика [113]. В кварцевые
ампулы было отобрано семь навесок родия по 10 мг. К трем
добавили по 0,1 мл эталонного раствора иридия и раствор
упарили досуха. К каждой отобранной пробе добавили по
0,15 мл концентрированной НС1 и несколько капель концентри-
рованной HNO.->, ампулы замораживали жидким воздухом и
4*
99
быстро запаивали. Затем ампулы нагревали до температуры
200—215° С, что приводило к растворению проб.
Подготовленные таким образом пробы облучали, для фоно-
вых измерений облучали пустую ампулу. Через две недели вы-
держивания проводили измерения и из полученных данных
рассчитывали содержание иридия в исходном материале.
В дальнейшем проанализированный таким образом родий слу-
жил эталоном и анализ уже проводили без какой-либо предва-
рительной химической подготовки.
Ослабление эффекта возмущения происходит при разбавле-
нии анализируемого материала веществом, слабо поглощающим
нейтроны. Для этого можно использовать порошкообразные
алюминий, графит и другие вещества [17]. Естественно, что и
анализируемый материал должен быть в порошкообразной фор-
ме. Иногда переводят пробу в раствор и производят разбавле-
ние водой. Хотя гомогенность при этом гарантирована, однако
облучать жидкие препараты обычно сложнее, чем твердые.
Основной недостаток методов разбавления — необходимость
предварительной обработки пробы и добавления разбавителя,
что может быть источником нежелательных примесей.
Чтобы ограничить влияние эффекта возмущения на резуль-
таты анализа, иногда прибегают к облучению пробы такой
массы, при которой коэффициент возмущения оказывается бли-
зок к единице. Обычно в качестве критерия принимают массу
пробы, при которой коэффициент Ев равен 0,90 или 0,95. Для
подобных оценок может быть использован метод, изложенный
выше. В работе [109] приведена таблица, в которой сопостав-
лены результаты расчета максимальной массы всех элементов,
полученных на основе различных предложенных методов.
При использовании сильных поглотителей нейтронов эти ве-
личины оказываются довольно малыми. Например, при Ев =
= 0,90 масса сферической пробы составляет для золота 2,9 мг,
для индия 0,29 мг и для серебра 1,9 мг. Из этих примеров
видно, что стремление ограничить эффект возмущения потока
нейтронов за счет применения проб малой массы приводит к
понижению концентрированной чувствительности определения.
Когда потеря чувствительности или отбор такой малой на-
вески нежелательны, можно использовать пробу достаточно
больших размеров с экспериментальной оценкой необходимой
поправки. Обычно для этого определяют изменение удельной
активности макрокомпонента в зависимости от массы пробы,
т. е. снимают кривую самоэкранироваиия. По этой кривой мож-
но определить поплавку для анализируемых проб.
Например, Ю. В. Яковлев и Н. Н. Догадкпн [114] при ана-
лизе индия определили, что удельная активность пробы массой
100 мг составляет только 42% удельной активности эталона
(11 мкг индия). Отсюда для навесок 100 мг получается коэф-
фициент 2,4, на который надо умножать активности препаратов
100
определяемых элементов в пробе, прежде чем сравнивать их с
активностями эталонов.
-Необходимо подчеркнуть, что в этом случае определяется
коэффициент самоэкранирования для макрокомпонента, который
совпадает с коэффициентами экранирования определяемых ком-
понентов только при одинаковой зависимости сечений активации
макрокомпонента и данного элемента от энергии нейтронов.
Поэтому при использовании индия, являющегося сильным по-
глотителем резонансных нейтронов, этот метод (облучение в
обычном потоке с кадмиевым отношением для золота, равным
2—3) может дать значительную погрешность для многих эле-
ментов. Это связано с тем, что в величину коэффициента само-
экранирования индия значительный вклад дает коэффициент
/Р. Однако у определяемых элементов с небольшим резонансным
интегралом активации коэффициент jp влияет меньше на коэф-
фициент экранирования j3. Коэффициент /э при этом в основном
определяется ослаблением тепловых нейтронов индием.
Насколько сильно различаются коэффициенты самоэкрани-
рования в случае Со (7Р = 49 барн} и Аи (7р=1560 барн), мож-
но видеть из табл. 8 [112]. Сравнение приведенных данных по-
казывает, что у Со коэффициенты /т и /э очень близки, однако
для Аи они сильно различаются.
Таблица 8
Коэффициенты самоэкранирования кобальта и золота
Проба Толщина или диаметр, мм /т 7Р
Кобальтовая фольга 0,032 0,96 0,73 0,94
0,127 0,87 0,55 0,85
Кобальтовая проволока 0,254 0,94 0,55 0,92
0,508 0,91 0,47 0,87
1,016 0,82 0,37 0,785
Золотая фольга 0,025 0,96 0,43 0,77
0,051 0,93 0,34 0,73
0,254 0,79 0,14 0,56
0,508 0,68 0,05 0,46
Так, для золотой фольги толщиной 0,508 мм использование /э
ьместо /т при расчете поправочного коэффициента для элемента
с низким резонансным интегралом активации может привести
к завышению конечных результатов в 1,5 раза.
При анализах сильных поглотителей нейтронов эффект
экранирования в пробе можно сильно уменьшить, если прово-
дить облучение отфильтрованным потоком нейтронов [115]. Для
этого пробу и эталон помещают в пенал, изготовленный из того
же вещества, что и материал пробы. Толщина стенок пенала
101
должна быть достаточной для полного удаления той компонен-
ты потока нейтронов, которая наиболее сильно поглощается
веществом пробы. Конечно, эта процедура связана с потерей
чувствительности, но выигрыш состоит в надежности результа-
тов !i возможности анализа проб неправильной формы.
Интерферирующие ядерные реакции
В активационном анализе идентификация исследуемого эле-
мента и его количественное определение предполагают наличие
однозначной связи между аналитическим радиоизотопом и ис-
ходным стабильным изотопом. Иными словами, аналитический
радиоизотоп должен возникать только по одной определенной
ядерной реакции. Однако в ядерных процессах это условие
реализуется не всегда. Возможны случаи, когда один и тот же
радиоизотоп образуется в результате различных ядерных реак-
ций, иногда сочетающихся с процессами радиоактивного рас-
пада. Эти реакции в отличие от основной получили название
интерферирующих (мешающих).
Иногда к интерферирующим реакциям относят также такие,
которые приводят к образованию других радиоизотопов, но с
тем же зарядом ядра, что и у аналитического радиоизотопа
(радиоизотопы одного элемента). Однако в противоположность
предшествующему случаю эту помеху обычно можно обнару-
жить с помощью соответствующих физических методов и учесть.
Интерферирующие реакции могут быть источниками значи-
тельных погрешностей, зависящих от условий активации. Рас-
смотрим интерферирующие реакции, которые возможны при
облучении потоками тепловых нейтронов, полученных в резуль-
тате замедления быстрых нейтронов.
Для характеристики величины возможной систематической
погрешности, возникающей по интерферирующей ядерной ре-
акции, применяют коэффициент интерференции, который опреде-
ляют следующим образом. Обозначим радиоактивные продукты
ядерных реакций следующими символами: В — радиоизотоп,
образующийся по основной реакции из определяемого элемен-
та; В* — радиоизотоп, образующийся по интерферирующей
ядерной реакции.
Предположим, что имеем самый неблагоприятный случай и
радиоизотопы В и В* идентичны, а звездочка означает только
способ образования радиоактивных ядер. Тогда коэффициент
интерференции будет равен отношению активностей радиоизо-
топов В и В*, образующихся в результате облучения равных
количеств интерферирующего и определяемого элементов:
А р *
= (<24)
102
Иначе говоря, коэффициент интерференции численно равен
кажущейся добавке к количеству определяемого элемента, воз-
никающей вследствие интерферирующей ядерной реакции из
равного количества мешающего элемента. Поэтому если изве-
стно содержание мешающего элемента и определен коэффициент
интерференции для данных условий облучения, то легко можно
рассчитать необходимую поправку.
Нетрудно видеть также, что величина 1п кладет предел чув-
ствительности определения исследуемого элемента, если ме-
шающий элемент присутствует во много больших количествах.
Когда масса последнего равна 1 г, а определяемого элемента
/п г, то интерферирующая и основная реакции дают равный
вклад в активность образующегося радиоизотопа. Естественно,
что минимальное количество определяемого элемента, которое
еще может быть достаточно точно определено, будет иметь
величину, близкую к хотя чувствительность метода в отсут-
ствие помехи может быть гораздо выше.
Коэффициент интерференции можно определить эксперимен-
тально. Для этого определенное количество элемента (0,1 — 1 г),
дающего помеху, и эталон исследуемого элемента облучают
заданное время в постоянно используемой зоне облучения.
Затем определяют активности аналитического радиоизотопа,
образующегося по основной и интерферирующей ядерным реак-
циям при пересчете на 1 г исходного элемента. В зависимости
от конкретных условий это определение выполняется радиохи-
мическим или инструментальным методом. Во избежание по-
грешности проба мешающего элемента не должна содержать
в качестве примеси определяемый элемент. Его присутствие
должно быть проконтролировано каким-либо подходящим спо-
собом. Выделяются два основных типа интерферирующих ядер-
ных реакций — первого и второго порядка.
Интерферирующие ядерные реакции первого порядка. При
облучении тепловыми нейтронами реактора протекает основная
реакция z 'А(п, у) z А. Однако присутствие в потоке некото-
рой доли быстрых нейтронов и жестких у-квантов может выз-
вать целый ряд интерферирующих ядерных реакций типа:
1) ^_,А(п, р)«А; 2)^s3A(n, ct)"A; 3)^+IA(n, 2л)« А;
4) Г’А(у, n)zM А; 5)^> 'А(у,< А.
Как видно, конечные продукты всех перечисленных реакций
совпадают с продуктом основной реакции и поэтому дают до-
полнительный вклад в активность аналитического радиоизотопа.
Правда, третья и четвертая реакции при постоянстве спектраль-
ного состава излучения особой опасности не представляют, так
как протекают на изотопах определяемого элемента. К тому же
из-за малых величин сечений этих реакций по сравнению с
основной их вклад в активность радиоизотопа z А обычно
очень мал.
ЮЗ
Что карается остальных реакций, то они приводят к образо-
ванию радиоизотопа А из ядер элементов, имеющих поряд-
ковый номер на одну или две единицы больше. Таким обра-
зом, два элемента, стоящие справа от определяемого, могут
оказывать влияние на результаты анализа. Поскольку энергия
у-квантов, возникающих при делении 2®U, не превышает 10,5 Мэв
[95], а порог реакции (у, р) для подавляющего большинства
элементов выше этой величины, то эта реакция крайне редко
может быть источником помех. Обычно основным источником
помех являются интерферирующие реакции па быстрых нейтро-
нах (первая и вторая реакции). Уровень помех зависит от ве-
личины плотности потока быстрых нейтронов в месте облучения.
Эти реакции приводят к систематическим погрешностям, глав-
ным образом при определении двух соседних элементов, нахо-
дящихся слева от элемента (элементов) с высокой концентра-
цией. Большое число интерферирующих ядерных реакций на
быстрых нейтронах для различных матриц суммировано в ра-
боте Кали [116].
Величину возможной систематической погрешности можно
оценить путем расчета или экспериментально. Расчет коэффи-
циента интерференции проводится согласно уравнению (2.22)
__ ^в* 0*Л4 СТВ* Фб
11 Лв 0Л4* ств Фт
Поскольку МжМ*, то окончательно получаем
._____®в* gB* Фб
*и п ’ ’ ~Z •
®В °В
(4-25)
(4-26)
Изотопный состав основного и интерферирующего элементов
и сечения соответствующих реакций известны [30—32], поэтому
для расчета /., в условиях эксперимента необходимо знать от-
ношение плотностей потоков быстрых и тепловых нейтронов.
В табл. 9 приведены величины коэффициентов /п, полученные
расчетным путем, для некоторых интерферирующих реакций
[106]. При расчете было принято Фб/Фт = 1 и время облучения
48
Экспериментальный путь оценки коэффициента интерферен-
ции более точен и не требует предварительного определения от-
ношения Фб/Фт. Для этого определенное количество элемента,
дающего помеху, облучают в тех же условиях, в которых впо-
следствии проводятся аналитические определения. Измерение
активности интерферирующего радиоизотопа позволяет затем
оценить и коэффициент /и. Однако чтобы полученный результат
был надежен, необходимо быть уверенным, что взятый препа-
рат не содержит примеси элемента, для которого оценивается
величина помехи.
104
Таблица 9
Коэффициент /и для некоторых интерферирующих реакций
Определяе- мый элемент Радиоактив- ный изотоп Интерферирующая реакция
Na 24Na 24Mg (п,р) 27 А1 (л, а) 2- Ю-s РЮ”3
К 42К 45Sc (п,а) 7-Ю-2
Мп 56Мп 56Fe (п.р) 59Со (п ,а) 3- IO-3 1-10-5
Си 64Си 64Zn (п ,р) 5-IO—3
Fe 59Fe 59Со (п,р) 62Ni (я,а) l-10-i 3-10“4
Р 32Р 32S (п,р) 35С1 (я, а) 7-10—1 1-10“2
Са 45Са 48Ti (п,а) З-Ю-3
Для контроля за чистотой препарата могут быть использо-
ваны различные методы, в том числе и активационные. Напри-
мер, Кемп и Смейле [117] для оценки помехи при определении
Sc брали 130—140 мг спектрально чистого титана. Для допол-
нительного контроля чистоты титана применили облучение в
тепловой колонне реактора, где плотность потока быстрых ней-
тронов ничтожно мала. Только убедившись в отсутствии примеси
Sc, пробу титана облучали в рабочем канале реактора и рас-
считывали
Коэффициент интерференции можно определить и при нали-
чии примеси основного элемента в препарате мешающего, если
использовать метод двукратного облучения — с фильтром из
Cd и без него [118]. Тогда по результатам двух облучений эта-
лонов основного и мешающего элементов оценивают
^B’^Cd ^в*
тп ^в (^Cd ')
(4.27)
где та и т„ — масса эталонов определяемого и мешающего
элементов; Лв—активность аналитического радиоизотопа в
эталоне определяемого элемента; А3, и Лв, — активность
этого же радиоизотопа в эталоне мешающего элемента после
облучения в полном и отфильтрованном потоках; Леа — кадмие-
вое отношение для определяемого элемента.
Метод двукратного облучения позволяет непосредственно по-
лучать концентрацию определяемого элемента по уравнению
с
Л°б1Ц 4б|Ц- . • 100»%,
Л-4 и
(4.28)
105
где ЛОбщ(Лэ) и Л'общИ'э) — активность аналитического радио-
изотопа в стандартных условиях в навеске пробы w (эталоне
та) соответственно для облучения в полном и отфильтрованном
потоках нейтронов.
Более того, этот метод позволяет одновременно определить
и концентрацию в пробе мешающего элемента са:
^С<1А>бщ. Л>бщ
LCd ~ 1
ш/нЛэ
100%.
(4.29)
Если аналитический радиоизотоп образуется по двум интер-
ферирующим реакциям (п, р) и (п, а), то уравнение (4.28)
остается в силе. Однако определение концентрации мешающих
элементов становится уже невозможным, и необходимо опреде-
ление одного из этих компонентов каким-либо независимым спо-
собом, что просто сделать, используя излучение радиоизотопов
этих элементов, образующихся по реакции (п, у).
Важно отметить, что метод двойного облучения не снимает
ограничений в отношении чувствительности определения, для
реализации которой требуется облучение в условиях с хорошо
термализованным нейтронным потоком. Например, при определе-
нии меди по реакции 63Cu(n, у)иСи в цинке имеет место интер-
ферирующая реакция 64Zn(n, p)64Cu [119]. В результате пре-
дельная чувствительность метода [3-10~8 г при Ф=3-10“ ней-
трон/(см2 • сек)] из-за интерференции не может быть реализо-
вана. Так, при потоке нейтронов с Еса = 30 порог определения
меди в пробе цинка массой 1 г составляет всего 1,5-10~4 г, но
при Еса = 8000 он уже достигает 7,5-10-7 г.
Иногда лучшие результаты можно получить, если в качестве
аналитического использовать другой радиоизотоп, образую-
щийся из определяемого элемента. Например, для определения
меди в цинке лучше воспользоваться реакцией 65Си(н,- у)66Сщ
поскольку сечение интерферирующей реакции 66Zn(n, p)66Cu
меньше, чем в предыдущем случае [120].
Особое место среди интерферирующих реакций занимает
деление, ибо при этом образуются радиоизотопы многих эле-
ментов, находящихся в середине периодической системы." Так,
деление 235U тепловыми нейтронами дает радиоизотопы элемен-
тов примерно от цинка до гадолиния.
Активность мешающего радиоизотопа связана с содержа-
нием урана соотношением
Аг = Л : 0.): Ф [ 1 - е-°93?о-л А| (4.30)
где Д’с—число атомов урана; 0 — изотопное содержание 23;,U;
т)дел—выход рассматриваемого радиоизотопа в продуктах де-
ления; Tz — его период полураспада; п:1Р! — сечение деления.
Используя уравнение (4.30), Кали [116] рассчитал наиболее
важные помехи, которые создает присутствие 1 мкг 235U в пробе
106
массой 1 г при облучении в течение 5 суток [Ф=1012 ней-
трон! (см2-сек)]. Поскольку деление 235U приводит к помехам
при определении большого числа элементов, то при активаци-
онном анализе на тепловых нейтронах требуется проведение
предварительного анализа на уран. Когда помехи от продуктов
деления урана велики, приходится прибегать к его удалению
перед облучением. При наличии в потоке быстрых нейтронов
происходит еще деление и ^Th.
Помеха может возникнуть и в результате реакции радиаци-
онного захвата, если получающийся радиоизотоп начинает це-
почку радиоактивных распадов, сопровождающихся изменением
заряда ядер.
Примером может служить следующая цепочка:
17«Yb(n, у) 177Yb ——177Lu ———> 177Hf.
' 1' 1 ,9 ч 6,8 дня
Поскольку дочерний 177Lu совпадает с аналитическим радио-
изотопом, образующимся по реакции 176Lu(n, y)177Lu, то в при-
сутствии иттербия возникают затруднения при определении
лютеция. Активность мешающего радиоизотопа А2 связана с
количеством исходного элемента и условиями анализа следую-
щим соотношением:
4 = ^(1_е-хМ +
______В__________________(б ^а*обл_________е ^**расп
Xj — А,*_______________________________]
-I------—------(1 — е~,’’,обл) (е-’/'/Расп — е~Л,/Расп)]
(4.31)
где ядернофизические величины оп, 0П, пги, относятся к ин-
терферирующей реакции, a Xt и Х2—постоянные распада мате-
ринского и дочернего изотопов. Для оценки величины помехи
надо определить содержание в пробе мешающего элемента. Это
можно сделать, облучив вместе с пробой его эталон и проведя
количественное определение по материнскому или какому-либо
другому радиоизотопу.
К этому случаю примыкают помехи от радиоизотопов естест-
венных радиоактивных рядов. Например, при определении
висмута по реакции
208Bi (п, у) 23 °Bi —------------> 2,0Ро------------> 206РЬ
5,01 дня 138 дней
создается помеха от части радиоактивной цепочки уранового
107
ряда
21<>РЬ Р ) 21O£3i Р , 210Ро а > 206РЬ.
21 год 5,01 дня 138 дней
Наилучший способ введения поправки в данном случае состоит
в прямом измерении естественной активности в исходной пробе
до ее облучения [121].
Интерферирующие ядерные реакции второго порядка. Реак-
ции этого типа можно представить такой последовательностью
процессов:
М д (п, v) м+1д f-распад М+1 « (л, 7)
/А—-------> Z А--------:----> Z + 1 Ж-----
Ф, а /.г ~ Ф, а,
М-2. распад у «
Z +1Л - * л- /Л.
В этой цепочке ядерных процессов изотопы А и z~i А пред-
ставляют собой стабильные изотопы естественных элементов
(Л1 = 0, л3=0). Если конечный продукт этой последовательности
м+2 А совпадает с аналитическим радиоизотопом определяемого
элемента, то возникает неразрешимая помеха. Нетрудно заме-
тить, что в зависимости от вида 0-распада в этой последова-
тельности помеха возможна от элемента, стоящего справа или
слева от определяемого. Накопление числа ядер N*4 конечного
радиоизотопа следует уравнению
4
N* = А01ф2ст1(у3Я.2 С,е^Л‘го5л; (4.32)
i=i
где N'oi — число атомов первичного изотопа.
Постоянная С\- определяется из выражения
' |'| д V -
4=1
где А = 7>-гФо.
Коэффициент интерференции в случае помех второго поряд-
ка с учетом эффекта выгорания рассчитывается по соотноше-
нию [122]
I„ = — = -^0*- .-----Ф(Т1М:Н ~ а-ф)---V С,е“Л^°бл (4.33)
Л-4 Л1403 (е-^ф?0бл _ е-л^обл)
Согласно уравнению (4.33), величина коэффициента интер-
ференции определяется условиями облучения и соответствую-
щими ядерными характеристиками и в зависимости от них ме-
няется в широких пределах. В самом неблагоприятном случае
коэффициент 1,л достигает значений порядка IO-1’. Следователь-
но, возможность помех от реакций второго порядка следует
иметь в виду при определении микроколичеств элемента, поряд-
108
ковый номер которого отличается на ± 1 от порядкового номера
макрокомпонента.
Всего имеется 104 случая, когда реакции второго порядка
могут иметь место, в 65 случаях помехи от них являются
существенными. Величина помехи может быть определена рас-
четным путем или экспериментально. Расчеты по уравнению
(4.33) довольно сложны и трудоемки, поэтому они выполняются
с помощью вычислительных машин. Такие расчеты были выпол-
нены и их результаты суммированы в работе [123]. Расчеты
можно значительно упростить, применив разложение в ряд для
экспоненциальных членов уравнения (4.33) {122].
Глава
5
ФОТОАКТИВАЦИОННЫЙ АНАЛИЗ
§ 1. Моноэнергетическое и тормозное у-излучение
В фотоактивационном анализе для воздействия на ядра эле-
ментов используются у-кванты достаточно высокой энергии
> 1 /Мэе). По энергетическому спектру источники у-излуче-
ния делятся на две группы. К первой из них относятся источ-
ники, спектр которых является линейчатым и состоит из одной
или нескольких моноэнергетических линий, а во вторую группу
входят источники со сплошным спектром.
Источниками моноэнергетических квантов служат процессы,
связанные с переходами между уровнями ядер, такие, как ра-
диоактивный распад, захват нейтронов и протонов и резонанс-
ная флуоресценция. Недостатки этих источников обычно со-
стоят в сравнительно низкой энергии излучения, невозможности
изменения энергии и слабой интенсивности. Для получения
моноэнергетического излучения с переменной энергией находит
применение процесс аннигиляции быстрых позитронов [124}.
Однако сложность оборудования и низкая интенсивность не
позволяют использовать такие источники в аналитических
целях.
Следует отметить, что наличие источника моноэнергетиче-
ского излучения, позволяющего менять энергию в области при-
мерно 2—30 Мэв и обладающего при этом достаточно высокой
интенсивностью, предоставило бы исключительно интересные
возможности для анализа. Вследствие порогового характера и
резонансного хода кривых возбуждения фотоядерных реакций
путем изменения энергии облучения было бы значительно проще
подбирать условия для избирательного определения исследуе-
мого компонента.
Поскольку такие источники отсутствуют, то для фотоактива-
ционного анализа наибольшее применение получили различные
ускорители электронов, дающие тормозное излучение со сплош-
ным энергетическим спектром. Интенсивность тормозного излу-
чения современных ускорителей достигает высоких значений,
имеется возможность регулировки максимальной энергии в ши-
роких пределах. Достигаемая на ускорителях максимальная
энергия (более 1 Гэе) лежит далеко за областью, представляю-
щей интерес для аналитических применений (1—45 Мэв).
110
В связи с важным значением тормозного излучения для
активационного анализа необходимо рассмотреть подробнее его
характеристики. Тормозное излучение получают путем ускоре-
ния электронов тем или иным способом до высоких энергий с
последующим направлением их на мишень из тяжелого метал-
ла (W. Pt, Au и др.). В процессе радиационного торможения
электронов в веществе мишени образуется поток квантов, имею-
Рис. 23. Нормированные спектры тормозного из-
лучения для разных энергий электронов.
щий непрерывное распределение по энергии. Спектральное Рас-
пределение квантов простирается практически от нулевой энер-
гии до максимальной кинетической энергии электронов.
Спектр тормозного излучения имеет сложную форму и не
поддается точному теоретическому определению. Предложено
несколько способов расчета, дающих приближенное решение
этой проблемы. Наилучшее согласие с экспериментальными ре-
зультатами дает выражение, предложенное Шиффом. Оно до-
вольно сложно и требует весьма трудоемких расчетов. Соот-
ветствующие расчеты были выполнены с помощью вычислитель-
ных машин, и полученные данные можно найти в работе [124].-
Спектр тормозного излучения зависит от энергии ускоренных
электронов, что видно из рис. 23, на котором приведено норми-
рованное распределение интенсивности для пяти значений энер-
гии электронов. С ростом энергии первичных электронов в
спектре тормозного излучения возрастает доля более жестких
у-квантов.
111
Теоретические расчеты спектра тормозного излучения выпол-
няются в предположении, что используется бесконечно тонкая
мишень. С мишенью конечной толщины, особенно в случае цик-
лических ускорителей, происходит деформация спектрального
распределения за счет процессов, приводящих к поглощению
и уменьшению энергии фотонов.
Имеет место также угловая зависимость спектра тормозного
излучения. На рис. 24 показано угловое распределение излуче-
80 40 0 40 80
Зткланение от оси пучка град
Рис. 24. Угловое распределение
тормозного излучения бетатро-
на (25 Мэв) по активации ме-
ди (1) и углерода (2).
ния бетатрона на 25 А4эв, измерен-
ное по наведенной активности в про-
бах графита и меди. Видно, что ак-
тивация графита (ЕПор= 18,6 Мэв)
указывает на более узкие размеры
пучка, чем активация меди (£’Пор =
= 10,8 Мэв). Иначе говоря, при пе-
ремещении от центра пучка к его
периферии происходит смягчение
спектра тормозного излучения. Это
значит, что реальный спектр тор-
мозного излучения, проходящий че-
рез образец, будет зависеть от
охватываемого им телесного угла.
Дополнительные искажения в ис-
ходный спектр тормозного излуче-
ния вносит рассеянное излучение от
мишени, защиты и различных уст-
ройств, находящихся в эксперимен-
тальном зале.
Итак, реальный спектр тормоз-
ного излучения в облучаемой пробе
зависит от многих факторов и его
выполнено лишь весьма приближенно,
невысокую точность в оценке сечении,
определение может быть
Если к этому прибавить
а также сложную зависимость сечений фотоядерных реакций от
энергии у-квантов, то становится очевидной практическая невоз-
можность применения в фотоактивационном анализе абсолют-
ного метода. Поэтому все определения с помощью тормозного
излучения проводятся по методу эталонов или мониторов.
Для получения наиболее точных результатов обычно стре-
мятся проводить облучение равномерным потоком активирую-
щего излучения. Однако пучок тормозного излучения электрон-
ных ускорителей обладает высоким градиентом плотности, ко-
торый оказывается тем выше, чем больше энергия электронов.
На рис. 25 показано угловое распределение тормозного излуче-
ния линейного ускорителя для нескольких значений энергии.
В принятых полярных координатах интенсивность излучения в
прямом направлении условно взята за единицу, а угловое
распределение выражено в долях интенсивности в прямом на-
112
правлении. Как следует из рис. 25, электроны с энергией 3 Мэв
дают довольно широкий пучок тормозного излучения с относи-
тельно небольшим градиентом. По мере увеличения энергии
пучок сжимается и соответственно возрастает градиент в рас-
пределении интенсивности. При энергии 20—30 Мэв интенсив-
Рис. 25. Угловое распределение интенсивности
тормозного излучения в полярных координатах
при энергии:
7 — 3 ЛЬь: 2 — 5 Мэв-, 3 — 10 Мэв-. 4 — 20 Л/эв.
ность излучения уже довольно резко спадает при отклонении от
центра пучка.
Для более наглядной характеристики величины градиента
можно отметить, что при энергии электронов 20 Мэв угловой
раствор пучка, ограниченного областями с вдвое меньшей ин-
тенсивностью, чем в центре пучка, составляет примерно 6°. Это
значит, что на расстоянии 1 м от мишени интенсивность пучка
падает вдвое при отклонении от его центра примерно на 5 см.
113
Облучение проб часто проводят близко к мишени (10—20 см),
что неизбежно повышает величину градиента. Для получения
•более равномерного дозного поля в зоне облучения применяют
корректировку с помощью фильтров, что связано с потерей
интенсивности, или циклическое перемещение пучка электронов
ло мишени.
Энергия, Мэв
Рис. 26. Энергетическое распределение плотности
потока у-квантоз тормозного излечения.
Для оценки аналитических возможностей различных ускори-
телей, а также для сопоставления данных, полученных в раз-
личных условиях, необходима единая мера интенсивности тор-
мозного излучения. В отличие ог нейтронного активационного
анализа, где плотность потока нейтронов [нейтрон! (см--сек)]
оказывается удобной и общепринятой единицей для характери-
стики условий облучения, в фотоактивационном анализе плот-
ность потока у-квантов редко используется для этих целей.
114
На рис. 26 показан спектр тормозного излучения на рас-
стоянии 1 .и от мишени для пяти различных энергий электронов,
который выражен в единицах плотности потока у-квантов, отне-
сенных к току ускорителя 1 мка и энергетическому интервалу
0,5 Мэв. Хорошо виден сложный характер зависимости плот-
ности потока у-квантов от максимальной энергии излучения.
Поэтому для абсолютной
характеристики пучков тор-
мозного излучения используют
различные параметры, кото-
рые можно сравнительно про-
сто измерить. Одни из них яв-
ляются косвенными и харак-
теризуют мощность использо-
ванного ускорителя, как, на-
пример, ток ускорителя или
мощность пучка электронов.
Для прямой характеристики
пучка тормозного излучения
чаще всего используют мощ-
ность дозы излучения (р/лшн).
С помощью ионизационных
камер, соответствующим обра-
Энергия, naS
Рис. 27. Зависимость мощности дозы
от максимальной энергии тормозного-
излучения на расстоянии 1 .и от ми-
шени при токе ускорителя 1 мка.
зом прокалиброванных, в указанных единицах легко измерить-
мощность дозы излучения, создаваемой ускорителем в той или
иной области пространства. Для взаимного сравнения ускори-
телей мощность дозы определяется на стандартном расстоянии
от мишени, равном 1 м. Для этих условий определено соотно-
шение между мощностью дозы и током ускорителя при разной
энергии тормозного излучения (рис. 27).
Однако общие характеристики мощности ускорителя еще
не дают точной оценки условий в зоне облучения, которую часто
стремятся приблизить к мишени, чтобы повысить плотность
потока излучения. Воспользоваться ионизационной камерой
для измерений уже затруднительно из-за малых размеров пучка.
Тогда определение мощности дозы можно осуществить по вы-
ходу какой-либо хорошо изученной фотоядерной реакции. На-
пример, в работе [125] для этой цели использовали реакцию
55Мп(у, п)54Мп, выход которой при 20 Мэв составляет 2,0X
X 106 люль Измерение абсолютной активности 54Мп,
образовавшейся в известном количестве марганца в данных
условиях, позволяет рассчитать дозу излучения по уравне-
нию (5.6).
Облучение анализируемых проб проводится при определен-
ной энергии тормозного излучения, которая должна иметь высо-
кую стабильность во времени. Для повышения избирательности
определений облучения проводят при различной энергии тормоз-
ного излучения.
115
Чтобы установить связь между показаниями регулирующей
системы и энергией тормозного излучения, часто требуется ка-
либровка шкалы энергий по порогам фотоядерных реакций.
Порог определяют либо по появлению нейтронов, освобождае-
мых в результате реакции (у, и), либо путем регистрации на-
веденной активности. Для калибровочных опытов используют
ядерные реакции, пороги которых с высокой точностью опре-
делены из масс-спектрометрических измерений и данных по
^-'распаду.
§ 2. Взаимодействие жесткого у-излучения
с веществом
Взаимодействие у-квантов с веществом сильно отличается
от взаимодействия тепловых нейтронов. Наиболее важным с
точки зрения активационного анализа является значительно
более слабое взаимодействие у-квантов с ядерными частицами.
Это приводит к малым величинам сечений фотоядерных реакций
и соответственно к более низкой общей чувствительности фото-
активационного анализа. Другое важное отличие — пороговый
характер всех фотоядерных реакций. Возбуждение ядер проис-
ходит только под действием достаточно жестких у-квантов
(£,, > 1 Мэв). Менее важным отличием является сильное взаи-
модействие у-квантов с электронами, вследствие чего при про-
хождении через вещество пучок у-квантов ослабляется главным
образом за счет взаимодействия с электронными оболочками
.атомов. В общем жесткое у-излучение обладает высокой про-
никающей способностью, поэтому можно облучать значитель-
ные по массе пробы без заметного ослабления потока у-квантов.
При взаимодействии у-квантов с атомными ядрами возмо-
жен целый ряд процессов: возбуждение более высоких уровней
ядра (у, у'), ядерные реакции типа (у, п), (у, р), (у, а),
(у, f) и др. Взаимодействие у-квантов с ядрами имеет ярко
выраженный резонансный характер. В области резонанса се-
чение, начиная с пороговой энергии, быстро растет с увеличе-
нием энергии у-квантов до некоторого максимального значения
и затем снова падает (рис. 28). Резонансное взаимодействие
наблюдается при энергии у-квантов 10—20 Мэв. Резонансная
энергия при которой наблюдается максимальное сечение,
закономерно уменьшается с ростом массового числа
Ет = 40,7 • Л!-0-2. (5.1)
Ширина резонансных кривых очень велика и находится в
пределах 6—12 Мэв; по этой причине явление получило наз-
вание гигантского резонанса. В последнее время обнаружено,
что у ряда ядер кривая возбуждения в области гигантского
резонанса имеет тонкую структуру и состоит из нескольких
пи ков.
116
Теории, которая правильно объясняла бы все имеющиеся
факты в области фотоядерных реакций, пока не существует.
Первым приближением в этом направлении явилась модель,
которая объясняет гигантский резонанс коллективными движе-
ниями в ядре протонов относительно нейтронов. Однако с по-
мощью такой простой модели удалось
имеющиеся факты, и потребовалось
ее дальнейшее развитие, которое на-
шло свое воплощение в одночастичной
и позднее многочастичной модели.
Серьезной трудностью для всех
коллективных моделей явилось объяс-
нение экспериментально наблюдаемо-
го высокого выхода фотопротонов в
реакциях (у, р) на средних и тяжелых
ядрах. Для объяснения этого факта
была развита модель прямого фото-
расщепления ядра, т. е. механизма
реакции без образования составного
ядра.
Из всех фотоядерных реакций наи-
меньшие величины пороговой энергии
и наибольшие сечения в большинстве
случаев свойственны реакции (у, и).
Реакции, сопровождающиеся вылетом
заряженных частиц, требуют обычно
более высокой энергии у-квантов и
объяснить далеко не все
Рис. 28. Функция возбужде-
ния реакции 12С(у, я) "С.
имеют много меньшие величины сечений. Это связано с необ-
ходимостью преодоления потенциального барьера, что умень-
шает вероятность протекания реакции и требует дополнитель-
ных затрат энергии. Влияние потенциального барьера возра-
стает с ростом заряда ядра. По этой причине для реакции (у,
р), которая в области легких ядер имеет примерно такие же
сечения и пороги, как и реакция (у, п), в области тяжелых ядер
ей свойственны уже много меньшие сечения и более высокие
пороги.
Реакция (у, i) с относительно низким значением пороговой
энергии и достаточно высоким сечением протекает только на
очень ограниченном числе ядер (U, Th и т. д.).
Активность радиоизотопа, образовавшегося по фотоядерной
реакции под воздействием моноэнергетического излучения, мо-
жет быть рассчитана по уравнению, аналогичному (2.24):
. 6,02 10-W) / г- х , г- х /, —).t /г
Л= ——------------Ф(Е7) о (£.,)(! — е °°л), (5.2)
где Ф(Еу)—плотность потока у-кзаптов с энергией Ev;
a(t7) —сечение реакции при энергии Еу.
117
Однако при облучении тормозным излучением в связи с его
сплошным характером и резонансным ходом кривых возбуж-
дения фотоядерных реакций уравнение активации принимает
вид
= 6,82 -^6 (j _ е_„обл) о(£) dE, (5 3)
£Пор
где £пор — пороговая энергия фотоядерной реакции; £Макс —
максимальная энергия тормозного излучения; Dy — мощность
дозы излучения; о(£) —функция возбуждения; Фо(£, £макс) —
спектральное распределение потока квантов, приходящихся на
единичный интервал энергии при мощности излучения 1 р.
Интеграл, в уравнении (5.3) не поддается точному анали-
тическому решению. А. М. Якобсон [126] показал, что при ап-
проксимации функции Фо(£, £манс) линейной зависимостью и
при условии, что спектр тормозного излучения охватывает прак-
тически всю резонансную кривую, можно прийти к следующему
выражению:
Е
макс
Dy J Фо(£, ^макс )o(£)d£^O(£m) Ojjht? (5-4)
Е
''пор
где Ф(£т)—плотность потока квантов при энергии, соответ-
ствующей максимуму сечения; оИнт= ) o(£)d£ — интегральное
F '
пор
сечение реакции.
Другой подход состоит в численном интегрировании [127].
Для этого рассматриваемую область энергии разбивают на ряд
интервалов (обычно по I Мэв). Путем расчета получают спектр
тормозного излучения в этой области. Используя имеющиеся
данные или принимая разумные предположения о форме кривой
возбуждения ядерной реакции, в каждом интервале получают
произведение средней плотности потока на среднее сечение и
проводят суммирование по всем интервалам. Расчет выходов
фотоядерных реакций указанным методом дал согласие с экс-
периментальными значениями в пределах ±40%, за исключе-
нием С, О, N и F, где расхождение оказалось много больше.
Для получения лучшего согласия нужно более точно знать
спектр тормозного излучения в условиях эксперимента и кри-
вую возбуждения рассматриваемой фотоядерной реакции.
Точное определение последней представляет сложную зада-
чу и с точки зрения активационного анализа не является
необходимым. Для расчета аналитических возможностей метода
в определенных экспериментальных условиях лучше всего
иметь интегральные кривые возбуждения, г. е. зависимость
выходов фотоядерных реакций от энергии тормозного излуче-
118
ния в стандартных условиях. Применительно к рассматривае-
мому случаю выход определяется выражением
£
макс
Г(£ыакс) = У Ф0(£,£макс)о(£)а'£. (5.5)
£пор
Согласно уравнению (5.5), выход реакции представляет
собой количество взаимодействий, которое происходит в еди-
Рис. 29. Изменение выходов реакций <у. ш с зарядом ядра при
энергии торхозного излучения 20 Л1эв.
пице количества вещества под воздействием единичной дозы
облучения. Для радиоизотопа выход равен
Г(£макс) = —7--------
(1 — е оол) т
(5.6)
119
где Dv — мощность дозы излучения; туг— количество изотопа,
моль; Ad — активность радиоизотопа. Иными словами, выход
радиоизотопа численно равен активности насыщения, которая
Рис. 30. Изменение выходов реакции
(у, р) с зарядом ядра при энергии тор-
мозного излучения 20 Мэв.
образуется в одном моле
изотопа под воздействием
тормозного излучения еди-
ничной мощности.
Выход большинства фо-
тоядерных реакций был
определен Ока и др. [125,
128] при энергии тормозно-
го излучения 20 Мэв. Одна-
ко гораздо чаще активацию
элементов тормозным излу-
чением выражают величи-
ной, пропорциональной вы-
ходу,— удельной актив-
ностью элемента. При этом
указывают условия облуче-
ния и измерения, что позво-
ляет путем простого пере-
счета перейти к другим ус-
ловиям.
На рис. 29 показано из-
(у, п) при 20 Мэв в зависимости
мерно возрастает от легких ядер
(от 103 до 107). Заниженные зна-
чения выходов получаются для
ядер с магическим числом прото-
нов и нейтронов (39К, 54Fe, 52Сг,
зэу, 58Nj; 114Sn, i24Sb).
Выходы реакций (у, р) при
той же энергии меняются уже бо-
лее сложным образом (рис. 30).
В области легких ядер выходы
реакций (у, р) равны выходам
реакций (у, и), но после дости-
жения максимума в районе ни-
келя они резко уменьшаются и
затем остаются почти постоянны-
ми. Для элементов с Z>50 вели-
чины выходов составляют менее
0,1% выходов реакции (у, п).
Выходы реакций (у, а), (у,
2п), (у, рп) и других обычно бо-
лее чем на порядок меньше выхо-
да реакции (у, п). Поэтому, как
менение выходов реакции
от заряда ядра. Выход равно-
к тяжелым на четыре порядка
Рис. 31. Изменение удельной ак-
тивности насыщения реакций
16O(v, п)15О (/). 13С(у. »И'С (2)
и ,4.\(у, п)’3.\г (3) с энергией тор-
мозного излучения (ток ускорите-
ля равен 100 мка).
120
правило, наибольшую чувствительность фотоактивационного
определения дают радиоизотопы, образующиеся по реакции
(у, п). В отдельных случаях для аналитических определений
пригодны и реакции других типов. Однако в большинстве слу-
чаев возможность протекания этих реакций следует рассматри-
вать скорее как источник интерферирующих радиоизотопов.
Так же, как и при нейтронноактивационном анализе, особые
затруднения возникают в присутствии делящихся элементов.
Для выбора оптимальных условий фотоактивационного ана-
лиза большой интерес представляют кривые активации элемен-
тов, т. е. изменение удельной активности (выхода) в зависи-
мости от максимальной энергии тормозного излучения при по-
стоянном токе ускорителя. На рис. 31 такие кривые приведены
для О, С и N [129]. Обращает на себя внимание то обстоятель-
ство, что до 30 Мэв кривые активации меняются индивидуаль-
но, а выше 30 Мэв идут параллельно друг другу.
§ 3. Источники у-излучения
Радиоизотопные источники. Из радиоизотопов с достаточно
большим периодом полураспада лишь некоторые имеют энер-
гию излучения выше 2 Мэв и совершенно отсутствуют у-излуча-
тели с энергией выше 3 Мэв. Первоначально в фотоактивацион-
ном анализе довольно широко использовался радиоизотоп 124Sb
(7'1/2 = 60 дней, Еу = 1,69, 2,09 Мэв и др.). Позднее нашел при-
менение 60Со (Г1/2 = 5 лет, Еу = 1,17 и 1,38 Мэв). Кобальтовые
источники в основном предназначаются для радиационных ис-
следований, но попутно они могут служить и для аналитических
целей [130].
Предложено также проводить фотоактивационные определе-
ния с помощью радиационного контура реактора [131]. В этом
контуре через активную зону реактора циркулирует эвтектиче-
ский сплав состава: 13% Sn, 25% In, 62% Ga. Большая часть
(около 99%) всей радиационной мощности контура обусловлена
у-излучением !1В"'1п (Т1/2 = 54 лшн, £v =2,09, 1,49 Мэв и др.).
Облучатель представляет собой шар диаметром 15 c.v, через ко-
торый проходит канал для размещения проб. Общая плотность
потока в центре шара составляет 4-1012 квант/(см2-сек). Воз-
можности радиоизотопных источников ограничены, главным об-
разом из-за низкой энергии излучения. По этой причине они
пригодны только для возбуждения изомерны?; уровней некото-
рых элементов и фотонейтронного определения 2Н и Be.
Электростатические ускорители. В этих установках исполь-
зуется принцип прямого ускорения электронов в постоянном
электрическом поле. Высокое напряжение на ускорительную
трубку подается от электростатического генератора. Ускорители
этого типа позволяют получать электроны с энергией до 5—
6 Мэв. Этот предел обусловлен утечкой заряда по воздуху и
пробоем изоляции.
Электростатические ускорители дают довольно мощные пучки
ускоренных электронов (порядка нескольких миллиампер) и
соответственно тормозное излучение высокой интенсивности. Од-
нако они весьма громоздки и требуют для установки большого
помещения. Поскольку ускорители других типов имеют меньшие
габариты часто при лучших параметрах пучка (ток, энергия),
электростатические ускорители не перспективны для аналитиче-
ских целей.
Бетатроны. Это довольно простой и доступный ускоритель
электронов на малые и средние энергии [132]. Ускорение элек-
тронов в нем происходит под действием вихревого электриче-
ского поля, индуцируемого переменным магнитным полем в ва-
куумной камере. Источник электронов — инжектор, на анод
которого в определенный момент времени подается короткий
импульс высокого напряжения; при этом в камеру впрыскива-
ются электроны. Под действием вихревого электрического поля
электроны начинают вращаться по окружности с постоянным
радиусом, все время увеличивая свою энергию. Это продол-
жается до тех пор, пока нарастает магнитное поле. В конце
ускоряющего периода электроны сбрасываются с орбиты и по-
падают на мишень, где и возникает тормозное излучение. Ре-
гулируя момент сброса электронов, можно плавно менять мак-
симальную энергию тормозного излучения.
Первые типы бетатронов, нашедшие аналитическое приме-
нение, позволяли ускорять электроны до 25—30 Мэв, достигая
мощности дозы излучения порядка 100—200 р! (мин-м). Позднее
были сконструированы сильноточные бетатроны с мощностью
тормозного излучения до 103 рЦмин-м) и более [133].
Для значительного повышения плотности потока тормозного
излучения (правда, в весьма ограниченном объеме) Моринага
предложил метод внутрикамерного облучения [134]. Для этого
пробу облучают в специальной системе внутри ускорительной
камеры бетатрона непосредственно за мишенью, в которой про-
исходит торможение ускоренных электронов.
Сама система представляет собой цилиндр, который вводят
внутрь ускорительной камеры (рис. 32). Внутренний торец за-
крыт тонкой пластинкой. В целом система герметична и позво-
ляет поддерживать нормальный вакуум в камере. Внутрь ци-
линдра пробу вводят с помощью специального держателя [135].
Мишень подвешивают на обращенной к пучку стороне цилиндра.
Плотность потока у-квантов через пробу возрастает в такой
системе благодаря близости к мишени, так как пучок тормоз-
ного излучения не успевает заметно расшириться и имеет малое
сечение. Распределение интенсивности тормозного излучения
в системе для внутрпкамерных облучений, определенное по ак-
тивации медных фолы, приведено на рис. 33 [136]. Видно, что
122
распределение по всем трем измерениям имеет сильный гра-
диент, а размеры пучка тормозного излучения на уровне, со-
ставляющем половину максимальной интенсивности, примерно
Рис. 32. Конструкция системы для внутрикамерных облучений:
/ — конвертер; 2— проба; 3 — платиновый колпачок; -/ — тормозное из-
лучение; 5 — уплотняющее кольцо.
равны 2X4X15 мм. Следовательно, объем активной зоны со-
ставляет всего около 0,13 с,и3. Экспериментальное определение
средней мощности дозы излучения во внутрикамерной системе
Расстояние, мм
Рис. 33. Распределение интенсивности тормозного
излучения в системе для внутрикамерных облучений,
определенное по активации меди:
а — по толщине пробы; б — по высоте пробы; в — по ходу
пучка.
дало значение порядка 1,1-104 р/мин вместо 60 р/мин, которое
развивает бетатрон на расстоянии 1 м от мишени [137].
Микротрон. Первое практическое применение для фотоакти-
вационного анализа микротрон получил, недавно [138]. Имею-
123
шиеся сейчас данные свидетельствуют о том, что это весьма
перспективный ускоритель для получения интенсивных пучков
жесткого тормозного излучения [139].
В микротроне электроны ускоряются высокочастотным элек-
трическим полем в однородном и постоянном магнитном поле.
Движение электронов в вакуумной камере микротрона проис-
ходит по окружностям, имеющим общую точку касания, в ко-
торой располагается ускоряющий резонатор. При каждом про-
хождении через резонатор электроны получают приращение
энергии и переходят на следующую орбиту с большим радиу-
сом. Электроны, ускоренные до заданной энергии, выводятся
через специальный канал.
Микротрон — ускоритель электронов на средние энергии (5—
50 Мэв), т. е. он перекрывает приблизительно ту же самую об-
ласть энергий, что и бетатрон. Однако по сравнению с последним
микротрон — более компактный и эффективный ускоритель, об-
ладающий высокой интенсивностью излучения. Имеющиеся сей-
час конструкции микротронов обеспечивают интенсивность
пучка тормозного • излучения до 104 р!мин. В перспективе воз-
можно создание и более мощных ускорителей.
Линейные ускорители. Этот тип ускорителя пока продол-
жает занимать лидирующее положение среди рассмотренных
ускорителей как в отношении интенсивности, так и макси-
мальной энергии излучения [140]. Здесь электроны ускоряются
в волноводе, в котором с помощью высокочастотного генера-
тора возбуждаются бегущие волны. Электроны, попавшие на
гребень бегущей волны, увлекаются ею, увеличивая свою энер-
гию. При совпадении скорости электрона со скоростью распро-
странения бегущей волны ускорение электрона будет проис-
ходить непрерывно. С помощью линейных ускорителей можно
получать электроны с энергией 1000 Мэв и более.
Линейные ускорители на средние энергии (30—45 Л1эа) '—
довольно сложные и громоздкие установки, но они обеспечи-
вают высокую интенсивность излучения, которая на два-три
порядка превышает интенсивность других ускорителей на tv
же энергию электронов.
§ 4. Методы фотоактивационного анализа
Фотонейтронный метод
В результате реакции (у, п) образуется поток нейтронов,
интенсивность которого пропорциональна содержанию ядер,
принимающих участие в реакции. Возникший поток нейтронов,
обычно небольшой по интенсивности, с помощью современных
методов ядерной физики можно надежно зарегистрировать па
фоне более интенсивного у-излучения [141]. Чаще всего для
этого применяют газонаполненные или сцинтилляционные счет-
124
чики, а также активацию некоторых элементов (In, Ag, Dy).
Метод, в. основе которого лежит измерение интенсивности по-
тока нейтронов, образующихся при облучении анализируемых
проб жестким у-излучением, получил название фотонейтрон-
пого.
Для большинства элементов фотопейтронный метод не об-
ладает высокой избирательностью, так как при достаточно
высокой энергии у-квантов реакция (у, /г) протекает практи-
чески на ядрах все?; элементов, причем в подавляющем числе
случаев пороги реакций очень близки. Однако изотопы неко-
торых элементов имеют небольшую величину энергии связи
нейтрона (табл. 10).
Таблица 10
Изотопы с наименьшим порогом реакции (у, п)
Изотоп Содержание изотопа, % Порог реак- ции. U?e [ Изотоп Содержание изотопа, % Порог реак- ции, .Мэв
9 Be 100 1,67 ,зС 1,108 4,95
2Н 1'0 .0,015 0,037 2,23 4,14 6Li 7,58 5,35
Таким образом, 9Ве и 2Н обладают самым низким значе-
нием порога реакции (у, п). По этой причине фотонейтронному
методу определения Ье и -Н пр
энергией у-излучения в области
2—4 Мэв не мешает присутствие
в пробе любых других элемен-
тов, и метод является для них
исключительно специфичным.
Правда, возможны взаимные по-
мехи, что часто приходится учи-
тывать в практической работе.
Схема установки для фото-
нейтроппогр анализа показана па
рис. 34. Источник у-излучения по-
мешают в центр цилиндрической
кюветы, содержащей анализируе-
мую пробу. Кювета и источник
находятся в свинцовом цилиндре,
который уменьшает влияние у-из-
лученпя на счетчики нейтронов.
Этот цилиндр помещен в блок
парафинового замедлителя со сч
использовании источника с
Рис. 34. Схема установки для фо-
тоиейтрочиого анализа:
1 — парафин: 2- свинцовый цилиндр;
3 — радиоизотопный источник ун-.впн-
тслз; -У— кювета с пробой; 5 — каналы
для счетчиков нейтронов; б—внеш-
няя зашита.
ми нейтронов. Для за-
щиты обслуживающего персонала вся установка снаружи экра-
нируется свинцом и бетоном.
Наибольшее применение фотонейтронный метод получил
для определения Be. В этом случае источником у-излучения
чаще всего служит радиоизотоп 124Sb, а для регистрации потока
нейтронов — пропорциональные борные или сцинтилляционные
счетчики. Получающаяся чувствительность довольно высока и
при активности источника 100 мкюри и массе пробы 100 г до-
стигает 1—2• 10-4%. В целом метод очень прост, быстр и дает
хорошую точность. Элементы с большими сечениями поглощения
тепловых нейтронов (Cd, В и др.) затрудняют анализ. Приме-
нение кадмиевого фильтра позволяет полностью устранить влия-
ние этих элементов, но за счет некоторой потери чувствитель-
ности (на 20—30%). Подробное описание фотонейтронного ме-
тода определения можно найти в работах [142, 143].
Предпринимаются попытки использовать для фотонейтрон-
ных определений электронные ускорители [144]. Это позволяет
повысить чувствительность и обеспечить большую безопасность
работы. Однако возникают проблемы, связанные со стабиль-
ностью интенсивности и энергии излучения.
Фатовозбуждение изомерных уровней
Под воздействием достаточно жестких фотонов некоторые
ядра по реакции А (у, у') А* переходят в метастабильное со-
стояние с достаточно большим периодом полураспада. Боль-
шинство изомерных уровней имеют энергию в интервале 0,14-
4-1 Мэв. Однако известно, что прямое возбуждение изомерных
уровней электромагнитным излучением не происходит. Поэтому
ядро должно быть возбуждено до более высокого энергетиче-
ского уровня, при высвечивании которого уже возможен ча-
стичный или полный переход на изомерный уровень. Распад
изомера совершается путем у-перехода, который часто сильно
конвертирован. Всего имеется около 40 стабильных изотопов,
принадлежащих 30 элементам, у которых период полураспада
изомерных состояний превышает 0,5 сен [131].
Кривые возбуждения реакций (у, у') тоже показывают
резонансный характер, причем максимум приходится на об-
ласть энергий вблизи порога реакции (у, и). В области гигант-
ского резонанса сечения реакций (у, у') имею! крайне малую
величину, но при энергии порядка 20 Мэв появляется второй
максимум.
При облучении у-квантами с энергией ниже порога основных
фотоядерных реакций метод фотоактивационного анализа, ос-
нованный на возбуждении изомерных уровней, вследствие не-
большого числа активирующихся элементов и больших различий
схем распада изомеров весьма специфичен и может быть осно-
вой быстрых и надежных инструментальных методик анализа
различных проб. Ограничения этого метода обусловлены ма-
лыми величинами сечений реакций (у, у'), которые имеют
значения порядка 10-31—КУ-32 см2, и малым выходом у-излу-
чения изомеров из-за конверсии. Это приводит к низкой чув-
ствительности анализа (табл. 11).
Таблица И
Чувствительность метода фотовозбуждения, мг
Эле- мент Изомер T / 1 ' 2 Еу, кэв Со- источ- ник* In-источник11 * Линейный ускоритель*”
Se ”mSe 17,5 сек 160 1 15 0,16
Вг 79mBr 4,78 сек 208 2 15 —
Sr 87ZnSr 2,08 ч 388 25 500 0,3
Y 89my 16,5 сек 908 — 3,4
Rh i03mRh 56 мин 40 — 8 2,0
Ag 44 — 40 сек 94-87 15 50 0,2
Cd HimCd 48,7 мин 150, 247 9 80 0,08
In 4,5 ч 335 1,1 2 0,02
Ba 13’™ Ba 2,57 мин 662 — — 1,9
Lu Пв’ПЩ 3,71 ч 89,1050 — — 0,96
Pt 195^?p^ 4,08 дня 31,130 — 1000 0,2
Au 197WAU 7,4 сек 130,277 — 2000 0,1
Hg j99m]^g 49 мин 159,372 — 4000 0,9
Hf 19 сек 160,217 — 30 0,1
’Источник активностью 80 ккюри [130].
*’Радиационный контур (Ф— 1-1012 квант/(см2 сек) [131].
’’’Линейный ускоритель ( Е~ = 4,2 Мэв, 1=50 мка) [145].
Сравнительно слабая активация элементов в указанных
условиях обусловливает необходимость использования наиболее
эффективного метода регистрации у-излучения изомеров для
достижения наивысшей чувствительности. Поэтому измерения
обычно выполняют с помощью сцинтилляционных кристаллов
с колодцем. Предельная чувствительность анализа, которая
может быть достигнута с мощным ускорителем при энергии
излучения вблизи порога реакции (у, п), составляет примерно
10 7—10~8 г. Такая чувствительность при анализе реальных
проб еще не достигнута, и в опубликованных исследованиях
метод фотовозбуждения преимущественно используется для
определения макрокомпонентов (более 0,1 %).
Можно отметить, что при облучении бериллия излучением
ускорителя получается источник нейтронов с достаточно высо-
кой плотностью потока. При этом оказывается, что облучение
многих элементов этими нейтронами из-за высоких сечений
реакции (и, у) дает на два-три порядка более высокую чувст-
вительность определения, чем в случае фотовозбужденчя изо-
мерных уровней [81]. Это обстоятельство приводит к другому
ограничению метода фотовозбуждепия, так как при энергии
у-излучения выше 2,2 Мэв возможно появление помех от про-
127
дуктов реакции (п, у), если исследуемые пробы содержат Be
или воду.
Гамма-активационный метод
В этом методе аналитические определения проводятся по
излучению радиоизотопов, образующихся по фотоядерным ре-
акциям с пороговой энергией выше 6 Мэв. Чаще всего облуче-
ние проводят при энергии тормозного излучения 20—30 Мэв,
но иногда верхний предел достигает 70 Мэв. Для повышения
избирательности определения часто приходится соответствую-
щим образом регулировать энергию ускорителя.
Среди методов фотоактивационного анализа именно у-акти-
иационный метод в последнее время привлек наибольшее вни-
мание и претерпел интенсивное развитие. Проделана большая
работа по исследованию общих характеристик метода — изуче-
нию наиболее перспективных фотоядерных реакций, оценке чув-
ствительности определения элементов, рассмотрению факторов,
определяющих избирательность и точность метода.
Так, Ока и др. [125, 128] провели широкие исследования про-
дуктов фотоактивации элементов тормозным излучением линей-
ного ускорителя на 20 Мэв. Аналогичные исследования, но при
разных значениях энергии тормозного излучения (12, 15 и
25 Мэв) проведены Андерсеном и др. [146]. Облучение некото-
рых элементов тормозным излучением линейного ускорителя
на 35 Мэв выполнено Дебруном и Альбером [147]. Лутц [127]
расчетным путем оценил удельные активности элементов для
энергий 25, 30 и 35 Мэв. Подробное исследование аналитиче-
ских возможностей бетатрона с внутрикамерным облучением
выполнил Р. А. Кузнецов [137, 148, 149].
Каталог сцинтилляционных спектров у-излучения радиоизо-
топов, образующихся под действием тормозного излучения с
энергией 30 Мэв, подготовлен Бейкером и др. [150]. Точные
данные по энергии этих радиоизотопов суммированы в [151,
152]. Анализ накопленных данных показывает, что при энергии
тормозного излучения выше 20 Мэв большая часть элементов
периодической системы активируется достаточно хорошо. В их
число входят такие важные элементы, как О, N, С и др. Со-
ответствующие оценки приводят к выводу, что для многих
элементов чувствительность может достигать Ю-'—10“80/0.
Наименее благоприятен у-активационный метод для следую-
щих элементов: Н, Li, Be, В, La, Dy, Lu, Bi.
§ 5. Основные области применения
фотоактивационного анализа
К середине 1970 г. по фотоактивационному анализу было
опубликовано около 200 работ [153], что свидетельствует о мед-
ленном развитии метода. Как показали недавние исследования,
128
фотоактивационный метод имеет достаточно хорошие аналити-
ческие характеристики и может быть полезен при решении
многих проблем анализа. Поэтому основная причина отстава-
ния этого направления, видимо, связана с отсутствием простых
в управлении, надежных в работе, относительно недорогих, по
достаточно мощных ускорителей.
Гамма-активационный метод, несомненно, занимает веду-
щее место в фотоактивационном анализе, что обусловлено ши-
роким кругом определяемых элементов и более высокой чув-
ствительностью. Именно этот метод находит преимущественное
применение в области малых концентраций. Конечно, по срав-
нению с активационным анализом на тепловых нейтронах ре-
актора у-активационный анализ имеет несколько меньшую
предельную чувствительность. Однако, по оценке Лутца [153],
по количеству элементов, для которых чувствительность пре-
вышает 10 7 г, электронный ускоритель со средним током
100 мка и £Торм -~30 Мэв равноценен ядерному реактору с
плотностью потока тепловых нейтронов 1 -1013 нейтрон/(см2Х
X сек).
Гамма-активационный анализ в противоположность тепловым
нейтронам позволяет успешно определять некоторые элементы
(С, N, О и др.). К тому же при действии жесткого у-излучения
значительно слабее проявляются эффекты, связанные с ослаб-
лением активирующего излучения веществом пробы.
Конечно, при у-активации возникают свои специфические
трудности. В частности, при определении малых компонентов
гораздо более существенной может оказаться проблема интер-
ферирующих ядерных реакций. Это объясняется не только тем,
что фотоядерные реакции могут протекать по нескольким ка-
налам, но и тем, что это излучение всегда сопровождается
потоком нейтронов, возникающих в реакциях (у, п). Однако
проблема интерферирующих реакций в у-активационном анализе
еще не получила глубокой разработки.
По своему составу образующиеся при фотоактпвации радио-
изотопы значительно отличаются от продуктов облучения тепло-
выми нейтронами. Среди них преобладают нозитронноактивные
радиоизотопы, часто с близкими схемами распада. Это обстоя-
тельство в определенной степени затрудняет применение инст-
рументального метода, и поэтому в случае у-активанионного
определения малых компонентов чаще приходится прибегать к
радиохимическому разделению.
Применение фотоактивационного анализа в настоящее время
идет по двум основным направлениям. Одно из них связано с
разработкой методик анализа некоторых легких элементов и
прежде всего О, С и N [153]. Основные усилия исследователей
здесь направлены на достижение по возможности наибольшей
чувствительности определения этих элементов в различных ма-
5 Р- А. Кузнецов
129
Таблица 12
Чувствительность и фотоактивационные характеристики некоторых
легких элементов
Эле- мент Фотоядерная реакция Е пор ’ Мэв ТЧ„ мин ‘ * £₽*• Мэв Чувствитель- ность**, мкг
с 12С (у, п) нс 18,6 20,7 0,97 0,04
N 14N (у, п) 13N 10,5 10,1 1,20 0,06
О 16О (у, п) 15,7 2,05 1,73 0,05
F 1’F (у, п) 18F 10,5 112 0,65 0,02
Р 31Р (у, п) зоР 12,4 2,56 3,30 0,015
S 32S (у, пр) 30Р 19,2 2,56 3,30 0,3
Fe 64Fe (у, п) 53Fe 13,8 8,9 2,84
Си 63Cu (у, л) 62Си 10,9 9,7 2,92 —
*Все радиоизотопы — практически чистые позитронные излучатели.
**Линейный ускоритель (fTopM = 28 Мэв, I = 50 мка) [ 154].
териалах. Хотя предел у-активационного определения С, N и О
довольно высок (табл. 12), но вследствие не очень благоприят-
ных фотоактивационных характеристик этих элементов (высокие
пороги, низкие сечения, чисто позитронный распад и т. д.) об-
ласть их инструментального определения ограничивается мате-
риалами, основные компоненты которых слабо активируются
при облучении, и пределом чувствительности порядка 10~3—
10~4 %. Причем результаты анализа часто находятся в сильной
зависимости от присутствия в пробе других примесей. Как рас-
ширение круга анализируемых на эти элементы материалов,
так и повышение чувствительности и надежности анализа тре-
бует применения химического выделения.
В области макроконцентраций фотоактивационный анализ
позволяет создавать весьма быстрые и достаточно точные
инструментальные методики для этих элементов, как, напри-
мер, определение С, О и N в природном угле [155] или изучение
стехиометрического состава соединений кислорода с иодом
[135].
Другое направление фотоактивационного анализа — экс-
прессные инструментальные методы анализа сравнительно вы-
соких (0,001 —100%) содержаний элементов [156]. В это на-
правление в меру своих возможностей дают вклад все методы
фотоактивационного анализа. Успехи в разработке быстрых
методик анализа часто представляют большое практическое
значение. При определении высоких содержаний на облучение
одной пробы часто достаточно всего несколько минут, следо-
вательно, с одним ускорителем при его непрерывной работе
можно обеспечить массовый анализ проб. Необходимо только,
чтобы аналитическая лаборатория была оснащена необходи-
мым количеством измерительной и анализирующей аппарату-
130
ры, работающей преимущественно в автоматическом режиме.
В'качестве примера можно привести методику у-активацион-
ного определения F во флюоритовой руде [157]. Инструменталь-
ная методика позволяет проводить анализ 200 проб за рабо-
чий день.
Фотоактивациониые методы анализа вещественного состава
горных пород и руд приобретают болыное значение для поиска
полезных ископаемых, аналитического контроля процессов пере-
работки и обогащения в горнорудной и перерабатывающей
промышленности [143, 158]. Для многих практически важных
элементов предложены экспрессные методики, которые обла-
дают достаточной чувствительностью и точностью.
Весьма перспективно применение фотоактивацноппых мето-
дов для анализа проб сравнительно простого состава, таких,
например, как сплавы. Избирательность метода к определяе-
мым компонентам может быть столь высокой, что становится
возможным экспрессный анализ со сравнительно простым обо-
рудованием при хорошей чувствительности и надежности [159,
160]. Возможна также автоматизация процесса аналитического
контроля. Фотоактивациоиный анализ позволяет также опре-
делять несколько компонентов в одной навеске, хотя это уже
требует более сложной методики анализа.
Помимо отмеченных выше основных направлений имеются
разрозненные и весьма разнородные попытки применения фо-
тоактивационного анализа для решения различных аналитиче-
ских проблем. Они часто дают хорошие примеры, иллюстриру-
ющие аналитические возможности фотоактивациоввого ана-
лиза.
Пока подавляющее число предложенных фотоактивацион-
ных методик рассчитано на одновременное определение в
пробе небольшого числа компонентов. Чаще всего анализ вклю-
чает один-два компонента и очень редко три. Однако возмож-
ности метода в целом, видимо, значительно шире, поэтому
ведутся предварительные исследования для разработки много-
компонентных методик при анализе объектов сложного соста-
ва [161].
5*
Глава
6
АКТИВАЦИОННЫЙ АНАЛИЗ
С ПРИМЕНЕНИЕМ ЗАРЯЖЕННЫХ ЧАСТИЦ
§ 1. Взаимодействие заряженных частиц с веществом
Для облучений могут быть использованы ядра различных
элементов, имеющих необходимую энергию. Источниками пото-
ков заряженных частиц служат либо некоторые ядерные про-
цессы, либо специальные установки, в которых ускоряются ионы
этих элементов. Практическое применение в активационном
анализе находят главным образом ядра (ионы) изотопов водо-
рода и гелия: протоны р, дейтроны d, ядра трития /, гелий-3
(3Не) и а-частицы. Проводятся первые исследования с тяжелы-
ми ионами [364].
При взаимодействии ускоренных ионов с ядрами элементов
протекают многообразные ядерные превращения, специфичные
для каждой используемой частицы. Тем не менее все методы
активационного анализа на заряженных частицах имеют и об-
щие черты. Прежде всего, следует отметить наличие порога
ядерных реакций, протекающих с их участием, который в основ-
ном связан с кулоновским барьером. Однако известно, что из-за
туннельного эффекта заряженная частица имеет некоторую ве-
роятность проникновения через барьер, которая непрерывно
уменьшается с падением энергии частиц. Следовательно, нет ка-
кого-то определенного значения энергии, ниже которого про-
никновение заряженной частицы в ядро полностью исключено.
Поэтому, исходя из чисто практических соображений, за порог
реакции с участием заряженных частиц принимают такую ве-
личину энергии, при которой вероятность туннельного эффекта
пренебрежимо мала. Эту величину принято называть эффек-
тивным барьером. Связь между кулоновским и эффективным
барьерами можно представить соотношением
СэФф = k5Uк <С 1).
Величина k,-, обусловливается типом заряженной частицы и при-
нятой вероятностью преодоления кулоновского барьера. На-
пример, для ионов 3Не [162] рекомендуется &б = 0,62. Для экзо-
энергетических реакций величина порога совпадает с эффектив-
ным барьером, а для эндоэнергетических определяется тем
процессом, который требует большей энергии частицы.
132
Функции возбуждения многих ядерных реакций с участием
заряженных частиц имеют общую форму (рис. 35): быстрый
рост при превышении пороговой энергии, достижение макси-
мума и после этого медленный спад. Уменьшение сечения на
последнем участке обычно связано с появлением конкуренции
со стороны других реакций, которые становятся энергетически
возможными. Плавный ход кривой возбуждения иногда нару-
шается резонансными пиками,
где наблюдается сильное воз-
растание сечения. Большинст-
во ядерных реакций, представ-
ляющих аналитический инте-
рес, имеют величину сечения
между 0,001 и 1 барн.
Большое влияние на анали-
тические характеристики мето-
да оказывает сильное взаимо-
действие заряженных частиц с
электронными оболочками ато-
мов при их движении в веще-
стве мишени. В результате ча-
стица быстро теряет энергию
на возбуждение и ионизацию
атомов. Поскольку потеря
Рис. 35. Функция возбуждения реак-
ции 2;iXa(d, p)24Na.
энергии в одном акте взаимо-
действия на большей части пути много меньше (в среднем око-
ло 35 эв), чем полная энергия частицы, а масса частицы много
больше массы электрона, однородные заряженные частицы оди-
наковой начальной энергии проходят в веществе примерно рав-
ный путь, практически не отклоняясь от первоначального на-
правления. Таким образом, величина пробега R3.4 пучка заря-
женных частиц в данном веществе пропорциональна их началь-
ной энергии.
