/








































































































































































































































































































































































































































Author: Вартанян Р.С.
Tags: фармакология общая терапия токсикология химическая технология химические производства химия биология
ISBN: 5-89481-218-6
Year: 2004




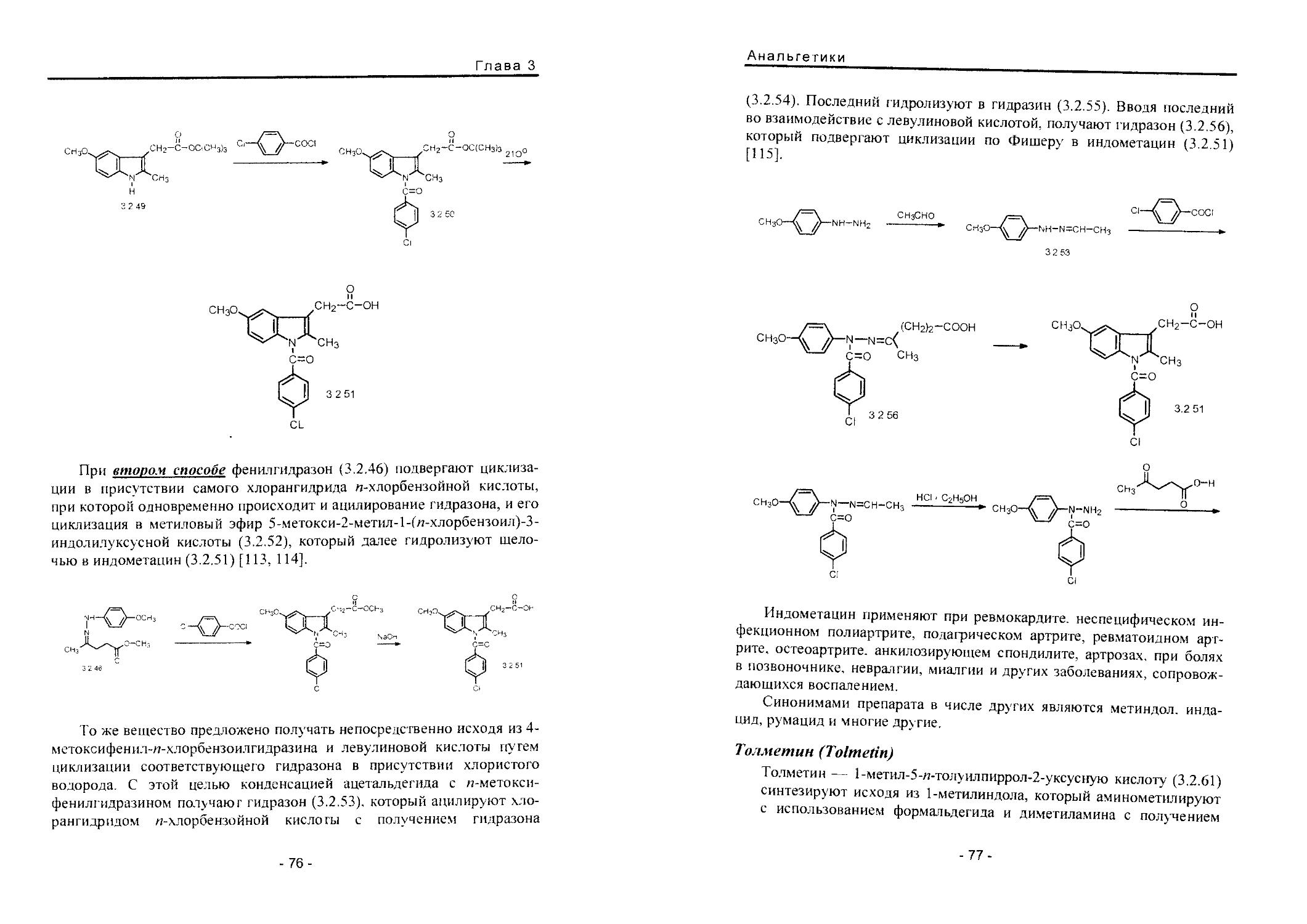
Text
Р.С. Вартанян
СИНТЕЗ ОСНОВНЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ
Медицинское информационное агентство
Р. С. Вартанян
СИНТЕЗ ОСНОВНЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ
Медицинское информационное агентство Москва - 2004
УДК 615.012
ББК 35.66 В18
Вартанян Р. С.
В18 Синтез основных лекарственных средств. — М.: Медицинское
информационное агентство, 2004. — 845 с.
ISBN 5-89481-218-6
В книге описан синтез более 700 лекарственных препаратов, в основном входящих в «Эссенциальный список лекарственных средств», рекомендуемый ВОЗ, под их генерическими названиями. Практически для всех описываемых препаратов приводятся ссылки на методы их получения (около 2200), а также их наиболее распространенные синонимы. Синтез различных групп лекарств, в основном, излагается в том порядке, в котором они традиционно приводятся в общепринятых курсах фармакологии. Практически все главы начинаются с общепринятого определения препаратов с данным типом активности, приводится краткая характеристика всей группы, классификация рассматриваемых препаратов, и далее описывается их синтез, каждый из которых завершается областью применения данного препарата.
Для фармацевтов, провизоров, химиков, биологов, токсикологов, технологов в области производства лекарственных препаратов и др. специалистов.
УДК 615.012
ББК 35.66
ISBN 5-89481-218-6
© Р.С. Вартанян, 2004
© ООО “Медицинское информационное агентство”. Оформление, 2004
Все права защищены. Никакая часть книги не может быть воспроизведена в какой бы то ни было форме без письменного разрешения владельцев авторских прав
Светлой памяти моего отца, учителя, научного руководителя, моего ближайшего друга и страстного болельщика, академика САРКИСА АМБАРЦУМОВИЧА ВАРТАНЯНА посвящаю
От автора
Настоящая книга, как впрочем, наверное, и любая другая, представляет собой попытку самовыражения и давно назревшей необходимости в систематизации тех сведений, которые накапливались в течение более чем тридцати лет моей работы в области синтеза лекарственных препаратов и, собственно, семи лет работы над самой книгой. На мой взгляд, результат может заполнить тот явный пробел, который имеется в литературе по этому вопросу.
Первоначальный план этой книги довольно сильно отличался от настоящего варианта. Предполагалось показать синтез препаратов в динамике. Для некоторых препаратов это был направленный синтез массива потенциально активных веществ, из которых в результате сотрудничества химиков, биологов, фармакологов, токсикологов, технологов и других людей самых разных специальностей впоследствии рождался препарат. Иногда новый препарат появлялся в результате приложения возможностей нового реагента или нового доступного исходного вещества.
Предполагалось вкратце затронуть историю создания хотя бы некоторых препаратов. Хотелось привести известные курьезные случаи при работе над ними, привести зачастую весьма любопытные истории рождения их названий и не менее интересные истории, связанные с изменением сферы применения препарата после прохождения клинических испытаний. Однако в какой-то момент времени я понял, что выхожу за рамки возможного формата для одного человека и что эта работа не сможет быть закончена в разумные сроки.
Поэтому*, в ряду нескольких альтернативных подходов, я остановился на предлагаемом, не беллетризованном варианте, в котором синтез различных групп лекарств, в основном, излагается в том порядке,
в котором они традиционно излагаются в общепринятых курсах фармакологии. Это было сделано с весьма определенной целью — гармонизовать предлагаемый материал с тем курсом фармакологии, который проходят будущие медики и фармацевты.
Практически все главы начинаются с общепринятого определения препаратов с данным типом активности, приводится краткая характеристика всей группы, классификация рассматриваемых препаратов, и далее описываются конкретные синтезы, каждый из которых завершается областью применения данного препарата.
Из многотысячного массива лекарств, имеющих хождение на фармацевтическом рынке, мною, в основном, описаны лекарственные препараты, которые входят в «Эссенциальный список лекарственных средств», рекомендуемый ВОЗ, под их генерическими названиями.
Конечно, практически для всех описываемых более чем 700 препаратов, что примерно вдвое больше чем в «Списке», приводятся ссылки на методы их получения (около 2200), а также их наиболее распространенные синонимы. Однако с целью избежать любого недопонимания изложение ведется только на основе названий генериков.
Самая большая глава — антибиотики — формально не подходит для изложения в книге с таким названием, но поскольку основное внимание в ней сосредоточено на описании синтетической части получения полусинтетических антибиотиков, считаю, что она обязательно должна была войти в настоящую книгу.
Несомненно, что после вышеуказанных сокращений текст был приведен к определенному алгоритму, и, используя весьма небольшой словарь (а именно, тот весьма ограниченный набор фраз, традиционно применяемый для описания синтезов любых химических соединений), оказалось практически невозможно представить описания самих синтезов менее однообразными, и я прекрасно осознаю, что собственно в этой части результат получился более чем тривиальным.
Однако весьма надеюсь на то, что семь лет, ушедшие на написание этой книги, были потрачены не зря и что она может заинтересовать тех, кто работает или планирует начать работу в данной увлекательнейшей области биологически активных соединений — синтеза лекарственных препаратов.
Р. С. Вартанян
20.07.2002г.
Список сокращений
б АПК — 6-аминопенициллановая кислота
АД — артериальное давление
АКТГ — адренокортикотропный гормон
АДФ — аденозиндифосфат
АМФ — аденозинмонофосфат
АПФ — ангиотензин-превращающий фермент
АТФ — аденозинтрифосфат
ГАМК — гамма-аминомасляная кислота
ДОФА — дигидроксифенилаланин
ЖКТ — желудочно-кишечный тракт
ЛПВП — липопротеиды высокой плотности
ЛПНП — липопротеиды низкой плотности
ЛПОНП — липопротеиды очень низкой плотности
ЛППП — липопротеиды промежуточной плотности
МАО — моноаминооксидаза
МБК — минимальная бактерицидная концентрация
МИК — минимальная ингибирующая концентрация
мРНК — мессенджерная РЖ
НПВС — нестероидные противовоспалительные средства
ПСБ — пенициллинсвязывающйе белки
тРНК — транспортная РЖ
ФИО — фактор некроза опухоли
ФСК — факторы, стимулирующие колонии
цАМФ — циклический аденозинмонофосфат
ЦНС — центральная нервная система
1 Глава 1
j Общие анестетики
В настоящее время термин «общая анестезия» (наркоз) в хирургической практике означает состояние организма с обратимой потерей сознания при контролируемом уровне депрессии нервной системы и включающем следующие компоненты: анальгезию (отсутствие боли), амнезию (отсутствие памяти), подавление рефлекторной деятельности (брадикардия, ларингоспазм) и снижение тонуса скелетной мускулатуры.
Соответственно, общими анестетиками считаются препараты, которые вызывают обезболивание, ослабляют рефлекторную и мышечную деятельность и. в конечном счете, вызывают потерю сознания.
Идеальный анестетик должен обладать всеми перечисленными выше свойствами, а также большой широтой терапевтического индекса и незначительными побочными эффектами.
В анестезиологии применяются препараты, которые блокируют или подавляют нейрологические импульсы, опосредованные центральной нервной системой (ЦНС). и позволяющие проводить хирургические, акушерские и диагностические процедуры безболезненно.
Общие анестетики подразделяются на два типа — ингаляционные (галотан, энфлуран, изофлуран, метоксифлуран, закись азота) и неингаляционные (барбитураты, кетамин и этомидат).
1.1. Ингаляционные анестетики
Задачей ингаляционной анестезии является достижение такой концентрации (парциального давления) препарата в мозге, которая достаточна для достижения требуемой степени анестезии. Для этого молекулы анестетика должны пройти путь из легких в мозг через разные биофазы, поэтому ингаляционные анестетики должны быть растворимы в крови и тканевых жидкостях.
Механизм, согласно которому ингаляционные анестетики проявляют свой эффект, неизвестен. Поскольку они не принадлежат к единому
-6-
Общие анестетики
химическому классу соединений, не выявлены также корреляции структура-активность. Ингаляционные анестетики неспецифичны, поскольку они взаимодействуют не со специальными рецепторами, и поэтому для них не существует специфических антагонистов. Взаимодействие ингаляционных анестетиков с клеточными структурами может быть описано только как Ван дер Вальсовские взаимодействия. Существует ряд гипотез, объясняющих действие ингаляционных анестетиков, однако ни одна из них не может адекватно описать весь спектр вызываемых ими эффектов.
Действие общих анестетиков может быть объяснено блокадой ионных каналов или определенными изменениями в механизмах высвобождения нейротрасмиттеров
Из ряда предложенных механизмов можно упомянуть 3 гипотезы:
1. Гидратная гипотеза. Молекулы анестетиков могут образовывать гидраты со структурированной водой, что может затормаживать функции мозга на соответствующих участках. Однако не выявлены корреляции между способностью образовывать гидраты и активностью ингаляционных анестетиков.
2. Гипотеза ионных пор. Анестетики блокируют ионные каналы путем взаимодействия с клеточными мембранами, уменьшая приток ионов Na+ и увеличивая приток ионов К+ в клетку, что приводит к развитию анестезии.
3. Гипотеза текучести мембран. Анестетики стабилизируют, скорее — фиксируют в определенном состоянии, мембраны клеток, затрудняя их способность к текучести, что вызывает изменения в работе ионных каналов.
Выбор конкретного анестетика или их комбинации производится в зависимости от типа медицинского вмешательства. Долгое время в качестве ингаляционных анестетиков широко применялись эфир, хлороформ, трихлорэтилен, этилхлорид или хлорэтан, а также циклопропан. Сегодня чаще всего в медицине используются следующие ингаляционные анестетики: галотан, энфлуран, изофлуран, метоксифлуран и закись азота.
Галотан (Halotane)
Галотан — 2-бром-2-хлор-1,1,1-трифторэтан (1.1.2) получают путем присоединения фтористого водорода к трихлорэтилену и при одновременном замещении атомов хлора в присутствии треххлористой сурьмы при 130 °C. Полученный при этом 2-хлор-1,1.1-трифторэтан
-7-
Глава 1
(1.1.1) подвергается далее бромированию при 450 °C с образованием галотана [1, 2, 3].
F Вг
I I F-C-C-H
< I
F CI
1 1 2
HZFZ ! SbCI3 130°С Br2 4500
CI2C=CH-CI ----------------► Р3С-СНг-С1 --------
1 1 1
Галотан является широко применяемым современным ингаляционным анестетиком. Он начинает действовать очень быстро, приятен для пациента и достаточно безопасен. Единственным препятствием к его применению является его гепатотоксичность. Применяется в хирургии при краткосрочных и длительных вмешательствах.
Наиболее распространенным синонимом галотана является фторо-тан.
Энфлурин (Enflurane)
Энфлуран — 2-хлор-1,1,2-трифторэтилдифторметиловый эфир (1.1.4) получают хлорированием на свету 2-хлор-1,1,2-трифтор-этилметилового эфира в 2-хлор-1,1,2-трифторэтилдихлорметиловый эфир (1.1.3) с последующим замещение атомов хлора на фтор в дихлорметильной группе фтористым водородом в присутствии пятихлористой сурьмы или смесью трехфтористой сурьмы с пятихлористой сурьмой [4, 5].
H2F2 / SbCI3
ci2 ? SbF3/sbci5
•С-С-О-СНз ---► F-C-C-O-C-H ------2----
II III
HF H F Cl
1 1 3
Cl F F
F-C-C-O-C-H i i i HF F
1 1 4
Энфлуран обладает практически всеми свойствами галотана и применяется по тем же показаниям. Абсорбируется хуже.
Синонимом препарата является этран.
Изофлуран (Isoflurane)
Изофлуран — 2-хлор-2-(дифторметокси)-1,1,1-трифторэтан (1.1.8) получают исходя из 2,2,2-трифторэтанола. С этой целью 2,2,2-трифторэтанол первоначально метилируют диметилсульфатом и полученный при этом метиловый эфир (1.1.5) подвергают хлорированию молекулярным хлором с получением 2-(дихлорметокси)-
-8-
Общие анестетики
1,1,1-трифторэтана (1.1.6). Далее взаимодействием (1.1.6) с фтористым водородом в присутствии пятихлористой сурьмы атомы хлора в последнем замещают на атомы фтора. Полученный эфир (1.1.7) вновь подвергают хлорированию молекулярным хлором с получением изофлурана [6, 7].
сг3-сн2-он +
CF3-CH2-OCHCI2
11 6
(СНзО^ЗОг
h2f2/ Sbci6
кон
CF3 —СН2 “’ОСНз
CF3 “CH2“OCHF2
1 1 7
CF3-CH— OCHF2
Cl
1 1.8
Изофлуран аналогичен по действию энфлурану, однако из-за несколько острого запаха его применение иногда вызывает определенные трудности.
Синонимом изофлурана является форан.
Метоксифлуран (Methoxyflurane)
Метоксифлуран — 2,2-дихлор-1,1-дифторэтилметиловый эфир (1.1.10) получают исходя из 1,1-дифтор-2,2,2-трихлорэтана, который подвергают дегидрохлорированию гидроокисью калия в 1,1-дихлор-2,2-дифторэтилен (1.1.9), к которому в присутствии гидроокиси калия присоединяют метанол [8].
Cl F
CI—с-с-н
I I Cl F
КОН
Cls /F СН3ОН / КОН F
С=С --------------► Н-С-С-ОСНз
Cl \ Cl F
1 1 10
Метоксифлуран является весьма мощным ингаляционным анестетиком, вызывающим отличное расслабление скелетной мускулатуры. Однако его относительно высокая растворимость, вызывающая медленный выход пациента из состояния анестезии, несколько ограничивает его применение. Другим недостатком метоксифлурана является то, что продуктами его биотрансформации являются ионы фтора, что может привести к развитию почечной недостаточности. Поэтому рекомендуется использовать метоксифлуран для анестезии при вмешательствах продолжительностью не более 2 ч.
-9-
Глава 1
Весьма часто встречающимся синонимом метоксифлурана является пентран.
Закись азота (Nitrous Oxide)
Закись азота (1.1.11) получают либо термическим разложением нитрата аммония, либо окислением аминосульфоновой кислоты азотной кислотой [9, 10, И].
nh4no3
n2o
2Н2О
1111
h2n, о
+ HNO3 но о
n2o + Н2О + H2so4
1.1 11
Закись азота, которую называют также веселящим газом, является слабым анестетиком. Обычно ее применяют совместно с гипнотиками, анальгетиками и миорелаксантами. Ввиду отсутствия у него какого-либо депрессивного влияния на дыхание его иногда называют идеальным анестетиком. Однако, согласно последним сообщениям, применение закиси азота в течение более 2 ч противопоказано, поскольку он вызывает резкое уменьшение уровня метионинсинтетазы, что, в свою очередь, может вызвать у пациентов резкое понижение уровня витамина В|2со всеми вытекающими последствиями.
1.2. Неингаляционные анестетики
Для введения пациента в наркоз в современной анестезиологии, как правило, сочетают несколько препаратов, применяемых как до начала ингаляционной анестезии, так и параллельно с этой процедурой; при этом кроме специальных средств используют и ряд соединений, лишь формально классифицируемых как неингаляционные анестетики и являющихся представителями других фармакологических классов соединений (анальгетиков, транквилизаторов, нейролептиков и др.).
Следует отметить, что при неингаляционной анестезии контроль за процессом и его регулирование осуществлять значительно труднее, чем при ингаляционной анестезии. Однако простота техники внутривенной анестезии, ее разнообразные комбинированные формы (нейро-лептанальгезия, атаралгезия, транквиланалгезия) делают эти варианты общей анестезии весьма полезными в клинике.
При общей анестезии в качестве краткодействующих специальных средств для неингаляционного наркоза применяются кетамин и этоми-
-10-
Общие анестетики
дат, а также ряд препаратов, принадлежащих к совершенно разным химическим классам, в том числе краткодействующие барбитураты — тиопентал и метогекситал, опиоидные анальгетики — морфин и фентанил, а также ряд бензодиазепиновых транквилизаторов — диазепам, лоразепам и мидазолам, т. е. препараты, которые упоминаются в данном разделе как неингаляционные анестетики, несмотря на то, что даже формально они таковыми не являются и не проявляют всех четырех, присущих анестетикам по определению, характерных свойств.
Кетамин (Ketamine)
Кетамин — 2-(о-хлорфенил)-2-(2-метиламино)циклогексанон (1.2.4) синтезируют исходя из 2-хлорбензонитрила, который вводят в реакцию с магнийбромциклогексаном с получением 1-(2-хлорбен-зоил)циклопентана (1.2.1). Последний бромируют молекулярным бромом в соответствующий бромкетон (1.2.2), который при взаимодействии с водным раствором метиламина образует метилиминопроизводное (1.2.3) с одновременным гидролизом третичного атома брома. При дальнейшем кипячении продукта реакции в декалине происходит перегруппировка с расширением цикла, и образуется кетамин. Предложены и другие объяснения механизма реакции трансформации метилиминопроизводного (1.2.3) в конечный продукт, в частности — через промежуточное образование эпоксисоединения, однако ни один из них нельзя считать удачным и достоверно доказанным [12, 13].
Кетамин — специальное средство для неингаляционного наркоза, используемое при кратковременных хирургических вмешательствах, которое вызывает состояние, называемое диссоциативной анестезией, и которое обеспечивает амнезию и анальгезию, сохраняя у пациента нормальное дыхание и мышечный тонус. Кетамин практически не имеет мышечнорасслабляющих свойств.
Премедикация морфином и скополамином, бензодиазепинами или б\тирофенонами понижает дисфорический эффект кетамина.
Синонимами препарата являются кетанест, кеталар и др.
-11 -
Глава 1
Этомидат (Etomidate)
Этомидат — этиловый эфир 1-(а-метилбензил)имидазол-5-карбо-новой кислоты (1.2.8) получают по следующей схеме, которая, очевидно, является частным случаем получения производных имидазола, взаимодействия а-аминокарбонильных соединений с тиоцианатами. Реакцией а-метилбензиламина с этиловым эфиром хлоруксусной кислоты получают М-этоксикарбонилметил-N-l-фенилэтиламин (1.2.5), который далее подвергают формилирова-нию муравьиной кислотой. Полученный при этом N-этоксикарбо-нилметил-Ы-формил-И-1-фенилэтиламин (1.2.6) далее подвергают С-формилированию этилформиатом в присутствии этилата натрия, и полученный продукт без выделения обрабатывают раствором тиоцианата калия в соляной кислоте. В результате происходящей при этом реакции тиоцианат иона с альдегидной карбонильной группой и одновременно происходящего гидролиза N-формамидной защиты происходит известная реакция гетероциклизации с получением 5-этоксикарбонил-2-меркапто-1-(1-фенилэтил)имидазола (1.2.7). Тиольную группу в последнем удаляют окислительным детиониро-ванием при взаимодействии со смесью азотной и азотистой кислот (азотная кислота в присутствии нитрита натрия), которое, по-видимому, протекает через стадию образования неустойчивой сульфиновой кислоты, которая чрезвычайно легко теряет SO2, что, в итоге, приводит к получению искомого этомидата [14, 15].
С С Н 2 ~ С СОС 5
нсоон
сно
- NH-CH2-COOC2rb
1 26
1 C2H5ONa/ НСООС2Н5
2 KSCN / HCI
1 HNOa/JNaNCy
2 Na2CO3
Этомидат — производное имидазола, структурно не похожее на другие анестетики, является средством для ингаляционного наркоза, длительность действия которого зависит от введенной дозы. Препарат не проявляет анальгетических свойств и обладает противосудорожной активностью. Поскольку его внутривенное введение вызывает быструю
-12-
Общие анестетики
потерю сознания, его можно классифицировать и как седативный гип-нотик. Из-за плохой растворимости в воде с pH выше 3 в клинике он применяется в виде растворов в пропиленгликоле, что вызывает болезненность при инъекциях. Более того, препарат вызывает послеоперационную тошноту и рвоту, что несколько ограничивает его применение. Скорость, с которой наступает потеря и затем восстановление сознания, несколько меньше таковой у барбитуратов.
Синонимом препарата является гипномидат и др.
В хирургической практике практически используются два барбитурата. тиопентал и метогекситал. Однако следует знать, что барбитураты являются гипнотиками и в терапевтических дозах имеют весьма слабые анальгетические и мышечнорасслабляющие свойства, которыми должен обладать общий анестетик.
Тиопентал (Thiopental)
Тиопентал — 5-этил-5-(1-метилбутил)-2-тиобарбитуровую кислоту (1.2.10) получают алкилированием этилмалонового эфира 2-бром-пентаном в присутствии этилата натрия. Полученный при этом этил-(1-метилбутил)малоновый эфир (1.2.9) вводят в реакцию гетероциклизации с тиомочевиной, также используя в качестве основания этилат натрия [16, 17].
Н?С3-(рН-0г + СНз
МсГ -п / (Ц№СЗ
Н5С2 О О U2H5 -————————*
НтС3-<рН С2Нб
СНз 1 2 9
Тиопента,! является барбитуратом ультракороткого действия, используется для гладкого и приятного для пациента осуществления анестезии. Выход из состояния анестезии при применении обычных терапевтических доз происходит уже через 15 мин после введения. Тиопентал имеет прямое дозозависимое угнетающее действие на миокард, на ЦНС и в меньшей степени действует на гладкую мускулатуру сосудов. Применяют для наркоза при непродолжительных хирургических операциях.
Барбитураты вообще и тиопентал в частности переводят в растворимую форму обработкой основаниями. Поэтому очень часто тиопентал поступает на рынок под названием тиопентал натрия. В данном случае образование соли происходит за счет атома серы в ентиольной форме.
- 13-
Глава 1
Наиболее распространенными синонимами тиопентала являются пентотал, трапанал, фармотал, интравал и др.
Метогекситал (Methohexital)
Метогекситал — 5-аллил-1-метил-5-(1-метил-2-пентинил)барбиту-ровую кислоту (1.2.15) синтезируют по классической схеме получения производных барбитуровой кислоты, а именно: взаимодействием производных малонового эфира с производными мочевины. Исходный аллил-(1-метил-2-пентинил)малоновый эфир (1.2.14) получают последовательным алкилированием самого малонового эфира, сначала 2-бром-З-гексином с получением (1-метил-2-пенти-нил)малонового эфира (1.2.13), а затем аллилбромидом. 2-бром-З-гексин (1.2.12), в свою очередь, получают исходя из реактива Нормана, синтезируемого из 1-бутина и этилмагнийбромида, взаимодействием с уксусным альдегидом и последующим бромированием полученного карбинола (1.2.11) трехбромистым фосфором. Вводя диалкилированный таким образом малоновый эфир (1.2.14) во взаимодействие с N-метилмочевиной, получают метогекситал (1.2.15) [18].
Н5С2-С=С-МдВг
О + сн3-с-н
Н5С2--С=С-СН-СНз
ОН
1.2.11
РВг3
НдСг-О'
О-С2Н5
Н5С2 -с=с-сн-сн3
Вг
1,2.12
СН2=СН— СН2-ВГ
н5с2 -с=с-сн-сн3
о о
HsC2—О О-С2Н5
н5с2-сес —сн сн2-сн=сн2
СН3 1.2.14
CH3NHCONH2
н5с2-с=с
СНз
НгС о
НгС^СН 1.2,15
Метогекситал также является барбитуратом ультракороткого действия и применяется по тем же показаниям, что и тиопентал. Имеет несколько меньшее время действия, чем тиопентал, однако в клинике эта разница слабо различима.
Синонимами препарата являются пентотал, интравал, фармотал, ра-вонал п др.
-14-
Общие анестетики
Как уже было отмечено, в практике анестезиологии в качестве вспомогательных веществ широко применяются также опиоидные анальгетики, в частности — морфин, фентанил, альфентанил и суфентанил.
морфин фентанил
СгН5^ Д^.СНзСНг—
N=N
суфентанил
альфентанил
Синтез этих соединений описывается в разд. 3.1. «Опиоидные анальгетики».
Кроме опиоидов для снятия беспокойства у пациентов во время анестезии очень часто применяются и бензодиазепины: диазепам, лоразепам и мидазолам, которые проявляют анксиолитический, седативный, противосудорожный эффекты, вызывают амнезию и мышечное расслабление.
диазепам лоразепан мидазолам
- 15-
Глава 1
Синтез первых двух описывается в разд. 5.1. «Бензодиазепины», где одновременно показан и синтез структурных аналогов мидазолама (алпразолам и др.).
Поскольку средства для наркоза относятся к разным классам химических соединений, общих закономерностей между их химическим строением и активностью не существует, Установлены лишь частные закономерности для отдельных рядов соединений (барбитураты, бензодиазепины и др.).
Список литературы
1. US Pat. 2.921.098 (1960).
2. Brit. Pat. 767.774 (1955).
3. Brit. Pat. 805.764 (1957).
4. US Pat. 3.469.011 (1969).
5. US Pat. 3.527.813 (1970).
6. US Pat. 3.535.388 (1970).
7. US Pat. 3.535.425 (1970).
8. Brit. Pat. 928.786(1960).
9. Archibald E. H The Preparation of Pure Inorganic Substances. New York: Wiley, 1932. — P. 246.
10. Schenk A. Handbook of Preparative Inorganic Chemistry. 2nd ed. New York: Academic Press, 1963. — Vol. 1. — P. 484.
11. US Pat. 2.11.276(1938).
12. US Pat. 3.254.124 (1966).
13. Belg. Pat. 634.208 (1963).
14. US Pat. 3.354.173 (1967).
15. Janssen P A. J. et al./'Arzneim Forsch. — 1971. Vol. — 21. — P. 1234.
16. US Pat. 2.153.729 (1939).
17. US Pat. 2.876.225 (1959).
18. US Pat. 2.872.448 (1959).
j Местные анестетики
Местные анестетики — это препараты, применяемые с целью временного и обратимого устранения болевой чувствительности определенных участков тела путем блокирования проведения импульсов в нервных волокнах.
В клинике местные анестетики применяются различными способами в самых разных случаях, требующих местного обезболивания начиная с простейших процедур по удалению небольшого участка поврежденного поверхностного слоя кожи и до сложнейших операций по пересадке органов. Местные анестетики широко применяются в клинике для обезболивания начиная со стоматологических процедур и до гинекологических вмешательств.
В терапевтических концентрациях местные анестетики обратимо блокируют нервную передачу, вызывают местную потерю чувствительности, устраняют при этом чувство боли и предотвращают мышечную активность.
В отличие от общих анестетиков эти препараты вызывают потерю чувствительности определенного участка без потери пациентом сознания.
Местные анестетики применяют для облегчения переносимости боли и болезненности, при раздражениях и зуде, связанных с нарушением целостности кожи и слизистых оболочек (порезы, укусы, раны, сыпи, аллергические состояния, грибковые инфекции, кожные язвы, трещины).
Их применяют при проведении офтальмологических процедур, таких как тонометрия, гониоскопия, удаление инородных тел, и при малых хирургических вмешательствах.
Местные анестетики широко применяются в хирургии, гинекологии и стоматологии.
В определенных случаях местные анестетики (лидокаин, прокаинамид) могут применяться в качестве антиаритмических средств.
Глава 2
Местные анестетики могут быть классифицированы как по принципиальным способам их клинического применения, так и на основании их принадлежности к определенному химическому классу соединений.
С медицинской точки зрения, согласно методам клинического применения, местную анестезию можно дифференцировать следующим образом.
Поверхностная анестезия
Местное применение препаратов этого ряда на слизистых оболочках носа, рта, гортани, трахеобронхиального древа, глаз, мочевого и желудочно-кишечного трактов (ЖКТ) вызывает поверхностную анестезию.
С этой целью применяют бензокаин, циклометикаин, гексилкаин, кокаин, лидокаин и тетракаин.
Инфильтрационная анестезия
Прямое введение местного анестетика в кожу или более глубоко расположенные ткани для хирургического вмешательства называют инфильтрационной анестезией.
С этой целью в основном применяют лидокаин, мепивакаин, бупи-вакаин, этидокаин и прокаин.
Проводниковая, или регионарная, анестезия
Введение местного анестетика в индивидуальный нерв или группу нервов при небольших хирургических вмешательствах с целью блокирования чувствительности, двигательной активности часто называют проводниковой, или регионарной, анестезией. Этот метод часто применяется при хирургических вмешательствах на плече, руке, шее, ноге.
Чаще всего с этой целью применяют лидокаин, мепивакаин и бупи-вакаин.
Спинномозговая анестезия
Спинномозговая анестезия заключается в введении местных анестетиков непосредственно в спинномозговую жидкость, что вызывает симпатическую блокаду, потерю чувствительности и мышечное расслабление путем воздействия анестетика на всем пути спинномозгового нерва. Метод применяется при больших хирургических вмешательствах.
Как правило, с этой целью применяют лидокаин, мепивакаин и бу-пивакаин.
-18-
Местные анестетики
Эпидуральная анестезия
Под эти термином понимают введение местных анестетиков в оболочку спинного мозга, в межпозвоночное пространство. Эпидуральная анестезия применяется при акушерских и гинекологических вмешательствах, не требующих очень быстрого развития анестезии.
С этой целью применяют лидокаин, мепивакаин, бупивакаин, эти-докаин и хлорпрокаин.
В качестве местного анестетика для офтальмологического вмешательства в клинике впервые в 1884 г. был применен алкалоид — кокаин. Сегодня в связи с опасностью развития наркомании и высокой токсичностью его клиническое применение сильно ограничено. Однако расшифровка его структуры, попытки его синтеза, выведения закономерностей структуры-активности, упрощения предполагаемой фармакофорной части молекулы явились одним из мощных стимулов для развития химии синтетических лекарственных средств. Первым синтетическим местным анестетиком, внедренным в клиническую практику в 1905 г., явился прокаин (новокаин). Позже были синтезированы тысячи соединений с аналогичными свойствами, однако лишь около 10-12 соединений нашли применение на практике. В 1947 г. был внедрен лидокаин, а в 1963 г. — бупивакаин (местный анестетик длительного действия).
Как агенты, блокирующие проводимость в аксонах и дендритах, местные анестетики отличаются от соединений, блокирующих нейрональную передачу в синапсах.
Предложен механизм действия местных анестетиков, согласно которому они являются блокаторами натриевых каналов.
Согласно этому механизму, молекулярными мишенями действия местных анестетиков являются вольтажзависимые натриевые каналы, имеющиеся во всех нейронах. Процесс местной анестезии соответствующими препаратами схематично можно представить следующим образом.
В состоянии покоя между аксоплазмой и внешней частью клетки существует определенный потенциал покоя. Этот потенциал покоя обеспечивается относительными концентрациями ионов Na’ и К* вдоль мембраны нерва.
При стимулировании нерва мембрана деполяризуется, в этом районе открываются натриевые каналы и ионы Na’устремляются в клетку. У пика деполяризации открываются выходные калиевые каналы. Последние покидают клетку, и клетка реполяризуется.
Этот процесс длится 1-2 мс, после чего нервная клетка, передав необходимый импульс, восстанавливает свой ионный градиент.
-19-
Глава 2
Полагают, что после введения в организм местного анестетика в виде водорастворимой соли, в зависимости от его рКа и pH тканевой жидкости, устанавливается равновесие между основной и катионной формой применяемого препарата. Полагают также, что только основная (незаряженная) форма препарата может пройти и проходит через соединительную ткань, окружающую нервное волокно через фосфолипиды плазматической мембраны в аксоплазму. В аксоплазме основание вновь ионизируется в той мере, которая определяется внутриклеточной pH. Предполагают, что далее препараты селективно связываются с внуз'ри-клеточной поверхностью натриевых каналов и блокируют вход ионов Na" в клетку. Это приводит к торможению процесса деполяризации, необходимого для распространения потенциала действия, к повышению порога электрического возбуждения нерва и, тем самым, к устранению чувства боли.
Поскольку процесс связывания анестетиков с ионными каналами обратим, то после прекращения введения препарат диффундирует обратно в сосудистую систему и метаболизируется, а функция нервной клетки полностью восстанавливается.
Механизм действия бензокаина несколько отличается от изложенного выше. Предположительно, он действует путем растворения в фосфолипидной мембране и растягивает ее. Это деформирует натриевые каналы, что, в свою очередь, своеобразным способом понижает натриевую проводимость.
Аналогичный механизм растягивания (изменение текучести) мембраны был предложен и для объяснения механизма действия общих анестетиков.
С химической точки зрения общие анестетики можно классифицировать как сложные эфиры н-аминобензойной кислоты и диалкиламиноалканолов или же как анилиды диалкилзамещенных по аминогруппе а-аминокислот.
При формализованном рассмотрении все применяемые местные анестетики состоят из трех частей: ароматического кольца (липофильная часть), промежуточной углеводородной цепи и аминной части (гидрофильная группа). Замещение в ароматическом кольце и в аминной части меняет как растворимость, так и степень связывания
-20-
Местные анестетики
части меняет как растворимость, так и степень связывания анестетиков с рецепторами, что, в свою очередь, определяет силу и продолжительность действия препаратов. Принято считать, что способность вызывать аллергические реакции, стабильность и, в ряде случаев, токсичность также обусловлены строением этой связующей цепочки, которая, в свою очередь, определяет место биотрансформации и инактивации препарата — либо ферментативным гидролизом в плазме (сложноэфирные анестетики), либо разложением в печени (аминоамидные анестетики).
Интересно, что ряд антигистаминных, антихолинергических и адренергических препаратов, имеющих аналогичную химическую структуру. также проявляют местноанестезирующие свойства. Возможно, что и они, взаимодействуя с внутренней аксоплазматической мембраной, уменьшают ионный поток и, в частности, поток ионов Na+ внутрь нервных клеток.
2.L Местные анестетики ряда аминоэфиров
Прокаин (Procaine)
Прокаин — 2-диэтиламиноэтиловый эфир 4-аминобензойной кислоты (2.1.1), больше известный под названием новокаин, получают двумя путями. Первый способ заключается в непосредственном взаимодействии этилового эфира 4-аминобензойной кислоты с 2-диэтиламиноэтанолом в присутствии этилата натрия. Второй способ синтеза исходит из 4-нитробензойной кислоты, которую взаимодействием с хлористым тионилом трансформируют в хлор-ангидрид (2.1.2) и далее этерифицируют 2-ди-этиламинотанолом, после чего нитрогруппу в полученном сложном эфире (2.1.3) восстанавливают в аминогруппу водородом в присутствии никеля Ренея [1, 2, 3, 4].
С2Н5
H2N // СООС2Н5 * HO-CH2~CH2-N
С2Н5
C2H5ONa /=\ С2Н5
--------* H2N—4 Л—coo— ch2-ch2-n
С2Н5
Н2 / Raney Ni
O2N
С2Н5
COO—CH2-CH2-N
C2H5
2 1 3
- 21 -
Глава 2
ho-ch2-ch2-n
p2H5
С2Н5
Прокаин является местным анестетиком с коротким периодом действия. Применяется для купирования болевого синдрома различного генеза, широко используется при инфильтрационной, проводниковой, эпидуральной и спинномозговой анестезии, для потенцирования действия основных препаратов при общей анестезии. Может вызывать аллергические реакции.
Наиболее распространенными синонимами прокаина являются новокаин, адрокаин. имплетол, мелкаин.
Хлорпрокаин (Chloroprocaine)
Хлорпрокаин — 2-диэтиламиноэтиловый эфир 2-хлор-4-амино-бензойной кислоты (2.1.5) является хлорированным в о-положении по отношению к карбонильной группе бензольного кольца аналогом прокаина. Синтез препарата осу ществляют прямым взаимодействием гидрохлорида хлорангидрида 4-амино-2-.хлорбензойной кислоты 12 1 44 и гидрохлорида 2-диетиламиноэтанола. Необходимый для синтеза гидрохлорид хлорангидрида 4-амино-2-хлорбензойной кислоты получают взаимодействием 2-хлор-4-аминобензойной кислоты с хлористым тионилом [5].
h2n ^~~"оон
Cl
SOCl2
НД. —л— COCI НС1
С2Н5
HO-CHj-CHj-N HCI
С2Н5
С2Н5
2 1 5
Хлорпрокаин показан при необходимости быстрого достижения эффекта обезболивания и используется для инфильтрационной анестезии, блокирования периферической нервной передачи, спинальной и эпидуральной анестезии.
-22-
Местные анестетики
Синонимом препарата является нескаин.
Тетракаин (Tetracaine)
Тетракаин — 2-диэтиламиноэтиловый эфир 4-бутиламинобензой-ной кислоты (2.1.6) также является структурным аналогом прокаина, в котором аминогруппа в бензольном кольце замещена бутильным радикалом. Методы, предложенные для его синтеза, повторяют вышеописанные методы синтеза прокаина или хлорпрокаина, но с использованием вместо 4-аминобензойной кислоты 4-бутилами-нобензойной кислоты. Предложен также вариант синтеза, исходящий непосредственно из прокаина (2.1.1). путем его непосредственного взаимодействия с масляным альдегидом и одновременным восстановлением водородом с использованием в качестве катализатора палладия на угле [6].
'СизСНаСНгСОН 2 н2, Pd-C
/=\ С2Н5
C4H9NH—4 A—COO—CH2~CH2~N
С2н5
2 1 6
Тетракаин — мощный местный анестетик длительного действия. В основном используется при спинномозговой анестезии.
Наиболее популярными синонимами тетракаина являются понтока-ин и бутилкаин.
Кокаин (Cocaine)
Кокаин — 3-3-бензоилокси-2()-метоксикарбонилтропан (2.1.13) фактически можно считать практическим и, в определенном смысле. идеологическим родоначальником анестетиков ряда аминоэфиров. Алкалоид кокаин был выделен в 1860 г. из листьев кокаинового куста (Erythroxylon coca), содержащего разные алкалоиды, производные экогонина (2.1.11), значительная часть которых приходится на долю кокаина. В 1898 г. была установлена его структура.
Большая часть добываемого из естественных источников кокаина получается полусинтетическим путем. С этой целью омылением смеси алкалоидов, извлеченных из листьев кокаина, получают экогонин (2.1 11), карбоксильную группу которого метилируют с получением метилового эфира экогонина (2.1.12). Полученный продукт далее бен-зоилируют по спиртовой гидроксильной группе с получением кокаина (2 1 13). Этот процесс соответствует превращениям, указанным на первой схеме синтеза кокаина.
-23-
Глава 2
Первый синтез кокаина был осуществлен в 1902 г. Из предложенных вариантов наиболее рациональными можно считать следующие две схемы его синтеза.
Первый способ синтеза исходит из калиевой соли этилового эфира ацетондикарбоновой кислоты, электролизом которой получают этиловый эфир сукцинилдиуксусной кислоты (2.1.7), которая при дальнейшем взаимодействии с метиламином образует 1-метил-2,5-дикарбэто-ксиметилиденпирролидин (2.1.8). Восстановление двух двойных связей в этом соединении приводит к получению 1-метил-2,5-дикарбэтокси-метилпирролидина (2.1.9). Последний подвергают внутримолекулярной циклизации в условиях реакции Дикмана с использованием в качестве конденсирующего средства этилата натрия с получением этилового эфира тропин-2-карбоновой кислоты (2,1.10). Восстановлением кетогруппы в последнем и последующим гидролизом карбэтосильной группы получают тропин-2-карбоновую кислоту' или экогонин (2.1.11). Метилирование карбоксильной группы последнего в сложный эфир (2.1.12) и дальнейшее ацилирование гидроксильной группы хлорангид-ридом бензойной кислоты приводит к рацемической смеси 3-бен-зоилокси-2-метоксикарбонилтропанов (2.1.3), из которой, пользуясь меньшей растворимостью, выделяют П,Ь-кокаин. Разделение оптических изомеров осуществляют через соли D-бром-камфор-Р-сульфоновой кислоты, однако при гидролизе происходит отщепление не только бромкамфорсульфонильной группы, но и бензоильной, в связи с чем после разделения осуществляют повторное бензоилирование [7].
Н5С2ООС - CH 2 N CH-CQOC2H5 СН3
C2HsONa
2 он;
СН3ОН
нс1
сн3
N
-24-
Местные анестетики
Согласно второму способу, исходят из тропанона, который подвергают метоксикабонилированию метиловым эфиром угольной кислоты в присутствии натрия с получением натриевой соли (2,1.14), которую после кислотного гидролиза, восстановления карбонильной группы полученного кетоэфира амальгамой натрия или электролитически ацилируют хлорангидридом бензойной кислоты с получением искомого продукта [8].
(СН3О)2СО
Na
1- н2со3
Как и в предыдущем случае, конечным продуктом является рацемат, из которого выделяют левовращающий изомер. Позднее были предложены и другие методы синтеза [9, 10].
Кокаин лишь в исключительных случаях используется как поверхностный анестетик в офтальмологии ввиду его сильного действия на ЦНС и быстрого возникновения к нему пристрастия.
2.2. Местные анестетики ряда аминоамидов
Лидокаин (Lidocaine)
Лидокаин — 2-(диэтиламино)-М-(2,6-диметилфенил)ацетамид (2.2.2) синтезируют исходя из 2,6-диметиланилина путем его взаимодействия с хлорангидридом хлоруксусной кислоты с получением а-хлор-2,6-диметилацетанилида (2.1.1) и дальнейшим его взаимодействием с диэтиламином [11].
С2Н.
H-N< СН3
С2«5 Л~К Я С2Н5
---------*• е y-NH-c-cH2N {
СгНь
СНз 2.2 2
Лидокаин — наиболее широко применяемый местный анестетик. Его отличная терапевтическая активность, быстрота достижения эффекта и достаточная продолжительность действия делают его пригодным
-25-
Глава 2
практически для любого клинического применения. Препарат стабилизирует клеточные мембраны, блокирует натриевые каналы, способствует выходу ионов К+ из клеток и ускоряет процесс реполяризации клеточных1 мембран. Применяют при терминальной инфильтрационной, проводниковой, эпидуральной и спинальной анестезии при оперативных вмешательствах в стоматологии, отоларингологии, акушерстве и гинекологии. Применяют также при желудочковой экстрасистолии и тахикардии, особенно в острой фазе инфаркта миокарда.
Синонимами препарата являются анестокаин, ксилокаин, нефлуан и многие другие.
Мепивакаин (Mepivacaine)
Мепивакаин — \-(2,6-диметилфенил)-1-метил-2-пиперидинкар-боксамид (2.2.3). Для его синтеза предложено два основных метода. Согласно первому способу, мепивакаин получают, вводя во взаимодействие этиловый эфир 1-метилпиперидин-2-карбоновой кислоты с 2,6-диметиланилиномагний бромидом, который получают, обрабатывая 2,6-диметиланилин этилмагнийбромидом [12, 13, 14].
2 2 3
Согласно второму способу, взаимодействием 2,6-диметиланилина с хлорангидридом пиридин-2-карбоновой кислоты первоначально получают 2,6-ксилидид а-пиколиновой кислоты (2.2.4), ароматическое пиридиновое кольцо которого восстанавливают в пиперидиновое водородом с использованием в качестве катализатора платины на угле.
Полуденный при этом 2,6-ксилидид а-пипеколиновой кислоты (2.2.5) метилируют в мепивакаин формальдегидом при одновременном восстановлении водородом с использованием в качестве катализатора палладия на угле [15].
H2/Pt—С / HCI
-26-
Местные анестетики
(СН2О)п/ H2Pd—С
2 2 3
Мепивакаин по своим свойствам сходен с лидокаином, однако обладает более продолжительным действием.
Синонимами мепивакаина являются карбокаин и эстрадурин.
Бупивакаин (Bupivacaine)
Бупивакаин — Ы-(2,6-диметилфенил)-1-бутил-2-пиперидинкарбокс-амид (2.2.7) химически сходен с мепивакаином и отличается лишь заменой в пиперидиновом кольце N-метильного заместителя на N-бутильный. Для его синтеза также предложено два метода. Первый способ исходит из а-пиколин-2,6-ксилидида (2.2.4). Алкилированием последнего бромистым бутилом получают соответствующую соль пиридиния (2.2.6). Эту соль восстанавливают водородом в пиперидиновое производное, используя в качестве катализатора окись платины с получением бупивакаина [13, 16].
С4Н9ВГ
Н2 Pto
СНз С4Н9
227
Второй способ исходит непосредственно из хлорангидрида пипе-ридин-2-карбоновой кислоты, которую вводят во взаимодействие с 2,6-диметиланилином. Образующийся при этом амид (2.2.8) алкилируют далее бромистым бутилом в бупивакаин [17, 18, 19].
С4Н9ВГ
227
Как лидокаин и мепивакаин, бупивакаин используется для инфильтрационной, спинальной и эпидуральной анестезии, блокирования
-27-
Г лава 2
нервной передачи. Наиболее важное отличительное его свойство — более длительное время действия. Препарат в основном применяют для оперативных вмешательств в урологии и нижнеторкальной хирургии длительностью до 3-5 ч; в абдоминальной хирургии — длительностью до 45-60 мин. Используют для блокады тройничного нерва, крестцового и плечевого сплетений, при вправлении вывихов, при эпидуральной анестезии при кесаревом сечении.
Наиболее распространенным синонимом бупивакаина является маркаин.
Этидокаин (Etidocaine)
Этидокаин — М-(2,6-диметилфенил)-2-(этилпропиламино)бутан-амид (2.2.12) также является анилидом а-диалкиламиноамино-кислоты, однако последовательность реакций для его получения несколько отличается от рассмотренных выше. На первой стадии синтеза 2,6-диметиланилин вводят во взаимодействие с хлорангид-ридом ос-бром масляной кислоты с получением броманилида (2.2.9). Далее с целью повышения выхода продукта последующей реакции нуклеофильного замещения галогена на пропиламинную группу осуществляют замещение атома брома в полученном амиде на атом йода. Полученное при этом йодопроизводное (2.2.10) с легкостью вступает во взаимодействие с пропиламином, образуя аминоамид (2.2.11), который далее повергают N-этилированию диэтилсульфатом в этидокаин [20, 21].
,соос2н5
CjHcONa
(C2H5O)SO2
Этидокаин по своим фармакологическим свойствам сходен с мепи-вакаином, однако, в определенной степени, обладает и свойствами мышечного релаксанта.
Синонимом препарата является дуранест и др.
-28-
Местные анестетики
Прилокаин (Prilocaine)
Прилокаин — 2-(пропиламино)-о-пропионтолуидид (2.2.14) по своему строению относится к той же группе, что и этидокаин, и структурно отличается от последнего тем, что во время синтеза вместо 2,6-диметиланилина использован о-толуидин вместо фрагмента масляной кислоты; в структуру препарата включен фрагмент пропионовой кислоты, а концевая пропилэтиламинная группа заменена на пропиламинную. С целью синтеза прилокаина о-толуидин вводят во взаимодействие с бромангидридом бромпропионовой кислоты, и полученный при этом бромпропионилтолуидид (2.2.13) далее вводят во взаимодействие с пропиламином с получением искомого прилокаина [22, 23].
о
Вг-С-СН-Вг
СНз
о
NH -с -CH -NH — С3Н7
СНз
По своим фармакологическим параметрам прилокаин сравним с лидокаином. однако по причине ряда токсических проявлений препарат на практике используется редко.
Известными синонимами прилокаина являются цитанест и ксило-нест.
2,3, Поверхностные анестетики
Бензокаин (Benzocaine)
Бензокаин — этиловый эфир 4-аминобензойной кислоты (2.3.1). Классическим и оптимальным способом получения бензокаина является восстановление этилового эфира 4-нитробензойной кислоты в бензокаин водородом, получаемым прямо в реакционной среде при взаимодействии железных опилок с разбавленными кислотами [24,25.26].
O2N
соос2н5
h2n —6 Л—соос2нь
23 1
Бензокаин применяется при поверхностной анестезии на коже или на слизистых оболочках в виде аэрозолей или мазей для облегчения бо
-29-
Г лава 2
лей при зуде и ожогах, порезах, укусах, и т. д. Начинает действовать через 15 30 с после применения и действует в течение 12-15 мин.
Синонимами препарата являются анестезин, дермопласт и др.
Циклометикаин (Cyclomthycaine)
Циклометикаин — этиловый эфир 3-(2-метилпиперидино)пропил-о-циклогексилоксибензойной кислоты (2.3.4) получают по нижеприведенной схеме. Алкилированием 2-метилпиперидина 3-хлорпро-панолом-1 получают 3-(2-метилпиперидино)пропанол-1 (2.3.2), спиртовую группу которого замещают на хлор с помощью хлористого тионила. Полученный при этом 3-(2-метилпиперидино)пропил-хлорид-1 (2.3.3) далее вводят во взаимодействие с 4-циклогексило-ксибензойной кислотой с получением циклометикаина [27, 28].
Циклометикаин также применяется при поверхностной анестезии на коже или на слизистых оболочках при порезах, укусах, а также при урологических обследованиях.
Известным синонимом препарата является сурфакаин.
Список литературы
1. Einhorn A et al.AAnn. 371, 125,131,142, 162 (1909).
2. US Pat. 812.554(1906).
3. Ger. Pat. 179.627 (1904).
4. Ger Pat. 194.748(1905).
5. US Pat. 2.460.139(1949).
6. US Pat. 1.889.645 (1932).
7. IVillstatter R. et al./'Ann. 434, 111 (1923).
8. Robinson J ' J. Chem. Soc., Ill, 762 (1917).
9. Tufariello J. et al.//Tetrahedron Letters. 1978, 1733.
10. Tufariello J. etal./J. Am. Chem. Soc. 101, 2435 (1979).
-30-
Местные анестетики
11.
12.
13
14
15.
16.
17.
18.
19
20.
21.
22.
23.
24.
25
26.
27
28.
US Pat. 2.441.498(1948).
US Pat. 2.799.679(1957).
Ekenstram B. et al./'Acta. Chem. Scand. 11. 1183 (1957).
Rinderknecht Я.'/Helv. Chim. Acta. 42, 1324 (1959).
US Pat. 4.110,331 (1977).
Brit. Pat. 869.978 (1959).
US Pat. 2.792.399(1957).
US Pat. 2.955.111 (1960).
TuUar В F /, J. Med. Chem. 14, 891 (1971).
US Pat. 3.812.147(1974).
US Pat. 3.862.321 (1975).
Brit. Pat. 839.943 (1958).
Lofgren N. et al,//Acta Chem. Scand. 14, 486, 490 (1960).
Limprichi A.JAnn. 303, 278 (1898).
Org. Syn. 8, 66(1928).
Org. Syn. Coll. Vol. I. 240 (2nd ed., 1941).
US Pat. 2.439.818(1948).
Me Elvain S et al.//J. Am. Chem. Soc. 68, 2592 (1946).
Глава 3
Анальгетики
Анальгетиками называются препараты, устраняющие или облегчающие чувство боли, которое сопутствует многим патологическим состояниям. Трудно перечислить все те ситуации, при которых необходимо применение анальгетиков. Это касается, например, мышечных или головных болей, при которых обычно используют анальгетики типа аспирина, не вызывающие никаких проблем, связанных с возможным развитием зависимости. Более интенсивные боли, возникающие во время и после хирургических вмешательств, как правило, устраняются введением опиоидных анальгетиков типа морфина или меперидина. К сожалению, даже весьма кратковременное применение этих анальгетиков может привести к развитию привыкания, к формированию лекарственной зависимости и толерантности.
При хронических болях, связанных с хроническими воспалительными реакциями (ревматоидный артрит и др.), пациенты могут годами применять анальгетики из ряда нестероидных противовоспалительных средств (НПВС).
Боль — очень важный защитный феномен, сопутствующий многим патологическим состояниям. Однако, выполняя свою сигнализирующую функцию, боль при чрезмерной интенсивности может, в свою очередь, усугубить течение основного заболевания* а в некоторых случаях — например, при тяжелой травме — способствовать развитию шока.
Проблема облегчения болевых ощущений так же стара, как само человечество. Наверное, с достаточной уверенностью можно утверждать, что именно выделение старейшего из известных болеутоляющих средств — морфина — из опийного мака в начале XIX столетия послужило импульсом для интенсивного развития химии, фармакологии, фармации.
Анальгетики делятся на две группы: опиоиды (морфиноподобные вещества), которые воздействуют преимущественно на ЦНС. и неопио-иды (НПВС или жаропонижающие анальгетики), влияющие преимущественно на периферическую нервную систему.
Опиоиды и неопиоиды различаются по многим параметрам, среди
- 32 -
Анальгетики
которых следует особенно выделить следующие: опиоиды являются более мощными анальгетиками, однако они не обладают противовоспалительным действием; опиоиды могут вызвать зависимость и толерантность, поэтому их применение должно быть кратковременным. Кроме того, неопиоидные анальгетики редко применяются в инъекционной форме.
Несмотря на то. что препараты обеих групп устраняют боль, их фармакологическое действие различно, в связи с чем указанные группы рассматриваются раздельно.
3.1. Опиоидные анальгетики
Опиоиды подразделяются на три большие подгруппы по действию на опиоидные рецепторы: агонисты, смешанные агонисты-антагонисты и антагонисты.
Опиоидные агонисты имеют сродство к опиоидным рецепторам, имитируя активность эндогенных опиоидных анальгетиков. Смешанные агонисты-антагонисты являются полусинтетическими производными морфина, которые проявляют агонистическую активность в отношении одних опиоидных рецепторов и антагонистическую — в отношении других. Опиоидные антагонисты связываются с опиоидными рецепторами, но не активируют их. Эти соединения не используются для анальгезии. Их терапевтическое значение заключается в том, чтобы устранить побочные эффекты, возникающие при абсолютной или относительной передозировке или непереносимости больным препаратов, а также для лечения случаев зависимости от опиоидов.
Агонисты включают природные алкалоиды опия (морфин, кодеин, смесь природных алкалоидов — пантопон и омнопон); их аналоги (гидрокодон и гидроморфон, оксикодон и оксиморфон); производные морфинана (леворфанол); ряд синтетических соединений производных фе-нилпиперидина (меперидин, промедол); 4-анилидопиперидины (фент-нил. суфентанил, альфентанил); производные дифенилгептана (метадон, пропоксифен).
Смешанные агонисты-антагонисты включают производные морфинана (налорфин, буторфанол), фенантрена (налбуфин), бензоморфана (пентазоцин, дезоцин), ориправина (бупренорфин).
Антагонистами являются налоксон, налтрексон.
Общепринято считать, что действие опиоидов опосредуется определенными рецепторами. Предполагается существование нескольких типов опиоидных рецепторов: ц, к, 8, ст. Некоторые из них, в свою оче
-33-
Глава 3
редь, подразделяются на подтипы. Не исключено, что опиоидные рецепторы локализованы в мембранном фрагменте синаптосомальной фракции, и, возможно, они являются гликопротеинами. Они подвержены конформационным изменениям в определенных условиях, что. вероятно, и предопределяет их селективное связывание с агонистами или антагонистами.
Опиоиды имеют различную химическую структуру, и их относительный анальгетический потенциал зависит от множества разных факторов, включая их сродство к специфическим местам связывания на рецепторах, от проявляемой активности на самих рецепторах и фармакокинетических особенностей.
Разные типы опиоидных рецепторов были постулированы именно для объяснения различного действия опиоидов.
Рецепторы, воздействие на которые вызывает реакцию организма, аналогичную таковой при введении морфина, — анальгезию, депрессию дыхания, миоз, расстройства ЖКТ и эйфорию — были названы ц-ре-цепторами. Рецепторы, воздействие на которые вызывает эффекты, аналогичные эффектам, вызываемым кетазоцином, — анальгезию, седативный эффект, миоз — были названы к-рецепторами. Анальгетические рецепторы, вызывающие также психотомиметические реакции: дисфорию галлюцинации, стимуляцию дыхательной и сердечно-сосудистой систем, мидриаз — характерные для соединений класса агонист-антагонист типа N-аллилнорметазоцина, были названы ст-рецепторами. Рецепторы, реагирующие на воздействие энкефалинов и вызывающие анальгезию и высвобождение гормона роста, были названы 5-рецепторами.
Несмотря на то, что многочисленные исследования подтверждают тот факт, что воздействие на разные рецепторы вызывает различный эффект, их точная природа и роль еще окончательно не выяснены.
Физиологическая роль эндогенной опиоидной системы не ограничивается лишь болью или анальгезией. Она однозначно играет роль в регуляции эндокринной, поведенческой, терморегуляционной, иммунной и гастроинтестинальной систем, равно как и участвует в механизмах развития привыкания и зависимости от опиоидов. Не исключено, что эндогенные опиоиды могут взаимодействовать со многими другими нейротрансмиттерными системами.
Концепция, согласно которой опиоиды вызывают анальгезию в результате взаимодействия с определенными рецепторами, была предложена много лет назад, однако до 1973 г. специфические места связывания опиоидов не были идентифицированы как рецепторы и не было
-34 -
Анальгетики
определено их распределение. Частота распределения мест связывания опиоидов значительно варьирует в различных регионах ЦНС и особенно высока в структурах мозга, ассоциированных с физиологическими функциями, связанными с действием опиоидов, что указывает на корреляцию между местом связывания и эффектом. Опиатные рецепторы найдены и вне ЦНС и, в частности, в вагусе и в ЖКТ.
Нейрохимические данные свидетельствуют, что опиоидные рецепторы в мозге ассоциированы с пресинаптическими структурами, функционируя путем уменьшения высвобождения нейротрансмиттеров.
Полагают, что взаимодействие агонистов с опиоидными р-рецепто-рами приводит к увеличению оттока из клетки ионов К и одновременно создает затруднения притоку ионов Са2+ внутрь клетки, что делает нейрон менее возбудимым.
Агонисты к-рецепторов непосредственно ингибируют вход ионов Са2’ в нейроны, просто уменьшая их поток через вольтажзависимые кальциевые каналы. Эти подтверждается фактами, указывающими, что повышенные концентрации ионов С а2' ослабляют действие морфина, а пониженные — усиливают. Действие морфина на восприятие боли отличается от действия местных анестетиков. Местные анестетики уменьшают и ослабляют восприятие боли, препятствуя передаче сигнала от места проявления боли. Опиоиды же очень мало воздействуют на аксональное проведение, скорее блокируя межнейрональную передачу болевых импульсов на разных уровнях интеграции ЦНС.
В мозге и других тканях были Обнаружены эндогенные олигопептиды связывающиеся с участками опиоидных рецепторов и действующие аналогично опиоидам. Первыми из них, которые были выделены и расшифрованы, являлись метэнкефалин и лейэнкефалин. Пептид с намного большей молекулярной массой и с аналогичной активностью — бета-эндорфин был найден в подмозговом и дуговидном придатках
Другой пептид, названный динорфином, был идентифицирован позднее. Эндогенным опиоидным пептидам приписывается и много других различных функций, включая участие в нейропередаче, однако механизм их опиоидного действия не ясен.
Опиоиды вызывают побочные реакции, которые ограничивают их применение. К ним относятся угнетение дыхания, тошнота, рвота, запоры, повышение уровня артериального давления, задержка мочеиспускания. потоотделение, зуд и другие, наиболее опасной из которых, конечно, является депрессия дыхания. Опиоиды вызывают зависимость и привыкание.
-35-
Глава 3
Агонисты
Наиболее широко в качестве агонистов в медицинской практике используются алкалоиды опия — морфин и кодеин. Однако широкое применение нашли и полусинтетические производные (гидроморфон, оксиморфон, гидрокодон, оксикодон), которые, в определенных случаях, оказываются более предпочтительными для применения, и чисто синтетические мощные соединения (метадон, меперидин, фентанил, суфентанил и др.).
Опиоидные агонисты действуют в первую очередь на р-рецепторы. Необходимо знать, что следует избегать применения соединений этого класса при черепно-мозговой травме, бронхиальной астме и других гипоксических состояниях, при острой алкогольной интоксикации, судорожных состояниях и острых болях со стороны органов брюшной полости.
Морфин (Morphine)
Морфин — 4,5-эпокси-17-метилморфин-7-ен-3,6-диол (3.1.19) является самым старым из известных анальгетиков. Практически его получают из опиума — высушенного млечного сока недозрелых головок опийного мака, анальгетические свойства которого известны более чем 3000 лет. Растение содержит большой ряд и других алкалоидов, которые подразделяются на группы фенантренов и бензилизохинолинов. Однако предложены и синтетические способы получения морфина. Один из предложенных виртуозных многостадийных способов синтеза морфина описывается ниже.
В рассматриваемом способе синтеза морфина исходят из 2,6-диоксинафталина (3.1.1), который взаимодействием с хлористым бензоилом трансформируют в монобензоат (3.1.2) и далее взаимодействием с азотистой кислотой переводят в 1-нитрозопризводное (3.1.3). Далее последовательным восстановлением нитрозогруппы водородом (с использованием в качестве катализатора палладия) и дальнейшим мягким окислением продукта треххлористым железом получают 6-бензилокси-1,2-нафтохинон (3.1.4). Последний с помощью четырехокиси серы восстанавливают в 6-бензилокси-1,2-нафтогидрохинон, который метилируют диметилсульфатом в 5,6-диметокси-2-бензоат (3.1.5), и далее щелочным гидролизом трансформируют в 5,6-диметокси-2-нафтол (3.1.6). Применяя последовательно те же стадии синтеза, а именно: нигрозиро-вание, восстановление и окисление теми же реагентами, получают 5,6-
-36-
Анальгетики
диметокси- 1,2-нафтохинон (3.1.8). Проведением реакции Кновенагеля с циануксусным эфиром в присутствии ферроцианида калия достигается окисление продукта конденсации. Продукт реакции (3.1.9) гидролизуется и декарбоксилируется далее в 5,6-диметокси-4-цианометил-1,2-нафтохинон (3.1.10), на основе которого далее ведется построение вначале фенантреновой, а далее морфинановой систем. С этой целью 5,6-диметокси-4-цианометил-1,2-нафтохинон (3.1.10) вводят в реакцию 4т2 циклоприсоединения с бутадиеном-1,3 с получением с умеренными выходами 3,4-диметокси-9,10-диоксо-13-цианометил-5,8,9,10,13,14-гекса-гидрофенантрена (3.1.11). При восстановлении полученного дикетона (3.1.11) водородом с использованием меднохромокисного катализатора был получен кетолактам (3.1.12). При обработке последнего алюмо-гидридом лития обе карбонильные группы (и кетонная, и амидная) подвергаются исчерпывающему восстановлению, и далее вторичный атом азота метилируется смесью формальдегида и муравьиной кислоты в рацемический метиловый эфир р-Д6-дигидродезоксикодеина (3.1.13). Обработкой последнего Ь(+)-дибензоилвинной кислотой выделяют искомый (+)метиловый эфир р-Аб-дигидродезоксикодеина. Последний подвергают гидратации в присутствии горячей разбавленной серной кислоты с получением метилового эфира р-дигидротебаинола (3.1.14). Энергичной обработкой гидроксидом калия в диэтиенгликоле происходит частичное деметилирование до Р-дигидротебаинола, окислением которого в системе wpew-бутилат калия — бензофенон получают р-дигидротебаинон (3.1.15). Полученный таким образом )3-дигидро-тебаинон (3.1.15) подвергают далее бромированию 3 молями брома в уксусной кислоте в (-)-1-бромкодеинон (3.1.17), который выделяют в виде 2,4-динитрофенил-гидразона. Очевидно, что на этой стадии синтеза происходит образование двойной связи при Ст-Cg и одновременно оксидного мостика при С4-С5. Более того, при этом происходит и обращение конфигурации при С14, т. е. изоморфинановая система изомеризуется в морфинановую. Дальнейшим восстановлением (-)-1-бромко-деинона (3.1.17) алюмогидридом лития получают кодеин (3.1.18), который деметилируют в искомый морфин (3.1.19) гидрохлоридом пиридина [1,2].
С6Н5СОС1
•ОСОС6Н5
.ОСОС6Н5
-37-
Г лава 3
[01
-38-
Анальгетики
На схеме структурная формула морфина (3.1.19) приведена в форме, позволяющей наглядно проследить последовательность происходящих трансформаций. Ниже приведено более принятое изображение морфина, согласно которому легче проследить те изменения, которые приводят к получению его практически ценных производных.
По сегодняшний день морфин является тем стандартным анальгетиком, с которым сравниваются все остальные, и продолжают разрабатываться альтернативные пути его синтеза [3, 4, 5, 6]. Тем не менее синтез морфина экономически не целесообразен, поскольку намного дешевле обходится его выделение из природного сырья.
Морфин является основным представителем и основным прототипом группы мощных опиоидных анальгетиков. Наиболее важное применение морфина заключается в его способности устранять боль. Его применяют в хирургии при премедикации больного для хирургических вмешательств до того, как начинается процедура общей анестезии. Широко применяется при инфаркте миокарда не только для облегчения болей, но также для успокоения пациента и даже для уменьшения потребности в кислороде. Используют при легочных отеках и некоторых формах диареи. Морфин назначают во всех тех случаях, когда действие НПВС недостаточно и требуется применение мощных опиоидных анальгетиков.
Сравнительно простые модификации молекулы морфина привели к получению ряда соединений, отличающихся по своей анальгетической активности.
Кодеин (Codeine)
Кодеин — 4,5-эпокси-17-метилморфин-7-ен-3-метокси-6-ол (3.1.20) является составной частью алкалоидов опийного мака. От морфина кодеин отличается тем, что гидроксильная группа при С3 в ароматическом кольце метилированна. Содержание кодеина в опии не удовлетворяет потребностям медицины, и поэтому кодеин получа-
-39-
Г лава 3
ют полусинтетическим путем из морфина путем селективного метилирования ароматической гидроксильной группы при С3. Обычные метилирующие агенты приводят к метилированию обеих гидроксильных ipynn. Селективное метилирование гидроксильной группы при С3 в ароматическом кольце можно осуществить при помощи диазометана, нитрозометилуретана или нитрозометилмоче-вины. Однако применение этих реагентов представляет определенные сложности для осуществления реакции в промышленных масштабах. Было предложено в качестве метилирующих средств применять хлорид триметилфениламмония или метилтолуолсульфо-нат диметиланилина в присутствии алкоголятов натрия. В основном кодеин получается метилированием 3-гидроксильной группы фенольного кольца морфина этилатом триметилфениламмония [7, 8].
[CgH5N(CH3)3]C2H5O
По своим свойствам кодеин близок к морфину, но его болеутоляющие свойства выражены слабее и препарат в меньшей степени вызывает привыкание. Препарат очень эффективен при оральном применении и используется для облегчения средних и умеренных болей. Часто используется в качестве противокашлевого средства.
Синонимами препарата являются кодил, акутус и др.
Героин (Heroin)
Героин — 3,6-диацетил-4,5-эпокси-17-метилморфин-7-ен (3.1.21) получают одновременным ацетилированием двух гидроксильных групп морфина уксусным ангидридом или ацетилхлоридом [9, 10].
(СН3СО)2О
-40-
Анальгетики
Обладая большей по сравнению с морфином растворимостью в липидах, он быстрее проходит через гематоэнцефалический барьер, однако действует так же, как и морфин, в который он трансформируется в мозге. Наркотическое действие, угнетение дыхания, токсичность, малая широта терапевтического действия, большая опасность привыкания обуславливают отсутствие каких-либо его преимуществ перед морфином.
Использование героина в медицине запрещено, поскольку нет таких терапевтических целей, которых нельзя было бы достигнуть другими препаратами.
Гидроморфон (Hydromorphone)
Гидроморфон — 4,5-эпокси-3-гидрокси-М-метил-6-оксоморфинан (3.1.22) является соединением родственным морфину и отличается отсутствием двойной связи в положении Су-С8 и наличием кетогруппы вместо гидроксильной при С6. Препарат получают изомеризацией морфина в присутствии палладиевого или платинового катализаторов [11, 12]. Другим способом получения является окисление дигидроморфина [13, 14].
Pd or Pt
Гидроморфон более растворим, чем морфин, и примерно в 8 раз активнее последнего при парентеральном приеме. Большая растворимость позволяет уменьшить объем инъекционной жидкости, что имеет значение при необходимости многократных инъекций. Начинает действовать быстрее, но время его действия короче, чем у морфина. Обладает большим седативным эффектом и меньшей способностью вызывать эйфорию. Гидро.морфон имеет то же применение, что и морфин. Побочные реакции аналогичны.
Синонимом препарата является дилаудид и др.
Оксиморфон (Oxymorphone)
Оксиморфон — 4,5-эпокси-3,14-дигидрокси-М-метил-6-оксоморфи-нан (3.1.26) химически сходен с гидроморфоном. Отличается от
-41 -
Глава 3
гидроморфона наличием гидроксильной группы при С(3. Препарат получают исходя из тебаина (3.1.23), который, окисляя пероксидом водорода в муравьиной кислоте, переводят в 14-гидроксикодеинон (3.1.24). Двойную связь в последнем восстанавливают водородом, трансформируя это соединение в оксикодон (3.1.25). Последний деметилируется бромистым водородом в оксиморфон [15, 16].
Н2О2
НС ООН
Н2 / Pd
НВг
Оксиморфон примерно в 10 раз активнее морфина. Вызываемые им эйфорический, равно как и рвотный, эффекты выражены значительно сильнее, чем у морфина. Оксиморфон проявляет также слабую противо-кашлевую активность. Побочные реакции аналогичны таковым у морфина. Препарат предназначен для облегчения умеренных и сильных болей при хирургических и гинекологических вмешательствах и послеоперационных болей.
Синонимом препарата является нуморфан и др.
Оксикодон (Oxycodone)
Оксикодон — 4,5-эпокси-3-метокси-14-гидрокси-М-метил-6-оксо-морфинан (3.1.25), синтез которого из 14-гидроксикодеинона (3.1.24) был описан выше, может быть получен и другими способами, например, окислением кодеина бихроматом калия в уксусной кислоте [17], также является структурным аналогом морфина и кодеина.
-42-
Анальгетики
В отличие от гидрокодона его применяют в качестве анальгетика в комбинации с другими препаратами, такими как аспирин или ацетаминофен. Оксикодон сходен с морфином по эффективности продолжительности действия и предпочтителен для орального применения.
Синонимами препарата являются роксикодон, проладон, перкдан, эутаген, оксикон и многие другие.
Гидрокодон (Hydrocodone)
Гидрокодон — 4,5-эпокси-3-метокси-Ы-метил-6-оксоморфинан (3.1.27) — соединение, химически родственное морфину и кодеину. Гидрокодон получают изомеризацией кодеина (3.1.20) над палладиевым или платиновым катализаторами [18]. Препарат предложено получать также гидрированием кодинона [19] и окислением дигидрокодеина [20].
Pd or Pt
Гидрокодон проявляет выраженные анальгетические и противокаш-левые свойства, что является его основным клиническим применением. Может вызвать зависимость и привыкание.
Синонимами препарата являются дикодид, детуссин, викодин и др.
Леворфанол (Levorphanol)
Леворфанол — (-)-З-гидро.кси-М-метилморфинан (3.1.35) является производным морфинана. Синтез леворфанола осуществляют исходя из циклогексанона путем его конденсации с циануксусной кис-
-43-
Глава 3
лотой по Кновенагелю, при которой происходит одновременное декарбоксилирование с образованием 1-циклогексенилацетонитрила (3 1 28) Восстановлением нитрильной группы последнего водородом в присутствии кобальта Ренея получают 2-(1-циклогексенил) этиламин (3.1.29). Полученный амин далее ацилируют хлорангидридом 4-метоксифенилуксусной кислоты (3.1.30) с образованием амида 2-(1-циклогексенил)-4-метоксифенилуксусной кислоты (3.1.31). Циклизация последнего хлорокисью фосфора приводит к получению 1-(4-метоксибензил)-3,4,5,б,7,8-гексагидрохинолина (3.1.32). Иминную связь восстанавливают водородом в присутствии никеля Ренея с получением 1-(4-метоксибензил)-1,2,3,4,5.6,7.8-октаагидрохино-лина (3.1.33), который метилируют формальдегидом в присутствии никеля Ренея в 1-(4-метоксибензил)-2-метил-1,2,3,4,5,6,7,8-окта~ агидрохинолин (3 1 34) На последней стадии синтеза в 1-(4-метоксибензил)-2-метил-1,2,3,4,5,6,7,8-октаагидрохинолин (3.1.34) подвергают циклизации с одновременным деметилированием в 3-гидрокси-М-метилморфинан — леворфанол (3.1.35), оптические антиподы которого далее разделяют с помощью (+)-винной кислоты [21,22].
соон
CH-CN
W2 z Ranay—Со
-44-
Анальгетики
ОСНз
Н2 Ranay—Ni
Правовращающий изомер не является анальгетиком, однако обладает противокашлевыми свойствами. Левовращающий изомер — левометорфан проявляет активность сходную с морфином, однако ряд побочных эффектов, таких как тошнота и рвота, а также способность вызывать запоры выражены слабее. В инъекционной форме он в 4-8 раз эффективнее морфина. Продолжительность действия также больше, чем у морфина. Препарат рекомендован для облегчения от умеренных до сильных болей при печеночных и почечных коликах, при инфаркте миокарда, серьезных травмах, при болях во время опухолевых заболеваний и для облегчения послеоперационных болей.
Синонимами препарата являются дроморан, леводроморан и др.
Метадон (Methadone)
Метадон — 6-диметиламино-4,4-дифенил-3-гептанон (3.1.37) получают алкилированием дифенилацетонитрила 1-диметиламино-2-пропилхлоридом в присутствии амида натрия. Полученный при этом 4-диметиламино-2,2-дифенилвалеронитрил (3.1.36) вводят далее во взаимодействие с этилмагнийбромидом и гидролизуют [23, 24, 25, 26]. Полученный в результате рацемат разделяют при помощи (т-)-винной кислоты, выделяя (-)-метадон [26, 27, 28, 29].
СНл рш
С(-6н-сн2-ь.< 3
СНз
1 С2Н§МдВг
2 Н?О
-45-
Глава 3
Метадон является синтетическим опиоидом действующим на ц-ре-цепторы и с качественными и количественными характеристиками аналогичными морфию,'. Принципиальная разница заключается в большей эффективности при оральном приеме и большей длительности действия. Кроме использования в качестве мощного анальгетика, препарат применяется и для лечения наркомании, поскольку замещает на рецепторе другие агонисты.
Синонимами препарата являются физептон, мефенон, долофин и др.
Меперидин (Meperidine)
Меперидин — этиловый эфир 1-метил-4-фенилпиперидин-4-карбо-новой кислоты (3.1.39) является синтетическим опиоидным анальгетиком. Его синтез осуществляют путем алкилирования бензилдианида Кт,К-бнс-(2-хлорэтил)-К-метиламином в присутствии амида натрия с образованием 1-метил-4-фенил-4-цианопиперидина (3.1.38) и последующим его кислотным этанолизом в меперидин [30, 31, 32].
с1сн2сн2
N-CHj CICH2CH2 /
NaNH2
CgHgOH I н4
3 1 3S
Меперидин относится к анальгетикам ряда фенилпиперидинов. Эти соединения также являются агонистами, хотя структурно сильно отличаются от морфина. Препарат проявляет и антихолинергическую активность. Как и морфин, он вызывает выброс гистамина и спазм гладкой мускулатуры. Практически неактивен при оральном применении. Большинство фармакологических свойств и показаний к применению сходны с таковыми у морфина, однако у препарата отсутствуют противо-кашлевые свойства. При парентеральном введении активность составляет примерно ’/8 таковой у морфина. Меперидин широко используется при премедикации и сбалансированной анальгезии. Предпочтителен для применения в акушерской практике по причине быстрого наступления анальгезии и короткого времени действия.
Наиболее часто употребляемыми синонимами являются петидин, долантин и демерол.
Промедо. i (Promedol)
Промедол — 1,2,5-триметил-4-фенил-4-пропионилоксипиперидин
-46-
Анальгетики
(3.1.45) также относится к анальгетикам, относящимся к ряду фе-нилпиперидинов, и в определенном смысле представляет собой «обращенный» меперидин, в котором, в отличие от меперидина, карбонильная группа присоединена к четвертому положению пиперидинового кольца через кислородный, а не через углеродный атом. Синтез этого соединения в принципе отличается от синтеза меперидина и основан на использовании 1,2,5-триметилпиперидин-4-она. Последний получается из диметилвинилэтинилкарбинола (3.1.40), продукта конденсации винилацетилена с ацетоном по реакции Фаворского, который далее подвергают дегидратации в винилизопро-пенилацетилен (3.1.41). Тройную связь в винилизопропенилацети-лене далее гидратируют в разбавленной серной кислоте в метаноле и в присутствии солей двухвалентной ртути (реакции Кучерова). Полученный при этом винилизопропенилкетон (3.1.42), в основном в виде метоксипроизводных продуктов присоединения метанола к активированным двойным связям, вводят в реакцию гетероциклизации с метиламином с получением 1,2,5-триметилпиперидин-4-она (3.1.43). Последний подвергают действию фениллития с получением 1,2,5-три.метил-4-фенилпиперидин-4-ола (3.1.44). Ацилированием последнего хлорангидридом пропионовой кислоты получают промедол [33].
НзС \
с-esc— сн =сн2
»3С ОН , 1 40
н?О'Н*Нд‘ + н CH3NH2
"-г- —1»нз<;=с-с-сн=сн—сн3 ----•
CgHgLi
3 1 43
HjC =с-с=с—-СИ =сн2
3 1 41
CHSCOCI
Препарат быстро всасывается и оказывает сильное анальгезирую-щее действие, как при парентеральном, так и при оральном приеме. Это соединение меньше угнетает дыхательный центр, чем морфин. Оказывает спазмолитическое действие на гладкую мускулатуру. Применяют в качестве болеутоляющего средства при хирургических вмешательствах травмах и заболеваниях, сопровождающихся болевыми ощущениями.
Синонимом препарата является тримеперидин.
-47-
Глава 3
Лоперамид (Loperamide)
Лоперамид — 1-(4-хлорфенил)-4-1идрокси-\т,К-диметил-сс,а-дифе-нил-1-пиперидин-бутирамид (3.1.55), предложенный в качестве анальгетика, получают путем алкилирования 4-(4-хлорфенил)-4-гидроксипиперидина (3.1.50) бромистым 1\т,М-диметил-(3,3-дифе-нилтеграгидро-2-фурилиден)аммонием (3.1.54) в присутствии основания.
Исходный 4-(4-хлорфенил)-4-гидроксипиперидин (3.1.50) получают взаимодействием 1-бензилпиперидин-4-она (3.1.48) с 4-хлорфенилмаг-нийбромидом и последующим дебензилированием продукта (3.1.49) путем восстановления водородом с использованием в качестве катализатора палладия на угле.
Исходный 1-бензилпиперидин-4-он (3.1.48) синтезируют по реакции Дикмана внутримолекулярной циклизацией К-бензил-И,К-ди(Р-карбетоксиэтил)амина (3.1.46), легко получаемого взаимодействием бензиламина с этилакрилатом, в 1-бензил-3-карбэтоксипиперидин-4-он (3.1.47) и последующего кислотного гидролиза и термического декарбоксилирования.
2 Н2С=СН-СООС2Н5
Бромистый Ы,М-диметил-(3,3-дифенилтетрагидро-2-фурилиден)ам-моний (3.1.54) синтезируют исходя из этилового эфира дифенилуксусной кислоты, который вводят во взаимодействие с окисью этилена в присутствии гидроксида натрия с получением 2.2-дифенилбутиро-лактона (3.1.51). При взаимодействии последнего с бромистым водородом в уксусной кислоте происходит раскрытие лактонного кольца с образованием 2,2-дифенил-4-броммасляной кислоты (3.1.52). Последнюю переводят в хлорангидрид (3.1.53) с помощью хлористого тионила и далее взаимодействием с водным раствором диметиламина циклизуют
-48-
Анальгетики
в искомый бромистый НМ-диметил-(3,3-дифенилтетрагидро-2-фурили-ден)аммоний (3.1.54). Вводя последний в реакцию с 4-(4-хлорфенил)-4-гидроксипиперидином (3.1.50), получают искомый лоперамид (3.1.55) [34, 35, 36].
NaOH
НВг
ВгСНгСНг -С-СООН
3 1 52
В настоящее время лоперамид чаще применяется в качестве препарата от диареи, а не в качестве анальгетика и даже переведен в список лекарств безрецептурного отпуска, поскольку его действие на ЦНС незначительно. Препарат снижает тонус и моторику гладкой мускулатуры кишечника вследствие связывания с опиатными рецепторами кишечника. Применяют для симптоматического лечения острой и хронической диареи различного генеза.
Синонимами препарата являются имодиум, диссенте, блоке, брек и др.
Дифеноксилат (Diphenoxylate)
Дифеноксилат — этиловый эфир 1-(3-циано-3,3-дифенилпропил)-4-фенилпиперидин-4-карбоновоЙ кислоты (3.1.58) также является препаратом ряда 4-фенилпиперидинов. На практике применяются два способа его получения. Первый способ заключается в алкилировании 2,2-дифенил-4-бромбутиронитрилом этилового эфира 4-фе-нилпиперидин-4-карбоновой кислоты (3,1.56), который, в свою очередь, получается путем, аналогичным синтезу меперидина, исходя из 1-бензил-4-фенил-4-цианопиперидина, Последний подвергают этанолизу в присутствии кислоты и последующему дебензилирова-
-49-
Глава 3
нию. Вторым способом является синтез, осуществляемый путем алкилирования дифенилацетонитрила этиловым эфиром 1-(2-хлор-этил)-4-фенилпиперидин-4-карбоновой кислоты (3.1.57), получаемой из этилового эфира 4-фенилпиперидин-4-карбоновой кислоты путем взаимодействия с Р-хлорэтанолом или же взаимодействием с этиленоксидом и последующим замещением на хлор образующейся в результате раскрытия эпоксидного кольца гидроксильной группы хлористым тионилом [37, 38].
3.1.56
3 1 57
Препарат является структурным аналогом меперидина и лоперамида, однако практически повторяет все фармакологические свойства лоперамида. Аналогично лоперамиду основным показанием для его практического применения является лечение диареи.
Синонимами препарата являются реасек, ломотил и др.
Фентанил (Fentanyl)
Фентанил — 1-фенэтил-4-К-пропиониланилинопиперидин (3.1.63) является весьма мощным внутримышечным или внутривенным анальгетиком. Синтез фентанила осуществляют исходя из 1-бен-зилпиперидин-4-она (3.1.48), который конденсируют с анилином с получением соответствующего основания Шиффа (3.1.59). Двойную связь в последнем восстанавливают алюмогидридом лития и полученный 1-бензил-4-анилинопиперидин (3.1.60) ацилируют ангидридом пропионовой кислоты. Полученный при этом 1-бензил-4-М-пропиониланилинопиперидин (3.1.61) подвергают дебензилированию водородом с использованием в качестве катализатора палладия на угле с выделением 4-\-пропиониланилинопиперидина
- 50 -
Анальгетики
ладия на угле с выделением 4-М-пропиониланилинопиперидина (3.1.62), который далее алкилируют 2-фенилэтилхлоридом с получением фентанила (3.1.63) [39, 40].
3 1 59
3 1 60
(C2HSCO)2O
Фентанил по анальгетическому действию превосходит морфин примерно в 100 раз. Оказывает угнетающее действие на дыхательный центр, замедляет сердечный ритм. Фентанил используется в анестезиологии как самостоятельно, так и в сочетании с дроперидолом, для ней-ролептанальгезии, при премедикации разными видами наркоза, при постоперационной анальгезии. В отличие от морфина не вызывает выброса гистамина. Применяют в условиях специализированных стационаров.
Синонимами препарата являются фентанест, лепрофен и др.
Альфентанил (Alfentanyl)
Альфентанил — 1Ч-[1-[2-(4-этил-4,5-дигидро-5-оксо-1Я-тетразол- 1-ил)этил]-4-(метоксиметил)-4-пиперидил]анилинопропионамид
(3.1.71) является следующим представителем препаратов этого же класса анилидопиперидинов, отличающихся от фентанила наличием второго заместителя в 4-м положении пиперидинового кольца и заменой фенильного радикала в арилэтильном заместителе при атоме азота пиперидинового кольца на ароматический гетероцикл — тетразолон. Синтез альфентанила заключается в алкилировании N-(4-
- 51 -
Глава 3
метоксиметил)-4-пиперидил)-пропионанилида (3.1.68) 1-(4-этил-4,5-дигидро-5-оксо-1Я-тетразол-1-ил)этил-2-хлоридом (3.1.66).
К-(4-метоксиметил)-4-пиперидил)-пропионанилид (3.1.68) получают исходя из 1-бензилпиперидин-4-она (3.1.48) путем его конденсации с анилином в присутствии циановодородной кислоты. Полученный при этом 4-анилино-4-циано-1-бензилпиперидин (3.1.64) далее подвергают алкоголизу с получением 4-анилино-4-карбэтокси-1-бензилпиперидина (3.1.65). который восстанавливают алюмогидри-дом лития в 4-анилино-4-гидроксиметил-1-бензилпиперидин, который в реакционной среде метилируют йодистым метилом до 4-ани-лино-4-метоксиметил-1-бензилпиперидина (3.1.66). Полученный продукт ацилируют с помощью пропионового ангидрида в 1-бензил-4-метоксиметил-4-К-пропионил-анилинопиперидин (3.1.67) и далее подвергают дебензилированию водородом с использованием в качестве катализатора палладия на угле в 4-метоксиметил-44М-пропионил-анилинопиперидин (3.1.68).
1-(4-этил-4,5-дигидро-5-оксо-1Я-тетразол-1-ил)этил-2-хлорид (3.1.66) синтезируют исходя из этилизоцианата и азида натрия. Полученный в результате реакции 2+3 циклоприсоединения 4-этил-4,5-дигидро-5-оксо-1Я-тетразол (3.1.69) далее подвергают алкилированию 1-бром-2-хлорэтаном с получением 1-(4-этил-4,5-дигидро-5-оксо-1Я-тетразол-1-ил) этил-2-хлорида (3.1.70), который и вводят в взаимодействие с 4-метокси-метил-4-К-пропиониланилинопиперидином (3.1.68) с получением альфентанила (3.1.71) [41, 42, 43].
HCN
3 1 48
С2Н50Н / Н
1 1Л1Н4
2 СН31 t
(CjHsCOkO*
- 52-
Анальгетики
N
3 1 67
сн2оснз
О н
C2HSvn Ar;.CH^H?CI
N=N
3 5 70
C2H5NCO * NaNg
N=N
BrCH2CH2CI
3.1.68 + 3.1.70
О
c2h5^n An-ch2ch2 I I N=N
3.1.71
Главное отличие альфентанила от фентанила заключается в более коротком времени его действия. Применяется в анестезиологической практике наряду с барбитуратами при кратковременных хирургических вмешательствах.
Синонимами препарата являются алфента, рапифен и др.
Суфентанил (Sufentanyl)
Суфентанил — и-(4-(метоксиметил)-1 -[2-(2-тиенил)этил]-К-фенил-пропанамид (3.1.72) — препарат аналогичный альфентанилу и отличается от него заместителем у азота пиперидинового кольца. Синтез во многом повторяет элементы синтеза альфентанила, и лишь на последней стадии алкилирование 4-метокс иметил-4-N-пропионил-анилинопиперидина (3.1.68) проводится метансульфо-натом 2-гидроксиэтилтиофена [44, 45, 46].
СН2СН2 -OSO2 -снг
-53-
Глава 3
Суфентанил превосходит фентанил по активности в 5 10 раз. Применяется в анестезиологической практике при хирургических вмешательствах.
Синонимом препарата является суфента и др.
Смешанные агонисты-антагонисты
Препараты этой группы проявляют и агонистическую, и антагонистическую активность. Принято считать, что агонистическая активность проявляется благодаря их взаимодействию с ц-рецепторами, а антагонистическая — на других (в частности, на к- и о-рецепторах). Механизм их действия мало понятен. Несмотря на то, что их действие может проявиться в виде анальгетического эффекта, определенного угнетения дыхания и в виде других проявлений, свойственных препаратам типа морфина, они могут блокировать и даже обращать эффекты агонистов, а также отменять абстинентный синдром у пациентов с зависимостью к опиоидам. Интересно, что может возникнуть толерантность к агонистическим свойствам этих препаратов, но не возникает толерантности к их антагонистическим свойствам. Зависимость может возникнуть и при их длительном применении.
Эта группа соединений используется для анальгезии в случаях умеренных и сильных болей. Они менее эффективны чем морфин, однако они не вызывают сильной депрессии дыхания при передозировках.
Налорфин (Nalorphine)
Налорфин — N-аллилнорморфин (3.1.75) получают исходя из морфина путем его полного ацетилирования, т. е. трансформацией в героин (3.1.21), с целью временной защиты гидроксильных групп и далее подвергают деметилированию. Для этого героин (3.1.21) повергают воздействию бромциана. Полученное при этом N-циано-производное (3.1.73) гидролизуют раствором соляной кислоты в деметилированный морфин — норморфин (3.1.74), вторичную аминную группу которого подвергают алкилированию аллилброми-дом [47, 48].
- 54 -
Анальгетики
Налорфин обладает более слабой, чем у морфина, анальгетической активностью, однако самостоятельного значения в качестве анальгетика не имеет. Его применяют в качестве антагониста наркотических анальгетиков. Он устраняет вызываемые агонистами опиатных рецепторов угнетение центра дыхания, брадикардию, рвоту и т. п.
Налорфин являлся первым соединением, использовавшимся при передозировках наркотиков, и в частности героина. Однако он проявляет ряд побочных эффектов, в том числе может вызвать зрительные галлюцинации, и поэтому в некоторых странах запрещен к применению.
Наиболее популярным синонимом препарата является наркан.
Пентазоцин (Pentazocine)
Пентазоцин — 1,2,3,4,5,6-гексагидро-6,11-диметил-3-(3-метил-2-бутенил)-2,6-метано-3-бензазоцин-8-ол (3.1.81) является производным бензоморфана. Синтез пентазоцина осуществляют исходя из 3,4-диметилпиридина. Подвергнув последний действию йодистого метила, получают 1,3,4-триметилпиридиний йодид (3.1.76), который вводят во взаимодействие с 4-метоксибензилмагний-хлоридом с получением 2-метоксибензил-3,4-диметил-1,2-дигид-ропиридина (3.1.77). Двойную связь при С5 полученного соединения восстанавливают водородом с использованием в качестве катализатора палладия с получением 2-метоксибензил-3,4-диме-тил-1,2,3,4-тетрагидропиридина (3.1.78). Последний подвергают внутримолекулярному алкилированию и одновременному деметилированию эфирной связи бромистоводородной кислотой, в результате чего образуется 2-гидрокси-2,5,9-триметилбензо-6-мор-фен (3.1.79), который подвергают N-деметилированию бромцианом с получением 2-гидрокси-5,9-диметилбензо-6-морфена (3.1.80). Алкилирование последнего 1-бром-3-метил-2-бутеном дает пентазоцин [49, 50J.
-55-
Г лава 3
3 1 77
1 BrCN
2 Ht
СНз
Br-CH2-CH=c(
СНз
Пентазоцин — слабый антагонист морфина. Он значительно слабее, чем налорфин или леваллорфан, но обладает сильно выраженным анальгетическим эффектом. Пентазоцин является первым анальгетиком агонист-антагонистического действия появившимся на фармацевтическом рынке. По анальгетической активности уступает морфину, однако в значительно меньшей степени угнетает дыхательный центр. В качестве агониста он, в основном, воздействует на к-рецепторы. При оральном применении по активности сравним с кодеином. Отмечены случаи возникновения толерантности. Пентазоцин применяют при болях различной интенсивности и для премедикации перед оперативным вмешательством.
Наиболее часто встречающимся синонимом препарата является фортран.
Налбуфин (Nalbuphine)
Налбуфин — 17-(циклобутилметил)-4,5а-эпоксиморфинан-3.6а,14-триол (3.1.85) синтезируют исходя из оксиморфона (3.1.26), кого-
- 56-
Анальгетики
рый после зашиты гидроксильных групп путем ацетилирования подвергают действию бромциана с получением N-цианопроизвод-ного, далее — гидролизу соляной кислотой с получением 14-гидро-ксидигидронорморфона (3.1.82). Трансформацию последнего в искомый налбуфин (3.1.85) осуществляют либо восстановлением карбонильной группы полученного 14-гидроксидигидронормор-фона боргидридом натрия (3.1.83) и дальнейшим алкилированием продукта циклобутилметилбромидом, либо ацилированием (3.1.82) хлорангидридом циклобутанкарбоновой кислоты (3.1.84) и дальнейшим одновременным восстановлением двух карбонильных групп в полученном соединении алюмогидридом лития в искомый продукт [51, 52].
1 (СН3СО)2О
2 BrCN
з HCI
3 i 83
3 1 85
Налбуфин является мощным анальгетиком и в качестве такового по активности равен морфинг'. По строению сходен с оксиморфоном и опиоидным антагонистом налоксоном. Побочные эффекты выражены значительно меньше, чем у налорфина. Налбуфин назначают в качестве препарата для облегчения переносимости умеренной и острой боли. Применяется в качестве дополнительного средства при сбалансированной анестезии, для пред- и послеоперационной анальгезии, при гинекологических вмешательствах.
Наиболее распространенным синонимом налбуфина является ну-баин.
-57-
Глава 3
Бупренорфин (Buprenorphine)
Бупренорфин — 17-(циклопропилметил)-ос-(1,1-диметилэтил)-4,5-эпокси-18,19- дигидро-3-гидрокси-6-метокси-а-метил-6,1 4-этеномор-финан-7-метанол (3.1.91) получают исходя из одного из алкалоидов морфина — тебаина (3.1.23). Синтез бупренорфина начинают на основе продукта реакции 4+2 циклоприсоединения тебаина и ме-тилвинилкетона. Результирующий продукт 7-ацетил-6,14-эндо-этано-тетрагидротебаин (3.1.86) далее восстанавливают водородом с использованием в качестве катализатора палладия на угле в 7-аце-тил-6,14-эндо-этано-тетрагидро-тебаин (3.1.87). Последний вводят во взаимодействие с /ирелг-бутил-магнийхлоридом с получением 6,14-эндо-этано-7-(2-гидрокси-3,3-диметил-2-бутил)-тетрагид-ротебаина (3.1.88). Полученный продукт деметилируют с помощью бромциана с получением 6,14-эндоэтано-7-(2-гидрокси-3,3-диметил-2-бутил)тет-рагидронор-тебаина (3.1.89). Ацилируя последний хлорангидридом циклопропанкарбоновой кислоты и дальнейшим восстановлением введенной карбонильной группы получают N-циклопропилметил-6,14-эндо-этано-7-(2-гидрокси-3,3-диметил-2-бутил)-тетрагидронор-тебаина (3.1.90). Конечный бупренорфин (3.1.91) получают селективным деметилированием метоксигруппы, связанной с ароматическим кольцом, при высокотемпературном воздействии на 3.1.90 гидроокисью калия [53, 54].
О
Н2С =СН-С-СН3
lCH3)3CMgCL
3 1 86
3 1 89
-58-
Анальгетики
Бупренорфин проявляет свойства анальгетика центрального действия, не угнетает дыхательный центр, не вызывает развития привыкания и лекарственной зависимости. В общем, препарат лишен дисфорического и психотомиметического эффектов. В отличие от остальных рассмотренных агонистов-антагонистов бупренорфин проявляет частичный агонистический эффект и на ц-рецепторах. Блокирует эффекты морфина примерно на 30 ч. Проявляет ряд некоторых уникальных эффектов, не свойственных соединениям этого ряда. Препарат применяют при болевых синдромах средней интенсивности различного генеза.
Наиболее распространенным синонимом является бупренекс.
Опиоидные антагонисты
Опиоидные антагонисты соединения имеют выраженную антагонистическую активность и, в отличие от смешанных агонистов-антагонистов, не проявляют агонистической активности.
Эффективность и сила опиоидных антагонистов варьируют в зависимости от типа опиоидных рецепторов — ц-, S, к, о, с которыми они взаимодействуют. Полной ясности о механизме их действия нет. Однако высказано предположение, что они антагонизируют действие эндогенных опиатных пептидов.
Эти соединения являются антагонистами и по отношению к агонистам-антагонистам. Они антагонизируют действие агонистов, смешанных агонистов-антагонистов. К ним не возникает зависимости и толерантности. К ним не возникает также привыкания и пристрастия. Их используют при передозировках опиоидных анальгетиков или при их непереносимости больными, а также при лечении наркомании.
Налоксон (Naloxone)
Налоксон — (-)-17-(аллил)-4,5-эпокси-3,14-дигидроксиморфинан-6-он (3.1.92) получают алкилированием 14-гидроксидигидронормор-финана (3.1.82) аллилбромидом [55, 56, 57, 58].
-59-
Г лава 3
Следует отметить, что N-аллильное замещение в ряду производных морфина, как правило, приводит к антагонистическим свойствам. Налоксон в несколько раз сильнее налорфина в качестве антагониста. Препарат блокирует опиатные рецепторы, кстраняет центральное и периферическое действие опиоидов, в том числе и угнетение дыхания. Налоксон применяют при передозировке наркотических анальгетиков.
Синонимами препарата являются наркан, талвин и др.
Налтрексон (Naltrexone)
Налтрексон — (-)-17-(циклопропилметил)-4,5-эпокси-3,14-дигидро-ксиморфинан-6-он (3.1.93) является N-циклопропилметильным производным оксиморфона (3.1.82). Одним из методов получения является путь, аналогичный синтезу налоксона и заключающийся в применении вместо аллилбромида циклопропилметилбромида [59].
[>— СН2-8г
Препарат не имеет агонистических свойств. По своим фармакологическим характеристикам налтрексон сходен с налоксоном, однако отличается от него по двум важным показателям — большей продолжительностью действия и тем. что его метаболит 6-р-налтрексол также является мощным антагонистом. Налтрексон потенциально гепатоток-сичен. Налтрексон применяют для блокады фармакологических эффектов опиоидов при их передозировке.
Синонимами препарата являются налотрекс, трексан и др.
-60-
Анальгетики
3.2. Нестероидные противовоспалительные средства, или жаропонижающие анальгетики
Огромное количество препаратов, принадлежащих к различным классам соединений, проявляют анальгетическое, жаропонижающее и противовоспалительное действие. При этом они лишены многих нежелательных эффектов, присущих опиоидным анальгетикам (депрессия дыхания, привыкание и т. п.). Их называют ненаркотическими анальгетиками, аспириноподобными веществами, жаропонижающими анальгетиками и др., чтобы отличить от опиоидов, и нестероидными противовоспалительными и жаропонижающими анальгетиками, чтобы отличить от глюкокортикоидов. Точный механизм действия этих препаратов окончательно не выяснен. Полагают, что он может быть связан с их способностью ингибировать синтез простагландинов, что уменьшает сенсибилизирующее влияние последних на чувствительные окончания, а это, в свою очередь, уменьшает эффект действия медиаторов и, в частности, брадикинина. Однако анальгетическая и противовоспалительная активность этих препаратов не всегда коррелирует с их способностью подавлять синтез простагландинов. Существуют и другие предположения о механизме действия ненаркотических анальгетиков. Эксперименты на животных указывают на то, что анальгетическое действие этого ряда препаратов является периферическим, однако не исключено, что ацетаминофен может иметь и центральное действие, блокируя передачу болевых импульсов.
В общем, неопиоидные анальгетики характеризуются тремя основными видами действия: анальгетическим, противовоспалительным и жаропонижающим; применяются для облегчения головной боли, миалгии, артралгии; не обладают седативным и снотворными эффектами. Эйфория, привыкание и лекарственная зависимость при их применении не возникают.
Нестероидные противовоспалительные и жаропонижающие анальгетики классифицируются как:
• производные арилкарбоновых кислот (аспирин, дифлусинал и др.);
• пиразолоны (фенилбутазон, метамизол и др.) и прочие (в частности, ацетаминофен и фенацетин);
• производные антраниловой кислоты (флуфенамовая кислота, мефенамовая кислота, меклофенамовая кислота);
• производные арилуксусной кислоты (диклофенак, фенклофенак);
-61 -
Г лава 3
• производные арилпропионовой кислоты (ибупрофен, кегопро-фен, напроксен, фенопрофен и др.);
• производные индолил/инденуксусной кислоты (индометацин, сулиндак и др.);
• оксикамы (пироксикам, изоксикам).
Производные салициловой кислоты
Аспирин (Aspirin)
Аспирин — ацетилсалициловая кислота (3.2.2) получается ацетилированием салициловой кислоты (3.2.1) ангидридом или хлорангид-ридом уксусной кислоты [60. 61, 62, 63].
соон
3.2.1
(СН3СО)2О
соон о l^s^O-С-СНз
3.2.2
Препарат оказывает анальгетическое, жаропонижающее и противовоспалительное действие, а также уменьшает агрегацию тромбоцитов. Полагают, что основным механизмом его действия является необратимое ацетилирование циклооксигеназы, в результате чего нарушается синтез простагландинов, простациклинов и тромбоксана. Вследствие этого уменьшается пирогенное влияние простагландинов на центры терморегуляции и на чувствительные нервные окончания, что приводит к понижению их чувствительности к болевым медиаторам. Необратимым нарушением синтеза тромбоксана А2 в тромбоцитах объясняют антиагрегантное действие аспирина.
Аспирин сегодня используется в количествах больших, чем любое другое лекарственное средство. Аспирин широко применяется при головных и невралгических болях, при ревматических состояниях, при болевых синдромах разной этиологии, для устранения болевых ощущений при менструациях. Его применяют при лихорадочных состояниях, профилактике и лечении тромбозов и эмболий, профилактики и лечении ишемических нарушений и мозгового кровообращения.
Аспирин является препаратом, имеющим наибольшее число синонимов. Синонимами препарата являются ацетосал, кислота ацетилсалициловая, цетосал и oipoMHoe количество других.
В медицинской практике используются и неацетилированные салицилаты.
-62-
Анальгетики
Дифлунисал (Diflunisal)
Дифлунисал — 2’.4’-дифтор-4-гидрокси-3-бифенилкарбоновую кислоту (3.2.5) синтезируют исходя из диазониевой соли, получаемой из 2,4-дифторанилина и изо-амил-нитрита, и анизола в присутствии солей одновалентной меди, осуществляя классическую схему получения биарилов Полученный при этом 4-(2,4-дифторфенил)анизол (3.2.3) деметилируют йодистым водородом в 4-(2,4-дифторфенил)-фенол (3.2.4), Последний вводят во взаимодействие с двуокисью углерода в присутствии основания по методу Кольбе с получением дифлунисала (3.2.5) [64. 65, 66, 67].
HI ' СН3СООН 0CH3 -----------1
СО2 / К2СОз
F СООН
325
Являясь ингибитором, дифлунисал (простагландин синтетазы) проявляет анальгетическое, жаропонижающее и противовоспалительное действие. Препарат применяют для долговременного и кратковременного симптоматического облегчения слабых и умеренных болей, при остеоартритах и ревматоидных артритах.
Синонимами препарата являются долобид, адомал, ноладол и др.
На практике применяются и другие производные салициловой кислоты в виде солей.
Салицилат магния (magnesium salicylate) и салицилат натрия (sodium salicylate) менее эффективны, чем соответствующие дозы аспирина, однако лучше переносятся больными, чувствительными к аспирину. Хопинмагнийтрисалицилат (cholinemagnesiumtrisahcylate) представляет собой смесь салицилата холина и салицилата магния с тем же действием, что и у аспирина, однако лучше переносится больными, у которых наблюдаются желудочно-кишечные явления при приеме аспирина.
-63-
Г лава 3
Пиразолоны
Значительную роль в качестве анальгетиков, противовоспалительных и жаропонижающих средств в медицине играют производные пира-золона. К ним относятся антипирин, бутадион, амидопирин, фенилпира-зон, сульфинпиразон, метамизол натрия (анальгин) и некоторые другие. По анальгезирующей и противовоспалительной активности они близки к производным салициловой кислоты. Хотя механизм их действия выяснен не полностью, полагают, что производные пиразолона так же. как и аспирин, ингибируют биосинтез простагландинов, а также уменьшают проницаемость капилляров и препятствуют развитию воспалительной реакции. Серьезным ограничением для широкого применения пиразолонов в медицине являются случаи возникновения при их применении агранулоцитоза.
Наиболее широко в медицине применяется метамизол натрия, хотя в некоторых странах он запрещен, а также комбинированные препараты на его основе и, в частности, баралгин, представляющий собой комбинированный препарат на основе анальгина со спазмолитиком 4'-(это-ксипиперидин)карбметоксибензофеноном и ганглиоблокатором 2,2-ди-фенил-4-пиперидилацетамидом.
Фенилбутазон (Phenylbutazone)
Фенилбутазон — 4-бутил-1,2-дифенил-3,5-пиразолидиндион (3.2.6) получают в одну' стадию взаимодействием гидразобензола с бутилмалоновым эфиром [68, 69].
ОС2Н5
С4Н9
3 2 58
Фенилбутазон применяется для снятия слабых и умеренных болей, при головной боли, ревматоидных артритах и остеоартритах.
Синонимами препарата являются алговерин, азолид, бутазолидин И др.
Сульфинпиразон (Sulfinpyrazone)
Сульфинпиразон — 1,2-дифенил-4-2-(фенилсульфинил)этил-3.5-
пиразолидиндион (3.2.8) является аналогом фенилбутазона; его по-
-64-
Анальгетики
лучают аналогичным способом — конденсацией гидразобензола с 2-(2-фенилтиоэтил)малоновым эфиром в пиразолдион (3.2.7) с последующим окислением тиолового эфира перекисью водорода в уксусной кислоте в сульфоксид — сульфинпиразон (3.2.8) [70, 71].
Сульфинпиразон применяется по тем же показаниям, что и фенилбутазон.
Синонимами препарата являются ангуран и энтурен.
Метамизол натрия (Metamizole sodium)
Метамизол натрия — 1 -фенил-2,З-диметил-4-метиламинопиразо-лон-5-К-метансульфонат натрия (3.2.16) получают многостадийным синтезом исходя из ацетоуксусного эфира и фенилгидразина. Их взаимодействие приводит к получению 1-фенил-З-метилпиразо-лона-5 (3.2.9). Метилированием последнего йодистым метилом получают 1-фенил-2,3-диметилпиразолон-5 (3.2.10). Это соединение самостоятельно используется в медицине в качестве жаропонижающего и противовоспалительного анальгетика под названием антипирин. Последний подвергают нитрозированию нитритом натрия в кислой среде с получением 1-фенил-2,3-диметил-4-нитрозопира-золона-5 (3.2.11). Восстановление нитрозопроизводного (3.2.11) разными восстановителями приводит к получению 1-фенил-2,3-диметил-4-аминопиразолона-5 (3.2.12). Последний вводят во взаимодействие с бензальдегидом с образованием легко выделяющегося
-65-
Глава 3
кристаллического 1 -фенил-2,З-диметил-4-бенилиденаминопиразо-лона-5 (3.2.13), который метилируют по иминному атому азота диметилсульфатом с получением четвертичной соли (3.2.14). Гидролизом полученной соли получают 1-фенил-2,3-диметил-4-метил-аминопиразолон-5 (3.2.15). Обработка последнего водным раствором смеси бисульфита натрия и формальдегида приводит к получению 5-М-метансульфоната натрия 1-фенил-2,3-диметил-4-метилами-нопиразолона (3.2.16) — искомого метамизол натрия [72, 73, 74, 75].
сн
о о
ЛА,.;1'*-:
NaNO2 Н
[Н]
3 2 12
32 14
СНз
NaHSO3/ СН2О
32 15
NaOSO2CH2
3216
Метамизол натрия обладает выраженным анальгетическим, жаропонижающим свойством и слабо выраженным противовоспалительным действием и очень удобен в случаях, когда необходимо быстро создать в крови высокую концентрацию препарата. Метамизол натрия применяют при болях различного происхождения (почечной и желчной колике, невралгии, миалгии, при травмах, ожогах, головной и зубной боли). При применении препарата возможны аллергические реакции, при длительном применении — гранулоцитопения.
Синонимами препарата являются дипирон, анальгин и многие другие.
-66-
Анальгетики
Производные я-аминофенола
Ацетаминофен (Acetaminophen)
Ацетаминофен — д-ацетаминофенол (3.2.80) получают взаимодействием п-амнофенола с уксусным ангидридом [76, 77].
но
nh2
(СН3СО)2О
3 2 17
В отличие от описанных НПВС ацетаминофен лишен противовоспалительных и противоревматических свойств. Недавно было показано, что он, как и аспирин, также ингибирует действие циклооксигеназы в мозге и даже мощнее, чем аспирин. С другой стороны, механизм анальгетического действия ацетаминофена не совсем ясен, поскольку он слабо действует на периферическую циклооксигеназу.
Ацетаминофен широко применяется в качестве анальгетика и жаропонижающего средства. Асетаминофен предназначен для умеренной анальгезии. Он так же эффективен, как и аспирин, и используется для анальгезии при головной боли (от слабой до умеренной), миалгии, артралгии, хронических болях при онкологических, послеоперационных болях и т. д.
Синонимами препарата являются параацетамол, тайленол и многие другие.
Производные антраниловой кислоты
Производные антраниловой кислоты являются прямыми структурными аналогами производных салициловой кислоты. Они обладают анальгезирующей, противовоспалительной и жаропонижающей активностью. По анальгезирующей и жаропонижающей активности они близки к пиразолонам, а по противовоспалительной активности превосходят салицилаты. Механизм действия этого ряда нестероидных противовоспалительных анальгетиков не окончательно выяснен.
Флуфенамовая кислота (Flufenamic acid)
Флуфенамовая кислота — М-(а,а,а-трифтор-.и-толил)антраниловая кислота (3.2.18) получается взаимодействием 2-хлорбензойной кислоты с 3-трифторметиланилином в присутствии поташа и медных опилок [78, 79].
-67-
Глава 3
а С ООН
CL
32 18
Флуфенамовая кислота показана при умеренных болях и при дисменорее, но ее не следует применять более одной недели вследствие возможной нефротоксичности, желудочно-кишечной токсичности и анемии. Часто применяют совместно с антикоагулянтом варфарином, действие которого усиливается при совместном применении с флуфе-намовой кислотой.
Синонимами препарата являются арлеф, флексокуган, ромазал и др.
Мефенамовая кислота (Mefenamic acid)
Мефенамовую кислоту — М-(2,3-ксилил)антраниловую кислоту (3.2.19) получают примерно аналогичным способом — взаимодействием калиевой соли 2-бромбензойной кислоты с 2,3-диметил-анилином в присутствии ацетата двухвалентной меди [80, 81].
(СН3СОО)2Си
соон
Применяют по тем же показаниям, что и флуфенамовую кислоту.
Синонимами препарата являются паркемед, понстан, понстел и др.
Меклофенамовая кислота (Meclofenamic Acid)
Меклофенамовую кислоту — К-(2,6-дихлор-.и-толил)антраниловую кислоту (3.2.20) получают аналогично флуфенамовой кислоте взаимодействием калиевой соли 2-бромбензойной кислоты с 2,6-ди-хлор-3-метиланилином в присутствии двубромистой меди в среде N-этилморфолина и диглима [82, 83].
-68-
Анальгетики
CuBr2
Препарат применяют по тем же показаниям, что и флуфенамовую кислоту.
Синонимом препарата является мовенс.
Нифлумиковая кислота (Niflumic acid)
Нифлумиковая кислота — 2-3-(1рифторметил)анилино никотиновая кислота (3.2.9) получается либо взаимодействием 2-хлор-никотиновой кислоты с 3-трифторметил-анилином [84, 85, 86], либо 2-аминоникотиновой кислоты с 1-бром-З-трифторметил-бензолом [87].
Си 1 К2СО3
Применяют по тем же показаниям, что и описанные выше препараты.
Синонимами препарата являются актол, флунир, форенол, нифлу-рил и др.
Производные пропионовой кислоты
Этот ряд противовоспалительных, анальгетических и жаропонижающих соединений (ибупрофен, напроксен, кетопрофен, фенопрофен) в равной мере можно называть как производными пропионовой кислоты, так и производными фенилпропионовой кислоты. Механизм их действия окончательно не ясен, однако полагают, что он также связан с подавлением активности простагландина синтетазы.
Ибупрофен (Ibuprofen)
Ибупрофен — 2-(4-изо-бутилфенил)пропионовую кислоту (3.2.23) можно синтезировать разными методами [88, 98]. Первым спосо-
-69-
Глава 3
бом (простейшим) является синтез ибупрофена, заключающийся в ацилировании ито-бутилбензола хлорангидридом уксусной кисло-1ы. Полученный при этом изо-бутилбензофенон (3.2.21) вводят во взаимодействие с цианидом натрия с получением оксинитрила (3.2.22). который под действием йодистоводородной кислоты в присутствии фосфора превращается в 2-(4-шо-бутилфенил) пропионовую кислоту (3.2.23). последовательно проходя стадии дегидратации, восстановления и гидролиза.
AICI3
HI I Р
СНз н3с-сн-сн2
NaCN
3 2 21
СНз H3C-CH-CH2
3 2 23
Второй способ синтеза ибупрофена заключается в хлорметилиро-вании г/зо-бутилбензола с получением 4-изо-бутилбензилхлорида (3.2.24). Взаимодействием последнего с цианидом натрия получают 4-дзо-бутилбензилиианид (3.2.25), который алкилируют в присутствии амида натрия йодистым метилом в 2-(4-изо-бутилбензил)-пропионитрил (3 2 26). Гидролиз последнего в присутствии основания приводит к ибупрофену (3.2.23).
СНз
НзС -C'i-CH2
+ СН2О НС!
ZnCl2
СНз H3C-CH-CH2
3 2 24
NaCN
1 NaNHz £сщ
СНз н3с-сн-сн2
он
----► 3 2 23
3 2 26
Ибупрофен является первым препаратом из производных пропионовой кислоты, разрешенных к клиническому использованию. Ибупрофен проявляет анальгетическое, жаропонижающее и противовоспалительное действие, которое сравнимо и даже превосходит таковое
- 70-
Анальгетики
аспирина и ацетаминофена. Переносится лучше аспирина, и случаев побочных реакций наблюдается мало. Применяют для лечения ревматоидного артрита, при различных формах суставных и внесуставных ревматоидных заболеваний, а также при болях, возникающих в результате воспалительных поражений периферической нервной системы, при обострении подагры, невралгии, миалгии, анкилозирующем спондилите, радикулите, травматическом воспалении мягких тканей и опорнодвигательного аппарата. Как вспомогательное средство используется при инфекционно-воспалительных заболеваниях ЛОР-органов, аднекси-те, первичной дисменорее, головной и зубной болях. Не рекомендуется больным с язвой желудка или перенесшим таковую.
Наиболее распространенными синонимами ибупрофена являются бру фен, ибуфен, мотрин, ребуген и др.
Напроксен (Naproxen)
Напроксен — 2-(6-метокси-2-нафтил)-пропионовая кислота (3.2.15) может быть получена методами синтеза, описанными для ибупрофена, а также методами, которые будут описаны ниже для синтезов фенопрофена (3.2.21) и кегопрофена (3.2.27) исходя из 2-ацетил-или 2-хлорметил-6-метоксинафталина [99, 100, 101].
сн3о
3 2 27
сн3
СН-СООН
Аналогично другим препаратам этого ряда напроксен проявляет анальгетическое, жаропонижающее и более длительное противовоспалительное действие. Вызывает ослабление и исчезновение болевого синдрома, в том числе и болей в суставах, уменьшение скованности и припухлости суставов. Применятся по тем же показаниям, что и ибупрофен.
Синонимами препарата являются напросин, пропаксен, апотекс.
Фенопрофен (Fenoprofen)
Фенопрофен — 2-(3-феноксифенил)пропионовую кислоту (3.2.32 ) получают синтезом, исходящим из 3-гидроксиацетофенона, который этерифицируют бромбензолом в присутствии поташа и медных опилок с получением 3-феноксиацетофенона (3.2.28). Карбониль-нчю группу последнего восстанавливают боргидридом натрия,
- 71 -
Глава 3
и полученный спирт (3.2.29) бромируют трехбромисым фосфором. Взаимодействием полученного бромопроизводного (3.2.30) с цианистым натрием получают 2-(3-феноксифенил)пропионитрил (3.2.31), который гидролизуют в искомый фенопрофен (3.2.32) [102, 103].
он
СН-СНз
3 2 29
СООН
СН-СНз
3 2 32
Химически и фармакологически фенопрофен сходен с вышеописанным рядом соединений. Применяют для лечения симптомов ревматоидного артрита и остеоартрита, однако фенопрофен проявляет и ряд нежелательных побочных эффектов.
Синонимами препарата являются налфон, диета и др.
Кетопрофен (Ketoprofen)
Кетопрофен — 2-(3-бензоил)пропионовую кислоту (3.2.37) синтезируют исходя из 3-метилбензофенона, который подвергают бромированию с получением 3-бром-метилбензофенона (3.2.33). Взаимодействием последнего с цианистым натрием получают 3-циано-метилбензофенон (3.2 34), который вводят во взаимодействие с диэтиловым эфиром угольной кислоты в присутствии этилата натрия. Полученное таким образом производное циануксусного эфира (3.2.35) алкилируют йодистым метилом, и полученный продукт (3.2.36) подвергают кислотному гидролизу с получением кетопрофена (3.2.37) [104,105, 106].
C2H5ONa I СО(ОС2Н£»2
3 2 35
-72-
Анальгетики
Кетопрофен применяют для облегчения слабых и умеренных болей, при ревматоидном артрите, остеоартрите, анкилозирующем спондилите, подагре, боли в позвоночнике, невралгии, миалгии. Применяют также при неосложненных травмах, в частности спортивных, при растяжении или разрыве связок и сухожилий мышц. Препарат проявляет ряд нежелательных побочных эффектов на почечную и печеночную функции, ЖКТ.
Наиболее распространенными синонимами являются алревмат, фас-тум, кеталгин, реупрофен и др.
Производные уксусной кислоты
В качестве противовоспалительных, анальгетических и жаропонижающих соединений в медицине кроме производных пропионовой кислоты весьма широко применяются и другие препараты и, в частности, производные уксусной кислоты (диклофенак, феклофенак, алклофенак и др.). Полагают, что их противовоспалительное, анальгетическое и жаропонижающее действие также обусловлено подавлением активности простагландин синтетазы.
Диклофенак (Diclofenac)
Диклофенак — 2-[(2,6-дихлорфенил)-амино]-фенилуксусную кислоту (3.2.42) синтезируют исходя из 2-хлорбензойной кислоты и 2,6-дихлоанилина. Взаимодействием последних в присутствии гидроокиси калия и меди получают М-(2,6-дихлорфенил)антра-ниловую кислоту (3.2.38), карбоксильную группу которой подвергают восстановлению алюмогидридом лития. Полученный при этом 2-[(2,6-дихлорфенил)-амино]-бензиловый спирт (3.2.39) подвергают далее хлорированию хлористым тионилом в 2-[(2,6-дихлорфенил)-аминофбензилхлорид (3.2.40) и далее, действуя цианидом натрия, превращают в 2-[(2,6-дихлорфенил)-амино]-бензилхцианид (3.2.41). Гидролиз нитрильной группы последнего приводит к диклофенаку (3.2.42) [107, 108].
SOCI2
-73-
Глава 3
NaCN
NaOH
Диклофенак обладает всеми свойствами, присущими ряду препаратов — производных пропионовой кислоты, а по силе противовоспалительного и анальгетического действия превосходит аспирин, анальгин и ибупрофен. Применяют при остром ревматизме, ревматоидном артрите, остеоартрите, анкилозируюшем спондилите, артрозах, при болях в позвоночнике, невралгии, миалгии. Редко вызывает побочные эффекты.
Наиболее распространенным синонимом является вольтарен.
Фенклофенак (Fenclofenac)
Фенклофенак — о-[(2,4-дихлорфенокси)фенил]уксусная кислота (3.2.45) получается синтезом, исходящим из 2,4-дихлорфенола и 2-хлорацетофенона. взаимодействием которых в присутствии гидроокиси натрия и порошкообразной меди получают соответствующий г-ацетил-З'д’-дихлор-дифениловый эфир (3.2.43). Последний вводится во взаимодействие с серой и морфолином с получением по методу Вильгеродта тиоамида (3.2.44), который далее гидролизуют в искомый фенклофенак [109, ПО].
Си / NaOH
Препарат применяется по тем же показаниям, что и диклофенак. Синонимом препарата является фленак.
- 74 -
Анальгетики
Индолил/инденуксусные кислоты
Препараты этого ряда (индометацин, толметин, сулиндак и др.) являются очень эффективными нестероидными противовоспалительными препаратами с сильно выраженной анальгетической активностью. Являются сильными ингибиторами биосинтеза простагландинов.
Индометацин (Indometacin)
Индометацин — 1-(л-хлорбензоил)-5-метокси-2-метилиндол-3-ук-сусную кислоту (3.2.51) предложено получать разными методами. Во всех случаях предлагаемые способы синтеза исходят из 4-ме-токсифенилгидразина. Согласно первому способу, осуществляют реакцию получения индолов из фенилгидразона (3.2.46) по Фишеру с использованием в качестве карбонильной компоненты метилового эфира левулиновой кислоты, а в качестве кислоты — хлористого водорода в этаноле с получением метилового эфира 5-метокси-2-метил-3-индолилуксусной кислоты (3.2.47). Продукт гидролизуют щелочью до 5-метокси-2-метил-3-индолилуксусной кислоты (3.2.48), из которой с использованием дарсте-бутилового спирта и хлорида цинка в присутствии дипиклогексилкарбодиимида получают трет-бутиловый эфир 5-метокси-2-метил-3-индолилуксусной кислоты (3.2.49). Последний подвергают ацилированию по индольному атому азота хлорангидридом ч-хлорбензойной кислоты в диметилфор-мамиде, используя в качестве основания гидрид натрия. Полученный трет-бутиловый эфир 1-(ч-хлорбензоил)-5-метокси-2-метил-3-индолилуксусной кислоты (3.2.50) далее подергают термическому расщеплению до соответствующей (3.2.51) [111, 112].
кислоты
индометацина
CH3Q—4 Л— NH—NHj
О
г-н
t-Нз v jj о
NH—<7 0СНэ
N
JL ^*\>3-СН3
СН3 J
3 2 46 °
3 2 43
2 (СНз)зОН / ZnCI2
-75-
Глава 3
При втором способе фенилгидразон (3.2.46) подвергают циклизации в присутствии самого хлорангидрида и-хлорбензойной кислоты, при которой одновременно происходит и ацилирование гидразона, и его циклизация в метиловый эфир 5-метокси-2-метил-1-(и-хлорбензоил)-3-индолилуксусной кислоты (3.2.52), который далее гидролизуют щелочью в индометацин (3.2.51) [113, 114].
То же вещество предложено получать непосредственно исходя из 4-метоксифенил-и-хлорбензоилгидразина и левулиновой кислоты путем циклизации соответствующего гидразона в присутствии хлористого водорода. С этой целью конденсацией ацетальдегида с п-метокси-фенилгидразином получают гидразон (3.2.53), который ацилируют хлорангидридом п-хлорбензойной кислоты с получением гидразона
- 76-
Анальгетики
(3.2.54). Последний гидролизуют в гидразин (3.2.55). Вводя последний во взаимодействие с левулиновой кислотой, получают гидразон (3.2.56), который подвергают циклизации по Фишеру в индометацин (3.2.51) [И5].
СНзСНО
/=\ (СН2)2-СООН
СН3О—d Л—N—N=C<
'-' с=0 СНз
HCI • с2н5он
Индометацин применяют при ревмокардите, неспецифическом инфекционном полиартрите, подагрическом артрите, ревматоидном артрите. остеоартрите, анкилозирующем спондилите, артрозах, при болях в позвоночнике, невралгии, миалгии и других заболеваниях, сопровождающихся воспалением.
Синонимами препарата в числе других являются метиндол. инда-цид, румацид и многие другие.
Толметин (Tolmetin)
Толметин — 1-метил-5-и-толуилпиррол-2-уксусную кислоту (3.2.61) синтезируют исходя из 1-метилиндола, который аминометилируют с использованием формальдегида и диметиламина с получением
- 77-
Г лава 3
2-диметиламинометил-1-метилиндола (3.2.57) Последний метилируют йодистым метилом с получением соответствующей четвертичной соли (3.2.58). Взаимодействием последней с цианистым натрием получают 1-метил-2-пирролил)ацетонитрил (3.2.59), который ацилируют по свободному a-положению индольного кольца хлоангидридом 4-метилбензойной кислоты с использованием хлористого алюминия. Полученный при этом 1-метил-5-и-голуилпиррол-2-ацетонитрил (3.2.60) подвергают далее щелочному гидролизу с получением толметина (3.2.61) [116, 117, 118].
Толмегин, как и все вышеописанные препараты, ингибирует синтез простагландинов и проявляет выраженные анальгетические, противовоспалительные и жаропонижающие свойства. Применяется для снятия слабых и умеренных болей при ревматоидных артритах и остеоартритах.
Синонимами препарата являются толектин, толмекс и др.
Сулиндак (Sulindac)
Су-линдак — 5-фтор-2-метил-1-[и-(метилсульфинил)бензилиден]-инден-3-уксусная кислота (3.2.67) получается многостадийным синтезом, исходящим из н-фторбензалдегида, конденсацией которого с ангидридом пропионовой кислогы в присутствии ацетата натрия получают 4-фтор-а-метилкоричную кислоту (3.2.62). Восстановлением двойной связи с водородом с использованием в качестве катализатора палладия на угле получают 4-фтор-а-метилдигидро-коричную кислоту (3.2.63). В присутствии полифосфорной кислоты последнюю циклизуют в 5-фтор-2-метил-3-инданон (3.2.64). Полученный кетон вводят в реакцию Кневенагеля с циану ксусной кислотой и далее декарбоксилируют до 5-фтор-2-метилиден-3-у ксусной кислоты (.3.2.65). Конденсацией последней с л-меркаптобен-зальдегидом в присутствии метилата натрия получают 5-фтор-2-
-78-
Анальгетики
метил-1-(4-метилтио-бензилиден)-3-инден-уксуснуто кислоту (3.2.66), атом серы в которой окисляют перйодатом натрия до искомого сульфоксида (3.2.67) — сулиндака [119, 120, 121, 122].
3 2 67
Сулиндак применяется для снятия слабых и умеренных болей, при ревматоидных артритах и остеоартритах.
Синонимами препарата являются супрол, имбарал и др.
Оксикамы
Оксикамы являются представителями еще одного ряда противовоспалительных, анальгетических и жаропонижающих соединений, механизмом действия которых, по всей вероятности, является угнетение синтеза простагландинов. Эти препарат способны ослаблять болевой синдром средней интенсивности.
Пироксикам (Piroxicam)
Пироксикам — 1,1-диоксид 4-гидрокси-2-.метил-М-2-пиридил~2Я-1,2-бензотиазин-З-карбокса.мида (3.2.78) получают исходя из сахарина (3.2.79). Описаны два способа синтеза сахарина. Обычно исходят из толуола, который сульфируют хлорсульфоновой кислотой с получением изомерных 4- и 2-толуолсульфохлоридов. Изомерные продукты разделяются вымораживанием. Жидкую фракцию 2-толу-
-79-
Г лава 3
олсульфохлорид (3 2.68) отделяют от кристаллического 4-толуол-сульфохлорида и вводят во взаимодействие с аммиаком с получением 2-толуолсульфонамида (3.2.69). Окислением последнего перманганатом калия или оксидом шестивалентного хрома в серной кислоте поле чают сахарин — имид о-сульфобензойной кислоты (3.2.70) [123.' 124, 125, 126].
CISO3H
3 2 69
3 2 70
Альтернативный путь получения сахарина исходит из метилового эфира о-амино-бензойной (антраниловой кислоты). Последний подвергают диазотированию азотистой кислотой, и полученную диазониевую соль (3.2.71) вводят во взаимодействие с двуокисью серы в присутствии двухлористой меди с получением метилового эфира о-сульфобензойной кислоты (3.2.72). Взаимодействием последней с молекулярным хлором получают метиловый эфир о-хлорсульфонилбензойной кислоты (3.2.73), взаимодействием которой с аммиаком получают метиловый эфир о-суль-фониламид бензойной кислоты (3.2.74). Последний в присутствии хлористого водорода циклизуется в сахарин (3.2.70).
^у^ЮООСНз
<Хч''''’,*У''ЧМН2
NaNO2/ HCI
^Х^СООСНз
Ci
SO2 / [CuCy
3 2 71
аСООСНз
so2—ОН
3 2 72
Cl2
0.СООСН3
Г
ЪО2~ CI
3 2 73
NH3
0.СООСН3
Г
^SO-—NH2
3 2 74
3 2 70
Взаимодействием сахарина с гидроокисью натрия осуществляют замещение имидного атома водорода сахарина на натрий с получением натриевой соли (3.2.75). Последнюю вводят во взаимодействие с метиловым эфиром хлоругольной кислоты с получением замещенного сахариновым фрагментом метилового эфира уксусной кислоты (3.2.76). Под действием метилата натрия в диметилсульфоксиде последний подвергается перегруппировке в 1,1-диоксид 3-метоксикарбонил-3,4-дш идро-2-
-80-
Анальгетики
7/-1,2-бензотиазин-4-он (3.2.77). Последний метилируют метилйодидом по атому азота с получением 3.2.78 Наконец, вводя полученный продукт во взаимодействие с 2-аминопиридином, получают пироксикам (3 2.79)
NaOh
С1СНгСООСН3
Как уже было отмечено, пироксикам также относится к нестероидным противовоспалительным средствам. Применяется при воспалительных и дегенеративных заболеваниях опорно-двигательного аппарата, сопровождающихся болевым синдромом. Препарат применяют при ревмокардите, неспецифическом инфекционном полиартрите, подагрическом артрите, ревматоидном артрите, остеоартрите, анкилозирующем спондилите, артрозах, при болях в позвоночнике, невралгии, миалгии и других заболеваниях, сопровождающихся воспалением.
Синонимами препарата являются фелден, дексикам, роксен и др.
Изоксикам (Isoxicam)
Изоксикам — 1,1-диоксид 4-гидрокси-2-метил-Кт-(5-метил-3-изо-ксазолил)-2Я-1,2-бензотиазин-3-карбокса.мида (3.2.80) получают аналогично пироксикаму, используя на последней стадии амидирования 1,1-диоксида 3-метоксикарбонил-3,4-дигидро-2-Я-1,2-бензо-тиазин-4-она (3.2.78) вместо 2-аминопиридина З-амино-5-метил-изоксазол [127, 128, 129, 130].
h2n
СНз
Применяют по тем же показаниям, что и пироксикам.
Синонимами препарата являются флоксикам и млаксикам.
- 81 -
Глава 3
Список литературы
1. Gales et al./ J. Am. Chem.Soc., 22. ^28, 1141,4839 (1950).
2. Gates, Tschudi.tS. Am. Chfem. Soc., 74.1109 (1952).
3. Gates, Tschudi'H. Am. Chem. Soc.. 78, 1380 (1956).
4. Ijima I. et al. 'J. Org. Chem., 43, 1462 (1978).
5. Bijsterveld E. J., Sinnige H. 77/Rec. Trav. Chim. 95, 24 (1976).
6. Beyerman H. C et al./''Rec. Trav. Chim. 97, 127 (1978).
7. Ger. Pat. 247.180 (1912).
8. Rodionow IV/ 'Bull. Soc. Chim. France, 39, 305 (1926).
9. Snyder H. R. et al.',J. Am. Chem. Soc., 80, 3708 (1958).
10. Small, Lutz. Chemistry of the Opium Alcaloids, 'Public Health reports. Washington. 1932. Suppl. No. 103.
11. Ger. Pat. 365.683 (1922).
12. Ger. Pat. 623.821 (1935).
13. Rappoport Z et al.'.'J. Org. Chem., 15., 1103 (1950).
14. US Pat. 2.628.962 (1953).
15. US Pat. 2.806.033 (1957).
16. Weiss H. D J. Am. Chem. Soc., 77, 5891 (1955).
17. Ger. Pat. 411.530(1925).
18. Ger. Pat. 623.821 (1935).
19. Mannich C. et al.'/Arch. Pharm. 258, 295 (1920).
20. Ger. Pat. 415.097(1925).
21 Schnider О et al./'Helv. Chim. Acta. 33, 1437 (1950).
22. Schnider O. etal.'Helv. Chim. Acta. 33, 1437 (1950).
23. Ger. Pat. 890.506 (1944).
24. Ehrhart G. et al.'/Ann. 561, 52 (1948).
25. Shultz H. et al.//J. Am. Chem. Soc. 69, 2454 (1947).
26. Easton R et al./ J. Am. Chem. Soc. 69, 2941 (1947).
27. US Pat. 2.644.010(1953).
28. US Pat. 2.983.757 (1961).
29. Larsen A. et al.''J. Am. Chem. Soc. 70, 4194 (1948).
30. US Pat. 2.167.351 (1939).
31. Ger. Pat. 679.281 (1937).
32. Smissman E. et al.71. Am. Chem. Soc. 81. 1201 (1959).
33. Назаров H. Н.иЖ. Общ. X., 26, 31 17 (1956).
34. Stokbroekx R Med. Chem. 16, 782 (1973).
35. US Pat. 3.714.159 (1973).
36 US Pat. 3.884.916(1975).
- 82 -
Анальгетики
37. US Pat. 2.898.340 (1959).
38. US Pat. 4.086.234 (1978).
39. US Pat. 3.141.823 (1964).
40. US Pat. 3.164.600(1965).
41. US Pat. 4.167.574 (1979).
42. Ger. Pat. 2.819.873 (1978).
43. Janssens F. et. al.//J. Med. Chem. 29, 2290 (1986).
44. Ger. Pat. 2.610.228 (1976).
45. US Pat. 3.998.834 (1976).
46. Daele van G. H. P. et al.//Arzneimittel-Forseh. 26. 1521 (1976).
47. US Pat 2.364.833 (1944).
48. US Pat. 2.891.954 (1959).
49. Belg. Pat. 611.000 91962).
50. Archer S et al./71. Med. Chem. 7, 123 (1964).
51. Brit. Pat. 1.119.270(1968).
52. US Pat. 3.393.197 (1968).
53. Brit. Pat. 1.136.214 (1968).
54. US Pat. 3.433.791 (1969).
55. US Pat. 3.254.088 (1966).
56. Ger. Pat. 1.183.508 (1962).
57. Brit. Pat. 939.287 (1963).
58. Olafson R. A et al.//Tetrahedron Letters. 1977. 1567.
59. US Pat. 3.332.950(1967).
60. Gerhardt C./Ann. 87. 149 (1853).
61. US Pat. 3.235.583 (1966).
62. US Pat. 2.731.492 (1956).
63. Ger. Pat. 2.635.540 (1959).
64. Hannah J. etal.AJ. Med. Chem. 2, 1093 (1978).
65. US Pat. 3.674.870(1972).
66. US Pat. 3.681.445 (1972).
67. US Pat. 3.714.226(1973).
68. US Pat. 2.562.830(1948).
69. Brit. Pat. 812.449 (1959).
70. US Pat. 2.700.671 (1955).
71. Pfister R. et al./ Helv. Chim. Acta. 44, 232 (1961).
72. Ger. Pat. 476.663 (1922).
73. Ger. Pat. 421.505 (1920).
74. Ger. Pat. 254.711 (1911).
75. Ger. Pat. 259.577 (1911).
-83-
Глава 3
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
ИЗ
114
Lumiere М etal Bull Soc Chun France [3] 33. 785 (1905)
I SPat 2 998 450 (1961)
Moffett R etal J Am Chem Soc 82 1605 )1960)
Fr Pat M 1 341 (1961)
US Pat 3 138 636(1964)
Ger Pat 1 163 846(1961)
LSPat 3 313 848 (1967)
Ger Pat 1 149 015 (1961)
Ger Pat 1 470 014 (1964)
Belg Pat 657 266 (1964)
US Pat 3 415 834 (1968)
US Pat 3 337 570 (1967)
Brit Pat 971 700(1964)
US Pat 3 228 831 (1966)
US Pat 3 385 886 (1968)
Brit Pat 1 9514 812 (1976)
LSPat 3 959 364 (1976)
Brit Pat 1 160 725 (1967)
LS Pat 43)21 478 (1977)
But Pat 1 535 690(1977)
Walker J etal Tetrahedron Letters 1977, 3707
Pinhey J et al / Tetrahedron Letters 1980, 965
Shion Г etal /J Org Chem 43, 2936 (1978)
Harrison J etal /J Med Chem 13, 203 (1970)
US Pat 3 904 682 (1975)
US Pat 4 009 197 (1977)
US Pat 3 600 437 (1971)
Fr Pat 2 015 718 (1971)
Fr Pat M 6 444 (1971)
LSPat 3 641 127 (1972)
Pinna G et al Farmaco Ed Sci 35, 684 (1980)
US Pat 3 558 690 (1971)
US Pat 3 778 470 (1973)
Bnt Pat 1 308 327 (1971)
US Pat 3 766 263 (1973)
US Pat 3 161 654 (1964)
Shen Г etal 'J Am Chem Soc 85. 488 (1963) }amamotoH Chem Pharm Bull 16, 17(1968) 'tamamoto H et al Chem Pharm Bull 16, 647 (1968)
- 84 -
Анальгетики
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
Germ Pat 1 795 674 (1968)
US Pat 3 752 826 (1973)
Carson J etal J Med Chem 14,646(1971)
Fr Pat 1 574 570 (1969)
Ger Pat 2 039 426 (1971)
US Pat 3 647 858 (1972)
US Pat 3 654 349 (1972)
Shuman R etal /J Org Chem 42, 1914 (1977)
Ger Pat 1 943 265 (1970)
US Pat 3 591 584(1971)
Lombardino J etal J Med Chem 15. 848 (1972)
Lombardino J etal. J Med Chem 16, 493 (1973)
Ger Pat 2 208 351 (1972)
US Pat 3 787 324 (1974)
Lombardino J etal nJ Med Chem 14, 973 (1971)
Zinnes H etal/J Med Chem , 25, 12 (1982)
Глава 4
Снотворные средства (гипнотики и седативные препараты)
Снотворные средства — это препараты, способствующие развитию и нормализации сна. Однако сон, вызываемый большинством препаратов, по своему течению отличается от естественного сна.
Около 100 лег бромиды, затем хлоральгидрат и в последующем барбитураты были единственными препаратами, способными облегчить состояние пациентов, страдающих бессонницей и пациентов с невротическими расстройствами.
Однако сегодня известны многие соединения различных химических классов, которые могут вызвать различную степень депрессии ЦНС, снимать беспокойство и вызывать сон — они могут быть классифицированы как гипнотики и седативные препараты. Эффекты, вызываемые этими препаратами, находятся в непосредственной зависимости от применяемых доз, проявляясь в различной степени депрессии ЦНС — от седации и сна до полной потери сознания.
Седация — это средняя степень депрессии ЦНС, в то время как гипноз — это степень депрессии ЦНС, близкая к естественному сну. С химической точки зрения снотворные — седативные и гипнотические препараты — классифицируются как барбитураты, бензодиазепиновые гипнотики и т п. За редким исключением любое из этих соединений может быть применено для достижения седативного эффекта или состояния сна. В настоящее время менее токсичные бензодиазепины, поэтому они все более вытесняют класс барбитуратов по причине возможности возникновения к ним хронической зависимости.
Препараты обоих классов в принципе являются депрессантами ЦНС, а вызываемые ими некоторые эффекты, если не все, очевидно связаны с воздействием на рецепторный комплекс у-аминомасляной кислоты (ГАМК).
-86-
Снотворные средства
4.1. Барбитураты
Действие барбитуратов на ЦНС выражается самым широким образом — от небольших изменений в поведении пациента, до проявления более выраженных эффектов, таких как седация, сон или общая анестезия, зависящих, как правило, от введенной дозы.
Фармакологическая основа подобной депрессии ЦНС весьма сложна по своей природе. Препараты действуют на различных участках ЦНС, вмешиваясь в передачу импульсов в синапсах и, в общем, тормозя передачу импульсов в спинной мозг.
Несмотря на то, что полный механизм действия барбитуратов в настоящее время выяснен не полностью, кажется весьма вероятным, что барбитураты усиливают ГАМК-индуцированный поток хлорид ионов в нейрональную ткань, в результате чего мембрана клетки гиперполяри-зуется
ДруI не эффекты барбитуратов на ЦНС, возможно, также связаны со свойством барбитуратов повышать пропускающую способность мембран
Высказываются предположения, что молекулы барбитуратов проникают в липидный слой мембран и понижают жесткость их структурной организации. Гипотетически возможно, что, как и некоторые другие препараты других классов, например, такие как общие анестетики, они также могут действовать путем изменения способности мембран клеток пропускать ионные потоки, воздействовать на высвобождение нейротрансмиттеров, изменять конформацию ферментов. Т. е. не исключено, что они действуют не по рецепторному механизму, а своим физическим прису гствием в мембране. Однако ни одна из выдвинутых гипотез не выдерживает строгой критики.
Клинически полезные барбитураты условно подразделяют на 4 группы.
1. Долгодействующие барбитураты (6-8 ч) — мефобарбитал, метарбитал, фенобарбитал.
2. Барбитураты средней продолжительности действия (4-6 ч) — амобарбитал, бутарбитал, талбутал.
3 Короткодейству ющие барбитураты (2-4 ч) — пентобарбитал, секобарбитал.
I Барбитураты ультракороткого времени действия (10-30 мин) — мезогекситал, тиопентал, тиамилал.
Несмотря на то, что данная классификация весьма удобна для практического медицинского персонала, следует учитывать, что продолжи-
- 87 -
Г лава 4
дельность времени действия препарата, особенно первых трех групп соединений, кроме структуры самого соединения зависит от различных факторов, таких как лекарственная форма, путь введения, патология, при которой применяется препарат, общее время лечения и т. д.
Барбитураты применяются в течение короткого времени для лечения бессонницы, поскольку к барбитуратам при ре!улярном применении (в среднем в течение около трех недель) может возникнуть толерантность.
Барбитураты применяются также для контроля острых конвульсивных состояний и для лечения различных форм эпилепсии. Препараты применяют для пред- и постоперационной седации, а также для седации в дневное время с целью устранить у пациента чувство беспокойства, нервозности и напряжения. Барбитураты применяются также для лечения кататоничесих и маниакальных реакций и в качестве средств, используемых в психоанализе (наркоанализ и наркотерапия).
Барбитураты ультракороткого времени действия применяются в анестезии.
Барбитураты — производные барбитуровой кислоты получают в результате конденсации производных малоновой кислоты с производными мочевины.
О о
С2н5-о
Они являются слабыми кислотами, образующими соли по второму положению кольца. В литературе описаны определенные закономерности, касающиеся корреляций структура-активность в этом ряду соединений.
Как правило, для проявления центрального депрессивного действия барбитураты должны содержать два заместителя при С5 гидрированного пиримидинового кольца. 5,5-диэтилпроизводное (производное барбитуровой кислоты) является слабым гипнотиком, тогда как барбитураты с одной этильной группой и другим заместителем (с более длинной углеводородной цепью) проявляют более сильный гипнотический эффеы
Более того, препараты с разветвленным алкильным заместителем имеют большую гипнотическую активность, чем с заместителями с нормальной углеводородной цепью.
-88-
Снотворные средства
Барбитураты с фенильной группой при С5 менее сильные гипноти-кн, чем соединения с алифатическим или алициклическим заместителем, однако они имеют выраженное антиэпилептическое и противосудорожное действие.
N-метилирование повышает липидную растворимость препаратов и уменьшает длительность действия препаратов. Оно может придать препарату также антиэпилептические свойства, тогда как метилирование по обоим атомам азота приводит к соединениям, вызывающим судороги.
Замещение атома кислорода во втором положении на серу (тиобарбитураты) вызывает выраженное повышение коэффициента распределения липид — вода у 5,5-дизамещенных барбитуратов
Эти соединения имеют большую силу в качестве гипнотиков, чем их кислородные аналоги при внутривенном введении, однако их малая водорастворимость и локализация в жировом депо делают их непригодными для орального применения в качестве гипнотиков. Их в основном используют в качестве внутривенных анестетиков (барбитураты ультракороткого действия).
Долгодействующие барбитураты
Фенобарбитал (Phenobarbital)
Фенобарбитал — 5-этил-5-фенилбарбитуровую кислоту или 5-этил-5-фенилгексагидропиримидин-2,4,6-трион (4.1.4) предложено синтезировать разными способами [1, 2, 3, 4]. !
Принципиального различия между ними нет. Первый способ заключается в том, что бензилцианид подвергают этанолизу в кислой среде с получением этилового эфира фенилуксусной кислоты, метиленовую группу которого подвергают ацилированию диэтиловым эфиром щавелевой кислоты с получением диэтилового эфира фенилоксобулан-диоевой кислоты (4.1.1), которая при нагревании легко теряет окись углерода с превращением в фенилмалоновый эфир (4.1.2). Алкилировав ние последнего эгилбромидом в присутствии этилата натрия приводит к образованию а-фенил-а-этилмалонового эфира (4.1.3), конденсацией которого с мочевиной получают фенобарбитал (4.1.4) [1].
< А— Сн -С = к С2Н50Н ' Н \\ С—-----------—-
СН2~СООС2Н5
СООС2Н5
СООС2Н5
СООС2Н5
сн ---------
I
СООС2Н5
-89-
Г лава 4
СООС2Н5
4 1 2
1 C2H5ONa
2 C2H5Br
^ООС2Н5 с-с2н5
СООС2Н5
4 1 3
H2NCONh2
Второй способ синтеза фенобарбитала заключается в получении а-фенилциануксусного эфира (4.1.5) конденсацией бензилцианида с ди-этиловым эфиром угольной кислоты в присутствии этилата натрия. Алкилированием эфира 4.1.5 этилбромидом получают а-фенил-а-этил-циануксусный эфир (4.1.6), который далее циклизуют в 4-имино-производное (4.1.7) Кислотным гидролизом последнего получают фенобарбитал (4.1.4) [2].
CHj-C=N
1 C2H5ONa
2 ОС(ОС2Н5)2
)°0С^ О2Н5В._
СН ---------------*
I
H2NCONH2
4 1 4
Фенобарбитал оказывает успокаивающее, снотворное и противосудорожное действие. Широко применяется при лечении эпилепсии, хореи и спастических параличей и применяется в качестве составной части большого числа комбинированных препаратов, в частности валокордина и корвалола.
Наиболее распространенным синонимами являются люминал, фе-немал, гипнотал и очень многие другие.
Мефобарбшпал (Мephobarbital)
Мефобарбитал — 5-этил-1-метил-5-фенилбарбитуровую кислоту (4.1.8) получают по одной из схем, используемых для синтеза фенобарбитала, но с применением метилмочевнны [5].
-90-
Снотворные средства
и Р2Н5
H-N'
СНз 418
Препарат используется в качестве седативного средства для снятия беспокойства и напряжения, а также при больших и малых эпилептических припадках.
Синонимами препарата являются барбифенал, энфенемал, метилфенобарбитал.
Метарбитал (Metharbital)
Метарбитал — 5,5-диэтил-1-метилбарбитуровую кислоту (4.1.9) получают конденсацией диэтилмалонового эфира с метилмочевиной
[6. 7].
Н—N'
u f2H5
с2н5
N О
СНз 419
Метарбитал, как и фенобарбитал и мефобарбитал, оказывает противосудорожное действие. Применяют при больших и малых эпилептических припадках.
Синонимами препарата являются эндиэмалум, гемонил, метабар-битал.
Барбитураты средней продолжительности действия
Амобарбитал (Amobarbital)
Амобарбитал — 5-этил-5-изо-амилбарбитуровую кислоту (4.1.10), как и все барбитураты, получают взаимодействием производных малоновой кислоты с производными мочевины. В частности, с целью получения амобарбитала а-этил-а-изо-амилмалоновый эфир вводят во взаимодействие с мочевиной (в присутствии этилата натрия) с получением амобарбитала (4.1.10) [8, 9].
- 91 -
Глава 4
о о
1 C2H5ONa
2 С2Н5Вг
СН3
СН3СНСН2СН2Вг
О О
С2Н5
H2NCONH2
СНз
С0Н50 ОС2Н5
С2Нб СН2СН2СНСНз
СНз
H-N
СН2СН2СНСН3
С2н5
4 1.10
о о
о^кГмэ н
Амобарбитал применяют как снотворное при различных видах бессонницы, а также в качестве успокаивающего и противосудорожного средства.
Наиболее часто используемыми синонимами являются барбамил, амитал и гипнамил.
Бутабарбитал (Butabarbital)
Бутабарбитал — 5-этил-5-изо-бутилбарбитуровую кислоту (4.1.11) также предложено получать аналогичным путем — конденсацией а-этил-а-изо-бутилмалонового эфира с мочевиной [9].
H2NCONH2
СНз о СНСН2СН3
С2Н5О ОС2Н5
С2Н5 СНСН2СН3 СНз
H-N
С2Н5
СГ N О I н
4 1 11
о о
Бутабарбитал также применяют как снотворное при различных видах бессонницы и в качестве успокаивающего средства.
Наиболее часто используемым синонимом является бутизон.
Талбутал (Talbutal)
Талбутал — 5-аллил-5-втор-бутилбарбитуровую кислоту (4.1.121 получают взаимодействием а-аллил-а-втор-бутилмалонового эфира с мочевиной [ 10).
-92-
С н о т в о рные средства
о?Нз
П снсн2сн3
н -n"N—сн2 —сн =сн2
O N О
Н
4 1 12
Препарат отличается от бутабарбитала тем, что вместо изобутиль-ного радикала в качестве одного из заместителей при С5 в бутабарбитале использован вторбутильный радикал.
Талбутал применяют в качестве седативного снотворного средства по тем же показаниям, что и бутарбитал.
Синонимами препарата являются профундол, лотустат и др.
Короткодействующие барбитураты
Пентобарбитал (Pentobarbital)
Пентобарбитал — 5-этил-5-(2-амил)барбитуровую кислоту (4.1.13 ) получают по схеме, аналогичной схеме получения амобарбитала, с той лишь разницей, что алкилирование а-этилмалонового эфира проводят не 1-бром-З-метилбутаном, а 2-бромпентаном с получением пентобарбитала (4.1.13) [И, 12, 13].
H-N
О с2н5
—СНС3Н7
•О 6нз
О^ N Н
4.1 13
В принципе пентобарбитал может рассматриваться как изомер амо-оарбитала. По действию он также схож с ним и отличается меньшей продолжительностью действия и лучшей переносимостью. Препарат применяют в качестве успокаивающего, а также как снотворное при кратковременной бессоннице.
Наиболее часто используемым синонимом препарата является нем-б\тал.
- 93 -
Глава 4
Секобарбитал (Secobarbital)
Секобарбитал — 5-аллил-5-(1-мегилбутил)барбитуровую кислоту (4.1.14) также получают по стандартной схеме взаимодействием а-аллил-а-(1-метнлбутил)-малонового эфира с мочевиной 114)
о?Нз
П снсн2сн2сн3
—сн2—сн =сн2
O N ХО н
4 1 14
Препарат применяют по тем же показаниям, что и пентобарбитал, — в качестве успокаивающего, а также как снотворное при кратковременной бессоннице
Наиболее часто используемыми синонимами являются барбосек, хинасед и др.
Барбитураты ультракороткого времени действия
Синтез и свойства метогекситала (1.2.15) и тиопентала (1.2.10) были описаны в гл. 1 «Общие анестетики».
О *ГН3
П СН-С=С-С2Н5 h-N'^l—СН2—СН =СН2
Со N ЧО
СН3
1 2 15
СНС3Н7 СНз
1 2 10
Из рассмотренных выше барбитуратов наиболее широко в медицине используются фенобарбитал, амобарбитал и бутабарбитал, а также метогекситал и тиопентал.
4.2. Бензодиазепины
Производные бензодиазепинов являются основным классом анксиолитиков или транквилизаторов — соединений для лечения состоя
-94-
Снотворные средства
ний общего беспокойства, и их синтез и свойства в качестве таковых будут обсуждены отдельно.
Однако, несмотря на то, что основной клинический эффект используемых в медицине бензодиазепинов в принципе качественно одинаков, определенные бензодиазепины применяются в несколько иных целях, чем препараты для снятия беспокойства.
В частности, такие представители ряда бензодиазепинов, как флуразепам, темазепам и триазолам используются в качестве гипнотиков, а клоназепам — в качестве противосудорожного препарата. Более того, в настоящее время самыми эффективными фармакологическими сред-щвами, используемыми для лечения нарушений сна, являются флуразепам, темазепам и триазолам.
Однако в малых дозах указанные гипнотики являются седативными препаратами. Полагают, что основное их действие заключается в устранении психического напряжения и наступающее при этом успокоение способствует развитию сна. Механизм действия бензодиазепинов связывают с их взаимодействием со специальными бензодиазепиновыми рецепторами, считая, что в результате такого связывания повышается аффинитет ингибируюшего нейротрансмиттера — ГАМК к соответствующим рецепторам, что и усиливает тормозное действие ГАМК.
Рассматриваемые флуразепам, темазепам и триазолам, очевидно, повышают ингибирующий эффект ГАМК на ЦНС.
Триазолам (Triazolam)
Триазолам — 8-хлор-6-(2‘-хлорфенил)-1-метил-4-Я-5-триазоло[4,3-а]-[1,4]бензодиазепин (4.2.4) получают по схеме, включающей ключевую стадию классического синтеза бензодиазепинов — взаимодействие о-аминобензофенонов с производными а-аминокислот. В данном случае взаимодействием 2-амино-2’,5-дихлорбензофенона с этиловым эфиром глицина получают 7-хлор-5-(2-хлорфенил)-2,3-дигидро-1-Я-1,4-бенодиазепин-2-он (4.2 1). Взаимодействием последнего с пятисернистым фосфором карбонильную группу трансформируют в тиокарбонильную с получением 7-хлор-5-(2-хлор-фенил)-2,3-дигидро-1-Я-1,4-бензодиазепин-2-тиона (4.2.2). Атом серы в полученном циклическом тиоамиде, в свою очередь, замещают взаимодействием с ацетилгидразидом с получением соответствующего ацетилгидразона (4.2.3), который при нагревании циклизуется в триазолам (4.2.4) [15. 16, 17, 18, 19, 20].
-95-
Глава 4
Триазолам — препарат, наиболее часто выписываемый пациентам при бессоннице.
Однако к нему очень быстро может развиться пристрастие, а также ряд других побочных эффектов: утренняя бессонница, беспокойство в дневное время и прочее.
Синонимами препарата являются нормисон, ремстан, ресгорил и др.
Темазепам (Temazepam)
Темазепам — 7-хлор-1,3-дигидро-3-гидрокси-1-метил-5-фенил-2Я-1,4-бензодиазепин-2-он (4.2.7) получают из промежуточного продукта синтеза оксазепама 7-хлор-5-фенил-1,3-дигидро-2Я-1.4-бензодиазепин-2-он-4-оксида (5.1.17) путем метилирования амидного атома азота в первом положении бензодиазепинового кольца диметилсульфатом с получением 1-метил-7-хлор-5-фенил-1,3-ди-гидро-2Я-1.4-бензодиазепин-2-он-4-оксида (4.2.5), который далее подвергают ацегоксилированию уксусным ангидридом с получением 1-метил-3-ацетокси-7-хлор-5-фенил-1,3-дигидро-2Я-1,4-бензоди-азепин-2-она (4.2.6). (При этом, очевидно, происходит превращение, аналогичное реакции Полоновского.)
Щелочной гидролиз последнего соединения (4.2.6), имеющий целью снятие ацетильной группы, приводит к искомому темазепаму (4.2.7) [21, 22. 23.24. 25, 26].
-96-
Снотворные средства
Темазепам является умеренно эффективным гипнотиком. По окончании курса его приема иногда может вновь возникнуть бессонница.
Наиболее часто используемым синонимом препарата является гал-цион.
Флуразепам (Flurazepam)
Флуразепам — 7-хлор-1-[2-(диэтиламино)этил]-5-(2’-фторфенил)-1.3-дигидро-2//-1,4-бензодиазепин-2-он (4.2.14) предложено получать многостадийным синтезом исходя из 2-амино-5-хлор-2|-фторбензофенона. Взаимодействием последнего с хлорангидридом бромуксусной кислоты получают 2-(бромацетил)амино-5-хлор-2'-фторбензофенон (4.2.8), реакцией которого с диэтиламином получают 2-(диэтиламиноацетил)амино-5-хлор-2|-фторбензофенон (4.2.9). Восстановлением обеих карбонильных групп в последнем алюмо-гидридом лития получают 2-(21-диэтиламино)этиламино-5-хлор-2‘-фторбензгидрол (4.2.10). Аминогруппу последнею ацилируют хлорангидридом фталимидоуксусной кислоты с получением фталимидопроизводного (4.2.11). Снятием защитной фталимидной группы гидразингидратом получают 2-(2'-диэтиламино)этиламиноацетил)-амино-5-хлор-2’-фторбензгидрол (4.2.12). Обработка последнего бромистоводородной кислотой приводит к внутримолекулярной дегидратации с замыканием в семичленный бензодиазепиновый цикл 7-хлор-1-[2-( диэтиламино )этил]-5-(2'-фторфенил)-1,3,4.5-тетрагидро-2Я-1.4-бензодиазепин-2-он (4.2.13). Наконец, окислением N4-C5 свя-
- 97 -
Г лава 4
зи последнего 2,3-дихлор-5,6-дициано-1,4-бензохиноном получают искомый флуразепам (4.2.14) [27, 28, 29, 30].
h
Вг—СН2 — C~Cl
СН2~Вг С=О
Флуразепам наиболее хорошо изученный гипнотик. Используется при трудностях со сном или засыпанием, частых или ранних просыпаниях. Побочные эффекты наблюдаются редко.
Наиболее распространенными синонимами являются далмадорм, далман, валдорм. фелисон и др.
4,3. Прочие гипнотики и седативные препараты
Еще до появления класса бензодиазепинов ряд эмпирически найденных соединений использовался в медицине в качестве гипнотиков. Среди них хлоральгидрат, параальдегид, этхловинол, этинамат, глуте-тимид. метиприлон.
-98-
Снотворные средства
Хлоральгидрат (Chloral Hydrate)
Хлоральгидрат — 2,2,2-трихлор-1,1-этандиол (4.3.1) получают либо хлорированием этанола, либо хлорированием ацетальдегида с последующим присоединением к полученному трихлоруксусному альдегиду молекулы воды [31].
сн3-с-н
(or СН3-СН2-ОН)
О
С13с—С-н
Н2О
он CI3C-C-OH н
431
Седативно-гипнотическое действие хлоральгидрата очевидно следует объяснять образованием трихлоэтанола, получаемого в результате его восстановления в тканях. Несмотря на то, что точный механизм действия хлоральгидрата неизвестен, препарат, очевидно, действует аналогично этанолу в ЦНС, т. е. повышая проницаемость мембран, что приводит к седации или сну.
Хлоральгидрат может применяться при бессоннице как альтернатива бензодиазепинам.
Синонимами препарата являются аквахлораль, хлорадорм, хлора-тол. ноктек и др.
Паральдегид (Paraldehyde)
Паральдегид — 2,4,6-триметил-1,3.5-триоксан (4.3.2) является примером ацетальдегида, который получают катализируемой кислотами полимеризацией ацетальдегида при средних и высоких температурах [32, 33].
НзС^О^СНз
СТ .О
сн3
4 3 2
Точный механизм действия паральдегида неизвестен, однако, наиболее вероятно, что действует он сам, а не продукты его биотрансформации. Показания к применению аналогичны хлоральгидрату.
Синонимами препарата являются эльдальдегид, параль и парааль-дегид.
-99-
Глава 4
Этхлорвинол (Ethchlorvinol)
Этхлорвинол — 1-хлор-3-этил-1-иентен-4-ин-3-ол (43.3) получают конденсацией ацетилена с 1-хлор-1-пентен-3-оном в жидком аммиаке [34, 35].
О
CI—СН=СН-С-С2Н5 + НС=СН
^=сн
CI—сн=сн-с-с2н5
433 0Н
Седативный гипнотик этхлорвинол имеет примерно ту же активность и токсичность, как и фенобарбитал, однако его гипнотический эффект достигается и исчезает быстрее. По ряду причин при лечении бессонницы используется значительно реже бензодиазепинов.
Синонимами препарата являются арвинол, ностел, плацидил и др.
Этинамат (Ethinamate)
Этинамат — карбамат 1-этинилциклогексанопа (4.3.4) получают конденсацией ацетилена с циклогексаноном и последующей трансформацией полученного карбинола в карбамат путем последовательного взаимодействия с фосгеном и далее с аммиаком [36, 37].
+ НС5СН
1 СОС12
2 NH3
434
Этинамат — гипнотик, который, однако, не имеет существенных преимуществ по сравнению с барбитуратами и бензодиазепинами и при лечении бессонницы используется значительно реже.
Синонимами препарата являются валамид. валмад, валамин и др.
Глутетимш) (Glutethmide)
Глутетимид — 2-этил-2-фенилглутаримид (4.3.6) получают присоединением (по Михаэлю) 2-фенилбутиронитрила к метилакрилату с последующим щелочным гидролизом нитрильной группы в соединении (4.3.5) в амидную и последующей кислотной циклизацией продукта в искомый глутегимид (4.3.6) [38, 39, 40. 41. 42].
- 100-
Снотверные средства
- НгС=СН-СООСН3
СН2-СН2—СООСНз
1 он
2 Н
4 3 5
Глутетимид — гипнотическое и седативное средство, предназначенное для лечения бессонницы. Гипнотический эффект примерно аналогичен таковому у пентобарбитала. Назначается пациентам, не переносящим барбитураты. Однако не имеет преимуществ перед бензодиазепинами и поэтому применяется редко.
Синонимом препарата является дориден.
Метиприлон (Methyprylon)
Метиприлон — 3,3-диэтил-5-метил-2.4-пиперидиндион (4.3.12) синтезирую!' исходя из диэтилацетоуксусного эфира, взаимодействием которого с метил формиатом в присутствии натрия получают 4-оксиметилен-2,2-диэгилацетоуксусный эфир (4.3.7). Реакцией с аммиаком последний трансформируется в 4-аминометилен-2,2-диэтилацетоуксусный эфир (4.3.8). Обработкой полученного продукта этилатом натрия осуществляют внутримолекулярное амидирование — циклизацию в 3,3-диэтил-1,2,3,4-тетрагидропиридин-2,4-дион (4.3.9). Восстановлением двойной связи последнего водородом над палладиевым катализатором получают 3,3-диэтил-пиперидин-2,4-дион (4.3.10), который вновь подвергают формилиро-ванию в 3.3-диэтил-5-гидроксиметилен-пиперидин-2,4-дион (4.3.11) взаимодействием с метилформиатом в присутствии натрия. Восстановлением введенной гидроксиметиленной группы водорода в метильную группу получают искомый метилприлон (4.3.12) [43, 44].
НСООСНз
ЦНз
437
i^sONa
Na НСООСНз
- 101 -
Глава 4
Нг Ranay— Ni
4 3 11
4 3 12
Как и все рассматриваемые в данной главе препараты, метиприлон предназначен для лечения бессонницы. Фармакологические эффекты метиприлона сходны с таковыми у барбитуратов. Однако с введением в медицинскую практику бензодиазепинов преимущество стало отдаваться последним.
Синонимами препарата являются ноктар, нолудан и др.
Список литературы
1. Ger. Pat. 247.952 (1911).
2. US Pat. 2.358.072(1944).
3. Chamberlain E. et al.//J. Am. Chem, Soc. 57. 352 (1935).
4. Pinhev J. et al./Tetr. Lett. 21. 965 (1980).
5. Ger. Pat. 537.366(1929).
6. Halpern О et al./T. Am. Pharm. Assoc. 38, 352 (1949).
7. Snyder H et al.,7J. Am. Chem. Soc. 75, 1881 (1953).
8. Brit. Pat. 191.008 (1922).
9. US Pat. 1.856.792 (1932).
10. Volwiler E.IEE Am. Chem. Soc. 47. 2236 (1925).
11. Volwiler E.'/J. Am. Chem. Soc. 52. 1676(1930).
12. Ger. Pat. 293.163 (1915).
13. Brit. Pat. 650.354 (1948).
14. US Pat. 1.954.429(1934).
15. Ger. Pat. 2.012.190(1970).
16. US Pat. 3.701.782 (1972).
17. US Pat. 3.987.052 (1976).
18 US Pat. 3.422.091 (1969).
19. Archer G. et al.,7J. Org. Chem. 29, 231 (1964).
20. Hester J. et al./ J. Med. Chem. 14. 1078 (1971).
21. Brit. Pat. 1.022.642(1962).
22. Brit. Pat. 1.022.645 (1962).
23. Bell S. etal.' J. Org. Chem. 27. 1691 (1962).
24. US Pat. 3.197.467 (1965).
- 102-
Снотворные средства
25. US Pat. 3.340.253 (1967).
26. US Pat. 3.374.225 (1968).
27. US Pat. 3.567.710(1971).
28. US Pat. 3.299.053 (1967).
29. Belg. Pat. 629.005 (1963).
30. Inaba S. et al.//J. Med. Chem. 8, 815 (1965).
31. Fai'-brother J 'Anal Profiles Drug Subs. 2. 85 (1973).
32. Kckule F. et al.//Ann. 162, 125 (1872).
33. US Pat. 2.864.827 (1958).
34. US Pat. 2.746.900 (1956).
35. McLamore W. et al./'J. Org. Chem. 20, 109 (1955).
36 US Pat. 2.816.910(1957)
37. Ger. Pat. 1. 021.843 (1953).
38. Ger. Pat. 950.193 (1951).
39. US Pat. 2.673.205 (1951).
40. Tagmann A. et al.//Helv. Chim. Acta. 35. 1541 (1952).
4 i. Salmon-Legagneur R. et al. /Compt. Rend. 234, 1060 (1952).
42. Salmon-Legagneur R. etal.'/Bull. Soc. Chim. France, 1953, 70.
43. US Pat. 2.680.116 (1954).
44. Ger. Pat. 930.206 (1953).
t Глава 5 l Анксиолитики 1 (транквилизаторы)
Многие заболевания сопровождаются беспокойством, тревожным состоянием. При этом развивается синдром, характеризующийся ощущениями беспомощности и безысходности, мрачными предчувствиями, астенией. Он может сопровождаться головными болями, повышенным потоотделением, тошнотой, тахикардией, сухостью во рту и т. д. Беспокойное состояние и тревога могут возникнуть как в результате невротических причин, так и иметь соматопсихическую природу, связанную с развитием патологий при таких заболеваниях, как болезни сердечнососудистой системы, новообразования, гипертония и заболевания ЖКТ. Препараты, применяемые для устранения беспокойства, напряжения, тревоги и страха и которые практически не снижают внимание, не действуют на психомоторную активность пациента, получили название анксиолитиков или транквилизаторов. Большинство из них обладает седативным и гипнотическим действием, и в высоких дозах их эффекты во многом схожи с воздействием барбитуратов. Однако основное преимущество данной группы перед барбитуратами заключается в значительно большем значении величины соотношения седативный.'гипно-тический эффекты. Иными словами, соотношение между дозами, снимающими напряжение, и дозами, вызывающими сон, у анксиолитиков значительно больше, чем таковое у барбитуратов. Основное применение транквилизаторов заключается в облегчении таких эмоциональных симптомов, как возбуждение, беспокойство, тревога, мышечное напряжение, повышенная двигательная активность, связанных с психо-невротическими или психосоматическими нарушениями. В качестве самостоятельных препаратов они мало приемлемы для купирования острых психотических состояний и используются в этих случаях в соче-
- 104 -
Анксиолитики
танпи с антипсихотическими средствами.
Применяемые в настоящее время анксиолитики в медицине делятся на 2 группы:
1) бензодиазепины — диазепам, хлордиазепоксид, хлоразепат, галазепам, лоразепам, мидазолам, алпразолам, оксазепам, празепам;
2) прочие анксиолитики, или анксиолитики небензодиазепиновой структуры, которые представлены мепробаматом, буспироном, хлормезаноном и гидроксизином.
5.1. Бензодиазепины
До открытия класса бензодиазепинов основными препаратами для коррекции психоэмоциональных расстройств использоватись седативные и гипнотические препараты и, в частности, фенобарбитал или глу-тетимид.
Весьма эффективными средствами для лечения невротических состояний оказались бензодиазепины — большая группа соединений, первый представитель которых — хлордиазепоксид был синтезирован в 1930-х годах и внедрен в медицинскую практику в конце 1950-х годов. В последующем в медицинскую практику были внедрены более десятка других производных бензодиазепинов. Все они проявляют почти сходную фармакологическую активность и терапевтическую эффективность, отличаясь лишь количественными показателями.
Анксиолитический эффект бензодиазепинов специфичен и уникален и отличается от седативных и гипнотических препаратов других классов.
Основными эффектами бензодиазепинов на ЦНС являются снятие беспокойства, тревоги, седативный эффект, расслабление скелетной мускулатуры и снотворное действие.
Они в меньшей степени, чем гипнотики и седативные средства, угнетают дыхательную систему и в меньшей степени вызывают зависимость.
Некоторые представители препаратов ряда бензодиазепинов обладают несколько иным спектром применения. Флуразепам, триазолам и темазепам используются в качестве снотворных средств, а карбамазепин — в качестве противосудорожного.
Бензодиазепины с выраженным анксиолитическим действием и отсутствием или слабо выраженным седативно-гипнотическим эффектом называют дневными транквилизаторами (медазепам).
-105-
Глава 5
С химической точки зрения бензодиазепины формально делятся на 2 главные группы:
1) простые 1,4-бензодиазепины (хлордиазепоксид, диазепам, лоразепам);
2) гетероциклические 1,4-бензодиазепины (алпразолам, мидазолам и др.).
Необходимым условием для проявления бензодиазепинами анксиолитической активности является наличие электроотрицательной группы в положении С? бензодиазепиновой системы. Наличие фенильной группы в положении С5 системы также увеличивает фармакологическую активность соединений.
Экспериментальные данные позволяют предположить, что механизм действия бензодиазепинов заключается в стимуляции бензодиазе-пин-ГАМК-рецепторного комплекса. Следовательно, бензодиазепины, как класс, потенцируют активность тормозных ГАМК-эргических систем мозга.
Хотя бензодиазепины непосредственно с ГАМК-рецепторами не реагируют, возможно, что они связываются со специфическим рецептором на хлоридном ионофоре. Это бензодиазепин-рецепторное взаимодействие, очевидно, вызывает аллостерические изменения ГАМК-рецепторов, что, в свою очередь, повышает ингибирующую активность ГАМК, которая выражается увеличением потока хлорид ионов через ГАМК-активированные ионные каналы.
Основное применение бензодиазепины нашли для симптоматического устранения чувства беспокойства, напряжения и раздражительности, связанных с неврозами, неврозоподобными состояниями, депрессией и психосоматическими расстройствами.
Бензодиазепины применяются для премедикации перед проведением оперативных вмешательств с целью достижения атараксии пациента, в качестве вспомогательных средств при лечении эпилепсии, при столбняке и других патологических состояниях, сопровождающихся гипертонусом скелетных мышц. Как уже было отмечено, некоторые бензодиазепины применяются в качестве снотворных (флуразепам, триазолам и темазепам) и даже противосудорожных средств (карбамазепин).
Диазепам (Diazepam)
Диазепам — 7-хлор-1,3-дигидро-1-метил-5-фенил-2/7-1,4-бензодиа-зепин-2-он (5.1.2), с химической точки зрения является простейшим из всего ряда рассматриваемых производных 1,4-бензодиазепин-2-
- 106 -
Анксиолитики
онов. Диазепам предложено получать различными способами, исходящими из 2-амино-5-хлорбензофенона. Первый способ заключается в непосредственной циклоконденсации 2-амино-5-хлор-бензофенона или 2-метиламино-5-хлорбензофенона с гидрохлоридом этилового эфира глицина. Амидный атом азота получаемого в этом варианте 7-хлор-1,3-дигидро-5-фенил-27/-1,4-бензодиазепин-2-она (5.1.1) метилируют диметилсульфатом, что приводит к получению диазепама (5.1.2).
HsNCHjCOOCnHg
(СНзСОгЗОр
Второй способ отличается от первого тем, что стадия метилирования азота осуществляется до реакции циклоконденсации. С этой целью исходный 2-амино-5-хлор-бензофенон первоначально тозилируют п-толуолсульфохлоридом, и далее натриевую соль полученного тозилата (5.1.3) алкилируют диметилсульфатом. Полученный в результате реакции 2-(толуолсульфонил)метиламино-5-хлорбензофенон (5.1.4) гидролизуют в кислой среде до 2-метиламино-5-хлорбензофенона (5.1.5), который подвергают циклоконденсации реакцией с гидрохлоридом этилового эфира глицина и получением целевого диазепама (5.1.2) [1, 2, 3, 4, 5].
- 107-
Глава 5
При третьем способе синтеза исходят из 2-метиламино-5-хлор-бензофенона (5.1 5), который ацилируют хлорангидридо.м хлоруксусной кислоты с образованием 2-хлорацетилметиламидо-5-хлорбензофенона (5.1.6). Взаимодействием последнего с уротропином замещают атом хлора в хлорацетильной части молекулы с получением гексаметилентетраминового производного — 2-аминоацетилметиламидо-5-хлорбен-зофенона, при гидролизе которого этанольным раствором хлористого водорода получают диазепам (5.1.2) [6, 7].
CICOCHaCt
1 (СНгЭбЩ
2 HCi
Предложен и четвертый способ получения диазепама, исходящий из 1-метил-3-фенил-5-хлор-2-аминометилиндола (5.1.12), окислением которого хромовым ангидридом получается диазепам. Синтез проводится исходя из 5-хлоранилина, действием на который азотистой кислоты получают диазониевую соль (5.1.7). Азосочетанием последней с а-бен-зилацетоуксусным эфиром в щелочной среде получают 4-хлорфенил-гидразон этилового эфира фенилпировиноградной кислоты (5.1.8), который в присутствии соляной кислоты по реакции Фишера трансформируется в этиловый эфир 5-хлор-3-фенилиндолил-2-карбоновой кислоты (5.1.9). Алкилируя полученный индол по атому азота диметилсульфатом, получают этиловый эфир 1-метил-5-хлор-3-фенилиндолил-2-карбоновой кислоты (5.1.10). Взаимодействием последнего с аммиаком получают соответствующий амид (5.1.11), восстановлением которого алюмогидридом лития получают 1-метил-3-фенил-5-хлор-2-амино-метилиндол (5.1.12). Далее используют практически ценное свойство хромового ангидрида размыкать индольное кольцо до соответствующего производного аминобензофенона (5.1.13), который в условиях реакции циклизуется в диазепам [8].