Знание соотношения энергия — пробег часто очень важно для
практических целей. Однако поскольку простое уравнение, ко-
торое связывало бы пробег и энергию и было справедливо для
разных частиц и веществ различного состава, отсутствует, то точ-
ные данные получают экспериментально или путем сложных
расчетов. Предложено также несколько эмпирических выраже-
ний для оценки величины пробега. Согласно работе [163], для
активационных применений достаточно точные результаты для
простых веществ (элемент) дает соотношение
р =24__________—
*\З.Ч „ 2 3/.
(6.1)
где /м — эффективный ионизационный потенциал вещества;
М— массовое число вещества; ZM и zm — относительный заряд
матрицы и активирующей частицы; Е3. ч — энергия частиц, Мэв.
133
Для определения величины пробега в веществах сложного
состава применяют правило Брэгга, которое в математической
форме записывается в виде уравнения
— . 4- , (6.2)
Rt Ri R2 Rn
где R; — пробег в веществе сложного состава; Rx, R2,...,Rn —
пробег в отдельных компонентах; w2,wn—доля каждого
простого компонента в веществе.
В целом пробег заряженных частиц в веществе мал и изме-
ряется долями миллиметра (табл. 13).
Таблица 13
Пробег протонов в некоторых металлах
£р> Мэв А1 Си Та
ММ мг/см2 ММ мг/см2 ММ
1 3,94 0,015 6,3 0,007 12 0,007
10 169 0,63 219 0,24 336 0,2
20 574 2,1 719 0,8 1042 0,63
Еще одно общее свойство методов облучения заряженными
частицами — одновременное протекание нескольких ядерных
реакций на одном изотопе. Как правило, реакции сопровожда-
ются изменением зарядового состояния облучаемых ядер, при-
чем для легких элементов значительную вероятность при уме-
ренной энергии активирующего излучения имеют реакции с из-
менением заряда на несколько единиц. Множественность кана-
лов ядерных реакций часто оказывается неблагоприятным фак-
тором, так как в результате облучения возникает сложная смесь
радиоизотопов, имеют место нежелательные помехи для опре-
деления исследуемых элементов. В результате появляются за-
труднения при выполнении анализов, возникает потребность в
применении более сложных процедур и методов.
Так, под действием протонов наиболее вероятно протекание
следующих реакций: % А(р, у) М г Мр> п) ?+1 А;
"А(р, 2я) MjA А; А(р, d)?-' А; % А(р, а) А. Если к
этому добавить, что перечисленные реакции относятся к одно-
му изотопу, а многие элементы состоят из нескольких изотопов,
то станет очевидным, что возможное количество образующихся
радиоактивных продуктов может быть значительным. Очень
часто это обстоятельство заставляет снижать максимальную
энергию активирующего излучения, чтобы повысить избиратель-
ность определения, что приводит к следующему: 1) активация
134
заряженными частицами оказывается наиболее благоприятной
для определения легких элементов, где кулоновский барьер
низок; 2) уменьшается чувствительность.
Мерой интенсивности потока заряженных частиц служит ток
пучка пли число частиц в единицу времени. При облучении
мишени достаточной толщины заряженные частицы в пучке дви-
жутся прямолинейно, постепенно теряя энергию, но с незначи-
тельным изменением полного числа частиц. Вследствие потери
энергии у частиц меняется вероятность вступления в реакцию,
пока энергия не упадет ниже пороговой. Эти особенности облу-
чения заряженными частицами приводят к специфической форме
уравнения активации.
При облучении тонкой мишени, в которой не происходит
заметной потери энергии частиц, уравнение активации имеет
следующий вид:
dA = F4cxo0dx (1 — е-Л/обл) t (6.3)
где dA— активность тонкой мишени, имп!сек-, F4 — поток ча-
стиц, частиц;сек.-, сх — концентрация атомов определяемого эле-
мента в мишени, атомАмг-, его — сечение реакции при энергии
облучения, см2-, dx— толщина мишени, мг)см2.
Потери энергии при прохождении через вещество для частиц
нерелятивистских скоростей даются уравнением
dE 2ле4г2 .. ,, . 4^з.ч
---=---------NVZ In — —
rfx Е3 ч-----/ср
(6.4)
где е — заряд электрона; Е3. ч—энергия частицы; г— заряд
частицы; Z — заряд ядер вещества; Л'г — число атомов в 1 смл
вещества; /ср — средняя энергия возбуждения атома.
Для образца сложного состава, учитывая аддитивность тор-
мозных способностей элементов, входящих в его состав, урав-
нение (6.4) можно написать в виде
dE
dx
2ne4Z2
4E
^з.ч
ср
(6-5)
Если величину dx из уравнения (6.5) подставить в уравнение
(6.3) и проинтегрировать, считая, что логарифмический член
мало меняется с энергией, так что можно ввести постоянное
среднее значение Е3.ч, то получим
/ — 71 \ /?макс
F4cx (1 — е ’ обл) J £3 ча (£) dE
Ad =----------------------------------. (6.6)
4Е
YNZiwdz2 In---
Др
Из-за трудностей, встречающихся при определении некото-
рых параметров, абсолютный метод анализа на основе уравне-
135
ния (6.6) мало практичен. Поэтому в активационном анализе
на заряженных частицах находят преимущественное применение
разные варианты метода эталонов (мониторов). Аналитические
определения с помощью заряженных частиц можно осуществить
двумя методами — путем облучения тонкой или толстой ми-
шени. В подавляющем большинстве случаев используется по-
следний метод.
Когда анализируемая проба и эталон настолько тонки, что
потерей энергии частицами можно пренебречь, а облучение и
измерение проводятся в идентичных условиях, то расчет кон-
центрации определяемого элемента проводится из соотношения
= сх
Лэ сэ
Однако при облучениях тонких слоев потоком заряженных ча-
стиц наблюдается потеря вещества мишени, обусловленная эф-
фектом ядер отдачи [164, 165]. В процессе упругого столкнове-
ния или ядерной реакции ядро мишени может приобрести
энергию, достаточную для вылета из тонкой мишени. Чтобы
компенсировать эти потери, для облучения берут несколько
(более трех) одинаковых тонких фольг, складываемых вместе.
После облучения крайние фольги отбрасываются, а средние
служат для аналитических определений. Понятно, что в ходе
облучения средние фольги теряют примерно столько же ядер,
сколько приобретают от соседних фольг, поэтому их состав
практически не меняется.
В методе толстого слоя толщина пробы (эталона) превы-
шает пробег заряженных частиц. Однако тогда из-за неболь-
ших размеров пучка облучение пробы и эталона неизбежно
должно осуществляться в разных опытах, поэтому в этой си-
туации наиболее удобен метод мониторов. Чаще всего для этой
цели измеряют ток пучка с помощью цилиндра Фарадея, иног-
да прибегают к монитору из подходящего материала, который в
виде очень тонкой фольги при облучении располагают перед
пробой (эталоном).
В последнем случае выражение для вычисления конечного
результата имеет вид
^Х ___ АдУ СХ (AZ) V \ /ср / э
^МЭ Гэ S (NZ)3 / 42; \
1п( ----— I
\ Ар ' х
Конечно, если анализируемая проба и эталон имеют строго оди-
наковый химический состав, то выражение (6.8) упрощается и
совпадает с уравнением (6.7). Однако в качестве эталонов
обычно используют стехиометрические соединения, состав кото-
рых сильно отличается от состава пробы. В таком случае, как
136
следует из уравнения (6.8), необходимо учесть различие в тор-
мозной способности вещества пробы и эталона. Для определе-
ния поправок разработаны экспериментальные методы, но они
сложны и приближенны [166].
Предложено несколько способов получения количественных
результатов, которые позволяют избежать необходимости введе-
ния поправок и упростить процедуру активационного анализа
с заряженными частицами по методу толстого слоя. Так, Рикси
и Хан [162, 164] показали, что для аналитических целей более
подходит не обычное сечение реакции, а некоторая величина,
которую они назвали средним сечением:
я я
| о (х) dx Jo (х) dx
о
где о(х) —функция изменения сечения с глубиной слоя; R3. ч —
величина пробега заряженных частиц определенной энергии в
веществе мишени. Поскольку имеет место связь пробег —
энергия, то для среднего сечения может быть выведена другая
форма уравнения (6.9):
F
_ П ИЗЛ
о = ——- f o(E)dE, (6.10)
/?2 J
изл £Пор
где о(Е)—функция возбуждения реакции; Еизл— энергия ак-
тивирующего излучения; ЕПОр— пороговая энергия реакции.
Важная особенность среднего сечения состоит в том, что
его величина слабо зависит от материала мишени и практиче-
ски постоянна для данной ядерной реакции и энергии облуче-
ния. Экспериментально было показано, что при переходе от
ядер с Z = 4 до Z = 95 величина о меняется только на 8% и
всего на 3% в интервале от Z = 4 до Z = 57.
Если воспользоваться параметром о, то интегрирование урав-
нения (6.3) дает выражение
Ах = F4c^R3 ,ч (1 — е~“обл). (6.Ц)
Уравнение (6.11) по форме практически совпадает с основным
уравнением активации и, следовательно, допускает применение
тех же методов получения количественных результатов, что и в
случае других методов активационного анализа. Однако для
того чтобы такой подход стал возможным, необходимо опреде-
лить величину о для данной реакции на каком-либо одном ве-
ществе. Конечно, для решения более широкого круга аналити-
ческих проблем желательно иметь функцию изменения о от
энергии заряженных частиц.
137
Величина а может быть рассчитана из экспериментальных
данных по уравнению (6.11). Оценку также можно сделать пу-
тем графического интегрирования функции возбуждения [урав-
нение (6.9)]. Был еще развит метод аналитического интегриро-
вания при аппроксимации определенных участков функции воз-
буждения прямыми линиями [162]. Тогда получаются простые
выражения, которые позволяют быстро и с приемлемой точно-
стью (7—14%) провести необходимые расчеты.
Для получения количественных результатов при активации
заряженными частицами Энгельманом [167] был предложен
метод «эквивалентной толщины». Если анализируемую пробу
с толщиной, превышающей пробег заряженных частиц, облучить
с тонким монитором, который содержит известное количество
определяемого компонента, то отношение активностей этого ком-
понента в пробе и мониторе будет равно
jVvS j Fxs (х) dx
-------------, (6.12)
4М
где Nv — количество примеси в единице объема пробы, г/см5',
тУ1 — количество определяемого компонента в мониторе, г; S —
облучаемая площадь пробы, см-; Fo — начальный поток частиц,
частиц/сек; Fx— поток частиц на глубине х, частиц/сек; х0 —
пробег частиц в анализируемом материале, см; о(х) —изменение
сечения с глубиной пробы; Ах и Лм— активность пробы и мо-
нитора. Уравнение (6.12) допускает количественное определе-
ние даже при различном составе пробы и монитора, однако для
хо
этого необходимо определить интеграл f Fxa(x)dx.
о
Чтобы облегчить аналитическую процедуру, вводят величину
х0
| Fxo (х) dx
где е:1[;Е — эквивалентная толщина.
Вновь введенный параметр е-)кв может быть измерен экспери-
ментально следующим образом. Между двумя мониторами по-
мещают тонкую фольгу из анализируемого материала, проводят
облучение и рассчитывают отношение активностей монитора.
Затем количество фолы между мониторами постепенно увели-
чивают, пока их общая толщина не превысит пробег заряжен-
ных частиц. Представляют графически зависимость отношения
активностей мониторов от толщины прокладки и определяют
площадь под получившейся кривой активации. Величина этой
площади численно равна эквивалентной толщине. Определив од-
138
нажды этот параметр для анализируемой пробы, можно прово-
дить количественные расчеты по уравнению
Nv = . —Дд__. (6.14)
-•1м ‘5тэкн
Если содержание примеси желательно выразить в виде относи-
тельной концентрации, то можно воспользоваться соотношением
сЛ. = —, (6.15)
^М^ЭКвРпр
где рпр—плотность анализируемого материала.
Метод эквивалентной толщины (в той форме, в какой он из-
ложен выше) имеет два недостатка: во-первых, анализируемый
материал для получения кривой активации должен быть досту-
пен в виде тонких фольг (пленок), а это не всегда возможно;
во-вторых, для каждого нового материала надо заново опре-
делять величину Гэкв-
Избежать обоих затруднений можно путем математического
перехода от одной матрицы к другой [165, 168]. Такие расчеты
выполняются с помощью вычислительных машин по специаль-
но составленной программе. Одновременно можно вычислить
эквивалентную толщину методом численного интегрирования
кривой активации для требуемой энергии облучения. Следова-
тельно, измерив однажды кривую активации для какой-либо
аналитической реакции при максимально возможной энергии
излучения на подходящей матрице путем математических рас-
четов, можно определить величину еЭ1;в для матрицы любого
состава и требуемого промежуточного значения энергии излуче-
ния. Например, при определениях углерода кривые активации
оказывается удобнее всего получать с помощью майларовой или
полиэтиленовой пленки, из которой изготовляется необходимое
количество мониторов [1651. Их собирают в стопку достаточной
толщины и облучают коллимированным пучком заряженных
частиц (1 см2). После облучения первый монитор отбрасывает-
ся, так как он не скомпенсирован относительно ядер отдачи.
Затем последовательно измеряют активность мониторов и после
введения поправок на распад рассчитывают отношение активно-
стей и строят кривую активации (рис. 36). Далее следуют уже
разобранные операции.
Наконец, Н. Н. Краснов [163] разработал метод получения
количественных результатов активационного анализа с приме-
нением заряженных частиц на основе экспериментально опре-
деленного выхода ядерной реакции. С учетом выхода реакции
уравнение активации на заряженных частицах принимает вид
Ad r/1,oli (1 — е-и°бл), (6.16)
Л
139
где /ион — сила тока пучка ионов, мка (Т'ч— 6,3* 10^^/^оп); Y
выход ядерной реакции, расп)(сек-мка • ч). Соотношение (6.16)
позволяет рассчитать выход реакции по измеренным в ходе
эксперимента параметрам (Л^, /Ион, Л>б.ч). Полученная величина
учитывает все каналы реакций, приводящих к данному изотопу
при облучении определяемого элемента.
W 38 36 34 32 30 28
Энереия1 МэВ
Рис. 36. Кривая активации углерода по реак-
ции 12С(а, ап) "С.
Экспериментальные величины выходов, естественно, удобнее
получить для простых веществ. Тогда для толстого слоя веще-
ства сложного состава уравнение активации принимает вид
= .7ион(1_е-^обл), (6.17)
Kk Pi
где kAim — активность радиоизотопа k, образующегося из эле-
мента i в матрице т; Wim — относительное содержание i-ro эле-
мента; /.fe— постоянная распада радиоизотопа k; рт и р,- — про-
ницаемость вещества матрицы и
/ АВ Г1 гд\
заряженных частиц! р= —I'f].
определяемого элемента для
Для вещества сложного со-
става величина рт определяется с помощью соотношения, ана-
логичного (6.2).
Знание величины выхода реакции позволяет определять со-
держание абсолютным методом по уравнению (6.17). Если при-
бегнуть к методу эталонов, но в качестве последнего использо-
вать любое подходящее вещество известного состава, процесс
получения количественных результатов упрощается при одно-
временном повышении точности:
Wim = wi3
!1Ai,„P3
kAi3pm
(6-18)
140
Как видно из уравнения (6.18), необходимость в определении
выхода отпадает, а абсолютные измерения заменяются относи-
тельными.
При полном поглощении пучка заряженных частиц практиче-
ски вся его энергия переходит в тепло, количество которого мо-
жет быть значительным. Так, мощность пучка протонов при токе
100 мка и энергии 20 Мэв составляет 2 кет. Нагревание пробы
при облучении может привести к ее разрушению или частичной
потере вещества. Поэтому успешно облучать интенсивными по-
токами заряженных частиц можно только тугоплавкие материа-
лы. Охлаждение пробы водой или применение специальных
вращающихся систем представляет частичное решение пробле-
мы. Тем не менее облучения при предельных токах (100—
1000 мка) используются довольно редко. Наиболее практич-
ный интервал токов ионных пучков лежит между 0,1 и 10 мка.
Необходимость ограничения тока пучка связана еще и с радиа-
ционными разрушениями материала пробы, которые также мо-
гут привести к нежелательным последствиям.
§ 2. Источники заряженных частиц
Радиоизотопные источники и ядерные реакции. Как было
отмечено ранее, некоторые радиоизотопы тяжелых элементов
испускают а-частицы, энергия которых лежит в интервале
44-9 Мэв. Из имеющихся а-излучателей наибольшее распро-
странение получил 210Ро (£«=5,3 Мэв, ТД/2= 140 дней). Источ-
ники приготовляют путем нанесения тонкого слоя 210Ро на ме-
таллический диск. Чтобы избежать потерь 210Ро с поверхности
источника за счет агрегатной отдачи, активный слой сверху
покрывают тонкой пленкой, что несколько уменьшает энергию
а-частиц. Активность источников лежит в интервале 0,14-1 кюри
(соответствует току ускорителя 0,0014-0,01 мка). Для умень-
шения энергии излучения применяют тонкие фильтры. Недо-
статок полониевого источника заключается в его относительно
небольшом периоде полураспада. В этом отношении более бла-
гоприятными характеристиками обладает 241 Ат (£а =5,49 Мэв,
Т1/2==458 лет).
Поток заряженных частиц может возникнуть в результате
различных ядерных реакций. Наибольший интерес представля-
ют реакции, протекающие с участием тепловых нейтронов. Это
следствие высоких сечений и плотностей потоков, позволяющих
получать потоки заряженных частиц большой интенсивности.
Фактически в аналитическую практику вошла только одна
реакция 6Li(n, а)3Н, получающиеся в ней ядра трития имеют
энергию 2,7 Мэв.
Подготовка к облучению состоит в тщательном смешивании
тонкоизмельченной пробы с порошкообразной литиевой солью.
В результате каждая частица анализируемого материала ока-
зывается окруженной частицами соли, которые при облучении
в реакторе становятся источниками ядер трития. Поскольку
пробег ядер трития такой энергии в твердых веществах состав-
ляет примерно 20—30 мкм, то и размеры частиц анализируемого
материала не должны превышать удвоенной величины про-
бега.
Источником заряженных частиц могут быть также упругие
столкновения быстрых нейтронов с ядрами некоторых легких
элементов. Доля уносимой ядром энергии нейтрона умень-
шается с увеличением массы ядра и угла столкновения. Наи-
большую энергию быстрый нейтрон передает протону при ло-
бовом столкновении, в этом случае энергия протона отдачи
оказывается равной начальной энергии нейтрона. Таким обра-
зом, облучая водородсодержащее соединение быстрыми нейт-
ронами реактора, можно получить поток протонов отдачи со
сплошным энергетическим спектром, простирающимся до
максимальной энергии нейтронов деления.
Число протонов отдачи, образующихся в 1 см3 вещества за
1 сек, определяется уравнением
iVp= JnpOJ£)®„(£)dE, (6.19)
6
где Np — число протонов отдачи; пр — плотность протонов в ве-
ществе; оя(Е)—сечение упругого рассеяния; Ф1г(£)—плот-
ность потока нейтронов.
Сечение упругого рассеяния уменьшается с ростом энергии
нейтронов и для нейтронов в области энергий 1—20 Мэв падает
от 6 до 0,5 барн [69]. Так как потери энергии протонами отдачи
в облучаемом веществе пропорциональны числу электронов в
1 см3 пе, то поток протонов Фр пропорционален отношению
Пр/tip. Это означает, что поток протонов будет заметно изме-
няться от одного водородсодержащего вещества к другому.
При облучении тяжелой воды нейтронами с энергией 1'4 Мэв
можно получить дейтроны отдачи, максимальная энергия кото-
рых достигает около 12,5 Мэв (пробег в воде 1,2 мм) [169].
Потоки заряженных частиц, получающиеся в ядерных реакциях
и процессах отдачи, невелики. Однако, поскольку они возни-
кают непосредственно в объеме анализируемой пробы (смеси),
общая масса облучаемого вещества оказывается большой.
Ускорители. В ядерной физике для получения заряженных
частиц с энергией 0,1—30 Мэв в основном применяются электро-
статические ускорители, циклотроны и линейные ускорители.
Первые два типа представляют собой основные инструменты
анализа на заряженных частицах. Электростатические ускорите-
ли уже упоминались в связи с получением потоков тормозного
излучения.
142
Циклотрон — резонансный ускоритель. Ионы набирают энер-
гию при многократном ускорении, двигаясь в постоянном маг-
нитном поле в резонанс с переменным электрическим полем вы-
сокой частоты. Магнитное поле, в котором находится ускори-
тельная камера, заставляет ионы двигаться по окружности,
радиус которой тем больше, чем выше скорость ионов. По до-
стижении необходимой энергии ионы попадают на исследуемую
пробу (внутреннее облучение). Пучок ионов может быть также
выведен из камеры циклотрона с помощью отклоняющей систе-
мы (внешнее облучение). Облучение на выведенном пучке
больше подходит для активационных определений, поскольку
при этом более просто решается проблема охлаждения, уста-
новки и извлечения проб в ходе анализа.
Излучение циклотрона импульсное. Средняя величина ион-
ного тока достигает 1 ма и более (1015—1016 частиц) сек). Хоро-
шо сфокусированный пучок ионов имеет круглое сечение пло-
щадью около 1 см2 (84]. Энергия заряженных частиц, получае-
мых на циклотроне, обычно лежит в интервале 10-4-15 Мэв, при
этом разброс энергии частиц в пучке не превышает 1%. Неко-
торые конструкции циклотронов допускают широкое изменение
энергии пучка, а также позволяют ускорять частицы разного
типа.
Циклотрон — весьма сложная и дорогая установка, требую-
щая специального помещения и квалифицированного обслужи-
вающего персонала. При всем этом он по сравнению с ядерным
реактором имеет очень малую производительность, так как из-за
малых размеров пучка одновременно можно облучать неболь-
шое число проб. Все это затрудняет широкое аналитическое
применение циклотронов. Делаются попытки разработать неболь-
шие и сравнительно простые конструкции циклотронов [170].
§ 3. Методы активационного анализа
на заряженных частицах
По-впдимому, наиболее подходящая основа для классифи-
кации методов активационного анализа с помощью заряжен-
ных частиц — тип используемой бомбардирующей частицы, от
которой зависят наиболее характерные ядерные реакции и осо-
бенности каждого метода.
Облучение протонами. Протоны могут вызвать ядерные реак-
ции следующих типов: (р, у), (р, п), (р, 2и), (р. d\. (р, а)
п др. Из них наибольшее применение для аналитических при-
менений нашли реакции (р, у), (р, п) и (р, а).
В результате реакции (р, у) образуется ядро с зарядом на
единицу больше исходного, конечное ядро довольно часто ока-
зывается стабильным. Для этого типа реакций очень характер-
ны резко выраженные резонансы. Сечение реакции (р, у) обыч-
но мало для всех элементов.
143
По реакции (р, п) образуется изобар исходного ядра, кото-
рый, как правило, радиоактивен и путем позитронного распада
опять переходит в исходное ядро. Из-за различия масс протона
и нейтрона эти реакции также эндоэнергетичны. Для реакций
(р, п) характерны высокие сечения.
Реакции (р, а) приводят к ядрам с зарядом на единицу
меньше, которые в большинстве своем оказываются стабильны-
ми. Сечения реакций (р, а) имеют высокие значения для лег-
ких ядер и уменьшаются для тяжелых.
Облучение дейтронами. Под действием дейтронов возможно
протекание реакций (d, р), (d, п), (d, a), (d, 2п), (d, t) и др.
Известно, что энергия связи нуклонов в дейтроне мала и состав-
ляет лишь 2,2 Мэв, в то время как средняя энергия связи нук-
лона в более тяжелых элементах равна примерно 8 Мэв. Поэто-
му ядерные превращения, вызываемые дейтронами, всегда силь-
но экзоэнергетичны и часто происходят при относительно низ-
кой энергии дейтронов.
Исследование ядерных реакций под действием дейтронов по-
казало, что реакция (d, р) имеет обычно более низкий порог,
чем остальные реакции. Этот факт обусловлен особым характе-
ром взаимодействия дейтронов с ядрами. При приближении к
ядру дейтрон попадает в его кулоновское поле, которое, не дей-
ствуя на нейтрон, отталкивает протон. Так как расстояние между
нуклонами в дейтроне велико, то нейтрон может проникнуть в
поле действия ядерных сил раньше, чем протон преодолеет
кулоновский барьер. При этом происходит «развал» дейтрона,
и если дейтрон имел небольшую энергию, то протон из-за куло-
новского отталкивания не сможет проникнуть внутрь ядра. Та-
кой механизм ядерной реакции получил название реакции
«срыва».
Реакция (d, р) является основной при энергиях дейтронов
порядка нескольких мегаэлектронвольт. Нетрудно видеть, что в
результате этой реакции образуются те же радиоизотопы, что и
по реакции (п, у). С ростом энергии дейтронов начинают играть
важную роль ядерные реакции других типов.
Облучение ядрами трития. Этот метод пока не получил су-
щественного развития. Единственным примером облучений яд-
рами трития в аналитических целях является реакция
|6О(/, n)18F. Источником ядер 3Н чаще всего служит реакция
BLi(n, сс)3Н. Так как кулоновский барьер ядер кислорода равен
3,2 Л1э8, а энергия получающихся ядер трития составляет
2,7 Мэв, то сечение реакции 16О(^, n)18F имеет довольно высокую
величину (о = 0,5 барн). Метод сопряженных реакций можно
применять для определения как кислорода, так и лития
[171, 172]. Делаются первые попытки получать пучки ядер три-
тия на ускорителях [173].
Облучение ядрами 3Не. Это сравнительно новый метод,
предложенный Марковичем и Махони в 1962 г. [174]. Уже в этой
144
работе была отмечена перспективность метода, особенно для
определения легких элементов, в связи со специфичным харак-
тером ядерных реакций, протекающих с участием ядер 3Не.
Из-за низкой энергии связи нуклонов в ядре 3Не (2,6 Мэв
на нуклон) многие ядерные реакции с их участием являются
экзоэнергетическими. Поэтому пороги реакций связаны преиму-
щественно с кулоновским барьером, а сечения реакций довольно
высоки. Под воздействием 3Не протекают разнообразные ядер-
ные реакции: (3Не, п), (3Не, р), (3Не, 2п), (3Не, ос), (3Не, 2а)
и т. д. Как правило, при этом образуются нейтронодефицитные
радиоизотопы.
Облучение а-частицами. Для ос-частиц сравнительно неболь-
шой энергии (4—20 Мэв) характерны реакции типа (а, п),
(ос. р) и (а, у). С ростом энергии становятся возможными более
сложные ядерные реакции. Поскольку энергия связи нуклонов
в ядре 4Не велика (около 7 Мэв на нуклон), большинство
ядерных реакций с участием а-частиц эндоэнергетические. Про-
дукты реакции (ос, р), как правило, стабильны, в то время как
реакция (а, п) часто дает радиоизотопы.
§ 4. Применение активационного анализа
на заряженных частицах
Не пытаясь дать полный обзор по применению заряженных
частиц в аналитических целях, отметим только основные тен-
денции, которые определяют развитие этого метода. Активация
заряженными частицами, получаемыми с помощью современ-
ных ускорителей, допускает разработку аналитических методик
для весьма широкого круга элементов. Тем не менее основные
усилия аналитиков, развивающих этот метод, направлены преж-
де всего на разработку методов определения легких элементов,
преимущественно на очень низком уровне концентраций.
Эта проблема имеет большое практическое значение. Изве-
стно, что нейтронный и фотоактивационный методы не дают ее
полного решения, тогда как активационный анализ на заряжен-
ных частицах как раз наиболее благоприятен для определения
именно легких элементов [26]. При этом важную роль играет
ряд моментов. Здесь и возможность выбора удобного канала
ядерной реакции, и благоприятные характеристики схем распа-
да продуктов активации. Пороги ядерных реакций на легких
элементах низки, поэтому, ограничив энергию заряженных ча-
стиц, можно избежать активации средних и тяжелых элемен-
тов. Сечения многих реакций достаточно велики.
Исследованию ядерных реакций различных заряженных ча-
стиц с легкими элементами посвящен ряд работ [175—177].
Особенно большое внимание было уделено методу активации с
помощью ядер 3Не [162, 164, 174]. Оценка чувствительности ак-
тивационного анализа на заряженных частицах показала, что
145
предел обнаружения легких элементов часто достигает 10-7%.
На основе активации заряженными частицами предложен ряд
высокочувствительных методик определения таких важных эле-
ментов, как бор, кислород, углерод, азот и другие, в различных
полупроводниковых материалах и чистых металлах. Однако воз-
можности активационного анализа на заряженных частицах не
ограничиваются простым определением общего количества ис-
следуемого компонента. Возможны самые различные примене-
ния этого метода, связанные главным образом с изучением по-
верхностных слоев образцов. Так, с помощью тонкого пучка
ионов (диаметром 0,05 мм) удалось осуществить сканирование
поверхности образца и изучить распределение кислорода по
ней [178]. Пучок ионов, энергии которых совпадают с одним из
резонансов исследуемого компонента, оказался полезен при изу-
чении распределения этого компонента по глубине тонкого по-
верхностного слоя [179].
Активационный анализ на заряженных частицах используется
и как инструментальный метод анализа сравнительно высоких
содержаний легких элементов (преимущественно Li, В, Be, F).
В большинстве случаев такие методы предназначены для мас-
сового анализа при поисках полезных ископаемых, для опера-
тивного контроля состава продуктов переработки и обогащения
[158]. Для этих целей наиболее пригодны радиоизотопные источ-
ники заряженных частиц.
Облучение заряженными частицами позволяет установить
изотопный состав элемента. Соответствующие методики пред-
ложены для водорода [169], лития и бора [180], кислорода [181],
кальция [182], причем изотопный состав часто может быть
определен в тонком поверхностном слое или в небольшом коли-
честве вещества. Определения характеризуются высокой чувст-
вительностью и точностью.
Следует обратить внимание еще на одну особенность анали-
тических определений с помощью заряженных частиц. Имеется
в виду гораздо более широкое использование мгновенного излу-
чения для аналитических целей, чем это имеет место для других
методов активационного анализа. Видимо, решающую роль
здесь играет тот факт, что поток заряженных частиц легко по-
глощается веществом пробы, которая, таким образом, служит
естественным фильтром, тогда как возникающие в ходе ядерного
взаимодействия нейтроны и у-кванты легко проникают через нее
и мог^т быть зарегистрированы без помех со стороны первич-
ного активирующего излучения. Различие в составе, энергии и
пространственном распределении активирующего и мгновенного
излучений делает возможной их раздельную регистрацию. По-
этому ядерные реакции, сопровождающиеся вылетом заряжен-
ных частиц, тоже находят важное аналитическое применение.
Глава
7
СПЕКТРОМЕТРИЧЕСКИЕ МЕТОДЫ
ИНСТРУМЕНТАЛЬНОГО
АКТИВАЦИОННОГО АНАЛИЗА
§ 1. Методы инструментального активационного анализа
Облучение анализируемой пробы вызывает сложный спектр
вторичного излучения (мгновенного или задержанного). Изуче-
ние качественного состава, энергетического распределения и не-
которых других характеристик этого излучения дает ключ к
идентификации ядерной реакции или радиоизотопа. Эти данные
в сочетании с параметрами активирующего излучения (тип,
энергия) позволяют установить элемент, ответственный за появ-
ление в исследуемом спектре излучения определенного типа и
энергии. Интенсивность этого излучения дает необходимую ко-
личественную информацию.
Методы анализа, основанные целиком на физических средст-
вах дискриминации, идентификации и количественного определе-
ния по вторичному излучению облученных элементов, объединя-
ются под общим названием «инструментальный активационный
анализ». В свою очередь, последний довольно четко распадается
на две группы. Одну из них составляют методы, связанные с
регистрацией энергетического распределения определенного вида
излучения; их удобнее обозначить как «спектрометрический ин-
струментальный активационный анализ». Другая группа вклю-
чает несколько разнородных методов, для которых введено
условное название «специальные методы инструментального
активационного анализа» (см. гл. 8). Часть этих методов при-
годна только для определения единичных элементов, а другая
чаще всего используется для повышения избирательности спек-
трометрического метода.
Спектрометрия ионизирующего излучения и прежде всего
у-излучения — наиболее гибкий и универсальный метод инстру-
ментального активационного анализа. Возможности спектро-
метрического метода определяются как свойствами ионизирую-
щего излучения, так и параметрами регистрирующей установки.
Из большого разнообразия методов спектрометрии ионизи-
рующего излучения [183, 184] наибольшее значение для актива-
ционного анализа приобрели системы, работающие на принципе
преобразования энергии исследуемого излучения в последова-
тельность электрических импульсов с последующим анализом по-
лучающегося амплитудного распределения. Для аналитических
147
целей наиболее важны следующие параметры спектрометров:
форма амплитудного распределения, линейность энергетической
шкалы, временное и энергетическое разрешение и другие.
Если с помощью многоканального спектрометра измерить
моноэнергетическое излучение
Рис. 37. а-Спектр 210Ро.
какого-либо радиоизотопа, то на
его выходе будет получено не-
которое распределение числа
отсчетов по каналам анализи-
рующей системы. Примером
может служить спектр a-излу-
чения 210Ро (рис. 37). При
этом пик на получившейся
спектрограмме имеет харак-
терную колоколообразную фор-
му, близкую к распределению
Гаусса. Ширина пика много
больше естественной ширины
линии и обусловлена разре-
шающей способностью спект-
рометра. Для характеристики
разрешения общепринятой ме-
рой служит ширина пика на
половине его высоты (см.
рис. 37). Разрешение спектро-
метра может быть выражено в
абсолютных (£1/2) и относительных (6г=£1/2/£Изл, где £изл—
энергия излучения) единицах.
Как правило, предел разрешению спектрометра кладет ста-
тистический характер процессов, происходящих в детекторе при
преобразовании энергии излучения в импульсе напряжения (то-
ка). В отдельных случаях становится заметным влияние шумов
электронных устройств. Разрешение спектрометра зависит от
энергии регистрируемого излучения и улучшается с ростом по-
следней согласно приблизительному соотношению £i/-? =а +
+ 6/ /£ИЗ Л» где а и b — постоянные величины, характерные для
данного спектрометра.
Реакция спектрометра на моноэнергетическое излучение, ко-
торая выражается на выходе в виде амплитудного распределе-
ния, прежде всего обусловливается характером взаимодействия
излучения с веществом детектора. Определенное влияние на
получающееся амплитудное распределение оказывают парамет-
ры детектора, конструкционные особенности спектрометра, фор-
ма и толщина источника, интенсивность и состав сопутствующе-
го излучения и некоторые другие факторы. От правильного уче-
та всех факторов зависят надежность оценки возможностей
проведения анализа в данной экспериментальной ситуации, а
также точность качественной и количественной интерпретации
полученных данных.
148
В активационном анализе наиболее важную роль играет
спектроскопия у-излучения, значительно реже прибегают к
спектроскопии заряженных частиц. Причины этого станут оче-
видны из дальнейшего текста. Имеются также единичные при-
меры спектрометрии нейтронов по времени пролета [185]. Из
имеющихся приборов для спектрометрических измерений наи-
большее распространение получили сцинтилляционные и полу-
проводниковые спектрометры.
Для определения энергии неизвестных линий спектрометр
должен быть прокалиброван по излучателям с известной энер-
гией. Калибровочные графики обычно имеют хорошую линей-
ность, хотя иногда отмечаются небольшие нарушения линейно-
сти чаще всего аппаратурного происхождения.
§ 2. Спектрометрия тяжелых заряженных частиц
Среди радиоизотопов, образующихся при различных спосо-
бах активации, лишь изредка появляются а-излучатели. Спек-
троскопия a-излучения, которая в настоящее время преимуще-
ственно выполняется с помощью поверхностно-барьерных детек-
торов, требует приготовления достаточно тонкого источника.
Поэтому «-спектрометрическому измерению обязательно долж-
но предшествовать радиохимическое выделение с нанесением
выделенного препарата на подходящую подложку в виде тон-
кого слоя. Очень часто для этого применяют электрохимическое
осаждение. Возможно, конечно, применение инструментального
варианта, но количество вещества и толщина слоя анализируе-
мой пробы должны быть малы. Из-за редкого появления
«-излучателей в продуктах активации их определение на фоне
Р- и у-излучения можно успешно осуществлять, не прибегая к
спектрометрическим измерениям. Примером такого случая мо-
жет служить определение Bi по дочернему 210Ро в некоторых
рудах [121].
Однако определение отношения 231Pa/238U оказалось удоб-
ным проводить путем облучения пробы тепловыми нейтронами
(при этом происходит реакция 231Pa(n, у)232Ра -j~y^*232U) с по-
следующим химическим выделением урана, электрохимическим
осаждением препарата и измерением а-спектра. Искомое отно-
шение после соответствующей калибровки определяется из отно-
шения числа отсчетов a-линий 238U (4,2 Мэв) и 232U
(5,3 Мэе) [186].
Довольно широкое распространение получила спектроскопия
тяжелых заряженных частиц (преимущественно протонов и
а-частиц), испускаемых в ходе ядерных реакций. Хотя облуче-
ние возможно различными видами излучения, но предпочтение
пока отдается заряженным частицам. Для таких определений
типичной является система, схематично представленная на
149
рис. 38 [187]. Для работы система устанавливается внутри уско-
рительной камеры источника ионов.
Ядерные реакции на заряженных частицах часто сопровож-
даются вылетом вторичных заряженных частиц другого типа,
чем исходные. Энергия вторичных частиц при определенной
энергии облучения — это величина, характерная для данной
ядерной реакции, которая допускает надежную идентификацию
Рис. 38. Схема экспериментальной установки для
аналитических исследований по методу спектро-
метрии мгновенных заряженных частиц:
1— цилиндр Фарадея; 2 — мишень; 3 — детектор; 4 —
предусилитель; 5 — основной усилитель; 6 — одноканаль-
ный анализатор; 7—счетное устройство; 8—многока-
нальный анализатор: 9 — интегратор тока.
взаимодействующих ядер. Большая часть вторичных частиц вы-
летает в направлении, противоположном движению пучка ионов,
поэтому детектор стремятся расположить должным образом.
Однако конечные размеры детектора и необходимость пропу-
стить пучок ионов к мишени приводят к компромиссному реше-
нию— детектор помещают под углом 150—165° по отношению
к направлению движения пучка ионов.
От мишени претерпевает упругое рассеяние часть ионов
первичного пучка, поэтому для их поглощения детектор закры-
вают тонкой пленкой из майлара. Чтобы подавление рассеян-
ных частиц было успешным, максимальная энергия пучка не
должна превышать некоторого предела. С помощью такой уста-
новки измеряют спектры вторичного излучения мишени, по
которым можно осуществить идентификацию ядер, вступающих
в реакцию, и проводить количественные расчеты (рис. 39).
Таким способом можно получить разнообразную аналитиче-
скую информацию [179, 181, 187]. Процесс измерения очень
прост и быстр. Учитывая, что в таких экспериментах энергия
150
активирующего излучения мала (0,5—1,5 Мэв), метод приго-
ден только к исследованию тонких поверхностных пленок на
содержание некоторых легких элементов. В результате появ-
ляется возможность изучения реакций и процессов в тонких
поверхностных слоях. Это может быть кинетика окисления ме-
таллов, сорбция в парах, диффузия в твердом теле и т. д.
Номер капала
Рис. 39. Спектр мгновенных а-частиц реакций (d, а) на
мишени из А120з, обогащенной lfO:
/ — реакция lfO(d, a)”N; 2 — реакция 1BO(d, a),6N.
Чувствительность метода очень высока. Например, при токе
пучка дейтронов 1 мка и энергии 730 кэв по реакции
18О(р, a)15N может быть надежно обнаружено 3.7-1 (У12 г изо-
топа 18О при длительности облучения всего 3,5 мин. Столь вы-
сокая чувствительность обусловлена низким фоном полупровод-
никовых детекторов и высоким выходом заряженных частиц. При
изучении изотопных эффектов можно определить отношение
isQ jeo непосредственно из получившегося спектра по отноше-
нию площадей пиков (см. рис. 39).
Для достаточно толстых слоев метод позволяет исследовать
распределение концентрации изучаемого компонента по глуби-
не поверхностного слоя [179]. Так, четко выраженный резонанс-
ный характер реакции 18О(р, a)15N (рис. 40) предоставляет
очень удобную возможность для таких исследований.
Если энергия протонов равна 629 кэв, то имеет место мак-
симальный выход a-частиц. При повышении энергии пучка об-
ласть резонансной энергии протонов перемещается в глубь про-
бы. Следовательно, основной процесс генерации a-частиц будет
происходить уже в тонком слое па некоторой глубине. Интен-
сивность a-частиц дает количество искомого компонента на этой
глубине. Меняя последовательно энергию излучения, можно
151
установить исследуемое распределение кислорода по глубине.
Разрешение высокое и составляет ~320 А для окисной пленки
на поверхности циркония.
20
i5
01—
500
I
S
/ 1 „ооооооооооооооо
1°°°°
0-0-0-°-°
550 600 650 70.Q
Знергия, кэВ
750
Рис. 40. Функция возбуждения реакции 180(р, a)I5N.
§ 3. Спектрометрия р-излучения
Подавляющее большинство радиоизотопов, образующихся
при всех методах активации, испытывает p-распад с испуска-
нием электронов или позитронов. Этот процесс, как правило,
сопровождается у-излучением, и в общей массе радиоизотопов
лишь небольшое число их являются чистыми р-излучателями.
Поскольку спектроскопия у-излучения имеет значительное
превосходство перед спектроскопией p-излучения, последняя
получила незначительное распространение. Основные ее недо-
статки вытекают из сплошного энергетического распределения и
небольшой проникающей способности р-частиц. Последнее свой-
ство требует приготовления для прецизионных измерений доста-
точно тонких источников.
Для идентификации р-излучателя необходимо определить
максимальную энергию спектра. В практике активационного
анализа для этой цели находят применение полупроводниковые
и сцинтилляционные бета-спектрометры и метод поглощения.
Идентификация и раздельное количественное определение не-
скольких компонентов в смеси любым из перечисленных выше
методов представляют сложную проблему и практически реали-
зуются при числе компонентов не более 3—4. Возникающие при
анализе сложных смесей р-излучателей затруднения обусловле-
на
ны низкой характерностью [3-спектров (сплошное распределе-
ние, несколько p-переходов у одного радиоизотопа, помехи со
стороны у-излучения). Поэтому бета-спектрометрия в актива-
ционном анализе находит преимущественное применение для
проверки радиохимической чистоты препаратов и идентифика-
ции р-излучателей с простой схемой распада (без сопровож-
дающего у-излучения), а также для раздельного определения
2—3 компонентов в смеси, сильно различающихся по макси-
мальной энергии р-спектра.
Метод поглощения
Пробег р-частиц в веществе пропорционален их начальной
энергии. Поэтому, определив максимальный пробег р-частиц
в нужном веществе путем последовательного увеличения тол-
Рис. 41. Зависимость между пробегом и энергией
Р--частиц.
щины фильтра между источником и детектором до постоянной
скорости счета, можно оценить максимальную энергию спектра
по приближенному соотношению [43]:
£р, макс = 1,92 Г Макс + 0,227?^ макс ;
0,05 < макс < 3,0 Мэв.
(7.1)
На рис. 41 связь между пробегом и £р, макс представлена гра-
фически.
Метод анализа кривых поглощения в своей наиболее про-
стой форме дает значение £р, макс с относительной погрешностью
10—15%, причем для достижения такой точности требуется до-
статочно высокий начальный уровень скорости счета — более
153
3-103 имп 'мин. Точность определения энергии методом погло-
щения можно повысить, используя более сложные и трудоемкие
методы [43, 184].
В настоящее время метод поглощения, как метод идентифи-
кации и анализа смесей 0-излучателей, по существу вытеснен
более совершенными методами спектроскопии с помощью сцин-
тилляционных или полупроводниковых спектрометров. Сейчас
практический интерес могут представлять лишь отдельные слу-
чаи, когда этот метод дает простое решение аналитической
проблемы.
Например, Винчестер [188] использовал метод поглощения
для избирательного определения К в силикатных минералах и
породах. Поскольку радиоизотоп 42К (Ер, макс = 3,55 Мэв, Ti/2 =
= 12,5 ч) обладает весьма жестким 0-излучением, то, используя
алюминиевый фильтр толщиной 700 мг/см2, можно дискримини-
ровать p-излучение многих других элементов (кроме As, Ga, In)
и определить К в интервале 0,14-5% с относительной погреш-
ностью 2%.
Сцинтилляционные и полупроводниковые бета-спектрометры
Для анализа сплошных p-спектров ни один тип спектромет-
ров не имеет особых преимуществ перед другим. Определение
Др, макс с помощью бета-спектрометров может производиться
в дифференциальном или интегральном режиме как из тонких,
так и из толстых источников. В зависимости от условий калиб-
ровка спектрометра производится по радиоизотопам с известной
Др, макс (сплошной спектр) или по электронам конверсии (моно-
энергетический спектр). Бета-спектрометры позволяют доста-
точно точно определять Др, макс с препаратом сравнительно не-
большой активности.
Интегральные методы. Галлик и др. [189] показали, что зави-
симость числа отсчетов от положения порога интегрального дис-
криминатора при измерении 0-излучателя с простой схемой
распада на сцинтилляционном спектрометре в основной своей
части представляет прямую линию. Если эту прямолинейную
часть продлить, то она пересечет ось абсцисс в точке, которая
характеризует максимальную энергию излучения. Положение
точки пересечения мало зависит от обратного отражения, тол-
щины препарата, расстояния источник — детектор.
Оценку энергии препарата осуществляют по калибровочно-
му графику с точностью до 2 отн. % для энергий выше 0,3 Мэв.
Большое удобство интегрального метода заключается в возмож-
ности использования для определений не только сухих препа-
ратов, но и растворов, которые наливают в небольшую кювету.
В случае смеси двух 0-излучателей кривая зависимости чис-
ла отсчетов от уровня дискриминации сначала криволинейна и
переходит в прямую линию, когда мягкий компонент полностью
154
дискриминируется. Суммарную кривую можно разложить на
компоненты. Экстраполируя прямую до оси ординат и вычитая
ее из обшей кривой, получают кривую для мягкого компонен-
та. Такой подход позволяет проводить качественный и количе-
ственный анализ простой смеси р-излучателей. Присутствие же-
сткого у-излучения может помешать анализу.
Другой подход к быстрой идентификации р-излучателей ин-
тегральным методом развит Трусевичем и др. [190]. С помощью
сцинтилляционного спектрометра измеряют число отсчетов ис-
следуемого препарата и эталоны (известный р-излучатель) при
нескольких фиксированных уровнях интегральной дискримина-
ции. Для каждого уровня находят отношение Ад Аф, где Аф —
число отсчетов для нижнего уровня дискриминации; Аф — число
отсчетов для гго уровня дискриминации. Аналогичную про-
цедуру выполняют для эталонного препарата. Затем строят
сравнительную кривую, откладывая для каждого уровня дис-
криминации в логарифмическом масштабе по оси абсцисс зна-
чения Аф./Аф для исследуемого препарата, а по оси ординат —
для эталонного. Угол наклона получившейся прямой (или одного
из участков двухкомпонентной кривой) позволяет найти по ка-
либровочному графику энергию излучения радиоизотопа в ис-
следуемом препарате.
Для идентификации одного р-излучателя с простой схемой
распада достаточно выполнить измерения только при двух уров-
нях дискриминации. Область идентифицируемых активностей
простирается до 10 fi—1(У 10 кюри. Для исследований пригодны
толстые пробы. Этим методом возможен также анализ смеси
Р-излучателей, но тогда число используемых уровней дискрими-
нации должно быть больше.
Дифференциальные методы. С тонким источником диффе-
ренциальный метод дает непосредственно спектр p-излучения с
его характерной формой (рис. 42). Если полученные данные
представить в специальных координатах, то на графике полу-
чится прямая линия [183]. Удобство графика Ферми — Кюри
(как называют эту систему построения) состоит в том, что точ-
ка пересечения прямой с осью ординат дает максимальную энер-
гию спектра. Если в анализируемом спектре присутствует не-
сколько р-излучателей разной энергии, то получившуюся слож-
ную кривую можно разложить таким образом, чтобы была вы-
делена прямая линия для каждого компонента. Относительное
содержание каждого р-излучателя можно определить по площа-
ди под соответствующей прямой.
Однако при использовании метода встречается и ряд специ-
фических трудностей, которые обусловлены искажениями формы
p-спектра под действием некоторых факторов. Чаще всего силь-
ное искажение спектра происходит вследствие рассеяния
р-частиц в препарате и отражения от подложки. Поэтому их
толщина должна быть меньше 1 мг'см2. Это значит, что подоб-
155
ный метод пригоден в основном для проверки радиохимической
чистоты препаратов, не содержащих большой массы носителя.
К значительному нарушению спектра может привести и об-
ратное рассеяние от сцинтиллятора. Для предотвращения рас-
сеяния электронов сцинтиллятору необходимо придать специаль-
ную форму. Поскольку фор-
Рис. 42. Форма р_-спектра радиоизото-
па 32Р:
1 — дифференциальный спектр; 2 — график
Ферми—Кюри.
ма спектра зависит также
от заряда ядра и степени
запрещенности р-перехода,
то для сохранения прямоли-
нейного характера графиков
Ферми — Кюри может по-
требоваться введение соот-
ветствующей поправки.
Прево и Кейт[191] пред-
ложили более простой и
достаточно точный метод
определения максимальной
энергии p-излучения. С по-
мощью полупроводникового
бета-спектрометра получа-
ют спектр препарата. Затем
в верхней части спектра выбирают две точки с энергией соот-
ветственно £1 (£1»6/10£ф,макс) и £2(£2^8/10£р,макс) и под-
считывают число отсчетов (Ni и N2). Тогда £р,макс можно рас-
считать из приближенного соотношения
макс —
[£г (ЛДЛД^ - £г]
[(ATM/T-U
(7.2)
Для однокомпонентного спектра погрешность в величине
£р , макс не превышает 2 отн. %.
Сплошной спектр p-излучения и широкая распространенность
Р-излучателей обусловливают низкую избирательность спектро-
метрического инструментального анализа на этой основе. В ре-
зультате область применения такого метода сильно сужается,
ограничиваясь определением макрокомпонентов в пробах про-
стого состава (2—3 компонента), к которым у-спектрометриче-
ский анализ по каким-либо причинам неприменим. При этом
для успешного определения необходимо значительное различие
£р, маке Для повышения избирательности часто приходится при-
бегать к помощи других параметров схем распада.
Примером может служить методика фотоактивационного
анализа фосфорно-сурьмяного катионита [192], основные ком-
поненты которого (О, Р и Sb) при активации образуют чистые
позитронные излучатели (см. табл. 12), причем 30Р имеет высо-
кую энергию £р, макс, a I20Sb — значительно отличающийся пе-
риод полураспада. Поэтому в принятой методике приводили два
измерения p-спектра на сцинтилляционном спектрометре (через
156
1 и 21 мин после окончания облучения в течение 1 мин при
Еторм=25 Мэв). Из первого спектра определяли число отсче-
тов 30Р по жесткой области и суммарное число отсчетов
зор _ц 1 5q_|_ 12osb по мягкой. Второе измерение дает число отсче-
тов только I20Sb (30Р и 15О распадаются). По этим данным,
определив экспериментально необходимые коэффициенты в опы-
тах с элементарными Р и Sb, можно рассчитать число отсчетов
для 15О.
§ 4. Спектрометрия у-излучения
За небольшим исключением, любой процесс неупругого ядер-
ного взаимодействия или радиоактивного распада сопровож-
дается испусканием у-квантов. Это излучение обусловлено пере-
ходами между разными энергетическими уровнями возбужден-
ного ядра, образовавшегося в результате ядерного взаимодейст-
вия или радиоактивного распада ядра-предшественника. Поэто-
му испускаемое у-излучение имеет строго определенное значе-
ние энергии и очень узкую естественную ширину линии. Эти
свойства у-излучения составляют хорошую основу для иденти-
фикации и избирательного определения элементов. Благоприят-
ным моментом является также высокая проникающая способ-
ность у-излучения, что позволяет проводить у-спектрометриче-
ский анализ с достаточно толстыми пробами. Сочетание перечис-
ленных свойств с широкой распространенностью процессов, со-
провождающихся у-излучением, обеспечивает у-спектрометриче-
скому методу ведущее положение в инструментальном актива-
ционном анализе.
Однако, чтобы в полной мере реализовать возможности, за-
ложенные в этом методе, необходимы адекватные системы реги-
страции. Хотя в этом направлении достигнут значительный про-
гресс, многие ограничения у-спектрометрического анализа все
же связаны с несовершенством существующих методов регист-
рации и анализирующей аппаратуры. Чтобы правильно подойти
к оценке возможностей и ограничений метода, необходимо крат-
ко рассмотреть взаимодействие у-излучения с веществом и ха-
рактеристики используемых гамма-спектрометров. Более под-
робное изложение этих вопросов можно найти в специальной
литературе [41, 183, 184, 193].
Взаимодействие у-излучения с веществом
В практике активационного анализа наибольшее значение
приобрели системы, основанные на ионизирующем действии
регистрируемого излучения. Как известно, у-кванты непосред-
ственно не вызывают ионизацию вещества, и поэтому их реги-
страция осуществляется через ионизирующее действие вторич-
ных заряженных частиц, которые возникают при взаимодейст-
157
вии у-кваптов с электронными оболочками атомов, полем атом-
ного ядра, нуклонами и т. д. В этих процессах у-квант может
передать заряженной частице (обычно электрону) всю или часть
своей энергии. Механизм поглощения и рассеяния у-квантов в
веществе можно представить тремя основными процессами:
фотоэффектом, комптоновским рассеянием и образованием пар.
Фотоэффект. Этим термином обозначают происходящий под
воздействием у-квантов процесс вырывания электрона из атома
с передачей ему всей энергии. Кинетическая энергия образовав-
шихся фотоэлектронов Ер, кИН равна энергии поглощенного
у-кванта за вычетом энергии связи электрона:
Ер, кин = Ет — Есв. (7.3)
Фотоэффект происходит только на связанных электронах,
находящихся на К- или Е-оболочке средних или тяжелых ядер.
Сечение фотоэффекта на К-оболочке приблизительно следует
зависимости
75
(Оф)/< = const ——, (7.4)
Е 11
v
где Z — заряд ядер атомов среды.
Образовавшийся возбужденный атом теряет энергию путем
испускания электронов Оже или характеристического рентгенов-
ского излучения. Если фотоэлектроны, рентгеновские кванты или
электроны Оже полностью теряют свою энергию в рабочем
объеме детектора, то сигнал на выходе будет соответствовать
полной энергии зарегистрированного у-кванта.
Комптоновское рассеяние. При взаимодействии у-квантов со
свободными или слабосвязанными электронами происходит
процесс рассеяния с передачей электрону только части энергии,
которая в зависимости от угла рассеяния может принимать
различные значения в области от нуля до максимальной вели-
чины, определяемой соотношением
Е
^макс =----—(7.5)
тпс-
2Е.
где т0—масса покоя электрона; с—скорость света (тсс2=
= 0,511 Мэв).
Комптоновское рассеяние играет отрицательную роль в
у-спектрометрических измерениях, так как создает непрерывное
амплитудное распределение, которое является фоном для
у-квантов более низких энергий. В детекторе у-квант может
претерпеть несколько последовательных актов рассеяния, в ре-
зультате чего он передаст всю свою энергию электронам отда-
чи. Тогда суммарный сигнал будет иметь амплитуду полного
поглощения.
158
Образование пар. Если энергия у-кванта выше 1,02 Мэв, то
при его взаимодействии с полем ядра становится возможным
процесс образования пар, т. е. исчезновения у-кванта с одно-
временным образованием пары частиц — электрона и позитрона.
Суммарная кинетическая энергия частиц равна Еу—2тас2.
После потери кинетической энергии на ионизацию атомов пози-
Рис. 43. Сечения различных процессов при взаимодействии
у-излучения с веществом:
/ — фотоэффект; 2 — комптоновское рассеяние; 3 — образование пар.
трон аннигилирует с образованием двух у-квантов, которые раз-
летаются в противоположных направлениях, имея энергию
0,511 Мэв каждый.
Величины сечений различных процессов взаимодействия
у-квантов с веществом детектора зависят от энергии регистри-
руемого излучения и материала детектора (рис. 43). Нетрудно
видеть, что кристаллы Nal(Tl) более эффективны для регистра-
ции у-излучения из-за большей величины сечения фотоэффекта
и меньшей роли комптоновского рассеяния. Однако Ge(Li)-де-
текторам свойственно более высокое разрешение.
Для обоих детекторов при малых энергиях преобладающую
роль играет фотоэффект. Но с ростом энергии сечение фото-
эффекта резко уменьшается и на уровне в несколько сот кило-
электронвольт становится сравнимым с сечением комптонов-
159
ского рассеяния, которое с ростом энергии у-квантов падает
значительно медленнее. Процесс образования пар имеет замет-
ное сечение только при энергии у-квантов выше 2 Мэв, и его
роль одинакова для обоих типов детекторов.
Гамма-спектрометры
Несомненно, что гамма-спектрометр относится к обязатель-
ной принадлежности любой лаборатории, занимающейся актива-
ционным анализом. Пока более распространен сцинтилляцион-
ный спектрометр, поскольку он прост в эксплуатации, легко до-
ступен и к тому же имеет длительную историю практического
применения для аналитических целей. Полупроводниковый
спектрометр — прибор для активационного анализа сравни-
тельно новый. Он, конечно, более сложен в эксплуатации и
дорог. Однако полупроводниковый спектрометр показывает от-
личные аналитические результаты. Надо надеяться, что про-
гресс в совершенствовании полупроводниковых детекторов и
связанной с ним электронной аппаратуры приведет к появле-
нию более простых, доступных и совершенных спектрометров,
удобных для рядовых аналитических применений.
Сцинтилляционный спектрометр. Однокристалльный спектро-
метр— наиболее простая и распорстраненная конструкция. Как
правило, в качестве сцинтиллятора выступают монокристаллы
Nal(Tl), которые обладают высокой конверсионной способно-
стью и имеют большую плотность (3,67 а/слг3) при высоком
эффективном атомном номере (2эфф = 50). Эти качества кри-
сталлов Nal(Tl) в сочетании с возможностью получения моно-
кристаллов больших размеров обусловливают высокую эффек-
тивность регистрации у-излучения при удовлетворительном
энергетическом разрешении. Так как среднее время высвечива-
ния сцинтилляций при комнатной температуре равно
2,5-10—7 сек, то кристаллы Nal(Tl) пригодны для измерения
довольно высоких уровней активности. Особенность кристаллов
Nal(Tl) состоит в их гигроскопичности, поэтому их упаковы-
вают в специальные герметичные контейнеры.
В гамма-спектрометрах кристаллы Nal(Tl) используются в
сочетании со спектрометрическими фотоумножителями. Эта
система обычно оформляется в виде отдельного выносного
блока, который включает в себя еще ряд дополнительных уст-
ройств: светонепроницаемый кожух, свинцовую защиту, держа-
тель источника и т. д. (рис. 44).
Поскольку при практическом использовании гамма-спектро-
метра уровень активности измеряемых источников может изме-
няться в значительных пределах, для подбора оптимальных
условий измерения служит стойка, которая позволяет изменять
расстояние источник—детектор. Увеличение расстояния умень-
шает абсолютную эффективность спектрометра.
160
Иногда в у-спектрометрических исследованиях применяют
коллиматоры — толстые пластины из свинца с небольшим от-
верстием посередине. Коллимация у-излучения уменьшает веро-
ятность ряда вторичных процессов, что упрощает получаю-
щийся спектр и облегчает его обработку. С другой стороны,
коллиматор сильно уменьшает абсолютную эффективность спек-
трометра, и поэтому его применение в случае слабоактивных
источников противопоказано.
Часто в спектрометрах ис-
пользуют кристаллы Nal(Tl) с
колодцем. Анализируемый препа-
рат в специальном контейнере по-'
мещают в колодец, что дает;
близкую к 4л геометрию измере-
ния и соответственно наиболее
высокую эффективность регист-
рации. Недостаток такого мето-
да — более сложная интерпрета-
ция результатов измерения, что
связано с протеканием ряда спе-
цифических процессов, главным
среди которых является суммиро-
вание. Объем колодца для боль-
ших кристаллов может достигать
нескольких десятков милли-
литров. ы’ 1
На выходе ФЭУ получаются
Рис. 44. Конструкция выносного
блока сцинтилляционного гамма-
спектрометра:
1 — защита; 2 — светонепроницаемый
кожух; 3 — держатель источника; 4 —
стоика; — источник; 6 — кристалл
Nal(Tl); 7 — ФЭУ; 6 — устройство с
делителем и катодным повторителем.
сигналы довольно большой ам-
плитуды, которые после неболь-
шого усиления поступают на вход
многоканального анализатора.
Оптимальное число каналов ана-
лизатора для регистрации ампли-
тудного распределения от сцин-
тнлляциошюго детектора нахо-
дится в пределах 100—512. Современный сцинтилляционный
спектрометр — сравнительно компактный и простой в работе.
Параметры однокристалльного сцинтилляционного спектро-
метра сильно зависят от размеров кристалла. Прежде всего с
ростом последнего при регистрации жесткого у-излучения (выше
0.5 Мэв) увеличивается эффективность спектрометра по пику
полного поглощения и уменьшается комптоновское распределе-
ние. Однако одновременно с ростом размеров кристалла ухуд-
шается разрешение и возрастает стоимость. Дополнительным
следствием является также увеличение фона спектрометра, кото-
рый примерно пропорционален объему кристалла.
Фон прибора представляет результат воздействия на сцин-
тиллятор трех основных компонентов: 1) космического излуче-
6 Р А. Кузнецов
161
ния; 2) излучения 48К, который присутствует в кристалле
Nal(Tl) в качестве примеси; 3) излучения естественных радио-
активных элементов, которые присутствуют в конструкцион-
ных материалах (упаковке кристалла, защите детектора
и т. д.).
Уровень фона, обусловленный космическим излучением в не-
экранированном детекторе, довольно велик, что объясняется
высокой эффективностью кристаллов Nal(Tl) к у-излучению и
сравнительно большим объемом используемых кристаллов. Для
подавления фона от космического излучения и других внешних
источников приходится сооружать вокруг детектора специаль-
ную защиту. Сталь толщиной 15—20 см или другой материал
эквивалентной толщины обеспечивает оптимальную защиту от
внешнего ионизирующего излучения.
Весьма важный источник фона — загрязненность вещества
кристалла, конструкционных материалов упаковки кристалла и
ФЭУ естественным радиоактивным 40К. Использование йоди-
стого натрия, стекла и других конструкционных материалов с
низким содержанием калия (менее 1 -10—4%) позволяет значи-
тельно уменьшить этот источник фона.
Например, по данным работы [194], фон кристалла Nal(Tl)
размером 9,6X3,6 см без защиты равен 7000 има/мин (между
0,09—2,5 Мэе). Помещение детектора в стальную защиту тол-
щиной 19 см снизило уровень фона до 480 имп[мину а введение
дополнительной защиты вокруг кристалла из трижды пере-
гнанной ртути толщиной 2,4 см — до 360 имп!мин. Показано,
что оставшаяся активность обусловлена 226Ra и 40К, которые
присутствуют в виде примесей в кристалле, его упаковке и
стекле ФЭУ. Кристалл перепаковали в контейнер с кварцевым
окошком, при этом фон понизился до 150 UMnjMUH.
Как показывает практика, наиболее оптимальные размеры
кристалла Nal(Tl) для универсального применения, т. е. для
регистрации излучения в широком интервале энергий
(0,14-3 Мэв), находятся в области 7,5X7,5 см. Однако в от-
дельных случаях может оказаться выгодным применить более
тонкий кристалл, так как это приводит к повышению избира-
тельности при регистрации мягкого у-излучения в присутствии
жесткого.
Полупроводниковый спектрометр. Можно без преувеличения
сказать, что гамма-спектрометр с Ge (Li)-детектором открыл
совершенно новые возможности для инструментального актива-
ционного анализа [195—197]. Этот факт прежде всего обуслов-
лен хорошим энергетическим разрешением, которое на порядок
превышает разрешение гамма-спектрометра с кристаллом
Nal(TI). Однако внедрение полупроводниковые; гамма-спектро-
метров в аналитическую практику привело к новым проблемам,
которые необходимо разрешать для успешной реализации по-
явившихся возможностей.
162
Порядковый номер Ge равен 32, плотность — о,33 г!смъ.
Современная технология пока не позволяет получать детекторы
столь же больших размеров, что и кристаллы Nal(Tl). Неболь-
шой заряд ядер Ge и недостаточные объемы детекторов при-
водят к относительно низкой эффективности полупроводниковых
спектрометров к у-излучению.
Рис. 45. Конструкция выносного блока спектрометра с Ge(Li)-де-
тектором:
1 — Ое(Ь1)-детектор в алюминиевом контейнере; 2 — трубка для заполнения
сосуда Дьюара жидким азотом; 3 — сосуд Дьюара; 4— предусилитель;
5 — трубка, по которой проба поступает в позицию для измерения; 6 — пнев-
магический регулятор положения пробы: " — транспортирующая трубка для
подачи проб в хранилище; 8— транспортирующая трубка для подачи проб
из хранилища: 9 — свинцовая защита; 10 — дверь в свинцовой защите;
11 — фотоэлектрическая система для контроля наличия пробы в счетной по-
зиции; р—давление.
Ge (Li)-детектор работает нормально при температуре жид-
кого азота и в глубоком вакууме. Обеспечение этих условий
усложняет конструкцию выносного блока спектрометра
(рис. 45). Обязательным элементом спектрометра становится
сосуд Дьюара, который поддерживает низкую температуру де-
тектора в течение длительных интервалов времени между двумя
заполнениями жидким азотом (от 6 до 15 дней в зависимости
от объема сосуда Дьюара).
6
163
Величина сигнала полупроводникового детектора довольно
мала (на уровне милливольт), поэтому к входным параметрам
предусилителя (входная емкость, уровень шумов) предъяв-
ляются весьма жесткие требования. Для понижения уровня
шумов предусилителя, которые могут положить предел разре-
шению спектрометра, его также приходится охлаждать до низ-
кой температуры.
Вследствие высокого энергетического разрешения полупро-
водникового детектора анализатор для одновременного измере-
ния у-излучения в широком энергетическом интервале (до
2—3 Мэв) должен иметь не менее 4000 каналов. Это довольно
сложный и дорогой прибор. При этом в случае анализа слож-
ных объектов количество получающейся информации настолько
велико, что ручная обработка спектров становится затрудни-
тельной и приходится прибегать к помощи электронных вычис-
лительных машин.
Так же, как и в случае сцинтилляционных детекторов, пара-
метры полупроводникового спектрометра зависят от размеров
Ge (Li)-детектора. Глубина дрейфа ионов лития, которая может
быть достигнута за приемлемый интервал времени, едва превы-
шает 1,0 см, что при площади около 10 см2 дает объем всего
10 см3. Детектор в виде такого диска носит название планар-
ного. Получение детекторов большего объема требует особого
подхода. Например, в цилиндре из Ge делают отверстие и осу-
ществляют дрейф лития со всех поверхностей детектора, кроме
одной. Это так называемые коаксиальные детекторы. Наиболь-
ший достигнутый объем коаксиального детектора составляет
около 200 см3. Правда, увеличение объема детектора связано
с некоторой потерей энергетического разрешения.
§ 5. Аппаратурная форма линии гамма-спектрометра
Форма амплитудного распределения, которое получается на
выходе гамма-спектрометра при регистрации моноэнергетиче-
ского у-излучения, является сложной и зависит от многих фак-
торов. Конечно, форму амплитудного распределения в первую
очередь определяют основные процессы взаимодействия у-пзлу-
чения с веществом детектора. С другой стороны, на форму
амплитудного распределения оказывает определенное влияние
ряд факторов, обусловленных протеканием вторичных про-
цессов в детекторе, особенностями конструкции гамма-спектро-
метра, составом измеряемой смеси радиоизотопов. Важное
значение имеют также вещественный состав, масса и геометри-
ческая форма пробы, излучение которой подвергается у-спек-
трометрическому анализу.
Если экспериментальные условия скорректированы таким
образом, чтобы влияние всех побочных факторов стало незна-
чительным, то типичная форма амплитудного распределения
164
при регистрации моноэнерге-
тического излучения будет
иметь вид, представленный на
рис. 46. В получившемся рас-
пределении четко выделяются
две области: пик, который обу-
словлен , процессами полного
поглощения энергии у-квантов
(фотоэффект и многократное
рассеяние), и непрерывное ам-
плитудное распределение, ко-
торое своим появлением в ос-
новном обязано комптоновско-
му рассеянию.
Пик полного поглощения
имеет фундаментальное значе-
днергия, кэб
Рис. 46. Сцинтилляционный спектр
у-излучения MMn [Nal(Tl) ЮОх
X10Q .иль поглотитель из Be
304 мг/см2, геометрия 40%].
ние для у-спектрометрического
анализа. Положение мак-
симума пика полного по-
глощения определяет
энергию регистрируемого
у-излучения, а его пло-
щадь или высота служат
мерой интенсивности из-
лучения данной энергии.
Ширина пика полного по-
глощения, которая опре-
деляется энергетическим
разрешением спектромет-
ра, также представляет
важную характеристику
амплитудного распреде-
ления. Данные рис. 47 по-
казывают изменение раз-
решающей способности
сцинтилляционного и по-
лупроводникового спект-
рометров с энергией реги-
стрируемого у-излучения
[195]. Значительное пре-
восходство Ge (Li)-детек-
торов, особенно планар-
ных, в энергетическом
разрешении перед кри-
сталлами Nal(Tl) совер-
шенно очевидно.
Пик полного погло-
щения отделен от края
Рис. 47. Энергетическое разрешение сцин-
тилляционного и полупроводниковых спек-
трометров:
1 — предельное теоретическое разрешение: 2 —
планарный Се(Ы)-детектор объемом 2 or"; 1--
коаксиальный Се(Ы)-детектор объемом 47 с.иД
4 — кристалл Nal(TI) размером 72x72 мм.