5 1 7
СН2-СН-СОСН3
СООС2Н5
- 108-
Анксиолитики
Диазепам оказывает анксиолитическое, седативное, снотворное, центральное миорелаксирующее, противосудорожное действие. Препарат подавляет чувство страха, тревоги и напряжения. Его применяют при нервном напряжении, возбуждении, беспокойстве, нарушениях сна, нейровегетативных расстройствах, психоневрозах, навязчивых неврозах, истерических или ипохондрических реакциях, фобиях.
Наиболее часто применяемыми синонимами являются седуксен, ре-ланиум, валиум, сибазон, апаурин и многие другие.
Празепам (Prazepam)
Празепам — 7-хлор-1-(циклопропилметил)-1.3-дигидро-5-фенил-2//-1,4-бенздиазепин-2-он (5,1.18) отличается от диазепама лишь заместителем при атоме азота в первом положении диазепиновой системы. Препарат получают по схеме синтеза, очень близкой схеме синтеза диазепама (5.1.2). При первом способе в качестве исходного используют тот же 2-амино-5-хлорбензофенон, который подвергают ацилированию .хлорангидридом циклопропанкарбоновой кислоты. Полученный при этом 2-циклопропилкарбониламино-5-хлор-
-109-
Глава 5
бензофенон (5.1.14) восстанавливают далее алюмогидридом лития в 2-циклопропилметиламино-5-хлорбензгидрол (5.1.15), и далее полученный продукт вновь окисляют двуокисью марганца в 2-цикло-пропилметила.мино-5-хлорбензофенон (5.1.16). Последний ацилируют хлорангидридом фтал и.м идо уксус ной кислоты. Фталимидную защиту в полученном продукте (5.1.17) удаляют обработкой гидразином. в ходе чего в условиях синтеза происходит внутримолекулярная реакция иминообразования, приводящая к получению искомого празепама (5.1.18) [9, 10].
LIALH4
HzNNHz
Согласно второй способу синтеза, более простому, исходят из 7-хлор-5-фенил-2.3-дигидро-1Я-1,4-бензодиазепин-2-она (5.1.1), который алкилируют циклопропил-метилбромидом в присутствии амида натрия в празепам (5.1.11) [И, 12].
Вг-СН2—
Фармакологические свойства препарата практически повторяют свойства диазепама, отличаясь лишь большей продолжительностью действия. Препарат применяют по тем же показаниям, что и диазепам.
-110-
Анксиолитики
Наиболее распространенным синонимом празепама является цен-тракс.
Галазепам (Halazepam)
Галазепам — 7-хлор-1-(2|,21,2'-трифтор-1-этил)-1,3-дигидро-5-
фенил-2Я-1.4-бензодиазепин-2-он (5.1.19) также отличается от диазепама заместителем при атоме азота в первом положении бензодиазепиновой системы, который в данном случае представлен 221,2‘-трифторэтильной группой и может быть получен по любой из приведенных выше схем [13, 14].
ср3 сн7
Фармакологические свойства последнего практически повторяют свойства диазепама, отличаясь меньшей продолжительностью действия. В основном применяют при беспокойных состояниях.
Наиболее распространенным синонимом является паксипам.
Хлордиазепоксид (Chlordiazepoxid)
Хлордиазепоксид — 7-хлор-2-метиламино-5-фенил-3//-1,4-бензо-диазепин-4-оксид (5.1.22) синтезируют также исходя из 2-амино-5-хлорбензофенона. Обычным путем — взаимодействием с гидроксиламином получают оксим 2-амино-5-хлорбензофенона (5.1.20), который под действием хлорангидрида хлоруксусной кислоты в уксусной кислоте легко циклизуется в 6-хлор-2-хлорметил-4-фенилхиназолин-3-оксид (5.1.21). Взаимодействие последнего с первичными аминами и, в частности, с метиламином приводит к интересной перегруппировке (с расширением цикла), и продуктом реакции оказывается 7-хлор-2-метиламино-5-фенил-ЗЯ-1,4-бензодиазепин-4-оксид (5.1.22) — хлордиазепоксид. Аналогичная перегруппировка с расширением цикла протекает и под действием щелочей и алкоголятов, однако необходимо отметить, что с диалкилам инам и реакция протекает с образованием ожидаемых
-111 -
Глава 5
продуктов замещения 2-диалкиламинометильных производных 6-хлор-4-фенилхиназолин-3-оксида.
Хлордиазепоксид был первым представителем анксиолитиков бензодиазепинового ряда внедренным в медицинскую практику [15, 16, 17].
nh2oh
С1СН2СОС'
Хлордиазепоксид применяют при лечении невротических состояний для уменьшения чувства страха, тревоги, напряженности. Он оказывает успокаивающее действие на ЦНС, вызывает мышечную релаксацию, обладает противосудорожной активностью. Часто применяется после операционных вмешательств.
Наиболее часто применяемыми синонимами являются элениум и либриум.
Оксазепам (Oxazepam)
Оксазепам — 7-хлор-1,3-дигидро-3-гидрокси-5-фенил-2Я-бен-зодиазепин-2-он (5.1.25) получают вышеописанным способом, открытым при синтезе хлордиазепоксида, но с применением в качестве нуклеофила не первичного амина, а простого неорганического основания. С этой целью 6-хлор-2-хлорметил-4-фенил-хиназолин-3-оксид (5.1.21) подвергают обработке гидроксидом натрия с получением 7-хлор-5-фенил-1.2-дигидро-ЗЯ-1.4-бензо-диазепин-2-он-4-оксида (5.1.23). Последний подвергают весьма любопытной реакции — ацетоксилированию ангидридом уксусной кислоты по третьему положению бензодиазепинового кольца, напоминающей реакцию Полоновского с получением 7-хлор-1.3-дигидро-3-ацетокси-5-фенил-2Я-бензодиазепин-2-она (5.1.24). Последу юшим гидролизом ацетильной гру ппы продукта подумают ок-
- 112 -
Анксиолитики
сазепам (5.1.25) [18, 19, 20, 21, 24].
Оксазепам по своим фармакологическим свойствам сходен с хло-диазепоксидом и диазепамом, однако оказывает несколько менее резкое действие, менее токсичен и проявляет менее выраженный миорелак-сантный эффект. Часто переносится лучше других транквилизаторов. Препарат применяют при неврозах, состояниях беспокойства, страха, напряженности, нарушениях процесса засыпания, психовегетативных расстройствах.
Наиболее распространенными синонимами являются нозепам и та-зепа.м.
Лоразепам (Lorazepam)
Лоразепам — 7-хлор-5-(о-хлорфенил)-1,3-дигидро-3-гидрокси-2Я-1,4-бензодиазепин-2-он (5.1.31) синтезируют по схеме, включающей элементы синтеза как хлордиазепоксида, так и оксазепама, исходя из 2-амино-2*,5-дихлорбензофенона. Взаимодействием последнего с гидроксиламином получают оксим (5.1.26), вводя который в реакции) с хлорангидридом хлоруксусной кислоты, в результате гетероциклизации получают 6-хлор-2-хлорметил-4-(2'-хлорфенил)хиназолин-3-оксид (5.1.27). Реакция последнего с метиламином, как и в случае с хлордиазепоксидом, приводит к перегруппировке с расширением цикла с образованием 7-хлор-2-метиламино-5-(2|-хлорфенил)-ЗЯ-1,4-бензодиазепин-4-оксида (5.1.28). Полученный бензодиазепин-4-оксид подвергают ацили-
- 113-
Глава 5
роваиию ангидридом уксусной кислоты по вторичному атому азота и далее гидролизуют соляной кислотой в 7-хлор-5-(21-хлорфенил)-1,2-дигидро-ЗЯ-1,4-бензодиазепин-2-он-4-оксид (5.1.29). При взаимодействии последнего с уксусным ангидридом происходит реакция типа перегруппировки Полоновского с получением 3-ацето-ксилированного бензодиазепина — 7-хлор-1,3-дигидро-3-ацетокси-5-(2'-хлорфенил)-2Я-бензодиазепин-2-она (5.1.30). продуктом гидролиза которого и является лоразепам (5.1.31) [18, 25, 26, 27, 28, 29].
С1СНгСОС1
Показания к применению такие же, как и у других транквилизаторов. а также используется при кардионеврозе, для предоперационной премедикации и в качестве вспомогательного средства при эндоскопических процедурах. Отличается несколько меньшей продолжительностью действия.
Наиболее распространенными синонимами являются ативан и та-вор.
Хлоразепат (Clorazepate)
Хлоразепат — 7-хлор-2,3-дигидро-2,2-дигидрокси-5-фенил-1Н-1,4-бензодиазепин-3-карбоновая кислота (5.1 34), применяемая в виде
- 114-
дикалиевой соли, получается по еше одной, представляющей самостоятельный интерес, схеме синтеза. В качестве исходного соединения используется 2-амино-5-хлорбензонитрил, который взаимодействием с фенилмагнийбромидом переводится в имин 2-амино-5-хлорбензофенона (5.1.32). Реакцией последнего с аминомалоновым эфиром получают продукт гетероциклизации — 7-хлор-1,3-дигид-ро-3-карбэтокси-5-фенил-2Я-бензодиазепин-2-он (5.1.33), при гидролизе сложноэфирной группы которого спиртовым раствором гидроксида калия получается дикалиевая соль (5.1 34) — хлоразепат [30, 31. 32]
кон
Препарат относится к транквилизаторам длительного действия и применяется по всем показаниям, по которым применяются другие транквилизаторы, а также в качестве вспомогательного средства при эпилептических припадках.
Наиболее распространенными синонимами являются транксен, нок-фан и др
Алпразолам (Alprazolam)
Алпразолам — 8-хлор-1-метил-6-фенил-4//-5-триазоло[4.3-а][1,4]бен-зодиазепин (5.1 39) является химическим аналогом триазолама (4 2 4) и отличается от него отсутствием атома хлора в о-положении 6-фенильного кольца. Для его синтеза может быть применена и применяется такая же схема синтеза, как и для синтеза триазолама. но исходят из 2-амино-5-хлорбензофенона [33, 34, 35]. Однако
-115-
Глава 5
предложен и нестандартный подход для получения алпразолама, исходящий из 2,6-дихлор-4-фенилхинолина, взаимодействием которого с гидразином получают 6-хлор-2-гидразино-4-фенилхинолин (5.1.35). Кипячение последнего с ортоуксусным эфиром в ксилоле приводит к гетероциклизации в производное триазола (5.1 36). Полученный продукт подвергают окислительному расщеплению с использованием перйодата натрия и диоксида рутения в системе аце-тон-вода с получением 2-[4-(3'-метил-1,2,4-триазоло)]-5-хлорбензо-фенона (5.1.37). Оксиметилированием последнего формальдегидом и последующим замещением полученной при этом гидроксильной группы трехбромистым фосфором даст 2-[4-(3|-метил-5|-бромме-тил-1,2,4-триазоло)]-5-хлорбензофенон (5.1.38). Замещением атома брома на аминогруппу аммиаком и следующей за этой реакцией спонтанной внутримолекулярной гетероциклизации получают алпразолам (5.1.39) [36, 37, 38].
Алпразолам является транквилизатором короткого действия, который применяют при беспокойных состояниях, панических расстройствах, депрессивном синдроме.
Наиболее распространенным синонимом препарата является кса-накс.
Как уже было отмечено, среди производных бензодиазепинов найдены препараты с выраженным анксиолитическим действием и отсутствующим или слабо выраженным седативно-гипнотическим эффектом, которые были названы дневными транквилизаторами. Представителем
- 116 -
Анксиолитики
дневных транквилизаторов является медазепам — препарат, отличающийся от диазепама лишь отсутствием карбонильной группы в семичленном азепиновом кольце.
Медазепам (Medazepam)
Медазепам - 7-хлор-2,3-дигидро-1-метил-5-фени_л-1Я-1,4-бензодиа-зепин (5.1.40) предложено получать разными способами. Первый способ заключается в восстановлении карбонильной группы в самом диазепаме (5.1.2) алюмогидридом лития [39, 40].
ыА1Н4
Второй способ получения медазепама заключается в первоначальном восстановлении алюмогидридом лития карбонильной группы в 7-хлор-5-фенил-2,3-дигидро-1Я-1,4-бензодиазепин-2-оне (5.1.1) — первом промежуточном продукте при синтезе диазепама, получаемом при циклоконденсации 2-амино-5-хлорбензофенона с этиловым эфиром глицина до 7-хлор-2,3-дигидро-5-фенил-1Я-1,4-бензодиазепина (5.1.41) и дальнейшем метилировании вторичного аминного атома азота последнего метилйодидом, используя в качестве основания гидрид натрия [41, 42].
1 NaH
2 CH3l
Третий способ получения медазепама заключается в новом способе получения 7-хлор-2,3-дигидро-5-фенил-1Я-1,4-бензодиазепина (5.1.41), заключающемся в гетероциклизации, происходящей при взаимодействии 1-(2,5-дихлорфенил)-1-фенилимина с этилендиамином. Исходный
-117-
Г лава 5
1-(2,5-дихлорфенил)-1-фенилимин (5.1.42) получают взаимодействием 2,5-дихлорбензонитрила с фенилмагнийбромидом [43].
Наконец, предложен четвертый способ получения медазепама исходя из 4-хлор-\-мети.т-анилина, который вводят во взаимодействие с этиленимином в присутствии хлористого алюминия с получением К-(4-хлорфенил)-М-метилэтилендиамина (5.1.43). Ацилируя последний бензоил хлоридом получают соответствующий амид (5.1.44), который циклизуют в искомый медазепам (5.1,40) с использованием хлорокиси фосфора [44, 45].
Предложены и разные модификации описанных способов [46, 47].
Медазепам является дневным транквилизатором, оказывает анксиолитическое, миорелаксирующее и противосудорожное действие. Препарат устраняет чувство тревоги, восстанавливает эмоциональное спокойствие и оказывает стабилизирующее воздействие на вегетативную нервную систему. Медазепам применяют при неврозах, психопатии, сопровождающейся возбуждением, напряженности, повышенной раз
- 118-
Д н К С ПОЛИТИКИ
дражительности. бессоннице, функциональных неврозах сердечно-сосх диетой системы.
Синонимами препарата являются нобриум, транквиракс, азепамид
и др
5.2. Анксиолитики небензодиазепиновой структуры
Мепробамат (Meprobamate)
Мепробамат — дикарба.мат 2-метил-2-пропил-1,3-пропандиола (S.2.2) получают взаимодействием 2-метилвалерианового альдегида с двумя молекулами формальдегида и дальнейшей трансформацией полученного 2-метил-2-пропилпропан-1,3-диола (5.2.1) в дикарба-маз путем последовательных реакций с фосгеном и аммиаком [48. 49. 50].
С3Н7-СН-СНО + СН2О
СНз сэн7-с-сн2- он сн2-он
52 1
1 cocl2
2 NH3 г
СН3
с-,н7 -с -сн2 -осомн2 ch2-oconh2
5 2 2
Мепробамат был предложен до введения в медицинскую практику бензодиазепинов. Точный механизм действия препарата неизвестен, однако его эффекты на ЦНС больше похожи на эффекты барбитуратов, чем бензодиазепинов, но с более коротким временем действия. После введения в практику бензодиазепинов препарат стал использоваться значительно реже Мепробамат применяется при лечении беспокойных состояний, связанных с ежедневными обычными бытовыми стрессами, в основном, как анксиолитик дневного действия.
Синонимами препарата являются цирпон, экванил, стензол, мепрон, милтахн. атраксин и др.
Буспирон (Buspirone)
Буспирон — 8-[4-[4-(2-пиримидинил)-1-пиперазинил]бутил]-8-аза-спиро[4,5]декан-7,9-дион (5.2.6) получают взаимодействием 1-(2-пиримидил)-4-(4-аминобутил)пиперазина(5.2.4) с 8-оксаспиро[4,5] декан-7,9-дионом (5.2.5). В свою очередь. 1-(2-пиримидил)-4-(4-аминобутил)пиперазин (5.2.4) получают взаимодействием 1-(2-пи-римцдил)пиперазина с 4-хлорбутиронитрилом с получением 4-(2-
- 119-
Глава 5
пиримидил)-1-(3-цианопропил)пиперазина (5.2.3), который восстанавливают водородом над никелем Ренея в (5.2.4) [51, 52, 53, 54, 55].
CI(CH2)3CN
Н2 ’ не—
Буспирон является весьма специфическим препаратом, который, возможно, явится представителем нового химического класса анксиолитиков — азаспиронов. Препарат проявляет равную бензодиазепинам активность в качестве анксиолитика, однако лишен противосудорожных и мышечно-расслабляющих свойств, характерных для бензодиазепинов. Кроме того, он не вызывает зависимости и привыкания.
Механизм его действия выявлен неокончательно. Он не воздействует на ГАМК-рецепторы, что имеет место при использовании бензодиазепинов, однако имеет большое сродство к серотониновым (5-НТ) и умеренное сродство к дофаминовым (D2) рецепторам. Буспирон эффективен в качестве анксиолитика. Среди побочных реакций буспирона встречаются головокружение, сонливость, головные боли, нервозность, утомляемость, слабость. Препарат предназначен для лечения беспокойных состояний, при которых наблюдается напряжение, мышечные боли, учащенное сердцебиение, головокружение, страх и г. п., то есть беспокойных состояний несколько иного оттенка, чем бытовые, и не совсем связанных с ежедневными обычными стрессами.
Синонимами препарата являются анизал, аксорен, буспар, буспи-нен, буспинол, народ, травин и др.
Гидроксизин (Hydroxyzine)
Гидроксизин — 2-[2-[4-(и-.хлор-а-фенилбензил)-1-пиперазинил]-этокси]этанол (5.2.6) получают алкилированием 1-(4-хлорбензгид-рил)пиперазина 2-(2-гидроксиэтокси)этилхлоридом [56. 57, 58. 59, 60, 61].
- 120-
Анксиолитики
Cl—СН2-СН2-О-СН2-СН2-ОН
Гидроксизин предназначен для симптоматического лечения беспокойства и напряжения, связанного как с неврозами, так и с органическими болезненными состояниями. Препарат обладает мышечнорасслабляющим, антигистаминным и анальгетическим, местноанестезирующим, противорвотным действием и имеет большую широту терапевтического действия. Применяют, главным образом, при премедикации и последующей обшей анестезии, в ходе которой препарат потенцирует действие меперидина и барбитуратов. Препарат часто применяют в педиатрии в качестве мягкого седативного средства.
Синонимами препарата являются атаракс, агиракс, дурракс, виста-рил и др.
Хлормезанон (Chlormezanone)
Хлормезанон — 2-(и-хлорфенил)тетрагидро-3-метил-4Я-1,3-тиазин-4-он 1,1-диоксид (5.2.8) получают при совместной конденсации меркаптопропионовой кислоты, метиламина и 4-хлорбензальдегида, очевидно, через промежуточную стадию образования 4-хлорбен-зилиденметиламина с получением аминотиоацеталя — 2-(п-хлор-фенил)-тетрагидро-3-метил-4Я-1,3-тиазин-4-она (5.2.7). Окисляя перманганатом калия атом серы, получают хлормезанон (5.2.8) [62, 63].
•н
СНз^Н2 * hS-CH2-CH2-COOH ------►
КМпО4 _
- 121 -
Глава 5
Хлормезанон улучшает эмоциональное состояние устраняя умеренное беспокойство и напряжение пациента Однако препарат обладает рядом побочных эффектов и поэтому, не имея особых преимуществ перед другими анксиолитиками, редко применяется на практике
Синонимами препарата являются транкопал, алинам, флексипирин и др
Список литературы
1 Sternbach L et al //J Org Chem 26 4936(1961)
2 US Pat 3 109 843 (1963)
3 US Pat 3 371 085 (1968)
4 Gates M J Org Chem 45, 1675 (1980)
5 Ishikura M et al//J Org Chem 47. 2456 (1982)
6 Ger Pat 2 016 084(1970)
7 Ger Pat 2 223 482 (1972)
8 Yamamoto И etal 'Chem Ber 101,4245 (1968)
9 US Pat 3 192 199(1965)
10 Fr Pat 1 394 287 (1965)
11 US Pat 3 192 206 (1965)
12 InabaS Chem Pharm Bull Г7, 1263 (1969)
13 Steinman M et al 7 J Med Chem 16, 1354 (1973)
14 LS Pat 3 429 874(1969)
15 US Pat 2 893 992 (1959)
16 Ger Pat 1 096 363 (1959)
17 Sternbach L et al / J drg Chem 26, 1111 (1961)
18 US Pat 3 176 009(1965)
19 US Pat 3 296 249 (1967)
20 LSPat 3 109 843 (1963)
21 Belg Pat 629 227 (1967)
22 BellS etal ZJ Org Chem 27,562 (1962)
23 BellS etal J Org Chem 27, 1691 (1962)
24 BellS etal J Org Chem 33 216(1968)
23 Belg Pat 621 819 (1963)
26 US Pat 3 296 249 (1967)
23 Brit Pat 1 057 492 (1967)
28 Childress A etal J Pharm Sci 53, 577 (1964)
29 Childress 1 etal 'J Med Chem 11, 457 (1968)
30 US Pat 3 516 988 (1970)
31 Ger Pat 1 518 764 (1965)
- 122-
Анксиолитики
Т2 33 34 35 36 27 38 39 40
41 42 43 44 45 46 47 48 49
50 51 52 53 54 55 56 57 58
59 60 61 62 63
Schmitt J etal Chim Ther 4,239 (1969)
I S Pat 3 987 052 (1976)
Ger Pat 2 012 190 (1970)
Hester J etal ''Tetrahedron Letters 1971. 1609
Walker 4 et al//J Med Chem 20, 1694(1977)
LSPat 3 709 898 (1972)
IS Pat 3 781 289 (1973)
Steinbach L etal J Org Chem 28.2456(1963)
LS Pat 3 109 843 (1963)
Belg Pat 620 773 (1963)
LSPat 3 131 Г8(1964)
Ger Pat 1 934 385 (1969)
Ger Pat 1 695 188 (1967)
Ger Pat 1 795 811 (1967)
InabaS etal "Chem Pharm Bui 20, 1628 (1972)
Mthahc M etal//J Heterocycl Chem 14, 941 (1977)
US Pat 2 724 720(1955)
Swiss Pat 373 026(1963)
Ludvig В et al/'J Am Chem Soc 73, 5779 (1951)
Ger Pat 2 057 845 (1970)
US Pat 3 976 776(1976)
LSPat 3 907 801 (1975)
US Pat 3 717 634(1973)
Wu 1 etal 'J Med Chem 15, 477 (1972)
US Pat 2 899 436(1959)
Ger Pat 1 049 383 (1954)
Ger Pat 1 061 786 (1954)
Ger Pat 1 068 262 (1954)
Ger Pat 1 072 624 (1954)
Ger Pat 1 075 116(1954)
Brit Pat 815 203 (1977)
Surrey 4 etal 'J Am Chem Soc 80,3469,3471 (1958)
j Глава 6
\ Антипсихотики (нейролептики)
Антипсихотики — препараты, обладающие определенным седативным эффектом, улучшающие настроение и успокаивающие поведение психотических больных, не вызывающие зависимость, были предложены для лечения психотических расстройств (устранение симптоматики психозов — бреда, галлюцинаций) или шизофренических пациентов. Препараты этой группы часто называются также нейролептиками. Раньше для них использовался также термин «большие транквилизаторы» для того, чтобы отличить их от «малых транквилизаторов» — анксиолитиков
Эти препараты вызывают эмоциональное спокойствие и весьма эффективны для лечения пациентов с острыми и хроническими симптомами. По существу, появление в медицинской практике этих препаратов спасло бесчисленное множество пациентов от необходимости жизни в закрытых стенах психиатрических клиник.
Первичными показаниями для антипсихотических средств является симптоматика при следующих расстройствах: шизофрения и ши-зофреноподобные расстройства, бредовые (параноидные) состояния, кратковременные психические расстройства, аффективные расстройства с психотической симптоматикой, психотические расстройства, развивающиеся вследствие основного соматического заболевания.
Должна быть проведена четкая грань между антипсихотиками, используемыми для лечения острых и хронических психозов, и анксиолитиками. предназначенными для лечения беспокойства и напряжения, связанного с психоневротическими или с психосоматическими расстройствами. Антипсихотические препараты обладают значительно более мощным воздействием на ЦНС, но не являются депрессантами ЦНС и, как правило, более токсичны Однако даже при длительном применении они не вызывают зависимости и привыкания — весьма важной проблемы, возникающей при длительном использовании анксиолитиков.
Антипсихотические препараты подразделяются на шесть химических групп, а неклассифицируемые препараты рассматриваются как прочие. Это препараты ряда.
- 124 -
А н т и п сихотики
1) фенотиазинов (хлорпромазин, промазин, трифлупромазин, анегофеназин, флуфеназин, перфеназин, перхлорфеназин, трифлуоперазин, мезоридазин, тиоридазин);
2) тиоксантенов (хлорпротиксен, тиотиксен);
3) бутирофенонов (галоперидол, трифлуперидол, дроперидол, флуанизон);
4) дигидроиндолонов(молиндон);
5) дибензоксазепинов (локсапин) и дибензодиазепинов (клозапин);
6) дифенилбутилпиперидинов (пимозид. флуспирилен и пенфлу-ридол);
7) прочие, которые включают сульпирид, препараты лития и некоторые другие.
Следует иметь в виду, что, несмотря на наличие количественных различий, несомненно имеющихся как в одной и той же группе антипсихотических средств, гак и в различных группах, существенных качественных различий между их фармакологическим действием нет, т. е. при применении в терапевтически эквивалентных дозах их клиническая эффективность практически одинакова. И выбор конкретного антипсихотического препарата в каждом конкретном случае в основном базируйся на попытках минимизировать побочные эффекты (например, выбрать препарат с меньшим седативным эффектом для лиц. имеющих дело с техникой, или препарат с меньшим гипотензивным эффектом для пациентов в возрасте).
Специфическая этиология психотических расстройств в настоящее время изучена недостаточно. Предполагается, однако, что первичной причиной психотического поведения может быть возникновение дисбаланса дофаминергических функций в ЦНС. Многие исследователи придерживаются точки зрения, что именно увеличение дофаминовой активности в специфических областях ЦНС и является причиной ненормального поведения
Механизм действия нейролептиков недостаточно ясен. Однако принято считать, что они являются антагонистами дофамина и дофаминомиметиков и их действие определенным образом связано с блокадой дофаминовых D-рецепторов, что проявляется в виде изменений в поведенческих реакциях. Более того, не исключено их блокирующее влияние и на серотониновые рецепторы и М-холинорецепторы. Не исключено также, что антипсихотические средства, по-видимому, нарушают процесс высвобождения и обратного нейронального захвата ряда биогенных аминов
- 125-
Глава 6
Кажется вероятным, что антипсихотические препараты, конкурентно связываясь с дофаминовыми рецепторами, блокируют действие дофамина на соответствующих рецепторных участках, тем самым понижая психотическую активность. Центральные дофаминовые рецепторы подразделяются на D,- и D2-, а согласно некоторым источникам, и D3-pe-цепгоры. Эти рецепторы имеют высокое сродство к дофамину, но отличаются чувствительностью к нейролептикам различных химических рядов. Например, препараты ряда фенотиазинов являются неселективными конкурентными D|- и В2-антагонистами. В отличие от фенотиазинов, антипсихотики ряда бутирофенона, такие как галоперидол, проявляют селективное действие только на 02-рецепторы.
Фармакологическое действие антипсихотических средств очень сложно. Кроме способности изменять поведение эти препараты имеют ряд других центральных и периферических эффектов.
Антипсихотики, или нейролептики, применяются для воздействия на пациента при острых и хронических психозах, как органической природы, так и индуцированных. Эти препараты применяются для контроля маниакальной фазы при маниакально-депрессивных психозах — для снятия беспокойства, страха, возбуждения, связанных с соматическими заболеваниями, для контроля агрессивности, тика и других неадекватных состояний.
6.1. Производные фенотиазинов
Производные фенотиазина являются неселективными, конкурентными Dr и В2-антагонистами и блокируют активность дофамина на соответствующих рецепторных участках. Кроме того, их действие выражается блокадой и серотониновых рецепторов, и «-адренорецепторов, и холинергических, никотиновых и мускариновых рецепторов.
Фенотиазины проявляют сложный фармакологический спектр действия и на центральную, и на периферическую нервную систему. Кроме того они действуют на эндокринную систему.
Каждое соединение этого ряда в определенной степени отличается от другого по своим качественным, но в основном по своим количественным показателям. Все они действуют на ЦНС, вызывая умеренный седативный и противорвотный эффект, действуют на процессы терморегуляции, скелетную мускулатуру, эндокринную систему, потенцируют действие анальгетиков.
В зависимости от типа замещения при атоме азота фенотиазинового кольца эта группа препаратов подразделяется на три подгруппы и, в частности, на подгруппы фенотиазинов с алифатической боковой це
-126 -
Антипсихотики
пью (хлорпромазин, промазин, трифлупромазин). а также производных пиперазина (ацетофеназин, флуфеназин, перфеназин, перхлорфеназин, трифлуоперазин) и пиперидина (мезоридазин, тиоридазин).
Препараты первой подгруппы (с алифатической боковой цепью) наряд) с выраженным антипсихотическим действием отличаются способностью вызывать заторможенность, вялость, интеллектуальную заторможенность. Седативное действие этих препаратов превосходит активность других препаратов фенотиазинового ряда.
Пиперазиновым производным присуще наличие стимулирующего компонента.
Препараты пиперидинового ряда обладают более слабой психотической активностью, но не имеют седативной компоненты.
Фенотиазины с алифатической боковой цепью и с пиперидиновыми заместителями имеют больший седативный эффект, чем пиперазиновые производные.
Весьма существенное влияние на активность этих соединений оказывает природа заместителя во втором положении фенотиазинового кольца, в качестве которого предпочтительнее иметь акцепторную гру пггу.
Фенотиазины имеют разнообразное применение в медицине. В основном они используются как антипсихотики. Несмотря на то, что они не излечивают болезнь, они позволяют уменьшить психотические симптомы до такой степени, что могут обеспечить больному лучший контакт с реальностью. Иногда фенотиазины применяются для снятия острого беспокойства и, в частности, панических реакций вызванных зависимостью к амфетаминам или диэтиламиду лизергиновой кислоты (ЛСД). Фенотиазины применяются также для снятия поведенческих проблем у детей, которые не поддаются лечению другими средствами. Иногда фенотиазины применяются в предоперационный период, поскольку снимают беспокойство, контролируют рвоту, икот) и понос, а также вызывают мышечное расслабление.
Промазин (Promazine)
Промазин — 10-(3-диметиламинопропил)фенотиазин (6.1.1) получают алкилированием фенотиазина 3-диметиламинопропилхло-ридом в присутствии амида натрия [1, 2, 3].
,снз NaNH2
с;—сн2-сн2-сн2-гД ----------->
СН3
X}
I СНз
СН2 ~СН2 ”Ch’2 ~
6 1 1
СНз
- 127-
Глава 6
В психиагрической практике промазин применяют при легких течениях психомоторного возбуждения \ больных шизофренией, при параноидных, маниакально-депрессивных состояниях, при неврозах, алкогольных психозах и др. Иногда применяют в анестезиологической практике.
Наиболее распространенными синонимами являются кальмизан, пропазин, залофен, спарин. протактил и др.
Хлорпромазин (Chlorpromazine)
Хлорпромазин — 2-хлор-10-(3-диметиламинопропил)фенотиазин
(6.1.2) получают по аналогичной схеме, но алкилированием 2-хлор-фенотиазина 3-диметиламино-пропилхлоридом [4, 5, 6].
СН2 - СН2 -СН2 - N<
СН3
6 1 2
В психиатрической практике хлорпромазин применяют при различных состояниях психомоторного возбуждения у больных шизофренией, при хронических параноидных, а также маниакально-депрессивных состояниях. при неврозах, алкогольных психозах и неврозах, сопровождающихся возбуждением, страхом, напряжением, бессонницей. Особенностью хлорпромазина, в сравнении с другими нейролептиками, является выраженный седативный эффект. Иногда применяют в анестезиологической практике для потенцирования наркоза. Препарат обладает также умеренным противовоспалительным действием.
Наиболее распространенными синонимами являются аминазин, мегафен, ларгактил, торазин, промпар и др.
Трифлупромазин (Triflupromazine)
Трифлупромазин — 2-трифторметил-10-(3-диметиламино пропил)-фенотиазин (6.1.3) также получают алкилированием 2-трифтор-метилфенотиазина 3-диметиламино-пропилхлоридом в присутствии амида натрия [7, 8, 9, 10, 11, 12].
сн2 - сн2 -сн2 -
СНз
6 1 3
-128 -
Антипсихотики
В психиатрической практике трифлупромазин применяют при психомоторных возбуждениях у больных шизофренией, параноидных состояниях, маниакально-депрессивных состояниях, при неврозах.
Наиболее распространенным синонимом является весприн.
Прох.юрперазин (Prochlorperazine)
Прохлорперазин — 2-хлор-10-[3-(4-метил-1-пиперазинил)пропил]-фенотиазин (6.1.4) получают алкилированием 2-хлорметилфено-тиазина (4-метил-1-пиперазинил)пропил-3-хлоридом в присутствии амида натрия или 2-хлор-10-[(3-хлорпропил)]фенотиазина 1-метил-пиперазином [13, 14, 15, 16].
СН2-СН2-СН2— N N- СНз
6 1 4
Подобно другим пиперазиновым производным фенотиазина прохлорперазин ослабляет психотическую симптоматику и оказывает стимулирующее действие.
Наиболее распространенными синонимом является метеразин.
Трифлуоперазин (Trifluoperazine)
Трифлуоперазин — 2-трифторметил-10-[3-(4-метил-1-пиперази-нил)пропил]-фенотиазин (6.1.5) синтезируют вышеописанным способом, алкилируя 2-трифторметилфенотиазин 4-метил-1-пиперази-нилпропилхлоридом [11, 17, 18, 19, 20].
Трифлуоперазин является одним из наиболее активных антипсихотических препаратов. Нейролептическому эффекту сопутствует умеренный стимулирующий эффект. Особенностью трифлуоперазина явля
- 129 -
Глава 6
ется то, чго больные вместо обычной скованности и слабости, характерной при применении других производных фенотиазина, становятся более оживленными. Препарат обладает сильным противорвотным действием. Его широко применяют в психиатрии для лечения шизофрении и других психических заболеваний.
Наиболее распространенными синонимами являются мобадид, трифтазнн, стелазин и кальмазин и др.
Флуфеназин (Fluphenazine)
Флуфеназин — 4-[3-[2-(трифторметил)фенотиазин-10-ил] пропил]-1-пиперазиноэтанол (6.1.8) получают любым из вышеописанных способов [21. 22, 23, 24, 25, 26, 27]. Алкилируя 2-трифторметилфе-нотиазин 4-формил-1-пиперазинилпропилхлоридом в присутствии амида натрия, синтезируют 2-трифторметил-10-[3-(4-формил-1-пи-перазинил)пропил]фенотиазин (6.1.6). Далее щелочным гидролизом удаляют N-формильную группу, получая 2-трифторметил-10-[3-(1-пиперазинил)пропил]фенотиазин (6.1.7). Последний аткилируют ацетатом 2-бромэтанола-1, и далее кислотным гидролизом снимают защитную ацетильную группу, получая флуфеназин (6.1.8) [27, 28].
1 вг-сн2-сн2-ососн3
Флуфеназин является весьма сильным антипсихотическим препаратом. Нейролептическому эффекту' сопутствует и стимулирующий эффект. Препарат обладает противорвотным действием. Его применяют в психиатрии для лечения различных форм шизофрении и других психических заболеваний.
Наиболее распространенными синонимами являются фторфеназин, модитен, дапотум, мотивал, пермитил и др.
- 130 -
Антипсихотики
Тиоридазин (Thioridazine)
Т иоридазин — 10-[2-( 1 -метил-2-пиперидил)этил]-2-(метилтио)фено-тиазин (6.1.9) синтезируют по аналогичной схеме, алкилируя 2-ме-тилтиофенотиазин 2-(2-хлорэтил)-1-метилпиперидином [29, 30].
н
ch2ch2ci
СНз
NaNH2
СН3
По антипсихотической активности тиоридазин уступает аминазину'. Он наиболее эффективен при психических и эмоциональных расстройствах, сопровождающихся страхом, напряжением и возбуждением. Препарат назначают при различных видах шизофрении, психозах, неврозах.
Наиболее распространенными синонимами являются сонапакс и меллерил.
Мезоридазин (Mesoridazine)
Мезоридазин — 10-[2-( 1 -метил-2-пиперидил)этил]-2-( метилсульфи-нил)фенотиазин (6.1.13) также синтезируют по аналогичной схеме, однако алкилируя окисленную форму 2-метилтиофенотиазина 2-ме-тилсульфонилфенотиазин 2-(2-хлорэтил)-1-метилпиперидином.
С этой целью 2-метилтиофенотиазин первоначально ацилируют уксусным ангидридом по атому азота с получением 10-ацетил-2-метил-тиофенотиазина (6.1.10). Полученное ацетильное производное далее окисляют пероксидом водорода в 10-ацетил-2-метилсульфонилфеноти-азин (6 1.11). Деацилированием последнего метанольным раствором поташа получают 2-метилсульфонилфенотиазин (6.1.12), который и алкилируют 2-(2-хлорэтил)-1-метилпиперидином в присутствии амида натрия с получением искомого мезоридазина (6.1.13) [31].
1СНчС0'2^
К2СОз СНЭОН
-131 -
Глава 6
сн3
NaNH2
Мезоридазин действует аналогично другим фенотиазиновым нейролептикам и показан при шизофрении, при возникновении поведенческих проблем, при психоневротических проявлениях, при остром и хроническом алкоголизме.
Синонимами препарата являются лиданил, серенгил и др.
6.2. Производные тиоксантенов
Структурно тиоксантены отличаются от фенотиазинов тем, что в центральном кольце трициклической системы атом азота замещен на углеродный, соединенный с боковой цепью двойной связью. Их фармакологическое действие практически сходно с соответствующими фенотиазиновыми аналогами. Они имеют тот же механизм действия и аналогичное влияние на ЦНС. Препараты этого ряда отличаются друг от друга количественными показателями.
Хлорпротиксен (Chlorprothixene)
Хлорпротиксен — 2-хлор-9-[(1-диметиламино)-3-пропилиден]тио-ксантен (6.2.7) предложено получать исходя из 2-хлортиоксантона (6.2.3). Исходный 2-хлортиоксантон (6.2.3) синтезируют исходя из 2-меркаптобензойной кислоты, взаимодействием которой с 1-бром-4-хлорбензолом получают 2-(4-хлорфенилтио)бензойную кислоту (6.2.1), которую реакцией с пятихлористым фосфором переводят в хлоран-гидрид (6.2.2) и далее с использованием треххлористого алюминия циклизуют в 2-хлортиоксантон (6.2.3) [32]. Альтернативным способом получения 2-хлортиоксантона (6.2.3) является получение 2-(4-хлорфенилтио)бензойной кислоты (6.2.1) взаимодействием 2-йодбен-зойной кислоты с 4-хлортиофенолом [33]. Полученный 2-хлортиоксантон (6.2 3) в качестве карбонильного компонента вводят во взаимодействие в реакции либо с 3-диметиламинопропилмагнийброми-дом [33], либо с аллилмагнийбромидом [34, 35, 36] с получением соответствующих третичных спиртов (6.2.4 или 6.2.5). Дегидратация первого осуществляется путем ацилирования третичной гидроксильной группы хлорангидридом уксусной кислоты и последую
- 132-
Антипсихотики
щим пиролизом образовавшегося ацетата, приводя к искомому хлорпротиксену (6.2.7).
Дегидратация третичного спирта (6.2.5) осуществляется в условиях хлорирования третичной спиртовой группы хлористым тионилом с образованием диена — 2-хлор-9-(3-пропен-1-илиден)тиоксантена (6.2.6), присоединение к которому диметиламина при высокой температуре приводит к получению искомого хлорпротиксена (6.2.7).
(CHafeNH
- 133 -
Глава 6
Хлорпротиксен обладает антипсихотическим и седативным действием. выраженной противорвотной активностью. Применяют при различных психозах, шизофрении, реактивных и невротических депрессиях с преобладающей тревожной симптоматикой, состояниях возбуждения, связанных со страхом и напряжением. В малых дозах может применяться как успокаивающее средство при неврозах.
Синонимами хлорпротиксена являются клотиксен и тарасан.
Тиотиксен (Thiothixene)
Гиотиксен — Х’,М-диметил-9-[3-(4-метил-1-пиперазинил)пропилиден] тиоксантен-2-сульфонамид (6.2.14) синтезируют исходя из 9//-тио-ксантена, взаимодействием которого с хлорсульфоновой кислотой получают 9Я-тиоксантен-2-сульфоновую кислоту (6.2.8). Последнюю трансформируют в 2-диметиламиносульфонил-9Н-тиоксантен (6.2.9) путем последовательных реакций с хлористым тионилом и диметиламином. Взаимодействием 2-диметиламиносульфонил-9//-тиоксантена (6.2.9) с бутиллитием и далее с метилацетатом получают 9-ацетил-2-диметиламиносульфонил-9Н-тиоксантен (6.2.10). Аминометилированием последнего диметиламином получают 9-(2-диметиламинопропи-ониил)-2-диметиламиносульфонил-9//-тиоксантен (6.2.11). Взаимодействием последнего с 1-N-метилпиперазином осуществляют замену диметилам ин ной группы в ацильном фрагменте молекулы на N-метил-пиперазиновую с получением продукта (6.2.12). Карбонильную группу последнего восстанавливают в спиртовую с помощью боргидрида натрия, и далее осу ществляют дегидратацию продукта (6.2.13) в искомый тиотиксен (6.2.14) с помощью хлорокиси фосфора [37, 38, 39,40].
1 SOCI2
2 (СНз)г^н
2 СН3СООСН^
SO2N(CH3)2
1 C4H9L1
СН2О <СН-фКН ст Xi ——
- 134-
Антипсихотики
POCI3
Препарат проявляет определенное химическое и фармакологическое сходство с пиперазиновыми производными фенотиазинового ряда и применяется при лечении психотических расстройств.
Показания к применению тиотиксена те же, что и для применения хлорпротиксена.
Синонимами препарата являются орбинамон, наван и др.
6.3. Производные бутирофенонов
Ряд различных соединениях пиперидинового и пиперазинового рядов замещенных по атому азота и-фторбутирофенонной группой проявляют значительную нейролептическую активность (галоперидол, триф-лу перидол, дроперидол, меторин).
Интерес к производным бутирофенона как в качестве антипсихотических средств, так и в анестезиологии весьма значителен. Они проявляют фармакологические эффекты и механизм действия, весьма сходные с фенотиазинами и тиоксантенами, т. е. блокируют дофаминергические рецепторы. Однако они являются более селективными по отношению к П2-рецепторам.
Трифлуперидол (Trifluperidol)
Трифлуперилол — 4-[4-(а,а,а-трифтор-.и-толил)-4-гидроксипипе-ридино]-4*-фтор-бутирофенон (6.3.3) получают взаимодействием 1-бензил-4-пиперидона (3.1.48) с реактивом Гриньяра, приготовленным из 1-трифторметил-З-бромбензола и магния, в результате чего получают 1-бензил-4-гидрокси-4-(3-трифторметилфенил)пи-перидин (6.3.1), восстановлением которого водородом в присутствии в качестве катализатора палладия на угле снимают бензильную защиту с получением 4-гидрокси-4-(3-трифторметилфенил)пипери-
-135-
Г лава 6
дина (6.3.2). Алкилированием атома азота последнего со-хлор-4-фторбутирофеноном получают трифлуперидол (6.3.3) [41, 42].
ВгМд
Pd-C
Необходимый для этого 4’-хлор-4-фтор-бутирофенон (6.3.4) получают ацилированием фторбензола хлорангидридом 4-хлормасляной кислоты.
о
ci—с-(сн2)3-С1
А1С|3
—, 0
F—Z У- С-СН2-СН2-СН2-С1
634
Трифлуперидол является мощным антипсихотическим препаратом. Он усиливает действие снотворных, наркотиков, анальгетиков. Оказывает противосудорожное и противорвотное действие.
Препарат применяют при психозах, сопровождающихся моторным и психическим возбуждением, при затяжных приступах периодической шизофрении, при состояниях, сопровождающихся тяжелой депрессией и бредом, при алкогольных психозах. По способности купировать маниакальное возбуждение превосходит другие нейролептики.
Синонимами препарата являются триперидол, психоперидол, три-седил и др.
Галоперидол (Haloperidol)
Г ало пери дол — 4-[4-(/г-хлорфенил)-4-гидроксипиперидино]-41 -фторбутирофенон (6.3.6) получают алкилированием 4-(4-хлорфе-нил)-4-гидроксипиперидина (6.3.7) 4'-хлор-4-фторбутирофеноном (6.3.4). Синтез 4-(4-хлорфенил)-4-гидроксипиперидина (6.3.7) осуществляют исходя из 2-(4-хлорфенил)пропена, взаимодействием которого с формальдегидом и хлоридом аммония получают проме-
- 136-
Антипсихотики
жуточный 4-метил-4-(4-хлорфенил)-1,3-оксазин (6.3.5) очевидно через стадии постулированные для протекания реакций типа реакции Принса. Обработка последнего соляной кислотой приводит к получению 4-(4-хлорфенил)-1,2,3,6-тетрагидропиридина (6.3.6) очевидно через стадию раскрытия гидрированного 1,3-оксазинового цикла, дегидратации и последующей рециклизациии. Присоединение к двойной связи полученного 4-(4-хлорфенил)-1,2,3,6-тетрагид-ропиридина (6.3.6) бромистого водорода и последующий щелочной гидролиз образующегося при этом 4-(4-хлорфенил)-4-бромпипери-дина позволяют получить 4-(4-хлорфенил)-4-гидроксипиперидин (6.3.7). взаимодействием которого с 4!-хлор-4-фторбутирофеноном (6.3.4) получают целевой галоперидол (6.3.6) [41,42, 43, 44, 45, 46].
636 637
634 + 637
Галоперидол является одним из наиболее активных современных нейролептиков. Его высокая антипсихотическая активность сочетается с умеренным седативным эффектом. Препарат эффективно купирует психомоторные возбуждения различного рода. Применяют при шизофренических психозах, маниакальных, параноидных бредовых состояниях, при депрессиях, при психомоторных возбуждениях различного генеза. при бреде и галлюцинациях различного происхождения.
Наиболее распространенными синонимами являются галдол, веза-дол, линтон и др.
Дроперидол (Droperidol)
Дроперидол — 1-[I-[3-(и-фторбензоил)пропил]-1,2,3,6-4-пиридил]-2-бензимидазолинон (6.3.11) синтезируют исходя из 1-бензил-З-карб-этоксипиперидин-4-она (3.1.47), который вводят во взаимодействие
- 137-
Глава 6
с о-фенилендиамином. Очевидно, первоначально образующееся в условиях реакции производное 1,5-бензодиазепина перегруппировывается в 1-(1-бензил-1,2,3.6-тетрагидро-4-пиридил)-2-бензимида-золон (6.3.9). Дебензилирование последнего водородом над палладиевым катализатором в 1-( 1,2,3,6-тетрагидро-4-пиридил)-2-бенз-имидазолон (6.3.10) и алкилированием последнего 4'-хлор-4-фтор-бутирофеноном (6.3.4) получают искомый дроперидол (6.3.11) [47, 48,49].
Нейролептик дроперидол оказывает антипсихотическое, седативное, противошоковое, действие. Потенцирует действие средств для наркоза. В психиатрической практике дроперидол применяют при психомоторном возбуждении, галлюцинациях. Основное применение препарат находит в анестезиологии для нейролептанальгезии в комбинации с фентанилом. Применяют как для премедикации, так и в процессе хирургических операций, а также в послеоперационном периоде.
Синонимами препарата являются таламонал, дролептан, лептофен, инновар и др.
Флуанизон (Fluanison)
Флуанизон — 4'-фтор-4-[4-(о-метоксифенил)-1-пиперазинил]-бути-рофеион (6.3.12) получают взаимодействием 1-(2-метоксифенил)-пиперазина с 4’-хлор-4-фторбутирофеноном (6.3.4) [50].
•с — C.t'2 —CHj -*СН2 ”С
Флуанизон является нейролептиком с седативными свойствами, у которого антипсихотическое действие выражено относительно слабо. Препарат применяют в качестве самостоятельного или дополнительного
- 138-
Антипсихотики
средства при психомоторном возбуждении при острой и хронической шизофрении, маниакально-депрессивном синдроме.
Синонимами препарата являются седаланд, меторин и др.
6.4. Производные дигидроиндолонов
Производные дигидроиндолонов структурно не принадлежат ни к одному из рассматриваемых выше классов препаратов. Однако механизм их действия, показания к применению и побочные эффекты весьма сходны с производными фенотиазинов.
Молиндон (Molindone)
Молиндон — 3-этил-6,7-дигидро-2-метил-5-(морфолиноэтил)-индол-4(5/7)-он (6.4.3) получают нитрозированием диэтилкетона азотистой кислотой или метилнитритом в нитрозодиэтилкетон (6.4.1). Восстановлением последнего цинком в уксусной кислоте в 2-амино-диэтилкетон в присутствии циклогександиона-1,3 получают 3-этил-2-метил-4,5.6,7-тетрагидроиндол-4-он (6.4.2). Аминометилированием последнего морфолином и формальдегидом получают молиндон (6.4.3) [51, 52].
сн гн /СНз
HNO2 or CH3ONO2 п
---------2---* н3сх А /СНз хсн2 с
б 41 NOH
Молиндон более активный антипсихотик, чем хлорпромазин. Его седативный эффект выражен слабее. Побочные эффекты также выражены слабее, чем у более мощных нейролептиков. Он способствует уменьшению спонтанных движений и агрессивности и показан для лечения проявлений психотических нарушений и, в частности, случаев хронической и острой шизофрении.
Синонимом препарата является мобан.
-139-
Глава 6
6.5. Производные дибензоксазепинов и дибензодиазепинов
Производные дибензоксазепинов и дибензодиазепинов также структурно не принадлежат ни к одному из вышеперечисленных классов препаратов. Однако механизм их действия, показания к применению и побочные эффекты аналогичны производным фенотиазинов.
Локсапин (Loxapine)
Локсапин — 2-хлор-11-(4-метил-1-пиперазинил)дибенз[Ь,Г][1,4]ок-сазепин (6.5.4) синтезируют исходя из 2-(4-хлорфенокси)анилина. Ацилированием последнего хлоругольным эфиром получают N-это-ксикарбонил-2-(4-хлорфенокси)анилин (6.5.1). который вводят во взаимодействие с N-метилпиперазином с получением уреида (6.5.2). Обработка последнего смесью хлорокиси фосфора и фосфорного ангидрида получают локсапин (6.5.3) [53, 54, 55].
С1СООС2Н6
651
6 53
Локсапин более выраженный активный антипсихотик, чем хлорпромазин. Его седативный эффект уступает эффекту хлорпромазина. Показания к применению и побочные эффекты соответствуют таковым у производных фенотиазинов. Локсапин используется для лечения проявлений психотических нарушений и, в частности, случаев хронической и острой шизофрении.
Синонимами препарата являются локсопак и локситан.
-140-
А н т и п с ихотики
Клозапин (Clozapin)
Клозапин — 8-хлор-11-(4-метил-1-пиперазинил)-5//-дибензо[Ь,е]-[1,4]диазепин (6.5.7) предложено получать двумя путями. Согласно первому способу, 4-хлор-2-нитроанилин в присутствии медных опилок арилируют метиловым эфиром о-хлорбензойной кислоты с получением соответствующего дифениламина (6.5.4). Взаимодействием последнего с N-метилпиперазином сложноэфирную группу в полученном полифункциональном дифениламине трансформируют в амидную (6.5.5). Далее водородом в присутствии никеля Ренея, восстанавливают нитрогруппу в полученном 4-хлор-2-нитро-2'-карб-(К'-метилпиперазино)амиде (6.5.5) до аминной. Взаимодействием полученного продукта (6.5.6)с хлорокисью фосфора осуществляют гетероциклизацию в искомый дибензодиазепин — клозапин (6.5.7) [56, 57, 58].
Н3С-М N-H
Н? / Raney—Ni
Второй способ синтеза клозапина исходит из 8-хлор-10,11-Дигидро-5Н-дибензо-[Ь,е]1,4-диазепин-11-тиона, который алкилируют по атому серы дибензодиазепинового кольца 4-нитробензилхлоридом в присутствии wpew-бутилата калия с получением N-метилпронзводного (6.5.8). Взаимодействием последнего с N-метилпиперазином получают искомый клозапин (6.5.7) [59].
|СН3}з~К
n S
- 141 -
Глава 6
Клозапин является нейролептиком с выраженным антипсихотическим и седативным действием. Не вызывает выраженного общего угнетения и экстрапирамидных расстройств. Препарат применяют при острых и хронических формах шизофрении, маниакальных состояниях, маниакально-депрессивном психозе, психомоторном возбуждении, различных психотических состояниях.
Синонимами препарата являются лепонекс. ипрокс и др.
6.6. Производные дифенилбутилпиперидинов
Представителями дифенилбутилпиперидинов являются пимозид, флуспирилен и пенфлуридол, которые относятся к мощным нейролептическим препаратам с выраженными антипсихотическими свойствами, близкими к галоперидол}. Основная особенность этого ряда препаратов — их пролонгированное действие. Механизм их действия однозначно не выяснен, однако понятно, что они блокирует дофаминергическую активность.
Пимозид (Pimozide)
Пимозид — 1-[1-[4,4-бис(н-фторфенил)бутил]-4-пиперидил]-2-бенз-импдазолинон (6.6.5) структурно очень напоминает дроперидол за исключением наличия двойной связи в пиперидиновом кольце и замене п-фторбутирофенонного радикала при атоме азота пиперидинового кольца на 4,4-бис(н-фторфенил)бутильный. Необходимый для синтеза пимозида, равно как и флуспирилена и пенфл}ридола, 4,4-бис-(щфторфенил)-бутилхлорид (бромид) (6.6.3) получают взаимодействием двух молей 4-н-фторфенилмагнийбромида с эфиром циклопропанкарбоновой кислоты, в результате чего образуется бис-(4-н-фторфенил)циклопропилкарбинол (6.6.1). Обработка последнего хлористым гионилом (трехбромистым фосфором) приводит к дегидратации и, естественно, к одновременному раскрытию циклопро-пильного кольца с образованием 1,1-бис-(4-фторфенил)-4-хлор(бром)-1 -бутена (6.6.2). Восстановление двойной связи последнего водородом над палладиевым катализатором приводит к получению 1,1-бис-(4-фюрфенил)бутил хлорида (бромида) (6.6.3) [60, 61, 62, 63].
MgBr
£>— СООС2Н5
SOCI2
- 142 -
Антипсихотики
Н2 / Pd-c
Собственно синтез пимозида заключается в алкилировании 4-(2-бензимидазолинон) пиперидина (6.6.4), получаемого восстановлением 1-(1.2,3,6-тетрагидро-4-пиридил)-2-бензимидазолона (6.3.10) водородом над никелем Ренея, ранее полученным 4.4-бмс-(4-фторфенил)бутилхло-ридом (6.6.3) в результате чего и образуется пимозид (6.6.5).
^2' Ranay-Mi
По спектру фармакологического действия пимозид близок к галоперидолу. Его применяют как в стационаре, так и в амбулаторных условиях для поддерживающей терапии больных, страдающих шизофренией, параноидными состояниями, психотическими и невротическими расстройствами с параноидными признаками. При острых психозах не-приюден, так как не обладает психомоторно-седативным действием. Препарат применяется при лечении пациентов, страдающих синдромом loyperra. Пимозид имеет ряд побочных эффектов, многие из которых сходны с таковыми у фенотиазинов.
Синонимом препарата является орап.
-143-
Глава 6
Флуспирилен (Fluspirilen)
Флуспирилен — 8-[4,4-бис(п-фторфенил)бутил]-1 -фенил-1,3,8-триазаспиро[4,5]декан-4-он (6.6.9) получают исходя из 1-бензил-4-анилино-4-нианопиперидина (3.1.64) путем его кислотного гидролиза в амид (6.6.6) и последующей гетероциклизации с помощью формамида 4-аминокарбонильной и 4-анилинной функциональных групп в имидазолоновый фрагмент, создав, тем самым, искомую спирогетероциклическую систему — 8-бензил-1-фенил-1,3,8-три-азаспиро[4,5]декан-4-он (6.6.7). Восстановлением водородом с использованием в качестве катализатора палладия на угле снимают с последнего защитную N-бензильную i руппу с получением 1-фе-нил-1,3,8-триазаспиро[4,5]декан-4-она (6.6.8). Алкилируя последний 1,1-£шс-(4-фторфенил)бутил бромидом (6.6.3), получают флуспирилен (6.6.9) [64, 65, 66].
3 1 4S
HCN
3 1 64
H2SO4
О
В первую очередь препарат применяют для поддерживающей терапии больных, страдающих хроническими психическими заболеваниями после лечения в стационаре. Удобен для применения в амбулаторной практике из-за отсутствия выраженного гипноседативного эффекта.
- 144 -
А нт и психотики
Синонимами препарата являются имап, редептин и др.
Пенфлуридол (Penfluridol)
Пенфлуридол — 4-(4-хлор-3-трифторметилфенил)-1-[4,4-5ис-(п-фторфенил)бутил]-4-пиперидинол (6.6.12) синтезируют реакцией Гриньяра между 1-карбметоксипиперидин-4-оно.м и 4-хлор-З-трифторметилфенилмагнийбромидом с получением 1-карбметокси-(4-хлор-3-трифторметилфенил)-4-пиперидинола (6.6.10). Щелочным гидролизом карбметоксильной группы последний превращают в (4-хлор-3-трифторметилфенил)-4-пиперидинол (6.6.11), алкилированием которого 1,1-бнс-(4-фторфенил)бутил бромидом (6.6.3) получают пенфлуридол (6.6.12) [67, 68, 69].
кон
6 6 10
По своему фармакологическому действию препарат близок к пимо-зиду, однако его действие значительно более длительное, что связано с медленным метаболизмом препарата. Пенфлуридол показан для поддерживающей терапии в амбулаторных условиях у пациентов, страдающих шизофренией, а также у пациентов в параноидном, психотическом и невротическом состояниях.
Синонимами препарата являются семап, лонгоперидол и др.
6.7. Прочие нейролептики
Сулъпирид (Sulpirid)
Сульпирид — Ы-[(1-этил-2-пирролидинилметил]-5-сульфамоил-0-анизамид (6.7.2) получают исходя из 5-аминосульфосалициловой кислоты. Метилируя последнюю диметилсульфатом, получают 2-метокси-5-аминосульфонилбензойную кислоту' (6.7.1), которую
-145-
Г лава 6
трансформируют в амид, используя в качестве аминной компоненты 2-аминомезил-1-этилпирролидин, а в качестве конденсирующего средства— карбонил-1,1'-бдс-имидазол [70, 71, 72, 73, 74].
6 7 2
Сульпирид обладает умеренной нейролептической активностью в сочетании с некоторым стимулирующим и тимолептическим эффектом. Не оказывает седативного действия. Оказывает противорвотное, умеренное каталептогенное и антисеротониновое действие. Способствует улучшению кровоснабжения желудка. Ускоряет восстановительные процессы в тканях. Препарат применяют при шизофрении, депрессии, мигрени, нарушениях поведенческих функций и язвенной болезни желудка и двенадцатиперстной кишки.
Синонимами препарата являются арминол, догматил, конфидан, эг-лонил и многие другие.
Препараты лития
Соли лития были предложены в медицине для лечения подагры и растворения почечных камней. Однако позже было установлено, что препараты лития способны купировать острое маниакальное возбуждение у людей и предупреждать аффективные приступы. Механизм действия препаратов лития выяснен неокончательно, однако ясно, что ионы лития влияют на транспорт ионов натрия в нервных и мышечных клетках, вследствие чего ионы лития выступают как антагонисты ионов натрия.
Лития карбонат (Lithium carbonat)
Лития карбонат получают взаимодействием солей лития с содой или поташом и далее труднорастворимую соль подвергают очистке [75].
2LiCI + Na2CO3= 2NaCI + Li2CO3
Наиболее распространенным препаратом лития является лития карбонат, который обладает антиманиакальным действием. Полагают, что литий изменяет транспорт ионов Na- в нейронах, влияя на внутриклеточное содержание катехоламинов, нормализует психическое состояние. не вызывая общей заторможенности.
-146-
Анти психотики
Препарат применяют при маниакальных состояниях различного генеза. профилактике и лечении аффективных психозов.
Синонимами препарата являются эскалит, камколит, каболит. лити-зин и др.
Список литературы
1. US Pat. 2.519.886 (1950).
2. Ger. Pat. 824.944 (1950).
3. Wirth 1F.//Arzneim.-Forscch. 8, 507 (1958).
4. US Pat. 2.645.640(1953).
5. Ger. Pat. 910.301 (1951).
6. Charpentier B. et aU/Compt Rend. 235, 59 (1952).
7. Ger. Pat. 1.095.836 (1956).
8. US Pat. 2.921.069(1960).
9. Brit. Pat. 813.961 (1959)
10 Duhm B. et al.//Z. Naturforsch. 13, 756 (1958).
11. Craig P. et al.C'J. Org. Chem. 22, 709 (1959).
12. Yale H. et al./J. Am. Chem. Soc. 79, 4375 (1957).
13. US Pat. 2.902.484 (1959).
14. Brit. Pat. 780.193 (1957).
15. Ger. Pat. 1.037.461 (1955).
16. Fr. Pat. 1.167.627 (1959).
17. US Pat. 2.921.069(1960).
18. Ger. Pat. 1.095.836 (1956).
19. Ger. Pat. 1.165.034 (1956).
20. Brit. Pat. 813.861 (1959).
21. US Pat. 3.058.979 (1962).
22. Ger. Pat. 1.095.836 (1956).
23. Brit. Pat. 829.246 (1960).
24. Brit. Pat. 833.474 (1960).
25. US Pat. 3.194.733 (1965).
26. Yale H. et al./7 J. Am. Chem. Soc. 82, 2039 (1960).
27. Anderson E. et al.//Arzneim.-Forsch. 12, 937 (1962).
28. USPat. 2.766.235 (1956).
29. US Pat. 3.239.514 (1966).
30. Bourquin J et al.//Helv. Chim. Acta. 41. 1072 (1958).
31. US Pat. 3.084.161 (1960).
32. Ger. Pat. 1.044.103 (1957).
33. USPat. 2.951.082 (1960).
- 147-
Г лава 6
34. US Pat. 3.116.291 (.1963).
35. Ger. Pat. 1.168.446 (1959)
36. Ger Pat. 1.418.517 (1959).
37. Ger. Pat. 1.470.157 (1963).
38. US Pat 3.310.553 (1967).
39. Belg. Pat. 647.066 (1964).
40. Muren J et al. 'J. Med. Chem. 13, 17 (1970).
41. Brit. Pat. 895.309(1962).
42. US Pat. 3.438.991 (1969).
43. Belg. Pat. 577.977 (1959).
44. Janssen P etal.UMed. Chem. 1, 281 (1959).
45. Brit. Pat. 1.141 664 (1966).
46. US Pat. 4.086.234 (1975).
47 Brit. Pat. 989.755 (1962).
48. US Pat. 3.141.823 (1964).
49 US Pat. 3.161.645 (1964).
50. US Pat. 2.997.472 (1961).
51. US Pat. 3.491.093 (1970).
52. Belg. Pat. 670.798 (1966).
53. US Pat. 3 412.193 (1968).
54. US Pat. 3.546.226(1970).
55. Schmutz J etal.'/Helv. Chim. Acta 50, 245 (1967).
56. Fr. Pat. 1.334.944 (1963).
57. US Pat. 3.539.573 (1970).
58. Brit. Pat. 980.853 (1961).
59. Hunziker F et al. /Helv. Chim. Acta 50. 1588 (1967).
60. Fr. Pat. М 3.695 (1963).
61. US Pat. 3.196.157 (1965).
62. Ger. Pat. 1.470.124 (1963).
63. Janssen P et aL'/Arzneim.-Forsch. 18. 261, 279, 282 (1968).
64. Belg. Pat. 633.914 (1963).
65 US Pat. 3.238.216(1966).
66. Ger. Pat. 1.470.125 (1963).
67. Ger. Pat. 2.040.231 (1970).
68. US Pat. 3.575.990(1971).
69. Sindelar J. et al.//Coll. Czech. Chem. Commun. 38, 3879 (1973).
70. Ger. Pat. 1.595.915 (1965).
71. Ger. Pat. 1.795.723 (1965).
72. US Pat. 3.342.826 (1969).
73. US Pat. 4.077.976(1978).
74. Ger. Pat. 2.903.891 (1979).
75. Caley M et al.' Inorg. Syn. 1, 1, (1939)
-148 -
4 Глава 7
j Антидепрессанты
Препараты, применяющиеся для лечения психических расстройств, сопровождающихся депрессией, называются антидепрессантами Это препараты, которые могут облегчить множество симптомов, ассоциированных с широким набором психосоматических расстройств, известных как депрессия.
Иными словами, антидепрессанты способны устранить или уменьшить ряд расстройств со стороны психоэмоциональной сферы, обозначаемых в психоневрологической практике как депрессивный синдром.
В свою очередь состояние, характеризуемое термином ^депрессия», включает аффективные расстройства, которые часто сопровождаются рядом других нарушений, включая беспричинную печаль, расстройств сна, изменения аппетита, различные психомоторные нарушения, потерю интереса к удовольствиям, ощущение своей бесполезности и довольно часто суицидальные мысли.
Существует достаточно приемлемая, хотя и не общепринятая, классификация депрессий, основанная на их этиологии. Выделяют следующие типы депрессий:
— эндогенные депрессии, характеризующиеся потерей интереса к удовольствиям, потерей либидо, малой подвижностью, нарушениями сна:
-149-
Глава 7
— невротические, или беспокойные, депрессии, характеризующиеся беспокойством, напряжением, «сверхреактивностью» по отношению к неожиданностям и потерям, раздражительностью и беспомощностью;
— ситуационная депрессия, обычно возникающая при воздействии внешних стрессовых факторов;
— маниакально-депрессивные нарушения, характеризующиеся чередующимися выраженными и неадекватными изменениями со стороны настроения.
Общим свойством всех антидепрессантов является их положительное влияние на аффективную сферу пациента, что сопровождается улучшением настроения и общего эмоционального состояния.
Несмотря на то, что первичные биохимические нарушения, ответственные за депрессию и маниакально-депрессивные состояния, остаются не полностью раскрытыми, некоторые факты позволяют предположить, чго депрессивные состояния могут быть вызваны дефицитом норэпинефрина (норадреналина) и серотонина. Большинство препаратов, применяемых для лечения этих заболеваний, действуют посредством воздействия на систему биогенных аминов мозга, приводя в действие тот пли иной механизм, способствующий увеличению их содержания в со-ответст вующих участках мозга.
Существует 4 класса антидепрессантов:
1) трициклические антидепрессанты (имипрамин, тримипрамин амитриптилин, доксепин, десипрамин, протриптиллин, нор-триптичлин амоксапин, мапротиллин);
2) антидепрессанты второго поколения, или атипичные антидепрессанты, — это разнородная в химическом отношении группа предложенных в последнее время препаратов (бупропион, тразодон, флуоксетин);
3) ингибиторы моноаминооксидазы (МАО) (фенелзин, изокар-боксазид, транилципромин);
4) амфетамины и другие стимуляторы ЦНС (декстроамфетамин, метилфенидат).
Наиболее часто и широко применяемыми препаратами для лечения эндогенных депрессий являются трициклические антидепрессанты. По своему клиническому действию они похожи на фенотиазиновые антипсихотики. Антидепрессанты второго поколения отличаются от первых спектром побочных эффектов. Ингибиторы МАО проявляют мень
- 150-
Анти депрессанты
шую клиническую эффективность, чем трициклические антидепрессанты, и их обычно применяют для лечения острых депрессий, не поддающихся успешному лечению трициклическими антидепрессантами. Наконец, некоторые стимуляторы ЦНС, большинство из которых является производными амфетамина, используются преимущественно в случаях умеренных депрессий, однако их малая эффективность в случаях острых депрессий и выраженная способность вызывать привыкание ограничивают их применение
При выборе препаратов для фармакотерапии депрессий следует учитывать как особенности самих препаратов, так и уровень тяжести и симптоматику заболевания.
7.1. Трициклические антидепрессанты
Наиболее часто используемые препараты — это трициклические антидепрессанты, получившие свое название исходя из общности их химического строения, а именно, системы, состоящей из двух бензольных колец, сочлененных с центральным семичленным кольцом, с присоединенной к центральному кольцу диалкиламиноалкильной группой. В тависимости от заместителей при концевом атоме азота в аминосо-дс-ржащей боковой цепи, последние, в свою очередь, подразделяются на фетичные (имипрамин, амитриптилин, тримипрамин, доксепин) и вторичные (десипрамин, нортриптилин, протиптилин) амины. Данная классификация весьма формальна и не основана на наиболее существенных струмурных различиях рассматриваемых препаратов, однако она принята в фармакологии.
1 рициклические антидепрессанты химически, фармакологически и токсикологически очень похожи на антипсихотики фенотиазинового ряда.
Механизм действия трициклических депрессантов окончательно не выяснен, и ни одна из предложенных сегодня гипотез не в состоянии полностью объяснить их антидепрессантый эффект.
Полагают, что трициклические антидепрессанты ингибируют обратный (нейрональный) захват норэпинефрина (норадреналина) и/или серотонина пресинаптическими нервными окончаниями, блокируя один из ведущих механизмов их инактивации, и тем самым увеличивают концентрацию указанных аминов, потенцируя их эффекты. Следует отметить, что. как правило, вторичные амины — представители трициклических антидепрессантов проявляют большую активность, блокируя ооратный нейрональный захват норэпинефрина, в то время как третичные амины больше влияют на обратный нейрональный захват серотонина.
-151 -
Глава 7
Не исключено также, что трициклические антидепрессанты блокируют пресинаптические а2-адренорецепторы, увеличивая тем самым количество высвобождаемого норэпинефрина и/или серотонина.
Трициклические антидепрессанты применяют для облегчения симптомов депрессии, особенно эндогенного типа, для контроля беспокойства, связанного с депрессивным состоянием, для лечения депрессий \ пациентов с маниакально-депрессивным психозом и т. п.
Третичные амины — представители трициклических антидепрессантов
Имипрамин (Imipramine)
Имипрамин — 5-[3-(диметиламино)пропил]-10,11-дигидро-5Я-ди-бенз[Ь,1]азепин (7.1.1) получают алкилированием 10,11-дигидро-5//-дибенз[Ь,Г]азепина 3-диметиламинопропилхлоридом в присутствии амида натрия [1, 2, 3].
сн3
а—сн2 -сн2 -сн2—n у
СНз
NaNHj
СНз
СНз
Имипрамин является основным представителем типичных трициклических антидепрессантов. Препарат действует путем блокады механизма обратного захвата биогенных аминов. Не ингибирует активность .МАО. Имипрамин уменьшает тоску. двигательную заторможенность, улучшает настроение, повышает психический и общий тонус организма. Его применяют при депрессивных состояниях различной этиологии, сопровождающихся моторной и илеаторной заторможенностью, при ночном недержании мочи у детей, при паркинсонизме.
Основными синонимами препарата являются тофранил, сурпликс, имизин и мелипрамин и др.
Тримипрамин (Trimipramine)
Тримипрамин — 5-[3-(диметиламино)-2-метилпропил]-10,11-дигидро-5Н-дибенз[Ь,Г]азепин (7.1.2) получают совершенно аналогично имип-рамину, но алкилируя 10,11-дигидро-5Я-дибенз[Ь,1]азепин 3-диметил-а.мино-2-мегилпропилхлоридом [4. 5].
-152 -
Антидепрессанты
N
I /Из
СН2-СН-СНг-N\
71 2 СНз СНз
Так же как и имипрамин, его применяют при депрессивных состояниях различной этиологии. По эффективности препарат аналогичен имипрамину.
Синонимом тримипрамина является сурмонтил.
Амитриптилин (Amitriptyline)
Амитриптилин — 5-(3-диметиламинопропилиден)-10,11-дигидро-дибенз цикло гептен (7.1.4), который отличается от имипрамина тем, что атом азота в центральной части трициклической системы заменен атомом углерода, связанным с боковой цепью двойной связью. Амитриптилин (7.1.4) синтезируют взаимодействием 10,11-дигид-ро-№Л’-диметт-5//-дибензо[а,(1]-циклоге1[тен-5-она с 3-диметилами-нопропилмагнийбромидом с последующей дегидратацией полученного третичного спирта (7.1.3) соляной кислотой [6, 7, 8, 9, 10, 11].
СНз
1
СНз
<
CHj
HCI
Альтернативным способом синтеза является взаимодействие 10,11-лигидро-1\’,М-диметил-5Я-дибензо[а,с1]-циклогептен-5-она с циклопро-пилмагний бромидом с получением 10,11-дигидро-К,1\т-диметил-5Я-Дибензо[а,с1]-циклогептен-5-циклопропил-5-ола (7.1.5). Взаимодействием последнего с бромистым водородом в уксусной кислоте осуществляют раскрытие циклопропильного кольца с получением 5-(3-бромпро-
-153 -
Глава 7
пилиден)-10,11-дигидро-5Я-дибензо[а,с1]циклогептена (7.1.6). Алкилируя последним диметиламин, получают амитриптиллин (7.1.4) [12, 13].
Амитриптиллин применяют при тревожно-депрессивных состояниях. Он переносится лучше, чем имипрамин.
Наиболее часто встречающимися синонимами являются триптизол и амиприн.
Доксепин (Doxepin)
Доксепин — (11 [16Я]-(3-ди.метиламинопропилиден)-6,11-дигидро-бенз[Ь.е]оксепин (7.1.11) получают по аналогичной схеме взаимодействием 6,11-дигидродибенз-[Ь,е]оксепин-11-она (7.1.9) с 3-диме-тиламинопропилмагнийбромидом с последующей дегидратацией полученного третичного спирта (7.1.10) соляной кислотой [14, 15, 16, 17].
на
Исходный 6,11-дигидродибенз[Ь,е]оксепин-11-он (7.1.9) синтезируют исходя из этилового эфира 2-феноксиметил бензойной кислоты (7.1.7), который легко получают взаимодействием этилового эфира 2-бромметилбензойной кислоты и фенола в присутствии основания.
- 154 -
Антидепрессанты
Полеченный эфир (7.1.5) гидролизуют до 2-феноксиметилбензойной кислоты (7.1.8), которую циклизуют в 6,11-дигидродибенз[Ь,е]оксепин-11-он (7.1.9) действием ангидрида трифторуксусной кислоты.
^z?x^CH2Br
№ОН
NaOH
7 1 7
Механизм действия доксепина также предположительно связан с влиянием на адренергическую передачу в ЦНС, в частности, с блокадой нейронального захвата норэпинефрина. Доксепин применяют при тревожно-депрессивных и беспокойных состояниях, неврозах, алкоголизме, органических заболеваниях ЦНС. психозах.
Наиболее часто встречающимися синонимами являются адапин и синекван.
Вторичные амины — представители трициклических антидепрессантов
Дезипрамин (Desipramine)
Дезипрамин — 10,11-дигидро-5-[3-(метила.мино)пропил]-5/7-дибенз-[Ь.1]азепин (7.1.13) отличается от имипра.мина наличием лишь одной дополнительной метильной группы при атоме азота боковой пропиламинной цепи. Предложенные способы синтеза дезипрамина весьма просты, и разница состоит лишь в способе введения вторичной метиламинной группы в структуру препарата.
Первый способ синтеза заключается в алкилировании 10,11-дигидро]-5Я-дибенз[Ь,Г]азепина 1-бром-З-хлорпропаном в присутствии амида натрия в хлорпроизводное (7.1.12) и последующем его взаимодействии с метиламином с получением дезипрамина (7.1.13) [18, 19, 20].
-155-
Г лава 7
NaNH2
<- Cl—СН2 -СН2 -СН2 -Вт -----1
7 1 13
Второй способ заключается в алкилировании 10,11-дигидро]-5Я-ди-бенз[Ь,Г]азепина 3-(К’-бензил-М-пропиламино)пропил хлоридом в присутствии амида натрия и последующим дебензилированием полученного продукта (7.1.14) путем восстановления водородом над палладиевым катализатором [21,22].
NaNHj
Н2/ Pd-C
Наконец, при третьем способе исходят из имипрамина (7.1.1), который подвергают деметилированию последовательным взаимодействием с хлоругольным эфиром с получением 5-[3-(И-карбэтокси-ЬН метил)амиинопропил]-10,11-дигидро-5Н-дибенз-[Ь,1]азепина (7.1.15), щелочной гидролиз которого приводит к дезипрамину (7.1.13) [23, 24].
С1СООС2Н5
кон
7 1 13
Антидепрессанты
Дезипрамин применяется при депрессивных состояниях различной этиологии и, в частности, при эндогенных депрессиях.
Синонимами являются норпрамин и пертофран.
Нортриптилин (Nortriptyline)
Нортриптилин — 5-(3-метиламинопропилиден)-10,11-дигидродибензцикл о гептен (7.1.17). Нортриптилин отличается от дезипрамина геми же признаками, которые отличают амитриптилин от имипра-мина, а именно: в нортриптилине атом азота в центральной части трициклической системы дезипрамина заменен атомом углерода, связанным с боковой цепью двойной связью.
Две предложенные схемы для синтеза нортриптилина основаны на N-деметилировании амитриптилина (7.1.4). Третья схема предполагает взаимодействие метиламина с 5-(3-бромпропилиден)-10,11-дигидро-5Я-дибенз[а.с1] циклогептеном (7.1.18).
Согласно первому способу, деметилирование осуществляют путем взаимодействия амитриптилина (7.1.4) с метилйодидом, что часто приводит к получению четвертичной аммониевой соли (7.1.16), взаимодействие которой с метиламином при относительно высокой температуре дает искомый нортриптилин (7.1.17) [25].
СН31
ch3nh2
I
7 1 17
Во втором способе предусмотрено взаимодействие амитриптилина 17.1.4) с хлоругольным эфиром, приводящее к замещению мезильной группы в аминной части молекула на этоксикарбонильную (7.1.18), с последующим гидролизом образовавшегося продукта в нортриптилин (7.1.17) [26].
- 157-
Глава 7
CICOOC2HE
7 1 17
Согласно третьему способу, нортриптилин получают взаимодействием метиламина с 5-(3-бромпропилиден)-10,11-дигидро-5Я-дибенз[а,с1] циклогептеном (7.1.6) [8].
Предложено еще несколько модификаций описанных выше способов получения нортриптилина [27, 28, 29, 30, 31, 32].
Нортриптилин — препарат с относительно коротким латентным периодом действия. Он практически не обладает седативным эффектом. Его применяют при маниакально-депрессивных психозах, при всех формах эндогенных депрессий, а также при больших депрессивных состояниях.
Наиболее распространенными синонимами нортриптилина являются авентил, нортрилен, мотивал, вивидил и памелор.
Протриптилин (Protriptyline)
Протриптилин — \’-метил-5Я-дибензо[а,с!]циклогептен-5-пропил-амин (7.1. 22) отличается от всех описанных выше препаратов тем, что в положение Ci0-Cn центрального семичленного кольца трициклического фрагмента молекулы введена двойная связь. В тоже время из положения С5 исключена свободная электронная пара, принадлежащая либо атому азота, либо экзоциклической двойной связи, что несомненно поменяло как архитектуру всей молекулы в целом, так и расположение в ней фармакофорных участков.
Протриптилин синтезируют алкилированием 5Я-дибензо[а.<1]цик-логептена 3-(М-формил-1Х-метиламино)пропилхлоридом (7.1.20), получаемым, согласно приведенной схеме, из соединения (7.1.19). Полученный полупродукт (7.1.21) подвергают щелочному гидролизу, что приводит к получению протриптилина (7.1.21) [33. 34, 35, 36, 37, 38].
-158-
Антидепрессанты
HO-CH2-CH2-CH2-NH —СНз
h2ncho
ho-ch2-ch2-ch2-n(
SOCI2
ci—ch2-ch2-ch2~n (
CH2-ch2-ch2-n(
CH2 -CH2 —CH2 —N H —CH3
7 1 22
Протриптилин является мощным антидепрессантом, механизм действия которого неясен. Он не является ингибитором МАО и не стиму-лир\ет ЦНС. Препарат начинает действовать намного быстрее и действует намного дольше, чем имипрамин или амитриптилин. Протриптилин не имеет седативных и транквилизирующих свойств. Препарат применяют в условиях клиники для лечения глубоких депрессий.
Наиболее распространенными синонимами являются конкордин, триптил и вивактил.
Мапротилин (Maprotiline)
Мапротилин — Т\*-метил-9.10-этаноантрацен-9(10/7)-пропиламин (7 1.22) получают реакцией 4-г2 циклоприсоединения 9-(3-метил-а.минопропил)антрацена с этиленом [39,40. 41].
Н2С=СН2 |Т|
СН2 - СН2 -СН2 -NH —СНз
CH2-CH2-CH2-NH —СНз
7 1 22
Мапрошлин часто называют тетрациклическим антидепрессантом. Этот «гибридный» препарат, имеющий как элементы классических трициклических антидепрессантов, так и элементы протриптилина, по своим фармакологическим и клиническим проявлениям больше напоминает имипрамин.
- 159-
Глава 7
Препарат нарушает нейрональный захват моноаминов в ЦНС, обладает умеренной транквилизирующей и холинолитической активностью. Значительно повышает настроение, уменьшает чувство страха. Мапро-ти.тин применяется при различных формах депрессий, сопровождающихся чувством страха, раздражительностью.
Синонимом препарата является людиомил.
7.2. Ингибиторы МАО
МАО — это сложная ферментная система, присутствующая практически во всех органах, которая катализирует дезаминирование или инактивацию различных природных биогенных аминов и, в частности, норэпинефрина (норадреналина), эпинефрина (адреналина) и серотонина. Ингибирование МАО повышает количество упомянутых, биогенных аминов в нервных окончаниях. Очевидно, ингибируя их дезаминирование. ингибиторы МАО повышают внутриклеточную концентрацию эндогенных аминов, что, по-видимому, и является причиной их антиде-прессантного действия.
Препараты, которые образуют стабильные комплексы с МАО и тем самым ингибируют ее действие, давно применяются в медицине в качестве антидепрессантов и называются ингибиторами МАО. (Не исключено. что ингибиторы МАО действуют не путем комплексообразования с ней, а путем образования ковалентных связей, необратимо инактивируя ее.)
С большой осторожностью, с химической точки зрения, эти препараты можно охарактеризовать как производные гидразина, что однозначно следует из рассмотрения структур фенелзина и изокарбоксазида и из параллельного рассмотрения вместе с ними структуры транилами-на, отвлекшись при этом от символа атомов азота и представив электронное строение циклопропана.
Механизм антидепрессивного действия этого ряда препаратов вероятно связан с торможением ими процесса окислительного дезаминирования нейромедиаторов (норэпинефрина, эпинефрина, дофамина и серотонина). участвующих в передаче нервного возбуждения в ЦНС.
Существенным недостатком этих препаратов является их высокая токсичность, связанная с тем, что они ингибируют не только МАО, но и ряд других неспецифических ферментов.
Ингибиторы МАО применяются для лечения острых эндогенных, экзогенных и реактивных депрессий, не поддающихся лечению трициклическими антидепрессантами, а также для контроля депрессивной фазы при маниакально-депрессивных психозах.
-160-
Антидепрессанты
Фенелзин (Phenelzine)
Фенелзин — 2-фенилэтилгидразин (7.2.1) получают взаимодействием 2-фенилэтилбромида с гидразином [42, 43, 44, 45].
СН2-СН2—Вг + H2N-NH2 ---------► CH2-CH2-NH-NH2
7 2 1
Фенелзин — ингибитор МАО, который применяют для лечения пациентов с депрессиями, характеризуемыми как атипичные, неэндогенные или невротические, у которых часто наблюдается смешанные беспокойство и депрессия или фобии. Фенелзин не является препаратом первого выбора, поэтому его используют при депрессивных состояниях, не поддающихся контролю другими лекарственными средствами.
Синонимом препарата является нардил.
Изокарбоксазид (Isocarboxazide)
Изокарбоксазид — 2-бензилгидразид 5-метил-З-изоксазолкарбо-новой кислоты (7.2.6) можно синтезировать исходя из апетонилапе-тона, при нитрозировании которого азотистой кислотой получается 5-метил-изоксазол-З-карбоновая кислота (7.2.2). Этерификацией последней получают этиловый эфир 5-метил-изоксазол-З-карбоно-вой кислоты (7.2.3). Синтезированный эфир (7.2.3) далее вводят во взаимодействие с бензилгидразином с получением изокарбоксазида (7.2.6) либо с гидразином с получением гидразида 5-метил-изо-ксазол-3-карбоновой кислоты (7.2.4). Вводя последний в реакцию с бензальдегидом, получают гидразон (7.2.5), который далее восстанавливают в изокарбоксазид (2.2.6) [46, 47].
О О соон II 11 HNO2 п Z Н3С СН2 CHj СНз *" 11 n НзС^О' 722 сно CONHNH2 [Ц CONHN=CH- — «V 724 725 С2Н5ОН'Н+ СООС2Н5 •“ 11 H2NNH2» Н3С О 723 ch2-nhnh2 -С') CONHNH-С р '=' L1ALH4 п if н3с^ол 726
Глава 7
Изокарбоксазид является мощным ингибитором МАО. Как и фенел-зин. изокарбоксазид используют при депрессивных состояниях, не поддающихся контролю другими препаратами.
Синонимом препарата является марплан.
Транилципромин (Tranylcypromine)
Транилципромин — (±)-транс-2-фенилциклопропиламин (7.2.10) в отличие от вышеописанных препаратов не является производным гидразина. Синтез его осуществляют из этилового эфира 2-фенил-циклопропанкарбоновой кислоты (7.2.7), которую получают взаимодействием стирола с диазоуксусным эфиром. Эфир 2-фенилцик-лопропанкарбоновой кислоты (7.2.7) гидролизуют щелочью в 2-фе-нилциклопропанкарбоновую кислоту (7.2.8), и выделяют транс-изомер, с которым и продолжают работу. Взаимодействием транс-изомера кислоты с хлористым тионилом получают хлорангидрид /нранс-2-фенилциклопропанкарбоновой кислоты (7.2.9), из которого взаимодействием с азидом натрия получают соответствующий азид кислоты, перегруппировывающийся в условиях реакции Курциуса в транилципромин (7.2.10) [48, 49].
7 2 7
1 NaN3
2 HCI t
Как и вышеописанные препараты, ингибитор МАО транилципромин также используют при депрессивных состояниях, не поддающихся контролю другими препаратами.
Синонимами препарата являются трансамин, пармодалин, парнат И др.
- 162 -
Антидепрессанты
7.3. Антидепрессанты второго поколения (атипичные антидепрессанты)
Это ряд химически неоднородных соединений, не классифицируемых и не относящихся ни к классу трициклических антидепрессантов, ни к классу ингибиторов МАО, проявляют весьма эффективную анти-депрессивную активность.
Весьма вероятно, что их действие также обусловлено способностью ингибировать захват норэпинефрина или серотонина. Однако ввиду разнородности группы возможный механизм действия каждого будет рассмотрен отдельно.
Амоксапин (Amoxapine)
Амоксапин — 2-хлор-11-(1-пиперазинил)-дибенз[Ь,1]оксазепин (7.3.2) является непосредственным аналогом нейролептика локсапина (6.5.3), отличаясь от последнего лишь отсутствием метильной группы в пиперазиновом фрагменте молекулы. С другой стороны, его можно отнести и к классу трициклических антидепрессантов, основное различие которых заключается в месте присоединения боковой цепи к центральному семичленному кольцу трициклической системы. Синтез амоксапина, как и локсапина, осуществляют исходя из 2-(4-хлорбензокси)анилина, который, как и в случае синтеза локсапина, ацилируют хлоругольным эфиром в (6.5.1) и далее трансформируют в уреид (7.3.1) взаимодействием с 1-карбэтокси-пиперазином. Циклизацией уреида (7.3.1) смесью пятиокиси и хлорокиси фосфора в дибензоксазепин и последующим щелочным гидролизом получают амоксапин (7.3.2) [50, 51, 52, 53].
Р2О5' РОС13
Антидепрессивное действие амоксапина сравнимо с имипрамином и амитриптилином. Препарат проявляет антагонистическую активность в отношении дофаминовых (D2) рецепторов.
-163-
Глава 7
Препарат больше предназначен для облегчения симптомов у пациентов с невротическими или реактивными депрессиями, чем с атипичными депрессиями. Обладает рядом серьезных побочных действий.
Синонимами препарата являются асендин, амоксан, моксадил и др.
Бупропион (Bupropion)
Бупропион — 1 -(3-хлорфенил)-2-[( 1,1 -диметилэтил)амино]-1 -пропанон (7.3.5) синтезируют исходя из 3-хлорбензонитрила, взаимодействием которого с этилмагнийбромидом получают 3-хлорпро-пиофенон (7.3.3). Бромируя последний молекулярным бромом, получают 3-хлор-а-бромпропиофенон (7.3.4), взаимодействием которого с третбутиламином получают бупропипон (7.3 5) [54, 55, 56, 57, 58].
Бупропион является а-аминокетоном, структурно родственным амфетаминам, проявляет уникальную активность, сравнимую с другими антидепрессантами. Полагают, что бупропион восстанавливает общее количество циркулирующего в организме норэпинефрина. Это соединение слабо ингибирует обратный захват дофамина, не проявляет антихо-линергической активности и ингибирует МАО.
Его эффективность в качестве антидепрессанта сравнима с таковой у трициклических антидепрессантов и с ингибитором захвата серотонина — флуоксетином. Препарат предпочтительно назначают при больших депрессиях с ослаблением психомоторной деятельности.
Синонимами препарата являются амфебугамон и веллбутрин.
Флуоксетин (Fluoxetin)
Флуоксетин — 3-[и-(трифторметил)-фенокси]-?4-метил-3-фенилпро-пиламин (7.3.6) получают взаимодействием п-трифторметилфенола с 3-(хлор)-Ы-метил-3-фенилпропиламином в присутствии поташа [59, 60].
Антидепрессанты
Cl
ch-ch2-ch2-nh-ch3
736
Флуоксетин — фенилпропиламин ингибирует обратный нейрональный захват серотонина, что предположительно, имеет прямое отношение к антидепрессантному действию. Это же соединение не воздействует или мало воздействует на обратный нейрональный захват норэпинефрина или дофамина. В дополнение препарат не связывается с холинергическими, гистаминергическими или а-адренергическими рецепторами, что, полагают, является причиной побочных эффектов трициклических антидепрессантов.
Эффективность флуоксетина при лечении пациентов с умеренными депрессиями сравнима с эффективностью трициклических антидепрессантов. Препарат способствует повышению настроения, устраняет чувство страха и напряжения. Он не обладает седативным эффектом. Флуоксетин применяют при депрессиях, а также при булемических неврозах. Выбор флуоксетина, тем не менее, предпочтителен в случаях, когда пациенту противопоказаны седативные, гипотензивные и ан-тихолинергические побочные эффекты, вызываемые другими антидепрессантами.
Синонимом препарата является прозак.
Тразодон (Trazodon)
Тразодон — 2-[3-[4-(.м-хлорфенил)-1-пиперазинил]пропил]-5-триа-золо[4,3-а]пиридин-3(2Я)-он (7.3.8) синтезируют исходя из 2-хлор-пиридина, взаимодействием которого с семикарбазидом получают з-триазол-3-он[4,3-а]пиридин (7.3.7). Алкилирование последнего 1-(3-хлорпропил)4-(3-хлорфенил)пиперазином дает тразодон (7.3.8) [61.62].
Hzt.-HN-
- 165-
Г лава 7
Полагают, что тразодон в терапевтических дозах ингибирует обратный нейрональный захват серотонина. Он не является ингибитором МАО, не является стимулятором ЦНС. Препарат мало воздействует на обратный нейрональный захват норэпинефрина или дофамина. В дополнение к отмеченному препарат не связывается с холинергическими, а-адренергическими рецепторами.
Синонимами препарата являются томбран, прагмарел, дезирел и др.
7.4. Амфетамины и другие стимуляторы ЦНС
Амфетамины — синтетические симпатомиметические амины являются мощными стимуляторами ЦНС, и некоторые из них, в частности декстроамфетамин (8.1.2.2) и метилфенидат (8.1.2.6), иногда применяются для лечения депрессивных состояний. Они повышают настроение, стимулируют двигательную активность, бдительность, позволяют лучше концентрироваться. Однако, в зависимости от дозы и личности пациента, могут вызвать и разную степень эйфории и часто приводят к зависимости и привыканию.
8 122
Число разрешенных к медицинскому применению амфетаминов весьма ограничено, и детально их синтез и свойства будут рассмотрены в гл. 8 «Стимуляторы ЦНС».
Список литературы
1 US Pat. 2.554.736 (1951).
2. Ger Pat. 829.167 (1950).
3. Gaefliger .4. et al. 'Helv. Chim. Acta. 37, 472 (1954).
4. Fr. Pat. 1.172.014 (1955).
5. Jacob R Compt. Rend. 252, 2117 (1961).
6. Brit. Pat. 858.187 (1959).
7. Brit. Pat. 858.188 (1959).
8. Hoffsommer R. et al.7J. Org. Chem. 27, 4134 (1962).
- 166-
Антидепрессанты
9. Hoffsommer R. et al.//J. Org. Chem. 28. 1751 (1963).
10. Hoffsommer R. et al./J. Med. Chem. 8, 555 (1965).
11. Belg. Pat. 584.061 (1960).
12. US Pat. 3.205.264 (1965).
13 Ger. Pat. 1.468.138 (1963).
14. US Pat. 3.420.851 (1969).
15. Ger. Pat. 1.232.161 (1961).
16. Stach R. et al.//Monatsh. 93, 896 (1962).
17. Bickelhaupt F. et al.//Monatsh. 95,485 (1964).
18. Fr. Pat. M 796 (1960).
19. Brit. Pat. 908.788 (1960).
20. Ger. Pat. 1.189.550(1960).
21. US Pat. 3.454.698 (1969).
22. US Pat. 3.454.554 (1969).
23. Ger. Pat. 1.288.599(1962).
24. Ger. Pat. 1.445.800(1962).
25. Ft. Pat. 1.345.936(1963).
26. Ger. Pat. 1.288.599 (1962).
27. Ger. Pat. 1.266.755 (1962).
28. Belg. Pat. 628.904 (1963).
29. Ger. Pat. 1.269.614 (1962).
30. US Pat. 3.281.469(1962).
31. US Pat. 3.215.739(1960).
32. US Pat. 3,372.196 (1968).
33. US Pat. 3.244.748 (1966).
34. US Pat. 3.271.451 (1966).
35. Belg. Pat. 617.967 (1962).
36. Ger. Pat. 1.287.573 (1963).
37. Ger. Pat. 1.468.212 (1962).
38. Engelhardt E. et al.//J. Med. Chem. 11, 325 (1968).
39. Ger. Pat. 1.518.691 (1965).US Pat. 3.399.201 (1968).
40. Wilhelm M et al.//Helv. Chim. Acta. 52, 1385 (1969).
41. US Pat. 3.000.903 (1961).
42. Votocek .4. /Coll. Czech. Chem. Commun. 4, 271 (1932).
43. Biel J. et al. /J. Am. Chem. Soc. 81, 2805 (1959).
44. US Pat. 2.908.688 (1959).
45. Gardner D et al./.'J. Med. Chem. 2, 133 (1960).
46. US Pat. 2.997.422 (1961).
47. US Pat. 4.016.204(1977).
-167 -
Г лава 7
48. Burger A et al. ''J. Am. Chem. Soc. 70, 2198 (1948).
49. USPat. 3.663.696 (1972).
50. Schmut-iz J etal.'.Helv. Chim. Acta. 50, 2455 (1967).
51. Fr Pat. 1.508.536(1968).
52. SchmultzJ et al..' Chim. Ther. 2, 424 (1967).
53. Get. Pat. 2.059.618 (1970).
54. Ger. Pat. 2.064.934 (1970).
55. Can. Pat. 977.778 (1970).
56 USPat. 3.819 706 (1974).
57. USPat. 3.885.046 (1975).
58. Ger. Pat. 2.500.110 (1975)
59. US Pat. 4.314.081 (1982).
60. USPat 3.381.009 (1968).
61. Ger. Pat. 1.645.947 (1966).
Глава 8
Стимуляторы ЦНС
Огромное число физиологически активных веществ оказывает стимулирующее действие на ЦНС, однако число используемых с этой целью лекарственных препаратов в медицине весьма ограничено.
Собственно стимуляторами ЦНС или психостимуляторами считаются вещества, повышающие бдительность и понижающие потребность во сне, т. е. способствующие временному бодрствованию, повышающие настроение и способность к адекватному восприятию действительности и внешних раздражителей, уменьшающие чувство усталости и повышающие физическую и умственную работоспособность. Это препараты, применяемые по жизненным показаниям.
Некоторые стимуляторы ЦНС, такие как амфетамины и метилфенидат, иногда используются для улучшения настроения у пациентов с депрессией. Однако в отличие от антидепрессантов, рассмотренных в гл. 7 «Антидепрессанты», эти соединения лишь повышают планку уровня возбудимости ЦНС и никак не действуют на депрессию, поэтому следует различать термины «антидепрессант» и «психостимулятор».
Стимуляторы ЦНС можно классифицировать следующим образом.
Психомоторные стимуляторы — соединения, оказывающие стимулирующее влияние преимущественно на функции головного мозга и активирующие психическую и физическую деятельность организма. К ним относятся метилксантины (кофеин, теофиллин, пентоксифиллин); амфетамины (декстроамфетамин, метамфетамин). а также метилфенидат и пемолин.
Стимуляторы дыхания, или аналептики, — соединения, которые наряду с определенной активацией психической и физической деятельности организма возбуждают преимущественно сосудодвигательный и дыхательный центры продолговатого мозга (док-сапрам, алмитрин).
Препараты, уменьшающие аппетит, или аноректики, — лекарственные средства, активирующие психическую и физическую деятельность организма, но преимущественно и акцентированно возбуждающие центр насыщения в гипоталамусе (фентермин, диэтил пропион).
-169-
Глава 8
Иногда с целью повышения умственной работоспособности применяются и ноотропы — препараты, улучшающие функциональное состояние мозга. Их действие связано с кровоснабжением и метаболизмом головного мозга.
8.1. Психомоторные стимуляторы
8.1.1. Метилксантины
Кофеин (Caffeine)
Кофеин — 1,3,7-триметилксантин (23.3.6) — наиболее широко используемый стимулятор ЦНС. Это алкалоид, содержащийся в листьях чая {Thea sinensis), в семенах кофе {Coffea arabica), в семенах какао {Theobroma cacao), в семенах кола {Cola acuminata) и других растениях, синтез которого будет описан в гл. 23 «Препараты для лечения заболеваний дыхательной системы».
СНз
23 3 6
Чашка кофе может содержать от 50 до 150 мг кофеина, напитки семейства кола — от 35 до 55 мг. К числу метилксантинов относятся теофиллин — 1,3-диметилксантин — основной (характерный) алколо-ид чая и теобромин — 3,7-диметилксантин (23.3.19) — основной алкалоид какао. В малых дозах кофеин является относительно слабым психостимулятором и используется для повышения внимания, а также облегчения головных болей, связанных с расстройствами мозгового кровообращения.
Кофеин обладает стимулирующим действием на дыхательный и сосудодвигательный центры, возбуждает центры блуждающих нервов. Он оказывает прямое стимулирующее влияние на миокард, может в больших дозах вызвать тахикардию и аритмию.
Кофеин оказывает двоякое действие на уровень артериального давления (АД). Центральным механизмом — путем возбуждения сосудодвигательного центра препарат повышает уровень АД. Прямым воздействием на гладкую мускулатуру сосудистой стенки способствует
-170-
Стимуляторы ЦНС
расширению сосудов.
На лиц с нормальным АД препарат практически не действует, однако при введении его на фоне гипотензии артериальное давление повышается (нормализуется). Под влиянием кофеина повышается секреция желез желудка.
Полагают, что стимулирующее действие кофеина связано с его способностью конкурентно связываться с рецепторами аденозина — фактора, уменьшающего процессы возбуждения в мозге. Замещение его кофеином приводит к стимулирующему эффекту, поскольку метилксан-тины и аденозин вызывают противоположно направленные эффекты. Согласно другой точке зрения, ингибируя фосфодиэстеразу, кофеин увеличивает концентрацию циклического аденозинмонофосфата (цАМФ), рассматриваемого как вторичный медиатор, при помощи которого осуществляются физиологические эффекты многих биологически активных веществ. В частности, под влиянием цАМФ усиливаются процессы гликогенолиза, стимулируются метаболические процессы в разных органах и тканях, в том числе и в ЦНС.
Кофеин применяют для стимулирования психической деятельности, при утомлении, мигрени, гипотензии.
Синонимы препарата не популярны. К ним относятся кафекон, коффан и некоторые другие.
8.1.2. Амфетамины, а также метилфенидат и пемолин
Амфетамины являются мощными синтетическими психостимуляторами с очень большим потенциалом привыкания. Они повышают бдительность и способность концентрироваться, временно улучшают настроение и стимулируют двигательную активность. Однако, в зависимости от дозы и, что очень важно, личности, они могут вызвать разную степень эйфории, .могут повышать АД, способствовать сокращению сфинктера мочевого пузыря и развитию мидриаза. Длительное применение амфетаминов часто приводит к раздражительности, бессоннице, потливости. При отмене препаратов может возникнуть депрессия. Попытки выйти нз депрессивного состояния с применением увеличенных доз тех же амфетаминов приводит к привыканию и зависимости с образованием порочного круга. Применение еще больших доз препарата вызывает эйфорию, галлюцинации и другие психотические эффекты с симптомами, весьма сходными с клиническими симптомами параноидной формы шизофрении.
-171 -
Глава 8
Полагают, что механизм действия амфетаминов заключается в их способности высвобождать из пресинаптических окончаний эпинефрин (адреналин) и дофамин, стимулирующие соответствующие рецепторы в ЦНС. Не исключено также, что они уменьшают нейрональный захват указанных аминов, а также ингибируют их расщепление МАО. Характерным для этого ряда соединений является и влияние на дыхательный центр, на расположенный в гипоталамусе центр насыщения, что приводит к подавлению чувства голода, позволяя использовать аналоги рассматриваемых соединений в качестве анорекзиков.
Наряду> с ЦИС соединения данной группы влияют йа нервную систему. Они опосредованно стимулируют а- и Р-адренорецепторы. Адреномиметические свойства этих соединений схожи со свойствами норэпинефрина (норадреналина), однако во много раз уступают ему по активности.
По химическому строению амфетамины весьма близки к эпинефрину (адреналину) и норэпинефрину (норадреналину), дофамину, отличаясь от них отсутствием гидроксильных групп в ароматическом кольце и алифатической цепи.
сн3 СЩ-С-NHR-i
R
эпинефрин
сн2-сн2-мн2
дофамин
амфетамин
Амфетамины используют при лечении нарколепсии, для повышения работоспособности, при утомлении, лечении синдрома отсутствия внимания у детей, а также при лечении ожирения. Эффекты амфетаминов могут потенцироваться трициклическими антидепрессантами, ингибиторами МАО, ацетазоламидом, кокаином, фуразолидоном, пропоксифеном, бикарбонатом натрия и другими препаратами, подщелачивающими мочу. Одновременно антагонистами амфетаминов являются препараты, подкисляющие мочу, — аскорбиновая и глутаминовая кислоты. фенотиазины, галоперидол, мефенамин. препараты лития, фруктовые соки.
Декстроамфетамин (Dextroamphetamine)
Декстроамфетамин — Э-2-амино-1-фенилпропан (8.1.2.2) предложено получать разными методами. Первый способ заключается в использовании методики реакции Лейкарта для взаимодействия метилбензилкетона и формиата аммония с получением формамида
- 172-
Стимуляторы ЦНС
(8.1 2.1), который гидролизуют в 2-амино-1-фенилпропан (8.1.2.2) соляной кислотой [1]. Аналогичная методика предложена с использованием формамида вместо формиата аммония [2].
HCOONHj
СН3 сн2-сн-мн2
8 122
Второй способ заключается в моноалкилировании аммиака 2-хлор-1-фенилпропаном [3].
сн3
СН2—CH—Cl
NH3
СН3 сн2-CH-nh2
8 122
Согласно третьему способу, 2-амино-1-фенилпропан (8.1.2.2) предложено получать реакцией Гофмана из амида а-бензилпропионовой кислоты [4, 5].
СНз ch2-ch-conh2
NaOBr
СН3
СН2—CH—nh2
8122
Полученный любым из перечисленных методов 2-амино-1-фенил-пропан (амфетамин) разделяют на изомеры с помощью D-винной кислоты, выделяя требуемый декстроамфетамин — В-2-амино-1-фенилпро-пан(8.1.2.2) [6, 7].
Декстроамфетамин является мощным стимулятором нервной системы, оказывающим свое действие путем высвобождения из пресинап-тических нервных окончаний дофамина и норэпинефрина, стимулируя тем самым центральные дофаминергические и норадренергические рецепторы. В определенных дозах препарат усиливает процесс возбуждения в ЦНС, уменьшает утомляемость, улучшает настроение и работоспособность, понижает потребность во сне, уменьшает аппетит. Декстроамфетамин следует применять с осторожностью и только по медицинским показаниям при лечении нарколепсии, последствий энцефалита и других заболеваний, сопровождающихся апатией, сонливостью, астенией, для временного повышения физической и умственной
- 173 -
Г лава 8
работоспособности, для лечения синдрома отсутствия внимания у детей, а также при лечении ожирения.
Синонимами препарата являются D-амфетамин, дексамфетамин, дексалон, темподекс, зенидекс и многие другие.
Метамфетамин (Methamphetamine)
Метамфетамин — ( ')-К-а-диметилфенилэтиламин (8.1.2.3) можно поле чать восстановлением (-)-эфедрина водородом с использованием в качестве катализатора палладия на угле [8].
Н2 / Pd-C
СНз сн2—CH—NHCH3
8 12 3
Другим способом получения метамфетамина является восстановление метилбензилкетона водородом в присутствии метиламина [9].
о сн2-с-сн3
CH3NH2/ н2
СНз ch2-ch-nhch3
8 12 3
Препарат повторяет свойства декстроамфетамина и применяется по тем же показаниям.
Синонимами препарата являются перитин, филопон, дзоксин, метам пеке и др.
Метилфенидат (Methylphenidat)
Метилфенидат — метиловый эфир а-фенил-2-пиперидилук-сусной кислоты (8.1.2.6), получают по следующей схеме. Арили-рованием бензилцианида 2-хлорпиридином в присутствии основания получают а-фенил-а-(2-пиридил)ацетонитрил (8.1.2.4). Сернокислым гидролизом нитрильной группы в кислотную и последующей ес этерификацией метанолом получают метиловый эфир а-фенил-а-(2-пиридил)пиридилуксусной кислоты (8.1.2.5). Пиридинный фрагмент в последнем восстанавливают в пиперидиновый водородом над платиной с получением метилфенидата (8.1.2.6) [10, 11, 12].
- 174 -
Стимуляторы ЦНС
8 1 2.4
1 H2SO4
2 СН3ОН / HCI
Метилфенидат является стимулятором ЦНС сходным с амфетаминами, однако в обычных дозах имеет более выраженное действие на умственную деятельность, чем на физическую или двигательную. В терапевтических дозах он не повышает кровяное давление, скорость дыхания или скорость работы сердца. Все эти эффекты, а также ряд других, связанных с общим возбуждением ЦНС (тремор, тахикардия, гиперпирексия, состояние замешательства), возникают при применении больших доз. Применяется для лечения средних депрессий и апатичных состояний, а также в качестве дополнительного средства при лечении синдрома отсутствия внимания у детей.
Синонимами препарата являются меридил, риталин и др.
Пемолин (Pemolin)
Пемолин — 2-амино-5-фенил-2-оксазолин-4-он (8.1.2.7) получают конденсацией этилового эфира миндальной кислоты с гуанидином [13, 14].
/=\ 9Н
СН-СООС2Н5 +
NH h2n-c-nh2
8 1 2.7
Пемолин — уникальный по структуре стимулятор ЦНС, проявляющий минимум симпатомиметических эффектов, фармакологически повторяет свойства амфетаминов и метилфенидата, но имеет меньший потенциал к привыканию, чем другие стимуляторы ЦНС. Препарат повышает бдительность и двигательную активность и вызывает слабую
- 175-
Глава 8
эйфорию, что, возможно, связано с увеличением дофаминергических передач в структурах ЦНС.
Пемолин применяют при нарколепсии и повышенной сонливости, а также при лечении синдрома отсутствия внимания у детей.
Синонимами препарата являются традон, дельтамин, волитал. феноксазол, антимеран, цилерт и др.
8.2. Стимуляторы дыхания, или аналептики
Аналептики — препараты, стимулирующие действие на дыхательный и сосудодвигательный центры продолговатого мозга.
Аналептики. в первую очередь, используются в качестве антагонистов при передозировке депрессантов (гипнотиков, наркотиков). Имея достаточно малую широту терапевтического действия, даже при небольшой передозировке они могут вызвать стимуляцию других образований ЦНС, вызывая ряд нежелательных побочных эффектов, таких, как стимуляция сердечно-сосудистой системы, гиперрефлексия, рвота, судороги.
Доксапрам (Doxapram)
Доксапрам — 1-этил-4-(2-морфолиноэтил)-3,3-дифенил-2-пирро-лидон (8.2.4) получают по следующей схеме. Дифенилацетонитрил в присутствии амида натрия алкилируют 1-этил-З-хлорпирроли-дином с получением (1-этил-3-пирролидинил)дифенилацетонитрила (8.2.1). Кислотным гидролизом нитрильной группы получают (1-этил-3-пирролидинил)дифенилуксусную кислоту (8.2.2). Взаимодействие последней с трехбромистым фосфором (хлористым тионилом, бромистым тионилом, уксусным ангидридом) приводит к перегруппировке с раскрытием пирролидинового кольца и последующим замыканием в пирролидиновое с получением 1-этил-4-(2-бромэтил)-3,3-дифенил-2-пирролидона (8.2.3). Замещением атома брома на морфолиновую группу получают доксапрам (8.2.4) [15, 16, 17, 18].
NaNHj
H2SO4
Р8г3
- 176 -
Стимуляторы ЦНС
823
8.2.4
Доксапрам увеличивает частоту и глубину дыхания. Препарат применяют при постнаркозной депрессии дыхания, при депрессиях дыхания вызванных применением лекарств.
Синонимами препарата являются доксаприл, допрам и др.
Алмитрин (Almitrin)
Алмитрин — 2,4-бис(аллиламино)-6-[4-[бис(лг-фторфенил)метил]-1-пиперазинил]-5-триазин (8.2.6) синтезируют взаимодействием 1-[бис-(и-фторфенил)метил]пиперазина с цианурил хлоридом с получением 2,4-дихлор-6-[4-[бис(и-фторфенил)метил]-1-пиперазинил]-з-триазина (8.2.5). Вводя последний в реакцию с аллиламином, получают алмитрин (8.2.6) [19, 20, 21, 22].
NaOh
H2N-CH2-CH=CH2
- 177-
Г лава 8
Алмитрин, как и доксапрам, увеличивает частоту и глубину дыхания. Кроме того полагают, что он перераспределяет легочное кровообращение, увеличивая его в альвеолах, что приводит к относительно лучшей вентиляции легких. Имеет более пролонгированное действие, чем доксапрам.
Синонимами препарата являются вектарион, дуксил и др.
8.3. Препараты, подавляющие аппетит (аноректики)
Группа структурно родственных амфетаминам препаратов, подавляющих аппетит и используемых при терапии ожирения, называется аноректиками. Соединения данной группы проявляют аналогичный амфетаминам спектр фармакологического и токсикологического действия и используются в качестве вспомогательных средств при лечении ожирения, осуществляемого при индивидуально составленной программе ограничения калорийности принимаемой пищи. Ни одно из используемых веществ не превышает по активности амфетамин, однако меньший потенциал зависимости делает их применение предпочтительным. Механизм действия этих препаратов сходен с таковым амфетаминов. Они активируют центр насыщения в гипоталамусе, тем самым уменьшая аппетит. Аноректики могут усиливать эффекты наркотиков, барбитуратов, алкоголя и других депрессантов ЦНС.
Фентермин (Phentermin)
Фентермин — а,а-диметилфенилэтиламин (8.3.4) отличается от амфетамина наличием дополнительной метильной группы в a-положении к аминогруппе. Синтез препарата осуществляют исходя из бензальдегида, конденсацией которого с 2-нитропропаном получают карбинол (8.3.1). Восстановлением нитрогруппы в последнем получают 2-амино-2-метил-1-фенилпропанол (8.3.2). Взаимодействием с хлористым тионилом гидроксильную группу последнего замешают на атом хлора с получением 2-амино-2-метил-1-фенилпро-пилхлорида (8.3.3). Восстановлением последнего водородом с использованием в качестве катализатора палладий на карбонате кальция получают фентермин (8.3.4) [23, 24].
C-IO
СНз
H-C-NO2
СН3
/==\ СН3
Л //~9н“'9“№г '—' ОН СНз
Н-21 Raney— Ni
. СНз
G У-СН-С-МН2 4—' ОН СНз
SOC12
832
- 178 -
Стимуляторы ЦНС
сн3 сн-с-nh2 Cl СНз
Н2/ Pd-СаСОз
СНз ch2-c-nh2
СНз
8 3.3
8.3.4
Действие препарата заключается в активации центра насыщения в гипоталамусе и уменьшении аппетита, что при ограничении калорийности пиши приводит к уменьшению массы тела.
Синонимами препарата являются ионамин, линия, липопилл, терамин И др.
Диэтилпропион (Diethylpropion)
Диэтилпропион — 1-фенил-2-диэтиламинопропанон (8.3.6) получают бромированием пропиофенона в а-бромпропиофенон (8.3.5) с последующим замещением атома брома на диэтиламинную группу [25, 26].
о
с-сн-сн3
Вг
HN(C2H5)2
z=\ О
/—\ 11
Л п— С—СН—СНз
N(C2H5)2
8.3 6
Диэтилпропион, в принципе, повторяет все характерные для амфетаминов фармакологические свойства и применяется для лечения ожирения с ограничением калорийности пищи.
Синонимами препарата являются амфепрамон, анорекс, аписат, ре-генон, тенуат. тепанил и др.
Список литературы
1. Бобранский Б. и др.,'ОКПХ, 14, 410 (1941).
2. Магдисон О. и др./'ЖОХ, И, 339 (1941).
3. Patrick Т. et al.//J. Am. Chem. Soc. 68, 1009 (1946).
4. iVoodruft E. et al./.'J. Am. Chem. Soc. 60, 465 (1938).
5. Buck J. 7J. Am. Chem. Soc, 54. 3661 (1932)
6. Brit. Pat. 508.757 (1939).
7. US Pat. 2.276.508 (1942).
8. Emde H. /Helv. Chim. Acta. 12, 365 (1929).
-179-
Глава 8
9. Ogata A.II}. Pharm. Soc. Japan. 451, 751 (1919).
10. US Pat. 2.507.631 (1950).
11. US Pat. 2.957.880(1960).
12. Panizzon A./'Uelv. Chim. Acta. 27, 1748 (1948).
13. US Pat. 2.892.753 (1959).
14. Traube W. et al./'Ber. 46, 2077 (1913).
15. US Pat. 3.192.230(1965).
16. US Pat. 3.192.206(1965).
17. Belg. Pat. 613.734 (1962).
18. Lunsford C. et al./7J. Med. Chem. 7, 302 (1964).
19. Fr. Pat. 2.019.646 (1969).
20. Ger. Pat. 1.947.332 (1970).
21. US Pat. 3.647.794(1972).
22. Brit. Pat. 1.256.513 (1969).
23. US Pat. 2.408.345 (1946).
24. US Pat. 2.590.079 (1952).
25. US Pat. 3.001.910(1961).
26. Hyde E. et al.//J. Am. Chem. Soc. 50, 2287 (1928).
I n
। Глава 9
j Противоэпилептические j средства
Эпилепсия — хроническое заболевание, проявляющееся пароксизмальными приступами, обусловленными патологическим возбуждением церебральных нейронов.
Эпилепсия сопровождается различной степенью нарушения сознания. Существуют судорожные и бессудорожные формы приступов эпилепсии, каждая из которых характеризуется своеобразной клинической картиной. Однако практически для всех разновидностей эпилепсии характерны определенные изменения в электроэнцефалограммах.
Припадки генерируются в эпилептогенном очаге головного мозга и могут ограничиваться подрагиванием конечностей. Если же судорожный разряд начинает распространяться и возбуждение охватывает оба полушария головонго мозга, то начинаются припадки. Выделяют большие эпилептические судорожные припадки (grand mal) и малые приступы эпилепсии (petit mal). Однако в самом общем случае судороги — это непроизвольные мышечные сокращения, которые могут возникнуть в результате патологических процессов как внутри, так и вне головного мозга. Они могут возникнуть под воздействием токсинов, травм, гипертермии, при передозировке лекарств или при отмене лекарственной зависимости.
Для лечения эпилепсии применяются различные препараты, в том числе барбитураты и бензодиазепины, которые используются для снятия острых судорожных состояний, возникающих и в результате иных, чем эпилепсия, причин.
Полагают, что в генезисе эпилепсии могут быть задействованы различные механизмы и на любой из них можно оказать лекарственное воздействие.
С клинической точки зрения формально противоэпилептические препараты в основном делятся на 2 категории:
1) эффективные при больших припадках (фенитоин, карбамазепин, фенобарбитал, примидон);
2) эффективные при малых припадках (этосуксимид, ацетазоламид. клоназепам, триметадион, вальпроевая кислота).
-181 -
Глава 9
Лечение каждого частного случая эпилепсии производят определенными препаратами, начиная с одного наименования, однако впоследствии для полного контроля над заболеванием иногда требуется второй, а нередко и третий, препарат. Частое изменение доз или препаратов не рекомендуется.
С химической точки зрения противоэпилептические препараты это'
• производные гидантоинов (фенитоин, мефенитоин и этотоин);
• барбитураты (фенобарбитал, мефобарбтал, метабарбтал. а также примидон);
• сукцинимиды (этосуксимид, метсуксимид, фенсуксимид);
• бензодиазепины (диазепам, хлордиазепоксид. клоназепам, лоразепам);
• оксазолидиндионы (триметадион, параметадион);
• вальпроевая кислота, карбамазепин, ацетазоламид.
Механизм действия противоэпилептических средств недостаточно ясен, поскольку не ясна и этнология эпилепсии. Очевидно, что у различных препаратов он различен. Одни препараты блокируют натриевые каналы, другие воздействуют на ГАМК-систему, усиливая ГАМК-зависи.мое торможение ЦНС, третьи изменяют внутриклеточное соотношение концентраций ионов К' и Са2+, четвертые блокируют N-метил-D-аспартатный (NMDA) рецептор, ответственный за высокочастотные разряды, возникающие при эпилепсии.
Способы синтеза противоэпилептических препаратов будут рассмотрены согласно их химической классификации.
9.1. Производные гидантоинов
Механизм действия гидантоинов окончательно не выяснен. Согласно одной из гипотез, гидантоины предупреждают высокочастотную активацию эпилептогенного очага, а также способствуют выведению из нервных клеток ионов Na', что снижает возбудимость нейронов и препятствует их активации при поступлении к ним импульсов из эпилептогенного очага.
Фенитоин (Phenytoin)
Фенитоин -- 5,5-дифенилимидазолидин-2,4-дион (9.1.1) предложено синтезировать двумя способами. Первый способ предполагает непосредственное взаимодействие бензила с мочевиной с получением искомого продукта (9.1.1) [1].
- 182-
Противоэпилептические средства
h2n -c-nh2
9 1 1
Второй способ заключается во взаимодействии бензофенона с цианидом натрия в присутствии карбоната аммония с одновременной циклизацией полученного продукта — карбоксиаминонитрила и его пере-группировкой в условиях реакции в фенитоин (9.1.1) [2].
По действию на ЦНС фенитоин можно считать отличным антиэпи-тептическим препаратом с незначительным седативным эффектом. Даже в больших дозах он не вызывает гипноза. Предполагается, что фенитоин способствует выведению ионов Na из нервных клеток, за счет чего снижается возбудимость нейронов, что препятствует их активации при поступлении импульсов из эпилептогенного очага. Кроме того, фенитоин уменьшает приток ионов К’ в ходе реполяризации. Не исключено. что именно в результате перераспределения этих ионных потоков фенитоин значительно замедляет распределение возбуждения в мозге.
Фенитоин применяют для лечения эпилепсии, главным образом при больших припадках.
Основными синонимами фенитоина являются дифенин, алепсин, дилантин и солантил.
Этотоин (Ethotoin)
Этогоин — 3-этил-5-фенилимидазолидин-2,4-дион (9.1.5) получают способом, примерно аналогичным вышеописанному и заключающемся во взаимодействии оксинитрила, полученного из бензальдегида (9.1.2), с мочевиной или гидрокарбонатом аммония с промежуточным образованием производного мочевины (9.1.3), который в кислых условиях циклизуется в 5-фенилгидантоин (9.1.4). Алкилирование последнего этилйодидом приводит к этотоину (9.1.5) [3, 4].
- 183 -
Г лава 9
сно
Этотоин менее активен и менее токсичен, чем фенитоин. Применяют по тем же показаниям, что и фенитоин, т. е. для контроля над большими и сложными припадками.
Основным синонимом препарата является пеганон.
9.2. Барбитураты
Эффективность барбитуратов в качестве антиэпилепттгческих средств может быть отнесена к их воздействию на возбудимость нейронов эпилепто1енно1о очага, а также ГАМК-ергическую передачу в ЦНС путем повышения ингибирующего действия ГАМК. Более того, барбитураты могут уменьшать возбуждающие эффекты глутамата в синапсах. В настоящее время не выяснено, который из этих предполагаемых механизмов более важен для проявления антиэпилептической активности.
Фенобарбитал (Phenobarbital)
Фенобарбитал — 5-этил-5-фенилбарбитуровая кислота (4.1.4), способы синтеза которого были уже описаны в гл. 4 «Снотворные средства (гипнотики и седативные препараты)». Препарат широко используется как самостоятельно, так и в составе различных комбинированных лекарственных средств.
- 184-
Противоэпилептические средства
Фенобарбитал заметно понижает возбудимость двигательных центров головного мозга, и поэтому его широко используют при лечении эпилепсии как при малых, так и при больших припадках, при лечении хореи и спастических параличей.
Синонимами препарата являются люминал, адонал, седонал и многие другие.
При эпилепсии применяются также такие барбитураты, как мефо-барбитал (4.1.8) и метарбитал (4 1.9), описанные выше.
п F5 ^-СгНб
H-N
O N О
СН3 4 1 9
Примидон (Primidone)
Примидон — 5-этил-5-фенилгексагидропиримидиндион-4,6 (9.2.1) получают взаимодействием диамида этилфенилмалоновой кислоты с формамидом [5, 6]. Альтернативными способами являются электролитическое восстановление фенобарбитала или каталитическое восстановление соответствующей 2-тиобарбитуровой кислоты [7].
О h-c-nh2
Примидон по химическому строению близок к фенобарбиталу с тем отличием, чго карбонильная группа в положении С2 заменена на метиленовую Эта модификация привела к получению препарата с сильным противосудорожным действием без выраженного снотворного эффекта.
Примидон применяют главным образом при больших припадках.
Основными синонимами являются гексамидин и милепсин.
- 185-
Глава 9
9.3. Сукцинимиды
Сукцинимиды — группа препаратов, производных имида янтарной кислоты, используемых при малых формах эпилепсии в отсутствие припадков.
Этосуксимид (Ethosuximide)
Этосуксимид — З-этил-З-метилпирролидин-2,5-дион (9.3.4) получают исходя из метилэтилкетона и циануксусного эфира, которые конденсируют по Кновенагелю, и к полученному продукту (9.3.1) присоединяют циановодород. После кислотного гидролиза и декарбоксилирования синтезированного динитрила (9.3.2) получают 2-метил-2-этилянтарную кислоту (9.3.3). Взаимодействием последней с аммиаком получают диаммониевую соль, при последующем нагревании которой происходит гетероциклизация в этосуксимид (9.3.4) [8. 9].
о
х
НзС^С2Н5
С2Н5О
Н3С СООС2Н5
)с=с< _ Н5С2 С =N
9 3.1
СН3 с2н5-с—сн~соос2н5 N=C C~N
9.3.2
н+ сн3
* С2Н5“С-СН2-СООН
СООН'
9.3.3
NH3
934
Этосуксимид — противосудорожный препарат, применяющийся при малых формах эпилепсии.
Выпускается также под названиями аксамид, суксилен, ронтон и пикнолепсин.
Фенсуксимид (Phensuximide)
Фенсуксимид — 1-метил-3-фенилпирролидин-2,5-дион (9.3.5) синтезируют взаимодействием фенилянтарной кислоты или ее ангидрида с метиламином [10, 11].
н-сн2—соон соон
снзын2
- 186 -
Противоэпилептические средства
Фенсуксимид так же, как и этосуксимид является противосудорожным препаратом, применяющимся при малых формах эпилепсии.
Выпускается также под названием милонтин.
9.4. Вальпроевая кислота
Вальпроевая кислота (Valproic acid)
Вальпроевая кислота — 2-пропилвалериановая кислота (9.4.3) получается алкилированием циануксусного эфира двумя молями про-пилбромида с получением дипропилциануксусного эфира (9.4.1). Гидролизом и декарбоксилированием карбэтоксильной группы получают дипропилапетонитрил (9.4.2), который гидролизуют в вальпроевую кислоту (9.4.3) [12, 13, 14, 15].
СгН^ОМа
СНз-СН2-СН2— Вг —2—*
9 —N _ _ NaOH
CH3 —СН2 —СН2 С СООС2Н5
СНз*-СН2 —СН2 9 4 1
СН3-СН2-СН2\
yCH-C=N сн3—сн2—сн2
NaOH
СН3-СН2-СН2\
усн-соон сн3-сн2-сн/
Вальпроевая кислота и ее соли являются новой группой противоэпилептических препаратов, отличающаяся от известных, как по химической структуре, так и по механизму действия. Предполагается, что она воздействует на метаболизм ГАМК. Показано, что вальпроевая кислота повышает уровень ГАМК в мозге путем конкурентного ингибирования ГАМК трансаминазы и дегидрогеназы янтарного полуют ьдеги да.
Препарат оказывает не только противосудорожное действие, но и улучшает психическое состояние больных.
Вальпроевая кислота, как и сукцинимиды, используется при эпилепсии в отсутствие припадков, а клиническая эффективность ее превышает таковую сукцинимидов.
Наиболее распространенными синонимами препарата являются делании и конву леке.
-187-
Г лава 9
9.5. Карбамазепин
Карбамазепин (Carbamazepine)
Карбамазепин — 5Я-дибенз[Ь,1]азепин-5-карбоксамид (9.5.2) синтезируют взаимодействием 5Я-дибенз[Ь,1]азепина с фосгеном с образованием 5-хлоркарбокси-5Я-дибенз[Ь,1]азепина (9.5.1) и последующим его взаимодействием с аммиаком с получением целевого карбамазепина (9.5.2) [16]. Альтернативным путем синтеза является непосредственное взаимодействие 5Н-дибенз[Ь,1]азепина с цианатом калия [17].
1 СОС1г
2 NHg
С=О
NH2 9 5 1
KOCN
Карбамазепин применяют главным образом при больших припадках При малых припадках недостаточно эффективен. Имеются данные о ряде серьезных побочных действий.
Синонимом препарата является тегретол.
9.6. Бензодиазепины
Бензодиазепины главным образом используются в медицине в качестве транквилизаторов. Однако они с успехом применяются и при эпилепсии для контроля состояния длительных судорог. Наиболее широко применяются диазепам (5.1.2) и хлордиазепоксид (5.1.22), синтез которых описан в гл. 5 «Анксиолитики (транквилизаторы)».
Третьим препаратом бензодиазепинового ряда, нашедшим применение при лечении эпилепсии, является клоназепам.
- 188-
Противоэпилептические средства
Клоназепам (Clonazepam)
Клоназепам — 5-(о-хлорфенил)-1.3-дигидро-7-нигро-2Я-1,4-бензо-диазепин-2'Он (9.6.5) получают по стандартной схеме получения производных 1,4-бензодиазепинов, но с той разницей, что акцепторную (в данном случае нитро) группу при С7 бензодиазепиновой системы вводят на последней стадии синтеза. Синтез клоназепама исходит из 2-хлор-2'-нитробензофенона, который восстанавливают в 2-хлор-2'-аминобензофенон (9.6.1) водородом над никелем Ренея. Аминопроизводное ацилируют бромангидридом бромуксусной кислоты бромацетамид (9.6.2) и далее взаимодействием с аммиаком переводят в аминоацетамид (9.6.3). Последний под действием пиридина циклизуют в 5-(2-хлорфенил)-2,3-дигидро-1Я-1,4-бензодиа-зепин-2-он (9.6.4). Нитрование полученного продукта в мягких условиях нитратом калия в серной кислоте приводит к клоназепаму (9.6.5) [18. 19,20,21,22,23].
HNO3
Препарат оказывает выраженное противосудорожное, а также центральное миорелаксирующее, анксиолитическое и седативное действие.
Клоназепам применяют при эпилепсии, сомнамбулизме, различных формах мышечного гипертонуса, бессоннице (особенно у больных с органическими поражениями головного мозга), психомоторном возбуждении.
Синонимами препарата являются клонопин и ривотрил.
- 189 -
Глава 9
9.7. Ацетазоламид
Ацетазоламид (Acetazolamide)
Ацетазоламид — 5-ацетамидо-1,3,4-тиадиазол-2-сульфонамид (9.7.5). Синтез ацетазоламида основывается на получении 2-амино-5-меркапто-1,3,4-тиадиазола (9.7 2), который получают взаимодействием тиоцианата аммония с гидразином с образованием гидра-зино-М,^'-бис-(тиомочевины) (9.7.1), которая под действием фосгена циклизуется в тиазол (9.7.2). Ацилируя последний ангидридом уксусной кислоты, получают 2-ацетиламино-5-мер-капто-1.3,4-тиадиазол (9.7.3). Продукт хлорируют в 2-ацетилами-но-5-меркапто-1,3.4-тиадиазол-5-сульфонилхлорид (9.7.4). который взаимодействием с аммиаком трансформируют в ацетазоламид (9.7.5) [24, 25].
^NH_$CN + Н2М~ГчН2
S S
»» н
H2N -’C-NH-NH-C-NH2
97 1
N----N (CH3CO)2O
fl fl --------------1
h2n s sh 972
N---N
CH-yCONH
97 3
CH3CONH S SO2CL
Ацетазоламид применяют при эпилепсии в отсутствие припадков, а также в сочетании с другими антиэпилептическими препаратами.
Наиболее распространенным синонимом препарата является диа-мокс.
9.8. Оксазолидины
Эта группа соединений представлена двумя препаратами, которые применяются только при малых формах эпилепсии в отсутствие припадков.
Триметадион (Trimethadione)
Триметадион — 3.5,5-триметилоксазолидин-2,4-дион (9.8.2) синтезируют метилированием диметилсульфатом 5.5-триметилоксазо-лидин-2.4-диона (9.8.1), который, в свою очередь, получают циклоконденсацией эфира 2-гидроксиизомасляной кислоты с мочевиной [26, 27,28].
- 190-
Противоэпилептические средства
СНз
H3C “С-СООС2Н5 он
h2n
CjHgONa
(CH3O)2SO2
СНз
982
Тримегадион применяют при малых формах эпилепсии, не поддающихся лечению другими препаратами.
Синонимами препарата являются триметин и троксидон.
Параметадион (Paramethadione)
Параметадион — 5-этил-3,5-диметилоксазолидин-2,4-дион (9.8.3), отличающийся от триметадиона лишь заменой одного метильного радикала на этильный, получают по совершенно аналогичной схеме исходя не из 2-гидроксиизомасляной кислоты, а из 2-гидрокси-2-метилмасляной кислоты [29].
СНз о----с2н5
сАгАо сн3 983
Параметадион также применяют при малых формах эпилепсии. Синонимом препарата является парадион.
Список литературы
1 Biltz Н,/Вег. 41, 1391 (1908).
2. US Pat. 2.409 754 (1946).
3. Pinner А 7Вег. 21, 2325 (1888).
4 US Pat. 2.793.157 (1946).
5. US Pat. 2.578.847 (1951).
6. Ger. Pat. 843.413 (1950).
7 Brit. Pat. 6661)27 (1952).
8 US Pat. 2.993.835 (1961).
9. Sircar G et al./ J. Chem. Soc. 1927, 1252.
10 US Pat. 2.643 258 (1953).
11 Long L et al.7J. Am. Chem. Soc. 73. 4895 (1951).
12 Fr. Pat. M 2.442 (1962).
-191 -
Глава 9
13. Brit. Pat. 980.279 (1963).
14. US Pat. 3.325.361 (1967).
15. Wiemannet A. al./'Bull. Soc. Chim. France 1958. 199.
16. US Pat. 2.948.718 (1960).
17. D.D.R. Pat. 133.052 (1977).
18. Strenbach L. et al.7J. Med. Chem. 6, 261 (1963).
19. US Pat. 3.116.203 (1963).
20. US Pat. 3.123.529 (1964).
21. US Pat. 3.121.114 (1964).
22. US Pat. 3.203.990(1965).
23. US Pat. 3.335.181 (1967).
24. US Pat. 2.554.816 (1951).
25. Robin R et al.'/J. Am. Chem. Soc. 72, 4890 (1950).
26. US Pat. 2.559.011 (1951).
27. US Pat. 2.575.692 (1951).
28. Spielman L //J. Am. Chem. Soc. 66, 1244 (1944).
29. US Pat. 2.575.693 (1951).
5 Глава 10
I Средства, применяемые
I при паркинсонизме
Паркинсонизм — дегенеративное, медленно прогрессирующее заболевание ЦНС, связанное с поражением базальных ганглиев и характеризующееся четырьмя основными симптомами: замедленностью, скованностью движений, ригидностью и тремором в покое. Этиология болезни неизвестна.
Наиболее вероятной причиной перечисленных выше двигательных нарушений может быть недостаток дофамина, который оказывает тормозящее влияние на регуляцию функций спинного мозга.
С другой стороны, в регуляции экстрапирамидной системы задействованы холинергические нейроны.
Больше века лечение паркинсонизма основывалось на использовании центральных антихолинергических веществ. Вплоть до недавнего времени при паркинсонизме использовались различные препараты алкалоидов белладонны, характерной особенностью которых является .холинолитическое действие, т. е. способность уменьшать чувствительность к ацетилхолину — медиатору холинергических синапсов.
На сегодняшний день накопилось достаточное количество фактов, позволяющих утверждать, что паркинсонизм является следствием дисбаланса между действием дофаминергической и холинергической систем и что лечение паркинсонизма должно заключаться либо в блокировании избыточной стимуляции холинергической системы, либо в нормализации функциональной активности дофаминергической системы.
Следовательно, одним из подходов фармакотерапии паркинсонизма может заключаться в устранении дефицита дофамина. Поскольку сам дофамин не проникает через гематоэнцефалический барьер, с этой целью использу ют прекурсор дофамина — леводопу либо препараты, высвобождающие дофамин, либо агонисты дофаминовых рецепторов, либо ингибиторы инактивации дофамина. С другой стороны, при лечении паркинсонизма следует применять антихолинергические препараты.
Исходя из вышесказанного, лечение паркинсонизма должно быть основано на применении 2 групп веществ:
1) средств, стимулирующих дофаминергические системы мозга;
2) средств, ингибирующих холинергические системы мозга.
- 193 -
Глава 10
10.1. Средства, влияющие на дофаминергические системы мозга
В медицинской практике применяются четыре типа дофаминергических препаратов, которые могут быть охарактеризованы как прекурсоры дофамина (леводопы), препараты, высвобождающие дофамин (амантадин), агонисты дофаминовых рецепторов (бромкриптин), ингибиторы инактивации дофамина (селегилин).
Прекурсоры дофамина позволяют создать повышенные концентрации дофамина. Другая группа — препараты, высвобождающие дофамин, были обнаружены случайно при создании противовирусного препарата амантадина. Препарат может быть полезен пациентам, у которых имеется депо дофамина. Третья группа — агонисты дофаминовых рецепторов — является группой вспомогательных препаратов и позволяет проводить лечение меньшими дозами леводопы. Наконец, четвертый тип препаратов, который представлен селегилином является ингибитором разновидности моноаминооксидаз (МАО-В) — фермента, обеспечивающего внутриклеточную инактивацию дофамина в пресинаптиче-ских нервных окончаниях.
Леводопа (Levodopa)
Леводопа — (-)-3-(3,4-дигидроксифенил)-Ь-аланин (10.1.1) представляет собой левовращающий изомер диоксифенилаланина, используемый в качестве предшественника дофамина. Существует несколько способов получения леводопы, как полусинтетических, заключающихся в микробиологическом гидроксилировании L-тиро-зина (10.1.1) [1, 2], так и чисто синтетических.
Окисление L-тирозина, предусматривающее селективное введение гидроксильной группы в положение С3 бензольного кольца тирозина, можно осуществить и чисто синтетическим путем, применяя в качестве окислителя смесь перекиси водорода и сульфата двухвалентного железа в воде при постоянной подаче кислорода [3].
Н2О2 / FeSO4
НО
ио -4 СНг-СН-СООН '—' nh2 10 1 1
Другой путь синтеза леводопы заключается в ацетилировании тирозина ацетилхлоридом в присутствии хлористого алюминия с последующим окислительным дезацилированием 3-ацетилтирозина (10.1.2) с использованием перекиси водорода в растворе гидроокиси натрия [4, 5, 6, 7].
- 194-
Средства, применяемые при паркинсонизме
но
CH3COCL/ ALCL3
СН3СО
но—4 Усн2—сн—соон nh2
10 1 2
Н2О2' NaOH
НО
10 1 1
Предложены пути синтеза леводопы, исходящие из ванилина [8, 9, 10, 11, 12, 13, 14]. Согласно одному из них, конденсацией ванилина с гидантоином, последующим восстановлением двойной связи в образующемся продукте (10.1.4) и его гидролизом получают рацемический дигидроксифенилаланин (ДОФА), из которого выделяют леводопу [8].
И
1 Нг' Pd-C
ЙЕС-----». D,L-Dopa -----» 10 1 1
Леводопх синтезируют также исходя из пипероналя, альдегидную группу которого восстанавливают водородом над никелем Ренея, получая пиперониловый спирт, или 3,4-метилендиоксифенилметанол (10.1.5). Действием хлористого водорода последний переводят 3,4-метилендиоксифенилметилхлорид (10.1.6). Реакцией этого соединения с ацетамидомалоновым эфиром получают (3,4-метилендиоксифенил-метил)ацетамидомалоновый эфир (10.1.7). Щелочной гидролиз и частичное декарбоксилирование последнего приводят к получению защищенного по двум гидроксильным группам и по аминогруппе продукту (10.1.8), гидролизом амидной группы которого с использованием фермента такадиастазы непосредственно выделяют исключительно L-3-(3.4-метилендиоксифенил)аланин (10.1.9). Снятием метилендиоксиза-шитной группы с помощью бромистого водорода получают леводопу (10.1.1) [15].
Ho. Ranay— Ni
- 195-
Г лава 1 О
Г Y=\ СООС2Н5 NaOH I /~“\
О—<Л сн2-с-соос2н5 -------► о—4 Л—сн2-сн-соон ►
NH-COCH3 NH-СОСНз
10 17 10 18
| )=Х НВг
о-< Л—сн2-с н-соон --------*-1011
'—' nh2
10 1 9
Среди многих попыток выправить дефицит дофамина при паркинсонизме введение пациенту непосредственного предшественника дофамина — леводопы считается весьма логичной терапией, поскольку леводопа проходит через гематоэнцефалический барьер, где превращается в дофамин, приводя его уровень в норму. Таким образом, леводопа устраняет или ослабляет проявления паркинсонизма. Леводопа относится к наиболее эффективным средствам при лечении паркинсонизма, исключая паркинсонизм, вызванный лекарственными средствами. К сожалению, он обладает рядом нежелательных, побочных эффектов.
Наиболее распространенными синонимами являются 1-дофа, мадо-пар, допар, синемет, лародопа и др.
Амантадин (Amantadine)
Амантадин — 1-адаманганамин (10.1.12) синтезируют исходя из адамантана. Последний бромируют в 1-бромадамантан (10.1.10), который при нагревании со смесью ацетонитрила и серной кислоты в условиях реакции Риттера трансформируется в 1-ацетиламиноадамантан (10.1.11). Гидролиз последнего щелочью приводит к амантадину (10.1 12) [16, 17].
CH3CN ' H2SO4
Амантадин является средством, повышающим концентрацию дофамина в синаптической щели путем его высвобождения из нейронов
- 196 -
Средства, применяемые при паркинсонизме
и угнетения процесса его обратного захвата. Амантадин — противовирусный препарат. Свойства облегчать симптомы паркинсонизма были выявлены у него случайно.
Лечение паркинсонизма комбинацией леводопы, антихолинергиче-ских препаратов и амантадина дает лучшие результаты, чем при применении любого из этих препаратов в отдельности.
Синонимами препарата являются мидантан и симметрел.
Бромокриптин (Bromocriptin)
Бромокриптин — 2-бромэргокриптин (10.1.13) является полусинте-тическим производным природного алкалоида спорыньи — эргок-риптина (производного лизергиновой кислоты), который получают бромированием эргокриптина N-бромсукцинимидом [18, 19].
Бромокриптин — дофаминомиметик, который является агонистом дофаминовых В2-рецепторов, обладает выраженной антипаркинсониче-ской активностью. Применяется для лечения всех фаз идиопатического и постэнцефалического паркинсонизма. Однако он обладает рядом нежелательных, побочных эффектов, вплоть до возникновения психических нарушений при длительном применении.
Наиболее распространенными синонимами являются парлодел, бромэргон и др.
Селегилин (Selegiline)
Селегилин — М-метил-Ы-(2-пропинил)~2-метил-1 -фенилэтиламин (10.1.14) получают алкилированием (-)метамфетамина (8.1.2.3) про-паргилбромидом [20, 21, 22, 23].
/==\ 9Нз Вг-СН2-С=СН 4 Л— СН2—CH-NH '—' СНз 8 123 /=\ сн3 CH2-CH-N —СН2-С£СН ' ' СНз 10 1 14
-197-
Глава 10
Препарат является избирательным ингибитором МАО-В, которая подавляет процессы инактивации дофамина и способствует повышению его уровня в головном мозге. Для лечения паркинсонизма препарат обычно применяют в комбинации с леводопой.
Наиболее распространенными синонимами препарата являются депренил, элдеприл, элдопан и др.
10.2. Антихолинергические препараты (центральные холиноблокаторы)
Первыми препаратами, использованными при лечении паркинсонизма. были алкалоиды (атропин и скополамин), и в течение многих лет они являлись единственными препаратами, используемыми с этой целью.
На сегодняшний день при лечении паркинсонизма эти алкалоиды применяются крайне редко и практически полностью заменены синтетическими препаратами, проявляющими центральные антихолинергические свойства (центральные холиноблокаторы). Они подавляют стимулирующие холинергические влияния путем угнетения холинорецепторов. Полагают, что на синтез, высвобождение и гидролиз ацетилхолина они не влияют. Их действие, таким образом, способствует уменьшению или устранению двигательных нарушений, связанных с поражением экстрапири-рамидной системы. Они уменьшают ригидность, в несколько меньшей мере — акинезию, а также мало влияют на тремор.
Терапевтическая ценность этих препаратов относительно невелика, и они применяются либо в комбинации с леводопой, либо в случаях слабо-выраженного паркинсонизма, в основном, с целью снятия ригидности.
Вместе с тем они вызывают ряд побочных действий, включая общую слабость головокружение и т. п.
Тригексифенидил (Trihexphenidyl)
Тригексифенидил — 1-циклогексил-1-фенил-З-пиперидинопропан-1-ол (10.2.2) получают взаимодействием 2-( 1-пиперидино)про-пиофенона (10.2.1) с циклогексилмагнийбромидом. Исходный 2-(1-пиперидино)пропиофенон (10.2.1), в свою очередь, получают аминометилированием бензофенона с использованием параформа и пиперидина [24, 25, 26, 27].
1022
- 198-
Средства, применяемые при паркинсонизме
Тригексифенидил — антипаркинсоническое средство, обладающее центральным и периферическим антихолинергическим действием, а также прямым релаксирующим действием на гладкие мышцы. Препарат уменьшает мышечную ригидность и общую скованность и относительно мало влияет на тремор. Применяется при паркинсонизме как в виде монотерапии, так и в комбинации с леводопой.
Наиболее распространенными синонимами являются паркопан. паркинсан и циклодол.
Проциклидин (Procyclidine)
Проциклидин — 1-циклогексил-1-фенил-3-пиирроли динопропан-1-ол (10.2.3) получают совершенно аналогичным путем, но исходя из 2-(1-пирролидино)пропиофенона [28, 29, 30, 31, 32].
10 2 3
По своим фармакологическим свойствам практически не отличается от тригексифенидила.
Наиболее распространенным синонимом препарата является кемад-рин.
Бипериден (Biperiden)
Бипериден — 1-(5-норборнен-2-ил)-1-фенил-3-пиперидинопропан-1-ол (10.2.4) также синтезируют по методу получения тригексифенидила, но взаимодействием 2-(1-пиперидино)пропиофенона (10.2.1) с 5-норборнен-2-илмагнийбромидом [33. 34].
10 24
Бипериден повторяет свойства указанных выше препаратов. Синонимом препарата является акинетон.
- 199-
Глава 10
Дифенгидрамин (Diphenhydramine)
Дифенгидрамин — 2-дифенилметокси-М.М-диметиламин (10.2.5) получают этерификацией 2-диметиламиноэтанола бензгидрилбро-мндом [35, 36, 37].
СНз
+ НО-ОН2-СН2—N
СНз
Дифенгидрамин также уменьшает мышечную ригидность и общую скованность и относительно мало влияет на тремор. В общем, трудно отдать предпочтение какому-либо из перечисленных препаратов.
Синонимами препарата являются бенадрил и бенилин.
Бензтропин (Benztropine)
Бензтропин — 3-дифенилметокси)тропан (10.2.6) получают взаимодействием тропина с дифенилдиазометаном [38].
Препарат используется для быстрого устранения острых дистонических реакций при паркинсонизме. Не устраняет тремор.
Синонимом препарата является когентин.
Этопропазин (Ethopropazine)
Этопропазин — 10-(2-диэтиламинопропил)фенотиазин (10.2.7) получают алкилированием фенотиазина 1-диэтиламино-2-пропилхло-ридом в присутствии амида натрия [39, 40].
-200-
Средства, применяемые при паркинсонизме
н
СНз C2Hg
+ Cl—ch-ch2-n(
С2Н5
NaNH2
1 / ‘
ch3-ch-ch2-n(
10 2 7 С2Н5
Этопропазин — производное фенотиазина с выраженной антихоли-нергической активностью, который эффективно уменьшает мышечную ригидность и общую скованность, включая тремор. Применяется как при паркинсонизме, так и в других случаях экстрапирамидных расстройств, в том числе и в случаях, вызванных фенотиазиновыми препаратами.
Синонимами препарата являются парсидол, лизиван, паркин и др.
Список литературы
i
1. Атао S. et al.'/Ann. Sankyo Res. Lab. 23. 249 (1971).
2. Sih C. J. et al.//J. Am. Chem. Soc. 91, 6204 (1969).
3. Waser E. et al.//Heh. Chim Acta. 4, 657 (1921).
4. Bretschneider H et al J, Helv. Chim Acta. 56. 2857 (1973).
5. Ger. Pat. 2.023.459 (1970).
6. Ger. Pat. 2.023.460 (1970).
7. Ger. Pat. 2.023.461 (1970).
8. USPat. 2.605.282 (1952)/
9. USPat. 4.005.127 (1977).
10. Ger. Pat 2.223 063 (1971).
11. Ger. Pat. 2.210.938 (1972).
12. USPat. 4.124.533 (1978).
13. Knowles W. et al.'/J. Am. Chem. Soc. 97, 2567 (1975).
14. Vineyard B. et al.z/J, Am. Chem. Soc. 99. 5946 (1977).
15. Yamada S. et al.//Chem. Pharm. Bull. 10, 680, 688, 693 (1963).
16. Stetter H. et al. ' Chem. Ber. 93, 226 (1960).
17. USPat. 3.310.469 (1967).
18. USPat. 3.752.814 (1973).
19. Ger. Pat. 1.926.045 (1969).
20. Ger. Pat. 1.568.277 (1966).
21. USPat. 4.564.706(1986).
22. Fr. Pat. M 2635 (1964).
23. Fowler J.‘,1. Org. Chem. 42, 2637 (1977).
-201 -
Г лава 1 О
24. US Pat. 2.680.115 (1954).
25. US Pat. 2.716.121 (1955).
26. US Pat. 2.682.543 (1954).
27. Brit. Pat. 750.156 (1956).
28. US Pat. 2.682.543 (1954).
29. US Pat, 2.891.890(1959).
30. US Pat. 2.826.590 (1958).
31. US Pat. 2.842.1 15 (1958).
32. Adamson J. et al./'J. Chem. Soc. 1951, 52.
33. US Pat. 2.789.1 10 (1957).
34. Ger. Pat. 1.007.067 (1953).
35. US Pat. 2.421.714 (1947).
36. US Pat. 2.427.878 (1947).
37. US Pat. 2.397.799 (1946).
38. US Pat. 2.595.405 (1952).
39. US Pat. 2.526.118 (1950).
40. US Pat. 2.607.773 (1952).
, Глава 11
f Адренергические
l (симпатомиметические)
} препараты
Адренергические препараты — это природные или синтетические соединения, частично или полностью повторяющие эффекты норэпинефрина (норадреналина), эпинефрина (адреналина) и дофамина и вызывающие биологический ответ, сходный с активацией симпатической нервной системы. Поскольку они воспроизводят стимуляцию симпатических нейронов, их также называют симпатомиметиками.
Не поддающаяся усилиям воли, симпатическая нервная система играет важную роль в регуляции сердечной деятельности, сосудистого юнуса, функциональной активности гладкой мускулатуры и железистого аппарата путем высвобождения в качестве медиаторов из периферических нервных окончаний и в синапсах ЦНС эндогенных адренергических веществ — катехоламинов.
Эти соединения, в большом количестве выделяются в организме в периоды физического или эмоционального стресса и играют огромною роль для адаптации организма к стрессовой ситуации.
В самом организме катехоламины (p-арилэтиламины, имеющие в положениях Сз и С4 ароматического кольца гидроксильные группы) — дофамин. норэпинефрин (норадреналин) и эпинефрин (адреналин) в основном вырабатываются надпочечниками из общего прекурсора — тирозина, который в организме, в первую очередь, гидроксилируется в метаположение ароматического кольца ферментом тирозингидроксилазой. Образующийся при этом ДОФА далее декарбоксилируется ферментом L-декарбоксилазой ароматических кислот с образованием дофамина. Далее дофамин гидроксилируется ферментом дофамин-р-гидроксилазой с образованием норэпинефрина (норадреналина). Наконец, концевая первичная аминная группа метилируется ферментом фенилэтаноламин-К-метил-трансферазой с образованием эпинефрина (адреналина).
тирозин
ДОФА
дофамин
-203 -
Глава 11
но
)=\ ?н
но -Z ch-ch2-nh2
норэпинефрин
(норадреналин)
НО )==\ ?н
но—ch-ch2-nh-ch3
эпинефрин
(адреналин)
Адренергические препараты используются ввиду их способности воздействовать на сердечно-сосудистую систему, вызывать бронхолитический эффект, стимулировать ЦНС, проявлять мидриатичесское и ано-рексигенное действие. С другой стороны, именно широкий спектр активности, обусловленный, очевидно, большим сродством к различным и не только к адренергическим рецепторам, ограничивает их применение ввиду ряда нежелательных побочных эффектов.
Адренергические или симпатомиметические препараты составляют большую группу веществ — препараты прямого действия, которые непосредственно взаимодействуют с адренергическими рецепторами. К ним относятся эпинефрин, фенилэфрин и изопротеренол, добутамин, тербуталин, альбутерол, метапротеренол, изоэтарин, клонидин, нафазо-лин. оксиметазолин, тетрагидрозолин, ксплометазолин.
Препараты непрямого действия проявляют симпатомиметический эффект, опосредованно вызывая высвобождение эндогенных катехоламинов. Симпатомиметическая активность этих препаратов, таким образом. зависит от наличия в организме катехоламинов. К ним относится тирамин — соединение, используемое, в основном, в качестве анализатора в экспериментальных исследованиях.
Наконец, некоторые препараты имеют двойное действие — прямое и непрямое. К ним относятся дофамин, эфедрин, фенилпропаноламин, метараминол, амфетамины.
Первичное взаимодействие между адреномиметиками и эффекторной клеткой осуществляется посредством адренергических рецепторов, исключительно широко представленных в мозге, в различных органах и тканях. Строение адренорецепторов неизвестно.
Концепция рецепторов, как известно, основывается на наличии определенных клеточных структур, которые ответственны за связывание биологически активных соединений.
Молекулярная структура лиганд — связывающий участок рецептора определяет специфику физиологического ответа организма. Связывание адренергического агониста, равно как и любого другого препарата, действующего путем субсграт-рецепторного взаимодействия, с соответствующим рецептором на поверхности мембраны вызывает каскад биохимиче
-204-
Адренергические препараты
ских реакций в клетке, в конечном счете приводящих к изменению ее функционально-метаболического состояния. Лекарство, таким образом, содержит информационное послание, передаваемое внутрь клетки и соответствующим образом распространяющееся, что и вызывает измеримые эффекты на уровне ткани или органа. Специфическое связывание препарата с рецептором активирует определенный биологический процесс, который можег завершиться, регуляцией ионных каналов, секрецией желез, изменением ферментной активности и т. д.
Каждый из адренергических препаратов в отдельности выявляет значительные качественные и количественные различия, как фармакодинамического. так и фармакокинетического характера, что обеспечивает разумное их терапевтическое применение.
Постулируют два основных класса рецепторных белков, связывающих адренергические препараты, которые исторически были определены как а- и p-рецепторы и далее были, в свою очередь, разбиты на четыре подтипа адренергических рецепторов -- аь а2: р, и р2.
Несмотря на некоторые исключения, активация агрецепторов, в общем, приводит к возбуждению, (З-рецепторы — ответственны за релаксацию тканей. Активация ^-рецепторов выражается в стимулирующем эффекте на сердце и почки; активация пресинаптических адренер! иче-ских aj-рецепторов, возможно, предполагает механизм обратной связи — ингибирование нейронального высвобождения норэпинефрина. В то же время стимуляция постсинаптических а2-рецепторов, аналогично сц-ре-цепторам, вызывает возбуждение тканей.
На основе анатомических, фармакологических, биохимических и иных критериев показано, что «.(-рецепторы присутствуют, главным образом, в эффекторных органах; а2-рецепторы расположены в адренергических нейронах, в пресинаптических участках; [3,-рецепторы расположены, преимущественно, в сердечных и почечных тканях; р2-рецепторы обнаружены во многих других органах (бронхах, сосудах, матке и др.).
Вариации ответов организма на различные адренергические препараты основываются на их относительной селективности при взаимодействии с различными рецепторами, которые исключительно неравномерно распределены в эффекторных структурах (сердце, сердечно-сосудистая система, легкие, мозг, периферическая нервная система и т. д.)
В общем, ответ эффекторного органа на эпинефрин (адреналин) и или норэпинефрин (норадреналин) непосредственно определяется типом адренорецептора, равно как и соотношением а- и [3-адрено-рецепторов.
-205-
Глава 11
Типичное фармакологическое действие адренергических препаратов заключается в следующем:
— стимуляция работы сердца — повышение частоты и силы сердечных сокращений;
— вазомоторные воздействия — вазодилатация, вазоконстрикция.
- регуляция эндокринного состояния — модуляция инсулина, ренина и ряда гормонов;
— регуляция метаболического состояния — усиленный гликогенолиз в печени и в мышцах, высвобождение жирных кислот из тканей;
— психомоторное возбуждение — со стороны ЦНС.
Спектр сердечно-сосудистых, дыхательных, гормональных, метаболических и нейропсихических ответов, которые могут быть вызваны адренергическими препаратами, в общем очень похож на многие из адаптивных реакций организма — на повышенную физическую активность и физический стресс.
Адренергические препараты с клинической точки зрения формально можно классифицировать и нижеприведенным образом, хотя при этом некоторые препараты могут одновременно фигурировать в разных группах:
• эндогенные (эпинефрин, норэпинефрин, дофамин) и синтетические катехоламины (изопротеренол, добутамин);
• вазопрессорные амины (метараминол, метоксамин, .мефентер-мин);
• противоотечные (тетрагидрозолин. фенилэфрин, нафазолин, эфедрин);
• бронхолитики (эфедрин, метапротеренол, изоэтарин, тербуталин);
• релаксанты гладкой мускулатуры (ритодрин, арлидин, изо-ксуприн);
• стимуляторы ЦНС (амфетамины).
С химической точки зрения адренергические препараты имеют весьма много общего и рассматриваются как замещенные фенилэтила-мины.
R3 п
. р а
Rd —Z X-CH-CH-NH-R
'—' Rz Ri
-206-
Адренергические препараты
Обращают на себя внимание корреляции между структурой симпа-томиметиков и проявляемой ими биологической активностью:
1. Симпатомиметическая активность максимальна, когда между ароматическим кольцом и аминогруппой имеется два атома углерода.
2. Чем меньше степень замещения при аминогруппе, тем больше селективность соединения для активации а-адренорецепторов, и, наоборот, увеличение объема заместителя при первичной аминогруппе придает большую селективность по отношению к [3-адренорецепторам.
3. Замещение при а-углеродном атоме предупреждает окислительную дезактивацию молекулы препарата моноаминооксидазой и, таким образом, намного увеличивает продолжительность его действия. В то же время замещение при а-углеродном атоме способствует непрямому действию препарата — способность к высвобождению эндогенных катехоламинов из нейрональных запасников.
4. Активность препарата в значительной степени зависит от наличия гидроксильных групп в положениях Cj или С4 ароматического кольца. Последнее условие необходимо для активации как а- так и p-адренорецепторов. Соединения с наличием гидроксильной группы только при С3 в ароматическом кольце выявляют высокое соотношение прямой/непрямой агонистической активности. Соединения с наличием гидроксильной группы только при С4 в ароматическом кольце, выявляют высокое соотношение непрямой/прямой активности. Фенилэтиламины. не содержащие гидроксильных групп в ароматическом кольце (не катехоламины), выявляют больший стимулирующий эффект на ЦНС. чем катехоламины.
7 7.1. Агонисты прямого действия
Средства, стимулирующие а-и Р-адренорецепторы
Эноогенные и синтетические катехоламины
Три главных эндогенных катехоламина — эпинефрин (адреналин), норэпинефрин (норадреналин) и дофамин — осуществляют функционирование симпатической нервной системы и присутствуют практически во всех частях организма. Кроме того, большое число других лекар
- 207 -
Глава 11
ственных средств проявляют свою активность путем модификации действия одного или более из этих эндогенных веществ таким образом, что катехоламины оказываются задействованы в проявлении результата довольно широкого набора лекарств. В медицине применяются и синтетические симпатомиметики прямого действия, которые широко используются при лечении многих патологий. Терапевтические показания к применению катехоламинов основаны на их сосудосуживающем, бронхолитическом и сердечностимулирующем действии.
Эпинефрин (Epinephrine)
Эпинефрин - 1_-1-(3,4-дигидроксифенил)-2-метиламиноэтанол (11.1.2) получают как из ткани надпочечников убойного скота [1. 2], так и синтетическим путем. Синтез эпинефрина осуществляют исходя из ю-хлор-3,4-дигидроксиацетофенона - - хлорацетилпирокатехи-на, взаимодействием которого с избытком метиламина получают ю-метиламино-3,4-дигидрокси)ацетофенон (11.1.1). Восстановлением последнего водородом над никелем Ренея, действием амальгамы алюминия или электролитическим восстановлением получают D,L-эпинефрин (11,1.2) [3, 4, 5. 6, 7, 8, 9], который разделяют на изомеры с использованием (-)винной кислоты [10].
СО-СНг-CL
CH3NH2
CO-CH2-NHCH3
Н2 / Raney—Ni
11 1 1
НО
НО
11 1 2
Эпинефрин, эндогенный катехоламин, больше известен под официальным английским названием адреналин. Эпинефрин — мощный агонист, как cz- так и p-адренергических рецепторов. Его действие весьма сложно и зависит не только от сравнительного распределения адренергических рецептивных участков в разных тканях и органах, но также от дозировки и путей введения. Природный изомер эпинефрина(-) в 50 раз более активен, чем (+)изомер.
Препарат прямо и несиецифично активирует как а-, так и р-адренер-гические рецепторы. Активация а-адренорецепторов приводит к сужению большинства сосудов. Активация р,-адренорецепторов увеличивает ско
-208 -
Адренергические препараты
рость работы сердца и силу сокращений сердечной мышцы Активация р:-адренорецепторов приводит к расширению бронхов и сосудов скелетных мышц. Повышается уровень глюкозы в крови, понижается внутриглазное давление.
Несмотря на то, что основное фармакологическое действие отражается на сердечно-сосудистой и дыхательной системах, полный спектр его эффектов указывает его физиологическую значимость в качестве системного нейрогормона, вовлеченного в активацию большого ряда защитных функций.
Фактически он является прототипом многих адренергических препаратов. и поэтому следует несколько подробнее рассмотреть его действие на отдельные органы системы. Типичная реакция на внутривенное введение эпинефрина — стремительный подъем преимущественно систолического АД. Подобное действие эпинефрина возникает благодаря комбинированному действию, т. е. во-первых, сокращению большинства сосудов и, во-вторых, стимуляции миокарда, выражающейся повышением силы сокращений и частоты работы сердца. Эпинефрин и другие симпатомиметические препараты с р2-адреноагонистическими свойствами ответственны за релаксацию бронхиальной мускулатуры и усиление бронходила 1ации. Более того, а-адренергическая агонистическая активность эпинефрина проявляется в виде сокращения сосудов легких и развития противоотечных эффектов.
Препарат применяется при анафилактических, аллергических и других реакциях гиперчувсгвительности. в качестве агента, повышающего АД при гипотензии, в качестве бронхолитика при отеках легких, в качестве противоотечного средства в ЛОР и офтальмологической практике, а также с целью усиления и пролонгирования действия местных анестетиков.
Эпинефрин используется для купирования приступов бронхиальной астмы, для выведения из анафилактического шока, при гипергликемической коме, аллергических реакциях. Препарат применяют в качестве местного сосудосуживающего средства и, в частности, в офтальмологии для понижения внутриглазного давления.
Имеется огромное количество синонимов эпинефрина — аднефрин, адренат. биоренин, эпринал, гемостатин, ниералин. синдернин и др. Однако основным синонимом эпинефрина является адреналин.
Норэпинефрин (Norepinephrin)
Норэпинефрин — Б-1-(3,4-дигидроксифенил)-2-аминоэганол (11.1.4) предложено получать двумя методами, исходящими из 3,4-дигидро-ксибензальдегида. Согласно первому способу, взаимодействием с цианистым водородом указанный альдегид трансформируется
- 209-
Глава 11
в циангидрин (11.1.3). который далее восстанавливают в норэпинефрин (11 1 5) [11, 12].
Второй способ заключается в конденсации диацетата того же альдегида с нитрометаном с получением (3,4-диацетоксифенил)-2-нитро-этанола (11.1.5). нитрогруппу которого далее подвергают восстановлению. и продукт (11 1.6) гидролизуют в искомый норэпинефрин (11.1.4) [4.9.13,14].'
CH3NO2
сн3соо
>=\ ?н [Н]
сн3соо-4 л— ch-ch2no2 —
11 1 5
он
сн—ch2-nh2
NaOH
11 1 4
1116
Норэпинефрин — основной нейротрансмиттер, продуцируемый и высвобождаемый адренергическими нейронами, в литературе также называйся и описывается как (-)норадреналин или левартеренол. Этот вазопрессорный катехоламин уменьшает и сопротивляемость, и емкость кровяны . сосудов путем стимулирования а-адренорецепторов и имеет прямое кардиостимулирующее действие, осуществляемое путем активации р]-адренорецепторов. Норэпинефрин проявляет значительно меньшую активность, чем эпинефрин в качестве препарата для расширения сосудов путем активации 02-адренергических рецепторов. Типичной реакцией на внутривенное введение норэпинефрина является повышение как систолического, так и диастолического АД,
Норэпинефрин применяют для усиления сердечных сокращений и тля повышения АД при резком его понижении, которое может возникнуть при хирургических вмешательствах и травмах.
Синонимами препарата являются артеренол, левартеренол. левофед и др.
-210-
Адренергические препараты
Средства, стимулирующие преимущественно Р-адренорецепторы
Изопротеренол (Isoproterenol)
Изопротеренол — 1-(3.4-дигидроксифенил)-2-гтзо-пропиламино-этано.ч (11.1.8) получают по схеме, аналогичной схеме получения эпинефрина. Взаимодействием ю-хлор-3,4-дигидрокси-ацетофенона (хлорацетилпирокатехола) с изопропиламином получают со-изопро-пиламино-3,4-дигидроксиацетофенон (11.1.7), восстановлением в котором карбонильной группы водородом с использованием в качестве катализатора палладия на угле получают изопротеренол (11.1.8) [И, 12].
Н2 / Pd-C
11 1 8
сн3
сн3
Изопротеренол является представителем симпатомиметических препаратов с высокой селективностью к p-адренорецепторам. Как уже было указано, наличие объемистой мзо-пропильной или трет-бутильной групп при атоме азота р-фенилэтила.минного скелета обычно придает соединениям большее сродство к p-адренергическим рецептивным участкам, чем к а-адренергическим. Изопротеренол лишен значительного а-адренергического агонистического действия. Активация Pi-адренергических рецепторов в сердце повышает положительное хронотропное и ионотропное действие. Периферическое сосудистое сопротивление повышается путем вызванного задействованным р2-адре-нергической системой расширением сосудов, в основном, в скелетной му скула гуре, но также в почечном и мезентериальном кровообращении
Это сложное, объединенное кардиостимулирующее и сосудорасширяющее действие выражается значительным повышением сердечного выброса и ударного объема. В результате активации р2-адреноре-непторов усиливается также бронходилатация.
-211 -
Глава 11
Изопротеренол применяют при бронхоспазме, астме, сердечной блокаде, шоке.
Синонимами препарата являются протеренол, изопреналин, изад-рин, норизадрин, новодрин и др.
Изоэтарин (Isoetharine)
Изоэтарин — 3,4-дигидрокси-сс-[1-(нзо-пропиламино)пропил]бен-зиловый спирт (11.1.11), отличающийся от изопротеренола наличием дополнительного радикала — этильной группы в боковой этиламинной цепи, получают бромированием 3,4-дибензилоксибути-рофенона молекулярным бромом с последующим взаимодействием полученного бромпроизводного (11.1.9) с изопропила.мином. Продукт (11.1.10) подвергают восстановлению водородом с использованием палладиевого катализатора, при котором одновременно восстанавливается и карбонильная группа и осуществляется снятие защитных бензильных групп с получением изоэтарина (11.1.11) [15].
с6н5сн?о
)=\ и сбн5сн2о-4 Л—С-СН2-С2Н5
С6Н5СН2С)
С6Н5СН2О —4 С_СН С2Нг
'—' Вг
СНз h2n-ch(
СНз
11 1 9
СбНбСНгО,
>=\ я ,снз
с6н5сн2о—А />—C-CH-NH-сну v-y I СНз
С2Н5
Н2, Pd-C
НО л Он СН3
но—4 Л—ch-ch-nh-ch(
I Сн3
ь2Н5
11 1 11
Изоэтарин — симпатомиметик прямого действия со сравнительно низкой селективностью к р2-адренорецепторам. Однако вызывает быстрое успокоение бронхоспазма, по сравнению с более селективными бронходилата горами.
Применяют при лечении хронических обструктивных заболеваний верхних дыхательных путей.
Синонимами препарата являются астмалитан и бронкосол.
Тербуталин (Terbutaline)
Тербу талин — сс-[(дареги-бутиламино)метил]-3,5-дигидроксибензи-ловый спирт (11.1.14), принципиально отличающийся от рассмотренных соединений расположением гидроксильных групп в бен-
- 212 -
Адренергические препараты
зольном кольце, получают бромируя 3,5-дибензилоксиацетофенон в соответствующий 3,5-дибензилоксибромацетофенон (11.1.12), который вводят во взаимодействие с М-бензил-Х-отреот-бутиламином с получением аминокетона (11,1.13). Восстановление последнею водородом над палладиевым катализатором приводит к тербуталину (11.1.14) [16, 17. 18].
:6Н5СН2о
С6Н5СН2О
С6Н5СН2О
СбН5СН2О 11112
CH^CeHs
H-N
С(СН3)з
C3H5O Н2О
п ,СгксбНэ
4 Л“С-СН2—N
/ С(СН3)3
СбН5СН2О И 1 13
Н2 Pd“С
Тербуталин — синтетический симпатомиметический амин, Является одним из наиболее селективных стимуляторов р2-аДренорецепторов прямого действия. Стимулирует [32-адренорецепторы гладкой мускулатуры бронхов, расслабляя их, и сравнительно мало действует на Pi-адренорецепторы сердца.
Применяют для купирования и профилактики бронхоспазма при бронхиальной астме, хроническом бронхите, эмфиземе легких и других бронхолегочных заболеваниях.
Синонимами препарата являются бретин и бриканил.
Метапротеренол (Metaproterenol)
Метапротеренол — а-[(нзо-пропиламино)метил]-3,5-дигидрокси-бензиловый спирт (11.1.15) практически является аналогом тербуталина, в котором дарещ-бутиламинная группа замещена на изо-пропиламинную. Синтез препарата осуществляется аналоючно синтезу тербуталина [19, 20. 21].
но
но
он
СН-СН2-NH
11 1 15
СН3
-сну
СНз
-213-
Глава 11
Метапротеренол менее селективен, чем тербуталин и альбутерол, однако широко используется при лечении хронических обструктивных заболеваний верхних дыхательных путей.
Синонимами препарата являются орсипреналин, метапрел и алю-пенг.
Фенилэфрин (Phenylephrine)
Фенилэфрин — 1-(3-гидроксифенил)-2-метиламиноэтанол (11.1.16), принципиальное отличие которого от эпинефрина заключается в отсутствии гидроксильной группы при С4 ароматического колыаа, получают по схеме аналогичной получению эпинефрина, однако с применением вместо (в-хлор-3,4-дигидроксиацетофенона оэ-хлор-3-гидроксиацетофенона [1 1, 22, 23].
но он сн— ch2-nhch3
и 116
Этот синтетический препарат имеет как химическое, так и фармакологическое сходство с норэпинефрином. Характерная особенность фенилэфрина — это отчетливо выраженная селективность по отношению к «-адренорецепторам, особенно «(-адренорецепторам. Хотя фенилэфрин и увеличивает сокращаемость сосудов, он практически не является кардиостимулятором.
Фенилэфрин применяют при гипотензии, пароксизмальной суправентрикулярной тахикардии, шоке, а также местно, в частности в виде носовых капель для снятия отеков.
Синонимами препарата являются алмефрин. дегест, неокседрин, метаокседрин и многие другие.
Ритодрин (Ritodrine)
Ритодрии — 4-гидрокси-а-[1-[(4|-гидроксифенэтил)амино]этил]-бензиловый спирт (11.1.19) отличается от эпинефрина уже несколькими штрихами и находится в рассматриваемом ряду из-за наличия лишь одной гидроксильной группы в ароматическом кольце фени-лэтиламинного фрагмента классических симпатомиметиков. Второе существенное отличие от всего рассматриваемого ряда заключается в замене традиционного концевого изо-пропил- или трет-бу гитаминного фрагмента на н-гидроксифенилэтиламинный. На
-214-
Адренергические препараты
конец, третье его отличие — наличие метильной группы у а-атома фени.тэтиламинной части симпатомиметиков роднит его с изоэта-рином.
Препарат синтезируют исходя из 4-бензилоксипропиофенона, под-ьергая его бромированию в 4-бензилокси-а-бромпропиофенон (11.1.17). Последний вводят во взаимодействие с 2-(4-бензилоксифенил)этила-мином с получением промежуточного продукта (11.1.18), который далее повергают дебензилированию водородом с использованием палладиево-10 катализатора с получением ритодрина (11.1.19) [24, 25].
СбНдСНгО
H2N ~СН2~СН2
ОСНгСбНэ
Ритодрин является селективным стимулятором [^-адренорецепторов преиму щественно мочеполовой системы. Применяют в качестве юколитического средства при угрозе преждевременного прерывания беременности, только в специализированных медучреждениях.
Синонимами препарата являются утопар и препар.
Альбутерол (Albuterol)
Альбутерол — 2-/иреот-бутиламино-1-(4-гидрокси-3-гидроксиметил-фенил)этанол (11 1.26) принципиально отличается от всех вышеописанных симпатомиметиков тем, что гидроксильная группа при Cj ароматического кольца заменена на гидроксиметильную. Препарат предложено получать двумя путями. При первом способе исходят из 4-гидроксиацетофенона, хлорметилированием которого получают 4-гидрокси-З-гидрокси.метилацетофенон (11.1.20). Последний ацетилируют в диацетилпроизводное (11.1.21) и далее премируют в соответствующий бромацетофенон (11.1.22). Его взаимодействием с Х-бензил-Х-шреда-бутиламином получают производное аминоацетофенона (11.1.23), ацетильные группы которого гидролизуют соляной кислотой, и полученный при этом проду кт (11.1.24) подвергают восстановлению — вначале боргид-ридом натрия для трансформации кетонной группы в спиртовую (11.1.251, затем водородом над палладиевым катализатором для
-215-
Глава 11
удаления бензильной группы с получением альбутерола (11.1.26) [26,27,28.29,30].
сн2с6н5
С1СН3)з
CH3COONa СН3СООН / (СНзСО)гО
СН3СОО-СН2
сн3соо-сн2
CH3COG—\-С*СН2-Вг
СНзСОО -сн2
>=\ £ СН2С6Н5
СНзСОО-4 Л-С~СН2 — N
C(CH3h
11 1 23
HCL
НО-сн2
и рНгСб'-1;
С~СН2 —N С(СН3)з
НО-СН2
NaBH4 _ ?=\ ?Н СН2С6Н5
-----* HO—G Л-СН-СН2—N
С(СН3)з
Н2 ' Pd-C
но-сн2 у=\ ?н
НО—\-CH-CH2-NH—С(СНз)з
11 1 26
Второй способ синтеза мало отличается от вышеприведенного и заключается в первоначальном получении 4-гидрокси-З-ацетоксибром-ацетофенона (11.1.27) путем ацилирования метилового эфира салициловой кислоты хлорангидридом бромуксусной кислоты. Последний также вводят во взаимодействие с М-бензил-М-отре/и-бутиламином, и полученный продукт (11.1.28) полностью гидрируют алюмогидридом лития в N-бензилзамешенный альбутерол (11.1.29), бензильную группу которого также удаляют восстановлением водородом над палладиевым катализатором в искомый альбутерол (11.1.26) [31].
НОСН2
11 1 29
СН2СбН5
N
С(СН3)з
Н2/ Pd-C
НО-СН2
ОН
н°~/ \-CH-CH2-NH—С(СН3)з
11 1 26
-216-
Адренергические препараты______________________________
Альбутерол является р2-адренергическим симпатомиметическим амином со сходными с тербуталином фармакологическими характеристиками. Почти не оказывает действия на ргадренорецепторы сердца. Оказывает выраженный бронхолитический эффект, предупреждая или копируя спазмы бронхов, снижает сопротивление в дыхательных путях, х величивает жизненную емкость легких.
Широко используется при острой и хронической бронхиальной астме и других заболеваниях дыхательных путей, протекающих со спастическим состоянием бронхов.
Синонимами препарата являются алопрол, вентолин, вольмакс, сальбутамол, сальбувент. спреор и др.
Добутамин (Dobutamine)
Добутамин — (±)(4-[2(41 -гидроксифенил)-1 -метилпропил]-3,4-дигид-роксифенилэтиламин (11.1.31) серьезно отличается по строению от всех рассмотренных выше препаратов и, главным образом, отсутствием гидроксильной группы у p-углеродного атома фенилэтила-минного фрагмента классических симпагомиметиков. Вторым существенным отличием от рассмотренных препаратов является присутствие в качестве концевого аминного фрагмента н-гидрокси-фенил-нзо-бутиламинного фрагмента.
Препарат получают взаимодействием 3,4-диметоксифенилэтил-2-амина с 1-(4-метоксифенил)-3-бутаноном с одновременным восстановлением образующейся при этом иминной связи и получением продукта (11.1 30), метоксильные связи которого расщепляют бромистым водородом, получая добутамин (11.1.31) [32, 33].
сн3о
сн3
СН3О—d б— CH2-CH2-NH-CH-CH2-CH2—(х Л— ОСНз
НВг
11 1 30
НО
н2/ Pd-C
11 1 31
-217-
Глава 11
Добутамин является препаратом, селективно активирующим pi-адренорецепторы сердца и одновременно [32-адренорецепторы сосудов и а,-адренорецепторы миокарда. Механизм ею действия весьма сложен.
Добутамин применяют в случаях, когда при острой сердечной недостаточности необходимо кратковременно усилить сокращения миокарда и, в частности, при декомпенсации сердечной деятельности, связанной с хирургическими вмешательствами на сердце или при его органических заболеваниях.
Синонимом добутамина является добутрекс.
Средства, стимулирующие преимущественно а-адренорецепторы
Описываемые далее адреномиметики прямого действия — сс-адре-нергические агонисты по своему строению резко отличаются от рассмотренных выше препаратов.
Клонидин (Clonidine)
Клонидин — 2-(2,6-дихлорфениламино)имидазолин (11.1.34) синтезируют исходя из 2,6-дихлоранилина, взаимодействием которого с гиоцианагом аммония получают М-(2.6-дихлорфенил)тиомоче-вину (11.1.32). Метилированием последней в (11.1.33) дальнейшим взаимодействием с этилендиамином получают клонидин (11.1.34) [34.35,36,37,38,39].
11 1 34
Клонидин — селективный сс2-адренергический агонист. Клонидин оказывает выраженное гипотензивное действие, связанное со снижением общего периферического сосудистого сопротивления, урежением частоты сердечных сокращений, уменьшением сердечного выброса.
- 218 -
Адренергические препараты
Механизм действия препарата обусловлен стимуляцией сс2-адреноре-цепгоров тормозных структур головного мозга и уменьшением симпа-шческой импульсации к сосудам и сердцу.
Препарат понижает системное кровяное давление и скорость работы сердца, стимулируя сс2-адренорецепторы в определенных частях ЦНС, и используется в основном в качестве антигипертензивного средства. Клонидин применяют при различных формах гипертонической болезни и для купирования гипертонических кризов. Используют также в офтальмологической практике при открытоугольной глаукоме.
Синонимами клонидина являются гемитон. катапресан, клофелин.
Противоотечные симпатомиметики
Наиболее популярными препаратами в качестве противоотечных средств для слизистых оболочек сегодня являются препараты нафазо-лин. оксиметазолин, тетрагидрозолин и ксилометазолин, являющиеся производными имидазолина. Это полностью ct-адренергические агонисты, которые вызывают осушивающий эффект путем сужения кровяных сосудов в слизистых.
Для снятия отеков слизистых оболочек в медицине используются также фенилэфрин и фенилпропаноламин, который является симпато-миметиком смешанного действия.
Нафазолин (Naphazoline)
Нафазолин — 2-(1-нафтилметил)-2-имидазолин (11.1.36) синтезируют исходя из (1-нафтил)-ацетонитрила, который взаимодействием с этанолом переводят в иминоэфир (11.1.35) и далее подвергают гетероциклизации в искомое производное имидазолина (11.1.36) взаимодействием с этилендиамином [40].
С2Н5ОН
/—\ NH
С. «г—СН2~С-ОСгНз
4 11 1 35
H2NCH2CH2NH2
N-j
СН2-^ 11 1 36
Нафазолин применяют при острых ринитах, связанных с простудой, аллергическими реакциями, острыми и хроническими воспалительными состояниями, в частности, при воспалениях гайморовых полостей, а также для остановки носовых кровотечений.
Синонимами препарата являются нафтизин, санорин, риназин, привин.
-219-
Глава 11
Оксиметазолин (Oxymetazoline)
Оксиметазолин — 6-тл£>едг-бутил-3(2-имидазолин-2-илметил)-2,4-диметилфенол (11 1.39) получают хлормегилированием 6-трет-бутил-2,4-диметилфенола и дальнейшей трансформацией полуденного хлорметилпроизводного (11 1.37) в нитрил (11.1 38). Взаимодействием последнего с этилендиамином получают оксиметазолин (11 1 39) [41, 42].
Оксиметазолин применяют по тем же показаниям, что и нафазолин. в основном при ринитах
Синонимами препарата являются африн и дурамист.
Ксилометазолин (Xylometazoline)
Ксилометазолин — 2-(4-тл/?еш-бутил-2.6-диметилбензил)-2-имида-золин (11.1.40) также получают в одну стадию — циклоконденсацией 4-/н£>етл-бутил-2,6-диметилбензилцианида с этилендиамином [43, 44]
(СН3)3С
H2NCH2CH2NH2
Ксилометазолин применяют при ринитах, ларингитах, синуситах, воспалении гайморовой полости, аллергических заболеваниях полости носа и горла.
Синонимами препарата являются галазолин и отривин
-220 -
Адренергические препараты
Тетрагидрозолин (Tetrahydrozoline)
Тетрагидрозолин — 2-(1,2.3.4-тетрагидро-1-нафталинил)-2-имида-золин (11.1.41) получают в одну стадию — гетероцикл изапней 1-циа-ногетралина с этилендиамином [45].
HZNCHZCHZNHZ
Тетрагидрозолин в основном применяют, в качестве глазных капель для сужения сосудов, а также местно при небольших воспалениях, укусах.
Основными синонимами препарата являются визин и тизин.
11.2. Агонисты непрямого действия
Единственное соединение непрямого действия, находящее практическое применение только в экспериментах, — это тирамин, который проявляет симпатомиметический эффект, вызывая высвобождение эндогенного норэпинефрина. Очень быстро инактивируется МАО. Клинического применения практически не имеет.
Тирамин (Tyramine)
Тирамин — 4-(2-аминоэтил)фенол (11.2.1) можно синтезировать различными путями и. в частности, декарбоксилированием тирозина [46, 47, 48]. Препарат выделяют также из тканей убойного скота.
но
НО-СН2 “СН2 “NHg
112 1
Синонимами препарата являются мидриал, утерамин и др.
11.3. Агонисты смешанного действия
Практически все обсуждаемые вещества смешанного действия имеют непрямое действие типа тирамина в дополнение к прямому — активации адренорецепторов и являются а- и [3-адреномиметиками непрямого (опосредованного) действия.
- 221 -
Г лава 11
Дофамин (Dopamine)
Дофамин — 2-(3.4-дигидроксифенил)-этиламин (11.3.1) в качестве лекарственного средства получают деметилированием 2-(3,4-диметокси-фенил)эгиламина(19.4.3) бромистым водородом [49, 50, 51].
СНоО
СН3О
сн2-ch2-nh2
НВг
НО
сн2- сн2- nh2
19 4 3
113 1
Дофамин найден во всех симпатических нейронах и ганглиях и в ЦНС. Как препарат в дополнение к стимуляции дофаминергических рецепторов дофамин опосредованно стимулирует и а- и p-адренорецепторы. Дофамин вызывает также высвобождение эндогенного норэпинефрина. Механизм действия основан на возбуждающем влиянии на р-адрено-рецепторы (в малых и средних дозах) и на а-адренорецеторы (в больших дозах). Оказывает положительное инотропное действие на сердце; улучшает кровоснабжение; избирательно расширяет почечные и брыжеечные кровеносные сосуды; не вызывает снижения артериального давления: незначительно увеличивает частоту сердечных сокращений.
Дофамин проявляет свое основное действие на сердечнососудистую систему, почки, а также брыжейку. Используется как временное средство для лечения гипотензии и циркуляторного шока, вызванного инфарктом миокарда, травмой, отказом почек, эндогенной септицемией. Главным показанием к применению препарата является шок различного генеза (кардиогенный, послеоперационный, инфекционно-токсический, анафилактический); тяжелая гипотония; угрожающая почечная недостаточность.
Синонимом дофамина является интропин.
Эфедрин (Ephedrine)
Эфедрин — [_-э£шш/7о-1-фенил-2-метиламинопроианол-1 (11.3.4) синтезируют разными путями исходя из бензальдегида. Согласно первому способу, бензальдегид конденсируют с нитроэтаном с получением 2-метил-2-нитро-1-фенилэтанола (11.3.2), который восстанавливают до 2-метил-2-амино-1-фенилэтанола (11.3.3). представляющего собой смесь изомеров, из которой дробной кристаллизацией выделяют необходимый 1-изомер, Метилированием последнего полу чают эфедрин (11.3.4) [52, 53]
- 222 -
Адренергические препараты
СНО
CJHgNOz
/=\ . 3
4 Л—CH-CH-NH2
' он
СН31
/=\ ?нз
4 Л—CH-CH-NHCH3 '—' он
11 34
Второй способ синтеза заключается в сбраживании глюкозы карболигазой дрожжей в присутствии бензальдегида, который при этом превращается в (-)-1-фенил-2-кетолропанол (11.3.5). Последний восстанавливают водородом в присутствии метиламина с получением искомого эфедрина (11.3,4) [54, 55].
сно
113 5
о и
-с-сн3
CH3NH?/ Н2/ Pt
/=\ СНз
Z h— CH-CH-NHCH3 '—' он
1134
Эфедрин — алкалоид, содержащийся в различных видах семейства эфедровых, и его до сих пор экстрагируют из Ephedra simca и Ephedra equisetinaL Ввиду наличия в нем двух асимметрических атомов, существует в четырех изомерных формах Псевдоэфедрин (d-изоэфедрин) — стереоизомер с фармакологическим действием, очень тонко отличающимся от эфедрина Фармакологическое действие эфедрина типично для некатехоламинных симпатомиметиков смешанного типа. Препарат стимулирует как а-, так и p-адренорецепторы и одновременно вызывает высвобождение норэпинефрина из синаптических нейронов. Его сосудосуживающая способность примерно в 100 раз уступает таковой у эпинефрина, однако продолжительность действия примерно в 10 раз больше. Препарат значительно менее токсичен, чем эпинефрин, что позволяет широко применять го в медицине.
Основное применение препарат находит при бронхиальной астме, аллергических заболеваниях, в качестве противоотечного для слизистых оболочек при ринитах, а также в качестве средства, повышающего давление при оперативных вмешательствах. Применяют местно в офтальмологии как сосудосуживающее средство для расширения зрачка.
Синонимами препарата являются эцифин, эфедрол, манадрин, каль-цидрини др.
Фенилпропаноламин (Phenylpropanolamine)
Фенилпропаноламин — В,Ь-э/д4шро-1-фенил-2-метиламинопропа-нол-1 (11.3.7) синтезируют из пропиофенона, нитрозируя его до иэо-нитрозопропиофенона (11 3.6). Восстановлением последнего
-223-
Глава 11
водородом в соляной кислоте с одновременным использованием двух катализаторов, палладия на угле и платины на угле, получают норэфедрин (11.3.7) [56, 57, 58, 59].
Фармакологическое действие фенилпропаноламина сходно с действием эфедрина. Этот симпатомиметик может вызвать временное повышение давления крови и используется по тем же показаниям, что и эфедрин, главным образом при простудных заболеваниях в составе различных комбинированных лекарств. Кроме того, он обладает слабым центральностимулирующим и анорексигенным действием.
Основным синонимом препарата является норэфедрин.
Метараминол (Metaraminol)
Метараминол — Г-1-(3-гидроксифени,з)-2-аминопропан-1-ол (11.3.11) предложено получать двумя путями. Первый способ (синтетический) исходит из 3-гидроксипропиофенона. Гидроксильную группу последнего защищают алкилированием хлористым бензилом с получением 3-бензилоксипропиофенона (11.3.8). Взаимодействием с бу-тилнитритом последний подвергают нитрозированию в изо-нитро-зокетон (11.3.9), который восстановлением водородом над никелем Ренея переводят в 1-(3-бензил-оксифенил)-2-аминопропан-1-ол (11.3.10), защитную бензильную группу последнего удаляют восстановлением водородом с использованием палладиевого катализатора и получением рацемического метараминола (11.3.11). Искомый L-изомер выделяют с помощью (+)-винной кислоты [60, 61]
НС CishgChaC /^Ч Й СсНэСН2С_ V/---CH2~C-b ► 11 3S C6HSCH2O 7=\ nh2 4 ^/“СН-СН-СНз ' ' OH 11 3 10 1? CjHgNC; /==\_" - Ra.e/--Ni_ C Cn2-CH3 • c Ci .j \OH -1 3 d HO H2/Pd-C ^=\ ^H2 * У-СН-СН-СНз ' ' OH 113 11
- 224 -
Адренергические препараты
Второй способ (полисинтетический) заключается в сбраживании D-глюкозы в присутствии 3-ацетоксибензальдегида с получением гидрокси-1-(3-гидроксифенил)-ацетона (11.3.12), карбонильную группу которого восстанавливают водородом над палладиевым катализатором в присутствии аммиака с получением метараминола (11.3.11) [62, 63, 64, 65J.
о
-с-СНз
NH3 н2, Ра-С
НО
nh2 СН-СН-СНз он
113 11
Метараминол — симпатомиметический амин прямого и непрямого действия, имеющий гемодинамические характеристики, сходные с норэпинефрином. Препарат обладает способностью поднимать как систолическое, так и диастолическое давление.
Препарат применяют при гипотензивном шоке, с целью поднятия давления, который может возникнуть при спинномозговой анестезии, при хирургических осложнениях, при травмах головного мозга.
Синонимами метараминола являются арамин, изофенилэфрин, ме-тарадин и др.
Амфетамины (Amphetamines)
Термин амфетамины обычно используется в отношении рацематам амфетамина, декстроамфетамина (8.1.2.2) и метамфетамина (8.1.2 3).
СНз
СН2~СН—ЫНз
СНз
СН2-СН-NHCH3
8 122
8 123
Как препараты смешанного типа действия амфетамины активируют адренергические рецепторы и одновременно высвобождают эндогенные катехоламины, норэпинефрин и дофамин из нейронов мозга и периферии. Симпатомиметические свойства на периферии весьма сходны с таковыми эфедрина. Амфетамин повышает систолическое и диастолическое давление и имеет слабо выраженное бронхолитическое действие. Эти эффекты более продолжительны, но менее выражены, чем у эпинефрина. Отличительной особенностью амфетаминов является их психостимулирующая активность. Большие дозы могут вызвать галлюци-
-225-
Глава 11
нации, психотическое состояние сходное с параноидальной шцзо френией В качестве симпатомиметика амфетамин иногда применяют при с дабост родовой деятельности
Синонимами амфетамина являются фенамин и бензедрин
Список литературы
1 lakaminel J Soc Chem Ind 20, 746, (1901)
2 Aldrich P / Am J Physiol 5,457,(1901)
3 Stolz 4 Ber 37,4149(1904)
4 Payne G Ind Chem 37 523 (1961)
5 Loewe G / Arzneimittel Forsch 4, 583 (1954)
6 Ger Pat 152 814 (1903)
Gei Pat 157 300(1903)
8 Ger Pat 222 451 () 908)
9 Tullar В J Am Chem Soc 70, 2067 (1948)
10 Flacher D //Z Physiol Chem 58 189 ((1908)
11 Ger Pat 723 278 (1942)
12 US Pat 2 308 232 (1943)
13 US Pat 2 774 789 (1956)
14 Pratesi et al / J Chem Soc 1959, 4062
15 Ger Pat 638 650 (1936)
16 Brit Pat 1 199 630(1967)
17 Belg Pat 704 932 (1968)
18 US Pat 3 937 838 (1976)
19 US Pat 3 341 594 (1967)
20 Ger Pat 1 275 069 (1960)
21 Belg Pat 611 502 (1961)
22 US Pat 1 932 347 (1934)
23 Bergmann M et al1 J Org Chem 16. 84 (1951)
24 Belg Pat 660 244 (1965)
25 US Pat 3 401 944 (1968)
26 LSPat 3 642 896 (1972)
27 S Afr Pat 67 05 591 (1968)
28 US Pat 3 644 353 (1972)
29 Collin D etal J Med Chem 13,674(1970)
30 Collin D etal J Med Chem 14. 89s (1971)
31 US Pat 3 70s 233 (19^2)
32 Ger Pat 2 317 710(1973)
33 LSPat 3 987 200 (19'76)
- 99P, .
Адренергические препараты
34 35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51 '2
53
54
55
56 5-7
58
59
60
61
62
63
64
65
Ger Pat 1 303 141 (.1961)
US Pat 3 202 660 (1965)
US Pat 3 236 857 (1966)
Belg Pat 653 933 (1964)
Brit Pat 1 016 514 (1962)
Brit Pat 1 034 938 (1964)
US Pat 2 161 938 (1939)
US Pat 3 147 275 (1964)
Ger Pat 1 117 588 (1961)
US Pat 2 868 802 (1959)
Ger Pat 1 049 387 (1957)
US Pat 2 731 471 (1956)
Barger G J Chem Soc 95, 1127 (1909)
leaserH TIeh Chim Acta 8,766(1925)
BuckR J Am Chem Soc 55. 3389 (1933)
BaxelerS Ann 513, 196(1934)
StiehlH GBer 69,2640 (1936)
Ger Pat 247 906(1909)
Spath E et al / Monatsh 41, 319 (1920)
Manske et al J Am Chem Soc 51,580, 1906(1929)
US Pat 1 956 950(1934)
Ger Pat 469 782(1926)
Nagai И etal / Arm 470, 157 (1929)
US Pat 3 028 429(1962)
Hartung W etal//J Am Chem Soc 51, 2262 (1929)
Hartung И etal ;J Am Chem Soc 74. 5927 (1952)
Ger Pat 555 404 (1930)
US Pat 1 951 229 (1934)
Ger Pat 571 229 (1930)
US Pat 1 948 162 (1934)
US Pat 1 995 709 (1935)
Brit Pat 396 951 (1932)
Глава 12
{ Адреноблокирующие
I препараты
Термин «адреноблокатор» относится к препаратам, способным конкурировать с катехоламинами и другими адреномиметиками за связывание с адренергическими рецепторами и тем самым блокировать эффекты симпатических нервов, вызванных либо их стимуляцией эндогенными симпатомиметиками, либо введенными извне экзогенными адренергическими препаратами. Процесс синтеза норэпинефрина (норадреналина) в организме истинные адреноблокаторы не затрагивает.
В соответствии с вызываемым в организме ответом адреноблокирующие препараты классифицируются как а-адреноблокаторы, р-адре-ноблокаторы и блокаторы адренергических нейронов.
а-адреноблокаторы вызывают расширение периферических сосудов, а некоторые из них расслабляют и гладкую мускулатуру.
В противоположность этому P-адреноблокаторы мало влияют на сосудистый тонус. Более того, p-адреноблокаторы предотвращают сосудорасширяющее действие эпинефрина. В таких органах, как сердце, которые регулируются преимущественно Р-адренорецепторами, р-адре-ноблокаторы противодействуют возбуждающему действию норэпинефрина.
В свою очередь, а- и p-адреноблокаторы подразделяются на селективные и не селективные. Неселективные (3-адреноблокаторы проявляют сродство как к р,-, так и к р2-адренорецептивным участкам. К препаратам этой категории относятся препранолол, надолол, тимолол и лабеталол (комбинированный а- и p-адреноблокатор). Селективными pt-блокаторами являются ацебутол, атенолол, эсмолол и метопролол, которые в терапевтических дозах преимущественно связываются с р|-адренорецептивными участками.
На сегодняшний день еще нет терапевтически полезных селективных р2-адреноблокаторов, хотя уже существует ряд экспериментальных соединений с выраженной р2-адреноблокирующей активностью.
Наиболее широко P-адреноблокаторы применяют при лечении стенокардии, гипертонической болезни, тахикардии, аритмии.
-228-
Адреноблокирующие препараты
Совершенно аналогично а-адреноблокаторы также подразделяются на селективные и неселективные. Примерами неселективных а-адреноблокаторов являются фентоламин и феноксибензамин, а также эргоалкалоиды — эрготамин и эргоновин. Несмотря на то, что их практическое применение в медицине не связано с их а-блокирующей способностью, исторически они были первыми исследованными а-адре-ноблокаторами. Селективные а-адреноблокаторы в терапевтических дозах выявляют большую степень сродства либо к «|-адренорецеп-тивным участкам (празозин, теразозин), либо к аг-адренорецептивным у часткам (иохимбин).
В медицинской практике а-адреноблокаторы — препараты, блокирующие at- и а2-адренорецепторы, — используются сравнительно редко. Наиболее важным эффектом a-адреноблокаторов является расширение периферических сосудов, в связи с чем их применяют при различных нарушениях периферического кровообращения, при геморрагическом и кардиогенном шоке, при котором типичным признаком является спазм артериол.
Последней группой адреноблокаторов, условно названных блокаторами адренергических нейронов, являются препараты, подавляющие синтез, хранение и высвобождение биогенных аминов (норэпинефрина, дофамина или серотонина) в нервных окончаниях.
К этому ряду препаратов относятся резерпин, гуанадрел, гуанети-дин и метирозин, и их используют главным образом в качестве антигипертензивных препаратов.
12.1. $-адреноблокаторы
Препараты, проявляющие обратимое конкурентное блокирующее действие на Р-адренорецептивные участки рецепторной системы и противодействующие эффектам катехоламинов, называются Р-адренобло-каторами.
Эти препараты селективно уменьшают кардиостимулирующее, сосудорасширяющее, бронхолитическое и метаболическое (гликогенолитическое и липолитическое) действия катехоламинов, высвобождаемых из адренергических нервных окончаний и надпочечников.
Введение Р-адреноблокаторов в медицину явилось одним из основных достижений фармакологии сердечно-сосудистой системы. Вначшге эти препараты использовались только при лечении эссенциальной гипертонии. В настоящее время они используются при лечении стенокардии. аритмиях, мигрени, инфаркте миокарда, глаукоме.
Глава 12
Их эффективность при .многих болезнях объясняется конкурентным блокированием p-адренорецепторов в автономной нервной системе, в принципе любым из примененных препаратов класса 1-арилокси-З-аминопропанолов-2, в результате чего понижаются скорость работы сердца и сила сердечных сокращений, замедляется атриовентрикулярная проводимость, понижается уровень ренина в плазме, понижается давление. Центральные эффекты p-адреноблокаторов выявляются и на уровне сосудодвигательного центра в гипоталамусе, в результате чего замедляется выход симпатических тонических импульсов.
В основную группу p-ареноблокирующих препаратов включены пропранолол, метопролол, надолол. атенолол, тимолол, ацебутол, пиндо-лоп, эсмолол и комбинированный а- и р-адреноблокатор — лабеталол.
В химической структуре Р-адреноблокаторов много общего. Практически все они являются производными 1-арилокси-З-аминопропа-нола-2. в положении С( которого обязательно наличие замешенной или незамещенной ароматической или гетероароматической группы, связанной эфирной связью с трехуглеродной цепью. Радикал R при атоме азота пропанового фрагмента должен быть представлен либо третичной бутильной группой (надолол, тимолол), либо изо-пропильной группой (все остальные препараты).
Ar-O-CH2-CH-CH2 —NH-R ОН
Следует отметить, что замещенная этаноламинная группа в структуре p-адреноблокаторов сходна с таковой у многих соединений с агонистической адренергической активностью (изопротеренол (11.1.9), альбутерол (11.1.21) и др.), и поэтому не исключено, что она может быть ответственной за высокое сродство рассматриваемых адреноблокаторов к Р-адренергическим рецепторам.
Левовращающие изомеры этих препаратов являются гораздо более мощными адреноблокаторами, чем правовращающие, однако все эти препараты производятся и используются в виде рацемических смесей.
Рассматриваемые препараты обратимо связываются с Р-адренер-гическими рецептивными участками и конкурентно предотвращают активацию этих рецепторов катехоламинами, высвобождаемыми симпатической нервной системой или вводимыми извне симпатомиметиками.
Как уже было отмечено, Р-адренорецепторы подразделяются на 3.-адренорецепторы, которые преимущественно находятся в сердечной мышце, и р?-адренореиепторы, находящиеся преимущественно в бронхиальной и сосудистой мускулатуре. Соответственно. Р-адрено-
-230-
Адреноблокирующие препараты
блокирующие вещества классифицируются по их селективности по отношению к этим рецепторам.
Соединения, проявляющие примерно равное сродство к рг и р2-ре-пепгорам независимо от дозы, классифицируются как неселективные блокаторы, например, надолол, пропранолол, пиндолол, тимолол и лабе-|элол (комбинированный а- и Р-адреноблокатор). Препараты, имеющие в терапевтических дозах большее сродство к Pi-рецепторам, чем к р2-ре-цепторам, например, ацебутол, атенолол, метопролол и эсмолол, называются селективными или кардиоселективными р-адреноблокаторами.
Важно отметить, что эта селективность не абсолютна и что она зависит от вводимых доз. При больших дозах селективность нивелируется и оба подтипа p-адренорецепторов ингибируются в равной мере. В дополнение к блокированию Р-адренорецепторов эти препараты воздействуют на сердечно-сосудистую систему и иным способом.
Пропранолол (Propranolol)
Пропранолол — 1-(гг?о-пропиламино)-3-(1-нафтокси)-2-пропанол (12.1.2) предложено получать двумя путями исходящими из одних и тех же исходных веществ. Первый способ заключается во взаимодействии 1-нафтола с эпихлоргидрином. Раскрытием эпоксидного кольца получают 1-хлор-3-(1-нафтокси)-2-пропанол (12.1.1), который далее вводят во взаимодействие с изо-пропиламином с получением пропранолола (12.1.2).
Второй способ синтеза, осуществляемый теми же реагентами, но уже в присутствии основания, заключается в первоначальном получении 3-(1-нафюкси)пропиленоксида (12.1.3), дальнейшее взаимодействие которого с изопропиламином, происходящее с раскрытием эпоксидного кольца, приводит к получению пропранолола (12.1.2) [1, 2, 3, 4, 5, 6].
-231 -
Глава 12
Пропранолол самый старый и наиболее широко используемый неселективный [3-адреноблокатор, который является прототипом этого ряда препаратов. Обладает антиангинальным, гипотензивным и анти-аритмическим действием. Пропранолол — сердечный депрессант, который действует и на механические, и электрофизиологические свойства миокарда. Он может блокировать атриовентрикулярную проводимость и автоматизм потенциалов синусного узла, а также адренергическую стимуляцию, вызванную катехоламинами, тем не менее препарат понижает сократимость миокарда, скорость работы сердца, давление крови и потребность миокарда в кислороде.
Все эти свойства и делают пропранолол и другие Р-адреноблокаторы полезными антиаритмиками и антиангинальными препаратами.
Пропранолол понижает кровяное давление у большинства пациентов с эссенциальной гипертонией. Этот эффект может быть вызван рядом возможных механизмов. В их числе понижение сердечного выброса, ингибирование высвобождения ренина, понижение симпатического выброса из ЦНС, ингибирование высвобождения норэпинефрина из симпатических постганглионарных нейронов и т. п.
Однако ни один из предложенных механизмов не объясняет адекватно антигипертензивную активность пропранолола и других р-блокаторов.
Пропранолол показан при лечении артериальной гипертонии, стенокардии, экстрасистолии, суправентрикулярной аритмии, вентрикулярной тахикардии, мигрени, гипертрофическом субаортальном стенозе и феохромоцитоме. Применяется также в послеострой фазе инфаркта миокарда.
Общепринятыми синонимами препарата являются анаприлин, обзи-дан, индерал и многие другие.
Метопролол (Metoprolol)
Метопролол — 1-(«зо-пропиламино)-3-[4|(2-метоксиэтил)фенокси)]-2-пропанол (12.1.5) получают взаимодействием 4-(2-метоксиэтил)-фенола с эпихлоргидрином в присутствии основания с выделением 1,2-эпокси-3-[4'(2-метоксиэтил)фенокси)]пропана (12.1.4), дальнейшее взаимодействие которого аналогично описанному выше с изо-пропиламином, происходящему с раскрытием эпоксидного кольца, приводит к получению метопролола (12.1,5) [7, 8].
;н3о -сн2-сн2
ОН
О
СН2С!
NaOH /^Х _
---► сн3о-сн2-сн2—4 п—о-сн2—*
CjH7NH2
-232-
Адреноблокирующие препараты
СН3О-СН2-СН2
12.1.5
СН3
о-сн2-сн-сн2 —NH-CH ОН СНз
В отличие от пропранолола, блокирующего как рг, так и р2-адре-норецепторы, метопролол проявляет кардиоселективное действие, т. е. в терапевтических дозах он блокирует ргадренорецепторы с незначительным воздействием на р2-адренорецепторы.
Метопролол применяют при умеренной гипертензии, острых состояниях, инфаркте миокарда, для предупреждения омертвления сердечно-сосудистых тканей, при стенокардии, тахикардии, экстрасистолии, для вторичной профилактики после перенесенного инфаркта.
Наиболее распространенными синонимами препарата являются ло-прессор, беталок и др.
В медицине широко используются и другие [3-адреноблокаторы, методы синтеза которых мало отличаются от вышеприведенных. Поэтому далее приводятся лишь их названия, структурные формулы, фармакологические свойства и синонимы.
Ацебутол (Acebutol)
Ацебутол — 3'-ацетил-4'-[2-гидрокси-3-(«зо-пропиламино)пропо-кси]бутиранилид (12.1.6) [9, 10].
СНз О-СН2-СН-СН2 — NH-CH CH3CO.Jx. он СНз
NH -СО-С3Н7
Ацебутол — селективный Pi-адреноблокатор. Обладает антианги-нальным, гипотензивным и антиаритмическим действием. Применяют при артериальной гипертонии, для профилактики приступов стенокардии. при нарушениях ритма сердца.
Синонимами препарата являются ацебутолол, секторль и др.
Атенолол (Atenolol)
Атенолол — 2-[41 [2-гидрокси-3-(«эо-пропиламино)пропокси]фенил]-ацетамид (12.1.7) [11. 12, 13].
-233 -
Глава 1 2
СНз
о-сн2 -сн -СН2 —NH-CH
X. ОН СНз
ф
ch2-conh2
Атенолол — селективный р|-адреноблокатор. или иначе — кардиоблокатор. Так же как и ацебутол, атенолол обладает антиангинальным, гипотензивным и антиаритмическим действием. Применяют при артериальной гипертонии, для профилактики приступов стенокардии, синусовой тахикардии, наджелудочковых тахиаритмий.
Синонимами препарата являются тенормин, калтен, ибинол и др.
Надолол (Nadolol)
Надолол — 1-(/и/!етл-бутиламино)-3-[(5,6,7,8-тетрагидро-г/ио6,7-дигид-рокси-1-нафтил)-окси]-2-пропанол (12.1.8) [14, 15, 16].
но
но
О-СН2 -CH -СН2 -NH -С(СН3):
X он
Надолол — неселективный p-адреноблокатор пролонгированного действия. Так же как и рассмотренные выше P-адреноблокаторы, препара 1 обладает антиангинальным, гипотензивным и антиаритмическим действием. Применяют при артериальной гипертонии, для профилактики приступов стенокардии, синусовой тахикардии.
Синонимами препарата являются коргард. солгол, корзид и др.
Пиндолол (Pindolol)
Пиндолол — 1-(индол-4-илокси)-3-(гтзо-пропиламино)-2-пропанол (12.1.9) [17, 18].
СНз
н
- 234 -
Адреноблокирующие препараты
Пиндолол, как и надолол, является неселективным Р-адренобло-катором. Препарат обладает антиангинальным, гипотензивным и антиаритмическим действием. Применяют при артериальной гипертонии, при стенокардии напряжения (профилактика приступов), наджелудочковой тахикардии, тахисистолической форме мерцания предсердий, наджелудочковой и желудочковой экстрасистолии.
Синонимами препарата являются карвискен, вискен и др.
Тимолол (Timolol)
Тимолол — 1-(дадеда-бутила.мино)-3-[(4-морфолино)-1,2,5-тиадиазол-3-ил)окси]-2-пропанол (12.1.10) [19, 20, 21, 22, 23, 24, 25, 26, 27].
О-СН2 -CH-сн2 -NH -С(СН3)з
ГЛ ОН
12 1.10
Тимолол — неселективный p-адреноблокатор, предупреждающий действие катехоламинов. При местном применении в виде глазных капель понижает внутриглазное давление. Применяют при хронической открытоугольной глаукоме, вторичной глаукоме, закрытоугольной глаукоме.
Синонимами препарата являются модукрин, тимакор, тимоптол, блокадрен, тимолид и др.
Лабеталол (Labetalol)
Лабеталол — 2-гидрокси-5-[1-гидрокси-2-[(1-метил-3-фенилпропа-нол)амино]этил] бензамид (12.1.12) предложено получать N-алки-лированием М-бензил-М-(4-фенил-2-бутил)амина 5-бромацетилса-лициламидом с получением аминокетона (12.1.11), который далее дебензилируют водородом с использованием палладий-платинового катализатора на угле в лабеталол (12.1.12) [28, 29, 30].
-235-
Глава 12
HjN “С
?=\_ ?н ? /=\
40 Cb — CH2“f'’—9Н~СН2“СН2 —4 /1
Структурно лабеталол отличается or других p-адреноблокаторов уже хотя бы тем, что является производным этаноламина, а не 1-арил-окси-З-аминопропанола-2, что не содержит арилоксильной группы, что является производным фенилпропиламина, а не изопропил- или трет-бутиламина, что амипоэтанольный фрагмент его соединен с ароматическим фрагментом не эфирной связью. В то же время ароматическая часть молекулы, в отличие от вышеприведенных типичных структур P-адреноблокаторов. достаточно функционализированна и представляет собой замещенный салициламид. Структурно препарат напоминает добутамин (11.1.31), однако, в принципе, малые структурные отличия делают его антагонистом добутамина. В фармакологическом плане он является селективным конкурентным блокатором а,- и неселективным блокатором Р-адренергических рецепторов, что приводит к снижению давления у гипертензивных пациентов.
Показанием для применения лабеталола является лечение эссенциальной гипертонии.
Основными синонимами препарата являются трандат, абетол, ами-пресс, прессалол и др.
12.2. си-адреноблокаторы
Соединения, способные блокировать а-адренергические рецептивные участки, предотвращая действие на них различных агонистов, называются а-блокаюрами.
Характерной особенностью а-адреноблокаторов является их способность уменьшать прессорный эффект фармакологических доз эпинефрина (адреналина).
В частности, постсинаптические а .-блокаторы действуют на «-рецептивные участки, расположенные на сосудистой гладкой мускулатуре, и противодействуют прессорному, сосудосуживающему эффекту эпинефрина и норэпинефрина. Кроме того, они проявляют прямой расслабляющий эффект на гладкую мускулатуру, что приводит к периферическому расширению сосудов, в итоге приводящее к понижению давления. Однако они проявляют также кардиостимулирующий эффект.
- 236 -
Адреноблокирующие препараты
что нередко становится причиной тахикардии.
Пресинапгические «г-рецептивные участки расположены на симпатических нервных окончаниях, и их блокада, очевидно, по механизму обратной связи повышает выброс эпинефрина из нервных окончаний. Подобное фармакологическое действие имеет весьма ограниченную клиническую полезность, однако является ценным лабораторным инструментом. Представителем селективных а2-адреноблокаторов является алкалоид иохимбин.
Клинически полезными а-блокатора.ми являются:
1) долгодействующие неконкурентные антагонисты (фенокси-бензамин), которые образуют прочные химические связи с «-рецептивными участками, в результате чего блокада а-рецептора может длиться днями и даже неделями;
2) обратимые конкурентные антагонисты — неселективные (фентоламин, толазолин) и агселективно действующие (празозин, тетразозин) — обратимо и конкурентно блокируют а-рецептивные участки; их действие может продолжаться несколько часов. В то же время, блокада ими а-рецепгоров может быть прервана и прекращена действием больших доз агониста, например норэпинефрина;
3) эргоалкалоиды (эрготамин, эргоновин) также проявляют определенную неселективную а-адреноблокирующую активность, однако, в первую очередь, проявляют спазмогенное действие на гладкую мускулатуру, вызывая сужение сосудов;
4) селективные а~адреноблокаторы, представителем которых является алкалоид иохимбин, имеют ограниченное клиническое применение.
До л го действующие неконкурентные антагонисты
Феноксибензамин (Phenoxybenzamine)
Феноксибензамин — Ы-(2-хлорэтил)-М-( 1 -метил-2-феноксиэтил)бен-зиламин (12.2.5) получают взаимодействием фенола с прониленокси-дом с получением 1-фенокси-2-пропанола (12.2.1), хлорированием которого хлористым тионилом получают 1-фенокси-2-пропилхлорид (12.2.2). Взаимодействие последнего с 2-аминоэтанолом приводит к получению 1-фенокси-2-(2-гидроксиэтил)аминопропана (12.2.3). Алкилированием вторичной аминогруппы последнего получают
-237 -
Глава 12
\-(2-гидроксиэтил)-К-( 1 -метил-2-феноксиэтил)бензиламин (12.2.4), гидроксильную группу которого хлорируют хлористым тионилом с получением феноксибензамина (12.2.5) [31].
СНз
г-сн -NH-СН2-Сг|2-Oh
12 2 3
CeHjCHiCI
и2МСЧч-н2ОН
SOCI2
/=\ СНз
О-СН2-СН-К1-СН2-СН2-С1
12 2 5
Феноксибензамин является галогеналкиламином, структурно родственным алкилирующим агентам, используемым в химиотерапии. Механизм его долговременной блокады а-адренорецепторов, вероятно, дол-кен бы 1 ь объяснен их необратимым алкилированием. Скорее всего, после кратковременного воздействия на а,- и а2-адренорецепторы наступает их необратимая блокада. Возможно, Р-хлорэтиламинный фрагмент в 1канях организма образует высокореакционноспособный этиле-нимониевый интермедиат, который затем необратимо алкилирует рецептор. Подобная блокада необратима и называется неравновесной рецепторной блокадой.
Феноксибензамин применяется при лечении феохромоцитомы, опухоли мозгового слоя надпочечников, при которой продуцируется большое количество эпинефрина, что ведет к значительному повышению давления.
Синонимом препарата является дибензилин.
Обратимые конкурентные антагонисты
Неселективные адреноблокаторы
Толазолин (Tolazoline)
Толазолин — 2-бензил-2-имидазолин (12.2.7) получают гетероциклизацией этилендиамина с этиловым эфиром иминофенилуксусной
- 238-
Адреноблокирующие препараты
кислоты (12.2.6) с получением целевого продукта (12.2.7) [32, 33, 34, 35]. Структура толазолина поразительно напоминает «-адренергические агонисгы — противоотечные симпатомиметики.
С2Н5ОН
NH CH2-C-OC2h5
H2NCH2CH2NH2
1226
Толазолин слабый, обратимый а-адреноблокатор — препарат, понижающий сопротивление периферических сосудов и повышающий венозную наполняемость. Однако он проявляет и р-адреномимети-ческую активность, заключающуюся в стимуляции работы сердца, которая выражается в виде тахикардии, и холинергическую активность, заключающуюся в стимуляции работы ЖКТ, и гистаминоподобную активное! ь, заключающуюся в стимуляции желудочной секреции.
Препарат показан для лечения устойчивых форм легочной гипертензии у новорожденных, в случаях, когда системная артериальная оксигенация не может быть достигнута обычным путем при тщательнейшем наблюдении профессионалов.
Синонимами препарата являются прискол, прискофен, имидалин 11 др
Фентоламин (Phentolamine)
Фентоламин — 2-[[К-(3|-гидроксифенил)-лара-толуидино]метил]-2-имидазолин (12.2.8) получают алкилированием 3-(4-метилани-лино)фенола 2-хлор.метилимидазолино.м [36, 37].
Фентоламин также является производным имидазолина, проявляющим прямое а-адреноблокирующее, мышечнорасслябляющее действие на гладкую мускулатуру, а также холиномиметическое, гистаминное и симпатомиметическое действие. Химическое варьирование структуры
-239 -
Глава 12
позволяет сделать некоторые из его свойств более выраженными. К примеру, вышеописанный толазолин — 2-бенил-2-имидазолин, структурный аналог фентоламина, имеет более выраженное мышечно-расслябляющее действие на гладкую мускулатуру, чем а-адренобло-кирующее.
Фентоламин свое действие проявляет, конкурируя с катехоламинами за связывание с а-адренорецепторами, и именно поэтому называется конкурентным блокатором, имеющим большое сродство, но малую активность к этим рецептивным участкам. Такая субстрат-рецепторная блокада понижает способность а-адренорецепторов к взаимодействию с симпатомиметическими аминами и, соответственно, уменьшае1 значимость ответа вызванного эндогенными или экзогенными аминами. Время блокады а-адренорецепторов фентоламином значительно меньше, чем у феноксибензамина.
Фентоламин применяют при расстройствах периферического кровообращения, в частности при начальных стадиях гангрены, при лечении трофических язв конечностей, пролежней, отморожений.
Синонимами препарата являются регитин и дибазин.
агселективные адреноблокаторы
Эти препарагь! являются периферическими коронарорасширяющи-ми препаратами с а-адреноблокирующей активностью и отличаются специфичностью только к ai-адренорецепторам. В отличие or рассмотренных выше феноксибензамина и фентоламина, они селективно блокируют а!-рецепторы и имеют малое сродство к а2-адренергическим рецепторам.
Известно, что норэпинефрин регулирует свое собственное высвобождение из адренергических нервных окончаний по механизму отрицательной обратной связи посредством а2-рецепторов на гюстсинаптической мембране. В то же время празозин и тетразозин — это единственные известные селективные агадреноблокаторы, которые в терапевтических дозах не блокируют а2-адренергические рецепторы. Таким образом, при их применении механизм обратной связи для высвобождения норэпинефрина не включается.
Празозин (Prazosin)
Празозин — 1-(4-амино-6,7-диметокси-2-хиназолинил)-4-(2-фуроил)-пиперазин (12.2.12) синтезируют исходя из 2-амино-4,5-диметокси-бензойной кислоты, которуто действием цианата натрия подвергают
-240-
Адреноблокирующие препараты
гетероциклизации в 2,4-дигидрокси-6,7-диметоксихиназолин (12.2.9). Замещая гидроксильные группы полученного соединения на атомы хлора реакцией с хлористым тионилом или смесью хлорокиси фосфора с пягихлористым фосфором, получают 2,4-дихлор-6,7-диметокси-хиназолин (12.2.10). Последующим взаимодействием с аммиаком атом хлора при С4 пиримидинового кольца замещают на аминогруппу, что приводит к получению 4-амино-2-хлор-6.7-димегоксихиназолина (12.2.11). Введением последнего в реакцию с 1-(2-фуроил)пипера-зином получают празозин (12.2 12) [38, 39, 40,41, 42,43, 44, 45,46.47].
Празозин показан для лечения средней или умеренной гипертензии. При применении препарата кровяное давление понижается без существенных изменений таких показателей работы сердца, как частота сокращений, коронарный поток, сердечный выброс.
Синонимами препарата являются минипресс и минизид.
Тетразозин (Tetrazosin)
Тетразозин — 1-(4-амино-6,7-диметокси-2-хиназолинил)-4-(2-тетра-гидрофуроил)-пиперазин (12.2.13) отличается от празозина лишь тем, что фурильный радикал заменен в нем на тетрагидрофуриль-ный. Синтез осуществляют по совершенно аналогичной схеме с использованием вместо 1-(2-фуроил)пиперазина 1-(2-тетрагидрофу-роил)пиперазина [48. 49. 50, 51].
122 13
- 241 -
Глава 1 2
Тетразозин применяют по тем же показаниям, что и празозин, однако он имеет то преимущество, что его можно принимать один раз в день.
Синонимами препарата являются геитрин и вазокард.
Эргоалкалоиды (алкалоиды спорыньи)
Эрготамин, эргоновин и ряд других алкалоидов выделены из спорыньи, являющейся продуктом грибкового заражения зерна, и в частности ржи. В ранней истории цивилизации и в средние века потребление зерна, зараженного спорыньей, оканчивалось гангреной конечностей, выкидышами, судорогами.
Алкалоиды спорыньи были первыми изученными адреноблокаторами. Несмотря на то. что большинство алкалоидов спорыньи проявляют о.-адреноблокирующую активность, их фармакология зачастую различна. Химически эрготамин и эргоновин являются производными лизергиновой кислоты.
Эрготамин (Ergotamine)
Эрготамин — 3|,б',181-трион,12|-гидрокси-2|-метил-5|-бензил)-(5'al-эрготамамн (12.2.14) получают микробиологическим синтезом [52].
Эрготамин в качестве а-адреноблокатора проявляет прямой сосудосуживающий эффект из-за чего и используется в медицине, в частности для прерывания острых атак головной мигрени. Препарат категорически противопоказан при хронических заболеваниях ввиду возможных проявлений таких побочных эффектов, как инициирование гангрены.
Синонимами препарата являются синерган, секотамин и др.
-242-
Адреноблокирующие препараты
Эргоновин (Ergonovine)
Эрюновин — 1-гидроксиметилэтиламид лизергиновой кислоты (12.2 15) получают этерификацией D-лизергиновой кислоты 2-ами-нопропаноло.м в диметил формамиде, прямой обработкой реакционной смеси фосгеном [53, 54, 55, 56, 57, 58, 59].
сн3
НгН - сн — сн2-он
DMF / СОС12
СНз
Эргоновин не проявляет значимой а-адреноблокируюшей активности. Однако, как и эрготамин, применяется в акушерско-гинекологической практике, для остановки послеродовых кровотечений.
Вероятнее всего механизмом его действия является прямое спазмогенное влияние на матку.
Синонимами препарата являются эргометрин и эрготрен.
Селективные ат-адреноблокаторы
Иохимбин (Yohimbine)
Иохимбин — метиловый эфир (±)-2а-гидроксииохимбан-la-карбоновой кислоты (12.2.16). Выделяют из растений ряда Corynanthe johimbe и Rauwolfia serpentina [60, 61]. Осуществлен также синтез иохимбина [62, 63, 64, 65, 66].
Иохимбин — селективный а2-адренергический антагонист. Химически сходен с алкалоидом резерпином. Являясь производным индоли-лалкиламина, селективно блокирует а2-адренергические рецепторы. На нервных окончаниях он ослабляет механизм отрицательной обратной связи высвобождения норэпинефрина. Препарат обладает симпато
- 243-
Глава 12
миметическим эффектом, но может вызвать и симпаголитическое действие. Очевидно, требуются дополнительные исследования, чтобы однозначно обрисовать его фармакологическое действие. В настоящее время нет четких показаний по его применению.
Синонимами препарата являются коримбин. валимбин и др.
12.3. Блокаторы адренергических нейронов
Блокаторы адренергических нейронов вызывают распад биогенных аминов в окончаниях нейронов. Эти препараты могут вмешаться в синтез, хранение и высвобождение норэпинефрина, дофамина и серотонина.
Резерпин (Reserpine)
Резерпин — метиловый эфир 2а,11-диметокси-3-(3,4,5-тримет-оксибензонлокси)-иохимбан-1-карбоновой кислоты (12 3.1). Резерпин является одним из алкалоидов, выделяемых из многолетних кустарников семейства Rauwolfia [67, 68, 69, 70, 71, 72]. Осуществлен также синтез резерпина [73, 74, 75, 76].
Резерпин вызывают распад норэпинефрина, дофамина и серотонина в окончаниях нейронов. Он ослабляет внутриклеточный захват биогенных аминов и уменьшает способность к их хранению в везикулах. Не исключено, что резерпин действует на мембраны везикул, необратимо ингибируя АТФА^ -зависимый процесс, ответственный за захват биогенных аминов в межнейрональные везикулы. Распад катехоламинов выражается в уменьшении количеств внутринейронального серотонина и дофамина.
Резерпин применяется при лечении гипертензии, однако не является лекарством выбора из-за большого числа побочных реакций. На его основе имеется много препаратов комбинированных с другими гипертензивными средствами, в частности, с диуретиками.
Резерпин выпускается под множеством названий, из которых можно выделить серпасил, бринердин, диупрес и др.
- 244 -
Адреноблокирующие препараты
Гуанетидин (Guanethidine)
Гуанетидин — Р-(1-азациклооктил)этилгуанидин (12.3.4) предложено получать по следующей простой схеме. Азоцин алкилируют хлорацетонитрилом с получением 1-азоцинилацетонигрила (12.3.2), который восстанавливают алюмогидридом лития в 1-(2-амино-этилуазонин (12.3.3). Взаимодействием последнего с S-метилтио-мочевиной получают гуанетидин (12.3.4) [77, 78. 79].
Cl—ch2-cn
^N —CH2-CN
lialh4
1232
NH ch3-s-c-nh2 ,N —CH2—CH2—NH2 -----------1
12 3 3
/ Nh
I N—CH2-CH2-NH-C-NH2
12 3 4
Гуанетидин не действует на эффекторные клетки, как это имеет место с адреноблокаторами. Он воздействует на конечные разветвления симпатических периферических нервных волокон и проникает в нейрон по тому же механизму обратного захвата, который возвращает норэпинефрин в окончания нейрона из синаптической области. Внутри нейрона гуанетидин аккумулируется и конкурирует за места складирования в грануле с норэпинефрином. С увеличением концентрации гуанетидина норэпинефрин замещается им, и, соответственно, в нейроне понижается количество трансмиттера способного к выбросу. В результате стимуляции нерва может быть выброшен сам гуанетидин, который, однако, сам не является стимулятором адренергического рецептора. Вдобавок к нарушению наличия запасов катехоламинов в адренергических нервных окончаниях препарат также воздействует на наличие запасов катехоламинов в таких органах, как сердце, селезенка и аорта. Поскольку препарат не проходит через гематоэнцефалический барьер, то он не действует на центральные симпатические нейроны.
Гуанетидин применяется при острой гипертензии, когда применение более общепринятых препаратов оказывается безуспешным. Это очень сильный и долгодействующий препарат, и часто его действие продолжается еще 2-3 дня после его применения.
Синонимами препарата являются октадин, исмелин. санотензин и др.
- 245-
Глава 12
Гуанадрел (Guanadrel)
Гуанадрел — (1.4-диоксиспиро[4,5]дец-2-илметил)гуанидин (12.3.8) получают, подвергая циклогексанон катализации 3-хлор-1,2-пропан-диоло.м с образованием 2-хлорме1ил-1,4-диоксиспиро[4,5]декана (12.3.5), которым далее алкилируют фталимид натрия. Полученное фталимидопроизводное (12.3.6) после щелочного гидразинолиза трансформируется в 2-аминометил-1,4-диоксиспиро[4,5]декан (12.3.7) и вводится во взаимодействие с S-метилтиомочевиной с получением искомого гуанадрела(12.3.8) [80, 81,82].
+ НО-СН2-СН-СН2-С1 он
12 3 5
Гуанадрел — блокатор адренергических нейронов, используемый при эссенциальной гипертензии. Механизм его действия и побочные реакции сходны с гуанетидином.
Препарат применяют для лечения гипертензии у пациентов, неадекватно реагирующих на тиазидные диуретики. Может быть использован в качестве дополнительного средства при тиазидном лечении для достижения оптимального уровня кровяного давления.
Синонимом препарата является гилорел.
Метирозин (Metyrosine)
Метирозин — (-)а-метилтирозин (12.3.11) предложено получать несколькими путями, наиболее простым из которых представляется синтез, исходящий из 4-метоксибензилацетона, который взаимодействием с цианистым калием в присутствии карбоната аммония переводят в гидантоин (12.3.9). Обработкой последнего йодистым водородом снимается защитная метильная группа в бензольном кольце и продукт (12.3.10) гидролизуется гидроокисью бария в ра-
- 246 -
Адреноблокирующие препараты
цемическую смесь а-метил-В,Ь-тирозина, из которой выделяется искомый L-изомер (12.3.11) [83].
KCN / (NH4)2CO3
СН3О
СНз
СН2-С-СООН nh2
Ва(ОН)2
12.3 11
Метирозин является .метилированным производным тирозина. Препарат конкурентно ингибирует действие тирозингидроксилазы и гем самым понижает образование эпинефрина и норэпинефрина.
Используется для лечения пациентов с феохромоцитомой в случаях, когда наблюдается повышение уровня катехоламинов.
Синонимом препарата является демсер.
' Список литературы
1. US Pat. 3.337.628 (1967).
2. US Pat. 3.520.919 (1970).
3. Brit. Pat. 994.918 (1963).
4. Brit. Pat. 994.918 (1963).
5. Belg. Pat. 640.312 (1964).
6. Belg. Pat. 640.313 (1964).
7. US Pat. 3.873.600(1975).
8. Ger. Pat. 2.106.209 (1971).
9. S. Afr. Pat. 68 08.345 (1969).
10. US Pat. 3.857.952 (1974).
11. Ger. Pat. 2.007.751 (1970).
12. US Pat. 3.663.607 (1972).
13. US Pat. 3.836.671 (1974).
14. Ger. Pat. 2.258.995 (1973).
15. US Pat. 3.935.267 (1976).
16. Ger. Pat. 2.421.549 (1974).
17. Swiss. Pat. 469.002 (1969).
- 247 -
Глава 12
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
Swiss Pat 472 404 (1969)
Ger Pat 1 925 955 (1970)
Ger Pat 1 925 956 (1969)
LSPat 3 655 663 (1972)
US Pat 3 657 237 (1972)
W'asson A et al ZJ Med Chem .15., 651 (1972)
US Pat 3 718 647 (1973)
US Pat 3 729 469 (1973)
US Pat 3 812 182 (1974)
US Pat 4 145 550(1979)
Ger Pat 2 032 642 (1971)
US Pat 4 012 444 (1977)
Clifton J et al /J Med Chem 25, 670 (1982)
LSPat 2 599 000(1952)
US Pat 2 161 938 (1939)
Ger Pat 615 527 (1934)
Ger Pat 687 196 (1938)
Ger Pat 842 063 (1945)
US Pat 2 503 059 (1950)
Urech E et al Helv Chim Acta 33, 1386 (1950)
US Pat 3 511 836 (1970)
LS Pat 3 635 979 (1972)
US Pat 3 663 706(1972)
Brit Pat 1 156 973 (1970)
Honkanen E et al J Heteroc>cl Chem I7, 797 (1980)
US Pat 3 935 213 (1976)
US Pat 4 062 844 (1977)
US Pat 4 138 561 (1979)
Belg Pat 861 821 (1977)
Belg Pat 861 822 (1977)
US Pat 4 026 894(1977)
Ger Pat 2 646 186 (1977)
Ger Pat 2 831 112 (1979)
US Pat 4 251 532 (1981)
Stoll A Helv Chim Acta 28, 1283 (1945)
Stoll 1 et al Heh Chim Acta 26, 956 (1943 )
US Pat 2 090 430(1937)
US Pat 2 447 214(1948)
US Pat 2 736 728 (1956)
US Pat 2 774 763 (1956)
LSPat 2 809 920 (1957)
- 248-
Адреноблокирующие препараты
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
US Pat 3 141 887(1964)
Hofmann К /Heh Chim Acta 37. 849 (1954)
Bader F J Am Chem Soc 76, 1695 (1954)
Tamelen Fan E et al /80, 5006 (1958)
Liljegren D et al 'J Org Chem 27, 377 (1962)
TamelenkanE et al 91, 7315 (1967)
Wenkert E et al J Am Chem Soc 100, 4894 (1978)
Smarmy a I et al / Heterocycles 14. 631 (1980)
Ger Pat 967 469 (1954)
US Pat 2 752 351 (1956)
LSPat 2 833 771 (1958)
US Pat 2 887 489 (1957)
US Pat 2 938 906 (1960)
Dorfman L et al//Helv Chim Acta 37, 59 (1954)
Ger Pat 1 088 062 (1957)
Woodward R et al //78, 2023 (1956)
Pearlman В '/J Am Chem Soc 101, 6404 (1979)
И ender P etal/ J Am Chem Soc 102, 6157 (1980)
US Pat 2 928 829 (1960)
US Pat 3 006 913 (1961)
US Pat 3 055 882 (1962)
Fr Pat 1 522 153 (1967)
S Afr Pat 67 06 328 (1968)
US Pat 3 547 951 (1970)
Stein G etal J Am Chem Soc 77, 700 (1955)
Potts К ZJ Chem Soc 1955, 1632
Saari A /J Org Chem 32,4074 (1967)
US Pat 2 868 818 (1959)
j Глава 13 j Холиномиметики
Холиномиметики, или холинергические препараты, — это препараты, которые вызывают эффекты сходные с реакцией, возникающей при введении ацетилхолина или стимуляции ганглиев парасимпатической нервной системы. Эти препараты имитируют действие эндогенно высвобождаемого ацетилхолина. Известно, что действие ацетилхолина в определенных органах может быть воспроизведено алкалоидом мускарином, в других органах алкалоидом — никотином. На этом основано подразделение холинорецепторов на так называемые мускариновые (М-холинорецепторы) и никотиновые (Н-холинорецепторы).
Холинорецепторы разной локализации имеют неодинаковую чувствительность к различным препаратам.
Более чем 10 миллиардов нейронов, составляющих нервную систему человека, сообщаются друг с другом посредством медиаторов.
Ацетилхолин, ряд аминов, определенные аминокислоты и пептиды и аденозин являются медиаторами в ЦНС. Под упомянутым термином амины следует подразумевать норэпинефрин, дофамин, серотонин, а также. с весьма большой долей вероятности, гистамин и норэпинефрин.
Медиаторными аминокислотами считают глутаминовую и аспарагиновую кислоты, которые возбуждают постсинаптические мембраны многих нейронов, а ГАМК и глицин, являющиеся ингибирующими трансмиттерами. Эндорфины, энкефалины и субстанция Р считаются пептидергическими трансмиттерами. Имеется много соединений имитирующих действие указанных медиаторов.
Общепринятым на сегодняшний день считается, что:
- - медиатором постганглионарных парасимпатических нервных окончаний гладкой мускулатуры, сердечной мышцы и экзокринных желез является ацетилхолин;
— медиатором постганглионарных симпатических нервных окончаний гладкой мускулатуры, сердечной мышцы и экзокринных желез (за исключением потовых) является норэпинефрин (норадреналин);
— медиатором всех окончаний двигательных нервов является ацетилхолин;
-250-
Холиномиметики
— медиа!орами в ЦНС являются ацетилхолин, норэпинефрин, дофамин, серотонин, гистамин, глутаминовая и аспарагиновая кислоты, ГАМК, глицин, аденозин и ряд пептидов.
Основным медиатором в парасимпатической части автономной нервной системы, которая иннервирует главным образом ЖКТ, глаз, сердце, дыхательный гракт, секрецию желез, является ацетилхолин. Весьма небольшое число болезненных состояний можно объяснить дисфункцией холинергических участков периферической автономной системы, хотя именно они являются ключевыми для сохранения всех нормальных функций организма.
Хотя сам ацетилхолин и является веществом, без которого невозможно представить нормальное функционирование организма в целом, два свойства делают его крайне неудобным для применения в качестве лекарственного средства. Первое— весьма короткое время действия из-за быстрого его расщепления холинэстеразами и второе, что более важно, — эго многогранность его действия, ввиду чего становится практически невозможным сделать его действие узконаправленным, для решения данной конкретной задачи. Однако ряд производных ацетилхолина более устойчивы к действию холинэстераз и могут проявить большую селективность действия.
Таким образом, холиномиметики — это препараты, которые имитируют действие эндогенно высвобождаемого ацетилхолина.
Классификации этих препаратов основываются на механизме их действия, которое проявляется либо прямой стимуляцией холинергических рецепторов эфирами холина или холиномиметическими алкалоидами. либо непрямым путем — ингибированием ацетилхолинэстераз — ферментов, ответственных за химическую деструкцию ацетилхолина в месте его действия. Последние, в свою очередь, подразделяются на обратимые ингибиторы холинэстеразы и необратимые ингибиторы холинэстеразы.
13.1. Холиномиметики прямого действия
Холиномиметики прямого действия являются препаратами, действующими путем прямой стимуляции холинергических рецепторов. Эти препараты делятся на препараты, стимулирующие мускариновые (М-холинорецепторы) или никотиновые (Н-холинорецепторы) рецепторы.
Препараты, эффективность которых в первую очередь связана со стимуляцией мускариновых рецепторов, включают холиновые эфиры, т. е. сам ацетилхолин и его структурные аналоги — метахолин, карба-хол, бетанехол и природные алкалоиды — мускарин и пилокарпин.
-251 -
Глава 13
Препараты тейсгвие которых основывается на стимх тяцни никоти новых рецепторов включают алка юиды — никотин и тобе тин
Холиновые эфиры
Препараты этою к тасса вктючаки ацеппхолин и его структурные аналоти метахотпн. бетанехол и карбахол Несмотря на го что указанные препараты способны непосредственно стимулировать все холинергические рецепторы их терапевтическая эффективность опосредована воздействием на мускариновые рецепторы (подтипы М, и М->) Соеди нения оттичаюгся то тько продолжительностью действия и, в некоторой степени се тективностью к рецепторам Ацетилхолин явдяется протоги пом всей труппы Для терапевтическою испо тьзования в настоящее время не предложено селективных Мг и М2-агонисюв
Ацетилхолин (Acetylcholine)
Ацетихолин — хлорид 2-ацетокси N \,\т-тримети тэти т аммония (13 1 1) весьма просто синтезировать раз тичными пх тями Согласно первому способу, 2-хлорэтанол вводят во взаимодействие с триме-гиламином, и полученный при этом гидрохлорид N,N,N-TpiiMeTnn-этил-2-эгатю ламина (В 1 1), называемый также холином, ацет и тируют антидридоут или хдорантидридом уксусной кислоты с получением ацетилхолина (13 12) Второй способ зактючается во взаимодействии гриметиламина с окисью этилена с ио течением гидроксида N А,А-триУтетидэтил-2-этаноламттна (13 13), который взаимодействием с хлористым водородом переводят в гидрохлорид (13 1 1) и далее ацетилируют вышеописанным способом Наконец, третий способ — ацетитхотин можно подучать также взаимодействием ацетата 2-хлор этанола с триэтиламнпом [1.2 3, 4, 3, 6. 7J
^CH3)tN
ci—chz - сн2-он
'СНД2К —CH2-CHZ-OH Ci
CH3CCCI
13 1 1
+ О
(CH^N — Ch -CHZ-O-C"CH3 С
1о 2
А
(CH / N
(CHj^N —СТ-Ц-СНЦ-СН ОН
13 1 3
- 252 -
X о л и н о м и м е т и к и
О
II (Chj^N + 01—СН2 "“СН2 “O-C-Ch s
13 2
Ацегилхонш представляет собой мо теку ту холина, ацетилированию но атому кислорода Ввиду наличия высокопотярной заряженной аммониевой группы ацешлхолин не проходил через липидные мембраны По этой причине вводимый извне препарат задерживается во вне-клеючном просгранс1ве и не проникае! через гематоэнцефалический барьер
Ацетилхолин не имеет терапевтической ценности в качестве препарата для внутривенного введения из-за многопланового действия и быстрой инактивации холинэстеразой Одновременно возможно возникновение коллапгоидного состояния, может резко понизиться АД, ос1анови!ься сердце Однако его применяют в виде i лазных капель тля вызывания миоза во время операций катаракты, 1де и проявтяеюя ею преимущество, способствующее быстрому послеоперационному восстановлению
Синонимом препарата является миоход
Метало, тн (Methacholine)
Метахотпн — хлорид 1-аиетокси-2-(\,1Х,\-лримегил)пропич аммония (Н 14), или иначе ацетил-Р-мегилхолин. можно получить любым из вышеописанных меюдов [8]
си3 о
(CHJ3N —Cr-^-CH —O-C-CHj ci
13 1 4
Малое структурное изменение наличие метильной группы с p-yi-теродного атома холина выражается в двух важных изменениях фармако югическо! о профиля мотекулы В отдичие от ацети <холина, yieiaxo-лин гидролизуется то 1ько ацетилхолинэстеразой, а скорость его гидролиза значительно меньше, чем у ацетхолина 1 тким образом действие метахолина зпачи!ельно продолжительнее, чем с аиетидхо т-на Ьо iee юго наличие мети юной группы у р у мерочною атома холина обеспечивает соединению большую селективность действия Метаконид непосредственно действует на мускариновые рецепторы главой мускх iaiyры желез п сердца и очень с табо дейсюует на никелиновые рецепторы автономных ганглиев скелешой муску татуры Эти две осо
- 253 -
Глава 1 3
бенности — длительность действия и повышенная селективность — являются основными различиями в фармакологическом действии метахолина и ацетилхолина. Препарат применяется только для диагностики бронхиальной гиперреактивности.
Синонимом препарата является провохолин.
Карбахол (Carbachol)
Карбахол — хлорид 2-карбамоилокси-М,М,М-триметилэтил аммония (13.1.7) получают взаимодействием 2-хлорэтанола с фосгеном с образованием хлоругольного эфира 2-хлорэтанола (13.1.5). Взаимодействием с аммиаком последний переводят в соответствующий амид (13.1.6), и далее взаимодействием с эквимольным количеством триметиламина получают карбахол (13.1.7) [9, 10, 11.12,13].
+ СОС12------Ci—CH;-CH2-O*C“Ci
13 1 5
NH, ? (СНз)зМ
---CI— СНг-СН2~О-С-НН2 ----------—
13 1 6
+ о
(CH3)3N —ch2-ch2-o-c-nh2 Cl
13 1 7
В отличие от ацетилхолина и метахолина, карбахол содержит вместо ацетильной карбаминовую функциональную группу, которая не так чувствительна к гидролизу холинэстеразами. Опытами in vitro показано, что скорость его гидролиза как минимум вдвое меньше, чем у ацетихо-лина.
Карбахол — мощный холиновый эфир, стимулирующий как мускариновые, так и никотиновые рецепторы, и проявляет все фармакодинамические свойства ацетилхолина, вызывая вдобавок расширение сосудов, понижение скорости сердцебиения. Он повышает тон и сокращаемость гладкой мускулатуры, стимулирует слюнные, глазные и потовые железы, автономные ганглии и скелетную мускулатуру. По этим причинам он, подобно ацетилхолину, ограниченно применяется в терапии, за исключением офтальмологической практики, и при послеоперационной атонии кишечника и мочевого пузыря. При закапывании в глаз сужает зрачок и снижает внутриглазное давление.
Применяют при острой и хронической глаукоме.
Синонимами препарата являются дорил и миостат.
- 254 -
Холиномиметики
Бетанехол (Betanechol)
Бетанехол — хлорид 2-карбамоилокси-1-(М,Т4,М-триметил)пропил аммония (13.1.8) предложено получать либо последовательным взаимодействием хлорида 1-(Х,\т,К-триметиламмоний)пропан-2-ола с фосгеном и аммиаком, либо совершенно аналогичным синтезу карбахола путем — последовательным взаимодействием 1-хлор-2-пропанола с фосгеном, аммиаком и далее с триметиламином с получением бетанехола (13.1.8) [14, 15].
* СНз
CH3)3N — СНг-СН — О-Н CI
1 соа2
2 NH3
СНз о (CH3)3N — сн2-сн— o-c-nh2 ci
13.1.8
СНз ci—сн2-сн-он
1 COCI2
2 NH3
СНз О
Cf—сн2-сн— 0-C-NH2
(CH3)3N
13.1.8
Бетанехол является препаратом, в котором скомбинированы структурные особенности метахолина и карбахола, т. е. препарат содержит в своей структуре как 0-метильную, так и карбаматную функциональные группы и, вполне логично, проявляет фармакологические свойства обоих препаратов. Он устойчив к гидролизу холинэстеразами и мало действует на никотиновые рецепторы автономных ганглий и нервно-мышечных соединений. Бетанехол имеет более селективное действие на мускариновые рецепторы ЖКТ и мочевого пузыря, чем другие холиновые эфиры.
Терапевтическое применение препарата основано на этом действии, и его применяют при лечении послеоперационной необструктивной задержки мочи и нейрогенной атонии мочевого пузыря. Ранее он использовался для лечения желудочно-кишечных заболеваний и болезни Альцгеймера.
Синонимами препарата являются дувоид, миотонии и урехолин.
Природные мускариновые алкалоиды
Мускарин (Muscarine)
Мускарин — хлорид З-метил-З-гидрокси-З-^.Т^Д’-триметиламмо-нийметплентетра-гидрофурана (13.1.14) был впервые выделен из ядовитых грибов — мухоморов Amanita muscaria Его можно синте-
-255-
Глава 13
зировать раз тичиыми спосооами из совершенно различных исходных веществ [16 17 1ь, 19 20 21 22 23 24] и в частное ги исходя из 2' диметил 3 карбоксиметилфурана вводя ею в реакцию Кхр нихса г е последовательным взаимодействием с гидразином и да тее с азотистой кисдотой в изоиропи товом спирте с по тхчснием уретана (13 19) кислотным гидроткзом которого по ту чают 2 5-димети 1 2Н фуранон-3 (В 1 10) Аллильным бромированием по с 1еднего потхчаюг 2-мети i-5-бромметил 211 фуранои 3 (В 1 И) ко торый вводя 1 во взаимодействие с диметиламином с по течением 2 мети 1-х диметиламинометил 2Я-фуранона 3 (13 1 12) Восстановление последнего приводит к получению 2 метил-3-гидрокси > диме-тиламинометилтеграгидрофурана (13 1 13) взаимодействием которого с хлористым метилом потхчаюг мхскарин (13 1 14) в виде смеси стереоизомеров
СН CL
Мускарин явтяется природным алкалоидом, встречающимся в ря те дикорастущих мухоморов Несмотря на то, что мхскарин не имеет тера певтическото значения он пре тставляет интерес из-за выраженных ток сических свойств и поскольку исторически он быт одним из первых систематически изхченных хотиномиметических веществ Это соединение тегто в основу классификации холинергических мускариновых рецепторов Действие мхекарина сходно с действием ацетилхолина на периферические автономные эффекторные органы и антатоптируется атропином В оттичие от ацетилхолина мхскарин не действует на нико тиновые рецепторы
Отрав тение грибами требует серьезного медицинского вмешательства. поскольку мускарин хорошо абсорбируется из АКТ и поэтому может привести к несчастным случаям Мускарин значительно ботее мощен чем аиетитхотии возможно из за большей устойчивости Нс будучи эфиром он не подвергается гидролизу холинэстеразами Геоа-певтического применения не имеет Отравление мускарином леч-тт су ть фатом атропина
- 25b -
Холиномиметики
Синонимов соединения практически нет
Пилокарпин (Pilocarpine)
Пилокарпин — 3-этил-4-(1-метил-5-имилазолилметил)тетрагидро-фуран-2 он (13 1 22) является алкалоидом, получаемым из листьев тропического растения Pilocarpus Jaborandi Его синтез осуществляют несколькими путями [25 26, 27, 28, 29, 30, 31, 32J, наиболее приемлемым из которых следует считать путь исходящий из 2-этип-З-карбокси-2-бутиролактона [25, 26, 27], который с помощью хлористого тионила превращают в хлорангидрид (13 1 15) и далее вводят во взаимодействие с диазометаном и этанолом, получая соответствующий этиновый эфир (реакция Арндта—Айсгерта), который гидролизуют в кислоту (13 1 16) Полученная кислота (13 1 16) действием хлористого тионила вновь переводится в хлорангидрид (13 1 17) Последняя обрабатывается диазометаном Но на сей раз промежуточно образующийся кетон обрабатывается хлористым водородом с получением хлоркетона (13 1 18) Взаимодействием последнего с фталимидом калия и последующим удалением фталимидной защитной группы кислотным гидролизом получают аминокетон (13 1 19), который вводят во взаимодействие с подкисленным раствором тиоциана!а калия с получением 3-этил-4-(2-меркапто-5 имидазо лилметил)тетрагидрофуран-2-она (13 1 20) Мягкое окисление последнею позволяет удалить меркаптогруппу из продукта (13 1 20) и получить 3-этил-4-(5-имидазолилметил)тетрагидрофуран-2-он (13 1 21) Алкилирование полученного продукта йодистым метилом приводит к образованию пилокарпина (13 1 22)
SOCI2
1 CH2N2 +
2 С2Н5ОН Н
С2Н5
О
13 1 17
СН2СОС1
1 CH2N2
2 НС!
СН2СОСН2С1
13 1 18
-257 -
Глава 13
CH2COCH2NH2
13.1 19
kncs/н
13.1 20
13 1 21
CH3I
13 1,22
Действие пилокарпина состоит в стимуляции мускариновых рецепторов и поэтому при систематическом введении сходно с действием ацетилхолина. Соединение отличается от ацетилхолина отсутствием какого-либо воздействия на никотиновые рецепторы, стимулирует ЦНС. Его эффекты блокируются атропином. Находит также терапевтическое применение в офтальмологии в качестве мистического средства.
Синонимами являются пилофрин, изоптокаприн, атмокаприн.
Природные никотиновые алкалоиды
Никотин (Nicotine)
Никотин — 1-метил-2-(3-пиридил)пирролидин (13.1.27) — алкалоид, который выделяют из растений рода Nicotiana (Nicotiana to-bacum, Nicotiana rustica и др.) можно синтезировать разными путями [33, 34, 35, 36]. В частности, предложено исходить из этилового эфира никотиновой кислоты, которую конденсируют с N-метилпир-ролидоном с получением 1-метил-2-никотиноил пирролидона-2 (13.1.23). Кислотный гидролиз последнего приводит к раскрытию пирролидинового кольца с промежуточным получением кислоты (13.1.24), которая в условиях реакции декарбоксилируется в у-ами-нокетон (13.1.25). Карбонильную группу последнего восстанавливают до спиртовой, и далее образовавшийся продукт (13.1.26) подвергают дегидратации в никотин (13.1.27).
СгНдОИа
сн-снг-ch2-nhch3 С СЮН
13 1 24
- 258-
Холиномиметики
|^'|[’СН2 —СН2 —СН2 -NHCH3
13.1.25
13 1 27
Никотин интенсивно изучался по разным причинам. Он использовался в фармакологии в экспериментальных целях для характеристики холинергических никотиновых рецепторов и для стимулирования и блокирования автономных ганглиев. На сегодня никотин является объектом внимания из-за его большого наличия в табаке, что является фактором риска для возникновения многих заболеваний. Никотин действует путем взаимодействия с периферическими холинергическими никотиновыми рецепторами на постсинаптической мембране в автономных ганглиях и нервно-мышечных соединениях, равно как и никотиновых рецепторов в ЦНС. В низких дозах, скажем при курении сигарет, никотин стимулирует рецепторы, вызывая деполяризацию мембраны и приток ионов Na+ или Са2+. В высоких дозах стимуляция сопровождается пролонгированной блокадой реполяризации. Это вызывает нереагирование рецептора на последующую стимуляцию ацетилхолином, высвобожденным из преганглионарных холинергических тканей, в результате чего и блокируется нервная передача. Это явление характеризуется как деполяризующая ганглиевая блокада.
Единственным терапевтическим применением никотина является его использование в составе жевательной резинки, в качестве временного средства при попытках бросить курить.
Синонимом препарата в виде салицилата является эндернол.
Лобелии (Lobeline)
Лобелии — 1-метил-2-(Р-гидрокси-р-фенилэтил)-б-фенацилпипе-ридин (13.1.33) является основным алкалоидом листьев Lobelia inflata. Синтез осуществляют конденсацией 2,6-диметилпиридина с двумя молями бензальдегида с образованием а,а'-дистирилпи-ридина (13.1.28) [37, 38, 39]. Исчерпывающее бромирование последнего и далее дегидробромирование полученного тетрабромпроизвод-ного (13.1.29) приводит к получению а,а'-дифенилэтинилпиридина (13.1.30). Гидратацией тройных связей продукта (13.1.30) получают а,а'-дифенацилпиридин (13.1.31). Взаимодействием последнего с метиловым эфиром н-толуолсульфокислоты получают N-метил-«ара-толуолсульфонат оца'-дифенацилпиридиния (13.1.32), который
- 259 -
Глава 13
осторожно восстанавливают водородом в искомый лобелии (13.1.33) в условиях одновременного применения платинового и палладиевого катализаторов. В результате получается продукт в виде рацемической смеси, из которой, при необходимости, можно выделить левовращающий изомер.
С&Ч5СН—СН
ВГ2
Вг Вг
CgHsCH "СН гч СН—СНС6Н5
Вг Вг
13 1 29
КОН
сбн5с=с
13 1 30
С — CCgHg
+ +2
Н2О / Н ' Нд
О
СбН5-С-СН2
о
СН2-С-С6Н5
р-СНзСбЩЗОзСНз
о с6н5-с-сн2
о
СНг-С-СбНб
СНз
13 1 32
р—CH3C6H4SO3
Н2/ Pt, Pd
о он
С6Н5-С-СН2 N сн2-сн — сен5 СНз
13 1 33
Действие лобелина во многих отношениях сходно с никотином, однако он слабее никотина в 50-100 раз. Он также является первичным стимулянтом и вторичным депрессантом симпатических ганглиев, парасимпатических ганглиев и надпочечников и др. Может применяться в качестве средства против курения.
Синонимами препарата являются лоброн, вентарон, юнилобин, ло-бетон и др.
13.2. Холиномиметики непрямого действия
Ингибиторы холинэстеразы являются очень важным классом соединений семейства холиномиметиков. Кроме своей терапевтической значимости, некоторые из них используются в качестве пестицидов
- 260 -
Холиномиметики
в сельском хозяйстве, а наиболее токсичные — в качестве химических отравляющих средств. Использование этих веществ основано на изменениях, которые происходят после инактивации холинэстеразы или псевдохолинэстеразы (менее специфичного фермента), т. е. эффектов, наблюдаемых в результате накопления ацетилхолина в нейрональноэффекторных соединениях. Ингибиторы холинэстеразы классифицируются как на основе их химической структуры, так и на типе их химического взаимодействия с ферментом, который определяет их временное действие.
Имеется 3 больших класса ингибиторов холинэстеразы:
1) карбаматы — физостигмин, неостигмин, пиридостигмин и ряд инсектицидов типа карбарила:
2) четвертичные амины — эдрофоний, амбеноний и демека-рий;
3) органофосфаты — изофлурофат, эхотиофат, инсектициды типа малатиона и паратиона, а также боевые отравляющие вещества типазомана.
Основываясь на разнице в длительности их ингибирующего эффекта, ингибиторы холинэстеразы могут быть классифицированы как обратимые и необратимые ингибиторы.
Обратимыми ингибиторами являются карбаматы и четвертичные амины. Необратимыми ингибиторами холинэстеразы являются органофосфаты
Обратимые ингибиторы холинэстеразы
Обратимые ингибиторы холинэстеразы образуют переходный комплекс с ферментом аналогично ацетилхолину. Эти соединения находятся в конкуренции с ацетилхолином за связывание с активными участками фермента. Химическая структура классических обратимых ингибиторов (физостигмина и неостигмина) говорит о сходстве с ацетилхолином. Эти соединения имеют большое сродство к ферменту и их ингибирующее действие обратимо. Эти ингибиторы отличаются от ацетилхолина тем, что не так легко расщепляются ферментами. Ферменты реактивируются намного медленнее, чем происходит последующий гидролиз ацетилхолина. Поэтому фармакологический эффект, вызываемый этими соединениями, обратим.
Эдрофоний также является обратимым ингибитором.
-261 -
Глава 13
Карбаматы
Физостигмин (Physostigmine)
Физостигмин — 1,3а.8-триметил-2,3,3а,8а-тетрагидропирроло[2,3-Ь]-индол-5-ил^-метилкарбамат (13,2.7) является алкалоидом, выделяемым из так называемых калабарских бобов — семян ядовитого африканского растения семейства бобовых Physostigma venosum. Синтетически физостигмин предложено получать разными путями [40, 41, 42], один из которых исходит из ивра-этоксиметиланилина, который вводят во взаимодействие с бромангидридом а-бромпро-пионовой кислоты в присутствии хлористого алюминия с получением 1,3-диметил-5-этоксииндолин-2-она (13.2.1). Взаимодействием последнего с хлорацетонитрилом в присутствии этилата натрия получают 1,3-диметил-5-этокси-3-цианометилиндолин-2-он (13.2.2). Нитрильную группу восстанавливают до аминной, которую далее метилируют с получением 1.3-диметил-5-этокси-3-(Р-метиламино-этил)индолин-2-она (13.2.3). Карбонильную группу последнего восстанавливают с получением аминоспирта (13.2.4), дегидратация которого приводит к получению 1,3а,8-триметил-2,3,3а,8а-тетрагидро-пирроло[2.3-Ь]-5-этоксииндола (13.2.5). Этоксильная защитная группа последнего удаляется действием бромистого водорода с получением соединения с фенольным гидроксилом (13.2.6), которое вводят во взаимодействие с метилизоцианатом с получением искомого физостигмина (13.2.7).
CH3NHCOO
13 2 6 СНз СНз
CH3NCO
-262-
Холиномиметики
Физостигмин легко абсорбируется из ЖКТ и других слизистых оболочек. Попав в кровоток, препарат легко пересекает и гематоэнцефалический барьер. Препарат инактивируется холинэстеразой плазмы. Физостигмин имеет минимальное прямое воздействие на холинергические рецепторы. Из-за способности проникать в ЦНС препарат применяется в качестве антидота при возникновении в организме токсических концентраций препаратов с антихолинергичес-кими свойствами, таких как атропин, антигистамины, фенотиазины и трициклические антидепрессанты. Его действие на организм, в общем, сходно с действием ацетилхолина и он применяется по тем же показаниям в офтальмологии для сужения зрачка и понижения глазного давления при глаукоме.
Синонимами препарата являются эзерин и мезитинон.
Неостигмин (Neostigmine)
Неостигмин — метилсульфонат ^.>;^-триметил-мета-(диметил-карбамоилокси)-фениламмония (13.2.9), который можно рассматривать как упрощенный аналог физостигмина, получают взаимодействием 3-диметиламинофенола с диметилкарбамоил хлоридом с получением диметилкарбамата (13.2.8) и последующим алкилированием диметилс>льфатом (13.2.9) [43].
Неостигмин — ингибитор холинэстеразы содержит четвертичный атом азота и, как следствие, из-за затрудненного прохождения через гематоэнцефалический барьер проявляет минимальную токсичность, связанную с ингибированием холинэстеразы в мозге. Наличие в молекуле четвертичного атома азота приводит и к другому значительному различию между физостигмином и неостигмином, а именно: неостигмин, кроме ингибирования холинэстеразы, имеет прямой стимулирующий эффект на холинергические рецепторы. Однако за исключением этих серьезных различий общее действие неостигмина аналогично действию физостигмина. Как и другие обратимые ингибиторы холинэстеразы, неостигмин проявляет мощное антикурареподобное действие. Это свойство неостигмина используется в анестезиологии для преодоления паралича скелетной мускулатуры вызванной курареподобными препаратами.
-263-
Г лава 13
Неостигмин в основном применяют при миастении, двигательных нарушениях после травм мозга, параличах, атрофии зрительного нерва, при лечении атонии кишечника и мочевого пузыря.
Синонимами препарата являются прозерин, простигмин, стигмосан и др.
Пиридостигмин (Pyridostigmine)
Пиридостигмин — диметилкарбамат З-гидрокси-1 -метил пиридиний бромида (13.2.11) синтезируют из 3-гидроксипиридина взаимодействием с диметиламинокарбамоилхлоридом с получением 3-(ди-метиламинокарбамоил)пиридина (13.2.10). Подвергая последний действию метилбромида, получают пиридостигмин (13.2.11) [44].
о
Cl—C-N(CH3)2
р
о—C-N(CH3)2
СН3Вг
Качественно фармакологические свойства пиридостигмина аналогичны свойствам неосзигмину.
Синонимом препарата является местинон.
Четвертичные амины
Эдрофоний (Edrophonium)
Эдрофоний — хлорид этил-(3-гидроксифенил)диметиламмония (13.2.13) получают взаимодействием 3-диметиламинофенола с этилбромидом с образованием бромида этил(.меота-гидроксифе-нил)диметиламмония (13.2.12), атом брома которого обменивают на хлор, взаимодействием с хлористым серебром с получением эдро-фония (13.2.13) [45].
(CHg^N
С2Н5Вг
СНз
С2Н5 — N
СНз'
AgCL
Фармакологически эдрофоний также схож с неостигмином, однако он начинает действовать быстрее и действует в течение более короткого промежутка времени.
Синонимом препарата является тенсилон.
-264-
Холиномиметики
Амбеноний (Ambenonium)
Амбеноний — хлорид [оксалилбис(иминоэтилен)]бис[орото-хлорбен-зил)диэтиламмония] (13.2.15) получают взаимодействием диэтилок-салата с двумя молями М,М-диэтилэтилендиамина с получением ок-салилбис(иминоэтилен)бис1Ч,М-диэтиламина (13.2.14), который алкилируют двумя молями 2-хлорбензилхлорида с получением амбенония (13.2.15) [46,47, 48].
°, fl
О О
/—X + (СгН5)гм-сн2-сн2-нн2 > (C2h5;2n -сн2-сн2—nh nh—ch2-ch2-n(c2h5;2
С2Н5-О O-CzHs
о о
+ 02^5 м tC2H5
СН2— N-CH2-CH2— NH NH—CH2-CH2-N—СН2
С2Н5 13 2 15 С2Н5
Фармакологические свойства амбенония сходны с неостигмином и пиридостигмином и достигаются путем обратимой инактивации холинэстеразы.
Синонимом препарата является мителаз.
Демекарий (Demecarium)
Демекарий — гидроксид МД^-декаметиленбисДмеотаДМ-метилкар-бамоилокси)-фенилтриметиламмония] (13.2.18) получают взаимодействием двух молей фосгена с 1,10-бг/с-(метиламино)-декано-(М,М'-диметилдекаметилен-1,10-диамином с получением бискарба-моилхлорида (13.2,16), который переводят в бискарбамоилэфир (13.2.17) взаимодействием с двумя молями натриевой соли 3-диме-тиламинофенола. Взаимодействием с метилбромидом последнего получают демекарий (13.2.18) [49].
2 СОС12
СНз сн3
+ H—N—(СН2)-|0—N"”H
СН3 СН3
Cl— CO-N-(CH2)ia— N-CO-CI
(CH3)2N
132 16
(CH3)2N
СНз СНз
.zJVzO-CO-N -(CHZ)1(J-N -CO—О
LJ 13 2 17
N(CH3)2
СНЗВГ
-265-
Г лава 13
,CH3)3N
СН3
СНз
CO-N-(CH2)io— N-CO—О
13218
N(CH3)3
2 Вг
Демекарий — диссимметричное соединение, содержащее две аммонийные и две карбаматные группы. Соединение является обратимым ингибитором холинэстеразы, но с более продолжительным действием, чем дру гие. Применяют для сужения зрачка, повышения внутриглазного давления при лечении глаукомы, а также для снятия атропинового мидриаза.
Синонимами препарата являются тосмилен и гуморосол.
Необратимые ингибиторы холинэстеразы
Органофосфаты
Вторым классом ингибиторов холинэстеразы являются фосфорорганические соединения общей формулы:
R,0 о
М
r2o X
Фосфорорганические соединения действуют путем комплексообразования с гидроксильной группой серина в эфирной части холинэстеразных ферментов с образованием ковалентной связи с атомом фосфора.
В отличие от быстрого гидролиза комплекса ацетилхолина с ферментом и несколько более замедленного гидролиза комплексов карбаматов. фосфорилированный фермент с водой реагирует очень медленно, приводя, в общем, к необратимому ингибированию работы фермента. При применении большинства фосфорорганических веществ в организме должен произойти новый синтез фермента, чтобы восстановить холинэстеразную активность ткани. Несмотря на то, что подобная активность называется необратимой, некоторые химические соединения, такие как оксимы, могут восстановить жизнедеятельность фермента. Однако фосфорилированный фермент может подвергнуться и процессу, определяемому как старение, при котором органофосфат теряет алкиль-ную группу, образуя более сильную необратимую связь с ферментом, чю уже делает фермент не способным к восстановлению оксимами.
- 266 -
Холиномиметики
Признаки и симптомы острой токсичности, возникающие при введении фосфорорганических антихолинэстеразных соединений, могут быть предсказаны с легкостью и объясняются гиперактивностью парасимпатической нервной системы, нервно-мышечных соединений, автономных ганглиев и холинергических нервов ЦНС. Смерть возникает по причине депрессии дыхания, вызванной депрессией ЦНС, паралича диафрагмы и межреберных мышц из-за аккумуляции избыточного количества ацетилхолина. Некоторые органофосфаты полезны в качестве медицинских препаратов, другие — в качестве инсектицидов и потенциальных химических боевых веществ по причине их высокой токсичности.
Изофлурофат (Isoflurophate)
Изофлурофат — ди-лзо-пропиловый эфир фторфосфорной кислоты (13.2.21) получают взаимодействием мзо-ггропилового спирта с треххлористым фосфором с получением ди-изо-нропилфосфита (13.2.19), который хлорируют в 13.2.20, и далее действием фтористого натрия обменивают атом хлора на фтор с получением изофлурофата (13.2.21) [50].
PCL3 (СН3)2СН -о О СЬ (СНз)2СН -о О NaF (СНз)2СН -о О
(СНз^СНОН ----► --1» >р([ ---► )Р\
(СНз)2СН -О Н (СНз)2СН -О CI (СН3)2СН -О F
132 19 13 2 20 13 2 21
Первое различие между изофлурофатом и средствами типа физостигмина заключается в постоянстве (инерционности) действия. Согласно описанному выше возможному механизму, изофлурофат вызывает необратимую инактивацию холинэстераз. Причем инактивируются как ацетилхолинэстераза, так и «неспецифические» холинэстеразы плазмы. Однако изофлурофат имеет большее сродство к последним. Изофлурофат используется шля лечения определенных типов глаукомы, когда миотики кратковременного действия непригодны.
Синонимами препарата являются флороприл, флуостигмин и диф-лу ПИЛ.
Экотаофат (Echothiophate)
Экотиофат — 8-(2-триметиламинозтил)-О,О-диэтилгиофосфат (13.2.23) получают взаимодействием диэтилхлорфосфорной кислоты с 2-ди-метиламнноэтилмеркаптаном с получением 8-(2-диметиламино-
_ 9R7 -
Глава 13
этил)-О,О-диэтилтиофосфата (13.2.22), который алкилируют ме-тилйодидом с получением экотиофата (13.2.23) [51].
с2н5—о о с2н5-0 О с
+ нз-сн2-сн2-4(сн3)г ----)рт —
С2н5-О Cl С2Н5-О 's-CH2-CH2-N(CH3;2
13 2 22
с2н5-о о W
С2н5- О 'S-CH2-CH2-N(CH3)3 |
13 2 23
Экотиофат является фосфорилтиохолином с фармакологическим действием, аналогичным таковому изофлурофата, однако спонтанное восстановление фосфорилированного фермента происходит быстрее, чем это происходит после применения изофлурофа-фата. Препарат применяют при различных формах глаукомы.
Синонимами препарата являются эходид и фосфолин йодид.
Другие фосфорорганические ингибиторы холинэстеразы
В настоящее время получены многие фосфорорганические соединения, применяемые в быту и сельском хозяйстве в качестве инсектицидов. Многие из них являются липорастворимыми соединениями, быстро и полностью адсорбируемыми практически всеми путями, включая кожу, дыхательный тракт и ЖКТ. Большинство фосфорорганических соединений подвергаются биотрансформации путем гидролиза сложно-эфирной группировки и выделяются с мочой. Малатион, широко используемый дома и в саду, малотоксичен для людей, поскольку легко гидролизуется. Однако его применение основано на том, что у насекомых гидролиз протекает значительно медленнее.
Фосфорорганические соединения типа зарин, зоман и табун являются одними из наиболее токсичных известных химических соединений, применяемых в качестве боевых отравляющих веществ. Они вызывают очень быстрое старение ферментов, не подвергающееся реактивации. Поэтому нет соответствующей терапии для лечения этих отравлений.
Лечение интоксикации фосфорорганическими соединениями включает искусственное дыхание, введение атропина — антагониста мускариновых рецепторов, введение пралидоксима, являющегося реактиватором холинэстеразы.
-268-
Холиномиметики
Пралидоксим (Pralidoxime)
Пралидоксим — 2-пиридинальдоксима метилхлорид (13.2.25) синтезируют взаимодействием пиридин-2-альдегида с гидроксиламином с получением пиридин-2-альдоксима (13.2.24), который далее вводят во взаимодействие с метилйодидом с получением искомого пралидоксима (13.2.25) [52, 53, 54, 55].
h2noh
CH3I
ch=n-oh
13.2.24
СНз
13.2.25
Пралидоксим — сильный нуклеофил. Он реактивирует фосфорилированный фермент путем двухстадийной реакции. Первая стадия — образование комплекса между оксиматионом и фосфорилированным ферментом. Вторая стадия — восстановление ферментной активности и образование фосфорилированного оксима. Высокая реактивирующая способность пралидоксима приписывается его способности сочетаться с отрицательно заряженной группой на поверхности фермента и высокой степени молекулярного соответствия между оксимом и фосфорилированной холинэстеразой. Кажется вероятным, что оксимы могут реагировать непосредственно с ингибитором, превращая его в безвредное соединение, а также реактивировать ингибированный фермент как в крови, так и в тканях. Применяют при отравлениях органофосфатами, параличе дыхательных мышц и, вообще, при холинергическом кризисе.
Синонимами препарата являются контратион и протопам.
Глава 13
Список литературы
1. Bayer A Ann. 14^, 235 (1867).
2. Nothnagel <V.//Arch. Pharm. 232. 265 (1894).
3. Fourneau E. et al., 'Bull. Soc. Chim. France. [4] 15, 544 (1914).
4. Ger. Pat. 801.210(1948).
5. USPat. 1.957.443 (1934).
6. USPat. 2.012.268 (1935).
7. US Pat. 2.013.536 (1935).
8. USPat. 2.040.146 (1936).
9. Ger. Pat. 539.329(1930).
10. Ger. Pat. 553.148 (1930).
11. Ger. Pat. 590.311 (1932).
12. Hayworth R. et al.,7J. Chem. Soc. 1947, 176.
13. USPat. 2.374.367 (1945).
14. US Pat. 2.322.375 (1943).
15. USPat. 1.894.162 (1933).
16. Kogi H. et al. 'Rec. Trav. Chim. 76, 109 (1957).
17. Kogi H et al.//Expenentia. 13, 137 (1957).
18. Cox A. et al.,/Helv. Chim. Acta. 41, 229 (1958).
19. Matsumoto H. et al.//Tetrahedron. 25, 5889 (1969).
20. Still W et al./'J. Org. Chem. 45. 3375 (1980).
21. IVhttmg B. et al.//Can. J. Chem. 50, 3322 (1972).
22. Mubarak A. et al.//Tetrahedron Letters. 21, 2453 (1980).
23. Mubarak A. et al.,7J. Chem. Soc. Perkin Trans I, 1982, 809.
24. PochetS. et al./'J. Org. Chem. 47, 193 (1982).
25. Preobrashenski N. et al./'Ber. 66. 1187 (1933).
26. Preobrashenski N. et al.//Ber. 66. 1536 (1933).
27. Preobrashenski К et al.' Ber. 68, 850 (1935).
28. DeGraw К//Tetrahedron. 28, 967 (1972).
29. Link K. et al.'/Helv. Chim. Acta. 55, 1053 (1972).
30. \oordam A. et al.//Rec. Trav. Chim. 98, 467 (1979).
31. Langenbeck HG/Angew. Chem. 60, 297 (1948).
32. Van Rossum J. et al.'716, 373 (1960).
33. Pinner N.//Ber. 26, 294 (1893).
34. Pictet J. et al./'Ber. 37, 1225 (1904).
35. Craig J."J. Am. Chem. Soc. 55, 2854 (1933).
36 Nakane M. et al.' J. Org. Chem. 43, 3922 (1978).
37. Schoppf C./'Angew Chem. 50, 786 (1937).
38. Wieland H et al.'/Ann. 473. 102 (1929).
-270-
Холиномиметики
39. Scheuing G. et al./'473.126 (1929).
40. Julian P et al./O. Am. Chem. Soc. 57, 755 (1935).
41. Harley-Mason J. et al.//J. Chem. Soc. 1954, 3651.
42. Wijnberg J. et al.//Tetrahedron. 34, 2399 (1978).
43. USPat. 1.905.990(1933).
44. US Pat. 2.572.579 (1951).
45. USPat. 2.647.924(1953).
46. US Pat. 3.096.373 (1963).
47. Ger. Pat. 1.024.517 (1954).
48. Phillips A ;’J. Am. Chem. Soc. 73, 5822 (1951).
49. USPat. 2.789.891 (1957).
50. USPat. 2.409.039(1946).
51. USPat. 2.911.430(1959).
52. USPat. 2.816.113 (1957).
53. USPat. 3.123.613 (1964).
54. USPat. 3.140.289 (1964).
55. USPat. 3.155.674(1964).
Глава 14
Антихолинергические препараты
Антихолинергические препараты — это соединения, проявляющие конкурентное блокирующее действие на холинергические рецепторы.
Указанная обширная группа препаратов может быть разбита на 3 подгруппы на основе их относительной специфичности к различным типам холинорецепторов.
Первая группа
Вторая группа
Третья группа
«классических» антихолинергических средств представлена антимускариновыми соединениями (атропин, пропантелин), которые блокируют действие ацетилхолина или вводимых холиномиметических препаратов на мускариновых (М-рецепторных) участках ЦНС и железистого аппарата, миокарда, гладкой мускулатуры.
антихолинергических препаратов представлена ганг-лиоблокаторами (мекамиламин. триметафан), которые ингибируют холинергическую передачу как в автономных парасимпатических, так и в симпатических ганглиях путем блокирования (никотиновых) Н-ре-цепторов. '
антихолинергических препаратов представлена ингибиторами передачи (тубокурарин, панкуроний), блокирующими никотиновые Н-рецепторы скелетной мускулатуры, и будет рассмотрена в гл. 15 «Мышечные релаксанты».
14Л. Антимускариновые препараты
М-холиноблокаторы являются конкурентными антагонистами ацетилхолина, а также других М-холиномиметиков в отношении постсинаптических М-холинорецепторов.
Парасимпатические холинергические рецепторы локализованы в гладкой мускулатуре сосудов, бронхов, ЖКТ, мочевого пузыря, сердце, мускулатуре глаз, большинстве экзокринных железах и в ЦНС.
- 272-
Антихолинергические препараты
М-холиноблокаторы проявляют широкий спектр фармакологических эффектов и применяются по самым различным показаниям. Они могут быть использованы:
— с целью вызвать мидриаз или циклоплегию при офтальмологических исследованиях;
— при предоперационной подготовке пациента с целью уменьшения слюноотделения и предотвращения брадикардии:
— для уменьшения секреции ЖКТ в случаях язвы желудка, спазмов и других желудочно-кишечных заболеваний;
— с целью уменьшения нософарингеальной и бронхиальной секреции при респираторных и аллергических заболеваниях;
— для предотвращения и облегчения двигательных нарушений;
— для лечения детского энуреза и уменьшения частоты мочеиспускания;
— для облегчения некоторых симптомов паркинсонизма;
— в качестве антидота при передозировке холинергических препаратов, при отравлениях антихолинэстеразными препаратами, фосфорорганическими инсектицидами и пестицидами.
Антимускариновые препараты классифицируются как:
• алкалоиды (атропин, гиосциамин, скополамин);
• антихолинергики ряда четвертичных аминов (анизотропии, клидиний, гликопирролат, гексоциклий, изопропамид, мепен-золат, метантелин, метскополамин, пропантелин);
• антипаркинсонические препараты ряда третичных аминов (бензтропин, бензпериден, этопропазин, орфенадрин, процик-лидин. тригексифенидил);
• спазмолитики ряда третичных аминов (дицикломин, оксибу-тинин, оксифенциклимин);
• мидриатики ряда третичных аминов (циклопентолат, тропикам ид).
Алкалоиды
Старейшими препаратами этой группы являются различные галено-вые препараты, выделяемые из красавки (Atropa belladonna), белены (Hyoscyamus niger) и дурмана (Datura stramonium). Все они получаются из растений, содержащих L-гиосциамин и несколько меньшие и варьируемые количества L-скополамина. В качестве блокатора мускариновых
-273 -
Глава 14
рецепторов L-гиосциамин намного активнее, чем D-гиосциамин, как на периферии, так и в ЦНС. Однако рацемическая смесь D.L-гиосциамина, более известная как атропин, предпочтительнее для большинства медицинских целей ввиду большей доступности.
Атропин и его аналог скополамин являются двумя наиболее важными антимускариновыми препаратами. Эти алкалоиды и родственные им соединения применяются в офтальмологии и анестезиологии, при сердечных и желудочно-кишечных заболеваниях, при паркинсонизме. Большое значение они имеют в качестве антидотов при антихолинэсте-разных интоксикациях.
Атропин и скополамин являются эфирами троповой кислоты с тропином и скопином соответственно. Скопин отличается от тропина лишь наличием эфирного кислородного мостика между атомами углерода С6 и С; тропина.
Описаны подтипы мускариновых рецепторов (М| и М2), которые активируются или блокируются различными веществами, однако оба типа мускариновых рецепторов активируются эндогенным нейротрансмиттером — ацетилхолином и блокируются атропином или скополамином. Несмотря на то, что атропин и скополамин являются обратимыми холиноблокирующими агентами, константы их диссоциации с М-рецеп-торами в несколько раз меньше, чем у ацетихолина.
Соответственно их действие является более продолжительным (до нескольких дней).
Неоднократно наблюдалось, что атропин более эффективен при блокировании эффектов экзогенно вводимого ацетилхолина и других парасимпатомиметиков, чем при блокировании эффектов, возникающих после стимуляции волокон парасимпатических и холинергических нервов. Причиной этого могут быть 2 фактора:
1) ацетихолин высвобождается после нервных импульсов в области очень близкой к М-рецепторам эффекторных клеток и, следовательно, действует более эффективно, чем ацетилхолин, поступающий путем циркуляции, и чем парасимпатические вещества, поступающие тем же путем;
2) в определенных органах, таких как мочевой пузырь, постганглионарные парасимпатические волокна в дополнение к ацетилхолину, могут высвобождаться и другие нейротрансмиттеры, например аденозингрифосфат, действие которых не блокируется атропином.
Алкалоиды белладонны имеют весьма широкий фармакологический спектр. В дополнение к их способности к блокированию М-рецепторов
-274-
Антихолинергические препараты
атропин и скополамин действуют и на другие рецепторы, проявляя соответствующие эффекты. Они могут блокировать никотиновые холинергические рецепторы, однако в дозах значительно выше чем те, которые применяются в клинике. Атропин проявляет также свойства местных анестетиков и блокатора гистаминовых (Н,) рецепторов. Атропин и скополамин практически полностью абсорбируются из ЖКТ и из конъюнктивы. Скополамин может абсорбироваться и через кожу.
Атропин (Atropin)
Атропин — О,Е-8-метил-8-азабицикло[3.2.1]окт-3-иловый эфир ос-гид-роксиметилфенилуксусной кислоты (14.1.4) можно синтезировать по обычной схеме синтеза тропановых алкалоидов. Конденсацией малеинового диальдегида с метиламином и ацетондикарбоновой кислотой получают тропенон (14.1.1), который является основным исходным веществом как для синтеза атропина, так и для синтеза скополамина. Карбонильную группу тропинона восстанавливают с получением тропенола (14.1.2), после чего гидрируют двойную связь между углеродными атомами С6 и С7 тропанового кольца с получением тропина (14.1.3). Этерификацией последнего тропо-вой кислотой получают искомый атропин (14.1.4) [1, 2, 3, 4, 5, 6].
сн-сно II сн-сно
о
CH3NH2 + HOOc-CHa-c-CHj-CDOH
СНзСООМа
LIALH4
о н-о-с-сн-с6н5
сн2-он
Атропин применяют при язвенной болезни, пилороспазме. холецистите, желчнокаменной болезни, спазмах кишечника и мочевых путей, бронхиальной астме.
Атропин часто применяется в процессе анестезии в хирургии. Основная цель — минимизировать секрецию в бронхах и в носоглотке,
Глава 14
которая может затруднить дыхание. В случаях, когда необходимо дополнительное седативное действие, предпочтителен скополамин.
В офтальмологической практике атропин применяют в диагностических целях — для расширения зрачка, и при острых воспалительных заболеваниях и травмах глаза.
Атропин часто применяют при простудных заболеваниях, для временного осушения носоглотки. В сочетании с другими средствами атропин применяется как антидот при отравлениях антихолинэстераз-ными агентами, такими как фосфорорганические инсектициды и нервно-паралитические газы. В этих случаях атропин устраняет или нивелирует многие токсические проявления являющиеся результатом действия высокой концентрации ацетилхолина.
Агропин первый эффективный препарат, предложенный для симптоматического лечения паркинсонизма. Показано, что в отличие от гастроинтестинальных спазмолитиков, разные синтетические вещества, проходящие через гематоэнцефалический барьер, такие как тригекси-фенидил, этопропазин, бензтропин, проциклидин, орфенадрин и бипе-риден, также эффективны при симптоматическом лечении паркинсонизма. Для его лечения общепризнанным препаратом является леводопа, однако исследования последних лет указывают на определенные ограничения при его применении.
Синонимами препарата являются атроптол, атропизол, О,Ь-гиос-циамин и др.
Скополамин (Scopolamine)
Скополамин — С-9-метил-3-окса-9-азатрицикло[3.2.1.0Г'4]нон-7-иловый эфир а-гидроксиметилфенилуксусной кислоты (14.1.6) можно синтезировать из тропенола (14.1.2) окислением двойной связи между углеродными атомами С6 и С? тропанового кольца с получением эпоксидного производного — скопина (14.1.5). Этерификацией последнего троповой кислотой получают скополамин (14.1.6) [7, 8].
[О]
О н-о-с-сн-с6н6 сн2—он
СНз
N
14 1 6 \ °
о-с-сн-с5н6 сн2-он
14 1 2
- 276 -
Антихолинергические препараты
Скополамин применяют практически по тем же показаниям, что п атропин, но с учетом того, что он оказывает седативный эффект и уменьшает двигательную активность, рекомендовано его применение и при симптомах паркинсонизма.
Синонимами препарата являются джосцин, осцин и др.
Антихолинергики ряда четвертичных аминов
В течение последних десятилетий для клинического использования при лечении язвы желудка, пилороспазма, гиперперистальтики был предложен ряд синтетических атропиноподобных веществ с большим спазмолитическим, но с меньшим антихолинергическим действием и проявляющих меньше побочных эффектов.
Большинство из этих препаратов действуют по одному из грех механизмов: мускариновой блокады, прямого подавления активности гладкой мускулатуры и блокады парасимпатических ганглиев, и ни один из них не лишен присущих агропину побочных эффектов. Список этих препаратов включает ряд четвертичных аммониевых солей (про-пантелин, метскополамин, анизоторопин, мепензолат, изопропамид, гликопирролат, клидиниум и гексоцицилиум).
Метскополамин (Methscopolamin)
Метскополамин — нитрат 7-(3-гидрокси-1-оксо-2-фенилпропокси)-9,9-диметил-3-окса-9-азонийтрицикло[3.2.1.0.2,4]нонана (14.1.7) получают взаимодействием скополамина (14.1.6) с метилбромидом (иногда с последующей заменой бромид иона на нитрат ион) взаимодействием с нитратом серебра [9, 10].
1.СН31
2.AgNO3
О-С-СН-С6Н5
сн2-он
О-С-СН-С6Н5 сн2-он
Метскополамин ингибирует мускариновое действие ацетилхолина на постганглионарных парасимпатических эффекторных участках. Препарат применяют при лечении язвы желудка.
-277 -
Г лава 14
Синонимами препарата являются памин и сколии.
Анизотропии (Anisotropin)
Анизотропии — метилбромид 2-пропилпентаноилтропиния (14.1.9) синтезируют ацилированием тропина (14.1.3) хлорангидридом 2-про-пилвалериановой кислоты с получением эфира (14.1.8) и дальнейшим его взаимодействием с метилбромидом с получением анизо-гропина (14.1.9) [11].
о
CL—С-СН-С3Н7
Анизотропии ингибирует секрецию желудочного сока и восстанавливает нормальную работу желудка. Применяют при лечении язвы же-лу дка.
Синонимом препарата является вальпин.
Пропантелин (Propantheline)
Пропантелин — Х-метилА’-(1-метилэтил)-К-[2-[(9Я-ксантен-9-илкарбонил)окси]этил]-2-пропанам.мония бромид (14.1.11) получают взаимодействием хлорангидрида ксантен-9-карбоновой кислоты с 2-ди-гузо-пропиламиноэтанолом с получением эфира (14.1.10), который взаимодействием с метилбромидом переводят в четвертичную соль — пропантелин (14.1.11) [12, 13].
сн(Сн3)г
СН3В'
СО-О—CH2-CH2-N — СНз Вг
14 111 СН(СНз)2
Фармакологическое действие пропангелина качественно сходно
-278-
Антихолинергические препараты
с атропином. На ЦНС действует слабее атропина. В отличие от атропина, он больше проявляет ганглиоблокирующее действие, чем анти-му скариновое. Более того, при передозировке он вызывает нервно-мышечную курареподобную блокаду. Применяют при лечении язвы желудка
Синонимами препарата являются норпант, пропантел, робанталин.
Мепензолат (Mepenzolat)
Мепензолат — 3-[(гидроксидифенилацетил)окси]-1,1 -диметил пипе-ридиния бромид (14.1.13) получают путем этерификации бензиловой кислоты 1-метил-З-хлорпиперидином и последующим взаимодействием полученного эфира (14.1.12) с метилбромидом [14, 15].
14 1 13
Мепензолат ингибирует мускариновое действие ацетилхолина на постганглионарных парасимпатических эффекторных участках. Применяют при лечении язвы желудка, воспалениях кишечника вместе с другими препаратами.
Синонимами препарата являются кантил и эфторон.
Клидиний (Clidinium)
Клидиний — бромид З-бензилоилокси-1-метилхинуклидиния (14.1.19) получают взаимодействием 3-гидроксихинуклидина (14.1.17) с хлор-ангидридо.м бензиловой кислоты с получением эфира (14.1.18), который далее алкилируют по атому азота метилбромидом с получением клидиния (14.1.19) [16].
Синтез самою 3-гидроксихинуклидина (14.1.17) осуществляют исходя из метилового эфира шо-никотиновой кислоты, которую взаимо-
-279-
Глава 14
действием с этиловым эфиром бромуксусной кислоты переводят в соль пиридиния (14.1.14). Восстановлением последней водородом с использованием платинового катализатора получают 1-карбэтоксиметил-4-карб-метоксипиперидин (14.1.15), из которого циклизацией по Дик.ману, используя в качестве основания калий или этила! калия, получают хинуклидин-3-он (14.1 16). Карбонильную группу последнего восстанавливают до спиртовой водородом над окисью платины с получением 3-гидроксихину клидина (14.1.17)
соосн,
Вг-СЬД- СООС2Н5
Н2 ' P1Q
I
CH2-COOC2H, 14 1 15
Н2 / РЮ
14.1.16 14.1.17
Клидиний ингибирует мускариновое действие ацетилхолина на постганглионарных парасимпатических эффекторных участках
Препарат применяют при лечении язвы желудка.
Синонимом препарата является кварзан
Гликопирролат (Glycopyrrolate)
Гликопирролат - 3-[(циклопентилгидроксифенилацетил)окси]-1,1-диметилпирролидиния бромид (14.1.22) получают исходя из метилового эфира а-циклопентил.манделовой кислоты ( 14.1.20) путем переэтерификации с использованием в качестве спиртового компонента З-гидрокси-1-метилпирролидина с получением эфира (14.1.21), который далее переводят в четвертичную соль взаимодействием с метилбромидом с получением гликопирролата (14.1.22). Исходный из метиловый эфир а-циклопентил-манделовой кислоты (14.1.20) получают взаимодействием циклопентилмагнийбромида с метиловым эфиром фенилглиоксиловой кислоты [17, 18]
О о н н
О-СНз
-280-
Антихолинергические препараты
СНгВг
14 1 22
Гликопирролат ингибирует секрецию желудочного сока и восстанавливает нормальную работу желудка. Препарат применяют при лечении язвы желудка, воспалениях кишечника, а также в качестве предоперационного средства для ингибирования избыточной секреции желудка.
Синонимом препарата является робинул.
Изопропамид (Isopropamid)
Изопропамид — (3-карбамоил-3,3-дифенилпропил)ди-изо-пропил-метиламмония йодид (14.1.25) синтезируют алкилированием дифенилацетонитрила ди-изо-пропиламиноэтилхлоридом в присутствии амида натрия с последующим гидролизом нигрильной группы образовавшегося соединения (14.1.23) в амид (14.1 24). Алкилированием последнего метилйодидом получают изопропамид (14.1.25) [19, 20. 21. 22].
СН(СН3)2
CI—сн2-сь2 -n(
СН(СНз)2
CH2-CH2-n(
,4,23 СН(СН3)2
C-CONH;, I СН(СН3)2
ch2-ch2-n(
14 1 24 СН(СНз)2
СН3Вг
C-CONH2^(j;H(CH3;2
Ch2—СН2—N—СН3 Вг
14 1 25 СН(СН3)2
Изопропамид ингибирует мускариновое действие ацетилхолина на постганглионарных парасимпатических эффекторных участках. Применяют при лечении язвы желудка, воспалениях кишечника.
Синонимами препарата являются дарбид и мобадид.
- 281 -
Г лава 14
Гексоциклий (Hexocycliutn)
Гексониклий — метилсульфат 4-(р-циклогексил-р-гидроксифен-этил)-1,1-пиперазиния (14.1.28) синтезируют, алкилируя 1-метилпи-перазин а-бромацетофеноном с получением 4-метил-1-фенанилпи-перазина (14.1.26). Вводя последний во взаимодействие с цикло-гексилмагнийбромидом получают 4-(Р-циклогексил-р-гидрокси-фенэтил)-1,1-пиперазин (14.1.27), алкилированием которого диме-гилсульфатом получают гексоциклий [23].
С-СН2Вг
(CH3O)2SO2
CH,OSOj
Гексоциклий ингибирует мускариновое действие ацетилхолина на постганглионарных парасимпатических эффекторных участках. Применяют при лечении язвы желудка.
Синонимом препарата является трал.
Антипаркинсонические препараты ряда третичных аминов
При лечении паркинсонизма применяются препараты, проявляющие центральные антихолинергические свойства. Полагают, что на синтез, высвобождение и гидролиз ацетилхолина они не влияют. Их лечебная эффективность проявляется в уменьшении или устранении двигательных нарушений, обусловленных поражением экстрапира-мидной системы. Они уменьшают ригидность, в несколько меньшей мере — акинезию и мало влияют на тремор.
Терапевтическая ценность этих препаратов относительно невелика, и они применяются либо в комбинации с леводопой, либо в случаях слабовыраженного паркинсонизма. К этим препаратам относятся описанные в гл. 10 «Средства, применяемые при паркинсонизме» тригек-сифенидил (10.2.2), проциклидии (10.2.3), бипериден (10.2.4), бензтро-пин( 10.2.6), этопропазин (10.2.7) и др.
-282-
Антихолинергические препараты
10 2 2
CH3-C.4-CH2-n(
10 2 7 С2Н5
Спазмолитики ряда третичных аминов
Синтетические спазмолитики ряда третичных аминов (дицикломин, оксибутинин, оксифенциклимин) проявляют прямое антиспастическое действие на гладкую мускулатуру и ингибирует мускариноподобное действие на нее ацетилхолина
Эти препараты проявляют более слабую, чем у атропина, антихоли-нергическую активность, однако значительно более выраженное антиспастическое действие. Их применяют для лечения, так называемого синдрома раздраженной толстой кишки и при запорах.
Дицикломин (Dicyclomin)
Дицикломин — диэтиламиноэтиловый эфир 1-циклогексилциклогексанкарбоновой кислоты (14.1,32) предложено получать двумя путями. Согласно первому способу, бензилцианид подвергают алкилированию 1,5-дибромпентаном с получением 1-циано-1-фенил-циклогексана (14 1.29). Последний подвергают алкоголизу с получением этилового эфира 1-фенил-1-циклогексанкарбоновой кислоты (14.1.30). которую подвергают переэтерификации с использованием в качестве спиртового компонента 2-диэтиламиноэтанола в присутствии натрия с получением 2-диэтиламиноэтилового эфира 1-фенилциклогексанкарбоновой кислоты (14.1.31), фенильную группу которой восстанавливают до циклогексильной водородом над окисью платины [24, 25].
- 283-
Глава 14
2г — (Crl2j5_'Br
ch2cn
14 1 29
НО-СН2 ~ СН2 -N(C2H5)2
,С0-0—Сн2 -сн2 -N(C2H5)2
Н2 i Pto
Второй способ синтеза дицикломина исходит из цианоциклогексана, который подвергают алкилированию циклогексилбромидом с получением 1-цианобициклогексана (14.1.33). Последний подвергают алкоголизу с получением этилового эфира 1-бициклогексанкарбоновой кислоты (14.1.34), которую подвергают переэтерификации 2-диэтилами-ноэтанолом в присутствии натрия [25].
слон н
------
14 1 33
НО-СН2- CH2-N(C2H5)2
14 1 34
14 1 32
Дицикломин ингибирует мускариновое действие ацетилхолина на постганглионарных парасимпатических эффекторных участках. В комбинации с другими препаратами применяют при лечении язвы желудка и при коликах у детей, для лечения синдрома раздраженной толстой кишки.
Синонимами препарата являются антиспас, бентил, дибент, формулекс.
Оксибутинин (Oxybutinin)
Оксибутинин — 4-диэтиламино-2-бутиноловый эфир а-фенилцик-логексангликолевой кислоты (14.1.35) получают либо реакцией Манниха с использованием пропаргилового эфира сс-фенил-а-цикло-гексангликолевой кислоты, параформа и диэтиламина, либо переэтерификацией метилового эфира а-фенил-а-циклогексангликоле-вой кислоты с использованием 1-ацетокси-4-диэтиламино-2-бутина в присзтствии метилата натрия [26].
- 284 -
Антихолинергические препараты
с 2^5
•Н
С2н5 CH3ONa сн3-соо-снг-с=с—N ------------
CjHg
Оксибутинин предназначен для облегчения неприятных симптомов при опорожнении кишечника и мочевого пузыря.
Синонимом препарата является дитропан.
Оксифенциклимин (Oxyphencyclimin)
Оксифенциклимин — 1,4,5,6-тетрагидро-1-метил-2-пиримидинме-таноловый эфир а-фенилциклогексангликолевой кислоты (14.1.37) получают этерификацией а-фенил-а-циклогексангликолевой кислоты 2-хлорметил-1-метил-1,4,5,6-тетрагидропиримидином (14.1.36) в присутствии йодистого калия. Исходный 2-хлорметил-1-.метил-1,4,5.6-тетрагидропиримидин (14.1.36), в свою очередь, получают взаимодействием метилового эфира иминохлоруксусной кислоты с 3-метиламинопропиламином [27, 28. 29].
NH
CI—СН2-С-ОСН3 + СН3-NH-CHz-СН2-СН2- КН2
14 1.37
Оксифенциклимин весьма широко применяют по тем же показаниям. что и дицикломин и оксибутинин.
Синонимами препарата являются орбигастрил, гастрисед, гастрикс, дарикон и др.
- 285-
Глава 14
Мидриатики ряда третичных аминов
Антихолинергические препараты ряда третичных аминов (цикло-пентолат, тропик-амид) применяются и местно в качестве мидриатиков с целью вызвать циклопегию и мидриаз. В первую очередь, они используются в качестве вспомогательных средств для исследования глаза и других диагностических процедур до, во время и после офтальмологических вмешательств.
Циклопентолат (Cyclopentolat)
Циклопентолат — 2-(диметиламино)этиловый эфир 1-гидрокси-циклопентан-а-фенилуксусной кислоты (14.1.39) получают этерификацией а-(1-гидроксициклопентил)фенилуксусной кислоты (14.1.38) 2-диметиламиноэтилхлоридом.
а-(1-гидроксициклопентил)фенилуксусную кислоту (14.1.38) получают взаимодействием натриевой соли фенилуксусной кислоты с циклопентаноном в присутствии изопропилмагнийбромида [30].
СНз
Cl—CHj-CHj-N
СНз
,СН3
N
СН3
Циклопентолат— эффективный мидриатик и циклоплегик с очень быстрым началом и относительно коротким временем действия. Применяется при офтальмоскопии и для вызывания предоперационного мидриаза.
Синонимами препарата являются мидрилат, цикложил. цикломид-рил, пентолайр и др.
Тропикамид (Tropicamid)
Тропикамид — 1\’-(4-пиридилмет ил)-\-этил-р-гидрокси-а-фенилпро-пиона.мид (14.1.41) получают взаимодействием О-ацетилпропил-хлорида с этил(4-пиридил-метил)амином с последующим кислотным гидролизом ацетильной группы в полученном амиде (14.1.40) [31].
- 286 -
Антихолинергические препараты
СНзСОО
3 14 140
НС!
Тропикамид, так же как и циклопентолат, применяется при офтальмоскопии, для получения предоперационного мидриаза и для тестирования узкое гольной глаукомы.
Синонимами препарата являются мидрин, мидриацил, мидриафайр, тропикацил.трипатар и др.
14.2. Ганглиоблокирующие вещества
Ганглиоблокирующие вещества — это соединения, селективно действующие на нервную передачу в автономных ганглиях. (Теоретически гашлиблокаторы могут упразднить всю автономную активность организма.)
Эти препараты классифицируют как деполяризующие и антидепо-зяризующие ганглиоблокаторы.
Деполяризующие гангзиоблокапюры, примером которых может служить никотин, вначале стимулируют постганглионарные рецепторы, а затем блокируют последующую активацию рецептора, предотвращая гем самым реполяризацию постсинапгической мембраны. Фармакологические эффекты никотина очень разнообразны и в большой степени зависят от дозы, экспозиции, физиологического состояния индивидуума.
Антидеполяризующие ганглиоблокаторы, к которым относятся клинически эффективные препараты мекамиламин и триметафан, действуют как конкурентные антагонисты ацетилхолина на постганглионарных рецептивных участках Их основное действие заключается в понижении сосу дистого тонуса, выраженном расширении сосудов и понижении периферической сопротивляемости. Венозное расширение вызывает застой крови, соответственно, уменьшение количества возвращаемой крови в сердце и понижение сердечного выброса. Оба этих эффекта выража-
-287-
Глава 14
ются в виде гипотензии. В то же время ганглиоб.токаторы вызывают ортостатическую гипотензию, что является крайне нежелательным побочным эффектом. Они являются средствами для понижения давления, однако используются редко из-за большого числа побочных эффектов, выражающихся в виде тахикардии, мидриаза, пониженной активности ЖКТ. задержки мочи, сухости во рту и т. п.
В течение 1950-х и в начале 1960-х годов ганглиоблокаторы были практически единственными веществами, используемыми для лечения общей гипертензии. В настоящее время они практически заменены более эффективными средствами и их клиническое применение весьма незначительно.
Мекамиламин (Mecamylamine)
Мекамиламин — М,2,3,3-тетраметилнорборнан-2-иламин (14.2.2) получают исходя из 2,3,3-триметилнорборнена-2, вводя его в реакцию Риттера с цианистоводородной кислотой в концентрированной серной кислоте с получением 2,3,3-триметилнорборнан-2-ил-фор-миламина (14,2.1), восстановление которого алюмогидридом лития приводит к мекамиламину (14.2.2) [32, 33].
+ HCN + H2so4
NH-CHO
СНз
14 2 1
LiALH4
NH—СНз
СНз
14 2 2
В настоящее время мекамиламин — единственный ганглиоблока-тор, применяемый при общей гипертензии, однако ввиду развития к нему привыкания и в связи с внедрением в медицинскую практику многих других антигипертензивных препаратов необходимость в нем отпала.
Синонимами препарата являются мевазин, инверзин и др.
Триметафан (Trimethaphan)
Т риметафан — D-3,4-( 1,3-дибензил-2-оксоимидазолидино)-1,2-три-метилентио-фания D-камфорсульфонат (14.2.12) является промежуточным продуктом синтеза биотина (витамина Н). Препарат полу-чают исходя из фумаровой кислоты, бромирование которой приводит к мезо-дибромянтарной кислоте (14.2.3). Взаимодействием последней с бензиламином получают 2.3-бис-(бензиламино)янтар-
-288-
Антихопинергические препараты
ную кислоту (14.2.4), обработкой которой фосгеном получают 1,3-дибензил-2-оксоимидазолидин-4,5-дикарбоновую кислоту (14.2.5). Дегидратацией последней получают соответствующее имидазолиновое производное янтарного ангидрида (14.2.6). Восстановлением последнего цинком в уксусной кислоте и последующей обработкой сероводородом получают 1,3-дибензил-2,5-диоксо-тетрагидротиено-[3,4]имидазолин (14.2.7), который вводят во взаимодействие с 3-это-ксипропилмагийбромидом. Полученный карбинол (14.2.8) подвергают кислотной дегидратации в (14.2.9), и далее восстанавливают образовавшуюся при этом двойную связь водородом с использованием в качестве катализатора никеля Ренея. Расщеплением эфирной связи в полученном продукте (14.2.10) с помощью бромистого водорода в уксусной кислоте получают бромид 3,4-(1,3-дибензил-2-оксоимидазолидино)-1,2-тримегилентиофания (14.2.11), обработкой которого D-камфорсульфнатом серебра получают триметафан (14.2.12) [34. 35, 36, 37, 38].
н соон 'с=с
НООС н
НООС СООН
с6н6-CH2-NH2
C6H5-CH2-NH Nh-СН2-С6Н5
н—с-^—н
НООС соон
СОС'2
1 Zn 1 СЧзСООН
2 H2S/ НС!
Вг—Мд—1CH2J3— ОС2Н5
СН;—СьН5
НВг' СНзСООН
-289-
Глава 14
СНз , СНз
•SO3A8
Триметафан применяют для контролируемого снижения давления в ходе хирургических вмешательств, для быстрой регутяции при резких повышениях давления, срочных вмешательств при отеке легких, при ишемической болезни сердца, в случаях, когда другие препараты не могут быть применены
Синонимом препарата является арфонад и др
Список литературы
1 Ladenburg 4 "Atm 217, 75 (1883)
2 Willstatter R Вег 31, 1537 (1898)
3 Willstatter R Ann 326, 23 (1903)
4 Schwenker E et al Z/Ber 99, 2407 (1966)
5 Robinson J J Chem Soc 111, 762 (1917)
6 Ger Pat 247 455 (1912)
7 Fodor G et al / /Chem & Ind 1956, 764
8 DoboP etal J Chem Soc 1959. 3461
9 US Pat 2 753 288 (1956)
10 Ger Pat 145 996(1902)
11 US Pat 2 962 499(1960)
12 US Pat 2 659 732 (1953)
13 Cusic 4 etal J Org Chem 16, 1921 (1951)
14 US Pat 2 918 408 (1959)
15 Biel M etal /J Am Chem Soc 77,2250 (1955)
16 US Pat 2 648 667(1955)
17 US Pat 2 956 062 (1960)
18 lunsfordH et al/4 Med Pharm Chem 2,523 (1960)
19 Bnt Pat 772 921 (1955)
20 Ger Pat 1 003 744 (1955)
21 Janssen P etal Arch Int Pharmacodyn Ther 103, 82 (1955)
22 US Pat 2 823 233 (1958)
-290-
Антихолинергические препараты
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
LSPat 2 907 765 (1959)
US Pat 2 474 796(1949)
Tilford C etal J Am Chem Soc 69, 2903 (1947)
Bnt Pat 940 540 (1961)
But Pat 795 758 (1956)
Ger Pat 1 058 515 (1956)
Faust T etal 1 Am Chem Soc 81,2214(1959)
US Pat 2 554 511 (1951)
US Pat 2 726 245 (1955)
LSPat 2 831 027 (1958)
Stain G etal 7J Am Chem Soc 78, 1514 (1956)
US Pat 2 489 238 (1949)
LSPat 2 519 720(1950)
US Pat 3 740 416(1973)
US Pat 4 130 713 (1978)
La^ielleS etal/'J Am Chem Soc 100, 1558 (1978)
। Глава 15
! Мышечные
I релаксанты
Мышечные релаксанты (миорелаксанты) — это большая группа химических соединений, обладающих способностью расслаблять скелетные мышцы.
На скелетную мускулатуру можно воздействовать довольно разнообразной и широкой группой веществ, действующих как на уровне нервно-мышечных соединений, так и на различных уровнях спинного мозга и ствола головного мозга.
Некоторые из них, воздействуя на передачу нервных импульсов в местах нервно-мышечных контактов, способны парализовать скелетную мускулатуру и используются преимущественно как вспомогательные вещества при анестезии в ходе малых хирургических вмешательств.
С другой стороны, имеются миорелаксанты, которые, действуя либо на передачу нервных импульсов в нервно-мышечных синапсах или непосредственно на сократительный механизм скелетной мускулатуры, либо на передачу импульсов на уровне спинного мозга, вызывают различную степень мышечного расслабления вплоть до полного блокирования скелетной мускулатуры. Последние используются для облегчения состояния пациента при мышечных спазмах, гиперрефлексии, гиперкинезах, связанных с воспалением, стрессом и рядом неврологических заболеваний.
В соответствии с локализацией и механизмом действия миорелаксанты могут быть классифицированы как:
• миорелаксанты периферического действия,
• мышечные релаксанты прямого действия,
• мышечные релаксанты центрального действия.
Активность миорелаксантов периферического действия проявля
-292-
Мышечные релаксанты
ется в области нервно-мышечных контактов, в результате чего ослабляется передача от окончаний двигательных нейронов к мембранам клеток скелетных мышц.
Последние, в свою очередь, включают блокаторы нервно-мышечной передачи, которые можно подразделить на анпгиоеполяризующие препараты (тубокурарин, атракурий, галламин) и деполяризующие препараты (сукцинилхолин).
Мышечные релаксанты прямого действия непосредственно блокируют процесс сокращения самих мышечных волокон. Из миотропных препаратов прямого действия в практической медицине используется только дантролен.
Широко применяются также мышечные релаксанты центрального действия (баклофен, циклобензаприн, карисопродол, метокарбамол, хлорфенезин, хлорзоксалон, орфенадрин и диазепам), которые подавляют передачу двигательных импульсов в межнейрональных синапсах ЦНС.
15.1. Блокаторы нервно-мышечной передачи
Это соединения, блокирующие передачу импульсов от двигательных нервных окончаний к скелетной мускулатуре.
Предполагается, что есть два механизма блокирования передачи нервных импульсов. Одна группа препаратов, родоначальником и типичным представителем которой является тубокурарин, называется антидеполяризующими препаратами. Конкурентно связываясь с соответствующим Н-холинорецепторным участком, они противодействуют действию ацетилхолина на постсинаптическую мембрану, предотвращая его деполяризующее действие и тем самым исключают возможность возбуждения мышечного волокна.
Следует иметь в виду, что из-за очень малой разницы в дозах, вызывающих необходимое мышечное расслабление и способствующих развитию паралича скелетной мускулатуры, небольшая передозировка курареподобных соединений может привести к серьезным нарушениям со стороны дыхательной функции и резкому понижению давления. Передозировку снимают путем введения антихолинэстеразных средств, которые, блокируя ацетилхолинэстеразу, повышают концентрацию ацетилхолина в синаптической щели, используют искусственное дыхание с применением кислорода, а при необходимости — средства, повышающие АД (левартеренол).
Другая группа препаратов, представителем которой является сукцинилхолин, называется деполяризующими препаратами. Соединения
-293-
Глава 15
указанной группы вызывают первоначальную активацию (деполяризацию) рецептора с последующей его длительной и стойкой блокадой, что приводит к задержке реполяризации, блокированию возможности последующей стимуляции рецептора и в итоге к нарушению проведения возбуждения с нерва на мышцу.
В отличие от недеполяризуюших веществ, эти препараты являются не конкурирующими антагонистами, а, наоборот, более устойчивыми агонистами, чем сам ацетилхолин.
Приемлемыми для практики антагонистами деполяризующих средств в настоящее время медицина не располагает.
Антидеполяризующие нервно-мышечные блокаторы
Впервые нервно-мышечные блокаторы были выделены из кураре — экстракта, получаемого из южно-американских растений вида Strych-nos и Chondodendron Сегодня в качестве антидеполяризующих или курареподобных препаратов, которые называются также антидеполяризующими, или конкурентными блокаторами, используются синтетические соединения, а также тубокурарин — алкалоид, выделяемый из кураре.
Тубокурарин и большинство синтетических курареподобных соединений содержат два или более четвертичных атома азота, расположенных на расстоянии примерно 1.0 ± 0,1 ши друг от друга, что представляется необходимым условием для связывания этого типа препаратов с никотиновыми холинорецепторами.
Эти препараты используются при оперативных вмешательствах, когда требуется релаксация скелетных мышц, в травматологии для репозиции отломков при вправлении сложных вывихов, при столбняке. Интересно, что курареподобные препараты расслабляют мышцы в определенной последовательности. В первую очередь расслабляется мускулатура лица, шеи, затем конечностей и туловища. В последнюю очередь выключаются дыхательные мышцы и диафрагма, что сопровождается остановкой дыхания.
Соединения этой группы включают тубокурарин, метокарин, гал-ламин, панкуроний, векуроний и атракурий.
Тубокурарин (Tubocurarine)
Тубокурарин — дихлорид 71,12'-дигидрокси-6,6|-диметокси-2,2,21.2 -тетраметилтубокурариния (15.1.1) получают из водных экстрактов растений родов Chondrodendron [1, 2, 3, 4, 5, 6].
- 294-
Мышечные релаксанты
Предложены методы синтеза тубокурарина [7. 8].
Тубокурарин применяют, главным образом, в анестезиологии в качестве миорелаксанта, вызывающего длительное расслабление мускулатуры во время операций. Малыми дозами препарата удается вызвать временное расслабление скелетной мускулатуры без существенного изменения основных функций организма, что, в частности, используют при эндотрахеальной интубации или в ортопедии при репозиции обломков, вправлений сложных вывихов и др.
Основными синонимами препарата являются тубарин, курарин.
Метокурин (Metocurine)
Метокурин — 6,617|,12|-тетраметокси-2,2,21,21-тетраметилтубоку-рариния дихлорид (15.1.2) получают метилированием двух гидроксильных групп тубокурарина метилхлоридом [9].
СН3С|
15 1 1 -------:
Метокурин применяют по тем же показаниям, что и тубокурарин.
Синонимом препарата является метубин.
Галламин (Galiamin)
Галламин — трийодид 1,2,3-трис(2-триэтиламиноэтокси)бензола
(.15.1.4) синтезируют исходя из пирогаллола, гидроксильные груп
- 295-
Глава 15
пы которого этерифицируются 2-диэтиламиноэтилхлоридом в присутствии амида натрия. Полученный при этом 1.2,3- грис(2-триэтил-аминоэтокси)бензол (15.1 3) далее алкилируется по всем трем атомам азота этилйодидом с получением галламина (15.1.4) [10, 11].
он
1 NaNH2
2 CL-CH2-CH2-CH2-N(C2H5)2
О —~СН2 —CHj “СН2 —N(C2Hg)2
—о—Сн^—СНг-CHj—NfCzHaig
О—СН2 ~СН2 —СН2 —N<£-2^5)2 151 3
О—СН2 ~СН2“СН2 ""М(С2Н5)з
+
О—СН2 ~СН2 “СН2 “N(C2H5)3
+
О—CH2-CH2-CH2-N(C2H5)3
15 1 4
3 Г
Галламин применяют по тем же показаниям, что и тубокурарин. Синонимом препарата является флакседил.
Панкуроний (Pancuronium)
Панкуроний — 1,1'-(За,17р-диацетокси-5а-андростан-2р,1бр-илен-)-бис-(1-метилпиперидиния) дибромид (15.1.8) получают исходя из 3,17-бмс-(ацетокси)-2,16-5а-андростадиена. Окислением последнего 3-хлорнадбензойной кислотой получают бис-эпоксисоединение (15.1.5), взаимодействием которого с пиперидином и дальнейшим гидролизом выделяют аминокетон (15.1.6). Кетогруппу образовавшегося соединения (15.1.6) восстанавливают боргидридом натрия до спиртовой с получением бис-аминоспирта (15.1.7), и дальнейшим ацетилированием спиртовых групп уксусным ангидридом и алкилированием обоих атомов азота метилбромидом получают целевой панкуроний (15.1.8) [12, 13, 14].
-296-
Мышечные релаксанты
Панкуроний является стероидным соединением, но гормональной активностью не обладает. Применяется в анестезиологии в качестве миорелаксанта, вызывающего длительное расслабление мускулатуры, при хирургических вмешательствах на грудной и брюшной полостях, в проктологии, офтальмологии, ортопедической практике, а также при операциях на сердце.
Синонимом препарата является павулон.
Векуроний (Vecuronium)
Векуроний — 1 -[(20,За,5а, 160,170)-3,17-бис(ацетокси)-2-( 1 -пипе-ридинил)-андростан-16-ил]-1-метилпиперидиния бромид (15.1.9) отличается от панкурония лишь гем, что в алкилированное состояние — четвертичную соль переводят лишь пиперидиновый заместитель у С|0 стероидного скелета [15, 16].
Векуроний применяют по тем же показаниям, что и панкуроний. Синонимом препарата является норкурон.
-297 -
Глава 15
Атракурий (Atracurium)
Атракурий — дибензолсульфонат 2,2’-[1,5-пентадиилбис[окси(3-оксо-3,1-пропандиил)]]бис[1-[(3,4-диметоксифенил)метил]-1,2,3,4-тетрагидро-6.7-диметокси-2-метилизохинолиния] (15.1.12). Синтез этого соединения осуществляют исходя из бис-акрилового эфира 1,5-пентандиола (15.1.10), получаемого из хлорангидрида акриловой кислоты и 1.5-пентандиола. К последнему по реакции Михаэля присоедиеняют две молекулы вторичного амина — тетрагидропапаверина с получением продукта (15.1.11), оба атома азота которого метилируют метилбензолсульфонатом с полхчением атракурия (15.1.12) [17, 18, 19].
СН3О
СНзО
15 1 10
*1 0
•N— Н2С-СН-С—О-(СН2)5~О—’С-СН-СН2 — N.
ОСНз
ОСНз
о
Атракурий применяют по тем же показаниям, что и тубокурарин. Синонимом препарата является тетракурий.
-298-
Мышечные релаксанты
Деполяризующие нервно-мышечные блокаторы
В отличие от недеполяризующих веществ, деполяризующие нервно-мышечные блокаторы, являются не конкурирующими антагонистами, а. наоборот, более устойчивыми агонистами, чем ацетилхолин. В принципе. они отличается от ацетилхолина только большей продолжительностью действия. Эти препараты взаимодействуют с тем же рецепторным участком, что и ацетилхолин. Но, поскольку эти препараты инактивируются медленнее, чем ацетилхолин, то и действуют на синапсе дольше, вызывая более устойчивую деполяризацию. Таким образом, блокируется процесс реполяризации рецептора, и наступает релаксация скелетных мышц.
Большое практическое значение в качестве нервно-мышечного блокатора в медицине имеет сукцинилхолин.
Сукцинилхолин (Succinylcholine)
Сукцинилхолин — дихлорид 2,2'-[(1,4-диоксо-1,4-бутандиил)бмс-(окси)]бмс^,К,Х-триметилэтиламмония] (15.1.14). который можно рассматривать как удвоенную молекулу ацетилхолина (диацетилхолин). получают взаимодействием дихлорангидрида янтарной кислоты с 2-диметиламиноэтанолом и дальнейшим переводом полученного бис-(2-диметиламиноэтил)сукцината (15.1.13) в четвертичную соль — сукцинилхолин (15.1.14) [20, 21, 22, 23, 24],
CHi-C-CI i
CH--C-CI м G
О
СН2-С-О—CH2“CH2-N(GH3)2 сн С|
2 HO-Cri2-CH2-N(CH3)2 ---► |
CH2-C-O—CH2-CH2-N(CH3)2 о
15 1 13
н +
СНг-С-О— CH2-CH2-N(CH3)3
СНг -с-0—СНг-СНг -N(CH3)3 о +
15 1 14
2 CI
Сукцинилхолин является единственным терапевтически используемым деполяризующим нервно-мышечным блокатором. В отличие от нетеполяризующи.х веществ, сукцинилхолин является не конкурирую
Глава 15
щим антагонистом, а, наоборот, более устойчивым агонистом, чем ацетилхолин. Таким образом, сукцинилхолин отличается от ацетилхолина только продолжительностью действия и, соответственно, действует дольше, вызывая более устойчивую деполяризацию. Таким образом блокируется процесс реполяризации, и наступает мышечное расслабление. При этом мышцы, вызывающие тонкие движения (глазные, головные лицевые, шейные) наиболее чувствительны и блокируются первыми, после чего блокируются мышцы конечностей и, наконец, наиболее устойчивые дыхательные мышцы. Восстановление происходит по окончании действия препарата.
Терапевтическое использование сукцинилхолина заключается в предотвращении у пациентов непроизвольных движений. Его применяют при кратковременных операциях, интубации трахеи и другие эндоскопических процедурах.
Синонимами препарата являются листенон, мидарин, сукострин, дитилин и др.
/5.2. Мышечные релаксанты прямого действия
Единственным препаратом такого типа, получившим широкое признание, является дантролен.
Дантролен (Dantrolen)
Дантролен — 1 -Ц[5-(4-нитрофенил)-2-фуранил]метилен]амино]-2,4-имидазолидин-дион (15.2.2) получают взаимодействием 4-нитрофе-нилдиазоний хлорида с фурфуролом с образованием 5-(4-нитро-фенил)-2-фуранкарбоксальдегида (15.2.1), который далее вводят во взаимодействие с 1-аминогидантоином с получением соответствующего гидразона—дантролена (15.2.2) [25, 26].
II И / CuCL2 /=\ NaNOj/HCL АА + - ° СН0
О2М —\\ й—NH2 ----------► °2n —\\ д—N2 CL ----------------*
-300-
Мышечные релаксанты
Дантролен является препаратом, который вызывает спастическое сокращение мышц, В отличие от других мышечных релаксантов, он непосредственно воздействует на сократительный механизм, вмешиваясь в процесс высвобождения ионов Са2+ из саркоплазматической сети. Это выражается в несогласованности механизма возбуждения-сокращения скелетной мускулатуры, что в большей степени отражается на волокнах быстрых, а не медленных мышц.
Дантролен используется для контроля проявлений клинической спастичности, возникающей в результате серьезных клинических случаев, таких как ранения, паралич, церебральный паралич и рассеянный склероз.
Синонимами препарата являются дантрий и данулен.
15.3. Мышечные релаксанты центрального действия
К сожалению, паралич скелетной мускулатуры, достигаемый применением курареподобных соединений, в большинстве случаев не является пригодным при общих состояниях спастичности, сопровождающихся поражениями ЦНС, а также при местных повреждениях и воспалениях. Конечно, нервно-мышечная блокада облегчает спазм, однако сопровождается потерей способности произвольным движениям.
В условиях мышечной спастичности бывают необходимы препараты, способные снять болевой мышечный спазм без утраты способности мышцы к произвольным сокращениям и без ухудшения мозговой функции.
Многие депрессанты ЦНС вызывают мышечное расслабление. Среди них следует отметить алкоголь и барбитураты, которые, однако, не используются для этой цели, поскольку вызывают значительную седацию и другие эффекты. Поиск селективных ЦНС-активных веществ, ответственных за возможное достижение мышечного расслабления, привел к получению ряда интересных соединений, предложенных клинике, ни одно из которых, однако, не смогло полностью удовлетворить необходимым требованиям. Тем не менее мышечные релаксанты, действующие посредством воздействия на ЦНС. широко используются при лечении растяжений, разрывов, артритов и других мышечных нарушениях. В их число входит большая гетерогенная группа химических соединений, которые воздействуют через спинной мозг и подавляют мо-носинаптические и полисинаптические рефлексы. Среди них баклофен, циклобензаприн, карисопродол, метокарбамол, хлорфенезин, хлорзок-салон. орфенадрин и диазепам.
- 301 -
Г лава 15
Баклофен (Baclofen)
Баклофен — 4-амино-3-(4'-хлорфенил)масляную кислоту (15.3.5) предложено получать двумя методами. Согласно первому способу, 4-хлорбензальдегид конденсируют с двумя молями ацетоуксусного эфира с получением продукта (15.3.1), который сначала подвергают щелочному расщеплению с получением 3-(4-хлорфенил)глутаровой кислоты (15.3.2). Дегидратацией последней получают ангидрид 3-(4-хлорфенил)глутаровой кислоты (15,3.3) и далее обработкой аммиаком — соответствующий глутаримид (15.3.4). Действием на последний щелочным раствором галогена (расщепление по Гофману) получают баклофен (15.3.6) [27, 28].
2 СН3
О-С2Н5
СгНбОМа
-СООС2Н5
-СООС2Н5
СОСНз
NaOH
СОСНз
(СН3СО)2О
1535
Второй способ синтеза баклофена исходит из этилового эфира 4-хлоркоричной кислоты. Присоединением к ней нитрометана в присутствии тритона В получают этиловый эфир р-(4-хлорфенил)-у-нитро-масляной кислоты (15.3.6), нитрогруппу которой восстанавливают водородом над никелем Ренея в этиловый эфир Р-(4-хлорфенил)-у-амино-масляной кислоты (15.3.7), который далее гидролизуют в целевой баклофен (15 3.5) [29].
•coccus
CH2-NO2 |И)
СН ---
1535
-302-
Мышечные релаксанты
Ьаклофен является замещенным аналогом ГАМК. Предполагается, ч'о его действие состоит во взаимодействии с ГАМК-рецепторами, что приводит к ингибированию высвобождения возбуждающих нейро ;рансмитт еров.
Показан при признаках мышечной спастичности, рассеянном склерозе и других спинальных расстройствах. Может быть полезен па-циешам с мышечными спазмами в результате повреждений спинного мозга.
Синонимом препарата является лиоресаль.
Цик. юбензаприн (Cyclobenzaprine)
Циклобензаприн — К,^т-диметил-3-(дибензо[а,б]циклогептен-5-или-ден)пропиламин (15.3.9) получают взаимодействием 5Я-дибензо[а,б]-циклогептен-5-она с 3-диметиламинопропилмагнийхлоридом и дальнейшей дегидратацией образующегося карбинола (15 3.S) в кислых условиях в циклобензаприн (15.3.9) [30, 31, 32].
1539
СНз
HCI
Циклобензаприн структурно схож с трициклическим антидепрессантами. Действует на уровне ствола головного мозга. Препарат применяют в качестве вспомогательного средства для облегчения мышечного спазма, связанного с острыми болезненными состояниями мышц.
Синонимом препарата является флексерил.
Кариспродол (Carisprodol)
Кариспродол — дикарбамат М-изо-пропил-2-метил-2-пропил-1,3-пропандиола (15.3.12) получают взаимодействием 2-метил-2-пропилпропандиола-1.3 с одним молем фосгена с получением хлоругольного эфира (15.3.10), из которого взаимодействием с изопро-
- 303 -
Г лава 15
пиламином получают карбамат (15.3,11). Вводя последний в реакцию либо с О-этилуретаном, либо с цианатом натрия, получают ка-риспродол (15.3.12) [33].
СНз СОС|2 СН3 H2NCH(CH3)2
С3Н7-С-СН2-О-Н ---------► С3Н7-С-СН2-О-СО-С1 *
сн2-о-н сн2-о-н
13.3.10
СНз
С-.Ц-- -с -СН2 - O-CO-HNCH(CH3)2
СН2-0-Н
13.3.11
C2H5OCONH2 or NaCNO
СНз
C3H7-C-CH2-O-CO-HNCH(CH3)2
CH2-NH2
13 3.12
Кариспродол подавляет .межнейрональную деятельность ретикулярной формации спинного мозга. Применяют в качестве вспомогательного средства при утрате гибкости скелетной мускулатуры, а также для облегчения болей, возникающих при этом.
Синонимами препарата являются рела, сома, карисома, санома.
Метокарбамол (Methocarbamol)
Метокарбамол — карбамат 3-(2-метоксифенокси)-1,2-пропандиола-1
(15.3.13) синтезируют последовательным действием фосгена и затем аммиака на 3-(2-метоксифенокси)пропандиол-1,2 [34, 35].
осн3
о-сн2-сн-сн2-он он
1.СОС12
2. NH3
OCH3
с />— о—сн2—сн-сн2—oconh2 '---' он
153.13
Метокарбамол подавляет мультисинаптические пути в спинном мозге. Применяют для облегчения спазмов и болей в скелетной мускулатуре, а также при лечении столбняка.
Синонимами препарата являются делаксин, форбаксин, робамол, робаксин, тресортил.
-304-
Мышечные релаксанты
Хлорфенезин (Chlorphenesin)
Хлорфенезин — 3-(4-хлорфенокси)-1,2-пропандиол (15.3.14) синтезируют аналогично описанному выше метокарбамолу исходя из 3-(4-хлорфенокси)-1,2-пропандиола [36, 37, 38].
15 3.14
о-сн2 —сн-сн2-oconh2 он
Хлорфенезин действует по невыясненному механизму. Применяют для облегчения болей в скелетной мускулатуре.
Синонимами препарата являются маолат и мусил.
Хлорзоксазон (Chlorzoxazon)
Хлорзоксазон — 5-хлор-2-бензоксазолинон (15.3.15) получают реакцией гетероциклизации при взаимодействии 2-амино-4-хлорфе-нола с фосгеном [39].
СОС12
15.3.15
Хлорзоксазон подавляет мультисинаптические пути в спинном мозге. Применяют для облегчения болей в скелетной мускулатуре.
Синонимами препарата являются оксирен и парафлекс.
Г лава 15
Список литературы
1. Dutcher J. et al.//J. Am. Chem. Soc. 68, 419 (1946).
2. Dutcher J. et al. '/J. Am. Chem. Soc. 74, 2221 (1952).
3. USPat. 2.409.241 (1946).
4. US Pat. 2.600.539 (1952).
5. Everett A etai.//J. Chem. Soc. D, 1970, 1020.
6. Codding P. et al. /J. Chem. Soc. D, 1972, 1174.
7. Naghaway J. et aJ.//J. Pharm. Sci. 68, 655 (1979).
8. Веронин А. и др./,'Доклады АН СССР. 122. 77 (1958).
9. USPat. 2.581.903 (1952).
10. USPat. 2.544.076(1951).
11. Ger. Pat. 817.756(1947).
12. Bucket W. et aV/Chim. Ther. 2, 186 (1967).
13. Bucket IF. et al.//J. Med. Chem. 16, 1116 (1973).
14. USPat. 4.177.190(1975).
15. Bucket IE et al.//J. Med. Chem. 16, 116 (1973).
16. Eur. Pt. Appl. 8824(1980).
17. USPat. 4.179.507(1979).
18. Ger. Pat. 2.655.833 (1976).
19. Stenkale J. et al.//Eur. J. Med. Chem. 16, 515 (1981).
20. Fusco R. et al./'/Gazz. Chim. Ital.//79, 129 (1949).
21. Tammelin L. ,'Acta Chem. Scand. 7, 185 (1953).
22. Walker J./A. Chem. Soc. 1950, 193.
23. Wang C. et al.''/Org. Prep. Proc. Int. 11. 93 (1979).
24. Austrian Pat. 171.411 (1952).
25. USPat. 3.415.821 (1968).
26. Snyder H. et al./ 'J. Med Chem. 10, 807 (1967).
27. USPat. 3.471.548 (1969).
28. USPat. 3.634.428 (1972).
29. Jap. Pat. 16.692 (’70) (1970).
30. Brit. Pat. 858.187 (1961).
31. Villani F. et al.7J, Med. Pharm. Chem. 5, 373 (1962).
32. Wintrop H. et al./'J. Org. Chem. 27, 230 (1962).
33. US Pat. 2.937.119(1960).
34. US Pat. 2.770.649 (1956).
35. Yale H. et al.// J. Am. Chem. Soc. 72, 3710 (1950).
36. USPat. 3.161.567(1964).
37. USPat. 3.214.336 (1965).
38. Brit. Pat. 628.497 (1948).
39. USPat. 2.895.877 (1959).
-306-
I
j Гл а в a 16
। Антигистаминные
а препараты
I
Препараты, которые конкурентно блокируют эффекты гистамина на соответствующих рецепторных участках, называются антигистаминными препаратами.
Открытие и синтез гистамина явились крупными достижениями в фармакологии, медицине и иммунологии. Этот природный мощный биогенный амин широко распространен практически во всех тканях млекопитающих и вовлечен в разные физиологические процессы. Реакция организма на гистамин характеризуется сокращением гладкой мускулатуры, признаками воспаления, сужением сосудов и симптомами, характерными для шока. Достоверно известно, что гистамин играет центральную роль при аллергических реакциях, реакциях гиперчувствительности и участвует в механизмах ответов организма на воспалительный процесс.
xnz^ch2-ch2-nh2 < н
гистамин
Гистамин синтезируется в тканях путем декарбоксилирования аминокислоты L-гистидина — процесса, катализируемого пиридоксаль-фосфатзависи.мым ферментом (L-гистидиндекарбоксилазой). Гистамин может поступать в организм с пищей, а также продуцируясь бактериями ЖКТ. Однако эти источники не создают дополнительных запасов гистамина, поскольку в организме экзогенный гистамин легко катаболизи-руется.
Гистамин распределяется и запасается в тучных клетках большинства органов, в которых он сохраняется в выделительных цитоплазматических гранулах в виде гепарин-протеазных матриц, составляя около 10% от их массы. Гистамин становится физиологически активным только после высвобождения из гранул.
-307 -
Глава 16
Гистамин обнаружен и в тканевых жидкостях — желудочном соке, крови, моче. Лишь 2 3 % гистамина выводятся из организма неизмененными. В основном, он метаболизируется двумя ферментными путями, включающими дезаминирование диаминоксидазой и метилирование гистамин N-метилтрансферазой.
После выделения из тканевых депо гистамин может вызывать огромное множество физиологических эффектов. Однозначно доказана ею роль в различных патологических процессах, связанных с острыми и хроническим аллергическими реакциями, а также реакциями гиперчувствительности. Вместе с тем функции эндогенного гистамина (в проведении нервной передачи, выделении желудочного сока, роста тканей, их восстановления) остаются не до конца выясненными.
Несмотря на то, что высвобождение эндогенного гистамина может быть вызвано разными причинами, полагают, однако, что важнейшим является иммунологический ответ организма. Принято считать, что во время анафилаксии и аллергии на поверхности тучных клеток и базофилов происходит специфическое взаимодействие иммуноглобулина Е с антигеном, в результате чего включается каскад биохимических событий, приводящих к дегрануляции с высвобождением гистамина.
Кроме указанных выше реакций антиген-антитело, которые играют критическую роль в патогенезе многих аллергических, анафилактических реакций и реакций гиперчувствительности, гистамин может быть выделен из тканевых депо и в результате физических воздействий, влияния так называемых либерантов гистамина — ряда химических веществ, различных препаратов и токсинов.
Существует большой класс соединений, которые способны высвобождать гистамин. Это могут быть ферменты и яды, морфин и d-ту-бокурарин, полимеры, например декстран. Более того, повреждения тканей (травмы, укусы, стресс) также могут вызвать высвобождение гистамина, по всей вероятности, как следствие высвобождения при этом эндогенного полипептида брадикинина. Действие всех вышеперечисленных веществ, как и ряда других, на организм может способствовать формированию анафилактических реакций.
Высвобождение гистамина блокируется разными ингибиторами ферментов и другими веществами (никотинамид).
Главное физиологическое действие гистамина проявляется на сердечно-сосудистой системе, несосудистой гладкой мускулатуре, экзокринных железах и надпочечниках.
Его наиболее важные фармакологические эффекты заключаются в расширении вен и капилляров, увеличении проницаемости капилляров, увеличении скорости работы сердца, сокращении несосудистой
- 308 -
Антигистаминные препараты
гладкой мускулатуры (сужение бронхов, перистальтики ЖКТ), стимуляции секреции желудочного сока, выделении катехоламинов из надпочечников.
Фармакологическое действие гистамина опосредуется двумя мембранными рецепторными связывающими участками, названными Нг и Hj-реиепторами. Нгрецепторы расположены в гладкой мускулатуре сосудов, бронхиол и ЖКТ, тогда как Н2-рецепторы найдены в стенках желудка, в миокарде и определенных сосудах.
Поэтому представляется весьма вероятным, что сокращение несосудистой гладкой мускулатуры является эффектом воздействия на Н -реиепторы, а выделение желудочного сока и увеличение частоты сердцебиений связано с активацией Н2-рецепторов; при этом расширение сосудов и повышение проницаемости капилляров являются результатом комбинированного воздействия на оба типа рецепторов.
Имеются также специфические различия в распределении рецепторов в различных тканях и у различных животных. Так, если мыши и крысы достаточно устойчивы к эффектам гистамина, то организм морских свинок и человека весьма чувствительны.
Антигистаминные препараты классифицируются как антагонисты Н|- и Н2-рецепторов и в количественном отношении Н|-антагонисты превалируют. Более того, термин «антигистаминный препарат» больше ассоциируется именно с Нгантагонистами. Н2-блокаторы проявляют специфический эффект на гистаминные рецепторные участки, расположенные в стенке желудка и значительно понижают секрецию соляной кислоты.
Аллергические заболевания представляют собой сложный набор нарушений с хроническими и острыми проявлениями, варьирующими от легких покраснений, сыпи и насморка до сильной, а возможно и фатальной. анафилаксии. Установлено, что около 10% населения могут быть подвержены какой-либо форме аллергии. Терапия, направленная на устранение источника аллергена, не всегда оказывается успешной. В ряде случаев бывает не выявлен и сам аллерген. Поэтому проводится симптоматическое лечение с применением Н.-антигистаминных препаратов.
Сам гистамин в настоящее время не имеет терапевтического значения и в клинике не применяется, хотя и была попытка его использования в качестве препарата для лечения ахлоргидрии — отсутствия в желудке соляной кислоты. Он может быть использован в малых дозах с диагностическими целями, т. е. для стимуляции желудочных желез для выяснения их способности генерировать соляную кислоту и иногда, при диагностике феохромоцитомы.
-309-
Г лава 1 6
16.1. Н/-антигистаминные препараты
Антигистаминные препараты были открыты в конце 1930-х годов. К 1950-м годам были предложены высокоэффективные антагонисты гистамина — трипеленнамин и дифенилгидрамин, которые инициировали широкие исследования в области синтеза этого типа препаратов.
Все эти соединения являются обратимыми, конкурентными антагонистами гистамина на Нгрецепторах и не проявляют существенной активности в отношении Н2-рецепторов. Антагонисты Нгрецепторов в разной степени блокируют эффекты гистамина в различных органах или системах и могут обеспечить защиту организма от аллергических или анафилактических реакций. Сами по себе они не выявляют значительной самостоятельной активности, и поэтому в терапевтическом плане их используют только для блокирования эффектов, возникающих в результате выделения гистамина. Иначе говоря, их эффекты заметны только при повышенной гистаминной активности. Более того, эти антигистаминные препараты только уменьшают высвобождение или метаболизм гистамина, но никак не влияют на его синтез.
Несмотря на четкие различия в относительной активности этих препаратов, они имеют сравнимые фармакодинамические свойства и терапевтическое применение при рассмотрении в качестве единой группы препаратов.
Наиболее распространенные Нгантигистаминные препараты структурно напоминают гистамин с участием замещенной этиламинной боковой цепи, однако они имеют два ароматических кольца и только формально могут быть представлены общей формулой:
Аг1\ | |
X-C-C-N
Ar2Z II \r2
где Ari и Аг2 являются карбоциклическими или гетероциклическими ароматическими кольцами, одно или оба из которых могут быть отделены от X атомом углерода;
X является кислородом, углеродом или азотом;
R; и R2 представляют собой алкильные заместители, обычно метильные группы.
Блокаторы Нггистаминных рецепторов принято группировать согласно их химическому строению:
-310-
Антигистаминные препараты_____________________________________
• производные этаноламина (дифенгидрамин, клемастин);
• производные этилендиамина (трипеленнамин, пириламин);
• алкиламины (хлорфенирамин, дексхлорфенирамин, бромфени-рамин);
• пиперазины (циклизин, меклизин, гидроксизин);
• фенотиазины (прометазин, тримепразин);
• пиперидины (ципрогептадин, дифенилпиралин);
• прочие, не поддающиеся определенной химической классификации (терфенадин, астемизол).
Их клиническая эффективность и проявляемые побочные эффекты значительно отличаются при переходе от группы к группе и от пациента к пациенту. Эти препараты предотвращают действие как эндогенного, гак и экзогенного гистамина, однако значительно более эффективны в отношении первого.
Все они действуют путем конкурентного связывания с Н|-рецепторами. Их применяют для облегчения симптомов аллергических заболеваний (аллергических ринитов, других аллергических реакций), для лечения анафилактических реакций, временного облегчения состояния бессонницы, в качестве вспомогательной терапии при лечении паркинсонизма и эстрапирамидных расстройств антипсихотиками, для облегчения кашля простудного, аллергического и другого происхождения, предотвращения и контроля тошноты и рвоты, в качестве вспомогательных препаратов при анальгезии послеоперационных болей и для предоперационной седации.
Аминоалкильные эфиры
Дифенгидрамин (Diphenhydramine)
Дифенгидрамин — К,М-диметил-(2-дифенилметокси)этиламин (16.1.1) синтезируют простым взаимодействием бензгидрилбромида и 2-ди-метиламиноэтанола [1, 2, 3].
* СНз
СН-Вг + HO-CH2-CH2-n(
\ СНз
> СН3
сн-о—ch2-ch2-n(
' СНз
/ 16 1 1
- 311 -
Г лава 1 6
Дифенгидрамин — один из основных представителей антигистаминных препаратов, блокирующих Нгрецепторы. Кроме антигистаминной активности дифенгидрамин оказывает местноанестезирующее действие, расслабляет гладкую мускулатуру, проявляет седативное и снотворное действие.
Дифенгидрамин применяют при симптомах аллергии, при лечении крапивницы, сенной лихорадки, сывороточной болезни и других аллергических заболеваний, а также в качестве успокаивающего и снотворного средства, как самостоятельно, так и в сочетании с другими препаратами.
Синонимами препарата являются димедрол, бенадрил, аллерган, вальдрен и многие другие.
Дименгидринат (Dimenhydrinate)
Дименгидринат — (16.1.2) является комплексным соединением №,М-диметил(2-дифенилметокси)этиламина — дифенгидрамина с 8-хлортеофиллином.
Блокируя Н|-рецептор, дименгидринат одновременно воздействует и на рвотный центр [4. 5].
Дименгидринат применяют для предупреждения и купирования морской и воздушной болезни, при тошноте и рвоте.
Синонимами препарата являются драмамин, дадалон, эмедил, тра-велин и др.
Клемастин (Clemastine)
Клемастин — 2-[2-[ 1 -(4-хлорфенил)-1 -фенилэтокси]этил]-1 -метил-пирролидин (16.1.4) получают взаимодействием 1-(4-хлорфенил)-1-фенилэтанола (16.1.3) с 2-(2-хлорэтил)-1-метилпирролидином, используя в качестве основания амид натрия. Исходный 1-(4-.хлор-фенил)-1-фенилэтанол (16.1.3) получают либо взаимодействием 4-хлорбензофенона с метилмагнийхлоридом, либо взаимодействием 4-хлорацетофенона с фенилмагнийбромидом [6, 7, 8].
-312-
А н т и г истаминные препараты
CH3MgCi
Клемастин применяют при симптомах аллергии, ринитах, отеке Квинке, анафилактическом шоке, сывороточной болезни, сенной лихорадке, аллергических дерматитах и дерматозах, хронической экземе.
Синонимами препарата являются тавегил, мекластин.
Эти лен ди амины
Трипеленнамин (Tripelennamine)
Трипеленнамин — Кт-бензил-\т|,К'-диметил-К-2-пиридилэтилен-диамин (16.1.6) синтезируют взаимодействием 2-бензиламинопири-дина (16 1.5) с 2-диметиламино-этилхлоридом в присутствии амида натрия. 2-бензиламинопиридин, в свою очередь, может быть легко получен восстановлением основания Шиффа, полученного конденсацией 2-аминопиридина с бензальдегидом [9, 10, 11].
НС ООН
СНз ci—ch2-ch2-n(
СНз
СНз
—ch2-ch2-n\
СНз
161 6
-313-
Глава 16
Препарат уменьшает аллергический ответ организма, вызванный гистамином. Трипеленнамин применяют при симптомах аллергии, ринитах, конъюнктивитах, аллергических и анафилактических реакциях.
Синонимами препарата являются пеламин и пирибензамин.
Пириламин (Pyrilamine)
Пириламин — М-(4-метоксибензил)-Хт' ,N '-диметил-К-2-пиридил-этилендиамин (16.1.7) синтезируют аналогичным путем, но с использованием 2-(4-метоксибензиламино)пиридина [10, 11, 12].
сн3о-4 Z/~CH2\
N
СН3
—сн2-сн2-м(
СНз
16 1 7
Пириламин также применяют при симптомах аллергии, ринитах.
Синонимами препарата являются вистосан, антисан и триаминик.
Хлорпирамин (Chlorpyramin)
Хлорпирамин — К-(4-хлорбензил)-Х|,Х|-диметил-Х-2-пиридил-этилендиамин (16.1.9) получают несколько иным путем, а именно: взаимодействием 2-бромпиридина с Х-(4-хлорбензил)-Х|,К|-диме-тилэтилендиамином (16.1.8). М-(4-хлорбензил)-М',Х’-диметилэти-лендиамин (16.1.8), в свою очередь, получают конденсацией 4-хлорбензальдегида с N.N-диметилэтилендиамином с последующим восстановлением иминной группы [13, 14, 15, 16, 17, 18, 19].
СНз h2n-ch2-ch2-n(
СНз
Н2 / Raney— Ni
сн3
СНз
- 314 -
А н т и г истаминные препараты
Хлорпирамин применяют при аллергических дерматозах, аллергическом рините и конъюнктивите, при медикаментозных аллергиях, в начальной стадии бронхиальной астмы, при экземе, нейродермите, контактных дерматитах и токсикодермии. Препарат оказывает также седативный эффект.
Синонимами препарата являются супрастин, хлортрипеленамин, синопен.
Алкиламины
Хлорфенирамин (Chlorpheniramine)
Хлорфенирамин — 3-(иора-хлорфенил)-3-(2-пиридил)пропилди-метиламин (16.1.12) предложено получать двумя путями. Первый способ исходит из 4-хлорбензилцианида, взаимодействием которого с 2-хлорпиридином в присутствии амида натрия получают 4-хлорфенил(2-пиридил)ацетонитрил (16.1.10). Алкилируя последний 2-диметиламиноэтилхлоридом в присутствии амида натрия, синтезируют у-(4-хлорфенил)-у-циано-Х,Х-диметил-2-пиридилпро-панамин ( 16.1.11), гидролиз и декарбоксилирование которого приводит к хлорфенирамину (16.1.12) [20].
NaNH2
NaNH2 СНз
2 Cl—CH2-CH2-n( СНз
CN
СНз chz-ch2-n<
СНз
16 1 11
H2SO4
СНз
СНз
Второй способ синтеза исходит из пиридина, который подвергают алкилированию 4-хлорбензилхлоридом с получением 2-(4-хлорбензил)-пиридина (16.1.13). Алкилируя последний 2-диметиламиноэтилхлоридом в присхтствии амида натрия, получают хлорфенирамин (16.1.12) 121].
-315-
Г лава 1 6
CH2-CI
NaNH2 ch3
2 CI—СН2-СН2-м(
СН3 -----------------» 16 112
Хлорфенирамин уменьшает аллергический ответ организма, вызванный гистамином. Препарат применяют при симптомах аллергии, ринитах, а также в составе многочисленных композиций с эфедрином и псевдоэфедрином, рекомендуемых применять при простудных заболеваниях, инфекциях верхних дыхательных путей, аллергических ринитах.
Синонимами препарата являются хлортриметон, гистаспан, трипо-лон, телдрин.
Дексхюрфенирамин (Dexchlorpheniramine)
Дексхлорфенирамин — D( Д-3-(яара-хлорфенил)-3-(2-пиридил)про-пилдиметиламин получают разделением рацемата, получаемого в результате синтеза хлорфенирамина (16.1.12) с помощью D-фе-нилянтарной кислоты [22, 23, 24].
Активность препарата примерно вдвое превышает таковую у хлорфенирамина.
Дексхлорфенирамин также применяют при симптомах аллергии, ринитах, дерматитах.
Синонимом препарата является поларамин.
Бро.чфенирамин (Brompheniramine)
Бромфенирамин — 3-(«ара-бромфенил)-3-(2-пиридил)пропилди-метиламин (16.1.14) является аналогом хлорфенирамина с той разницей, что атом хлора в бензольном кольце заменен на атом брома. Соответственно, его синтез осуществляют по аналогичной схеме [22, 23].
сн3
сн3
-316-
Антигистаминные препараты
Бромфенирамин также применяют при симптомах аллергии, ринитах, дерматитах Активность препарата примерно такая же, как и у хлорфенирамина.
Синонимами препарата являются диметан, бромбей, спентан. вел-тан и др.
Пиперазины
Цитизин (Cyclizine)
Циклизин — 1-(дифенилметил)-4-метилпиперазин (16.1.15) получают алкилированием 1-метилпиперазина бенгидрилбромидом [25, 26].
H-N N-CH3
Циклизин проявляет антигистаминное и антихолинергическое действие и применяется при рвоте и поносе. Точный механизм его действия не выяснен
Синонимами препарата являются марезин и мигрил.
Меклизин (Meclizine)
Меклизин — 1-[(4-хлорфенил)метил]-4-[(3-.метилфенил)фенил]-пиперазин (16.1.16) получают восстановительным аминированием смеси 3-метилбензальдегида с 1-(4-хлорбензгидрил)пиперазином, с использованием водорода над никелем Ренея [27, 28, 29].
-317-
Глава 16
Меклизин активно воздействует на рвотный центр и применяется при рвоте и поносе.
Синонимами препарата являются антиверт, боноин, ламин, рокли-зин. вертол.
Гидроксизин (Hydroxyzine)
Гидроксизин — 2-[2-[4-[(4-хлорфенил)фенилметил]-1-пиперази-нил]этокси]этанол (16.1.17) получают алкилированием 1-(4-хлор-бензгидрил)пиперазина 2-(2-гидроксиэтокси)этилхлоридом [30, 31, 32,33,34.35].
CI—сн2-сн2—о—сн2-сн2-он
Гидроксизин — антигистаминной препарат с М-холиноблоки-рующими свойствами и выраженным влиянием на ЦНС. Препарат подавляет подкорковые участки ЦНС, включая лимбическую систему и ретикулярную формацию. Потенцирует действие наркотических анальгетиков, проявляет седативный эффект. Применяют в качестве симптоматического средства при атопическом дерматозе, в качестве седативного средства до и после операционных вмешательств, для предупреждения рвоты и поноса, для купирования ажитации и эмоциональных расстройств.
Синонимами препарата являются атаракс, дурракс, вистарил.
Фенотиазины
Прометазин (Promethazine)
Прометазин — 10-(2-диметиламинопропил)фенотиазин (16.1.18) полз чают алкилированием фенотиазина 1-диметиламино-2-пропил-хлоридом [36. 37].
- 318-
Антигистаминные препараты
। н
СНз
Cl—ch-ch2-n( СНз СНз
NaNH2
I СНз
сн-сн2— СНз СНз
16 1 8
Являясь производным фенотиазина, прометазин по строению и фармакологическим свойствам напоминает хлорпромазин. Препарат проявляет сильною антигистаминную активность, а также выраженное действие на ЦНС. Потенцирует действие снотворных и анальгезирую-ших препаратов.
Прометазин применяют при лечении аллергических заболеваний: крапивницы, сывороточной болезни, сенной лихорадке, дерматозах, а также при ревматизме с выраженным аллергическим компонентом, при аллергических осложнениях, вызванных антибиотиками и другими лекарственными средствами, для усиления действия анальгетиков и местных анестетиков.
Синонимами препарата являются аллерган, фенэрган, пипольфен, протазин и др
Тримепразин (Trimeprazine)
Тримепразин — 10-(3-диметиламино-2-метилпропил)фенотиазин (16.1.19) получают алкилированием фенотиазина 1-диметиламино-2-метилпропилхлоридом [38J.
.S
N
I /Из
СН2-СН-СН2—N\
СНз снз
161.19
Тримепразин применяют при лечении зуда при дерматитах как аллергического, так и неаллергического происхождения.
Синонимом препарата является темарил.
- 319-
Глава 16
Пиперидины
Ципрогептадин (Cyproheptadine)
Ципрогептадин — 4-(дибензо[а,б]циклогептен-5-илиден)-1-метил-пиперидин (16,1.21) получают взаимодействием 1-метил-4-магний-хлорпиперидина с 5/7-дибензо[а.с1]циклогептен-5-оном с получением карбинола (16.1.20), дегидратация которого в кислой среде приводит к получению ципрогептадина (16.1.21) [39, 40].
MgCl
СНз
Ципрогептадин обладает антианафилактической активностью, связанной со способностью тормозить высвобождение гистамина и других медиаторов из тучных клеток Главным образом препарат применяют для лечения приступов бронхиальной астмы, аллергических бронхитов, ринитов и аллергических кожных реакций, а также для вспомогательной терапии при анафилактических реакциях.
Синонимами препарата являются периактин и вимикон.
Терфенадин (Terfenadine)
Терфенадин — а-(4-трет-бутилфенил)-4-(гидроксидифенилметил)-1-пиперидино-бутанол (16.1.24) предложено получать разными путями. Согласно одному из них, бензил-4-магнийхлорпиперидин вводят в реакцию с бензофеноном с получением (1-бензил-4-пи-перидил)дифенилкарбинола (16.2.22), который далее подвергают дебензилированию восстановлением водородом с использованием в качестве катализатора палладия на угле с получением (4-пипе-ридил)дифенилкарбинола (16.2.22). Последний алкилируют либо 1-(4-/нреш-бутилфенил)-4-хлорбутанолом с получением терфенади-на (16.1.24), либо алкилирование осуществляют (4-трет-оутплфе-нил)-3-хлорпропиофеноном с получением продукта (16.1.25), карбонильную группу которого восстанавливают до спиртовой, получая целевой терфенадин (16.1.24) [41, 42, 43. 44. 45. 46].
- 320 -
Антигистаминные препараты
сн2
СбН5
Н2 ' Pd-C
Терфенадин отличается от других антигистаминных препаратов не только химической структурой, но и тем, что его действие начинается через 1-2 ч и продолжается около 12 ч, достигая пика действия через 3 4 ч. Препарат показан для облегчения симптомов, связанных с сезонными аллергическими ринитами и конъюнктивитами, при ангионевротическом отеке, кожных аллергических реакциях, а также в составе комплексной терапии при бронхиальной астме.
Синонимами препарата являются селдан. гистадин, трексил и др.
Астемизол (Astemizole)
Астемизол — 1-[(4-фторфенил)метил]-М-[1-[2-(4-метоксифенил)-Э1ил]-4-пиперидинил]-бензимидазол-2-амин (16.1.31) получают многостадийным синтезом исходя из 1-карбэтокси-4-аминопи-перидина и 2-нитроизотиоцанобензола, при взаимодействии которых получается производное тиомочевины (16.1.26). Нитрогруппу последнего восстанавливают, и далее осуществляют S-метилирование продукта, который в условиях реакции подвергается внутримолекулярной циклизации в производное бензимидазола — N-[l-[2-(4-карбэгокси)]-4-пиперидинил]бензимидазол-2-амин (16.1.28). Последний алкилируют 4-фторбензилхлоридом в 1-[(4-фгорфенил)-
- 321 -
Глава 16
метил]-К'-[1-[2-(4-карбэтокси)]-4-пиперидинил]бензимидазол-2-амин (16.1,29). Карбэтоксильную группу полученного соединения (16.1 29) гидролизуют бромисто-водородной кислотой с получением незамещенного по атому азота производного пиперидина (16.1.30), алкилирование которого 2-(4-метоксифенил)этилметансульфонатом приводит к астемизолу (16.1.31) [47, 48].
CI—С-кцСгНзЬ
Н2/ Pd-C
СНЭ1 /СН3ОН
16 1 31
Астемизол применяют для профилактики и лечения сезонного острого и хронического аллер! ического ринита, аллергического конъюнктивита. крапивницы, отека Квинке, других аллергических состояний и дерматитов.
Синонимами препарата являются гисманал. гистазол др.
- 322 -
Анти гистаминные препараты
Дифенилпирал ин (Diphenylpyraline)
Дифенилпиралин — 4-дифенилметокси-1-метилпиперидин (16.1.32) получают алкилированием 4-гидрокси-1-метилпиперидина бензгид-рилбромидом [49. 501.
N-CH3
Дифенилпиралин — антигистаминный препарат с антихолинерги-ческим и седативным действием.
Препарат предназначен для симптоматического лечения сезонных аллергий и аллергических реакций, а также в качестве вспомогательного средства для терапии анафилактических реакций.
Синонимами препарата являются арбид, темпил, гистрил, гисприл и др.
16.2. Антагонисты Н^-рецепторов
Антагонисты Н2-рецепторов практически полностью блокируют секрецию соляной кислоты в желудке в ответ на большинство стимулов, Эги препараты играют огромную роль для лечения язвы желудка, связанной с гиперсекрецией, поскольку проявляют способность уменьшать как объем желудочного секрета, так и общую кислотность, и активность пепсина
Препараты этого ряда применяют для лечения язвы желудка и двенадцатиперстной кишки и при гиперсекреторных состояниях.
Традиционные, или Н|-антигистаминные, препараты блокируют многие эффекты, вызванные гистамином, однако они оказываются не в состоянии противостоять событиям, опосредованным Н2-рецепто-рами, в частности, избыточной секреции желудочного сока. В 1977 г. оыз предложен антагонист Н2-рецепторов — циметидин, революцини-зировавший лечение язвы желудка. Впоследствии был предложен ранитидин, а позже — уже препараты с минорными структурными И фармакологическими различиями — фамотидин и низатидин.
Антаншисты ЬЬ-рецепторов обратимо и конкурентно ингибируют действие гистамина на Н2-рецепторах. Они являются чистыми антаго
- 323-
♦Глава 16
нистами, поскольку не действуют на Hi-рецепторы, [3-адренорецепторы или мускариновые рецепторы. Более того, они не воздействуют в сколько-нибудь значительной мере на синтез, высвобождение и биотрансформацию гистамина.
Структура циметидина представляет собой метилимидазольное кольцо с серосодержащей боковой цепью с цианогуанидинной группой. Казалось, что наличие имидазольного кольца в циметидине, имеющееся и в структуре гистамина, должно являться определяющим в проявлении Н2-блокирующей активности, однако создание ранитидина, фамотидина и низатидина. содержащих фурановое и тиазольное кольца вместо имидазольного, показало неверность высказанного предположения.
Циметидин (Cimetidine)
Циметидин — 1-циано-2-метил-3-[2-[[5-метилимидазол-4-ил)метил]-тио]этил]-гуанидин (16.2.5) синтезируют по следующей схеме. Взаимодействием 2-хлорацетоуксусного эфира с двумя молями формамида получают 4-карбэтоксн-5-метилимидазол (16.2.1). Восстановлением карбэтоксильной группы последнего натрием в жидком аммиаке получают 4-гидроксиметил-5-метилимидазол (16.2.2), Гидрохлорид полученного спирта вводят во взаимодействие с гидрохлоридом 2-меркаптоэтиламина, с получением дигидрохлорида 4-(2-аминометил)-тиометил-5-метил-имидазола (16.2.3). Последний вводят во взаимодействие с М-цианимидо-8,8-диметилдитиокар-бонатом с получением производного тиомочевины (16.2.4), которое взаимодействием с метиламином трансформируют в циметидин (16.2.5) [51, 52, 53, 54, 55, 56, 57, 58].
СООС2Н5
N " ij Na ' NH3
CH2-S—CH2-Ch2-NH2 n
CH3—S
^=N-CN
CH3-S
-324-
А н т и гистаминные препараты
N-CN
CH2-S— CH2-CH2-NH-C-NH-CH3
Н
16 2 5
Циметидин является представителем первого поколения антигистаминных препаратов, блокирующих Н2-рецепторы. Основным фармакологическим эффектом циметидина является угнетение секреции желудочного сока, связанное с блокадой Н2.рецепторов стенок желудка. Он подавляет продукцию соляной кислоты, как базальную, так и стимулированную пищей, гистамином, гастрином, одновременно понижая активность пепсина.
Циметидин применяется при лечении язвенной болезни желудка и двенадцатиперстной кишки и других состояниях, сопровождающихся повышением кислотности и избыточной секрецией желудочного сока. Используют для профилактики поражений и кровотечений верхних отделов жкт.
Синонимами препарата являются гагамет, цинамет, беломет.
Ранитидин (Ranitidine)
Ранитидин — М[2-[[[5-димтиламино)метил]-2-фуранил]метил]тио]-этил]-М'-метил-2-нитро-1,1-этендиамин (16.2.8) синтезируют исходя из фурфурилового спирта, который повергают аминометилированию с использованием диметиламина и параформа с получением 5-(диметиламинометил)фурфурилового спирта (16.2.6). Далее взаимодействием с гидрохлоридом 2-меркаптоэтиламина получают продукт замещения гидроксильной группы — 5-диметиламино-мегил-2-(2‘-аминоэтил)тиометилфуран (16.2.7). Вводя последний во взаимодействие с К-метил-1-метилтио-2-нитроэтенамином, получают ранитидин (16.2.8) [59. 60, 61, 62, 63, 64].
|Г~—п П---П hS-CH2-CH2-NH->
|1 П + сн2о + (Ch3)2Nh ----► II П ... ;
T'Xhn-OH -CH2^XO'Xh2-Oh
16.2.6
- 325-
Глава 16
CH3S
)ch=ch-no2
CH3NH
(CH3)2N-СН2 О СН2—S—CH2“CH2~NH2
16.2.7
I] II ch-no2
(CH3)2N -СНг^О^СНг-З—ch2-ch2-nh-c-nhch3
16.2.8
Ранитидин -- препарат второго поколения блокаторов Н2-рецеп-торов. Так же как и циметидин, ранитидин подавляет, как базальную, так и стимулированную пищей, гистамином, гастрином и ацетилхолином продукцию соляной кислоты. Одновременно снижает активность пепсина и применяется при лечении язвенной болезни желудка и двенадцатиперстной кишки, а также других состояниях, сопровождающихся повышенной кислотностью ЖКТ.