165
непрерывного амплитудного распределения небольшим прова-
лом, величина которого в энергетических единицах составляет
около 250 кэв. Непрерывное распределение в сложных спектрах
играет роль помехи, и поэтому при идентификации пиков и ко-
личественной обработке спектров его приходится тщательным
образом учитывать.
Приведенный на рис. 46 сцинтилляционный у-спектр Г;4Л1п
получен в какой-то степени в идеализированных условиях. При
выполнении рядовых анализов создать такие условия, как пра-
вило, не удается, а это приводит к некоторой деформации
амплитудного распределения, появлению в нем дополнительных
деталей. Ответственность за изменение формы амплитудного
распределения могут нести как внутренние (тип, размеры и
форма детектора), так и внешние (размеры, масса и состав
пробы, конструкционные особенности спектрометра и т. д.)
факторы.
Влияние вторичных процессов и конструкции
гамма-спектрометров
Эффекты утечки вторичных частиц и квантов. Чтобы погло-
щенный квант дал вклад в пик полного поглощения, необходи-
мым условием является полная потеря энергии вторичными ча-
стицами и квантами в рабочем объеме детектора. Однако вто-
ричное излучение имеет определенную вероятность покинуть
рабочий объем детектора, унося с собой часть или всю полу-
ченную энергию. Вероятность утечки обычно оказывается тем
выше, чем ближе к поверхности детектора был поглощен пер-
вичный у-квант.
Утечка вторичных электронов приводит к нарушению сим-
метрии пика полного поглощения и дает дополнительный вклад
в непрерывное распределение. Этот эффект проявляется тем за-
метнее, чем меньше размеры детектора и выше энергия
у-квантов.
Как уже упоминалось, процесс фотопоглощения сопровож-
дается характеристическим рентгеновским излучением вещества
детектора. Так же, как и электроны, кванты рентгеновского из-
лучения, возникшие вблизи поверхности детектора, могут выйти
за пределы его рабочего объема. Тогда выходной импульс бу-
дет соответствовать энергии Еу—£рецт. Этот эффект наиболее
существен для кристаллов Nal(Tl), поскольку рентгеновское из-
лучение иода имеет энергию 28 кэв. Поэтому в сцинтилляцион-
ных спектрометрах утечка рентгеновских квантов иода приводит
к появлению двух пиков с энергиями Еу и Еу —Ереит. При малой
энергии первичного у-излучения эти пики могут быть разреше-
ны, а при высокой энергии они сливаются вместе, приводя к
увеличению асимметрии пика полной энергии. Следует отметить,
что утечка рентгеновских квантов иода заметно проявляется
166
только в области низких энергий у-квантов, так как именно в
этом случае значительная часть первичного излучения претер-
певает фотопоглощение в поверхностном слое детектора и рент-
геновские кванты имеют большую вероятность покинуть рабочий
объем детектора.
Весьма специфичные процессы имеют место в случае жест-
ких у-квантов, которые взаимодействуют с веществом детектора
с образованием пары электрон — позитрон. Последний при ан-
нигиляции дает два у-кванта. При этом возможны три варианта
дальнейшего хода событий: либо поглощение обоих аннигиля-
ционных квантов, либо одного из них, либо уход обоих кван-
тов. В соответствии с этими возможностями в амплитудном
распределении появляются три пика с энергиями Еу, Е.,—т0с2
и Еу—2т0с2.
Обратное рассеяние. Первичное у-излучение может претер-
певать комптоновское рассеяние на различных частях гамма-
спектрометра с последующей регистрацией детектором. В этом
случае над непрерывным распределением появляется пик об-
ратного рассеяния. Интенсивность пика обратного рассеяния
зависит от многих факторов: положения источника, конструкции
держателя и массы радиоактивного препарата, размеров кри-
сталла, конструкции и материала защиты гамма-спектрометра,
коллимации излучения [41, 198].
Отношение энергии рассеянного у-кванта к энергии первич-
ного излучения находится по формуле
=-----------!--------- (7.6)
Е Е ’ 4
V 1 4-------‘V’ (1 — cos брас)
m(,c-
где брас — угол между направлением падающего и рассеянного
квантов.
Очевидно, что в кристалл могут попасть только кванты, рас-
сеянные на 90—180°. Поскольку в этом интервале углов Ерас
слабо меняется с ростом 0, спектр импульсов от обратного рас-
сеяния имеет форму более или менее оформленного несиммет-
ричного пика с энергией в области 150—300 кэв.
Интенсивность обратного рассеяния сильно зависит от рас-
стояния источник — кристалл и оказывается тем меньше, чем
меньше это расстояние. Заметный вклад в обратное рассеяние
дает наличие около источника значительного количества рас-
сеивающих материалов, в качестве которых могут выступать
подложка, держатель или основа источника. Последнее обстоя-
тельство очень существенно, так как в практике активационного
анализа очень часто для измерений используют непосредствен-
но облученную пробу, которая может иметь значительную мас-
су и большие размеры. Ввести поправку на обратное рассеяние
в этом случае достаточно сложно [198].
167
Значительное уменьшение обратного рассеяния дают исполь-
зование детекторов больших размеров и коллимация у-излуче-
ния. Интенсивность обратного рассеяния также тем меньше,
чем выше порядковый номер материала защиты и больше
внутренние размеры защитной камеры, в которой размещается
детектор.
Поскольку пик обратного рассеяния затрудняет определение
у-линий с энергией 100—300 кэв, то всегда следует принимать
все возможные меры, чтобы уменьшить его интенсивность.
Флуоресценция. Под действием р- и у-излучения в конст-
рукционных материалах и защите гамма-спектрометра может
возникнуть характеристическое рентгеновское излучение. Обыч-
но свинцовая защита является источником рентгеновского из-
лучения с энергией 72 кэв. Для его подавления поверхность
свинца покрывают двумя-тремя слоями материалов в порядке
уменьшения заряда ядер. Материалы подбирают таким обра-
зом, чтобы они имели высокое сечение для поглощения флуо-
ресценции предшествующего слоя. В качестве покрытий обычно
используют последовательные слои следующих материалов:
Та—Sn—Си или Cd—Си, толщина которых равна 0,1 — 1 мм.
Влияние геометрии. Форма амплитудного распределения за-
висит от расстояния источник — детектор. С изменением рас-
стояния меняется телесный угол и соответственно средняя
длина пробега у-квантов в детекторе. От последней зависит ве-
роятность событий фотопоглощения и многократного рассеяния,
а следовательно, и эффективность детектора. По этой причине
спектры анализируемых проб и эталонов измеряют в одинако-
вых геометрических условиях. Возможно использование п раз-
ной геометрии, но тогда требуется предварительная калибровка
используемых позиций для приведения к одинаковым условиям
измерения.
Влияние схем распада радиоизотопов
и интенсивности источников излучения
Схема распада радиоизотопа и интенсивность источника так-
же могут влиять на форму получающегося спектра; в одних
случаях они приводят к появлению в спектре новых пиков, в
других дают дополнительный вклад в непрерывное амплитудное
распределение.
Тормозное излучение. Большинство радиоизотопов, полу-
чающихся при различных методах активации, оказываются
Р-- или р+-излучателями. Так как сцинтилляционные и полу-
проводниковые детекторы чувствительны к этим видам излуче-
ния, необходимо использовать поглотители соответствующей
толщины, помещаемые между источником и детектором. В про-
цессе поглощения р~- и р+-частицы теряют энергию на иониза-
цию атомов и тормозное излучение. Поскольку спектр тормоз-
168
ггого излучения сплошной, то, будучи зарегистрирован детекто-
ром, он дает вклад в непрерывное амплитудное распределение.
Доля тормозного излучения в энергетических потерях
0-частиц, как правило, мала и для нормального спектра связа-
на с максимальной энергией излучения £р, макс и атомным но-
мером поглотителя Z следующим соотношением:
Люрм = , макс» (7.7)
где/?п примерно равно 0,33-10“3 Мэе-1.
Обычно тормозное излучение при использовании поглотите-
лей из легких элементов мало по интенсивности и слабо влияет
на форму спектра. Однако имеются два случая, когда роль
тормозного излучения может оказаться весьма существенной.
Во-первых, тормозное излучение заметно искажает спектр тех
радиоизотопов, у которых велика интенсивность 0-перехода
сразу на основной уровень, т. е. при малом выходе у-излучения.
Во-вторых, с мешающим действием тормозного излучения при-
ходится сталкиваться при инструментальном спектрометриче-
ском активационном анализе материалов, облучение которых
приводит к образованию чистых 0_-излучателей. Тормозное из-
лучение, возникающее при поглощении 0 -излучения матрицы,
создает иногда очень сильные помехи для определения слабых
у-излучателей.
Следует отметить, что точная форма и интенсивность тормоз-
ного излучения источника зависят от различных эксперимен-
тальных факторов, таких, как материал конструкций, находя-
щихся вблизи источника, размеры и состав исследуемой пробы
и т. д. Это затрудняет введение поправки на тормозное излуче-
ние, когда в этом возникает необходимость.
Суммирование. Если в ходе регистрации одного у-кванта
детектором будет зарегистрирован второй у-квант, то на выходе
спектрометра появится импульс, амплитуда которого будет
соответствовать суммарной энергии, потерянной обоими у-кван-
тами в рабочем объеме детектора. За эффект суммирования
полностью ответствен детектор, который фиксирует события,
следующие друг за другом в пределах его временной разре-
шающей способности, как одно событие.
Суммирование может быть обусловлено эффектом истинных
совпадений, когда совместно регистрируются два каскадных
у-кванта, или случайного попадания двух независимых у-кван-
тов в детектор в пределах его временного разрешения. Сумми-
рование может быть связано не только с первичным излучением
источника, но и с рассеянным и тормозным излучением. Ре-
зультатом суммирования может быть появление в конечном
спектре отдельных пиков, когда два каскадных или случайных
у-кванта полностью передают свою энергию детектору, или
увеличение непрерывного амплитудного распределения при ча-
стичной передаче энергии.
169
Отличить одиночный пик от пиков случайного или истинного
суммирования позволяют следующие свойства: 1) скорость сче-
та одиночного пика меняется линейно с абсолютной эффектив-
ностью спектрометра, в то время как пик суммирования —
квадратично; 2) скорость счета случайных совпадений меняется
квадратично с интенсивностью источника, а истинных совпаде-
ний—линейно. Суммирование в целом играет отрицательную
роль, так как заметно усложняет и без того сложную аппара-
турную линию спектрометра. Особенно велико влияние сумми-
рования в условиях высокой геометрии и при работе с высоко-
активными источниками излучения.
Позитронные излучатели. Аннигиляция позитрона приводит
к появлению двух у-квантов с энергией 511 кэв, которые дают
соответствующий пик в спектре. Аннигиляционное излучение
имеет высокий выход и часто используется в количественных
определениях по позитронно-активным радиоизотопам. Однако,
поскольку энергия аннигиляционного излучения у всех у-излу-
чателей одинакова, для идентификации радиоизотопа прихо-
дится прибегать к анализу кривых распада или к измерению
совпадений аннигиляционного излучения с каким-либо другим
характеристическим излучением радиоизотопа.
Основная часть случаев аннигиляции происходит после пол-
ной потери энергии позитроном. Поэтому при измерении у-из-
лучателей по аннигиляционному излучению особое значение
приобретают толщина и форма источника и геометрические ус-
ловия. Чтобы избежать возможных ошибок, следует либо окру-
жить источник достаточно толстым поглотителем (лучше из
элемента с низким порядковым номером), либо применять источ-
ники одинаковых толщины и состава в стандартных геометриче-
ских условиях. Для позитронно-активных радиоизотопов суще-
ственное значение имеет эффект суммирования аннигиляцион-
ного излучения с квантами совпадающего во времени у-излуче-
ния. Суммирование с у-квантами обратного рассеяния дает до-
полнительный пик в области около 0,7 Мэв.
§ 6. Качественный и количественный анализы спектров
Как показано выше, реакция спектрометра на моноэнерге-
тическое у-излучение имеет сложный характер. Это приводит
к тому, что регистрация у-излучения какого-либо компонента,
как правило, происходит при мешающем воздействии других
компонентов, присутствующих в исследуемом радиоактивном
препарате. Поэтому извлечение надежной и точной качествен-
ной и количественной информации из полученного эксперимен-
тально амплитудного распределения требует достаточно слож-
ных и трудоемких методов обработки. Решение задачи оказы-
вается тем сложнее, а получающиеся результаты менее точными
и надежными, чем больше число компонентов (у-линий) в исход-
170
ной смеси и чем выше различия в их интенсивности (для слаоых
компонентов), чем ниже характеристики спектрометра. Обра-
ботка результатов у-спектрометрических измерений в настоя-
щее время представляет собой достаточно обширную и в извест-
ной мере самостоятельную дисциплину, рассмотрению которой
посвящены отдельные монографии и обширные обзоры [199,
200].
В общем виде задача определения энергии и интенсивности
отдельных моноэнергетических линий из амплитудного распре-
деления сводится к решению интегрального уравнения вида
#(У) = ]ЛИ£?)К(£?, (7.8)
где N(V)—измеренное амплитудное распределение; /1!l(£v)—
исходный спектр у-излучения; /((£v, V')—функция отклика
спектрометра, т. е. вероятность для у-кванта с энергией £v про-
извести импульс с амплитудой V.
Для решения интегрального уравнения (7.8) с целью опре-
деления числа у-линий, их энергии и интенсивности предложено
несколько методов. При небольшом числе компонентов можно
использовать сравнительно простые способы и приемы, а для
анализа многокомпонентных спектров часто приходится прибе-
гать к достаточно сложным методам обработки, выполняемым
с помощью вычислительных машин.
Наиболее простое и корректное решение уравнения (7.8)
получается, когда обеспечены условия независимости, аддитив-
ности и стабильности функций отклика спектрометра при реги-
страции суммы моноэнергетических линий. Удовлетворение этих
требований предполагает известную идеальность характеристик
спектрометра. Поскольку в действительности положение дел об-
стоит далеко не всегда должным образом, то появляется неко-
торая неопределенность в конечных результатах, что затрудняет
идентификацию компонентов и влияет на точность количествен-
ных оценок. Положение усугубляется статистическим характе-
ром регистрируемых сигналов и различных процессов, которые
имеют место в детекторе и анализирующей системе.
Определение энергии у-линий и идентификация
радиоизотопов из простых спектров
Для перевода амплитудных распределений в энергетические
спектрометр необходимо должным образом прокалибровать. Эту
операцию выполняют путем измерения радиоизотопов с простым
спектром излучения, энергия которого хорошо известна [201].
Чтобы получить исходные данные для построения калибровоч-
ного графика, необходимо точно определить положение макси-
мума пика полного поглощения эталонной у-линии. Число заре-
гистрированных отсчетов в пике полного поглощения должно
быть достаточно большим (более 104 отсчетов), чтобы умень-
171
шить неопределенность в положении максимума распределения
из-за статистических колебаний числа отсчетов в каналах ана-
лизатора. При этом число каналов, приходящихся на один пик
полного поглощения, должно быть достаточно велико (более 5).
Оценка положения максимума пика в измеренном амплитуд-
ном распределении может быть выполнена визуально. Несколь-
ко более точно локализует максимум пика пересечение каса-
тельных к сторонам пика полного поглощения. Наиболее же
точные результаты дают различные варианты метода линеари-
зации [202] или расчетные методы [201]. Погрешность в оцен-
ке положения максимума пика из однократно измеренного спек-
тра может составлять только 0,2—0,3 ширины канала.
По данным эталонных измерений строят градуировочный
график спектрометра (зависимость номер канала — энергия
пика), который обычно представляет собой прямую линию. Вы-
сокую линейность амплитудной характеристики имеют
Ge (Li)-детекторы, и наблюдаемые отклонения от линейности
связаны с работой усилителя и электронных систем многока-
нального анализатора. Для сцинтилляционного спектрометра
отклонение от линейности достигает нескольких процентов (в
интервале 0,14-3 Мэв). Градуировку спектрометра можно
представить и в математической форме путем обработки экс-
периментальных данных соответствующими методами.
Процедура определения энергии пиков в спектре исследуе-
мого источника у-излучения уже имеет обратный порядок по
отношению к процессу калибровки и может быть сравнительно
просто и с хорошей точностью осуществлена для достаточно
интенсивных и четких пиков. Для них требуется тщательная
оценка номера канала, который соответствует максимуму пика,
с последующим определением энергии из калибровочного графи-
ка. В благоприятных условиях определение энергии пика пол-
ного поглощения из однократно измеренного спектра может
быть осуществлено с погрешностью порядка 0,05 £i/2 [184],
где Д1/2 — разрешение спектрометра для у-излучения с энергией
Следовательно, минимальная погрешность в определении
энергии пика (в области около 1 Мэв) будет составлять для
полупроводникового спектрометра 0,1 кэв и для сцинтилляци-
онного — 3 кэв.
Для сравнения можно указать, что прецизионные измерения
с Ge (Li)-спектрометром дают погрешность всего 5—20 эв в ши-
роком интервале энергий у-излучений [201]. Правда, для полу-
чения результатов с такой точностью необходимы прецизионная
аппаратура, многократные и длительные измерения спектров в
наиболее благоприятных условиях и сложные методы обработки
получающихся данных.
Конечно, в рядовой аналитической работе действует ряд
ограничивающих и неблагоприятных факторов, которые препят-
ствуют достижению столь высокой точности при определении
172
энергии. Как правило, реально достижимая точность составляет
0,3—0,5 кэв для полупроводниковых [200] и 6—10 кэв для
сцинтилляционных спектрометров. Первая из указанных величин
вполне удовлетворяет практическим потребностям.
Однако в трудных ситуациях возможно дальнейшее увеличе-
ние погрешности в определении энергии пиков полного поглоще-
ния. Так, если интенсивность неизвестной линии в анализируе-
мом спектре мала, то на точности локализации максимума пика
сказываются статистические колебания числа отсчетов в кана-
лах анализатора. Положение еще более усугубляется, если сла-
бый пик расположен на непрерывном амплитудном распределе-
нии от интенсивных линий с более высокой энергией.
Максимум пика полного поглощения может несколько изме-
нить свое положение, если он попадет на склон комптоновского
распределения или какой-либо более интенсивной линии. Воз-
можно смещение линий спектра при высокой загрузке спектро-
метра. Последний фактор особенно сильно сказывается на
пиках, расположенных в низкоэнергетической части спектра.
На точность определения энергии у-излучения оказывает
влияние стабильность работы спектрометра. Так, по данным ра-
боты [195], дрейф калибровочной шкалы полупроводникового
спектрометра, работающего в нетермостатированных условиях,
достигает 10 кэв за 24 ч. Следовательно, во время проведения
экспериментов требуется контроль за калибровкой спектромет-
ра, который может выполняться путем эпизодических измерений
эталонных источников или постоянно специальными системами
стабилизации [203].
При анализе препаратов со сложным спектром у-излучения
очень велика роль разрешения спектрометра. Кроме уже отме-
ченной потери точности низкое разрешение приводит к дополни-
тельным затруднениям в определении энергии даже интенсив-
ных линий в результате эффектов наложения.
Когда энергия пика установлена, можно попытаться иден-
тифицировать радиоизотоп, который ответствен за его появле-
ние в измеренном спектре. При этом чем точнее определена
энергия пика, тем проще и надежнее идентификация радиоизо-
топа, которую осуществляют простым сравнением измеренных
значений энергий пиков с табличными данными по энергии
у-переходов [30, 32, 204, 205]. Идентификацию заметно облег-
чают таблицы, в которых у-переходы расположены в порядке
возрастания энергии [30, 32], а для сцинтилляционных измере-
ний полезны спектры отдельных радиоизотопов [32, 206]. При
идентификации всегда следует иметь в виду способ образова-
ния радиоизотопа и использованный метод активации.
Из-за различий в разрешении и точности определения энер-
гии пиков возможности сцинтилляционных и полупроводнико-
вых спектрометров для надежной идентификации существенно
различны. Так, при погрешности в величине энергии пиков менее
173
1 кэв (полупроводниковый спектрометр) редко возникают за-
труднения в идентификации радиоизотопов, образующихся при
облучении тепловыми нейтронами, так как различия в энергиях
у-переходов обычно превышают несколько килоэлектронвольт.
В случае же сцинтилляционных спектрометров возникают опре-
деленные затруднения и не всегда идентификация может быть
однозначной.
В сомнительных случаях для повышения надежности иден-
тификации, как правило, требуется привлечь некоторые допол-
нительные сведения. Для этого, например, проверяют, соответ-
ствует ли полуширина разрешению спектрометра в данном энер-
гетическом интервале. Если полуширина пика будет больше,
чем необходимо, то это свидетельствует о том, что пик состоит
из двух или большего числа у-линий близкой энергии. Этот же
факт подтверждает искажение формы пика.
Весьма эффективное средство повышения надежности иден-
тификации представляет измерение периода полураспада пика.
Наличие в измеренном спектре набора пиков, обусловленных
распадом радиоизотопа со сложным у-спектром, может также
служить дополнительным критерием идентификации. При иден-
тификации переходов следует помнить о возможности помех со
стороны пиков обратного отражения, суммирования, утеч-
ки и т. д.
Выше была рассмотрена процедура определения энергии
достаточно интенсивных пиков и идентификации радиоизотопов
в сравнительно простых смесях. Обработка сложных спектров,
определение энергии и идентификации слабых пиков, а также
получение количественной информации требуют применения
более сложных методов, а часто и вычислительных машин, о
чем речь пойдет ниже.
Получение количественных результатов
из простых спектров
Мерой интенсивности регистрируемого излучения может слу-
жить либо число отсчетов в максимуме пика полного поглоще-
ния W,-m, либо общее число отсчетов в пике Ns (площадь пика),
накопленное за определенную длительность измерения. По-
скольку форма пика близка к кривой Гаусса, то указанные
величины связаны соотношением
А^= 1,064Е1/2А^. (7.9)
Для спектров, представленных в графической форме, пло-
щадь пика можно оценить обычными способами. Однако на вы-
ходе многоканального анализатора измеренный спектр выра-
жается в виде числа отсчетов, накопленных в каналах в тече-
ние периода измерения. Изображение такого спектра имеет
форму гистограммы (рис. 48).
174
Для примера взят очень простой случай, когда достаточно
интенсивный пик расположен на равномерно возрастающем
фоне. Если каналы, ограничивающие пик с обеих сторон, обо-
Рис. 48. Участок у-спектра с одиночной линией на вы-
ходе многоканального анализатора:
1 — средняя линия фона в методе трапеции; 2 — нижняя гра-
ница в определениях по методу калиброванной доли пика.
значить соответственно
и /гв, то общее число отсчетов, кото-
рое содержится в каналах между ними, равно
Ng = 2
(7.Ю)
где Nj — число отсчетов в i-канале. Чтобы определить чистую
площадь пика, надо вычесть фон из величины NG. Учесть собст-
венный фон прибора можно обычным порядком из предвари-
тельных измерений. Проблема оказывается много сложнее,
когда пик расположен на непрерывном амплитудном распреде-
лении от более жестких линий.
Если в области более высоких энергий расположен одиноч-
ный пик, площадь которого можно оценить без затруднений, то
его вклад в площадь под пиком более мягкого у-излучателя
рассчитывают из выражения .¥'ф =&ЛЧЖ, где А'^ф— вклад жест-
кого компонента в площадь под пиком мягкого компонента;
N-ьж —число отсчетов в пике жесткого компонента; k — коэф-
175
финиент пропорциональности, определяемый экспериментально
по эталонному спектру жесткого компонента. Когда число более
жестких компонентов равно двум и более, то такой способ опре-
деления поправки на непрерывное распределение становится
мало практичным. Тогда предполагают, что в области пика
фон меняется линейным образом и необходимую поправку мож-
но рассчитать как площадь получающейся трапеции (рис. 48)
на основе числа отсчетов в каналах анализатора, расположен-
ных до и после рассматриваемого пика. Чтобы результаты были
точнее, вычисляют среднее значение числа фоновых отсчетов
для нескольких каналов, лежащих по обеим сторонам пика.
Тогда число фоновых отсчетов в области пика будет равно
(Л'фн + ЛГфв) ., , ,
Л?2ф 2 (^в Ч- 1 )• (7-11)
Теперь можно рассчитать чистую площадь пика:
Ns = Ne — N^. (7.12)
Для аппроксимации линии фона в области пика предложены
и другие методы, но сравнение их показывает, что метод трапе-
ции, будучи самым простым, все же входит в число наиболее
точных [200]. Однако метод трапеции дает хорошие результа-
ты только при условии, что число отсчетов в пике превышает
число отсчетов фона (ЛТ>.Меф). В противном случае точность
результатов быстро падает с уменьшением отношения
и для получения минимальной погрешности приходится подби-
рать оптимальное число суммируемых каналов и более точно
учитывать изменение фонового спектра.
Широкое распространение для количественных расчетов по-
лучил метод калиброванной доли пика [207, 208], который тоже
предполагает прямолинейный ход фона под пиком полного по-
глощения. Отличие этого метода от метода трапеции состоит в
том, что подсчитывается не вся площадь пика, а только ее опре-
деленная часть (см. рис. 48). Граничные каналы, которые мы
обозначим k' и k', выбираются симметрично относительно кана-
ла, соответствующего максимуму пика. Тогда для оценки числа
отсчетов имеем уравнение
, 2в / N х
~2 Н ) (7J3)
‘=*н
Точность получающихся результатов оказывается тем выше,
чем более строго выполняется условие линейности фона и по-
стоянства формы пика и, конечно, чем больше число отсчетов
в пике и меньше величина, поправки. Что касается линейности
фона, то это условие довольно хорошо соблюдается в случае
спектров от Ge (Li)-детекторов, поскольку пик полного погло-
176
щения занимает сравнительно небольшую энергетическую об-
ласть па непрерывном амплитудном распределении. Ширина
пиков в сцинтилляционных спектрах много больше, и поэтому
вероятность попадания на криволинейный участок амплитудного
распределения значительно возрастает [209—211]. Как пока-
зано Стерлинским [210], систематическая погрешность, обус-
ловленная кривизной фоновой линии, может быть обнаружена
при сопостазлении числа отсчетов в калиброванной площади
пика в исследуемом спектре и спектре эталона при увеличении
числа суммируемых каналов от единицы до разумной предель-
ной величины.
Джуле показал, что при высокой загрузке спектрометра воз-
можно искажение формы пика, которое при использовании ме-
тода калиброванной части пика приведет к погрешности в ко-
нечном результате [212]. Уменьшить влияние этого источника
погрешности можно путем соответствующего выбора числа ка-
налов, вклад которых суммируется при расчетах.
Определение величины .\'( по уравнению (7.13) имеет тот
недостаток, что статистические колебания величин jV/; и Nhs
при неблагоприятном соотношении пик — фон оказывают силь-
ное влияние на точность получающегося результата, как это
можно видеть из выражения для среднего квадратического от-
клонения [210, 211]:
s =
+(*. *,+1)г(А'».+№.).
(7.14)
Это затруднение можно обойти, если для количественных
расчетов использовать не величину однократно калиброванной
доли пика , а сумму величин, получающихся при последова-
тельном увеличении числа суммируемых каналов [211]:
N?
(7.15)
Хотя введенная величина N's не имеет особого физического
смысла, но ее конечное значение пропорционально регистрируе-
мой активности, и поэтому она вполне может быть использована
для количественных расчетов.
Расчет содержаний по данным, полученным из зарегистри-
рованного у-спектра, может быть осуществлен любым из рас-
смотренных ранее методов (см. гл. 3 § 2). Хотя в большинстве
случаев прибегают к методу эталонов (мониторов), у-спектро-
метрические измерения допускают достаточно точные определе-
ния абсолютной активности радиоизотопа, которую в случае
177
достаточно большого периода полураспада изотопа можно рас-
считать по уравнению
Ad
sfsfк^изм
(7.16)
где Nz —число отсчетов в пике (или его определенной части);
е. — абсолютная эффективность регистрации; fB и fK—-выход и
Рис. 49. Эффективность пика пол-
ного поглощения (1—3) и процес-
са образования пар с утечкой
обоих аннигиляционных кван-
тов (4, 5):
1 — кристалл Nal(Tl) размером 72Х
X 72 мм; 2, 4 — коаксиальный Ge(Li)-
детектор объемом 47 см1; 3, 5 — пла-
нарный Ое(Ы)-детектор объемом
1,8 см1.
коэффициент конверсии у-перехо-
да соответственно. Величина е
может быть получена путем рас-
чета или калибровки [202, 206].
Наиболее просто калибровку
спектрометра можно провести с
помощью радиоизотопов с изве-
стной абсолютной активностью.
Если для этих экспериментов при-
менить радиоактивный ряд, на-
чинающийся с 228Th и находя-
щийся в равновесии, то калибро-
вочный график получается в ре-
зультате однократного измере-
ния [200].
Очень интересно сопоставле-
ние полупроводникового и сцин-
тилляционного спектрометров по
эффективности регистрации у-из-
лучения (рис. 49). Как видно, со-
отношение эффективностей яв-
ляется обратным по отношению
к энергетическому разреше-
нию— сцинтилляционные спект-
рометры значительно превосхо-
дят полупроводниковые. Правда,
узкая линия обеспечивает полу-
проводниковому спектрометру
лучшее отношение сигнал/шум,
что важно при обнаружении сла-
бых линий в присутствии высоко-
го уровня помех. Однако при ре-
гистрации интенсивности линий
с заданной погрешностью полу-
проводниковый спектрометр тре-
бует более длительных измере-
ний, что возможно только для
достаточно долгоживущих радиоизотопов при слабоактивном
препарате. С другой стороны, этот фактор заметно снижает про-
изводительность полупроводниковых спектрометров.
178
Выше были рассмотрены наиболее простые методы получе-
ния количественной информации. Они дают хорошие результа-
ты по четко разрешенным пикам с большим числом отсчетов.
В менее благоприятных ситуациях (многокомпонентность спект-
ра, малая интенсивность или наложение отдельных пиков
и т. д.) требуются более сложные методы расчетов, для прове-
дения которых, как правило, нужны электронные вычислитель-
ные машины (ЭВМ).
Обработка сложных спектров
Когда исследуемый препарат содержит значительное число
радиоизотопов, каждый из которых является источником одной
или нескольких у-линий, то это приводит к соответствующему
осложнению получающегося на выходе спектрометра ампли-
тудного распределения. При этом со всей полнотой начинают
проявляться отрицательные последствия ограниченной разре-
шающей способности и сложной функции отклика гамма-спект-
рометров. В результате в амплитудном распределении появ-
ляются пики, площади которых частично или полностью пере-
крываются, и ложные пики, обусловленные различными побоч-
ными эффектами, кроме того, происходит нивелирование слабых
пиков, которые маскируются статистическими колебаниями не-
прерывного амплитудного распределения от более жестких ли-
ний. В этих условиях процесс извлечения необходимой инфор-
мации из амплитудного распределения становится более труд-
ным, а получающиеся результаты менее точными и часто неодно-
значными. В наибольшей степени эти ограничения проявляются
в случае сцинтилляционных спектрометров. Чтобы убедиться в
справедливости этого положения, достаточно даже беглого
взгляда на рис. 50, на котором представлены спектры одной и
той же облученной нейтронами пробы, полученные с помощью
сцинтилляционного и полупроводникового спектрометров [195].
Различие в разрешении существенно влияет на возможность
применения спектрометра того или иного типа для измерения
сложных спектров и приводит к некоторой специфике в подхо-
де к обработке результатов. Для спектров, полученных с по-
мощью полупроводниковых спектрометров, удовлетворительные
результаты даже в случае сложных спектров дают те простые
методы, которые были рассмотрены выше. Что касается сцин-
тилляционных спектрометров, то их применимость к анализу
сложных спектров довольно ограничена, а обработка спект-
ров много труднее.
Одним из специфичных методов обработки сложных спект-
ров, который обычно применяется к сцинтилляционным спект-
рам, является метод последовательного вычитания [207, 213,
179
214]. При разложении суммарного спектра на отдельные ком-
поненты этим методом прежде всего определяют энергию наи-
более жесткой линии или идентифицируют радиоизотоп, кото-
рому она принадлежит. Затем, используя эталонную форму
линии (спектр радиоизотопа), проводят вычитание ее из сум-
марного спектра. Методика осуществления этой операции мо-
Рис. 50. у-Спектр пробы морской воды, измеренный с помощью
сцинтилляционного [Nal(Tl) размером 72x120 лги] (/) и полу-
проводникового [Ge(Li)-детектор объемом 20 си3] (2) спектро-
метров.
жет быть различной: графическое разложение [207], инверси-
онное введение эталонного спектра в анализатор с контролем
по электроннолучевому индикатору [215], обработка на ЭВМ
[214]. Операцию вычитания последовательно применяют ко
всем линиям спектра до его полного разложения на отдельные
составляющие.
Необходимые для разложения эталонные спектры получают
либо расчетным путем, что бывает редко, либо эксперименталь-
но с помощью эталонных источников моноэнергетического у-из-
лучения. Методом интерполяции рассчитывают формы линий
180
для промежуточных значений энергии. Условия получения эта-
лонных спектров должны совпадать с условиями измерения ис-
следуемых источников.
В ходе последовательного вычитания происходит накопление
погрешностей, что в конечном счете сказывается на точности
результатов, получаемых для низкоэнергетических компонентов.
К дальнейшему увеличению неопределенности результатов при-
водит присутствие в спектре тормозного излучения, учесть кото-
рое довольно трудно. Определенные трудности представляет
разложение спектров, содержащих перекрывающиеся пики. Не-
стабильность анализатора — также существенный фактор, влия-
ющий на конечные результаты разложения.
В заключение можно указать на два случая, когда метод
вычитания оказывается очень полезным. Прежде всего имеется
в виду возможность применения его для вычитания спектра
фона из суммарного измеренного спектра. При определениях
по короткоживущим изотопам этот метод позволяет ввести по-
правку на присутствие долгоживущих активностей [216]. Ана-
лизируемую пробу измеряют сразу же после облучения и полу-
ченный спектр фиксируют. Затем через некоторый промежуток
времени, который необходим для распада короткоживущих ра-
диоизотопов, повторно измеряют спектр пробы и вычитают его
из полученного ранее. Разностный спектр представляет собой
спектр короткоживущих радиоизотопов. Этот метод применим
только при значительном различии периодов полураспада ко-
роткоживущих и долгоживущих радиоизотопов.
Применение вычислительных машин
для анализа у-спектров
Активация пробы сложного состава с последующим измере-
нием на детекторе высокого разрешения может дать обширную
информацию в виде спектра с большим количеством пиков (до
сотни), фиксированного в памяти многоканального анализатора
(до 104 каналов). К этому следует добавить данные по этало-
нам, условиям облучения и измерения. Значителен также объем
вспомогательной информации, касающейся параметров схем
распада радиоизотопов, эталонных спектров, сведений по кали-
бровке спектрометра и т. д.
Поскольку конечным результатом аналитического определе-
ния должны быть сведения по составу пробы и содержанию от-
дельных компонентов в ней, то вся наличная информация долж-
на быть соответствующим образом свернута. Ручная обработка
таких объемов информации трудоемка и длительна, поэтому для
хскорения процесса обработки приходится прибегать к помощи
(быстродействующих ЭВМ.
181
Тогда вывод данных из многоканального анализатора осу-
ществляют в форме, удобной для ее последующего введения в
ЭВМ (магнитная лента, перфолента, перфокарты и т. д.).
Иногда устанавливают прямую связь анализатора с вычисли-
тельным центром и даже прибегают к включению ЭВМ в состав
измерительного комплекса.
Свертка аналитических данных на ЭВМ осуществляется по
программам, основанным на различных математических мето-
дах: наименьших квадратов, итераций, преобразований Фурье,
матричного и т. д. Наибольшее распространение пока имеет
первый из упомянутых методов [200]. Описание математиче-
ских методов и используемых программ не входит в задачу дан-
ной монографии и ниже будут затронуты только некоторые
практические аспекты, связанные с обработкой полученных дан-
ных на ЭВМ.
Сглаживание. При анализе спектров с помощью ЭВМ пер-
вой операцией часто становится сглаживание исходных данных,
которое сводится к некоторому усреднению статистических коле-
баний числа отсчетов в каналах путем соответствующей матема-
тической обработки. Сглаживание осуществляют таким обра-
зом, что получающийся спектр сохраняет все основные парамет-
ры исходного, но он более удобен для последующей обработки
и позволяет более четко выявлять слабые пики [217]. При сгла-
живании небольшой участок спектра аппроксимируют степен-
ным полиномом, который подгоняют к экспериментальным дан-
ным методом наименьших квадратов.
При таком подходе сглаженное значение Ni в канале рас-
считывается из соотношения
_ , /=+«
Ni~ 2 am,jNi+h
/==—ш
(7.17)
где константа ат, j и нормирующая константа qm находятся
из таблиц [200]. Величина 2т + 1 представляет собой число
точек, по которым производится сглаживание, и для получения
наилучших результатов требует оптимизации. Процесс сглажи-
вания можно повторить несколько раз, что положительно влияет
на форму спектра.
Определение положения пика. Простейший способ локализа-
ции пика с помощью ЭВМ состоит в сканировании спектра в
поисках каналов, где выполняются условия:
Nk-2 <Nk — p VN~k;
Nk+2<Nk-p^N~k,
182
где р — экспериментально выбираемая константа (обычно
/?=1). Этот метод недостаточно чувствителен к слабым пикам и
не может разделять двойные пики.
Более надежен метод первой производной спектра, которая
меняет свой знак с положительного на отрицательный при пере-
ходе через максимум пика. Этот метод применяют к предвари-
тельно сглаженным спектрам, и местоположение пика от-
мечается при выполнении следующих условий:
Л(/г)<0;
У1 (& Д- и) < 0 для п = 1, 2, . . ., г;
Yr(k — п) > 0 для п = 1, 2, . . ., I,
где У] — первая производная; г и / — верхний и нижний гранич-
ный каналы.
Наилучшие результаты дает метод второй производной, ко-
торый позволяет не только обнаружить и локализовать пик на
большом фоне, но и отличить действительно одиночный пик от
края непрерывного распределения или двойного пика. Суть ме-
тода состоит в том, что вторая производная от сглаженного
спектра отлична от нуля только внутри пика. Однако из-за ста-
тистической природы исходных данных разброс значений второй
производной очень велик и для обнаружения слабых пиков при-
ходится прибегать к сглаживанию спектра второй производной.
Определение площади пика. Когда пик обнаружен, пытаются
получить необходимые данные о его площади путем подбора
методом наименьших квадратов параметров в уравнении, опи-
сывающем пик. Наиболее часто считают форму пика полного
поглощения гауссинианом, расположенным на линейном фоне.
Тогда соответствующая функция будет иметь вид
Nt = Де 2s2 4-5/+С, (7.18)
где s2 — дисперсия кривой Гаусса; km— положение максимума
пика; А, В и С— постоянные. Площадь пика равна Л^=2,5Д«.
Если форма пика или величина Ер2, полученная при первом
определении, указывает на два или более совместных пика, то
они могут быть определены раздельно путем решения методом
наименьших квадратов системы уравнений типа (7.18).
Для определения площади пика могут быть применены и те
простые методы, которые были рассмотрены ранее. Получив
данные о положении пика и его площади, можно без труда
перейти к рассмотрению энергий линий, идентификации изото-
пов и к проведению необходимых количественных расчетов.
183
Качественный анализ спектров на ЭВМ. Примером может
служить методика качественного анализа у-спектров, предло-
женная Адамсом и Дамсом [218] применительно к аналитиче-
ским определениям на основе облучения тепловыми нейтронами.
Для получения спектров использован Ое(Ы)-спектрометр. По-
ложение пиков в спектре определяли визуально по электронно-
лучевому индикатору многоканального анализатора. Однако эта
операция может быть выполнена ЭВМ по специальной програм-
ме. Положение пика, выраженное номером канала анализатора,
конвертируется в энергию пика по калибровке спектрометра, ко-
торая записана в памяти ЭВМ в форме степенного полинома
вида
— А + Bkin -|- С^т 4- ... Сп_ ikm,
(7.19}
где Еу—энергия пика; km — номер канала, на который прихо-
дится максимум пика; А, В, С — постоянные (получены мето-
дом наименьших квадратов из данных калибровочных измере-
ний). При этом учитывается дрейф энергетической шкалы спек-
трометра, которая периодически контролируется путем повтор-
ных измерений источников 241Ат (59,5 кэв) и 60Со (1332,4 Мэв).
Для идентификации радиоизотопов проводится сравнение
полученных энергий пиков с энергиями у-переходов, хранящи-
мися в запоминающем устройстве ЭВМ. Набор хранящихся све-
дений о всех радиоизотопах, образующихся при облучениях
нейтронами, весьма широк. Прежде всего он включает наиболее
точные значения энергий до 5 основных у-переходов и периода
полураспада радиоизотопа. В памяти ЭВМ записаны также не-
которые необходимые дополнительные сведения: энергии менее
интенсивных у-переходов, удельные активности элементов при
стандартном потоке нейтронов и условия измерения эталонов,
абсолютные интенсивности переходов, способы образования ра-
диоизотопов.
Связь пика с каким-либо основным переходом радиоизотопа
считается установленной, если энергия пика в пределах довери-
тельного интервала совпадает с энергией перехода. При иденти-
фикации принимаются во внимание радиоизотопы, чей период
полураспада превышает 0,04 Zpa(.n. В рассматриваемой работе
доверительный интервал для четких пиков оценивается из соот-
ношения
АЕу = Q,5EV + 0,7,
(7.20)
где АЕу — доверительный интервал, кэв-, Еу — энергия пика,
Мэв. Для слабых или плохо фиксированных пиков доверитель-
ный интервал устанавливается более широким.
184
Программа составлена таким образом, что при неоднознач-
ной идентификации ЭВМ перечисляет всех возможных кандида-
тов и тогда требуется привлечение дополнительных сведений
для окончательного решения. Проверяется также возможность
появления отдельных пиков за счет вторичных процессов (пиков
утечки, суммирования, рентгеновское излучение). Оставшиеся
неидентифицировавные слабые пики сравниваются с энергиями
малоинтенсивных переходов (хранятся во вспомогательном за-
поминающем устройстве) радиоизотопов, наличие которых в
ПУСК
Чтение входных данных: условия
облучения и измерения. Точное или-*
приблизительное положение пиков
Определение энергии пиков
Чтение основных данных: радиоизо-
топы, Т^.-, способ образования, актив- ,
ность насыщения, основные у-перехо'
ды
Сравнение
у которых
энергии пиков с энергиями переходов,
1 .м 4>0,04 /расп.
Установление надежности идентификации. Отклоне-
ние невозможных случаев
Отбор пиков утечки, суммирования и рентгеновского
излучения
I Выдача результатов предварительной
I идентификации
4
Установление возможных компонентов источника.
Проверка генетически связанных радиоизотопов
Расчет чувствительности для принятых условий
анализа
Выдача списка обнаруженных радио-
изотопов
Чтение вспомогательных данных:
у-переходы слабой интенсивности
Отбор малоинтенсивных пиков обнаруженных радио-
изотопов и случаев неоднозначной идентификации
Выдача списка обнаруженных радио-
изотопов, Т112, способы образования
Выдача данных о радиоизотопах, энер-
гии у-переходов, энергии пиков
Выдача списка неидентифицированных
пиков
4
СТОП
Рис. al. Схема качественной идентификации пиков с помощью ЭВМ.
185
источнике уже установлено по данным основного запоминаю-
щего устройства.
Наконец, результаты идентификации выдаются в виде выход-
ных данных, которые перечисляют обнаруженные пики, их энер-
гии и радиоизотопы, к которым они отнесены. Отдельно отме-
чаются неидентифицированные пики. Схема идентификации пи-
ков с помощью ЭВМ показана на рис. 51.
Полная обработка у-спектров на ЭВМ. Применение ЭВМ до-
пускает полную автоматизацию процесса обработки полученных
данных с выдачей на выходе требуемой аналитической инфор-
мации. Полная программа обработки имеющихся данных ис-
пользует отдельные элементарные операции, которые были рас-
смотрены выше. Дополнительно включаются сведения об усло-
виях анализа и необходимые операции с ними. Поскольку про-
цесс накопления данных и их обработка могут быть разделены
во времени, то иногда на ЭВМ обсчитывают одновременно ре-
зультаты многих определений.
Программа полностью автоматической обработки результа-
тов активационного анализа включает следующие основные ста-
ПУСК
Ввод: данные, считываемые____ ____Ввод: перечисление основных
с магнитной ленты параметров программы
Преобразование записи магнитной ленты
Выбор требуемого спектра
Картотека данных для -^Внутренняя калибровка по энергии
градуировки |
Результаты измерения мо- -^-Введение поправки на поток нейтронов
нитора |
Определение фона
Ф
Определение площадей пиков. Разре- ч-Значения энергии
шение дублетов у-переходов |
Г Картотека
Определение весовых количеств-^—Содержание элемен- радиоизо-
| тов в эталонах топов
Введение поправки на холостой опыт^Данные по 1
облучению
чистого филь- *--'
тра
I
Переход к концентрации элементов
в воздухе I
Выдача конечных результатов |
СТОП
Рис. 52. Схема обработки на ЭВМ результатов аналитических определении
186
дни: 1) качественный анализ спектров; 2) расчёт площадей
пиков; 3) расчет содержания обнаруженных элементов; 4) выда-
чу окончательных данных о содержании элементов в исследуе-
мой пробе. Примером того, как такая программа реализуется
на практике, может служить работа Дамса и др. [219] по опре-
делению содержания элементов в аэрозолях атмосферы.
При отборе проб воздух прокачивают через специальный
фильтр. После облучения тепловыми нейтронами реактора из-
меряется у-спектр фильтра на Ge (Li)-спектрометре. Результаты
измерений фиксируются на магнитной ленте, которая впослед-
ствии вводится в считывающее устройство ЭВМ. На одной ленте
записываются данные до 100 определений, обработка которых
по заданной программе выполняется отдельно (рис. 52). Дли-
тельность расчетов 100 спектров, зарегистрированных с по-
мощью 4096-каналыюго анализатора, не превышает 1 ч.
§ 7. Применение у-спектрометрического метода
для инструментального активационного анализа
Широкое внедрение у-спектрометрии в практику активаци-
онного анализа дало исключительно плодотворные результаты,
которые нашли свое выражение в разнообразных аналитических
методах, рассчитанных на определение одного или нескольких
элементов и выполняемых быстро, с малыми затратами труда
и без разрушения пробы. Инструментальный у-спектрометри-
ческпй активационный анализ — исключительно гибкий метод,
он может быть приспособлен для проведения аналитических
определений в самых различных условиях.
В зависимости от длительности высвечивания возбужденных
уровней или периодов полураспада радиоизотопов, которые слу-
жат основой для аналитического определения, появляются не-
которые особенности в методике инструментального у-спектро-
метрического активационного анализа. По этому признак}' мож-
но выделить следующие случаи анализа: 1) по мгновенному из-
лечению; 2) по очень короткоживущим изотопам; 3) по коротко-
жив'.щим изотопам; 4) по средне- и долгоживущим изотопам.
Надо заметить, что перечисленные варианты существенно раз-
личаются не только по методике анализа, но и по своим анали-
тическим характеристикам.
Анализ по мгновенному ^-излучению
В процессе многих ядерных взаимодействий происходит
испускание характеристического у-излучения, длительность вы-
свечивания которого очень мала (менее 10~9 сек). Понятно, что
его регистрация возможна только при одновременном облучении
187
пробы потоком активирующего излучения. Однако при этом тре-
буется исключить возможность воздействия активирующего из-
лучения на детектор у-излучения. Наиболее просто это условие
можно выполнить при облучении заряженными частицами, ко-
торые легко поглощаются веществом анализируемой пробы. До-
стоинства анализа по мгновенному излучению состоят в быстро-
те получения аналитических результатов и возможности опреде-
ления элементов, которые не дают радиоизотопов с благоприят-
ными аналитическими характеристиками.
Облучение заряженными частицами. Метод регистрации
мгновенного у-излучения при облучении заряженными части-
цами во многом аналогичен методу спектроскопии мгновенных
заряженных частиц (см. § 1 этой главы) с тем отличием, что
высокая проникающая способность и изотропия у-излучения по-
зволяют значительно упростить методику анализа. Энергия
испускаемого моноэнергетического у-излучения зависит от энер-
гетического баланса ядерной реакции и способа распада состав-
ного ядра и в предельном случае достигает весьма высокой ве-
личины (для реакции Чл(р, у)8Ве £7= 17,6 Мэв). Выход мгно-
венного у-излучения в реакциях заряженных частиц высок, но
спектр излучения часто сложен.
Успешные аналитические определения путем регистрации
мгновенного у-излучения возможны при небольшой энергии за-
ряженных частиц (не выше 2—5 Мэв), так как с их ростом силь-
но увеличивается выход нейтронов, что значительно осложняет
проблему защиты детектора. При низкой энергии активирую-
щего излучения возможно определение только легких элементов
в тонких слоях вещества. Предел обнаружения лежит несколь-
ко выше, чем в случае спектроскопии мгновенных заряженных
частиц, из-за более высокого фона детектора, взаимных помех
со стороны у-излучения разных реакций и влияния вторичного
и рассеянного излучения. При использовании ускорителя предел
обнаружения находится на уровне около 10 4 % и редко опу-
скается ниже [220]. Наиболее обстоятельно исследованы воз-
можности метода при облучении протонами с энергией 0,5 Мэв
[221]. Весьма полезные сведения могут быть получены с прото-
нами резонансных энергий [222].
Рассмотрим некоторые практические примеры использования
этого метода. Так, для определения фтора в опаловом стекле
по реакции 19F(р, ay) |6О (£v=6,14 и 7,12 Мэв) Рабин и др.
[223] использовали протоны с энергией 1,4 Мэе. Кусочки стекла
(1,8X1.8X0,3 см) облучали протонами при токе пучка 1 мка и
регистрировали у-излучение с энергией 6—7 Мэв с помощью
сцинтилляционного счетчика. Эталоном служил NaF, так как он
имеет примерно tv же самую тормозную способность, что и
материал пробы. Помех со стороны других элементов, входящих
в состав опалового стекла, замечено не было. Измеренная кон-
центрация фтора в пробах была порядка 3%.
188
И. X. Лемберг и др. [181] проводили определения по реак-
ции 18О(а, ny)2'Ne (Е?=350 кэв). Анализируемую пробу нано-
сили в виде тонкого слоя на медную пластинку, покрытую нике-
лем. Облучение а-частицами с энергией 4,6 Мэв (источник —
циклотрон) длительностью 5—10 мин при токе пучка 10 8 а и
измерении мгновенного у-излучения сцинтилляционным спек-
трометром обеспечивает чувствительность примерно 2 10 ‘‘ г.
Регистрация мгновенного у-излучения при облучении а-ча-
стицами 238Рц (Ю мкюри) была использована для определения
изотопного состава Li и В [180]. Этот метод с изотопными
источниками a-излучения оказался полезен также при анализе
некоторых руд на В, Be и F [158].
Облучение нейтронами. Под действием нейтронов мгновен-
ное у-излученне возникает в процессах радиационного захвата
медленных нейтронов и неупругого рассеяния быстрых нейтро-
нов [224]. Источниками нейтронов при определениях по мгно-
венному излучению могут служить нейтронные генераторы, не-
которые типы радиоизотопных источников и пучки нейтронов,
выведенные из активной зоны реакторов. При этом должна
быть обеспечена защита детектора как от первичного нейтрон-
ного излучения источника, так и от сопутствующего ему пер-
вичного и вторичного у-излучения. Вообще проблема снижения
уровня фона детектора при регистрации мгновенного у-излуче-
ния представляет сложную задачу и часто высокий уровень ме-
шающей активности оказывает сильное влияние на аналитиче-
ские характеристики метода (чувствительность, правильность и
точность).
Поскольку сечения реакций (и, у) выше, чем реакций не-
упругого рассеяния, то первый метод, как правило, демонстри-
рует более высокую чувствительность, и поэтому исследованию
возможностей именно этого метода было уделено основное
внимание [225, 226]. Энергия возбуждения ядра в результате
реакции (п, у) практически равна энергии связи захваченного
нейтрона и, следовательно, в большинстве случаев составляет
6—8 /Изе. При распаде возбужденного уровня обычно испу-
скается сложный спектр у-излучения, энергетический интервал
которого заключен между 0,01 —10 Мэв. Сложность спектра,
т. е. количество испускаемых при распаде у-линий, имеет тен-
денцию к возрастанию при переходе от легких к тяжелым эле-
ментам. У последних нередки случаи, когда распад возбужден-
ных состояний дает до 10—25 у-линий разной энергии и интен-
сивности [70]. Сложность получающихся спектров предопреде-
ляет необходимость применения детекторов высокого разре-
шения.
В первых исследованиях для регистрации у-излучения радиа-
ционного захвата использовались магнитные спектрометры, но
они обладают низкой эффективностью. Значительный толчок
развитию этого направления дало появление Ge (Li)-детекторов
189
[227, 228]. Сцинтилляционные спектрометры дают удовлетвори-
тельные результаты только в сравнительно благоприятных си-
туациях [229].
Согласно уравнению (2.17), регистрация мгновенного у-из-
лучения должна давать наилучшие результаты для элементов
с высокими сечениями радиационного захвата нейтронов Gd,
Eu, Sm, В, Cd и т. д. [225, 227]. При плотности потока нейтро-
нов 106 нейтрон/(см2 сек) и спектрометре с Ge (Li)-детектором
объемом 20 см3 достигаемая чувствительность для отмеченных
выше элементов при навеске 1 г и времени облучения 10 мин
составляет около 101 % [225]. Однако перечисленные элементы
имеют мягкое у-излучение, что заметно снижает избиратель-
ность их определения в присутствии других компонентов. Более
высокая специфичность и проникающая способность жесткого
у-излучепия создают лучшие условия для определения эле-
ментов, у которых наиболее интенсивные переходы радиацион-
ного захвата лежат выше 3 Л1эв [226]. В эту группу входят
следующие элементы: Cl, Sc, Ti, V, Cr, Мп, Fe, Со, Ni, Си, Zn,
А и и Hg.
Плотность потока выведенных пучков тепловых нейтронов
достигает 108 нейтрон/(см2-сек), что указывает на более высо-
кие потенциальные возможности метода, чем отмеченные выше.
Однако даже с учетом этого факта предельная чувствитель-
ность анализа по мгновенному излучению все же значительно
уступает предельной чувствительности анализа по излучению
радиоизотопов, правда, несомненное преимущество первого из
них состоит в более высокой скорости определений.
Схематично система для анализа по мгновенному излуче-
нию радиационного захвата на выведенном из реактора пучке
нейтронов показана на рис. 53. Для этого метода значительные
трудности представляют подавление и учет интерферирующего
излучения, которое приводит к значительному повышению уров-
ня фона детектора. Этот эффект может быть связан как с инду-
цированием мгновенного излучения (радиационного захвата и
пеупругого рассеяния) в различных конструкционных и вспо-
могательных материалах, так и с рассеянием нейтронов пробой
и деталями конструкции. Рассеянные нейтроны в основном по-
глощаются защитой, давая при этом излучение радиационного
захвата, и в небольшой степени попадают в детектор.
Положение усложняет и то обстоятельство, что в этом мето-
де одновременно регистрируется у-излучение, возникающее в
процессах радиационного захвата и пеупругого рассеяния ней-
тронов и при радиоактивном распаде образовавшихся радиоизо-
топов. Для исключения последнего источника помех предложено
модулировать поток нейтронов [225, 230]. Что касается различ-
ных компонентов фона, то их вклад можно оценить из спектров,
полученных в различных условиях: а) облучение пробы полным
потоком нейтронов; б) облучение пробы с Cd-фильтром в пуч-
190
не; в) облучение без пробы, но с Cd-фильтром; г) полный по-
ток нейтронов в отсутствие пробы.
Наибольшее значение метод спектрометрии мгновенного
у-излучения, вызываемого нейтронами, приобрел в ядерной гео-
физике для исследования вещественного состава пород [231].
В лабораторных условиях этот метод ввиду свойственных ему
Рис. 53.
венному
1 — пучок
роков из
Схема установки для анализа по мгно-
у-излучению радиационного захвата:
нейтронов; 2—проба; <3 — поглотитель нейт-
r'LiF; 4— Се(1Д)-детектор; 5 —свинцовая за-
щита.
ограничений находит применение сравнительно редко. Он слу-
жит преимущественно для определения элементов, которые при
облучении нейтронами не дают радиоизотопов с подходящими
периодами полураспада.
Е. М. Лобанов и др. [232] предложили использовать у-спект-
ры захвата для диагностики минералов. Образцы минералов по-
мещают в коллимированный пучок тепловых нейтронов, выве-
денных из горизонтального канала реактора типа ВВР-С. Для
регистрации у-излучения захвата используется сцинтилляцион-
ный гамма-спектрометр. Изучение спектров 10 различных мине-
ралов показало, что получающиеся спектры в каждом случае
имеют характерную форму. Поэтому сравнение измеренных
спектров с эталонными позволяет быстро идентифицировать ис-
следуемый минерал. Это избавляет от необходимости проводить
подробный химический анализ.
191
Комар и др. [233] рассмотрели возможность анализа биоло-
гических проб по у-излучению радиационного захвата.
С помощью Ge (Li)-детектора и пучка нейтронов 2 -107 ней-
трон/(см2 сек) оказалось возможным определять С1, К, В, N
в пробах крови; Са, Р, С1 и N в костях; N и S в волосах. Перед
анализом пробы высушивали, поскольку требовалось удалить Н,
излучение которого (£7 = 2,23 Мэв) мешало определениям. От-
мечены также трудности в проведении количественных опреде-
лений, так как по условиям анализа необходима эталонная
проба того же состава, что и исследуемая. Можно воспользо-
ваться методом внутреннего монитора, но это требует либо
добавления к пробе вещества, содержащего ртуть, либо неза-
висимого способа определения хлора в пробах.
Анализ по ^-излучению очень короткоживущих
радиоизотопов
До сравнительно недавнего времени радиоизотопы с перио-
дом полураспада в интервале 1 мсек—1 сек не находили ана-
литического применения. Однако успехи в развитии методики
активационного анализа позволили провести первые исследова-
ния аналитических возможностей радиоизотопов с такими ко-
роткими периодами полураспада. Поскольку транспортировка
пробы при этом практически исключена, приходится прибегать
к методике, в какой-то степени аналогичной методике работы с
мгновенным излучением. Исследуемая проба располагается в
пучке активирующего излучения, а детектор вторичного у-излу-
чения непосредственно около нее. Однако режим непрерывного
облучения с одновременной регистрацией заменяется импульс-
ным режимом с разделенными во времени процессами облуче-
ния и измерения.
Как источники импульсных потоков заряженных частиц на-
ходят применение циклотроны [234], а быстрых нейтронов —
нейтронные генераторы [235]. Лишь на самом краю указанного
интервала периодов полураспада можно прибегнуть к помощи
быстрых транспортирующих систем и тогда отпадает необходи-
мость в импульсном режиме работы источника активирующего
излучения. Недавно предложены конструкции пневмотранспорт-
ных систем с временем переноса челнока с пробой из зоны об-
лучения к гамма-спектрометру около 0,1—0,2 сек [216].
Для очень короткоживущих изотопов доля ядер, находя-
щихся в радиоактивном состоянии даже при облучении до насы-
щения, крайне мала, и для повышения чувствительности прихо-
дится прибегать к многократному повторению циклов облуче-
ние— измерение с суммированием спектра в памяти многока-
нального анализатора. Пусть общая длительность одного цикла
складывается из длительности отдельных стадий 7’ = /Обл +
.192
+ ^ЯГп + /„лм, тогда зарегистрированное за п циклов число от-
счетов в пике исследуемого компонента будет равно [234]
— —<ТЛГ-4Е (1 — е-?/обл) е~’WPacn ( 1 — е“?7и;;м) X
(7-21)
где Ф — плотность потока активирующего излучения; NA — чис-
ло исходных атомов активирующегося изотопа; е — абсолютная
эффективность регистрации.
При определении по столь короткоживущим изотопам необ-
ходимо с высокой точностью выдерживать очень короткие ин-
тервалы времени. Например, для определения натрия по реак-
ции 23Na(d, p)24mNa (7'1/2 = 20 мсек, £v=0,47 Мэв) был исполь-
зован следующий режим: = /II3M = 20 мсек и /расп=1 мсек.
При этом полное время одного анализа в многоцикличном ре-
жиме составляет 5—10 мин.
Для этого метода существенное значение имеет форма им-
пульса активирующего излучения [235]. Изменение формы
импульса в ходе анализа может быть источником погрешности.
С другой стороны, показано, что импульс в виде прямоуголь-
ного треугольника дает по сравнению с прямоугольным импуль-
сом более высокий уровень активности при одинаковой дозе
активирующего излучения [236].
Возможности аналитического применения очень короткожи-
вущих изотопов исследованы еще слабо, но некоторые предва-
рительные выводы уже можно сделать. Количество элементов,
для которых этот метод благоприятен, видимо, будет очень
мало. Достигнутый уровень чувствительности невысок и не пре-
вышает 10~3%. В системе с неподвижной пробой велик уро-
вень помех от интерферирующего излучения, что сказывается на
точности получаемых результатов.
Спектрометрический анализ по ^-излучению
короткоживущих изотопов
К настоящему времени инструментальный активационный
анализ по у-излучению короткоживущих изотопов получил ши-
рокое распространение. Среди достоинств метода выделяются
экспрессность анализа и возможность определения некоторых
практически важных элементов, у которых только короткоживу-
щие изотопы позволяют получать хорошие аналитические ре-
зультаты. Существенным фактором оказывается также широкое
распространение короткоживущих изотопов с благоприятными
активационными характеристиками и параметрами схем распа-
да. Так, при облучении тепловыми нейтронами 33 элемента дают
короткоживущие изотопы [66] и 29 элементов при облучении
7 Р. А. Кузнецов
193
жестким тормозным излучением [137, 148] или быстрыми
(14 Мэв) нейтронами [101]. Чувствительность определения по
короткоживущим изотопам достаточно высока и, например, при
облучении тепловыми нейтронами [Ф = 3,8-10й нейтрон! (см2 X
Хсек)] в большинстве случаев составляет 10~8—10~п г [66].
Особенность методик активационного анализа по коротко-
живущим изотопам состоит в том, что они предусматривают од-
новременное определение в пробе небольшого числа компонен-
тов (1—3). Очень часто режим анализа (энергия облучения,
длительность интервалов времени на разных стадиях анализа
и т. д.) устанавливается таким образом, чтобы обеспечить наи-
более оптимальное сочетание аналитических характеристик ме-
тода (чувствительность, избирательность, точность и т. д.).
Однако оптимизация режима проводится, как правило, в расче-
те на определение только одного компонента в пробе. Но это
не очень серьезное ограничение, поскольку быстрый распад ко-
роткоживущих компонентов позволяет проводить повторные
анализы одной и той же пробы. Понятно, что повторные анали-
зы могут быть оптимизированы уже в отношении других ком-
понентов. При анализе по короткоживущим изотопам каждая
проба проходит основные аналитические стадии (облучение и
измерение), как правило, индивидуально и с небольшим вре-
менным интервалом между ними.
Короткоживущие изотопы находят применение при опреде-
лении в широком интервале концентраций — от макроколичеств
до области малых концентраций (преимущественно Ю !—
10-5%). В последнем случае инструментальный анализ возмо-
жен только тогда, когда основные компоненты не мешают опре-
делениям.
Для элементов, радиоизотопы которых имеют периоды полу-
распада между 1 —100 сек, часто применяют цикличный метод
анализа. При этом методика анализа может быть оптимизиро-
вана как в отношении длительности одного цикла [237], так и
числа циклов [238].
Сложную проблему при работе с короткоживущими изото-
пами представляет введение поправок на мертвое время. Так,
при измерениях короткоживущих изотопов, активность которых
при этом быстро уменьшается, возникает необходимость при-
бегать к высоким начальным скоростям счета для получения
хорошей статистической точности. Однако многоканальные ана-
лизаторы имеют большое мертвое время, которое к тому же свя-
зано с номером канала зависимостью типа
т* = то + ^ть (7.22)
где тй — мертвое время при регистрации импульса в А-м кана-
ле; то — минимальное мертвое время; п — мертвое время, при-
ходящееся па один канал.
194
Обстоятельный анализ проблемы введения поправок на
мертвое время многоканального анализатора при регистрации
спектра у-излучения пробы, активность которой заметно ме-
няется за время измерения, выполнен О. К- Николаенко и др.
[239]. Остановимся на самом общем случае. Пусть активность
пробы составляют п компонентов с постоянными распада Xi,
л2 Измерения проводятся па /-канальном анализаторе,
имеющем измеритель доли просчетов (индикатор загрузки) или
счетчик живого времени.
Тогда поправка на просчеты для i-го компонента будет
равна
аг = ------------ "f r'^Dtdt. (7.23)
1 — е ^/пзм q
В свою очередь, величина £)( определяется из выражения
Dt = 1----------------!-----------, (7.24)
1 -/ л
1 +--------Х 1
' 1 — Do 10
где Do и Df—доля просчетов в начале измерения и момент вре-
мени /; Di о—парциальная доля просчетов /-го компонента в
начале измерения. Для величины Din действительно выра-
жение
i
Di0 = У «/ДВ,
(7.25)
где п,-;о — скорость счета /-го компонента в /-м канале в началь-
ный момент времени. Величины Din можно определить следую-
щим образом: в ходе регистрации спектра несколько раз заме-
ряют мгновенную долю просчетов (число замеров должно соот-
ветствовать количеству компонентов) и после подстановки их в
уравнение (7.24) получают систему уравнений, которую ре-
шают.
Как видно, проблема введения поправок на просчеты много-
канального анализатора в целом достаточно сложна и требует
знания состава компонентов (постоянных распада). Лишь в
частном случае одного компонента эта задача имеет сравни-
тельно простое решение [240, 241]. Применение ЭВМ позволяет
ускорить и облегчить соответствующие расчеты [239].
Спектрометрический анализ по ^-излучению
средне- и долгоживущих изотопов
Длительность анализа по радиоизотопам этих групп обычно
довольно велика, особенно если при этом стремятся к достиже-
нию предельной чувствительности. Прежде всего, конечно, за-
7* 195
траты времени определяются длительностью облучения, которое
может занимать от нескольких минут до недели. Часто перед
измерением пробы должны быть выдержаны для распада неже-
лательных короткоживущих компонентов. К тому же иногда
проводят несколько последовательных измерений спектра
у-излучения пробы на протяжении достаточно длительного ин-
тервала времени. Такой подход позволяет выявить большее чис-
ло компонентов в пробе и обеспечивает более надежную их
идентификацию. Длительность измерений спектра при слабой
активности пробы также может быть значительной, но ее пре-
дел не превышает нескольких часов.
Преимущества спектрометрического активационного анализа
по у-излучению средне- и долгоживущих радиоизотопов состоят
в высокой чувствительности и возможности определения многих
компонентов из одной пробы. Поэтому основное применение
этот метод находит в тех случаях, когда есть потребность в вы-
сокрй чувствительности определения одного или нескольких
компонентов, а задержка во времени существенной роли не
играет. Однако, несмотря на большой интервал времени, кото-
рый иногда проходит между поступлением пробы на облучение
и получением конечного результата, трудоемкость анализа в
целом оказывается низкой, поскольку анализ выполняется без
разрушения пробы и она не требует особых дополнительных опе-
раций. Более того, одновременно может быть облучено значи-
тельное количество проб, которые после определенной выдерж-
ки последовательно измеряются. Тогда даже при длительном
облучении и выдержке затраты времени на одну пробу могут
оказаться сравнительно небольшими. Если при этом еще воз-
можно определять несколько компонентов, то анализ в расчете
на один элемент становится уже довольно быстрым и эконо-
мичным.
Из-за необходимости длительных облучений анализ по сред-
не- и долгоживущим изотопам выполняется преимущественно с
помощью реактора.
Длительность облучений на ускорителях, как правило, не
превышает 20—30 мин, поэтому с их помощью анализ возможен
только по радиоизотопам, период полураспада которых не пре-
вышает 10 ч.
Для средне- и долгоживущих радиоизотопов процессы облу-
чения и измерения могут быть разделены достаточным проме-
жутком времени, который может быть использован для транс-
портировки проб. Это позволяет проводить активационные
определения в лабораториях, которые не располагают собст-
венным мощным источником активирующего излучения.
Наиболее удачны для у-спектрометрического активационного
анализа пробы со слабоактивирующейся основой. Например,
при облучении тепловыми нейтронами имеется ряд элементов и
их соединений, которые активируются в незначительной степе-
196
ни. К этой группе можно отнести такие элементы, как Be, С, Н,
О, N и др.
Спектрометрический анализ можно применять и к пробам,
макрокомпоненты которых при облучении дают только коротко-
живущие изотопы. Выдержка пробы перед измерением позво-
ляет избавиться от помех. Длительность выдержки, естественно,
зависит от периода полураспада наиболее долгоживущего ра-
диоизотопа и степени активации микрокомпонентов. Этот фак-
тор накладывает некоторые ограничения на период полураспа-
да радиоизотопов, которые могут быть использованы для ана-
литических определений. Обычно радиоизотопы с Д/г^Ю мин
мало влияют на возможности спектрометрического метода. При
облучении тепловыми нейтронами этому условию удовлетворяют
такие элементы, как Al, Mg, Ti, V и некоторые другие.
Определенные особенности имеет анализ проб, облучение
которых приводит к образованию чистых р_-излучателей. По-
скольку детекторы гамма-спектрометров чувствительны к ^-из-
лучению, то оно должно быть отфильтровано. Для этого между
детектором и источником помещают пластинку из алюминия
или оргстекла, как правило, достаточна пластинка толщиной
1,5 см. Однако поглощение ^“-излучения не решает проблему
до конца, так как при этом возникает тормозное излучение до-
статочно высокой интенсивности, которое заметно мешает опре-
делениям. Величина помехи зависит от степени активации ос-
новы и максимальной энергии ^--излучения. При облучении те-
пловыми нейтронами достаточно долгоживущие чистые р_-излу-
чатели дают следующие элементы: Si, Р, Tl, S, Са.