Синонимами препарата являются зантак, азантак. раниплекс. рани-дил и др.
Фамотидин (Famotidine)
Фамотидин — 3-[[(2-((аминометил)амино-4-тиазолил)метил]тио]-М-(аминосульфонил)пропанимидамид (16.2.13) синтезируют исходя из 8-(2-аминотиазол-4-ил-метил)изотиомечевины (16.2.9), которую получают взаимодействием 1,3-дихлорацетона с двумя молекулами тиомочевины, при котором одновременно с образованием ^азольного кольца происходит замещение атома хлора в промежуточно образующемся 2-амино-5-хлорметилтиазоле. Взаимодействием последнего с 2-хлорпропионитрилом получают 8-(2-аминотиазол-4-ил-метил)-2-цианоэтан (16.2.10), который, в свою очередь, вводят в реакцию с бензоилизтиоцианатом. Полученное в результате этой реакции производное бензоилтиомочевины (16.2.11) подвергают сначала S-метилированию метилйодидом и далее расщеплению аммиаком до 3-[[(2-((аминометил)амино-4-тиазолил)-метил1тио]этил-цианиида (16.2.12). Последовательным метанолизом нитрильной группы и последующим взаимодействием полученного иминоэфира с сульфонамидом получают фамотидин (16.2.13) [65, 66, 67. 68. 69, 70]'.
- 326 -
А н т и гистаминные препараты
ci—ch2-ch2-cn
16 2 10
1 K2CO3
2 CH3I
3 NH3
1 СИ3ОН / HCI
HsN I 2 tNH2i2Sa^ Jl~D n-so2-nh2
N CH2-S—ch2-ch2-cn ^=N'^fN'^'CH2-S —ch2-CH2-C-NH2
H2o h2n
'62'2 16213
Так же как и ранитидин, фамотидин относится к препаратам второго поколения блокаторов Н2-рецепторов и так же, как и два других вышеописанных препарата, применяется при лечении язвенной болезни желудка и двенадцатиперстной кишки и других состояниях, сопровождающихся повышенной кислотностью ЖКТ.
Синонимами препарата являются фамодил, гастридин, пепиид и др.
Низатидин (Nizatidine)
Низатидин — К-[2-[[[2-[(диметиламино)метил]4-тиазолил]метил]-гио]-этил]-2-нитро-1,1-этендиамин (16.2.15 ). По своему химическому строению низатидин является некоторым гибридом структур ранитидина и фамотидина, где использованы боковая цепь ранитидина и несущий гетероцикл — 2-аминотиазол. Соответственно, синтез последнего также представляет собой определенную комбинацию подходов, примененных при получении обоих препаратов прото типов. Исходным соединением служит 2-(диметиламино-метил)-4-гидроксиметилтиазол, из которого последовательным
-327-
Глава 1 6
взаимодействием с гидрохлоридом 2-меркаптоэтиламина и далее с М-ме1ил-1-метилтио-2-нптроэтенамином получают искомый низа-гидин (16.2.15) [71, 75].
1 HS-CH--сн2-nh2
2 С Н 3N Н
'CH=O-N0-
I~X i?"'10-
ICH3)2NCH2 CH2-S— CH2-C42-Nri-C-NHCH3
Так же как и другие вышеописанные препараты, низатидин применяется при лечении язвенной болезни желудка, двенадцатиперстной кишки и других состояниях, сопровождающихся повышенной кислотностью ЖКТ.
Синонимами препарата являются аксид и пульвулес.
Список литературы
1. US Pat. 2.397.799 (1946).
2. US Pat. 2.421.714(1947).
3. US Pat. 2.427.878 (1947).
4. US Pat. 2.499.058 (1950).
5. US Pat. 2.534.813 (1950).
6. Brit. Pat. 942.152 (1963).
7. Fr. Par. M1313 (1961).
8. Ebnother A. et al.'/Helv. Chim. Acta. 59, 2462 (1976).
9. US Pat. 2.4067594 (1946).
10. US Pat. 2.502.151 (1950).
11. Huttrer C. et al./-J. Am. Chem. Soc. 68. 1999 (1946).
12. Bovet et al.' C. R. Soc. Biol. 138, 99 (1944).
13. US Pat. 2.569.314 (1951).
14. US Pat. 2.607.778 (1952).
15. Swiss. Pai. 264,754 (1950).
16. Swiss. Pat. 266,234 (1950).
17. Swiss. Pat. 266,235 (1950).
18. Brit. Pat. 651.596(1951).
19. Vaughan J. et al./4. Org. Chem. 14, 228 (1949).
20. US Pat 2.567.245 (1951).
-328 -
А н т и г и стаминные препараты
21. US Pat 2.676.964 (1954).
22. US Pat. 3.061.517 (1962).
23. US Pat. 3.030.371 (1962).
24. Brit. Pat. 834.984 (1958).
25. US Pat. 2.630.435 (1953).
26. Baltzly R et al.//J. Org. Chem. 14, 775 (1949).
27. US Pat. 2.709.169(1955).
28. Bnt. Pat. 705.979 (1954).
29. Belg. Pat. 502.889 (1951).
30. US Pat. 2.899.436(1959).
31. Ger. Pat. 1.049.383 (1953).
32 Ger. Pat. 1.061.786(1953).
33. Ger. Pat. 1.068.262 (1953).
34. Ger. Pat. 1.072.624 (1953).
35. Ger. Pat. 1.075.116(1953).
36. US Pat. 2.530.451 (1950).
37. US Pat. 2.607.773 (1952).
38. US Pat. 2.837.518 (1958).
39. US Pat. 3.014.911 (1961).
40. Engelhardt E. et al.., J. Med. Chem. 8, 829 (1965).
41. US Pat. 3.878.217 (1975).
42. Ger. Pat. 2.303.305 (1973).
43. Ger. Pat. 2.303.306(1973).
44. Ger. Pat. 2.503.362 (1973).
45. Brit. Pat. 1.412.605 (1972).
46. Carr A et al.//Arzneimittel-Forsch. 32, 1157 (1982).
47. US Pat. 4.219.559(1980).
48. Eur. Pat. Appl. 5318 (1979).
49. US Pat. 2.479.843 (1949).
50. Ger. Pat. 934.890(1951).
51. US Pat. 3.894.151 (1975).
52. US Pat. 3.950.333 (1976).
53. US Pat. 4.000.302(1976).
54. Ger. Pat. 2.320.131 (1973).
55. Ger. Pat. 2.344.779(1973).
56. Belg. Pat. 804.144 (1974).
57. Kairisalo P. et al.,. Arch. Pharm. (Wienheim). 316. 688 (1983).
58, Brimblecombe R. et al.'/J. Int. Med. Res. 3, 86 (1975).
- 329-
Глава 16
59. Ger. Pat. 2.734.070 (1977).
60. US Pat. 4.128.658 (1978).
61. Fr. Pat. 2.384.765 (1978).
62. US Pat. 4.399.293 (1978).
63. US Pat. 4.399.294(1978).
64. Belg. Pat. 888.747 (1980).
65. US Pat. 4.283.408 (1981).
66. Ger. Pat. 2.951.675 (1979).
67. Ger. Pat. 3.008.056(1980).
68. Brit. Pat. 2.052.478 (1980).
69. Brit. Pat. 2.055.800 (1979).
70. Belg. Pat. 882.071 (1980).
71. US Pat. 4.375.547 (1983).
72. Eur. Pat. Appl. 49618 (1982).
f Гл а в a 17
I Кардиотонические
I препараты
Препараты, увеличивающие силу сокращений миокарда и тем самым улучшающие его работоспособность и эффективность, называются кардиотоническими средствами. Это определение традиционно и весьма долгое время относилось к группе препаратов, называемых сердечными гликозидами, однако в последнее время клинике предложены новые кардиотонические препараты (негликозидные кардиотонические средства, являющиеся ингибиторами фосфодиэстеразы — амринон и мил-ринон), проявляющие сходные свойства. Кардиотонические средства иногда называют положительными инотропными препаратами, т. е. веществами, усиливающими силу мышечных сокращений, в данном случае — усиливающими силу сокращений миокарда.
Кардиотонические препараты предназначены для лечения сердечной недостаточности.
Сердечная недостаточность — весьма распространенное заболевание Оно может быть определено, как неспособность сердца прокачать достаточное количество крови для снабжения кислородом и питательными веществами органы и ткани, что ведет к появлению усталости, одышки, отеков. Причиной возникновения сердечной недостаточности чаще всего являются артериальная гипертензия и ишемическая болезнь сердца. Она может проявляться как в острой форме, в виде резкого снижения сердечного выброса с симптомами нарушения кровообращения, так и в хронической форме, выражающейся болями в сердце.
В дополнение к инотропным средствам при лечении сердечной недостаточности применяются также диуретики (гл. 21 «Диуретики»), которые увеличивают выведение ионов Na’ и воды из организма, снижают объем циркулирующей крови, понижают нагрузку на сердце и устраняют отеки; сосудорасширяющие средства (вазодилататоры), которые способствуют уменьшению венозного и артериального давления. что снижает сосудистый тонус и. соответственно, нагрузку на сердце и уменьшает его потребность в кислороде. Иногда применяются и адреномиметики — эпинефрин (адреналин), норэпинефрин (норадре
- 331 -
Глава 17
налин), изопротеренол, тербуталин, альбутерол, обладающие способностью увеличивать силу сердечных сокращений и стимулирующие повышение сердечного выброса. Однако они одновременно повышают скорость сердечного ритма, обладают аритмогенным действием, а также увеличивают потребность миокарда в кислороде, что нежелательно и может привести к усилению ишемии. Иногда применяют также дофамин и леводопу. Последний в организме, несомненно, превращается в дофамин, являясь, таким образом, оральной формой дофамина в виде пролекарства.
Все указанные симпатомиметики ограниченно используются для кратковременной (24 ч и менее) поддержки сердца.
Оптимальная терапия сердечной недостаточности часто требует одновременного применения двух или более из перечисленных выше гру пп препаратов.
17.1. Сердечные гликозиды
Для лечения сердечной недостаточности широко применяются гликозиды, выделяемые из листьев различных видов наперстянки Digitalis lanta (наперстянка шерстистая), Digitalis purpurea (наперстянка пурпуровая) и строфанта Strophantus КотЬе (строфанта Комбе), а также ряда других растений (ландыш, обвойник, олеандр, морозник, желтушник, джут, харг и др.), проявляющие прямое воздействие на миокард и усиливающие силу его сокращений.
Основным свойством сердечных гликозидов является их избирательное действие на сердце, причем главным эффектом следует считать усиление систолы, что создает наиболее экономный режим для работы сердца: сильные систолические сокращения сменяются периодами отдыха (диастолы), способствующими восстановлению энергетических ресурсов миокарда.
Общий кардиодинамический эффект сердечных гликозидов довольно сложен, поскольку7 является комбинацией их прямого действия на миокард и непрямого действия, изменяющего электрофизиологические свойства сердца (автоматизм, проводимость и возбудимость).
Есть основания предполагать, что сердечные гликозиды, равно как и другие инотропные вещества, действуют на сокращаемость сердца путем воздействия на процесс переноса ионов Са" через мембрану миокардиоцитов. Воздействие на электрическую проводимость клеточных мембран опосредовано изменениями в транспорте ионов Na’, FC и Са“', что является результатом их непрямого ингибирующего действия на Na’-, К*-АТФазу клеточных мембран.
- 332 -
Кардиотонические препараты
Сердечные гликозиды применяются для лечения острой хронической сердечной недостаточности, при определенных видах сердечных аритмий, кардиогенном шоке. Они рассматриваются в качестве единой группы, поскольку имеют сходные фармакологические характеристики. Они могут иметь один и тот же агликон, но остатки различных сахаров, и наоборот. При выборе препарата этого ряда следует учитывать не только его активность,х но и быстроту наступления эффекта, что сильно зависит от физико-химических свойств этого ряда препаратов, которые принято подразделять на полярные и неполярные. К полярным (гидрофильным) гликозидам обычно относят строфантин, который применяют внутривенно и эффект которого наблюдается уже через 5—10 мин после введения. К неполярным гликозидам относят дигитоксин и дигоксин, которые применяют в основном перорально или ректатьно и эффект которых наблюдается через 2—4 ч после приема.
Выбор препарата и способ его введения зависят от показаний. При острой сердечно-сосудистой недостаточности и внезапной декомпенсации прибегают к внутривенному введению строфантина или корглюко-на. При хронической сердечной недостаточности перорально применяют дигитосин или дигоксин.
То, что обычно экстрагируется из обоих видов Digitalis, представляет собой уже частично гидролизованные гликозиды.
Природными гликозидами, содержащимися в Digitalis purpurea, являются пурпуреагликозид А и пурпуреагликозид В, которые под действием ферментов расщепляются на дигитоксин и глюкозу или, соответственно. гитоксин и глюкозу, а при действии кислот полностью гидролизуются до агликонов дигитоксигенина и, соответственно, гито-ксигенина, а также дезоксисахаров дигитозы и глюкозы.
В Digitalis lanta содержатся гликозиды — лантозиды А. В и С. Лантозид А при щелочном гидролизе отщепляет ацетильную группу и превращается в пурпуреагликозид А. лантозид В — в пурпуреагликозид В. Структура двух лантозидов — лантозида В и лантозида С — отличается лишь гем, что в лантозиде В имеется гидроксильная группа при С)о его генина (дитоксигенин), в то время как в генине лантозида С (дигоксигенин) имеется дополнительная гидроксильная группа и при С|2 генина.
Продукт, который обычно экстрагируется из обоих видов Digitalis, предщавляет собой уже частично гидролизованные гликозиды — дигитоксин и дигоксин. Дигоксин выделяют только из Digitalis lanta.
Смесь сердечных гликозидов, выделяемых из семян Strophantus Kombe. содержит в основном К-строфантин-Р и К-строфантозид.
- 333-
Глава 1 7
К-строфантин-0 состоит из агликона сгрофантидина и сахарного остатка, состоящего из цимарозо-р-П-глюкозы. К-строфантозид имеет сахарный остаток из трех единиц — цимарозо-р-В-глгокозо-а-П-глюкозы. Использование термина «строфантин» в общем относится ко всем гликозидам этого ряда.
Углеводная часть различных сердечных гликозидов может быть моно-, ди- , три- и тетрасахаридом, а агликон (генин) — стероидом с определенными структурными особенностями. Главная роль сахарных остатков, очевидно, заключается в способствовании растворению гени-нов.
Для агликонов сердечных гликозидов характерны следующие структурные особенности: сочленение колец А и В — цис, колец В и С — транс, колец С и D — цис, в положении 17Р находится бутенолидный фрагмент. Большинство соединений отосится к 5р ряду. За одним исключением гидроксильная группа в положении Сз также имеет р-кон-фигурацию. Физиологически активные соединения обязательно содержат гидроксильную группу в положении 14(3. Группы ОН и СО в положениях 11, 12, 16 и 19, по-видимому, оказывают меньшее влияние на активность.
Главная роль сахарных остатков, которыми этерифицирована гидроксильная группа при Сз, очевидно заключается в способствовании растворению генинов.
Простейшие сердечные генины — дигитоксигенин, гитоксигенин и строфангидин являются агликонами важнейших гликозидов Digitalis lanta. Digitalis purpurea и Strophantus Kombe.
дигитоксигенин
нс
гитоксигенин строфантидин
Дигитоксин (Digitoxin)
Дигитоксин — 3р,14р-дигидрокси-5р-кард-20(22)енолид-3-триди-гитоксид (17.1.1) является гликозидом, выделяемым из листьев различных видов наперстянки. Из 10 кг листьев выделяется около 6 г дигитоксина [1, 2, 3, 4, 5, 6, 7. 8, 9].
- 334 -
Кардиотонические препараты
Дигитоксин применяют при хронической сердечной недостаточности, тахиаритмической форме мерцания предсердий, пароксизмальной мерцательной аритмии, пароксизмальной суправентрикулярной тахикардии.
Синонимами препарата являются кардигин, кордален, кристодигин, пуродпгини др.
Дигоксин (Digoxin)
Дигоксин — Зр,140-дигидрокси-5р-кард-2О(22)-енолид-3-ригиток-сид (17.1.2) также является гликозидом, выделяемым из различных видов наперстянки. От дигитоксина отличается тем, что в положении С|й стероидного скелета содержит дополнительную гидроксильную группу. Продукт экстрагируют из листьев Digitalis lanta, Digitalis onentalis или Scrophulariaceae [10, 11, 12. 13, 14, 15, 16].
он
-335-
Глава 17
Дигоксин оказывает сильное систолическое действие и замедляет сердечный ритм. По сравнению с дигитоксином, быстрее выводится из организма. Применяют при хронической сердечной недостаточности при декомпенсированных клапанных пороках сердца, перегрузке миокарда при артериальной гипертонии, тахикардии желудочковой фибрилляции и других аналогичных состояниях.
Синонимами препарата являются цедоксин, ланакордин, ланоксин и др.
Строфантин (Strophantinum)
Строфантин состоит из смеси гликозидов и, в основном, К-стро-фантина-Р — Зр,5,14-тригидрОкси-19-оксо-5р-кард-20(22)-енолид-З-П-цимаро-Р-П-гликозида (17.1.3) и К-строфантозида — 30,5,14-тригидрокси-19-оксо-5р-кард-20(22)-енолид-3-В-цимаро-р-Е)-глюко-a-D-гликозида (17.1.4). Препарат, в основном, получают выделением из семян Strophantus КотЬе [17, 18].
Строфантин применяют при острой сердечно-сосудистой недостаточности и, в частности, после инфаркта миокарда, при хронической сердечной недостаточности, сердечной декомпенсации, суправентрикулярной тахикардии и мерцательной аритмии.
Синонимами препарата являются комбетин, строфопан и др.
17.2. Другие положительные инотропные препараты
Нет никакого сомнения в том, что вышеприведенные гликозиды являются наиболее удовлетворительными инотропным соединениями. Однако по ряду причин некоторым пациентам препараты дигиталиса могут быть противопоказаны. В качестве инотропного агента интенсивно изучался теофиллин, однако он оказался непригодным для долговременного применения.
-336 -
Кардиотонические препараты
В последние годы в качестве иноторпных средств нашли применение производные ряда бипиридинов — амринон и минринон.
Механизм действия этих препаратов не совсем понятен. Однако весьма вероятно, что они ингибируют клеточную фосфодиэстеразу миокарда, что приводит к повышению клеточного уровня цАМФ, который в свою очередь способствует сокращениям клеток миокарда. Ясно, что эти препараты не являются агонистами p-адренорецепторов и их эффект не опосредован ингибированием Na+-, КГ-АТФазы. Одновременно они увеличивают поток ионов Са2’ в клетки. Их применяют для кратковременного контроля за пациентами, неадекватно реагирующими на сердечные гликозиды, диуретики и коронарорасширяющие средства.
Амринон (Amrinone)
Амринон — 3-амино-5-(4-пиридинил)-2(1Я)-пиридинон (17.2.4) можно синтезировать исходя из пиридин-4-уксусной кислоты, взаимодействием которой с комплексом диметилформамид — хлорокись фосфора получают 2-(4-пиридил)-3-диметил-аминоакролеин (17 2.1). Взаимодействием последнего с амидом цианукеусной кислоты получают 3-циано-5-(4-пиридил)-2(1Я)-пиридинон (17.2.2). Гидролизом цианогруппы последнего получают 3-карбамил-5-(4-пиридил)-2(1Я)-пиридинон (17.2.3). Вводя последний в перегруппировку Гофмана (с применением брома в гидроокиси натрия), получают амринон (17.2.4).
Альтернативным путем синтеза, также исходящим из 3-циано-5-(4-пиридцл)-2(1Я)-пиридинона (17.2.2), является его кислотный гидролиз до соответствующей кислоты — 3-карбокси-5-(4-пиридил)-2(1Я)-пиридинона (17.2.5), нитрование которого азотной кислотой в присутствии серной кислоты приводит к 3-нитро-5-(4-пиридил)-2(1Я)-пириди-
- 337-
Глава 17
нону (17.2.6). Восстановлением нитрогруппы последнего водородом получают искомый амринон (17.2.4) [19. 20].
3-циано-5-(4-пиридил)-2(1Л/)-пиридинон (17.2.2) предложено получать также конденсацией 2-(4-пиридил)малонового диальдегида с амидом циануксусной кислоты [21].
Амринон является производным бипиридина и занял весьма полезную нишу в арсенале инотропных средств дчя кратковременного применения в тех случаях, когда организм неадекватно отвечает на гликозидные препараты и коронарорасширяющие средства. Препарат уникален тем, что вызывает расширение сосудов и не вызывает аритмии или признаков миокардиальной аритмии.
Амринон применяют при кратковременном лечении сердечной недостаточности, не подвергающейся лечению другими препаратами.
Синонимами препарата являются инокор и винокарм.
Милринон (Milrinone)
Милринон — 1,6-дигидро-2-метил-6-оксо-(3,4’-бипиридин)-5-кар-бонитрил (17.2.7) является метильным аналогом промежуточного продукта синтеза амринона (17.2.2).
- 338 -
Кардиотонические препараты
CN
Милринон обладает всеми свойствами амринона, лучше переносится при оральном приеме и является более сильным препаратом, однако по ряду причин не рекомендован к применению в США.
Синонимом препарата является примакор.
Список литературы
1. Cloetta Н //Arch. Exp. Pathol. Pharmacol. 112. 261 (1926).
2. Windaus A. et al.//Ber. 58. 2503 (1925).
3. US Pat. 2.449.673 (1948)
4. US Pat. 2.557.916 (1951).
5. US Pat. 2.615.884 (1952).
6. Ger. Pat. 646.930(1933).
7. Hungarian Pat. 155.252 (1968).
8. Hungarian Pat. 156.753 (1968).
9. Ger. Pat. 2.006.926(1969).
10. Smith S '-'J. Chem. Soc. 1930. 508.
11. Brit. Pat. 337.091 (1929).
12. Indian Pat. 62.497 (1958).
13. Hungarian Pat. 149.778 (1959).
14. Hungarian Pat. 151.897 (1964).
15. Hungarian Pat, 156.753 (1968).
16. Ger. Pat. 2.225.039 (1972).
П. Ger. Pat. 721.001 (1937).
18. Ger. Pat. 737.540 (1937).
19. US Pat. 4.004.012 (1977).
20. US Pat. 4.072.746(1978).
21. Brit. Pat. 2.070.008 (1981).
| Гл а в a 18
। Антиаритмические препараты
Препараты, применяющиеся для предотвращения и лечения нарушений нормальной скорости и ритма сокращений сердца, называются антиаритмическими препаратами.
Аритмии возникают из-за нарушений в образовании электрических импульсов и их проводимости в сердце или обеих причин одновременно.
Ритм сердца регулируется ацетилхолином и норэпинефрином (норадреналином).
В норме ритм сердечной деятельности зависит от активности пейс-мейкерных клеток (водителей ритма) синоатриального узла. При нарушении их функции нарушается сердечный ритм, что клинически проявляется разной симптоматикой. Аритмия может быть связана и с появлением эктопических очагов, генерирующих импульсы с большей частотой, чем нормальные водители ритма.
Ритм сокращений сердца зависит от очень многих параметров: состояния водителей ритма и проводящей системы, кровоснабжения миокарда и других факторов, и, следовательно, аритмии могут возникнуть в силу различных причин вызванных нарушениями генерирования электрических импульсов или их проведения. Они могут быть вызваны пороками сердца, ишемией миокарда, электролитными и кислотноосновными изменениями, нарушениями иннервации сердца, интоксикацией организма и т. п.
Препараты, применяющиеся для лечения аритмий, могут воздействовать на проводящие системы сердца, его возбудимость, автоматизм, величину эффективного рефрактерного периода, адренергическую и холинергическую иннервацию сердца. Соответственно, нарушенный ритм сердечных сокращений могут восстанавливать соединения самых различных химических классов.
Как уже было отмечено, аритмии возникают из-за нарушений в образовании электрических импульсов и их проводимости в сердце, осу
- 340 -
д н т и а ритмические препараты
ществляемой посредством переноса ионов \а, К+, Са2’ через клеточные мембраны, или обеих причин одновременно. Поэтому механизм действия многих антиаритмических препаратов заключается в блокировании натриевых или кальциевых ионных каналов миокарда, что пролонгирует время, необходимое этим каналам для восстановления после активации, и что в свою очереХь действует на проводящие системы сердца, его возбудимость, автоматизм и т. п.
Основываясь на тонком понимании механизма возникшей тахикардии. требующей хорошего знания электрофизиологии сердца, и знании воздействия каждой группы лекарств на данный механизм, в большинстве случаев можно достаточно точно подобрать конкретный препарат для конкретного пациента.
Классификация антиаритмических препаратов может быть основана на разных принципах (например, по месту действия препарата). Это могут быть вещества, действующие непосредственно на миокард и проводящую систему самого сердца, или вещества, влияющие на эфферентную иннервацию сердца. Их можно рассматривать как группы препаратов, эффективных при наджелудочковых аритмиях или эффективных при желудочковых аритмиях.
Однако более или менее общепринятой на сегодняшний день классификацией препаратов, применяемых для лечения тахиаритмий, является классификация, основанная на особенностях их воздействия на электрофизиологические или биохимические процессы в миокарде.
В соответствии с этим антиаритмические препараты принято подразделять на 4 основные группы. К первой группе относят препараты, блокирующие натриевые каналы миокарда (хинидин, прокаинмид, ди-зопирамид, лидокаин, токаинид, фенитоин, мексилетин, флекаинид, энкаинид). Препараты, блокирующие действие на сердце эндогенных катехоламинов, которые имеют определенное значение в патогенезе аритмий, относят ко второй группе антиаритмических препаратов (пропранолол). К третьей группе относят препараты с не совсем ясным механизмом действия, которые одновременно оказывают и адреноблокирующее действие и. в определенной степени, влияют на трансмембранный перенос ионов, возбудимость и проводимость и, в итоге, пролонгируют потенциал действия в предсердно-желудочковом пучке и волокнах Пуркинье (амиодарон, бретилий). Наконец, четвертая группа антиаритмических препаратов представлена антиангннальными средствами — блокаторами кальциевых каналов (верапамил).
В свою очередь, основываясь на специфических особенностях влияния различных веществ в пределах одной группы (в частности, первой группы), их подразделяют на подгруппы. Некоторые исследователи
- 341 -
Г лава 18
придерживаются системы разделения антиаритмических препаратов на пять групп без деления на подгруппы.
Препараты группы I
За редким исключением, все препараты, относящиеся к данной группе, являются местными анестетиками, образующими комплексы с липопротеинами мембран клеток миокарда и тем самым блокирующими проводимость натриевых каналов клеточной мембраны для потока ионов Na+ внутрь и способствующими выводу ионов К из клеток миокарда, что, в итоге, приводит к слабому подавлению деполяризации клеток миокарда, сокращению времени их реполяризации и уменьшению скорости проведения возбуждения. Этот ряд препаратов пролонгирует потенциал действия и увеличивает эффективный рефрактерный период миокарда. В миокарде, главным образом в желудочках, подавляется автоматизм эктопических очагов.
Подгруппа IA
Препараты этой подгруппы замедляют скорость проведения возбуждения, уменьшают возбудимость волокон Пуркинье, подавляют автоматизм эктопических участков и увеличивают эффективный рефрактерный период. Они проявляют прямое и опосредованное антихо-линергическое действие. К этой подгруппе относятся антиаритмиче-ские препараты хинидин, прокаинамид, дизопирамид. Препараты этой подгруппы применяются при лечении нарушений синусового ритма, пароксизмальной, суправентрикулярной и вентрикулярной аритмии, профилактики артериальной фибрилляции и преждевременных сокращений сердца.
Хинидин (Quinidin)
Хинидин — (5-винил-2-хинуклидинил)-(6-метокси-4-хинолил)-метанол (18.1 1) является декстроизомером алкалоида хинина и является одним из четырех наиболее важных алкалоидов, выделяемых из коры хинного дерева [1, 2, 3]. Хинидин представляет собой вторичный спирт, радикалами в котором являются 5-мето-ксихинолиновое кольцо и 3-винилхинуклидин. Хинидин отличается о г хинина только конфигурацией углеродного атома карбонильной группы, и его предложно было получать изомеризацией хинина (37.1.1.47) [4]. Предложены также способы синтеза хинина [5, 6. 7].
- 342 -
А и т и а р и тмические препараты
37 1 1 47
18 1 1
Хинидин проявляет все фармакологические свойства хинина, включая аинтималярийное, жаропонижающее и др. Хинидин назначают при разных видах ариший для предотвращения тахикардии и фибрилляции предсердия и. в частности, для профилактики мерцательной аритмии, пароксизмальной суправентрикулярной тахикардии, экстрасистолии и желудочковой тахикардии. Однако он является токсичным препаратом. и его применяют относительно редко.
Препарат выпускается также под названиями кардиохин, дурахин, хинидекс и др.
Прокаинамид (Procainamide)
Прокаинамид — 4-амино-Х-[2-(диэтиламино)этил]бензамид (18.1.3) получают взаимодействием хлорангидрида 4-нитробензойной кислоты с К,Х'-диетилэгилендиамином с дальнейшим восстановлени-ем нитрогруппы в полученном 4-нитро-Ы-[2-(диэтиламино)этил]-бензамиде (18.1.2) в аминогруппу [8, 9].
С2н5
H2N -CH2-CH2-n(
Сгн5
С2Н5
сгН5
h2n
18 1 3
Химическое отличие прокаинамида от прокаина заключается в замене эфирной группы па амидную. Действие прокаинамида качественно сходно с действием прокаина. Его влияние на работу сердца идентично влиянию хинидина. Прокаинамид предпочтительнее прокаина в качестве антиаритмика, поскольку, в отличие от прокаина, он лучше адсорбируется при оральном применении и поскольку труднее гидролизуется ютеразами плазмы, имеет большую продолжительность действия.
-343-
Глава 18
Прокаинамид предназначен для лечения пароксизмальной предсердной тахикардии, предсердной фибрилляции, преждевременных желудочковых сокращений и желудочковой тахикардии. Для быстрого достижения терапевтических концентраций парентеральное введение прокаинамида предпочтительнее хинидина.
Синонимами препарата являются амидопрокаин, кардиоритмин, но-вокаинамид, пронестил и др.
Дизопирамид (Disopyramid)
Дизопирамид — а-(2-диизопропиламиноэтил)-а-фенил-2-пиридин-ацетамид (18.1.6) получают арилированием бензилцианида 2-хлор-пиридином в присутствии амида нагрия с последующим алкилированием полученного а-фенил-а-(2-пиридил)ацетонитрила (18.1.4) 2-ди-мзо-пропиламиноэтилхлоридом с использование амида натрия. Сернокислый гидролиз полученного нитрила (18.1.5) приводит к полу чению а-(2-диизопропила.миноэтил)-а-фенил-2-пиридинаце-тамида — диизопирамида [10, 11, 12].
NaNHs
СН(СН3)2
Ci—ch2-ch2-n(
СН(СНз)2
СН(СНз)2
-ch2-n(
СН(СН3)2
18 1 5
Н2ЗО4
Дизопирамид структурно не относится ни к одному из известных классу антиаритмиков, однако являясь препаратом группы блокаторов натриевых каналов IA класса, оказывает мембраностабилизируюшее действие, увеличивает эффективный рефрактерный период и продолжительность потенциала действия в предсердиях и желудочках. Он вызывает снижение сократимости и возбудимости миокарда, замедление проводимости, подавление автоматизма синусового узла. Дизопирамид показан для предотвращения и восстановления наджелудочковой и же
-344 -
днтиаритмические препараты
лудочковой экстрасистолии и тахикардии с целью предотвращения мерцания и трепетания предсердий, профилактики аритмий.
Препарат выпускается также под названиями дикорантил, димодан, напамид, норпас. ритмилен, ритмодан и др.
Подгруппа IB
Препараты подгруппы IB увеличивают электрический порог возбудимости желудочков во время диастолы, подавляют автоматизм и диастолическую деполяризацию, уменьшают продолжительность рефрактерного периода и отличаются от препаратов подгруппы IA тем, что если первые блокируют открытые натриевые каналы, то препараты подгруппы IB, в основном, блокируют неактивные натриевые каналы. Это означает следующее: так как они быстро элиминируют с нормальных, открытых натриевых каналов, то на здоровые участки миокарда оказывают слабое влияние. В зоне ишемии миокарда гипоксия вызывает деполяризацию клеточных мембран, и возникают аритмогенные очаги. При этом многие натриевые каналы инактивируются и становятся чувствительными к препаратам этого класса, которые улучшают проводимость и уменьшают время реполяризации этих клеток сердца. Препараты подгруппы IB мало воздействуют на мышцу предсердий, атриовентрикулярную проводимость, сокращаемость миокарда, сердечный выброс и систолическое артериальное давление. Препараты этой подгруппы — лидокаин, токаинид, мексилетин являются местными анестетиками, однако применяются при острых вентрикулярных аритмиях, таких, которые могут возникнуть при инфаркте миокарда, хирургических вмешательствах и катетеризации сердца, интоксикациях сердечными гликозидами. Фенитоин, не относящийся к ряду местных анестетиков и являющийся противосудорожным средством, ограниченно применяется только в качестве орального средства, заменяющего лидокаин при пароксизмальной тахикардии, вызванной интоксикацией.
Лидокаин (Lidocain)
Лидокаин является 2-диэтиламино-21,61-диметилацетанилидом (2.2.3).
Синтез лидокаина описан в гл. 2 «Местные анестетики».
сн3
у ° ZC2H5
О— NH— C-CH2~N\
Сгн5
СН3 223
-345-
Глава 18
Лидокаин является прототипом антиаритмических препаратов подгруппы IB и наиболее широко применяется для лечения и предотвращения вентрикулярной эктопической активности при инфаркте миокарда.
Аналогично прокаинамиду, лидокаин является амидом с местноанестезирующим действием. Обычно лидокаин вводится внутривенно для кратковременной терапии желудочковой экстрасистолии, тахикардии, особенно в острой фазе инфаркта миокарда, профилактики фибрилляции желудочков при остром инфаркте миокарда, аритмии по жизненным показаниям и при аритмиях, которые могут возникать при хирургических манипуляциях на сердце.
Синонимами препарата являются лидопен, ксилокаин, ксилокард и др.
Токаинид (Tocainid)
Токаинид — 2-амино-2',б'-диметилпропионанилид (18.1.8) получают взаимодействием 2,6-диметиланилина с бромангидридом 2-бромпропионовой кислоты и последующим замещением атома брома в полученном амиде (18.1.7) на аминогруппу [13, 14, 15, 16].
Вг-С-СН-СНз Вг
Токаинид применяют для подавления симптомов вентрикулярной аритмии и тахикардии, при преждевременных сердечных сокращениях.
Синонимом препарата является тонокард.
Мексилетин (Mexiletin)
Мексилетин — 1-метил-2-(21,2|-диметилфенокси)этиламин (18.1.11). Синтез мексилетина осуществляют взаимодействием натриевой соли 2,6-диметилфенола с хлорацетоном с получением 1-(2,6-диме-тилфенокси)-2-пропанона (18.1.9). Взаимодействием последнего с гидроксиламином получают соответствующий оксим (18.1.10). Восстановлением оксиминогруппы в последнем водородом над никелем Ренея получают мексилетин (18.1.11) [17, 18, 19, 20].
о
01—сн2-с-сн3
СНз 18 19
nh2oh
- 346 -
Антиаритмические препараты
СНз ?н
о-сн2—С-СНз
Н2 / Raney'Ni
СНз 18.1 10
СНз 18 1 11
Мексилетин показан при желудочковой экстрасистолии и желудочковой тахикардии, фибрилляции желудочков (в том числе в остром периоде инфаркта миокарда).
Синонимом препарата является мекситил.
Фенитоин (Phenytoin)
Синтез противосудорожного препарата фенитоина (9.1.1) описан в гл. 9 «Противоэпилептические средства».
н
9 1 1
Основные эффекты фенитоина на сердце сходны с таковыми лидокаина. По существу, его применение ограничено, и он, в основном, применяется только в качестве орального средства, заменяющего лидокаин при пароксизмальной тахикардии, в частности, вызванной интоксикацией препаратами дигиталиса.
Синонимами препарата являются дилантин и дифенилан.
Подруппа IC
Препараты этой подгруппы также являются блокаторами натриевых каналов. Они в значительной мере подавляют деполяризацию клеток миокарда и незначительно сокращают время их реполяризации, подавляют автоматизм синусовых узлов. Эти препараты отличаются от рассмотренных выше тем, что уменьшают проводимость и увеличивают рефрактерный период желудочков. В эту подгруппу включены флекаи-нид и энкаинид. Указанные препараты применяются для предотвращения и регуляции суправентрикулярной тахикардии и предсердной фибрилляции у пациентов с нормальной или близкой к нормальной желудочковой функцией, желудочковых аритмиях.
- 347-
Глава 18
Флекаинид (Flecainid)
Флекаинид — Хт-(2-пиперидилметил)-2,5-бмс-(2,2,2-трифторэто-кси)бензамид (18.1.14) получают исходя из 2,5-дигидроксибен-зойной кислоты. Взаимодействием последней с трифторэтилфтор-метилсульфонаюм осуществляют исчерпывающее 2,2,2-трифтор-этоксилирование всех трех гидроксильных групп с получением 2,2,2-трифторэтилового эфира 2,5-бис-(2,2,2-трифторэтокси)бензой-ной кислоты (18.1.12). Взаимодействием последнего с 2-аминоме-тилпиридином получают соответствующий амид (18.1.13), восстановлением пиридинового кольца которого водородом получают флекаинид (18.1.14) [21. 22, 23, 24].
СОО-СН2СР3
О-СН2СГ3
0-CH2CF3
18 1 13
О-Crt2CP3
18 1 14
Химически флекаинид является аналогом прокаинамида, в положения С2 и С5 бензольного кольца которого введены 2,2,2-трифторэто-ксильные группы, а боковая диаминоэтильная цепь завершается в пиперидиновом кольце
Эти изменения значительно меняют фармакологические свойства прокаинамида, однако флекаинид сохраняет местноанестезирующие свойства.
Аналогично другим местным анестетикам, флекаинид применяется при вентрикулярных аритмиях по жизненным показаниям.
Синонимом препарата является тамбокор.
Энкаинид (Encainid)
Энкаинид — М-(2-пиперидилэтил)-4-метокси)бензамид (18.1.15) получается ацилированием 2-(1-метил-2-пиперидилэтил)анилина хлорангидридом 4-метоксибензойной кислоты. Химическая структура энкаинида значительно отличается от других местных анестетиков и антиаритмиков [25, 26, 27].
- 348-
д н т и а ритмические препараты
18 1 15
Клиническое применение энкаинида в первую очередь связано с наличием серьезной желудочковой тахиаритмии, однако, как и флекаинид, он бывает достаточно эффективен и при наджелудочковой аритмии и применяется по жизненным показаниям.
Синонимом препарата является энкаид.
Препараты группы II
Препараты этой группы представляют собой р-адреноблокаторы, антиаритмическая активность которых связана с устранением действия не сердце адренергической иннервации и циркулирующего адреналина. Поскольку все Р-блокагоры уменьшают стимулирующие симпатические нервные импульсы катехоламинов на сердце, уменьшая трансмембранный перенос ионов Na*, скорость проведения возбуждения, то при этом уменьшается синусовая скорость и сокращаемость миокарда, подавляется автоматизм синусовых узлов и ингибируются и наджелудочковая, и желудочковая тахиаритмии.
Возможно, что p-адреноблокаторы регулируют ритм сердца и успокаивают ишемию также и понижением потребности сердца в кислороде.
Их применяют при аритмиях, связанных с нервным стрессом, при инфаркте миокарда, тиреотоксикозе, сопровождающихся повышением адренергической активности. Более того, многие антиаритмические препараты сами могут вызвать аритмию, особенно у пациентов с ишемической болезнью сердца. Исключение составляют рассматриваемые Р-адреноблокаторы.
В силу вышеизложенного практически все Р-адреноблокаторы могут быть применены в качестве антиаритмиков.
Однако в практической медицине группа пока представляется одним препаратом — пропранололом. Появились публикации и по использованию в качестве антиаритмика атенолола. Противопоказаний по использованию p-блокаторов наряду с другими антиаритмиками не имеется.
- 349 -
Глава 1 8
Пропранолол (Propranolol)
Синтез пропранолола — 1-изопропиламино-3-(1-нафтилокси)пропан-2-ола (12.1.3) описан в гл. 12 «Адреноблокирующие препараты».
о—сн
он СН3
2-сн-ch2-nh-ch( СНз
12 1 3
Пропранолол изучен наиболее детально и в эксперименте, и в клинике. Его применяют при вентрикулярной тахикардии, при аритмиях, вызванных передозировкой препаратов дигиталиса или возникших в результате тиреотоксикоза либо избыточной катехоламинной активности. Несмотря на то, что имеется много 0-адреноблокаторов, пропранолол считается препаратом первого выбора, хотя другие блокаторы кальциевых блокаторов могут оказаться не меиее эффективными.
Пропранолол урежает ритм сердечных сокращений, увеличивает эффективный рефрактерный период предсердно-желудочкового узла и угнетает его проводимость, уменьшает автоматизм клеток сердца, снижает возбудимость и сократимость миокарда. Препарат применяют при суправентрикулярных и желудочковых аритмиях.
Синонимами препарата являются анаприлин, детенсол, индерал, новапранол и др.
Препараты группы III
Препараты этой группы оказывают антиаритмическое действие, замедляя реполяризацию и увеличивая продолжительность потенциала действия и эффективного рефрактерного периода во всех отделах сердца. Препарат этой группы — амиодарон блокирует некоторые ионные каналы и а- и 0-адренорецепторы сердца. Их используют при неэффективности других антиаритмических средств при желудочковых аритмиях, не поддающихся лечению другими препаратами, по жизненным показаниям.
Амиодарон (Amiodarone)
Амиодарон — 2-бутил-3-бензофуранил-4-[2-(диэтиламино)этокси]-3,5-дийодфенил кетон (18.1.21) получают по следующей схеме. Бензофуран ацилируют ангидридом масляной кислоты в присутствии фосфорной кислоты с получением 2-бутироилбензофенона
-350-
Антиаритмические препараты
(18.1.16). Восстановлением карбонильной группы в последнем по Вольфу—Кижнеру с использованием гидразингидрата полу'чают 2-бутилбензофуран (18.1.17). Последний ацилируют хлорангидридом 4-метоксибензойной кислоты с получением 2-бутил-3-(4-метокси-бензоил)бензофурана (18.1.18) и далее подвергают деметилированию гидрохлоридом пиридина с получением 2-бутил-3-(4-гидрокси-бензоил)-бензофурана (18.1.19). Полученный продукт йодируют в присутствии йодида калия с получением 2-бутил-3-бензофуранил-4-(2-гидрокси-3,5-дийод-фенил) кетона (18.1.20), который далее вводят во взаимодействие с 2-диэтиламиноэтилхлоридом с получением амиодарона (18.1.21) [28, 29].
н3РО4
(С3Н7СО)2О --------
H2NNH2
18.1 18
(C2H5)2NCH2CH2CI
18 21
Химически амиодарон совершенно отличается от других антиарит-миков и содержит в качестве заместителя два атома йода и диэтиламино этанольную группу, и, в целом, молекула амиодарона весьма напоминает стру ктуру тироксиноподобных молекул.
- 351 -
Глава 1 8
Антиаритмическое действие амиодарона связано с его способностью блокировать К+, Na', Са2+ каналы и неконкурентно блокировать а- и 0-адренорецепторы сердца, пролонгировать потенциал действия и эффективного рефрактерного периода клеток предсердий, атриовентрикулярного соединения и желудочков сердца, что сопровождается снижением автоматизма синусного узла, замедлением атриовентрикулярной проводимости.
Клиническое применение амиодарона ограничено из-за его высокой токсичности, которая заключается в сердечной блокаде, брадикардии (вызванной сердечной недостаточностью), нарушении функций щитовидной железы, нейропатии, повышенной светочувствительности, что значительно ограничивает применение амиодарона. Он показан только при терапии весьма серьезных тахиаритмий, таких как повторяющаяся желудочковая фибрилляция и гемодинамически нестабильная желудочковая тахикардия, и только под наблюдением врача в условиях клиники.
Синонимами препарата являются кордарон, ритмарон и др.
Бретилий (Bretilium)
Бретилий — тозилат К-(о-бромбензил;-\-эгил-\:.К-лимегиламмония (18.1.22) получают взаимодействием о-бромбензилтозилата с этил-диметиламином [30].
Сг|3 n-czh5 СНз
СНз 2— n-c2h5 СНз
18 1 22
Препарат плохо абсорбируется при оральном применении и применяется только в виде внутривенных или внутримышечных инъекций. Однако, аналогично многим другим четвертичным аммониевым солям, препарат инициирует выброс нейрональных катехоламинов, что может вызвать тахикардию, повысить кровяное давление и т. п.
Бретилий обладает симпатолитическим действием, связанным с блокированием выделения норэпинефрина (норадреналина) из преси-наптических нервных окончаний. Препарат имеет и прямое действие непосредственно на ишемические миоциты. Бретилий — средство неотложной помощи, который используется в ситуациях желудочковой тахикардии и фибрилляции желудочков, главным образом в остром периоде инфаркта миокарда, когда применение иных препаратов и про-
- 352 -
Антиаритмические препараты
цед\р оказывается безуспешным. Он должен применяться только в случаях неотложной помощи и требует большой осторожности.
Синонимами препарата являются вретилол, орнид и др.
Препараты группы IV
Препараты этой группы, являясь блокаторами кальциевых каналов, ингибируют медленный трансмембранный ток ионов Са2+ в клетки проводящей системы сердца в ходе деполяризации, что вызывает замедление атриовентрикулярной проводимости и увеличение эффективного рефрактерного периода атриовентрикулярных узлов, что в итоге приводит к релаксации гладкой мускулатуры сердца и восстанавливает нормальный синусовый ритм при суправентрикулярных тахикардиях.
На сегодняшний день группа представлена одним препаратом — блокатором кальциевых каналов — верапамилом, который, в основном, используется в качестве антиангинального средства, а также препарата для контроля за гипертензией.
BepancLMici (Verapamil)
Синтез верапамила 5-[(3,4-диметоксифенэтил)метиламино]-2-(3,4-диметоксифенил)-изопропилвалеронитрила (19.3.15) будет описан в гл. 19 «Антиангинальные препараты».
193 15
В качестве антиаритмического средства верапамил применяют и для лечения суправентрикулярных аритмий, таких как пароксизмальная предсердная тахикардия, для контроля над фибрилляцией предсердий. Подавляя вход кальция в клетку, верапамил оказывает отрицательное инотропное действие, и поэтому его нельзя комбинировать с Р-адрено-блокаторами или хинидином, так как при этом усиливается его отрицательный инотропный эффект.
В качестве антиаритмика верапамил в основном применяется при желудочковых аритмиях, однако в настоящее время он постепенно вытесняется аденозином.
Синонимами препарата являются изоптин, калан, финоптин, фали-кард, манидон и многие другие.
- 353 -
Г лава 1 8
Список литературы
1 Turner R , Woodward R , 'The Alkaloids. 3. 1-63 (1953).
2. US Pat. 2.878.252 (1959).
3. G^r. Pat. 2.156.725 (1971)
4. Doering W et al.//J. Am. Chem. Soc. 69, 1700 (1949).
5. Gutzwiller J. et al./J. Am. Chem. Soc. 92. 204 (1970).
6. Gutzwiller J. et al.Z/Helv. Chim. Acta. 56, 1494 (1973).
7. Gutzwiller J et al. 7J. Am. Chem. Soc. 100, 576(1978).
8. Baltzy R et al.//J, Am. Chem. Soc. 64, 2231 (1942).
9. Yamazaki M et al., 'J. Pharm. Soc. Japan. 73. 294 (1953).
10 US Pat. 3.225.054 (1962).
11. Ger. Pat. 1.470.2156(1962)
12. Adelstein J et al.//J. Med. Chem. 16, 309 (1973).
13 Ger. Pat. 2.235 745 (1972).
14. Brit. Pat. 1.461.602 (1974).
15. Ger. Pat. 2.400.540(1974).
16 Byrnes E 'J. Med. Chem. 22, 1171 (1979).
17. US Pat. 3 954.872 (1976).
18. US Pat. 3.659.019 (1972).
19. Fr Pat 1.551.055 (1968).
20. S.Afr. Pat 69 03.772 (1970).
21. US Pat. 3.900.481 (1975).
22. US Pat. 3.655.728 (1972).
23. US Pat. 4.005.209 (1977).
24. Banitt E. et al./'J. Med. Chem. ^0. 821 (1977).
25. Ger. Pat. 2.210.154 (1972).
26. US Pat. 3.931.195 (1976).
27. Dvkstra S et al.//J. Med. Chem. 16, 1015 (1973).
28. Fr. Pat. 1.339.389 (1962).
29. US Pat. 3.248.401 (1966).
30. US Pat. 3.038.004 (1962).
I Глава 19
k
। Антиангинальные
| препараты
Антиангинальными препаратами являются лекарственные средства, применяемые для купирования и профилактики патологических состояний, связанных с коронарной недостаточностью и объединенных термином «ишемическая болезнь сердца».
К ишемической болезни относятся стенокардия («грудная жаба») и инфаркт миокарда.
Стенокардия возникает в результате дисбаланса между потребностью и снабжением кислородом ишемической области миокарда. Следовательно, теоретически при стенокардии необходимы препараты либо понижающие потребность миокарда в кислороде, либо улучшающие его снабжение кислородом. Этого можно достичь либо уменьшением нагрузки на сердце, либо путем снижения системного венозного и артериального давления (нитраты и нитриты), либо частичным подавлением адренергической иннервации сердца (Р-адреноблокаторы), либо угнетением транспорта ионов Са2* в клетки миокарда, поскольку сокращение гладких мышц сосудов контролируется концентрацией ионов Са2+ в цитоплазме (блокаторы кальциевых каналов). Фактически результирующим эффектом перечисленных выше рядов препаратов становится снижение потребности сердца в кислороде.
Таким образом, основными препаратами, используемыми во время терапии ишемии миокарда и для облегчения болей при стенокардии, являются нитраты и нитриты (нитроглицерин, динитрат изосорбида и тетранитрат пентаэритрита), вещества подавляющие адренергические системы сердца — |3-адреноблокаторы (атенолол, метопролол, пропранолол и надолол) и блокаторы кальциевых каналов (верапамил, дилтиа-зем, нифедипин и никардипин), а также некоторые более старые препараты и, в частности, папаверин и дипиридамол.
- 355-
Глава 19
19.1. Нитраты и нитриты
Еще более 100 лет назад было замечено, что амилнитрат и нитроглицерин облегчают боли при стенокардии, и поскольку нитриты и нитраты имеют свойство расширять сосуды, умеренно увеличивая кровоток к миокарду, стало принятым считать, что расширение коронарных сосудов облегчает состояние больного стенокардией.
Кажется вероятным, что механизм действия всех рассматриваемых органических нитратов и нитритов должен быть аналогичен. Они проявляют прямой релаксирующий эффект на сосудистую гладкую мускулатуру, в результате чего наступает общее расширение сосудов, при котором доминирует расширение вен, а расширение артерий происходит в меньшей степени. В результате уменьшается сосудистое сопротивление, что приводит к уменьшению нагрузки на сердце и к снижению потребности миокарда в кислороде, и состояние гипоксии устраняется. Более того полагают, что нитроглицерин и другие нитраты уменьшают размеры ишемического поражения, что связывают с улучшением кровоснабжения миокарда.
Механизм действия нитратов выяснен не до конца. На основании имеющихся фактов можно с определенной долей вероятности утверждать, что в клетках гладкой мускулатуры нитраты трансформируются в нитриты и затем высвобождают NO. Последний, в свою очередь, взаимодействует с гунилатциклазой, вызывая повышенный синтез гуанозин 3',5'-монофосфата (циклический GMP). В результате активируется GMP-зависмая протеинкиназа, и, как следствие, происходит меньшее фосфорилирование мышечных белков, а дефосфорилированные мышечные белки имеют меньшую способность к сокращению, итоговым результатом чего становится снижение потребности сердца в кислороде.
Таким образом, нитраты расслабляют всю гладкую мускулатуру, включая систему печени, мочевого пузыря и бронхиол. Однако наиболее активная релаксация происходит в кровеносных сосудах.
Тем не менее селективное расширение коронарных сосудов не может полностью объяснить лечебное действие коронарорасширяющих средств. При стабильных формах стенокардии ввиду повреждения сосудов образуются жесткие сосудистые структуры, не способные к расширению. Более того, коронарное кровообращение само отвечает на ишемию расширением сосудов. Не поврежденные атеросклерозом сосуды увеличивают кровоток к нормальному миокарду путем расширения и, соответственно, уменьшают поток крови к ишемической области. Это явление называется «сосудистым воровством» и имеет скорее отрицательное, чем положительное значение.
- 356 -
Антиангинальные препараты
В качестве антиангинальных препаратов лекарственные формы нитратов представлены как быстродействующими препаратами, используемыми для снятия острых приступов стенокардии, так и препаратами пролонгированного действия, которые применяются для предотвращения приступов стенокардии. Нитроглицерин — глицерилтринитрат и амил-нитрит являются препаратами с быстрым началом, но с коротким периодом действия. Другие органические нитраты, используемые для более пролонгированного действии, представляют из себя тетранитрат эритрита, динитрат изосорбида и тетранитрат пентаэритрита. Из органических нитритов в медицине применяется амилнитрит.
В последние 10 лет коронарорасширяющие средства зарекомендовали себя как первичные соединения в терапии сердечной недостаточности.
Нитроглицерин (Nitroglycerine)
Нитроглицерин — 1,2,3-пропантриолтринитрат (19.1.1) получают нитрованием глицерина азотной кислотой [1, 2, 3].
сн2-он сн—он сн2—он
HNO3/ Н^Рд
сн2 -ono2 сн—ono2 CH2""ONO2
19 1 1
Препарат вызывает уменьшение нагрузки на сердце за счет расширения периферических вен, уменьшает потребность миокарда в кислороде и способствует перераспределению коронарного кровотока в области миокарда со сниженным кровообращением.
Нитроглицерин применяют преимущественно для купирования острых и хронических приступов стенокардии, при инфаркте миокарда.
Синонимами препарата являются тринитроглицерин, гринитрол, тринитрин и многие другие.
Тетранитрат пентаэритрита (Pentaerythriol tetranitrat)
Тетранитрат пентаэритрита — 2,2-бис(гидроксиметил)-1,3-пропан-диолтетранитрат (19.1.2) также получают реакцией нитрования, но уже 2,2-бис(гидроксиметил)-1,3-пропандиола — пентаэритрита азотной кислотой [4].
НО-СН2 СН2~ОН HNO3/ НгЭОд o2no-ch2 сн2—ono2
?с< --------->с<
НО-СН2 СН2-ОН ОгЫО-СН/ ch2-ono2
19 1 2
-357-
Глава 19
Тетранитрат пентаэритрита применяют при хронической сердечной недостаточности. Препарат предупреждает наступление приступов стенокардии, облегчает их течение
Синонимами препарата являются нитропентон, нитринал, вазокор, вазолат, пентилан, эринит и многие другие.
Июсорбиддинитрат (Isosorbiddinitrat)
Изосорбиддинитрат — 1,4:3,6-диангидросорбит-2.5-динитрат (19.1.4) получают внутримолекулярной дегидратацией D-сорбита в изосорбид (19.1.3) с использованием иара-толуолсульфокислоты с после-дующм нитрованием двух гидроксильных групп азотной кислотой [5, 6, 7].
снг- он н-с-он он-с-н Н-С-ОН н-с-он
сн2-он
HNO3/ H2SO1
19 1 4 0N02
Изосорбиддинитрат также применяют при хронической сердечной недостаточности, для предупреждения приступов стенокардии. Действует продолжительно.
Синонимами препарата являются изордил, метронитрон, вазкардин и др.
19.2. Препараты ряда $-адреноблокаторов
Имея свойство понижать потребность сердца в кислороде, р-адре-ноблокаторы и, в частности, атенолол, метопролол, пропранолол и надолол рекомендуются для лечении хронической стенокардии, которая часто развивается после перенесенного инфаркта миокарда. Уменьшение потребности сердца в кислороде Р-адреноблокаторами достигается понижением скорости работы сердца, уменьшением кровяного давления и сокращаемости миокарда. Следует отметить, что острые приступы стенокардии лучше всего снимаются нитроглицерином. Однако, несомненно, терапия p-адреноблокаторами комплементарна терапии нитратами. Более того, нередко нитраты противодействуют некоторым нежелательным эффектам Р-адреноблокаторов, поэтому наиболее часто применяется комбинированная терапия нитратами и р-адреноблока-торами.
-358 -
Ан тиангинальные препараты
Пропранолол (Propranolol)
Пропранолол — 1-(мдо-пропиламино)-3-(1-нафтокси)-2-пропанол (.12.1.2). Синтез препарата описан в гл. 12 «Адреноблокирующие препараты».
СН3
О-СН2-СН-СН2 —NH -СН он 'СНз
12.1.2
Пропранолол — неселективный Р-адреноблокатор, который действует и на механические и электрофизиологические свойства миокарда. Препарат понижает сократимость миокарда, скорость работы сердца, давление крови и потребность миокарда в кислороде. Эти свойства делаю! пропранолол и другие p-адреноблокаторы полезными антианги-нальными препаратами.
Пропранолол показан при лечении гипертонии, стенокардии, суправентрикулярной аритмии, вентрикулярной тахикардии, мигрени, гипертрофическом субаортальном стенозе и феохромоцитоме. Применяется в послеострой фазе инфаркта миокарда.
Общепринятыми синонимами препарата являются анаприлин. обзи-дан, индерал. новапранол, дефенсол.
Метопролол (Metoprolol)
Метопролол — 1-(мзо-пропиламино)-3-[4'(2-метоксиэтил)фенокси)]-2-пропанол (12.1.5). Синтез препарата описан в гл. 12.
/=\ СН3
СН3О-СН2-СН2—4 /)—О-СН2-СН-СН2— NH-CH
N' ОН СНз
12.1.5
В отличие от пропранолола, блокирующего как рг, так и р2-адре-норецепторы, метопролол проявляет кардиоселективное действие, т. е. в терапевтических дозах он блокирует р,-адренорецепторы с незначительным воздействием на р2-адренорецепторы.
Метопролол применяют при инфаркте миокарда, для предупреждения омертвления сердечно-сосудистых тканей, при стенокардии.
- 359 -
Глава 1 9
Наиболее распространенным синонимом препарата является ло-прессор.
Ацебутол (Acebutol)
Ацеб\ тол — 3 '-ацетил-4 ,-[2-гидрокси-3-(гтзо-пропилампно)пропокси]-
бутиранилид (12.1,6). Синтез препарата также описан в гл. 12.
СНз
О-СН2-СН-СН2—NH-CH
ОНзСО^Х. он СНз
12 1 6
МН~СО~СзН7
Применяется для профилактики стенокардии.
Синонимом препарата является сектрал.
Атенолол (Atenolol)
Атенолол — 2-[4' [2-гидрокси-3-(мзо-пропиламино)пропокси]фенил]-ацетамид (12.1.7). Синтез препарата описан в гл. 12.
СНз
сн2-сомн2
Препарат применяется для профилактики стенокардии.
Синонимом препарата является тенормин.
Надолол (Nadolol)
Надолол — 1-(т/?е/и-бутиламино)-3-[(5,6.7,8-тетрагидро-г/ио6,7-Ди-гидрокси-1-нафтил)-окси]-2-прпанол (12.1.8). Синтез препарата описан в гл. 12.
но
но
O-CH2-CH-CH2-NH-C(CH3)3
X он
12 18
- 360 -
днтиангинальные препараты
Препарат применяется для профилактики хронической стенокардии.
Синонимом препарата является коргард.
19.3. Блокаторы кальциевых каналов
Эта группа препаратов разрабатывалась в качестве коронарорас-ширяюших средств и некоторое время использовалась в качестве таковой, пока не было выяснено, что они ингибируют сокращающее действие кальция на гладкую мускулатуру и сердечную мышцу и местом их действия являются кальциевые каналы на поверхности клетки, служащие для проведения ионов Са2+ в клетку. Первоначально их стали называть антагонистами кальция, однако позже для этого класса соединений предпочтение было отдано названию блокаторы кальциевых каналов.
Химически блокаторы кальциевых каналов представляют довольно разнообразную группу соединений, что может свидетельствовать о разнообразных рецептивных участках как на поверхности клеточной мембраны, так и внутри нее. Верапамил, который можно рассматривать как производное бензилцианида, является одним из старейших и до сих пор активно используемым соединением этого класса. Дилтиазем является тио-диазепином, а нифедипин и никардипин — производным дигидро-пирирдина.
Сегодня является очевидным факт, что сокращение гладких мышц сосудов контролируется концентрацией ионов Са2+ в цитоплазме. Основной принцип действия блокаторов кальциевых каналов заключается в том. что они нарушают проникновение ионов Са2‘ в мышечные клетки сердца и сосудов. Уменьшение поступления ионов С а2* в клетки миокарда приводит к уменьшению возможности использования энергии фосфатных связей для механической работы сердца. В результате снижаются сила сердечных сокращений и работа сердца, что, в свою очередь, приводит к уменьшению потребности сердца в кислороде.
Предложены два механизма, регулирующих клеточную концентрацию кальция. Первый из них называется механизмом электромеханического сопряжения. При этом полагают, что вольтажзависимые кальциевые каналы раскрываются в ответ на деполяризацию мембраны, и в результате внеклеточные ионы Са2’ устремляются в клетку. Второй из предложенных механизмов независим от деполяризации мембраны. Он предполагает высвобождение ионов Са*’ из саркоплазматической сумочки, что вызывает поток внеклеточных ионов Са2
- 361 -
Глава 1 9
в клетку через не связанные с напряжением кальциевые каналы. Независимо от механизма проникновения ионов Са2+ в клетку, их повышенная концентрация приводит к связыванию с кальмадулином. В свою очередь Са2+-кальмадулиновый комплекс инициирует фосфорилирование легких цепей миозина путем активации киназы легких цепей. Взаимодействие фосфорилированных легких цепей миозина с актином в свою очередь вызывает сокращение гладкой мускулатуры.
Блокаторы кальциевых каналов могут блокировать поток кальция в клетку по любому из приведенных механизмов. Однако вольтажзави-симые каналы отвечают на меньшие концентрации кальция, чем воль-тажнезависимые. Отсюда следует, что соотношение вольтажзависи-мых'вольтажнезависимых каналов определяет селективность ответов вен и артерий. В клинически применяемых дозах блокаторы кальциевых каналов расслабляют гладкую мускулатуру артерий и мало действуют на вены. Необходимо отметить, что процесс возбуждения-сокращения в сердечных миоцитах больше зависит от потока внутрь клетки Na\ а не Са2+ ионов. Следовательно, в дозах, которые релаксируют гладкую мускулатуру. блокаторы кальциевых каналов относительно мало влияют на сокращаемость сердца.
Дилтиазем (Diltiazem)
Дилтиазем — ацетат 5-[2-(диэтиламино)этил]-г/ис-2,3-дигидро-3-гид-рокси-2-(4-метоксифенил)-1,5-бензотиазепин-4(5Я)-она (19.3.10) получают по следующей схеме. Конденсацией в условиях реакции Дарзана 4-метоксибензальдегида с метилхлорацетатом в присутствии метилата натрия получают метиловый эфир 3-(4-мето-ксифенил)глипидиловой кислоты (19.3.5). Раскрытием эпоксидного цикла последнего взаимодействием с 2-аминотиофенолом получают метиловый эфир 2-гидрокси-3-(2'-аминофенилтио)-3-(4'’-метокси-фенил)пропионовой кислоты (19.3.6). Гидролиз полученного соединения щелочью приводит к получению соответствующей кислоты (19.3.7) в виде рацемической смеси, из которой взаимодействием с (+)-а-фенилэтиламиио.м получают трео-(+)-2-гидрокси-3-(2|-ами-нофенилтио)-3-(4"-метоксифенилпропионовую кислоту (19.3.8). Кипячение последней в смеси уксусный ангидрид/диметилформа-мид'пиридин приводит к замыканию тиазепинового цикла и одновременному ацилированию гидроксильной группы с образованием (+)-г/гю-2-(4-метоксифенил-3-ацетокси-2,3-дигидро-1.5-бензотиазе-пин-4-(5/Г)-она (19.3.9). Алкилирование последнего 2,2-диметил-аминоэтил-хлоридом приводит к дилтиазему (19.3.10) [8, 9, 10, 11, 12, 13. 14, 15].
-362 -
днтиангинальные препараты
NaOH
1937
(CH3CO)2O>DMF/Py>
CICH2CH2N(CH3)2
N(CH3)2
Дилтиазем уменьшает трансмембранное поступление ионов С а2’ в клетки сердечной мышцы и гладкой мускулатуры сосудов. Вызывает расширение коронарных и периферических сосудов, увеличивает коронарный кровоток, предотвращая развитие спазма коронарных артерий, снижает повышенное АД и уменьшает тахикардию.
Препарат показан при стабильной и нестабильной стенокардии (в том числе и после инфаркта миокарда), при артериальной гипертонии.
Общепринятыми синонимами препарата являются изоптин, ипро-вентарил и др.
Верапамил (Verapamil)
Верапамил — 5-[(3,4-диметоксифенэтил)метиламино]-2-(3,4-диме-токсифенил)изопропилвалеронитрил (19.3.15) получают по схеме, исходящей в качестве основного исходного вещества из 3,4-диме-токсифенилацетонитрила. Сам синтез конечного продукта (19.3.15) сводится к алкилированию 2-(3,4-диметоксифенил)-3-метилбути-ронитрила (19.3.11) 1\т-[2-(3,4-диметоксифенил)-этил]-М-3-хлорпро-пил)-К-метиламином (19.3.14).
- 363-
Гпава 19
Исходный 2-(3,4-диметоксифенил)-3-метилбутиронитрил (19.3.11) получают алкилированием 3,4-Диметоксифенилацетонитрила изопро-пилхлоридом в присутствии амида натрия. Алкилирующий агент — N-[2-(3,4-диметоксифенил)-этил]-М-3-хлорпропил)-М-метила.мин (19.3.14) также получают исходя из 3,4-диметоксифенилацетонигрила последовательным восстановлением в 3,4-диметоксифенилэтиламин (19.3.12) с последующим метилированием в М-метил-М-3,4-диметоксифенилэти-ламин (19.3.13). Далее полученный М-[2-(3,4-диметоксифенил)-этил)-М-метиламин (19.3.12) алкилируют 1-хлор-З-бромпропаном в искомый N-[2-(3,4-диметоксифенил)-)тил]-К-3-хлорпропи;1)-М-метиламин (19.3.14), которым и алкилируют 2-(3.4-диметоксифенил)-3-метилбутиронитрила (19.3.11) в конечный продукт—верапамил (19.3.15) [16, 17, 18, 19].
н2м ~сн2 - сн2
O-CHj
снэ|
19 3 12
н-ь-сн2-сн2 СНз
Вг-СНгСНгСН, -с
О-СНз
Cl—CH2-CH2-CH2-N-CH2-CH2-4 Л—О-СН3 сн3 '—'
1S 3.11
19.3 14
,0-СНз
СН3- О СН3х^СН3 (
>=\ <ГН /={
СНз-O-Z у— С-СН2-СН2-СН2—N-CH2-CH2~4 У-О-СНз ' C=N СНз ' '
193 15
Верапамил обладает антиаритмической, антиангинальной и гипотензивной активностью. Снижает потребность миокарда в кислороде за счет снижения сократимости миокарда и урежения частоты сердечных сокращений. Вызывает расширение коронарных артерий и увеличение коронарного кровотока. Снижает тонус гладкой мускулатуры, периферических артерий и общее периферическое сосудистое сопротивление.
-364 -
днтиангинапьные препараты
Оказывает антиаритмическое действие при наджелудочковых аритмиях.
Верапамил применяют для профилактики приступов стенокардии, артериальной гипертонии, при лечении и профилактике наджелудочковых аритмий (пароксизмальная наджелудочковая тахикардия, мерцание предсердий, трепетание предсердий, экстрасистолия).
Синонимами препарата являются изоптин, калан, финоптин, фали-кард, манидон и многие другие.
Нифедипин (Nifedipin)
Нифедипин — диметиловый эфир 1,4-дигидро-2,6-диметил-4-(2‘-нитрофенил)-3,5-пири-диндикарбоновой кислоты (19.3.16) получают синтезом Ганча, исходящего из двух молей [3-дикарбонильного соединения — метилового эфира ацетоуксусной кислоты, альдегида 2-нитробензальдегида и аммиака. Последовательность промежуточных стадий синтеза точно не установлена [20, 21, 22, 23].
Нифедипин вызывают расслабление гладкой мускулатуры сосудов, расширяет коронарные и периферические артерии, снижает периферическое сопротивление, АД, улучшают снабжение сердца кислородом.
Нифедипин применяют для профилактики приступов стенокардии, их копирования, при гипертонии, в составе комбинированной терапии хронической сердечной недостаточности.
Синонимами препарата являются адалат, коринфар, прокардиа, ни-фекор.
Никардипин (Nicardipine)
Никардипин — 1,4-дигидро-2,6-диметил-4-(3-нитрофенил)-метил-2-| метилфенилметил)-амино]этиловый эфир 3,5-пиридидинкарбоно-вой кислоты (19.3.17) синтезируют аналогично нифедипину с той лишь разницей, что в реакции Ганча с о-нитробензальдегидом одновременно используются два разных p-дикарбонильных соединения. При этом одно из них в виде енаминной формы ацетоуксусного эфира одновременно используется и в качестве аминной компонен
-365-
Глава 19
ты Реакция гетероциклизации осу шестляется взаимодействием метилового эфира Р-аминокротоновой кислоты с 2-метил-2-бензил-аминоэтиловым эфиром ацетоуксусной кислоты [24. 25, 26, 27].
Никардипин расслабляет гладкую мускулатуру сосудов, снижает сопротивление коронарных и периферических сосудов, увеличивает кровоток в сосудах головного мозга, вызывает умеренный и стойкий гипотензивный эффект, уменьшает потребность миокарда в кислороде.
Препарат применяют при артериальной гипертонии, при хронической стабильной стенокардии, для профилактики стенокардии и при нарушениях мозгового кровообращения по ишемическому типу.
Синонимами препарата являются нердипин, карден и др.
19.4. Прочие препараты
Современные препараты, применяющиеся при лечении стенокардии. были описаны выше. Однако имеются весьма старые препараты и подходы для ее лечения и, в частности, использование папаверина и дипиридамола, которые применяются и сегодня.
Папаверин (Papaverin)
Папаверин — 1-вератрил-6,7-диметоксиизохинолин 19.4.7) предложено синтезировать исходя из вератрола.
Вератрол подвергают хлорметилированию с получением 3,4-диме-токсибензилхлорида (19.4.1). Взаимодействием последнего с цианистым калием получают 3,4-диметоксибензилцианид (19.4.2). Подвергая полученный 3,4-диметоксибензилцианид восстановлению водородом над никелем Ренея получают гомовератриламин (19.4.3), а кислотным гидролизом 3,4-диметоксифенилуксусную кислоту (19.4.4). Взаимодействием полученных соединений синтезируют соответствующий амид (19.4.5). Циклизацией последнего по Бишлеру—Напиральскому с использованием хлорокиси фосфора получают 3,4-дигидропапаверин (19.4.6). который дегидрируют в искомый папаверин нагреванием в тетралине при высокой температуре [28, 29, 30, 31].
- 366 -
Днтиэнгинэльныб препараты
KCN
19 4 2
СН2-CN
IS 4 3
CH3°\rJ^:VCH2"Cl сн3о'л=^>
Папаверин является алкалоидом сырого опия, однако не относится к соединениям класса морфина и не обладает анальгетическими свойствами. Он является периферическим вазодилататором и непосредственно во’.действует на сосуды. Вызывает расширение коронарных, мозговых, легочных артерий. Способствует увеличению церебрального кровотока и снижению церебрального сосудистого сопротивления. В терапевтических дозах он понижает кровяное давление. В больших дозах может вызвать аритмию. Несмотря на отсутствие однозначного терапевтического эффекта, тем не менее его продолжают широко использовать при итрхшениях периферического и мозгового кровообращения и в качестве ДОронарорасширяющего средства вообще, а также при спазмах периферических сосудов и сосудов головного мозга, бронхов.
Синонимами препарата являются папавин, церебид, миобид, наварен вазоснан и многие другие.
- 367 -
Глава 19
Дипиридамол (Dipyridamol)
Дипиридамол — 2.21,211,2111 -[(4,8-дипиперидинопиримидо[5,4-с1] пиримидин-2, 6-диил)-диимино]-тетраэтанол (19.4.13) синтезируют исходя из 5-нитрооротовой кислоты (19.4.8), легко получаемой, в свою очередь, нитрованием 2,4-дигидрокси-6-метилпиримидина, который обычно получают конденсацией мочевины с ацетоуксусным эфиром. Восстановлением нитрогруппы в 5-нитрооротовой кислоте различными восстановителями получают 5-аминооротовую кислоту (19.4.9), которая взаимодействием с мочевиной или с цианатом калия приводит к получению 2,4,6,8-тетрагидроксипиримидо[5,4-<1]-пиримидина (19.4.10). Подвергая последний действию смеси хлорокиси фосфора и пятихлористого фосфора получают 2.4,6,8-тетра-х.порпиримидо[5,4-б]пиримидина (19.4.11). Взаимодействием полученного тетрахлорида с пиперидином замещают атомы хлора при С4 и С8 гетеропикличской системы пиперидином с получением 2,6.-дихлорпиримидо-4,8-дипиперидино[5,4-б]пиримидина (19.4.12). Вводя полученный продукт во взаимодействие с диэтаноламином получают дипиридамол (19.4.13) [32, 33].
hfyCHjCHjOH <2
Дипиридамол усиливает коронарное кровообращение, улучшает снабжение миокарда кислородом, потенцирует активность аденозина и препятствует его метаболизации. Препарат тормозит агрегацию тромбоцитов, блокирует фосфодиэстеразу, улучшает микроциркуляцию и препятствует образованию тромбов.
Препарат показан при хронической коронарной недостаточности, профилактики и лечении тромбозов.
Синонимами препарата являются ангинал, курантил, стенокор. тром-пресантин и многие другие.
- 368-
АнтиангинальНые препараты
Список литературы
1. Sobrero Я./, Ann. 64, 398 (1847)
2. Williamson E.//Ann. 92. 305 (1854).
3. DiCarlo /-'./Drug Metab. Rev. 4, 1 (1975).
4. US Pat. 2.370.437 (1945).
5. Goldberg £.//Acta Physiol. Scand. 15. 173 (1948).
6. Krantz J. et al./'J. Pharmacol. Exper. Ther. 67, 187 (1939).
7. Ger. Pat. 2.623.800(1976).
8. Ger. Pat. 1.805.714 (1968).
9. USPat. 3.562.257 (1968).
10. US Pat. 4.416.819(1983).
11. US Pat. 4.438.035 (1984).
12. US Pat. 4.552.695 (1985).
13. Ger. Pat. 3.415.035 (1984).
14. Nagao T. et al.'/Chem. Pharm. Bui. 21, 92 (1973).
15. Kugita H. et al.//Chem. Pharm. Bui. 19. 595 (1971).
16. US Pat. 3.261.859 (1966).
17. Belg. Pat. 615.861 (1962).
18. Ramuz Я./'Helv. Chim. Acta. 58, 2050 (1975).
19. Theodore L. et al.//J. Org. Chem. 52, 1309(1987).
20. US Pat. 3.485.847 (1969).
21. US Pat. 3.644.627(1972).
22. Brit. Pat. 1.173.862(1968).
23. S.Afr. Pat. 68 01.482(1968).
24. Belg. Pat. 811.324 (1974).
25. USPat. 3.985.758 (1976).
26. Iwanami M. et al./7Chem. Pharm. Bui. 27, 1426 (1979).
27. Shibamtma T et al.//Chem. Pharm. Bui. 28, 2809 (1980).
28. Pictet A. et al.//Compt. Rend. 149, 210 (1909).
29. Pictet A. et al./'Ber. 42, 2943 (1909).
30. Браз Г. и др./,ЖПХ. 26, 337 (1953).
31. Goldberg A/.//Chem. Prod. Chem. News. 17, 371 (1954).
32. Brit. Pat. 807.826(1956).
33. USPat. 3.031.450(1962).
Глава 20
Гипол ипидем ические средства
Препараты, понижающие концентрацию липопротеидов в плазме путем ингибирования их выработки в организме либо путем их вывода из плазмы, называются гиполипидемическими или противосклеротиче-скими препаратами.
Атеросклероз — это состояние организма, характеризующееся повышенным содержанием в плазме крови атерогенных липопротеидов, отложением липидов, в том числе холестерина, в виде сложных эфиров внутри стенок артериальной системы, выражающееся в постепенном затруднении кровообращения. Наиболее подходящее название для этой болезни — липопротеинемия. Клинически это проявляется в виде ишемической болезни сердца, инсульта, нарушения мозгового кровообращения, периферической ишемии.
Атеросклероз гораздо более сложное заболевание, чем простая констатация факта высокой концентрации липопротеидов в крови и сужения сосудов.
Основной механизм развития атеросклеротического процесса не совсем ясен. Кажется весьма вероятным, что развитию атеросклероза предшествуют метаболические нарушения синтеза, транспорта и утилизации липидов. Липиды, такие как триглицериды и сложные эфиры холестерина, циркулируют в крови в виде частиц (липопротеидов), покрытых гидрофильной оболочкой, состоящей из фосфолипидов и свободного холестерина. Холестерин транспортируется частицами различного размера, составленными из триглицеридов, эфиров холестерина и фосфолипидов, каждая из которых играет весьма определенную роль.
Липопротеиды разделяют на пять основных типов. Самыми крупными частицами являются хиломикроны, затем идут липопротеиды очень низкой плотности (ЛПОНП), липопротеиды промежуточной плотности (ЛППП), липопротеиды низкой плотности (ЛПНП), и, нако-
-370 -
Г и п о л ипидемические средства
ней, самыми маленькими являются липопротеиды высокой плотности (ЛПВП). Разделение вышеперечисленных холестеринсодержащих комплексов осуществляют с помощью ультрацентрифугирования.
Таким образом, хиломикроны являются самыми крупными частицами с наименьшей плотностью, которые образуются в клетках эпителия тонкого кишечника и состоят из экзогенных триглицеридов (жиров), в отношении которых выполняют транспортные функции, а также эфиров холестерина.
ЛПОНП образуются в печени и включают, главным образом, эндогенные триглицериды и сложные эфиры холестерина с ненасыщенными жирными кислотами. В организме они подвергаются липолизу с образованием короткоживущих ЛППП, содержащих примерно равные количества триглицеридов и сложных эфиров холестерина. Последние вновь подвергаются липолизу, переходя ьЛПНП, в которых уже преобладают сложные эфиры холестерина.
ЛПВП образуются в печени и кишечнике в результате катаболизма хиломикронов и ЛПОНП и по сравнению с другими липопротеидами содержат значительно больше сложных эфиров холестерина с ненасыщенными жирными кислотами, а также фосфолипидов и специфических белков. Однако, в отличие от атерогенных ЛПНП и ЛПОНП, которые метаболизируясь высвобождают холестерин, который в виде сложных эфиров отлагается в тканях, хиломикроны и ЛПВП не атерогенны. Существует прямая связь между концентрацией в плазме и крови ЛПНП и выраженностью атеросклеротических изменений в средних и крупных артериях.
Вероятное течение процесса образования атеросклеротических бляшек, очевидно, кратко можно описать следующим образом. В течение времени, в основном с годами, в определенной точке сосуда, где по какой-то причине имеется турбулентность потока крови, происходит повреждение эндотелия. Тромбоциты начинают притягиваться к поврежденному участку. Комбинированное действие эндотелиального роста и вызванного тромбоцитами фактора роста привлекает к этому участку макрофаги, вызывая воспалительную реакцию, приводящую к гипертрофии средней мышцы артерии, что сужает сосуд по всей его длине с образованием бляшки. Эндотелий никогда не восстанавливается полностью и может явиться участком образования тромбов в суженном сосуде.
Тактика понижения уровня холестерина в организме в основном заключается либо в выведении его избыточного количества из плазмы, либо в ингибировании синтеза ЛПНП и ЛПОНП. Соответственно, гипо-липидемические средства подразделяются на препараты, усиливающие
-371 -
Глава 20
катаболизм и выведение из организма атерогенных липопротеидов и липидов (колестипол и холестирамин), и средства, препятствующие образованию атерогенных липопротеидов — фибраты (клофибрат, гемфиброзил), природные соединения — статины (ловастатин, мевастатин и их аналоги), а также пробукол и никотиновая кислота. Кроме того, иногда применяют препараты, снижающие уровень холестерина в организме с необъясненными до конца механизмами действия — декстротироксин и неомицин.
Однако, наряду с приведенными выше отрицательными свойствами, присущими холестерину, следует иметь в виду, что весьма определенная его часть в организме расходуется и на превращение в адренальные, половые гормоны и витамин D.
20.1. Препараты, удаляющие желчные кислоты
z Одним из путей катаболизма холестерина в организме является его трансформация в желчные кислоты, важнейшими из которых являются холевая, дезоксихолевая. литохолевая и холановая кислоты.
холестеринхолевая кислота
.соон
.соон
.соон
дезоксихолевая кислота
литохолевая кислота
холановая кислота
При связывании желчных кислот и их удалении из организма нарушается баланс холестерин — желчные кислоты и начинается компенсаторное увеличение трансформации холестерина в желчные кисло™, в результате чего уменьшается содержание самого холестерина. Препараты, удаляющие холестерин в виде нерастворимых производных желч
- 372-
Гиполипидемические средства
ных кислот, стимулируют трансформацию организмом холестерина в желчные кислоты. Результирующим эффектом является общее понижение количества холестерина в организме.
В настоящее время доступны два препарата, связывающих желчные кислоты в ЖКТ и выводящих их из организма — колестипол и холестирамин. Оба являются высокомолекулярными соединениями — анионообменными смолами. Они отличаются друг от друга по химическому строению, однако имеют один и тот же механизм действия. Будучи нерастворимыми в воде они не абсорбируются из ЖКТ и связывают желчные кислоты на своих четвертичных аммониевых участках, выделяя их в связанном с ионообменной смолой виде с фекалиями.
Колестипол (Colestipol)
Колестипол (20.1.1) представляет собой гидрохлорид продукта сополимеризации эпихлоргидрина с диэтилентриамином, содержащим различное число кватернизованных атомов азота. Точной формулы продукта сополимеризации нет, но его примерное строение можно выразить следующим образом (20.1.1) [1, 2, 3, 4J.
А
H2N-СН2-'СН2 -NH-CH? —сн2-nh2 + ci—сн2—сн—сн2 --------►
-ЧНН— CH2-CH2-NH-CH2-CH2-NH-CH2-CH-CH2 — NH—CH2—CHj-NH-CHj-CHj-NH-)- mHCI OH n
20 1 1
Колестипол связывает желчные кислоты, которые выделяются, в связанном с ионообменной смолой виде. Препарат понижает уровень общего холестерина и холестеринсодержащих ЛПНП в плазме крови, не изменяя уровень ЛПВП.
Колестипол применяют при гиперхолестеринемии (в том числе при атеросклерозе и артериальной гипертонии).
Синонимами препарата являются холестабил, холестид, лестид и др.
Холестирамин (Cholestyramine)
Холестирамин (20.1.2) является сополимером дивинилбензола и стирола, который подвергают хлорметилированию и вводят во взаимодействие с триэтиламином [5].
- 373 -
Г лава 20
Сн —СН2
(CH3)3N
20 1 2
Холестирамин, аналогично колестиполу, связывает желчные кислоты, которые выделяются в связанном с ионообменной смолой виде, и применяется по тем же показаниям.
Синонимами препарата являются колибар, кванталан, квестран и др.
20.2. Препараты, ингибирующие синтез холестерина
К препаратам этого ряда относятся клофибрат и гемфиброзил, относящиеся к производным феноксикарбоновых кислот — фибраты, пробу кол, являющийся замещенным бис-меркапто-фенолом, а также природные соединение — ловастатин, мевастатин и их аналоги. Поскольку действие всех этих препаратов трудно объяснить единым механизмом, то они будут рассмотрены по отдельности.
Клофибрат (Clofibrate)
Клофибрат — этиловый эфир 2-(4-хлорфенокси)-изо-масляной кислоты (20.2.2) получают этерификацией 2-(4-хлорфенокси)-изо-масляной кислоты (20.2.1) этиловым спиртом. Последнюю, в свою очередь, полу чают одностадийным синтезом взаимодействием 4-хлор-
- 374-
Гиполипидемические средства
фенола, ацетона и хлороформа в присутствии щелочи, очевидно, путем первоначального образования хлорэтона — трихлор-отредг-6} лилового спирта, который в условиях реакции этерифицируется в (4-хлорфенокси)трихлор-дгде«-бутиловый эфир и далее гидролизуется в искомую кислоту' (20.2.1) [6, 7, 8].
СН3 NaOh
У=О + СЧС13 ------------
сн/
С2Н5ОН ‘ н
20 2 2
сн3 о-с-соос2н5
сн3
Это производное арилоксиизомасляной кислоты оказалось наиболее активным из серии соединений, найденных в начале 1960-х годов и понижающих общую концентрацию липидов и холестерина в плазме. Конкретный участок воздействия клофибрата неоднозначен. Быстро и полностью трансформируясь в организме в парахлорфеноксимасля-ную кислоту, он повышает активность липопротеинлипазы, которая внутри сосудов повышает скорость превращения определенного количества ЛПОНП в ЛППП и далее в ЛПНП. Клофибрат непосредственно уменьшает синтез в печени и распределение ЛПОНП.
Клофибрат понижает и концентрацию триглицеридов в плазме. В общем, замечено, что клофибрат снижает уровень холестерина на 5-10 %, а уровень триглицеридов — на 20-25 %. Уровень протеинов высокой плотности при этом не меняется.
Препарат, в основном, применяют при наличии большого количества ЛПОНП.
Синонимами препарата являются мисклерон, аколестол, атеросол, лиситерол, регелан и др.
Гемфиброзил (Gemfibrozil)
Гемфиброзил — 2,2-диметил-5-(2,5-диметилфенокси)валериановую кислоту (20.2.4) получают либо гидролизом этилового эфира 2,2-диметил-5-(2.5-диметилфенокси)валериановой кислоты (20.2.3). по-
-375-
Г лава 20
лучаемого алкилированием этилового эфира 2,2-диметилвалери-ановой кислоты 3-(2.5-диметилфеноксипропилбромидом-1 в присутствии диизопропиламида лития, либо окислением соответствующего альдегида (20.2.4) [9, 10, 11].
СЧ3 (I C3H7)2NLi
Н—С —СООС-Н5 -------------
СНз
СНз -(СН2)з— С-сон
СНз
20 2 S
СНз
О-(СН2)з—С-СООН
СНз
20 2 4
С химической точки зрения в определенном смысле гемфиброзил родственен клофибрату и имеет аналогичное фармакологическое применение. Основное действие гемфиброзила, как и клофибрата, заключается в значительном понижении уровня протеинов очень низкой плотности в плазме. При этом имеет место и небольшое понижение уровня протеинов низкой плотности и увеличение образование ЛПВП.
Препарат применяют при гиперлипопротеинемиях, не корригирующихся специальной диетой и физической нагрузкой
Синонимами препарата являются лопид, иполипид, нормолин и др.
Пробукол (Probucol)
Пробу кол — бис(3,5-ди-дгредг-бутил-4-гидроксифенил)меркаптол ацетона (20.2.6) получают тиокетализанией ацетона 2,Ь-ш\-трет-бутил-4-меркаптофенолом в присутствии хлористого водорода [12, 13, 14, 15. 16].
(СНз)зС
СНз
>=0 сн/
(СН3)3С С{СНз)з
20 2 6
- 376 -
Гиполипидемические средства
Внедренный сравнительно недавно в качестве антигиперхолестере-мического препарата, пробукол химически отличается от других препаратов. Он представляет собой одото-ди-/идедг-бутил, замещенный бис-меркаптофенол. Механизм действия пробукола неясен. Будучи весьма липофильным соединением он легко распределяется в жировую ткань, и в результате около 20 % его максимальной концентрации в крови еще сохраняется в течение 6 месяцев.
Пробукол понижает уровень общего холестерина, главным образом за счет ЛПНП, не воздействуя на триглицериды и ЛПОНП. Высказано предположение, что он ингибирует синтез самого холестерина и усиливает выведение желчных кислот. При его применении уменьшается фракция протеинов низкой плотности, однако еще значительнее уменьшается фракция протеинов большой плотности. С эпидемиологической точки зрения это вредно, поскольку понижение концентрации протеинов большой плотности означает меньшее выведение холестерина из тканей. Однако в любом случае пробукол снижает уровень холестерина в плазме на 10-15 %. Более того показано, что пробукол способствует уменьшению зоны некроза при ишемии миокарда.
Препарат показан при нарушениях липидного обмена, связанных с повышением уровня холестерина и ЛПНП, когда диета и физические нагрузки не оказывают достаточного гипохолестеринемического эффекта.
Синонимами препарата являются бифенабид и лорелко.
Ловастатин (Lovastatin) и Мевастатин (Mevastatin)
Ловастатин — (1S,3R,7S, 8S,8aR)-1,2,3,7,8,8а-гексагидро-3.7-диме-тил-8-[-[2К,4К)-тетрагидро-4-гидрокси-6-оксо-2/7-пиран-2-ил]этил]-1-нафтенил (Б)-2-метилбутират (20.2.7), выделенный из Monascus rubber [17] и Aspergillus terreus [18], а также (lS,3R,7S,8S,8aR)-l,2,3,7,8,8a-reKcaraflpo-7-MeTwi-8-[-[2R, 4R)-TerparH4po-4-rHflpoKCH-6-оксо-2//-пиран-2-ил]этил]-1-нафтенил (8)-2-метилбутират (20.2.8), выделенный из Pemcillium citrinum [19, 20], а также из Penicillium brevicompactum [21].
Ловастатин и мевастатин являются препаратами новой генерации, внедренными в практику для лечения гиперлипохолистеринемии, и представляют из себя уникальные соединения. Ловастатин выделен из грибков Aspergillus terreus. Химически аналогичное соединение — мевастатин, отличающийся лишь отсутствием метильной группы при С3 нафталиновой системы, — выделено из грибков Penicilinium citrinum.
- 377-
Г лава 20
20 2 7
Эти гиполипидемические средства являются конкурентными ингибиторами фермента, лимитирующими скорость синтеза холестерина в печени на стадии левалоновой кислоты.
Препараты считаются наиболее эффективным для снижения уровня холестерина, триглицеридов, ЛПОНП в плазме и умеренно повышают содержание ЛПВП. Препараты применяют при гиперлипопро-теинемиях, не корригирующихся специальной диетой и физической нагрузкой.
Синонимами ловастатина являются ловалип, мевакор, мевинакор, сивлор; мевастатина — CS-500, ML-236 В.
Никотиновая кислота (Nicotinic Acid)
Никотиновая кислота — пиридин-3-карбоновая кислота (20.2.9) в промышленности получается нагреванием под давлением параальдегида — тримера ацетальдегида с аммиаком, приводящим к получению 2-метил-5-этилпиридина, окислением которого азогной кислотой получают искомый продукт [22. 23, 24, 25].
20 2 9
Никотиновая кислота, или витамин РР, в больших дозах понижает уровень холестерина и триглицеридов в плазме. Несмотря на весьма проблематичные побочные эффекты, она имеет долгую и успешную историю в качестве антигиперлипопротеинемического препарата. Причем в этом плане ее применение не связано с применением в качестве
- 378-
Гиполипидемические средства
витамина. Ее действие сводится к понижению выработки протеинов очень низкой плотности, что очевидно связано с ингибированием липолиза в жировых тканях. Важно отметить, что никотиновая кислота не воздействует на общий синтез холестерина в организме. Никотиновая кислота или никотиновая кислота в сочетании с препаратами, удаляющими желчные кислоты, могут снизить уровень холестерина и триглицеридов на 10-30 %.
Препарат применяют при пеллагре (авитаминоз РР), атеросклерозе, заболеваниях печени, язвенной болезни желудка и двенадцатиперстной кишки, длительно не заживающих ранах и язвах.
Синонимами препарата являются никонацид, пернивит, энзикол, ниацин и др.
20.3. Прочие препараты
Декстротироксин (Dextrothyroxine)
Декстротироксин — П-З.З'Дб'-тетрайодтиронин (25.1.10). Синтез этого препарата приведен в гл. 25 «Тиреоидные гормоны и антитиреоидные препараты».
। i
I I
25 1 10
Декстротироксин ускоряет распад холестерина и липопротеидов, активируя катаболизм холестерина в печени, в результате чего холестерин более интенсивно трансформируется в желчные кислоты. Снижает уровень ЛПНП в плазме и ЛПОНП в жировой ткани.
Препарат рекомендован для лечения гиперлипопротеинемии.
Синонимами препарата являются холоксин, лизолипин, натексин, травенон и др.
Неомицин (Neomycin)
Неомицин является комплексом аминогликозидных антибиотиков (неомицин А, неомицин В, неомицин С), вырабатываемых актиномицетом Streptomyces fradiae и родственными микроорганизмами [26,27,28.29,30].
- 379 -
Г лава 20
К примеру неомицин В является 0 2 6 диамино-2,б-дидезокси-a-D-г люкопиранозип-( 1 —>4)-О-[О-2,6-диамино-2 б-дидезокси-P-L идопира-нозил(1—>3)-Р-В-рибофу ранозил-(1—>5)]-2-дезокси-Г)-стрептамином (20 3 1)
Неомицин обладает широким спектром антибактериального дейст вия Эффективен как в отношении ряда грамположительных, так и гра-мотрицательных микроорганизмов Однако он способен связываться с холестерином и желчными кислотами В сочетании с другими препаратами, удаляющими желчные кислоты, или никотиновой кислотой неомицин способен блокировать абсорбцию холестерина и желчных кислот, что значительно понижает уровень холестерина в плазме
Синонимами препарата являются мианин, неоспорин, эндомиксин, кортиспорин, тописпорин и др
Список литературы
1 Ger Pat 1 927 336 (1969)
2 Ger Pat 2 053 585 (1971)
3 US Pat 3 692 895 (1971)
4 US Pat 3 803 237 (1974)
5 US Pat 3 383 281 (1968)
6 US Pat 3 262 850(1966)
7 Bnt Pat 860 303 (1961)
- 380-
Гилолипидемические средства
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
Julia М etal Bull Soc Chim Fr 1956,777
Ger Pat 1 925 423 (1969)
US Pat 3 674 836(1972)
US Pat 4 126 637 (1978)
US Pat 3 862 332 (1975)
Ger Pat 1 767 443 (1968)
US Pat 3 576 883 (1971)
Fr Pat 1 561 853 (1969)
Neuworth M etal/7 Med Chem 13,722 (1970)
Endo A ) J Antibiot 32, 852 (1979)
US Pat 4 231 938(1980)
US Pat 3 983 140(1975)
Ger Pat 2 524 3S5 (1976)
Brovin A et al ' J Chem Soc Perkm Trans I, 1976, 1165
US Pat 2 905 688 (1959)
US Pat 3 983 140(19^5)
Ger Pat 2 256 508 (1972)
NenzA etal Hydrocarbon Process 47,(11) 139 (1968)
US Pat 2 799 620(1957)
US Pat 2 848 365 (1958)
US Pat 3 005 815 (196D
US Pat 3 022 228(1962)
US Pat 3 108 996 (1963)
? Глава 21 t Диуретики
Диуретики — это препараты, увеличивающие выделение из организма избыточной воды и солей, которые аккумулируется в тканях, с мочой. '
Избыточное количество внеклеточной жидкости в организме образуется в результате неспособности почек выводить ионы Na’ с такой скоростью, чтобы вместе с ними выделилось достаточное количество воды. Поэтому эффективность диуретика, в первую очередь, зависит от ею способности выводить из организма ионы Na , поскольку этому сопутствует выведение осмотически эквивалентного количества воды из тканевых жидкостей. Исключение составляют диуретики классифицируемые как ингибиторы карбонангидразы.
Основное терапевтическое применение диуретиков заключается в уменьшении общей отечности, коррекции специфического ионного дисбаланса, уменьшении кровяного давления, уменьшении скорости образования внутриглазной жидкости, уменьшении давления в легочных клиньях.
Диуретики являются препаратами, широко используемыми в медицине при самых различных патологиях и, в первую очередь, для снятия отеков, при лечении гипертензии, при сердечной недостаточности, ги-перкальцийурии. глаукоме и некоторых формах эпилепсии, циррозе печени, нефрозе.
Принципиальный участок действия диуретиков на выделение отечных жидкостей из организма — это нефронные отделы почек. В основном диуретики увеличивают выделение почками воды и солей, путем подавления реабсорбции некоторых основных ионов (в основном, ионов Na+ и СТ), однако, в определенной степени, увеличивается также и выделение ионов К+, Са2’, Mg2’ и гидрокарбонат ионов.
Диуретическим эффектом обладает большое количество химически разнородных соединений. Согласно предполагаемому механизму действия, диуретические препараты классифицируются по 5 группам:
-382-
Диуретики
1) осмотические диуретики (маннитол, глицерин, мочевина, изосорбид);
2) диуретики, подавляющие активность карбонангидразы (ане-1 азоламид, метазоламид, дихлорфенамид);
3) иазиды (хлортиазид, гидрохлортиазид. хлорталидон, индапа-мид и др.);
4) петельные диуретики (буметанид. этакриновая кислота, фуросемид);
5) калийсберегаюшие диуретики (амилорид, спиронолактон, канренон, триаметерен).
21.1. Осмотические диуретики
Осмотические диуретики являются единственной группой соединений, действие которой не связано с взаимодействием с соответствующими рецепторами или прямым блокированием какого-либо почечного транспортного механизма. Фармакологическая активность этой группы зависит исключительно от осмотического давления, вызываемого наличием молекул препарата в растворе. Находясь во внеклеточной жидкости. эти соединения вызывают переход внутриклеточных молекул воды во внеклеточное пространство и препятствуют ее проникновению во внутрь клетки. Найдена линейная зависимость между величиной диуретического эффекта и концентрацией осмотических диуретических препаратов.
Наряду с глицерином, мочевиной и изосорбидом, основным и главным препаратом этой группы является маннитол.
Маннитол (Mannitol)
Маннитол — В(-)-маннит (21.1.1) получают восстановлением D(-)-маннозы [1, 2. 3, 4].
сно сн2он
но-с-н но-с-н
но-с-н 1 Н1» но-с-н н-с-он * н-с-он н-с-он н-с-он
сн2он СН2ОН
21 1 1
Маннитол — наиболее употребляемый сегодня препарат из группы осмотических диуретиков. Повышает осмотическое давление в почечных канальцах, вызывая уменьшение реабсорбции воды в нефронах.
- 383 -
Г лава 21
В результате выделяется большое количество свободной воды, усиливается выведение натрия, как правило без значительного выделения ионов К . Маннитол используется как дополнительное средство для предотвращения олигурии и анурии. Может быть использован и для понижения внутриглазного давления в пре- и постоперационном периоде при офтальмологических процедурах, а также при отеках мозга.
Синонимами препарата являются осмосал, осмитрол, ренитрол и др.
21.2. Диуретики, подавляющие активность карбонангидразы
Препараты данной группы ингибируют активность карбонангидразы — фермента, который катализирует в организме обратимую реакцию взаимодействия воды с двуокисью углерода с образованием угольной кислоты. Механизм действия этой группы препаратов недостаточно ясен. Однако ингибирование активности карбонангидразы приводит к уменьшению образования угольной кислоты и увеличению выделения с мочой бикарбонат ионов и ионов Na+ и К , что в итоге приводит к значительному усилению процесса выведения воды из организма.
Показаниями к применению ингибиторов карбонангидразы являются отеки при легочно-сердечной недостаточности, глаукома (открытоугольная, вторичная и предоперационная узкоугольная главкома), малые эпилептические припадки, предменструальное высокое давление и острая горная болезнь. Полагают, что при глаукоме действие препаратов. возможно, связано с угнетением карбонангидразы ресничного тела и уменьшением секреции водянистой жидкости глаза. При лечении малых форм эпилепсии действие препаратов, возможно, связано с угнетением карбонангидразы мозга, в результате чего уменьшается выделение спинномозговой жидкости.
Из препаратов, подавляющих активность карбонангидразы, на практике применяются ацетазоламид, метазоламид, дихлорфенамид.
Ацетазоламад (Acetazolamide)
Ацетазоламид — 5-ацетамидо-1,3,4-тиадиазол-2-сульфонамид (9.7.5) синтезируется по схеме, приведенной в гл. 9 «Противоэпилептиче-ские средства».
N—N А А CH3CONH SO2NH2
975
-384 -
Диуретики
Ацетазоламид является ароматическим сульфонамидом, используемым в качестве ингибитора карбонангидразы. Препарат способствует выработке щелочной мочи с повышенной концентрацией бикарбонат, Na* и К+ ионов. Ингибируя карбонангидразу, препарат подавляет реабсорбцию ионов Na* в обмен на ионы Н’, усиливает выделение бикарбонат и Na’ ионов и уменьшает выделение ионов СГ. При этом в почках удерживаются ионы СГ для покрытия недостатка бикарбонат ионов и сохранения ионного баланса. Электролитный состав жидкости, выделяемой почками, у пациентов, принимающих ингибиторы карбонангидразы, характеризуется повышенным содержанием ионов Na’ и К+ и бикарбонат ионов и умеренным увеличением количества воды. Моча становится щелочной, и концентрация бикарбонат ионов в плазме понижается.
Ацетазоламид — слабый диуретик с ограниченным применением при отеках, связанных с сердечной недостаточностью, при глаукоме, малых эпилептических припадках, горной болезни.
Синонимами препарата являются мидамор, модамид, цетазол, диа-мокс. диакарб и др.
Метазоламид (Methazolamide)
Метазоламид — ^(4-метил-2-сульфамоил-1,3,4-тиадиазол-2-ин-5-илидеи)ацетамид (21.2.3) получают из промежуточного продукта синтеза ацетазоламида — 2-ацетиламино-5-.меркапто-1,3,4-тиадиазола (9.7.3). Последний бензилируют бензилхлоридом по меркаптогруппе с получением 2-ацетиламино-5-бензилтио-1,3,4-тиадиазола (21.2.1). Дальнейшее метилирование продукта йодистым метилом приводит к образованию М-(4-метил-2-бензилтио-1,3,4-тиадиазол-2-ин-5-илиден)ацетамида (21.2.2). Окислением и одновременным хлорированием последнего в водном растворе уксусной кислоты и взаимодействием полученного при этом хлорсульфопроизводного с аммиаком получают (21.2.3) [5, 6, 7].
0 СбН5СН2С1 $ % /=\ СН3|
21 2 1
Н3С-С—X'
1 Cl2 / CH3COOH / Н2О
2 ИНз
СНз
N-N
21 2 2
21 2 3
- 385-
Глава 21
Действие препарата сходно с таковым ацетазоламида, и он применяется для понижения внутриглазного давления при лечения открытоугольной и вторичной глаукомы, перед хирургическим вмешательством при острой открытоугольной глаукоме.
Синонимом препарата является нептазан и др.
Дахлорфенамид (Dichlorphenamide)
Дихлорфенамид — 4,5-дихлорбензол-1,3-дисульфонамид (21.2.6) получают довольно простым путем, исходящим из 2-хлорфенола. 2-хлорфенол подвергают сульфохлорированию хлорсульфоновой кислотой с получением 4-гидрокси-5-хлорбензол-1,3-дисульфохло-рида (21.2.4). Гидроксильную группу последнего замещают на атом хлора с использованием пятихлористого фосфора с получением 4,5-дихлорбензол-1,3-дисульфохлорида (21.2.5), взаимодействием которого с аммиаком получают искомый дихлорфенамид (21.2.6) [8].
cisOaH
CI
Cl—SOj ——он
ЗО2 ~С1
21 2 4
РС15
21 2 5
NH3
21 2 6
Как и описанные выше препараты, дихлорфенамид применяется для выведения избыточной воды при повышенном внутриглазном давлении и применяется для лечения открыгоугольной и вторичной глаукомы, а также в случаях, когда требуется понизить внутриглазное давление перед хирургическим вмешательством при острой открытоугольной глаукоме.
Синонимом препарата является даранид и др.
21.3. Тиазидные диуретики
Наибольшая в количественном отношении группа диуретиков класса тиазидов структурно родственна антибактериальным препаратам класса сульфонамидов, однако эти соединения в целом не проявляет
- 386 -
Диуретики
антибактериальной активности.
Препараты этой группы представляют собой производные бензотиадиазина, как правило замещенные при С7 бензольного кольца сульфонамидной группой и атомом хлора или другой электроноакцепторной группой (трифторметильной) при С6. Гидрогенизованное тиадиазиновое кольцо бензотиадиазиновой системы допускает введение разнообразных заместителей в положение С3, что позволило создать значительное число препаратов и сделать определенные корреляции между структурой и активностью в этом ряду соединений. В частности установлено, что в большинстве случаев, восстановление двойной связи при С3-С4 препаратов ряда тиазидов увеличивает их диуретическую активность.
Точный механизм действия неизвестен, однако качественно тиазидные диуретики действуют одинаково, и их отличия в целом носят количественный характер.
Участком воздействия тиазидных диуретиков являются дистальные канальцы нефронов. Препараты этой группы ингибируют реабсорбцию ионов Na' и СГ, а также К* и Mg2+ и вызывают усиленное их выведение из организма вместе с осмотически эквивалентным количеством воды. Тиазиды эффективны и при ацидозе или алкалозе, ингибируют карбонангидразу in vitro и понижают АД у гипертензивных пациентов. Антигипертензивный эффект тиазидов может быть объяснен их диуретическим действием, г. е. уменьшением объема циркулирующей крови. Не исключено также, что антигипертензивное действие тиазидов происходит вследствие их спазмолитического действия на стенки сосудов, возможно, в результате изменения содержания ионов Na+ внутри мышечных волокон. Под действием тиазидов меняется и реактивность сосудистой системы, снижаются прессорные реакции на сосудосуживающие вещества (адреналин и др.). Большинство побочных действий тиазидов бывает связано с гипотензией или с нарушениями электролитного баланса, например I ипонатриемия, гипокалиемия или гипомагнеземия. Примерно у 1 % пациентов, пользующихся тиазидными диуретиками, возникает кожная сыпь. Тиазиды используются для лечения гипертензии либо самостоятельно, либо в сочетании с другими антигипертензивными препаратами.
На практике наиболее часто применяются гидрохлортиазид, хлор-тиазид, бентиазид. бендрофлуметиазид, гидрофлуметиазид, политиазид, трихлометиазид. а также родственные тиазидам, но таковыми не являющиеся, хлорталидон, метолазон и индапамид.
- 387-
Глава 21
Хлортиазид (Chlorthiazide)
Хлортиазид — 1.1-диоксид 6-хлор-2Я-1.2.4-бензотиадиазин-7-суль-фонамида (21.3.3), как и все тиазидные диуретики, получают примерно по одной и той же схеме. 3-хлоранилин (либо 3-трифторме-тиланилин) подвергают сульфохлорированию хлорсульфоновой кислотой с получением 4,6-сульфохлоридо-З-хлоранилина (21.3.1), взаимодействием которого с аммиаком получают 4,6-сульфонами-до-3-хлоранилин (21.3.2). Нагревание последнего с формамидом приводит к получению хлортиазида (21.3.3) [9, 10, 11].
2 CISO3H
2 NH3
HCONH2
H2NSo2 x45sszJ4'so2nh2
21 3 2
Cl
H2NSO2
Диуретическое действие хлортиазида. как и других препаратов этого ряда, обусловлено уменьшением реабсорбции ионов Na" и СГ почками при одновременном усиленном их выведении из организма.
Препарат оказывает сильное диуретическое действие как при ацидозе, так и при алкалозе. Применяют при артериальной гипертонии, при отечных синдромах различного генеза, застойных явлениях при сердечно-сосудистой недостаточности, нефрозах и нефритах, токсикозах. Особенно рекомендуется применение препарата при гипертонической болезни. В ряде случаев препарат понижает и внутриглазное давление.
Синонимами препарата являются клотрид, диупрес, диурил и др.
Гидрохлортиазид (Hydrochlorthiazide)
Гидрохлортиазид — 1,1-диоксид 6-хлор-3.4~дигидро-2Н-1,2,4-бен-зотиадиазин-7-сульфонамида (21.3.4) получают либо циклизацией 4,б-сульфонамидо-3-хлоранилина (21.3.2) с использованием пара-форма, при котором одновременно происходит восстановление двойной связи в положении С^-С4. либо препарат получают восстановлением этой же двойной связи в хлортиазиде (21.3.3) формальдегидом. Эго небольшое изменение в структуре повышает актив-
-388-
ность препарата по сравнению с хлорзиазидом, улучшает его абсорбируемость при оральном применении [12, 13, 14. 15, 16, 17].
H2NSO2
21 32
NH2
so2nh2
(Ch2O)n
СН2О
CI
H2NSO2
Гидрохлортиазид является одним из самых широко используемых препаратов этого ряда и применяется по тем же показаниям, что и хлортиазид. Гидрохлортиазид вызывает меньшее ингибирование карбонан-гидразы, но вызывает в 5-Ю раз больший диурез ионов Na+, чем хлортиазид, в одной и той же дозе.
Синонимами препарата являются хлорзид, диаква, эсидрикс, гидро-диурил, гидрозид, гипотиазид, новогидразид, урозид и др.
Бендрофлуметиазид (Bendroflum ethiazide)
Бендрофлуметиазид — 1,1-диоксид 3-бензил-6-(трифторметил)-3,4-дигидро-2//-1,2,4-бензотиадиазин-7-сульфонамида (21.3.6) получают по совершенно аналогичной схеме получения вышеописанных препаратов, используя в качестве карбонильного компонента фенилуксусный альдегид или его ацеталь, а в качестве о-аминосуль-фонамидного компонента 2,4-дисульфонамидо-5-трифторметилани-лин (21.3.5) [18, 19. 20,21].
f3c
h2nso2
21 3 5
NH2
SO2NH2
Бендрофлуметиазид может применяться по тем же показаниям, что и вышеописанные препараты, однако в основном он показан в качестве вспомогательного средства для снятия отеков, связанных с сердечной недостаточностью, циррозом печени, и при отеках, вызванных применением кортикостероидов.
Синонимами препарата являются синесалин, доцидразин, тенсио-норм, апринокс, натуретин и др.
-389-
Глава 21
Политиазид (Polythiazide)
Политиазид — 1,1-диоксид 2-метил-3-(2,2,2-трифторэтилтиометил)-6-хлоро-3,4-дигидро-2Я-1,2,4-бензогиадиазин-7-сульфонамида (21.3.8) также получают по аналогичной схеме, конденсируя 4-аминосуль-фонил-5-хлоро-2-метиламиносульфониланилин (21.3.7) с диметилацеталем 2.2,2-трифторэтилтиоацетальдегида [22].
h2nso2 •^5:x>^so2nhch3
21 3 7
СН3О
)сН~СН2—SCH2CF3 сн3о
н
F3C ^,СН2 -SCH2CF3
.XsJL ,N-CH3
H2NSO2 s
02
21 3 8
Политиазид выявляет более выраженный антигипертензивный эффект, чем политиазид, и самостоятельно может применяться по тем же показаниям, что и вышеописанные препараты. Однако его в основном применяют в составе комбинированных лекарств, предназначенных для понижения давления, в частности, минизида, представляющего собой комбинацию празозина и политиазида.
Синонимами препарата являются дренусил, нефрил, ренис и др.
Число диуретиков ряда тиазидов весьма значительно и не исчерпывается вышеприведенными. К широко используемым препаратам относятся гидрофлуметазид, трихлорметазид, метилциклотиазид, циклотиазид, бензотиазид, диазоксид и др., методы синтеза которых и фармакологическое действие которых практически аналогичны вышеприведенным.
Гидрофлу.иетиазид (Hydroflumethiazid)
Гидрофлуметиазид — 1,1-диоксид 3,4-дигидро-6-трифторметил-2Я-
1,2,4-бензотиадиазин-7-сульфонамида (21.3.9) [21, 23, 24, 25].
F3C
H2NSO2
Синонимами препарата являются леодрин, диуритенс, гидренокс, риосил, ронтил, диукардин, салурон, салутензин и др.
-390-
Диуретики
Трихлорметиазид (Trichlormethiazid)
Трихлорметиазид — 1,1-диоксид 3,4-дигидро-3-(дихлорметил)-6-хлор-2/7-1,2,4-бензотиадиазин-7-сульфонамида (21.3.10) [26. 27, 28, 29.30,31].
N^-CHClj
h2nso2
4^24 ,N-H
О2
21.3 10
Синонимами препарата являются эсмарин, эсмалорид. анатран, кар-вакрон, интромен, санамирон, метагидрин, наква, триазид и др.
Менипииклотиазид (Methylcyclothiazid)
Метилциклотиазид — 1,1-диоксид 3,4-дигидро-3-(хлорметил)-
6-.хлор-2/7-1,2.4-бензотиадиазин-7-сульфонамида (21.3.11) [23].
н
.N ^снг-о
H2NSO2
21 3 11
,N-CH3
°2
Синонимами препарата являются тиазидил, эндурон и др.
Циклотиазид (Cyclothiazid)
Циклотиазид — 1,1-диоксид 3,4-дигидро-3-(5-норборнен-2-ил)-6-хлор-2Н-1,2.4-бензотиадиазин-7-сульфонамида (21.3.12) [32, 33,
Синонимами препарата являются диампрес, циклотериам, ангидрон и др.
- 391 -
Глава 21
Бензтиазид (Benzthiazid)
Бензтиазид — 1,1-диоксид 3,4-дигидро-3-(бензилтиометил)-б-хлор-2//-1,2,4-бензотиадиазин-7-сульфонамида (21.3.13) [35, 36, 37].
Синонимами препарата являются дитериам, дитид, регулон, аква-таг, гидрекс и др.
Диазоксид (Diazoxid)
Диазоксид — 1,1 -диоксид 3-метил-7-хлор-2Я-1,2,4-бензотиадиазина (21.3.14) [38,39, 40].
21 3 14
Синонимами препарата являются гипертоланум, гиперстат, эуде-мин, проглицем и др.
Препараты, родственные тиазидным диуретикам
Диуретические препараты — метолазон, хлорталидон и индапа-мид— относятся к диуретикам и антигипертензивным препаратам. Химически они не относятся к тиазидам, однако являются сульфонамидными производными и имеют, в определенном смысле, структурное сходство и сходный механизм действия, обычный для тиазидов, за исключением того, что они не ингибируют карбонангидразу. Поэтому, чисто формально, они рассматриваются в одной группе с тиазидными диуретиками.
Способность метолазона, хлорталидона и индапамида удалять из организма отечные жидкости практически одинакова с тиазидными диуретиками. Эти препараты применяются для снятия отеков, связанных с печеночными, почечными или сердечными заболеваниями, а также для лечения общей гипертензии либо самостоятельно, либо в сочетании с другими препаратами.
- 392-
Диуретики
Метолазон (Metolazone)
Метолазон — 7-хлоро-1,2,3,4-тетрагидро-2-метил-4-оксо-3-о-толил-6-хиназолинсульфонамид (21.3.20) получают исходя из 5-хлор-2-мегиланилина. Аминогруппу последнего ацилируют хлоругольным эфиром с получением 5-хлор-1\’-этоксикарбонил-2-метиланилина (21.3.15). Далее продукт обычным путем, т. е. последовательным взаимодействием с хлорсульфоновой кислотой и аммиаком, трансформируют в 4-сульфонамидо-5-хлор-М-этоксикарбонил-2-метил-анилин (21.3.16). Метильную группу' последнего окисляют перманганатом калия с получением 5-сульфона.мидо-4-.\лор-,\'-этокси-карбонил антраниловой кислоты (21.3.17). Обработкой хлористым тионилом последняя циклизуется в соответствующий ангидрид (21.3.18). Действием о-толуидина последний превращается в 2-ами-но-5-аминосульфонил-4-хлор-о-толуолбензамид (21.3.19). Наконец, взаимодействием последнего с диметилацеталем уксусного ангидрида получают метолазон (21.3.20) [41, 42, 43, 44, 45, 46, 47].
С1СООСгН5
21 3 16
NH -СООСгНд
соон
21 3 17
21 3 18
Н3С -СН(ОСН3)2
Метолазон действует на дистальные канальцы, увеличивая выделение воды, ионов Na*, К и СГ. Препарат показан для лечения отеков, вызванных сердечной недостаточностью и почечными нарушениями, включая нефротический синдром.
Синонимами препарата являются диуло, матеникс, зароксолин.
- 393-
Глава 21
Хлорталидон (Chlortalidone)
Хлорган идон — 2-хлор-5-(1-гидрокси-3-оксо-1-изоиндолинил)-бензолсульфамид (21.3.26) предложено получать разными путями, исходящими из 2'-карбокси-4-хлорбензофенона (21.3.21), который легко получается при ацилировании хлорбензола фталевым ангидридом в присутствии хлористого алюминия. Согласно первому способу, полученный бензофенон (21.3.21) подвергают нитрованию азотной кислотой с получением 2'-карбокси-3-нитро-4-хлорбен-зофенона (21.3.22). Нитрогруппу в полученном соединении восстанавливают двухлористым оловом до 2'-карбокси-3-амино-4-хлор-бензофенона (21,3.23). Далее последовательным диазотированием и взаимодействием с двуокисью серы в присутствии двухлористой меди получают соответствующий сульфохлорид (21.3.24). Под действием хлористого тионила последний циклизуется во фталид (21.3.25), который под действием водного аммиака перегруппировывается в производное изоиндолина с одновременным замещением атома хлора в сульфогруппе на аминогруппу, в результате чего получается хлорталидон (21.3.26) [48, 49].
1 HNO2
2 SO2 I СН3СООН / CuCi2
SOCL2
Второй способ синтеза также исходит из 21 2-карбокси-4-хлорбензофенона (21.3.21), который при восстановлении цинком в уксусной кислоте трансформируется в 3-(4'-хлорфен ил) фтал ид (21.3.27). Сульфохлорированием последнего получают соответствующий сульфо-
- 394 -
Диуретики
хлорид (20.3.28), который под действием пятихлористого фосфора хлорируется в 3-(4|-хлорфенил-3|-хлорсульфо)-3-хлорфталид (21.3.25). Последний вышеописанным способом под действием водного аммиака перегруппировывается в хлорталидон (21.3.26) [50].
Zn, СН3СООН
NH3
Третий способ синтеза, исходящий также из 2'-карбокси-4-хлор-бензофенона (21.3.21), заключается в непосредственной циклизации > казанного карбоксибензофенона в 3-(4'-хлорфенил)фталимидин (21.3.29). Последовательным сульфохлорированием и аминированием последнего получают 2-хлор-5-(3-оксо-1-изоиндолинил)-бензолсульфамид (21.3.29), который различными окислителями, такими как кислород либо перекись водорода в щелочи, или хромовая кислота, в уксусной кислоте окисляют в хлорталидон (21.3.26) [51, 52].
- 395 -
Гпава 21
По активности хлорталидон весьма близок к бензотиадиазиду (21.3.13) и показан в качестве самостоятельного препарата или в комбинации с другими антигипертензивными средствами для понижения АД, а также в качестве вспомогательного средства для лечения отеков, вызванных сердечной недостаточностью и почечными нарушениями, включая нефротический синдром.
Синонимами препарата являются гигротон, новаталидон, уридон и др.
Индапамид (Indapamide)
Индапамид — 4-хлоро-1Ч-(2-метил-1-индолинил)-3-сульфамоил-бензамид (21.3 33) синтезируют исходя из 2-метилиндолина, нитро-зированием которого получают 2-метил-1-нитрозоиндолин (21.3.31). Восстановление последнего алюмогидридом лития приводит к получению 1-амино-2-метилиндолина (21.3.32). Ацилирование последнего хлорангидридом З-сульфонил-амино-4-хлорбензойной кислоты приводит к (21 3.33) [53, 54].
CHj
NaNO2
21 3 33
Индапамид является производным бензолсульфонамидов, и его механизм действия аналогичен таковому тиазидов. Препарат преднагначен для понижения АД, а также в качестве вспомогательного средства для лечения отеков, вызванных сердечной недостаточностью.
Синонимом препарата является лозол и др.
21.4. Петельные диуретики
Наиболее мощный диуретический эффект оказывают петельные диуретики, которые резко ингибируют реабсорбцию ионов \а и СГ из почечных канальцев в корковых и мозговых частях и восходящего уча
-396-
Диуретики
стка петли нефрона. Как при оральном, так и при внутривенном введении они вызывают быстрое увеличение выделения из почек ионов Na" и СГ и увеличение объема выделяемой мочи.
Одновременно с увеличением выделением с мочой ионов Na' и СГ наблюдается увеличение выделения ионов К , FT, Mg2' и Са2+. Кислотность мочи при этом повышается, концентрация ионов аммония падает. При этом может возникнуть гипохлоремический алкалоз. Наиболее широко применяемыми петельными диуретиками являются буметанид (производное моносульфамоил метаниламида), этакриновая кислота (производное арилоксиуксусной кислоты) и фуросемид (производное моносульфамоилантраниловой кислоты), которые имеют большую диуретическую эффективность, чем тиазиды. Буметанид, этакриновая кислота и фуросемид используются при лечении отеков, связанных с острой и хронической сердечной недостаточностью, цирроза печени, нефротическом синдроме и почечных заболеваниях. Они часто применяются у пациентов, не переносящих тиазиды. Их используют также при лечении хронической гипертензии как самостоятельно, так и в сочетании с другими антигипертензивными препаратами. Эффективность и безопасность буметанида и этакриновой кислоты при хронической ышертензии не доказана. Петельные диуретики эффективны при лечении острой гиперкалиемии.
Буметанид (Bumetanide)
Буметанид — 3-бутиламино-4-фенокси-5-сульфамоилбензойную кислоту (21.4.6) синтезируют исходя из 4-хлорбензойной кислоты. На первой стадии синтеза ее подвергают сульфохлорированию хлорсульфоновой кислотой с получением 4-хлор-З-хлорсульфо-нилбензойной кислоты (21.4.1), которая далее нитруется азотной кислотой в 4-хлор-3-хлорсульфонил-5-нитробензойную кислоту (21.4.2). Взаимодействием последней с аммиаком получают 5-ами-носульфонил-4-хлор-З-нитробензойную кислоту (21.4.3), которая взаимодействием с фенолятом натрия трансформируется в 5-ами-носульфонил-З-нитро-5-феноксибензойную кислоту (21.4.4). Восстановление нитрогруппы в последней водородом с использованием в качестве катализатора палладия на угле получают 3-амино-5-аминосульфонил-5-феноксибензойную кислоту (21.4.5). Наконец взаимодействием последней с бутиловым спиртом в присутствии серной кислоты получают искомый буметанид (21.4.6) [55, 56. 57, 58, 59].
-397-
Глава 21
С4Н9ОН / H2SO4
0,>N
C4H9NH
21 4 6
Буметанид применяют для снятия отеков, связанных с сердечной недостаточностью, при болезнях печени и почек, включая нефротический синдром, при асцитах, гипертензии.
Синонимом препарата является бумекс и др.
Этакриновая кислота (Ethacrynic acid)
Этакриновая кислота — [2,3-дихлоро-4-(2-метиленбутирил)фено-кси]уксусн\ю кислоту (21,4.9) получают исходя из 2,3-дихлор-феноксиуксусной кислоты. Последнюю ацилируют хлорангидридом масляной кислоты с получением 4-бутироил-2,3-дихлорфенокси-уксусной кислоты (21.4,7), которую далее аминометилируют в условиях реакции Манниха с использованием диметиламина и формальдегида. Полученный продукт (21.4.8) далее подвергается термическому расщеплению с образованием ненасыщенного кетона — этакриновой кислоты (21.4.9) [60, 61, 62].
А1С!з
О /=4 СН2О/iCrl3)2NH
С2Н£-СИ2'-С-4 \-о~сн2-соон -------------------’
С) С! 21 4 7
Н О i и с2н5-с-с сн2 I
о-сн2-соон
21 4 8
Эгакриновая кислота является сильным диуретиком, который назначают при отеках, связанных с сердечной недостаточностью, при оте
-398-
диуретики
ках почечного происхождения, не поддающихся действию других диуретиков, при отеках мозга и легких.
Синонимами препарата являются урегит, эдекрин и др.
Фуросемид (Furosemide)
Фуросемид — 4-хлоро-М-фурфурил-5-сульфамоилантраниловая кислота (21.4.11) получается довольно-таки простым синтезом, исходящим из 2,4-дихлорбензойной кислоты, которую последовательным взаимодействием с хлорсульфоновой кислотой и аммиаком переводят в 5-аминосульфонил-4,6-дихлорбензойную кислоту (21.4.10). Взаимодействие последней с фурфуриламином приводит к фуросемиду (21.4.11) [63, 64, 65, 66].
! CISOjH
2 NH3
21 4 10
HjN -СН2
Фуросемид — высокоэффективный и быстродействующий диуретик, действие которого, как и всех рассмотренных петельных диуретиков. связано с блокадой реабсорбции ионов в восходящем колене петли нефрона. Препарат применяют при отечном синдроме различного генеза, при отеках легких, мозга, хронической почечной недостаточности, некоторых формах гипертонического криза, при отравлениях барбитуратами и другими соединениями, выделяющимися преимущественно с мочой.
В ряде случаев фуросемид оказывается более эффективным, чем дру гие диуретики. Кроме диуретического эффекта он также расширяет периферические сосуды. Часто назначается в комбинации с другими антигипертензивными препаратами.
Синонимами препарата являются лазикс, лазизикс. франил, уросе-мид и многие другие.
21.5. Калийсберегающие диуретики
Калийсберегающие диуретики отличаются от других диуретиков тем, что увеличивают выведение из организма ионов Na+ и одновременно уменьшают выведение ионов К\ В общем, при использовании в качестве самостоятельных средств, препараты этого класса не являются мощными диуретиками, и специально их применяют лишь в случаях гиперкалиемии. В основном их используют в комбинации с другими диуретиками с целью увеличения диуреза и предупреждения развития
-399-
Глава 21
гипокалиемии. Ввиду совершенно различного строения и наличия специфических особенностей свойства препаратов этого ряда (спиронолактон, гриамтрен и амилорид) будут рассмотрены по отдельности.
Спиронолактон (Spironolactone)
Спиронолактон — 7-ацетат у-лактона 17-гидрокси-7-меркато-3-оксо-17-а-прегн-4-ен-21-карбоновой кислоты (21.5.8). Спиронолактон в промышленности получают двумя путями, исходящими из андростенолона — Зр-гидрокси-5-андростен-17-она. Согласно первому способу, андростенолон подвергают этинилированию ацетиленом по Норману с использованием амида натрия в жидком аммиаке с получением 17а-этинил-ЗР-,17Р-дигидрокси-5-андростена (21.5.1). Последовательным взаимодействием последнего с метил-магнийбромидом и двуокисью углерода получают соответствующую пропиоловую кислоту (21.5.2). Восстановлением тройной связи в последней водородом с использованием в качестве катализатора палладия на карбонате кальция получают соответствующее производное акриловой кислоты (21.5.3), которую без выделения обрабатывают кислотой, что приводит к циклизации в производное непредельного лактона (21.5.4). Двойную связь в последнем восстанавливают водородом с использованием в данном случае в качестве катализатора палладия на угле. Полученный при этом предельный лактон (21.5.5) подвергают окислению по Оппенауеру, в непредельное кетопроизводное — 4-андростен-3,17-дион (21.5.6). В результате дальнейшего окисления продукта (21.5.6) хлоранилом получают диенон (21.5.7), присоединением тиоуксусноп кислоты к которому получают искомый спиронолактон (21.5.8) [67, 68, 69, 70, 71].
-400-
Диуретики
Второй способ синтеза исходит из 4-андростен-З,17-диона (21.5.6), который подвергают этинилированию пропаргиловым спиртом в присутствии треш-бутилата калия с получением 17р-гидрокси-17а-(3-гид-роксипропинил)-4-андростен-3-он (21.5.9), тройную связь в котором полностью восстанавливают водородом с использованием в качестве катализатора комплекса трифенилфосфина с хлористым родием с получением 17р-гидрокси-17а-(3-гидроксипропил)-4-андростен-3-она (21.5.10). Окислением последнего оксидом шестивалентного хрома в пиридине получают лактон (21.5.6), который аналогично описанному выше окисляют хлоранилом в (21.5.9) и далее вводят во взаимодействие с тиоук-сусной кислотой с получением искомого спиронолактона (21.5.8) [72].
Спиронолактон является калийсберегающим диуретиком, отличающимся по механизму действия от других препаратов этого класса.
Препарат является конкурентным антагонистом альдостерона и его действие наиболее эффективно, когда уровень циркулирующего в организме альдостерона высок.
-401 -
Глава 21
он СОСН2ОН о--- /
альдостерон
Альдостерон является минералокортикостероидом, участвующим в регуляции электролитного баланса в организме. Альдостерон уменьшает выведение из организма ионов Na+, увеличивая их реабсорбцию, и увеличивает секрецию ионов IC в почечных канальцах. Являясь конкурентным антагонистом альдостерона, спиронолактон, блокирует рецепторы альдостерона и тем самым повышает выведение с мочой ионов Na+, СГ и соответствующего эквивалентного количества воды, сохраняя при этом количество ионов К* в организме.
Спиронолактон используегся как индивидуально, так и в сочетании с тиазидами, поскольку уменьшает калийурез, вызванный тиазидовыми диуретиками. Препарат применяется при отечном синдроме, вызванном хронической сердечной недостаточностью, циррозе печени, гиперальдостеронизме, гипокалиемии, вызванной другими диуретиками.
Синонимами препарата являются альдактон, верошпирон и др.
Триамтерен (Triamterene)
Триамтерен— 2,4,7-триамино-6-фенилптеридин (21.5.13) получают по следующей схеме. Взаимодействием гуанидина с динитрилом малоновой кислоты получают 2,4,6-триаминопиримидин (21.5.11). Последний подвергают нитрозированию взаимодействием с азотистой кислотой, в результате чего получают 5-нитрозо-2,4,б-триами-нопиримидин (21.5.12), который конденсацией с бензилцианидом в присутствии метилата натрия циклизуется в триамтерен (21.5.13) [73, 74’ 75, 76].
NH
х h2n nh2
NEC, ,CEN
CH3ONa HNq2 НгМ^ЛЧ^Д'Щг
NH2
21 5 11
NH2
21 S 12
-402-
диуретики
2 CH3ONa
21 5 13
Триамтерен является производным пиразина, ингибирующим реабсорбцию ионов Nar без увеличения выделения ионов КТ Препарат проявляет примерно такой же результирующий эффект, как и спиронолактон, однако действует не по механизму конкурентного связывания с альдостероновыми рецепторами. Его действие не связано с влиянием на секрецию альдостерона или антагонизма к нему, а является результатом прямого воздействия на почечные канальцы.
Этот калийсберегающий диуретик вызывает умеренное увеличение выделения ионов Na и бикарбонат ионов в мочу и понижает выделение ионов К+ и аммония. Мало влияет на объем мочи.
Препарат рекомендуется в сочетании с другими диуретиками для лечения отеков, вызываемых обычными причинами обусловленных не-дос 1 аточностыо кровообращения, циррозом печени, нефротическим синдромом.
Синонимами препарата являются диазид, ревитен, птерофен и др.
Амилорид (Amyloride)
Амилорид — М-амидино-3,5-диамино-6-хлорпиразинкарбоксамид (21.5.18) синтезируют исходя из 5,6-диаминоурацила, который взаимодействием с глиоксалем трансформируют в производное пи-разинопиримидана (21.5.14), и далее действием сильной щелочи расщепляют с получением З-аминопиразин-2-карбоновой кислоты (21.5.15). Последнюю этерифипируют в соответствующий метиловый эфир (21.5.16) и далее последовательно обрабатывают хлористым сульфурилом и аммиаком с получением метилового эфира 3,5-диамино-6-хлорппразин-2-карбоновой кислоты (21.5.17). Взаимодействие последней с гуанидином приводит к амилориду (20.5.18) [77.78. 79, 80, 81, 82].
yHG оно
21 S 14 ОН
ьаон ^оон СНзОН/н +
21 5 15
-403 -
Глава 21
СООСНз
nh2
21 5 16
SO2CI2
2
СООСНз
NH2
21 5 17
NH
х
nh2
21 5 18
Ам и ториц также является калийсберегающим диуретиком, проявляющим умеренную активность Не является антагонистом альдостерона Он ингибирует реабсорбцию ионов Na+ и уменьшает выведение ионов К Амилорид редко используется индивидуально, а как правило, в сочетании с тиазидами или петельными диуретиками Используется, в основном, в сочетании с тиазидными диуретиками при сердечной недостаточности и гипертензии, особенно в тех случаях, когда необходимо предотвращение гипокалиемии
Синонимами препарата являются арумил, диурсан, модамид, фру-мит и др
Список литературы
1 USPat 2.642 462 (1953)
2 USPat 2 749 371 (1956)
3 LSPat 2 759 024(195)
4 Маккее М et al / Chem Commun 1980, 930
5 Young J etal/J Am Chem Soc 78,4649 (1956)
6 USPat 2 783 241 (1957)
т Paia M ' Farmaco Ed Sci 13,650(1958)
8 USPat 2 835 702 (1958)
9 USPat 2 809 194 (1957)
10 USPat 2 937 169(1960)
11 Novella EH Am Chem Soc 79, 2028 (1957)
12 USPat 3 163 645 (1964)
13 USPat 3 164 588 (1965)
14 USPat 3 025 292 (1962)
15 I S Pat 3 043 840(1962)
16 Siemens de G etal Expenentia 14,463 (1958)
17 Ger Pat 1 163 332 <1964)
18 US Pat 3 265 573 (1966)
19 LS Pat 3 392 168 (1968)
20 Bnt Pat 863 474(1961)
-404-
Диуретики
21
22
23
24
25
26
27
28
29
30
31
22
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
Holarege С et al / J Am Chem Soc 81. 4807 (1959) USPat 3 009 911 (1961)
Close W etal/ J Am Chem Soc 82. 1132 (1960) }aleR et al '-'J Am Chem Soc 82. 2042 (1960) Novello E et al "J Org Chem 25, 970 (1960) Brit Pat 949 373 (1960)
Ger Pat 1 147 233 (1960)
Stevens de G et al lExperiemia 16, 113 (1960) Sherlock M et al /'Expenentia 16. 184 (1960) Bnt Pat 954 023 (1960)
USPat 3 264 292(1960)
LSPat 3 275 625 (1966)
Bnt Pat 915 236(1961)
Whitehead C etal Org Chem 26, 2814 (1961)
USPat 3 111 517 (1963)
USPat 3 440 244(1969)
Ger Pat 1 137 740(1962)
USPat 3 345 365 (1967)
USPat 2 986 573 (1961)
Raffa 1 etal Farmaco Ed Set 17,244 (1962)
USPat 3 360 518 (1967)
USPat 3 357 111 (1971)
USPat 3 761 480(1972)
Ger Pat 1 620 740(1966)
Ger Pat 2 131 622 (1971)
Ger Pat 2 035 657(1970)
Shetty В et al 7J Med Chem 13, 886 (1970)
USPat 3 055 904(1962)
GrafW et al /'Heh Chim Acta 42. 1085 (1959) USPat 4 188 330(1980)
Eur Pat Appl 51215(1981)
Eur Pat Appl 51 217(1981)
USPat 3 565 911 (1971)
Fr Pat 2 003 311 (1969)
Ger Pat 1 964 503 (1970)
Ger Pat 1 964 504 (1970)
USPat 3 806 534(1974)
US Pat 3 634 583 (1972)
FeitP etal /J Med Chem. 14. 432 (1971)
USPat 3 255 241 (1966)
Belg Pat 612 755 (1962)
-405-
Глава 21
62 Schultz Е et ai. 'J. Med. Pharm. Chem. 5, 660 (1962).
63. US Pat. 3.058.882 (1962).
64. Ger. Pat. 1.122.541 (1962).
65. Ger. Pat. 1.213.846(1970).
66. Sturm K. et al."Chem. Ber. 99, 328 (1966).
67 US Pat. 3.013.012 (1961).
68. Ger. Pat. 1.121.610 (1959).
69 USPat. 3.137.690 (1964)
70. Celia J. et al,/J. Org. Chem. 24, 1109 (1959).
71. Dodson R et al.AJ. Am. Chem. Soc. 81, 1224 (1959).
72. Ger. Pat. 2.327.448 (1973).
73. US Pat. 3.081.230 (1963).
74. Spickett R. et al.'/J. Chem. Soc. 1954, 2887.
75. Pachterl//!. Org. Chem. 28, 1191 (1963).
76. Os deneM et al. /J. Med. Chem. 10, 431 (1967).
77. US Pat. 3.313.813 (1967).
78. Belg. Pat. 639.386(1963).
79. Brit. Pat. 1.066.855 (1963).
80. Bicking J et al., /J. Med. Chem. 8, 638 (1965).
81. Cragoe E et al.,'J. Med. Chem. И), 66 (1967).
82. Smith R. et al. 'J. Am. Chem. Soc. 101. 191 (1979).
I Глава 22
} Антигипертензивные i препараты
л
Препараты, применяемые для лечения гипертонической болезни, а также симптоматических гипертоний, называются антигипертензивными препаратами.
Гипертензия — синдром, характеризуемый повышенным АД, которое зависит от множества факторов. В число основных факторов, определяющих уровень АД, входят параметры работы сердца и сосудов, а также обьем, вязкость и электролитный состав циркулирующей крови.
Нормальная величина давления варьирует в зависимости от пола и возраста. Более того, различные медицинские школы по-разному определяют само значение этой величины. Этиология 90-95 % случаев этого заболевания неизвестна, и эти случаи называют первичной или эссенциальной гипертензией, а их лечение носит паллиативный характер и направлено на снижение систолического или диастолического давления и, в общем, достаточно эффективно позволяет контролировать АД пациента в течение длительного времени. При этом действие антигипертензивных веществ может быть направлено на различные звенья физиологической системы, регулирующей АД.
Остальные 5-10 % случаев гипертензии возникают по причине стеноза почечных артерий или сужения аорты, синдрома Кушинга и феохромоцитомы. Гипертензия, возникшая вследствие указанных причин, называется вторичной гипертензией.
Основными участками, контролирующими АД организма, являются ЦНС. симпатические ганглии, адренергические нервные окончания, ).задкая мускулатура сосудов, почки и артериолы и. наконец, ренин-ангиотензиновая система.
С целью снижения АД можно воздействовать на гладкую мускулатуру сосудов гидралазином, диазоксидом, миноксидилом, нитропруссидом натрия, диуретиками, блокаторами кальциевых каналов, которые расслабляют гладкую мускулатуру сосудов и тем самым понижают и систолическое, и диастолическое давление.
-407-
Глава 22
I fa автономные ганглии с целью снижения давления можно воздействовать н-холиноблокаторами (ганглиоблокаторами), мекамиламином и триметафаном.
С целью снижения АД путем воздействия на адренергическую систему можно:
— стимулировать а-адренорецепторы (клонидин, гуанабенз, гу-аифацин, метилдопа), что приведет к уменьшению симпатической импульсации к сосудам и сердцу и в итоге уменьшит сердечный выброс и скорость сердца, а следовательно, снизит АД;
— блокировать а i-адренорецепторы (празозин, тетразозин), наиболее существенным эффектом чего явится расширение периферических сосудов, что приведет к снижению давления;
— блокировать Р-адренорецепторы (пропранолол, атенолол, надолол и др.), что уменьшит сердечный выброс и периферическое сопротивление сосудов, результатом чего также станет понижение давления.
С той же целью снижения давления можно воздействовать на ренин-ангиотензиновую систему ингибиторами ангиотензин-превра-щающего фермента (АПФ) — каптоприл, эналаприл. Эти препараты блокирует работу АПФ, результатом чего подавляется образование ангиотензина II и устраняется его сосудосуживающее действие на артериальные и венозные сосуды. На почки и артериолы с целью снижения давления можно воздействовать диуретиками. Наконец, с целью снижения давления можно воздействовать на гладкую мускулатуру сосудов блокаторами кальциевых каналов (верапамил, дилтиазем, нифедипин).
Основываясь на механизме действия, антигипертензивные препараты могут быть разделены на 8 классов:
• диуретики.
• Р-адреноблокаторы.
• центрально действующие симпатолитики,
• периферически действующие симпатолитики,
• блокаторы кальциевых каналов,
• миотропные гипотензивные препараты,
• ингибиторы АПФ,
• активаторы калиевых каналов
-408-
Анти гипертензивные препараты
В зависимости от остроты гипертензии, к лечению антигипертензивными препаратами стратегически приступают в определенном порядке. Разумеется, этот порядок должен быть гибок и должен обеспечивать альтернативные подходы, но обязательно следует придерживаться некоторых общих принципов.
В качестве первого шага при слабой гипертензии попытку снижения давления начинают с приема диуретиков или p-адреноблокаторов, или малых доз ингибиторов ЛПФ. При гипертензии от слабой до умеренной для лечения рекомендуется использовать Р-адреноблокаторы или ингибиторы АПФ, или клонидин, гуанабенз, гуанфацин, или метил-допа, или празозин, тетразозин, или блокаторы кальциевых каналов, или резерпин. При гипертензии от умеренной до сильной рекомендуется использовать гидралазин и большие дозы ингибиторов ангиотензин конвертирующего фермента. При сильной гипертензии используют гуа-нетидин или гуанадрел, а также миноксидил. Наконец, в неотложных случаях гипертензии рекомендуется использовать нитропруссид натрия, диазоксид, триметафан или лабеталол.
Общепринятым принципом антигипертензивной терапии является одновременное использование нескольких препаратов, воздействующих на основные участки, контролирующие АД, и в основном предполагает использование сочетания диуретиков, адреноблокаторов, ингибиторов АПФ или блокаторов кальциевых каналов.
22.1. Диуретики
Терапия гипертензии предполагает широкое использование диуретиков, включая тиазидные диуретики, родственные им метолазон (21.3.20) и индапамид (21.3.26), петельный диуретик фуросемид (21.4.11), а также калийсберегающие диуретики — спиронолактон (21.5.S), триаметрен (21.5.13), амилорид (21.5.18).
Молекулярный механизм действия диуретиков в качестве антигипертензивных средств недостаточно ясен, однако применение диуретиков вызывает значительное увеличение количества воды и электролитов, выделяемых с мочой, что приводит к уменьшению объема внеклеточной жидкости и плазмы. А это, в свою очередь, приводит к снижению сердечного выброса, основного параметра, ответственного за снижение АД и венозного возврата крови. Постепенно сердечный выброс восстанавливается, но гипотензивный эффект сохраняется, возможно, из-за уменьшения периферического сопротивления сосудов. Не исключено также, что диуретики каким-то образом снижают сосудистую активность норадреналина или других факторов давления в организме,
-409-
Глава 22
Методы синтеза тиазидных диуретиков, применяемых при гипертензии описаны в предыдущей главе.
Тиазидные диуретики
Наиболее широко применяются хлортиазид (21.3.3), гидрохлортиазид (21.3.4), бендрофлуметиазид (21.3.6), политиазид (21.3.8), гидро-флутиазид (21.3.9), трихлорметазид (21.3.10), метилциклотиазид (21.3.11), циклотиазид (21.3.12), бензтиазид (21.3.13).
н
FjC __.СНг-ЗСНгСРз
I П '
As. A, ,n~ch3
n2nsa2 s
Ог
21 ЗЭ
21 3 8
н
Cky?sy N у CHjC! H2NSO2 -^X^s-n~СНз
Ог
Препараты, родственные тиазидам
К этим препаратам относятся метолазон (21.3.20), хлорталидон (21.3.26), индапамид (21.3.33), а также петельные диуретики буметанид (21.4.6), этакриновая кислота (21.4.9), фуросемид (21.4.11).
СН3 SO2Nh2
C4H9NH
о-сн2-соон
21 3 33
•410-
Анти гипертензивные препараты
Калийсберегающие диуретики
К этим препаратам относятся спиронолактон (21.5.8), триаметрен (21.5.13), амилорид (21.5.18)
о NH
21.5 18
Эффекты этих препаратов практически одинаковы. Эту группу препаратов характеризуют 3 основных побочных эффекта: 1) гиперурице-мия, 2) гиперглицемия и 3) нарушение электролитного баланса, характеризуемого гиперкалиемией, гипохлоремией и метаболическим алкалозом.
Фуросемид является более эффективным, однако не самым мощным антигипертензивным препаратом по сравнению с тиазидами.
22.2. ^-адреноблокаторы
Наиболее часто используемыми антиадренергическими препаратами при терапии гипертензии являются Р-адреноблокаторы. Несмотря на многолетнее их использование, механизм их действия понят не полностью. Однозначно ясно лишь то, что они являются конкурентными антагонистами адреналина и норадреналина на сердечных р-адреноре-цепторах.
Полагают, что так же, как и в случае диуретиков, использование p-адреноблокаторов приводит к понижению сердечного выброса. Снижается скорость работы сердца и общее периферическое сопротивление сосудов.
Так же как и в случае диуретиков, сердечный выброс постепенно восстанавливается, но гипотензивный эффект сохраняется. Наибольшая способность понижать давление выявлена у уникального Р-адрено-блокагора — лабеталола, который сочетает в себе неизбирательное р-адре-ноблокирующее действие как на рг, так и на р2-рецепторы с одновременной блокадой агрецепторов.
В отличие от других адреноблокаторов, лабеталол понижает кровяное давление больше путем снижения сопротивления периферических сосудов, чем путем подавления функции миокарда. При этом на
-411 -
Глава 22
ряду со снижением давления практически не изменяется скорость сердца.
В настоящее время для терапии гипертензии применяются 8 наиболее широко используемых в медицине Р-адреноблокаюров, синтез которых был описан в гл. 12 «Адреноблокирующие препараты», — пропранолол (12.1.3), метопролол (12.1.5), ацебугол (12.1.6 ), атенолол (12.1.7), надолол (12.1.8), пиндолол (12.1.9), тимолол (12.1.10) и лабеталол (12.1.12).
12 1 2
/=\ СНз
СН3О -СН2-СН2—М ff-О-СН2-СН-СН2—Mh'CH
'—' он сн3
,СН3
О-СН2-СН-СН2 — NH-CH
СНзСО^Х. он СНз
Т у 1216
NH-CO-C3H7
СНз
О-СН2-СН-СН2—NH-CH
1 ОН СНз
ch2-conh2
о-сн2-сн-снг -NH-C^afa он
"2 1 ю
Наиболее часто побочными эффектами при применении р-адре-ноблокаторов являются чувство усталости, похолодание конечностей, а также повышение уровня триглицеридов и липопротеидов в крови.
-412-
Анти гипертензивные препараты
22.3. Центрально действующие адренергические препараты (симпатолитики)
Стимуляция а-адренергических рецепторов в определенных участках ЦНС приводит к гипотензии.
Механизм действия препаратов обусловлен стимуляцией аг-адренорецепторов тормозных структур головного мозга. Полагают, что взаимодействие этих препаратов с а2-адренорецепторами выражается в угнетении нейронов вазомоторного центра продолговатого мозга и снижении активности гипоталамуса, что приводит к уменьшению симпатической импульсации к сосудам и сердцу. В итоге умеренно уменьшаются сердечный выброс и скорость сердца, и, следовательно, понижается АД.
Малочисленные клинически полезные антигипертензивные препараты этого ряда, такие как клонидин, гуанабенз и гуанфацин, очевидно, действуют одинаково путем воздействия на а2-адренорецепторы.
Метилдопа, рассматриваемая в ряду с перечисленными выше препаратами, в организме трансформируется в а-мегилнорадреналин, который, стимулируя а2-адренорецепторы, тормозит симпатическую им-пульсацию. вызывая понижение АД.
Клонидин (Clonidine)
Синтез препарата (11.1.34) описан в гл. И «Адренергические (симпатомиметические) препараты».
CI
11 1 34
Клонидин селективный а2-адренергический агонист, который оказывает выраженное гипотензивное действие, связанное со снижением общего периферического сосудистого сопротивления, урежением частоты сердечных сокращений, уменьшением сердечного выброса. В комбинации с оральными диуретиками клонидин является препаратом выбора при лечении разных степеней гипертензии
Клонидин применяют при различных формах гипертонической болезни и для купирования гипертонических кризов. Используют также в офтальмологической практике при открытоугольной глаукоме.
Синонимами клонидина являются гемитон, катапресан, клофелин.
. 414 -
Глава 22
Гуанабенз (Guanabettz)
Гуанабенз — [(2,6-дихлорбензилиден)амино]гуанидин (22 3.1) синтезируют в отну стадию взаимодействием 2,6-дихлорбензальдегида с аминогуанидином [1, 2, 3]
NH
•СНО + H2N-NH-C-NH2
Z Йн
/-CH=NH—NH-C-NH2
22 3 1
Гуанабенз а2-адренергический агонист, оказывающий выраженное гипотензивное действие, связанное со снижением общего периферического сосудистого сопротивления, урежением частоты сердечных сокращений, уменьшением сердечного выброса.
Применяется как самостоятельно, так и в комбинации с оральными диуретиками при лечении разных степеней гипертензии.
Синонимом препарата является витенсин.
Гуанфацин (Guanfacin)
Гуанфацин — 1Ч-амидино-2-(2,6-дихлорфенил)ацетамид (22.3.2) также получают весьма простым синтезом — взаимодействием хло-рангидрида или эфира, 2,6-дихлорфенилуксусной кис-лоты с гуанидином [4, 5, 6, 7].
CI С(
NH
H2N—C-NH2
Гуанфацин действует по тому же механизму, что и клонидин и гуанабенз, и применяется по тем же показаниям
Синонимом препарата является эсту лик.
Метилдопа (Methyldopa)
Метилдопа — (-)-3-(3,4-дигидроксифенил)-2-метилаланин (22.2.5) предложено получать несколькими методами, отличающимися лишь небольшими деталями. Один из них исходит из 3,4-димето-кснфенилацетона. который вводят в реакцию Штреккера—Зелинского с использованием цианида калия и карбоната аммония с по-
-414-
А ч т и г ипертензивные препараты________________________________
лучением 4-метил-4-(3,4-диметоксибензил)гидантоина (22.3.3), который далее гидролизуют в присутствии гидроокиси бария в (±)-3-(3.4-димегоксифенил)-2-метилаланин (22.3.4). Последний подвергают ацетилированию по аминогруппе, и далее рацемическую смесь разделяют с использованием (-)-1-фенилэтиламина. Выделенный изомер гидролизуют бромистоводородной кислотой с одновременным удалением и метокси-, и ацетильной группы с получением искомою (-)-3-(3,4-дигидроксифенил)-2-метилаланина (22.3.5) [8, 9, 10] Предложены альтернативные методы синтеза [11, 12, 13].
Сг!3О
2=\ сн3
СНзО—Z сн2-С-COOri
' ' nh2
22 3 4
\__ 9Нз
НО—сн2-с—соон
NH2
22 3 5
Метилдопа представляет собой а-метилированное производное леводопы, оказывает гипотензивное действие вследствие снижения общего периферического сосудистого сопротивления и уменьшения работы сердца. Антигипертензивное действие метилдопы предполагает биотрансформацию метилдопы в метилнорадреналин (метилнорэпинефрин), который действует в качестве «фальшивого нейротрансмиттера». В настоящее время общепринята точка зрения, согласно которой действие метилдопы осуществляется посредством ЦНС. где метилнорэпи-нефрин. являясь мощным стимулятором а-адренорецеп горов продолговатого мозга, тормозит сосудодвигательный центр.
Препарат применяют при артериальной гипертонии, гипертонических кризах
Синонимами препарата являются альдомет, допегит, мопатил и др.
22.4. Периферически действующие симпатолитики (а-адреноблокаторы)
Как уже было отмечено (см. гл. 12 «Адреноблокирующие препараты»), характерной особенностью а-адреноблокаторов является их способность уменьшать прессорный эффект адреналина (эпинефрина). В частноеги. постсинаптические а,-блокаторы действуют на а-рецеп-
Глава 22
тивные участки, расположенные на сосудистой гладкой мускулатуре и противодействуют прессорному, сосудосуживающему, эффекту' адреналина (эпинефрина) и норадреналина (норэпинефрина). Кроме того, они проявляют прямой расслабляющий эффект на гладкую мускулатуру, что приводит к периферическому расширению сосудов, в итоге вызывающему понижение давления. Наверное, наиболее важным эффектом а-адреноблокаторов является расширение периферических сосудов. Однако они проявляют также кардиостимулирующий эффект, что нередко становится причиной тахикардии.
Попытки лечения гипертензии такими препаратами, как фентоламин (12.2.1) и феноксибензамин (12.2.6), блокирующими а-адренер-гические рецепторы, оказались безрезультатными при лечении гипертензии как по причине большого числа сопутствующих им побочных эффектов, так и по причине развития к ним толерантности.
Однако ряд оц-селективных адреноблокаторов и блокаторов адренергических нейронов применяются при лечении гипертензии.
Обратимые конкурентные а, -селективные адреноблокаторы
Препараты с а-адреноблокирующей активностью, отличающиеся специфичностью только к ai-адренорецепторам, представляют собой периферические коронарорасширяющие средства. Празозин и тетразо-зин — селективные а (-адреноблокаторы, которые в терапевтических дозах применяют для понижения АД.
Празозин (Prazosin)
Синтез препарата (12.2.12) описан в гл. 12.
Празозин показан для лечения средней или умеренной гипертензии. При применении препарата кровяное давление понижается без существенных изменений таких показателей работы сердца, как скорость, коронарный поток, сердечный выброс.
Празозин показан при слабой и умеренной гипертензии.
Синонимами препарата являются минипресс и минизид.
-416-
д нти гипертензивные препараты
Тетразозин (Tetrazosin)
Синтез препарата ^12.2,13) описан в гл. 12.
Тетразозин применяют по тем же показаниям, что и празозин, однако он имеет го преимущество, что его можно принимать один раз в день.
Синонимами препарата являются геитрин и вазокард.
Блокаторы адренергических нейронов
Блокаторы адренергических нейронов вызывают распад биогенных аминов в окончаниях нейронов. Эти препараты могут вмешаться в синтез, хранение и высвобождение норэпинефрина, дофамина и серотонина.
Резерпин (Reserpine)
Синтез препарата (12.3.1) описан в гл. 12.
Резерпин вызывает распад норэпинефрина, дофамина и серотонина в окончаниях нейронов. Он ослабляет внутриклеточный захват биогенных аминов и уменьшает способность к их хранению в везикулах.
Резерпин применяется при лечении гипертензии, однако не является лекарством выбора из-за большого числа побочных реакций. Тем не менее на его основе имеется много комбинированных с другими гипертензивными средствами, в частности, с диуретиками препаратов.
Резерпин выпускается под множеством названий, среди которых можно выделить его синонимы серпасил, бринердин, диупрес и др.
-417-
Глава 22
Гуанетидин (Guanethidine)
Синтез препарата (12.3.2) описан в гл. 12.
С1 иН
N—ch2-ch2-nh-c-nh2
12 3.4
Гуанетидин не воздействует на эффекторные клетки. Он проникает в нейрон, где аккумулируется и замещает норэпинефрин. В результате стимуляции нерва может быть выброшен сам гуанетидин, который, однако, не является стимулятором адренергического рецептора.
Гуанетидин применяется при острой гипертензии, когда применение более общепринятых препаратов оказывается безуспешным. Это — мощный долгодействующий антигипертензивный препарат, однако он действует на кровяное давление пациента исключительно в ортостатическом положении и не действует в лежачем.
Гуанетидин очень сильный и долгодействующий препарат, и часто его действие продолжается еще 2-3 дня после его отмены.
Синонимами препарата являются октадин, исмелин, санотензин и др.
Гуанадрел (Guanadrel)
Синтез препарата (12.3.8) описан в гл. 12.
12 3.8
ch2-nh-c-nh2
NH
Гуанадрел — блокатор адренергических нейронов, используемый при эссенциальной гипертензии. Механизм его действия и побочные реакции сходны с гуанетидином.
Препарат применяют для лечения гипертензии у пациентов, неадекватно реагирующих на тиазидные диуретики. Может быть использован в качестве дополнительного средства при тиазидном лечении для достижения оптимального уровня кровяного давления.
Синонимом препарата является гилорел.
-418-
Антигипертензивные препараты
22.5. Блокаторы кальциевых каналов
В дополнение к применению в качестве антиангинальных и антиаритмических средств блокаторы кальциевых каналов используются для лечения слабой и умеренной гипертензии. Эти препараты препятствуют проникновению в клетки гладких мышц периферических сосудов ионов Са2’ и вызывают расслабление периферических сосудов, что приводит к снижению АД. В клинически применяемых дозах блокаторы кальциевых каналов расслабляют гладкую мускулатуру артерий и мало действуют на вены. В дозах, которые релаксируют гладкую мускулатуру, блокаторы кальциевых каналов относительно мало влияют на сокращаемость сердца.
При антигипертензивной терапии наиболее часто используются дилтиазем (19.3.10), верапамил (19.3.15) и нифедипин(19.3.16), которые кажутся равно эффективными препаратами для лечения гипертензии. Методы их синтеза уже были предложены в гл. 19 «Антиангинальные препараты».
Дилтиазем (Diltiazem)
Синтез препарата (19.3.10) описан в гл. 19.
Дилтиазем уменьшает трансмембранное поступление ионов Са2+ в клетки сердечной мышцы и гладкой мускулатуры сосудов. Вызывает расширение коронарных и периферических сосудов. Увеличивает коронарный кровоток, предотвращая развитие спазма коронарных артерий. Снижает повышенное АД и уменьшает тахикардию.
Препарат показан при стабильной и нестабильной стенокардии (в том числе и после инфаркта миокарда), при артериальной гипертонии.
-419-
Глава 22
Верапамил (Verapamil)
Синтез препарата (19.3.15) описан в гл. 19.
р-СНз
СН3~О СН3ххСН3 \ сн
СНз-О—У- С-СН2-СН2-СН2—N-CH2-CH2—С Л— О-СНз
'--' C=N СНз '-'
193 15
Верапамил обладает антиаритмической. антиангинальной и гипотензивной активностью. Снижает потребность миокарда в кислороде за счет снижения сократимости миокарда и урежения частоты сердечных сокращений. Вызывает расширение коронарных артерий и увеличение коронарного кровотока. Снижает тонус гладкой мускулатуры, периферических артерий и общее периферическое сосудистое сопротивление. Оказывает антиаритмическое действие при наджелудочковых аритмиях.
Верапамил применяют для профилактики приступов стенокардии, артериальной гипертонии, при лечении и профилактике наджелудочковых аритмий.
Нифедипин (Nifedipin)
Синтез препарата (19.3.16) описан в гл. 19.
н
I
193 16
Нифедипин вызывает расслабление гладкой мускулатуры сосудов, расширяет коронарные и периферические артерии, снижает периферическое сопротивление, АД, улучшают снабжение сердца кислородом.
22.6. Миотропные гипотензивные препараты
Препараты этого класса (гидралазин и нитропруссид натрия) понижают АД, главным образом, непосредственным спазмолитическим действием на гладкую мускулатуру артериол, что приводит к понижению
-420 -
д н т и г и пертензивные препараты
сопротивления периферических сосудов вызывая их расширение. Диастолическое давление обычно понижается больше, чем систолическое.
Гидралазин (Hydralazin)
Гидралазин — 1-гидразинонафталазин (22.6.4) получают окислительным хлорированием фталида с одновременным гидролизом продукта, в результате чего получают гидроксифталид (22.6.1), который взаимодействием с гидразином переводят в фталазон (22.6.2). Подвергая последний действию хлорокиси фосфора, получают 1-хлорфталазин (22.6.3), замещая в котором атом хлора гидразином, получают искомый гидралазин (22.6.4) [14, 15, 16].
РОС|3
Гидралазин проявляет антигипертензивный эффект путем прямого расслабления гладкой мышцы сосудов. Причем препарат воздействует на артериальные сосуды с минимальным действием на венозные. В результате понижается сопротивление периферических сосудов, и кровяное давление понижается (диастолическое больше, чем систолическое).
Препарат существенно не влияет на несосудистую гладкую мускулатуру или сердечные ткани. Гомеостатические циркуляторные рефлексы остаются нативными, вызванная гипотензия активирует сердечнососудистые рефлексы, выражающиеся в увеличении работы сердца, силы и объема сердечного выброса. Поэтому препарат наиболее эффективно используется в сочетании с Р-блокаторами.
Сам по себе гидралазин используется при гипертензии, однако не является лекарством выбора даже при мягких формах.
Синонимами препарата являются апрессин, гипатол, депрессан и др.
-421 -
Глава 22
Нитропруссид натрия (Nitroprusside natrii)
Нитропруссид натрия —Na2Fe(CN)5NO (22.6.7) получают последовательными реакциями, включающими взаимодействие ферроцианида калия с азотистой кислотой с получением нитропруссида калия (22.6.5). дальнейшим переводом его в нитропруссид меди (22 6.6), и далее взаимодействием последнего с карбонатом натрия получают нитропруссид натрия (22.6.7).
2K4Fe(CN)6 + 4HNO2 + 2H2SO4 = K2Fe(CN)5NO + 2NO + 2(CN)2 + 2K2SO4 + 4H2O
22 6 5
K2Fe(CN)5NO + CuSO4 = CuFe(CN)5NO + K2SO4
22 6 6
CuFe(CN)5NO + Na2CO3 = Na2Fe(CN)5NO + CuCO3
22 6 7
Нитропруссид натрия — мощный, мгновенно действующий внутривенный препарат, используемый для понижения кровяного давления при гипер(ензивных кризах. Гипотензивный эффект вызывается периферической вазодилатацией в результате прямого воздействия как на артериальные, так и на венозные сосуды.
Побочные эффекты также наступают очень быстро, однако продолжаются очень недолго по причине весьма короткого времени полужизни препарата. Нитропруссид нагрия биотрансформируется в цианид и тиоцианат, вследствие чего при передозировке может появиться тиоцианатная или цианидная интоксикация.
Наличие таких препаратов, как диазоксид и нитропруссид натрия, значительно увеличило возможность резкого снижения АД в экстренных случаях, которое, однако, должно производиться под постоянным контролем медицинского персонала.
Синонимами препарата являются ниприд, нипрутон и др.
22.7. Ингибиторы АПФ
Ингибиторы АПФ проявили себя в качестве весьма эффективных антигипертензивных препаратов, начавших вытеснять Р-адренобло-каторы, особенно при монотерапии гипертензии.
Эти препараты ингибируют АПФ, который осуществляет гидролиз неактивного ангиотензина I в активный ангиотензин II. являющийся активным эндогенным вазопрессорным веществом.
-422-
А н т и г и пертензивные препараты______________________________
Ahi иотензин I является прогормоном, который образуется в результате действия ренина на пептидный субстрат, вырабатываемый печенью.
Ренин, в свою очередь, является протеолитическим ферментом, продуцируемым почками и контролирующим физиологические функции других органов.
Секреция самого ренина контролируется нервной системой и, возможно, недавно открытым сердечным пептидным гормоном.
Ангиотензин I относительно неактивен и активируется превращением в ангиотензин II АПФ. Рецепторы мембран клеток гладкой мускулатуры артериол и коры надпочечников (секреция альдостерона) специфично стимулируются ангиотензином II. В результате повышается периферическое сопротивление сосудов, повышается скорость работы сердца, увеличивается сердечный выброс и происходит задержка воды и ионов Na . В свою очередь, индуцированное повышение давления путем обратной связи вызывает уменьшение выделение ренина.
Существует гипотеза, что в основе этиологии эссенциальной гипертензии во всех случаях лежит нарушение ренин-ангиотензиновой системы Однако, несмотря на всю кажущуюся привлекательность этого предположения, для ее признания в качестве единственной причины повышения АД еще нет достаточных доказательств.
В то же время, независимо от того, вызвана ли гипертензия повышением уровня ренина или другими причинами, у гипертоников ингибиторы АПФ вызывают понижение как систолического, так и диастолического АД, и их действие усиливается диуретиками.
Препараты ряда ингибиторов АПФ (каптоприл, эналаприл) являются эффективными антигипертензивными препаратами, применяющимися для лечения всех типов гипертензии как самостоятельно, так и в комбинациях с другим препаратами, а также для лечения сердечной иедостато ч ности.
Каптоприл (Captopril)
Каптоприл — 1-[(28)-3-меркапто-2-метилпропионил]-Ь-пролин (22.7.4) получают следующими путями; при первом способе — прямым ацилированием L-пролина хлорангидридом 3-ацетилтио-2-метилпропионовой кислоты (22.7.2), получаемого из 3-аце-тилтио-2-метилпропионовой кислоты (22.7.1), которую, в свою очередь, получают взаимодействием метакриловой и тиоуксусной кислот. Образовавшийся в результате взаимодействия L-пролина с хлорангидридом З-ацетилтио-2-метилпропионовой кислоты 1-(3-ацетилтио-2-П-метилпропаноил)-Ь-пролин (22.7.3) далее под-
-423 -
Глава 22
веруют аммонолиз) аммиаком с получением искомого капюпри-ла (22 7 4).
сн3 н2с=с-соон
FH3
CHj-CO-S— Н2С -сн —соон
SOCI2
22 У 1
H;.-CO-SH
сосн
н-s—ч2с-сн-со
1
22 7 4 СН3
Второй способ отличается от первого лишь тем, что ацилированию хлорангидридом З-ацетилгио-2-метилнропионовой кислоты (22.7.2) подвергают предварительно защищенный по карбоксильной группе L-пролин - трет-буч иловый эфир L-нролина (22.7.6) Последний получают по следующей схеме: I.-пролин ацилируют хлор-бепзиловым эфиром с получением N-бензилоксикарбонил L-пролина (22.7.7), который вводят во взаимодействие с изобутиленом с целью получения трет-бутипового эфира N-бензилоксикарбонил — L-пролина (22.7.8). Воссоновлением последнею водородом с использованием в качестве катализатора палладия на угле получают трет-бутиловып эфир L-пролина (22 7.9). После ацилирования последнего хлорангидридом (22.7.2) получают /ире/и-бутиловый эфир 1-(3-ацетилтио-2-метил-пропаноил)-Ь-пролина (22.7.10), из которого последовательно удаляются защитные группы С помощью трифторуксусной кислоты I идролизуется сложноэфирная часть молекулы с получением 1-(3-ацетилгио-2-ме1Илпропаноил)-П-пролина (22.7 3). из диастереомеров которою после разделения отбираеюя 1-(3-анетилтио-2-Е)-метил-пропанои.1)-1 -иролин, и далее аналогично вышеописанному его подвер! аюг аммонолизу с получением каптоприла (22 7.4) [17, 18, 19. 20. 21, 22, 23]
- 424 -
Антигипертензивные препараты
СНз-СО-S—Н2С -сн-со
СООС(СН;)з
22 7 3 -----»• 22 7 4
СНз 22 7 10
Каптоприл является наиболее изученным веществом из ряда ингибиторов АПФ. предложенным в качестве антигипертензивного средства. Препарат блокирует АПФ, в результате чего подавляется образование ангиотензина II и устраняется его вазоконстрикторное действие на артериальные и венозные сосуды. При этом снижается общее сосудистое периферическое напряжение, и уменьшаемся АД.
Препарат применяют при гипертонии и хронической сердечной недостаточности.
Синонимами препарата являются капотен, каприл и др.
Эналаприл (Enalapril)
Эналаприл - (S)- 1-[N-[1 -(этоксикарбо нил)-3-фенилпропил]-Е-ала-нил]-1.-пролин (22.7.12) получают взаимодействием бензиловою эфира дипептида I-аланил -L-пролина с этиловым эфиром 3-бен-зоилакриловой кислоты с образованием продукта (22 7.1). восстановлением которого водородом с использованием в качестве катализатора никеля Ренея удаляется защитная бензильная группа и получаезся искомый эналаприл (22.7.12) [24]. Предложены и аль-1ернативные пути синтеза [25, 26. 27, 28, 29].
СН=СН-СООС2Н5
СООСН2С6Н5
22 7 12
-425-
Г лава 22
1 ак же как и каптоприл, эналаприл селективно подавляет ренин-ашиотензин-альдостероновую систем}, ингибирует АИФ, предотвращав превращение ангиотензина I в ангиотензин II.
Применяют при гипертонии и при хронической сердечной недостаточное! и.
Синонимами препарата являются вазотек, ренитек, наприлен и др.
22.8. Активаторы калиевых каналов
Минокснди.1 (Minoxidil)
Миноксидил - - 6-амино-1.2-дигидро-1-гидрокси-2-имино-4-пипе-ридинопиримидии (22.8.5) получают исходя из барбитуровой кислоты, взаимодействием которой с хлорокисью фосфора получают 2,4,6-трихлорпиримидин (22.8.1). Действием аммиака последний переводят в 2,4-диамино-6-хлорпиримидин (22.8.2). Далее полученный 2,4-диамино-6-хлорпиримидин (22.8.2) вводят в реакцию с 2,4-дихлорфенолом в присутствии гидроокиси калия с получением 2,4-лиа.мино-6-(2,4-дихлорфеноксн)-пиримидина (22 8.3). Окисляя последний 3-хлор-пербензойной кислотой, получают 2,4-диамино-6-(2,4-дихлорфенокси)пиримидин-3-оксид (22.8.4), 2,4-дихлорфено-ксильную группу в котором при высокой температуре замещают на пиперидиновую с получением миноксидила (22.8.5) [30. 31. 32, 33].
РОСЩ
22 8 1
Миноксидил — периферический вазодилататор, непосредственно расслабляющий гладкую мускулатуру сосудов и тем самым понижаю
-426 -
Антигипертензивные препараты
щий и сис1 одическое, и диастолическое давление. Его действие связываю! с активацией калиевых каналов. Открытие калиевых каналов вызывает гиперполяризацию клеток гладкой мускулатуры, что. в свою очередь, снижает поступление внутрь клеток ионов Са“’, необходимых для поддержания сосудистого тонуса. Однако при применении миноксидила одновременно с гипотензией может возникнуть тахикардия, повышается секреция ренина, а также происходит задержка воды и ионов \а .
Вследствие серьезных потенциальных побочных действий препарат показан только при острой гипертензии, не поддающейся лечению другими препаратами, и обязательно в сочетании с двумя другими антиги-пергензивными средствами.
Синонимом препарата является лонитен.
Диазоксид (Diazoxid)
Диазоксид — 7-хлор-3-метпл-2-Я-1,2.4-бепзотиадиазин-1,1-диоксид (21.3.14) получают конденсацией 2-аминосульфонил-4-.\лоранилина с гриэгилортоацегатом [34, 35, 36].
о2
(С2Н5О)зССН3
21 3 14
Диазоксид является недиуретическим производным тиазидов, резко снижающим кровяное давление путем прямого расслабления гладких мышц артериол, возможно, в результате активации калиевых каналов клеток гладкой мускулатуры артериол. При этом препарат мало влияет на венозную систему и сердце. В дополнение к гипотензивному действию диазоксид вызывает резкое повышение уровня глюкозы в крови вследствие ингибирования высвобождения инсулина из поджелудочной же )езы К его неблагоприятным эффектам относят также задержку в организме ионов Na и воды, повышение содержания в крови мочевой кислоты.
Препарат применяется в случаях необходимости неогложного снижения давления при острой гипертензии. При эссенциальной гипертен-JHii диазоксид не применяется.
( ннонимом препарата является гиперстат.
-427 -
Глава 22
Список литературы
1. Ger. Pat 1.802.364 (1967).
2. Baum Т et al. ,'Experientia. 25. 1066 (1969).
3. Bnt. Pat. 1.019.120(1966).
4. Fr Pat. 1.584.670 (1969).
5. US Pat. 3.632 645 (1972).
6. Ger. Pat 1.793.483 (1968)
7. Bream J. et al.- Aizneimittel-Forsch. 25, 1477 (1975).
8. US Pat. 2.868.818(1959).
9. Bnt. Pat. 936.074 (1960).
10. Tristram E et al.'/J. Org. Chem. 29, 2053 (1964).
11. US Pat. 3.158.648 (1964).
12. Tristram E et al.''J. Org. Chem 29, 1424 (1964).
13. Slates H et al., J. Org. Chem. 33. 1209 (1968).
14. US Pat. 2.484.029 (1949).
15. Ger. Pat. 848.818 (1945).
16. Druey H. et al.''Helv. Chim. Acta. 34, 204 (1951).
17. US Pat. 4.046.889 (1976).
18. US Pat. 4.105.776 (1978).
19. US Pat. 4.154.840 (1979).
20. US Pat. 4.154.935 (1979).
21. Ger. Pat. 2.703.828 (1977).
22. Ondetti M et al.,/Science. 196, 441 (1977).
23. Nam D et al. 'J. Pharm. Sei. 73, 1843 (1984).
24. US Pat. 4.442.030(1984).
25. Patched A. et al./Nature. 288, 280 (1980)
26. IHvratt.W et al.>"J. Org. Chem. 49, 2816 (1984).
27. US Pat. 4.374.829 (1983).
28. US Pat. 4.472.380(1984).
29. Eur. Pat. Appl. 12.401 (1979).
30. US Pat. 3.382 247(1968).
31. US Pat. 3.461.461 (1969).
32. US Pat. 3.644.364 (1972).
33. McCall J et al. J. Org. Chem. 40, 3304 (1975).
34. US Pat. 3.345.365 (1967).
35. US Pat. 2.986.573 (1961).
36. Raffa I et al., Farmaco. Ed. Set. 17, 244 (1962).
-428 -
1 Гл а в а 23
Препараты для лечения заболеваний дыхательной f системы
Заболевания дыхательной системы могут возникнуть в результате воздействия на нее инфекций, действия чужеродных атентов разной природы, в том числе пыли, химических веществ, курения, переохлаждения, 1енетических факторов, в результате острых поражений или реакций т иперчувствительности. Терапия заболеваний дьтхательной системы в обшем сводится к восстановлению соответствующих физиологических функций. В частности, антибиотики устраняют инфекции, поразившие респираторный тракт, глюкокортикоиды устраняют его воспаление, бронходидата горы (бронхолитики) расслабляют гладкую мускулатуру бронхиол и раскрывают заблокированные для прохождения воздуха участки и т. и
Особое место среди легочных заболеваний занимает астма, которая является клиническим синдромом, характеризующимся повышенной возбудимостью тт сужением дыхательных путей и. следовательно, явлениями одышки, затрудненным дыханием и кашлем. Пациенты, страдающие астмой, могут проявлять признаки хронического бронхита иди эмфиземы легких.
Поскольку в основном молекулярные механизмы этих натолотиче-ских изменений недостаточно изучены, терапия астмы, легочных заболеваний тт других заболеваний дыхательной системы в основном направлена на предупреждение и устранение сопутствующих этим заболеваниям симптомов.
Соответственно, препараты для лечения заболеваний дыхательной системы можно рассматривать как иротивоотечные препараты сосудосуживающего действия, применяемые как носовые капли, противокаш-левьте и отхаркивающие средства, а также как бронхолитики и другие препараты, применяемые для лечения бронхиальной астмы, такие как мегидксаптины, антихолннергические препараты, адренергические препараты, ингибиторы высвобождения медиаторов аллергии и кортикостероиды.
-429 -
Г лава 23
23.1. Противоотечные сосудосуживающие препараты
Сосудосуживающие препараты, как правило а-адреномиметики, применяются как временные успокаивающие средства при острых ринитах вирусного или аллергического происхождения, синуситах, евста-хеигах. При местном применении в виде капель или аэрозолей они сужают артериолы слизистой оболочки носа, приводя к снижению отека, гиперемии, экссудации
С этой целью часто применяют симпатомиметики с выраженным противоотечным действием, такие как нафазолин (11.1.36), гетрагидро-золин (11.1 37), ксилометазолин (11.1.38), оксиметазолин (11.1.40) и др., синтез и свойства которых были описаны в гл. 11 «Адренергические (симпатомиметические) препараты».
снз
11 1 41
23.2. Противокашлевые препараты и отхаркивающие средства
Заболевания дыхательной системы как правило сопровождаются кашлем — защитным механизмом, посредством которого инородные вещества, раздражители и слизь выделяются из дыхательного тракта.
Противокашлевые препараты могут действовать на уровне «кашлевого центра» в продолговатом мозге, а также на различных участках трахеобронхиального древа. Препараты, проявляющие противокашле-аой эффект, делятся на две группы. Это препараты центрального действия, наркотические протвокашчевые средства или опиаты, к которым О!носятся кодеин и гидрокодон, а также разнородная группа препаратов, проявляющих и центральный, и периферический эффекты, подавляющие кашель, называемая ненаркотическими противокашлевыми средствами (декстромегорфан, бензонатат).
- 430-
Препараты для лечения заболеваний дыхательной системы
Наркотические противокашлевые средства
Кодеин (Codeine)
Синтез препарата (3.1.20) был описан в гл. 3 «Анальгетики».
СН3О о ОН
3 1 20
Препарат подавляет кашлевой рефлекс в кашлевом центре в про-до.поватом мозге, и его используют при остром и хроническом рефлекторном кашле
Гидрокодон (Hydrocodone)
Синтез препарата (3.1.27) был описан в гл. 3.
сн3
сн3о хо о 3 1 27
Гидрокодон — мощный противокашлевой препарат, который широко применяется для подавления кашлевого рефлекса. Он входит в состав эффективных противокашлевых коммерческих препаратов в комбинациях с гуайфенезином (энтусс), с гоматропином (гикодан), с фенилпропаноламином (гикомин), фенилтолоксамином (туссио-некс) и псевдоэфедрином и гуайфензином (туссенед).
Ненаркотические противокашлевые средства
Декстрометорфан (Dextrometorphan)
Декстрометорфан — (9а.13а,14а)-3-метокси-17-метилморфинан (23.2 1) получают исходя из (±)-3-гидрокси-М-метилморфинана путем замещения фенольной гидроксильной группы на метоксильную
-431 -
Г лава 23
с использованием фенилтриметиламмоний хлорида и метилата натрия в метаноле. Полученный рацемический продукт (±)-3-метокси-N-метилморфинан разделяют на изомеры с помощью D-винной кислоты с получением декстрометорфана [1, 2].
СНз
1— N
СН3
но
Препарат обладает выраженным противокашлевым эффектом и минимальным действием на ЦНС, не вызывает привыкания.
Синонимами препарата являются коффекс, робидекс, седатусс и др.
Бензонатат (Benzonatat)
Бензонатат — и-бутиламинобензоат 2,5,8,11,14,17,20,23,26-нона-октакозан-28-ола (23.2.2) получают переэтерификацией этилового эфира 4-бутиламинобеизойной кислоты монометиловым эфиром нонаэтиленгликоля. Препарат является структурным аналогом местного анестетика тетракаина [3, 4].