Во многих других случаях активация основных компонен-
тов пробы в той или иной степени создает помехи для спектро-
метрического анализа. Результатом такого влияния может быть
значительное ухудшение аналитических характеристик метода.
Когда уровень помех становится очень большим, то инструмен-
тальный вариант уже не дает удовлетворительного решения ана-
литической задачи и требуется прибегнуть к помощи радиохими-
ческого разделения.
Глава
8
СПЕЦИАЛЬНЫЕ МЕТОДЫ
ИНСТРУМЕНТАЛЬНОГО
АНАЛИЗА
§ 1. Анализ кривых распада
Период полураспада радиоизотопа — величина постоянная
и характерная и поэтому может быть использована для целей
идентификации. Чтобы определить период полураспада какой-
либо регистрируемой активности, надо проследить за ее измене-
нием во времени, т. е. снять кривую распада. Поскольку распад
радиоизотопа следует экспоненциальному закону [см. урав-
нение (2.15)], то представление кривой распада в полулогариф-
мическом масштабе дает для однокомпонентной системы пря-
мую линию, наклон которой определяет период полураспада.
Если исследуемый препарат содержит смесь нескольких
радиоизотопов с разными периодами полураспада, то изменение
активности во времени уже будет следовать выражению
Av = У Aofte
Й=1
(8-1)
где Az — суммарная активность препарата; Ао/; — активности
отдельных радиоизотопов в начальный момент времени; А*— по-
стоянная распада А-го компонента; /расп — длительность распа-
да; п — число радиоизотопов.
Регистрируя через определенные промежутки времени Г,
число отсчетов за интервал времени tj, получают набор точек
вида
Ni= V (!_е-ЧЛ) (/=1,2,. . ., т), (8.2)
fe=i
где .Vj — число отсчетов, зарегистрированное в /-м измерении;
Л'о,; — исходное число радиоактивных ядер й-го компонента;
е — эффективность. Анализируя эти данные различными мето-
дами. можно определить постоянные распада (периоды полурас-
пада) и начальную активность каждого из компонентов. Полу-
ченные значения периодов полураспада позволяют найти по та-
блицам, какие радиоизотопы содержатся в исследуемом пре-
парате.
198
Наиболее простой способ разложения кривых распада — гра-
фический. Например, кривую распада двухкомпонентной смеси
анализируют следующим образом (рис. 54). Полученные точки
наносят па график в полулогарифмическом масштабе. Как мож-
но видеть, получившаяся кривая по мере распада короткоживу-
щего изотопа переходит в прямую
распаду долгоживущего изотопа.
Прямую линию экстраполируют
до оси ординат и по ее наклону
определяют период полураспада
долгоживущего компонента. Вы-
читая затем число отсчетов дол-
гоживущего изотопа из суммар-
ной кривой распада, получают
данные по распаду короткожи-
вущего изотопа. Представленные
графически в полулогарифмиче-
ском масштабе эти данные дают
прямую линию с большим накло-
ном, по величине которого опре-
деляют период полураспада ко-
роткоживущего компонента. Точ-
ки пересечения полученных ли-
ний с осью ординат дают началь-
ные активности радиоизотопов.
Анализ смеси, включающей не-
сколько компонентов, про-
изводится аналогичным об-
лииию, которая соответствует
Рис. 54. Графический анализ кри-
вой распада активности пробы,
состоящей из двух радиоактив-
ных компонентов:
1 — измеренная кривая распада; 2 —
кривая распада долгоживущего ком-
понента (Т'1/2 = Ю мин)-, 3— кривая
распада короткоживущего компонен-
та мин).
разом.
Однако графический анализ кривых распада можно успеш-
но применять только тогда, когда выполнен целый ряд условий.
Прежде всего необходимо, чтобы компоненты не имели слишком
близких величин периодов полураспада. По оценке Монка и др.
[242] надежное расчленение радиоизотопов возможно только при
различии величин периодов полураспада не менее чем в 1,5 раза.
Для получения точных и надежных результатов надо иметь до-
статочно большое число точек на кривой распада, которая к то-
му же должна быть прослежена в течение нескольких периодов
полураспада наиболее долгоживущего из определяемых радио-
изотопов. И наконец, требуется довольно высокий начальный
уровень активности, чтобы позволить измерение кривой распа-
да с необходимой статистической точностью в течение достаточ-
но длительного времени. Последнее условие предъявляет жест-
кие требования к стабильности счетной аппаратуры, воспроиз-
водимости условий измерения и т. д.
Пз этих условий вытекают и ограничения метода: трудоем-
кость, длительность получения конечных результатов, низкая
избирательность. Как правило, анализ кривых распада приме-
199
няется только при определениях по радиоизотопам со сравни-
тельно небольшими периодами полураспада (T’i/2<3 дня) и для
смесей, состоящих не более чем из трех-пяти компо-
нентов.
Серьезный недостаток метода в применении к задачам акти-
вационного анализа заключается в сильной зависимости чув-
ствительности н точности определения компонентов от колеба-
ний в составе и относительной интенсивности радиоизотопов,
образующихся при облучении исследуемой пробы. Очевидно,
что при анализе кривых распада более долгоживущие компонен-
ты будут фоном для более короткоживущих. Статистические
колебания этого фона определяют минимальную активность
короткоживущих компонентов, которая может быть надежно за-
регистрирована, и заметно уменьшают точность получаемых ре-
зультатов. С другой стороны, необходимость выдерживания про-
бы до полного распада более короткоживущих компонентов уве-
личивает длительность анализа и приводит к соответствующему
уменьшению активности более долгоживущих компонентов, что
также сказывается на статистической точности результатов.
В практике активационного анализа часты случаи, когда ка-
чественный состав получающейся при облучении пробы смеси
радиоизотопов известен. Эти сведения могут вытекать из спо-
соба и условий облучения (или измерения), а также из дополни-
тельных источников, связанных с предшествующей историей про-
бы, или могут быть получены в результате предварительных
экспериментов (анализ кривой распада, спектрометрические из-
мерения, радиохимическое разделение и т. д.). Знание состава
радиоизотопов (точнее постоянных распада) значительно облег-
чает получение количественной информации из результатов из-
мерений активности исследуемой пробы, проведенных последо-
вательно через определенные интервалы времени. Одновременно
повышается точность результатов и сокращаются затраты вре-
мени на проведение анализа, методика которого к тому же мо-
жет стать более простой.
Одна из часто применяемых методик при известном соста-
ве радиоактивных компонентов состоит в проведении серии пос-
ледовательных измерений спада активности пробы со временем
с последующим получением системы уравнений на основе соот-
ношения (8.2). Понятно, что решение этой системы будет воз-
можно при достаточном числе измерений (m^n), при этом дли-
тельность и момент отдельных измерений должны быть оптими-
зированы с учетом известных постоянных распада относительно
предельной точности и чувствительности определений. Надо от-
метить, что оптимизация может быть распространена и на ре-
жим облучения, что благоприятным образом скажется на воз-
можностях метода. Конечно, при таком подходе нет необходи-
мости каждый раз решать получающуюся систему уравнений, а
можно вывести готовые выражения, подстановка в которые ре-
200
зультатов измерения сразу же даст число отсчетов или содер-
жание для определяемых компонентов.
Другой способ получения количественных результатов из из-
мерений спада активности пробы при известных компонентах
состоит в использовании номограмм [78]. Хотя предварительное
построение номограммы иногда требует длительных расчетов,
получение конечных результатов по ним осуществляется быстро
путем простого графического построения. Оба метода, упомяну-
тые выше, применимы к простым системам с числом компонен-
тов не выше 4—5.
Обработка совокупности экспериментальных данных вида
(8. 2) может производиться и математическими средствами пре-
образований Фурье и наименьших квадратов при участии ЭВМ
[200,243]. Причем метод преобразований Фурье позволяет рас-
считывать как периоды полураспада компонентов, так и их на-
чальные активности. Метод наименьших квадратов применяется
только для обработки систем с известным составом компо-
нентов [200].
Хотя зависимость активности радиоизотопа от длительности
распада сравнительно редко используется для целей их иден-
тификации, она часто выступает как важное вспомогательное
средство в повышении избирательности активационного анализа.
Наибольшее значение кривые распада активности пробы при-
обретают тогда, когда по тем или иным причинам избиратель-
ность применяемых методов регистрации низка. Например, облу-
чение жестким тормозным излучением многих важных элемен-
тов приводит к образованию чистых позитронно-активных ра-
диоизотопов, для измерения которых наиболее подходит анниги-
ляционное у-излучение. Однако последнее имеет одинаковую
энергию у всех радиоизотопов, поэтому только обработка кри-
вой распада позволяет расчленить активность пробы на отдель-
ные составляющие, не прибегая к радиохимическому разделе-
нию.
Некоторые методы, основанные на- интегральной дискрими-
нации по энергии излучения (чаще всего р-излучения), типу ча-
стиц (счетчики нейтронов), корреляции событий во времени (ме-
тод совпадений) и т. д., часто нуждаются в дополнительной ин-
формации, извлекаемой из кривых распада. Особое значение
анализ кривых распада приобретает при работе с короткожи-
вущими изотопами.
Однако даже при использовании гамма-спектрометров высо-
кого разрешения последовательное снятие спектров через опре-
деленные интервалы времени позволяет получить ценную анали-
тическую информацию (в плане числа компонентов, надежности
и точности результатов, чувствительности определения). Для
сцинтилляционных спектрометров такой способ исследования
у-излучения анализируемой пробы по существу является нор-
мальной практикой.
201
Анализ кривых распада, полученных в режиме интегрального
счета без какой-либо дискриминации излучения (счетчик Гей-
гера— Мюллера, сцинтилляционные счетчики и т. д.), теперь
используется довольно редко. В таком варианте он полезен толь-
ко при определениях основных компонентов пробы (при содер-
жании более 0,01%), и лишь в особо благоприятных случаях
граница сдвигается в область более низких концентраций. Иног-
да метод анализа кривых распада применяют для контроля ра-
диохимической чистоты выделенных препаратов. При этом метод
довольно чувствителен к посторонним радиоактивным примесям
с отличающимся периодом полураспада, присутствие которых
нарушает линейный ход кривой распада, изображенной в полу-
логарифмическом масштабе. Однако, если облучение какого-
либо элемента приводит к образованию двух или более радио-
изотопов, то способность метода к выявлению слабых посторон-
них радиоактивных примесей падает.
Последовательные измерения спада активности пробы могут
выполняться вручную. Однако при массовых анализах эта до-
вольно утомительная операция может быть передана автомати-
ческому устройству, которое само меняет препараты, задает
длительность измерения согласно программе и фиксирует ре-
зультаты. При анализе короткоживущих активностей исполь-
зуют специальный режим работы многоканального анализатора,
когда кривая распада фиксируется в каналах анализатора,
переключаемых через заданные интервалы времени.
§ 2. Метод совпадений и антисовпадений
Принципы метода и основные приборы
Корреляция регистрируемых событий во времени — важное
средство повышения избирательности инструментального акти-
вационного анализа*. Возможны два основных режима работы
систем, основанных на этом принципе. В одном из них система
настраивается на регистрацию совпадающих во времени собы-
тий, в то время как несовпадающие отбрасываются (метод сов-
падений). В другом, наоборот, подавляются совпадающие во
времени события, а регистрируются как основной сигнал несов-
падающие (метод антисовпадений). Для работы в режиме сов-
падений или антисовпадений регистрирующая система должна
содержать два детектора. Если ввести третий детектор, то ока-
зывается возможным одновременное сочетание режимов совпа-
дений и антисовпадений в работе одной системы.
Для целей ядерной физики был разработан ряд конструкций,
работающих в режимах совпадений или антисовпадений
* Имеет место также пространственная корреляция, по она не рассмат-
ривается, так как ее роль в аналитических применениях незначительна.
202
[184, 244]. Возможности применения многих из них для целей
активационного анализа были рассмотрены и испытаны. В конеч-
ном итоге в практике активационного анализа более или менее
закрепились четыре основных типа систем: 1) простые системы
совпадений; 2) системы совпадений с суммированием; 3) сис-
темы антисовпадений; 4) универсальные системы совпадений —
Рис. о5. Схема универсального спектрометра сов-
падений и антисовпадений:
1 — основные
3 — источник;
для анализа
детекторы; 2— вспомогательный детектор;
4— ФЭУ; 5 — блок электронных устройств
время-амплитудных соотношений; 6 — мно-
гоканальный анализатор.
антпсовпадений. Поскольку последняя система включает основ-
ные элементы предшествующих, она и будет служить примером
(рис. 55).
Рассматриваемая система включает два основных детектора,
в качестве которых в зависимости от решаемой задачи могут
выступать как полупроводниковые, так и сцинтилляционные
детекторы. Вспомогательный детектор — всегда сцинтилляцион-
ный и может быть изготовлен из монокристалла Nal(Tl) или
сцинтиллирующего пластмассового блока (реже используется
жидкий сцинтиллятор). Сигналы от всех детекторов поступают
к набору электронных устройств, функции которых могут быть
весьма разнообразны: 1) усиление; 2) согласование длительно-
сти задержки; 3) формирование и преобразование; 4) ампли-
тудная дискриминация; 5) отбор совпадающих во времени сиг-
налов; 6) исключение совпадающих во времени сигналов;
7) сложение амплитуд; 8) линейное пропускание с амплитуд-
ной дискриминацией; 9) линейное пропускание в режиме сов-
падений или антисовпадений. Регистрирующее устройство мо-
жет быть простым счетным прибором или многоканальным ана-
лизатором.
Приведенная на рис. 55 универсальная система позволяет
использовать различные сочетания детекторов в разнообразных
режимах работы. Ниже вкратце отмечены только те из них,
которые оказались наиболее полезны для целей активационно-
го анализа.
205
Интегральные системы совпадений. В простые системы сов-
падений входят только два детектора (вспомогательный отсут-
ствует). Как правило, используются совпадения между каскад-
ными у-квантами, хотя возможны другие сочетания, например,
р—у-совпадения. Простые системы совпадений допускают не-
сколько режимов работы; самый простой из них — регистрация
одновременных событий без дополнительной дискриминации.
Тогда импульсы от детекторов после усиления и формирования
поступают в специальное электронное устройство — схему сов-
падений, на выходе которого сигнал появляется только в том
случае, если импульсы разделены небольшим интервалом вре-
мени. Одна из важнейших характеристик схемы совпадений —
разрешающее время, представляющее максимальный интервал
времени между импульсами, в пределах которого они регистр! -
руются как совпадающие (одновременные). Если импульсы от
детекторов следуют друг за другом через интервал времени,
превышающий разрешающее время схемы совпадений, то выход-
ной сигнал отсутствует. Выходные сигналы схемы совпадений
регистрируются счетным устройством.
Поскольку радиоактивный распад—статистический процесс,
то даже при облучении каждого детектора отдельным источни-
ком излучения будут наблюдаться импульсы, разделенные мень-
шим интервалом времени, чем разрешающее время спектромет-
ра совпадений. Эти импульсы будут давать случайные совпаде-
ния.
Скорость счета случайных совпадений связана с активностью
препарата, который не дает каскадных у-квантов, и параметра-
ми системы совпадений следующим соотношением:
псл = 2тсЛ1елевсолсов (8.3)
где nf..i — скорость счета случайных совпадений; тс — разрешаю-
щее время схемы совпадений; А? — суммарная активность пре-
парата; ед и ев— эффективности; о>А и ®в— телесные углы де-
текторов А и В. Таким образом, для уменьшения помех от слу-
чайных совпадений при высоком уровне активности анализи-
руемой пробы необходима система совпадений с минимальным
разрешающим временем.
Однако имеется предел временному разрешению спектромет-
ра совпадений. Сравнительно просто с помощью полупроводни-
ковых и сцинтилляционных детекторов можно получить разре-
шающее время порядка 2-10—8 сек (быстрые совпадения), хотя
иногда находят применение и установки с разрешающим време-
нем около Ю ° сек (медленные совпадения).
Нежелательные совпадения могут дать также у-кванты, пре-
терпевшие в одном из детекторов частичную потерю энергии
(комптоновское рассеяние, процесс образования пар с уходом
квантов аннигиляции) и зарегистрированные другим детектором.
204
Этот процесс может иметь высокую вероятность, так как, стре-
мясь получить наивысшую геометрию, детекторы часто распола-
гают вплотную. Для уменьшения этого эффекта между детекто-
рами помещают свинцовый коллиматор, в отверстие которого
вводится анализируемый препарат [245]. Возможны и другие
решения этой проблемы, но они обычно связаны с уменьшением
общей эффективности спектрометра совпадений.
Если в измеряемом препарате есть каскадный излучатель с
активностью Дк, то скорость счета истинных совпадений будет
определяться уравнением
«совн = ЛкЕЛевсол®в, (8-4)
где Псовп — скорость счета истинных совпадений.
Дифференциальные системы совпадений. Метод интеграль-
ного счета совпадений не свободен от помех со стороны других
каскадных излучателей. Поэтому прибегают к дополнительной
дискриминации регистрируемых импульсов по амплитуде. Для
этого в цепь детекторов вводят одноканальные дифференциаль-
ные анализаторы, которые настраиваются на регистрацию кас-
кадного у-излучения исследуемого радиоизотопа. Выходные сиг-
налы от дифференциальных анализаторов поступают в схему
совпадений. При этом счетное устройство получает сигнал ре-
Г'ис1рации 1о,1ько or iex у-кванюв, импульсы or которых пона-
дают в заранее выбранные энергетические интервалы анализа-
торов и совпадают в пределах разрешающего времени спектро-
метра совпадений.
Такой режим работы позволяет избавиться от помех со сто-
роны несовпадающего излучения и многих каскадных излучате-
лей. Однако если последние имеют более высокую энергию кас-
кадного излучения, то часть непрерывного амплитудного рас-
пределения от них попадает в выбранные энергетические интер-
валы и дает мешающие истинные совпадения. Другой недостаток
подобной системы состоит в том, что она может быть исполь-
зована только для одновременного определения одного компо-
нента в пробе.
Спектрометр совпадений в режиме линейного пропускания.
Этого типа спектрометры представляют более широкие возмож-
ности в отношении числа одновременно определяемых компо-
нентов. В этой конструкции каналы спектрометра совпадений
выполняют несколько различные функции. Импульсы с одного
детектора после усиления и дискриминации (а иногда и без нее)
поступают в схему линейного пропускания. Если одновременно
с ним сюда поступит импульс с другого детектора, то он без
изменения формы и амплитуды пройдет на вход многоканаль-
ного анализатора, где и будет зафиксирован. Несовпадающие
импульсы через схему линейного пропускания не проходят, поэ-
тому в памяти многоканального анализатора получается энер-
205
гетический спектр совпадающего излучения, к которому можно
применить обычные методы обработки.
Спектрометры суммарных совпадений. Если импульсы кас-
кадного излучения от обоих детекторов после согласования по
амплитуде сложить, то получается импульс суммарной амплиту-
ды, величина которой равносильна регистрации излучения с сум-
марной энергией каскадных переходов. Суммарный импульс,
пройдя через одноканальный дифференциальный анализатор,
настроенный на область суммарной энергии каскадов, по-
падает в схему совпадений, работающую в режиме ли-
нейного пропускания. Тогда через это электронное устройство
к многоканальному анализатору пропускаются совпадающие
импульсы от одного из детекторов. В получающемся спектре ос-
таются только совпадающие пики полного поглощения, включая
и пик суммирования, но практически без непрерывного ампли-
тудного распределения. Однако ограниченность таких приборов
состоит в том, что они настроены на регистрацию излучения од-
ного определенного изотопа, тогда как в другом режиме с тем
же оборудованием можно получить более обширную информа-
цию. К тому же спектрометр суммарных совпадений не свобо-
ден от помех со стороны каскадных излучателей с достаточно
высокой энергией каскадных переходов [244].
Спектрометр антисовпадений. Ранее было отмечено, что серь-
езные затруднения при обработке спектров, полученных с одним
детектором, часто вызывает непрерывное амплитудное распре-
деление. Поэтому конструкции спектрометров, позволяющие
уменьшить этот эффект при сохранении энергетического разре-
шения и эффективности регистрации, представляют большой ин-
терес [246]. Наибольшее распространение получили системы с
основным детектором, установленным внутри вспомогательного.
Последний регистрирует у-кванты, которые были рассеяны
основным детектором и, следовательно, потеряли в нем только
часть своей энергии. Импульсы от обоих детекторов поступают
в схему антпсовпадений, которая пропускает па вход многока-
нального анализатора импульсы от основного детектора только
в отсутствие запрещающего импульса от вспомогательного (за-
щитного) детектора. В результате такого отбора исключается
значительная часть непрерывного амплитудного распределения,
а также уменьшается фон спектрометра.
Одна из возможных конструкций спектрометра антисовпаде-
ний приведена на рис. 56 [247]. Основной Ge (Li)-детектор по-
мещают внутрь пластмассового сцинтиллятора размером
62X58 см. Источник вводят внутрь системы и располагают
прямо перед детектором. В режиме антпсовпадений непрерывное
амплитудное распределение 137Cs уменьшается в 10 раз при не-
значительном ослаблении пика полной энергии (менее 2%).
В данной конструкции в режиме антпсовпадений интенсивность
естественного фона спектрометра также снижена в 10 раз.
206
Кроме того, из принципа работы спектрометра антисовпаде-
пий следует, что излучение радиоизотопов с каскадными перехо-
дами тоже будет подавляться. Например, пики полного поглоще-
ния 60 Со (Ev = 1,17 и 1,33 Мэв) уменьшаются в спектре антисов-
падений примерно в 6,5 раза, а их непрерывное распределение
падает в 55 и 45 раз. Чтобы избежать потери информации, в па-
мяти многоканального
анализатора, разделен-
ной на две части (по
2048 каналов), одновре-
менно фиксируется
спектр антисовпадений и
совпадений.
Универсальный спек-
трометр совпадений —
антисовпадений. Если
внутрь вспомогательного
сцинтиллятора ввести вто-
рой детектор, то полу-
чается система, которая
может работать в раз-
личных режимах в зави-
симости от решаемой ана-
литической задачи (см.
рис. 55). Наиболее инте-
ресен режим совпадений
между основными детек-
торами и антисовпадений
по отношению к вспомо-
гательному детектору.
С помощью Ge (Li)-детек-
торов объемом по 70 см3
и пластмассового сцин-
тиллятора размером 72Х
Х72 см получено умень-
шение непрерывного ам-
плитудного распределе-
ния от источника несов-
Рис. 56. Устройство спектрометра антисов-
падений:
/ — ФЭУ; 2 — пластмассовый сцинтиллятор; 3 —
система для ввода источника в спектрометр;
4 — вакуумная трубка с Ое(Ы)-детектором; 5 —
криостат: 6‘— парафин с бором; 7 — свинец.
падающего излучения бо-
лее чем па 4 порядка величины {245].
При этом для каскадного излучателя с низкой энергией у-
квантов выигрыш в избирательности составляет 800. Однако
имеет место значительное уменьшение эффективности регистра-
ции, особенно для жестких у-квантов (в 100 раз для 1 Мэв).
Поэтому эта система дает небольшое увеличение чувствительно-
сти инструментального анализа для узкого круга радиоизото-
пов, которые образуются при активационном анализе на тепло-
вых нейтронах.
207
Аналитические возможности метода совпадений
Распространенность каскадных у-излучателей и универсаль-
ность метода у — у-совпадений. Наилучшие результаты метод
у — у-совпадепий дает при определении каскадных у-излучате-
лей в присутствии интенсивного у-излучения, которое не дает
истинных совпадений. Следовательно, повышение избиратель-
ности достигается ценой некоторой потери универсальности ана-
лиза. Более того, стремясь избежать помех от других каскад-
ных излучателей, прибегают к помощи различных дополнитель-
ных средств, которые еще больше сокращают число одновремен-
но определяемых компонентов. Имеющийся опыт показывает,
что, как правило, установка совпадений специально настраи-
вается на определение только одного компонента пробы, т. е.
метод совпадений рассматривается как вспомогательное сред-
ство при решении частных аналитических задач.
Однако соответствующий анализ схем распада радиоизотопов,
получающихся при различных способах активации, показывает,
что каскадные у-излучатели весьма распространены. Наиболее
детальный анализ возможностей метода совпадений выполнен
применительно к облучению тепловыми нейтронами [248, 249].
Оказывается, что в этом случае образуется 88 каскадных у-излу-
чателей с периодом полураспада более 3 ч, которые являются
продуктами активации 50 элементов. Еще больше каскадных
у-излучателей, если включить в их число позитронно-активные
радиоизотопы, получается при облучении быстрыми нейтронами,
жестким тормозным излучением и заряженными частицами.
Из этих данных вытекает, что сфера приложения метода сов-
падений достаточно широка, но для извлечения наиболее пол-
ной информации спектрометр совпадений должен работать в ре-
жиме линейного пропускания без введения дополнительной
дискриминации в управляющем канале. Обработку полученных
спектров совпадений можно проводить обычными методами. Та-
кой подход может оказаться особенно полезен в случае значи-
тельных помех от несовпадающего излучения (тормозное излу-
чение, радиоизотопы без каскадных переходов).
Избирательность. Как отмечено выше, каскадные у-излуча-
тели распространены достаточно широко. Очень часто в каска-
де испускается несколько у-квантов. Поэтому раздельное опре-
деление каскадных излучателей в пробах сложного состава тре-
бует применения детекторов с высоким разрешением или прив-
лечения дополнительных методов, способствующих повышению
избирательности анализа (чаще всего прибегают к анализу кри-
вых распада и иногда к регистрации задержанных совпадений).
Избирательность метода совпадений по отношению к источ-
никам несовпадающего излучения определяется уровнем интен-
сивности этого излучения и разрешающим временем спектромет-
ра совпадений. Характеризуя избирательность совпадений от-
208
ношением скорости счета помехи к скорости счета сигнала, бу-
дем иметь
G==_2«i_ (8.5)
^совп
Предполагая наиболее простой случай — измерение одного кас-.
кадного излучателя в присутствии радиоизотопов, не имеющих
каскадных переходов, из уравнений (8. 3) и (8. 4) будем иметь
для интегральных измерений (еаь—еа’й евь = едг)
2тсД?
Gcobh = —(8.6)
Итак, с ростом интенсивности мешающего излучения избира-
тельность определения падает по квадратичному закону. Это
обстоятельство диктует необходимость предельного повышения
временного разрешения спектрометра совпадений и ограничи-
вает его применимость к пробам со сравнительно небольшой
суммарной активностью.
Интересно сравнить, каков же получается выигрыш в из-
бирательности метода совпадений ' относительно простого инте-
грального режима измерения. Из аналогичных соображений из-
бирательность счетного режима с детектором А равна
Г. __ - /й -7\
счет - • 1 )
Тогда выигрыш в избирательности с методом совпадений сос-
тавляет
= 2тсДг. (8.8)
Осчет
Для получения необходимой избирательности относительно
каскадных излучателей приходится вводить дополнительную
дискриминацию по энергии излучения. Для этого случая изби-
рательность метода для искомого компонента х следует из со-
отношения
СОВП, X
2тсД2е.ivetjv -у Лкие/1иеВн
(8.9)
где Дкл- — активность искомого каскадного излучателя; Л,:п —
активность интерферирующего каскадного излучателя; е — до-
ля излучения соответствующего источника, которое попадает в
выбранные энергетические интервалы спектрометра совпадений.
Как следует из уравнения (8.9), интерферирующий каскад-
ный радиоизотоп с более жестким у-излучением может давать
помехи через непрерывное амплитудное распределение, прохо-
дящее через выбранные энергетические интервалы спектрометра.
Поэтому достижение максимальной избирательности определе-
ния требует правильного выбора энергии регистрируемого кас-
209
кадпого излучения, если есть такая возможность, и ширины ка-
налов анализаторов. Эти выводы легко распространяются на
более широкий круг интерферирующих каскадных излучателей.
Следует также отметить, что источниками помех могут быть
процессы рассеяния первичного излучения и образования пар с
утечкой аннигиляционных квантов в детекторах спектрометра
совпадений. Конструкция спектрометра должна предусматри-
вать минимальную возможность попадания вторичных квантов
в другой детектор, так как это приводит к появлению совпадаю-
щих импульсов.
Чувствительность. Эффективность регистрации у-излучения
спектрометром совпадений меньше, чем спектрометром с одним
детектором, как это следует из уравнения (8.4). Поэтому приме-
нение метода совпадений, как правило, связано с уменьшением
предельной чувствительности активационного анализа. Допол-
нительными неблагоприятными факторами может оказаться по-
ниженный выход или значительная конверсия одного из компо-
нентов каскадного перехода.
Понижение уровня фоновой активности в режиме совпаде-
ний не всегда компенсирует потерю эффективности, так как влия-
ние первого фактора на чувствительность в грубом приближе-
нии пропорционально корню квадратному, а второго — прямо
пропорционально. Значит, падение эффективности в 10 раз тре-
бует для повышения чувствительности уменьшения фоновой ак-
тивности более чем в 100 раз. Повышение избирательности ме-
тода совпадений обычно связано с необходимостью установле-
ния более узких границ энергетических интервалов, а это непо-
средственно сказывается на эффективности. Замена сцинтилля-
ционных детекторов полупроводниковыми также связана со зна-
чительной потерей эффективности.
Общая активность измеряемой пробы не должна быть боль-
шой, что может потребовать соответствующего изменения режи-
ма активации или уменьшения массы пробы. В конечном счете
это тоже сказывается на концентрационной чувствительности
определения.
В отдельных случаях метод совпадений позволяет повысить
чувствительность инструментального анализа, но выигрыш одна-
ко сравнительно невелик и не превышает одного порядка. По-
скольку основное достоинство метода совпадений состоит в бо-
лее высокой избирательности, свое основное применение он на-
ходит как метод инструментального анализа в области сред-
них и высоких концентраций.
Аналитические возможности спектрометров антисовпадений
Спектрометры антисовпадений сохраняют основные характе-
ристики спектрометра с одним детектором — разрешение и эф-
фективность. Уменьшение фона и непрерывного амплитудного
210
распределения облегчает обработку результатов измерений и
повышает надежность и чувствительность определения по у-из-
лучению, которое не входит в каскадные переходы. Однако для
измерения каскадных излучателей спектрометры антисовпаде-
ний мало пригодны. Как показано Купером [250], спектрометр
антисовпадений дает улучшение чувствительности для 2/3 ра-
диоизотопов, образующихся при активации тепловыми нейтро-
нами.
В спектрометрах антисовпадений с источником, размещае-
мым внутри вспомогательного детектора вблизи основного (см.
рис. 56), нельзя измерять препараты с высоким уровнем актив-
ности. Предел лежит где-то около 1 мккюри и задается уровнем
допустимой загрузки в канале защитного детектора.
§ 3. Аналитическое применение запаздывающих нейтронов
Амиель и Пенсах предложили метод активационного опреде-
ления U и Th, а также некоторых других элементов путем ре-
гистрации запаздывающих нейтронов, испускаемых при распаде
отдельных радиоизотопов [251]. Поскольку число элементов, ко-
торые приводят к таким радиоизотопам, очень мало, а нейтроны
могут быть зарегистрированы без помех со стороны других ви-
дов излучения, анализ оказывается весьма специфичным.
Последовательность ядерных процессов в этом случае выгля-
дит следующим образом. При облучении некоторых элементов
образуются радиоизотопы, известные как предшественники из-
лучателей нейтронов. После |3-распада предшественника дочер-
нее ядро оказывается в столь возбужденном состоянии, что для
него становится энергетически возможным испускание нейтро-
на. Поскольку распад возбужденного состояния происходит прак-
тически мгновенно, период полураспада нейтронного излучения
совпадает с периодом полураспада радиоизотопа — предшест-
венника. Эти радиоизотопы — короткоживущие и имеют период
полураспада в пределах от долей секунды до десятков секунд.
Поэтому длительность облучения мала, а задержка перед из-
мерением должна быть короткой. Регистрацию нейтронов обыч-
но проводят с помощью газонаполненных детекторов с BF3,
которые располагают в блоке парафина. Импульсы от счет-
чиков усиливаются и регистрируются счетным устройством,
управляемым таймером, задающим время задержки между
концом облучения и началом счета. Для понижения уровня
фона парафиновый блок окружают защитой из смеси парафина
с бором и слоем кадмия. Такая установка может обеспечить
эффективность регистрации запаздывающих нейтронов в преде-
лах 5—10%.
При делении U и Th возникает несколько предшественников
запаздывающих нейтронов, период полураспада которых состав-
ляет от 0,2 до 60 сек. Поэтому большое значение для точности
211
определения этих элементов имеет правильный выбор продол-
жительности облучения, выдержки и измерения. По данным ра-
боты [251], оптимален следующий режим: /об.-, = 60 сек, гр;1(.п=
= 20 сек и 0,3м = 60 сек.
Определение урана. Тепловые нейтроны вызывают только
деление 235 U и поэтому служат специфичным средством для оп-
ределения урана. При Ф=1013 нейтрон/(см2-сек) и указанном
выше режиме 1 мкг U дает 1,2-104 нейтронов, что обеспечивает
достаточно высокую чувствительность, которая, например, при
анализе хондритов оказалась равной 3-10~8 г [252]. Точность ана-
лиза хорошая, и для пробы с содержанием LJ~10 мкг относи-
тельная погрешность составляет 1%. При наличии в потоке
быстрых нейтронов возможны помехи со стороны Th и больших
количеств О.
Если, с другой стороны, общее количество U в пробе извест-
но, то можно рассчитать относительное содержание изотопов
238 U и 235 U. В смешанном потоке нейтронов требуется двукрат-
ное облучение пробы — с кадмиевым фильтром и без него. От-
носительная погрешность при определении изотопного состава
менее 0,5%.
Определение тория. Из-за небольшой величины сечения де-
ления 232Th быстрыми нейтронами чувствительность много хуже,
чем для U. Так, 1 мкг Th после облучения в потоке с Ф =
= 10’3 нейтрон/(см2 сек) испускает только 412 нейтронов. Основ-
ная трудность при определении Th — обычное присутствие U в
ториевых рудах. Поэтому требуется облучение пробы с кад-
миевым фильтром и без него с введением поправки на U.
Определение кислорода, лития, азота и фтора. Некоторые
реакции на легких ядрах приводят к образованию радиоизотопа
17N (7'1/2 = 4,14 сек), который тоже принадлежит к предшествен-
никам запаздывающих нейтронов. На быстрых нейтронах про-
текают реакции 17О(щ p)17N и 18О(«, d)17N. Однако небольшая
распространенность тяжелых изотопов кислорода и малые ве-
личины сечений обусловливают низкий выход 17N. Так, облуче-
ние естественного кислорода до насыщения в потоке нейтронов
деления с Ф=1013 нейтрон/(см2-сек) дает интегральный выход
нейтронов, равный 80 нейтрон/мг.
Последовательные реакции 6Li(п, а)3Н и 18O(L a)17N дают
более высокий выход изотопа l7N. Облучение раствора 1 мг
6Li в обычной воде потоком тепловых нейтронов с
Ф=1013 нейтрон/(см2-сек) уже дает 4-Ю4 нейтронов. Исполь-
зуя воду, обогащенную 18О, можно повысить выход нейтронов.
Изотоп 17N образуется также по реакции p)17N, но ее се-
чение несколько ниже сечения реакции 18О(/, a)17N. Последо-
вательные реакции при соответствующем подборе условий мож-
но использовать для определения Li или О, или же N.
Аналитическое применение находят и реакции с образова-
нием изотопа 17N, протекающие под действием тормозного из-
212
-лучения: 18О(у, p),7N и 19F(y, 2 p)17N. С помощью мощного
ускорения электронов (£торм = 60 Мэв, / = 50 мка) чувстви-
тельность доходит для F до 0,09-10 *’ г и для О до 0,4- 10~б г
[253]. Высокая проникающая способность жесткого тормозного
излучения и нейтронов позволяет определять О в толстых слит-
ках металлов [254], но при условии, что содержание F и деля-
щихся элементов в них мало.
§ 4. Дейтерий и бериллий в качестве пороговых детекторов
Амиель и др. [255, 256] воспользовались фотоядерной реак-
цией 2Н(у, /г)1!-! для дискриминации у-излучателей. Поскольку
порог этой реакции равен 2,23 Мэв, то дейтериевая мишень со
счетчиком нейтронов реагирует только на кванты с энергией
выше этого предела. Конструкция измерительной установки ана-
логична рассмотренной в предыдущем параграфе. Отличие ее
состоит только в том, что в середину парафинового блока уста-
навливают полиэтиленовый сосуд с 1 л тяжелой воды (обогаще-
ние до 98%) •
Измеряемый препарат вводят в центр сосуда, а образую-
щиеся нейтроны регистрируют борными счетчиками. Подобная
система нечувствительна к высокому уровню у-излучения, энер-
гия которого ниже пороговой. Однако поскольку она анало-
гична интегральному дискриминатору, то для идентификации
радиоизотопов необходим дополнительный критерий, в качестве
которого пригоден только анализ кривых распада. При этом
процесс идентификации сравнительно прост в связи с неболь-
шим числом радиоизотопов со столь жестким у-излучением.
Некоторые помехи анализу могут возникнуть в присутствии
сильных поглотителей (В, Cd и др.) или, наоборот, источников
нейтронов. В качестве последних могут выступать пред-
шественники запаздывающих нейтронов и реакция (у, п) на Be
(порог 1,67 Мэе). Все предшественники — короткоживущие,
поэтому небольшая задержка (1—3 мин) перед измерением до-
статочна для их полного распада. Величину помехи от фотоядер-
ной реакции на Be можно оценить экспериментально, если за-
менить тяжелую воду обычной.
В качестве примера приложения счета фотонейтронов можно
привести методики для Na [255], S и Са [256]. В ходе анализа
пробу отбирают в полиэтиленовую ампулу и облучают в оеак-
торе [Ф = 5—7-Ю12 нейтрон/(см2сек)]. Длительность облуче-
ния составляет для S 10 мин, для Са — 17 мин и 1 ч для Na.
Измерения активности выполняют с фотонейтронным счетчи-
ком. Фон счетчика мал (около 10 ими!мин), низка также
эффективность, которая составляет 1 -104 распад'отсчет для
37S [11 имп!(мин-мг)] и 2-Ю4 распад'отсчет для 49Са
[440 имп) (мин- мг)]. В скобках приведена удельная скорость сче-
та. При этом чувствительность равна 0,3 мкг (Na), 0,8 мг (Са)
213
и 0,19 мг (S). Возможны помехи от других жестких излучате-
лей (56Мп, 140La, 78Ga), которые, как правило, можно обнару-
жить из кривых распада.
Относительная погрешность лежит в пределах 1—2%. Испы-
тания правильности, проведенные на пробах с известным содер-
жанием определяемых элементов, показали отсутствие систе-
матической погрешности.
Если в счетчике 2Н заменить на Be, то это будет равносильно
снижению уровня дискриминации до 1,67 Мэв. Это приведет
к расширению круга определяемых элементов, но при одновре-
менном снижении избирательности.
§ 5. Применение счетчика Черенкова
Под действием очень быстрых частиц в прозрачных твердых,
жидких или газообразных веществах возникает излучение в ви-
димой области спектра, которое может быть зарегистрировано
ФЭУ. Поэтому устройство счетчика Черенкова схоже с устрой-
ством сцинтилляционного счетчика с той лишь разницей, что
в качестве вещества, в котором вызывается свечение, исполь-
зуется обычная дистиллированная вода или плексиглас.
Счетчик Черенкова относится к пороговым детекторам, кото-
рые чувствительны к достаточно жесткому излучению. Это свой-
ство счетчика позволяет использовать его для измерения радио-
изотопов с очень жестким Р~-излучением в присутствии мягкого
излучения высокой интенсивности. Поскольку число таких ра-
диоизотопов мало, метод достаточно избирателен, причем для
повышения избирательности можно применить фильтры и сня-
тие кривой распада.
Первоначально Лукенс и Лаш [257] предложили проводить
измерения с помощью счетчика Черенкова при определении кис-
лорода по реакции ieO(n, p)16N. Несмотря на уникальность схе-
мы распада изотопа 16N, образование высокого уровня мешаю-
щей активности при облучении в реакторе препятствует приме-
нению других методов физической дискриминации. Так, при об-
лучении в реакторе Triga, где плотность потока нейтронов с
энергией выше 9,6 Мэв равна 109 нейтрон/(см2-сек) [общая
плотность потока 3,5-1012 нейтрон (см2-сек)], некоторые пробы
(масса 1 г) через 3 мин после облучения имели интенсивность
излучения примерно 100 мр/ч. В присутствии излучения такой
интенсивности сцинтилляционный детектор (при высокой геомет-
рии) нс может нормально работать.
Все радиоизотопы с очень высокой энергией р-'-пзлучеяия
весьма короткоживущие, поэтому только с появлением быстрых
транспортирующих систем оказалось возможным несколько рас-
ширить сферу приложения счетчика Черенкова в области акти-
вационного анализа. Лукенс [258] подробно исследовал возмож-
ности этого метода для определения Be, Li, В, О, Е и Mg при
214
облучении потоком нейтронов реактора, работающего в им-
пульсном или стационарном режиме (табл. 14).
Таблица 14
Чувствительность определения Be, Li, В, О, F и Mg при использовании
счетчика Черенкова
Эле- мент Радио- изотоп T\ сек £&. Мэв Интенсив- ность перехода, 0/ /0 Эффек- тивность, 0 ' /0 Удельная скорость счета*, имп/(сек-г) Чувстви- тель- ность* *, мкг
Режим А Режим Б
Ве 6Не 0,8 3,51 100 2,9-10-4 * * 6,6-10« 15
Li 8Li 0,84 13 100 25 1,3-1011 1,9-109 8- 10“4
В 8Li 0,84 13 100 25 8,8-107 В 1,3-10» 1,1
О 16N 7,14 10,4 26 12 2,7-10^ 7,0-104 370
F 16N 7,14 10,4 26 12 1,2-10» 3,1-Ю7 0,83
Mg 26Na 1,04 6,7 100 — 6,4-105 — 160
Режим А: импульсный интегральный поток тепловых нейтронов 1,8 -К)14 неитрон/с.и2,
нейтронов деления — 3,5 • 1014 нейтрон/см2.
Режим Б: стационарный, плотность потока тепловых нейтронов 2,5 • 1О': ней-
трон /{См- • сек), нейтронов деления — 5,9 • 10'- нейтрон/(см2 • сек).
* В расчете на конец облучения.
** Режим работы реактора импульсный, минимальная скорость счета принята рав-
ной 100 ими/сек.
В ixd4cCi.Dc ди 1 СК 1 ир2. р -ИЗдуЧСНйл с».'1уЖИ1 uivCni (72X72 AtAt)
в форме цилиндра с углублением, в которое вводится облучен-
ная проба. Стенки углубления покрыты фильтром из алюминия,
меди и полиэтилена, который поглощает все [5 -частицы с энер-
гией менее 3 Мэв. Импульсы от ФЭУ, который сочленен с люси-
том, поступают в многоканальный анализатор, работающий в
режиме многоканального счетчика. При измерениях переклю-
чение каналов происходит через 0,4 сек, в результате в памяти
многоканального анализатора фиксируется кривая распада ре-
гистрируемой активности, анализ которой позволяет выявить от-
дельные компоненты.
В использованной системе время транспортировки равно
0,6 сек. Даже с учетом этой задержки видно, что рассматривае-
мый метод очень благоприятен для определения лития, особен-
но при работе реактора в импульсном режиме. Однако для дру-
гих элементов получающаяся чувствительность много хуже. Отт
носителыюе стандартное отклонение результатов при импульс-
ном режиме реактора составляет 2% •
§ 6. Метод треков деления
Прайс с сотр. [259] предложили метод определения деля-
щихся ядер по трекам, которые образуют продукты деления при
движении в веществе. Первоначально этот метод разрабаты-
вался применительно к проблеме определения возраста по трекам
спонтанного деления урана, но впоследствии получил широкое
215
распространение как исключительно чувствительный метод ко-
личественного определения ядер, способных к вынужденному
делению.
Было показано, что продукты деления при движении в твер-
дых телах оставляют за собой узкую непрерывную область ра-
диационного разрушения (трек), которую можно сделать види-
мой путем избирательного химического травления. В качестве
травящих агентов в зависимости от исследуемого объекта ис-
пользуют неорганические кислоты (HF, HNO3, НС1 и др), ще-
лочи (NaOH и КОН) и некоторые другие реагенты [260—262].
Длительность травления находится в пределах от нескольких
секунд до нескольких часов.
В процессе травления реагент проникает в разрушенную об-
ласть, растворяет вещество в ней и таким образом увеличи-
вает размеры начального трека до такой степени, что он ста-
новится видимым в оптический микроскоп. До травления треки
можно наблюдать только с помощью электронного микроскопа,
так как их размеры равны примерно 10 нм.
Треки деления после протравливания имеют ряд характер-
ных признаков, которые позволяют отличить их от дефектов и
дислокаций в твердом теле. Прежде всего они имеют специфи-
ческую форму, которая зависит от исследуемого материала, хи-
мического реагента и времени травления. Как дополнительные
факторы при идентификации могут выступать данные по глуби-
не треков, не превышающей 20 мкм, закономерности распреде-
ления подозрительных образований и высокотемпературному
отжигу.
Определение формы треков после травления, подбор химиче-
ского реагента, его концентрации и длительности травления
обычно проводят на детекторах, которые были подвергнуты об-
лучению продуктами деления 252 Cf. Можно прибегнуть также
к делению урана, если облучить нейтронами детектор в контак-
те со слоем урана.
Для количественных определений этим методом проводят
облучение пробы излучением (тепловые или быстрые нейтроны,
жесткое тормозное излучение), которое вызывает деление ядер
тяжелых элементов (U, Th и др.). Детектором индуцированных
продуктов деления может служить как вещество пробы, так и
подходящий материал, прикладываемый перед облучением к ее
ровной поверхности. Второй способ практически оказался более
удобен и поэтому получил более широкое распространение, де-
текторами при этом служат слюда, стекла и некоторые органи-
ческие полимерные материалы (лавсан, майлар и др.).
После травления подсчитывают число треков, приходящихся
на единицу поверхности детектора. Тогда для облучения тепло-
выми нейтронами связь между концентрацией способных к деле-
нию ядер и плотностью треков имеет вид:
Ртр ~ ^делФинт^дел^тр^эФФ» (8.10)
216
где ртр — плотность треков, трек!см2-, — концентрация деля-
щихся ядер, атом/см3; ФШ1Т — интегральный поток нейтронов,
нейтронам2-, Одел — сечение деления, см2\ етр — эффективность
регистрации треков; /?зфф— эффективный пробег продуктов де-
ления. см~'.
Величины /?эфф и етр для данного типа детектора определяют-
ся в отдельных опытах [261]. С учетом постоянных величин кон-
центрацию определяемых ядер рассчитывают из уравнения
ЛГде.л = ^-^-, (8.11)
ФИнт
где k — постоянная величина.
Интегральный поток нейтронов можно определить с по-
мощью подходящего монитора. При этом возможны два подхо-
да. Один из них состоит в облучении известного количества ура-
на или золота с последующей оценкой Фпнт по радиоактивным
продуктам. В другом случае мишень с тонким слоем урана [262]
или стекло с известным содержанием урана [263] облучается
в контакте с детектором треков деления. Можно воспользоваться
и методом внутреннего эталона, тогда известное количество
урана добавляется либо прямо к порошкообразной пробе, либо
к аликвоте раствора, полученного в результате химической обра-
ботки пробы.
Основное пппмепенне метод тоеков деления находит в иссто-
дозаниях по геохимии и космохимии урана [260, 264]. Предель-
ная чувствительность обнаружения урана этим методом весьма
высока и достигает И!1 %.
Глава
9
РАДИОХИМИЧЕСКИЕ МЕТОДЫ
АНАЛИЗА
§ 1. Особенности и методика радиохимических разделений
Радиохимический вариант имеет важное значение для ак-
тивационного анализа в целом. Прежде всего радиохимические
методы незаменимы в тех случаях, когда чисто инструменталь-
ный подход не дает удовлетворительного решения. Число таких
случаев достаточно велико, и они крайне важны. Здесь, в пер-
вую очередь, можно отметить анализ проб с сильноактивирую-
щейся основой. Радиохимические методы широко используются,
когда требуются надежные определения при концентрациях,
близких к предельной чувствительности активационного метода.
Важную роль радиохимические методы играют при определе-
ниях, включающих большое число элементов. Радиохимическое
выделение оказывается необходимым и при определении эле-
ментов, для которых избирательность инструментальных мето-
дов низка. /Можно еще упомянуть применение химических мето-
дов в тех случаях, когда возникает необходимость предвари-
тельной подготовки пробы перед облучением (концентрирование,
удаление нежелательных компонентов и т. д.). Наконец, к по-
мощи радиохимического метода часто еще прибегают, когда
отсутствует необходимое ядернофизическое оборудование, кото-
рое достаточно дорого и сложно в эксплуатации.
Для радиохимических разделений используют обычные ме-
тоды аналитической химии. Однако выполнение радиохимиче-
ских разделений имеет некоторые особенности, которые выте-
кают главным образом из того факта, что носителем каче-
ственной и количественной информации служит активность ра-
диоизотопов. Поэтому добавление в разумных пределах ста-
бильных изотопов (а иногда и радиоактивных, но с другой схе-
мой распада) не влияет на результат конечного определения.
Следовательно, путем введения носителя можно проводить раз-
деление в оптимальных концентрационных условиях, а по вы-
ходу носителя (индикатора) определять химический выход.
Обычно раствор носителей определяемых элементов добав-
ляют к анализируемой пробе перед ее разложением, которое
выполняют каким-либо подходящим способом [265,266]. В боль-
шинстве случаев для этого находят применение обработка раст-
ворами неорганических кислот или их смесью и сплавление с
218
солями щелочных элементов. На выбор методики разложения
могут оказать влияние и особенности решаемой аналитической
задачи (необходимость удаления основы на первой стадии, оп-
ределение летучих элементов и т. д.).
Для элементов, имеющих более чем одно стабильное валент-
ное состояние, необходимо создать условия, которые обеспечи-
вают полный химический обмен между введенным носителем и
аналитическим радиоизотопом. В противном случае возможна
значительная потеря активности и химический выход носителя
не будет соответствовать выходу радиоизотопа. В конечном сче-
те это приведет к искажению результатов анализа.
Для обеспечения полного обмена между носителем и радио-
изотопом необходимо соблюдать несколько общих правил [116]:
1. Носители вводить до разложения пробы.
2. Носители должны быть в виде растворимого соединения,
которое не осаждается реагентами, используемыми для разло-
жения пробы.
3. Когда возможно, проводить цикл окислительно-восстано-
вительных реакций.
4. Комплексообразующие реагенты, которые образуют устой-
чивые ковалентные связи с определяемыми элементами, должны
отсутствовать в растворах, используемых на этой начальной
стадии.
5. Исследовать возможные потери на этой стадии радиоизо-
топа путем улетучивания, образования коллоидов или любым
другим путем.
Количества вводимых носителей зависят в основном от ме-
тодов разделения, которые используются в последующих ста-
диях. Если схема химического разделения предусматривает при-
менение метода осаждения, то количество носителя лежит в
пределах 5—30 мг.
При многоэлементных схемах анализа часто нежелательно
вводить носители всех элементов в полном количестве при
разложении пробы, так как это может вызвать некоторые за-
труднения при выполнении отдельных начальных операций.
В таких случаях сначала можно ввести носитель в количестве
нескольких микрограммов с добавлением полного количества
после завершения предварительных стадий.
Некоторые методы химического анализа позволяют прово-
дить разделение и радиохимическую очистку радиоизотопов в
безносителыюм состоянии. К ним относятся экстракция, ионооб-
менная и экстракционная хроматография и некоторые другие
методы. Разделение в безносительном состоянии имеет опреде-
ленные достоинства, основные из которых — небольшие объемы
растворов, простота подготовки препаратов к измерению, воз-
можность приготовления тонких источников для спектроскопии
слабо проникающих излучений и т. д. В результате уменьшают-
ся расход реактивов и затраты времени на проведение анализа.
219
Однако полное отсутствие носителей имеет и нежелатель-
ную сторону, так как слишком велика вероятность потери ра-
диоизотопов из-за адсорбции, образования коллоидов,
улетучивания и т. д. По-видимому, введение на микрограммовом
(1 —100 мкг) уровне — оптимальный вариант.
Процедура переведения пробы в раствор очень важна, так
как на этой стадии могут происходить потери радиоизотопов.
При определении Sb, As, Hg, Se и некоторых других элементов
возможны потери вследствие улетучивания. Так, при темпера-
туре 200° С за 15 мин из раствора НС1 и H2SO4 теряется по
крайней мере 30% следующих элементов: Sb, As, В, Ge, Hg и
Se. При тех же температуре и времени из раствора НС1 и НС1О4
теряется более 99% Сг, Os, Re, Ru, Sn.
Вообще практика показывает, что соляную кислоту следует
избегать, если растворение происходит в системе без обратного
холодильника, так как возможны потери многих элементов. Для
растворения проб лучше использовать азотную и хлорную кис-
лоты или их смесь. Например, из кипящей смеси HNO3+HCIO4
не наблюдается никаких потерь Sb, As, Cd, Сг, Со, Си, Fe,.
Pb, Mo, Se, Ag, Sr и Zn; отмечена только частичная потеря Hg.
Хорошие результаты дает раствор HNO3 + HCIO4 + H2SO4. но
его применение противопоказано, когда присутствуют большие
количества Cd и РЬ. Из горячей H2SO4 теряются некоторые ме-
таллоиды (особенно фосфор), а из НСЮ4 при 200°С наблю-
дается потеря некоторых летучих окислов.
Часто для перевода пробы в раствор применяют сплавление
с различными реагентами. Хотя проблема потерь в этом случае
подробно еще не исследована, лучшими реагентами для сплав-
ления признаны перекись натрия и смесь NaOH + KNO3 (25:1).
Перевод носителя и радиоактивного изотопа в одинаковое
химическое состояние можно осуществить двумя методами:
1) ввести в раствор носитель во всех возможных для данного
элемента формах с последующим переводом всех их в одно
валентное состояние какой-нибудь окислительно-восстановитель-
ной реакцией;
2) ввести в раствор носитель в одном валентном состоянии
с последующим проведением цикла окислительно-восстанови-
тельных реакций с тем, чтобы носитель прошел все возможные
валентные формы и окончательно оказался в одной из них.
Из практических соображений часто используют упрощенный
вариант последней процедуры, применяя обработку каким-ни-
будь сильным окислителем, который переводит все определяемые
элементы в их высшее окисленное состояние. В большинстве
случаев для этого подходит нагревание с концентрированной
НС1О4; имеется мало элементов, которые не окисляются ею
до их высшей валентности. Кипячение с НС1О4 имеет еще то
преимущество, что при этом разрушаются многие комплексные
ионы, а это благоприятствует полноте обмена. Однако, применяя
220
НС104, следует помнить, что для одних элементов это может
привести к потерям, а для других—затруднить обмен между
различными валентными формами [267].
Из раствора, получающегося после разложения пробы, выде-
ляют определяемые элементы, используя различные методы,
имеющиеся в арсенале аналитической химии. Все химические
методы разделения основываются на распределении разделяе-
мых элементов между двумя гетерогенными фазами: жидкость
— жидкость, жидкость — твердая фаза, жидкость — газ, твердая
фаза — газ. Операция разделения может осуществляться в
статических и динамических условиях. Последние обычно поз-
воляют получать более чистые разделения в одной операции.
Наиболее важные характеристики основных методов разделе-
ния и особенности их использования в активационном анализе
рассматриваются ниже.
Случаи применения радиохимических методов весьма разно-
образны, и их условно можно классифицировать следующим об-
разом.
1. Удаление мешающего компонента.
2. Выделение определяемого компонента.
3. Выделение группы элементов.
4. Выделение нескольких групп элементов.
5. Полное радиохимическое разделение большого числа
элементов.
Случаи 1, 3 и 4 предполагают, что конечное определение вы-
полняется с помощью какого-либо инструментального метода.
В случаях 2 и 5 имеется принципиальная возможность получе-
ния препарата в форме, пригодной для измерения наиболее чув-
ствительными интегральными методами. Однако тогда необхо-
димо создать такие условия, чтобы получить полное выделение
каждого определяемого элемента в отдельную фракцию с высо-
кой степенью радиохимической чистоты. Эффективность выделе-
ния характеризуется химическим выходом, который для радио-
изотопа равен отношению
<9Л)
Ло
где ЛЛК и Л.г0— активности радиоизотопа в конечной фракции и
исходной смеси. Применительно к выделенной фракции анало-
гичные выражения могут быть написаны для любого радиоизо-
топа, входящего в исходную смесь. Тогда избирательность дан-
ной аналитической процедуры к определяемому элементу в при-
сутствии мешающего радиоизотопа равна отношению
GX11M = ^, (9.2)
Дм
где г]м — доля мешающего радиоизотопа, попадающая во фрак-
цию определяемого элемента.
221.
Поскольку для аналитических определений пригодны только
такие методы, которые обеспечивают т|.( ~ 1, то
Gx„M = ёоч = —. (9.3)
Пи
В этом случае избирательность метода совпадает с обычно ис-
пользуемым в радиохимии фактором очистки. Таким образом,
последний равен отношению исходной активности мешающего
элемента к его активности, попадающей в конечную фракцию
определяемого элемента.
Когда на конечной стадии используется интегральный метод
измерения, радиохимическое выделение должно обеспечивать
выполнение условия
(9.4)
£оч
где Лм0 — активность мешающего радиоизотопа в исходной
смеси. Поскольку Ах и Дм0 могут колебаться от одного опреде-
ления к другому, для подстановки в уравнение (9. 4) берут ми-
нимальное значение Лх, а для Лм0—максимальное. Если в ис-
ходной смеси присутствует несколько радиоактивных элементов,
то принятая методика выделения должна обеспечивать выпол-
нение условия (9. 4) для каждого из них.
Когда фактор очистки, достигаемый при одностадийном раз-
делении относительно какого-либо элемента, недостаточен, при-
меняют несколько последовательных стадии. Тогда общий фак-
тор очистки будет равен
£общ =---------------• (9.5)
Основное преимущество радиохимического метода перед ин-
струментальным состоит в высокой избирательности, которая
при многостадийных разделениях по существу ничем не ограни-
чена. Это важное обстоятельство, поскольку часто возникает си-
туация, когда необходимо раздельно определять компоненты,
различающиеся по уровню активности на много порядков.
Примером может служить определение микрокомпонентов в
сильноактивирующейся матрице. Предельно обнаруживаемый
уровень активности составляет примерно 10~12 кюри [268], а ак-
тивность матрицы может достигать 10 кюри. Следовательно,
для получения надежных результатов при содержаниях, близких
к пределу обнаружения, нужен метод с крайне высокой избира-
тельностью, способный обнаруживать слабый сигнал на фоне
столь сильной помехи. Для радиохимического метода достиже-
ние фактора очистки порядка 1013 и выше вполне возможно,
хотя и потребуется довольно сложная методика разделения и
очистки. Однако это — экстремальный пример, и в большинстве
случаев достаточны методы с фактором очистки 105—108.
Для сравнения можно отметить, что для инструментального
метода допустимый интервал регистрируемых активностей пе-
рекрывает примерно пять порядков (104 имп!сек составляет
верхний предел допустимой загрузки для многоканального ана-
лизатора и 0,1 имп/сек— минимально регистрируемый уровень
скорости счета). Он может быть несколько расширен при благо-
приятном сочетании периодов полураспада (за счет выдержи-
вания) и путем применения некоторых методических приемов
(схемы совпадений и антисовпадений, облучение в фильтрах
и т. д.). И все же возможности этого метода в плане повышения
избирательности более ограничены, чем у радиохимического
метода.
В результате радиохимической обработки получаются раз-
личные фракции определяемых элементов, которые должны
иметь достаточную радиохимическую чистоту, чтобы гаранти-
ровать точность результатов. При этом конечная форма
фракции должна соответствовать используемому методу изме-
рения.
Поскольку при разделении и радиохимической очистке эле-
ментов для ускорения анализа и повышения избирательности
часто не добиваются полного (100%-ного (выхода, необходимо
определить поправку на химический выход. Возможны три ос-
новных способа определения химического выхода. Прежде все-
го— это различные физико-химические методы. Часто исполь-
зуется гравиметрический метод, когда элемент переводится в со-
ответствующую весовую форму и по количеству его в конечном
осадке, зная количество введенного носителя, рассчитывают хи-
мический выход. Наиболее простой способ состоит в нанесении
определяемого элемента в весовой форме на измерительную ми-
шень и определении массы осадка. При гравиметрическом методе
определения химического выхода количество носителя должно
быть более 5 мг. При использовании более чувствительных мето-
дов определения, таких, как колориметрия, микротитрование и
др., количество носителя может быть менее 1 мг. При этом хими-
ческий выход может определяться из отдельной аликвоты раст-
вора.
Другой способ — введение в анализируемый раствор перед
разделением известного количества радиоактивного индикатора
определяемого элемента со схемой распада, отличающейся ог
схемы распада аналитического радиоизотопа. После радиохими-
ческого выделения, используя различие схем распада, по выходу
радиоактивного идикатора определяют химический выход эле-
мента. Для этого, например, можно использовать различия в ти-
пе распада, энергии излучения и периоде полураспада.
Последний метод состоит в повторном облучении выделен-
ного и измеренного препаратов [269]. Так как количество эле-
мента в препарате после добавления известного количества но-
сителя велико, облучение может быть коротким, а отсутствие
223'
мешающих элементов позволяет непосредственно измерять ак-
тивность. Поэтому, используя короткое облучение, транспор-
тировку препарата и эталона с помощью пневмопочты и быстрое
измерение, химический выход можно определить всего за не-
сколько минут.
§ 2. Метод осаждения и соосаждения
В методе осаждения в исходный раствор вводят специфиче-
ский реагент, который образует с нужным элементом трудно-
растворимое соединение, выделяющееся в виде самостоятельной
твердой фазы. Получившийся осадок отделяют от раствора цен-
трифугированием или фильтрованием. Метод осаждения — ос-
нова классических методов разделения. Он достаточно хорошо
изучен и обстоятельно изложен в обширной литературе
[266, 270]. Довольно широко метод осаждения применяют в ра-
диохимической практике. Большое достоинство метода — приме-
нимость его к подавляющему числу элементов периодической
системы, поскольку многие из них в определенных условиях об-
разуют труднорастворимые соединения. Исключение составляют
некоторые газообразные элементы.
При проведении активационного анализа концентрации опре-
деляемых элементов в растворе после разложения пробы обыч-
но оказываются слишком низкими, чтобы могло быть достигну-
то произведение растворимости, поэтому добавление изотопных
носителей обязательно. При использовании осаждения как ос-
новного метода разделения активированных элементов проведе-
ние анализа обычно полностью совпадает с общим ходом ана-
лиза, изложенным ранее (см. § 4 гл. 3).
Недостаток метода состоит в трудности разделения некото-
рых близких по свойствам элементов (редкоземельных, щелоч-
ных и др.). Однако более серьезный недостаток метода осажде-
ния при использовании его для решения радиохимических за-
дач— соосаждение вместе с определяемым элементом посторон-
них радиоактивных элементов. В весовом отношении количество
соосадившегося элемента обычно ничтожно, однако он может
при этом обладать достаточной активностью, чтобы исказить ре-
зультаты активационного определения.
Соосаждение может быть обусловлено объемным распределе-
нием микрокомпонента между осадком и раствором и повер-
хностной адсорбцией. В пределах каждого из основных типов
имеется ряд различных процессов, приводящих к соосаждению
[271]. Многообразие процессов приводит к тому, что соосажде-
ние— обычное явление, которое наблюдается при осаждении
какого-либо соединения из раствора, содержащего микрокомпо-
иепты.
Более часто соосаждение обусловлено адсорбцией радиоак-
тивных элементов на поверхности осадков и окклюзией. В этом
.224
случае количество соосадившейся микропримеси зависит от ве-
личины и природы поверхности осадка. Особенно сильно адсор-
бируют микропримеси аморфные осадки; адсорбционная спо-
собность кристаллических осадков много меньше. Поэтому пе-
рекристаллизация осадков часто способствует удалению адсор-
бированных или захваченных примесей. Уменьшить адсорбцию
можно путем проведения осаждения из относительно разбав-
ленного раствора или подбора pH среды. Осаждение лучше
проводить из горячего раствора.
Адсорбция радиоактивного элемента на осадках становится
ничтожной, если его концентрация в растворе повышена вве-
дением носителя, который в этом случае является удерживаю-
щим. Введение удерживающего носителя желательно не только
для увеличения радиохимической чистоты осадка, но и для
уменьшения потерь активности элементов, выделение которых
предусмотрено последующими стадиями анализа.
Из-за соосаждения посторонних радиоактивных элементов
с выделяемым осадком необходим целый ряд операций радио-
химической очистки. Эти операции могут включать повторное
осаждение в присутствии удерживающих носителей, дистилля-
цию, экстракцию и т. д. Необходимость проведения операций
радиохимической очистки сильно увеличивает трудоемкость и
длительность анализа. Для повышения избирательности выделе-
ния обычно применяют наиболее специфичные реакции, хотя и
не всегда количественные. Поэтому при использовании метода
осаждения в качестве основного метода разделения химический
выход обычно составляет 30—70%, т. е. он недостаточно высок,
что приводит к значительным потерям активности и соответст-
вующему понижению чувствительности анализа. Кроме того,
требуются дополнительные затраты труда и времени для пере-
вода элемента в подходящую форму при определении химическо-
го выхода.
Если в качестве конечного метода измерения используется
интегральный |3-счет, то необходима строгая стандартизация ус-
ловий регистрации. Это также требует некоторых дополнитель-
ны?; операций по подготовке препарата и эталона к измерению.
К этому надо добавить, что самопоглошение [3-излучения в ве-
ществе препарата ведет к уменьшению измеряемой активности,
а выделение радиоизотопов с большой массой носителя пре-
пятствует применению 4л бета-счетчиков, являющихся наиболее
эффективными детекторами низких активностей.
Однако достижение высокой радиохимической чистоты не яв-
ляется обязательным, если конечное определение проводится с
помощью спектрометрических методов. Чаще всего для этого
используют сцинтилляционную у-спектромстрию. При этом так-
же отсутствуют жесткие требования к конечному препарату.
В результате разделение и подготовительные операции требуют
меньше времени и анализ становится более быстрым.
8 Р. А. Кузнецов
225
Явление соосаждения не только имеет отрицательный эф-
фект, но и находит полезное практическое применение. Можно
отметить три основных направления в использовании соосажде-
ния: концентрирование микроэлементов, удаление мешающих
радиоизотопов, предварительное неспецифическое выделение оп-
ределяемого радиоизотопа из исходного раствора.
Соосадителями обычно служат гидроокиси многовалентных
металлов (Fe3+, А13+ и др.), которые образуют аморфные осад-
ки. Возможно применение некоторых других малорастворимых
неорганических соединений (в частности, сульфидов), а также
органических соосадителей.
На степень соосаждения радиоэлементов оказывают влияние
многие факторы: химическая природа соосадителя и исследуе-
мого элемента, состав и pH раствора, присутствие комплексо-
образующих реагентов и т. д. Механизм соосаждения достаточно
сложен и поэтому этот процесс не всегда поддается эффективно-
му контролю.
§ 3. Ионообменная хроматография
Ионный обмен и типы ионитов
Разделения методом ионообменной хроматографии приобре-
ли большое значение в аналитической химии и радиохимии
[272—274]. С помощью этого метода удалось решить много важ-
ных и интересных аналитических проблем, таких, как разделение
щелочных и редкоземельных элементов, что трудно осуществить
другими методами.
Особенно четко проявляются достоинства ионообменной хро-
матографии при разделении элементов, находящихся в раство-
рах в микроконцентрациях или в безносительном состоянии.
Поэтому весьма разнообразное применение находит ионооб-
менная хроматография для решения различных радиохимиче-
ских проблем, таких, как выделение радиоактивных элементов
из облученных мишеней без носителя, производство радиоак-
тивных индикаторов и т. д.
В основе метода лежит способность ряда нерастворимы?: ор-
ганических и неорганических соединений, содержащих активные
группы с подвижными ионами, к обмену этих ионов на ионы
различных электролитов, вводимых в раствор. В обычной радио-
химической практике методом ионообменной хроматографии
разделяют малые количества исследуемых ионов в растворах
электролитов с высокой концентрацией. В этих условиях сорб-
ция каждого иона будет определяться только обменом данного
иона с ионами электролита. При этом ионный обмен, если он не
сопровождается рядом побочных эффектов — гидролизом, окис-
лительно-восстановительными реакциями и т. д., — полностью
обратим.
226
Процесс обмена может быть представлен в виде реакции
Mc + Gp^Mp + Gc,
где Л1 и G — обменивающиеся ионы (заряды опущены), а ин-
дексы «с» и «р» соответственно обозначают смолу и раствор. К
этой реакции можно применить закон действующих масс
Кобм
[G]c [М]р
[GJP [Af]c ’
(9.6)
где Л’обм — константа обмена, а величины в скобках представ-
ляют активности ионов в смоле и растворе. Константы обмена
могут служить для характеристики сродства ионита к различ-
ным ионам. Но из-за трудностей в определении активностей
ионов в смоле и растворе для представления сорбируемости ио-
нов используют экспериментально определяемые коэффициенты
распределения, которые равны отношению концентраций дан-
ного иона в равновесных фазах. В областях линейной изотермы
этот коэффициент не зависит от концентрации иона:
, (9.7)
mp
где D4. — весовой коэффициент распределения; те — количество
попа на 1 г сухой смолы; тр — количество иона в 1 мл раство-
ра. Иногда пользуются объемным коэффициентом распределе-
ния который связан с Dw уравнением
Dv = Г>а,рс, (9.8)
где рс — плотность слоя смолы (количество сухой смолы в 1 мл).