C4HSNH—4 fl— СООС2Н6 ’ но- 1СН2СН2О)8-СН, С4Н<ЛН—4 СОС— (СН2СН.2О)9-СНЭ
Предполагается, что он действует по двум механизмам: селективной анестезией раздраженных рецепторов в легких и одновременным подавлением кашлевого центра.
Синонимами препарата являются тессалон, вентуссин и др.
Отхаркивающие средства
При вязком сухом секрете бронхиальных желез кашель можно уменьшить либо рефлекторно, путем увеличения секреции желез слизистой оболочки бронхов или путем повышения активности мерцательного эпителия и усиления сокращения мышц бронхов (гуайфенезин, йодистый калий, терпингидрат), либо разжижением секрета муколитиками — препаратами, уменьшающими вязкость мокроты путем деполимеризации полисахаридов, входящих в ее состав. Наиболее широко при этом
- 432 -
Препараты для лечения заболеваний дыхательной системы
применяется ацетилцистеин.
Гуайфенетн (Guaifenesin)
Гуайфенезин — 3-(о-метоксифенокси)-1,2-пропандиол (23.2.3) предложено получать взаимодействием гваякола с 3-хлорпропан-1,2-диолом или с глицидолом [5, 6. 7, 8, 9, 10].
—"ОН
ОН
Гуайфенезин способствует выделению секрета из желез слизистой оболочки бронхов, облегчая кашель при простудных заболеваниях, бронхитах, бронхиальной астме.
Синонимами препарата являются робитуссин, лотуссин и др.
А цетил цистеин (A cetylcysteine)
Ацетилцистеин — N-ацетил-Г-цистеин (23.2.4) получают взаимодействием гидрохлорида L-цистеина с уксусным ангидридом в присутствии ацетата натрия [И, 12, 13].
(СН3СО)2О
HS—сн2—СН—СООН ----------* HS-CH2-CH-COOH
NH, НС! CHjCOONa СНзССД
23 2 4
Ацетилциетеин является препаратом, применяющимся при кашле с целью уменьшения вязкости мокроты.
Синонимами препарата являются флумуцетин, мукомист и др.
23.3. Бронходилататоры (бронхолитики)
Метилксантины
Ксантины принадлежат к семейству соединений, содержащих пуриновую циклическую систему — одну из наиболее важных гетероциклических систем, встречающихся в природе, получаемой сочленением пиримидинового и имидазольного колец.
Три наиболее известных природных метилксантина это теофиллин, теобромин и кофеин. Метилксантины проявляют сходный спектр биологической активности.
-433 -
Г лава 23
Главными источниками этих соединений являются чай, какао и кофе соответственно. В качестве бронхолитика наибольший интерес представляет теофиллин.
Теофиллин (Theophyllin)
Теофиллин — 1,3-диметилксантин (23.3.5) содержится в небольшом количестве в чайных листьях Синтетически его получают методом Траубе — общим методом, предложенным для получения пуриновых оснований. В данном случае взаимодействием N.N-диметил-мочевины с циануксусным эфиром в присутствии уксусного ангидрида получают цианацетилметилмочевину (23.3.1), которую циклизуют в 6-амино-1.3-диметилурацил (23.3.2). Полученное соединение действием азотистой кислоты трансформируют в 5-нитрозо-6-амино-1,3-диметилурацил (23.3.3). Восстановлением нитрозогруппы в последнем получают 5,6-диамино-1,3-диметилурацил (23.3.4), последующим взаимодействием которого с формамидом получают искомый теофиллин (23.3.5) [14, 15, 16, 17].
NHCHj
NaOH
23 3 3
HCONH2
23 3 4
23 3 5
Механизм действия теофиллина в качестве бронхолитика неизвестен Однако предложено несколько гипотез, основанных на его структурном сходстве с аденозином и 3',5'-цАМФ.
Аденозин является эндогенным медиатором, который, взаимодействуя с мембранными рецепторами, может вызывать сокращение бронхов. Теофиллин же препятствует этому взаимодействию, предотвращая вызванный этим субстратрецепторным взаимодействием бронхоспазм.
Полагают, что теофиллин может ингибировать фосфодиэстеразу, что. в свою очередь, может привести к повышению уровня клеточного
-434 -
Препараты для лечения заболеваний дыхательной системы циклического аденозинмонофосфата и. следовательно, к расслаблению гладкой мускулатуры дыхательных путей. Однако теофиллин не является мощным ингибитором фосфодиэстеразы и, в принципе, необходимые для этого концентрации могут быть недостижимы in vivo.
С другой стороны, теофиллин ингибирует обратный захват катехоламинов, что может повысить уровень циклического аденозинмонофосфата и тем самым вызвать бронхолитический эффект.
Наконец, ;еофил.тин является блокатором аденозиновых рецепторов. и именно это его свойство может быть ответственно за его бронхолитическое действие.
Несмотря на то, что последний механизм может быть основным в случае с теофиллином, некоторые ксантины, будучи, в общем, лишенными способности связываться с аденозиновыми рецепторами, выявляют такую же, если не большую, бронхолитическую активность, как у теофиллина.
Теофиллин и другие метилксантины проявляют фармакологический эффект и на ряд других систем. Конечно, наиболее выраженным их эффектом является расслабление гладкой мускулатура дыхательных путей. Однако теофиллин является стимулятором ЦНС, понижает артериальное кровяное давление, повышает диурез, проявляет кардиотоническую активность, оказывает определенное действие на ЖКТ. Перечисленные эффекты являются наиболее часто встречающимися побочными действиями при применении теофиллина в качестве бронхолитика.
Действие на ЦНС прямо зависит от дозы введенного препарата и может появляться усталостью, беспокойством, тремором и даже конвульсиями при относительно больших дозах.
На сердечно-сосудистую систему теофиллин действует, проявляя положительный ионотропный и хронотропный эффекты на сердце, что, вероятно, могло бы быть вызвано повышенным притоком ионов Са2, смодулированным циклическим аденозинмонофосфатом и действием на специфические сердечные фосфодиэстеразы. В желудочно-кишечной системе метилксантины одновременно стимулируют секрецию и желудочной кислоты, и пищеварительных ферментов.
Теофиллин уменьшает сократительную активность гладкой мускулатуры, расширяет бронхи и кровеносные сосуды, уменьшает легочное сосудистое сопротивление, стимулирует дыхательный центр, повышает частоту и силу сердечных сокращений. Препарат применяют при бронхиальной астме — для предупреждения приступов и для систематического лечения. Теофиллин применяют также для симптоматического лечения бронхоспастнческого синдрома другой этиологии (хронические
-435-
Глава 23
обструктивные заболевания легких, хронический бронхит, эмфизема лег ких).
На его основе создано большое число комбинированных препаратов, например аминофиллин или эуфиллин (сочетание теофиллина с этилендиамином), дифиллин, окстрифиллин.
Синонимами теофиллина являются адофиллин. астмофиллин. тео-цин и многие дру гие.
Антихолинергические препараты
Антихолинергические препараты, в частности атропин (14.1.4) или скополамин (14.1.6), веками использовались и для лечения обструктивных легочных заболеваний. Ингибируя действие ацетилхолина на гладкую мускулатуру дыхательной системы, такие препараты предотвращают бронхоспазм, возникающий в результате разрядки вагусного нерва. Однако они воздействуют на многие ткани и системы и. следовательно, выявляют широкий спектр побочных эффектов. В настоящее время их редко применяют для лечения кашля, возникшего от определенных раздражителей. Однако четвертичное производное атропина — ипратропий бромид часто используется при хронических бронхитах, эмфиземе легких и астме.
Ипратропий бромид (Ipratropium bromide)
Ипратропий бромид — За-гидрокси-8-изопропил-1аЯ,5аЯ-тропа-ния бромид (23.3.6) получают взаимодействием N-изо пропил норагропина с бромистым метилом [18, 19, 20].
Препарат оказывает бронхолитическое действие за счет уменьшения холинергических влияний на бронхиальную мускулатуру (М-холи-ноблокирующее действие). Устраняет спазм бронхов. Применяется для
-436 -
□ репараты для лечения заболеваний дыхательной системы
лечения и профилактики бронхиальной астмы легкой и средней степени, особенно сопутствующей заболеваниям сердечно-сосудистой системы.
Синонимами препарата являются атровент, итроп, ипрафен и др.
Ингибиторы высвобождения медиаторов аллергии
Медиаторы аллергии (гистамин, лейкотриены и др.) участвуют в формировании бронхоспазма. Блокируя высвобождение медиаторов аллергии у аллергических субъектов, некоторые препараты, в частности кромолин, являющийся прототипом препаратов этого типа, блокируют и ранние и поздние фазы реакции организма в ответ на экспозицию аллергенов. Эти препараты не имеют бронхорасширяющих свойств, однако ингибируют вызванный антигенами или другими причинами бронхоспазм.
Кромолин (Cromolyn)
Кромолин — 5,5'[2-гидрокситриметилен)диокси)]бис-4-оксо-4Я-1-бензопиран-2-карбоновую кислоту' (23.3.9) получают взаимодействием 2,6-дигидроксиацетофенона с эпихлоргидрином, при котором происходит как реакция замещения атома хлора в эпихлоргидрине, так и раскрытие эпоксидного кольца с образованием бис-продукта (23.3.7). Циклизацией последнего в бис-хромоновое производное с по.мошью диэтилоксалата в (23.3.8) и последутощим щелочным гидролизом сложноэфирных групп продукта получают искомый кромолин (23.3.9) [21, 22, 23].
СООС2Н5
СООС2Н5
Кромолин стабилизирует мембраны тучных клеток, угнетая высво-оождение из них медиаторов аллергии и активацию эозинофилов, ней-
-437-
Глава 23
грофилов, тромбоцитов и макрофагов, которые участвуют в формировании бронхоспазма.
Кромолин отличается от большинства медикаментов, применяемых при обструктивных заболеваниях дыхательных путей, тем, что исполь-зу ется только в качестве профилактического средства.
Препарат применяют при бронхиальной астме, а также для профилактики сезонных, постоянных и вызываемых физической нагрузкой приступов астмы и при аллергических ринитах.
Синонимами препарата являются интал. оптикром, аллергоспазмин, ломупрей и многие другие.
Кортикостероиды
Многие кортикостероиды используются в качестве ингаляционных средств при бронхиальной астме.
Кортикостероиды не являются бронходилататорами, и принято считать. что их действие просто сводится к противовоспалительному иммунодепрессивному эффекту, что весьма положительно сказывается на течении бронхиальной астмы.
Они широко используются при обструктивных заболеваниях дыхательных путей, однако не применяются при острых приступах бронхиальной астмы.
Множество побочных реакций, вызываемых ими при систематическом применении, в определенной степени ограничивает их применение в качестве средств для лечения бронхиальной астмы.
При обструктивных легочных заболеваниях, в основном используются преднизолон (27.1.33), метилпреднизолон (27.1.38), дексаметазон (27.1.51) оеклометазон (23.3.10) и флунизолид (23.3.11). Синтез и детальное описание некоторых из них будут приведены в гл. 27 «Кортикостероиды».
-438-
Препараты для лечения заболеваний дыхательной системы
Адренергические препараты (агонисты р2-рецепторов, симпатомиметики)
Одним из наиболее важных применений симпатомиметиков — агонистов р2-адренорецепторов является лечение обструктивных заболеваний дыхательных путей. Весьма вероятно, что эти препараты действуют путем повышения уровня циклического аденозинмонофосфата, избы-Iочное количество которого, очевидно, образуется в результате активации аденилатциклазы. Результатом действия агонистов р2-адреноре-цешоров является расслабление гладкой мускулатуры, в данном случае гладкой мускулатуры бронхов, и, одновременно, ингибирование высвобождения аллергогенных медиаторов.
Одними из первых агонистов p-адренорецепторов, применяемых ранее и в настоящее время в качестве бронхолитика, являются эпинефрин или адреналин, изопротеренол и, особенно, эфедрин. При лечении и профилактике обструктивных заболеваний дыхательных путей используются также и другие агонисты p-адренорецепторов, такие как изоэтарин. тербуталин, альбутерол, метапротеренол, а также описываемые в настоящем разделе фенотерол (23.3.16), пирбутерол (23.3.22), прокжерол (23.3.25).
Син tea и фармакологические свойства первых шести из рассматриваемых препаратов — эпинефрина (адреналина) (11.1.2). изопротеренола (11.1.8), альбутерола (11.1.26), тербуталина (11.1.12), метапро-теренола (11.1.15), изоэтарина (11.1.11), а также эфедрина (11.3.4) — были описаны в гл. 11 ^Адренергические (симпатомиметические) препарат ы».
но
СН-Сн2-'^~СН{СН3/г он
1118
но
>=v <^5
НО—й сН“ск”NH-CH(CH3k
'—' он
-439-
Глава 23
-Crl2-NM-C(CHjh
СНз
Ch—СН—NH—СН OH
3
1134
Фенотерол (Fenoterol)
Фенотерол — 3,5-дигидрокси-а[[(-я-гидрокси-а-метилфенэгил)-амино]метил]-бензиловый спирт (23.3.16) синтезируют исходя из 3,5-диацетоксиацетофенона, бромированием которого получают 3,5-диацетоксибромацетофенон (23.3.12). Последний вводят во взаимодействие с 2-бензиламино-1-(4-метоксифенил)-пропаном с получением соответствующего третичного амина (23.3.13). Гидролизом ацетильных групп в последнем и удалением защитной бензильной группы восстановлением водородом с использованием в качестве катализатора палладия на угле получают вторичный амин (23.3.14). Подвергая последний действию бромистоводородной кислоты расщепляют эфирную связь в бензольном кольце с по-л^чением фенольного производного (23.3.15). Наконец, восстановлением карбонильной группы в последнем водородом получают искомый фенотерол (23.3.16) [24, 25, 26].
СН2-СбН5 h-n-ch-ch2 СНз
-440 -
Препараты для лечения заболеваний дыхательной системы
н-с
Н-0 23 3 15
Фенотерол — препарат, избирательно стимулирующий р2-адре-норепепторы. Препарат расширяет бронхи и кровеносные сосуды, оказывает выраженное токолитическое действие, уменьшает сократительную активность и понижает тонус матки. Основное применение находит при преждевременных родах.
Синонимами препарата являются беродуал, беротек, дуовент и др.
Пирбутерол (Pirbuterol)
Пирбутерол — ,1-диметилэтил]амино]метил]-3-гидрокси-
2.6-пиридиндиметанол (23.3.22) синтезируют исходя из 3-гидро-ксипиридина, последовательно подвергая его гидроксиметилированию и далее алкилированию бензилхлоридом по ароматической гидроксильной группе с получением 3-бензилокси-2,6-бг/с-(гидро-ксиметил)пиридина (23.3.17). Селективным окислением 6-гидро-ксиметильной группы двуокисью марганца получают 3-бензилокси-2-гидроксиметилппперидин-б-альдегид (23.3.18). Конденсацией последнего с нитрометаном получают соответствующий нитрометил-карбинол (23.3.19), нитрогруппу которого восстанавливают до аминной водородом, с использованием в каестве катализатора никеля Ренея с получением а.миноспирта (23.3.20). Алкилированием аминогруппы последнего трет-о\тилбромидом получают вторичный амин (23.3.21), снятием защитной бензильной группы которого восстановлением водородом получают пирбутерол (23.3.22) [27, 28, 29.30,31,32,33].
1 неон
2 СдНбСНгВг^
№02
ZJ;^x_xOCH2C6H5
А I
н-с n снгОн о 23 3 18
CH3NO2
—СНд-
ОСН2С5Н5
сн2он
ши к, ^•'OCHoCgHg
Н2 Raney—ГФ
H2N—СН2—Сг* N СН2ОН1
(СНз)зСВг
-441 -
Г лава 23
^гх^осн2с6н5 (CHjjjCNH-CHi-CH^N^C^OH он
Н2 Pd—С
(CH3)3CNH-CH2-CH
он
23 3 22
он
СН2ОН
23 3 Л
Этот относительно селективный агонист р2-адренорецепторов структурно весьма похож на альбутерол и проявляет сходные с ним брохолитические свойства Применяется в качестве ингаляционного средства при течении бронхиальной астмы
Синонимами препарата являются эксирел и максайр
Прокатеро i (Procaterol)
Прокатерол 5-[1 гидрокси-2-[(1-метилэти 1)амино]бу гил] 8-гид-рокси 2(1Н)хинолон (23 3 25) получают ацилированием 8-гидрокси-2(1Я)хинопона хлорангидридом 2-броммасляной кислоты по пятому положению хинолиновой системы с получением соединения (23 3 23) Подвергая последнее действию изопропиламина, получают аминокетон (23 3 24), карбонильную группу которого восстанавливают боргидридом натрия с получением прокатерола (23 3 2о) [34 35,36 37,38]
о
С2НО-СН-С-CI А1С10
Вг
(CH3)2CH-NH2
23 3 24
NaBH4
23 3 25
Гак же как и пирбу герол прокатерол проявляет сходные с альбутеролом и брохолитические свойства с несколько большей продолжительностью действия Его рекомендовано применять в качеслве ингаляционного средства при лечении бронхиальной астмы
Синонимами препарата являются онсукил масакин, прокадил, меп-тин и др
-442-
Препараты для лечения заболеваний дыхательной системы
1 2
3 4
5 6
7 8
9 10 И
12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38
Список литературы
I S Pat 2 676 177 (1954)
SchniderO etab Heh Chim Acta 34,2211 (1951)
LSPat 2 714 608 (1955)
US Pat 2 714 609 (1955)
Bnt Pat 628 497 (1948)
Span Pat 212 920 (1954)
Ger Pat 3 106 995 (1982)
I S Pat 4 390 732(1983)
Marie E J Chem Soc 10L 305 (1912)
1'aleH etaPJ Am Chem Soc 72,3710(1950)
LS Pat з 091 569 (1963)
US Pat 3 184 505 (1965)
Smith H et al J Org Chem 26, 820 (1961)
Ger Pat 834 105 (1949)
Traube IV /Вег 33, 3035 (1900)
Гринберг А Ж П X 13, 1461 (1940)
Гебнер 4 и др//Ж Общ X 16, 179 (1946)
US Pat 3 505 337 (1967)
S Afr Pat 67 07 766(1970)
Schulz IV et al n Arzneimittel-Forsch 26. 960 (1976)
Bnt Pat 1 144 905 (1966)
LSPat 3 419 378 (1968)
Baker G et al 'J Med Chem 16, 87 (1973)
US Pat 3 341 593 (1967)
Ger Pat 1 286 047 (1962)
Belg Pat 640 433 (1962)
US Pat 3 700 681 (1972)
US Pat 3 763 173 (1973)
US Pat 3 772 314 (1973)
4 S Pat 3 786 160(1974)
US Pat 3 948 919 (1976)
LSPat 4 031 108 (1977)
Ger Pat 2 204 195 (1972)
Belg Pat 833 841 (1975)
LSPat 4 026 897 (1977)
loshizaki S et al' Chem PharmBul 28, 3441 (1980) \akava*aK et al/О Med Chem 19.1138 (1976) loshizaki S et al //J Med Chem 20, 1103 (1977)
-443-
‘ Глава 24
« Антикоагулянты,
} антиагреганты,
, тромболитики, гемостатики
Препараты, препятствующие тромбообразованию и предотвращающие свертывание или образование новых сгустков крови, называются антикоагулянтами.
Препараты, уменьшающие агрегацию тромбоцитов крови, называются антиагрегантами
Препараты, ускоряющие лизис уже образованных сгустков крови, называются тромболитиками, или фибринолитиками
Препараты, способствующие уменьшению и остановке кровотечения, называются гемостатическими препаратами.
Коагуляционныый и фибринолитический процессы являются очень важными защитными физиологическими механизмами организма, и только очень тонко отрегулированное взаимодействие между ними обеспечивает равновесное гомеостатическое состояние сосудистой системы.
В нормальных условиях образование микроскопических сгустков крови часто бывает необходимо для восстановления поврежденной части сосудов. При этом поврежденный сосуд восстанавливается путем обновления его эндотелиальной поверхности, а образующийся в этом случае нерастворимый сгусток далее эффективно удаляется фибринолитической системой путем его протеолитического расщепления на растворимые фрагменты.
Процесс образования сгустков крови и последующего их лизиса представляет собой очень сложную картину, зависящую о г множества веществ — факторов коагуляции (фибриноген, протромбин, тромбопластин. кальций, ангигемофильный фактор и др). присутствующих в плазме, клетках крови и в меньшей степени в других тканях.
Процесс агрегации тромбоцитов и его hhi ибнрование весьма выражено регулируется системой тромбоксан—простациклин. Тромбоксан Ат усиливает агрегацию, а простациклин (простагландин 12) препятствует агрегации.
-444-
A
н т и к о агулянты, антиагреганты, тромболитики, гемостатики
Стимуляторами агрегации являются также простагландин Е2, коллаген сосудистой стенки, тромбин, аденозиндифосфат, серотонин и кате-коламины.
Ингибиторами агрегации являются простагландин Е,, аденозинмонофосфат, аденозин, метилксантины, антагонисты серотонина, гепарин и др.
Нарушение эндогенного контроля над коагуляционным и фибринолитическим процессами может привести к тяжелым последствиям.
С одной стороны, нелимитируемое инициирование коагуляции может привести к тромбозам и. соответственно, к ишемии, инфарктам или смерти. С другой стороны, сбой механизмов коагуляции может привести к геморрагии. Соответственно, в зависимости от характера нарушения, при котором могут возникнуть клинические проблемы разной сложности, оба состояния требуют коррекции. С этой целью применяют антикоагулянты, антиагреганты, тромболитики и гемостатики.
24.1. Антикоагулянты
Антикоагулянты предотвращают развитие процесса коагуляции крови. Терапия антикоагулянтами, в первую очередь, направлена на предотвращение образования тромбов в сосудах, являющегося основной причиной смерти при тромбоэмболических заболеваниях.
Антикоагулянты подразделяются на антикоагулянты прямого действия, т е. влияющие на факторы свертывания непосредственно в крови, и непрямого действия, т. е. влияющие на синтез факторов свертывания крови в печени. С другой стороны, антикоагулянты классифицируют как парентеральные и оральные препараты. Единственным представителем парентеральных антикоагулянтов является гепарин К оральным антикоагулянтам относится ряд производных кумарина (дикумарол, этилбискумацетат, варфарин, фенпрокумон и аценокумарол) и инданона (фениндион, анизиндион).
Антикоагулянты прямого действия, или парентеральные антикоагулянты
К первому типу антикоагулянтов прямого действия относится гепарин.
Гепарин (Heparin)
Гепарин — природный антикоагулянт, образующийся в организме.
Источниками коммерческого гепарина являются слизистая свиного кишечника и бычьи легкие [1, 2, 3, 4, 5].
-445-
Глава 24
Гепарин является смесью природных сульфатированных мукополисахаридов и, в основном, содержится в гранулах тучных клеток. Особенно много гепарина содержится в печени и легких. Лизосомы тс иных клеток содержат протеазы и гликозидазы, которые очевидно разрушают содержащийся в них гепарин — протеоглюкан, образуя разнообразные сульфатированные олигосахариды, которые и представляют собой гепарин, присутствующий во внеклеточном пространстве, а в виде очищенных образцов используемый в клинике. Гепарин активен только при парентеральном введении. Наиболее часто его применяют внутривенно.
Гепарин - гетерогенная смесь сульфатированных полисахаридов, построенных из повторяющихся единиц D-глюкозамина, D-глюко-роновой и L-идуроновой кислот. Собственно коммерческий гепарин является смесью ряда соединений с разной длиной цепи с молекулярной массой от 5000 до 30 000. Моносахариды, образующие гепарин, модифицированы либо N-ацетильными, либо N- или О-сульфатными группами и соединены гликозидными связями, образуя полимеры типа 24 1.6 с разной длиной цепи. Основными моносахаридами, образующими гепарин, являются 6-сульфат 2-дезокси-2-сульфамино a-D-глюкозы (24 1.1), 2-сульфат a-L-идуроновой кислоты (24.1.2), 2-ацетамидо-2-дезокси a-D-глюкозы (24.1.3), p-D-глюкороновая кислота (24.1.4) и a-L-идуроковая кислота (24.1.5). Эти сахара присутствуют в коммерческом гепарине в убывающем порядке (24.1.1) > (24.1.2) > (24.1.3) > (24.1.4) > (24.1.5). Ввиду наличия в молекулах сульфатных и карбоксильных рупп гепарин является сильнокислым соединением, которое частично нейтрализуется замещением кислых атомов водорода в сульфатных группах ионами Na'.
NhSOa
24 1 1
-446 -
А н ти к о агулянты, антиагреганты, тромболитики, гемостатики
Полагают, что гепарин действует путем нейтрализации ряда активирующих факторов свертывания крови, нарушая переход протромбина в тромбин.
Гепарин применяют для предотвращения тромбообразования при инфаркте миокарда, при тромбозах и эмболиях, для поддержания жидкого состояния крови в аппаратах искусственного кровообращения и гемодиализа.
Синонимами препарата являются артевен, гепален, лепаран, ликве-мин, пангеприн, ветрен и многие другие.
Антагонист гепарина
Антагонистом гепарина, использующимся при его передозировках, является протамин — смесь протеинов, которую выделяют из спермы рыб. Взаимодействуя с гепарином, он инактивирует его путем образования нерас творимого комплекса.
К антикоагулянтам прямого действия относится и цитрат натрия, который используется для стабилизации крови при ее консервации. Полагают, что его антикоагулянтное действие заключается в связывании ионов Са* '. необходимых для превращения протромбина в тромбин.
Антикоагулянты непрямого действия, или энтеральные антикоагулянты
Наиболее широко используемые в клинике энтеральные антикоагулянты являются структурными производными 4-гидроксикумарина — соединения, которое был выделено из сладкого клевера и явившегося причиной фатального геморрагического диатеза у скота в 1920-х годах, так называемой «болезни сладкого клевера». После выявления того факта, что именно кумарины способны подавлять синтез протромбина, были развернуты интенсивные исследования в области синтеза кумариновых производных, в результате чего в медицину были внедрены ди-кумарол (бисгидроксикумарин), этилбискумацетат, варфарин. фенпро-кумон и аценокумарол.
Их терапевтическое действие зависит от способности подавлять образование в печени ряда функциональных факторов свертывания крови. Эти факторы описаны как витамин К-зависимые факторы, поскольку их оиосинтез гепатоцитами частично связан с метаболизмом гепатического витамина К. Оральные антикоагулянты эффективны только in vivo, поскольку принцип их действия заключается в угнетении в печени синтеза протромбина, проконвертина и других факторов свертывания крови. Их иногда условно называют антагонистами витамина К.
-447 -
Глава 24
Дикумарол (Dicoumarol)
Дикумарол - 3,з’-метилен-бис-(4-гидроксикумарин) (24.1.8) получают исходя из 4-гидроксикумарина (24.1.7), который, в свою очередь, получают из метилового эфира салициловой кислоты путем его циклизации в хромоновое производное с использованием натрия или метилата натрия либо из о-оксиацетофенона, путем взаимодействия с диэтилкарбонатом в присутствии этилата натрия. Конденсацией полученного 4-гидроксику.марина в качестве фенольного компонента с формальдегидом получают дикумарол [6, 7, 8, 9].
Na
CzHgONa
О
+ I
с2н5о СС2Н5
Препарат применяют в целях профилактики и лечения тромбозов, тромбофлебитов, тромбоэмболий, для предупреждения тромбообразо-ваиия в послеоперационном периоде.
Синонимами препарата являются бисгидроксикумарин, дикумол, кромолин и др.
Этилбискумацетат (Ethyl Biscoumacetate)
Этилбискумацетат — этиловый эфир бмс-(4-гидрокси-3-ку.маринил)-уксусной кислоты (24.1.9) получают аналогично из 4-гидроксикумарина, но с использованием вместо формальдегида глиоксиловой кислоты, этилглиоксилата или его полуацеталя [10, 11. 12, 13].
он
24 1 7
ОН
С2Н5О-СН-СООС2Н5
-448-
д н т и к о агулянты, антиагреганты, тромболитики, гемостатики
Препарат применяют по тем же показаниям, что и дикумарол.
Синонимами препарата являются неодикумарин, этилдикумарол, тримексан. дикумацил и др.
Варфарин (Warfarin)
Варфарин — 3-(а-ацетонилбензил)-4-гидроксикумарин (24.1.10) синтезируют по реакции Михаэля присоединением бензальацетона к 4-гидроксикумарину (24.1.7) в присутствии пиридина [14, 15, 16, 17. 18. 19].
24 1 7
Варфарин показан в качестве антикоагулянта .для предотвращения и лечения глубокого венозного тромбоза и легочной эмболии.
Синонимами препарата являются кумадин, панварфин, софраин, варнерин и др.
А ценокумарол (Acenocoumarol)
Аценокумарол — 3-(а-ацетонил-и-нитробензил)-4-гидроксикума-рин (24.1.11) получают по схеме, совершенно аналогичной получению варфарина, но с использованием л-нитробен-зальацетона [20].
Препарат применяют по тем же показаниям для предотвращения и лечения тромбозов и легочной эмболии.
Синонимом препарата является синтром.
Фенпроку.мон (Phenprocoumon)
Фенпрокумон — 3-(а-этилбензил)-4-гидроксикумарин (24.1.14) получают ацилированием натриевой соли диэтилового эфира (1-фенилпропил)малоновой кислоты хлорангидридом ацетилсалици-
-449-
Глава 24
левой кислоты с получением соединения (24.1.12), которое под действием .метилата натрия циклизуется в 3-(а-этилбензил)-2-карбэто-кси-4-гидроксикумарин (24.1.13). Щелочным гидролизом последнего и дальнейшим декарбоксилированием получают фенпрокумон (24.1.14) [21, 22, 23, 24, 25. 26, 27, 28].
ОС2Н5
СОСНз
C2H5ONa
24 1 13
NaOH
24 1 12
Фенпрокумон применяют по тем же показаниям, что и все описанные выше препараты.
Синонимами препарата являются маркумар и ликвамар.
Фениндион (Phenindion)
Фениндион — 3-фенилиндан-1,3-дион (24.1.16) предложено получать двумя путями. Первый способ заключается в конденсации бензальдегида с фталидом в присутствии этилата натрия. Очевидно образующийся при этом фенилметиленфталид (24.1.15) в условиях реакции перегруппировывается в искомый фениндион (24.1.16). Второй способ заключается в конденсации фенилуксусной кислоты с фталевым ангидридом с образованием и выделением фенилме-тиленфталица (24.1.15), который далее в присутствии алкоголята натрия перегруппировывают в фениндион [29].
CoHjONa
-450-
Антикоагулянты, антиагреганты, тромболитики, гемостатики
Аналогично производным кумарина фениндион, соединение класса индандпонов. действует путем изменения биосинтеза коагулирующих протеинов в печени.
Препарат применяют в целях профилактики и лечения тромбозов, громбофлебитов, тромбоэмболий. Однако вследствие вызываемых им ряда побочных эффектов, таких как полиурия, полидипсия, тахикардия и др., редко используется в практической медицине.
Синонимами препарата являются пиндион. биндан, гедулин, индон, фенилин,ректадион.
Анизиндион (Anisindione)
Анизиндион — 3-(«-метоксифенил)индан-1,3-дион (24.2.11) отлича-егся от фениндиона лишь наличием «-метоксильной группы в фе-"нильном кольце. Его синтезируют совершенно аналогичными способами. что и фениндион. но с использованием «-метоксибензальдегида или «-метоксифенил} ксусной кислоты [30, 31, 32].
Препарат применяют по тем же показаниям, что и фениндион. Синонимами препарата являются унидон и мирадон.
24.2. Антиагреганты
Ингибиторы агрегации тромбоцитов крови имеют большое практическое значение и в значительной степени регулируются системой тромбоксан-простациклин.
Тромбоксан А2 увеличивает, а простациклин (простагландин 12) препятствует агрегации тромбоцитов крови.
Стимуляторами агрегации являются также простагландин Е;. коллаген сосудистой стенки, тромбин, аденозиндифосфат, серотонин и катехоламины.
-451 -
Глава 24
Ингибиторами агрегации являются также простагландин Et, аденозинмонофосфат, аденозин, мегилксантины, антагонисты серотонина, гепарин и др.
Практическое значение в медицине в качестве ингибиторов агрегации тромбоцитов крови приобрели такие нестероидные противовоспалительные жаропонижающие анальгетики, как аспирин, индометацин, ибупрофен и др., которые, блокируя циклооксигеназу, предотвращают трансформацию арахидоновой кислоты в тромбоксан А2. Другие ингибиторы агрегации тромбоцитов крови, такие как коронарорасширяющий препарат дипиридамол, а также тиклопидин, модулируют активацию тромбоцитов.
Аспирин (Aspirin)
Синтез и свойства препарата (3.2.2) описаны в гл. 3 «Анальгетики».
соон о
Х.о-с-сн3
3.2.2
Аспирин является ингибитором циклооксигеназы, что выражается в нарушении синтеза тромбоксана А2 и простациклина (простагландина 12), которые являются функциональными антагонистами. Причем синтез тромбоксана А2 подавляется в большей степени при использовании аспирина в малых дозах. Применение аспирина уменьшает риск инфаркта миокарда и повышает выживаемость больных с инфарктом миокарда. Снижает опасность инсульта при нарушениях мозгового кровообращения.
Сульфинпиразон (Sulfinpyrazone)
Синтез препарата (3.2.8) описан в гл. 3.
328
-452-
Антикоагупянты, антиагреганты, тромбопитики, гемостатики
Сульфинпиразон используется в медицине в качестве нестероидного противовоспалительного жаропонижающего анальгетика, однако полагают, что он ингибирует циклооксигеназу тромбоцитов. Кроме того возможно также, что его действие связано и с действием на мембрану тромбоцитов и уменьшением количества высвобождаемых при этом аденозиндифосфата и серотонина, способствующих агрегации тромбоцитов.
В огличие от аспирина он не воздействует на лиц, не имеющих нарушений системы агрегации.
Индометацин (Indometacin)
Синтез и свойства индометацина (3.2.51) описаны в гл. 3.
3 2 51
Индометацин так же, как и аспирин обратимо ингибирует действие циклооксигеназы, блокируя образования тромбоксана А2.
Дипиридамол (Dipiridamol)
Синтез препарата (19.4.13) был описан гл. 19 «Антиангинальные препараты».
N сн2-сн2-он
НО- СН2 —CH2-N N у
но-сн2—сн2 м
194 13
Дипиридамол известен в качестве коронарорасширяюшего средства, однако он обладает и определенной антиагрегантной активностью.
-453 -
Глава 24
Его предложено применять для предупреждения тромбообразования после протезирования клапанов сердца в сочетании с варфарином. Механизм антиагрегантного действия дипиридамола не совсем ясен, а эффективность сомнительна.
Тиклопидин (Ticlopidin)
Тиклопидин — 5-(о-хлорбензил)-4,5,6,7-тетрагидротиено[3,2-с]пи-ридин (24 2 1) предложено получать разными способами [33, 34, 35, 36, 37. 3S, 39]. Первый способ заключается в N-алкилировании 4,5,6,7-тетрагидротиено[3,2-с]пиридина 2-хлорбензилхлоридом.
Согласно второму способу, тиено[3.2-с]пиридин подвергают N-ал-килированию 2-хлорбензилхлоридом, и полученную таким образом соль пиридиния (24.2.2) далее восстанавливают боргидридом натрия до искомого гиклоиидина
CH-CI
№ВН4 ---► 24 2 1
Наконец, третий способ синтеза заключается в алкилировании тиофена окисью этилена с получением 2-(2’-гидрокси)этилтиофена (24.2 3), взаимодействием которого с н-толуолсульфохлоридом получа-Ю1 соответствующий тозилат (24.2.4) Замещением тозильной группы в последнем с использованием 2-хлорбензиламина получают амин (24 2.5), который в условиях реакции хлорметилирования циклизуется в искомый тиклопидин (24 2.1).
24 2 3
сн2-сн2-он
СНз
SO2~CI
-454 -
д н г и к о агулянты, антиагреганты, тромболитики, гемостатики
24 2 5
Тиклопидин подавляет агдезию тромбоцитов и обладает антиагре-гашной активностью. Предполагается, что его действие связано с дей-сгвием на мембрану тромбоцитов и уменьшением количества высвобождаемых аденозиндифосфата и серотонина, способствующих агрегации тромбоцитов.
В широкомасштабных клинических испытаниях тиклопидин выявил ряд преимуществ по сравнению с аспирином.
Синонимами препарата являются тиклид, анагрегал, тиклозан и др.
24.3. Фибринолитики (тромболитики)
Фибринолитики — это соединения, способные растворять уже образовавшиеся тромбы. Принцип их действия заключается либо в активации-физиологической системы фибринолиза, активируя фибринолитический фермент плазмин (фибринолизин), стимулирующего лизис и выведение существующих сгустков крови, либо в восполнении недос-1аюшего фибринолизина. (Гепарин и оральные антикоагулянты неэффективны в плане уменьшения размера уже существующих фибриновых сгусгков.)
С целью активации эндогенного фибринолитического механизма обычно вводят активаторы плазминогена. Наиболее часто применяется такие препараты, как стрептокиназа и урокиназа.
Полагают, что стрептокиназа взаимодействуя с плазминогеном, вызывает изменение конформации плазминогена, в результате чего разрываекя ряд пептидных связей в плазминогене и образуется плазмин
Считается также, что в отличие от стрептокиназы, урокиназа непосредственно расщепляет плазминоген в плазмин, который и вызывает разрушение фибрина и других составляющих сгустков крови.
Однако их применение связано с определенным риском возникновения кровотечений из-за возможного разру шения в плазме фибриногена и дру гих коагулирующих факторов.
Принципиально новым типом фибринолиз иков является активатор 1качевою плазминотена — альтеплаза.
-455-
Глава 24
Стрептокиназа (Streptokinase)
Стрептокиназа является белком, продуцируемым определенными штаммами гемолитического стрептококка группы С, и является первым клинически полезным фибринолитиком [40, 41. 42, 43, 44, 45]. В отличие от других активаторов плазминогена, стрептокиназа не является ферментом и сама не расщепляет какие-либо связи в молекуле плазминогена. Она создает эквимолекулярное соединение с плазминогеном, образуя комплекс стрептокиназа-плазминоген. В плазминогенной части полученного комплекса при этом возникают определенные конформационные изменения, приводящие к разрыву некоторых пептидных связей и трансформирующие этот комплекс в комплекс стептокиназа-плазмин, или свободный плазмин, которые и расщепляют фибрин.
Препарат имеет время полужизни в плазме 15-30 мин и используется внутривенно для лечения пациентов с острыми массивными эмболиями легких и тромбов вен. а также при инфарктах миокарда.
Недавно предложен ряд производных стрептокиназы, в частности ацетилированных, которые разрабатываются для применения в качестве фибринолитиков.
Синонимами препарата являются кабикиназа, стрептаза и др.
Урокиназа (Urokinase)
Урокиназа — фермент, экстрагируемый из человеческой мочи или клеток почек человека [46, 47. 48, 49, 50, 51, 52, 53, 54, 55]. который непосредственно расщепляет весьма определенную пептидную связь (Arg-560-Val561) в молекуле плазминогена, трансформируя таким образом его в плазмин.
Показания к применению такие же, как и у стрептокиназы. Синонимом препарата является аббокиназа и др.
Альтеплаза (Alteplase)
Альтеплаза является препаратом человеческого активатора тканевого плазминогена (t-PA) и представляет собой гликопротеин молекулярной массы 68 000, синтезируемый сосудистыми эндотелиальными клетками.
Молекулы t-PA впервые были выделены из среды культивируемых клеток меланомы человека [56, 57, 58], но в настоящее время методами генной инженерии получают также генетически рекомбинантную фор-м\ rt-PA.
-456-
Анти коагулянты, антиагреганты, тромболитики, гемостатики
Являясь главным эндогенным промотором фибринолиза, t-PA свя-зываекя с фибрином и так же, как и урокиназа, расщепляет пептидную связь Arg-560 — Val-561 в молекуле фибрин-связанного плазминогена, превращая последний в активную молекулу плазмина, который вызывает расщепление фибриновых сгустков. Его действие локализуется в тромботических областях, и, соответственно, вероятность возникновения системного фибринолиза при его применении намного ниже, чем та, которая может возникнуть при применении стрептокиназы и урокиназы.
Обе формы (t-PA и rt-РА) имеют очень короткое время полураспада (около 3 мин) и быстро выделяются из организма, в основном, печенью. Поэтому они применяются только инфузионным методом. Препарат можно назначать пациентам, которым противопоказана стрептокиназа (например, пациентам, недавно перенесшим стрептококковую инфекцию).
Синонимом препарата является активаза и др.
24.4. Гемостатики (прокоагулянты)
Гемостатики являются средствами, применяемыми для остановки кровотечений местно или при их резорбтивном действии.
' Иногда после травм, хирургических вмешательств, маточных кровотечений, при циррозе печени, передозировке фибринолитиков и из-за других причин активность системы фибринолиза в организме может повыситься настолько, что может стать причиной кровотечений.
Гемостатики, применяемые в настоящее время в медицине действуют либо путем замещения дефицита факторов свертывания (антиге-мофильный фактор), который получают из человеческой плазмы, либо повышением концентрации эндогенных факторов свертывания в плазме (десмопрессин) или ингибированием естественного механизма фибринолиза (аминокапроновая кислота, транексамовая кислота).
Системные гемостатики
Аминокапроновая кислота (Aminocaproic Acid)
Аминокапроновая кислота — 6-аминогексановая кислота (24.4.1) получается гидролизом s-капролактама при высокой температуре [56,*57, 58, 59, 60,61.62].
h2n-сн2-сн2-сн2-сн2-сн2-соон
24 4 1
- 457 -
Глава 24
Поскольку связывание плазминогена или плазмина с фибриногеном или фибрином опосредуется лизиновыми группами, имеющимися в структурах фибрина и фибриногена, аминокапроновая кислота, являясь структурным аналогом лизина и отличаясь от него отсутствием лишь одной аминогруппы, выступает в качестве конкурирующего ингибитора для связывания плазмин(оген)а с фибрином. Аминокапроновая кислота сдвигает гомеостатический баланс в сторону коагуляции, восстанавливая при этом активность фибринолитического механизма.
Аминокапроновая кислота, не являясь прокоагулянтом, как таковая используется при хирургических вмешательствах и различных патологических состояниях, сопровождающихся повышением фибринолитической активности крови и тканей. Препарат используется для остановки кровотечений.
Синонимами препарата являются афибрин, капрацид, эпсиламин. кофламин и др.
Транексамовая кислота (Tranexamic acid)
Транексамовая кислота — /и/?анс-4-(а.минометил)циклогексанкар-боновую кислоту (24.4.5) синтезируют исходя из 4-метил-бензонитрила, окислением метильной группы которого получают мононитрил терефталевой кислоты (24.4.2). Нитрильную группу в последнем восстанавливают водородом с использованием в качестве катализатора никеля Ренея. Бензольное кольцо полученной при этом 4-аминометилбензойной кислоты (24.4.3) восстанавливают до циклогексильного водородом с использованием в качестве катализатора платины с получением изомерной смеси 4-аминометил-циклогексанкарбоновой кислоты (24.4.4), из натриевых солей которой кристаллизацией выделяют требуемый транс-изомер (24.4.5) [63, 64, 65, 66, 67, 68].
24 4 4
24 4 5
Транексамовая кислота также рассматривается в качестве структурного аналога лизина. Полагают, что она работает по тому же механизму,
-458-
Антикоагупянты, антиагреганты, тромболитики, гемостатики
что и аминокапроновая кислота, однако активнее последней в 6-10 раз. Препарат ингибирует действие активатора плазмина и плазминогена, обладает гемостатическим действием. Его применяют при кровотечениях или риске кровотечений на фоне усиления фибринолиза (злокачественные новообразования, послеродовые кровотечения, желудочно-кишечные кровотечения, гематурия и т. п.)
Синонимами препарата являются угурол, циклокапрон, амкацид. транекс и др.
Антигемофильный фактор (Antihemophilic Factor)
Антигемофильный фактор — белок, превращающий протромбин в тромбин и замещающий дефицит эндогенного гемофильного фактора, получают переработкой человеческой плазмы [69. 70].
Препарат применяют при лечении классической гемофилии А, для остановки кровотечений.
Синонимами препарата являются гемофил Т, моноклат, криоблин и др.
Десмопрессин (Desmopressin)
Десмопрессин - - 8-П-аргининвазопрессин-1-(3-меркаптопропио-новая кислота) (24.4.6) является структурным аналогом вазопрессина. Препарат получают многостадийным синтезом, методами специфичными для пептидной химии, и поэтому его синтез здесь не рассматривается [71, 72, 73, 74].
Mep-Tyr-Phe-GIn-Asn-Cys-Pro-D-Arg-Gly-NH 2
24 4 6
Наряду с основным использованием препарата в качестве антидиуретика при лечении диабета его применяют и при лечении классической гемофилии А.
Синонимом препарата является DDAVP.
Местные прокоагулянты
Тромбин (Thrombin)
Для остановки местных кровотечений часто применяют тромбин — препарат естественного тромбина, катализирующий превращение фибриногена в фибрин.
-459-
Г лава 24
Тромбин получают из бычьей плазмы [75, 76, 77] и применяет дтя остановки кровотечения из открытых сосудов, когда невозможно применить другую технику
Препарат применяют для остановки мелких кровотечений
Синонимом препарата является тромбостат и др
Абсорбирующий жечатин (Gelatine Absorbable)
Абсорбирующий желатин применяется в виде стерильных пленок губок или пудры для наружного применения Он иногда применяется и орально при желудочных кровотечениях, холя и весьма ограниченно
Синонимами препарата являются гельфоам, гельфипьм и др
Микрофибриллярный коллагеновый гемостат (Microfibrillar Collagen Hemostat)
Микрофибриллярный коллагеновый гемостат получают из бычьего колпагена
Его исподьзуюг при хирургических вмешательствах в качестве вспомогательного средства при кровотечениях когда другие процедуры неэффективны и непрактичны
Синонимом препарата является авитен и др
Окисленная целлюлоза (Oxidized Cellulose)
Окисленная целлюлоза это хирургическая марля обработанная диоксидом азота При контакте с тканевыми жидкостями образует иску сел венные сгустки, обеспечивающие механический гемостаз
Синонимами препарата являются новоцел, оксицел, сурджицел И др
Список литературы
1 US Pat 2 884 358 (1959)
2 US Pat 2 989 438 (1961)
3 LSPat 3 016 331 (1962)
4 LS Pat 4 119 754(1978)
5 Bnt Pat 1 S39 332 (1976)
6 US Pat 3 419 578 (1968)
7 Bnt Pat 1 144 905 (1966)
8 Ger Pat 1 543 579 (1966)
-460-
Антикоагулянты, антиагреганты, тромболитики, гемостатики
9
10 И 12 13
14 Ь
16 П 18
19 20 21
22 23
24 2 о 26
27 28 '29
30 31 32 33
34 Зэ 36 37 38
39 40
41
42 43
44 45 46
47 48 49
Barker G etal J Med Chem 16,87 (1973)
US Pat 2 482 510(1949)
US Pat 2 482 511 (1949)
I SPat 2 482 512 (1949)
Stahmann 4 et al ‘ J Am Chem Soc 65, 2285 (1943)
US Pat 2 427 578 (1947)
I S Pat 2 777 859 (1957)
LS Pat 2 765 321 (1956)
US Pat 3 077 481 (1963)
US Pat 3 239 529 (1966)
West E etaP/J Am Chem Soc 83,2676 (1961)
LSPat 2 648 682 (1953)
LSPat 2 701 804 (1955)
LSPat 2 872 457 (1959)
LSPat 3 239 529(1966)
LSPat 2 723 276 (1955)
JW/tJetal Monatsh 87,218 (1956)
Schroder H etal /J Am Chem Soc 79,3291 (1957)
PohlL/J Med Chem 18,513 (1975)
IalenteE etal 'J Med Chem 21, 141,213 (1978)
DieckmannJ Ber 47 1439(1914)
US Pat 2 899 358 (1959)
Koelsch C //J Am Chem Soc 58, 1331 (1936)
Horeau J etal Bull Soc Chim France 1948,53 Majfrand J et al Z/Eur J Med Chem 9 483 (1974) Ger Pat 2 530 516 (1975)
I S Pat 4 127 580(1978)
US Pat 4 174 448 (1979)
Ger Pat 2 048 372 (1971)
LSPat 3 758 506 (1973)
Thutllier G et al ', Eur J Med Chem 9,625 (1974)
US Pat 2 666 729 (1954)
US Pat 2 701 227 (1955)
US Pat 3 063 913 (1962)
US Pat 3 063 914 (1962)
LSPat 3 107 203 (1963)
US Pat 3 138 542 (1964)
I S Pat 2 961 382 (1961)
LSPat 2 989 440(1961)
US Pat 3 081 236 (1963)
L’S Pat 3 256 158 (1966)
- 461 -
Глава 24
50. USPat. 4.160.697(1979).
51 USPat. 4.169.764 (1979).
52. Sobel G etal.'/Am. J. Physiol. 171 768 (1952).
53. White W et al.//Biochemistry. 5, 2160(1966).
54 LesukA et al.//Science 147,880 (1965).
55. Astrup T et al.//Proc. Soc. Exp. Biol. Med. 81, 675 (1952).
56 4strup T et al.'/Nature. 159, 681 (1947).
57. Astrup T et al.//Nature. 170, 929 (1952).
58. Tagnon H et al./Proc. Soc. Exp. Biol. Med. 70, 359 (1949).
59. Galat A et al. 7J. Am. Chem. Soc. 68, 2729 (1946).
60. Eck J.'-'Org. Syn. Coll. Vol. II. 28 (1943).
61. Meyers C et al./-Org. Syn. Coll. Vol. IV, 39 (1963).
62 Garamaise E et al., 'Can. J. Chem. 34, 743 (1956).
63. Einhorn A -'Justus Liebigs Ann. Chem. 310, 194 (1900).
64. Levine M et al./'J. Org. Chem. 24, 115 (1959).
65. Ger. Pat. 1.443.755 (1964).
66. Bnt. Pat. 1 202.189(1969).
67. Bnt. Pat 1.409.938 (1973).
68. US Pat. 3.499.925 (1970).
69 Hershgold A et al./ 'J. Lab. Clin. Med. 77, 185 (1971).
70. Legaz M et al.,'Methods Enzymol 45B, 83 (1976).
71. Fr. Pat. 1.540.536 (1968).
72. US Pat. 3.497.491 (1970).
73. Huguenm R et al.//Helv. Chim. Acta. 49, 695 (1966).
74. ZaoralM et al Coll. Czech. Chem. Commun. 32, 1250 (1970).
75. Seegers IF et al.,/J. Biol. Chem. 126,91 (1938).
76 Seegers W et al, 'J. Biol. Chem. 146. 511 (1942).
77 USPat. 2.398.077(1946).
• Глава 25
\ Тиреоидные гормоны
\ и антитиреоидные препараты
Эндогенные йодсодержащие тиреоидные гормоны — L-тироксин и L-трийодтиронин продуцируются щитовидной железой, которая посредством двух указанных йодсодержащих гормонов проявляет выраженный метаболический контроль практически над всеми клетками организма Путем контролирования скорости окислительных клеточных процессов эти гормоны участвуют в регуляции роста и развития организма, в формировании мозга и костной ткани, воздействуют на активность ЦНС, на сердечно-сосудистую систему, ЖКТ, метаболизм углеводов, жиров и белков, влияют на регуляцию температуры тела, мышечную активность, водно-электролитный баланс и воспроизводство, играя весьма важную роль для нормального физического и умствен-но! б разни гия В отличие от многих других гормонов, они воздействуют не на отдельные органы, а проявляют диффузный эффект на весь организм в целом.
Сишез. хранение и высвобождение тиреоидных гормонов щитовидной железой в основном регулируется тиреотропным гормоном, а йодиды, необходимые для их синтеза, обычно поступают в организм с пищей.
Болезни, связанные с щитовидной железой, являются результатом либо избыточного продуцирования тиреоидного гормона — гиперти-реоиди iM, либо его недостаточности — гипотиреоидизм. В обоих случаях результатом может быть зоб.
Основным клиническим применением тиреоидных гормонов явля-егся течение гипотиреоидизма.
Эта болезнь характеризуется понижением или отсутствием секреции эндогенных тиреоидных гормонов. При возникновении в детском возрасте она клинически может быть описана как кретинизм (детский гипотиреоидизм), а у взрослых как микседема (взрослый гипотиреоидизм), проявляющаяся в снижении умственной и физической работоспособности, угнетении обменных процессов в организме и отеками. Поскольку тиреоидная функция не восстанавливается, клинический эф-фек! используемых препаратов проявляется лишь в течение применения
-463-
Глава 25
тиреоидных гормонов. Применение Т1феоидных гормонов при гипотиреоидизме представляет собой заместительную терапию и не вылечивает самое заболевание.
В настоящее время для лечения гипотиреоидизма применяются очень небольшое число различных препаратов, таких как препараты щитовидной железы животных, а также синтетические препараты.
Используются тиреоидин — высушенный тиреоид, который готовится из бычьих, овечьих и свиных тиреоидных желез, а также тирогло-булин (пролоид) — очищенный экстракт свиных тиреоидных желез, синтетические препараты — левотироксин и лиотиронин, а также лот-рикс — смесь синтетических левотироксина и лиотиронина в соотношении 4:1.
При гиперфункции щитовидной железы секреция избыточного количества тиреоидных гормонов приводит к гипертиреоидному состоянию (базедова болезнь, зоб). При этом заболевании применяют препараты, угнетающие продукцию тиреотропного гормона в передней доле гипофиза (дийодтирозин), в щитовидной железе (пропилтиоурацил, метилтиоурацил, метимазол, карбимазол), а также препараты, разрушающие клетки фолликулов щитовидной железы (радиоактивный йод).
25.1. Препараты для лечения гипотиреоидизма
Гипотиреоидизм (микседема) возникает при сбоях работы щитовидной железы в выработке тиреоидного гормона. Лечение заключается в замещении этого гормона вышеуказанными препаратами. Предпочтительно лечение левотироксином, который представляет собой 3.5,3',5'-тетрайодтиронин. Используются также препарат лиотиронин — L-3.5.5'-трийодтиронин, а также лотрикс — смесь синтетических левотироксина и левотиронина в соотношении 4:1. Из препаратов животного происхождения используют тиреоидин, а также тиреоглобулин (пролоид).
Левотироксин (Levothyroxin)
Левотироксин — 1.-3-[4-(4-гидрокси-3,5-дийодо-фенокси)-3,5-ди-йодфенил]аланин (25.1.10) получают многостадийным синтезом исходя из 4-гидрокси-3-йодо-5-нитробензальдегида. Взаимодействием последнего с бензолсульфохлоридом в пиридине получают соответствующий бенюлсульфонат (25.1.1), бепзолсульфонильную группу в котором с легкостью замещают на 4-метоксифенилоксильную взаимодействием с 4-мегоксифенолом. Полученный 3-йодо-4-(4-
-464-
Тиреоидные гормоны и антитиреоидные препараты
метоксифенокси)бензальдегид (25.1.2) далее вводят во взаимодействие с а-ацетилглицином в присутствии ацетата натрия по типу-реакции Кневенагеля, при которой образующееся илиденовое соединение циклизуется в производное оксазолона (25.1.3). Оксазолоновое кольцо последнего размыкают взаимодействием с метилатом натрия с получением искомого производного кротоновой кислоты (25.1.4). Нитрогруппу в последнем восстанавливают до аминогруппы водородом в присутствии в качестве катализатора никеля Ренея, с получением соответствующего амина, и далее последовательным диазотированием и замещением диазогруппы на йод получают ме-т иловый эфир а-ацетамидо-3,5-дийодо-4-(4-метоксифенокси)кро-тоновой кислоты (25.1.6). Далее, подвергая полученное соединение одновременному воздействию йодистоводородной кислоты и фосфора в уксусной кислоте, восстанавливают двойную связь в полученном производном кротоновой кислоты и снимают метоксильную защиту с фенольного кольца. При этом одновременно гидролизуется ацетильная группа при атоме азота и получается D,L-3,5-дийодтиронин (25.1.7). Аминогруппу в последнем вновь защищают формилированием муравьиной кислоты в присутствии уксусного ангидрида с получением П,Ь-М-формил-3,5-дийодтиронина. Разделение изомеров в полученном рацемате осуществляют с использованием бруцина с получением П-(-)-К-формил-3,5-дийодтиронина Г-(+)-М-фор.мил-3,5-дийодтиронина (25.1.8). Защитную формильную группу в последнем вновь гидролизуют с использованием бромистоводородной кислоты с получением ГД-Д-З,5-дийодтиронина (25.1.9), который повергают прямому йодированию с использованием йода в присутствии йодистого калия в водном метиламине с получением искомого левотироксина [1, 2, 3, 4].
JH3CONH-CH2-COOH
-465-
Г лава 25
Н2 Rare,— Ni
1 NaNOj H2SO4
2 KI >2 <КН2)гСО
l2Z KI / CH3NH2
Racemate resolution
—CH—СООН NH2
25 1 10
Эффекты препарата сильно зависят от доз. В малых дозах левотироксин обладает анаболическим действием. В средних дозах стимулирует рост и развитие тканей, метаболизм белков жиров и углеводов, повышает функциональную активность ЦНС и сердечно-сосудистой системы, почек и печени. В больших дозах препарат тормозит тиреотропную активность гипофиза и угнетает функцию щитовидной железы.
Левотироксин применяют при гипотиреоидизме, микседеме, тиреотоксикозах, эритроидных состояниях, кретинизме.
Синонимами препарата являются элтроксин, левоид. нороксин, син-троид И др.
Лиотиронин (Liothyronine)
Лиотиронин — 1_-3-[4-(4-гидрокси-3-йодо-фенокси)-3,5-дийодфе-нил]аланин (25.1.11) получают по совершенно аналогичной схеме с использованием при прямом йодировании Ь-(+)-3,5-дийодтиро-нина (25.1.9) одного эквивалента йода [5, 6, 7, 8, 9].
- 466 -
Тиреоидные гормоны и антитиреоидные препараты
Лиотиронин обладает всеми свойствами левотироксина, однако он оказывает более быстрый эффект и меньше связывается с белками крови. Показания к применению лиотиронина такие же, как у левотироксина — гипотиреозы, эутиреоидный зоб, тиреоидит, однако считается, что его применение более целесообразно на первой стадии лечения.
Синонимами препарата являются тибон. циномел, тетроксин и др.
25.2. Препараты для лечения гипертиреоидизма
Гипертиреоидизм возникает вследствие избыточного производства тиреоидных гормонов по разным причинам. Лечение возникающего при этом тиреотоксикоза (базедовой болезни} заключается в применении лекарств, ингибирующих избыточный синтез гормонов, в применении радиоактивного йода с целью вывода из строя фолликулов шитовидной железы с избыточной активностью или ее удаления.
Препараты, используемые при гипертиреоидизме можно классифицировать как препараты, угнетающие синтез тиреотропного гормона в передней доле гипофиза, и к которым относятся дийодтирозин и йод, а также препараты, угнетающие синтез тиреоидных гормонов в щитовидной железе (пропилтиоурацил, метилтиоурацил, метимазол, карби-мазол).
Наиболее пригодны в этом плане последние препараты, классифицируемые как тиоамиды. Они сходны в химическом отношении и содержат в качестве функциональной группы остаток тиомочевины — тионамидную группу. Наиболее предпочтительны пропилтиоурацил и метимазол, хотя широко используются и метилтиоурацил и карби-мазол.
Тиоамиды являются восстановителями. Они ингибируют синтез тиреоидных гормонов путем ингибирования пероксидазной ферментной системы, катализирующей окисление поступающих в организм с пищей йодид ионов в йод, необходимый для йодирования производных тирозина. Таким образом они уменьшают концентрацию свободного йода, необходимого для взаимодействия с производными тирозина, а также могут блокировать реакцию окислительного сочетания моно- и дийод-гирозинов с образованием L-тироксина и Т-трийодтиронина
Для лечения тиреотоксикоза иногда применяют препараты, нарушающие поглощение йода щитовидной железой, и в частности перхлорат калия
В некоторых случаях рекомендуется применять препараты радиоактивного йода, например йодотоп — (Nalljl). Последний кумулируется в щитовидной железе, включается в состав L-тироксина и L-трийод-
- 467-
Г лава 25
тиронина и в результате последующего радиоактивного распада и вызываемого вследствие этого слабого P-излучения разрушает клетки фолликулов шитовидной железы, что приводит к постепенному уменьшению секреции тиреоидного гормона.
Дийодтирозин (Diiodothyrosinе)
Дийодтирозин — 3,5-дийодтирозин (25.2.1) получают прямым йодированием тирозина йодом в присутствии йодида натрия в водном этилам ине либо в смеси уксусной и соляной кислот с добавлением перекиси водорода [10, И, 12].
Дийодтирозин не обладает выраженной гормональной активностью. Однако он тормозит выработку тиреотропного гормона передней доли гипофиза, активирующего деятельность щитовидной железы.
Препарат применяют при гипертиреоидных формах эндемического и спорадического зоба, при диффузном токсическом зобе и других заболеваниях, сопровождающихся тиреотоксикозом.
Синонимами препарата являются дитирин, йодоглобин и др.
Пропилтиоурацил (Propylthiouracil)
Пропилтиоурацил — 6-пропил-2-тио-2,4-(1Н,ЗН)-пиримидиндион
(25.2.2) получают конденсацией пропионилуксусного эфира с тиомочевиной в присутствии этилата натрия [13].
он
H2NCONH2
C2H5ONa
С3Н7 N SH
25 2 2
Препарат обладает выраженным тиреостатическим эффектом и вызывает уменьшение синтеза тироксина в щитовидной железе. Препятствует процессу йодирования тиреоглобулина, снижает образование активной формы йода в шитовидной железе, блокирует систему пероксидаз.
-468 -
Тиреоидные гормоны и антитиреоидные препараты
Пропилтиоурацил применяют при гипертиреозе, тиреотоксических кризах, при гиреодектомии.
Синонимами препарата являются пропицил и тиреостат.
Метилтиоурацил (Methylthiouracil)
Мепштиоурацил — 6-метил-2-тио-2.4-( 1Н,ЗЯ)-ииримидиндион (25.2.3) получают совершенно аналогично конденсацией пропионилуксусного эфира с тиомочевиной в присутствии этилата натрия [14].
он
25 2 3
Метилтиоурацил применяют по тем же показаниям, что и пропилено) рацил.
Синонимами препарата являются муркаин и метиоцил.
Метимазол (Methimazol)
Метимазол — 1-метил-2-имидазолтиол (25.2.5) получают взаимодействием диэтилацеталя аминоуксусного альдегида с метилизотиоцианатом с последующим гидролизом ацетальной группы образующегося производного дизамещенной мочевины (25.2 4) раствором серной кислоты, при которой одновременно происходит реакция циклизации с образованием имидазольного кольца искомого мегимазола [15, 16].
н h2SC>4
+ Ch3~N=C=S --------► CH3-NH-C~NH-CH2CH(OC2H5)2 -----------►
25 2 4
СНз
Метимазол также нарушает синтез тироксина и трийодтиронина непосредственно в щитовидной железе, и его применяют по тем же показаниям. что и пропилтиоуранил и метилтиоурацил для лечения гипер-ф) нкции щитовидной железы у больных с базедовой болезнью.
Синонимами препарата являются меразолил. тиамазол. метотирин. гимидазол и др.
-469-
Глава 25
Карбимазол (Carbitnazol)
Карбимазол. — этиловый эфир 3-метил-2-тиоимидазолин-1 -карбоновой кислоты (25.2.7) получают одновременным взаимодействием этиленацеталя бромуксусного альдегида с метиламином и изоцианатом калия с образованием З-метил-2-имидазолтиона (25.2.6), который далее подвергают ацилированию по атому азота этиловым эфиром хлоругольной кислоты с получением искомого продукта (25.2.7) [17, 18, 19].
Си
У—СН2-Зг + СН-ЛН2 + KSCN о
н
сн.
С1СООС2Н5
сн3
25 2 7
СООС2Н5
Показания к применению карбимазола такие же, как и у всех перечисленных выше препаратов.
Синонимами препарата являются карботироид и неомерказол.
Список литературы
1. Nahm Н. et al./'Chem. Вег. 96, 1 (1963).
2. Chalmers К et al.//J. Chem. Soc. 1949, 3424.
3. Ger. Pat. 1.067.826(1955).
4. Ger. Pat. 1.077.673 (1958).
5. US Pat. 2.784.222 11957).
6. US Pat. 2,823.164 (1958).
7. US Pat. 2.993.928 (1961).
8. Brit. Pat. 671.070 (1949).
9. Roche L. et al.//Bull. Soc. Chim. France. 4, 462 (1957).
10. Bernes J. et al./7J. Chem. Soc. 1950, 2824.
11. Jurd L.'/J. Am. Chem. Soc. 77, 5747 (1955).
12. US Pat. 2.835.700 (1958).
13. Anderson G. et al./,'J. Am. Chem. Soc. 67, 2197 (1945).
14. List R,/Justus Liebigs Ann. Chem. 236, 1 (1886).
15. Wohl A. et al./'Ber. 22, 1354v(1889).
16. Jones R. et al., /J. Am. Chem. Soc. 71,4000 (1949).
17. US Pat. 2.671.088 (1954).
18. US Pat. 2.815.349(1957).
19. Baker JJ. Chem. Soc. 1958, 2387.
-470-
5 Глава 26
\ Инсулин и синтетические i гипогликемические средства
Препараты, применяемые с целью снижения уровня глюкозы в крови, называются гипогликемическими средствами. Соответственно, вещества, вызывающие увеличение уровня глюкозы в крови, называются гипергликемическими средствами.
Изменения в уровне содержания глюкозы в крови могут быть вышины различными причинами, основной из которых является сахарный диабет.
Сахарный диабет — метаболическое заболевание, связанное с высоким уровнем сахара в крови и сопровождаемое также нарушением метаболизма углеводов, липидов и белков. Наиболее частым биохимическим нарушением при сахарном диабете является кетоацидоз.
При лечении сахарного диабета используются инсулин и другие гипогликемические средства.
В зависимости от состояния организма диабет классифицируется по двум типам: инсулинзависимый (тип I), при котором абсолютно подавлена выработки эндогенного инсулина самим организмом, и инсулинне-зависимый (тип II). возникающий либо по причине недостаточности выработки самого инсулина, либо ввиду сбоев работы инсулиновых рецепторов, что часто является результатом других нарушений в организме и, в частности, ожирения.
26Л. Инсулин
Гормон поджелудочной железы инсулин является специфическим антидиабетическим средством, особенно при диабете типа I. Человеческий инсулин представляет собой двухцепочечный белок с молекулярной массой около 6000. состоящий из 51 аминокислоты (цепь А — 21 аминокислота, цепь В — 30 аминокислот), связанной двумя дисульфидными мостиками.
-471 -
Г лава 26
nh2s-------------------s NH2 nh2
Glv-IIau-Va'-Glu-GIu-Cys (jys-Thr-Ser-ileu-C/S-Ser^eu-Tyr-Glu-Leu-GIU'Asp-T/r Gys-Asp- NH2
Nh2NH2 S—-------------------S S— ' 1 S
III I
Pne Val-Asp-Glu-His-Leu-CyS-Gi^-Ser-His--eu-Vai-Glu-Aia-Leu-TyT-Leu-Val-Cys-GlU’Gly-Arg-Gly-Phe-Phe-Tyr-Thr-Pro-Lys-Thr
Свиной инсулин отличается от человеческого лишь аминокислотой в положении 30, а бычий — в положениях 8, 10 и 30.
Инсулин был открыт в 1921 г. Он был выделен из поджелудочной железы млекопитающих [1, 2, 3, 4, 5, 6, 7, 8. 9]. В настоящее время осуществлен синтез инсулина человека и некоторых животных (свиней и крупного рогатого скота) [10, И, 12], а также предложены способы его получения методами генной инженерии [13, 14, 15, 16].
В организме инсулин синтезируется Р-клетками островков Лангерганса поджелудочной железы. Скорость его образования изменяется в зависимости от типа принятой пищи, желудочно-кишечных гормонов и нейронального контроля. Циркулирующий в организме инсулин имеет время биологической полужизни около 5 мин. Быстро разлагается ферментами и выделяется из крови печенью и почками.
Механизм гипогликемического действия инсулина окончательно не выяснен. Однако принято считать, что инсулин действует, связываясь со специфическими рецепторами на поверхности инсулпнчувствительных тканей, таких как скелетные и сердечные мышцы, жировая ткань, лейкоциты.
Инсулин понижает содержание сахара в крови путем превращения глюкозы в гликоген. Применение инсулина при сахарном диабете приводит к снижению уровня сахара в крови и накоплению в тканях гликогена. Уменьшение глюкозы в крови устраняет гликозурию и. соответственно, уменьшает повышенные диурез и жажду, нормализует углеводный, белковый и жировой обмен, снимает явления диабетической комы.
Инсулин эффективен при инсулинзависимом сахарном диабете. Поскольку инсулин разрушается пищеварительными ферментами, способ его введения исключительно парентеральный. Обычно подкожно, реже внутримышечно или внутривенно.
Имеются препараты инсулина пролонгированного действия, что обеспечивает его медленное всасыванием из места введения.
Препараты инсулина понижают уровень глюкозы в крови.
Инсулин применяют при инсулинзависимом и инсулиннезависимом сахарном диабете.
-472 -
Инсулин и синтетические гипогликемические средства
26.2. Синтетические гипогликемические препараты
Для больных с диабетом типа II, у которых эндогенная секреция инсулина в некоторой степени функционирует, предложен ряд весьма эффективных гипогликемических препаратов.
Шесть наиболее широко применяемых сегодня препаратов подразделяю! ся на две группы. Препараты первой генерации —• производные сульфонилмочевины были наиболее популярны в ранние 1980-е годы и включают толбутамид, ацетогексамид, толазамид и хлорпропамид.
Препараты второй генерации — производные гуанидина, такие как глибурид и глипизид, вошли в медицинскую практику после 1984 г. Все они имеют весьма общие химическое строение и механизм действия и отличаются строением боковой цепи и. соответственно, активностью и фармакокинетическими особенностями.
Считается, что действие сульфаниламидов заключается в повышении секреции инсулина. Предполагается также, что гипогликемический эффек! этих препаратов связан и с подавлением высвобождения глюкагона — гормона, продуцируемого а-клетками островков Лангерганса поджелудочной железы ц, представляющего собой полипептид, состоящий из остатков 29 аминокислот. Влияние глюкагона на углеводный обмен проявляется гипергликемией, что связано с усилением гликоге-ногенеза (синтез глюкозы из неуглеродных прекурсоров) в печени.
Важной особенностью всех рассматриваемых препаратов является возможность их перорального применения.
Многие из лекарственных препаратов, применяемых при различных заболеваниях, являются антагонистами оральных гипогликемических препаратов (кортикостероиды, тиреоидные гормоны, тиазидные диуретики, фуросемид и оральные контрацептивы). В то же время имеются препараты, потенцирующие их действие (фенилбутазон, клофибрат, дикумарол и салицилаты).
Толбутамид (Tolbutamid)
Толбутамид — 1-бутил-З-и-толуолсольфонилмочевина (26.2.2) получается в одну стадию взаимодействием и-толуолсулъфаниламида (в виде натриевой соли) с бутилизоцианатом [17, 18, 19. 20].
Сн3 Л—SO2-MH2 + С4НЭ-Н=С=О
26.2 2
-473-
Глава 26
Толбутамид является одним из широко применяемых противодиа-бетических средств. Его действие предположительно связывается со стимулирующим действием на Р-клетки поджелудочной железы, вследствие чего происходит интенсивное выделение инсулина. Препарат применяют при сахарном диабете типа II средней тяжести без выраженных микрососудистых осложнений.
Синонимами препарата являются мебенол, орамид, орабет, толбу-тон, бутамид, растинон и др.
Хюрпропамид (Chlorpropamid)
Хлорпропамид — 1-(н-хлорфенилсульфонил)-3-пропилмочевина (26.2.3) получается совершенно аналогичным способом — взаимодействием льхлорбензолсульфаниламида с пропилизоцианатом [21, 22,23, 24,25, 26].
о
SO2-NH-C-NH-C3H7
26 2 3
Показания к применению и механизм действия также сходен с таковым для всей рассматриваемой группы соединений, т. е. связан со стимуляцией секреции инсулина при наличии функционирующих тканей поджелудочной железы.
Препарат назначают при инсулиннезависимом сахарном диабете стабильного течения.
Синонимами препарата являются диабиниз, хлороназ и др.
Ацетогексамид (Acetohexamid)
Ацетогексамид — 1-(и-ацетил фенилсульфонил)-3-циклогексил-мочевина (26.2.6) также получается по аналогичной схеме — взаимодействием н-хлорбензолсульфаниламида с циклогексилизоцианатом. Необходимый для этой цели н-ацетилбензолсульфаниламид получают диазотированием я-аминоацетофенона в присутствии двуокиси серы и хлорида одновалентной серы с получением сульфохлорида (26.2.4). который далее взаимодействием с аммиаком переводят в сульфонамид (26.2.5). Взаимодействием последнего с циклогексилизоцианатом получают ацетогексамид (26.2.6) [27, 28, 29,30.31]
-474-
Инсупин и синтетические гипогликемические средства
26 2 8
Показания к применению и механизм действия ацетогексамида аналогичен таковым у всей рассматриваемой группы соединений.
Синонимами препарата являются цикламид, аглирал и др.
Толазамид (Tolazamid)
Голазамид — 1-(гексагидро-1//-азепин-1-ил)-3-(«-толуилсульфо-нил)мочевина (26.2.8). Сохраняя структурное сходство с препаратами первого поколения, толазамид отличается от всех рассмотренных выше наличие^ семикарбазидной группы вместо остатка мочевины и азепиновой вместо циклогексильной.
Синтез препарата осуществляют взаимодействием этил-(и-толу-о.чсульфонил)карбамата (26.2.7), который, в свою очередь, получают из н-голуолсульфонамида и этилового эфира хлоругольной кислоты с 1-аминоазепином [32. 33, 34, 35].
Препарат, также являясь производным сульфонилмочевины I генерации, обладает стимулирующим действием на р-клетки поджелудочной железы и тем же спектром действия, что и вся группа рассматриваемых соединений.
Толазамид применяют при инсулиннезависимом сахарном диабете без выраженных микрососудистых осложнений.
Синонимами препарата являются норглицин, толиназ и др.
-475-
Глава 26
Глибурид (Gliburide)
Глибурид — 1-[4-[2-(5-хлоро-2-метоксибензамидо)э1ил]-фенил-сульфонил]-3-циклогексил-мочевина (26.2.11), являясь препаратом второго поколения, отличается от рассмотренных выше более сложным строением сульфаниламидной части молекулы, в которую введена дополнительная фармакофорная группировка.
Синтез препарата осуществляют исходя из хлорангидрида 2-мето-кси-5-хлорбензойной кислоты, которую трансформируют в амид (26.2.9) взаимодействием с 2-фенилэтила.мином. Последний последовательно подвергают сульфохлорированию хлорсульфоновой кислотой и далее амидированию аммиаком с получением сульфонамида (26.2.10). Полученный сульфонамид вводят во взаимодействие с циклогексилизоцианатом с получением искомого глибурида (26.2.11) [36. 37, 38, 39].
1 CISO3H
2 NH3
Препарат принадлежит к производным сульфонилмочевины II генерации. Подобно всем рассматриваемым оральным гипогликемическим препаратам, он является стимулятором р-клеток поджелудочной железы, а с другой стороны, повышает чувствительность к инсулину и степень его связывания с клетками-мишенями. Одновременно препарат отличается лучшей переносимостью. Гипогликемизирующий эффект наступает при значительно меньших дозах, чем при применении препаратов I генерации.
Препарат применяют при сахарном диабете типа И средней тяжест и без выраженных микрососудистых осложнений.
Синонимами препарата являются глибенкламид, манинил, дигабен, даонил и многие другие.
Глипизид (Glipizid)
Глипизид — 1-циклогексил-3-[[и-[2-(5-метилпиразинкарбоксамидо)-этил]фенил]сульфонил]-мочевина (26.2.13) отличается от глибурида
- 476 -
Инсулин и синтетические гипогликемические средства
строением амидной части молекулы, в которой фрагмент 2-метокси-5-хлорбензойной кислоты заменен на фрагмент 6-метилпиразин-карбоновой кислоты. Метод синтеза препарата также альтернативен вышеприведенному. В данной схеме синтеза сначала 6-метил-пиразинкарбоновую кислоту взаимодействием с тионилхлоридом переводят в соответствующий хлорангидрид, который далее подвергают действию 4-(2-аминоэтил)бензолсульфонамида с получением соответствующего амида (26.2.12). Далее полученный сульфонамид по традиционной схеме вводят во взаимодействие с цнк-логексилизоцианатом с получением искомого глипизида (26.2.13) [40.41.42].
сн3
сн2—сн2
26 2 13
Показания к применению и механизм действия также сходны с таковыми глибурида.
Синонимами препарата являются глибиниз. минидаб, глюкотрол и др.
Список литературы
1. Banting F. et aL//J. Lab. Clin. Med. 7, 251 (1921-22).
2. Лбе/Л./'Proc. Nat. Acad. Sci. USA, 12, 132 (1926).
3. Lens /.'/Biochim. Biophys. Acta. 2, 76 (1948).
4. Sanger F. et al./'/Biochem. J. 49, 463 (1951).
5. Sanger F. et al.'.Biochem. J. 53, 353, 366 (1953).
6. Ryle A et al. 'Biochem. J. 60, 541 (1955).
7. Sanger F ’/Science. 129, 1340 (1959).
8. Nicol D. et al. Nature. 187, 483 (1960).
9. Hodgkin D. "Verh. Schweiz. Naturforsch. Ges. 150. 93 (1970).
10. Katsoyannis P. et al. J. Am. Chem. Soc. 88. 164, 166 (1966).
-477-
Глава 26
11. Katsoyannis Р. et aL Science, 177. 623 (1972).
12. Katsoyannis P et al. Recent Progr. Horm. Res. 23, 505 (1967).
13. Hsiung H. et al./-Nucleic Acid Res. 6, 1371 (1979).
14. Hsiung H et al.''Nucleic Acid Res. 7, 2199 (1979).
15. Hsiung H et aU/Nucleic Acid Res. 8, 5753 (1980)
16. Johnson /.//Science. 219, 632 (1983).
17. US Pat. 2.968.158 (1961).
18. Ger. Pat. 974.062(1955).
19. Ger. Pat 1.066.575 (1959).
20. Brit. Pat. 808.071 (1959).
21. US Pat. 3.013.072 (1961).
22. US Pat. 3.349.124(1967).
23. Ruschig H et al."'Arzneim. Forsch. 8. 448 (1958).
24 Bnt. Pat. 853.555 (1960).
25. Marshall J et al./-J. Org. Chem. 23, 927 (1958).
26. Bauer et al./-J. Org. Chem. 31, 3440 (1960).
27. Ger. Pat. 1.177.631 (1967).
28. Ger. Pat. 1.135.891 (1960).
29. US Pat. 3.320.312 (1967).
30. Bnt. Pat. 912.789(1962).
31. Marshall J J. Med. Chem. 6, 60 (1963).
32. US Pat. 3.063.903 (1962).
33. Bnt. Pat. 887.886 (1960).
34. Ger. Pat. 1.196.200(1961).
35. Wright J et al.'J. Med. Pharm. Chem. 5, 815 (1962).
36. Aumuller W. et alUArzneim.-Forsch. 16, 1640 (1966).
37. Ger. Pat. 1.283.837 (1967).
38. Ger. Pat. 1.301.812 (1965).
39. Belg. Pat. 684.652 (1966).
40. Ger. Pat. 2.012.138(1969).
41. US Pat. 3.669.966(1969).
42. Amborgi Г. et al.'Arzneim.-Forsch. 21 200 (1971).
-• Глава 2 7
1 Кортикостероиды
Термином «кортикостероиды» обозначаются стероидные гормоны, секретируемые корой надпочечников. К ним относятся глюкокортикоиды (гидрокортизон, 11-дегидрокортикостерон, кортикостерон), минералокортикоиды (альдостерон. 11-дезоксикортикостерон, 11-дезокси-17-оксикортикостерон) и половые стероидные гормоны (андростерон, андростендион. эстрон, прогестерон). Образование этих гормонов находится под прямым контролем полипептидного адренокортикотропного гормона (АКТГ, кортикотропина) вырабатываемого передней долей гипофиза. Человеческий АКТГ состоит из 39 аминокислот и имеет молекулярную массу 4500. Он отличается от АКТГ животных аминокислотным составом на участке 29-33.
Глюкокортикоиды являются эндогенными соединениями, влияющими на метаболизм углеводов, липидов и протеинов и проявляющих противовоспалительное, десенсибилизирующее и антиаллергическое действие. Они являются иммунодепрессантами, а также обладают противошоковым и антитоксическим действием.
Минералокортикоиды являются эндогенными соединениями, воздействующими на жидкостный и электролитный баланс в организме.
Однако спектр биологических свойств многих кортикостероидов, как правило, намного шире спектра свойств, присущих как «чистым» глюкокортикоидам, так и «чистым» минералокортикоидам по определению.
Природные стероидные гормоны в организме синтезируются из холестерина. Стадией, лимитирующей скорость, является окисление боковой цепи холестерина с образованием прегненолона и изокапронового альдегида.
Все кортикостероиды являются производными циклопентанофенан-трена с кетогруппами при С\ и С2о и ненасыщенной связью между С4 и С*. (обозначаемой как А4), а также обязательным наличием аксиальной Р-СО-СН2ОН боковой цепи при Ci7. Друг от друга они отличаются при-су гствием или наличием кето- или [3-гидроксильной групп при Си, а также при Сг и,'или С18. Эти отличия определяют главные фармаколо-I ические свойства этих соединений и их прекурсоров.
- 479-
Г лава 27
Стероиды обратимо связываются в плазме с двумя протеинами: кортикостероидсвязывающим глобулином, являющимся специфическим а2-глобулином. и альбумином, который имеет неспецифическую активность и слабовыраженное сродство к стероидам.
Не связанные с плазмой свободные стероиды проникают в целевые клетки путем пассивной диффузии и связываются с цитоплазматическим растворимо-связанным протеином (акцепторным участком), образуя комплекс стероид—протеин. Последний и проникает в ядро, где взаимодействует со стероидными рецепторами на хроматине.
Кортикостероиды не вылечивают болезни, но широко используются при различных состояниях, когда бывает необходимо использовать их противовоспалительные, иммуносупрессорные и минералокортикоидные свойства. Кроме того, они применяются для заместительной терапии у пациентов с недостаточностью надпочечников. Кортикостероиды могут применяться по жизненным показаниям при астме, острых аллергических реакциях и отторжении трансплантатов. Эффективны при неинфекционных гранулематозных заболеваниях, таких как саркоцид, коллагеновые сосудистые заболевания, ревматоидные артриты, лейкемии. При лечении ряда дерматологических и офтальмологических заболеваний стероиды широко применяются в составе лосьонов, мазей и т. п.
Кортикостероиды являются очень мощными препаратами, и при их применении практически невозможно избежать побочных реакций. Поэтому их следует применять в самых малых эффективных дозах и очень короткое время. Рекомендуется их применение не обрывать резко и их введение прекращать постепенно.
27.1. Глюкокортикоиды
Глюкокортикоиды — это соединения, которые в первую очередь действуют на метаболизм углеводов, белков и жиров и, в определенной степени, на электролитный и водный баланс в организме. Фактически они действуют на все ткани и органы и могут изменять иммунный ответ
-480 -
Кортикостероиды
организма на воздействия различного типа. Конечным результатом их действия является индукция (анаболизм) или подавление (катаболизм) синтеза протеинов. Они воздействуют на сердечно-сосудистую систему. ЖКТ. скелетную мускулатуру, кожу и соединительную ткань, кровь и эндокринную систему. Прямым показанием к их применению является острая и хроническая недостаточность надпочечников. Их используют при коллагенозах, ревматоидных артритах, ревматизме, экземе, нейродермитах и других кожных заболеваниях, при аллергиях, трансплантации тканей, бронхиальной астме и многих других заболеваниях.
Связь между химическим строением и биологическим действием кортикостероидов весьма сложна. Однако ряд обобщений сделать можно.
Для проявления глюкокортикоидной активности в структуре прегнановой системы очень важно наличие гидроксильных групп в положениях С,।р и С|7«. В то же время для проявления адекватной минералокортикоидной активности необходимо наличие кислородной функции при С|। и С|8, или же требуется отсутствие кислородных функций одновременно и при Сц,'и при Ср. В общем, кажется вероятным, что глюкокортикоидное связывание с рецепторными участками должно происходить с участием С) iP-гидроксильной группы и С|7р-СО-СН2ОН бокового фрагмента стероидной системы. Присутствие в молекуле других объемистых аксиально ориентированных P-заместителей, как правило, препятствует связыванию стероидной молекулы с рецепторами, в то время как аналогичные экваториальные a-заместители больших трудностей не создают.
Основными эндогенными глюкокортикоидами являются гидрокортизон и кортизон. Получены и применяются многочисленные синтетические аналоги природных глюкокортикостероидов, которые оказались более эффективными и в настоящее время почти полностью заменили кортизон (преднизолон, преднизон, дексаметазон и др.).
Глюкокортикоиды применяются орально, внутримышечно, внутривенно, ингаляционно, в виде мазей, кремов и т. п.
Гидрокортизон (Hydrocortison)
Гидрокортизон — lip,17а,21-тригидроксипрегн-4-ен-3,20-дион (27.1.8) предложено получать разными методами из разных исходных соединений со стероидным скелетом. Согласно первому способу. гидрокортизон получают исходя из декстропрегненолона. С этой целью двойную связь между С16 и С]7 декстропрегненолона подвергают окислению с использованием перекиси водорода в ще
-481 -
Глава 27
лочи с получением эпоксида (27 1.1). Взаимодействием последнего с бромистоводородной кислотой осуществляют раскрытие эпоксидного кольца с получением 16-бром-17-гидроксидекс1ропрегнено-лона (27.1.2). Полученное бромпроизводное подвергают дебромированию водородом с использованием в качестве катализатора палладия на угле, и далее вторичную гидроксильную группу подвергают этерификации муравьиной кислотой в присутствии и-то-луолсульфокислоты с получением З-формилокси-17-гидрокси-декстропрегненолона (27.1.3). Далее полученный З-формилокси-17-гидроксидекстропрегненолон подвергают бромированию молекулярным бромом, в результате чего бромируется и двойная связь между С4 и С5 и ацетильная метильная группа с получением триб-ромпроизводного (27.1 4). Взаимодействием продукта с йодидом натрия осуществляют дегалогенирование полученного вицинального дибромида, в ходе которого одновременно происходит и смещение двойной связи в положение между углеродными атомами Cs и С6 с получением бромкетона (27.1.5). Взаимодействием последнего с ацетатом калия и далее с уксусным ангидридом в присутствии п-толуолсульфокислоты получают диацетат (27.1.6). С учетом того факта, что в отличие от ацетатов, формиаты легко окисляются и дают те же продукты, что и соответствующие спирты, полученный диацетат окисляют по Оппенауэру, используя изопропилат алюминия и циклогексанон в качестве акцептора водорода. При этом одновременно происходит изомеризация двойной связи в первоначальное положение между С4 и С5 с образованием устойчивого сопряженного винилкетона, и далее ацетильные защиты обеих гидроксильных групп гидролизуют с использованием гидроокиси калия с получением 17-гидрокси-11-дезоксикортикостерона (27.1.7). Последний подвергают микробиологическому окислению по положению С,। с получением искомого гидрокортизона (27.1.8). Подобными реакциями микробиологического окисления с применением самых различных микроорганизмов возможно осуществить гидроксилирование стероидов в различные положения, применяя в качестве исходного легко доступный прогестерон [1, 2, 3. 4, 5].
-482 -
Кортикостероиды
Второй способ, интересный с точки зрения химика-синтетика, получения гидрокортизона заключается в использовании в качестве исходного вещества прогестерона. На первой стадии синтеза прогестерон подвергают микробиологическому окислению, аналогичному вышеописанному, с получением 11а-гидроксипрогестерона (27.1.9). Полученную таким образом гидроксильную группу окисляют оксидом шестивалентного хрома в уксусной кислоте с получением 11-кетопрогестерона (27.1.10). Последний вводят в реакцию с диэтилоксалатом в присутствии этила<а натрия с получением соответствующего а-кетоэфира в виде натриевого енолята (27.1.11), который подвергают бромированию двумя эквивалентами брома с получением дибромкетона (27.1.12). Полученный дибромкетон подвергают перегруппировке Фаворского и далее гидролизуют с получением ненасыщенной кислоты (27.1.13). Затем карбонильную группу в положении Сз кетализируют с использованием этиленгликоля в присутствии п-толуолсульфокислоты, при которой происходит миграция двойной связи в положение между углеродными атомами С5 и С6 с образованием кеталя (27.1.14). Полученный продукт восстанавливают алюмогидридом лития. При этом до спиртовых групп восстанавливаются как карбоксильная, так и кетогруппа Си с образованием диола (27.1.15). Далее последовательно снимается кетальная защита, в ходе чего в кислых условиях двойная связь вновь мигрирует обратно в первоначальное положение между С4 и С5, и первичная гидроксильная группа ацилируется ангидридом уксусной кислоты в пиридине с получением продукта (27.1.16). Двойную связь в последнем окисляют с использованием перекиси водорода в присутствии четырех-окиси осмия в N-метилморфолине с получением ацетата гидрокортизона (27 1.17). Гидролизом ацетильной группы в последнем гидроокисью калия получают гидрокортизон (27.1.8) [6, 7].
-483-
Глава 27
Гидрокортизон можно получать, также используя в качестве исходного ацетат кортизона. С этой целью ацетат кортизона обрабатывают семикарбазидом, в ходе чего получают дисемикарбазон (27.1.18) в результате взаимодействия по обеим карбонильным группам при С3 и С20. Далее карбонильную группу при Сц восстанавливают боргидридом калия до спиртовой, в ходе чего одновременно удаляется ацетильная группа при гидроксиле у С;, с получением семикарбазона (27.1.19). Снятием семикарбазидной защиты с помощью азотистой кислоты получают гидрокортизон (27.1.8).
Гидрокортизон применяется как в виде свободного спирта (речь идет о гидроксильной группе при С21). так и в виде ацетата, сукцината, фосфата [8].
-484 -
Кортикостероиды
2N-NH-C-NH2,
NaNOyJ HCI
27 1 a
Гидрокортизон оказывает противошоковое, противоаллергическое, противовоспалительное действие. Повышает содержание глюкозы в крови, увеличивает .выведение калия и уменьшает выделение натрия из организма. Проявляет антиметаболическое действие и уменьшает синтез гистамина в организме.
Препараты гидрокортизона применяются при острых воспалениях, шоке, первичной или вторичной почечной недостаточности, недостаточности надпочечников, при язвенных колитах, ревматоидных, подагрических. псориатрических артритах, при коллагеновых и дерматологических заболеваниях, аллергических состояниях, офтальмологических и желудочно-кишечных заболеваниях, при заболеваниях дыхательных пу тей.
Синонимами препарата являются кортеф, гидрокортон и др.
Кортизон (Cortison)
Кортизон — 17а,21-дигидроксипрегн-4-ен-3,11,20-трион (27.1.26) также предложено получать разными способами из соединений, изначально имеющих стероидный скелет молекулы.
Первый способ, весьма схожий с вышеизложенным методом для получения гидрокортизона, исходящим из прогестерона, заключается в том, что прогестерон подвергают микробиологическому окислению с получением 11а-гидроксипрогестерона (27.1.9). Далее гидроксильную группу окисляют оксидом шестивалентного хрома в уксусной кислоте с получением 11-кетопрогестерона (27.1.10). Последний вводят в реакцию с диэтилоксалатом в присутствии этилата натрия с получением
-485-
Глава 27
соответствующего «-кетоэфира в виде натриевого енолята (27.1.11), который подвергают бромированию двумя эквивалентами брома с получением дибромкетона (27.1.12). Полученный дибромкетон подвергают перегруппировке Фаворского, но продукт далее не гидролизуют и выделяют ненасыщенную кислоту в виде метилового эфира (27.1.20). Взаимодействием последнего с пирролидином получают диенамин (27.1.21). который подвергают восстановлению алюмогидридом лития, в результате чего кетогруппа при Сн трансформируется в гидроксильную. а карбметоксильная — в первичную спиртовую с образованием соединения (27,1.22). Кислотным гидролизом последнего и дальнейшим ацетилированием получают ацетат (27.1.23), гидроксильную группу при Си в котором окисляют оксидом шестивалентного хрома до кетонной с получением соединения (27.1.24). Последнее подвергают действию чесырехокиси осмия, и полученный при этом осмат окисляют четырех-окисыо марганца в N-метилморфолине с получением ацетата кортизона (27.1 25). Гидролиз ацетильной группы в последнем бикарбонатом калия приводит к получению кортизона (27.1.26) [6. 9. 10].
-486 -
Кортикостероиды
Второй способ получения кортизона исходит из ацетата дигидрокортизона. Подвергая последний монобромированию молекулярным бромом получают 4-бромпроизводное ацетата дигидрокортизона (2Т 1.27). Подвергая последнее действию семикарбазида, осуществляют отщепление бромистого водорода, и одновременно получают семикарбазон по кетогруппе при С3 с образованием продукта (27.1.28). Далее 21-О-ацетилкортизон (27.1.29) выделяют из семикарбазона с помощью пировиноградной кислоты. Гидролизом ацетильной группы в последнем бикарбонатом калия получают искомый кортизон (27.1.26) [11, 12, 13].
КНСОз I СНзОН^
Кортизон применяют при воспалительных процессах, аллергии, недостаточности надпочечников.
Синонимами препарата являются кортизан, корто, адресон, кортад-рен и др.
Преднизон (Prednison)
Преднизон — 17а,21-дигидроксипрегна-1,4-диен-3,11,20-трион
(27.1.30) отличается от кортизона наличием дополнительной двойной связи между С| и С2. Предложены различные пути его синтеза. В одном из них. как и в случае синтеза кортизона, исходят из ацета-
-487-
Глава 27
та дигидрокортизона. Однако в данном случае последний подвергают дибромированию молекулярным бромом с получением 2,4-дибромпроизводного ацетата дигидрокортизона (27.1.30). Дегидробромированием последнего лутидином и дальнейшим гидролизом ацетильной группы бикарбонатом калия получают искомый преднизон (27.1.31) [14, 15] Преднизон получают также микробиологическим дегидрированием кортизона [16, 17].
Преднизон применяется по тем же показаниям, что и кортизон: при оспалительных процессах, аллергии, недостаточности надпочечников, днако он в несколько раз активнее последнего и меньше влияет на митральный обмен.
Синонимами препарата являются кортансил, декортизил, паракорт и др.
чеднизолон (Prednisolon)
Преднизолон 11 [3,17«,21 -тригидроксипрегна-1,4-диен-3,20-дион
(27 1.33). Структурно преднизолон отличается от преднизона тем, что кетогруппа в положении Сг1 преднизона заменена гидроксильной. Синтез преднизолона осуществляют либо микробиологическим дегидрированием гидрокортизона по связи С(-С2 [16, 17, 18, 19]. либо исходя из 21-ацетокси-11р,17а-дигидрокси-5а-прегнан-3,20-диона, который подвергают дибромированию молекулярным бромом в уксусной кислоте в положения С2 и С4, и далее полученный дибромид (27.1.32) дегидробромируют нагреванием в коллиди-1е с получением ацетата преднизолона по положению С2). Гидролиз юследнего приводит к образованию преднизолона (27.1.33) [14].
- 488-
Кортикостероиды
Преднизолон также оказывает противошоковое, противоаллергическое. противовоспалительное, иммуносупрессивное действие. Повышает содержание глюкозы в крови, увеличивает выведение калия п \ меньшает выделение натрия из организма.
Преднизолон применяют по тем же показаниям, что и все кортикостероиды: при ревматизме, полиартрите, бронхиальной астме, нейродермитах, экземе.
Синонимами препарата являются антизолон, декор тин, кортолон, прекорталон и многие другие.
Метилпреднизолон (Methylprednisolon)
Метилпреднизолон — 11[3,17а,21-тригидрокси-6о.-метилпрегна-1,4-диен-3,20-дион (27.1.38) отличатся от преднизолона наличием метильной группы в положении С6 стероидного скелета молекулы. Кажущееся весьма простым различие в структуре требует самостоятельного подхода к синтезу. Синтез исходит из гидрокортизона (27.1.8), карбвнильные группы которого первоначально подвергают кетализации этиленгликолем в присутствии следов кислоты, при которой двойная связь в положении С4-С5 изомеризуется положение С; Сй с получением диэтленкеталя (27.1.34). Продукт окисляют до эпоксида (27.1.35) с использованием надбензойной кислоты. Далее полученный эпоксид вводят во взаимодействие с метилмаг-нийбромидом, и последующим снятием кетальной защиты восстановлением водородом получают 5-гидрокси-6-метилпроизводное дигидрокортизона (27.1.36). Полученный [1-гидроксикетон дегидратируют с помощью щелочи, и далее образовавшийся ба-метил-кортизон (27.1.37) подвергают микробиологическому дегидрированию по положению С|-С2 с получением искомого метилпреднизолона (27.1.38) [20, 21,22,23, 24, 25].
-489-
Глава 27
та дитидрокорт изона. Однако в данном случае последний подвергают дибромированию .молекулярным бромом с получением 2,4-дибромпроизводного ацетата дигидрокортизона (27.1.30). Дегидробромированием последнего лутидином и дальнейшим гидролизом ацетильной группы бикарбонатом калия получают искомый преднизон (27.1.31) [14, 15]. Преднизон получают также микробиологическим дегидрированием кортизона [16, 17].
Преднизон применяется по тем же показаниям, что и кортизон: при воспалительных процессах, аллергии, недостаточности надпочечников, однако он в несколько раз активнее последнего и меньше влияет на минеральный обмен.
Синонимами препарата являются кортансил, декортизил, паракорт и др.
Преднизолон (Prednisolon)
Преднизолон - 11р.17а.21-тригидроксипрегна-1,4-диен-3,20-дион (27.1.33). Структурно преднизолон отличается от преднизона тем, что кетогруппа в положении Си преднизона заменена гидроксильной. Синтез преднизолона осуществляют либо микробиологическим дегидрированием гидрокортизона по связи С|-С2 [16. 17, 18, 19]. либо исходя из 21-ацетокси-11р, 17а-дигидрокси-5а-прегнан-3,20-диона, который подвергают дибромированию молекулярным бромом в уксусной кислоте в положения С2 и С4, и далее полученный дибромид (27.1.32) дегидробромируют нагреванием в коллидине с получением ацетата преднизолона по положению С,, Г идролиз последнего приводит к образованию преднизолона (27.1.33) [14].
-488 -
Кортикостероиды
Преднизолон также оказывает противошоковое, противоаллергическое. противовоспалительное, иммуносупрессивное действие Повышает содержание глюкозы в крови, увеличивает выведение калия и уменьшает выделение натрия из организма.
Преднизолон применяют по тем же показаниям, что и все кортикостероиды: при ревматизме, полиартрите, бронхиальной астме, нейродермитах, экземе.
Синонимами препарата являются антизолон, декоргин, кортолон, прекорталон и многие другие.
Метилпредиизолон (Мethylprednisolon)
Метилпреднизолон — 11 р, 17а,21 -тригидрокси-ба-метилпрегна-1,4-диен-3,20-дион (27.1.38) отличатся от преднизолона наличием метильной группы в положении Се стероидного скелета молекулы. Кажущееся весьма простым различие в структуре требует самостоятельною подхода к синтезу. Синтез исходит из гидрокортизона (27.1.8), карбонильные группы которого первоначально подвергают кетализации этиленгликолем в присутствии следов кислоты, при которой двойная связь в положении С4-С5 изомеризуется положение С5"С6 с получением диэтиленкеталя (27.1.34). Продукт окисляют до эпоксида (27.1.35) с использованием надбензойной кислоты. Далее полученный эпоксид вводят во взаимодействие с метилмаг-нийбромидом, и последующим снятием кетальной защиты восстановлением водородом получают 5-гидрокси-6-метилпроизводное дигидрокортизона (27.1.36). Полуденный [3-гидроксикетон дегидратируют с помощью щелочи, и далее образовавшийся ба-метил-кортизон (27.1.37) подвергают микробиологическому дегидрированию по положению С,-С2 с получением искомого метилпреднизолона (27.1.38) [20, 21, 22, 23, 24, 25].
-489-
Глава 27
Метилпреднизолон, являясь аналогом преднизолона, оказывает более продолжительный эффект, чем преднизолон и кортизон, и практически не обладает минералокортикостероидной активностью и лучше переносится.
Синонимами препарата являются метипред, медронметризон и др.
Дексаметазон (Dexamethason)
Дексаметазон — 9а-фтор-16а-метил-11 р, 17,21-тригпдроксипрегна-1.4-диен-3,20-дион (27.1.51), или. проше, 9а-фтор-1 би-метилпреднизолон. Характерным отличительным признаком дексаметазона является наличие в его молекуле атома фтора при С9 стероидного кольца.Многостадийный синтез дексаметазона осуществляют исходя из За-ацетокси-16-прегнен-11,20-диона, взаимодействием которого с метилмагнийбромидом в присутствии бромида лития получают За-гидрокси-16а-метилпрегнан-11,20-дион (27.1.39). Далее осуществляют введение 17а-гидроксильной группы. С этой целью взаимодействием с уксусным ангидридом в присутствии и-то-луолсульфокислоты получают З-ацетокси-17-енолацетат (27.1.40), который эпоксидируют надбензойной кислотой в 27.1.41, и далее продукт гидролизуют щелочью с получением оксикетона (27.1.42). Введение второй гидроксильной группы при СД осуществляют последовательным бромированием метильной группы молекулярным бромом, обменом атома брома на йод и взаимодействием йодида с ацетатом калия с получением соответствующего ацетоксикетона (27.1.43). Гидроксильную группу при С3 в последнем окисляют до карбонильной оксидом шестивалентного хрома в пиридине с получением 3,11,20-трикетона (27.1.44), который вновь подвергают бромированию молекулярным бромом, но уже в положение С4. Дегидрогалогенирование последнего осуществляют с помощью семикарбазида, в результате чего образуется ненасыщенный трикетон (27.1.45). Во избежание образования семикарбазонов по кетогруппам в положениях С3 и С2о конечный продую' обрабатывают пировиноградной кислотой. Далее специально получают семикарбазоны по кетогруппам в положениях С3 и С20. и не вступающую в реакцию образования семикарбазона кетогруппу в положении Clt восстанавливают до спиртовой с помощью боргидрида натрия. После снятия защитных семикарбазонных групп получают 21-О-ацетокси-16р-метилгидрокортизон (27.1.46). Полученный продукт подвергают дегидратации путем последовательной трансформации в мезилат и дальнейшим отщеплением метансульфокислоты с использовани-
-490-
Кортикостероиды
ем метансульфохлорида в пиридине. Полученный при этом продукт содержит двойную связь в положении С9-Си (27.1.47). Взаимодействием последнего с гипобромной кислотой получают бромгидрин (27.1.48). Действием ацетата калия последний трансформируют в эпоксид (27.1.49). Далее взаимодействием с фтористоводородной кислотой осуществляют раскрытие эпоксидного кольца, при котором образуется фторгидрин (27.1.50). Наконец, микробилогическим дегидрированием последнего по связи С,-С2 и одновременным деацетилированием получают дексаметазон (27.1.51) [26, 27, 28].
1 ВГ2
3 СНэСОССОН
CHjSo^i р,
-491 -
Глава 27
Дексаметазон применяют по гем же показаниям, чго и все кортикостероиды. однако он проявляет значительно более сильное противовоспалительное и противоаллергическое действие.
Препарат применяют при циркуляторном коллапсе (шоке во время или после хирургической операции), травме, потере крови, инфаркте миокарда, ожогах. При тяжелых инфекциях — токсемии, сосудистом коллапсе при менингококковой инфекции, септицемии, дифтерии, брюшном тифе, перитоните. При экстренных аллергических состояниях — астматическом статусе, отеке гортани, острой анафилактической реакции на лекарственные препараты, пирогенных реакциях.
Синонимами препарата являются декакорт, декардан, гексадрол, новометазон и др
Бетаметазон (Betamethason)
Бетаметазон — 9а-фтор-1бр-метил-11р,17,21-тригидроксипрегна-1.4-диен-3.20-дион (27.1.49), или, проще, 9а-фтор-1бр-метилпред-низолон (27.1.52). Как видно из химического названия препарата, бетаметазон отличается от дексаметазона лишь ориентацией метильной группы при С|6. Предложенный метод синтеза отличается от метода рядом деталей и последовательностью осуществления реакций кроме первой стадии, а именно- введения метильной группы в положение С16 стероидного кольца. Синтез бетаметазона, как и дексаметазона, осуществляют исходя из За-ацетокси-16-прегнен-11.20-диона, однако метильную группу в положение С,6 стероидного кольца вводят не с использованием метилмагнийбромида, а путем его взаимодействия с диазометаном, с последующим восстановлением двойной связи между углеродными атомами Си-Сг стероидного кольца водородом с использованием в качестве катализатора палладия на угле, за счет чего и обеспечивается соответствующая p-ориентация вводимой метильной группы [29].
- 492 -
Кортикостероиды
Являясь изомером дексаметазона, бетаметазон в основном применяется в качестве препарата местного применения при дерматитах, экземе, а также в качестве средства против зуда.
Синонимами препарата являются бетакорт, целестон, суперкортен
и др
Триамицинолон (Triamicinolon)
Т риамипинолон — 9а-фтор-11 р, 16а, 17,21 -тетрагидроксипрегна-1,4-диен-3,20-дион (27.1.61) по химическому строению триамици-нолон отличается от дексаметазона тем, что а-метильная группа при С|6 заменена на гидроксильную. Синтез препарата осуществляют исходя из 21-О-анетата гидрокортизона (27.1.17). С этой целью на первой стадии синтеза обе карбонильные группы исходного подвергают кегализации с помощью этиленгликоля. Далее гидроксильные группы в полученном дикетале (27.1.53) замещают на хлор с помощью хлористого тионила, и продукт подвергают дегидрохлорированию с помощью щелочи, в ходе чего i идролизуется и 21-О-ацетильная группа. Вновь ацетилируя эту гидроксильную группу, уксусным ангидридом получают триен (27.1.54). Взаимодействием последнего четырехокисью осмия получают вицинальный диол (27.1.55). Вторичная гидроксильная группа при С|6 последнего подвергается ацетилированию уксусным ангидридом в пиридине с получением диацетата (27.1.56). Обработкой продукта N-бромацетамидом в хлорной кислоте получают бромгидрин (27.1 57). который действием ацетата калия трансформируется в эпоксид (27.1.58) Раскрытием эпоксидного кольца фтористоводородной кислотой получают соответствующее 9-фтор-11-тидрокси производное (27.1.59). Микробиологическим дегидрированием последнего связь С|-С2 окисляют в двойную с получением ацетата триамицинолона (27.1.60), гидролизом ацетильных групп которого получают искомый гриамицинолон (27.1.61) [30, 31, 32].
-493 -
Глава 27
Триамипинолон по фармакологическому действию близок к дексаметазону и в некоторых случаях лучше переносится.
С инонимами препарата являются ледеркорт, кенакорт, делсолон и др.
27.2. Минералокортикоиды
Минералокортикоиды играют большую роль в регуляции электролитного и водного баланса особенно в почках, где они способствуют реабсорбции ионов Na и воды из мочи.
Основным эндогенным минералокортикоидом является альдостерон, который, однако, не используется в клинике. Широко применяется дезоксикортикостерон.
В огличие от глюкокортикоидов, минералокортикоиды действуют на углеводный обмен незначительно. Противовоспалительными и анти-аллергическими свойствами не обладают.
Их применяют при хронической недостаточности надпочечников, а также с целью повышения тонуса и работоспособности мышц.
Корреляции между химической струк1урой и действием минералокортикоидов весьма сложны, однако ряд частных заключений сделать можно.
-494-
Кортикостероиды
В частности, следует отметить, что поскольку 11-дезоксикорти-кос герои не проявляет глюкокортикоидной активности, но является мощным минералокортикоидом и имеет только два потенциально реакционно-способных заместителя, которые могут взаимодействовать с рецептором. то это взаимодействие должно осуществляться посредством С-.-кего и или СГР-СО-СН2ОН групп. В то же время замечена необходимость либо одновременного наличия кислородных функций при Сии C|S, либо одновременного отсутствия кислородных функций и при Си, и при С г в структуре прегнановой системы.
Фторирование в положение С9а повышает минералокортикоидную активность и Сц-гидрокси (гидрокортизон), и С1Гдезокси (дезоксикортикостерон) соединений.
Альдостерон (Aldosteron)
А.тьдог. iepoH — 11р.21-дигидроксипрегн-4-ен-2,18,20-трион (27.2.4) синтезируют исходя из 21-О-ацетилкортикостерона, взаимодействием которого с хлористым нитрозилом в пиридине получают нит-ри| (27.2.1). Под действием фотохимического облучения последний трансформируется в оксим (27 2.2), гидролиз которого азотистой кислотой приводит к получению полуацеталя (27.2.3), являющегося ацетатом искомого альдостерона. Щелочной гидролиз ацетильной группы последнего приводит к искомому альдостерону (27.2.4) [33].
-495-
Глава 27
По ряду причин альдостерон практически не используется в качестве терапевтического средства для корректировки электролитных нарушений при недостаточности надпочечников, с этой целью чаще используются дезоксикортикостерон и флудрокортизон.
Дезоксикортикостерон (Desoxycorticosteron)
Дезоксикортикостерон — ацетат 21-гидроксипрегн-4-ен-3,20-диона (27.2.6) предложено получать различными методами. Первым способом (простейшим) является йодирование прогестерона по метильной группе, с последующим взаимодействием полученного йодпроизводного (27.2.5) с ацетатом калия, что приводит к получению искомого дезоксикортикостерона в виде ацетата (27.2.6) [34].
Второй способ синтеза ацетата дезоксикортикостерона (27.2.6) исходит из 3-гидрокси-5.16-прегнадиен-20-она. Первоначальным актом синтеза является защита гидроксильной группы при С3. Ее подвергают формилированию муравьиной кислотой с получением формиата (27.2.7). Взаимодействием последнего с изопропенилацетатом в присутствии н-толуолсульфоислоты получают енолацетат (27.2.8). Обработав полученный енолацетат йодсукцинимидом, который взаимодействует исключительно по енолацетатной двойной связи, получают йодкетон, взаимодействие которого с ацетатом калия приводит к образованию ацетата (27.2.9). Далее двойную связь при С16-Сг восстанавливают водородом с использованием в качестве катализатора палладия на угле, и получают продукт (27.2.10). Окислением последнего изопропилатом алюминия в присутствии циклогексанона получают ацетат дезоксикортикостерона (27.2.6) [35, 36].
27 2 7
- 496 -
Кортикостероиды
Дезоксикортикостерон вызывает усиленную реабсорбцию ионов Na* и выброс ионов К из почечных канальцев, что приводит к повышению гидрофильности тканей. Способствует увеличению объема плазмы и повышению АД. Повышает тонус и улучшает работоспособность мышц.
Препара! применяют при недостаточности функции коры надпочечников. миастении, астении, адинамии, обшей мышечной слабости.
Синонимами препарата являются перкортен, докаболин, корти-грон и др.
Флудрокорт из он (Fludrocortison)
Флудрокортизон — 9а-фтор-11р,17а,21-тригидроксипрегн-4-ен-3,20-дион (27.2.14) предложено получать из ацетата гидрокортизона (27.1.17). На первой стадии синтеза осуществляют дегидратацию молекулы гидрокортизона с помощью хлорокиси фосфора в пиридине с получением продукта с двойной связью в положении С9-Сц (27.2.11). Полученную двойную связь эпоксидируют путем первоначальной трансформации в бромгидрин с использованием N-бром-ацетамида и дальнейшим дегидробромированим с помощью ацетата натрия получают 21-О-ацетокси-9б-11Р-эпокси-17а-гидрокси-4-прегнен-3,20-дион (27.2.12). Раскрытие эпоксидного кольца в последнем, как и в вышеописанных случаях, осуществляют с помощью фтористоводородной кислоты, в результате чего получается
-497 -
Г лава 27
21 О-анетат флудрокортизона (27 2 13) Гидролизом ацетильной гр\ппы последнею ацетатом калия получают флудрокортизон (27 2 14) [37]
1 CH3CONH9
2 CHjCOONa
Препарат обладает практически той же активностью что и дезоксикор гикостерон, и даже вытесняет последний из медицинского применения
Основным синонимом препарата является флоринеф
Список литературы
1 US Pat 2 649 401 (1953)
2 US Pat 2 638 023 (1953)
3 US Pat 2 794 816 (1957)
4 Julian P et al //J Am Chem Soc 72, 5145 (1950)
5 Sondheuner F et al ZZJ Am Chem Soc 78. 816 (1956)
6 US Pat 2 769 823 (1956)
7 Hogg J et al J Am Chem Soc 77. 4436 (1955)
8 Olixeto E et al Z/J Am Chem Soc 78, 1736 (1956)
9 US Pat 2 602 769 (1952)
10 Applezvveig V //Steroid Drugs Vol 62 (Nev,-York, Toronto, London 1962)
11 Applet eig A / Steroid Drugs Vol 1, 14 (New-York, Toronto, London 1962)
12 Kendall E et al z J Biol Chem 166. 345 (1946)
-498 -
Кортикостероиды
13
14
15
16
17
18
19
20
21
22
23
24
23
26
27
28
29
30
31
32
33
34
3^
36
37
38
39
40
41
42
Kendall Е et al J Biol Chem 173, 2П (1948) IS Pat 2 897 216 (1959)
Applezsxetg Ni Steroid Drugs Vol 1, 66 (New-York, Toronto, London 1962)
LSPat3 134 718 (1964)
Wettstein A etal/TIelv Chim Acta 39, 734 (1956)
Ger Pat 1 135 899 (1960)
Nobile A et al J Am Chem Soc 77, 4184 (1955)
US Pat 2 897 218 (1959)
Speero G etal/T ChemSoc 78, 6213 (1956)
Speero G et al 1 J Chem Soc 79, 1515 (1957)
US Pat 3 053 832 (1962)
US Pat 4 041 055 (1977)
Fried J et al /J Am Chem Soc 81, 1235 (1959) Arth G et al / J Am Chem Soc 80, 3160 (1958) Ger Pat 1 113 690 (1958)
ApplezsseigN Steroid Drugs Vol 1 72 (New-York, Toronto, London 1962)
US Pat 3 164 618 (1965)
US Pat 2 789 118 (1956)
Bernstein S et al NJ Am Chem Soc 78,5693 (1956) Bernstein S et al /J Am Chem Soc 81 1689(1959) Barton D etal/T Am Chem Soc 82 2641 (1960) Ringold H et al /J Am Chem Soc 80, 250 (1958) Sondheimer F et al J Am Chem Soc 79, 5034 (1957) It all M et al J Am Chem Soc 77, 5665 (1955) Fried J et al ' J Am Chem Soc 76, 1455 (1954) Hirschmann R et al/J Am Chem Soc 78, 4956 (1956) Ger Pat 1 035 133 (1956) Ger Pat 1 028 572 (1957) US Pat 2 894 007 (1959) Bnt Pat 792 224 (1954)
i? Глава 28
? Женские
| половые гормоны
Среди многочисленных регуляторов деятельности организма, гормонов, огромное значение имеют два стероидных гормона: эстрадиол — женский половой гормон и тестостерон — мужской половой гормон. Присутствуя в организме в ничтожных количествах, они регулируют половую дифференциацию и воспроизводство, а также действуют на функционирование многих других физиологических систем. Несмотря на огромное сходство химической структуры, по физиологическому действию они весьма различны.
Двумя основными классами женских половых гормонов являются эстрогены и прогестины, которые образуются в яичниках, в меньших количествах — корой надпочечников, в период беременности — плацентой. Совместно они выполняют весьма важные функции по развитию вторичных женских половых признаков, контроля беременности, контроля овуляторного и менструального циклов и модуляции большого числа метаболических процессов.
Одним из наиболее важных областей применения синтетических эстрогенов и прогестинов является их использование в качестве оральных контрацептивов. Эти препараты могул представлять собой фиксированные комбинации, содержащие постоянные количества ктрогена (например, этинилэстрадиола) и прогестина (например, норэтиндрона). комбинации, содержашие постоянное количество эстрогена с варьирующими дозами прогестина, или же таблетки, состоящие только из прогестина в постоянной дозе.
-500-
Женские половые гормоны
28.1. Эстрогены
Эстрогенами называются соединения природного или синтетического происхождения, координирующие системную регуляцию во время овуляторного цикла, включая регуляцию репродуктивного тракта, грудей, слизистых и других тканей. Они также играют важную роль в развитии некоторых опухолей, и, в частности, эстрогены, равно как и антиэстрогены, используются также при лечении опухолей груди и простаты.
Эстрогены — общий термин, используемый для описания соединений, обладающих эстрогенной активностью. Однако они, в свою очередь, могут быть подразделены на две группы: эстрогены, являющиеся функционализированными производными стероидной структуры, и соединения, не содержащие стероидного кольца.
Существует три природных человеческих эстрогена: 17Р-эстрадиол, наиболее мощный эстроген, который трансформируется в более слабый метаболит эстрон и который, в свою очередь, метаболизируется в эстриол.
17Р-эстрадиол — главный эстроген, синтезируемый и выделяемый нормальными женскими яичниками. Его окисленный аналог — эстрон в значительно меньшей степени также выделяется пременопаузальными яичниками. Гормональное действие эстрогенов на целевые ткани основывается на сложном механизме, включающем их взаимодействие со специфическими эстрогеновыми цитоплазматическими рецепторами. После связывания с последними происходит конформационная перестройка, в результате которой эстроген-рецепторный комплекс проникает в ядро, где в последствии диссоциирует и возвращается в нативное состояние.
Эстрогены применяются при недостаточной функции яичников.
Заместительная эстрогональная терапия используется в агонадаль-ном, менопаузальном и гипоталамическом и аменоррагическом состояниях (т. е. в случаях первичного гипогонадизма и гормональной терапии у постменопаузных женщин). Эстрогены рекомендуются также при других клинических эндокринных нарушениях, если установлен гипоэ-строгенизм.
Наиболее широко эстрогены используются в комбинации с прогестинами в составе ряда оральных контрацептивных препаратов. Часто используются для предупреждения и лечения остеопороза и в качестве препаратов заместительной терапии для устранения ряда симптомов при наступлении менопаузы. Эстрогены используются также в качестве антагонистов андрогена в случаях андрогенчувсгвительных форм рака.
- 501 -
Глава 28
Из препаратов стероидной структуры в медицине широко применяют эстрон, эстрадиол, эстриол и этинилэстрадиол.
Из препаратов нестероидной структуры применяют синэстрол. ди-этилстильбесгрол, хлортрианизен. Для исследовательских целей многие препараты можно получать из мочи беременных женщин или беременных животных. Препаративное же получение часто осуществляют исходя из растительных стеринов - — сапонина, эргостерина и стигмастерина и др. или же из холестерина либо желчных кислот предварительной деструкцией боковой цепи с образованием соответствующих прекурсоров для дальнейших синтезов.
Осуществлены и полные синтезы всех главных стероидных гормонов. В качестве одного из примеров полных синтезов будет приведен лишь относительно простой синтез эстрона.
Эстрон (Estron)
Эстрон — 3-гидроксиэстра-1.3.5(10)-триен-17-он (28.1.9) был получен синтетическим путем различными методами. Согласно первому способу, наиболее простой и одной из первых схем синтеза, происходит следующее. Конденсацией 3-метоксифенилацетиена с би-ииклогексан-1,5-дионом по Фаворскому был получен соответствующий карбинол (28.1.1). Тройная связь последнего была восстановлена водородом над палладиевым катализатором с получением третичного спирта (28.1.2), который был далее дегидратирован в кислых условиях в соединение (28.1.3). Внутримолекулярным алкилированием последнего в присутствии безводного хлористого алюминия был получен тетрациклический кетон (28.1.4), который конденсацией с бензальдегидом был трансформирован в енон (28.1.5). Последний был метилирован в P-положение относительно кетогруппы йодистым метилом в присутствии mpew-бутилата калия, и полученное соединение (28.1.6) было подвергнуто озонолизу с образованием дикарбоновой кислоты (28.1.7). Циклизация последней в производное циклопентанона привело к получению метилового эфира искомого эстрона (28.1.8), деметилированием фенольного гидроксила в котором бромистоводородной кислотой был получен искомый эстрон (28.1.9) [1,2].
-502 -
Женекие половые гормоны
ЧВг
Второй способ синтеза исходит из доступного андростенолона (ди-гидро эпиандростерона) (28.1.10), который, в свою очередь, получают из ацетата дибромхолестерина. Восстановлением двойной связи в андро-стенолоне водородом с использованием в качестве катализатора палладия на угле получают кетоспирт (28.1.11), который далее оксидом шестивалентного хрома окисляют в дикетон (28.1.12). Последний подвергают бромированию молекулярным бромом в уксусной кислоте с получением дибромида (28.1.13). Продукт подвергают дегидробромированию нагреванием в коллидине с получением диенона (28.1.14). При нагревании последнего при температуре около 530 °C происходит отщепление молекулы метана с ароматизацией кольца А и образованием искомого эстрона (28.1.9).
- 503 -
Г лава 2&
Аналогичный подход к синтезу эстрона продемонстрирован с использованием в качестве исходного 1,4,6-андростатриен-3.17-диона (28.1.15), который при нагревании до 600 °C также ароматизируется с выбросом молекулы мегана с образованием 8,9-дигидроэквиленина. Двойная связь последнего в положении С,,С'г была восстановлена водородом над палладиевым катализатором с получением искомого эстрона (28.1 9).
Предложены и другие методы синтеза получения эстрона с использованием в качестве исходных других доступных функционализированных производных стероидов, выделяемых из природных источников и далее модифицированных [3, 4, 5, 6, 7, 8, 9. 10, 11, 12. 13. 14. 15, 16, 17. 18, 19, 20, 21, 22].
Как уже было отмечено, эсгрон является фолликулярным гормоном, необходимым для развития женского организма от периода половою созревания до климактерического периода. Препарат применяют при недостаточности функции яичников, при климактерических или постка-страционных расстройствах, бесплодии, переношенной беременности, слабости родовой деятельности, после гинекологических операций.
Синонимами препарата являются фолликулин, кетоксиэстрин, теле-стрин, бестрон и многие другие.
Эстрадиол (Estradiol)
Эстрадиол — эстра-1,3.5(10)-1риен-3,17Р-диол (28.1.17) проще всего получить восстановлением кетогруппы эстрона различными восстановителями и. в частности, боргидридом калия [23, 24].
КВНд
- 504 -
Женские половые гормоны
Альтернативным путем получения эстрадиола является способ, основанный на использовании ацетата андростенолона (З-ацетокси-5-андростен-17-она) (28.1.18). Восстановлением кетогруппы последнего водородом с использованием в качестве катализатора никеля Ренея и дальнейшим ацилированием образовавшейся гидроксильной группы хлористым бензоилом получают диэфир (28.1.19). Последний подвергают ряду превращений, и. в частности, восстанавливают двойную связь в положении С5-С6 водородом над платиновым катализатором, далее подвергают мягкому шелочному гидролизу в метаноле ацетильную защитную группу при С3, окисляют полученную гидроксильную группу до кетонной. с использованием оксида шестивалентного хрома, и далее водной щелочью гидролизуют бензоильную защиту гидроксильной группы при С|7 с получением кетоспирта (28.1.20). Последний, аналогично методам уже описанным при получении эстрона, подвергают бромированию молекулярным бромом с получением дибромида (28.1 21). Далее продукт подвергают дегидробромированию нагреванием в коллидине с получением диенона (28.1.22). При нагревании последнего при температуре около 325 °C в тетралине происходит отщепление молекулы метана с ароматизацией кольца А и образованием искомого эстрадиола (28.1.17) [25, 26]. Эстрадиол получают также другими методами [27, 28].
Эстрадиол образуется в организме женщины вместе с эстроном и при применении в виде эфиров кислот оказывает сильное и продолжительное эстрогенное действие. Применяется по тем же показаниям, что и эстрадиол.
Синонимами препарата являются делэстроген, эстравал, ретестин, деладиол и др.
- 505 -
Глава 28
Эстриол (Estriol)
Эстриол — эстра-1,3,5(10)-гриен-3.16а.17[3-триол (28.1.25} предложено получать из метилового эфира эстрона (28.1.8). Исходный метиловый эфир эстрона взаимодействием с г/эо-пропенилаце-татом в присутствии и-толуолсульфокислоты переводят в соответствующий енолацетат (28.1.23). Далее полученный енолацетат окисляют до эпоксида с применением надбензойной кислоты. Образовавшийся при этом эпоксид (28.1 24) подвергают восстановлению алюмогидридом лития с получением эстриола (28.1.25) [29, 30, 31, 32, 33].
Эстриол значительно менее активен, чем эстрадиол, однако обладает селективным свойством стимулировать кровоснабжение и восстановление эпителия гениталий. Кроме того, при его применении уменьшаются психические симптомы климактерического синдрома. Применяется в период пременопаузы и менопаузы, при атрофии кожи и признаках дегенерации гениталий и т. п.
Синонимами препарата являются овестин, гормонин и др.
Этинилэстрадиол (Ethinylesradiol)
Этинилэстрадиол — 17а-этинил-1,3,5(10)-эстратриен-3,17Р-диол
(28.1.26) получают либо конденсацией эстрона с ацетиленом в присутствии гидроокиси калия (реакция Фаворского), либо взаимодействием ацетиленида натрия в жидком аммиаке с эстроном [34, 35. 36].
- 506 -
Женские половые гормоны
нс=сн
Фактически этинилэстрадиол отличается по строению от эстрадиола наличием этинильной группы у Ср. Однако такое различие в строении приводит к существенному повышению эстрогенной активности препарата.
Синонимами препарата являются прогинон, бинордин, гестрол, ов-ранет, оврат и многие другие.
Гексэстрол (Hexestrol)
Гексэстрол — 4,4'-(1.2-диэтилэтилен)дифенол (28.1.29) представляет собой производное а,р-дифенилэтана и является синтетическим эстрогеном. Гексэстрол получают реакцией димеризации типа реакции Вюрца 1 -бром-1 -(4-метокси-фенил)пропана (28 1.27) в присутствии натрия, магния, алюминия или железа. Исходный 1-бром-1-(4-метоксифенил)пропан (28 1.27), в свою очередь, получают присоединением бромистого водорода к 4-метокси-1-пропенилбензолу. Последующим снятием метоксильных защитных групп с образовавшегося продукта димеризации (28.1.28) йодистоводородной кислотой получают гексэстрол (28.1.29) [37, 38, 39, 40. 41,42, 43].
СН3О
нс=нс-сн3
НВг
СН3О
Вг
нс—сн2—сн3
Na
28 1 28
28 1 27
28 1 29
Значительно отличаясь по строению от стероидных эстрогенных гормонов, препарат проявляет все присущие этим гормонам биологические свойства.
-507-
Глава 28
Гексэстрол применяют по тем же показаниям, что и эстрон. Его применяют также при раке предстательной железы или ее гипертрофии у мужчин
Синонимами препарата являются, синэстрол, циклоэстрол и др.
Ju)mu.icmiLib6ecmpo:i (Diethylstilbestrol)
Диэтилстильоесгрол — шраис-аД-диэтилЛД^сгильбендиол (28.1.34) предложено получать разными методами. Согласно первому способу, дезоксианизоин алкилируют йодистым этилом в присутствии этилата натрия, и полученный кетон (28.1.30) вводят в реакцию Гриньяра с этилмагнийбромидом с получением карбинола (28.1.31) Дегидратацией последнего перегонкой в присутствии и-толуол-сульфокислоты получают диметиловый эфир стильбестрола (28.1.32), метильные группы которого снимаются нагреванием при высокой температуре с гидроокисью калия с образованием диэтилстильбэст-рола (28.2 33) [44],
^2Н5 С—Сг!
ОН С2г
ОСН
TosOH
Второй способ получения диэтилстильбестрола заключается в следующем: 4-гидрокси-пропиофенон подвергают пинаконовому восстановлению с использованием амальгамы натрия, в результате чего получают соответствующий пиакон (28.1.34). Далее полученный пинакон подвергают действию соляной кислоты с получением продукта пинако-линовой перегруппировки — кетона (28.1.35). Кетогруппу последнего восстанавливают с использованием натрия в изоамиловом спирте, и в результате получают соответствующий карбинол (28.1.36). Полученный карбинол обрабатывают соляной кислотой, одновременно осуществляя и дегидратацию, и перегруппировку в диэтилстильбэстрол (28.1.33) [45. 46]
- 508 -
Женекие половые гормоны
он
Диэтилстильбестрол является производным стильбена и отличается от синестрола наличием двойной связи с трансконфигурацией двух фенильных групп. По эстрогенной активности препарат превосходит и эстрон, и гексэстрол.
Синонимами препарата являются дистильбен, менопакс, стильфо-строл, гилостерон, антигестил и очень многие другие.
28.2. Антиэстрогены
Как следует из самого названия, антиэстрогенами являются соединения, подавляющие эффекты эстрогенов. Это синтетические вещества нестероидной структуры, весьма вероятно, действующие путем блокады эстрогенных рецепторов, подавляя тем самым эффекты эст-poi енов.
Однако в малых дозах, связываясь с теми же рецепторами, они оказывают умеренный эстрогенный эффект и только в больших дозах оказывают антиэстрогенное действие. Их применяют как препараты, улучшающие функциональную деятельность яичников в основном при бесплодии у женщин, а также при лечении эстроген-рецептор позитивного рака грудной железы. Их применяют также при андрогенной недостаточности и олигоспермии у мужчин.
Основными антиэстрогенными препаратами, применяемыми в ме-тицинской практике, являются синтетические препараты кломифен и тамоксифен, относящиеся к ряду диэтилстильбесгрола.
- 509 -
Глава 28
Кломифен (Clomiphen)
Кломифен — 2-[и-(2-хлор-1,2-дифенилвинил)фенокси]триэтиламин (28.2.4) предложено синтезировать из 4-гидроксибензофенона, взаимодействием которого с 2-диэтиламнноэтилхлоридом в присутствии щелочи получают 4-(2-диэтиламиноэтокси) бензофенон (28.2.1). Последний вводят в реакцию Гриньяра с бензилмагнийхлоридом с получением соответствующего карбинола (28.2.2). Дегидратацией последнего хлористым водородом получают 2-[и-1,2-дифенил-винил)фенокси] триэтиламин (28.2.4), винильный атом водорода которого замещают на атом хлора с использованием N-хлорсук-цинимида с получением кломифена (28.2.4) [47, 48].
Кломифен действует путем усиления фолликулярного роста, вызывая овуляцию. Основным показанием для применения кломифена является индукция овуляции у неовулируюших женщин, у которых еще сохраняется некоторая выработка эстрогена. Кломифен используется при бесплодии для повышения репродуктивноеги женщин-олигоовуляторов. имеющих три. четыре овуляционных цикла в год, приводя к нормальной ежемесячной овуляции.
-510-
Женские половые гормоны
Синонимами препарата являются кломнд, серофен, клостильбегит и др.
Тамоксифен (Tamoxifen)
Тамоксифен — (2)-2-[л-(1,2-дифенил-1-бутенил)фенокси]1\т,М-диметилэтиламин (28.2.8) предложено синтезировать исходя из а-эгилдезоксибензоина. Взаимодействием последнего с 4-мето-ксимагнийбромидом получают соответствующий карбинол (28.2.5). формальной дегидратацией которого получают производное стильбена (28.2.6), и дальнейшим нагреванием с гидрохлоридом хинидина в качестве деметилирующего реагента получают 2-[я-(1,2-дифенил-1-бутенил)фенол] (28.2.7). Фенольный гидроксил далее алкилируют диметилам иноэтилхлоридом с использованием в качестве основания этилата натрия с получением смеси Е и Z изомеров искомого продукта, из которой дробной кристаллизацией из петролейного эфира выделяют искомый Z изомер тамоксифена (28.2.8).
28 2 7
HsC2\
n-ch2-ch2-ci HsCj'’
28 2 8
Альтернативным путем получения тамоксифена является непосредственное введение в реакцию с а-этилдезоксибензоином в качестве реактива Гриньяра 4-(2-диметиламиноэтокси)фенилмагнийбромида, дальнейшей дегидратацией образовавшегося карбинола (28.2.9) и последующим разделением смеси Е и Z изомеров [49, 50, 51, 52, 53, 54, 55, 56].
- 511 -
Глава 28
Тамоксифен является конкурентным ингибитором эстрадиола. Он используется при паллиативном лечении эстроген-рецептор позитивном раке груди. Используется в сочетании с другими химиотерапевтическими средствами.
Синонимами препарата являются нолвадекс, кессар и др.
28.3. Прогестины
Термин «прогестин» объединяет прогестерон и другие природные или синтетические соединения с аналогичным прогестерону физиологическим действием
Прогестерон является гормоном, вырабатывающимся стероидогенными тканями — желтым телом и плацентой. Прогестерон подготавливает эндометрий к имплантации яйцеклетки, предупреждает овуляцию, способствует увеличению железистой ткани молочных желез.
Его структура больше напоминает структуру мужского полового гормона тестострона, отличаясь от него лишь заместителем при Ср (ацетильная группа вместо гидроксильной), чем структуру женских половых гормонов — эстрона или эстролов. Прогестерон считается гормоном беременности, поскольку вырабатывается в течение всего периода беременности, уменьшает возбудимость и сократимость матки и одновременно препятствует созреванию новых яйцеклеток. Этот результат его действия (предотвращение образования яйцеклеток) равносилен временной стерильности женщины. Обнаружение этою факта привело к созданию нового метода предохранения от беременности путем поддержания в организме искусственного гормонального состояния
- 512 -
Женские половые гормоны
псевдобеременности.
Прогестины применяются при различных нарушениях менструального цикла, при функциональных маточных кровотечениях разного происхождения, в качестве контрацептивов. Прогестиновая терапия применяется и при лечении эндометриоза и эндометриальной карциноме. Прогестерон не эффективен при оральном применении вследствие интенсивной метаболизации, поэтому применяется парентеральным либо трансвагинатьным путем.
Получен ряд активных синтетических прогестинов для использования в качестве оральных контрацептивов и обладающих большей, чем у прогестерона, активностью и продолжительностью действия. Более того, сделаны определенные заключения относительно модификации структуры прогестерона с целью получения новых прогестинов. Показано. что прогестерон значительно теряет свою характерную биологическую активность при введении в положение С)7 гидроксильной группы. В то же время этерификация этой гидроксильной группы длиннопепо-чечной жирной кислотой, такой как капроновая, приводит к образованию долгодействующего при парентеральном введении прогестина — капроната окипрогестерона.
Весьма эффективными прогестинами являются Ср этинильные производные — линэстренол, норгестрел, норэтиндрон, которые обеспечивают высокоэффективную контрацепцию.
Прогестерон (Progesteron)
Прогестерон — прегн-4-ен-3,20-дион (28.3.1) получают окислением прегненолона изопропилатом алюминия в присутствии циклогексанона в качестве акцептора протонов [57, 58, 59, 60, 61, 62, 63]. Сам прегненолон получают последовательным окислением и дальнейшим расщеплением боковой цепи стигмастерина — стерина растительного происхождения, выделяемого из соевых бобов.
Прогестерон применяют в качестве контрацет ива. при аменорее, пременопаузальном синдроме, бесплодии, недонашивании беременности, ановуляторных маточных кровотечениях.
- 513 -
Глава 28
Синонимами препарата являются прогестасерт, синергон, цикло-гест, протес гол. пролу тон и многие другие.
Гидроксипрогестерон (Hydroxyprogesteron)
Гидроксипрогестерон — 17а-гидроксипрегн-4-ен-3,20-дион (28.3.6) синтезируют исходя из дегидропрегненолона (28.3.2). Сам дегидропрегненолон получают последовательной деструкцией и окислением боковой спирокегальной группы диосгенина-агликона — одного из сапонинов растительного происхождения, выделенного из Dtscorea Двойную связь при С|6~С|7 дегидропрегненолона окисляют перекисью водорода в присутствии щелочи до эпоксидной (28 3.3). Взаимодействием полученного эпоксида с бромистым водородом в уксусной кислоте получают бромгидрин (28.3.4). Гидроксильную группу при С3 стероидной системы формилируют муравьиной кислотой, и далее восстановлением водородом над палладиевым катализатором удаляют атом брома при С16 с получением продукта (28.3.5). Гидроксильную группу при С,- последнего ацилируют ангидридом уксусной кислоты или других карбоновых кислот (например, капроновой) в присутствии п-толуолсульфо-кислоты, и далее окисляют формильную группу при С3 изопропила-том алюминия в присутствии циклогексанона, при которой одновременно происходит изомеризация двойной связи из положения С5-С6 в положение С4-С5 с получением искомого эфира гидроксипрогестерона, в данном случае ацетата (28.3.6), в каком виде они и применяются в медицинской практике [64, 65]. Предложены и альтернативные пути синтеза [66, 67, 68. 69, 70, 71, 72].
- 514 -
Женские половые гормоны
По биологическим свойствам препарат сходен с прогестероном, однако ботее стоек в организме и действует медленнее, оказывая пролонгированный эффект. Применяют при менструальных расстройствах, аменорее, угрожающем выкидыше и прочих патологических процессах, связанных с недостаточностью желтого тела.
Синонимами препарата являются неолютин, сигинон, капростерон и др-
Медроксипрогестерон (Medroxyprogesteron)
Медроксипрогестерон — 17а-гидрокси-6а-метилпрегн-4-ен-3,20-дион (28 3.37) отличается от гидроксипрогестерона наличием в положении С6 дополнительной метильной группы, которую вводят различными методами [73, 74, 75, 76, 77].
Применяется по тем же показаниям, что и вышеописанные препараты. Синонимами препарата являются амен, курретаб, провера и др.
Мегестрол (Megestrol)
Мегестрол — ацетат 17а-гидрокси-6а-метилпрегна-4,6-диен-3,20-диона (28.3.3) является продуктом дегидрирования медроксипрогестерона (28.3.7) хлоранилом (тетрахлор-л-бензохиноном) в присутствии /7-толуолсульфокислоты, в результате чего образуется дополнительная двойная связь в положении Сб~С7, и дальнейшим ацетилированием продукта (28.3.8) в искомый мегестрол (28.3.9) уксусным ангидридом в присутствии и-толуолсульфокислоты [78, 79].
- 515 -
Глава 28
Являясь представителем класса прогестинов, препарат применяется при различных формах рака и, в частности, рака груди, почек и др.
Синонимами препарата являются мегестат, ниагестин, мегейс и др.
Норэтиндрон (Norethindrone)
Норэтиндрон — 17а-этинил-17Р-гидроксиэстр-4-ен-3-он (28.3.12) получают исходя из 19-нор-4-андростен-3,17-диона (28.3.10), который, в свою очередь, получают частичным восстановлением литием в жидком аммиаке ароматической части З-О-метилового эфира эстрона и одновременно кетогруппы при С|7 в спиртовую, которую далее окисляют обратно в кетогруппу оксидом шестивалентного хрома в уксусной кислоте. Сопряженную с двойной связью карбонильную группу при С3 далее трансформируют в этиловый эфир соответствующего енола (28.3.11) с помощью ортомуравьиного эфира. Продукт далее этинилируют ацетиленом в присутствии третбутилата калия. После гидролиза соляной кислотой образующегося О-калиевого производного, при котором гидролизуется и енольная группа, получают норэтиндрон (28.3.12) [80, 81, 82, 83, 84, 85].
Препарат может применяться и в виде эфиров карбоновых кисло г и, в частности, ацетата.
Препарат вызывает переход слизистой оболочки матки в секреторную фазу, что способствует развитию оплодотворенной яйцеклетки. Уменьшает возбудимость и сократимость мускулатуры матки. Применяется при аменорее, маточных кровотечениях, бесплодии, невынашивании беременности, миоме, мастопатии, раке эндометрия и ряде других патолог ий.
- 516 -
Женские половые гормоны
Синонимами препарата являются концеплан. норфор. бревинор и многие другие.
Норгестрел (Norgestrel)
Норгестрел — (±)13-этил-17а-этинил-17р-гидроксиэстр-4-ен-3-он (28.3 13) отличается от норэтиндрона наличием этильной группы при С|3 стероидной системы, и его синтезируют этинилированием 3-ги-дрокси-18-.метил2.5|0-эстрадиен- 17-она [86, 87. 88. 89. 90. 91, 92].
Наличие этильной группы при С13 стероидной системы делает его менее активным, чем прогестерон, однако сохраняет активность при оральном приеме, что обеспечивает эффективную контрацепцию.
Синонимами препарата являются ановлар, норлест, миллигинон, лоэстрин и др.
Этистерон (Ethisteron)
Этистерон — 17-этинил-17-гидроксиандростен-4-ен-3-он (28.3.14) получают этинилированием андростенолона ацетиленом и последующим окислением гидроксильной группы при С, стероидной системы [93. 94].
НзС он
Этистерон отличается от норэтиндрона наличием двух метильных группы при С10 и Св стероидной системы. Это делает его менее активным, чем прогестерон, однако препарат сохраняет активность при оральном применении, чем обеспечивается высокоэффективная контрацепция.
Синонимами препарата являются аменореи, оралутон. прегнорал и др.
-517-
Г лава 28
Список литературы
1. Johnson W et al.//J. Am. Chem. Soc. 72. 1426 (1950).
2. Johnson W. et al./'J. Am. Chem. Soc. 74, 2832 (1952).
3. Anner G. et al.//Helv. Chim. Acta. 31, 2173 (1948).
4. Anner G. et al.//Helv. Chim. Acta. 32, 1957 (1949).
5. Anner G. et al.//Helv. Chim. Acta. 33, 1379 (1950).
6. Торгов И. и др.//ДАН СССР. 135, 73 (1960).
7. Sih Ch. et al. /J. Am. Chem. Soc. 87, 2765 (1965).
8. Kalvoda J. et al./,Helv. Chim. Acta. 46, 1361 (1963).
9. Dryden H. et al.//86, 742 (1964).
10. Morand P. et al.//Chem. Rev. 68, 85 (1968).
11. Pelluz L. et al.//Angew. Chem. 72. 725 (1960).
12. Velluz L. et aL/Angew. Chem. 77. 185 (1965).
13. Smith H. et al./'J.Chem.Soc. 1963. 5072.
14. Smith H. et al./7Experientia. 19, 177 (1963).
15. Torgov I. et al.//Tetrahedron Lett. 1963, 1553.
16. Torgov I. et al.'/Tetrahedron Lett. 1964. 171.
17. Fieser L., Fieser M.//Steroids. New York: Reinhold, 1959. P. 495.
18. Windholz T. et al./'Angew. Chem. Int. Ed. 3, 353 (1964).
19. Bartlett P. et al.//J. Am. Chem. Soc. 95, 7501 (1973).
20. Danishefsky S. et al.//J. Am. Chem. Soc. 98, 4975 (1976).
21. Kametani T. et al.//J. Am. Chem. Soc. 99, 3461 (1977),
22. Grieco P. et al./'J. Org. Chem. 45, 2247 (1980).
23. US Pat. 2.096.744(1937).
24. Ger. Pat. 698.796(1932).
25. Inhoffen H. etal.'/Ber. 74, 1911 (1941).
26. US Pat. 2.361.847 (1944).
27. Eder U. et al./,Her. 109, 2948 (1976).
28. Oppolzer IF. et al./<Helv. Chim. Acta. 63. 1703 (1980).
29. Gallaher F./;J. Am. Chem. Soc. 76, 2943 (1954).
30. Butenandt A. et al.'-'Z. Naturforsch. 1, 82 (1946).
31. Huffmann M. et al.//Science, 100. 312 (1944).
32. Huffmann M. et al. /J. Am. Chem. Soc. 69, 1835 (1947).
33. LIS Pat. 1.967.351 (1930).
34. Inhoffen H. et al. /Ber. 21, Ю24 (1938).
35. Ger. Pat. 702.063 (1938).
36. Ger. Pat. 516.444(1937).
37. Dodds E. et al.//Proc. R. Soc. London, Ser. В 218. 253 (1940).
38. Bernstein S. et al./.'J. Am. Chem. Soc. 62, 2871 (1940).
39. Butt-Hoi N. et al.'-'L Org. Chem. 14, 1023 (1949).
-518-
Женские половые гормоны
—• “ ммммм*"^^ммммм^ммм^*м<мм^мма
40. US Pat. 2.357.985 (1944).
41. Brit. Pat. 523.320 (1938).
42. Fr. Pat. 855.879 (1939).
43. Kharash M. et al. "J. Am. Chem. Soc. 65, 491 (1943).
44. Dodds E. et al.//Nature. 141,247 (1938).
45. US Pat. 2.421.401 (1947).
40. Ger. Pat. 715.542 (1939).
47. US Pat. 2.914.563 (1959).
48. Belg. Pat. 782.321 (1971).
49. Bnt. Pat. 1.013.907 (1963).
50. Brit. Pat. 1.064.629(1966).
51. Ger. Pat. 1.468.088 (1963).
52. Bedford G. et al., /Nature. 212, 733 (1966).
53. US Pat. 4.536.516(1985).
54. Robertson D. et al./'J. Org. Chem. 47, 2387 (1982).
55. Miller R. et al.-7J. Org. Chem. 50, 2121 (1985).
56. Al-Hassan M. et al.//Synth. Commun. 17, 1247 (1987).
57. US Pat. 2.379.832(1945).
58. Oppenauer R.o'Rec. Trav. Chim. Pays-Bas Belg. 56, 137 (1937).
59. US Pat. 2.232.438 (1941).
60. US Pat. 2.420.489 (1947).
61. Heyl F. et al.//J. Am. Chem. Soc. 72, 2617 (1950).
62. Stomp G. et al./-J. Am. Chem. Soc. 65, 491 (1943).
63. Johnson J. et al.//J. Am. Chem. Soc. 93. 4332 (1971).
64. RingoldH. et 1. J. Am. Chem. Soc. 78, 816 (1956).
65. US Pat. 2.802.839 (1947).
66. US Pat. 4.041.055 (1977).
67. US Pat. 2.648.662(1953).
68. US Pat. 2.777.843 (1957).
69. US Pat. 2.786.857 (1957).
70. US Pat. 2.813.060 (1977).
71. US Pat. 3.000.883 (1961).
72 Cutler F. et al.,-J. Org. Chem. 24, 1629(1959).
73. US Pat. 3.147.290 (1964).
74. Ellis B. et al,''J. Chem. Soc. 1957- 4092.
75. US Pat. 3.061.616(196).
76. Ger. Pat. 1.097.986(1957).
77. Babcock J. et al.' J. Am. Chem. Soc. 80, 2904 (1958).
78 US Pat. 2.891.079 (1959).
79. Ringold H. et 1. J. Am. Chem. Soc. 81, 7312 (1959).
80. US Pat. 2.774.122 (1956).
- 519-
Глава 28
81. US Pat. 2.774.777 (1956).
82. Djerassi C etal 'J. Am. Chem. Soc. 76, 4092 (1954).
83 Ringold H et 1. J. Am. Chem. Soc. 78, 2477 (1956).
84. L'eberwasser H. et al. /Helv. Chim Acta. 34, 344 (1963).
85. Onken D et al./Die Pharmazie. 25, 3 (1970).
86. Bnt. Pat. 1.041.279 (1961).
87. Bnt. Pat. 1.041.280 (1961).
88. Ger. Pat. 2 030.056 (1970).
89. Buzby G et al./'J. Med. Chem. 9. 782 (1966).
90. Smith H et al.,/J. Chem Soc. 1963, 5072.
91. Smith H et al./ J. Chem Soc. 1964. 4472.
92 Smith H et al.VExpenentia. 19, 394 (1963).
93. USPat. 2 272 131 (1942).
94. USPat. 4 041.055 (1977).
Глава 29
" Мужские половые гормоны : и анаболические стероиды
Гермин «андрогены» относится к ряд» природных и синтетических соединений, проявляющих маскулинизирующее и анаболическое действие тестостерона — основного физиологического эндогенного андрогенного I ормона.
Андрогены, или, как часто их определяют, мужские половые гормоны или анаболические стероиды, и, в частности, тестостерон в мужском организме вырабатываются мужскими половыми железами. Андрогены имеют маскулинизирующий, анаболический и ростостимулирующий эффекты. Под их влиянием развиваются половые органы, вторичные половые признаки, контролируется сперматогенез. Тестостерон способствует также синтезу белка в организме (анаболическое действие), увеличивает реабсорбцию воды и ряда ионов в почках. Физиологический эффект тестостерона и других андрогенов проявляется на протяжении всей жизни мужчины, начиная с внутриутробной, в частности с маскулинизации урогенитального тракта. Мужской организм вырабатывает ежедневно от 2,5 до 10 мг тестостерона и в наибольшем количестве ранним утром Андрогены играют весьма важную роль в сперматогенезе и созревании спермы, в процессе роста мускульной массы, тембре звучания голоса и т. д. Следует отметить, что женский организм также вырабатывает андрогены, но в значительно меньшем количестве, чем у мужчин. Использование андрогенных стероидов в медицинской практике показано в качестве заместительной терапии у мужчин при недостаточной функции мужских половых желез, половом недоразвитии. импотенции, климактерических и других нарушениях, а у женщин — при раке молочной железы и яичников, при климактерических расстройствах.
Как и все стероидные препараты, андрогены являются функционализированными производными циклопентанофенаптрена.
Было проведено много исследований, имеющих целью создать препараты с высоким андрогенным и малым анаболическим потенциалом, однако в большинстве своем они оказались безуспешными. Будучи структурно родственными тестостерону, они проявляют как андроген
- 521 -
Глава 29
ную. так и анаболическую активность, что часто используется спортсменами для наращивания массы мышц.
В то же время все используемые в настоящее время анаболики имеют андрогенную активность.
29.1. Андрогены
В практической медицине в качестве андрогенов широко используются эфиры тестостерона (пропионат, ципионат и энантат) и синтетический аналог тестостерона — метилтестостерон.
Андрогенный эффект тестостерона во многих тканях зависит от степени его превращения в дигидротестостерон ферментом 5-а-редук-газой в целевых тканях. Считается, что все стероиды, включая тестостерон. проявляют свой эффект путем связывания с соответствующими рецепторами в целевых тканях. Показано, что сродство к рецепторам 5-а-дигидротестостерона к андрогенным рецепторам примерно в 10 раз превышает таковое у тестостерона. Показано также, что связывание андрогенов с соответствующими рецепторами приводит к синтезу в организме некоторых специфических белков, т. е. их применение всегда сопровождается и анаболическим действием.
С разными целями были получены многочисленные синтетические аналоги андрогенов, в том числе и с целью пролонгирования андрогенною эффекта, и с целью увеличения абсорбируемости препаратов при разных путях введения. Модификации тестостерона включали этерификацию 17-р-гидроксильной группы и, в частности, получение сложных эфиров тестостерона метилированием (метил тестостерон, дромстано-лон), деметилированием (нандролон), введением атомов галогена в стероидный скелет (флуоксиместерон), заменой циклогексанового кольца А стероидной структуры на тетрагидропирановое (оксандролон), введением дополнительного гетероциклического кольца в стероидную структуру (станозолол).
Андрогены используются при терапии андро! енодефицитных состояний, таких как гипофункция яичек, евнухизм и евнухоидизм, кастрация, импотенция, климактерические состояния, а также при раке моточной железы и яичников у женщин до 60 лет.
Тестостерон (Testosteron)
Тестостерон - 17Р-гидроксиандрост-4-ен-3-он (29.1.5) предложено получать разными путями из различных исходных соединений, включая холестерин, но, однако, чаще всего исходят из ацетата анд-ростеполона. С этой целью кетогруппу при С)7 стероидной системы
- 522 -
Мужские половые гормоны
ацетат-андростенолон восстанавливают до гидроксильной либо с помошью боргидрида натрия, либо алюмогидридом лития, либо водородом над никелем Ренея, но всегда с получением 17Р-гид-роксисоединения. В рассматриваемом варианте синтеза восстановление боргидридом натрия или водородом над никелем Ренея приводит к получению ЗР-ацетокси-5-андростен-17Р-ола (29.1.1). Полеченную в результате восстановления гидроксильную группу далее подвергают ацилированию хлористым бензоилом в пиридине с получением диэфира (29.1.2). После чего с учетом разницы в кислотной части двух присутствующих в молекуле сложноэфирных групп, а также давно замеченного факта, что эфиры — производные 17-гидроксигруппы гидролизуются труднее, чем эфиры — производные 3-гидроксигруппы, селективным гидролизом гидроокисью калия в этаноле снимается ацетильная защита гидроксильной группы при С3, и полученный спирт (29.1.3) окисляется до кетона (29.1.4) изопропилатом алюминия в присутствии циклогексанона в качестве акцептора водорода, в ходе которого одновременно происходит изомеризация двойной связи из положения С5-Сб в положение С4-С5. Дальнейшим гидролизом щелочью сохранившейся сложноэфирной части молекулы получают искомый тестостерон (29.1.5) [1, 2, 3, 4, 5, 6]. При необходимости перевода его в соответствующий сложный эфир (пропионат, энантат, ципионат и некоторые другие эфиры тестостерона) далее осуществляют необходимое ацилирование.
В лечебных целях препараты (эфиры) тестостерона применяется при половом недоразвитии, функциональных нарушениях в половой системе, гипертрофии предстательной железы, климактерических рас
-523 -
Глава 29
стройствах у мужчин, некоторых видах бесплодия, обусловленных нарушением сперматогенеза, посткастранионном синдроме, эндокринной импотенции. Тестостерон иногда применяют для лечения женщин с опухолями половых органов и молочных желез, при климактерических расстройствах, когда противопоказаны эстрогенные препараты. При пероральном применении сам тестостерон неактивен, поэтому, в основном, применяется в виде эфиров карбоновых кислот (пропионат, энантат, ципионат и некоторые другие эфиры тестостерона).
Синонимами препарата являются гистерон, малоген, менопакс, тес-торал и др., а синонимами эфиров тестостерона являются андронат, де-потест, эверон и многие другие.
Мепгилтестостерон (Methyltestosteron)
Метилтестостерон — 17Р-гидрокси-17а-метиландрост-4-ен-3-он (29.1.7) также предложено синтезировать из андростенолона, вводя его во взаимодействие с метилмагниййодидом с получением соответствующего третичного спирта (29.1.6) и дальнейшим окислением гидроксильной группы при С3 в кетонную оксидом шестивалентного хрома. При этом в условиях реакции происходит одновременная изомеризацией двойной связи с получением искомого метилтесто-стерона (29.1.7) [7, 8, 9, 10].
CH3Mgl
Метилтестостерон, являясь синтетическим аналогом тестостерона, обладает всеми свойствами тестостерона, оказывая стимулирующее действие на развитие мужских половых органов и вторичных половых признаков, однако не разрушается ферментами ЖКТ и поэтому может применяться орально. Применяется по тем же показаниям, что и тестостерон при половом недоразвитии, функциональных нарушениях в половой сфере, климаксе у мужчин и связанными с этим сосудистыми нервными расстройствами. Препарат применяют также при дисфункциональных маточных кровотечениях в преклимактерическом и климактерическом периодах у женщин, при раке молочной железы и яичников.
- 524-
Мужские половые гормоны
Синонимами препарата являются андрорал, тесюрал. оравирон и др.
29.2. Антагонисты андрогенов
Существуют и антагонисты андрогенов — препараты, снижающие продукцию андрогенов мужскими половыми железами и подавляющие сперматогенез путем блокады рецепторов андрогенов. К ним относятся ципротерон и флутамид.
Ципротерон (Cyproteron)
Ципротерон — ацетат 6-хлор-17а-гидрокси-1а,2а-метиленпрегна-4,6-диен-3.20-диона (29.2.14) получают из ацетата 17а-гидрокси-прогестерона (28.3.36). Дегидрированием последнего хлоранилом (теграхлор-н-безохиноном), в результате чего образуется дополнительная двойная связь в положении С6-С- (29.2.8). и дальнейшим дегидрированием двуокисью селена получают соответствующий дивпнилкетон — 17а-ацетокси-1,4,6-прегна-триен-3,20-дион (29.2.9). Подвергая последний воздействию диазометана, осуществляют реакцию 1,3-диполярного присоединения по С,-С2 двойной связи стероидной системы с получением производного дигидропиразола (29.2.10). Последний под действием хлорной кислоты в диоксане разлагается с выбросом молекулы азота и образованием циклоиро-панового производного (29.1.11). Далее перекисью бензоила селективно окисляют двойную связь в положении С6-С7, и образовавшийся эпоксид (29.2.12) подвергают действию хлористого водорода в уксусной кислоте, в результате чего происходит образование хлоргидрина и его последующая дегидратация и одновременно раскрытие циклопропанового кольца с образованием соединения (29.2 13). Нагревая последнее с коллидином, в результате внутримолекулярного алкилирования осуществляют циклизацию в циклопропановое кольцо с получением ципротерона (29.2.14) [11, 12].
- 525-
Глава 29
Ципротерон применяют для контроля полового влечения у мужчин в случае сексуальных отклонений, а также при неоперабельной карциноме предстательной железы, У женщин препарат применяется при тяжелых случаях андроген изации, а у детей — при идиопатической преждевременной половой зрелости.
Синонимами препарата являются ципростат, андрокур, дайан и др.
Флупгамид (Flutamid)
Флутамид — 4’-нигро-3'-трифторметилизобутиранилид (29.2.15), несгероидный антагонист андрогенов, получают ацилированием 4-нитро-З-трифторметиланилина хлорангидридом изомасляной кислоты [13. 14, 15].
Nh2
о СНз
CI—с-сн-сн3
.—, О СН?
/—\ II I
O/J—4 Л—NH-C-CH-
F3C~ 29 2 15
СНз
Флутамид — нестероидный препарат, обладающий антиандроген-ным действием. Препарат блокирует связывание андрогенов клетками тканей-мишеней, препятствуя действию андрогенов Механизм его действия, возможно, связан также с торможением транспорта дигидротестостерона. Препарат способствует уменьшению размеров и плотности предстательной железы и уменьшает количество метастазов при раке последней, в связи с чем и применяется при паллиативном лечении рака предстательной железы.
Синонимами препарата являются фугерел, эулекин и др.
- 526 -
Мужские половые гормоны
29.3. Анаболики
В резулыате поиска новых андрогенных соединений были получены очень близкие по структуре тестостерону вещества с более выраженной. и даже избирательной, анаболической активностью. Их наибо-iee характерным свойством является способность стимулировать синтез белка в opi аиизме, улучшать азотистый обмен, задерживать выделение из организма азота, серы, фосфора, калия и кальция, в результате чего увеличивается масса мышц, развивается костная ткань и улучшается общее сосюяиие организма, улучшается аппетит.
Анаболические стероиды используют при кахексии, астении, после пулевой icpaiinn, при остеопорозе, для стимуляции регенераторных процессов
Нандролон (Nandrolon)
Нандролон — 17[3-гидроксиэстр-4-ен-3-он (29.1.7) получают исходя из эстрадиола (28.1.17). Фенольную гидроксильную группу последнею подвергают метилированию диметилсульфатом в присутствии гидроокиси натрия с получением соответствующего метилового эфира (29 3 1), и далее ароматическое кольцо восстанавливают литием в жидком аммиаке с получением эфира енола (29.3.2)
1 идролиз эфира енола смесью соляной и уксусной кислот приводит 14 образованию кетогруппы и одновременной изомеризации двойной связи из положения С5-С|0 в положение С4- С; с получением искомого нан 1ролона (29.1 3) [16. 17, 18, 19]. При необходимости использования
- 527-
Глава 29
в виде эфиров кислот продукт ацилируют производными соответст-вмоших кислот [20j.
Нандролон способствует созданию мышечной массы юла, усиливает процесс развития костной ткани.
Основными показаниями к применению нандролона, как и других анаболических стероидов, являются нарушения белкового анаболизма, астения, заболевания, сопровождающиеся потерей белка, недостаточность надпочечников, стероидный диабет, продолжительное неподвижное состояние.
Синонимами препарата, используемого в виде эфиров кислот, являются ретаболил. феноболин, эубодин и многие друз ие.
Флуоксиместерон (Fluoxymesteron)
Флуоксиместерон — 9-фтор-110,170-дигидрокси-17а-метиландросг-4-ен-З-он (29.3.8) получают исходя ит 11 Р-гидрокси-4-андростен-3,17-диона, который взаимодействием с пирролидином переводят в диенамин (29.3.4). Далее подвергают действию метилмагниййодида с получением после гидролиза 110,170-дигидрокси-17а-метил-андрост-4-ен-З-она (29.3.5). Дегидратацией последнего путем селективного тозилирования вторичной гидроксильной группы при Сп п-толуолсу льфохлоридом и последующим действием щелочи получают диен (29 3 6). двойную связь которого при Со-С,, трансформируют в эпоксидную последовательным действием М-бромапетамида во влажном растворителе (источник НОВг) и щелочью (29.3.7). Дальнейшим действием фтористого водорода осуществляют раскрытие эпоксидного кольца с получением искомого флуоксиместерона (29.3.8) [21, 22. 23, 24, 25, 26, 27, 28, 29. 30].
Фтуоксиместерон применяется по тем же показаниям, что и нанд-
ро.тон
- 528 -
Мужские половые гормоны
Синонимами препарата являются ультандрен, галостерин, галоте-стин И др
Оксандролон (Oxandiolon)
Оксандролон — 17Р-гидрокси-17а-метил-2-окса-5-андростан-3-он (29.3 10) получают окислением С|-С2 двойной связи 17Р-гидрокси-17а-мегил-1-андросген-3-она смесью тетра-ацетата свинца и четы-рехокиси осмия с расщеплением кольца А стероидной системы и получением альдегидокислоты (29.3.9). При восстановлении альдегидной группы последнего боргидридом натрия в результате внутримолекулярной циклизации непосредственно образуется лактон (29 3.10), представляющий собой искомый оксандролон [31, 32].
PbtCHjCOOIt OsOj,
Оксандролон применяется по тем же показаниям, что и нандролон. Синонимами препарата являются вазором, анавар и др.
Станозол (Stanozol)
Станозол - 17а-метил-5а-андростаио[3,2-с]пиразол-17Р-ол (29.3.13) получают исходя из продукта восстановления двойной связи при С4 С, в метилтестостероне, представляющего собой самостоятельный интерес в качестве анаболического препарата местанолона (29 3.1 1). Местанолон подвергают форматированию этилформиа-том в присутствии этилата натрия с получением 2-формильного (оксиметиленового) производного (29.3.12), которое взаимодействием с гидразином с легкостью циклизуется в искомый станазол (29.3 13), представляющий собой конденсированную с пиразолом стероидную систему [33. 34].
Станозол применяется по тем же показаниям, что и нандролон. Синонимами препарата являются стромба, винстрол и др.
- 529-
Глава 29
Список литературы
1. USPat. 2.308.833 (1943).
2. USPat. 2.308.834 (1943).
3. US Pat. 2.379.832 (1936).
4. USPat. 2.387.469 (1945).
5. USPat. 2 236.574 (1941).
6. USPat. 2.341.110(1939).
7. USPat. 2.143.453 (1939).
8. US Pat. 2.374.369 (1945).
9. USPat. 2.374.370(1945).
10. Ruzicka L. Helv. Chim. Acta. 18, 1487 (1935).
11. Ger. Pat. 1.289.991 (1965).
12. USPat. 2.234.093 (1966).
13. Ger. Pat. 2.130.450 (1972).
14. Ger. Pat. 2.261.293 (1973).
15. USPat. 3.847.988 (1974).
16. USPat. 2 698.855 (1955).
17. USPat. 2.774.777 (1956).
18. Wilds A et al.''J. Am. Chem. Soc. 75, 5366 (1953).
19. Djerassi C. et al., 'J. Am Chem. Soc. 76, 4092 (1954).
20. USPat. 2.998.423 (1961).
21. US Pat. 2.793.218 (1957).
22. USPat. 2.813.881 (1957).
23. US Pat. 2.837.517 (1958).
24. US Pat. 3.029.263 (1962).
25. Ger. Pat. 1.037.447 (1955).
26. Heyl W et al.//J. Am. Chem. Soc. 75, 1918 (1953).
27. Bernstein S. et al.'/J. Org. Chem. 19, 41 (1954).
28. Fried J et al.Am. Chem. Soc. 75, 2273 (1953).
29. Fried J etal., J. Am. Chem. Soc. 76, 1455 (1954).
30. Herr M et al.' J. Am. Chem. Soc. 78, 501 (1956).
31. USPat. 3.128.823 (1964).
32. Pappo R. et al./''Tetrahedron Lett. 1962, 365.
33. Clinton R etal. J. Am. Chem. Soc. 81, 1513 (1959).
34. Clinton R et al. 'J. Am. Chem. Soc. 83, 1478 (1961).
i Глава 30
I Антинеопластики
Рак — это заболевание, встречающееся у людей и животных, при котором нарушается структура и нормальное функционирование тканей организма. Точная этиология большинства видов рака неизвестна. Однако хорошо известно, что инфекции, факторы окружающей среды (химические вещества, инородные частицы, радиация), генетические факторы, могут индуцировать трансформацию нормальной клетки в неопластическую, т. е. размножающуюся и функционирующую ненормально.
Рак может быть охарактеризован следующими параметрами.
Клетки начинают безудержно размножаться, поскольку нарушаются механизмы контроля роста. Клетки перестают дифференцироваться. Клетки начинают проявлять инвазивность и приобретают способность к метастазированию, т. е. выявляться в тканях, удаленных от места первоначальной локализации. Клетки начинают усиленный синтез макромолекул из нуклеозидов и аминокислот.
Лечение рака включает хирургическое вмешательство, радиацию, иммунотерапию и химиотерапию неопластическими препаратами. До настоящего времени химиотерапия применяется в качестве дополнения к хирургическому вмешательству или радиационной терапии с целью удалить возможные оставшиеся метастатические клетки. Тем не менее некоторые виды опухолей в настоящее время лечат, в первую очередь, химиотерапевтическими средствами.
Как уже отмечалось, лечение пациентов больных раком зависит от успешного удаления или разрушения всех раковых клеток в организме. В настоящее время даже при наилучшем детектировании, вылечивается только определенный процент диагностированных пациентов.
Причиной этого является не плохая диагностика, а то что рак часто распространяется за пределы его локализации, делая лечение местной формы неадекватным. Итак, хирургическое вмешательство, химиотерапия и облучение являются тремя составляющими лечения рака, однако только химиотерапия эффективно вылечивает системное заболевание.
-531 -
Г лава 30
Химиотерапия рака в общем неспецифична. Под этим подразумевается, что препарат убивает не только раковые клетки, но и нормальные клетки. Ввиду этой неспецифичности вырабатываются специальные стратегии для повышения потенциала уничтожения раковых клеток и понижения токсичного воздействия на нормальные ткани. Опыт десятилетий показал, что рост опухолевых клеток происходит значительно интенсивнее, чем рост клеток той ткани, из которой они образовались. Парадоксально, однако, что в отличие от ожидаемого нормальные ткани часто регенерируются быстрее, чем раковые. Поэтому после цитоин! и-бируюшего эффекта определенных препаратов нормальные ткани после химиотерапии могут восстанавливаться.
Препараты, применяемые при лечении рака, подразделяются на шесть групп: антиметаболиты, алкилирующие агенты, антибиотики, препараты, выделенные из растений, гормоны и группа веществ, которая не укладывается в вышеперечисленную классификацию и потому рассматривается в разд. «Прочие».
30.1. Антиметаболиты
Антиметаболиты являются структурными аналогами обычных клеточных метаболитов, таких как фолиевая кислота, пиримидины и пурины, которые будучи введены в организм начинают имитировать структуру обычных метаболитов. Конкурируя с природными пуринами и пиримидинами в метаболических схемах, они вмешиваются в синтез нуклеиновых кислот, включаясь в их состав вместо обычных метаболитов. Это приводит к образованию клеточных продуктов, которые уже не могут нормально функционировать. Тем самым нарушаются клеточные процессы деления и размножения клеток.
Кроме того, являясь структурными аналогами природных веществ, антиметаболиты могут действовать не только внедрением в метаболический процесс и образованием «фальшивых» нефункциональных метаболитов, но и ингибированием каталитических функций определенного фермента или ферментной системы.
Антиметаболиты подразделяют на три группы: антагонисты фолиевой кислоты (метотрексат), производные пурина (меркаптопурин, тиогуанидин). производные пиримидина (фторурацил, флоксуридин, цитарабин).
30.1.1. Антагонисты фолиевой кислоты
Антагонисты фолиевой кислоты и. в частности, метотрексат действуют путем конкурентного связывания с ферментом дигидрофолатре-
- 532-
Антинеопластики
дуктазой вместо фолиевой кислоты. Последняя является общим исходным соединением для ферментнокатализируемых реакций переноса метильной фуппы. Фолаты являются переносчиками одноуглеродных групп (метилирующих групп), необходимых в ходе синтеза пурина и пиримидинтимидилата и, в частности, для метилирования дезокси уридин монофосфата в дезокситимидин монофосфат. Сродство дигид-рофолагредуктазы к антиметаболитам намного больше, чем к обычным субстратам — фолиевой кислоте и ее восстановленным формам. Из-за выраженного сродства дигидрофолатредуктазы к метотрексату даже большие дозы фолиевой кислоты, вводимой с ним одновременно, оказываются бесполезными для предотвращения эффектов метотрексата.
фолиевая кислота
дигидрофолиевая кислота
метотрексат
Метотрексат (Methotrexat)
Метотрексат — М-[«-[[(2,4-диамино-6-пиеридинил)метил]метил-амино]-бензоил]-Е-(±)-глютаминовую кислоту (30.1.1 8) получают взаимодействием \-(4-метиламинобензоил)глютаминовой кислоты (30.1.1.3) с 2-амино-4-гидрокси-6-бромметил-птеридином (30.1.1.7). Для этой цели М-(4-метиламинобензоил)глюта.миновую кислоту (30.1.1.3) синтезируют исходя из хлорангидрида 4-нитробензойной кислоты, которую вводят во взаимодействие с L-глютаминовой кислотой с получением К-(4-нитробензоил)глютаминовой кислоты (30.1.1.1). нитрогруппу которой восстанавливают до аминогруппы с использованием водорода над никелем Ренея с получением N-(4-аминобензоил)глютаминовой кислоты (30.1.1.2). Последнюю подвергают восстановительному метилированию с использованием
- 533-
Г лава 30
формальдегида и водорода с получением М-(4-метиламинобензоил)-глют аминовой кислоты (30.1.1.3).
Второй фрагмент молекулы метотрексата — 2-амино-4-гидрокси-6-бромметилпюридин (30.1.1.7) получают исходя из 2,4,6-триаминопи римидина (30.1.1 4). который легко получается взаимодействием динитрила малоновой кислоты с гуанидином. Последний нитрозируют безводной азо(истой кислотой в 2,4,6-триамино-5-нитрозопиримидин (30 1.1 5) и далее восстанавливаю г боргидридом натрия в 2,4,5,6-тетра-аминопиримидин (30 1.1 6). Действуя на последний 1.2-дибромпропионовым альдегидом, продуктом присоединения брома к акролеину, получают 2-амино-4-гидрокси-6-бромметилптеридин (30.1.1 7) Алкилированием полученным бромидом аминного атома азота в Ы-(4-мети-ламинобензоил)глютаминовой кислоте (30.1.1.3) синтезируют метотрексат (30.1.1.8) [1,2, 3,4. 5, 6, 7, 8].
соон
С н2 сч2 HnN-CH соон
соон СНо
,—> О СНг
o2n-4 У-c-nh—сн
'—' соон
30111
Н2 Raney—Ni
СООН
30 1 1 2
СН2О/ Zn-NgOH
СООН СНг ,__ О СН?
/—\ II I -
HN—\\ п—C—NH—“СИ СНз '-- СООН
30 1 1 3
30116
6г
Вг-сн2-сн-сно
- 534 -
днтинвоплэстики
30 1 , з + 30 1 1 7
30 1 1 8
Метотрекса! показан для лечения осгрой лимфатической лейкемии, хориокарциномы, неходжкинской лимфомы, саркомы костей, опухолей головы, шеи. груди и легких.
Синонимами препарата являются фармигрексат. ледертрексат, эма-тексат, макстрекс, фолекс, мексат и др.
30.1.2. Производные пурина
Среди многих синтезированных аналогов пурина меркаптопурин и тиогуанин оказались наиболее эффективными при химиотерапии рака.
Эти соединения ингибируют синтез пуриновых нуклеотидов, которые состоят из пуриновых оснований и фосфорилированной рибозы. Оба соединения должны быть трансформированы в нуклеотиды путем присоединения фосфорибозильного фрагмента.
Мер кап топур ин (Mercaptopurin)
Меркапгопурин — 6-ггуринтиол получают исходя из мочевой кислоты (30.1.2.5), которую синтезируют из барбитуровой кислоты (30 1.2.1) Барбитуровая кислота (30.1.2.1) легко получается конденсацией мочевины с малоновым эфиром и далее нитрозируется азотистой кислотой. Нитрозопроизводное (30.1.2.2) восстанавливается водородом, получаемым взаимодействием олова с соляной кислотой в амин (урамил) (30.1.2.3) и далее вводится во взаимодействие с изоциановой кислотой с получением псевдомочевой кислоты (30 1.2.4). Последняя при нагревании в присутствии соляной кислоты циклизуется в мочевую кислоту (30.1 2.5). При действии пятихлористого фосфора на мочевую кислоту образуется 2,6,8-три-хлорпурин (30 1.2.6). Три атома хлора в трихлорпурине сильно отличаются по отношению к замещению нуклеофилами. Атом хлора при С6 намного более активен, чем атом хлора в положении С2, а последний более активен, чем атом хлора при С8, что позволяет последовательно манипулировать ими. Взаимодействием 2,6,8-три-х.юрпурина (30.1,2 6) с аммиаком замещается атом хлора при С6 с получением дихлорпроизводного (30.1.2.7), которое далее восста-
-535-
Глава 30
навливается йодистым водородом до гипоксантина (30.1.2.8). Дей-сгвием пентасульфида фосфора последний трансформируется в меркаптопурин (30.1.2.9) [9, 10, 11, 12, 13. 14, 15].
О 0 0
(“С-~МН2 +
hno2
Sn ' HCI
HI
P2S5
Меркаптопурин в организме превращается в активную форму препарата — нуклеотид 6-тиоинозин-5-фосфат. Нуклеотид 6-тиоинозин-5-фосфат методом отрицательной обратной связи ингибирует первый шаг в синтезе инозин-5-фосфата, препятствуя его превращению в адениновые или гуаниновые нуклеотиды, необходимые для синтеза ДНК. Таким образом, меркаптопурин ингибирует синтез и взаимопревращения пуриновых нуклеотидов, что ведет к торможению синтеза ДНК в пролиферирующих клетках во время клеточного цикла.
Меркаптопурин применяют при лимфобластомном и миелобла-стомном лейкозах и лечении нейролейкемии.
Синонимами препарата являются изимпур, классен. пуринэтол и др.
Тиогуанин (Thioguanine)
Тиогуанин — 2-аминопурин-6-тиол (30.1.2.12) получают исходя их 2,8-дихлор-6-гидроксипурина (30.1.2.7), в котором действием аммиака замешают второй атом хлора при С2 на аминогруппу с получением 2-амино-8-хлор-6-гидроксипурина (30.1.2.7), который далее восстанавливается йодистым водородом до 2-амино-
- 536 -
Антинеопластики
лурин-6-ола (30.1.2.11). Замену гидроксильной группы на меркаптогруппу в положении С6 осуществляют взаимодействием с пентасульфидом фосфора с получением тиогуанина (30.1.2.12) [14, 16, 17, 18, 19. 20].
Тиогуанин в организме конвертируется в активную форму — 6-тио-гуанин-5-фосфат.
Главный механизм его действия заключается во включении его три-фосфатной формы в ДНК, замещении гуанинового нуклеотида и ингибировании синтеза ДНК.
Меркаптопурин имеет значение при лечении и взрослых, и детей в качестве препарата поддерживающей терапии. Тиогуанин может иметь определенное клиническое применение и в комбинации с другими препаратами при острой миелогенной терапии.
Синонимом препарата является ланвис и др.
30.1.3. Производные пиримидина
В качестве потенциальных противораковых препаратов синтезировано большое число фторированных аналогов пиримидина. Наиболее глубоко изучены фторурацил и флоксуридин. Возможно, что наиболее важным механизмом действия фторированных пиримидинов является ингибирование синтеза тимидилат синтетазы и, таким образом, воздействие на процесс выработки ДНК. Это действие сопровождается образованием активного метаболита, 5-фтордезоксиуридиномонофосфата как из фтору рацила, так и флоксуридина. 5-фтордезоксиуфидиномоно-фосфат образует комплекс с восстановленным фолат И5,№0-метилен-тетрагидрофолатом. Этот комплекс ингибирует тимидилат синтетазу и понижает метилирование 2-дезоксиуридиновой кислоты для образования тимидиловой кислоты. Возникающий при этом дефицит тимидина вызывает повреждение и гибель клеток.
Считается также, что трифосфорилированная форма фторированных аналогов пиримидина может быть включена в РНК и воздействовать на синтез протеинов.
- 537-
Глава 30
Фторурацил (Fluorouracil)
Фторурацил — 5-фторурацил (30.1.3.3) получают конденсацией этилового эфира фторуксусной кислоты с этилформиатом в присутствии этилата калия с получением гидроксиметиленфторуксусного эфира (30.3.1) и дальнейшей его циклизацией взаимодействием с S-метил-изо-тио.мочевиной в 2-метилтио-4-гидрокси-5-фторпири-мидин и последующим гидролизом соляной кислотой во фторурацил (30.1.3.3) [21, 22]. Альтернативным путем синтеза 5-фторура-цила является прямое фторирование урацила фтором или трифтор-метилгипофторидом [23, 24, 25, 26, 27, 28].
f-ch2-COOC2Hs ♦ НСООС2Н5
СгН5ОК
SCH3 h2n-c=nh
HCI
FZ / CF3OF
о
н
30 1 34
Фторурацил действует путем ингибирования синтеза пиримидинов и, соответственно, образования ДНК.
Применение фторурацила показано при лечении карциномы головы и шеи, ободочной и прямой кишок, груди, желудка, мочевого пузыря, поджелудочной железы, при актиничных и солнечных кератитах.
Синонимами препарата являются эффлудерм, флудикс, флуоробла-стнн, ару.мел. бентон, лнфрил и др.
Флоксуридин (Floxuridine)
Флоксуридин — 5-фтор-1-(2-дезоксирибофуранозил)-пиримидин-2,4-(1Я,ЗЯ)-дион (30.1.3.5) является пиримидиновым нуклеозидом, получаемым взаимодействием фторурацила (30.1.3.3) с 2-дезокси-рибофуранозилбромидом в присутствии серебряных или ртутных солей [29, 30, 31. 32, 33].
- 538-
д н т инеопластики
н
30 1 33
Ag or Нд
Флоксуридин используется методом инфузионного введения в печеночную артерию при метастазах в печени.
Синонимом препарата является FUDR и др.
Цитарабин (Cytarabin)
Цитарабин — 4-амино-1-р-арабинофуранозил-2(1Я)пиримидон (30.1.3.8) получают исходя из l-p-D-арабинофуранозилурацила путем предварительного ацилирования гидроксильных групп с получением триацетилпроизводного (30.1.3.6), дальнейшей заменой карбонильной группы в положении 4-пиримидинового кольца на тиокарбонильную с использованием пентасулъфида фосфора и, наконец, замещением меркаптогруппы в (30.1.3.7) на аминогруппу с использованием аммиака и одновременным гидролизом ацетильных защитных групп с получением цитарабина (30.1.3.8) [34, 35, 36, 37.38].
30 1 3 6
Цитарабин так же, как и вышеописанные препараты, действует путем ингибирования синтеза пиримидинов и, соответственно, образования ДНК в клетках.
Цитарабин является антиметаболитом эффективным при лечении лейкемии. Аналогично другим пиримидиновым антиметаболитам цитарабин должен быть «активирован» путем его первоначальной трансформации в соответствующий нуклеотид. Активной формой препарата
-539-
Глава 30
является цитарабин трифосфа! Цитарабин используется при всех видах лейкемии.
Синонимами препарата являются цитозин, арабинозид. ара-С.
30.2. Алкилирующие агенты
Алкилирующие агенты — высокореакционоспособные соединения, способные к образованию ковалентных связей с нуклеофильными участками (амино-, гидрокси-, сульфгидрил- и карбоксигруппами, азотистыми гетероциклами) таких молекул, как нуклеиновые кислоты, фосфаты, аминокислоты и протеины. Как правило, алкилирующие препараты, применяемые в медицине, алкилируют положение N7 гуанина. Однако алкилирование в принципе может происходить и происходит и по Об или N3 гуанина, по N(, N3 или N-. аденина либо по N3 цитозина. При этом многие составляющие клеток, включая ДНК, РНК, протеины, компоненты мембран и др., также алкилируются. Однако, согласно рабочей гипотезе, основной цитотоксический эффект этих соединений может быть объяснен их способностью к связыванию именно с нуклеотидами ДНК. Это приводит к неправильному считыванию информации с ДНК, и, как правило, клетка погибает из-за вмешательства в репликацию ДНК и митоза. Бифункциональные алкилирующие агенты имеют возможность образовывать межцепочечные связи в ДНК вызывая кросссвязывание двух цепочек ДНК и считаются более цитотоксичными, чем монофункциональные алкилирующие агенты.
Схематический механизм действия алкилирующих препаратов на примере самого простого из них — мехлорэтамина можно пояснить следующей схемой.
_Ch2"~Ch2~Cl
СН3-Н
СР 2~ СгЦ—Cl
-540-
Антинеопластики
Несмотря на то, что алкилирующие агенты выявляют общий механизм действия, их клиническое применение варьирует в зависимости от разницы в фармакокинетике, метаболизме, липидной растворимости, способности проникать в клеточные мембраны и токсичности. Они мо-г>т быть классифицированы как азотсодержащие горчичные производные (мехлорэтамин, хлорамбуцил, мелфалан, циклофосфамид, ифосфа-мид), производные этиленимина (тиотепа), нитрозомочевины (карму-стин, лому стин, стрептозоцин), алкулсульфонаты (бусульфан), производные платины (цисплатин, карбоплатин).
30.2.1. Азотсодержащие горчичные производные
Мехлорэтамин (Mechlorethatnine)
Мехлорэтамин — бис-(2-хлорэтил)метиламин (30.2.1.2) получают взаимодействием метиламина с окисью этилена с получением бис-(2-гидроксиэтил)метиламина (30.2.1.1). который взаимодействием с хлористым тионилом трансформируется в искомый мехлорэтамин [39, 40,41].
ch3nh2
,0
сн2-сн2-он
CH3-N
сн2-сн2-он
SOCI2
302 1 1
CH2-CH2-CI CH3-N
СН2—CH2—Cl
30 2 1 2
Мехлорэтамин широко используется внутривенно в комбинации с другими препаратами при болезни Ходжкина, лимфосаркоме, лейкемии. бронхогенной карциноме.
Синонимами препарата являются азотоиприт. хлорэтамин, хлорэта-зид, мустин и многие другие.
Х-юрамбуии.1 (Chlorambucil)
Хлорамбуцил — 4-[н-[бис(2-хлорэтил)амино]фенил]масляную кислоту (30.2.1.7) получают исходя из ацетанилида и янтарного ангидрида. На первой стадии синтеза ацетанилид ацилируют ангидридом янтарной кислоты с получением 4-(4-ацетаминофенил)-4-кетомасляной кислоты (30.2.1.3). Кетогруппу в последней восстанавливают водородом в метаноле растворе с использованием в качестве катализатора палладия на угле. В результате получают метиловый эфир 4-(4-ацетаминофенил)-масляной кислоты (30.2.1.4). Обработка последнего щелочью с целью гидролиза как амидной.
- 541 -
Глава 30
так и эфирной части молекулы приводит к получению 4-0-аминофе-нил)масляной кислоты (30.2.1.5), взаимодействие которой с окисью этилена приводит к получению 4-[н-[бис(2-гидро-ксиэтил)амино]фенил]масляной кислоты (30.2.1.7). Замещением всех гидроксильных групп в последней с помощью хлорокиси фосфора и дальнейшей обработкой водой с целью гидролиза до кислоты промежуточно образовавшегося хлорангидрида получают хлорамбуцил (20 2 1.7) (42, 43, 44, 45, 46].
СНз-СО-NH
О
С-СН2“СН2-СООН
302 1 3
H2/Pd—С СН3ОН
(СН2)з"~СООСНз
302 1 4
КаОЧ
(СН2)з—СООН
1 POCI;
но-сн2-сн2
N но-снг-снг7
(СН2)з“СООН
30 2 1 6
2 Н2О
CI—сн2-сн2
ci—сн2—сн/
(СН2)з-СООН
Хлорамбуцил хорошо адсорбируется при оральном применении. Используется при хронической лимфатической лейкемии и множественной миеломе.
Синонимами препарата являются лейкеран, амбохлорин, хлорбутин и др.
Мелфалан (Melphalan)
Мелфалан Ь-3-[н-[бис(2-хлорэтил)амино]фенил]аланин (30.2.1.13) является структурным аналогом хлорамбуцила, в котором фрагмент масляной кислоты заменен на фрагмент аминокислоты — аланина. Синтез препарата осуществляют исходя из L-фенилаланина, нитрованием которого азотной кислотой получают 4-нитро-1_-фенил-аланин (30.2.1.8). Взаимодействием последнего со спиртом в присутствии хлористого водорода получают хлоргидрат этилового эфира 4-нитро-С-фенилаланина (30.2.1.9), аминогруппу в котором защищают путем перевода во фталимид взаимодействием с янтар-
- 542 -
Антии еопластики
ным ангидридом (30.2.1.10). Нитрогруппу в последнем восстанавливают до аминогруппы водородом с использованием в качестве катализатора палладия на карбонате кальция. Полученный ароматический амин (30.2.1.11) далее вводится во взаимодействие с окисью этилена с получением бис(2-гидоксиэтил)-амино производного (30.2.1.12). Гидроксильные группы в последнем замещаются на атомы хлора взаимодействием с хлористым тионилом, после чего обработкой соляной кислотой снимается фталимидная защита с получением мелфалана (30.2.1.13) [47, 48, 49, 50].
/=\ ОгНфн HCI
~ * OsN—V. л—СН2-СН-СООН ------------•
'-' NH;
Н2 / Pd-СаСОз
30 2 1 12
1 SOCI2
2 НС!
30 2 1 13
сн2-сн-соон nh2
Мелфалан применяется внутривенно и орально для лечения множественной миеломы, рака груди, шеи, яичников.
Синонимом препарата является алкеран.
Под названием сарколизин, или рацемелфалан, широко применяется также рацемическая форма этого препарата — D,L-3-[h-[6hc(2-хлорэтил)амино]фенил]аланин.
Циклофосфомид (Cyclophosphamide)
Циклофосфамид — 2-[бис(2-хлорэтил)амино]тетрагидро-2Я-1,3,2-оксазафосфорин 2-оксид (30.2.1.15) получают взаимодействием бис(2-хлорэтил)амина с хлорокисью фосфора с получением N,N-
- 543-
Глава 30
бис(2-хлорэтил)дихлорфосфорамида (30.2.1.14), который дальнейшим взаимодействием с 3-аминопропанолом трансформируется в циклофосфамид (30.2.1.15) [51, 52, 53, 54, 55, 56, 57, 58, 59].
СН2-СН2“С1
H-N
СН2-СН2~С1
+ РОС13
С1^>° ch2-ch2-cj
Cr^N
Ch2-CH2-CI
30 2 1 14
-ОН
Н2С h2C^c,NH2
____»
С0-р<° СН2-СН2-С!
I—-N
сн2~сн2—CI
30 2 1 15
Характерное химическое строение препарата придает ему избирательную противоопухолевую активность. Находясь в крови он практически не активен, однако при проникновении в опухолевые клетки под действием содержащихся в них относительно больших количеств фосфамидаз он разлагается с высвобождением собственно цитостатического вещества — бис(2-хлорэтил)амина. Это означает, чго алкилирующее действие этого препарата узко направленно именно на опухолевые клетки. Циклофосфамид является одним из наиболее часто применяемых химиотерапевтических средств.
Циклофосфамид применяется и внутривенно, и орально. Используется при хронической лимфатической лейкемии, болезни Ходжкина, лимфомы Буркиттса, множественной миеломе, раке груди, шеи и яичников и др.
Синонимами препарата являются эндоксан, циклостин, цитоксан, циклофосфан и др.
И фосфамид (Ifosfatnide)
Ифосфамид — 3-(2-хлорэтил)-2-[(2-хлорэтил)амино]тетрагидро-277-1,3,2-оксазафосфорин-2-оксид (30.2.1.17), который можно рассматривать как соединение, изомерное циклофосфамиду (30.2.1.15) и аналогичное ему по действию, получают взаимодействием N-(2-хлорэтил)-Хт-(3-гидроксипропил)амина с хлорокисью фосфора с получением 3-(2-хлорэтил)-2-хлортетрагидро-2Я-1,3,2-оксазафосфорин 2-оксида (30.2,1.16), который далее вводят во взаимодействие с N-(2-хлорэтил)амином с получением искомого ифосфамида (30.2.1.17) [60. 61. 62].
- 544 -
Анти неопластики
Ифосфамид — экспериментальный препарат, аналог циклофосфамида.
Синонимами препарата являются голоксан и митоксан.
30.2.2. Производные этиленимина
Этиленимины являются высокореакционноспособными алкилирующими реагентами. Они алкилируют ДНК по положению N7 гуанина аналогично мехлорэтамину. Этиленимины оказывают цитостатическое действие и угнетают развитие пролиферирующей, в том числе злокачественной, ткани. Они нарушают обмен нуклеиновых кислот и блокируют митотическое деление ткани.
Их применяют при раке яичников, молочной железы, неоперабельных опухолях, прочих рецидивах и метастазах. В медицине в основном применяется триэтилен-фосфортриамид — тиотепа (30.2.2.1), однако предложены и применяются и другие препараты этого ряда — бензотеф (30.2.2.2), дипин (30.2.2.3), тиодипин (30.2.2.4), фосфемид (30.2.2.5) и др.
ЗС 2 2 -
ЗС 2 2 5
Тиотепа (Thiotepa)
Тиотепа — трис(1-азиридинил)фосфина сульфид (30.2.2.1) получают взаимодействием этиленимина с хлортиоокисью фосфора [63, 64].
30 22 1
Триэтилентиофосфорамид используется внутривенно при ранних стадиях карциномы печени, яичников и груди. Препарат применяют при хроническом лимфолейкозе, лимфогранулематозе, лимфосаркоматозе.
- 545 -
Г лава 30
Его применяют для уменьшения числа рецидивов и метастазов, при комплексном лечении различных форм рака.
Синонимами препарата являются теспамин, тилфосфамид, тиофо-сил и др.
30.2.3. Алкилсульфонаты
Единственным препаратом ряда алкилсульфонатов, нашедшим медицинское применение, является бусульфан.
Бусульфан (Busulfan)
Бусульфан — 1,4-бутандиолдиметансульфонат (30.2.3.1) получают взаимодействием бутандиола с метансульфонилхлоридом [65, 66].
СН2~СНг—ОН сн2-сн2-он
2 CH3-SO2-CI
СН2—СН2—O~SO2—СН3
CH2-CH2-O-SO2-CH3
30 2 3 1
Бусульфан является бифункциональным алкилирующим агентом, слабосвязывающимся с ДНК, однако не выявляет способности к кросссвязыванию Бусульфан избирательно алкилирует положение N7 гуанина. а также алкилирует сульфгидрильные группы глутатиона и цистеина. В отличие от других алкилирующих агентов, он мало действует на лимфоциты и намного меньше проявляет иммуносупрессивные свойства Имеет сильные цитотоксические свойства и способность убивать стволовые клетки. Может применяться орально. Используется в основном при хронической миелогенной лейкемии и полицитемии. В отличие от других алкилирующих агентов не увеличивает случаев вторичной лейкемии.
Синонимами препарата являются цитосульфан, лейкосульфан, мие-лосан. митостан и др.
30.2.4. Нитрозомочевины
Следующую группу антинеопластических препаратов в клинике представляют нифозомочевины (ломустин. кармустин, стрепгозоцин). Имеются и дрхгие препараты этого ряда (нимустин, семустин и др.), отличающиеся лишь группой, обозначенной R на нижеприведенной схеме Предполагается, что в организме нитрозомочевины расщепляются на Р-хлорэтанол и на алкилизоцианаты. Образовавшийся р-хлор-
- 546 -
Анти неопластики
этанол является высокореакционноспособным алкилирующим агентом, а алкилизоцианаты — карбамоилирующими агентами для протеинов, которые также проявляют определенную цитотоксическую активность.
Вероятная схема распада в организме нитрозомочевин на возможные активные компоненты приводится ниже
о
R—NH-С-N—СН2—СН2-С| ----► R-N=C=O + Cl—CH2-CH2-N=N-OH
N=O
Cl—CH2~CH2—OH
JJo.nyqmuH (Lomustine)
Ломустин — 1-(2-хлорэтил)-3-циклогексил-1-нитрозомочевину (30.2.4.3) получают взаимодействием этаноламина с циклогексилизоцианатом. в результате чего образуется 1-(2-гидроксиэтил)-3-цик-логексилмочевина (30.2.4 1). Взаимодействием последней с хлористым тионилом гидроксильную группу в нем замещают на атом хлора с получением 1-(2-хлорэтил)-3-циклогексилмочевины (30.2.4.2). Последнюю нитрозируют в безводных условиях с применением безводной муравьиной кислоты и нитрита натрия и получают ломустин (30.2.4.3) [67,68]
h2n-сн2-сн2-он
30 24 2
HCOOH / NaNO2
О
Cl—CH2-CH2-N—С—NH O=N
SOCI2
30 24 3
Как и другие нитрозомочевины, ломустин действует в качестве агента алкилирующего ДНК, а также ингибирует различные ключевые ферментативные реакции путем кабамоилирования белков. Ломустин применяется только орально. Применяется при новообразованиях ЦНС, опухолях мозга, глотки, гортани, лимфогранулематозе, неходжкинской лимфоме, раке легких и ЖКТ.
Синонимами препарата являются белустин, CCNU и др.
-547-
Глава 30
Кармустин (Сarm ustine)
Кармустин — 1,3-бис(2-хлорэтил)-1-нитрозомочевипа (30.2.4 4) получается нитрозированием 1,3-бис(2-хлорэтил)мочевины грехоки-сью азота [68, 69].
II II
Cl—СН2-СН2—NH-C-NH— СН2-СН2-С1 ---CI—СН2-СН2—NH-C-N-CH2~CH2~CI
N=O 30 2 4 4
Механизм действия препарата аналогичен механизму действия ло-мустина.
Кармустин применяется только внутривенно. Применяется при неходжкинской лимфоме, множественной миеломе и опухолях мозга.
Синонимами препарата являются кармубрис, нитрумон, BICNU.
Стрептозоцин (Streptozocine)
Стрептозонин — 2-дезокси-2-[[(метилнитрозамино)карбонил]-амино]-О-глюкоза (30.2.4.5) является антибиотиком, выделенным из продуктов жизнедеятельности Streptomyces achromogenes [70, 71, 72].
N=O I
NH-C-N-СНз
о
30 24 5
Несмотря на то, что препарат, являясь антибиотиком лишь формально, относится к ряду нигрозомочевин, отсутствие характерной хлорэтильной группы придает ему свойства отличные от других нитрозосоединений. Он проявляет алкилирующие (метилирующие свойства), однако не способен к кросс-связыванию цепей ДНК. Он не обладает также карбамоилирующей активностью присущей другим нитрозомочевинам вследствие происходящей быстрой внутримолекулярной реакции циклизации гликозилизоцианата, получаемого в ходе трансформаций в организме стрептозоцина. Однако препарат ингибирует синтез аминокислот. необходимых для построения белков раковой клеткой.
-548-
Антинеопластики
Стрептозоцин применяется только внутривенно. Назначается, в основном, при раке поджелудочной железы и при карциноидах.
Синонимом препарата является заносар и др.
30.2.5. Производные платины
Цисплатин (Cisplatin)
Цисплатин — г/ис-диаминодихлорплатина (30.2.5.Г) получается восстановлением гексахлорплатината калия гидразином до тетра-хлорплатината шия. взаимодействием которого с аммиаком получают цисплатин (30 2.5.1) [73, 74, 75, 76, 77].
K2[PtCI4]
NH3/ NH4CI
H3N МНз
>pt< Cl' CI
302 5 1
Цисплатин является неорганическим платиносодержащим препаратом с алкилирующими свойствами. Вызывает кросс-связывание цепей ДНК и РНК. В частности показано, что цисплатин, как и другие алкилирующие агенты, связывается преимущественно с положением N7 двух соседних дезоксигуанилатов в ДНК, что препятствует ее репликации. Используется только внутривенно Высокоактивен по отношению к карциноме яичек и яичников, головы, шеи, селезенки легких и др.
Синонимами препарата являются платинол, платибластин, плати-некс, неоплатин и др.
Карбоплатин (Carboplatin)
Карбоплатин — г/ис-диамино-( 1,1 -циклобутандикарбоксилат)пла-гины-(П) получают исходя из цисплатина последовательным взаимодействием с раствором нитрата серебра и далее с циклобутан-1,1-дикарбоновой кислотой с получением искомого карбоплатина (30.2 5.2) [78,79].
AgNOj
CIS—[(NH3)2Pt(H201J(N03)2
30 2 5 1
- 549-
Глава 30
Среди большого массива производных платины, карбоплатин является единственным соединением, вошедшим в медицинскую практику и оминающимся от цисплатина замещением двух атомов хлора на фрагмент циклобутан-1.1-дикарбоновой кислоты. Подобно цисплатину карбоплатин также реагирует с ДНК с образованием как внутренних, так и внешних кросс-связей. Спектр активности и показания к применению практически аналогичны цисплатину.
Синонимом препарата является параплатин и др.
30.3. Антибиотики
Ряд антибиотиков обладает выраженными цитостатическими свойствами, они весьма эффективны при лечении определенных опухолей. К их числу относятся актиномицин, антрациклины — даунорубицин и доксорубицин, блеомицин и др. Некоторые из них, кроме митоксан-трона, являющегося чисто синтетическим препаратом, получаются микробиологическим путем, для некоторых разработаны и синтетические пути синтеза, которые приводятся в литературных источниках. В данном разделе будет рассмотрено лишь их строение и показания к применению.
Дактиномицины
Дактиномицин (Dactinomycin)
Дактиномицин — 2-амино-1,9-бис-(2.9-диизопропил-6.10,13-триметил-1.48,11,14-пентаоксо-7-окса-3,10.13,17а-тетрааза-5-бицикло[ 14.3.0]-нонадецилкарбамоил)-4,6-диметил-ЗЯ-феноксазин-3-ои (30.3.1) является одним из первых антибиотиков, выделенных из культуральной жидкости микроорганизмов рода Streptomyces parvulus [80, 81, 82. 83, 84. 85, 86, 87].
СНз сн3
30 3 1
- 550 -
Антинеопластики
Дактиномицин является хромонопептидом с феноксазиновым кольцом и двумя циклическими полипептидами, присоединенными по карбоксильным группам в положениях 2- и 9-феноксазинового кольца.
В общем дактиномицины проявляют ингибирующий эффект как на грамположительные, так и на грамотрицательные бактерии, а также на грибки. Однако дактиномицин проявляет и выраженное неопластическое действие. Он образует комплекс с ДНК за счет связывания гуанинцитозиновых сегментов, в результате чего блокируется ДНК-зависимый синтез РНК. Другим цитотоксическим эффектом дактиномицина является вызываемые им нарушения скручивания ДНК. Кроме того, дактиномицин ингибирует топоизомеразу II. Может вводиться внутривенно. Применяется при опухолях Вильмса, саркоме Юинга и Капоши, лимфоме и др.
Синонимами препарата являются актиномицин D, космоген и др.
Блеомицины
Блеомицин (Bleomycin)
Блеомицины — стереоизомеры 6-амино-М-[2-[[4-[[1-[[[2-[4-(амино-карбонил)[2,4'-битиазол]-2’-ил]этил]амино]карбонил]-2-гидрокси-пропиламино-2-гидрокси-1,3-диметил-4-оксобутил]амино-1-[[[2-0-[3-О-(аминокарбонил)-а.-П-маннопиранозил-а.-Ь-глюкопиранозил]-окси]-17/-имидазол-4-илметил]-2-оксоэтил]-2-[1-(2,3-диамино-3-ок-сопропил)-4-оксо-азетидинил]-5-метил-4-пиримидинкарбоксамидов (30.3 2).
- 551 -
Г лава 30
Блеомицины — комплекс не менее 16 гликопептидных антибиотиков, полученных из рода Streptomyces verticilus. отличающихся значением R [88, 89, 90, 91, 92, 93, 94]. Блеомицины проявляют противоопухолевою. антивирусную и антибактериальную активность. Связываясь с ДНК, они вызывают нарушения в скручивании как одной, гак и двух цепей ДНК. В меньшей степени они ингибируют синтез РНК и белков. Вводятся внутривенно или внутримышечно. Применяется при лимфомах. карциномах и саркомах.
Синонимами препарата являются бленоксан. блеоцин и др.
Антрациклины
Доксорубицин и даунорубицин — антибиотики, полученные из микроорганизмов родаStreptomycespeucetius Структура этих анграцик-линов включает аминосахаридный остаток даунозамин с планарным нафтаценхиноновым ядром. Доксорубицин отличается о г даунорубици-на наличием одной гидроксильной группой при Сч. Предложен ряд механизмов, согласно которым антрациклины проявляют цитотоксичность. Они вызывают расслоение ДНК, участвуют в окислительно-восстановительных реакциях, хелатируют двухвалентные катионы и взаимодействуют с клеточными мембранами, изменяя их функции. Применяются при острой лейкемии, лимфоме, раке груди и яичников и прочих солидных опухолях
Доксорубицин (Doxorubicin)
Доксорубицин — 7,8,9,10-тетрагидро-6,7,9.11-тетрагидрокси-9-гидроксианетил-4-метокси-5,12-нафтаценхинон-7-(3-амино-5-метил-2,3-дидезокси-Ь-ликсопиранозид) (30.3.3), который выделен из культуральной жидкости Streptomyces peucetms var caesius [95, 96] и позже синтезирован [97, 98, 99, 100]
30 3 3
- 552 -
Антинеопластики
Доксорубицин является одним из наиболее эффективных неопластических препаратов и. в основном, применяется в комбинациях с другими препаратами для лечения солидных опухолей.
Препарат применяется при лейкемиях, различных саркомах, практически при всех видах рака, нейробластоме, лейкозах, лимфомах.
Синонимами препарата являются адриацин, адриабластин и др.
Даунорубицин (Daunorubicin)
Даунорубицин — 9-ацетил-7,8,9,10-тетрагидро-6,7,9,11-тетра1ид~ рокси-4-метокси-5.12-нафтаценхинон-7-(3-а.мино-5-метил-2.3-диде-зокси-П-ликсопиранозид) (30.3.4) выделен из культуральной жидкости Streptomyces peucetius [101, 102, 103. 104] и позже был синтезирован [105, 106, 107, 108].
30 3 4
Механизм действия даунорубицина и показания к применению такие же, как и у доксорубицина.
Синонимами препарата являются даунобластин, рубомицин, церу-бИдИН
Митомицин (Mitomycin)
Митомицин — карбамат 6-амино-1,1а,2,8,8а.8Ь.-гексагидро-8-гидро-ксиметил)-8а-метокси-5-метилазирино[1,2-а]индол-4,7-диона (30.3.6).
о NH
30 3 6
- 553 -
Глава 30
Митомицин является антибиотиком, выделенным из продуктов жизнедеятельности микроорганизмов рода Streptoniyces caepitosus, и содержит хиноновый, уретановый и азиридиновый фрагменты [109, 110, 111, 112, 113, 114], который позже был синтезирован [115, 116].
Он также является алкилирующим агентом и ингибирует синтез ДНК. Вводится внутривенно в сочетании с другими химиотерапевтическими препаратами для лечения карциномы поджелудочной железы, груди, легких простаты головы, шеи и др.
Синонимом препарата является аметицин и др.
30.4. Соединения, выделенные из растений
Винбластин и винкристин являются алкалоидами, полученными из растений рода баравинок (Vinca rosea). Эти соединения вызывают задержку деления клеток в метафазе и ингибируют сборку микротубул и, соответственно, сбой митотического веретенообразования. Ингибируют синтез нуклеиновых кислот и белков. Винбластин и винкристин отличаются лишь заместителем при атоме азота индольного фрагмента молекул и применяются в комбинации с другими химиотерапевтическими средствами. В основном применяются при лейкозах, миеломах, саркомах, раке различных органов и при лимфоме.
Винбластин (Vinblastine)
Винбластин — [3aR-[3aa,4p,5p,5ap,9(3R‘.5S*,7R*,9S*),10bR*.13aa]]-
метил-4-(аиетил-окси)-9-[5-этил-1,4.5,6,7.8,9,10-октагидро-5-гидро-
R = —СООСН3
ксп-9-(.метоксикарбонпл)-2Я-3,7-метаноазациклоу ндецино-[5.4-Ь]-
- 554 -
Антинеопластики
индол-9-ил]-За-этил-За,4,5,5а,6,11,12,13а-октагидро-5-гидрокси-8-мегокси-6-метил-1Я-индолизино[8,1-cd]-карбазол-5-карбоксил ат (30.4.1) выделяют из Vinca rosea [117. 118, 119, 120].
Винбластин подавляет рост клеток на стадии метафазы, действует на метаболизм аминокислот, в частности на уровне включения глутаминовой кислоты в лимоннокислый цикл и превращения в мочевину, а также ингибирует синтез белка и нуклеиновых кислот.
Винбластин применяется при острой лимфобластной лейкемии, болезни Ходжкина, неходжкинских лимфомах, нейробластоме, саркоме и других опухолевых заболеваниях.
Синонимами препарата являются велбан, экзал и др.
Винкристин (Vincristine)
Винкристин — [ЗаК-СЗааЛР^р^арДОЮЗ’ДОЗ’), 1 ObR*, 13аа]]-метил-4-(ацетил-окси)-9-[5-этил-1,4,5,6,7,8,9,10-октагидро-5-гидро-кси-9-(метоксикарбонил)-2//-3,7-метаноазаниклоундецино-[5,4-Ь]-индол-9-ил]-За-этил-За,4,5,5а,6,11,12,13а-октагидро-5-гидрокси-8-меюкси-6-формил-1Я-индолизино[8,1^]-карбазол-5-карбоксилат (30.4.2) также выделяют из Vinca rosea [121, 122, 123]. Предложены и полу синтетические способы получения препарата [124, 125, 126].
А—ОСОСНз СООСНз
R = —СООСНз
Механизм действия винкристина и показания к применению такие же. как и у винбластина.
Синонимами препарата являются лейрокристин, онковин и др.
-555 -
Глава 30
Эпиподофиллотоксины
Этопозид и тенипозид являются синтетическими производными экстракта американского растения мандрагора (майское яблоко). Механизм их действия выяснен не однозначно, однако они действуют на фермент топоизомеразу II, что вызывает нарушения в скручивании ДНК. Кроме того, препараты ингибируют синтез ДНК и РНК, а также транспорт нуклеозидов к клеткам. Цитотоксическое действие на нормальные клетки наблюдается только в очень больших дозах. Препараты проявляют значительную активность при лимфоме, лейкемии и саркоме Капоши, при опухолях яичка.
Этопозид (Etoposide)
Этопозид — [5К-(5а,5аР,8аа.9р)]-9-[4,6-0-этилиден-р-0-глюкопи-ранозил)окси]-5,8.8а,9-тетрагидро-5-(4-гидрокси-3.5-диметоксифе-нил)фуро[3|,41:6,7]-нафто[2,3-<1]-1,3-диоксол-6(5а//)-он (30.4.5) получают исходя из 4|-дезметилэпиподофиллотоксина. фенольную группу которого предварительно ацилируют бензиловым эфиром хлоругольной кислоты с получением 4|-карбобензиллокси-4|-дезме-тилэпипо-дофиллотоксина (30.4 3). Далее гидроксильную группу в положении С9 этого соединения эгерифицируют 4,6-О-эгилиден-2,3-ди-О-ацетил—P-D-глюкопиранозой в присутствии эфирата трехфтористого бора получением соответствующего глюкопиранозида (30.4.4). Удалением ацетильных групп в глюкопиранозильной части молекулы с помощью ацетата цинка в метилате натрия, а также снятием карбобензоксильной защиты, восстановлением водородом с использованием в качестве катализатора палладия на угле получают искомый этопозид (30.4.5) [127, 128].
- 556 -
Антинеопластики
30 4 5
Этопозид применяют при герминогенных опухолях, раке яичников, желудка и легких, болезни Ходжкина и неходжкинских лимфомах как при моно-, так и при комбинированной терапии.
Синонимом препарата является вепезид и др.
Тенипозид (Teniposide)
Тенипозид -- [5К-(5а,5ар,8аа,9р)]-9-[4,6-О-(2-тиенилмегилен)-|3-Г)-глюкопиранозил)окси]-5,8,8а,9-тетрагидро-5-(4-гидрокси-3.5-диме-токсифенил)фуро[3 ‘,4':6,7]-нафто[2,3-с1]-1,3-диоксол-6-(5а/7)-он (30.4.9) синтезируют примерно по аналогичной схеме исходя из 41-бензиллокси-4'-дезметилэпиподофиллотоксина (30.4.6), который этерифицируют 2,3,4,6-тетра-О-ацетил-р-В-глюкозой в присутствии эфирата трехфтористого бора с получением гликозида (30,4.7).
1 ZnsCHjCOO?
2 з2 ^d-C
Ацетильные и бензоильные защитные группы в последнем удаляют последовательным применением ацетата цинка в мезилате натрия и дальнейшим восстановлением водородом, и получают диол (30.4.8). Последний далее трансформируется в соответствующий ацеталь 2-фор-
- 557 -
Глава 30
милгиофена, являющийся искомым тенипозидо.м (30.4.9) [128, 129, 130, 131. 132J
ZnCI2
30 4 9
Тенипозид применяют по тем же показаниям, что и этопозид, однако препарат проявляет активность, превосходящую таковую этопозида в 5-10 раз.
Синонимами препарата являются эпиподофиллотоксин, вумон и др.
30.5. Гормональные препараты
Гормональные препараты успешно применяются при комплексном лечении злокачественных новообразований. Причем опухоли могут быть как гормонально зависимые, так и гормонально чувствительные. I ормонально зависимые опухоли регрессируют при исключении гормонального воздействия. Следовательно, возможно уменьшить и даже предотвратить развитие гормонально зависимых опухолей лекарствами. В частности, антиэстрогенный препарат тамоксифен предотвращает стимуляцию эстрогенами опухолевых клеток рака груди. То же касается и аминоглутетимида — ингибитора синтеза кортикостероидов надпочечниками.
Гормонально чувсгвительные опухоли могут регрессировать и часто приостанавливают свой рост при введении определенных гормонов. Основными гормнальночувстви тельными формами рака являются карцинома груди, простаты, лимфомы и некоторые виды карциномы.
Гормональными препаратами, ингибирующими рост определенных человеческих опухолей, являются стероиды, включающие андрогены, эстрогены, прогестины и кортикостероиды, причем используются только глюкокортикоиды Более того, ни кортизол, ни кортизон не используются при лечении злокачественных новообразований, а используются нреднион. преднизолон, метилпреднизолон и дексаметазон.
- 558 -
Антинеопластики
I ормональные препараты не вылечивают рак. однако проявляют выпаженное паллиативное действие, за исключением цитотоксического действия глюкокортикоидов на лимфоидные клетки. Речь идет, в частности. о преднизоне, который применяется при лечении лимфомы и определенных лейкемий при комбинированной терапии. Точный ме-ханшм действия стероидов до конца не выяснен.
Андрогены
Андрогены — производные тестостерона (29.1.3), метилтестостерон (29.1 7). флуоксиместерон (29.3.8), тестолактон (30.5.1) наиболее часто используются для паллиативного лечения рака груди у постменопаузальных женщин, которым показана гормональная терапия. Точный механизм противоопухолевого эффекта андрогенов неизвестен. Однако полазают, что андрогены блокируют рост клеток путем ингибирования транспорта природного гормона роста в клетку. Более того, андрогены могул ингибировать синтез эстрогенов, вызывая тем самым истощение запасов эстрогенов.
Эстрогены
Эстрогены — эстрон (28.1.9), эстрадиол (28.1.17), этинилэстрадиол (2s 1.2о). диэтилстильбестрол (28 1.33), хлортрианизен (30.5.2) используются дтя паллиативного лечения постменопаузального рака груди, рака простаты и рака груди у мужчин. Считается весьма вероятным, что механизм их действия сходен с механизмом действия андрогенов.
78 1 33
- 559 -
Глава 30
Прогестины
Прогестины — стероидные соединения родственные прогестерона — капроат гидроксипрогестерона (28.3.6), ацетат медроксипрогестерона (28.3.7) и ацетаг мегестрола (28.3.9) применяются при паллиативном лечении карциномы груди и почечных опухолях. Прогеоины могут иметь прямое местное воздействие на клетки и одновременно понижать количество лютеинизирующего гормона.
Кортикостероиды
Кортикостероиды — синтетические стероидные препараты, получаемые из природного гормона гидрокортизона, и, в частности, преднизон часто используются при комбинированной терапии для лечения острой и хронической лимфоцитной лейкемии, ходжкинской и не.ход-жкинской лимфомах, множественной миеломе и раке груди. Кортикостероиды оказывают противоопухолевый эффект, связываясь с кортикостероидными рецепторами, присутствующими во многих раковых лимфоидных клетках, что приводит к ингибированию и транспорта глюкозы, и фосфорилирования, что уменьшает количество энергии, необходимой для митоза и синтеза протеинов, что, соответственно, приводит к лизису клеток.
27 1 31
Негормопальные препараты
В дополнение к гормональным препаратам в химиотерапии рака широко используются также пять других препаратов, не являющихся стероидами, но имеющих прямое отношение к данному разделу Ими
-560-
Антинеопластики
являются аминоглутетимид, флугамид, митотан, тамоксифен, лейпро-лнд.
Аминоглутетимид (Aminoglutethimide)
Аминоглттетимид — (±)-2-(4-аминофенил)-2-этилглу гаримид (30.5.4) получают двумя путями. Первый способ исходит из глутетимида (4 3 6). нитрованием которого нитрующей смесью получают 2-^4-нитрофенил)-2-этилглутаримид (30.5.3). Восстановлением нитрогруппы в последнем водородом с использованием в качестве катализатора никеля получают искомый аминоглутетимид (30 5.4).
НМОз H2SO4
Н2< Ni
30 5 4
Второй способ синтеза исходит из 2-фенилбутиронитрила, нитрованием которого в аналогичных условиях получают 2-(4-нитрофенил)-бутиронитрил (30.5.5). Последний в условиях реакции Михаэля в присуди вии гидроокиси бензилтриметиламмония присоединяют к метилакрилату, и продукт подвергают кислотному гидролизу смесью уксусной и серной кислот, при котором происходит циклизация в 2-(4-нит-рофенил)-2-этилглутаримид (30.5.3), который аналогично вышеописанному и восстанавливают водородом в искомый продукт — аминоглутетимид (30,5.4) [133, 134].
HNO3
30 5 5
СН2=СН-СООСН3
РЬСН2М(ОНз;з ОН
СНзСООН , H2SO4
Н2/ N1
н
30 5 4
Препарат блокирует трансформацию холестерина в прегненолон и андрогенов в эстрогены в надпочечниках и тем самым полностью подавляет выработку всех стероидных гормонов. Аминоглутетимид ис
-561 -
Г лава 30
пользуется при паллиативном лечении постменопаузальной карциномы груди и карциноме простаты.
Синонимами препарата являются ориметен. цитадрен и др.
Флутамид (Flutamide)
Синтез флутамида (29.2.15) описан в гл. 29 «Мужские половые гормоны и анаболические стероиды».
/=\ ° с1-13
O2N—4 У-ЫН-С-СН-СНз
F3C 29 2 15
Препарат блокирует связывание андрогенов клетками тканей-мишеней и препятствует биологическому действию андрогенов, в том числе в клетках опухоли предстательной железы.
Используется при карциноме простаты в сочетании с лейпролидом.
Митотан (Mitotane)
Митотан — 1,1-дихлор-2-(о-хлорфенил)-этан (30.5.8) получают алкилированием хлорбензола 1-(2-хлорфенил)-2,2-дихлорэтаном (30.5.7) в присутствии серной кислоты. Необходимый для этой цели 1-(2-хлорфенил)-2,2-дихлорэтан (30.5.7), в свою очередь, получают взаимодействием 2-хлофенилмагнийбромида с дихлоруксусны.м альдегидом [135].
H2SO4
CI
Митотан — производное инсектицида ДДТ очень быстро понижает уровень кортикостероидов и их метаболитов в крови и моче и используется при неоперабельной метастатической карциноме простаты.
Синонимом препарата является лизодрен и др.
Тамоксифен (Tamoxiphen)
Синтез тамоксифена (28.2.8) был описан в гл. 28 «Женские половые гормоны».
- 562-
дн7и неопластики
Препарат блокирует рецепторы эстрогенов, тормозя прогрессирование опухолевого заболевания, стимулируемого эстрогенами.
Тамоксифен используется при паллиативном лечении рака груди у пре- и постменопаузальных женщин.
Лейпролид (Leuprolide)
Лейпролид — 5-оксо-Е-пролил-Ь-гистидил-Ь-триптофил-Ь-серил-Т-шрозил-О-лейцил-Ь-лейцил-Ь-аргинил^-этил-Т-пролинамид
(30.5.9) получают синтетическим путем [136. 137, 138, 139].
pyroGlu-His-Trp-Ser-Tyr-D-Leu-Leu-Arg-Pro-NHEt
Нонапепгид лейпролид является синтетическим аналогом декапептида - гонадотропин высвобождающего гормона, превосходит по активности естественный гормон и значительно снижает уровень тестостерона и дигидротестостерона у мужчин и эстрогенов у женщин. При этом имеет место также ингибирование фолликулостимулирующего гормона и лютеинизирующего гормона.
Лейпролид используется при раке простаты, когда орхиэктомия или эстрогенная терапия противопоказаны пациенту.
Синонимом препарата является люпрон.
30.6. Прочие антинеопластические препараты
Рассматриваемые ниже препараты классифицируются как прочие, носко шку либо по механизму их действия, либо по принадлежности к определенному химическому классу их трудно было включить в под-Разделы, изложенные согласно традиционно принятой классификации антинеоплас гических средств.
-563 -
Глава 30
Гидроксимочевина (Hydroxyurea)
Гидроксимочевина (30.6.1) получается взаимодействием цианата натрия с гидроксиламином. При этом используется гидрохлорид гидроксиламина и основная ионообменная смола [140, 141].
. ,. о
amberlit и
NaOCN + NH2OH х HCI -----------* HO-NH-C~NH2
30 6 1
Гидроксимочевина действует путем подавления дегидрофосфатре-дуктазы - фермента, который восстанавливает рибонуклеотиды в дезоксирибонуклеотиды, необходимые для синтеза ДНК.
Препарат резко понижает количество белых кровяных клеток при острых лейкемиях. Применяется при лейкемиях, меланомах и карциномах.
Синонимами препарата являются литалир, гидреа, онкокарбид и др.
Митоксантрон (Mitoxantrone)
Митоксантрон — 1,4-дигидрокси-5.8-онс[[2-[(2-гидроксиэтил)ами-но)эгил]амино]-9,10-антрацендион (30.6.3) структурно родственен антибиотику доксорубицину. Его синтезируют исходя из дантрона (1,8-дигидроксиантрахинона), который действием азотной кислоты и далее смесью сульфида и тиосульфата натрия в щелочи трансформируется в 1.4,5,8-гетрагидроксиантрахинон (30.6.2). Взаимодействием последнего с 2-аминоэтиламиноэтанолом в присутствии хлоранила (2,3,5.6-геграхлорбензохинона-1,4) получают искомый митоксантрон (30.6.3) [142, 143, 144, 145].
он о он
о
1 HNO-J
2 Na2S, Na2SO3/ NaOH
30 6 2
/СНг-СНг-ОН
HN
Vh2-CH2-NH2
ОН О NH—СН2—СН2—NH—СН2—СН2—ОН
ОН О nh—сн2—сн2—nh—сн2—сн2—он
30 6 3
- 564 -
Анти неопластики
Механизм его действия однозначно не выяснен, однако полагают, что митоксантрон фу нкционирует, связываясь с ДНК и тем самым пару шает процесс скручивания цепей Используется внутривенно для лечения острой нелимфатической лейкемии, рака груди и т п.
Синонимом препарата является новантрон.
Дакарбазин (Dacarbazine)
Дакарбазин — 5-(3,3-диметил-1-триазено)имидазол-4-кабоксамид (30 6 5) получают диазотированием 5-аминои.мидазол-4-карбокс-амида азотистой кислотой, в результате чего получается 5-диазоими-дазол-4-карбоксамид (30.6.4). Взаимодействием последнего с диметиламином получают искомый дакарбазин (30.6.5) [146].
CONH? CONH2 CONh?
N if NaNOz ' H2SO4» N—nJ (CH3)2NH N—
Ч'ън; N n2+ ^xn'^'n=n-n(
H ' H CH3
30 6 4 30 6 6
Принято считать дакарбазин пока еще первым, но тем не менее представителем ряда триазеновых производных Показано, что он является алкилирующим агентом, причем препарат ингибирует синтез РНК и протеинов в большей степени, чем ДНК.
Дакарбазин применяется внутривенно при болезни Ходжкина, саркоме мягких тканей и метастатической меланоме.
Синонимом препарата является дитипен.
Прокарбазин (Procarbazine)
Прокарбазин — 1-метил-2-(«-изопропилкарбамоилбензил)-гидразин (30.6 8) синтезируют исходя из 1,2-б«с-(бензилоксикарбонил)-1-ме-тилгидразина, который алкилируют метиловым эфиром 4-бромме-тилбензойной кислоты в присутствии гидрида натрия с получением 1.2-бг/с-(бензилоксикарбонил)-1-метил-2-(л-карбметокси)бензил гидразина (30.6.6) Гидролизом карбметоксильной группы последнего гидроокисью натрия, трансформацией полученной карбоксильной группы в хлорангидридную, и, наконец, взаимодействием продукта с изопропиламином получают 1,2-бг/с-(бензилоксикарбонил)-1-ме-гил-2-(/г-изопропилкарбамоил)бензилгидразин (30.6.7). Гидролизом бензилоксисарбонильных групп в последнем бромистым водородом в уксусной кислоте получают искомый прокарбазин (30.6.8) [147, 148.149,150].
- 565 -
Г лава 30
CO-0-Cri2C6>-i=;
I I
СН3 —N-----
6t-CH
NaH
СООСН3
I NaCH
2 SOCb
3 (СНЭ<СНПН2
свн5сн2-о-co со-о-снгС6Н5
CH3-N---N—CH2
CONHCH(CH3)2
HBr' CH3COOH
30 6 7
CH3NHNH-CH2
CONHCH(CH3)2
30 6.8
Первоначально прокарбазин был синтезирован в качестве ингиби-юра МАО Однако впоследствии выяснилась его способность выступать в роли алкилирующего агента и ингибировать синтез ДНК, РНК и протеинов. Вместе с тем существует точка зрения, согласно которой прокарбазин накапливается в опухолевых тканях и генерирует в клетках перекисные и гидроперекисные радикалы, чго имитирует эффект ионизирующей радиации. Препарат используется при злокачественных новообразованиях лимфатических тканей, опухолях мозга, легких, болезни Ходжкина.
Синонимом препарата является натулан.
Амсакрин (Amsacrine)
Амсакрин — 4-(9-акридиниламино)-3-метоксифенил-М-метансуль-фона.мид (30.6.11) получают сульфонирированием 4-нитро-лг-ани-зидина метансульфохлоридом с получением сульфонамида (30.6.9), нигрогруппу в котором восстанавливают до аминогруппы водородом с получением 4-амино-З-метоксифенил-М-метансульфонамида (30.6.10). Взаимодействием последнего с 9-хлоракридином получают амсакрин (30.6.11) [151. 152, 153].
СНз—SO2-CI
NH-SO2~ СН3
30 6 9
Fe / HCI
- 566-
днтинеот-пасчики
30 6 10
Амсакрин — препарат, проходящий интенсивные испытания при острой лейкемии и лимфоме. Является цитотоксическим препаратом, связывающимся с ДНК, с выраженной специфичностью к паре аденозин-i ирозин. тем самым ингибирующим синтез ДНК. Предполагается его использование при острой лейкемии.
Синонимом препарата является амсидил.
Аспарагиназа (Asparaginase)
Аспарагиназа является ферментом, гидролизующим L-аспарагин до L-аспарювой кислоты, чем и вызывает истощение запасов L-acna-рагина, ингибируя тем самым синтез протеинов и нуклеиновых кислот. Эффективен при острой лимфоцитной лейкемии [154].
Синонимом препарата является элспар.
, Список литературы
1 Seeger D et al. 'J. Am. Chem. Soc. 71, 1753 (1949).
2. Piper J. et al.. 'J. Heterocycl. Chem. 11, 279 (1974).
3. Chaykowsky M et aU'J. Med. Chem. 17, 1212 (1974).
4. Ger. Pat. 2.741.270(1977).
5. US Pat. 4.057.548(1977).
6. US Pat. 4.067.867 (1978).
?. US Pat. 4.080.325 (1978).
8. Ger. Pat. 2.741.383 (1977).
9. Brit. Pat. 713.286 (1951).
10. US Pat. 2.721.886 (1955).
H. US Pat. 2.724.711 (1955).
12. US Pat. 2.933.498 (1960).
13. Ehon G et al.' J. Am. Chem. Soc. 74, 411 (1952).
14. US Pat. 2.697.709 (1954).
15 Beaman A. et al.' J. Am. Chem. Soc. 83, 4042 (1961).
- 567-
Г лава 30
16. US Pat. 2.884.697 (1959).
17. US Pat. 2.800.473 (1957).
18. USPat. 3.019.224 (1962).
19. USPat. 3.132.144 (1964).
20. Elion G. et al./'J. Am. Chem. Soc. 77, 1676 (1955).
21. USPat. 2.802.005 (1957).
22. Duschinsky R. et al.7J. Am. Chem. Soc. 79, 4559 (1957).
23. US Pat. 3 682.917 (1972).
24. US Pat. 3 846.429 (1974).
25 US Pat. 3.954.758 (1976).
26. Ger. Pat. 2.149.504 (1971).
27. Ger. Pat. 2.719.245 (1977)
28. Ger. Pat. 2.726.258 (1978).
29 Hoffer M. et al. 'J. Am. Chem. Soc. 81, 4112 (1959).
30. USPat. 2.885.396(1959).
31. USPat. 2.970.139 (1961).
32. USPat. 2.949.451 (1960).
33. USPat. 3 041.335 (1962).
34. USPat. 3.116.282 (1963).
35. Roberts И-' et al.//J. Org. Chem. 32. 816 (1967).
36. Formageot H et al./'Tetrahedron Lett. 1966, 3499.
37. Hessler E et al., J. Org. Chem. 41, 1828 (1976).
38. Claesen C et al./ Tetrahedron, 26, 3859 (1985).
39. Prelog V. et al./ Coll. Czech. Chem. Commun. 7, 93 (1935).
40. Hanby E. et al.-'/J. Chem. Soc. 1947, 513.
41. 4brams J et al. J. Soc. Chem. Ind. (London) 68. 280 (1949).
42. Brit. Pat. 727.336 (1955).
43. US Pat. 2.944.079(1960).
44. US Pat. 3.043.301 (1962).
45. US Pat. 3.046.301 (1962).
46. Everett A etal.//J. Chem. Soc. 1953, 2390.
47. USPat. 3.032.584 (1962).
48. USPat. 3.032.585 (1962).
49. Bergel L. et al. 'J. Chem. Soc. 1954. 2409.
50. Bergel L. et al. J. Chem. Soc. 1955, 1223.
51. Ger Pat. 1 057.119 (1956).
52. US Pat. 3.018.302 (1962).
53. Arnold H. et aL.Naturwiss. 45. 64 (1957).
54. Arnold H. et al. 'Nature. 181. 931 (1958).
55. ArnoldH et al. Angew. Chem. 70. 539 (1958).
56. Karie I et al. J. Am. Chem. Soc. 99, 4803 (1977).
- 568 -
Литинеопластики
S7 Adamaik D. et al."Angew. Chem. Int. Ed, 16, 330 (1977).
58. Kwashima T et al."J. Org. Chem. 43, 1111 (1978).
59. Sato T. et al. ’ J. Org. Chem. 48, 98 (1983).
60 Fr. Pat. 1.530.962 (1968).
61. USPat. 3.732.340(1973).
62. Brassfield H. et al./'J. Am. Chem. Soc. 97, 4143 (1975).
63USPat. 2.670.347(1954).
64. Japan Pat. 55 218 (1955).
65. Bnt Pat. 700.677 (1950).
66. USPat. 2.917.432 (1959).
67. Johnston T et al/'T. Med. Chem. 9, 892 (1966).
68. Johnston T. et al.,,J. Med. Chem. 6, 669 (1963).
69. Ger. Pat. 2.528.365 (1975).
70. f'avra J et al.' Antibiot. Ann. 1959-1960. 230.
71. Ger Pat. 1.090.823 (1960).
72. USPat. 3.027.300(1962).
73. Rosenberg B. et al./'Nature (London), 222, 385 (1969).
74. Ger. Pat. 2.906.700 (1979).
75. Kauffman G et al./7Inorg. Synth. 7, 239 (1963).
76. Ger. Pat. 2.318.020 (1972).
77. Ger. Pat. 2.329.485 (1972).
78. US Pat. 4.140.707 (1979).
79. Harrison R et aL'/Inorg. Chim. Acta. 46, L15 (1980).
80. USPat. 2.378.876 (1962).
81. Manaker J et al.,', Antibiot. Ann. 1954-55, 853.
82. Bullock J et al.,7J. Chem. Soc. 1957. 3280.
83. Brockmann H. et al.,'/J. Chem. Soc. 1964, 384, 435.
84. Brockmann H. etal.//Ber. 101, 1312 (1968).
85 Meienhojer J //J. Am. Chem. Soc. 92, 3771 (1970).
86. Tanaka T et al.//Bull. Chem. Soc. Japan, 53, 1352 (1980).
87. Nakajima K. et al./'Pept. Chem. 19, 143 (1981).
88. Takita T et al./''J. Antibiot. 21, 79 (1968).
89. Takita T et al./,'J. Antibiot. 22, 237 (1969).
90. Takita T et al. "J. Antibiot. 25, 755 (1972).
91. Takita T et al. J. Antibiot. 31, 801 (1978).
92. Takita T etal. Tetrahedron, 23, 521 (1982).
93. 4oyagii et al.-', J. Am. Chem. Soc. 104. 5537 (1982).
94. Saito S etal. J. Antibiot. 36, 92 (1983).
95. US Pat. 3.590.028 (1971).
96. Bnt. Pat. 1.161.278 (1968).
97. Arcamone F. etal. Ger. Pat. 1.917.874 (1969).
- 569 -
Глава 30
98. Лгсатопе F. et al.//Tetrahedron Letters, 1969, 1007.
99. Acton E. et al. J. Med. Chem. Г7. 659 (1974).
100. US Pat. 4.012.448 (1977).
101. Gassinelli G. et alGGiom. Microbiol. 11, 167 (1963).
102. DiMarco A et al. (Nature, 201. 706 (1964).
103. US Pat. 4.012.284 (1977).
104. US Pat. 3.997.662 (1976).
105. Marsh P. et al../Chem. Commuu. 1967, 973.
106. Yamaguchi T. et al./'Carbohyd. Res. 59. 343 (1977).
107. Wovkulich P. etal.-', J. Am. Chem. Soc. 103, 3956 (1981).
108. Acton E. et al.7/J. Med. Chem. 17. 659 (1974).
109. Hata T. et al '/J. Antibiot. 9A. 141 (1956).
110. WakakiS. et al.,, Antibiot. Chemoter. 8, 228 (1958).
111. Bnt. Pat. 830.874 (1960).
112. US Pat. 3 660.578 (1972).
113. US Pat. 3.042.582 (1972).
114. Lefemine D et. al."J. Am. Chem. Soc. 84, 3184 (1962).
115. Fukuyama T. et al.' Tetrahedron Letters, 1977. 4295.
116. Kashi ). J. Nat. Prod. 42, 549 (1979).
117. Svoboda G. et al.FT Farm. Sci. 51, 707 (1962).
118. US Pat. 3.097.137 (1963).
119. US Pat. 3.225.030(1965).
120. US Pat. 4.070.358 (1978).
121. Belg. Pat. 867.670(1978).
122 Fr. Pat. 2.210.392 (1972).
123. Bnt Pat. 1.382.460 (1972).
124. US Pat. 3.899.493 (1975).
125. Rahman A. et al.'/Tetrahedron Letters. 1976. 2351.
126. Mangeney P et al.'/J. Am. Chem. Soc. 101. 2243 (1979).
127. US Pat. 3.524.844 (1970).
128. Keller-Juslen C. et al./"J, Med. Chem. 14. 936 (1971).
129. S. Air. Pat. 66 07,585 (1968).
130. US Pat. 3.524.844 (1970).
131. Fr. Pat. 1.518.706 (1966).
132. Fr. Pat. 2.320.104 (1975).
133 US Pat. 2.848.455 (1958).
134. Finch N etal. 'Experientia, 31. 1002 (1975).
135 Haller В et al.’J. Am. Chem. Soc. 67. 1591 (1945).
136. Ger. Pat. 2.446.005 (1975).
13"7. US Pat. 4.005.063 (1977).
- 570 -
дмтинеопластики
138. Vilchez-Martinez J et al.//Biochem. Biophys. Res. Commun. 59. 1226(1974).
139. Fujino M. et al. /Biochem. Biophys. Res. Commun. 60. 406 (1974).
140. Hantzsch .4., Ann. 299. 99 (1898).
141. US Pat. 2.705.727 (1955).
142. US Pat. 4.197.249 (1980).
143. Ger. Pat. 2.835.661 (1979).
144. Zee-Cheng R. et al./ZJ. Med. Chem. 21, 291 (1978).
145. Murdock К et al.//J. Med. Chem. 22, 1024 (1979).
146. Shealy J et al./-'J. Org. Chem. 27, 2150 (1962).
147. US Pat. 3.520.926(1970).
148. Bnt. Pat. 968.460(1962).
149. Zeller P et al./'Experientia. 19, 129 (1963).
150. Belg. Pat. 618.638 (1962).
151. Cain B. et al.,', J. Med. Chem. 18, 1110 (1975).
152. Cain B. et al., 'J. Med. Chem. 20, 987 (1977).
153. Denny W et al./'J. Med. Chem. 21, 5 (1977).
154. Ho D. et al.', J. Biol. Chem. 245, 3708 (1970).
Глава 31
' Иммунофармакологические f препараты
Способность организма самостоятельно защищать себя от определенных болезней называется иммунитетом.
Иммунитет к определенным заболеваниям может быть как врожденным, так и приобретенным. Большинство типов иммунитета либо приобретается в течение жизни в ответ на заражение различными микроорганизмами (активно приобретенный иммунитет), либо достигается специальной, направленной выработкой в организме антител в 01вет на искусственное заражение заранее убитыми или ослабленными микроорганизмами (пассивно приобретенный, искусственный, иммунитет), что обычно достигается вакцинацией.
Иммунизация, или селективное усиление иммунного ответа организма, является одним из способов борьбы с инфекционными заболеваниями путем вакцинации, т. е. выработки антител против конкретных, специфических патогенов.
Иммунной реакцией несомненно является и воспаление. В этом плане препараты, описанные в других главах как антигистаминные средства, НПВС, антисеротониновые препараты и многие другие, можно формально ,акже отнести к иммунофармакологическим. Однако в этой главе будут рассмотрены лишь препараты, имеющие прямое действие только на клетки, обладающие иммунными функциями, такие как лимфоциты, клетки плазмы и подтипы этих клеток. Следует отметить, что продукты жизнедеятельности самих этих клеток, такие как лн.мфо-кины, интерфероны и интерлейкины, сами по себе являются весьма важными иммунофармакологическими препаратами. Иммунная система обладает огромным количеством антител, распознающих «свои>. и «чужие» молекулы Она играет огромную роль при аутоиммунных болезнях, при реакциях гиперчувствительности организма на определенные раздражители и при отторжении трансплантатов.
- 572-
Иммунофармакологические препараты
Иммхнная система жизненно необходима не только для защиты организма от чужеродных тел как органического и неорганического происхождения. но также от собственных клеток, трансформировавшихся в чужеродные. Она служит также для удаления больных, мертвых или инородных клеток, а также, по всей вероятности, служит основной защитой организма ог рака, подавляя многие возникающие очаги новообразований и часто не дает образоваться метастазам
Различные лекарственные средства способны влиять на специфические иммунные реакции. Они могут как повышать общую сопротивляемость организма или его неспецифический иммунитет, так и подавлять имму иные реакции организма.
Таким образом, контроль над болезнями иммунологическими средствами означает либо генерирование в организме необходимого иммунитета. либо подавление нежелательных иммунных реакций.
Очевидно, чю иммунофармакологические препараты имеют огромное тначение при болезнях иммунной системы, при трансплантации органов, при вирусных инфекциях и, в частности, при лечении СПИДа.
31.1. Иммуностимуляторы
Повышение общей сопротивляемости организма наблюдается под влиянием ряда известных препаратов— иммуностимуляторов (кофеин, фенамин, метилурацил), витаминов (ретинол, аскорбиновая кислот, витамины группы В), производных нуклеиновых кислот, а также и специальных. созданных для згой цели препаратов природного или генно-инженерного происхождения.
К ним относятся такие протеины, как лимфокины, и в частности интерлейкин-2, являющийся гликопротеином, состоящим из 133 аминокислот, а также так называемые факторы, стимулирующие колонии (ФСК) макрофагов или гранулоцитов, вырабатываемые некоторыми клетками, к которым относится ФСК-мульти или интерлейкин-3; грану-лоцитоз стимулирующий фактор, являющийся негликозилированным протеином; макрофагоз стимулирующий фактор, являющийся высоко-гликозилированным гомодимерным протеином; фактор стимулирующий и гранулоцитоз и макрофагоз, являющийся мономерным гликопротеином, — все эти факторы стимулируют пролиферацию, дифференциацию и функционирование целевых клеток на различных стадиях развития.
Для этой же пели предложены препараты, названные факторами, вызывающими некроз опухолей (ФНО-а и ФНО-[3).
- 573 -
Г лава 31
В качестве иммуностимуляторов широко используются и интерфероны — семейство гликопротеинов, вырабатываемых в макрофагах (а-ин1ерфероны). вырабатываемые в макрофагах и фибробластах (|3-ин-терфероны), вырабатываемые в лимфоцитах (у-интерфероны), названные так из-за способности взаимодействовать с вирусными РНК и воз-действоваать на синтез белка, В настоящее время в медицине используются коммерчески доступные а-, Р- и у-интерфероны.
В качестве чисто синтетического иммуностимулятора применяется практически только левамизол, первоначально предложенный в качестве противоглистного средства и широко применяющийся в качестве такового и в настоящее время.
Левамизол (Levantizol)
Левамизол — 2.3,5.6-тетрагидро-6-фенилимидазо[2.1-Ь]тиазол (31 1.4) предложено получать разными путями. Первый способ исходит из а-бромацетофенона, взаимодействием которого с 2-имино-1,3-тиазо-лидином получают 3-фенацил-2-имино-1,3-тиазолидин (31.1.1). Подвергая продукт действию уксусного ангидрида, получают 3-фен-ацил-2-ацетилимино-1,3-тиазолидин (31.1.2). Кетонную группу в последнем восстанавливают до спиртовой с использованием бор-гидрида натрия, и образовавшуюся гидроксильную группу в (31.1.3) замещают на хлор действием хлористого тиснила. Кипячением продукта в уксусном ангидриде замыкается имидазольный цикл с образованием продукта (31.1.4).
31 1 3
1 SOCI2
2 (СН3СО)2О
31 1 4
Второй способ, представляющий несколько иной подход, был реализован при использовании в качестве исходного вещества окиси стирола. Взаимодействием последней с 2-имино-1,3-тиазолидином получают 3-(2-фенил-2-гидроксиэтил)-2-имино-1,3-тиазолидин (31.1.5).
- 574 -
мм мунофармакологические препараты___________________
Поспелова тельной обработкой которого хлористым тионилом и далее уксусным ангидридом получают искомый левамизол (31.1 4).
Наконец, исходя из той же окиси стирола, предлагается третий способ получения продукта. Окись стирола вводят во взаимодействие с азиридином с получением 2-азиридино-1-фенилэтанола-1 (31.1.6). Обработкой последнего изотиоцианатом калия или тиомочевиной получают 3-(2-фенил-2-гидроксиэтил)-2-имино-1,3-тиазолидин (31.1.5), после-девате юной обработкой которого аналогично вышеописанному хлористым тионилом и далее уксусным ангидридом получают искомый левамизол (31.1 4) [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12].
SOCfe (СНзСО)гО
Левамизол обладает иммуномодулирующей активностью. Полагают. что препарат регулирует клеточные механизмы иммунной системы и механизм его действия может быть связан с активацией и пролиферативным ростом Г-лимфоцитов, увеличением числа моноцитов и активацией макрофаюв, а также с увеличение подвижности и хемотаксиса нейтрофильных гранулоцитов. Левамизол оказывает также антигельминтное действие.
Он повышает общую сопротивляемость организма, восстанавливает измененные функции Т-лимфоцитов и фагоцитов. Препарат может выполнять и функции иммуномодулятора, усиливая слабую реакцию клеточного иммунитета, ослабляет сильную и не действует на нормальную Реакцию
Левамизол показан при первичных и вторичных иммунодефицитных состояниях, аутоиммунных заболеваниях, хронических и рециди-виРующи\ инфекциях, аденокарциноме толстой кишки, гельминтозах 11 Ревматоидном артрите.
Синонимами препарата являются декарис, тетрамизол и др.
- 575-
Глава 31
31.2. Иммунодепрессанты
Наряд} с иммуностимуляторами в медицинской практике необходимы препараты, подавляющие иммуногенез, продукцию антител, что особенно важно при трансплантации различных тканей и органов, когда организм вырабатывает антитела, вызывающие гибель пересаженных тканей, а также при лечении некоторых аутоиммунных заболеваний.
Иммунодепрессивной активностью обладают вещества различных фармакологических групп — глюкокортикоиды, цитостатики, антибиотики (циклоспорин).
Глюкокортикоиды
Многие синтетические производные глюкокортикоидов широко применяются в качестве иммунодепрессантов. Глюкокортикоиды (кортизон, преднизолон, метилпреднизолон, бетамешзон, дексаметазон, триамгшинолон и др.) обычно применяются в сочетании с другими иммунодепрессантами. особенно в случаях, сопровождающихся воспалениями. Иммунодепрессивное действие глюкокортикоидов связывают с понижением уровня лимфоцитов, эозинофилов и базофилов в крови, подавлением распознавания антигена, угнетением фазы пролиферации лимфоцитов.
Цитотоксические препараты
Предположительно любое цитотоксическое вещество, разрушающее костный мозг и лимфоидную ткань, может быть использовано в качестве иммуносупрессора. Среди этих препаратов наиболее широко, и главным образом при аутоиммунных заболеваниях, используются винкристин, метотрексат и цитарбаин. Однако их применение следует считать экспериментальным. Только метотрексат достаточно обоснованно признан в качестве первичного препарата для лечения ревматоидных артритов.
Кроме того, один из серии аналогов меркаптопурина — азатиоприн, предложенный в качестве цитотоксического препарата, оказался более эффективным как иммуносупрессор.
Азатиоприн (Azathioprin)
Азатиоприн — 6-[(1-метил-4-нитроимидазол-5-ил)тио]пурин (31.2.1) получают гегероарилированием SH-i руппы 6-меркаптопурина (30.1.2.9) 5-хлор-1-метил-4-нитроимидазолом в присутствии ацетата натрия в качестве слабого основания [13].
- 576 -
Фактически азатиоприн является пролекарством, поскольку в организме он превращается в меркаптопурин. Возможно, что именно это является причиной его преимущества в качестве иммуносупрессора перед меркаптопурином.
Механизм действия азатиоприна в качестве цитотоксического препарата не отличается от механизма действия других антиметаболитов.
Азатиоприн является основным препаратом, применяемым при трансплантациях, особенно при пересадке почки. Сегодня во многих случаях вместо азатиоприна используют циклоспорин. Однако азатиоприн полезен в комбинации с цикоснорином и даже предпочтителен в определенных случаях.
Синонимами препарата являются азумек, имуран и др.
Циклофосфамид (Cyclophosphamide)
Синтез и свойства препарата (30.2.1.15) описаны в гл. 30 «Антинеопластики.».
СН2-СН2-С1 i-'-N
N'H CH2-CH2-CI
30 2 1 15
Кроме применения в качестве алкилирующего средства при химиотерапии рака, циклофосфамид является уникальным препаратом в качестве иммуносупрессора. Во-первых, он является наиболее мощным из всех подобных препаратов. Во-вторых, он разрушает пролиферирующие клетки, а также, по-видимому, алкилирует определенную часть оставшихся клеток. Наконец, его действие на Т-клетки таково, что несмотря на общий супрессорный эффект, он может в определенных обстоятельствах подавлять ответ этих клеток на антигены.
Циклофосфамид успешно применяется при трансплантации костей. В малых дозах препарат эффективен при аутоиммунных нарушениях.
- 577-
Глава 31
Циклоспорин A (Cyclosporin А)
Циклоспорин А — [К-[К*,К*-(Е)]]-циклический-(Е-аланил-Ц-аланил-Х-метил-Е-лейцил-Х-метил-Е-лейцил-Х-метил-Е-валил-З-гидрокси-Х,4-диметил-Е-2-амино-6-октеноил-Е-а аминобутирил-Х-метилгли-цил-Х-метил-Ь-лейцил-Е-валил-Х-метил-Е-лейцин (31.2.2), который экстрагируются из культуральной жидкости продуктов жизнедеятельности гриба Tolypocladium inflatum [14, 15. 16, 17] и который предложено получать синтетическим путем [18, 19].
СНзх^Н
I
сн3х^.снэ н сн2
сн сн-.. ^сн-,но. сн сн3 сн2 сн сн 3 сн2 СНз
CH3-N-CH-C-о=с 10 о
СН—СН2“СН9
СН3—N 8 н
N-CH-C-N-CH-C-сн311 о сн31 о
f? ? н о 6
N-СН-С-N—СН2 н 2 О 3 с=о | ; N—СН3
О 5 h oj
II I II 4 I
О—C—CH—N—C—CH“N—C—CH—N—C—CH—N—C—CH
CH3 CH3 CH2 CH3 СН СН2 Ан снз снз Ан
СНз^СНз СНз^СНз
31 2 2
Циклоспорин А является мощным иммуносупрессивным препаратом, предназначенным для предотвращения отторжения при пересадке почек, печени или сердца.
С открытием циклоспоринов началась новая эра в развитии иммунофармакологии. Циклоспорины продуцируются мицелиальными грибами Tolypocladium inflatum, Tricoderma polysporum и Cylindrocarpon lucidum, выделенными из почв.
Циклоспорин А является первым препаратом, воздействующим на специфическую линию клеток защитных сил организма. Он подавляет именно 4-клетки, в отличие обычных цитотоксиков, воздействующих на все линии клеток одновременно. Циклоспорин А значительно облегчает «принятие» трансплантатов и повышает возможности для лечения заболеваний аутоиммунной системы
Все циклоспорины (А, В. С. .. U, V. W) являются олигопептидами, состоящими из И аминокислот, замкнутых в цикл. Все составляющие являются известными аминокислотами за исключением первой, которая
- 578-
Иммунофармакологические препараты
никогда ранее не была выделена из природных источников. Все они являются L-аминокислотами за исключением аминокислот в положениях 3 и 8. За счет возникающих водородных связей структура циклоспорина достаточно жесткая. Циклоспорины А, В, С, ...U, V, W отличаются лишь второй аминокислотой.
Осуществлен полный синтез как самого циклоспорина А, так и ряда других циклоспоринов. Синтезировано также много структурных аналогов и выявлены некоторые закономерности в плане структура-активность. Выяснено, что активность препарата обуславливается всей циклической структурой, а не отдельными ее фрагментами. Вместе с тем ясно, что структура аминокислоты в положении 1 является важным фактором, определяющим активность. Несмотря на то, что молекула ошосительно велика, циклоспорин проникает через клеточные мембраны с легкостью. Возможно, что для циклоспорина не существует соо!ве|С1вуюших «узнающих?) рецепторов. Однако существует цитозольный циклоспорин-связывающий белок, известный как цитофилин, с молекулярной массой около 15 000. Циклофилины обнаружены пре-имущес1венно в Т-клетках, однако они найдены и в других тканях, в частности в мозге и почках. Их точное назначение и функции неизвестны. Предполагается, что они контролируют РНК при синтезе лим-фокинов Поскольку механизм действия циклоспорина все еще интенсивно исследуется, необходимо отметить, что он не цитотоксичен в общем смысле этого слова, поскольку не подавляет функции костного мозга.
Несмотря на то, что циклоспорин применяется не очень давно, он зарекомендовал себя как препарат номер один при трансплантациях.
Циклоспорин изучается также в качестве средства для лечения ряда аутоиммунных заболеваний, включая диабет, множественный склероз. миастению, ревматоидный артрит, псориаз. Препарат оказался весьма эффективным также и при лечении шистосомиаса, малярии, филарияза.
Синонимами препарата являются сандиммун и неорал.
Глава 31
Литература
1. US Pat. 3.274.209 (1966).
2. Raemaekers A. et al. J. Med. Chem. 9. 545 (1966).
3. Spicer L et al./,'J. Org. Chem. 33, 1350 (1968).
4. Raemaekers A et al. '/Tetrahedron Letters. 1967. 1467.
5. Bullock W et al. 7J. Med. Chem. 11, 169 (1968).
6. Ger. Pat. 1.076.651 (1966).
7. Brit. Pat. 1.076.109(1965).
8. US Pat. 3.565.907 (1971).
9. US Pat. 3.579.530 (1971).
10. US Pat. 3.478.047 (1969).
11. US Pat. 4.070.363 (1978).
12. US Pat. 4.107.170 (1978).
13. US Pat. 3.056.785 (1962).
14. US Pat. 4.IP.118 (1978).
15. Ger. Pat. 2.455.559 (1974).
16. Ruegger A. et al. 7HeIv. Chim. Acta. 59, 1075 (1976).
17. Kobel H. et al./dEur. J. Appl. Microbiol. Biotechnol. 14, 237 (1982).
18. A'enger R / Angew. Chem. 97, 88 (1985).
19. Wenger R.i Transplant. Proc. 15(4), Suppl. 1, 2230 (1983).
20. Enr. Pat. Appl. 34 567(1981).
J Гл а в a 32
; Антибиотики
Препараты, используемые для лечения инфекционных заболеваний, называются антибиотиками, противоинфекционными средствами, антимикробными или химиотерапевтическими препаратами. Несмотря на то. что все эти термины, в принципе, взаимозаменяемы, тем не менее, первые три — антибиотики, противоинфекционные средства и антимикробные препараты, — в основном, употребляются при описания лекарств, используемых для лечения инфекционных заболеваний, в то время как термин «химиотерапевтические препараты» больше ассоциируется с препаратами, применяемыми для лечения рака.
Собственно антибиотиками называются природные соединения, продуцируемые микроорганизмами и способные ингибировать рост болезнетворных микробов, бактерий и некоторых простейших микроорганизмов.
Полу синтетическими антибиотиками считаются продукты частичного химического видоизменения антибиотиков, выделенных из природных источников.
Таким образом, антибиотиками являются соединения, продуцируемые микроорганизмами и имеющие способность убивать или ингибировать рост бактерий и других микроорганизмов. Это определение вносит определенную разницу между антимикробными препаратами, продуцируемыми микроорганизмами, и полностью синтетическими соединениями. Это различие носит чисто академический характер, и сегодня слово антибиотик довольно часто используется для определения антимикробных препаратов вообще. Следует отметить, что существуют продуцируемые микроорганизмами соединения с противогрибковым и противоопухолевым действием, которые также классифицируются как антибиотики.
- 581 -
Глава 32
Основной концепцией антимикробного действия является избира-тезьная токсичность, заключающаяся в том, что рост инфекционного организма ингибируется или он уничтожается определенным препаратом без повреждения клеток хозяина. Все используемые в клинической практике антимикробные препараты являются селективно токсичными по отношению к микроорганизмам. Высокая избирательная токсичность антибиогиков на микроорганизмы объясняется особенностями организации микробных клеток, принципиально отличающихся от клеток млекопитающих. Природа и степень этой селективности и определяет, является ли данный антимикробный препарат в общем не токсичным по отношению к клеткам млекопитающих или же он все-таки проявляет определенную токсичность и по отношению к определенным тканям млекопитающих.
Антимикробные препараты проявляют антибактериальный эффект посредством одного из следующих механизмов:
— ингибирование синтеза клеточных стенок микроорганизмов (р-лактамные антибиотики, ванкомицин, циклосерин);
— ингибирование синтеза протеинов микроорганизмов (аминогликозиды, эритромицин, клиндамицин, хлорамфеникол и тетрациклины);
- - ингибирование синтеза нуклеиновых кислот или их функций в микроорганизмах (сульфонамиды, триметоприм, метронидазол, хинолоны и рифампин);
— ингибирование или изменение функций внешних и/или цитоплазматических мембран микроорганизмов (полимиксин).
Рациональный подход антимикробной терапии инфекций основан па выделении и идентификации инфицирующего организма и определении его чувствительности к антимикробному препарату. Наиболее широко используются ш vitro тесты, такие как диффузия в агаре и определение минимальной подавляющей концентрации в жидкой среде.
В методе диффузии в агаре бумажный диск, содержащий определенное количество антимикробного препарата, помещается на поверхность плас 1 инки, покрытой агаром, содержащим стандартное количество бактерий. После инкубации в течение определенного времени измеряются диаметры чистой зоны вокруг дисков, которые отмечают отсутствие роста бактерий. Диаметры интерпретируются как чувствительный, промежуточный и резистентный по отношению к конкретному препарату после сравнения со стандартом. Недостатком метода следует считать возможность только качественного определения
- 582 -
Антибиотики
ингибиру юшей активности.
Качественная чувствительность определяется и методом определения минимальной подавляющей концентрации в жидкой среде. Наблюдают серию двукратных разбавлений препарата в растворе, содержащим стандартизованное количество микроорганизмов. Результаты выражаются индексом минимальной ингибирующей концентрации (МИК), являющейся минимальной концентрацией препарата, ингибирующего видимый рост после инкубации в течение ночи. Минимальная бактерицидная концентрация (МБК) определяется по отсутствию бактериального роста на агаровых пластинках и реинкубированных в течение еще одной ночи. Это наименьшая концентрация препарата, уничтожающего как минимум 99,9 % бактериального содержимого теста. Данные МИК и МБК должны быть скоррелированны с концентрацией препарата, достигаемой в плазме и других тканях, а также в жидкостях организма.
Противомикробные препараты могут быть классифицированы как бактериостатические (например, тетрациклины, сульфонамиды) и как бактерицидные (например, пенициллины).
Бактериостатические препараты ингибируют бактериальный рост, но не уничтожают эти организмы в клинически достижимых концентрациях. Следует ожидать, что МБК таких препаратов значительно выше чем МИК.
Бактерицидные препараты вызывают гибель микробных клеток и их лизис в клинически достижимых концентрациях. Для этих препаратов МБК близка или равна МИК.
Лечение бактериостатиками останавливает бактериальный рост, позволяя таким образом нейтрофилам и другим защитным силам организма вывести патоген.
13 процессе использования антибиотиков по отношению к ним можег наблюдаться резистентность. Резистентность бактерий к антимикробным препаратам может быть охарактеризована и классифицирована по двум признакам: внутренняя и приобретенная резистентность. Внутренняя резистентность микроорганизма является генетическим свойством микроорганизма, закодированным в хромосомах и распространенным на все линии данного вида микроорганизмов. Приобретенная резистентность означает, что данная линия бактериального вида приобрела способность сопротивляться данному антимикробному препарату.
Приобретенная резистентность подразумевает изменения в ДНК оактерий таким образом, что появляются новые характерные штрихи новою фенотипа. Резистентность при этом достигается двумя путями:
- 583-
Г лава 32
мутацией хромосом в бактериях или приобретением новых участков ДНК (плазмид), кодирующих функцию резистентности.
Биохимический механизм внутренней и приобретенной резистентности одинаков и может быть объяснен как результат одной из следующих 4 причин:
1. инактивация или модификация лекарств ферментами бактерий;
2. образование такого барьера проницаемости, чтобы npenapai не мог достигнуть целевого участка его действия;
3. изменение самой цели таким образом, чтобы препарат не мог связаться или воздействовать на нее;
4. выработка измененного метаболического пути, который позволяет миновать действие препарата.
Если организм подвергается одновременному действию двух антимикробных препаратов, результирующим эффектом может оказаться аддитивный эффект — синергизм или антагонизм.
Препараты считаются аддитивно действующими, когда активность лекарств в комбинации равна сумме их независимых активностей. Общий эффект двух антимикробных препаратов может быть меньше (антагонизм) или больше (синергизм) суммарного эффекта. Синергизм может возникнуть в результате различных механизмов действия, таких как последовательная блокада общего метаболического пути или повышения проницаемости бактериальных клеток.
Существует одновременно несколько подходов к классификации антибиотиков, определяемых, главным образом, профессиональными интересами исследователей.
Антибиотики, в частности, классифицируются по:
— принципу их биологического происхождения (например, антибиотики, вырабатываемые определенными микроорганизмами);
— механизму их биологического действия (например, антибиотики, ингибирующие синтез клеточной стенки, антибиотики, нарушающие функции мембран, антибиотики подавляющие синтез нуклеиновых кислот);
— принципу спектра их биологического действия (например, противобактериальные антибиотики узкого спектра действия, активные преимущественно в отношении грамположитель-ных организмов, противобактериальные антибиотики широ
- 584 -
Антибиотики
кого спектра действия, противотуберкулезные антибиотики, противогрибковые антибиотики, противоопухолевые антибиотики и противоамебные антибиотики);
— принципу их химического строения (например, Р-лактамные антибиотики. тетрациклины, аминогликозиды, макролиды и др.).
32.1. ^-лактамные антибиотики
Р-лактамные антибиотики рассматриваются в виде четырех групп, которые включают пенициллины, цефалоспорины, монобактамы и кар-бапенемы. Все они содержат четырехчленное Р-лактамное кольцо, необходимое для проявления антибактериальной активности, р-лактамное кольцо сочленено с пятичленным тиазолидиновым кольцом в пенициллинах и с шестичленным дигидротиазиновым кольцом в цефалоспоринах. В карбапенемах Р-лактамное кольцо также сочленено с пятичленным кольцом, однако карбоциклическим. Монобактамы представляют собой моноциклическую р-лактамную структуру, в которой к атому азота присоединена боковая сульфогруппа.
Penicillins
Cephalosporins
Carbapenems
Monobactams
- 585 -
Г лава 32
Основным механизмом действия р-лактамных антибиотиков является ингибирование синтеза клеточных стенок бактерий, что и вызывают их быструю гибель. Первичным актом их действия является включение работы аутолитических ферментов, которые далее разрушают клеточные стенки и вызывают лизис бактерий.
Клеточная стенка бактерий структура уникальная и не имеет аналогов в клетках млекопитающих. Клеточная стенка защищает бактериальную клетку от лизиса, который может произойти из-за разницы осмотических давлений между цитоплазмой и окружающей средой.
Основной составляющей клеточной стенки бактерий является смешанный полимер, известный как муреин, или пептидогликан. Пептидогликан представляет собой длинные полисахаридные цепи, кросс-связанные короткими соединяющими пептидами. Полисахаридные цепи составлены из двух варьируемых аминосахаров — N-ацетилглюкоз-амина и N-ацетилмураминовой кислоты. Например, в Staphylococcus aureus (золотистый стафилококк) тетрапептид, состоящий из L-аланина, D-глютаминовой кислоты, L-лизина и D-аланина, присоединен к каждой из единиц N-ацетилмураминовой кислоты, образуя боковые цепи гликановых цепей. Многие из этих тетрапептидов кросс-связаны друг с другом либо непосредственно, либо короткими пептидными цепями. В Staphylococcus aureus L-лизин одного из тетрапептидов связан пентаглициновой цепью с D-аланином другой. Такая структура придает определенную жесткость стенкам бактерий. Пептидогликановый слой грамотрицательных бактерий тоньше, чем грамположительных, и имеет меньше кросс (поперечных) связей.
Синтез пептидогликана клеточных стенок бактерий может быть разделен на 3 стадии, согласно тому, где происходит реакция.
Первая стадия происходит в цитоплазме, в результате чего синтезируется прекурсорная единица — уридиндифосфо-К’-ацетимурамил пентапептид и такой антиботик, как, например, циклосерин — препарат, который наиболее часто используется при лечении туберкулеза, блокирует синтез клеточных стенок именно на этой стадии путем конкурентного ингибирования стадии внедрения аланина в пентапептид.
Реакции, относящиеся ко второй стадии, происходят тогда, когда прекурсорная единица перемещается вдоль цитоплазматической мембраны. В первой реакции N-ацетилмурамил-пентапептидный участок присоединяется (посредством пирофосфатного мостика) к несущему фосфолипиду, связанному с цитоплазматической мембраной. Затем присоединяется N-ацетилглюкозамин, образуя дисахарид-пентапептид-Р-Р-фосфолипид. Затем происходит дальнейшая модификация пента-
- 586 -
антибиотики
пептидной цепи, например, присоединение пентаглицина в случае того же Staphylococcus aureus Модифицированный дисахарид последовательно отщепляется от ме.мбраносвязанного фосфолипида и затем связывается с наращивающейся частью уже имеющегося пептидогликана. Эта реакция опосредуется ферментом пептидогликансинтетазой. Основные повторяющиеся единицы пептидогликана, таким образом, собираются. образуя гликопептидные полимеры. Этот процесс может нарушаться таким антибиотиком, как ванкомицин, который ингибирует пепти до г л иканс интетазу.
Третья стадия (конечная) синтеза клеточных стенок происходит вне стенок цитоплазматической мембраны. При этом линейные гликопептидные полимеры превращаются в кросс (поперечно) связанные посредством реакции транспептизации. Фермент транспептидаза, являющийся мембраносвязанным, связывает пентапептидные боковые цепи путем замещения терминальных D-аланинов.
Как уже было отмечено, Р-лактамные антибиотики вмешиваются в биосинтез основной составляющей клеточной стенки бактерий — пептидогликана. Вследствие того, что в клетках человека и других млекопитающих этот процесс не имеет места, Р-лактамные антибиотики относительно малотоксичны для человека.
р-.гаьтамные антибиотики специфично связываются с рядом белков цитоплазматической мембраны и известных как пенициллинсвязываю-щие белки (ПСБ). Эти белки являются ферментами, вовлеченными в реакции транспептизации при расформировании клеточных стенок в ходе роста и разделения.
К примеру, Escherichia coli имеет шесть ПСБ. ПСБ-la и ПСБ-lb, которые являются транспептидазами, включенными в синтез пептидогликана. ПСБ-2 необходимы для поддержания «стержневой» формы бактерий Селективное ингибирование этого фермента вызывает выработку иных, «нестержневых», форм бактерий, которые, в конечном счете, подвергаются лизису. ПСБ-3 необходимы для формирования перегородок в ходе деления. Селективное ингибирование этого фермента приводит к образованию неких волокнистых форм бактерий, содержащих множество единиц стержневой формы, не способных отделиться одна «г другой, что в результате также приводит к их гибели. Различные Р-лактамные антибиотики имеют селективное сродство к одной или нескольким ПСБ. Инакгивирование определенных (высокомолекулярных) (КБ (ПСБ-la. -1b, -2 или -3) вызывает гибель клеток. В отличие от них. инактивация низкомолекулярных ПСБ (ПСБ-4, -5 и -6) не является ic-тальной для бактерий.
- 587-
Глава 32
Резистентность патогенных микроорганизмов к р-лактамным антибиотикам может возникнуть по одному или сразу нескольким из нижеприведенных механизмов:
— неспособности препарата достигнуть непосредственно места своего действия;
— изменению функций ПСБ, что выражается в понижении сродства к препарату;
— инактивации препарата бактериальными ферментами.
Р-лактамные антибиотики должны проникнуть через внешнее покрытие клетки, чтобы достигнуть целевых ПСБ на поверхности мембраны. В грамположительных бактериях клеточная слепка является единственным слоем, покрывающим цитоплазматическую мембрану. У некоторых их видов с внешней стороны клеточной стенки имеется также и полисахаридная капсула. Однако ни одна из описанных структур не может явиться барьером для прохождения таких маленьких молекул, как Р-лактамы. Поэтому предположение, что причиной возможной резистентности является неспособность достижения Р-лактамны.ми антибиотиками целевых ПСБ, вряд ли следует считать возможным механизмом резистентности для грамположительных бактерий.
Грамотрицагельные бактерии имеют более сложную структуру поверхности клетки. Пептидогликановый слой опять-таки является внешним по отношению к цитоплазматической мембране. Однако у них кроме нее имеется еще одна полисахаридная внешняя мембрана. Эта внешняя мембрана, построенная из липополисахаридов и липопротеинов, может представлять собой серьезный барьер для прохождения гидрофильных молекул. Прохождение через эту мембрану Р-лактамных антибиотиков возможно только через трансмембранные каналы — порины, образуемые протеинами. Показано, что р-лактамные антибиотики диффундируют через пориновые каналы и легкость этого процесса варьирует в зависимости от их размера, заряда и гидрофильных свойств. Соответственно, предположение считать возможным механизмом резистентности для грамотрицательных бактерий неспособность достижения целевых ПСБ Р-лактамны.ми антибиотиками также маловероятно.
Вторым механизмом резистентности к Р-лактамным антибиотикам может явиться изменение целевых ПСБ, выражающееся в понижении их сродства к Р-лактамной молекуле.
- 588-
Антибиотики
Наконец, наиболее важным механизмом резистентности к р-лактамным антибиотикам является выработка бактериями р-лактамаз. р-лактамазы разрывают C-N-связь в Р-лактамном кольце антибиотиков. А поскольку ее наличие является абсолютным требованием для взаимодействия с ПСБ, разрыв Р-лактамного кольца приводит к поте-ре антибактериальной активности.
Cj шествует много Р-лактамаз, которые могут быть классифицированы по-разному: типу субстрата, расположению в генах (хромосомы или плазмиды), месту выработки. Некоторые из этих ферментов прямо гидролизуют пенициллины (пенициллиназы), другие гидролизуют цефалоспорины (цефалоспориназы), третьи распространяются на более широкий спектр субстратов. Некоторые бактерии обладают свойством индуцировать синтез р-лактамаз. Синтез Р-лактамаз. который в обычном состоянии подавлен, индуцируется в присутствии некоторых р-лактамных антибиотиков.
Таким образом, р-лактамные антибиотики могут различными путями ингибировать процесс синтеза клеточных стенок бактерий и тем самым вызывать их быструю гибель.
32.1.1 . Пенициллины
Пенициллин был открыт в 1928 г. Александром Флемингом, заметившим, что контаминация плесенью одной из его экспериментальных культур — стафилококков вызывала лизис бактерий. Поскольку плесень относилась к роду Penicillium, он назвал эту антибактериальную субстанцию пенициллин.
Примерно через десятилетие группа исследователей Оксфордского университета выделила сырую субстанцию, состоящую из нескольких низкомолекулярных веществ — пенициллинов (F, G, К, О, V, X). Пенициллин-G (бензилпенициллин), наиболее активный из них, в 1941 г был предложен для клинических исследований.
о
r-c-nh___^S-^СНз
юь о соон
пенициллин
-589-
Глава 32
Препараты группы пенициллина эффективны при инфекциях, вызванных грамположительными бактериями (стрептококками, стафилококками, пневмококками и др.), спирохетами и другими патогенными микроорганизмами. Препаразы этой группы неэффективны в отношении вирусов, микобактерий туберкулеза, грибов и большинства грамот-онцательных микроорганизмов.
Получение пенициллина — весьма важная веха в развитии микробиологии. химии и медицины, знаменующая создание мощной антибиотической промышленности и формирование современной биотехнологии.
Были предприняты и осуществлены попытки химического синтеза пенициллинов, однако эти способы практического значения не приобрели.
Весьма важный прогресс в развитии пенициллинов произошел в 1959 г. когда пенициллиновое ядро — 6-аминопенициллановая кислота (6-AIIK). отщепленная от боковой цепи, была выделена из культуры Penici’.lium chrysogenum
М-Хсн’
о соон
6-аминопенициллановая кислота
- 590 -
д н '< и б иотики
Е последующем ацилирование 6-АПК производными различных кис Ю1 привело к созданию большого числа полисинтетических пенициллинов.
В настоящее время пенициллин (бензилпенициллин, пенициллин-G) з огромных количествах (десятки тысяч тонн) выпускается микробиологической промышленностью. Пенициллин могут вырабатывать мно-г не виды грибов Penicilhum, а также некоторые виды грибов Aspergillus В промышленных условиях получают культуральные жидкости с содержанием более 30 мг мл пенициллина. Примерно 2/3 выпускаемого пенициллина используется для получения 6-АПК. При этом, несмотря на возможность чисто химического дезацилирования, наиболее перспективным способом получения 6-АПК считается ферментативный метод гидролиза молекулы бензилпенициллина с участием иммобилизованной пенициллинамидазы — фермента, выделяемого практически из ьсех пенициллинпродупирующих грибов. Следует отметить, что сама 6-АПК практически лишена антибиотических свойств. Однако ее ацилированием различными производными кислот было получено более 50 000 полусинтетических пенициллинов, из которых в настоящее время лишь чуть больше 30 применяются в медицине.
При вариациях ацильной части боковой цепи молекулы пенициллинов отмечены значимые изменения свойств полученных соединений. Выяснилось, что именно боковая ацильная часть молекулы определяет антимикробный спектр, чувствительноегь к р-лактамазам и определяет фармакокинетические особенности конкретного пенициллина. Особенностью некоторых полусинтетических пенициллинов (метициллина, оксациллина, клоксациллина) является их эффективность в отношении штаммов микроорганизмов (стафилококков), резистентных к бензилпенициллину. Более того, некоторые полусинтетические пенициллины (ампициллин) активны и в отношении большинства грамотрицательных микроорганизмов.
С химической точки зрения первым типом полусинтетических пенициллинов, несомненно, следует считать относительно простые пенициллины производные 6-АПК и ароматических или арилоксикарбоновых кислот (бензилпенициллин, феноксиметилпенициллин, метициллин, нафициллин).
Другим типом полусинтетических пенициллинов следует считать пенициллины — производные гетероилкарбоновых кислот, в которых боковая ацильная группа представлена ароматическим гетероциклическим кольцом (оксациллин, клоксациллин, диклоксациллин).
Следующим типом полусинтетических пенициллинов следует считать пенициллины, в которых в роли ацильного радикала выступает
- 591 -
Г лава 32
аминокислота, в основном а-аминофенилуксусная (фенилглинин) или и-окси-а-аминофенилуксусная, — соответственно ампициллин и амоксициллин.
Наконец четвертым типом замещения в боковой ацильной части пенициллинов следует считать замещение производными дикарбоновых кислог(карбенциллин,тикарциллин).
Все пенициллины используются в качестве натриевых или калиевых солей.
Бензилпенициллин (Benxylpenicillin)
Бензилпенициллин — [28-(2а,5а,бР)]-3,3-диметил-7-оксо-б-(фе-
нилацетамидо)-4-тиа-1-аза-бицикло[3.2.0]-гептан-2-карбоповую кислоту (32.1.1.1) получают биотехнологическим способом с использованием в качестве продуцента гриба вида Pemcillium chrysogenum и в качестве прекурсора — фенилуксусную кислоту. Для медицинского применения используются натриевые или калиевые соли, для чего экстракт культуральной жидкости обрабатывается водным раствором соответствующей щелочи, и коммерческим продуктом являются соответствующие лиофилизованные соли [1. 2, 3, 4, 5]. Предложен и чисто синтетический способ получения бензилпенициллина [6, 7].
32 1 1 1
Бензилпенициллин, или пенипиллин-G, имеет узкий антимикробный спектр. Он активен по отношению к грамположительным бактериям (стафилококк, стрептококк, пневмококк), возбудителю дифтерии, палочке сибирской язвы. Грамогрицательные бактерии к нему резистентны. Бензилпенициллин расщепляется желудочной кислотой и разрушается стафилококковой пенициллиназой.
Бензилпенициллин является лекарством выбора при инфекциях, вызванных чувствительными организмами. Последние включают стрептококковые инфекции (за исключением энтерококков), гонококки, менингококки, не продуцирующие |3-лак1амазу анаэробы. Бензилпенициллин применяют при крупозной и очаговой пневмонии, гнойных инфекциях кожи и мягких тканей и слизистых оболочек, перитоните, цистите, сифилисе, дифтерии и других инфекционных заболеваниях.
- 592 -
Антибиотики
Синонимами препарата являются мегациллин, традониллин, бициллин. суграциллин и др.
Феноксиметилпенициллин (Phenoxymethylpenicillin)
Феноксиметил пенициллин — [28-(2о.,5а,бР)]-3,3-диметил-7-оксо-6-(феноксиацетамидо)-4-тиа-1-азабицикло-[3.2.0]-гептан-2-карбоновую кислоту (32.1.1.2) также получают биотехнологическим способом с использованием в качестве продуцента гриба вида Penicillium chrysogenum, а в качестве прекурсора феноксиуксусной кислоты [8, 9, 10, 11]. Как и для бензилпенициллина, предложен и синтетический способ получения феноксиметилпенициллина [6, 12, 13, 14, 15].
32 1 1 2
Феноксиметилпенициллин, или пенициллин-V, кислотоустойчив и используется вместо пенициллина-G при необходимости перорального применения. Активен в отношении грамположительных (стафилококк, стрептококк, пневмококк) и грамотрицательных (менингококк, гонококк) кокков, спирохет, клостридий, коринебактерий.
Феноксиметилпенициллин применяют при бронхите, пневмонии, ангине, скарлатине, гонорее, сифилисе, гнойных заболеваниях кожи и мягких тканей и других инфекционных заболеваниях.
Синонимами препарата являются беромицин. викен, изоциллин, кристален, феноспен, утициллин и др.
Метициллин (Methicillin)
Метициллин — [28-(2и,5а,6|3)]-3,3-диметил-7-оксо-6-(2,6-димето-ксибензамидо)-4-тиа-1-азабицикло[3.2.0]-гептан-2-карбоновую кислоту (32.1.1.3) получают ацилированием 6-АПК хлорангидридом 2,6-ди-метоксибензойной кислоты в присутствии триэтиламина [15, 16, 17]
ОСНз
32 1 1 3
- 593-
Г лава 32
Подобно другим полусинтетическим пенициллинам, метициллин оказывает антибактериальное действие, сходное с бензилпенициллином. Его основным отличием от бензилпенициллина является то, что он не инактивируется ферментом пенициллиназой и поэтому' эффективен в отношении возбудителей, продуцирующих этот фермент (стафилококков).
Препарат применяют при инфекциях, вызванных устойчивыми к бензилпенициллину стафилококками (септицемии, пневмонии, эмпиемы, остеомиелит, абсцессы, раневые инфекции и др.).
Синонимами препарата являются цинопенил, целбенин, стафциллин и др.
Нафциллин (Nafcillin)
Нафциллин — [25-(2а,5а,6|3)]-3,3-диметил-7-оксо-6-(2-этокси-1-
нафтамидо)-4-тиа-1-азабицикло[3.2.0]-гептан-2-карбоновую кислоту' (32.1.1.4) получают ацилированием 6-АПК хлорангидридом 2-этокси-1-нафтойной кислоты в присутствии триэтиламина [18. 19, 20].
32 1 1 4
Препарат эффективен против грамположительных кокков и стафилококков, продуцирующих пенициллиназу. Применяют по тем же показаниям, что и метициллин.
Синонимами препарата являются нафцил, наллпен, юнипен и др.
Другим типом полусинтетических пенициллинов, несомненно, следует считать пенициллины производные гетероилкарбоновых кислот, в которых в роли радикала в боковой ацильной группе представлено ароматическое гетероциклическое кольцо, как правило изоксазольное. в третьем положении которого, в свою очередь, имеется замещенный или незамещенный фенильный радикал (оксациллин, клоксациллин, диклоксациллин). Эти резистентные к пенициллиназе пенициллины (оксациллин. клоксациллин, диклоксациллин) активны по отношению к пенициллину-G и резистентным стафилококкам. Их антимикробный спектр ограничивается т рамположительными микроорганизмами.
- 594 -
Антибиотики
Резистентные к пенициллиназе пенициллины являются лекарством выбора при инфекциях резистентных к пенициллину-G, Staph aureus или коагулазотрицательным стафилококкам. Они эффективны также при инфекциях, вызванных неэнтерококковыми видами стрептококков, включая стрептококки групп А, В, С и G, а также пневмококки.
Оксациллин (Oxacillin)
Оксациллин — [25-(2а,5а,6|3)]-3,3-диметил-7-оксо-6-(5-метил-3-фенил-4-изоксазолкарбоксамидо)-4-тиа-1-азабицикло[3.2.0]-гептан-2-карбоновую кислоту' (32.1.1.10) получают взаимодействием хлорангидрида 5-метил-3-фенил-4-изоксазолкарбоновой кислоты (32.1.1.9) с 6-АПК в присутствии бикарбоната натрия. Сам же хлорангидрид 5-метил-3-фенил-4-изоксазолкарбоновой кислоты (32.1.1.9) получают по следующей схеме.
Взаимодействием бензальдегида с гидроксиламином получают бензальдоксим (32.1.1.5). окисленим которого хлором получают хлорангидрид бензгидроксиминовой кислоты (32.1.1.6). Последний вводят во взаимодействие с ацетоу ксусным эфиром в присутствии этилата натрия с получением этилового эфира 5-метил-3-фенил-4-изоксазолкарбоновой кислой! (32.1.1 7). Щелочным гидролизом полученного эфира получают соответствующую кислоту (32.1.1.8), взаимодействием которой с хлористым тионилом получают требуемый для ацилирования хлорангидрид [21,22, 23,24].
NhjOn
NdOH
SOCI-
32 1 1 10
- 595 -
Глава 32
По спектру антимикробного действия оксациллин аналогичен бензилпенициллину. Однако он сочетает в себе устойчивость к пенициллиназе со стойкостью в кислой среде, что позволяет применять его не только внутримышечно, но и перорально. Применяют при инфекциях, вызванных пенициллиназаобразующими стафилококками, устойчивыми к бензил- и фепоксиметилпенициллина.м (септицемии, пневмонии, абсцессы, эмпиемы, остеомиелит, инфицированные ожоги, раневые инфекции и др.)
Синонимами препарата являются криптоциклин, люиипен, опто-циллин, тотоциллин и др.
Клоксациллин (Cioxacillin)
Клоксациллин — [28-(2а,5а,бР)]-3,3-диметил-7-оксо-б-[(5-мегил-3-(о-хлорфенил)-4-изоксазолкарбоксамидо)]-4-тиа-1-азабицикло[3.2.0]-гептан-2-карбоновую кислоту (32.1.1.11) получают по вышеприведенной схеме исходя и о-хлорбензальдегида [21, 22, 23. 25].
32 1 1 11
По механизму действия и по показаниям препарат аналогичен оксациллину
Синонимами препарата являются ампиклокс, орбенин, тотаклокс, тегапен и др.
Диклоксациллин (Dicloxacillin)
Диклоксациллин — [28-(2с1,5а,бР)]-3,3-диметил-7-оксо-6-[(5-ме-тил-3-(2,6-дихлорфенил)-4-изоксазолкарбоксамидо)]-4-тиа-1-азаби-цикло[3.2.0]-гептан-2-карбоновую кислоту' (32.1.1.12) также получают по приведенной выше схеме, используя в качестве исходного 2.6-дихлорбензальдегид [26, 27, 28].
- 596 -
Антибиотики
По механизму действия, по антибактериальном} спектр}' и по показаниям препарат существенно не отличается от оксациллина и клокса-циллина.
Синонимами препарата являются диклоцил, новапен, диклекс И др.
Следующим типом полусинтетических пенициллинов следует считать пенициллины, в которых в роли ацильного радикала выступает аминокислота, в основном а-аминофенилуксусная или п-окси-а-амино-фенилуксусная (ампициллин, амоксициллин).
Антимикробный спектр аминопенициллинов сходен с пеницилли-ном-G та исключением того, что они воздействуют и на ряд грамотри-цагельных микроорганизмов. Оба аминопенициллина разрушаются стафилококковой пенициллиназой.
Ампициллин (Ampicillin)
Ампициллин — [28-[2а,5а,бр(8*)]]-3,3-диметил-7-оксо-6-(2-амино-2-фенилацетамидо)-4-тиа-1-азабицикло[3.2.0]-гептан-2-карбоновую кислоту (32.1.1.16) предложено получать различными способами, ио сути сводящимися к различным способам защиты аминогруппы в исходном фенилглицине Первый способ (один из наиболее широко применяемых) заключается в использовании бензилового эфира хлоругольной кислоты (карбобензоксихлорида). Взаимодействием последнего с фенилглицином первоначально получают карбо-бепзоксифенилглицин (32.1.1.13). Обработкой последнего этиловым эфиром хлоругольной кислоты в присутствии триэтилам ина получают смешанный ангидрид (32.1.1.14) с защищенной аминогруппой, который с легкостью вступает во взаимодействие с 6-АПК в присутствии бикарбоната натрия с получением натриевой соли N-кар-бобензокси-защищенного ампициллина (32.1.1.15). Удалением кар-бобензокси защитной группы гидрогенолизом с использованием в качестве катализатора палладия на карбонате бария получают искомый ампициллин (32.1.1.16) [29, 30, 31. 32, 33, 34. 35].
о
С1С—О—С Н 2
- 597-
Глава 32
32 1 1 16
Второй способ получения ампициллина аналогичен вышеописанному и отличается способом защиты аминогруппы в исходном фенилг-липине. С этой целью используется ацетоуксусный эфир, взаимодействием которого с натриевой солью фенилглинина получают промежуточный аминокротоновый эфир (32.1.1.17), Последовательной трансформацией последнего в смешанный эфир (32.1.1.18) и последующим взаимодействием с 6-АПК в присутствии бикарбоната натрия получают ампициллин (32.1.1.16) в виде натриевой соли [36].
сн3
р
ci—с-о-с2н5
32 1 1 17
(C2H5)3N
Чанс Оз
32 ' 1 36
Предложен также третий способ прямого ацилирования 6-АПК хлораш идридом гидрохлорида фенилглицина [37].
32 1 1 16
-598-
Антибиотики
Ампициллин обладает широким спектром действия и эффективен при инфекциях, вызванных различными чувствительными организмами — активен в отношении грамположительных и грам отрицательных кокков, кишечной палочки, сальмонелл, шигелл. энтерококков, листерий, некоторых штаммов гемофильной палочки. Ампициллин является лекарством выбора при инфекциях вызванных Р-лактамаз отрицательными видами Н influenzae, L. monocytogenes и энтерококков. Препарат применяют при бронхите, пневмонии, дизентерии, сальмонеллезе, коклюше, пиелонефрите, эндокардите, сепсисе и т. д.
Синонимами препарата являются амблосин, бинотал, люципен, то-танен, амфипен, ампицил, пенберин и многие другие.
Амоксици-LiuH (Amoxycillin)
Амоксициллин — [28-[2а,5о',бР(8’)]]-3,3-диметил-7-оксо-6-[[ами-но-(4-гидроксифенил)-ацетил]амино]-4-тиа-1-азабицикло[3.2.0]-геп-(ан-2-карбоновую кислоту (32.1.1.21) предложено получать двумя путями. Первый способ, предполагающий енаминовую защиту аминогруппы 4-гидроксифенилаланина, исходит из натриевой соли 4-i идроксифенилаланина, взаимодействием которой с ацетоуксусным эфиром получают енамин — натриевую соль а-(4-гидро-ксифенил)-а-(карбметоксипропен-2-ил-амино)уксусной кислоты (32 1.1.19). Взаимодействием полученного аминокротоната с этиловым эфиром хлоругольной кислоты в N-метилморфолине получают соответствующий смешанный эфир (32.1.1.20), который вводят во взаимодействие с триметилсилиловым эфиром 6-АПК [38, 39].
СН, О-С2Н5
о
CI—с-о-сгн5
СНз
КаНСОз
COOSi(CH3^
32 1 1 21
-599-
Глава 32
Второй способ предполагает непосредственное взаимодействие хлорангидрида хлоргидрата П-(-)-2-(4-гидроксифенил)глицина с триметилсилиловым эфиром 6-АПК [40].
нэ
СН-СЭС1 hH2 но
Необходимый для этой цели триметилсилиловый эфир 6-АПК, в свою очередь, получают взаимодействием триметилхлорсилилана с 6-АПК в присутствии триэтиламина.
Амоксициллин обладает широким спектром действия аналогичным ампициллину. Показания к применению такие же. как и у ампициллина.
Синонимами препарата являются амоксикан, амоксил, ларогид, ро-бамокс. тримокс, вимокс, утимокс и др.
Несомненно, к этой же группе соединений следует от нести и аналоги ампициллина, замещенные по аминному фрагменту фенилглицина (азлоциллин, мезлоциллин, пиперациллин).
А 1лоци.1лин (Azlocillin)
Азлоциллин — (2S,5R,6R)-3,3-flHMeTiui-7-OKCO-6-[(R)-2-(2-oKCO-имидазолидин-1-карбоксамидо)-2-фенилацетамидо]-4-гиа-1-азаби-цикло[3.2.0]гептан-2-карбоновую кислоту (32.1.1.24) предложено получать по следующей схеме. 2-имидазолидинон ацилируют фосгеном с получением 1-хлоркарбонил-2-имидазолидинона (32.1.1.22). Полученный 1-хлоркарбонил-2-имидазолидинон (32.1.1.22) вводят в реакцию с П(-)-а-фенилглицином с получением К’-(2-оксоимида-золидин-1-карбоксамидо)-фенилглицина (32.1.1.23). Взаимодействием последнего с 6-.АПК в присутствии триэтиламина получают искомый азлоциллин (32.1.1.24) [41,42, 43].
ЧаНСОз
-600-
Антибиотики
Азлоциллин активен в отношении грамположительных и грамотри-цательных аэробных и анаэробных микроорганизмов. Высокоэффективен в 01 ношении синегнойной палочки, включая штаммы, резистентные к карбенциллину и аминогликозидам. Разрушается Р-лактамазами. Применяется при таких бактериальных инфекциях, как пиелонефрит, уретрит, цистит, эндометрит, холангит, холецистит, сепсис, перитонит, эндокардит, менингит, пневмония, инфекции кожи и мягких тканей, инфицированные ожоги и т. д.
Синонимами препарата являются секуропен и азлин.
Мезлоциллин (Mezlocillin)
Мезлоциллин — (25,5К,6К)-3.3-ди.метил-7-оксо-6-[(К)-2-[3-ме-
1 илсульфонил)-2-оксоимидазолидин-1-карбоксамидо]-2-фенил-ацетамидо]-4-тиа-1-азабицикло[3.2.0]-гептан-2-карбоновую кислой (32.1.1.27) получают ацилированием ампициллина (32.1.1.16) 3-хлоркарбонил-1-метансульфонил-2-имидазолидино-ном (32.1.1.26) в присутствии триэтиламина. Необходимый для этой цели 3-хлоркарбонил-1-метансульфонил-2-имидазолидинон (32.1.1.26) получают сульфонилированием 2-имидазолидинона ме-гансульфохлоридом с получением 1-метансульфонил-2-имидазо-лидинона (32.1.1.25) и последующим его взаимодействием с фосгеном [44, 45, 46].
- 601 -
Г лава 32
Аналогично азлоциллину мезлоциллин применяется при инфекциях мочеполового тракта, гинекологических инфекциях, интраабдоминаль-ных инфекциях, инфекциях кожи и дыхательного тракта.
Синонимами препарата являются байпен, мезлин. оптоциллин.
Пиперациллин (Piperacillin)
Пиперациллин — (25,5К.6К)-3.3-ди.метил-7-оксо-6-[(2К)-2-[(4-этил-2,3-диоксо-1-пиперазинил) формамидо]-2-фенилацетамидо]-4-тиа-1-азабицикло[3.2.0]-гептан-2-карбоновую кислоту (32.1.1.30) также предложено получать ацилированием ампициллина (32.1.1.16). но уже 1-хлоркарбонил-4-этилпиперазин-2,3-дионом (32.1.1.29). Необходимый для этой цели 1-хлоркарбонил-4-этилпиперазин-2,3-дион (32.1,1 29) получают взаимодействием N-этил-этилендиамина с ди-этилоксалатом с образованием 4-этилпиперазин-2,3-диона (32.1.1.28) и далее ацилированием фосгеном после первоначального силилиро-вания продукта по атому азота триметилхлорсиланом [47, 48. 49, 50].
Cjf-ig—Nh-СН4~СН — NHj
- 602 -
Антибиотики
Область применения препарата аналогична таковой азлоциллина и мезлоциллина
Синонимами препарата являются пиприл, пипрацил, авоцин и др.
Наконец четвертым типом замещения в боковой ацильной части пенициллинов следует считать замещение производными дикарбоновых кислот (карбенциллин, гикарциллин).
Карбенциллин (Carbencillin)
Карбенциллин — [28-(2и,5о',6р)]-3,3-дшметил-7-оксо-6-(2-карбокси-2-фенилацетамидо)-4-тиа-1-азабицикло[3.2.0]-гептан-2-карбоновую кислоту (32.1.1.32) предложено получать прямым ацилированием 6-АПК в присутствии бикарбоната натрия хлорангидридом моно-бензилового эфира фенилмалоновой кислоты с получением бензилового эфира карбенциллина (32.1.1.31), гидрогенолизом которого водородом с использованием в качестве катализатора палладия либо на угле, либо на карбонате кальция получают искомый продукт (32.1.1.32) [51, 52, 53, 54, 55, 56, 57, 58].
ЬаКСОз
Pd-C
32 1 1 32
Карбенциллин обладает широким спектром антибактериальной ак-ивносги в отношении грамотринательных и грамположительных мик-poopi анизмов. Однако применение препарата при инфекциях, вызванных грамположительными микроорганизмами, нецелесообразно. Его применяют при заболеваниях, вызванных грамотрицательными микро-ор|анизмами. чувствительными к этому антибиотику, — инфекциях мочевых путей, септицемии, эндокардите, менингите, остеомиелите, перитоните, гнойном отите, раневых инфекциях, инфицированных ожогах и т и.
Синонимами препарата являются кариндапен, пиопен, геопен, гри-пенин и др
- 603 -
Глава 32
Тикар ц илл ин (Ticarcillin)
Тикарпиллин — [28-(2сс,5а,бР)]-3,3-диметил-7-оксо-6-[2-карбокси-2-(3-тиенил)ацетамидо]-4-тиа-1-азабицикло[3.2.0]-гептан-2-карбоно-вую кислоту (32.1.1.34) также предложено получать прямым ацилированием 6-АПК в присутствии гидроокиси натрия, но уже хлорангидридом 3-тиенилмалоновой кислоты (32.1.1.33) с получением тикарциллина [52, 53, 54, 55, 56, 57, 59J.
С ООН он-соон
соон
H-CO-CI
КаНСОз
По антимикробному спектру действия и областям применения ти-карциллин сходен с карбенциллином.
Синонимами препарата являются бетабактил, тикар, тиметин и др.
Ингибиторы р-лактамазы
Клавулановая кислота и сулбактам
(Clavulanic acid and Sulbactam)
Добавление ингибиторов р-лактамазы, таких как клавулановая кислота (32.1.1.35) или сулбактам (32.1.1.36), к пенициллинам либо к аминоиенициллинам широкого спектра действия значительно расширяет их антимикробный спектр.
СН7ОН
32 1 1 35
32 1 1 36
Клав} лановую кислоту выделяют из Streptomyces clavuhgerus [60. 61. 62, 63, 64, 65, 66]. а сулбактам — сульфон пенициллановой кислоты, получают из 6-АПК [67, 68, 69]. Оба соединения обладают весьма слабыми антибактериальными свойствами и действуют, ообразуя необратимые комплексы с р-лактамазой, которые инактивируют фермент, в результате чего р-лактамный антибиотик успевает разрушить
- 604 -
Антибиотики
микроорганизм. В настоящее время применяется ряд комбинированных препаратов, содержащих различные сочетания антибиотиков и ингибиторов р-лактамазы.
32.1.2 . Цефалоспорины
Из продуктов ферментации гриба Cephalosporium асгетотсит был выделен ряд антибиотиков — цефалоспоринов Р, N, С. Мажорным компонентом смеси является цефалоспорин С — амид о'-аминоадипиновой и 7-аминоцефалоспорановой кислот.
о
НООС-СН-(СН2)з-С-NH
NH2
СН2-О-СОСН3 соон
цефалоспорин С
Химический или ферментативный гидролиз этого соединения позволяет получать большие количества 7-аминоцефалоспорановой кислоты, ацилированием аминогруппы которой производными различных кислот аналогично ряду полусинтетических пенициллинов был создан ряд полусинтетических р-лактамных цефалоспориновых антибиотиков, которых в настоящее время насчитывается около 25 000, из которых около 100 внедрено в медицинскую практику. Причем, в отличие от пенициллинов, полусинтетические цефалоспорины получают как расширением спектра различных кислот, которыми ацилируется 7-ами-ноцефалоспорановая кислота, так и модификациями внутри аминоцефа-лоспоранового ядра (R| и R2).
соон соон
^-аминоцефалоспорановая киск/та
полусинтетические цефалоспорины
Цефалоспориновое ядро состоит из Р-лактамното кольца, сочлененного с шестичленным дигидротиазиновым кольцом. В отличие от пенициллинового ядра цефалоспориновое ядро значительно более устойчиво
- 605 -
Глава 32
к Р-.тактамазам. Более того, оно имеет больше зчастков для возможных модификаций Модификации R( в ацильной боковой цепи изменяют антибактериальную активность, тогда как модификации R2 ассоциируются с изменениями в фармакокинетике и метаболических параметров препарата
Радикалы R, в ацильной боковой цепи в принципе не отличаются или мало отличаются от применяемых в синтезе пенициллинов.
В то же время модификации R2 довольно существенны, и их можно определить как следующие:
О сн2-о-с-сн.
uGOH
це фал от ин цефзроксим цефепим цефотаксим цефокситин
цефпиром цефапирин цефтазидим
СООН r2
цефак.тор цефалексин цефтизоксим цефиксим цефрадин нефадроксил
цефазотин цефамандол цефтриаксон це( цефоперазон
ютетан
- 606 -
Антибиотики
соон
цефотетан моксал актам цефаклор
Бо iee того, последний из приведенных препаратов — моксалактам содержит дигидрооксазиновое кольцо вместо общего для всех цефалоспоринов дигидротиазинового кольца. Некоторые цефалоспорины содержат дополнительную метоксильную группу в положении С- фрагмента лмипоцефалоспорановой кислоты (цефотетан. цефаклор).
Цефалоспорины классифицируются по четырем группам или подразделяются на четыре поколения на основе спектра их активности. Соединения одного поколения отличаются друг от друга главным образом фармакокинешческими особенностями, хотя у них могут быть и различия по отношению к отдельным микроорганизмам.
Цефалоспорины I поколения
Цефалоспорины II поколения
Цефалоспорины III поколения
(цефалотин. цефалоридин, цефалексин, цефапирин, цефазолин, цефад-роксил, цефради и др.) обладают высокой биологической активностью в отошении стафилококков, стрептококков, пневмококков, многих видов энтеробактерий.
(цефуроксим, цефамандол, цефокситин, цефотетан, цефаклор и др.) характеризуются высокой активностью в отношении грамотрицатель-ных микроорганизмов, устойчивы к действию Р-лактамаз. Не оказывают заметного действия на энтерококки.
(цефотаксим, цефтизоксим, цефтриаксон, цефазидим, цефоперазон и ряд других) отличаются высокой антимикробной активностью против энтеробактерий, в том числе устойчивых к другим антибиотикам. Эта группа устойчива к действию Р-лак-
-607-
Глава 32
Цефалоспорины IV поколения
та.маз. образуемых грамотрицательными бактериями Однако они умеренно активны по отношению к стафилококкам.
(цефепим и цефпиром) активны в отношении широкого спектра грамположительных и грамотрица-тельных аэробов. Обладают необычно малым сродством к р-лак-тамазам и способны очень быстро проникать в периплазматическое пространство.
Цефалоспорины I поколения
Цефалоспорины I поколения (цефалотин, цефазолин, цефапирин, цефрадин, цефалексин и цефадроксил) обладают высокой биологической активностью в отношении стафилококков, стрептококков, пневмококков, многих видов энтеробактерий, в том числе Escherichia coli. Proieus mirabilis. Они гидролизуются многими р-лактамазами, продуцируемыми грамотрицательными бактериями и поэтому имеют относительно узкий спектр активности по отношению к грамотрицательиым бактериям. Цефалоспорины I поколения являются альтернативой пенициллинам для лечения стафилококковых и неэнтерококковых инфекций у пациентов, не переносящих пенициллин. Они широко используются при профилактике сердечно сосудистых, ортопедических и прочих хирургических вмешательствах.
Цефалотин (Cefalothin)
Цефалотин — (6К-транс)-3-[(ацетилокси)метил]-8-оксо-7-[(2-тие-нилацегил)амино]-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (32.1.2.1) получают прямым взапмодействим хлорангидри-да 2-тиенилуксусной кислоты с 7-аминоцефалоспорановой кислотой в присутствии бикарбоната натрия [70, 71, 72, 73, 74].
- 608 -
Антибиотики
Цефалотин антибиотик с широким спектром антимикробного действия -- активен в отношении грамположительных (стафилококки, стрептококки, пневмококки, палочки дифтерии, сибирской язвы) и гра-мотрицагельных микроорганизмов (менингококки, гонококки, шигеллы. сальмонелла, кишечная и гемофильная палочки, клебсиеллы), неэффективен в отношении синегнойной палочки, микобактерий туберкулеза, анаэробных микроорганизмов.
Цефалотин применяют при бактериальных инфекциях нижних дыхательных п\ гей, мочевыводящих путей, кожи и мягких тканей, костей и суставов, сепсисе, перитоните, остеомиелите, мастите, раневых, послеоперационных и прочих инфекций.
Синонимами препарата являются кефлин, сеффин, коаксин и др.
Цефапирин (Cefapirin)
Цефапирии — (6К-транс)-3-[(ацетилокси)метил]-8-оксо-7-[[(4-пиридинилтио)ацетил]амино]-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (32.1.2.4) получают ацилированием 7-амино-цефалоспорановой кислоты хлорангидридом 4-пиридилтиоуксусной кислоты (32.1.2.3), получаемого последовательным взаимодействием 4-хлорпиридина с меркаптоуксусной кислотой в присутствии щелочи с образованием 4-пиридилтиоуксусной кислоты (32.1.2.2) и дальнейшим переводом полученной кислоты в хлорангидрид взаимодействием с пятихлористым фосфором. Альтернативным способом получения цефапирина является ацилирование 7-амино-цефалоспорановой кислоты бромангидридом бромуксусной кислоты с получением бромацетильного производного (32.1.2.5), который далее вводят во взаимодействие с 4-меркаптопиридином в присутствии триэтиламина с получением искомого цефапирина (32.1.2.4) [75, 76, 77, 78].
S—сн2-соон
PCI5
h2—O“COCnj СООН
- 609-
Глава 32
С ООн
р Br-CH2-CO—C-NH
32
СН2-О-СОСН3 соон
Спектр действия и показания к применению цефапирина такие же, как и у цефаллотина.
Синонимами препарата являются бристосеф, цефарин. цефатрексил, цефадил и др.
Цефазолин (Cefazolin)
Цефазолин — (6В-транс)-3[[(-5метил-1,3,4-тиадиазол-2-ил)тио]метил]-8-оксо-7-[( 1 W-тетразол-1 -илацетил)амино]-5-тиа-1 -азабицикло[4.2.0]-окт-2-ен-2-карбоновую кислоту (32.1.2.7) получают взаимодействием 7-аминоцефалоспорановой кислоты со смешанным ангидридом (32.1.2.6) — результатом взаимодействия тетразолилуксусной кислоты с пивалиновой (триметилуксусной) кислотой. Дальнейшим его взаимодействием с 2-меркапто-5-метил-1,3,4-тиадиазолом осуществляют замещение 3-ацетоксильной группы на меркаптотиадиа-зольную с получением цефазолина (32.1.2.7) Существенным структурным отличием данного препарата от рассмотренных ранее цефалоспоринов I поколения является замена 3-ацетоксиметильной группы на 1,3,4-тиадиазол-2-илтиометильную [79, 80, 81].
соон
32 1 2 7
-610-
Антибиотики
Спектр действия цефазолина и показания к применению такие же, как и у цефаллотина.
Синонимами препарата являются цефацидал, кефзол, пефамезин. цефазил, золин, ансеф и многие другие.
Цефалексин (Cefalexin)
1 Цефалексин — [6К-[6а,7Р(К*)]]-3-метил-8-оксо-7-[(аминофенил-ацетил)амино]-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновую кислоте (32.1.2,10) в определенной степени можно считать аналогом ампициллина, поскольку ацильным фрагментом, вводимым в структуру 7-аминоцефалоспорановой кислоты, как и в случае ампициллина. является фенилглициновый фрагмент. Кроме того, структура данного препарата, а также ряда других цефалоспоринов, рассматриваемых ниже, несколько упрощена заменой 3-ацетоксиметильной группы на метильную. Синтез цефалексина осуществляют исходя из цефалоглицина (32,1.2.9), который получают взаимодействием 7-а.миноцефалоспорановой кислоты со смещенным ангидридом, полученным взаимодействием N-карбобензоксиглицина и изобутилового эфира хлоругольной кислоты в присутствии триэтиламина. Снятием с полученного продукта (32.1.2.8) N-карбобензокси защитной группы восстановлением водородом с использованием в качестве катализатора палладия на угле получают цефалоглицин (32 1.2.9) в виде внутренней соли. Восстановлением последнего водородом, но уже с использованием в качестве катализатора палладия на сульфате бария осуществляют деацетоксилирование в третьем положении 7-аминоцефалоспорановой кислоты с получением искомого цефалексина (32.1.2.10) [82, 83, 84, 85, 86].
1 'Cr3,2CH-CO-3l .
н2/ Ро—ВаБОд
- 611 -
Глава 32
Цефалексин имеет широкий спектр антимикробного действия: активен в отношении грамположительных и грамотрицательных микроорганизмов. Неэффективен в отношении синегнойной палочки, микробактерий туберкулеза, анаэробных микроорганизмов. Применяется при бактериальных инфекциях верхних и нижних дыхательных путей, мочевыводящих путей, кожи и мягких тканей.
Синонимами препарата являются цепорексин. орацеф, кефлекс. це-фазал и др.
Цефрадин (Cephradin)
Цефрадин — [6К-[6а,7р(К*)]]-3-метил-8-оксо-7-[(амино-1,4-цик-логексадиен-1-илацетил)амино]-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (32.1.2.13) является близким аналогом цефалексина и отличается тем, что фенильный радикал в фенилглицине частично гидрирован в 1,4-циклогексадиенильный. Синтез препарата осуществляют исходя из фенилглицина, который частично восстанавливают литием в жидком аммиаке с получением 1,4-цикло-гексадиенилглицина (32.1.2.11), аминную группу в последнем защищают взаимодействием с метиловым эфиром ацетоуксусной кислоты в присутствии метилата натрия. Полученную при этом соль (32.1.2.12) трансформируют в смешанный ангидрид взаимодействием с этиловым эфиром хлоругольной кислоты в триэтила-мине и вводят во взаимодействие с дезацетоксилированной 7-ами-ноцефалоспорановой кислотой с получением искомого цефрадина (32.1.2.13) [87, 88. 89].
сн—со—он
NH2
:2Н5-СО-С1 / (C2H5;3N
32 12 11 СООН
Спектр действия и показания к применению цефрадина такие же, как и у цефалексина.
Синонимами препарата являются велосеф, сефрил, цефро и др.
-612-
Антибиотики
Цефадроксил (Cefadroxil)
Цефадроксил — [6К-[6а,7Р(Я‘)]]-3-метил-8-оксо-7-[[амино(4-гидро-ксифенил)ацетил]амино]-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбо-новую кислоту (32.1.2.14), являющуюся аналогом цефалексина и отличающуюся лишь наличием гидроксильной группы в четвертом положении фенильного кольца фенилглицина, получают по схеме, аналогичной схеме синтеза цефрадина [90, 91, 92, 93, 94, 95, 96].
’ СгН3-С&-С1 ’
СООН
Цефадроксил имеет широкий спектр антимикробного действия -активен в отношении грамположительных и грамотрицательных микроорганизмов. Как и все вышеописанные препараты, действует бактерицидно путем нарушения процесса восстановления мембран бактерий.
Синонимами препарата являются бидоцеф, цефадрил, дурасеф, ультрацеф и др.
Цефалоспорины II поколения
Цефалоспорины II поколения (цефуроксим, цефамандол, цефони-цид, цефоранид) характеризуются большей, по сравнению с цефалоспоринами I поколения, активностью в отношении грамотрицательных бактерий. Они имеют большую р-лактамазную устойчивость и более широкий спектр активности по отношению к грамотрицательным организмам, однако не оказывают заметного действия на энтерококки, Pseudomonas aeruginosa. Кроме того к цефалоспоринам II поколения относят и цефокситин, получаемый из цефамицина С. цефотетан, полу-синтетическое производное органомицина-G, а также цефаклор). Цефокситин и цефотетан применяются при смешанных аэробоанаэробных инфекциях. Их отличительной химической особенностью является наличие дополнительной метоксильной группы в положении С7 фрагмента аминоцефалоспорановой кислоты, а отличительной химической осо-оенностью цефаклора является еще и отсутствие характерной для всех Цефалоспоринов замещенной гидроксиметильной грзппы в положении Ц Фрагмента аминоцефалоспорановой кислоты.
- 613 -
Г лава 32
Цефуроксим (Cefuroxim)
Цефуроксим — карбамат (2)-моно(О-метилоксима) (6R,7R)-7-[2-(2-фурил)глиоксиламидо]-3-(гидроксиметил)-8-оксо-5-тиа-1-азабицик-ло[4.2.0]окт-2-ен-2-карбоновой кислоты (32.1 2 18) получают исходя из 2-аиетилфурана Окислением последнего азотистой кислотой получают 2-фурилглиоксалевую кислоту (32.1.2.15), взаимодействием которой с О-метилгидроксиламином получают соответствующий оксим — см«-2-.метоксимино-2-(2-фурил)уксу сную кислоту (32.1 2.16). которую далее взаимодействием с хлорангидридом щавелевой кислоты в диметилформамиде трансформируют в смешанный ангидрид и вводят во взаимодействие с бензгидриловым эфиром 7-аминоцефалоспорановой кислоты. Полученный продукт (32 1.2 17) подвергают ферментативному гидролизу в щелочной среде, при котором не затрагивается бензгидрильная зашита, а гидролизуется лишь уксуснокислая часть молекулы в положении С3 фрагмента аминоцефалоспорановой кислоты. Образовавшийся продукт со свободной гидроксиметильной группой (32.1.2.18) подвергают действию хлорсульфоизоцианата с промежуточным получением соо1ветствующего N-хлорсульфонилуретана (32.1.2.19), который гидролизуется водой до уретана (32.1.2.20). Наконец, снятием бенз-гидрильной зашиты трифторуксусной кислотой получают искомый цефуроксим (32.1.2.21) [97, 98, 99, 100, 101, 102. 103, 104, 105, 106, 107, 108, 109].
NaNO? НС
32 1 2 16
СООСН(СбНб'2
С ОСН
COOCHjCgHg 2
:гдС осн.
-614-
Антибиотики
СООН
32 1 2 21
Другим. кажущимся несколько более простым, способом получения цефуроксима является прямое ацилирование 7-амино-З-аминокарбо-нилоксимегил-З-цефем-4-карбоновой кислоты (32 1.2 22) — продукта, выделяемого из культуральной жидкости Stretomyces lactamdurans хлорангидридом сг/н-2-метоксимино-2-(2-фурил)уксусной кислоты, получаемым взаимодействием соответствующей кислоты с пятихлористым фосфором [110, 111].
соон
32 ’ .
’ РС16
СООН
32 1 2 21
Цефоруксим действует бактерицидно. Имеет широкий спектр антимикробного действия. Устойчив к действию р-лактамаз. Высокоактивен в отношении грамотрицательных микроорганизмов (кишечная и гемофильная палочки, сальмонеллы, шигеллы, энтеробактер, гонококки). Активен также в отношении грамположительных микроорганизмов (стафилококки, стрептококки). Не активен в отношении различных ви-'юз Pseudomonas, большинства штаммов энтерококков, многих штаммов Enterobacter cloacae, метициллинрезистентных стафилококков и Listeria H'onocvtogenes.
Препарат применяют при бактериальных инфекциях, вызванных чувствительными к препарату микроорганизмами. Это могут быть абдоминальные и гинекологические инфекции, сепсис и менингит, эндо-ьардиг. инфекции мочевыводящих и дыхательных путей, костей, суставов кожи и мягких тканей. Широко используется при пневмониях, а также бактериальном менингите у детей, при послеоперационных инфекционных осложнениях.
Синонимами препарата являются цефтин, зинацеф, курокси.м, ке-фокс и многие другие.
-615-
Глава 32
Цефамандол (Cefamandol)
Цефамандол — 7-О-манделамидо-3-[[( 1-метил-1 Н-те гразол-5-ил)-тио]метил]-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновую кислоту (32.1.2.25) получают из 7-аминоцефалоспорановой кислоты. Предварительно защитив свободную аминогруппу в 7-аминоцефалоспорановой кислоте формилированием муравьиной кислотой в присутствии уксусного ангидрида получают 7-формамидоце-фалоспорановую кислоту (32.1.2.23), ацетоксильную группу в которой замешают взаимодействием с 1-метил-1,2,3.4-тетразол-5-тио-лом, и после снятия N-формильной зашиты соляной кислотой получают 7-амино-3-( 1-метил-1,2,3-4-тетразол-5-ил)-тиомегил-3-це-фем-4-карбоновую кислоту (32.1.2.24). Взаимодействием последней со смешанным ангидридом, полученным из миндальной кислоты и фосгена получают искомый цефамандол (32.1.2.25) [112, ИЗ, 114, 115, 116, 117, 118, 119].
ПСООЬ
(СП3СС1/Э
COCH
2 HCI
Фармакологическое действие и показания к применению цефаман-дола аналогичны таковым цефуроксима и цефамандола.
Синонимами препарата являются мандокеф, кефандол, кефадол и многие дру! ие.
Цефоницид (Cefonicid)
Цефоницид — 7-О-манделамидо-3-[[(1-сульфометил-1//-тетразол-5-ил)тио]метил]-8-оксо-5-гиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоно-вая кислота (32.1.2.26) структурно родственна цефамандолу и отличайся наличием сульфогруппы при метильном заместителе тетра-
-616-
Антибиотики
зольного кольца. Препарат получают методами, аналогичными ме-юдам синтеза цефамандола [120, 121, 122, 123].
32 1.2 26
Фармакологическое действие, антимикробный спектр и показания к применению аналогичны таковым цефамандола.
Синонимом препарата является моноцид.
Цефоранид (Ceforanid)
Цефоранид — 7-[2-(а-амино-о-толил)ацетамидо]-3-[[(карбокси-метил-1//-тетразол-5-ил)-тио]метил]-8-оксо-5-тиа-1-азабицикло[4.2.0]-окт-2-сн-2-карбоновая кислота (32.1.2.27) также структурно родственна цефамандолу и отличается тем, что ацилирующей кислотой является о-аминометилфенилуксусная кислота, а также наличием карбоксильной группы при метильном заместителе тетразольного кольца. Препарат также получают методами, аналогичными методам синтеза цефамандола [124. 125, 126, 127, 128].
32 1 2 27
Фармакологическое действие, антимикробный спектр и показания к применению аналогичны таковым цефамандола.
Синонимом препарата является прецеф.
К препаратам II поколения цефалоспоринов относятся также цефок-ентин и цефотетан, являющиеся цефамицинами и принципиально отличающиеся от других цефалоспоринов наличием метоксильной группы
- 617-
Глава 32
в положении 7 цефалоспориновой системы, что сильно повышает их устойчивость по отношению к Р-лактамазам.
Цефокситин (Cefoxitin)
Цефокситин — карбамат 3-(гидроксиметил)-8-оксо-7-метокси-7-[(2-тиенилацетил)амино]-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоно-вой кислоты (32.1.2.30) предложено получать различными методами. При первом способе исходят из цефамицина С — 7p-(D-5-амино-5-карбоксивалер-амидо)-3-аминокарбонилгидроксиметил-7-ме-токси-З-цефем-4-карбоновой кислоты, в которой изначально присутствует метоксильная группа при С7, и задача получения искомого препарата сводится, собственно, к переамидированию.
СН3О
HOOC-CH-(CH2)3-CO-NH---Iх >
NH2 c/-n'^xch2-o-co-nh2
соон
цефамицин С
При втором способе синтез препарата начинают исходя из 7-аминоцефалоспорановой кислоты, в положение С7 которой необходимо внедрить метоксильную группу.
В одном из примеров синтеза цефокситина из цефамицина С первоначально осуществляют защиту свободной аминогруппы с помощью толуолсульфохлорида, и продукт выделяют в виде хорошо кристаллизующейся соли с дициклогексиламином (32.1.2.28). Далее с помощью метилхлорме!илового эфира этерифицируезся карбонильная группа в положении 2 цефалоспориновой системы, полученное соединение (32.1.2.29) подвергается действию 2-(2-тиенил)ацетилхлорида и далее с целью снятия эфирной защиты с карбоксильной функции — действию раствора хлористого водорода в метаноле с получением искомого цефокситина (32.1.2.30) [129].
1 TosCl
-618-
Антибиотики
Третий способ синтеза цефокситина исходит из 7-амино-цефалоспорановой кислоты, а точнее из ее бензгидрилового эфира (32 1 2.31). который получают предварительным тозилированием аминогруппы исходной 7-аминоцефалоспорановой кислоты, этерификацией карбоксильной группы дифенилдиазометаном и последующим снятием тозильной защиты.
Действием азотистой кислоты полученный продукт диазотируется с получением дифенилметилового эфира 7-диазоцефалоспорановой кислоты (32.1.2.32). Последовательным действием на полученное соединение азидом триэтиламмония в дихлорметане и далее бромазидом получают дифенилметиловый эфир 7-бром-7-азидоцефалоспорановой кислоты (32.1.2.33). Обработка последнего метанолом в присутствии борфторида серебра осу ществляют замещение атома брома, и получают дифенилметиловый эфир 7-.метокси-7-ази-доцефалоспорановой кислоты (32.1.2.34). Полученный азид восстанавливают водородом в присутствии окиси платины в качестве катализатора с получением дифенилметилового эфира 7-метокси-7-аминоцефалоспорановой кислоты (32.1.2.35). Ацилируя последнюю 2-(2-тиенил)ацетилхлоридом, получают дифенилметиловый эфир 7-метокси-7-[2-(2-тиенил)-ацетамидо]цефалоспора-новой кислоты (32.1.2.36), сложноэфирную часть которой гидролизуют с помощью трифторуксусной кислоты, и далее полученную кислоту действием бикарбоната калия переводят в калиевую соль (32.1.2.37). Полученный продукт далее гидролизуют ферментом ацетилэстеразой Cttrusi до калиевой соли 3-гидроксиметил-7-.меюкси-7-[2-(2-тиенил)-ацетамидо]-3-цефем-4-карбоновой кислоты (32.1.2.38). Способом уже ранее описанным, а именно первоначальным действием хлорсульфони-лизоцианатом и далее гидролизом водой, полученное соединение (32.1.2 38) трансформируют в искомый цефокситин (32.1.2.20) [130, 131, 132. 133, 134, 135. 136, 137, 138].
, k 1 tCjHshNHNj
\z-sx. NaNGj >iCi * at43
C^N''^!:XH;-O~CaC'43
СС.ЭН COOCH.Ceh-.^ COOCrttCsHsh
32' 2 31 32 1 2 32
-619-
Глава 32
COOCn CgHjn
2 KrlCO;
CH3C
CH3O
соон
32 1 2 30
Фармакологическое действие, антимикробный спектр и показания к применению аналогичны таковым цефамандола.
Синонимами препарата являются мефокситин, бетацеф, цефокси-нол, тифокс.
Цефотетан (Cefotetan)
Цефотетан — 7р-[(карбамоилкарбоксилатометилен)-1,3-дитиетан~2-ил]-карбоксамидо-7-метокси-3-(1-метилтетразол-5-ил)-тиометил-3-цефем-4-карбоповая кислота (32.1.2.43) получают по следующей схеме. Вначале осуществляют S-алкилирование тринатриевой соли 4-карбокси-3-гидрокси-5-меркаптоизотиазола (32.1.2.41) 7Р-бром-ацетамидо-7сх-метоксицефалоспорановой кислотой, которую получают по вышеприведенной схеме (32.1.2.31) -> (32.1.2 37) с той лишь разницей, что ацилирование на стадии (32.1.2.35) -> (32.1.2.36) осуществляют не 2-(2-тиенил)ацетилхлоридом. а бромангидридом бромуксусной кислоты.
Далее при взаимодействии полученного продукта (32.1.2.42) с 1-ме-тил-1.2,3,4-тетразол-5-тиолом в присутствии бикарбоната натрия одновременно с ожидаемым замещением в условиях реакции происходит
- 620 -
Антибиотики
и перегруппировка с раскрытием изотиазоль но го кольца и получением производного карбамоилкарбокси.татометилен-1,3-литиетана — цефо-гетена (32.1.2.43) [139, 140].
Фармакологическое действие, антимикробный спектр и показания к применению иефотетана аналогичны таковым у цефамандола.
Синонимами препарата являются цефотен, апатеф, цепан, дарвилен и др.
Цефаклор (Cefaclor)
Цефаклор — (6К,7К)-7-[(К)-2-амино-2-фенилацетамидо]-3-хлор-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2 карбоновая кислота (32.1.2.48) принципиально отличается от рассматриваемых и применяющихся в медицинской практике антибиотиков цефалоспоринового ряда всех поколений наличием вместо замещенной метиленовой группы в положении 3 цефалоспораиовой кислоты атома хлора. Синтез препарата осуществляется исходя из наиболее доступного антибиотика этого ряда — цефалотина (32.1.2.1), карбоксильную группу которого защищают этерификацией взаимодействием с 4-нитробензилбромидом в триэтиламине с получением 4 нитробензилового эфира 7-(2-тиенилацетамидо)-цефалоспорано-вой кислоты (32.1.2.40). Взаимодействием последней с этилксанто-тенатом калия замещают ацетоксильную группу в положении 3 це-фалоспорановой системы, и получают соответствующее S-производное (32.1.2.41). Восстановлением последнего цинком в муравьиной кислоте продукт десулъфируют, и получают 4-нитробен-зиловый эфир 3-экзо-метилен-7-(2тиенилацетамидо)-цефем-4-кар-боновой ислоты (32.1.2.42). Экэометиленовую группу окисляют озоном и образовавшееся дикарбонильное производное переводят
- 621 -
Глава 32
в енольную форму (32.1.2.43) действием сернистого ангидрида. Далее гидроксильную группу действием хлористого тионила в диме-тилформамиде замещают на атом хлора с получением 4-нитро-бензилового эфира 3-хлор-7-(2-тиенилацетамидо)-3-цефем-4-карбо-новой кислоты (32,1.2.44) Полученный продукт подвергают дезацилированию действием смеси пиридина с пятихлористым фосфором в изобутаноле с получением гидрохлорида 4-нитро-бензплового эфира 7-амино-3-хлор-3-цефем-4-карбоновой кислоты (32.1.2.45). Последний ацилируют N-защишенным производным фенилглицина — М-(от/тет-бутоксикарбонил)-П-а-фенилглицином в присутствии К-этоксикарбонил-2-этокси-1,2-дигидрохинолина в тетрагидрофуране с получением продукта (32.1.2.46). Трет-бутоксикарбонильную защиту последнего удаляют кипячением ацетонитриле в присутствии п-толуолсульфокислоты. Наконец, восстановлением водородом с полученного тозилата (32.1.2.47) снимают 4-нитробензильную защитную группу с использованием цинка и соляной кислоты в димегилформамиде с получением цефаклора (32.1.2.48) [141, 142, 143].
С ОСС Н /2 С>2
СООСН2С5Н4НО2
Zn нсоон
1 DMF 1 SOCI2 2 HCI
СООСм2СбН4МО2
COOCH2C6H4NO2
COOH
C6H5“CH“NHCOO(CH3)3
Cl
COOCH2C6H4NO^
COQCH2C6H4NO2 .
32 1 2 45
-622-
Антибиотики
соон
32 1 2 48
Фармакологическое действие, антимикробный спектр и показания к применению нефаклора аналогичны таковым у цефамандола.
Синонимами препарата являются панорал, альфатил, дистаклор. па-нацеф, цеклор и др.
Цефалоспорины III поколения
Цефалоспорины III поколения (цефотаксим, цефтизоксим, цефтриаксон. цефтазидим, цефоперазон и моксалактам) отличаются высокой ан1имикробной активностью против энтеробактерий, в том числе устойчивых к другим антибиотикам. Они относительно более устойчивы к гидролизу р-лактамазами и имеют самый широкий спектр грамотри-цателыюй активности. Для них характерна повышенная антибиотическая активность в отношении Escherichia coh, ряда штаммов Proteus Enterobcter. Вместе с тем они обладают умеренной активностью в отношении стафилококков и используются при полирезистентных гра-могрицательных инфекциях при лечении бактериального менингита и гонореи. Следует отметить, что моксалактам содержит дигидрооксазиновое кольцо вместо общего для всех цефалоспоринов дигидротиазинового кольца. Таким образом, это не цефалоспорин, не цефамицин и не пенициллин, однако по спектру фармакологического действия является соединением, родственным всем трем перечисленным выше антибиотикам. и классифицируется как цефалоспорин III поколения. С химической точки зрения их объединяет то, что практически все они являются производными 7-аминоцефалоспориновой кислоты, ацилированной по аминогруппе 2-(2-амино-4-тиазолил)-2-метоксиминоуксусной кислотой. По фармакологическому действию цефалоспорины III поколения. в основном, отличаются друг от друга фармакокинетическими особенностями, а также некоторыми признаками по отношению к Pseudomonas aeruginosa и Staphylococcus aureus.
Цефотаксим (Cefotaxim)
Цефотаксим — ct-O-метилоксим ацетата (6R.7R) -7-[2-(2-амино-4-зиазо.тил)глиоксилами-до]-3-(гидроксиметил)-8-оксо-5-тиа-1-азаби-
-623-
Глава 32
цикло[4.2.0]окт-2-ен-2-карбоновой кислоты (32.1.2.56) получают ацилированием 7-аминоцефалоспорановой кислоты в присутствии дициклогексилкабодиимида 2-(2-амино-4-тиазолил)-2-метоксими-ноуксусной кислотой, защищенной по аминогруппе тритильной зашитой (32.1.2.54) После снятия тритильной зашиты с образовавшегося продукта (32.1.2.55) разбавленной муравьиной кислотой получают искомый цефотаксим (32.1.2.56). Необходимый для данного синтеза, а также для синтеза ряда других антибиотиков цефалоспоринового ряда этиловый эфир 2-(2-амино-4-тиазолил)-2-мего-ксиминоу ксусной кислоты получают исходя и ацетоуксусного эфира. Нитрозированием последнего азотистой кислотой получают изонитрозоацетоуксусный эфир (32.1.2.49), О-метилированием гидроксильной группы которого диметилсульфатом в присутствии поташа получают этиловый эфир 2-ацетил-2-метоксиминоуксусной кислоты (32.1.2.50). Бромируя полученный метиловый эфир оксима молекулярным бромом в хлористом метилене в присутствии п-толуолсульфокислоты получают 4-бром-2-метоксиминоацетоук-сусный эфир (32.1 2.51). Взаимодействием последнего с гиомочеви-ной по классической схеме получения тиазолов из о-бром-карбонильных соединений и тиоамидов получают этиловый эфир 2-(2-амино-4-тиазолил)-2-мегоксиминоу ксусной кислоты (32.1.2.52) Взаимодействием последнего с трифенилхлорметаном в присутствии триэтиламина осуществляют тритильную защиту аминогруппы с получением этилового эфира 2-(2-тритиламино-4-тиазолил)-2-ме-токсиминоуксусной кислоты (32.1.2.53). который гидролизуют в кислоту (32.1.2.54) с помощью гидроксида натрия. Полученную кислоту (32.1.2.54), как уже было отмечено, используют для ацилирования 7-аминоцефалоспорановой кислоты в присутствии дицик-логексилкарбодиимида с получением тритилированного цефаток-сима - - а-О-метилоксима ацетата 7-[2-(2-тритиламино-4-тиазолил)-глиоксиламидо]-3-(гидроксиметил)-8-оксо-5-тиа-1-азабпцикло[4.2.0]-окт-2-ен-2-карбоновой кислоты (32 1.2.55). Наконец, снятием с синтезированного продукта (32.1.2.55) тритильной зашиты с помощью разбавленной муравьиной кислоты получают цефотаксим (32.1.2.56) [144, 145, 146, 147, 148].
сн3АД-2н5 NOH N-OCH-
32 ” 2 49 32 1 2 50
- 624 -
Антибиотики
Цефотаксим обладает широким спектром антимикробного действия Действует бактерицидно. Высокоактивен в отношении грамотрицательных микроорганизмов, устойчивых к другим антибиотикам, — £ Coh, Citobacter, Proteus mirabilis, Proteus indole, Providenaa, Klebsiella, Serratia. некоторые штаммы Pseudomonas, Haemophilus mfluenzale Цефотаксим менее активен в отношении стрептококков, пневмококков, менингококков, гонококков, бактероидов. Устойчив по отношению к большинству Р-лактамаз грамположительных и грамотрицательных микроорганизмов.
Препарат применяют при бактериальных инфекциях тяжелого течения, вызванных чувствительными к препарату микроорганизмами, — перитоните, сепсисе, абдоминальных инфекциях, инфекциях органов малою таза, нижних дыхательных и мочевыводящих путей, костей, сус-1авов, кожи, мягких тканей, инфицированных ранах и ожогах.
Синонимами препарата являются клафоран, заривиз и др.
Цефтизоксим (Ceftizoxim)
Цефтизоксим — а-О-метилоксим (6К,7К)-7-[2-(2-амино-4-тиазо-лил)глиоксиламидо]-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-кар-боновая кислот)' (32.1.2.64) получают по схеме синтеза приводимой ниже, которая исходит из 4-нитробензилового эфира З-гидрокси-7-(2-фенилацета.мидо)-3-цефем-4-карбоновой кислоты (32 1.2.57), по-лучаемой с использованием набора методов, применяемых при синтезе цефаклора (32.1.2.48). Восстановлением двойной связи при С3-С4 в исходном 4-нитробензиловом эфире 3-гидрокси-7-(2-фе-нилацетамидо)-3-цефем-4-карбоновой кислоты (32.1.2.57) борги дридом натрия в метаноле пол) чают 4-нитробензиловый эфир 3-гидрокеи-7-(2-фенилацетамидо)-3-цефем-4-карбоновой кислоты
-625-
Г лава 32
(32 1 2.58), гидроксильную группу в котором ацилируют ангидридом уксусной кислоты в пиридине с получением ацетата (32.1.2.59). Действуя на последний триэтила.мином. отщепляют молекулу уксусной кислоты с получением 4-нитробензилового эфира 7-(2-фе-нилацегамидо)-3-цефем-4-карбоновой кислоты (32.1.2.60). Действием на последний пятихлористым фосфором в пиридине и последующим метанолизом дезацилирую! амидный фрагмент молекулы, и получают 4-нитробензиловый эфир 7-амино-3-цефем-4-карбо-новой кислоты (32 1 2.61). Предварительным силилированием аминогруппы последнего триметилсилилацетамидом и дальнейшим ацилированием хлорангидридом 2-(2-формамидо-4-тиазолил)-2-ме-токсиминоуксу сной кислоты, получаемого взаимодействием с хлорокисью фосфора в диметилформамиде непосредственно в условиях реакции, получают 4-нитробензиловый эфир а-О-метилоксима 7-[2-(2-формамамидо-4-тиазолил)глиоксиламидо]-8-оксо-5-тиа-1-аза-бицикло[4 2.0]окт-2-ен-2-карбоновой кислоты (32 1.2.62). Восстановлением последнего водородом с использованием в качестве катализатора палладия на угле снимают 4-нитробензильную защиту с карбоксильной группы и получают кислоту (32.1.2 63). Наконец, гидролизом формамидной части молекулы хлористым водородом в метаноле получают искомый цефтизоксим (32.1.2.64) [149,150,151].
СООСНгСбЩНО?
32 1 2 57
(СН3СО)20( Ру
COOCH2CsH4NO2
COOCH2C8H4NO2
32 1 2 58
1 PCI5 / Ру
COOCH2CSH4NO2
32 1 2 60
1 CH,CONHSi(CH3?3
Pd-C
-626-
Антибиотики
32 1 2 63
на i сн3он
32 1 2 64
Цефгизоксим применяют при бактериальных инфекциях нижних дыхательных путей, инфекциях мочевыводящих путей, костей, суставов, кожи, мягких тканей, абдоминальных инфекциях.
Синонимами препарата являются цефтикс и эпосерин.
Цефтриаксон (Ceftriaxon)
Цеф1риаксон — 7-[[(2-амино-4-тиазолил)-2-(2)-(метоксимино)аце-т ил]амино]-8-оксо-3-[[(1,2,5,6-тетрагидро-2-метил-5,6-диоксо-1,2,4-триазин-3-ил)тио]метил]-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбо-новая кислота (32 1.2.72) получают ацилированием 7-амино-З-[[(2 5-дигидро-6-гидрокси-2-метил-1,2,4-триазин-5-он-3-ил)тио]ме-1ип]-3-цефем-4-карбоновой кислоты (32.1 2.70), защищенной по аминогруппе хлорацетильной группой, хлорангидридом 2-(4-тиазо-1ил)-2-метоксиминоуксусной кислоты, а именно 2-(2-хлорацетами-де-4-гиазолил)-2-метоксиминоацетилхлоридом (32.1.2.67). Последний получают исходя из этилового эфира 2-(2-амино-4-тиазолил)-2-метоксиминоуксусной кислоты (32.1.2.52), аминную группу которо-I о защищают ацилированием хлорангидридом хлоруксусной кислоты в диме гилацет амиде с получением этилового эфира 2-(2-хлор-ацетамидо-4-тиазолил)-2-метоксиминоуксусной кислоты (32.1.2.65). Гидролизом сложноэфирной части последнего гидроокисью калия до кислоты (32 1.2 66) и ее дальнейшим взаимодействием с пяти-хлеристым фосфором в присутствии триэтиламина получают 2-(2-хлорацегамидо-4-тиазолил)-2-метоксиминоацетилхлорид (32.1.2.67). Параллельно осуществляется синтез 7-амино-3-[[(2,5-дигидро-6-i и ’рокси-2-метил-1,2.4-триазин-5-он-3-ил)тио]метил]-3-цефем-4-кар-боновой кислоты (32 1.2.70) С этой целью метилгидразин вводится во в ;аимодействие с тиоцианатом калия с получением 1-амино-1-метилгио.мочевины (32.1.2.68), которую вводят в реакцию с диме-тилоксалаюм в присутствии метилата натрия с получением продукта геюроциклизации — 2,5-дигидро-6-гидрокси-2-метил-3-мерка-
-627 -
Глава 32
пто-1,2,4-триазин-5-она (32.1.2.69). Взаимодействием последнего с 7-аминоцефалоспорановой кислотой осуществляют замещение апетоксильной ipynnw последней на 2,5-дигидро-6-гидрокси-2-метил-1,2,4-триазин-5-он-3-тиольную с получением 7-амино-З-[[(2,5-дигидро-6-гидрокси-2-метил-1,2,4-триазин-5-он-3-ил)тио]метил]-З-цефем-4-карбоновой кислоты (32.1.2.70). которую ацилируют ранее полученным хлорангидридом (32.1.2.67) в тетрагидроферане в присутствии гидроокиси натрия с получением N-хлорацетил-замещенного производного искомого продукта (32.1.2.71). Снятие хлорацетильной защиты с последнего осуществляют: последовательным взаимодействием продукта (32.1.2.71) с тиомочевиной в присутствии бикарбоната натрия, при котором получают новое тиазольное производное. Дальнейшим расщеплением образовавшегося вторичного гетероароматического амина муравьиной кислотой полу'чают цефтриаксон (32.1.2.72) [152, 153, 154, 155, 156].
ССОСНз
s I
JI Crt СООСЬз СНэО№
CH3-NH-4rt2 + -SCN --------*• НгМ N' ------------------------
NH2
32 1 2 68
32 1 2 70
-628-
Антибиотики
Препарат обладает широким спектром антимикробного действия, который включает большинство клинически значимых микроорганизмов: грамположительных, грамотрицательных, аэробных и анаэробных, в том числе синегнойную палочку. Устойчив по отношению к большинству р-лактамаз грамположительных и грамотрицательных бактерий. Применяют при перитоните, сепсисе, менингите, холангите, эмпиеме желчного пузыря, пневмонии, абсцессе легкого, пиелонефрите, инфекциях костей, суставов, кожи, мягких тканей, абдоминальных и гинекологических инфекциях, инфицированных ранах и ожогах.
Основным синонимом препарата является ропефин.
Цефтазидим (Ceftazidim)
Цефтазидим — внутренняя соль 1-[[7-[[(2-амино-4-тиазолпл)[(1-карбокси-1-метилэтокси)имино]ацетил]амино]-2-карбокси-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-3-ил]метил]пиридин-2-карбоновой кислоты (32.1.2.82). Как и в случае синтеза цефтриаксона, получение цефтазидима предусматривает предварительный синтеза двух составляющих. В качестве цефалоспоринового фрагмента используется дигидрохлорид 7-амино-3-(1-пиридинометил)цеф-3-ен-кар-боновой кислоты (32.1.2.80), а ацильной — модифицированная структура (32.1,2.77), представляющая собой на сей раз не производное 2-(2-амино-4-тиазолил)-2-метоксиминоуксусной кислоты, а производное 2-(2-амино-4-тиазолил)-2-(2-гаре/и-бу токсикарбонил-2-пропоксимино)уксусной кислоты, которую получают по следующей схеме. Нитрозированием ацетоуксусного эфира получают изо-нитрозоацетоуксусный эфир (32.1.2.49), который подвергают хлорированию хлористым сульфурилом в хлористом метилене с получением 4-хлор-2-гидроксиминоацетоуксусного эфира (32.1.2.73). Взаимодействием последнего с тимочевиной по классической схеме получения тиазолов взаимодействия а-галогнкарбонильных соединений с тиоамидами получают этиловый эфир (7.)-2-(2-аминоти-азо.т-4-ил)-2-гидроксиминоуксусной кислоты (32.1.2.74). Аминную группу в последнем защищают взаимодействием с трифенилхлор-метаном в диметилформамиде в присутствии триэтиламина с получением этилового эфира (2)-2-(2-тритиламино-тиазол-4-ил)-2-гид-роксиминоуксусной кислоты (32.1.2.75). Гидроксильную группу в полученном соединении алкилируют /л/яте-бутиловым эфиром а-бромизомасляной кислоты в диметилсульфоксиде в присутствии поташа с получением этилового эфира (2)-2-2-(щ/?еш-оутокси-карбонил-проп-2-окси.мино-2-(2-тритиламинотиазол-4-ил) уксусной кислоты (32.1.2.76). Этоксикарбонильую группу в последнем гид
- 629-
Глава 32
ролизуют гидроокисью натрия, и после подкисления реакционной смеси выделяют соответствующую кислоту (32.1.2.77), которую да-пее взаимодействием с пятихлористым фосфором переводят в хлорангидрид (32.1 2.78) для использования в качестве ацилирующего реагента
Второй небходимый фрагмент — дихлоргидрат 7-амино-3-(1-пиридинометил)цеф-3-ен-карбоновой кислоты (32.1.2.80) получают из цефалоридина (32.1.2.79) — цефалоспоринового антибиотика, самостоятельно использующегося в медицине и получающегося в виде внутренней соли взаимодействием цефалотина (32.1.2.1) с пиридином путем замещения в нем ацетоксильной группы на пиридиновую. Обрабатывая цефалоридин сначала триметил-хлорсиланом в присутствии диметила-нилина и далее пятихлористым фосфором и последующим взаимодействием с 1,3-бутандиолом, осуществляют дезацилирование, получая искомую 7-амино-3-(1-пиридинометил)цеф-3-ен-карбоновую кислоту (32.1.2.801. Последнюю ацилируют ранее полученным хлорангидридом (32.1.2.78) с получением продукта (32.1.2.81), обработкой которого смесью муравьиной и соляной кислот снимают обе защитные группы (трифенилметильную и mpem-бутильную) с получением цефтазидина (32.1.2 82) в виде дигидрохлорида [157, 158, 159, 160, 161, 162].
- 630-
Антибиотики
32 1 2 80
Как и большинство описанных выше антибиотиков цефалоспоринового ряда III поколения, цефтазидим обладает широким спектром антимикробного действия, который включает большинство клинически значимых микроорганизмов: грамположительных, грамотрицательных, аэробных и анаэробных. Устойчив по отношению к большинству Р-лактамаз грамположительных и грамотрицательных бактерий.
Препарат применяют при лечении большинства бактериальных инфекций тяжелого течения.
Синонимами препарата являются фортум, цефтим, стацеф, тази-пеф.
Цефоперазон (Cefoperazon)
Цефоперазон — (6К,7К)-7-[(К)-2-(4-этил-2,3-диоксо-1-пиперазин-карбоксамидо)-2-(н-гидроксифенил)ацетамидо]-3-[[(1-метил-1//-тет-разол-5-ил)тио]метил]-8-оксо-5-тиа-1-азабицикло[4.2.0]окт-2-ен-2-карбоновая кислота (32.1.2.84) получается ацилированием 7-амино-3-( 1-мет ил-1,2.3,4-тетразол-5-ил)-тиометил-3-цефем-4-карбоновой кислоты (32.1.2.24) смешанным ангидридом, полученным из хлор-} сольного эфира и а-(4-этилпиперазин-2,3-дион-1-карбониламино)-4-гидроксифенилуксусной кислоты (32.1.2.83), в свою очередь получаемой из 4-этилпиперазин-2,3-дион-1-карбоновой кислоты (32.1.1 29) и натриевой соли 4-гидроксифенилглицина [163, 164, 165, 166, 167, 168].
- 631 -
Глава 32
1ССООС2Н5 DMF C6H5N CH-jj?
Цефоперазон также обладает широким спектром антимикробного действия, который включает большинство клинически значимых микроорганизмов: грамположительных, грамотрицательных, аэробных и анаэробных. Устойчив по- отношению к большинству р-лактамаз грамположительных и грамотрицательных бактерий.
Цефоперазон применяют при бактериальных инфекциях нижних дыхательных, мочевыводящих и половых путей, инфекциях костей, суставов, кожи, мягких тканей, абдоминальных и гинекологических инфекциях.
Синонимами препарата являются цефазон, цефобид, цефобис и многие другие.
Моксалактам (Moxalactam)
Моксалактам — 7Р[2-карбокси-2-(4-гидроксифенил)ацетамидо]-7а-метокси-3-(1 -метилтетр азол-5-ил)-тиометил-1-окса-детиа-3-цефем-4-карбоновая кислота (32.1.2.98) получается многостадийным синтезом исходящим из б-АПК, ацилированием которой бензоилхло-ридом в присутствии триэтиламина получают 6-бензоилпенициллин (32 1.2.85). Карбоксильную группу последнего защищают взаимодействием с дифенилдиазометаном и получением 3-дифенил-метилового эфира 6-бензоилпенициллина (32.1.2.86). Окисление последнего молекулярным хлором в щелочных условиях получают S-оксид 3-дифенилметилового эфира 6-бензоилпениниллина
- 632-
Антибиотики
(32.1.2.87). При взаимодействии последнего с трифенилфосфином вместо ожидаемого раскисления сульфоксида в сульфид происходит раскрытие тиазинового цикла с выбросом атома серы, одновременным его замещением кислородом и последующим образованием циклического иминоэфира (32.1.2.88). Хлорированием двойной связи последнего хлором и последующей обработкой полученного ди-хлорпроизводного бикарбонатом натрия получается хлораллильное производное (32.1.2.89), взаимодействием которого с йодистым калием получают продукт замещения хлора на йод — йодаллильное производное (32.1.2.90), и, наконец, гидролизом которого в диметилсульфоксиде в присутствии оксида одновалентной меди получают соответсвующий аллиловый спирт (32.1.2.91). Нагреванием последнего в присутствии эфирата трехфтористого бора продукт рециклизуется с образованием оксазинового кольца и обратной трансформацией циклического иминоэфира в амидную форму (32.1.2.92). Подвергая полученный продукт с экзоциклической двойной связью хлорированию и дальнейшей обработкой продукта хлорирования 1,7-диазабицикло-[4.5.0]ундец-6-еном в качестве основания, получают 3-хлорметильное производное (32.1.2.93). Действуя на последний /ире/и-бутилгипохлоритом, очевидно с целью получения N-хлорпроизводного, и далее метилатом лития, после подкисления и обработки тиосульфатом натрия получают дифенилметиловый эфир 7р-бензоиламидо-7а-метокси-3-(хлорметилил)-1-окса-детиа-З-цефем-4-карбоновой кислоты (32.1.2.94). Вводя последний во взаимодействие с натриевой солью 5-меркапто-1-метилтетразола, получают дифенилметиловый эфир 7Р-бензоиламидо-7а-метокси-3-(1-метилтетразол-5-ил)-тиометил -1-окса-детиа-З-цефем-4-карбоновой кислоты (32.1.2.95). Дебензоилированием последнего последовательной обработкой пятихлористым фосфором в пиридине и далее метанолом и диэтиламином получают дифенилметиловый эфир 7Р-амино-7а-метокси-3-(1-метилтетразол-5-ил)-тиометил-1-окса-детиа-3-цефем-4-карбоновой кислоты (32.1.2.96). Ацилируя последний смешанным ангидридом, получаемым из монодифенилметилового эфира (4-гидроксифенил)малоновой кислоты и окса-лилхлорида в присутствии триэтиламина, получают ди-дифенил-метиловый эфир по обеим карбоксильным группам искомого продукта (32.1.2.97). Наконец, снятием указанной дифенилметильной защиты с обеих карбоксильных групп кипячением с трифторуксусной кислотой в толуоле получают искомый моксалактам (32.1 2.98) [168, 169, 170, 171, 172, 173].
- 633-
Глава 32
OMSO I HjO CgjQ
32 2 90
H =h2-OH SF» 'c^4. ,гучн=
COOCHfCeHsn
32 ‘ 2 91
СООСН(СбН5)2
32 1 2 92
1 (СНз)зОС11 CH-OL!
COOCH(C8H5)2
J2 I 2 93
HS
CH3
COOCH(CSH5)2 32 1 2 94
' PC 5 Pv 2 Ch3QH
CCOCH'C6HS/2 <1пз
32 t £ 96
~V J— CH-COOK COOCHiCeHsJj
(СгНл!гМН Ci—CO-CO-Cl
CF3COOH
Как уже было отмечено и как видно из схемы синтеза, моксалактам содержит дигидрооксазиновое кольцо вместо общего для всех цефалоспоринов дигидротиазинового кольца, и, таким образом, это соединение формально не может быть причислено ни к цефалоспоринам, ни к це-фамицинам, ни к пенициллинам, однако по спектру фармакологическо
- 634 -
Ан тибиотики
го действия оно, являясь соединением, родственным всем трем перечисленным выше антибиотикам, классифицируется как цефалоспорин III поколения.
Моксалактам устойчив к действию Р-лактамаз — пенициллиназ и цефалоспориназ, продуцируемых грамотрицательными и грамполо-жительными бактериями. Многие штаммы ряда микроорганизмов, обладающие множественной устойчивостью к другим антибиотикам — полисинтетическим пенициллинам, цефалоспоринам и аминогликозидам, чувствительны к моксалактаму.
Препарат применяют при инфекциях органов дыхания, мочевых путей, брюшной полости, гинекологических инфекциях, инфекциях костей, суставов, кожи, мягких тканей, при гонорее.
Синонимами препарата являются латамоксеф, фестамоксин, мокса-цеф, моксам и многие другие.
Цефалоспорины IV поколения
Цефалоспорины IV поколения (цефепим, цефпиром) обладают еще более широким спектром действия и активны против большинства грамположительных и грамотрицательных аэробных и анаэробных микроорганизмов. По спектру действия они похожи на цефалоспорины III поколения — цефотаксим и цефтриаксон, химическим гибридом которых формально они являются, сочетая в себе аминотиазолилметоксииминоацетамидо групп) в положении 7 и четвертичный атом азота, входящий в гетероциклическую систему в положении 3, что придает их молекулам свойства цвиттериона, однако они превосходят по активности препараты III поколения против Pseudomonas aeroginosa, Staphylococcus aureus, в том числе некоторых метициллинорезистентных штаммов. В терапевтически значимых концентрациях они действуют на Streptococcus faecalis Препараты активны против Enterobacter cloacae, Klebsiella pneumoniae. Они очень быстро транспортируются в пери-плазматическое пространство, и к ним практически не обнаружена резистентность чувствительных штаммов энтеробактерий.
Цефепим (Cefepim)
Цефепим — хлорид {6В.-[6а,7|3(2)]}-1-[(7-{[(2-амино-4-тиазолил)-(метоксимино)ацетил]-амино}-2-карбокси-8-оксо-5-тиа-1-азабицик-ло[4.2.0]окт-2-ен-3-ил)метил-1-метил]пирролидиния (32.1.2.99) получают комбинацией методов, описанных для синтеза цефалоспоринов III поколения, и, в частности, цефалоридина (32.1.2.79) и цефтазидима (32.1.2 82) [174, 175, 176].
-635-
Глава 32
32 1 2 99
Цефепим применяется при бактериальных инфекциях, вызванных чувствительными к препарату микроорганизмами, при септицемии, бактериемии, осложненных инфекциях верхних и нижних отделов мочевыделительной системы, пневмонии, абсцессах легкого, эмпиемы плевры, лихорадке у больных с нейтропенией, инфекционных поражениях кожи и мягких тканей.
Синонимами препарата являются максипим, цепим, цепимекс и др.
Цефпиром (Cefpirom)
Цефпиром — {6К-[6а,7р(2)]}-1-[(7-{[(2-амино-4-тиазолил)-(ме
токсимино)ацетил]амино}-2-карбокси-8-оксо-5-тиа-1-азабицикло-[4.2.0]окт-2-ен-3-ил)метил-1-метил]пирролидиния хлорид (32.1.2.100) также получают методами, описанными для синтеза цефалоспоринов III поколения, и в частности цефтазидима (32.1.2.82) [177, 178].
nh2 s^n
32 1 2 100
Препарат устойчив по отношению к широкому спектру р-лактамаз. Спектр активности аналогичен таковому цефалоспорина III поколения цефотаксима (32.1.2.56), однако цефпиром более активен по отношению к некоторым стафилококкам, энтерококкам, а также некоторым энтеро
- 636 -
Антибиотики
бактериям.
Синонимами препарата являются цефрон, цедиксен и др.
32.1.3 . Пенемы и карбапенемы
Кроме пенициллинов и цефалоспоринов, образуемых мицеллиаль-ными грибами, к группе Р-лактамных антибиотиков относятся и вещества, вырабатываемые стрептомицетами и называемые пенемами. Этот класс антибиотиков химически сходен с производными пенициллановой кислоты, однако отличается от них наличием внутрикольцевой, сопряженной с карбоксильной группой, двойной связи, что придает им определенное сходство и с цефалоспорановой кислотой, наличием вместо двух метильных групп во втором положении пятичленного кольца S-алкильного заместителя в и отсутствием аминогруппы в шестом положении.
Пенемы SCH-29482 (R=CH2CH3) и SCH-34343 (R=CH2CH2OCONH2) пока еще не нашли применения в медицине.
СНз
Представителями другого класса антибиотиков являются карбапенемы, отличающиеся от пенемов отсутствием атома серы в пенемовом кольце:
• тиенамицин (R = CH2CH2NH2),
• оливановая кислота (R = СН = CHNH2),
• имипенем (R = CH2CH2NHC = NH).
СНз
о соон
Имипенем (Imipenem)
Имипенем — [^-[5а,6а(К*)]]-6-(1-гидроксиэтпл)-3-[[2-[(имино-
метил)амино]этил]тио]-7-оксо-1-азабицикло[3.2.0]гепт-2-ен-2-карбо-
новая кислота (32.1.3 1) является единственным карбапенемом, ис-
- 637 -
Г лава 32
пользующимся в настоящее время в клиниках. Его получают из (иенамицина, выделенного из Streptomyces cattleya взаимодействием с гидрохлоридом метилового эфира иминомуравьиной кислоты [179, 180, 181. 182].
NH
х
Н ОСг!3
О СООН
32 1 3 1
В отличие от пенициллинов и цефалоспоринов, имеющих боковую аминоацильную группу, присоединенную к Р-лактамному кольцу, в имипенеме имеется гидроксиэтильная боковая цепь. У соединения наблюдается значительная резистентность к гидролизу Р-лактамазами, очевидно благодаря транс-конфигурации боковой цепи, в то время как у пенициллинов и цефалоспоринов боковая цепь имеет тщс-конфи-гурацию.
Имипенем обладает широким спектром антимикробного действия, который включает большинство клинически значимых микроорганизмов: грамположительных. грамотрицательных, аэробных и анаэробных. Усюйчив по отношению к большинству Р-лактамаз грамположительных и грамотрицательных бактерий. Применяют при бактериальных инфекциях нижних дыхательных, мочевыводящих и половых путей, инфекциях костей, суставов, кожи, мягких тканей, интраабдоминальных и гинекологических инфекциях, бактериальных септицемиях, эндокардитах.
Имипенем подвергается ферментной инактивации в почках. Чтобы обойти эту проблему, он используется в соотношении 1:1 в комбинации с циласгатином — натриевой солью [R-[R’.S’-(Z)]]-7-[(2-aMHHO-2-чарбоксиэтил)тио]-2-(2.2-диметилциклопропил)карбонил]ами-но]-2-ге-теновой кислоты (32.1.3.6), которая ингибирует метаболизм имипенема в почках. Эта комбинация двух соединений и применяется в медицине под названием примаксин.
Циластатин (Cilastatin)
Циластатин — (2)-7-[2-амино-2-карбэтоксиэтил)тио]-2-[[(2,2-диме-гилцикло-пропил)карбонил]амино]-2-гептеновую кислоту (32.1.3.6) получают исходя из этилового эфира 1,3-ди-тиан-2-карбоновой кислоты (защищенной по альдегидной группе 1,3-пропандитиолом этилового эфира глиоксиловой кислоты), который алкилируют 1,5-
-638 -
Анти биотики
дибромпентаном в присутствии амида натрия в смеси растворителей диметилформамидтолуол с получением этилового эфира 7-бром-2-[2-(1,3-дитиано)]гептановой кислоты (32.1.3.2). Окислительный гидролиз последнего N-бромсукцинимидом в смеси растворителей ацетонитрил-вода приводит к получению этилового эфира 7-бром-а-кетогептановой кислоты (32.1.3.3). Кислотным гидролизом последнего бромистым водородом в уксусной кислоте получают 7-бром-а-кетогептановую кислоту' (32,1.3.4). Последнюю вводят во взаимодействие с амидом 2,2-диметилциклопропанкар-боновой кислоты с получением соответствующего енамида — (Z)-7-бром-2-(2.2-диметилциклопропанкарбоксамидо)-2-гептеновой кислоты (32.1.3.5). Полученный продукт используют для S-алкилирования L-пистеина, в результате чего получают искомый циласта-тин (32.1.3.6) [183, 184].
S СООС2Н5
Вг—,Сг2-5”Вг * Д
s /снгъ-ег
Cr,3CN I HjO
и
Вт—(CHjfe-c-coocjHj
32 1 з з
О 'C-M2J5-C-COOH
32 ' 3 4
СН3
я А"СНз НгМ-С—М
н
CH2-SH H2N--C"H соон
СН2“S—(.СНгИ h2n--c-h
СООН
Н СООН СНз
А~снз
о Н
32 1 3.6
Циластатин используют при лечении заболеваний, вызванных по-лирезистентными грамотрицательными микроорганизмами, и серьезных смешанных инфекциях, включая заражение бактериями Staphylococcus aureus. Ввиду его мощной активности против анаэробных бактерий имипенем эффективен при монотерапии интраабдоминальных инфекций. Применяется при инфекционных заболеваниях нижних дыхатель
-639-
Глава 32
ных путей, мочеполового тракта, гинекологических инфекциях, бактериальных септицемиях, инфекциях костей, кожи и т. д,
32.1.4 . Монобактамы
Некоторые виды микроорганизмов, в частности Chromobacterium violaceum, в процессе жизнедеятельности могут синтезировать своеобразные Р-лактамные антибиотики, имеющие моноциклические структуры — монобактамы. Примером таких монобактамов являются нокарди-цины, в частности нокардицин А.
он
.-. N О
/-—\ н и
НООС—СН—СН2—СН2—О-< A—C-C-HN. ОН '-'
о сн—с он
соон'--'
нокардицин А
Первым полностью синтетическим моноциклическим Р-лактамным антибиотиком является азтреонам. Антимикробная активность этого препарата в основном проявляется в отношении широкого спектра аэробных грамотрицательных бактерий. Он устойчив к р-лактамазам и не индуцирует их образование. Механизм его действия идентичен таковому других р-лактамных антибиотиков в отношении грамотрицательных бактерий. Под влиянием азтреонама инактивируются ПСБ.
Азтреонам (Aztreonam)
Азтреонам — (2)-2[[[(2-амино-4-тиазолил)[[(28.38)-2-метил-4-оксо-1-сульфо-3-азетидинил]карбамоил]метилен]амино]окси]-2-метилпро-пионовую кислоту (32.1.4.9) получают исходя из треот-бутил-оксикарбонилтреонина, взаимодействием которого с О-бензиловым эфиром гидроксиламина в присутствии дициклогексилкарбодиими-да в качестве дегидратирующего агента с добавлением 1-гидрокси-бензгриазола получают бензиловый эфир соответствующей гидроксамовой кислоты (32.1.4.1). Подвергая последний действию трифенилфосфина и диэтилового эфира азодикарбоновой кислоты, осуществляют циклодегидратацию продукта в (38-траис)-М-бензил-окси-З-треот-бутилоксикарбониламино-4-метилазетидинон (32.1.4.2). Дебензилироанием последнего путем восстановления водородом
-640-
Антибиотики
с использованием в качестве катализатора палладия на угле получают (38-транс)-М-гидрокси-3-отреот-б\тилоксикарбониламино-4-метилазетидинон (32.1.4,3). Гидроксильную группу в последнем убирают восстановлением треххлористым титаном с получением азетидинона (32.1.4.4). Снятием треот-бутилоксикарбонильной зашиты с последнего с помощью трифторуксусной кислоты и последующим ацилированием полученного продукта бензиловым эфиром хлоругольной кислоты получают (38-транс)-бензилоксикарбонил-а.мино-4-метилазетидинон (32.1.4.5). Сульфируя последний комплексом трехокиси серы с диметилформамидом получают соответствующую N-сульфокислоту. Переводом полученной N-сульфо-кислоты в калиевую соль взаимодействием с гидрофосфатом калия и, наконец, обменом катиона калия на тетрабутиламмониевый катион, взаимодействием с сульфатом тетрабутиламмония получают продукт (32.1.4.6). Восстановлением последнего водородом с использованием в качестве катализатора палладия на угле получают З-амино-4-метилмонобактамовую кислоту (32.1.4.7). Ацилируя последнюю (2)-2-2-(дифенилметилоксикарбонил-проп-2-оксимино-2-(2-амино-тиазол-4-ил) уксусной кислотой в присутствии дициклогексил карбодиимида и 1-гидроксибензтриазола, получают дифенилметиловый эфир искомого азтреонама (32.1.4.8), который гидролизуют до азтреонама (32.1.4.9) с помощью трифторуксусной кислоты [185, 186, 187].
он
(СН3)зСО—СО-NH-CH-СН-СНз + соон
он (СНз>зСО—СО—NH—СН—СН—СНз СО-NH—О-СН2
(Свн3 Р 02HfcOCC-N=N-COOC2i0
1 ClCOOCH2CeH5
2 CF3COOH
1 SO3/Dh/F
2 К2НРОд +
3 (C4Hg)4N HSO4
32 1 44
32 1 4 5
-641 -
Глава 32
CeH5CH2O-l
•со—NH
ЗОз^СДЧдЦИ
32 1 4 6
СНз
Н2 / Pd-C
32 1 4 7
СН3-С-СООСН(С6Н5)
СН3 32 1 48
СРзСООН
СН3-С-СООСН(С6Н6) СНз
32 1 4 9
СНз-С-СООН СНз
Полагают, что метильная группа в положении 4 повышает стабильность р-лактамного кольца по отношению к большинству р-лактамаз, и в то же время препарат не индуцирует образование р-лактамаз, как это происходит при применении цефалоспоринов и имипенема.
Аминоацильная боковая цепь препарата, аналогичная таковой у це-фтазидина, ответственна за высокую активность по отношению к гра-мотрицательным аэробным бактериям. Спектр действия азтреонама весьма близок к антимикробному спектру аминогликозидов, и в большинстве случаев он является их потенциальным заменителем.