Отношение коэффициентов распределения называют факто-
ром разделения dpa3^DxID2, он служит для характеристики воз-
можной легкости и чистоты разделения. Чем больше фактор
разделения, тем быстрее и эффективнее могут быть разделены
ионы.
В аналитической практике сейчас находит применение весьма
широкий ассортимент ионитов, различающихся как по своей
природе, так и по ионообменным свойствам и различным харак-
теристикам [275]. Прежде всего по знаку обменивающихся ионов
все иониты распадаются на катиониты и аниониты. Природа
соединений, составляющих основу ионита, приводит к наличию
неорганических и органических ионитов. Некоторые иониты
проявляют чисто ионный механизм обмена, другие же по-
казывают определенную избирательность по отношению к от-
дельным ионам. Среди последних особо выделяются внутриком-
плексные ионообменные соединения. В пределах перечисленных
основных классов ионитов возможно деление на мелкие группы
по характеру основности (кислотности), виду функциональных
групп и т. д.
8:
227
Для аналитических разделений наиболее часто используются
органические иониты, неорганические — значительно реже. Среди
первых наиболее важны быстродействующие и универсальные
сильнокислые катиониты и сильноосновные аниониты. Другие
типы ионитов используются эпизодически.
Избирательность ионного обмена
Ионообменные разделения основываются на различии срод-
ства ионита к разделяемым ионам, что находит отражение в ве-
личинах коэффициентов распределения. Сродство ионита к ионам
является сложной функцией многих параметров, которую практи-
чески невозможно выразить в виде точных математических со-
отношений. Поэтому в большинстве случаев используют эмпири-
ческий подход к определению необходимых условий разделения,
который состоит в экспериментальном определении коэффициен-
тов распределения разделяемых ионов и последующей оценке
фактора разделения. Однако, зная действие различных пара-
метров, часто довольно точно можно предсказать влияние из-
менения условий на сорбируемость ионов.
Прежде всего ионит определенного типа способен к сорбции
только катионов или анионов. Правда, иногда наблюдаются
случаи аномального ионного обмена, но они крайне редки.
При сорбции ионов одинакового заряда сродство ионита за-
висит от строения и свойств ионита, от характеристик ионов и
состава и температуры раствора. Если иметь в виду монофунк-
циональные сильноосновные аниониты и сильнокислотные ка-
тиониты, то можно отметить некоторые эмпирические и приб-
лиженные закономерности. В разбавленных водных раство-
рах при нормальной температуре сродство ионита к ионам
возрастает следующим образом с изменением некоторых (Ьак
торов: а) при увеличении заряда иона; б) при увеличении атом-
ного номера элементов для катионов; в) при уменьшении ра-
диуса иона; г) при увеличении процента сшивки и обменной ем-
кости ионита. Влияние температуры на сродство довольно нез-
начительно и чаще всего наблюдается уменьшение сродства с
ее увеличением.
Надо отметить, что рассмотренные факторы, кроме знака за-
ряда, не дают больших значений факторов разделения ионов,
и обычно значительное повышение избирательности ионообмен-
ных разделений связано с использованием комплексообразова-
ния. Это может быть сделано двумя путями. Один из них со-
стоит в использовании комплексообразующих ионитов, которые
проявляют высокую избирательность к определенного типа
ионам [273, 276].
Другой подход основывается на введении в раствор комп-
лексообразующих реагентов. Избирательность ионообменного
выделения в этом случае обеспечивается подбором специфичного
228
комплексообразующего реагента, его оптимальной концентра-
ции, а также применением соответствующего ионита. Большое
значение в методе комплексообразования имеет pH среды,
поэтому его величина представляет дополнительный фактор
в обеспечении высокой избирательности ионообменного раз-
деления.
Важно отметить еще два фактора, которые позволяют опре-
деленным образом воздействовать на избирательность ионного
обмена. Так, на избирательность значительное влияние оказы-
вает проведение ионообменных разделений в неводных раство-
рителях. Причем это влияние в зависимости от ряда других
факторов может сказаться как в увеличении коэффициентов
распределения, так и в их уменьшении. Изменения в величинах
коэффициентов распределения при этом могут быть весьма зна-
чительными. Наконец, на избирательность ионообменного выде-
ления элементов с несколькими устойчивыми валентными сос-
тояниями можно воздействовать через изменение валентного
состояния.
Кинетика ионного обмена
Для получения высокой чистоты и количественного выхода
определяемых элементов разделение методом ионообменной хро-
матографии необходимо проводить в равновесных условиях или
близких к ним. Поэтому скорость протекания различных стадий
ионообменного процесса определяет длительность аналитическо-
го разделения. Обстоятельный анализ кинетики ионного обмена
дан в книге Ю. А. Кокотова и В. А. Пасечника [277].
Можно выделить три основных стадии ионообменного про-
цесса: 1) стадия массопереноса, которая включает перемещение
обменивающихся ионов из раствора к поверхности зерен иони-
та и обратно, осуществляемое совместно диффузией и конвек-
цией; 2) стадия массопередачи, которая обеспечивает перемеще-
ние обменивающихся ионов внутри зерен ионита (как правило,
за счет диффузии); 3) стадия собственно ионообменного про-
цесса.
Будучи ионной реакцией, последняя стадия обычно проте-
кает весьма быстро. Поэтому стадии массопереноса и массо-
передачи оказываются лимитирующими в кинетике ионообмен-
ных процессов. В связи с тем, что основной процесс на этих ста-
диях составляет диффузия ионов, различные факторы, воздей-
ствующие на скорость диффузии, в конечном счете определяют
кинетику ионообменного разделения. Так, повышение темпера-
туры и уменьшение размеров зерен ионита приводят к более
быстрому установлению равновесия. Скорость диффузии зави-
сит также от заряда и размеров ионов и уменьшается с их уве-
личением. Для органических ионитов рост степени сшивки при-
водит к уменьшению скорости диффузии ионов.
229
В целом скорость ионообменных процессов достаточно ве-
лика, что позволяет осуществлять с их помощью довольно быст-
рые разделения. Однако иногда отмечаются случаи замедления
отдельных стадий ионообменного процесса, что, в свою очередь,
ухудшает аналитические характеристики метода' разделения.
Например, замедленность ионообменной стадии часто отмечается
у комплексообразующих ионитов. Ионы многих металлов обра-
зуют с этими ионитами инертные комплексы, взаимодействие
которых с вытесняющими ионами при нормальной температуре
происходит крайне медленно. Явно замедленной кинетикой обла-
дают неорганические иониты. Замедление кинетики ионного
обмена часто имеет место в неводных средах.
Иногда аномалии в ход ионообменного разделения могут
внести и химические процессы в растворе. Например, медлен-
ность установления равновесия между различными анионными
формами As54- в растворах HF приводит к появлению двух пи-
ков на кривой вымывания [278]. При повышении температуры
мышьяк дает только один пик.
Разделение элементов методом ионообменной
хроматографии
Для аналитических целей ионный обмен преимущественно
находит применение в форме ионообменной хроматографии. При
этом разделение осуществляется при пропускании анализируе-
мого раствора, а затем элюента (вымывающего раствора) че-
рез колонку, заполненную соответствующим ионитом. Наиболее
просто таким способом можно разделять элементы, находящиеся
в растворе в различной ионной форме (катионной или анион-
ной). Для ионов одинакового заряда достижение высокой чи-
стоты выделяемых фракций оказывается более сложным и тре-
бует достаточно четкого подбора условий разделения.
Теория хроматографических разделений. При внесении раз-
деляемых ионов в хроматографическую колонку условия подби-
рают таким образом, чтобы разделяемые ионы сорбировались
верхним слоем ионита, образуя узкие зоны. Затем через колон-
ку пропускают элюент с такой скоростью, чтобы обмениваю-
щиеся ионы успевали войти в подвижное равновесие. Под дейст-
вием потока жидкости зоны сорбированных элементов сме-
щаются вниз по колонке, в конце которой происходит их после-
довательное выделение. Поскольку в процессе прохождения
зоны по колонке происходит большое число актов сорбция — де-
сорбция, то в результате достигается высокая эффективность
разделения.
В силу статистического характера процессов сорбция — де-
сорбция кривые вымывания элементов обычно имеют типичную
колоколообразную форму, которая в равновесных условиях близ-
ка к гауссовой кривой (рис. 57). Если выходная кривая несим-
230
метрична, то это свидетельствует либо о неравновесных условиях
разделения, либо о перегрузке колонки.
Объем раствора, при котором наблюдается максимальная
концентрация вымываемого элемента, является характеристи-
кой адсорбционной способности его иона в данных условиях и
связан с коэффициентом распределения простым соотноше-
нием
у = i + Dv = i Dwpc, (9.9)
где г7— объем раствора, соответствующий максимуму кривой
вымывания н выраженный в объемах колонки; i — свободная
доля объема колонки (обычно 0,4).
Объем злюатц
Рис. 57. Форма кривой вымывания двух компонентов.
Эффективность или чистоту разделения можно охарактери-
зовать величиной перекрытия зон разделяемых элементов
(рис. 58), которая для первой зоны численно будет равна <712 =
= Ani2lml (для второй <?2i = Ami/m2) при условии, что граница
раздела зон проходит через точку равного содержания обоих
элементов. Нетрудно видеть, что обратная величина 1/q при
Ш1 = т2 (условие, которое легко выполняется при введении носи-
телей) равна фактору очистки.
Величина перекрытия зон определяется фактором разделе-
ния элементов и числом теоретических тарелок. Последнюю ве-
личину можно оценить, исходя из кривой вымывания
iVT T 8 (и Ао1/е)2, (9.10)
где jVt. т — число теоретических тарелок; Avi/e — ширина зоны
на уровне
Число теоретических тарелок зависит от длины колонки и
степени зернения смолы. Последняя определяет эффективную
высоту теоретических тарелок, которая в предельном случае
соответствует диаметру частиц смолы. Однако в реальных ус-
ловиях из-за неоднородности упаковки смолы, высокой скорости
протекания раствора и некоторых других причин эффективная
высота теоретических тарелок оказывается во много раз больше.
231
В целом чем длиннее колонка и мельче размер зерен смолы,
тем выше число теоретических тарелок.
Следовательно, чтобы получить необходимую чистоту разде-
ления, требуется так подобрать условия, чтобы обеспечить либо
большое различие коэффициентов распределения, либо достаточ-
ное число теоретических тарелок. При заданной степени чисто-
ты разделения требуемое число теоретических тарелок быстро
возрастает с уменьшением фактора разделения (рис. 59).
Рис. 58. Перекрытие выходных кривых двух ком-
понентов.
В активационном анализе радиохимическая процедура часто
должна обеспечивать фактор очистки выше 104. Поскольку ис-
пользовать колонки длиннее 10 см нежелательно из-за значи-
тельного увеличения времени разделения, то при разработке
аналитических методик особое внимание следует уделять нахож-
дению условий, при которых фактор разделения превышает 2.
Чем выше фактор разделения, тем короче может быть исполь-
зуемая колонка, а скорость вымывания — увеличена. Когда
производится разделение одновременно нескольких элементов,
для выделения каждого из них должны быть подобраны опти-
мальные условия.
Согласно типу процессов, обеспечивающих избирательность
разделения, возможны два метода ионообменной хроматогра-
фии— вытеснительный и комплексообразующий.
Вытеснительная ионообменная хроматография. В этом ме-
тоде используют вытеснение сорбированных ионов ионами элек-
тролита, пропускаемого через колонку. Скорость смещения зоны
тем выше, чем меньше сорбируемость вытесняемых ионов в дан-
ной среде. Поэтому при пропускании электролита через колон-
ку, на которой сорбированы ионы разных типов, они будут вы-
мываться в порядке возрастания сорбируемости.
При разделении микроколичеств элементов, когда их количе-
ство составляет менее 1 % емкости ионита в колонке, могут быть
приняты некоторые упрощающие предположения, что позволяет
рассчитать изменение коэффициентов распределения с концен-
232
трацией электролита. В этих условиях можно считать, что отно-
шение коэффициентов активностей обменивающихся ионов по-
стоянно. Если при этом проникновение электролита в смолу
Рис. 59. Зависимость числа теоретических таре-
лок от чистоты разделения и отношения v2/vi.
ничтожно, то можно написать
Dw = const----!--, (9.П)
где [G] — концентрация электролита; Zj и г2 — заряды ионов
электролита и микрокомпонента.
Если зависимость коэффициента распределения от концен-
трации электролита представить в логарифмической шкале, то
233
Рис. 60. Зависимость коэф-
фициента распределения от
концентрации электролита:
1 — для ионов с меньшим за-
рядом: 2 — для ионов с боль-
шим зарядом.
получится прямая линия, тангенс угла наклона которой равен
—Zi!Z2.
Нетрудно видеть, что в этих условиях фактор разделения
двух ионов не может быть увеличен при изменении концен-
трации электролита, так как их коэффициенты распределения
меняются одинаково. Однако увеличение концентрации электро-
лита приводит к ускорению вымывания ионов и уменьшению
рабочих объемов растворов.
Что касается разделения ионов разной валентности, то фак-
тор разделения будет зависеть от концентрации электролита,
так как наклон соответствующих пря-
мых на графике lg£>ir— lg[G] имеет
разную величину (рис. 60). Эти пря-
мые пересекаются в точке /; при этой
концентрации электролита ионы раз-
делить нельзя. Видно, что при вымы-
вании электролитом с концентрацией
ниже, чем в точке I, первым будет
вымываться ион с меньшим зарядом.
При концентрации электролита выше,
чем в точке I, будет наблюдаться об-
ратный порядок.
Однако уравнение (9.11) справед-
ливо только в идеализированных ус-
ловиях, а в реальных ситуациях ход
зависимости коэффициентов распреде-
ления от концентрации вытесняющего
иона часто сложнее. Например, в раз-
бавленных растворах соляной кислоты щелочные элементы на
катионите по порядку величины коэффициентов распределения
располагаются в соответствии с ростом атомного веса, а в кон-
центрированных растворах соляной кислоты наблюдается об-
ратный порядок. Такая же картина наблюдается и в группе
щелочйоземелцных элементов. Этот факт, по-видимому, объяс-
няется изменением гидратации ионов, которая уменьшается с
ростом ионной силы раствора.
Вообще в чистом виде вытеснительную ионообменную хро-
матографию используют сравнительно редко и главным обра-
зом для разделения щелочных элементов. При этом для
ускорения вымывания элементов с более высокими величинами
D„ часто прибегают к последовательному повышению концен-
трации электролита в элюенте (рис. 61).
Комплексообразующая ионообменная хроматография. В этом
методе основную роль при разделении играет способность ионов
к комплексообразованию с функциональными группами ионита
или комплексообразующими реагентами, вводимыми в элюент.
Комплексообразующая ионообменная хроматография — более
эффективный метод разделения, чем вытеснительный.
234
Возможны два основных варианта комплексообразующей
ионообменной хроматографии. Когда из-за близости химических
свойств элементов избирательность комплексообразования не
очень велика, для разделения применяют элюат с одним ком-
плексообразующим реагентом постоянной или постепенно изме-
няющейся концентрации. Иногда весьма эффективным средст-
вом является изменение величины pH водного раствора.
Рис. 61. Разделение щелочных элементов (ионит—
вольфрамат циркония, колонка размером
2X150 .mm).
Возможность успешного разделения определяется природой
разделяемых ионов, типом ионита и устойчивостью комплексных
соединений. Используя этот вариант комплексообразующей
ионообменной хроматографии, можно достаточно быстро и эф-
фективно разделять такие близкие по химическим свойствам
элементы, как редкоземельные. Так, при разделении какой-либо
пары редкоземельных элементов с помощью катионита и ком-
плексообразующего реагента фактор разделения определяется
следующим соотношением:
d,(9.12)
где 7С1 и Кя — константы обмена ионов; К'п и Kt! —константы
нестойкости их комплексов.
Более благоприятен случай, когда избирательность комплек-
сообразования велика и возможен перевод одного из первона-
чально сорбированных ионов полностью в несорбируемое со-
стояние, в то время как остальные ионы остаются сорбирован-
ными на ионите. Если, например, элементы сорбированы на
катионите в виде положительных ионов, то можно подобрать
специфичный комплексообразующий реагент, который меняет
знак соответствующего иона на нейтральный или отрицатель-
ный.
235
На анионитах металлы сорбируются в виде отрицательных
комплексов и разрушение этих комплексов при введении ком-
плексообразующего реагента, дающего более устойчивые ней-
тральные или положительные комплексы с соответствующим
металлом, или при изменении условий приводит к высоко спе-
цифичному выделению. Количество реагентов, способных обра-
зовывать в растворах комплексы со многими или отдельными
элементами, весьма велико, и соответственно может быть пред-
ложено много различных вариантов групповых и поэлементных
разделений.
Однако среди большого количества комплексообразующих
сред наибольшего внимания заслуживают растворы неорганиче-
ских кислот, что обусловлено следующими причинами.
1. К настоящему времени изучена сорбируемость почти всех
элементов периодической системы в растворах важнейших неор-
ганических кислот (НС1, HF, HNO3, НС1О4 и др.) на сильноос-
новных анионитах и сильнокислотных катионитах.
2. Многие элементы показывают очень высокую сорбируе-
мость на сильноосновных анионитах в растворах некоторых не-
органических кислот.
3. Во многих случаях успешное разделение сорбированных
элементов может быть достигнуто простым изменением кон-
центрации кислоты.
4. Кислоты из выделенных фракций удаляются простым упа-
риванием, что иногда упрощает процедуру подготовки фракции
к измерению активности.
§ 4. Экстракция
Основы метода и классификация экстракционных систем
Экстракция — один из основных методов разделения и выде-
ления в химическом анализе [279, 280]. Достоинства экстракции
заключаются в быстроте выполнения аналитических операций,
простоте аппаратуры, высокой избирательности при правильно
выбранных условиях, возможности автоматизации процесса раз-
деления и т. д. Экстракционный метод отличается большой уни-
версальностью, поскольку находит применение для выделения
почти всех элементов периодической системы. Экстракция как
аналитический метод обладает большой гибкостью и с одинако-
вым успехом позволяет выделение индивидуального элемента
с высокой степенью чистоты и групповые разделения, дает воз-
можность осуществлять концентрирование элементов и решать
задачу удаления сильноактивных макрокомпонентов и многие
другие проблемы.
Экстракционные методы разделения основаны на распреде-
лении вещества между двумя несмешивающимнся растворите-
лями. Обычно используется экстракция веществ из водных рас-
236
творов с помощью органических растворителей. Если в обеих
фазах растворенное вещество имеет одну и ту же химическую
форму, то оно будет распределяться между растворителями в
соответствии с законом Нернста, т. е. при равновесии отношение
концентраций растворенного вещества в обеих фазах при дан-
ной температуре будет постоянным.
Однако во многих случаях распределяющиеся вещества испы-
тывают химические взаимодействия с различными компонентами
в каждой из фаз. Многообразие химических процессов, кото-
рые могут иметь место при этом, приводит к наличию много-
численных экстракционных систем. По типу экстрагируемых
соединений экстракционные системы можно разделить па не-
сколько групп.
1. Простые вещества с ковалентной связью. В эту группу
входят такие вещества, как I2, HgCl2, GeCl4 и др. Они хорошо
экстрагируются различными растворителями.
2. Внутрикомплексные соединения. Это обширная и очень
важная в практическом отношении экстракционная система, ко-
торая обстоятельно рассмотрена в книгах Стары [281] и Ю. А. Зо-
лотова [282]. Для проведения экстракционных разделений в этом
случае используют определенного типа органические соединения,
способные образовывать циклические соединения при участии
иона металла. Сюда входят такие экстракционные реагенты,
как дитиокарбамипаты, дитизон, 8-оксихинолин, р-дикетоны,
оксимы и др. Образующиеся внутрикомплексные соединения
обладают хорошей растворимостью в органических растворите-
лях типа углеводородов, бензола, хлороформа и плохо раство-
римы в воде.
3. Координационно-несольватированные соли. Некоторые ор-
ганические соли (типа солей тетрафениларсония или тетрафе-
нилфосфония), а также соли крупных органических соединений
(типа Ее(Фен)|+, где Фен—1,10-фенатролин), которые вслед-
ствие больших размеров не имеют координационных связей с
молекулами воды, хорошо экстрагируются растворителями в
присутствии крупных однозарядных анионов (типа перхлората
и перрената).
4. Минеральные кислоты (НО, НВг, HI, HNO3, НСЮ4,
H2SO4 и др.). Они экстрагируются высокоосновными полярными
растворителями (реагентами) типа простых и сложных эфиров,
кетонов, спиртов, а также аминов с высоким молекулярным
весом.
5. Комплексные металлокислоты. Экстракция комплексов
металлов с галогенидными, роданидными и подобными ионами
(типа HFeCl4, HAuC14, HSbCl4 и т. д.), кислородсодержащими
растворителями или аминами находит широкое применение в
анализе.
6. Координационно-сольватированные соли. Некоторые рас-
творители образуют смешанные комплексные соединения с неор-
237
ганическими молекулами. Типичный пример — экстракция ура-
нил нитрата трибутилфосфатом.
7. Гетерополикислоты. Эти соединения экстрагируются кис-
лородсодержащими растворителями.
8. Разные соединения. Многообразие механизмов, по кото-
рым протекает экстракционный процесс, очень велико и не под-
дается однозначной классификации. Поэтому вне отмеченных
выше групп остается экстракция еще некоторых соединений, ко-
торые и образуют сборную группу.
Экстракция — обратимый процесс, который в случае внутри-
комплексных соединений можно представить уравнением
М"+ + п(НА)0^(МАя)о + пН+
с константой экстракции
[МА„]О[Н+]" п
ех [М"+] [НА]" '
Для характеристики экстракционных систем широко исполь-
зуется коэффициент распределения
(9-14)
который представляет отношение общей концентрации элемента
в органической фазе к общей концентрации в водной фазе при
равновесии.
Избирательность экстракционных разделений
Для оценки эффективности экстракционного выделения слу-
жит фактор извлечения, который определяется как доля от об-
щего количества элемента, перешедшая в органическую фазу.
Фактор извлечения связан с коэффициентом распределения со-
отношением
где v и По — объемы водной и органической фаз.
Тогда эффективность экстракционного разделения двух ком-
понентов будет характеризовать фактор обогащения (аналог
фактора очистки), который при однократной экстракции равен
Если условия подобраны так, что v = v0 и £)vl»l, то при
Псг'С! имеем
(9.17)
238
Отсюда следует, что для обеспечения высокой избирательно-
сти экстракционного разделения двух элементов необходимо
подобрать условия, обеспечивающие достаточно полное извле-
чение определяемого элемента в органическую фазу (Dci>10)
при наименьшей экстракции второго компонента. Например,
для двух радиоизотопов с одинаковой исходной активностью,
чтобы получить фактор обогащения более 100 (загрязнение
вторым компонентом менее 1%), необходима экстракционная
методика с Z)ci> Ю и Z)c2^ 10~2.
Высокая избирательность экстракционного метода разделе-
ния получила всеобщее признание. Эта особенность экстракции
обусловлена наличием большого числа факторов, изменяя ко-
торые, можно воздействовать определенным образом на экс-
тракционное поведение разделяемых элементов [283—285].
Успех экстракционного разделения прежде всего связан с
правильным выбором экстракционной системы и органического
растворителя. Интересные возможности для обеспечения изби-
рательности открывает экстракция с использованием смесей
растворителей. Во многих случаях смесь двух растворителей
или реагентов приводит к значительному увеличению коэффи-
циентов распределения по сравнению с суммой коэффициентов
распределения для индивидуальных растворителей и реагентов
(синергетический эффект). В других случаях, наоборот, приме-
нение смеси растворителей или реагентов приводит к резкому
уменьшению коэффициентов распределения (антагонистический
эффект).
Один из важнейших факторов, позволяющих простым спосо-
бом воздействовать на избирательность экстракционного разде-
ления,— выбор значения pH водного раствора. Особенно силь-
ное влияние оказывает изменение pH в случае экстракции внут-
рикомплексных соединений. Величину pH подбирают в этом
случае так, чтобы экстрагировался один из определяемых эле-
ментов, а остальные элементы, дающие менее прочные соедине-
ния с данным комплексообразующим реагентом, оставались бы
в водном растворе. Повышая pH, создают условия для экстрак-
ции следующего элемента; таким образом, в благоприятном
случае можно последовательно выделить ряд элементов из ис-
ходной смеси. Если определяемый элемент образует с комплек-
сообразующим реагентом менее прочное соединение, чем ме-
шающие элементы, то последние могут быть предварительно
извлечены при более низком pH раствора; после повышения pH
среды экстрагируется уже определяемый элемент. Выбор опти-
мальной кислотности водного раствора очень важен и в случае
экстракции ионных ассоциатов и других комплексов.
Когда изменением pH не удается обеспечить необходимой
избирательности экстракционного разделения, применяют мас-
кирующие реагенты. Значительное повышение избирательности
метода в этом случае получают в результате связывания при-
239
сутствующих в растворе мешающих элементов в неэкстрагируе-
мые соединения. Возможность использования данного маски-
рующего реагента в конкретной экстракционной системе опре-
деляется константами образования комплексов элементов как
с маскирующим, так и с экстрагирующим реагентом. В некото-
рых случаях дальнейшее увеличение избирательности экстрак-
ции можно получить, используя комбинации маскирующих реа-
гентов.
При экстракции внутрикомплексных соединений наиболее ча-
сто в качестве маскирующих используются такие реагенты, как
тартраты, цитраты, фториды и ЭДТА.
Для элементов, имеющих несколько устойчивых валентных
состояний, хороший метод увеличения избирательности экстрак-
ции—изменение окислительного состояния их ионов в анализи-
руемом растворе. Изменяя валентное состояние, можно либо
предотвратить образование экстрагируемых комплексов, либо
перевести элемент в экстрагируемое соединение. При этом, ко-
гда элемент находится в неэкстрагируемом соединении, из вод-
ного раствора извлекают все экстрагирующиеся элементы. По-
том, изменив валентное состояние, в тех же условиях проводят
избирательную экстракцию определяемого элемента. Возможны
и другие варианты использования валентного состояния для
повышения избирательности экстракции.
Валентное состояние элементов изменяют, добавляя в рас-
твор подходящие окислители или восстановители, выбор которых
зависит от химических свойств разделяемых элементов и осо-
бенностей данной экстракционной системы.
Для повышения чистоты разделения используют также такие
методические приемы, как реэкстракция или несколько после-
довательных циклов экстракции или реэкстракции. Последняя
обычно состоит в переводе экстрагированного элемента из орга-
нической фазы в свежий водный раствор. Подбирая состав
водной фазы, можно либо удалить мешающие элементы, либо
избирательно реэкстрагировать определяемый элемент. Избира-
тельность реэкстракции можно обеспечить введением соответ-
ствующих комплексообразующих реагентов, подбором pH вод-
ного раствора, изменением валентности и т. д.
Иногда повышению чистоты разделения способствует про-
мывка экстракта (или реэкстракта) небольшими порциями све-
жего раствора, содержащего вес необходимые компоненты в
оптимальной концентрации. При промывке как примеси, так и
основной компонент перераспределяются между фазами. При
оптимальных условиях большая часть извлекаемого элемента,
который имеет высокий коэффициент распределения, остается
в экстракте, в то время как значительная часть загрязнений,
обладающих значительно меньшими коэффициентами распреде-
ления, переходит в свежий раствор. Промывка несколькими пор-
циями свежего раствора может обеспечить практически полное
240
удаление загрязнений при пренебрежимо малых потерях основ-
ного компонента. В результате достигается высоко избиратель-
ное выделение.
Следует еще отметить применение высаливателсй, присутст-
вие которых в растворе увеличивает коэффициенты распределе-
ния многих комплексных соединений. Для заметного увеличения
экстракции обычно требуется создание в растворе высокой кон-
центрации неорганических солей. Использование высаливателя
для улучшения экстракции определяемого элемента может так-
же способствовать извлечению мешающих элементов. Поэтому
требуется подобрать такой высаливатель, при котором полу-
чается наилучший фактор обогащения для определяемого эле-
мента.
При активационном анализе высокая концентрация неорга-
нических солей в водной фазе может получиться при растворе-
нии основы пробы. Введение же посторонних солей в раствор
не всегда целесообразно, так как в дальнейшем их присутствие
может представить значительные трудности при выполнении
последующих этапов анализа.
Значительными возможностями для обеспечения высокой из-
бирательности выделения обладает обменная экстракция внут-
рикомплексных соединений [286]. При этом используется вытес-
нение определяемым элементом в ходе экстракционного разде-
ления элемента, который в составе хелатного комплекса входит
в экстрагент. Избирательность выделения обусловливается раз-
личиями в константах устойчивости комплексов и их константах
распределения.
Непосредственное отношение к избирательности экстракцион-
ного разделения имеет явление взаимного влияния элементов,
которое может проявляться в форме соэкстракции и подавления
экстракции [287]. Эти явления могут быть существенными и ока-
зывать большое влияние на результат экстракционного разде-
ления.
Обнаруженные случаи соэкстракции по механизму протекаю-
щих процессов распадаются на три основные группы. К первой
группе относятся случаи соэкстракции, когда катионы элемента,
которые нормально не экстрагируются в данных условиях, об-
разуют экстрагирующуюся ионную пару с комплексным анио-
ном, содержащим основной элемент. По этому механизму отме-
чена в основном соэкстракции щелочных, щелочноземельных
и редкоземельных элементов.
Во второй группе можно объединить случаи соэкстракции,
которые обусловлены образованием в водной фазе слабо диссо-
циированных смешанных соединений, способных экстрагиро-
ваться. Примером может служить увеличение коэффициента
распределения вольфрама при экстракции анилином из кислых
растворов в присутствии больших количеств молибдена и ва-
надия.
24!
Последняя, наиболее изученная группа-—соэкстракция эле-
ментов при извлечении комплексных металлогалогенидных кис-
лот кислородсодержащими растворителями. Правда, в данном
случае соэкстракция выражается в увеличении коэффициента
распределения микроэлемента в присутствии макрокомпо-
нента.
Так, при экстракции диизопропиловым эфиром коэффициент
распределения цинка в присутствии макроколичеств железа
возрастает в 20 раз. Этот тип соэкстракции объясняется образо-
ванием смешанных ионных ассоциатов в органической фазе.
Такие реакции оказываются возможными только в случае рас-
творителей с низкой диэлектрической проницаемостью и относи-
тельно невысокой донорской способностью. К ним относится
большинство простых эфиров.
При использовании растворителей с относительно высокой
диэлектрической проницаемостью и значительной донорской
способностью часто имеет место подавление экстракции.
Эти явления подчеркивают необходимость тщательного ана-
лиза используемой экстракционной системы и проверки имею-
щихся литературных данных в разрабатываемой методике раз-
деления. Тем не менее можно отметить, что соэкстракция не
играет такой большой роли при экстракционных разделениях,
как соосаждение в методе осаждения. Поэтому можно заклю-
чить, что экстракция — исключительно гибкий метод разделения,
имеющий достаточно разнообразных средств для создания весь-
ма избирательных методик разделения и выделения большин-
ства элементов периодической системы.
При разработке схем анализа смесей сложного химического
состава и выборе специфичных условий выделения отдельного
элемента большую помощь оказывают таблицы, показывающие
зависимость коэффициентов распределения от условий экстрак-
ции. Такие таблицы составляют для определенного комплексо-
образующего реагента и растворителя, иногда в систему вводят
и маскирующий реагент.
Кинетика экстракции
Разделения экстракционным методом обычно проводят в рав-
новесных условиях. Поэтому время, которое необходимо затра-
тить на выполнение разделения, непосредственно зависит от ско-
рости протекания экстракционных процессов. В большинстве
экстракционных разделений равновесие устанавливается всего
за несколько минут, однако имеется целый ряд случаев, когда
для достижения равновесия требуется несколько часов и даже
дней. Обстоятельно рассмотрена кинетика экстракции в рабо-
тах [279, 288].
Время достижения равновесия в экстракционной системе за-
висит от скорости протекания двух стадий: а) скорости химиче-
242
ских реакций, приводящих к образованию экстрагируемых соеди-
нений; б) скорости переноса этих соединений из одной фазы
в другую (массопередача).
Хотя в зависимости от свойств определенной экстракционной
системы процессом, определяющим кинетику экстракции, может
стать как первая, так и вторая стадия, в большинстве случаев
медленное установление равновесия определяется химическими
реакциями в водной и органической фазах.
Исследования также показали, что при экстракции ионных
ассоциатов равновесие в общем устанавливается быстрее, чем
при экстракции внутрикомплексных соединений. В последнем
случае имеется ряд факторов, влияющих на скорость протекания
химических реакций. Прежде всего следует отметить влияние
концентрации экстракционного реагента. С ростом концентра-
ции увеличивается скорость экстракции. К такому же эффекту
приводит и увеличение pH водной фазы. Введение в раствор
маскирующих реагентов часто приводит к значительному замед-
лению скорости экстракции.
Что касается скорости массопередачи, то она определяется
скоростью диффузионных процессов в водной и органической
фазах и скоростью перехода комплексных соединений через гра-
ницу раздела. В свою очередь, эти процессы зависят от таких
факторов, как размеры и форма комплексного соединения, вяз-
кость растворителя, наличие поверхностно-активных веществ
и т. д.
Большое значение для установления равновесия в экстрак-
ционной системе имеют продолжительность контакта фаз и ин-
тенсивность перемешивания. Эти операции должны обеспечить
установление равновесия. Однако не следует увеличивать без
необходимости продолжительность и интенсивность контакта.
Помимо того что в результате увеличивается время, затрачивае-
мое на разделение, при этом могут протекать побочные процес-
сы, затрудняющие получение необходимых результатов. Такими
процессами при длительном контакте могут быть разложение
реагента и растворителя, изменение валентного состояния эле-
мента, изменение состава экстрагирующегося соединения.
Скорость установления равновесия в экстракционной систе-
ме зависит также от перемещения одной фазы относительно
другой. Поэтому слишком интенсивное перемешивание не спо-
собствует ускорению экстракции, так как оно приводит лишь
к быстрому движению всей смеси без заметного увеличения
относительного перемещения фаз. В результате энергичного пе-
ремешивания может сильно увеличиться степень дисперсности
одной фазы в другой вплоть до образования эмульсий. Эти
процессы также снижают относительную подвижность фаз.
Обычно для достижения равновесия достаточно бывает просто
переворачивать делительную воронку в течение нескольких
минут.
243
§ 5. Экстракционная хроматография
Основу этого метода составляет экстракция [289]. Однако
процесс разделения осуществляется в динамичных условиях с
помощью хроматографической колонки, заполненной носителем
с фиксированной на нем неподвижной фазой. Вторая подвиж-
ная фаза заполняет свободный объем колонки.
По сравнению с обычной методикой экстракционных разде-
лений экстракционная хроматография обладает рядом до-
стоинств: 1) динамичность хроматографических процессов поз-
воляет разделять близкие по свойствам соединения; 2) методика
разделений крайне проста, так как отпадает процедура разде-
ления фаз; 3) иногда имеется возможность использовать более
широкий ассортимент сред и растворителей (сильные окислите-
ли, плохо расслаивающиеся растворители и т. д.).
В зависимости от того, какая фаза фиксирована на носителе,
выделяются два варианта экстракционной хроматографии. Исто-
рически первььм был развит метод с фиксированной водной фа-
зой, который первоначально получил название распределитель-
ной хроматографии. Метод с фиксированной органической фа-
зой появился позже и стал называться экстракционной (распре-
делительной) хроматографией с обращенной фазой.
Для оценки чистоты разделения и положения пика на кри-
вой вымывания для обоих методов экстракционной хромато-
графии сохраняют свою силу положения теории, рассмотренные
в связи с описанием ионообменной хроматографии. Объем элю-
ента, который необходимо пропустить через колонку для дости-
жения максимума на выходной кривой, рассчитывается по урав-
нению
^макс ~ ^додв “Г" ^с^Ф> (9-18)
ГД6 ^’подв объем подвижной фазы (свободный объем колонки);
Dc — коэффициент распределения; V&— объем фиксированной
фазы. Число теоретических тарелок может быть оценено по урав-
нению (9.10).
Тогда высота эффективной теоретической тарелки (ВЭТТ)
получается делением длины колонки на WT. т- С увеличением £><
ВЭТТ уменьшается и стремится к постоянной величине. После-
довательное уменьшение объема неподвижной фазы сначала
приводит к уменьшению, а затем к увеличению ВЭТТ. Этот эф-
фект объясняют тем, что при малых количествах неподвижной
фазы она не полностью покрывает поверхность носителя. Насы-
щение поверхности соответствует минимуму ВЭТТ, а при даль-
нейшем увеличении неподвижной фазы происходит соединение
отдельных зерен носителя в более крупные агрегаты. На ВЭТТ
оказывает также влияние состав водной и органической фаз.
Важную роль в разделениях методом экстракционной хро-
матографии играют вещества, выступающие в качестве носите-
-244
лей неподвижной фазы. Они должны удовлетворять целому
ряду требований: прочно удерживать на своей поверхности вод-
ную или органическую фазу; быть инертными, чтобы никакие
химические или адсорбционные процессы по возможности не
могли иметь места; быть устойчивыми в применяемых раство-
рителях; допускать получение в форме, удобной для заполнения
колонок.
В качестве носителей водной фазы обычно используют цел-
люлозу, силикагель и иониты. Наиболее полно отвечают отме-
ченным требованиям иониты. Заполнение колонки производят
взвесью носителя в водном растворе или органическом раство-
рителе, насыщенном водой. Промывают колонку органическим
растворителем для удаления избытка водной фазы. Для нане-
сения разделяемой смеси на колонку применяют либо насыще-
ние им небольшого количества носителя, либо растворение ее
в органическом растворителе. Носитель или растворитель затем
переносят в колонку и пропускают экстрагент.
Экстракционная хроматография с фиксированной водной фа-
зой имеет два серьезных ограничения. Прежде всего, сопря-
жено с известными трудностями изменение состава водной фа-
зы, что в конечном счете лишает метод необходимой гибкости.
Не очень удобны методы внесения смесей в колонки, что часто
затрудняет или ухудшает разделение.
Этих недостатков лишена экстракционная хроматография с
фиксированной органической фазой. В качестве носителя орга-
нической фазы служат силиконированный силикагель, фторо-
пласт-3, фторопласт-4, поливинилхлорид, полиэтилен и некото-
рые другие материалы. Нанесение экстрагента на носитель осу-
ществляют двумя способами. Чаще всего порошок носителя
смешивают с раствором известного количества экстрагента в
летучем растворителе и последний испаряют при непрерывном
перемешивании. В другом способе экстрагент пропускают через
колонку, предварительно заполненную сухим носителем, и его
излишек удаляют водным раствором, который впоследствии ис-
пользуется для разделений.
Для заполнения колонок применяют носитель не только в по-
рошкообразной форме, но и в виде пористых блоков. Так, из
фторопласта-4 удается приготовить упругие пористые блоки, ко-
торые плотно вводятся в колонку подходящего диаметра. Из
пористых блоков можно изготовить колонки большого диаметра
и такой пористости, чтобы растворы элюата протекали без до-
полнительного давления.
В ходе разделений экстрагент постепенно смывается с колон-
ки и это происходит тем быстрее, чем растворимее экстрагент в
водном растворе. Чтобы избежать ухудшения разделения из-за
уменьшения количества экстрагента, приходится предварительно
насыщать нм используемые водные растворы. Однако обычно
предпочитают применять малорастворимые в водном растворе
245
экстрагенты, число которых довольно ограничено (трибутилфос-
фат, триоктилфосфиноксид, ди-2-этилгексилфосфорная кислота,
триоктиламин, трикаприлметиламмоний).
Емкость колонок, используемых в экстракционной хромато-
графии с обращенными фазами, зависит от характера экстра-
гента и обычно довольно высока. Для примера в табл. 15 приве-
дена емкость колонок из фторопласта-4 для разных элементов-
и экстрагентов.
Таблица 15
Емкость колонок из фторопласта-4
Марка носителя Экстрагент Элемент Емкость, MS,'МЛ объема колонки
Полимеризационный порошок ТБФ Fe3+ 26
ТБФ Ga3+ 16
Отожженный и размолотый фторопласт, ТБФ U« + 85
размер зерен 0,1—0,2 мм МИБК Mos+ 1
Блочный фторопласт Д2ЭГФК U« + 30
ТБФ 28-
Нанесение разделяемых элементов на колонку не вызывает
затруднений, поскольку осуществляется в водном растворе. Од-
нажды подготовленная колонка может использоваться много-
кратно. При необходимости заменить' экстрагент можно про-
мыть колонку ацетоном или диэтиловым эфиром, после чего на-
нести на нее новый экстрагент.
Поскольку экстрагент на носителе фиксирован в виде тонкой
пленки, диффузия в органической фазе протекает крайне быст-
ро, что ускоряет процесс установления равновесия. Поэтому раз-
деление элементов с сильно различающимися коэффициентами
распределения можно проводить при высокой скорости протека-
ния раствора [порядка 12—18 мл/(см2 мин)].
При разработке экстракционно-хроматографических методик
разделения можно пользоваться литературными данными по
экстракции элементов.
§ 6. Специальные методы радиохимического разделения
Рассмотренные выше метод осаждения, ионный обмен и экс-
тракция относятся- к основным методам химического разделения.
Именно эти методы наиболее широко используются для прове-
дения радиохимических разделений в ходе активационного опре-
деления широкого круга элементов. Однако помимо них в ра-
диохимическом варианте активационного анализа находят при-
менение многие другие методы.
246
Пока что большинство из них лишь эпизодически привле-
каются для решения сравнительно узких задач активационного
анализа и поэтому здесь они условно отнесены к специальным
методам радиохимического разделения. Такая роль этих мето-
дов вытекает либо из методических особенностей, которые за-
трудняют их применение в активационных определениях, либо
из ограниченности аналитических возможностей. Некоторые из
этих методов, хотя и являются перспективными, пока не полу-
чили широкого распространения.
Ниже рассматриваются лишь некоторые из этих методов, ко-
торые, как представляется, приобрели важное значение для па-
диохнмического варианта активационного анализа.
Дистилляция
Перевод исследуемого элемента при соответствующих усло-
виях из твердого тела или раствора в газовую фазу достаточно
широко используется в препаративной химии и аналитической
практике. Способностью образовывать летучие соединения или
переходить в газообразную форму обладает значительное число
элементов периодической системы. Однако часто для этого тре-
буются весьма специфические условия (высокие температуры,
агрессивные среды и т. п.), которые трудно реализовать в ана-
литической работе *. Поэтому дистилляционные методики в прак-
тике активационного анализа используются для радиохимиче-
ского выделения небольшого числа элементов, которые могут
быть переведены в газовую фазу в сравнительно мягких усло-
виях и с применением простой аппаратуры.
Дистилляция — довольно селективный метод выделения эле-
ментов. Конечно, в первую очередь, это связано с тем, что весь-
ма ограниченное число элементов показывает способность к пе-
реходу в летучее состояние в обычно используемых условиях.
Однако достоинство метода дистилляции состоит еще в том,
что для каждого из таких элементов можно подобрать достаточ-
но специфичные условия выделения. Это достигается различны-
ми путями в зависимости от химических свойств выделяемого
элемента. Например, благородные газы легко извлекаются при
простом растворении или расплавлении пробы. Среди металлов
уникальной способностью к дистилляции в элементарной форме
обладает ртуть, которую можно отогнать непосредственно из
твердой пробы. В элементарной форме могут быть отогнаны и
галогены.
В других случаях большое значение имеет среда, из которой
производится дистилляция. Так, галогеноводородные кислоты
* Сейчас интенсивно развивается газовая хроматография неорганических
веществ (290]. Однако в активационном анализе этот метод разделения ка-
кого-либо существенного распространения еще не получил.
247
(НС1, HBr и HI) можно отогнать просто из среды H2SO4. Неко-
торые элементы летучи в виде соединений с галогенами (As, Hg,
Ge, Sn, Sb и др.) и поэтому для их дистилляции требуется вве-
дение в анализируемый раствор галогенсодержащих соединений.
Из окислительной среды в виде четырехокисей выделяются Ru
и Os.
На избирательность выделения в некоторых случаях можно
повлиять, регулируя температуру в дистилляционной установке.
Определенные возможности в этом плане открывают такие хими-
ческие операции, как изменение валентного состояния при осу-
ществлении дистилляции или применение дополнительной очист-
ки в газовой фазе. Например, мышьяк легко отгоняется в виде
AsCl3, но AsC15 уже обладает крайне низкой летучестью. Подоб-
ная ситуация имеет место и в случае сурьмы.
Дистилляционный метод разделения элементов требует опре-
деленной квалификации исполнителя, плохо поддается автома-
тизации и часто продолжителен во времени. Обычно дистилля-
ционное выделение занимает 30—60 мин. Однако в благоприят-
ных случаях это время может быть сокращено до нескольких
минут.
Применение дистилляции для проведения радиохимических
разделений при выполнении активационного анализа различных
объектов весьма разнообразно. Так, дистилляция — очень эф-
фективный метод в тех случаях, когда она может быть исполь-
зована для удаления макрокомпонентов облученной пробы, осо-
бенно если ее можно применить
Таблица 16
Примеры дистилляционных методик
удаления основы
Основа Летучее соединение Основа Летучее соединение
Sb SbCl3 Графит со2
As AsCl3 Se Se2Clo
Ge GeCl4 Si SiF4
на первой стадии радиохимиче-
ской обработки. В результате
уменьшается радиационная
опасность при сильноактивиру-
ющейся основе и появляется
возможность применения эко-
номичных полумикрохимиче-
ских методик разделения в
дальнейшем. Типичные приме-
ры дистилляционного удале-
ния основы приведены в
табл. 16 [291].
Дистилляцию применяют и
в качестве группового метода выделения летучих компонентов.
Выделенная группа может быть подвергнута у-спектрометриче-
ско.му анализу или дальнейшему радиохимическому разделению.
Дистилляционным методом иногда выделяют отдельные эле-
менты в радиохимически чистом виде. Наиболее важные дистил-
ляционные методики для активационных определений суммиро-
ваны в работах [17, 116, 266].
Летучесть элементов имеет и нежелательную сторону, так
как является источником потерь при выполнении различных
операций, требующихся по ходу анализа. Потери могут иметь
248
место на стадии подготовки пробы к анализу, во время облуче-
ния, в ходе различных радиохимических операций. Особенно
велика вероятность потерь в тех операциях, которые связаны с
нагреванием, обработкой кислотами и упариванием. Поэтому
для получения точных результатов при определении легколету-
чих элементов требуются особые меры предосторожности.
Например, Съёостранд [292] при определении Hg и As в био-
логических и органических материалах применял следующую
методику. Анализируемые пробы запаивали в кварцевые ампу-
лы. После облучения в реакторе ампулу тщательно мыли и по-
мещали в трубку из полимерного материала, запаянную с
одного конца. В эту трубку заливали смесь кислот и ее запаи-
вали со второго конца. Затем трубку сдавливали до разрушения
кварцевой ампулы и быстро встряхивали. После вскрытия труб-
ки содержимое переносили в дистилляционный аппарат. В ра-
боте [293] облученную ампулу замораживали жидким азотом,
разрушали и пробу в твердом виде отбирали для дальнейшей
работы.
Электрохимические методы
Электрохимические методы позволяют производить осажде-
ние определяемого элемента в контролируемых условиях на по-
верхности электрода. Электролитическое осаждение может быть
осуществлено как с использованием внешних источников тока,
так и без них. Большей частью элементы осаждаются на элек-
троде в виде металлов или окисей (гидроокисей).
Для электрохимических методов разделения свойственны
простота и малая трудоемкость .методики, практическое отсутст-
вие соосаждення. Конечные препараты, полученные электроли-
тическим методом, часто имеют малую толщину рабочего слоя,
что очень важно при измерении и спектроскопии излучений с
низкой проникающей способностью. Упрощается процедура оп-
ределения химического выхода, поскольку количество осадивше-
гося элемента можно определить по разности массы электрода
до и после разделения.
Электрохимическое осаждение используется для выделения
сравнительно небольшого числа элементов. Методика выделения
довольно длительна и не всегда обеспечивает количественный
выход. Избирательность электролитического осаждения в целом
не столь высока, чтобы обеспечить высокий фактор очистки, но
применение различных приемов часто позволяет сравнительно
простыми средствами достичь удовлетворительных результатов.
Большей частью электрохимическое осаждение применяется на
конечной стадии радиохимического выделения, когда основная
масса мещающих компонентов уже удалена.
Имеется целый ряд электрохимических методов, которые иг-
рают важную роль в аналитической химии [266] и радиохимии
[6, 267, 271]. Ниже будут отмечены только те из них, которые в
249
той или иной степени были использованы в исследованиях по
активационному анализу.
Электроосаждение под действием внешнего напряжения. При
подаче потенциала на два электрода, погруженных в анализи-
руемый раствор, происходят разряд ионов и их осаждение на
поверхности электрода. Металлы обычно выделяются на катоде.
Избирательность выделения обеспечивается подбором потенциа-
ла, плотности тока, состава электролита и материала электрода.
При электролизе водных растворов с катодом из благородных
металлов на нем осаждаются только те элементы, потенциал
выделения которых лежит выше потенциала выделения во-
дорода.
Так, Моустри и др. [294] проводили электрохимическое выде-
ление Ag при активационном анализе цинка. Облученную пробу
растворяли в смеси H2SO4 + HNO3. После введения в раствор
сульфаминовой кислоты электролиз выполняли при напряжении
1,5 в в специальной электролитической ячейке. Осадок серебра
растворяли в HNO3, в раствор вводили неактивный цинк и элек-
тролиз повторяли. Получен фактор очистки 104—105.
Метод внутреннего электролиза. В этом методе выделение
элемента происходит под действием потенциала, возникающего
при погружении разнородных металлов в раствор электролита.
В качестве катода используется Pt, а анода — один из следую-
щих металлов: Al, Zn, Cd, Fe, Pb. Избирательность выделения
определяется материалом анода, присутствием комплексообра-
зующих реагентов и величиной pH раствора электролита.
А. А. Кист и др. [295] с помощью внутреннего электролиза
выделяли медь при нейтронноактивационном определении в био-
логических объектах. Облученную пробу растворяли в царской
водке в присутствии 5 мг носителя меди. После упаривания вво-
дили электролит (0,2 мл H2SO4+I мл уксусной кислоты, разбав-
ление водой до 20 мл). Раствор переносили в электролизер (ка-
тод Pt, анод А1). Процесс проводили в течение 1 ч при темпе-
ратуре 90—95° С. Катод промывали водой и ацетоном, высуши-
вали, взвешивали и отправляли на измерение. По данной
методике химический выход достигает 50%, коэффициент очист-
ки выше 10® (относительно 24Na и 32Р).
А. И. Свиридова и др. [296] рассмотрели возможность груп-
пового электрохимического выделения. Методом внутреннего
электролиза с Pt-катодом и Al-анодом возможно одновременное
выделение Ag, Си, Hg, Bi, Аи и других металлов. Показано, что
если в исследуемый раствор ввести носитель меди и проводить
электроосаждение, то происходит совместное выделение индика-
торных количеств Hg, Ag, Bi и Au. Выход этих элементов ра-
стет пропорционально количеству выделившейся меди, но по
абсолютной величине не совпадает с выходом меди (оказывается
выше, как для Ag, или ниже, как для Bi). Удобным групповым
носителем может служить серебро.
250
Цементация. Самопроизвольное выделение некоторых метал-
лов на подложке из металлов с более отрицательным электро-
химическим потенциалом (цементация) не дает высокого вы-
хода [296]. Получающийся коэффициент очистки низок. Поэтому
метод не представляет интереса для активационного анализа.
Амальгамный метод
Для некоторых избирательных выделений находит примене-
ние металлическая ртуть или ее амальгамы. Химической основой
выделения может служить либо процесс изотопного обмена,
либо восстановительное амальгамирование. В первом случае
водный или органический раствор, содержащий радиоактивные
ионы определяемого металла *М"+, приводят в контакт с раз-
бавленной жидкой амальгамой того же металла Ма. В резуль-
тате изотопного обмена между амальгамой и раствором осу-
ществляется реакция
Равновесие будет смещено вправо, если [Ма] »[М*''+], что
без труда реализуется при определении малых количеств ком-
понентов.
Во втором случае используется процесс восстановления иона
с последующим растворением амальгамой
— *М"+ + вМа Д: *Ма + ВМ"1+.
п
Здесь равновесие смещается вправо, когда элемент-восстанови-
тель ЕМ менее благороден, чем определяемый элемент М. Вос-
становителем может служить сама ртуть или амальгама како-
го-либо из металлов. При восстановлении ртутью происходит
избирательное выделение только благородных металлов (Au, Pt)
[297]. Введение комплексообразующих реагентов (ионов CN~)
позволяет проводить избирательное выделение Hg методом изо-
топного обмена [298].
Скорость амальгамного обмена очень велика, и равновесие
при нормальной температуре, но при интенсивном перемешива-
нии устанавливается за 1—3 мин. Химический выход составляет
90—100% [298]. При изменении состава водной среды для неко-
торых элементов можно обеспечить избирательную реэкстракцию.
Измерение активности можно проводить непосредственно в
капле ртути или в растворе после реэкстракции. Выделение
этим методом может быть очень быстрым. Например, при оп-
ределении Ag облучают заранее подготовленный раствор, тогда
затраты времени на извлечение пробы из челнока пневмопочты,
выполнение выделения и подготовку для измерения составляют
всего 70 сек. Поэтому в качестве аналитического радиоизотопа
оказалось возможным использовать 110Ag с периодом полурас-
пада 24,2 сек.
Глава
10
НЕКОТОРЫЕ ПРАКТИЧЕСКИЕ АСПЕКТЫ
РАДИОХИМИЧЕСКОГО МЕТОДА
§ 1. Планирование радиохимических разделений
Аналитическая практика постоянно выдвигает перед актива-
ционным анализом новые задачи, значительное число которых
требует применения радиохимического разделения. К тому же
имеет место непрерывное стремление совершенствовать уже
имеющиеся методы в плане повышения производительности и
упрощения аналитических процедур, снижения стоимости анали-
зов при сохранении и даже повышении чувствительности опреде-
ления и точности результатов.
Поскольку возможны самые различные аналитические ситуа-
ции, связанные как с составом и числом определяемых элемен-
тов, так и с разнообразием исследуемых объектов, то выбор
оптимальной схемы радиохимического анализа часто представ-
ляет собой сложную проблему. Для ее корректного решения
необходим большой объем предварительных сведений, касаю-
щихся анализируемой пробы и определяемых элементов. Проб-
лема усложняется необходимостью учета ядернофизических
свойств всех компонентов, входящих в состав пробы. Наконец,
в конкретной методике анализа радиохимическое разделение
должно быть согласовано с конечным радиометрическим мето-
дом, так как именно полная процедура определяет аналитиче-
ские возможности метода в целом [299].
В связи со сложностью задач, отсутствием необходимых дан-
ных и закономерностей применение вычислительных машин для
оптимального планирования радиохимических разделений ока-
зывается невозможным. Выбор радиохимической схемы анализа
полностью зависит от квалификации исполнителя, от которого
требуются глубокое знание химии элементов, большой анали-
тический и радиохимический опыт работы, обстоятельное зна-
комство с теорией и методическими особенностями разных ме-
тодов разделения и их возможностями. Большую помощь в пла-
нировании радиохимических разделений оказывают различные
источники аналитической и химической информации [270, 300],
а также обширный опыт, накопленный непосредственно в обла-
сти активационного анализа [27, 28]. Очень полезные сведения
можно извлечь из сводных данных по поведению большинства
элементов в ионообменных, экстракционных и других процессах.
252
Периодически публикуемые обзоры дают представление о со-
стоянии того или иного аналитического метода и тенденциях его
развития.
Если говорить о тенденциях в использовании различных ме-
тодов разделения в активационном анализе, то в последние годы
значительное распространение получили методы, основанные
на экстракции и ионном обмене, которые сильно потеснили
преобладавший до этого метод осаждения. Средн различных
возможных способов проведения разделений особый интерес вы-
зывают колоночные методы, обычно отличающиеся простотой
исполнения и хорошей чистотой разделения.
С помощью колонок, работающих на основе экстракционных,
ионообменных, изотопных и других процессов, можно решать
самые различные радиохимические задачи. Они применяются с
одинаковым успехом как для групповых разделений, так и для
выделения отдельных элементов в радиохимически чистом виде.
Особо следует отметить возможность успешного разделения
близких по свойствам элементов. Многоэлементные разделения
могут выполняться путем последовательного пропускания смеси
элементов либо в разных средах через ряд отдельных колонок
с различными наполнителями, либо сразу через несколько ко-
лонок, соединенных в серию.
Вследствие большой трудоемкости аналитических операций
при разработке радиохимических разделений желательно полу-
чить нужный результат за одну стадию [299]. Однако это не все-
гда обеспечивает высокую радиохимическую чистоту получен-
ного препарата, и поэтому требуется высокоизбирательный
метод измерения. Когда же необходим высокий фактор очистки,
приходится проводить несколько последовательных химических
операций.
При этом руководствуются следующими правилами [116].
Требуемая степень радиохимической чистоты достигается более
быстро и эффективно, когда в отдельных стадиях используются
различные химические процедуры. Так, последовательное приме-
нение нескольких различных методик разделения обычно оказы-
вается эффективнее, чем многократное повторение какой-либо
отдельной методики. Другое правило — обязательное включение
в радиохимическую схему высокоспецифичных методик выделе-
ния определяемого элемента. Наконец, все операции радиохи-
мического разделения желательно отрабатывать таким образом,
чтобы они были количественными. При этом увеличивается об-
щий химический выход и соответственно возрастает чувстви-
тельность.
Иногда приходится планировать проведение радиохимиче-
ских разделений в весьма специфических условиях. Так, химиче-
ская обработка сильноактивных проб должна проводиться в за-
щитной камере с дистанционным манипулятором. Это, несом-
ненно, затрудняет выполнение операций, поэтому они должны
253
быть по-возможности простыми, допускать дистанционное управ-
ление и обеспечивать эффективное удаление сильноактивных
компонентов.
Например, при определении галлия в вольфраме активность
основы достигает 20 кюри [301]. Поэтому облучение пробы про-
водится в кварцевой ампуле с притертой пробкой, что облегчает
операцию извлечения пробы после облучения. Для выделения
галлия используется простая экстракционная методика в экс-
тракторе с дистанционным управлением. После сброса высокой
активности основы органическая фаза с извлеченным галлием
передается в соседнюю лабораторию, где в более удобных усло-
виях проводится дополнительная радиохимическая очистка.
В аналогичной ситуации при активационном анализе арсенида
галлия однократная экстракция диэтилдитиокарбаминатных
комплексов хлороформом при рН = 7-4-9 позволила выделить
группу элементов (Мп, Си, Со, Сг, Zn, Cd, Ni, Те, Fe) с высоким
фактором очистки (около 107) [302].
При разработке конкретного метода предварительно схема
радиохимического анализа намечается на основе имеющегося
опыта в области аналитических разделений и наличных литера-
турных данных. Однако намеченная таким образом схема ана-
лиза требует тщательной проверки, так как механическое пере-
несение какой-либо методики на новый объект или использова-
ние литературных данных в имеющейся ситуации редко дает
сразу удовлетворительные результаты. Поэтому оказываются
необходимыми большие усилия по тщательной проверке каждой
операции, потребность в которой возникает в ходе радиохими-
ческого анализа.
В радиохимической практике для подобных целей очень эф-
фективно используются радиоактивные индикаторы. С помощью
радиоактивных индикаторов элементов, поведение которых необ-
ходимо исследовать, можно быстро проверить общую пригод-
ность намеченной схемы радиохимического анализа или ее
отдельной стадии, уточнить условия разделения, оценить хими-
ческий выход и фактор очистки.
Например, для изучения эффективности выбранного анали-
тического метода к необлученной анализируемой пробе добав-
ляют известное количество радиоактивного индикатора и носи-
тель исследуемого элемента. Пробу переводят в раствор и
выполняют необходимые аналитические операции. После изме-
рения активности индикатора в конечной фракции можно рас-
считать химический выход. Аналогичным способом проверяют
чистоту разделения, только в этом случае к пробе добавляют
известное количество радиоактивного индикатора возможной
помехи и определяют долю активности, попавшую в конечную
фракцию исследуемого элемента. Проведя такую операцию для
всех элементов, которые могут быть потенциальными источника-
ми радиоактивных загрязнений данной фракции, можно оценить
254
общую эффективность метода химического выделения опреде-
ляемого элемента.
Поскольку традиционный путь разработки схемы радиохими-
ческого анализа достаточно сложен и трудоемок, а также тре-
бует высокой квалификации исполнителя, Гирарди и др. [303]
сделали первую попытку упростить этот процесс путем приме-
нения механических средств или вычислительных машин. В свя-
зи с исключительной важностью этой проблемы на методологи-
ческих принципах, положенных в основу этой работы, следует
остановиться подробнее.
В принятой модели процесс радиохимического анализа пред-
ставляется как последовательность элементарных стадий, в каж-
дой из которых в результате распределения между фазами
происходит разделение радиоизотопов на две группы. Следова-
тельно, проблема планирования радиохимической схемы анализа
состоит в таком выборе числа и последовательности элементар-
ных стадий, которые обеспечивают оптимальное решение постав-
ленной задачи. Однако, учитывая крайнюю сложность проблемы
и обилие самой разнообразной аналитической информации, та-
кую задачу в полном объеме с помощью механических средств
или вычислительных машин пока решить невозможно. Поэтому
приходится принять ряд упрощающих предположений.
1. В качестве основы для разделений выбирается хромато-
графический метод преимущественно с неорганическими сор-
бентами.
2. Устанавливается определенный фиксированный набор
сред, которые могут быть использованы для разделений. Воз-
можность изменения среды и объемов раствора с целью улуч-
шения разделения исключается.
3. Считается, что разделяемые ионы существуют в растворе
при крайне низкой концентрации и поэтому они не взаимодей-
ствуют друг с другом.
Принятая элементарная стадия разделения состоит в пропу-
скании анализируемой смеси, содержащейся в 5 мл соответст-
вующего раствора, через хроматографическую колонку (7Х
ХЗО.члг) с выбранным сорбентом и в последующем пропускании
30 мл того же самого раствора. Для набора необходимой инфор-
мации было изучено поведение большого числа элементов в дан-
ной элементарной стадии, представляющей определенную ком-
бинацию сорбент — среда (табл. 17). Результаты этих исследо-
ваний составляют библиотеку химической информации [304]. Для
примера на рис. 62 показано поведение элементов в системе
гидратированная пятиокись сурьмы — 7 М HNO3.
Для планирования разделений всю информацию по поведе-
нию элементов представляют с помощью только четырех кате-
горий: 1) вымывается свыше 95%; 2) сорбируется свыше 95%,;
3) частично сорбируется (между 5 — 95%); 4) поведение неоп-
ределенно или неизвестно. Само планирование может быть вы-
255.
Таблица 17
Список использованных сорбентов и сред
Сорбент Шифр Среда Шифр
Гидратированная двуокись марганца HMD I М HC1 1 с
Безводная двуокись марганца AMD 6Л1 НС1 6С
Гидратированная пятиокиеь сурьмы НАР 12 М HC1 12С
Кислая окись алюминия ААО 0,1 /И HNO:1 при 65° С 0,1 м
Двуокись олова TDO 1 М HNO3 1 м
Фосфат циркония ZPH 7 М HNO3 7 М
Сульфид меди CUS 14Л4 HNO3 14 Af
Хлорид меди сис 1 м нсю4 1 Р
Оксалат церия сох 6 M НС1О4 6Р
Анионит Дауэкс-1 AER 6М HF 6F
Катионит Дауэкс-50 CER 1 М H2SO4 1 S
полнено либо на основе перфокарт, либо путем использования
вычислительных машин.
Рис. 62. Поведение элементов в системе гидратированная пятиокиеь
сурьмы — 7 Л4 HNO3:
/ — сорбируется более 95%: 2 — сорбируется частично (затемненная доля круж-
ка указывает степень сорбируемости элемента); 3— вымывается более 95%;
4—поведение невоспроизводимо (поведение остальным элементов не исследо-
валось).
Система перфокарт. Имеющаяся химическая информация
для каждого элемента переносится на специальную перфокарту
(рис. 63), где с помощью отверстий фиксируется поведение эле-
256
. Кузнецов
Рис. 63. Перфокарта с записанной химической информацией (расшифровку обозначений см. в табл. 17).
мента в различных элементарных стадиях. Перфокарта состоит
из двух симметричных половин. На левой половине отверстие
означает, что элемент вымывается в данной системе сорбент —
среда, а на правой половине отверстие уже означает сорбцию.
В других случаях отверстие не пробивается. Чтобы определить
условия, в которых возможно разделение двух элементов Мг и
М;, берут их перфокарты, одну из них переворачивают и скла-
дывают вместе. При этом плоскость «вымывание» на одной
карточке совпадает с плоскостью «сорбция» на другой. Сквоз-
ные отверстия будут указывать элементарные стадии, в которых
поведение этих элементов противоположно. Именно эти стадии
можно использовать для разделения.
Аналогичный прием может быть применен для выбора усло-
вий групповых разделений. С помощью перфокарт можно ре-
шать и более сложные проблемы, такие, как полное разделение
трех радиоизотопов или отделение одного элемента от ряда дру-
гих. Однако это требует довольно длительного манипулирова-
ния перфокартами, а получающееся решение дает удовлетвори-
тельный результат только тогда, когда удается подобрать усло-
вия для одностадийного разделения. Двустадийное разделение
нежелательно, так как при этом несколько изменяются стан-
дартные условия. Для решения проблемы в такой ситуации
можно пойти на упаривание раствора после первой стадии с по-
следующим растворением в новом стандартном растворе или
применить последовательное соединение колонок.
Планирование с помощью вычислительных машин. Примене-
ние вычислительных машин дает возможность более полно ис-
пользовать данные химической библиотеки. Программа, напи-
санная на языке FORTRAN IV для вычислительной машины
IBM. 360/65, позволяет установить последовательность элемен-
тарных стадий, в которых один ион или группа ионов может
быть отделена от других мешающих ионов. Программа состоит
из ряда логических операций над матрицей химической инфор-
мации, в которой поведение различных ионов относительно всех
пар сорбент — среда (кратко пара С/S) закодировано специаль-
ным способом.
Поскольку разработка методики планирования химических
разделений с помощью вычислительных машин находится еще
в начальной стадии, ограничимся только рассмотрением общей
последовательности логических операций на примере самого про-
стого случая. Пусть имеем набор п элементов, из которого нуж-
но выделить один элемент Мь
Следуя логической процедуре, использованной в методе пер-
фокарт, рассмотрим поведение всех исследованных элементов в
каждой паре С/S. Когда в данной паре С/S некоторые элементы
ведут себя неопределенным образом (частично сорбируются или
поведение неизвестно), то эта пара отвергается. Если обнару-
живается пара С/S, в которой поведение всех исследуемых эле-
258
ментов однозначно, выполняется следующая операция, которая
сравнивает поведение выделяемого элемента с поведением ос-
тальных. Если обнаруживается, что поведение первого не сов-
падает с поведением остальных, то в этой паре CIS возможно
разделение в одной стадии. В противном случае программа про-
должается дальше, пока не будут исследованы все пары C/S.
10 s
10°
250 500
750 1000 1250 1500
Энергия, кэб
Рис. 64. Определение меди в пробе нержавеющей стали:
/ — первичный у-спектр пробы; 2 — у-спектр раствора после сорбции
“Мп и 187W; 3 — у-спектр сорбента после распада 56Мп и 187W.
Если в результате окажется, что одностадийное решение не
дает полного разделения, то можно рассмотреть варианты с ча-
стичным решением (неполное отделение от мешающего элемен-
та) или двустадийное разделение. Количество возможных вари-
антов решений тогда сильно возрастает. С помощью вычисли-
тельной машины можно решать и более сложные задачи типа:
1) выделение определенной группы элементов из исходной сме-
си, 2) полное разделение нескольких элементов.
В качестве практического примера возьмем разработку мето-
дики определения меди в пробах нержавеющей стали. Образо-
вание 187W при облучении тепловыми нейтронами препятствует
измерению активности 64Си. Применение метода перфокарт для
выбора методики отделения вольфрама от меди показывает, что
это может быть сделано в 15 случаях. Из них была выбрана ме-
тодика пропускания раствора пробы в 1 М НСЮ4 через колон-
ку с гидратированной двуокисью марганца. Результат разделе-
ния виден на рис. 64. Вместе с 64Си выделился 56Мп, который
только частично сорбируется в выбранных условиях. Для пол-
9* 259
ного исключения марганца вместе с вольфрамом система перфо-
карт дает три возможных решения. К более широкому набору
вариантов приводит обработка с применением вычислительной
машины.
§ 2. Субстехиометрическое выделение элементов
Упростить радиохимический вариант при одновременном по-
вышении избирательности позволяет метод субстехиометриче-
ского выделения элементов [5]. Сущность метода состоит в том,
что, используя различные химические средства, из общей массы
исследуемого элемента выделяют точно определенное количество
и измеряют его активность. Тогда справедливо следующее со-
отношение, связывающее полную активность элемента Ах с из-
меренной активностью фракции ах:
Ал = ах-^, (10.1)
где wox и wBX — общее и выделенное количества элемента. По-
добную операцию можно выполнить и с эталоном. Тогда из
уравнения (10.1) при условии швж = швя и щОх = ^оэ, что легко
осуществить введением одинакового количества носителя, по-
лучаем
тх = т3 • -2^-, (10.2)
вэ
где тх и /иэ — количества определяемого элемента в пробе и эта-
лоне до введения носителя (mx^wox и тэ4Сшоэ).
Последнее соотношение — основа применения субстехиомет-
рического метода выделения в активационном анализе. Оно по-
казывает, что при соблюдении отмеченных выше условий отпа-
дает необходимость в определении химического выхода.
Для субстехиометрического выделения в анализируемый рас-
твор вводят реагент, который вступает в соединение (комплекс)
с определяемым элементом. Однако количество добавляемого
реагента должно быть меньше, чем это требуется по стехиомет-
рическому соотношению. В результате часть элемента переходит
в новое соединение (комплекс), часть остается в исходном виде.
Используя различия химического состояния элемента в новом
соединении и в непрореагировавшем остатке, можно осущест-
вить их разделение. Для этого пригодны самые различные мето-
ды: экстракция, ионный обмен, осаждение и т. д.
В наиболее общем виде подход для определения условий, не-
обходимых для субстехиометрического выделения, сформулиро-
ван Г. А. Пережогиным и И. П. Алимариным [305]. Обозначим
исходный раствор как первую фазу, а вновь добавляемую или
образующуюся при разделении фазу — как вторую. Согласно
тому, в какой фазе окажется новое соединение, возможны два
260
варианта субстехиометрического выделения. По первому вари-
анту это соединение количественно переходит во вторую фазу,
по второму — остается в исходном растворе. Первый вариант
имеет определенные преимущества перед вторым и поэтому на-
ходит наиболее широкое применение. Именно этот вариант и
будет рассмотрен ниже.
Пусть М — преобладающая форма определяемого элемента,
благоприятная для образования с реагентом R комплекса MR,
тогда конечный результат обеих стадий субстехиометрического
выделения можно представить в виде соответствующих равно-
весий и констант:
M1 + R1^(MR)1, KMR = _®_; (10.3)
(MR)1^(MR).2> DMr = (10.4)
[MR]1
где Лмн — константа устойчивости комплекса MR; Dmk — кон-
станта двухфазного распределения комплекса (для простоты
заряды опущены, а стехиометрические отношения приняты рав-
ными единице).
Основное условие субстехиометрического выделения состоит
в том, что используемое определенное количество реагента пол-
ностью вступает во взаимодействие с выделяемым элементом.
Доля реагента, которая по тем или иным причинам окажется
вне этого комплекса, вызовет систематическую погрешность в
результатах определения. Обозначим через бц долю реагента,
входящую в равновесных условиях в состав комплекса с опреде-
ляемым элементом. Поскольку практически целесообразно из-
влечение 50% определяемого элемента, условие субстехиометри-
ческого выделения будет выражено соотношением
^MRCMPMR > ^R (Vl'V-2) (10 5)
2 ~f" %MRCM 1 — $R
где cM — начальная концентрация элемента M, а щ и v2— объ-
емы фаз. Следовательно, для субстехиометрпческого выделения
необходимо подобрать реагент, образующий достаточно устой-
чивый комплекс с определяемым элементом, и обеспечить усло-
вия для его количественного перехода во вторую фазу.
Уравнение (10.5) справедливо для идеализированных усло-
вий, т. е. в отсутствие других элементов (реагентов) и форм
комплексов элемента М с реагентом R, при достижении равно-
весия и т. д. В реальных ситуациях положение может быть бо-
лее сложным. Например, для повышения избирательности или
для предотвращения гидролиза в раствор вводят дополнитель-
ный реагент X, который может взаимодействовать с определяе-
261
мым элементом. Тогда условие субстехиометрического выделе-
ния принимает вид [306]:
.. 26„ (vr'v.,)
, (Ю.6)
С1 Sr)(1—ам)
где ам — доля элемента М, входящего в равновесных условиях
в состав комплекса MX. Если конкурирующий реагент образует
очень прочный комплекс MX, то концентрация реагента не дол-
жна превышать половины концентрации определяемого эле-
мента.
В исходном растворе возможно присутствие элемента G, ко-
торый также образует комплекс с реагентом R. Тогда субстехио-
метрическое выделение возможно при условии [307]:
т - ( ^MRDMR , <\
/7Zg 77------ д-----------г 1 тм, (Ю.7)
26r \ J
где mG и /т?м — исходные количества конкурирующего и опреде-
ляемого элементов; bR'— доля конкурирующего элемента, во-
шедшего в состав комплекса с реагентом. Конечно, при актива-
ционных определениях условие (10.7) можно реализовать путем
введения носителя элемента М (®м2>^м) • Однако такой под-
ход предполагает, что в исходном растворе количества элемен-
тов М и G крайне малы.
Для субстехиометрических выделений наиболее часто исполь-
зуется экстракция. Тогда для некоторых систем ион водорода
выступает как конкурирующий элемент [307]. Поэтому субсте-
хиометрическое выделение оказывается возможным только то-
гда, когда концентрация иона водорода в растворе меньше
некоторой величины (пороговый pH). Для общего случая, когда
элемент М образует комплекс вида MR„, условие для порого-
вого pH будет иметь вид
рН> 1g-Д- - (-----------l)lg-^-------Llg/<3K-lgCM, (10.8)
1 — оГ1 \ п ) V п
к
где Кл,; — константа экстракции.
Следует отметить, что субстехиометрический метод — это спо-
соб разделения определяемого элемента на две части, одна из
которых имеет точно определенную величину, а не метод отде-
ления от посторонних элементов. Поэтому избирательность экс-
тракционного субстехиометрического выделения определяется
избирательностью самого экстракционного метода. Однако фак-
тор обогащения может быть выше, чем при экстракционном
разделении с избытком реагента. Величина фактора обогащения
при субсте.хио.метрическом выделении [308] равна
262
где
Kmg
(/<MRn£,MRjm
(^gr^grJ"
При m = n=l получается более простое выражение
gcy6 = у(/<мо+ 1). (10.10)
Если сравнить это выражение с фактором обогащения при
избытке экстракционного реагента [уравнение (9.16)], то можно
показать, что при Кмс<=1 эти величины совпадают, но при
Лмс.>1 gcy6>goG- Если Kmg<1, ТО в лучшем случае goi-,=-l,
тогда как gryo= 1/2. В последнем случае для повышения изби-
рательности можно воспользоваться теми же приемами, что и
в обычной экстракции. При активационных определениях необ-
ходимость повышения избирательности выделения возникает
тогда, когда конкурирующий элемент не препятствует субсте-
хиометрическому выделению определяемого элемента из-за ма-
лого содержания, но создает затруднения при измерениях ак-
тивности.
При проведении активационного анализа с использованием
субстехиометрического выделения основные стадии следующие:
1) облучают одновременно пробу и эталон;
2) при переводе в раствор к пробе и эталону добавляют оди-
наковое количество носителя;
3) из обоих растворов в стандартных условиях проводят
субстехиометрическое выделение определяемого элемента;
4) измеряют активности выделенных фракций.
В качестве примера рассмотрим активационное определение
золота в пробах метеоритов, пород и минералов [309]. Облучен-
ную пробу обрабатывают царской водкой в присутствии 1 мг
носителя золота. Полученный раствор упаривают почти досуха,
приливают 10 мл 0,1 и. НС1, фильтруют в делительную воронку
и экстрагируют 2 мл 1,5-10—3 М раствора хлорида тетрафенил-
арсония в хлороформе. Экстракт центрифугируют для удаления
капелек воды и измеряют активность 198Аи на сцинтилляцион-
ном гамма-спектрометре. Аналогичным образом обрабатывают
эталон. Количественный расчет проводится по уравнению (10.2).
Обрусник и Адамек [310] развили метод вытеснительного суб-
стехиометрического выделения, который позволяет повысить из-
бирательность выделения определяемого элемента в присутст-
вии мешающих элементов. Принцип метода заключается в вы-
теснении определяемого элемента из его комплекса элементом,
образующим с реагентом более устойчивый комплекс. Проте-
кающие процессы можно представить следующей реакцией;
c;MR,„ + mG mGR? + qM.
(10.11)
263
При обеспечении условий субстехиометрического выделения для
такой реакции в водной фазе получают определенное количество
элемента М.
Последовательность операций в случае вытеснительного суб-
стехиометрического выделения можно рассмотреть на примере
активационного определения индия в граните и галлии [311].
К облученной пробе добавляют определенное количество носи-
теля и производят растворение. После установления определен-
ной величины pH в раствор вводят комплексообразующие реа-
генты для связывания некоторых мешающих элементов. Индий
экстрагируют раствором стехиометрического количества дити-
зона в СС14. Экстракт три раза промывают 0,1 М раствором бу-
ры для удаления возможного избытка дитизона и некоторых
нестойких дитизонатов. После этого экстракт встряхивают с бу-
ферным раствором, содержащим субстехиометрическое количе-
ство ртути.
Поскольку Hg образует с дитизоном более прочный ком-
плекс, чем индий, происходит вытеснение последнего из ком-
плекса. Затем измеряют активность индия, перешедшего в вод-
ную фазу. Аналогичную процедуру проводят и с эталоном.
Ю. В. Яковлев и О. В. Степанец [312, 313] соединили вытес-
нительное выделение с методикой экстракционной хроматогра-
фии. Для разделения использовали колонку размером 4Х100лш,
заполненную фторопластом-4. Неподвижной фазой служил рас-
твор диэтилдитиокарбамината цинка в СС14. В порядке возра-
стания константы устойчивости дитиокарбаминатных комплек-
сов рассматриваемые элементы располагаются в следующей
последовательности: Hg2+>Cu2+>Cd2+>Zn2+.
Методика выделения Cd и Си при активационном анализе
иттрия следующая. Облученную пробу переводят в раствор в
присутствии носителей Cd и Си. Причем количество Cd должно
быть достаточным для полного вытеснения Zn из его дптиокар-
баминатного комплекса. Предварительно определяемые элемен-
ты отделяют от макрокомпонента обычной экстракцией их ди-
тиокарбаминатных комплексов хлороформом при рН = 5-?6 в
присутствии цитрата калия. Органическую фазу упаривают,
комплексы разрушают при обработке смесью H2SO4+HNO3.
Остаток в водном растворе пропускают через колонку. При этом
Си образует узкую зону в верхней части колонки, а остальную
часть занимает Cd. Колонку промывают водой для удаления
избытка Cd. Затем 50% Cd из колонки вытесняется соответст-
вующим количеством меди при пропускании раствора Cu(NO3)2.
Элюат собирают для измерения.
Остаток кадмия удаляют из колонки при пропускании до-
бавочного количества меди. Для выделения 50% Си, находя-
щейся на колонке, используется водный раствор Hg(NO3)2.
К смеси эталонов определяемых элементов применяли анало-
гичную методику обработки.
264
Как было отмечено выше, при субстехиометрическом выде-
лении какого-либо элемента могут возникнуть помехи со сто-
роны элементов, образующие более прочные и хорошо экстра-
гирующиеся комплексы с реагентом. Однако в случае актива-
ционного анализа такая ситуация может быть использована
для группового выделения элементов [314, 315]. При этом обес-
печиваются субстехиометрическое выделение одного из опреде-
ляемых элементов и практически 100%-ный выход для осталь-
ных. Следовательно, в любом случае отсутствует необходимость
определения химического выхода, но для конечного определе-
ния нужен спектрометрический метод.
Так, при определении Au, Hg и Си в биологических пробах
проводили субстехиометрическое выделение меди при экстрак-
ции диэтилдитиокарбаминатов из раствора 2 н. H2SO4 +
4-0,1 н. НСЮ4 (ряд экстрагируемое™ Au3+>Hg2+>Cu2+) [315].
Такая операция возможна, если носители Au и Hg составляют
только 1% общего количества носителя меди. Тогда после экс-
трагирования раствором диэтилдитиокарбамината цинка в ССЦ
в экстракте оказывается стехиометрическое количество Си и
100% Ag и Hg.
§ 3. Быстрые радиохимические методики
Вследствие неизбежных затрат времени на разложение про-
бы и выполнение операций выделения и очистки радиохимиче-
ский вариант, как правило, находит применение при определе-
ниях по радиоизотопам с Л/г^ЗО мин. В области более коротко-
живущих изотопов несомненное преимущество остается за ин-
струментальным методом.
Однако иногда избирательность последнего оказывается не-
достаточной для решения данной аналитической проблемы, а
определяемый элемент не образует других радиоизотопов с бла-
гоприятным периодом полураспада. Например, при активаци-
онном анализе биологических проб успешному применению ин-
струментального метода для определения кобальта и магния по
короткоживущим изотопам (60тСо и 27Mg) мешает сильная ак-
тивация натрия и некоторых других компонентов [316]. В по-
добных ситуациях могут оказаться полезными быстрые радио-
химические методики, которые позволяют осуществить необхо-
димое разделение за сравнительно короткое время.
Вообще говоря, радиохимия накопила значительный опыт по
выделению короткоживущих изотопов. В ее арсенале имеются
настолько быстрые методики, что оказывается возможным выде-
ление радиоизотопов с периодом полураспада в несколько секунд
[317, 318]. Однако опыт радиохимии может быть использован
в практике активационного анализа лишь частично, поскольку
анализируемые пробы имеют сложный состав, что затрудняет
их разложение, на которое требуется определенное время.
265
Особенность быстрых радиохимических методик состоит в
том, что они предусматривают выделение только одного (редко
двух) элемента. Быстрота разделения играет решающую роль,
поэтому используемые химические методы не обязательно отли-
чаются высокой избирательностью и количественным выходом.
Поскольку затраты времени на дополнительную очистку неже-
лательны, методика выделения не всегда дает препараты вы-
сокой радиохимической чистоты и поэтому на конечной стадии
приходится применять методы регистрации, обладающие до-
статочно высокой избирательностью (обычно сцинтилляционный
гамма-спектрометр).
Схема анализа с быстрым радиохимическим разделением
примерно следующая. Пробу и эталон (монитор) транспорти-
руют на облучение и обратно с помощью пневмопочты. Облу-
ченную пробу быстро растворяют, используя подходящие реа-
генты. Для проб, плохо растворяющихся в воде или кислотах,
часто применяют плавку с перекисью натрия в никелевом тиг-
ле. Тонкоизмельченную пробу вносят в тигель, в котором уже
расплавлено несколько граммов Na2O2, тигель нагревают на го-
релке до красного каления и быстро охлаждают, погружая в
холодную воду. Затем плав обрабатывают кислотой. Таким об-
разом удается переводить в раствор пробы горных пород и био-
логические пробы в течение 1 мин. После этого следуют хими-
ческое выделение и измерение на сцинтилляционном гамма-
спектрометре. Химический выход обычно определяют после из-
мерения активности раствора.
Для быстрых выделений используют различные химические
методы. Так, при определении родия в горных породах по
104"'Rh (7’i/2=4,4 мин) Стил и Мейнке [319] воспользовались
экстракцией. Пробу облучали в реакторе 5 мин. По поступле-
нии в лабораторию пробу вносили в 10-кратное количество
Na2O2, предварительно нагретой в никелевом тигле. Смесь
сплавляли 1 мин, плав охлаждали и растворяли в 20 мл кон-
центрированной НО. Вводили 5 мг RhCl3, 1 мл 10%-ного ра-
створа винной кислоты и 8 мл пиридина и раствор фильтровали
в делительную воронку. Промывали тигель и фильтр 5 мл
6 п. НС1. Затем к раствору добавляли 15 мл 12 н. NaOH. После
встряхивания в течение 1 мин пиридиновый слой отделяли.
Экстракт делили на две части. Одна часть поступала на изме-
рение активности 104mRh, а другую использовали для определе-
ния химического выхода родия спектрофотометрическим методом.
Очень быстрое выделение может дать изотопный обмен. На-
пример, при определении магния в крови после обработки про-
бы концентрированной HNO3 и НС1О4 к полученному раствору
добавляли заранее подготовленный осадок MgNH4PO4 • 6 Н2О
[320]. После встряхивания в течение 2 мин осадок отделяли
и направляли на измерение. Вся химическая процедура требо-
вала около 5 мин.
266
Еще более быстрая методика предложена А. М. Вассерма-
ном и др. [138] при у-активационном определении кислорода в
чистых материалах. Облученную па микротроне пробу протрав-
ливают, помещают в графитовую капсулу, которую затем на-
гревают в специальной системе импульсным нагревом (6—
12 сек) до 3000° С в токе аргона. Проба плавится с восстанов-
лением окислов до СО, уносимой током аргона. При прохож-
дении через окись меди, нагретой до 600° С, происходит окисле-
ние до СО2, которая поглощается щелочью. Измерение активно-
сти 15О (7'1/2==2,1 мин) производится непосредственно в погло-
тителе с помощью двух сцинтилляционных счетчиков, включен-
ных на совпадения. Между концом облучения и началом изме-
рения проходит всего 1,5—2 мин.
Это одна из самых быстрых радиохимических методик, при-
мененных для активационного определения, а период полу-
распада, равный 2 мин, представляет нижнюю границу распро-
странения радиохимического метода.
§ 4. Автоматизация радиохимических разделений
Обычно радиохимические разделения весьма трудоемки, тре-
буют выполнения значительного числа ручных операций, боль-
шого внимания и часто высокой квалификации исполнителя.
Отмеченные обстоятельства обрекают квалифицированного ана-
литика на выполнение в ходе анализа значительного числа ру-
тинных операций и препятствуют широкому использованию ра-
диохимических методов при серийных анализах. Поэтому по-
стоянно предпринимаются попытки упрощения радиохимических
процедур и разработки систем с частичной или полной автома-
тизацией радиохимических разделений [321].
Для облегчения работы с микроколонками, используемыми
для разделения активированных элементов методом ионооб-
менной хроматографии, А. И. Калинин и Р. А. Кузнецов [63]
предложили простое автоматическое устройство. Оно состоит из
датчика для счета капель, который может быть основан на
изменении проводимости цепи при прохождении капли между
двумя платиновыми проволочками (или на применении фото-
элементов), и управляющего устройства (рис. 65).
Система работает следующим образом. Колонку подготав-
ливают к работе, затем управляющему устройству задают чис-
ло капель, которое необходимо пропустить при выделении дан-
ной фракции, и на колонку подают сжатый воздух для продав-
ливания элюента. Когда необходимое число капель будет про-
пущено, управляющее устройство с помощью трехходового кра-
на, запускаемого соленоидом, отключает колонку от источника
сжатого воздуха и продавливание элюента прекращается. Одно-
временно включается сигнал, означающий, что выделение окон-
чено. Колонку подготавливают для вымывания следующей
267
фракции, и снова контроль за ходом выделения осуществляет
управляющее устройство.
Гуд и др. [322] для проведения групповых экстракционных
разделений использовали автоматическую экстракционную
ячейку, схематическое устройство которой показано на рис. 66.
Рис. 65. Схематичное устройство авто-
матизированной микроколонки:
/ — датчик счетчика капель; 2— тефлоновая
вата; 3 — слой смолы; 4 — колонка; 5 — элю-
ент; 6 — пробка; 7 — соленоид с краном; S'-
сигнализатор; 9 — управляющее устройство;
1и— тефлоновая чашечка; р — давление.
В этой системе можно
выделить четыре основ-
ные части: экстракцион-
ную ячейку, распредели-
тельную систему, враща-
ющийся столик с пробир-
ками для сбора выделен-
ных фракций и блок уп-
равления.
Экстракционная ячей-
ка включает в себя сте-
клянный цилиндр, ме-
шалку и пару Pt-электро-
дов, впаянных в нижней
части ячейки. Система
приспособлена только для
работы с экстрагентами
тяжелее воды. Перемеши-
вание фаз производит
мешалка. Конец трубки
специальной конструкции
погружен в органическую
фазу. При вращении мо-
тора центробежной силой
органическую фазу заса-
сывает в отверстие в
трубке и поднимает на-
верх, где она вытекает в
водную фазу. При разде-
лении фаз с помощью
Pt-электродов замеряется
электропроводность ра-
створа. При попадании
электродов в водную фа-
зу электропроводность возрастает, что приводит к срабатыва-
нию реле, управляющего нижним краном.
Распределительная система подготавливает следующую пор-
цию реагента для введения в экстракционную ячейку. Это со-
кращает время между последовательными стадиями экстрак-
ционного разделения, так как отбор необходимого количества
реагента, выполняемый автоматически, происходит во время
осуществления в экстракционной ячейке предшествующей ста-
дии. Здесь уместно отметить, что одна распределительная ячей-
268
ка может обслуживать несколько экстракционных ячеек, а для
выполнения разделения по принятой схеме каждая экстракцион-
ная ячейка соединена с несколькими распределительными ячей-
ками с разными реагентами.
Работа системы сбора
фракций не требует коммен-
тариев. Управляющее уст-
ройство по заданной про-
грамме производит все опе-
рации, предусмотренные хо-
дом анализа.
Разделение с применени-
ем автоматической экстрак-
ционной ячейки протекает
следующим образом. Пере-
веденная в раствор проба
переносится в экстрактор, и
система запускается в рабо-
ту. Из распределительной
системы под давлением по-
ступает первый экстрагент,
после перемешивания в те-
чение заданного времени
экстрагент удаляется. Опе-
рацию экстракции с тем же
экстрагентом повторяют еще
дважды, и экстракты соби-
раются в одной пробирке.
По выделении первой
группы элементов в экст-
ракционную ячейку вводят-
ся необходимые реагенты и
устанавливается опреде-
ленная величина pH. Снова
вводится экстрагент соглас-
но схеме анализа, и экст-
ракция следующей группы
элементом производится Рис. 66. Автоматическая экстракционная
стандартным образом. ячейка:
Предусмотрев необходи- ‘3 Z'с“дшалка?а"™;
мое количество стадий И ПО-ЭК|:тРакциЯннаД, ячейк£Н « — управляющее уст-
о роиство; 7 — Pt-контакты; 8 — мотор вращающе-
доорав УСЛОВИЯ ДЛЯ каждой гося стола; 9 — пробирки для сбора фракций,
из них, получают ряд фрак-
ций, содержащих опреде-
ленные группы элементов. В частности, в рассмотренной работе
25 определяемых элементов разбивали на 6 групп (см. рис. 77).
Самсалом и др. [323] была разработана весьма совершен-
ная система для автоматизированного радиохимического раз-
209
деления большого числа элементов на несколько групп мето-
дами ионообменной и экстракционной хроматографии (рис. 67).
Основу системы составляет серия последовательно соединенных
хроматографических колонок.
Для подачи растворов в колонки в ходе разделения служит
система насосов. Отдельный насос представляет собой стеклян-
ный цилиндр, в котором плотно ходит поршень из тефлона.
Поршни всех насосов перемещаются синхронно.
Рис. 67. Схема автоматизированной хроматогра-
фической системы:
Н — насос с промывным раствором; С — спираль для
смешивания растворов; К — колонка.
Разделение проводится следующим образом. Заранее подго-
товленные колонки закрепляют в держателе и соединяют труб-
ками из тефлона друг с другом и с системой насосов. В каждый
цилиндр насоса вводится необходимое количество соответству-
ющего элюента. Анализируемый раствор переносят в начало
серии колонок и включают двигатель.
Насосы начинают подавать рабочие растворы в системе в
соответствующих местах. Для перемешивания растворов, по-
ступивших от предшествующей колонки и насоса, на входе ко-
лонки имеется спираль из тефлона. Смесь разделяемых эле-
ментов последовательно проходит через всю серию колонок. Со-
став растворов, поступающих от насосов, и наполнение колонок
подобраны так, чтобы обеспечить сорбцию на каждой колонке
небольшой группы элементов (см. ниже рис. 75). Полная схема
предусматривает разделение 40 элементов на 11 групп.
Все разделение завершается за 5 мин. Колонки с сорбиро-
ванными элементами извлекают из системы и после небольшой
промывки направляют на измерение у-спектров. В держатели
вставляют свежие колонки, насосы заполняют рабочими ра-
створами, и система готова к следующему разделению.
Глава
11
АНАЛИТИЧЕСКИЕ ХАРАКТЕРИСТИКИ
АКТИВАЦИОННЫХ МЕТОДОВ
§ 1. Чувствительность
Порог определения активационным методом. Рассмотрению
чувствительности активационного анализа посвящено значи-
тельное число специальных и особенно прикладных исследова-
ний, в результате которых показана его принадлежность к са-
мым чувствительным методам современной аналитической хи-
мии. Однако требования науки и техники к определению крайне
низких содержаний компонентов в различных объектах продол-
жают расти, что заставляет постоянно развивать техническую
базу и совершенствовать методику уже существующих методов,
а также искать принципиально новые способы анализа. Отсюда
естественным образом вытекает необходимость рассмотрения до-
стигнутого уровня чувствительности наиболее перспективных
активационных методов и возможных путей дальнейшего про-
гресса в этом направлении.
Процедура активационного определения четко распадается
на две основные стадии: активацию и измерение. Поскольку
они следуют друг за другом, выполняются с помощью специ-
альных устройств и в значительной степени определяются раз-
ными факторами, то их влияние на чувствительность определе-
ния можно рассматривать раздельно.
Стадия активации в общем виде может быть представлена
следующей последовательностью процессов:
А —В-----------—-> Е.
аА
С D
По этой схеме взаимодействие активирующего излучения с
изотопом А определяемого элемента приводит к образованию
аналитического радиоизотопа В, который в процессе радиоак-
тивного распада переходит в продукт Е. Кроме того, изотоп А
может иметь несколько каналов ядерных реакций с образова-
нием ряда продуктов, объединенных под общим символом С.
271
С другой стороны, радиоизотоп В также может взаимодейство-
вать с активирующим излучением, давая продукт D. На прак-
тике сочетание различных процессов может быть и иным, но
мы остановимся только на этом наиболее распространенном
случае.
Максимальная чувствительность аналитической процедуры
достигается тогда, когда обеспечиваются условия для перевода
наибольшей доли ядер А в аналитически активное состояние
[324]. Применительно к активационным методам эта доля, т. е.
отношение jVb/Мдо, в зависимости от ядернофизических констант
и условий облучения меняется согласно соотношению:
_УВ =-________________ [1 _ е' ('- : Ф<1в-фД\с)'обл1 е-фаАс'обл л 1
*А0 (* + Фав-ФаАС) 1 Л •'
где Nb — число ядер В к моменту времени /Обл! ЛГАО— исходное
число ядер А; Оас = СТд + ос. При облучении тепловыми нейтро-
нами, когда для ядер А воз-
можна только одна реакция
(п, у), имеем ос=0. Следует
Рис. 68. Кривые накопления аналитиче-
ского (/—6) и выгорания исходного (7)
изотопов в зависимости от длительности
облучения потоком тепловых нейтронов
плотностью 1014 (/, 2, 3, 6, 7) и
1016 (4, 5) нейтрон/(см2 • сек) при сле-
дующих параметрах:
Од =100 барн (1—7)\ og=10 барн (/—3, 5-7)
и 1000 барн (4); сек (Л» 1 4 (Л,
3 дня (3—5) и 1 год (6’),
практически равен 1,
простую форму, широко известную
[уравнение (2.23)].
а само уравнение
заметить, что оценка отно-
шения tVbAVao при %=
= Фстас—Фов требует осо-
. бого подхода [111].
Согласно уравнению
(11.1), при постоянной плот-
ности потока отношение
jVb/;Vao для аналитического
изотопа сначала растет с
увеличением длительности
облучения, достигает макси-
мального значения и затем
падает из-за выгорания изо-
топа А (рис. 68). Если в
ходе облучения соблюдают-
ся условия /.>Фп.- и
лЗ>Ф(УВ, то пользуются про-
стым правилом, что для до-
стижения близкой к макси-
мальной чувствительности
достаточно облучение в те-
чение 5—10 Tt/2. При этом
выгорание ядер А оказы-
вается ничтожным и послед-
ний член в уравнении (Н.1)
принимает наиболее
в виде уравнения активации
272
Таблица 18
Некоторые предельные параметры активационных определений
Метод активации zw>) I'nmiiJDh ‘ф rulna ‘о о е Фа/?.
г—0 1 -6‘9=’О 01 '/*./. 1 Г 7", / 2=.3 дня (?.=2,7-10—» <<•«-') Z'i/2=1 год (Х=2,2-10—" сек~')
у-Кванты (более 10 Мэв) 10Ы 1 Щ-12 1,5- Ю-li 5,3- I0-s 3,7-10—~ 4,5- 10-е
Заряженные 1014 5 5- Ю-I» 7,2-10—9 2,6-10—6 1,8-10-* 2,3-10—2
частицы Тепловые нейтроны 101* 104 10-е 1,5-10-5 5,3-Ю-3 Х~Фа X Фа
Быстрые нейтроны 10“ о 2-10-и 2,910—12 1,1-10—» 7,4-10~8 2,0-10—*
В табл. 18 суммированы предельные практически используе-
мые параметры для различных случаев активационного анали-
за. Из приведенных данных следует, что в большинстве акти-
вационных определений условия л^>Фодс и 7.^>Фов реализу-
ются и при достижении насыщения имеет место равенство
JVB _ Фад _ Фоа7’-/2
Л\о ~ X ~ ^0.693
Полученное соотношение показывает, что при однократном об-
лучении до насыщения в постоянном потоке доля аналитически
активных ядер пропорциональна произведению параметров ис-
ходного и получающегося изотопов (стдЛ/г). Это свидетельст-
вует о значительной роли сечения и периода полураспада в
формировании чувствительности активационных методов.
Как отмечено выше (§ 4 гл. 4), особое место занимает об-
лучение в мощных потоках тепловых нейтронов изотопов с вы-
соким сечением активации, поскольку здесь для достаточно дол-
гоживущих радиоизотопов уже достигается условие Х^Фодс
или Л^Фов. Тогда оптимальное время облучения нельзя опре-
делять на основе отмеченного выше простого правила и следует
пользоваться уравнением (11.1). Видимо, интересно будет по-
ставить эту проблему в более общем плане, задавшись вопро-
сом, как повлияет на характеристики активационных методов
значительное увеличение плотности потока активирующего из-
лучения.
Основываясь на уравнении (11.1) и используя обычный
прием математического анализа, нетрудно получить выражение
273
для оптимально» длительности облучения /маис, при которой
достигается максимум отношения Мв/ЛДо'-
In
А -у в„Ф
ПдСФ
А, + пвФ - - (ТЛСФ
(Н-2)
Анализ уравнений (11.1) и (11.2) приводит к выводу, что
повышение плотности потока активирующего излучения весьма
благоприятно для аналитических характеристик любого акти-
вационного метода, поскольку
Плотно-сть потока нейтронов,
' нейтрон/(см г- сек) (
Рис. 69. Длительность облучения (/),
необходимая для достижения макси-
мума отношения A'b/jV.-io (2).
К сожалению, практической
перспективы в обозримом будущем не предвидится. Прежде
при этом сокращается дли-
тельность облучения до полу-
чения максимума отношения
/Vb/iVao с одновременным ро-
стом самого отношения.
Этот вывод на конкретном
примере облучения тепловыми
нейтронами для аЛ=103 барн,
ов= 1 барн и 7'1/2= 1 день ил-
люстрируется кривыми рис. 69.
Как видно, в выбранных усло-
виях оптимальная длитель-
ность облучения при плотно-
сти потока 1021 нейтрон/ (см2Х
Хсек) составляет несколько
секунд, а доля аналитически
активных ядер достигает 95%.
реализации столь заманчивой
всего отсутствуют технические возможности превышения до-
ступных в настоящее время плотностей потоков на несколько
порядков величины. С другой стороны, не следует забывать о
пропорциональном росте активности основы, что может создать
значительные затруднения при обработке облученной пробы.
Для облучений быстрыми нейтронами и жесткими квантами
реально ожидать повышения предельных потоков на 1—2 поряд-
ка с соответствующим уменьшением порога определения. Од-
нако даже при более значительном увеличении плотности для
этих методов будут сохраняться условия /.А>Ф<тлс и л2>Фов.
При активации заряженными частицами чувствительность ме-
тода ограничивается не плотностью потока активирующего из-
лучения, а мощным выделением тепла в анализируемой пробе
при облучении. Поэтому здесь дальнейший прогресс скорее бу-
дет связан с совершенствованием способов отвода тепла от
пробы.
Что касается потоков тепловых нейтронов, то в перспективе
ожидается достижение плотностей порядка 2—3 • 10|е нейтрон/
/(см2-сек). Однако это будут, видимо, уникальные установки.
274
Следовательно, для ряда методов активационного анализа
в принципе можно ожидать в будущем некоторого повышения
чувствительности за счет совершенствования источников активи-
рующего излучения, но на этом пути возникают значительные
трудности технического и методического характера. Поэтому
параметры, приведенные в табл. 18, еще длительное время бу-
дут предельными и будут задавать чувствительность активаци-
онных определений на стадии облучения.
Как было показано ранее, наиболее высокое значение отно-
шения Л^в/Л'ло достигается для долгоживущих радиоизотопов,
что соответственно требует длительных облучений. Однако ре-
альная длительность облучения имеет предел, связанный с ус-
ловиями работы источника активирующего излучения и анали-
за. Так, длительные облучения до нескольких недель могут про-
водиться в реакторах. С другой стороны, длительные облучения
более нескольких часов на ускорителях технически трудны и
экономически нерациональны. Как правило, нежелателен и зна-
чительный разрыв между началом и концом анализа. Однако
ограничение длительности облучения соответственно снижает
предельную чувствительность метода.
Стадии активации и измерения обычно разделены некото-
рым интервалом времени /Расп, в течение которого происходит
уменьшение числа ядер аналитического радиоизотопа. Поэтому
необходимо стремиться к минимальной величине задержки по
отношению к периоду полураспада радиоизотопа (/Pa<ii<CT>/2).
Для инструментального варианта минимальная длительность
задержки определяется скоростью транспортировки пробы к из-
мерительной установке и составляет доли секунды. Однако ча-
сто для повышения избирательности инструментального анализа
возникает необходимость специально выдерживать пробу перед
измерением.
В радиохимическом варианте нужны определенные затраты
времени на выполнение операций разделения. Задержка небла-
гоприятным образом сказывается на чувствительности опреде-
ления по короткоживущим изотопам.
На стадии измерения требуется зарегистрировать предельно
возможное число ядер, находящихся в аналитически активном
состоянии. Поэтому для активационных методов длительность
измерения должна определяться по тому же принципу, что и
длительность облучения (/ПЗлг^5—ЮТфг). Однако из-за ряда
факторов практическая длительность измерения не превышает
10 ч.
Тогда, если пренебречь возможной задержкой, доля ядер,
давших вклад в аналитический сигнал за время измерения, бу-
дет равна
(И.З)
NАО ЛАО
275
где Nr* — число ядер, претерпевших распад за время измере-
ния. Эту зависимость для конкретных условий иллюстрируют
кривые рис. 70.
Из приведенных выше соотношений следует, что для опре-
делений в условиях ограниченной длительности облучения и из-
мерения (соответственно не более 6 дней и 10 ч) наибольшую
чувствительность обеспечивают среднеживущие радиоизотопы
(1 ч<:7|/2с;2—3 дня). Прав-
Рис. 70. Доля ядер, которые дают
вклад в аналитический сигнал при
следующих условиях анализа:
Ф—1П14 нейтрон/{см2 сек); <7д=10ав =
= 100 барн; <обл=й днсй;«изм=1 ч (!) и
10 ч (2).
да, при определениях по ко-
роткоживущим изотопам мо-
жет быть использован циклич-
ный анализ, т. е. последова-
тельность из нескольких цик-
лов облучение — измерение с
накоплением числа отсчетов.
Однако такой подход возмо-
жен только на инструменталь-
ной основе, т. е. при сохра-
нении целостности пробы. Ме-
тодика цикличного анализа
более сложна, а число циклов
ограничено (не более 10). По-
скольку цикличный анализ
применяется для определений
только по радиоизотопам с
периодом полураспада менее
60 сек, с его помощью не удается приблизиться к чувствительно-
сти определений по среднеживущим изотопам (конечно, при
других равных параметрах).
На стадии измерения чувствительность определяется возмож-
ностью получения надежной информации при регистрации сла-
бого аналитического сигнала обычно в присутствии помех, при-
чины которых могут быть весьма разнообразны. В случае ра-
диометрических методов минимальное число распадов ядер
аналитического радиоизотопа, которое представляет предел об-
наружения, зависит от параметров детектора излучения и усло-
вий измерения.
Между выходным сигналом (числом отсчетов) и числом рас-
падов, которые совершились за период измерения, существует
связь типа
'«отсчет ксчет'*В ,
где £сч<-т — коэффициент счетности; Nотсчет ЧИСЛО ОТСЧСТОВ ИЗ’
мерительного устройства. В свою очередь коэффициент &счет
представляет собой произведение целого ряда параметров: эф-
фективности детектора, геометрии системы проба—детектор,
поглощения и рассеяния излучения в веществе пробы и вспо-
276
могательных материалах, выхода излучения, химического вы-
хода.
Значительное влияние на предел обнаружения оказывают
фон измерительного устройства и интерференция со стороны
других радиоизотопов. Обстоятельно проблема измерения ма-
лых активностей с использованием различных измерительных
устройств рассмотрена в работах [268, 325]. Применительно к
проблемам активационного анализа оценка достигаемой чув-
ствительности при использовании различных измерительных
устройств сделана А. К. Лаврухиной и др. [326]. Показано, что
в сопоставимых условиях с помощью низкофонового 4 л бета-
счетчика можно обнаружить на два порядка меньшие количе-
ства элементов, чем с помощью сцинтилляционного гамма-спект-
рометра или торцового бета-счетчика. Продолжая это сравне-
ние, видимо, интересно рассмотреть радиохимический вариант
с измерением препаратов на самой чувствительной установке
и инструментальный вариант, основанный на получившем ши-
рокое распространение гамма-спектрометре с Ge (Li)-детекто-
ром.
По-видимому, наибольшую чувствительность регистрации
излучения могут дать 4 л бета-счетчики с экраном антисовпаде-
ний, которые обладают низким фоном (около 1 имп]мин) и вы-
сокой эффективностью (около 0.5—1). Однако применение это-
го детектора требует трудоемкого радиохимического выделения
с высоким фактором очистки и сложной процедуры подготовки
препарата к измерению.
Будем считать порогом количественного определения сиг-
нал, измеряемый с 25%-ной относительной погрешностью. Тог-
да, принимая фон установки постоянным и хорошо известным
из предшествующих опытов, можно рассчитать порог опреде-
ления (см. § 3 гл. 3). Соответствующие оценки для нескольких
элементов при облучении тепловыми нейтронами приведены в
табл. 19.
Полученные оценки показывают превосходство радиохимиче-
ского подхода перед инструментальным в плане чувствительно-
сти определения. Однако нелишне напомнить, что сопоставле-
ние сделано в некоторых идеализированных условиях, которые
предполагают отсутствие помех со стороны каких-либо других
компонентов анализируемой пробы. На практике же приходится
сталкиваться с затруднениями, которые обусловлены либо про-
теканием интерферирующих реакций, либо влиянием излучения
других радиоизотопов на измерение активности аналитического
радиоизотопа.
В отношении первых можно заметить, что их влияние ска-
зывается сильнее в случае применения высокочувствительного,
но неизбирательного конечного метода регистрации. Это легко
понять, поскольку при высокой чувствительности начинает про-
являться действие весьма слабых эффектов (интерферирующие
277
Таблица 19
Порог определения V, Мп, As, Р и Cs с 25 %-ной относительной погрешностью
активационным анализом на тепловых нейтронах*
Эле- мент Радио- изотоп Г,/2 Ежт, барн /:т • Мэв (ВЫХОД, ° о) -VB- распад/г Порог определения, е
4 л бета- счетчик •* * Ge (Ы)-спект- рометр* * *
V 51V 3,8 мин 4,5 1,43 (99) 1,3-1015 1-Ю — 9
Мп 5вМп 2,6 ч 13 0,85 (99) 1,1-104 7- 10-1в 2-10—13
As 76As 1,1 ДНЯ 6,7 0,56 (38) 1 ,7-104 6- Ю-le 3-10-13
Р 3ip 14,5 дня 0,19 — 3,5- low 3- 10-1“ —
Cs 134Cs 2,1 года 30 0,61 (95) 2,7-1015 4-10-и 1-10-11
* Плотность потока 10!4 нейтрон/(см: сек), длительность облучения до 6 дней,
а измерения до 10 ч.
** Эффективность составляет 0.5 и фон 1 имп /мин, длительность задержки I
*** Детектор объемом 43 см\ данные по эффективности и фону при защите из 10 см
свинца взяты из работы [327].
реакции с очень малым сечением и протекающие на элементах
с низкой концентрацией). С другой стороны, интегральное из-
мерение не позволяет выделить излучение аналитического ра-
диоизотопа в присутствии других радиоизотопов того же са-
мого элемента, часть из которых может образоваться по интер-
ферирующим реакциям из соседних элементов. Естественно, что
эти затруднения прежде всего относятся к радиохимическому
варианту.
В свою очередь, чувствительность инструментального вари-
анта подвержена сильному влиянию со стороны других радио-
активных компонентов пробы. Прежде всего это влияние осу-
ществляется через комптоновское распределение. Однако не ме-
нее важным фактором может оказаться и временная разрешаю-
щая способность анализирующей аппаратуры, что ограничивает
суммарную активность пробы. Поэтому часто реальная чувст-
вительность инструментального варианта оказывается на не-
сколько порядков ниже предельно возможной.
Следует заметить, что в большинстве практических приме-
нений для определения малых концентраций рабочая область
активационного анализа находится между 10~6—10',! г
ПО1—10~9%). Только в отдельных случаях достигнут предел
порядка 10 1—10'13 г. Следовательно, приведенные в табл. 19
оценки чувствительности определения указывают на некоторые
еще не реализованные потенциальные возможности метода.
И этот прогресс может осуществляться преимущественно с при-
менением достаточно специфичного и тонкого радиохимического
выделения, которое предшествует длительному измерению на
высокочувствительной установке.
Чувствительность активационного анализа на тепловых ней-
тронах. Наиболее высокую абсолютную и концентрационную
278
чувствительность обеспечивает облучение тепловыми нейтрона-
ми реактора. Это результат благоприятного сочетания ряда
факторов: высоких сечении и потоков, возможности облучения
достаточно больших навесок в течение длительного времени (до
нескольких недель). Некоторое представление о возможностях
метода можно получить из табл. 20 [18], которая показывает,
что в указанных условиях для 52 элементов чувствительность
превышает 10"9 г. При увеличении потока нейтронов и приме-
нении эффективных методов регистрации еще более 10 элемен-
тов могут перейти этот предел.
Таблица 20
Чувствительность активационного анализа на тепловых нейтронах*
Элемент
Eu, Dy
Мп, Со, Rh, Ag, In, Sm, Ho, Lu, Re, Ir, Au
Na, Sc, V, Cu, Ga, As, Br, Kr, Pd, Sb, I, La, Pr, Tb,
Tm, Yb, W, Hg, Th
Al, Cl, Ar, K, Cr, Zn, Ge, Se, Rb, Sr, Y, Nb, Cd, Cs,
Gd, Er, Hf, Ta, Os, U
P, Ni, Mo, Ru, Sn, Те, Xe, Ba, Ce, Nd. Pt, T1
Mg, Si, Ca, Ti, Bl
S, Fe, Zr
Pb
Менее 10 13
10-v2_]0-u
10-и—ю-w
10-10—10-’
10-»—10“’
10-*-10—7
10-1—10-’
10-‘
* ф=] . l()r- нейтрон/(см2 • сек), /O67 = lo0 ч.
* * При критерии 10 расп/сек.
Активационный анализ на тепловых нейтронах неблагоприя-
тен для легких элементов: Н, Не, С, N, О, Ne. Кроме того, еще
два легких элемента (Li и В) из-за очень короткого периода
полураспада аналитического радиоизотопа требуют специальной
методики анализа. Сравнительно низкую чувствительность об-
лучения тепловыми нейтронами имеет еще для шести элементов:
F' S, Ca, Fe, Zr, Pb.
Чувствительность у-активационного анализа. В дополнение
к уже отмеченным факторам чувствительность у-активацион-
ного анализа зависит от максимальной энергии тормозного из-
лучения. Для повышения концентрационной чувствительности
можно применять большие навески. Длительность облучений не
превышает нескольких часов.
Распределение элементов по чувствительности при использо-
вании бетатрона с внутрикамерным облучением показано в
табл. 21 [149]. Эти оценки получены при энергии тормозного
излучения 26,3 Мэв и средней мощности дозы излучения 1,1 X
Х104 р!мин. Поскольку средняя навеска в данных эксперимен-
279
Таблица 21
Чувствительность ^-активационного анализа
Элемент Предел обнаруже- ния, мкг*
С, Р, Си, Zn, Ga, Br, Rb, Ag, Sb, Pr N, O, F, Mg, Cl, K, Sc, Fe, Se, Sr, Zr, Cd, Те, Ba, Sm, Ho, Hf Al, Si, S, Ti, Cr, Ni, Ge, Mo, Ru, Pd, In, Sn, I, Cs, Ce, Nd, Gd, Tb, Er, Ta, Au, Hg Na, As, Y, Nb, Rh, Eu, Tm, W, Re, Os, Ir, Pt, Tl, Pb Са, V, Mn, Yb H, Li, Be, B, La, Lu, Bi Менее 1 1 — 10 10—100 100—1000 Более 1000 Не активируются
* Рассчитан по интенсивности пиков полного поглощения, измеренных с помощью
гамма-спектрометра с кристаллом Nal(Ti) 70X40 л/.u. Время облучения не более 30 мин,
время измерения не более 10 мин.
тальных условиях составляет 2 г, то предельная концентрацион-
ная чувствительность достигает около 10~5%. Применение мощ-
ных ускорителей с более высокой максимальной энергией, уве-
личение длительности облучения и измерения могут повысить
Заря! ядер атомай матрицы
Рис. 71. Чувствительность опреде-
ления кислорода в различных ма-
трицах при облучении ионами 2Не
(10 Мэв) по реакциям:
1 — 16O(3He, p)lsF: 2 — 16О(3Не, anV’O:
3 — ,6О(3Нс. а)"О: 4 — 'ОГНс. 2а)"С.
них. Показано, что для Be, В,
чувствительность на 2—3 поряд-
ка. Это приведет к снижению
предела обнаружения примерно
до 10-7—10-8% [125, 154].
Чувствительность активаци-
онного анализа на заряженных
частицах. При активации заря-
женными частицами появляется
новый фактор, влияющий на кон-
центрационную чувствительность.
Пропорционально увеличению
пробега частиц в веществе про-
бы с ростом энергии и порядко-
вого номера макрокомпонента по-
вышается и чувствительность
анализа по методу толстого слоя
(рис. 71). Поскольку активацион-
ный анализ на заряженных ча-
стицах находит основное приме-
нение для определения малых ко-
личеств легких элементов, то и
оценки возможностей метода вы-
полнены преимущественно для
С, N, О, F облучение разными
типами заряженных частиц дает предельные значения около
10-7-10~8 % [175].
280
§ 2. Точность активационного анализа
Источники погрешности
Как количественный метод активационный анализ обладает
следующими особенностями. Прежде всего — это косвенный ме-
тод, так как в эксперименте измеряется не сама определяемая
величина (масса, концентрация), а активность (скорость счета),
которая связана с ней соответствующим уравнением (см. гл. 3,
§ 2). Измеряемая величина — активность—имеет дискретный
характер и следует специфичной форме распределения, которая
только в определенных условиях переходит в нормальное (см.
гл. 3, § 1). Наконец, активационный анализ в целом относится
к многостадийным методам (см. гл. 3, § 3). Обычно в обяза-
тельные стадии входят отбор пробы, облучение, измерение и
количественный расчет. В радиохимическом варианте добавля-
ется стадия радиохимического разделения и очистки.
Причем все основные стадии анализа состоят из нескольких
операций, каждая из которых — потенциальный источник по-
грешности. Часть этих операций — общая с другими аналити-
ческими методами, другая — специфична для активационного
анализа. К первым, например, относятся такие операции, как
взвешивание, измерение объема раствора и т. д. Как пра-
вило, не эти стадии вносят основную погрешность в конечный
результат, так как относительное среднее квадратическое от-
клонение для них обычно не превышает 2%. Их влияние стано-
вится заметным только в случае очень малых навесок или
объемов.
Для активационного анализа специфичны следующие источ-
ники погрешности:
1) градиент потока нейтронов;
2) возмущение потока нейтронов;
3) интерферирующие ядерные реакции и процессы;
4) неконтролируемые потери активности в радиохимических
операциях за счет неполного обмена, адсорбции, улетучивания
и т. д.;
5) процесс измерения активности (самопоглощение излуче-
ния, воспроизводимость геометрических условий, стабильность
измерительной аппаратуры и т. д.);
6) статистическая погрешность определения активности
(статистический характер измеряемой величины, влияние фона
прибора и мешающих активностей).
Отмеченные возможные источники погрешности в той или
иной степени оказывают влияние на точность получаемых ре-
зультатов. Некоторые из них могут привести к односторонним
и значительным смещениям результатов от истинного значе-
ния. Путь к получению надежных результатов состоит в пра-
вильном планировании хода анализа, тщательном учете и огра-
281
ничении всех возможных источников погрешностей и аккурат-
ном выполнении аналитического определения.
Многие затруднения при получении точных результатов воз-
никают вследствие того, что активационный анализ в целом не
относится к методам, свободным от мешающего влияния со-
става пробы. Это влияние может осуществляться через возму-
щение потока активирующего излучения, интерферирующие ре-
акции, неоднородность состава, самопоглощение регистрируемо-
го излучения, сильную активацию основы и т. д. В каждом кон-
кретном случае нужны тщательный анализ возможных путей,
влияния основы на конечные результаты и разработка соот-
ветствующей методики анализа или определение необходимых
поправок.
Сравнительно легко это можно сделать при анализе проб
с постоянным и простым составом (по основным компонентам),
как, например, при анализе некоторых материалов высокой чи-
стоты. Однако в случае проб переменного состава учет влияния
матрицы может оказаться более трудным делом.
Для анализа источников погрешности и представления ко-
нечных результатов активационных определений применимы
общепринятые методы математической статистики [48, 49]. Ди-
сперсия случайной погрешности косвенных измерений определя-
ется по уравнению
+РЧ2*2 + • • -+РМ2*2, (ил)
\ дхг ) х' X, дх.> / х‘ \ дхп J хп
где s2 — генеральная дисперсия используемой методики; s2 —
генеральная дисперсия отдельной аналитической операции. Ког-
да генеральные дисперсии не известны, их можно заменить
выборочными с соответствующим переходом к распределению
Стьюдента. Выборочную дисперсию данной методики можно
оценить, используя результаты многократных определений.
Вклад отдельных факторов в общую дисперсию исследуют с
помощью дисперсионного анализа.
Точность метода эталонов
В методе эталонов концентрацию определяемого компонента
сх обычно рассчитывают по уравнению
с,;
п.
п
(П-5)
w
где пх и п;1 — скорости счета аналитического радиоизотопа в
пробе и эталоне; гпх— количество элемента в эталоне; щ— мас-
са пробы. Расчет но уравнению (11.5) предполагает, что облу-
чение пробы и эталона, а затем измерение активности прово-
дятся совершенно в идентичных условиях. Однако из-за разли-
282
чий в форме нахождения и в количественном отношении воз-
никают неизбежные различия в условиях облучения, обработки
и измерения.
Как правило, облучение и измерение эталона проводятся в
условиях, исключающих интерференцию со стороны матрицы
и посторонних радиоизотопов. Количество элемента в эталоне
подбирается оптимальным для того, чтобы получить наимень-
шую относительную погрешность при измерении активности.
Различные промежуточные операции сведены к минимуму. Од-
нако для пробы ситуация может быть гораздо более сложной
и для приведения условий анализа пробы к эталонным прихо-
дится вводить целый ряд поправок, определяемых эксперимен-
тально или путем расчета.
В полной форме уравнение для определяемой концентрации
(после сокращения постоянных величин) выглядит следующим
-образом:
(И.6)
Отношения идентичных параметров в уравнении (11.6) можно
заменить поправочными коэффициентами, тогда, введя еще по-
правку на химический выход и мертвое время, получим
Сх = 2^ . Л2 M^?)feHa(.fepacilfexllMA!T , (11.7)
Л, W ° м
где ks — поправка, учитывающая различия в эффективности
регистрации; —отношение эффективных сечений; k&~ от-
ношение средних плотностей потоков; knac — отношение факто-
ров насыщения; kvatn—поправка на распад; /гХПм—поправка
на химический выход; kx*—поправка па мертвое время.
Поскольку в методе эталонов облучение эталона и пробы
проводится одновременно, то kliac равно 1 и может быть исклю-
чено из дальнейшего рассмотрения. Без затруднений и с незна-
чительной погрешностью можно рассчитать fepaPIT. В случае у-
измерений сравнительно просто можно обеспечить условия, ког-
да k* равно 1. Коэффициенты £Х11М и kxK определяются в ходе
эксперимента; нередки случаи, когда они оказывают значитель-
ное влияние на конечный результат.
Наибольшие трудности вызывает определение коэффициен-
тов и йф. Способы оценки этих поправок и их характер
специфичны для каждого метода активационного анализа, по-
этому они подробно были рассмотрены ранее.
283
Согласно уравнениям (11.4) и (11.7), относительное среднее
квадратическое отклонение в определении концентрации равно
1/ ^rn Д- "4~ $rnl ' - ^rkSrk * "4“
х 1 х •> э Е а
+ s;fc_ + «;А.расп ч- s;A.xiiM + Sr\ . (11.8)
Следовательно, для повышения точности результатов активаци-
онного анализа необходимо добиваться уменьшения парциаль-
ных относительных погрешностей и прежде всего тех из них,
которые дают наибольший вклад в величину src^.
Когда коэффициенты k£ и &ф близки к единице и отсутст-
вуют интерферирующие ядерные реакции, метод эталонов дает
наиболее точные результаты.
Точность м&тода мониторов
В этом разделе будет рассмотрен только метод мониториро-
вания по активации какого-либо элемента. Тогда концентрацию
определяемого компонента рассчитывают по уравнению
с = —— . Пмэ Отэ J 9)
«э пмх W
где пмэ и «мх — скорость счета монитора, облученного соответ-
ственно одновременно с эталоном и пробой. Это уравнение
предполагает, что монитор имеет всегда одну и ту же массу.
Условия анализа при этом должны быть идентичными.
Возможны два варианта мониторирования: монитор нахо-
дится вне пробы (внешний монитор); монитором служит один
из компонентов пробы (внутренний монитор). Применение каж-
дого из этих вариантов мониторирования оказывает определен-
ное влияние на точность результатов, поэтому их следует рас-
смотреть раздельно.
Метод внешнего монитора. Уравнение (11.9) можно запи-
сать в форме
где
^э = -м^/пэ. (Н-10>
«э
Величина имеет смысл калибровочной постоянной, которая
может быть определена экспериментально с высокой точностью
в опытах с многократным облучением и измерением эталона и
монитора в принятых условиях анализа. По результатам этих
экспериментов можно оценить среднее квадратическое отклоне-
ние величины &мэ. Очевидно, что погрешность результатов ана-
284
лиза проб с благоприятным составом (матрица не влияет на
результат анализа) будет иметь величину такого же порядка.
Поэтому, если погрешность калибровочной постоянной слишком
велика, необходимо выявить причины, вызывающие рассеяние
результатов.
Если написать для £мэ развернутое выражение, то получим
/, _ ПМЭ Гм Ом Фм v/
_ Ж
П3 тк еэ оэ фэ
(1 — e~?‘M<o6jl) (ц л)
( J _ е~^э^обл) е~^"э^расп
Заменив отношения идентичных факторов соответствующими
коэффициентами, придем к выражению
__^МЭ_
\мэ —
п.
—— kEk' ^ф^рааЛнас-
(П-12)
Поправки на химический выход и разрешающее время здесь
не включены, так как калибровочные опыты обычно проводятся
в условиях, когда они равны единице. Следует напомнить, что
в общем случае определяемый элемент и элемент-монитор не
совпадают друг с другом, а это приводит к соответствующим
различиям ядернофизических параметров. В результате почти
все коэффициенты в уравнении (11.12) отличны от единицы и
зависят от экспериментальных условий.
Коэффициент k3 отражает отличия в эффективности регист-
рации излучения монитора и эталона, а его среднее квадра-
тическое отклонение — воспроизводимость условий измерения.
Измерения монитора и эталона (пробы) могут проводиться с
помощью одной измерительной установки, но тогда необходима
определенная последовательность во времени; измерения могут
выполняться и одновременно, но на разных установках. В по-
следнем случае необходим контроль за стабильностью устано-
вок, что может быть выполнено путем периодического измерения
эталонного радиоактивного препарата. Практика показывает,
что при тщательно отработанной методике измерения можно
достигнуть высокой воспроизводимости величины k, (sr =С 1 %).
В методе мониторирования особого внимания заслуживает
коэффициент ka Поскольку эффективные сечения для монито-
ра и эталона имеют разную зависимость от энергии активи-
рующего излучения, то изменения его энергии или спектраль-
ного распределения могут явиться источником серьезных по-
грешностей. Как правило, метод мониторов может успешно при-
меняться только тогда, когда имеется уверенность в высокой
стабильности этих параметров.
Для обеспечения постоянства коэффициента k® необходимо
либо облучение в строго равномерном потоке излучения, либо
285
жесткая фиксация геометрических условий при облучении в
неравномерном потоке. Конечно, пространственное распределе-
ние активирующего излучения при этом не должно изменяться
в течение всей серии определений.
Вследствие различий постоянных распада метод мониторов
чувствителен к временным факторам—длительности облучения,
распада и измерения. Влияние этих факторов сказывается тем
сильнее, чем больше различие в постоянных распада и чем бо-
лее короткоживущие изотопы используются для определений.
При правильно отработанной методике анализа точная фикса-
ция требуемых интервалов времени в целом не представляет осо-
бых затруднений даже при ручном управлении ходом анализа.
Однако, когда задаваемые интервалы времени сравнительно не-
велики, лучше использовать специальные автоматические системы.
В связи с различиями постоянных распада монитора и опре-
деляемого элемента важным моментом оказывается стабиль-
ность интенсивности активирующего излучения в течение облу-
чения. Наибольшей нестабильностью, если не приняты специаль-
ные меры, отличаются источники, основанные на ускорении за-
ряженных частиц. У таких источников обычно наблюдаются хао-
тические колебания интенсивности излучения около среднего
значения, что часто связано с колебаниями питающего напря-
жения. Реже встречаются другие формы нестабильности. От ам-
плитуды колебаний, временного их распределения и соотноше-
ния постоянных распада будет зависеть интегральная актив-
ность монитора н эталона и, следовательно, коэффициент АПас-
Лучший путь устранения этого источника погрешности—стаби-
лизация интенсивности источника. Если это невозможно, то сле-
дует подобрать монитор с постоянной распада, как можно бли-
же к постоянной распада определяемого элемента.
Однажды определенное значение коэффициента может
быть использовано длительное время, если условия анализа со-
храняются постоянными. Для проверки справедливости послед-
него положения проводят эпизодические определения величины
Амэ. В случае выхода полученной величины за пределы довери-
тельного интервала требуется тщательная проверка всех усло-
вий анализа.
При аналитических определениях все условия калибровоч-
ных опытов по-возможности должны быть воспроизведены. Это
прежде всего относится к объемам препаратов и геометриче-
ским условиям, а также к интервалам времени, энергии активи-
рующего излучения и некоторым другим факторам. В методе
мониторов любые изменения указанных параметров сразу же
скажутся на конечном результате и возникнет необходимость
введения соответствующей поправки или корректировки схемы
анализа. Так же как и в методе эталонов, здесь трудно учесть
влияние матрицы. В остальном действие экспериментальных
факторов такое же, как и в случае калибровочных опытов.
286
Метод внутреннего монитора. Для метода эталонов и внеш-
него монитора значительные трудности представляет точное оп-
ределение коэффициента k&, который зависит от градиента по-
тока, формы пробы и величины возмущения потока активирую-
щего излучения веществом пробы. Чтобы в какой-то степени
обойти это затруднение, был развит метод внутреннего монито-
ра, когда в качестве монитора используется один (или несколь-
ко) компонент анализируемой пробы.
Требования, предъявляемые к внутреннему монитору, такие
же, как и в случае внешнего монитора. Дополнительное усло-
вие состоит в специфичности схемы распада, которая необходи-
ма для того, чтобы излучение монитора можно было измерить
с необходимой точностью в присутствии излучения других ком-
понентов пробы (в инструментальном варианте анализа). При-
менение метода внутреннего монитора возможно в следующих
случаях.
1. В пробе имеется компонент, содержание которого извест-
но или постоянно во всей серии анализируемых проб.
2. При переменном составе определяемых компонентов сум-
марная масса их известна.
3. Проба находится в такой форме, что можно приготовить
гомогенную смесь с монитором, специально добавляемым к
TT Aq
В любом случае для количественных расчетов необходимо
знать отношение удельной активности исследуемого элемента к
активности определенного количества элемента-монитора. Это
может быть достигнуто несколькими путями. Обычный путь со-
стоит в добавлении известного количества исследуемого эле-
мента к одной из проб анализируемого вещества. Этот способ
обычно носит название метода внутреннего эталона. Другой
подход заключается в определении содержания исследуемого
элемента в анализируемом веществе каким-либо независимым
методом. Навеска этого вещества затем используется в качест-
ве эталонной пробы.
. Иногда для калибровочных целей могут быть использованы
некоторые соединения с определенным стехиометрическим отно-
шением исследуемого элемента и элемента-монитора.
В одном из вариантов метода внутреннего монитора методи-
ка анализа сводится к следующим операциям [328]. Отбира-
ются две навески исходного вещества, находящегося в порош-
кообразной форме. К одной из навесок добавляют известное
количество определяемого элемента и смесь тщательно переме-
шивают. Масса эталона должна быть мала по сравнению с на-
веской (m3<^w), а анализируемый материал должен содержать
компонент, который может служить монитором.
После облучения и измерений рассчитывают отношение
удельных скоростей счета монитора в обеих навесках, что
287
дает поправку, учитывающую различия в средних плотностях
потока активирующего излучения
% . %
w' w"
(11.13)
Индекс «'» относится к первой, а «"» — ко второй навеске.
Зная эту поправку и имея данные измерений активности оп-
ределяемого элемента в обеих навесках, можно получить коли-
чественные результаты по следующему соотношению:
пхтэ
Шх ~----------------
t . ш''
I п а —----п
|_ ш'
Нами был разработан другой вариант метода внутреннего
монитора [329]. Если проводится анализ пробы с переменным
содержанием определяемых компонентов, но с постоянной и из-
вестной суммарной массой, то возможен следующий подход.
Предварительно одним из отмеченных выше методов попарно
определяют отношения удельных скоростей счета компонентов,
что дает коэффициенты а,ц. После облучения пробы и измере-
ния скоростей счета компонентов можно написать следующую
систему уравнений:
i = 1, 2, 3, . . ., п,
/=1,2,3, . . ., п, (U.15)
тс mj J 1
1 b
n
V Щ. = mv,
i= i
где n — число компонентов; т, — содержание отдельного ком-
понента; /Пу — общая масса определяемых компонентов. По-
скольку система (11.15) включает п уравнений с п неизвест-
ными, то ее'решение дает содержание отдельных компонентов.
Особенно простой случай — исследование соединений с пере-
п
меппым составом, когда J] int = w представляет собой общую
i=i
массу пробы.
Частный случай этого метода — определение стехиометриче-
ских отношений компонентов. Для примера рассмотрим двух-
компонентную систему. Из калибровочных опытов известно от-
ношение удельных активностей компонентов ai2. Тогда для про-
бы с известным отношением этих компонентов справедливо
п, п* т. Пл
-----: —— — ос12 или —- — —1.
тх т.> " ai2n2
(11.16)
Весовое отношение можно заменить молярным, тогда имеем
иД,
х = .
а12п.2Л'11
(11.17)
288
Точность активационного анализа на тепловых
нейтронах реактора
У каждого метода активационного анализа имеются свои
особенности, которые специфичным образом влияют на сходи-
мость и правильность получаемых результатов. Поэтому обсуж-
дение этих аналитических характеристик должно проводиться
индивидуально для каждого метода. Следует напомнить, что
отдельные источники погрешности основных методов активаци-
онного анализа уже были затронуты ранее в соответствующих
разделах.
Поскольку облучение тепловыми нейтронами реактора — ве-
дущий метод активационного анализа, то исследованию источ-
ников погрешности общей точности этого метода было уделено
большое внимание. Так, Кали и др. [330] критически рассмот-
рели эту проблему в целом, систематизировали источники по-
грешности и оценили их вклад в суммарную погрешность ана-
лиза. Результаты их оценок можно видеть в табл. 22. Конечно,
t
Т а б л и ц а 22
Источники погрешности активационного анализа на тепловых нейтронах
I I
Общие источники погрешности Частные источники погрешности относительное среднее квад- ратическое отклонение, %
Химическая Взвешивание проб (более 1 мг) 0,5
обработка Взвешивание эталонов 0,03—0,1 мг 2
Определение выхода 2
Облучение Экранирование (поправка менее 50%) 4
Депрессия потока 2
Другие факторы, обусловливающие изменение по- 2
тока Абсолютное значение плотности потока тепловых 5
нейтронов Величина кадмиевого отношения 2
Длительность облучения более 1 мин Мало
менее 1 мин 3
Негомогенность потока нейтронов 1
Ядерные Период полураспада 2—10
константы Параметры схем распада (выходы, коэффициенты 2—50
конверсии и т. д.) Сечение активации 5—30
Измерение Калибровка детектора 3
активности Скорость счета (менее 103 имп/сек) 4
Герметические факторы 1
Другие Интерферирующие ядерные реакции Не оцени-
ИСТОЧНИКИ валось
х/а 10 Р- А. Кузнецов
289
приведенные данные следует рассматривать как ориентировоч-
ные и погрешности конкретной методики анализа могут не-
сколько отличаться от указанных величин. Общая оценка дала
следующие величины относительного среднего квадратического
отклонения: 1) для метода эталонов 7%; 2) для абсолютного
метода 10—60%.
Гирарди и др. [331] тщательно изучили возможности абсо-
лютного метода анализа. Было показано, что с уточненными
данными по сечениям реакций и параметрам схем распада при
измерении на сцинтилляционном гамма-спектрометре, прокали-
брованном по эффективности, относительная погрешность со-
ставляет 10—20%. Правда, указанная величина получена толь-
ко для отдельных элементов и в модельных опытах, т. е. без
учета влияния матрицы, при облучении в хорошо термализован-
ном потоке нейтронов совместно с монитором из кобальта
(1%-ный сплав с алюминием). По существу эти результаты
относятся к одному из вариантов метода мониторов.
Критический анализ метода мониторов при облучениях теп-
ловыми нейтронами был выполнен Гирарди и др. [332], а так-
же Н. А. Дубинкой и Л. Л. Пелекисом [333]. Специфичную
проблему в данном случае представляет влияние вариаций
спектра нейтронов на правильность и сходимость получаемых
результатов.
Различия в спектре нейтронов могут возникнуть по несколь-
ким причинам: из-за возмущения нейтронного потока веществом
пробы или при облучении в непосредственной близости от ис-
следуемых проб других объектов, сильно поглощающих нейтро-
ны; при облучении серии проб в разных каналах реактора;
вследствие перестройки активной зоны реактора и т. д. Изме-
нение спектра нейтронов сказывается на величине коэффициен-
та k„ [см. уравнение (11.12)], поскольку определяемый эле-
мент и монитор могут иметь разный ход зависимости эффектив-
ного сечения от соотношения тепловых и резонансных нейтро-
нов в общем потоке. Напомним, что для облучений в реакторе
<T=oTfT + Zpfp, где fT и fp соответственно доля тепловых и резо-
нансных нейтронов в общем потоке.
Если калибровочные опыты сделаны в потоке с одним спект-
ральным составом (fv' = b), а облучение проб проводится в дру-
гих условиях (fy"=£b), то появляется смещение результатов,
величина которого равна отношению
...........................
ka _ т + Vp + Vp _ т Пт P qT ц । । g)
ka ат^т + %^р Otfv + Ipfp , Zp £ + _!pe
/ т i * / p Qp
°T
290
где индекс «'» относится к калибровочным условиям, а —
к условиям анализа (звездочкой отмечены параметры мони-
тора).
Большинство аналитических облучений проводится в пото-
ках, в которых доля резонансных нейтронов лежит в пределах
0,01—0,1. Представление о величине смещения результатов для
изотопов с разной величиной отношения /р/(Тт, которое возникает
при увеличении или уменьшении доли резонансных нейтронов
в два раза, можно получить из табл. 23 [333].
Таблица 23
Величина поправки, возникающая при изменения спектра нейтронов,
для изотопов с разной величиной отношения /Р/ат
Изотоп 7Р ат feA а Изотоп д ат feA а
fP=0.2 fp=0,05 /₽=<>•2 fp=0,05
33Na 0,457 1,06 0,97 ’5 As 8,75 0,70 1,28
55Мп 0,88 1,01 0,99 i”Au 15,9 0,63 1,43
*3Си 0,975 1,00 1,00 127J 25,5 0,58 1,55
38 7п 1,72 094 1,04
* Монитор изготовлен из меди, калибровка сделана при fp=0,I.
Из приведенных данных следует, что в указанных условиях
менее чувствительны к изменению спектра нейтронов те эле-
менты, которые имеют близкую к монитору величину отноше-
ния Ip/fs?. В противном случае смещение результатов может до-
стигнуть значительной величины. Отсюда со всей очевидностью
вытекает вывод о необходимости подбора монитора с величи-
ной отношения /р/(Тт, близкой к аналогичному отношению для
определяемых элементов. Однако при одновременном определе-
нии многих элементов с одним монитором это условие выпол-
нить практически невозможно. Поэтому метод одного монитора
применим главным образом тогда, когда число определяемых
компонентов мало, или поток нейтронов хорошо термализован
(fp 0,01), или же его спектральный состав поддерживается с
достаточно высокой стабильностью.
Чтобы обойти ограничения, свойственные методу одного мо-
нитора, Декорте и др. [334] развили метод трех мониторов.
Идея метода состоит в том, чтобы путем облучения и после-
дующего измерения активности трех элементов с различным от-
ношением получить необходимые данные для расчета сред-
них величин /т и /р. Зная последние значения, а также отно-
»/2 Ю* 291
шения для всех определяемых элементов, по уравнению
(11.18) не представляет труда рассчитать значения коэффици-
ентов k$ для новых условий облучения. В качестве мониторов
в упомянутой работе предложено использовать Со, In и Au.
Применение метода внутреннего монитора позволяет более
точно учесть средний поток в пробе и в определенной степени
контролировать эффект возмущения потока нейтронов веществом
пробы. Можно полагать, что более эффективным средством в
последнем случае может оказаться метод нескольких монито-
ров.