Азтреонам используют при лечении инфекций мочевыводящих путей, желчного тракта, остеомиелитов, гонореи, интраабдоминаль-ных. гинекологических инфекций, при инфекциях костей, кожи и других, вызванных аэробными грамотрицательными микроорганизмами. У пациентов с выявленными или подозреваемыми смешанными инфекциями он должен быть применен в сочетании с другими препаратами, такими как клиндамицин, метронидазол, нафциллин или ванкомицин.
-642-
Антибиотики
32.2. Макролидные антибиотики
Макролидные антибиотики. используемые в настоящее время в клинике, включают в себя прототип этих соединений — эритромицин, выделенный из вида Streptomyces erythreus, и два относительно новых препарата — кларитромицин и азитромицин. Они относятся к группе антибиотиков, известных под названием макролиды, названных так, поскольку они содержат макроциклическое лактонное кольцо (14-членное в эритромицине и кларитромицине, являющимся по сути 6-мето-ксиэритромицином, и 15-членное в азитромицине благодаря наличию в кольце дополнительного атома азота), к которому присоединены остатки дезоксисахаров.
Известны также макролиды с 12-членным лактонным кольцом, получившие название патулолиды, и макролиды с 16-членным лактонным кольцом, названные изенамицинами.
В группу макролидных антибиотиков, продуцируемых в основном сгрептомицетами, сегодня входит около 100 соединений.
Макролиды имеют важное клиническое значение. Существуют прямые показания к их применению, и одновременно они являются альтернативой пенициллинам у пенициллиналлергичных лиц. Макролиды на сегодняшний день считаются одними из самых безопасных антибиотиков.
Макролиды (эритромицин и др.) ингибируют синтез бактериальных белков Основные механизмы синтеза протеинов у человека и у бактерий идентичны. Однако имеется существенная разница, позволяющая конкретному антибиотику проявлять селективную токсичность по отношению к бактерии.
Как известно, первым шагом в синтезе протеинов является транскрипция генетического кода с ДНК на мессенджерную РНК (мРНК) — процесс, зависимый от РНК полимеразы (транскриптазы). Участок нуклеотидов в мРНК отражает порядок нуклеотидов в ДНК и, таким образом, содержит информацию, определяющую последовательность, в которой должны быть соединены аминокислоты для образования соответствующего белка.
Синтез белка происходит на рибосомах, которые можно представить как некие станки, на которых собираются белки из разных аминокис тот.
Бактерии содержат 80-S рибосом, которые состоят из двух неравных составляющих: большой 50-S субъединицы и малой 30-S субъединицы. Эти две субъединицы имеют разные функции мРНК связывается с 30-S субъединицей, в то время как 50-S субъединица служит для при
-643 -
Глава 32
соединения аминокислот и участком для поддержания растущей пептидной цепи. Эти участки, известные как акцепторные (А) и донорные (Р) участки, соответственно и находятся очень близко друг к другу.
Аминокислоты доставляются к комплексу рибосома — мРНК транспортной РНК (тРНК). Для каждой аминокислоты, которая должна быть включена в синтезируемый белок, существует специфическая тРНК. Каждая тРНК, в свою очередь, специфична по отношению к одному нуклеотидному участку (нуклеотидному триплету или кодону) в мРНК. Таким образом, тРНК является двухконцевой молекулой, в которой один конец специфичен по отношению к одной аминокислоте, а другой — специфичен к участку из трех нуклеотидов в мРНК. Ами-ноацильная тРНК связывается с участком А на 50-S субъединице. Рост пептидной цепи осуществляется переносом и связыванием пептидной цепи от участка Р на участок А посредством катализа пептидилтрансфе-разой. После образования пептидной связи происходит сложный процесс «транслокации» тРНК с удлиненной пептидной цепью: она переносится от участка А к участку Р, и 30-S субъединица передвигается на один кодон вдоль мРНК. Участок А освобождается и становится готовым принять другую аминоацильную тРНК, предопределяемую следующим кодовым триплетом на мРНК. Такой процесс наращивания продолжается до завершения построения белковой цепи. Макролиды ингибируют синтез бактериальных белков, связываясь с бактериальной 50-S субъединицей и тем самым предотвращая рост пептидной цепи, скорее всего, вмешиваясь на стадии транслокации.
В то же время эти препараты не связываются с рибосомами млекопитающих, что и является причиной их селективной токсичности. Макролиды могут проявлять себя и как бактериостатики, и как бактерициды в зависимости от концентрации препарата, чувствительности микроорганизмов, скорости их роста и, собственно, величины колонии. Антибактериальная активность макролидов зависит от кислотности среды. Большая активность наблюдается в нейтральной и основной средах по сравнению с кислой. В частности, эритромицин инактивируется в кислой среде желудка. Макролиды имеют относительно широкий спектр действия и активны в отношении грамположительных и грамотрицательных микроорганизмов, актиномицетов, микоплазм, спирохет, хламидий, риккетсий, определенных микобактерий и др.
Эритромицин (Erythromycin)
Эритромицин — (3R*,4S*, 5S‘,6R‘,7R’,9R*.l lR’,12R*,13S*,14R*)-4-[(2,6-дидезокси-3-С-метил-3-0-метил-а-Б-рибо-гексопиранозил)-ок-си]-14-этил-7,12,13-тригидрокси-3,5,7,9,11,13-гексаметил-6-[[3,4,6-
- 644 -
Антибиотики
гридезокси-3-(диметиламино)-Р-0-кснло-гексопиранозил]окси]окса-циклотетрадекан-2,10-дион (32.2.1), более точно называемый эритромицином А, впервые был выделен в 1952 г. из культуральной жидкости микроорганизмов вида Streptomyces erytherus. В культуральной жидкости в минорных количествах обнаруживаются также эритромицины В и С. Эритромицин В отличается от эритромицина А тем, что в положении 12 вместо гидроксильной группы находится атома водорода, а эритромицин С отличается от эритромицина А тем, что в положении 3 вместо кладинозы к макроциклу присоединен остаток другого углевода — микарозы.
В настоящее время эритромицин А производится только микробиологическим путем с использованием активных штаммов микроорганизмов вида Saccharopolospora erythraea [188, 189, 190, 191].
эритромицин А
эритромицин В
эритромицин С
Как уже было отмечено, эритромицин и другие антибиотики, обсуждаемые в этой главе, ингибируют синтез бактериальных белков.
Эритромицин ингибирует синтез протеинов бактерий путем обратимого связывания с их 50-S рибосомальной субъединицей и тем самым блокирует образование новых пептидных связей. Эритромицин классифицируется как бактериостатический антибиотик. Однако он может проявлять и бактерицидное действие против некоторых видов микробов при определенных концентрациях.
Бактериальная резистентность к эритромицину может возникать по двум возможным механизмам: невозможностью преодоления покрытия клетки, что в частности имеет место в случае с микроорганизмом Enierobacteriaceae. или в случае наличия метилированного аденина в 23-S рибосомальной РНК 50-S субъединицы, что понижает сродство эритромицина к ней.
-645-
Г лава 32
Эритромицин действует на грамположительные (стафилококки, продуцирующие и не продуцирующие пенициллиназу, стрептококки, пневмококки, клостридии и некоторые грамотрицательные микроорганизмы — гонококки, бруцеллы, гемофильная и коклюшная палочки, легионеллы), микоплазмы, хламидии, спирохеты, риккетсии. К эритромицину устойчивы кишечная и синегнойная палочки, а также палочки шигеллы, сальмонеллы и др.
Эритромицин применяют при таких бактериальных инфекциях, как дифтерия, коклюш, трахома, ангина, скарлатина, отит, синусит, холецистит, пневмония, гонорея и др.
Эритромицин является альтернативой пенициллину для лечения инфекций, вызванных чувствительными организмами. Препарат является лекарством выбора при ряде пневмоний, дифтерии, энтеритов и т. п.
Синонимами препарата являются илозон. меромицин, эритропед и многие другие.
Кларитромицин (Claritromycin)
Кларитромицин — (2R’.3S*,4S*, 5R’,6R’,8R’,10R’,l lR’,12S’,13R*)-3-(2,6-дидезокси-3-С-3-О-диметил-сх-Ь-рмбо-гексопиранозилокси-6-ме-токси-9-okco-I 1,12-дигидрокси-2,4,6,8,10,12-гексаметил-5-(3,4,6-три-дезокси-3-диметиламино-[3-П-ксшго-гексопиранозилокси)циклопен-тадекан-13-олид (32.2.2) является полусинтетическим аналогом эритромицина А, в котором гидроксильная группа при С6 замещена на метоксильную [192, 193].
-646-
Антибиотики
Кларитромицин лучше абсорбируется и меньше чем эритромицин раздражает ЖКТ. Полагают, что его активность в 2-4 раза превосходит таковую у эритромицина по отношению к ряду стрептококков и стафилококков и некоторым другим микроорганизмам. Показан для лечения бактериальных бронхитов, пневмонии, кожных и половых инфекций. Полакают, что кларитромицин является наиболее активным макролидом при течении атипичных микобактерий.
Синонимом препарата является биаксин и др.
А з и. тром ииии (A zitromycin)
Азитромицин — (2R’,3S*,4R*, 5R’, ,8R’,10R’,llR’,12S*,13S*,14S*)-3-
[(2,6-дидезокси-3-С-3-0-диметил-сх-Ь-рмбо-гексопиранозилокси)-2-этил-3,4,10-тригидрокси-3,5,6,8,10,12,14-гепта-метил-11-(3,4,6-три-дезокси-3-диметиламино)-Р-П-ксило-гексопиранозил окси-1-окса-6-аза-циклопентадекан-15-он (32.2.3) также является макролидным антибиотиком, получаемым полусинтетическим путем [194, 195].
Его активность в 2-4 раза меньше чем у эритромицина по отношению к ряду стрептококков и стафилококков и некоторым другим микроорганизмам, однако он активнее других макролидов по отношению к определенным анаэробным организмам. Подобно другим макролидам азитромицин активен по отношению к патогенам респираторного тракта и пагогенам. передаваемым половым путем. Применяют для лечения бактериальных бронхитов, пневмонии, кожных и половых инфекций.
Синонимом препарата является зитромакс и др.
-647-
Г лава 32
32.3. Тетрациклины
Тетрациклины представляют собой семейство химически сходных соединений, которые объединяет наличие четырех сочлененных в гидронафтаценовую систему колец, а разницу в активности определяют различные заместители базовой структуры.
Первый антибиотик тетрациклинового ряда — хлортетрациклин, выделенный из культуральной жидкости жизнедеятельности Streptomyces aureofacines, был введен в медицинскую практику в 1948 г. Впоследствии в течение с 1950 по 1972 г. в медицинскую практику были внедрены еще 6 препаратов тетрациклинового ряда:
• окситетрациклин, выделенный из Streptomyces rimosus,
• тетрациклин (полусинтетический),
• демеклоциклин, выделенный из мутантного вида Streptomyces aureofacines,
• метациклин (полусинтетический),
• доксициклин (полусинтетический),
• миноциклин (полусинтетический).
Предложены и методы синтеза антибиотиков тетрациклинового ряда, которые, однако, представляют чисто академический интерес и не имеют практического значения.
Несмотря на некоторые различия этих препаратов в плане фармакокинетических особенностей, их широкий спектр антимикробной активности во многом очень схож.
Тетрациклины, как и макролиды, ингибируют синтез протеинов бактерий. Существенным элементом этого процесса является энергозависимый перенос препарата через цитоплазматическую мембрану, что приводит к их аккумуляции в клетке. В клетке препараты обратимо связываются с 30-S рибосомальной субъединицей бактерий. Этот процесс блокирует присоединение аминоацильной тРНК к мРНК 30-S рибосоме, что и приводит к ингибированию синтеза протеинов. Селективная токсичность тетрациклинов заключается в различной способности их проникновения в бактериальные клетки и клетки млекопитающих, у кото
-648-
Антибиотики
рых отсутствует соответствующая система транспорта. Антимикробный спектр всех тетрациклинов практически одинаков. Отличия проявляются лишь в степени активности по отношению к тем или иным микроорганизмам. Тетрациклины активны по отношению к огромному разнообразию микроорганизмов, включая грамположительные, грамотрица-тельные, аэробные и анаэробные. Они активны в отношении спирохет, микоплазм, риккетсий, хламид и некоторых протозойных инфекций. Однако они не активны по отношению к стрептококковым инфекциям, синегнойной палочке и некоторым другим. Резистентность к тетрациклинам проявляется в виде пониженной способности бактерий аккумулировать антибиотик внутри клетки. Этот процесс опосредуется плазмидами. Резистентность по отношению к какому-либо тетрациклину, как правило, обозначает резистентность и ко всем остальным.
Тетрациклины являются препаратами выбора по отношению к широкому ряду инфекций, включая хламидиальные, риккециозные и др.
Хлортетрациклин (Chlortetracyclin)
Хлортетрациклин — 7-хлор-4-диметиламино-1,4,4а,5,5а,6,11,12а-октагидро-3,6,10,12,12а-пентагидрокси-6-метил-1,11 -диоксо-2-наф-таценкарбоксамид (32.3.1) получают биосинтетическим путем в результате жизнедеятельности микроорганизма актиномицета (Streptomyces aureofaciens) [196, 197, 198, 199, 200, 201].
32 3 1
Хлортетрациклин — антибиотик широкого спектра действия, оказывающий бактериостатический эффект в отношении грамположительных (стафилококки, в том числе продуцирующие пенициллиназу, стрептококки, пневмококки, клостридии, листерии, палочка сибирской язвы) и грамотрицательных (гонококки, коклюшная палочка, кишечная палочка, энтеробактер, клебсиелла, сальмонелла, шигелла) микроорганизмов, а также риккетсий, хламидий, микоплазм, спирохет. Устойчивы к препарату синегнойная палочка, протей, серрации. большинство штаммов Bacteroides fragilis, большинство грибов, мелкие вирусы. Препарат применяют при пневмонии, бронхите, эмпиеме легких, ангине, холецистите, коклюше, эндокардите, эндометрите, кишечных инфекци
-649-
Глава 32
ях, простатите, сифилисе, гонорее, коклюше, бруцеллезе, остеомиелите, гнойных инфекциях мягких тканей и других, вызванных чувствительными к препарату микроорганизмами.
Синонимами препарата являются ауреомицин, биомицин, ксанто-мицин и др.
Окситетрациклин (Oxytetyracyclin)
Окситетрациклин — 4-диметиламино-1,4,4а,5,5а,6,11,12а-октагидро-3,6,10,12,12а-гексагидрокси-6-метил-1,11 -диоксо-2-нафтаценкарбокс-амид (32.3.2) получают биосинтетическим путем в результате жизнедеятельности актиномицета Streptomyces rimosus [202, 203, 204, 205].
32.3.2
По антибактериальному спектру препарат близок к хлортетрацик-лину. Применяют по тем же показаниям. Относится к тетрациклинам короткого времени действия.
Синонимами препарата являются тетрамицин, оксимицин и др.
Тетрациклин (Tetracyclin)
Тетрациклин — 4-диметиламино-1,4,4а,5,5а,6,11,12а-октагидро-3,6,10,12,12а-пентагидрокси-6-метил-1,11-диоксо-2-нафтаценкарбо-ксамид (32.3.3) получают восстановлением хлортетрациклина водородом, используя в качестве катализатора палладий на угле. Однако его можно получать и микробиологическим путем с использованием актиномицета Streptomyces viridifaciens, а также определенного мутанта Streptomyces aureofaciens [206, 207, 208, 209, 210, 211, 212, 213,214].
32 3 1
Н2/ Pd-C
32 3 3
-650-
Антибиотики
По антибактериальному спектру окситетрациклин близок к хлор-тетрациклину. Препарат также относится к тетрациклинам короткого времени действия, и его применяют по тем же показаниям, что и хлор-тетрациклин.
Синонимами препарата являются акроминин, бициклин, циклопар, саркоциклин и многие другие.
Демеклоциклин (Demeclocyclin)
Демеклоциклин — 7-хлор-4-диметиламино-1,4,4а,5,5а,6,11,12а-октагидро-3,6.10,12,12а-пентагидрокси-1,11 -диоксо-2-нафтаценкар-боксамид (32.3.4) продуцируется мутантным штаммом Streptomyces aureofaciens, у которого нарушен механизм переноса метильных групп, и, таким образом, демеклоциклин, или деметилхлортетра-циклин, отличается от хлортетрациклина, окситетрациклина и тетрациклина отсутствием метильной группы при С6 гидронафтаценовой системы. В результате получается антибиотик более устойчивый к кислотам и щелочам по сравнению с метильными гомологами [215, 216, 217, 218, 219, 220, 221].
32 3.4
По антибактериальному спектру демеклоциклин мало отличается от всего ряда тетрациклинов, однако время его полураспада несколько больше, чем у описанных выше антибиотиков с коротким временем действия, поэтому его относят к тетрациклинам среднего или промежуточного времени действия. Применяют демеклоциклин по тем же показаниям, что и другие антибиотики тетрациклинового ряда.
Синонимами препарата являются ледермицин, биотетрицин, дециклин, декломицин.
Метациклин (Methacyclin)
Метациклин — 4-димегиламино-1,4,4а,5,5а,6,11,12а-октагидро-3,6,10.12,12а-пентагидрокси-6-метилен-1,11-диоксо-2-нафтаценкар-боксамид (32.3.6) получают исходя из окситетрациклина (32.3.2), взаимодействием которого с комплексом серный ангидрид — пиридин в результате реакции окисления и одновременно сульфирова-
-651 -
Глава 32
ния получают промежуточное нафтаценсульфотетрагидрофурано-вое производное (32.3.5), которое под действием фтористоводородной кислоты расщепляется с образованием метациклина (32.3.6) [222, 223, 224, 225].
Метациклин применяют по тем же показаниям, что и другие антибиотики тетрациклинового ряда. В некоторых случаях он лучше переносится, чем другие тетрациклины.
Синонимами препарата являются рондомицин, метамицин, адрами-цин и др.
Доксициклин (Doxycyclin)
Доксициклин — 4-диметиламино-1,4,4а,5,5а,6,11,12а-октагидро-3,5,10,12,12а-пентагидрокси-6-метил-1,11 -диоксо-2-нафтаценкарбо-ксамид (32.3.7) — препарат, являющийся изомером тетрациклина и отличающийся от тетрациклина расположением одной гидроксильной группы. Формально же доксициклин можно считать результатом переноса С6-гидроксильной группы тетрациклина в положение С5. Доксициклин предложено получать двумя путями исходя из окситетрациклина (32.3.2). Первый способ синтеза предполагает дегидроксилирование окситетрациклина по положению С6 путем восстановления третичной гидроксильной группы водородом с использованием в качестве катализатора родия на угле [226, 227].
32 3 2
32.3 7
Второй способ имеет аналогию с получением метациклина, а именно: он предполагает стадию окисления гомоаллильной системы, но с использованием в качестве окислителя N-хлорсукцинимида, при кото-
-652-
Антибиотики
ром образуется нафтацентетрагидрофурановое производное (32.3.8) и которое под действием фтористоводородной кислоты расщепляется с образованием 11а-хлор-6-экзомтиленового производного (32.3.9). Восстановительным дехлорированием последнего тиосульфатом натрия промежуточно получают метациклин (32.3.6), к метиленовой группе которого в условиях проведения радикальных реакций осуществляют присоединение тиофенола с получением производного (32.3.10). Последнее восстанавливают водородом над никелем Ренея в качестве катализатора, при котором происходит восстановительная десульфуризация, и получается доксициклин [225, 228, 229, 230].
/Доксициклин применяют по тем же показаниям, что и другие антибиотики тетрациклинового ряда, однако он относится к долгодействующим тетрациклинам. В некоторых случаях он более активен по отношению к ряду микроорганизмов, лучше переносится, чем другие тетрациклины.
Синонимами препарата являются азудоксат, кодидоксал, эфтапан, вибрамицин и др.
Миноциклин (Minocyclin)
Миноциклин — 4,7-б«с-(диметиламино)-1,4,4а,5,5а.6,11,12а-ок-таги дро-3,10.12,12а-тетра-гидрокси-1,11 -диоксо-2-нафтаценкарбо-ксамид (32.3.17) получают исходя из 6-деметилтетрациклина (32.3.11), получаемого в результате жизнедеятельности штамма Streptomyces aureofaciens, у которого нарушен механизм переноса метильных групп, или же из обычного штамма того же микроорганизма. но при добавлении в среду для развития данного акгиноми-цета таких соединений, как этионин, D-норлейцин или D-метионин,
-653-
Глава 32
являющихся антиметаболитами метионина — основного донора метильных групп при микробиологическом синтезе молекулы тетрациклина. Гидрогенолизом упомянутого 6-деметилтетрациклина ; 32.3.11) водородом с использованием в качестве катализатора палладия на угле получают 4-диметиламино-1,4,4а,5,5а,6,11,12а-октагидро-3,10,12,12а-тетрагидрокси-1,11 -диоксо-2-нафтаценкарбо-ксамид (32.3 12), который нитруют в положение 9 нитратом калия в жидкой фтористоводородной кислоте с получением нитросоединения (32.3.13). Последнее восстанавливают до соответствующего аминосоединения (32.3.14) водородом над двуокисью платины. Полученный а.минофенол (32,3.14) далее нитруют азотной кислотой в присутствии серной кислоты с получение1М 7-нитро-9-амино-4-диметил амино-1,4,4а,5,5а,6,11.12а-октагидро-3,10,12,12а-тетрагидро-кси-1.11-диоксо-2-нафтаценкарбоксамида (32.3.15). Последний подвергают диазотированию действием бутилнитрита в серной кислоте, и полученное диазопроизводное (32.3.16) восстанавливают водородом с использованием палладия на угле. При этом продукт деазотируется, и одновременно нитрогруппа восстанавливается до аминогруппы, которую подвергают исчерпывающему метилированию формальдегидом в миноциклин (32.3.17) [231, 232, 233, 234, 235, 236. 237].
1 Н2 / Pd—С
2 СН2О
32 3 16
32 3 17
-654-
Антибиотики
Миноциклин применяют по тем же показаниям, что и другие анти-био^ики тетрациклинового ряда. В некоторых случаях он хуже переносится, чем другие тетрациклины, и, в частности, действует на вестибулярный аппарат. Кроме того, как это следует уже из схемы синтеза, он значительно дороже других тетрациклинов, получаемых чисто микробиологическим путем.
Синонимами препарата являются клиноцин, миноцин, вектрин и др.
32.4. Аминогликозиды
Аминогликозиды являются соединениями, содержащими два или более аминосахаров, связанных гликозидными связями с аминоцикли-тольным кольцом (агликоном). Шестичленное аминоциклитольное кольцо является либо стрептидином (1,3-дигуанидино-2,4,5,6-тетрагидро-ксициклогексан), как в стрептомицине, либо 2-дезоксистрептамином 11,3-диамино-4,5,6-тригидроксициклогексан), как во всех других ами-ногтикозидах.
стрептидин
2-даоксистрепта.нин
2-дезоксистрептаминовые антибиотики могут далее дифференциро-ваться по числу и типу сахаров, присоединенных к аминоциклитольно-му кольцу. Так, неомицины, включающие сам неомицин и паромомицин. имеют три сахарных остатка (две аминогексозы и одну' неаминную пентозу), присоединенные к 2-дезоксистрептамину. Аналоги канамици-ка (тобрамицин, амикацин) и гентамицина (сисомицин, нетилмицин) имеют по две аминогексозы, присоединенные к центральному агликону. Последние два ряда отличаются типом 3-аминогексозы: для ряда кана-мицинов -- это канозамин. для ряда гентамицинов — это гаросамин. Вариации внутри самих рядов аминогликозидов обусловлены разницей в боковых цепях аминосахаров и агликона.
Из 8 применяемых в настоящее время аминогликозидов 5 получены нт разных видов Streptomyces
• стрептомицин (выделен из Streptomyces gnseus),
• неомицин (выделен из Streptomyces fradiae),
• паромомицин (выделен из Streptomyces rimosus).
- 655-
Г лава 32
• канамицин (выделен из Streptomyces kanamyceticus),
• тобрамицин (выделен из Streptomyces tenebrarius).
Гентамицин выделен из Micromonospora purpurea. Он состоит из смеси примерно равных количеств 3 соединений: гентамицина Сь С1а и С2. Амикацин и нетилмицин являются полусинтетическими препаратами. Амикацин получается химической модификацией кана.мицина. Нетилмицин является полусинтетическим производным шизомицина, выделенного из Micromonospora inyoensis.
Аминогликозидные антибиотики являются бактерицидными. Они ингибируют синтез протеинов и приводят к неправильному считыванию генетического кода. Общим элементом в процессе, приводящему к летальному исходу бактерий, является активный перенос препарата из окружающей среды в бактериальную клетку, что приводит к большой аккумуляции препарата в клетке, намного превосходящей таковую в ее окружении. Аминогликозиды легко диффундируют сквозь внешнюю мембрану бактерий и проходят в периплазматическое пространство. Первичным внутриклеточным участком действия аминогликозидов является бактериальная рибосома. Очевидно, существует как минимум 2 разных типа рибосомального связывания: один — специфический для стрептомицина и другой — участвующий в связывании с другими аминогликозидами. Стрептомицин связывается с 30-S рибосомальной субъединицей. Другие аминогликозиды связываются к разным участкам как ЗО-S, так и 50-S рибосомальных субъединиц и не могут конкурировать со стрептомицином за связывание с ЗО-S рибосомой.
Связывание аминогликозидов с рибосомами проявляется как непосредственным ингибированием синтеза протеинов, так и неправильным считыванием генетического кода с матрицы мРНК, что в результате приводит к внедрению неправильных аминокислот в полипептидные цепи. Однако ни один из этих эффектов полностью не объясняет бактерицидный эффект аминогликозидов. Аминогликозиды являются препаратами, используемыми преимущественно против аэробных и некоторых грамотрицательных микроорганизмов, Staphylococcus aureus и микобактерий. Эти антибиотики не активны по отношению к анаэробным микроорганизмам.
Бактериальная резистентность по отношению к аминогликозидам может быть объяснена изменениями в связывающих участках на рибосомах, ухудшением внутриклеточного транспорта и дезактивацией препаратов микробными ферментами.
Стрептомицин связывается со специфическим белком (S[2) на 30-S субъединице рибосомы. Изменение этого белка в результате мутации
- 656 -
Антибиотики___________________________________________________
делает рибосомы неспособными к связыванию со стрептомицином, что и придает организму резистентность. Мутационная резистентность к стрептомицину встречается весьма часто. Гентамицин, тобрамицин, нетилмицин и амикацин связываются со многими участками на обеих субъединицах рибосом, и поэтому мутационная резистентность к ним не носит общего характера.
Второй механизм резистентности — это затрудненный транспорт в клетку, который приводит к резистентности ко всем аминогликозидам. Этот тип резистентности не носит общего характера среди грамотрицательных аэробных и некоторых других микроорганизмов. Поскольку транспорт аминогликозидов через цитоплазматическую мембрану является кислородзависимым процессом, особенно анаэробные бактерии всегда проявляют резистентность по отношению к этим бактериям.
Наиболее значимым механизмом резистентности является опосредованная плазмидами выработка ферментов, которые фосфорилируют, аденилируют или ацетилируют специфические амино- или гидроксильные группы в молекуле аминогликозидов. Эти ферменты не вырабатываются вне клеток. Они обнаруживаются в периплазматической области. Как только препарат проникает через внешнюю мембрану и достигает периплазматической области, он подвергается изменению ферментом. Видоизмененный препарат наряду с неизмененным конкурирует за проникновение в клетку, но оказывается неспособным связываться с рибосомами. Как следствие, вторая энергозависимая фаза захвата аминогликозидов ингибируется. Найдено около 20 подобных ферментов.
В свою очередь аминогликозиды отличаются по своей способности противостоять ферментной инактивации:
— гентамицин и тобрамицин чувствительны к одним и тем же ферментам;
— нетилмицин несколько более устойчив к этим модификациям ферментов;
— амикацин наиболее устойчив к описанным в настоящее время ферментам;
— гентамицин, тобрамицин, нетилмицин и амикацин эффективны для лечения аэробных инфекций и некоторых грамотрицательных бацилл.
При серьезных инфекциях, вызванных Pseudomonas aeruginosa, эти препараты используются в комбинации с антибиотиками широкого спектра действия — пенициллином, цефтазидином, имипенемом или
- 657-
Глава 32
азтреонамом. Стрептомицин является лекарством выбора при лечении туляремии, чумы и бруцеллеза (в комбинации с тетрациклином). Он не используется для лечения других грамотрицательных бактериальных инфекций из-за высокой вероятности возникновения резистентности, которая может развиться благодаря всего лишь одной мутации. Однако следует особенно отметить, что практически все антибиотики ряда аминогликозидов не метаболизируются в организме, накапливаются в почках и обладают определенной ото- и нефротоксичностью.
Стрептомицин (Streptomycin)
Стрептомицин — ш/шнс-2,4-дигуанидино-3,5,6-тригидроксицикло-гексил-5-дезокси-2-0-(2-дезокси-2-метиламино-а-Ь-глюкопиранозил)-З-С-гидроксиметил-р-Ь-ликсо-пентофуранозид (32.4.1) выделяют из культуральной жидкости жизнедеятельности актиномицета Streptomyces griseus [238, 239, 240, 241, 242, 243, 244, 245, 246, 247].
32 4 1
Стрептомицин обладает широким спектром антибактериальной активности. Эффективен в отношении большинства грамотрицательных и некоторых грамположительных бактерий; стафилококков, стрептококков, пневмококков, гонококков, менингококков, возбудителей дизентерии, бруцеллеза, туберкулеза, туляремии, чумы и др.
Препарат применяют при различных заболеваниях, вызванных чувствительными к нему микроорганизмами, — бактериальном эндокардите, перитоните, менингите, инфекциях мочевых путей, кишечных инфекциях, туляремии, чуме, бруцеллезе и т. п.
Синонимами препарата являются стрептан, стрептокол и др.
-658-
Антибиотики
Неомицин (Neomycin)
Неомицин является комплексом антибиотиков (неомицины А, В, С, D, Е и F), образующихся в результате жизнедеятельности актиномицета Streptomyces fradiae. Причем неомицин А, называемый также неамином, является 2-дезокси-4-0-(2,6-диамино-2,6-дидезокси-а-П-глюкопиранозил)-П-стрептамином (32.4.2) и не обнаруживает антибиотических свойств. В то же время неомицин В — 0-2,6-диамино-2,6-дидезокси-а-П-глюкопиранозил(1—>4)-О-[О-2,6-диа-мино-2.6-дидезокси-Р-Ь-идопиранозил-(1—>3)-Р-О-/щбо-фуранозил-(1—>5)]-2-дезокси-П-стрептамин (32.4.3), отличающийся от неомицина А наличием второго гликозидного остатка, уже проявляет мощную антибактериальную активность. Неомицин С (32.4.4) отличается от неомицина В ориентацией аминометильной группы в неозаминной части молекулы [248, 249, 250, 251, 252, 253, 254, 255].
Неомицин, как и стрептомицин, обладает широким спектром антибактериальной активности. Эффективен в отношении большинства грамотрицательных и некоторых грамположительных бактерий, стафилококков, пневмококков, гонококков, менингококков, возбудителей Дизентерии. В отношении стрептококков малоактивен. Антибиотический эффект неомицина в отношении многих видов бактерий выше, чем } стрептомицина. В то же время чувствительные к неомицину микроорганизмы приобретают устойчивость к нему в меньшей степени, чем к стрептомицину.
Препарат применяют при различных желудочно-кишечных заболеваниях. вызванных чувствительными к нему микроорганизмами, в том
- 659-
Глава 32
числе и энтеритах, вызванных устойчивыми к другим антибиотикам микробами. Однако ввиду высокой ото- и нефротоксичности его предпочитают применять местно при гнойных заболеваниях кожи, инфицированных ранах, конъюнктивитах, кератитах и др.
Синонимами препарата являются фармицетин, софрамицин. тутто-минин и др.
Паромомицин (Paromomycin)
Паромомицин — 0-2-амино-2-дезокси-а-П-глюкопиранозил(1—>4)-0-[0-2,6-диамино-2,6-дидезокси-р-Ь-идопиранозил-(1-»3)-Р-0-/шбо-фуранозил-(1—>5)]-2-дезокси-П-стрептамин (32.4.5) с химической точки зрения отличается от неомицина В лишь заменой 6-аминогруппы в глюкопиранозильной части молекулы на гидроксильную и выделен из культуральной жидкости жизнедеятельности ак-тиномицетов Streptomyces rimosus [256, 257, 258].
Антибактериальная активность и показания к применению паромомицина аналогичны таковым неомицина. Кроме того он рекомендуется при лечении острых и хронических форм кишечного амебиоза.
Синонимами препарата являются аминозидин, катенулин, кресто-мицин, гидроксимицин, мономицин, зигомицин и др.
Канамицин (Kanamycin)
Канамицин — О-3-амино-3-дезокси-а-О-глюкопиранозил-(1-»6)-О-[6-дезокси-6-амино-а-П-глюкопиранозил-(1->4)]-2-дезокси-В-стреп-тамин (3.4.6) выделен из культуральной жидкости жизнедеятельности актиномицета Streptomyces kcmamyceticus, который продуцирует три антибиотика — канамицин А, В и С [259, 260, 261, 262].
-660-
Антибиотики
Канамицин А сходен со стрептомицином и неомицинами и обладает широким спектром антимикробного действия. Активен в отношении большинства грамположительных, а также грамотрицательных микро-ор!анизмов (стафилококков, кишечной палочки, клебсиеллы, палочки Фридлендера, протея, шигеллы, сальмонеллы).
Применяют при лечении сепсиса, менингита, остеомиелита, перитонита, пневмонии, пиелонефрите, пиелоцистите, раневых инфекциях, послеоперационных гнойных осложнениях, вызванных чувствительными к препарату микроорганизмами.
Синонимами препарата являются кармицин, камаксин, резистоми-цин и многие другие.
Тобрамицин (Tobramycin)
Тобрамицин — 0-3-амино-3-дезокси-а-0-глюкопиранозил-(1—>6)-О-[2.6-амино-2,3,6-три-дезокси а-Т)-рибо-глюкопиранозил-(1->4)-2-дезокси-П-стрептамин (3.4.7) выделен из культуральной жидкости жизнедеятельности актипомпцета Streptomyces tenebrarius [263, 264, 265,266,267,268,269, 270].
- 661 -
Глава 32
Тобрамицин высокоактивен в отношении грамотрицательных мик-роор1анизмов (синегнойной и кишечной палочек, клебсиеллы, серра-ции. провиденсии, энтеробактер, протея, сальмонеллы, шигеллы), а также грамположительных микроорганизмов (стафилококков, в том числе устойчивых к пенициллину и некоторым цефалоспоринам), некоторых штаммов стрептококков.
Применяют при бактериальных инфекциях тяжелого течения: перитоните, сепсисе, менингите, остеомиелите, эндокардите, пневмонии, эмпиеме плевры, абсцессе легкого, гнойных инфекциях кожи и мягких тканей, инфекциях мочевыводящих путей, вызванных чувствительными к препарату микроорганизмами.
Синонимами препарата являются небицин, обрацин и др.
Гентамицин (Gentamycin)
Гентамицин — это комплекс антибиотиков, выделенных из культуральной жидкости жизнедеятельности актиномицета Micromonospora purpurea, который состоит из смеси примерно равных количеств 3 соединений: гентамицина С|, С,а и С2 [271, 272, 273. 274, 275,276, 277,278].
32 4 8
гентамицин С, (R = R, = СН3) гентамицин С2 (R = СН3, R| = Н) гентамицин Ct (R = R) = Н)
Гентамицин обладает широким спектром биологического действия и высокоактивен в отношении штаммов стафилококков, устойчивых к пенициллинам и другим антибиотикам, многих грамотрицательных микроорганизмов — синегнойной палочки, клебсиеллы, энтеробактер, сальмонеллы, шигеллы, протея.
- 662-
Антибиотики
Применяют при пиелонефрите цистите, пневмонии, эмпиеме плевры, перитоните, сепсисе, менингите, гнойных инфекциях кожи и мягких (канеи инфекциях ран, ожогов и прочих, вызванных чувствительными к npenapaiy микроорганизмами. Гентамицин является препаратом выбора при бактериальных инфекциях тяжелого течения, вызванных неустановленным возбудителем.
Синонимами препарата являются гарамицин, генгацилин, рибоми-цин и мно! ие другие.
Амикацин (Amikacin)
Амикацин — О-3-амино-3-дезокси-а-В-глюкопиранозил-(1-»4)-О-[6-амино-6-дезокси-а-О-глюкопиранозил-(1->6)]-ГГ-(4-амино-Е-2-ги-1роксибутирил)-2-дезокси-Ь-стрептамин (3.4.10) является полусинте-тическим антибиотиком, получаемым из канамицина (3.4.6), первичною аминогрупп) в котором предварительно защищают ацилированием М-(бензоилоксикарбонилокси)сукцинимидом в диметилфор-мамиде. после чего полученный продукт (32.4.9) обрабатывают эфиром полученным из N-i идроксисукцинимида и бензилоксикарбо-ниламнно-а-Г-(-)гидроксимасляной кислоты, в результате чего селективно ацилируется 4-аминогруппа стрептаминовой части молекулы. Дальнейшим снятием двух бензилоксикарбониламинных защитных групп традиционным способом, восстановлением водородом с использованием в качестве катализатора палладия на угле получают искомый амикацин (32.4.10) [279, 280, 281, 282, 283. 284,285, 286].
32 4 9
32 4 10
- 663 -
Глава 32
Амикацин высокоэффективен в отношении грамотрицательных микроорганизмов (синегнойной и кишечной палочек, клебсиеллы, сер-рации, провиденсии, энтеробактер, протея, сальмонеллы, шигеллы), а также грамположительных микроорганизмов (стафилококков, в том числе устойчивых к пенициллину и некоторым цефалоспоринам), некоторых штаммов стрептококков.
Применяют при бактериальных инфекциях тяжелого течения: перитоните, сепсисе, менингите, остеомиелите, эндокардите, пневмонии, эмпиеме плевры, абсцессе легкого, гнойных инфекциях кожи и мягких тканей, инфекциях мочевыводящих путей, вызванных чувствительными к препарату микроорганизмами.
Синонимами препарата являются амикин, биклин, новамин и др.
Нетилмицин (Netiltnycin)
Нетилмицин — 0-3-дезокси-4-С-метил-3-(метиламино)-р-Ь-араби-нопиранозил(1—>4)-О-[2,6-диамино-2,3,4,6-тетрадезокси-а-О-г7ш/е/2о-гекс-4-енопиранозил-(1-»6)-2-дезокси-\т3-этил-Ь-стрептамин (3.4.10) также является полусинтетическим антибиотиком, который получают в две стадии из другого известного антибиотика — сизомици-на (32.4.11), продуцируемого культурой Micromonospora inyoensis. На первой стадии синтеза взаимодействием сизомицина с уксусным альдегидом при весьма определенной кислотности среды (pH 5) удается селективно получить имин по 3-аминогруппе 2-дезокси-стрптаминовой части молекулы. Полученный имин далее гидрируется цианоборгидридом натрия до этиламинопроизводного — не-тилмицина (32.4.12) [287, 288, 290, 291].
1 pH 5
2 СНзСНО
3 NaBH3CN
32.4 12
Нетилмицин также высокоэффективен в отношении грамотрицательных микроорганизмов (синегнойной и кишечной палочек, клебси-
-684-
Антибиотики
еллы. серрации, провиденсии, энтеробактер, протея, сальмонеллы, ши-геллы). а также некоторых грамположительных микроорганизмов (стафилококков, некоторых штаммов стрептококков).
Применяют при бактериальных инфекциях тяжелого течения, вызванных чувствительными к препарату микроорганизмами.
Синонимами препарата являются нетиллин, зетамицин и др.
32.5. Линкозамиды
Линкозамиды — линкомицин и клиндамицин — являются представителями очень небольшой группы препаратов, представляющих собой аминокислоту, связанную с аминосахаром.
Линкозамиды связываются с 50-S рибосомальными субъединицами бактерий и ингибируют синтез протеинов. Они ингибируют также действие пептидил. Линкозамиды являются бактериостатическими антибиотиками, однако при достижении определенных концентраций в плазме они проявляют и бактерицидное действие против некоторых бактерий. Линкозамиды высокоактивны против анаэробных инфекций, таких как Peptococcus, Peptostreptococcus, Actinomyces, Propionibacerium и Clostridium fringens, некоторых видов Peptococcus и Clostridium.
Практически все аэробные грамотрицательные бактерии резистентны к линкозамидам.
Резистентность к линкозамидам может иметь место из-за невозможности препаратов проходить через клеточную оболочку бактерий или изменений в рибосомальных участках связывания.
Наиболее часто линкозамиды используются при лечении анаэробных инфекций, таких как интраабдоминальные и женские инфекции.
Линкозамиды являются хорошей альтернативой р-лактамным антибиотикам для лечения инфекций, вызванных золотистой палочкой или стрептококками. Полезны при лечении остеомиелитов и септических артритов ввиду больших концентраций, достигаемых в костях.
Линкомицин (Lincomycin)
Линкомицин — 6,8-дидезокси-6-отрянс-(1-метил-4-пропил-Ь-2-пир-ролидинкарбоксамидофЬметилтио-П-э/шотро-аЛЭ-гаэакто-октопи-ранозид (32.5.1) является первым линкозамидом, нашедшим применение в клинической практике и который был выделен в 1962 г. из культуральной жидкости жизнедеятельности актиномицета Streptomyces lincolnensis [292, 293, 294, 295].
-665-
Глава 32
Линкомицин оказывает антибактериальное действие в отношении грамположительных микроорганизмов (стафилококки, стрептококки, пневмококки, палочка дифтерии, клостридии). Применяется при бактериальных инфекциях тяжелого течения: сепсисе, остеомиелите, септическом эндокардите, пневмонии, абсцессе легкого, раневых инфекциях, гнойном менингите. Линкомицин является антибиотиком резерва при инфекциях, вызванных штаммами стафилококка и другими грамполо-жительными микроорганизмами, резистентными к пенициллину и другим антибиотикам.
Синонимами препарата являются линкоцин, мицивин, альбиотик и др.
Клиндамицин (Clindamycin)
Клиндамицин — метил-[7-хлор-6,7,8-тридезокси-6-шрамс-( 1 -метил-4-пропил-1--2-пирроллидин-карбоксамидо)-1-тио-Ь-тре<э-с(-В-г<ма-кшо-октопиранозид] (32.5.2), который является 7(8)-хлоро-7-дезок-сипроизводным линкомицина, получают замещением гидроксильной группы линкомицина (32.5.1) при С- обработкой трифенилдихлоридом в ацетонитриле (реактив Райдона), при котором одновременно происходит и обращение конфигурации при данном атоме углерода [296, 297, 298, 299, 300, 301, 302, 303].
(СзНызР СН3СМ С1г
Он превосходит базовое соединение линкомицина по всем положительным параметрам, включая абсорбцию в ЖКТ, антибактериальную
-666 -
Антибиотики
активность и антимикробный спектр.
Синонимами препарата являются клеоцин, клинимицин, далацин и др.
32.6. Хлорамфеникол
Хлорамфеникол — В-/и/?ео-2,2-дихлор-М-[р-гидрокси-а-(гидрокси-метил) л-нитрофенилацетамид (32.6.7) впервые был выделен в 1947 г. из культуральной жидкости жизнедеятельности актиномицета Streptomyces venezuelae, однако в настоящее время он производится только синтетическим путем. При использовании синтетического рацемата без предвари гельного разделения на D-mpeo и Ъ-трео формы препарат называется синтомицином. Предложено два основных пути синтеза хлорамфеникола. Первый способ исходит из 4-нитроацетофенона, бромированием которого молекулярным бромом получают ю-бром-4-нитроацетофенон (32.6.1). Последний переводят в ©-амино-4-нитроацетофенон (32.6.2) последовательным получением четвертичной соли с уроторопином и последующим ее расщеплением до амина хлористым водородом. Полученный аминокетон ацилируют ангидридом уксусной кислоты с получением <о-ацетамидо-4-нитроацетофенона (32.6.3), и продукт подвергают оксиметилированию параформом с получением а-ацетамидо-р-гид-рокси-4-нитропропиофенона (32.6.4). Восстановлением карбонильной группы в последнем изопропилатом алюминия в изопропиловом спирте получают D, П-от/?ео-2-ацетамидо-1-(4-нитрофенил)-1,3-пропандиол (32.6.5). гидролизом ацетильной группы в котором соляной кислотой получают О,Ь-трео-2-амино-1(4-нитрофенил)-1,3-пропандиол. Полученную рацемическую смесь аминов обрабатывают камфор-В-сульфо-кислотой, полученные энантиомерные соли разделяют, и после щелочного гидролиза отобранной соли получают В(-)-от/?ео-2-амино-1-(4-нитрофенил)-1,3-пропандиол (32.6.6). Ацилированием аминогруппы последнего метиловым эфиром дихлоруксусной кислоты получают искомый хлорамфеникол (32.6.7) [304, 305, 306, 307, 308, 309, 310, 311, 312].
' (CHWN4
2 НС!
О ИНСОСНз
снэсо)го
OaN
О
С—СН2—NHCOCH3
(СН2О)п
O2N
с-сн-сн2-он
А1[ОСН(СН3)2]з
32 6 3
32 6 4
- 667-
Глава 32
Второй способ синтеза исходит из коричного спирта, взаимодействием которого с бромноватистой кислотой получают 2-бром-1-фенил-1,3-пропандиол (32.6.8). гидроксильные группы в котором защищают кетальной защитой взаимодействием с ацетоном с получением 5-бром-2.2-диметил-4-фенил-1,3-диоксана (32.6.9). Реакцией полученного бромида с аммиаком получают изомерную смесь D,L-/wp(?o-5-a.MHHo-2,2-диметил-4-фенил-1,3-диоксанов, обработкой которой D-винной кислотой, разделением полученных солей и последующим щелочным гидролизом отобранной соли получают В-(-)-«рео-5-амино-2,2-диметил-4-фенил-1,3-диоксан (32.6.10). Ацилированием последнего метиловым эфиром дихлоруксусной кислоты получают О-(-)-щ/?ео-5-дихлор-ацетамидо-2,2-диметил-4-фенил-1,3-диоксан (32.6.11). Далее осуществляют нитрование бензольного кольца, в ходе которого расщепляется и 1,3-диоксановый цикл с получением динитрата В-(-)-щрео-2-дихлор-ацетамидо-1-(4-нитро-фенил)-1,3-пропандиола (32.6.12). Восстанавливая нитрогруппы в последнем сульфатом двухвалентного железа получают искомый хлорамфеникол (32.6.7) [313, 314, 315].
Вг2 Н?О
-eSC'4 ------► 32 6 ”
Cl2CH-COOCh3
NOj
32 6 12
Хлорамфеникол ингибирует синтез протеинов в бактериях и в меньшей мере в эукаротических клетках. Препарат хорошо диффундирует в бактериальную клетку, где обратимо связывается с 50-S рибосомальной субъединицей. Это предотвращает присоединение аминокислотного окончания тРНК к связывающим участкам 50-S рибосомы Связывание аминоацильной тРНК с 30-S субъединицей при этом не нарушается. Клетки млекопитающих, содержащие 80-S рибосомы, не затрагиваются хлорамфениколом. Однако препарат ингибирует синтез митохондриальных протеинов в клетках млекопитающих, возможно, по причине сходства митохондриальных и бактериальных рибосом.
-668-
Антибиотики
Хлорамфеникол имеет широкий спектр антимикробной активности, включающий грамположительные, грамотрицательные, аэробные и анаэробные бактерии, спирохеты, микоплазмы, хламидии и др. Однако он может вызвать выраженное угнетение кроветворения, сопровождающееся ретикулоцитопенией, гранулоцитопенией, а в тяжелых случаях — апластической анемией.
Резистентность к хлорамфениколу обычно объясняется наличием плазмид, определяющих выработку хлорамфеникол ацетилтрансферазы. Этот фермент ацетилирует препарат, делая его неспособным связываться с 50-S субъединицей бактериальной рибосомы.
Хлорамфеникол является потенциально токсичным препаратом и имеет немного показаний к применению. Он является препаратом выбора для лечения тифозной лихорадки, применяется для лечения абсцесса мозга. До недавнего времени он являлся лекарством выбора при терапии бактериальных менингитов у детей (в комбинации с ампициллином). Однако в настоящее время для этих целей предпочтительнее цефалоспорины III поколения. Хлорамфеникол является эффективной альтернативой при ряде инфекций в тех случаях, когда препараты первого выбора по той или иной причине не могут быть использованы. Однако препарат никогда не должен применяться при инфекциях, которые надежно могут быть вылечены другими антимикробными препаратами.
Синонимами препарата являются левомицетин, аминдан. аквамице-зин, хлормицетин, ортохлор, опулетс, лейкомицин и очень многие другие.
32.7. Прочие антибиотики
Спектиномицин (Spectinomycin)
Спектиномицин — 4а,7,9-тригидрокси-2-метил-6,8-бг<с-(метиламино)-пергидроксипирано-[2,3-Ь]бензодиоксан-4-он (32.7.1). выделенный из продуктов жизнедеятельности актиномицетов Streptomyces spectabilis, является аминониклитольным, но не аминогликозидным антибиотиком, поскольку не содержит ни аминосахаридной части, ни гликозидной связи [316, 317. 318. 319. 320, 321, 322. 323, 324].
н
32 7 1
- 669-
Г лава 32
Аналогично стрептомицину спектиномицин связывается с рибосомальной ЗО-S субъединицей микроорганизмов и ингибирует синтез протеинов, однако при этом не происходит неправильного считывания генетического кода. Несмотря на широкий спектр активности, спек-тиноминин используется только при гонококковых инфекциях. Дру-ine грамотрицательные бактерии начинают проявлять резистентность в ходе лечения. Препарат эффективен в отношении большинства штаммов гонококков, а также ряда других грамотрицательных микроорганизмов.
Применяют для лечения острого гонорейного уретрита и проктита у мужчин и острого гонорейного проктита у женщин, вызванного чувствительными к препарату штаммами гонококков.
Синонимами препарата являются актиноспектоцин, спектам, тога-мицин и др.
Ванкомицин (Vancomycin)
Ванкомицин (32.7.2) был выделен в 1956 г. из продуктов жизнедеятельности актиномицегов Streptomyces onentalis (в настоящее время Nocardia oriemalis]. Основываясь на его химическом строении и составе, ванкомицин классифицируется как гликопептидный антибиотик. Его молекулярная масса значительно больше, чем у любого другого из практически применяемых антибиотиков [325, 326, 327, 328, 329, 330]
-670-
Антибиотики
Ванкомицн ингибирует синтез клеточных стенок бактерий. В отличие от р-лактамных антибиотиков, которые ингибируют третью стадию синтеза пептидогликана, ванкомицин действует на второй стадии построения клеточной стенки бактерий. Ванкомицин ингибирует ту реакцию, в которой повторяющаяся единица клеточной стенки отделяется от цитоплазматического мембраносвязанного фосфолипида и связывается с \же существующим пептидогликаном. Препарат также повреждает протопласты путем воздействия на цитоплазматические мембраны и пу гем ингибирования синтеза РНК. Ванкомицин активен только в отношении грамположительных бактерий. Он является наиболее мощным из известных антибиотиков в отношении и Staphylococcus aureus, и Staphylococcus epidermidus, включая метицицлин- и цефалоспоринрези-стентные штаммы.
Резистентность грамотрицательных организмов, таких как микобактерии, грибы, вирусы, простейшие, к ванкомицину возникает из-за барьера непроницаемости для препарата, обеспечиваемого их внешней мембраной. Резистентность к ванкомицину среди грамположительных организмов встречается редко.
Ванкомицин применяют при бактериальных инфекциях тяжелого течения, вызванных чувствительными к препарату микроорганизмами, при неэффективности пенициллинов и цефалоспоринов или непереносимости их больными при сепсисе, эндокардите, пневмонии, абсцессе легких, остеомиелите, менингите, энтероколите. Ванкомицин является лекарством выбора при инфекциях, вызванных метицицлинрезистент-ными видами Staphylococcus aureus, Staphylococcus epidermidus и другими коагулазотрицательными стафилококками, при эндокардитах, дифтеропдных инфекциях, применяется для лечения тяжело больных пациентов с колитом, вызванным Clostridium difficile.
Синонимом препарата является ванкоцин.
Рифампицин (Rifampicin)
Рифампицин — 5,6,9,17,19,21 -гексагидрокси-23 -мето кси-2,4,12,16,18, 20,22-гептаметил-8-(Т\т-(4-метил-1-пиперазинил)-формимидоил]-2.7-(эпоксипентадека-1,11,13-триенимино)-нафто-[2,1-Ь]фуран-1,11(2/7)-диона-21-ацетат (32.7.8).
Рифампицин является полусинтетическим производным рифампицина В — макролактамного антибиотика — одного из не менее чем пяти антибиотиков из смеси рифампицинов А, В, С, D и Е, которые называются рифампициновым комплексом (введенным в медицинскую практику в 1968 г.) и продуцируются актиномицетами Streptomyces
- 671 -
Г лава 32
mediteranei (Nocardia mediteranei). Синтез рифампицина начинается с того, что водный раствор рифампицина в условиях реакции окисляется в новое производное рифампицин S (32.7.4) с промежуточным образованием рифампицина О (32.7.3). Восстановлением хиноновой структуры последнего водородом с использованием в качестве катализатора палладия на угле получают рифампицин SV (32.7.5). Подвергая полученный продукт аминометилированию смесью формальдегида и пирролидина, получают З-пирролидинометилрифампинин SV (32.7.6). Окисляя последний тетраацетатом свинца до енамина и последующим гидролизом водным раствором аскорбиновой кислоты, получают 3-фор-милрифампицин SV (32.7.7). Вводя последний во взаимодейстие с 1-ами-но-4-метилпиперазином, получают искомый рифампицин (32.7.8) [331, 332. 333, 334].
H2N-N N-СН3
-672 -
Антибиотики
Рифампицин является антибиотиком широкого спектра действия и отличается от рифампицина SV (32.7.5), также применяемого в медицине в качестве самостоятельного препарата, большей эффективностью при приеме внутрь и более широким антибактериальным спектром действия
Рифампицин проявляет бактерицидный эффект путем ингибирования синтеза РНК. Препарат ингибирует ДНК-зависимую РНК-поли-меразу, предотвращая начало развития цепи, а не ее удлинение. Рифампицин не связывается с РНК-полимеразой ядра клеток млекопитающих и не воздействует на соответствующий синтез РНК. Он может ингибировать митохондриальный синтез РНК, однако концентрации, требуемые для этого, в несколько сот раз превышают таковые, необходимые для синтеза РНК.
Рифампицинн высокоактивен в отношении Mycobacterium tuberculosis, в ряду атипичных микобактерий по отношению к Mycobacterium kansasii. Mycobacterium marinum и к большинству видов Mycobacterium scrofulaceum и Mycobacterium xenopt. Чувствительность других микобактерий варьирует. Рифампицин проявляет активность и в отношении Mycobacterium lepare.
Кроме микобактерий, рифампицин проявляет активность и в отношении широкого ряда организмов. Он высокоактивен в отношении Sta-phyilococcus aureus, неэнтерококковых видов Streptococcus и Listeria monocytogenes. Среди грамотрицательных видов к рифампицину очень чувствительны Neisseria meningitidis, Haemophilus influenzae и Legionella. Устойчивы E coli и Proteus mirabilis. Анаэробные кокки, виды Clostridium и Bacteroids часто оказываются чувствительны к рифампицину.
Рифампицин является наиболее эффективным лекарством для лечения легочной и нелегочной форм туберкулеза, включая туберкулезный менингит. Всегда должен применяться в сочетании с другими препаратами.
Синонимами препарата являются рифадин, римактан, рифапиам, римактазид и др.
Полимиксины
Полимиксины являются группой родственных полипептидных антибиотиков, вырабатываемых спорообразующими почвенными бактериями Bacillus polymyxa и Bacillus circulans и отличающихся аминокислотным составом. Было определено пять разных полимиксинов — полимиксин А, В, С, D и Е, отличающихся аминокислотным составом и дифференцированных добавочными буквенными обозначениями
- 673-
Глава 32
и названиями — полимиксин В (аэроспорин), полимиксин Е (колистин). Известно, что одни штаммы Bacillus polymyxa в процессе развития образуют лишь полимиксин А и С, другие синтезируют полимиксин В и D. Позже был выделен полимиксин М, сульфометильное производное полимиксина Е.
В структуре всех этих антибиотиков присутствует треонин и а,у-ди-аминомасляная кислота. Особенность группы полимиксинов в том, что в их составе имеется 4-5 свободных у-аминных групп а,у-диами-номасляной кислоты, что придает им свойства катионных детергентов, способных образовывать комплексы с фосфолипидами клеточных мембран. По антибиотическому действию все полимиксины сходны.
Полимиксин В (Polymyxine В)
Полимиксин В — М-[3-амино-1-[[1-[[3-амино-1-[[6,9,18-трис-(2-аминоэтил)-15-бензил-3-(1-гидроксиэтил)-12-изобу тил-2,5,8,11,14, 17,20-гептаоксо-1,4,7,10,13,16,19-гептаазациклотрис-21 -ил]-карбамо-ил]-пропил]-карбамоил]-2-гидроксипропил]-карбамоил]-пропил]-6-метилоктанамид (32.7.9) [335, 336, 337].
Полимиксин Е (Polymyxine Е)
Полимиксин Е — М-[3-амино-1-[[1-[[3-амино-1-[[6,9,18-трис-(2-аминоэтил )-3-(1-гидроксиэтил)-12,15-диизобутил-2,5,8,11,14,17,20-гептаоксо- 1,4,7,10,13,16.19-гептаазациклотрис-21 -ил]-карба.моил]-пропил]-карбамоил]-2-гидроксипропил]-карбамоил]-пропил]-6-ме-тил-октанамид (32.7.10) [338].
-674-
ДНТИбИОТИКИ
В принципе, при физиологических pH полимиксины являются катионными поверхностно-активными соединениями и проявляют свое бактерицидное действие путем взаимодействия с фосфолипидной компонентой цитоплазматической мембраны чувствительных бактерий и тем самым нарушают осмотическую целостность их клеточной мембраны, способствуя выведению многих компонентов, в том числе ионов К+, из цитоплазмы в окружающую среду. Детальный биохимический механизм их действия пока неясен.
Практически все полимиксины исключительно активны против аэробных грамотрицательных микроорганизмов. В частности, они активны против Pseudomonas aeruginosa, Escherichia coli, Klebisella pneumoniae, Enterobacter, Salmonella, Shigella Haemophilus и др. He действуют на кокковые аэробные (стафило-, стрепто-, пневмо-, гоно- и менингококки) и на анаэробные микроорганизмы, а также на большинство штаммов Proteus, на возбудителей туберкулеза и дифтерии.
Эти препараты применяют при желудочно-кишечных заболеваниях (колиты, энтероколиты, при острой и хронической дизентерии, при сепсисе, менингите, пневмонии, инфекциях мочевых путей и других заболеваниях, вызванных Pseudomonas aeruginosa, когда другие антибиотики неэффективны. В виде мазей с успехом используются при лечении некоюрых форм экзем, фурункулов, гидраденитов и других кожных заболеваний.
Парентеральное применение полимиксинов ограничено ввиду их нейро- и нефротоксических эффектов. Они не являются лекарствами выбора для какой-то конкретной инфекции. Однако их применяют при серьезных, угрожающих жизни инфекциях, например бактеремии, вы-шанной. как уже было отмечено, некоторыми штаммами Pseudomonas aeruginosa.
- 675-
Глава 32
Бацитрацины
Бацитрацины также являются полипептидными антибиотиками, выделенными из к}латеральной жидкости жизнедеятельности Bacillus hcheniformis Выделено десять индивидуальных бацитрацинов А, Аь В, С, D, Е, Fb F2, F^, G Однако сам препарат, называемый бацитрацин и применяемый в медицине, является смесью полипептидных антибиотиков Все бацитрацины, являясь полипептидами, содержат в своем составе и тиазол иновое кольцо
Бацитрацин A (Bacitracin А)
Бацитрацин А — М-[[2-(1-амино-2-метилбутил)-4,5-дигидро-4-тиа-золил]карбонил] бацитрацин F (32 7 11) составляет основную часть выделенных фракций [338, 339, 340, 341]
СО— Leu—G'u—I Isu—Ly s—Orn—I leu
NH—CO \
NH2 HOOC-Asp—Asp—His—Phe
NH2
32 7 11
Бацитрацин -- бактерицидный препарат и ингибирует образование линейных пептидогликановых цепей, являющихся основным компонентом клеточных стенок бактерий К препарату чувствительно большинство грамположительных бактерий, включая стафилококки, стрептококки, а также Clostridium dijficile В то же время препарат почти не действует на грамотрицательные бактерии Наиболее часто препарат применяется наружно при местном лечении гнойных инфекций глаз и кожи Показания для внутримышечного введения препарата крайне ограничены ввиду его нефротоксичности Он может быть использован как препарат крайнего случая у детей со стафилококковой пневмонией, с эмпиемой, устойчивой ко всем другим антибиотикам
Антибиотики
Список литературы
1
2
3
4
5
6
7
8
9
10 И
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
Fleming A Bnt J Exp Pathol 10, 226 (1929)
С letterbuck Р et al / Biochem J 26, 1907 (1932)
Chain E etal /Lancet 2,226(1940)
Abraham E et al //Lancet 2, 177 (1941)
US Pat 3 024 169 (1962)
LSPat 3 159 617(1964)
Bachi M et al J Chem Soc Perkm Trans I, 1980, 11
US Pat 2 479 295 (1946)
IS Pat 2 479 296(1946)
US Pat 2 562 410(1946)
US Pat 2 941 995 (1960)
Sheehan J etal /J Am Chem Soc 79, 1262 (1957)
Sheehan J etal /J Am Chem Soc 81, 3089 (1959)
Sheehan J etal /J Am Chem Soc 84, 2983 (1962)
Glombitza V Ann 673. 166(1964)
US Pat 2 951 839 (1960)
Doyle F etal 'J Chem Soc 1962. 1457
US Pat 3 157 639 (1964)
US Pat 3 248 386 (1966)
Bnt Pat 880 400 (1964)
US Pat 2 996 501 (1961)
Bnt Pat 905 778 (1961)
Bnt Pat 958 478 (1963)
Doyle F etal 'Nature 192, 1183 (1961)
Dovle F etal /J Chem Soc 1963, 5838,
US Pat 2 239 50T(1966)
Bnt Pat 978 299 (1962)
Belg Pat 657 504 (1964)
Bnt Pat 893 049 (1959)
Bnt Pat 902 703 (1961)
US Pat 2 985 648 (1961)
Ger Pat 1 139 844 (1960)
Ger Pat 1 545 534 (1965)
IS Pat 2 985 648 (1961)
Doyle F etal J Chem Soc 1962, 1440
Bnt Pat 991 586 (1964)
US Pat 3 140 282 (1964)
- 677-
Г лава 32
38 39 40
41 42
43 44
45
46 47 48 49
50 51
52 53
54
55 56 57 58
59 60 61
62 63
61
65 66 67
68 69 ”0 71
"'3
‘4
^5 76
LSPat 4 128 547 (1978)
Bnt Pat 1 339 605 (1972)
Ger Pat 2 611 286(1976)
Fr Pat 2 100 682 (1971)
US Pat 3 933 795 (1976)
Ger Pat 2 104 579 (1971)
Ger Pat 2 152 967 (1971)
Ger Pat 2 152 968 (1971)
Ger Pat 2 318 955 (1973)
Ger Pat 2 519 400(1975)
Ger Pat 2 824 610(1978)
Bnt Pat 1 508 062 (1975)
USPat 4 087 424(1978)
USPat 3 142 673 (1964)
US Pat 3 282 926(1966)
USPat 1 492 291 (1970)
Ger Pat 1 295 558 (1964)
Ger Pat 1 770 225 (1968)
Bnt Pat 1 004 670(1964)
Bnt Pat 1 197 973 (1967)
Belg Pat 646 991 (1964)
Ger Pat 2 244 556 (1972)
Bnt Pat 1 508 97'' (1975)
Ger Pat 2 517 316 (1975)
Ger Pat 2 560 074(1975)
Bnt Pat 1 543 563 (1976)
US Pat 4 490 294(1984)
USPat 4 490 295 (1984)
Brown A etal J Antibiot 29, 668 (1976)
Belg Pat 867 859 (1978)
US Pat 4 234 579 (1980)
Volkman R et al 7J Org Chem 47, 3344 (1982)
Belg Pat 618 663 (1962)
LSPat 3 218 318 (1965)
Fi Pat 1 384 197 (1965)
Chainette R etal J Am Chem Soc 84,3401 (1962)
Ratcliffe R etal Tetrahedron Lett 1973.4649
LSPat 3 422 100(1969)
L S Pat 3 503 967(1970)
LS Pat 3 578 661 (1971)
Crast L etal J Med Chem 16. 1413 (1973)
-678-
Антибиотики
79 I S Pat 3 516 997(1970)
«о S Afr Pat 68 04 5]3 (1970)
81 k .mune 4 et al / J Antibiot 23, 131 (1970)
82 I 8 Pat 3 507 861 (1970)
S' LSPat 3 275 626 (1966)
84 R>anC etal J Vied Chem 12, 310 (1968)
8' Fr Pat 1 524 225 (1967)
86 Bnt Pat 1 174 335 (1967)
8” ( S Pat 3 485 819 (1969)
88 Get Pat 1 931 722 (1969)
89 Dolfim E et al / J Med Chem 14, 117 (1971)
90 LSPat 3 489 752 (1970)
91 LS Pat 3 98s 741 (1968)
92 Bnt Pat 1 240 687 (1968)
93 Bnt Pat 1 532 682 (1977)
94 Get Pat 2 216 113 (1972)
93 LSPat 3 816 253 (1974)
96 I S Pat 4 504 657 (1985)
97 Bnt Pat 1 453 049 (1974)
98 Ger Pat 1 931 722 (1969)
9'9 Bnt Pat 1 453 049 (1974)
100 Ger Pat 2 439 880 (1974)
101 Ger Pat 2 462 376(1974)
102 Ger Pat 2 204 060 (1972)
103 Ger Pat 2 223 375 (1972)
104 Ger Pat 2 265 234(1972)
105 USPat 3 966 717 (1976)
106 USPat 3 971 778 (1976)
107 LSPat 3 974 153 (1976)
108 Ger Pat 2 706 413 (1977)
109 USPat 4 267 320 (1981)
110 USPat 4 075 061 (1978) 111 LSPat 4 164 447(1976) 112 LSPat 3 641 021 (1972) 113 USPat 3 840 531 (1974) 114 US Pat 3 974 153 (1976) 115 US Pat 3 903 278 (1975) 116 Ger Pat 2 018 600(1970) 1Г Ger Pat 2 06э 621 (1970) 118 Get Pat 2 730 579 (197'?) 119 Ger Pat 2 312 99^(1973)
-679-
Глава 32
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
Ger Pat 2 611 270(1976)
US Pat 4 048 311 (1977)
US Pat 4 093 723 (1978)
US Pat 4 159 373 (1979)
US Pat 4 100 346(1978)
US Pat 4 172 196(1979)
US Pat 4 182 863 (1980)
Ger Pat 2 538 804(1976)
Gottstain W etal J Amtibtot 29, 1226(1976)
Ger Pat 2 456 528 (1974)
Ger Pat 2 318 829(1973)
Ger Pat 2 365 582 (1973)
Ger Pat 2 129 675(1971)
Ger Pat 2 143 331 (1971)
Ger Pat 2 203 653 (1972)
Ger Pat 2 258 278 (1972)
US Pat 3 775 410(1973)
US Pat 3 843 641 (1974)
Hiraoka T etal Heterocycles (Japan) 8,719(1977)
Ger Pat 2 824 559 (1978)
Iwanami M etal Chem Pharm Bull 28, 2629 (1980)
US Pat 3 925 372 (1975)
Ger Pat 2 408 698 (1974)
Chainette R etal 71 Med Chem 18, 403 (1975)
Ger Pat 2 702 501 (1977)
Ger Pat 2 708 439 (1977)
Ger Pat 2 256 736(1976)
US Pat 4 098 888 (1978)
US Pat 4 152 432 (1979)
US Pat 4 427 674(1984)
US Pat 4 463 002(1984)
Ger Pat 2 810 922 (1978)
Ger Pat 2 922 036(1979)
US Pat 4 431 806(1984)
US Pat 4 327 210(1982)
But Pat 2 022 090(1979)
Reiner R etal /J Antibiot 33 783 (1980)
Ger Pat 2 921 316(1979)
US Pat 4 258 041 (1981)
US Pat 4 525 587 (1985)
Bnt Pat 2 025 398 (1979)
- 680 -
антибиотики
161
162
163
164
165
166
167
168
169
170
171
172
174
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
Ger Pat 3 037 102 (1981)
LSPat 4 329 453 (1982)
US Pat 4 110 327(1978)
Ger Pat 3 519 400(1975)
Bnt Pat 1 508 062(1975)
Bnt Pat 2 508 071 (1976)
US Pat 4 410 522 (1983)
Belg Pat 837 682 (1976)
Ger Pat 2 713 370(1977)
US Pat 4 138 486(1979)
US Pat 4 180 571 (1979)
Nansada M et al dJ Med Chem 22, 758 (1979)
Narisada M et al //Heterocycles 7, 839 (1977)
US Pat 4 406 899(1983)
Ger Pat 3 307 550(1983)
Naito T et al /'J Antibiot 39, 1092 (1986)
US Pat 4 609 653 (1986)
Seibert G etal '/Arzneimittel-Forsch 33, 1084(1983)
US Pat 4 194 047(1980)
Ger Pat 2 652 679(1976)
Leanza И et al/'J Med Chem 22, 1435 (1979)
Shinkat 1 et al //Tetrahedron, 23. 4903 (1982)
bur Pat Appl 48 301 (1982)
Eur Pat Appl 48 025 (1982)
Ger Pat 3 104 145 (1981)
Bnt Pat 2 071 650(1981)
Neth Pat Appl 8 100 571 (1981)
US Pat 2 653 899 (1953)
US Pat 2 823 203 (1958)
Bnt Pat 1 121 056(1957)
Ger Pat 2 758 942(1977)
US Pat 4 331 803 (1982)
Eur Pat Appl 41 355 (1981)
US Pat 4 517 359 (1985)
Belg Pat 892 357 (1982)
US Pat 2 482 055 (1949)
US Pat 2 609 329 (1949)
US Pat 2 899 422 (1959)
US Pat 2 987 449 (1961)
US Pat 3 050 446(1962)
Duggat В Ann N Y Acad Sci 51, 177 (1948)
- 681 -
Г пава 32
202. US Pat. 2.516.080(1950).
203. Finlay A. et al.//Science. 111. 85 (1950).
204. Regna P et al./'Ann. N. Y. Acad. Sci. 53, 221 (1950).
205. Regna P. et al.//J. Am. Chem. Soc. 73, 4211 (1951).
206. US Pat. 2.699.054 (1955).
207. US Pat. 3.005.023 (1961).
208. Boothe J et al.'/J. Am. Chem. Soc. 75, 4621 (1953).
209. Conover L. et al.'/J. Am. Chem. Soc. 75, 4622 (1953).
210. US Pat. 2.734.018 (1956).
211. US Pat. 2.712.517 (1955).
212. US Pat, 2.886.595 (1959).
213. US Pat. 3.019.173 (1962).
214. US Pat. 3.301.899 (1967).
215. US Pat. 2.878.289 (1959).
216. US Pat. 3.012.946(1961).
217. US Pat. 3.019.172 (1962).
218. US Pat. 3.050.446(1962).
219. US Pat. 3.154.476(1963).
220. Ger. Pat. 1.041.213 (1957).
221. McCormic J. et al./'J. Am. Chem. Soc. 79. 4561 (1957).
222. US Pat. 2.984.686(1961).
223, US Pat. 3.026.354 (1962).
224. Blackwood R. et al/J. Am. Chem. Soc. 83, ТПЪ (1961).
225. Blackwood R et al.. 'J. Am. Chem. Soc. 85. 3943 (1963).
226. US Pat. 3.019.260(1962).
227. Ger. Pat. 1.082.905 (1958).
228. US Pat. 3.200.149(1965).
229. Ger. Pat. 1.793.556(1961).
230. Ger. Pat. 1.298.522 (1961).
231. US Pat. 3.148.212 (1964).
232. US Pat. 3.226.436 (1965).
233. US Pat. 3.345.410 (1967).
234. Ger. Pat. 1.245.942 (1962).
235. Church R. et al./ 'J. Org. Chem. 36, 723 (1971).
236. Martell A. et al.//J. Med. Chem. 10, 44 (1967).
237. Bernardi J. et al.//Farmaco Ed. Sci. 30, 736 (1975).
238. US Pat. 2.449.866(1948).
239. US Pat. 2.765.302 (1956).
240. US Pat. 2.868.779 (1959).
241. US Pat. 2.493.489 (1950).
242. Fried J et al/ J. Biol. Chem. 168, 391 (1947).
-682-
антибиотики
243. Fried J etal.//J. Biol. Chem. 174, 57 (1948).
244. Fried J et al./'J. Am. Chem. Soc. 69, 1549 (1947).
245. Fried J. et al.'/J. Am. Chem. Soc. 71, 2452 (1949).
246. Fried J. et al.,7}. Am. Chem. Soc. 71, 135 (1949).
247. Peck R. et al.//J. Am. Chem. Soc. 70, 3968 (1948).
248. US Pat. 2.799.620(1957).
249. US Pat. 2.848.365 (1958).
250. US Pat. 3.108.996 (1963).
251. US Pat. 3.005.815 (1961).
252 US Pat. 3.022.228 (1962).
253. Waksman S. et al.//Science. 109, 305 (1949).
254. PeckR. et al.//J. Am. Chem Soc. 71, 2590 (1949).
255. Regna P et al.//J. Am. Chem. Soc. 72, 1045 (1950).
256. US Pat. 2.916.485 (1959).
257. US Pat. 2.895.876(1959).
258. Brit. Pat. 880.035 (1959).
259. US Pat. 2.931.798 (1960).
260. US Pat. 2.936.307(1960).
261. US Pat. 2.967.177 (1961).
262. US Pat. 3.032.547 (1962).
263. US Pat. 3.691.279 (1972).
264. Ger. Pat. 2.514.985 (1975).
265. Ger. Pat. 2.361.115 (1973).
266. Ger. Pat. 2.533.985 (1974).
267. Okutani T. et al.//J. Am. Chem. Soc. 99, 1278 (1977).
268. Tagaki У et al./O. Antibiot. 26, 403 (1973).
269. Tanabe T. et alUTetrahedron Lett. 1977, 3607 .
270. Tagaki У. et al.//Bull. Chem. Soc. Japan, 49. 3649 (1979).
271. US Pat. 3.091.572 (1963).
272. US Pat. 3.136.704(1964).
273. Weinstein M. et al.//Antimicrob. Ag. Chemoter. 1963, 1.
275. Maehr H et al.//J. Am. Chem. Soc. 89, 6787 (1967).
276. Nagabushan A. et al.//J. Org. Chem. 40, 2830 (1975).
277. Meyer zu Reckendorf W. et al.//Ber. 105, 2546 (1972).
278. Chmielewski M et al.//Carbohyd. Res. 70, 275 (1979).
279. Kawaguchi H. et al.//J. Antibiot. 25. 695 (1972).
280. Ger. Pat. 2.234.315 (1972).
281. US Pat. 3.781.269 (1973).
282. US Pat. 3.974.137(1976).
283. Ger. Pat. 2.432.644 (1974).
284. Ger. Pat. 2.716.533 (1977).
-683-
Глава 32
285
286
287
288
289
290
291
292
293
294
295
296
297
298
299
300
301
302
303
304
305
306
307
308
309
310
311
312
313
314
315
316
317
318
319
320
321
322
323
324
325
Ger Pat 2 818 822 (1978)
Ger Pat 2 818 992 (1978)
Ger Pat 2 462 485 (1974)
Ger Pat 2 437 160(1974)
USPat 4 029 882 (1977)
USPat 4 002 742 (1973)
Wright J Chem Commun 1976, 206
USPat 3 086 912 (1963)
USPat 3 155 580 (1964)
USPat 3 380 992 (1968)
USPat 4 091 204(1978)
USPat 3 418 414(1968)
US Pat 3 475 407 (1969)
US Pat 3 509 127(1970)
USPat 3 627 887 (1971)
USPat 3 544 551 (1970)
US Pat 3 513 155 (1970)
Birkenmeyer H etal /J Med Chem 13 616 (1970)
Magerlein В et al //Antimicrob Ag Chemoter 1966, 727 US Pat 2 483 871 (1949)
USPat 2 483 884(1949)
USPat 2 483 885 (1949)
USPat 2 483 892 (1949)
USPat 2 687 434(1954)
USPat 2 651 661 (1953)
USPat 2 786 870(1957)
Long L etal/J Am Chem Soc 71, 2469 (1949)
RebstockM et al //J Am Chem Soc 71, 2458 (1949)
Belg Pat 539 991 (1955)
Belg Pat 558 378 (1957)
Ger Pat 1 016 718 (1953)
USPat 3 206 360(1965)
USPat 3 234 092 (1966)
USPat 3 272 706(1966)
USPat 3 819 485 (1974)
Mason J et al //Antibiot & Chemoter 11, 118(1961) Bergy M et al //Antibiot & Chemoter 11, 661 (1961)
Sinclair A et al Antimicrob Ag Chemoter 1961, 503 Hoeksema H et al' J Am Chem Soc 84, 3212 (1962)
Wiley P et aU J Am Chem Soc 85, 2652 (1963) USPat 3 067 099 (1962)
- 684-
Антибиотики
326
327
328
329
330
331
332
333
334
335
336
337
338
339
340
341
342
Eur Pat Appl 145 484 (1984)
Williamson M et al //J Am Chem Soc 84. 3212 (1962)
Mai shall F et al //J Med Chem 8, 18 (1965)
Wenngs W et al /J Chem Soc Perkin Trans I 1972, 443
Smith К et al '/J Chem Soc Perkin Trans I 1974, 2369 USPat 3 342 810(1967)
Ger Pat 2 846 321 (1978)
Ger Pat 1 089 513 (1959)
Maggi M et al /'Chemotherapia 11, 285 (1966)
USPat 2 565 057 (1951)
US Pat 2 595 605 (1952)
USPat 2 771 397 (1956)
USPat 2 915 432 (1959)
USPat 2 498 165 (1950)
USPat 2 828 246 (1951)
Johnson В et al//Science 102. 376 (1945)
Anker H etal/J Bactenol 55, 249 (1948)
I Глава 33
t Антимикробные препараты
33.1. Сульфонамидные препараты и триметоприм
Сульфонамидные препараты являются группой синтетических антимикробных препаратов широкого спектра действия в отношении как грамположительных, так и грамотрицательных микроорганизмов, введенных в медицинскую практику еще до открытия пенициллинов.
Сульфонамидные препараты являются производными сульфаниламида (и-аминобензолсульфонамида), который является структурным аналогом н-аминобензойной кислоты — компонента, необходимого бактериям для синтеза фолиевой кислоты, — предшественника синтеза пурина, нуклеиновых кислот и, особенно, ДНК.
п-аминобепзойная кислота
сульфаниламид
Клетки животных не способны сами синтезировать фолиевую кислоту, и она должна поступать в их организм с пищей.
Вместе с тем большинство бактерий не способны утилизировать фолиевую кислоту экзогенного происхождения и самостоятельно осуществляют необходимый для жизнедеятельности синтез фолиевой кислоты. Эта разница между бактериальными и животными клетками
- 686-
Антимикробные препараты
и является причиной избирательной токсичности сульфонамидов.
Сульфаниламид, имея структуру близкую к структуре п-аминобен-зойной кислоты, конкурирует с п-аминобензойной кислотой за включение в молекулу фолиевой кислоты и, в итоге, занимая место л-амино-бензойной кислоты, «мешает» биосинтезу фолиевой кислоты. В результате «введенные в заблуждение» ферменты конструируют «ложную» молекулу фолиевой кислоты, не способную выполнять жизненные функции истинной фолиевой кислоты.
соон
«ложная фолиевая кислота»
Таким образом, сульфонамиды являются бактериостатическими препаратами, которые ингибируют рост бактерий путем вмешательства в микробный синтез фолиевой кислоты. Точнее, сульфонамиды блокируют биосинтетический путь синтеза фолиевой кислоты, конкурентно ингибируя опосредованное ферментом дигидроптероат синтетазой превращение и-аминобензойной кислоты в фолиевую кислоту, что позволяет считать их антиметаболитами.
В настоящее время в медицинской практике применяются различные су тьфаниламидные препараты, выбор которых зависит от различных факторов и, в первую очередь, от типа возбудителя и течения заболевания, скорости всасывания препарата из ЖКТ и скорости его выделения, его способности проникать в различные органы и ткани и т. п.
Сульфонамиды имеют широкий спектр антимикробной активност, включая Staphylococcus aureus, неэнтерококковые виды Streptococcus, Listcia monocytogenes, Nocardia, Neisseria. Haemophilias influenzae, эн-терические грамотрицательные виды Eschrrichia coh и Proteus mirabilis и некоторые виды анаэробных бактерий. В первую очередь, сульфона-'ш.:ы используются для лечения неосложненных инфекций мочеполо-В'чо тракта, при инфекциях, вызванных Nocaria asteroides, стрептококковых фарингитов, менингококковых заболеваниях, токсоплазмозов И тр.
При длительном применении к ним возникает резистентность. Бак-териатьная резистентность к сульфонамидам может развиться в резуль
- 687-
Глава 33
тате мутаций, выражающихся либо в перепроизводстве /г-аминобен-зойной кислоты, либо в изменениях в самой дигидропротероат синтетазе, которая становится менее чувствительной к препаратам. Резистентность может быть также опосредована плазмидами, кодирующими выработку дигидропротероат синтетазы или же пониженной проницаемостью мембраны бактериальных клеток для препарата.
Четкой классификации сульфаниламидных препаратов в настоящее время нет. Однако их группируют как системного (резорбтивного действия) и местного применения. Их подразделяют на короткодействующие (сульфацетин, сульфадиазин, сульфамеразин, сульфаметазин, сульфаметизол. сульфизоксазол); средней длительности действия (сульфаметоксазол, сульфапиридин); долгодействующие (сульфаметок-сипиридазин, сульфамтер), которые, однако, более не применяются в качестве самостоятельных препаратов вследствие весьма редких, но, тем не менее, встречающихся реакций гиперчувствительности. В число препаратов для местного применения входят препараты дня офтальмологического применения (сульфацетамид, сульфизоксазол), вагинального применения (сульфабензамид. сульфацетамид, сульфатиазол, сульфизоксазол) и наружного применения (мафенид, сульфадиазин серебра). Наконец, в эту группу входит и сульфасалазин (фталилсульфатиазол) — препарат, действующий в просвете кишечника, но плохо всасываемый из ЖКТ.
В химическом отношении, за редкими исключениями (например, фталазол), сульфаниламидные препараты можно представить следующей простой формулой:
h2n
Синтезировано огромное число соединений этого типа, главным образом, путем взаимодействия 4-ацетиламинобензолсульфохлорида с различными аминами, преимущественно ароматического и гетероаро-матического рядов, с последующим снятием защитной ацетильной группы с аминной части молекулы либо взаимодействием 4-амино-бензолсульфонамида с соответствующими ароматическими или гете-роароматическими галогенпроизводными. Ввиду простоты исполнения преимущественно пользуются первым методом. При выведении корреляции структура-активность было показано, чго свободная «-аминогруппа в бензольном кольце необходима для проявления антибактери
- 688 -
Антимикробные препараты
альной активности и может быть замещена только такими группами, которые позволяют обратную ее трансформацию в свободную аминогруппу. Наличие дополнительных заместителей в о- и и-положениях бензольного кольца снижает активность данного ряда соединений в качестве антибактериальных средств. Замещение одного из атомов водорода при атоме азота в сульфонамидной части молекулы приводит к значительным изменениям и в активности, и в растворимости соединений. Более того, природа заместителя при сульфонильном радикале определяет антимикробную активность и фармакокинетические особенности каждого отдельно взятого соединения. Таким образом, при наличии легкодоступного и многотоннажного продукта — 4-аие-зиламинобензолсульфохлорида, получаемого взаимодействием ацети-ланилина с хлорсульфоновой кислотой, или же при наличии 4-а.ми-побензолсульфонамида, получаемого дальнейшим взаимодействием 4-ацтиламинобензолсульфохлорида с аммиаком с последующим гидролизом задача синтеза новых потенциальных сульфонамидов практически сводится к получению новых гетероароматических аминов. Этими путями получены рассматриваемые ниже наиболее широко применяемые сульфонамиды.
Сульфацетин (Sulfacetin)
Сульфацетин — \|-(1-этил-1.2-дигидро-2-оксо-4-пиримидинил)-сульфаниламид (33.1.5) получают взаимодействием 4-ацетилами-нобензолсульфохлорида с 1-этилцитозином (33.1.3) с последующим восстановительным дезацилированием ацетанилидной части молекулы (33.1.4) с использованием системы цинк-гидроокись натрия в искомый сульфацетин. 1-этилцитозин (33.1.3), в свою очередь, получают исходя из 3-этиламинопропионитрила, взаимодействием которого с циановой кислотой (цианат калия — соляная кислота) на первом этапе синтеза получают 1-(2-цианоэтил)-1-этилмочевину (33.1.1). Последняя в присутствии метилата натрия легко циклизуется в 1-этил-5,6-дигидроцитозин, который с целью последующего окисления ординарной связи Cs-C6 выделяют в виде гидробромида (33.1.2). Оптимальным окислителем для этой цели оказался бром, и при использовании в качестве растворителя нитробензола был получен гетероароматический амин — 1-этилцитозин (33.1.3), который был трансформирован в искомый сульфацетин описанным выше путем — взаимодействием с 4-ацетиламинобензолсульфохло-ридом и последующим снятием защитной ацетильной группы с аминной части молекулы [1, 2, 3, 4].
-689-
Глава 33
NC—CH2-CH2-NH-C2H5
О 1 CH3ONa
KNCO HCI H?N—C 2 HBr
---------‘n-C^5 -------------------------
NC—CH2-CH/
33 1 1
Zn NaOH
CH3-c-NH
33 1 5
Препарат эффективен при инфекциях, вызванных стрептококками, гонококками, пневмококками, гонококками, стафилококками, а также кишечной палочкой.
Сульфацетин применяют при пневмонии, церебральном менингите, стафилококковом и стрептококковом сепсисе и других инфекционных заболеваниях.
Синонимом препарата является реноквид.
Сульфадиазин (Sulfadiazin)
Сульфадиазин — М^-З-пиримидинилсульфаниламид (33.1.7) синтезируют взаимодействием 4-ацтиламинобензолсульфохлорида с 2-ами-нопиримидином с получением ацетанилидного производного (33.1.6), последующий гидролиз которого щелочью приводит к получению искомого с^льфадиазина [5, 6, 7, 8].
о __
CH3-C-NH—SOj-CI т
33 1 в
33 1 7
Так же как и сульфацетин, препарат эффективен при инфекциях, вызванных стрептококками, гонококками, пневмококками, гонококками, стафилококками, а также кишечной палочкой.
- 690-
Антимикробные препараты
Сульфадиазин применяют при пневмонии, церебральном менингите. стафилококковом и стрептококковом сепсисе и других инфекционных заболеваниях, однако он является препаратом выбора при нокар-диозе. Ввиду малой растворимости и определенной нефротоксичности препарат не рекомендуют при инфекциях мочеполового тракта.
Синонимами препарата являются ренковид, фламазин, стеринор, тер-фонил и др.
В виде серебряной соли (сульфадиазин серебра) применяют в качестве наружного антибактериального средства, в основном для лечения ожоговых ран. Полагают, что наличие в молекуле препарата иона серебра способствует усилению антимикробного и ранозаживляющего действия.
Сульфамеразин (Sulfamerazin)
Сульфамеразин — К'-(4-метил-2-пиримидинил)сульфаниламид (33.1.12) также получают описанным выше способом — взаимодействием 4-ацтиламинобензолсульфохлорида с 2-амнно-4-мегил-пиримидином (33.1.10), который, в свою очередь, получают по традиционной схеме синтеза производных пиримидина. Конденсацией ацетоуксусного эфира с гуанидином получают 4-метил-2-амино-ниримидин-6-он (33.1.8). Взаимодействием последнего с хлорокисью фосфора получают 4-метил-2-амино-6-хлорпиримидин (33.1.9). Далее восстановлением водородом с использованием в качестве катализатора палладия на угле удаляют атом хлора при С„ пиримидинового кольца, Полученный 4-метил-2-аминопиримидин (33.1.10) вводят во взаимодействие с 4-ацтиламинобензолсульфохлоридом с получением ацетанилидного производного (33.1.11), последующий гидролиз которого щелочью приводит к получению искомого сульфамеразина [8. 9, 10].
1 ак же как и все рассматриваемые сульфаниламиды, препарат эффективен при инфекциях, вызванных стрептококками, гонококками,
-691 -
Глава 33
пневмококками, гонококками, стафилококками, а также кишечной палочкой.
Синонимами препарата являются досульфин, полагин, ромезин и др.
Сульфаметазин (Sulfamethazine)
Сульфаметазин — М'-(4,6-диметил-2-пиримидинил)сульфаниламид (33.1.13) также предложено получать описанным выше способом — взаимодействием 4-ацетиламинобензолсульфохлорида с 2-амино-4.6-диметилпиримидином, который, в свою очередь, получают конденсацией ацетилацетона с гуанидином и последующим гидролизом ацетиламинной группы щелочью [11, 12, 13, 14, 15].
33 1 13
Препарат применяют при пневмококковых, стафилококковых и стрептококковых инфекциях, а также при сепсисе, гонорее и других инфекционных заболеваниях.
Синонимами препарата являются сульфадимезин, сульфадимидин.
Сульфаметизол (Sulfamethizole)
Сульфаметнзол — №-(5-метил-1,3,4-тиадиазол-2-ил)сульфанила-мид (33.1.15) предложено получать двумя путями. Согласно первому способу, 5-амино-2-метил-1,3,4-тиадиазол вводят во взаимодействие с 4-нитробензхолсульфохлоридом с получением нитропроизводного (33.1.14), который восстанавливают с помощью железных стружек в уксусной кислоте до искомого сульфаметизола.
02М—к о— SO2-Cl
33 1 14
Fe Н +
33 1 15
N-N
-692-
Антимикробные препараты
Второй способ получения сульфаметизола заключается во взаимодействии 4-ацетиламинобензолсульфохлорида с тиосемикарбазоном ацетальдегида последующей окислительной циклизации продукта (33 1.16) в замещенный 1,3,4-тиадиазол в присутствии феррицианида калия в щелочи с одновременным снятием защитной ацетильной группы [16. 17].
S" и /=\ Лн <-з1г-е(СМб
- * СНт—С—\Н“A
№СН"*СНз
33 1 16
Препарат обладает антибактериальной активностью в отношении стрептококков пневмококков, стафилококков, менингококков и гонококков, кишечной палочки, возбудителя дизентерии и др. Малотоксичен. В основном применяют при острых неосложненных инфекциях мочеполового тракта, вызванных чувствительными микроорганизмами. Ввиду его быстрого вывода из организма почками, уровень препарата в плазме остается низким, и поэтому препарат не используется для лечения инфекций локализованных вне мочеполового тракта. Более предпочтительным препаратом является сульфизоксазол.
Синонимами препарата являются уросол, руфол, тиосульфил и др.
Сульфизоксазол (Sulfizoxazole)
Сульфизоксазол — №-(3,4-диметил-5-изоксазолил)сульфаниламид (33.1.19) синтезируют, вводя во взаимодействие 4-ацстилами-нобензолсульфохлорид и 5-амино-3,4-диметилизокс-азол (33.1.17). который, в свою очередь, получают гетероциклизацией 2-метил-ацетилацетонитрила с гидроксиламином и последующим кислотным гидролизом соляной кислотой защитной ацетильной группы в полученном продукте (33.1.18) [18, 19].
н2кюн
33 1 17
СН3-C-NH
- 693-
Глава 33
CH3 CH3
CH3-C-NH—Z~"\— so2 — NH*^o-
CH3 CH3
h2nO-s--nh^
33 1 18
33 1 19
Как и все рассматриваемые сульфаниламиды, препарат эффективен при инфекциях, вызванных стрептококками, гонококками, пневмококками, гонококками, стафилококками, а также кишечной палочкой. Однако после орального применения около 90 % его связывается с протеинами плазмы, и препарат распределяется по большинству тканей и тканевым жидкостям, вследствие чего он является препаратом выбора для многих системных инфекций.
Синонимами препарата являются гантрисин. фултрексин, сульфазан. сульфолар и др.
Сульфаметоксазол (Sulfamethoxazole)
Сульфаметоксазол — №-(5-метил-3-изоксазолил)сульфаниламид (33.1.20) получают по совершенно аналогичной схеме, но с использованием в качестве гетероциклической компоненты З-амино-5-ме-тилизоксазола [20].
33 1 20
Так же как и сульфизоксазол, препарат эффективен при инфекциях, вызванных стрептококками, гонококками, пневмококками, гонококками, стафилококками, а также кишечной палочкой. В отличие от сульфи-зоксазола, после орального применения лишь около 70 % его связывается с протеинами плазмы, и препарат распределяется по большинству тканей и тканевым жидкостям. Однако, поскольку его выделение происходит значительно медленнее, чем у сульфизоксазола, он не требует частого введения и также является препаратом выбора для многих системных инфекций. Более того, он является составной частью комбинированного препарата, называемого бактрим, бисептол и др., который будет рассмотрен ниже с фиксированным соотношением с триметопримом.
Синонимами препарата являются гантонол, синоми, сульфизоме-зол и др.
- 694 -
Антимикробные препараты
Сг. 1ьфапиридин (Sulfapyridin)
Сульфапиридин — К'-(2-пиридил)сульфаниламид (33.1.21) также пол>чают по аналогичной схеме исходя из 4-ацетила.минобен-золсулъфохлорид и 2-аминопиридина [21, 22, 23].
H2N—4 Л—so2-nh— '—' IM-
33 1 21
Как и все сульфаниламиды, препарат обладает антибактериальной активностью в отношении стрептококков пневмококков, стафилококков. менингококков и гонококков, кишечной палочки, возбудителя дизентерии и др. Является препаратом длительного действия.
Синонимами препарата являются бациллопирин, плюразол, сульфидин, гиасептол.
G гьфасалазин (Sulfasalazin)
Q льфасалазин — 5-[п-[(4,6-диметил-2-пиридинил)сульфамоил]-фенилазо]салициловая кислота (33.1.22) является производным описанного выше с^лфапиридина и одним из тех немногих сульфаниламидов, в которых свободная аминогруппа в бензольном кольце модифицирована; получают реакцией азотосочетания диазониевой соли, полученной взаимодействием сульфапиридина (33.1.21) с азоте той кислотой, с салициловой кислотой в щелочи [24, 25].
1 NaNO2
НООС
/ КОН
НООС
33 1 22
Препарат обладает антибактериальной активностью в отношении некоторых кокков и кишечной палочки. Одновременно оказывает выраженный лечебный эффект у больных неспецифическим язвенным ко-титом. что объясняется отщеплением в организме 5-аминосалициловой кислоты и сулфапиридина. обладающих противовоспалительным и ан-шбактериальными свойствами. Применяют при язвенном колите, хроническом ревматоидном артрите и острых, и подострых воспалительных заболеваниях.
Синонимами препарата являются салазопирин, салазосульфапири-•ин и др.
-695-
Г пава 33
Фталилсульфатиазол (Phtalylsulfathiazole)
Фталилсульфатиазол — (Н4-фталоилсульфаниламидо>-тиазол (33.1.24) также является одним из тех сульфаниламидов, в которых свободная аминогруппа в бензольном кольце модифицирована, чем, очевидно, и объясняется локализация места их действия. Препарат получают взаимодействием сулфатиазола (33.1.23), синтезируемого по стандартной схеме исходя из 4-ацетиламинобензолсульфохлорида и 2-аминотиазола, с фгалевым ангидридом [26, 27].
33 1 24
Как и другие сульфаниламиды, препарат обладает антибактериальной активностью в отношении стрептококков, пневмококков, стафилококков, менингококков и гонококков, кишечной палочки, возбудителя дизентерии и др. Фталазол медленно всасывается из ЖКТ, и его основная масса при пероральном применении задерживается в кишечнике, где он медленно расщепляется. При этом, как и в случае с сульфасалазином, создается высокая концентрация сульфаниламида в кишечнике, чем и объясняется его большая активность в отношении кишечных инфекций. Препарат применяют при дизентерии, колитах, гастроэнтеритах и при оперативных вмешательствах на кишечнике для предупреждения гнойных осложнений.
Синонимами препарата являются фталазол, энтерозол. сульфазол, талазол и многие другие.
Сульфадоксин (Sulfadoxin)
Сульфадоксин — 4.5-диметокси-6-сульфанила.мидопиримилдин (33.1.33) синтезируется по стандартной схеме исходя из 4-ацетил-аминобензолсульфохлорида и 4-амино-5,6-диметоксипиримидина. Однако любопытен сам синтез 4-амино-5,6-диметоксипиримидина (33.1.31), исходящий из метилового эфира метоксиуксусной кислоты. Взаимодействием последнего с ди.метиловым эфиром щавелевой кислоты в присутствии метилата натрия синтезируют метоксипроизводное (33.1.25), пиролизом которого получают диметиловый эфир метоксималоновой кислоты (33.1.26). Взаимодействием последнего с аммиаком получают диамид метоксималоновой кислоты (33.1.27). Гетероциклизацией последнего взаимодействием с фор
- 696-
Антимикробные препараты
мамидом в присутствии этилата натрия получают 4.6-диокси-5-мегоксипиримидин (33.1.28), который далее трансформируют в 4,6-дихлор-5-метоксипиримидин (33.1.28). Подвер1ая полеченный 4,6-дихлор-5-метоксипиримидин (33.1.29) действию аммиака, получают 4-амино-6-хлор-5-метоксипиримидин (33.1.30), и далее взаимодействием полученного соединения с метилатом натрия получают искомый 5,6-диметокси-5-аминопиримидин (33.1.31). Взаимодействием последнего с 4-ацетиламинобензолсульфохлоридом и последующим гидролизом ацетильной группы в 33.1.32 получают сульфадоксин [28, 29, 30, 31].
сн3оос-соссн3
ОСН3 OCr-j
СН3ООС-СО-СН-СООСН3 -------* СН3ООС—СН-СООСН3
33 • 25 33'26
нсомиг ' О2Н5очэ
РОСГз
СНзОЫа
По антибактериальному действию препарат аналогичен другим сульфаниламидам, однако обладает весьма длительным, пролонгированным, действием. Время его полужизни составляет от 120 до 200 ч. Сульфадоксин применяют при инфекционных заболеваниях, вызванных чувствительными к сульфаниламидным препаратам микроорганизмами, — инфекциях органов дыхания, желчевыводящих и мочевых путей гнойных инфекциях различной локализации, при остеомиелите, синусите и других инфекциях. Применяется в комбинации с противомалярийными препаратами.
Синонимами препарата являются сульформетоксин, фанасил, фан-си дар.
Сульфален (Sulfalen)
Сульфален — З-метокси-2-сульфаниламидопиразин (33.1.41) также, как и другие сульфаниламиды, синтезируется по стандартной схеме исходя из 4-ацетиламинобензолсульфохлорида, который вводят во взаимодействие с З-амино-2-метоксипиразином, синтез кото-
-697-
Г лава 33
рого предложено осуществлять двумя технологически реализуемыми путями. Первый способ заключается в бромировании 2-ами-нопиразина с использованием в качестве растворителя уксусной кислоты с получением 3,5-дибром-2-аминопиразина (33.1.34). Взаимодействием последнего с метилатом натрия получают 3-мето-кси-5-бром-2-аминопиразин (33.1.35). Восстановлением водородом с использованием в качестве катализатора палладия на угле атом брома продукта в положении С5 замещают на атом водорода с получением З-метокси-2-аминопиразина (33.1.36). Согласно второму способу, З-метокси-2-аминопиразин (33.1.36) предложено получать исходя из З-гидроксипиразин-2-карбоксамида. Взаимодействием последнего с хлорокисью фосфора замещают гидроксильную группу на хлор, и одновременно подвергают дегидратации карбоксамидную группу с получением З-хлор-2-цианопиразина (33.1 37). Далее взаимодействием с метилатом натрия получают 3-метокси-2-цианопиразин (33.1.38) Цианогруппу в последнем гидролизуют щелочью в присутствии перекиси водорода до карбоксамидной с получением З-метокси-2-карбоксамидопиразина (33.1.39). Полученный продукт подвергают перегруппировке Гофмана действием гипохлорита натрия с получением искомого З-метокси-2-амино-пиразина (33.1.36). Взаимодействием последнего с 4-ацетиламино-бензолсульфохлоридом и последующим гидролизом ацетильной группы щелочью в 33.1.40 получают сульфален [32, 33, 34].
CH3ONa
НгС>2' NaOH
CO~NH2
ОСНз
33 1 3?
33 1 38
-698-
Антимикробные препараты
Сульфален также является бактериостатическим сульфаниламидом очень длительного действия с широким спектром антимикробной активности. Применяют по тем же показаниям, что и сульфадоксин. Время его полужизни также составляет около 150 200 ч. Препарат в меньшей степени, чем другие сульфаниламиды, связывается с протеинами плазмы, что обеспечивает его высокую концентрацию в крови в свобо-юй форме. Поэтому при профилактическом применении (при хронических заболеваниях легких, ревматизме, болезнях почек и мочевыводя-ших путей) сульфален можно назначать даже один раз в неделю в один прием.
Синонимами препарата являются келфизин, сульфаметопиразин, сульфаметоксипиразин и др.
Сульфаметоксипиридазин (Sulfamethoxypyridazine)
Сульфаметоксипиридазин — №-(6-метокси-3-пиридазинил)сульфа-ниламид (33.1.43) получают замещением атома хлора в 6-хлор-3-(4-аминобензолсульфониламидо)пиридазине на метоксигруппу (33.1.42) с использованием метилата натрия. Исходный же 6-хлор-3-(4-ами-нобензолсульфониламидо)-пиридазин (33.1.42), в свою очередь, получают взаимодействием 4-аминобензолсульфаниламида с легкодоступным 3,6-дихлорпиридазином [35, 36].
о-
К2СО3
-----* H2N-A_/“SO2‘
CH3ONa /=\
.Gl --------» HjN—d Л—SO?-
Ci
33 1 42
Препарат обладает антибактериальной активностью в отношении некоторых кокков и кишечной палочки. Препарат обладает длительным действием Применяют для лечения пневмонии, бронхитов, тонзиллитов. гнойных отитов и менингитов, гнойных инфекций мочеполового тракта, дизентерии и др.
Синонимами препарата являются сульфапиридазин, суфалекс, рета-сульфон и многие другие.
Сульфацетамид (Sulfacetamid)
Сульфацетамид — N’-ацетилсульфаниламид (33.1.44) получают либо прямым алкилированием ацетамида 4-аминобензолсульфо.хло-ридом, либо взаимодействием 4-аминобензолсульфонамида с уксусным ангидридом и последующим селективным восстановительным дезацилированием полученного ацетамида (33.1.45) с использованием системы цинк-гидроокись натрия [37, 38].
-699-
Глава 33
h2n—< Л—so2-ci
h2n-co-ch3
(CH3CO)2O
h2n—6 Л—so2—nhcochj
331 44
H2N ~< Л—SO2-NH2
ChaCONH—а Л— SO2-NHCOCH3
33 1 45
Препарат обладает антибактериальной активностью в отношении стрептококков, пневмококков, стафилококков, менингококков и гонококков. Применяют для лечения пневмонии, гнойных трахеобронхитов, инфекций мочеполового тракта, при гонорейных заболеваниях глаз новорожденных и взрослых и др.
Синонимами препарата являются альбуцид, цетамид, пронтамид, себизон и др
Сулъфабензамид (Sulfabenzamid)
Сульфабензамид — N’-бензоилсульфаниламид (33.1.46) получают аналогично сульфацетамиду [38, 39, 40, 41].
h2n—6 A—so2-nhcoc6h5
33 1 46
Применяют по тем же показаниям, в основном в виде мазей, при вагинальных инфекциях.
Синонимом препарата является сульфабензид.
Мафенид (Maphenid)
Мафенид — и-(аминометил)-бенолсульфамид (33.1.48) — препарат, структурно несколько отличающийся от всех рассмотренных выше препаратов тем. что аминогруппа в параположении к сульфонамидной отдалена от бензольного кольца на одну метиленовую группу. Синтез препарата осуществляют исходя из N-бензилацетамида, последовательным взаимодействием которого с хлорсульфоновой кислотой и далее с аммиаком получают 4-ацетамидометил)-бензолсульфонамид (33.1.47). Гидролизом последнего шелочью получают мафенид [42, 43, 44].
- 700 -
антимикробные препараты
1 CISOsH
Л-*. 2 NH;, /Г\ NaOH л-ъ
Ch3CONHCH2—-------* CH3CONHCHg—SO2-Nrt2 -► h^-ch,,—so2-nh2
33 1 47 33 1 48
Синонимами препарата являются марфанил, мезудин, амбамид, сеп-тицид и др.
В настоящее время наиболее широко используются сульфизоксазол, сульфаметоксазол, сульфадиазин, сульфаметизол и трисульфапиримидины (смесь сульфа.меразина, сульфаметазина и сульфадиазина). Первые два из отмеченных препаратов используются наиболее широко. Долгодействующий сульфонамид — сульфадиоксин применяется только в комбинации с пириметамином (антагонистом фолиевой кислоты) для профилактики и лечения тропической лихорадки.
В комбинации с пириметамином или триметопримом сульфаниламиды активны в отношении некоторых протозойных инфекций, включая Toxoplasma, Plasmodium falciparum и Pneumocystis carinii.
Диаминомиримидины
Диаминомиримидины — триметоприм и пириметамин — являются антибактериальными синтетическими препаратами, ингибиторами ди-гидрофолатредуктазы, которые используются как самостоятельно, так и в комбинации с сульфаниламидами, в частности, с сульфаметоксазолом (котримоксазолом, бактримом, бисептолом, сульфатримом и многими другими).
Собственно диаминомиримидины (триметоприм, пириметамин) были предложены в качестве лечебных и профилактических средств против малярийных инфекций. Показано, что все сильные ингибиторы дигидрофолатредуктазы могут вывести из строя малярийный паразит с относительно малыми последствиями на хозяина.
Близкое структурное сходство этих препаратов, а также структурное сходство между птеридиновым кольцом фолиевой кислоты и диа-минопиримидиновым фрагментом пириметамина скорее всего является причиной сродства этих соединений к рецептивному участку дигидро-фолагредуктазы.
Все эти соединения являются ингибиторами дигидрофолатредуктазы в бактериях, плазмодиях и человека. К счастью, они имеют зна-
-701 -
Г лава 33
чительно большее сродство к бактериальным и протозойным дигид-рофолатредуктазам. Пириметамин, jc примеру, ингибирует дигидрофол атредуктазу паразита при концентрациях в несколько сотен раз ниже, чем это требуется для ингибирования дигидрофолатредуктазы человека. Это и является основой их селективной токсичности. Селективная токсичность может быть повышена при снабжении организма хозяина фолиевой кислотой, которую паразит не в состоянии использовать.
Таким образом, триметоприм в организме действует путем вмешательства в действие дигидрофолатредуктазы — фермента, восстанавливающего дигидрофолиевую кислоту в тетрагидрофолиевую. Эта процесс необходим для биосинтеза живыми организмами пуринов и, соответственно, ДНК. Восстановление дигидрофолиевой кислоты в тетрагидрофолиевую у людей также катализируется дигидрофолатредукта-зой. Однако триметоприм имеет во много тысяч раз больший ингибирующий эффект в отношении бактериальных ферментов, чем в отношении аналогичных ферментов млекопитающих, что и является основным преимуществом триметоприма.
Различные сульфонамиды ингибируют одну из стадий этого биосинтетического пути внедрением вместо n-аминобензойной кислоты в дигидрофолиевую кислоту сульфаниламида. Последовательная блокада одного и того же биосинтетического пути двумя препаратами — сульфаниламидом и триметопримом одновременно — отличается высокой степенью синергичности по отношению к широкому спектру микроорганизмов.
В сочетании триметоприма с сульфометоксазолом проявляется очень сильный его эффект по отношению ко многим микроорганизмам. Причем оптимальным считается сочетание сульфаметоксазола и триметоприма 20:1. Это соотношение в плазме достигается применением препаратов, в которых это соотношение равно 5:1. Конечно, эти соотношения можно широко варьировать, однако наличие триметоприма в смеси с сульфаниламидом является залогом успеха при лечении.
Триметоприм (Trimethoprim)
Триметоприм — 2,4-диамино-543\4\5'-триметоксибензил)пири-мидин (33.1.51) предложено получать различными путями. Первый способ синтеза исходит из этилового эфира 3,4,5-триметоксидигид-рокоричной кислоты, формилированием которой этилформиатом с использованием в качестве основания натрия получают енол полуальдегида З'Л^б’-триметоксибензилмалонового эфира (33.1.49), который вводят в реакцию гетероциклизации с гуанидином с полу-
- 702-
Антимикробные препараты
чением 2-амино-4-гидрокси-5-(31,4',5|-триметоксибензил)пирими-дина (33.1.50). Последовательным замещением гидроксильной группы в последнем на хлор с применением хлорокиси фосфора и далее на аминогруппу с использованием аммиака получают искомый триметоприм [45, 46, 47].
Na
Г1СООС2Н5 ------
СН3О /=\ соос2н5
СНзО—4 У-сн2-^ у-* сн-ок
СНз° 33 1 49
NH х HjN NH2
33 1 50
1 POCI3
2 NH3
СНз° 33 151
Все остальные способы синтеза исходят из 3,4,5-триметоксибенз-альдегида. Согласно второму способу, конденсируя 3,4,5-тримето-ксибензальдегид с 3-этокси- или 3-анилинопропионитрилом получают соответствующее бензилиденовое производное (33.1.52), непосредственное взаимодействие которого с гуанидином приводит к получению тримеюприма [48, 49, 50, 51].
(NHCgHj)
Согласно третьему способу, триметоприм предложено получать также конденсацией 3,4,5-триметоксибензальдегида с динитрилом малоновой кислоты по Кневенгелю с получением илиденового производного (33.1.53), которое частично восстанавливают водородом с использованием в качестве катализатора палладия на угле до енамина (33.1.54), который взаимодействием с гуанидином трансформируется в триметоприм [52, 53].
гя3о
35 1 53
-703-
Глава 33
NH
Наконец,, триметоприм предложено получать четвертым способ бом, предусматривающим в качестве первого шага также конденсацию по Кневенгелю 3,4,5-триметоксибензальдегида, но уже с эфиром циа-нуксусной кислоты с получением илиденового производного (33.1.55). Двойную связь в последнем восстанавливают водородом над палладием на угле с получением З'д'д’-триметоксибензилциануксусного эфира (33.1.56). Вводя последний в реакцию гетероциклизации с гуанидином, получают искомый триметоприм [54, 55].
1 РОС1з
2 Н2 / Pd-C
□осгн5
СН3О
33 1 51
Триметоприм имеет широкий спектр антимикробной активности. Он в 20-100 раз более активен в отношении большинства бактериальных видов, чем сульфаметоксазол. Триметоприм активен по отношению к таким грамположительным аэробным бактериям, как Staphylococcus aureus, Staphylococcus epidermidis, к различным видам Streptococcus и Listeria monocytogenes. Против видов Nocardia триметоприм уступает сульфонамидам.
Триметоприм активен по отношению к таким грамотрицательным аэробным бактериям, как большинство Е. coli, Enterobacter, Proteus, Klebsiella, Providencia, Morganella. Serratia marcescens, Citrobacter, Salmonella, Shigella, Yersinia enterocolitica, чувствительным к триметоприму.
Триметоприм активен также по отношению к Legionella, Acinetobacter, l-'ibrio. Aeromonas, Pseudomonas maltophila, Pseudomonas cepacia, однако Pseudomonas aeruginosa резистентна по отношению
- 704 -
Антимикробные препараты
к триметоприму.
Haemophilus influenzae и Haemophilus ducreyi чувствительны к триметоприму. Патогенные Neisseria (менингококки и гонококки) и Branha-mella catarrhalis умеренно резистентны к триметоприму, однако очень чувствительны к сочетанию триметоприм-сульфаметоксазол. Анаэробные бактерии в общем резистентны к триметоприму, в то время как сочетание триметоприм-сульфаметоксазол воздействует и на них. К этому сочетанию чувствительны и все виды Pneumocystis carinii.
Бактериальная резистентность к триметоприму может возникать по ряду причин: неспособности препарата проникнуть сквозь мембранную оболочку (Ps. aeruginosa); наличие дигидрофолатредуктазы, не чувствительной к ингибированию триметопримом; перепроизводство дигидрофолатредуктазы и мутации, выражающейся в тиминовой зависимости, когда организму требуется экзогенный тимин для синтеза ДНК, т. е. обход метаболической блокады, вызываемой триметопримом.
Резистентность к комбинации триметоприм-сульфаметоксазол всегда ниже чем к любому из этих препаратов, используемых по отдельности.
Эта комбинация препаратов, коммерческими названиями которых являются котримоксазол, бактрим, бисептол, сульфатрим и многие другие, применяется при лечении инфекций дыхательных путей, инфекциях мочеполового тракта, желудочных инфекциях, хирургических инфекциях, энтеритах, менингитах и прочих болезнях.
Пириметамин (Pyrimethamin)
Пириметамин — 2,4-диамино-5-(4|-хлорфенил)-6-этилпиримидин (33.1.60) получают исходя из 4-хлорбензилцианида, конденсацией которого с метиловым эфиром пропионовой кислоты в присутствии метилата натрия получают Р-кетонитрил (33.1.58). Взаимодействием последнего с ортомуравьиным эфиром получают метоксимети-леновое производное (33.1.59), гетероциклизацией которого в пиримидин с использованием в качестве бинуклеофила гуанидина получают искомый пириметамин (33.1.60) [55, 56, 57, 58, 59, 60].
о
CI
сн2-с=ч
С2Н5 0—CH3
CH3ONa
НС(ОСН3)з
33 1 58
-705-
Глава 33
С2Н5\
33 1 59
Пириметамин, являясь антагонистом фолиевой кислоты, оказывает противомикробное действие в отношении возбудителей малярии, обладает споронтонидным действием. Препарат эффективен также в отношении возбудителя токсоплазмоза. Применяется для профилактики малярии и лечения токсоплазмоза.
33.2. Хинолоны
Хинолоны являются группой структурно сходных антимикробных препаратов, проявляющих высокую активность против многих микроорганизмов.
Формально первым представителем нового класса антимикробных препаратов, называемых препаратами ряда хинолонов, будучи производным нафтиридина, явилась налидиксовая кислота (невиграмон), которая была синтезирована в 1962 г. и была предложена для лечения инфекций мочеполового тракта. Основной спектр ее действия включает грамотрицательные бактерии. Препарат эффективен также в отношении кишечной палочки, протея, клебсиелл, шигелл, сальмонелл. В последующие годы был синтезирован и ряд других химически сходных соединений, таких как оксолиновая кислота (собственно производное хинолона) и пиноксацин (производное циннолина), однако все они имели относительно узкий антимикробный спектр.
налидиксовая кислота
оксолиновая кислота
Огромный прогресс был достигнут в 1980-х годах, и он был связан с введением в положение Со молекулы 4-хинолонов атома фтора, а в положение С- — пиперазинового фрагмента. Введение атома фтора в указанное положение резко увеличило активность препаратов в отношении грамположительных микроорганизмов и расширило их спектр действия,
- 706 -
Аитимикробные препараты______________________________________
включив в него и грамотрицательные микроорганизмы. Введение пипе-разинового фрагмента в положении С7 обеспечило активность группы препаратов и в отношении Pseudomonas aeruginosa. Заместители при атоме азота хинолоновой структуры и в пиперазиновом кольце могут варьировать от препарата к препарату.
Все фторхинолоны, применяемые на практике (ципрофлоксаин, эноксапин, норфлоксацин, офлоксацин), имеют примерно одинаковый антимикробный спектр действия, включающий большинство аэробных грамотрицательных и некоторые грамположительные бактерии. Фторхинолоны высокоактивны против большинства энтеробактерий, включая Escherichia coli, Enterobacter, Proteus mirabilis, Proteus vulgaris, Morganella morganii, Providencia, Citobacter и Serratia. Они активны также в отношении Pseudomonas aeruginosa, включая штаммы, устойчивые к другим антибактериальным препаратам. К фгорхинолонам чувствительно большинство штаммов Acinetobacter аэробных грамотрицательных микроорганизмов. Фторхинолоны высоко активны против большинства грамотрицательных бактериальных патогенов ЖКТ: Shigella, Salmonella, Yersinia enterocolitica, Aeromonas species и Vibrio species. Трамотрицательные коккобактерии Haemophilus influenzae, Haemophilus ducreyi и трамотрицательные кокки Neisseria meningitides, Neisse-ria gonorrhoeae, Moraxella также очень чувствительны к фторхи-нолонам.
Фторхинолоны активны и в отношении большинства грамположительных бактерий, Staphylococcus aureus, Staphylococcus epidermidus, хотя применяемые при этом концентрации должны быть несколько выше чем для грамотрицательных бактериальных патогенов.
Фторхинолоны являются мощными бактерицидными препаратами, которые изменяют структуру и функции бактериальной ДНК, воздействия на активность ферментной ДНК-гиразы (топоизомеразы II). Этот фермент ответственен за отрицательные суперспиральные скручивания (отрицательные суперспирали) в ковалентно закрытую циркулярную ДНК, а также за разрыв и повторное соединение (катенация, декатенация) ДНК-витков, замкнутых связями в цепь. ДНК-гираза способна расположить бактериальную ДНК, имеющую примерную длину' в 1300 pm, скажем в Escherichia coli, внутри клетки размером от 2 до 3 pm. Этот фермент необходим для репликации, восстановления, транскрипции определенных оперонов ДНК. ДНК-гираза состоит из двух А и двух В схбъединиц. Хинолоны воздействуют непосредственно на работу субъединицы А.
Как уже было отмечено, препараты этого ряда имеют сходный ан-шмикробный спектр, включающий большинство аэробных грамотрица-
- 707-
Глава 33
тельных и некоторые грамположительные бактерии. Специфическая разница в активности этих препаратов отмечается по отношению к некоторым конкретным микроорганизмам, их относительной токсичности, фармакокинетическим особенностям и т. п. Например ципрофлоксацин и норфлоксацин имеют сходный антимикробный спектр, однако в зависимости от типа микроорганизмов норфлоксацин может оказаться слабее от 2 до 8 раз.
Выявлены два механизма резистентности по отношению к фторхи-нолонам: изменение субъединицы А ДНК-гиразы и понижение проницаемости внешней мембраны бактерий. Резистентность опосредуется хромосомами, а не плазмидами бактерий. Возникновение резистентности во время применения препаратов встречается крайне редко.
Ввиду своих фармакокинетических особенностей (выраженной биодоступности при пероральном применении, распределения по тканям и проникновения в них, широкого спектра антибактериальной активности и др.) фторхинолоны имеют значительный потенциал для лечения инфекций практически любой анатомической локализации. Фторхинолоны весьма эффективны при лечении инфекций дыхательных путей, мочеполового тракта, костей, кожи и мягких тканей и др.
Налидиксовая кислота (Nalidixic Acid)
Налидиксовая кислота — 1-этил-1,4-дигидро-7-метил-4-оксо-1,8-нафтиридин-3-карбоновая кислота (33.2.4) получают синтезом, предусматривающим на первой стадии взаимодействие 2-амино-6-метилпиридина и этоксиметиленмалонового эфира, в результате чего получается продукт замещения (33.2,1), который при нагревании циклизуется в этиловый эфир 4-гидрокси-7-метил-1,8-нафтиридин-3-карбоновой кислоты (33.2.2). Гидролизом последнего щелочью получают соответствующую кислоту (33.2.3). Алкилированием последней этилйодидом в присутствии гидроксида калия получают налидиксовую кислоту [60, 61. 62. 63, 64].
33 2 1
ОН
C2b5t ‘ КОН
-708-
Антимикробные препараты
Налидиксовая кислота действует бактерицидно или бактериостатически в зависимости от чувствительности микроорганизма и концентрации. Препарат эффективен в отношении грамотрицательных микроорганизмов: кишечной палочки, сальмонелл, шигелл, протея, палочки Фридлендера. Применяют при пиелонефрите, цистите, уретрите, простатите, инфекциях ЖКТ.
Синонимами препарата являются неграм, невиграмон, уралгин, уро-грам, винтрон и многие другие.
Оксолиновая кислота (Oxolinic acid)
Оксолиновая кислота — 5-этил-5,8-дигидро-8-оксо-1-диоксоло[4,5-3]-хинолин-7-карбоновая кислота (33.2.9), в принципе, получается по аналогичной налидиксовой кислоте схеме синтеза, однако исходящей в качестве ароматической аминной компоненты не из 2-ами-но-6-метилпиридина, как в случае налидиксовой кислоты, а из 3,4-метилендиоксианилина (33.2.6). Последний получают, восстанавливая водородом 3,4-метилендиокси-1-нитробензол (33.2.5), используя в качестве катализатора двуокись платины. Сам 3,4-метилен-диокси-1-нитробензол (33.2.5), в свою очередь, получают нитрованием 1,2-метилендиоксибензола азотной кислотой. Далее взаимодействием полученного 3,4-метилендиокси-анилина (33.2.6) и этоксиметиленмалонового эфира получают продукт замещения (33.2.7), который при нагревании циклизуется в этиловый эфир 4-гидрокси-6,7-метилендиоксихинолин-З-карбоновой кислоты (33.2.8). Гидролизом последнего щелочью в диметилформамиде и непосредственной обработкой продукта этилйодидом получают искомую оксоли-новую кислоту [65, 66, 67].
HNO3
33 2 5
зз 2 е
I КаОН DMF
2 С2Н51
Как и налидиксовая кислота, препарат эффективен в отношении грамотрицательных микроорганизхмов и применяется по тем же показаниям.
- 709 -
Г лава 33
Синонимами препарата являются нидантин, продоксол, околин, уроксол и др.
Циноксацин (Cinoxacin, Azolinic acid)
Циноксацин — 1 -этил-1,4-дигидро-4-оксо[1,3]-диоксоло[4,5^]цин-нолин-3-карбоновая кислота (33.2.14) синтезируется по иной схеме, которая исходит из 2-амино-4,5-мет илендиоксиацетофенона (33.2.10), получающегося воссгановлением водородом над платиновым катализатором 4,5-метилендиокси-2-нитроацетофенона. В условиях диазотирования последнего происходит спонтанная гетероциклизация в 4-гидрокси-6,7-мегилендиоксициннолин (33.2.11), очевидно в результате наличия в условиях реакции значительного количества енольной формы ацетофенона (33.2.10). Полученный циннолин (33.2 11) далее подвергают бромированию молекулярным бромом в присутствии ацетата калия и получают 3-бром-4-гидрокси-6,7-метилендиоксициннолин (32.2.12). Взаимодействием последнего с цианидом одновалентной меди в димет илформамиде атом брома в нем замещают на цианогруппу с получением З-циано-4-гидрокси-6,7-метилендиоксициннолина (33.2.13). Полученный продукт алкилируют по положению 1 этилйодидом, используя в качестве основания гидрид натрия, а цианогруппу гидролизуют до карбоксильной смесью соляной и уксусной кислот с получением искомого цинок-сацина [68, 69]
№NO2 ' НС,
Br2 СНзСООК
Cu2(CN)2 DMF
1 С2Н5' , МН OfvF
2 НС СН3СООН
Препарат эффективен в отношении грамотрицательных микроорганизмов и применяется по тем же показаниям, что и налидиксовая и оксолиновые кислоты.
Синонимами препарата являются цинобактин, носсацин, уронорм И др.
- 710 -
Антимикробные препараты
Норфлоксацин (Norfloxacin)
Норфлоксацин — 1-этил-6-фтор-1,4-дигидро-4-оксо-7-(1-пипера-зинил)-3-хинолинкарбоновая кислота (33.2.18) является первым представителем из серии фторированных хинолонов и одновременно первым препаратом, содержащим пиперазиновый заместитель из вошедших в медицинскую практику производных хинолонов. Способ его синтеза в принципе не отличается от способа синтеза предложенного для синтеза налидиксовой и оксолиновой кислот. Взаимодействием З-хлор-4-фторанилина и этоксиметиленмалонового эфира, получают продукт замещения (33.2.15), который при нагревании в дифениловом эфире циклизуется в этиловый эфир 6-фтор-7-хлор-1,4-дигидро-3-хинолин-4-он-карбоновой кислоты (33.2.16). Непосредственной обработкой продукта этил-йодидом в присутствии триэтиламина и последующим гидролизом щелочью получают 1-этил-6-фтор-7-хлор-1.4-дигидро-3-хинолин-4-он-карбоновую кислоту (33.2.17). Вводя последнюю во взаимодействие с пиперазином, получают норфлоксацин (33.2.18) [70, 71, 72. 73, 74, 75].
„СН С2Н5О
1 С2Н5ВГ (CjHgbh
2 NaO^
Норфлоксацин обладает широким спектром бактерицидного действия Высокоактивен в отношении большинства грамотрицательных и некоторых грамположительных микроорганиз.мов. К препарату не чувствительны анаэробные бактерии, малочувствительны энтерококки и акинетобактер. Препарат применяют при бактериальных инфекциях мочевыводящих путей, предстательной железы, ЖКТ, гонореи, диареи путешественников.
Синонимами препарата являются нороксин, баразан, фулграм, ба-цидал и др.
- 711 -
Г лава 33
Ципрофлоксацин (Ciprofloxacin)
Ципрофлоксацин — 1 -циклопропил-6-фтор-1,4-дигидро-4-оксо-7 -(1-пиперазинил)-3-хинолинкарбоновая кислота (33.2.19) получают по совершенно аналогичной схеме синтеза с использованием на стадии алкилирования вместо этилйодида циклопропилбромида [76, 77. 78].
33 2 19
Ципрофлоксацин обладает широким спектром антимикробного действия. Высокоактивен против грамотрицательных микроорганизмов: синегнойной, гемофильной и кишечной палочек, шигелл, сальмонелл, менингококка, гонококка, некоторых разновидностей энтерококков. Препарат активен также в отношении многих штаммов стафилококков, кампилобактера, легионелл, микоплазм, хламидий, микобактерий. К препарату резистентны Ureaplasma urealyttcum, Clostridium difficile, X'ocardia asteroids. Препарат применяют при инфекциях мочевых путей, дыхательных путей, желчевыводящих путей, инфекционно-воспалительных заболеваниях брюшной полости и органов малого таза, костей, суставов, кожи.
Ципрофлоксацин эффективен также при бактериальных простатитах, неосложненной гонорее, остеомиелитах и легочных инфекциях. Он эффективен при лечении острых инфекционных диареях, включая диарею путешественников, и энтеритах. Побочные реакции при его применении встречаются редко.
Синонимами препарата являются ципроквин, ципролет, ципропан, ципросан. ципроцинол и многие другие.
Эноксацин (Enoxacin)
Эноксацин — 1-эгил-6-фтор-1,4-дигидро-4-оксо-7-( 1-пиперазинил)-1,8-нафтиридин-З-карбоновая кислота (33.2.24) отличается от норфлоксацина исключительно тем, что в положении 8 норфлоксацина атом углерода замещен атомом азота, т. е. препарат принадлежит не ряду хинолонов, а ряду нафтиридинов. Синтез препарата осуществляется исходя из 2.6-дихлор-З-нитропиридина, взаимодействием которого с N-этоксикарбонилпиперазином получают продукт заме
-712-
Антимикробные препараты
щения атома хлора в положении 2 пиридинового кольца (33.2.20). Последовательным замещением в нем атома хлора в положении С6 на аминогруппу аммиаком, ацилированием полученной аминогруппы уксусным ангидридом и, наконец, восстановлением нитрогруппы в положении Сз пиридинового кольца водородом получают 6-амино-3-ацетиламино-2-(4-этоксикарбонилпиперазинил)пиридин (33.2.21). С целью введения в положение С3 атома фтора прибегают к реакции Шимана. С этой целью свободную аминогруппу диазотируют амилнитритом, и полученную диазониевую соль обрабатывают борфтористоводородной кислотой. Образовавшийся фторборат диазония подвергают пиролизу с получением З-фтор-6-ацетилами-но-2-(4-этоксикарбонилпиперазинил)пиридина. Наконец, снятием ацетильной защиты аминной группы в положении С6 получают 3-фтор-6-амино-2-(4-этоксикарбонилпиперазинил)пиридин (33.2.22). Полученный амин (33.2.22) вводят во взаимодействие с этоксиметиленмалоновым эфиром, в результате чего получается производное аминометиленмалонового эфира, нагревание которого приводит к получению этилового эфира 6-фтор-1,4-дигидро-4-оксо-7-(4-это-ксикарбонил-1-пиперазинил)-1,8-нафтиридин-3-карбоновой кислоты (33.2.23). Алкилированием последнего этилйодидом и дальнейшим гидролизом двух карбэтоксильных групп получают эноксацин [79, 80, 81. 82].
’ NH3
2 (СНзСО^О
3 hz Pd—С
В плане антибактериального спектра и области применения эноксацин, в принципе, повторяет все свойства норфлоксацина. Однако он лучше абсорбируется из ЖКТ и имеет более продолжительное время полу жизни.
Синонимами препарата являются бактидан, флумарк и др.
-713-
Глава 33
Офлоксацин (Ofloxacin)
Офлоксацин — (+) 9-фтор-3-метил-10-(4-метил-1-пиперазинил)-7Я-пиридо-(1,2,3-бе)-1,4-бензоксазин-7-оЬсо-6-карбоновая кислота (32.2.30) имеет несколько более усложненную, по сравнению с вышеописанными препаратами, структур), главная особенность которой состоит в наличии метилзамещенного оксиэтиленового мостика между атомом азота и положением С8 хинолоновой системы, а также наличием метильного заместителя в положении С4 пиперазинового кольца. Синтез препарата осуществляют исходя из 2,3,4-трифторнитробензола, взаимодействием которого с гидроокисью калия получают 2-гидрокси-3,4-дифторнитробензол (33.2.25). Вводя последний во взаимодействие с хлорацетоном в присутствии йодистого калия и поташа, получают соответствующий эфир гидроксиацетона (33.2.26). Исчерпывающим восстановлением последнего водородом над никелем Ренея в одну стадию получают искомое производное дифторбензоксазина (33.2.27), минуя отдельное и, пожалуй, невозможное, да и ненужное, последовательное выделение амина, далее внутреннего имина и. наконец, целевого продукта. По уже многократно описанной выше схеме синтезированный вторичный гетероциклический амин (33.2.27) вводят во взаимодействие с этоксиметиленмалоновым эфиром, и полученное аминомети-лен.малоновое производное (33.2.28) циклизуют в пиридобен-зоксазиновую систем). Однако, в отличие от ранее описанных случаев, где данная реакция осуществлялась в высокотемпературном режиме, в данном случае реакция осуществлялась с применением полифосфорной кислоты. Полученный при этом этиловый эфир 9,10-дифтор-3-метил-7Я-пиридо(1,2,3-йе)-1,4-бензоксазин-7-оксо-6-карбоновой кислоты подвергают гидролизу до соответствующей кислоты (33.2.29). Наконец, вводя последнюю во взаимодействие с N-метилпиперазином, осуществляют замещение атома фтора в положении С)0 пиридобензоксазиновой системы с получением офлоксацина (32.2.30) [84, 85, 86].
- 714 -
Антимикробные препараты
Офлоксацин также обладает широким спектром антимикробного действия. Высокоактивен против грамотрицательных микроорганизмов: синегнойной, гемофильной и кишечной палочек, шигелл, сальмонелл, хламидий. Применяют при инфекциях дыхательных путей, уха, горла, носа, кожи, мягких тканей, костей, суставов, инфекционно-воспалительных заболеваниях органов брюшной полости, почек, мочевыводящих путей, органов малого таза, гениталий, гонореи.
Синонимами препарата являются таривид, флобанин и др.
33.3. Нитрофураны
Некоторые производные нитрофурана обладают выраженной антимикробной активностью. Все эти соединения характеризуются тем, что являются производными 5-нитрофурфурола, полученными взаимодействием с различными соединениями, формально содержащими гидра-зинную функциональную группу. Нитрофураны эффективны в отношении грамположительных и грамотрицательных микроорганизмов, а также трихомонад и лямблий. В низких концентрациях препараты действуют бактериостатически, в высоких — бактерицидно. Наиболее эффективны против некоторых штаммов Escherichia coll, Klebsiella, Enterobacter и Citobacter. Следует отметить, что они бывают эффективны п в отношении некоторых микроорганизмов, устойчивых к антибиотикам и сульфонамидам. Наиболее широко используются нитрофура-зон, фуразолидон, нитрофурантоин. Производные нитрофурана используют как орально, так и в качестве наружных средств. Препараты этого ряда аккумулируются в моче и желчи. Скорость абсорбции весьма зависима от кристаллической формы препарата. Нитрофураны используются преимущственно в качестве антисептиков для наружного применения (чшрофуразон). а также для лечения инфекций мочевыводящих путей и кишечника. Точный механизм действия этих препаратов не установ--1ен Однако кажется вероятным, что они ингибируют ряд бактериальных ферментных систем, скорее всего путем повреждения их ДНК. По-Дащют, что фермент нитроредуктаза трансформирует эти препараты
-715-
Глава 33
। короткоживущие промежуточные радикалы, которые взаимодействует с ДНК бактерий, нарушая процесс их скручивания. Редко встречаю-цаяся резистентность может возникнуть в результате мутаций, связан-тых с потерей активности фермента нитроредуктазы.
(1итрофураюн (Nitrofurazon)
Нитрофуразон — семикарбазон 5-нитрофурфурола (33.3.1) получают взаимодействием 5-нитрофурфурола с семикарбазидом [87. 88].
о
h2n-nh—с—nh2
ззз 1
Нитрофуразон является эффективным препаратом, действующим на ряд грамположительных и грамотрицательных микроорганизмов (стафилококки, стрептококки, дизентерийная палочка, кишечная палочка, палочка паратифа и др.). Применяется, в основном, наружно для лечения и предупреждения гнойно-воспалительных процессов и внутрь для лечения бактериальной дизентерии. Нитрофуразоном орошают раны, накладывают влажные повязки. Его применяют в виде капель, практически при всех гнойных процессах, требующих назначения антибактериальных препаратов.
Синонимами препарата являются фурациллин, фурацин, антибио-птал, ваброцид и др.
Нитрофурантоин (Nitrofurantoin)
Нитрофурантоин— 1-(5-нитрофурфурилиденамнно)гидантоин (33.3.5) синтезируют исходя из гидразиноуксусной кислоты (33.3.2), получаемой взаимодействием хлоруксусной кислоты с гидразином. Взаимодействием гидразиноуксусной кислоты с цианатом калия получают семикарбазидоуксусную кислоту' (33.3.3), которая при нагревании циклизуется в 1-аминоидантоин (33.3.4). Взаимодействием последнего с диацетилацеталем 5-нитрофурфурола получают искомый нитрофурангоин [89, 90, 91, 92, 93].
h2n-nh2
H2N-NH-CH2-COOH
КПСС1
- 716 -
Антимикробные препараты
33 3 4
О2Ы
СН(О-СОСН3)2
о
33 3 5
Так же как и нитрофуразон, нитрофурантоин является эффективным препаратом, действующим на ряд грамположительных и грамотрицательных микроорганизмов (стафилококки, стрептококки, дизентерийная палочка, кишечная палочка, палочка паратифа и др.). В основном препарат применяют при лечении инфекционных заболеваний мочевых п\тей (пиелиты. пиелонефриты, циститы, уретриты).
Синонимами препарата являются фурадонин, итуран, фенурин, >ролонг, цистофуран, нитрофурин и многие др)гие.
Фуразолидон (Furazolidone)
Фуразолидон — 3-(5-нитрофурфурилиден)амино-2-оксазолидинон (33 7.8) получают исходя из 2-гидразиноэтанола, взаимодействием которого с диэтилоксалатом получают З-амино-2-оксазолидон. Взаимодействием последнего с бензальдегидом получают соответствующий гидразон (33 3.7). Очисткой последнего и последующим взаимодействием с 5-нитрофурфуролом получают фуразолидон [94, 95, 96. 97].
(CjHgO^CO
h2n-nh-сн2-сн2-он --------------
Фуразолидон, как и рассмотренные выше производные нитрофурана. эффективен против грамположительных и грамотрицательных микроорганизмов. Однако он обладает и противотрихомонадной акгивно-
- 717 -
Г лава 33
стью и эффективен при лямблиозе. По сравнению с нитрофуразоном и нитрофурантоином, фуразолидон более активен в отношении грамотрицательных микроорганизмов и одновременно менее токсичен. Фуразолидон применяют по тем же показаниям, что нитрофуразон и нигро-фурантоин, как внутрь, так и местно.
Синонимами препарата являются диафурон, фуроксан. итифур. ва-гинол, медарон и др.
Список литературы
1. US Pat. 3.375.247 (1968).
2. Ger. Pat. 1.620.140 (1966).
3. Neth. Pat. Appl. 6.610.815 (1967).
4. Doub L. et al.//J. Med. Chem. 13, 242 (1970).
5. US Pat. 2.407.966(1946).
6. US Pat. 2.407.966(1946).
7. Brit. Pat. 557.055 (1943).
8. Roblin V et al./,'J. Am. Chem. Soc. 62, 2002 (1940).
9. US Pat. 2.407.966 (1946).
10. Sprague G. etal.//J. Am. Chem. Soc. 63, 3028 (1941).
11. Brit. Pat. 546.158 (1942).
12. Brit. Pat. 552.887 (1943).
13. US Pat. 3.119.818 (1964).
14. Caldwell et al.//J. Am. Chem. Soc. 63, 2188 (1941).
15. Roblin Г. et al.'ZJ. Am. Chem. Soc. 64, 567 (1942).
16. US Pat. 2.358.031 (1944).
17. US Pat. 2.447.702 (1948).
18. US Pat. 2.430.094 (1947).
19. Ger. Pat. 819.855 (1944).
20. US Pat. 2.888.455 (1959).
21. VS Pat. 2.275.354 (1942).
22. Brit. Pat. 512.145 (1939).
23. Winterbottom Am. Chem. Soc. 62, 160 (1940).
24. US Pat. 2.396.145 (1946).
25. Doraswamy G. et al.//J. Indian Chem. Soc. 23 (278 (1946).
26. US Pat. 2.324.013 (1943).
27. US Pat. 2.324.015 (1943).
28. US Pat. 3.132.139(1964).
29. Belg. Pat. 618.639 (1962).
30. Grussner .4. et al.' Monatsh. Chem. 96, 1676 (1965).
- 718 -
литимикробные препараты
31. Bretschneider A. et al./7Monatsh. Chem. 96, 1661 (1965).
32. Brit. Pat. 928.151 (1963).
33. US Pat. 3.098.069 (1963).
34. Camerino B. et al.VGazz. Chim. Ital. 90, 1815 (1960).
35. US Pat. 2.712.012 (1954).
36. VS Pat. 2.833.761 (1958).
37. US Pat. 2.411.495 (1946).
38. Crossley i\'. et al.//J. Am. Chem. Soc. 61, 2950 (1939).
39. US Pat. 2.240.496(1941).
40. Brit. Pat. 541.958 (1942).
41. Siebenmannei M. al.,/J. Am. Chem. Soc. 61, 2950 (1939).
42. US Pat. 2.288.531 (1942).
43. Miller G. et al.,7J. Am. Chem. Soc. 62, 2099 (1940).
44 Bergeim L. et al.,7J. Am. Chem. Soc. 66, 1459 (1944).
45. US Pat. 2.909.522 (1959).
46. Bnt. Pat. 875.562 (1958).
47. Ger. Pat. 1.103.931 (1958).
48. US Pat. 3.049.544 (1962).
49. Ger. Pat. 1.303.544 (1960).
'0 Ger. Pat. 1.445.176 (I960).
51. Ger. Pat. 1.795.586 (1974).
52. Ger. Pat. 2.443.080 (1974).
53. Brit. Pat. 1.445.254 (1974).
54. Ger. Pat. 2.165.362 (1970).
55. Ger. Pat. 2.258.238 (1971).
56 US Pat. 2.576.939(1951).
57. US Pat. 2.602.794 (1952).
58. US Pat. 2.680.740 (1954).
59 Russel R. et al.'VJ. Am. Chem. Soc. 73, 3763 (1951).
60. Longeman H. et al.A'Ber. 87, 435 (1954).
61. US Pat. 3.149.104 (1964).
62. Brit. Pat. 1.338.023 (1971).
63. Belg. Pat. 612.258 (1962).
64. Lesher G et al./'J. Med. Pharm. Chem. 5, 1063 (1962).
65. US Pat. 3.287.458 (1966).
66. Ger. Pat. 2.103.805 (1971).
M. Kaminsky F. et al.. J. Med. Chem. 11, 160(1968).
68 Ger. Pat. 2.005.104(1970).
69. US Pat. 3.669.965 (1972).
70. US Pat. 4.146.719(1979).
71. Ger. Pat. 2.804.097 (1979).
-719-
Г лава 33
72. Belg. Pat. 863.429 (1978).
73. Ger. Pat. 2.840.910 (1979).
74. US Pat. 4.292.317 (1981).
75. Koga H. et al./7J. Med. Chem. 23, 1358 (1980).
76. Ger. Pat. 2.142.854 (1983).
77. US Pat. 4.670.444 (1981).
78. Grohe K. et al.'/Ann. 1987 29.
79. Eur. Pat. Appl. 9425 (1980).
80. US Pat. 4.352.803 (1982).
81. US Pat. 4.359.578 (1982).
82. Matsumoto J. et al./.'J. Med. Chem. 27. 292 (1984).
83. Eur. Pat. Appl. 47.005 (1982).
84. US Pat. 4.382.892 (1983).
85. Egava H. et al./'Chem. Pharm. Bull. 34, 4098 (1986).
86. Atarashi S. et al./'Chem. Pharm. Bull. 35, 1896 (1987).
87. US Pat. 2.416.234 (1947).
88. US Pat. 2.927.110(1960).
89. US Pat. 2.610.181 (1950).
90. US Pat. 2.779.786(1957).
91. US Pat. 2.898.335 (1959).
92. US Pat. 2.927.110 (1960).
93. Swirska A. et al.'/Przem. Chem. 11, 306 (1955).
94. US Pat 2.759.931 (1956).
95. US Pat. 2.742.462 (1956).
96. Brit. Pat. 735.136 (1955).
97. Gever G. et al./'J. Am. Chem. Soc. 77, 2277 (1955).
Глава 34
1 Антимикобактериальные । препараты
Патогенными организмами, вызывающими весьма серьезные заболевания у человека, являются микобактерии Mycobacterium tuberculosis, Mycobacterium avium, Mycobacterium leprae, Mycobacterium kansasii, Mycobacterium fortuitum-M, Mycobacterium chelonae и некоторые другие. Характерной особенностью микобактерий является высокое содержание в них (около 40 % от собственного веса) липидов, главным образом на внешней стенке бактерий.
Типичные микобактерии вызывают, как правило, туберкулез, атипичные — некоторые другие заболевания. Микобактерии группы I — фотохромогены вызывают заболевания, связанные с пигментацией; микобактерии группы II — скотохромогены вызывают лимфадениты у де1ей; микобактериальные организмы группы III вызывают определенные легочные заболевания, и, наконец, микобактерии группы IV (Mycobacterium fortuitum) вызывают редко встречающиеся легочные и нелегочные заболевания, не поддающиеся лечению обычным набором антитуберкулезных препаратов и требующие лечения антибиотиками широкого спектра. Особняком стоит проказа (лепроз, болезнь Хансена) вызываемая микроорганизмом Mycobacterium leprae
34Л. Антитуберкулезные препараты
Туберкулез является инфекцией, вызываемой микобактериями Mycobacterium tuberculosis, наиболее часто поражающей легкие и характеризующейся такими симптомами, как острое воспаление, некроз тканей и часто развитием открытых полостей.
В некоторых случаях патоген проникает в лимфу или кровь, и инфекция может распространиться и по другим тканям организма. Современная терапия туберкулеза весьма эффективна, однако может быть трудна и длительна. Патоген быстро приобретает резистентность к терапии одним препаратом. Более того, многие штаммы приобрели резистентность и к би-, и даже к мультилекарственной терапии, поэтому антитуберкулезные препараты, как правило, используются в виде комбинации двух, а иногда и трех препаратов.
-721 -
Г лава 34
Препараты, используемые при терапии туберкулеза, весьма отличаются по активности и токсичности и делятся на 2 группы.
Препараты I группы объединяют лекарственные средства с высшим уровнем эффективности при относительно низкой токсичности. В эту группу входят изониазид, этамбутол, пиразинамид, а также антибиотики — рифампицин и стрептомицин.
Подавляющее большинство пациентов может быть успешно вылечено этими препаратами. Иногда по причине микробной резистентности и'или в связи с пациентзависимыми причинами может возникнуть необходимость в применении препаратов II группы. Сюда включены этио-намид, антибиотики (циклосерин, капреоминин. канамицин), а также структурно очень простой препарат и и-аминосалициловая кислота. Указанные препараты несколько более токсичны, чем препараты I груп-ппы и имеют определенные ограничения по применению.
Химиотерапия туберкулеза должна включать применение двух или более эффективных препаратов для предотвращения умножения числа резистентных мутантов. Лечение должно продолжаться достаточно долго. чтобы предотвратить рецидив медленно растущих внутриклеточных организмов.
Препараты I группы
Изониазид (Isoniazid)
Изониазид — гидразид изоникотиновой кислоты (34.1.1) получают взаимодействием этилового эфира изоникотиновой кислоты с гидразином [1,2. 3, 4. 5].
о
С-ОС2Н5
о
С- NH-Ь1Н2
34.1 1
Изониазид является гиразидом изоникотиновой кислоты, введенным в медицинскую практику для лечения туберкулеза в 1953 г. Изониазид проявляет бактерицидное действие на Mycobacterium tuberculosis Он ингибирует синтез миколиевой кислоты — важной составляющей микобактериальной клеточной стенки. Миколиевая кислота присуща только микобактериям, и именно это является причиной селективной токсичности препарата по отношению к этим микроорганизмам. Существующие в природе резистентные к изониазиду мутанты встре-
-722 -
днтимикобактериапьные препараты
«I I-”*™*™ ' " . -I II - .
чаются редко, и в спонтанно растущей популяции туберкулезных бацилл их можно наблюдать в соотношении примерно один на 105-106 организмов. Большие популяции микроорганизмов порядка 1О9-1О10 банили в открытых легочных полостях, уже содержат значительное число резистентных мутантов. Если при лечении применять только изониазид, то будет наблюдаться увеличение числа мутантов, которые могут стать доминантным фенотипом. Переход от чувствительных, в основном, к нечувствительным микроорганизмам в ходе лечения называется вторичной, или приобретенной, резистентностью, которая может возникнуть в течение нескольких недель.
Изониазид является наиболее важным лекарством для лечения легочной и нелегочной форм туберкулеза. Он активен по отношению как к внутриклеточным, так и внеклеточным организмам. Для предотвращения вторичной резистентности изониазид должен применяться с другим эффективным препаратом (обычно рифампином).
Синонимами препарата являются тубазид, андразид, ниазид, пири-дизин и многие другие.
Эта.чбутол (Ethambutol)
Этамбутол — (+)-М.]9|-этиленбис-(2-аминобутан-1-ол) (34.1.4) предложено получать несколькими путями. Согласно первому способу, нитропропан подвергают оксиметилированию формальдегидом, и далее нитрогруппу в полученном 2-нитробутаноле (34.1.2) восстанавливают водородом до аминогруппы с получением рацемического (±)2-аминобутанола. Применением L( г) винной кислоты из рацемата выделяют (+)2-аминобутанол (34.1.3). Взаимодействием последнего с 1,2-дихлорэтаном в присутствии гидроокиси натрия получают этамбутол (43.1.4) [6, 7, 8, 9, 0, 11, 12, 13, 14, 15].
СН-.О Н2 ' Raney—Nf / NaOH
CHt-CH2-CH2-NO2 -► CH3“ Crt2-CH--NO2 —---► CH3-CH2~CH-NH2 ------►
CH2-OH CH2-OH
34.1 2 34 1 3
CH3~CH2-CH-NH~CH2-CH2"NH-CH-CH2-CH2“CH3 6h2-OH HO-CH2
34 1,4
Второй способ синтеза заключается в получении (+)2-аминобу-танола (34.1.3) путем восстановления водородом гидрохлорида этилово
-723-
Г лава 34
го эфира L-2-аминомасляной кислоты с одновременным использованием в качестве катализаторов никеля Ренея и окиси платины. При этом однозначно получается чистый (+)2-аминобутанол. Взаимодействием последнего с 1,2-дихлорэтаном в присутствии гидроокиси натрия получают искомый этамбутол (34.1.4) [16, 17].
Н2! Raney—N>. PtO
СН3—СН2—СН—СООС2Н5 ------------------------► ch3-ch2-ch-nh2
мн2 CH2—ОН
34 1 3
Третий способ синтеза весьма любопытен и, очевидно, напоминает реакцию Риттера, которую проводят в присутствии хлора. Способ заключается во взаимодействии 1-бутена и ацетонитрила в присутствии хлора, в результате чего, очевидно, происходит 1,4-присоединение хлора к ироду кту реакции Риттера с образованием промежуточного дихлорида (33.1.5), который гидролизуют водой с получением N-[l-(xnop-метил)-пропил]-ацета.мида (33.1.6). Кипячением последнего с соляной кислотой получают рацемический (±)2-аминобутанол, из которого вышеописанным способом с использованием L(+) винной кислоты выделяют (т)2-аминобутанол (34.1.3). Взаимодействием последнего с 1,2-дихлорэтаном в присутствии гидроокиси натрия получают искомый этамбутол (34.1.4) [18, 19,20,21].
н2с нс
C^-er2-CH=CH: + Сн3<Л ’ с.2 -СНз-СНг-СН-СНг-О -СНЭ-СН2-СН-СН2-С| --► 34 1 3
N=c-CH: -iN-C-CH3
Cl О
34 5 5 34 1 6
Этамбутол был открыт в 1961 г. Препарат имеет бактериостатическое действие по отношению к Mycobacterium tuberculosis, однако точный механизм его действия неизвестен. Препарат ингибирует перенос микотиновой кислоты в клеточную стенку Mycobacterium smegmatis, чем и объясняется его избирательная токсичность. Этамбутол активен только по отношению к микобактериям Mycobacterium tuberculosis, Mycobacterium kansasii, Mycobacterium scrofulaceum Чувствительность других микобактерий к этамбутолу весьма различна. Все остальные бактерии практически резистентны по отношению к нему. Случаи первичной резистентности к этамбутолу единичны Вторичная резистентность возникает, когда препарат используется самостоятельно без параллельного применения другого эффективного антитуберкулезного препарата, например изониазида или рифампина. Механизм резистент
- 724 -
Антимикобактериальные препараты
ности к препарату неизвестен.
Синонимами этамбутола являются диамбутол, клобутол, гибистал, тубетол и многие другие.
Пиразинам ид (Pyrazitiamid)
Пиразинамид — пиразинкарбоксамид (34.1.11) синтезируют исходя из хиноксалина (34.1.7), который получают взаимодействием о-фе-нилендиамина с глиоксалем. Окисляя последний перманганатом калия, получают пиразин-2,3-дикарбоновую кислоту (34.1.8). Декарбоксилированием последней при нагревании получают пиразин-2-карбоновую кислоту (34.1.9). Этерификацией полученной кислоты метанолом в присутствии хлористого водорода и дальнейшим взаимодействием полученного эфира (34.1.10) с аммиаком получают пиразинамид [22. 23, 24, 25, 26, 27, 28].
34 1 7
341 а
.N^COOH СНзОН ! НС|
34 1 10
NH3
34 1 11
34 1 9
Пиразинамид был синтезированный в 1952 г. Он является азаанало-1ОМ никотинамида, однако уступает ему по активности Точный механизм его действия неизвестен. Препарат проявляет гепатотоксичность.
Синонимами препарата являются декса.мбутол, .миамбутол, эсанбу-тол. эбутол и др.
Рифампицин (Rifampicin)
Рифампицин — ацетат 5,6,9,17,19,21-гексагидрокси-23-метокси-2,4,12,16.18,20,22-гептаметил-8-[К-(4-метил-1-пиперазинил)-форми-мидоил]-2,7-(эпоксипентадека-1,11,13-триенимино)-нафто-[2,1 -Ь]фу -ран-1.11(2Н)диона-21 (32.7.8). Рифампицин является полусинтети-ческим производным рифампицина В, продуцируемого актиноми-цетами Streptomyces mediteranei (Nocardia mediteranei) и введенного в медицинскую практику в 1968 г. Синтез рифампицина был описан в гл. 32 «Антибиотики».
- 725-
Г лава 34
Рифампицин проявляет бактерицидный эффект путем ингибирования синтеза РНК. Препарат ингибирует ДНК-зависимую РНК полимеразу, предотвращая начало развития цепи, а не ее наращивание. Рифампицин не связывается с РНК полимеразой ядра клеток млекопитающих и не воздействует на соответствующий синтез РНК. Он может ингибировать митохондриальный синтез РНК, однако концентрации, требуемые для этого, в несколько сот раз превышают таковые, необходимые для синтеза РНК.
Рифампицин высоко активен в отношении Mycobacterium tuberculous, в ряду атипичных микобактерий — по отношению к Mycobacterium kansasti, Mycobacterium mannum и к большинству видов Mycobacterium scrofulaceum и Mycobacterium xenopi Чувствительность других микобактерий варьирует. Рифампицин проявляет активность и в отношения Mycobacterium lepare
Кроме микобактерий, рифампицин проявляет активность и в отношении широкого ряда организмов. Он высокоактивен в отношении Staphylococcus aureus, неэнтерококковым видам Streptococcus и Listeria monocytogenes Среди грамотрицательных видов к рифампицину очень чувствительны Neisseria meningitidis, Haemophilus influenzae и Legionella Устойчивы E coh и Proteus mirabihs Анаэробные кокки, виды Clostndtum и Bacteroids часто оказываются чувствительны к рифампицину
Рифампицин является наиболее эффективным лекарством для лечения легочной и нелегочной форм туберкулеза, включая туберкулезный менингит Всегда должен применяться в сочетании с другими препарата ми
Синонимами препарата являются рифадин. римактан, рифапиам, римакгазид и др.
- 726 -
Антимикобактермальные препараты
Стреппюмицин (Streptomycin)
Стрептомицин — щраяс-2,4-дигуанидино-3,5,6-тригидроксицик-логексил-5-дезокси-2-О-(2-дезокси-2-метиламино-а-Е-глюкопирано-зил)-3-С-гидроксиметил-р-Ь-ликсо-пентофуранозид (32.4.1) выделяют из культуральной жидкости жизнедеятельности актиномицета Streptomyces griseus Его синтез был описан в гл. 32 «Антибиотики».
32 4 1
Стрептомицин обладает широким спектром антибактериальной активности. Является первым клинически эффективным препаратом, примененным для лечения туберкулеза.
Синонимами препарата являются стрептан, стрептокол и др.
Препараты II группы
Этионамид (Etionamid)
Этионамид — 2-(этил)изоникотинтиоамид (34 1.18) — производное изоникотиновой кислоты получают по следующей схеме. Конденсацией диэтилоксалата с метилэтилкетоном в присутствии этилата натрия получают этиловый эфир пропионилпировиноградной кислоты (34.1.12). Конденсацией последней с цианацетамидом осуществляют гетероциклизацию с получением 3-циано-4-карбэтокси-6-этил-2-пири-дона (34.1.13), гидролизом которого соляной кислотой получают 4-карбокси-6-этил-2-пиридон (34.1.14). Взаимодействием последнего со смесью хлорокиси и пятиокиси фосфора синтезируют
-727 -
Г лава 34
хлорангидрид 6-этил-2-хлоризоникотиновой кислоты, дальнейшей обработкой которого этиловым спиртом получают эгиловый эфир 6-этил-2-хлоризоникотиновой кислоты (34.1.15). Восстановлением последней водородом с использованием палладиевого катализатора убирают атом хлора в положении 2 пиридинового кольца с получением этилового эфира 6-этил-изоникотиновой кислоты (34.1.15). Взаимодействием последнего с аммиаком и дальнейшей дегидратацией полученного при этом амида 6-этилизоникотиновой кислоты пятиокисью фосфора получают нитрил 6-этилизоникотиновой кислоты (34.1 17). Наконец, вводя последний во взаимодействие с сероводородом, получают этионамид [29, 30. 31, 32].
СООС2Н6
СООС2Н6
о с,н5-с-сн3
S II C-NHo
34 1 18
СгНбОНа
Этионамид активен по отношению к Mycobacterium tuberculosis и Mycobacterium leprae, но не оказывает влияния на другие микроорганизмы. Препарат усиливает фагоцитоз в очаге туберкулезного воспаления, что способе гвуег его рассасыванию. Однако препарат часто вызывает побочные эффекты, связанные с ЖКТ, и гепатотоксические эффекты примерно у 5 % пациентов.
Синонимами препарата являются трекатил, этимид, тиомид, тубер-мин,тубероид и др.
- 728 -
Антимикобактериальные препараты
Циклосерин (Cycloserin)
Циклосерин — 4-амино-З-изоксалидинон (34.1.19) может быть получен как биотехнологическим путем в результате жизнедеятельности актииомицетов Streptomyces garyphalus, Streptomyces orchida-ceus и Streptomyces lavenduale, так и синтетическим путем, исходящим из метилового эфира D-серина, взаимодействием которого с пятихлористым фосфором замешают гидроксильную группу' серина на атом хлора и дальнейшим взаимодействием полученного продукта (34.1.19) с гидроксиламином осуществляют гетероцикли-запию в искомый циклосерин (34 1.20) [33, 34. 35, 36, 37, 38, 39, 40, 41.42].
PCI5 NHZ-OH
СН2-СН-СООС2Н5 ------► СН2-СН-СООС2Н5 ---------
ОН NH2 C1 NHZ
34 1 19
Циклосерин обладает широким спектром антибактериального действия, действуя как на грамположительные, так и на грамотрицательные бактерии, а также на микобактерии туберкулеза, устойчивые к препаратам I группы. Препарат действует бактерицидно, нарушая синтез клеточной стенки микобактерий. Принятым объяснением этому является его химическое сходство с D-аланином, что позволяет ему конкурентно угнетать активность ферментов D-аланинрацемазы и D-аланинсинте-тазы. в результате чего нарушается образование дипептида О-аланил-Б-аланина, необходимого для построения клеточной стенки бактерий. Циклосерин применяют для лечения пациентов с хроническими формами туберкулеза, у которых лечение препаратами 1 группы оказалось безрезультатным.
Синонимами препарата являются цикловальдин, цикломицин. се-ромицин и др.
Капреомицин (Capreotnycin)
Капреомицин — полинептидный антибиотик (34.1.21), выделенный из культуральной жидкости продуктов жизнедеятельности Streptomyces capreolus, представляет собой комплекс из минимум четырех микробиологически активных составляющих, лишь частично охарактеризованных. Капреомицины IA и IB представлены ниже.
-729-
Глава 34
2 HSO4
2
CHNHCONh2
+ NH —C-CO-MH-C
+ NH3 CO CO
H3N(CH2)3CHCH2CONHCH2-CH2-CH nh
NH CH2 +
CO-CH-NHSCO-CH-NH3
CH2-R
34 1 21
капреомицин IA R- OH капреомицин IB R = H
Капреомицин имеет выраженное подавляющее действие против Mycobacterium tuberculosis и Mycobacterium bovis. Большинство, штаммов Mycobacterium kansasii также чувствительны к капреоминину, однако другие, нетуберкулезные штаммы, не чувствительны к нему. Препарат часто используется при необходимости применения парентеральной терапии, путем глубоких внутримышечных инъекций. Капреомицин менее токсичен, чем канамицин и имеет несколько больший бактериостатический эффект.
Синонимами препарата являются каиромицин, капастат, отослал и др.
Канамицин (Kanamycin)
Канамицин — 0-3-амино-3-дезокси-а-П-глюкопиранозил-(1—>6)-О-[6-дезокси-6-амино-а-П-глюкопиранозил-(1->4)]-2-дезокси-П-стреп-тамин (32.4.6) выделен из культуральной жидкости жизнедеятельности актиномицета Streptomyces kanamyceticus, который продуцирует зри антибиотика (канамицины А, В и С) и описан в гл. 32.
h2n
32 4 6
-730-
Антимикобактериальные препараты
Канамицин А сходен со стрептомицином и неомицинами и обладает широким спектром антимикробного действия. Активен в отношении большинства грамположительных, а также грамотрицательных микроорганизмов (стафилококков, кишечной палочки, клебсиеллы, палочки Фридлендера, протея, шигеллы, сальмонеллы).
Препарат применяют при лечении сепсиса, менингита, остеомиелита. перитонита, пневмонии, пиелонефрите, пиелоцистите, раневых инфекциях, послеоперационных гнойных осложнениях, вызванных чувствительными к препарату микроорганизмами. Канамицин применяют для лечения туберкулеза легких и других органов при устойчивости к другим противотуберкулезным препаратам.
Синонимами препарата являются кармицин, камаксин, резистоми-цин и .многие другие.
п-аминоса.тциловаякислота (n-aminosalicylic acid)
я-аминосалициловая кислота — 5-амино-2-гидроксибензойная кислота (34.1.22) получается по реакции Кольбе непосредственным взаимодействием .м-аминофенола с бикарбонатом калия и с двуокисью углерода при нагревании и небольшом давлении в 5-10 атмосфер [43, 44, 45, 46, 47].
КНСО3
со2
он
34 1.22
л-аминосалициловая кислота является бактериостатиком, ингибирующим большинство туберкулезных микобактерий. По туберкулостатической активности уступает изониазиду и стрептомицину. Препарат нефро- и гепатотоксичен. Применяется редко.
Синонимом препарата является апацизин,
34.2. Препараты для лечения лепроза
Поврежденная кожа, потеря ею болевой чувствительности и увеличение нервов являются тремя основными признаками лепроза — заболевания, вызываемого микобактериями Mycobacterium leprae. Заболевание крайне заразно. Дети и мужчины более чувствительны к болезни, чем женщины. Инкубационный период может длиться несколько лет, что затрудняет раннюю диагностику и лечение лепроза, или проказы.
- 731 -
Глава 34
Химиотерапия лепрозадо 1982 г заключалась в применении дапсо-на. который давал хорошие клинические результаты. Однако, вследствие возникновения первичной и втрричной резистентности в результате его долговременного применения, в настоящее время стало обязательным применение определенного сочетания препаратов — лечение дап-соном сопровождается применением рифампицина и клофазимина. Допускается и применение зтионамида.
Дапсон (Dapson)
Дапсон - 4,4-диаминодифенилсульфон (34.2 3) получают исходя либо из 4-хлорнитробензола. либо из натриевой соли 4-ацетами-добезолсульфоновой кислоты. Взаимодействием 4-хлорнитробензола с сульфидом натрия получают 4,4|-динитродифенилтио-эфир (34.2.1), окислением атома серы в котором дихроматом калия в серной кислоте получают 4,41-динитродифенилсульфон (34.2.2). Восстановлением нитрогрупп в последнем двухлористым оловом в соляной кислоте получают искомый дапсон Предложено также предварительно осуществлять восстановление нитрогрупп до аминогрупп. защищать их ацетильной защитой, окислить атом серы до сульфона дихроматом калия и затем гидролизом убрать защитные ацетильные группы [48, 49, 50]
34 2 1
К2СГ2О7 / H2SO4
SnCL / НС!
34 2 2
34 2 3
Иной подход к синтезу далсона осуществлен исходя из 4-ацетами-добезолсульфоновой кислоты, взаимодействием которой с 4-хлорнитробензолом при высоких температурах получают 4-ацетамидо-4'-нитродифенилсульфон (34.2.4). Восстановлением низрогрупп в последнем дву хлористым оловом в соляной кислоте и одновременным гидролизом в условиях реакции ацетильной группы получают искомый дапсон [51, 52. 53].
-732-
Антимикобактериальные препараты
Cr'^ONh
SrC,?-1 nCl
Дапсон, впервые предложенный в 1941 г . обладает как бактерицидной, так и бактериостатической активностью в отношении Mycobacterium leprae и Mycobacterium tuberculosis Предложен также для лечения больных герпетиоформным дерматитом. Полагают, что механизм ею действия заключается в конкурентном ингибировании фермента дигидропротеатсинтетазы, блокирующей синтез фолиевой кислоты у микроорганизма, поскольку его также можно рассматривать в качестве аналога н-аминобензойной кислоты.
Клофазимин (Clofazimine)
Клофазимин — 2-(л-хлоранилино)-5-(«-хлорфенил)-3,5-дигидро-3-(изопропилимино)-феназин (34.2.6) синтезируют, окисляя 2-(л-хлор-анилино)анилин раствором хлористого железа в воде, что приводит к получению 2-(п-хлоранилино)-5-(л-хлорфенил)-3,5-дигидро-3-ими-нофеназина (34.2.5) Взаимодействием последнею с первичными аминами, в частности с изопропиламином, происходит формальное замещение атома водорода в иминной части молекулы на алкильную группу вводимой аминогруппы, в данном случае не изопропильную, с получением искомого препарата — клофазимина [54, 55, 56].
Клофазимин является замещенным иминофеназином и впервые был предложен для лечения лепроза в 1962 г., однако вошел в медицинскую практику в конце 1980-х годов. Механизм его действия не окончательно понят, однако есть предположение, что он может ингибировать образование матриц с ДНК, что приводит к задержке роста микобактерий Клофазимин проявляе! бактерицидный эффект промежуточный между дапсоном и рифампицином.
- 733 -
Глава 34
1
2
3
4
5
6
7
8
9
10 И
12 В
14 1*>
16 Г
18
19
20
21
22
23
24
25
26
27
28
29
30 ч 32
31 U
35
36 3-7 18
39
Список литераторы
LSPat 2 596 069 (1952)
LSPat 2 830 994 (1958)
Ger Pat 1 116 667 (1956)
Meyer H et al / A-lonatsh Chem 33 393 (1912)
Lrbanski 1 etal '/Roc? Chem 27, 161 (1953)
I S Pat 3 176 040 (1965)
Belg Pat 600 640(1961)
Belg Pat 613 545 (1962)
Wilkinson R et al / J Am Chem Soc 83, 2212 (1961)
Wilkinson R etal 'J Med Pharm Chem 5,835 (1962)
LSPat 3 553 257 (1971)
LSPat 3 769 347 (1973)
Ger Pat 2 205 269 (1972)
I SPat 3 944 616(1976)
Fr Pat 2 351 090 (1974)
Ger Pat 2 446 320 (1974)
Bnt Pat 1 469 014(1974)
USPat 3 944 617 (1976)
I S Pat 3 944 618 (1976)
LSPat 3 944 619 (1976)
Bnt Pat 1 541 290(1976)
Ger Pat 623 257 (1934)
Bnt Pat 451 304 (1939)
LSPat 2 149 279 (1939)
I S Pat 2 677 641 (1954)
I SPat 2 780 069(1957)
HallS etal J Am Chem Soc 62, 664 (1940)
Kushner 4 etal J Am Chem Soc 74. 3617 (1952)
Bnt Pat 800 251 (1957)
Libermann S etal Compt Rend Acad Sei 242, 2409 (1956) Libermann S et al 'Bull Soc Chim France 1958, 687 Johnson К etal J \m Chem Soc 117, 5009 (1995) Plattner P etal Helv Chim Acta 40, 1531 (1957)
Smart J etal Expenentia 13, 291 (1957)
USPat 2 772 280 (1954)
USPat 2 840 565 (1958)
LS Pat 2 773 878 (1956)
LSPat 2 789 983 (1957)
I S Pat 2 845 437 (1958)
- 734 -
Антимикобактериапьные препараты
40 41 42
43 44 45
46
47 48 49
50 51
52 S3 54 55
56
USPat 3 124 590(1964)
Bnt Pat 768 007 (1957)
Stammer В etal J Am Chem Soc 77. 2346 (1955)
USPat 2 540 104(1951)
USPat 2 844 625 (1958)
Ger Pat 50 835 (1889)
Sheehan J / J Am Chem Soc 70, 1665 (1948)
Erlenmeyer E etal /Helv Chim Acta 31, 988 (1948)
USPat 2 235 889 (1945)
Fromm E et al Ber 41, 2264 (1908)
Raiziss G etal /J Am Chem Soc 61,2763 (1939)
Ferry C etal /Organic Synth 22,32 (1942)
Bnt Pat 510 127(1938)
USPat 2 227 400(1940)
Barry I etal /Nature. П9. 1013 (1957)
USPat 2 948 726(1960)
Barry I et al J Chem Soc 1958 859
Глава 35
{ Противогрибковые препараты
Грибки, по сравнению с бактериями или вирусами, являются более сложными организмами. Они имеют рибосомы, компоненты клеточных стенок, ядерную мембрану, поэтому антибактериальные антибиотики, как правило, неэффективны против патогенных грибков.
Грибковые инфекции (микозы) встречаются реже чем бактериальные или вирусные. Однако это утверждение может оказаться неверным для некоторых географических районов, благоприятных для существования и роста специфических грибковых патогенов. Некоторые грибковые инфекции могут располагаться на поверхности тела и лишь доставлять локальное беспокойство, другие могут быть системными, угрожающими жизни. Причем одни и те же микроорганизмы (например, Candida) могут из поверхностной локализации перейти и на внутренние органы, приводя к системным заболеваниям с серьезными осложнениями. Грибковые (микотические) инфекции доставляют много беспокойства и, как правило, трудно вылечиваются.
Грибковые инфекции условно делятся на три категории, дерматофитные, кожно-слизистые и системные.
Наиболее распространенными являются дерматофитные грибковые инфекции, включающие кожу, волосы и ногти. Большинство инфекций может быть вылечено применением таких наружных средств, как толнафтаг, ундециленовая кислота, галопрогин, клотримазол и ми-коназол Для глубоких инфекций, в частности инфекций ложа ногтя, орально назначают гризеофульвин. В настоящее время в качестве препарата для лечения хронических дерматофитов широко используется кетоконазол.
Кожно-стизистые инфекции, вызываемые в основном грибком Candida albicans, включают участки влажной кожи и слизистые мембраны (т. е. ЖКТ. перианальные и вульвовагинальные области). Для их лечения наружно применяются амфотерицин В, миконазол. клотримазол и нистатин. При хронических инфекциях орально применяется кетоконазол.
- 736 -
Противогрибковые препараты
Системные грибковые заболевания встречаются реже, однако пред-ciaBiHioi собой серьезною проблему, поскольку являются хроническими по своей природе и трудно поддаются диагностированию и лечению.
В соответствии с вышеописанным противогрибковые препараты характеризуются как дерматофитные, кожно-слизистые и системные, и, соответственно, их рассматривают как:
• противогрибковые препараты для лечения системных и поверхностных инфекций,
• противогрибковые препараты для лечения только поверхностных инфекций,
• противогрибковые препараты для лечения только системных инфекций.
35.1. Противогрибковые препараты для лечения системных и поверхностных инфекций
К противогрибковым препаратам для лечения как системных, гак и поверхностных инфекций относятся амфотерицин В, нистатин, кетоконазол и миконазол
Амфотерицин В (Amphotericin В)
Амфотерицин В — [1R(1R^3S\5R\6R\9R\11R\15S*,16R',17R’, 18S’,19E*,21E',23*,25E‘',27E*,29E*,31E*,33R’,35S,,36R’.37S*)]-33-[3-амино-3.6-дидезокси-Р-0-манопиранозил)-окси]-1,3,5.6,9,11,17,37-октагидрокси-15.16,18-триметил-13-оксо-14,39-диоксабицикло-[33 3 1]-нонатриакоита-19,21,23,25,27,29,31-гептаен-36-карбоновая кислота (35.1.1.) является большим полиенновым антибиотиком, получаемым из культуральной жидкости жизнедеятельности актиномицета Streptomyces nodosus [1, 2, 3, 4. 5]
- 737 -
Глава 35
Соединение имеет широкий спектр антигрибковой активности, включая Candida albicans, Leishmania brasiliensis, Mycobacterium leprae, Histoplasma capsulaium, Blastomyces dermatitidus и Coccidioides immitis. Имеет фунгистатическую и фунгицидную активность в зависимости от используемой дозы.
Противогрибковая активность амфотерицина В проявляется по причине его связывания со стеролами, в частности с эргостеролом, в клеточной мембране чувствительных грибков. Это взаимодействие создает поры в мембранах и увеличивает проницаемость мембран в отношении малых молекул, понижая таким образом функцию мембран в качестве осмотического барьера и делая клетки более чувствительными к разрушению. Амфотерицин В активен по отношению к растущим клеткам и клеткам, находящимся в состоянии покоя. Однако соединение невысокоселективно и взаимодействует и с клетками хозяина — млекопитающего.
Несмотря на многие побочные реакции, амфотерицин В остается первым препаратом для лечения острых, сильных системных грибковых инфекций. Применяется при генерализованных грибковых инфекциях: кандидомикозе, аспергиллезе, гистоплазмозе, криптококкозе, кокцидо-микозе, бластомикозе, легочных микозах.
Синонимами препарата являются амфоциклин, фунгизон, фунгилин и др.
Нистатин (Nystatin)
Нистатин — стереоизомерные 33-[3-амино-3,6-дидезокси-р-П-ма-нопиранозилокси]-1,3,4,7,9,11,17,37-октагидрокси-15,16,18-триме-тил-13-оксо- 14,39-диоксабицикло-(33.3.1 )-нонатриаконта-19,21,25, 27,29,31-гексаен-36-карбоновая кислота (35.1.2) [6, 7, 8, 9, 10, 11, 12, 13, 14].
- 738-
Противогрибковые препараты
Нистатин был выделен в 1949 г. из продуктов жизнедеятельности актиномицетов Streptomyces noursei и был первым из найденных антимикотических антибиотиков. Это полиеновый антибиотик, структурно сходный с амфотерицином В. Имеет широкий спектр активности. Механизм противогрибкового действия сходен с механизмом действия амфотерицина В. Применяется для профилактики и лечения кандидных инфекциий кожи и слизистых мембран. С профилактической целью применяют для предупреждения и развития кандидомикоза при длительном лечении препаратами пенициллина и антибиотиками других групп, особенно при пероральном применении антибиотиков тетрациклинового ряда, левомицетина и др.
Синонимами препарата являются биофанал, моронал, ниспорил, фа-зигин, кандекс и др.
Имидазолы
Для лечения грибковых инфекций оказались весьма пригодными некоторые производные имидазола. К ним относятся кетоконазол, ми-коназол, клотримазол, эконазол и бутоконазол и др.
Противогрибковая активность производных имидазолов в настоящее время объясняется их способностью селективно повышать проницаемость мембран грибковых клеток путем вмешательства в биосинтез стеринов, в частности эргостерина, ингибируя его синтез, и изменением липидного состава мембраны. Однако, в отличие от амфотерицина В, производные бензимидазола активны только по отношению к растущим клеткам. Клетки хозяина при этом не подвергаются действию препаратов, поскольку для своей жизнедеятельности млекопитающие используют экзогенные стеролы.
Кетоконазол ((Ketoconazol)
Кетоконазол — 1-ацетил-4-[4-[2-(2,4-дихлорфенил)-2-(1//-ими-
дазол- 1 -илметил)-1,3-диоксолан-4-илметил]фенил]пиперазин (35.1.6) синтезируют исходя из 2.4-дихлорфнацилбромида, кетализацией которого с помощью глицерина получают г/ис-2-(2,4-дихлорфенил)-2-бромэтил-4-гидроксиметил-1,3-диоксолан (35.1.3). Ацилируя гидроксильную группу последнего бензоил хлоридом и далее алкилируя полущенным соединением имидазол, получают производное (35.1.4). Далее щелочным гидролизом удаляют бензоильную группу и взаимодействием с метансульфохлоридом получают мезилат (35.1.5). Наконец алкилируя последний 1-ацетил-4-(4-гидрокси-фенил (пиперазином, получают кетокеназол [15, 16, 17, 18].
-739-
Глава 35
35 1 6
Кетоконазол имеет широкий спектр противогрибковой активности, включая многие кандидные инфекции. Обладает фунгицидной и фунги-статической активностью в отношении дерматофтов, дрожжевых грибков, диморфных грибков и эумицетов. Активен также в отношении стафилококков и стрептококков. Эффективен при хронических заболеваниях, при лечении грибковых инфекций ЖКТ, половых органов, кожи, волос и ногтей. Используется и в составе шампуней при лечении и профилактике плесневых микозов, себорейного дерматита, перхоти.
Синонимом препарата является низорал и др
Миконазол (Miconazol)
Миконазол — 1-[2.4-дихлор-р-[(2,4-дихлорбензил)окси]фенэтил]-имидазол (35.1.8) так же, как и кетоконазол, синтезируют исходя из 2,4-дихлорфнацилбромида, взаимодействием которого с имидазолом получают 1 -(2,4-дихлорбензоилметил)-имидазоло[2,4-дихлоро-ы-( 1-имндазолил)ацетофенон] (35.1.6) Восстановлением карбонильной группы в последнем боргидридом натрия получают 1-(2,4-дихлорфенил)-2-(1-имидазолил)-этанол (35.1.6), алкилированием
- 740-
Противогрибковые препараты
гидроксильной группы которого 2.4-дихлорбензилбромидом с использованием в качестве основания такого мощного основания, как гидрид натрия, получают миконазол [19, 20, 21].
н
ch4Z^~ch2~n3
С1 3516
NaBHj
Миконазол предназначен в основном для внешнего применения при кандидных и дерматофитных инфекциях кожи и вагинальном кандидозе. а также при острых внутренних микозах.
Синонимами препарата являются акнидазил, дактар, дермонистар и др.
35.2. Противогрибковые препараты для лечения поверхностных инфекций
Гризеофульвин (Griseofulvin)
Гризеофульвин — 7-хлоро-2',4,6-триметокси-б'-метилспиро[бензо-фуран-2(ЗЯ),1 ‘-И-циклогексенфЗ1,4-дион (35.2.1) — антибиотик, продуцируемый плесневым грибком Penicillium patulum [22, 23, 24, 25, 26, 27*28, 29].
35 2 1
Гризеофульвин является противогрибковым средством для лечения поверхностных инфекций, оказывающим фунгистатическое действие на
- 741 -
Глава 35
разные виды дерматофитов (трихофиты, микроспорумы, ахорионы, эпидермофитоны). Антимикотическая активность объясняется способностью ингибировать клеточный митоз грибков, вызывая образование многоядерных дефектных клеток. Спектр антимикотической активности гризеофульвина включает дерматофитные инфекции кожи, ногтей и скальпа. Препарат неэффективен в отношении глубоких системных микотических инфекций. Неэффективен при кандидомикозах. К нему может развиться резистентность.
Препарат, в основном, применяют при лечении больных, страдающих фавусом, трихофитией и микроспорией волосистой части головы и гладкой кожи, поражением нощей, красным эпидермофитоном. Гри-зеофульвин применяется только в виде оральных лекарственных форм.
Синонимами препарата являются фульцин, ликуден, грифульвин и др.
Имидазолы
Эконазол (Econazol)
Эконазол — 1-[2,4-дихлор-р-[(4-хлорбензил)окси]фенэтил]-имида-зол (35.2.2) является аналогом миконазола, отличающимся отсутствием одного атома хлора в бензильной части молекулы и синтезируемого аналогичным путем лишь с использованием на последней стадии 4-хлорбензилхлорида вместо 2,4-дихлорбензилбромида [19, 20, 21].
35 2 2
Эконазол применяется наружно, только накожно, при лечении лишаев и кандидозов, вызванных чувствительной к препарату флорой (Frichophiton rubrum, Trichophiton mentagrophytes, Trichophiton tonsurans, Microsporum canis, Microsporum audouini, Microsporum gypseum, Candida albicans). При местном применении препарат вызывает гибель грибков в течение 3 дней.
Синонимами препарата являются певарил, экостатин, дермазол и др.
-742-
Противогрибковые препараты
Сулкоиазол (Sulconazol)
Схлконазол — 1-[2,4-дихлор-р-[(4-хлорбензил)тио]фенэтил]-имида-зол (35.2.2) является аналогом эконазола, отличающегося от последнего заменой эфирного кислородного мостика, соединяющего 4-хлорбензильную часть молекулы с фенэтилимидазольной, на тио-эфирный. Соответствующие изменения в синтезе препарата заключаются в замещении гидроксильной гуппы в 1-(2,4-дихлор-фенил)-2-(1-имидазолил)-этаноле (35.1.6) на атом хлора с использованием хлористого тионила и последующем взаимодействии полученного хлорида с 4-хлорбензилмеркаптаном с получением сулконазола [30. 31].
1 soci2
2 HS-СН2
сн2
Так же как и эконазол, сулконазол применяется наружно, только накожно, по тем же пок-заниям, что и эконазол
Синонимами препарата являются экселдерм, сулкозил и др.
Бутоконазол (Butoconazol)
Бутоконазол — 1-[4-(4-хлорфенил)-2-[(2,6-дихлорфенил)тио]бу-тил]-1Я-имидазол (35.2.6) синтезируют исходя из 4-хлорбен-зилмагний бромида, взаимодействием которого с эпихлоргидрином получают 4-(4|-хлорфенил)-1-хлорбутанол-2 (35.2.4), взаимодействием которого с имидазолом в присутствии натрия получают 4-(4'-хлорфенил)-1-(1Я-имидазол-ил)бутанол-2 (35.2.5). Замещая в последнем гидроксильную группу на хлор с использованием хлористого тионила и далее взаимодействием с 2,6-дихлортиофенолом получают бутоконазол [32, 33].
9
CH2~MgBr
О
Сн2-С1
СН2-СН2-СН-СН2“С1 он
35 2 4
- 743 -
Г лава 35
1 SOCI2
Бутоконазол является фунгостатиком и его формально можно отнести к классу имидазолов, но лишь по признаку наличия в структуре препарата имидазольного кольца. Полагают, что он, как и миконазол, эконазол и другие «чистые» представители ряда имидазолов, также ингибирует биосинтез эргостерина в цитоплазматической мембране грибков, однако вполне вероятно, что это не есть единственный механизм его действия. Препарат эффективен при вагинальных инфекциях, вызванных каидидами разного типа. Применяется в качестве только наружно средства и только вагинально.
Синонимами препарата являются фемстат, листовин и др.
Терконазол (Terconazol)
Т ерконазол — 1-[4-[[2-(2,4-дихлорфенил)-2-( 1Н-1,2,4-т риазол-1 -ил-метил)-1,3-диоксолан-4-ил]метокси]фенил]-4-(1-метилэтил)-пипера-зин (35.2.13) химически весьма напоминает кетоконазол с той лишь разницей, что вместо имидазольного кольца в структуру препарата включено триазольное кольцо, а пиперазиновое кольцо вместо ацетильной замещено изопропильной группой. Синтез препарата осуществляют исходя из 2,4-дихлорацетофенона, взаимодействием которого с глицерином в присутствии л-толуолсульфокислоты получают кеталь — 2-(2,4-дихлорфенил)-2-метил-4-гидроксимезил-1,3-диоксолан (35.2.7). Бромируя последний молекулярным бромом по метильной группе, и далее ацилированием свободной гидроксильной группы хлористым бензоилом получают 2-(2.4-дихлорфенил)-2-бромметил-4-бензоилоксиметил-1.3-диоксолан (35.2.8). Взаимодействием последнего с 1,2,4-триазолом в присутствии натрия и последующим гидролизом защитной бензоильной группы гидроокисью натрия получают 2-(2,4-дихлорфенил )-2-( 1Я-1,2,4-триазол-1 -илметил)-4-гидроксиметил-1,3-диоксолан (35.2.9). Обработкой последнего метаисульфохлоридом получают мезилат (35.2.10).
уН2-ОК
СН-ОН
СН2-СН
-744-
Противогрибковые препараты
/т—N
1 N N
Н
2 NaOH
35 2 9
CH3SO2C1
35 2 1С
Второй необходимый компонент синтеза — 1-изопропил-4-(4-гидроксифенил)пиперазин (35.2.12) получают исходя из 4-(4-метокси-фенилпиперазина). Восстанавливая последний водородом в присутствии ацетона и используя в качестве катализатора палладий на угле, получают 1-изопропил-4-(4-метоксифенил)пиперазин (35.2.11). Обработкой полученного продукта концентрированной бромистоводородной кислотой снимают защиту с фенольного гидроксила с получением 1-изопропил-4-(4-гидроксифенил)пиперазина (35.2.12)
35 2 12
Наконец, взаимодействием мезилата (35.2.10) с полученным 1-изо-пропил-4-(4-гидроксифенил)пиперазином (35.2.12) получают искомый терконазол [34, 35. 36, 37].
CHt—OSOzCH-:
/Ч
Терконазол эффективен для фунгистатического действия на многие штаммы Candida и дерматофитов. Точный механизм его действия неясен, однако, подобно другим описанным выше азолам, препарат ингибирует действие фермента ланостерол 1-деметилазы цитохрома Р-450 чувствительных грибков, вызывая уменьшение количества эргостерина, необходимого микроорганизму для конструирования мембраны и приобретения соответствующей проницаемости. Препарат применяется только наружно и только вагинально для лечения вульвовагинальных кандидозов.
Синонимами препарата являются теразол, теркоспор и др.
-745-
Глава 35
Клотр аназол (Clotrimazol)
Клотримазол — 1-(о-хлор-а,а-дифенилбензил)имидазол (35.2. 15) получают взаимодействием 2-хлортрифенилметилхлорида (35.2.14) с имидазолом в присутствии триэтила.мина [38, 39, 40, 41, 42].
35 2 14
Н
35 2 15
Исходный 2-хлортрифенилметил.хлорид предложено получать различными путями. В частности, хлорированием на свету 2-хлортолуола получают 2-хлортрихлорметилбензол (35.2.16), взаимодействием которого с бензолом в присутствии хлористого алюминия получают 2-хлор-трифенилметилхлорид (35.2.14).
' А1С13
35 2 14
Альтернативным путем получения 2-хлортрифенилметилхлорида являекя реакция Гриньяра между 2-хлорбензофеноном и фенилмаг-нийбромидом и замещением гидроксильной группы в полученном 2-хлортрифенилмегилкарбиноле (35.2.17) на хлор с помощью хлористого тионила.
35 2 17
- 746 -
Противогрибковые препараты
И, наконец, взаимодействием пятихлористого фосфора 2-хлор-бензофеноном получают 2'-хлор-1,1-дихлордифенилметан (35.2.18), алкилированием последним бензола в присутствии хлористого алюминия также получают 2-хлортрифенилметилхлорид (35.2.14).
РС15
Клотримазол также формально относится к производным имидазола лишь по признаку наличия в структуре молекулы препарата имидазольного кольца. Полагают, что он, как и миконазол, эконазол и другие «чистые» представители ряда имидазолов, также ингибирует биосинтез эргостерина в цитоплазматической мембране грибков.
По фармакологическому действию клотримазол очень схож с мико-назолом. Имеет широкий спектр противогрибковой активности. Эффективен в отношении дерматофитов, а также оказывает противомикробное действие в отношении стрептококков и стафилококков. Эффективен и в отношении трихомонад. Препарат очень широко применяется для лечения поверхностных инфекций как накожно, так и вагинально.
Синонимами препарата являются канестен, эмпепид, лотримин, ми-коспорин и др.
Прочие
Нафтифин (Naftiflne)
Нафтифин — (Е)-?4-метил-М-(3-фенил-2-пропенил)-1-нафталинметанамин (35.2.19) получают алкилированием М-метил-(1-нафтилме-тил)амина кротилхлоридом в присутствии соды [43,44,45, 46,47,48].
На2С03
35 2 19
-747 -
Г лава 35
Нафтифин является первым представителем нового класса противогрибковых препаратов, классифицируемых как аллиламины. и разрешенного к применению только наружно и только накожно в качестве препарата с широким спектром действия в отношении дерматофитных и кандидных инфекций. Согласно первоначально полученным данным, препарат превосходит по активности эконазол. Более того, препарат не имеет местнораздражающего действия. Полагают, что фунгицидная активность препарата основывается на его способности ингибировать грибковый фермент сквален эпоксидазу и тем самым понижать концентрацию эргостерола. Соответствующий фермент млекопитающих при этом ингибируется значительно меньше.
Синонимами препарата являются экзодерил, нафтин и др.
Галопрогин (Haloprogin)
Галопрогин — 3-йодо-2-пропинил-2,4,5-трихлорфениловый эфир (35.2.21) получают замещением на йод смесью йода и йодистого калия, медного производного 2,4,5-трихлорфенилпропарги.пового эфира (35.2.20), получаемого по стандартной методике из пропаргилбро-мида и 2,4,5-трихлорфенола в присутствии гидроокиси натрия [49, 50, 51].
Галопрогин применяют в качестве наружного средства при умеренных дерматофитных инфекциях (опоясывающий лишай), он эффективен при кожных кандидных инфекциях.
Синонимами препарата являются галотекс, мицилан, миканден и др.
Толнафтат (Tolnaftat)
Толнафтат — 0-(2-нафтил)-К-.метил-1'<-(3-толил)-тиокарбамат (35.2.23) получают взаимодействием эквимольных количеств 2-нафтола и тиофосгена с получением продукта монозамещения в фосгене (35.2.22), который далее вводят во взаимодействие с N-метил-З-толуидином с получением искомого толнафтата [52, 53, 54, 55].
- 748 -
Противогрибковые препараты
Голнафтат применяют в качестве наружного средства при умеренных дерматофитных инфекциях (опоясывающий лишай), он неэффективен при кандидных инфекциях.
Синонимами препарата являются тинатокс, тонофтал, тимопед, ти-надерм.тинактин и др.
Ундециленовая кислота (Undecylenic acid)
Ундециленовая кислота — 10-ундеценовая кислота (35 2.24) получается пиролизом при 400 "С и низком давлении (50мм) оксипроизводного олеиновой кислоты — рицинолиевой кислоты, глицерид которой образует главную составляющую касторового масла [56, 57]
CHj-|CH2'5-CH-CH2-CH = CH-(CHz).-COOH ► Н2С=СН-(СН2)8-СООг| ♦ СНз—(СН2)5-СН=О он
35 2 24
Ундециленовая кислота, равно как и ундециленат цинка весьма эффективны в качестве наружного средства при умеренных дерматофитных инфекциях и дрожжевых дерматитах, но неэффективны при опоясывающем лишае и при кандидных инфекциях.
Синонимами препарата являются бензодем, микоцид, ундетин и др.
Натамицин (Natamycin)
Натамицин — смесь стереоизомерных 22-[(3-амино-3,6-дидезокси-Р-О-маннопиранозил)окси]-1,3,26-тригидрокси-12-метил-10-оксо-6, 11,28-триоксатрицикло[22.3.1.0.5 ']-октакоза-8,14,16,18,20-ленген-25-карбоновых кислот (35.2.25). как и амфотерицин и нистатин, является полиеновым антибиотиком и выделяется из продуктов жизнедеятельности актиномицетов Streptomyces natalensis [58, 59, 60].
Спектр его активности несколько более узок, чем амфотерицина и нистатина, но одновременно препарат и менее токсичен. Особенно вы
-749-
Глава 35
раженная активность отмечена в отношении некоторых штаммов Fusarium и Cefalosporium Натамицин является препаратом для лечения поверхностных грибковых инфекций и применяется только в офтальмологических целях.
Синонимами препарата являются пимафугии, пимарицин, теннеце-тин и др.
35.3. Противогрибковые препараты для лечения системных инфекций
Флуцитозин (Flucytosin)
Флуцитозин — 5-фторцитозин (35.3.3) предложено получать исходя из фторурацила (30.1.3.3). При первом способе, подвергая последний действию хлорокиси фосфора в диметиланилине, получают 2,4-дихлор-5-фторпиримидин (35.1.1), взаимодействием которого с аммиаком получают продукт замещения атома хлора в положении 4 пиримидинового кольца — 4-амино-2-хлор-5-фторпиримидин (35.3.2). Гидролизом хлорвинильного фрагмента в последнем раствором соляной кислоты получают искомый флуцитозин [61, 62, 63, 64].
РОС13
ЗС 1 3 3
нгО/ на
Альтернативным вторым способом синтеза является получение флуцитозина из прекурсора фторурацила — из 5-фтор-2-метилтио-ураиила (30.1.3.2) примерно по аналогичной схеме. Обработкой 5-фтор-2-метилтиоурацила (30.1.3.2) с пятихлористым фосфором получают 4-хлор-5-фтор-2-метилтиопиримидин (35.3.4), который взаимодействием с аммиаком трансформируют в 4-амино-5-фтор-2-метилтиопирими-дин (35.3.5). Гидролизом метилтиовинильного фрагмента в последнем действием концентрированной бромистоводородной кислоты получают искомый флуцитозин [65, 66, 67].
30 1 3 2 35 3 4 35 3 5
НВг
-750-
Противогрибковые препараты
Флуцитозин является фторированным производным пиримидина. Его спектр активности уже чем у амфотерицина В. Однако он проявляет синергическое действие при использовании в комбинации с амфотерицином В. В чувствительных грибках флуцитозин трансформируется в 5-фторурацил, который, в свою очередь, превращается в 5-фтордезо-ксих ридиловую кислоту — ингибитор тимидилат синтетазы и, соответственно, синтеза ДНК. В процесс может быть вовлечен и трифосфат 5-фторурацила, вызывающий образование дефектных РНК. Механизм высокоселективен, поскольку клетки млекопитающих не в состоянии превращать большие количества флуцитозина в 5-фторурацил.
Флуктозин используется в качестве самостоятельного препарата, однако, как правило, его используют в комбинации с амфотерицином для лечения определенных системных грибковых инфекций, в частно-С1И для лечения подкожных хромобластомикозов. Препарат интенсивно используется при лечении системных инфекций мочеполовых путей, вызванных различными штаммами Candida.
Синонимами препарата являются анкобон, анкотил и др.
Список литературы
1. Gold W. et al / 'Antibiot. Ann. 1955-1956, 579.
2. J andeputte J et al.//Antibiot. Aim. 1955-1956, 587.
3. Dutcher J. et al.//Antibtot. Ann. 1955-1956, 866.
4. Walters A et al.//J. Am. Chem. Soc. 79, 5076 (1957).
5. US Pat. 2.908.611 (1959).
6. Hazen E. et al.//Science, 112, 423 (1950).
7. Hazen E. et al.//Proc. Soc. Exp. Biol. Med. 76, 93 (1951)
8. Cohen R. et al./.'Arch. Pediatrics, 69, 414 (1952).
9. US Pat. 2.786.781 (1957).
10. US Pat. 2.797.183 (1957).
11. US Pat. 2.832.719(1958).
12. US Pat. 3.517.100(1970).
13. ZelinskiJ. et al.//J. Antibiot. 41, 1289 (1988).
14. Seneca //.//Antibiot. Ann. 1955-1956, 697.
15. Ger. Pat. 2.804.096(1978).
16. US Pat. 4.144.346(1979).
17. US Pat. 4.223.036(1980).
18. Heers J. et al.'/J. Med. Chem. 22, 1003 (1979).
19. Ger. Pat. 1.940.388 (1970).
20. US Pat. 3.717.655 (1973).
-751 -
Г лава 35
21. Godefroi Е. et al. / J. Med. Chem. 12, 784 (1969).
22. Brit. Pat. 784.618 (1955).
23. USPat. 2.900.304 (1959).
24. USPat. 3.038.839 (1962).
25. USPat. 3.069.328 (1962).
26. USPat. 3.069.329 (1962).
27. Oxford A et al. Biochem. J. 33, 240 (1939).
28. Grove J et al.,'Chem. & Ind. (London), 1951,219.
29. Grove J et al./, J. Chem. Soc. 1952, 3977.
30. Ger. Pat. 2.541.833 (1976).
31. USPat. 4.038.409 (1977).
32. USPat. 4.078.071 (1978).
33. lialker К et al.', J. Med. Chem. 21. 840 (1978).
34. USPat. 4.144.346(1979).
35. USPat. 4.223.036(1980).
36. Heers J. et al.- J. Med. Chem. 26, 611 (1983).
37. Heers J. et ab/J. Med. Chem. 22, 1003 (1979).
38. Ger. Pat. 1.617.481 (1967).
39. Ger. Pat. 1.670.976 (1968).
40. USPat. 3.660.577 (1972).
41. USPat. 3.705.172 (1972).
42. S.Afr. Pat. 68 05.392 (1972).
43. Ger. Pat. 2.716.943 (1977).
44. Ger. Pat. 2.809.211 (1978).
45. Belg. Pat. 853.976 (1977).
46. US Pat. 4.282.251 (1981).
47. Span. Pat. 504.432 (1982).
48. Loibner H. et al./7Terahedron Letters. 25, 2535 (1984).
49. US Pat. 4.038.409(1977).
50. Japan Pat. 64 19.791 (1964).
51. Seki S. et al. Agr. Biol. Chem. (Tokyo), 27, 150 (1963).
52. USPat. 3.334.126 (1967).
53. Bnt. Pat. 967.897 (1962).
54. Fr. Pat. 1.337.797 (1963).
55. Noguchi N et al.'/J. Pharm. Soc. Japan, 88, 335 (1968).
56. Kraft F '-Ber. 10. 2034 (1877).
57. Perkins G. et al. J. Am. Chem. Soc. 49, 1070 (1927).
58. Struyk et al.,' Antibiot. Ann. 1957-1958, 878.
59. Bnt Pat. 844.289 (1960).
60. Brit. Pat. 846.933 (1960).
61. USPat. 3.040.026 (1962).
- 752-
Противогрибковые препараты
62. USPat. 3.185.690 (1967).
63. Dunschinsky R. et al./, J. Am. Chem. Soc. 79, 4559 (1957).
64. Undheim К et aI.,7Acta. Chem. Scand. 23, 294 (1969).
65. US Pat. 3.945.038 (1967).
66. US Pat. 2.802.005 (1957).
67. US Pat. 3.368.938 (1968).
68. Ger. Pat. 2.716.943 (1977). (32)
69. Ger. Pat. 2.809.211 (1978).
70. Belg. Pat. 853.976 (1977).
71. US Pat. 4.282.251 (1981).
72. Span. Pat. 504.432 (1982).
73. Loibner H. et al.//Terahedron Letters. 25, 2535 (1984).
74. US Pat. 4.038.409 (1977).
75. Japan Pat. 64 19.791 (1964).
76. Seki S et al./.'Agr. Biol. Chem. (Tokyo). 27, 150 (1963).
77. USPat. 3.334.126(1967).
78. Brit. Pat. 967.897 (1962).
79. Fr. Pat. 1.337.797 (1963).
80. Noguchi N. et al./'J. Pharm. Soc. Japan, 88, 335 (1968).
81. Kraft F.IIKec. 10, 2034 (1877).
82. Perkins G et al./'J. Am. Chem. Soc. 49, 1070 (1927).
J Глава 36
5 Противовирусные । препараты
Вирусы служат причиной большего числа болезней, чем любая другая группа паразитов. Они могут вызвать слепоту, глухоту, паралич, умственную отсталость, различные врожденные дефекты и, по крайней мере у некоторых растений и животных, рак. Среди наиболее известных вирусных заболеваний следует назвать корь, свинку, оспу, ветряную оспу, грипп, полиомиелит, желтую лихорадку. Имеются подозрения, что вирусы являются причиной рассеянного склероза, болезни Ходжкина, болезни Дауна и даже, возможно, шизофрении.
Однако, несмотря на значительный прогресс, достигнутый в области лечения бактериальных инфекций, достижения химиотерапии вирусных заболеваний значительно скромнее. Имеется лишь небольшой набор доступных препаратов для лечения весьма ограниченного числа вирусных заболеваний.
Вирус — это одна из величайших загадок биологии. Его очень трудно дифференцировать в качестве объекта, относящегося к живой или неживой природе. Место вируса, по-видимому, находится где-то посередине между молекулой и живым организмом. Одновременно его можно считать и самым мелким организмом, и самой большой молекулой. Большую часть времени у вируса нельзя обнаружить никаких признаков жизни; в таком состоянии он может оставаться годами. Однако в любой момент он может «воскреснуть»; для этого нужна лишь восприимчивая клетка, которую вирус мог бы заразить.
Каждый вирус способен заражать только ограниченный ассортимент клеток. Это позволяет разделять их на 3 основные группы;
1) вирусы, поражающие растения;
2) вирусы, поражающие животных, в том числе и человека;
3) вирусы, поражающие одноклеточные организмы (микробы или бактерии).
Вирусы являются значительно более простыми организмами, чем бактерии, и построены из двух веществ — белка и нуклеиновой кислоты. Простую вирусную частицу можно считать единичной нуклеопро
-754-
Противовирусные препараты
теидной молекулой, образованной из молекулы нуклеиновой кислоты, химически связанной с объемистой белковой молекулой. Причем белковая молекула играет роль защитной оболочки. Таким образом, вирус схематично можно описать как некий нуклеиновокислотный вкладыш, защищенный белковым покрытием. Вирус может содержать либо РНК, либо ДНК, но никогда не содержит обе кислоты вместе. Тип нуклеиновых кислот является основой для одной из классификаций вирусов. Вире сы являются облигативными внутриклеточными паразитами, которые после внедрения в клетку, т. е. после ее инфицирования, используют многие биохимические системы клетки хозяина. Внедрение в живую клетку и участие в ее сложных интимных механизмах, как будто он является частью ее хромосомного аппарата при относительной простоте собственного строения, с одной стороны, создают огромные трудности для нахождения избирательно токсичных по отношению к вирусам препаратов, с другой стороны — делают вирус мощным орудием для исследований. В частности, в процессе размножения вирусы в основном используют аппарат биосинтеза клеток организма хозяина, модифицируя его определенным образом. Поэтому очень трудно подобрать соединения с клинически полезной противовирусной активностью, которые не повреждали бы нормальный клеточный метаболизм неинфицированных клеток хозяина и, тем самым, не оказывали бы на него токсического действия. Тем не менее имеются определенные противовирусные препараты, обладающие определенной избирательностью действия в отношении зараженных вирусом клеток и подавляющих репликативный цикл вирусов.
Несмотря на то, что точный механизм инфицирования весьма специфичен для каждого типа вирусов, общую схему инфицирования можно представить следующим образом. Вирус абсорбируется на поверхности клетки хозяина, по всей вероятности путем электростатических взаимодействий. Не исключено, что многие вирусы присоединяются к определенным вирус-специфичным рецепторам. Затем вирус проникает в мембрану клетки хозяина и там высвобождает из белковой оболочки нуклеиновую кислоту, утрачивая при этом свою индивидуальность как вирус. Далее вирусная нуклеиновая кислота начинает вести себя так, как будто она представляет собой функциональную часть клетки хозяина. Начинаются репликация и транскрипция вирусного генома либо в цитоплазме, либо в ядре клетки хозяина. В результате описанных событий производится значительное количество вирусной нуклеиновой кислоты и белка для генерации нового поколения вирионов. При этом репликационный механизм клетки хозяина полностью выключается, и, раз начавшись, описанный цикл многократно повторяется.
- 755-
Глава 36
Химиотерапия вирусных инфекций достигается 3 различными подходами, включающими:
1) профилактическую иммунизацию;
2) использование эндогенных противовирусных веществ, например интерферона, который, как полагают, связываясь со специфическими рецепторами на поверхности клеточной мембраны, инициирует внутри клетки синтез РНК и белков, проявляющих противовирусный эффект;
3) применением антивирусных препаратов.
Первыми препаратами, предложенными в качестве противовирусных средств, явились йдоксуридин и цитарабин, а также, несколько позже, видарабин, названные антивирусными препаратами первого поколения. Они имеют ограниченное клиническое применение из-за низкого терапевтического индекса (отношения эффективности к токсичности)- Эти препараты воздействуют непосредственно на репликацию вирусов, однако ингибируют и определенные функции клеток хозяина. Позже были предложены амантадин, ацикловир, рибавирин и зидовудин. В настоящее время для лечения СПИДа предложен ряд новых препаратов — рибавирин, амплиген, дидезоксицитидин и фоскарнет.
Исследования в области синтеза противовирусных препаратов только недавно позволили сделать значительный шаг в области лечения заболеваний, вызванных вирусом простого герпеса, и совсем недавно для лечения СПИДа. Эти препараты действуют путем ингибирования процесса размножения вирусных клеток.
В настоящее время в качестве противовирусных препаратов используются амантадин, видарабин, трифлуридин, йдоксуридин, ацикловир, рибавирин и зидовудин. Анализ механизмов действия существующих и применяемых противовирусных препаратов позволяет заключить, что они могут повышать резистентность клетки к вирусу (интерфероны); угнетать адсорбцию вируса на клетке, проникновение его в клетку, процесс его «депротеинизации» в клетке (амантадин); они также являются атиметаболитами, ингибирующими синтез нуклеиновых кислот. Клиническая «полезность» этих пиримидиновых и пуриновых препаратов напрямую зависит от их способности селективно блокировать синтез вирусных нуклеиновых кислот, не затрагивая при этом синтез нуклеиновых кислот клеток хозяина.
Амантадин (Amantadin)
Амантадин — 1-адамантанамин (36.1.3) получают исходя из адамантана, бромированием которого молекулярным бромом вначале по-
- 756 -
Противовирусные препараты
думают 1-бромадамантан (36.1.1). Вводя последний в реакцию Риттера, получают 1-ацетиламиноадамантан (36.1.2). Гидролизом последнего гидроксидом натрия получают амантадин [1, 2, 3,4, 5, 6].
Амантадин является первичным амином — производным адамантана. Влияет на миксовирусы, которые относятся к РНК-co держащим вирусам. Препарат имеет очень узкий спектр действия и показан только при лечении и профилактике гриппа А. Используется также при лечении паркинсонизма. Точный механизм антивирусного действия амантадина не полностью понятен. Полагают, что он является блокатором ионных каналов. Предполагается также, что амантадин ингибирует абсорбцию вирусных частичек в клетку хозяина, что выражается в сбоях при проникновении вируса в клетку или ингибировании «процесса раздевания» вируса. Основное применение — профилактика гриппа типа А2.
Синонимами препарата являются симметрел, вирегит, мантадан И др.
Ацикловир (Acyclovir)
Ацикловир — 2-амино-1,9-дигидро-9-[(2-гидроксиэтокси)метил]-6Я-пурин-6-он (36.1.5). Согласно первому способу, его получают алкилированием гуанина 1-бензоилокси-2-хлорметоксиэтаном в три-этиламине. Гидроксильная и аминогруппы гуанина при этом предварительно защищаются триметилсилильной защитой обработкой гексаметилдисилазаном. После гидролиза полученного при этом продукта водой выделяется 9-(2-бензоилоксиметоксиметил)гуанин (36.1.4). Обработкой последнего метанольным раствором аммиака удаляют защитную бензоильную группу с гидроксиэтоксиметиль-ного фрагмента с получением ацикловира.
I СНз)з&1^Н
ЫНз ' ОЬзСН
-757-
Глава 36
он
сн2—о-сн2-сн2-он
36 1 5
Второй способ получения ацикловира исходит из 2,6-дихлор-пурина, алкилированием которого тем же 1-бензоилокси-2-хлормето-ксиэтаном, но уже в системе триэтиламин — диметилформамид получают 2,6-дихлор-9-(2-бензоилоксиэтоксиметил)пурин (36.1.6). Обработкой последнего метанольным раствором аммиака замещают оба атома хлора на аминогруппы, а последующим диазотированием с использованием нитрита натрия в разбавленной уксусной кислоте, селективно замещают одну из двух аминогрупп, а именно: аминогруппу в положении С6 пуриновой системы на гидроксильную группу. Наконец, обработкой продукта метанольным раствором аммиака снимают и бензоильную защиту с синтезированного 9-(2-бензоилоксиэтоксиметил)гуанина (36.1.4) с получением ацикловира [7, 8, 9, 10,11].
с о-о- сн2-сн2-о- снг-CI
1 NH3
2 NaNOj/ СНгСООН
3 Nh3 CH3OH
СНг—O-CH2-CH?~0—СО
36 1 8
сн2—О-СН2-СН2-ОН
36 1 5
Как видно из химической формулы, ацикловир является нуклеозидным аналогом гуанозина, замещенным в боковой цепи не традиционным циклическим сахарным остатком, а 2-гидроксиэтоксиметильной ациклической боковой цепью.
Ацикловир обладает противовирусной активностью в отношении вируса простого герпеса типа 1 и 2, вируса опоясывающего лишая, вируса Эпштейна—Барр и цитомегаловируса.
Механизм противовирусной активности ацикловира заключается в его трансформации в трифосфат и последующем ингибировании син
-758-
Противовирусные препараты
теза ДНК-вируса. Его действие высокоселективно. Ацикловир проникает в инфицированные вирусом клетки и фосфорилируется до монофосфата тимидин киназой, индуцированной вирусом герпеса. Неинфи-цированные клетки не используют ацикловир в качестве субстрата. Далее монофосфат последовательно трансформируется в дифосфат и затем в трифосфат, который и является ингибитором вирусной ДНК полимеразы, а также включается в вирусную ДНК, где действует в процессе обрыва цепей, предотвращая таким образом дальнейшее удлинение цепи молекулы ДНК и, соответственно, репликацию ДНК-вируса. Ацикловир применяется при простом герпесе, при поражении глаз, гениталий и герпетических поражениях другой локализации, при опоясывающем лишае, ветряной оспе.
Синонимами препарата являются зовиракс, цикловиран, сифивирал и др.
Видарабин (Vidarabin)
Видарабин — 9-В-арабинофуранозил-6-амино-9-И-пурин (36.1.10) получают как микробиологическим путем из культуральной жидкости жизнедеятельности актиномицетов Streptomyces antibioticus NRRL 3238, так и синтетическим путем. Согласно первому способу, препарат было предложено получать из ацетонида-р-В-кси-длофуранозид аденина — 9-(3',5'-О-изопропилиден-Р-В-ксидло-фуранозид)аденина, взаимодействием которого с метансульфохлоридом был получен мезилат 9-(3|,5|-0-изопропилиден-2 -О-метан-сульфонил-Р-Н-ксидлофуранозид) аденина (36.1.7). Длительным кипячением последнего в 90%-й уксусной кислоте с полученного соединения снимается ацетонильная защитная группа с получением продукта (36.1.8). Взаимодействие последнего с метилатом натрия приводит к получению эпоксида — 9-(2',3'-ангидро-Р-люксо-фуранозил)аденина (36.1.9). Наконец кипячением полученного эпоксида с ацетатом или бензоатом натрия в системе диметилфор-мамид — вода расщепляют эпоксидный цикл с получением соответствующего дигидроксипроизводного видарабина [12, 13].
Cr13ONe
- 759 -
Г лава 36
CH3COONa / DMF / Н20
36 1 10
Второй способ синтеза видарабина, разработанный позже, заключается в алкилировании 6-бензамидопурина 2,3,5-три-О-бензил-В-ара-бинофуранозил хлоридом с использованием натрия в жидком аммиаке. При этом одновременно происходит и дебензоилирование по положению 6 пуриновой системы и де-О-бензилирование гидроксильных групп фуранозильного фрагмента молекулы с получением видарабина [14].
nhcoc6h5
ССН2С6Н5
NH3 ' Na
ОН
36 1 10
Видарабин (арабинозид аденина) является стереоизомером аденозина. Этот аналог пуринового нуклеозида проявляет селективную активность против вируса герпеса. В его структуре остаток рибозы замещен арабинозой. Подобно ацикловиру он превращается в моно-, ди-и трифосфат в инфицированных вирусом клетках, ингибируя таким образом ДНК полимеразу и, соответственно, препятствуя синтезу ДНК-вируса примерно в 20-40 раз больше, чем у клеток хозяина. С легкостью метаболизируется в менее активное, но опять-таки противовирусное соединение — арабинозилгипоксантин. С успехом применяется при герпетическом энцефалите, при осложненном опоясывающем лишае. Применяется в виде глазных лекарственных форм при герпетическом кератоконъюнктивите.
Синонимом препарата является Вира-А.
Идоксуридин (Idoxuridin)
Идоксуридин — 5-йод-1-(2-дезоксирпбофуранозил)пиримидин-2,4-(1 Я.3/7)-дион (36.1.14) получают по следующей схеме. 5-йодурацил
- 760-
Противовирусные препараты
ацилируют уксусным ангидридом с получением 1-анетил-5-йодурацила (36.1.11 ). Обработкой последнего ацетатом двухвалентной ртути получают 5-йодомономеркурийурацил (36.1.12), взаимодействием которого с 1-бромдидезокси-П-рибофуранозил-3,5-бис(и-толуолсульфонатом) получают дитозильное производное (36.1.13). Гидролизом тозильных защитных групп с помощью гидроксида натрия и дальнейшей обработкой полученного вещества уксусной кислотой с целью выделения целевого продукта получают искомый идоксуридин [15, 16, 17, 18, 19].
Нд-ОСОСНз12
36 1 13
1. NaOH
2 СН3СООН
36 1 14
Идоксуридин является аналогом тимидина— йодированным производным дезоксиуридина. Идоксиуридин действует на ДНК-вирусы и не действует на РНК-вирусы. Основное действие идоксуридина заключается в ингибировании репликации ДНК-вируса путем внедрения в сам вирус. При системном введении этот нуклеозид фосфорилируется как вирусной, так и клеточной тимидин киназой в активное трифосфорили-рованное соединение, которое ингибирует синтез как вирусной, так и клеточной ДНК. Поскольку препарат затрагивает клетки млекопитающих и, кроме того, обладает тератогенным, мутагенным и иммуносупрессивным действием, его назначение ограничивается только в качестве препарата для наружного применения. Используется в основном в офтальмологии при герпетической инфекции глаз (кератитах).
Синонимами препарата являются герплекс, стоксил, идувиран и др.
- 761 -
Г лава 36
Трифлуридин (Trifluridin)
Трифлуридин — 5-трифторметил-1-(2-дезоксирибофуранозил)-пиримидин-2,4-(1Н.ЗН)-дион (36.1.22) получают исходя из 5-три-фторметилурацила. Последний получают по следующей схеме. В начале из трифторацетона получают оксинитрил (36.1.15), ацетилированием которого получают соответствующий трифторацетат (36.1.16). Пиролиз последнего приводит к трифторметилакрилонит-рилу (36.1.17). Присоединением к последнему сухого бромистого водорода в метаноле, в процессе которого происходит и метанолиз нитрильной группы, получают бромид (36.1.18). Взаимодействием бромида с мочевиной получают ключевое соединение (36.1.19), при кислотном гидролизе которого происходит гетероциклизация в дигидропиримидин (36.1.20). Бромированием полученного дигидропиримидина молекулярным бромом и последующим дегидробромированием полученного продукта (36.1.21) нагреванием в диме-тилформамиде получают 5-трифтор-метилурацил (36.1.22). Последний вводят во взаимодействие с 2-дезокси-В-рибоз-1-фосфатом с использованием фермента нуклеозид фосфорилазы, либо обработкой гекаметилдисилазаном и далее трихлорметилсиланом получают 2,4-три.метилсилилокси-5-трифторметил пиримидин (36.1.23). Применение гексаметилдислазана, который сам и не образует триметилсилиловых эфиров, обусловлено тем, что использование сочетания этих двух реагентов приводит к получению оптимальных выходов тримтилсилиловых эфиров. Взаимодействием полученного пиримидинового производного с 3,5-бис(4-нитробензоатом)-2-дезо-ксирибофуранозил хлоридом в присутствии ацетата двухвалентной ртути получают соответствующий дитриметилсилилоксинуклеозид, обработкой которого водным раствором йодистого калия снимают защитные группы. Полученный продукт подвергается предварительной очистке путем хроматографирования, и далее обработкой метанольным раствором диизопропиламина, с фуранозильной части снимается 4-нитро-бензоильная защита с получением искомого трифлуридина [20, 21, 22, 23].
- 762 -
Противовирусные препараты
36 1 20
Трифлуридин также является галогенированным производным тимидина и применяется при кератоконъюнктивитах. По действию и области применения аналогичен идоксурдину. Так же как и идоксуридин, трифлуридин превращается в грифосфат, ингибируя ДНК полимеразу.
Синонимом препарата является вироптик.
Зидовудин (Zidovudin)
Зидовудин — 3*-азидо-3'-дезокситимидин (36.1.26) получают исходя из 1-(2'-дезокси-5'-0-тритил-Р-В-ликсозил)тимина, обработкой которого метане}льфо.хлоридом в пиридине получают соответствующий мезилат (36.1.24). Замещением мезильной группы на азидную с использованием азида лития в диметилформамиде получают продукт (36.1.25) с обращенной конфигурацией при С3 фуранозиль-ного кольца. Кипячением последнего в 80%-й уксусной кислоте с него удаляется тритильная защита с получением зидовудина [24, 25,26. 27,28].
CH3OSO2CI
UN3
СН3СООН
36 1 24
- 763-
Глава 36
Зидовудин — антиретровирусный препарат, клинически активный против ВИЧ-1 и предназначен для лечения ВИЧ-инфицированных пациентов
Зидовудин — аналог тимидина, который ингибирует репликацию вирусов СПИДа. Он также превращается в моно-, ди- и трифосфаты теми же клеточными ферментами, которые катализуют фосфорилирование тимидина и тимидиновых нуклеозидов. Далее зидовудин-трифосфат включается вирусной реверс-транскриптазой в концевой фрагмент растущей цепи вирусной ДНК и тем самым вызывает обрыв цепи вирусной ДНК в инфицированных вирусом клетках. Зидовудин был разрешен для лечения больных СПИДом. Он значительно продлевает жизнь больных, однако обладает рядом токсических эффектов.
Синонимами препарата являются азидотимидин и ретровир.
Рибавирин (Ribavirin)
Рибавирин — 1-Р-Н-рибофуранозил-1Я-1,2,4-триазол-3-карбокс-амид (36.1.28) получают вводя во взаимодействие метиловый эфир 1,2.4-триазол-З-карбоновой кислоты с О-1,2,3,5-тетраацетил-|3-В-рибофуранозой с получением метилового эфира 1-0-1,2.3,5-тет-раацетил-Р-В-рибофуранозил-1,2,4-триазол-3-карбоновой кислоты (36.1.27), обработкой которого аммиачным раствором метанола осуществляют одновременно дезацетилирование углеводной части и амидирование карбалкоксильной части продукта с получением рибавирина [29, 30, 31, 32, 33, 34, 35. 36, 37].
SH3CO-Q
О-СОСНз
чн3 СН3ОН
Рибавирин является синтетическим аналогом нуклеозидов. Эффективен против многих ДНК- и РНК-вирусов: вирусов гриппа, герпеса. Механизм его действия не совсем ясен. Однако вполне вероятно, что он не является одим и тем же для всех вирусов. Препарат испытывался и на ряде больных СПИДом с получением различных результатов.
Синонимом препарата является виразол.
-764-
Противовирусные препараты
Список литературы
1. US Pat. 3.310.469 (1967).
2. US Pat. 3.152.180 (1964).
3. Brit. Pat. 925.728 (1963).
4. Belg. Pat. 646.581 (1964).
5. Stetter H. et al./Der. 93, 226 (1960).
6. Gerzon K. et al./'.'J. Med. Chem. 6. 760 (1963).
7. Belg. Pat. 833.006(1975).
8. Ger. Pat. 2.539.963 (1976).
9. US Pat. 4.199to4574 (1980).
10. Matsumoto H. et al.'/Chem. Pharm. Bull. 36, 1153 (1988).
11. Schaeffer H. et al./TNature, 272, 583 (1978).
12 Reist E. et al.//J. Org. Chem. 27, 3274 (1962).
13. Lee W. et al.''J. Am. Chem. Soc. 82, 2648 (1960).
14. Reist E. et al./'J. Org. Chem. 29, 3725 (1964).
15. Fr. Pat. 1.336.866(1962).
16. Brit. Pat. 1.024.156(1963).
17. Prusoff W.t Biochim. Biophys. Acta. 32, 295 (1959).
18. Cheong K. et al./.'J. Biol. Chem. 235, 1441 (1960).
19. Chang F et al. 7J. Med. Chem. 6, 428 (1963).
20. US Pat. 3.531.464(1970).
21. US Pat. 3.201.387 (1965).
22. Heidelberger C. et al.'/J. Am. Chem. Soc. 84, 3597 (1962).
23. Heidelberger C. et al. 7J. Med. Chem. 7, 1 (1964).
24. Lin T.-Sh.H. Med. Chem. 21, 109 (1978).
25. Horowitz J. etal.//J. Org. Chem. 27, 3045 (1962).
26. Horowitz J. et al.//'J. Org. Chem. 29, 2076 (1964).
27. Glinski R. et al..-.'J. Org. Chem. 38, 4299 (1973).
28. Chu C. et al./'Tetrahedron. 29, 5349 (1988).
29. Ger. Pat. 2.220.246(1972).
30. Ger. Pat. 2.441.823 (1974).
31. Ger. Pat. 2.511.828 (1975).
32. US Pat. 3.976.545 (1976).
33. US Pat. 4.138.547 (1979).
34. Witkowski Med. Chem. 15. 1150 (1972).
35. Witkowski J о J. Carbohyd. Nucl. 5, 363 (1978).
36. Ito Y et al./ Tetrahedron Letters. 1979, 2521.
37. Schmidt R et aU'Berr. 114.2825 (1981).
-765-
: Глава 3 7
1 Препараты для лечения ; протозойных инфекций
Наиболее распространенными протозойными инфекциями, вызываемыми патогенными простейшими, являются малярия, лейшманиоз и трипаносомоз, а также трихомониаз, амебиаз, гиардиоз и токсоплазмоз. Все виды протозы являются одноклеточными организмами, приспосабливаемыми к различным условиям намного более гибко, чем бактерии. Они имеют довольно сложный жизненный цикл и поэтому существуют во многих формах, которые требуют разных подходов для лечения больных, зараженных протозойными инфекциями. Протоза — типичный паразит, который оккупирует клетку хозяина, размножается в ней и разрушает ее.
Предупреждение лечение протозойных заболеваний заключается в контроле над распространением заболеваний, улучшении санитарно-гигиенических условий жизни, проведении вакцинаций и, собственно, лечении. Следует иметь в виду, что малярия распространяется комарами. в частности посредством укуса самцами Anofeles mosquito; лейшманиоз распространяется зараженными песчинками; трипаносомоз распространяется мухой цеце; амебиаз и гиардиоз распространяются через пищу и воду, а токсоплазмоз — через мясные продукты и зараженных кошек.
Для борьбы с протозойными паразитами используется очень много химиотерапевтических препаратов различного типа.
37Л. Антималяр ийные препараты
Малярия является наиболее распространенным из заболеваний, вызываемых протезами. Возбудителями малярии являются плазмодии
- 766-
Препараты для лечения протозойных инфекций
(Plasmodium falciparum, Plasmodium vivax, Plasmodium malarias и Plasmodium ovale). Малярийный плазмодий имеет 2 цикла развития — бесполый, который происходит в органиме зараженного человека (шизогония), и половой, происходящий в теле самого комара (спорогония). При укусе комаром в организм человека попадают спорозиты, образующиеся в крови комара из мужских и женских гематоцитов. Последние внедряются в клетки печени, где образуют первичные тканевые шизонты, которые растут и делятся, трансформируясь в мерозоиты. Мерозоиты далее выделяются в кровь человека и проникают в эритроциты, где происходит их дальнейшее развитие. При созревании в эритроцитах шизонты вновь делятся и трансформируются в мерозоиты. Последние периодически высвобождаются из оккупированных эритроцит-ных клеток и поражают новую группу эритроцитов, начиная описанный процесс вновь. Этот процесс длится 3-4 дня, и момент разрушения эритроцитов и выхода мерозоитов в кровь проявляется в виде приступа малярийной лихорадки и называется периэритроцитарной формой малярии. Однако при заражении плазмодиями Plasmodium vivax. Plasmodium malariae и Plasmodium ovale возможен и другой ход развития событий, называемый экзоэритроцитарной формой малярии, когда паразиты, находящиеся в мерозоитной стадии развития, остаются или вновь внедряются в клетки печени. Это возобновляет эритроцитарный цикл развития плазмодия и наступление рецидивов. При тропической малярии параэритроцитарные формы отсутствуют.
Химиотерапия малярии заключается в воздействии на различные стадии жизненного цикла паразита. Соответственно, антималярийные препараты подразделяются на 3 группы:
1) воздействующие на эритроцитарную стадию жизненного цикла,
2) разрушающие экзоэритроцитарную (или гепатическую) стадию,
3) воздействующие на обе стадии одновременно.
Старейшими препаратами, применяемыми против малярии, являются хинин и хинидин. В настоящее время для лечения малярии применяют аминохинолины, такие как хлороквин и его аналоги, преимущественно для воздействия на паразита в ходе эритроцитарной стадии, и примахин — для воздействия на паразита в экзоэритроцитарной стадии. В последнее время начали применяться мефлохин и природное соединение хингошу. а также различные антибиотики в сочетании с антима-лярийными препаратами
- 767-
Глава 37
3 7.1.1. Препараты, эффективные против эритроцитарной стадии заражения плазмодиями
4-аминохинолины и хинолинметанолы
В эритроцитарной стадии заражения плазмодиями применяются 2 класса препаратов: 1) 4-аминохинолины и 2) хинолинметанолы. 4-аминохинолины (хлороквин, амодиаквин и гидроксихлороквин) являются синтетическими соединениями. Их наиболее важной структурной характеристикой является тип заместителя при С7 и С.( хинолинового кольца. Показана необходимость аминного заместителя при С4 хинолинового кольца, которая может варьировать с сохранением антималярий-ной активности этого типа соединений в целом, однако необходимым условием для проявления антималярийной активности является наличие атома хлора при С7 хинолинового кольца. Прототипом этой группы соединений является хлороквин.
Другая группа препаратов, используемых во время эритроцитарной стадии малярийной инфекции, представлена хинолинметанолами. Эта группа включает мефлохин и хингошу, а также хинные алкалоиды, получаемые из коры хинного дерева, из которых только хинин все еще продолжает использоваться при лечении малярии.
Хлороквин (Chloroquin)
Хлороквин — 7-хлор-4-(4-диэтиламино-1-метилбутиламиио)хино-лин (37.1.3) получают взаимодействием 4,7-дихлорхинолина (37.1.1.1) с 4-диэтиламино-1-метилбутиламином (37.1.1.2) при 180 °C [1,2, 3].
СНз С2Н5
Н2Ч-СН-(СН2)з-Ч\
С2Н5
С2Н5
(CH2)3-n(
С2Н5
37 1 1 3
37 1 I 2
Для осуществления синтеза необходимого для этой цели 4.7-ди-хлорхинолина (37.1.1.1) предложено несколько способов, исходящих из 3-хлоранилина. Первый способ заключается во взаимодействии 3-хлор-анилина с этоксиметиленмалоновым эфиром с получением (3-хлор-анилино)метиленмалонового эфира (37.1.1.4), высокотемпературная гетероциклизапия которого приводит к получению этилового эфира
- 768 -
Препараты для лечения протозойных инфекций
7-хлор-4-гидроксихинолин-3-карбоновой кислоты (37.1.1.5). Гидролизом последнего гидроокисью натрия получают 7-хлор-4-гидроксихи-нолин-3-карбоновую кислот} (37.1.1.6), которая нагреванием при 250 -270 °C декарбоксилируется с образованием 7-хлор-4-гидроксихинолина (37.1.1.7). Обработкой последнего хлорокисью фосфора получают один из искомых компонентов для синтеза хлороквина — 4,7-дихлорхинолин (37.1.1.1) [4, 5].
37 1 1 ’
37 1 1 1
Второй способ для получения 4,7-дихлорхинолина (37.1.1.1) заключается во взаимодействии 3-хлоранилина с диэтиловым эфиром ща-велевоуксусной кислоты в присутствии уксусной кислоты с получением соответствующего имина (37.1.1.8), который при нагревании до 250 °C гетероциклизуется в этиловый эфир 7-хлор-4-гидроксихинолин-2-кар-боновой кислоты (37.1.1.9) с примесью 5-хлор-4-гидроксичинолин-2-карбоновой кислоты (37.1.1.10). которая отделяется от основного продукта путем кристаллизации из уксусной кислоты. Щелочным гидролизом этилового эфира 7-хлор-4-гидроксихинолин-2-карбоновой кислоты (37.1.1.9) и дальнейшим высокотемпературным декарбоксилированием полученной при этом кислоты (37.1.1.11) получают 7-хлор-4-гидроксихинолин (37.1.1.7). Взаимодействием последнего с хлорокисью фосфора по вышеописанной схеме получают 4,7-дихлор-хинолин (37.1.1.1) [6].
СН
-769-
Г лава 37
Наконец, третий способ из предложенных вариантов получения 4,7-дихлорхинолина (37.1.1.1) заключается во взаимодействии 3-хлор-анилина с этиловым эфиром формилуксусной кислоты с получением имина (37.1.1.12), который при нагревании непосредственно циклизуется в 7-хлор-4-гидроксихинолин (37.1.1.7), взаимодействием которого с хлорокисью фосфора по уже неоднократно описанной схеме получают 4.7-дихлорхинолин (37.1.1.1) [7].
Второй компонент, необходимый для синтеза хлороквина, — 4-ди-этиламино-1-метилбутиламин (37.1.1.2) также предложено получать различными путями. Согласно первому способу, алкилированием ацетоуксусного эфира 2-диэтиламиноэтилхлоридом получают 2-диэтил-а.миноэтилацетоуксусный эфир (37.1.1.13), кислотным гидролизом которого соляной кислотой с одновременным декарбоксилированием получают 1-диэтиламино-4-пентанон (37.1.1.14). Восстановительным аминированием последнего водородом и аммиаком с использованием в качестве катализатора никеля Ренея получают 4-диэтиламино-1-метилбутиламин (37.1.1.2) [8].
С2н5
CI—СН2-СН2-М\
С2Н5
сн3
С2Н5 сн~сн2-сн2-м( с2н5 ОС2Но
371 1 12
О С2Н5
CH3-C-CH2-CH2-CH2-N\
С2Н5
Н2 NH3/[Raney—Ni)
СН3 С2Н5
Н2М-СН-(CH2;3-n{
С2Н5
37 1 1 2
Вторым способом, предложенным для получения 4-диэтиламино-1-метилбут илам ина (37.1,1.2), является путь, исходящий из 3-ацетил-бугиролактона (37.1.1.15), который, в свою очередь, получают взаимодействием ацетоуксусного эфира с этиленоксидом. Кислотным гидролизом сложноэфирной группы в 3-ацетилбутиролактоне (37.1.1.15) с одновременным декарбоксилированием получают 1-бром-
- 770 -
Препараты для лечения протозойных инфекций
4-пептанон (37.1.1.16). Взаимодействием последнего с диэтиламином получают 1-диэтиламино-4-пентанон (37.1.1.14), восстановительным аминированием которого водородом и аммиаком с использованием в качестве катализатора никеля Ренея получают 4-диэтиламино-1-метилбутиламин (37.1.1.2) [9].
С С2н5
СНз-С-сНг-СНз-СНг-м^
С2и5
37 1 1 14
Нг, МНз [Rsney—Ni]
сн3 С2Н5
h2n-ch—(CH2hj-M\
C2H5
37112
Хлороквин является лекарством выбора для профилактики и лечения острых форм малярии, вызванной Plasmodium vivax, Plasmodium malariae, Plasmodium ovale, а также чувствительных форм Plasmodium falciparum Механизм его действия не совсем ясен, однако имеется несколько гипотез, объясняющих его антималярийную активность. Хлороквин и его аналоги ингибируют синтез нуклеиновых кислот паразита, воздействуя на матричную функцию ДНК. Это происходит путем преимущественного связывания препарата водородными связями с пуриновыми фрагментами и последующим внедрением молекулы хлороквина между упорядочненно уложенными парами оснований в спирали ДНК паразита. Таким образом хлороквин предотвращает транскрипцию и трансляцию, значительно ограничивая синтез ДНК и РНК паразитом. Селективная токсичность хлороквина по отношению именно к малярийным плазмодиям приписывается также способности паразит ирован-ных красных кровяных клеток концентрировать этот препарат в количествах, примерно в 25 раз превосходящих таковую в нормальных эритроцитах.
Существует и другая гипотеза. Хлороквин имеет большое сродство к тканям паразита и концентрируется в его цитоплазме. Как слабое основание он повышает pH внутриклеточных лизосом и эндосом, а для заражения паразитами клеток млекопитающих требуется значительно более кислая среда этих органелл. Как следствие, при применении хлороквина ингибируются рост и развитие паразитов.
Итак, главным качеством хлороквина, которым он превосходит все другие противомалярийные препараты, является влияние на эригроци-
-771 -
Глава 37
тарные шизонты (гематошизотропное действие). Однако хлороквин обладает и амебицидным действием. У него обнаружены также иммунодепрессивные и противоаритмические свойства.
Препарат применяют при всех видах малярии, для химиопрофилактики, а также при внекишечном амебиазе и амебном абсцессе печени.
Синонимами препарата являются нивахин, хингамин, делагил, ре-зохин, атрохин и др.
Гидроксихлороквин (Hydroxychloroquin)
Г идроксихлороквин — 7-хлор-4-[4-[этил(2-гидроксиэтил)амино]-1 -метилбутиламинохинолин (37.1.1.19) получают по схеме, близкой к получению хлороквина. Взаимодействием 1-.хлор-4-пентанона с 2-этиламиноэтанолом получают соответствующий аминокетон (37.1.1.17), который подвергают восстановительному аминированию в условиях, аналогичных описанным выше, с получением 4-[этил(2-гидроксиэтил)амино]-1-метилбутиламина (37.1.1.18). Вводя последний во взаимодействие с 4,7-дихлорхинолином (37.1.1.1), получают искомый гидроксихлорокв ин [10, 11 ].
О С2Н5 ^2 |Лапеу-Ь>)
СН3—С—CHj—CHj—C.Hj Сн2—CH2~Oh
37 1 ' '1
С2Н5
С Н 2—О Н 2C“i
Гидроксихлороквин, как и хлороквин, также применяется для лечения острых форм малярии, вызванной Plasmodium vivax, Plasmodium malariae, Plasmodium ovale, а также чувствительных форм Plasmodium falciparum. Он так же эффективен и так же безопасен, как и хлороквин, однако не имеет явных преимуществ перд ним. Единственным преимуществом является несколько лучшая переносимость. Его применение несколько более ограничено, чем применение хлороквина.
Синонимами препарата являются плаквенил, квенсил, торемонил и др.
Амодиаквин (Amodiaquin)
Амодиаквин — 4-[(7-хлор-4-хинолил)амино]-а-диэтиламино-о-крезол (37.1.1.21) получают взаимодействием 4,7-дихлор.хинолина
- 772 -
Препараты для лечения протозойных инфекций
(37.1.1.1) с 4-аминофенолом с получением 7-хлор-4-(4-гидрокси-фениламино)хинолина (37.1.1.20), который далее вводят в реакцию аминометилирования с использованием формальдегида и диэтиламина с получением амодиаквина [12, 13, 14, 15].
Амодиаквин также является структурным аналогом хлороквина и гид-роксихлороквина и существенных различий в активности не выявляет. Поэтом)' его применяют реже, чем описанные выше препараты.
Синонимами препарата являются флавохин и камохин.
Хинин (Quinine)
Хинин — (5-винил-2-хинуклидинил)-(6-метокси-4-хинолил)метанол
(37.1.1.47) выделяют из коры хинного дерева [16].
37 1 1 47
Начиная с XVII в. хинная кора применялась в Европе как противолихорадочное средство, а затем как средство для лечения малярии. Еще в 1820 г. из коры хинного дерева были выделены два алкалоида — хинин и цинхонин, представляющие собой неконденсированные бигетероциклы, сочетающие в себе два гетероциклических ядра — хинолина и хинуклидина. Причем хинин является метоксильным производным цинхонина и левовращающим изомером хинидина. Его каркас состоит из хинолинового кольца, в положении 4 которого посредством метиленового мостика присоединено хинуклидиновое кольцо. Метоксигруппа при С6 хинолинового кольца и винильная группа в хинуклидиновом кольце повышают активность соединения, однако не являются абсо
- 773 -
Глава 37
лютно необходимыми для проявления соединениями этого ряда противомалярийной активности. Хинин приобрел огромное значение как базовая структура для синтеза многочисленных соединений с противомалярийной активностью. Синтез препарата весьма сложен и был осуществлен разными путями, но в принципе сводится к сложноэфир-ной конденсации эфиров 6-метоксихинолиновой (хининовой) кислоты (37.1.1.27) и N-бензоилгомомерохинена (37.1.1.42), которые могут быть получены разными путями, и последующей последовательной обработке полученного продукта гипобромидом и этилатом натрия с целью создания хинуклидинового фрагмента.
Одним из методов получения этилового эфира хининовой кислоты (37.1.1.27) следует считать первый способ, осуществляемый по следующей схеме. Взаимодействим н-анизидина и ацетоуксусного эфира в присутствии серной кислоты получают 6-метоксилепидин (37.1.1.22), гидроксильную группу в котором замешают на атом хлора взаимодействием со смесью хлорокиси и пятихлористого фосфора с получением 2-хлор-4-метил-6-метоксихинолина (37.1.1.23). Восстановлением последнего водородом с использованием в качестве катализатора палладия убирают атом хлора при С2, и получают 4-метил-6-метоксихинолин (37 1 1 24). Конденсацией последнего с бензальдегидом получают 2-(6-метокси\инолинил-4)-стирол (37.1.1.25), окислением двойной связи в котором с помощью перманганата калия получают 6-метокси-хинолиновую кислоту — цинхонин (37.1.1.26). которую далее обычным способом переводят в эфир (37.1.1.27).
Вторым способом получения этилового эфира хининовой кислоты (37.1.1.27) является метод, исходящий из H-N-метилацетанизидина и ди-этилоксалата. в результате взаимодействия которых получают «-N-ме-тилацетанизид щавелевоуксусной кислоты (37.1.1.28). Гетероциклизация последнего в кислых условиях приводит к получению N-метил-З-кето-4-карбэтокси-6-метоксихинолина (37.1.1.29), взаимодействием
-774-
Препараты для лечения протозойных инфекций
которого со смесью хлорокиси и пятихлористого фосфора получают 2-хлор-4-карбэтокси-6-метоксихинолин (37.1.1.30). Восстанавливая последний водородом с использованием в качестве катализатора палладия, получают искомый этиловый эфир 6-метоксихинолиновой кислоты (37.1.1.27).
СООС21ч«
I
СООС jr- 5
37 1 1 30
37 1 1 27
Синтез N-бензоилгомо.мерохинена (37.1 1.42) осуществляют исходя из 7-оксиизохинолина (37.1.1.32). получающегося взаимодействием аминоацегаля с м-оксибензальдегидом с промежуточным выделением имина (37 1.1.31). который далее в присутствии серной кислоты циклизуется в исходный 7-оксиизохинолин (37.1.1.32). Полученный 7-оксиизохинолин аминометилируют смесью формальдегида и пиперидина с получением 7-оксм-8-пиперидинометилизохинолина (37.1.1.33). Восстанавливая последний водородом с использованием в качестве катализатора палладия, удаляют пиперидиновый фрагмент с получением 7-окси-8-метилизохинолина (37.1.1.34), который далее с целью гидрирования пиридинового фрагмента вновь восстанавливают водородом, на сей раз используя в качестве катализатора окись палладия, и получают 7-окси-8-метилтетрат идроизохинолин (37.1.1.35). N-ацилируя последний уксусным ангидридом и подвергая продукт дальнейшему гидрированию водородом с использованием в качестве катализатора платины, получают смесь стереоизомерных Х-ацетил-7-окси-8-метилдекагид-роизохино.шнов, из которых выделяется z/wc-изомер (37.1.1.36), и с помощью оксида шестивалентного хрома его окисляют до Ы-ацетил-7-кето-8-метилдекагидроизохинолина (37.1.1.37). Взаимодействием последнего с этилнитритом в присутствии этилата натрия расщепляют метилциклогексаноновый фрагмент с образованим N-ацетил-Ю-окси-минодигндрогомомерохннена (37.1.1.38), который далее водородом в присутствии платинового катализатора восстанавливается в амин
-775-
Глава 37
(37.1.1.39). Подвергая последний исчерпывающем} метилированию йодистым метилом, пол} чают четвертичную соль (37.1.1.40), расщеплением которой концентрированной щелочью при нагревании получают рацемический гомомерохинен (37.1.1.41). Последовательной этерификацией и дальнейшим бензоилированием продукта получают этиловый эфир N-бензоилгомомерохинена (37.1.1.42).
СгН5О
)сн~сн^\н^
Н2 Pd
Н2 &dO
’ (CHjCo^o
2 Нг Pt
37 1 1 42
Далее этиловый эфир N-бензоилгомомерохинена (37.1.1.42) в при-с}тствии этилата натрия конденсируют с этиловым эфиром 6-мето-ксихинолиновой кислоты (37.1.1.27) с получением производного хино-токсина (37.1.1 43). Кипячением с соляной кислотой осуществляют гидролиз карбэтоксильной и бензоильной групп и одновременно декарбоксилирование с получением соединения (37.1.1.44). Обработкой последнего гипобромитом натрия получают N-бромпроизводное (37.1.1 45),
- 776 -
Препараты для лечения протозойных инфекций
взаимодействием которою с этилатом натрия реализуют ключевой момент данного синтеза — трансформацию пиперидинового производного в хинукидиновое (37 1.1.46). Восстанавливая кетогруппу в последнем алюмогидридом лития, получают искомый хинин (37.1.1.47) [17, 18, 19, 20,21,22].
C^gONa
СьНбСО
СООС2Н5
сьн3со
HCI
37 4 1 43
37 1 1 46
Хинин по типу действия является противомалярийным препаратом, сходным с хлороквином, однако уступает ему по активности.
Аналогично хлороквину, хинин присоединяется к плазмодиевой ДНК, вмешиваясь в синтез нуклеиновых кислот и предотвращая их репликацию и транскрипцию. Хинин подавляет также огромное число ферментных систем и поэтому характеризуется как общий протоплаз-мический яд. Этот факт хорошо согласуется с действием хинина на мембраны, его местноанестезирующим и кардиодепрессивным действиями.
При оральном применении хинин эффективно действует в комбинации с пириметамином, сульфадиазином и'или тетрациклином при лечении неосложненных случаев хлороквинрезистентных форм Plasmodium falciparum. Ввиду многих побочных реакций, его применение крайне ограничено. Единственным показанием к применению в на
- 777-
Г лава 37
стоящее время являются формы малярии, резистентные к синтетическим препаратам.
Синонимами препарата являются бронхопульмин, никоприв, хин-нам и др
Меф. юхин (Mefloquin)
Мефлохин — В,Р-эритро-а-2-пиперидил-2,8-бис’-(трифторметил)-4-хинолинметанол (37.1.1.53) предложено получать разными путями, исходящими из 2-трифтор.метиланилина. Согласно первому способу, гетероциклизацией продукта взаимодействия 2-трифтор-метиланилина с трифторацетоуксусным эфиром получают 2,8-бис-(трифторметил)-4-гидроксихинолин (37.1.1.48). Реакцией продукта с трехбромистым фосфором гидроксильную группу' в положении 4 хинолинового кольца замещают на атом брома с получением 2,8-бис-(трифторметил)-4-бромхинолина (37.1.1.49). Далее взаимодействием последнего с бутиллитием получают литийорганическое производное — 2,8-бис-(трифторметил)-4-литийхинолин (37.1.1.50). Реакцией последнего с двуокисью углерода получают 2,8-бис-(трифторметил)-4-хинолинкарбоновую кислоту (37.1.1.51). Вводя полученную кислоту во взаимодействие с 2-литпйпиридином, получают кетон (37.1.1.52). Восстанавливая в последнем одновременно как кетогруппу, так и пиридиновое кольцо водородом с использованием в качестве катализатора платины, получают искомый мефлохин [23].
37 1 1 51
Н2/ Pt
37 1 1 53
Второй способ синтеза мефлохина исходит из описанного выше
2,8-ог?с-(трифторметил)-4-литийхинолина (37.1.1.50), который вводят во
- 778 -
Препараты для лечения протозойных инфекций
взаимодействие с 2-формилпиридином с получением а-2-пиридил-2,8-бг<с-(трифторметил)-4-метанолхинолина (37.1.1.54). Пиридильную группу в последнем также восстанавливают аналогично вышеописанному, и в результате получают искомый мефлохин [24].
37 1 1 50
Н2 ! РЮ
CF3
37 1 1 S3
Наконец, третий способ синтеза мефлохина также исходит из 2-трифторметиланилина, однако в данном случае его вводят во взаимодействие с хлоральгидратом и гидроксиламином с получением изонит-розоацетил(2-грифторметил)анилида (37.1.1.55), который при нагревании в присутствии серной кислоты циклизуется в 7-трифтормети-лизатин (37.1.1.56) (рекция Зандмейера). Далее взаимодействием полученного 7-трифторметилизатина (37.1.1.56) с 1,1,1-трифторацетоном в присутствии щелочи по реакции Фридландера получают описанную выше 2,8-бис-(трифторметил)-4-хинолин карбоновую кислоту (37.1.1.51). Взаимодействием с гидроокисью лития последнюю переводят в литиевую соль, которую вводят во взаимодействие с реактивом Гриньяра, полученным из 2-бромпиридина и магния — 2-магнийбромпиридином. Полученный при этом кетон (37.1.1.52) вновь восстанавливают водородом в присутствии платинового катализатора с получением искомого мефлохина [25].
О
СРз-С-ОНз ; МаОН
CF3
37 1 1 53
-779-
Г лава 37
Мефлохин является аналогом хинина и отличается от него тем, что боковая цепь при С, хинолинового кольца содержит вместо хинуклиди-нового фрагмента пиперидиновый, а положения С; и С8 замещены трифторметильными группами.
Этот антималярийный препарат был создан для лечения и профилактики хлороквинрезистентных форм малярии, вызванной Plasmodium falciparum. Клинически не активен против экзоэритроцитарных форм Plasmodium vivax, однако весьма активен против кровяных шизонтоци-дов Plasmodium vivax и Plasmodium falciparum и, соответственно, предназначен для лечения слабых и умеренных форм малярии, вызванной указанными плазмодиями.
Синонимом препарата является лариам.
Хингошу (Quinghaosu)
Хингошу — октагидро-3,6,9-триметил-3,12-эпокси-12//-пирано-(4,3-ди)-1,2-бензодиоксепин-10-(3/7)-он (37.1.1.57) выделяют из растения Artemisia атта [26]. Препарат предложено также синтезировать [27].
Хингошу является последним принципиальным открытием в этой области и является гетероциклическим соединением, не имеющим в своей структуре атомов азота. Препарат взят из китайской народной медицины. Его выделяют из растения Artemisia атта. Удивительно, что соединение, совершенно отличающееся по структуре от описываемых в настоящей главе, проявляет совершенно одинаковое терапевтическое действие. Основной интерес к хингошу обусловлен тем, что он активен в отношении резистентных форм малярии, вызванной Plasmodium falciparum, и даже его церебральных форм.
Синонимами препарата являются артемизин, артемизинин и др.
-780-
Препараты для лечения протозойных инфекций
37.1.2 . Препараты, эффективные против гепатической (экзоэритроцитарной) стадии заражения плазмодиями
8-аминохинолины
Примахин (Primaquin)
Примахин — 8-[(4-амино-1-метилбутирил)амино]-6-метоксихино-лин (37.1.2.4) получают из 6-метокси-8-нитрохинолина (37.1.2.1), который синтезируют по реакции Скраупа из 4-метокси-2-нитро-анилина и глицерина в присутствии серной кислоты. Нитрогруппу в последнем восстанавливают с получением 6-метокси-8-амино-хинолина (37.1.2.2). Алкилируя аминогруппу последнего 4-бром-1-фталимидопентаном, получают 8-[(4-фталимидо-1-метилбутирил)-аминофб-метоксихинолин (37.1.2.3), гидразинолизом которого с последнего снимается фталимидная защита с получением примахина [28, 29].
h2n-nh2
сн3
37 1 24
Перенос боковой цепи из четвертого положения хинолинового кольца в восьмое полностью меняет спектр активности соединения. В отличие от 4-замещенных аминохинолинов, примахин практически не действует на эритроцитарные формы малярийного паразита. Его активность ограничена тканевыми формами паразита у млекопитающих и в самих комарах. Это делает примахин особенно ценным препаратом, позволяющим радикальное излечение и одновременно профилактику. что обычно не достигалось при использовании эритроцитарных препаратов.
-781 -
Глава 37
Место действия примахина — митохондрии малярийного паразита. Кажется вероятным, что примахин вмешивается в процессы переноса электронов, вызывая повреждение митохондриальных ферментных систем. Это выражается в набухании и вакуолизации митохондрий паразита. Митохондрии хозяина при этом не затрагиваются.
Примахин является наиболее эффективным и наименее токсичным препаратом из всего ряда доступных 8-аминохинолинов. Используется в основном при лечении экзоэритроцитарных форм малярии, вызванной Plasmodium vivax и Plasmodium ovale. Он действует также на половые формы плазмодиев, которые при применении препарата гибнут в организме человека.
Примахин применяется при лечении и профилактике отдаленных рецидивов трехдневной, четырехдневной и тропической малярии.
Синонимом препарата является авлон и др.
37.1.3 . Препараты, эффективные против гепатической и эритроцитарной форм заражения плазмодиями
Бигу аниды и диаминопиримидины оказались активными соединениями как в отношении экзоэритроцитарных, так и эритроцитарных форм плазмодий. Хлоргу'анид (37.1.2.3) в качестве противомалярийного препарата был внедрен в медицинскую практику в результате работы над большой серией производных гуанидина. Предполагается, что в организме он трансформируется в активное дигидротриазиновое соединение.
Несколько позже в результате интенсивных исследований антиме-габолитов фолиевой кислоты был предложен пириметамин (33.1.60). Триметоприм (33.1.51) — результат более поздних исследований. Близкое структурное сходство этих препаратов с птеридиновым фрагментом фолиевой кислоты несомненно определяет их сродство к рецептивному участку дигидрофолатредуктазы.
Н7М
Оигидротриазиновый метаболит хлоргуанида
хлоргуанид
пириметамин триметоприм
- 782 -
Препараты для лечения протозойных инфекций
Все эти соединения являются ингибиторами дигидрофолатредук-тазы в бактериях, плазмодиях и человека. К счастью, они имеют значительно большее сродство к бактериальным и протозойным дигид-рофолатредуктазам. Пириметамин, к примеру, ингибирует паразитную дигидрофолатредуктазу при уровнях в несколько сотен раз ниже, чем это требуется для ингибирования дигидрофолатредуктазы человека. Это и является основой их селективной токсичности. Селективная токсичность может быть повышена при дополнительном снабжении организма хозяина фолиевой кислотой, которую паразит не в состоянии использовать. Собственно первоначально диаминопиримидины (триметоприм, пириметамин) были предложены в качестве лечебных и профилактических средств против малярийных инфекций. Показано, что все сильные ингибиторы дигидрофолатредуктазы могут вывести из строя малярийный паразит с относительно малыми последствиями для хозяина.
Как уже было отмечено, бнгуаниды и диаминопиримидины активны как в отношении экзоэритроцитарных, так и эритроцитарных форм плазмодий. Каждый из этих препаратов в отдельности может применяться для профилактики, однако наибольший эффект достигается при их комбинированном применении с сульфонамидами.
Было показано, что некоторые сульфоны и сульфонамиды могут представлять интерес в качестве средств для лечения малярии. Экспериментальное изучение выявило выраженный синергизм между сульфонамидами и хлоргуанидом и пириметамином.
Хлоргуанид (Chloroguanid)
Хлоргу'анид — Х|-(4-хлорфенил)-КР-изопропи.пбигуанид (37.1.3.2) получают исходя из 4-хлоранилина и дицианамида натрия, взаимодействием которых получают (4-хлорфенил)-дициандиамид (37.1.3.1). Вводя последний в реакцию с изопропиламином, получают искомый хлорнуанид [30, 31, 32].
37 1 32
В условиях in vitro хлоргуанид неактивен, однако в организме он трансформируется в активное дигидротриазиновое соединение.
Хлоргуанид активен как в отношении экзоэритроцитарных, так и эритроцитарных форм плазмодий. Препарат наиболее полезен для
-783-
Глава 37
подавляющей терапии. Применяется при профилактике малярии, которую следует начинать за 2 недели до входа в малярийную зону и продолжать прием в течение 8 недель.
Синонимами препарата являются бигуанид, бигумал, палудрин, прогуанид и др.
Пириметамин (Pyrimethamin)
Пириметамин — 2,4-диамино-5-(4'-хлорфенил)-6-этилпиримидин (33 1.60), синтез которого был описан в гл. 33 «Антимикробные препараты».
Этот мощный ингибитор дигидрофолатредуктазы используется для профилактики и лечения малярии, вызванной плазмодиями Plasmodium vivax, Plasmodium malanae, Plasmodium ovale, включая Plasmodium falciparum
Пириметамин, являясь антагоистом фолиевой кислоты, оказывает противомикробное действие в отношении возбудителей малярии и одновременно обладает споронтоцидным действием. Препарат эффективен также в отношении возбудителя токсоплазмоза. Применяется для профилактики малярии и лечения токсоплазмоза.
Препарат может применяться только в профилактических целях, однако вследствие быстрого развития к нему резистентности, а также с учетом того факта, что наибольший эффект достигается при его комбинированном применении с сульфадоксином, выпускается также комбинированный препарат под названием фансидар, содержащий пириметамин — сульфадоксин в соотношении 1:20.
Комбинация пириметамина, сульфонамида и хинина является препаратом выбора при острых атаках малярии при ее хлороквинрезист-нтных формах.
Пириметамин в комбинации с сульфадиазином или трисульфапиримидином является препаратом выбора при токсоплазмозе.
Синонимом этого комбинированного препарата является дараприм.
- 784 -
Препараты для лечения протозойных инфекций
37.1.4 . Прочие антималярийные препараты
Малярийный паразит чувствителен ко многим разным группам лекарств, вследствие чего в зависимости от конкретных случаев используются различные комбинации препаратов.
Хинакрин (Quinacrine)
Хинакрин — 6-хлор-9-(4-диэтиламино-1-метилбутиламино)-2-мето-ксиакридин (37.1.4.3) синтезируют исходя из 6,9-дихлор-2-метоксиакридина (37.1.4.2) и уже описанного ранее 4-диэтиламино-1-метилбутиламина (37.1.1.2). Необходимый для этого синтеза 6,9-ди-хлор-2-метоксиакридин (37 1.4.2) получают в две стадии. С этой целью первоначальным взаимодействием 2,4-дихлорбензойной кислоты и л-анизидина в присутствии медной пыли и поташа получают 2-(4-метоксианилино)-4-хлорбензойную кислоту (37.1.4.1), которую реакцией с хлорокисью фосфора переводят в требуемый 6,9-дихлор-2-метоксиакридин (37.1.4.2) [33, 34, 35. 36, 37, 38].
Си ' К2СО3
37 1 4 1
СНз С2Н5
H2N-CH-(Ch2)3-N(
37 1 1 2 °2Н5
С2Н5
-N<
С2Н5
37 1 43
Хинакрин — производное акридина, химически и клинически очень сходное с 4-аминохинолинами. Он являлся основным лекарством для профилактики и терапии малярии в ходе Второй мировой войны. Сейчас он редко используется для лечения малярии, однако применяется для лечения амебиаза.
Синонимами препарата являются мепакрин. атабрин, акрисуксин и др.
При лечении устойчивых форм малярии используется также тетрациклин в комбинации с пириметамином, сульфонамидами и сульфонами и дапсон, который широко используется при лечении лепроза, как правило в сочетании с пириметамином.
- 785-
Г лава 37
37.2. Препараты для лечения амебиаза
Амебиазом считается инфицирование организма протезами Entamoeba histolytica, которые чаще всего поражают толстый кишечник, однако могут поражать и легкие, печень, мозг и другие органы. При поражении ЖКТ болезнь может протекать без клинических симптомов, с умеренно выраженными клиническими симптомами (диарея, судороги, метеоризм), а также с симптомами острой амебной дизентерии, сопровождаемой кровавой диареей, рвотой, лихорадкой и дегидратацией организма. Присутствие протозойных микроорганизмов Entamoeba histolytica в других органах может вызвать некроз печени, амебный гепатит, абсцесс легких. Описаны случаи поражения сердца (вызывающие перикардит) и мозга (приводящие к абсцессу мозга).
Микроорганизм передается от одного лица к другому посредством амебных цист — формы существования амеб, в которых протеза имеет наибольшую сопротивляемость воздействию извне. Цисты не повреждаются желудочным соком, поэтому проходят в кишечник, где могут развиться в трофозоиты, которые поражают слизистую кишечника, абсорбируются и могут далее проникать в другие органы. Entamoeba histolytica является причиной амебиаза имеет довольно сложный жизненный цикл в организме хозяина. Выделяемые в неизмененном виде цисты являются источником для новых заражений. Имеется много препаратов, применяемых для лечения амебиаза. Однако ни один из них не может считаться совершенно эффективным, поскольку этот микроорганизм очень резистентен.
Препараты, применяемые при лечении амебиаза, могут быть охарактеризованы по принципу их основного места действия. Например, некоторые препараты (йодохинол, дилоксанид, паромомицин) активны только против амеб, локализованных в просвете кишечника, и применяются при лечении асимптомных или умеренных формах амебиаза. Друше (эметин, хлороквин) активны против амеб, локализованных в стенке кишечника и других тканях и применяются при амебной дизентерии и гепатических абсцессах. Третьи (метронидазол, тинидазол) активны против амеб как кишечной, так и внекишечной локализации.
Йодохинол (lodoquinol)
Иодохинол — 5,7-дийод-8-хинолинол (37.2.2) получают йодированием 8-оксихинолина (37.2.1) смесью йодид калия/йодат калия. Исходный 8-окснхинолин (37.2.1) получают исходя из 2-аминофенола и глицерина в присутствии серной кислоты и нитробензола (синтез Скраупа) [39, 40].
- 786 -
Препараты для лечения протозойных инфекций
NH2 он
СН2-СН-СЧ2 он он он
H2SO4
37 2 1
KI / кю3
I
ОН
37 2 2
Йодохинол является амебоцидом, применяемым против Entamoeba histolytica, и активен как против цист, так и против трофозоитов, локализованных в просвете кишечника; считается препаратом выбора при лечении асимптомных или умеренных форм амебиаза. Механизм действия препарата неизвестен.
Иодохинол применяется при заболеваниях кишечным амебиазом средней тяжести.
Синонимами препарата являются дихинол, йодоксин, дийодоквин, амебаквин и др.
Дилоксанид (Diloxanide)
Дилоксанид — 2,2-дихлор-М-(4-фуроилоксифенил)-Ы-метилаце-тамид (37.2.4) получают ацилированием 2,2-,тихлор-УЦ4-гидрокси-фенил)-К-метилацетамида (37.2.3) хлорангидридом-2-фуранкарбо-новой кислоты. Сам 2,2-дихлор-М-(4-гидроксифенил)-Т<-метилаце-тамид (37.2.3) получают N-ацилированием 4-гидрокси-Кт-метилани-лина либо хлорангидридом дихлоруксусной кислоты, либо весьма оригинальным способом — с использованием циангидрина хлораля [41,42,43].
Дилоксанид также активен против цист и против трофозоитов, локализованных в просвете кишечника, и применяется при лечении носи
- 787-
Глава 37
телей Entamoeba histolytica, а также асимптомных или умеренных форм амебиаза. В стадии перехода к трофозоитам препарат менее активен, чем йодохинол.
Синонимом препарата является фурамид.
Паромомицин (Paromomycin)
Паромомицин (32.4.5) был описан гл. 32 «Антибиотики».
Препарат рекомендуется при лечении острых и хронических форм кишечного амебиаза, а также при лечении кишечных бактерий Salmonella и Shigella.
Синонимами препарата являются аминозидин, катенулин, кресто-мицин, гидроксимицин, мономицин, зигоминин и др.
Эметин (Emetine)
Эметин — 3-этил-2.3,4,6,7,11Ь-гексагидро-9,10-диметокси-2-( 1,2,3, 4-тетрагидро-6,7-диметокси-1-изохинолилметил)-17/-бензо[а]хино-лизин (37.2.8) является природным соединением. Традиционно алкалоид эметин извлекался из растения ипекакуанна (Бразильский корень) и использовался в качестве основного средства для лечения амебиаза, лейшманиоза, дизентерии. Предложены различные пути синтеза эметина, каждый из которых исходит из гомовератрилами-на — 2-(3,4-диметоксифенил)этиламина [44, 45, 46, 47, 48].
-788 -
Препараты для лечения протозойных инфекций
При совместном каталитическом гидрировании этилового эфира р-(а‘-циано)пропилглутаровой кислоты и гомовератриламина водородом происходит реакция восстановительного аминирования с выбросом аммиака и промежуточным образованием вторичного амина (37.2.5), который в условиях реакции подвергается внутримолекулярной циклизации с получением 1-[2-(3,4-диметоксифенил)-этил]-4-карбэтоксиме-тил-6-этилпиперидона-2 (37.2.6). Взаимодействием полученного лактама с хлорокисью фосфора осуществляют гетероциклизацию последнего в производное бензохинолизина (37.2.7). Дальнейшим взаимодействием продукта с гомовератриламином получают соответствующий амид. Последний действием хлорокиси фосфора циклизуют в изохинолиновое производное, пиридиновое кольцо в котором далее гидрируют водородом до рацемической смеси продуктов, из которой выделяют искомый эметин.
37 2 8
Препарат обладает прямым амебицидным эффектом против трофозоитов Entamoeba histolytica в тканях и не активен против цист, как в просвете и стенке кишечника, так и в других органах. Механизм действия эметина заключается в блокировании синтеза протеинов в эукариотических, но не прокариотических клетках, ингибируя процесс по
- 789 -
Глава 37
строения полипептидной цепи. Причем синтез протеинов ингибируется в клетках паразитов и млекопитающих, но не в бактериях.
В качестве средства для лечения амебиаза эметин в настоящее время применяется лишь в случаях резистентности к другим препаратам.
Синонимами препарата являются ипецин и метилцефалин.
Хлороквин (Chloroquin)
Синтез и свойства хлороквина (37.1.1.3) описаны в настоящей главе в разд. 37.1.
Хлороквин широко применяется также при лечении амебиаза.
Метронидазо. i (Metronidazole)
Метронидазол — 2-метил-5-нитроимидазол-1-этанол (37.2.10) получают нитрованием 2-метилимидазола с получением 2-метил-5-нитроимилазола (37.2. 9), который дальнейшим взаимодействием с 2-хлорэтанолом или этиленоксидом с легкостью трансформируется в искомый метронидазол [49, 50].
н
I
Crl2“CH2—он
37 2 10
Метронидазол является синтетическим противопротозойным препаратом. впервые внедренным в медицинскую практику в 1960 г. Препарат подавляет развитие трихомонад, гиардий, амеб, лямблий, бактероидов, фезобактерий и некоторых других. Эффективен в отношении облигатных анаэробных бактерий. Неактивен в отношении аэробных микроорганизмов.
Метронидазол с легкостью проникает через клеточные оболочки как аэробных, так и анаэробных бактерий. Далее активация и действие препарата требе ют протекания определенных восстановительных про
- 790 -
Препараты для лечения протозойных инфекций
цессов, которые имеют место в анаэробных организмах и некоторых бактериях в анаэробных условиях. При этом нитрогруппа препарата подвергается восстановлению белками с низким редокс потенциалом. Эта реакция понижает концентрацию неизмененного метронидазола и вызывает градиент, позволяющий большему количеству препарата проникнуть в бактерию. Некий нестабильный и неидентифицированный промежуточный продукт восстановления метронидазола взаимодействует с бактериальной ДНК, вызывая гибель протозойного паразита. Механизм резистентности к метронидазолу выяснен неокончательно. Метронидазол проявляет высокую бактерицидную активность по отношению к большинству анаэробов {Protococcus, Peptostreptococcus, Clostridium Bacteroides и Fusobacterium), а также некоторых неспорооб-разуюших грамположительных организмов {Actinomyces, Eubacterium, Bifidobacterium, Propionibacterium). Неактивен по отношению к аэробным инфекциям. Высокоактивен против анаэробных протозойных инфекций, включая Trichomonas vaginalis, Entamoeba histolytica, Giardia lamblia и Balantidium coli.
Метронидазол является лекарством выбора при амебиазах, вагинальном трихомониазе и трихомонадном уретрите у мужчин, лямблиозе, амебной дизентерии, анаэробных инфекциях, вызванных чувствительными к препарату микроорганизмами.
Синонимами препарата являются флагил, протостат, трихопол и вагимид.
Тинидазол (Tinidazol)
Тинидазол — 1-2-(этилсульфонил)этил-2-метил-5-нитроимидазол (37.2.12) получают также из 2-метил-5-нитроимидазола (37.2.9), который взаимодействием с 2-эгоксисульфонил я-толуолсульфо патом (37.2,11) трансформируется в искомый тинидазол. Необходимый для этой цели 2-этоксисульфонил-и-толуолсульфонат (37.2.11), в свою очередь, получается тоизилированием 2-этилсульфонилэта-нола и-толуолсульфохлоридом [51, 52. 53].
С2Н=-ЗО2-СЬ2-СН;-ОН • --->. c!h5_so2-c,'2-ch2-c-so2-^^-chj ------У--------
37 2
C2H5““SO2”"CH2’"CH2
37 2 12
-791 -
Г лава 37
По механизму действия и по показаниям к применению тинидазол очень похож на метронидазол. Эффективен также в отношении амеб, трихомонад, лямблий, при остром язвенном гингивите, послеоперационных анаэробных инфекциях. Его применяют для лечения практически всех протозойных инфекций.
Синонимами препарата являются тинапорт, тинимед и тинисан.
37.3. Препараты для лечения лейшманиоза
Группа трипаносомных протозойных паразитов рода Leishmania вызывает ряд болезней, известных как лейшманиоз. Болезнь в основном распространена в тропиках и субтропиках и переносится специфическими кровососущими насекомыми. Различают кожный лейшманиоз (возбудитель Leishmania tropica) и висцеральный лейшманиоз (возбудитель Leishmania donovani). При местном лечении кожного лейшманиоза применяют акрихин и мономицин.
При лечении висцерального лейшманиоза (калаазар) широко применяют препарат пятивалентной сурьмы — стибиоглюконат натрия Следует отметить, что органические соли трехвалентной сурьмы (литиевая соль антимониляблочной кислоты — антиомалин, производное арилсульфоновой кислоты — антимосан, производное /?-аминофе-нилстилбоновой кислоты - стибамин и др.) долгое время являлись традиционными и эффективными препаратами для лечения лейшманиоза. Недавно было показано, что соли пятивалентной сурьмы более удобны для применения. Полагают, однако, что в организме пятивалентная сурьма восстанавливается до трехвалентной. Для лечения лейшманиоза применяются также амфотерицин В. метронидазол и пентамидин.
Стибиоглюконат натрия (Sodium Stibogluconate)
Стибиоглюконат натрия — тринатриевую соль 2,4:2|,4|-0-(окси-дисгибилидин)-8Ь,БЬ -диоксида-П-глюконовой кислоты (37.3.1) получают действием глюконовой кислоты на свежеосажденную сурьмяную кислоту и нейтрализацией полученного раствора гидроокисью натрия, после чего искомый продукт — стибиоглюконат натрия - - осаждают спиртом [54]. Стибиоглюконат натрия является препаратом выбора для лечения лейшманиоза. Полагают, что он ингибирует гликолитический фермент (фосфофруктокнназу). участвующий в цикле Кребса, происходящем в ор!анизме паразиюв рода Leishmania в результате чего паразитический микроорганизм перестает вырабатывать необходимую для своею существования энергию и пог пбает.
- 792 -
Препараты для лечения протозойных инфекций
н сн-о-н сн-о-н
2 СЧ-О-Н сн-о-н сн-о-н
I
соон
NaOH
HSb(OH)6 -------
— 3
н н
сн-о-н н-о-сн
сн-о-н н-о-сн
1 1 4.
СН-ОХ ОН О О-СН 3Na
СН-О—;Sb-O-Sb^O-CH
сн-о/ ю-сн
I I
соо 'оос
37 3 1
Синонимом препарата является солюсурмин и др.
37.4. Препараты для лечения трипаносомоза
Трипаносомиаз — болезнь, больше известная под названием сонная болезнь, выражается хроническим сном, головными болями, нарушением координации движений, апатией, потерей интеллекта и, при отсутствии лечения, смертью. В Африке трипаносомиаз переносится мухой цеце, инфицированной трипаносомами (Trypanosoma gambtence и Trypanosoma rhodesience). В Южной Америке болезнь также переносится кровососущими насекомыми. Известна группа диамидиновых соединений, активных против трипаносом. Наиболее широко используемым для этой цели соединением является пентамидин. Нередко применяют ни-футрифокс и более старый препарат — сурамин.
Пентамидин (Pentamidine)
Пентамидин — 4,41-(пентметилендиокси)дибензамидин (37.4.2) получают взаимодействием 4-гидроксибензонитрила с 1,5-дибром-пентаном в присутствии гидроксида натрия с получением (,5-бис-(4-цианофенокси)пентана (37.4.1). Последовательным взаимодействием последнего со спиртовым раствором хлористого водорода с промежуточным получением иминоэфира и далее со спиртовым раствором аммиака получают искомый пентамидин [55, 56, 57]
Вг—’В'
NaOH
2 NHh С2Н5ОН
- 793-
Г лава 37
О—(СН2)5—О
NH и c-nh2
37 4 2
Препарат уничтожает микроорганизмы, которые вызывают трипаносомоз и лейшманиоз. Полагают, что пентамидин может действовать либо путем ингибирования процесса окислительного фосфорилирования, либо путем ингибирования процесса метаболизма глюкозы, либо ингибированием активности дигидрофолатредуктазы, либо взаимодействия с ДНК или нуклеотидами паразита.
Синонимами препарата являются ломидин, небупент и др.
Нифуртимокс (Nifurtimox)
Нифуртимокс — 1,1 -диоксид-4-[(5-нитрофурилиден)амино]-3-ме-тилтиоморфолин (37.4.7) получают по следующей схеме синтеза. Взаимодействием 2-меркаптоэтанола с пропиленоксидом в присутствии гидроокиси калия получают (2-гидроксиэтил)-(2-гидрокси-пропилсульфид (37.4.3), внутримолекулярной дегидратацией которого с помощью бисульфата калия получают 2-метил-1,4-окситиан (37.4.4). Окислением последнего пероксидом водорода получают 2-метил-1,4-окситиан-4,4-диоксид (37.4.5), который при взаимодействии с гидразином трансформируется в 4-амино-З-метилтетра-гидро-1,4-тиазин-1,1-диоксид (37.4.6). Вводя последний во взаимодействие с 5-нитрофурфуролом, получают соответствующий гидразон — искомый нифутримокс [58, 59].
HQ-CH2-CH2—SH
НО-'СН2~С’-’2 S hO-CH-CHj1 сн3
KHSO4
h2n-nh2
37 4 6
37 4 3
37 4 7
- 794 -
Препараты для лечения протозойных инфекций
Нифуртимокс является препаратом выбора при острой форме южно-американской формы трипаносомоза — болезни Чагаса. Полагают, что препарат действует путем образования реакционноспособных радикалов (супероксид, гидропероксид, гидроксил) в паразитах, что приводит к потере паразитом каталазы и глутатион пероксидазы и повышению у последнего чувствительности к пероксиду водорода, что нарушает его нормальную жизнедеятельность.
Синонимом препарата является лампит и др.
Сурамин (Suramin)
Сурамин — гексанатриевая соль ]8,8Скарбонил-гж’-[имино-3,1-фениленкарбонилимино-4-метил-3,1-фенилен]карбонилимино]-бнс-1,3,5-нафталинтрисульфоновой кислоты (37.4.13) может быть получена взаимодействием 1-аминонафталин-3,6,8-трисульфоновой кислоты с хлорангидридом 4-метил-З-нитробензойной кислоты с получением нитробензоильного производного (37.4.8). Нитрогруппу в последнем восстанавливают активированным железом в аминопроизводное (37.4.9), которое ацилируют .м-нитробензоилхлоридом с получением нового нитропроизводного — .н-нитробензоил-(4-ме-тил-3-аминобензоил)-1-аминонафталин-3,6,8-трисульфоновой кислоты (37.4.10). Последний вновь восстанавливают в амин (37.4.11) гем же способом. Взаимодействием полученного продукта с фосгеном получают [8,8'-карбонил-бнс-[имино-3,1-фениленкарбонили-мино-4-метил-3,1-фенилен]карбонилимино]-бис-1,3,5-нафталинтрисульфоновую кислоту (37.4.12), обработкой которой гидроксидом натрия получают сурамин [60, 61, 62, 63].
HO3S
Fe
- 795 -
Глава 37
HO3s
COCI2
Сурамин — очень старый препарат, полученный в 1920-х годах при исследованиях некоторых ядов и красителей (трипановый красный и голубой, афридол фиолетовый) в качестве препаратов с трипаносоми-цидной активностью. Точный механизм действия препарата не выяснен. Однако полагают, что сурамин абсорбируется в трипаносомах, где, возможно. обратимо связывается с белками. В настоящее время препарат используется редко при лечении ранних форм сонной болезни.
Синонимами препарата являются антрипол, натримин, германии и др.
37.5. Препараты для лечения других протозойных инфекций
Протозойной инфекцией считается также лямблиоз, который вылечивается хинакрином. Иногда в случаях лямблиоза применяют метронидазол и фуразолидон. Другой распространенной протозойной инфекцией является трихомониаз, для лечения которого применяют описанные выше метронидазол и тинидазол. Лечение токсоплазмоза осуществляют сульфаниламидами и хлоридином.
- 796-
Препараты для лечения протозойных инфекций
Список литературы
1. USPat. 2.233.970(1941).
2. Ger. Pat. 683.692 (1937).
3. Drake N. et al.//J. Am. Chem. Soc. 68. 1214 (1946).
4. Price C. et al.,/'J. Am. Chem. Soc. 68. 1204 (1946).
5. Ger. Pat. (DDR). 53.065 (1966).
6. Surrey A. et al.//J. Am. Chem. Soc. 68, 113 (1946).
7. USPat. 2.478.125 (1949).
8. Ger. Pat. 486.079(1924).
9. Elderfield R. et al./, J. Am. Chem. Soc. 68. 1579 (1946).
10. USPat. 2.546.658 (1951).
11. Surrey A. et al.7J Am. Chem. Soc. 72, 1814 (1950).
12. USPat. 2.474.819 (1949).
13. US Pat. 2.474.821 (1949).
14. Burckhalter G. et al.'7J. Am. Chem. Soc. 70, 1363 (1948).
15. Nalaraj an M. et al./7Arzneimittel-Forsch. 22, 1230 (1972).
16. Jucker H. et al.//Ulmann’s Enzyklopadie der Technischen Chemie.
3, 213 (1953).
17. Woodward R. et al./7J. Am. Chem. Soc. 66, 849 (1944).
18. Woodward R. et al.//J. Am. Chem. Soc. 67, 860 (1945).
19. Taylor et al./, J. Am. Chem. Soc. 94. 6218 (1972).
20. Gutzwiller et al./'J. Am. Chem. Soc. 100. 576 (1978).
21. Grethe G. et al.,7J. Am. Chem. Soc. W0, 589 (1978).
22. Imanishi T. et al., Chem. Pharm. Bull. 30, 1925 (1982).
23. Ohnmacht C et al., 'J. Med. Chem. 14, 926 (1971).
24. Ger. Pat. 2.940.443 (1979).
25. Ger. Pat. 2.806.909 (1978).
26. Jing-Ming L. et al.,/'Acta Chim. Sinica. 37. 129 (1979).
27. Schmid G. et al.'/J. Am. Chem. Soc. 105, 624 (1983).
28. Elderfield R. et al./'J. Am. Chem. Soc. 68, 1524 (1946).
29. Eldertield R. et al.//J. Am. Chem. Soc. 77, 4816 (1955).
30. Curd R et al./'J. Chem. Soc. 1946, 729.
31. CurdR. etal.,.J. Chem. Soc. 1948, 16.30.
32. Ainly H. et al.' J. Chem. Soc. 1949, 98.
33 Ger. Pat. 553.072 (1934).
34. Ger. Pat. 571.499(1934).
35. USPat. 2.1 13.357 (1938).
36. USPat. 1.782.727 (1930).
37. USPat. 1.889.704 (1932).
38. Wingler A. /Angew. Chem. 61, 49 (1949).
- 797-
Глава 37
39 Ger. Pat. 411.050(1925).
40. Papesch J J. Am. Chem. Soc. 58. 1314 (1936)
41. Bnt. Pat. 767.148 (1954).
42. Bnt Pat 786.806 (1955).
43. US Pat. 2.912.438(1959).
44. Евстигнеева P и др./,'ДАН СССР, 75, 539 (1950).
45. Tamelen Van E et al.//J. Am. Chem. Soc. 91, 7359 (1969).
46. Kametam T et al.'/J. Chem. Soc. Perkin. Trans. I, 1979, 1211.
47. TakanoS et al.,/J. Org. Chem, 43. 4169 (1978).
48. FujiiT et aU/Tetrahedron. 36, 1539 (1980).
49. US Pat. 2.944.061 (1960).
50. Cossar К et al.//Arzneimittel-Forsch. 16, 23 (1966).
51. US Pat. 2.376.311 (1968).
52. Ger. Pat. 1.745.780 (1967).
53. Miller M et al.,'J. Med. Chem. 13, 849 (1970).
54. Bose J et al./«'Indian. J. Pharm. И., 155 (1949).
55. Bnt. Pat. 507.565 (1939).
56. US Pat. 2.394.003 (1946).
57. Ashley J et al.'«J. Chem. Soc. 1942, 103.
58. Ger. Pat. 1.170 957(1964).
59. US Pat. 3.262.930(1966).
60. Bnt. Pat. 224.849 (1923).
61. Dressel J /'J. Chem. Ed. 38, 620 (1961).
62 Fourneau E et al.'/Compt. Rend. 178, 675 (1924).
63 Hey mann B.i1 Angew. Chem. 37, 585 (1924).
!»
J Глава 38
‘ Антигельминтные j препараты
Гельминтные инфекции представляют большую проблему для жизни миллионов людей на Земле, особенно в тропиках и субтропиках. В зависимости от типа и локализации возбудителя гельминтоз может протекать бессимптомно или же явиться причиной анемии, поражения кровеносных сосудов, печени, глаз. Различают кишечные и внекишеч-ные гельминтозы. Возбудители классифицируются как круглые черви (нематоды) и плоские черви, к которым причисляют ленточных червей (цестоды) и сосальщиков (трематоды). Большинство нематодных инфекций локализуется в кишечном тракте, однако некоторые из них могут проникать в иные органы, включая сердце, печень, легкие, мышцы и др., из которых их удаление значительно сложнее. Цестодные инфекции обычно локализуются в ЖКТ, но известны случаи их проникновения в циркуляционную систему. Трематоды вызывают хроническое инфицирование, называемое шистосомоз, при котором поражаются кровеносные сосуды и нарушаются функции и структуры различных органов печени, кишечника, мочеполовою тракта.
Антигельминтные препараты предназначены для искоренения гельминтов и выведения их из организма хозяина, и их подразделяют на:
— средства, нарушающие нервно-мышечную координацию гельминтов;
— средства, действующие на энергетические процессы гельминтов. в частности на метаболизм глюкозы;
— средства, действующие на ферментную систему гельминтов, кладку и высиживание яиц гельминтами и т. п.
Большинство антигельминтных препаратов предназначены для воздействия на определенных гельминтов. Исторически в качестве лекарственных средств при гельминтозах применяли галогензамещенные углеводороды, фенол и родственные им соединения: сульфаниламиды, производные хинолина, нафтохинона, фенотиазина, ряд природных со
- 799-
Глава 38
единений, выделенных из листьев полыни и папоротника, эфирных масел (производных пинена), алкалоидов (ареколиновой группы), алкалоидов ряда эметина и многие другие. Однако в настоящее время эссенциальными препаратами для лечения гельминтоза являются нижеописанные лекарственные средства.
Мебендазол (Mebendazol)
Мебендазол — метил-[5-(бензоил)-1Я-бензимидазол-2-ил] карбамат (38.1.5) является производным бензимидазола, который получают путем взаимодействия 3,4-диаминобензофенона (38.1.3) с N-мето-ксикарбонил-Б-метилтиомочевиной (38.1.4) [1, 2].
38 1 3
CHjS\
C=N-СООСНз h2nz
38 1 4
Необходимые реагенты получают следующим способом. Нитрованием 4-хлорбензофенона азотной кислотой при температуре ниже 5 °C получают 4-хлор-З-нитробензофенон (38.1.1), атом хлора в котором замешают на аминогруппу нагреванием до 125 °C с раствором аммиака в метаноле с получением 4-амино-З-нитробензофенона (38.1.2). Восстановлением нитрогруппы в последнем водородом с использованием в качестве катализатора палладия на угле получают 3,4-диамино-бензофенон (38.1.3).
Pd-C
Второй реагент — N-метоксикарбонил-Б-метилтиомочевину (38.1.4) получают взаимодействием метилового эфира хлорутольной кислоты с S-метилтиомочевииой.
CH3S\
C=N-H
h2nz
+ Cl—СООСНз
CH3SX
C=N-COOCH3
HzNZ
31 1 4
- 800 -
Антигельминтные препараты
Точный механизм действия мебендазола окончательно не выяснен, но кажется вероятным, что он вызывает необратимое ингибирование захвата и утилизации глюкозы организмом паразита, тормозит образование в их организме аденозинтрифосфата, вызывая тем самым гликогеновое истощение и последующую гибель паразитов.
Мебендазол применяется для лечения энтеробиоза, аскаридоза, ан-килостомидоза, стронголоидоза, трихоцефалеза, трикуриаза. аскаридоза, трихострондиоза, анциклостомиаза и смешанных генльминтозов. Его применение в течение 3 дней 2 раза в день в дозах по 100 мг вызывает полное излечение от 90 до 100 % пациентов.
Синонимами препарата являются вермокс, мебутар, панфуган и многие другие.
Альбендазол (Albendazol)
Альбендазол — метил-[5-(пропилтио)-1Я-бензимидазол-2-ил]карба-мат (38.1.8) также получают гетероциклизацией производного фе-нилеидиамина в производное бензимидазола. С этой целью 3-хлор-6-нитроацетанилид вводят во взаимодействие с пропилмеркаптаном с получением З-пропилтио-6-нитроацетанилида (38.1.6). Восстановлением нитрогруппы в последнем водородом также с применением в качестве катализатора палладия на угле получают 4-(про-пилтио)-о-фенилендиамин (38.1.7). Взаимодействием полученного производного о-фенилендиамина с цианамидом и далее с метиловым эфиром хлоругольной кислоты получают искомый альбендазол [3].
no2
NH-СОСН3
C3H7SH
NH-COCH3
38 1 6
NOZ Hz/Pd-C
38 1 7
1 H2NCN
2 CICOOCH3
C3H7S
38 1 8
N
NH-C00CH3
N
1
Производное бензимидазола — альбендазол — препарат широкого антигельминтного спектра. Проявляет антигельминтный эффект против чувствительных цестод и нематод путем блокирования процесса захвата глюкозы паразитами, выражающееся истощением запасов гликогена
-801 -
Глава 38
и последующего понижения уровня аденозинтрифосфата, вследствие чего паразиты обездвиживаются и погибают. Препарат применяется при заражении Acaris lumbricoides, Ancylostoma duodenale, Necaior ameri-canus, Enterobius vermicularis и Trichuris trichiura
Синонимом препарата является СКФ 62979 и др.
Тиабендазол (Thiabendazol)
Тиабендазол — 2-(4‘-тиазолил)бензимидазол (38.1.9) также получают аналогичным путем — гетероциклизацией, происходящей при взаимодействии о-фенилендиамина с 1.3-гиазол-4-карбоновой кислотой [4, 5, 6].
н
38 1 9
Тиабендазол — антигельминтный препарат широкого спектра действия. Хотя детальный механизм его действия выяснен неокончательно, кажется очевидным, что его действие опосредовано ингибированием специфического для гельминтов фермента— фумаратредуктазы.
Тиабендазол активен по отношению к большинству нематодных инфекций, включая Angyostrongylus cantonesis, Strongyloides stercoralis, Trichinella spiralis, Toxocara cams, Toxocara cati, Ancylostoma caninum, Ancylostoma braziliense, Ancylostoma duodenale, Dracunculus medinesis, Capillaria philippinesis, а также при лечении Acaris cantonesis и Shisto-soma stercoralis.
Синонимами препарата являются минтезол, минзолум и др.
Ниридазол (Niridazol)
Ниридазол — 1-(5-нитро-2-тиазолил)-2-имидазолидинон (38.1.11) получают взаимодействием 2-амино-5-нитротиазола с 2-хлорэтил-изоцианатом с получением дизамещенной мочевины (38.1.10), нагреванием которой осуществляют внутримолекулярную реакцию алкилирования с образованием искомого имидазолидинонового производного - ниридазола [7, 8, 9, 10, 11].
- 802 -
Антигельминтные препараты
Ниридазол проявляет шистосомицидное и амебицидное действия. Механизм действия неясен. Кажется вероятным предположение, что, концентрируясь в организме паразита, он вызывает ингибирование активации фосфорилазы, выражающейся в истощении запасов гликогена, а также ингибирует сперматогенез паразитов, воздействует на продукцию яиц.
Показан для лечения болезней, вызванных Dracunculus meddinesis, а также Shistosoma Haematobium и Shistosoma mansoni. Относится к третичным препаратам и используется только в отсутствие препаратов выбора.
Синонимом препарата является амбилар и др.
Пиперазин (Piperazin)
Пиперазин (38.1.12) является многотоннажным продуктом органического синтеза, который получают исходя из этаноламина путем нагревания последнего в среде аммиака при температуре 150-220 °C и давлении 100-250 атмосфер. Применяют в виде солей, как правило в виде адипината [12, 13].
NH3 !-------X
H2N—СН2—СН2—ОН ------► H-N N-H
38 1.12
Пиперазин является альтернативным препаратом и применяется для лечения при заражении различными видами нематод, в частности при энтеробиозе и аскаридозе. Препарат вызывает паралич нематод, блокируя ацетилхолиновую передачу. Это вызывает отлипание паразитов от слизистых стенок и выделение их из организма. Аскаридоз требует лечения в течение двух дней, энтеробиоз — семи.
Синонимами препарата являются таснон, увилон, антепар, бексин и многие другие.
Диэтилкарбамазин (Diethylcarbamazine)
Диэтилкарбамазин — \,\-лиэтил-4-метил-1 -пиперазинкарбоксамид (38.1.13) получают ацилированием 1-метилпиперазина диэтилкар-бамоилхлоридом [14, 15. 16].
N-C-CI
H-N Д-СНз
С2Н5\ О /------Ч
N-C—-N N-CH3
С2Н5/ \!
38 1 13
- 803-
Г лава 38
Диэтилкарбамазин является производным пиперазина. Механизм его действия выяснен не полностью. Однако весьма вероятно, что он вызывает понижение мышечной активности и даже паралич у гельминтов. Препарат вызывает быстрое исчезновение паразитов вида Brugia malayi, Loa loa и Wuchereria bancrofti, а также применяется при заболеваниях, вызванных Onchocerca volvulus и Mansonella strep-tocerca.
Синонимами препарата являются гетразан. нотезин, баносид и др.
Празиквантел ((Praziquantel)
Празиквантел — 2-(циклогексилкарбонил)-1,2,3,6.7,11Ь-гекагидро-4//-пиразино[2,1-а]изохинолин-4-он (38.1.15) представляет собой производное пиразинохинолина, который предложено получать двумя путями [17, 18, 19]. Согласно первому способу, 1-амино-метил-1,2,3,4-тетрагидроизохинолин алкилируют хлоруксусной кислотой, и далее полученный амин ацилируют хлорангидридом цик-логексанкабоновой кислоты с получением 1-(К-карбоксиметил-М-циклогексилкарбониламинометил)-1,2,3,4-тетра-гидро-изохинолина (38.1.14), нагреванием которого при 150 °C получают искомый празиквантел.
1 С1Сн2СООН
38 1 ’4
38 1 15
Второй способ синтеза исходит из изохинолина, взаимодействием которого со смесью хлорангидрид циклогексанкабоновой кислоты'цианистый калий получают дигидропроизводное изохинолина (38.1.16), восстановлением которого водородом над никелем Ренея получают продукт восстановления (переамидирования) — амид-1-(М-циклогек-силкарбониламинометил)-1.2,3,4-тетрагидроизохинолин (38.1.17). Ацилированием последнего хлорангидридом хлоруксусной кислоты получают хлорацегильное производное (38.1.18), при нагревании которого в присутствии диэтиламина в результате внутримолекулярного алкилирования получается искомый продукт — празиквантел.
- 804 -
Антигельминтные препараты
38 1 15
Празиквантел — антигельминтный препарат широкого спектра действия. Кажется вероятным, что он воздействует на клеточную мембрану шистосом путем повышения проницаемости и следовательно потока в нее ионов С а2 с последующим параличом мышц паразита. В результате присоски паразита перестают действовать, и он выводится из организма хозяина. Препарат показан при лечении всех форм шистосомоза у людей. Является лекарством выбора при лечении Shistosoma mansoni, Shistosoma haematobium, Shistosoma japonicum, а также Shistosoma inter-calatum и Shistosoma mekongi.
Синонимами препарата являются дронцит, билтрицид, цезол.
Пирантел (Pyrantel)
Пирантел — 1,4,5.6-тетрагидро-1-метил-2-[/иранс-2-(2-тиенил)ви-нил]-пиримидин (38.1.22) — производное тетрагидропиримидина, получают исходя из 3-(2-тиенил)-акрилонитрила (38.1.19, который получают по реакции Кновенагеля конденсацией фурфурола с циануксусной кислотой. Кислотным гидролизом последнего получают 3-(2-тиенил)акриламид (38.1.20). Вводя последний во взаимодействие с пропансульфоном, получают иминоэфир (18.1.21), взаимодействием которого с N-метил-триметилендиамином получают искомый пирантел [20, 21, 22, 23].
Cn3COONh4/
02
s
36 1 19
- 805-
Глава 38
O-CH2-CH2-CH2-SO2ti
38 ' 2<
СН3
HN-СН2-СН2-СН2-МН2
Пирантел является высокоэффективным антигельминтным средством при энтеробиозе, аскаридозе, анкилостомидозе и некаторозе. Антигельминтный эффект проявляется в виде мощного холиномимегическо-го воздействия на мышечные клетки нематод путем связывания с их холинергическими рецепторами, что приводит к ингибированию нейромышечной передачи в организме паразитов, сокращению их мускулатуры, и. в результате, к выделению гельминтов из ЖКТ хозяина. Вылечивает 85-100 % случаев аскаридоза и 90-100 % случаев энтеро-био-за. Является лекарством выбора при заражениях Enterobius vermi-culuris, Acaris lumbricoides, а также Ancylostoma duodenale, Necator americanus.
Синонимами препарата являются гелмекс, комбантрин, антиминт и др.
Левамизол (Levamisol)
Левамизол — (-)-2,3,5,6-теграгидро-6-фенилимидазо[2,1-Ь]тиазол (38.1.27) является производным имидазотиазола и является L-изомером DL-тетрамизола (±)-2.3,5,6-тетрагидро-6-фенилимидазо [2,1-Ь]тиазола (18.1.26), который предложено получать различными способами. Первый способ исходит из а-бромацетофенона, взаимодействием которого с 2-имино-1,3-тиазолидином получают продукт N-алкилирования 1-фенацил-2-имино-1,3-тиазолидин (38.1.23). Ацилированием продукта уксусным ангидридом получают 2-аце-тилиминопроизводное (38.1.24). Восстановление кетонной карбонильной группы в последнем боргидридом натрия приводит к получению ключевого продукта синтеза — 1-(2-фенил-2-гирокси-этил)-2-ацетилимино-1,3-тиа-золидина (38.1.25). Замещением в последнем гидроксильной группы на хлор с помощью хлористого тионила и последующей обработкой продукта уксусным ангидридом осуще-сгвляют реакцию гетероциклизашш в рацемическую смесь (±)-2, 3.5.6-тетрагидро-6-фенилимидазо[2,1-Ь]тиазолов (38.1.26), называемую также гетрамизолом. Обработкой последней D-10-камфорсульфокислотой выделяют искомый L-изомер —левамизол [24, 25].
-806-
Антигельминтные препараты
Следующие способы получения левамизола отличаются лишь методами получения 1 -(2-фенил-2-гироксиэтил)-2-имино-1,3-тиазолидина (38.1.28). Второй способ исходит из взаимодействия окиси стирола с 2-имино-1,3-тиазолидином и последующей обработкой полученного продукта (38.1.28) хлористым тионилом и далее уксусным ангидридом, что приводит к получению тетрамизола [25].
1 SOCI2
2 (СНзСО^О
Третий способ заключается в первоначальном взаимодействии окиси стирола с этиленимином с получением М-(2-фенил-2-1 идроксиэтил)этиленимина (38.1.29), взаимодействием которого с тиоцианатом калия или с тиомочевиной получают 1-(2-фенил-2-гироксиэтил)-2-имино-1,3-тиазолидин (38.1.28). Далее уже описанным выше путем (обработкой продукта сначала хлористым тионилом, а затем уксусным ангидридом) получают тетрамизол [26, 27, 28, 29, 30, 31].
1 SOCI2
2 (СНзСО)2О
38 1 26
- 807 -
Глава 38
Принципиальным отличием четвертого способа получения тетра-мизола от вышеописанных заключается в осуществлении реакции гетероциклизации в присутствии гидроокиси натрия вместо уксусного ангидрида, что делает более понятным возможный механизм реализуемой гетероциклизации. В предлагаемом методе 1-(2-фенил-2-гироксиэтил)-2-имино-1,3-тиазолидин (38.1.28) получают путем взаимодействия окиси стирола с этанолам ином, дальнейшим замещением гидроксильной группы на хлор в полученном производном диэтаноламина (38.1.30) хлористым тионилом, частичным кислотным гидролизом образующегося при этом дихлорида (38.1.31) до первичного хлорида (38.1.32) и, наконец, взаимодействием последнего с тиомочевиной получают 1-(2-фе-нил-2-гироксиэтил)-2-имино-1,3-Тиазолидин (38.1.28). Взаимодействием последнего с хлорсульфоновой кислотой получают 1-(2-фенил-2-хлор-этил)-2-имино-1,3-тиазолидин (38.1.33), который циклизуется в искомый тетрамизол при обработке щелочью [32, 33, 34, 35, 36].
h2n-ch2-ch2-oh
он I
CH-CH2’“NH~CH2-'CH2-OH
38.1 30
SOCI2
38 1 31
Н2О / HCI
S ri2N~C-Nb2
CISO3H
38 1 33
Наконец, предложен еще и пятый способ получения 1-(2-фенил-2-гироксиэгил)-2-имино-1,3-тиазол идина (38.1.28), заключающийся во взаимодействии 2-амино-1-фенилэтанола с 2-бромэтилизотиоцианатом, приводящим непосредственно к образованию ключевого 1-(2-фенил-2-гироксиэтил)-2-имино-1.3-тиазолидина (38.1.28), который трансформируют в тетрамизол последовательным взаимодействием с хлористым тионилом и далее с уксусным ангидридом [37].
-808-
Антигельминтные препараты
SCN-CH2-CH2-3f
1 SOCl2
2 (СН3со)гО
36 1 28
Левамизол является препаратом выбора при аскаридозе. Многочисленные исследования указывают на то, что однократный прием дозы левамизола вылечивает от 90 до 100 % пациентов с аскаридозом, в частности зараженных Ancylostoma duodenale. Препарат менее эффективен при анкилостомидозе и стронгилоидозе. Однако препарат не эффективен при заражении Necator americanus. Кажется вероятным, что препарат оказывает ганглиостимулирующий эффект на ткани паразита как в парасимпатических, так и в симпатических участках. Более того предполагается, что препарат оказывает иммуномодулирующий эффект на организм хозяина.
Синонимами препарата являются декарис, соласкил, эргамизол, трамизол, иммунол и др.
Никлозамид (Niclosamid)
Никлозамид — 2',5-дихлор-4'-нитросалициланилид (38.1.34) получают взаимодействием 5-хлорсалициловой кислоты с 2-хлор-4-нитроанилином в присутствии треххлористого фосфора [38, 39, 40].
РСЦ
Никлозамид является производным салициламида и является эффективным антигельминтным препаратом. Его действие заключается в ингибировании митохондриального окислительного фосфорилирования как у млекопитающих, так и у паразитов. Одновременно он ингибирует захват глюкозы и кислорода паразитом. В терапевтических дозах практически не проявляет фармакологических эффектов на организм хозяина.
Никлозамид эффективен против кишечных цестод, таких как Diphyllobothrium latum, Taenia saginata. Taenia solium, Dipilidium camnum, Hymenolepis diminuta и Hymenolepis diminuta, но эффективен
- 809 -
Глава 38
против нематод. Препарат эффективен при одноразовом применении в количестве 2 г.
Синонимами препарата являются иомесан, иклоцид, тредемин и др.
Бефениум (Bephenium)
Бефениум — З-гидрокси-2-нафтоат бензилдиметил(2-феноксиэтил)-аммония (38.1.37) получают взаимодействием натриевой соли 3-ги-дрокси-2-нафтойной кислоты с хлоридом бензилдиметил(2-фено-ксиэтил)аммония (38.1.36). Последний, в свою очередь, получают из хлористого бензила и М-(2-феноксиэтил)диметиламина (38.1.35), который синтезируют взаимодействием фенолята натрия с 2-диме-тиламиноэтилхлоридом [41,42].
/СНз С!—Ch2-CH2-N
хсн3
38 1 36
38 1 37
Препарат проявляет антигельминтное действие, действуя в качестве холинергического агониста и вызывая паралич мускулатуры паразитов, что способствует их удалению из кишечника. Применяют при лечении аскаридоза, анкилостомидоза, энтеробиоза, трихостронгилоидоза, три-хоцефалеза. Однократное применение дозы бефениума вылечивает от 80 до 100 % пациентов, зараженных Ancylostoma duoclenale Менее эффективен по отношению к Necator americanus.
Синонимом препарата является аклопар и др.
- 810 -
Антигельминтные препараты
Битионол (Bithionol)
Битионол — 2,2'-тиобис(4,6-дихлорфенол) (38.1.38) получают взаимодействием 2,4-дихлорфенола, растворенного в четыреххлористом углероде с однохлористой серой в присутствии хлористого алюминия [43, 44].
он он
38 1 38
Битионол представляет собой бггс-дихлорфенол и структурно схож с гексахлорофеном. Является антигельминтным препаратом выбора для лечения при заражении человека Fasciola hepatica, а также альтернативой препарату празиквантел, применяемому для лечения легочного и церебрального парагонимиаза.
Точный механизм действия неясен, однако кажется вероятным, что он ингибирует окислительное фосфорилирование в Paeagonumus westermani.
Синонимами препарата являются актамер, битин, превенол и др.
Список литературы
1. Ger. Pat. 2.029.637 (1970).
2. US Pat. 3.657.267 (1972).
3. US Pat. 3.915.986 (1975).
4. US Pat. 3.017.415 (1962).
5. US Pat. 3.262.939(1966).
6. US Pat. 3.274.208 (1966).
7. Brit. Pat. 986.562 (1963).
8. Ger. Pat. 2.033.611 (1970).
9. Ger. Pat. 2.117.050 (1971).
10. Belg. Pat. 632.989 (1963).
11. Lambert C et al.//Experientia. 20, 452 (1964).
12. US Pat. 2.799.617 (1957).
13. Bnt. Pat. 767.826 (1957).
14. US Pat. 2.467.893 (1949).
15. US Pat. 2.467.895 (1949).
16. Kushner A et al.-"J. Org. Chem. 13, 151 (1948).
- 811 -
Г лава 38
п
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
Ger Pat 2 362 539 (1977)
LSPat 4 01 1 411 (1977)
FrehelD etal /Heterocycles 20, 1731 (1983)
Belg Pat 658 987 (1965)
LS Pat 3 >02 661 (1970)
S A.fr Pat 68 00 516(1968)
Austin Л et al//"Nature 212, 1273 (1966)
US Pat 3 274 209(1966)
Raesmaekers 4 et al /J Med Chem 9, 545 (1966)
Ger Pat 1 795 651 (1966)
IS Pat 3 679 725 (1972)
LSPat 3 4”8 047 (1969)
Brit Pat 1 076 109(1965)
Bnt Pat 1 131 799(1966)
Bnt Pat 1 131 800(1966)
Ger Pat 2 233 481 (1972)
Ger Pat 2 264 911 (19^2)
LSPat 3 855 234 (1974)
US Pat 4 070 363 (1978)
US Pat 4 107 170(1978)
Ger Pat 2 034 081 (1970)
US Pat 3 079 297(1963)
US Pat 3 113 067(1963)
Bnt Pat 824 345 (1959)
US Pat 2 918 401 (1959)
Ger Pat 1 117 600(1961)
Ger Pat 583 055 (1933)
LSPat 2 849 494 (1958)
Указатель лекарственных препаратов
BICNU 548 Аквамицетин 669
CCNU 547 Акватаг 392
CS-500 378 Аквахлораль 99
DDAVP 459 Акинетон 199
1),к-гиосциамин 276 Аклопар 810
D-амфетамин 174 Акнидазил 741
FUDR 539 Аколестод 375
ML-236 В 378 Акрисуксин 785
н-аминосалициловая кислота 731 Акромицин 651
1-дофа 196 Аксид 328
Аббокиназа 456 Аксорен 120
Абетол 236 Актамер 811
Авентил 158 Активаза 457
Авлон 782 Актиномицин D 551
Агиракс 121 Актол 69
Аглирал 475 Акуту с 40
Адалат 365 Алговерин 64
Адапин 155 Алепсин 189
Адомал 63 Алинам 122
Адофиллин 436 Алкеран 543
Адрам ицин 652 Аллерган 312
Адреналин 204 Аллергоспазмин 438
Адресон 487 Алмефрин 214
Адриабластин 553 Алмитрин 177
Адриацин 553 Алопрол 217
Адрокаин 22 Алпразолам 115
Азантак 326 Алревмат 73
Азатиоприн 576 Алфента 53
Азепамид 119 Альбендазол 801
Азидотимидин 764 Альбиотик 666
Азитромицин 647 Альбутерол 215
Азлин 601 Альбуцид 700
Азлоциллин 600 Альдактон 402
Азолид 64 Альдомет 415
Азотоиприт 541 Альдостерон 495
Азтреонам 640 Альтеплаза 456
Азудоксат 653 Ачьфатил 623
Азумек 577 Альфентанил 51
-813-
Указатель лекарственных препаратов Указатель лекарственных препаратов
Алюпент 214 Амфотерицин В 737 Апринокс 389 Ацето! ексамид 474
Амантадин 196 Амфоциктин 738 Арабинозид 540 Ацетосал 62
Амбамит 701 Анавар 529 Ар амин 225 Ацикловир 757
Амбеноний 265 Анагрегал 455 Ара-С 540 Байпен 602
Амбитар 803 Анальгин 66 Арвинот 100 Баклофен 302
Амбпосин 599 Анаприлин 232 Арлеф 68 Бактидан 713
Амбохюрин 542 Анатран 391 Арминол 146 Баносид 804
Амебаквин 787 Ангидрон 391 Артевен 447 Баразан 711
Амен 515 Ангинал 368 Артемизин 780 Барбамил 92
Аменореи 517 Андразид 723 Артемизиним 780 Барбифенал 91
Л медицин 554 Андрокур 526 Артеренол 210 Барбосек 94
Амидопрокаин 344 Андронат 524 Арумил 404 Бацидал 711
Амикацин 663 Андрорал 525 Арфонад 290 Бациллопирин 695
Амикин 664 Анестезин 30 Асендин 164 Бацитрацин 676
Амиторид 403 Анестокаин 26 Аспарагиназа 567 Бексин 803
Аминазин 128 А низал 120 Аспирин 62 Белу стин 547
am индан 669 Анизиндион 451 Act емизол 321 Бенадрил 200
Аминогтутет имид 561 Анизотропии 278 Астмалитан 212 Бендрофтуметиазид 389
Аминозидин 660 Анкобон 751 Атабрин 785 Бензедрин 226
Аминокапроновая кислота 457 Анкот ил 751 Атаракс 121 Бензилпенициллин 592
Амиодарон 350 Ановлар 517 Атеноло 1 233 Бензодем 749
Амипре< с 236 Анорекс 179 Атеросол 375 Бензокаин 29
Амитал 92 Ансеф 611 Ативан 114 Бензоната! 432
Амитриптилин 153 Анте пар 803 Атмокаприн 258 Бензтиазид 392
Амкацид 459 Антибиоптал 716 Атраксин 119 Бензтропин 200
Амобарбитал 91 Антиверт 318 Атракурий 298 Бенилин 200
Амо щаквин -?72 Антигемофильный фактор 459 Атровент 437 Бентил 284
Амексан 164 Антигестил 509 Атропизол 276 Бентон 538
Амоксапин 163 Антизолон 489 Атропин 275 Беродуал 441
Амоксикан 600 Антимеран 176 Атроптот 276 Беромицин 593
Амоке ит 600 Антиминт 806 Атрохин 772 Беротек 441
Амоксициттин 599 Антисан 314 Ауреомицин 650 Бестрон 504
Ампиклокс 596 Антиспас 284 Афибрин 458 Бетабактит 604
Ампицит 599 Антрипот 796 Африн 220 Бетакорт 493
Ампициттин 597 Антуран 65 Ацебутол 233 Беталок 233
Амринон 33 7 Апатеф 621 Аценокхмарот 449 Бетаметазон 492
Амсакрин 566 Апаурин 109 Ацетазо там ид 384 Бетанехол 255
Амсидит 567 Апацизин 731 Ацетаминофен 67 Бетацеф 620
Амфебу гамон 164 Аписат 179 Ацетилсалициловая кислота 62 Бефениум 810
Амфепрамон 179 Апотекс 71 Ацетилхолин 252 Биаксин 647
Амфипен 599 Апрессин 421 Ацегилцистеин 433 Бигу анид 784
- 814 -
- 815-
Указатель лекарственных препаратов
Указатель лекарственных препаратов
Ьигу мал 784 Бупропион 164
Бидоцеф 613 Буспар 120
Биклин 664 Буспинен 120
Билтрицид 805 Буспинол 120
Биндан 451 Буспирон 119
Бинордин 507 Бусульфан 546
Бинотал 599 Бу габарбитал 92
Биомицин 650 Бутазолидин 64
Биотетрицин 651 Бу там ид 474
Биофанал 739 Бутизон 92
Ьинериден 199 Бутилкаин 23
Бисгидроксику марин 448 Бутоконазо i 743
Битин 811 Ваброцид 716
Битионол 811 Вагимид 791
Бифенабид 377 Вагинол 718
Бицик ши 651 Вазкардин 358
Бленоксан 552 Вазокард 242
Блеомицин 551 Вазокор 358
Блеоцин 552 В азол ат 358
Блокадрен 235 Вазором 529
Блоке 49 Вазоспан 367
Боноин 318 Вазотек 426
Бревинор 517 Валамид 100
Брек 49 Валамин 100
Бретилий 352 Валцорм 98
Бретин 213 Валимбин 244
Бриканил 213 Валиум 109
Бринердин 244 Валмад 100
Бристосеф 610 Вальдрен 312
Бромбей 317 Вальпин 278
Бромкриптин 194 Вальпроевая кислота 187
Бромфенирамин 316 Ванкоцин 671
Бромэргон 197 Варнерин 449
Бронкосол 212 Варфарин 449
Ьронхопульмин 778 Везадол 137
Бруфен 71 Вектарион 178
Бумекс 398 Вектрин 6з5
Бу метан ид 397 Векуроний 297
Бупивакаин 27 Велбан 555
Бупренекс 59 Веллбутрин 164
Бупренорфин 58 Велосеф 612
Велтан 317 Галоперидол 136
Вентолин 217 Галопрогин 748
Вентуссин 432 Галостерин 529
Вепезид 557 Галотан 7
Верапамил 353 Галотекс 748
Вермокс 801 Галотестин 529
Верошпирон 402 Галцион 97
Вертол 318 Гантонол 694
Весприн 129 Гантрисин 694
Ветрен 447 Гарамицин 663
Вибрамицин 653 Г астридин 327
Вивактил 159 Гастрикс 285
Вивидил 158 Гастрисед 285
Видарабин 759 Гедулин 451
В из ин 221 Геитрин 242
Викен 593 Гексадрол 492
Викодин 43 Гексамидин 185
Вимикон 320 Гексоциклий 282
Вимокс 600 Гексэстрол 507
Винбластин 554 Гелмекс 806
Винкристин 555 Гемитон 219
Винокарм 338 Гемонил 91
Винстрол 529 Гемофил Т 459
Винтрон 709 Гемфиброзил 375
Вира-А 760 Гентамицин 662
Виразол 764 Гентацилин 663
Вирегит 757 Геопен 603
Вироптик 763 Гепален 447
Вистарил 121 Гепарин 445
Вистосан 314 Германии 796
Витенсин 414 Героин 40
Бициллин 593 Герплекс 761
Волитал 176 Гестрол 507
Вольмакс 217 Г етразан 804
Вольтарен 74 Гигротон 396
Вретилол 353 Г идралазин 421
By мои 558 Г идреа 564
Галазепам 111 Гидре кс 392
Галазолин 220 Гидренокс 390
Г алдо т 137 Гидродиурил 389
Гал ламин 294 Гидрозид 389
- 816 -
-817-
Указатель лекарственных препаратов
Указатель лекарственных препаратов
Г идрокодон 43 Гуайфенезин 433 Де падиол 505 Диклофенак 73
Гидроксизин 120 Дадалон 312 Де лакеин 304 Диклоцил 597
Гидроксимицин 660 Дайан 526 Делсолон 494 Дикодид 43
Г идроксимочевина 564 Дактар 741 Дельтамин 176 Дикорантит 345
Г ндроксипрогестерон 514 Дактиномицин 550 Делэстроген 505 Дикумарол 448
Гидроксихтороквин 772 Далацин 667 Демекарий 265 Дикумацил 449
Г идроморфон 41 Далмадорм 98 Демеклоциклин 651 Дику мол 448
Г идрофлу ме гиазид 390 Далман 98 Демерол 46 Дилантин 183
Г идрохлортиазид 388 Дантрий 301 Демсер 247 Дилаудид 41
Г илорел 246 Дантролен 300 Депакин 187 Дилоксанид 787
Гипатот 421 Данулен 301 Депотест 524 Дилтиазем 362
Гиперстат 427 Даонил 476 Де прени i 198 Димедрол 312
Гипертоланум 392 Дапотум 130 Депрессан 421 Дименгидринат 312
Г ипнамил 92 Дапсон 732 Дерм азол 742 Диметан 317
Гипномидат 13 Даранид 386 Дермонистар 741 Димодан 345
Г ипнотал 90 Дараприм 784 Дермопласт 30 Дипиридамол 368
Г ипотиазид 389 Дарбид 281 Десмопрессин 459 Дипирон 66
Гисманат 322 Дарвилен 621 Де гуссин 43 Диссенте 49
Гисприл 323 Дарикон 285 Дециклин 651 Диета 72
Гистадин 321 Даунобластин 553 Джосцин 277 Диетаклор 623
Гистазо I 322 Даунорубицин 553 Дзоксин 174 Дистильбен 509
Г истаспан 316 Дегест 214 Диабиниз 474 Дитериам 392
Г истерон 524 Дезипрамин 155 Диазепам 106 Дитид 392
Гистрит 323 Дезирел 166 Диазид 403 Дитилин 300
Г либенкламид 476 Дезоксикортикостерон 496 Диазоксид 392 Дитирин 468
Глибиниз 477 Декакорт 492 Диакарб 385 Дитицен 565
Глибурид 476 Де кардан 492 Диаква 389 Дитропан 285
Г тикопирролат 280 Декарис 575 Диамбутол 725 Диукардии 390
Г типизид 476 Декломицин 651 Диамокс 190 Диуло 393
Глутетимид 100 Декортизил 488 Диафу рон 718 Диу прес 244
I люкотрол 477 Декортин 489 Дибазин 240 Диурил 388
Г олоксан 545 Дексалон 174 Дибензилин 238 Диуритенс 390
Гормонин 506 Дексамбутол 725 Дибент 284 Диурсан 404
Гризеофу львин 741 Дексаметазон 490 Дигабен 476 Дифенгидрамин 200
Грипенин 603 Дексамфетамин 174 Дигитоксин 334 Дифенилан 347
Грифульвин 742 Дексикам 81 Дигоксин 335 Дифенилпиралин 323
Гуанабенз 414 Декстроам фетам и н 172 Дизопирамид 344 Дифенин 183
Гуанадрел 246 Декстромегорфан 431 Дийодоквин 787 Дифеноксилат 49
Гуанетидин 245 Декстротироксин 379 Дийодтирозин 468 Дифлу нисал 63
Iуанфацин 414 Дексхлорфенирамин 316 Диклекс 597 Дифлупил 267
Гзморосол 266 Де лаги л 772 Диклоксациллин 596 Дихинол 787
-818-
-819-
Указатель лекарственных препаратов
Указатель лекарственных препаратов
Дихторфенамид 386 Заривиз 625 Имплетот 22 Канестен 747
ДИЦИКТОМИН 283 Зароксо шн 393 Имуран 577 Кантил 279
Диэтилкарбамазин 803 Зенидекс 174 Инверзин 288 Капастат 730
Диэтилпропнон 179 Зетамицин 665 Ин дон 451 Капотен 425
Диэтилс 1 и льбестро л 508 Зигомицин 660 Индапамид 396 Капрацид 458
Добутамин 217 Зидовудин 763 Индацид 77 Капреомицин 729
Добутрекс 218 Зинацеф 615 Индерал 232 Каприл 425
Догматил 146 Зигромакс 647 Индометацин 75 Капромицин 730
Доксапрам 176 Зовиракс 759 Инновар 138 Капростерон 515
Доксаприл 177 Ибинол 234 Инокор 338 Каптоприл 423
Доксепин 154 Ибупрофен 69 Интал 438 Карбамазепин 188
Доксициклин 652 Ибуфен 71 Интравал 14 Карбенциплин 603
Доксорубицин 552 Йдоксуридин 760 Интромен 391 Карбимазол 470
Долантин 46 Идувиран 761 Интропин 222 Карбокаин 27
Долобид 63 Изадрин 212 Иомесан 810 Карбоплатин 549
Долофин 46 Изимпур 536 Ионамин 179 Карботироид 470
Допар 196 Изокарбоксазид 161 Йохимбин 243 Карвакрон 391
Долетит 415 Изоксикам 81 Ипецин 790 Карвискен 235
Дойрам 177 Изониазид 722 Ипопипид 376 Карден 366
Дориден 101 Изопреналин 212 Ипратропий бромид 436 Кардигин 335
Дорит 254 Изопропамид 281 Ипрафен 437 Кардиоритмин 344
Досу тьфин 692 Изопротеренол 211 Ипрокс 142 Кардиохин 343
Дофамин 222 Изоптин 353 Исмелин 245 Кариндапен 603
Доцидразин 389 Изоптокаприн 258 Итифур 718 Карисома 304
Драмамин 312 Изордил 358 Итроп 437 Кариспродол 303
Дрену сил 390 Изосорбиддинитрат 358 Иту ран 717 Кармицин 661
Дролептан 138 Изофенитэфрин 225 Ифосфамид 544 Кармубрис 548
Дроморан 45 Изофлуран 8 Иодоглобин 468 Кармустин 548
Дронцит 805 Изофлурофат 267 Иодоксин 787 Катапресан 219
Дроперидол 137 Изоциллин 593 Йодохинол 786 Катену тин 660
Дувоид 255 Изоэтарин 212 Кабикиназа 456 Кванталан 374
Дуксил 178 Иклоцид 810 Каболит 147 Кварзан 280
Дуовент 441 Итозон 646 Калан V53 Квенсил 772
Дурамист 220 Имап 145 Калте н 234 Квестран 374
Дуранест 28 Имбарал 79 Кальмизан 128 Келфизин 699
Дурасеф 613 Имидалин 239 Кальцидрин 223 Кемадрин 199
Дурахин 343 Имизин 152 Камакеин 661 Кенакорт 494
Дурракс 121 Имипенем 637 Камколит 147 Кессар 512
Закись азота 10 Имипрамин 152 Камохин 773 Кета тар И
Заносар 549 Им му НОЛ 809 Канамицин 660 Кеталгин 73
Зантак 326 Имодиу м 49 Кандекс 739 Кетамин 11
-820-
- 821 -
Указатель лекарственных препаратов
Указатель лекарственных препаратов
Кетанест 11
Кетоконазо i 739
Кетоксиэстрнн 504
Кетопрофен 72
Кефатот 616
Кефантот 616
Кефзол 611
Кефтекс 612
Кефлин 609
Кефокс 615
Клавулановая кис юта 604
Кларитромицин 646
Классен 536
Клафоран 625
Кпемастин 312
Ктеоцин 667
Ктидиний 279
Клиндамицин 666
Клинимицин 667
Ктиноцин 655
Кд обvтот 725
Клозапин 141
Ктоксаци 1лин 596
Кломид 511
Кломифен 510
Клоназепам 189
Клонидин 218
К юнопин 189
Ктостильбе! ит 511
Ктотиксен 134
Кютрид 388
Клотримазол 746
Клофазимин 733
Клофетин 219
Ктофибрат 374
Коаксин 609
Когентин 200
Ко теин 39
Кодидоксал 653
Кодит 40
Кокаин 23
Колестипол 373
Колибар 374
Комбетин 336
Комбантрин 806
Конвулекс 187
Конкордии 159
Контратион 269
Конфидан 146
Концеппан 317
Коргард 234
Кордален 335
Кордарон 3>2
Корзид 234
Коримбин 244
Коринфар 365
Кортадрен 487
Кортансил 488
Кортизан 487
Кортизон 485
Кортиспорин 380
Кортитрон 497
Корто 487
Кортолон 489
Космоген 551
Кофламин 458
Коффекс 432
Крестомицин 660
Криобтин 459
Криптоциклин 596
Кристален 593
Кристодигин 335
Кромолин 437
Ксанакс 116
Ксантомицин 650
Ксилокаин 26
Ксилокарт 346
Ксиломета зо пин 220
Ксипонест 29
Кумадин 449
Курантил 368
Курарин 295
Куроксим 615 Лизиван 201
Курретаб 515 Лизодрен 562
Лабе га. юл 235 Лизотипин 379
Лазизикс 399 Ликвамар 450
Лазикс 399 Ликвемин 447
Ламин 318 Ликуден 742
Лампит 795 Линил 179
Ланвис 537 Линкомицин 665
Чаноксин 336 Линкоцин 666
Ларгактит 128 Линтон 137
Лариам 780 Лиоресаль 303
Чародопа 196 Лиотиронин 466
Ларотид 600 Липопилл 179
Латамоксеф 635 Лиситерол 375
Левамизол 574 Листенон 300
Левартеренол 210 Лис говин 744
Чеводопа 194 Литалир 564
Леводроморан 45 Литизин 147
Левоид 466 Лития карбонат 146
Левомицетин 669 Лифрил 538
Чеворфанот 43 Лобелии 259
Левотироксин 464 Ловалип 378
Чевофед 210 Ловастатин 377
Чедеркорт 494 Лозол 396
Ледермицин 651 Локсапин 140
Ле тертрексат 535 Локситан 140
Лейкеран 542 Локсопак 140
Лейкомицин 669 Ломидин 794
Лейкосу тьфан 546 Ломотил 50
Лейпротит 563 Ломупрен 438
Лейрокристин 555 Ломустин 547
Леодрин 390 Лонгоперидот 145
Лепаран 447 Лонитен 427
Лепонекс 142 Лоперамид 48
Лепрофен 51 Лопид 376
Лептофен 138 Лопрессор 233
Лестид 373 Лоразепам 113
Либртм 112 Лорелко 377
Лиданил 132 Лотримин 747
Лидокаин 25 Лотуссин 433
Лидопен 346 Лоту стат 93
- 822 -
- 823-
Указатель лекарственных препаратов
Указатель лекарственных препаратов
Лоэстрин 517 Мезитинон 263
Лю ДНОМ ИТ 160 Мезтин 602
Пюминал 90 Мезлоциллин 601
Люпрон 563 Мезоридазин 131
Люципен 596 Мезудин 701
Мадопар 196 Мекам илам ин 288
Максайр 442 Мекластин 313
Максипим 636 Меклизин 317
Макстрекс 535 Меклофенамовая кислота 68
Малоген 524 Мексат 535
Манадрин 223 Мексилетин 346
Мандокеф 616 Мекситил 347
Манидон 353 Мелипрамин 152
Манинип 4'76 Мел каин 22
Маннито т 383 Меллерил 131
Мантадан 757 Метфалан 542
Маотат 305 Менопакс 509
Мапротилин 159 Мепакрин 785
Марезин 317 Мепензолат 279
Маркаин 28 Меперидин 46
Марку мар 450 Мепивакаин 26
Марплан 162 Мепробамат 119
Марфанил 701 Мепрон 119
Матеникс 393 Мептин 442
Мафенид У)0 Меразолил 469
Мебендазол 800 Меридит Г5
Мебенот 474 Меркаптолу рин 535
Мебутар 801 Меромицин 646
Мевазин 288 Местинон 264
Мевакор 378 Метабарбитат 91
Мевастатин 377 Метагидрин 391
Мевинакор 378 Метадон 45
Мегафен 128 Метазоламид 385
Меганит тин 593 Метамизол натрия 65
Мегейс 516 Метаминин 652
Мегест ат 516 Ме1ампекс 174
Мегестрол 515 Метамфетамин Г4
Медазепам 117 Метаоксе трин 214
Медарон 718 Мегапрел 214
Медроксипрогестерон 515 Метапротерено i 213
Медронметризон 490 Метарадин 255
Метарам ино л 224 Миацин 380
Метарбитал 91 Мигрил 317
Метахолин 253 Мидамор 385
Метаниктин 651 Мидантан 197
Метеразин 129 Мидарин 300
Метилдопа 414 Мидриал 221
Метилпреднизолон 489 Мидриафайр 287
Мети ттестостерон 524 Мидриацил 287
Метилтиоу ранил 469 Мидрилат 286
Метилфенидат 174 Мидрин 287
Мегилфенобарбитал 91 Миелосан 546
Метилцефалин 790 Миканден 748
Метилциклотиазид 391 Миконазол 740
Метимазол 469 Микоспорин 747
Метиндот 77 Микоцид 749
Метиоцит 469 Милепсин 185
Мегипред 490 Миллигинон 517
Метиприлон 101 Милонтин 187
Метирозин 246 Милринон 338
Метициллин 593 Милтау н 119
Метогекситал 14 Минидаб 477
Метокарбамол 304 Минизид 241
Метокарин 294 Минипресс 241
Метокспфлуран 9 Миноксидил 426
Метокурин 295 Миноциклин 653
Метотазон 393 Миноцин 655
Метопролол 232 Минтезол 802
Меторин 139 Миобид 367
Метотирин 469 Миостат 254
Метотрексат 533 Миотонии 255
Метронидазол 790 Миохол 253
Метронитрон 358 Мисклерон 375
Метскопотамин 277 Мите лаз 265
Метубин 295 Митоксан 545
Мефенамовая кислота 68 Митоксантрон 564
Мефенон 46 Митомицин 553
Мефлохин 778 Мито с тан 546
Мефобарбитал 90 Митотан 562
Мефокситин 620 Мицивин 666
Мехторэтамин 541 Мицилан 748
Миамб\ гот 725 Млаксикам 81
- 824 -
-825-
Указатель лекарственных препаратов
Указатель лекарственных препаратов
Мобадид...............130
Мобан.................139
Мовенс.................69
Модамид...............385
Модитен...............130
Моду крин.............235
Моксадил..............164
Моксалактам...........632
Моксам................635
Моксацеф..............635
Молиндон..............139
Моноклат..............459
Мономицин.............660
Моноцид...............617
Мопатил...............415
Моронал...............739
Морфин.................36
Мотивал...............130
Мотрин.................71
Мтиоцил...............469
Мукомист..............433
Муркаин...............469
Мусил.................305
Мускарин..............255
Мустин................541
Наван.................135
Надолол...............234
Наква.................391
Налбуфин...............56
Налидиксовая кислота..708
Наллпен...............594
Налоксон...............59
Налорфин...............54
Налотрекс..............60
Налтрексон.............60
Налфон.................72
Нандролон.............527
Налам ид..............345
Наприлен..............426
Напроксен..............71
Напросин...............71
Нардил..................161
Наркан...................55
Народ...................120
Натамицин...............749
Натексин................379
Натримин................796
Натулан.................566
Натуретин...............389
Нафазолин...............219
Нафтизин................219
Нафтин..................748
Нафтифин................747
Нафцил..................594
Нафциллин...............594
Небицин.................662
Небу пент...............794
Невиграмон..............709
Неграм..................709
Нембутал.................93
Неодикумарин............449
Неокседрин..............214
Неолютин................515
Неомерказол.............470
Неомицин................379
Неоплатин...............549
Неорал..................579
Неоспорин...............380
Неостигмин..............263
Нептазан................386
Нердипин................366
Нескаин..................23
Нетиллин................665
Нетилмицин..............664
Нефлуан..................26
Нефрил..................390
Ниагестин...............516
Ниазид..................723
Ниацин..................379
Нивахин.................772
Нидантин................710
Низатидин...............327
Низорал................740
Никардипин.............365
Никлозамид.............809
Никонацид..............379
Никоприв...............778
Никотин................258
Никотиновая кислота....378
Ниприд.................422
Нипрутон...............422
Ниридазол..............802
Ниспорил...............739
Нистатин...............738
Нитринал...............358
Нитроглицерин..........357
Нитропентон............358
Нитропруссид натрия....422
Нитрофуразон...........716
Нитрофурантоин.........716
Нитрофурин.............717
Нитрумон...............548
Нифедипин..............365
Нифекор................365
Нифлумиковая кислота....69
Нифлурил................69
Нифуртимокс............794
Нобриум................119
Новамин................664
Новантрон..............565
Новапен................597
Новаталидон............396
Новогидразид...........389
Новодрин...............212
Новокаин................22
Новокаинамид...........344
Новометазон............492
Нозепам................113
Ноктар.................102
Ноктек..................99
Ноктран................115
Ноладол.................63
Нол ваде кс............512
Нолу дан...............102
Норадреналин...........204
Норгестрел.............517
Норглицин..............475
Норизадрин.............212
Норку рон..............297
Норлест................517
Нормисон................96
Нормолин...............376
Нороксин...............466
Норпант................279
Норпас.................345
Норпрамин..............157
Нортрилен..............158
Нортриптилин...........157
Норфлоксацин...........711
Норфор.................517
Норэпинефрин...........209
Норэтиндрон............513
Норэфедрин.............224
Ностел.................100
Нотезин................804
Нубаин..................57
Нуморфан................42
Обзидан................232
Обрацин................662
Овестин................506
Овранет................507
Оврат..................507
Огостал................730
Околин.................710
Оксазепам..............112
Оксандролон............529
Оксациллин.............594
Оксибу тинин...........284
Оксикодон...............42
Оксикон.................43
Оксиметазолин..........220
Оксимицин..............650
Оксиморфон..............41
Оксирен................305
- 826 -
- 827 -
Указатель лекарственных препаратов
Указатель лекарственных препаратов
Окситетрациклин........650
Оксифенциклимин........285
Оксолиновая кислота....709
Октадин................245
Онковин................555
Онкокарбид.............564
Онсукил................442
Оптикром...............438
Оптоциллин.............596
Опулетс................669
Орабет.................474
Оравирон...............525
Оралутон...............517
Орамид.................474
Орап...................143
Оранеф.................612
Орбенин................596
Орбигастрил............285
Орбинамон..............135
Ориметен...............562
Орнид..................353
Орсипреналин...........214
Ортохлор...............669
Осмитрол...............384
Осмосал................384
Осцин..................277
Отривин................220
Офлоксацин.............714
Павацен................367
Павулон................297
Паксипам...............111
Палудрин...............784
Памелор................158
Памин..................278
Панацеф................623
Панварфин..............449
Пангеприн..............447
Панкуроний.............296
Панорал................623
Панфуган...............801
Папаверин..............366
Папавин.................367
Параальдегид.............99
Парацетамол..............67
Парадион................191
Паракорт................488
Параль...................99
Паральдегид..............99
Параметадион............191
Параплатин..............550
Парафлекс...............305
Паркемед.................68
Паркин..................201
Паркинсан...............199
Паркопан................199
Парлодел................197
Пармодалин..............162
Парнат..................162
Паромомицин.............660
Парсидол................201
Певарил.................742
Пеганон.................184
Пеламин.................314
Пемолин.................175
Пенберин................599
Пентазоцин...............55
Пентамидин..............793
Пентилан................358
Пентобарбитал............93
Пентолайр...............286
Пентотал.................14
Пентран..................10
Пенфлуридол.............145
Пепцид..................327
Периактин...............320
Перитин.................174
Перкдан..................43
Перкортен...............497
Пермитил................130
Пернивит................379
Пертофран...............157
Петидин..................46
Пилокарпин.............257
Пилофрин...............258
Пимарицин..............750
Пимафугин..............750
Пимозид................142
Пиндион................451
Пиндолол...............234
Пиопен.................603
Пиперазин..............803
Пиперациллин...........602
Пипольфен..............319
Пипрацил...............603
Пиприл.................603
Пиразинамид............725
Пирантел...............805
Пирбутерол.............441
Пирибензамин...........314
Пиридизин..............723
Пиридостигмин..........264
Пириламин..............314
Пириметамин............705
Пироксикам..............79
Плаквенил..............772
Платибластин...........549
Платинекс..............549
Платинол...............549
Плацидил...............100
Плюразол...............695
Полагин................692
Поларамин..............316
Полимиксин В...........674
Полимиксин Е...........674
Политиазид.............390
Понстан.................68
Понстел.................68
Прагмарел..............166
Празепам...............109
Празиквантел...........804
Празозин...............240
Пралидоксим............269
Превенол...............811
Прегнорал...............517
Преднизолон.............488
Преднизон...............487
Прекорталон.............489
Препар..................215
Прессалол...............236
Прецеф..................617
Привин..................219
Прилокаин................29
Примахин................781
Примидон................185
Прискол.................239
Прискофен...............239
Пробу кол...............376
Провера.................515
Провохолин..............254
Прогестасерт............514
Прогестерон.............513
Прогестол...............514
Прогинон................507
Проглицем...............392
Прогуанид...............784
Продоксол...............710
Прозак..................165
Прозерин................264
Прокадил................442
Прокаин..................21
Прокаинамид.............343
Прокарбазин.............565
Прокардиа...............365
Прокатерол..............442
Проладон.................43
Про лутон...............514
Промазин................127
Промедол.................46
Прометазин..............318
Промпар.................128
Пронестил...............344
Пронтамид...............700
Пропазин................128
Пропаксен................71
- 828 -
- 829 -
У к а з а т е ль лекарственных препаратов
Указатель лекарственных препаратов
Пропантел 279 Ренитек 426 Ру фот 693 Синопен 315
Пропан гелии 278 Ренитрол 384 Салазопирин 695 Синтроид 466
Проппттиотрацит 468 Ренковид 691 Салазосу тьфапиридин 695 Синдром 449
Пропицит 469 Ресторит 96 Салу рои 390 Синэстрол 508
Пропранотот 231 Регабо тил 528 Салу тензин 390 Сифивирал 759
Простигмин 264 Ретасульфон 699 Сальбу вент 217 Сколин 278
Протазин 319 Ретестин 505 Сальбутамол 217 Скопотамин 276
Протактил 128 Ретровир 764 Санамирон 391 Сотантил 183
Прогеренол 212 Ре> профен 73 Сандимму н 579 Сотаскил 809
Протонам 269 Рибавирин 764 Санома 304 Солгол 234
Протостат 791 Рибомицин 663 Санорин 219 Солюсурмин 793
Протриптилин 158 Ривотрит 189 Санотензин 245 Сома 304
Профт ндол 93 Римактазид 673 Саркоциктин 651 Сонапакс 131
Прохторперазин 129 Римант ан 673 Себизон 700 Софраин 449
Проциктидин 199 Риназин 219 Седаланл 139 Софрамицин 660
Психоперидот 136 РиосИТ 390 Седатусс 432 Спарин 128
Птерофен 403 Риталин 175 Седу ксен 109 Спектиномицин 669
Пу тьв\ тес 328 Ритмарон 352 Секобарбитал 94 Спентан 317
Псринэтол 536 Ритмитен 345 С екотамин 242 Спиронолактон 400
П\ родш ин 335 Ритмодан 345 Секторль 233 Спреор 217
Равонал 14 Ритодрин 214 С ектрат 360 Станозот 529
Раниди 1 326 Рифадин 673 Секуропен 601 Стафциллин 594
Раниплекс 326 Рифампицин 671 Сетдан 321 Стацеф 631
Рани I и 1ин 325 Рифапиам 673 С е теги тин 197 Стелазин 130
Расгинон 474 Робаксин 304 Семап 143 Стензол 119
Peaceк 50 Робамокс 600 Серентит 132 Стенокор 368
Ребмен 71 Робамот 304 Серомицин 729 Стеринор 691
Ревитен 403 Робанталин 279 Серофен 511 Сгибиоглюконат натрия 792
Pei етан 375 Робидекс 432 Серпаси т 244 Стигмосан 264
Регенон 179 Робинут 281 Сефрит 612 Стильфострол 509
Регитин 240 Робитуссин 433 Сеффин 609 Стоксил 761
Реп юн 392 Роктизин 318 Сибазон 109 Стрептаза 456
Редептин 145 Роксен 81 Сив тор 378 Стрептан 727
Резерпин 244 Роксикодон 43 Сигинон 513 Стрептозоцин 548
Резистомицин 661 Ромазал 68 Сим метрел 197 Стрептокиназа 456
Реюхин 7'72 Ромезин 692 Синекван 757 СтрептокоI 658
Рента 1,ион 451 Рондомицин 652 Синемет 196 Стрептомицин 658
Ре та 304 Ронти1 390 Синерган 242 Стромба 529
Ре танит м 109 Роцефин 629 Синергон 514 Строфантин 336
Ремстан 96 Рубомицин 553 € инесалин 389 Строфопан 336
Ренне 790 Румацид 77 С иноми 694 Су грацилтин 593
831 -
-830-
Указатель лекарственных препаратов
Указатель лекарственных препаратов
Схкострин 300 Суфентанил 53 Т есторал 524 Т иоридазии 131
СсКЦИНИ 1ХОТИН 299 Тавегил 313 Тестостерон 522 Тиосульфил 693
Сулбактам 604 Тавор 114 Тетрагидрозолин 221 Тиотепа 545
Сулиндак 78 Гагамет 325 Тегразозин 241 Тиотиксен 134
Сулкозил 743 Тазепам ИЗ Тетракаин 23 Тиофосил 546
Сулконазол 743 Тазицеф 631 Тетракурий 298 Тирамин 227
Сульпирид 145 Тайленол 67 Тетрамизол 575 Тиреостат 469
Сульфабензамид 700 Гал азол 696 Тетрамицин 650 Тифокс 620
Сульфадиазин 690 Таламонал 138 Тетранитрат пентаэритрита 557 Тобрамицин 667
Сульфадимезин 692 Талбутал 92 Тетрациклин 650 Токаинид 346
Сульфадимидин 692 Талвин 60 Тетроксии 467 Толазамид 475
Сульфадоксин 696 Талофен 128 Т иабендазол 802 Толазолин 238
Су льфазан 694 Тамбокор 348 Тиазидил 391 Толиназ 475
Сульфизоксазол 693 1амоксифен 511 1 иамазол 469 Толбутамид 473
Сульфазол 696 Тарасан 134 Т иасептот 695 Толбутон 474
Сульфален 697 Таривид 715 Тибистал 725 Толектин 78
Сульфамеразин 691 Таснон 803 Тибон 467 Толмекс 78
Су льфаметазин 692 Тегапен 596 Тизин 147 Толметин 77
Сульфаметизол 692 Тегретол 188 Тикар 604 Толнафтат 748
С\ тьфаметоксазол 694 Телдрин 316 Тикарциллин 604 Томоран 166
Су тьфаметоксипиразин 699 Телестрин 504 Тиклид 455 Тонокард 346
Cv тьфамегоксипиридазин 699 Темазепам 96 Тиклозан 455 Тонофтал 749
Су льфаметопиразин 699 Гемарил 319 Тиклопидин 454 Тописпорин 380
Сульфапиридазин 699 Темподекс 174 Тилфосфамид 546 Торазин 128
Сульфапиридин 695 Тенипозид 557 Тимакор 235 3 оремонил 772
Сульфасалазин 695 Геннецетин 750 Тиметин 604 Тосмилен 266
Су пьфаиетамид 699 Тенормин 234 Тимидазол 469 Тотаклокс 596
Сульфацетин 689 Тенсилон 264 Тимолид 235 Тотален 599
Сульфидин 695 1енсиоиорм 389 Тимолол 235 Тотоциллин 596
Сульфизомезол 694 Генуат 179 Тимопед 749 Т офранил 152
Сульфинпиразон 64 Теофиллин 434 Тимоптол 235 Травелин 312
Сульфолар 694 Теоцин 436 Тинадерм 749 Травенон 379
Су тьформетоксин 697 Тепанил 179 Тинактин 749 Травин 120
Су перкортен 493 Теразол 745 Тинапорт 792 Традон 176
Су прастин 315 Терамин 179 Тинатокс 749 Традоциллин 593
Су прол 79 Тербуталин 212 Тинидазоп 791 Тразодон 765
Сурамин 795 Терконазот ’’44 Тинимед 792 Трал 282
Сурмонтил 153 Геркоспор 745 Тинисан 792 Трамизот 809
Сурпликс 152 Терфенадин 320 Гиогуанин 536 Грандат 236
Суфалекс 699 Теспамин 546 Т и ом ид 728 Транекс 459
Суфента 54 Тессалон 432 Т иопентал 73 Транексамовая кислота 458
- 832-
- 833 -
Указатель лекарственных препаратов
Трани щипромии 162 Трихопол 791
Транквиракс 119 1роксидон 191
Транкопал 122 Тромбин 459
Транксен 115 Гро мбо стат 460
Трансамин 162 Тромпресантин 368
Трапанал 14 Тропикамид 286
Тредемин 810 Тропикацит 287
Т рекати п 728 Тубазид 723
Трексан 60 Ту барин 295
Трексил 321 Тубермин 728
Тресортил 304 Тубероид 728
Т риази 1 391 Тубетол 725
Триазолам 95 Губокурарин 294
Триаминик 314 Туттомицин 660
Триамицпнопон 493 Увилон 803
Триамтерен 402 У гл ро п 459
Тригексифенидип 198 Утьтандрен 529
Гримексан 449 Утьтрацеф 613
Тримеперидин 47 Ундециленовая кислота 749
Тримепразин 319 Ундетин 749
Триметадион 190 Унидон 451
Триметафан 288 Уралгин 709
Триметин 191 Урегит 399
Триметоприм 702 Урехопин 255
Тримипрамин 152 Уридон 396
Тримокс 600 Урограм 709
Тринитрин 357 Урозид 389
Тринитро! пиперин 357 Урокиназа 456
Тринитро 1 357 У ролонг 717
Трипатар 287 Уронорм 710
Трипе геннамин 313 Уросемид 399
Триперидоп 136 Уросоп 693
Триполон 316 Утерамин 221
Триптизол 154 Утимокс 600
Трипти 1 159 \тициллин 593
Триседи। 136 Утопар 215
Трифлхперидот 135 Фазигин 739
1 рифлупромазин 128 Фаликард 353
Трифт\рипин 762 Фамотидин 326
Трифтазин 130 Фа мод ил 3,21
Т рихлормет иазид 391 Фанасип 697
- 834 -
Указатель лекарственных препаратов
Фансидар 697 Фпакседил 296
Фармитрексат 535 Фламазин 691
Фармицетин 660 Флекаинид 348
Фармотал 14 Флексерил 303
Фас гх м 73 Флексипирин 122
Фетден 81 Флексокутан 68
Фетисон 98 Фленак 74
Фемстат 744 Флобацин 715
Фенамин 226 Флоксикам 81
Фенелзин 161 Флоксуридин 538
Фенемал 90 Флоринеф 498
Фениндион 450 Флороприл 267
Фенилбхтазон 64 Флумарк 713
Фенилин 451 Флхнир 69
Фенилпропаноламин 223 Флуоксетин 164
Фенилэфрин 214 Флу оксиместерон 528
Фенитоин 347 Флуспирилен 144
Фенклофенак 74 Флу гам ид 526
Фенобарбитал 89 Флуфенамовая кислота 67
Феноболин 528 Флу цитозин 750
Феноксазол 176 Флуанизон 138
Феноксибензамин 237 Флу ДИКС 538
Феноксиметилпенициллин 593 Флудрокортизон 497
Феноспен 593 Флумуцетин 433
Фенотерол 440 Флуоробластин 538
Фенпрокхмон 449 Флуостигмин 267
Фенопрофен 71 Флуразепам 97
Фенсуксимид 186 Флуфеназин 130
Фентанест 51 Фо леке 535
Фентанил 50 Фолликулин 504
Фентермин 178 Форан 9
Фентоламин 239 Форбаксин 304
Фенурин 717 Форенол 69
Фенэрган 319 Форму леке 284
Фестамоксин 635 Фортрал 56
Физе птон 46 Фортум 631
Физостигмин 262 Фосфо ЛИН йодид 268
Филопон 174 Франил 399
Финоптин 353 Фрумил 404
Флавохин 773 Фталазол 696
Ф гаг ил 791 Фталилсудьфатиазол 696
- 835 -
Указатель лекарственных препаратов
Указатель лекарственных препаратов
Фторотан 8 Хлорпромазин 128
Фторурацил 538 Хлорпропамид 474
Фторфеназин 130 Хлорпротиксен 132
Фугерел 526 Хлорталидон 394
Фуиграм 711 Хлортетрациклин 649
Фу ттрексин 694 Хлортиазид 388
Фульцин 742 Хзортриметон 316
Фунгизон 738 Хлортрипеленамин 315
Фунгилии 738 Хлорфенезин 305
Фурадонин 717 Хлорфенирамин 315
Фуразолидон 717 Хторэтазид 541
Фурамид 788 Хлорэтамин 541
Фурацитин 716 Холеслабил 373
Фурацин 716 Холее гид 373
Фу роксан 718 Холестирамин 373
Фуросемид 399 Холоксин 379
Хинакрин 785 Цедиксен 637
Хинасед 94 Цедоксин 336
Хингамин 772 Цезол 805
Хингошу ~80 Цеклор 623
Хинидекс 343 Цечбении 594
Хинидин 342 Целестон 493
Хинин ^73 Центраке 111
Хиннам 778 Це пан 621
Хзорадорм 99 Цепим 636
Хторазепат 114 Цепимекс 636
Хлоральгидраг 99 Цепорексин 612
Хлорамбу цил 541 Церебид 367
Хлорамфеникол 667 Церубидин 553
Хлоратол 99 Цетазол 385
Хлорбутин 542 Цетамид 700
Хлоргуанид 783 Цел осад 62
Хзордиазепоксид 111 Цефадил 610
Хлорзид 389 Цефадрил 613
Хлорзоксазон 305 Цефадроксил 613
Хлормезанон 121 Цефазал 612
Хлормицетин 669 Цефазопин 610
Хлороквин 768 Цефазон 632
Хлороназ 474 Цефаклор 621
Хлорпирамин 314 Цефалексин 611
Хлорпрокаин 22 Цефалотин 608
Цефамандол Цефапирин Цефарин Цефатрексил Цефацидал Цефепим Цефобид Цефобис Цефоксинот Цефокситин Цефоницид Цефоперазон Цефоранид Цефотаксим Цефотен Цефотетан Цефпнром Цефрадин Цефро Цефрон Цефтазидим Цефтизоксим Цефтикс Цефтим Цефтин Цефтриаксон Цефуроксим Цикламид Циклизин Циклобензаприн Цикловальдин Цикловиран Циклогест Циклодол Цикложил Циктокапрон Циклометикаин Цикломидрил Цикломицин Циклопар Цикдопентотат
616 Циклосерин 609 Циклоспорин А
610 | Цикюсгин
610 Циклотериам
611 1 Циклотиазид
635 Циклофосфамид
632 Циклофосфан
632 Циклоэстрол
620 Циластатин
618 Цилерт
616 Циметидин
631 Цинамет
617 Цинобактин
623 Циноксацин
621 Циномел
620 Цинопенил
636 Ципрогептадин
612 Ципроквин
612 Ципролег
637 Ципропан
629 1 Ципросан
625 I Ципростат
627 Ципротерон
631 Ципрофлоксацин
615 Ципроцинол
627 Цирпон
614 Цисплатин
475 1 Цислофуран
317 Цитадрен
303 Цитанест
729 Цитарабин
759 Цитозин
514 Цитоксан
199 Цитосульфан
286 Эбутол
459 Эверон
30 1 Эглонил
286 Эдекрин
729 Эдрофоний
651 Эзерин
286 Экванил
729 578 544 391
391 543
544 508
638
176
324
325
710
710
467 594
320
712 712
712
712 526
525
722
712
119
549
717
562
29
539
540
544
546
725
524
146 399
264
263
119
- 836-
- 837-
Указатель лекарственных препаратов
Экзал 555 Эритропед 646
Экзотерил 748 Эсанбутол 725
Эконазол 742 Эсидрикс 389
Экостатин 742 Эсхалит 147
Экотиофат 267 Эсмалорид 391
Эксе тдерм 743 Эсмарин 391
Эксирет 442 Эстравал 505
Этдеприл 198 Эстрадиол 504
Элдопан 198 Эстраду рин 27
Элениум 112 Эстриол 506
Э зспар 567 Эстрон 502
Элгроксин 466 Эсту лик 414
Эльдальдегид 99 Этакриновая кислота 398
Эматексат 535 Этамбутоз 723
Эмедил 312 Этидокаин 28
Эмегин 788 Этилбискумацетат 448
Эмпецнд 747 Эгилдикумарол 449
Эналаприл 425 Этимид 728
Эндиэмалсм 91 Этинамат 100
Эндоксан 544 Этинилэстрадиол 506
Эндо МИ КС ин 380 Этионамид 727
Эндурон 391 Этистерон 517
Энзико1 379 Этомидат 12
Энкаид 349 Этопозид 556
Энкаинид 348 Этопропазин 200
Эноксацин 712 Этотоин 183
Энгерозол 696 Этран 8
Эн г) рен 65 Этхлорвинол 100
Энфенемал 91 Эуболин 528
Энфту ран 8 Эудемин 392
Эпинефрин 208 Эулекин 526
Эпиподофил зотоксин 558 Эутаген 43
Эпосерин 627 Эуфиллин 436
Энеи 1ЭМИН 458 Эфедрин 222
Эргамизол 809 Эфедрол 223
Эрюметрин 243 Эфтапан 653
Эргоновин 243 Эфторон 279
Эрготамин 242 Эффлудерм 538
Эрготрен 243 Эходид 268
Эринит 358 Эцифин 223
Эритромицин 644 Юнипен 594
- 838 -
Содержание
От автора................................................... 3
Список сокращений .......................................... 5
Глава 1 Общие анестетики.................................... 6
1 1 Ингаляционные анестетики.......................... 6
1 2 Неингаляционные анестетики....................... 10
Глава 2 Местные анестетики................................. 17
2 1 Местные анестетики ряда аминоэфиров.............. 21
2 2 Местные анестетики ряда аминоамидов.............. 25
2 3 Поверхностные анестетики......................... 29
Гтава 3 Анальгетики...................................... 32
3 1 Опиоидные анальгетики............................ 33
3 2 Нестероидные противовоспалительные средства, или жаропонижающие анальгетики.................... 61
Глава 4 Снотворные средства (гипнотики и седативные препара гы)................................................ 86
4 1 Барбитураты...................................... 87
4 2 Бензодиазепины................................... 94
4 3 Прочие гипнотики и седативные препараты.......... 98
Глава 5 Анксиолитики (транквилизаторы).................... 104
5 1 Бензодиазепины.................................. 105
5 2 Анксиолитики небензодиазепиновой структуры......... 119
Глава 6 Антипсихотики (нейролептики).................... 124
6 1 Производные фенотиазинов........................ 126
6 2 Производные тиоксантенов........................ 132
6 3 Производные бутирофеноиов....................... 135
6 4 Производные дигидроиндолонов.................... 139
6 5 Производные дибензоксазепинов и дибензодиазепинов ...................................... 140
6 6 Производные дифенилбутилпиперидинов............. 142
6 7 Прочие нейролептики............................. 145
Глава 7 Антидепрессанты................................... 149
7 1 Трициклические антидепрессанты.................. 151
7 2 Ингибиторы МАО.................................. 160
7 3 Антидепрессанты второго поколения (атипичные антидепрессанты)................................. 163
7 4 Амфетамины и другие стимуляторы ЦНС............. 166
-839-
Содержание
Глава 8. Стимуляторы ЦНС................................. 169
8.1. Психомоторные стимуляторы..................... 170
8.1.1. Метилксантины.......................... 170
8.1.2. Амфетамины, а также метилфенидат и пемолин........................................ 171
8.2. Стимуляторы дыхания, или аналептики........... 176
8.3. Препараты, подавляющие аппетит (аноректики)... 178
Глава 9. Противоэпилептические средства.................. 181
9.1. Производные гидантоинов....................... 182
9.2. Барбитураты................................... 184
9.3. Сукцинимиды................................... 186
9.4. Вальпроевая кислота........................... 187
9.5. Карбамазепин.................................. 188
9.6. Бензодиазепины.............................. 188
9.7. Ацетазоламид.................................. 190
9.8. Оксазолидины.................................. 190
Глава 10. Средства, применяемые при паркинсонизме.........193
10.1. Средства, влияющие на дофаминергические системы мозга....................................... 194
10.2. Антихолинергические препараты (центральные холиноблокаторы)............................... 198
Глава 11. Адренергические (симпатомиметические) препараты.................................................203
11.1. Агонисты прямого действия.....................207
11.2. Агонисты непрямого действия................. 221
11.3. Агонисты смешанного действия................. 221
Глава 12. Адреноблокирующие препараты.................... 228
12.1, Р-адреноблокаторы............................ 229
12.2. а-адреноблокаторы............................ 236
12.3. Блокаторы адренергических нейронов........... 244
Глава 13. Холиномиметики..................................250
13.1. Холиномиметики прямого действия...............251
13.2. Холиномиметики непрямого действия.............260
Глава 14. Антихолинергические препараты...................272
14.1. Антимускариновые препараты....................272
14.2. Ганглиоблокирующие вещества...................287
Глава 15. Мышечные релаксанты............................ 292
15.1. Блокаторы нервно-мышечной передачи........... 293
- 840 -
Содержание
15.2. Мышечные релаксанты прямого действия...........300
15,3. Мышечные релаксанты центрального действия......301
Глава 16. Антигистаминные препараты....................... 307
16.1. Н.-антигистаминные препараты.................. 310
16.2. Антагонисты 1Ь-рецеиторов..................... 323
Глава 17. Кардиотонические препараты...................... 331
17.1. Сердечные гликозиды............................332
17.2. Другие положительные инотропные препараты......336
Глава 18. Антиаритмические препараты.......................340
Глава 19. Антиангинальные препараты........................355
19.1. Нитраты и нитриты............................. 356
19.2. Препараты ряда [3-адреноблокаторов............ 358
19.3. Блокаторы кальциевых каналов.................. 361
19.4. Прочие препараты.............................. 366
Глава 20. Гиполипидемические средства..................... 370
20.1. Препараты, удаляющие желчные кислоты...........372
20.2. Препараты, ингибирующие синтез холестерина.....374
20.3. Прочие препараты.............................. 379
Глава 21. Диуретики....................................... 382
21.1. Осмотические диуретики........................ 383
21.2. Диуретики, подавляющие активность карбонангидразы...................................... 384
21.3 Тиазидные диуретики.......................... 386
21.4. Петельные диуретики........................... 396
21.5, Калийсберегаюшие диуретики.................... 399
Глава 22. Антигипертензивные препараты.................... 407
22.1. Диуретики..................................... 409
22.2. [3-адреноблокаторы.......................... 411
22.3. Центрально действующие адренергические препараты (си.мпатолитики).................................... 413
22.4. Периферически действующие симпатолитики (а-адреноблокаторы)...................................415
22.5. Блокаторы кальциевых каналов...................419
22.6. Миотропные гипотензивные препараты............ 420
22.7. Ингибиторы АПФ.................................422
22.8. Активаторы калиевых каналов....................426
Глава 23. Препараты для лечения заболеваний дыхательной системы....................................................429
- 841 -
Содержание
23 1 Противоотечные сосх досуживающие препараты..... 430
23 2 Противокашлевые препараты и отхаркивающие
средства....................................... 430
23 3 Бронходилататоры (бронхолитики)............... 433
Глава 24 Антикоагулянты, антиагреганты, тромболитики гемостатики...............................................444
24 1 Антикоагу тянты................................445
24 2 Антиагреганты..................................451
24 3 Фибринопитики(тромболитики)....................455
24 4 Гемостатики (прокоагулянты)....................457
Глава 25 Тиреоидные гормоны и антитиреоидные препараты.... 463
25 1 Препараты для лечения гипотиреоидизма.......... 464
25 2 Препараты для лечения гипертиреоидизма......... 467
Г лава 26 Инсу тин и синтетические гипогликемические средства......................................... 471
26 1 Инсулин........................................ 471
26 2 Синтетические гипогликемические препараты....... 473
Глава 27 Кортикостероиды................................. 479
27П Глюкокортикоиды................................. 480
27 2 Минералокортикоиды............................. 494
Глава 28 Женские половые гормоны......................... 500
28 1 Эстрогены...................................... 501
28 2 Антиэстрогены.................................. 209
28 3 Прогестины..................................... 512
Глава 29 Мужские половые гормоны и анаболические стероиды................................................. 521
29 1 Андрогены ..................................... 522
29 2 Анта! онисгы андрогенов........................ 525
29 3 Анаболики...................................... 527
Г тава 30 Антинеопластики................................ 531
30 1 Антиметабопиты................................. 532
30 1 1 Антагонисты фолиевой кислоты............. 532
30 1 2 Производные пурина....................... 535
30 13 Производные пиримидина.................... 537
30 2 Алкилирующие агенты............................ 540
30 2 1 Азотсодержащие горчичные произволные...... 541
30 2 2 Производные этиленимина.................. 545
30 2 3 Алкилсхльфонаты...........................546
-842-
Содержание
30 2 4 Нитрозомочевины......................... 546
30 2 5 Производные платины..................... 549
30 3 Антибиотики ................................ 5з0
30 4 Соединения выделенные из растений............. 554
30 5 Гормональные препараты........................ 558
30 6 Прочие антивеопластические препараты.......... 563
Глава 31 Иммунофармакологические препараты.............. 572
31 1 Иммуностимуляторы............................. 573
31 2 Иммунодепрессанты............................. 576
Т лава 32 Антибиотики................................. 581
32 1 ^-лактамные антибиотики....................... 585
32 1 1 Пенициллины............................. 589
32 1 2 Цефалоспорины........................... 605
32 1 3 Пенемы и карбапенемы.................... 637
32 1 4 Монобактамы............................. 640
32 2 Макролидные антибиотики....................... 643
32 3 Тетрациклины.................................. 648
32 4 Аминогликозиды................................ 655
32 5 Линкозамиды................................... 665
32 6 Хлорамфеникол................................ 667
32 7 Прочие антибиотики........................... 669
Г лава 33 Антимикробные препараты....................... 686
33 1 Сульфонамидные препараты и триметоприм........ 686
33 2 Хинолоны...................................... 706
33 3 Ни грофу раны................................. 715
Глава 34 Антимикобактериальные препараты................ 721
34 1 Анти туберкулезные препараты.................. 721
34 2 Препараты для лечения лепроза................. 731
Т шва Зд Противогрибковые препараты......................736
Зд 1 Противогрибковые препараты для лечения системных и поверхностных инфекций.................... 737
Зт 2 Противогрибковые препараты для лечения поверхностных инфекций..................................741
35 3 Противогрибковые препараты для лечения системных инфекций...................................... 750
Глава 36 Противовирусные препараты...................... 754
Гтава 3" Препараты для течения протозойных инфекций..... 766
1 Антималярийные препараты..................... 766
-843-
Содержание
37.1.1 . Препараты, эффективные против эритроцитарной стадии заражения плазмодиями.................. 768
37.1.2 . Препараты, эффективные против гепатической (экзоэритроиитарной) стадии заражения плазмодиями............................................. 781
37.1.3. Препараты эффективные против гепатической и эритроцитарной форм заражения плазмоди-
ями....................................... 782
37.1.4 . Прочие антималярийные препараты........ 785
37.2. Препараты для лечения амебиаза................ 786
37.3. Препараты для лечения лейшманиоза............. 792
37.4. Препараты для лечения трипаносомоза........... 793
37.5. Препараты для лечения других протозойных инфекций.............................................796
Глава 38. Антигельминтные препараты...................... 799
Указатель лекарственных препаратов....................... 813
Р.С. Вартанян
Синтез основных лекарственных средств
Руководитель научно-информационного отдела, кандидат мед. наук А.С. Макарян
Главный редактор, канд. мед. наукД.Д Проценко
Зам. главного редактора С.А. Зайцева Ответственный за выпуск Е.Д. Броун Компьютерная верстка Е.Б. Родина Дизайн обложки С. С. Филиппов
Изд. лиц. №064889 от 24.12.96. Подписано в печать 15.01.2004.
Формат 60x90/16. Бумага офсетная. Гарнитура «Times New Roman» Объем 53 печ. л. Тираж 1000 экз.
ООО «Медицинское информационное агентство»,
119048, Москва, М. Трубецкая ул., д. 8 (ММА им. И.М. Сеченова).
Телефакс 245-86-20, 242-91-10
E-mail: miapubl@mail.ru http://www.medagency.ru
Интернет-магазин: www.medkniga.ru