Ряд исследователей [335—336] проводили активационный
анализ некоторых эталонных проб с хорошо установленным
содержанием определяемых компонентов, в результате чего по-
казано согласие получаемых результатов с сертификатными в
пределах относительной погрешности 5—10%. Причем в каче-
стве эталонных были использованы пробы разного состава:
сплавы, стали, некоторые породы и т. д.
Интересные результаты были получены при сравнении пра-
вильности и сходимости некоторых аналитических методов, ис-
пользуемых для определения малых концентраций элементов
[337]. Эти исследования были проведены при участии лабо-
раторий нескольких стран и таким образом, чтобы по возмож-
ности воспроизвести условия рядовых анализов. Статистическая
обработка полученных результатов позволила оценить величину
относительной погрешности отдельного определения (при
95 %-ном доверительном интервале) для каждого из исследован-
ных методов: а) эмиссионной спектрографии (40%); б) поляро-
графии (25%); в) нейтронного активационного анализа (20%);
г) абсорбционной спектрофотометрии (10%).
Хотя эти данные в общем ориентировочные, так как они по-
лучены без учета влияния матрицы, тем не менее ясно, что ак-
тивационный анализ на тепловых нейтронах имеет хорошую
точность по сравнению с другими методами анализа малых кон-
центраций. Если при этом учесть, что этот метод для многих
элементов обладает высокой чувствительностью и не нуждается
в поправке на холостой опыт, то становится очевидной принад-
лежность активационного анализа на тепловых нейтронах к наи-
более надежным и лучшим методам определения малых коли-
честв элементов.
Точность активационного анализа на быстрых нейтронах
Эта проблема рассматривается на примере определения кис-
лорода в различных объектах с использованием нейтронного ге-
нератора. Ввиду особого практического значения анализу кис-
лорода этим методом было уделено большое внимание. Обстоя-
тельно исследованы источники погрешности и разработаны ме-
тоды и системы для получения надежных результатов. Основ-
292
ные результаты этих исследований во многом типичны и со-
храняют свое значение для других случаев применения актива-
ционного анализа на быстрых нейтронах.
Определение кислорода проводится по реакции 16O(n, p)I6N
(£пор = 9.6 Мэв). Радиоизотоп 16N (Г1/г=7,4 сек) имеет очень
жесткое у-излучение (6,1 и 7,1 Мэв). Ввиду особой специфично-
сти схемы распада 16N другие радиоизотопы, образующиеся при
облучении быстрыми нейтронами большинства элементов пе-
риодической системы, практически не мешают определению
кислорода.
К неблагоприятным характеристикам нейтронных генерато-
ров, которые несколько затрудняют проведение аналитических
определений с их помощью, относятся следующие: 1) высокий
градиент потока нейтронов во всех направлениях; 2) неста-
бильность потока нейтронов от одного облучения к другому и
даже в ходе одного облучения; 3) умеренная плотность потока
быстрых нейтронов. В силу действия последнего фактора для
достижения достаточно высокой концентрационной чувствитель-
ности (около 10_3% и менее) приходится облучать пробы боль-
шой массы (до 100 г). Такие пробы имеют значительные разме-
ры и вследствие градиента потока активируются неравномерно.
Отмеченные особенности нейтронного генератора как источника
нейтронов претъявляют особые требования к воспроизводимости
геометрических условий при облучении и измерении, а также
надежности методов контроля за интенсивностью нейтронного
потока.
В рассматриваемом случае важный фактор представляет ко-
роткий период полураспада 16N, который приводит к необходи-
мости использовать короткие интервалы времени на всех ста-
диях анализа. Поэтому успешное выполнение аналитических
определений оказывается возможным только с помощью авто-
матизированной системы с быстрой пневмопочтой и управляю-
щим устройством, которое точно выдерживает временной режим
анализа.
Предложены два основных типа систем: одноканальные, поз-
воляющие транспортировку только пробы (эталона), и двухка-
нальные, которые приспособлены к одновременному облуче-
нию и транспортировке пробы и эталона (монитора). Посколь-
ку в применении каждой из этих систем имеется своя специ-
фика, они будут рассмотрены раздельно.
В одноканальной системе облучение пробы и эталона неиз-
бежно должно производиться в разных опытах. Поэтому для
контроля за интенсивностью потока нейтронов прибегают к раз-
личным методам мониторирования. Возможны следующие вари-
анты: измерение активности кислородсодержащего препарата,
расположенного неподвижно вблизи мишени нейтронного гене-
ратора; применение органических сцинтилляторов для регистра-
10 Р. А, Кузнецов
293
ции быстрых нейтронов по протонам отдачи; использование
счетчика тепловых нейтронов с замедлителем; измерение актив-
ности воды, охлаждающей мишень [338].
При потоке нейтронов, постоянном в течение облучения, пер-
вые три метода дают совпадающие результаты, а последний,
если не приняты специальные меры для стабилизации скорости
течения воды, значительно увеличивает разброс результатов.
Методы простого суммирования числа отсчетов оказываются не-
удовлетворительными из-за нарушения пропорциональности
между измеренной дозой нейтронов и наведенной активностью
16N. В случае переменной интенсивности нейтронного потока
для регистрации импульсов от счетчика нейтронов необходимо
использовать интегрирующую цепочку с постоянной времени,
равной среднему времени жизни радиоизотопа 16N (10,6 сек)
[339].
Правильно отработанная методика мониторирования дает
возможность получить простыми средствами удовлетворитель-
ную точность; как правило, относительная погрешность при
этом лежит в пределах 5—10% при анализах самых разнооб-
разных проб. Повышение точности определений, когда статисти-
ческая погрешность измерения активности мала, возможно
только при более тщательной отработке методики контроля за
условиями анализа. Как показано Андерсом и Бриденом [339],
а также Моттом и Оранджем [340], основную ответственность
за рассеяние результатов несут следующие факторы: 1) недо-
статочно четкая воспроизводимость положения пробы при облу-
чении и измерении; 2) неоднородность проб; 3) локальные ко-
лебания потока нейтронов. Последний фактор обусловлен не-
стабильностью работы ионного источника, неравномерностью
распределения и «выгорания» трития по площади мишени, сме-
щениями пучка ионов и другими причинами.
Вращение пробы при облучении и измерении, а также тща-
тельная упаковка проб перед облучением уменьшают действие
отмеченных факторов, в результате чего получена относитель-
ная погрешность около 2,0% [339]. При анализе небольших
проб (0,8—1 г) хорошая точность была достигнута при исполь-
зовании в качестве монитора воды, охлаждающей мишень. Ско-
рость пропускания воды стабилизирована с погрешностью 0,5%
[34!].
Удовлетворительные результаты при сравнительно простой
методике дает метод внутреннего монитора. Так, при опреде-
лении кислорода в магнии, нержавеющей стали и титане ме-
тодом внутреннего монитора была получена относительная по-
грешность, равная 3—5% [342]. Основной источник рассеяния
результатов—большое различие в периодах полураспада мо-
нитора и изотопа 16N.
Достаточно точные результаты можно получить и методом
внешнего монитора, если последний тесно связан с условиями
294
облучения пробы. Так, Картуннен и Гарднер [343] как мони-
тор использовали муфту из кислородсодержащего вещества
(плексиглас), которая надевается на полиэтиленовую ампулу
с пробой. Длина муфты достаточна, чтобы полностью пере-
крыть пробу. В ходе облучения проба вращается, что позволяет
облучать пробу и монитор в хорошо усредненном потоке нейт-
ронов. После облучения муфта вручную снимается с ампулы и
они одновременно измеряются на двух сцинтилляционных спект-
рометрах с вращающимися держателями. По этой методике точ-
ная фиксация интервалов времени не требуется. Получена от-
носительная погрешность порядка 1%.
И. П. Лисовский и Л. А. Смахтин [344] для мониторирова-
ния использовали активацию ампулы из нержавеющей стали,
в которой облучается проба. Мониторирование основано на ре-
акции 56Fe(«, р)56Мп. Получены результаты, аналогичные пред-
шествующим.
Заметно повысить точность определений позволяет метод,
многократных облучений. Применение для анализа одной про-
бы нескольких циклов облучение — измерение (обычно от 4 до
10 циклов) не только уменьшает статистическую погрешность
измерения активности leN, но и приводит к усреднению резуль-
тата за счет случайных изменений положения пробы при облу-
чении и измерении от цикла к циклу [345, 346].
Повышение точности результатов возможно также с приме-
нением двухканальной системы. Для анализа кислорода пред-
ложено два основных типа таких систем: без вращения и с
вращением проб. Последние наиболее сложны по устройству.
Наличие двух транспортирующих каналов позволяет проводить
одновременное облучение, а затем транспортировку и измере-
ние пробы и эталона (монитора).
В системах без вращения одновременно облучаются проба
и кислородсодержащий монитор. Проба располагается ближе
к мишени нейтронного генератора, а монитор—непосредствен-
но за пей. Обычно проба и монитор имеют дискообразную
форму. В такой системе исключаются погрешности за счет об-
щей нестабильности потока нейтронов в течение облучения и
частично в объеме зоны облучения. По данным работы [346]
относительная погрешность, получаемая с такими системами,
составляет около 2%.
Практически полностью исключить все источники случайных
погрешностей, кроме статистической погрешности в измерении
активности, удалось с помощью двухканальных вращающихся
систем. Эти системы позволяют одновременно облучать пробы и
эталон в хорошо усредненном потоке нейтронов. Так, авторами
работы [340] этим методом при измерениях пробы и эталона
на сцинтилляционных спектрометрах, имеющих кристаллы
Nal(Tl) с колодцем, и при использовании идентичных проб и
эталонов и некоторой дефокусировки пучка ионов нейтронного
10* 295
генератора была получена относительная погрешность порядка
0,33%. Полученная точность почти полностью определяется ста-
тистической погрешностью в измерении активности 16N.
Сечения взаимодействия быстрых нейтронов с ядрами ато-
мов имеют много меньшие величины, чем в случае тепловых
нейтронов. Поэтому эффект ослабления потока быстрых нейтро-
нов веществом пробы обычно не оказывает заметного влияния
на получаемые результаты. Однако поскольку для достижения
высокой концентрационной чувствительности приходится при-
бегать, к достаточно большим пробам, возникает опасение,
что этот факт начнет оказывать влияние на конечные резуль-
таты.
Ослабление потока быстрых нейтронов обусловлено тремя
процессами: поглощением, упругим и неупругим рассеянием.
Первые два процесса выводят нейтроны из пучка, а в резуль-
тате неупругого рассеяния нейтрон теряет значительную долю
первоначальной энергии и уже не может вызвать аналитиче-
скую ядерную реакцию.
Наиболее простой способ ограничения влияния эффекта эк-
ранирования на результаты определений состоит в использова-
нии эталонов того же состава, массы и формы, что и анализи-
руемые пробы. Однако этот способ не всегда оказывается воз-
можным, и часто в качестве эталонов находят применение раз-
личные вещества, в которых содержание кислорода хорошо из-
вестно. Обычно это соединения с точно определенным стехио-
метрическим составом. Например, как материал для изготовле-
ния эталонов широко используются различные органические
соединения: полиметилметакрилат [344, 345, 347], смесь стеа-
риновой кислоты с парафином [348] и т. д. Эталонам придают
такую же форму и размеры, что и у анализируемых проб. Это
позволяет избежать поправки на градиент потока нейтронов,
которая может быть весьма существенной.
Проведенные оценки показывают, что при анализе веществ с
малой плотностью различием в степени ослабления потока
нейтронов и у-излучения радиоизотопа I6N веществом пробы
можно пренебречь. Однако при определении кислорода в ме-
таллах с высокой плотностью, особенно в случае больших на-
весок, требуется введение соответствующих поправок.
Расчетный способ оценки поправки на ослабление потока
быстрых нейтронов предложен в работах [349, 350]. Отмечен-
ные особенности методики анализа делают расчетные способы
очень сложными, поэтому приходится прибегать к некоторым
упрощениям и помощи вычислительных машин. Результаты
оценок коэффициентов экранирования быстрых нейтронов и са-
мопоглощения у-излучения радиоизотопа l6N показаны в
табл. 24.
Из приведенных данных видно, что величина даже при
довольно больших пробах мало отличается от единицы и слабо
296
зависит от порядкового номера элемента-матрицы. Ослабление
у-излучения веществом пробы оказывается более значительным.
Для корректного введения поправки на последний эффект пред-
ложены методы расчета [349, 350].
Таблица 24
Коэффициенты /б и у для проб разного состава
Материал пробы if, Jv Материал пробы if, /у
Железо* 0,926 0,885 Вольфрам* 0,918 0,629
Медь* 0,934 0,869 Алюминий** 0,95 —
Молибден* 0,943 0,826 Плексиглас** 0,93 —
* Толщина пробы 1 см.
** Толщина пробы 0,3 см.
Для кислорода возможны помехи со стороны отдельных эле-
ментов. Основная интерферирующая реакция — 19F(n, a)16N.
Экспериментально определенный коэффициент интерференции
при энергии нейтронов 14 Мэв равен 0,42 [351]. Для определе-
ния поправки на фтор можно использовать другие ядерные ре
акции. Однако во многих объектах, представляющих особый
практический интерес, содержание фтора ничтожно, и поэтому
помехи с его стороны отсутствуют. Влияние реакции
1!В(/2, р)!1Ве, которая дает радиоизотоп иВе (Т =13,5 сек)
с жестким у-излучением (£v=6,81 и 7,99 Мэв), много слабее.
При определении низких содержаний кислорода (<10~3%)
необходимо учитывать наличие на поверхности проб окисного
слоя, толщина которого зависит от природы материала и спо-
соба подготовки его к анализу [349]. Так, поверхностный слой
пробы стали или алюминия может содержать соответственно
до 5 и 2,5 мкг'см2 кислорода.
Проблема поверхностных загрязнений тесно связана со спо-
собом подготовки пробы к облучению. Здесь возможны два
варианта: облучение в упаковке из подходящего материала
или без упаковки. В последнем случае поверхность легко мо-
жет быть загрязнена при хранении, транспортировке и других
промежуточных операциях. Нельзя также полностью исключить
возможность помех со стороны процесса окисления поверхности
пробы или адсорбции влаги.
Упаковка в герметичную капсулу предохраняет пробу от
возможных внешних загрязнений, позволяет упаковывать в
инертной атмосфере, допускает анализ порошкообразных ве-
ществ и т. д. Однако материал упаковки должен удовлетворять
ряду требований: 1) практически не содержать примеси кисло-
297
рода, 2) быть инертным, 3) обладать достаточной механической
прочностью.
Особенно трудно удовлетворить первое требование. Поэтому
при облучении в ампулах требуется поправка на холостой опыт,
но при этом нужно убедиться, что эта поправка постоянна. Ра-
дикальный способ состоит в удалении упаковки перед измере-
нием, но это требует 3—5 сек, а иногда исключает возможность
повторного анализа.
Уменьшить величину окисной пленки позволяют такие прие-
мы, как подготовка поверхности пробы к анализу в бескисло-
родной среде и замена рабочего газа в пневмопочте азотом или
инертным газом. Эффективным средством может быть травле-
ние поверхности облученной пробы химическим способом. При
анализе алюминия для удаления поверхностного слоя достаточ-
ной толщины применялась обработка раствором NaOH
(160 г/л) при температуре 90—95° С в течение всего 5 сек [352J.
При попытке достигнуть предельной чувствительности опре-
деления кислорода в некоторых матрицах сталкиваются с силь-
ным увеличением погрешности, обусловленной мертвым време-
нем и перегрузкой регистрирующей аппаратуры. Например,
при интенсивности потока нейтронов 5-109 нейтрон'сек облуче-
ние алюминия и меди дает интегральную скорость счета более
105 имп!сек [349]. При такой загрузке происходит смещение
энергетической шкалы измерительной установки, что приводит
к изменению эффективности регистрации у-излучения 16N.
Точность у-активационного анализа
По точности получаемых результатов и источникам погреш-
ности у-активационный анализ имеет много общего с активаци-
онным анализом на быстрых нейтронах. Это обстоятельство в
целом обусловлено некоторыми общими свойствами активирую-
щего излучения в обоих случаях: высокой проникающей спо-
собностью, значительным градиентом плотности и, как правило,
нестабильностью интенсивности. Основным способом получения
количественных результатов также является метод мониторов.
Учитывая эту общность, видимо, нет необходимости останавли-
ваться на всех возможных источниках погрешности и можно
ограничиться рассмотрением только тех из них, которые пред-
ставляются специфичными для данного метода.
В отличие от активационного анализа на быстрых нейтро-
нах, где энергия начальных нейтронов зависит от используемой
ядерной реакции и практически постоянна, источники тормоз-
ного излучения позволяют менять энергию в широких пределах.
Поэтому возникает проблема точной установки желаемой энер-
гии, поскольку в ходе экспериментов часто приходится прово-
дить облучения при разных уровнях предельной энергии. Кро-
298
ме того, заданная энергия в ходе облучений должна поддержи-
ваться постоянной. Так как в определенных энергетических ин-
тервалах ход зависимости удельной активности элементов от
энергии тормозного излучения резко различен, то в методе мо-
ниторирования это может привести к ухудшению точности опре-
делений.
Наиболее простой способ исключения этого источника по-
грешности состоит в применении в качестве монитора опреде-
ляемого элемента или элемента, имеющего аналогичный ход
интегральной кривой возбуждения. Возможно также использо-
вание различных систем стабилизации энергии излучения.
Предварительные оценки показывают, что при стабилизации
максимальной энергии тормозного излучения в пределах
±15 кэв этот фактор не оказывает влияния на точность полу-
чаемых результатов.
Как уже отмечалось выше, жесткое тормозное излучение об-
ладает высокой проникающей способностью, поэтому при облу-
чении небольших проб эффект ослабления практически можно
не учитывать. Однако для достижения максимальной концен-
трационной чувствительности иногда приходится прибегать к
весьма большим навескам (до 100—200 г), но тогда эффект
экранирования становится заметным.
Известно, что ослабление моноэнергетического у-излучения
веществом подчиняется закону
Фх = Фпе~ИА',
(11.19)
где Ф(| и ФЛ-—исходная плотность потока у-квантов и плот-
ность на глубине х см; р — линейный коэффициент поглощения,
слг± Тогда уравнение активации (5.3) с учетом поглощения
тормозного излучения в случае облучения однородной пробы
параллельным пучком принимает вид:
6,02 lO-^SpR, 1 £м?кс
Л =------—( Ф0(Е, ENakX)e-^*dEdx, (Ц.20)
0 £пор
где ц(£)—функция изменения линейного коэффициента погло-
щения с энергией у-излучения; <$ — площадь пробы; р — плот-
ность вещества пробы. Аналитическое решение приведенного
выражения невозможно, и расчет может быть выполнен только
численным интегрированием.
Однако, как показал Лутц [353], в условиях резонансного
хода функции возбуждения функцию р(£) с достаточной для
практических целей точностью можно заменить постоянной ве-
личиной ц(£т), где Ет— резонансная энергия рассматривае-
мой ядерной реакции. Тогда поправочный коэффициент ослаб-
299
ления, определяемый как отношение удельной активности дан-
ного элемента в пробах его удельной активности в отсутствие
поглощения, равен
(1_е-^(£т)г)
/торм- (И.21>
И (Am) L
где I — толщина пробы, см; ц(Ет)—линейное сечение погло-
щения вещества пробы при энергии Ет, см~*. Однако теорети-
ческая оценка ослабления в реальных условиях значительно
затрудняется из-за градиента потока активирующего излучения
и угловой зависимости спектрального распределения
'1>о(Д f.MaKc). Экспериментально поправку на ослабление тор-
мозного излучения можно определить с помощью двух монито-
ров, устанавливаемых до и после пробы.
В определениях при умеренных энергиях тормозного излу-
чения (до 25 Мэв) очень малую роль играют интерферирую-
щие реакции, поскольку выход ядерных реакций с вылетом за-
ряженных частиц низок. Однако при увеличении энергии до
35—60 Мэв роль таких реакций возрастает. Специфичным ис-
точником помех может оказаться поток нейтронов, которые
всегда сопровождают жесткое тормозное излучение. Однако,
поскольку основное свое применение у-активациоиный, анализ
пока находит в области концентраций выше 10-5%, интерфери-
рующие реакции редко играют существенную роль.
Вообще говоря, анализу общей точности у-активационного
анализа уделено мало внимания. Имеются только данные по
оценке сходимости и правильности метода применительно к не-
которым конкретным системам. В большинстве случаев относи-
тельное стандартное отклонение лежит в пределах 54-20%.
Анализ источников погрешности при использовании бетатро-
на с внутрикамерным облучением был сделан Р. А. Кузнецо-
вым [136]. Для этого случая характерны малые размеры зоны
облучения, сильный градиент плотности и значительная неста-
бильность излучения. Поэтому единственную возможность по-
лучения надежных количественных результатов в таких усло-
виях предоставляет метод мониторов.
Указанные особенности пучка излучения предъявляют жест-
кие требования к анализируемым пробам. Очевидной является
необходимость выдерживать с высокой точностью размеры и
форму облучаемых проб. Более того в статических условиях
можно анализировать только однородные пробы, иначе полу-
ченные результаты не будут характеризовать среднее содержа-
ние исследуемого компонента в пробе. Неоднородные пробы
перед облучением необходимо измельчить и гомогенизи-
ровать.
Порошкообразные пробы упаковываются в небольшие кю-
веты, выточенные из алюминия. Эти кюветы вводятся в специ-
альном держателе в систему для внутрикамерных облучений.
300
Все устройство сконструировано таким образом, чтобы обеспе-
чить хорошую воспроизводимость положения пробы и монитора
в зоне облучения.
Перечень исследованных факторов, методика исследования
и полученные результаты приведены в табл. 25. При выполне-
нии каждого опыта проводилось не менее 30 определений. Ана-
лиз полученных данных показывает, что в данном случае основ-
ным фактором, определяющим точность результатов, является
операция упаковки пробы.
Таблица 25
Влияние различных факторов на точность определений
Номер опыта Изучаемый фактор Методика исследования Относи- тельное стандарт- ное откло- нение, %
1 Стабильность установки и вое- Периодические измерения 0,5
2 производимое^ геометрических условий при измерении Воспроизводимость геометри- активности эталонного пре- парата Облучение двух медных 1,1
3 ческих условий при облучении и погрешность мониторирования Влияние нестабильности энец- мониторов Облучение медного и по- 1.3
4 гии излучения Влияние нестабильности ин- лиэтиленового мониторов Облучение буры с медным 1,6
5 тенсивности излучения Влияние операции упаковки пробы в контейнер монитором. Ряд навесок, ди- сперсионный анализ 2,6
На результаты определений могут влиять величина навески
и состав пробы. Для изучения этих факторов были приготовле-
ны смеси из сахарозы и окиси в разных соотношениях. Из каж-
дой смеси отбирали по две пробы, которые упаковывали в кю-
веты, и проводили определение удельной активности углерода
при облучении с медным монитором. Оказалось, что при значи-
тельном изменении величины навески (244—728 мг) и состава
пробы (содержание свинца меняется в пределах 0—80%)
удельная активность не показывает значимого отклонения от
среднего значения.
Точность активационного анализа на заряженных
частицах
Основные особенности этого метода были рассмотрены в
гл. 6, здесь же будет сделано несколько дополнительных заме-
чаний, касающихся точности метода при определении крайне
низких содержаний элементов по методу толстого слоя.
301
Небольшая глубина активируемого слоя обостряет пробле-
му поверхностных загрязнений, особенно это относится к опре-
делениям кислорода. Поэтому для получения надежных дан-
ных о содержании определяемой примеси во внутренних ча-
стях пробы необходимо прибегать к травлению. Эксперимен-
тально показано, что толщина удаляемого слоя должна быть
не менее 25 лг/ои [354]. Источники загрязнений поверхности проб
могут быть самыми различными. Причем посторонние примеси
могут попасть на поверхность даже в ходе облучения при сорб-
ции паров масла от паромасляных насосов или через процесс
ядер отдачи с поверхности мониторов или покрывающих
фольг [355].
Для негомогенных проб представительность результатов
активационного анализа на заряженных частицах будет весьма
низкой, поскольку активация пробы неравномерна. Это прежде
всего связано с изменением величины сечения по мере проник-
новения пучка вглубь пробы, возможен также градиент в рас-
пределении заряженных частиц по сечению пучка [355].
В методах анализа с помощью заряженных частиц важ-
ную роль играют интерферирующие реакции, которые могут
давать аналитический радиоизотоп даже на ядрах элемен-
тов, отстоящих на несколько зарядовых единиц от опреде-
ляемого.
Более наглядно оценить проблемы, встречающиеся в ходе
активационного анализа на заряженных частицах, можно на
конкретном примере, скажем, определения малых содержаний
кислорода при облучении ядрами 3Не. Именно этот метод рас-
сматривается как один из наиболее перспективных путей реше-
ния этой важной аналитической задачи [170, 177, 355]. Перечень
аналитических и интерферирующих реакций приведен в
табл. 26 [177].
Таблица 26
Аналитические и интерферирующие ядерные реакции
при определении кислорода путем облучения ядрами 3Не
Аналитические реакции Интерферирующие реакции Пороговая энергия, Abe
«ОрНе, р) 18F 19F(3He, ос) 18F ЭКЗО
16О (3Не, n)'“Ne—i8F 20Ne(3He, ар) 18F 3,1
20Ne (3Не, а n)38Ne t38F 9,1
23Na (3Не, 2а) ,8F 0,4
2ТА1 («Не, За) '“F 11,6
29Si («Не, HX)i«F 11,«
3»Si («Не, i«N) 18F 11,5
302
Как видно, при определении кислорода возможна помеха
со стороны ряда элементов. Однако степень помех различна
п зависит от аналитической ситуации. Например, дискримина-
цию многих помех можно осуществить, если выбрать энергию
активирующего излучения ниже порога интерферирующей ре-
акции. Однако таким способом нельзя избавиться от помех со
стороны фтора, так как реакция 19F(3He, ct)18F экзоэнергетиче-
ская.
Следовательно, определение кислорода может быть осуще-
ствлено либо при содержании фтора ниже некоторого предела
(этот случай часто встречается на практике), либо при введе-
нии поправки на его присутствие, но это возможно в ограничен-
ном интервале концентраций фтора и требует независимого
определения его содержания. При значительной помехе со
стороны фтора можно попытаться разрешить проблему при
переходе к другому методу активации, при котором эта помеха
уже не существенна.
Следует особо остановиться па последних двух реакциях,
поскольку это несколько необычный источник помех. Действи-
тельно, интерферирующие реакции со столь значительным изме-
нением заряда ядра представляют довольно редкую проблему
в других методах активационного анализа. Исключение состав-
ляет только реакция деления. По существу пассматпиняемые
реакции есть реакции деления легких ядер, так как они приво-
дят к двум ядрам примерно равной массы. Сечение такой реак-
ции мало, по помеха может быть значительной, если требуется
определение малых количеств кислорода в кремнии [177].
К неблагоприятным факторам активационного анализа на
ядрах 3Не следует отнести и то обстоятельство, что важнейшие
продукты активации — чистые позитронные излучатели. Это
исключает возможность спектрометрического инструментально-
го анализа и требует применения радиохимического выделения
lhF или анализа кривых распада. Достоинства и нетостатки
каждого из этих вариантов были обсуждены ранее. Дополни-
тельные затруднения для инструментального анализа может
представить сильная активация основы. Эта помеха наиболее
важна при анализе матриц из легких элементов, так как в слу-
чае тяжелых элементов можно установить энергию активирую-
щего излучения ниже эффективного барьера.
В заключение можно отметить, что имеющиеся многочис-
ленные оценки точности предлагаемых методик дают значения
относительной погрешности, лежащие в пределах 5—25%. По-
грешность уменьшается до 2—5% при определении сравни-
тельно высоких содержаний. При тщательном планировании
аналитической методики можно обеспечивать правильность
получаемых результатов. Это подтверждается сопоставлением
результатов активационных определений с данными других ме-
тодов. Альтернативный путь для проверки правильности ме-
303
тода состоит в применении для анализа перекрестных реак-
ций [355].
Сущность метода заключается в определении одного и того
же компонента в исследуемой пробе путем активационного ана-
лиза на разных заряженных частицах. Поскольку при этом ме-
няется вклад интерферирующих реакций, совпадение результа-
тов этих определений будет свидетельствовать об отсутствии
помех. Однако при расхождении результатов никаких опреде-
ленных выводов без дополнительных исследований сделать
нельзя.
§ 3. Избирательность активационного анализа
Термин «избирательность» призван характеризовать спо-
собность данного метода к точному определению нужного эле-
мента в исследуемой пробе.
Количественно избирательность инструментального метода
можно выразить коэффициентом избирательности, который ра-
вен отношению интенсивностей сигналов, даваемых в принятых
условиях анализа интерферирующим и определяемым элемен-
тами при равном их содержании,
L _ (1 - е~?'“М (1 - е'"Х"М е^расп
nsx М71аг0Л*71 (1 — е~Л*'°бл) (1 — е~''х*ям) е~л?расп
где и nsx — удельное число отсчетов интерферирующего и
определяемого элементов. По смыслу коэффициент избиратель-
ности аналогичен коэффициенту интерференции, но в противо-
положность последнему имеет более общее значение, так как
учитывает все возможные каналы помех от интерферирующего
элемента.
Если используется радиохимический вариант, то коэффи-
циент избирательности будет равен
Lt
Lt
ёоч ’
(11.23}
где gO4 — фактор очистки. В уравнении (11.23) величина L,
включает избирательность метода конечного определения. Если
используется какой-либо интегральный метод регистрации, то
избирательность задается процедурой химического разделения,
а также отношением Оц/ол.
Для проб сложного состава, когда возможна интерферен-
ция от многих элементов, уровень помех при определении дан-
304
ного элемента будет равен
Q __ _ ^-LfCi (JI 24^
nsxmx mx cx
где tnt — весовое количество, a c, — концентрация i-го компо-
нента. Таким образом, уровень помех показывает вклад от ин-
терферирующих элементов в долях концентрации определяемого
элемента в пробе. Иными словами, уровень помех в данной ана-
литической ситуации показывает кажущуюся концентрационную
добавку, возникающую в результате влияния интерферирую-
щих элементов на конечный результат, т. е. представляет собой
величину систематической погрешности в определении концен-
трации с.т.
Следовательно, для правильного определения сх, когда ве-
личина 0п оказывается большой, требуется либо внести изме-
нения в методику анализа, чтобы снизить уровень помех ниже
определенного уровня (Оп<СО,1), либо прибегнуть к введению
соответствующей поправки. Однако возможности последнего
способа ограничены, так как возникает необходимость в оцен-
ке вклада от интерферирующих элементов, что далеко не всег-
да представляет простую проблему. К тому же введение по-
правки неизбежно увеличивает погрешность величины сх, так
что эга операция nacio теряет смысл при Gn> 10.
Рассмотрим роль основных параметров в формировании об-
щей избирательности метода, для чего обратимся к уравнению
(11.22). Влияние атомного веса и содержания активирующе-
гося изотопа очевидно и не требует пояснений.
Большое значение для коэффициента избирательности
имеет отношение сечений активации интерферирующего и опре-
деляемого элементов. Для получения высокой избирательности
желательно подобрать метод активации и энергию активирую-
щего излучения, чтобы обеспечить минимум отношения оп/оЛ-.
Однако методы активации редко позволяют достигнуть решения
проблемы только таким путем. Довольно часты такие ситуа-
ции, когда, наоборот, отношение оп/ох оказывается неблаго-
приятным.
Определенная возможность для достижения необходимой
избирательности возникает при различиях в периодах полурас-
пада интерферирующего и аналитического радиоизотопов. Ес-
ли Тх>Тп, то необходимо длительное облучение и выдержива-
ние пробы перед измерением. В противном случае время облу-
чения должно быть коротким, длительность задержки мини-
мальной. Первый случай предпочтительнее с точки зрения ос-
новных аналитических характеристик (чувствительности и точ-
ности). В целом подбор временного режима эффективен толь-
ко при значительных различиях в периодах полураспада.
Наконец, фундаментальное значение для избирательности
имеет метод регистрации, который определяет отношение еи/ег,
305
т. е. отношение эффективностей регистрации интерферирую-
щего и аналитического радиоизотопов. Высокая избиратель-
ность достигается тогда, когда возможно создать условия для
регистрации аналитического излучения с высокой эффектив-
ностью (ех~ 1) при полной дискриминации интерферирующего
излучения (£„ = 0). Решение этой проблемы складывается из
двух моментов: специфичности схемы распада и энергетическо-
го разрешения измерительного устройства.
Под специфичностью схемы распада понимается такое соче-
тание параметров, которое в принципе создает возможность
для однозначной идентификации и точного количественного
определения аналитического радиоизотопа. Хотя среди большой
массы образующихся при активации радиоизотопов отсутст-
вуют радиоизотопы с совершенно идентичными схемами распа-
да, однако в отношении специфичности они далеко не равно-
ценны.
Весьма избирательно определение по радиоизотопам, кото-
рые распадаются уникальным образом. Примером может слу-
жить исключительно избирательный метод определения урана
по трекам деления, образующимся при облучении тепловыми
нейтронами. Уникален распад некоторых изотопов с испуска-
нием запаздывающих нейтронов или радиоизотопа 16N, кото-
рый имеет наиболее жесткое у-излучение среди радиоизотопов,
образующихся при облучении быстрыми нейтронами. Правда,
некоторую конкуренцию последнему может составить радиоизо-
топ пВе, но выход жестких у-квантов у него мал.
В таких случаях высокая избирательность обеспечивается
сравнительно простыми средствами без применения сложных
измерительных устройств с высоким энергетическим разреше-
нием. Поскольку избирательность здесь достигается за счет
уникальности схемы распада, эти методы применимы только к
очень небольшому числу элементов.
Высокой специфичностью обладают схемы распада, сопро-
вождающиеся испусканием моиоэнергетического излучения
(у, а). В активационном анализе наибольшее распространение
получила у-спектроскопия, что связано с такими благоприят-
ными характеристиками у-излучения, как узкая ширина линии,
высокий выход, достаточно большая проникающая способ-
ность. К тому же распад подавляющего большинства радио-
изотопов, образующихся при всех видах активации, сопровож-
дается испусканием у-излучения. По у-излучению в принципе
возможна однозначная идентификация радиоизотопа и очень
избирательное определение.
К сожалению, практической реализации таких возможностей
в какой-то степени препятствуют ограниченное энергетиче-
ское разрешение имеющихся детекторов и некоторые особен-
ности взаимодействия у-излучения с веществом (комптои-
эффект).
306
Повысить избирательность позволяют также такие особен-
ности схем распада, как каскадное у—у-излучение или времен-
ная корреляция между разными видами излучений. Хорошие
результаты часто дает одновременное использование каких-ли-
бо двух параметров схем распада, например регистрация моно-
энергетического излучения в сочетании с периодом полураспада
и т. д.
Низкой избирательностью характеризуются методы, осно-
ванные на регистрации 0-излучения радиоизотопов.
Когда имеется конкретная аналитическая проблема, необ-
ходимо так планировать методику анализа, чтобы оптималь-
ным образом достичь желаемого результата. Часто это де-
лается на основе накопленного опыта, некоторых предвари-
тельных сведений и имеющихся возможностей. Однако выбор
наиболее корректного метода анализа далеко не всегда оказы-
вается простым делом. Наилучшие результаты могут быть по-
лучены с помощью ЭВМ, в которые вводятся необходимые све-
дения об определяемом компоненте и ориентировочном составе
исследуемой пробы, а также соответствующие ядернофизиче-
ские данные. Обработка этой информации по заданной про-
грамме может дать обоснованные решения относительно выбо-
ра метода активации, способа измерения аналитического ра-
диоизотопа и оптимального режима при облучении и изме-
рении.
Оптимизация условий инструментального анализа (подбор
^обл, ^и.зм, ^раси, e.v. ен) относительно избирательности (minZ.)
для какого-либо одного компонента в пробе с примерно изве-
стным составом может быть выполнена обычными математиче-
скими методами на основе соотношения (11.22). Однако сле-
дует помнить, что избирательность тесно связана с правильно-
стью метода и поэтому такая процедура оптимизации даст ус-
ловия с наименьшей систематической погрешностью, но режим
при этом не обязательно будет оптимальным с точки зрения
сходимости и чувствительности. Поскольку условия, которые
обеспечивают наивысшую для данной методики сходимость и
чувствительность, совпадают, их оптимизацию можно прово-
дить на основе соотношения (3.24) пли аналогичного ему
[356, 357].
При 'оптимизации условий предпочтение обычно отдают схо-
димости (чувствительности), но при этом, чтобы процедура не
оказалась непригодной в отношении правильности (избиратель-
ности), необходимо ввести дополнительное условие вида
s>rB (s — среднее квадратическое отклонение, гв — смеще-
ние). Если есть возможность определения поправки на смеще-
ние, то последнее условие отпадает.
Вообще говоря, строгая оптимизация условий анализа
имеет смысл только при массовых определениях одного компо-
нента в матрице, состав которой меняется в небольших преде-
307
лах. Такие аналитические задачи часто возникают в ходе про-
изводственной деятельности и редко в научно-исследователь-
ской работе.
Однако если разрабатывается метод анализа большого чис-
ла элементов в одной пробе, то способ и условия активации по
необходимости не должны быть избирательными и проблему
можно решить в основном путем повышения избирательности
конечного определения. В этом случае значительно чаще нахо-
дят применение радиохимические методы.
Ни радиохимический, ни инструментальный подход не дает
необходимой избирательности, когда интерферирующие реак-
ции приводят к образованию радиоизотопа, тождественного с
аналитическим. Поскольку сечения интерферирующей и основ-
ной реакций показывают разную зависимость от энергии акти-
вирующего излучения, то изменение условий облучения часто
позволяет подавить или оценить помеху.
Нейтронный активационный анализ. Облучение тепловыми
нейтронами приводит к активации значительного числа эле-
ментов, шкала избирательности для которых практически сов-
падает с распределением по величине сечения активации. По-
этому наиболее благоприятный случай — определение элемен-
тов с высоким сечением активации в матрице из неактивирую-
щихся или слабоактивирующихся элементов.
Как правило, в результате реакции (п, у) образуются ней-
троноизбыточные радиоизотопы, обычно испытывающие [М-рас-
пад с последующим испусканием у-квантов [30]. Среди важней-
ших радиоизотопов лишь несколько испытывают чистый
Р -распад (без сопровождающего у-излучения), регистрация
которого обеспечивает низкую избирательность инструменталь-
ного метода. Очень редко образуются позитронные излучатели,
поэтому их аннигиляционное излучение можно довольно изби-
рательно регистрировать методом совпадений.
При анализе проб сложного состава на содержание боль-
шого числа микроэлементов эффективны только гамма-спек-
трометры с высоким разрешением или радиохимический ва-
риант. Многие несложные аналитические задачи часто могут
быть решены и с помощью сравнительно простых средств.
Распределение по избирательности может быть изменено
при облучении отфильтрованным потоком нейтронов. Исполь-
зование фильтров из Cd, In, Sm, В и Li повышает избиратель-
ность примерно для 34 элементов, имеющих высокий резонанс-
ный интеграл активации: Li, Cl, Sc, Мп, Со, Ga, As, Вг, Sr, Mo,
Rh, Pd, Ag, In, Sb, Те, I, La, Nd, Sm, Eu, Lu, Hf, Ta, W, Re,
Ir, Pt, Au, Hg, Tl, Th, U, Pu [96]. Выигрыш в избирательности
может достигать примерно одного порядка, но одновременно с
общей потерей чувствительности. <
Избирательность активационного анализа на быстрых ней-
тронах в целом много хуже, так как реакции обычно имеют не-
308
сколько каналов, различия в величинах сечений ядерных реак-
ций меньше, а получающиеся радиоизотопы часто оказываются
позитронно-активными.
Гамма-активационный анализ. Облучение тормозным излу-
чением с достаточно высокой энергией вызывает активацию
значительного числа элементов [125, 127, 137, 148]. Среди про-
дуктов активации преобладают нейтронодефицитные радио-
изотопы, которые испытывают позитронный распад. Целый ряд
важных элементов дают чистые позитронные излучатели (ПС,
13N, 15О, 18F, 30Р и др.). Распределение образующихся радио-
изотопов по энергии наиболее интенсивного у-излучения и по
периоду полураспада показано на рис. 72 [149].
Обращает на себя внимание такая особенность в распреде-
лении у-излучения, как заметное концентрирование радиоизо-
топов в области энергий 511 кэв (аннигиляционное излучение).
Эта особенность продуктов у-активации имеет большое значе-
ние для избирательности инструментального у-активационного
анализа по следующим причинам: 1) у-спектрометрический
анализ не применим к смеси позитронно-активных радиоизото-
пов; 2) у-линии с энергией менее 0,51 Мэв приходится изме-
рять на фоне комптоновского распределения от аннигиляцион-
ного излучения, которое обычно имеет высокий выход и ин-
тснсиыюсть\ 3) в результате совпзденьй эннигиляционыого
излучения с другими у-квантами велика вероятность образова-
ния линий суммирования, особенно если измерения проводятся
при высокой геометрии.
Таким образом, инструментальный у-активационный анализ
имеет наиболее высокую избирательность для радиоизотопов
с жестким излучением (С1, К, Si и др.). Избирательность опре-
деления элементов по радиоизотопам с энергией менее 0,5 Мэв
заметно хуже. Особые затруднения возникают при определении
элементов, дающих чистые позитронные излучатели.
Для обеспечения избирательности инструментального опре-
деления в случае, когда у-спектрометрия оказывается недоста-
точно эффективной, приходится использовать различные физи-
ческие средства: p-спектрометрию, анализ кривых распада,
схемы совпадений и антисовпадений, облучение при разной
энергии тормозного излучения и т. д.
Каждый из этих методов не является высокоизбирательным,
и поэтому их применение ограничено определенными концентра-
ционными соотношениями исследуемого и интерферирующих
элементов.
Все фотоядерные реакции экзоэнергетичны и имеют доволь-
но высокие пороги. Гистограмма распределения основных фото-
ядерных реакций по величине порога показана на рис. 73. Сле-
довательно, уменьшая энергию тормозного излучения, можно
исключить протекание реакций с самыми высокими порогами.
309
ы
о
О
2fi
”L0
i '
О
89my
о о
?SmAl ?3Мд
Wmttf
77п>*в
59 Ос
^ТА °
°<0°
10'
О
34m
Cl
О
2SAl
О
^Mo
о
52y
о
&AI
о
*«5С
о
о
^Ni
119тс,
9tmMs о
4SmSe
О
1^,
В1
is0 °0п^к Г1
Мр .
7 Ъ
^°Рг
о
20ч mPb
о
**s₽
о
e90e
о
83Zr
rsmn.
о Sl
>-* 80d
’Zr?"V«XfSJZ ХЭСг
да^г
7V> |\ 3‘,mC‘ ^Г, 87mSp
iV3SrtiCu 12086^ O3S^ OHSmln
< °^S> МУА°<^Ъ
---U,----------L-----
i°52mEu
О
' °о£и
^^ГЧО
9llb
оо
8”»
о
™Се
iO4
ssa.
о
’ . ..
s9^4rs,
iOs
^6‘“
Период полураспаду сек
Рис. 72. Диаграмма распределения у-излучения продуктов фотоактивации по энергии н периоду полураспада.
Это сокращает количество активирующихся элементов и таким
образом облегчает инструментальный анализ.
Однако из гистограммы видно,
сплошного характера тормозного
в случае помех от небольшого
числа реакций с наиболее высо-
кими порогами. Для реакций с
порогом ниже 14 Мэв этот ме-
тод уже мало эффективен. К
этому следует добавить, что
уменьшение энергии тормозного
излучения ускорителя сопровож-
дается значительным падением
активации элементов, а значит, и
чувствительности. Например,
уменьшение энергии излучения
от 26 до 15 Мэв приводит к па-
дению удельной активности эле-
ментов на порядок или больше.
Отсутствует возможность по-
вышения избирательности опре-
деления путем изменения энер-
пти о6лх,’чсния в области ВЫСО-
ких энергий (выше 25 Мэв), где
кривые возбуждения фотоядер-
ных реакций идут практически
что такой подход (с учетом
излучения) полезен только
Рис. /3. Гистограмма распреде-
ления фотоядерных реакций по ве-
личине порога.
параллельно друг другу.
Из-за ограниченной избирательности инструментальный
у-активационный анализ может найти широкое применение
только в области концентраций выше 10^3%. Мощные ускори-
тели электронов позволяют достигнуть более высокой чувстви-
тельности (до 10“6—10~8%). Однако для ее реализации прихо-
дится прибегать к помощи более избирательного метода —
радиохимическому выделению определяемых компонентов.
§ 4. Многоэлементный активационный анализ
Из предшествующего изложения следует, что активацион-
ный анализ, если рассматривать его в совокупности всех мето-
дов, обладает универсальными возможностями в том смысле,
что он позволяет определять практически все имеющиеся в при-
роде элементы. Достигаемая чувствительность, как правило,
весьма высока и редко уступает чувствительности других мето-
дов, способных к определению столь же широкого кругд эле-
ментов. Нет и ограничений диапазона определяемых концентра-
ций, который может простираться от предела обнаружения до
зн
максимально возможной концентрации (100%). Однако полная
реализация всех возможностей активационного анализа может
потребовать достаточно сложной схемы анализа с включением
в нее различных режимов и способов активации и регистрации
вторичного излучения, а также радиохимического разделения.
Отмеченная универсальность активационного анализа поз-
воляет устанавливать содержание большого числа элементов в
пробах сложного состава, т. е. осуществлять многоэлементный
активационный анализ. Пока что основное свое применение
многоэлементные схемы находят для определения малых ком-
понентов в различных природных и промышленных объектах.
При этом особую ценность многоэлементному активационному
анализу придает тот факт, что можно получить данные о содер-
жании большого числа элементов из одной небольшой пробы,
а это очень важный момент, когда исследуемый материал до-
ступен в ограниченном количестве или является ценным и даже
уникальным.
Примером могут служить полупроводниковые материалы и
вещества высокой чистоты. Некоторые природные объекты мо-
гут быть получены в весьма небольших количествах (космиче-
ская пыль, метеориты, микровключения в минералы и т. д.),
также как и пробы, которые необходимо брать из живого орга-
низма. Однако даже при исследовании материалов, доступных в
значительных количествах, многоэлементный анализ имеет свои
преимущества в виде обширной информации и более высокой
точности определения корреляционных отношений в содержа-
нии компонентов. Немаловажными обстоятельствами могут
оказаться быстрота и экономичность определений в расчете на
один компонент.
Схема многоэлементного активационного анализа может
основываться целиком на радиохимическом или инструменталь-
ном варианте либо на комбинации некоторого количества ра-
диохимических операций с у-спектрометрическим окончанием.
Если к этим вариантам добавить возможность включения в схе-
му анализа различных методов активации, то многообразие от-
крывающихся путей для проведения многоэлементного актива-
ционного анализа становится очевидным.
Из отдельных методов активационного анализа наилучшими
возможностями в отношении чувствительности и числа опреде-
ляемых элементов обладает активационный анализ на тепло-
вых нейтронах реактора. Именно поэтому почти все опублико-
ванные к настоящему времени схемы активационного анализа,
включающие достаточно большое число элементов (более 15),
основаны на облучении тепловыми нейтронами. Лишь единич-
ные элементы имеют неблагоприятные ядерные характеристики,
и их определение этим методом затруднено. Однако в таких
случаях можно воспользоваться активацией быстрыми нейтро-
нами, жесткими фотонами и заряженными частицами.
312
Первые схемы многоэлементного активационного анализа
предусматривали полное радиохимическое разделение определяе-
мых компонентов, поскольку в тот период физические средства
дискриминации ионизирующих излучений еще не достигли та-
кого уровня, как это имеет место в настоящее время. Первона-
чально эти схемы основывались на методе осаждения с исполь-
зованием в той или иной степени элементов классической серо-
водородной схемы анализа. Однако серьезные недостатки по-
следней применительно к многоэлементному активационному
анализу привели к тому, что сейчас эти схемы имеют только
исторический интерес. Позднее были предложены схемы, ис-
пользующие новые прогрессивные методы разделения, такие,
как экстракция и ионный обмен.
У схем с полным радиохимическим разделением есть свои
достоинства — возможность выделения любого элемента из лю-
бой матрицы. Существенный фактор представляет также воз-
можность проведения конечных измерений с помощью простой
и высокочувствительной аппаратуры. Однако серьезными недо-
статками схем с полным радиохимическим разделением яв-
ляются сложность, трудоемкость и длительность. Необходи-
мость затраты времени на перевод вещества пробы в раствор
и выполнение радиохимических операций накладывают опреде-
ленные ограничения на период полураспада радиоизотопов, ко-
торые могут бы 1 ь использованы для определений. Практически
в многоэлементных схемах предусматривается выделение ра-
диоизотопов, имеющих период полураспада от нескольких ча-
сов и выше.
Совершенствование ядернофизических средств регистрации
и анализа ионизирующих излучений оказало большое влияние
на развитие многоэлементного активационного анализа, кото-
рое пошло преимущественно либо по пути использования чисто
инструментального варианта, либо сочетания его с радиохими-
ческим подходом.
По причинам, отмеченным ранее, многоэлементный инстру-
ментальный анализ возможен только с помощью гамма-спек-
трометров высокого разрешения. Напомним, что спектрометры
с Ge (Li)-детекторами, как правило, имеют энергетическое
разрешение 3—5 кэв в области 1 Мэв, а точность определения
энергии достигает ±0,5 кэв. Что же дают такие характеристи-
ки для инструментального нейтронноактивационного анализа
сложных объектов? Однако прежде всего следует отметить, что
в этих случаях инструментальный подход сталкивается с за-
труднениями двух типов: 1) наложением линий близких энер-
гий и наличием непрерывного амплитудного распределения;
2) значительными различиями в уровне наведенной активности
компонентов. Роль первого фактора уже была обсуждена и не
требует дополнительных комментариев. Второй фактор связан
с величиной временного разрешения и допустимой загрузкой
1 1 Р. А. Кузнецов
313
электронных систем спектрометра. Из-за влияния этого факто-
ра приходится ограничивать скорость счета излучения (актив-
ность пробы), и таким образом ставится предел концентрацион-
ной чувствительности определения слабоактивируюгцихся ком-
понентов в присутствии сильноактивирующихся. Часто этот
фактор приводит к падению предельной чувствительности ме-
тода на несколько порядков.
Влиянием отмеченных факторов объясняется то положение,
что методика многоэлементного активационного анализа часто
оказывается достаточно длительной и сложной. Обычный путь
получения наиболее полной аналитической информации состоит
в комбинировании разных режимов облучения и измерения,
что требует двух-трех облучений пробы и длительных измере-
ний у-излучения пробы с последующей обработкой данных на
ЭВМ. Типичным примером может служить схема анализа аэро-
золей (рис. 74), предложенная Дамсом и др. [358]. Эта схема
предусматривает определение 33 элементов и требует двукрат-
ного облучения пробы (короткого 5 мин и более длительного
2—5 ч) при различной плотности потока тепловых нейтронов.
Измерения активности пробы с помощью Ge (Li)-спектрометра
(детектор объемом 30 аи3) проводятся через заданные интер-
валы времени. К характерным особенностям подобных схем
относится то, что конечные результаты могут быть получены
только через 20—30 дней, а длительность измерений может
быть большой (более часа).
Возможности инструментального варианта могут быть не-
сколько расширены, если дополнительно прибегнуть к мето-
дам, позволяющим повысить избирательность анализа. Мож-
но упомянуть метод совпадений для выделения каскадных из-
лучателей или применение фильтров при облучении в реакторе.
Применение разных способов активации в сочетании с ядер-
нофизическими средствами дискриминации, видимо, также
представляет перспективное направление в развитии много-
элементного инструментального активационного анализа. Пер-
вой попыткой, где достаточно широко использован такой под-
ход, является работа Шмитта и др. [359] по определению 14 ос-
новных элементов в горных породах и каменных метеоритах.
Схема анализа одной пробы включает многократные облуче-
ния разными видами активирующего излучения (быстрыми и
тепловыми нейтронами, тормозным излучением) в различных
режимах с последующим измерением у-излучения проб сцин-
тилляционным и полупроводниковым спектрометрами.
Хотя инструментальный многоэлементный анализ имеет за-
метные успехи, отмеченные выше причины создают все же зна-
чительные трудности для дальнейшего прогресса. Принци-
пиальные ограничения чисто радиохимического или инструмен-
тального метода применительно к многоэлементному актива-
ционному анализу привели к развитию схем, сочетающих оба
314
метода. Именно на этом пути оказалось возможным создание
схем, позволяющих определение максимального числа элемен-
тов с наибольшей точностью и чувствительностью при умерен-
ной продолжительности анализа.
Рис. 74. Схема инструментального активационного анализа аэрозолей.
Среди многочисленных возможных сочетаний инструмен-
тального и радиохимического методов можно более или менее
четко выделить следующие основные случаи.
И
315
1. Радиохимический метод используется очень ограниченно
только для удаления мешающих инструментальному анализу
сильноактивных компонентов. Этот вариант часто встречается
при анализе биологических проб и горных пород, где требуется
удаление главным образом сильноактивного 24Na.
2. Схемы с равноценным сочетанием радиохимического и
инструментального методов. Такие схемы строятся на основе
радиохимического разделения определяемых элементов на не-
сколько групп, удобных для последующего спектрометрическо-
го анализа. При правильном выборе схемы радиохимического
анализа и использования специфичных методов разделения
оказывается возможным создание достаточно быстрых и эко-
номичных аналитических методик для активационного опреде-
ления большого числа элементов в самых разнообразных
объектах.
3. Расширенное использование инструментального метода в
сочетании с групповым разделением. Чаще всего дополнительно
проводят анализ той же пробы инструментальным методом по
короткоживущим изотопам.
Исторически первыми стали развиваться схемы с группо-
вым разделением, которые разрабатывались в расчете на при-
менение в качестве конечного метода измерения сцинтилля-
ционного гамма-спектрометра. Эти приборы и сейчас сохра-
няют свое значение как наиболее доступные и распространен-
ные. Сравнительно низкое разрешение этих спектрометров
предъявляет соответствующие требования к составу и количе-
ству радиоизотопов в группе. Число радиоизотопов в одной
группе должно быть мало (не более 6—7), при этом нельзя
также сочетать радиоизотопы с близкими энергиями у-излу-
чения.
Пионером в области развития групповых методов разделе-
ния является Самсал. Им предложено несколько вариантов
своей схемы группового многоэлементного анализа, разрабо-
танной применительно к анализу биологических проб. Послед-
ний вариант представлен на рис. 75 [323]. Эта схема преду-
сматривает разделение 40 элементов на 13 групп методами
дистилляции, ионообменной и экстракционной хроматографии.
Разделение осуществляется с помощью автоматической систе-
мы, которая была описана ранее (см. гл. 10, § 4).
В ходе анализа облученную навеску биологйческой пробы
переводят в раствор при обработке H2SO4 + H2O2 в дистилля-
ционной установке. Затем добавляют НВг и отгоняют летучие
компоненты. Эта операция занимает 6—7 мин. Оставшийся
после дистилляции раствор переносят в начало серии из 10
хроматографических колонок и проводят разделение па груп-
пы, которое требует 5 мин. Выделенные группы элементов ока-
зываются сорбированными на колонках, которые разъединяют,
промывают; затем элементы измеряют на сцинтилляционном
316
гамма-спектрометре. Для большинства элементов химический
выход в соответствующей группе составляет 95—100%.
Схема группового разделения, в которой используются
преимущества микроколоночной ионообменной хроматографии,
предложена А. И. Калининым и др. [360] для анализа кремния
Проба* носители,
обработка Н280^нг0г,
до б. НВ г, дистилляция
Дистиллят
As, Cl, Ge, Hg,
Re, Ru,Sb,Se,Sn
Доб. H202, duc-
тилляция
Поглотитель
NaOH
Br, Os, I
Bio-Rati AG2*8
Au, Ag, Ir, MO,
Pa, V, W-
0,5 h. HBr
Bio-Rad AG 2*8
CO
Экстракция
Д2ЭГФК
HP, Np, SC, Zr,
Tb^Lu
Na0H*CH3C00Na
5h. NaOH
Bio-Rad Chelex 100 (Nd")
Ba, Ca, Mg, Sr
Экстракция
Д2ЭГФК
Al,Ti,La-^Cd
бн. HN03
pH=3
Bio-Rad KCF-1
Cs, K, Rb
T 12H.LWI
J"
Bio-Rad AG 2*8
Юн. LiCl
T
1
Bio-Rad Chelex ЮО(нД)
Co, Cr, Си, Мп, Nl
Bio-Rad AC 7x4
Fe, Ca, in
Bio-Rad Chelex 700(АГ3)
P
Рис. 75. Схема группового радиохимического разделения элементов при акти-
вационном анализе биологических проб.
и кварца высокой чистоты. Пробы переводят в раствор при
обработке смесью HNO3 + HF. После полного удаления Si ана-
лизируемый раствор последовательно пропускают через шесть
микроколонок, заполненных соответствующими ионитами
(рис. 76). Элюенты подобраны таким образом, чтобы обеспе-
чить сорбцию на одной колонке небольшого числа элементов.
Сорбированные элементы затем вымывают и измеряют на сцин-
тилляционном гамма-спектрометре. Колонки работают полу-
317
автоматически. Схемой предусматривается разделение 25 эле-
ментов на 7 групп; полное время химической обработки, начи-
ная с операции травления пробы, составляет 4 ч. Кстати ска-
Анализируемый раствор
Упаривание досуха
Ш3 + НС10^
АВ-17
СЮ^-форма
1M(NHZ),CS
--------*- Аи
Упаривание досуха
0,1н.НБ
КУ-г *16
Р+-форма
Ун. НС1
Упаривание досуха
8н. НС1
Восстановление серой
8h.HCI + 1h.HF
Си, Со, Са HCl,HzD,
In,Zn,Fe, 1h.NHi>0H
Cd
АВ-17
с1~-форма
АВ-17
СГ-форма
Зн.НСЮ^ Sb, Та,
Sn,Mo
Упаривание досуха
As,P,W
Mn,Ni,Cr, Ун. PCI
Ад,РЗЭ
АВ-17
он~-форма
Упаривание
Zh.NH^CL
Ca, Sr,
Ва
0,1 М трилонБ
рН=10
к.у-г*1б
nhI-форма
Na, К,
Kb, Cs
Рис. 76. Схема группового радиохимического разделения элементов
методом ионообменной хроматографии при активационном анализе
кремния и кварца.
зать, эта схема может быть изменена, и тогда она позволит
выделять каждый определяемый элемент в индивидуальном
виде.
Для активационного анализа стекол Гуд и др. [322] приме-
нили экстракционную схему группового разделения (рис. 77).
318
Она предусматривает разделение 25 элементов на 6 групп по-
следовательной экстракцией ТТА, ДДДК и ТБФ при контроли-
руемом pH. Разделение выполняет экстрактор специальной
конструкции, который автоматически выполняет все операции,
Оргхраза - Водная фаза
Рис. 77. Схема группового экстракционного разделения
элементов при активационном анализе стекол (ТТА — те-
ноилтрифторацетон, ДДДК — диэтиламмоний диэтилдитио-
карбаминат, ТБФ — трибутилфосфат).
требующиеся по ходу анализа (см. гл. 10, § 4). Химический
выход элементов составляет 95%.
Весьма знаменательным является то обстоятельство, что
в последнее время предложено несколько схем группового ра-
319
диохимического разделения, рассчитанных на использование
для конечных измерений Ое(Ы)-спектрометра. Естественно, что
с прибором такого типа количество элементов в группе может
быть увеличено, а процедура разделения упрощена. Правда,
применение Ge (Li)-детектора связано с понижением эффек-
тивности регистрации, по этой причине в некоторых случаях
стали предусматривать отдельное выделение малоактивных эле-
(1роба,
обработка ррч
Органа- Экстракция ,(СгН5)г0
ческая ।---------------—-----
фаза т
Удаление эфира,
осаждение NaOH
Осадок^ |
Экстракция
Органа- (CzHsM из 6н.НС1
ческая I ~1
фаза " !
te,Ge,Sb, Сброс
Аи,Са,Мо
б
13 НС1
I
Дистилляция
из НВг+Нг50^, У1Рг
। ^Дистиллят
Осаждение Осаждение
сульфидов сульфидов
I
Осаждение As, Sb, Hg,
' (ПН„)з(РМопОм) Re,0s,Se
стВорение 1
HCL + HHO. '
Дауэкс AG1х8
зн. на
Со, Sc Zn
Рис. 78. Схема группового радиохимического разделения элементов при
активационном анализе каменных метеоритов.
ментов с измерением этой группы на сцинтилляционном спек-
трометре. Такой подход позволяет наиболее рационально ис-
пользовать все имеющиеся в распоряжении аналитика химиче-
ские и физические средства для получения наибольшего эф-
фекта.
Лауль и др. [62] при определении 17 элементов в каменных
метеоритах использовали радиохимическое выделение трех
групп элементов и индивидуально Tl, Cs и Zn (рис. 78). Боль-
шие группы измеряются на полупроводниковом спектрометре,
остальные фракции (кроме Т1) на сцинтилляционном. Резуль-
таты обрабатываются на ЭВМ. Поскольку аналитический ра-
диоизотоп таллия (204Т1) —чистый р -из.тучатель, то необходим
р-счет.
Филби и др. [361] изучили возможности, которые представ-
ляет радиохимическое удаление наиболее мешающего компо-
320
нента (24Na) для анализа многих элементов в горных породах.
Как оказалось, отсутствие 24Na дает возможность включить в
анализ ряд радиоизотопов с периодом полураспада в интерва-
ле 1—50 ч. Схема анализа состоит из четырех облучений раз-
ной длительности с последующим многократным измерением
у-активности проб с помощью Се(Ы)-спектрометра (рис. 79).
Рис. 79. Комбинированная схема активационного анализа горных пород.
После одного из облучений проводится разложение пробы
сплавлением с LiBO2 и затем сорбция 24Na гидратированной
пятиокисью сурьмы. Предложенная схема позволяет определять
32 элемента, но полная длительность анализа достигает трех
месяцев.
Наибольшее число одновременно определяемых элементов
было достигнуто в схемах, которые сочетают широкое использо-
вание физических средств с групповым радиохимическим раз-
делением. Такой подход был использован Моррисоном и др.
[362] в схеме многоэлементного активационного анализа гор-
ных пород. Эта схема предусматривает проведение измерений
321
с помощью Ge (Li)-спектрометра, а в отдельных случаях — при-
менение системы совпадений — антисовпадений.
Серия коротких облучений позволяет определить 10 элемен-
тов (Al, V. Mg, In, Мп, Ni, К, Dy, Cl, Na). Отдельную навеску
(1 г) облучают 8 ч тепловыми нейтронами реактора [Ф =
Проба,
носители 8с, Zn, La, As, В г.
Обработка НР+'Нг80^,
дистилляция
Остаток, растворение
6 8н. НС1, сорбция Sb2O5
истиллят
7SAs,8ZBr
Дау экс 1*S
S5Zn,S9mZn,
™Sb,™Sb,
^7Hg,1^Hg
£
о,5н. на
] Сорбе нт
VzTa
53Fe,BOCo,nCu,7zQa1
Z33Np, 187W, 9sMo
Упаривание, раство-
рение в 8н. на.
Экстракция ТБФ
Органи- --------------
ческая 1
фаза '
181 Hf, 97Zr, wSc,
wScS7Sc,175Hf™Pa
I
«К, seRb, mCs, ^Sc,131Ba,
в58г™Се,™Ш,™8т,™Еи,
15*Eu, 153&d, 16aTb, 1wLa, 1BSHo,
mTm,169Yb,17W7Ca
Рис 80. Схема группового радиохимического разделения при актива-
ционном анализе горных пород.
= 7-10й нейтрон!(слг2• сек)}, выдерживают 15 ч. После перевода
пробы в раствор производится радиохимическое разделение
элементов на группы методами сорбции, экстракции и ионного
обмена согласно схеме анализа (рис. 80). Общее число элемен-
тов, которое может быть определено этим методом в различ-
ных горных породах, составляет 45, причем область опреде-
ляемых концентраций простирается от 5 10 до 10%.
Колеман [363] сравнил точность инструментального анализа
с полупроводниковым и сцинтилляционным гамма-спектромет-
рами и анализа с групповым радиохимическим разделением.
Исследование проводилось на примере стекол и включало
322
определение 20 элементов. Как оказалось, в данном конкрет-
ном случае метод группового разделения позволяет оценивать
содержание всех элементов (инструментальными методами не-
которые из них не обнаруживались) при общей точности, кото-
рая выше точности инструментального метода. Наиболее пло-
хие результаты были получены с Ge (Li)-спектрометром (объ-
ем 20 см3, разрешение 7 кэв для £v=l,33 Мэе), что связано с
его низкой эффективностью.
В заключение, видимо, следует отметить, что традиционное
соперничество между радиохимическим и инструментальным
методами в области многоэлементного анализа наилучшим об-
разом разрешается в схемах, предусматривающих творческое
сочетание этих методов.
СПИСОК ЛИТЕРАТУРЫ
1. Старосельская-Никитина О. А. История радиоактивности и возникно-
вение ядерной физики. М., Изд-во АН СССР, 1963.
2. Hevesy G. V., Levi Н. In: Hevesy G. Adventures in Radioisotope Rese-
arch. V. 1. N. Y., Pergamon Press, 1962, p. 47.
3. Seaborg G. T., Levingood .1. J. Artifical radioactivity as a test for minute
treaces of elements. — J. Amer. Chem. Soc., 1938, v.’60, p. 1784.
4. Meinke W. W. Activation analysis utilizing fast radiochemical separa-
tions. — In: Radioisotopes in the Physical Sciences and Industry. IAEA,
Vienna, 1960, v. 2, p. 277.
5. Stary J., Ruzicka J. Substoichiometric determination of traces of metals. —
Taianta, 1964, v. 11, p. 697.
6. Вдовенко В. M. Современная радиохимия. М., Атомиздат, 1969.
7. Гринберг А. А. Радиоактивные индикаторы в химии комплексных сое-
динении.— «Успехи химии», 1940, т. 9, с. 771.
8. Айдаркин Б. С. и др. К методике определения бериллия в рудах по фо-
тонейтронам.— «Тр. Радиевого ин-та», 1957, т. 5, вып. 2, с. 89.
9. Алимарин И. П., Яковлев Ю. В. Ядернофизические методы анализа. —
«Атомная энергия», 1969, т. 26, с. 127.
10. Руденко Н. П. Радиохимические методы анализа. — «Ж. аналит. хи-
мии», 1967, т. 22, с. 1736.
11. Алимарин И. П. Современное состояние и некоторые перспективы раз-
вития методов аналитической химии. — «Ж. аналит. химии», 1970, т. 25,
с. 599.
12. Моррисон Дж., Скогербоу Р. Общие аспекты определения следов эле-
ментов.— В кн.: Физические методы анализа следов элементов. Пер. с
англ. М., «Мир», 1967, с. 9.
13. Гамбарян Р. Г., Иванов И. Н., Штань А. С. Современный активацион-
ный анализ. — В кн.: Радиационная техника. М., Атомиздат, 1967, вып. 1,
с. 61.
14. Реформатский И. А. Горячие и изотопные лаборатории. Изд. 2-е. М.,
Атомиздат, 1971.
15. Виноградов А. П. Проблема чистоты материалов. — В кн.: Методы оп-
ределения и анализа редких элементов. М.. Изд-во АН СССР, 1961, с. 5.
16. Горюнова Н. А. и др. Задачи и пути развития аналитической химии по-
лепроводниковых материалов. — «Тр. комиссии по аналитической хи-
мии». 1968. т. 16, с. 3.
17. Боуэн Г., Гиббонс Д. Радиоактивационныц анализ. Пер. с англ. Под ред.
И. П. Алимарина. М.. Атомиздат, 1968.
18. Winchester J. W. Radioactivation analysis in inorganic geochemistry.—
In: Progress in inorganic chemistry V. 2. Cambridge, 1960, p. 1.
19. Showalter D., Schmitt R. Application of activation analysis to geochemi-
cal, meleoritic and Lunar studies. — In: Advances in activation analysis.
V. 2. London, Academic Press. 1972, p. 185.
20. Кист А. А.. Лобанов E. M. Активационный анализ в биологии и меди-
324
пине. — В кн.: Активационный анализ биологических объектов. Ташкент,
Изд-во ФАН УзССР, 1967. с. 130.
21. Lux F. Application of activation analysis in biochemistry. — Z. analyt.
Chem.. 1969, Bd. 243. S. 107.
22. Jervis R. E, Application of activation analysis in forensic medicine.—
Isotopes and Radiat. Technol.. 1969. v. 6. No. 1, p. 57.
23. Савосии С. И. и др. Основные направления использования типовой ап-
паратуры активационного анализа при исследовании горных пород и
руд. — В кн.: Ядерная геофизика. М„ Атомиздат, 1968, вып. 1, с. 20.
24. Rhodes J. R. Applications of neutron activation to on-stream analysis. —
Isotopes and Radiation Technol., 1969. v. 6, No. 4, p. 359.
25. Fite L. E., Wainerdi R. E. Automated nuclear activation analysis in geo-
chemistry.— Miner. Sci. and Engng, 1970, v. 2. No. 3, p. 3.
26. Пронман И. M. Современное состояние активационных методов опреде-
ления газовых примесей в металлах и полупроводниковых материалах.—
В кн.: Методы определения и исследования состояния газов в металлах.
М., «Наука». 1968, с. 134.
27. Activation analysis: a bibliography through 1971. NBS Technical Note 467,
Washington, 1972.
28. AALC — File Activation Analysis Literature Cards. Budapest, Akademiai
Kiado.
29. Neutron Activation Analysis Abstracts. Ed. Sutherland, London, Sci. and
Technol. Agency, 1972.
30. Медине И. В. Справочные таблицы для нейтронноактивационного ана-
лиза. Рига. «Зинатне». 1969.
31. Алиев А. И. и др. Ядернофизические константы для нейтронного актива-
ционного анализа. М., Атомиздат, 1969.
.32 . Маслов И. А., Лукницкий В. А. Справочник по нейтронному активаци-
онному анализу. Л., «Наука». 1971.
33. Мухии К. М. Введение в ядерную физику. Изд. 2-е. М„ Атомиздат, 1965.
34. Фриндлер Г., Кеннеди Дж., Миллер Дж. Ядерная химия и радиохимия.
Пер. с англ. М., «Мир», 1967.
35. Sorantin Н., Millner Р. Nomenclature of radioisotopes. — Mikrochim. acta,
1966. v. 6, p. 1153.
36. Джелепов Б. С., Пекер Л. К. Схемы распада радиоактивных ядер
А<100. М — Л.. «Наука», 1968.
37. Джелепов Б. С.. Пекер Л. К., Сергеев В. О. Схемы распада радиоак-
тивных ядер А^ЮО. М.—Л., «Наука», 1963.
38. De Goeij J., Houtman J. P., Konij J. B. Errors in activation analysis due
to variating in isotopic ratio. — Radiochim. acta, 1966, v. 5, p. 117.
39. Крыженкова H. А., Лобанов E. M., Кист А. А. Об уравнении активации
при переменном потоке. — В кн.: Нейтронноактивационный анализ. Таш-
кент, ИзД-во ФАН УзССР, 1971, с. 3.
40. Меднис И. В. Вычисление величины наведенной активности при прерыви-
стом облучении. — В кн.: Методы и применения нейтронноактивацион-
ного анализа. Рига, «Зинатне», 1969, с. 21.
41. Абрамов А. И., Казанский Ю. А., Матусевич Е. С. Основы эксперимен-
тальных методов ядерной физики. М.. Атомиздат, 1970.
42. Калашникова В. И., Козодаев М. С. Детекторы элементарных частиц.
М.. «Наука», 1966.
43. Лукьянов В. Б. Измерение и идентификация бета-радиоактпвных препа-
ратов. М.. Госатомиздат, 1963.
44. Матвеев В. В., Хазанов Б. И. Приборы для измерения ионизирующих
излучений. Изд. 2-е. AL, Атомиздат, 1972.
45. Егиазаров Б. Г.. Корытко Л. А., Сельдяков Ю. П. Измерительная тех-
ника в инструментальном нейтронноактивационном анализе. М., Атом-
издат. 1972.
46. Акимов Ю. К. и др. Быстродействующая электроника для регистрации
ядерных частиц. М., Атомиздат, 1970.
325
47. Гольданский В. И., Куценко А. В., Подгорецкий М. И. Статистика от-
счетов при регистрации ядерных частиц. М., Физматгиз, 1959.
48. Налимов В. В. Применение математической статистики при анализе ве-
щества. М., Физматгиз, 1960.
49. Доерфель К. Статистика в аналитической химии. Пер. с нем. Под ред.
В. В. Налимова. М., «Мир», 1969.
50. Гунне X. Э., Пелекис Л. Л. Роль статистического характера излучения
в выборе критерия чувствительности иейтронноактивационного анализа.—
В кн.: Нейтронноактивационный анализ. Рига, «Зинатне», 1966, с. 5.
51. Currie L. A. Limits for qualitative detection and quantitative determina-
tion. — Anal. Chem, 1968, v. 40, p. 586.
52. Sterlinski S. The lower limit of detection for short-lived radioisotopes.—
Nucleonika, 1966, v. 11, No. 6, p. 441.
53. Хуснутдинов P, И., Лобанов E. M., Минралиев Г. Г. О чувствительности
активационного метода анализа. — В кн.: Активационный анализ Таш-
кент, Изд-во ФАН УзССР. 1971, с. 9.
54. Коренман И. М. Аналитическая химия малых концентраций. М., «Хи-
мия», 1967.
55. Brune D., Wenzel Н. Application of a liquid helium cryostat in neutron
activation analysis of fluids. — Analyt. Chem., 1970, v. 42, p. 511.
56. Алимарин И. П., Миклашанский А. 3., Яковлев Ю. В. Neutron activation
analysis of rare earth impurities in metallic uranium. — J. Radioanalyt.
Chem., 1970, v. 4, p. 45.
57. Лобанов E. M. и др. К вопросу определения золота в рудах и горных
породах. — В кн.: Радиационные эффекты в конденсированных средах.
Ташкент, Изд-во ФАН УзССР, 1964, с. 95.
58. Smathers J., Duffey D., Lakshmann S. Chelate enhancement of the sensi-
tivity for magnesium in neutron activation analysis. — Analvt chim. acta,
1969, v. 46, p. 9.
59. Василевская Л. С,, Назаренко В. А. Условия работы при анализе ве-
ществ высокой частоты. — В кн.: Аналитическая химия редких металлов
и полупроводниковых материалов. М., Моск, дом науч.-тех. пропаганды,
1970 (материалы семинара).
60. Зильберштейн X, И, и др. Спектральный анализ чистых веществ. Л.,
«Химия», 1971.
61. Kuncir J. е. a. Miltielement standart for routine instrumental activation
analysis of trace elements in rocks and tektites.—J. Radioanalvt. Chem.,
1970, v. 5, p. 369.
62. Laul J. C. e. a. An activation analysis technique for determining groups
of trace elements in rocks and chondrites. — J. Radioanalvt. Chem 1970,
v. 4, p. 241.
63. Калинин А. И. Радиоактивашюнное определение примесей в кремнии
и двуокиси кремния с применением хроматографии. Диссертация, 1965.
64. Еголин Я. Г. и др. Пневмотранспортные устройства для ядерно-актпва-
ционного анализа. — В кн.: Тр. седьмой конф, по ядерной электронике.
Т. 2, ч. 1. М., Атомиздат, 1969, с. 13.
65. Guinn V. Р. Reactor-produced short-lived radioisotopes used in neutron
activation analysis. — In: Production and use of short-lived radioisotopes
from, reactors. V 2. Vienna, IAEA, 1963, p. 3.
66. Леушкина Г. В. и др. Использование короткоживущих изотопов в ак-
тивационном анализе. — В кн.: Активационный анализ. Ташкент, Изд-во
ФАН Уз ССР, 1971, с. 23.
67. Progress in Nuclear Energy. Ser. IX, v. 10. Eds Stewart D. C., Elion H. A.
Oxford — London — New York, Pergamon Press, 1970.
68. Nagy L. C., Giber J. Adsorption phenomena in the determination of trace
impurities bv activation analysis. — Period, polvtechn. Chem. Engng. 1968,
v. 12, No. 1', p. 37.
69. Власов H. А. Нейтроны. Изд. 2-е. M., «Наука», 1971.
70. Грошев И. В. и др. (сост.) Атлас спектров гамма-лучей радиационного
захвата тепловых нейтронов. М., Атомиздат, 1958.
326
/1. Weber G., Guillaume N. Gamma-ray spectra and sensitivities for 2.8 Mev
neutron activation analysis. — J, Radioanalyt. Chem., 1970, v. 5. p. 25.
j'2. Бак M. А., Шиманская H. С. Нейтронные источники. M., Атомиздат, 1969.
73. Фрадкин Г. М. Изотопные источники нейтронов. М., Атомиздат, 1963.
74. Межиборская X. Б. Активационным анализ с -Г,2С{ (реферат). — «Атом-
ная техника за рубежом», 1971, ,\° 1, с. 35.
75. Дрынкин В. И., Лейпунская Д. И. Нейтронный активационный анализ об-
разцов горных пород с помощью изотопных нейтронных источников. М.,
ОНТИ ВИЭМС, 1968.
76. Downs W. Е. i2iSb—Be-source of thermal neutrons. — Nucl. Appl., 1968,
v. 5. No. 2, p. 55.
77. Stewart D. C., Horwitz E. P., Younquist С. H. ’-“Cm—Be-source of neut-
rons.— Nucl. Appl. Techn., 1970, v. 9, No. 6, p. 875.
78. Бланков E. Б. и др. Нейтронный активационный анализ в геологии и
геофизике. М„ «Наука», 1972.
79. Bibby D. М., Oldham G., Ware A. R. Tagets of neutron generators.—
Nucl. Energy, 1970, v. 11, No. 3. p. 68.
80. Neider R. e. a. Aktivirungsanalvse mit schnellen Neutronen. Materialprii-
fung, 1969. Bd. 11, Nr. 1. S. 28.'
81. Otvos J. W. e. a. Photoactivation and photoneutronen activation analysis.—
Nucl. Instrum, and Methods. 1961, v. 11, p. 187.
82. Wilkniss P. E. Use of a 60 Mev Linac for fast and variable energy neut-
ron activation analysis. — Analyt. Chem., 1969, v. 41, p. 421.
83. «Атомная энергия». 1971. т. 30, вып. 5, с. 486.
84. Bruninx Е, The cvclotron, a new role for an old nuclear physics tool. —
Kerntechnik, 1970. Bd. 12, Nr. 5/6, S. 226.
85. Guinn V. P. Reactors as neutron sources. — In: Adv. Activ. Analysis.
London. Academic Press. 1969. v. 1, p. 37.
86. Климов A. H. Ядерная физика и ядерные реакторы. М„ Атомиздат, 1971.
87. Юз Д. Нейтронные исследования на ядерных котлах. Пер. с ашл. AL,
Изд-во иностр, лит., 1954.
88. Файнберг С. М. Высокопоточные стаипионарные исследовательские ре-
акторы и их перспективы. — «Атомная энергия», 1970, т. 28, с. 162.
89. Stephens К. G., Walt R., Williams G. Н. Measurement of Neutron Flu-
xes.— In: Programming and Utilization of Research Reactors. V. 3, Lon-
don. IAEA, 1962, p. 63.
90. Столярова E. Л. Нейтронные спектрометры и их применение в приклад-
ных задачах. М._ Атомиздат, 1969.
91. Romanko J., Dungan W. Е. Specification and measurement of reactor
neutron spectra. — In: Neutron Dosimetry. V. I. Vienna, IAEA, 1963, p. 153.
92. Buchanan J. D. Activation analysis with a Triga reactor. — Proc. Inter-
nal. Conf. Modern Trends in Activation Analysis. Texas, 1961, p. 72.
93. Cole T. E., Weinberg A. M. Technology of research reactors. — Ann. Rev.
Nucl. Sci., 1962, v. 12, p. 221.
94. Алексеев Б. И. и др. Экспериментальное изучение характеристик реакто-
ра РГ-1М.— «Атомная энергия», 1972, т. 32, с. 315.
95. Гордеев И. В. и др. Ядернофизические константы (Справочник). Под
ред. А. И. Красина. Изд. 2. М., Госатомиздат, 1963.
96. Дамбург Н. А., Пелекис Л. Л. О некоторых возможностях активацион-
ного анализа на резонансных нейтронах. — «Ж. аналит. химии», 1971,
т. 26, с. 1869.
97. Borg D. С. е. о. Selective radioactivation and multiple coincidence spect-
rometry.— Internal. J. Appl. Rad. and Isotopes, 1961, v. 11, No 1, p. 10.
98. Гамбарян P. Г., Штань А. С. Нейтронно-резонансный анализ элемент-
ного состава вещества. — «Атомная энергия», 1968, т. 25. с. 111.
99. Гамбарян Р. Г. и др. Спектрально-резонансный нейтронный анализ. —
В кн.: Прикладная ядерная спектроскопия. Вып. 1. М., Атомиздат,
1970, с. 59.
100. Perdijon J. Experimental sensitivities in neutron activation and gamma
spectrometry with 150 kv accelerator.— Anal. Chem., 1967, v. 39, p. 448.
327
101. Штань А. С. и др. Чувствительность активационного анализа с исполь-
зованием реакций на 14 Мэв нейтронах.— В кн.: Радиационная тех-
ника. М„ Атомиздат, 1970, вып. 5, с. НО.
102. Mathur S. С., Oldham G. Interferences encountered in 14 Mev neutron
activation analysis. — Nucl. Energy, sept. — oct., 1967, p. 136.
103. Schulze W. Fast neutrons in activation analysis. — L'Analyse par Radio-
activation et ses applications aux sciences biologiques. Paris, 1964, p. 85.
104. Oldham G., Darrall K. G. The use of threshold energies in 14—Mev neut-
ron activation analysis.— Internat. J. Appl. Radiat. and Isotopes 1969,
v. 20, No. 1, p. 29. '
105. Green D. E. Activation analysis with a low’ power research reactors. V. 3.
London, IAEA, 1962, p. 315.
106. Маслов И. А. Планирование активационного анализа в условиях боль-
шого по величине потока нейтронов.— In: Radiochemical Methods of
Analysis. Vienna, IAEA, 1965, y. 1, p. 41.
107. Пелекнс Л. Л. Относительный и абсолютный способы нейтронноактива-
ционного анализа. — В кн.: Методы и применение нептронноактивацион-
ного анализа. Рига, «Зинатне», 1969, с. 5.
108. Kamemoto Y., Yamagishi S. Further studies of the neutron shielding of
metals in thermal neutron. — Bull. Chem. Soc. Japan, 1964, v. 37, p. 664.
109. Дамбург H. Д., Пелекис Л. Л., Протасова Л. Ф. Уменьшение потока и
его учет в нейтронноактивационном анализе. — В кн.: Нейтронноактива-
ционный анализ. Рига, «Зинатне», 1966, с. 15.
ПО. Hogdahl О. Т. Neutron absorption in pile neutron activation analysis. —
In: Radiochemical Methods of Analysis. V. I. Vienna, IAEA, 1965, p. 23.
111. Жарков В. А. О влиянии эффектов самоэкранирования и возмущения
нейтронного потока на процесс активации образцов тепловыми нейтро-
нами.— В кн.: Радиационная техника. Вып. 3. М., Атомиздат, 1969, с. 3.
112. Reynolds S. A., Mullins W. Т. Neutron flax perturbation in activation ana-
lysis.— Internat. J. Appl. Rad. and Isotopes, 1963, v. 14, p. 421.
113. Gijbels R., Hoste J. The determination of traces of iridium in rhodium
by neutron activation analysis. — Anal. chim. acta, 1967, v. 39, p. 89.
114. Яковлев Ю. В., Догадкин H. Н. Использование радиоактивации для оп-
ределения примесей в веществах высокой чистоты. — In; Radiochemical
Methods in Analysis. Vienna, IAEA, 1965, v. 1, p. 187.
115. Лобанов E. M. и др. Определение тантала и вольфрама в боре актива-
цией тепловыми нейтронами реактора. — В кн.: Активационный анализ
чистых материалов. Ташкент, Изд-во ФАН УзССР, 1968, с. 16.
116. Cali J. Р. Neutron activation analysis. — In: Trace Analysis of Semicon-
ductor Materials. Oxford—London — New York. Pergamon Press,
1964, p. 6.
117. Kemp D. M., Smales A. A. The determination of vanadium in rocks and
meteorites bv neutron activation analysis. — Anal. chim. acta, 1960,
v. 23, p. 397.'
118. Тушкова P. Я. и др. Активационный анализ на фоне помех от порою-
вых реакций. — В кн.: Активационный анализ. Ташкент, Изд-во ФАН
АН УзССР, 1971, с. 47..
119. Durham R. И., Navalkar М. Р„ Ricci Е. Threshold reaction interference
in neutron activation analysis. — In: Proc. Internat. Conf. Modern Trends
in Activation Analysis, Texas. 1961, p. 67.
120. Tomura K., Highchi H. The determination of copper in zinc by neutron
activation analysis using the Cu-65 (n. y) Cu-66 reaction.--Analyt. chim.
acta, 1967, v. 37, p. 33.
121. Лобанов E. M., Янковский А. В. Активационное определение висмута в
минеральном сырье. — «Ж. аналит. химии», 1966, т. 21, с. 1264.
122. Maenhaut W., Opde Beeck J. P. Interference bv second order reactions in
activation analysis.— J. Radioanalyt. Chem., 1970, v. 5. p. 115.
123. Opde Beeck J, P. A compilation of second order reaction interferences.—
J. Radioanalyt. Chem., I960, v. .3, p. 431; 1970, v. 4, p. 137.
328
124. Богданкевич О. В., Николаев Ф. А. Работа с пучком тормозного излу-
чения. М., Атомиздат, 1964.
125. Oka Y. е. a. Gamma-ray spectrometric study of the photoactivation pro-
ducts with 20 Mev bremsstrahlung. — J. Nucl. Science and Technology,
1967. v. 4 (7), p. 346.
126. Якобсон A. M. Расчет чувствительности гамма-активационного анали-
за.— В кн.: Трхды ин-та металлургии им. А. А. Байкова Вып 8 М..
Изд-во АН СССР, 1961, с. 178.
127. Lutz G. I. Calculation of sensitivities in photon activation analysis. —•
Analyt. Chem., 1969, v. 41, p. 424.
128. Oka Y. e. a. The research of yields of (у. P) reactions induced with
20 Mev bremsstrahlung. — Bull. Chem. Soc. Japan 1968 v. 41, No. 2,
p. 380.
129. Engelmann Ch. Analyses de tres faibles traces d'elements legers dans les
Materiaux de tres haute purete par activation dans les photons y.—
Internal. J. Appl. Radiat and Isotopes, 1967, v. 18, p. 569.
130. Veres A., Pavlicsek I. Nuclear photoactivation analysis by means of on
80 kCi 6,’Co y-radiation source. — J. Radioanalyt. Chem., 1969, v. 3, p. 25.
131. Абрамс И. А., Пелекис Л. Л. Гамма-активационный анализ при помощи
гамма-лучей с энергией ниже порога фотоядерных реакций. — В кн.:
Методы я применение нейтронноактивационного анализа. Рига, «Зинат-
ие», 1969, с. 71.
132. Ананьев Л. М., Воробьев А. А., Горбунов В. И. Индукционный ускори-
тель электронов — бетатрон. М„ Госатомиздат, 1961.
133. Воробьев А. А., Москалев В. А. Сильноточный бетатрон и стереобета-
трон. М„ Атомиздат. 1969.
134. Моринага X. Получение радиоизотопов на бетатроне по методу внутри-
камерного облучения. — В кн.: Получение и применение радиоактивных
изотопов. Избр. докп иностр ученых М.. Госатомиздат, 1962, с. 20.
135. Кузнецов Р. А., Грушко Ю. С., Котельников Л. А. Фотоактивационное
определение кислорода в соединениях с иодом. — «Радиохимия», 1970,
т. 12, с. 531.
136. Кузнецов Р. А. Аналитические возможности бетатрона с внутрикамер-
ным облучением. IX’. Точность и источники погрешности. «Радиохимия»,
1972, т. 14, № 6, с. 876.
137. Кузнецов Р. А. Аналитические возможности бетатрона с внутрикамер-
ным облучением. I. Элементы Н—Вг. — «Радиохимия», 1970, т. 12, с. 908.
138. Вассерман А. М. и др. Метод выделения 15О при активационном опре-
делении кислорода в чистых веществах. — «Ж. аналит. химии», 1969,
т. 24, с. 1710.
139. Капица С. П., Мелехин В. Н. Микротрон. М., «Наука», 1969.
140. Коломенский А. А. Состояние и перспективы развития электронных уско-
рителей. — В кн.: Тр. Междунар. конф, по электромагнитным взаимодей-
ствиям при низких и средних энергиях. Т. 4. Дубна, 1967, с. 3,
141. Аллен В. Д, Регистрация нейтронов. Пер. с. англ. Под ред. Б. В. Рыба-
кова. М., Госатомиздат, 1962.
142. Межиборская X. Б. Фотонейтронный метод определения бериллия. М„
Госатомиздат, 1961.
143. Берзин А. К., Сулин В. В. Фотоядерные методы анализа вещественного
состава образцов горных пород и руд. — В кн.: Состояние и перспективы
ядерногеофизических методов поиска и разведки полезных ископаемых.
М„ «Недра», 1969, с. 202.
144. Аббосов О. и др. Определение бериллия по реакции (у, п) на линейном
ускорителе. — «Изв АН ЛатвССР, сер. физ. и техн, наук», 1970, № 4,
с. 27.
145. Кодири С., Старчик Л. П. Определение элементов по реакции (у, у')- —
«Заводск. лаборатория», 1970, т. 36, с. 191.
146. Andersen G. Н. е. a. Photonuclear activation analysis of biological mate-
rials for various elements. — In: Nucl. act. techniques in the life scien-
ces, Vienna, IAEA, 1967, p. 99.
329
147. Dcbrun J., Albert Ph. Irradiation de quelques elements naturels dans les
photons de 35 Mev, applications en analyse par activation. — Bull. Soc.
chim. France, 1969, No. 3, p. 1020.
148. Кузнецов P. А. Аналитические возможности бетатрона с внутрикамер-
ныч облучением. II. Элементы Rb—U.--«Радиохимия», 1971,
т. 13, с. 473.
149. Кузнецов Р. А. Аналитические возможности бетатрона с внутрикамер-
ным облучением. III. Избирательность и чувствительность. — «Радиохи-
мия», 1971, т. 13, с. 475.
150. Baker С. A., Wood D. A., Hunter G. J. A cataloque of 30 Mev gamma
activation products.—AERE — R5547 (1967); AERE —R5818 (1968).
151. Adams F., Dams R. A compilation of precisely determined gamma-transi-
tion energies of radionuclides produced by reactor irradiation.—J. Radio-
analyt. Chem., 1969, v. 3, p. 99.
152. Galatanu V., Grecescu M. Tables of gamma ravs emited by radionuclides
produced through photonuclear reactions.—J. Radioanalyt' Chem., 1972,
v. 10, p. 315.
153. Lutz G. J. Photon activation analysis a review.—Analvt Chem. 1971,
v. 43, p. 93.
154. Engelmann Ch. Determination of oxygen, carbon, nitrogen and certain
other impurities by gamma ray activation. — In: Radiochemical Methods of
Analysis. V. I. Vienna, IAEA, 1965, p. 341.
155. Азимов С. А. и др. Одновременное гамма-активационное определение
углерода, кислорода и азота в природном угле. — «Тр. Ташк. ун-та»,
1970, вып. 388, с. 97.
156. Азимов С. А., Миллер Р. А., Хакимов М. Возможности экспрессного гам-
ма-активационного анализа. — «Тр. Ташк. ун-та», 1970, вып. 388, с. 5.
157. Азимов С. А., Миллер Р. А., Хакимов М. Гамма-активационное опреде-
ление содержания фтора в флюоритовой руде. — «Тр. Ташк. ун-та»,
1970, вып. 388, с. 92.
158. Плаксин И. Н., Старчик Л. П. Ядернофизические методы контроля ве-
щественного состава. М., «Наука», 1966.
159. Гедеонов А. Д., Кузнецов Р. А. Фотоактпвационный анализ Ga—Nb-
сплавов. — «Радиохимия», 1971, т. 13, с. 783.
160. Гвахария В. В., Токви И. Г., Кузнецов Р. А. Фотоактивационное опреде-
ление кремния в марганцевых рудах, продуктах переработки и спла-
вах.— В кн.: Марганец. Тбилиси, Груз. НИИ науч.-техн, информации,
1970. 2 (23), с. 90.
161. Миллер Р. А., Хакимов М. Гамма-активационный анализ многокомпо-
нентных систем. — «Тр. Ташк. ун-та», 1970, вып. 388, с. 168.
162. Ricci Е., Hahn R. L. Rapid calculation of sensitivities interferences and
optimum bombarding energies in He-3 activation analysis. Analyt. Chem.,
1968, v. 40, p. 54.
163. Краснов H. H. Физические основы активационного анализа на заряжен-
ных частицах. — «Атомная энергия», 1969, т. 26, с. 284.
164. Ricci Е., Hahn R. L. Sensitivities for activation analysis of 15 light ele-
ments with 18 Mev He-3 particles. — Analyt. Chem., 1967, v. 39, p. 794.
165. Rook H. L., Schweikert E. A. Ultra-trace determination of oxigen and car-
bon bv charged particle activation analysis. — Analvt Chem., 1969,
v. 41, p. 958.
166. Odeblad E. Activation analysis with charged particles. — Acta Radiol.,
1956, v. 45, p. 402.
167. Engelmann Ch. Activation analysis of some light elements in various ma-
terials using charge particles. — In; Radiochem. Methods in Analysis.
V. I. Vienna, IAEA, 1965, p. 405.
168. Задворный А. С. и др. Определение содержания кислорода в бериллии
активацией протонами. — «Ж. аналит. химии», 1970, т. 25, с. 346.
169. Pretorius R., Wainerdi R. Е. Determination of deuterium in heavy water
by secondary deutron activation. — Taianta, 1970, v. 17, p. 51.
170. Александрова Г. И., Веников Н. И., Шманенкова Г. И. Использование
330
ускоренных ионов Не-3 для анализа высокочистых материалов. — В кн.:
Аналитическая химия редких металлов и полупроводниковых материа-
лов. М._ Моск, дом науч.-техн, пропаганды, 1970, с. 194. [Материалы
семинара.]
171. Osmond R. G., Smales A. A. The determination by radioactivation of the
oxxgen content of powdered metals with particular reference to beryl-
lium.— Analyt. chim. acta, 1954, v. 10, p. 117.
172. Nikolow K-, Todorovsky D. On the non-dcstructive neutron activation
method for the determination of lithium based on a secondury nuclear re-
action with tritons. — Isotopenpraxis. 1969, v. 5, p. 408.
173. Barrandon J. N. e. a. Utilisation des deutons et des tritons de faibles
energies pour le dosage du carbone et de I’oxygene. — J. Radioanalyt.
Chem., 1970. v. 4, p. 115.
174. Markowitz S. S., Mahony J. D. Activation analysis for oxygen and other
elements bv He-3 induced nuclear reactions. — Analyt. Chem., 1962,
v. 34, p. 329.
175. Engelmann Ch. Contribution a 1’etude de la determination de Be, В, C,
N. О et F par activation au moyen de p, d. 3H et a. — J. Radiodnal.
Chem., 1971, v. 7, p. 89, 281.
176. Краснов H. И. и др. Выходы изотопов 13N, пС, 18F при облучении уг-
лерода и кислорода заряженными частицами. — «Атомная энергия»,
1969, т. 27, с. 125.
177. Nozaki Т., Yatsurugi Y., Akiyama N. Charge particle activation analysis
for carbon, nitrogen and o.xvgen in semiconductor silicon. — .1. Radioanalyt.
Chem., 1970, v. 4, p. 87.
178. Price P. B., Bird J. R. Proton microanalysis. — Nucl. Instrum, and Met-
hods, 1969, v. 69, No. 2, p. 277.
179. Скакун H. А. и др. Использование ядерной реакции 18О р, a) 15N для
изучения окисления металлов. — «Атомная энергия», 1971, т. 30. с. 456.
180. Джемардьян Ю. А„ Михаилов Г. И., Старчик Л. П. Изотопный анализ
Li и В по гамма-квантам, сопровождающим ядерные реакции
7Li (а, а') 7*Li, 10В (а, р) 13*С.— «Радиохимия», 1970, т. 12, с. 787.
181. Лемберг И. X., Гришин А. Б., Гусинский Г. М. Определение lsO по реак-
ции 18О (а, п у) 21Ne.— «Заводск. лаборатория», 1966, т. 32, с. 1499.
182. Мещеряков Р. П., Столбов Ю. М. Некоторые результаты измерений изо-
топного состава кальция в природных веществах. — «Геохимия», 1967,
№ 10, с. 1118.
183. Альфа-, бета- и гамма-спектроскопия. Пер. с англ. Вып. 1. Под ред.
К. Зигбана. М., Атомиздат, 1969.
184. Столярова Е. Л. Прикладная спектрометрия ионизирующих излучений.
Под ред. Г. Д. Латышева. М„ Атомиздат, 1964.
185. Peisach М., Pretorius R. Determination of deuterium in gases by neutron
time-of-flight spectrometry. — Analyt. Chem., 1967, v. 39, p. 650.
186. Rosholt J. N., Szabo B. j. Determination of protactinium by neutron acti-
vation and alpha spectrometry. — Proc. Internal. Conf. Modern Trends in
Activation Analysis. Washington, 1969, p. 327.
187. Amsel G., Samuel D. Microanalysis of stable isotopes of oxygen bv means
of nuclear reactions. — Analyt. Chem.. 1967, v. 39, p. 1689.
188. Winchester J. W. Determination of potassium in silicate minerals and
rocks neutron activation analysis. — Analyt. Chem., 1961, v. 33, p. 1007.
189. Gallic Y., Ledrand J., Grinberg B. Methode rapide d'identification des
emetteurs-p. Internat. J. Appl. Rad. and. Isotopes, 1962. v. 11, No. 2/3, p. 92.
190. Трусевич В., Жарновецки К., Пшона С. Быстрый метод идентификации
бета-излучателей.—«Атомная техника за рубежом». 1969, № 3. с. 44.
191. Prevo С. Т., Cate J. L. A practical solid-state beta spectrometer. — Nucl.
Instrum, and Methods, 1967, v. 55. No. 1, p. 173.
192. Котельников Л. А., Кузнецов P. А. Фотоактивациониый анализ фосфор-
но-сурьмяного катионита. — «Вестник ЛГУ», 1970, № 22, вып. 4. с. 137.
193. Акимов Ю. К. и др. Полупроводниковые детекторы ядерных частиц и
их применение. М., Атомиздат, 1967.
331
194. Miller С. E. e. a. Reduction of background of Nal(Tl) detectors. — Nu-
cleonics, 1956, v. 14. No. 4, p. 40.
195. Girardi F., Gazzi G, Gamma — ray spectroscopy by means of Ge(Li) de-
tectors in activation analysis. — In: Adv. in Activation Analysis. V. I.
London, 1969, p. 137.
196. Булатов Б. П. Состояние и перспективы развития современных полупро-
водниковых детекторов ионизирующих излучений. — «Атомная энергия»,
1970, т. 30, с. 285.
197. Heath R. L. Gamma - ray spectrometry and automated data system for
activation analysis. — Proc. Internat. Conf. Modern Trends in Activation
Analysis. Washington. 1969, p. 959.
198. Егоров К). А. Сцинтилляционный метод спектрометрии гамма-излучения
и быстрых нейтронов. М„ Госатомиздат, 1962.
199. Маталин Л. А., Наран Ж., Чубаров С. И. Методы регистрации и обра-
ботки данных в ядерной физике и технике. М., Атомиздат, 1968.
200. Quittner Р., Wainerdi R. Е. Computer evaluation of Nal(Ti) and Ge(Li)
gamma-ray spectra.—Atomic Energy Rev., 1970, v. 8, No. 2, p. 361.
201. Helmer R. G., Greenwood R. C., Gehrke R. J. Precise comparison and
measurement of gamma — ray energies with a Ge(Li) detector. — Nucl.
Instrum, and Methods, 1970, v. 77, p. 141; 1971, v. 96, p. 173.
202. Вартанов H. A„ Самойлов П. С. Практические методы сцинтилляцион-
ной гамма-спектрометрии. Под ред. В. В. Матвеева. М„ Атомиздат,
1964.
203. Волков А. Н., Штраннх И. В. Системы стабилизации спектрометров
ядерного излучения. — В кн.: Прикладная ядерная спектроскопия.
Вып. 1. М., Атомиздат, 1970. с. 175.
204. Dams R., Adams F. A compilation of gamma — transition energies of ra-
dionuclides produced by reactor irradiation. — J. Radioanalvt. Chem.,
1971, v. 7, No. 1. p. 127. "
205. Pagden M. H., Pearson G. J., Bewers J. M. An isotope catalogue for in-
strumental activation analysis. — J. Radioanalvt. Chem., 1971, v. 8, p. 127,
373; v. 9, p. 101.
206. Crouthamel С. E. Applied Gamma — Ray Spectroscopy. Ed. 2. Oxford,
Pergamon Press, 1970.
207. Covell D. F. Determination of gamma — ray abundance directly from the
total absorption peak. — Analyt. Chem., 1959, v. 31, p. 1785.
208. Liebscher K., Smith H. Quantitative interpretation of gamma — ray
spectra. — Analyt. Chem., 1968, v. 40, p. 1999.
209. Лобанов E. M. н др. Некоторые вопросы интерпретации гамма-спектров
при анализе многокомпонентных смесей. — В кн.; Активационный анализ
горных пород и других объектов. Ташкент Изд-во ФАН УзССР,
1967, с. 56.
210. Sterlinski S. Analysis of digital data from a multichanal analyzer in acti-
vation analysis. — Analyt. Chem., 1968, v. 40, p. 1995.
211. Sterlinski S. Features of the modified Covell method for computation of
totla absorption peak areas in complex gamma-ray spectra. — Analyt.
Chem.. 1970, v. 42. p. 151.
212. Yule H. P. Computation of Ge(Li)-detectors peak areas for activation ana-
lysis.— Analyt. Chem., 1968. v. 40. p. 1480.
213. Курочкин С. С. Обработка составных гамма-спектров no методу норми-
ровки— вычитания. — В кн.: Тр. Союзного и.-и. ин-та приборостроения.
Вып. 5. М„ Атомиздат, 1967, с. 27.
214. Furr А. К., Robinson Е. L., Robins С. Н. A spectrum stripping technique
for qualitative activation analysis using monoencrgetjc gamma spectra. —
Nucl. Instrum, and Methods. 1968. v. 63. p. 205.
215. Lee W. Direct estimation of gamma-ray abundances in radionuclide mix-
tures.— Analyt. Chem., 1959, v. 31, p. 800.
332
216. Wiernik IW., Amiels S. Use ol very short lived nuclides in nondestructive
activation analvsis with a fast shuttle rabbit.—J. Radioanalvt. Chetn.,
1969, v. 3, p. 393.
217. Yule H. P. Mathematical smoothing of gamma-rav spectra. — Nucl.
Instrum, and Methods, 1967, v. 54, No. 1, p. 61.
218. Adams F., Dams R. Computer—assisted qualitative analysis of gamma-ray
spectra. — J. Radioanalyt. Chem., 1971, v. 7, p. 329.
219. Dams R. e. a. Nondestructive neutron activation analysis of air pollution
particles.— Analyt. Chem., 1970, v. 42, p. 861.
220. Pierce T. B., Smales A. A. Some recent Harwell work on activation analv-
sis. — Z. analyt. Chem., 1966. Bd. 221, S. 80.
221. Pierce T. B., Peck R. F., Cuff D. R. Analytical applications of a 0,5—
Mev Cockroft — Walton set, based on the measurement of prompt gamma-
radiation. --Analyst, 1967, v. 92, No. 1092, p. 143.
222. Васильев С. С. и др. Использование реакции (р, у) для определения
содержания легких элементов в тонких поверхностных слоях образцов.—
«Атомная энергия», 1971, т. 30, с. 307.
223. Rabin S., Passel Т. О., Baily L. Е. Chemical analysis of surface by nuc-
lear methods.— Analyt. Chem., 1957, v. 29, p. 736.
'224 . Егиазаров Б. Г. Нейтронно-радиационные аналитические методы и прибо-
ры.— В кн.: Ядерное приборостроение. Вып. 14. М„ Атомиздат, 1971,
с. 63.
.‘22 5. Isenhour Т. L., Morrison G. Н. Modulation technique for neutron capture
gamma-rav measurements in activation analvsis.-- Analvt. Chem., 1966,
v. 38, p. 162.
226. Duffey D., El Kady A. Analytical sensitivities and energies of thermal —
neutron — capture gamma — rays.— Nucl. Instrum, and Methods, 1970,
v. 80. No. 1, p. 149.
227. lombard S. M.. Isenhour T. L. Neutron capture gamma — ray activation
analysis using Ge (Li)-detectors.—Analyt. Chem.. 1968, y. 40, p. 1990.
228. Демидов A. M., Говор Л. И., Иванов В. А. Использование германиевого
детектора для нейтронно-радиационного анализа содержания элементов
и изотопов.— «Атомная энергия». 1970, т. 28, с. 115.
229. Лобанов Е. М., Арипов Г., Аллабергенов Б. Р. Об идентификации спект-
ров гамма-лучей радиационного захвата многокомпонентных систем.
В кн.: Активационный анализ элементного состава геологических объек-
тов. Ташкент, Изд-во ФАН УзССР. 1967, с. 38.
230. Lombard S. М. е. a. Neutron capture gamma-ray activation analysis. De-
sign of apparaturs for trace analvsis.— Internal. J. Appl. Rad. and Isoto-
pes, 1968. v. 19, p. 15.
231. Хайдаров А. А. Ядерно-геофизические методы анализа горных пород и
руд. Ташкент, Изд-во ФАН УзССР. 1969.
232. Лобанов Е. М.. Романов М. М., Хайдаров А. А. Определение ртути в
рудах и концентратах с помощью спектрометрии радиации нейтронного
захвата.— В кн.: Радиационные эффекты в конденсированных средах.
Ташкент, Изд-во ФАН УзССР. 1964, с. 111.
233. Comar D. е. a. The use of neutron capture gamma radiations for the ana-
lysis of biological samples. — In: Proc. Internal. Conf. Modern Trends in
Activation Analysis. Washington, 1969. p. 114.
234. Chen N. S., Fremlin J. H. Activation analysis by pulsed cyclotron beams.—
Nucl. Instrum and Methods. 1970, v. 85, No. 1. p. 61.
235. Golanski A. Contribution to 14 Mev neutron activation analysis using
half-lives between 0.55 and 873 msec. - J. Radioanalvt. Chem., 1969, v. 3,
p. 161.
236. Sterlinski S. The dependence of activation upon the pulse form in a pulsed
neutron generator. — J. Radioanalyt. Chem., 1971, v. 7, p. 351.
237. Николаенко О. К., Штань А. С. О цикличном активационном анализе по
короткоживущим изотопам. — «Заводск. лаборатория», 1967. т. 33,
с. 1102.
333
238. Лобанов Е. М., Малышев А. И., Чанышева Т. И. Цикличный способ об-
лучения и измерения активности при анализе по короткоживущим изото-
пам.— В кн.: Активационный анализ элементного состава геологических
объектов. Ташкент, Изд-во ФАН УзССР. 1967, с. 14.
239. Николаенко О. К., Сидоров А. В., Штань А. С. Введение поправок на
«мертвое время» многоканального анализатора в активационном анализе
по короткоживущим изотопам.— В кн.: Радиационная техника. Вып. 5.
М., Атомиздат, 1970, с. 120.
240. Меднис И. В., Пелекис Л. Л. Определение начальной скорости счета при
измерении гамма-спектров короткоживущих изотопов на многоканальных
анализаторах. — «Изв. АН ЛатвССР, сер. физ. и техн, наук», 1970, № 1,
с. 3.
241. Wiernik М. Comparison of several methods proposed for correction of
dead-time losses in the gamma—ray spectrometry of very short-lived nuc-
lides.— Nucl. Instrum, and Methods. 1971, v. 95, No. 1, p. 13.
242. Monk R. G., Mercer A., Dounham T. Analysis of decay curves.— Analyt.
Chem., 1963. v. 35, p. 178.
243. Proc. Symp. on Applications of Computers to nuclear and radiochemistry.
Washington, NAS-NS-3107, 1963.
244. Вартанов H. А., Самойлов П. С. Прикладная сцинтилляционная гамма-
спектрометрия. Под ред. В. В. Матвеева. М., Атомиздат, 1969.
245. Cooper J. A. Radioanalytical applications of gamma — gamma coincidence-
techniques with Ge (Li ('-detectors. — Analyt. Chem., 1971, v. 43, p. 838.
246. Шепилов В. И. Полупроводниковые гаммы-спектрометры с подавлением
вклада комптоновского рассеяния.— «Приборы и техника эксперимента»,
1972, № 2, с. 7.
247. Cooper J. A., Rancitelli L. A., Perkins R. W. An anticoincidence — shielded
Ge(Li) gamma—ray spectrometer and its application to radioanalytical
chemistry pioblems.— J. Radioanalyt. Chem.. 1970, v. 6, p. 147.
248. Pagden M. H., Sutherland J. C. Resolving power of gamma—rav coinci-
dence spectrometry using Ge(Li)-detectors and its application to multiple-
radioisotope analysis.— Analyt. Chem., 1970, v. 42, p. 383.
249. Schulze W. Application of coincidence methods in activation analysis.— In:
Proc. Internat. Conf. Modern Trends in Activation Analysis. Texas, 1965,
p. 272.
250. Cooper J. A. Evaluation of Ge (Li) Compton suppression spectrometers for
non-destructive radiochemical analysis.— J. Radioanalyt. Chem., 1970, v. 6,
p. 177.
251. Амиел С., Пейсах M. Аналитическое применение задержанных нейтро-
нов.— «Атомная энергия», 1963, т. 14, с. 353.
252. Amiel S., Gilat J. Uranium content of chondrites by thermal neutron acti-
vation and delayed neutron counting.— Geochim. at cosmochim. acta, 1967,
v. 31, p. 1499.
253. Engelmann Ch., Scherle A. C. Determination de I'oxygene et du fluor au
moyen de I’activation par les photons gamma et le comtage des neutron
retardes emis par 17N.— J. Radioanalyt. Chem., 1970. v. 6, p. 235.
254. Дорош M. M., Мазюкевич H. П.. Шкода-Ульянов В. А. Определение
кислорода фотонейтронным методом.— В кн.: Электронные ускорители.
М., «Энергия», 1968, с. 561.
255. Amiel S., Peisach М. Radioactive determination of natrium by- counting
fotoneutrons.— Analyt. Chem.. 1963. v. 35. p. 1072.
256. Amiel S., Juliano J. O. Fast and non-destructive determination of S and
Ca by radioactivation and counting fotoneutrons.— Analyt. Chem., 1965.
v. 37, p. 346.
257. Lukens H. R., Lasch J. E. Use of the Cherenkov counter in the determina-
tion of oxygen by neutron activation analysis.— Internat. J. Appl. Rad.
and Isotopes, 1964, v. 15, p. 759.
258. Lukens H. R. A neutron activation analysis method for the determination
Be, Li, B, F and Pb. — J. Radioanalyt. Chem., 1968, v. 1, p. 349.
334
259. Price P. B., Walker R. M. Fission tracks in micas and dating minerals. —
.1. Geophys. Research.. 1963. v. 68, p. 4847.
260. Шуколюков Ю. А. Деление ядер урана в природе. М„ Атомиздат, 1970.
261. Fleischer R. L. Distribution of uranium in stone meteorite bv fission
trecks.— Geochirn. et cosmochim. acta, 1968, v. 39. p. 999.
262. Берзина А. Г. и др. Дозиметрия нейтронных потоков по следам от ос-
колков деления урака.— «Тр. ВНИИЯГГ», 1969, вып. 7. с. 215.
263. Fisher D. F. Homogenized fission track determination of uranium in whole
geologic samples.— Analyt. Chem., 1970, v. 42, p. 414.
264. Берзина И. F. Использование следов от осколков деления тяжелых эле-
ментов в веществах для решения практических задач.— «Тр. ВНИИЯГ1»,
1969. вып. 5, с. 298.
265. Долежал Я., Повондра П„ Шульцек 3. .Методы разложения горных пород
и минералов. Пер. с чешского. М., «Мир», 1968.
266. Гиллебрандт В. Ф. и др. Практическое руководство по неорганическому
анализу. Пер. с англ. Изд. 3-е. М„ «Химия», 1966.
267. Лаврухина А. К., Малышева Т. В., Павлоцкая Ф. И. Радиохимический
анализ. М„ Изд-во АН СССР. 1963.
268. Дементьев В. А. Измерение малых активностей радиоактивных препара-
тов. М.. Атомиздат, 1967.
269. Kamemoto Y., Yamagashi Sh. The reactivation analysis of aluminium —
the activation analysis of aluminium with special reference to yield deter-
mination.— Bull. Chem. Soc. Japan, 1963, v. 36, p. 1411.
270. Ефремов Г. В. Литература по неорганическому химическому анализу.
Л., Изд-во ЛГУ. 1964.
2/1. Радиохимия и химия ядерных процессов. Под ред. А. Н. Мурина. Л.,
Госхимиздат, 1960.
272. Самуэльсон О. Ионообменные разделения в аналитической химии. Пер. с
англ. М.. «Химия», 1966.
273. Тремийон Б. Разделения па ионообменных смолах. Пер. с фрагнц. М.,
«Мир». 1967.
274. Егоров Е. В., Макарова С. Б. Ионный обмен в радиохимии. Под ред.
А. В. Матвеева. М„ Атомиздат, 1971.
275. Лурье А. А. Сорбенты и хроматографические носители. М„ «Химия»,
1972.
276. Херинг Р. Хелатообразующие понообменники. Пер. с англ. М.. «Мир»,
1970.
277. Кокотов Ю. А., Пасечник В. А. Равновесие п кинетика ионного обмена.
Л., «Химия». 1970.
278. Калинин А. И., Кузнецов Р. А., Осипчук И. Л. Ионообменное поведение
мышьяка в растворах плавиковой кислоты.—«Вестник ЛГУ». 1968, №16,
с. 155.
279. Моррисон Дж., Фрайзер Г. Экстракция в аналитической химии. М., Гос-
химиздат. 1960.
280. Золотов Ю. А., Кузмин Н. М. Экстракционное концентрирование. М„
«Химия», 1971.
281. Стары И. Экстракция хелатов. Пер. с англ. М., «Мир», 1966.
282. Золотов Ю. А. Экстракция внутрикомплексных соединений. М.. «Наука»,
1968.
283. Золотов Ю. А., Петрухин О. М. Теория и практика экстракции.—
«Ж. ВХО им. Д. И. Менделеева». 1964, т. 9, № 2, с. 145.
284. Руденко Н. П. Выделение радиоактивных изотопов без носителя экстрак-
цией органическими растворителями. — «Тр. комиссии по аналитической
химии», 1963, т. 14, с. 11.
285. Вдовенко В. М. Экстракция как метод выделения и изучения радиоак-
тивных элементов.— «Ж. пеорг. химии». 1958, т. 3. с. 14о.
'286 . Спиваков Б. Я.. Золотов Ю. А. Обменная экстракция внутрикомплексных
соединений. — «Ж. аналит. химии», 1970, т. 25, с. 616.
335
287. Золотов Ю. А., Голованов В. И. Взаимное влияние элементов при эк-
стракции комплексных кислот.— «Ж. аналит. химии», 1970, т. 25, с. 610.
288. Золотов Ю. А., Алнмарнн И. П., Бодня В. А. Кинетика экстракции.—
«Ж. аналит. химии», 1964, т. 19, с. 28.
289. Катыхин Г. С. Применение колоночной распределительной хроматогра-
фии для разделения неорганических веществ.— «Ж. аналит химии» 1972.
т. 27, с. 849.
290. Анвар Б. И., Другое Ю. С. Газовая хроматография в анализе неоргани-
ческих веществ.— «Ж. аналит. химии», 1971, т. 26, с. 1180.
291. Gebauhr W. A separation process for general use’in activation analvsis.—
Radiochim. acta, 1965, v. 4, No. 4. p. 191.
292. Sjostrand B. Simultaneous determination of Hg and As in biological and.
organic materials by activation analysis.—Analyt. Chem. 1964 v. 36.
p. 814.
293. Samsahl K. Radiochemical method for determination of As, Br, Hg, Sb-
and Se in neutron — irradiated biological material.— Analyt. Chem. 1967.
v. 39, p. 1480.
294. Mousty F., Fouarge J., Duyckaerts G. Le dosage de traces d’argent dans
le zinc extra-pur par activation neutronique.— Analyt. chim. acta, 1966.
v. 36, p. 478.
295. Кист А. А., Свиридова А. И., Лобанов E. M. Упрощенное активационное
определение меди в биологических объектах.— В кн/. Активационный
анализ биологических объектов. Ташкент, Изд-во ФАН УзССР 1967,
с. 35.
296. Свиридова А. И. н др. Простейшие методы группового электрохимиче-
ского выделения в активационном анализе биологических объектов.—
В кн/. Активационный анализ биологических объектов. Ташкент Изд-во
ФАН УзССР, 1967, с. 42.
297. Monnier R., Loepfe Е. Separations radiochimiques rapides par echange sur
le mercure. — Analyt. chim. acta, 1968, v. 41, p. 475.
298. Loepfe E., Monnier D., Haerdi W. Schnelle isotopische Ternnugsmethode
zum selektiven Nachweis von kurzlebigen Radioisotopen.— Z. analvt.
Chem., 1966, Bd. 221, S. 109.
299. Zaduban M. Kriterien und Moglichkeiten einstufiger radiochemischer Tren-
nungsmethoden fur Routinanalysen.— Mikrochim. acta, 1970, Bd. 3, S. 433.
300. Терентьев А. П., Яновская Л. А. Химическая литература ц пользование
ею. Изд. 2-е. М., «Химия», 1967.
301. Salamon A. Activation analysis on gallium in tungsten.—J. Radioanalyt.
Chem.. 1970, v. 4, p. 81.
302. Догадкин H. H. Радиоактивашюнный анализ веществ с высоким сече-
нием активации и высоким сечением захвата нейтронов. Автореферат
дисс. М„ ГЕОХИ, 1969.
303. Girardi F., Gazzi G., Di Cola G. The assessment of radiochemical separa-
tion procedures by means of computational techniques —J. Radioanalyt.
Chem., 1970, v. 6. p. 359.
304. Girardi F., Pietra R., Sabbioni E. Radiochemical separation by retention
on ionic precipitate adsorption tests on 11 materials.— J. Radioanalyt.
Chem.. 1970, v. 5, p. 141.
305. Пережогнн Г. А., Алимарнн И. П. К теории субстехиометрического вы-
деления элементов. 1. Вывод условия субстехиометрического выделения
элементов.— «Ж. аналит. химии», 1970, т. 25, с. 862.
306. Пережогнн Г. А. К теории субстехиометрического выделения элементов.
2. Условие экстракционного субстехиометрического выделения.— «Ж. ана-
лит. химии», 1970, т. 25, с. 1041.
307. Пережогнн Г. А. К теории субстехиометрического выделения элементов.
3. Влияние конкурирующих элементов па экстракционное субстехиомет-
рическое выделение.— «Ж. аналит. химии», 1970, т. 25, с. 1048.
308. Пережогнн Г. А. К теории субстехиометрического выделения элементов.
4. Чувствительность и избирательность экстракционного субстехиометри-
ческого выделения.— «Ж. аналит. химии», 1970, т. 25, с. 1245.
336
309. Пережогин Г. А., Алимарин И. П. Нейтронноактивационное определение
золота в горных породах и метеоритах.— «Ж. аналит. химии» 1965 т 20,
с. 793.
310. Obrusnik I.. Adamek A. Replacement subsloichiometrv and its application
in activation analysis.— Taianta, 1968, v. 13, p. 433.
311. Obrusnik I. Determination of In and Sn by activation analvsis using repla-
cement substoichiometry.— Taianta, 1969, v. 16, p. 563.
312. Яковлев Ю. В., Степанец О. В. Субстехиометрическое активационное
определение кадмия и меди в иттрии с использованием вытеснительной
экстракционной хроматографии. — «Ж. аналит. химии», 1970, т. 25. с. 578.
313. Степанец О. В., Яковлев Ю. В., Алимарин И. П. Активационное опреде-
ление примесей в иттрии и молибдене с использованием экстракционной
вытеснительной хроматографии.—«Ж. аналит. химии», 1970, т. 25. с. 1906.
314. Elek A. .Bogancs J., Szabo Е. Substoichiometrv for multielement separation
by metal chelate extraction and its application analysis.— J. Radioanalyt.
Chem., 1970, v. 4. p. 281.
315. Kukuba F., Simkova M. Application of the group substoichiometric separa-
tion of Au. Hg and Cu diethyldithiocarbamates to determing them by
means of activation analysis.— .1. Radioanalyt. Chem., 1970. v. 4, p. 271.
316. Кист А. А. и др. Экспрессное радиохимическое выделение при актива-
ционном анализе биологических объектов.— Тр. 1-го Всесоюз. координа-
ционного совещания по активационному анализе. Ташкент Изд-во ФАН
УзССР. 1964, с. 148.
-317. Неггглап G., Denschlag О. Rapid chemical separations.— Annual. Rev.
Nucl. Sei., 1969, v. 19. p. 1.
318. Зайцева H. Г. Методы быстрого выделения радиоактивных элементов.—
В кн.: Радиохимические методы определения микроэлементов. М.— Л.,
••Наука». 1965. с. 16.
3!С|. Steel Е I , Mpinkp W W Determination of rhodium bv thermal neutron
activation analvsis using gamma-rav spectrometrv.— A’nalvt chim. acta,
1962, v. 26, p. 269.
320. Meinke W. W. Application of rapid radiochemical separations to the acti-
vation -analysis of biological samples.— [.’Analyse par radioactivation et
applications aux sciences biologiques. Paris, Prcsse L'niversitaire, 1964,
p. 145.
321. Firardi F. Radiochemical separations for activation analysis.— In: Proc.
Internat. Conf. Modern Trends in Activation Analvsis. Washington, 1959,
p. 577.
-322. Goode G. C., Baker C. W., Brooke N. M. An automated solventextraction
technique for radiochemical group separations: application to the analysis
of glass fragments bv thermalneutron activation.— Analyst, 1966, v. 94,
No. 1122. p. 728.
323. Samsahl K., Wester P. O., Landstrom O. An automatic group separation
sxstem for the simultaneous determination of a great number of elements
in biological materials.— Analyt. Chem., 1968, v. 40. p. 181.
324. Алимарин И. П. Успехи и проблемы определения следов примесей в чи-
стых веществах.— «Ж. аналит. химии», 1963, т. 18, с. 1412.
325. Watt D. Е., Ramsden D. High Sensitivity Counting Techniques. Oxford —
London - - New York, Pergamon Press, 1964.
326. Лаврухина А. К. и др. Изучение распределения редких элементов меж-
ду различными фазами каменных метеоритов методом нептронноактива-
пионного анализа.— Acta chim. acad. scient. Hungary, 1968, v. 57, No. 4,
p. 353.
327. Lewis C. R., Shafrir N. H. Low level Ge(Li) gamma-ray spectrometry in
marine radioactivity studies.— Nucl. Instrum, and Methods. 1971, v. 93,
p. 317.
328. Leliart G., Hoste J., Eakhaut Z. Neutron activation analysis using an in-
ternal standart.— Nature, 1958, v. 182, p. 600.
329. Малышев К. В. Фотоактивационный анализ оксинитрида циркония. Дип-
ломная работа. ЛГУ. 1969.
337
330. Cali J. P., Weiner J. R. The accuracy radioactivation analysis.— In: Proc.
Internal. Conf. Modern Trends in Activation Analysis. Texas, 1965, p. 253.
331. Girardi F., Guzzi G., Pauly J. Activation analysis by absolute gamma-ray
counting and direct calculation of weights from nuclear constants.— Ana-
lyt. (.hem., 1964, v. 36, p. 1558.
332. Girardi F., Gazzi G., Pauly J. Reactor neutron activation analysis by the
single comparator method.-—Analyt. Chem., 1965, v. 37, p. 1085.
333. Дубинская H. А., Пелекис Л. Л. Применение мониторов в нейтронноак-
тивационном анализе.— В кн.: Методы и применение нейтронноактива-
ционного анализа. Рига, «Зинатне», 1969, с. 35.
334. De Corte F., Speecke A., Hoste J. Reactor neutron activation analysis by
a triple comparator method. - J. Radioanalyt. Chem., 1969, v. 3, p. 205.
335. Morris D. F. Neutron — activation anahsis.— Metallurg Reviews 1962,
v. 7, No. 26, p. 241.
336. Malvano R. Neutron activation analysis in metallurgy.— Atomprexis 1965,
v. 11, No. 6, p. 309.
337. Cook G. B., Crespi M. B., Minczewsky J. International comparison of ana-
lytical methods for nuclear materials. 1. Accuracy and precision of some
techniques in routine trace analysis.— Taianta, 1963, No. 10, p. 917.
338. Idding F. A. A study of flux monitoring for instrumental neutron activa-
tion analysis.— Analyt. chim. acta, 1964, v. 31, p. 206.
339. Anders O. U., Briden D. W. A rapid nondestructive method of precision
oxygen analysis by neutron activation.— Analyt. Chem., 1965, v. 36, p. 287,
340. Mott W. E., Orange J. M. Precision activation analysis with 14 Mev neut-
rons.— Analyt. Chem.. 1965, v. 37. p. 1338.
341. Mathew P. 3., Pohl К. P. A simple cheap continuous flux monitor for
14 Alev neutrons.— Analyt. chim. acta. 1970, v. 51, p. 336.
342. Twitty B. L., Fritz К. M. Internal standard techniques for determination
of oxygen in magnesium, steel and titanium by activation analysis.— Ana-
lyt. Chem., 1967, v. 39, p. 527.
343. Karttunen E., Gardner D. G. A simplified precision system for oxygen ana-
Ixsis bv fast neutron activation.— Internal. J. Appl. Radiat and Isotopes,
1'969, \< 20, p. 801.
344. Лисовский И. П„ Смахтин Л. А. Прецизионная система для определения
кислорода методом активации быстрыми нейтронами.— «Атомная энер-
гия», 1970, т. 29, с. 450.
345. Николаенко О. К., Штань А. С. Учет эффектов самоэкранирования и са-
поглощения при анализе металлов на кислород методом активации бы-
стрыми нейтронами.— В кн.: Радиационная техника. Вып. 4, М., Атомиз-
дат, 1970, с. 175.
346. Van Grieken R., Speecke A., Hoste J. On the precision of oxygen determi-
nations in steel by 14 Mev neutron activation.—Analyt. chim. acta, 1970,
v. 52, p. 275.
347. Brune D., Jirlow K. Determination of oxygen in aluminium by means of
14 Mev neutrons with an account of flux attenuation in the sample.—
J. Radioanal. Chem., 1969, v. 2, p. 49.
348. Perdijon J. Procede de fabrication d'etalons pour le dosage de 1’oxygene-
par activation au moyen de neutrons de 14 Mev.— Nucl. Instrum, and
.Methods, 1969, v. 75, p. 331.
349. Николаенко О. К., Штань А. С. Определение низких содержаний кисло-
рода в металлах методом активации нейтронами с энергией 14 Д1эн. —
В кн.: Радиационная техника. М., Атомиздат, 19/0, с. 183.
350. Nargolwalla S. S., Crambes М. R., Suddueth J. F. Photon selfabsorption
corrections for the minimization of sistematic errors in 14 Mev neutron
activation analysis.— Analyt. chim. acta, 1970, v. 49, p. 425.
351. Morgan J. W., Ehmann W. D. Precise determination of oxygen and seiicon
in chondritic ineteorites by 14 Mev neutron activation with a single trans-
fer system. — Analyt. chim. acta, 1970, v. 49, p. 287.
352. Dugain F., Audre M,, Speecke A. Determination of the oxygen content in
338
aluminium by 14 Mev neutron activation effects of surface removal after
irradiation.— Radiochim. Radioanal. Letters, 1970, v. 4, p. 35.
353. Lutz G, J. Self-shielding corrections in photon activation analysis.— In:
Proc. Internal. Conf. Modern Trends in Activation Analvsis. Washington,
1969, p. 829.
354. Александрова Г. И. и др. Определение содержания кислорода в герма-
нии и кремнии при активации ионами Не-3.—В кн.: Методы определения
и исследования состояния газов в металлах. М„ «Наука». 1968, с. 102.
355. Schweikert Е. A., Rook Н. L. Determination of oxygen in silicon in the
sub-part-per-million range bv charged-particle activation analvsis.— Ana-
lyt. Chem., 1970, v. 42. p. 152’5.
356. Гордадзе Г. П. О выборе оптимального временного режима проведения
активационного анализа.— «Сообщения АН ГрсзССР» 1967, т 48, № 3,
с. 543.
357. Егиазаров Б. Г., Зюбко В. А., Селютин Р. П. Выбор аналитической мето-
дики в инструментальном активационном анализе с учетом чувствитель-
ности и статистической точности измерений.— «Атомная энергия», 1969,
т. 27, с. 460.
358. Dams R. е. a. Nondestructive neutron activation analysis of air pollu-
tion particles.— Analyt. Chem., 1970, v. 42, p. 861.
359. Schmitt R. A., Linn T. A,, Wakita H. The determination of fourteen com-
mon elements in rocks via sequental instrumental activation analysis.—
Radiochim. acta, 1970, v. 13. p. 200.
360. Калинин А. И., Кузнецов P. А., Моисеев В. В. Радиоактивашюнный ана-
лиз двуокиси кремния с применением ионообменной хроматографии.—
В кн.: Радиохимические методы определения микроэлементов. Л., «Нау-
ка», 1964, с. 161.
361. Filby R. Н., Haller W. A., Shah К. R. Determination of 32 elements in
mrk Iv. npiitron actuation analysis and high resolution gamma-ray spect-
rometrv.— J. Radioanalyt. Chem.. 1970, v. 5. p. 277.
362. Morrison G. H. e. a. Multielement neutron activation analysis of rock
using chemical group separation and high resolution gamma-spectromet-
ry— Analyt. Chem.. 1969. v. 41. p. 1633.
363. Colemann R. F. The precision of multielement techniques in activation aria-
hsis.— In: Proc. Internat. Conf. Modern Trends in Activation Analysis.
Washington. 1969. p. 1262.
364. Шукуров Я. Активационный анализ веществ с помощью ускоренных
тяжелых ионов.— В кн.' Активационный анализ. Ташкент, Изд-во ФАН
УзССР, 1971, с. 203.
ПРЕДМЕТНЫЙ УКАЗАТЕЛЬ
Абсолютный метод 36
Активационный анализ 9, 20
— — на заряженных частицах 6, 132
Амальгамный метод 251
Анализ кривых распада 198
Бета-распад 21
Бетатрон 122
Возмущение потока нейтронов 93
Выгорание изотопа 90
Выход ядерной реакции 19, 115, 139
— химический 56, 223
Гамма-активационный анализ 128
Гамма-спектрометр 160
— полупроводниковый 162
— сцинтилляционный 160
Градиент потока 65, 89, ИЗ
Детекторы газонаполненные 29
— полупроводниковые 30
— пороговые 213
— сцинтилляционные 29
Дистилляция 247
Избирательность активационного ана-
лиза 303
— гамма-активационного анализа 308
—• ионного обмена 228
— метода совпадений 208
— нейтронного активационного ана-
лиза 307
— радиохимического варианта 221
— экстракции 238
Излучение задержанное 20
— мгновенное 15, 20, 187
— гамма 23
Измерительные установки 31
Инструментальный вариант 6, 53. 147
Интерферирующие ядерные реакции
— — — второго порядка 108
-------- - первого порядка 103
Ионный обмен 226
340
Источники заряженных частиц 141
— нейтронов 62
— погрешности 281
— тормозного излучения 110
Кадмиевое отношение 80
Каналы ядерной реакции 16
Кинетика ионного обмена 229
— экстракции 242
Комптоновское рассеяние 158
Коэффициент экранирования 94
Кулоновский барьер 17
Линейный ускоритель 70
Мертвое время 31, 35,
Метод антисовпадений 202, 206
— мониторов 37
— совпадений 202, 208
— эквивалентных толщин 138
— эталонов 37, 136
Микротрон 70, 123
Многоэлементный активационный
анализ 310
Монитор 37
— внешний 38, 284
— внутренний 38, 287
Нейтроны быстрые 58, 73, 86
— запаздывающие 211
'— резонансные 58, 73, 81, 98
— тепловые 50, 58, 73, 76, 94
Нейтронные генераторы 67
Нейтронный активационный анализ
6, 77
Нейтронный размножитель 71
Обработка гамма-спектров 171
— — простых 171
---сложных 179
— -- с помощью ЭВМ 182
Образование пар 159
Осаждение и соосаждение 7, 224
Период полураспада 21
Поверхностное травление 10, 55, 298
Подготовка проб 45
Порог определения 39
Предел обнаружения 39
— — критический 39
— определения 39
Радиоактивный распад 20
Радиационный захват 60
Радиоизотопы 5, 20
— короткоживущие 51. 193
— очень короткоживущие 192
— средне- и долгоживущие 52, 195
Радиохимический вариант 7, 54, 218
Радиохимическое разделение 252
--- автоматизированное 267
— — быстрое 8, 265
— — субстехиометрическое 8, 260
Разрешение временное 30
— энергетическое 31, 148
Сечение ядерной реакции 18, 59
Спектр нейтронов реактора 73
Спектрометрия альфа-излучения 149
— бета-излучения 152
— гамма-излучения 157
Статистика измерений радиоактивно-
сти 33
Схема распада 21, 24
Тормозное излучение ПО, 168
Точность активационного анализа 10
--------на быстрых нейтронах 292
— — — на заряженных частицах 301
—-------на тепловых нейтронах ре-
актора 289
— гамма-активационного анализа 298
— метода мониторов 284
— метода эталонов 9Я9
Треки деления 215
Уравнение активации для переменно-
го потока 27
— — заряженными частицами 13&
— — при прерывистом облучении 28
— — простое 25. 26
— — тормозным излучением 118
Фактор очистки 222
Фотоактивашюнный анализ 6, 128
Фотовозбуждение изомерных уровней
126
Фотонейтронный метод 8, 124
Фотоэффект 158
Функция возбуждения 19, 133
Хроматография 230
— ионообменная 7, 226
— экстракционная 244
Электростатические ускорители 7, 121';
Электрохимическое выделение 249
Экстракция 7, 236
Эталоны 49
Эффективность регистрации 32
— детектора 31
Циклотрон 143
’i УБСТБИТСЛЬИОСТЬ 2KTIIB ацИОНИогл-
анализа 9. 271
--------на заряженных частицах.
280
— — — на тепловых нейтронах 278-
— гамма-активационного анализа 2791
Ядерные реакции 15
— — гамма-квантов 116
— — заряженных частиц 143
— — нейтронов 59, 78
— — пороговые 17
Ядерный реактор 5, 71
ОГЛАВЛЕНИЕ
Предисловие .......................................................... 3
Глава 1. Введение......................................................5
§ 1. Исторический обзор................................................5
§ 2. Основные особенности и области применения активационного анализа 9
Глава 2. Теоретические основы активационного анализа . 15
§ 1. ЯдерныЬ реакции.................................................15
§ 2. Радиоактивный распад...................................- 20
§ 3. Уравнение активации..............................................24
Глава 3. Методика активационного анализа..............................29
§ 1. Измерение ионизирующих излучений.................................29
§ 2. Методы получения количественных результатов . .... 36
§ 3. Оценка пределов обнаружения......................................39
§ 4. Общий ход активационного анализа.................................44
Глава 4. Нейтронный активационный анализ..............................58
§ 1. Основные виды взаимодействий нейтронов с атомными ядрами . 58
§ 2. Источники нейтронов..............................................62
§ 3. /Методы нейтронного активационного анализа.......................77
$ 4. Некоторые особенности и источники погрешности при облучении
тепловыми нейтронами.............................................89
Глава 5. Фотоактивационный анализ.....................................ПО
§ 1. Моноэнергетическое и тормозное у-излучение...................ПО
§ 2. Взаимодействие жесткого у-излучения с веществом . . . .116
§ 3. Источники у-излучения ..........................................121
§ 4. Методы фотоактивационного анализа............................... 124
§ 5. Основные области применения фотоактивационного анализа . . 128
Глава 6. Активационный анализ с применением заряженных частиц 132
§ 1. Взаимодействие заряженных частиц с веществом................132
§ 2. Источники заряженных частиц.................................141
§ 3. Методы активационного анализа на заряженных частицах . . 143
§ 4. Применение активационного анализа на заряженных частицах . . 145
Глава 7. Спектрометрические методы инструментального активацион-
ного анализа....................................................147
§ 1. Методы инструментального активационного анализа .... 147
§ 2. Спектрометрия тяжелых заряженных частиц.....................149
§ 3. Спектрометрия (3-излучения..................................152
§ 4. Спектрометрия у-излучения...................................157
§ 5. Аппаратурная форма линии гамма-спектрометра.................164
§ 6. Качественный и количественный анализы спектров..............170
§ 7. Применение у-спсктрометрического метода для инструментального
активационного анализа ..................................... . 187
Глава 8. Специальные методы инструментального анализа - 198
§ 1. Анализ кривых распада...........................................198
342
§ 2. Метод совпадений и антпсовпадений..............................202
§ 3. Аналитическое применение запаздывающих нейтронов . . . .211
§ 4. Дейтерий и бериллий в качестве пороговых детекторов . . .213
§ 5. Применение счетчика Черенкова..................................214
§ 6. Метод треков деления...........................................215
Глава 9. Радиохимические методы анализа.............................218
§ 1. Особенности и методика радиохимических разделений .... 218
§ 2. Метод осаждения и со^саждения..................................224
§ 3. Ионообменная хроматография.....................................226
§ 4. Экстракция . . ’.........................................236
§ 5. Экстракционная хроматография *.................................244
§ 6. Специальные методы радиохимического разделения .... 246
Глава 10. Некоторые практические аспекты радиохимического метода 252
§ 1. Планирование радиохимических разделений........................252
§ 2. Схбстехиометрнческое выделение элементов.......................260
§ 3. Быстрые радиохимические методики...............................265
§ 4. Автоматизация радиохимических разделений.......................267
Глава 11. Аналитические характеристики активационных методов 271
§ 1. Чувствительность...............................................271
§ 2. Точность активационного анализа................................281
§ 3. Избирательность активационного анализа.........................304
§ 4. Многоэлементный активационный анализ...........................311
Список литературы ...............................................394
Предметный указатель................................................340
1
Рафаил Алексеевич Кузнецов
АКТИВАЦИОННЫЙ АНАЛИЗ
Редактор В. А. Подошвина
Художественный редактор А. Т. Кирьянов
Переплет художника Г. А. Жегнна
Технический редактор Н. А. Власова
Корректор О. Р. Харламова
Сдано в набор 13/IX 1973 г.
Подписано к печати 13/ХП 197В г.
Т-14877 Формат 60х90’/|в Бум. типографская № 2
Усл. печ, л. 21,5. Уч.-изд. л. 22.98. «раж 2259.
Цена 2 р. 50 к. Зак. изд. 70058. Зав. тип. 538.
Атомиздат, 103031, Москва, К-31, ул. Жданова, 5.
Московская типография № 6 Союзполиграфпрома
при Государственном комитете Совета
Министров СССР по делам издательств.
полиграфии и книжной торгоЖ-ти
109088. Москва, Ж-88. Южнопортовая ул., 24.